Область техники, к которой относится изобретение
Изобретение относится к конъюгатам, содержащим аматоксины и антитела, в частности аматоксины, связанные с антителами, содержащими специфические цистеиновые остатки.
Уровень техники
Аматоксины являются циклическими пептидами, состоящими из 8 аминокислот, содержащихся в грибах Amanita phalloides (см. фиг. 1). Аматоксины специфически ингибируют ДНК-зависимую РНК-полимеразу II клеток млекопитающих и за счет этого также транскрипцию и биосинтез белка пораженных клеток. Ингибирование транскрипции в клетке приводит к остановке роста и пролиферации. Несмотря на то, что данная связь не является ковалентной, комплекс между аманитином и РНК-полимеразой II очень прочный (KD=3 нМ). Диссоциация аманитина из фермента (энзима) - очень медленный процесс, вследствие чего маловероятно восстановление поврежденной клетки. Когда ингибирование транскрипции продолжается слишком долго, в клетке произойдет запрограммированная гибель клеток (апоптоз).
Использование аматоксинов в качестве цитотоксических фрагментов для терапии опухолей уже исследовалось в 1981 году путем связывания антитела анти-Thy 1.2 с α-аманитином при помощи линкера, присоединенного к индольному кольцу Trp (аминокислота 4, см. фиг. 1) посредством диазотирования (Davis & Preston, Science 1981, 213, 1385-1388). У Davis & Preston сайт присоединения определен в положении 7'. У Morris & Venton также показано, что замещение в положении 7' приводит к образованию производного, которое поддерживает цитотоксическую активность (Morris & Venton, Int. J. Peptide Protein Res., 1983, 21 419-430).
В заявке на патент ЕР 1859811 А1 (опубликованной 28 ноября 2007 года) описаны конъюгаты, в которых γ-атом углерода аминокислоты 1 аматоксина β-аманитина связывается напрямую, т.е. без линкерной структуры, с альбумином или моноклональным антителом НЕА125, ОКТ3 или РА-1. Кроме того, было показано ингибирующее воздействие данных конъюгатов на пролиферацию клеток рака молочной железы (MCF-7), клеток лимфомы Беркитта (Raji) и клеток Т-лимфомы (Jurkat). Было предложено использование линкеров, включая линкеры, содержащие такие элементы, как амид, сложный эфир, простой эфир, тиоэфир, дисульфид, мочевину, тиомочевину, углеводородные фрагменты и аналогичные соединения, но такие конструкции фактически не были показаны и не были предоставлены более подробные сведения, например, о сайтах присоединения на аматоксинах.
В патентных заявках WO 2010/115629 и WO 2010/115630 (обе опубликованы 14 октября 2010 года) описаны конъюгаты, в которых антитела, такие как антитела анти-ЕрСАМ, например, гуманизированное антитело huHEA125, связаны с аматоксинами посредством (i) γ-атома углерода аминокислоты 1 амотоксина, (ii) 6' атома углерода аминокислоты 4 аматоксина или (iii) посредством δ-атома углерода аминокислоты 3 аматоксина, в каждом случае либо напрямую, либо через линкер между антителом и аматоксинами. Предложенные линкеры содержат такие элементы, как амид, сложный эфир, простой эфир, тиоэфир, дисульфид, мочевину, тиомочевину, углеводородные фрагменты и аналогичные соединения. Кроме того, было показано ингибирующее воздействие данных конъюгатов на пролиферацию клеток рака молочной железы (клеточная линия MCF-7), клеток карциномы поджелудочной железы (клеточная линия Capan-1), клеток рака толстой кишки (клеточная линия Colo205) и клеток холангиокарциномы (клеточная линия OZ).
Недостаток подходов, указанных выше, состоит в том, что связь с лизиновыми остатками в белковых доменах связывания с мишенью, например, антителами, является неспецифической и приводит к образованию конъюгатов смешанного состава с варьирующимися соотношениями содержания лекарственного средства к антителу (DAR), которые невозможно удовлетворительным образом контролировать. Например, в типичном антителе человека IgG1 находится от 70 до 100 лизиновых аминокислотных остатков. Обычно DAR, равный приблизительно 4, получают посредством реакции между соответствующим образом активированными конструкциями аматоксина и лизиновыми остатками. Кроме того, наблюдается очень неоднородная смесь позиций сочетания, при этом некоторые приводят к образованию конъюгатов с более высокой эффективностью и некоторые - с гораздо меньшей эффективностью. В данном случае не может быть обеспечена какая-либо определенная степень контроля.
Аналогичным образом, использование цистеинов, полученных посредством восстановления дисульфидных связей в молекулах антител, с последующим связыванием с токсинами, содержащими реагирующую с тиолом группу (тиол-реагирующую), приводит к образованию неоднородных смесей, так как в человеческом IgG1 присутствует 32 цистеина, и восемь из них доступны для связывания после восстановления четырех межцепочечных дисульфидных связей.
Можно заметить, что цитотоксическая активность и, следовательно, терапевтическая эффективность конъюгатов токсин-антитело увеличиваются в зависимости от получаемого DAR. Тем не менее, при увеличении DAR уменьшается переносимость. Таким образом, для оптимизации профиля конъюгата токсин-антитело крайне желательно контролировать как количество токсинов, связываемых с антителом (т.е. DAR), так и конкретное место (места) конъюгации токсина (токсинов). Только при таких обстоятельствах может быть обеспечена точная настройка доступного терапевтического окна и воспроизводимость результатов.
В ответ на эту ситуацию был разработан ряд способов специфического и контролируемого образования конъюгатов лекарственное средство-антитело, включая, например, сайт-специфическую конъюгацию с неприродными аминокислотами, которые были введены в последовательности антител дикого типа (см. Axup et al., Proc. Natl. Acad. Sci. U.S.A. 109 (2012) 16101).
В альтернативном подходе используют конструкции антител, содержащие одиночные цистеиновые остатки, полученные посредством мутагенеза последовательностей антител дикого типа. В оптимальном случае DAR, равное 2, может быть получено посредством мутагенеза одного аминокислотного остатка в легкой или тяжелой цепи IgG, имеющего две копии такой мутированной цепи, и DAR, равное 1, может быть получено посредством мутагенеза одного аминокислотного остатка в легкой или тяжелой цепи фрагмента моновалентного антитела, такого как фрагмент Fab, имеющий одну копию любой такой мутированной цепи.
В WO 2006/034488 описаны антитела, сконструированные посредством замены одной или нескольких аминокислот исходного антитела на несшитые, высоко реакционноспособные цистеиновые аминокислоты. В заявке описаны различные позиции как в легкой, так и в тяжелой цепи, в которых может происходить такое замещение.
В еще одной заявке WO 2011/005481 описано значительное количество позиций, которые могут быть использованы для замещения аминокислотных остатков дикого типа в последовательностях антител на реакционноспособные аминокислотные остатки, в частности на цистеиновые остатки.
В WO 2008/070593 описан аналогичный подход, в котором произведена замена аминокислотных остатков в части Fc на антителе, которые участвуют в связывании с Fc гамма-рецептором, включая замены цистеиновых остатков.
Несмотря на тот факт, что в этой области уже проделана большая работа, не было еще осуществлено связывание аматоксинов определенным образом путем управления соотношением DAR, например использованием специфических цистеинов для такого определенного связывания. Кроме того, представляется, что не выработано полное понимание того, какие позиции аминокислот следует использовать для таких конъюгаций на основе цистеина. В частности, до сих пор отсутствует информация о конъюгатах аматоксинов и антител, полученных посредством использования сконструированных цистеиновых остатков. Тем не менее, в свете высокой токсичности аматоксинов чрезвычайно важно выявить конструкции, которые проявляют высокую токсичность в отношении интересующей клетки-мишени, при этом показывая отличную стабильность и переносимость. До настоящего времени данная цель еще не достигнута.
Цель изобретения
Таким образом, по-прежнему необходим экономичный и надежный способ синтеза конъюгатов, содержащих аматоксины и антитела, в частности аматоксины, связанные с антителами, содержащими специфические цистеиновые остатки, при этом такие конъюгаты являются высокотоксичными по отношению к клеткам-мишеням и одновременно стабильными и хорошо переносимыми. Кроме того, задачей настоящего изобретения является выявление позиций в цепях антител, которые могут быть мутированы из исходного аминокислотного остатка в цистеиновый остаток, что приведет к образованию таких высокотоксичных, стабильных и хорошо переносимых конъюгатов.
Раскрытие изобретения
Настоящее изобретение разработано на основании неожиданно установленного факта, что можно выявить ограниченное число специфических аминокислотных остатков дикого типа, которые могут быть мутированы на отдельные непарные цистеиновые остатки, что приводит к образованию высокотоксичных, стабильных и хорошо переносимых конъюгатов с аматоксинами.
