ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение относится к области фармацевтики, в частности к хинолинил-содержащему соединению, его фармацевтической композиции и применению.
УРОВЕНЬ ТЕХНИКИ
Рецептор фактора роста эндотелия сосудов (VEGFR) является представителем семейства рецепторов тирозинкиназы. Он вызывает протекание ряд биохимических и физиологических процессов и, в конечном итоге, способствует образованию новых кровеносных сосудов за счет связывания со своим лигандом, фактором роста эндотелия сосудов (VEGF). В нормальных кровеносных сосудах, поддерживается баланс между факторами ангиогенеза и ингибиторами ангиогенеза. В процессе роста опухоли, высокая экспрессия VEGFR и VEGF нарушает этот баланс и способствует образованию новых кровеносных сосудов опухоли. Исследования показали, что высокая специфическая экспрессия рецептора и образование новых кровеносных сосудов являются предпосылками для роста опухоли. Рост и метастазирование злокачественных опухолей должны поддерживаться соответствующим питанием и выведением шлаков с помощью окружающих новых кровеносных сосудов. Следовательно, высокая экспрессия VEGFR и VEGF тесно связана с плотностью микрососудов и пролиферацией и метастазированием опухоли. Поскольку VEGFR-2 в основном распределяется в эндотелиальных клетках сосудов, он может прямо и косвенно подавлять рост опухоли и метастазирование, не затрагивая нормальные клетки, путем блокирования активности VEGFR для достижения идеального противоопухолевого эффекта. В настоящее время, для лечения опухолей одобрен Управлением по контролю за качеством пищевых продуктов и лекарственных препаратов США (FDA) ряд ингибиторов VEGFR. Выпускаемые фармацевтическими фирмами низкомолекулярные ингибиторы VEGFR включают кабозантиниб (XL184, BMS-907351), сунитиниб, сорафениб и вандетаниб (ZD6474). Около двадцати низкомолекулярных ингибиторов VEGFR проходят фазу клинических испытаний, в том числе ваталаниб (PTK787/ZK222584), AMG-706, пазопаниб и другие подобные ингибиторы. Поиск низкомолекулярных ингибиторов с высокой активностью в отношении VEGFR-2 стал чрезвычайно актуальной целью исследований в области терапии опухолей.
Гистондеацетилазы (HDAC) также тесно связаны с возникновением опухолей. Их ингибиторы могут снижать порог апоптоза опухолевых клеток, обладают обширной противоопухолевой активностью и могут применяться в комбинации с различными противоопухолевыми лекарствами. Гистондеацетилазы человека подразделяют на четыре категории: первая категория включает HDAC1, HDAC2, HDAC3 и HDAC8, вторая категория включает HDAC4, HDAC5, HDAC7 и HDAC9; HDAC6 и HDAC10 содержат два каталитических центра и их классифицируют как IIa; HDAC11 относится к классу IV, и его каталитический центр содержит аминокислотные остатки, общие для HDAC класса I и класса II, имеющие ионы цинка в каталитических центрах всех одиннадцати подтипов HDAC. Ингибиторы HDAC уже давно применяют в качестве препаратов, стабилизирующих настроение, и противоэпилептических препаратов в психиатрии и неврологии. Их применяют при лечении нейродегенеративных заболеваний, таких как болезнь Альцгеймера, болезнь Хантингтона и болезнь Паркинсона. Еще одним важным применением ингибиторов HDAC является их использование в качестве противоопухолевых препаратов. В настоящее время, Управлением по контролю за качеством пищевых продуктов и лекарственных препаратов США (FDA) одобрено два ингибитора HDAC для лечения Т-клеточной лимфомы кожи, а именно вориностат (SAHA) и ромидепсин (FK228). Известно много других ингибиторов HDAC, таких как PXD-101 (фаза II), LBH589 (фаза III), MS-275 (фаза II) и другие подобные ингибиторы HDAC, которые проходят стадии клинических испытаний.
Результаты фазы I клинического исследования, опубликованные в J.Clin.Oncol, ведущем международным журналом по клинической онкологии, показали, что ингибиторы HDAC могут эффективно способствовать положительной динамике у пациентов с солидными опухолями в прогрессирующей стадии с резистентностью к ингибиторам VEGFR. Несмотря на то, что комбинированные препараты позволяют эффективно решать проблемы невосприимчивости и резистентности к лекарственному средству, возникающие в случае применения препаратов, нацеленных на одну мишень, вызванные гетерогенностью и адаптируемостью опухолевых клеток, тем не менее, существуют также проблемы, такие как различия в фармакокинетических свойствах разных препаратов, неблагоприятные межлекарственные взаимодействия и плохое соблюдение пациентом предписанного режима терапии. Ожидается, что разработка противораковых лекарственных средств, нацеленных на несколько мишеней, позволит преодолеть упомянутые выше недостатки комбинированных лекарственных средств, и одновременно решить проблему невосприимчивости опухоли и резистентности к воздействию лекарственных средств, нацеленных на одну мишень.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В одном аспекте, в изобретении предлагается новое хинолинил-содержащее соединение или его фармацевтически приемлемая соль, сольват, активный метаболит, полиморф, меченное изотопом соединение, изомер или пролекарство, которое может применяться в качестве ингибитора тирозинкиназы и/или ингибитора гистондеацетилазы.
В другом аспекте, в изобретении предлагается фармацевтическая композиция.
В другом аспекте, в изобретении предлагается применение упомянутого выше соединения или его фармацевтически приемлемой соли, сольвата, активного метаболита, полиморфа, меченного изотопом соединения, изомера или пролекарства.
В настоящем изобретении предлагается хинолинил-содержащее соединение, представленное общей формулой (I) или (II), или его фармацевтически приемлемая соль, сольват, активный метаболит, полиморф, меченное изотопом соединение или изомер,
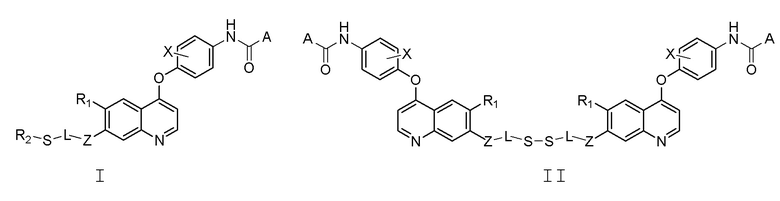
где,
A представляет собой 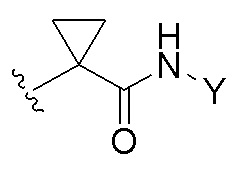 или -NR3R4;
или -NR3R4;
X представляет собой водород, галоген или замещенный или незамещенный C1-8 алкил;
Y представляет собой замещенный или незамещенный C3-8 циклоалкил, C3-8 гетероциклил, C6-20 арильную группу или C2-20 гетероарил;
Z представляет собой -O- или -S-;
L представляет собой линейный -(CH2)n-, n представляет собой целое число от 3 до 10, где, необязательно, один или более -CH2- заменяют на один или более из -NR5-, -(CO)-, -(CS)- и -CR5R6-, и/или, необязательно, один или более из -CH2CH2- могут быть заменены на -CH=CH-; или -(CO)NR7R8;
R2 представляет собой замещенный или незамещенный водород, C1-8 алкил, C1-8 алкокси, C1-8 алкилсульфанил, C1-8 галогеналкил, C3-8 циклоалкил, C3-8 гетероциклил, C6-20 арил, C2-20 гетероарил, -(CO)R9, -(CS)R9, или R2 соединен с одним из -CH2- в группах L, в результате чего R2, S и -CH2- вместе образуют C3-8 гетероциклическую группу или C2-20 гетероарильную группу;
R3 и R4 независимо представляют собой замещенный или незамещенный водород, C1-8 алкил, C1-8 алкоксил, C1-8 алкилсульфанил, C1-8 галогеналкил, C3-8 циклоалкил, C3-8 гетероциклил, C6-20 арил или C2-20 гетероарил;
R5 и R6 независимо представляют собой замещенный или незамещенный водород, C1-8 алкил, C1-8 алкоксил, C1-8 алкилсульфанил, C1-8 галогеналкил, гидроксил, меркапто, карбоксил, амино или циано;
R7 и R8 независимо представляют собой замещенный или незамещенный водород или C1-8 алкил;
R9 представляет собой замещенный или незамещенный C1-8 алкил, C1-8 алкоксил, C1-8 алкилсульфанил, C1-8 галогеналкил, C1-6 алкилсульфонил, C1-6 алкиламино, C3-8 циклоалкил, C3-8 гетероциклил, C6-20 арил, C2-20 гетероарил, C1-6 алкилен C3-8 циклоалкил, C1-6 алкилен C3-8 гетероциклил, C1-6 алкилен C6-20 арил, C1-6 алкилен C2-20 гетероарил, гидроксил, меркапто, нитро, амино, циано, или R9 соединен с любым из -CH2- в группе L, в результате чего -(CO)R9 или -(CS)R9 вместе с S, -CH2- образует C3-8 гетероциклил или C2-20 гетероарил.
Заместители для упомянутых выше групп могут быть выбраны из галогена, C1-8 алкила, C2-8 алкенила, C2-8 алкинила, C1-8 галогеналкила, C1-8 алкоксила, C1-8 алкилсульфанила, C3-8 циклоалкила, C3~8 гетероциклила, C6-20 арила, C2-20 гетероарила, C1-6 алкилэфирной группы, C1-6 алкилацила, C1-6 алкиламино, C1-6 алкилсульфонила, амино, гидроксила, меркапто, карбоксила, нитро, амидо или циано.
В приведенной выше формуле (I) или (II), группы, представленные с помощью X, Y, Z, L, R1 - R9 и их необязательные заместители включают, но этим не ограничивая, следующие группы.
Водород, который может быть обозначен как -H, и может быть также заменен на дейтерий или тритий.
Галоген может включать фтор, хлор, бром и йод.
C1-8 алкил может включать метил, этил, н-пропил, изопропил, 2-метил-1-пропил, 2-метил-2-пропил, 2-метил-1-бутил, 3-метил-1-бутил, 2-метил-3-бутил, 2,2-диметил-1-пропил, 2-метил-1-пентил, 3-метил-1-пентил, 4-метил-1-пентил, 2-метил-2-пентил, 3-метил-2-пентил, 4-метил-2-пентил, 2,2-диметил-1-бутил, 3,3-диметил-1-бутил, 2-этил-1-бутил, н-бутил, изобутил, вторбутил, третбутил, н-пентил, изоамил, неопентил, третамил, гексил, гептил, октил и другие подобные алкилы.
C1-8 алкокси может быть представлен как -OC1-8 алкил, где C1-8 алкил включает определенные выше группы, например, C1-8 алкокси включает метокси, этокси, н-пропокси и другие подобные алкоксильные группы.
C1-8 алкилсульфанил может быть представлен как -SC1-8 алкил, где C1-8 алкил включает определенные выше группы, например, C1-8 алкилсульфанил может включать метилтио, этилтио и другие подобные группы.
C1-8 галогеналкил может быть представлен как группа, в которой любое количество атомов водорода в C1-8 алкиле замещено с помощью галогена, где группы, включенные в C1-8 алкил и в галоген, перечислены выше; например, C1-8 галогеналкил может включать -CF3.
C1-6 алкилен представляет собой двухвалентную функциональную группу с двумя способными к замещению связями, который может включать линейный алкилен и разветвленный алкилен, и линейный алкилен может быть выражен как -(CH2)m-, где m представляет собой от 1 до 6, и может включать, например, метилен, этилен и другие подобные алкилены.
C3-8 циклоалкил может быть выражен в виде неароматического насыщенного карбоциклического кольца, включая только одно кольцо из углеродных атомов (с одним кольцом) и два кольца из углеродных атомов (с двумя кольцами). Например, C3-8 циклоалкил может включать 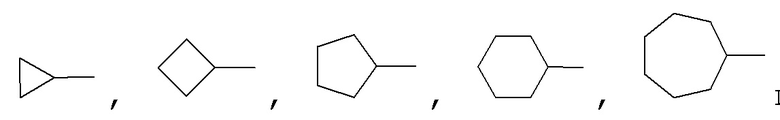 или другие подобные циклоалкилы.
или другие подобные циклоалкилы.
C3-8 гетероциклическая группа может быть представлен как группа, полученная в результате замены любого числа кольцевых атомов в C3~8 циклоалкиле на гетероатомы, такие как O, S, N, P, Si, и другие гетероатомы, где C3-8 циклоалкил включает группы, определенные выше. Например, C3-8 гетероциклическая группа может включать оксиранил, sulfiethanyl, азаэтил, азетидинил, оксобутанил, тиобутирил, тетрагидрофуранил, пирролидинил, оксазолидинил, тетрагидропиразолил, пирролинил, дигидрофуранил, дигидротиенил, пиперидинил, тетрагидропиранил, тетрагидро-тиопиранил, морфолинил, пиперазинил, дигидропиридинил, тетрагидропиранил, дигидропиранил, тетрагидропиранил, дигидротиопиранил, азациклогептил, оксациклогептил, тиациклогептил, оксаазабицикло[2,2,1]гептил, азаспиро[3,3]-гептил и другие подобные гетероциклические группы.
C6-20 арил может включать моноциклическую арильную группу, бициклическую арильную группу или полициклическую арильную группу. Например, он может включать фенил, бифенил, нафтил, фенантрил, антрил, азуленил и другие подобные арилы.
C2-20 гетероарил может представлять собой ненасыщенную группу, содержащую любое количество гетероатомов, таких как O, S, N, P и Si, в качестве кольцевых атомов. Число углеродных атомов в гетероариле может составлять 2-20, например, 2, 3, 4, 5, 6, 7, 8, 9, 10 или больше чем 10. Например, C2-20 гетероарил может включать пирролил, фурил, тиенил, имидазолил, оксазолил, пиразолил, пиридил, пиримидинил, пиразинил, хинолинил, изохинолинил, тетразолил, триазолил, триазинил, бензофуранил, бензотиенил, индолил, изоиндолил и другие подобные гетероарилы.
Гидроксильная группа может быть представлена как -OH.
Меркаптогруппа может быть представлена как -SH.
Нитрогруппа может быть представлена как -NO2.
Цианогруппа может быть представлена как -CN.
Карбоксильная группа может быть представлена как -COOH, и атом H в карбоксильной группе может быть также замещен с помощью заместителя с образованием соответствующей эфирной группы, которая может представлена как -COORa. Ra может представлять собой заместители, описанные в общей формуле (I), например, эфирная группа, замещенная с помощью C1-8 алкила, может быть представлена как -COOC1-8 алкильная группа, и где C1-8 алкильная группа определена выше.
Предпочтительно, чтобы эфирная группа представляла собой C1-6 алкильную эфирную группу, и C1-6 алкильная группа может включать все группы с числом углеродных атомов 1-6 в приведенном выше определении "C1-8 алкила".
Сульфонильная группа может быть представлен как -S(O)2Ra, Ra может представлять собой заместители, описанные в общей формуле(I). Например, сульфонильная группа, замещенная с помощью C1-8 алкильной группы, может быть представлена как -S(O)2 C1-8 алкил, где C1-8 алкил определен выше.
Предпочтительно, чтобы сульфонильная группа представляла собой C1-6 алкилсульфонил, и C1-6 алкильная группа может включать все группы с числом углеродных атомов от 1 до 6 в упомянутом выше определении "C1-8 алкила".
Ацильная группа может быть представлена как -CORa, Ra может представлять собой заместители, описанные в общей формуле (I). Например, ацильная группа, замещенная с помощью C1-8 алкильной группы, может быть представлена как -COC1-8 алкил, где C1-8 алкил определен выше.
Предпочтительно, чтобы ацильная группа представляла собой C1-6 алкилацил, и C1-6 алкильная группа может включать все группы с числом углеродных атомов от 1 до 6 в упомянутом выше определении "C1-8 алкила".
Аминогруппа может быть представлена как -NH2, -NHRa или -N(Ra)2, и Ra может представлять собой заместители, описанные в общей формуле (I). Например, аминогруппа, замещенная с помощью или -N(C1~8 алкил)2, где C1-8 алкил определен выше.
Предпочтительно, чтобы аминогруппа представляла собой C1~6 алкиламино, и C1-6 алкильная группа может включать все группы с числом углеродных атомов от 1 до 6 в упомянутом выше определении "C1-8 алкила".
В амидогруппе, аминогруппа определена выше.
В приведенных выше определениях, в случае изменений числа углеродных атомов, приведенные выше определения изменяются только в соответствии с изменением числа углеродных атомов, и это не приводит к изменению определения типа группы. Например, "C1-6 алкил" может включать все группы, соответствующие числу углеродных атомов от 1 до 6 в определении "C1-8 алкила", такие как метил, этил и н-пропил, изопропил, н-бутил, изобутил, вторбутил, третбутил, н-пентил, изопентил, неопентил и н-гексил и другие подобные алкилы. "C1-4 алкил" может включать все группы, соответствующие числу углеродных атомов от 1 до 4 в определении "C1-8 алкила", такие как метил, этил и н-пропил, изопропил, н-бутил, изобутил, вторбутил и третбутил.
В приведенных выше определениях, атомы в каждой группе, такие как C, H, O, N, S, и другие подобные атомы, могут быть независимо заменены на их изотопы. Например, водород может быть заменен на дейтерий, тритий, и C1-8 алкил может быть заменен на дейтерированный C1-8 алкил, в том числе, но этим не ограничивая, на дейтерированный метил, дейтерированный этил, дейтерированный н-пропил, дейтерированный изопропил, дейтерированный н-бутил, дейтерированный изобутил, дейтерированный вторбутил, дейтерированный третбутил, и так далее.
Кроме того, в приведенной выше формуле (1), заместители для групп включают, но этим не ограничивая, водород, дейтерий, фтор, хлор, бром, метил, этил, пропил, изопропил, н-бутил, вторбутил, третбутил, метокси, этокси, пропокси, изопропокси, -CN, -CF3, -NH2, -NH(C1-4 алкил), -N(C1-4 алкил)2, -CO2C1-4 алкил, -CO2H, -NHC(O)C1-4 алкил, -SO2C1-4алкил, -C(O)NH2, -C(O)NH(C1~4алкил), -C(O)N(C1-4алкил)2, циклопропил, циклобутил, циклопентил, циклогексил, пирролидинил, пиразолил, пиперидинил, пиридил, пиперазинил, триазинил, фуранил, тиофуранил, морфолинил, тиоморфолинил, фенил, нафтил, дифенил, терфенил, и другие подобные заместители.
В некоторых вариантах осуществления изобретения, общая формула (I) или (II) представлена следующей формулой (I-1), (I-2), (II-1) или (II-2):
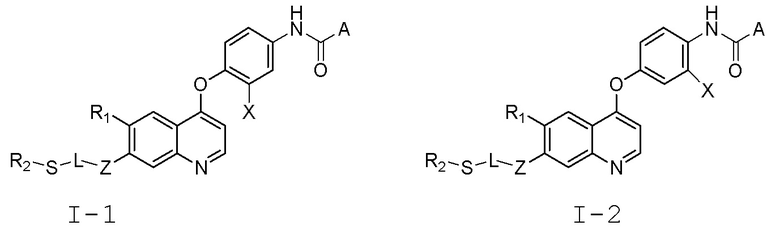
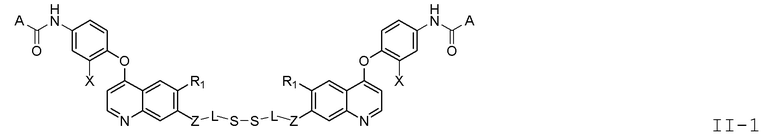
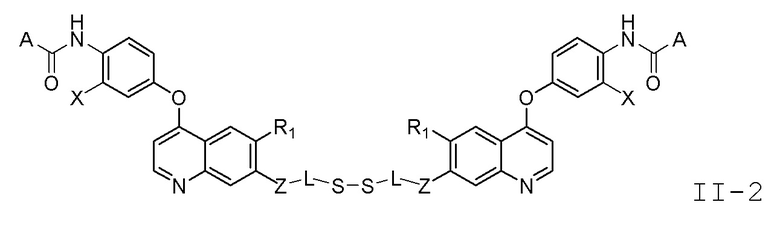
где, X представляет собой водород, F или Cl.
В некоторых вариантах осуществления изобретения, символ "A" представляет собой одну из следующих групп:
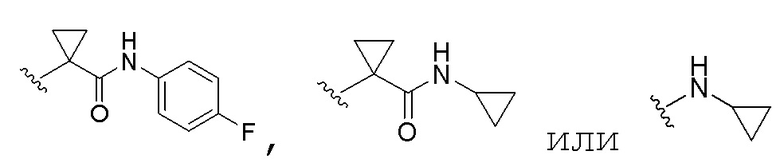 .
.
В некоторых вариантах осуществления изобретения, символ "L" представляет собой одну из следующих групп: -CH2-CO-NH-(CH2)p-*, p представляет собой целое число от 3 до 6, и один или более из -CH2CH2- могут быть необязательно заменены на -CH=CH-, и "*" обозначает конец, присоединенный к группе "Z"; или
линейный -(CH2)o-, где "o" представляет собой целое число от 5 до 7, и один или более из -CH2CH2- могут быть необязательно заменены на -CH=CH-.
В некоторых вариантах осуществления изобретения, символ "L" представляет собой одну из следующих групп:
*-CH2CH=CH(CH2)q-, *-(CH2)2CH=CH(CH2)q- или *-(CH2)3CH=CH(CH2)q-, q представляет собой целое число от 1 до 4, где "*" обозначает конец, присоединенный к группе "Z".
В некоторых вариантах осуществления изобретения, R1 представляет собой C1-4 алкоксил (например, метокси или этокси) или -(CO)NH2.
В некоторых вариантах осуществления изобретения, когда R2 присоединен к -CH2- в группе L, так что R2, S, -CH2- вместе образуют C3-8 гетероциклил или C2-20 гетероарил, гетероциклил или гетероарил могут также содержать гетероатомы, которые не являются S, такие как O, N, S, и другие гетероатомы; предпочтительно, чтобы образованная структура представляла собой гетероциклил или гетероарил, содержащий от 4 до 8 кольцевых атомов.
В некоторых вариантах осуществления изобретения, R2 представляет собой замещенный или незамещенный водород, C1-4 алкил или -(CO)R9. R9 представляет собой замещенный или незамещенный C1-4 алкил, C1-4 алкоксил, C1-4 галогеналкил, C3-6 циклоалкил, C3-6 гетероциклил, C6-12 арил, C3-12 гетероарил, C1-4 алкилен C3-6 циклоалкил, C1-4 алкилен C3-6 гетероциклил, C1-4 алкилен C6-12 арил, C1-4 алкилен C3-12 гетероарил, гидроксил, меркапто, нитро, амино или циано; и заместители, если таковые имеются, выбирают из F, Cl, Br, метила, этила, н-пропила, изопропила, -NH2, гидроксила, карбоксила, меркапто или циано.
Предпочтительно, чтобы R9 представляет собой замещенный или незамещенный метил, этил, н-пропил, изопропил, н-бутил, изобутил, вторбутил, третбутил, метокси, этокси, метилтио, -(CH2)m-C3-6 гетероциклил, -(CH2)m-C3-12 гетероарил, фенил, нафтил или бифенил, и m представляет собой целое число от 1 до 3; более предпочтительно, чтобы кольцевые атомы гетероциклила и гетероарила содержали, по меньшей мере, один атом N.
В некоторых вариантах осуществления изобретения, когда R9 присоединен к -CH2- в группе L, так что -(CO)R9 или -(CS)R9 и S, -CH2- вместе образуют C3-8 гетероциклил или C2-20 гетероарил, гетероциклил или гетероарил могут также содержать гетероатомы, которые не являются S, такие как O, N, S, и другие гетероатомы; предпочтительно, чтобы образованная структура представляла собой гетероциклил или гетероарил, содержащий от 4 до 8 кольцевых атомов.
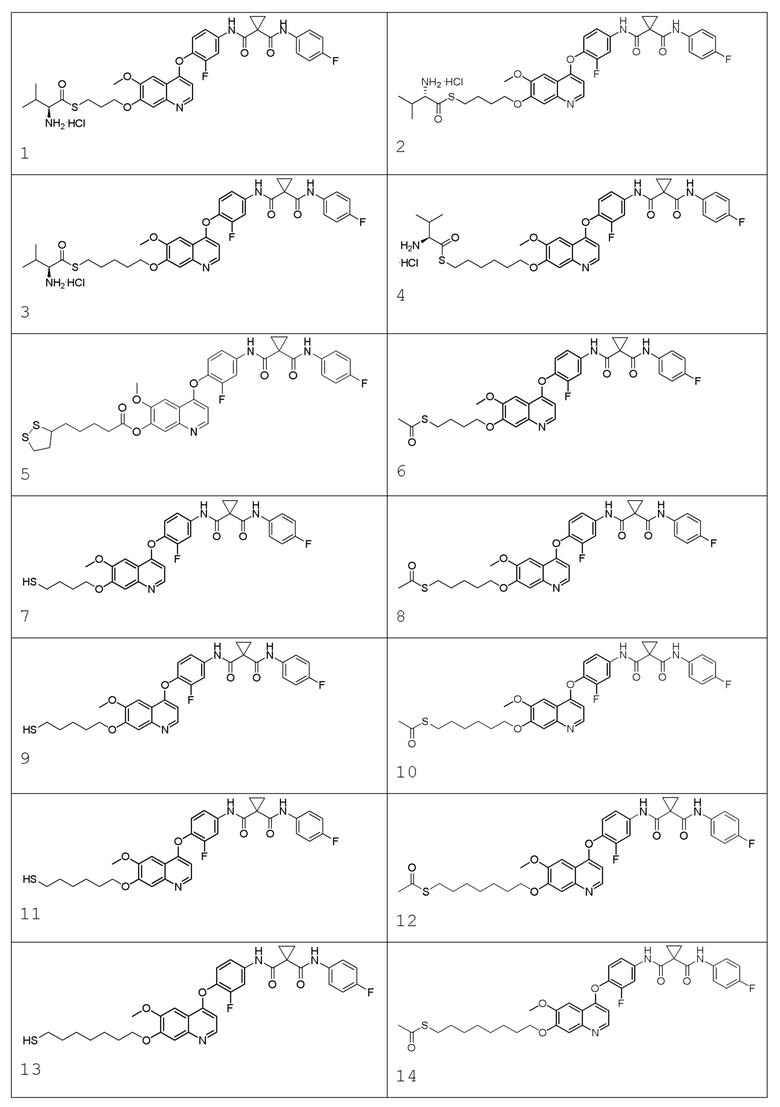
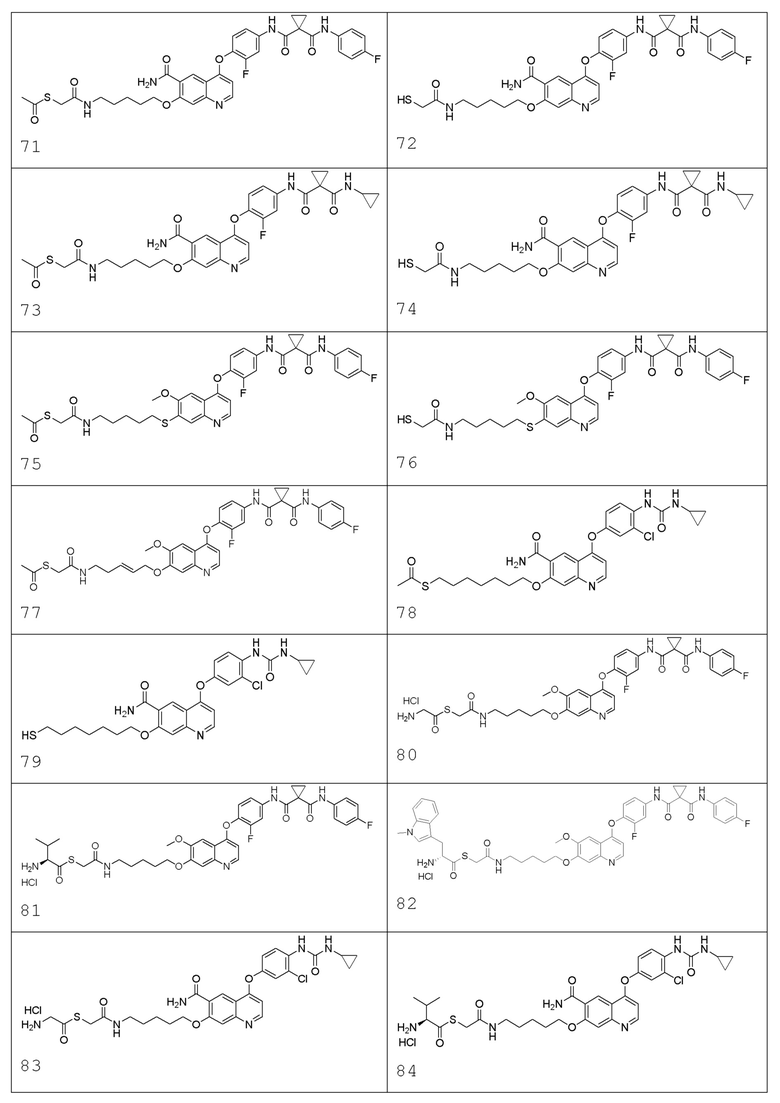
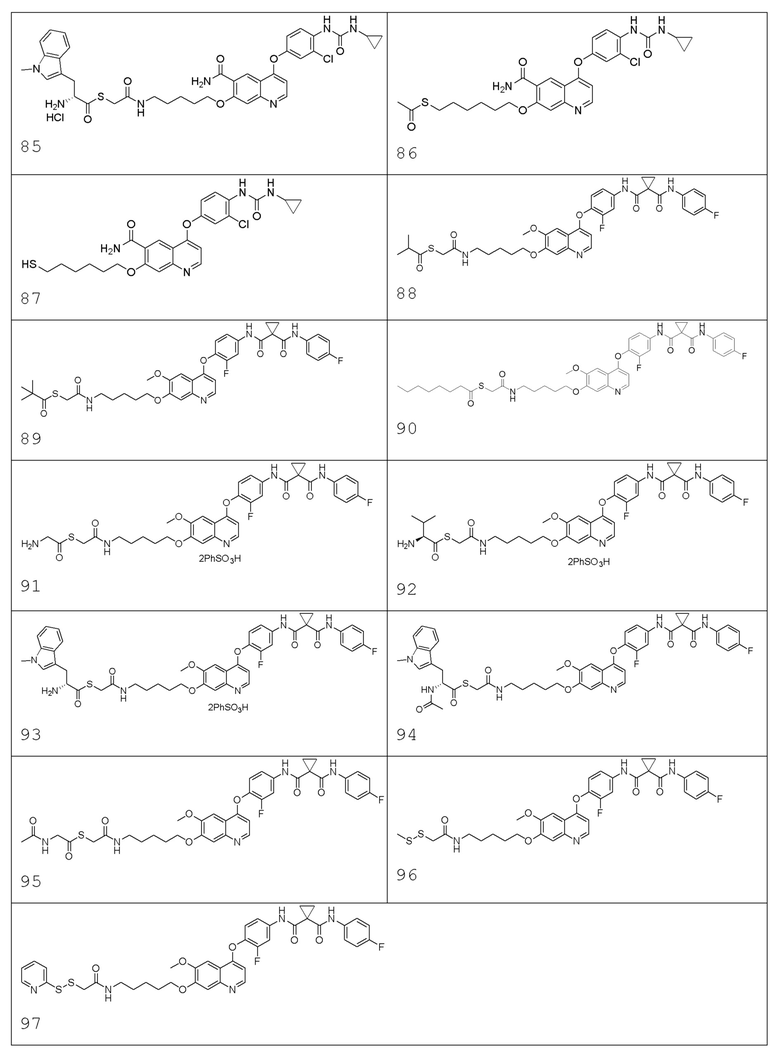
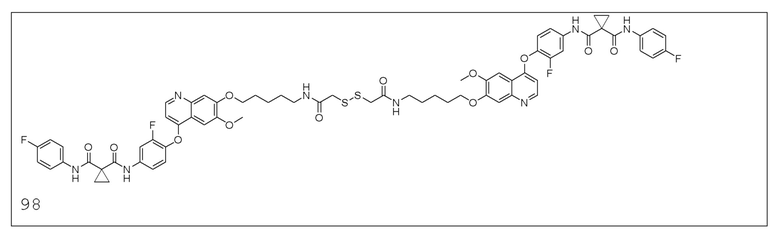
В некоторых вариантах осуществления изобретения, соединение по изобретению выбирают из следующих соединений:
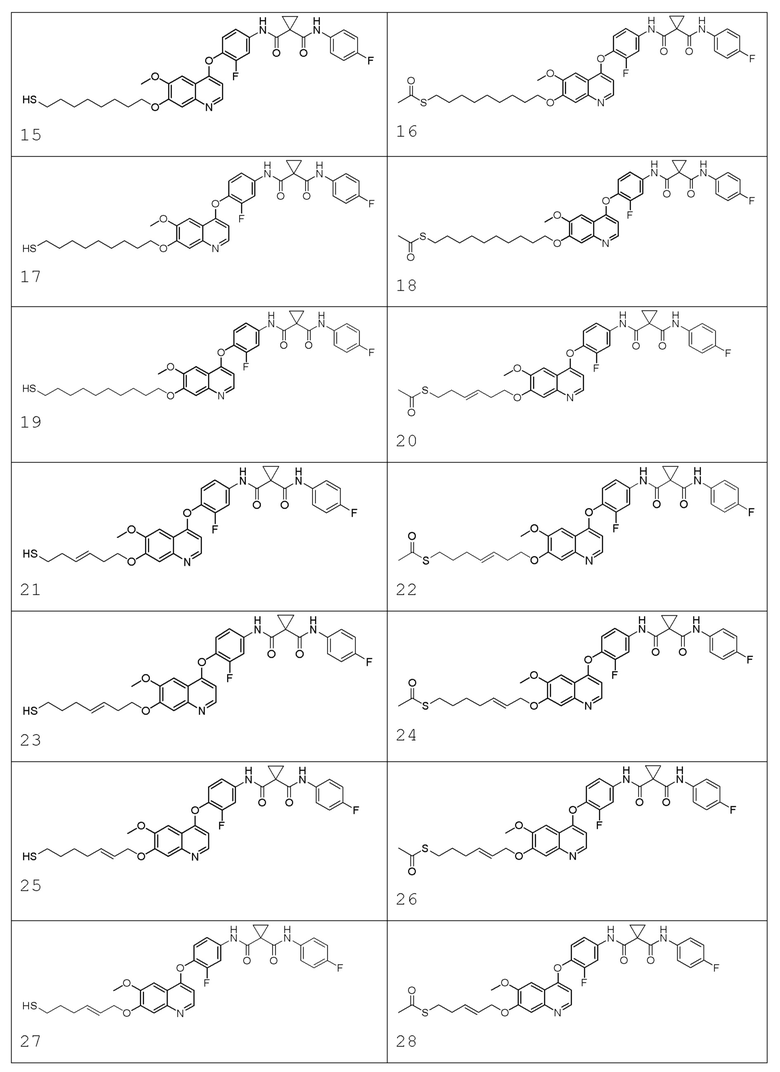
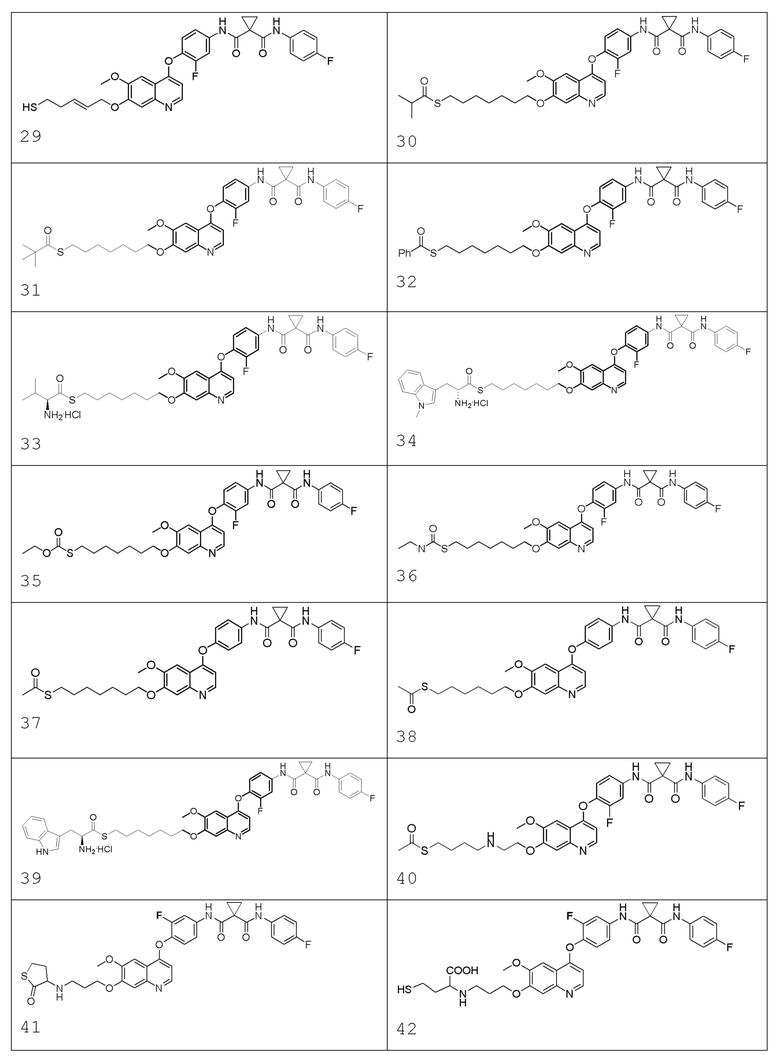
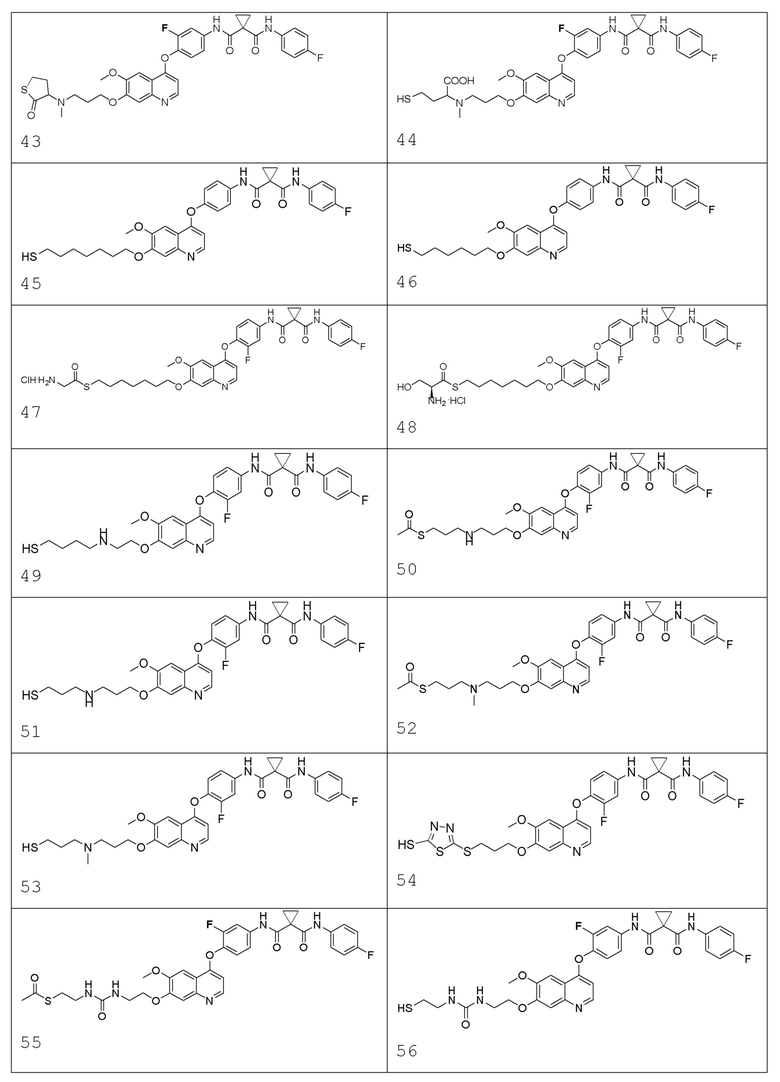
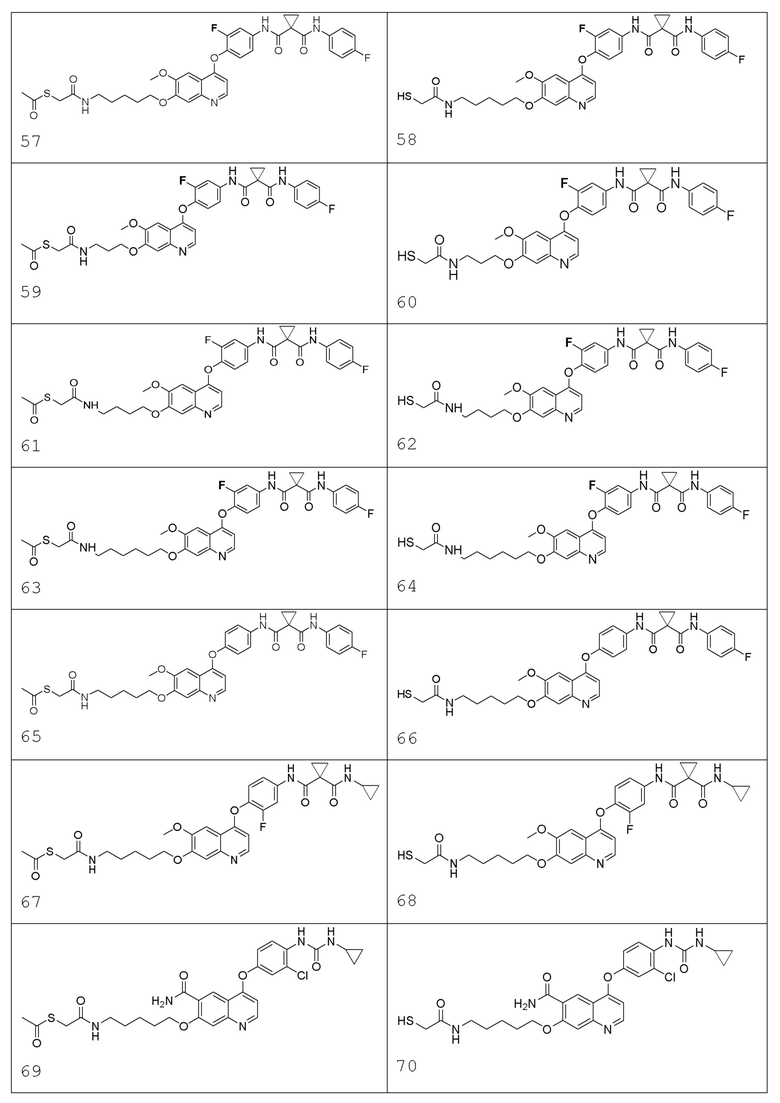
В настоящем изобретении также предлагается фармацевтическая композиция, содержащая соединение, описанное в любом одном из приведенных выше технических решениях, или его фармацевтически приемлемую соль, сольват, активный метаболит, полиморф, меченное изотопом соединение, изомер и фармацевтически приемлемый носитель.
