УРОВЕНЬ ТЕХНИКИ
Toll-подобные рецепторы (TLR) представляют собой семейство трансмембранных белков, которые распознают структурно консервативные молекулы, происходящие и уникальные для патогенов, называемые патоген-ассоциированными молекулярными паттернами (PAMPS). Как таковые, TLR функционируют в иммунной системе млекопитающих в качестве передовых сенсоров патоген-ассоциированных молекулярных паттернов, обнаруживая присутствие вторгшихся патогенов (Takeuchi and Akira 2010 Cell 140:805-820). Участие TLR в сигнальных иммунных клетках вызывает биосинтез выбранных цитокинов (например, интерферонов I типа), индукцию костимулирующих молекул и повышенную способность к презентации антигена. Это являются важными молекулярными механизмами, активирующими врожденный и адаптивный иммунный ответ. Соответственно, агонисты и антагонисты TLR находят применение при модулировании иммунных ответов. Агонисты TLR обычно используют для стимуляции иммунных ответов, тогда как антагонисты TLR обычно используют для ингибирования иммунных ответов (Gosu et al 2012. Molecules 17:13503-13529).
Геном человека содержит 10 известных TLR, из которых TLR3, TLR7, TLR8 и TLR9 распознают нуклеиновые кислоты и продукты их разложения. Распределение TLR7, TLR8 и TLR9 ограничено эндосомальными компартментами клеток, и они преимущественно экспрессируются в клетках иммунной системы. В конфигурации активированного димерного рецептора, TLR7 и TLR8 распознают одноцепочечную РНК на одном лиганд-связывающем участке и продукты деградации рибонуклеозидов гуанозин и уридин, соответственно (а также низкомолекулярные лиганды с родственными структурными мотивами) во втором лиганд-связывающем участке (Zhang et al 2016 Immunity 45(4);737-748: Tanji et al 2015 Nat Struct Mol Biol 22: 109-115).
Были идентифицированы некоторые низкомолекулярные агонисты TLR7 или TLR8. Эти агонисты могут быть сгруппированы в пуриноподобные молекулы, такие как 7-тиа-8-оксогуанозин (TOG, изаторибин) или соединения на основе имидазохинолина, такие как имиквимод. Пока что Имиквимод является единственным одобренным агонистом TLR7, продаваемым в виде 5% крема (Aldara). Он обеспечивает примерно 80% 5-летнего клиренса поверхностных базальноклеточных карцином, которые являются наиболее распространенным видом рака во всем мире, что демонстрирует важность агонистов TLR7 в иммунотерапии рака. Функциональная экспрессия TLR7, по-видимому, ограничена специфическими иммунными клетками. Вовлечение TLR7 в плазмоцитоидные дендритные клетки приводит к индукции интерферона α/β, который играет важную роль в контроле адаптивного иммунного ответа (Bao and Liu 2013 Protein Cell 4:40-5).
Вовлечение TLR8 в миелоидные дендритные клетки, моноциты и дендритные клетки, происходящие из моноцитов, индуцирует выраженный провоспалительный цитокиновый профиль, характеризующийся повышенным продуцированием фактора некроза опухоли-α, интерлейкина-12 и IL-18 (Eigenbrod et al J Immunol, 2015, 195,1092-1099).
Низкомолекулярные агонисты TLR также исследовались на предмет использования в качестве адъювантов вакцин (Dowling, ImmunoHorizons 2018, 2(6) 185-197).
Таким образом, практически все основные типы моноцитарных и дендритных клеток могут быть активированы агонистами TLR7 и TLR8, чтобы стать высокоэффективными антигенпрезентирующими клетками, способствуя тем самым эффективному врожденному и адаптивному иммунному ответу. Большинство типов антигенпрезентирующих клеток экспрессируют только один из этих двух рецепторов, соответственно, малые молекулы с мощной агонистической активностью против обоих рецепторов TLR7 и TLR8 являются потенциально более эффективными иммунными адъювантами, чем только агонисты TLR7.
Таким образом, низкомолекулярный агонист TLR7/TLR8 (TLR7/8) с двойной биологической активностью может обеспечить дополнительные преимущества по сравнению с более селективным агонистом TLR7 и вызывать врожденные иммунные ответы в более широком диапазоне антигенпрезентирующих клеток и других ключевых типов иммунных клеток, включая плазмоцитоидные и миелоидные дендритные клетки, моноциты и В-клетки (van Haren et al 2016 J Immunol 197:4413-4424; Ganapathi et al 2015 Plos One 10(8).e0134640). Такие мощные двойные агонисты TLR7/8 также могут быть эффективными при стимулировании эффективных противоопухолевых ответов при раке (Singh et al 2014 J. Immunol 193 4722-4731: Sabado et al 2015 Cancer Immunol Res 3 278-287, Spinetti et al 2016 Oncoimmunol 9;5(11):e1230578: Patil et al 2016 Mini Rev Med Chem 16:309-322).
Несмотря на успех Имиквимода (Aldera) в лечении поверхностной базальноклеточной карциномы, остается потребность не только в более мощных агонистах TLR7, но и в сбалансированных, мощных агонистах TLR7/8 для расширения возможностей лечения пациентов с различными видами рака. Этими вариантами лечения могут быть местные введения, которые будут доставлять лекарство непосредственно к опухоли, ограничивая при этом системные побочные эффекты. Альтернативно, системно вводимые агонисты TLR7 или агонисты TLR7/8 будут иметь преимущество, заключающееся в возможности достижения труднодоступных для введения опухолей, а также множественных опухолей, через системный кровоток.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение представляет, частично, соединение формулы (I), включая формулы (Ia) и (Ib), в совокупности, соединение по изобретению или его фармацевтически приемлемую соль. Такие соединения активируют TLR7 человека (hTLR7), а также активируют TLR8 человека (hTLR8), тем самым влияя на биологические функции. В некоторых вариантах осуществления, изобретение представляет соединения, которые являются двойными агонистами, селективными в отношении как TLR7, так и TLR8 (агонисты TLR7/8). В другом варианте осуществления, изобретение представляет соединения, которые являются агонистами, селективными в отношении TLR7. Другой вариант осуществления представляет фармацевтические композиции и лекарственные средства, содержащие соединения по изобретению или их фармацевтически приемлемые соли, отдельно или в комбинации с дополнительными противораковыми терапевтическими агентами.
Настоящее изобретение также представляет, частично, способы получения соединений, фармацевтически приемлемых солей и композиций по изобретению, и способы применения вышеизложенного отдельно или в комбинации с дополнительными противораковыми терапевтическими агентами.
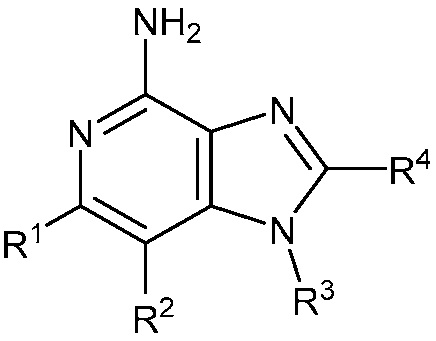
В одном аспекте, изобретение представляет соединение Формулы (I):
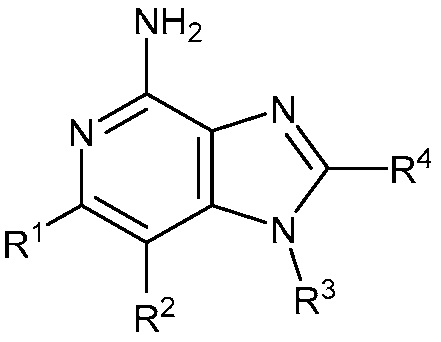
(I)
или его фармацевтически приемлемую соль, где
R1 и R2 независимо являются C1-3 алкилом; или
R1 и R2 объединены с образованием 5-7-членного карбоциклического кольца, где указанное карбоциклическое кольцо может быть насыщенным или ненасыщенным;
R3 является
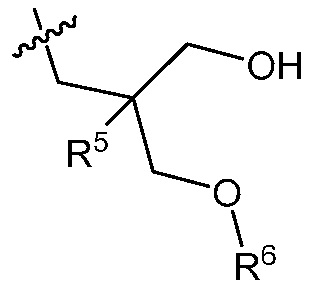 ;
;
R4 является C1-6 алкилом, или (CH2)nO(CH2)mCH3, где C1-6 алкил или любой атом углерода (CH2)nO(CH2)mCH3 группы замещен от 0 до 3 атомами галогена, насколько позволяет валентность;
R5 является C1-3 алкилом, или OC1-3 алкилом, где C1-3 алкил замещен от 0 до 3 F;
R6 является H, или C1-3 алкилом, где C1-3 алкил замещен от 0 до 3 F;
m равен 0-2; и
n равен 1-3.
В другом аспекте, изобретение представляет фармацевтическую композицию, содержащую соединение по изобретению в соответствии с любой из описанных в настоящем документе формул или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель или эксципиент. В некоторых вариантах осуществления, фармацевтическая композиция содержит два или несколько фармацевтически приемлемых носителя и/или эксципиента.
В другом аспекте, изобретение также представляет терапевтические способы и применения, включающие введение соединения по изобретению или его фармацевтически приемлемой соли.
В другом аспекте, изобретение представляет способ лечения аномального роста клеток, в частности, рака, у субъекта, нуждающегося в этом, включающий введение субъекту терапевтически эффективного количества соединения по изобретению или его фармацевтически приемлемой соли. Соединения по настоящему изобретению можно вводить в виде отдельных агентов или их можно вводить в комбинации с другими противораковыми терапевтическими агентами, включая агенты стандарта клинической практики, подходящие для конкретной формы рака. Оно также включает применение соединения по изобретению или его фармацевтически приемлемой соли в производстве лекарственного средства для лечения аномального роста клеток, в частности, рака, у субъекта, нуждающегося в этом.
В другом аспекте, изобретение представляет соединение по изобретению или его фармацевтически приемлемую соль для применения в качестве лекарственного средства, в частности, лекарственного средства для лечения аномального роста клеток, такого как рак.
В еще одном аспекте, изобретение представляет применение соединения по изобретению или его фармацевтически приемлемой соли для производства лекарственного средства для лечения аномального роста клеток, такого как рак, у субъекта.
В другом аспекте, объем изобретения включает фармацевтически приемлемые соли соединений по изобретению. Соответственно, фраза «или его фармацевтически приемлемая соль» подразумевается в описании всех соединений, описанных в настоящем документе, если прямо не указано иное.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Как представлено, изобретение относится к соединению формулы (I), представленному выше.
Настоящее изобретение может быть легче понято со ссылкой на следующее подробное описание дополнительных вариантов осуществления изобретения и включенные в него примеры. Следует понимать, что используемая в настоящем документе терминология предназначена только для целей описания конкретных вариантов осуществления и не предназначена для ограничения. Кроме того, следует понимать, что, если конкретно не указано в настоящем документе, терминология, используемая в настоящем документе, должна иметь свое традиционное значение, известное в соответствующей области техники.
Используемые в настоящем документе формы единственного числа «a», «an» и «the» включают ссылки во множественном числе, если не указано иное. Например, «а» заместитель включает один или несколько заместителей. Термин «примерно» означает наличие значения, попадающего в пределы принятого стандарта ошибки среднего, при рассмотрении его специалистом в данной области техники.
Термин «алкил», используемый в настоящем документе, означает одновалентную углеводородную группу с прямой или разветвленной цепью формулы -CnH(2n+1). Неограничивающие примеры включают метил, этил, пропил, бутил, 2-метилпропил, 1,1-диметилэтил, пентил и гексил.
В некоторых случаях, количество атомов углерода в углеводородной группе (например, алкиле) обозначается префиксом «Cx-Cy» или «Cx-y», где x является минимальным, и y является максимальным количеством атомов углерода в группе. Так, например, «(C1-C6)алкил» или «C1-6 алкил» относится к алкильному заместителю, содержащему от 1 до 6 атомов углерода.
Термин «галоген», используемый в настоящем документе, относится к фториду, хлориду, бромиду или йодиду.
Термин «галогеналкил», используемый в настоящем документе, относится к алкильной группе, которая замещена, по меньшей мере, одним галогеновым заместителем. Если более чем один атом водорода замещен атомом галогена, галогены могут быть одинаковыми или разными. Неограничивающие примеры включают фторметил, дифторметил, трифторметил и 2,2,2-трифторэтил.
Используемый в настоящем документе термин «биотерапевтический агент» означает биологическую молекулу, такую как антитело или слитый белок, которая блокирует передачу сигналов лиганда/рецептора на любом биологическом пути, который поддерживает сохранение и/или рост опухоли или подавляет противоопухолевый иммунный ответ.
Используемый в настоящем документе термин «химиотерапевтический агент» представляет химическое соединение, применимое для лечения рака. Химиотерапевтические агенты, применимые в способах лечения по настоящему изобретению, включают цитостатические и/или цитотоксические агенты. Химиотерапевтические агенты включают сам агент или любую его фармацевтически приемлемую соль, сокристалл или сольват. Химиотерапевтические агенты дополнительно описаны в настоящем документе.
Используемые в настоящем документе следующие термины используются взаимозаменяемо и означают любое одно или несколько терапевтических средств, отличных от соединения по настоящему изобретению, которые используются или могут быть использованы для лечения рака: «дополнительный противораковый терапевтический агент» или «дополнительный химиотерапевтический агент» или «дополнительный терапевтический агент».
Биотерапевтический агент и химиотерапевтический агент оба являются примерами дополнительного противоракового терапевтического агента.
Используемый в настоящем документе термин «цитотоксический агент» относится к агенту, который оказывает цитотоксическое и/или цитостатическое действие на клетку, и «цитостатический эффект» относится к ингибированию пролиферации клетки.
Используемый в настоящем документе термин «цитостатический агент» относится к агенту, который оказывает цитостатическое действие на клетку, тем самым ингибируя рост и/или размножение определенного подмножества клеток (т.е. опухолевых клеток).
Используемый в настоящем документе термин «иммуномодулирующий агент» относится к агенту, который стимулирует иммунный ответ посредством продуцирования цитокинов и/или антител и/или модулирования функции Т-клеток, тем самым ингибируя или снижая рост подмножества клеток (т.е. опухолевых клеток) прямо или косвенно, позволяя другому агенту быть более эффективным.
«Состоит по существу из» и варианты, такие как «состоит по существу из» или «состоящий по существу из», используемые в описании и формуле изобретения, указывают на включение любых перечисленных элементов или группы элементов, а также на необязательное включение других элементов, аналогичной или другой природы, чем перечисленные элементы, которые существенно не изменяют основные или новые свойства указанной схемы дозирования, способа или композиции. В качестве неограничивающего примера, агонист OX40, который состоит по существу из указанной аминокислотной последовательности, может также включать одну или несколько аминокислот, включая замены одного или нескольких аминокислотных остатков, которые существенно не влияют на свойства связывающего соединения.
Используемый в настоящем документе термин «эффективная дозировка» или «эффективное количество» лекарственного средства, соединения или фармацевтической композиции, является количеством, достаточным для воздействия на любой один или несколько полезных или желаемых, включая биохимические, гистологические и/или поведенческие симптомы, заболевания, его осложнения, и промежуточные патологические фенотипы, возникающие в процессе развития заболевания. Для терапевтического применения, «терапевтически эффективное количество» относится к такому количеству вводимого соединения, которое в некоторой степени облегчит один или несколько симптомов заболевания, подвергаемого лечению. Что касается лечения рака, терапевтически эффективное количество относится к такому количеству, которое оказывает действие (1) уменьшения размера опухоли, (2) ингибирования (то есть, замедления до некоторой степени, предпочтительно, остановки) метастазирования опухоли, (3) ингибирования до некоторой степени (то есть, замедления до некоторой степени, предпочтительно, прекращения) роста опухоли или инвазивности опухоли, (4) ослабления до некоторой степени (или, предпочтительно, устранения) одного или нескольких признаков или симптомов, связанных с раком, (5) уменьшения дозы других лекарственных средств, необходимых для лечения заболевания, и/или (6) усиления действия другого лекарственного средства, и/или (7) замедления прогрессирования заболевания у пациента.
Эффективную дозировку можно вводить за одно или несколько введений. Для целей настоящего изобретения, эффективная дозировка лекарственного средства, соединения или фармацевтической композиции является количеством, достаточным для проведения профилактического или терапевтического лечения, прямо или косвенно. Как понятно в клиническом контексте, эффективная дозировка лекарственного средства, соединения или фармацевтической композиции может быть достигнута или не достигнута в сочетании с другим лекарственным средством, соединением или фармацевтической композицией.
«Фармацевтическая композиция» относится к смеси одного или нескольких соединений по изобретению или их фармацевтически приемлемой соли, сольвата, гидрата или пролекарства, в качестве активного ингредиента и, по меньшей мере, одного фармацевтически приемлемого носителя или эксципиента. В некоторых вариантах осуществления, фармацевтическая композиция содержит два или несколько фармацевтически приемлемых носителей и/или эксципиентов. В других вариантах осуществления, фармацевтическая композиция дополнительно содержит, по меньшей мере, один дополнительный противораковый терапевтический агент.
В одном аспекте, изобретение представляет фармацевтическую композицию, содержащую соединение по изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель или эксципиент. В некоторых вариантах осуществления, фармацевтическая композиция содержит два или несколько фармацевтически приемлемых носителей и/или эксципиентов.
В некоторых вариантах осуществления, фармацевтическая композиция дополнительно содержит, по меньшей мере, один дополнительный противораковый терапевтический агент. В некоторых таких вариантах осуществления, комбинация обеспечивает аддитивное, большее, чем аддитивное, или синергетическое противораковое действие.
«Опухоль» применительно к субъекту, у которого диагностирован или подозревается рак, относится к злокачественному или потенциально злокачественному новообразованию или массе ткани любого размера и включает первичные опухоли и вторичные новообразования. Солидной опухолью является аномальный рост или масса ткани, которая обычно не содержит кист или жидких областей. Примерами солидных опухолей являются саркомы, карциномы и лимфомы. Лейкозы (рак крови) обычно не образуют солидных опухолей (National Cancer Institute, Dictionary of Cancer Terms).
«Опухолевая масса» или «опухолевая нагрузка» относится к общему количеству опухолевого материала, распределенного по всему телу. Опухолевая масса относится к общему количеству раковых клеток или общему размеру опухолей по всему телу, включая лимфатические узлы и костный мозг. Опухолевая масса может быть определена различными способами, известными в данной области техники, такими как, например, использование штангенциркуля, или в организме с использованием методов визуализации, например, ультразвука, сканирования костей, компьютерной томографии (КТ) или магнитно-резонансной томографии (МРТ).
Термин «размер опухоли» относится к общему размеру опухоли, который можно измерить как длину и ширину опухоли. Размер опухоли можно определить различными способами, известными в данной области техники, такими как, например, измерение размеров опухоли при удалении у субъекта, например, с помощью штангенциркуля, или во время нахождения в организме с использованием методов визуализации, например, сканирования костей, УЗИ, компьютерной томографии или МРТ.
Используемый в настоящем документе термин «субъект» относится к человеку или животному. Если субъектом является человек, субъект также может называться «пациент».
Термин «лечить» или «лечение» рака, используемый в настоящем документе, означает введение соединения по настоящему изобретению субъекту, страдающему раком или у которого диагностирован рак, для достижения, по меньшей мере, одного положительного терапевтического эффекта, такого как, например, уменьшение количества раковых клеток, уменьшение размера опухоли, снижение скорости инфильтрации раковых клеток в периферические органы или снижение скорости метастазов опухоли или роста опухоли, обращение вспять, облегчение, ингибирование прогрессирования или профилактика нарушения или состояния, к которому применяется этот термин, или одного или нескольких симптомов такого нарушения или состояния. Термин «лечение», используемый в настоящем документе, если не указано иное, относится к акту лечения, такому, как «лечение» определенное непосредственно выше. Термин «лечение» также включает адъювантное и неоадъювантное лечение субъекта.
Для целей настоящего изобретения благоприятные или желаемые клинические результаты включают, но не ограничены этим, один или несколько из следующих: снижение пролиферации (или уничтожение) неопластических или раковых клеток; ингибирование метастазов или неопластических клеток; сокращение или уменьшение размера опухоли; ремиссия рака; уменьшение симптомов, возникающих в результате рака; повышение качества жизни больных раком; уменьшение дозы других лекарственных средств, необходимых для лечения рака; замедление прогрессирования рака; излечение рака; преодоление одного или нескольких механизмов резистентности рака; и/или продление выживаемости больных раком. Положительные терапевтические эффекты при раке могут быть измерены несколькими способами (см., например, W. A. Weber, Assessing tumor response to therapy, J. Nucl. Med. 50 Suppl. 1:1S-10S (2009). Например, в отношении ингибирование роста опухоли (Т/С), в соответствии со стандартами National Cancer Institute (NCI), Т/С меньше или равное 42% является минимальным уровнем противоопухолевой активности. Т/С <10% считается высоким уровнем противоопухолевой активности, при этом T/C (%)=средний объем леченной опухоли/средний объем контрольной опухоли x 100.
В некоторых вариантах осуществления, лечение, достигаемое соединением по изобретению, определяется в отношении любого из следующих: частичный ответ (PR), полный ответ (CR), общий ответ (OR), выживаемость без прогрессирования (PFS), выживаемость без заболевания (DFS) и общая выживаемость (OS). PFS, также называемая «время до прогрессирования опухоли», означает период времени во время и после лечения, в течение которого рак не растет, и включает в себя количество времени, в течение которого пациенты испытывают CR или PR, а также количество времени, в течение которого пациенты имеют стабильное заболевание (SD). DFS относится к периоду времени во время и после лечения, в течение которого пациент остается без заболевания. OS относится к пролонгации ожидаемой продолжительности жизни по сравнению с наивными или не леченными субъектами или пациентами. В некоторых вариантах осуществления, ответом на комбинацию по изобретению является любой из PR, CR, PFS, DFS, OR или OS, который оценивают с использованием критериев оценки ответа Response Evaluation Criteria in Solid Tumors (RECIST) 1.1.
Термин «добавка» используют для обозначения того, что результат комбинации двух соединений, компонентов или таргетных агентов не превышает суммы каждого соединения, компонента или таргетного агента по отдельности.
Термин «синергия» или «синергический» используют для обозначения того, что результат комбинации двух соединений, компонентов или таргетных агентов больше, чем сумма каждого соединения, компонента или таргетного агента по отдельности. Это улучшение заболевания, состояния или нарушения, подвергаемого лечению, является «синергическим» эффектом. «Синергическим количеством» является количество комбинации двух соединений, компонентов или таргетных агентов, которое дает синергетический эффект, такой, как «синергический» определен в настоящем документе.
При определении синергетического взаимодействия между одним или двумя компонентами, оптимальный диапазон для эффекта и диапазоны абсолютных доз каждого компонента для эффекта могут быть окончательно измерены путем введения компонентов в различных диапазонах доз и/или соотношениях доз пациентам, нуждающимся в лечении. Однако наблюдение синергии в моделях in vitro или моделях in vivo может предсказать эффект у людей и других видов, и модели in vitro или модели in vivo существуют, как описано в настоящем документе, для измерения синергетического эффекта. Результаты таких исследований также можно использовать для прогнозирования диапазонов соотношений эффективных доз и концентраций в плазме, а также абсолютных доз и концентраций в плазме, необходимых для людей и других видов, например, путем применения фармакокинетических и/или фармакодинамических способов.
Схема лечения соединением по изобретению, которая эффективна для лечения онкологического больного, может варьироваться в зависимости от таких факторов, как состояние болезни, возраст и вес пациента, а также от способности терапии вызывать противораковый ответ у субъекта. Хотя вариант осуществления любого из аспектов изобретения может быть неэффективен для достижения положительного терапевтического эффекта у каждого субъекта, он должен быть эффективным для статистически значимого количества субъектов, что определяется любым статистическим тестом, известным в данной области техники, таким как t-критерий Стьюдента, тест хи-квадрат, U-критерий Манна-Уитни, критерий Крускала-Уоллиса (H-критерий), критерий Джонкхиера-Терпстра и тест Уилкона.
Термины «схема лечения», «протокол дозирования» и «схема дозирования» используют взаимозаменяемо для обозначения дозы и времени введения каждого соединения по изобретению, отдельно или в комбинации с другим терапевтическим агентом.
«Улучшение» означает уменьшение или улучшение одного или нескольких симптомов при лечении комбинацией, описанной в настоящем документе, по сравнению с отсутствием введения комбинации. «Улучшение» также включает укорочение или уменьшение продолжительности симптома.
«Аномальный рост клеток», используемый в настоящем документе, если не указано иное, относится к росту клеток, который не зависит от нормальных регуляторных механизмов (например, потеря контактного ингибирования). Аномальный рост клеток может быть доброкачественным (не раковым) или злокачественным (раковым).
Термины «рак» или «злокачественный» относятся к любому злокачественному и/или инвазивному росту или опухоли, вызванной аномальным ростом клеток. Рак включает первичный рак, возникающий в определенном месте тела, метастатический рак, который распространился из места, где он возник, в другие части тела, рецидив исходного первичного рака после ремиссии и второй первичный рак, который является новым первичным раком у пациента с историей предшествующего рака другого типа, отличающегося от второго первичного рака. Рак включает солидные опухоли, названные по типу образующих их клеток, рак крови, костного мозга или лимфатической системы. Примеры солидных опухолей включают саркомы и карциномы. Рак крови включает лейкоз, лимфому и миелому. Дополнительные примеры рака включают бластомы и актинический кератоз. Рак также включает первичный рак или метастазы локализации, выбранной из группы, состоящей из полости рта, пищеварительной системы, дыхательной системы, кожи, молочной железы, половой системы, мочевыделительной системы, зрительной системы, нервной системы, эндокринной системы и лимфомы.
Если не указано иное, все ссылки в настоящем документе на соединения по изобретению включают ссылки на их соли, сольваты, гидраты и комплексы, а также на сольваты, гидраты и комплексы их солей, включая их полиморфы, стереоизомеры и меченные изотопами варианты.
Соединения по изобретению могут существовать в форме фармацевтически приемлемых солей, таких как, например, кислотно-аддитивные соли и основно-аддитивные соли соединений одной из представленных в настоящем документе формул. Используемый в настоящем документе термин «фармацевтически приемлемая соль» относится к тем солям, которые сохраняют биологическую эффективность и свойства исходного соединения. Фраза «фармацевтически приемлемые соли», используемая в настоящем документе, если не указано иное, включает соли кислотных или основных групп, которые могут присутствовать в соединениях формул, описанных в настоящем документе.
Например, соединения по изобретению, которые являются основными по своей природе, способны образовывать широкий спектр солей с различными неорганическими и органическими кислотами. Хотя такие соли должны быть фармацевтически приемлемыми для введения животным, на практике часто желательно сначала выделить соединение по настоящему изобретению из реакционной смеси в виде фармацевтически неприемлемой соли, а затем просто превратить последнюю обратно в соединение в виде свободного основания путем обработки щелочным реагентом и затем превратить последнее свободное основание в фармацевтически приемлемую кислотно-аддитивную соль. Кислотно-аддитивные соли основных соединений по настоящему изобретению могут быть получены обработкой основного соединения по существу эквивалентным количеством выбранной минеральной или органической кислоты в среде водного растворителя или в подходящем органическом растворителе, таком как метанол или этанол. После выпаривания растворителя получают желаемую твердую соль. Желаемую кислую соль также можно осадить из раствора свободного основания в органическом растворителе путем добавления к раствору подходящей минеральной или органической кислоты.
Кислоты, которые можно использовать для получения фармацевтически приемлемых кислотно-аддитивных солей таких основных соединений, включают такие, которые образуют нетоксичные кислотно-аддитивные соли, т.е. соли, содержащие фармакологически приемлемые анионы, такие как гидрохлорид, гидробромид, гидройодид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, изоникотинат, ацетат, лактат, салицилат, цитрат, кислый цитрат, тартрат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат и памоатн.
Примеры солей включают, но не ограничиваются ими, ацетат, акрилат, бензолсульфонат, бензоат (например, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат и метоксибензоат), бикарбонат, бисульфат, бисульфит, битартрат, борат, бромид, бутин-1,4-диоат, эдетат кальция, камзилат, карбонат, хлорид, капроат, каприлат, клавуланат, цитрат, деканоат, дигидрохлорид, дигидрофосфат, эдетат, эдисилат, эстолат, эзилат, этилсукцинат, формиат, фумарат, глуцептат, глюконат, глутамат, гликолят, гликоллиларсанилат, гептаноат, гексин-1,6-диоат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, γ-гидроксибутират, йодид, изобутират, изотионат, лактат, лактобионат, лаурат, малат, малеат, малонат, миндалят, мезилат, метафосфат, метансульфонат, метилсульфат, моногидрофосфат, мукат, напсилат, нафталин-1-сульфонат, нафталин-2-сульфонат, нитрат, олеат, оксалат, памоат (эмбонат), пальмитат, пантотенат, фенилацетаты, фенилбутират, фенилпропионат, фталат, фосфат/дифосфат, полигалактуронат, пропансульфонат, пропионат, пропиолат, пирофосфат, пиросульфат, салицилат, стеарат, субацетат, суберат, сукцинат, сульфат, сульфонат, сульфит, таннат, тартрат, теоклат, тозилат и валерат.
Иллюстративные примеры подходящих солей включают органические соли, полученные из аминокислот, таких как глицин и аргинин, аммиак, первичные, вторичные и третичные амины и циклические амины, такие как пиперидин, морфолин и пиперазин, и неорганические соли, полученные из натрия, кальция, калия, магния, марганца, железа, меди, цинка, алюминия и лития.