Таким образом, в одном аспекте настоящее изобретение относится к конъюгату общей формулы:
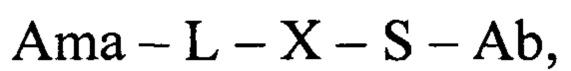
где Ama - аматоксин, L - линкер, X - фрагмент, полученный посредством связывания тиольной группы с тиол- реагирующей группой, S - атом серы аминокислотного остатка цистеина, и Ab - последовательность антитела или функциональный фрагмент антитела, содержащие указанный цистеиновый остаток, при этом указанный цистеиновый остаток (i) расположен в домене антитела, выбранном из CL, CH1, CH2 и СН3; (ii) расположен в позиции, в которой последовательность зародышевой линии, имеющая гомологию, наиболее близкую к последовательности указанного домена антитела, содержит аминокислотный остаток, отличный от цистеина; и (iii) расположен в позиции, которая подвергается воздействию растворителя.
В еще одном аспекте настоящее изобретение относится к конъюгату общей формулы:
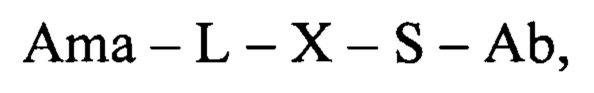
где Ama - аматоксин, L - линкер, X - фрагмент, полученный посредством связывания тиольной группы с тиол-реагирующей группой, S - атом серы аминокислотного остатка цистеина, и Ab - последовательность антитела или функциональный фрагмент антитела, содержащие указанный цистеиновый остаток, где указанный цистеиновый остаток выбран из перечня 118Cys с тяжелой цепью, 239Cys с тяжелой цепью и 265Cys с тяжелой цепью, особенно 118Cys с тяжелой цепью и 265Cys с тяжелой цепью.
В третьем аспекте настоящее изобретение относится к способу синтеза конъюгата общей формулы:
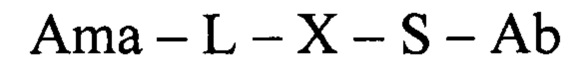
реакцией соединения Ama - L - X', где X' - тиол-реагирующая группа, с антителом Ab - SH, где группа -SH - тиол аминокислотного остатка цистеина, и Ab - последовательность антитела, содержащая указанный цистеиновый остаток, при этом указанный цистеиновый остаток выбран из перечня: 118Cys с тяжелой цепью, 239Cys с тяжелой цепью и 265Cys с тяжелой цепью, особенно 118Cys с тяжелой цепью и 265Cys с тяжелой цепью.
В четвертом аспекте настоящее изобретение относится к набору, содержащему (i) соединение Ama - L - X', где X' - тиол-реагирующая группа, и (ii) антитело Ab - SH, где группа -SH - тиол аминокислотного остатка цистеина, и Ab - последовательность антитела, содержащая указанный цистеиновый остаток, при этом указанный цистеиновый остаток выбран из перечня: 118Cys с тяжелой цепью, 239Cys с тяжелой цепью и 265Cys с тяжелой цепью, особенно 118Cys с тяжелой цепью и 265Cys с тяжелой цепью.
В пятом аспекте настоящее изобретение относится к способу синтеза соединения Ama - L - X', где X' - малеимидная группа, включающему в себя этап (а) реакции аматоксина, содержащего нуклеофильную группу, с соединением Y-L-X'', в которой
Y - уходящая группа, и
X'' - защищенная малеимидная группа.
В шестом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей конъюгат по настоящему изобретению.
В седьмом аспекте настоящее изобретение относится к способу лечения заболевания, ассоциированного с клетками, являющимися мишенью, включающему этап контактирования указанных клеток с конъюгатом по настоящему изобретению, в котором указанное антитело является специфичным для указанной мишени.
Краткое описание чертежей
На фиг. 1 показаны структурные формулы различных аматоксинов. Цифры, выделенные жирным шрифтом (от 1 до 8), обозначают стандартную нумерацию восьми аминокислот, образующих аматоксин. Также показаны стандартные обозначения атомов в аминокислотах 1, 3 и 4 (греческие буквы от α до γ, греческие буквы от α до δ и цифры от 1' до 7', соответственно).
На фиг. 2 показан схематический вид молекулы IgG1 и позиции аминокислотных остатков, мутированных до цистеиновых остатков и для связывания с токсинами. НС-А118С; HC-S131C; HC-S239C; HC-D265C; НС-Е269С; HC-N297C; НС-А327С; HC-I332C; остатки HC-D265 и HC-N297 участвуют в связывании Fc γ-рецептора; замещение помешает связыванию рецептора и последующему поглощению в рецептор-положительных клетках.
На фиг. 3 показан схематический вид векторов экспрессии для образования тяжелых и легких цепей трастузумаба в соответствии с примером 1.
На фиг. 4(A) показаны развернутые масс-спектры антитела трастузумаб НС-А118С и его конъюгата: (а) развернутый масс-спектр антитела трастузумаб НС-А118С; (b) развернутый масс-спектр конъюгата лекарственное средство-антитело (ADC) трастузумаб НС-А118С-30.0880; на фиг. 4(B) показаны развернутые масс-спектры антитела трастузумаб НС-A118C - легкой цепи (а) и тяжелой цепи (b); на фиг. 4(C) показаны развернутые масс-спектры легкой цепи (а) и тяжелой цепи (b) ADC трастузумаб НС-А118С-30.0880.
На фиг. 5(A) показан масс-спектр интактной Her-30.0643, определенный посредством жидкостной хроматографии с масс-спектрометрией (LC/MS): соотношение лекарственное средство-антитело = 3,7 аматоксинов на IgG; на фиг. 5(B) показаны масс-спектры легкой цепи Her-30.0643, определенные посредством жидкостной хроматографии с масс-спектрометрией (LC/MS); на фиг. 5(C) показаны масс-спектры тяжелой цепи Her-30.0643, определенные посредством жидкостной хроматографии с масс-спектрометрией (LC/MS).
На фиг. 6 показано связывание четырех примеров мутантов цистеина с антителом-мишенью на клетках SKOV-3 по сравнению с исходным антителом (два разных продукта экспрессии).
На фиг. 7(A) показан анализ различных конструкций на мембране для блоттинга, окрашенных антиаманитиновым антителом: дорожки 1 и 3: голые антитела трастузумаб; дорожки 2 и 4: антитело трастузумаб, конъюгированное с конструкцией аманитина Her-30.0643 через лизиновое связывание; дорожка 5: голое антитело трастузумаб с мутацией НС-А118С; дорожка 6: антитело трастузумаб с мутацией НС-А118С, конъюгированное с конструкцией аманитина Her-30.1619 через цистеиновое связывание с 118 С (бромацетамидовая связь, стабильный линкер); дорожка 7: антитело трастузумаб с мутацией НС-А118С, конъюгированное с конструкцией аманитина Her-30.1704 через цистеиновое связывание с 118С (бромацетамидовая связь, расщепляемый линкер); дорожка 8: антитело трастузумаб с мутацией НС-А118С, конъюгированное с конструкцией аманитина Her-30.0880 через цистеиновое связывание с 118С (малеимидная связь, стабильный линкер); дорожка 9: антитело В, конъюгированное с конструкцией аманитина Her-30.1699 через цистеиновое связывание с 118С (малеимидная связь, расщепляемый линкер); верхний гель: денатурирующие и восстанавливающие условия; время воздействия: 60 с; нижний гель: денатурирующие и невосстанавливающие условия; время воздействия: приблизительно 5 с; на фиг. 7(B) показан анализ различных конструкций с использованием (i) окрашивания белков Кумасси (верхняя половина); и (ii) вестерн-блоттинг антитела анти-аманитина (нижняя половина) в денатурирующих и восстанавливающих условиях (левая сторона): и денатурирующих и невосстанавливающих условиях (правая сторона).
На фиг. 8(A) и (В) показаны результаты анализов цитотоксичности WST-I с использованием Fc γ рецептор-положительных клеток ТНР-1 и различных конъюгатов с различными цистеиновыми мутантами антитела трастузумаб: на фиг. 8(A): время инкубации 120 ч; на фиг. 8(B): время инкубации трастузумаба 72 ч; на обеих фигурах две кривые в верхней правой части графика получены из (ADCs) конъюгатов антитело-лекарственное средство на основе мутантов HC-D265C и HC-N297C, т.е. мутантов остатков, которые, как известно, участвуют в связывании с Fc γ-рецептором; остальные графики получены из ADCs на основе мутантов НС-А118С; HC-S239C; НС-Е269С; НС-А327С и HC-I332C.
На фиг. 9 показаны результаты исследования переносимости конъюгата Ab трастузумаб НС-А118С (стабильный линкер, малеимидная связь) у мышей. ПМТ: потеря массы тела; н/о*: не определено из-за недостаточного связывания; н/о**: не определено из-за смертельности при более низкой дозе; н/о***: не определено из-за отсутствия материала; Т НС-A118C-30.0880 не показывает потерю веса при 37,5 мг/кг.
