ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001] Настоящее изобретение относится к области терапевтических воздействий на заболевания и нарушения, которые связаны с повышенными уровнями липидов и липопротеинов. Более конкретно, настоящее изобретение относится к применению ингибиторов PCSK9 в лечении пациентов с высоким риском возникновения сердечно-сосудистых заболеваний, имеющих гиперхолестеринемию и установленное коронарное заболевание сердца (CHD) или эквивалентные риски возникновения CHD, которые резистентны к терапии посредством максимально переносимой дозы статинов.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
[0002] Гиперхолестеринемия, в частности, повышение уровней холестерина липопротеинов низкой плотности (LDL) (LDL-C), представляет основной риск развития атеросклероза и коронарного заболевания сердца (CHD) (Sharrett et al., 2001, Circulation 104:1108-1113). Холестерин липопротеинов низкой плотности определяется как главная мишень снижающей холестерин терапии и принимается в качестве валидной суррогатной конечной точки терапии. Многочисленные исследования продемонстрировали, что снижение уровней LDL-C снижает риск возникновения CHD, при этом существует сильная прямая зависимость между уровнями LDL-C и явлениями CHD; при снижении уровня LDL-C на каждый 1 ммоль/л (~40 мг/дл) смертность от сердечно-сосудистых заболеваний (CVD) и заболеваемость сердечно-сосудистыми заболеваниями снижается на 22%. Большие снижения LDL-C приводят к большему снижению явлений, а сравнительные данные интенсивного и стандартного лечения статинами указывают на то, что чем ниже уровень LDL-C, тем большей является польза для пациентов с очень высоким риском возникновения сердечно-сосудистых (CV) заболеваний.
[0003] Современные лекарственные препараты, снижающие уровень LDL-C, включают статины, ингибиторы всасывания холестерина (например, эзетимиб [EZE]), фибраты, ниацин и секвестранты желчных кислот. Статины назначают чаще всего, поскольку они показали более высокую способность к снижению уровня LDL-C и уменьшению риска возникновения явлений CHD. Однако многие пациенты с риском возникновения сердечно-сосудистых заболеваний (CVD) имеют плохо контролируемый уровень холестерина липопротеинов низкой плотности (LDL-C), несмотря на терапию статинами.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0004] В настоящем изобретении предложены способы лечения гиперхолестеринемии. В частности, способы по настоящему изобретению применимы для лечения пациентов с высоким риском развития сердечно-сосудистых заболеваний, имеющих гиперхолестеринемию и установленное коронарное заболевание сердца (CHD) или эквивалентные риски возникновения CHD, которые резистентны к терапии посредством максимально переносимой дозы статинов.
[0005] Согласно одному аспекту способы по настоящему изобретению включают введение одной или нескольких доз ингибитора PCSK9 пациенту с высоким риском возникновения сердечно-сосудистых заболеваний, имеющему гиперхолестеринемию и установленное коронарное заболевание сердца или эквивалентные риски возникновения CHD, который резистентен к терапии посредством максимально переносимой дозы статинов (т.e. гиперхолестеринемия, резистентна к терапии посредством максимально переносимой дозы статинов в отсутствие ингибитора PCSK9 с другой гиполипидемической терапией или без нее). Согласно определенным вариантам осуществления настоящего изобретения ингибитор PCSK9 вводят пациенту с высоким риском возникновения сердечно-сосудистых заболеваний в качестве дополнительной терапии к используемой пациентом терапии статинами.
[0006] Согласно другому аспекту способы по настоящему изобретению включают выбор пациента с высоким риском возникновения сердечно-сосудистых заболеваний, который осуществляет прием согласно схеме терапии, предусматривающей ежедневную дозу статина (например, терапии посредством максимально переносимой дозы статинов), и введение пациенту одной или нескольких доз ингибитора PCSK9 совместно с (т. е. "наряду с") терапией статинами.
[0007] Один аспект настоящего изобретения предусматривает способ лечения пациента с высоким риском возникновения сердечно-сосудистых заболеваний, имеющего гиперхолестеринемию и установленное коронарное заболевание сердца (CHD) или эквивалентные риски возникновения CHD, который резистентен к терапии посредством максимально переносимой дозы статинов с другой липид-корригирующей терапией или без нее, включающий введение пациенту одной или нескольких доз ингибитора пропротеиновой конвертазы субтилизин/кексинового типа 9 (PCSK9), где у пациента проявляется резистентность гиперхолестеринемии, несмотря на лечение при помощи терапии посредством максимально переносимой дозы статинов с другой липид-корригирующей терапией или без нее в отсутствие ингибитора PCSK9.
[0008] Другой аспект настоящего изобретения предусматривает способ снижения уровня холестерина липопротеинов низкой плотности (LDL-C) у пациента с высоким риском возникновения сердечно-сосудистых заболеваний, имеющего гиперхолестеринемию и установленное коронарное заболевание сердца (CHD) или эквивалентные риски возникновения CHD, который резистентен к терапии посредством максимально переносимой дозы статинов с другой липид-корригирующей терапией или без нее, включающий введение пациенту одной или нескольких доз ингибитора пропротеиновой конвертазы субтилизин/кексинового типа 9 (PCSK9), где у пациента проявляется резистентность уровня LDL-C, несмотря на лечение при помощи терапии посредством максимально переносимой дозы статинов с другой липид-корригирующей терапией или без нее в отсутствие ингибитора PCSK9.
[0009] Другой аспект настоящего изобретения предусматривает способ лечения гиперхолестеринемии у пациента с высоким риском возникновения сердечно-сосудистых заболеваний, имеющего гиперхолестеринемию и установленное коронарное заболевание сердца (CHD) или эквивалентные риски возникновения CHD, который резистентен к терапии посредством максимально переносимой дозы статинов с другой липид-корригирующей терапией или без нее, включающий введение пациенту одной или нескольких доз ингибитора пропротеиновой конвертазы субтилизин/кексинового типа 9 (PCSK9), где у пациента проявляется резистентность гиперхолестеринемии, несмотря на лечение при помощи терапии посредством максимально переносимой дозы статинов с другой липид-корригирующей терапией или без нее в отсутствие ингибитора PCSK9.
[0010] Другой аспект настоящего изобретения предусматривает способ обеспечения улучшения в отношении уровня в сыворотке крови одного или нескольких липидных компонентов у пациента с высоким риском возникновения сердечно-сосудистых заболеваний, имеющего гиперхолестеринемию и установленное коронарное заболевание сердца (CHD) или эквивалентные риски возникновения CHD, который резистентен к терапии посредством максимально переносимой дозы статинов с другой липид-корригирующей терапией или без нее, включающий введение пациенту одной или нескольких доз ингибитора пропротеиновой конвертазы субтилизин/кексинового типа 9 (PCSK9), где у пациента проявляется резистентность липидного компонента, несмотря на лечение при помощи терапии посредством максимально переносимой дозы статинов с другой липид-корригирующей терапией или без нее в отсутствие ингибитора PCSK9. В определенных аспектах настоящее изобретение обеспечивает снижение уровня в сыворотке крови липидного компонента, выбранного из группы, состоящей из LDL-C, Apo B, не-HDL-C, общего холестерина, Lp(a) и триглицеридов. В определенных аспектах настоящее изобретение обеспечивает повышение уровня в сыворотке крови липидного компонента, выбранного из группы, состоящей из HDL-C и ApoA-1.
[0011] В определенных аспектах установленное CHD определяют по CHD в анамнезе, включая одно или несколько из следующего: острый инфаркт миокарда (MI), бессимптомный MI, нестабильная стенокардия, процедура реваскуляризации коронарных сосудов и клинически значимое CHD, диагностированное с помощью инвазивного или неинвазивного тестирования.
[0012] В определенных аспектах эквивалентные риски возникновения CHD включают один или несколько из следующих четырех критериев: 1) заболевание периферических артерий (PAD); 2) ишемический инсульт; 3) хроническое заболевание почек (CKD) и/или 4) известные случаи сахарного диабета в анамнезе и 2 или более дополнительных факторов риска, включая: a) гипертензию в анамнезе, b) лодыжечно-брахиальный индекс ≤0,90 в анамнезе, c) микроальбуминурию или макроальбуминурию в анамнезе или анализ мочи с помощью индикаторной полоски при скрининговом визите с уровнем белка >2+, d) предпролиферативную или пролиферативную ретинопатию или лазерное лечение ретинопатии в анамнезе и e) известные случаи раннего развития CHD в семейном анамнезе.
[0013] В определенных аспектах ингибитор PCSK9 представляет собой антитело или его антигенсвязывающий фрагмент, которые специфически связывают PCSK9. В некоторых аспектах антитело или его антигенсвязывающий фрагмент содержат определяющие комплементарность участки (CDR) тяжелой и легкой цепей из пары аминокислотных последовательностей вариабельного участка тяжелой цепи/вариабельного участка легкой цепи (HCVR/LCVR), выбранной из группы, состоящей из SEQ ID NO: 1/6 и 11/15. В некоторых аспектах антитело или его антигенсвязывающий фрагмент содержат аминокислотные последовательности CDR тяжелой и легкой цепей, имеющие SEQ ID NO: 12, 13, 14, 16, 17 и 18. В некоторых аспектах антитело или его антигенсвязывающий фрагмент содержат HCVR с аминокислотной последовательностью под SEQ ID NO:11 и LCVR с аминокислотной последовательностью под SEQ ID NO:15. В некоторых аспектах антитело или его антигенсвязывающий фрагмент содержат аминокислотные последовательности CDR тяжелой и легкой цепей, имеющие SEQ ID NO: 2, 3, 4, 7, 8 и 10. В некоторых аспектах антитело или его антигенсвязывающий фрагмент содержат HCVR с аминокислотной последовательностью под SEQ ID NO:1 и LCVR с аминокислотной последовательностью под SEQ ID NO:6. В некоторых аспектах антитело или его антигенсвязывающий фрагмент связывается с тем же эпитопом на PCSK9, что и антитело, содержащее аминокислотные последовательности CDR тяжелой и легкой цепей, имеющие SEQ ID NO: 12, 13, 14, 16, 17 и 18 или SEQ ID NO: 2, 3, 4, 7, 8 и 10. В некоторых аспектах антитело или его антигенсвязывающий фрагмент конкурируют за связывание с PCSK9 с антителом, содержащим аминокислотные последовательности CDR тяжелой и легкой цепей, имеющие SEQ ID NO: 12, 13, 14, 16, 17 и 18 или SEQ ID NO: 2, 3, 4, 7, 8 и 10.
[0014] В определенных аспектах антитело или его антигенсвязывающий фрагмент, которые специфически связывают PCSK9, вводят пациенту в дозе, составляющей приблизительно 75 мг, с частотой один раз каждые две недели. В некоторых аспектах дозу, составляющую приблизительно 75 мг, продолжают вводить, если уровень LDL-C у пациента, измеренный после пяти или более доз составляет <70 мг/дл. В некоторых аспектах введение дозы, составляющей приблизительно 75 мг, прекращают, если уровень LDL-C у пациента, измеренный после пяти или более доз остается ≥70 мг/дл, и при этом антитело или его антигенсвязывающий фрагмент, которые специфически связывают PCSK9, впоследствии вводят пациенту в дозе, составляющей приблизительно 150 мг, с частотой один раз каждые две недели. В некоторых аспектах антитело или его антигенсвязывающий фрагмент, которые специфически связывают PCSK9, вводят пациенту в дозе, составляющей приблизительно 150 мг, с частотой один раз каждые две недели.
[0015] В определенных аспектах ингибитор PCSK9 вводят пациенту совместно с терапией посредством максимально переносимой дозы статинов. В некоторых аспектах терапия посредством максимально переносимой дозы статинов предусматривает аторвастатин в ежедневной дозе, составляющей от приблизительно 40 мг до приблизительно 80 мг. В некоторых аспектах терапия посредством максимально переносимой дозы статинов предусматривает розувастатин в ежедневной дозе, составляющей от приблизительно 20 мг до приблизительно 40 мг. В некоторых аспектах терапия посредством максимально переносимой дозы статинов предусматривает симвастатин в ежедневной дозе, составляющей приблизительно 80 мг. В некоторых аспектах ингибитор PCSK9 вводят пациенту совместно с другой липид-корригирующей терапией, где другая липид-корригирующая терапия предусматривает терапевтическое средство, выбранное из группы, состоящей из эзетимиба, фибрата, ниацина, омега-3 жирной кислоты и смолы, связывающей желчные кислоты.
[0016] В определенных аспектах способ обеспечивает улучшение по меньшей мере одного ассоциированного с гиперхолестеринемией параметра, выбранное из группы, состоящей из: (a) снижения уровня холестерина липопротеинов низкой плотности (LDL-C) у пациента по меньшей мере на 40%; (b) снижения уровня аполипопротеина B100 (ApoB) у пациента по меньшей мере на 35%; (c) снижения уровня холестерина, не относящегося к холестерину липопротеинов высокой плотности (не-HDL-C), у пациента по меньшей мере на 35%; (d) снижения уровня общего холестерина у пациента по меньшей мере на 20%; (e) повышения уровня холестерина липопротеинов высокой плотности (HDL-C) у пациента по меньшей мере на 1%; (f) снижения уровня триглицеридов у пациента по меньшей мере на 1%; (g) снижения уровня липопротеина a (Lp(a)) у пациента по меньшей мере на 10% и (h) повышения уровня алипопротеина А-1 у пациента по меньшей мере на 1%.
[0017] Другие варианты осуществления по настоящему изобретению будут очевидны из обзора последующего подробного описания.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0018] Фигура 1A представляет собой графическое отображение плана исследования для ODYSSEY COMBO I. Фигура 1B представляет собой графическое отображение плана исследования для ODYSSEY COMBO II. LDL-C, холестерин липопротеинов низкой плотности; LLT, липид-корригирующая терапия; NCEP ATP III, III доклад группы экспертов по лечению взрослых в рамках Национальной образовательной программы по холестерину; PO, перорально; Q2W, каждые 2 недели; R, рандомизация; SC, подкожно; TLC, терапевтические изменения образа жизни.
[0019] Фигура 2 представляет собой график, демонстрирующий процентное изменение среднего значения, определенного методом наименьших квадратов (+/- SE), рассчитанного уровня LDL-C от исходного уровня для плацебо и алирокумаба в зависимости от времени для анализа ITT - популяции ITT в исследовании ODYSSEY COMBO I. Примечание: Средние значения, определенные методом наименьших квадратов, и стандартные ошибки (SE) получены с помощью анализа на основе MMRM (модели со смешанными эффектами для повторных измерений). Модель включает фиксированные категориальные эффекты группы лечения, страты рандомизации согласно IVRS, момента времени, взаимосвязи лечение-момент времени, взаимосвязи страта-момент времени, а также непрерывные фиксированные ковариаты значения исходного уровня LDL-C и взаимосвязи исходный уровень LDL-C-момент времени.
[0020] Фигура 3 представляет собой график, демонстрирующий процентное изменение среднего значения, определенного методом наименьших квадратов (+/- SE), рассчитанного уровня LDL-C от исходного уровня для плацебо и алирокумаба в зависимости от времени для анализа в ходе лечения - популяции mITT в исследовании ODYSSEY COMBO I. Период лечения для оценки эффективности заканчивается на дату последней инъекции+21 день. Примечание: Средние значения, определенные методом наименьших квадратов, и стандартные ошибки (SE) получены с помощью анализа на основе MMRM (модели со смешанными эффектами для повторных измерений). Модель включает фиксированные категориальные эффекты группы лечения, страты рандомизации согласно IVRS, момента времени, взаимосвязи лечение-момент времени, взаимосвязи страта-момент времени, а также непрерывные фиксированные ковариаты значения исходного уровня LDL-C и взаимосвязи исходный уровень LDL-C-момент времени.
[0021] Фигура 4 представляет собой график уровней LDL-C в зависимости от времени для пациентов, получавших лечение алирокумабом с/без повышения дозы на 12 неделе в исследовании ODYSSEY COMBO I.
[0022] Фигура 5 представляет собой график процентного снижения уровня LDL-C от исходного уровня на 24 неделе для различных демографических подгрупп алирокумаба и плацебо в исследовании ODYSSEY COMBO I.
[0023] Фигура 6 представляет собой график, демонстрирующий процентное изменение среднего значения, определенного методом наименьших квадратов (+/- SE), рассчитанного уровня LDL-C от исходного уровня в зависимости от времени для популяции ITT в исследовании ODYSSEY COMBO II. Примечание: Средние значения, определенные методом наименьших квадратов, и стандартные ошибки (SE) получены с помощью анализа на основе MMRM (модели со смешанными эффектами для повторных измерений). Модель включает фиксированные категориальные эффекты группы лечения, страты рандомизации согласно IVRS, момента времени, взаимосвязи лечение-момент времени, взаимосвязи страта-момент времени, а также непрерывные фиксированные ковариаты значения исходного уровня LDL-C и взаимосвязи исходный уровень LDL-C-момент времени.
[0024] Фигура 7 представляет собой график достигнутых концентраций LDL-C на 24 неделе по децильным значениям исходного уровня LDL-C у пациентов для групп алирокумаба и эзетимиба в исследовании ODYSSEY COMBO II.
[0025] Фигура 8 представляет собой график концентраций LDL-C в зависимости от момента времени исследования в соответствии со статусом увеличения дозы (анализ в отношении группы пациентов, сформированной в зависимости от назначенного лечения) для групп алирокумаба и эзетимиба в исследовании ODYSSEY COMBO II. LS=метод наименьших квадратов; SD=стандартное отклонение; SE=стандартная ошибка.
[0026] Фигура 9 представляет собой таблицу данных процентного изменения от исходного уровня до 24 недели уровня LDL-C в разных подгруппах (анализ ITT) групп алирокумаба и эзетимиба в исследовании ODYSSEY COMBO II. BMI=индекс массы тела; CI=доверительный интервал; CKD=хроническое заболевание почек; LS=метод наименьших квадратов; MI=инфаркт миокарда; SC=подкожно; TG=триглицерид.
[0027] Фигура 10 представляет собой графическое отображение плана исследования для ODYSSEY LONG TERM. Беседы по телефону отмечены курсивом, и их выполняют каждые 4 недели между визитами в исследовательский центр до окончания визитов в период двойного слепого лечения. Отметки в плане исследования являются следующими: HeFH: гетерозиготная семейная гиперхолестеринемия; LDL-C: холестерин липопротеинов низкой плотности; LLT: липид-корригирующая терапия; Q2W: каждые две недели и SC: подкожно.
[0028] Фигура 11 представляет собой график, демонстрирующий процентное изменение среднего значения, определенного методом наименьших квадратов (+/- SE), рассчитанного уровня LDL-C от исходного уровня для плацебо и алирокумаба в зависимости от времени для популяции ITT в исследовании ODYSSEY LONG TERM во время анализа с предварительно заданными параметрами. Примечание: Средние значения, определенные методом наименьших квадратов, и стандартные ошибки (SE) получены с помощью анализа на основе MMRM (модели со смешанными эффектами для повторных измерений). Модель включает фиксированные категориальные эффекты группы лечения, страты рандомизации согласно IVRS, момента времени, взаимосвязи лечение-момент времени, взаимосвязи страта-момент времени, а также непрерывные фиксированные ковариаты значения исходного уровня LDL-C и взаимосвязи исходный уровень LDL-C-момент времени.
[0029] Фигура 12 представляет собой ряд графиков, показывающих процентное изменение среднего значения, определенного методом наименьших квадратов (SE), от исходного уровня до 24 недели для уровня LDL-C в популяции с не-HeFH и популяции с HeFH исследования ODYSSEY LONG TERM (A), а также той же самой популяции пациентов HeFH, разделенных в зависимости от исходных уровней LDL-C (< 160 мг/дл и ≥160 мг/дл (B) и < 190 мг/дл и ≥190 мг/дл. (C)). Все пациенты получали фоновое лечение статином (на максимально переносимом уровне). Подгруппа пациентов также получала дополнительную липид-корригирующую терапию.
[0030] Фигура 13 представляет собой график, демонстрирующий различие процентного изменения среднего значения, определенного методом наименьших квадратов (SE), от исходного уровня до 24 недели в разных подгруппах исследования ODYSSEY LONG TERM. Отметки в графике являются следующими: CKD: хроническое заболевание почек; TG: триглицериды. Все пациенты получали фоновое лечение статином (на максимально переносимом уровне). Подгруппа пациентов также получала дополнительную липид-корригирующую терапию.
[0031] Фигура 14 представляет собой график, демонстрирующий различие процентного изменения среднего значения, определенного методом наименьших квадратов (SE), от исходного уровня до 24 недели в группах применения конкретного статина и LLT в исследовании ODYSSEY LONG TERM. Все пациенты получали фоновое лечение статином (на максимально переносимом уровне). Подгруппа пациентов также получала дополнительную липид-корригирующую терапию.
[0032] Фигура 15 представляет собой график, демонстрирующий процентное изменение среднего значения, определенных методом наименьших квадратов (+/- SE), рассчитанного уровня LDL-C от исходного уровня для плацебо и алирокумаба в зависимости от времени для популяции ITT в исследовании ODYSSEY LONG TERM. Значения, приведенные выше точек данных, указывают среднее значение, определенное методом наименьших квадратов, абсолютных уровней холестерина LDL, значения, приведенные ниже точек данных, указывают % изменение среднего значения, определенного методом наименьших квадратов, от исходного уровня. Значения в приведенной ниже таблице указывают количество пациентов со значениями уровня холестерина LDL, доступными для анализа ITT в каждый момент времени, т.e. измерения уровня холестерина LDL, полученные как у пациентов, получающих лечение, так и у пациентов после окончания лечения (для пациентов, которые прекратили исследуемое лечение, но вернулись в клинику для проведения обследования). Недостающие данные учитывали с помощью модели со смешанными эффектами для повторных измерений.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0033] Перед описанием настоящего изобретения необходимо понимать, что настоящее изобретение не ограничивается описанными конкретными способами и условиями экспериментов, поскольку такие способы и условия могут варьировать. Также необходимо понимать, что терминология, используемая в данном документе, предназначена лишь для целей описания конкретных вариантов осуществления и не предполагает ограничительный характер, поскольку объем настоящего изобретения будет ограничиваться лишь прилагаемой формулой изобретения.
[0034] Если не определено иное, все технические и научные термины, используемые в данном документе, имеют такое же значение, которое обычно понимается специалистом в области, к которой принадлежит настоящее изобретение. Используемый в данном документе термин "приблизительно", при использовании со ссылкой на конкретное описываемое числовое значение, означает, что значение может отличаться от описываемого значения не более чем на 1%. Например, используемое в данном документе выражение "приблизительно 100" включает 99 и 101 и все значения между ними (например, 99,1, 99,2, 99,3, 99,4 и т. д.).
[0035] Хотя любые способы и материалы, подобные или эквивалентные тем, которые описаны в данном документе, можно использовать при осуществлении настоящего изобретения на практике, ниже описаны предпочтительные способы и материалы. Все публикации, упомянутые в данном документе, включены в данный документ посредством ссылки во всей своей полноте.
Гиперхолестеринемия и установленное коронарное заболевание сердца или эквивалентные риски возникновения CHD, которые резистентны к терапии посредством максимально переносимой дозы статинов
[0036] Настоящее изобретение в целом относится к способам и композициям для лечения пациентов с высоким риском возникновения сердечно-сосудистых заболеваний, имеющих гиперхолестеринемию и установленное CHD или эквивалентные риски возникновения CHD, которые резистентны к статинами, т. e. гиперхолестеринемия, резистентна к схеме терапии, предусматривающей ежедневную максимально переносимую дозу статина. Используемое в данном документе выражение "резистентный" в отношении гиперхолестеринемии означает, что концентрация холестерина липопротеинов низкой плотности (LDL-C), концентрация общего холестерина и/или концентрация триглицеридов в сыворотке крови пациента не снижаются до общепризнанного, допустимого с медицинской точки зрения уровня (с учетом относительного риска возникновения у пациента ишемической болезни сердца) после по меньшей мере 4 недель схемы терапии, предусматривающей стабильную ежедневную дозу статина. Например, выражение "пациент, имеющий гиперхолестеринемию, резистентную к статину" включает пациентов с концентрацией LDL-C в сыворотке крови больше приблизительно 70 мг/дл, 100 мг/дл, 130 мг/дл, 140 мг/дл или более (в зависимости от исходного риска возникновения у пациента заболевания сердца) после того, как пациент получал лечение согласно схеме со стабильными ежедневными дозами статинов в течение по меньшей мере 4 недель.
[0037] Согласно определенным вариантам осуществления у пациента с высоким риском возникновения сердечно-сосудистых заболеваний, который поддается лечению способами по настоящему изобретению, имеется гиперхолестеринемия (например, концентрация LDL-C в сыворотке крови больше или равна 70 мг/дл), несмотря на прием стабильной ежедневной дозы статина (с другой гиполипидемической терапией или без нее) в течение по меньшей мере 4 недель, 5 недель, 6 недель или более. В определенных вариантах осуществления гиперхолестеринемия у пациента с высоким риском возникновения сердечно-сосудистых заболеваний резистентна к терапии посредством максимально переносимой дозы статина (также обозначаемой в данном документе как "схема терапии с ежедневной максимально переносимой дозой статинов").
[0038] Используемое в данном документе выражение "терапия посредством максимально переносимой дозой статинов" означает схему терапии, предусматривающую введение ежедневной дозы статина, которая является максимально переносимой дозой для определенного пациента. "Максимально переносимая доза" означает самую высокую дозу статина, которую можно вводить пациенту без возникновения недопустимых нежелательных побочных эффектов у пациента. Терапия посредством максимально переносимой дозы статинов включает без ограничения, например, 40-80 мг аторвастатина ежедневно, 20-40 мг розувастатина ежедневно или 80 мг симвастатина (если пациент уже принимает эту дозу на протяжении >1 года). Однако пациенты, не способные переносить приведенные выше дозы статинов, могли получать ежедневно более низкую дозу аторвастатина, розувастатина или симвастатина, при условии, что была допустимая причина для отказа от применения более высоких доз. Некоторые примеры допустимых причин для приема пациентом более низкой дозы статинов включают неблагоприятные эффекты при более высоких дозах, пожилой возраст, низкий индекс массы тела (BMI), региональные практики, местный листок-вкладыш, сопутствующие лекарственные препараты, опасения по поводу когнитивных нежелательных явлений и сопутствующие состояния, такие как нарушенная толерантность к глюкозе/нарушение уровня глюкозы натощак, повышенный уровень глюкозы в крови, гликированный гемоглобин, заболевание печени или повышенный уровень ферментов печени и мышечные симптомы и/или повышенный уровень креатинфосфокиназы.
[0039] Настоящее изобретение также включает способы лечения пациентов с высоким риском возникновения сердечно-сосудистых заболеваний, имеющих гиперхолестеринемию и установленное CHD или эквивалентные риски возникновения CHD, которые резистентны к терапии посредством максимально переносимой дозы статинов, включающие ежедневное введение других статинов, таких как церивастатин, питавастатин, флувастатин, ловастатин и правастатин.
Выбор пациентов
[0040] В настоящем изобретение предусмотрены способы и композиции, применимые для лечения пациентов с высоким риском возникновения сердечно-сосудистых заболеваний, имеющих гиперхолестеринемию и установленное CHD или эквивалентные риски возникновения CHD, которые резистентны к схеме терапии с ежедневной максимально переносимой дозой статинов.
[0041] Установленное CHD определяют как документально подтвержденный случай CHD в анамнезе, включая одно или несколько из следующего: острый инфаркт миокарда (MI), бессимптомный MI, нестабильная стенокардия, процедура реваскуляризации коронарных сосудов (например, чрескожное коронарное вмешательство [PCI] или шунтирование коронарных артерий [CABG]) и клинически значимое CHD, диагностированное с помощью инвазивного или неинвазивного тестирования (такого как коронарография, электрокардиограмма с нагрузкой на беговой дорожке, стресс-эхокардиография или радионуклидная визуализация).
[0042] Эквивалентные риски возникновения CHD включают один или несколько из следующих 4 критериев: 1) заболевание периферических артерий (PAD); 2) ишемический инсульт; 3) хроническое заболевание почек и/или 4) известные случаи сахарного диабета в анамнезе и 2 или более дополнительных факторов риска, включая: a) гипертензию в анамнезе (установленную по лекарственным препаратам против гипертензии), b) лодыжечно-брахиальный индекс ≤0,90 в анамнезе, c) микроальбуминурию или макроальбуминурию в анамнезе или анализ мочи с помощью индикаторной полоски при скрининговом визите (неделя -2) с уровнем белка >2+, d) предпролиферативную или пролиферативную ретинопатию или лазерное лечение ретинопатии в анамнезе, e) известные случаи раннего развития CHD в семейном анамнезе (CHD у отца или брата в возрасте до 55 лет; CHD у матери или сестры в возрасте до 65 лет).
[0043] Как используется в данном документе, при заболевании периферических артерий (PAD) должен удовлетворяться один из следующих критериев [a, b или c]: a) текущая перемежающаяся хромота (мышечный дискомфорт в нижней конечности, проявляющийся при физических нагрузках, который возникает повторно и облегчается при покое в течение 10 минут) предполагаемого атеросклеротического происхождения вместе с лодыжечно-брахиальным индексом < 0,90 в любой ноге в состоянии покоя, или b) перемежающаяся хромота в анамнезе вместе с эндоваскулярной процедурой или хирургическим вмешательством на одной или обеих ногах по причине атеросклеротического заболевания, или c) критическая ишемия конечностей в анамнезе вместе с тромболизисом, эндоваскулярной процедурой или хирургическим вмешательством на одной или обеих ногах по причине атеросклеротического заболевания.
[0044] Используемый в данном документе документально подтвержденный ишемический инсульт включает ишемический инсульт с очаговым ишемическим неврологическим дефицитом, который продолжался более 24 часов, как считается, имеющий атеротромботическое происхождение. Компьютерную томографию (CT) или магнитно-резонансную томографию (MRI) можно выполнять для исключения кровоизлияния и неишемического неврологического заболевания.
[0045] Используемое в данном документе хроническое заболевание почек (CKD) определяют с помощью расчетной скорости клубочковой фильтрации (eGFR), составляющей ≥30‒<60 мл/мин./1,73 м2 в течение 3 месяцев или более.
[0046] Согласно определенным вариантам осуществления пациент с высоким риском возникновения сердечно-сосудистых заболеваний может быть выбран на основании наличия одного или нескольких дополнительных факторов риска, выбранных из группы, состоящей из возраста (например, старше 40, 45, 50, 55, 60, 65, 70, 75 или 80 лет), расовой принадлежности, национального происхождения, пола (мужчина или женщина), особенностей физических нагрузок (например, регулярно занимается физическими упражнениями, не занимается физическими упражнениями), других предшествующих патологических состояний (например, диабет II типа, высокое кровяное давление и т. д.) и текущего статуса приема лекарственных препаратов (например, текущий прием бета-блокаторов, ниацина, эзетимиба, фибратов, омега-3 жирных кислот, смол, связывающих желчные кислоты и т. д.).
[0047] Согласно настоящему изобретению пациенты с высоким риском возникновения сердечно-сосудистых заболеваний могут быть выбраны на основании комбинации одного или нескольких из вышеупомянутых критериев выбора или терапевтических характеристик.
Введение ингибитора PCSK9 в качестве дополнительной терапии к терапии посредством максимально переносимой дозы статинов
[0048] Настоящее изобретение включает способы, где пациенту с высоким риском возникновения сердечно-сосудистых заболеваний, имеющему гиперхолестеринемию и установленное CHD или эквивалентные риски возникновения CHD, который резистентен к схеме терапии со стабильной ежедневной максимально переносимой дозой статинов в отсутствие ингибитора PCSK9, вводят ингибитор PCSK9 согласно определенному дозированному количеству и определенной частоте дозирования, и при этом ингибитор PCSK9 вводят пациенту в качестве дополнения к схеме терапии статинами. Например, согласно определенным вариантам осуществления, если у пациента с высоким риском возникновения сердечно-сосудистых заболеваний имеется гиперхолестеринемия и установленное CHD или эквивалентные риски возникновения CHD, резистентные, несмотря на схему терапии с постоянной ежедневной максимально переносимой дозой статинов, включающую, например, 40-80 мг аторвастатина, то пациенту с высоким риском возникновения сердечно-сосудистых заболеваний можно вводить ингибитор PCSK9 в определенном количестве и с определенным интервалом дозирования, при этом пациент продолжает следовать своей схеме терапии со стабильной ежедневной дозой статинов.
[0049] Способы по настоящему изобретению включают дополнительные схемы терапии, в которых ингибитор PCSK9 вводят в качестве дополнительной терапии к той самой схеме терапии со стабильной ежедневной максимально переносимой дозой статинов (т. е., к тому самому дозированному количеству статинов), которую получал пациент с высоким риском возникновения сердечно-сосудистых заболеваний до получения ингибитора PCSK9. В других вариантах осуществления ингибитор PCSK9 вводят в качестве дополнительной терапии к схеме терапии с ежедневной максимально переносимой дозой статинов, предусматривающей статин в количестве, которое больше или меньше дозы статинов, которую пациент получал до получения ингибитора PCSK9. Например, после начала схемы лечения, предусматривающей ингибитор PCSK9, вводимый с определенной частотой дозирования и в определенном дозированном количестве, ежедневная доза статина, вводимая или назначаемая пациенту, может (a) оставаться той же самой, (b) повышаться или (c) снижаться (например, дозу повышают или дозу снижают) по сравнению с ежедневной дозой статинов, которую пациент с высоким риском возникновения сердечно-сосудистых заболеваний принимал до начала схемы терапии ингибитором PCSK9, в зависимости от терапевтических потребностей пациента.
Терапевтическая эффективность
[0050] Способы по настоящему изобретению приведут в результате к обеспечению улучшения в отношении уровня в сыворотке крови одного или нескольких липидных компонентов, выбранных из группы, состоящей из LDL-C, ApoB, не-HDL-C, общего холестерина, HDL-C, ApoA-1, триглицеридов и Lp(a). Например, согласно определенным вариантам осуществления настоящего изобретения введение фармацевтической композиции, содержащей ингибитор PCSK9, пациенту с высоким риском возникновения сердечно-сосудистых заболеваний, имеющему гиперхолестеринемию и установленное CHD или эквивалентные риски возникновения CHD, который резистентен к схеме терапии со стабильной ежедневной максимально переносимой дозой статинов (например, введение ингибитора PCSK9 в добавление к терапии посредством максимально переносимой дозы статинов, которую принимает пациент), приведет в результате к среднему процентному снижению холестерина липопротеинов низкой плотности (LDL-C) от исходного уровня в сыворотке крови, составляющему по меньшей мере приблизительно 40%, 41%, 42%, 43%, 44%, 45%, 46%, 47%, 48%, 49%, 50%, 51%, 52%, 53%, 54%, 55%, 56%, 57%, 58%, 59%, 60% или больше; среднему процентному снижению ApoB от исходного уровня, составляющему по меньшей мере приблизительно 35%, 36%, 37%, 38%, 39%, 40% или больше; среднему процентному снижению не-HDL-C от исходного уровня, составляющему по меньшей мере приблизительно 35%, 36%, 37%, 38%, 39%, 40%, 41%, 42%, 43%, 44%, 45%, 46%, 47%, 48%, 49%, 50% или больше; среднему процентному снижению общего холестерина от исходного уровня, составляющему по меньшей мере приблизительно 20%, 21%, 22%, 23%, 24%, 25%, 26%, 27%, 28%, 29%, 30%, 31%, 32%, 33%, 34%, 35% или больше; среднему процентному повышению HDL-C от исходного уровня, составляющему по меньшей мере приблизительно 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10% или больше; среднему процентному повышению ApoA-1 от исходного уровня, составляющему по меньшей мере приблизительно 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10% или больше; среднему процентному снижению триглицеридов от исходного уровня, составляющему по меньшей мере приблизительно 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10% или больше, и/или среднему процентному снижению Lp(a) от исходного уровня, составляющему по меньшей мере приблизительно 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, 20%, 21%, 22%, 23%, 24%, 25% или больше.
Ингибиторы PCSK9
[0051] Способы по настоящему изобретению включают введение пациенту с высоким риском возникновения сердечно-сосудистых заболеваний терапевтической композиции, содержащей ингибитор PCSK9. Используемый в данном документе "ингибитор PCSK9" означает любое средство, которые связывается или взаимодействует с PCSK9 человека и ингибирует нормальную биологическую функцию PCSK9 in vitro или in vivo. Неограничивающие примеры категорий ингибиторов PCSK9 включают низкомолекулярные антагонисты PCSK9, пептидные антагонисты PCSK9 (например, молекулы "пептид-ассоциированных антител"), а также антитела или антигенсвязывающие фрагменты антител, которые специфически связывают PCSK9 человека.
[0052] Термин "пропротеиновая конвертаза субтилизин/кексинового типа 9 человека", или "PCSK9 человека", или "hPCSK9", используемый в данном документе, относится к PCSK9 с последовательностью нуклеиновой кислоты, представленной под SEQ ID NO:197, и аминокислотной последовательностью под SEQ ID NO:198 или ее биологически активному фрагменту.
[0053] Термин "антитело", используемый в данном документе, предназначен для обозначения молекул иммуноглобулинов, содержащих четыре полипептидные цепи, две тяжелые (H) цепи и две легкие (L) цепи, соединенные между собой дисульфидными связями, а также их мультимеров (например, IgM). Каждая тяжелая цепь содержит вариабельный участок тяжелой цепи (в данном документе обозначен аббревиатурой HCVR или VH) и константный участок тяжелой цепи. Константный участок тяжелой цепи содержит три домена: CH1, CH2 и CH3. Каждая легкая цепь содержит вариабельный участок легкой цепи (в данном документе обозначен аббревиатурой LCVR или VL) и константный участок легкой цепи. Константный участок легкой цепи содержит один домен (CL1). Участки VH и VL можно дополнительно подразделять на участки гипервариабельности, называемые участками, определяющими комплементарность (CDR), чередующиеся с более консервативными участками, называемыми каркасными участками (FR). Каждый VH и VL состоит из трех CDR и четырех FR, расположенных от амино-конца до карбокси-конца в следующем порядке: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. В различных вариантах осуществления настоящего изобретения FR антитела к PCSK9 (или его антигенсвязывающей части) могут быть идентичными последовательностям зародышевой линии человека или могут быть изменены естественным или искусственным путем. Аминокислотная консенсусная последовательность может быть определена на основании анализа "бок-о-бок" двух или более CDR.