Соединения по изобретению, которые включают основную группу, такую как аминогруппа, могут образовывать фармацевтически приемлемые соли с различными аминокислотами в дополнение к кислотам, упомянутым выше.
Альтернативно, полезные соединения, которые являются кислыми по своей природе, могут быть способны образовывать основные соли с различными фармакологически приемлемыми катионами. Примеры таких солей включают соли щелочных или щелочноземельных металлов и, в частности, соли натрия и калия. Все эти соли получают обычными методами. Химические основания, которые используют в качестве реагентов для получения фармацевтически приемлемых основных солей по настоящему изобретению, представляют собой основания, которые образуют нетоксичные основные соли с кислыми соединениями по настоящему изобретению. Эти соли могут быть получены любым подходящим способом, например обработкой свободной кислоты неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидроксид щелочного металла или гидроксид щелочноземельного металла и подобные. Эти соли также могут быть получены обработкой соответствующих кислых соединений водным раствором, содержащим желаемые фармакологически приемлемые катионы, и последующим выпариванием полученного раствора досуха, предпочтительно при пониженном давлении. Альтернативно, их также можно приготовить путем смешивания низших алканольных растворов кислых соединений и желаемого алкоксида щелочного металла вместе с последующим выпариванием полученного раствора досуха тем же способом, что и ранее. В любом случае, предпочтительно использовать стехиометрические количества реагентов для обеспечения полноты реакции и максимального выхода желаемого конечного продукта.
Химические основания, которые могут быть использованы в качестве реагентов для получения фармацевтически приемлемых основных солей соединений по изобретению, которые являются кислыми по своей природе, являются такими, которые образуют нетоксичные основные соли с такими соединениями. Такие нетоксичные основные соли включают, но не ограничены ими, соли, полученные из таких фармакологически приемлемых катионов, как катионы щелочных металлов (например, калия и натрия) и катионов щелочноземельных металлов (например, кальция и магния), аммония или водорастворимые аддитивные соли амина, такие как N-метилглюкамин (меглюмин), низший алканоламмоний и другие основные соли фармацевтически приемлемых органических аминов.
Могут также образовываться полусоли кислот и оснований, например, гемисульфатные и гемикальциевые соли.
Обзор подходящих солей см. в Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (Wiley VCH, 2002). Способы получения фармацевтически приемлемых солей соединений по изобретению и взаимопревращающихся форм соли и свободного основания известны специалистам в данной области техники.
Соли по настоящему изобретению могут быть получены способами, известными специалистам в данной области техники. Фармацевтически приемлемая соль соединений по изобретению может быть легко получена путем смешивания вместе растворов соединения и желаемой кислоты или основания, в зависимости от ситуации. Соль может осаждаться из раствора и собираться фильтрованием или может быть выделена выпариванием растворителя. Степень ионизации в соли может варьироваться от полностью ионизированной до почти неионизированной.
Специалистам в данной области техники должно быть понятно, что соединения по изобретению в форме свободного основания, имеющие основную функциональную группу, могут быть превращены в кислотно-аддитивные соли путем обработки стехиометрическим избытком соответствующей кислоты. Кислотно-аддитивные соли соединений по изобретению могут быть повторно превращены в соответствующее свободное основание обработкой стехиометрическим избытком подходящего основания, такого как карбонат калия или гидроксид натрия, обычно в присутствии водного растворителя и при температуре между примерно 0°С и 100°С. Форма свободного основания может быть выделена обычными способами, такими как экстракция органическим растворителем. Кроме того, кислотно-аддитивные соли соединений по настоящему изобретению могут быть взаимозаменяемы за счет использования преимущества различной растворимости солей, летучести или кислотности кислот или путем обработки ионообменной смолой с соответствующим содержанием. Например, на обмен может влиять реакция соли соединений изобретения с небольшим стехиометрическим избытком кислоты с более низким pK, чем кислотный компонент исходной соли. Это превращение обычно проводят при температуре примерно от 0°С до точки кипения растворителя, используемого в качестве среды для процедуры. Подобные обмены возможны с основно-аддитивными солями, как правило, через посредничество формы свободного основания.
Соединения по изобретению могут существовать как в не сольватированной, так и в сольватированной формах. Когда растворитель или вода прочно связаны, комплекс будет иметь четко определенную стехиометрию, не зависящую от влажности. Однако, когда растворитель или вода слабо связаны, как в канальных сольватах и гигроскопичных соединениях, содержание воды/растворителя будет зависеть от влажности и условий сушки. В таких случаях не стехиометрия будет нормой. Термин «сольват» используют в настоящем документе для описания молекулярного комплекса, включающего соединение по изобретению и одну или несколько молекул фармацевтически приемлемого растворителя, например, этанола. Термин «гидрат» используется, когда растворителем является вода. Фармацевтически приемлемые сольваты в соответствии с изобретением включают гидраты и сольваты, в которых растворитель кристаллизации может быть изотопно замещен, например D2O, d6-ацетон, d6-ДМСО.
Также в объем изобретения включены такие комплексы, как клатраты, комплексы включения лекарственное средство-хозяин, в которых, в отличие от вышеупомянутых сольватов, лекарственное средство и хозяин присутствуют в стехиометрических или не стехиометрических количествах. Также включены комплексы лекарственного средства, содержащие два или несколько органических и/или неорганических компонентов, которые могут находиться в стехиометрических или не стехиометрических количествах. Образовавшиеся комплексы могут быть ионизированными, частично ионизированными или не ионизированными. Обзор таких комплексов см. в J Pharm Sci, 64 (8), 1269 1288 by Haleblian (August 1975), описание которого полностью включено в настоящий документ посредством ссылки.
Изобретение также относится к пролекарствам соединений представленных в настоящем документе формул. Таким образом, некоторые производные соединений по изобретению, которые могут иметь небольшую фармакологическую активность или не иметь ее, при введении пациенту могут быть превращены в соединения по изобретению, например, путем гидролитического расщепления. Такие производные называются «пролекарствами». Дополнительную информацию об использовании пролекарств можно найти в «Pro drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T Higuchi and W Stella); «Bioreversible Carriers in Drug Design», Pergamon Press, 1987 (ed. E B Roche, American Pharmaceutical Association) и Guarino, V.R; Stella, V.J.: Biotech Pharm. Aspects 2007 5 (Pt2) 133-187, описание которых полностью включено в настоящее описание посредством ссылки.
Пролекарства в соответствии с изобретением могут быть получены, например, путем замены соответствующих функциональных групп, присутствующих в соединениях по изобретению, определенными группами, известными специалистам в данной области техники как «пролекарства», как описано, например, в «Design of Prodrugs» by H Bundgaard (Elsevier, 1985), описание которой полностью включено в настоящий документ посредством ссылки.
Некоторые неограничивающие примеры пролекарств по изобретению включают:
(i) если соединение содержит карбоксильную функциональную группу (COOH), ее сложный эфир, например, замена водорода на (C1-C6)алкил;
(ii) если соединение содержит спиртовую функциональную группу (ОН), ее простой эфир, например, замещение водорода на (C1-C6)алканоилоксиметил или группу фосфатного эфира; и
(iii) если соединение содержит первичную или вторичную амино функциональную группу (NH2 или NHR, где R ≠ H), ее амид, например, замена одного или обоих атомов водорода подходящей метаболически лабильной группой, такой как амид, карбамат, мочевина, фосфонат, сульфонат и т.д.
Дополнительные примеры замещающих групп в соответствии с приведенными выше примерами и примеры других типов пролекарств можно найти в вышеупомянутых ссылках.
Наконец, некоторые соединения по изобретению могут сами действовать как пролекарства других соединений по изобретению.
Также в объем изобретения включены метаболиты соединений описанных в настоящем документе формул, т.е. соединения, образующиеся in vivo при введении лекарственного средства.
Соединения представленных в настоящем документе формул могут иметь асимметричные атомы углерода. Связи углерод-углерод соединений по изобретению могут быть изображены в настоящем документе сплошной линией (  ), сплошным клином (
), сплошным клином (  ) или пунктирным клином (
) или пунктирным клином (  ). Использование сплошной линии для обозначения связей с асимметрическими атомами углерода означает, что включены все возможные стереоизомеры (например, определенные энантиомеры, рацемические смеси и т.д.) на этом атоме углерода. Использование сплошного или пунктирного клина для изображения связей с асимметричными атомами углерода означает, что предполагается включить только показанный стереоизомер. Возможно, что соединения по изобретению могут содержать более одного асимметрического атома углерода. В этих соединениях использование сплошной линии для обозначения связей с асимметрическими атомами углерода означает, что все возможные стереоизомеры должны быть включены, а стереоцентр присоединен. Например, если не указано иное, предполагается, что соединения по изобретению могут существовать в виде энантиомеров и диастереомеров или в виде рацематов и их смесей. Использование сплошной линии для изображения связей с одним или несколькими асимметричными атомами углерода в соединении по изобретению и использование сплошного или пунктирного клина для изображения связей с другими асимметрическими атомами углерода в том же соединении означает, что смесь диастереомеров присутствует.
). Использование сплошной линии для обозначения связей с асимметрическими атомами углерода означает, что включены все возможные стереоизомеры (например, определенные энантиомеры, рацемические смеси и т.д.) на этом атоме углерода. Использование сплошного или пунктирного клина для изображения связей с асимметричными атомами углерода означает, что предполагается включить только показанный стереоизомер. Возможно, что соединения по изобретению могут содержать более одного асимметрического атома углерода. В этих соединениях использование сплошной линии для обозначения связей с асимметрическими атомами углерода означает, что все возможные стереоизомеры должны быть включены, а стереоцентр присоединен. Например, если не указано иное, предполагается, что соединения по изобретению могут существовать в виде энантиомеров и диастереомеров или в виде рацематов и их смесей. Использование сплошной линии для изображения связей с одним или несколькими асимметричными атомами углерода в соединении по изобретению и использование сплошного или пунктирного клина для изображения связей с другими асимметрическими атомами углерода в том же соединении означает, что смесь диастереомеров присутствует.
Соединения по изобретению, которые имеют хиральные центры, могут существовать в виде стереоизомеров, таких как рацематы, энантиомеры или диастереомеры.
Стереоизомеры соединений представленных в настоящем документе формул могут включать цис- и транс-изомеры, оптические изомеры, такие как (R) и (S) энантиомеры, диастереомеры, геометрические изомеры, вращательные изомеры, атропоизомеры, конформационные изомеры и таутомеры соединений по изобретению, включая соединения, проявляющие более одного типа изомерии; и их смеси (такие как рацематы и диастереомерные пары).
Также включены кислотно-аддитивные соли или основно-аддитивные соли, где противоион является оптически активным, например, d-лактат или l-лизин, или рацемическим, например, dl-тартрат или dl-аргинин.
При кристаллизации любого рацемата возможны кристаллы двух различных типов. Первым типом является упомянутое выше рацемическое соединение (истинный рацемат), в котором образуется одна гомогенная форма кристалла, содержащая оба энантиомера в эквимолярных количествах. Вторым типом является рацемическая смесь или конгломерат, где две формы кристаллов образуются в эквимолярных количествах, каждая из которых содержит один энантиомер.
Соединения по изобретению могут проявлять явления таутомерии и структурной изомерии. Например, соединения могут существовать в нескольких таутомерных формах, включая енольную и иминовую форму, и кето и енаминовую форму и геометрические изомеры и их смеси. Все такие таутомерные формы входят в объем соединений по изобретению. Таутомеры существуют в виде смесей таутомерного набора в растворе. В твердой форме обычно преобладает один таутомер. Хотя может быть описан один таутомер, настоящее изобретение включает все таутомеры соединений представленных формул.
Кроме того, некоторые из соединений по изобретению могут образовывать атропоизомеры (например, замещенные биарилы). Атропоизомерами являются конформационные стереоизомеры, которые возникают, когда вращение вокруг простой связи в молекуле предотвращается или значительно замедляется в результате пространственных взаимодействий с другими частями молекулы, и заместители на обоих концах одинарной связи не симметричны. Взаимное превращение атропоизомеров происходит достаточно медленно, чтобы их можно было разделить и выделить в заданных условиях. Энергетический барьер для термической рацемизации может быть определен пространственными препятствиями для свободного вращения одной или нескольких связей, образующих хиральную ось.
Если соединение по изобретению содержит алкенильную или алкениленовую группу, возможны геометрические цис/транс (или Z/E) изомеры. Цис/транс изомеры могут быть разделены обычными способами, хорошо известными специалистам в данной области техники, например, хроматографией и фракционной кристаллизацией.
Обычные методы получения/выделения индивидуальных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата (или рацемата соли или производного) с использованием, например, хиральной жидкостной хроматографии высокого давления (ЖХВД) или сверхкритической жидкостной хроматографии (СЖХ).
Альтернативно, рацемат (или рацемический предшественник) может быть подвергнут взаимодействию с подходящим оптически активным соединением, например, спиртом, или, в случае, когда соединение содержит кислотную или основную группу, кислотой или основанием, таким как винная кислота или 1-фенилэтиламин. Полученную диастереомерную смесь можно разделить хроматографией и/или фракционной кристаллизацией, и один или оба диастереоизомера превратить в соответствующие чистые энантиомеры способами, хорошо известными специалисту в данной области техники.
Хиральные соединения по изобретению (и их хиральные предшественники) могут быть получены в энантиомерно обогащенной форме с использованием хроматографии, обычно ВЭЖХ, на асимметричной смоле с подвижной фазой, состоящей из углеводорода, обычно гептана или гексана, содержащего от 0 до 50% изопропанола, обычно от 2 до 20% и от 0 до 5% алкиламина, обычно 0,1% диэтиламина. Концентрация элюата дает обогащенную смесь.
Стереоизомерные конгломераты могут быть разделены обычными способами, известными специалистам в данной области техники; см., например, «Stereochemistry of Organic Compounds» by E L Eliel (Wiley, New York, 1994), описание которой полностью включено в настоящее описание посредством ссылки.
Энантиомерная чистота соединений, описанных в настоящем документе, может быть описана по показателю энантиомерного избытка (эи), который указывает на степень, в которой образец содержит один энантиомер в большем количестве, чем другой. Рацемическая смесь имеет эи 0%, в то время как отдельный полностью чистый энантиомер имеет эи 100%. Точно так же, диастереомерная чистота может быть описана по показателю диастереомерного избытка (ди).
Настоящее изобретение также включает изотопно-меченые соединения, которые идентичны соединениям, указанным в одной из приведенных формул, но с тем отличием, что один или несколько атомов заменены атомом, имеющим атомную массу или массовое число, отличное от атомной массы или массового числа, обычно встречающегося в природе.
Изотопно-меченые соединения по изобретению, как правило, могут быть получены обычными методами, известными специалистам в данной области техники, или процессами, аналогичными описанным в настоящем документе, с использованием соответствующего изотопно-меченого реагента вместо не меченого реагента, используемого в других случаях.
Примеры изотопов, которые могут быть включены в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как, но не ограничиваясь ими, 2H, 3H, 13C, 14C, 15N, 18O, 17O, 32P, 35S, 18F и 36Cl. Некоторые изотопно-меченые соединения по изобретению, например те, в которые включены радиоактивные изотопы, такие как 3H и 14C, можно использовать в анализах распределения лекарственного средства и/или субстрата в тканях. Изотопы трития, т.е. 3H, и углерода 14, т.е. 14C, особенно предпочтительны из-за простоты их получения и обнаружения. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2H, может обеспечить определенные терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличение периода полужизни in vivo или снижение требований к дозировке, и, следовательно, в некоторых обстоятельствах может быть предпочтительным. Изотопно-меченые соединения по настоящему изобретению, как правило, могут быть получены путем проведения процедур, описанных на схемах и/или в приведенных ниже примерах и примерах получения, путем замены не изотопно-меченого реагента, изотопно-меченным реагентом.
Соединения по изобретению, предназначенные для фармацевтического применения, можно вводить в виде кристаллических или аморфных продуктов или их смесей. Их можно получить, например, в виде твердых слоев, порошков или пленок такими способами, как осаждение, кристаллизация, сушка вымораживанием, сушка распылением или сушка выпариванием. Можно использовать микроволновую или радиочастотную сушку.
В другом варианте осуществления изобретение представляет соединение формулы (Ia)
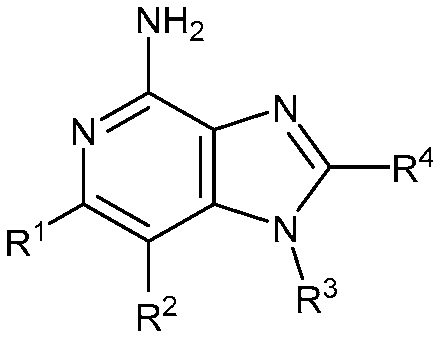
(Ia)
или его фармацевтически приемлемая соль, где
R1 и R2 независимо являются C1-2 алкилом; или
R1 и R2 объединены с образованием 5-7-членного карбоциклического кольца, где указанное карбоциклическое кольцо может быть насыщенным или ненасыщенным;
R3 является
 ;
;
R4 является C3-5 алкилом или (CH2)nO(CH2)mCH3;
R5 является C1-2 алкилом;
m равно 1; и
n равно 1.
В другом варианте осуществления, в изобретении представлено соединение формулы (Ia), или его фармацевтически приемлемая соль, где R1 и R2 независимо являются C1-2 алкилом.
В другом варианте осуществления, в изобретении представлено соединение формулы (Ia), или его фармацевтически приемлемая соль, где R1 и R2 объединены с образованием 5-7-членного карбоциклического кольца, где указанное карбоциклическое кольцо может быть насыщенным или ненасыщенным.
В другом варианте осуществления, в изобретении представлено соединение формулы (Ia), или его фармацевтически приемлемая соль, где
R1 и R2 объединены с образованием 5-7-членного карбоциклического кольца, где указанное карбоциклическое кольцо может быть насыщенным; и
R4 является (CH2)nO(CH2)mCH3.
В другом варианте осуществления, в изобретении представлено соединение формулы (Ia), или его фармацевтически приемлемая соль, где карбоциклическим кольцом является циклопентил.
В другом варианте осуществления, в изобретении представлено соединение формулы (Ia), или его фармацевтически приемлемая соль, где карбоциклическим кольцом является циклогексил.
В другом варианте осуществления, в изобретении представлено соединение формулы (Ia), или его фармацевтически приемлемая соль, где
R1 и R2 объединены с образованием 5-7-членного карбоциклического кольца, где указанное карбоциклическое кольцо может быть ненасыщенным; и
R4 является C3-5 алкилом.
В другом варианте осуществления, в изобретении представлено соединение формулы (Ia), или его фармацевтически приемлемая соль, где карбоциклическим кольцом является фенил.
В другом варианте осуществления, в изобретении представлено соединение формулы (Ib)
(Ib)
или его фармацевтически приемлемая соль, где
R1 и R2 независимо являются C1-3 алкилом; или
R1 и R2 объединены с образованием 5-7-членного карбоциклического кольца, где указанное карбоциклическое кольцо может быть насыщенным или ненасыщенным;
R3 является
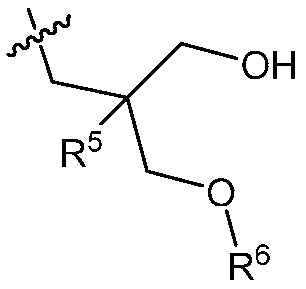 ;
;
R4 является C1-6 алкилом, или (CH2)nO(CH2)mCH3, где указанный C1-6 алкил или любой атом углерода (CH2)nO(CH2)mCH3 группы замещен от 0 до 3 галогенами, насколько позволяет валентность, где галогеном является F;
R5 является C1-3 алкилом или OC1-3 алкилом, где C1-3 алкил замещен от 0 до 3 F;
R6 является H или C1-3 алкилом, где C1-3 алкил замещен от 0 до 3 F;
m равно 0-2; и
n равно 1-3.
В другом варианте осуществления, в изобретении представлено соединение формулы (Ib), или его фармацевтически приемлемая соль, где
R1 и R2 независимо являются C1-2 алкилом; или
R1 и R2 объединены с образованием 5-7-членного карбоциклического кольца, где указанное карбоциклическое кольцо может быть насыщенным или ненасыщенным;
R3 является
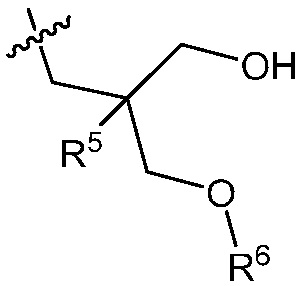 ;
;
R5 является C1-3 алкилом или OC1-3 алкилом, где C1-3 алкил замещен от 0 до 2 F; и
R6 является H.
В другом варианте осуществления, в изобретении представлено соединение формулы (Ib), или его фармацевтически приемлемая соль, где R5 является C1-2 алкилом.
В другом варианте осуществления, в изобретении представлено соединение формулы (I), (Ia), (Ib), или его фармацевтически приемлемая соль, где R4 является н-пропилом, н-бутилом или н-пентилом.
В другом варианте осуществления, в изобретении представлено соединение формулы (I), (Ia), (Ib), или его фармацевтически приемлемая соль, где R4 является -CH2-O-CH2CH3.
В другом варианте осуществления, в изобретении представлено соединение формулы (I), (Ia), (Ib), или его фармацевтически приемлемая соль, где R5 является метилом или этилом.
В другом варианте осуществления, в изобретении представлено соединение формулы (I), (Ia), (Ib), или его фармацевтически приемлемая соль, где R6 является H.
Другим вариантом изобретения является один или несколько из каждого примера, описанного в настоящем документе, и включает, но не ограничен ими, соединения, выбранные из:
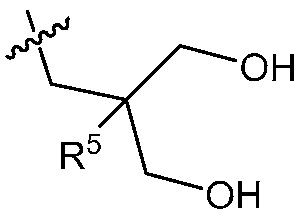
2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диола;
2-((4-амино-2-(этоксиметил)-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диола;
2-((4-амино-2-(этоксиметил)-7,8-дигидроциклопента[b]имидазо[4,5-d]пиридин-1(6H)-ил)метил)-2-метилпропан-1,3-диола;
2-((4-амино-2-(этоксиметил)-6,7,8,9-тетрагидро-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диола;
3-(4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-2-(метоксиметил)-2-метилпропан-1-ола;
(R)-3-(4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-2-(метоксиметил)-2-метилпропан-1-ола;
(S)-3-(4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-2-(метоксиметил)-2-метилпропан-1-ола;
2-((4-амино-6,7-диметил-2-(2,2,2-трифторэтил)-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диола;
2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-этилпропан-1,3-диола;
2-((4-амино-2-бутил-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диола;
2-((4-амино-2-бутил-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диола; и
2-((4-амино-2-пентил-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диола;
или его фармацевтически приемлемую соль.
Другим вариантом изобретения является один или несколько из каждого примера, описанного в настоящем документе, и включает, но не ограничен ими, соединения, выбранные из:
2-((4-амино-2-бутил-6,7,8,9-тетрагидро-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диола;
2-((4-амино-2-бутил-7,8-дигидроциклопента[b]имидазо[4,5-d]пиридин-1(6H)-ил)метил)-2-метилпропан-1,3-диола;
2-((4-амино-2-этил-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диола; и
2-((4-амино-2-пропил-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диола;
или его фармацевтически приемлемую соль.
Другой вариант осуществления изобретения представляет фармацевтическую композицию, содержащую соединения формулы (I) и любые их варианты осуществления или их фармацевтически приемлемую соль, а также фармацевтически приемлемый носитель или эксципиент.
Другой вариант осуществления изобретения представляет способ лечения рака у субъекта, нуждающегося в этом, включающий введение субъекту терапевтически эффективного количества соединений формулы (I) и любых их вариантов осуществления или их фармацевтически приемлемой соли.
Рак, подлежащий лечению, включает плоскоклеточную карциному, базальноклеточные карциномы, миелому, мелкоклеточный рак легкого, немелкоклеточный рак легкого, глиому, лимфому Ходжкина, неходжкинскую лимфому, острый миелоидный лейкоз (AML), множественную миелому, рак желудочно-кишечного (тракта), рак почки, рак яичников, рак печени, лимфобластный лейкоз, лимфоцитарный лейкоз, колоректальный рак, рак эндометрия, рак почки, рак простаты, рак щитовидной железы, меланому, хондросаркому, нейробластому, рак поджелудочной железы, мультиформную глиобластому, рак шейки матки, рак головного мозга, рак желудка, рак матки, рак мочевого пузыря, включая не мышечно-инвазивный рак мочевого пузыря, гепатому, рак груди и рак головы и шеи.
Более конкретные примеры рака, подлежащего лечению, включают базальноклеточные карциномы, мелкоклеточный рак легкого, немелкоклеточный рак легкого, неходжкинскую лимфому, рак яичников, колоректальный рак, рак почки, рак простаты, рак щитовидной железы, меланому, рак поджелудочной железы, рак мочевого пузыря (не мышечно-инвазивный рак мочевого пузыря), гепатому, рак груди и рак головы и шеи.
Другой вариант осуществления изобретения относится к лечению рака, выбранного из базальноклеточной карциномы, рака яичника, меланомы, не мышечно-инвазивного рака мочевого пузыря, рака груди и рака головы и шеи.
Другой вариант осуществления изобретения относится к лечению меланомы, рака желудочно-кишечного тракта, рака груди, рака яичников и рака головы и шеи.
Другой вариант осуществления изобретения касается лечения рака желудочно-кишечного тракта. Такие желудочно-кишечные виды рака включают рак ротовой полости, пищевода, желудка, желчевыделительной системы, поджелудочной железы, тонкой кишки, толстой кишки, прямой кишки и заднего прохода.
Другой вариант осуществления изобретения относится к лечению не мышечно-инвазивного рака мочевого пузыря.
Другой вариант осуществления изобретения относится к соединениям формулы (I) и любым их вариантам осуществления или их фармацевтически приемлемым солям для применения при лечении рака у субъекта, нуждающегося в этом.
Другой вариант осуществления изобретения относится к соединениям формулы (I) и любым их вариантам осуществления или их фармацевтически приемлемым солям для применения при лечении рака, где указанное лечение включает введение дополнительного терапевтического агента.
В другом аспекте изобретение относится к способу ингибирования пролиферации раковых клеток у субъекта, включающему введение субъекту соединения по изобретению или его фармацевтически приемлемой соли в количестве, эффективном для ингибирования пролиферации клеток.
В другом аспекте, изобретение относится к способу ингибирования инвазивности раковых клеток у субъекта, включающему введение субъекту соединения по изобретению или его фармацевтически приемлемой соли в количестве, эффективном для ингибирования инвазивности клеток.
В другом аспекте, в изобретении представлен способ индукции апоптоза в раковых клетках у субъекта, включающий введение субъекту соединения по изобретению или его фармацевтически приемлемой соли в количестве, эффективном для индукции апоптоза.
В другом аспекте, изобретение относится к способу ингибирования метастазирования раковых клеток у субъекта, включающему введение субъекту соединения по изобретению или его фармацевтически приемлемой соли в количестве, эффективном для ингибирования клеточного метастазирования.
В другом аспекте, изобретение относится к способу ингибирования ангиогенеза у субъекта, включающему введение субъекту соединения по изобретению или его фармацевтически приемлемой соли в количестве, эффективном для ингибирования ангиогенеза.
В другом аспекте изобретение относится к способу лечения рака у субъекта, нуждающегося в этом, включающему введение субъекту терапевтически эффективного количества соединения по изобретению или его фармацевтически приемлемой соли. Указанный способ также включает введение соединения по изобретению, по меньшей мере, с одним дополнительным терапевтическим агентом.
Изобретение также относится к способам профилактики инфекционного заболевания у субъекта, нуждающегося в этом, включающим введение фармацевтической композиции в количестве, достаточном для профилактики инфекционного заболевания у указанного субъекта. То есть, в некоторых вариантах осуществления, настоящее изобретение относится к профилактическим вакцинам. В некоторых вариантах осуществления, субъект-млекопитающее подвергается риску воздействия инфекционного агента. «Профилактика» инфекционного заболевания означает защиту субъекта от развития инфекционного заболевания. В некоторых вариантах осуществления, профилактика инфекционного заболевания дополнительно включает защиту субъекта от заражения инфекционным агентом (например, защиту субъекта от развития острой или хронической инфекции). Кроме того, в настоящем изобретении представлены способы облегчения симптома инфекционного заболевания у субъекта-млекопитающего, нуждающегося в этом, включающие введение фармацевтической композиции в количестве, достаточном для облегчения симптома инфекционного заболевания у указанного субъекта. То есть, в некоторых вариантах осуществления, настоящее изобретение относится к терапевтическим вакцинам. В некоторых вариантах осуществления, субъект остро или хронически инфицирован инфекционным агентом. Инфекционное заболевание может быть вирусным (например, вирусы гепатита, герпеса или папилломы человека), бактериальным, грибковым или паразитарным заболеванием. В некоторых вариантах осуществления, фармацевтическая композиция дополнительно содержит вирусный, бактериальный, грибковый или паразитарный антиген. «Облегчение» симптома инфекционного заболевания означает улучшение симптома, предпочтительно, уменьшение степени заболевания.
Терапевтические способы и использование
Изобретение дополнительно относится к терапевтическим способам и применениям, включающим введение соединений по изобретению или их фармацевтически приемлемых солей отдельно или в комбинации с другими терапевтическими агентами или паллиативными агентами.
В одном аспекте, изобретение представляет способ лечения аномального роста клеток у субъекта, нуждающегося в этом, включающему введение субъекту терапевтически эффективного количества соединения по изобретению или его фармацевтически приемлемой соли.