В некоторых вариантах осуществления фармацевтической композиции по настоящему изобретению, фармацевтическая композиция может представлять собой любую обычно применяемую лекарственную форму, такую как пероральная лекарственная форма и инъекционная лекарственная форма, в том числе, но этим не ограничивая, пероральные лекарственные формы, парентеральные лекарственные формы, лекарственные формы для местного применения, ректальные лекарственные формы и другие подобные лекарственные формы. Например, фармацевтическая композиция может представлять собой таблетки, капсулы, пилюли, порошки, препараты с замедленным высвобождением, растворы и суспензии для перорального введения; стерильные растворы, суспензии или эмульсии для парентерального введения; и мази, кремы, гели, и так далее для местного применения; или суппозитории для ректального введения.
В настоящем изобретении также предлагаются упомянутые выше соединения или их фармацевтически приемлемые соли, сольваты, активные метаболиты, полиморфы, меченные изотопом соединения, изомеры или пролекарства, и упомянутые выше фармацевтические композиции для применения для получения лекарственного средства для лечения заболеваний, связанных с протеинкиназами и/или гистондеацетилазой.
В настоящем изобретении также предлагаются упомянутые выше соединения или их фармацевтически приемлемые соли, сольваты, активные метаболиты, полиморфы, меченные изотопом соединения, изомеры или пролекарства, и упомянутые выше фармацевтические композиции для применения для получения лекарственного стредства для лечения заболеваний, связанных с протеинкиназами и/или гистондеацетилазой.
В некоторых вариантах осуществления, протеинкиназы могут включать следующие категории: ALK, AXL, BTK, CDK11, c-Met, KDR, VEGFR2, RET, PDGFR-α, PDGFR-β, c-KIT, Flt3 , MEK1, MEK2, CSF1R, EPHA2, MKNK2, TIE2, TRKA, SRC, PLK4, RON, EGF1R, HER2, HER3, HER4, PDGFR-α, c-fms, FLT1, Src, Frk, Btk, CsK, Abl, Fes , Fps, Fak, AcK, Yes, Fyn, Lyn, Lck, Hck, Fgr, Yrk, PDK1, TAK1, Tie-1, YSK4, TRK B, TRK C, SLK, PKN2, MST1R, MAP4K, DDR2.
В некоторых вариантах осуществления, гистондеацетилаза может включать HDAC1, HDAC2, HDAC3, HDAC4, HDAC5, HDAC6, HDAC7, HDAC8, HDAC9, HDAC10 и HDAC11.
В некоторых вариантах осуществления, гистондеацетилаза может включать HDAC6.
В некоторых вариантах осуществления, связанные с протеинкиназами и/или гистондеацетилазами заболевания могут включать псориаз, цирроз печени, диабет, нейродегенеративные заболевания, опухоли, иммунопатологические заболевания, сердечно-сосудистые заболевания, и другие подобные заболевания.
Нейродегенеративные заболевания могут включать болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, и другие подобные заболевания. Опухоли могут включать рак легких, рак костей, рак поджелудочной железы, рак кожи, рак головы и шеи, меланому кожи или внутриглазную меланому, рак матки, рак яичников, рак прямой кишки, рак анального канала, рак желудка, рак толстой кишки, рак молочной железы, рак маточной трубы, рак эндометрия, рак шейки матки, рак влагалища, рак вульвы, болезнь Ходжкина, рак пищевода, рак тонкой кишки, рак эндокринной системы, рак щитовидной железы, рак паращитовидной железы, саркому мягких тканей, рак уретры, рак полового члена, рак предстательной железы, хронический или острый лейкоз, рак мочевого пузыря, рак почки или рак мочеточника, рак печени, новообразования центральной нервной системы, опухоли позвоночника, аденомы гипофиза, стромальные опухоли желудочно-кишечного тракта, колоректальный рак, немелкоклеточный рак легких, мелкоклеточный рак легких, мастоцитоз, глиому, саркому, лимфому и другие подобные заболевания.
В настоящем изобретении также предлагается способ лечения связанных с протеинкиназами и/или гистондеацетилазами заболеваний. Способ включает стадию введения эффективного количества упомянутого выше соединения или его фармацевтически приемлемой соли, сольвата, активного метаболита, полиморфа, меченного изотопом соединения или изомера, или упомянутой выше фармацевтической композиции субъекту, нуждающемуся в таком лечении.
В настоящем изобретении также предлагается способ ингибирования тирозинкиназы и/или гистондеацетилазы, включающий контактирование тирозинкиназы и/или гистондеацетилазы с эффективным количеством упомянутого выше соединения или его фармацевтически приемлемой соли, сольвата, активного метаболита, полиморфа, меченного изотопом соединения или изомера, или упомянутой выше фармацевтической композиции.
Предлагаемые в настоящем изобретении хинолинил-содержащие соединения обладают двойственными молекулярными функциями, могут быть использованы в качестве нацеленного на несколько мишеней ингибитора тирозикиназы/гистондеацетилазы, обладающего одновременно действием двух типов ингибиторов. Эти соединения характеризуются высокой биологической активностью и высокими фармакокинетическими свойствами, в частности, они могут найти применение при лечении опухолей.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, то все используемые в изобретении научные и технологические термины имеют значения, которые являются общепринятыми для специалистов в данной области. Если не указано иное, то содержание всех цитируемых в изобретении патентов, заявок на патенты и публикаций включено в настоящее изобретение путем ссылки на них. В случаях, когда в тексте приводится фирменное наименование, оно относится к соответствующему продукту или его активному ингредиенту.
Следует иметь в виду, что приведенное выше краткое описание и последующее подробное описание изобретения приводятся только в качестве примеров и предназначены только для разъяснения, и они не накладывают никаких ограничений на объект изобретения. Следует отметить, что если в тексте изобретения четко не указано иное, то форма единственного числа, используемая в описании и формуле изобретения, включает в себя и форму множественного числа указанного элемента. Также следует отметить, что, если не указано иное, то использование союза "или" означает "и/или". Кроме того, термин "включающий" и другие формы, такие как "состоящий" и "содержащий" являются неограничивающими.
Определения стандартных химических терминов можно найти в литературе, в том числе в учебном пособии Carey and Sundberg's "Advanced Organic Chemistry 4th Ed, Vol A (2000) and B (2001), Plenum Press, New York. Если не указано иное, то используются традиционные для данной области техники методы, например, масс-спектрометрия, ядерный магнитный резонанс (ЯМР), высокоэффективная жидкостная хроматография (HPLC), химия белков, биохимия, технология рекомбинантных ДНК и фармакологические методы. Если в изобретении специально не приведены конкретные определения, то используемые в этом изобретении соответствующие термины и лабораторные приемы и методы аналитической химии, синтетической органической химии, а также медицинской и фармацевтической химии являются хорошо известными для специалистов в данной области. Для химического синтеза, химического анализа, получения лекарственного средства, приготовления лекарственной формы, доставки лекарственного средства и лечения пациентов могут использоваться стандартные методы. Стандартные методы могут использоваться для получения рекомбинантной ДНК, синтеза олигонуклеотидов и культивирования клеток тканей и трансформации (например, электропорации, инфицирования липидов). Например, может быть использован набор с инструкциями, предлагаемый фирмой-производителем, или методы проведения реакции и выделения и очистки продуктов реакции могут быть осуществлены в соответствии со способами, известными в данной области техники, или в соответствии со способом, описанным в настоящем изобретении. В общем случае, упомянутые выше методы и способ могут быть осуществлены традиционными методами, хорошо известными в данной области техники и описанными в различных обзорных публикациях или более конкретных публикациях. Эти публикации описаны и цитируются в настоящем изобретении.
В случае, когда заместитель описывают обычной химической формулой, написанной слева направо, этот заместитель также включает в себя химически эквивалентные заместители, образуемые при написании структурной формулы справа налево. Например, формула CH2O эквивалентна формуле OCH2.
Термин "замещенный" означает, что любой один или несколько атомов водорода на конкретном атоме заменены на заместитель, при условии, что соблюдается правило валентности для конкретного атома, и соединение после замещения является стабильным. В случае, когда заместителем является оксо (то есть =O), это означает, что заменены два атома водорода, и оксо не может присутствовать в ароматической группе.
В случае, когда какая-либо переменная (например, R) встречается более одного раза в составе или структуре соединения, ее определение в каждом случае является независимым. Так, например, если группа замещена с помощью 0-2 заместителей R, то группа может быть необязательно замещена максимум с помощью двух заместителей R, и R имеет независимые варианты выбора в каждом случае. Кроме того, комбинации заместителей и/или их вариантов является разрешенными только в том случае, если такие комбинации приводят к стабильным соединениям.
Используемое в изобретении обозначение Cm-n относится к части, содержащей m-n углеродных атомов. Например, группа "C1-6" означает, что часть имеет 1-6 атомов углерода, то есть группа содержит 1 атом углерода, 2 атома углерода, 3 атома углерода ... 6 атомов углерода. Следовательно, например, "C1-6алкил" относится к алкилу, содержащему 1-6 атомов углерода, то есть алкильную группу выбирают из метила, этила, пропила, изопропила, н-бутила, изопропила, н-бутила, вторбутила, третбутила… и так далее. Диапазон числовых значений в описании изобретения, например "1-6" относятся к каждому целому числу в данном диапазоне. Например, "1-6 атомов углерода" означает, что группа может иметь 1 атом углерода, 2 атома углерода, 3 атома углерода, 4 атома углерода, 5 атомов углерода или 6 атомов углерода.
Термин "член" относится к числу скелетных атомов, образующих кольцо. Например, пиридин представляет собой шестичленное кольцо, а пиррол является пятичленным кольцом.
Термин "фармацевтически приемлемый" относится к тем соединениям, материалам, композициям и/или лекарственным формам, которые одобрены на основе проведения достоверного медицинского исследования и подходят для использования при контакте с тканями человека и животных без проявления чрезмерной токсичности, раздражения, аллергических реакций или других проблем или осложнений заболевания, и которые соответствуют приемлемой величине соотношения польза/риск.
Термин "фармацевтическая композиция" относится к биологически активному соединению, необязательно смешанному, по меньшей мере, с одним фармацевтически приемлемым химическим компонентом или веществом. Фармацевтически приемлемый химический компонент или вещество представляет собой "носитель", который способствует введению соединения в клетки или ткани. Он включает, но этим не ограничивая, стабилизаторы, разбавители, суспендирующие вещества, загустители и/или вспомогательные вещества.
Термин "фармацевтически приемлемая соль" относится к соли, которая сохраняет биологическую эффективность указанного соединения в форме свободной кислоты и свободного основания и не оказывает неблагоприятного воздействия на биологические или другие аспекты. Если не указано иное, то соли в настоящем изобретении могут включать соли металлов, соли аммония, соли, образованные с органическими основаниями, соли, образованные с неорганическими кислотами, соли, образованные с органическими кислотами, соли, образованные с аминокислотами, обладающими основными или кислотными свойствами, и другие подобные соли. Неограничивающие примеры солей металлов включают соли щелочных металлов, такие как натриевая соль, калиевая соль и соли других подобных металлов; соли щелочноземельных металлов, такие как соль кальциевая соль, магниевая соль, бариевая соль и соли других подобных металлов; соль алюминия и соли других подобных металлов. Неограничивающие примеры солей, образованных с органическими основаниями, включают соли, образованные с триметиламином, триэтиламином, пиридином, пиколином, 2,6-лутидином, этаноламином, диэтаноламином, триэтаноламином, циклогексиламином, дициклогексиламином и с другими подобными органическими основаниями. Неограничивающие примеры солей, образованных с неорганическими кислотами, включают соли, образованные с хлористоводородной кислотой, бромистоводородной кислотой, азотной кислотой, серной кислотой, фосфорной кислотой и с другими подобными кислотами. Неограничивающие примеры солей, образованных с органическими кислотами, включают соли, образованные с муравьиной кислотой, уксусной кислотой, трифторуксусной кислотой, фумаровой кислотой, щавелевой кислотой, яблочной кислотой, малеиновой кислотой, винной кислотой, лимонной кислотой, янтарной кислотой, метансульфоновой кислотой, бензолсульфоновой кислотой, п-толуолсульфоновой кислотой и с другими подобными кислотами. Неограничивающие примеры солей, образованных с аминокислотами, обладающих основными свойствами, включают соли, образованные с аргинином, лизином, орнитином и с другими подобными аминокислотами. Неограничивающие примеры солей, образованных с аминокислотами, обладающих основными свойствами, включают соли, образованные с аспарагиновой кислотой, глутаминовой кислотой и с другими подобными аминокислотами.
Фармацевтически приемлемые соли могут быть синтезированы из исходных соединений, содержащих кислотные радикалы или основания, традиционными химическими методами. Обычно такие соли получают взаимодействием этих соединений в форме свободной кислоты или основания со стехиометрическим количеством подходящего основания или кислоты в воде или органическом растворителе или в их смеси. Обычно предпочтительными являются неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил.
Термин "сольват" относится к физическому агрегату, образованному соединением по настоящему изобретению с одной или несколькими молекулами растворителя. Этот физический агрегат включает, в разной степени, ионы и ковалентные связи, такие как водородные связи. Было показано, что этот сольват может быть выделен, например, в случае, когда одна или несколько молекул растворителя смешиваются в кристаллической решетке. "Сольват" включает как фазу растворителя, так и выделяемый сольват. Существует множество примеров соответствующих сольватов, в том числе этанольные сольваты, метанольные сольваты и другие подобные сольваты. "Гидрат" представляет собой сольват, в которой в качестве растворителя используются молекулы воды (H2O). Одно или более соединений в настоящем изобретении могут быть приготовлены, при желании, в форме сольватов. Получение сольватов хорошо известно. Например, в публикации M. Caira et al., J. Pharmaceutical Sci., 93 (3), 601-611 (2004) описано получение сольвата противогрибкового лекарственного средства флуконазола, то есть получение сольвата при использовании этилацетата и воды. В публикациях E.C. van Tonder et al, AAPS PharmSciTech., 5(1), article 12 (2004); и AL Bingham et al, Chem. Commun., 603-604 (2001) также описаны аналогичные методы получения сольватов и гидратов. Типичный неограничивающий метод получения заключается в растворении соединения по настоящему изобретению в идеальном растворителе (органическом растворителе или воде или в их смешанном растворителе) при температуре выше нормальной температуры, затем охлаждении и выдерживании во времени для кристаллизации. Затем кристаллы разделяют стандартными методами. Путем использования метода ИК-спектроскопии может быть подтверждено наличие растворителя (воды), который образует сольват (гидрат) в кристалле.
Термин "активный метаболит" относится к активному производному соединения, образующемуся в результате метаболизма соединения.
Термин "полиморфы" относится к соединениям по настоящему изобретению, которые имеют различные формы кристаллической решетки.
Термин "меченное изотопом соединение" относится к меченному изотопом соединению по настоящему изобретению. Например, изотопы в соединении по настоящему изобретению могут включать различные изотопы элементов, таких как H, C, N, O, P, F, S, например 2H, 3H, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F и 36S.
Термин "стереоизомеры" относится к изомерам, образующимся в результате различного расположения в пространстве атомов в молекуле. Соединения по настоящему изобретению содержат такие элементы структуры, как центры асимметрии или хиральные центры и двойные связи. Вследствие этого, соединения по настоящему изобретению могут включать оптические изомеры, геометрические изомеры, таутомеры, атропоизомеры и другие изомеры. Все эти изомеры и их индивидуальные изомеры, рацематы и другие формы изомеров включены в объем настоящего изобретения. Например, в случае оптических изомеров, оптически активные (R)- и (S)-изомеры и D и L изомеры могут быть получены методами хирального разделения, хирального синтеза или методами с использованием хиральных реагентов или другими традиционными методами. Например, оптический изомер может быть превращен в диастереомеры путем взаимодействия с соответствующими оптически активными веществами (такими как хиральные спирты или хлорангидрид кислоты Мошера), выделен и превращен (например, путем гидролиза) в соответствующий индивидуальный изомер. В качестве другого примера, он может быть также выделен методом колоночной хроматографии.
"Фармацевтические композиции" в изобретении могут быть приготовлены хорошо известным в фармацевтике методом, и они могут быть введены или нанесены различными способами, в зависимости от того, требуется ли местное или системное лечение, и от области, подвергаемой лечению. Они могут быть введены местно (например, трансдермально, на кожу, в глаза и слизистые оболочки, включая интраназальную, вагинальную и ректальную доставку), введены ингаляционно (например, путем ингаляции или инсуффляции порошка или аэрозоля, в том числе через распылители, интратрахеально, интраназально), введены перорально или парентерально. Парентеральное введение включает внутривенную, интраартериальную, подкожную, интраперитонеальную или внутримышечную инъекцию или инфузию; или интракраниальное введение, такое как интратекальное или интрацеребровентрикулярное введение. Они могут быть введены парентерально в форме разовой болюсной дозы или, например, в форме инфузии при постоянном разряжении в шприце. Фармацевтическая композиция в изобретении включает, но этим не ограничивая, следующие формы: таблетки, пилюли, порошки, пастилки, порошки для приготовления раствора, крахмальные капсулы, эликсиры, суспензии, эмульсии, растворы, сиропы, аэрозоли (твердые или растворенные в жидком носителе); например, мази, мягкие и твердые желатиновые капсулы, суппозитории, стерильные растворы для инъекций и порошки в стерильной упаковке.
Термин "лечение" относится к терапевтическому лечению и предотвращению заболевания, или к профилактическим мерам, где цель состоит в том, чтобы предотвратить или замедлить (облегчить) нежелательные физиологические изменения или заболевания, такие как развитие или распространение рака. Применительно к целям настоящего изобретения, положительные или ожидаемые клинические результаты, обнаруживаемые или не обнаруживаемые, включают, но этим не ограничивая, облегчение симптомов, снижение степени тяжести заболевания, стабилизацию (то есть отсутствие ухудшения) состояния, отсрочку или замедление прогрессирования заболевания, улучшение или облегчение (частичное или полное) состояния. По сравнению с ожидаемой выживаемостью (без проведения лечения), "лечение" также подразумевает продление периода времени выживаемости. Пациенты, нуждающиеся в лечении, включают тех пациентов, которые уже имеют болезненное состояние или заболевание, и тех пациентов, которые имеют склонность к возникновению у них болезненного состояния или заболевание, или тех пациентов, которые хотят предотвратить болезненное состояние или заболевание.
Термин "введение" включает способ, с помощью которого соединение вводят субъекту для достижения требуемого воздействия. Примеры способа введения, которые могут быть использованы, включают инъекцию (подкожную, внутривенную, парентеральную, интраперитонеальную, интратекальную), местное введение, пероральное введение, ингаляцию, ректальное введение и трансдермальное введение.
Термин "эффективное количество" включает количество, которое является эффективным для достижения требуемого результата в пределах необходимой дозы и периода времени. Эффективное количество соединения может зависеть от таких факторов, как состояние субъекта, возраст и масса тела, а также от способности соединения вызывать требуемый ответ у субъекта. Режим дозирования может быть скорректирован для обеспечения наилучшего терапевтического ответа. Эффективное количество также представляет собой количество, при котором положительный терапевтический эффект соединения превышает любые из его токсических или вредных эффектов (таких как побочные эффекты).
Используемые в изобретении фразы "системно вводимое" и "периферически вводимое" означает введение соединения, лекарственного средства или другого вещества, для того чтобы оно проникло в организм пациента и, вследствие этого, было подвергнуто метаболическому процессу и другим аналогичным процессам.
Фраза "терапевтически эффективное количество" обозначает количество соединения по настоящему изобретению, которое позволяет (i) проводить лечение или предотвращать конкретное заболевание или состояние, (ii) ослаблять, улучшать или устранять один или несколько симптомов конкретного заболевания или состояния, или (iii) предотвращать или замедлять проявление одного или нескольких симптомов конкретного заболевания или состояния, описанных в изобретении.
Термин "субъект" относится к животным, таким как млекопитающие, включая, но этим не ограничивая, приматов (таких как люди), коров, овец, коз, лошадей, собак, кошек, кроликов, крыс, мышей и других подобных животных. В некоторых вариантах осуществления, субъектом является человек.
Фармацевтическая композиция может быть приготовлена в виде лекарственной формы с разовой дозой, и каждая доза может содержать приблизительно от 0,1 до 1000 мг, например, приблизительно от 5 до 1000 мг или приблизительно от 100 до 500 мг активного ингредиента. Термин "лекарственная форма с разовой дозой" относится к физически разделенному единичному элементу разовой дозы, подходящему для использования у людей и других млекопитающих, и каждый единичный элемент содержит заданное количество активного вещества, которое рассчитано для достижения требуемого терапевтического эффекта, смешанное с подходящим фармацевтическим носителем.
ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Для того чтобы задачи, технические решения и положительные эффекты настоящего изобретения стали более понятными, ниже будут дополнительно описаны технические решения в примерах вариантов осуществления настоящего изобретения.
В настоящем изобретении, соединения, описанные в настоящем изобретении, могут быть получены представленными далее методами. Представленные далее методы и примеры служат для иллюстрации этих методов. Эти методы и примеры никоим образом не следует рассматривать в качестве ограничения настоящего изобретения. Описанные в изобретении соединения также могут быть синтезированы с использованием стандартных методов синтеза, известных специалистам в данной области техники, или методы, известные в данной области техники, и методы, описанные в изобретении, могут быть использованы в комбинации.
В настоящем изобретении, соединения, описанные в настоящем изобретении, могут быть получены представленными далее методами. Представленные далее методы и примеры служат для иллюстрации этих методов. Эти методы и примеры никоим образом не следует рассматривать в качестве ограничения настоящего изобретения. Описанные в изобретении соединения также могут быть синтезированы с использованием стандартных методов синтеза, известных специалистам в данной области техники, или методы, известные в данной области техники, и методы, описанные в изобретении, могут быть использованы в комбинации.
Химические реакции в вариантах осуществления настоящего изобретения проводят в подходящем растворителе, и растворитель должен соответствовать химическим превращениям, описанным в настоящем изобретении, и применяемым реагентам и материалам. Для того чтобы получить соединения по настоящему изобретению, специалисты в данной области иногда вынуждены изменять или выбирать метод синтеза или схемы реакций применительно к существующим вариантам осуществления.
Важным соображением при планировании любого пути синтеза в данной области является выбор подходящей защитной группы для реакционноспособной функциональной группы (такой как аминогруппа в настоящем изобретении). Для квалифицированных специалистов-практиков в области органического синтеза авторитетным руководством в этом отношении является монография Greene and Wuts, Protective Groups In Organic Synthesis, Wiley and Sons, 1991. Содержание всех публикаций, процитированных в настоящем изобретении, включено в настоящее изобретение путем ссылок на них.
Протекание описанных в изобретении реакций может постоянно контролироваться любым подходящим методом, известным в данной области. Например, образование продукта можно контролировать с помощью широко применяемого метода, такого как спектроскопия ядерного магнитного резонанса (например, 1H или 13C), инфракрасная спектроскопия, спектрофотометрия (например, УФ-видимый диапазон спектра), масс-спектрометрия и так далее, или такого как хроматография, такая как высокоэффективная жидкостная хроматография (HPLC) или тонкослойная хроматография.
Пример синтеза 1
N-(3-фтор-4-((7-гидроксил-6-метоксихинолин-4-ил)окси)-фенил)-N-(4-фторфенил)циклопропил-1,1-диформамид
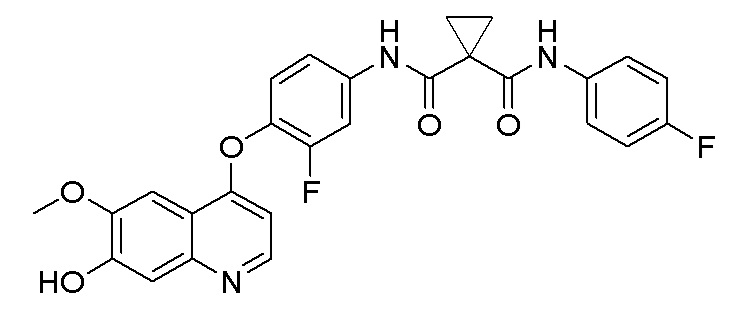
Стадия A. 7-(Бензилокси)-4-(2-фтор-4-нитрофенокси)-6-метоксихинолин
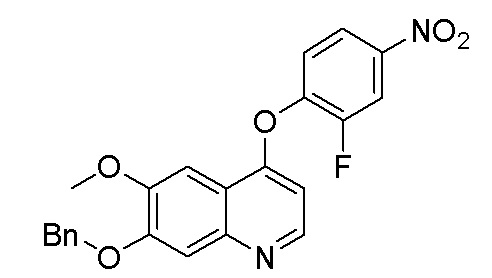
Растворяли 29,9 г (100 ммоль, 1,0 экв) 7-(бензилокси)-4-хлор-6-метоксихинолина и 23,4 г (150 ммоль, 1,5 экв) 2-фтор-4-нитрофенола в 10 мл DMF. Затем добавляли 41,4 г (300 ммоль, 3,0 экв) порошка карбоната калия при комнатной температуре. Затем перемешивали реакционный раствор при комнатной температуре в течение ночи. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), реакционную систему выливали в 550 мл воды, экстрагировали с помощью 600 мл этилацетата, и органическую фазу последовательно промывали два раза водой и насыщенным солевым раствором, органическую фазу сушили над безводным сульфатом натрия, испаряли досуха, и остаток очищали колоночной хроматографией на силикагеле с получением продукта (18,9 г, выход=45%).
Стадия B. 4-(4-Амино-2-фторфенокси)-6-метоксихинолин-7-ол
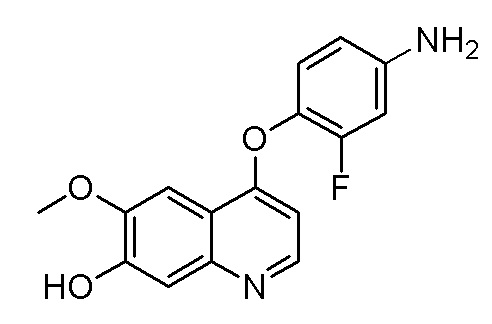
Растворяли 18,0 г (42,9 ммоль, 1,0 экв) 7-(бензилокси)-4-(2-фтор-4-нитрофенокси)-6-метоксихинолина и 0,9 г Pd/C в 180 мл метанола. В атмосфере водорода при давлении 0,3 МПа, три раза проводили замену газа. Реакционную смесь перемешивали в течение ночи при комнатной температуре. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), реакционную систему фильтровали под вакуумом. Осадок на фильтре промывали метанолом, и органическую фазу испаряли досуха при пониженном давлении с получением продукта (10,9 г, выход=85%).
Стадия C. N-(3-фтор-4-((7-гидроксил-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диформамид
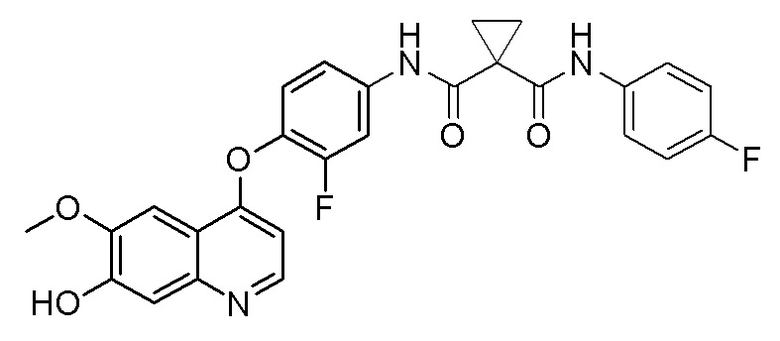
9,0 г (30 ммоль, 1,0 экв) 4-(4-амино-2-фторфенокси)-6-метоксихинолин-7-ола, 8,0 г (36 ммоль, 1,2 экв) 1-((4-(фторфенил) формамидо)циклопропан-1-карбоновой кислоты и 13,8 г (36 ммоль, 1,2 экв) HATU растворяли в 200 мл дихлорметана. Затем добавляли 19,5 г (150 ммоль, 5,0 экв) диизопропилэтиламина. Затем реакционную смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), реакционную систему выливали в 300 мл воды, и добавляли 150 мл DCM для проведения экстракции. Органическую фазу последовательно промывали два раза водным раствором лимонной кислоты и насыщенным солевым растворов, и органическую фазу сушили над безводным сульфатом натрия и испаряли досуха. Остаток очищали колоночной хроматографией на силикагеле с получением продукта (9,5 г, выход=63%).
LC-MS: m/z=506 [M+H] +.
Пример синтеза 2. N-(4-фторфенил)-N-(4-((7-гидроксил-6-метоксихинолин-4-ил)окси)фенил)циклопропан-1,1-диформамид
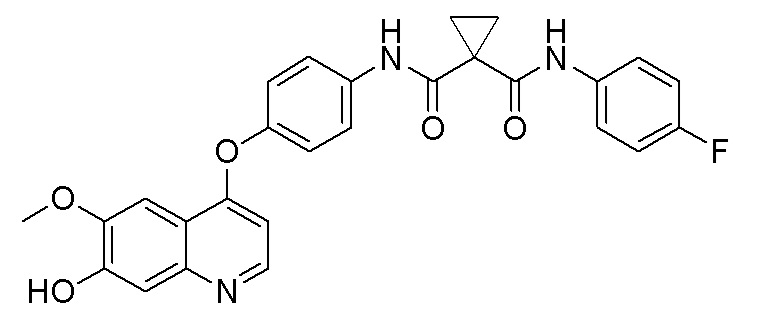
Метод синтеза был аналогичен методу синтеза, описанному в примере синтеза 1, за исключением того, что 2-фтор-4-нитрофенол заменяли на 4-нитрофенол, получая целевое соединение (3,2 г, выход=65%).
LC-MS: m/z=488 [M+H] +.
Пример синтеза 3. N-циклопропил-N-(3-фтор-4-((7-гидроксил-6-метоксихинолин-4-ил)окси)фенил)циклопропан-1,1-диформамид
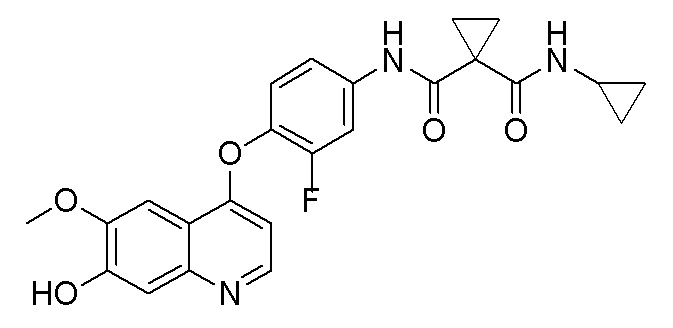
Метод синтеза был аналогичен методу синтеза, описанному в примере синтеза 1, за исключением того, что 4-фторанилин заменяли на циклопропиламин, получая целевое соединение (5,8 г, выход=53%).
1H ЯМР (400 МГц, DMSO-d6) δ 10,91 (с, 1H), 10,16 (с, 1H), 8,41 (с, 1H), 7,91-7,88 (м, 2H), 7,51-7,34 (м, 3H), 7,29 (с, 1H), 6,35 (д, J=4,0 Гц, 1H), 3,96(с, 3H), 2,71-2,66 (м, 1H), 1,36 (с, 4H), 0,65-0,64 (м, 2H), 0,63-0,62 (м, 2H).
LC-MS: m/z=452 [M+H] +.
Пример синтеза 4. N-(4-((6-аминоформил-7-гидроксихинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)циклопропан-1,1-диформамид
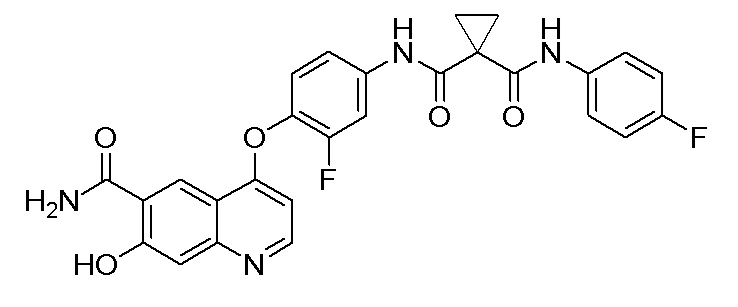
Стадия A. 7-(Бензилокси)-4-(2-фтор-4-(1-((4-фторфенил)-аминоформил)циклопропан-1-карбоксамидо)фенокси)хинолин-6-метилформиат
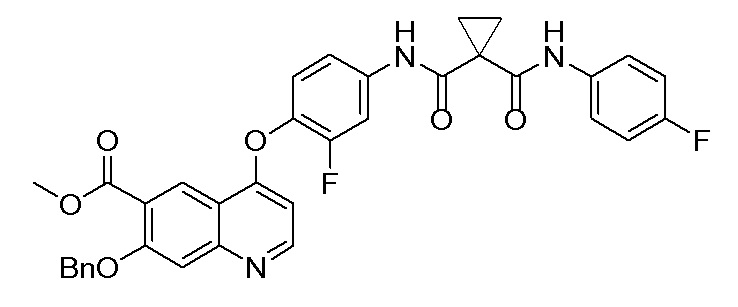
Стадия синтеза была аналогичной стадии синтеза, описанной в примере синтеза 1, за исключением того, что 7-(бензилокси)-4-хлор-6-метоксихинолин заменяли на метиловый эфир 7-(бензилокси)-4-хлорхинолин-6-карбоновой кислоты, получая целевое соединение (32,0 г, выход=65%).
LC-MS: m/z=624 [M+H] +.