На фиг. 10 показаны результаты исследования переносимости различных конъюгатов аматоксин-трастузумаб на яванских макаках: (А) случайная конъюгация лизина, стабильный линкер: (i) 0,3 мг/кг; (В) случайная конъюгация лизина, стабильный линкер: (i) 0,3 мг/кг; (С) конъюгат Ab трастузумаб НС-А118С со стабильным линкером / малеимидовой связью: (i) 0,3 мг/кг; (ii) 1,0 мг/кг; (iii) 3,0 мг/кг; (iv) 10,0 мг/кг; (D) конъюгат Ab трастузумаб HC-D265C со стабильным линкером / малеимидовой связью: (i) 0,3 мг/кг; (ii) 1,0 мг/кг; (iii) 3,0 мг/кг; (iv) 10,0 мг/кг.
На фиг. 11 показаны результаты анализа инкорпорации 5-бромдезоксиуридина (BrdU) для определения жизнеспособности клеток после инкубации в течение 72 часов с различными ADCs к HER2 и HER2-позитивными раковыми клетками SKOV-3.
На фиг. 12 показаны результаты анализа инкорпорации 5-бромдезоксиуридина для определения жизнеспособности клеток после инкубации в течение 72 часов с различными ADCs к HER2 и HER2-позитивными раковыми клетками NCI-N87.
На фиг. 13 показаны результаты анализа инкорпорации 5-бромдезоксиуридина для определения жизнеспособности клеток после инкубации в течение 120 часов с различными ADCs к ПСМА (PSMA) и ПСМА-позитивными клетками рака простаты С4-2.
На фиг. 14 показаны результаты модели подкожного HER2-положительного ксенотрансплантата SKOV-3, обработанного конъюгатом антитела анти-HER2 с лекарственным средством.
Осуществление изобретения
Прежде чем подробно описать настоящее изобретение, следует понимать, что настоящее изобретение не ограничивается частной методологией, протоколами и реагентами, описанными в настоящем документе, и они могут отличаться от описанных. Также следует понимать, что применяемая в настоящем описании терминология использована только для описания частных вариантов осуществления изобретения и не предназначена для ограничения объема настоящего изобретения, который будет ограничен только прилагаемой формулой изобретения. Если не определено иное, все технические и научные термины, используемые в настоящем описании, имеют значение, обычно понимаемое специалистом с обычной квалификацией в данной области техники.
Преимущественно используемые в настоящем описании термины имеют определения, указанные в «Многоязычном глоссарии биотехнологических терминов: (IUPAC Recommendations)», Leuenberger, H.G.W, Nagel, В. and  , H. eds. (1995), Helvetica Chimica Acta, CH-4010 Basel, Switzerland).
, H. eds. (1995), Helvetica Chimica Acta, CH-4010 Basel, Switzerland).
В настоящем описании и в последующей формуле изобретения, если по контексту не требуется иное, подразумевается, что слово «содержать» и такие словоформы, как «содержит» и «содержащий», включают указанный комплект, состав, этап или группу комплектов, составов или этапов, при этом также могут быть представлены любой дополнительный комплект, состав или этап или группа комплектов, составов или этапов, включая варианты осуществления изобретения, где отсутствует дополнительный комплект, состав или этап или группа комплектов, составов или этапов. В таких последних вариантах осуществления изобретения термин «содержащий» используется взаимозаменяемо с термином «состоящий из».
В тексте настоящего описания приводятся ссылки на несколько документов. Каждый из процитированных в настоящем описании документов (включая все патенты, патентные заявки, научные публикации, технические условия изготовителя, инструкции, представления последовательности номеров доступа GenBank и т.д.), указанные выше или ниже, полностью включен посредством ссылки в полном объеме, насколько это возможно, по соответствующему патентному праву. Никакое положение в настоящем описании не должно толковаться как признание того факта, что изобретение не может быть противопоставлено такому описанию с более ранним приоритетом в силу предшествующего изобретения.
Настоящее изобретение основано на неожиданно установленном факте, что в исходном антителе можно выявить определенное число специфических аминокислотных остатков, которые могут быть мутированы на отдельные непарные цистеиновые остатки, что приведет к образованию высокотоксичных, стабильных и хорошо переносимых конъюгатов с аматоксинами.
Таким образом, в одном аспекте настоящее изобретение относится к конъюгату общей формулы:
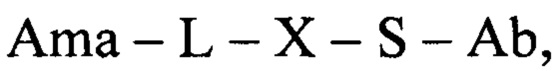
где Ama - аматоксин, L - линкер, X - фрагмент, полученный связыванием тиольной группы с тиол-реагирующей группой, S - атом серы аминокислотного остатка цистеина, и Ab - последовательность антитела или функциональный фрагмент антитела, содержащие указанный цистеиновый остаток, при этом указанный цистеиновый остаток (i) расположен в домене антитела, выбранном из CL, CH1, CH2 и СН3; (ii) расположен в позиции, в которой последовательность зародышевой линии, имеющая гомологию, наиболее близкую к последовательности указанного домена антитела, содержит аминокислотный остаток, отличный от цистеина; и (iii) расположен в позиции, которая подвергается воздействию растворителя.
В еще одном аспекте настоящее изобретение относится к конъюгату общей формулы:
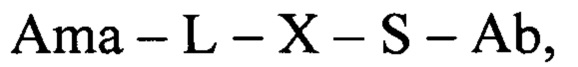
где Ama - аматоксин, L - линкер, X - фрагмент, полученный посредством связывания тиольной группы с тиол-реагирующей группой, S - атом серы аминокислотного остатка цистеина, и Ab - последовательность антитела или функциональный фрагмент антитела, содержащие указанный цистеиновый остаток, где указанный цистеиновый остаток выбран из перечня: 118Cys с тяжелой цепью, 239Cys с тяжелой цепью и 265Cys с тяжелой цепью, в особенности 118Cys с тяжелой цепью и 265Cys с тяжелой цепью.
В контексте настоящего изобретения термин «аматоксин» включает в себя все циклические пептиды, состоящие из 8 аминокислот, выделенные из рода Amanita и описанные у Wieland, Т. и Faulstich Н. (Wieland Т, Faulstich Н., CRC Crit Rev Biochem, 1978 Dec, 5(3): 185-260) и, кроме того, включает все его химические производные; а также все его полусинтетические аналоги; а также все его синтетические аналоги, сконструированные из структурных блоков в соответствии с основной структурой природных соединений (циклических, 8 аминокислот), а также все синтетические или полусинтетические аналоги, содержащие негидроксилированные аминокислоты вместо гидроксилированных аминокислот, а также все синтетические или полусинтетические аналоги, в которых тиоэфирсульфоксидный фрагмент замещен сульфидом, сульфоном или атомами, отличными от серы, например, атомом углерода, как в карбоаналоге аманитина, в каждом случае при этом любое такое производное или аналог обладают функциональной активностью посредством ингибирования РНК-полимеразы II млекопитающих.
Функционально аматоксины определяются как пептиды или депсипептиды, которые ингибируют РНК-полимеразу II млекопитающих. Предпочтительными аматоксинами являются аматоксины, имеющие функциональную группу (например, карбоксильную группу, аминогруппу, гидроксигруппу, тиольную группу или захватывающую тиол группу), которые могут реагировать с молекулами-линкерами или связывающимися с мишенью фрагментами, в соответствии с определением выше. Аматоксинами, которые особенно пригодны для конъюгатов по настоящему изобретению, являются α-аманитин, β-аманитин, γ-аманитин, ε-аманитин, аманин, аманинамид, амануллин и амануллиновая кислота, показанные на фиг. 1, а также их соли, химические производные, полусинтетические аналоги и синтетические аналоги. Особенно предпочтительными аматоксинами для использования в настоящем изобретении являются α-аманитин, γ-аманитин и ε-аманитин, особенно α-аманитин.
В контексте настоящего изобретения «линкер» относится к структуре, соединяющей два компонента, при этом каждый из них прикреплен к одному концу линкера. Если линкер представляет собой связь, прямое связывание аматоксина с антителом может уменьшить способность аматоксина взаимодействовать с РНК-полимеразой II. В частных вариантах осуществления изобретения линкер увеличивает расстояние между двумя компонентами и уменьшает стерические затруднения между этими компонентами, например, в данном случае между антителом и аматоксином. В частных вариантах осуществления изобретения линкер имеет непрерывную цепочку, состоящую из от 1 до 30 атомов (например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 30, 26, 27, 28, 29 или 30 атомов) в его основной цепи, т.е. длина линкера определяется как наименьшее соединение, измеряемое числом атомов или связей между фрагментом аматоксина и антителом, при этом одна сторона основной цепи линкера реагировала с аматоксином, а другая сторона доступна для реакции или реагировала с антителом. В контексте настоящего изобретения предпочтительно, чтобы линкером был С1-20-алкилен, С1-20-гетероалкилен, С2-20-алкенилен, С2-20-гетероалкенилен, С2-20-алкинилен, С2-20-гетероалкинилен, циклоалкилен, гетероциклоалкилен, арилен, гетероарилен, аралкилен или гетероаралкиленовая группа, необязательно замещенная. Линкер может содержать один или несколько структурных элементов, таких как карбоксамид, сложный эфир, простой эфир, тиоэфир, дисульфид, мочевина, тиомочевина, углеводородные фрагменты и аналогичные соединения. Линкер также может содержать комбинации двух или более таких структурных элементов. Каждый из таких структурных элементов может присутствовать в линкере более одного раза, например, два, три, четыре, пять или шесть раз. В некоторых вариантах осуществления изобретения линкер может содержать дисульфидную связь. Следует понимать, что линкер должен быть присоединен либо в ходе одного этапа, либо в ходе двух или более последующих этапов к аматоксину и антителу. Для этой цели линкер должен иметь две группы, предпочтительно на проксимальном и дистальном концах, которые могут (i) образовывать ковалентную связь с группой, присутствующей в одном из связываемых компонентов, предпочтительно активированную группу на аматоксине или связывающемся с мишенью пептиде, или (ii), которая активирована или может быть активирована для образования ковалентной связи с группой на аматоксине. Соответственно, предпочтительнее, чтобы на дистальном и проксимальном концах линкера находились химические группы, которые образуются в результате такой реакции связывания, например, сложный эфир, простой эфир, уретан, пептидная связь и т.д.