В другом аспекте, изобретение представляет способ лечения аномального роста клеток у субъекта, нуждающегося в этом, включающий введение субъекту некоторого количества соединения по изобретению или его фармацевтически приемлемой соли в сочетании с некоторым количеством дополнительного терапевтического агента (например, противоракового терапевтического агента), количество которых в совокупности является эффективным при лечении указанного аномального роста клеток.
В другом аспекте, изобретение представляет соединение по изобретению или его фармацевтически приемлемую соль для применения при лечении аномального роста клеток у субъекта.
В еще одном аспекте, изобретение представляет применение соединения по изобретению или его фармацевтически приемлемой соли для лечения аномального роста клеток у субъекта.
В другом аспекте, изобретение представляет фармацевтическую композицию для лечения аномального роста клеток у субъекта, нуждающегося в этом, где фармацевтическая композиция содержит соединение по настоящему изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель или эксципиент.
В другом аспекте, изобретение представляет соединение по изобретению или его фармацевтически приемлемую соль для применения в качестве лекарственного средства, в частности, лекарственного средства для лечения аномального роста клеток.
В еще одном аспекте, изобретение представляет применение соединения по изобретению или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения аномального роста клеток у субъекта.
В частых вариантах осуществления способов, представленных в настоящем документе, аномальным ростом клеток является рак. Соединения по изобретению могут быть введены в виде отдельных агентов, или могут быть введены в комбинации с другими противораковыми терапевтическими агентами, в частности, с агентами стандарта клинической практики, подходящими для конкретного вида рака.
В некоторых вариантах осуществления, представленные способы приводят к одному или нескольким из следующих эффектов: (1) ингибирование пролиферации раковых клеток; (2) ингибирование инвазивности раковых клеток; (3) индуцирование апоптоза раковых клеток; (4) ингибирование метастазирования раковых клеток; или (5) ингибирование ангиогенеза.
В некоторых вариантах осуществления, соединение по изобретению вводят в качестве терапии первой линии. В других вариантах осуществления, соединение по изобретению вводят в качестве терапии второй (или более поздней) линии.
Дозированные формы и схемы
На введение соединений по изобретению может влиять любой способ, который обеспечивает доставку соединений к месту действия. Эти способы включают пероральные пути, интрадуоденальные пути, парентеральную инъекцию (включая внутривенную, подкожную, внутримышечную, внутрисосудистую или инфузионную), местное и ректальное введение.
Схемы дозирования могут быть скорректированы для обеспечения оптимального желаемого ответа. Например, можно вводить один болюс, можно вводить несколько разделенных доз с течением времени или доза может быть пропорционально уменьшена или увеличена в соответствии с требованиями терапевтической ситуации. Особенно предпочтительно составлять парентеральные композиции в виде стандартной дозированной формы для простоты введения и однородности дозировки. Единичная дозированная форма, используемая в настоящем документе, относится к физически дискретным единицам, подходящим в качестве единичных доз для млекопитающих, подлежащих лечению; каждая единица содержит заранее определенное количество активного соединения, рассчитанное для получения желаемого терапевтического эффекта в сочетании с требуемым фармацевтическим носителем. Спецификация стандартных дозированных форм по изобретению диктуется и напрямую зависит от (а) уникальных характеристик химиотерапевтического агента и конкретного терапевтического или профилактического эффекта, который должен быть достигнут, и (b) ограничений, существующих в области составления композиций такого активного соединения для лечения чувствительности у индивидуумов.
Таким образом, специалисту в данной области техники будет понятно, основываясь на представленном в настоящем документе описании, что доза и схема дозирования корректируются в соответствии со способами, хорошо известными в области терапии. То есть, максимально переносимая доза может быть легко установлена, и также может быть определено эффективное количество, обеспечивающее определяемую терапевтическую пользу у пациента, а также временные требования для введения каждого агента для обеспечения определяемой терапевтической пользы у пациента. Соответственно, несмотря на то, что в настоящем документе приведены примеры определенных доз и схем введения, эти примеры никоим образом не ограничивают дозы и схемы введения, которые могут быть предоставлены пациенту при осуществлении настоящего изобретения.
Следует отметить, что значения дозировки могут варьироваться в зависимости от типа и тяжести состояния, подлежащего облегчению, и могут включать однократные или многократные дозы. Кроме того, следует понимать, что для любого конкретного субъекта конкретные схемы дозирования должны корректироваться с течением времени в соответствии с индивидуальными потребностями и профессиональным мнением лица, вводящего или контролирующего введение композиций, и что диапазоны дозировок, указанные в настоящем документе, являются только примерными и не предназначены для ограничения объема или практики заявленной композиции. Например, дозы можно корректировать на основе фармакокинетических или фармакодинамических параметров, которые могут включать клинические эффекты, такие как токсические эффекты и/или лабораторные показатели. Таким образом, настоящее изобретение охватывает индивидуальное повышение дозы, как определено специалистом в данной области техники. Определение подходящих дозировок и схем введения химиотерапевтического агента хорошо известно в соответствующей области техники, и специалисту в данной области техники должно быть понятно, что они охватываются, как только будут предоставлены идеи, описанные в настоящем документе.
Количество вводимого соединения по изобретению будет зависеть от субъекта, которого лечат, тяжести нарушения или состояния, скорости введения, расположения соединения и усмотрения лечащего врача. Однако эффективная доза находится в диапазоне от примерно 0,001 до примерно 100 мг на кг массы тела в сутки, предпочтительно, от примерно 1 до примерно 35 мг/кг/сутки, в виде однократной или разделенной дозы. Для человека массой 70 кг, это будет составлять от около 0,05 до около 7 г/сутки, предпочтительно от около 0,1 до около 2,5 г/сутки. В некоторых случаях, уровни доз ниже нижнего предела вышеуказанного диапазона могут быть более чем достаточными, в то время как в других случаях могут использоваться еще большие дозы, не вызывающие каких-либо вредных побочных эффектов, при условии, что такие большие дозы сначала разделяются на несколько малых доз для введения в течение суток.
Составы и пути введения
Используемый в настоящем документе термин «фармацевтически приемлемый носитель» относится к носителю или разбавителю, который не вызывает значительного раздражения организма и не отменяет биологическую активность и свойства вводимого соединения.
Фармацевтически приемлемый носитель может включать любой обычный фармацевтический носитель или эксципиент. Выбор носителя и/или эксципиента будет в значительной степени зависеть от таких факторов, как конкретный способ введения, влияние носителя или эксципиента на растворимость и стабильность, и природа дозированной формы.
Подходящие фармацевтические носители включают инертные разбавители или наполнители, воду и различные органические растворители (такие как гидраты и сольваты). Фармацевтические композиции могут, при желании, содержать дополнительные ингредиенты, такие как ароматизаторы, связующие вещества, эксципиенты и подобные. Таким образом, для перорального введения, таблетки, содержащие различные эксципиенты, такие как лимонная кислота, могут использоваться вместе с различными разрыхлителями, такими как крахмал, альгиновая кислота и некоторые сложные силикаты, и со связующими агентами, такими как сахароза, желатин и аравийская камедь. Примеры эксципиентов включают, без ограничения, карбонат кальция, фосфат кальция, различные сахара и типы крахмала, производные целлюлозы, желатин, растительные масла и полиэтиленгликоли. Кроме того, для таблетирования часто используют смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые композиции аналогичного типа также можно использовать в мягких и твердых желатиновых капсулах с наполнителем. Таким образом, неограничивающие примеры материалов включают лактозу или молочный сахар и полиэтиленгликоли с высокой молекулярной массой. Когда для перорального введения желательны водные суспензии или эликсиры, содержащееся в них активное соединение можно комбинировать с различными подсластителями или ароматизаторами, красителями или пигментами и, при желании, с эмульгаторами или суспендирующими агентами вместе с разбавителями, такими как вода, этанол, пропиленгликоль, глицерин или их комбинации.
Фармацевтическая композиция может быть, например, в форме, пригодной для перорального введения, в виде таблетки, капсулы, пилюли, порошка, составов с замедленным высвобождением, суспензии в растворе, для парентерального введения, в виде стерильного раствора, суспензии или эмульсии, для местного введения, в виде мази или крема, или для ректального введения, в виде суппозиториев.
Типовые формы для парентерального введения включают растворы или суспензии активных соединений в стерильных водных растворах, например, водных растворах пропиленгликоля или декстрозы. При желании такие дозированные формы могут быть соответствующим образом забуферены.
Фармацевтическая композиция может быть представлена в виде стандартных дозированных форм, пригодных для однократного введения точных дозировок.
Фармацевтические композиции, подходящие для доставки соединений по изобретению, и способы их получения будут очевидны специалистам в данной области. Такие композиции и способы их получения можно найти, например, в «Remington’s Pharmaceutical Sciences», 19th Edition (Mack Publishing Company, 1995), описание которого полностью включено в настоящее описание посредством ссылки.
Соединения по изобретению можно вводить перорально. Пероральное введение может включать проглатывание, так что соединение попадает в желудочно-кишечный тракт, или может применяться трансбуккальное или подъязычное введение, при котором соединение попадает в кровоток непосредственно изо рта.
Составы, подходящие для перорального введения, включают твердые составы, такие как таблетки, капсулы, содержащие порошки, жидкости или порошки, пастилки (включая наполненные жидкостью), жевательные таблетки, мульти- и наночастицы, гели, твердый раствор, липосомы, пленки (включая мукоадгезив), капсулы, спреи и жидкие составы.
Жидкие составы включают суспензии, растворы, сиропы и эликсиры. Такие составы могут быть использованы в качестве наполнителей в мягких или твердых капсулах и обычно включают носитель, например воду, этанол, полиэтиленгликоль, пропиленгликоль, метилцеллюлозу или подходящее масло, и один или несколько эмульгаторов и/или суспендирующих агентов. Жидкие составы также могут быть приготовлены восстановлением твердого вещества, например, из пакетика.
Соединения по изобретению также можно использовать в быстро растворяющихся и быстро распадающихся дозированных формах, таких как формы, описанные в Expert Opinion in Therapeutic Patents, 11 (6), 981 986 by Liang and Chen (2001), описание которых включено в настоящий документ посредством ссылки полностью.
Для таблетированных дозированных форм, в зависимости от дозы, лекарственное средство может составлять от 1% масс. до 80% масс. дозированной формы, более типично, от 5% масс. до 60% масс. дозированной формы. В дополнение к лекарственному средству, таблетки обычно содержат разрыхлитель. Примеры разрыхлителей включают гликолят крахмала натрия, карбоксиметилцеллюлозу натрия, карбоксиметилцеллюлозу кальция, кроскармеллозу натрия, кросповидон, поливинилпирролидон, метилцеллюлозу, микрокристаллическую целлюлозу, гидроксипропилцеллюлозу, замещенную низшим алкилом, крахмал, прежелатинизированный крахмал и альгинат натрия. Как правило, разрыхлитель составляет от 1% масс. до 25% масс, предпочтительно, от 5% масс. до 20% масс. дозированной формы.
Связующие вещества обычно используют для придания когезивных свойств составу для таблеток. Подходящие связующие вещества включают микрокристаллическую целлюлозу, желатин, сахара, полиэтиленгликоль, натуральные и синтетические камеди, поливинилпирролидон, прежелатинизированный крахмал, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу. Таблетки также могут содержать разбавители, такие как лактоза (моногидрат, высушенный распылением моногидрат, безводный и подобные), маннит, ксилит, декстроза, сахароза, сорбит, микрокристаллическая целлюлоза, крахмал и дигидрат двухосновного фосфата кальция.
Таблетки также могут необязательно включать поверхностно-активные вещества, такие как лаурилсульфат натрия и полисорбат 80, и глиданты, такие как диоксид кремния и тальк. Поверхностно-активные агенты, если они присутствуют, обычно находятся в количестве от 0,2% масс. до 5% масс. таблетки, и глиданты обычно составляют от 0,2% масс. до 1% масс. таблетки.
Таблетки также обычно содержат смазывающие агенты, такие как стеарат магния, стеарат кальция, стеарат цинка, стеарилфумарат натрия и смеси стеарата магния с лаурилсульфатом натрия. Смазывающие вещества обычно присутствуют в количествах от 0,25% масс. до 10% масс., предпочтительно, от 0,5% масс. до 3% масс. таблетки.
Другие традиционные ингредиенты включают антиоксиданты, красители, ароматизаторы, консерванты и вещества, маскирующие вкус.
Типовые таблетки содержат до примерно 80% масс. лекарственного средства, от примерно 10% масс. до примерно 90% масс. связующего агента, от примерно 0% масс. до примерно 85% масс. разбавителя, от примерно 2% масс. до примерно 10% масс. разрыхлителя и от примерно 0,25% масс. до примерно 10% масс. смазывающего агента.
Смеси для таблеток могут быть спрессованы напрямую или с помощью валика для образования таблеток. Смеси для таблеток или части смесей альтернативно могут быть влажными, сухими или гранулированными из расплава, затвердевшими из расплава или экструдированными перед таблетированием. Окончательный состав может включать один или несколько слоев и может быть с покрытием или без покрытия; или инкапсулированным.
Состав таблеток подробно обсуждается в «Pharmaceutical Dosage Forms: Tablets, Vol. 1», by H. Lieberman and L. Lachman, Marcel Dekker, N.Y., N.Y., 1980 (ISBN 0 8247 6918 X), описание которого полностью включено в настоящее описание посредством ссылки.
Твердые составы для перорального введения могут быть приготовлены для немедленного и/или модифицированного высвобождения. Составы с модифицированным высвобождением включают отсроченное, пролонгированное, импульсное, контролируемое, таргетное и запрограммированное высвобождение.
Подходящие составы с модифицированным высвобождением описаны в патенте США № 6,106,864. Подробности других подходящих технологий высвобождения, таких как высокоэнергетические дисперсии и осмотические частицы и частицы с покрытием, можно найти в Verma et al, Pharmaceutical Technology On line, 25(2), 1 14 (2001). Использование жевательной резинки для достижения контролируемого высвобождения описано в WO 00/35298. Описание этих ссылок включено в настоящий документ посредством ссылки во всей своей полноте.
Соединения по изобретению можно также вводить непосредственно в кровоток, в мышцу или во внутренний орган. Подходящие средства для парентерального введения включают внутривенное, внутриартериальное, внутрибрюшинное, подоболочечное, внутрижелудочковое, внутриуретральное, интрастернальное, внутричерепное, внутримышечное и подкожное введение. Подходящие устройства для парентерального введения включают игольчатые (включая микроигольчатые) инъекторы, безыгольные инъекторы и методы инфузии.
Составами для парентерального введения обычно являются водные растворы, которые могут содержать эксципиенты, такие как соли, углеводы и буферные агенты (предпочтительно, с pH от 3 до 9), но для некоторых применений они могут быть более подходящим образом составлены в виде стерильного не водного раствора или в виде высушенной формы для использования в сочетании с подходящим носителем, таким как стерильная апирогенная вода.
Приготовление парентеральных составов в стерильных условиях, например, путем лиофилизации, может быть легко осуществлено с использованием стандартных фармацевтических методик, хорошо известных специалистам в данной области техники.
Растворимость соединений по изобретению, используемых при приготовлении парентеральных растворов, может быть увеличена за счет использования соответствующих методов составления, таких как включение агентов, повышающих растворимость.
Составы для парентерального введения могут быть составлены для немедленного и/или модифицированного высвобождения. Составы с модифицированным высвобождением включают отсроченное, пролонгированное, импульсное, контролируемое, таргетное и запрограммированное высвобождение. Таким образом, соединения по изобретению могут быть составлены в виде твердой, полутвердой или тиксотропной жидкости для введения в виде имплантированного депо, обеспечивающего модифицированное высвобождение активного соединения. Примеры таких составов включают стенты, покрытые лекарственным средством, и микросферы PGLA.
Соединения по изобретению можно также вводить местно на кожу или слизистую оболочку, то есть дермально или трансдермально. Типовые составы для этой цели включают гели, гидрогели, лосьоны, растворы, кремы, мази, присыпки, повязки, пены, пленки, кожные пластыри, облатки, имплантаты, губки, волокна, бинты и микроэмульсии. Можно также использовать липосомы. Типовые носители включают спирт, воду, минеральное масло, жидкий вазелин, белый вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. Могут быть включены усилители проникновения; см., например, J Pharm Sci, 88 (10), 955 958 by Finnin and Morgan (October 1999). Другие способы местного введения включают доставку посредством электропорации, ионтофореза, фонофореза, сонофореза и инъекций с микроиглами или без игл (например, Powderject™, Bioject™ и т.д.). Описания этих ссылок включено в настоящий документ посредством ссылки во всей полноте.
Составы для местного введения могут быть составлены для немедленного и/или модифицированного высвобождения. Составы с модифицированным высвобождением включают отсроченное, пролонгированное, импульсное, контролируемое, таргетное и запрограммированное высвобождение.
Соединения по изобретению можно также вводить интраназально или путем ингаляции, как правило, в виде сухого порошка (либо отдельно, либо в виде смеси, например, в сухой смеси с лактозой, либо в виде частиц смешанного компонента, например, в смеси с фосфолипидами, такими как фосфатидилхолин) из ингалятора для сухого порошка или в виде аэрозольного спрея из баллона под давлением, насоса, спрея, распылителя (предпочтительно распылителя, использующего электрогидродинамику для получения мелкодисперсного тумана) или небулайзера, с использованием или без использования подходящего пропеллента, такого как 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3-гептафторпропан. Для интраназального применения, порошок может включать биоадгезив, например, хитозан или циклодекстрин.
Контейнер под давлением, насос, спрей, распылитель или небулайзер содержат раствор или суспензию соединения по изобретению, содержащую, например, этанол, водный этанол или подходящий альтернативный агент для диспергирования, солюбилизации или пролонгированного высвобождения активного агента, пропелленты в качестве растворителя и необязательное поверхностно-активное вещество, такое как сорбитантриолеат, олеиновая кислота или олигомолочная кислота.
Перед применением в составе сухого порошка или суспензии, лекарственный продукт микронизируют до размера, подходящего для доставки путем ингаляции (обычно менее 5 микрон). Это может быть достигнуто с помощью любого подходящего метода измельчения, такого как спирально-струйное измельчение, струйное измельчение в псевдоожиженном слое, обработка сверхкритической жидкостью с образованием наночастиц, гомогенизация под высоким давлением или сушка распылением.
Капсулы (изготовленные, например, из желатина или HPMC), блистеры и картриджи для использования в ингаляторе или инсуффляторе могут быть составлены таким образом, чтобы содержать порошковую смесь соединения по изобретению, подходящую порошковую основу, такую как лактоза или крахмал, и модификатор производительности, такой как лейцин, маннит или стеарат магния. Лактоза может быть безводной или в форме моногидрата, предпочтительно, последнее. Другие подходящие эксципиенты включают декстран, глюкозу, мальтозу, сорбит, ксилит, фруктозу, сахарозу и трегалозу.
Подходящий состав раствора для использования в распылителе, использующем электрогидродинамику для получения мелкодисперсного тумана, может содержать от 1 мкг до 20 мг соединения по изобретению на одно нажатие, и рабочий объем может варьироваться от 1 мкл до 100 мкл. Типовой состав включает соединение по изобретению, пропиленгликоль, стерильную воду, этанол и хлорид натрия. Альтернативные растворители, которые можно использовать вместо пропиленгликоля, включают глицерин и полиэтиленгликоль.
Подходящие ароматизаторы, такие как ментол и левоментол, или подсластители, такие как сахарин или сахарин натрия, могут быть добавлены к тем составам по изобретению, которые предназначены для ингаляционного/интраназального введения.
Составы для ингаляционного/интраназального введения могут быть составлены для немедленного и/или модифицированного высвобождения с использованием, например, поли(DL-молочнокислой согликолевой кислоты (PGLA). Составы с модифицированным высвобождением включают отсроченное, пролонгированное, пульсирующее, контролируемое, таргетное и запрограммированное высвобождение.
В случае ингаляторов с сухим порошком и аэрозолей, единица дозировки определяется с помощью клапана, который подает отмеренное количество. Единицы в соответствии с изобретением обычно устроены так, чтобы вводить отмеренную дозу или «впрыскивание», содержащее желаемое количество соединения по изобретению. Общая суточная доза может быть введена в виде разовой дозы или, чаще, в виде разделенных доз в течение суток.
Соединения по изобретению можно вводить ректально или вагинально, например, в форме суппозитория, пессария или клизмы. Какао-масло является традиционной основой для суппозиториев, но при необходимости могут использоваться различные альтернативы.
Составы для ректального/вагинального введения могут быть составлены для немедленного и/или модифицированного высвобождения. Составы с модифицированным высвобождением включают отсроченное, пролонгированное, импульсное, контролируемое, таргетное и запрограммированное высвобождение.
Соединения по изобретению можно также вводить непосредственно в глаз или ухо, как правило, в форме капель микронизированной суспензии или раствора в изотоническом стерильном солевом растворе с отрегулированным рН. Другие составы, подходящие для глазного и ушного введения, включают мази, биоразлагаемые (например, рассасывающиеся гелевые губки, коллаген) и не биоразлагаемые (например, силиконовые) имплантаты, пластины, линзы и системы частиц или везикул, такие как ниосомы или липосомы. Полимер, такой как поперечно-сшитая полиакриловая кислота, поливиниловый спирт, гиалуроновая кислота, целлюлозный полимер, например, гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза или метилцеллюлоза, или гетерополисахаридный полимер, например, гелановая камедь, может быть включен вместе с консервантом, таким как хлорид бензалкония. Такие составы также могут быть доставлены с помощью ионофореза.
Составы для глазного/ушного введения могут быть составлены для немедленного и/или модифицированного высвобождения. Композиции с модифицированным высвобождением включают отсроченное, пролонгированное, импульсное, контролируемое, таргетное или запрограммированное высвобождение.
Другие технологии
Соединения по изобретению можно комбинировать с растворимыми макромолекулярными соединениями, такими как циклодекстрин и его подходящие производные, или полимеры, содержащие полиэтиленгликоль, для улучшения их растворимости, скорости растворения, маскировки вкуса, биодоступности и/или стабильности для применения в любом из вышеупомянутых путей введения.
Например, обнаружено, что лекарственные комплексы циклодекстрина обычно применимы для большинства дозированных форм и путей введения. Могут быть использованы как комплексы включения, так и не включения. В качестве альтернативы прямому комплексообразованию с лекарственным средством, циклодекстрин можно использовать в качестве вспомогательной добавки, т.е. в качестве носителя, разбавителя или солюбилизатора. Наиболее часто для этих целей используют альфа, бета и гамма циклодекстрины, примеры которых можно найти в публикациях РСТ №№ WO 91/11172, WO 94/02518 и WO 98/55148, описание которых включено в настоящий документ посредством ссылки полностью.
Количество вводимого активного соединения будет зависеть от субъекта, которого лечат, тяжести нарушения или состояния, скорости введения, расположения соединения и усмотрения лечащего врача. Однако, эффективная доза обычно находится в диапазоне от примерно 0,001 до примерно 100 мг на кг массы тела в сутки, и часто от примерно 0,01 до примерно 35 мг/кг/сутки в виде однократной или разделенной дозы. Для человека массой 70 кг это будет составлять от примерно 0,07 мг/сутки до примерно 7000 мг/сутки, чаще примерно от 10 мг/сутки до примерно 1000 мг/сутки. Иногда дозировка составляет около 10, 20, 30, 40, 50, 60, 75, 100, 125, 150, 175, 200, 225, 250, 275, 300, 325, 350, 375, 400, 425, 450, 475, 500, 525, 550, 575, 600, 625, 650, 675, 700, 750, 800, 900 или 1000 мг/сутки. Иногда дозировка составляет от примерно 10 мг/сутки до примерно 1000 мг/сутки, от примерно 10 мг/сутки до примерно 750 мг/сутки, от примерно 10 мг/сутки до примерно 600 мг/сутки, от примерно 10 мг/сутки до примерно 300 мг/сутки, от примерно 10 мг/сутки до примерно 150 мг/сутки, от примерно 20 мг/сутки до примерно 750 мг/сутки, от примерно 20 мг/сутки до примерно до 600 мг/сутки, от примерно 20 мг/сутки до 330 мг/сутки, от примерно 20 мг/сутки до примерно 150 мг/сутки, от примерно 50 мг/сутки до примерно 750 мг/сутки, от примерно 50 мг/сутки до примерно 600 мг/сутки, от примерно 50 мг/сутки до примерно 300 мг/сутки, от примерно 50 мг/сутки до примерно 150 мг/сутки, от примерно 75 мг/сутки до примерно 750 мг/сутки, от примерно 75 мг/сутки до примерно 600 мг/сутки, от примерно 75 мг/сутки до примерно 300 мг/сутки или от примерно 75 мг/сутки до примерно 150 мг/сутки.
В некоторых случаях, уровни дозировки ниже нижнего предела вышеуказанного диапазона могут быть более чем достаточными, в то время как в других случаях могут использоваться еще большие дозы, не вызывающие каких-либо вредных побочных эффектов, при этом такие большие дозы обычно делятся на несколько меньших доз для введения в течение суток.
Составный набор
Поскольку может быть желательным введение комбинации активных соединений, например, с целью лечения конкретного заболевания или состояния, в объем настоящего изобретения входит вариант, в котором две или несколько фармацевтических композиций, по меньшей мере, одна из которых содержит соединение по изобретению, могут быть удобно объединены в виде набора, подходящего для совместного введения композиций. Таким образом, набор по изобретению включает две или несколько отдельных фармацевтических композиций, по меньшей мере, одна из которых содержит соединение по изобретению, и средства для раздельного хранения указанных композиций, такие как контейнер, разделенная бутылка или разделенный пакет из фольги. Примером такого набора является известная блистерная упаковка, используемая для упаковки таблеток, капсул и подобных.
Набор по изобретению особенно подходит для введения различных дозированных форм, например пероральных и парентеральных, для введения отдельных композиций с различными интервалами дозирования, или для титрования отдельных композиций относительно друг друга. Для облегчения соблюдения схемы, набор обычно включает инструкции по применению и может быть снабжен запоминающим устройством.
Комбинированная терапия
Используемый в настоящем документе термин «комбинированная терапия» относится к введению соединения по изобретению вместе с, по меньшей мере, одним дополнительным фармацевтическим или лекарственным агентом (например, противораковым терапевтическим агентом) либо последовательно, либо одновременно.
Как отмечено в настоящем документе, соединения по изобретению можно использовать в комбинации с одним или несколькими дополнительными противораковыми терапевтическими агентами. Эффективность соединений по изобретению при некоторых опухолях может быть повышена путем комбинирования с другими одобренными или экспериментальными способами лечения рака, например, радиационной терапией, хирургией, химиотерапевтическими агентами, таргетными терапиями, агентами, которые ингибируют другие сигнальные пути, которые разрегулированы в опухолях, и другими агентами, усиливающими иммунитет, такими как антагонисты PD 1 или PD L1 и подобными.
При использовании комбинированной терапии, один или несколько дополнительных противораковых терапевтических агентов можно вводить последовательно или одновременно с соединением по изобретению. В одном варианте осуществления, дополнительное противораковое терапевтическое средство вводят млекопитающему (например, человеку) перед введением соединения по изобретению. В другом варианте осуществления, дополнительный противораковый терапевтический агент вводят млекопитающему после введения соединения по изобретению. В другом варианте осуществления, дополнительное противораковое терапевтическое средство вводят млекопитающему (например, человеку) одновременно с введением соединения по изобретению.
Изобретение также относится к фармацевтической композиции для лечения аномального роста клеток у млекопитающего, включая человека, которая содержит количество соединения по изобретению, как определено выше (включая гидраты, сольваты и полиморфы указанного соединения или их фармацевтически приемлемые соли) в комбинации с одним или несколькими (предпочтительно, от одного до трех) дополнительными противораковыми терапевтическими агентами.
Классы дополнительных химиотерапевтических агентов, которые можно вводить в комбинации с соединением по настоящему изобретению, включают, но не ограничены ими: алкилирующие агенты, антиметаболиты, ингибиторы киназы, растительные алкалоиды веретенного яда, цитотоксические/противоопухолевые антибиотики, ингибиторы тоизомеразы, фотосенсибилизаторы, антиэстрогены и селективные модуляторы эстрогеновых рецепторов (SERM), антипрогестероны, негативные регуляторы эстрогеновых рецепторов (ERD), антагонисты эстрогеновых рецепторов, агонисты рилизинг-гормона лютеинизирующего гормона; агонист рецептора IL-2 (рекомбинантные цитокины или агонисты цитокиновых рецепторов); и антисмысловые олигонуклеотиды или производные олигонуклеотидов, которые ингибируют экспрессию генов, вовлеченных в аномальную пролиферацию клеток или рост опухоли.
Другие дополнительные химиотерапевтические агенты включают не только таксаны или платиновые агенты, но и HER2-таргетные агенты, например, трастузумаб.
В другом варианте осуществления, такие дополнительные противораковые терапевтические агенты включают соединения, полученные из следующих классов: ингибиторы митоза, алкилирующие агенты, антиметаболиты, противоопухолевые антибиотики, антиангиогенные агенты, ингибиторы топоизомеразы I и II, растительные алкалоиды, растительные алкалоиды веретенного яда, ингибиторы KRAS; ингибиторы МСТ4; ингибиторы MAT2a; ингибиторы alk/c-Met/ROS (включая кризотиниб или лорлатиниб); ингибиторы mTOR (включая темсиролимус или гедатолисиб); ингибиторы src/abl (включая бозутиниб); ингибиторы циклинзависимой киназы (CDK) (включая палбоциклиб, PF-06873600); ингибиторы erb (включая дакомитиниб); ингибиторы PARP (включая талазопариб); ингибиторы SMO (в т.ч. гласдегиб); ингибиторы EGFR T790M; ингибиторы PRMT5; ингибиторы TGFβR1; ингибиторы фактора роста; ингибиторы клеточного цикла, модификаторы биологического ответа; ингибиторы ферментов; и цитотоксические агенты.