Стадия B. 7-(Бензилокси)-4-(2-фтор-4-(1-((4-фторфенил)-аминоформил)циклопропан-1-карбоксамидо)фенокси)хинолин-6-карбоновая кислота
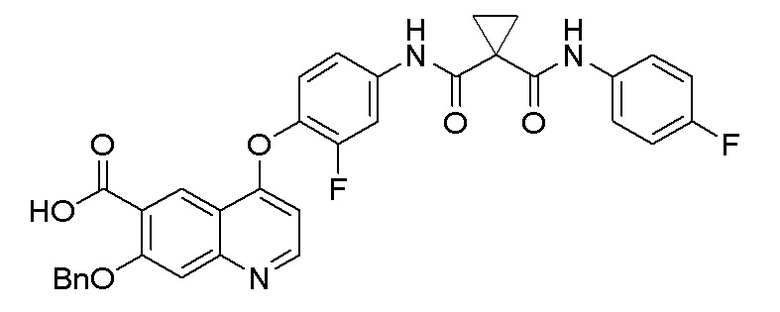
15,0 г (24,0 ммоль, 1,0 экв) 7-(бензилокси)-4-(2-фтор-4-(1-((4-фторфенил)аминоформил)циклопропан-1-карбоксамидо)- фенокси)хинолин-6-метилформиата растворяли в 150 мл метанола, и медленно добавляли в реакционную систему 4N водный раствор гидроксида натрия (15 мл) на ледяной бане. После добавления, проводили реакцию при комнатной температуре в течение 1 часа, и затем добавляли 1N водный раствор хлористоводородной кислоты на ледяной бане для доведения величины рН до нейтрального значения. Собирали твердое вещество фильтрацией под вакуумом и сушили его естественным образом с получением неочищенного продукта, который непосредственно использовали на следующей стадии (13,6 г, выход=93%). LC-MS: m/z=610 [M+H] +.
Стадия C. N-(4-((7-(бензилокси)-6-аминокарбамоилхинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)циклопропан-1,1-диформамид
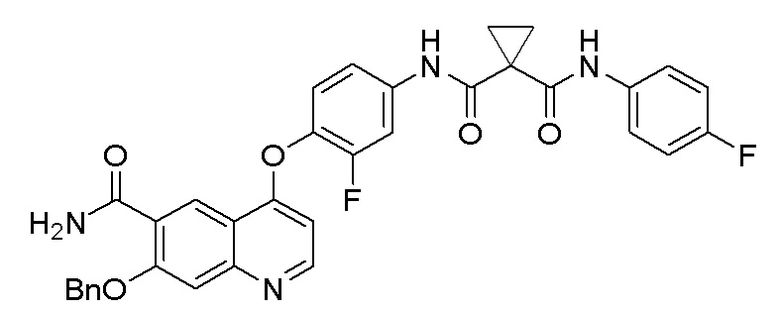
9,0 г (14,8 ммоль, 1,0 экв) 7-(бензилокси)-4-(2-фтор-4-(1-((4-фторфенил)аминоформил)циклопропан-1-карбоксамидо)фенокси)-хинолин-6-карбоновой кислоты и 11,3 г (29,6 ммоль, 2,0 экв) HATU растворяли в 1N растворе аммиака в дихлорметане (100 мл). Колбу герметизировали, и реакционный раствор перемешивали при комнатной температуре в течение ночи. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), реакционную систему выливали в 300 мл воды. Органическую фазу последовательно промывали два раза водным раствором лимонной кислоты и насыщенным солевой раствор, и органическую фазу сушили над безводным сульфатом натрия, испаряли досуха. Остаток очищали колоночной хроматографией на силикагеле с получением продукта (5,2 г, выход=58%). LC-MS: m/z=609 [M+H] +.
Стадия D. N-(4-((6-аминоформил-7-гидроксихинолин-4-ил)-окси)-3-фторфенил)-N-(4-фторфенил)циклопропан-1,1-диформамид
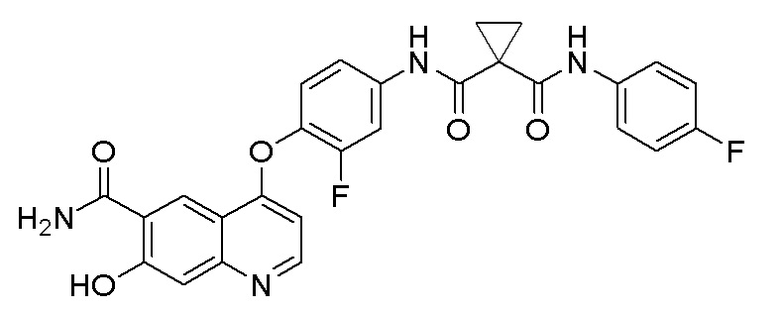
5,0 г (8,22 ммоль, 1,0 экв) N-(4-((7-(бензилокси)-6-аминоформилхинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)- циклопропан-1,1-диформамида и 0,5 г Pd/C растворяли и 60 мл метанола, и в атмосфере водорода при давлении 0,3 МПа три раза газ заменяли. Реакционную смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), реакционную систему фильтровали под вакуумом. Осадок на фильтре промывали метанолом, и органическую фазу испаряли досуха при пониженном давлении с получением продукта (3,6 г, выход=85%).
1H ЯМР (400 МГц, DMSO-d6) δ 12,99 (с, 1H), 10,44 (с, 1H), 10,03 (с, 1H), 9,01 (с, 1H), 8,83 (уш.с, 1H), 8,63 (д, J=4,0 Гц, 1H), 8,17(уш.с, 1H), 7,93 (дд, J=8,0, 4,0 Гц, 1H), 7,67--7,59 (м, 2H), 7,51--7,45 (м, 1H), 7,44--7,38 (м, 1H), 7,17 (кв, J=8,0 Гц, 2H), 6,39 (д, J=4,0 Гц, 1H), 1,48 (с, 2H), 1,47 (с, 2H).
LC-MS: m/z=519 [M+H] +.
Пример синтеза 5. N-(4-((6-карбамоил-7-гидроксихинолин-4-ил)окси)-3-фторфенил)-N-циклопропилциклопропан-1,1-дикарбоксамид
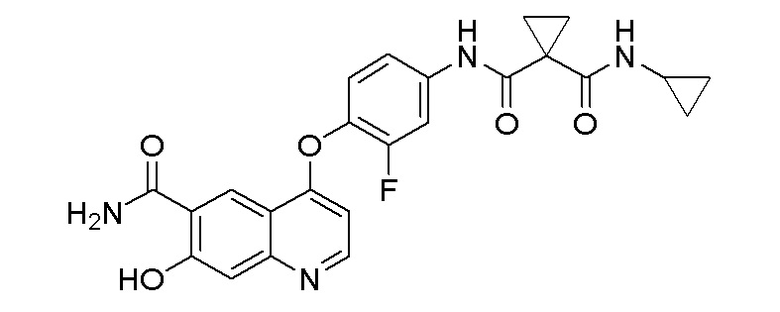
Метод синтеза был аналогичен методу синтеза, описанному в примере синтеза 4, за исключением того, что 4-фторанилин заменяли на циклопропиламин, получая целевое соединение (2,7 г, выход=62%).
1H ЯМР (400 МГц, DMSO-d6) δ 12,99 (с, 1H), 10,94 (с, 1H), 9,00 (с, 1H), 8,82 (с, 1H), 8,63 (д, J=4,0 Гц, 1H), 8,15 (с, 1H), 7,94-7,91 (м, 2H), 7,52-7,44 (м, 2H), 7,34 (с, 1H), 6,39 (с, d, J=4,0 Гц, 1H), 2,71-2,51 (м, 1H), 1,36 (с, 4H), 0,66-0,56 (м, 2H), 0,52-0,39 (м, 2H).
LC-MS: m/z=465 [M+H] +.
Пример 1. S-(3-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)-циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)-окси)пропил)-(S)-2-амино-3-метилтиобутирата гидрохлорид
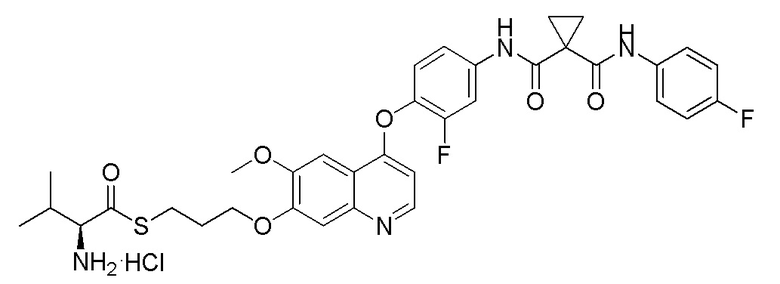
Стадия A. (S)-2-(N-третбутоксикарбонил)-3-метилтиомасляная кислота
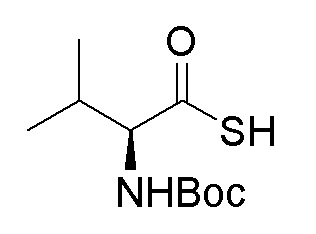
Добавляли 1 г (4,6 ммоль, 1,0 экв) N-третбутоксикарбонил-L-валина и 1,19 г (9,2 ммоль, 2,0 экв) DIPEA в 15 мл тетрагидрофурана и растворяли путем перемешивания с использованием магнитной мешалки, и затем охлаждали до 0~5°C на бане с ледяной водой. Растворяли 470 мг (4,6 ммоль, 1,0 экв) уксусный ангидрид в 5 мл THF, и затем медленно добавляли его по каплям в реакционную колбу. После добавления, перемешивали с помощью магнитной мешалки в течение 30 минут, и затем добавляли в реакционную колбу 389 мг (6,9 ммоль, 1,5 экв) NaSH. Удаляли баню с ледяной водой, и проводили реакцию в течение 10 часов при комнатной температуре. После завершения реакции, испаряли THF, затем добавляли 30 мл воды, и доводили величину рН pH 4 с помощью 10% раствора KHSO4. Экстрагировали два раза этилацетатом (50 мл × 2), собирали органическую фазу, промывали органическую фазу один раз насыщенным солевым раствором, сушили органическую фазу над безводным сульфатом натрия, и испаряли органическую фазу при центрифугировании с получением продукта в виде темно-желтой жидкости (483 мг, выход=45%), которую затем непосредственно использовали на следующей стадии.
Стадия B. N-(4-((7-(3-бромпропокси)-6-метоксихинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)циклопропан-1,1-диформамид
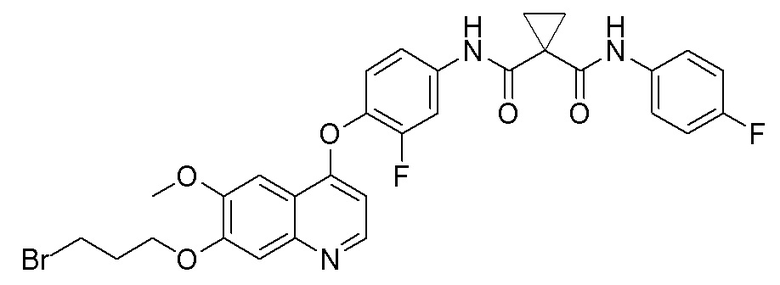
505 мг (1 ммоль, 1,0 экв) N-(3-фтор-4-((7-гидроксил-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диметилформамида (по поводу метода синтеза смотрите Пример синтеза 1) и 732 мг (3 ммоль, 3,0 экв) 1,3-дибромпропана растворяли в 10 мл N, N-диметилформамида. Добавляли 414 мг (3,0 ммоль, 3,0 экв) порошка карбоната калия при комнатной температуре. После добавления, реакционную систему перемешивали в течение ночи при комнатной температуре. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), реакционную систему выливали в 150 мл воды, и затем экстрагировали с помощью 200 мл этилацетата. Органическую фазу промывали два раза последовательно водой и насыщенным солевым раствором. Органическую фазу сушили над безводным сульфатом натрия и испаряли досуха. Остаток очищали колоночной хроматографией на силикагеле с получением продукта (600 мг, выход=95%).
Стадия C. S-(3-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)пропил)-(S)-2-((третбутоксикарбонил)амино)-3-метил-тиобутират
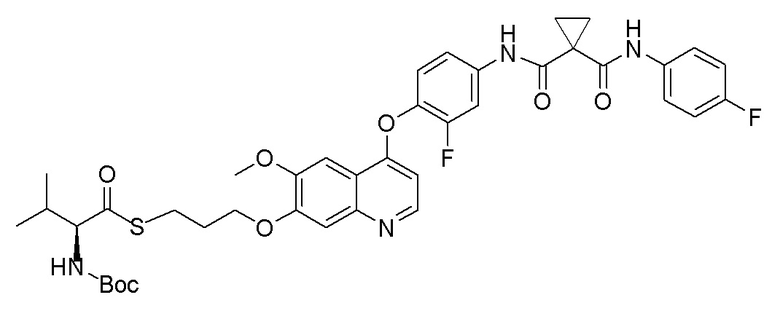
500 мг (0,80 ммоль, 1,0 экв) N-(4-((7-(3-бромпропокси)-6-метоксихинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)-циклопропан-1,1-диформамида и 332 мг (2,4 ммоль, 3,0 экв) порошка карбоната калия растворяли в 20 мл ацетона. Добавляли 560 мг (2,4 ммоль, 3,0 экв) (S)-2-(N-третбутоксикарбонил)-3-метилтиомасляной кислоты при комнатной температуре. Проводили реакцию при комнатной температуре в течение 4 часов. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), растворитель испаряли досуха. К остатку добавляли 150 мл воды, и затем экстрагировали с помощью 250 мл этилацетата. После проведения экстракции, органическую фазу промывали два раза последовательно водой и насыщенным солевым раствором, и затем органическую фазу сушили над безводным сульфатом натрия, испаряли досуха. Остаток очищали колоночной хроматографией на силикагеле с получением продукта (401 мг, выход=63%).
1H ЯМР (400 МГц, CDCl3) δ 10,08 (с, 1H), 8,62 (с, 1H), 7,81 (д, J=12,0 Гц, 1H), 7,60 (с, 2H), 7,54-7,47 (м, 2H), 7,34 (с, 1H), 7,30 (с, 1H), 7,25 (т, J=8,6 Гц, 1H), 7,08 (т, J=8,6 Гц, 2H), 6,50 (с, 1H), 5,12 (с, 1H), 4,30-4,01 (м, 3H), 4,07 (с, 3H), 3,13 (с,2H), 2,30-2,00 (м, 3H), 1,82 (с, 2H), 1,68 (с, 2H), 1,50 (с, 9H), 1,02 (д, J=6,7 Гц, 3H), 0,90 (д, J=6,7 Гц, 3H).
Стадия D. S-(3-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)пропил)-(S)-2-амино-3-метилтиобутирата гидрохлорид
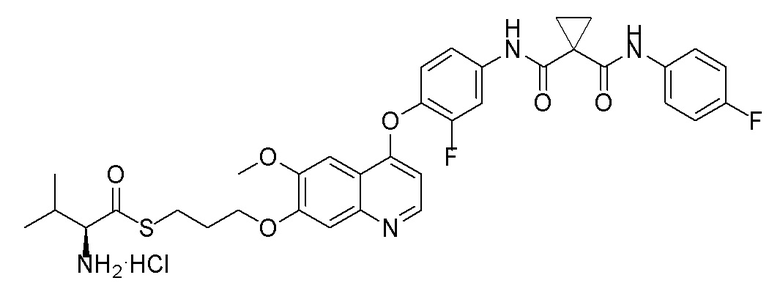
40 мг (0,14 ммоль, 1,0 экв) S-(3-((4-(2-фтор-4-(1-((4-фторфенил)аминоформил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)пропил)-(S)-2-((третбутоксикарбонил)-амино)-3-метилтиобутирата растворяли в 10 мл этилацетата. Пропускали через систему хлористый водород и проводили реакцию в течение 30 минут при комнатной температуре. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), реакционный раствор концентрировали и испаряли досуха. Остаток суспендировали в 15 мл эфира при перемешивании. После фильтрации под вакуумом, твердое вещество споласкивали эфиром и затем сушили под вакуумом с получением продукта (35 мг, выход=95%).
1H ЯМР (400 МГц, d-DMSO) δ 10,54 (с, 1H), 10,01 (с, 1H), 8,81 (с, 1H), 8,53 (с, 2H), 8,00 (д, J=14,1 Гц, 1H), 7,76 (с, 1H), 7,73 (с, 1H), 7,70-7,62 (м, 3H), 7,61 (с, 1H), 7,56 (д, J=9,1 Гц, 1H), 7,17 (т, J=8,9 Гц, 2H), 6,90 (с, 1H), 4,25 (т, J=8,0 Гц, 2H), 4,19 (с, 1H), 4,06 (с, 3H), 3,27-3,16 (м, 2H), 2,25-2,10 (м, 3H), 1,51 (д, J=10,3 Гц, 4H), 1,00 (дд, J=11,5, 7,0 Гц, 6H).
LC-MS: m/z=679 [M+H] +.
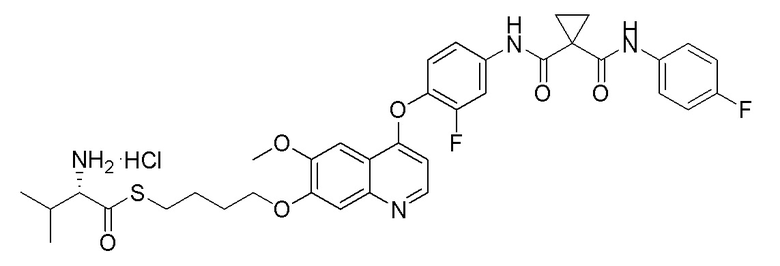
Пример 2. S-(3-((4-(2-Фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)бутил)-(S)-2-амино-3-метилтиобутирата гидрохлорид
Целевое соединение получали (37 мг, 94%) в соответствии с синтезом, описанным в примере 1, заменяя 1,3-дибромпропан на 1,4-дибромбутан.
1H ЯМР (400 МГц, d-DMSO) δ 10,55 (с, 1H), 10,03 (с, 1H), 8,82 (д, J=6,1 Гц, 1H), 8,54 (с, 2H), 8,01 (д, J=13,2 Гц,1H), 7,75 (д, J=11,5 Гц, 2H), 7,72-7,60 (м, 4H), 7,57 (д, J=8,8 Гц, 1H), 7,18 (т, J=8,9 Гц, 2H), 6,92 (с, 1H), 4,29 (с, 2H), 4,17 (с, 1H), 4,06 (с, 3H), 3,14 (м, 2H), 2,25 (с, 1H), 2,04-1,90 (м, 2H), 1,83-1,60 (м, 2H), 1,52 (д, J=9,6 Гц, 4H), 1,01 (дд, J=10,1, 7,0 Гц, 6H).
LC-MS: m/z=693 [M+H] +.
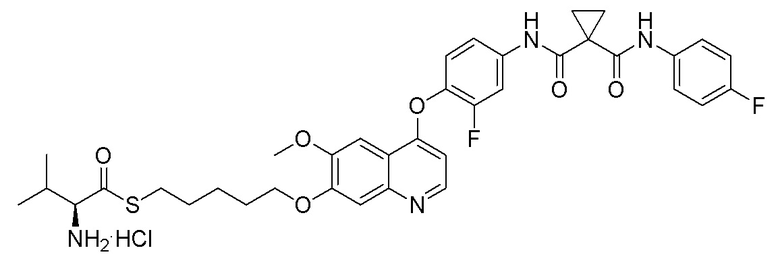
Пример 3. S-(3-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)пентил)-(S)-2-амино-3-метилтиобутират гидрохлорид
Целевое соединение получали в соответствии с синтезом, описанным в примере 1, заменяя 1,3-дибромпропан на 1,5-дибромпентан (38 мг, 96%).
1H ЯМР (400 МГц, d-DMSO) δ 10,56 (с, 1H), 10,04 (с, 1H), 8,82 (д, J=6,3 Гц, 1H), 8,56 (с, 2H), 8,01 (д, J=13,1 Гц, 1H), 7,77 (с, 2H), 7,71-7,61 (м, 3H), 7,58 (д, J=9,0 Гц, 1H), 7,18 (т, J=8,9 Гц, 2H), 6,92 (с, 1H), 4,25 (т, J=10,0 Гц, 2H), 4,15 (с, 1H), 4,07 (с, 3H), 3,08-2,95 (м, 2H), 2,26-2,19 (м, 1H), 1,92-1,80 (м, 2H), 1,75-1,58 (м, 2H), 1,6-1,46 (м, 6H), 1,07-0,93 (дд, J=10,1, 7,0 Гц, 6H).
LC-MS: m/z=707 [M+H] +.
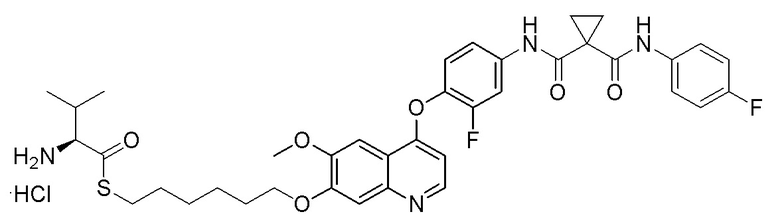
Пример 4. S-(3-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)гексил)-(S)-2-амино-3-метилтиобутирата гидрохлорид
Целевое соединение получали (20 мг, 85%) в соответствии с синтезом, описанным в примере 1, заменяя 1,3-дибромпропан на 1,6-дибромгексан.
1H ЯМР (400 МГц, d-DMSO) δ 10,53 (с, 1H), 10,01 (с, 1H), 8,80 (с, 1H), 8,48 (с, 2H), 8,00 (д, J=13,8 Гц, 1H), 7,75 (с, 2H), 7,66 (дд, J=24,3, 10,3 Гц, 4H), 7,56 (д, J=8,6 Гц, 1H), 7,18 (т, J=8,8 Гц, 2H), 6,89 (с, 1H), 4,24 (т, J=13,8 Гц, 2H), 4,16 (с, 1H), 4,05 (с, 3H), 3,10-2,95 (м, 2H), 2,28-2,08 (м, 1H), 1,88-1,77 (м, 2H), 1,64-1,52 (м, 2H), 1,51-1,40 (м, 8H), 1,00 (дд, J=11,4, 6,8 Гц, 6H).
LC-MS: m/z=721 [M+H] +.
Пример 5. 4-(2-фтор-4-(1-((4-фторфенил)карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил 5-(1,2-дитиолан-3-ил)пентаноат
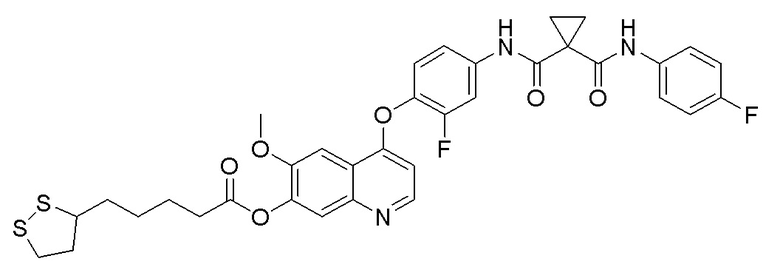
250 мг (0,50 ммоль, 1,0 экв) N-(3-фтор-4-((7-гидроксил-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диформамида, 153 мг (0,75 ммоль, 1,5 экв) липоевой кислоты и 151 мг (1,50 ммоль, 3,0 экв) триэтиламина растворяли в 10 мл дихлорметана. Добавляли 144 мг (0,75 ммоль, 1,5 экв) 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида и 101 мг (0,75 ммоль, 1,5 экв) 1-гидроксилбензотриазола. После добавления, перемешивали и проводили реакцию при комнатной температуре в течение ночи. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), реакционную систему выливали в 100 мл воды, экстрагировали с помощью 150 мл этилацетата. Органическую фазу промывали два раза последовательно водой и насыщенным солевым раствором, и затем сушили над безводным сульфатом натрия, испаряли досуха. Остаток очищали колоночной хроматографией на силикагеле с получением продукта (180 мг, выход=52%).
LC-MS: m/z=694 [M+H] +.
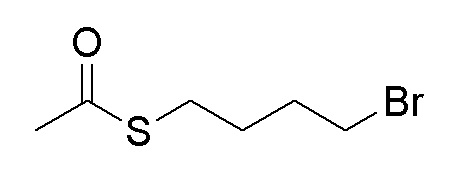
Пример 6. S-(3-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)пропил) (S)-2-амино-3-метилтиобутирата гидрохлорид
Стадия A. S-(4-бромбутил)-ацетилтиоэфир
1 г (8,77 ммоль, 1,0 экв) тиоацетата калия и 5,66 г (26,3 ммоль, 3,0 экв) 1,4-дибромбутана добавляли в 15 мл ацетона. После добавления, реакционную смесь перемешивали при комнатной температуре в течение 3 часов. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), фильтровали реакционный раствор. Белое твердое вещество споласкивали дихлорметаном, и органически фильтрат отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с получением продукта (1,25 г, выход=68%).
1H ЯМР (400 МГц, CDCl3) δ 3,44 (т, J=6,6 Гц, 2H), 2,93 (т, J=7,2 Гц, 2H), 2,36 (с, 3H), 2,02-1,90 (м, 2H), 1,78 (м, 2H).
Стадия B. S-(4-((4-(2-Фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)бутил)-ацетилтиоэфир
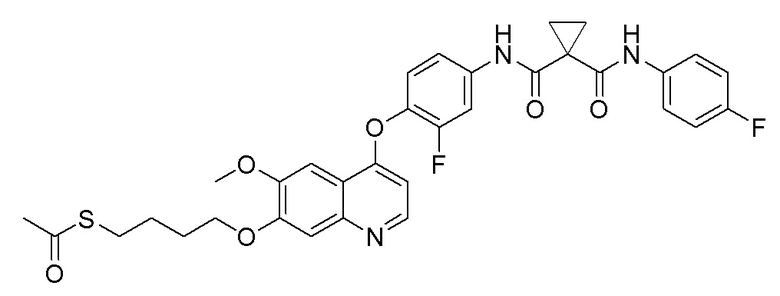
505 мг (1 ммоль, 1,0 экв) N-(3-фтор-4-((7-гидроксил-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диметилформамида и 422 мг (2 ммоль, 2,0 экв) S-(4-бромбутил)-ацетилтиоэфира растворяли в 10 мл N, N-диметилформамида и добавляли 414 мг (3,0 ммоль, 3,0 экв) порошка карбоната калия при комнатной температуре. После добавления, реакционную смесь перемешивали в течение ночи при комнатной температуре. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), реакционную систему выливали в 150 мл воды. Экстрагировали раствор с помощью 200 мл этилацетата, и органическую фазу затем промывали два раза водой и насыщенным солевым раствором. Органическую фазу сушили над безводным сульфатом натрия и испаряли досуха, и остаток очищали колоночной хроматографией на силикагеле с получением продукта (497 мг, выход=78%).
LC-MS: m/z=636 [M+H] +.
Пример 7. N-(3-фтор-4-((7-(4-меркаптобутокси)-6-метокси-хинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диформамид
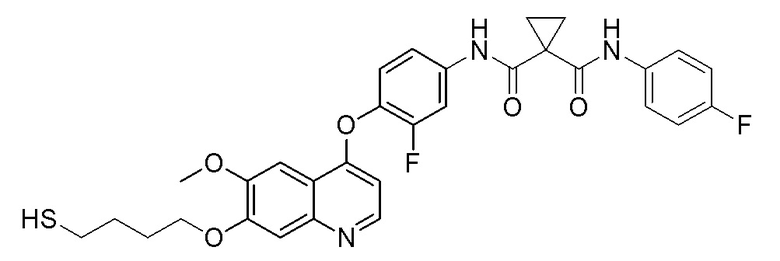
200 мг (0,80 ммоль, 1,0 экв) S-(4-((4-(2-фтор-4-(1-((4-фторфенил)аминоформил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)бутил)-ацетилтиоэфира растворяли в 10 мл метанола и затем добавляли 560 мг (2,4 ммоль, 10,0 экв) боргидрида натрия на ледяной бане. Выдерживали реакционную смесь на ледяной бане в течение 10 минут, затем подогревали до комнатной температуры и проводили реакцию в течение 2 часов. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), растворитель испаряли досуха. К остатку добавляли 100 мл водного раствора лимонной кислоты (10%) и экстрагировали с помощью 120 мл этилацетата. Органическую фазу последовательно промывали два раза водой и насыщенным солевым раствором, затем органическую фазу сушили над безводным сульфатом натрия, испаряли досуха. Остаток очищали колоночной хроматографией на силикагеле с получением продукта (55 мг, выход=29%).
LC-MS: m/z=594 [M+H] +.
Пример 8. S-(5-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)пентил)-ацетилтиоэфир
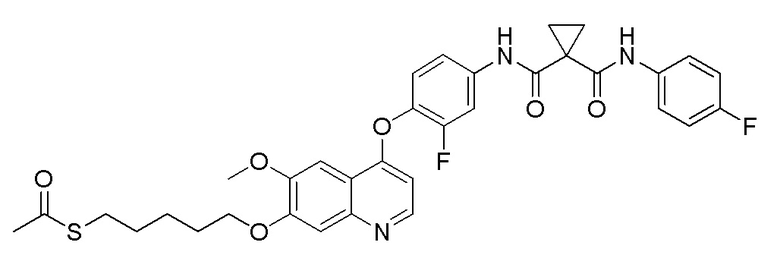
Получали целевое соединение (380 мг, 86%) в соответствии с синтезом, описанным в примере 6, заменяя 1,4-дибромбутан на 1,5-дибромпентан. LC-MS: m/z=650 [M+H] +.
1H ЯМР (400 МГц, CDCl3) δ 9,86 (с, 1H), 9,47 (с, 1H), 8,50 (д, J=4,1 Гц, 1H), 7,86 (д, J=11,8 Гц, 1H), 7,68-7,52 (м, 3H), 7,48-7,36 (м, 2H), 7,34-7,17 (м, 1H), 7,07 (с, 2H), 6,43 (с, 1H), 4,20 (с, 2H), 4,06 (с, 3H), 3,05 (с, 2H), 2,94 (с, 3H), 2,37 (с, 4H), 1,98 (с, 2H), 1,67 (м, 4H).
Пример 9 N-(3-фтор-4-((7-(5-меркаптопентилокси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил) циклопропан-1,1-диформамид
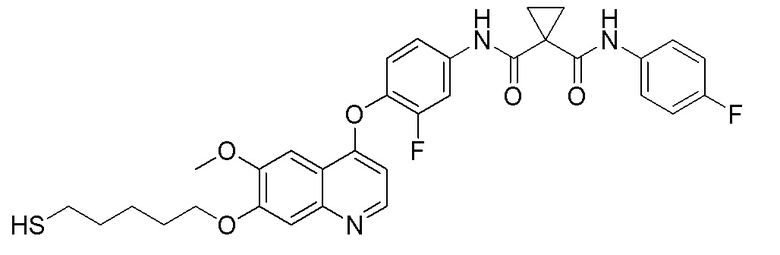
Получали целевое соединение (28 мг, 26%) в соответствии с синтезом, описанным в примере 7.
LC-MS: m/z=608 [M+H] +.
1H ЯМР (400 МГц, CDCl3) δ 9,73 (с, 1H), 9,29 (с, 1H), 8,51 (д, J=5,2 Гц, 1H), 7,84 (д, J=11,8 Гц, 1H), 7,65-7,54 (м, 3H), 7,44 (с, 1H), 7,37 (д, J=8,8 Гц, 1H), 7,26 (т, J=8,6 Гц, 1H), 7,08 (т, J=8,6 Гц, 2H), 6,42 (д, J=5,1 Гц, 1H), 4,23 (д, J=6,1 Гц, 2H), 4,07 (с, 3H), 2,62 (дд, J=14,5, 7,3 Гц, 2H), 2,47-2,38 (м, 2H), 2,02-1,97 (м, 2H), 1,76 (дд, J=14,1, 6,9 Гц, 2H), 1,71-1,62 (с, 4H).
Пример 10. S-(6-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)гексил)-ацетилтиоэфир
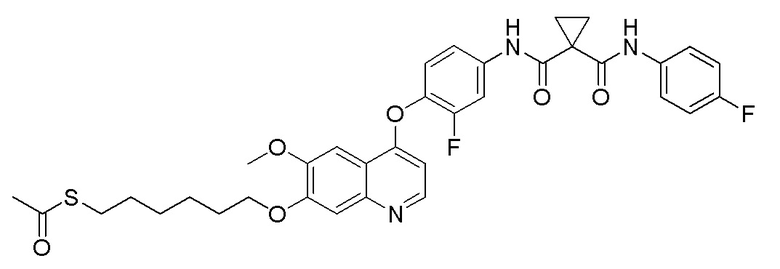
Получали целевое соединение (320 мг, 81%) в соответствии с синтезом, описанным в примере 6, заменяя 1,4-дибромбутан на 1,6-дибромгексан.
LC-MS: m/z=664 [M+H] +.
Пример 11. N-(3-фтор-4-((7-(6-меркаптогексилокси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диформамид
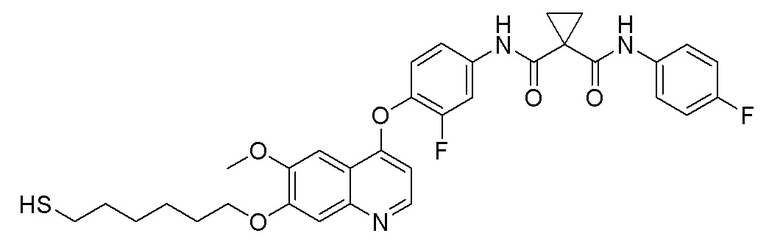
Получали целевое соединение (20 мг, 35%) в соответствии с синтезом, описанным в примере 7.
LC-MS: m/z=622 [M+H] +.
Пример 12. S-(7-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)гептил)-ацетилтиоэфир
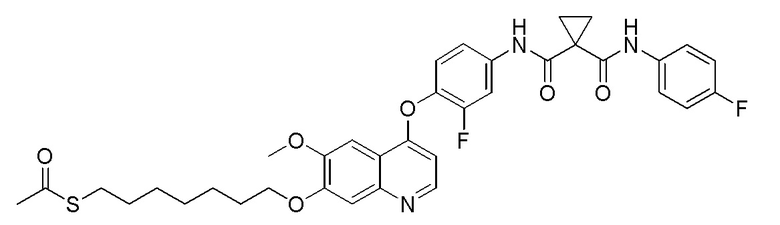
Получали целевое соединение (280 мг, 85%) в соответствии с синтезом, описанным в примере 6, заменяя 1,4-дибромбутан на 1,7-дибромгептан.
LC-MS: m/z=678 [M+H] +.
Пример 13. N-(3-фтор-4-((7-(7-меркаптогептилокси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диформамид
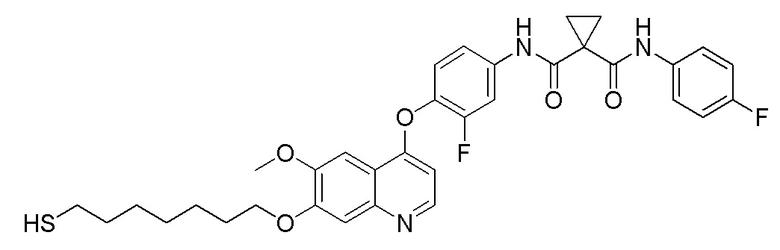
Получали целевое соединение (50 мг, 31%) в соответствии со стадиями синтеза, описанными в примере 7.
1H ЯМР (400 МГц, CDCl3) δ 10,20 (с, 1H), 8,63 (с, 1H), 8,48 (д, J=4,8 Гц, 1H), 7,80 (с, 1H), 7,78 (д, J=10,1 Гц, 1H), 7,58 (с, 1H), 7,51-7,41 (м, 3H), 7,32 (с, 1H), 7,22 (т, J=8,5 Гц, 1H), 7,05 (т, J=8,6 Гц, 2H), 6,42 (д, J=5,0 Гц, 1H), 4,17 (д, J=6,3 Гц, 2H), 4,05 (с, 3H), 2,54 (д, J=7,4 Гц, 2H), 1,99-1,89 (м, 2H), 1,83-1,75 (м, 2H), 1,70-1,56 (м, 4H), 1,52 (с, 2H), 1,42 (с, 4H).
Пример 14. S-(8-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)октил)-ацетилтиоэфир
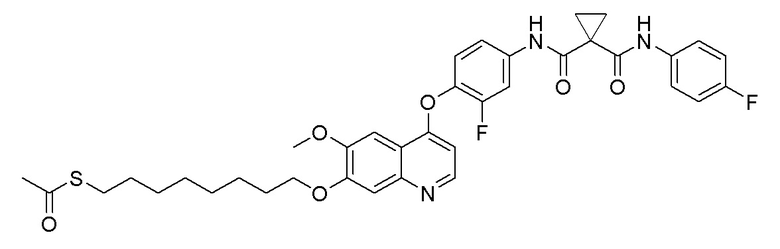
Получали целевое соединение (300 мг, 83%) в соответствии с синтезом, описанным в примере 6, заменяя 1,4-дибромбутан на 1,8-дибромоктан.
LC-MS: m/z=692 [M+H] +.
Пример 15. N-(3-Фтор-4-((7-(8-меркаптооктилокси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диформамид
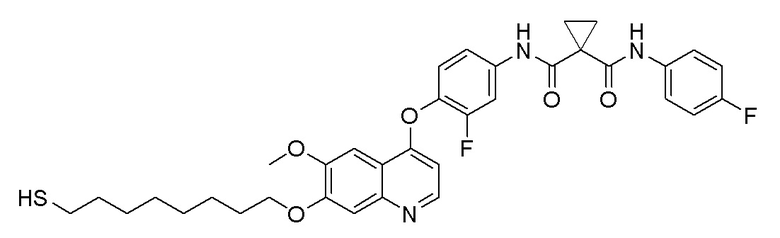
Получали целевое соединение (22 мг, 35%) в соответствии с синтезом, описанным в примере 7.
1H-ЯМР (400 МГц, CDCl3) δ 10,09 (с, 1H), 8,51 (д, J=4,7 Гц, 1H), 8,33 (с, 1H), 7,81 (дд, J=12,4, 1,9 Гц, 1H), 7,60 (с, 1H), 7,49 (дд, J=9,0, 4,8 Гц, 2H), 7,44 (с, 1H), 7,34-7,23 (м, 2H), 7,10 (т, J=8,6 Гц, 2H), 6,43 (д, J=5,1 Гц, 1H), 4,27-4,16 (м, 2H), 4,08 (с, 3H), 2,57 (дд, J=14,7, 7,5 Гц, 2H), 2,01-1,95 (м, 2H), 1,85 (дд, J=7,6, 4,8 Гц, 2H), 1,71-1,24 (м, 12H).