В частных вариантах осуществления изобретения линкер L представляет собой линейную цепочку длиной от 1 до 20 атомов, независимо выбранных из С, О, N и S, особенно от 2 до 18 атомов, более конкретно от 5 до 16 атомов и еще более конкретно от 6 до 15 атомов. В частных вариантах осуществления изобретения по меньшей мере 60% атомов в линейной цепи являются С-атомами. В частных вариантах осуществления изобретения атомы в линейной цепи связаны одинарными связями.
В частных вариантах осуществления изобретения линкер L представляет собой алкилен, гетероалкилен, алкенилен, гетероалкенилен, алкинилен, гетероалкинилен, циклоалкилен, гетероциклоалкилен, арилен, гетероарилен, аралкилен или гетероаралкиленовую группу, содержащие от 1 до 4 гетероатомов, выбранных из N, О и S, где указанный линкер необязательно замещен.
Термин «алкилен» относится к двухвалентным насыщенным углеводородным группам с прямой цепью, имеющим от 1 до 20 атомов углерода, включая группы, имеющие от 1 до 10 атомов углерода. В некоторых вариантах осуществления изобретения алкиленовыми группами могут быть низшие алкиленовые группы. Термин «низший алкилен» относится к алкиленовым группам, имеющим от 1 до 6 атомов углерода, и в некоторых вариантах осуществления изобретения от 1 до 5 или от 1 до 4 атомов углерода. Примеры алкиленовых групп включают, но не ограничены, метилен (-CH2-), этилен (-CH2-CH2-), н-пропилен, н-бутилен, н-пентилен и н-гексилен.
Термин «алкенилен» относится к двухвалентным группам с прямой цепью, имеющим от 2 до 20 атомов углерода, при этом по меньшей мере одна из углерод-углеродных связей является двойной, при этом другие связи могут быть одинарными, а также двойными. Термин «алкенилен», используемый в настоящем описании, относится к группам, имеющим от 2 до 20 атомов углерода, причем по меньшей мере одна из углерод-углеродных связей является тройной, при этом другие связи могут быть одинарными, двойными, а также тройными. Примеры алкениленовых групп включают в себя этенилен (-СН=СН-), 1-пропенилен, 2-пропенилен, 1-бутенилен, 2-бутенилен, 3-бутенилен и аналогичные соединения. Примеры алкиниленовых групп включают в себя этинилен, 1-пропинилен, 2-пропинилен и т.д.
При использовании в настоящем описании термин «циклоалкилен» подразумевает двухвалентное кольцо, являющееся частью любой стабильной моноциклической или полициклической системы, при этом такое кольцо имеет от 3 до 12 атомов углерода, но не содержит гетероатомов, и при этом такое кольцо является полностью насыщенным, и термин «циклоалкенилен» подразумевает двухвалентное кольцо, являющееся частью любой стабильной моноциклической или полициклической системы, при этом такое кольцо имеет от 3 до 12 атомов углерода, но не содержит гетероатомов, и при этом такое кольцо является, по меньшей мере частично, ненасыщенным (но за исключением любых ариленовых колец). Примеры циклоалкиленов включают в себя, помимо прочего, циклопропилен, циклобутилен, циклопентилен, циклогексилен и циклогептилен. Примеры циклоалкениленов включают в себя, помимо прочего, циклопентенилен и циклогексенилен.
При использовании в настоящем описании термины «гетероциклоалкилен» и «гетероциклоалкенилен» подразумевают двухвалентное кольцо, являющееся частью любой стабильной моноциклической или полициклической системы, при этом такое кольцо имеет от 3 до 12 атомов, и при этом такое кольцо состоит из атомов углерода и по меньшей мере одного гетероатома, в частности по меньшей мере одного гетероатома, независимо выбранного из группы, состоящей из N, О и S, причем гетероциклоалкилен относится к такому кольцу, которое является полностью насыщенным кольцом, и гетероциклоалкенилен относится к такому кольцу, которое является, по меньшей мере частично, ненасыщенным кольцом (но за исключением любых ариленовых или гетероариленовых колец).
Термин «арилен» означает бивалентное кольцо или кольцевую систему, являющуюся частью любой стабильной моноциклической или полициклической системы, при этом такое кольцо или кольцевая система имеет от 3 до 20 атомов углерода, но не имеет гетероатомов, и такое кольцо или кольцевая система состоит из ароматического фрагмента, определяемого правилом «4n+2» π электронов, включая фенилен.
При использовании в настоящем описании термин «гетероарилен» означает бивалентное кольцо или кольцевую систему, являющуюся частью любой стабильной моноциклической или полициклической системы, при этом такое кольцо или такая кольцевая система имеет от 3 до 20 атомов углерода, и такое кольцо или кольцевая система состоит из ароматического фрагмента, определяемого правилом «4n+2» π электронов и содержит атомы углерода и один или несколько гетероатомов азота, серы и/или кислорода.
В контексте настоящего изобретения термин «замещенный» подразумевает, что один или несколько атомов водорода, присутствующие в основной цепи линкера, замещены атомами, выбранными из указанной группы (групп), при условии, что нормальная валентность указанного атома или нормальная валентность соответствующего атома группы не превышена и что замещение приводит к образованию стабильного соединения. Термин «необязательно замещенный» подразумевает, что линкер либо не замещен, либо замещен в соответствии с указанным в настоящем описании определением на один или несколько заместителей в соответствии с указанным в настоящем описании определением. Когда заместителем является группа кето (или оксо, то есть =O), тио- или иминогруппа или аналогичная, то замещаются два атома водорода на основной цепи линкера. Примеры заместителей включают, например, алкил, алкенил, алкинил, циклоалкил, гетероциклоалкил, арил, гетероарил, аралкил, гетероаралкил, ацил, ароил, гетероароил, карбоксил, алкокси, арилокси, ацилокси, ароилокси, гетероароилокси, алкоксикарбонил, галоген, (тио) сложный эфир, циано, фосфорил, амино, имино, (тио)амидо, сульфгидрил, алкилтио, ацилтио, сульфонил, сульфат, сульфонат, сульфамоил, сульфонамидо, нитро, азидо, галогеналкил, включая перфторалкил (такой как трифторметил), галогеналкокси, алкилсульфанил, алкилсульфинил, алкилсульфонил, алкилсульфониламино, арилсульфонамино, фосфорил, фосфат, фосфонат, фосфинат, алкилкарбокси, алкилкарбоксиамид, оксо, гидрокси, меркапто, амино (необязательно моно- или дизамещенный, например, алкилом, арилом или гетероарилом), имино, карбоксамид, карбамоил (необязательно моно- или дизамещенный, например, алкилом, арилом или гетероарилом), амидино, аминосульфонил, ациламино, ароиламино, (тио) уреидо, (арилтио)уреидо, алкил(тио)уреидо, циклоалкил(тио)уреидо, арилокси, аралкокси или -O(CH2)n-ОН, -O(CH2)n-NH2, -O(CH2)nCOOH, -(CH2)nCOOH, -C(O)O(CH2)nR, -(CH2)nN(H)C(O)OR или N(R)S(O)2R, где n равно 1-4, и R независимо выбран из водорода, -алкила, -алкенила, -алкинила, -циклоалкила, -циклоалкенила, -(С-связанный-гетероциклоалкил), -(С-связанный-гетероциклоалкенил), -арил и -гетероарил, при этом допускаются несколько степеней замещения. Специалистам в данной области техники будет понятно, что заместители, такие как гетероциклоалкил, арил, гетероарил, алкил и т.д., или функциональные группы, такие как -ОН, -NHR и т.д., сами могут быть замещены при необходимости. Специалистам в данной области также будет понятно, что сами замещенные фрагменты также могут быть замещены при необходимости.
В частных вариантах осуществления изобретения линкер L содержит фрагмент, выбранный из одного из следующих фрагментов: дисульфид ((-S-S-), простой эфир (-O-), тиоэфир (-S-), амин (-NH-), сложный эфир (-O-С(=O)- или -С(=O)-О), карбоксамид (-NH-C(=O)- или -C(=O)-NH-), уретан (-NH-С(=O)-O- или -O-C(=O)-NH-) и фрагмент мочевины (-NH-C(=O)-NH-).