В другом варианте осуществления, такие дополнительные противораковые терапевтические агенты включают соединения, полученные из антиангиогенного агента, включая, например, ингибиторы тирозинкиназы/рецептора фактора роста эндотелия сосудов (VEGF) (VEGFR) (включая сунитиниб, акситиниб, сорафениб и тивозаниб), ингибиторы TIE-2, ингибиторы PDGFR, ингибиторы ангиопоэтина, ингибиторы PKCβ, ингибиторы COX-2 (циклооксигеназы II), интегрины (альфа-v/бета-3), ингибиторы ММР-2 (матричной металлопротеиназы 2) и ингибиторы ММР-9 (матричной металлопротеиназы 9). Предпочтительные антиангиогенные агенты включают сунитиниб (Sutent™), бевацизумаб (Avastin™), акситиниб (Inlyta™), SU 14813 (Pfizer) и AG 13958 (Pfizer). Дополнительные антиангиогенные агенты включают ваталаниб (CGP 79787), пегаптаниб октанатрий (Macugen™), вандетаниб (Zactima™), PF-0337210 (Pfizer), SU 14843 (Pfizer), AZD 2171 (AstraZeneca), ранибизумаб (Lucentis™), Neovastat™ (AE 941), тетратиомолибдата (Coprexa™), AMG 706 (Amgen), VEGF Trap (AVE 0005), CEP 7055 (Sanofi-Aventis), XL 880 (Exelixis), телатиниб (BAY 57-9352) и CP-868,596 (Pfizer). Другие антиангиогенные агенты включают энзастаурин (LY 317615), мидостаурин (CGP 41251), перифозин (KRX 0401), тепренон (Selbex™) и UCN 01 (Kyowa Hakko). Другие примеры антиангиогенных агентов включают целекоксиб (Celebrex™), парекоксиб (Dynastat™), деракоксиб (SC 59046), люмиракоксиб (Preige™), вальдекоксиб (Bextra™), рофекоксиб (Vioxx™), игуратимод (Careram™), IP 751 (Invedus), SC-58125 (Pharmacia) и эторикоксиб (Arcoxia™). Другие антиангиогенные агенты включают эксисулинд (Aptosyn™), сальсалат (Amigesic™), дифлунисал (Dolobid™), ибупрофен (Motrin™), кетопрофен (Orudis™), набуметон (Relafen™), пироксикам (Feldene™), напроксен (Aleve™, Naprosyn™), диклофенак (Voltaren™), индометацин (Indocin™), сулиндак (Clinoril™), толметин (Tolectin™), этодолак (Lodine™), кеторолак (Toradol™) и оксапрозин (Daypro™). Другие антиангиогенные агенты включают ABT 510 (Abbott), апратастат (TMI 005), AZD 8955 (AstraZeneca), инциклинид (Metastat™) и PCK 3145 (Procyon). Другие антиангиогенные агенты включают ацитретин (Neotigason™), плитидепсин (aplidine™), циленгтид (EMD 121974), комбретастатин А4 (CA4P), фенретинид (4 HPR), галофугинон (Tempostatin™), Panzem™ (2-метоксиэстрадиол), PF-03446962 (Pfizer), ребимастат (BMS 275291), катумаксомаб (Removab™), леналидомид (Revlimid™), скваламин (EVIZON™), талидомид (Thalomid™), Ukrain™ (NSC 631570), Vitaxin™ (MEDI 522) и золедроновую кислоту (Zometa™).
В другом варианте осуществления, такие дополнительные противораковые терапевтические агенты включают соединения, полученные из гормональных агентов и антагонистов. Примеры включают, когда антигормональные агенты регулируют или ингибируют действие гормонов на опухоли, такие как антиэстрогены и селективные модуляторы рецепторов эстрогена (SERM), и селективные деструкторы рецепторов эстрогена (SERD), включая тамоксифен, ралоксифен, дролоксифен, 4-гидрокситамоксифен, триоксифен, кеоксифен, LY117018, онапристон, торемифен (Фарестон) и фулвестрант. Примеры также включают ингибиторы ароматазы, которые ингибируют фермент ароматазу, который регулирует выработку эстрогена в надпочечниках, и включают такие соединения, как 4(5)-имидазолы, аминоглютетимид, мегестрола ацетат, экземестан, форместан, фадрозол, ворозол, летрозол и анастрозол; и антиандрогены, такие как флутамид, нилутамид, бикалутамид, лейпролид, флуридил, апалутамид, энзалутамид, циметидин и гозерелин.
В другом варианте осуществления такие дополнительные противораковые терапевтические агенты включают соединения, полученные из ингибиторов сигнальной трансдукции, таких как ингибиторы протеинтирозинкиназ и/или серин/треонинкиназ: ингибитор сигнальной трансдукции (например, ингибирующий средства, посредством которых регуляторные молекулы, управляющие основными процессами клеточного роста, дифференциации и выживания, коммуницируют внутри клетки). Ингибиторы сигнальной трансдукции включают малые молекулы, антитела и антисмысловые молекулы. Ингибиторы сигнальной трансдукции включают, например, ингибиторы киназы (например, ингибиторы тирозинкиназы или ингибиторы серин/треонинкиназы) и ингибиторы клеточного цикла. Более конкретно, ингибиторы сигнальной трансдукции включают, например, ингибиторы фарнезилпротеинтрансферазы, ингибитор EGF, ErbB-1 (EGFR), ErbB-2, pan erb, ингибиторы IGF1R, MEK (включая биниметиниб (Mektovi™)), ингибиторы c-Kit, ингибиторы FLT-3, ингибиторы K-Ras, ингибиторы PI3 киназы, ингибиторы JAK, ингибиторы STAT, ингибиторы Raf киназы, BRAF (включая энкорафениб (Braftovi™)), ингибиторы Akt, ингибитор mTOR, ингибиторы P70S6 киназы, ингибиторы пути WNT и мультитаргетные ингибиторы киназы.
В другом варианте осуществления, такие дополнительные противораковые терапевтические агенты включают доцетаксел, паклитаксел, паклитаксел, связанные с белком частицы, цисплатин, карбоплатин, оксалиплатин, капецитабин, гемцитабин или винорелбин.
В другом варианте осуществления, такие дополнительные противораковые терапевтические агенты включают соединения, полученные из эпигенетического модулятора, примеры которых включают ингибитор EZH2 (включая PF-06821497), SMARCA4, PBRM1, ARID1A, ARID2, ARID1B, DNMT3A, TET2, MLL1/2/3, NSD1/2, SETD2, BRD4, DOT1L, HKMTsanti, PRMT1-9, LSD1, UTX, IDH1/2 или BCL6.
В другом варианте осуществления, такие дополнительные противораковые терапевтические агенты включают соединения, которые являются иммуноонкологическими агентами, включая иммуномодулирующие агенты.
В другом варианте осуществления, рассматриваются комбинации с образраспознающими рецепторами (PRR). PRR являются рецепторами, которые экспрессируются клетками иммунной системы и которые распознают различные молекулы, ассоциированные с патогенами и/или повреждением или гибелью клеток. PRR участвуют как во врожденном иммунном ответе, так и в адаптивном иммунном ответе. Агонисты PRR можно использовать для стимуляции иммунного ответа у субъекта. Существует несколько классов молекул PRR, включая toll-подобные рецепторы (TLR), RIG-I-подобные рецепторы (RLR), рецепторы, подобные нуклеотидсвязывающему домену олигомеризации (NOD) (NLR), лектиновые рецепторы С-типа (CLR) и белок стимулятор генов интерферона (STING).
Белок STING функционирует и как датчик цитозольной ДНК и как адаптерный белок в передаче сигналов интерферона 1 типа. Термины «STING» и «стимулятор генов интерферона» относятся к любой форме белка STING, а также к вариантам, изоформам и видовым гомологам, сохраняющим, по меньшей мере, часть активности STING. Если не указано иное, например, посредством конкретной ссылки на STING человека, STING включает все виды млекопитающих с нативной последовательностью STING, например, STING человека, обезьяны и мыши, также известный как - TMEM173.
«Агонист STING», используемый в настоящем документе, означает любую молекулу, которая при связывании со STING (1) стимулирует или активирует STING, (2) усиливает, увеличивает, способствует, индуцирует или продлевает активность, функцию или присутствие STING, или (3) усиливает, повышает, способствует или индуцирует экспрессию STING. Агонисты STING, применимые в любом способе лечения, лекарственных средствах и применениях по настоящему изобретению, включают, например, лиганды нуклеиновой кислоты, которые связывают STING.
Примеры агонистов STING, которые можно использовать в способах лечения, лекарственных средствах и применениях по настоящему изобретению, включают различные иммуностимулирующие нуклеиновые кислоты, такие как синтетическая двухцепочечная ДНК, циклический ди-GMP, циклический GMP-AMP (cGAMP), синтетические циклические динуклеотиды (CDN), такие как MK-1454 и ADU-S100 (MIW815), и малые молекулы, такие как WO2019027858, WO20180093964, WO2017175156, WO2017175147.
Терапевтические антитела могут обладать специфичностью против множества различных антигенов. Например, терапевтические антитела могут быть направлены на опухолеассоциированный антиген, так что связывание антитела с антигеном способствует гибели клетки, экспрессирующей антиген. В другом примере, терапевтические антитела могут быть направлены на антиген на иммунной клетке, так что связывание антитела предотвращает снижение активности клетки, экспрессирующей антиген (и тем самым способствует активности клетки, экспрессирующей антиген). В некоторых ситуациях, терапевтическое антитело может функционировать с помощью нескольких различных механизмов (например, оно может как i) способствовать гибели клеток, экспрессирующих антиген, так и ii) предотвращать снижение антигеном активности иммунных клеток при контакте с клеткой, экспрессирующей антиген).
В другом варианте осуществления, такие дополнительные противораковые терапевтические агенты включают антитела, которые будут блокирующими или ингибирующиими на мишени: CTLA-4 (включая ипилимумаб или тремелимумаб), PD-1 или PD-L1 (включая атезолизумаб, авелумаб, цемиплимаб, дурвалумаб, ниволумаб или пембролизумаб), LAG-3, TIM-3 или TIGIT.
В другом варианте осуществления, такие дополнительные противораковые терапевтические агенты включают антитела, которые являются агонистами 4-1BB, OX40, GITR, ICOS или CD40.
В другом варианте осуществления, противораковой терапией может быть терапия CAR-T-клетками.
Примеры терапевтического антитела включают: анти-OX40 антитело, анти-4-1BB антитело, анти-HER2 антитело (включая конъюгат анти-HER2 антитело-лекарственное средство (ADC)), биспецифическое анти-CD47/анти-PD-L1 антитело и биспецифическое анти-P-кадгерин/анти-CD3 антитело. Примеры цитотоксических агентов, которые могут быть включены в ADC, включают антрациклин, ауристатин, доластатин, комбретастатин, дуокармицин, димер пирролобензодиазепина, димер индолино-бензодиазепина, энедиин, гелданамицин, майтанзин, пуромицин, таксан, алкалоид барвинка, камптотецин, тубулизин, гемиастерлин, сплайcеостатин, пладиенолид и их стереоизомеры, изостеры, аналоги или производные. Типовые иммуномодулирующие агенты, которые могут быть включены в ADC, включают ганцикловир, этанерцепт, такролимус, сиролимус, воклоспорин, циклоспорин, рапамицин, циклофосфамид, азатиоприн, микофенолгат мофетил, метотрекстрат, глюкокортикоид и его аналоги, цитокины, факторы роста стволовых клеток, лимфотоксины, фактор некроза опухоли (TNF), гемопоэтические факторы, интерлейкины (например, интерлейкин-1 (IL-1), IL-2, IL-3, IL-6, IL-10, IL-12, IL-15, IL-18 и IL-21), колониестимулирующие факторы (например, гранулоцитарный колониестимулирующий фактор (G-CSF) и гранулоцитарно-макрофагальный колониестимулирующий фактор (GM-CSF)), интерфероны (например, интерфероны-альфа, -бета и -гамма), фактор роста стволовых клеток, обозначенный как «фактор S1», эритропоэтин и тромбопоэтин или их комбинацию.
Дополнительные примеры терапевтических антител могут включать следующие антигены, где типовые антитела, направленные на антиген, также включены ниже (в квадратных скобках/скобках после антигена). Антигены, указанные ниже, также могут упоминаться в настоящем документе как «антигены-мишени» или подобные. Антигены-мишени для терапевтических антител в данном документе включают, например: 4-1BB (например, утомилумаб); 5Т4; А33; альфа-фолатный рецептор 1 (например, мирветуксимаб соравтанзин); Alk-1; BCMA [например, см. US9969809]; BTN1A1 (например, см. WO2018222689); CA-125 (например, абаговомаб); карбоангидраза IX; CCR2; CCR4 (например, могамулизумаб); CCR5 (например, леронлимаб); CCR8; CD3 [например, блинатумомаб (CD3/CD19 биспецифический), CD3/P-кадгерин биспецифический, CD3/BCMA биспецифический] CD19 (например, блинатумомаб, MOR208); CD20 (например, ибритумомаб тиуксетан, обинутузумаб, офатумумаб, ритуксимаб, ублитуксимаб); CD22 (инотузумаб озогамицин, моксетумомаб пазудотокс); CD25; CD28; CD30 (например, брентуксимаб ведотин); CD33 (например, гемтузумаб озогамицин); CD38 (например, даратумумаб, изатуксимаб), CD40; CD-40L; CD44v6; CD47 (например, Hu5F9-G4, CC-90002, SRF231, B6H12); CD52 (например, алемтузумаб); CD56; CD63; CD79 (например, полатузумаб ведотин); CD80; CD123; CD276/B7-H3 (например, омбуртамаб); CDH17; CEA; ClhCG; CTLA-4 (например, ипилимумаб, тремелимумаб), CXCR4; десмоглеин 4; DLL3 (например, ровальпитузумаб тезирин); DLL4; Е-кадгерин; EDA; EDB; EFNA4; EGFR (например, цетуксимаб, депатуксизумаб мафодотин, нецитумумаб, панитумумаб); EGFRvIII; эндосиалин; EpCAM (например, оппортузумаб монатокс); FAP; Фетальный ацетилхолиновый рецептор; FLT3 (например, см. WO 2018/220584); GD2 (например, динутуксимаб, 3F8); GD3; GITR; GloboH; GM1; GM2; HER2/neu [например, маргетуксимаб, пертузумаб, трастузумаб; адо-трастузумаб эмтанзин, трастузумаб дуокармазин [см. US8828401]; HER3; HER4; ICOS; IL-10; ITG-AvB6; LAG-3 (например, релатлимаб); Lewis-Y; LG; Ly-6; M-CSF [см. US7326414]; MCSP; мезотелин; MUC1; MUC2; MUC3; MUC4; MUC5AC; MUC5B; MUC7; MUC16; Notch1; Notch3; Nectin-4 (например, энфортумаб ведотин); OX40 [см. US7960515]; P-кадгереин [см. WO2016/001810]; PCDHB2; PDGFRA (например, оларатумаб); антиген клеток плазмы; PolySA; PSCA; PSMA; PTK7 [см. US9409995]; Ror1; SAS; SCRx6; SLAMF7 (например, элотузумаб); SHH; SIRPa (например, ED9, Effi-DEM); STEAP; TGF-beta; TIGIT; TIM-3; TMPRSS3; предшественник TNF-альфа; TROP-2 (например, сацитузумаб говитекан); TSPAN8; VEGF (например, бевацизумаб, бролуцизумаб); VEGFR1 (например, ранибизумаб); VEGFR2 (например, рамуцирумаб, ранибизумаб); Wue-1.
Типовые визуализирующие агенты, которые могут быть включены в ADC, включают флуоресцеин, родамин, люминофоры на основе комплексов лантанидов, и их производные, или радиоизотоп, связанный с хелатирующим агентом. Примеры флуорофоров включают, но не ограничиваются ими, изотиоцианат флуоресцеина (FITC) (например, 5-FITC), амидит флуоресцеина (FAM) (например, 5-FAM), эозин, карбоксифлуоресцеин, эритрозин, Alexa Fluor® (например, Alexa 350, 405, 430, 488, 500, 514, 532, 546, 555, 568, 594, 610, 633, 647, 660, 680, 700 или 750), карбокситетраметилродамин (TAMRA) (например, 5,-TAMRA), тетраметилродамин (TMR) и сульфородамин (SR) (например, SR101). Примеры хелаторов включают, но не ограничены ими, 1,4,7,10-тетраазациклододекан-N, N',N'',N'''-тетрауксусную кислоту (DOTA), 1,4,7-триазациклононан-1,4,7-триуксусную кислоту (NOTA), 1,4,7-триазациклононан, 1-глутаровую кислоту-4,7-уксусную кислоту (дефероксамин), диэтилентриаминпентауксусную кислоту (DTPA) и 1,2-бис(о-аминофенокси)этан-N, N,N',N'-тетрауксусную кислоту) (BAPTA).
Примеры терапевтических белков, которые могут быть включены в ADC, включают токсин, гормон, фермент и фактор роста.
Примеры биосовместимых полимеров, которые могут быть включены в ADC, включают водорастворимые полимеры, такие как полиэтиленгликоль (PEG) или его производные, и биосовместимые полимеры, содержащие цвиттерион (например, полимер, содержащий фосфорилхолин).
Примеры биосовместимых полимеров, которые могут быть включены в ADC, включают антисмысловые олигонуклеотиды.
Изобретение также относится к использованию радиации в сочетании с любым противораковым терапевтическим агентом, вводимым в настоящем изобретении. Более конкретно, соединения по изобретению можно вводить в сочетании с дополнительными терапиями, такими как радиационная терапия и/или химиотерапия.
Химический синтез
Следующие схемы и письменные описания представляют общие подробности относительно получения соединений по изобретению.
Соединения по изобретению могут быть получены любым способом, известным в данной области техники, для получения соединений аналогичной структуры. В частности, соединения по настоящему изобретению могут быть получены способами, описанными со ссылкой на следующие схемы, или конкретными способами, описанными в примерах, или способами, аналогичными тем и другим.
Специалисту в данной области техники будет понятно, что экспериментальные условия, изложенные на следующих схемах, иллюстрируют подходящие условия для осуществления показанных превращений и что может быть необходимо или желательно варьировать точные условия, используемые для получения соединений формулы (I) и соединения формулы (I), например, соединения формул (I), (Ia) или (Ib) и подобные.
Кроме того, специалисту в данной области техники будет понятно, что может быть необходимо или желательно на любой стадии синтеза соединений изобретения защитить одну или несколько чувствительных групп, чтобы предотвратить нежелательные побочные реакции. В частности, может быть необходимо или желательно защитить амино- или спиртовые группы. Защитные группы (PG), используемые при получении соединений по изобретению, можно использовать обычным образом. См., например, те, что описаны в «Greene’s Protective Groups in Organic Synthesis» by Theodora W Greene and Peter G M Wuts, third edition, (John Wiley and Sons, 1999), в частности в главе 7 («Protection for the Amino Group») и 2 («Protection for the Hydroxyl Group, Including 1,2- and 1,3-Diols»), включенной в настоящее описание посредством ссылки, в которой также описаны способы удаления таких групп.
Все производные формулы (I) могут быть получены способами, описанными в общих способах, представленных ниже, или их обычными модификациями. Настоящее изобретение также охватывает любой один или несколько из этих способов получения производных формулы (I) в дополнение к любым новым промежуточным соединениям, используемым в настоящем документе. Специалисту в данной области должно быть понятно, что следующие реакции можно нагревать термически или при микроволновом облучении, или в условиях проточной химии.
Кроме того, следует понимать, что может быть необходимо или желательно проводить превращения в порядке, отличном от порядка, описанного на схемах, или модифицировать одно или несколько превращений для получения желаемого соединения по изобретению.
В соответствии с первым способом, соединения формулы (I) могут быть получены из соединений промежуточного соединения (i), как показано на схеме 1.
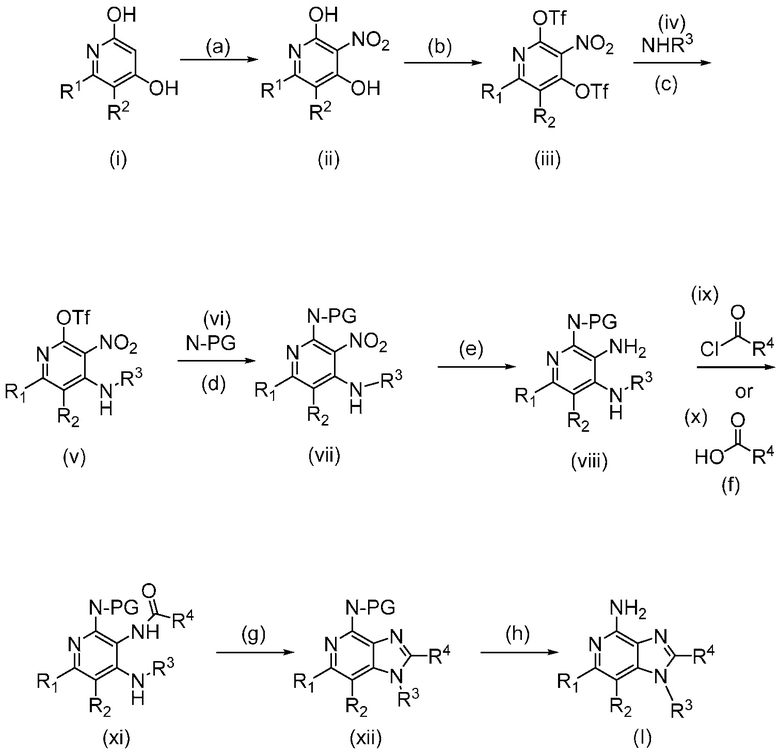
Схема 1
Возможные PG включают 4-метоксибензил, N, N-бис-(4-метоксибензил), трет-октил или другую подходящую аминозащитную группу; N, N-бис-(4-метоксибензил) чаще всего используют для соединений формулы (I).
Промежуточные соединения (i), (iv), (vi), (ix), (x) являются коммерчески доступными или могут быть синтезированы специалистами в данной области техники в соответствии с литературой или препаратами, описанными в настоящем документе. Для обсуждаемых здесь схем, когда соединения формулы (I) имеют хиральные центры, соответствующие энантиомеры могут быть разделены посредством хирального разделения рацемата, по необходимости. Кроме того, когда R3 содержит защитную группу, такую как кеталь или силил, при необходимости могут быть использованы подходящие условия снятия защиты, такие как метансульфоновая кислота в дихлорметане/метаноле/воде.
Промежуточное соединение (ii) может быть получено из промежуточного соединения (i) в соответствии со стадией (а), реакцией нитрования. В типовых способах используют подходящий нитрующий агент и подходящий органический или неорганический растворитель. Предпочтительные условия включают использование азотной кислоты в серной кислоте при 10°С.
Промежуточное соединение (iii) может быть получено из промежуточного соединения (ii) в соответствии со стадией (b), реакцией образования трифлата. В типовых способах используют трифторметансульфоновый ангидрид с подходящим органическим или неорганическим основанием в подходящем органическом растворителе при пониженных температурах. Предпочтительные условия включают использование триэтиламина в дихлорэтане при 0°С.
Промежуточное соединение (v) может быть получено из промежуточного соединения (iii), в соответствии со стадией (c), реакцией нуклеофильного ароматического замещения с промежуточным соединением (iv). В типовых способах используют подходящее органическое или неорганическое основание в подходящем органическом растворителе при КТ или повышенных температурах, либо термически, либо в условиях микроволнового облучения, либо в условиях проточной химии. Предпочтительные условия включают использование триэтиламина в дихлорэтане.
Промежуточное соединение (vii) может быть получено из промежуточного соединения (v) в соответствии со стадией (d), реакцией нуклеофильного ароматического замещения с промежуточным соединением (vi). В типовых способах используют подходящее органическое или неорганическое основание в подходящем органическом растворителе при КТ или повышенных температурах либо термически, в условиях микроволнового облучения, либо в условиях проточной химии. Например, использование бис(4-метоксибензил)амина с триэтиламином в дихлорэтане при 50°С при термическом нагреве или использование трет-октиламина с триэтиламином в толуоле при 75°С.
Промежуточное соединение (viii) может быть получено из промежуточного соединения (vii) в соответствии со стадией (e), стадией нитровосстановления. В типовых способах используют условия гидрирования с использованием подходящего источника водорода и подходящего катализатора гидрирования в подходящем органическом растворителе при КТ или при повышенных температурах, при термическом нагреве, при микроволновом облучении или в условиях проточной химии, или использование подходящего металла и подходящего донора водорода или протона в подходящем органическом растворителе. Предпочтительные условия включают использование цинковой пыли и формиата аммония в метаноле.
Промежуточное соединение (xi) может быть получено из промежуточного соединения (viii) в соответствии со стадией (f), стадии образования амидной связи с промежуточным соединением (ix) или (x). Типовые способы включают использование промежуточного соединения (ix) с подходящим органическим или неорганическим основанием в подходящем органическом растворителе, или использование промежуточного соединения (x) с подходящим амидным связующим реагентом и подходящим органическим или неорганическим основанием в подходящем органическом растворителе. В типовых условиях используют триэтиламин и дихлорметан при использовании промежуточного соединения (ix). В типовых условиях используют 50% масс. раствор ангидрида пропилфосфоновой кислоты в этилацетате, триэтиламине и этилацетате при использовании промежуточного соединения (х).
Промежуточное соединение (xii) может быть получено из промежуточного соединения (xi) в соответствии со стадией (g) образования имидазольного кольца. В типовых способах используют основные условия с использованием подходящего органического или неорганического основания при повышенных температурах, либо термически, либо в условиях микроволнового облучения, либо в условиях проточной химии; альтернативно, кислые условия с использованием подходящей органической или неорганической кислоты при повышенных температурах либо термически, либо в условиях микроволнового облучения, либо в условиях проточной химии. Альтернативно, подходящий дегидратирующий агент в подходящем органическом растворителе при повышенных температурах либо термически, либо в условиях микроволнового облучения, либо в условиях проточной химии. Предпочтительные условия включают использование гидроксида натрия в этаноле при 80°С при термическом нагревании, или трифенилфосфина и триэтиламина в четыреххлористом углероде при 80°С при термическом нагревании.
Соединения формулы (I) могут быть получены из промежуточного соединения (xii) в соответствии со стадией (h), удаления PG и соединений, содержащихся в R3, если они присутствуют, например, силила или кеталя. Типовые способы снятия защиты включают подходящую органическую или неорганическую кислоту в подходящем органическом растворителе при КТ или при повышенных температурах, либо термически, либо в условиях микроволнового облучения, либо в условиях проточной химии. Предпочтительные условия включают термический нагрев метансульфоновой кислоты в дихлорметане при 45°С с последующим добавлением метанола и воды.
Альтернативно, соединения формулы (I) могут быть получены из промежуточного соединения (iii), как показано на схеме 2.
Схема 2
Возможные амино-PG включают 4-метоксибензил, N, N-бис(4-метоксибензил), трет-октил, где трет-октил наиболее часто используется в примерах.
Уходящей группой (LG) является функциональная группа, помогающая конкретной реакции, и она включает OH, Cl, Br, I, OMs, OTs и OTf, где OH чаще всего используется в примерах.
Промежуточные соединения (ix), (x), (xiii), (xv) являются коммерчески доступными или могут быть синтезированы специалистами в данной области техники в соответствии с литературой или способами получения, описанными в настоящем документе.
Промежуточное соединение (xiv) может быть получено из промежуточного соединения (iii) в соответствии со стадией (i), реакцией нуклеофильного ароматического замещения с промежуточным соединением (xiii). В типовых способах используют подходящее органическое или неорганическое основание в подходящем органическом растворителе при КТ или повышенных температурах, либо термически, либо в условиях микроволнового облучения, либо в условиях проточной химии. Предпочтительные условия включают использование триэтиламина в дихлорэтане.
Промежуточное соединение (xvi) может быть получено из промежуточного соединения (xiv) в соответствии со стадией (j), реакцией нуклеофильного ароматического замещения с промежуточным соединением (xv). В типовых способах используют подходящее органическое или неорганическое основание в подходящем органическом растворителе при КТ или повышенных температурах, либо термически, либо в условиях микроволнового облучения, либо в условиях проточной химии. Предпочтительные условия включают использование бис(4-метоксибензил)амина с триэтиламином в дихлорэтане при 50°С при термическом нагреве или использование трет-октиламина с триэтиламином в толуоле при 75°С.
Промежуточное соединение (xvii) может быть получено из промежуточного соединения (xvi) в соответствии со стадией (k), стадией тандемного нитровосстановления и дебензилирования. В типовых способах используют условия гидрирования с использованием подходящего источника водорода и подходящего катализатора гидрирования в подходящем органическом растворителе при КТ или при повышенных температурах, при термическом нагреве, в условиях микроволнового излучения или в условиях проточной химии. Предпочтительные условия включают использование формиата аммония и 30% палладия на угле в этаноле при 55°С.
Промежуточное соединение (xviii) может быть получено из промежуточного соединения (xvii) в соответствии со стадией (l), стадии образования амидной связи с промежуточным соединением (ix) или (x). Типовые способы включают использование промежуточного соединения (ix) с подходящим органическим или неорганическим основанием в подходящем органическом растворителе, или использование промежуточного соединения (x) с подходящим амидным связующим реагентом и подходящим органическим или неорганическим основанием в подходящем органическом растворителе. Предпочтительные условия включают использование промежуточного соединения (ix) с триэтиламином и дихлорметаном при 0°С.