[0054] Термин "антитело", используемый в данном документе, также включает антигенсвязывающие фрагменты целых молекул антител. Термины "антигенсвязывающая часть" антитела, "антигенсвязывающий фрагмент" антитела и им подобные, используемые в данном документе, включают любой встречающийся в природе, получаемый ферментативным путем, синтетический или полученный с помощью методик генной инженерии полипептид или гликопротеин, которые специфически связывают антиген с образованием комплекса. Антигенсвязывающие фрагменты антитела могут быть получены, например, из целых молекул антител при помощи любых подходящих стандартных методик, таких как протеолитическое расщепление или рекомбинантные методики генной инженерии, включающие манипуляцию с ДНК, кодирующей вариабельные и необязательно константные домены антитела, и ее экспрессию. Такая ДНК известна и/или легко доступна, например, из коммерческих источников, библиотек ДНК (в том числе, например, библиотек "фаг-антитело") или ее можно синтезировать. ДНК можно секвенировать и с ней можно проводить химические манипуляции или манипуляции при помощи методик молекулярной биологии, например, для расположения одного или нескольких вариабельных и/или константных доменов в подходящей конфигурации или для введения кодонов, создания цистеиновых остатков, модификации, добавления или удаления аминокислот и т. д.
[0055] Неограничивающие примеры антигенсвязывающих фрагментов включают: (i) Fab-фрагменты; (ii) F(ab')2-фрагменты; (iii) Fd-фрагменты; (iv) Fv-фрагменты; (v) одноцепочечные молекулы Fv (scFv); (vi) dAb-фрагменты и (vii) минимальные распознающие единицы, состоящие из аминокислотных остатков, имитирующих гипервариабельный участок антитела (например, выделенный участок, определяющий комплементарность (CDR), такой как пептид CDR3), или пептид c ограниченной конформационной свободой FR3-CDR3-FR4. Другие сконструированные молекулы, такие как домен-специфические антитела, однодоменные антитела, антитела с удаленным доменом, химерные антитела, CDR-привитые антитела, диатела, триатела, тетратела, минитела, нанотела (например, моновалентные антитела, бивалентные антитела и т. д.), иммунопрепараты на основе модульного белка с малым размером молекул (SMIP) и вариабельные домены IgNAR акулы, также включены в выражение "антигенсвязывающий фрагмент", используемое в данном документе.
[0056] Антигенсвязывающий фрагмент антитела, как правило, будет содержать по меньшей мере один вариабельный домен. Вариабельный домен может быть любого размера или аминокислотного состава и будет, как правило, содержать по меньшей мере один CDR, который прилегает к или находится в рамке считывания с одной или несколькими каркасными последовательностями. В антигенсвязывающих фрагментах, имеющих домен VH, связанный с доменом VL, домены VH и VL могут располагаться относительно другу друга в любом подходящем порядке. Например, вариабельный участок может быть димерным и содержать димеры VH-VH, VH-VL или VL-VL. Альтернативно, антигенсвязывающий фрагмент антитела может содержать мономерный домен VH или VL.
[0057] Согласно определенным вариантам осуществления антигенсвязывающий фрагмент антитела может содержать по меньшей мере один вариабельный домен, ковалентно связанный по меньшей мере с одним константным доменом. Неограничивающие иллюстративные конфигурации вариабельных и константных доменов, которые можно обнаружить в антигенсвязывающем фрагменте антитела по настоящему изобретению, включают: (i) VH-CH1; (ii) VH-CH2; (iii) VH-CH3; (iv) VH-CH1-CH2; (v) VH-CH1-CH2-CH3; (vi) VH-CH2-CH3; (vii) VH-CL; (viii) VL-CH1; (ix) VL-CH2; (x) VL-CH3; (xi) VL-CH1-CH2; (xii) VL-CH1-CH2-CH3; (xiii) VL-CH2-CH3 и (xiv) VL-CL. В любой конфигурации вариабельных и константных доменов, в том числе любых иллюстративных конфигурациях, изложенных выше, вариабельные и константные домены могут быть либо непосредственно связаны друг с другом, либо могут быть связаны при помощи целого или части шарнирного или линкерного участка. Шарнирный участок может состоять по меньшей мере из 2 (например, 5, 10, 15, 20, 40, 60 или более) аминокислот, которые приводят к образованию гибкой или полугибкой связи между прилегающими вариабельными и/или константными доменами в одной молекуле полипептида. Кроме того, антигенсвязывающий фрагмент антитела по настоящему изобретению может содержать гомодимер или гетеродимер (или другой мультимер) из любых конфигураций вариабельных и константных доменов, изложенных выше, в нековалентной ассоциации друг с другом и/или с одним или несколькими мономерными доменами VH или VL (например, при помощи дисульфидной(дисульфидных) связи(связей)).
[0058] Как и в случае с целыми молекулами антител антигенсвязывающие фрагменты могут быть моноспецифическими или мультиспецифическими (например, биспецифическими). Мультиспецифический антигенсвязывающий фрагмент антитела будет, как правило, содержать по меньшей мере два различных вариабельных домена, где каждый вариабельный домен способен специфически связываться с отдельным антигеном или с другим эпитопом того же самого антигена. Любой формат мультиспецифических антител, в том числе форматы иллюстративных биспецифических антител, раскрытых в данном документе, могут быть адаптированы для применения в контексте антигенсвязывающего фрагмента антитела по настоящему изобретению при помощи стандартных методик, доступных из уровня техники.
[0059] Константный участок антитела важен с точки зрения способности антитела прикреплять комплемент и опосредовать клеточнозависимую цитотоксичность. Таким образом, изотип антитела может быть выбран на основании того, требуется ли антителу опосредовать цитотоксичность.
[0060] Термин "человеческое антитело", используемый в данном документе, предназначен для включения антител, имеющих вариабельные и константные участки, полученные из последовательностей иммуноглобулинов зародышевой линии человека. Человеческие антитела по настоящему изобретению при этом могут включать аминокислотные остатки, не кодируемые последовательностями иммуноглобулинов зародышевой линии человека (например, мутации, вводимые случайным или сайт-специфичным мутагенезом in vitro или соматической мутацией in vivo), например, в CDR и, в частности, CDR3. Однако, термин "человеческое антитело", используемый в данном документе, не предназначен для включения антител, в которых последовательности CDR, полученные из зародышевой линии другого вида млекопитающего, такого как мышь, привиты на каркасные последовательности человека.
[0061] Термин "рекомбинантное человеческое антитело", используемый в данном документе, предназначен для включения всех человеческих антител, которые получены, экспрессированы, созданы или выделены рекомбинантными способами, таких как антитела, экспрессируемые с помощью рекомбинантного вектора экспрессии, трансфицированного в клетку-хозяина (описанные подробнее далее), антитела, выделенные из комбинаторной библиотеки рекомбинантных человеческих антител (описанные подробнее далее), антитела, выделенные из животного (например, мыши), которое является трансгенным по генам иммуноглобулинов человека (см., например, Taylor et al. (1992) Nucl. Acids Res. 20:6287-6295), или антитела, полученные, экспрессированные, созданные или выделенные любыми другими способами, которые включают соединение последовательностей генов человеческих иммуноглобулинов с другими последовательностями ДНК. Такие рекомбинантные человеческие антитела имеют вариабельные и константные участки, полученные из последовательностей иммуноглобулинов зародышевой линии человека. Однако согласно определенным вариантам осуществления такие рекомбинантные человеческие антитела подвергают in vitro мутагенезу (или в случае использования животного, трансгенного по последовательностям человеческого Ig, in vivo соматическому мутагенезу) и, таким образом, аминокислотные последовательности участков VH и VL рекомбинантных антител представляют собой последовательности, которые, хотя происходят из последовательностей VH и VL зародышевой линии человека и связаны с ними, могут не встречаться в природе в репертуаре антител зародышевой линии человека in vivo.
[0062] Человеческие антитела могут встречаться в двух формах, что связано с гетерогенностью шарнирных участков. В одной форме молекула иммуноглобулина содержит стабильную четырехцепочечную конструкцию массой примерно 150-160 кДа, в которой димеры удерживаются вместе посредством дисульфидной связи, которая связывает тяжелые цепи. Во второй форме димеры не соединены дисульфидными связями, связывающими цепи, и образуется молекула массой приблизительно 75-80 кДа, состоящая из ковалентно связанных легкой и тяжелой цепей (полуантитело). Эти формы крайне сложно разделить даже после аффинной очистки.
[0063] Частота появления второй формы в различных изотипах интактных IgG обусловлена, без ограничения, структурными различиями, связанными с изотипом шарнирного участка антитела. Единственная аминокислотная замена в шарнирном участке шарнира человеческого IgG4 может значимо снизить появление второй формы (Angal et al. (1993) Molecular Immunology 30:105) до уровней, обычно наблюдаемых при использовании шарнира человеческого IgG1. Настоящее изобретение охватывает антитела с одной или несколькими мутациями в шарнирном участке, участке CH2 или CH3, что может быть предпочтительным, например, при получении, для повышения выхода предпочтительной формы антитела.
[0064] Термин "выделенное антитело", используемый в данном документе, означает антитело, которое было идентифицировано и отделено и/или извлечено по меньшей мере от/из одного компонента своего естественного окружения. Например, антитело, которое было отделено или извлечено по меньшей мере от/из одного компонента организма, или от/из ткани или клетки, в которых антитело изначально присутствует или продуцируется естественным путем, представляет собой "выделенное антитело" для целей настоящего изобретения. Выделенное антитело также включает антитело in situ в рекомбинантной клетке. Выделенные антитела представляют собой антитела, которые были подвергнуты по меньше мере одной стадии очистки или выделения. Согласно определенным вариантам осуществления выделенное антитело может практически не содержать другого клеточного материала и/или химических соединений.
[0065] Термин "специфически связывает" и т. п. означает, что антитело или его антигенсвязывающий фрагмент образуют комплекс с антигеном, который является сравнительно устойчивым в физиологических условиях. Способы определения наличия специфического связывания антитела с антигеном хорошо известны из уровня техники и включают, например, равновесный диализ, поверхностный плазмонный резонанс и т. п. Например, антитело, которое "специфически связывает" PCSK9, как используется в контексте настоящего изобретения, включает антитела, которые связывают PCSK9 или его часть с KD, составляющей менее приблизительно 1000 нМ, менее приблизительно 500 нМ, менее приблизительно 300 нМ, менее приблизительно 200 нМ, менее приблизительно 100 нМ, менее приблизительно 90 нМ, менее приблизительно 80 нМ, менее приблизительно 70 нМ, менее приблизительно 60 нМ, менее приблизительно 50 нМ, менее приблизительно 40 нМ, менее приблизительно 30 нМ, менее приблизительно 20 нМ, менее приблизительно 10 нМ, менее приблизительно 5 нМ, менее приблизительно 4 нМ, менее приблизительно 3 нМ, менее приблизительно 2 нМ, менее приблизительно 1 нМ или менее приблизительно 0,5 нМ, измеренной в анализе поверхностного плазмонного резонанса. Однако выделенное антитело, которое специфически связывает PCSK9 человека, характеризуется перекрестной реактивностью к другим антигенам, таким как молекулы PCSK9 от других (не относящихся к человеку) видов.
[0066] Антитела к PCSK9, применимые для способов по настоящему изобретению, могут содержать одну или несколько аминокислотных замен, вставок и/или делеций в каркасных участках и/или участках CDR вариабельных доменов тяжелой и легкой цепей по сравнению с соответствующими последовательностями зародышевой линии, из которых были получены антитела. Такие мутации можно легко определить сравнением аминокислотных последовательностей, раскрытых в данном документе, с последовательностями зародышевой линии, доступными, например, из публичных баз данных последовательностей антител. В настоящем изобретении предусмотрены способы, включающие применение антител и их антигенсвязывающих фрагментов, которые получены из любых аминокислотных последовательностей, раскрытых в данном документе, где одна или несколько аминокислот в одном или нескольких каркасных участках и/или участках CDR мутированы в соответствующий(соответствующие) остаток(остатки) последовательности зародышевой линии, из которой антитело получено, или в соответствующий(соответствующие) остаток(остатки) последовательности другой зародышевой линии, или в консервативную аминокислотную замену соответствующего(соответствующих) остатка(остатков) зародышевой линии (такие изменения последовательностей называются в данном документе собирательно как "мутации зародышевой линии"). Специалист в данной области, исходя из последовательностей вариабельных участков тяжелой и легкой цепей, раскрытых в данном документе, может легко получить множество антител и антигенсвязывающих фрагментов, которые содержат одну или несколько отдельных мутаций зародышевой линии или их комбинации. Согласно определенным вариантам осуществления все остатки каркасных участков и/или CDR в доменах VH и/или VL обратно мутированы в остатки, встречающиеся в исходной последовательности зародышевой линии, из которой произошло антитело. Согласно другим вариантам осуществления только определенные остатки обратно мутированы в остатки исходной последовательности зародышевой линии, например, мутированы только остатки, расположенные в первых 8 аминокислотах FR1 или в последних 8 аминокислотах FR4, или мутированы только остатки, расположенные в CDR1, CDR2 или CDR3. Согласно другим вариантам осуществления один или несколько остатков каркасных участков и/или CDR мутированы в соответствующий(соответствующие) остаток(остатки) последовательности другой зародышевой линии (т. е. последовательности зародышевой линии, которая отличается от последовательности зародышевой линии, из которой изначально произошло антитело). Кроме того, антитела по настоящему изобретению могут содержать любую комбинацию из двух или более мутаций зародышевой линии в пределах каркасных участков и/или участков CDR, например, где определенные отдельные остатки мутированы в соответствующий остаток последовательности конкретной зародышевой линии, в то время как определенные другие остатки, которые отличаются от последовательности исходной зародышевой линии, сохраняются или мутированы в соответствующий остаток последовательности другой зародышевой линии. Сразу после получения антитела и антигенсвязывающие фрагменты, которые содержат одну или несколько мутаций зародышевой линии, могут быть легко протестированы в отношении одного или нескольких требуемых свойств, таких как улучшенная специфичность связывания, повышенная аффинность связывания, улучшенные или усиленные биологические антагонистические или агонистические свойства (в случае необходимости), сниженная иммуногенность и т. п. Применение антител и антигенсвязывающих фрагментов, полученных при помощи этого общего способа, охвачено настоящим изобретением.
[0067] В настоящем изобретении также предусмотрены способы, включающие применение антител к PCSK9, содержащих варианты любых из аминокислотных последовательностей HCVR, LCVR и/или CDR, раскрытых в данном документе, с одной или несколькими консервативными заменами. Например, настоящее изобретение включает применение антител к PCSK9 с аминокислотными последовательностями HCVR, LCVR и/или CDR, например, с 10 или менее, 8 или менее, 6 или менее, 4 или менее и т. д. консервативными аминокислотными заменами относительно любой из аминокислотных последовательностей HCVR, LCVR и/или CDR, раскрытых в данном документе.
[0068] Термин "поверхностный плазмонный резонанс", используемый в данном документе, относится к оптическому феномену, который обеспечивает возможность анализа взаимодействий в реальном времени посредством выявления изменений концентраций белков в биосенсорной матрице, например, при помощи системы BIAcore™ (Biacore Life Sciences division of GE Healthcare, Пискатауэй, Нью-Джерси).
[0069] Термин "KD", используемый в данном документе, относится к равновесной константе диссоциации взаимодействия определенного антитела и антигена.
[0070] Термин "эпитоп" относится к антигенной детерминанте, которая взаимодействует со специфическим антигенсвязывающим сайтом в вариабельном участке молекулы антитела, известном как паратоп. Один антиген может иметь более одного эпитопа. Таким образом, различные антитела могут связываться с различными областями на антигене и могут иметь различные биологические эффекты. Эпитопы могут быть конформационными или линейными. Конформационный эпитоп образован пространственно сближенными аминокислотами из различных сегментов линейной полипептидной цепи. Линейный эпитоп образован смежными аминокислотными остатками в полипептидной цепи. При определенных условиях эпитоп может включать фрагменты сахаридов, фосфорильных групп или сульфонильных групп антигена.
[0071] Согласно определенным вариантам осуществления антитело к PCSK9, используемое в способах по настоящему изобретению, является антителом с pH-зависимыми характеристиками связывания. Используемое в данном документе выражение "pH-зависимое связывание" означает, что антитело или его антигенсвязывающий фрагмент проявляют "сниженное связывание с PCSK9 при кислом pH по сравнению с нейтральным pH" (для целей настоящего изобретения оба выражения могут использоваться взаимозаменяемо). Например, антитела "с pH-зависимыми характеристиками связывания" включают антитела и их антигенсвязывающие фрагменты, которые связывают PCSK9 с более высокой аффинностью при нейтральном pH, чем при кислом pH. В определенных вариантах осуществления антитела и антигенсвязывающие фрагменты по настоящему изобретению связывают PCSK9 по меньшей мере в 3, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100 раз или более высокой аффинностью при нейтральном pH, чем при кислом pH.
[0072] Согласно данному аспекту настоящего изобретения антитела к PCSK9 с pH-зависимыми характеристиками связывания могут характеризоваться одной или несколькими аминокислотными вариациями относительно исходного антитела к PCSK9. Например, антитело к PCSK9 с pH-зависимыми характеристиками связывания может содержать одну или несколько замен на гистидин или вставок из гистина, например, в одном или нескольких CDR исходного антитела к PCSK9. Таким образом, согласно определенным вариантам осуществления настоящего изобретения представлены способы, включающие введение антитела к PCSK9, которое содержит аминокислотные последовательности CDR (например, CDR тяжелой и легкой цепи), которые идентичны аминокислотным последовательностям CDR исходного антитела к PCSK9, за исключением замены одной или нескольких аминокислот в одном или нескольких CDR исходного антитела на остаток гистидина. Антитела к PCSK9 с pH-зависимым связыванием могут характеризоваться, например, 1, 2, 3, 4, 5, 6, 7, 8, 9 или более заменами на гистидин, либо в пределах одного CDR исходного антитела, либо распределенными в нескольких (например, 2, 3, 4, 5 или 6) CDR исходного антитела к PCSK9. Например, настоящее изобретение включает применение антител к PCSK9 с pH-зависимым связыванием, содержащих одну или несколько замен на гистидин в HCDR1, одну или несколько замен на гистидин в HCDR2, одну или несколько замен на гистидин в HCDR3, одну или несколько замен на гистидином в LCDR1, одну или несколько замен на гистидин в LCDR2 и/или одну или несколько замен на гистидин в LCDR3 исходного антитела к PCSK9.
[0073] Как используется в данном документе, выражение "кислый pH" означает pH 6,0 или меньше (например, меньше приблизительно 6,0, меньше приблизительно 5,5, меньше приблизительно 5,0 и т. п.). Выражение "кислый pH" включает значения pH, составляющие приблизительно 6,0, 5,95, 5,90, 5,85, 5,8, 5,75, 5,7, 5,65, 5,6, 5,55, 5,5, 5,45, 5,4, 5,35, 5,3, 5,25, 5,2, 5,15, 5,1, 5,05, 5,0 или меньше. Как используется в данном документе, выражение "нейтральный pH" означает pH от приблизительно 7,0 до приблизительно 7,4. Выражение "нейтральный pH" включает значения pH, составляющие приблизительно 7,0, 7,05, 7,1, 7,15, 7,2, 7,25, 7,3, 7,35 и 7,4.
Получение человеческих антител
[0074] Из уровня техники известны способы получения человеческих антител в трансгенных мышах. Любые такие известные способы можно использовать в контексте настоящего изобретения для создания человеческих антител, которые специфически связываются с PCSK9 человека.
[0075] При помощи технологии VELOCIMMUNE® (см., например, US 6596541, Regeneron Pharmaceuticals) или любого другого известного способа получения моноклональных антител изначально выделяют высокоаффинные химерные антитела к PCSK9 с человеческим вариабельным участком и мышиным константным участком. Технология VELOCIMMUNE® включает получение трансгенной мыши с геномом, содержащим вариабельные участки человеческих тяжелой и легкой цепей, функционально связанные с эндогенными мышиными локусами константного участка так, что мышь продуцирует антитело, содержащее человеческий вариабельный участок и мышиный константный участок в ответ на антигенную стимуляцию. ДНК, кодирующую вариабельные участки тяжелой и легкой цепей антитела, выделяют и функционально связывают с ДНК, кодирующей константные участки человеческих тяжелой и легкой цепей. Затем ДНК экспрессируют в клетке, способной экспрессировать полностью человеческое антитело.
[0076] Как правило, на мышь, полученную при помощи VELOCIMMUNE®, воздействуют антигеном, представляющим интерес, и из мышей, которые экспрессируют антитела, извлекают лимфатические клетки (такие как B-клетки). Лимфатические клетки можно сливать с линией клеток миеломы с получением иммортализированных линий клеток гибридомы, и такие линии клеток гибридомы подвергают скринингу и отбирают для идентификации линий клеток гибридомы, которые продуцируют антитела, специфические к антигену, представляющему интерес. ДНК, кодирующую вариабельные участки тяжелой цепи и легкой цепи, можно выделить и связать с константными участками требуемых изотипов тяжелой цепи и легкой цепи. Такой белок-антитело можно получать в клетке, такой как клетка CHO. Альтернативно, ДНК, кодирующую антиген-специфические химерные антитела или вариабельные домены легкой и тяжелой цепей, можно ь выделить непосредственно из антиген-специфических лимфоцитов.
[0077] Изначально выделяют высокоаффинные химерные антитела с человеческим вариабельным участком и мышиным константным участком. Антитела характеризуют и выбирают по требуемым характеристикам, в том числе по аффинности, селективности, эпитопу и т. д., при помощи стандартных процедур, известных специалистам в данной области. Мышиные константные участки заменяют требуемым человеческим константным участком с получением полностью человеческого антитела по настоящему изобретению, например, IgG1 или IgG4 дикого типа или модифицированного IgG1 или IgG4. В то время как выбранный константный участок может варьировать в зависимости от конкретного применения, у вариабельного участка сохраняются характеристики высокоаффинного связывания с антигеном или специфичности к мишеням.
[0078] Как правило, антитела, которые можно применять в способах по настоящему изобретению, характеризуются высокой аффинностью, как описано выше, при измерении связывания с антигеном либо иммобилизованным на твердой фазе, либо находящимся в жидкой фазе. Мышиные константные участки заменяют требуемыми человеческими константными участками с получением полностью человеческих антител по настоящему изобретению. В то время как выбранный константный участок может варьировать в зависимости от конкретного применения, у вариабельного участка сохраняются характеристики высокоаффинного связывания с антигеном или специфичности к мишеням.
[0079] Конкретные примеры человеческих антител или антигенсвязывающих фрагментов антител, специфически связывающих PCSK9, которые можно применять в контексте способов по настоящему изобретению, включают любое антитело или антигенсвязывающий фрагмент, которые содержат три CDR тяжелой цепи (HCDR1, HCDR2 и HCDR3), содержащиеся в вариабельном участке тяжелой цепи (HCVR) с аминокислотной последовательностью, выбранной из группы, состоящей из SEQ ID NO: 1 и 11 или в значительной степени подобной им последовательности с по меньшей мере 90%, по меньшей мере 95%, по меньшей мере 98% или по меньшей мере 99% идентичностью последовательности. Альтернативно, конкретные примеры человеческих антител или антигенсвязывающих фрагментов антител, специфически связывающих PCSK9, которые можно применять в контексте способов по настоящему изобретению, включают любое антитело или антигенсвязывающий фрагмент, которые содержат три CDR тяжелой цепи (HCDR1, HCDR2 и HCDR3), содержащиеся в вариабельном участке тяжелой цепи (HCVR) с аминокислотной последовательностью, выбранной из группы, состоящей из SEQ ID NO: 37, 45, 53, 61, 69, 77, 85, 93, 101, 109, 117, 125, 133, 141, 149, 157, 165, 173, 181 и 189 или в значительной степени подобной им последовательности с по меньшей мере 90%, по меньшей мере 95%, по меньшей мере 98% или по меньшей мере 99% идентичностью последовательности. Антитело или антигенсвязывающий фрагмент могут содержать три CDR легкой цепи (LCVR1, LCVR2, LCVR3), содержащиеся в вариабельном участке легкой цепи (LCVR) с аминокислотной последовательностью, выбранной из группы, состоящей из SEQ ID NO: 6 и 15 или в значительной степени подобной им последовательности с по меньшей мере 90%, по меньшей мере 95%, по меньшей мере 98% или по меньшей мере 99% идентичностью последовательности. Альтернативно, антитело или антигенсвязывающий фрагмент могут содержать три CDR легкой цепи (LCVR1, LCVR2 и LCVR3), содержащиеся в вариабельном участке легкой цепи (LCVR) с аминокислотной последовательностью, выбранной из группы, состоящей из SEQ ID NO: 41, 49, 57, 65, 73, 81, 89, 97, 105, 113, 121, 129, 137, 145, 153, 161, 169, 177, 185 и 193 или в значительной степени подобной им последовательности с по меньшей мере 90%, по меньшей мере 95%, по меньшей мере 98% или по меньшей мере 99% идентичностью последовательности.
[0080] В определенных вариантах осуществления настоящего изобретения антитело или его антигенсвязывающий фрагмент содержат шесть CDR (HCDR1, HCDR2, HCDR3, LCDR1, LCDR2 и LCDR3) из пар аминокислотных последовательностей вариабельных участков тяжелой и легкой цепей (HCVR/LCVR), выбранных из группы, состоящей из SEQ ID NO: 1/6 и 11/15. Альтернативно, в определенных вариантах осуществления настоящего изобретения антитело или антигенсвязывающий фрагмент содержат шесть CDR (HCDR1, HCDR2, HCDR3, LCDR1, LCDR2 и LCDR3) из пар аминокислотных последовательностей вариабельных участков тяжелой и легкой цепей (HCVR/LCVR), выбранных из группы, состоящей из SEQ ID NO: 37/41, 45/49, 53/57, 61/65, 69/73, 77/81, 85/89, 93/97, 101/105, 109/113, 117/121, 125/129, 133/137, 141/145, 149/153, 157/161, 165/169, 173/177, 181/185 и 189/193.
[0081] В определенных вариантах осуществления настоящего изобретения антитело к PCSK9 или его антигенсвязывающий фрагмент, которые можно применять в способах по настоящему изобретению, имеют аминокислотные последовательности HCDR1/HCDR2/HCDR3/LCDR1/LCDR2/LCDR3, выбранные из SEQ ID NO: 2/3/4/7/8/10 (mAb316P) и 12/13/14/16/17/18 (mAb300N) (см. публикацию заявки на выдачу патента США № 2010/0166768).
[0082] В определенных вариантах осуществления настоящего изобретения антитело или его антигенсвязывающий фрагмент содержат пары аминокислотных последовательностей HCVR/LCVR, выбранные из группы, состоящей из SEQ ID NO: 1/6 и 11/15. Альтернативно, в определенных вариантах осуществления настоящего изобретения антитело или антигенсвязывающий белок содержат пары аминокислотных последовательностей вариабельных участков тяжелой и легкой цепей (HCVR/LCVR), выбранные из группы, состоящей из SEQ ID NO: 37/41, 45/49, 53/57, 61/65, 69/73, 77/81, 85/89, 93/97, 101/105, 109/113, 117/121, 125/129, 133/137, 141/145, 149/153, 157/161, 165/169, 173/177, 181/185 и 189/193.
Фармацевтические композиции и способы введения
[0083] В настоящем изобретении предусмотрены способы, которые включают введение пациенту с высоким риском возникновения сердечно-сосудистых заболеваний ингибитора PCSK9, где ингибитор PCSK9 содержится в фармацевтической композиции. Фармацевтические композиции по настоящему изобретению составляют с подходящими носителями, наполнителями и другими средствами, которые обеспечивают надлежащий перенос, доставку, переносимость и т. п. Множество подходящих составов можно найти в справочнике, известном всем химикам-фармацевтам: Remington's Pharmaceutical Sciences, Mack Publishing Company, Истон, Пенсильвания. Эти составы включают, например, порошки, пасты, мази, желе, воски, масла, липиды, везикулы, содержащие (катионные или анионные) липиды (такие как LIPOFECTIN™), конъюгаты ДНК, безводные абсорбционные пасты, эмульсии типа "масло воде" и "вода в масле", эмульсии карбовакс (полиэтиленгликоли с различной молекулярной массой), полужидкие гели и полужидкие смеси, содержащие карбовакс. См. также Powell et al. "Compendium of excipients for parenteral formulations" PDA (1998) J Pharm Sci Technol 52:238-311.
[0084] Известны различные системы доставки и их можно использовать для введения фармацевтической композиции по настоящему изобретению, например, инкапсулирование в липосомы, микрочастицы, микрокапсулы, рекомбинантные клетки, способные экспрессировать мутантные вирусы, опосредованный рецепторами эндоцитоз (см., например, Wu et al., 1987, J. Biol. Chem. 262:4429-4432). Способы введения включают без ограничения внутрикожный, внутримышечный, интраперитонеальный, внутривенный, подкожный, интраназальный, эпидуральный и пероральный пути. Композицию можно вводить любым удобным способом, например инфузией или болюсной инъекцией, абсорбцией через эпителиальные или кожно-слизистые покровы (например, слизистую ротовой полости, слизистую прямой кишки и кишечника и др.), и можно вводить совместно с другими биологически активными средствами.
[0085] Фармацевтическую композицию по настоящему изобретению можно доставлять подкожно или внутривенно при помощи стандартной иглы и шприца. Кроме того, что касается подкожной доставки, при доставке фармацевтической композиции по настоящему изобретению широко используется устройство для доставки в виде шприца-ручки. Такое устройство для доставки в виде шприца-ручки может быть многоразового и одноразового использования. В устройстве для доставки в виде шприца-ручки многоразового использования, как правило, используется заменяемый картридж, который содержит фармацевтическую композицию. После введения всей фармацевтической композиции из картриджа и опустошения картриджа пустой картридж можно легко утилизировать и заменить новым картриджем, который содержит фармацевтическую композицию. Затем устройство для доставки в виде шприца-ручки можно использовать повторно. В устройстве для доставки в виде шприца-ручки одноразового использования заменяемый картридж отсутствует. Вместо этого устройство для доставки в виде шприца-ручки одноразового использования выпускают предварительно наполненным фармацевтической композицией, содержащейся в резервуаре устройства. После того как резервуар опустошается от фармацевтической композиции, устройство целиком утилизируют.
[0086] Многочисленные устройства для доставки в виде шприца-ручки или автоинжектора многоразового использования используются при подкожной доставке фармацевтической композиции по настоящему изобретению. Примеры включают без ограничения AUTOPEN™ (Owen Mumford, Inc., Вудсток, Великобритания), шприц-ручку DISETRONIC™ (Disetronic Medical Systems, Бургдорф, Швейцария), шприц-ручку HUMALOG MIX 75/25™, шприц-ручку HUMALOG™, шприц-ручку HUMALIN 70/30™ (Eli Lilly and Co., Индианаполис, Индиана), NOVOPEN™ I, II и III (Novo Nordisk, Копенгаген, Дания), NOVOPEN JUNIOR™ (Novo Nordisk, Копенгаген, Дания), шприц-ручку BD™ (Becton Dickinson, Франклин-Лэйкс, Нью-Джерси), OPTIPEN™, OPTIPEN PRO™, OPTIPEN STARLET™ и OPTICLIK™ (Sanofi-Aventis, Франкфурт, Германия), при этом упомянуты лишь несколько из них. Примеры устройств для доставки в виде шприца-ручки одноразового использования, используемых при подкожном введении фармацевтической композиции по настоящему изобретению, включают без ограничения шприц-ручку SOLOSTAR™ (Sanofi-Aventis), FLEXPEN™ (Novo Nordisk) и KWIKPEN™ (Eli Lilly), автоинжектор SURECLICKTM (Amgen, Таузанд-Окс, Калифорния), PENLETTM (Haselmeier, Штутгарт, Германия), EPIPEN (Dey, L.P.) и шприц-ручку HUMIRATM (Abbott Labs, Эббот-Парк, Иллинойс), при этом упомянуты лишь несколько из них.
[0087] В определенных ситуациях фармацевтическую композицию можно доставлять в системе с контролируемым высвобождением. В одном варианте осуществления можно использовать насос (см. Langer, выше; Sefton, 1987, CRC Crit. Ref. Biomed. Eng. 14:201). В другом варианте осуществления можно использовать полимерные материалы; см. Medical Applications of Controlled Release, Langer and Wise (eds.), 1974, CRC Pres., Boca Raton, Florida. Согласно еще одному варианту осуществления систему с контролируемым высвобождением можно поместить вблизи от мишени, на которую направлена композиция, при этом требуется намного меньшее количество, чем системная доза (см., например, Goodson, 1984, в Medical Applications of Controlled Release, supra, vol. 2, pp. 115-138). Другие системы с контролируемым высвобождением описаны в обзоре Langer, 1990, Science 249:1527-1533.
[0088] Инъекционные препараты могут включать лекарственные формы для внутривенных, подкожных, внутрикожных и внутримышечных инъекций, капельных инфузий и т. п. Эти инъекционные препараты можно получать при помощи известных способов. Например, инъекционные препараты можно получать, например, растворением, суспендированием или эмульгированием антитела или его соли, описанных выше, в стерильной водной среде или масляной среде, обычно используемых для инъекций. Водной средой для инъекций является, например, физиологический солевой раствор, изотонический раствор, содержащий глюкозу и другие вспомогательные средства и т. п., которые можно использовать совместно с подходящим солюбилизирующим средством, таким как спирт (например, этанол), многоатомный спирт (например, пропиленгликоль, полиэтиленгликоль), неионогенное поверхностно-активное вещество [например, полисорбат 80, HCO-50 (полиоксиэтиленовый (50 моль) аддукт гидрогенизированного касторового масла)] и т. п. В качестве масляной среды используют, например, кунжутное масло, соевое масло и т. п., которые можно использовать совместно с солюбилизирующим средством, таким как бензилбензоат, бензиловый спирт и т. п. Полученным таким способом инъекционным составом предпочтительно наполняют подходящую ампулу.
[0089] Преимущественно, фармацевтические композиции для перорального или парентерального применения, описанные выше, получают в лекарственных формах в стандартной дозе, подходящей для подбора дозы активных ингредиентов. Такие лекарственные формы в стандартной дозе включают, например, таблетки, пилюли, капсулы, инъекции (ампулы), суппозитории и т. п.
Дозировка
[0090] Количество ингибитора PCSK9 (например, антитела к PCSK9), вводимого субъекту с высоким риском возникновения сердечно-сосудистых заболеваний согласно способам по настоящему изобретению, как правило, представляет собой терапевтически эффективное количество. Как используется в данном документе, фраза "терапевтически эффективное количество" означает дозу ингибитора PCSK9, которая в результате приводит к выявляемому улучшению (по меньшей мере на приблизительно 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75% или более от исходного уровня) одного или нескольких параметров, выбранных из группы, состоящей из уровней LDL-C, ApoB, не-HDL-C, общего холестерина, HLDL-C, триглицеридов и Lp(a).
[0091] В случае антитела к PCSK9 терапевтически эффективное количество может составлять от приблизительно 0,05 мг до приблизительно 600 мг, например, приблизительно 0,05 мг, приблизительно 0,1 мг, приблизительно 1,0 мг, приблизительно 1,5 мг, приблизительно 2,0 мг, приблизительно 10 мг, приблизительно 20 мг, приблизительно 30 мг, приблизительно 40 мг, приблизительно 50 мг, приблизительно 60 мг, приблизительно 70 мг, приблизительно 75 мг, приблизительно 80 мг, приблизительно 90 мг, приблизительно 100 мг, приблизительно 110 мг, приблизительно 120 мг, приблизительно 130 мг, приблизительно 140 мг, приблизительно 150 мг, приблизительно 160 мг, приблизительно 170 мг, приблизительно 180 мг, приблизительно 190 мг, приблизительно 200 мг, приблизительно 210 мг, приблизительно 220 мг, приблизительно 230 мг, приблизительно 240 мг, приблизительно 250 мг, приблизительно 260 мг, приблизительно 270 мг, приблизительно 280 мг, приблизительно 290 мг, приблизительно 300 мг, приблизительно 310 мг, приблизительно 320 мг, приблизительно 330 мг, приблизительно 340 мг, приблизительно 350 мг, приблизительно 360 мг, приблизительно 370 мг, приблизительно 380 мг, приблизительно 390 мг, приблизительно 400 мг, приблизительно 410 мг, приблизительно 420 мг, приблизительно 430 мг, приблизительно 440 мг, приблизительно 450 мг, приблизительно 460 мг, приблизительно 470 мг, приблизительно 480 мг, приблизительно 490 мг, приблизительно 500 мг, приблизительно 510 мг, приблизительно 520 мг, приблизительно 530 мг, приблизительно 540 мг, приблизительно 550 мг, приблизительно 560 мг, приблизительно 570 мг, приблизительно 580 мг, приблизительно 590 мг или приблизительно 600 мг антитела к PCSK9.
[0092] Количество антитела к PCSK9, содержащееся в отдельных дозах, можно выражать в миллиграммах антитела на килограмм массы тела пациента (т. е. мг/кг). Например, антитело к PCSK9 можно вводить пациенту в дозе, составляющей от приблизительно 0,0001 до приблизительно 10 мг/кг массы тела пациента.