В частных вариантах осуществления настоящего изобретения линкер L содержит несколько m групп, выбранных из следующего перечня: алкилен, алкенилен, алкинилен, циклоалкилен, гетероалкилен, гетероалкенилен, гетероалкинилен, гетероциклоалкилен, арилен, гетероарилен, аралкилен и гетероаралкиленовая группа, при этом каждая группа может быть необязательно независимо замещена, линкер дополнительно содержит несколько n фрагментов, независимо выбранных из одного из следующих фрагментов: дисульфид ((-S-S-), простой эфир (-O-), тиоэфир (-S-), амин (-NH-), сложный эфир (-O-С(=O)- или -С(=O)-O), карбоксамид (-NH-C(=O)- или - C(=O)-NH-), уретан (-NH-C(=O)-O- или -O-C(=O)-NH-) и фрагмент мочевины (-NH-C(=O)-NH-), при этом m=n+1. В частных вариантах осуществления изобретения m равно 2 и n равно 1 или m равно 3 и n равно 2. В частных вариантах осуществления изобретения линкер содержит 2 или 3 незамещенные алкиленовые группы и 1 или 2, соответственно, фрагмента дисульфида, простого эфира, тиоэфира, амина, сложного эфира, карбоксамида, уретана или мочевины, связывающих незамещенные алкиленовые группы.
В частных вариантах осуществления изобретения С-атомы в линейной цепи независимо входят в необязательно замещенные метиленовые группы (-CH2-). В некоторых из таких вариантов осуществления изобретения необязательные заместители независимо выбраны из галогена и С1-6-алкила, в частности метила.
В частных вариантах осуществления изобретения линкер L является стабильным линкером.
В контексте настоящего изобретения термин «стабильный линкер» относится к линкеру, который является стабильным (i) в присутствии ферментов, в частности лизосомальных пептидаз, таких как катепсин В, и (ii) во внутриклеточной восстановительной среде.
В частных вариантах осуществления изобретения стабильный линкер не содержит (i) ферментативно расщепляемую субструктуру, в частности не-дипептидную последовательность, расщепляемую катепсином В, и/или (ii) дисульфидную группу. В таких частных вариантах осуществления изобретения линкер имеет длину до 12 атомов, в частности от 2 до 10, более конкретно от 4 до 9 и, наиболее предпочтительно от 6 до 8 атомов.
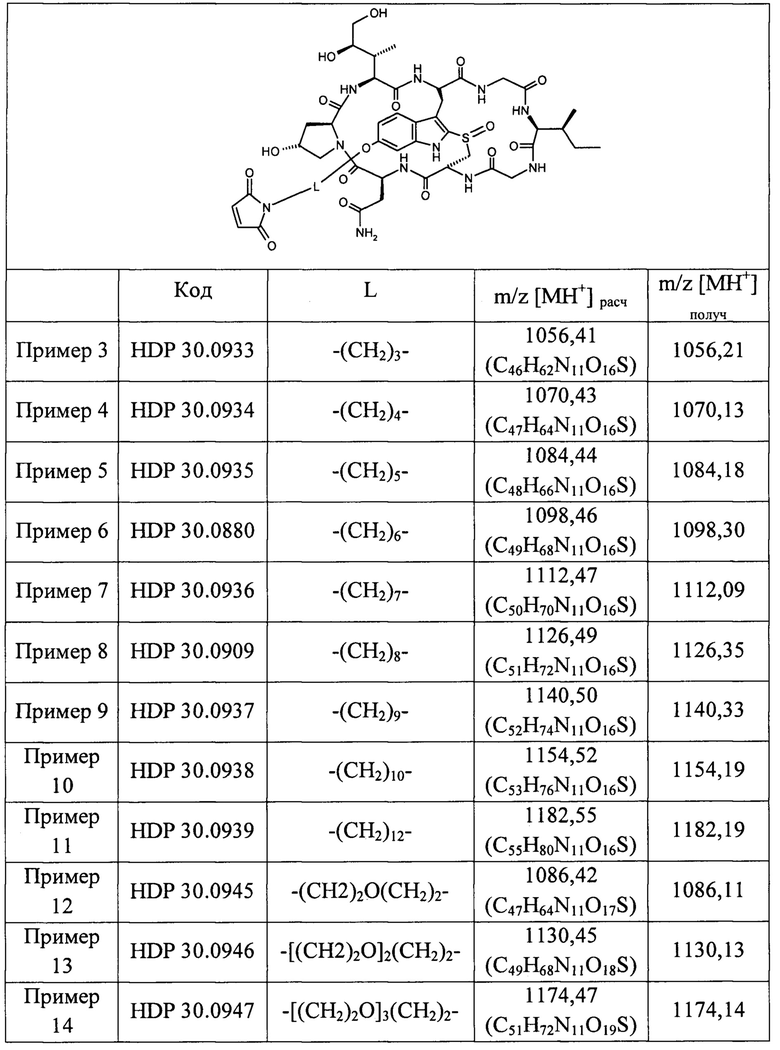
В частных вариантах осуществления изобретения фрагмент L-X-S, присутствующий в общей формуле раздела [0043] (абзац 4 на с. 12), выбран из следующей группы фрагментов:
(сторона аматоксина) -(CH2)2-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)3-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)4-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)5X-S- (сторона антитела);
(сторона аматоксина) -(CH2)6-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)7-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)8-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)9-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)10-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)11-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)12-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)16-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)2-O-(CH2)2-O-(CH2)2-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)2-O-(CH2)2-O-(CH2)2-O-(CH2)2-X-S- (сторона антитела); и
(сторона аматоксина) -(CH2)2-O-(CH2)2-O-(CH2)2-O-(CH2)2-O-(CH2)2-Х-S- (сторона антитела).
В других частных вариантах осуществления изобретения линкер является расщепляемым линкером.
В контексте настоящего изобретения термин «расщепляемый линкер» относится к линкеру, который расщепляется (i) каким-либо ферментом или (ii) в восстановительной среде.
В контексте настоящего изобретения термин «линкер, который расщепляется … каким-либо ферментом» относится к линкеру, который может быть расщеплен каким-либо ферментом, в частности лизосомальной пептидазой, такой как катепсин В, что приводит к высвобождению внутрь клетки нагрузки токсина, конъюгированного к целевому антителу после интернализации (см. Dubowchik et al., Bioconjug Chem., 13 (2002) 855-69). В частных вариантах осуществления изобретения расщепляемый линкер содержит дипептид, выбранный из: Phe-Lys, Val-Lys, Phe-Ala, Val-Ala, Phe-Cit и Val-Cit, в частности, где расщепляемый линкер дополнительно содержит п-аминобензил (РАВ) между дипептидами и аматоксином.
В других частных вариантах осуществления изобретения линкер является восстанавливаемым линкером.
В контексте настоящего изобретения термин «восстанавливаемый линкер» относится к линкеру, который может быть расщеплен во внутриклеточной восстановительной среде, в частности линкер, который содержит дисульфидные группы, что приводит к высвобождению внутрь клетки нагрузки токсина, конъюгированного к целевому антителу после интернализации внутриклеточной восстановительной средой (см. Shen et al. (1985) J. Biol. Chem. 260, 10905-10908).
В других частных вариантах осуществления изобретения линкер является расщепляемым линкером, в частности (i) линкером, расщепляемым ферментом, в частности линкером, содержащим дипептид, в частности дипептид, расщепляемый катепсином В, или (ii) восстанавливаемым линкером, в частности линкером, содержащим дисульфидную группу. В некоторых таких вариантах осуществления изобретения такой расщепляемый линкер имеет длину до 20 атомов, в частности от 6 до 18, более конкретно от 8 до 16 и наиболее предпочтительно от 10 до 15 атомов.
В частных вариантах осуществления изобретения линкер L во фрагменте L-X-S, присутствующем в общей формуле раздела [0043] (абзац 4 на с. 12), выбран из следующей группы фрагментов:
(сторона аматоксина) -(CH2)2-S-S-(CH2)2-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)3-S-S-(CH2)2-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)2-S-S-(CH2)3-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)3-S-S-(CH2)3-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)4-S-S-(CH2)4-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)2-CMe2-S-S-(CH2)2-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)2-S-S-CMe2-(CH2)2-X-S- (сторона антитела);
(сторона аматоксина) -(CH2)3-S-S- (сторона антитела);
(сторона аматоксина) -CH2-C6H4-NH-Cit-Val-CO(CH2)5-X-S- (сторона антитела)
(сторона аматоксина) -CH2-C6H4-NH-Ala-Val-CO(CH2)5-X-S- (сторона антитела);
(сторона аматоксина) -CH2-C6H4-NH-Ala-Val-CO(CH2)2-X-S- (сторона антитела);
(сторона аматоксина) -CH2-C6H4-NH-Ala-Phe-CO(CH2)2-X-S- (сторона антитела);
(сторона аматоксина) -CH2-C6H4-NH-Lys-Phe-CO(CH2)2-X-S- (сторона антитела);
(сторона аматоксина) -CH2-C6H4-NH-Cit-Phe-CO(CH2)2-X-S- (сторона антитела);
(сторона аматоксина) -CH2-C6H4-NH-Val-Val-CO(CH2)2-X-S- (сторона антитела);
(сторона аматоксина) -CH2-C6H4-NH-Ile-Val-CO(CH2)2-X-S- (сторона антитела);
(сторона аматоксина) -CH2-C6H4-NH-His-Val-CO(CH2)2-X-S- (сторона антитела);
(сторона аматоксина) -CH2-C6H4-NH-Met-Val-CO(CH2)2-X-S- (сторона антитела);
(сторона аматоксина) -CH2-C6H4-NH-Asn-Lys-CO(CH2)2-X-S- (сторона антитела); и
где -NH- и -СО-, расположенные по краям дипептидных последовательностей, представляют собой амино и карбонильные фрагменты линкера, образующие амидные связи с карбокси- и аминоконцом дипептида, соответственно.