Промежуточное соединение (xix) может быть получено из промежуточного соединения (xviii) в соответствии со стадией (m) образования имидазольного кольца. В типовых способах используют основные условия с использованием подходящего органического или неорганического основания при повышенных температурах, либо термически, при микроволновом облучении, либо в условиях проточной химии; кислых условиях с использованием подходящей органической или неорганической кислоты при повышенных температурах либо термически, в условиях микроволнового облучения, либо в условиях проточной химии, либо подходящего дегидратирующего агента в подходящем органическом растворителе при повышенных температурах, либо термически, в условиях микроволнового излучения, либо в условиях проточной химии. Предпочтительные условия включают использование гидроксида натрия в этаноле при 75°C при термическом нагреве.
Промежуточное соединение (xxi) может быть получено из промежуточного соединения (xix) в соответствии со стадией (n), реакцией нуклеофильного замещения или реакцией Мицунобу с промежуточным соединением (xx). Типовые способы включают подходящее органическое или неорганическое основание в подходящем органическом растворителе при КТ или повышенных температурах, либо термически, либо в условиях микроволнового облучения, либо в условиях проточной химии. Альтернативно, когда LG является гидроксильной группой, обработкой подходящим фосфином и подходящим азодикарбоксилатом (или комбинацией обоих в одном реагенте) в подходящем органическом растворителе при КТ или повышенных температурах, либо термически, либо в условиях микроволнового излучения, либо в условиях проточной химии. Предпочтительные условия, где LG является гидроксильной группой, включают использование цианометилентрибутилфосфорана в толуоле при 90°С или 100°С при термическом нагреве.
Соединения формулы (I) могут быть получены из промежуточного соединения (xxi) в соответствии со стадией (о), удалением PG и любой PG, содержащейся в R3, если она присутствует, например, кеталя или силана. Типовые способы включают подходящую органическую или неорганическую кислоту в подходящем органическом растворителе при КТ или при повышенных температурах либо термически, либо в условиях микроволнового облучения, либо в условиях проточной химии. Предпочтительные условия включают метансульфоновую кислоту в гексафторизопропаноле или трифторуксусную кислоту в дихлорметане с последующим добавлением метанола.
При осуществлении синтеза соединений по изобретению, специалист в данной области будет контролировать реакции с помощью обычных способов, которые включают тонкослойную хроматографию (ТСХ), жидкостную хроматографию/масс-спектроскопию (ЖХМС) и ядерный магнитный резонанс (ЯМР).
Специалисту в данной области также понятно, что соединения по изобретению могут быть получены в виде смесей диастереомеров или геометрических изомеров (например, цис и транс замещения в циклоалкановом кольце). Эти изомеры могут быть разделены стандартными хроматографическими методами, такими хроматография с нормальными фазами на силикагеле, препаративная жидкостная хроматография высокого давления с обращенной фазой или сверхкритическая жидкостная хроматография. Специалисту в данной области техники также понятно, что некоторые соединения по изобретению являются хиральными и, таким образом, могут быть получены в виде рацемических или скалемических смесей энантиомеров. Доступно несколько способов разделения энантиомеров, которые хорошо известны специалистам в данной области техники.
ПРИМЕРЫ
Если не указано иное, реакции проводят в атмосфере азота. Хроматографию на силикагеле проводят с использованием силикагеля 250-400 меш с использованием азота под давлением (~10-15 ф./кв.д.) для пропускания растворителя через колонку («флэш-хроматография»). Там, где указано, растворы и реакционные смеси концентрируют роторным испарением под вакуумом.
1H и 19F спектры ядерного магнитного резонанса (ЯМР) во всех случаях соответствуют предполагаемым структурам. Характерные химические сдвиги (δ) даны в частях на миллион в сторону слабого поля от тетраметилсилана (для 1H-ЯМР) с использованием обычных сокращений для обозначения основных пиков: например с, синглет; д, дублет; т, триплет; кв, квартет; м, мультиплет; ш, широкий. Для обычных растворителей используют следующие сокращения: CDCl3, дейтерохлороформ; d6-ДМСО, дейтеродиметилсульфоксид; и CD3OD, дейтерометанол. При необходимости таутомеры могут быть зарегистрированы в данных ЯМР; и некоторые обмениваемые протоны могут быть невидимы.
Масс-спектры, МС (m/z), регистрируют с использованием либо ионизации электрораспылением (ИЭР), либо химической ионизации при атмосферном давлении (ХИАД). В соответствующих случаях, и если не указано иное, представленные данные m/z относятся к изотопам 19F, 35Cl, 79Br и 127I.
Номенклатура написана в соответствии с описанием IUPAC (International Union of Pure and Applied Chemistry, созданного в Perkin Elmers Chemdraw 18.0.
В приведенных здесь неограничивающих примерах и примерах получения применяются следующие сокращения:
AcOH является уксусной кислотой;
водн. является водным;
Bn является бензилом;
ш является широким;
tBu является трет-бутилом;
°C является градусом Цельсия;
CO2 является двуокисью углерода;
CMBP является цианометилентрибутилфосфораном;
Cs2CO3 является карбонатом цезия;
ДХЭ является дихлорэтаном;
ДХМ является дихлорметаном; метиленхлоридом;
ДИПЭА/ДИЭА является N-этилдиизопропиламином, N, N-диизопропилэтиламином;
ДМА является диметилацетамидом;
ДМФ является N, N-диметилформамидом;
ДМСО является диметилсульфоксидом;
эи является энантиомерным избытком;
EtOAc является этилацетатом;
EtOH является этанолом;
Et3N является триэтиламином;
г является граммом;
HCO2NH4 является формиатом аммония;
HCl является хлористоводородной кислотой;
ГФИП является 1,1,1,3,3,3-гексафторизопропанолом;
HNO3 является азотной кислотой;
ЖХВД является жидкостной хроматографией высокого давления;
H2O является водой;
H2SO4 является серной кислотой;
Ч или ч является часом;
IPA/iPrOH является изопропанолом;
л является литром;
ЖХМС является жидкостной хроматографией/масс спектрометрией;
LiAlH4 является алюмогидридом лития;
LiOH является гидроксидом лития;
M является молярным;
MeCN является ацетонитрилом;
MeI является метилйодидом;
MeOH является метанолом;
мг является миллиграммом;
MgSO4 является сульфатом магния;
MHz является мена Герцем;
мин является минутами;
мл является миллилитром;
ммоль является миллимолем;
моль является молем;
МС m/z является пиком масс спектра;
MsOH является метансульфоновой кислотой;
NaH является гидридом натрия;
NaHCO3 является гидрокарбонатом натрия;
NaOH является гидроксидом натрия;
Na2SO4 является сульфатом натрия;
NH3 является аммиаком;
NH4OH является гидроксидом аммония;
NH(PMB)2 является бис(4-метоксибензил)амином;
ЯМР является ядерным магнитным резонансом;
Pd/C является палладием на угле;
pH является водородным показателем;
ч./млн. является частями на миллион;
ф./кв.д. является фунтами на квадратный дюйм;
Rt является временем удержания;
КТ является комнатной температурой;
TBDMS является третбутилдиметилсилилом;
TBSCl является хлоридом третбутилиметилсилила;
ТБМЭ/МТБЭ является трет-бутилдиметиловым эфиром;
ТЭА является триэтиламином;
Tf2O является трифторметансульфоновым ангидридом;
ТФК является трифторуксусной кислотой;
ТФУА является трифторуксусным ангидридом;
ТГФ является тетрагидрофураном;
ТСХ является тонкослойной хроматографией;
TsOH является п-толуолсульфоновой кислотой;
Zn является цинком;
мкл является микролитром;
мкмоль является микромолем
Хиральное разделение используют для разделения энантиомеров некоторых промежуточных соединений во время получения соединений по изобретению. Когда такое разделение проведено, разделенные энантиомеры обозначают как ENT-1 или ENT-2 в соответствии с порядком их элюирования. Для соединений с двумя хиральными центрами, стереоизомеры в каждом стереоцентре разделяют в разное время. Обозначение ENT-1 или ENT-2 промежуточного соединения или примера относится к хиральному центру разделения, выполненного на этой стадии. Признано, что когда стереоизомеры в хиральном центре разделены в соединении с двумя или несколькими центрами, разделенные энантиомеры являются диастереоизомерами друг друга. Обозначения ENT-1 или ENT-2 используют в настоящем документе для согласованности и относятся к разделенному хиральному центру. В качестве примера, но не ограничения, примеры 6 и 7 имеют хиральный центр. Энантиомеры разделяют на последней стадии. Хиральный центр изображен как две возможности, но неизвестно, какой пример является каким энантиомером. Следовательно, обозначения (R) и (S) не связаны ни с примером 6, ни с примером 7. Если разделение происходит на промежуточном продукте в этих примерах получения, после того как смесь подвергается процедурам разделения, хиральный центр обозначается как «абс» около этого центра, с пониманием того, что разделенные энантиомеры могут не быть энантиомерно чистыми, и конкретная ориентация этой связи не показана, поскольку энантиомер не был подтвержден. Как правило, обогащенный энантиомер в каждом хиральном центре составляет >90% выделенного материала. Предпринимаются также усилия по повышению энантиомерной чистоты в центре до >98% смеси и даже >99%.
Оптическое вращение энантиомера можно измерить с помощью поляриметра. Согласно наблюдаемым данным его вращения (или конкретным данным его вращения), энантиомер с вращением по часовой стрелке обозначен как (+)-энантиомер, и энантиомер с вращением против часовой стрелки обозначен как (-)-энантиомер. Рацемические соединения обозначаются либо отсутствием нарисованной или описанной стереохимии, либо наличием (+/-) рядом со структурой; в этом последнем случае указанная стереохимия является относительной (а не абсолютной) конфигурацией заместителей соединения.
При использовании препаративной ТСХ или хроматографии на силикагеле специалист в данной области техники может выбрать любую комбинацию растворителей для очистки желаемого соединения.
Пример (1): 2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)пропан-1,3-диол трифторацетат
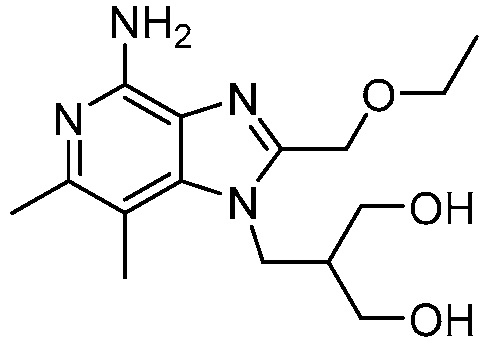
Стадия 1: Синтез 4-(бензиламино)-5,6-диметил-3-нитропиридин-2-ил трифторметансульфоната
В круглодонную колбу под азотом добавляют 5,6-диметил-3-нитропиридин-2,4-диол (7,56 г, 41,05 ммоль) в дихлорметане (300 мл). Туда добавляют триэтиламин (12,5 г, 123 ммоль, 17,2 мл) и реакционную смесь охлаждают до 0°C. Трифторуксусный ангидрид (23,2 г, 82,1 ммоль, 13,8 мл) добавляют по каплям в течение 8 мин. Реакционную смесь перемешивают при 0°C в течение 1,5 ч. Туда добавляют бензиламин (4,84 г, 45,2 ммоль, 4,93 мл) и реакционную смесь нагревают до КТ и перемешивают в течение 3 ч. Реакционную смесь промывают водой (2×100 мл) и насыщенным раствором соли (1×100 мл). Органические слои сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле (Гептан:Этилацетат 0-50% градиент) с получением указанного в заголовке соединения. Выход: 12,4 г, 30,5 ммоль, 74,3%. ЖХМС m/z 406,2 [M+H]+. 1H ЯМР (400 MГц, CDCl3) δ 7,34-7,44 (м, 3H), 7,28-7,31 (м, 2H), 5,24 (шс, 1H), 4,31 (д, J=5,07 Гц, 2H), 2,47 (с, 3H), 2,12-2,20 (м, 3H).
Стадия 2: Синтез N4-бензил-5,6-диметил-3-нитро-N2-(2,4,4-триметилпентан-2-ил)пиридин-2,4-диамина
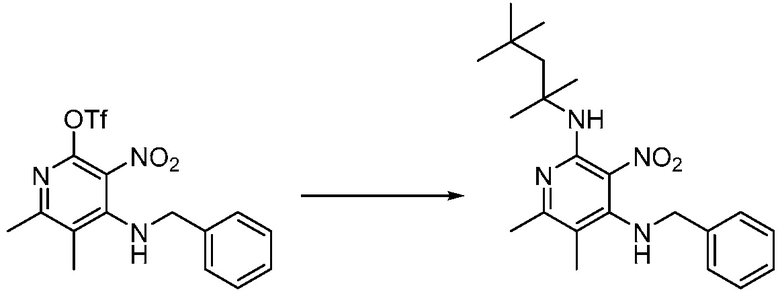
В круглодонную колбу загружают добавленный 4-(бензиламино)-5,6-диметил-3-нитропиридин-2-ил трифторметансульфонат (12,4 г, 30,5 ммоль) и толуол (100 мл). Триэтиламин (4,63 г, 45,7 ммоль, 6,38 мл) добавляют, затем трет-Октиламин (5,91 г, 45,7 ммоль, 7,34 мл). Реакционную смесь нагревают при 75°C в течение 16 ч. трет-Октиламин (5,91 г, 45,7 ммоль, 7,34 мл) добавляют, и реакционную смесь нагревают при 75°C в течение 48 ч. Раствор концентрируют на Celite® и очищают хроматографией на силикагеле (Гептан:Этилацетат 0-50% градиент) с получением указанного в заголовке соединения в виде красного масла. Выход: 8,93 г, 23,2 ммоль, 76,2%. ЖХМС m/z 386,4 [M+H]+. 1H ЯМР (400 MГц, CDCl3) δ 8,76 (с, 1H), 8,38 (шс, 1H), 7,32-7,39 (м, 2H), 7,27-7,31 (м, J=3,12, 3,12 Гц, 3H), 4,46 (д, J=4,29 Гц, 2H), 2,33 (с, 3H), 2,14 (с, 3H), 1,99 (с, 2H), 1,56 (с, 6H), 0,98 (с, 9H).
Стадия 3: Синтез 5,6-диметил-N2-(2,4,4-триметилпентан-2-ил)пиридин-2,3,4-триамина
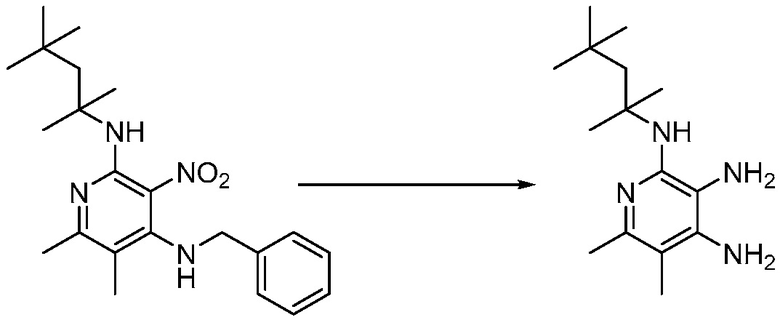
В круглодонную колбу с N4-бензил-5,6-диметил-3-нитро-N2-(2,4,4-триметилпентан-2-ил)пиридин-2,4-диамином (8,90 г, 23,2 ммоль) и этанолом (150 мл) добавляют формиат аммония (14,6 г, 231 ммоль). Палладий на угле (200 мг, 30% Pd) добавляют, и реакционную смесь перемешивают при 55°C в течение 2 ч. Реакционную смесь затем охлаждают до КТ и фильтруют через Celite® и фильтрат концентрируют. Остаток перемешивают в этилацетате в течение 1 ч, затем твердые вещества удаляют фильтрацией через Celite®. Фильтрат концентрируют с получением указанного в заголовке соединения в виде оранжевой камеди. Выход: 5,6 г, 21,2 ммоль, 91,5%. ЖХМС m/z 265,3 [M+H]+. 1H ЯМР (600 MГц, CDCl3) δ 8,62 (с, 1H), 2,40 (с, 3H), 1,98 (с, 3H), 1,66 (с, 2H), 1,29 (с, 6H), 1,05 (с, 9H).
Стадия 4: Синтез 2-(этоксиметил)-6,7-диметил-N-(2,4,4-триметилпентан-2-ил)-1H-имидазо[4,5-c]пиридин-4-амина
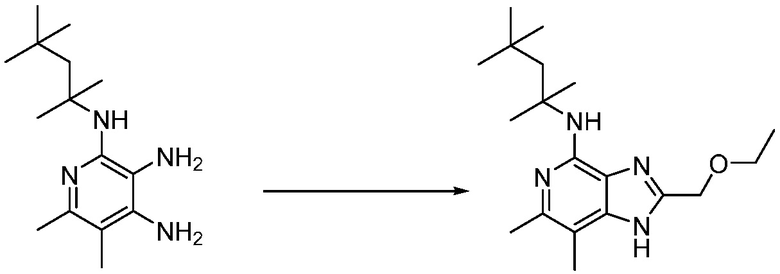
Раствор 5,6-диметил-N2-(2,4,4-триметилпентан-2-ил)пиридин-2,3,4-триамина (5,60 г, 21,2 ммоль) и дихлорметана (100 мл) охлаждают до 0°C. Туда добавляют 2-этоксиацетилхлорид (2,73 г, 22,2 ммоль, 2,44 мл), затем триэтиламин (3,21 г, 31,8 ммоль, 4,43 мл). Реакционную смесь перемешивают при 0°C в течение 1,5 ч. Реакционную смесь разбавляют дихлорметаном (100 мл) и органические слои промывают водой (2×50 мл). Органические слои сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток разбавляют этанол (100 мл) и добавляют гидроксид натрия (5,08 г, 127 ммоль, 8,47 мл, 15N). Реакционную смесь нагревают при 75°C в течение 16 ч. Реакционную смесь охлаждают до КТ и разбавляют этилацетатом и промывают водой (2 x). Объединенные водные слои промывают этилацетатом. Объединенные органические слои сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле (Гептан:Этилацетат, 0-100% градиент) с получением указанного в заголовке соединения. Выход: 1,40 г, 4,21 ммоль, 19,8%. ЖХМС m/z 333,1 [M+H]+. 1H ЯМР (400 MГц, CDCl3) δ 9,10 (шс, 1H), 4,96 (шс, 1H), 4,73 (с, 2H), 3,65 (кв, J=7,02 Гц, 2H), 2,42 (с, 3H), 2,25 (с, 3H), 2,07 (с, 2H), 1,59 (с, 6H), 1,29 (т, J=7,02 Гц, 3H), 0,99 (с, 9H).
Стадия 5: Синтез 1-((2,2-диметил-1,3-диоксан-5-ил)метил)-2-(этоксиметил)-6,7-диметил-N-(2,4,4-триметилпентан-2-ил)-1H-имидазо[4,5-c]пиридин-4-амина
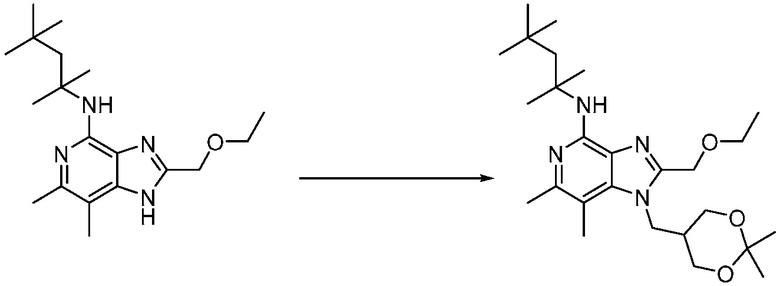
К раствору 2-(этоксиметил)-6,7-диметил-N-(2,4,4-триметилпентан-2-ил)-1H-имидазо[4,5-c]пиридин-4-амина (125 мг, 0,376 ммоль), (2,2-диметил-1,3-диоксан-5-ил)метанола (68,7 мг, 0,470 ммоль) в толуоле (2 мл) добавляют цианометилентрибутилфосфоран (136 мг, 0,564 ммоль, 0,564 мл, 1M в толуоле), и реакционную смесь нагревают при 90°C в течение 1,5 ч, затем охлаждают до КТ и перемешивают в течение 16 ч. Цианометилентрибутилфосфоран (136 мг, 0,564 ммоль, 0,564 мл, 1M в толуоле) добавляют, и реакционную смесь перемешивают при 90°C в течение 1,5 ч. Реакционную смесь абсорбируют на силикагель и очищают хроматографией на силикагеле (Гептан:Этилацетат 0-100% градиент) с получением указанного в заголовке соединения. Выход: 72 мг, 0,156 ммоль, 42%. ЖХМС m/z 461,3 [M+H]+. 1H ЯМР (400 MГц, CDCl3) δ 5,13 (с, 1H), 4,79 (с, 2H), 4,62 (д, J=7,81 Гц, 2H), 4,02 (дд, J=2,93, 12,29 Гц, 2H), 3,60 (кв, J=7,02 Гц, 2H), 3,51 (д, J=10,93 Гц, 2H), 2,43 (с, 3H), 2,38 (с, 3H), 2,06 (с, 2H), 1,90-1,98 (м, 1H), 1,58 (с, 6H), 1,47 (с, 6H), 1,24 (т, J=7,02 Гц, 3H), 0,99 (с, 9H).
Стадия 6: Синтез Примера 1: 2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)пропан-1,3-диола трифторацетата
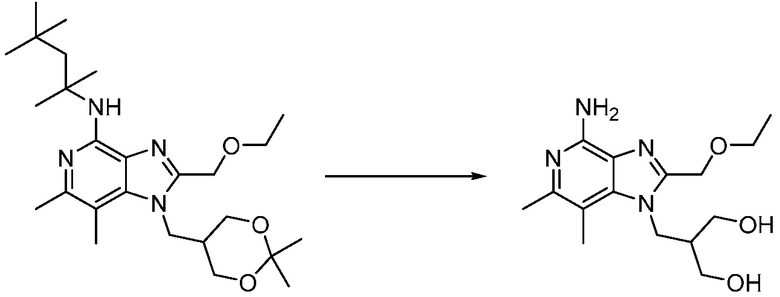
Раствор 1-((2,2-диметил-1,3-диоксан-5-ил)метил)-2-(этоксиметил)-6,7-диметил-N-(2,4,4-триметилпентан-2-ил)-1H-имидазо[4,5-c]пиридин-4-амина (72 мг, 0,16 ммоль) в 4:1 смеси дихлорметана:трифторуксусной кислоты (5 мл) перемешивают при КТ в течение 30 мин. Метанол (10 мл) добавляют, и реакционную смесь перемешивают при КТ в течение 1,5 ч. Реакционную смесь концентрируют, и остаток растворяют в диметилсульфоксиде (1 мл) и очищают ЖХВД с обращенной фазой. (Колонка: Waters Sunfire C18 19×100, 5u; Подвижная фаза A: 0,05% ТФК в воде (об./об.); Подвижная фаза B: 0,05% ТФК в ацетонитриле (об./об.); Градиент: выдержка при 95,0% H2O/5,0% Ацетонитрил в течение 1,0 мин, 95,0% H2O/5,0% Ацетонитрил линейный до 0% H2O/100% Ацетонитрил за 9,0 мин, выдержка при 0% H2O/100% Ацетонитрил до 10,0 мин. Поток: 25 мл/мин.). Выход: 18,1 мг, 0,043 ммоль, 27% ЖХВД время удержания: 1,17 мин (Колонка: Waters Atlantis® dc18 4,6×50, 5u; Подвижная фаза A: 0,05% ТФК в воде (об./об.); Подвижная фаза B: 0,05% ТФК в ацетонитрил (об./об.); Градиент: 95,0% H2O/5,0% Ацетонитрил линейный до 5% H2O/95% Ацетонитрил за 4,0 мин, выдержка при 5% H2O/95% Ацетонитрил до 5,0 мин. Поток: 2мл/мин.); ЖХВД m/z 309,4 [M+H]+.
Пример (2): 2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диол
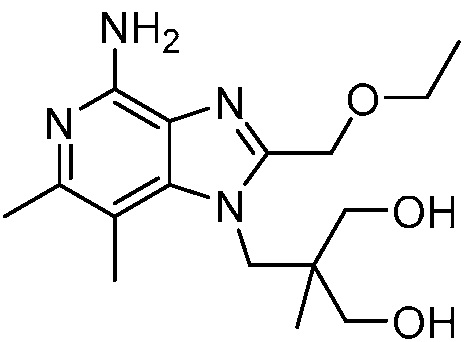
Стадия 1: Синтез 5,6-диметил-3-нитропиридин-2,4-диола
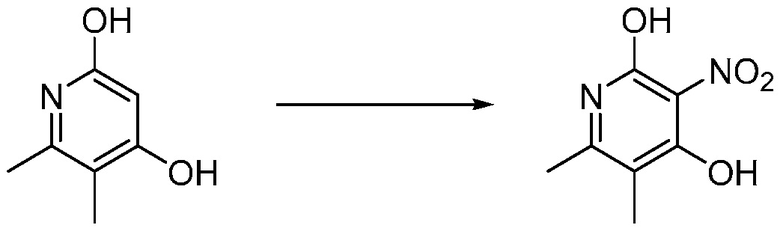
К 5,6-диметилпиридин-2,4-диолу (40,0 г, 287 ммоль) (Org. Lett., 2003, 5 (25), pp 4779-4782) при 18°C добавляют концентрированную серную кислоту (174 мл). Реакционную смесь охлаждают до 0°C, этот момент азотную кислоту (68-70%, 45,8 мл) добавляют в течение 1,5 ч, сохраняя внутреннюю температуру ниже 10°C. После завершения добавления, реакционную смесь перемешивают при 10°C в течение 30 мин. Эту реакционную смесь объединяют со второй реакцией из 40 г 5,6-диметилпиридин-2,4-диола. Объединенную реакционную смесь выливают в ледяную воду (2 л). Желтый осадок собирают фильтрацией и промывают водой (5×200 мл) и МТБЭ (5×100 мл). Собранное твердое вещество сушат под вакуумом с получением указанного в заголовке соединения в виде желтого твердого вещества. Объединенный Выход: 50 г, 271,7 ммоль, 47% выход на основе 80 г исходного пиридина. 1H ЯМР (400 MГц, ДМСО-d6) 12,34 (шс, 1H), 11,90 (шс, 1H), 2,21 (с, 3H), 1,90 (с, 3H).
Стадия 2: Синтез N2,N2-бис(4-метоксибензил)-5,6-диметил-3-нитро-N4-((2,2,5-триметил-1,3-диоксан-5-ил)метил)пиридин-2,4-диамина
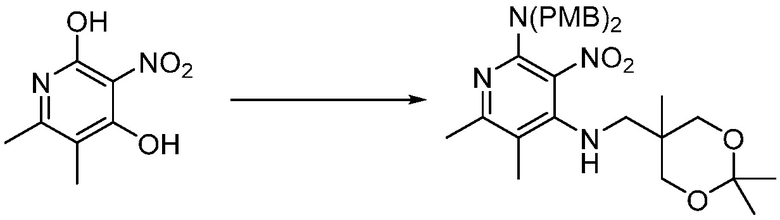
К раствору 5,6-диметил-3-нитропиридин-2,4-диола (70,0 г, 380,1 ммоль) в дихлорэтане (1,4 л), охлажденному до 0°C, добавляют триэтиламин (80,8 г, 798 ммоль). Трифторметансульфоновый ангидрид (220 г, 779 ммоль) добавляют в течение 30 мин при 0°C. Реакционную смесь перемешивают при 0°C в течение 1,5 ч. Триэтиламин (42,3 г, 418 ммоль) добавляют, затем порциями добавляют (2,2,5-триметил-1,3-диоксан-5-ил)метанамин (72,6 г, 456 ммоль) (получен из Organic & Biomolecular Chemistry, 14(2), 483-494; 2016). Реакционную смесь перемешивают при 0°C в течение 20 мин, затем перемешивают при 15°C в течение 18 ч. Реакционную смесь охлаждают до 0°C, в этот момент добавляют триэтиламин (115 г, 1,14 моль), затем бис(4-метоксибензил)амин (127 г, 494 ммоль). Реакционную смесь затем перемешивают при 50°C в течение 12 ч. Растворитель удаляют, и остаток очищают колоночной хроматографией на силикагеле (Петролейный эфир:Этилацетат градиент 0-10%). Фракции продукта собирают и выпаривают до 10% объема. Твердые вещества собирают фильтрацией, и фильтровальную лепешку промывают петролейным эфиром (3×50 мл). Фильтрат концентрируют и очищают колоночной хроматографией на силикагеле (Петролейный эфир:Этилацетат градиент 0-10%). Фракции продукта собирают и выпаривают до 10% объема. Твердые вещества собирают фильтрацией и фильтровальную лепешку промывают петролейным эфиром (3×20 мл), с получением указанного в заголовке соединения в виде желтого твердого вещества. Выход: 98 г, 173,6 ммоль, 45,7%. ЖХМС m/z 564,9 [M+H]+. 1H ЯМР (400 MГц, CDCl3) δ 7,05 (д, J=8,78 Гц, 4H), 6,80 (д, J=8,78 Гц, 4H), 6,47 (т, J=6,15 Гц, 1H), 4,34 (с, 4H), 3,79 (с, 6H), 3,54-3,67 (м, 4H), 3,42 (д, J=6,02 Гц, 2H), 2,35 (с, 3H), 2,21 (с, 3H), 1,43 (с, 3H), 1,41 (с, 3H), 0,83 (с, 3H).