Виды комбинированной терапии
[0093] Как описано в других частях данного документа, способы по настоящему изобретению могут включать введение пациенту с высоким риском возникновения сердечно-сосудистых заболеваний ингибитора PCSK9 совместно с ранее назначенной пациенту схемой терапии со стабильной ежедневной максимально переносимой дозой статинов. Согласно определенным вариантам осуществления настоящего изобретения дополнительные лекарственные средства, помимо статина, можно вводить пациенту совместно с ингибитором PCSK9. Примеры таких дополнительных лекарственных средств включают, например, (1) средство, которое ингибирует всасывание холестерина и/или реабсорбцию желчных кислот (например, эзетимиб); (2) средство, которое повышает катаболизм липопротеинов (такое как ниацин); и/или (3) активаторы фактора транскрипции LXR, который играет роль в выведении холестерина, такого как 22-гидроксихолестерин.
Схемы введения
[0094] Согласно определенным вариантам осуществления настоящего изобретения субъекту в течение определенного периода времени (например, наряду со схемой терапии с ежедневным введением статина) можно вводить несколько доз ингибитора PCSK9 (т.е. фармацевтической композиции, содержащей ингибитор PCSK9). Способы по данному аспекту настоящего изобретения включают последовательное введение субъекту с высоким риском возникновения сердечно-сосудистых заболеваний нескольких доз ингибитора PCSK9. Как используется в данном документе, выражение "последовательное введение" означает, что каждую дозу ингибитора PCSK9 вводят субъекту в разный момент времени, например, в разные дни, разделенные предварительно определенным интервалом (например, часами, днями, неделями или месяцами). В настоящем изобретении предусмотрены способы, которые включают последовательное введение пациенту с высоким риском возникновения сердечно-сосудистых заболеваний одной начальной дозы ингибитора PCSK9, затем одной или нескольких вторичных доз ингибитора PCSK9, и затем необязательно одной или нескольких третичных доз ингибитора PCSK9.
[0095] Термины "начальная доза", "вторичные дозы" и "третичные дозы" относятся к временной последовательности введения отдельных доз фармацевтической композиции, содержащей ингибитор PCSK9. Таким образом, "начальная доза" представляет собой дозу, которую вводят в начале схемы лечения (также обозначается как "исходная доза"); "вторичные дозы" представляют собой дозы, которые вводят после начальной дозы; и "третичные дозы" представляют собой дозы, которые вводят после вторичных доз. Все из начальной, вторичных и третичных доз могут содержать одинаковое количество ингибитора PCSK9, но, как правило, они могут отличаться друг от друга по частоте введения. Однако согласно определенным вариантам осуществления количества ингибитора PCSK9, содержащегося в начальной, вторичных и/или третичных дозах, отличаются друг от друга (например, корректируются в сторону повышения или понижения при необходимости) в ходе курса лечения. Согласно определенным вариантам осуществления две или больше (например, 2, 3, 4 или 5) доз вводятся в начале схемы лечения в виде "ударных доз", затем последующие дозы вводятся с меньшей частотой (например, "поддерживающие дозы").
[0096] Согласно иллюстративным вариантам осуществления настоящего изобретения каждую вторичную и/или третичную дозу вводят через 1-26 (например, 1, 1½, 2, 2½, 3, 3½, 4, 4½, 5, 5½, 6, 6½, 7, 7½, 8, 8½, 9, 9½, 10, 10½, 11, 11½, 12, 12½, 13, 13½, 14, 14½, 15, 15½, 16, 16½, 17, 17½, 18, 18½, 19, 19½, 20, 20½, 21, 21½, 22, 22½, 23, 23½, 24, 24½, 25, 25½, 26, 26½ или больше) недель после непосредственно предшествующей дозы. Выражение "непосредственно предшествующая доза", как используется в данном документе, означает дозу антигенсвязывающей молекулы в последовательности из нескольких введений, которую вводят пациенту перед введением ближайшей следующей дозы в последовательности, без промежуточных доз.
[0097] Способы по данному аспекту настоящего изобретения могут включать введение пациенту с высоким риском возникновения сердечно-сосудистых заболеваний любого количества вторичных и/или третичных доз ингибитора PCSK9. Например, в определенных вариантах осуществления пациенту вводят только одну вторичную дозу. Согласно другим вариантам осуществления пациенту вводят две или более (например, 2, 3, 4, 5, 6, 7, 8 или больше) вторичных доз. Аналогично, согласно определенным вариантам осуществления пациенту вводят только одну третичную дозу. Согласно другим вариантам осуществления пациенту вводят две или более (например, 2, 3, 4, 5, 6, 7, 8 или больше) третичных доз.
[0098] Согласно вариантам осуществления, включающим несколько вторичных доз, каждую вторичную дозу можно вводить с той же частотой, что и остальные вторичные дозы. Например, каждую вторичную дозу можно вводить пациенту с высоким риском возникновения сердечно-сосудистых заболеваний через 1-2, 4, 6, 8 или более недель после непосредственно предшествующей дозы. Аналогично, в вариантах осуществления, включающих несколько третичных доз, каждую третичную дозу можно вводить с той же частотой, что и остальные третичные дозы. Например, каждую третичную дозу можно вводить пациенту через 1-2, 4, 6, 8 или более недель после непосредственно предшествующей дозы. Альтернативно, частота, с которой вторичные и/или третичные дозы вводят пациенту, может варьировать на протяжении схемы лечения. Частота введения может корректироваться врачом на протяжении курса лечения в зависимости от потребностей отдельного пациента после клинического обследования.
[0099] Настоящее изобретение включает схемы введения, предусматривающие вариант с повышением (также обозначается в данном документе как "изменение дозы"). Как используется в данном документе, выражение "вариант с повышением дозы" означает, что после получения определенного числа доз ингибитора PCSK9 в случае, если пациент не достиг определенного снижения одного или нескольких терапевтических параметров, впоследствии дозу ингибитора PCSK9 повышают. Например, в случае использования схемы терапии, предусматривающей введение пациенту с высоким риском возникновения сердечно-сосудистых заболеваний антитела к PCSK9 в дозах по 75 мг с частотой один раз в две недели, если через 8 недель (т. е. после 5 доз, вводимых на 0 неделе, 2 неделе, 4 неделе, 6 неделе и 8 неделе) пациент не достиг концентрации LDL-C в сыворотке крови меньше 70 мг/дл, то дозу антитела к PCSK9 повышают, например, до 150 мг, которую вводят впоследствии один раз в две недели (например, начиная с 12 недели).
[00100] В определенных вариантах осуществления антитело к PCSK9 вводят субъекту в дозе, составляющей приблизительно 150 мг, каждые две недели, например, в количестве по меньшей мере шести доз.
[00101] В определенных вариантах осуществления антитело к PCSK9 вводят субъекту в дозе, составляющей приблизительно 75 мг, каждые две недели, например, в количестве по меньшей мере шести доз.
[00102] В некоторых вариантах осуществления антитело вводят субъекту в дозе, составляющей приблизительно 75 мг, каждые две недели в течение 12 недель и дозу сохраняют на уровне 75 мг каждые две недели в случае, если на 8 неделе значение уровня LDL-C у субъекта составляет меньше 70 мг/дл.
[00103] Согласно другим вариантам осуществления антитело вводят субъекту в дозе, составляющей приблизительно 75 мг, каждые две недели в течение 12 недель, и дозу повышают до приблизительно 150 мг каждые две недели в случае, если на 8 неделе значение уровня LDL-C у субъекта было больше или равнялось 70 мг/дл.
ПРИМЕРЫ
[00104] Следующие примеры изложены для того, чтобы обеспечить обычных специалистов в данной области полным раскрытием и описанием того, как осуществлять и применять способы и получать и применять композиции по настоящему изобретению, и они не предполагают ограничение объема того, что авторы настоящего изобретения рассматривают в качестве своего изобретения. Были приложены усилия для обеспечения точности в отношении используемых чисел (например, количеств, температуры и т. п.), однако, необходимо учитывать некоторые экспериментальные ошибки и отклонения. Если не указано иное, то части являются частями по массе, молекулярная масса является средней молекулярной массой, температура представлена в градусах Цельсия, а давление является атмосферным или близким к атмосферному.
Пример 1. Получение человеческих антител к PCSK9 человека
[00105] Человеческие антитела к PCSK9 получали, как описано выше в патенте США № 8062640. Иллюстративный ингибитор PCSK9, используемый в следующем примере, представляет собой человеческое антитело к PCSK9, обозначаемое "mAb316P", также известное как "алирокумаб". mAb316P имеет следующие характеристики аминокислотной последовательности: вариабельный участок тяжелой цепи (HCVR), содержащий SEQ ID NO:1, вариабельный домен легкой цепи (LCVR), содержащий SEQ ID NO:6; определяющий комплементарность участок 1 тяжелой цепи (HCDR1), содержащий SEQ ID NO:2, HCDR2, содержащий SEQ ID NO:3, HCDR3, содержащий SEQ ID NO:4, определяющий комплементарность участок 1 легкой цепи (LCDR1), содержащий SEQ ID NO:7, LCDR2, содержащий SEQ ID NO:8, и LCDR3, содержащий SEQ ID NO:10.
Пример 2. Эффективность и безопасность алирокумаба, полностью человеческого моноклонального антитела PCSK9 ("mAb316P"), у пациентов с высоким риском возникновения сердечно-сосудистых заболеваний, имеющих гиперхолестеринемию, резистентную к максимально переносимым дозам статинов
ВВЕДЕНИЕ
[00106] Целью исследований COMBO I и II была оценка эффективности и безопасности алирокумаба (ʺmAb316Pʺ) в качестве дополнительной терапии к терапии посредством стабильной максимально переносимой ежедневной дозы статинов, с другой липид-корригирующей терапией или без нее, у пациентов, имеющих гиперхолестеринемию на фоне высокого риска возникновения сердечно-сосудистых заболеваний. COMBO I представляло собой плацебо-контролируемое исследование, и пациенты могли получать другие виды липид-корригирующей терапии в дополнение к терапии статинами; в COMBO II сравнивали алирокумаб с эзетимибом у пациентов, получающих терапию статинами (но без других видов липид-корригирующей терапии). Каждое из исследований COMBO было рандомизированным, двойным слепым и плацебо-контролируемым. В обоих исследованиях использовали начальную дозу алирокумаба, составляющую 75 мг каждые 2 недели (Q2W; вводили в виде 1 мл раствора посредством автоинжектора). У пациентов с уровнями LDL-C ≥70 мг/дл после 8 недель лечения дозу повышали слепым способом на 12 неделе до 150 мг алирокумаба Q2W (также автоинжектор на 1 мл).
ЦЕЛИ ИССЛЕДОВАНИЯ
[00107] Первичной конечной точкой эффективности для каждого исследования являлось различие между режимами лечения в процентном изменении уровня LDL-C от исходного уровня до 24 недели.
[00108] Ключевыми вторичными конечными точками для каждого исследования были: процентное изменение рассчитанного уровня LDL-C от исходного уровня до 24 недели в популяции анализа в ходе лечения с применением всех значений уровня холестерина липопротеинов низкой плотности (LDL-C) в течение периода лечения для оценки эффективности (оцениваемая величина ITT); процентное изменение рассчитанного уровня LDL-C от исходного уровня до 12 недели (оцениваемая величина ITT); процентное изменение рассчитанного уровня LDL-C от исходного уровня до 12 недели (оцениваемая величина анализа у пациентов, получающих лечение); процентное изменение уровня аполипротеина (Apo) B от исходного уровня до 24 недели (оцениваемая величина ITT); процентное изменение уровня Apo B от исходного уровня до 24 недели (оцениваемая величина анализа у пациентов, получающих лечение); процентное изменение уровня холестерина, не относящегося к холестерину липопротеинов высокой плотности (не-HDL-C), от исходного уровня до 24 недели (оцениваемая величина ITT); процентное изменение уровня не-HDL-C от исходного уровня до 24 недели (оцениваемая величина анализа у пациентов, получающих лечение); процентное изменение уровня общего холестерина от исходного уровня до 24 недели (оцениваемая величина ITT); процентное изменение уровня Apo B от исходного уровня до 12 недели (оцениваемая величина ITT); процентное изменение уровня не-HDL-C от исходного уровня до 12 недели (оцениваемая величина ITT); процентное изменение уровня общего холестерина от исходного уровня до 12 недели (оцениваемая величина ITT); процентное изменение рассчитанного уровня LDL-C от исходного уровня до 52 недели (оцениваемая величина ITT); доля пациентов, достигающих рассчитанного уровня LDL-C <70 мг/дл (1,81 ммоль/л) на 24 неделе (оцениваемая величина ITT); доля пациентов, достигающих рассчитанного уровня LDL-C <70 мг/дл (1,81 ммоль/л) на 24 неделе (оцениваемая величина анализа у пациентов, получающих лечение); процентное изменение уровня липопротеина (a) (Lp(a)) от исходного уровня до 24 недели (оцениваемая величина ITT); процентное изменение уровня HDL-C от исходного уровня до 24 недели (оцениваемая величина ITT); процентное изменение уровня триглицеридов натощак (TG) от исходного уровня до 24 недели (оцениваемая величина ITT); процентное изменение уровня Apo A1 от исходного уровня до 24 недели (оцениваемая величина ITT); процентное изменение уровня Lp(a) от исходного уровня до 12 недели (оцениваемая величина ITT); процентное изменение уровня HDL-C от исходного уровня до 12 недели (оцениваемая величина ITT); процентное изменение уровня TG натощак от исходного уровня до 12 недели (оцениваемая величина ITT) и процентное изменение уровня Apo A1 от исходного уровня до 12 недели (оцениваемая величина ITT). Apo, аполипопротеин; HDL-C, холестерин липопротеинов высокой плотности; ITT, популяция, сформированная в зависимости от назначенного лечения; LDL-C, холестерин липопротеинов низкой плотности; Lp(a), липопротеин (a); TG, триглицериды.
ПЛАН ИССЛЕДОВАНИЯ
[00109] COMBO I представляло собой принадлежащее к фазе 3, рандомизированное, двойное слепое, плацебо-контролируемое, параллельно-групповое, мультицентровое, 52-недельное исследование. В данном исследовании оценивали эффективность и безопасность алирокумаба в качестве дополнительной терапии к стабильным, максимально переносимым ежедневным дозам статина, с другими видами стабильной липид-корригирующей терапии или без них, у пациентов с высоким риском, имеющих неконтролируемую гиперхолестеринемию (фигура 1A).
[00110] COMBO II представляло собой принадлежащее к фазе 3, рандомизированное, двойное слепое, контролируемое по активному препарату, параллельно-групповое, международное 104-недельное исследование. В исследование включали пациентов с высоким риском, имеющих неконтролируемую гиперхолестеринемию, принимающих терапию посредством стабильной максимально переносимой ежедневной дозы статинов. В отличие от COMBO I с режимом лечения, предусматривающим плацебо, в COMBO II включали режим лечения активным препаратом (эзетимиб) с планом исследования, предусматривающим два плацебо (фигура 1B), и пациентам не разрешали получать какой-либо из видов липид-корригирующей терапии кроме статина и их лечения, назначенного при рандомизации.
ВЫБОР ПАЦИЕНТОВ
[00111] У всех пациентов в COMBO I и II была гиперхолестеринемия и установленное CHD или эквивалентные риски возникновения CHD, с уровнем LDL-C, резистентным к максимально переносимой ежедневной дозе статина. В COMBO I отдельно пациентам также разрешали получать другие виды липид-корригирующей терапии наряду со статином при условии, что и статин, и другие виды липид-корригирующей терапии принимали при стабильной дозе в течение по меньшей мере 4 недель (6 недель для фенофибрата) до скринингового визита; в COMBO II требовалось, чтобы доза статина была стабильной в течение по меньшей мере 4 недель до скринингового визита и не разрешали получать другие виды липид-корригирующей терапии. При скрининге пациенты имели либо уровень LDL-C ≥70 мг/дл (≥1,81 ммоль/л) с документально подтвержденным CVD, либо уровень LDL-C ≥100 мг/дл (≥2,59 ммоль/л) без документально подтвержденного случая CVD в анамнезе.
[00112] ʹМаксимально переносимую дозуʹ статина определяли как либо 20 мг или 40 мг розувастатина ежедневно, либо 40 мг или 80 мг аторвастатина ежедневно, либо 80 мг симвастатина ежедневно (если пациент уже принимает эту дозу на протяжении >1 года). Однако пациенты, не способные переносить вышеприведенные дозы статинов, все еще подходили для включения в исследование, если они ежедневно принимали более низкую дозу аторвастатина, розувастатина или симвастатина при условии, что исследователь имел документально подтвержденную причину отказа от применения более высокой дозы.
COMBO I
[00113] Критерии включения в исследование COMBO I были следующими. Пациенты, имеющие гиперхолестеринемию и установленное коронарное заболевание сердца (CHD) или эквивалентные риски возникновения CHD (определения см. ниже), которые резистентны к максимально переносимой ежедневной дозе статина с другой липид-корригирующей терапией или без нее (LLT), обе при стабильной дозе в течение по меньшей мере 4 недель до скринингового визита (и 6 недель до скринингового визита в случае фенофибрата).
[00114] Определение максимально переносимой дозы (допустимо любое из следующего): 20 мг или 40 мг розувастатина ежедневно, 40 мг или 80 мг аторвастатина ежедневно или 80 мг симвастатина ежедневно (если пациент уже принимает эту дозу на протяжении >1 года). Пациентам, не способным принимать любую из вышеприведенных доз статинов, назначали лечение с ежедневной дозой аторвастатина, розувастатина или симвастатина, которая считалась подходящей для пациента, исходя из решения или опасений исследователя. Некоторые примеры допустимых причин для пациента, принимающего более низкую дозу статинов, включают: неблагоприятные эффекты при высоких дозах, пожилой возраст, низкий индекс массы тела (BMI), региональные практики, местный листок-вкладыш, сопутствующие лекарственные препараты и сопутствующие состояния, такие как нарушенная толерантность к глюкозе/нарушенный уровень глюкозы натощак.
[00115] Установленное CHD определяют как документально подтвержденный случай CHD в анамнезе, включая одно или несколько из следующего: острый инфаркт миокарда (MI), бессимптомный MI, нестабильная стенокардия, процедура реваскуляризации коронарных сосудов (например, чрескожное коронарное вмешательство [PCI] или шунтирование коронарных артерий [CABG]) и клинически значимое CHD, диагностированное с помощью инвазивного или неинвазивного тестирования (такого как коронарография, электрокардиограмма с нагрузкой на беговой дорожке, стресс-эхокардиография или радионуклидная визуализация).
[00116] Эквивалентные риски возникновения CHD включают один или несколько из следующих 4 критериев: 1) документально подтвержденное заболевание периферических артерий (PAD) (должен удовлетворяться один из следующих критериев [a, b или c]): a) текущая перемежающаяся хромота (мышечный дискомфорт в нижней конечности, проявляющийся при физических нагрузках, который возникает повторно и облегчается при покое в течение 10 минут) предполагаемого атеросклеротического происхождения вместе с лодыжечно-брахиальным индексом < 0,90 в любой ноге в состоянии покоя, или b) перемежающаяся хромота в анамнезе вместе с эндоваскулярной процедурой или хирургическим вмешательством на одной или обеих ногах по причине атеросклеротического заболевания, или c) критическая ишемия конечностей в анамнезе вместе с тромболизисом, эндоваскулярной процедурой или хирургическим вмешательством на одной или обеих ногах по причине атеросклеротического заболевания; 2) документально подтвержденный ишемический инсульт с очаговым ишемическим неврологическим дефицитом, который продолжался более 24 часов, как считается, имеющий атеротромботическое происхождение. Компьютерную томографию (CT) или магнитно-резонансную томографию (MRI) необходимо было провести для исключения кровоизлияния или неишемического неврологического заболевания; 3) документально подтвержденное хроническое заболевание почек (CKD), определенное с помощью расчетной скорости клубочковой фильтрации (eGFR), составляющей ≥30‒<60 мл/мин./1,73 м2 в течение 3 месяцев или более, включая скрининговый визит; и/или 4) известные случаи сахарного диабета в анамнезе и 2 или более дополнительных факторов риска, включая: a) гипертензию в анамнезе (установленную по лекарственным препаратам против гипертензии), b) документально подтвержденный лодыжечно-брахиальный индекс ≤0,90 в анамнезе, c) документально подтвержденные случаи микроальбуминурии или макроальбуминурии в анамнезе или анализ мочи с помощью индикаторной полоски при скрининговом визите (неделя -2) с уровнем белка >2+, d) документально подтвержденный случай предпролиферативной или пролиферативной ретинопатии или лазерное лечение ретинопатии в анамнезе, e) известные случаи раннего развития CHD в семейном анамнезе (CHD у отца или брата в возрасте до 55 лет; CHD у матери или сестры в возрасте до 65 лет).
[00117] Пациентов, которые соответствовали всем вышеперечисленным критериями включения, исследовали в отношении перечисленных ниже критериев исключения.
[00118] Критериями исключения, относящимися к методологии исследования, были следующие. Пациенты без установленного CHD или эквивалентных рисков возникновения CHD; уровень LDL-C <70 мг/дл (<1,81 ммоль/л) при скрининговом визите у пациентов с документально подтвержденным сердечно-сосудистым заболеванием [CVD] в анамнезе); уровень LDL-C <100 мг/дл (<2,59 ммоль/л) при скрининговом визите у пациентов без документально подтвержденного CVD в анамнезе); пациенты, не принимающие стабильную дозу липид-корригирующей терапии (включая статин) в течение по меньшей мере 4 недель и/или фенофибрат в течение по меньшей мере 6 недель, в зависимости от обстоятельств, до скринингового визита и от момента скрининга дo рандомизации; текущий прием статина, отличного от симвастатина, аторвастатина или розувастатина; симвастатин, аторвастатин или розувастатин не принимается ежедневно или принимается не в указанной дозе; ежедневные дозы превышают 80 мг аторвастатина, 40 мг розувастатина или 40 мг симвастатина (за исключением пациентов, принимающих 80 мг симвастатина в течение >1 года, подходящих для включения в исследование); применение фибратов, отличных от фенофибрата, в пределах 6 недель до скринингового визита или между скрининговым визитом и визитом рандомизации; применение нутрицевтических продуктов или безрецептурных терапевтических средств, которые могут воздействовать на уровни липидов, не вводимых при стабильной дозе/количестве на протяжении по меньшей мере 4 недель до скринингового визита или между скрининговым визитом и визитом рандомизации; применение продуктов на основе красного ферментированного риса в пределах 4 недель до скринингового визита или между скрининговым визитом и визитом рандомизации; пациенты, которые проходили плазмаферез в течение 2 месяцев до скринингового визита (неделя -2), или собираются пройти его во время исследования; недавно перенесенные (в течение 3 месяцев до скринингового визита или между скрининговым визитом и визитом рандомизации) MI, нестабильная стенокардия, приведшая к госпитализации, PCI, CABG, неконтролируемая сердечная аритмия, инсульт, транзиторная ишемическая атака, реваскуляризация сонной артерии, эндоваскулярная процедура или хирургическое вмешательство при заболевании периферических сосудов; предусмотрено проведение запланированных PCI, CABG, реваскуляризации сонной артерии или периферических сосудов во время исследования; систолическое кровяное давление (BP) >160 мм рт. ст. или диастолическое BP >100 мм рт. ст. при скрининговом визите или визите рандомизации; сердечная недостаточность III или IV класса согласно Нью-Йоркской кардиологической ассоциации (New York Heart Association) (NYHA) в анамнезе в течение последних 12 месяцев; известный случай геморрагического инсульта в анамнезе; возраст < 18 лет или совершеннолетия при скрининговом визите, в зависимости от того, что является большим; пациенты, которых предварительно не проинструктировали в отношении гипохолестеринемической диеты до скринингового визита; впервые диагностированный (в течение 3 месяцев до визита рандомизации) или плохо контролируемый (уровень гликированного гемоглобина [HbA1c] >8,5% при скрининговом визите) диабет; наличие какого-либо клинически значимого неконтролируемого эндокринного заболевания, которое, как известно, влияет на уровни липидов или липопротеинов в сыворотке крови (примечание: пациенты, получающие заместительную терапию гормонами щитовидной железы, могут быть включены в том случае, если дозировка была стабильной на протяжении по меньшей мере 12 недель до скрининга и между скрининговым визитом и визитом рандомизации, и уровень тиреотропного гормона (TSH) находится в пределах нормального диапазона Центральной лаборатории при скрининговом визите); случай бариатрической хирургии в анамнезе в течение 12 месяцев до скринингового визита; нестабильная масса, определенная по изменению >5 кг в пределах 2 месяцев до скринингового визита; известные случаи гомозиготной или гетерозиготной семейной гиперхолестеринемии (FH) в анамнезе; известные случаи потери функции пропротеиновой конвертазы субтилизин/кексинового типа 9 PCSK9 (т. е. генетическая мутация или вариация последовательности) в анамнезе; применение системных кортикостероидов, если только не применяемых в качестве заместительной терапии при заболевании гипофиза/надпочечников при стабильной схеме в течение по меньшей мере 6 недель до визита рандомизации (примечание: стероидные терапевтические препараты для местного, внутрисуставного, назального, ингаляционного и офтальмического применения не рассматриваются в качестве ʹсистемныхʹ и разрешены); применение непрерывной заместительной гормональной терапии эстрогеном или тестостероном, кроме случаев, когда схема была стабильной в последние 6 недель до скринингового визита (неделя -2) и не запланировано изменение схемы во время исследования; рак в анамнезе в течение последних 5 лет, за исключением излеченного до той меры, до которой это возможно, базально-клеточного рака кожи, плоскоклеточного рака кожи или рака шейки матки in situ; известный положительный результат анализа на HIV в анамнезе; пациенты, которые получали любые экспериментальные лекарственные средства, отличные от подготовительных плацебо-наборов для алирокумаба в течение 1 месяца или 5 периодов полувыведения, в зависимости от того, что является большим; пациенты, которые прежде участвовали в каком-либо клиническом испытании с применением терапии, предусматривающей алирокумаб или любое другое антитело к PCSK9; пациенты, которые отзывают согласие во время периода скрининга (пациенты, которые не желают продолжать или не в состоянии вернуться).
[00119] Дополнительными критериями исключения были условия/ситуации такие как: любое клинически значимое отклонение, идентифицированное во время скрининга, которое по решению исследователя или любого соисследователя сделало бы невозможным безопасное завершение исследования или ограничило бы оценку конечных точек, такое как основные системные заболевания или пациенты с короткой ожидаемой продолжительностью жизни; рассматриваемые исследователем или любым соисследователем как не подходящие для данного исследования по какой-либо причине, например, пациенты, которые, как считается, неспособны соответствовать определенным требованиям протокола, таким как запланированные визиты; пациенты, которые, как считается, неспособны вводить или переносить долговременные инъекции по мнению пациента или исследователя; исследователь или любой соисследователь, фармацевт, координатор исследования, другой персонал исследования или подобные, непосредственно участвующие в проведении протокола и т. д.; наличие каких-либо других условий (например, географических, социальных и т. п.), фактических или предполагаемых, которые, как считает исследователь, сдерживали бы или ограничивали бы участие пациента в течение всего срока исследования.
[00120] Лабораторные показатели в течение периода скрининга (не включая обследований при рандомизации): положительный результат исследования на поверхностный антиген вируса гепатита B или антитело к вирусу гепатита C (подтвержденный повторным тестированием); положительный тест на человеческий хорионический гонадотропин (hCG) в сыворотке крови или тест мочи на наличие беременности (включая 0 неделю) у женщин репродуктивного возраста; уровень триглицеридов >400 мг/дл (>4,52 ммоль/л) (допускается 1 повторное оценивание); уровень eGFR <30 мл/мин./1,73 м2; уровень аланинаминотрансферазы (ALT) или аспартатаминотрансферазы (AST) >3 x верхняя граница нормы (ULN) (допускается 1 повторное оценивание); уровень креатинфосфокиназы (CPK) >3 x ULN (допускается 1 повторное оценивание); уровень тиреотропного гормона (TSH) < нижней границы нормы (LLN) или >ULN.
[00121] Критерии исключения, относящиеся к фоновой терапии, были следующими. Все противопоказания для видов фоновой терапии или предупреждения/меры предосторожности по применению (при необходимости), представленные в соответствующей национальной инструкции по применению продукта.
[00122] Критерии исключения, относящиеся к алирокумабу, были следующими. Известная гиперчувствительность к терапевтическим средствам на основе моноклональных антител; беременные или кормящие грудью женщины; женщины детородного возраста, не защищенные высокоэффективным способом контроля рождаемости и/или не желающие или не способные пройти тест на наличие беременности. Женщины детородного возраста должны иметь подтвержденный отрицательный результат теста на наличие беременности при скрининговом визите и визите рандомизации. Они должны применять эффективный способ контрацепции на протяжении всего исследования и согласиться повторно проходить тест мочи на наличие беременности во время обозначенных визитов. Используемые способы контрацепции должны соответствовать критериям высокоэффективного способа контроля рождаемости согласно примечанию к руководству доклинических исследований безопасности для проведения клинических испытаний на людях и регистрационному свидетельству фармацевтических препаратов. У женщин постклимактерического возраста менструации должны отсутствовать на протяжении по меньшей мере 12 месяцев.
COMBO II
[00123] Критерии включения в исследование COMBO II были следующими. Пациенты, имеющие гиперхолестеринемию и установленное CHD или эквивалентные риски возникновения CHD (определения см. ниже), которые резистентны к максимально переносимой стабильной ежедневной дозе статина в течение по меньшей мере 4 недель до скринингового визита.
[00124] Определение максимально переносимой дозы (допустимо любое из следующего): 20 мг или 40 мг розувастатина ежедневно, 40 мг или 80 мг аторвастатина ежедневно и 80 мг симвастатина ежедневно (если пациент уже принимает эту дозу на протяжении >1 года - см. критерий исключения). Пациентам, не способным принимать любую из вышеприведенных доз статинов, должны были назначить лечение с ежедневной дозой аторвастатина, розувастатина или симвастатина, которая считалась подходящей для пациента, исходя из решения или опасений исследователя. Некоторые примеры допустимых причин для пациента, принимающего более низкую дозу статинов, включают: неблагоприятные эффекты при высоких дозах, пожилой возраст, низкий BMI, региональные практики, местный листок-вкладыш, сопутствующие лекарственные препараты и сопутствующие состояния, такие как нарушенная толерантность к глюкозе/нарушенный уровень глюкозы натощак.
[00125] Установленное CHD включает одно или несколько из следующего: острый MI; бессимптомный MI; нестабильная стенокардия; процедура реваскуляризации коронарных сосудов (например, PCI или CABG); клинически значимое CHD, диагностированное с помощью инвазивного или неинвазивного тестирования (такого как коронарография, электрокардиограмма с нагрузкой на беговой дорожке, стресс-эхокардиография или радионуклидная визуализация).
[00126] Эквивалентные риски возникновения CHD включают один или несколько из следующих 4 критериев: 1) документально подтвержденное PAD (должен удовлетворяться один из следующих критериев [a, b, или c]): a) текущая перемежающаяся хромота предполагаемого атеросклеротического происхождения вместе с лодыжечно-брахиальным индексом, равным или меньше 0,90 в любой ноге в состоянии покоя, или b) перемежающаяся хромота в анамнезе вместе с эндоваскулярной процедурой или хирургическим вмешательством на одной или обеих ногах по причине атеросклеротического заболевания, или c) критическая ишемия конечностей в анамнезе вместе с тромболизисом, эндоваскулярной процедурой или хирургическим вмешательством на одной или обеих ногам по причине атеросклеротического заболевания; 2) документально подтвержденный предшествующий ишемический инсульт с очаговым ишемическим неврологическим дефицитом, который продолжался более 24 часов, как считается, имеющий атеротромботическое происхождение. CT или MRI необходимо было провести для исключения кровоизлияния или неишемического неврологического заболевания; 3) документально подтвержденное CKD, определенное с помощью eGFR ≥30‒<60 мл/мин./1,73 м2 в течение 3 месяцев или более, включая скрининговый визит; и/или 4) известные случаи сахарного диабета в анамнезе и 2 или более дополнительных факторов риска: a) гипертензия в анамнезе (установленная по лекарственным препаратам против гипертензии), b) документально подтвержденный лодыжечно-брахиальный индекс ≤0,90 в анамнезе, c) документально подтвержденные случаи микроальбуминурии или макроальбуминурии в анамнезе или анализ мочи с помощью индикаторной полоски при скрининговом визите (неделя -3) с уровнем белка >2+, d) документально подтвержденный случай предпролиферативной или пролиферативной ретинопатии или лазерное лечение ретинопатии в анамнезе, e) известные случаи раннего развития CHD в семейном анамнезе CHD у отца или брата в возрасте до 55 лет; CHD у матери или сестры в возрасте до 65 лет).
[00127] Пациентов, которые соответствовали всем вышеперечисленным критериями включения, исследовали в отношении перечисленных ниже критериев исключения.
[00128] Критериями исключения, относящимися к методологии исследования, были следующие. Пациенты без установленного CHD или эквивалентных рисков возникновения CHD; уровень LDL-C <70 мг/дл (<1,81 ммоль/л) при скрининговом визите у пациентов с документально подтвержденным CVD в анамнезе; уровень LDL-C <100 мг/дл (<2,59 ммоль/л) при скрининговом визите у пациентов без документально подтвержденного CVD в анамнезе; изменение дозы статина или схемы дозирования от скрининга до рандомизации; текущий прием статина, который не представляет собой симвастатин, аторвастатин или розувастатин; симвастатин, аторвастатин или розувастатин не принимаются ежедневно или принимаются не в указанной дозе; ежедневные дозы превышают 80 мг аторвастатина, 40 мг розувастатина или 40 мг симвастатина (за исключением пациентов, принимающих 80 мг симвастатина в течение >1 года, подходящих для включения в исследование); применение ингибитора всасывания холестерина (т. e. эзетимиба), омега-3 жирной кислоты (при дозах ≥1000 мг ежедневно), никотиновой кислоты, секвестранта, связывающего желчные кислоты, или продуктов на основе красного ферментированного риса в течение последних 4 недель до скринингового визита или между скрининговым визитом и визитом рандомизации; применение фибратов в последние 6 недель до скринингового визита; применение нутрицевтических продуктов или безрецептурных терапевтических средств, которые могут воздействовать на уровни липидов, не вводимых при стабильной дозе/количестве на протяжении по меньшей мере 4 недель до скринингового визита или между скрининговым визитом и визитом рандомизации; пациенты, которые проходили плазмаферез в течение 2 месяцев до скринингового визита или собираются пройти его во время исследования; недавно перенесенные (в течение 3 месяцев до скринингового визита или между скрининговым визитом и визитом рандомизации) MI, нестабильная стенокардия, приведшая к госпитализации, PCI, CABG, неконтролируемая сердечная аритмия, инсульт, транзиторная ишемическая атака, реваскуляризация сонной артерии, эндоваскулярная процедура или хирургическое вмешательство при заболевании периферических сосудов; предусмотрено проведение запланированных PCI, CABG, реваскуляризации сонной артерии или периферических сосудов во время исследования; систолическое кровяное давление (BP) >160 мм рт. ст. или диастолическое BP >100 мм рт. ст. при скрининговом визите или визите рандомизации; сердечная недостаточность III или IV класса согласно NYHA в анамнезе в течение последних 12 месяцев; известный случай геморрагического инсульта в анамнезе; возраст <18 лет или совершеннолетия при скрининговом визите, в зависимости от того, что является большим; пациенты, которых предварительно не проинструктировали в отношении гипохолестеринемической диеты до скринингового визита; впервые диагностированный (в течение 3 календарных месяцев до визита рандомизации) или плохо контролируемый (HbA1c >9% при скрининговом визите) диабет; наличие какого-либо клинически значимого неконтролируемого эндокринного заболевания, которое, как известно, влияет на уровни липидов и липопротеинов в сыворотке крови (примечание: пациенты, получающие заместительную терапию гормонами щитовидной железы, могут быть включены в том случае, если доза была стабильной в течение по меньшей мере 12 недель до скрининга и между скрининговым визитом и визитом рандомизации, и уровень TSH находится в пределах нормального диапазона Центральной лаборатории при скрининговом визите); случай бариатрической хирургии в анамнезе в течение 12 месяцев до скринингового визита; нестабильная масса, определенная по изменению >5 кг в пределах 2 месяцев до скринингового визита; известные случаи гомозиготной или гетерозиготной FH в анамнезе; известные случаи потери функции PCSK9 (т. е. генетическая мутация или вариация последовательности) в анамнезе; применение системных кортикостероидов, если только не применяемых в качестве заместительной терапии при заболевании гипофиза/надпочечников при стабильной схеме в течение по меньшей мере 6 недель до визита рандомизации (примечание: стероидные терапевтические препараты для местного, внутрисуставного, назального, ингаляционного и офтальмического применения не рассматриваются в качестве ʹсистемныхʹ и разрешены); применение непрерывной заместительной гормональной терапии эстрогеном или тестостероном, кроме случаев, когда схема была стабильной в последние 6 недель до скринингового визита и не запланировано изменение схемы во время исследования; рак в анамнезе в течение последних 5 лет, за исключением излеченного до той меры, до которой это возможно, базально-клеточного рака кожи, плоскоклеточного рака кожи или рака шейки матки in situ; известный положительный результат анализа на HIV в анамнезе; пациенты, которые получали любые экспериментальные лекарственные средства, отличные от подготовительных плацебо-наборов для алирокумаба в течение 1 месяца или 5 периодов полувыведения, в зависимости от того, что является большим; пациенты, которые прежде проходили лечение по меньшей мере с помощью 1 дозы алирокумаба или любого другого моноклонального антитела к PCSK9 в других клинических исследованиях; пациенты, которые отзывают согласие во время периода скрининга (пациенты, которые не желают продолжать или не в состоянии вернуться).