LC-MS: m/z=650 [M+H] +.
Пример 16. S-(9-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)нонил)-ацетилтиоэфир
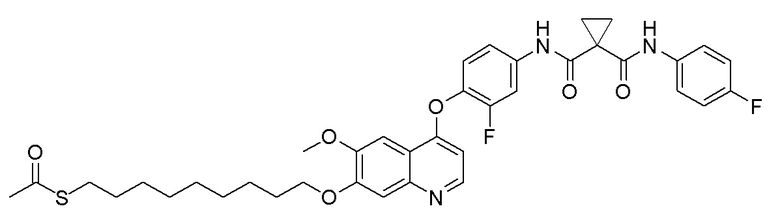
Получали целевое соединение (320 мг, 81%) в соответствии с синтезом, описанным в примере 6, заменяя 1,4-дибромбутан на 1,9-дибромнонан.
1H-ЯМР (400 МГц, CDCl3) δ 10,01 (с, 1H), 8,47 (д, J=5,3 Гц, 1H), 8,29 (с, 1H), 7,76 (дд, J=12,0, 2,3 Гц, 1H), 7,56 (с, 1H), 7,45 (дд, J=9,0, 4,7 Гц, 2H), 7,39 (с, 1H), 7,30-7,18 (м, 2H), 7,06 (т, J=8,6 Гц, 2H), 6,38 (д, J=5,2 Гц, 1H), 4,18 (т, J=6,9 Гц, 2H), 4,04 (с, 3H), 2,90-2,83 (м, 2H), 2,32 (с, 3H), 1,98-1,89 (м, 2H), 1,80 (дд, J=7,7, 4,9 Гц, 2H), 1,62 (дд, J=7,7, 4,8 Гц, 2H), 1,58-1,27 (м, 12H).
LC-MS: m/z=706[M+H] +.
Пример 17. N-(3-Фтор-4-((7-(9-меркаптононилокси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диформамид
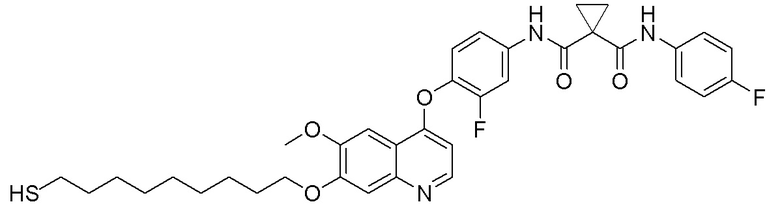
Получали целевое соединение (38 мг, 32%) в соответствии с синтезом, описанным в примере 7.
1H ЯМР (400 МГц, CDCl3) δ 10,08 (с, 1H), 8,51 (д, J=5,2 Гц, 1H), 8,41 (с, 1H), 7,80 (д, J=11,8 Гц, 1H), 7,60 (с, 1H), 7,54-7,40 (м, 3H), 7,34-7,23 (м, 2H), 7,10 (т, J=8,6 Гц, 2H), 6,43 (д, J=5,1 Гц, 1H), 4,23 (т, J=6,8 Гц, 2H), 4,08 (с, 3H), 2,56 (дд, J=14,8, 7,5 Гц, 2H), 2,02-1,94 (м, 2H), 1,89-1,80 (м, 2H), 1,73-1,23 (м, 14H).
LC-MS: m/z=664 [M+H] +.
Пример 18. S-(10-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)децил)-ацетилтиоэфир
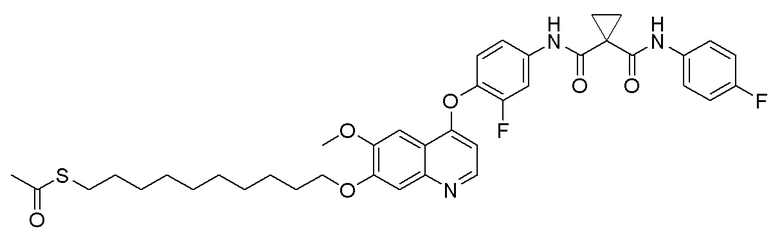
Получали целевое соединение (265 мг, 76%) в соответствии с синтезом, описанным в примере 6, заменяя 1,4-дибромбутан на 1,10-дибромдекан.
LC-MS: m/z=720 [M+H] +.
Пример 19. N-(3-фтор-4-((7-(10-меркаптодецилокси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диформамид
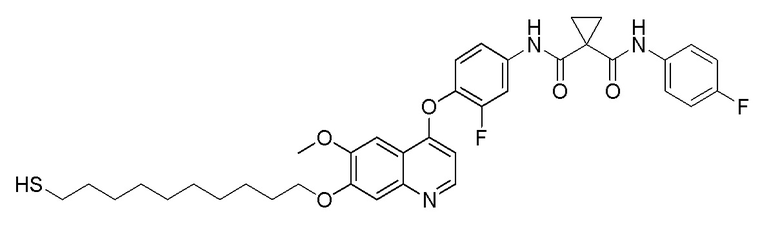
Получали целевое соединение (42 мг, 29%) в соответствии с синтезом, описанным в примере 7.
1H-ЯМР (400 МГц, CDCl3) δ 10,08 (с, 1H), 8,48 (д, J=5,5 Гц, 1H), 8,37 (с, 1H), 7,78 (д, J=13,3 Гц, 1H), 7,57 (с, 1H), 7,51-7,37 (м, 3H), 7,35-7,17 (м, 2H), 7,07 (т, J=8,3 Гц, 2H), 6,46-6,33 (м, 1H), 4,18 (т, J=6,7 Гц, 2H), 4,05 (с, 3H), 2,53 (дд, J=14,7, 7,7 Гц, 2H), 2,01-1,91 (м, 2H), 1,85-1,77 (м, 2H), 1,67-1,20 (м, 16H).
LC-MS: m/z=678 [M+H]+.
Пример 20. (E)-S-(6-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-формамидо)фенокси)-6-метоксихинолин-7-ил)окси)гексил-3-ен-1-ил)ацетилтиоэфир
Стадия A. (E)-1,6-дибром-3-гексен
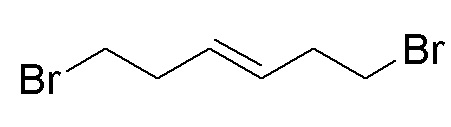
5,0 г (7,4 ммоль, 1,0 экв) 4-бром-1-бутена и 311 мг (0,37 ммоль, 0,05 экв) 1,3-бис(2,4,6-триметилфенил)-2-(имидазолидинилиден)(дихлорбензилиден)(трициклогексилфосфин)- рутения (катализатора Граббса второго поколения) добавляли в 5 мл толуола, продували три раза азотом, перемешивали при 90°C в течение 3 часов, и затем опять продували три раза азотом. Перемешивали при 90°C в течение ночи. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), отгоняли растворитель при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением продукта (2,70 г, выход=61%).
1H ЯМР (400 МГц, CDCl3) δ 5,65-5,56 (м, 2H), 3,50-3,42 (м, 4H), 2,67-2,60 (м, 4H).
Стадия B. (E)-6-бром-3-гексенацетилтиоэфир
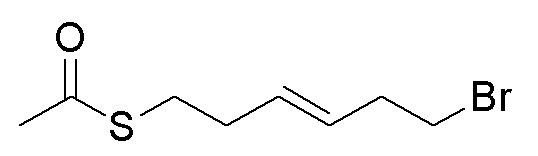
0,35 г (3,07 ммоль, 1,0 экв) тиоацетата калия и 1,95 г (26,3 ммоль, 3,0 экв) (E)-1,6-дибром-3-гексена добавляли в 15 мл ацетона. Затем реакционную систему перемешивали при комнатной температуре в течение 3 часов. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), фильтровали и споласкивали белое твердое вещество дихлорметаном. Органический фильтрат подвергали дистилляции при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с получением продукта (0,47 г, выход=65%).
Стадия C. (E)-S-(6-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбамоил)фенокси)-6-метоксихинолин-7-ил)окси)гексил-3-ен-1-ил)ацетилтиоэфир
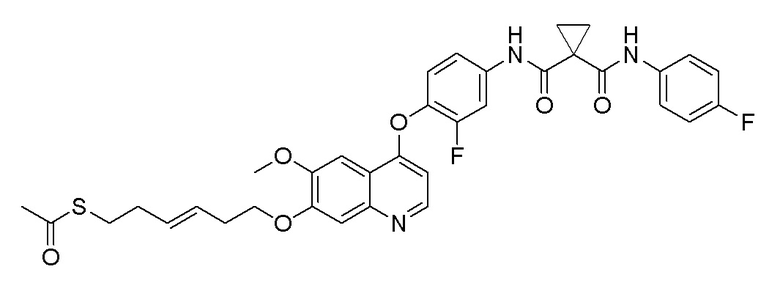
250 мг (0,5 ммоль, 1,0 экв) N-(3-фтор-4-((7-гидроксил-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диметилформамидаи 237 мг (1 ммоль, 2,0 экв) (E)-6-бром-3-гексен ацетилтиоэфира растворяли в 10 мл N, N-диметилформамида, затем добавляли 207 мг (1,5 ммоль, 3,0 экв) порошка карбоната калия при комнатной температуре. Реакционную систему перемешивали при комнатной температуре в течение ночи. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), реакционную систему выливали в 100 мл воды, затем экстрагировали с помощью 200 мл этилацетата. Органическую фазу последовательно два раза промывали водой и насыщенным солевым раствором, и затем сушили над безводным сульфатом натрия, испаряли досуха, и остаток очищали колоночной хроматографией на силикагеле с получением продукта (140 мг, выход=42%).
1H ЯМР (400 МГц, CDCl3) δ 10,18 (с, 1H), 8,60 (с, 1H), 8,50 (д, J=5,2 Гц, 1H), 7,80 (д, J=12,1 Гц, 1H), 7,60 (с, 1H), 7,49 (дд, J=8,6, 4,7 Гц, 2H), 7,43 (с, 1H), 7,32 (с, 1H), 7,24 (т, J=8,5 Гц, 1H), 7,08 (т, J=8,5 Гц, 2H), 6,43 (д, J=5,0 Гц, 1H), 5,63 (м, 2H), 4,21 (т, J=6,8 Гц, 2H), 4,07 (с, 3H), 2,95 (т, J=7,4 Гц, 2H), 2,70-2,63 (м, 2H), 2,40-2,28 (м, 5H), 1,82 (м, 2H), 1,67 (м, 2H).
Пример 21. (E)-N-(3-фтор-4-((7-((6-меркаптогексил-3-ен-1-ил)окси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)-циклопропан-1,1-диформамид
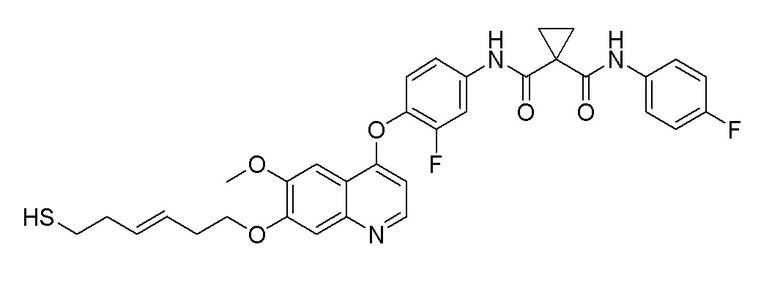
Гидролиз проводили в соответствии с синтезом, описанным в примере 7, целевое соединение (150 мг, 36%) получали из соединения примера 20.
1H ЯМР (400 МГц, CDCl3) δ 10,11 (с, 1H), 8,52 (д, J=5,3 Гц, 2H), 8,42 (с, 1H), 7,81 (д, J=12,3 Гц, 1H), 7,60 (с, 1H), 7,49 (дд, J=8,9, 4,8 Гц, 3H), 7,31 (с, 1H), 7,29-7,21 (м, 1H), 7,10 (т, J =5,5 Гц, 2H), 6,44 (д, J=4,9 Гц, 1H), 5,76-5,53 (м, 2H), 4,28-4,18 (м, 2H), 4,07 (с, 3H), 2,83-2,55 (м, 4H), 2,50-2,38 (м, 2H), 1,87-1,80 (м, 2H), 1,75-1,66 (м, 2H).
Пример 22. (E)-S-(7-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбамоил)фенокси)-6-метоксихинолин-7-ил)окси)гептил-4-ен-1-ил)ацетилтиоэфир
Стадия A. 1-пентенилацетилтиоэфир
Добавляли 2,0 г (17,5 ммоль, 1,0 экв) тиоацетата калия и 3,10 г (21,0 ммоль, 1,2 экв) 5-бром-1-пентена в 15 мл ацетона, затем перемешивали при комнатной температуре в течение 3 часов. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), фильтровали и споласкивали белое твердое вещество дихлорметаном. Затем органический фильтрат подвергали дистилляции при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с получением продукта (1,54 г, выход=61%).
Стадия B. N-(4-((7-(1-бутенилокси)-6-метоксихинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)циклопропан-1,1-диформамид
400 мг (0,79 ммоль, 1,0 экв) N-(3-фтор-4-((7-гидроксил-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диметилформамида и 213 мг (1,58 ммоль, 2,0 экв) 4-бром-1-бутена растворяли в 10 мл N, N-диметилформамида. Добавляли 327 мг (2,37 ммоль, 3,0 экв) порошка карбоната калия при комнатной температуре. После добавления, реакционную смесь перемешивали в течение ночи при комнатной температуре. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), реакционную систему выливали в 100 мл воды, экстрагировали с помощью 100 мл этилацетата. Органическую фазу затем два раза промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и испаряли досуха. Остаток очищали колоночной хроматографией на силикагеле с получением продукта (260 мг, выход=59%).
1H ЯМР (400 МГц, CDCl3) δ 10,21 (с, 1H), 8,66 (с, 1H), 8,50 (д, J=4,0 Гц, 1H), 8,05 (с, 1H), 7,81 (дд, J=4,0, 4,0 Гц, 1H), 7,60 (с, 1H), 7,52-7,49 (м, 2H), 7,44 (с, 1H), 7,34-7,28 (м, 2H), 7,25 (т, J=8,0 Гц, 1H), 7,09 (т, J=8,0 Гц, 2H), 6,42 (д, J=4,0 Гц, 1H), 6,02-5,95 (м, 1H), 5,25 (д, J=12,0 Гц, 1H), 5,18 (д, J=12,0 Гц, 1H), 4,28 (т, J=4,0 Гц, 2H), 4,08 (с, 3H), 2,75 (кв, J=12,0 Гц, 2H), 1,88-1,76 (м, 2H), 1,71-1,64 (м, 2H).
Стадия C. (E)-S-(7-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбамоил)фенокси)-6-метоксихинолин-7-ил)окси)гептил-4-ен-1-ил)ацетилтиоэфир
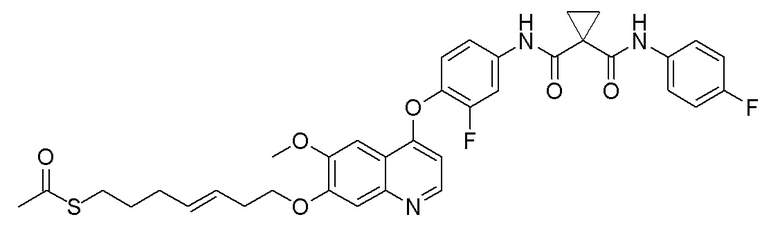
100 мг (0,18 ммоль, 1,0 экв) N-(4-((7-(1-бутенокси)-6-метоксихинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)-циклопропан-1,1-диформамида, 150 мг (1,8 ммоль, 10,0 экв) 1-пентенилацетилтиоэфира и 15 мг (0,018 ммоль, 0,1 экв) 1,3-бис- (2,4,6-триметилфенил)-2-(имидазолидинилиден)(дихлор-бензилиден)(трициклогексилфосфин)рутения (катализатора Граббса второго поколения) добавляли в 5 мл толуола. Продували три раза азотом, и реакционную систему перемешивали при 90°C в течение 4 часов, затем опять продували три раза азотом и перемешивали при 90°C в течение ночи. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением продукта (20 мг, выход=17%).
1H ЯМР (400 МГц, CDCl3) δ 10,25 (с, 1H), 8,72 (с, 1H), 8,50 (д, J=4,0 Гц, 1H), 7,79 (д, J=8,0, 1H), 7,81 (т, J =4,0 Гц, 1H), 7,61 (с, 1H), 7,52-7,49 (м, 2H), 7,36-7,27 (м, 1H), 7,25 (т, J=8,0 Гц, 1H), 7,08 (т, J=8,0 Гц, 2H), 6,45 (д, J=4,0 Гц, 1H), 5,59-5,56 (м, 1H), 5,25 (м, 1H), 4,25 (т, J=8,0 Гц, 2H), 3,96 (с, 3H), 2,59 (м, 2H), 2,56-2,48 (м, 5H), 1,58-1,52 (м, 2H), 1,50 (с, 6H).
Пример 23. (E)-N-(3-фтор-4-((7-((7-меркаптогептил-3-ен-1-ил)окси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)-циклопропан-1,1-диформамид
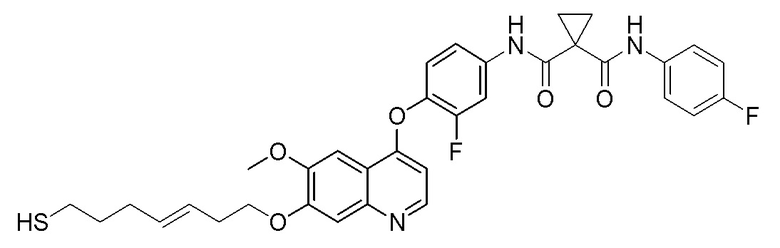
Гидролиз проводили в соответствии с синтезом, описанным в примере 7, целевое соединение (15 мг, 53%) получали из соединения примера 22.
LC-MS: m/z=634 [M+H]+.
Пример 24. (E)-S-(7-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбамоил)фенокси)-6-метоксихинолин-7-ил)окси)гептил-5-ен-1-ил)ацетилтиоэфир
Стадия A. 1-Гексенилацетилтиоэфир
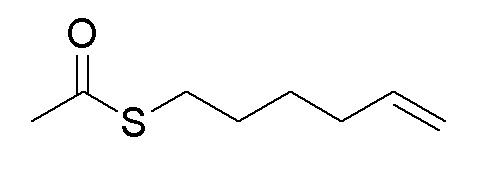
1 г (0,88 ммоль, 1,0 экв) тиоацетата калия и 1,71 г (11,1 ммоль, 1,2 экв) 6-бром-1-гексена добавляли в 15 мл ацетона. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), фильтровали и споласкивали белое твердое вещество дихлорметаном. Органический фильтрат подвергали дистилляции при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с получением продукта (0,81 г, выход=63%).
Стадия B. (E)-S-(7-бромгептил-5-ен-1-ил)ацетилтиоэфир
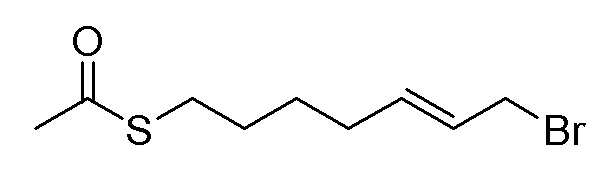
100 мг (0,63 ммоль, 1,0 экв) 1-гексенилацетилтиоэфира, 230 мг (1,89 ммоль, 3,0 экв) 3-бромпропена и 40 мг (0,063 ммоль, 0,1 экв), 1,3-бис(2,4,6-триметилфенил)-2-(имидазолидинилиден)-(дихлорбензилиден)(трициклогексилфосфин)рутения (катализатора Граббса второго поколения) добавляли в 2 мл дихлорметана, и продували три раза азотом. Реакционную смесь перемешивали в течение 3 часов при кипячении с обратным холодильником. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), растворитель отгоняли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с получением продукта (60 мг, выход=38%).
Стадия C. (E)-S-(7-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбамоил)фенокси)-6-метоксихинолин-7-ил)окси)гептил-5-ен-1-ил)ацетилтиоэфир
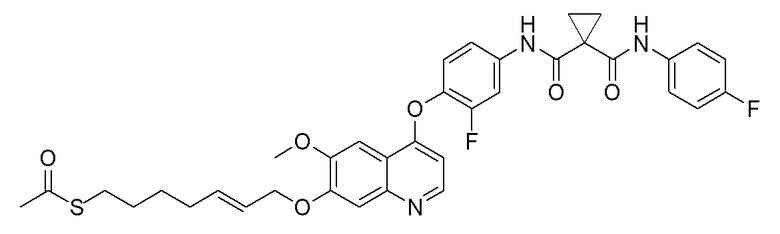
50 мг (0,1 ммоль, 1,0 экв) N-(3-фтор-4-((7-гидроксил-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диформамида и 50 мг (0,20 ммоль, 2,0 экв) (E)-S-(7-бромгептил-5-ен-1-ил)ацетилтиоэфира растворяли в 1 мл N, N-диметилформамида. Затем добавляли 35 мг (0,25 ммоль, 2,5 экв) порошка карбоната калия при комнатной температуре. Реакционную систему перемешивали в течение ночи при комнатной температуре. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), реакционную систему выливали в 100 мл воды, и затем экстрагировали с помощью 50 мл этилацетата. Органическую фазу промывали два раза последовательно водой и насыщенным солевым раствором, и затем сушили над безводным сульфатом натрия, испаряли досуха. Остаток очищали колоночной хроматографией на силикагеле с получением продукта (25 мг, выход=38%).
1H ЯМР (400 МГц, CDCl3) δ 10,08 (с, 1H), 8,51 (д, J=5,2 Гц, 1H), 8,44 (с, 1H), 7,81 (д, J=11,6 Гц, 1H), 7,61 (с, 1H), 7,55-7,42 (м, 3H), 7,32 (м, 1H), 7,28-7,24 (м, 1H), 7,12-7,08 (м, 2H), 6,44 (д, J=5,0 Гц, 1H), 5,99-5,84 (м, 2H), 4,75 (д, J=5,8 Гц, 2H), 4,09 (с, 3H), 2,91 (т, J=7,1 Гц, 2H), 2,36 (с, 3H), 2,16 (д, J=7,1 Гц, 2H), 1,67-1,61 (м, 2H), 1,55-1,52 (м, 2H), 1,30 (м,4H).
LC-MS: m/z=676 [M+H] +.
Пример 25. (E)-N-(3-фтор-4-((7-((7-меркаптогептил-2-ен-1-ил)окси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)-циклопропан-1,1-диформамид
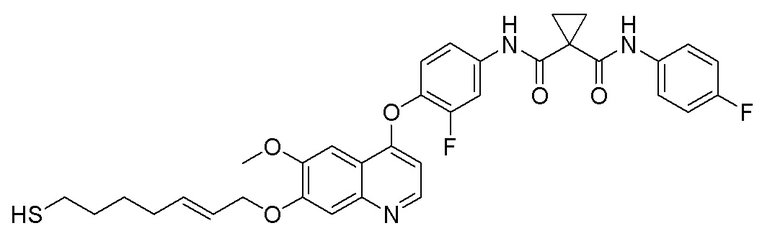
Гидролиз проводили в соответствии с синтезом, описанным в примере 7, целевое соединение (39 мг, выход=48%) получали из соединения примера 24.
1H ЯМР (400 МГц, CDCl3) δ 10,13 (с, 1H), 8,59-8,42 (м, 2H), 7,81 (д, J=11,8 Гц, 1H), 7,61 (с, 1H), 7,51-7,47 (м, 3H), 7,32 (с, 1H), 7,25-7,23 (м, 1H), 7,11-7,07 (м, 2H), 6,44 (д, J=5,2 Гц, 1H), 5,98-5,83 (м, 2H), 4,75 (д, J=5,8 Гц, 2H), 4,08 (с, 3H), 2,89 (уш.с, 1H), 2,57-2,52 (м, 2H), 2,16-2,15 (м, 2H), 1,66 (м, 2H), 1,62-1,44 (м, 2H), 1,29 (м, 4H).
LC-MS: m/z=634 [M+H]+.
Пример 26. (E)-S-(6-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбамоил)фенокси)-6-метоксихинолин-7-ил)окси)гексил-4-ен-1-ил)ацетилтиоэфир
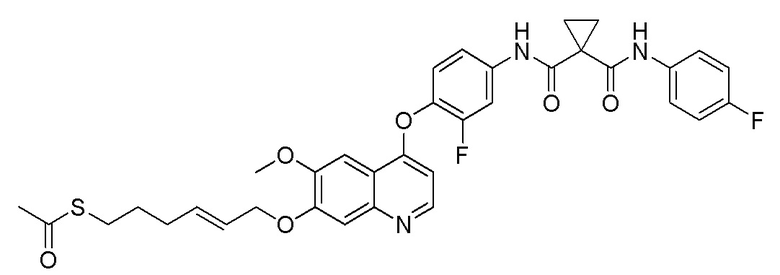
Получали целевое соединение (180 мг, выход=71%) в соответствии с синтезом, описанным в примере 24, заменяя 6-бром-1-гексен на 5-бром-1-пентен.
1H ЯМР (400 МГц, CDCl3) δ 10,08 (с, 1H), 8,51 (д, J=5,2 Гц, 1H), 8,40 (с, 1H), 7,83-7,79 (м, 1H), 7,62 (с, 1H), 7,54-7,41 (м, 3H), 7,31-7,22 (м, 2H), 7,13-7,08 (м, 2H), 6,44 (д, J=4,4 Гц, 1H), 5,98-5,86 (м, 2H), 4,75 (д, J=5,2 Гц, 2H), 4,09 (с, 3H), 2,92 (т, J=7,2 Гц, 2H), 2,37 (с, 3H), 2,24-2,2 (м, 2H), 1,85-1,83 (м, 2H), 1,76 (с, 4H).
LC-MS: m/z=662 [M+H]+.
Пример 27. (E)-N-(3-фтор-4-((7-((6-меркаптогексил-2-ен-1-ил)окси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)-циклопропан-1,1-диметиламид
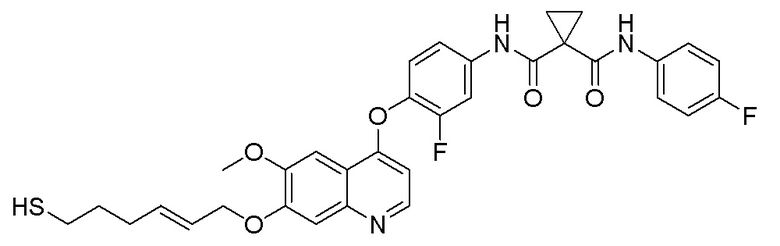
Гидролиз проводили в соответствии с синтезом, описанным в примере 7, целевое соединение (42 мг, выход=37%) получали из соединения примера 26.
1H ЯМР (400 МГц, CDCl3) δ 10,08 (с, 1H), 8,50 (с, 1H), 8,43 (с, 1H), 7,80 (д, J=11,2 Гц, 1H), 7,61 (с, 1H), 7,48-7,46 (м, 3H), 7,26 (д, J=8,2 Гц, 2H), 7,09 (м, 2H), 6,44 (с, 1H), 5,92-5,89 (м, 2H), 4,75 (с, 2H), 4,08 (с, 3H), 2,58-2,56 (м, 2H), 2,27-2,25 (м, 2H), 2,07 (с, 1H), 1,90-1,72 (м, 2H), 1,34 (с, 4H).
LC-MS: m/z=620 [M+H] +.
Пример 28. (E)-S-(5-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбамоил)фенокси)-6-метоксихинолин-7-ил)окси)пентил-3-ен-1-ил)ацетилтиоэфир
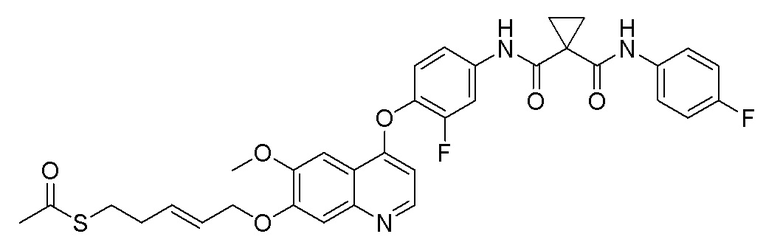
Получали целевое соединение (190 мг, выход=75%) в соответствии с синтезом, описанным в примере 24, заменяя 6-бром-1-гексен на 4-бром-1-бутен.
LC-MS: m/z=648 [M+H] +.
1H-ЯМР (400 МГц, CDCl3) δ 10,06 (с, 1H), 8,47 (с, 1H), 8,23 (с, 1H), 7,78 (д, J=11,0 Гц, 1H), 7,58 (с, 1H), 7,50-7,39 (м, 3H), 7,33-7,17 (м, 2H), 7,07 (т, J=8,5 Гц, 2H), 6,42 (д, J=4,6 Гц, 1H), 5,97-5,82 (м, 2H), 4,78-4,67 (м, 2H), 4,05 (с, 3H), 2,96 (т, J=7,2 Гц, 2H), 2,44-2,27 (м, 5H), 1,86-1,75 (м, 2H), 1,68-1,56 (м, 2H).
Пример 29. (E)-N-(3-фтор-4-((7-((5-меркаптогептил-2-ен-1-ил)окси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)-циклопропан-1,1-диформамид
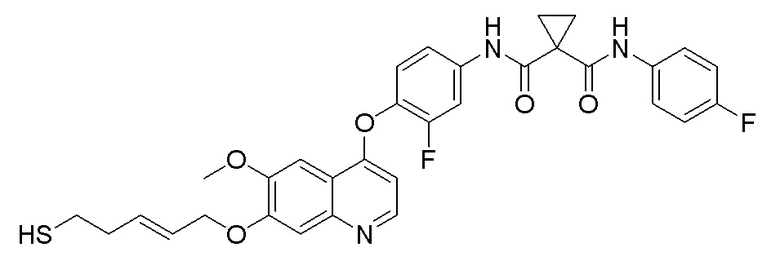
Гидролиз проводили в соответствии с синтезом, описанным в примере 7, целевое соединение (52 мг, выход=48%) получали из соединения примера 28.
LC-MS: m/z=606 [M+H] +.
1H-ЯМР (400 МГц, CDCl3) δ 10,07 (с, 1H), 8,50 (д, J=5,1 Гц, 1H), 8,23 (с, 1H), 7,80 (д, J=12,1 Гц, 1H), 7,60 (с, 1H), 7,51-7,43 (м, 3H), 7,35-7,19 (м, 2H), 7,09 (т, J=8,1 Гц, 2H), 6,44 (д, J=5,8 Гц, 1H), 6,00-5,87 (м, 2H), 4,80-4,74 (м, 2H), 4,08 (с, 3H), 2,65 (дд, J=14,5, 7,6 Гц, 2H), 2,52-2,41 (м, 2H), 1,98-1,55 (м, 4H).
Пример 30. S-(7-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)гептил)-2-метилтиопропионат
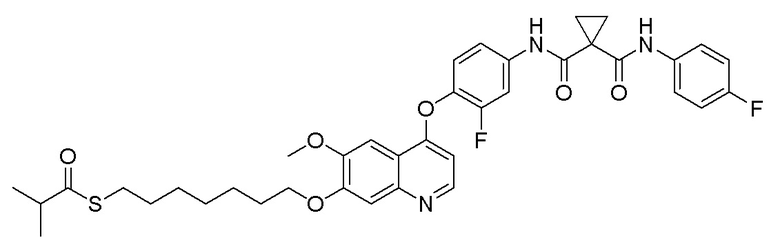
N-(3-фтор-4-((7-(7-меркаптогептилокси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диформамид (100 мг, 0,157 ммоль, из примера 13) и триэтиламин (50 мг, 0,494 ммоль) растворяли в DCM (15 мл). Добавляли изобутирилхлорид (100 мг, 0,943 ммоль) на ледяной бане и проводили реакцию при низкой температуре в течение 3 часов. После завершения реакции, систему промывали два раза насыщенным водным раствором NaHCO3 и один раз насыщенным водным раствором NaCl. Органическую фазу сушили, концентрировали и очищали колоночной хроматографией с получением целевого соединения (32 мг, 29%).
1H ЯМР (400 МГц, CDCl3) δ 10,07 (с, 1H), 8,59-8,35 (м, 2H), 7,79 (дд, J=12,0, 2,2 Гц, 1H), 7,60 (с, 1H), 7,53-7,45 (м, 2H), 7,43 (с, 1H), 7,27-7,23 (м, 2H), 7,11-7,07 (м, 2H), 6,43 (д, J=5,2 Гц, 1H), 4,21 (т, J=6,8 Гц, 2H), 4,07 (с, 3H), 2,90 (т, J=7,2 Гц, 2H), 1,99-1,94 (м, 2H), 1,65 (м, 5H), 1,50-1,41 (м, 6H), 1,30 (с, 4H), 1,23 (д, J=6,8 Гц, 6H).
LC-MS: m/z=706 [M+H] +.
Пример 31. S-(7-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)гептил)-2,2-диметилтиопропионат
Целевое соединение (88 мг, 85%) получали в соответствии с синтезом, описанным в примере 30.
1H ЯМР (400 МГц, CDCl3) δ 10,03 (с, 1H), 8,51 (д, J=5,2 Гц, 1H), 8,35 (с, 1H), 7,85-7,74 (м, 1H), 7,60 (с, 1H), 7,54-7,40 (м, 3H), 7,29-7,19 (м, 2H), 7,10 (т, J=8,6 Гц, 2H), 6,43 (д, J=4,8 Гц, 1H), 4,22 (т, J=6,6 Гц, 2H), 4,08 (с, 3H), 2,88 (т, J=7,2 Гц, 2H), 2,03-1,93 (м, 2H), 1,85-1,83 (м, 2H), 1,68-1,55 (м, 6H), 1,45 (с, 4H), 1,27 (с, 9H).
LC-MS: m/z=720 [M+H] +.
Пример 32. S-(7-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)гептил)тиобензоат
Целевое соединение (62 мг, 59%) получали в соответствии с синтезом, описанным в примере 30.
1H ЯМР (400 МГц, CDCl3) δ 10,07 (с, 1H), 8,53 (д, J=5,3 Гц, 1H), 8,46 (с, 1H), 8,03-8,01 (м, 2H), 7,81 (дд, J=12,0, 2,2 Гц, 1H), 7,61-7,59 (м, 2H), 7,52-7,47 (м, 5H), 7,28-7,24 (м, 2H), 7,13-7,08 (м, 2H), 6,45 (д, J=5,0 Гц, 1H), 4,24 (т, J=6,8 Гц, 2H), 4,08 (с, 3H), 3,13 (т, J=7,2 Гц, 2H), 2,02-1,96 (м, 2H), 1,86-1,82 (м, 2H), 1,79-1,74 (м, 2H), 1,69-1,66 (м, 2H), 1,63-1,44 (м, 2H), 1,31 (с, 4H).
LC-MS: m/z=740 [M+H] +.
Пример 33. S-(7-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)гептил)(S)-2-амино-3-метилтиобутирата гидрохлорид
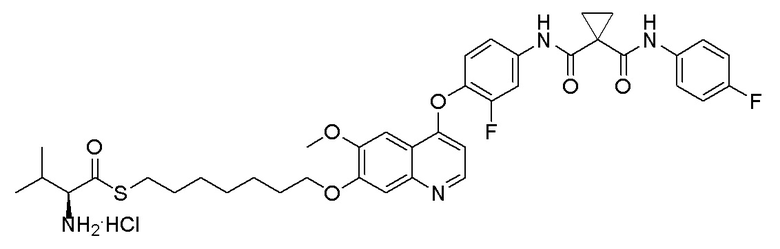
Стадия A. S-(7-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)гептил)(S)-2-((третбутоксикарбонил)амино)-3-метил-тиобутират
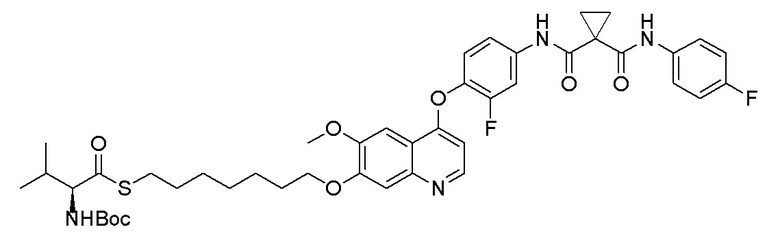
Boc-L-валин (85 мг, 0,376 ммоль) растворяли в DCM (15 мл), затем добавляли 4-диметиламинопиридин (20 мг, 0,376 ммоль) и 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (123 мг, 0,628 ммоль) на ледяной бане, и проводили реакцию при низкой температуре в течение 10 мин. Затем добавляли в реакционную систему N-(3-фтор-4-((7-(7-меркаптогептилокси)-6-метокси-хинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диформамид (100 мг, 0,157 ммоль, из примера 13), и проводили реакцию в течение ночи при комнатной температуре. После завершения реакции, систему промывали два раза насыщенным водным раствором NaHCO3 и один насыщенным водным раствором NaCl, органическую фазу сушили, концентрировали и очищали колоночной хроматографией с получением целевого соединения (110 мг, 85%).
1H ЯМР (400 МГц, CDCl3) δ 10,06 (с, 1H), 8,52 (д, J=4,0 Гц, 1H), 8,39 (с, 1H), 7,81 (с, 1H), 7,61 (д, J=10,1 Гц, 1H), 7,51-7,41 (м, 2H), 7,45 (с, 1H), 7,38-7,26 (м, 2H), 7,11 (т, J=8,6 Гц, 2H), 6,43 (д, J=4,0 Гц, 1H), 4,30-4,26 (м, 1H), 4,23 (т, J=4,0 Гц, 2H), 4,08 (с, 3H), 2,93 (т, J=4,0 Гц, 2H), 2,35-2,25 (м, 1H), 2,20-1,93 (м, 2H), 1,88-1,83 (м, 2H), 1,81-1,74 (м, 2H), 1,70-1,60 (м, 4H), 1,59-1,40 (м, 13H), 1,04 (д, J=4,0 Гц, 3H), 0,91 (д, J=4,0 Гц, 3H).