Стадия 3: Синтез N2,N2-бис(4-метоксибензил)-5,6-диметил-N4-((2,2,5-триметил-1,3-диоксан-5-ил)метил)пиридин-2,3,4-триамина
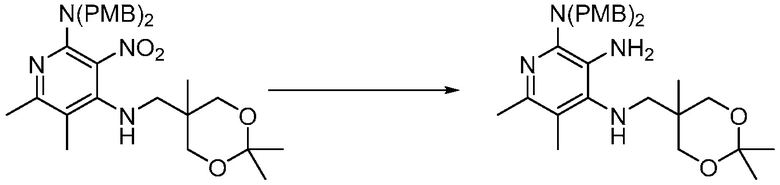
К раствору N2,N2-бис(4-метоксибензил)-5,6-диметил-3-нитро-N4-((2,2,5-триметил-1,3-диоксан-5-ил)метил)пиридин-2,4-диамина (57,0 г, 100,9 ммоль) в метаноле (673 мл) добавляют формиат аммония (63,7 г, 1,01 моль) и затем порошок цинка (66,0 г, 1,01 моль). Реакционную смесь перемешивают в течение 10 мин при 15°C. Реакционную смесь фильтруют через Celite®, и фильтрат концентрируют. Остаток растворяют в этилацетате и медленно добавляют воду с образованием белого осадка. Водный слой экстрагируют этилацетатом. Объединенные органические слои промывают насыщенным раствором соли, сушат над безводным сульфатом натрия, фильтруют и концентрируют с получением указанного в заголовке соединения в виде коричневого масла. Используют без дальнейшей очистки. Выход: 50,0 г, 96,4 ммоль, 95,5%. ЖХМС m/z 535,0 [M+H]+.
Стадия 4: Синтез N-(2-(бис(4-метоксибензил)амино)-5,6-диметил-4-(((2,2,5-триметил-1,3-диоксан-5-ил)метил)амино)пиридин-3-ил)-2-этоксиацетамида
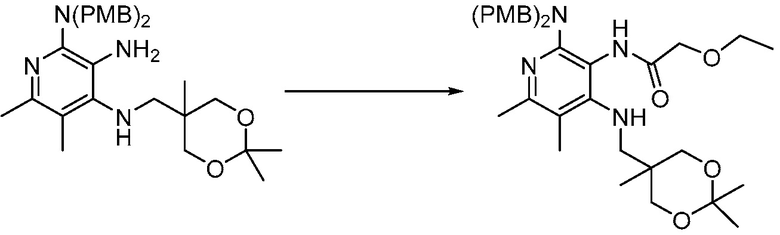
К раствору N2,N2-бис(4-метоксибензил)-5,6-диметил-N4-((2,2,5-триметил-1,3-диоксан-5-ил)метил)пиридин-2,3,4-триамина (100 г, 192,8 ммоль) в дихлорметане (1 л) добавляют триэтиламин (97,5 г, 964 ммоль, 134 мл). Реакционную смесь охлаждают до 0°C,в этот момент 2-этоксиацетилхлорид (37,8 г, 308 ммоль) добавляют по каплям. Ледяную баню удаляют, и реакционную смесь перемешивают при 25°C в течение 16 ч. Растворитель удаляют, и продукт используют без дальнейшей очистки. Выход: 150 г, 192,8 ммоль, признан количественным.
Стадия 5: Синтез 2-(этоксиметил)-N, N-бис(4-метоксибензил)-6,7-диметил-1-((2,2,5-триметил-1,3-диоксан-5-ил)метил)-1H-имидазо[4,5-c]пиридин-4-амина
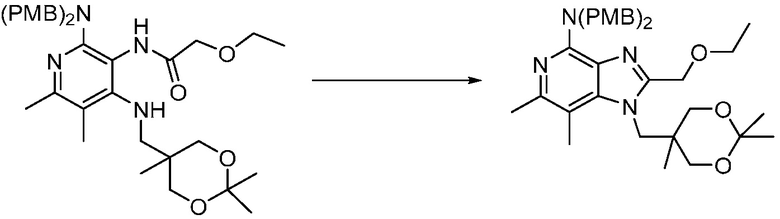
К раствору неочищенного N-(2-(бис(4-метоксибензил)амино)-5,6-диметил-4-(((2,2,5-триметил-1,3-диоксан-5-ил)метил)амино)пиридин-3-ил)-2-этоксиацетамида из предыдущей реакции в этаноле (3,45 л, охлажденный до 0°C) добавляют гидроксид натрия (64,4 мл, 15N водный). После добавления, реакционную смесь нагревают до кипения с обратным холодильником в течение 16 ч. Реакционную смесь охлаждают до 15°C, в этот момент образуется белый осадок. Твердые вещества фильтруют, и фильтровальную лепешку промывают водой и МТБЭ. Белые твердые вещества растворяют в этилацетате (1 L) и органический слой промывают насыщенным раствором соли, сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле (Этилацетат:Петролейный эфир градиент 0-45%) с получением продукта. Этот продукт объединяют с дополнительными 7,2 г продукта из другой партии, и перемешивают в течение 20 мин в 2:1 растворе петролейного эфира:МТБЭ. Твердые вещества фильтруют и сушат в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества. Объединенный выход: 94,52 г, 157 ммоль, 76% выход. ЖХМС m/z 603,0 [M+H]+. 1H ЯМР (600 MГц, ДМСО-d6) δ 7,16 (д, J=8,22 Гц, 4H), 6,83 (д, J=8,80 Гц, 4H), 4,82-5,27 (м, 6H), 4,36-4,80 (м, 4H), 3,70 (с, 6H), 3,51-3,68 (м, 2H), 3,40-3,46 (м, 2H), 2,41 (с, 3H), 2,34 (с, 3H), 1,39 (с, 3H), 1,34 (с, 3H), 1,07 (т, J=7,04 Гц, 3H), 0,56 (с, 3H).
Стадия 6: Синтез Примера (2): 2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диола
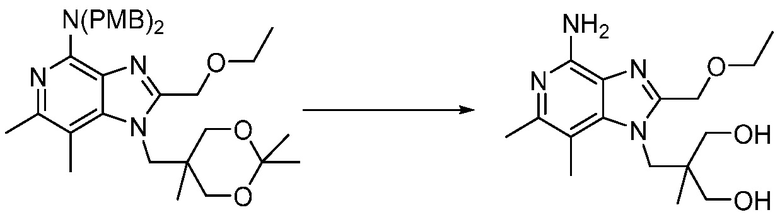
К раствору 2-(этоксиметил)-N, N-бис(4-метоксибензил)-6,7-диметил-1-((2,2,5-триметил-1,3-диоксан-5-ил)метил)-1H-имидазо[4,5-c]пиридин-4-амина (477 мг, 0,791 ммоль) и дихлорметана (3 мл) в пробирке добавляют концентрированную хлористоводородную кислоту (7,91 ммоль, 0,66 мл) по каплям. Пробирку закрывают крышкой, и реакционную смесь перемешивают при КТ в течение 2 ч, затем нагревают при 50°C в течение 1 ч. Дополнительную концентрированную хлористоводородную кислоту (4,8 ммоль, 0,40 мл) добавляют, и реакционную смесь продолжают нагревать при 50°C в течение 30 мин. Реакционную смесь затем охлаждают до КТ и перемешивают в течение 16 ч. Воду (2 мл) добавляют, и водный слой промывают дихлорметаном (2×3 мл). Водный слой доводят до pH 9 твердым карбонатом натрия. Реакционную смесь затем нагревают до кипения с обратным холодильником и охлаждают до 4°C. Твердые вещества фильтруют и промывают водой (5 мл) и простым эфиром (5 мл) и сушат под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества. Выход: 194 мг, 0,602 ммоль, 76,0%. ЖХМС m/z 323,3 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ 5,76 (с, 2H), 4,91-5,09 (м, 1H), 4,69-4,90 (м, 2H), 4,27-4,59 (м, 3H), 3,39-3,56 (м, 2H), 3,29-3,37 (м, 1H, предполагаемый, частично закрыт H2O), 3,14-3,29 (м, 2H), 2,95-3,13 (м, 1H), 2,41 (с, 3H), 2,29 (с, 3H), 1,06-1,14 (м, 3H), 0,48 (с, 3H).
Пример (3): 2-((4-амино-2-(этоксиметил)-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диол
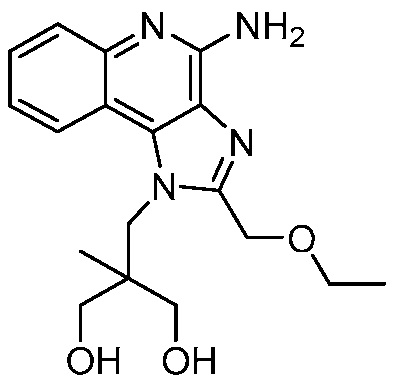
Получают по методике, аналогичной 2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диолу, начиная с хинолин-2,4-диола. Получают 18,5 мг. ЖХМС m/z 345,4 [M+H]+. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 0,58 (с, 3 H) 1,15 (т, J=7,02 Гц, 3 H) 3,07-3,77 (м, 7 H) 4,58-5,26 (м, 5 H) 7,52 (т, J=8,0 Гц, 1 H) 7,71 (т, J=8,0 Гц, 1 H) 7,79 (д, J=8,0 Гц, 1 H) 8,74 (д, J=8,0 Гц, 1 H) 9,19 (шс, 1 H)
Пример (4): 2-((4-амино-2-(этоксиметил)-7,8-дигидроциклопента[b]имидазо[4,5-d]пиридин-1(6H)-ил)метил)-2-метилпропан-1,3-диол
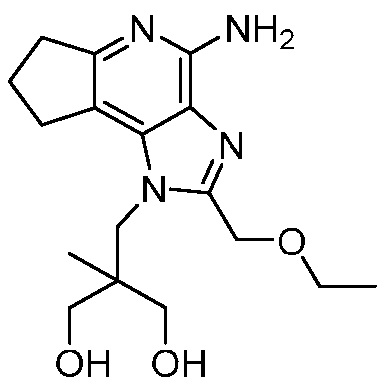
Получают по методике, аналогичной 2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диолу, начиная с 6,7-дигидро-5H-циклопента[b]пиридин-2,4-диола, получают 60 мг. ЖХМС время удержания: 0,715 мин (Колонка: ACQUITY UPLC BEH C18 50*2,1 мм, 1,7 мкм; Подвижная фаза A: 0,05% NH4OH в воде (об./об.); Подвижная фаза B: Ацетонитрил; Градиент: выдержка при 100% H2O в течение 0,10 мин, 100% H2O до 0% H2O/100% Ацетонитрил за 0,90 мин, выдержка при 0% H2O/100% Ацетонитрил в течение 0,2 мин. Поток: 1,0 мл/мин.). ЖХМС m/z 335,3 [M+H]+. 1H ЯМР (CDCl3, 400 MГц), характеристические пики: δ 5,1-5,3 (м, 2H), 4,8-5,0 (м, 2H), 4,1-4,8 (м, 3H), 3,5-3,8 (м, 6H), 3,1-3,3 (м, 2H), 2,9-3,0 (м, 2H), 2,1-2,2 (м, 2H), 1,26 (т, 3H, J=7,0 Гц), 0,65 (с, 3H).
Пример (5): 2-((4-амино-2-(этоксиметил)-6,7,8,9-тетрагидро-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диол, формиат
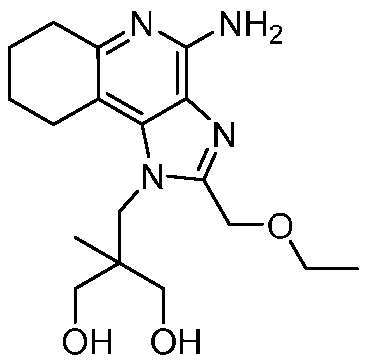
Получают по методике, аналогичной 2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диолу, начиная с 6,7-дигидро-5H-циклопента[b]пиридин-2,4-диола, получают 34 мг. ЖХМС время удержания: 0,621 мин (Колонка: ACQUITY UPLC BEH C18 50*2,1 мм, 1,7 мкм; Подвижная фаза A: 0,05% NH4OH в воде (об./об.); Подвижная фаза B: Ацетонитрил; Градиент: 95% H2O/5% Ацетонитрил до 0% H2O/100% Ацетонитрил в течение 1 мин, выдержка при 0% H2O/100% Ацетонитрил в течение 0,2 мин. Поток: 1,0 мл/мин.). ЖХМС m/z 349,2 [M+H]+. 1H ЯМР (Метанол-d4, 400 MГц), характеристические пики: δ 8,4-8,6 (м, 1H), 4,63 (с, 2H), 3,59-3,68 (м, 2H), 3,1-3,6 (м, 5H, предполагаемый, частично закрыт растворителем), 2,8-3,0 (м, 3H), 1,72-2,06 (м, 4H), 1,22 (т, 3H, J=7,0 Гц), 0,60 (с, 3H).
Примеры (6) и (7): (R)-3-(4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-2-(метоксиметил)-2-метилпропан-1-ол и (S)-3-(4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-2-(метоксиметил)-2-метилпропан-1-ол
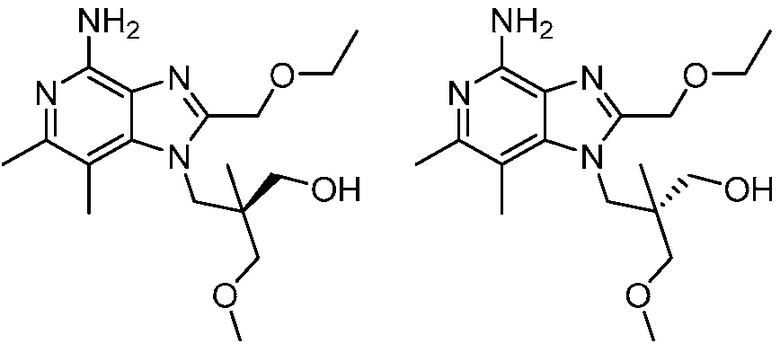
Стадия 1: Синтез (2,2,5-триметил-1,3-диоксан-5-ил)метанола
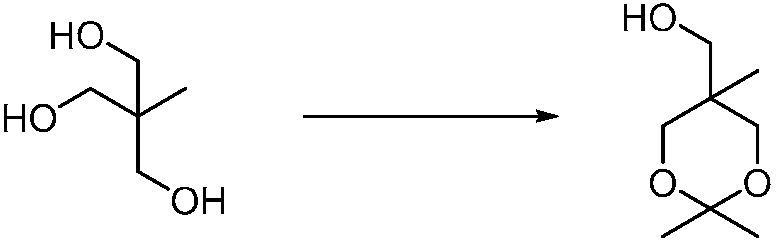
К перемешиваемому раствору 2-(гидроксиметил)-2-метилпропан-1,3-диола (50,0 г, 416,2 ммоль) и 2,2-диметоксипропана (65,0 г, 624,0 ммоль) добавляют ацетон (46 мл) и п-толуолсульфоновую кислоту (3,96 г, 20. 8 ммоль). Реакционную смесь нагревают при 30°C в течение 12 ч. Реакционную смесь охлаждают до КТ, и добавляют раствор бикарбоната натрия (12 г) в воде (400 мл). Водный слой экстрагируют этилацетатом, и органический слой промывают насыщенным раствором соли, сушат над безводным сульфатом натрия, фильтруют и концентрируют с получением указанного в заголовке соединения в виде белого твердого вещества. Выход: 52,9 г, 330,2 ммоль, 79,3%. 1H ЯМР (400 MГц, ДМСО-d6) δ 4,58 (т, J=5,40 Гц, 1H), 3,53-3,57 (м, 2H), 3,41-3,45 (м, 2H), 3,33-3,36 (м, 2H, частично закрыт растворителем), 1,32 (с, 3H), 1,26 (с, 3H), 0,74 (с, 3H).
Стадия 2: Синтез 5-(метоксиметил)-2,2,5-триметил-1,3-диоксана
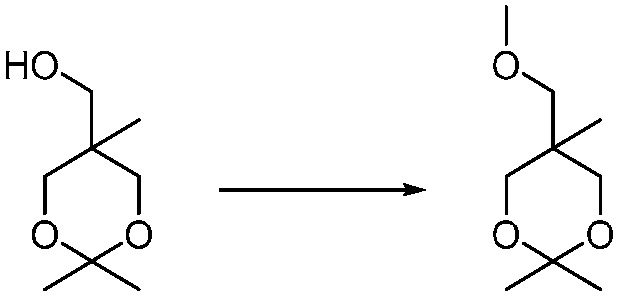
К суспензии гидрида натрия (33,8 г, 844 ммоль, 60% дисперсия в минеральном масле) в толуоле (1,35 л), охлажденной до 0°C, добавляют (2,2,5-триметил-1,3-диоксан-5-ил)метанол (67,6 г, 421,9 ммоль) и реакционную смесь перемешивают при 0°C в течение 10 мин и затем перемешивают при 40°C в течение 18ч. Реакционную смесь охлаждают до 0°C, добавляют метилйодид (135 г, 951,1 ммоль), и реакционную смесь перемешивают при 15°C в течение 60 ч. Реакционную смесь разбавляют водой (300 мл) и водный слой экстрагируют петролейным эфиром (3×100 мл). Объединенный органический слой концентрируют с получением указанного в заголовке соединения в виде желтого масла. Выход: 60,0 г, 344,4 ммоль, 81,6%. 1H ЯМР (400 MГц, ДМСО-d6) δ 3,51-3,57 (м, 2H), 3,43-3,49 (м, 2H), 3,28 (с, 2H), 3,25 (с, 3H), 1,33 (с, 3H), 1,27 (с, 3H), 0,79 (с, 3H).
Стадия 3: Синтез 2-(метоксиметил)-2-метилпропан-1,3-диола
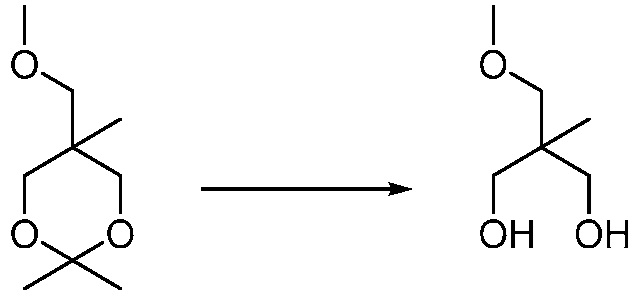
К суспензии 5-(метоксиметил)-2,2,5-триметил-1,3-диоксана (55,0 г, 315,7 ммоль) в метаноле (316 мл), охлажденной до 0°C, добавляют хлористоводородную кислоту (31,6 мл, 3M водную). Реакционную смесь перемешивают при 25°C в течение 30 мин. Реакционную смесь концентрируют и к остатку добавляют воду (100 мл). Водный слой экстрагируют петролейным эфиром (3×200 мл) и затем лиофилизируют с получением указанного в заголовке соединения в виде желтого масла. Выход: 32,2 г, 239,8 ммоль, 75,9%. 1H ЯМР (400 MГц, CDCl3) δ 3,65-3,69 (м, 2H), 3,62 (с, 2H), 3,54-3,58 (м, 2H), 3,37 (с, 2H), 3,34 (с, 3H), 0,81 (с, 3H).
Стадия 4: Синтез 3-((трет-бутилдиметилсилил)окси)-2-(метоксиметил)-2-метилпропан-1-ола
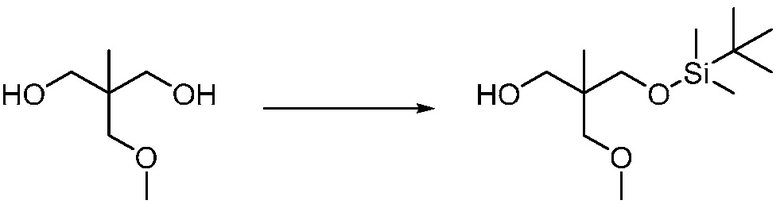
К раствору 2-(метоксиметил)-2-метилпропан-1,3-диола (515 мг, 3,84 ммоль) в тетрагидрофуране (25 мл), охлажденному до 0°C, добавляют гидрид натрия (144 мг, 3,61 ммоль, 60% дисперсия в минеральном масле). Реакционную смесь перемешивают при 0°C в течение 15 мин. трет-Бутилдиметилсилилхлорид (579 мг, 3,84 ммоль) в тетрагидрофуране (5 мл) добавляют по каплям. Реакционную смесь перемешивают при 0°C в течение 30 мин и при КТ в течение 3 ч. Реакционную смесь затем разбавляют метанолом (1 мл), насыщенным водным раствором бикарбоната натрия (10 мл) и водой (15 мл). Водный слой экстрагируют дихлорметаном (2×40 мл). Объединенные органические слои сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле (Гептан:Этилацетат, градиент 0-60%) с получением указанного в заголовке соединения, Выход: 446 мг, 1,80 ммоль, 46,8%. 1H ЯМР (400 MГц, CDCl3) δ 3,55-3,64 (м, 4H), 3,37 (д, J=3,90 Гц, 2H), 3,35 (с, 3H), 0,90-0,91 (с, 9H), 0,82 (с, 3H), 0,05-0,08 (с, 6H).
Стадия 5: Синтез 1-(3-((трет-бутилдиметилсилил)окси)-2-(метоксиметил)-2-метилпропил)-2-(этоксиметил)-6,7-диметил-N-(2,4,4-триметилпентан-2-ил)-1H-имидазо[4,5-c]пиридин-4-амина
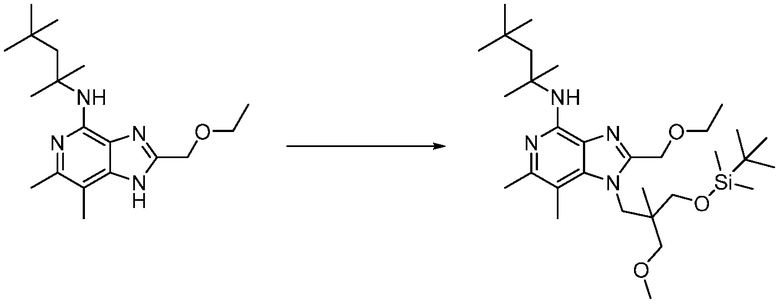
К раствору 2-(этоксиметил)-6,7-диметил-N-(2,4,4-триметилпентан-2-ил)-1H-имидазо[4,5-c]пиридин-4-амина (69 мг, 0,21 ммоль) и 3-((трет-бутилдиметилсилил)окси)-2-(метоксиметил)-2-метилпропан-1-ола (111 мг, 0,42 ммоль) в толуоле (1 мл) добавляют цианометилентрибутилфосфоран (125 мг, 0,519 ммоль, 0,519 мл, 1,0M в ацетонитриле). Реакционную смесь перемешивают при 100°C в течение 16 ч. Реакционную смесь абсорбируют на силикагель и очищают хроматографией на силикагеле (Гептан:Этилацетат, градиент 0-60%) с получением указанного в заголовке соединения. Выход: 69 мг, 0,12 ммоль, 59%. ЖХМС m/z 563,7 [M+H]+. 1H ЯМР (400 MГц, MeOD) δ 4,38-4,78 (м, 4H), 3,44-3,65 (м, 4H), 3,02-3,22 (м, 2H), 2,47 (с, 3H), 2,41 (с, 3H), 2,08 (шс, 2H), 1,56 (с, 6H), 1,25-1,35 (м, 4H), 1,19 (т, J=6,83 Гц, 3H), 0,94 (с, 9H), 0,92 (с, 9H), 0,69 (с, 3H), 0,07 (с, 6H).
Стадия 6: Синтез Примера (6) и (7) (R)-3-(4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-2-(метоксиметил)-2-метилпропан-1-ола и (S)-3-(4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-2-(метоксиметил)-2-метилпропан-1-ола
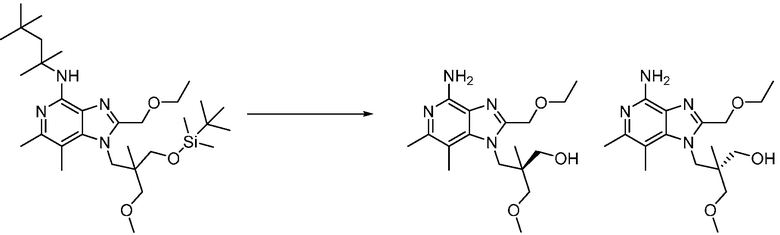
К раствору 1-(3-((трет-бутилдиметилсилил)окси)-2-(метоксиметил)-2-метилпропил)-2-(этоксиметил)-6,7-диметил-N-(2,4,4-триметилпентан-2-ил)-1H-имидазо[4,5-c]пиридин-4-амина (69 мг, 0,12 ммоль) в гексафторизопропаноле (5,0 мл) добавляют метансульфоновую кислоту (70,7 мг, 0,735 ммоль, 0,048 мл). Реакционную смесь перемешивают в течение 5ч при КТ. Реакционную смесь разбавляют насыщенным водным раствором бикарбоната натрия и промывают этилацетатом (1 x) и дихлорметаном (1 x). Органические слои объединяют, сушат над безводным сульфатом натрия, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле (Дихлорметан:Метанол, 0-30% градиент). Продукт собирают, концентрируют и фильтруют через нейлоновый диск. Растворитель удаляют с получением 41 мг рацемата. Рацемат затем растворяют в 1 мл этанола и очищают сверхкритической жидкостной хроматографией. (Колонка: Phenomenex Lux Cellulose 4 5 мкм 21×250 мм); Подвижная фаза A: Метанол об./0,2% Гидроксид аммония (об./об.); Подвижная фаза B: CO2 (об./об.); Градиент: 70,0% CO2/30,0% Метанол об./0,2% Гидроксид аммония изократный, в течение 5 мин. Поток: 75 мл/мин. Обратное давление: 120 бар), с получением энантиомера 1 в качестве Примера (6): 13,8 мг, 0,041 ммоль, 33,6%, 99% эи, и энантиомера 2 в качестве Примера (7): 13,1 мг, 0,039 ммоль, 31,9%, 99% эи. Время удержания СЖХ энантиомера 1 в качестве Примера (6): 3,04 мин, m/z 337,5 [M+H]+, время удержания СЖХ энантиомера 2 в качестве Примера (7): 4,15 мин, m/z 337,5 [M+H]+ (Колонка: Phenomenex Lux Cellulose 4 5 мкм 4,6×100 мм; Подвижная фаза A: Метанол об./0,2% Гидроксид аммония (об./об.); Подвижная фаза B: CO2 (об./об.); Градиент: 60,0% CO2 40,0% Метанол об./0,2% Гидроксид аммония изократный, в течение 5 мин. Поток: 1,5 мл/мин. Обратное давление: 120 Bar). Конкретная стереохимия Примера (6) и Примера (7) не определена, но каждый энантиомер имеет 99% эи, как указано выше.
Пример (8): 2-((4-амино-6,7-диметил-2-(2,2,2-трифторэтил)-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диол, трифторацетат
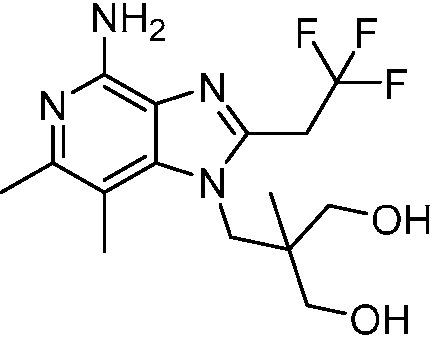
Стадия 1: Синтез N-(2-(бис(4-метоксибензил)амино)-5,6-диметил-4-(((2,2,5-триметил-1,3-диоксан-5-ил)метил)амино)пиридин-3-ил)-3,3,3-трифторпропанамида
К раствору 3,3,3-трифторпропионовой кислоты (15,1 мг, 0,118 ммоль, 0,0104 мл) добавляют N2,N2-бис(4-метоксибензил)-5,6-диметил-N4-((2,2,5-триметил-1,3-диоксан-5-ил)метил)пиридин-2,3,4-триамин (60 мг, 0,11 ммоль) и триэтиламин (22,7 мг, 0,224 ммоль, 0,031 мл). Ангидрид пропилфосфоновой кислоты (143 мг, 0,224 ммоль, 0,101 мл, 50% в этилацетате) добавляют, и реакционную смесь перемешивают при КТ в течение 1 ч. Воду добавляют, и водный слой промывают дважды этилацетатом. Органические слои объединяют и сушат над безводным сульфатом натрия, фильтруют и концентрируют. Неочищенное соединение применяют на следующей стадии без дальнейшей очистки или анализа, выход: 60 мг, 0,093 ммоль, 83%.