[00129] Дополнительными критериями исключения были условия/ситуации такие как: любое клинически значимое отклонение, идентифицированное во время скрининга, которое по решению исследователя или любого соисследователя сделало бы невозможным безопасное завершение исследования или ограничило бы оценку конечных точек, такое как основные системные заболевания; пациенты с короткой ожидаемой продолжительностью жизни; рассматриваемые исследователем или любым соисследователем как не подходящие для данного исследования по какой-либо причине, например, пациенты, которые, как считается, неспособны соответствовать определенным требованиям протокола, таким как запланированные визиты; пациенты, которые, как считается, не способны вводить или переносить долговременные инъекции по мнению пациента или исследователя; исследователь или любой соисследователь, фармацевт, координатор исследования, другой персонал исследования, непосредственно участвующие в проведении протокола и т. д., или их родственник; наличие каких-либо других условий (например, географических, социальных и т. п.), фактических или предполагаемых, которые, как считает исследователь, сдерживали бы или ограничивали бы участие пациента в течение всего срока исследования.
[00130] Лабораторные показатели в течение периода скрининга (не включая обследований при рандомизации): положительный результат исследования на поверхностный антиген вируса гепатита B или антитело к вирусу гепатита C; положительный тест на бета-hCG в сыворотке крови или тест мочи на наличие беременности (включая 0 неделю) у женщин репродуктивного возраста; уровень триглицеридов >400 мг/дл (>4,52 ммоль/л) (допускается 1 повторное оценивание); уровень eGFR <30 мл/мин./1,73 m2; уровень ALT или AST >3 x ULN (допускается 1 повторное оценивание); CPK >3 x ULN (допускается одно повторное оценивание); TSH <LLN или >ULN (допускается одно повторное оценивание).
[00131] Критериями исключения, которые относятся к активным препаратам сравнения и видам обязательной фоновой терапии, были следующие. Все противопоказания по отношению к эзетимибу или предупреждения/меры предосторожности по применению (при необходимости), представленные в соответствующей национальной инструкции по применению продукта; все противопоказания по отношению к фоновому лечению статинами или предупреждение/мера предосторожности по применению (при необходимости), представленные в соответствующей национальной инструкции по применению продукта.
[00132] Критерии исключения, относящиеся к алирокумабу, были следующими. Известная гиперчувствительность к моноклональному антителу или любому компоненту лекарственного продукта; беременные или кормящие грудью женщины; женщины детородного возраста, не защищенные высокоэффективным способом(способами) контроля рождаемости и/или не желающие или не способные пройти тест на наличие беременности. Женщины детородного возраста должны иметь подтвержденный отрицательный результат теста на наличие беременности при скрининговом визите и визите рандомизации. Они должны применять эффективный способ контрацепции в течение всего срока исследуемого лечения и на протяжении 10 недель после последнего приема исследуемого лекарственного препарата (инъекции или капсулы, в зависимости от того, что наступило позже) и согласиться на повторный тест на наличие беременности во время обозначенных визитов. (Используемые способы контрацепции должны соответствовать критериям высокоэффективного способа контроля рождаемости согласно «Примечанию к руководству доклинических исследований безопасности для проведения клинических испытаний на людях и регистрационному свидетельству фармацевтических препаратов». У женщин постклимактерического возраста менструации должны отсутствовать на протяжении по меньшей мере 12 месяцев).
ПРОЦЕДУРЫ ИССЛЕДОВАНИЯ
[00133] Пациенты, соответствующие критериям включения, вступали в период скрининга, который продолжался до 2 (COMBO I) или 3 (COMBO II) недель вплоть до рандомизации. Во время скрининга пациенты оформляли информированное согласие, проводилась дополнительная оценка критериев включения/исключения, проводился сбор информации о пациентах и пациентов обучали применению автоинжекторного устройства. Кроме того, получали показатели жизненно важных функций, выполняли электрокардиограмму в 12 отведениях и собирали образцы крови натощак и мочи для проведения анализа. AE оценивали от момента скринингового визита на протяжении всего исследования.
COMBO I
[00134] Пациентов, подходящих для включения в исследование, рандомизировали (2:1, алирокумаб:плацебо) со стратификацией по 1) предыдущему случаю инфаркта миокарда (MI) или ишемического инсульта в анамнезе и 2) интенсивности лечения статином для обеспечения равновесия между режимами лечения по этим факторам. После рандомизации пациенты входили в период двойного слепого лечения длительностью 52 недель. В дополнение к существующей терапии статином и другим существующим видам липид-корригирующей терапии, в соответствующем случае, пациенты, рандомизированные в группу алирокумаба, получали подкожно (SC) дозу 75 мг каждые 2 недели (Q2W), вводимую в виде одной инъекции объемом 1 мл с применением автоинжектора, с момента рандомизации до 12 недели. Пациенты, рандомизированные в группу плацебо, получали SC инъекцию 1 мл плацебо с помощью идентичного автоинжектора.
[00135] На 12 неделе у пациентов, рандомизированных в группу алирокумаба, повышали дозу до 150 мг Q2W, если уровень LDL-C на 8 неделе был ≥70 мг/дл (1,81 ммоль/л). Для сохранения "ослепления" пациента и исследователя не информировали об уровнях LDL-C на 8 неделе (или каких-либо значениях уровня липидов после рандомизации); сохранение или повышение дозы происходило автоматическим и слепым методом. Дозу алирокумаба 150 мг Q2W также вводили в виде 1 мл раствора в автоинжекторе.
[00136] Запланированные обследования пациентов в исследовательском центре в течение периода лечения включали обследования при рандомизации и затем на 4, 8, 12, 16, 24, 36 и 52 неделях (визит окончания лечения) (фигура 1A). После периода лечения наступал 8-недельный период последующего наблюдения.
COMBO II
[00137] Пациентов, подходящих для включения в исследование, рандомизировали (2:1, алирокумаб:эзетимиб) со стратификацией по 1) предыдущему случаю MI или ишемического инсульта в анамнезе, 2) интенсивности лечения статином и 3) географическому региону для обеспечения равновесия между режимами лечения по этим факторам. После рандомизации пациенты входили в период лечения длительностью 104 недель по типу двойного слепого исследования, предусматривающего два плацебо. Пациентов рандомизировали либо в группу с 75 мг алирокумаба SC Q2W плюс плацебо вместо эзетимиба перорально (PO) ежедневно, либо в группу с плацебо вместо алирокумаба SC Q2W плюс 10 мг эзетимиба PO ежедневно. На 12 неделе у пациентов, рандомизированных в группу алирокумаба, повышали дозу до 150 мг Q2W, если уровень LDL-C на 8 неделе был ≥70 мг/дл (1,81 ммоль/л).
[00138] Запланированные обследования пациентов в исследовательском центре проходили с регулярными интервалами от момента рандомизации до недели 104 (визит окончания лечения) (фигура 1B). После периода лечения наступал 8-недельный период последующего наблюдения.
COMBO I и II
[00139] В обоих исследованиях пациентов просили продолжать придерживаться постоянной диеты (диеты для терапевтического изменения образа жизни в соответствии с III докладом группы экспертов по лечению взрослых в рамках Национальной образовательной программы по холестерину или эквивалентной диеты), и ежедневная доза статина должна была быть стабильной на протяжении всего срока исследования от момента скрининга до визита последующего наблюдения. Изменение статина (и в случае COMBO I другой фоновой липид-корригирующей терапии) разрешали только при определенных обстоятельствах.
ОЦЕНКИ БЕЗОПАСНОСТИ
[00140] Оценки безопасности в ходе обоих исследований были следующими. Безопасность оценивали по сообщениям о нежелательных явлениях (AE) (включая установленные сердечно-сосудистые события), лабораторным анализам и измерению жизненно важных функций. Поскольку эффекты снижения уровня LDL-C при ингибировании PCSK9 наряду с приемом статина неизвестны, ряд AE определяли как представляющие особый интерес и отслеживали, включая отклонение уровня аланинаминотрансферазы, аллергические явления, гемолитическую анемию, беременность, передозировку исследуемым лекарственным средством, неврологические явления, офтальмологические явления и локальные реакции в месте инъекции. AE определяли как представляющие особый интерес на основании обзора предыдущих клинических исследований, переписки с регулирующими органами и комитетом по мониторингу данных, применению моноклонального антитела в данном исследовании и пути введения лекарственного средства.
[00141] Анализы безопасности были следующими. AE (включая установленные сердечно-сосудистые события), параметры лабораторных исследований и показатели жизненно важных функций сообщались описательно, на основании популяции для оценки безопасности (все рандомизированные пациенты, которые получали по меньшей мере 1 дозу или частичную дозу исследуемого лечения). Анализ безопасности был сосредоточен на периоде появления возникших в ходе лечения АЕ, определенном как время от момента первой дозы двойного слепого исследования до последней дозы двойного слепого исследования экспериментального продукта+70 дней (10 недель).
[00142] Безопасность Случаи возникших в ходе лечения нежелательных явлений (TEAE), сообщенные пациентом или отмеченные исследователем, серьезные нежелательные явления (SAE), TEAE, приводящие к прекращению лечения, AE, представляющие особый интерес (локальные реакции в месте инъекции, аллергические явления, выбранные неврологические явления и сердечно-сосудистые явления с экспертным подтверждением), случаи PCSA (потенциально клинически значимых отклонения) в параметрах лабораторных исследований, специфический анализ в отношении диабета или нарушенного контроля глюкозы и пациенты с 2 последовательными уровнями LDL-C < 25 мг/дл (0,65 ммоль/л).
COMBO I: СТАТИСТИЧЕСКИЕ МЕТОДЫ
Определение размера выборки
[00143] Конечный общий размер выборки составлял 306 при рандомизационном соотношении 2:1 (алирокумаб 204: плацебо 102).
Combo I: популяции для анализа
[00144] Популяцией для анализа первичной эффективности была популяция, сформированная в зависимости от назначенного лечения (ITT), определенная как все рандомизированные пациенты, которые имели оцениваемую первичную конечную точку, другими словами, имеющие доступное значение рассчитанного уровня LDL-C на исходном уровне и по меньшей мере одно доступное значение рассчитанного уровня LDL-C в пределах одного из периодов анализа вплоть до 24 недели (включая все рассчитанные уровни LDL-C для пациентов, получающих лечение, и пациентов, не получающих лечение).
[00145] Популяцией для анализа вторичной эффективности была модифицированная популяция, сформированная в зависимости от назначенного лечения (mITT), определенная как все рандомизированные пациенты, которые принимали по меньшей мере одну дозу или часть дозы экспериментального медицинского продукта (IMP) двойного слепого исследования и которые имели доступное значение рассчитанного уровня LDL-C на исходном уровне и по меньшей мере одно значение в пределах одного из периодов анализа вплоть до 24 недели во время периода лечения для оценки эффективности. Период лечения для оценки эффективности определяли как время от момента первого введения IMP двойного слепого исследования вплоть до 21 дня после последней инъекции препарата двойного слепого исследования.
[00146] Популяция для оценки безопасности включала всех рандомизированных пациентов, которые получали по меньшей мере одну дозу или часть дозы IMP двойного слепого исследования.
Combo I: анализы эффективности
[00147] Первичные анализы конечных точек эффективности проводили с применением подхода ITT (на основании популяции ITT, определенной выше), включая все данные по уровням липидов, независимо от того, продолжал ли пациент терапию или нет. Это соответствовало оцениваемым величинам ITT, определенным для первичных и ключевых вторичных конечных точек. Кроме того, анализы также проводили с применением подхода на основе пациентов, получающих лечение (на основании популяции mITT, определенной выше), включая данные по уровням липидов, собранным в течение периода лечения для оценки эффективности. Это соответствовало оцениваемым величинам анализа у пациентов, получающих лечение, для ключевых вторичных конечных точек.
[00148] С помощью подхода ITT анализировали всех пациентов, независимо от соблюдения ими схемы лечения; с помощью него оценивали пользу стратегии лечения, и он отражает максимально возможный эффект для популяции пациентов. С помощью подхода на основе пациентов, получающих лечение, анализировали эффект лечения, ограниченный периодом, в ходе которого пациенты фактически получали лечение. С помощью него оценивали пользу, которую позволило бы достичь лечение у пациентов, соблюдающих схему лечения до рассматриваемого момента времени.
[00149] Анализы эффективности выполняли в соответствии с лечением согласно рандомизации.
[00150] Все измерения, запланированные или незапланированные, натощак или не натощак, относили к периодам анализа с целью обеспечения оценки для моментов времени с 4 недели до 52 недели.
[00151] Принимая во внимание анализ первичной эффективности (подход ITT), процентное изменение рассчитанного уровня LDL-C от исходного уровня до 24 недели анализировали с применением подхода модели со смешанными эффектами для повторных измерений (MMRM). Применяли все данные после исходного уровня, доступные для периодов анализа с 4 недели до 52 недели, и недостающие данные учитывали при помощи MMRM. Модель включала фиксированные категориальные эффекты группы лечения (плацебо в сравнении с алирокумабом), страты рандомизации (согласно IVRS), момента времени (4 неделя - 52 неделя), взаимосвязи лечение-момент времени и взаимосвязи страта-момент времени, а также непрерывные фиксированные ковариаты значения исходного уровня LDL-C и взаимосвязи значение исходного уровня-момент времени. Данная модель обеспечивала скорректированные относительно исходного уровня оценки средних значений, определенных методом наименьших квадратов (средние значения LS), на 24 неделе для обеих групп лечения с их соответствующим 95% доверительным интервалом. Для сравнения группы алирокумаба с группой плацебо использовали соответствующее высказывание для тестирования различий данных оценок при 5% альфа-уровне значимости.
[00152] Была определена иерархическая процедура для тестирования ключевых вторичных конечных точек при контроле множественности сравнений (с применением приведенного выше порядка ключевых вторичных конечных точек). Первая ключевая вторичная конечная точка представляла собой процентное изменение рассчитанного уровня LDL-C от исходного уровня до 24 недели с применением подхода на основе пациентов, получающих лечение.
[00153] Непрерывные вторичные переменные, предположительно имеющие нормальное распределение (т. e. уровни липидов, отличных от TG и Lp(a)), анализировали с применением такой же модели MMRM, как для первичной конечной точки. Непрерывные конечные точки, предположительно имеющие ненормальное распределение (т. e. уровни TG и Lp(a)), анализировали с применением подхода множественного восстановления для обработки пропущенных значений с последующим применением модели устойчивой регрессии с конечной точкой, представляющей интерес, в качестве переменной отклика с применением M-оценки (с применением процедуры SAS ROBUSTREG), с группой лечения, стратой рандомизации (согласно IVRS) и соответствующим значением(значениями) исходного уровня в качестве эффектов для сравнения эффектов лечения. Обеспечивали объединенную оценку среднего значения в обеих группах лечения, а также различия данных оценок с их соответствующими SE, 95% CI и p-значением (посредством процедуры SAS MIANALYZE).
[00154] Бинарные вторичные конечные точки эффективности анализировали с применением подхода множественного восстановления для обработки пропущенных значений, с последующим применением стратифицированной логистической регрессии, при этом группа лечения выступала в качестве основного эффекта, а соответствующее значение(значениями) исходного уровня в качестве ковариаты, при стратификации по факторам рандомизации (согласно IVRS). Обеспечивали объединенные оценки соотношения шансов в сравнении с плацебо, 95% CI и p-значение (посредством процедуры SAS MIANALYZE).
Combo I: анализы безопасности
[00155] Анализы безопасности были описательными, их проводили на популяции для оценки безопасности согласно фактически полученному лечению. Для анализа TEAE и PCSA анализ безопасности был сосредоточен на периоде появления TEAE, определенном как время с момента первой дозы IMP двойного слепого исследования до 70 дней после последней инъекции двойного слепого исследования. Анализы лабораторных данных и определение показателей жизненно важных функций с течением времени выполняли для периода вплоть до 21 дня после последней инъекции IMP.
COMBO I: РЕЗУЛЬТАТЫ
Учет пациентов
[00156] Из 316 рандомизированных пациентов два пациента в группе алирокумаба не проходили лечение, и, следовательно, их не включали в популяцию для оценки безопасности. Пять рандомизированных пациентов не включали в популяцию ITT (отсутствует значение уровня LDL-C в пределах одного из периодов анализа до 24 недели). Семь рандомизированных пациентов исключили из популяции mITT (пациенты, исключенные из популяции ITT, и пациенты с отсутствием значения уровня LDL-C в пределах одного из периодов анализа вплоть до 24 недели в ходе периода лечения для оценки эффективности).
Таблица 1 Популяции для анализа
Для остальных популяций данные пациентов сводили в таблицу в соответствии с их лечением, назначенным при рандомизации.
[00157] В группе алирокумаба среди 191 пациентов, которые получили по меньшей мере одну инъекцию после 12 недели, 32 (16,8%) пациента получали автоматическое повышение дозы на 12 неделе от 75 мг алирокумаба Q2W до 150 мг Q2W слепым способом.
COMBO I: распределение для исследования
[00158] Из 640 пациентов, которых подвергли скринингу в отношении пригодности для включения в исследование, 316 затем рандомизировали в группу алирокумаба (209) или плацебо (107). Из 316 рандомизированных пациентов 2 (алирокумаб) не проходили лечение; 231 пациент (73,1%) завершил 52-недельное исследуемое лечение (75 - плацебо; 156 - алирокумаб); 311 (98,4%) составляли популяцию ITT (106 - плацебо; 205 - алирокумаб, 5 - пациенты с пропущенными значениями уровней LDL-C); 309 пациентов составляли популяцию анализа у пациентов, получающих лечение (105 - плацебо; 204 - алирокумаб) и 314 пациентов составляли популяцию для оценки безопасности (107 - плацебо; 207 - алирокумаб).
[00159] На протяжении 52-недельного периода двойного слепого исследования, соблюдение схемы лечения исследуемым лекарственным препаратом (подтверждение получения ≥80% запланированных инъекций) было аналогичным в группах лечения (98% - алирокумаб; 99% - плацебо). На основании уровней LDL-C на 8 неделе, составлявших ≥70 мг/дл, 32/191 пациентов (16,8%) получали свою дозу алирокумаба, повышенную до 150 мг, подкожно каждые 2 недели на 12 неделе (среди пациентов по меньшей мере с 1 инъекцией после 12 недели).
[00160] Введение IMP двойного слепого исследования преждевременно прекратили до наступления 52 недели 32 (29,9%) рандомизированных пациента в группе плацебо и 51 (24,4%) рандомизированный пациент в группе алирокумаба. Основными причинами для прекращения исследуемого лечения были "другие причины" и нежелательные явления. Эти "другие причины" включали следующее: пациент не соответствовал критериям в отношении завершения лечения вследствие либо пропущенной/находящейся за пределами интервала инъекции 50 недели или находящегося за пределами интервала визита 52 недели (17 пациентов), исключение субъекта не было указано конкретно (8 пациентов), закрытие исследовательского центра (4 пациента), внезапная смерть/смерть (3 пациента) (не было решения исследователя прекратить лечение до наступления смерти), разные причины (2 пациента).
COMBO I: обобщенные характеристики популяции
[00161] Демографические характеристики, характеристики заболеваний и параметры уровней липидов на исходном уровне были похожи у группы алирокумаба при сравнении с группой плацебо. Демографические характеристики на исходном уровне изложены в таблице 2. Лекарственный препарат для контроля уровня липидов и уровни липидов на исходном уровне изложены в таблице 3. Определение CHD включало острый инфаркт миокарда (MI), бессимптомный MI, нестабильную стенокардию, реваскуляризацию коронарных сосудов или клинически значимое CHD, диагностированное с помощью инвазивного или неинвазивного тестирования. Эквивалентные риски возникновения CHD включали заболевание периферических артерий, ишемический инсульт, хроническое заболевание почек (расчетная скорость клубочковой фильтрации от ≥30 дo <60 мл/мин./1,73 м2 в течение ≥3 месяцев) или сахарный диабет совместно с 2 или более дополнительными факторами риска (гипертензия, лодыжечно-брахиальный индекс ≤0,90, микро- или макроальбуминурия, анализ мочи с помощью индикаторной полоски с уровнем белка >2+, ретинопатия или раннее развитие CHD в семейном анамнезе [<55 лет у отца/брата или <65 лет у матери/сестры]).
[00162] В группе алирокумаба были численно большими доли пациентов-женщин и пациентов с диабетом 2 типа. Аналогично, более высокая процентная доля пациентов в группе плацебо получали другие виды LLT в дополнение к статину (49,5% в сравнении с 38,3% в группе алирокумаба). Однако эти отличия между группами лечения не оказались статистически значимыми.
[00163] Всех пациентов кроме одного лечили статином, 57,6% получали статин в высокой дозе (от 40 дo 80 мг аторвастатина ежедневно или от 20 дo 40 мг розувастатина ежедневно) и 8,2% получали эзетимиб в дополнение к статину. Среднее значение (SD) рассчитанного уровня LDL-C на исходном уровне составляло 102,2 (31,6) мг/дл (2,646 (0,820) ммоль/л). Примерно 78% рандомизированной популяции имели коронарное заболевание сердца (CHD) в анамнезе (таблица 5).
[00164] Период проведения инъекций был аналогичным для всех групп лечения со средним значением периода проведения, составлявшим 46 недель. В группе алирокумаба среди 191 пациентов, которые получили по меньшей мере одну инъекцию после 12 недели, 32 (16,8%) пациента получали автоматическое повышение дозы на 12 неделе от 75 мг алирокумаба Q2W до 150 мг Q2W слепым способом.
Таблица 2 - Демографические характеристики на исходном уровне
(N=209)
(N=107)
Все пациенты получали фоновую терапию с максимально переносимой дозой статина ± другую LLT. †p-значения сравнения данных на исходном уровне у групп лечения представлены с целью наглядности, в виде бланка для скрининга, с применением точного критерия Фишера для качественных данных и асимптотического однофакторного ANOVA для баллов по критерию Уилкоксона (критерий Краскела-Уоллиса) для непрерывных данных.
Таблица 3 - Лекарственный препарат для контроля уровня липидов и уровни липидов на исходном уровне
(N=209)
(N=107)
Таблица 4 Характеристики заболеваний и другие соответственные данные на исходном уровне - рандомизированная популяция
(N=107)
(N=209)
(N=316)
[00165] Было 4 пациента без полностью документально подтвержденного CHD или эквивалентные рисков возникновения CHD.
Таблица 5 Медицинский анамнез, представляющий особый интерес: сердечно-сосудистые нарушения в анамнезе и факторы риска - рандомизированная популяция
(N=107)
(N=209)
(N=316)
a в соответствии с пунктами, предварительно перечисленными в e-CRF.
b диагностированное с помощью инвазивного или неинвазивного тестирования.
PAD определено как перемежающаяся хромота вместе с лодыжечно-брахиальным индексом ≤ 0,90 или вместе с процедурой реваскуляризации/хирургией периферических сосудов или критическая ишемия конечностей вместе с процедурой реваскуляризации/хирургией периферических сосудов или тромболизисом.
Таблица 6 Медицинский анамнез, представляющий особый интерес: другой медицинский анамнез, представляющий особый интерес - рандомизированная популяция
(N=107)
(N=209)
(N=316)
Примечание: один пациент может учитываться в нескольких категориях
Таблица 7 Фоновая LMT при рандомизации - рандомизированная популяция
(N=107)
(N=209)
(N=316)
Статин в высокой дозе соответствует 40-80 мг аторвастатина ежедневно или 20-40 мг розувастатина ежедневно.
Таблица 8 Параметры эффективности в отношении липидов на исходном уровне - обобщенные результаты количественной оценки в стандартных единицах - рандомизированная популяция
(N=107)
(N=209)
(N=316)
[00166] Сбор данных по измеренным уровням LDL-C не планировали в начальном протоколе, и он был добавлен в измененной версии. Следовательно, значения измеренного уровня LDL-C доступны для меньшего количества пациентов по сравнению со значениями рассчитанного уровня LDL-C.
COMBO I: дозировка и длительность
[00167] Период проведения инъекций был аналогичным для всех групп лечения со средним значением периода проведения, составлявшим примерно 46 недель. В группе алирокумаба среди 191 пациентов, которые получили по меньшей мере одну инъекцию после 12 недели, 32 (16,8%) пациента получали автоматическое повышение дозы на 12 неделе от 75 мг алирокумаба Q2W до 150 мг Q2W слепым способом.
COMBO I: первичная конечная точка эффективности
[00168] Анализ ITT включает все значения уровня LDL-C, полученные у пациентов, получающих лечение, и у пациентов, не получающих лечение, вплоть до 52 недели. Анализ первичной конечной точки (процентное изменение рассчитанного уровня LDL-C от исходного уровня до 24 недели) проводили на основе модели MMRM в отношении популяции ITT с использованием оценок средних значений, определенных методом наименьших квадратов, на 24 неделе. Для шестнадцати (7,8%) пациентов в группе алирокумаба и 9 (8,5%) пациентов в группе плацебо не получили значение рассчитанного уровня LDL-C на 24 неделе. Эти пропущенные значения учитывали при помощи модели MMRM.
[00169] Результаты анализа первичной конечной точки представлены в таблице 9, в ммоль/л и мг/дл.
[00170] Наблюдали статистически значимое процентное снижение уровня LDL-C от исходного уровня до 24 недели в группе алирокумаба (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -48,2%) по сравнению с группой плацебо (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -2,3%) (различие для среднего значения, определенного методом наименьших квадратов, в сравнении с плацебо -45,9%, p<0,0001).
[00171] В группе алирокумаба снижение уровня LDL-C от исходного уровня наблюдали от 4 недели до 52 недели.
[00172] Небольшое уменьшение снижения уровня LDL-C в зависимости от времени наблюдали в группе алирокумаба (среднее значение, определенное методом наименьших квадратов, в сравнении со значением исходного уровня на 52 неделе составило -42,5, по сравнению с -48,2 на 24 неделе), вероятно, вследствие большего числа случаев прекращения лечения на 52 неделе по сравнению с 24 неделей и, как результат, более высокого числа значений уровня LDL-C у пациентов после окончания лечения, включенных на 52 неделе (8,8% на 52 неделе в сравнении с 3,9% на 24 неделе).
[00173] Действительно, эффект лечения является стабильным от 24 недели до 52 недели, когда рассматривают только уровни LDL-C у пациентов, получающих лечение (см. фигуру 3), тогда как небольшое уменьшение наблюдают, когда также включают уровни LDL-C у пациентов после окончания лечения (см. фигуру 2).
Таблица 9 Процентное изменение от исходного уровня для рассчитанного уровня LDL-C и другие параметры уровней липидов на 24 неделе (анализы ITT или у пациентов, получающих лечение, как указано)
(SE) %
-39,3
-43,6
Таблица 10 Рассчитанный уровень LDL-C в зависимости от времени - анализ ITT - популяция ITT
(N=106)
(N=205)
Примечание: средние значения, определенные методом наименьших квадратов, стандартные ошибки (SE) и p-значение, полученные из анализа на основе MMRM (модели со смешанными эффектами для повторных измерений). Модель включает фиксированные категориальные эффекты группы лечения, страты рандомизации согласно IVRS, момента времени, взаимосвязи лечение-момент времени, взаимосвязи страта-момент времени, а также непрерывные фиксированные ковариаты значения исходного уровня LDL-C и взаимосвязь значение исходного уровня LDL-C-момент времени.
Модель MMRM и описание исходного уровня проводили в отношении пациентов со значением на исходном уровне и значением после исходного уровня по меньшей мере в одном из периодов анализа, используемых в модели.
COMBO I: ключевые вторичные конечные точки эффективности
[00174] В таблице 11 обобщены результаты анализа ключевых вторичных конечных точек в иерархическом порядке. Все ключевые вторичные конечные точки являются статистически значимыми в соответствии с процедурой иерархического тестирования вплоть до конечной точки уровня HDL-C на 24 неделе (оцениваемая величина ITT) включительно. Статистическая значимость не была достигнута для конечной точки уровня TG натощак на 24 неделе (оцениваемая величина ITT).
Таблица 11 - Ключевые вторичные конечные точки эффективности
[00175] Анализ у пациентов, получающих лечение, процентного изменения уровня LDL-C от исходного уровня до 24 недели показывает результаты, которые согласуются с результатами анализа ITT, при этом различие для среднего значения, определенного методом наименьших квадратов, в сравнении с плацебо составило -49,9% в анализе у пациентов, получающих лечение, в сравнении с - 45,9% в анализе ITT. Действительно, у небольшого числа пациентов были значения уровня LDL-C, собранные после окончания лечения (т. е. больше чем через 21 день после последней инъекции) на 24 неделе: 5 пациентов (4,7%) в группе плацебо и 8 пациентов (3,9%) в группе алирокумаба.
[00176] В анализе ITT наблюдали статистически значимое процентное снижение уровня LDL-C от исходного уровня до 12 недели (т. e. до возможного повышения дозы) в группе алирокумаба (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -46,3%) по сравнению с группой плацебо (среднее значение, определенное методом наименьших квадратов, по сравнению с исходным уровнем +1,1%) (различие для среднего значения, определенного методом наименьших квадратов, в сравнении с плацебо составило -47,4%, p < 0,0001). Результаты, полученные в анализе у пациентов, получающих лечение, были подобными.
[00177] У тридцать девяти (19,5%) пациентов наблюдали два последовательных значения рассчитанного уровня LDL-C <25 мг/дл. Причин для какого-либо особого беспокойства относительно безопасности у этих 39 пациентов не наблюдали.
Таблица 12 Число (%) пациентов с 2 последовательными значениями уровня LDL-C < 25 мг/дл (<0,65 ммоль/л) в течение периода лечения- популяция для оценки безопасности
(N=107)
(N=207)
Знаменатель (/N) в пределах группы лечения представляет собой число пациентов в группе лечения, которые имели по меньшей мере два значения рассчитанного уровня LDL-C, определенные с промежутком по меньшей мере 21 день в период оценки эффективности.
1 2 последовательных значения берутся во внимание, если их разделяет по меньшей мере 21 день.
2 Первое значение рассчитанного уровня LDL-C <25 или <15 мг/дл среди первых 2 последовательных значений рассчитанного уровня LDL-C <25 или <15 мг/дл для пациента.
COMBO I: обобщенные результаты в отношении эффективности
[00178] На 24 неделе процентное изменение от исходного уровня у рассчитанного уровня LDL-C в популяции ITT было значимо больше в группе алирокумаба (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -48,2%) по сравнению с группой плацебо (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -2,3%) (различие для среднего значения, определенного методом наименьших квадратов, в сравнении с плацебо составило -45,9%, p < 0,0001) (см. таблицу 9). Анализ у пациентов, получающих лечение, процентного изменения уровня LDL-C от исходного уровня до 24 недели показывает результаты, которые согласуются с результатами анализа ITT, при этом различие для среднего значения, определенного методом наименьших квадратов, в сравнении с плацебо составило -49,9% в анализе у пациентов, получающих лечение (p<0,0001) (см. таблицу 9).
[00179] На 12 неделе, перед возможным повышением дозы от 75 мг дo 150 мг, статистическая значимость также достигалась в популяции ITT (среднее значение, определенное методом наименьших квадратов, в сравнении со значением исходного уровня -46,3% и+1,1% для групп алирокумаба и плацебо соответственно, различие для среднего значения, определенных методом наименьших квадратов, в сравнении с плацебо составило -47,4%; p <0,0001) (см. таблицу 11).
[00180] В группе алирокумаба снижение уровня LDL-C от исходного уровня наблюдали от 4 недели до 52 недели (см. таблицу 10). Небольшое уменьшение с течением времени снижения уровня LDL-C наблюдали в группе алирокумаба (среднее значение, определенное методом наименьших квадратов, в сравнении со значением исходного уровня на 52 неделе составило -42,5% в сравнении с -48,2% на 24 неделе), вероятно, вследствие большего числа случаев прекращения лечения на 52 неделе по сравнению с 24 неделей и, как результат, более высокого числа значений уровня LDL-C после исходного уровня, включенных на 52 неделе (8,8% на 52 неделе в сравнении с 3,9% на 24 неделе). Результаты измеренного уровня LDL-C (количественное определение бета-липопротеинов) согласовывались с результатами рассчитанного уровня LDL-C. Результаты анализа чувствительности первичной конечной точки с применением метода анализа паттернов с помощью смешанной модели согласовывались с результатами анализа ITT. Более конкретно, в группе ITT объединенная оценка для изменения среднего значения, определенного методом наименьших квадратов (SE), от исходного уровня на 24 неделе (%) составляла -44,2 (2,1 для группы алирокумаба и -1,5 (3,0) для группы плацебо. Различие % для среднего значения, определенного методом наименьших квадратов (SE), для алирокумаба в сравнении с плацебо составляло -42,7 (3,7) (95% CI составлял от -49,9 дo -35,4; p< 0,0001). Анализ чувствительности проводили для дополнительной оценки влияния недостающих данных на первичную конечную точку. В данном анализе пропущенные значения рассчитанного уровня холестерина LDL на протяжении периода ʺполучения леченияʺ многократно замещали с применением модели, присваивающей недостающие данные случайным образом, а пропущенные значения рассчитанного уровня холестерина LDL на протяжении периоде после завершения лечения многократно замещали с применением случайного выбора из нормального распределения, при этом среднее значение было равно собственному значению субъекта на исходном уровне.
[00181] На фигуре 4 показаны уровни LDL-C в зависимости от времени для пациентов, получавших лечение алирокумабом с повышением дозы/без повышения дозы на 12 неделе. У пациентов с повышением дозы уровень LDL-C был снижен дополнительно в среднем на 22,8% (SD 27,1) на 24 неделе по сравнению с 12 неделей. Среди 32 пациентов (16,8%) с повышением дозы алирокумаба на 12 неделе (на основании уровня LDL-C на 8 неделе ≥70 мг/дл) достигнутый уровень LDL-C был сопоставим на 24, 36 и 52 неделях с уровнем, наблюдаемым среди пациентов, у которых не осуществляли повышения дозы (уровень LDL-C на 8 неделе <70 мг/дл).
[00182] Относительные доли пациентов, достигающих уровня LDL-C <70 мг/дл (<1,81 ммоль/л) на 24 неделе, в популяции ITT (75,0%) и популяции из пациентов, получающих лечение (77,5%), были значимо больше в случае применения алирокумаба, а не плацебо (9,0% и 8,0%, соответственно, p < 0,0001).
[00183] Все ключевые вторичные конечные точки, включая уровни Apo B, не-HDL-C, общего-C, Lp(a), HDL-C в различные моменты времени, были статистически значимыми в соответствии с процедурой иерархического тестирования, за исключением конечной точки уровня TG натощак на 24 неделе (оцениваемая величина ITT) (см. таблицу 11). Значимые снижения от исходного уровня до 24 недели после терапии алирокумабом (в сравнении с плацебо) наблюдали для уровня не-HDL-C (-39,1 [1,8] % алирокумаб, -1,6 [2,5] % плацебо), аполипопротеина B (-36,7 [1,6] % алирокумаб, -0,9 [2,3] % плацебо), общего холестерина (-27,9 [1,3] % алирокумаб, -2,9 [1,8] % плацебо) и липопротеина (a) (-20,5 [2,0] % алирокумаб, -5,9 [2,8] % плацебо) (все p < 0,0001 для алирокумаба в сравнении с плацебо). Не наблюдали значимых изменений в уровнях триглицеридов, в то время как значимое и направленное в другую сторону увеличение уровня HDL-C (аполипопротеин A1) наблюдали в группе алирокумаба; 3,5 (1,1) % в сравнении с -3,8 (1,5) % для плацебо (p < 0,0001; см. таблицу 9).
[00184] Процентные изменения уровня LDL-C от исходного уровня на 24 неделе в соответствии с демографическими характеристиками и подгруппами пациентов для алирокумаба в сравнении с плацебо изложены на фигуре 5. 95% доверительные интервалы всех различий процентного снижения уровня LDL-C в случае алирокумаба и плацебо в пределах каждого уровня подгрупп, определенных по возрасту, полу, расе, этнической принадлежности, интенсивности фоновой терапии статинами, другой терапии LLT (в дополнение к статину) и случаев инфаркта миокарда или ишемического инсульта в анамнезе, находятся полностью слева от линии отсутствия эффекта. Эффект лечения наблюдали на каждом уровне в пределах каждой подгруппы. Кроме того, тесты на наличие взаимосвязи с возрастом, полом, расой, этнической принадлежностью, интенсивностью фоновой терапии статинами не достигали значимости на уровне 5% и, следовательно, демонстрировали согласованность эффекта лечения в пределах данных подгрупп. Однако для пациентов с другой терапией LLT (в дополнение к статину) и пациентов с инфарктом миокарда или ишемическим инсультом в анамнезе показан относительно более высокий эффект лечения (p-значение взаимосвязи 0,012 и 0,018 соответственно; см. фигуру 5).
[00185] Два последовательных значения рассчитанного уровня LDL-C <25 мг/дл (< 0,65 ммоль/л) наблюдали у 39 (19,5%) пациентов. Причин для какого-либо особого беспокойства относительно безопасности у этих пациентов не наблюдали.
COMBO I: обобщенные результаты относительно безопасности
[00186] Процентные доли пациентов, которые испытывали возникшие в ходе лечения нежелательные явления (TEAE), серьезные TEAE, TEAE, приводящие к смерти, и TEAE, приводящие к прекращению лечения, были аналогичными у групп лечения. Данные безопасности, собранные до последнего визита пациента на 52 неделе, изложены далее в таблице 13.
[00187] Из 209 пациентов, изначально рандомизированных в группу алирокумаба, 2 не получали лечения, это означает, что популяция для оценки безопасности содержала 207 пациентов в случае алирокумаба. Все рандомизированные пациенты с режимом лечения, предусматривающим плацебо, получали предназначенное им лечение, следовательно, популяция для оценки безопасности составляла 107 пациентов в случае плацебо.