Стадия B. S-(7-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)гептил)-(S)-2-амино-3-метилтиобутирата гидрохлорид
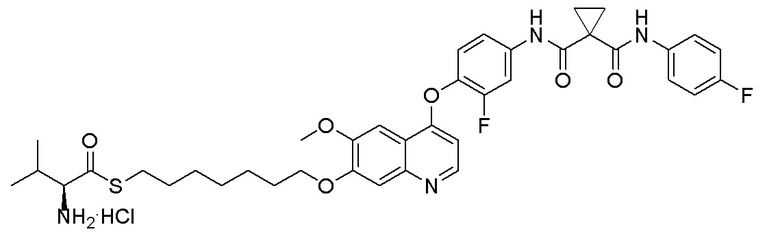
Целевое соединение (65 мг, 87%) получали в соответствии со стадией D, описанной в примере 1.
1H ЯМР (400 МГц, DMSO-d6) δ 10,57 (с, 1H), 10,04 (с, 1H), 8,82 (д, J=6,4 Гц, 1H), 8,63 (уш.с, 1H), 8,03 (д, J=6,4 Гц, 2H), 7,81-7,55 (м, 6H), 7,18 (м, 2H), 6,90 (д, J=6,4 Гц, 1H), 4,24 (т, J=13,8 Гц, 2H), 4,16 (с, 1H), 4,05 (с, 3H), 3,10-2,95 (м, 2H), 2,28-2,08 (м, 1H), 1,88-1,77 (м, 2H), 1,64-1,52 (м, 2H), 1,51-1,40 (м, 10H), 1,00 (дд, J=11,4, 6,8 Гц, 6H).
Пример 34. S-(7-((4-(2-фтор-4-(1-((4-фторфенил)амино-формил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)гептил)-(R)-2-амино-3-(1-метил-1H-индол-3-ил)тио-пропионата гидрохлорид
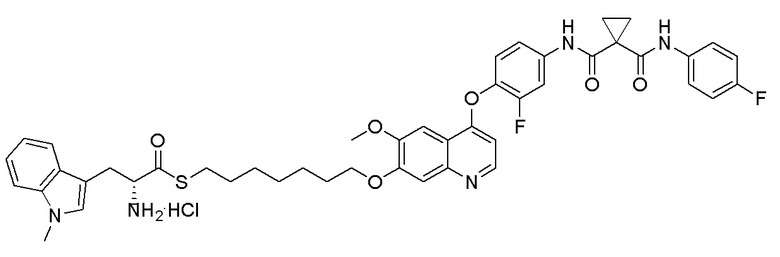
Целевое соединение (51 мг, 59%) получали в соответствии с синтезом, описанным в примере 33.
1H ЯМР (400 МГц, DMSO-d6) δ 10,56 (с, 1H), 10,04 (с, 1H), 8,82 (д, J=6,4 Гц, 1H), 8,63 (уш.с, 3H), 8,07-7,95 (м, 1H), 7,78-7,76 (м, 2H), 7,71-7,53 (м, 5H), 7,43 (д, J=8,2 Гц, 1H), 7,25 (с, 1H), 7,26-7,16 (м, 3H), 7,07 (т, J=7,0 Гц, 1H), 6,92 (д, J=6,4 Гц, 1H), 4,42 (м, 1H), 4,25 (т, J=6,4 Гц, 2H), 4,06 (с, 3H), 3,77 (с, 3H), 2,92-2,86 (м, 2H), 2,53 (т, J=4,0 Гц, 2H), 1,89-1,87 (м, 2H), 1,58-1,22 (м, 12H).
Пример 35. O-этил-S-(7-((4-(2-фтор-4-(1-((4-фторфенил)-аминоформил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)гептил)метилтиокарбонат
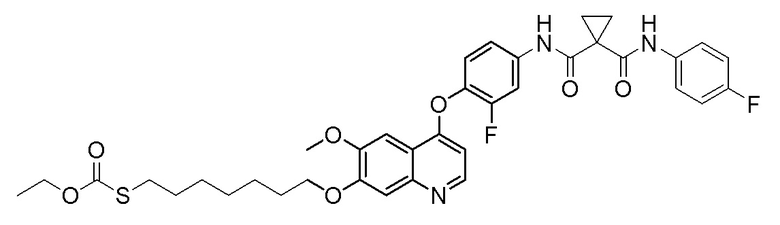
Получали целевое соединение (50 мг, 31%) в соответствии со стадиями, описанными в примере 30, заменяя изобутирилхлорид на этилхлорформиат.
LC-MS: m/z=708 [M+H] +.
Пример 36. N-этил-S-(7-((4-(2-фтор-4-(1-((4-фторфенил)-аминоформил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)гептил)тиокарбамат
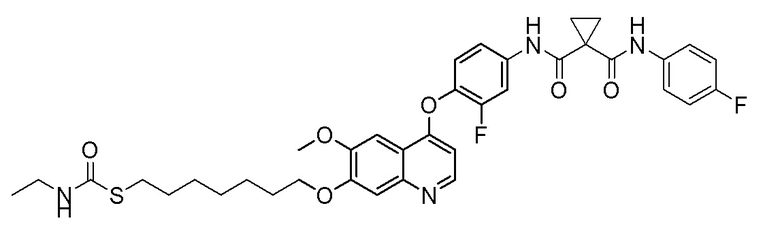
Получали целевое соединение (48 мг, 32%) в соответствии с синтезом, описанным в примере 30, заменяя изобутирилхлорид на этилизоцианат.
LC-MS: m/z=707[M+H] +.
Пример 37. S-(7-((4-(4-(1-((4-фторфенил)карбамоил)-циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)-окси)гептил)тиоацетат
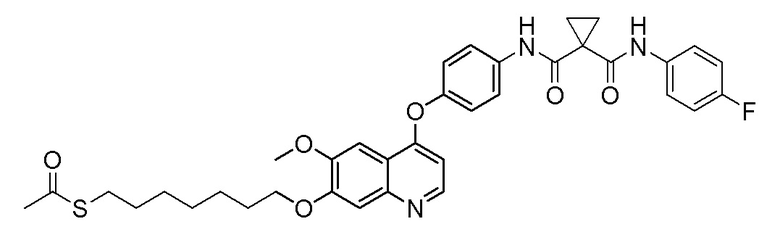
Стадия A. S-(6-бромгептил)-ацетилтиоэфир
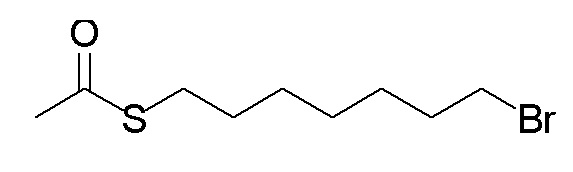
Целевое соединение (1,58 г, выход=72%) получали в соответствии со стадией A, описанной в примере 6.
Стадия B. S-(7-((4-(4-(1-((4-фторфенил)карбамоил)-циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)-окси)гептил)тиоацетат
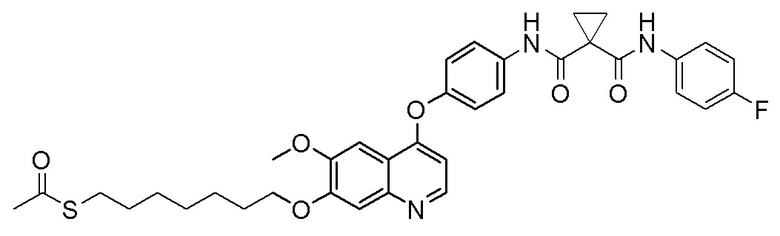
487 мг (1 ммоль, 1,0 экв) N-(4-фторфенил)-N-(4-((7-гидрокси-6-метоксихинолин-4-ил)окси)фенил)циклопропан-1,1-диформамида (по поводу метода синтеза смотрите пример синтеза 2) и 506 мг (2 ммоль, 2,0 экв) S-(7-бромгептил)-ацетилтиоэфира растворяли в 10 мл N, N-диметилформамида. Добавляли 414 мг (3,0 ммоль, 3,0 экв) порошка карбоната калия при комнатной температуре. Реакционную систему перемешивали в течение ночи при комнатной температуре. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), реакционную систему выливали в 150 мл воды и экстрагировали с помощью 200 мл этилацетата. Органическую фазу промывали два раза последовательно водой и насыщенным солевым раствором, и затем сушили над безводным сульфатом натрия, испаряли досуха. Остаток очищали колоночной хроматографией на силикагеле с получением продукта (485 мг, выход=74%).
1H ЯМР (400 МГц, CDCl3) δ 9,48 (с, 1H), 8,86 (с, 1H), 8,54 (с, 1H), 7,67 (д, J=8,4 Гц, 2H), 7,55-7,50 (м, 4H), 7,20 (д, J=8,8 Гц, 2H), 7,08-7,06 (м, 2H), 6,51 (д, J=5,4 Гц, 1H), 4,20 (с, 2H), 4,06 (с, 3H), 2,92 (т, J=7,2 Гц, 2H), 2,36 (с, 3H), 1,99-1,97 (м, 2H), 1,78-1,76 (м, 2H), 1,70-1,61 (м, 2H), 1,55-1,52 (м, 4H), 1,30 (м, 4H).
LC-MS: m/z=660[M+H] +.
Пример 38. S-(7-((4-(4-(1-((4-фторфенил)карбамоил)-циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)-окси)гексил)тиоацетат
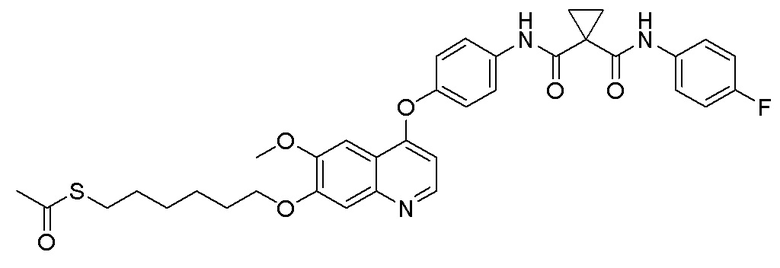
Получали целевое соединение (88 мг, 69%) в соответствии синтезом, описанным в примере 37.
1H ЯМР (400 МГц, CDCl3) δ 9,46 (с, 1H), 8,86 (с, 1H), 8,54 (с, 1H), 7,66 (д, J=8,4 Гц, 2H), 7,57-7,52 (м, 4H), 7,20 (д, J=8,8 Гц, 2H), 7,09-7,01 (м, 2H), 6,50 (д, J=5,4 Гц, 1H), 4,20 (с, 2H), 4,06 (с, 3H), 2,93 (т, J=7,2 Гц, 2H), 2,36 (с, 3H), 1,98-1,96 (м, 2H), 1,78-1,76 (м, 2H), 1,74-1,62 (м, 2H), 1,54-1,52 (м, 2H), 1,31 (м, 4H).
LC-MS: m/z=646[M+H] +.
Пример 39. S-(7-((4-(2-фтор-4-(1-((4-фторфенил)карбамоил)-циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)-окси)гептил)-(S)-2-амино-3-(1H-индол-3-ил)тиопропионата гидрохлорид
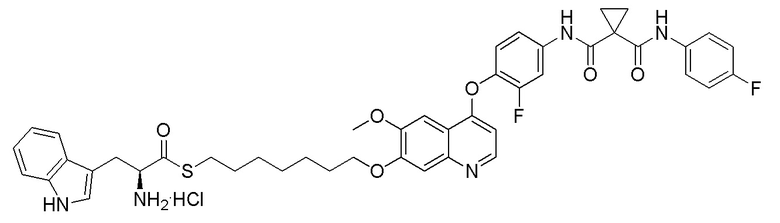
Целевое соединение (81 мг, 63%) получали в соответствии с синтезом, описанным в примере 33.
LC-MS: m/z=856[M+H] +.
Пример 40. S-(4-((2-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)этил)амино)бутил)тиоацетат
Стадия A. S-(4,4-диметоксибутил)ацетилтиоэфир
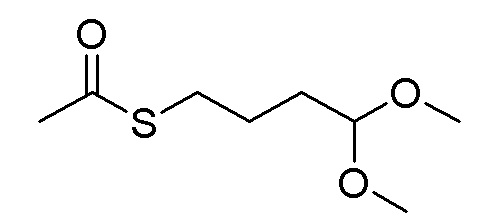
4-бром-1,1-диметоксибутан (1,97 г, 10 ммоль) растворяли в ацетоне (15 мл), добавляли тиоацетат калия (1,14 мг, 10 ммоль) при комнатной температуре, и реакцию проводили при комнатной температуре в течение 2 часов. После завершения реакции, реакционный раствор фильтровали. Осадок на фильтре промывали с помощью DCM, и органическую фазу сушили, концентрировали и очищали колоночной хроматографией с получением целевого соединения (1,72 г, 89%).
Стадия B. S-(4-карбонилбутил)ацетилтиоэфир
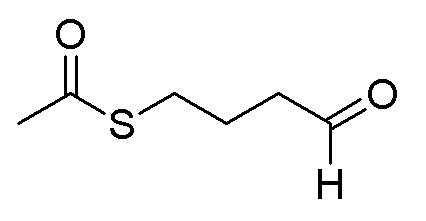
S-(4,4-диметоксибутил)ацетилтиоэфир (500 мг, 2,6 ммоль) растворяли в THF (5 мл), добавляли 6N раствор хлористоводородной кислоты (1,5 мл) на ледяной бане и проводили реакцию при комнатной температуре в течение 45 минут. После завершения реакции, реакционную систему промывали два раза насыщенным водным раствором NaHCO3, один раз насыщенным водным раствором NaCl. Органическую фазу сушили и концентрировали с получением целевого соединения (320 мг, 85%), которое непосредственно использовали на следующей стадии.
Стадия C. N-(4-((7-(2-аминоэтокси)-6-метоксихинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)циклопропан-1,1-диформамид
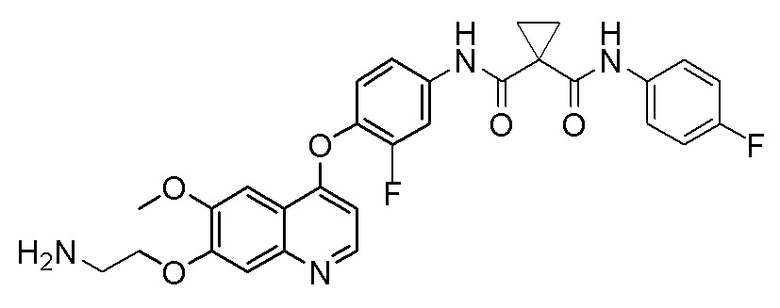
505 мг (1 ммоль, 1,0 экв) N-(3-фтор-4-((7-гидроксил-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диметилформамида и 174 мг (1,5 ммоль, 1,5 экв) 2-хлор-этиламина гидрохлорида растворяли в 10 мл N, N-диметилформамида, и затем добавляли 414 мг (3,0 ммоль, 3,0 ммоль, 3,0 экв) порошка карбоната калия при комнатной температуре. Реакционную систему перемешивали при комнатной температуре в течение ночи. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), реакционную систему выливали в 150 мл воды и экстрагировали с помощью 200 мл этилацетата. Органическую фазу два раза промывали водой и насыщенным солевым раствором, и затем сушили над сульфатом натрия и испаряли досуха. Остаток очищали колоночной хроматографией на силикагеле с получением продукта (320 мг, выход=58%).
LC-MS: m/z=549 [M+H] +.
Стадия D. S-(4-((2-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)этил)амино)бутил)тиоацетат
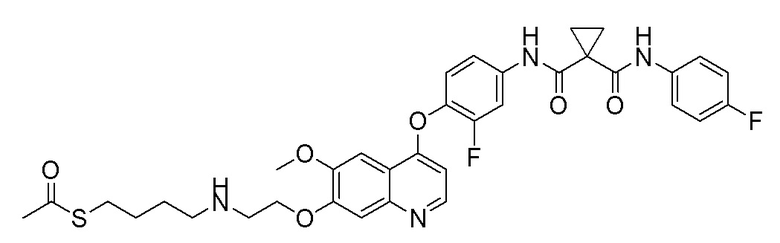
S-(4-карбонилбутил)ацетилтиоэфир (292 мг, 2,0 ммоль) растворяли в THF (3 мл), и добавляли N-(4-((7-(2-аминоэтокси)-6-(метоксихинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)циклопропан-1,1-диформамид (548 мг, 1,0 ммоль) вместе с одной каплей уксусной кислоты, затем выдерживали при комнатной температуре в течение 1,5 часов. После завершения реакции, в систему добавляли триацетилборгидрид натрия (870 мг, 4,1 ммоль) и выдерживали при комнатной температуре в течение 5 часов, затем промывали два раза насыщенным водным раствором NaHCO3 и один раз насыщенным водным раствором NaCl. Затем органическую фазу сушили, концентрировали и очищали колоночной хроматографией с получением целевого соединения (351 мг, 52%).
LC-MS: m/z=679[M+H] +.
Пример 41. N-(3-фтор-4-((6-метокси-7-(3-((2-оксотетрагидротиофен-3-ил)амино)пропокси)хинолин-4-ил)окси)фенил)-N-(4-фтор-фенил)циклопропан-1,1-дикарбоксамид
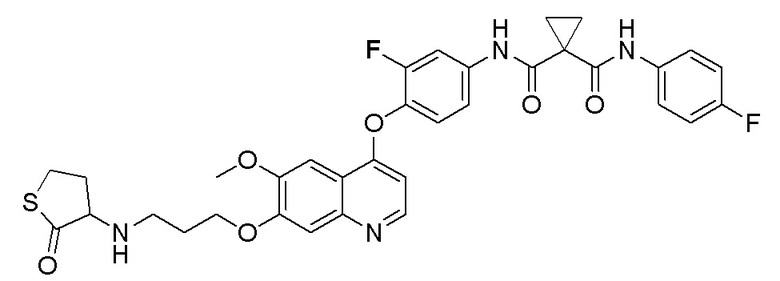
Стадия A. N-(4-((7-(3,3-диметоксипропокси)-6-метокси-хинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)циклопропан-1,1-диформамид
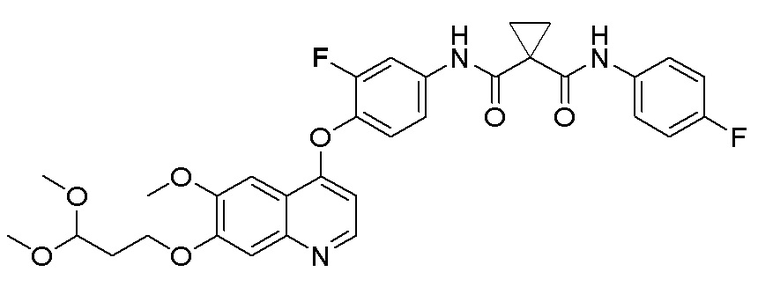
Получали целевое соединение (120 мг, 78%) в соответствии со стадией C, описанной в примере 20, заменяя (E)-6-бром-3-гексенилацетилтиоэфир на 3-бром-1,1-диметоксипропан.
LC-MS: m/z=608[M+H] +.
Стадия B N-(3-фтор-4-((6-метокси-7-(3-оксопропокси)-хинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-дикарбоксамид
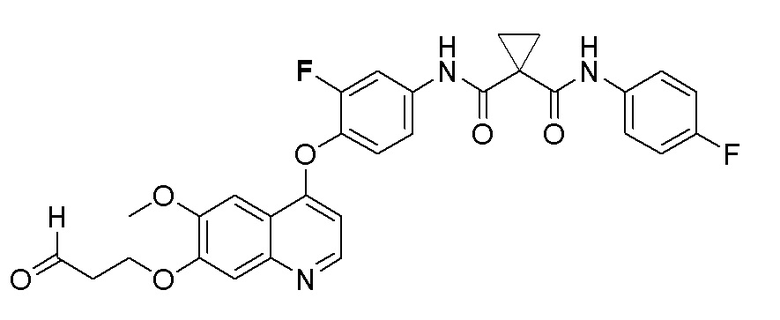
N-(4-((7-(3,3-диметоксипропокси)-6-метоксихинолин-4-ил)-окси)-3-фторфенил)-N-(4-фторфенил)циклопропан-1,1-диформамид (100 мг, 0,164 ммоль) растворяли в THF (5 мл), добавляли 2N хлористоводородную кислоту (1 мл) на ледяной бане и проводили реакцию при комнатной температуре в течение 1 часа. После завершения реакции, систему промывали два раза насыщенным водным раствором NaHCO3 водный раствор и один раз насыщенным водным раствором NaCl. Органическую фазу затем сушили и концентрировали с получением целевого соединения (80 мг, 87%).
LC-MS: m/z=562[M+H] +.
Стадия C. N-(3-фтор-4-((6-метокси-7-(3-((2-оксотетрагидротифен-3-ил)амино)пропокси)хинолин-4-ил)окси)фенил)-N-(4-фтор-фенил)циклопропан-1,1-дикарбоксамид
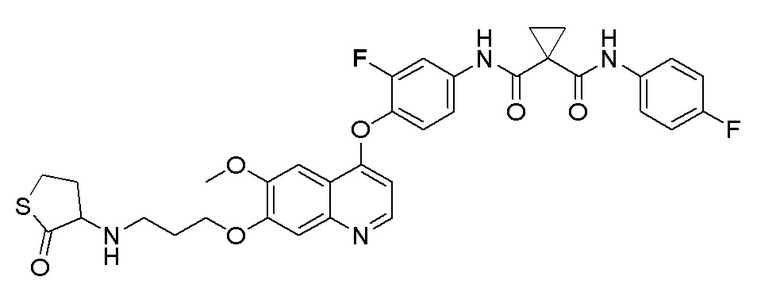
N-(3-фтор-4-((6-метокси-7-(3-((2-оксопропокси)хинолин-4-ил)окси)фенил)-N-(4-фтор-фенил)циклопропан-1,1-дикарбоксамид (100 мг, 0,164 ммоль) растворяли в THF (3 мл), добавляли гомоцистеин- тиолактон (25 мг, 0,164 ммоль) при комнатной температуре, и добавляли каплю уксусной кислоты, после чего проводили реакцию при комнатной температуре в течение 1 часа. После завершения реакции, добавляли в систему триацетилборгидрид натрия (174 мг, 0,82 ммоль), проводили реакцию при комнатной температуре в течение 3 часов. Промывали реакционную систему два раза насыщенным водным раствором NaHCO3 и один раз насыщенным водным раствором NaCl. Органическую фазу затем сушили, концентрировали и очищали колоночной хроматографией с получением целевого соединения (46 мг, 39%).
LC-MS: m/z=663[M+H] +.
Пример 42. (3-((4-(2-фтор-4-(1-((4-фторфенил)карбамоил)-циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)-окси)пропил)гомоцистеин
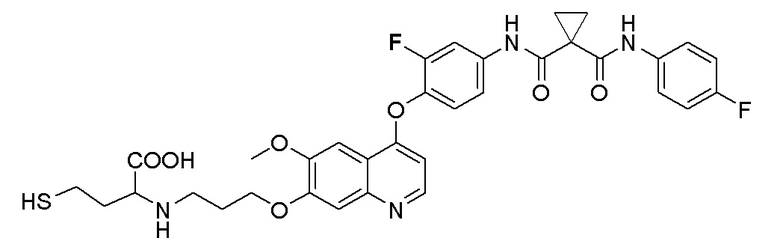
N-(3-фтор-4-((6-метокси-7-(3-((2-гомоцистеинлактон-3-ил)амино)пропокси)хинолин-4-ил)окси)фенил)-N-(4-фторфенил)-циклопропан-1,1-диформамид (20 мг, 0,03 ммоль) растворяли в THF (1 мл), и добавляли 1N раствор гидроксида натрия (0,1 мл) на ледяной бане. Раствор выдерживали при комнатной температуре и проводили реакцию в течение 10 минут. После завершения реакции, величину pH реакционной системы доводили до нейтрального значения 1N водным раствором хлористоводородной кислоты. Затем систему подвергали экстракции с помощью DCM, и органическую фазу сушили и концентрировали с получением целевого соединения (12 мг, 60%).
LC-MS: m/z=681[M+H] +.
Пример 43. N-(3-фтор-4-((6-метокси-7-(3-(метил((2-оксогидротиофен-3-ил)амино)пропокси)хинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-дикарбоксамид
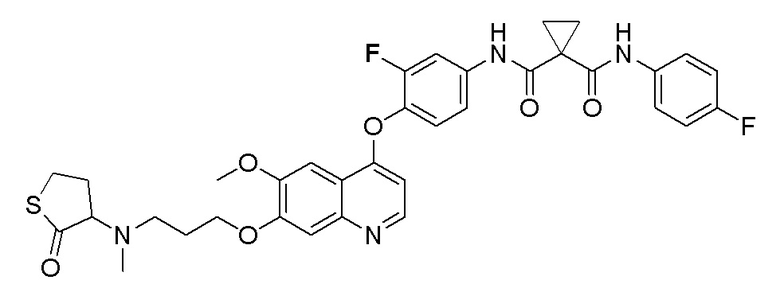
N-(3-фтор-4-((6-метокси-7-(3-((2- оксогидротиофен-3-ил)-амино)пропокси)хинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-дикарбоксамид (20 мг, 0,03 ммоль) растворяли в THF (1 мл), и добавляли при комнатной температуре водный раствор формальдегида (0,1 мл), добавляли каплю уксусной кислоты и проводили реакцию при комнатной температуре в течение 1 часа. После завершения реакции, добавляли в систему триацетилборгидрид натрия (32 мг, 0,15 ммоль) и выдерживали при комнатной температуре в течение 1,5 часов. Затем промывали систему два раза насыщенным водным раствором NaHCO3 и один раз насыщенным солевым раствором. Органическую фазу сушили, концентрировали и очищали препаративной тонкослойной хроматографией с получением целевого соединения (6 мг, 29%).
LC-MS: m/z=677[M+H] +.
Пример 44. N-(3-фтор-4-((6-метокси-7-(3-((2-гомоцистеин лактон-3-ил)амино)пропокси)хинолин-4-ил)окси)фенил)-N-(4-фтор-фенил)циклопропан-1,1-диформамид
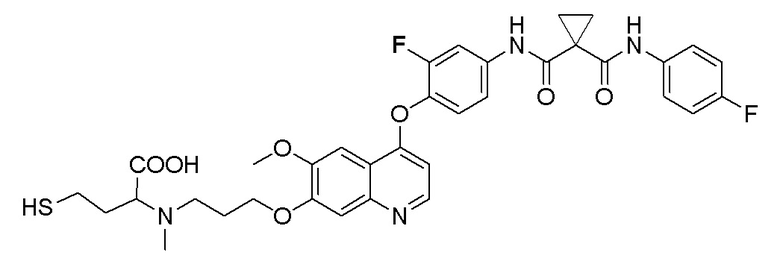
Проводили гидролиз в соответствии со стадией, описанной в примере 42, и целевое соединение (5 мг, 71%) получали из соединения примера 43.
LC-MS: m/z=695[M+H] +.
Пример 45. N-(4-фторфенил)-N-(4-((7-((7-меркаптогептил)-окси)-6-метоксихинолин-4-ил)окси)фенил)циклопропан-1,1-диформамид
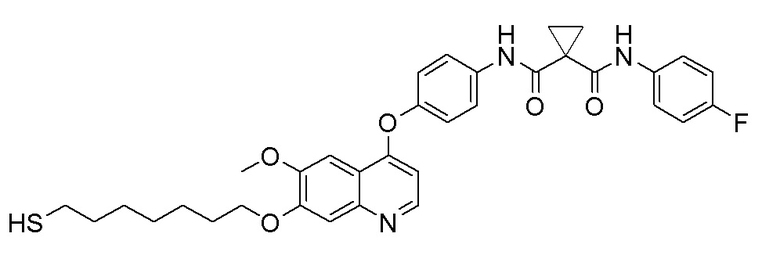
S-(7-((4-(4-(1-((4-фторфенил)карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)окси)гептил)-тиоацетат (50 мг, 0,03 ммоль) растворяли в THF (2,5 мл), и добавляли 1N водный раствор гидроксида натрия (0,1 мл) при комнатной температуре. Реакцию проводили при комнатной температуре в течение 0,5 часа. Величину pH реакционного раствора доводили до нейтральной величины 1N водным раствором хлористоводородной кислоты, и затем промывали два раза насыщенным солевым раствором. Органическую фазу сушили, концентрировали и очищали препаративной тонкослойной хроматографией с получением целевого соединения (32 мг, 68%).
LC-MS: m/z=618[M+H] +.
Пример 46. N-(4-фторфенил)-N-(4-((7-((6-меркаптогексил)-окси)-6-метоксихинолин-4-ил)окси)фенил)циклопропан-1,1-диформамид
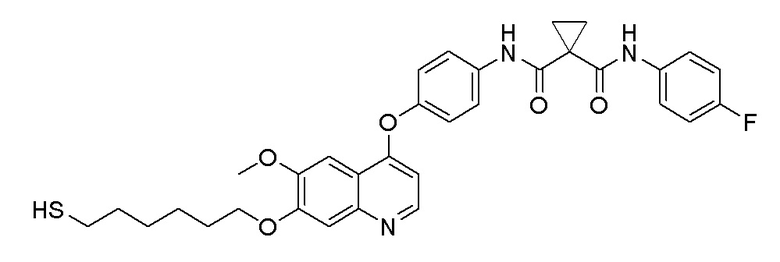
Проводили гидролиз в соответствии со стадией, описанной в примере 45, и целевое соединение (5 мг, 71%) получали из соединения примера 38.
LC-MS: m/z=604[M+H] +.
Пример 47. S-(7-((4-(2-фтор-4-(1-((4-фторфенил)карбамоил)-циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)-окси)гептил) 2-аминоацетилтиоэфира гидрохлорид
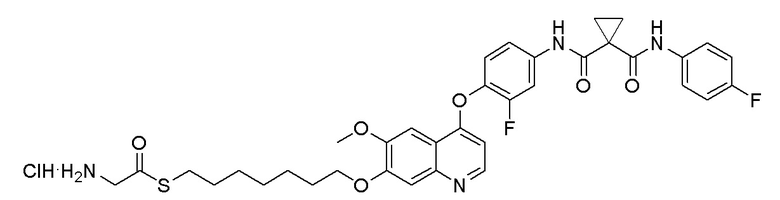
В соответствии со стадиями конденсации с аминокислотой и уделения Boc-защитной группы, описанными в примере 33, целевое соединение (18 мг, 71%) получали из соединения примера 13.
LC-MS: m/z=730[M+H] +.
Пример 48. S-(7-((4-(2-фтор-4-(1-((4-фторфенил)карбамоил)-циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)-окси)гептил)-(S)-серинтиоэфира гидрохлорид
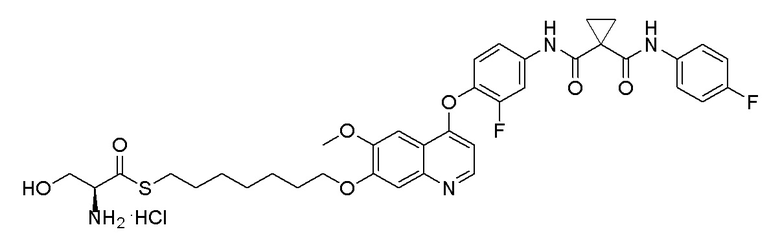
В соответствии со стадиями конденсации с аминокислотой и уделения Boc-защитной группы, описанными в примере 33, целевое соединение (18 мг, 71%) получали из соединения примера 13.
LC-MS: m/z=760[M+H] +.
Пример 49. N-(3-фтор-4-((7-(2-((4-меркаптобутил)амино)-этокси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)-циклопропан-1,1-диформамид
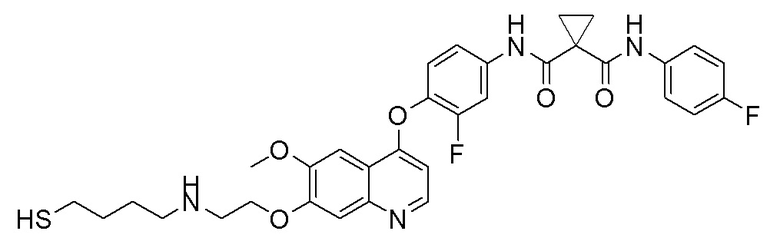
В соответствии с методикой проведения гидролиза, описанной в примере 45, целевое соединение (49 мг, 64%) получали, используя соединение примера 40 в качестве исходного реагента.
LC-MS: m/z=637[M+H] +.
Пример 50. S-(3-((3-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)пропил)амино)пропил)тиоацетат
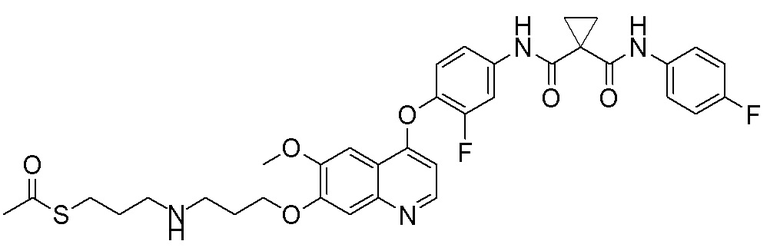
Целевое соединение (46 мг, 58%) получали в соответствии с синтезом, описанным в примере 40.
LC-MS: m/z=679[M+H] +.
1H ЯМР (400 МГц, CDCl3) δ 10,23 (с, 1H), 8,58 (с, 1H), 8,52 (д, J=5,0 Гц, 1H), 7,82 (д, J=12,5 Гц, 1H), 7,64 (с, 1H), 7,49 (м, 2H), 7,45 (с, 1H), 7,37-7,22 (м, 2H), 7,19-7,15 (м, 2H), 6,85 (уш.с, 1H), 6,46 (д, J=5,0 Гц, 1H), 4,34 (кв, J=4,0 Гц, 2H), 4,09 (с, 3H), 3,59 (кв, J=4,0 Гц, 2H), 2,35 (с, 3H), 2,30 (с, 2H), 2,16 (м, 2H), 2,08 (с, 4H), 1,84 (м, 2H), 1,68 (м, 2H).
Пример 51. N-(3-фтор-4-((7-(3-((3-меркаптопропил)амино)-пропокси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)-циклопропан-1,1-диформамид
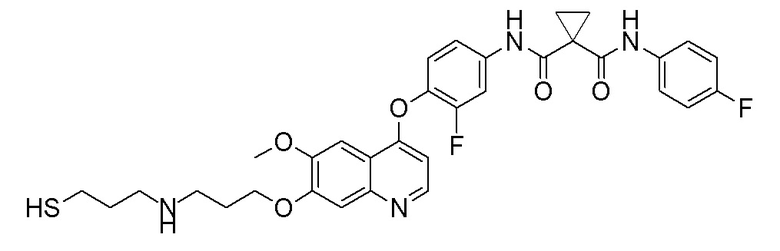
В соответствии с методикой проведения гидролиза, описанной в примере 45, целевое соединение (57 мг, 54%) получали из соединения примера 50.
LC-MS: m/z=637[M+H] +.
1H ЯМР (400 МГц, CDCl3) δ 10,31 (с, 1H), 8,63 (д, J=15,4 Гц, 2H), 7,90 (д, J=10,9 Гц, 1H), 7,72 (с, 1H), 7,65-7,56 (м, 3H), 7,47-7,29 (м, 3H), 7,23-7,11 (м, 2H), 6,92 (с, 1H), 6,55 (с, 1H), 4,42 (с, 2H), 4,18 (с, 3H), 3,67 (с, 2H), 2,27-2,20 (м, 8H), 1,92 (с, 2H), 1,77 (с, 2H).
Пример 52. S-(3-((3-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)пропил)(метил)амино)пропил)тиоацетат
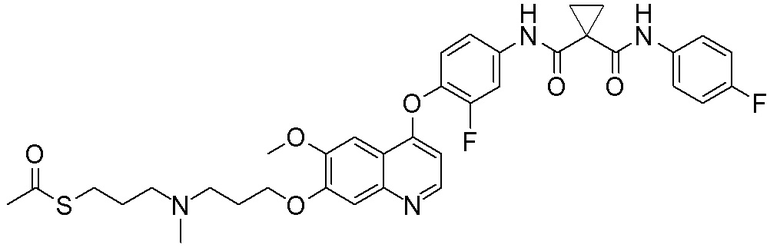
S-(3-((3-((4-(2-фтор-4-(1-((4-фторфенил)карбамоил)-циклопропан-1-карбоксамидо)фенилокси)-6-метоксихинолин-7-ил)окси)пропил)амино)пропил)тиоацетата (50 мг, 0,074 ммоль) (синтезированного в примере 50) растворяли в THF (1,5 мл), добавляли при комнатной температуре водный раствор формальдегида (0,1 мл), Pd/C (47 мг, 0,22 ммоль), и затем три раза продували H2. Проводили реакцию при комнатной температуре в течение 3,0 часов, и затем фильтровали под вакуумом. Органическую фазу концентрировали, и затем очищали препаративной тонкослойной хроматографией с получением целевого соединения (12 мг, 24%).
LC-MS: m/z=693[M+H] +.
Пример 53. N-(3-фтор-4-((7-(3-((3-меркаптопропил)(метил)-амино)пропокси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фтор-фенил)циклопропан-1,1-диформамид
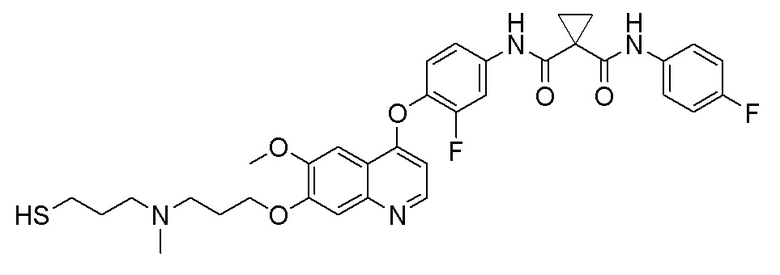
Проводили гидролиз в соответствии с методикой, описанной в примере 45, получая целевое соединение (7 мг, 48%).
LC-MS: m/z=651[M+H] +.