В контексте настоящего изобретения термин «фрагмент, полученный вследствие связывания тиольной группы с тиол-реагирующей группой» относится к структуре, полученной вследствие (i) нуклеофильного замещения уходящей группы Y, присутствующей в тиол-реагирующей группе, атомом серы цистеинового остатка, например, бромацетамидной группы, иодацетамида, 4,6-дихлор-1,3,5-триазин-2-иламиногруппы, алкилсульфона или гетероарилсульфона; (ii) добавления HS-группы цистеинового остатка к активированной двойной связи тиол-реагирующей группы, например, малеимиду, или (iii) дисульфидного обмена активированного дисульфида или метанитиосульфоната с атомом серы цистеинового остатка, например, пиридин-2-тиола, 5-нитропиридин-2-тиола или метансульфина в качестве уходящей группы; или (iv) любой иной химической реакции, которая приводит к образованию стабильной связи между атомом серы цистеинового остатка и реакционноспособным фрагментом, являющимся частью тиол-реагирующей группы.
Первичный фрагмент, полученный в результате связывания тиоловой группы, может быть необязательно дополнительно дериватизирован, например, сукцинимидилтиоэфир, полученный из малеимида, может быть гидролизован до тиоэфиров сукцинаминовой кислоты следующих общих структур
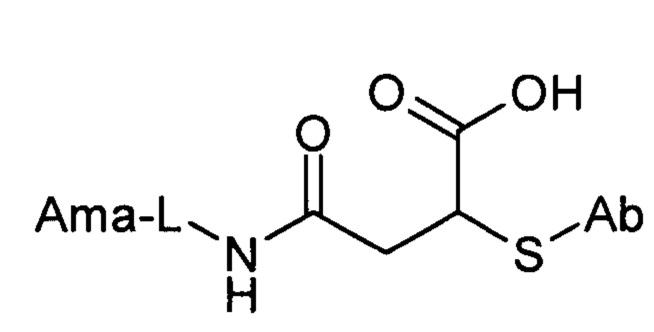 или
или 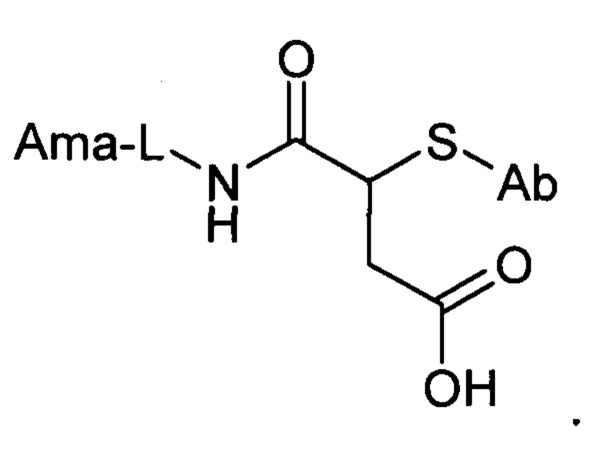
В контексте настоящего изобретения термин «тиол-реагирующая группа» относится к группе, которая избирательно реагирует с тиольной группой свободного цистеина антитела, в частности при значении рН в диапазоне от 6,0 до 8,0, более конкретно при значении рН в диапазоне от 6,5 до 7,5. В частности, термин «избирательно» означает, что менее 10% реакций связывания молекулы, содержащей тиол-реагирующую группу, с антителом, содержащим по меньшей мере один свободный цистеиновый остаток, представляют собой реакции связывания с нецистеиновыми остатками антитела, такими как остатки лизина, в частности менее 5%, более конкретно менее 2%.
В частных вариантах осуществления изобретения фрагмент, полученный вследствие связывания тиольной группы с тиол-реагирующей группой, выбран из следующих вариантов: тиолзамещенный ацетамид; тиолзамещенный сукцинимид; тиолзамещенная сукцинаминовая кислота; тиолзамещенный гетероарил, в частности тиолзамещенный бензотиазол, тиолзамещенный фенилтетразол и тиолзамещенный фенилоксадиазол; и дисульфид, где один атом серы получают из цистеинового остатка антитела. В частных вариантах осуществления изобретения фрагмент, полученный вследствие связывания тиольной группы с тиол-реагирующей группой, является тиолзамещенным сукцинимидом.
Термин «антитело или функциональный фрагмент антитела», используемый в настоящем описании, относится к молекулам иммуноглобулина и иммунологически активным частям молекул иммуноглобулина, т.е. к молекулам, которые содержат антигенсвязывающий сайт, который иммуноспецифически связывает антиген, т.е. частям антитела, содержащим по меньшей мере антигенсвязывающий фрагмент антитела. Также в изобретение включены аналогичные иммуноглобулину белки, которые выбирают с помощью методов, включающих в себя, например, фаговый дисплей для специфического связывания с молекулой-мишенью, например, с белком-мишенью Her-2/neu или ЕрСАМ. Молекулы иммуноглобулина по изобретению могут иметь любой тип (например, IgG, IgE, IgM, IgD, IgA и IgY), класс (например, IgG1, IgG2, IgG3, IgG4, IgA1 и IgA2) или подкласс молекулы иммуноглобулина. В частных вариантах осуществления изобретения антитело представляет собой IgG1. «Антитела и их антигенсвязывающие фрагменты», пригодные для использования в настоящем изобретении, включают, но не ограничены ими, поликлональные, моноклональные, моновалентные, биспецифические, гетероконъюгатные, мультиспецифические, человеческие, гуманизированные (в частности с привитыми гипервариабельными участками (CDR)), деиммунизированные или химерные антитела, Fab фрагменты, F(ab')2 фрагменты, фрагменты, полученные посредством библиотеки экспрессии Fab, нанотела, анти-идиотипические (анти-Id) антитела (включая, например, антитела против Id к антителам по изобретению), финомер-антитела (FynomAbs) (Brack et al., Mol. Cancer Ther. 13 (2014) 2030) и эпитопсвязывающие фрагменты любого из вышеуказанных соединений, которые содержат по меньшей мере одну из позиций каркаса тяжелой цепи по настоящему изобретению.
В некоторых вариантах осуществления изобретения антигенсвязывающие фрагменты являются антигенсвязывающими фрагментами антител человека по настоящему изобретению и включают, помимо прочего, Fab, Fab' и F(ab')2, Fd и другие фрагменты, содержащие по меньшей мере часть тяжелой цепи антитела, которые содержат по меньшей мере одну из позиций каркаса тяжелой цепи по настоящему изобретению. Такие антигенсвязывающие фрагменты антител могут содержать только вариабельный домен (домены) в сочетании со всей или частью следующих участков: шарнирная область, домены CL, CH1, CH2 и СН3. Также в изобретение включены такие антигенсвязывающие фрагменты, которые также включают в себя любую комбинацию вариабельного домена (доменов) с шарнирной областью, доменами CL, CH1, CH2 и СН3.
Антитела, используемые в изобретении, могут быть получены из любого животного материала, включая птиц и млекопитающих. Предпочтительнее, чтобы антитела были получены из материала человека, грызунов (например, мышей, крыс, морских свинок или кроликов), кур, свиней, овец, коз, верблюдов, коров, лошадей, ослов, кошек или собак. Особенно предпочтительно, чтобы антитела были получены из материала человека или мышей. При использовании в настоящем описании «человеческие антитела» включают в себя антитела, имеющие аминокислотную последовательность человеческого иммуноглобулина, и включают в себя антитела, выделенные из библиотек иммуноглобулина человека или из животного материала, трансгенные для одного или нескольких человеческих иммуноглобулинов, и которые не экспрессируют эндогенные иммуноглобулины, как описано, например, в патенте США №5,939,598 Kucherlapati & Jakobovits.