Таблица 13 Нежелательные явления и значения лабораторных показателей безопасности (популяция для оценки безопасности)
Все пациенты получали фоновую терапию с максимально переносимой дозой статина ± другая LLT
(n=207)
(n=107)
*Запросы в компанию MedDRA (CMQ). **Один пациент в группе плацебо и 1 пациент в группе алирокумаба перенесли по 2 явления, которые были положительно подтверждены экспертом: несмертельный инфаркт миокарда и процедура реваскуляризации коронарных сосудов по причине ишемии. TEAE представляют собой нежелательные явления, которые развились, ухудшились или стали серьезными на протяжении периода TEAE (время от первой до последней инъекции исследуемого лечения+70 дней). SAE=серьезные нежелательные явления; TEAE=возникшие в ходе лечения нежелательные явления; ULN=верхняя граница нормы.
Наиболее часто описываемыми и заслуживающими внимания SOC с более высокой частотой в группе алирокумаба по сравнению с группой плацебо были: "инфекции и инвазии": 37,2% в группе алирокумаба в сравнении с 27,1% в группе плацебо; ʺнарушение со стороны метаболизма и питанияʺ: 10,1% в группе алирокумаба в сравнении с 5,6% в группе плацебо; и "нарушения со стороны сердца": 8,2% в группе алирокумаба в сравнении с 4,7% в группе плацебо, хотя заметного дисбаланса для конкретного явления отмечено не было, заслуживающим внимание является то, что 9 явлений ишемии произошли в группе алирокумаба в сравнении с 3 в группе плацебо. Что касается результатов экспертного заключения, 5 пациентов перенесли-сосудистые явления, которые были положительно подтверждены экспертом (2,4%), в сравнении с 2 пациентами в группе плацебо (1,9%).
[00188] Среди SOC с более низкой частотой, SOC "нарушения со стороны органа зрения" наблюдали у 4,3% пациентов в группе алирокумаба в сравнении с 0,9% в группе плацебо без дисбаланса в отношении конкретного события.
[00189] Два (1,0%) и 3 (2,8%) случая смерти произошли в течение периода лечения в группе алирокумаба (инфаркт миокарда и эмболия легочной артерии) и плацебо (аденокарцинома пищевода, деменция и внезапная сердечная смерть) соответственно. Кроме того, одна смерть после завершения лечения (обострение заболевания коронарных артерий) произошла в группе алирокумаба.
[00190] Установленные CV явления включают все положительно установленные CV AE. Категории экспертного заключения являются следующими: смерть от CHD, несмертельный MI, смертельный и несмертельный ишемический инсульт, нестабильная стенокардия, при которой требуется госпитализация, застойная сердечная недостаточность, при которой требуется госпитализация, и процедура реваскуляризации коронарных сосудов по причине ишемии [PCI, CABG].
[00191] О SAE сообщали 12,6% пациентов в группе алирокумаба и 13,1% в группе плацебо в течение периода TEAE. Отсутствовала конкретная клиническая картина из числа предпочтительных терминов SAE, о них сообщали по отдельности, при этом частота была низкой.
[00192] Никакой конкретной клинической картины не было отмечено среди TEAE, приведших к окончательному прекращению лечения.
[00193] Среди явлений, представляющих интерес, не было обнаружено какого-либо конкретного признака TEAE, относящихся к неврологическим явлениям и нейрокогнитивным нарушениям.
[00194] Четырнадцать пациентов (11 (5,3%) в группе алирокумаба и 3 (2,8%) в группе плацебо) испытывали локальную реакцию в месте инъекции, возникшую в ходе лечения. Среди общих аллергических реакций дисбаланс отмечали для астмы с 5 (2,4%) случаями, включая 2 серьезных случая, в группе алирокумаба по сравнению с отсутствием случаев в группе плацебо.
[00195] Пять (7,4%) пациентов в группе алирокумаба и 1 (2,7%) пациента в группе плацебо, о которых сообщалось, что они имеют нарушенный контроль глюкозы на исходном уровне, классифицировали как имеющих диабет в периоде TEAE. Ни один пациент, которого классифицировали как имеющего нормальный статус на исходном уровне, не приобретал диабет.
[00196] Дисбаланс в отношении значений PCSA отмечали для снижения уровня гемоглобина от исходного уровня ≥20 г/л (6,3% в группе алирокумаба в сравнении с 3,9% в группе плацебо) и в отношении новых случаев значений уровня эритроцитов ≥6×1012/л (2,5% в группе алирокумаба в сравнении с отсутствием случаев в группе плацебо). Никаких других отклонений, связанных с PCSA не наблюдали.
[00197] Уровни LDL-C <25 мг/дл (на основании 2 последовательным измерениям с промежутком ≥21 день) наблюдали у 39 пациентов, которых лечили алирокумабом, 9 из которых имели уровень LDL-C <15 мг/дл. Общий профиль нежелательных явлений оказался аналогичным у пациентов без таких низких уровней LDL-C, хотя наблюдалась несколько более высокая частота миалгии (7,7% у пациентов с уровнем LDL-C <25 мг/дл в сравнении с 3,4% в группе алирокумаба в целом, 3,7% в группе плацебо), остеоартрита (7,7% в сравнении с 3,9% и 4,7%), артралгии (5,1% в сравнении с 3,9% и 7,5%) и протрузии межпозвонкового диска (5,1% в сравнении с 1,0% и 0%).
[00198] Антитела к алирокумабу обнаруживали в общей сложности у 18 пациентов (из 296 оцениваемых пациентов; 6,1%), при этом антитела к алирокумабу обнаруживали на исходном уровне в общей сложности у пяти пациентов, впоследствии рандомизированных в группу алирокумаба (3/197; 1,5%) или плацебо (2/99; 2,0%). У 13/197 (6,6%) пациентов, получавших лечение алирокумабом, наблюдали незначительный положительный результат теста на антитела, возникший в ходе лечения, при этом титры не превышали 240. У семи из этих пациентов антитела наблюдались временно и исчезали, несмотря на продолжающееся лечение алирокумабом. Медиана времени до обнаружения антител к алирокумабу составляла 12 недель; у положительных в отношении антитела пациентов не наблюдали никаких специфических клинических явлений. При этом наличие антител к алирокумабу не оказывало видимого эффекта на безопасность и эффективность.
COMBO I: общие результаты
[00199] Для данной популяции пациентов с HeFH и высокими исходными уровнями LDL-C, несмотря на максимально переносимую дозу статина (с другой LLT или без нее), были сделаны следующие наблюдения: 1) вводимый самостоятельно алирокумаб обеспечивал значимо большее снижение уровней LDL-C в сравнении с плацебо на 24 неделе; 2) абсолютное среднее снижение уровней LDL-C от исходного уровня составляло -90,8 мг/дл на 24 неделе в случае алирокумаба в сравнении с -15,5 мг/дл в случае плацебо; 3) средние достигнутые уровни LDL-C составляли 107 мг/дл на 24 неделе в случае алирокумаба в сравнении с 182 мг/дл в случае плацебо; 4) 32% пациентов из группы алирокумаба достигали уровня LDL-C <70 мг/дл, несмотря на исходный уровень LDL-C >190 мг/дл; 5) 57% пациентов из группы алирокумаба достигали уровня LDL-C <100 мг/дл на 24 неделе; и 6) алирокумаб в основном хорошо переносился и TEAE в основном были сравнимы с группой плацебо.
COMBO II: СТАТИСТИЧЕСКИЕ МЕТОДЫ
Определение размера выборки
[00200] Конечный общий размер выборки составлял 660 при рандомизационном соотношении 2:1 (алирокумаб 440: эзетимиб 220).
Combo II: временные рамки анализов
[00201] Анализ первой стадии включал конечные точки эффективности вплоть до 52 недели (окончательный анализ эффективности) и промежуточный анализ безопасности, который выполняли с использованием всех данных безопасности, полученных до общей даты завершения исследования (последний визит пациента на 52 неделе). Анализ данных по липидам после 52 недели был описательным. Эти результаты представлены в данном документе.
[00202] Анализ второй стадии (окончательный анализ) будет проводиться в конце исследования и будет состоять из окончательного анализа конечных точек эффективности вплоть до 104 недели и окончательного анализа безопасности.
Combo II: популяции для анализа
[00203] Популяцией для анализа первичной эффективности была популяция, сформированная в зависимости от назначенного лечения (ITT), определенная как все рандомизированные пациенты, которые имели оцениваемую первичную конечную точку, другими словами, имеющие доступное значение рассчитанного уровня LDL-C на исходном уровне и по меньшей мере одно доступное значение рассчитанного уровня LDL-C в пределах одного из периодов анализа вплоть до 24 недели (включая все рассчитанные уровни LDL-C для пациентов, получающих лечение, и пациентов, не получающих лечение).
[00204] Популяцией для анализа вторичной эффективности была модифицированная популяция, сформированная в зависимости от назначенного лечения (mITT), определенная как все рандомизированные пациенты, которые принимали по меньшей мере одну дозу или часть дозы экспериментального медицинского продукта (IMP) двойного слепого исследования и которые имели доступное значение рассчитанного уровня LDL-C на исходном уровне и по меньшей мере одно значение в пределах одного из периодов анализа вплоть до 24 недели во время периода лечения для оценки эффективности. Период лечения для оценки эффективности определяли как период времени от первого введения IMP двойного слепого исследования (капсулы или инъекции, в зависимости от того, что наступает раньше) до дня последней инъекции+21 день или дня приема последней капсулы+3 дня, в зависимости от того, что наступает раньше.
[00205] Популяция для оценки безопасности включала всех рандомизированных пациентов, которые получали по меньшей мере одну дозу или часть дозы IMP двойного слепого исследования.
Combo II: анализы эффективности
[00206] Первичные анализы конечных точек эффективности проводили с применением подхода ITT (на основании популяции ITT, определенной выше), включая все данные по уровням липидов, независимо от того, продолжал ли пациент терапию или нет. Это соответствовало оцениваемым величинам ITT, определенным для первичных и ключевых вторичных конечных точек. Кроме того, анализы также проводили с применением подхода на основе пациентов, получающих лечение (на основании популяции mITT, определенной выше), включая данные по уровням липидов, собранным в течение периода лечения для оценки эффективности. Это соответствовало оцениваемым величинам анализа у пациентов, получающих лечение, для ключевых вторичных конечных точек.
[00207] С помощью подхода ITT анализировали всех пациентов, независимо от соблюдения ими схемы лечения; с помощью него оценивали пользу стратегии лечения, и он отражает максимально возможный эффект для популяции пациентов. С помощью подхода на основе пациентов, получающих лечение, анализировали эффект лечения, ограниченный периодом, в ходе которого пациенты фактически получали лечение. С помощью него оценивали пользу, которую позволило бы достичь лечение у пациентов, соблюдающих схему лечения до рассматриваемого момента времени.
[00208] Анализы эффективности выполняли в соответствии с лечением согласно рандомизации.
[00209] Все измерения, запланированные или незапланированные, натощак или не натощак, относили к периодам анализа с целью обеспечения оценки для моментов времени с 4 недели до 104 недели.
[00210] Принимая во внимание анализ первичной эффективности (подход ITT), процентное изменение рассчитанного уровня LDL-C от исходного уровня до 24 недели анализировали с применением подхода модели со смешанными эффектами для повторных измерений (MMRM). Применяли все данные после исходного уровня, доступные для периодов анализа с 4 недели до 52 недели, и недостающие данные учитывали при помощи MMRM. Модель включала фиксированные категориальные эффекты группы лечения (эзетимиб в сравнении с алирокумабом), страты рандомизации (согласно IVRS), момента времени (от 4 недели до 52 недели), взаимосвязи лечение-момент времени и взаимосвязи страта-момент времени, а также непрерывные фиксированные ковариаты значения исходного уровня LDL-C и взаимосвязь значение исходного уровня-момент времени. Данная модель обеспечивала скорректированные относительно исходного уровня оценки средних значений, определенных методом наименьших квадратов (средние значения LS), на 24 неделе для обеих групп лечения с их соответствующим 95% доверительным интервалом. Для сравнения группы алирокумаба с группой эзетимиба использовали соответствующее высказывание для тестирования различий данных оценок при 5% альфа-уровне значимости.
[00211] Была определена иерархическая процедура для тестирования ключевых вторичных конечных точек при контроле множественности сравнений (с применением приведенного выше порядка ключевых вторичных конечных точек). Первая ключевая вторичная конечная точка представляла собой процентное изменение рассчитанного уровня LDL-C от исходного уровня до 24 недели с применением подхода на основе пациентов, получающих лечение.
[00212] Непрерывные вторичные переменные, предположительно имеющие нормальное распределение (т. e. уровни липидов, отличных от TG и Lp(a)), анализировали с применением такой же модели MMRM, как для первичной конечной точки. Непрерывные конечные точки, предположительно имеющие ненормальное распределение (т. e. уровни TG и Lp(a)), анализировали с применением подхода множественного восстановления для обработки пропущенных значений с последующим применением модели устойчивой регрессии с конечной точкой, представляющей интерес, в качестве переменной отклика с применением M-оценки (с применением процедуры SAS ROBUSTREG), с группой лечения, стратами рандомизации (согласно IVRS) и соответствующим значением(значениями) исходного уровня в качестве эффектов для сравнения эффектов лечения. Обеспечивали объединенную оценку среднего значения в обеих группах лечения, а также различия данных оценок с их соответствующими SE, 95% CI и p-значением (посредством процедуры SAS MIANALYZE).
[00213] Бинарные вторичные конечные точки эффективности анализировали с применением подхода множественного восстановления для обработки пропущенных значений, с последующим применением стратифицированной логистической регрессии, при этом группа лечения выступала в качестве основного эффекта, а соответствующее значение(значениями) исходного уровня в качестве ковариаты, при стратификации по факторам рандомизации (согласно IVRS). Обеспечивали объединенные оценки соотношения шансов по сравнению с эзетимибом, 95% CI и p-значение (посредством процедуры SAS MIANALYZE).
Combo II: анализы безопасности
[00214] Анализы безопасности были описательными, их проводили на популяции для оценки безопасности согласно фактически полученному лечению. Для анализа TEAE и PCSA анализ безопасности был сосредоточен на периоде TEAE, определенном как время с момента введения первой дозы IMP двойного слепого лечения (капсулы или инъекции, в зависимости от того, что наступит раньше) до 70 дней после последней инъекции двойного слепого исследования. Анализы лабораторных данных и определение показателей жизненно важных функций с течением времени выполняли для периода вплоть до 21 дня после последней инъекции IMP.
COMBO II: РЕЗУЛЬТАТЫ
Учет пациентов
[00215] Из 720 рандомизированных пациентов все пациенты проходили лечение и, следовательно, их включали в популяцию для оценки безопасности.
[00216] Тринадцать рандомизированных пациентов (12 в группе алирокумаба и 1 в группе эзетимиба) не включали в популяцию ITT (отсутствует значение уровня LDL-C в пределах одного из периодов анализа до 24 недели).
[00217] Двадцать одного рандомизированного пациента исключили из популяции mITT (пациенты, исключенные из популяции ITT, и пациенты с отсутствием значения уровня LDL-C в пределах одного из периодов анализа вплоть до 24 недели в время периода лечения для оценки эффективности).
[00218] В группе алирокумаба среди 446 пациентов, которые получили по меньшей мере одну инъекцию после 12 недели, 82 (18,4%) пациента получали автоматическое повышение дозы на 12 неделе от 75 мг алирокумаба Q2W до 150 мг Q2W слепым способом.
Таблица 14 Популяции для анализа
Для остальных популяций данные пациентов сводили в таблицу в соответствии с их лечением, назначенным при рандомизации.
Combo II: распределение для исследования
[00219] Распределение для исследования, воздействие и анализы безопасности оценивали с использованием всех данных вплоть до общей даты завершения исследования (определенной как дата последнего визита пациента на 52 неделе). Следовательно, данный анализ первой стадии включает данные после 52 недели для некоторых пациентов. Результаты, представленные в данном документе, доступны из данного анализа первой стадии и включают данные эффективности вплоть до 52 недели, данные безопасности вплоть до 52-102 недели (включая все данные, собранные до последнего визита пациента на W52).
[00220] Ни один из рандомизированных пациентов не завершил 104-недельный период лечения в ходе двойного слепого исследования, и 612 (85,0%) рандомизированных пациентов (N=406 при режиме лечения с алирокумабом и N=206 при режиме лечения с эзетимибом) продолжали лечение во время даты завершения анализа первой стадии.
[00221] Прием IMP двойного слепого исследования преждевременно прекращали до 104 недели 73 (15,2%) рандомизированных пациента в группе алирокумаба (включая 71 пациента, которые прекратили до 52 недели) и 35 (14,5%) рандомизированных пациентов в группе эзетимиба (включая 33 пациентов, которые прекратили до 52 недели).
[00222] Основными причинами для прекращения исследуемого вида лечения были нежелательные явления и другие причины. Категория ʺдругиеʺ представляла наиболее часто сообщаемые причины и включала следующее: пациент не соответствовал критериям включения/исключения (3 пациента), исключение субъекта по личным причинам (14 пациентов), внезапная смерть/смерть (5 пациентов) (не было решения исследователя прекратить лечение до наступления смерти), утрата контакта для последующего наблюдения (1 пациент). В этом анализе первой стадии окончательные результаты доступны для первичной конечной точки эффективности на 24 неделе и ключевых вторичных конечных точек эффективности, оцениваемых на 12 неделе, 24 неделе и 52 неделе. На 24 неделе первичная конечная точка эффективности была доступна для 428 (91,6%) в группе алирокумаба и 221 (92,1%) в группе эзетимиба. На 52 неделе первичная конечная точка эффективности была доступна для 414 (88,7%) в группе алирокумаба и 211 (87,9%) в группе эзетимиба.
[00223] Первичная конечная точка отсутствовала для 58 пациентов (39 пациентов (8,4%) в группе алирокумаба и 19 пациентов (7,9%) в группе эзетимиба). При визите на 24 неделе причины наличия пропущенных данных были следующими: 29 образцов не были собраны из-за преждевременного прекращения исследования; 8 образцов были собраны за пределами периодов анализа; 9 образцов отсутствовали, хотя визит 24 недели был совершен; 12 образцов были собраны, но измерения не могли быть осуществлены (липемия, недостаточное количество, уровень TG > 400 мг/дл [> 4,52 ммоль/л], утеря образца).
COMBO II: Демографические данные и характеристики на исходном уровне
[00224] Демографические характеристики, характеристики заболеваний и параметры уровней липидов на исходном уровне в основном были аналогичны у группы алирокумаба при сравнении с группой эзетимиба. Для группы алирокумаба (N=479) возраст (среднее число лет (SD)) составлял 61,7 (9,4), процентная доля мужчин составляла 75,2% (N=360), процентная доля представителей белой расы составляла 84,3% (N=404), процентная доля негроидной или афро-американской расы составляла 4,4% (N=21), процентная доля других рас составляла 11,3% (N=54), и BMI (среднее значение, кг/м2 (SD)) составлял 30,0 (5,4). Для группы эзетимиба (N=241) возраст (среднее число лет (SD)) составлял 61,3 (9,2), процентная доля мужчин составляла 70,5%(N=170), процентная доля представителей белой расы составляла 85,5% (N=206), процентная доля негроидной или афро-американской расы составляла 2,9% (N=7), процентная доля других рас составляла 11,6% (N=28), и BMI (среднее значение, кг/м2 (SD)) составлял 30,3 (5,1).
[00225] Исходные характеристики групп алирокумаба и эзетимиба изложены в таблицах 15-19. Среднее значение (SD) исходного уровня LDL-C составляло 107,3 (35,7) мг/дл (2,778 (0,926) ммоль/л). Уровень HbA1c (среднее значение (SD), %) составлял 6,05 (0,75) для группы алирокумаба и 6,07 (0,77) для группы эзетимиба. Примерно 90% рандомизированной популяции имели CHD в анамнезе (т. e. по меньшей мере одну из 5 приведенных ниже подкатегорий CHD), а остальные пациенты имели эквивалентные риски возникновения CHD (таблица 16), за исключением 2 пациентов в группе алирокумаба, которые не имели полностью документально подтвержденное CHD или эквивалентные риски возникновения CHD. В целом 30% имели диабет в анамнезе. Всех пациентов кроме одного лечили статином, 66,7% получали статин в высокой дозе (от 40 дo 80 мг аторвастатина ежедневно или от 20 дo 40 мг розувастатина ежедневно), и 2,1% ежедневно получали 80 мг симвастатина.
[00226] Суммарно подвергли скринингу 1112 пациентов с высоким риском возникновения сердечно-сосудистых заболеваний, 720 из которых подходили для включения в исследование и желали принять участвовать в исследовании. Из них 479 случайным образом распределили в группу алирокумаба и 241 - в группу эзетимиба. Средний (SD) возраст составлял 61,6 (9,3) года, 73,6% участников были мужчинами, 90,1% имели коронарное заболевание сердца, 30,7% имели диабет 2 типа, среднее значение (SD) рассчитанной концентрации LDL-C на исходном уровне составляло 107,3 (35,7) мг/дл, и 66,7% получали лечение статином в высокой дозе (т. e. 40/80 мг/день аторвастатина или 20/40 мг/день розувастатина). Пятнадцать пациентов принимали 80 мг симвастатина. Характеристики на исходном уровне были сбалансированы между двумя группами.
Таблица 15 Характеристики заболеваний и другие соответственные данные на исходном уровне - рандомизированная популяция
(N=241)
(N=479)
(N=720)
Таблица 16 Медицинский анамнез, представляющий особый интерес: сердечно-сосудистые нарушения в анамнезе и факторы риска - рандомизированная популяция
(N=241)
(N=479)
(N=720)
a в соответствии с пунктами, предварительно перечисленными в e-CRF.
b диагностированное с помощью инвазивного или неинвазивного тестирования.
PAD определено как перемежающаяся хромота вместе с лодыжечно-брахиальным индексом ≤ 0,90 или вместе с процедурой реваскуляризации/хирургией периферических сосудов или критическая ишемия конечностей вместе с процедурой реваскуляризации/хирургией периферических сосудов или тромболизисом.
Таблица 17 Медицинский анамнез, представляющий особый интерес: другой медицинский анамнез, представляющий особый интерес - рандомизированная популяция
(N=241)
(N=479)
(N=720)
Примечание: один пациент может учитываться в нескольких категориях
Таблица 18 Фоновая LMT при рандомизации - рандомизированная популяция
(N=241)
(N=479)
(N=720)
Статин в высокой дозе соответствует 40-80 мг аторвастатина ежедневно или 20-40 мг розувастатина ежедневно.
Таблица 19 Параметры эффективности липидного обмена на исходном уровне - обобщенные результаты количественной оценки в стандартных единицах - рандомизированная популяция
(N=241)
(N=479)
(N=720)
[00227] Сбор данных по измеренным уровням LDL-C не планировали в начальном протоколе, и он был добавлен в измененной версии. Следовательно, значения измеренного уровня LDL-C доступны для меньшего количества пациентов по сравнению со значениями рассчитанного уровня LDL-C.
COMBO II: дозировка и длительность
[00228] Период проведения инъекций (алирокумаб или плацебо) и приема капсул (эзетемиб или плацебо) был аналогичным среди групп лечения со средним значением периода проведения, составлявшим примерно 57 недель.
[00229] В группе алирокумаба среди 446 пациентов, которые получили по меньшей мере одну инъекцию после 12 недели, 82 (18,4%) пациента получали автоматическое повышение дозы на 12 неделе от 75 мг алирокумаба Q2W до 150 мг Q2W слепым способом.
[00230] Среднее значение (SD) срока проведения инъекций составляло 58,0 (18,7) недели (при среднем значении [SD] числа инъекций 26,6 [8,8]) при режиме лечения с алирокумабом и 57,7 (19,0) недели (число инъекций 26,6 [9,0]) при режиме лечения с эзетимибом. На момент проведения данного анализа 84,8% пациентов при режиме лечения с алирокумабом и 85,5% при режиме лечения с эзетимибом еще продолжали лечение (активное или плацебо). Восемьдесят двум (18,4%) пациентам при режиме лечения с алирокумабом повышали дозу на 12 неделе до схемы дозирования 150 мг Q2W, поскольку их уровень LDL-C на 8 неделе составлял ≥70 мг/дл.
COMBO II: первичная конечная точка эффективности
[00231] Анализ ITT включает все значения рассчитанного уровня LDL-C, полученные у пациентов, получающих лечение и во время перерывов в лечении вплоть до 52 недели. Анализ первичной конечной точки (процентное изменение рассчитанных уровней LDL-C от исходного уровня до 24 недели) представлен на основе модели MMRM в отношении популяции ITT с использованием показателей средних значений, определенных методом наименьших квадратов, на 24 неделе. Для тридцати девяти (8,4%) пациентов в группе алирокумаба и 19 (7,9%) пациентов в группе эзетимиба не были получены значения рассчитанных уровней LDL-C на 24 неделе. Эти пропущенные значения учитывали при помощи модели MMRM.
[00232] Результаты анализа первичной конечной точки представлены в таблице 20, в ммоль/л и мг/дл.
[00233] Наблюдали статистически значимое снижение процентного изменения уровня LDL-C от исходного уровня до 24 недели в группе алирокумаба (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -50,6%) по сравнению с группой эзетимиба (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -20,7%) (различие для среднего значения, определенного методом наименьших квадратов, в сравнении с эзетимибом, составляющее -29,8%, p<0,0001).
[00234] В группе алирокумаба снижение уровня LDL-C от исходного уровня наблюдали с 4 недели до 52 недели (см. фигуру 6 и таблицу 20). Снижение уровня LDL-C сохранялось вплоть до 52 недели в группе алирокумаба (среднее значение, определенное методом наименьших квадратов, в сравнении со значением исходного уровня на 52 неделе составляло -49,5%, в сравнении с -50,6% на 24 неделе).
Таблица 20 Процентное изменение от исходного уровня рассчитанных уровней LDL-C на 24 неделе: MMRM - анализ ITT - популяция ITT и популяция пациентов, получающих лечение
Рассчитанные уровни холестерина LDL
(N=240)
(N=467)
Таблица 21 Рассчитанные уровни LDL-C в зависимости от времени - анализ ITT - популяция ITT
(N=240)
(N=467)
Примечание: средние значения, определенные методом наименьших квадратов, стандартные ошибки (SE) и p-значение, полученные из анализа на основе MMRM (модели со смешанными эффектами для повторных измерений). Модель включает фиксированные категориальные эффекты группы лечения, страты рандомизации согласно IVRS, момента времени, взаимосвязи лечение-момент времени, взаимосвязи страта-момент времени, а также непрерывные фиксированные ковариаты значения исходного уровня LDL-C и взаимосвязи исходный уровень LDL-C-момент времени.
Модель MMRM и описание исходного уровня проводили в отношении пациентов со значением на исходном уровне и значением после исходного уровня по меньшей мере в одном из периодов анализа, используемых в модели.
COMBO II: ключевые вторичные конечные точки эффективности и дополнительные анализы
[00235] В следующей таблице обобщены результаты анализа в отношении ключевых вторичных конечных точек в иерархическом порядке. Все ключевые вторичные конечные точки являются статистически значимыми в соответствии с процедурой иерархического тестирования вплоть до конечной точки уровня HDL-C на 24 неделе (оцениваемая величина ITT) включительно.
[00236] Статистическая значимость не достигалась для уровней TG натощак на 24 неделе (оцениваемая величина ITT), и следовательно процедуру тестирования прекращали, p-значения для этой конечной точки представлены только для наглядности.
Таблица 22
[00237] Анализ процентного изменения уровня LDL-C от исходного уровня до 24 недели у пациентов, получающих лечение, показывает результаты, которые полностью согласуются с результатами анализа ITT, при этом различие для среднего значения, определенного методом наименьших квадратов, в сравнении с эзетимибом составило -30,6% в анализе в ходе лечения по сравнению с -29,8% в анализе ITT.
[00238] Следующие ключевые конечные точки, в том числе уровни Apo B, не-HDL-C, общего-C, Lp(a), HDL-C в различные моменты времени, а также доля пациентов, достигающих их целевых уровней LDL-C на 24 неделе, были статистически значимыми в соответствии с процедурой иерархического тестирования.
[00239] Большинство пациентов с высоким риском возникновения сердечно-сосудистых заболеваний, получавших алирокумаб на фоне приема статинов, достигали их целевых уровней LDL-C. Доля пациентов, достигавших рассчитанного уровня LDL-C <70 мг/дл (1,81 ммоль/л) на 24 неделе, была значительно выше в группе алирокумаба, чем в группе эзетимиба (объединенная оценка для доли, составляющей 77,0%, в группе алирокумаба в сравнении с 45,6% в группе эзетимиба, p<0,0001).
[00240] Отличие процентного изменения в уровне TG натощак от исходного уровня до 24 недели в анализе ITT не было статистически значимым. Среднее значение, определенное методом наименьших квадратов, в сравнении со значением исходного уровня составляло -13,0% в группе алирокумаба и -12,8% в группе эзетимиба (различие для среднего значения, определенного методом наименьших квадратов, в сравнении с эзетимибом составило -0,3%, p=0,9117).
[00241] У сто пяти (22,8%) пациентов наблюдали два последовательных значения рассчитанных уровней LDL-C <25 мг/дл. Никакого особого беспокойства относительно безопасности не наблюдали у этих 105 пациентов.
Таблица 23 Число (%) пациентов с 2 последовательными значениями уровня LDL-C < 25 мг/дл (<0,65 ммоль/л) в течение периода лечения- популяция для оценки безопасности
(N=241)
(N=479)
Знаменатель (/N) в пределах группы лечения представляет собой число пациентов в группе лечения, которые имели по меньшей мере два значения рассчитанного уровня LDL-C, определенные с промежутком по меньшей мере 21 день в период оценки эффективности.
1 2 последовательных значения берутся во внимание, если их разделяет по меньшей мере 21 день.
2 Первое значение рассчитанного уровня LDL-C <25 или <15 мг/дл среди первых 2 последовательных значений рассчитанного уровня LDL-C <25 или <15 мг/дл для пациента
Таблица 24 - Процентное изменение от исходного уровня до 24 недели уровней LDL-C и вторичных параметров уровней липидов (анализ у пациентов, получающих лечение)
(n=464)
(n=235)
COMBO II: обобщенные результаты в отношении эффективности
[00242] На 24 неделе процентное изменение от исходного уровня у рассчитанного уровня LDL-C в популяции ITT было значимо больше в группе алирокумаба (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -50,6%) по сравнению с группой эзетимиба (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -20,7%) (различие для среднего значения, определенного методом наименьших квадратов, в сравнении с эзетимибом составило -29,8%, p < 0,0001) (таблица 20). Анализ процентного изменения уровня LDL-C от исходного уровня до 24 недели у пациентов, получающих лечение, показывает результаты, которые согласуются с результатами анализа ITT, при этом различие для среднего значения, определенного методом наименьших квадратов, в сравнении с эзетимибом составило -30,6% в анализе в ходе лечения (p<0,0001) (таблица 24).
[00243] Динамика изменений концентраций LDL-C при режимах лечения с алирокумабом и эзетимибом от исходного уровня до 52 недели показана на фигуре 6. Средние концентрации LDL-C быстро падали в первые 4 недели, но в большей степени при режиме лечения с алирокумабом. В группе алирокумаба снижение уровня LDL-C от исходного уровня наблюдалось с 4 недели и сохранялось вплоть до 52 недели (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем на 52 неделе составляло -49,5% в сравнении с -50,6% на 24 неделе) (таблица 21).
[00244] Распределение значений LDL-C на исходном уровне и достигнутых значений на 24 неделе показано на фигуре 7. Медианное значение составило 40 мг/дл для алирокумаба в сравнении с 70 мг/дл для эзетимиба. Среднее значение достигнутых уровней LDL-C на 24 неделе составило 51,6 (1,4) мг/дл в случае алирокумаба и 82,5 (2,0) мг/дл в случае эзетимиба; эти отличия сохранялись до 52 недели.
[00245] Динамика изменений концентраций LDL-C в соответствии со статусом повышения дозы при режиме лечения с алирокумабом показана на фигуре 8. Только у 18% процентов пациентов в группе алирокумаба дозу повышали до 150 мг SC Q2W. Эти пациенты характеризовались намного более высокими значениями LDL-C на исходном уровне по сравнению с пациентами, которым не потребовалось повышение дозы (140,4 [47,4] мг/дл в сравнении с 101,1 [29,7] мг/дл), и снижения в процентах уровней LDL-C, достигнутых на 12 и 24 неделях, были более низкими. Повышение дозы на 12 неделе приводило к дополнительному снижению, составляющему 12,4%. Более того, абсолютное снижение уровней LDL-C к 24 неделе было немного большим в группе с повышенной дозой (63,5 в сравнении с 56,9 мг/дл) относительно остальной части группы алирокумаба.
[00246] Что касается ключевых вторичных конечных точек эффективности, статистически значимые снижения наблюдали для Apo B (22,4%), Lp(a) (21,7%) и не-HDL-C (22,9%) (для всех P<0,0001), и наблюдали 8,1% повышение уровня HDL-C на 24 неделе при режиме лечения с алирокумабом по сравнению с эзетимибом (P<0,0001) (таблица 22). Статистически значимое отличие не достигалось для уровней TG натощак на 24 неделе (снижение от исходного уровня на 13,5% у группы алирокумаба и 13,3% у группы эзетимиба) и, следовательно, процедуру тестирования в отношении данной конечной точки прекращали. Эффективность алирокумаба в сравнении с эзетимибом была стабильной у нескольких подгрупп в популяции ITT (фигура 9). В таблице 22 способ количественного определения бета-липопротеинов для определения процентного изменения уровня LDL-C от исходного уровня до 24 недели представлял собой анализ чувствительности, проводимый у 180 пациентов в группе эзетимиба и 361 пациента в группе алирокумаба.
COMBO II: Обобщенные результаты относительно безопасности
[00247] Частоты возникших в ходе лечения нежелательных явлений (TEAE) в пределах в среднем 58 (19)-недельного последующего наблюдения показаны в таблице 25. Общая процентная доля пациентов, которые испытывали по меньшей мере одно TEAE, составляла 71,2% при режиме лечения с алирокумабом и 67,2% при режиме лечения с эзетимибом. TEAE, приведшие к смерти, встречались у 2 (0,4%) пациентов при режиме лечения алирокумабом (причина обоих была связана с нарушением сердечной деятельности) и у 4 (1,7%) пациентов при режиме лечения эзетимибом (причина двух была связана с нарушением сердечной деятельности). Аналогичные процентные доли субъектов в обеих группах испытывали серьезное нежелательное явление (18,8% в группе алирокумаба в сравнении с 17,8% в группе эзетимиба). Более высокая доля пациентов в группе алирокумаба испытывали TEAE, что приводило к прекращению лечения (7,5% в сравнении с 5,4%), при этом не наблюдалось определенной схемы в уровне предпочтительного термина (таблица 25).
[00248] Отсутствовали признаки дисбаланса в TEAE на уровне класса поражений систем и органов, за исключением немного более высокой частоты инфекций и инвазий в случае алирокумаба (27,1% в сравнении с 25,3% в группе с эзетимибом), нарушений со стороны скелетно-мышечной и соединительной ткани (19,6% в сравнении с 17,0%) и нарушений со стороны желудочно-кишечного тракта (15,0% в сравнении с 13,7%) (таблица 25). Установленные сердечно-сосудистые явления были также более частыми в группе алирокумаба (4,8%, n =23; в сравнении с 3,7%, n=9) за счет более высоких частот несмертельных инфарктов миокарда и реваскуляризаций коронарных сосудов.
[00249] Частоты возникших в ходе лечения локальных реакций в месте инъекции оказалась немного более высокой при режиме лечения с алирокумабом (2,5% в сравнении с 0,8% в группе с эзетимибом) (таблица 25). Реакции характеризовались легкой интенсивностью, за исключением одной с умеренной интенсивностью, и ни одна из них не была серьезной. Два явления привели к прекращению в группе алирокумаба. У небольшой процентной доли пациентов в обеих группах развилась миалгия.
[00250] Отклонения в показателях лабораторных исследований были редки и встречались с аналогичной частотой в обеих группах, за исключением уровней аланинтрансаминазы и аспартаттрансаминазы, которые чаще встречались в группе алирокумаба, и нарушенного контроля глюкозы, который был менее частым в группе алирокумаба (таблица 25).
[00251] Сто пять (22,8% из 460) пациентов при режиме лечения с алирокумабом и ни один при режиме лечения с эзетимибом не характеризовался двумя последовательными значениями уровней LDL-C, составлявшими <25 мг/дл, в течение периода лечения. Частоты TEAE в этой группе были подобны таковым в группе эзетимиба, за исключением назофарингита, который чаще встречался в группе алирокумаба (таблица 25).
Таблица 25 - TEAE и параметры лабораторных исследований (популяция для оценки безопасности) через 52 недели в соответствии с классом поражений систем и органов и предпочтительным термином (в том числе наблюдаемые у пациентов с двумя значениями уровней LDL-C <25 мг/дл при лечении с алирокумабом)
(n=479)
(n=241)
LDL-C ≤25 мг/дл
(n=105)
COMBO II: общие результаты
[00252] В данной популяции пациентов с высоким риском возникновения CV, которые имели плохо контролируемые уровни LDL-C при максимально переносимой терапии статинами, были сделаны следующие наблюдения: 1) вводимый самостоятельно алирокумаб приводил к значительно большему снижению уровней LDL-C, чем эзетимиб через 24 недели (различие для среднего значения, определенного методом наименьших квадратов,- 29,8%); 2) вводимый самостоятельно алирокумаб характеризовался хорошим соблюдением режима лечения и хорошо переносился; 3) 77% пациентов, получавших алирокумаб, достигли целевого уровня LDL-C, составляющего <0,81 ммоль/л (70 мг/дл), к 24 неделе; снижение уровней LDL-C, составляющее ~50% в сравнении с исходным уровнем, сохранялось до 52 недели в случае алирокумаба; 4) средние уровни LDL-C, составляющие 1,4 ммоль/л (53,3 мг/дл), достигались к 52 неделе в случае алирокумаба; 5) для примерно 80% пациентов не требовалось повышение дозы алирокумаба до 150 мг Q2W, что свидетельствует от том, что 75 мг Q2W может быть достаточно для большинства пациентов; и 6) TEAE наблюдались с аналогичной частотой при режимах лечения с алирокумабом и эзетимибом.