Пример 54. N-(3-фтор-4-((7-(3-((5-меркапто-1,3,4-тритиазол-2-ил)тио)пропокси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-дикарбоксамид
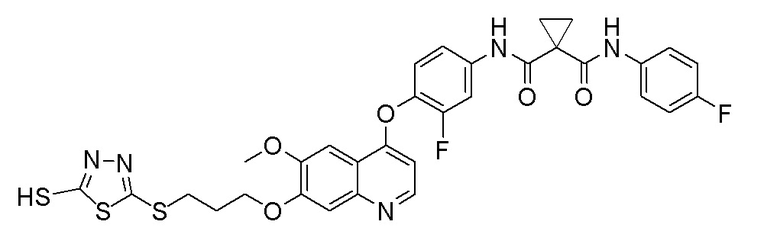
120 мг (0,8 ммоль, 5,0 экв) 1,3,4-тритиазол-2,5-дитиофенола растворяли в 10 мл N, N-диметилформамида, и добавляли при комнатной температуре 64 мг (1,6 ммоль, 10,0 экв) NaH. Проводили реакцию при перемешивании в течение 10 минут. Затем добавляли 100 мг (1 ммоль, 1,0 экв) N-(4-((7-(3-бромпропокси)-6-метоксихинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)-циклопропан-1,1-диформамида (синтезированный на стадии B примера 1). Проводили реакцию в течение ночи при комнатной температуре. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), реакционную систему выливали в 150 мл воды и затем экстрагировали с помощью 200 мл этилацетата. Промывали последовательно органическую фазу два раза водой и насыщенным солевым раствором. Органическую фазу затем сушили над безводным сульфатом натрия и испаряли досуха. Остаток очищали колоночной хроматографией на силикагеле с получением продукта (14 мг, выход=13%).
LC-MS: m/z=696[M+H] +.
1H ЯМР (400 МГц, CDCl3) δ 9,99 (с, 1H), 8,82 (с, 1H), 8,42 (с, 1H), 7,82-7,73 (м, 2H), 7,55 (с, 1H), 7,53 (м, 2H), 7,37 (д, J=7,7 Гц, 1H), 7,18 (кв, J=8,0 Гц, 1H), 7,06 (т, J=8,0 Гц, 2H), 6,20 (с, 1H), 4,57 (с, 2H), 4,30 (уш.с, 1H), 4,15 (с, 3H), 3,41 (с, 2H), 2,48 (с, 2H), 1,83 (с, 2H), 1,72 (с, 2H).
Пример 55. S-(2-(3-(2-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенилокси)-6-метокси-хинолин-7-ил)(окси)этил)уреидо)этил)этантиолат
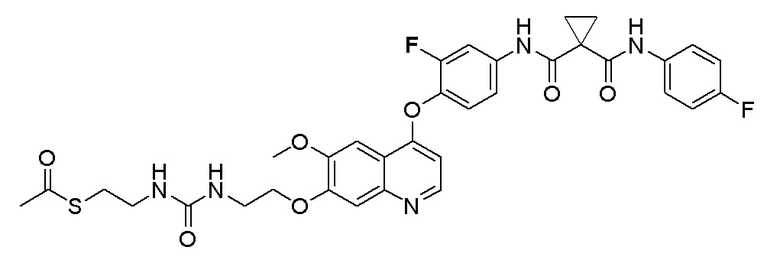
Стадия A. N-(4-((7-(2-(3-(2-хлорэтил)уреидо)этокси)-6-метоксихинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)-циклопропан-1,1-диформамид
При 0°C добавляли триэтиламин и трифосген (54 мг, 0,183 ммоль) к 2-хлорэтиламина гидрохлориду (64 мг, 0,547 ммоль) в DCM, проводили реакцию при этой температуре в течение 1 часа, и добавляли по каплям реакционную смесь в N-(4-((7-(2-аминоэтокси)-6-метоксихинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)циклопропан-1,1-диформамид в DCM, проводили реакцию при комнатной температуре в течение 1 часа, останавливали реакцию путем добавления воды, экстрагировали 3 раза с помощью DCM, промывали объединенную органическую фазу один раз насыщенным солевым раствором и сушили над безводным сульфатом натрия, концентрировали путем дистилляции при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением продукта (65 мг).
Стадия B. S-(2-(3-(2-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенилокси)-6-метокси-хинолин-7-ил)(окси)этил)уреидо)этил)тиоацетат
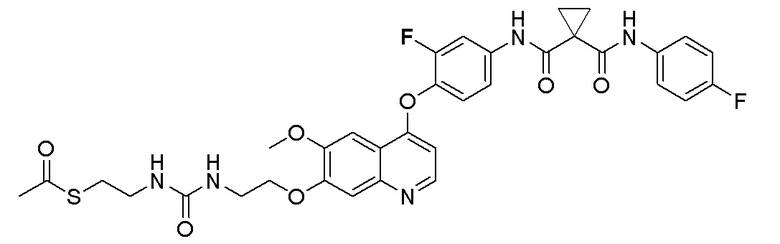
К N-(4-((7-(2-(3-(2-хлорэтил)уреидо)этокси)-6-метокси-хинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)циклопропан-1,1-диформамиду (60 мг, 0,092 ммоль) в ацетоне добавляли тиоацетат калия (21 мг, 0,183 ммоль), нагревали до 40°С, и проводили реакцию в течение 3 часов. Охлаждали до комнатной температуры, концентрировали путем дистилляции при пониженном давлении и очищали колоночной хроматографией на силикагеле с получением продукта (17 мг).
1H ЯМР (400 МГц, CDCl3) δ 10,13 (с, 1H), 9,94 (с, 1H), 9,50 (с, 1H), 8,50 (с, 1H), 7,91-7,77 (м, 1H), 7,70-7,37 (м, 5H), 7,24-7,22 (м, 2H), 7,07 (с, 2H), 6,42 (с, 1H), 4,28 (с, 2H), 4,03 (с, 3H), 3,82-3,47 (м, 4H), 3,06 (м, 2H), 2,43 (с, 3H), 1,30 (с, 4H). LC-MS: m/z=694 [M+H] +.
Пример 56. N-(3-фтор-4-((7-(2-(3-(2-меркаптоэтил)уреидо)-этокси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)-циклопропан-1,1-диформамид
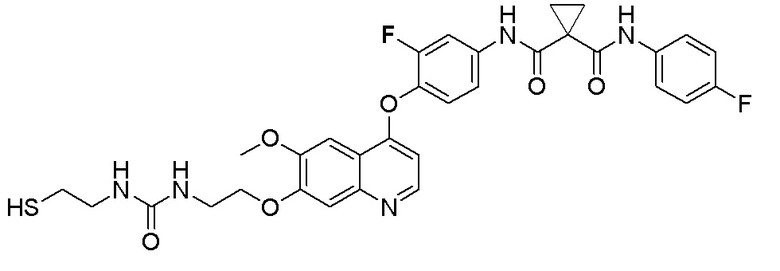
Проводили гидролиз в соответствии с методикой, описанной в примере 45, получая целевое соединение (8 мг, 54%).
LC-MS: m/z=652[M+H] +.
Пример 57. S-(2-((5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)пентил)амино)-2-оксо-этил)тиоацетат
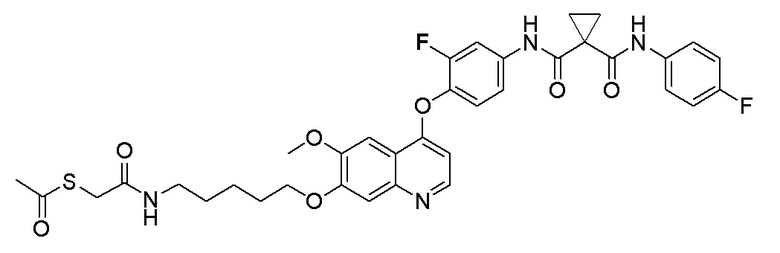
Стадия A. Третбутил (5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)пентил)аминокарбоксилат
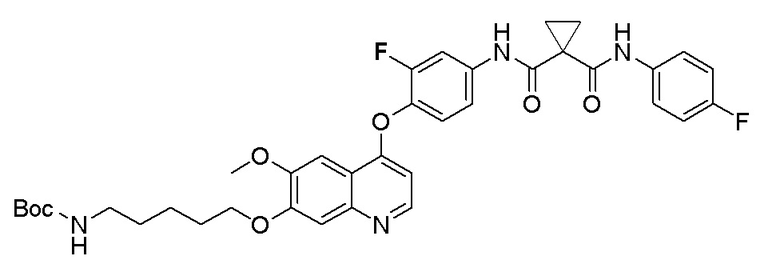
При 0°С к N-(3-фтор-4-((7-гидрокси-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диформамиду (505 мг, 1,0 ммоль), третбутил (5-гидроксипентил) карбамату (406 мг, 2,0 ммоль) и трифенилфосфину (786 мг, 3,0 ммоль) в DCM медленно по каплям добавляли диизоэфир азодикарбоксилата (606 мг, 3,0 ммоль). Проводили реакцию при комнатной температуре в течение 5 часов. Добавляли воду для остановки реакции, экстрагировали 3 раза с помощью DCM, промывали объединенную органическую фазу один раз насыщенным солевым раствором, и сушили над безводным сульфатом натрия. Органическую фазу концентрировали путем дистилляции при пониженном давлении и очищали колоночной хроматографией на силикагеле с получением продукта (380 мг, 55%).
Стадия B. N-(4-((7-((5-аминопентил)окси)-6-метоксихинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)циклопропан-1,1-диформамид
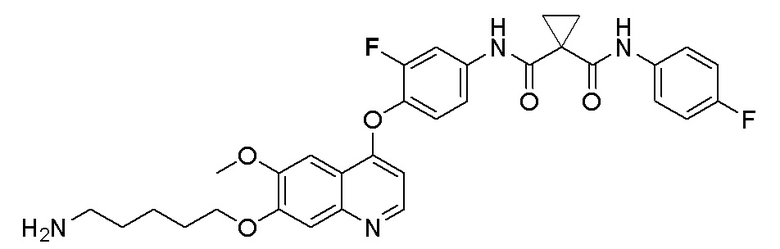
К третбутил (5-((4-(2-фтор-4-(1-((4-фторфенил)карбамоил)-циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-ил)-окси)пентил)карбамату (200 мг, 0,29 ммоль) в DCM добавляли трифторуксусную кислоту (0,5 мл), и проводили реакцию при комнатной температуре в течение 3 часов, затем останавливали реакцию насыщенным водным раствором бикарбоната натрия. Реакционный раствор экстрагировали 3 раза с помощью DCM, и объединенную органическую фазу промывали один раз насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали путем дистилляции при пониженном давлении с получением неочищенного продукта (130 мг, 76%), который непосредственно использовали в следующей реакции.
Стадия C. S-(2-((5-((4-(2-Фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)пентил)амино)-2-оксо-этил)тиоацетат
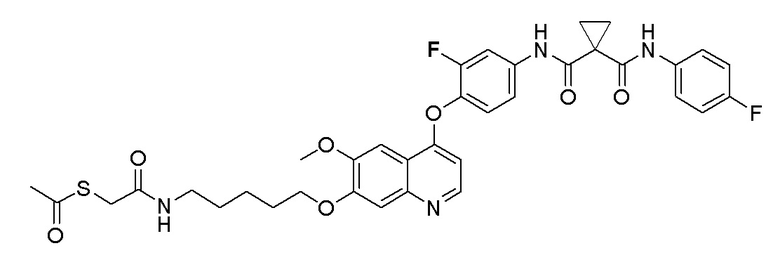
При 0°С к N-(4-((7-((5-аминопентил)окси)-6-метоксихинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)циклопропан-1,1-диформамиду (100 мг, 0,17 ммоль) и 2-(тиоацетат)уксусной кислоте (34 мг, 0,25 ммоль) в смешанном растворе DMF/DCM добавляли DIEA (84 мкл, 0,507 ммоль) и HATU (130 мг, 0,25 ммоль), и затем подогревали до комнатной температуры и проводили реакцию в течение 2 часов, останавливали реакцию водой, экстрагировали 3 раза с помощью DCM и промывали объединенную органическую фазу один раз насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали путем дистилляции при пониженном давлении, очищали колоночной хроматографией на силикагеле с получением продукта (60 мг, 50%).
1H ЯМР (400 МГц, CDCl3) δ 10,06 (с, 1H), 8,52 (с, 1H), 8,34 (с, 1H), 7,81 (д, J=12,4 Гц, 1H), 7,61 (с, 1H), 7,50-7,46 (м, 3H), 7,26-7,24 (м, 2H), 7,12-7,08 (м, 2H), 6,45 (с, 1H), 6,33 (с, 1H), 4,23 (с, 2H), 4,08 (с, 3H), 3,57 (с, 2H), 3,33-3,31 (м, 2H), 2,43 (с, 3H), 1,86-1,84 (м, 2H), 1,79-1,76 (м, 4H), 1,67 (с, 4H). LC-MS: m/z=707 [M+H] +.
Пример 58. N-(3-фтор-4-((7-((5-(2-меркаптоацетиламино)-пентил)окси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фтор-фенил)циклопропан-1,1-диформамид
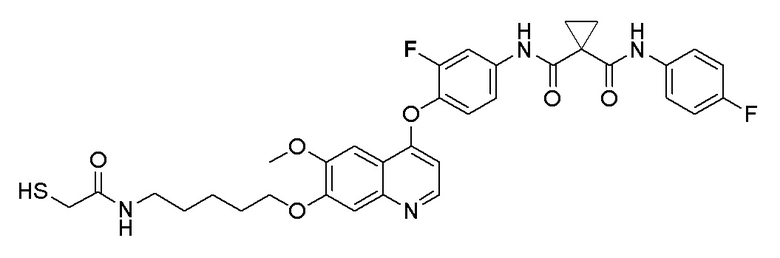
Проводили гидролиз в соответствии с методикой, описанной в примере 45, получая целевое соединение (12 мг, 67%) из соединения примера 57.
LC-MS: m/z=665[M+H]+.
Пример 59. S-(2-((5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)пентил)амино)-2-оксо-этил)тиоацетат
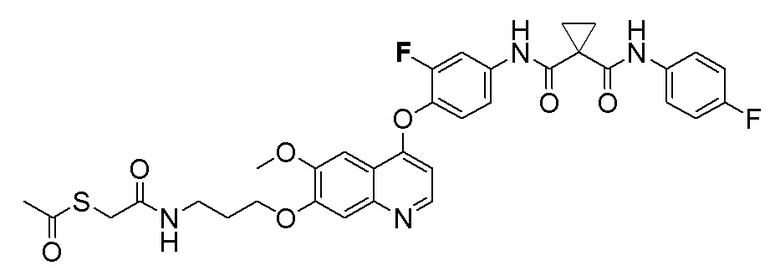
Получали целевое соединение (86 мг, 58%) в соответствии с синтезом, описанным в примере 57.
LC-MS: m/z=679[M+H]+.
Пример 60. N-(3-фтор-4-((7-((5-(2-меркаптоацетиламино)-пентил)окси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фтор-фенил)циклопропан-1,1-диформамид
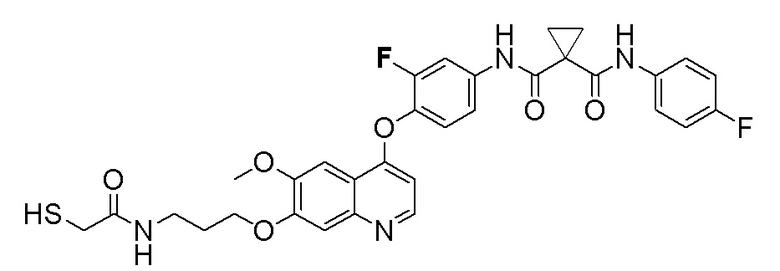
Проводили гидролиз в соответствии с методикой, описанной в примере 45, получая целевое соединение (35 мг, 39%) из соединения примера 59.
LC-MS: m/z=637[M+H]+.
Пример 61. S-(2-((5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)пентил)амино)-2-оксо-этил)тиоацетат
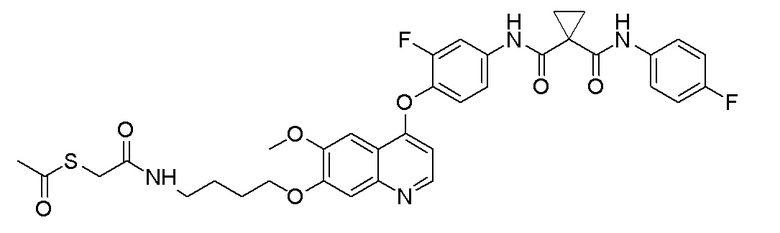
Получали целевое соединение (49 мг, 56%) в соответствии с синтезом, описанным в примере 57.
LC-MS: m/z=693[M+H]+.
Пример 62. N-(3-фтор-4-((7-((5-(2-меркаптоацетиламино)-пентил)окси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фтор-фенил)циклопропан-1,1-диформамид
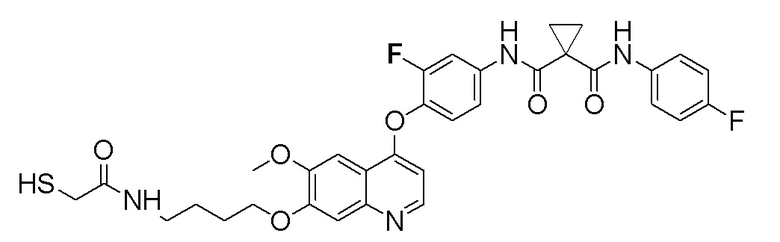
Проводили гидролиз в соответствии с методикой, описанной в примере 45, получая целевое соединение (60 мг, 71%) из соединения примера 61.
LC-MS: m/z=651[M+H] +.
Пример 63. S-(2-((5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)пентил)амино)-2-оксо-этил)тиоацетат
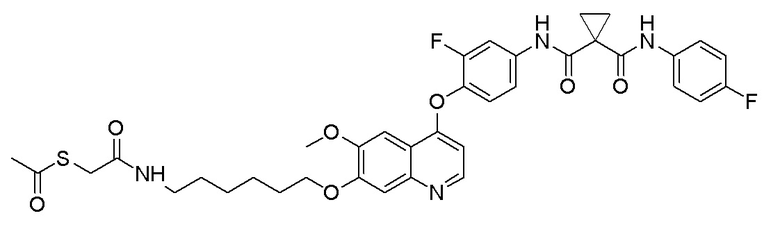
Получали целевое соединение (120 мг, 64%) в соответствии с синтезом, описанным в примере 57.
LC-MS: m/z=721[M+H] +.
Пример 64. N-(3-фтор-4-((7-((5-(2-меркаптоацетиламино)-пентил)окси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фтор-фенил)циклопропан-1,1-диформамид
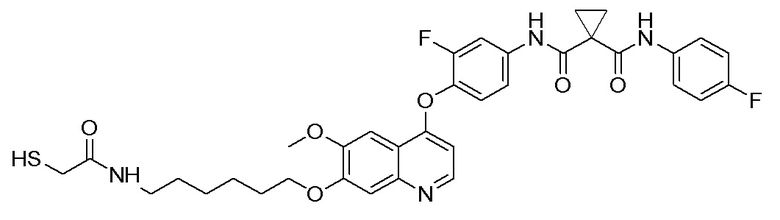
Проводили гидролиз в соответствии с методикой, описанной в примере 45, получая целевое соединение (52 мг, 72%) из соединения примера 63.
LC-MS: m/z=679[M+H] +.
Пример 65. S-(2-((5-((4-(4-(1-((4-фторфенил)карбамоил)-циклопропан-1-карбоксамидо)фенокси)-6-(метоксихинолин-7-ил)-окси)пентил)амино)-2-оксо-этил)тиоацетат
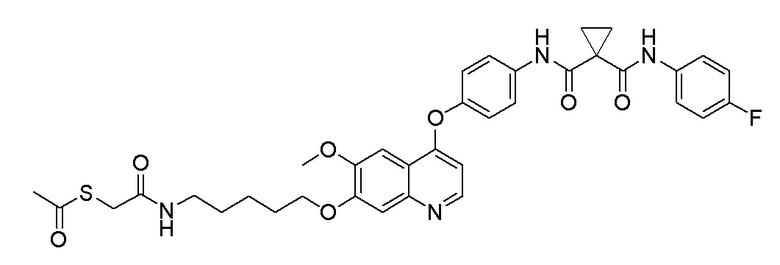
Получали целевое соединение (92 мг, 52%) в соответствии с синтезом, описанным в примере 57.
LC-MS: m/z=689 [M+H] +.
Пример 66. N-(4-фторфенил)-N-(4-((7-((5-(2-меркапто-ацетамидо)пентил)окси)-6-метоксихинолин-4-ил)окси)фенил)-циклопропан-1,1-диформамид
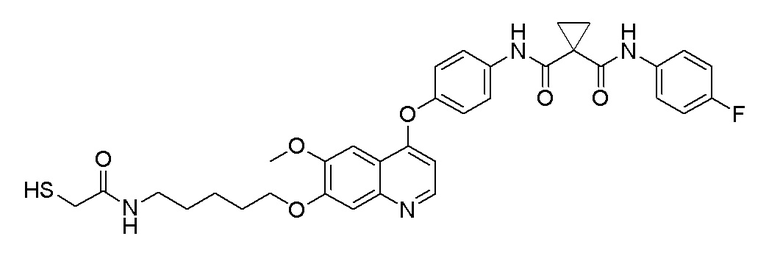
Проводили гидролиз в соответствии с методикой, описанной в примере 45, получая целевое соединение (32 мг, 46%) из соединения примера 65.
LC-MS: m/z=647[M+H] +.
Пример 67. S-(2-((5-((4-(4-(1-(циклопропилкарбамоил)-циклопропан-1-карбоксамидо)-2-фторфенокси)-6-метоксихинолин-7-ил)окси)пентил)амино)-2-оксо-этил)тиоацетат
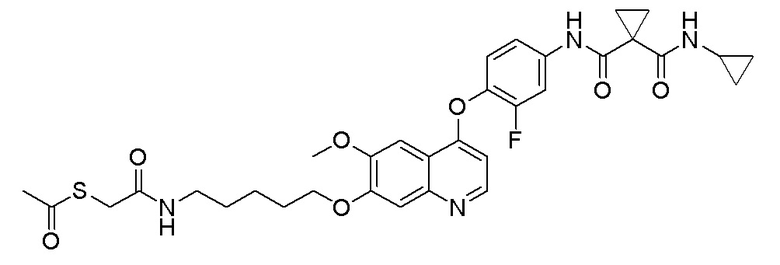
Получали целевое соединение (150 мг, 79%) в соответствии с синтезом, описанным в примере 57.
1H ЯМР (400 МГц, CDCl3) δ 11,35 (с, 1H), 8,50 (кв, J=4,0 Гц, 1H), 7,84 (дд, J=4,0, 8,0 Гц,1H), 7,62 (с, 1H), 7,42 (с, 1H), 7,36-7,30 (м, 2H), 7,24 (кв, J=8,0 Гц, 1H), 6,03-5,92 (м, 1H), 4,24 (кв, J=4,0 Гц, 2H), 4,08 (с, 3H), 3,43-3,25 (м, 2H), 3,02(с, 2H), 2,80-2,70 (м, 1H), 2,19-1,92 (м, 5H), 1,84-1,75 (м, 2H), 1,71-1,56 (м, 4H), 1,38-1,25 (м, 2H), 0,89 (дд, J=4,0, 8,0 Гц, 2H), 0,58 (дд, J=4,0, 8,0 Гц, 2H).
LC-MS: m/z=653 [M+H] +.
Пример 68. N-циклопропил-N-(3-фтор-4-((7-((5-(2-меркапто-ацетамидо)пентил)окси)-6-метоксихинолин-4-ил)окси)фенил)-циклопропан-1,1-диформамид
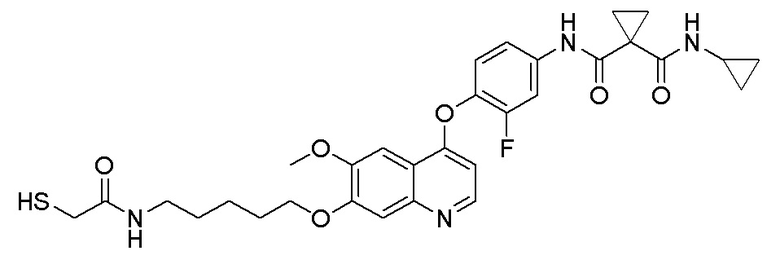
Проводили гидролиз в соответствии с методикой, описанной в примере 45, получая целевое соединение (22 мг, 38%) из соединения примера 67.
LC-MS: m/z=611[M+H] +.
Пример 69. S-(2-((5-((6-карбамоил-4-(3-хлор-4-(3-циклопропилмочевина)фенокси)хинолин-7-ил)окси)пентил)амино)-2-оксо-этил)тиоацетат
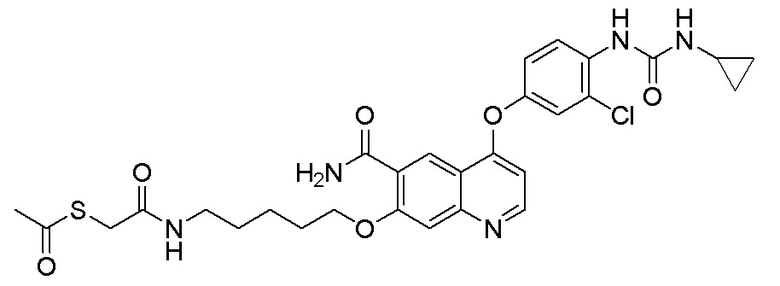
Стадия A. 5-Бромпентиламина гидробромидная соль
Медленно добавляли 5-аминопентанол (20 г, 194 ммоль) в 40% водный раствор бромистого водорода (200 мл) при комнатной температуре, и проводили реакцию при 100°С в течение 4 часов. Удаляли растворитель путем дистилляции при пониженном давлении, и воду в остатке поглощали метанолом (150 мл). Опять отгоняли растворитель при пониженном давлении. Полученный остаток суспендировали в DCM, и твердое вещество собирали фильтрацией под вакуумом и споласкивали его DCM, и сушили естественным образом с получением продукта (35 г, 75%).
Стадия B. S-(2-((5-бромпентил)амино)-2-оксо-этил)тиоацетат
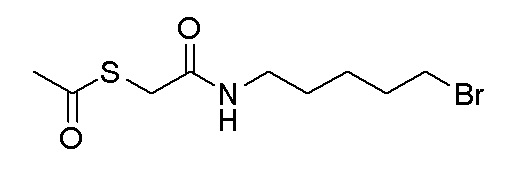
На ледяной бане, добавляли DMF (1 мл) и оксалилхлорид (2,5 мл, 30 ммоль) к 2-(ацетилтио)уксусной кислоте (2,68 г, 20 ммоль) в DCM, подогревали до комнатной температуры и проводили реакцию в течение 2 часов. Удаляли растворитель путем дистилляции при пониженном давлении. Остаток разбавляли с помощью 100 мл DCM, и растворитель опять отгоняли при пониженном давлении. Остаток непосредственно использовали на следующей стадии реакции (3,0 г).
К остатку в DCM (50 мл) добавляли 5-бромпентиламина гидробромид (2,47 г, 10 ммоль), и к реакционной системе медленно добавляли по каплям DIEA (9 мл, 50 ммоль) на ледяной бане. Температуру повышали до комнатной температуры, и проводили реакцию в течение 1 часа. Реакцию останавливали путем добавления воды, и реакционный раствор промывали 30% водным раствором лимонной кислоты (100 мл x 2). Органическую фазу промывали один раз насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали путем дистилляции при пониженном давлении и очищали колоночной хроматографией на силикагеле с получением продукта (1,31 г, 46%).
1H ЯМР (400 МГц, CDCl3) δ 6,30 (уш.с, 1H), 3,55 (с, 2H), 3,44 (кв, J=4,0 Гц, 2H), 3,33-3,23 (м, 2H), 2,45 (с, 3H), 1,93-1,86 (м, 2H), 1,57-1,46 (м, 4H).
Стадия C. 4-(3-хлор-4-(3-циклопропилмочевина)фенокси)-7-гидроксихинолин-6-карбоксамид
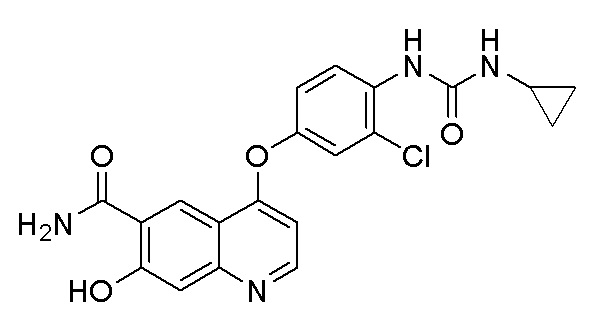
На ледяной бане, к 4-[3-хлор-4-(циклопропиламино- карбонил)аминофенокси]-7-метокси-6-хинолинкарбоксамиду (1,39 г, 3,37 ммоль) в DCM (30 мл) добавляли BBr3 (5 мл, 52 ммоль). После добавления, проводили реакцию на ледяной бане в течение 5 часов. Растворитель отгоняли при пониженном давлении, и реакционный раствор промывали насыщенным водным раствором бикарбоната натрия (100 мл x 2), и органическую фазу промывали один раз насыщенным солевым раствором. Сушили над сульфатом натрия, концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле с получением продукта (0,89 г, 66%).
Стадия D. S-(2-((5-((6-карбамоил-4-(3-хлор-4-(3-цикло-пропилмочевина)фенокси)хинолин-7-ил)окси)пентил)амино)-2-оксо-этил)тиоацетат
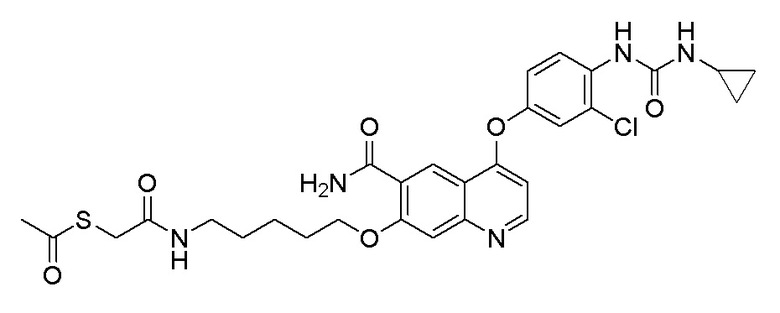
При комнатной температуре, к 4-(3-хлор-4-(3-циклопропил-мочевина)фенокси)-7-гидроксихинолин-6-карбоксамиду (300 мг, 0,73 ммоль) в DMF добавляли карбонат калия (201 мг, 1,46 ммоль) и S-(2-((5-бромпентил)амино)-2-оксоэтил)тиоацетат (206 мг, 0,73 ммоль). Проводили реакцию при комнатной температуре в течение 3 часов, добавляли воду, и экстрагировали 3 раза с помощью DCM. Объединенную органическую фазу промывали один раз насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали путем дистилляции при пониженном давлении с получением неочищенного продукта. Остаток очищали колоночной хроматографией на силикагеле с получением продукта (130 мг, 29%).
1H ЯМР (400 МГц, CDCl3) δ 9,25 (с, 1H), 8,66 (с, 1H), 8,42 (д, J=8,0 Гц, 1H), 7,92 (с, 1H), 7,83 (с, 1H), 7,53 (с, 1H), 7,21 (с, 1H), 7,11 (д, J=8,0 Гц, 1H), 6,55-6,48 (м, 3H), 5,84 (уш.с, 1H), 4,31 (с, 2H), 3,58 (с, 2H), 3,33 (с, 2H), 2,69 (с, 1H), 2,43 (с, 3H), 2,01 (с, 2H), 1,69-1,53 (м, 4H), 0,95-0,82 (м, 2H), 0,79-0,71 (м, 2H). LC-MS: m/z=636 [M+Na] +.
Пример 70. 4-(3-хлор-4-(3-циклопропилмочевина)фенокси)-7-((5-(2-меркаптоацетамидо)пентил)окси)хинолин-6-формамид
Проводили гидролиз в соответствии с методикой, описанной в примере 45, получая целевое соединение (22 мг, 38%) из соединения примера 69.
LC-MS: m/z=572 [M+H] +.
Пример 71. S-(2-((5-((6-карбамоил-4-(2-фтор-4-(1-((4-фторфенил)карбамоил)циклопропан-1-метиламидо)фенокси)хинолин-7-ил)окси)пентил)амино)-2-оксо-этил)тиоацетат
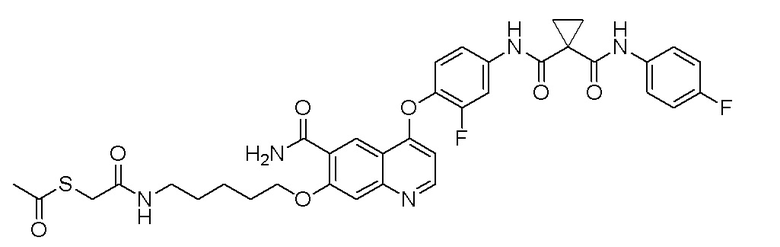
Получали целевое соединение (100 мг, 21%) в соответствии с синтезом, описанным в примере 69.
1H ЯМР (400 МГц, CDCl3) δ 10,11 (с, 1H), 9,46 (с, 1H), 9,23 (с, 1H), 8,64 (д, J=4,0 Гц, 1H), 7,98 (с, 1H), 7,87 (д, J=8,0 Гц, 1H), 7,53-7,49 (м, 3H), 7,18 (кв, J=8,0 Гц, 1H), 7,03 (кв, J=8,0 Гц, 1H), 6,53-6,42 (м, 2H), 6,39(д, J=4,0 Гц, 1H), 4,30 (кв, J=4,0 Гц, 2H), 3,58 (с, 2H), 3,33 (т, J=4,0 Гц, 2H), 2,44 (с, 3H), 2,08-1,95 (м, 2H), 1,74 (с, 4H), 1,68-1,55 (м, 4H).
LC-MS: m/z=720 [M+H] +.
Пример 72. N-(4-((6-карбамоил-7-((5-(2-меркаптоацетамидо)-пентил)окси)хинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)-циклопропан-1,1-диформамид
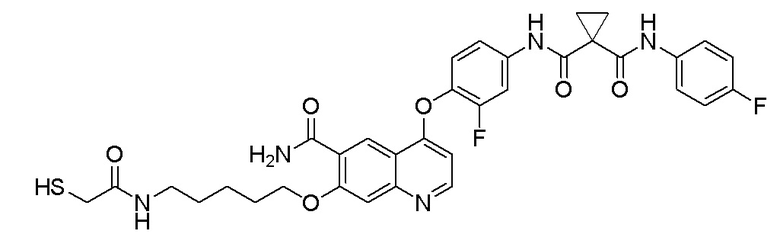
Проводили гидролиз в соответствии с методикой, описанной в примере 45, получая целевое соединение (18 мг, 43%) из соединения примера 71.
LC-MS: m/z=678 [M+H] +.
Пример 73. S-(2-((5-((6-карбамоил-4-(4-(1-(циклопропанил-карбамоил)циклопропан-1-карбоксамидо)-2-фторфенокси)хинолин-7-ил)окси)пентил)амино)-2-оксо-этил)тиоацетат
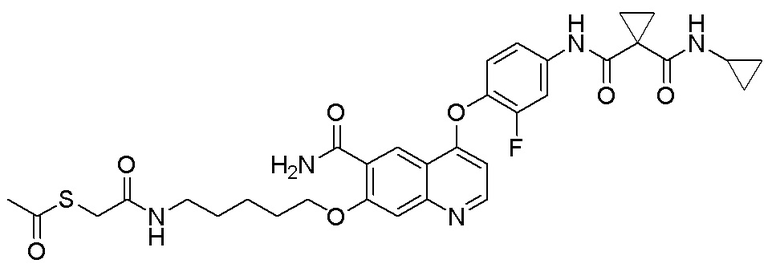
Получали целевое соединение (100 мг, 29%) в соответствии с синтезом, описанным в примере 69.
1H ЯМР (400 МГц, CDCl3) δ11,23 (с, 1H), 9,32 (с, 1H), 8,68 (д, J=8,0 Гц, 1H), 7,88-7,80 (м, 2H), 7,54 (с, 1H), 7,34 (т, J=8,0 Гц, 1H), 7,23 (т, J=8,0 Гц, 1H), 6,44 (д, J=4,0 Гц, 1H), 6,33 (уш.с, 1H), 6,09 (с, 1H), 5,95 (с, 1H), 4,33 (т, J=4,0 Гц, 2H), 3,57 (с, 2H), 3,34 (кв, J=4,0 Гц, 2H), 2,76 (с, 1H), 2,45 (с, 3H), 2,09-2,00 (м, 2H), 1,85-1,78 (м, 2H), 1,68-1,61 (м, 6H), 0,89-0,84 (м, 2H), 0,59-0,54 (м, 2H).
LC-MS: m/z=666 [M+H] +.
Пример 74. N-(4-((6-карбамоил-7-((5-(2-меркаптоацетамидо)-пентил)окси)хинолин-4-ил)окси)-3-фторфенил)-N-циклопропил-циклопропан-1,1-диформамид
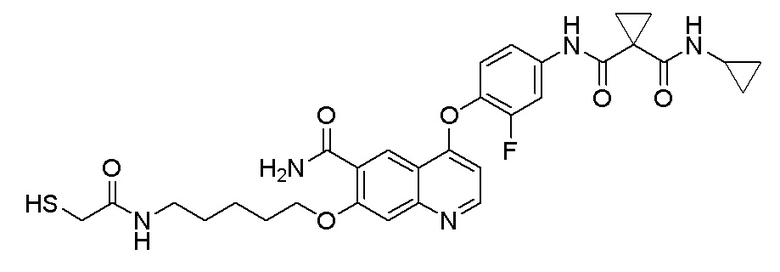
Проводили гидролиз в соответствии с методикой, описанной в примере 45, получая целевое соединение (14 мг, 28%) из соединения примера 73.
LC-MS: m/z=624 [M+H] +.
Пример 75. S-(2-((5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)сульфанил)пентил)амино)-2-оксоэтилтиоацетат
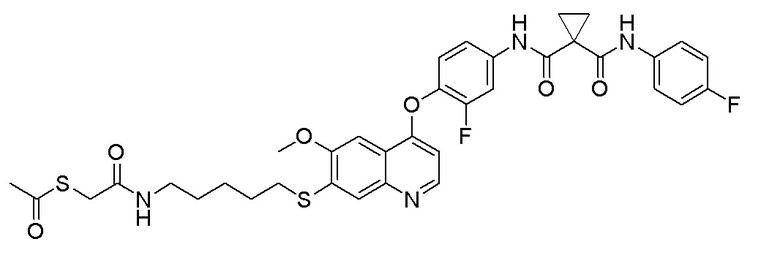
Стадия A. Третбутил (5-бромпентил) карбамат
При комнатной температуре, к раствору 5-бромпентиламина гидробромида (2,07 г, 8,38 ммоль) в дихлорметане (30 мл) добавляли дитретбутилдикарбонат (2,35 г, 10,89 ммоль), затем медленно добавляли по каплям триэтиламин (1,69 г, 16,76 ммоль) на ледяной бане. Проводили реакцию при комнатной температуре в течение 4 часов, и затем промывали 20% водным раствором лимонной кислоты. Органическую фазу сушили, концентрировали и очищали колоночной хроматографией с получением продукта (1,93 г, 87%).