Стадия 2: Синтез N, N-бис(4-метоксибензил)-6,7-диметил-2-(2,2,2-трифторэтил)-1-((2,2,5-триметил-1,3-диоксан-5-ил)метил)-1H-имидазо[4,5-c]пиридин-4-амина
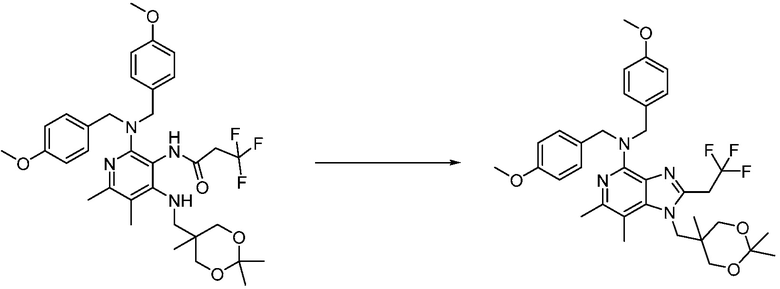
К раствору N-(2-(бис(4-метоксибензил)амино)-5,6-диметил-4-(((2,2,5-триметил-1,3-диоксан-5-ил)метил)амино)пиридин-3-ил)-3,3,3-трифторпропанамида (40 мг, 0,062 ммоль) добавляют четыреххлористый углерод (1 мл). Туда добавляют триэтиламин (18,8 мг, 0,186 ммоль, 0,026 мл) и трифенилфосфин (48,8 мг, 0,186 ммоль). Реакционную смесь нагревают при 80°C в течение 16 ч. Реакционную смесь затем охлаждают до КТ и фильтруют через Celite®, и фильтровальную лепешку промывают этилацетатом. Фильтрат концентрируют, и остаток очищают хроматографией на силикагеле (Гептан:Этилацетат, градиент 0-30%) с получением указанного в заголовке соединения. Выход: 10 мг, 0,016 ммоль, 26%. 1H ЯМР (400 MГц, CDCl3) δ 7,27-7,30 (м, 4H, предполагаемый, частично закрыт остаточным CHCl3), 6,82 (д, J=8,59 Гц, 4H), 5,25-5,37 (м, 2H), 4,83-5,01 (м, 3H), 4,33-4,58 (м, 2H), 3,79 (с, 6H), 3,76-3,84 (м, 1H, предполагаемый, частично закрыт пиком при 3,79 ч./млн.), 3,60-3,74 (м, 2H), 3,48-3,56 (м, 1H), 3,19-3,27 (м, 1H), 2,45 (с, 3H), 2,44 (с, 3H), 1,50 (с, 3H), 1,48 (с, 3H), 0,63 (с, 3H). ЖХМС m/z 627,5 [M+H]+.
Стадия 3: Синтез Примера (8): 2-((4-амино-6,7-диметил-2-(2,2,2-трифторэтил)-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диола, трифторацетата
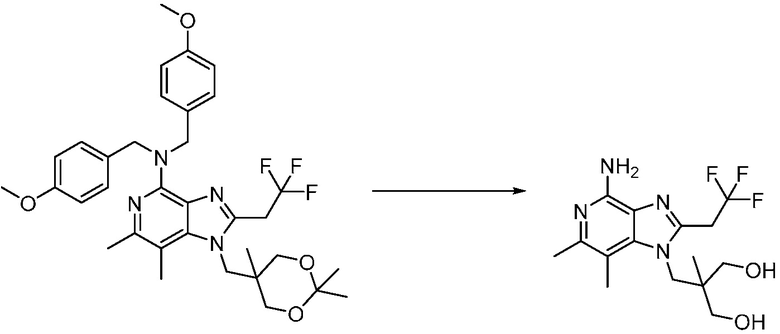
К раствору N, N-бис(4-метоксибензил)-6,7-диметил-2-(2,2,2-трифторэтил)-1-((2,2,5-триметил-1,3-диоксан-5-ил)метил)-1H-имидазо[4,5-c]пиридин-4-амина (10 мг, 0,016 ммоль) в гексафторизопропаноле (0,5 мл) добавляют метансульфоновую кислоту (2 капли). Реакционную смесь перемешивают при КТ в течение 3 ч. Реакционную смесь затем концентрируют и остаток разбавляют 1 мл диметилсульфоксида и очищают ЖХВД с обращенной фазой (Колонка: Waters Sunfire C18 19×100, 5u; Подвижная фаза A: 0,05% ТФК в воде (об./об.); Подвижная фаза B: 0,05% ТФК в ацетонитриле (об./об.); Градиент: 95,0% H2O/5,0% Ацетонитрил линейный до 70% H2O/30% Ацетонитрил за 8,5 мин до 0% H2O/100% MeCN до 9,0 мин, выдержка при 0% H2O/100% Ацетонитрил от 9,0 до 10,0 мин. Поток: 25 мл/мин.) с получением указанного в заголовке соединения, выход: 5,5 мг, 0,012, 75%; ЖХВД время удержания: 1,31 мин. (Колонка: Waters Atlantis® dc18 4,6×50, 5u; Подвижная фаза A: 0,05% ТФК в воде (об./об.); Подвижная фаза B: 0,05% ТФК в ацетонитриле (об./об.); 95,0% H2O/5,0% Ацетонитрил линейный до 5% H2O/95% Ацетонитрил за 4,0 мин, выдержка при 5% H2O/95% Ацетонитрил до 5,0 мин. Поток: 2 мл/мин.). ЖХВД m/z 347,5 [M+H]+.
Пример (9): 2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-этилпропан-1,3-диол, трифторацетат
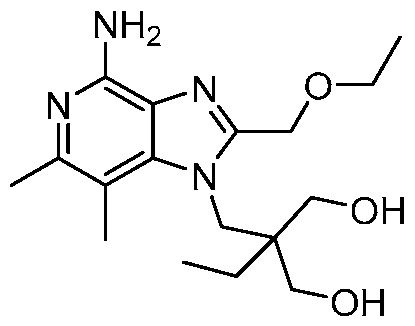
Получают по методике, аналогичной Примеру (2), с применением (5-этил-2,2-диметил-1,3-диоксан-5-ил)метанола (Polymer Chemistry, 8(3), 592-604; 2017), на стадии 5. Продукт выделяют ЖХВД с обращенной фазой (Колонка: Waters Sunfire C18 19×100, 5u; Подвижная фаза A: 0,05% ТФК в воде (об./об.); Подвижная фаза B: 0,05% ТФК в ацетонитриле (об./об.); Градиент: 95,0% H2O/5,0% Ацетонитрил линейный до 45% H2O/55% Ацетонитрил за 8,5 мин до 0% H2O/100% MeCN до 9,0 мин, выдержка при 0% H2O/100% Ацетонитрил от 9,0 до 10,0 мин. Поток: 25мл/мин.) с получением указанного в заголовке соединения, выход: 25,2 мг, 0,056 ммоль, 46,7%; ЖХВД время удержания: 1,35 мин (Колонка: Waters Atlantis® dc18 4,6×50, 5u; Подвижная фаза A: 0,05% ТФК в воде (об./об.); Подвижная фаза B: 0,05% ТФК в ацетонитриле (об./об.); 95,0% H2O/5,0% Ацетонитрил линейный до 5% H2O/95% Ацетонитрил за 4,0 мин, выдержка при 5% H2O/95% Ацетонитрил до 5,0 мин. Поток: 2 мл/мин.); ЖХВД m/z 337,5 [M+H]+.
Пример (10): 2-((4-амино-2-бутил-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диол
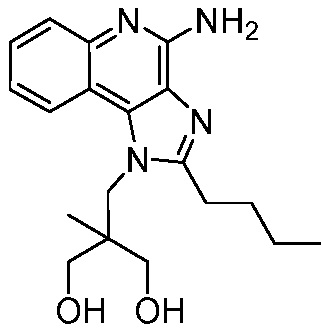
Получают по методике, аналогичной Примеру (1) 2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диолу, начиная с хинолин-2,4-диола на стадии 1 и применяя валероилхлолрид на стадии 4. Получают 37,8 мг. ЖХВД время удержания: 1,69 мин (Колонка: Waters Atlantis® dc18 4,6×50, 5u; Подвижная фаза A: 0,05% ТФК в воде (об./об.); Подвижная фаза B: 0,05% ТФК в ацетонитриле (об./об.); Градиент: 95,0% H2O/5,0% Ацетонитрил линейный до 5% H2O/95% Ацетонитрил за 4,0 мин, выдержка при 5% H2O/95% Ацетонитрил до 5,0 мин. Поток: 2 мл/мин.). ЖХВД m/z 343,5 [M+H]+.
Пример (11): 2-((4-амино-2-бутил-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диол
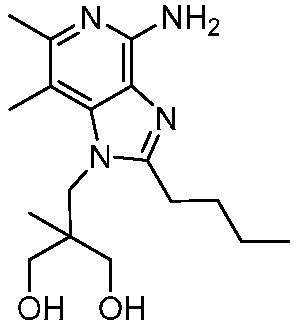
Получают по методике, аналогичной 2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диолу, применяя валероилхлорид в Примере 1, стадия 4. Получают 260 мг. 1H ЯМР (ДМСО-d6, 400 MГц) δ 5,56 (с, 2H), 4,8-5,0 (м, 2H), 4,3-4,5 (м, 1H), 4,1-4,3 (м, 1H), 3,1-3,3 (м, 3H), 3,0-3,1 (м, 1H), 2,8-3,0 (м, 2H), 2,40 (с, 3H), 2,28 (с, 3H), 1,6-1,7 (м, 2H), 1,3-1,4 (м, 2H), 0,91 (т, 3H, J=7,2 Гц), 0,45 (с, 3H). ЖХМС m/z 321,2 [M+H]+.
Пример (12): 2-((4-амино-2-пентил-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диол
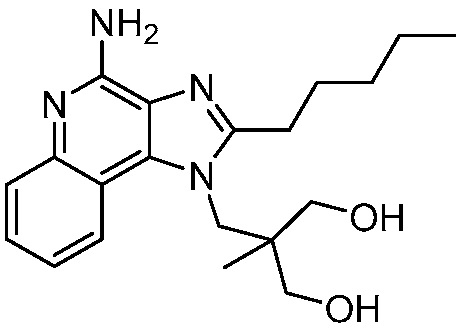
Получают по методике, аналогичной Примеру (1) 2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диолу, начиная с хинолин-2,4-диола на стадии 1 и применяя гексаноилхлорид на стадии 4. Получают 31,2 мг. ЖХВД время удержания: 1,88 мин (Колонка: Waters Atlantis® dc18 4,6×50, 5u; Подвижная фаза A: 0,05% ТФК в воде (об./об.); Подвижная фаза B: 0,05% ТФК в ацетонитриле (об./об.); Градиент: 95,0% H2O/5,0% Ацетонитрил линейный до 5% H2O/95% Ацетонитрил за 4,0 мин, выдержка при 5% H2O/95% Ацетонитрил до 5,0 мин. Поток: 2 мл/мин.). ЖХВД m/z 357,5 [M+H]+.
Пример (13): 2-((4-амино-2-бутил-6,7,8,9-тетрагидро-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диол, формиат
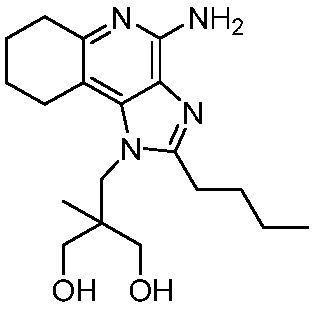
Стадия 1: Синтез 3-нитро-5,6,7,8-тетрагидрохинолин-2,4-диола
В 2 л колбу добавляют серную кислоту (275 мл). Реакционную смесь охлаждают на ледяной бане и 5,6,7,8-тетрагидрохинолин-2,4-диол (65 г, 390 ммоль) добавляют порциями в течение 15 минут. Реакционную смесь перемешивают в течение еще 10 минут. Азотную кислоту (39,6 мл, 885 ммоль) добавляют порциями со скоростью, сохраняющей внутреннюю температуру реакции ниже 30°C. Реакционную смесь перемешивают при КТ в течение еще 2 часов. Реакционную смесь медленно выливают в лед (2 л), твердые вещества фильтруют и промывают водой. Твердые вещества сушат под вакуумом при 50°C. Выход: 52,2 г, 390 ммоль, 63%. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 12,25 (шс, 1 H), 11,75 (шс, 1 H), 2,48 (м, 2 H), 2,34 (м, 2 H), 1,66 (м, 4 H). ЖХМС m/z 211,2 [M+H].
Стадия 2: Синтез 2,4-дихлор-3-нитро-5,6,7,8-тетрагидрохинолин
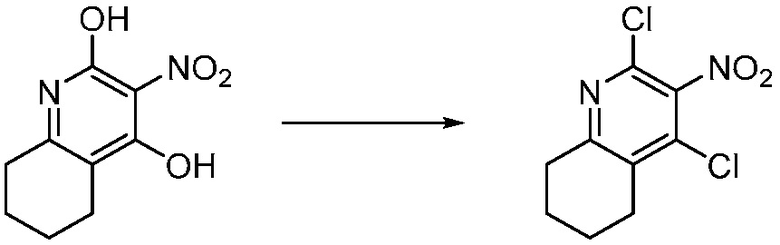
В 500 мл колбу добавляют 3-нитро-5,6,7,8-тетрагидрохинолин-2,4-диол (13,1 г, 53,0 ммоль), дихлорэтан (70,0 мл), оксихлорид фосфора (V) (65,1 г, 425 ммоль, 40,0 мл). Реакционную смесь нагревают при 80°C в течение 16 часов. Реакционную смесь затем охлаждают до КТ и выпаривают, затем выпаривают из толуола (2x). Остаток фильтруют через слой диоксида кремния с дихлорметаном, и элюент выпаривают с получением коричневого/оранжевого воскообразного твердого вещества. Выход: 9,2 г, 37,2 ммоль, 70%. 1H ЯМР (400 MГц, CDCl3) δ 2,92-3,02 (м, 2H), 2,77-2,85 (м, 2H), 1,85-1,95 (м, 4H). GCMS m/z 246,0. Дополнительный продукт получают в аналогичных условиях.
Стадия 3: Синтез 2-хлор-3-нитро-N-((2,2,5-триметил-1,3-диоксан-5-ил)метил)-5,6,7,8-тетрагидрохинолин-4-амина
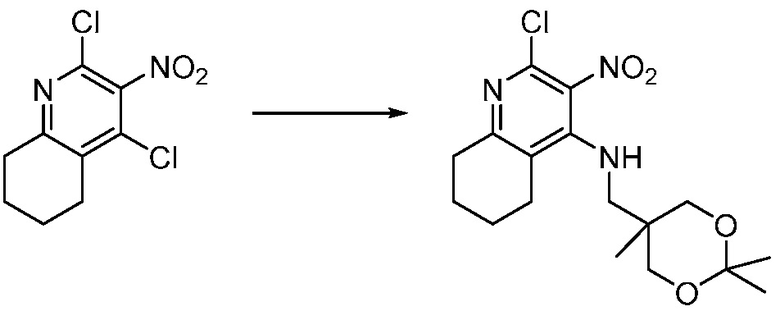
К 2,4-дихлор-3-нитро-5,6,7,8-тетрагидрохинолину (10,9 г, 44,5 ммоль) и диметилацетамиду (55 мл, содержит 20% воды) добавляют триэтиламин (9 г, 89,0 ммоль, 12,4 мл) и затем (2,2,5-триметил-1,3-диоксан-5-ил)метанамин (12,7 г, 80,1 ммоль), и реакционную смесь нагревают при 35°C в течение 16 часов. Реакционную смесь охлаждают до 0°C, добавляют воду (70 мл), реакционную смесь перемешивают в течение 45 мин. Твердые вещества фильтруют и промывают водой с получением указанного в заголовке соединения в виде оранжевого твердого вещества. Выход: 14,78 г, 39,96 ммоль, 89,8%. 1H ЯМР (400 MГц, CDCl3) δ 5,57 (шс, 1H), 3,67-3,78 (м, 4H), 3,21 (д, J=4,68 Гц, 2H), 2,83 (т, J=5,66 Гц, 2H), 2,48 (т, J=5,66 Гц, 2H), 1,78-1,92 (м, 4H), 1,47 (д, J=5,85 Гц, 6H), 0,87 (с, 3H). ЖХМС m/z 370,4 [M+H].
Стадия 4: Синтез N2,N2-бис(4-метоксибензил)-3-нитро-N4-((2,2,5-триметил-1,3-диоксан-5-ил)метил)-5,6,7,8-тетрагидрохинолин-2,4-диамина
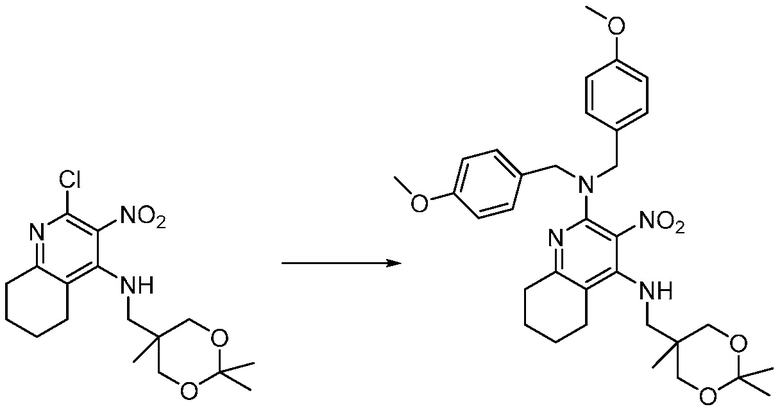
К 2-хлор-3-нитро-N-((2,2,5-триметил-1,3-диоксан-5-ил)метил)-5,6,7,8-тетрагидрохинолин-4-амину (12,98 г, 35,09 ммоль) добавляют бис(4-метоксибензил)амин (27,1 г, 105 ммоль) и изопропанол (65 мл). Реакционную смесь кипятят с обратным холодильником в течение 51 часа, затем перемешивают при КТ в течение 16 часов. Реакционную смесь разбавляют дихлорметаном (100 мл) и фильтруют через Celite®. Твердые вещества промывают дихлорметаном (50 мл). Органические слои концентрируют и разбавляют этанолом (40 мл) и перемешивают при КТ в течение 16 ч. Твердые вещества фильтруют, и колбу промывают этанолом (60 мл). Твердые вещества промывают этанолом (30 мл) и сушат под вакуумом с получением желтого твердого вещества. Выход: 14,5 г, 24,6 ммоль, 70,0%. 1H ЯМР (400 MГц, CDCl3) δ 7,07-7,12 (м, 4H), 6,76-6,84 (м, 4H), 6,39 (т, J=5,66 Гц, 1H), 4,30 (с, 4H), 3,79 (с, 6H), 3,55-3,67 (м, 4H), 3,45 (д, J=5,85 Гц, 2H), 2,73 (т, J=6,44 Гц, 2H), 2,66 (т, J=5,85 Гц, 2H), 1,72-1,86 (м, 4H), 1,44 (с, 3H), 1,41 (с, 3H), 0,82-0,88 (м, 3H). ЖХМС m/z 591,4 [M+H].
Стадия 5: N2,N2-бис(4-метоксибензил)-N4-((2,2,5-триметил-1,3-диоксан-5-ил)метил)-5,6,7,8-тетрагидрохинолин-2,3,4-триамин
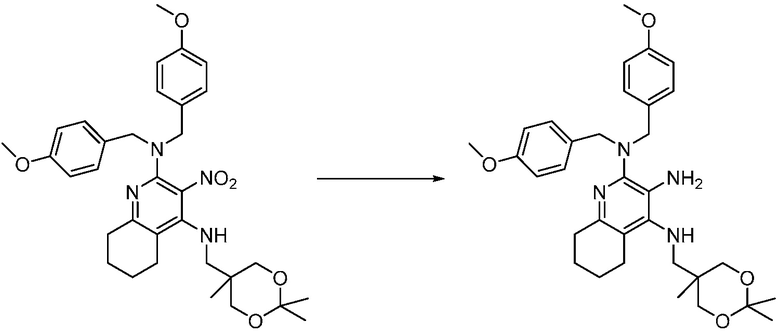
К N2,N2-бис(4-метоксибензил)-3-нитро-N4-((2,2,5-триметил-1,3-диоксан-5-ил)метил)-5,6,7,8-тетрагидрохинолин-2,4-диамину (7,2 г, 12,2 ммоль) добавляют метанол (40,6 мл), формиат аммония (3,84 г, 60,9 ммоль), и порошок цинка (3,99 г, 60,9 ммоль). Реакционную смесь перемешивают в течение 25 минут. Реакционную смесь фильтруют через слой Celite®, и фильтрат концентрируют. Остаток растворяют в этилацетате, промывают водой, насыщенным раствором соли, и органический слой сушат над безводным сульфатом натрия. Реакционную смесь фильтруют и концентрируют с получением указанного в заголовке соединения. Выход: 6,83 г, 12,19 ммоль, 100%, 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,21 (д, J=8,59 Гц, 4 H), 6,81 (д, J=8,59 Гц, 4 H), 4,15 (с, 4 H), 3,99 (с, 2 H), 3,76-3,83 (м, 8 H), 3,63-3,70 (м, 2 H), 3,21 (шс, 2 H), 2,74 (шт, J=5,66 Гц, 2 H), 2,55 (шт, J=5,46 Гц, 2 H), 1,74-1,87 (м, 4 H), 1,48 (с, 3 H), 1,46 (с, 3 H), 0,93 (с, 3 H) ЖХМС m/z 561,5 [M+H].
Стадия 6: Синтез N-(2-(бис(4-метоксибензил)амино)-4-(((2,2,5-триметил-1,3-диоксан-5-ил)метил)амино)-5,6,7,8-тетрагидрохинолин-3-ил)пентанамида
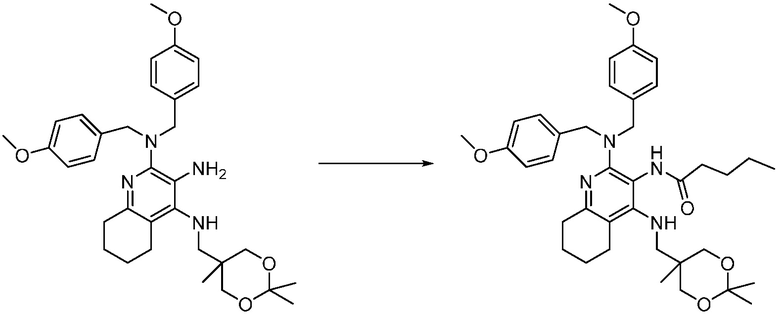
К N2,N2-бис(4-метоксибензил)-N4-((2,2,5-триметил-1,3-диоксан-5-ил)метил)-5,6,7,8-тетрагидрохинолин-2,3,4-триамину (6,84 г, 12,19 ммоль) добавляют дихлорметан (60,9 мл), воду (30,5 мл) и бикарбонат натрия (2,56 г, 30,5 ммоль). Валероилхлорид (1,62 г, 13,4 ммоль, 1,59 мл) добавляют по каплям в течение 1 минуты и затем перемешивают в течение 55 минут. Органический слой отделяют, и водный промывают дихлорметаном. Объединенные органические слои промывают насыщенным раствором соли, сушат над безводным сульфатом натрия, фильтруют и концентрируют с получением указанного в заголовке соединения, которое используют без дальнейшей очистки. Выход: 7,86 г, 12,19 ммоль. ЖХМС m/z 645,7 [M+H].
Стадия 7: Синтез 2-бутил-N, N-бис(4-метоксибензил)-1-((2,2,5-триметил-1,3-диоксан-5-ил)метил)-6,7,8,9-тетрагидро-1H-имидазо[4,5-c]хинолин-4-амина
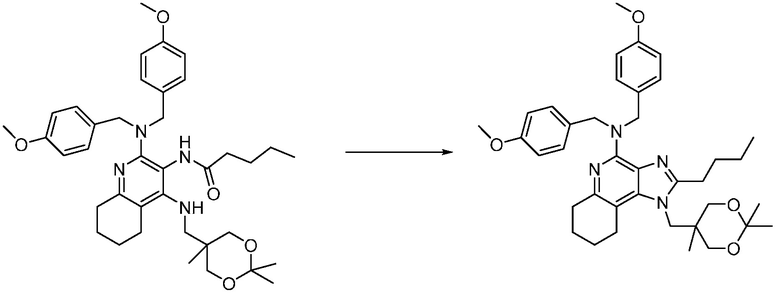
К N-(2-(бис(4-метоксибензил)амино)-4-(((2,2,5-триметил-1,3-диоксан-5-ил)метил)амино)-5,6,7,8-тетрагидрохинолин-3-ил)пентанамиду (7,86 г, 12,19 ммоль) добавляют этанол (122 мл) и гидроксид натрия (4,88 г, 60,9 ммоль, 3,22 мл, 50% масс. раствор в воде). Реакционную смесь нагревают при 100°C в течение 48 часов. Реакционную смесь охлаждают до КТ и твердые вещества фильтруют, промывают этанолом и сушат с получением указанного в заголовке соединения. Выход: 6,7 г, 10,7 ммоль, 87,7%. 1H ЯМР (400 MГц, ХЛОРОФОРМ-d) δ ч./млн. 7,26 (д, J=8,59 Гц, 4 H), 6,81 (д, J=8,59 Гц, 4 H), 4,96-5,40 (м, 4 H), 4,39-4,72 (м, 2 H), 3,79 (с, 6 H), 3,41-3,69 (м, 4 H), 2,63-3,13 (м, 6 H), 1,84 (шс, 4 H), 1,69 (квин, J=7,51 Гц, 2 H), 1,57 (шс, 4 H), 1,46 (с, 6 H), 1,35 (дкв, J=14,83, 7,41 Гц, 2 H), 0,89 (т, J=7,41 Гц, 3 H), 0,57 (с, 3 H). ЖХМС m/z 627,7 [M+H].
Стадия 8: Синтез 2-((4-амино-2-бутил-6,7,8,9-тетрагидро-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диола
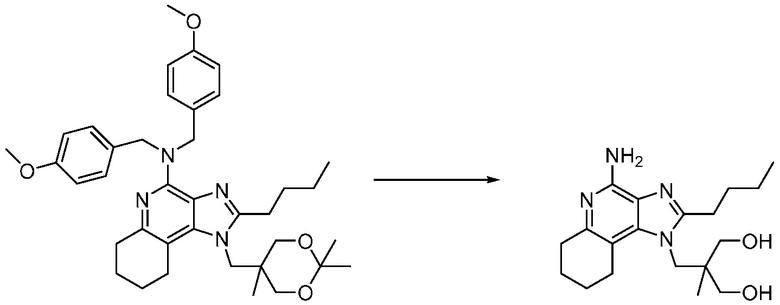
К 2-бутил-N, N-бис(4-метоксибензил)-1-((2,2,5-триметил-1,3-диоксан-5-ил)метил)-6,7,8,9-тетрагидро-1H-имидазо[4,5-c]хинолин-4-амину (6,70 г, 10,69 ммоль) добавляют толуол (32,4 мл) и концентрированную хлористоводородную кислоту (21 мл). Реакционную смесь нагревают при 60°C в течение 3,25 часов. Водный слой промывают толуол, нагревают до 60°C и доводят до pH 10 твердым карбонатом натрия. Реакционную смесь затем перемешивают при 60°C в течение 1 часа и 40 минут, затем охлаждают до КТ. Твердые вещества фильтруют, промывают водой и сушат под вакуумом при 40°C с получением Примера (13). Выход: 2,44 г, 7,04 ммоль, 65,9% Выход. 1H ЯМР (400 MГц, ДМСО-d6) δ ч./млн. 5,67 (шс, 2 H), 4,78 (шс, 2 H), 4,07-4,45 (м, 2 H), 2,71-3,30 (м, 8 H), 2,66 (шс, 2 H), 1,53-1,93 (м, 6 H), 1,35 (м, J=7,34, 7,34, 7,34, 7,34, 7,34 Гц, 2 H), 0,91 (т, J=7,41 Гц, 3 H), 0,43 (с, 3 H). ЖХМС m/z 347,3 [M+H].
Пример (14): 2-((4-амино-2-бутил-7,8-дигидроциклопента[b]имидазо[4,5-d]пиридин-1(6H)-ил)метил)-2-метилпропан-1,3-диол
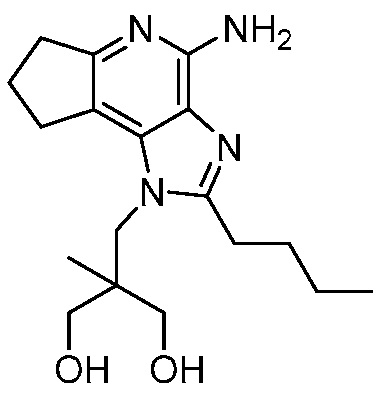
Получают по методике, аналогичной Примеру 13, начиная с 6,7-дигидро-5H-циклопента[b]пиридин-2,4-диола. Получают 336 мг (42,5% выход). Очищают ЖХВД (Колонка: Welch Xtimate 75*40 мм*3 мкм; Подвижная фаза A: 0,05% NH4OH в воде (об./об.); Подвижная фаза B: Ацетонитрил; Градиент: 80% A до 40% A/60% B за 10 мин, выдержка при 0% H2O/100% Ацетонитрил в течение 4 мин. Поток: 25 мл/мин.). ЖХМС m/z 333,1 [M+H]+. 1H ЯМР (400 MГц, ДМСО) δ 5,72 (с, 2H), 4,80 (шс, 2H), 4,14 (с, 2H), 3,25 (шс, 3H), 2,99-3,34 (м, 3H), 2,88 (шс, 2H), 2,62-2,78 (м, 2H), 2,02 (предполагаемый квин, J=7,34 Гц, 2H), 1,64-1,75 (м, 2H), 1,35 (предполагаемый квд, J=7,47, 14,74 Гц, 2H), 0,91 (т, J=7,28 Гц, 3H), 0,50 (с, 3H).