Пример 3. Долговременные безопасность и переносимость алирокумаба у пациентов с высоким риском возникновения сердечно-сосудистых заболеваний, имеющих гиперхолестеринемию, резистентную к гиполипидемической терапии: рандомизированное двойное слепое плацебо-контролируемое исследование
ВВЕДЕНИЕ
[00253] Данное исследование было предпринято для оценки долговременных безопасности и переносимости алирокумаба у пациентов с высоким риском возникновения сердечно-сосудистых заболеваний без целевого уровня LDL-C. Данная популяция без целевого уровня LDL-C на оптимизированной LMT представляет группу с наивысшим риском с хорошо идентифицируемой нереализованной медицинской потребностью, которую можно решить путем добавления алирокумаба к их видам терапии, модифицирующим уровень LDL-C. Сообщали о двух видах результатов: (1) промежуточный анализ с предварительно заданными параметрами выполняли, когда все пациенты достигали периода в исследовании в один год и примерно 25 процентов пациентов достигали периода в исследовании в 18 месяцев; и (2) окончательный анализ популяции для оценки безопасности, когда все пациенты завершали исследование.
ЦЕЛИ ИССЛЕДОВАНИЯ
[00254] Главной целью исследования была оценка долговременных безопасности и переносимости алирокумаба у пациентов с высоким риском возникновения сердечно-сосудистых заболеваний, имеющих гиперхолестеринемию, резистентную к их гиполипидемической терапии.
[00255] Вторичными целями исследования были: 1) оценка эффекта алирокумаба на уровни холестерина липопротеинов низкой плотности (LDL-C) через 24 недели лечения по сравнению с плацебо; 2) оценка эффективности алирокумаба на уровни LDL-C в другие моменты времени; 3) оценка эффекта алирокумаба на уровни аполипопротеина B (Apo B), холестерина, не относящегося к холестерину липопротеинов высокой плотности (не-HDL-C), общего холестерина (общий-C), липопротеина a (Lp [a]), холестерина липопротеинов высокой плотности (HDL-C), триглицеридов (TG) и аполипопротеина A-1 (Apo A-1) через 24 недели лечения и в другие моменты времени по сравнению с плацебо; 4) оценка появления антител к алирокумабу и 5) оценка фармакокинетики (PK) алирокумаба.
ПЛАН ИССЛЕДОВАНИЯ
[00256] Это было рандомизированное, двойное слепое, плацебо-контролируемое, несбалансированное (2:1, алирокумаб:плацебо), параллельно-групповое, многоцентровое, многонациональное исследование, оценивающее долговременные безопасность и переносимость алирокумаба у пациентов с высоким риском возникновения сердечно-сосудистых заболеваний, имеющих гиперхолестеринемию, которые были резистентны к максимально переносимой ежедневной регистрируемой дозе статина с другой гиполипидемической терапией или без нее. См. фигуру 10. У пациентов проводили стратификацию в соответствии с популяцией с heFH, MI или ишемическим инсультом в анамнезе, лечением статинами и географическим регионом. Пациентов с высоким сердечно-сосудистым риском определяли как 1) характеризующихся heFH (у которых могли быть или отсутствовать CHD/эквивалентные риски возникновения CHD) или 2) не имеющих предыдущего диагноза heFH, но имеющих гиперхолестеринемию вместе с установленным CHD или эквивалентными рисками возникновения CHD. Пациенты должны иметь гиперхолестеринемию и быть резистентными (т. е., с уровнем LDL-C ≥70 мг/дл [≥1,81 ммоль/л]), несмотря на терапию посредством максимально переносимой ежедневной регистрируемой дозы статина с другой гиполипидемической терапией или без нее при стабильной дозе на протяжении по меньшей мере 4 недель (6 недель для фенофибрата) до проведения скрининга.
Описание протокола
[00257] Пациентов рандомизировали по алирокумабу, получаемому по 150 мг каждые 2 недели. Схему лечения можно было при необходимости корректировать в ходе исследования, путем изменения протокола, для введения более низкой дозы с помощью схемы титрации.
[00258] Исследование состояло из 3 периодов: скрининга, двойного слепого лечения и последующего наблюдения. Период скрининга длился до 3 недель, включая промежуточный визит, во время которого пациента (или другое обозначенное лицо, такое как супруг, родственник и т. д.) обучали самостоятельному инъецированию/инъецированию с использованием плацебо. Оценки пригодности проводили, чтобы обеспечить основание для рандомизации пациентов в исследование. Период двойного слепого лечения представлял собой период лечения в ходе рандомизированного двойного слепого исследования длительностью 18 месяцев. Первую инъекцию во время двойного слепого периода осуществляли в исследовательском центре в день рандомизации и как можно скорее после рандомизации в исследование. Последующие инъекции осуществлялись пациентом (самостоятельная инъекция) или другим обозначенным лицом (таким как супруг, родственник и т.д.) в предпочитаемом пациентом местонахождении (дома...). Период последующего наблюдения представлял собой период после окончания периода двойного слепого лечения длительностью 8 недель.
[00259] У пациентов, которые достигали 2 последовательных рассчитанных уровней LDL-C < 25 мг/дл (0,65 ммоль/л) в ходе исследования, продолжали наблюдение и лечение.
[00260] Терапия статинами и другая гиполипидемическая терапия (при определенных условиях) должна быть стабильной (включая дозу) в течение первых 24 недель периода двойного слепого исследования, кроме исключительных обстоятельств, при которых особая озабоченность (включая без ограничений предупреждение об уровне триглицеридов, переданное центральной лабораторией) оправдывает такие изменения, на усмотрение исследователя. Начиная с 24 недели фоновую гиполипидемическую терапию можно было модифицировать только при определенных условиях.
[00261] Пациентам следовало придерживаться постоянной диеты (диета NCEP-ATPIII TLC или эквивалентная) на протяжении всего срока исследования от проведения скрининга. Диетолог или персонал исследовательского центра, обученный соответствующим образом, проверяли диету пациента во время скринингового визита и перидиочески на протяжении исследования. В таблице 26 представлен обзор TLC-диеты в случае высоких уровней холестерина.
Таблица 26
Общее содержание жиров 25% - 35% общего количества калорий*
Насыщенные жиры* < 7% общего количества калорий
Полиненасыщенные жиры до 10% общего количества калорий
Мононенасыщенные жиры до 20% общего количества калорий
Углеводы† 50% - 60% общего количества калорий*
Белок ~15% общего количества калорий
Холестерин < 200 мг/день (5,172 ммоль/†день)
Растительные стерины 2 г
Растворимые волокна, такие как псиллиум 10 г - 25 г
* ATP III допускает повышение общего содержания жиров до 35 процентов общего количества калорий и снижение углеводов до 50 процентов для лиц с метаболическим синдромом. Любое повышение потребления жиров должно производиться в форме либо полиненасыщенного, либо мононенасыщенного жира. Транс-жирные кислоты представляют собой другой тип жиров, которые повышают уровень LDL, их потребление следует поддерживать на низком уровне.
†Углеводы должны происходить преимущественно из пищи, богатой сложными углеводами, в том числе злаков, в первую очередь, цельных злаков, фруктов и овощей.
[00262] Пациенты с сердечно-сосудистыми явлениями от момента рандомизации до конечного визита последующего наблюдения имели пакет, который готовили и посылали для экспертного подтверждения в независимый Комитет по клиническим явлениям.
[00263] Пациентов подвергали тестированию цветового зрения периодически на протяжении всего исследования. Офтальмологическое подысследование проводили на подгруппе из исследуемой популяции.
Срок участия в исследовании
[00264] Срок исследования включал до 3 недель периода скрининга, 18 месяцев периода лечения с двойным слепым исследованием и 8 недель периода последующего наблюдения. Таким образом, срок исследования для пациента составлял примерно 20 месяцев. Концом исследования для пациента был последний запланированный протоколом визит или нормализация/стабилизация всех SAE и AESI, в зависимости от того, что наступило последним. Конец исследования определяли как последний визит периода последующего наблюдения для каждого пациента.
ВЫБОР ПАЦИЕНТОВ
[00265] Исследование было рассчитано на рандомизацию примерно 2100 пациентов при рандомизационном соотношении 2:1 следующим образом: алирокумаб - примерно 1400 пациентов; плацебо - примерно 700 пациентов.
[00266] Пациентов, удовлетворяющих следующим критериям, рассматривали для включения в исследование: 1) пациентов с гетерозиготной семейной гиперхолестеринемией (heFH)* с установленным коронарным заболеванием сердца (CHD) или эквивалентными рисками возникновения CHD или без них, которые были резистентны к максимально переносимой стабильной ежедневной дозе статина** на протяжении по меньшей мере 4 недель до скринингового визита (неделя -3) с другой гиполипидемической терапией (LMT) или без нее; или 2) пациентов с гиперхолестеринемией и установленным CHD или эквивалентными рисками возникновения CHD (определения см. ниже), которые были резистентны к максимально переносимой стабильной ежедневной дозе статина** на протяжении по меньшей мере 4 недель до скринингового визита (неделя -3) с другой липид-корригирующей терапией (LMT) или без нее.
[00267] *Диагноз heFH должен быть поставлен либо на основании генотипирования, либо по клиническим критериям. Для тех пациентов, которых не генотипировали, клинический диагноз мог быть основан либо на критериях WHO/Голландских критериях диагностики гетерозиготной семейной гиперхолестеринемии (Dutch Lipid Clinical Network) с показателем > 8 пунктов, либо на диагностических критериях регистра Саймона Брума с критерием для определенной FH.
[00268] ** Определение максимально переносимой дозы (любое из следующего допустимо): 1) 20 мг или 40 мг розувастатина ежедневно; 2) 40 мг или 80 мг аторвастатина ежедневно; 3) 80 мг симвастатина ежедневно (если уже принимает эту дозу на протяжении >1 года); 4) пациентов, не способных переносить ни одну из вышеперечисленных доз статинов, следует лечить ежедневной дозой аторвастатина, розувастатина или симвастатина, которая считается подходящей для пациента, исходя из решения и опасений исследователя. Некоторые примеры допустимых причин для пациента, принимающего более низкую дозу статинов, включают без ограничения: неблагоприятные эффекты при высоких дозах, пожилой возраст, низкий индекс массы тела, региональные практики, местный листок-вкладыш, сопутствующие лекарственные препараты, сопутствующие состояния, такие как нарушенная толерантность к глюкозе/нарушение уровня глюкозы натощак. Причину(ы) следует подтверждать документально.
[00269] Документально подтвержденное CHD в анамнезе включает одно или несколько из следующего: i) острый инфаркт миокарда (MI); ii) бессимптомный инфаркт миокарда; iii) нестабильную стенокардию; iv) процедуру реваскуляризации коронарных сосудов (например, чрескожная коронарная ангиопластика [PCI] или шунтирование коронарных артерий [CABG]); и/или v) клинически значимое CHD, диагностированное при помощи инвазивных или неинвазивных обследований (таких как коронарография, электрокардиограмма с нагрузкой на беговой дорожке, стресс-эхокардиография или радионуклидная визуализация).
[00270] Эквивалентные риски возникновения CHD включают один или несколько из следующих 4 критериев: i) документально подтвержденное заболевание периферических артерий (должен удовлетворяться один из следующих критериев [a, b или c]): текущая перемежающаяся хромота (мышечный дискомфорт в нижней конечности, который является как повторяющимся, так и проявляющимся при физических нагрузках и облегчаемым при покое в течение 10 минут) предполагаемого атеросклеротического происхождения вместе с лодыжечно-брахиальным индексом < 0,90 в каждой ноге в состоянии покоя, или b) перемежающаяся хромота в анамнезе (мышечный дискомфорт в нижней конечности, который является как повторяющимся, так и проявляющимся при физических нагрузках и облегчаемым при покое в течение 10 минут) вместе с эндоваскулярной процедурой или хирургическим вмешательством по отношению к одной или обеим ногам по причине атеросклеротического заболевания, или c) критическая ишемия конечностей в анамнезе вместе с тромболизисом, эндоваскулярной процедурой или хирургическим вмешательством по отношению к одной или обеим ногам по причине атеросклеротического заболевания; ii) подтвержденный предшествующий ишемический инсульт с очаговым ишемическим неврологическим расстройством, которое продолжалось более 24 часов, предположительно атеротромботического происхождения. Следует провести CT или MRI для исключения кровоизлияния и неишемического неврологического заболевания; iii) подтвержденное умеренное хроническое заболевание почек (CKD), как определено на основании 30 ≤ eGFR <60 мл/мин./1,73 м2 в течение 3 месяцев или более, включая скрининговый визит; iv) известные случаи сахарного диабета в анамнезе и 2 или более дополнительных факторов риска (перечисленных ниже): a гипертензия в анамнезе (установленная по приему медицинских препаратов против гипертензии), b документально подтвержденный лодыжечно-брахиальный индекс ≤0,90 в анамнезе, c документально подтвержденная микроальбуминурия или макроальбуминурия в анамнезе или анализ мочи с помощью индикаторной полоски при скрининговом визите (неделя -3) с уровнем белка >2+, d документально подтвержденная предпролиферативная или пролиферативная ретинопатия или лазерное лечение ретинопатии в анамнезе, e известные случаи раннего развития CHD в семейном анамнезе (CHD у отца или брата в возрасте до 55 лет; CHD у матери или сестры в возрасте до 65 лет).
[00271] Согласно диагностическим критериям регистра Саймона Брума для гетерозиготной семейной гиперхолестеринемии, определенная семейная гиперхолестеринемия характеризуется уровнями общего-C >6,7 ммоль/л (260 мг/дл) или холестерина LDL выше 4,0 ммоль/л (155 мг/дл) у ребенка <16 лет или уровнями общего-C >7,5 ммоль/л (290 мг/дл) или холестерина LDL выше 4,9 ммоль/л (190 мг/дл) у взрослого. (Уровни либо перед проведением лечения, либо наивысшие во время проведения лечения) плюс наличие сухожильных ксантом у пациента, или у ближайшего родственника (родителя, родных брата или сестры, ребенка), или у родственника 2-й степени (дедушки или бабушки, дяди, тети) или основанное на ДНК доказательство мутации рецептора LDL или присутствие семейного дефективного Apo B-100.
[00272] Согласно диагностическим критериям регистра Саймона Брума для гетерозиготной семейной гиперхолестеринемии, возможная семейная гиперхолестеринемия характеризуется уровнями общего-C >6,7 ммоль/л (260 мг/дл) или холестерина LDL выше 4,0 ммоль/л (155 мг/дл) у ребенка <16 лет или уровнями общего-C >7,5 ммоль/л (290 мг/дл) или холестерина LDL выше 4,9 ммоль/л (190 мг/дл) у взрослого. (Уровни либо перед проведением лечения, либо наивысшие во время проведения лечения) и по меньшей мере одно из следующего: инфаркт миокарда в возрасте до 50 лет у родственника 2-й степени или в возрасте до 60 лет у ближайшего родственника в семейном анамнезе, или повышенные уровни холестеринов >7,5 ммоль/л (290 мг/дл) у взрослого ближайшего родственника или родственника 2-й степени или >6,7 ммоль/л (260 мг/дл) у ребенка или родных брата или сестры в возрасте до 16 лет в семейном анамнезе.
[00273] Критерии WHO (Голландские диагностические критерии гетерозиготной семейной гиперхолестеринемии) для постановки диагноза гетерозиготной семейной гиперхолестеринемии (heFH) изложены в таблице 27.
Таблица 27
[00274] Пациентов, которые соответствовали всем вышеперечисленным критериями включения, исследовали в отношении перечисленных ниже критериев исключения.
[00275] Критериями исключения, относящимися к методологии исследования, были: 1) отсутствие установленного CHD в анамнезе или эквивалентные риски возникновения CHD или отсутствие диагноза heFH на основе генотипирования или клинических критериев; 2) LDL-C <70 мг/дл (<1,81 ммоль/л) при скрининговом визите (неделя -3); 3) отсутствие приема стабильной дозы LMT (включая статин) на протяжении по меньшей мере 4 недель и/или фенофибрата на протяжении по меньшей мере 6 недель, с учетом конкретных обстоятельств, перед скрининговым визитом (неделя -3) и от момента скрининга до рандомизации; 4) текущий прием статина, не представляющего собой cимвастатин, аторвастин или розувастатин; 5) прием симвастатина, аторвастина или розувастатина не осуществляется ежедневно или не осуществляется при зарегистрированной дозе; 6) ежедневные дозы превышают 80 мг аторвастина, 40 мг розувастатина или 40 мг симвастатина (за исключением пациентов, принимающих 80 мг симвастатина на протяжении более одного года, подходящих для включения в исследование); 7) применение фибратов, отличных от фенофибрата, в течение 6 недель до скринингового визита (неделя -3) или между скрининговым визитом и визитом рандомизации; 8) применение нутрицевтических продуктов или безрецептурных терапевтических средств, которые могут воздействовать на уровни липидов, не вводимых при стабильной дозе на протяжении по меньшей мере 4 недель до скринингового визита (неделя -3) или между скрининговым визитом и визитом рандомизации; 9) применение продуктов на основе красного ферментированного риса в течение 4 недель до скринингового визита (неделя -3) или между скрининговым визитом и визитом рандомизации; 10) пациент, который проходил плазмаферез в течение 2 месяцев до скринингового визита (неделя -3) или собирается пройти его; 11) недавние (в пределах 3 месяцев до скринингового визита [неделя -3] или между скрининговым визитом и визитом рандомизации) MI, нестабильная стенокардия, приведшая к госпитализации, неконтролируемая сердечная аритмия, CABG, PCI, операция или стентирование сонной артерии, апоплексический удар, транзиторная ишемическая атака (TIA), эндоваскулярная процедура или хирургическое вмешательство при заболевании периферических сосудов; 12) предусмотрено проведение запланированных PCI, CABG, реваскуляризация сонной артерии или реваскуляризации периферических сосудов во время исследования; 13) сердечная недостаточности III или IV класса согласно Нью-Йоркской кардиологической ассоциации (New York Heart Association) (NYHA) в анамнезе в течение последних 12 месяцев; 14) систолическое кровяное давление >180 мм рт. ст. или диастолическое кровяное давление >110 мм рт. ст. при скрининговом визите или визите рандомизации; 15) известный случай геморрагического инсульта в анамнезе; 16) возраст < 18 лет или совершеннолетия при скрининговом визите (неделя -3), в зависимости от того, что является большим; 17) известное активное заболевание оптического нерва в анамнезе; 18) пациенты, которых предварительно не проинструктировали в отношении гипохолестеринемической диеты до скринингового визита (неделя -3); 19) известная гомозиготная FH в анамнезе; 20) известная потеря функции PCSK9 (т. е. генетическая мутация или вариация последовательности) в анамнезе; 21) применение системных кортикостероидов, если только не применяемых в качестве заместительной терапии при заболевании гипофиза/надпочечников с постоянным режимом в течение по меньшей мере 6 недель до рандомизации. Примечание: стероидные терапевтические препараты для местного, внутрисуставного, назального, ингаляционного и офтальмического применения не рассматриваются в качестве ʺсистемныхʺ и разрешены; 22) применение непрерывной заместительной гормональной терапии, если только режим не был постоянным последние 6 недель до скринингового визита (неделя -3) и не планируется менять режим во время исследования; 23) рак в анамнезе в течение последних 5 лет, за исключением излеченного до той меры, до которой это возможно, базально-клеточного рака кожи, плоскоклеточного рака кожи или рака шейки матки in situ; 24) известный положительный результат анализа на HIV в анамнезе; 25) состояния/ситуации, такие как: a) любое клинически значимое отклонение, идентифицированное во время скрининга, которое по мнению исследователя или любого соисследователя сделало бы невозможным безопасное завершение исследования или ограничило бы оценку конечных точек, такое как основные системные заболевания, пациенты с короткой ожидаемой продолжительностью жизни, b) пациенты, которые, как считает исследователь или любой соисследователь, не подходят для данного исследования по какой-либо причине, например: i) пациенты, которые, как считается, неспособны соответствовать определенным требованиям протокола, таким как запланированные визиты; ii) пациенты, которые, как считается, неспособны вводить или неспособны переносить долговременные инъекции по мнению пациента или исследователя; iii) исследователь или любой соисследователь, фармацевт, координатор исследования, другой персонал исследования, непосредственно участвующие в проведении протокола и т. п, или их родственник; iv) наличие каких-либо других условий (например, географических, социальных и т. п.), фактических или предполагаемых, которые, как считает исследователь, сдерживали бы или ограничивали бы участие пациента в течение всего срока исследования; 26) пациент, который прежде проходил лечение по меньшей мере с помощью одной дозы алирокумаба или любого другого моноклонального антитела к PCSK9 в других клинических исследованиях; 27) пациент, который получал любые экспериментальные лекарственные средства, отличные от подготовительных плацебо-наборов для алирокумаба в течение 1 месяца или 5 периодов полувыведения, в зависимости от того, который является большим; 28) пациент, который отзывает согласие во время периода скрининга (пациент, который не желает продолжать или не в состоянии вернуться); 29) лабораторные показатели в течение периода скрининга (не включая лабораторные исследования при рандомизации): A) положительный результат исследования на поверхностный антиген вируса гепатита B и/или антитело к вирусу гепатита C (подтвержденный повторным тестированием), B) уровень триглицеридов (TG) >400 мг/дл (>4,52 ммоль/л) (допускается 1 повторное лабораторное исследование); C) положительный результат теста сыворотки крови или мочи на наличие беременности у женщин детородного возраста; D) уровень eGFR <30 мл/мин./1,73 м2 в соответствии с MDRD-уравнением с 4 переменными; E) HbA1c >10%; F) ALT или AST >3 x ULN (допускается 1 повторное лабораторное исследование); G) CPK >3 x ULN (допускается 1 повторное лабораторное исследование).
[00276] Врачи зачастую оценивают стадию сердечной недостаточности согласно системе функциональной классификации от Нью-Йоркской кардиологической ассоциации (NYHA). Эта система связывает симптомы с повседневной деятельностью и качеством жизни пациента, как показано в таблице 28.
Таблица 28
[00277] Критериями исключения, которые относятся к активным препаратам сравнения и/или видам обязательной фоновой терапии, были 30) все противопоказания по отношению к видам фоновой терапии или предупреждения/меры предосторожности по применению (при необходимости), представленные в соответствующей национальной маркировке продукта.
[00278] Критериями исключения, которые относятся к современному уровню знаний относительно алирокумаба, были: 31) известная гиперчувствительность к терапевтическим средствам на основе моноклональных антител; 32) беременные или кормящие грудью женщины; 33) женщины детородного возраста, не защищенные высокоэффективным способом(способами) контроля рождаемости и/или не желающие или не способные пройти тест на наличие беременности. Примечание: женщины детородного возраста должны иметь подтвержденный отрицательный результат теста сыворотки крови на наличие беременности при скрининге и теста мочи на наличие беременности при визите рандомизации. Они должны применять эффективные способы контрацепции на протяжении всего исследования и согласиться повторно проходить тест мочи на наличие беременности во время обозначенных визитов. Используемые способы контрацепции должны соответствовать критериям высокоэффективного способа контроля рождаемости согласно "примечанию к руководству доклинических исследований безопасности для проведения клинических испытаний на людях в отношении фармацевтических препаратов (CPMP/ICH/286/95)". У женщин постклимактерического возраста менструации должны отсутствовать на протяжении по меньшей мере 12 месяцев.
ИССЛЕДУЕМЫЕ ВИДЫ ЛЕЧЕНИЯ
[00279] Стерильный лекарственный продукт (IMP) алирокумаба вводили в концентрации 150 мг/мл в виде объема 1 мл, залитого в шприц. На протяжении периода двойного слепого лечения вводили алирокумаб или плацебо подкожно в виде 1 мл инъекции каждые 2 недели, начиная с 0 недели, продолжая вплоть до последней инъекции (т. е. 76 недели), которая проводилась за 2 недели до окончания периода двойного слепого лечения. В идеальном случае IMP следовало вводить каждые две недели подкожно в примерно одинаковое время дня, однако, было допустимо проводить ее в периоде интервала, составляющего ± 3 дня.
[00280] Следующие классы лекарственных средств идентифицировали как неисследуемые медицинские продукты (NIMP), поскольку данный медицинский препарат представлял собой либо фоновую терапию, либо медицинский препарат для возможной неотложной терапии: статины (розувастатин, аторвастатин, симвастатин); ингибиторы абсорбции холестерина (эзетимиб); секвестранты, связывающие желчные кислоты (например, холестирамин, колестипол, колесевелам); никотиновая кислота; фенофибрат; омега-3 жирные кислоты (≥ 1000 мг ежедневно).
[00281] Пациентов рандомизировали, чтобы вводить либо плацебо, либо алирокумаб во время периода лечения в ходе двойного слепого исследования. Рандомизационное соотношение алирокумаб:плацебо составляло 2:1. При рандомизации проводили стратификацию в соответствии с популяцией с heFH (да, нет), острым или бессимптомным MI или ишемическим инсультом в анамнезе (да, нет), лечением статинами (аторвастатин, 40-80 мг ежедневно, или розувастатин, 20-40 мг ежедневно, в противопоставление симвастатину с любой ежедневной дозой, аторвастатину с дозой ниже 40 мг ежедневно или розувастатину с дозой ниже 20 мг ежедневно) и регионом (Северная Америка, Западная Европа, Восточная Европа и остальные страны мира).
[00282] Сопутствующий медицинский препарат представлял собой любое лечение, получаемое пациентом одновременно с исследованием (до визита последующего наблюдения). В течение исследования прием сопутствующих терапевтических препаратов должен быть сведен к минимуму. Однако, если это считалось необходимым для благополучия пациента и при малой вероятности препятствия действию IMP, их можно было давать на усмотрение исследователя при стабильной дозе (по возможности). Кроме конкретной информации, относящейся к сопутствующим медицинским препаратам, предоставленной в данном разделе, будет разрешен и должен быть зафиксирован любой другой сопутствующий медицинский препарат(препараты). Если пациент имел уровень LDL-C ≥160 мг/дл (4,14 ммоль/л) при скрининговом визите (неделя -3) и проходил лечение только статином, т. е. без дополнительной LMT, то исследователю необходимо сообщить о причине, по которой пациент не проходил вторую LMT. Нутрицевтические продукты или безрецептурные терапевтические средства, которые могут оказывать влияние на уровни липидов, разрешали лишь в том случае, если их применяли при стабильной дозе в течение по меньшей мере 4 недель до скринингового визита, во время периода скрининга и продолжали принимать в течение первых 24 недель периода двойного слепого лечения. После визита на 24 неделе модификацию этих нутрицевтических продуктов или безрецептурных терапевтических средств разрешали, но в целом ее следовало избегать. Примеры таких нутрицевтических продуктов или безрецептурных терапевтических средств включали омега-3 жирные кислоты в дозах <1000 мг, растительные станолы, такие как встречающиеся в бенеколе, льняном масле, и псиллиум.
[00283] Пациенты принимали максимально переносимые ежедневные регистрируемые дозы статинов с другой гиполипидемической терапией во время исследования или без нее. Начиная от скринингового визита (неделя -3) и вплоть до окончания первых 24 недель периода двойного слепого лечения (24 неделя), не следует менять фоновую гиполипидемическую терапию. На протяжении этого времени не должны происходить коррекция дозы, прекращение или начало получения других статинов или другой гиполипидемической терапии, кроме чрезвычайных случаев, исключительных обстоятельств, при которых особая озабоченность (включая без ограничений предупреждение об уровне триглицеридов, переданное центральной лабораторией) оправдывает такие изменения, на усмотрение исследователя.
[00284] Не были разрешены следующие виды терапии на протяжении исследования (включая период скрининга вплоть до визита последующего наблюдения): фибраты, кроме фенофибрата; продукты на основе красного ферментированного риса и статин, кроме симвастатина, аторвастатина или розувастатина.
ОЦЕНКА БЕЗОПАСНОСТИ
[00285] Безопасность оценивали по следующим параметрам: регистрация нежелательных явлений (включая установленные сердечно-сосудистые явления); результаты стандартных лабораторных тестов (гематологический анализ, биохимический анализ и анализ мочи); результаты печеночной пробы (уровни ALT, AST, щелочной фосфатазы [ALP] и общий билирубин); уровень креатинфосфокиназы (CPK); анализ на антитело к вирусу гепатита C (если положительный, то затем подтверждали повторным тестированием); уровень витамина E (альфа-токоферол) и других жирорастворимых витаминов; уровень кортизола (при необходимости, с уровнями ACTH, подтвержденными повторным анализом, и, при необходимости, с последующей стимуляционной пробой на ACTH); оценка половых гормонов; электрокардиограмма (ЭКГ); показатели жизненно важных функций (систолическое и диастолическое кровяное давление и частота сердцебиений); физикальный осмотр (включая нейрологическое исследование); проверка цветового зрения (в виде скринингового теста для более тщательного офтальмологического тестирования при необходимости). Параметры безопасности (нежелательные явления [включая установленные сердечно-сосудистые явления], данные лабораторных анализов, показатели жизненно важных функций и ЭКГ) оценивали на протяжении всего исследования.
[00286] Над пациентами, которые достигнут 2 последовательных рассчитанных уровней LDL-C <25 мг/дл (0,65 ммоль/л), будут продолжать наблюдение.
[00287] У пациентов, которые имеют титр антитела к алирокумабу, составляющий 240 или выше, при визите последующего наблюдения, будут отбирать дополнительную пробу(пробы) на антитела через 6-12 месяцев после последней дозы и впоследствии приблизительно каждые 3-6 месяцев до тех пор, пока титр не опустится ниже 240.
[00288] Конечные точки безопасности, которые оценивали в данном испытании, представляли собой сердечно-сосудистые явления; аллергические явления; местную переносимость в месте инъекции; другие нежелательные явления (включая гемолитическую анемию); результаты лабораторных тестов: анализа мочи, гематологического теста (подсчет количества эритроцитов, распределение эритроцитов по объему (RDW), количество ретикулоцитов, уровень гемоглобина, гематокрит, количество тромбоцитов, подсчет количества лейкоцитов с дифференциальным подсчетом лейкоцитов), стандартных биохимических тестов (уровни натрия, калия, хлоридов, бикарбонатов, кальция, фосфора, азота мочевины, креатинина, мочевой кислоты, общего белка, альбумина, LDH, γ-глютамилтрансферазы [γGT]), уровень витамина E (альфа-токоферола) и других жирорастворимых витаминов, уровень кортизола (и, при необходимости, уровни ACTH, подтвержденные повторным анализом, и, при необходимости, последующая стимуляционная проба на ACTH), оценка половых гормонов, анализ на антитело к вирусу гепатита C, печеночная проба (уровни ALT, AST, ALP и общий билирубин) и уровень CPK; показатели жизненно важных функций, включая частоту сердцебиений и кровяное давление; а также ЭКГ в 12 отведениях.
ПРОЦЕДУРЫ ИССЛЕДОВАНИЯ
[00289] Для всех визитов после дня 1/0 недели (визит рандомизации) был допустим временной интервал, состоящий из определенного количества дней. Период интервала для визитов в недели 12 и 24 составлял ± 3 дня, в неделю 52 он составлял ± 5 дней, а для всех остальных визитов в исследовательский центр он составлял ± 7 дней в течение периода двойного слепого лечения и периода последующего наблюдения. Для визита рандомизации (день 1/0 неделя) был допустим период интервала, составляющий +3 дня, а для скринингового визита, во время которого проходило обучение инъекции (неделя -1), он составлял ± 7 дней. Для всех визитов после телефонного звонка был допустим период интервала, составляющий ± 7 дней. Для всех визитов после дня 1/визита рандомизации, если дата одного визита была изменена, то следующий визит происходил в соответствии с исходной схемой.
ОФТАЛЬМОЛОГИЧЕСКОЕ ПОДЫССЛЕДОВАНИЕ
[00290] Несмотря на доказательства того, что отклонения в органе зрения, отмеченные во время токсикологических исследований на крысах, не были обусловлены введением алирокумаба, у подгруппы пациентов в протоколе проводили тщательные офтальмологические обследования, чтобы подтвердить безопасность алирокумаба в отношении глаз.
[00291] Главной целью была оценка клинической безопасности алирокумаба в отношении глаз у пациентов с высоким риском возникновения сердечно-сосудистых заболеваний, имеющих гиперхолестеринемию, резистентную к их LMT.
[00292] Пациентов подвергали офтальмологическим обследованиям у офтальмологов/оптометристов в период скрининга. Если пациенты считались пригодными для основного исследования и удовлетворяли дополнительным критериям соответствия для подысследования, тогда пациентов повергали офтальмологическим обследованиям каждые 6 месяцев в течение 18-месячного периода двойного слепого лечения.
[00293] Первичной конченой точкой были нежелательные явления, связанные с офтальмологическими отклонениями (c применением SMQ ʺнарушения со стороны зрительного нерваʺ, ʺнарушения со стороны сетчаткиʺ и ʺнарушения со стороны роговицыʺ), на протяжении срока исследования.
[00294] Предполагалось, что примерно 270 пациентов примут участие в данном подысследовании. Все субъекты должны были соответствовать критериям пригодности для основного исследования и дополнительным критериям пригодности, кратко изложенным для данного подысследования, чтобы быть пригодными для данного подысследования.
[00295] Единственным дополнительным критерием включения было подписывание письменного информированного согласия.
[00296] Дополнительные критерии исключения для данного подысследования представляли собой: пациента, который отозвал согласие на офтальмологическое подысследование во время периода скрининга (пациент, который не желает продолжать); пациента, который не имеет возможности завершить все офтальмологические обследования во время периода скрининга; пациента с активным заболеванием оптического нерва (например, невритом зрительного нерва), обнаруженным во время офтальмологических обследований в период скрининга. Примечание - пациент c контролируемой открытоугольной глаукомой может подходить для данного исследования; а также пациент с активным хориоретинальным заболеванием, обнаруженным во время офтальмологических обследований в период скрининга.
Статистические методы
Определение размера выборки
[00297] При оценке безопасности размер выборки, составляющий 2100 пациентов (рандомизационное соотношение 2:1, т. е. алирокумаб:1400 и плацебо:700), обеспечивает возможность сбора данных по долговременной безопасности в обширной базе данных (по меньшей мере 1000 пациентов, испытывающих воздействие алирокумаба в течение минимум 12 месяцев, из которых примерно 900 пациентов испытывали воздействие алирокумаба в течение 18 месяцев).
[00298] При таком размере выборки и следующих допущениях: срок набора пациентов в исследование составляет 16 месяцев, 44% пациентов набирают в первые 9 месяцев, и частота выбывания из исследования составляет 25% и 35% в 12 месяцев и 18 месяцев соответственно, ожидаемое количество пациентов, которые испытывали воздействие алирокумаба в течение минимум 12 месяцев и 18 месяцев лечения, приведено ниже.
[00299] Ожидаемое количество пациентов, которые испытывали воздействие алирокумаба, согласно временным рамкам анализа
a Анализ первой стадии планировали, когда примерно 600 пациентов завершили 18-месячный период двойного слепого лечения.
[00300] Кроме того, размер выборки, составляющий 1400 пациентов, которых лечили с помощью алирокумаба, обеспечивает возможность обнаружения нежелательного явления при частоте ≥0,002 с достоверностью 95% в группе алирокумаба. Предполагалось, что от 122 до 142 пациентов, испытывавших воздействие алирокумаба, будут оценивать в офтальмологическом подысследовании в течение по меньшей мере 12 месяцев, с учетом размера выборки, составляющего 270 пациентов, и допуская, что показатель прекращения составит 25% за 12 месяцев. Это обеспечит возможность обнаружения офтальмологического явления при истинном возникновении от 0,021 до 0,024 в группе алирокумаба с достоверностью 95%.
Временные рамки анализов
[00301] Анализ первой стадии включал конечные точки окончательной эффективности вплоть до 52 недели (включая первичную конечную точку эффективности на 24 неделе) и промежуточный анализ безопасности, который проводили в отношении всех данных безопасности до общей даты завершения исследования (даты, когда примерно 600 рандомизированных пациентов завершили 18 месячный период двойного слепого лечения). Анализ данных по липидам после 52 недели был описательным. Результаты представлены в данном документе.
[00302] Анализ второй стадии (окончательный анализ) будет проводиться в конце исследования и будет состоять из окончательного анализа данных по эффективности в моменты времени, которые следуют после моментов времени, проанализированных при анализе первой стадии и окончательном анализе безопасности.
Популяции для анализа
[00303] Популяцией для анализа первичной эффективности была популяция, сформированная в зависимости от назначенного лечения (ITT), определенная как все рандомизированные пациенты, которые имели оцениваемую первичную конечную точку эффективности, другими словами, имеющие доступное рассчитанное значение исходного уровня LDL-C и по меньшей мере одно доступное рассчитанное значение уровня LDL-C с одним из периодов анализа до 24 недели (включая все рассчитанные уровни LDL-C у пациентов, получающих лечение, и пациентов, не получающих лечение).
[00304] Популяцией для анализа вторичной эффективности была модифицированная популяция, сформированная в зависимости от назначенного лечения (mITT), определенная как все рандомизированные пациенты, которые принимали по меньшей мере одну дозу или часть дозы двойного слепого исследуемого медицинского продукта (IMP) и которые имели доступное рассчитанное значение уровня LDL-C на исходном уровне и по меньшей мере одно доступное рассчитанное значение уровня LDL-C в пределах одного из периодов анализа до 24 недели в ходе периода лечения для оценки эффективности. Период лечения для оценки эффективности определяли как время от момента первого введения IMP двойного слепого исследования вплоть до 21 дня после последней инъекции препарата двойного слепого исследования.