Стадия B. S-(5-((третбутоксикарбонил)амино)пентил)-тиоацетат
При комнатной температуре, тиоацетат калия (0,43 г, 3,76 ммоль) добавляли к третбутил(5-бромпентил)карбамату (1,0 г, 3,76 ммоль) в DCM, и проводили реакцию при комнатной температуре в течение 2 часов. Фильтровали и споласкивали осадок на фильтре с помощью DCM. Фильтрат собирали и концентрировали путем дистилляции при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с получением продукта (0,66 г, 66%).
LC-MS: m/z=262 [M+H] +.
Стадия C. Третбутил (5-меркаптопентил)карбамат
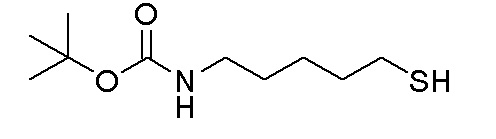
На ледяной бане, добавляли 1N водный раствор гидроксида натрия (3,80 мл, 3,80 ммоль) к S-(5-((третбутоксикарбонил)-амино)пентил)тиоацетату (0,66 г, 2,53 ммоль) в метаноле (30 мл), и затем проводили реакцию на ледяной бане в течение 10 минут. Повышали температуру до комнатной температуры, и проводили реакцию в течение 1 часа, добавляли DCM и воду для проведения экстракции. Органическую фазу промывали один раз насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали путем дистилляции при пониженном давлении, и остаток непосредственно использовали в следующей реакции (0,49 г, 89%).
LC-MS: m/z=220 [M+H] +.
Стадия D. 4-(2-фтор-4-(1-((4-фторфенил)карбамоил)-циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-трифторметансульфонат
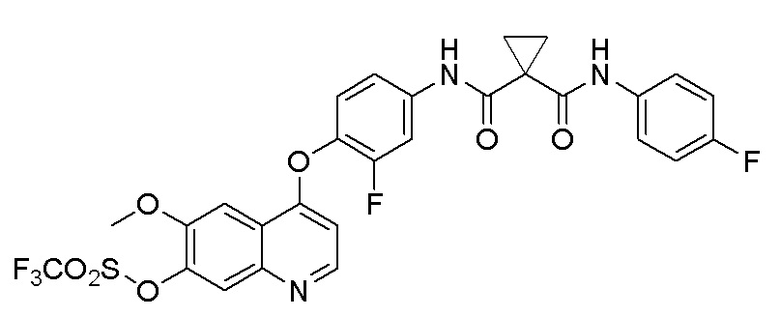
При 0°С, к N-(3-фтор-4-((7-гидрокси-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диформамиду (505 мг, 1,0 ммоль) и карбонату калия (414 мг, 3,0 ммоль) в DMF, добавляли трифторметансульфоновый ангидрид (423 мг, 1,5 ммоль), и проводили реакцию в течение 5 часов при комнатной температуре. Добавляли воду для остановки реакции, экстрагировали 3 раза с помощью DCM, и объединенную органическую фазу промывали 3 раза насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали путем дистилляции при пониженном давлении и очищали колоночной хроматографией на силикагеле с получением продукта (380 мг, 60%).
LC-MS: m/z=638 [M+H] +.
Стадия E. Третбутил (5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)сульфанил)пентил)карбамат
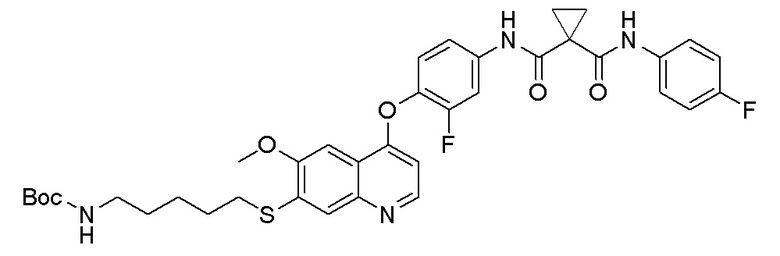
К 4-(2-фтор-4-(1-((4-фторфенил)карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метоксихинолин-7-трифторметан-сульфонату (505 мг, 1,0 ммоль) в диоксане добавляли третбутил (5-меркаптопентил)карбамат (414 мг, 3,0 ммоль) и DIEA (423 мг, 1,5 ммоль), и три раза продували азотом, затем добавляли Pd2(dba)3 (414 мг, 3,0 ммоль) и ant-Phos (414 мг, 3,0 ммоль), опять три раза продували азотом, повышали температуру до 120°С, и проводили реакцию в течение ночи. Добавляли воду для остановки реакции, и з раза экстрагировали с помощью DCM. Объединенную органическую фазу промывали один раз насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали путем дистилляции при пониженном давлении и очищали колоночной хроматографией на силикагеле с получением продукта (380 мг, 60%).
LC-MS: m/z=707 [M+H] +.
Стадия F. S-(2-((5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)сульфанил)пентил)амино)-2-оксоэтил)тиоацетат
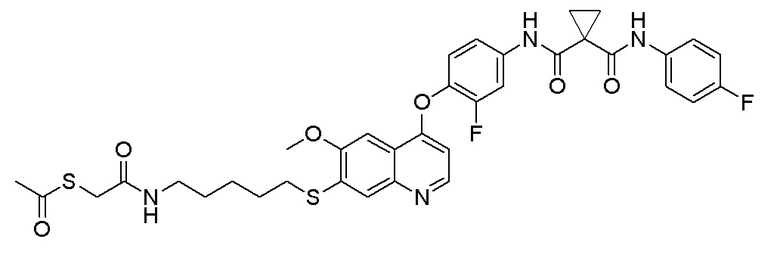
Целевое соединение (35 мг, 24%) получали в соответствии со стадиями B и C, описанными в примере 57.
1H ЯМР (400 МГц, CDCl3) δ10,05 (с, 1H), 8,54 (д, J=8,0 Гц, 1H), 8,23 (с, 1H), 7,84-7,78 (м, 2H), 7,57 (с, 1H), 7,53-7,46 (м, 2H), 7,27 (т, J=12Hz, 1H), 7,15-7,04 (т, J=8,0 Гц, 2H), 6,46 (д, J=8,0 Гц, 1H), 6,32 (уш.с, 1H), 4,10 (с, 3H), 3,57 (с, 2H), 3,29 (кв, J=8,0 Гц, 2H), 3,10 (т, J=8,0 Гц, 2H), 2,46 (с, 3H), 1,90-1,81 (м, 4H), 1,68-1,57 (м, 6H).
LC-MS: m/z=723 [M+H] +.
Пример 76. N-(3-фтор-4-((7-((5-(2-меркаптоацетамидо)-пентил)сульфанил)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фтор-фенил)циклопропан-1,1-диформамид
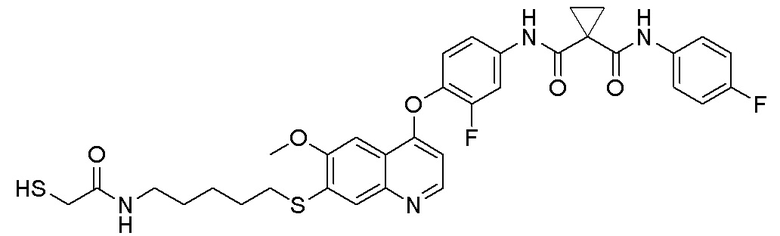
Проводили гидролиз в соответствии с методикой, описанной в примере 45, получая целевое соединение (40 мг, 30%) из соединения примера 75.
LC-MS: m/z=681 [M+H] +.
Пример 77. N-(3-фтор-4-((7-((5-(2-сульфанилацетамидо)-пентил)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)-циклопропан-1,1-диформамид
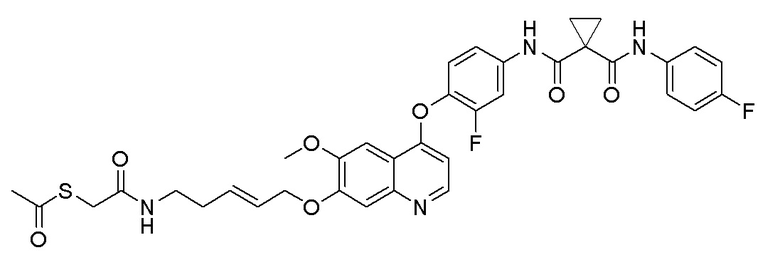
Целевое соединение (32 мг, 43%) получали в соответствии с синтезом, описанным в примере 24.
LC-MS: m/z=705 [M+H] +.
Пример 78. S-(7-((6-карбамоил-4-(3-хлор-4-(3-циклопропил-мочевина)фенокси)хинолин-7-ил)окси)гептил)тиоацетат
Получали целевое соединение (41 мг, 46%) в соответствии с синтезом, описанным в примере 69, заменяя S-(2-((5-бромпентил)-амино)-2-оксоэтил)тиоацетат на S-(7-бромгептил)-ацетилтиоэфир.
LC-MS: m/z=585 [M+H] +.
Пример 79. 4-(3-хлор-4-(3-циклопропилмочевина)фенокси)-7-((7-меркаптогептил)окси)хинолин-6-карбамоил
Проводили гидролиз в соответствии с методикой, описанной в примере 45, получая целевое соединение (22 мг, 38%) из соединения примера 78.
LC-MS: m/z=543 [M+H] +.
Пример 80. S-(2-((5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)пентил)амино)-2-оксоэтил)2-аминотиоацетата гидрохлорид
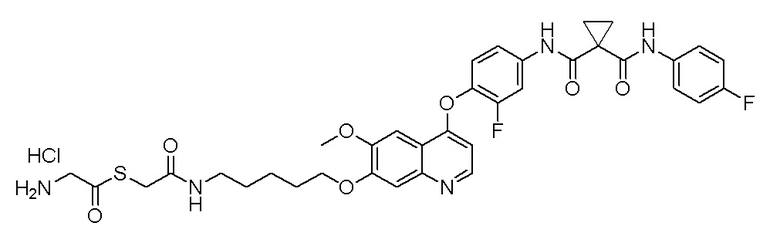
В соответствии со стадиями конденсации аминокислоты и удаления Boc-защитной группы, описанными в примере 33, для получения целевого соединения (42 мг, 62%) использовали соединение примера 58.
LC-MS: m/z=722 [M+H] +.
Пример 81. S-(2-((5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)пентил)амино)-2-оксо-этил)-(S)-2-амино-3-метилтиобутирата гидрохлорид
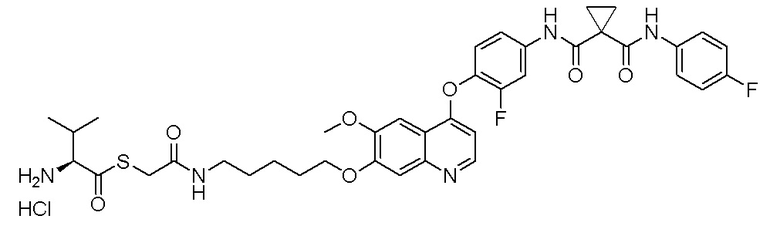
В соответствии со стадиями конденсации аминокислоты и удаления Boc-защитной группы, описанными в примере 33, для получения целевого соединения (58 мг, 58%) использовали соединение примера 58.
LC-MS: m/z=764 [M+H] +.
Пример 82. S-(2-((5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)пентил)амино)-2-оксоэтил)-(R)-2-амино-3-(1-метил-1H-индол-3-ил)тиопропионата гидрохлорид
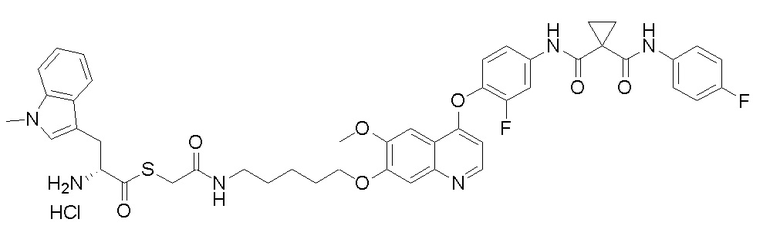
В соответствии со стадиями конденсации аминокислоты и удаления Boc-защитной группы, описанными в примере 33, для получения целевого соединения (80 мг, 69%) использовали соединение примера 58.
LC-MS: m/z=901 [M+H] +.
Пример 83. S-(2-((5-((6-карбамоил-4-(3-хлор-4-(3-циклопропилмочевина)фенокси)хинолин-7-ил)окси)пентил)амино)-2-оксоэтил)-2-аминотиоацетат гидрохлорид
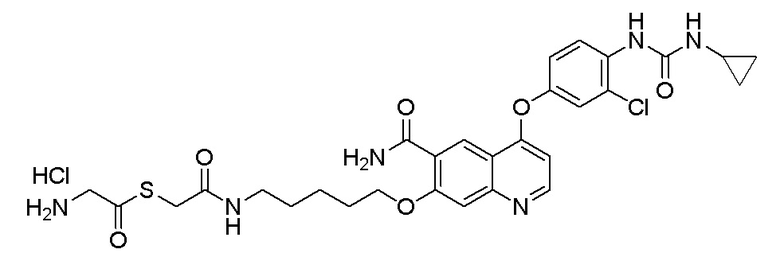
В соответствии со стадиями конденсации аминокислоты и удаления Boc-защитной группы, описанными в примере 33, для получения целевого соединения (59 мг, 67%) использовали соединение примера 70.
LC-MS: m/z=629 [M+H] +.
Пример 84. S-(2-((5-((6-карбамоил-4-(3-хлор-4-(3-циклопропилмочевина)фенокси)хинолин-7-ил)окси)пентил)амино)-2-оксоэтил)-(S)-2-амино-3-метилтиобутирата гидрохлорид
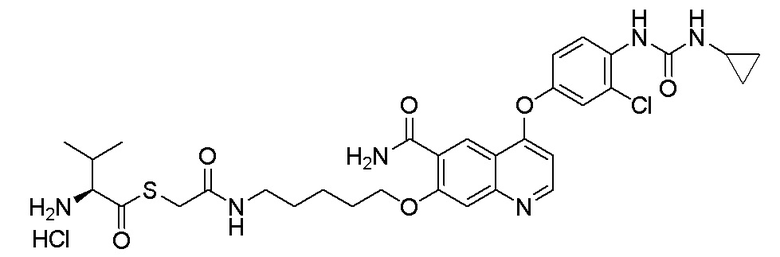
В соответствии со стадиями конденсации аминокислоты и удаления Boc-защитной группы, описанными в примере 33, для получения целевого соединения (49 мг, 36%) использовали соединение примера 70.
LC-MS: m/z=671 [M+H] +.
Пример 85. S-(2-((5-((6-карбамоил-4-(3-хлор-4-(3-циклопропилмочевина)фенокси)хинолин-7-ил)окси)пентил)амино)-2-оксоэтил)-(R)-2-амино-3-(1-метил-1H-индол-3-ил)тиопропионата гидрохлорид
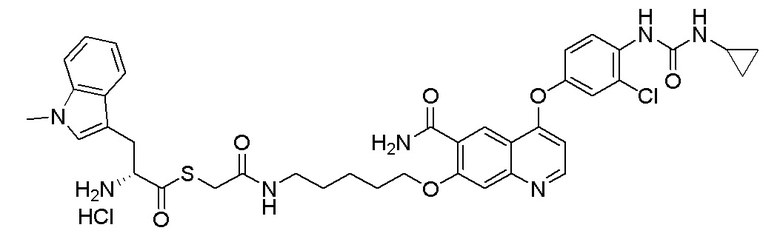
В соответствии со стадиями конденсации аминокислоты и удаления Boc-защитной группы, описанными в примере 33, для получения целевого соединения(63 мг, 51%) использовали соединение примера 70.
LC-MS: m/z=772 [M+H] +.
Пример 86. S-(7-((6-карбамоил-4-(3-хлор-4-(3-циклопропил-мочевина)фенокси)хинолин-7-ил)окси)гексил)тиоацетат
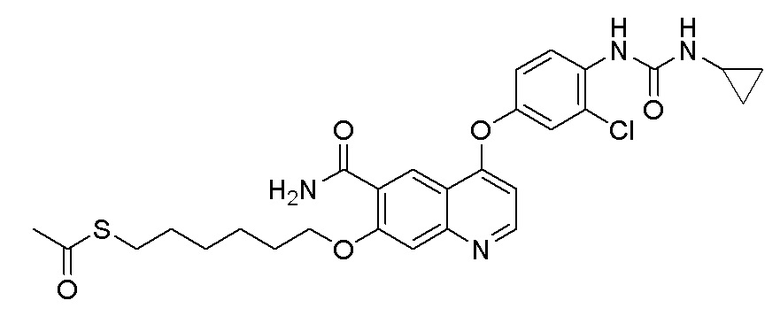
Получали целевое соединение (52 мг, 49%) в соответствии со стадиями, описанными в примере 69, заменяя S-(2-((5-бромпентил)амино)-2-оксоэтил)тиоацетат на S-(7-бромгексил)-ацетилтиоэфир.
1H ЯМР (400 МГц, CDCl3) δ9,31 (с, 1H), 8,70 (д, J=4,0 Гц, 1H), 8,45 (д, J=8,0 Гц, 1H), 7,95 (с, 1H), 7,80 (с, 1H), 7,65 (с, 1H), 7,25 (д, J=4,0 Гц, 1H), 7,13 (дд, J=8,0, 4,0 Гц, 1H), 6,53 (д, J=8,0 Гц, 1H), 6,25 (с, 1H), 5,47 (уш.с, 1H), 4,34 (т, J=8,0 Гц, 2H), 2,94 (т, J=4,0 Гц, 2H), 2,74-2,67 (м, 1H), 2,37 (с, 3H), 2,03-1,95 (м, 2H), 1,72-1,48 (м, 6H), 0,97-0,92 (м, 2H), 0,83-0,76 (м, 2H).
LC-MS: m/z=571 [M+H] +.
Пример 87. 4-(3-Хлор-4-(3-циклопропилмочевина)фенокси)-7-((7-меркаптогексил)окси)хинолин-6-карбамоил
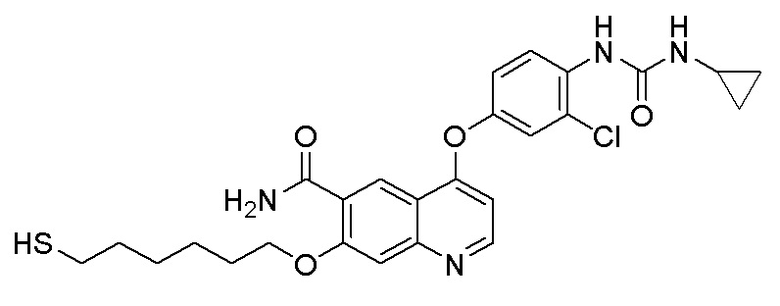
В соответствии со стадией гидролиза, описанной в примере 45, получали целевое соединение (45 мг, 40%) из соединения примера 86.
LC-MS: m/z=529 [M+H] +.
Пример 88. S-(2-((5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)оксо)фенил)амино)-2-оксоэтил)2-метилтиопропионат
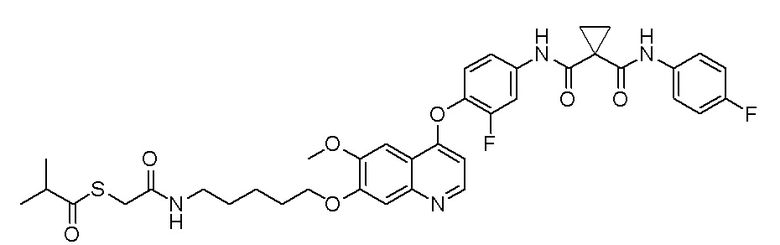
В соответствии со стадиями конденсации аминокислоты и удаления Boc-защитной группы, описанными в примере 33, для получения целевого соединения (80 мг, 69%) использовали соединение из примера 58.
1H ЯМР (400 МГц, CDCl3) δ 10,05 (уш.с, 1H), 8,71 (уш.с, 1H), 7,83 (дд, J=4,0, 8,0 Гц,1H), 7,60 (с, 1H), 7,58-7,47 (м, 3H), 7,44 (уш.с, 1H), 7,22 (т, J=8,0 Гц, 1H), 7,08 (т, J=8,0 Гц, 2H), 6,53-6,40 (м, 2H), 4,14-3,95 (м, 5H), 3,58 (с, 2H), 3,53 (с, 1H), 3,29 (кв, J=8,0 Гц, 2H), 2,84 (дт, 1H), 2,00-1,90 (м, 2H), 1,87-1,80 (м, 2H), 1,77-1,68 (м, 2H), 1,66-1,57 (м, 2H), 1,57-1,45 (м, 2H), 1,26(с, 3H), 1,25(с, 3H).
LC-MS: m/z=735 [M+H] +.
Пример 89. S-(2-((5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)оксо)фенил)амино)-2-оксо-этил)2,2-диметилтио-пропионат
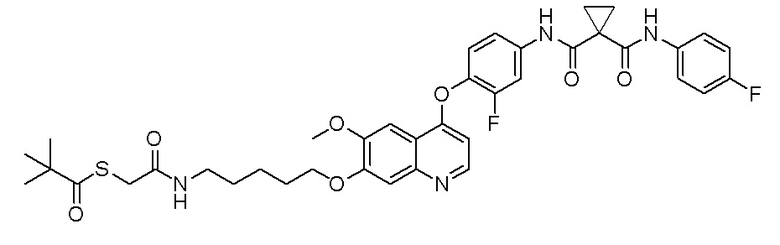
На ледяной бане, к N-(3-фтор-4-((7-((5-(2-меркапто-ацетиламино)пентил)окси)-6-метоксихинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропан-1,1-диформамиду (300 мг, 0,45 ммоль, полученному в примере 58) и триэтиламину (136 мг, 1,35 ммоль) в DCM добавляли пивалоилхлорид (82 мг, 0,68 ммоль). Проводили реакцию при комнатной температуре в течение 2 часов, добавляли воду и экстрагировали 3 раза с помощью DCM. Объединенные органические фазы промывали один раз насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного продукта, который затем очищали колоночной хроматографией на силикагеле с получением продукта (220 мг, 65%).
1H ЯМР (400 МГц, CDCl3) δ 10,20 (уш.с, 1H), 8,54 (уш.с, 1H), 7,85 (дд, J=4,0, 8,0 Гц,1H), 7,68-7,57 (м, 2H), 7,55-7,35 (м, 3H), 7,27 (т, J=8,0 Гц, 1H), 7,09 (т, J=8,0 Гц, 2H), 6,53(д, J=8,0 Гц, 1H), 6,44 (уш.с, 1H), 4,25-4,01 (м, 5H), 3,56 (с, 2H), 3,29 (кв, J=8,0 Гц, 2H), 2,02-1,90 (м, 2H), 1,88-1,82 (м, 2H), 1,75-1,45 (м, 6H), 1,31(с, 9H).
LC-MS: m/z=749 [M+H] +.
Пример 90. S-(2-((5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)оксо)фенил)амино)-2-оксо-этил)тиокаприлат
В соответствии со стадий конденсации аминокислоты, описанной в примере 33, целевое соединение (64 мг, 73%) получали из соединения примера 58.
1H ЯМР (400 МГц, CDCl3) δ 10,20 (с, 1H), 8,77 (с, 1H), 8,50 (с,1H), 7,82 (д, J=8,0 Гц, 1H), 7,61 (с, 1H), 7,55-7,41 (м, 3H), 7,34 (д, J=8,0 Гц, 1H), 7,25 (т, J=8,0 Гц, 1H), 7,07 (т, J=8,0 Гц, 2H), 6,52-6,40(м, 2H), 4,22 (т, J=8,0 Гц, 2H), 4,07 (с, 3H), 3,57 (с, 2H), 3,31 (кв, J=8,0 Гц, 2H), 2,63 (т, J=8,0 Гц, 2H), 2,05-1,90 (м, 2H), 1,86-1,77 (м, 2H), 1,75-1,55 (м, 6H), 1,45-1,20 (м, 10H), 0,89(т, J=8,0 Гц, 3H).
LC-MS: m/z=791 [M+H] +.
Пример 91. S-(2-((5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)пентил)амино)-2-оксо-этил)2-аминотиоацетата бензолсульфонат
В соответствии со стадиями конденсации аминокислоты и удаления Boc-защитной группы, описанными в примере 33 (заменяли газообразный хлористый водород на бензолсульфоновую кислоту), целевое соединение (42 мг, 62%)получали из соединения примера 58.
LC-MS: m/z=722 [M+H] +.
Пример 92. S-(2-((5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)пентил)амино)-2-оксо-этил)-(S)-2-амино-3-метилтиобутирата бензолсульфонат
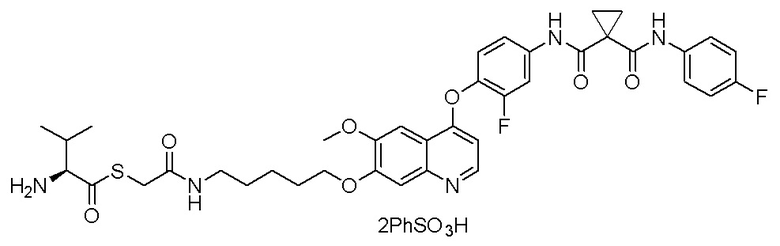
В соответствии со стадиями конденсации аминокислоты и удаления Boc-защитной группы, описанными в примере 33 (заменяли газообразный хлористый водород на бензолсульфоновую кислоту), целевое соединение (58 мг, 53%) получали из соединения примера 58.
LC-MS: m/z=764 [M+H] +.
Пример 93. S-(2-((5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)пентил)амино)-2-оксоэтил)-(R)-2-амино-3-(1-метил-1H-индол-3-ил)тиопропионата бензолсульфонат
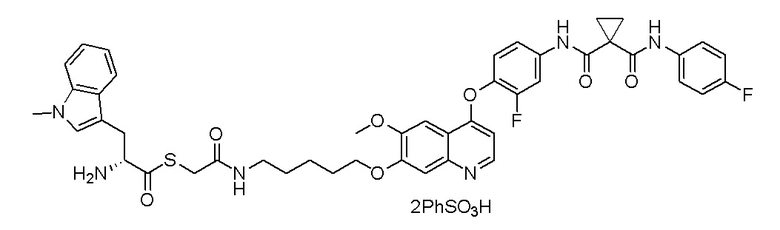
В соответствии со стадиями конденсации аминокислоты и удаления Boc-защитной группы, описанными в примере 33 (заменяли газообразный хлористый водород на бензолсульфоновую кислоту), целевое соединение (80 мг, 69%) получали из соединения примера 58.
LC-MS: m/z=865 [M+H] +.
Пример 94. S-(2-((5-((4-(2-Фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)пентил)амино)-2-оксоэтил)-(R)-2-ацетиламино-3-(1-метил-1H-индол-3-ил)тиопропионат
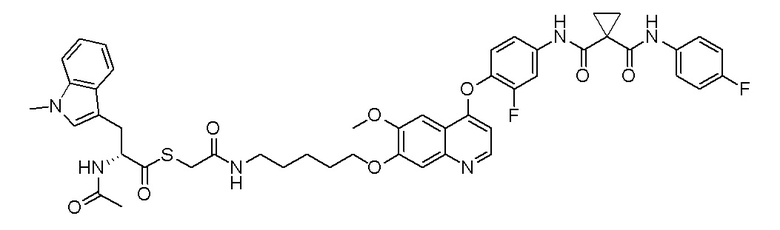
Стадия A. N-ацетил-1-метил-D-триптофан
5,0 г (22,9 ммоль, 1,0 экв) 1-метил-D-триптофана растворяли в 50 мл THF, и добавляли 10 мл водного раствора бикарбоната натрия (80,3 ммоль, 3,5 экв) при комнатной температуре, и затем добавляли по каплям 2,48 г уксусного ангидрида (22,9 ммоль, 1,0 экв). После добавления, реакционную смесь интенсивно перемешивали в течение 3 часов, и постоянно контролировали протекание реакции методом тонкослойной хроматографии (TLC). После завершения реакции, реакционный раствор концентрировали с удалением растворителя, и величину рН остатка доводили до pH 5 водным раствором лимонной кислоты, и осаждали белое твердое вещество. Твердое вещество собирали фильтрацией, промывали водой и сушили с получением целевого продукта (5,10 г, выход=86%).
LC-MS: m/z=261[M+H] +.
Стадия B. S-(2-((5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)пентил)амино)-2-оксо-этил)-(R)-2-ацетил-амино-3-(1-метил-1H-индол-3-ил)тиопропионат
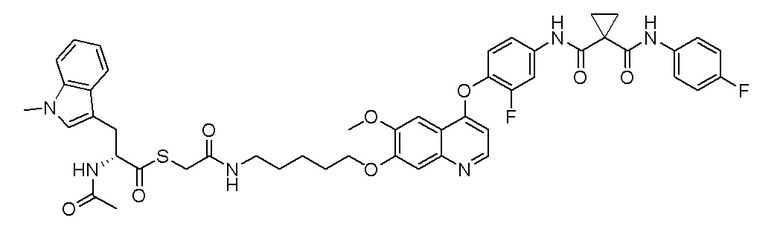
В соответствии со стадий конденсации аминокислоты, описанной в примере 33, целевое соединение (78 мг, 63%) получали из соединения примера 58.
LC-MS: m/z=907 [M+H] +.
1H ЯМР (400 МГц, CDCl3) δ 8,47 (д, J=4,0 Гц, 1H), 7,88 (дд, J=4,0, 8,0 Гц, 1H), 7,64-7,55 (м, 3H), 7,53-7,45 (м, 2H), 7,41-7,32 (м, 2H), 7,28 (д, J=8,0 Гц, 1H), 7,16-7,05 (м, 3H), 7,03-6,95 (м, 2H), 6,57 (д, J=4,0 Гц, 1H), 4,20 (т, J=8,0 Гц, 2H), 4,02 (с, 3H), 3,73 (с, 3H), 3,37-3,33 (м, 7H), 1,95 (с, 3H), 1,69 (с, 4H), 1,67 -1,56(м, 6H).
Пример 95. S-(2-((5-((4-(2-фтор-4-(1-((4-фторфенил)-карбамоил)циклопропан-1-карбоксамидо)фенокси)-6-метокси-хинолин-7-ил)окси)пентил)амино)-2-оксо-этил)-2-ацетил-аминотиоацетат
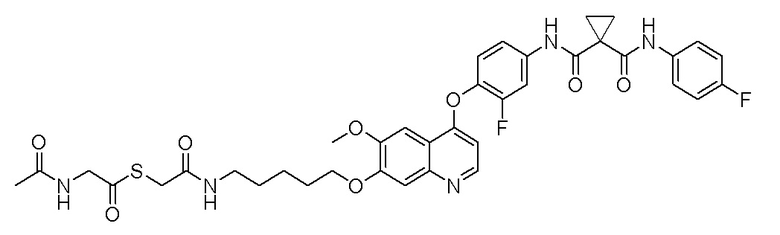
В соответствии со стадиями, описанными в примере 92, заменяли N-ацетил-1-метил-D-триптофан на ацетилглицин, получали целевое соединение (105 мг, 78%) из соединения примера 58.
LC-MS: m/z=764 [M+H] +.
Пример 96. N-(3-фтор-4-((6-метокси-7-((5-(2-(метил-дисульфанил)ацетиламино)пентил)окси)хинолин-4-ил)окси)фенил)-N-(4-фторфенил)циклопропил-1,1-диформамид
300 мг (0,45 ммоль, 1,0 экв) N-(3-фтор-4-((7-((5-(2-меркаптоацетиламино)пентил)окси)-6-метоксихинолин-4-ил)окси)-фенил)-N-(4-фторфенил)циклопропан-1,1-диформамида и 68 мг (0,54 ммоль, 1,2 экв) метил метилтиосульфоната растворяли в 5 мл этанола. Проводили реакцию при комнатной температуре в течение 3 часов и постоянно контролировали протекание реакции методом тонкослойной хроматографии (TLC). После завершения реакции, реакционный раствор концентрировали с удалением растворителя, и остаток очищали колоночной хроматографией с получением продукта (180 мг, выход=56%).
LC-MS: m/z=711 [M+H] +.
1H ЯМР (400 МГц, CDCl3) δ 10,09(с, 1H), 8,77 (с, 1H), 8,47 (д, J=4,0 Гц, 1H), 7,80 (дд, J=4,0, 8,0 Гц, 1H), 7,61 (с, 1H), 7,55-7,47 (м, 2H), 7,41 (с, 1H), 7,33 (с, 1H), 7,25 (т, J=8,0 Гц, 1H), 7,09 (т, J=8,0 Гц, 2H), 6,58(с, 1H), 6,42 (д, J=8,0 Гц, 1H), 4,22 (т, J=8,0 Гц, 2H), 4,07 (с, 3H), 3,46 (с, 2H), 3,40(кв, J=4,0 Гц, 2H), 2,49 (с, 3H), 2,05-1,95 (м, 2H), 1,86-1,79 (м, 2H), 1,75 -1,55(м, 6H).
Пример 97. N-(3-фтор-4-((6-метокси-7-((5-(2-(пиридин-2-дисульфанил)ацетамидо)пентил)оксо)хинолин-4-ил)оксо)фенил)-N-(4-фторфенил)циклопропан-1,1-диформамид
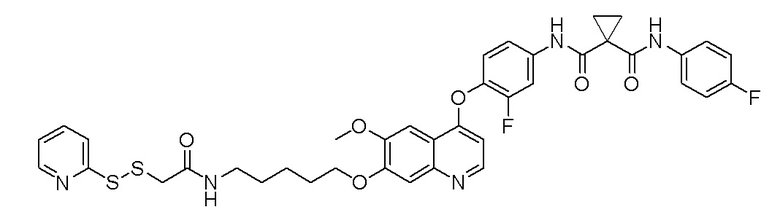
В атмосфере азота, уксусную кислоту (23 мг, 0,38 ммоль) добавляли к раствору 2,2'-дитиодипиридина (165 мг, 0,76 ммоль) в этаноле (3 мл), и добавляли по каплям при комнатной температуре раствор N-(4-((7-((5-аминопентил)окси)-6-метоксихинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)циклопропан-1,1-диформамида (250 мг, 0,38 ммоль, синтезированный в соответствии со стадиями, описанными в примере 58) в этаноле (1 мл). Проводили реакцию при комнатной температуре в течение 5 часов. Затем добавляли насыщенный водный раствор бикарбоната натрия, экстрагировали 3 раза с помощью DCM, объединенные органические фазы затем промывали один раз насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали путем дистилляции при пониженном давлении и очищали колоночной хроматографией на силикагеле с получением продукта (32 мг, 11%).
LC-MS: m/z=774 [M+H]+.
Пример 98. N, N'-((((((((2,2'-дисульфанилдиилбис(ацетил))-бис(мочевинадиил))бис(пентил-5,1-диил)) Бис(окси))бис(6-метоксихинолин-7,4-диил))бис(окси))бис(3-фтор-4,1-фенил))-бис(N-(4-фторфенил)циклопропан-1,1-диформамид)
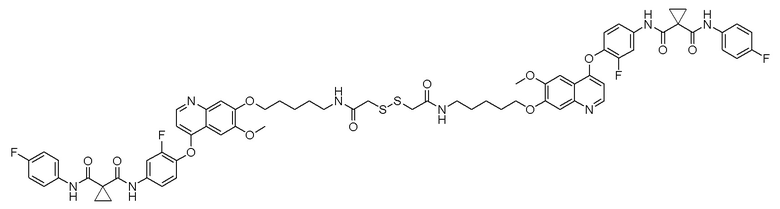
Стадия A. 2,2'-Тиодиацетилхлорид
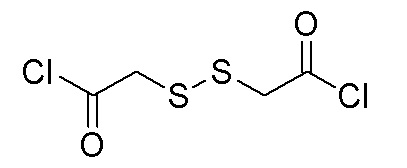
77 мг (0,42 ммоль, 1,0 экв) 2,2-дитиодиуксусной кислоты растворяли в 10 мл дихлорметана и добавляли 150 мг (1,27 ммоль, 3,0 экв) тионилхлорида при комнатной температуре, и затем добавляли 1 каплю N, N-диметилформамида. Перемешивали при комнатной температуре в течение 30 минут. После завершения реакции, что определялось методом тонкослойной хроматографии (TLC), реакционную систему концентрировали досуха и непосредственно использовали на следующей стадии реакции (93 мг, выход=100%).
Стадия B. N, N'-((((((((2,2'-дисульфанилдиилбис(ацетил))-бис(мочевинадиил))бис(пентил-5,1-диил)) бис(окси))бис(6-метоксихинолин-7,4-диил))бис(окси))бис(3-фтор-4,1-фенил))бис-(N-(4-фторфенил)циклопропан-1,1-диформамид)
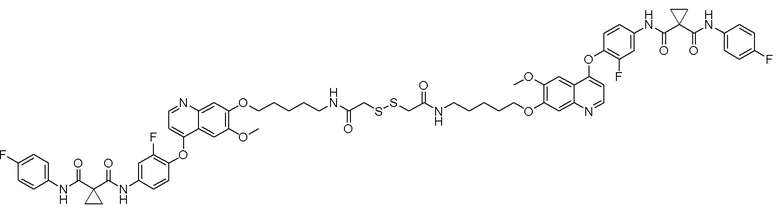
При 0°С, к N-(4-((7-((5-аминопентил)окси)-6-метокси-хинолин-4-ил)окси)-3-фторфенил)-N-(4-фторфенил)циклопропан-1,1-диформамиду (100 мг, 0,17 ммоль) (синтезированному в соответствии со стадией B, описанной в примере 57) в DCM последовательно добавляли Et3N (70 мкл, 0,51 ммоль), 2,2'-тиодиацетилхлорид (77 мг, 1,0 ммоль). Реакционный раствор подогревали до комнатной температуры и проводили реакцию в течение 30 минут, и затем добавляли воду для остановки реакции. Водную фазу экстрагировали один раз с помощью DCM, и объединенную органическую фазу промывали один раз насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали путем дистилляции при пониженном давлении и очищали колоночной хроматографией на силикагеле с получением продукта (32 мг, 18%).
LC-MS: m/z=1327[M+H] +.