При использовании в настоящем описании считается, что антитело или функциональный фрагмент антитела «специфически связывается» с антигеном, если он имеет константу диссоциации KD к указанному антигену как мишени 100 мкМ или менее, предпочтительно 50 мкМ или менее, предпочтительно 30 мкМ или менее, предпочтительно 20 мкМ или менее, предпочтительно 10 мкМ или менее, предпочтительно 5 мкМ или менее, предпочтительнее 1 мкМ или менее, предпочтительнее 900 нМ или менее, предпочтительнее 800 нМ или менее, предпочтительнее 700 нМ или менее, предпочтительнее 600 нМ или менее, предпочтительнее 500 нМ или менее, предпочтительнее 400 нМ или менее, предпочтительнее 300 нМ или менее, предпочтительнее 200 нМ или менее, еще предпочтительнее 100 нМ или менее, еще предпочтительнее 90 нМ или менее, еще предпочтительнее 80 нМ или менее, еще предпочтительнее 70 нМ или менее, еще предпочтительнее 60 нМ или менее, еще предпочтительнее 50 нМ или менее, еще предпочтительнее 40 нМ или менее, еще предпочтительнее 30 нМ или менее, еще предпочтительнее 20 нМ или менее и еще предпочтительнее 10 нМ или менее.
В контексте настоящей заявки термины «молекула-мишень» и «эпитоп-мишень», соответственно, относятся к антигену и эпитопу антигена, соответственно, который специфически связан антителом или функциональным фрагментом антитела. Предпочтительнее, чтобы молекула-мишень представляла собой опухолеассоциированный антиген, в частности антиген или эпитоп, который присутствует на поверхности одного или нескольких типов опухолевых клеток или опухолеассоциированных клеток в более высокой концентрации и/или в другой пространственной конфигурации по сравнению с поверхностью неопухолевых клеток. Предпочтительнее, чтобы указанный антиген или эпитоп присутствовал на поверхности одного или нескольких типов опухолевых клеток или опухолевых стромальных клеток, но не на поверхности неопухолевых клеток. В частных вариантах осуществления изобретения антитело специфически связывается с эпитопом HER-2/neu или адгезивной молекулой эпителиальной клетки (ЕрСАМ). В других вариантах осуществления изобретения указанный антиген или эпитоп предпочтительно экспрессируется на клетках, вовлеченных в аутоиммунные заболевания. В некоторых таких вариантах осуществления изобретения антитело специфически связывается с эпитопом IL-6 рецептора (IL-6R). В других вариантах осуществления изобретения указанный антиген или эпитоп предпочтительно экспрессируется на клетках, вовлеченных в воспалительное заболевание.
В частных вариантах осуществления изобретения антитело или функциональный фрагмент антитела специфически связывается с эпитопом, присутствующем на опухолевой клетке, в частности, при этом антитело специфически связывается с эпитопом рецептора 2 эпидермального фактора роста человека (HER2).
В частных вариантах осуществления изобретения антитело представляет собой трастузумаб или НЕА125, или фрагмент антитела, содержащий антигенсвязывающий фрагмент трастузумаба или НЕА125.
В частных вариантах осуществления изобретения более одной молекулы аматоксина связано с одним антителом или с одним функциональным фрагментом антитела. Увеличение числа аматоксинов на один конъюгат также увеличит токсичность. Однако при увеличении одновременно уменьшится переносимость. Соответственно, в частном варианте осуществления изобретения соотношение антитела или функционального фрагмента антитела к аматоксину находится в диапазоне от одного антитела или одного функционального фрагмента антитела на 1-4 молекулы аматоксина, в частности на 1,5-3,5 молекулы аматоксина, более конкретно на 1,8-2,5 молекулы аматоксина, более конкретно около 2 молекул аматоксина. Для расчета соотношения в случае димеров антитела, такого как IgG, димер рассматривается как один фрагмент.
В частных вариантах осуществления изобретения антитело или его антигенсвязывающий фрагмент выбирают из диатела, тетратела, нанотела, химерного антитела, деиммунизированного антитела, гуманизированного антитела или антитела человека.
В частных вариантах осуществления изобретения антигенсвязывающий фрагмент выбирают из группы, состоящей из Fab, F(ab')2, Fd, Fv, одноцепочечного Fv и дисульфид-связанного Fvs (dsFv).
В контексте настоящего изобретения термины «118Cys с тяжелой цепью», «239Cys с тяжелой цепью» и «265Cys с тяжелой цепью» относятся к позициям в тяжелой цепи последовательностей антител IgG1 человека, при этом нумерация осуществляется в соответствии с системой нумерации ЕС согласно Edelman et al., Proc. Natl. Acad. Sci. USA; 63 (1969) 78-85. Например, в том случае, когда начинают с Herceptin® (трастузумаб) в качестве исходной последовательности IgG1 человека, необходимо выполнить одну из следующих мутаций: HC-Ala118Cys; HC-Ser239Cys или HC-Asp265Cys.
В третьем аспекте настоящее изобретение относится к способу синтеза конъюгата общей формулы:
реакцией соединения Ama - L - X', где X' - тиол-реагирующая группа, с антителом Ab - SH, где группа -SH - тиол аминокислотного остатка цистеина, и Ab - последовательность антитела, содержащая указанный цистеиновый остаток, при этом указанный цистеиновый остаток выбран из перечня: 118Cys с тяжелой цепью, 239Cys с тяжелой цепью и 265Cys с тяжелой цепью, особенно 118Cys с тяжелой цепью и 265Cys с тяжелой цепью.
В четвертом аспекте настоящее изобретение относится к набору, содержащему (i) соединение Ama - L - X', где X' - тиол-реагирующая группа, и (ii) антитело Ab - SH, причем группа -SH - тиол аминокислотного остатка цистеина, и Ab - последовательность антитела, содержащая указанный цистеиновый остаток, при этом указанный цистеиновый остаток выбран из перечня: 118Cys с тяжелой цепью, 239Cys с тяжелой цепью и 265Cys с тяжелой цепью, особенно 118Cys с тяжелой цепью и 265Cys с тяжелой цепью.
В частных вариантах осуществления изобретения тиол-реагирующую группу X выбирают из бромацетамида, иодоацетамида, метилсульфонилбензотиазола, 4,6-дихлор-1,3,5-триазин-2-иламиногруппы, метилсульфонилфенилтетразола или метилсульфонилфенилоксадиазола, пиридин-2-тиола, 5-нитропиридин-2-тиола, метантиосульфоната и малеимида.
Таким образом, в частных вариантах осуществления настоящего изобретения X' представляет собой малеимидовую субструктуру, в которой нуклеофильная группа сконструированного цистеинового остатка может присоединяться к двойной связи малеимида.
В пятом аспекте настоящее изобретение относится к способу синтеза соединения Ama - L - X', где X' - малеимидная группа, включающему в себя этап (а) реакции аматоксина, содержащего нуклеофильную группу, с соединением Y-L-X'', где
Y - уходящая группа, и
X'' - защищенная малеимидная группа.
В частном варианте осуществления изобретения способ включает в себя дополнительный этап (b) удаления защитной группы из X''.
В предпочтительном варианте осуществления изобретения этап (а) проводят в щелочных условиях, в котором уходящую группу Y выбирают из Br, I, тозилата или мезилата и в котором X'' является стабильной по отношению к щелочным условиям.
В частном варианте осуществления изобретения X'' представляет собой аддукт Дильса - Альдера, полученный в результате реакции малеимида с 1,3-диеном. В частном варианте осуществления изобретения этап (b) включает в себя удаление защитной группы при помощи ретро-реакции Дильса-Альдера.
В более частном варианте осуществления изобретения X'' представляет собой аддукт Дильса - Альдера, получаемый на этапе (а) реакцией малеимида с циклопентадиеном, фураном или 2,5-диалкилфураном, и при этом этап (b) удаления защиты проводят в полярном апротонном растворителе при повышенной температуре.
Более конкретно X'' представляет собой экзо-аддукт Дильса - Альдера, получаемый на этапе (а) по реакции малеимида с 2,5-диметилфураном, и при этом этап (b) удаления защиты проводят в диметилсульфоксиде или N-метилпирролидоне (N-МП) при температуре от 80°С до 120°С.
В шестом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей конъюгат по настоящему изобретению.
В еще одном аспекте настоящее изобретение относится к конъюгату по настоящему изобретению для использования в качестве лекарственного средства.
В седьмом аспекте настоящее изобретение относится к способу лечения заболевания, ассоциированного с клетками, являющимися мишенью, включающему этап: контактирования указанных клеток с конъюгатом по настоящему изобретению, в котором указанное антитело или указанный функциональный фрагмент антитела является специфичным для указанной мишени.
В еще одном аспекте настоящее изобретение относится к конъюгату по настоящему изобретению для применения в лечении заболевания у пациента, в частности в котором заболеванием является рак, в частности рак, выбранный из группы, состоящей из рака молочной железы, рака поджелудочной железы, холангиокарциномы, колоректального рака, рака легких, рака предстательной железы, рака яичников, рака предстательной железы, рака желудка, рака почек, злокачественной меланомы, лейкемии и злокачественной лимфомы.
При использовании в настоящем описании термин «пациент» означает любое млекопитающее или птицу, которым может быть проведено лечение конъюгатами антител с токсинами, описанными в настоящем патенте. Предпочтительнее, чтобы «пациент» был выбран из группы, состоящей из лабораторных животных (например, мышей или крыс), домашних животных (включая, например, морских свинок, кроликов, цыплят, свиней, овец, коз, верблюдов, коров, лошадей, ослов, кошек или собак) или приматов, включая людей. Особенно предпочтительно, чтобы «пациентом» был человек.