[00305] Популяция для оценки безопасности включала всех рандомизированных пациентов, которые получали по меньшей мере одну дозу или часть дозы IMP двойного слепого исследования.
Анализы эффективности
[00306] Первичные анализы конечных точек эффективности проводили с применением подхода ITT (на основании популяции ITT, определенной выше), включая все данные по уровням липидов, независимо от того, продолжал ли пациент терапию или нет. Это соответствовало оцениваемым величинам ITT, определенным для первичных и ключевых вторичных конечных точек. Кроме того, анализы также проводили с применением подхода на основе пациентов, получающих лечение (на основании популяции mITT, определенной выше), включая данные по уровням липидов, собранным в течение периода лечения для оценки эффективности. Это соответствовало оцениваемым величинам анализа у пациентов, получающих лечение, для ключевых вторичных конечных точек.
[00307] С помощью подхода ITT анализировали всех пациентов, независимо от соблюдения ими схемы лечения; с помощью него оценивали пользу стратегии лечения, и он отражает максимально возможный эффект для популяции пациентов. С помощью подхода на основе пациентов, получающих лечение, анализировали эффект лечения, ограниченный периодом, в ходе которого пациенты фактически получали лечение. С помощью него оценивали пользу, которую позволило бы достичь лечение у пациентов, соблюдающих схему лечения до рассматриваемого момента времени.
[00308] Анализы эффективности выполняли в соответствии с лечением согласно рандомизации.
[00309] Все измерения, запланированные или незапланированные, натощак или не натощак, относили к периодам анализа с целью обеспечения оценки для моментов времени от 4 недели до 78 недели.
[00310] Принимая во внимание анализ первичной эффективности (подход ITT), процентное изменение рассчитанного уровня LDL-C от исходного уровня до 24 недели анализировали с применением подхода модели со смешанными эффектами для повторных измерений (MMRM). Применяли все данные после исходного уровня, доступные для периодов анализа с 4 недели до 52 недели, и недостающие данные учитывали при помощи MMRM. Модель включала фиксированные категориальные эффекты группы лечения (плацебо в сравнении с алирокумабом), страты рандомизации (согласно IVRS), момента времени (4 неделя - 52 неделя), взаимосвязи лечение-момент времени и взаимосвязи страта-момент времени, а также непрерывные фиксированные ковариаты значения исходного уровня LDL-C и взаимосвязи значение исходного уровня-момент времени. Данная модель обеспечивала скорректированные относительно исходного уровня оценки средних значений, определенных методом наименьших квадратов (средние значения LS), на 24 неделе для обеих групп лечения с их соответствующими SE и 95% доверительными интервалами. Для сравнения группы алирокумаба с группой плацебо использовали соответствующее противопоставляющее утверждение для тестирования отличий данных оценок при 5% альфа-уровне значимости.
[00311] Была определена иерархическая процедура для тестирования ключевых вторичных конечных точек при контроле множественности сравнений (с применением приведенного выше порядка ключевых вторичных конечных точек). Первая ключевая вторичная конечная точка представляла собой процентное изменение рассчитанного уровня LDL-C от исходного уровня до 24 недели с применением подхода на основе пациентов, получающих лечение.
[00312] Непрерывные вторичные переменные, предположительно имеющие нормальное распределение (т. e. уровни липидов, отличных от TG и Lp(a)), анализировали с применением такой же модели MMRM, как для первичной конечной точки. Непрерывные конечные точки, предположительно имеющие ненормальное распределение (т. e. уровни TG и Lp(a)) анализировали с применением подхода множественного восстановления для обработки пропущенных значений с последующим применением модели устойчивой регрессии с конечной точкой, представляющей интерес, в качестве переменной отклика с применением M-оценки (с применением процедуры SAS ROBUSTREG), с группой лечения, типическими группами рандомизации (согласно IVRS) и соответствующим(соответствующими) значением(значениями) исходного уровня в качестве эффектов для сравнения эффектов лечения. Обеспечивали объединенную оценку среднего значения в обеих группах лечения, а также различия данных оценок с их соответствующими SE, 95% CI и p-значением (посредством процедуры SAS MIANALYZE).
[00313] Бинарные вторичные конечные точки эффективности анализировали с применением подхода множественного восстановления для обработки пропущенных значений, с последующим применением стратифицированной логистической регрессии, при этом группа лечения выступала в качестве основного эффекта, а соответствующее значение(значениями) исходного уровня в качестве ковариаты, при стратификации по факторам рандомизации (согласно IVRS). Обеспечивали объединенные оценки соотношения шансов в сравнении с плацебо, 95% CI и p-значение (посредством процедуры SAS MIANALYZE).
Анализы безопасности
[00314] Анализы безопасности были описательными, их проводили на популяции для оценки безопасности согласно фактически полученному лечению. Анализ безопасности был сосредоточен на периоде появления TEAE, который определяли как время от первой дозы инъекции двойного слепого исследования вплоть до 70 дней после последней инъекции двойного слепого исследования. TEAE, которое развилось, ухудшилось или стало серьезным, или PCSA, возникшие после первой инъекции пациента в открытом расширенном исследовании (LTS13643), не считались произошедшими в период появления TEAE. Период появления TEAE ограничивали общей датой завершения исследования.
РЕЗУЛЬТАТЫ I- АНАЛИЗА С ПРЕДВАРИТЕЛЬНО ЗАДАННЫМИ ПАРАМЕТРАМИ
Учет пациентов
[00315] Проводили промежуточный анализ с предварительно заданными характеристиками, когда все пациенты достигали периода лечения в один год и примерно 25 процентов пациентов достигали периода лечения в 18 месяцев. Из 2341 рандомизированного пациента три пациента не проходили лечение и, следовательно, их не включали в популяцию для оценки безопасности.
[00316] Тридцать одного рандомизированного пациента не включали в популяцию ITT (отсутствует значение LDL-C в пределах одного из периодов анализа до 24 недели).
[00317] Сорок одного рандомизированного пациента исключили из популяции mITT (пациенты, исключенные из популяции ITT, и пациенты с отсутствием значения LDL-C в пределах одного из периодов анализа до 24 недели в ходе периода лечения для оценки эффективности).
Таблица 29 Популяции для анализа
Для остальных популяций данные пациентов сводили в таблицу в соответствии с их лечением, назначенным при рандомизации.
Распределение для исследования
[00318] На дату завершения в данном анализе с предварительно заданными параметрами 607 (25,9%) пациентов полностью завершили 18-месячный период двойного слепого лечения (определенный как осуществление воздействия по меньшей мере 76 недель и визит на 78 неделе), включая по меньшей мере 400 пациентов в группе алирокумаба, по договоренности с органами здравоохранения во время консультации относительно встречи по окончанию 2 фазы: 405 пациентов (26,1%) в группе алирокумаба и 202 пациента (25,6%) в группе плацебо.
[00319] В группе алирокумаба 1550 из 1553 действительно получали алирокумаб. 23% (n=349) этих пациентов полностью прошли 78 недель, 20% (n=311) прекратили лечение, а 57% (n=890) еще проходят лечение. Пациенты из группы алирокумаба, которые прекратили лечение, поступили так из-за плохого соблюдения протокола (n=54), нежелательного явления (n=98) или по другой причине (n=159). Популяции для оценки безопасности ITT для группы алирокумаба состояли из 1530 и 1550 пациентов соответственно. В группе плацебо все 788 получали плацебо. 22% (n=176) этих пациентов полностью прошли 78 недель, 19% (n=146) прекратили лечение, а 59% (n=466) еще проходят лечение. Пациенты из группы плацебо, которые прекратили лечение, поступили так из-за плохого соблюдения протокола (n=34), нежелательного явления (n=44) или по другой причине (n=67). Популяции для оценки безопасности ITT для группы алирокумаба состояли из 780 и 788 пациентов соответственно. Пациентов из популяции ITT учитывали во всех конечных точках эффективности вплоть до 52 недели. Пациенты в популяции для оценки безопасности получали либо алирокумаб, либо плацебо в течение по меньшей мере 52 недель.
[00320] Распределение для исследования, анализы воздействия и безопасности оценивали с использованием всех данных на момент общей даты завершения исследования, и они, следовательно, включали данные после 52 недели и вплоть до 78 недели или вплоть до визита последующего наблюдения. Окончательные результаты первичной конечной точки эффективности (на 24 неделе) и основные вторичные конечные точки эффективности (оцененные вплоть до 52 недели) предоставлены в данном анализе первой стадии.
[00321] Всего было 525 (22,4%) рандомизированных пациентов, которые завершили данный период исследуемого лечения вплоть до даты завершения в данном анализе с предварительно заданными параметрами, т. е. когда делали последнюю инъекцию IMP (76 неделя) и участвовали в последнем визите в целях лечения (78 неделя) в течение 21 дня после последней инъекции IMP и по меньшей мере 525 дней после рандомизации.
[00322] Двойное слепое введение IMP преждевременно прекратили 455 (19,4%) рандомизированных пациентов (310 (20,0%) пациентов в группе алирокумаба и 145 (18,4%) пациентов в группе плацебо). Для 1358 (58,0%) рандомизированных пациентов лечение продолжалось во время даты завершения анализа первой стадии.
[00323] В целом, основными причинами для прекращения исследуемого лечения были "другие причины - другое" и "нежелательное явление". Эти "другие причины" включали следующее: субъект отказался от продолжения (93 пациента), пациент не отвечал критериям eCRF в отношении завершения лечения вследствие одного из пропущенного/находящегося за пределами интервала визита 78 недели (71 пациент), смерти (6 пациентов) (при этом отсутствовало решение исследователя прекратить лечение до наступления смерти), разнообразных причин (5 пациентов), утраты контакта для последующего наблюдения (4 пациента), закрытия исследовательского центра (3 пациента) и переезда субъекта (2 пациента).
[00324] Кроме того, среди этих 455 пациентов, которые преждевременно прекратили лечение, 219 (14,1%) рандомизированных пациентов преждевременно прекратили прием двойного слепого IMP до визита 52 недели в группе алирокумаба и 111 (14,1%) в группе плацебо.
[00325] Наконец, 139 (5,9%) пациентов приняли участие в необязательном офтальмологическом подысследовании (вместо запланированных 270 пациентов).
[00326] В этом анализе первой стадии окончательные результаты доступны для первичной конечной точки эффективности в 24 неделю и ключевых вторичных конечных точек эффективности, оцениваемых в 12 неделю, 24 неделю и 52 неделю. На 24 неделе первичная конечная точка эффективности была доступна для 1384 (90,5%) в группе алирокумаба и 708 (90,8%) в группе плацебо.
[00327] Первичная конечная точка эффективности была пропущена для 218 пациентов. При визите на 24 неделе причины наличия пропущенных данных были следующими: 108 образцов не были собраны из-за преждевременного прекращения исследования; 47 образцов были собраны за пределами периодов анализа; 10 образцов не были собраны; 51 образец был собран, но измерения нельзя было произвести (недостаточное количество, TG>400 мг/дл (4,52 ммоль/л) и т. д.); и 2 образца не были собраны у пациентов, которых рандомизировали, но лечение которых не проводили.
Демографические данные и характеристики на исходном уровне
[00328] В общей сложности в данном исследовании рандомизировали 2341 пациента (1553 в группе алирокумаба в сравнении с 788 в группе плацебо). Демографические характеристики, характеристики заболеваний и параметры уровней липидов на исходном уровне были похожи у группы алирокумаба при сравнении с группой плацебо. Для группы алирокумаба (N=1553) возраст (среднее число лет (SD)) составлял 60,4 (10,4), процент мужчин составлял 63,3% (N=983), процент представителей белой расы составлял 92,8% (N=1441), и BMI (среднее значение, кг/м2 (SD)) составлял 30,2 (5,7). Для группы плацебо (N=788) возраст (среднее число лет (SD)) составлял 60,6 (10,4), процент мужчин составлял 60,2% (N=474), процент представителей белой расы составлял 92,6% (N=730), и BMI (среднее значение, кг/м2 (SD)) составлял 30,5 (5,5).
[00329] 17,7% пациентов (17,8% (N=276) из пациентов группы алирокумаба и 17,6% (N=139) из группы плацебо) имели heFH с установленным CHD, или эквивалентными рисками возникновения CHD, или без них. Включали 82,3% пациентов с гиперхолестеринемией несемейного происхождения с установленным CHD или эквивалентными рисками возникновения CHD. Подавляющее большинство в рандомизированной популяции (90,6%) имело коронарное заболевание сердца (CHD) или эквивалентные риски возникновения CHD. CHD регистрировали у 68,6% пациентов. 91,5% пациентов классифицировали как имеющих очень высокий риск (см. примечание ниже).
[00330] Демографические характеристики, характеристики заболеваний и параметры уровней липидов на исходном уровне были похожи у группы алирокумаба при сравнении с группой плацебо. Среднее значение (SD) рассчитанного исходного уровня LDL-C составляло 122,4 (42,2) мг/дл (3,171 (1,092) ммоль/л). Что касается фоновой гиполипидемической терапии, 1032 (44,1%) пациентов принимали статины в высокой дозе при рандомизации (т. е. аторвастатин в дозе 40-80 мг или розувастатин в дозе 20-40 мг ежедневно) и 334 (14,3%) получали эзетимиб в дополнение к статину.
[00331] Обзор исходных характеристик групп алирокумаба и плацебо изложен в таблице 34.
[00332] Период проведения инъекций был аналогичным среди групп лечения с медианным значением проведения, составлявшим 68 недель. К дате завершения анализа (7 мая 2014 года) 607 (25,9%) пациентов полностью завершили 18-месячный период двойного слепого лечения (т. е. завершили по меньшей мере 76 недель воздействия и визит на 78 неделе), включая по меньшей мере 400 пациентов в группе алирокумаба, по договоренности с органами здравоохранения во время консультации относительно встречи по окончанию 2 фазы: 405 пациентов (26,1%) в группе алирокумаба и 202 пациента (25,6%) в группе плацебо.
Таблица 30 Характеристики заболеваний и другие соответственные исходные данные - рандомизированная популяция
(N=788)
(N=1553)
(N=2341)
Таблица 31 Медицинский анамнез, представляющий особый интерес: сердечно-сосудистые нарушения в анамнезе и факторы риска - рандомизированная популяция
(N=788)
(N=1553)
(N=2341)
a в соответствии с пунктами, предварительно перечисленными в e-CRF.
b диагностированное с помощью инвазивного или неинвазивного тестирования.
PAD определено как перемежающаяся хромота вместе с лодыжечно-брахиальным индексом ≤ 0,90 или вместе с процедурой реваскуляризации/хирургией периферических сосудов или критическая ишемия конечностей вместе с процедурой реваскуляризации/хирургией периферических сосудов или тромболизисом.
Очень высокий риск возникновения CV определен как пациенты с heFH, которые имеют CHD или эквивалентные риски возникновения CHD, или пациенты с не-FH.
Высокий риск возникновения CV определен как пациенты с heFH, которые не имеют CHD или эквивалентные риски возникновения CHD.
[00333] Примечание: 219 пациентов никогда не имели ни коронарного заболевания сердца в медицинском анамнезе, ни эквивалентных рисков коронарного заболевания сердца в медицинском анамнезе. 200 из них представляли собой пациентов с heFH (относятся к категории высокого риска возникновения CV). Остальные 19 представляли собой пациентов с не-FH без установленного CHD в анамнезе или эквивалентных рисков возникновения CHD (т. е. имели критерий исключения E01). По согласию на уровне программы этих 19 пациентов с отклонением учитывали в категории с очень высоким риском возникновения CV.
[00334] Далее обобщены данные по гиполипидемической терапии (LMT), включающей статин, в анамнезе в момент скрининга: 1245 (53,2%) пациентов не принимали аторвастатин в дозе 40-80 мг, розувастатин в дозе 20-40 мг или симвастатин в дозе 80 мг ежедневно. Самой частой причиной была региональная практика и местный вариант инструкции по применению. Среди 656 (28,0%) пациентов, которые не принимали статин в высокой дозе или симвастатин в дозе 80 мг ежедневно из-за региональной практики/местного варианта инструкции по применению, 375 (57,2%) пациентов принимали симвастатин в дозе 40 мг.
Таблица 32 Фоновая LMT при рандомизации - рандомизированная популяция
(N=788)
(N=1553)
(N=2341)
Статин в высокой дозе соответствует 40-80 мг аторвастатина ежедневно или 20-40 мг розувастатина ежедневно.
Примечание: два пациента, по одному в каждой группе, не получали статин на момент рандомизации и получали IMP (вплоть до 10 недели в случае одного пациента и вплоть до 74 недели в случае другого пациента).
Таблица 33 Параметры эффективности липидного обмена на исходном уровне - обобщенные результаты количественной оценки в стандартных единицах - рандомизированная популяция
(N=788)
(N=1553)
(N=2341)
Примечание: измеренный уровень LDL-C оценивали посредством способа количественного определения бета-липопротеинов.
Таблица 34 Обзор исходных характеристик для групп алирокумаба и плацебо
(n=1553)
(n=788)
среднее значение(SD), ммоль/л [мг/дл]
[122,7 (42,6)]
[121,9 (41,4)]
среднее значение (SD), мг/дл
[152,6 (46,6)]
[152,0 (45,8)]
Все пациенты на фоне терапии с максимально переносимой дозой статинов ± другой липид-корригирующей терапией. †Пациенты должны были получать либо 20-40 мг розувастатина, 40-80 мг аторвастатина ежедневно, либо 80 мг симвастатина ежедневно, если только они не являются непереносимыми, и/или соответствующую другую дозу, которая назначалась в соответствии с решением исследователя.
‡Статин в высокой дозе: 40-80 мг аторвастатина или 20-40 мг розувастатина ежедневно.
a Высокая доза статина: 40-80 мг аторвастатина, 20-40 мг розувастатина ежедневно или 80 мг симвастатина ежедневно.
Дозировка и длительность
[00335] Период проведения инъекций был аналогичным среди групп лечения с медианным значением проведения, составлявшим 68 недель. Следует отметить, что 817 (35,0%) пациентов получали лечение в течение по меньшей мере 76 недель (543 (35,0%) пациентов в группе алирокумаба и 274 (34,6%) пациентов в группе плацебо). Из этих 817 пациентов 607 пациентов также совершили визит 78 недели ко времени даты завершения анализа первой стадии.
Первичная конечная точка эффективности
[00336] Анализ ITT включает все значения уровня LDL-C, полученные у пациентов, получающих лечение, и у пациентов, не получающих лечение, вплоть до 52 недели. Анализ первичной конечной точки (процентное изменение рассчитанных уровней LDL-C от исходного уровня до 24 недели) представлен на основе модели MMRM в отношении популяции ITT с использованием показателей средних значений, определенных методом наименьших квадратов, на 24 неделе. Для 146 (9,5%) пациентов в группе алирокумаба и 72 (9,2%) пациентов в группе плацебо не было получено рассчитанное значение LDL-C на 24 неделе. Эти пропущенные значения учитывали при помощи модели MMRM.
[00337] Результаты анализа первичной конечной точки представлены в таблице 35, в виде ммоль/л и мг/дл.
[00338] Наблюдали статистически значимое процентное снижение уровня LDL-C от исходного уровня до 24 недели в группе алирокумаба (среднее значение, определенное методом наименьших квадратов, среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -61,0%) по сравнению с группой плацебо (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем 0,8%) (различие для среднего значения, определенного методом наименьших квадратов, в сравнении с плацебо составило -61,9%, p<0,0001).
[00339] В группе алирокумаба стабильное снижение уровня LDL-C от исходного уровня наблюдали с 4 недели до 52 недели. Значения рассчитанных уровней LDL-C, изменение от исходного уровня и процентное изменение от исходного уровня для этого периода представлены в таблице 36 и на фигуре 11 (анализ ITT всех доступных измерений за 52-недельный период наблюдений), и они демонстрируют заметное отличие между группами, которые лечили с помощью алирокумаба и плацебо.
Таблица 35 Процентное изменение от исходного уровня у рассчитанных уровней LDL-C на 24 неделе: MMRM - анализ ITT - популяция ITT
(N=780)
(N=1530)
Модель MMRM и описание исходного уровня проводили в отношении пациентов со значением на исходном уровне и значением после исходного уровня по меньшей мере в одном из периодов анализа, используемых в модели.
За p-значением следует '*', если оно является статистически значимым согласно фиксированному иерархическому подходу, применяемому для обеспечения строгого контроля oбщего показателя ошибки I рода на уровне 0,05
Таблица 36 Рассчитанные уровни LDL-C в зависимости от времени - анализ ITT- популяция ITT
(N=780)
(N=1530)
Примечание: средние значения, определенные методом наименьших квадратов, стандартные ошибки (SE) и p-значение, полученные из анализа на основе MMRM (модели со смешанными эффектами для повторных измерений). Модель включает фиксированные категориальные эффекты группы лечения, страты рандомизации согласно IVRS, момента времени, взаимосвязи лечение-момент времени, взаимосвязи страта-момент времени, а также непрерывные фиксированные ковариаты значения исходного уровня LDL-C и взаимосвязи исходный уровень LDL-C-момент времени.
Модель MMRM и описание исходного уровня проводили в отношении пациентов со значением на исходном уровне и значением после исходного уровня по меньшей мере в одном из периодов анализа, используемых в модели.
[00340] Следует отметить, что в рамках проверки предположений модели была проведена оценка однородности эффекта лечения для всех исходных уровней липидов и показана значимая взаимосвязь лечение-исходный уровень LDL-C (p<0,0001). Для дальнейшего исследования этой взаимосвязи ниже представлен эффект лечения в отношении первичной конечной точки эффективности согласно 4 классам исходных уровней LDL-C. Показан сниженный эффект лечения в сравнении с плацебо для более высоких исходных уровней LDL-C (от -75,0% в случае пациентов с исходным уровнем LDL-C < 100 мг/дл до -41,3% в случае пациентов с уровнем LDL-C ≥ 160 мг/дл). Это наблюдение, по-видимому, обусловлено изменениями в группе плацебо, в которой процентное изменение уровня LDL-C от исходного уровня до 24 недели снижается с возрастанием категорий исходного уровня LDL-C. И наоборот, процентное изменение уровня LDL-C от исходного уровня до 24 недели стабильно у всех различных подкатегорий уровней LDL-C в пределах группы алирокумаба.
Таблица 37 Процентное изменение от исходного уровня у рассчитанных уровней LDL-C на 24 неделе: анализ подгрупп в соответствии с рассчитанными уровнями LDL-C на исходном уровне - анализ ITT
Процентное изменение от исходного уровня у рассчитанных уровней LDL-C на 24 неделе (%)
(N=780)
(N=1530)
Модель включает фиксированные категориальные эффекты группы лечения, страты рандомизации cогласно IVRS, фактор подгруппы, момент времени и взаимосвязи лечение-момент времени, страта-момент времени, фактор подгруппы-момент времени, группа лечения-фактор подгруппы и группа лечения-фактор подгруппы-момент времени для исходного уровня LDL-C в качестве фактора подгруппы.
Что касается других факторов подгруппы, модель также включает непрерывные фиксированные ковариаты значения исходного уровня LDL-C и взаимосвязь значение исходного уровня LDL-C-момент времени.
Число соответствует числу пациентов со значением на исходном уровне и значением после исходного уровня по меньшей мере в одном из периодов анализа, используемых в модели.
p-значение представлено только для наглядности.
Ключевые вторичные конечные точки эффективности
[00341] В таблице 38 обобщены результаты анализа ключевых вторичных конечных точек в иерархическом порядке. Все ключевые вторичные конечные точки эффективности являются статистически значимыми в соответствии c процедурой иерархического тестирования.
Таблица 38
Таблица 39 Процентное изменение от исходного уровня параметров уровня липидов на 24 неделе (популяция ITTa)
(N=780)
(N=1530)
Сокращения: CI, доверительные интервалы; HDL, липопротеин высокой плотности; ITT, популяция, сформированная в зависимости от назначенного лечения; LDL, липопротеин низкой плотности; LS, метод наименьших квадратов; LLT, липид-корригирующая терапия; SD, стандартное отклонение; SE, стандартная ошибка.
a Первичный и вторичный анализы эффективности проводили с применением подхода ITT, включающего пациентов со значением рассчитанного уровня холестерина LDL на исходном уровне и по меньшей мере одним рассчитанным значением уровня холестерина LDL в ходе лечения или во время перерывов в лечении в пределах одного из периодов анализа вплоть до 24 недели. Средние значения, определенные методом наименьших квадратов, SE и p-значение, полученные из анализа на основе модели со смешанными эффектами для повторных измерений. p-значения являются статистически значимым согласно фиксированному иерархическому подходу, применяемому для обеспечения контроля oбщего показателя ошибки 1 рода на уровне 0,05.
b Объединенная оценка скорректированного среднего значения (SE) показана для липопротеина (a) и триглицеридов.
C Уровень холестерина LDL измеряли посредством количественного определения бета-липопротеинов.
Таблица 40 Процентное изменение от исходного уровня параметров уровня липидов на 24 неделе - анализ у пациентов, получающих лечениеa
(N=777)
(N=1523)
Сокращения: CI, доверительные интервалы; HDL, липопротеин высокой плотности; LDL, липопротеин низкой плотности; LS, метод наименьших квадратов; LLT, липид-корригирующая терапия; SE, стандартная ошибка.
a В анализ у пациентов, получающих лечение, включали только пациентов, которые получали исследуемое лечение. Из анализа у пациентов, получающих лечение, исключали любых пациентов, не имеющих доступного значения рассчитанного уровня холестерина LDL в период от первой дозы исследуемого лечения вплоть до 21 дня после последней инъекции.
b Объединенная оценка скорректированного среднего значения (SE) показана для липопротеина (a) и триглицеридов.
Р-значения, полученные из анализа на основе модели со смешанными эффектами для повторных измерений.
Таблица 41 Процентное изменение от исходного уровня рассчитанных уровней LDL-C на 24 неделе (метод анализа паттернов с помощью смешанной моделиa)
(N=780)
(N=1523)
Сокращения: CI, доверительные интервалы; LDL, липопротеин низкой плотности; LS, метод наименьших квадратов; LLT, липид-корригирующая терапия; SE, стандартная ошибка.
a Этот анализ чувствительности был проведен для дополнительной оценки влияния недостающих данных на первичную конечную точку: в данном подходе недостающие значения рассчитанного уровня холестерина LDL в течение периода "получения лечения" подвергали множественному восстановлению с применением модели, присваивающей недостающее случайным образом, и недостающие значения рассчитанного уровня холестерина LDL в течение периода после завершения лечения подвергали множественному восстановлению с применением случайного выбора из нормального распределения, при этом среднее значение равно собственному значению субъекта на исходном уровне.
[00342] Анализы для каждой ключевой вторичной конечной точки подробно описаны в данном документе.
[00343] Анализ процентного изменения рассчитанного уровня LDL-C от исходного уровня до 24 недели у пациентов, получающих лечение, показывает результаты, которые согласуются с результатами анализа ITT, при этом различие для среднего значения, определенного методом наименьших квадратов, в сравнении с плацебо составило -63,5% в анализе в ходе лечения в сравнении с -61,9% в анализе ITT. Действительно, несколько пациентов имели значения уровня LDL-C, собранные после завершения лечения (т. е. более чем через 21 день после последней инъекции на 24 неделе: 37 пациентов (2,4%) в группе алирокумаба в сравнении с 20 (2,6%) в группе плацебо. Процентное изменение от исходного уровня параметров уровней липидов на 24 неделе в популяции ITT и в популяции пациентов, получающих лечение, можно увидеть в таблице 39 и таблице 40, соответственно. Метод анализа паттернов с помощью смешанной модели процентного изменения от исходного уровня у рассчитанных уровней LDL-C на 24 неделе можно видеть в таблице 41.
[00344] Анализ ITT и анализ процентного изменения рассчитанного уровня LDL-C от исходного уровня до 12 недели у пациентов, получающих лечение, также показали согласующиеся результаты.
[00345] На 24 неделе эффект лечения в отношении измеренных уровней LDL-C является сравнимым с эффектом в отношении рассчитанных уровней LDL-C. Результаты в отношении измеренных уровней LDL-C будут исследоваться дополнительно, в том числе корреляция между рассчитанными и измеренными уровнями LDL-C.
[00346] Анализ ITT проводили в отношении уровней Apo B, не-HDL-C и общего-C на 12 и 24 неделях. Анализ у пациентов, получающих лечение, также проводили на 24 неделе для уровней Apo B и не-HDL-C. Результаты анализа ITT и анализа в ходе лечения были аналогичными для этих 2 параметров уровней липидов. Группа алирокумаба характеризовалась значительным снижением уровней не-HDL-C, Apo B и Lp(a) относительно плацебо на 24 неделе.
[00347] Согласно анализу ITT на 24 неделе 80,7% пациентов с очень высоким риском возникновения CV или высоким риском возникновения CV достигали рассчитанного уровня LDL-C < 70 мг/дл или < 100 мг/дл соответственно в группе алирокумаба в сравнении с 8,5% в группе плацебо (p < 0,0001). Что касается анализа пациентов, получающих лечение, на 24 неделе, 82,8% пациентов с очень высоким риском возникновения CV или высоким риском возникновения CV достигали рассчитанного уровня LDL-C < 70 мг/дл или < 100 мг/дл соответственно в группе алирокумаба в сравнении с 8,5% в группе плацебо.
[00348] Долю пациентов, достигающих рассчитанного уровня LDL-C <70 мг/дл, исследовали при анализе ITT и анализе пациентов, получающих лечение, на 24 неделе. 79,3% пациентов из группы алирокумаба достигали этого уровня при анализе ITT (81,2% при анализе пациентов, получающих лечение) в сравнении с 8,0% в группе плацебо при анализе ITT (8,0% в анализе пациентов, получающих лечение).
Первичная конечная точка эффективности в соответствии со статусом подгруппы
[00349] Как описано далее, анализировали сравнения первичной конечной точки эффективности у различных подгрупп, включающих статус HeFH, демографические группы, медицинский анамнез и пациентов, которые достигали последовательных уровней LDL-C <25 мг/дл.
[00350] Сравнение процентного изменения уровней LDL-C от исходного уровня до 24 недели у пациентов, которые имеют гетерозиготную семейную гиперхолестеринемию (ʺHeFHʺ), и пациентов, не имеющих ее, представлено в таблице 43 и на фигурах 12A-C. На фигуре 12B показано сравнение популяций пациентов с HeFH, которые имеют исходные уровни LDL-C, составляющие <160 мг/дл или ≥ 160 мг/дл; на фигуре 12C показано сравнение популяций пациентов с HeFH, которые имеют исходные уровни LDL-C, составляющие <190 мг/дл или ≥ 190 мг/дл. Все пациенты получали фоновое лечение, включающее максимально переносимые дозы статинов и необязательно виды дополнительной липид-корригирующей терапии. Снижение процентного изменения уровней LDL-C от исходного уровня до 24 недели было статистически значимым в группах алирокумаба по сравнению с группами плацебо, независимо от того, имели ли пациенты HeFH (см. фигуру 12A). Снижение процентного изменения уровней LDL-C от исходного уровня до 24 недели также было статически значимым в группах алирокумаба по сравнению с группами плацебо, независимо от того имели ли пациенты с HeFH уровни LDL-C ≥ 160 мг/дл или <160 мг/дл (см. фигуру 12B) или уровни LDL-C ≥ 190 мг/дл или <190 мг/дл (см. фигуру 12C).
[00351] Отличие, наблюдаемое для процентного изменения средних значений, определенных методом наименьших квадратов (SE), от исходного уровня до 24 недели для уровней LDL-C у пациентов, получавших алирокумаб (по сравнению с пациентами, получавшими плацебо), также согласовывалось в случае пациентов с уровнями PCSK9, которые были меньше или больше медианного значения общей или свободной PCSK9. Медианные значения уровней общей PCSK9 составляли 637 нг/мл в группе алирокумаба и 640 нг/мл в группе плацебо. Уровни общей PCSK9 определяют как уровни всей циркулирующей PCSK9, которая включает PCSK9, которая является свободной (несвязанной), а также PCSK9, которая связана антителами к PCSK9 (и является неактивной). В случае пациентов с уровнями общей PCSK9 ниже медианного значения процентное изменение от исходного уровня рассчитанных уровней LDL-C было значимо больше в группе алирокумаба (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -60,1%) по сравнению с группой плацебо (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -2,8%) (различие для среднего значения, определенного методом наименьших квадратов, в сравнении с плацебо составило -57,3%, p < 0,0001). В случае пациентов с уровнями общей PCSK9, равными и превышающими медианное значение, процентное изменение от исходного уровня рассчитанных уровней LDL-C было значимо больше в группе алирокумаба (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -62,1%) по сравнению с группой плацебо (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем 4,1%) (различие от среднего значения, определенного методом наименьших квадратов, в сравнении с плацебо составило -66,0%, p < 0,0001). Процентное изменение уровней LDL-C от исходного уровня до 24 недели показывало значимо большее снижение у субъектов с уровнями общей PCSK9, равными или превышающими медианное значение, по сравнению с субъектами, у которых уровни общей PCSK9 ниже медианного значения (p-значение взаимосвязи 0,0003).
[00352] Аналогично, в случае пациентов с уровнями свободной PCSK9 ниже медианного значения процентное изменение от исходного уровня рассчитанных уровней LDL-C было значимо больше в группе алирокумаба (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -59,4%) по сравнению с группой плацебо (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -1,6%) (различие для среднего значения, определенного методом наименьших квадратов, в сравнении с плацебо составило -58,0%, p < 0,0001). В случае пациентов с уровнями свободной PCSK9, равными и превышающими медианное значение, процентное изменение от исходного уровня рассчитанных уровней LDL-C было значимо больше в группе алирокумаба (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -62,7%) по сравнению с группой плацебо (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем 3,0%) (различие для среднего значения, определенного методом наименьших квадратов, в сравнении с плацебо составило -66,0%, p < 0,0001). Процентное изменение уровней LDL-C от исходного уровня до 24 недели показывало значимо большее снижение у субъектов с уровнями свободной PCSK9, равными или превышающими медианное значение, по сравнению с субъектами, у которых уровни свободной PCSK9 ниже медианного значения (p-значение взаимосвязи 0,0015).
[00353] Снижение процентного изменения уровней LDL-C от исходного уровня до 24 недели, наблюдаемое в данном исследовании, было также статистически значимым в группах алирокумаба по сравнению с группами плацебо как у мужчин, так и у женщин. У мужчин процентное изменение от исходного уровня рассчитанных уровней LDL-C было значимо больше в группе алирокумаба (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -65,5%) по сравнению с группой плацебо (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -0,7%) (различие для среднего значения, определенного методом наименьших квадратов, в сравнении с плацебо составило -64,7%, p < 0,0001). У женщин процентное изменение от исходного уровня рассчитанных уровней LDL-C было значимо больше в группе алирокумаба (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -53,4%) по сравнению с группой плацебо (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем 3,2%) (различие для среднего значения, определенного методом наименьших квадратов, в сравнении с плацебо составило -56,6%, p < 0,0001). Процентное изменение уровней LDL-C от исходного уровня до 24 недели показало статистически большее снижение у мужчин, чем у женщин (p-значение взаимосвязи 0,0014).
[00354] Снижение процентного изменения уровней LDL-C от исходного уровня до 24 недели, наблюдаемое в исследовании ODDYSEY LONG TERM, также было статистически значимым в группах алирокумаба по сравнению с группами плацебо у различных подгрупп, включающих подгруппы, обусловленные расой, возрастом, BMI, умеренным хроническим заболеванием почек, статусом диабета и исходными уровнями TG натощак (см. фигуру 13), различными терапиями статинами и липид-корригирующими терапиями (LLT) (см. фигуру 14). Все пациенты получали фоновое лечение статином (на максимально переносимом уровне). Подгруппа пациентов также получала дополнительную липид-корригирующую терапию.
[00355] Два последовательных рассчитанных значения уровня LDL-C <25 мг/дл (< 0,65 ммоль/л) наблюдали у 562 (37,4%) пациентов. Проводили анализ в отношении частоты TEAE (PT) в сопоставлении со всей группой алирокумаба для оценки выраженных дисбалансов. TEAE, которые представляли собой AESI, которые встречались у пациентов с двумя последовательными рассчитанными значениями уровня LDL-C <25 мг/дл (< 0,65 ммоль/л), сравнивали в отношении их частоты в общей группе алирокумаба. Наконец, были пересмотрены теоретические AE, чтобы убедиться, что не существует сообщений о таких терминах, и в случае присутствия их дополнительно исследовали на данных на уровне пациента. Никакого особого беспокойства относительно безопасности на основании такого анализа у этих пациентов не наблюдали.
Таблица 42 Число (%) пациентов с 2 последовательными уровнями LDL-C < 25 мг/дл (<0,65 ммоль/л) в течение периода лечения - популяция для оценки безопасности (во время анализа с предварительно заданными параметрами)
(N=788)
(N=1550)
Знаменатель (/N) в пределах группы лечения представляет собой число пациентов в группе лечения, которые имели по меньшей мере два значения рассчитанного уровня LDL-C, определенные с промежутком по меньшей мере 21 день в период оценки эффективности.
1 2 последовательных значения берутся во внимание, если их разделяет по меньшей мере 21 день.
2 Первое значение рассчитанного уровня LDL-C <25 или <15 мг/дл среди первых 2 последовательных значений рассчитанного уровня LDL-C <25 или <15 мг/дл для пациента
Таблица 43 Процентное изменение от исходного уровня рассчитанных уровней LDL-C на 24 неделе: в соответствии со статусом heFH при рандомизации - популяция ITT
Процентное изменение от исходного уровня у рассчитанных уровней LDL-C на 24 неделе (%)
(N=780)
(N=1530)
Модель включает фиксированные категориальные эффекты группы лечения, страты рандомизации согласно IVRS, фактор подгруппы, момент времени и взаимосвязи лечение-момент времени, типическая группа-момент времени, фактор подгруппы-момент времени, группа лечения-фактор подгруппы и группа лечения-фактор подгруппы-момент времени для исходного уровня LDL-C, а также фиксированные ковариаты значения исходного уровня LDL-C и взаимосвязь значение исходного уровня LDL-C-момент времени.