Пример (15): 2-((4-амино-2-этил-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диол
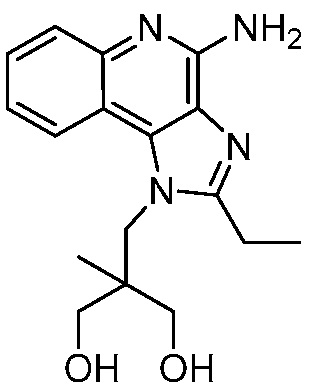
Получают по методике, аналогичной 2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диолу, начиная с хинолин-2,4-диола. Получают 301 мг. Очищают ЖХВД (Колонка: Phenomenex Gemini NX-C18 150*30 мм*5 мкм; Подвижная фаза A: 0,05% NH4OH в воде (об./об.); Подвижная фаза B: Ацетонитрил; Градиент: 95% A до 55% A/45% B за 7 мин, выдержка при 0% H2O/100% Ацетонитрил в течение 2 мин. Поток: 30 мл/мин.) 1H ЯМР (400MГц, ДМСО-d6) d 8,51 (д, J=8,1 Гц, 1H), 7,58 (д, J=8,3 Гц, 1H), 7,38 (т, J=7,5 Гц, 1H), 7,18 (т, J=7,4 Гц, 1H), 6,43 (с, 2H), 4,98 (шс, 2H), 4,78 (шс, 1H), 4,45 (шс, 1H), 3,19 (шс, 4H), 3,02 (кв, J=7,4 Гц, 2H), 1,34 (т, J=7,4 Гц, 3H), 0,55 (с, 3H). ЖХМС m/z 315,1 [M+H]+.
Пример (16): 2-((4-амино-2-пропил-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диол
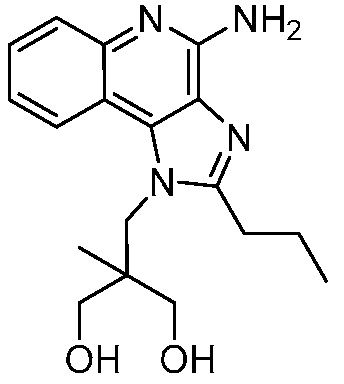
Получают по методике, аналогичной Примеру 13, начиная с хинолин-2,4-диола. Получают 15,8 мг. Очищают ЖХВД (Колонка: Waters XBridge C18 19×100, 5 мкм; Подвижная фаза A: 0,03% NH4OH в воде (об./об.); Подвижная фаза B: 0,03% NH4OH Ацетонитрил; Градиент: 95% A до 50% A/50% B за 8,5 мин, выдержка при 0% H2O/100% Ацетонитрил в течение 1 мин. Поток: 25 мл/мин;). ЖХВД QC (Колонка: Waters Atlantis dC18 4,6×50, 5 мкм; Подвижная фаза A: 0,05% ТФК в воде (об./об.); Подвижная фаза B: 0,05% ТФК Ацетонитрил; Градиент: 95% A до 5% A/95% B за 4,0 мин, выдержка при 5% H2O/95% Ацетонитрил в течение 1 мин. Поток: 2 мл/мин.; время удержания: 1,38 мин). ЖХМС m/z 329,5 [M+H]+.
Пример (17): 2-((4-амино-2-(2-метоксиэтил)-7,8-дигидроциклопента[b]имидазо[4,5-d]пиридин-1(6H)-ил)метил)-2-метилпропан-1,3-диол
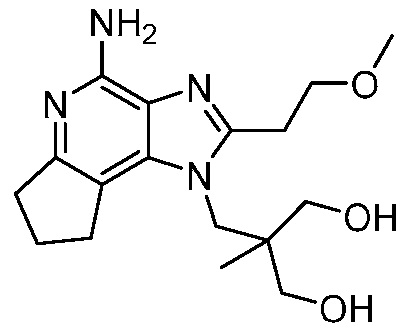
Пример 17 получают по методике, аналогичной 2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диолу, начиная с 6,7-дигидро-5H-циклопента[b]пиридин-2,4-диола. Получают 70 мг. Очищают ЖХВД (Колонка: Phenomenex Gemini NX-C18 75*30 мм*3 мкм; Подвижная фаза A: 0,05% NH4OH в воде (об./об.); Подвижная фаза B: Ацетонитрил; Градиент: 100% A до 70% A/30% B за 7 мин, выдержка при 0% H2O/100% Ацетонитрил в течение 2 мин. Поток: 30 мл/мин.). ЖХМС m/z 335,1 [M+H]+. 1H ЯМР (400 MГц, ДМСО) δ 5,87 (шс, 2H), 4,81 (шс, 2H), 4,17 (шс, 2H), 3,71 (предполагаемый т, J=6,72 Гц, 2H), 3,30 (шс, 3H), 3,20-3,28 (м, 4H), 3,23 (с, 3H), 2,68-2,79 (м, 2H), 2,03 (квин, J=7,27 Гц, 2H), 0,50 (с, 3H). 1 протон не наблюдается (закрыт).
Пример (18): 2-((4-амино-2-(2-метоксиэтил)-6,7,8,9-тетрагидро-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диол
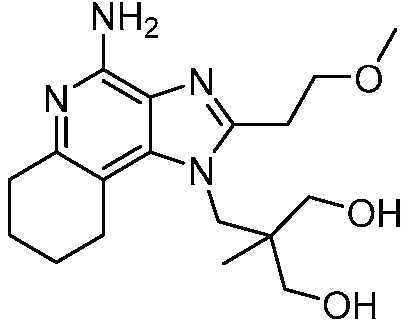
Пример (18) получают по методике, аналогичной 2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диола, начиная с 5,6,7,8-тетрагидро-хинолин-2,4-диола. Получают 68 мг. Очищают ЖХВД (Колонка: Phenomenex Gemini NX-C18 75*30 мм*3 мкм; Подвижная фаза A: 0,05% NH4OH в воде (об./об.); Подвижная фаза B: Ацетонитрил; Градиент:100% A до 60% A/40% B за 7 мин, выдержка при 0% H2O/100% Ацетонитрил в течение 2 мин. Поток: 30 мл/мин.). 1H ЯМР (400MГц, ДМСО-d6) δ 5,70 (с, 2H), 4,82 (шс, 2H), 4,29 (шс, 2H), 3,69 (шс, 2H), 3,30-2,72 (м, 11H), 2,66 (шс, 2H), 1,74 (шс, 4H), 0,43 (с,3H). ЖХМС m/z 349,2 [M+H]+.
Пример (19): 2-((4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диол
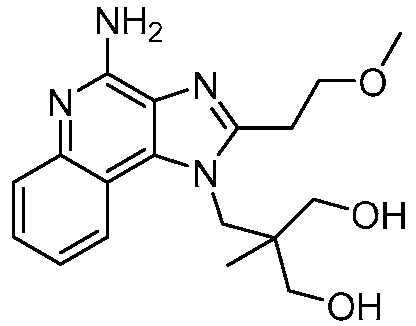
Пример (19) получают по методике, аналогичной 2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диолу, начиная с хинолин-2,4-диола. Получают 108 мг. Очищают ЖХВД (Колонка: Phenomenex Gemini NX-C18 75*30 мм*3 мкм; Подвижная фаза A: 0,05% NH4OH в воде (об./об.); Подвижная фаза B: Ацетонитрил; Градиент: 97% A до 57% A/43% B за 7 мин, выдержка при 0% H2O/100% Ацетонитрил в течение 2 мин. Поток: 25 мл/мин.;) ЖХМС m/z 345,3 [M+H]+. 1H ЯМР (400MГц, ДМСО-d6) δ=8,52 (д, J=8,3 Гц, 1H), 7,58 (дд, J=1,1, 8,3 Гц, 1H), 7,43-7,34 (м, 1H), 7,23-7,14 (м, 1H), 6,45 (с, 2H), 5,00 (т, J=4,8 Гц, 2H), 4,76 (шс, 1H), 4,53 (шс, 1H), 3,79 (шс, 2H), 3,43 (шд, J=5,8 Гц, 3H), 3,30-3,24 (м, 4H), 3,19 (шс, 2H), 0,55 (с, 3H).
Биологическое тестирование
Описанные примеры тестируют на биологическую активность в функциональных клеточных анализах с использованием клеток HEK293, стабильно сверхэкспрессирующих TLR7 или TLR8 человека. Анализы проверяют способность каждого примера стимулировать секрецию интерферона-альфа (IFNα) в первичных мононуклеарных клетках крови человека (PBMC).
Функциональные анализы клеток hTLR7 и hTLR8
Для определения способности каждого примера активировать toll-подобный рецептор 7 человека (hTLR7) или toll-подобный рецептор 8 человека (hTLR8) используют клеточные репортерные системы. Клетки HEK293, стабильно сверхэкспрессирующие либо hTLR7, либо hTLR8 вместе с репортерным геном, содержащим оптимизированный секретированный эмбриональный ген щелочной фосфатазы (SEAP), под контролем минимального IFN-b промотора, слитого с пятью сайтами связывания NF-κB и AP-1, получают от Invivogen (HEK-Blue™ hTLR7, кат. № Hkb-htlr7; HEK-Blue™ hTLR8, кат. № Hkb-htlr8). Стимуляция hTLR7 или hTLR8 в этих клетках активирует NF-κB и AP-1 и индуцирует продуцирование SEAP, которое можно количественно определить с помощью реагента для обнаружения щелочной фосфатазы.
Клетки поддерживают в среде Игла, модифицированной по Дульбекко (DMEM) (содержащей фетальную телячью сыворотку (FBS), инактивированную нагреванием (10%), глутамакс (2 мМ), пенициллин/стрептомицин, бластицидин (10 мкг/мл), зеоцин (100 мкг/мл) и нормоцин (100 мкг/мл)). В первый день анализа образцы получают с использованием 11-точечных полулогарифмических серийных разведений из 10 мМ маточного раствора ДМСО, и 50 нл помещают в 384-луночные планшеты Viewplates (PerkinElmer, кат. № 6007480). Положительный контрольный агонист TLR7/8 и отрицательный контроль (ДМСО, без примера) также помещают в планшет для анализа и используют для определения доли эффекта в процессе анализа. После ресуспендирования в среде для анализа DMEM, содержащей инактивированную нагреванием FBS (10%), глутамакс (2 мМ) и пенициллин/стрептомицин, в предварительно приготовленные планшеты добавляют 10000 клеток/20 мкл/лунку. Планшеты инкубируют в течение ночи (16-20 часов) при 37°С в среде с 5% CO2. Предварительно смоченные крышки Microclime (Labcyte, LLS-0310) используют для предотвращения испарения. На второй день анализа, реагент для обнаружения QUANTI-Blue™ готовят путем восстановления порошка QUANTI-Blue™ (InvivoGen, Rep-qb1) 100 мл стерильной воды и выдерживают для уравновешивания до 37°С в течение 15 минут. В каждую лунку добавляют по 20 мкл реагента для обнаружения QUANTI-Blue™, и планшеты инкубируют при комнатной температуре в течение 180 мин. В конце инкубации планшеты считывают с помощью планшетного ридера Envision (Perkin Elmer), измеряющего поглощение при 650 нм.
Используя положительный (агонист TLR7/8 ) и отрицательный (ДМСО) контроли, долю (%) эффекта рассчитывают для каждого примера с использованием следующего уравнения:
% эффекта=100 - [100 * {(Пример - Положительный контроль) / (Отрицательный контроль - Положительный контроль)}]
% эффекта при каждой концентрации каждого примера рассчитывают с использованием пакета программного обеспечения ABase (IBDS) и относят к количеству SEAP, продуцированной в лунках положительного и отрицательного контроля, содержащихся в каждом планшете для анализа. Концентрации и значения % эффекта для каждого примера подгоняют с использованием 4-параметрической логистической модели в ABase, и рассчитывают концентрацию каждого примера, которая дает 50% ответ (EC50).
Анализ INFα из мононуклеарных клеток периферической крови (PBMC)
Для определения способности каждого примера индуцировать высвобождение интерферона-альфа (IFNα) из свежевыделенных мононуклеарных клеток периферической крови (РВМС) используют анализ гомогенной флуоресценции с временным разрешением (HTRF). Цельную кровь человека собирают у здоровых доноров путем пункции вены в соответствии с протоколами Pfizer (протокол № GOHW RDP-01), одобренными Shulman Institutional Review Board. 50 мл образца венозной крови человека от отдельных доноров гепаринизируют добавлением в коническую пробирку, содержащую 714 единиц Heparin Sodium Injection MDV (Fresenius Kabi, кат. № 70041), с последующим осторожным переворачиванием пробирки несколько раз. Затем кровь переносят в колбу, коническую пробирку промывают 40 мл PBS, содержащего 2 мМ EDTA (PBS-EDTA), и промывочную жидкость добавляют в колбу с кровью при осторожном перемешивании. 30 мл разбавленной крови добавляют в 3 отдельные пробирки histopaque (Sigma, кат. № A0561), нанося непосредственно на фритту. Затем пробирки Histopaque центрифугируют в течение 15 минут при 1000 g в настольной центрифуге. После разделения по плотности, избыточную верхнюю фазу плазмы отсасывают в пределах ~5 мл выше границы раздела фаз, и оставшуюся плазму вместе с мутной границей раздела фаз, содержащей РВМС, осторожно декантируют в новую коническую пробирку. 15 мл PBS-EDTA добавляют в пробирку histopaque и осторожно взбалтывают для удаления оставшихся РВМС, прилипших к стенке пробирки, и эту промывку добавляют к существующим РВМС в пробирке. Объем пробирки доводят до 40 мл с помощью PBS-EDTA, и пробирки вращают при 250×g в течение 12 минут при комнатной температуре. После аспирации супернатанта, осадок осторожно ресуспендируют с 10 мл PBS-EDTA и снова центрифугируют при 250×g в течение 12 минут. Полученный супернатант декантируют, и осадок ресуспендируют в 20 мл лизирующего буфера АСК (ThermoFisher, кат. № A10492-01) с последующей инкубацией при комнатной температуре в течение 5 минут. Объем каждой пробирки доводят до 50 мл добавлением PBS-EDTA, и пробирки вращают при 177×g в течение 12 минут при комнатной температуре. Осадок РВМС снова ресуспендируют в 10 мл PBS без ЭДТА, и пробирки последний раз вращают при 177×g в течение 10 минут. Супернатант декантируют, и РВМС ресуспендируют в среде для анализа (базовая среда RPMI с 10% инактивированной нагреванием FBS, 2 мМ глутамакса и пенициллина/стрептомицина).
Каждый пример готовят для анализа с использованием 11-точечных полулогарифмических серийных разведений из исходного раствора 2,5 мМ ДМСО, и 400 нл помещают в 384-луночный планшет Viewplate (Perkin Elmer, кат. № 6007480). Положительные и отрицательные контроли (описанные ранее) также помещают в планшет для анализа и используют для определения доли эффекта в процессе анализа. РВМС подсчитывают и высевают при плотности 100000 клеток/100 мкл/лунку; планшеты накрывают предварительно смоченной крышкой microclime для предотвращения испарения и инкубируют в течение 24 часов при 37°С в атмосфере с 5% CO2. По окончании инкубации, крышку microclime снимают, и планшеты вращают при 1000 об/мин в течение 5 минут. 16 мкл кондиционированной среды из клеточного планшета переносят в отдельный 384-луночный планшет малого объема (Greiner One, 784080). Уровни IFNα определяют количественно с использованием набора HTRF® (Cisbio, кат. № 62HIFNAPEG) в соответствии с инструкциями производителя. Набор содержит два разных специфических антитела, одно из которых помечено D2 (акцептор), и другое помечено криптатом (донор), и буфер для обнаружения. Исходные растворы антител разводят 1:20 в буфере для обнаружения. Для одного 384-луночного планшета 62,5 мкл исходного раствора антител D2 и 62,5 мкл исходного раствора антител криптата добавляют к 2,375 мл буфера для обнаружения и хорошо перемешивают. В каждую лунку, содержащую кондиционированную среду, полученную из соответствующей лунки планшета Viewplate, добавляют по 4 мкл смеси антител. Планшеты небольшого объема герметично закрывают и инкубируют в течение 24 часов при комнатной температуре. Сигнал HTRF считывают с помощью планшетного ридера Envision с несколькими метками (Perkin Elmer) с использованием возбуждения 330 нм и испускания 615 нм и 665 нм. Результаты рассчитывают как (отношение 665 нм/615 нМ)*10000, и необработанные данные преобразовывают в концентрацию IFNα (пг/мл) с использованием стандартной кривой цитокинов. Как обсуждалось выше, положительный (агонист TLR7/8) и отрицательный (ДМСО) контроли используют для расчета доли (%) эффекта для каждого примера с использованием следующего уравнения:
% эффекта=100 - [100 * {(Пример - Положительный контроль) / (Отрицательный контроль - Положительный контроль)}]
% эффекта при каждой концентрации для каждого примера рассчитывают с использованием набора программ ABase (IBDS) и относят к количеству IFNα, продуцируемого в лунках положительного и отрицательного контроля, содержащихся в каждом планшете для анализа. Концентрации и значения % эффекта для каждого примера подгоняют с использованием 4-параметрической логистической модели в ABase, и концентрацию примера, которая вызывает 50% ответ (EC50), рассчитывают и представляют в таблице 1 как среднее геометрическое значение EC50, если Пример тестируют более одного раза. Пустая ячейка в таблице 1 указывает на то, что для этого примера в этом конкретном анализе не было получено никаких данных.
Таблица 1
Все публикации и патентные заявки, цитируемые в описании, полностью включены в настоящее описание посредством ссылки. Специалистам в данной области техники будет очевидно, что в них могут быть внесены определенные изменения и модификации без отклонения от сущности или объема прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ СОЕДИНЕНИЯ И ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2785126C2 |
| ФАРМАЦЕВТИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2654942C2 |
| ПРОИЗВОДНОЕ ПИРИМИДИНА И ПЯТИЧЛЕННОГО АЗОТСОДЕРЖАЩЕГО ГЕТЕРОЦИКЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКИЕ ПРИМЕНЕНИЯ | 2019 |
|
RU2775229C1 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ - 759 | 2008 |
|
RU2481348C2 |
| ИМИДАЗОПИРИМИДИНЫ КАК ИНГИБИТОРЫ EED И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2836176C2 |
| ПРОИЗВОДНОЕ ПИРАЗОЛА, ПРИГОДНОЕ В КАЧЕСТВЕ ИНГИБИТОРА РI3К | 2016 |
|
RU2710549C2 |
| Макроциклический ингибитор тирозинкиназы и его применение | 2019 |
|
RU2798231C2 |
| СОЕДИНЕНИЯ ГЕТЕРОАРИЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ТКБ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2742122C2 |
| ПРОИЗВОДНЫЕ 4-АМИНО-ИМИДАЗОХИНОЛИНА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ АГОНИСТАМИ TLR7 И/ИЛИ TLR8 | 2015 |
|
RU2698902C2 |
| ИНГИБИТОРЫ МУТАЦИИ HER2 | 2022 |
|
RU2834124C2 |
Группа изобретений относится к области фармацевтической химии и включает соединение формулы (I) и фармацевтическую композицию на его основе, где в формуле (I) R1 и R2 независимо являются C1-3 алкилом; или R1 и R2 объединены с образованием 5-7-членного карбоциклического кольца, где указанное карбоциклическое кольцо может быть насыщенным или ненасыщенным; R3 является 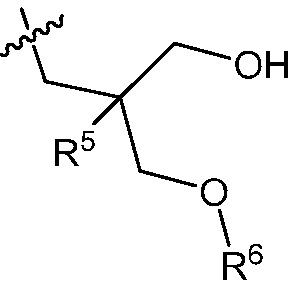 ; R4 является C1-6 алкилом или (CH2)nO(CH2)mCH3, где C1-6 алкил или любой атом углерода (CH2)nO(CH2)mCH3 группы замещен от 0 до 3 галогенами, насколько позволяет валентность; R5 является C1-3 алкилом или OC1-3 алкилом, где C1-3 алкил замещен от 0 до 3 F; R6 является H или C1-3 алкилом, где C1-3 алкил замещен от 0 до 3 F; m равно 0-2; n равно 1-3. Технический результат - соединение формулы (I), обладающее агонистической активностью в отношении TLR7/TLR8. 3 н. и 18 з.п. ф-лы, 1 табл., 19 пр.
; R4 является C1-6 алкилом или (CH2)nO(CH2)mCH3, где C1-6 алкил или любой атом углерода (CH2)nO(CH2)mCH3 группы замещен от 0 до 3 галогенами, насколько позволяет валентность; R5 является C1-3 алкилом или OC1-3 алкилом, где C1-3 алкил замещен от 0 до 3 F; R6 является H или C1-3 алкилом, где C1-3 алкил замещен от 0 до 3 F; m равно 0-2; n равно 1-3. Технический результат - соединение формулы (I), обладающее агонистической активностью в отношении TLR7/TLR8. 3 н. и 18 з.п. ф-лы, 1 табл., 19 пр.
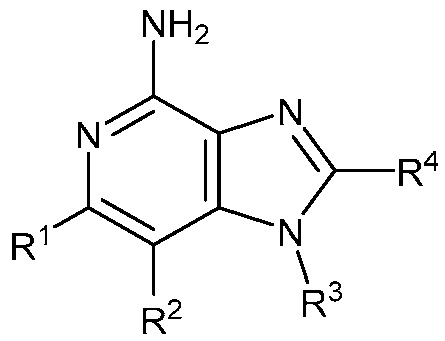 (I)
(I)
1. Соединение формулы (I)
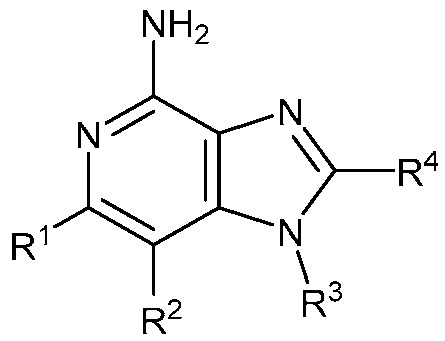 (I)
(I)
где
R1 и R2 независимо являются C1-3 алкилом; или
R1 и R2 объединены с образованием 5-7-членного карбоциклического кольца, где указанное карбоциклическое кольцо может быть насыщенным или ненасыщенным;
R3 является
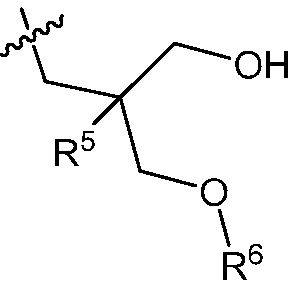 ;
;
R4 является C1-6 алкилом или (CH2)nO(CH2)mCH3, где C1-6 алкил или любой атом углерода (CH2)nO(CH2)mCH3 группы замещен от 0 до 3 галогенами, насколько позволяет валентность;
R5 является C1-3 алкилом или OC1-3 алкилом, где C1-3 алкил замещен от 0 до 3 F;
R6 является H или C1-3 алкилом, где C1-3 алкил замещен от 0 до 3 F;
m равно 0-2; и
n равно 1-3.
2. Соединение по п. 1, отличающееся тем, что
R1 и R2 независимо являются C1-2 алкилом; или
R1 и R2 объединены с образованием 5-7-членного карбоциклического кольца, где указанное карбоциклическое кольцо может быть насыщенным или ненасыщенным;
R3 является
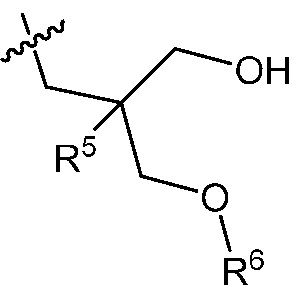 ;
;
R4 является C3-5 алкилом или (CH2)nO(CH2)mCH3;
R5 является C1-2 алкилом;
R6 является H;
m равно 1; и
n равно 1.
3. Соединение по п. 2, отличающееся тем, что R1 и R2 независимо являются C1-2 алкилом.
4. Соединение по п. 2, отличающееся тем, что R1 и R2 объединены с образованием 5-7-членного карбоциклического кольца, где указанное карбоциклическое кольцо может быть насыщенным или ненасыщенным.
5. Соединение по п. 4, отличающееся тем, что R1 и R2 объединены с образованием 5-7-членного карбоциклического кольца, где указанное карбоциклическое кольцо может быть насыщенным.
6. Соединение по п. 5, отличающееся тем, что карбоциклическим кольцом является циклопентил.
7. Соединение по п. 5, отличающееся тем, что карбоциклическим кольцом является циклогексил.
8. Соединение по п. 4, отличающееся тем, что
R1 и R2 объединены с образованием 5-7-членного карбоциклического кольца, где указанное карбоциклическое кольцо может быть ненасыщенным; и
R4 является C3-5 алкилом.
9. Соединение по п. 8, отличающееся тем, что карбоциклическим кольцом является фенил.
10. Соединение по п. 1, отличающееся тем, что
R1 и R2 независимо являются C1-3 алкилом; или
R1 и R2 объединены с образованием 5-7-членного карбоциклического кольца, где указанное карбоциклическое кольцо может быть насыщенным или ненасыщенным;
R3 является
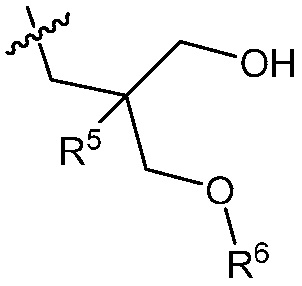 ;
;
R4 является C1-6 алкилом или (CH2)nO(CH2)mCH3, где указанный C1-6 алкил или любой атом углерода (CH2)nO(CH2)mCH3 группы замещен от 0 до 3 галогенами, насколько позволяет валентность, где галогеном является F;
R5 является C1-3 алкилом или OC1-3 алкилом, где C1-3 алкил замещен от 0 до 3 F;
R6 является H или C1-3 алкилом, где C1-3 алкил замещен от 0 до 3 F;
m равно 0-2; и
n равно 1-3.
11. Соединение по п. 10, отличающееся тем, что
R1 и R2 независимо являются C1-2 алкилом; или
R1 и R2 объединены с образованием 5-7-членного карбоциклического кольца, где указанное карбоциклическое кольцо может быть насыщенным или ненасыщенным;
R3 является
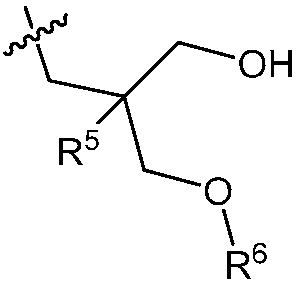 ;
;
R5 является C1-3 алкилом или OC1-3 алкилом, где C1-3 алкил замещен от 0 до 2 F; и
R6 является H.
12. Соединение по п. 11, отличающееся тем, что R5 является C1-2 алкилом.
13. Соединение по п. 1, выбранное из:
2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диола;
2-((4-амино-2-(этоксиметил)-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диола;
2-((4-амино-2-(этоксиметил)-7,8-дигидроциклопента[b]имидазо[4,5-d]пиридин-1(6H)-ил)метил)-2-метилпропан-1,3-диола;
2-((4-амино-2-(этоксиметил)-6,7,8,9-тетрагидро-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диола;
3-(4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-2-(метоксиметил)-2-метилпропан-1-ола;
(R)-3-(4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-2-(метоксиметил)-2-метилпропан-1-ола;
(S)-3-(4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-2-(метоксиметил)-2-метилпропан-1-ола;
2-((4-амино-6,7-диметил-2-(2,2,2-трифторэтил)-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диола;
2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-этилпропан-1,3-диола;
2-((4-амино-2-бутил-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диола;
2-((4-амино-2-бутил-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диола; и
2-((4-амино-2-пентил-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диола.
14. Соединение по п. 1, представляющее собой 2-((4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)метил)-2-метилпропан-1,3-диол.
15. Соединение по п. 1, представляющее собой 2-((4-амино-2-(этоксиметил)-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диол.
16. Соединение по п. 1, представляющее собой 2-((4-амино-2-(этоксиметил)-7,8-дигидроциклопента[b]имидазо[4,5-d]пиридин-1(6H)-ил)метил)-2-метилпропан-1,3-диол; или
2-((4-амино-2-(этоксиметил)-6,7,8,9-тетрагидро-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диол.
17. Соединение по п. 1, представляющее собой 3-(4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-2-(метоксиметил)-2-метилпропан-1-ол;
(R)-3-(4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-2-(метоксиметил)-2-метилпропан-1-ол; или
(S)-3-(4-амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-c]пиридин-1-ил)-2-(метоксиметил)-2-метилпропан-1-ол.
18. Соединение по п. 1, представляющее собой 2-((4-амино-2-бутил-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диол; или
2-((4-амино-2-пентил-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диол.
19. Фармацевтическая композиция, обладающая агонистической активностью против TLR7/TLR8, содержащая эффективное количество соединения по любому из пп. 1-18 и фармацевтически приемлемый носитель или эксципиент.
20. Соединение по п. 1, выбранное из:
2-((4-амино-2-бутил-6,7,8,9-тетрагидро-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диола;
2-((4-амино-2-бутил-7,8-дигидроциклопента[b]имидазо[4,5-d]пиридин-1(6H)-ил)метил)-2-метилпропан-1,3-диола;
2-((4-амино-2-этил-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диола; и
2-((4-амино-2-пропил-1H-имидазо[4,5-c]хинолин-1-ил)метил)-2-метилпропан-1,3-диола.
21. Фармацевтическая композиция, обладающая агонистической активностью против TLR7/TLR8, содержащая эффективное количество соединения по п. 20 и фармацевтически приемлемый носитель или эксципиент.
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Токарный резец | 1924 |
|
SU2016A1 |
| Gorden K | |||
| B | |||
| et al., Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8 | |||
| The journal of immunology, 2005, vol.174, no.3, p.1259-1268 | |||
| ИМИДАЗОХИНОЛИНЫ С ИММУНОМОДУЛИРУЮЩИМИ СВОЙСТВАМИ | 2008 |
|
RU2475487C2 |
| ОКСИЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, СПОСОБНЫЕ МОДУЛИРОВАТЬ БИОСИНТЕЗ ЦИТОКИНОВ | 2004 |
|
RU2412942C2 |