Оценка биологической активности
1. Ингибирующее действие соединений в отношении HDAC6
Для обнаружения ингибирующего действия соединений на активность HDAC6 использовали метод флуоресцентного анализа HDAC6. Конкретная методика анализа выглядит следующим образом.
Готовят 10 мМ исходного раствора соединения в DMSO. Берут 10 мкл исходного раствора и разбавляют с помощью 90 мкл DMSO с получением рабочий раствор с содержанием 1 мМ. Последовательно разбавляют в три раза рабочий раствор соединения с получением растворов с 11 концентрациями, включая раствор DMSO для отрицательного контроля. Добавляют 3 мкл каждой концентрации соединения к 197 мкл реакционного буфера (20 мМ Hepes pH 8,0, 137 мМ NaCl, 2,7 мМ KCl, 1 мМ MgCl2, 0,05% BSA, 0,5 мМ TCEP), хорошо перемешивают, и добавляют 10 мкл в 384-луночный планшет. В конечной реакционной системе, концентрации соединения составляли от 10 мкМ до 0,51 нМ. Готовят вторые экземпляры растворов, и добавляют 10 мкл раствора 3XHDAC (BPS, Cat.50006, 0,3 нМ) в каждую лунку, затем инкубируют при 23°C в течение 20 минут. Затем добавляют 3-кратное количество раствора субстрата (Anaspec, Cat.61855, 15 мкМ), центрифугируют для перемешивания и инкубируют при 23°C в течение 90 минут. Добавляют 30 мкл смеси трипсин/SAHA (20 мМ Hepes pH 8,0, 100 мМ NaCl, 10 мМ SAHA, 0,01 мг/мл трипсина), инкубируют при 23°C в течение 60 минут для прерывания реакции. И наконец, считывают данные флуоресценции с помощью планшет-ридера Envision (возбуждение при длине волны 390 нм, испускание при длине волны 460 нм). Высокое значение при 430 нм указывает на высокую активность киназы, в то время как низкое значение при 430 нм указывает на то, что активность киназы была ингибирована. И наконец, анализируют данные с помощью программного обеспечения XLfit5 и рассчитывают для соединения величину IC50. Вориностат (SAHA) использовали в качестве соединения для положительного контроля. Результаты представлены в таблице 1.
2. Ингибирующее действие соединений в отношении VEGFR2
Проводили исследование воздействия соединений на VEGFR2 (KDR) методом анализа по сдвигу пятна. Начальная испытуемая концентрация соединения составляла 1000 нМ, использовали трехкратное разведение, получали растворы с 10 концентрациями и проводили эксперимент в двух параллельных испытаниях. Нинтеданиб (Selleckchem, Cat. S1010) использовали в качестве соединения для положительного контроля. Методика эксперимента может быть кратко описана следующим образом: киназу VEGFR2 (Carna, Cat.08-191) при конечной концентрации 0,5 нМ и испытуемое соединение смешивали в лунках планшета Optiplate-384F и инкубировали в течение 10 минут при комнатной температуре. Затем добавляли АТФ при конечной концентрации 95 мкМ и 3 мкМ киназного субстрата 22 (Gill Biochemical (glbiochem), Cat.112393). После смешивания, проводили реакцию при комнатной температуре в течение 30 минут. Добавляли раствор для обнаружения остановки реакции и определяли степень преобразования с помощью Caliper EZ Reader II.
Анализ данных:
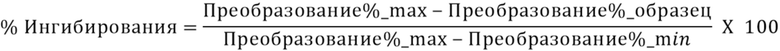
где, Преобразование%_образец относится к скорости преобразования считывания образца; Преобразование%_min: средняя величина лунок с отрицательным контролем, которая представляет собой скорость преобразования считывания лунок без ферментной активности; Преобразование %_max: средняя величина лунок с положительным контролем, представляет собой скорость преобразования считывания лунок без ингибирования с помощью соединений.
Аппроксимация кривой доза-ответ: откладывается логарифм концентрации по оси X и процент ингибирования по оси Y. Кривую доза-ответ аппроксимировали с помощью программы для анализа GraphPad Prism 5 с переменным углом наклона log (ингибитор) - ответ с получением величины ингибирующей активности (IC50) для каждого соединения. Результаты представлены в таблице 2.
3. Исследование фармакокинетики
Самок крыс линии SD подразделяли на группы, по 3 крысы в каждой группе, и вводили им внутрижелудочно соединения примеров 10, 12, 24, 57, 61, 89, 90, 93, 94, 96 (10 мг/кг). Соблюдали режим голодания в течение ночи перед введением соединения, и период голодания составлял от 10 часов до введения и до 4 часов после введения соединения. Кровь собирали через 0,25, 0,5, 1, 2, 4, 8 и 24 часа после введения соединения. Для анестезии изофлураном использовали аппарат для анестезии мелких животных. Затем 0,3 мл цельной крови собирали из нижней части венозного сплетения и помещали в пробирку с противосвертывающим средством гепарином. Образец центрифугировали при 4°C, 4000 об/мин в течение 5 минут, плазму переносили в центрифужную пробирку и хранили при -80°C до момента проведения анализа. Образец плазмы экстрагировали методом осаждения белка, а экстракт обрабатывали и анализировали методом жидкостной хроматографии с тандемной масс-спектрометрией (LC/MS/MS). Результаты исследования фармакокинетики представлены в таблице 3.
Несмотря на то, что для иллюстрации настоящего изобретения были раскрыты предпочтительные варианты осуществления, тем не менее, для специалистов в данной области техники является очевидным, что могут быть сделаны различные модификации, дополнения и замены в пределах сущности и объема настоящего изобретения, определяемых формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2681849C2 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ с-MET | 2013 |
|
RU2619130C2 |
| ПРОИЗВОДНОЕ АЦИЛТИОМОЧЕВИНЫ ИЛИ ЕГО СОЛЬ, И ЕГО ПРИМЕНЕНИЕ | 2009 |
|
RU2503664C2 |
| ДЕСТРУКТОРЫ БЕЛКА MDM2 | 2017 |
|
RU2743432C2 |
| НОВОЕ ПРОИЗВОДНОЕ ПИРИДИНА И ПРОИЗВОДНОЕ ПИРИМИДИНА (3) | 2006 |
|
RU2362771C1 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОАРИЛОМ ПИРИДИНЫ И СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2756743C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| ФАРМАЦЕВТИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ Axl | 2011 |
|
RU2573834C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| БИАРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛОК-БЕЛКОВОГО ВЗАИМОДЕЙСТВИЯ YAP/TAZ-TEAD | 2021 |
|
RU2830596C1 |
Группа изобретений относится к фармацевтической химии и включает соединения формулы (I) или (II), конкретные соединения, указанные в формуле изобретения, или их фармацевтически приемлемые соли, стереоизомеры, их применение и способ лечения и ингибирования на их основе. Где в формулах (I) или (II) A представляет собой 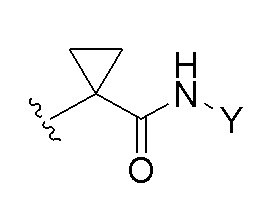 или -NR3R4; X представляет собой водород или галоген; Y представляет собой C3-8 циклоалкил или C6-20 арил, замещенный галогеном; Z представляет собой -O- или -S-; L представляет собой линейный -(CH2)n-, n представляет собой целое число от 3 до 10, где, необязательно, один или более из -CH2- заменены на один или более -NR5-, -(CO)- и -CR5R6-, и/или, необязательно, один или более из -CH2CH2- могут быть заменены на -CH=CH-; R1 представляет собой C1-6 алкокси или -(CO)NR7R8; R2 представляет собой водород, C1-8 алкилсульфанил или -(CO)R9; R3 и R4 независимо представляют собой водород или C3-8 циклоалкил; R5 и R6 независимо представляют собой водород, C1-8 алкил или карбоксил; R7 и R8 независимо представляют собой водород; R9 представляет собой C1-4 алкил, необязательно замещенный одной или несколькими группами, выбранными амино, гидроксила, NHC(O)C1-4алкила; фенил, C1-4алкиленC3-12гетероарил, где гетероарил содержит один атом N и необязательно замещен C1-4алкилом, и C1-4алкилен замещен группой, выбранной из амино и NHC(O)C1-4алкила. Технический результат – производные хинолинила в качестве ингибиторов VEGFR-2 и/или HDAC6. 6 н. и 10 з.п. ф-лы, 3 табл., 98 пр.
или -NR3R4; X представляет собой водород или галоген; Y представляет собой C3-8 циклоалкил или C6-20 арил, замещенный галогеном; Z представляет собой -O- или -S-; L представляет собой линейный -(CH2)n-, n представляет собой целое число от 3 до 10, где, необязательно, один или более из -CH2- заменены на один или более -NR5-, -(CO)- и -CR5R6-, и/или, необязательно, один или более из -CH2CH2- могут быть заменены на -CH=CH-; R1 представляет собой C1-6 алкокси или -(CO)NR7R8; R2 представляет собой водород, C1-8 алкилсульфанил или -(CO)R9; R3 и R4 независимо представляют собой водород или C3-8 циклоалкил; R5 и R6 независимо представляют собой водород, C1-8 алкил или карбоксил; R7 и R8 независимо представляют собой водород; R9 представляет собой C1-4 алкил, необязательно замещенный одной или несколькими группами, выбранными амино, гидроксила, NHC(O)C1-4алкила; фенил, C1-4алкиленC3-12гетероарил, где гетероарил содержит один атом N и необязательно замещен C1-4алкилом, и C1-4алкилен замещен группой, выбранной из амино и NHC(O)C1-4алкила. Технический результат – производные хинолинила в качестве ингибиторов VEGFR-2 и/или HDAC6. 6 н. и 10 з.п. ф-лы, 3 табл., 98 пр.
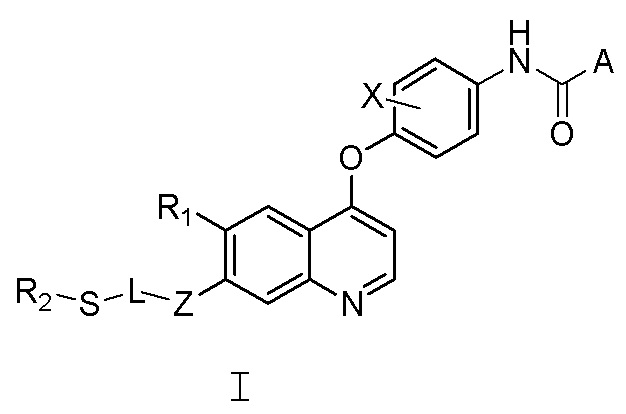
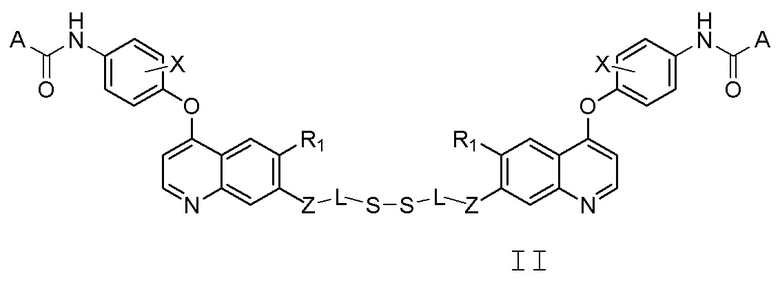
1. Хинолинил-содержащее соединение, представленное общей формулой (I) или (II), или его фармацевтически приемлемая соль или стереоизомер,
где A представляет собой  или -NR3R4;
или -NR3R4;
X представляет собой водород или галоген;
Y представляет собой C3-8 циклоалкил или C6-20 арил, замещенный галогеном;
Z представляет собой -O- или -S-;
L представляет собой линейный -(CH2)n-, n представляет собой целое число от 3 до 10, где, необязательно, один или более из -CH2- заменены на один или более -NR5-, -(CO)- и -CR5R6-, и/или, необязательно, один или более из -CH2CH2- могут быть заменены на -CH=CH-;
R1 представляет собой C1-6 алкокси или -(CO)NR7R8;
R2 представляет собой водород, C1-8 алкилсульфанил или -(CO)R9;
R3 и R4 независимо представляют собой водород или C3-8 циклоалкил;
R5 и R6 независимо представляют собой водород, C1-8 алкил или карбоксил;
R7 и R8 независимо представляют собой водород;
R9 представляет собой C1-4 алкил, необязательно замещенный одной или несколькими группами, выбранными амино, гидроксила, NHC(O)C1-4алкила; фенил, C1-4алкиленC3-12гетероарил, где гетероарил содержит один атом N и необязательно замещен C1-4алкилом и C1-4алкилен замещен группой, выбранной из амино и NHC(O)C1-4алкила.
2. Соединение по п. 1, где общая формула (I) или (II) представлена формулами (I-1), (I-2), (II-1) или (II-2):
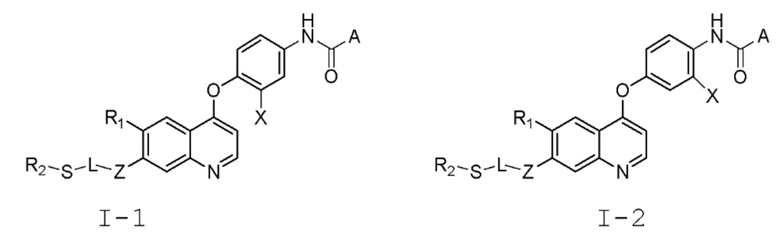
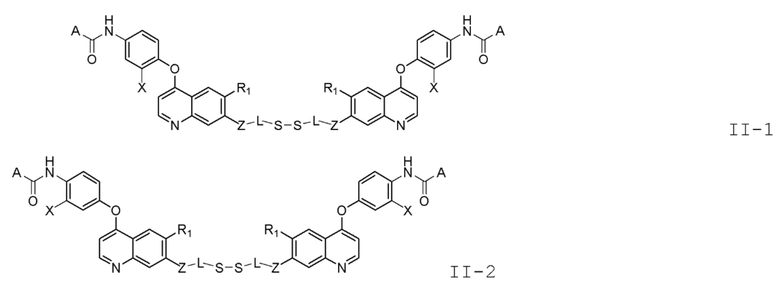
где X представляет собой водород, F или Cl; или его фармацевтически приемлемая соль или стереоизомер.
3. Соединение по п. 1 или 2, где A представляет собой следующую группу:
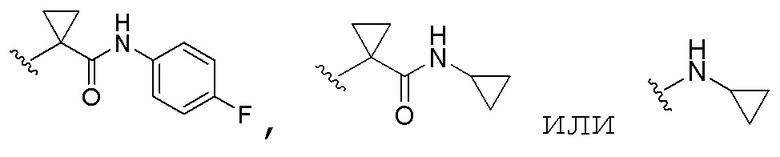 ; или его фармацевтически приемлемая соль или стереоизомер.
; или его фармацевтически приемлемая соль или стереоизомер.
4. Соединение по любому одному из пп. 1-3, где L представляет собой одну из следующих групп: -CH2-CO-NH-(CH2)p-*, p представляет собой целое число от 3 до 6, и один или более из -CH2CH2- могут быть необязательно заменены на -CH=CH-, и * обозначает конец, соединенный с группой Z; или
линейный -(CH2)o-, где o представляет собой целое число от 5 до 7 и один или более из -CH2CH2- могут быть необязательно заменены на -CH=CH-; или его фармацевтически приемлемая соль или стереоизомер.
5. Соединение по любому одному из пп. 1-4, где L представляет собой одну из следующих групп:
*-CH2CH=CH(CH2)q-, *-(CH2)2CH=CH(CH2)q- или *-(CH2)3CH=CH(CH2)q-, q представляет собой целое число от 1 до 4, где * обозначает конец, присоединенный к группе Z; или его фармацевтически приемлемая соль или стереоизомер.
6. Соединение по любому одному из пп. 1-5, где R1 выбирают из C1-4 алкоксила или -(CO)NH2; или его фармацевтически приемлемая соль или стереоизомер.
7. Соединение по любому одному из пп. 1-6, где R2 представляет собой водород, C1-8 алкилсульфанил или -(CO)R9;
где R9 представляет собой C1-4 алкил, необязательно замещенный одной или несколькими группами, выбранными амино, гидроксила, NHC(O)C1-4алкила; фенил, C1-4алкиленC3-12гетероарил, где гетероарил содержит один атом N и необязательно замещен C1-4алкилом и C1-4алкилен замещен группой, выбранной из амино и NHC(O)C1-4алкила.
8. Соединение по п. 7, где гетероарил содержит один атом N; или его фармацевтически приемлемая соль или стереоизомер.
9. Соединение, которое выбирают из следующих соединений:
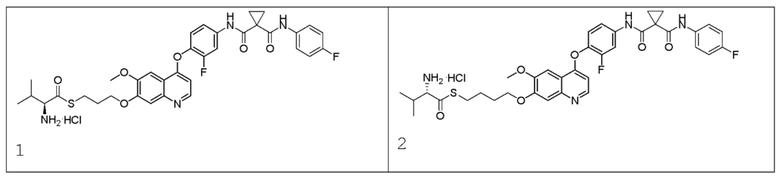
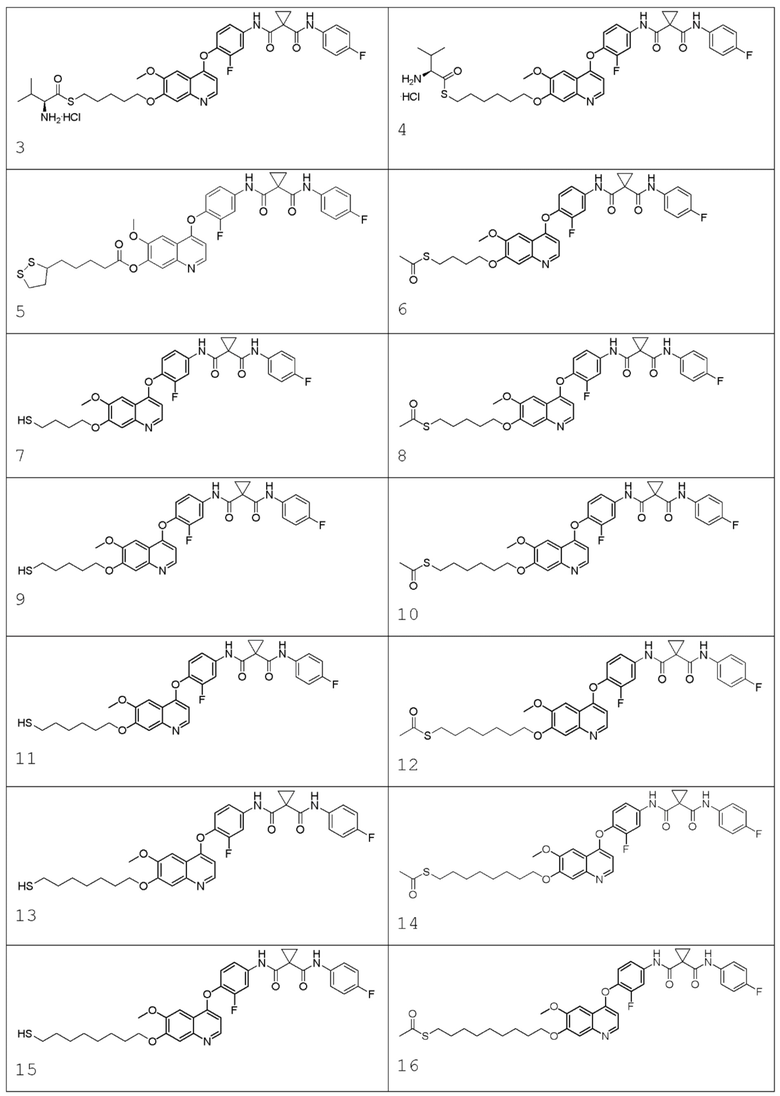
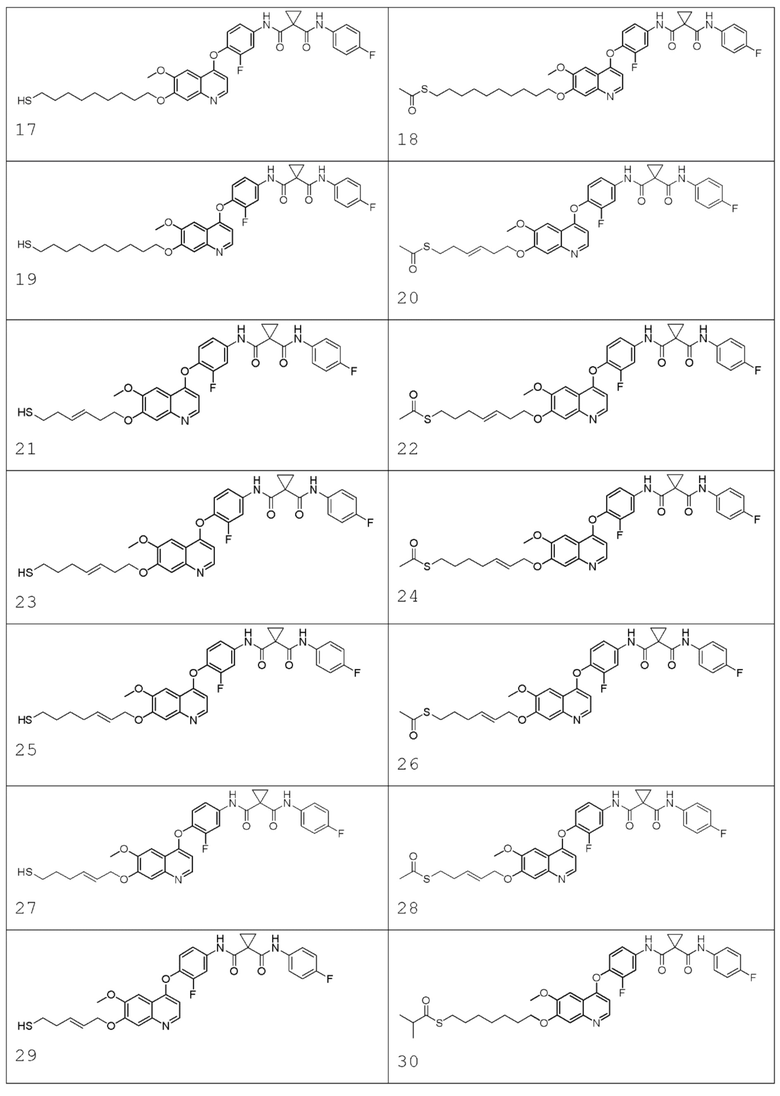
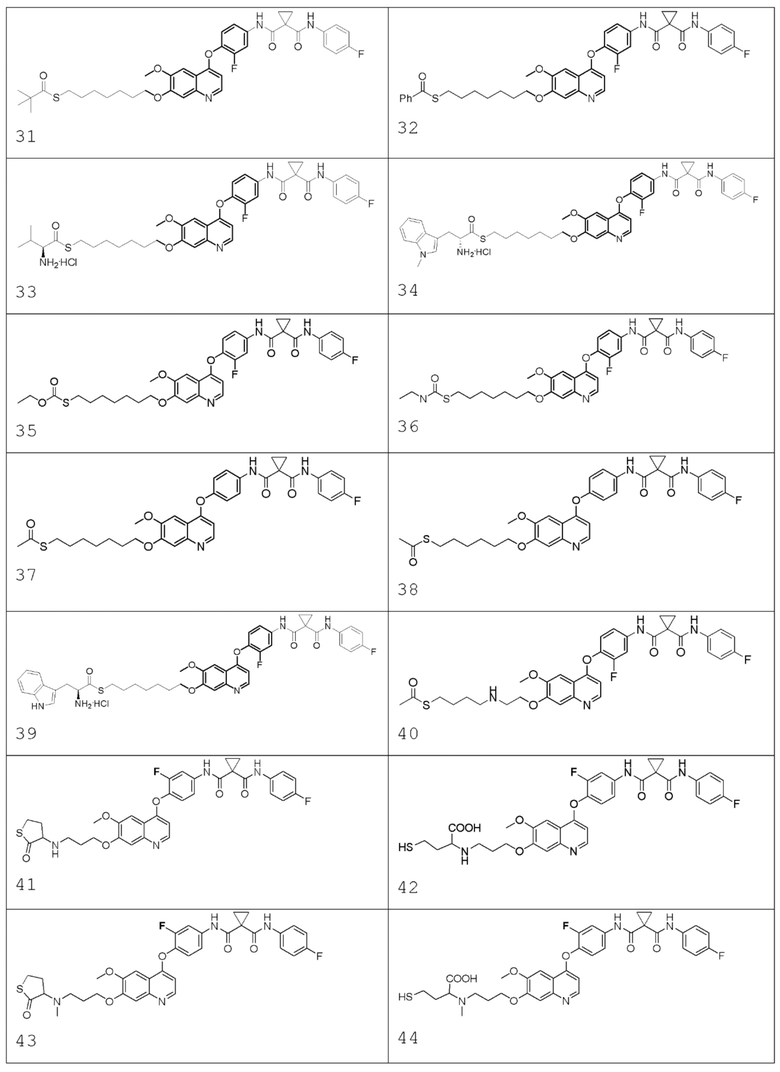
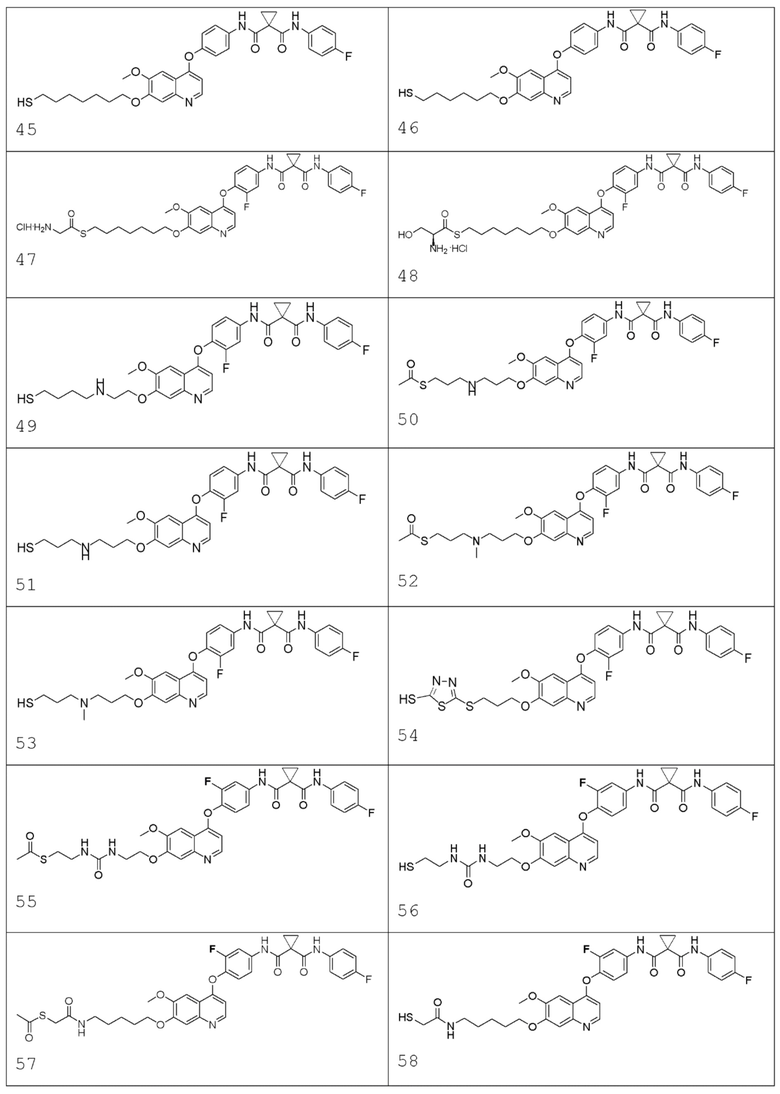
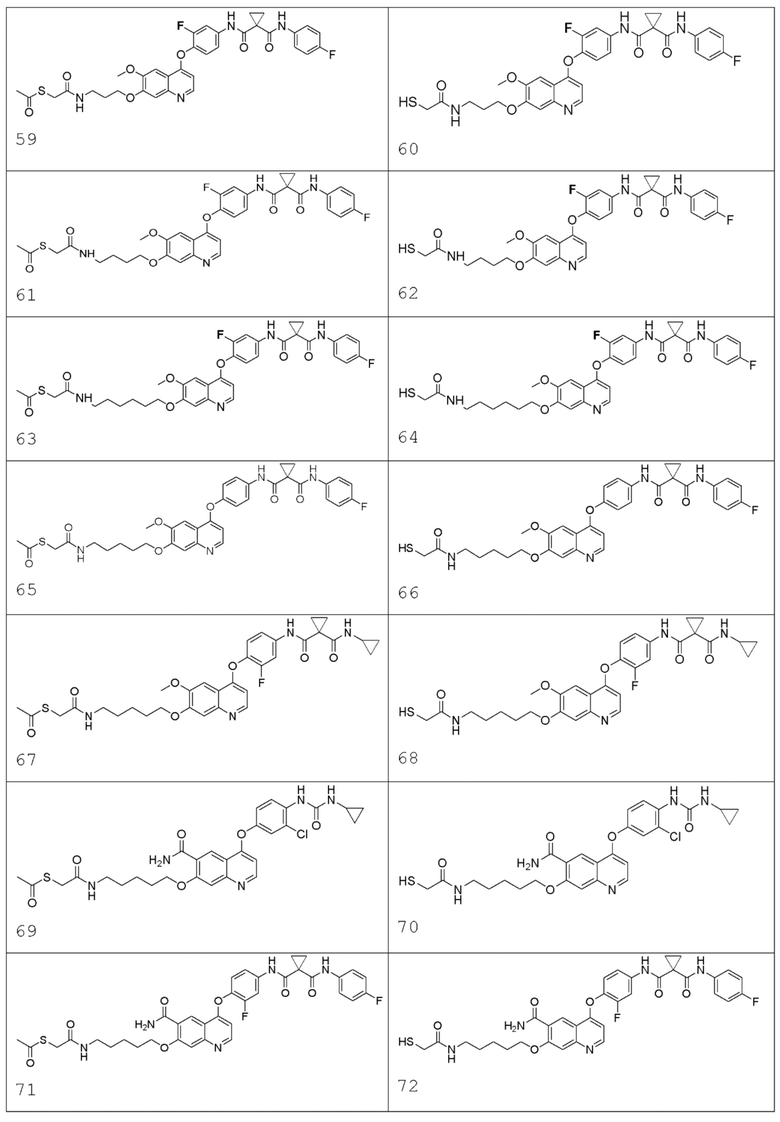
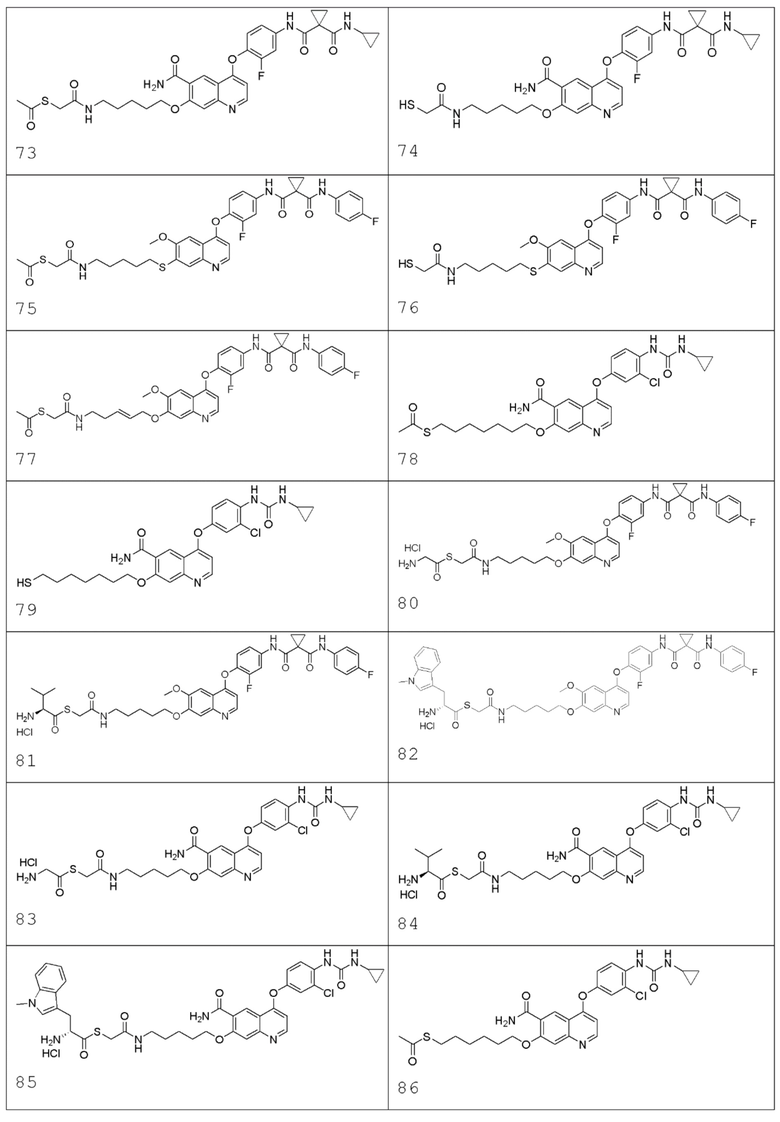
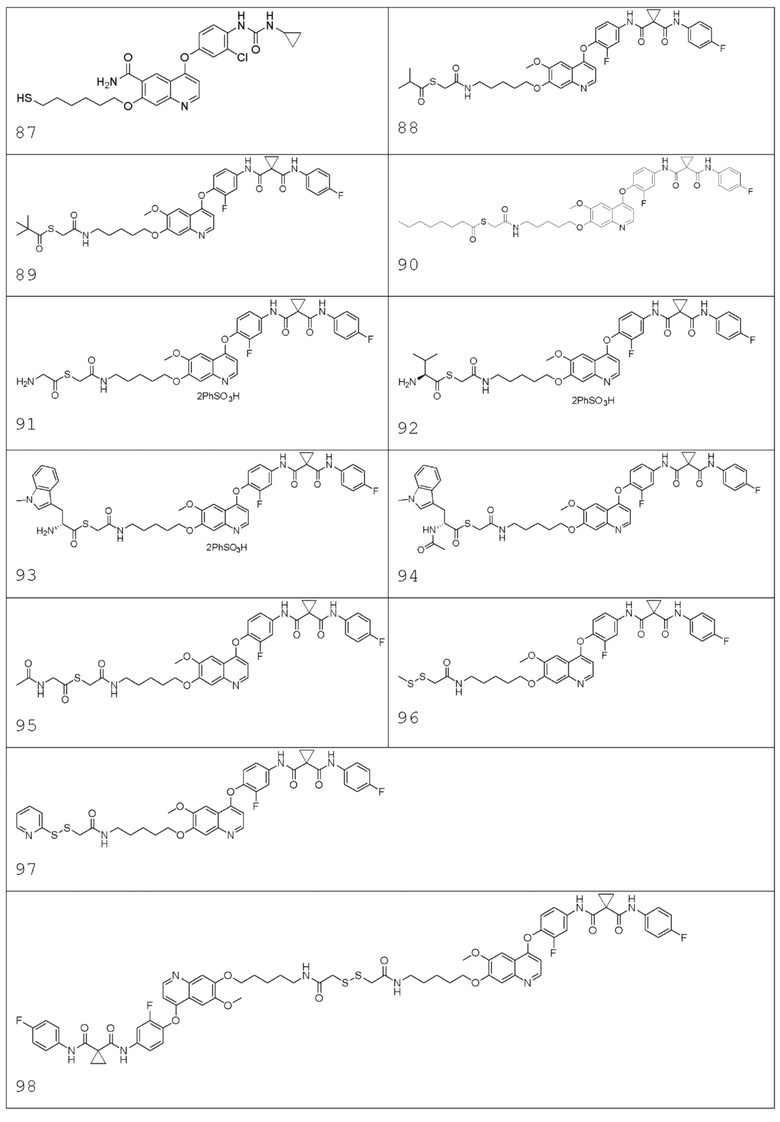
или его фармацевтически приемлемая соль или стереоизомер.
10. Применение соединения или его фармацевтически приемлемой соли или стереоизомера по любому одному из пп. 1-9 для получения лекарственного средства для лечения заболеваний, связанных с VEGFR-2 и/или HDAC6.
11. Применение по п. 10, где заболевания включают псориаз, цирроз печени, диабет, нейродегенеративные заболевания, опухоли, иммунопатологические заболевания и сердечно-сосудистые заболевания.
12. Применение соединения или его фармацевтически приемлемой соли или стереоизомера по любому одному из пп. 1-9 для получения ингибитора VEGFR-2 и/или ингибитора HDAC6.
13. Применение по любому одному из пп. 10-12, где гистондеацетилаза представляет собой HDAC6.
14. Способ лечения заболеваний, связанных с VEGFR-2 и/или HDAC6, включающий стадию введения соединения или его фармацевтически приемлемой соли или стереоизомера по любому из пп. 1-9 субъекту, нуждающемуся в таком лечении.
15. Способ по п. 14, где заболевания включают псориаз, цирроз печени, диабет, нейродегенеративные заболевания, опухоли, иммунопатологические заболевания и сердечно-сосудистые заболевания.
16. Способ ингибирования тирозинкиназы и/или гистондеацетилазы, включающий контактирование VEGFR-2 и/или HDAC6 с эффективным количеством соединения или его фармацевтически приемлемой соли или стереоизомера по любому из пп. 1-9.
| ХИНОЛИЛ-СОДЕРЖАЩЕЕ СОЕДИНЕНИЕ ГИДРОКСАМОВОЙ КИСЛОТЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, А ТАКЖЕ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ АНОМАЛЬНОЙ АКТИВНОСТЬЮ ПРОТЕИНКИНАЗЫ И/ИЛИ ГИСТОНДЕАЦЕТИЛАЗЫ | 2011 |
|
RU2573633C2 |
| Зефирова О.Н | |||
| и др., Об истории возникновения и развития концепции биоизостеризма | |||
| Вестник Московского университета, Серия 2 | |||
| Химия, 2002, т | |||
| Зубчатое колесо со сменным зубчатым ободом | 1922 |
|
SU43A1 |
| Способ модулирования для радиотелефона | 1921 |
|
SU251A1 |
| Станок для придания концам круглых радиаторных трубок шестигранного сечения | 1924 |
|
SU2019A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |
| CN 105131082 A, 09.12.2015 | |||
| Heider U | |||
| et al., Histone deacetylase | |||