Число соответствует числу пациентов со значением на исходном уровне и значением после исходного уровня по меньшей мере в одном из периодов анализа, используемых в модели.
p-значение представлено только для наглядности.
Обобщенные результаты в отношении эффективности
[00356] На 24 неделе процентное изменение от исходного уровня у рассчитанного уровня LDL-C в популяции ITT было значимо больше в группе алирокумаба (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем -61,0%) по сравнению с группой плацебо (среднее значение, определенное методом наименьших квадратов, в сравнении с исходным уровнем 0,8%) (различие для среднего значения, определенного методом наименьших квадратов, в сравнении с плацебо составило -61,9%, p < 0,0001) (см. таблицу 35). Анализ процентного изменения уровня LDL-C от исходного уровня до 24 недели у пациентов, получающих лечение, показывает результаты, которые согласуются с результатами анализа ITT, при этом различие для среднего значения, определенного методом наименьших квадратов, в сравнении с плацебо составило -63,5% в анализе в ходе лечения (p<0,0001). Действительно, на 24 неделе небольшое число пациентов (всего 2,5% пациентов) имели значения уровня LDL-C, собранные после завершения лечения (т. е. более чем через 21 день после последней инъекции). На 24 неделе эффект лечения в отношении измеренных уровней LDL-C (среднее значение, определенное методом наименьших квадратов, в сравнении со значением исходного уровня -61,3%) является сравнимым с эффектом в отношении рассчитанных уровней LDL-C (анализ ITT).
[00357] В группе алирокумаба стабильное снижение уровня LDL-C от исходного уровня наблюдали с 4 недели до 52 недели.
[00358] Оценивали однородность эффекта лечения для всех исходных уровней липидов, и была показана значимая взаимосвязь лечение-исходный уровень LDL-C (p<0,0001). По-видимому, данное наблюдение обусловлено изменениями в группе плацебо. Действительно, изменение уровня LDL-C от исходного уровня до 24 недели стабильно у всех различных подкатегорий уровней LDL-C в пределах группы алирокумаба. Процентное изменение среднего значения, определенного методом наименьших квадратов (SE), от исходного уровня до 24 недели для уровней LDL-C у пациентов в группе алирокумаба было статистически значимым и стабильным для целого ряда исходных значений уровня LDL-C (таблица 37).
[00359] Все ключевые вторичные конечные точки эффективности были статистически значимыми в соответствии c процедурой иерархического тестирования.
[00360] Два последовательных рассчитанных значения уровня LDL-C <25 мг/дл (< 0,65 ммоль/л) наблюдали у 562 (37,4%) пациентов. Причин для какого-либо особого беспокойства относительно безопасности у этих пациентов не наблюдали.
Обобщенные результаты относительно безопасности
[00361] Для целей данного исследования TEAE представляют собой нежелательные явления, которые развились, ухудшились или стали серьезными на протяжении периода TEAE (время от первой дозы двойного слепого лечения до семидесяти дней после последней дозы). Во время анализа с предварительно заданными параметрами процентные доли пациентов, которые испытывали TEAE, серьезное TEAE и TEAE, приведшие к прекращению лечения, были аналогичными в разных группах лечения (см. таблицу 44).
[00362] Для обеих групп лечения наиболее часто (≥ 10%) сообщалось о SOC, которые представляли собой "инфекции и инвазии" (45,5% в группе алирокумаба в сравнении с 46,1% в группе плацебо), "нарушения со стороны костно-мышечной и соединительной тканей" (27,2% в группе алирокумаба в сравнении с 28,6% в группе плацебо), "нарушения со стороны желудочно-кишечного тракта" (18,6% в группе алирокумаба в сравнении с 18,8% в группе плацебо), "нарушения со стороны нервной системы" (17,0% в группе алирокумаба в сравнении с 17,8% в группе плацебо), "общие нарушения и нарушения в месте введения дозы" (15,4% в группе алирокумаба в сравнении с 17% в группе плацебо), "травмы, отравления и осложнения, вызванные проведением процедур" (13,4% в группе алирокумаба в сравнении с 14,2% в группе плацебо) и "нарушения со стороны дыхательной системы, органов грудной клетки и средостения" (11,0% в группе алирокумаба в сравнении с 10,9% в группе плацебо). В таблице 45 перечислен ряд TEAE для выбранных классов поражений систем и органов. В таблице 46 перечислены процентные доли различных TEAE в группах, которые лечили с помощью алирокумаба и плацебо, а также специально перечислены TEAE, наблюдаемые у пациентов, которых лечили алирокумабом (с помощью указания процентной доли пациентов), с двумя последовательными измерениями уровня LDL-C, составляющими менее 25 мг/дл. Все пациенты получали фоновое лечение статином (на максимально переносимом уровне). Подгруппа пациентов также получала дополнительную липид-корригирующую терапию. Пациенты, которые достигали уровней LDL-C, составляющих менее 25 мг/дл, в двух или более последовательных периодах составляли 36,3% популяции пациентов, которых лечили алирокумабом (562 пациентов из 1550). Определенные представляющие интерес лабораторные значения (аланинаминотрансфераза > 3 x ULN, аспартатаминотрансфераза > 3 x ULN и креатинкиназа >3 x ULN) показаны в таблице 47. Несколько пациентов из группы алирокумаба (31 из 1461, 2,1%) имели сниженные уровни витамина E в плазме, которые были ниже внешней границы нормы, в течение периода TEAE (по сравнению с 1 пациентом из 738 в группе плацебо, 0,1%).
[00363] Не было обнаружено никакого дисбаланса относительно частоты TEAE (предпочтительный термин). Для обеих групп лечения наиболее часто (≥ 5% в каждой группе) сообщалось о следующих TEAE: назофарингит (12,6% в группе алирокумаба в сравнении с 12,7% в группе плацебо), инфекция верхних дыхательных путей (7,0% в сравнении с 8,0%), реакция в месте инъекции (5,7% в сравнении с 4,3%), грипп (5,4% в сравнении с 5,5%), диарея (5,3% в сравнении с 5,1%), инфекция мочевыводящих путей (5,2% в сравнении с 6,2%), бронхит (5,2% в сравнении с 4,7%) и головная боль (4,8% в сравнении с 5,6%). Большинство реакций в месте инъекции (возникшие в ходе лечения локальные реакции в месте инъекции) были легкой или умеренной интенсивности, при этом первая реакция происходила в первые три месяца терапии (57 дней в случае алирокумаба в сравнении со 100 днями в случае плацебо) у большинства пациентов.
[00364] TEAE, представляющие особый интерес, включали: возникшие в ходе лечения локальные реакции в месте инъекции; явления, связанные с общими аллергическими реакциями; неврологические явления; нейрокогнитивные нарушения; установленные сердечно-сосудистые явления; нейрокогнитивные нарушения и гемолитическая анемия. Преобладание данных явлений в группах алирокумаба и плацебо изложено в таблице 48. Преобладание специфических нейрокогнитивных нежелательных явлений в группах алирокумаба и плацебо изложено в таблице 49.
[00365] Сообщалось о девятнадцати (19) случаях смерти на протяжении исследования (11 (0,7%) в группе алирокумаба в сравнении с 8 (1,0%) в группе плацебо). Если сфокусироваться на TEAE, приведших к смерти, сообщалось о 7 (0,5%) случаях смерти в группе алирокумаба в сравнении с 8 (1,0%) в группе плацебо. Согласно экспертному заключению основная причина смерти была связана с сердечно-сосудистой системой в обеих группах лечения (на протяжении периода TEAE 5 (0,3%) случаев смерти по причине сердечно-сосудистой патологии в группе алирокумаба в сравнении с 5 (0,6%) в группе плацебо).
[00366] О SAE сообщали 394 пациента (255 (16,5%) пациентов в группе алирокумаба и 139 (17,6%) пациентов в группе плацебо) в течение периода TEAE. Никакого дисбаланса между группами не наблюдалось.
[00367] Никакой конкретной клинической картины не было отмечено среди TEAE, приведших к окончательному прекращению лечения.
[00368] Среди явлений, представляющих интерес, не было обнаружено никаких конкретных сообщений относительно TEAE, связанных с неврологическими явлениями и офтальмологическими явлениями (в том числе в офтальмологическом подысследовании). Относительно нейрокогнитивных нарушений 18 (1,2%) наблюдали в группе алирокумаба в сравнении с 4 (0,5%) в группе плацебо, они включали в основном амнезию, нарушения памяти и состояние спутанности сознания. О случаях гемолитической анемии не сообщалось.
[00369] Что касается общей аллергической реакции, аналогичные доли пациентов сообщали по меньшей мере об одном таком явлении в группе алирокумаба (140 [9,0%]) в сравнении с группой плацебо (71 [9,0%]).
[00370] 124 (5,3%) пациента (90 (5,8%) пациентов из группы алирокумаба и 34 (4,3%) пациента из группы плацебо) испытывали возникшие в ходе лечения локальные реакции в месте инъекции. Большинство из этих явлений были слабой или умеренной интенсивности.
[00371] В соответствии с поисковым анализом в отношении ʺдиабета и нарушенного контроля глюкозыʺ в общей сложности 36 (6,3%) пациентов в группе алирокумаба и 10 (3,6%) пациентов в группе плацебо, которые были классифицированы как имеющие "нарушенный контроль глюкозы" на исходном уровне, определили как диабетиков в периоде TEAE. Два пациента, которых классифицировали с нормальным статусом на исходном уровне, стали диабетиками (по одному в каждой группе лечения).
[00372] Отсутствовал дисбаланс в отношении PCSA на основе параметров лабораторных исследований, ECG и жизненно важных функций.
Таблица 44 - Возникшие в ходе лечения нежелательные явления (во время анализа с предварительно заданными параметрами)
Все пациенты на фоне терапии с максимально переносимой дозой статинов ± другой липид-корригирующей терапией.
(n=1550)
(n=788)
a Результаты относительно безопасности из анализа первой стадии (52 недель для всех пациентов, продолжающих лечение, и включающие ~600 пациентов, которых подвергали воздействию в течение 78 недель).
bTEAE представляют собой нежелательные явления, которые развились, ухудшились или стали серьезными на протяжении периода TEAE (время от первой до последней инъекции исследуемого лечения+70 дней).
Таблица 45 - TEAE в соответствии с классом поражений систем и органов (≥2%) в любой группе (во время анализа с предварительно заданными параметрами)
Все пациенты на фоне терапии с максимально переносимой дозой статина ± другой LLT.
(n=1550)
(n=788)
Таблица 46 - TEAE, перечисленные в соответствии с подгруппой пациентов (во время анализа с предварительно заданными параметрами)
% (n) пациентов
(n=1550)
(n=788)
Сокращения: CHD, коронарное заболевание сердца; CV, сердечно-сосудистый; MI, инфаркт миокарда; LDL, липопротеин низкой плотности; LLN, нижняя граница нормы; LLT, липид-корригирующая терапия; SAE, серьезные нежелательные явления; TEAE, возникшие в ходе лечения нежелательные явления; ULN, верхняя граница нормы.
a Выбор предпочтительных терминов основан на запросе в компании MedDRA ʺдиабетʺ и включает предпочтительные термины ʺсахарный диабетʺ, ʺсахарный диабет без адекватного контроляʺ, ʺскоротечный сахарный диабет 1 типаʺ, ʺинсулинорезистентный диабетʺ, ʺинсулинозависимый сахарный диабет 2 типаʺ, ʺсахарный диабет 1 типаʺ, ʺсахарный диабет 2 типаʺ, ʺнедостаточность питания связанная с сахарным диабетомʺ и ʺконтроль сахарного диабетаʺ и исключает ʺпониженный уровень глюкозы в кровиʺ.
Таблица 47 - Лабораторные значения, представляющие интерес (во время анализа с предварительно заданными параметрами)
% (n) пациентов
Таблица 48 - Нежелательные явления, представляющие особый интерес (во время анализа с предварительно заданными параметрами)
Все пациенты на фоне терапии с максимально переносимой дозой статинов ± другой липид-корригирующей терапией.
(n=1550)
(n=788)
† Установленные CV явления Установленные CV явления включают все положительно установленные CV AE. Категории экспертного заключения являются следующими: смерть от CHD, несмертельный MI, смертельный и несмертельный ишемические инсульты, нестабильная стенокардия, при которой требуется госпитализация, застойная сердечная недостаточность, при которой требуется госпитализация, и процедура реваскуляризации коронарных сосудов по причине ишемии [PCI, CABG]. ‡Запросы в компанию MedDRA (CMQ)a. Выбор предпочтительных терминов основан на стандартизованном запросе MedDRA, в том числе 'демиелинизация' (в широком смысле+в узком смысле), 'периферическая нейропатия' (в широком смысле+в узком смысле) и 'Синдром Гийена-Барре' (в широком смысле+в узком смысле), с исключением предпочтительных терминов 'острый респираторный дистресс-синдром', 'астения', 'остановка дыхания' и 'дыхательная недостаточность'.
Таблица 49 - Нейрокогнитивные нежелательные явления(во время анализа с предварительно заданными параметрами)
Все пациенты на фоне терапии с максимально переносимой дозой статинов ± другой липид-корригирующей терапией.
(n=1550)
(n=788)
a Выбор предпочтительных терминов основан на запросе в компанию MedDRA.
РЕЗУЛЬТАТЫ II- ОКОНЧАТЕЛЬНЫЙ АНАЛИЗ
Хотя все первичные и вторичные конечные точки эффективности были достигнуты к моменту промежуточного анализа с предварительно заданными параметрами, исследование продолжали до завершения, что дало возможность провести анализ популяции для оценки безопасности полностью.
Обобщенные характеристики популяции для оценки безопасности
Средний показатель воздействия исследуемого лекарственного средства составлял 70 недель для 2338 пациентов, включенных в анализ безопасности (1550 в группе алирокумаба и 788 в группе плацебо), что дает 2061 пациенто-год воздействия 150 мг алирокумаба каждые две недели. Общий средний показатель соблюдения схемы исследуемого лечения (т.е. процентная доля дней, в которые пациент вводил инъекции согласно запланированному режиму введения доз) составлял 98,0% и 97,6% в группе алирокумаба и плацебо соответственно. Среднее значение срока последующего наблюдения (вне зависимости от соблюдения схемы лечения) в популяции для оценки безопасности составляло 80,9 недели для алирокумаба и 80,1 недели для плацебо. Показатели прекращения исследуемого лечения составляли 28% для алирокумаба и 25% для плацебо.
Эффективность
Стабильное снижение уровня холестерина LDL от исходного уровня наблюдали от 4 до 78 недели в группе алирокумаба (фигура 15). Результаты для измеренных уровней холестерина LDL согласовывались с результатам для рассчитанных уровней холестерина LDL. Процентное снижение холестерина LDL было немного ниже на 78 неделе (52%) в сравнении с 24 неделей (61%) в анализе в отношении группы пациентов, сформированной в зависимости от назначенного лечения; на это наблюдение влияли значения, собранные после досрочного прекращения лечения.
Безопасность
Процентные доли пациентов, которые испытывали возникшие в ходе лечения нежелательные явления, были сходными в обеих группах лечения (81% в случае алирокумаба в сравнении с 83% в случае плацебо) (таблица 50). Возникшие в ходе лечения нежелательные явления, приведшие к прекращению приема исследуемого лекарственного средства, встречались у 7,2% пациентов, получавших алирокумаб, и 5,8% пациентов, получавших плацебо. Относительно конкретных нежелательных явлений отличия между группой алирокумаба и группой плацебо наблюдались по частотам реакций в месте инъекции (5,9% в сравнении с 4,2% соответственно), миалгии (5,4% в сравнении с 2,9% соответственно), нейрокогнитивных нарушений (1,2% в сравнении с 0,5% соответственно) и офтальмологических явлений (2,9% в сравнении с 1,9% соответственно; таблица 50).
Среди пациентов в группе алирокумаба 575 (38% от общего количество) имели два последовательных рассчитанных уровня холестерина LDL, составляющих менее 25 мг/дл. Частоты возникших в ходе лечения нежелательных явлений для этих пациентов были сопоставимы с частотами по всей группе алирокумаба.
Таблица 50. AE, представляющие интерес, и лабораторные значения показателей безопасности (анализ безопасности)
Пациенты с максимально переносимой терапией статинами ± другой LLT
(N=1550)
(N=788)
P-значения рассчитаны с использованием точного теста Фишера и без поправки на множественные сравнения. Предоставлено только для наглядности.
Общее резюме
Данное исследование представляет собой наиболее масштабное и продолжительное двойное слепое исследование ингибитора PCSK9. Данный анализ предусматривает приблизительно 1900 пациенто-лет двойного слепого воздействия на пациентов с использованием 150 мг алирокумаба Q2W. У пациентов с высоким риском возникновения CV, принимающих максимально переносимую дозу статина ± другую липид-корригирующую терапию, были сделаны следующие наблюдения: 1) лечение алирокумабом при самостоятельном введении приводило к значимо большему снижению уровней LDL-C, чем плацебо, на 24 неделе (различие для среднего значения, определенного методом наименьших квадратов, составило -61,9%); эффективность оставалась стабильной на протяжении всего лечения, и на 78 неделе алирокумаб снижал уровни LDL-C от исходного уровня на дополнительные 56 процентов в сравнении с плацебо; 2) 79% пациентов в группе алирокумаба достигали целевых уровней LDL-C, составляющих <0,81 ммоль/л (70 мг/дл) на 24 неделе; 3) снижения уровней LDL-C наблюдали у пациентов в группе алирокумаба с 4 недели, и пациенты в группе алирокумаба достигали средних уровней LDL-C, составляющих 1,4 ммоль/л (53,1 мг/дл), на 52 неделе; и 4) TEAE встречались с аналогичной частотой при режиме лечения с алирокумабом и плацебо, а также подгруппы пациентов, которых лечили алирокумабом (n=562), с двумя последовательными уровнями LDL-C, составляющими <25 мг/дл.
Объем настоящего изобретения не ограничен конкретными вариантами осуществления, описанными в данном документе. Так, различные модификации настоящего изобретения, помимо описанных в данном документе, будут очевидны специалистам в данной области техники исходя из изложенного выше описания и прилагаемых фигур. Такие модификации находятся в пределах объема прилагаемой формулы изобретения.
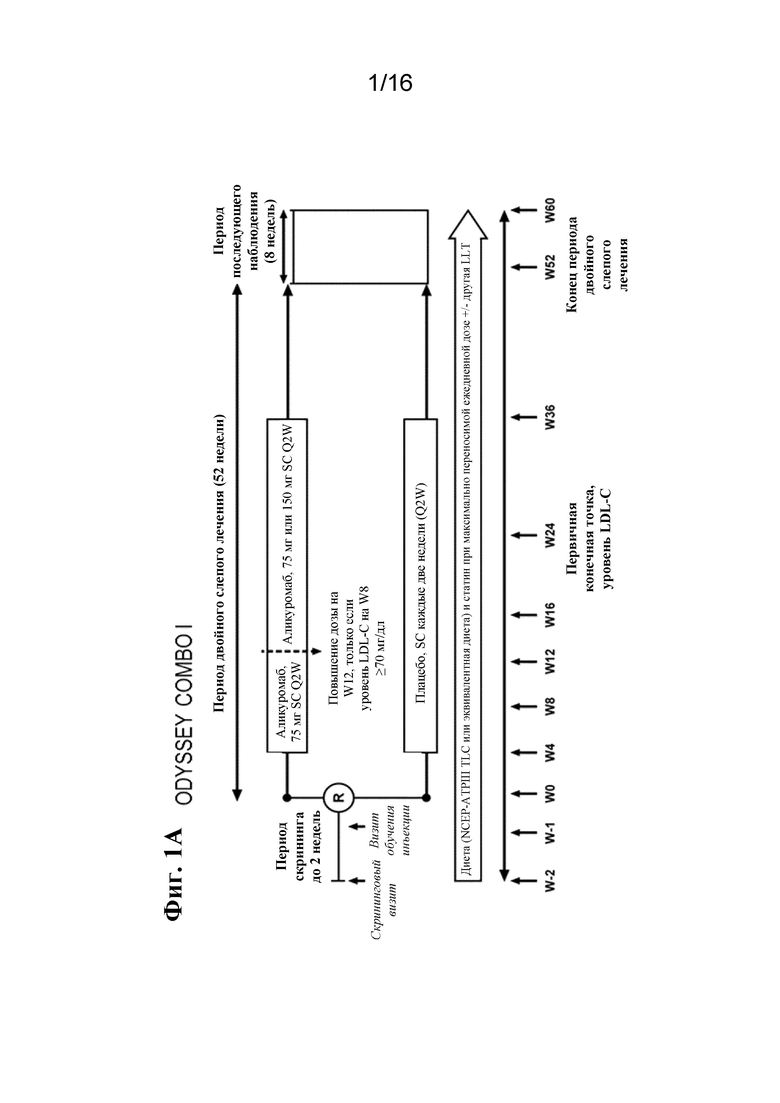
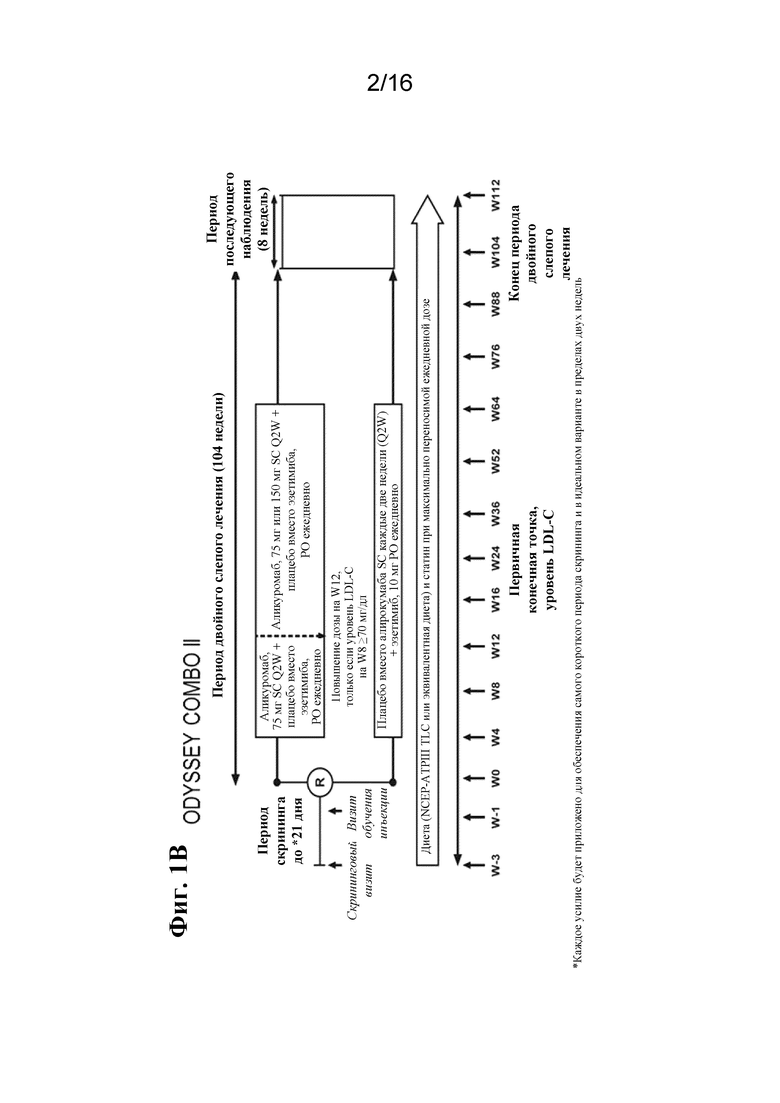




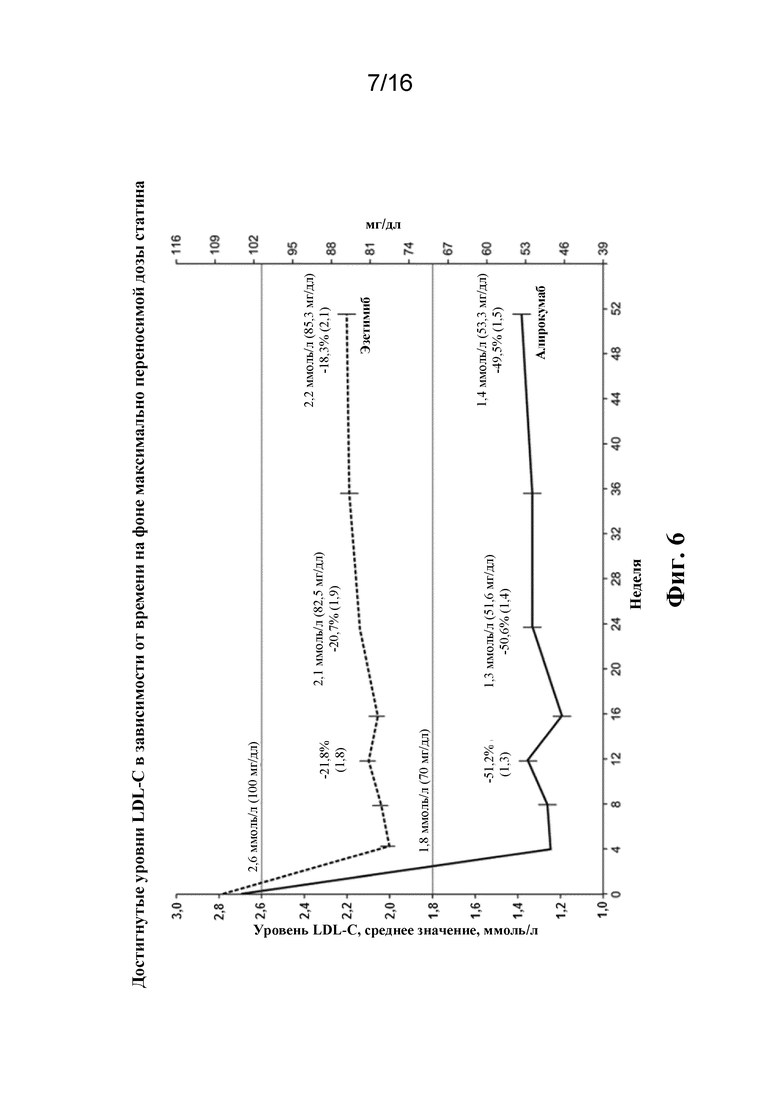
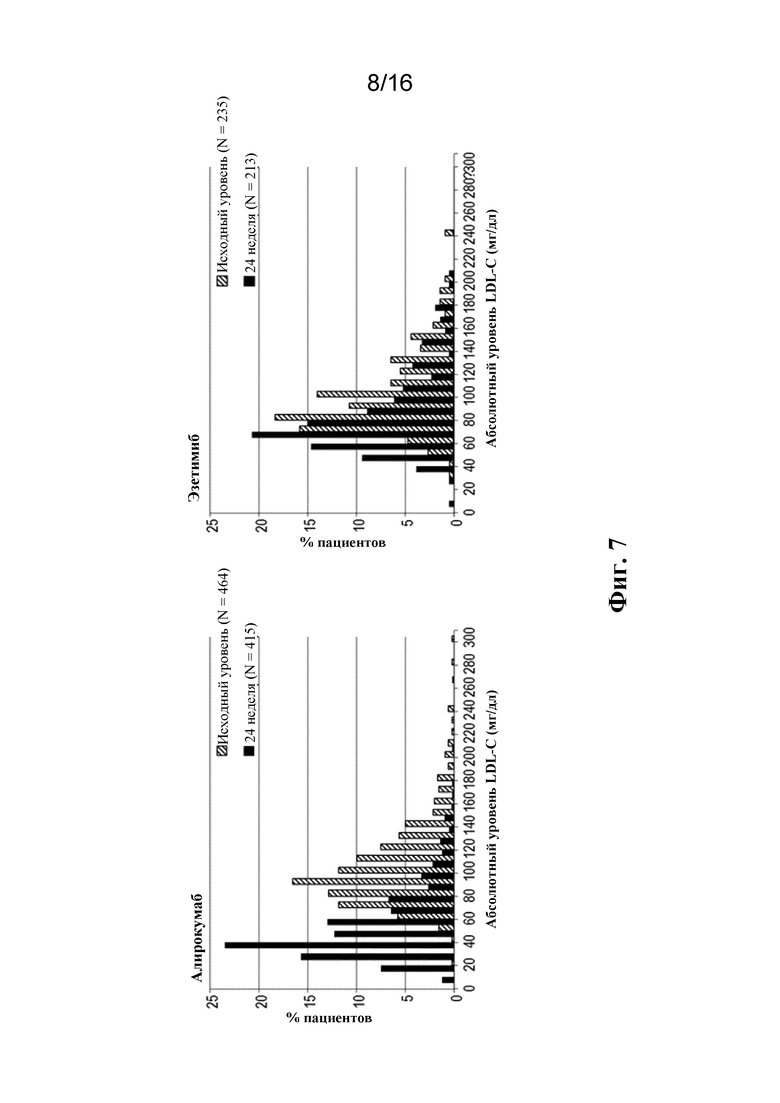







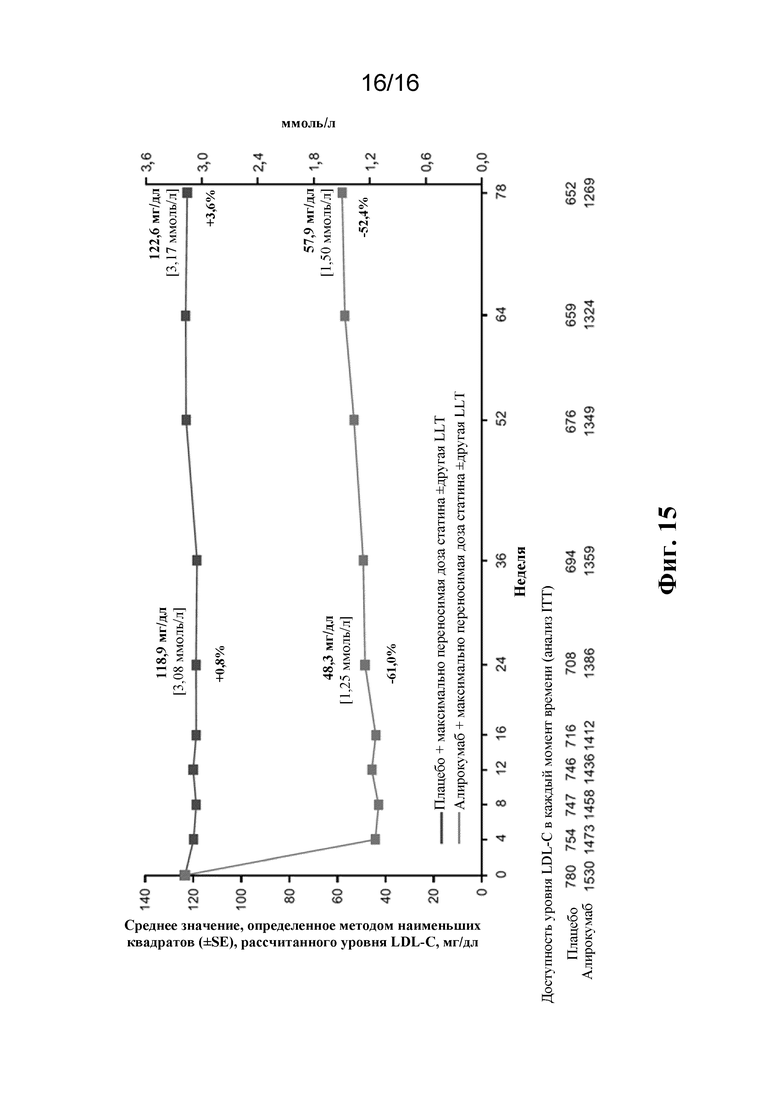
Изобретение относится к лечению гиперхолестеринемии. Раскрыто применение антитела или его антигенсвязывающего фрагмента, которые специфически связывают пропротеиновую конвертазу субтилизин/кексинового типа 9 (PCSK9) человека, содержащих три CDR тяжелой цепи: SEQ ID NO: 2, 3 и 4, и три CDR легкой цепи: SEQ ID NO: 7, 8 и 10, для лечения пациента, который имеет гиперхолестеринемию и уровень холестерина липопротеинов низкой плотности (LDL-C) выше 70 мг/дл, несмотря на лечение при помощи максимально переносимой терапии статинами; и установленное коронарное заболевание сердца (CHD) или эквивалентные риски возникновения CHD, где одна или несколько начальных доз в 75 мг антитела или его антигенсвязывающего фрагмента вводятся пациенту и где одна или несколько дополнительных доз в 75 мг антитела или его антигенсвязывающего фрагмента вводятся пациенту приблизительно через каждые две недели, если уровень LDL-C ниже 70 мг/дл; или одна или несколько дополнительных доз в 150 мг антитела или его антигенсвязывающего фрагмента вводятся пациенту через каждые две недели, если уровень LDL-C превышает или равен 70 мг/дл. Изобретение обеспечивает лечение гиперхолестеринемии, которая резистентна к терапии статинами. 11 з.п. ф-лы, 15 ил., 50 табл., 3 пр.
1. Применение антитела или его антигенсвязывающего фрагмента, которые специфически связывают пропротеиновую конвертазу субтилизин/кексинового типа 9 (PCSK9) человека, содержащих три CDR тяжелой цепи, изложенных под SEQ ID NO: 2, 3 и 4, и три CDR легкой цепи, изложенных под SEQ ID NO: 7, 8 и 10, для лечения гиперхолестеринемии у пациента, который имеет
(a) уровень холестерина липопротеинов низкой плотности (LDL-C) выше 70 мг/дл, несмотря на лечение при помощи максимально переносимой терапии статинами; и
(b) установленное коронарное заболевание сердца (CHD) или эквивалентные риски возникновения CHD,
где одна или несколько начальных доз, составляющих 75 мг, антитела или его антигенсвязывающего фрагмента вводятся пациенту, и где
(i) одна или несколько дополнительных доз, составляющих 75 мг, антитела или его антигенсвязывающего фрагмента вводятся пациенту приблизительно через каждые две недели, если уровень LDL-C ниже, чем 70 мг/дл; или
(ii) одна или несколько дополнительных доз, составляющих 150 мг, антитела или его антигенсвязывающего фрагмента вводятся пациенту через каждые две недели, если уровень LDL-C превышает или равен 70 мг/дл.
2. Применение по п. 1, где антитело или его антигенсвязывающий фрагмент содержат аминокислотную последовательность вариабельного участка тяжелой цепи (HCVR), изложенную под SEQ ID NO: 1, и аминокислотную последовательность вариабельного участка легкой цепи (LCVR), изложенную под SEQ ID NO: 6.
3. Применение по п. 1 или 2, где пациент получал самую высокую дозу статина, которую можно ввести пациенту без возникновения недопустимых неблагоприятных побочных эффектов, и не получал никакую другую липид-корригирующую терапию.
4. Применение по п. 1 или 2, где пациент получал (a) самую высокую дозу статина, которую можно ввести пациенту без недопустимых неблагоприятных побочных эффектов у пациента, и (b) по меньшей мере одну другую липид-корригирующую терапию.
5. Применение по любому из пп. 1-4, где пациент имеет один или более эквивалентных рисков возникновения CHD, выбранных из группы, состоящей из:
1) заболевания периферических артерий (PAD);
2) ишемического инсульта;
3) хронического заболевания почек; и
4) случая сахарного диабета в анамнезе.
6. Применение по п. 5, где пациент имеет сахарный диабет 2 типа.
7. Применение по п. 5, где пациент имеет гипертензию.
8. Применение по любому из пп. 1-7, где каждая из начальных и дополнительных доз представляет собой раствор для инъекций объемом 1 мл, содержащий 75 мг или 150 мг антитела или его антигенсвязывающего фрагмента.
9. Применение по любому из пп. 1-8, где антитело или его антигенсвязывающий фрагмент вводятся путем подкожной инъекции.
10. Применение по любому из пп. 1-9, где антитело или его антигенсвязывающий фрагмент находятся в предварительно заполненном устройстве для доставки в виде шприца-ручки, картридже устройства для доставки в виде шприца-ручки многоразового использования или устройстве для доставки в виде автоинжектора.
11. Применение по любому из пп. 1-10, где антитело или его антигенсвязывающий домен вводят пациенту в комбинации с максимально переносимой терапией статинами.
12. Применение по любому из пп. 1-11, где при введении субъекту антитела или его антигенсвязывающего фрагмента достигается одно или несколько из следующего:
(i) снижение уровня LDL-C у пациента по меньшей мере на 40%;
(ii) снижение уровня аполипопротеина B100 (ApoB) у пациента по меньшей мере на 35%;
(iii) снижение уровня холестерина, не относящегося к холестерину липопротеинов высокой плотности (не-HDL-C), у пациента по меньшей мере на 35%;
(iv) снижение уровня общего холестерина у пациента по меньшей мере на 20%;
(v) повышение уровня холестерина липопротеинов высокой плотности (HDL-C) у пациента по меньшей мере на 1%;
(vi) снижение уровня триглицеридов у пациента по меньшей мере на 1%;
(vii) снижение уровня липопротеина a (Lp(a)) у пациента по меньшей мере на 10% и
(viii) повышение уровня аполипопротеина A1 у пациента по меньшей мере на 1%.
| СПОСОБ ОПТИМИЗАЦИИ ОТБОРА СВЕТЛЫХ ТОПЛИВНЫХ ДИСТИЛЛЯТОВ В ПРОЦЕССЕ ПЕРВИЧНОЙ ПЕРЕГОНКИ НЕФТИ | 2019 |
|
RU2706070C1 |
| RU 2011129316 A, 20.01.2013 | |||
| WO 2013039969 A1, 21.03.2013. | |||