При использовании в настоящем описании, слова «лечить», «лечение» или «терапия» заболевания или нарушения означает выполнение одного или нескольких из следующего: (а) уменьшение тяжести нарушения; (b) ограничение или предотвращение развития симптомов, характерных для нарушения (нарушений), в отношении которых проводят лечение; (с) подавление усиления симптомов, характерных для нарушения (нарушений), в отношении которых проводят лечение; (d) ограничение или предотвращение рецидива нарушения (нарушений) у пациентов, которые ранее имели нарушение (нарушения); и (е) ограничение или предотвращение повторного возникновения симптомов у пациентов, у которых ранее имелись симптомы этого нарушения (нарушений).
При использовании в настоящем описании лечение может включать в себя введение конъюгата или фармацевтической композиции по настоящему изобретению пациенту, при этом «введение» включает в себя введение in vivo, а также введение непосредственно в ткань ex vivo, например, в венозные трансплантаты.
В частных вариантах осуществления изобретения используют терапевтически эффективное количество конъюгата по настоящему изобретению.
«Терапевтически эффективным количеством» является количество терапевтического средства, достаточное для достижения намеченной цели. Эффективное количество данного терапевтического вещества будет варьироваться в зависимости от таких факторов, как природа вещества, путь введения, размер и вид животного, которому вводят терапевтический агент, и цель введения. Эффективное количество в каждом отдельном случае может быть эмпирически определено квалифицированным специалистом в соответствии с существующими способами в данной области техники.
В еще одном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей аматоксин по настоящему изобретению, а также содержащей один или несколько фармацевтически приемлемых разбавителей, носителей, эксципиентов, наполнителей, связующих, лубрикантов (смазывающих веществ), глидантов (скользящих веществ), дезинтегрантов, адсорбентов и/или консервантов.
Термин «фармацевтически приемлемый» означает утвержденный регулирующим ведомством федерального правительства или правительства штата или указанный в Фармакопее США или другой общепризнанной фармакопее для применения у животных и более конкретно у людей.
В частных вариантах осуществления изобретения фармацевтическую композицию применяют в форме системно вводимого лекарственного средства. Лекарственное средство включает в себя парентеральные препараты, которые включают в себя, помимо прочего, инъекционные и инфузионные препараты. Инъекционные препараты производят либо в форме ампул, либо в виде так называемых готовых к применению инъекционных форм, например, готовых к применению шприцов или одноразовых шприцов и, кроме того, в виде флаконов с прокалываемой пробкой для многократного извлечения препарата. Введение инъекционных препаратов может быть выполнено в виде подкожного (п/к), внутримышечного (в/м.), внутривенного (в/в) или внутрикожного (в/к) введения. В частности, возможно получить соответственно подходящие инъекционные составы в виде суспензии кристаллов, растворов, систем дисперсных наночастиц или коллоидных частиц, таких как, например, гидрозоли.
Инъекционные препараты могут быть дополнительно получены в виде концентратов, которые можно растворять или диспергировать водными изотоническими разбавителями. Инфузионный состав также может быть приготовлен в виде изотонических растворов, жирных эмульсий, липосомальных составов и микроэмульсий. Аналогично инъекционным препаратам, инфузионные составы также могут быть приготовлены в виде концентратов для разбавления. Инъекционные составы также можно применять в виде постоянных инфузий как в стационарной, так и в амбулаторной терапии, например, посредством мини-насосов.
К парентеральным лекарственным составам могут быть добавлены, например, альбумин, плазма, средство для увеличения объема, поверхностно-активные вещества, органические разбавители, вещества, влияющие на рН, комплексообразующие вещества или полимерные вещества, в частности в качестве веществ, влияющих на адсорбцию конъюгатов антител с токсинами по изобретению в белки или полимеры, или они также могут быть добавлены с целью снижения адсорбции конъюгатов антител с токсинами по изобретению в такие материалы, как инъекционные инструменты или упаковочные материалы, например, пластик или стекло.
Аматоксины по настоящему изобретению, содержащие антитело, могут быть связаны с микроносителями или наночастицами в препаратах для парентерального введения, например, с мелкодисперсными частицами на основе поли(мет)акрилатов, полилактатов, полигликолятов, полиаминокислот или полиэфируретанов. Составы для парентерального введения также могут быть модифицированы как депо-составы, например, на основе «принципа использования нескольких компонентов», если конъюгаты антител с токсинами по изобретению вводят в тонкодисперсной, диспергированной и суспендированной форме, соответственно, или в виде суспензии кристаллов в лекарственном средстве, или на основе «принципа использования одного компонента», если конъюгат антител с токсинами по изобретению заключен в фармацевтическую форму, например, в таблетку или палочку, которая впоследствии имплантируется. Такие имплантаты или лекарственные депо-средства в однокомпонентных и многокомпонентных составах часто состоят из так называемых биоразлагаемых полимеров, таких как, например, полиэфиры молочной кислоты и гликолевой кислоты, полиэфируретаны, полиаминокислоты, поли(мет)акрилаты или полисахариды.
Адъюванты и носители, добавляемые при производстве фармацевтических композиций по настоящему изобретению, приготовленные в виде препаратов для парентерального введения, предпочтительно включают в себя aqua sterilisata (стерилизованную воду); вещества, влияющие на значение рН, такие как, например, органические или неорганические кислоты или основания, а также их соли; буферные вещества для регулирования значений рН; вещества для регулирования изотоничности, такие как, например, хлорид натрия, гидрокарбонат натрия, глюкоза и фруктоза; тензиды и поверхностно-активные вещества, соответственно, и эмульгаторы, такие как, например, неполные эфиры жирных кислот полиоксиэтиленсорбитана (например, Tween®) или, например, сложные эфиры жирных кислот полиоксиэтиленов (например, Cremophor®); жирные масла, такие как, например, арахисовое масло, соевое масло или касторовое масло; синтетические сложные эфиры жирных кислот, например, этилолеат, изопропилмиристат и нейтральное масло (например, Miglyol®), а также полимерные адъюванты, такие как, например, желатин, декстран, поливинилпирролидон, добавки, увеличивающие растворимость органических растворителей, такие как, например, пропиленгликоль, этанол, N,N-диметилацетамид, пропиленгликоль; или комплексообразующие вещества, такие как, например, цитрат и мочевина; консерванты, такие как, например, гидроксипропиловый эфир и метиловый эфир бензойной кислоты, бензиловый спирт; антиоксиданты, такие как, например, сульфит натрия; и стабилизаторы, такие как, например, этилендиаминтетрауксусная кислота (EDTA).
При изготовлении фармацевтических композиций по настоящему изобретению в виде суспензий в предпочтительном варианте осуществления изобретения добавляют загустители для предотвращения осаждения конъюгатов антител с токсинами по изобретению или тензиды и полиэлектролиты для обеспечения ресуспендируемости осадка и/или комплексообразующие вещества, такие как, например, этилендиаминтетрауксусная кислота. Также возможно получить комплексы активного ингредиента с различными полимерами. Примерами таких полимеров являются полиэтиленгликоль, полистирол, карбоксиметилцеллюлоза, Pluronics® или сложный эфир жирной кислоты и полиэтиленгликоль сорбита. Конъюгаты антител с токсинами по изобретению также могут быть введены в жидкие составы в форме соединений включения, например, с циклодекстринами. В частных вариантах осуществления изобретения в качестве дополнительных адъювантов могут быть добавлены диспергирующие вещества. Для производства лиофилизатов могут быть использованы поддерживающие вещества, например, маннит, декстран, сахароза, альбумин человека, лактоза, ПВП или виды желатина.
ПРИМЕРЫ
Далее настоящее изобретение объяснено более подробно при помощи неограничивающих примеров:
Пример 1
Конструирование мутантов цистеина и условия связывания
1.1 Получение антитела
На фиг. 2 показан схематический вид молекулы IgG1 и позициий аминокислотных остатков, мутированных до цистеиновых остатков, и для связывания токсинов. Все антитела были получены в эукариотических клетках Expi293 (Life Technologies) путем транзиентной трансфекции векторами экспрессии, кодирующими тяжелые и легкие цепи (фиг. 3). Последовательности генов с мутациями для Cys замещений синтезировали при помощи GeneArt и вводили в экспрессирующие плазмиды стандартными методами молекулярного клонирования на основе ферментов эндонуклеазы и лигазы. Результаты экспериментов по клонированию проверяли посредством ферментативного рестрикционного анализа и секвенирования (GATC Biotech, Германия). Для трансфекции клетки Expi293 культивировали в колбах Эрленмейера, вращаемых со скоростью 125 об/мин и при 8% CO2 до плотности приблизительно 3,0×106 клеток на мл. Комплексы реагентов ДНК и ПЭИ (PEI, полиэтиленимин) получали в среде Opti-MEM с отношением тяжелых и легких цепей 2:3. После добавления комплексов ДНК : ПЭИ к питательной среде клетки Expi293 инкубировали в течение 24 часов. Клетки центрифугировали при 460 g и комнатной температуре в течение 15 мин и заменяли питательную среду для обеспечения продолжительного изготовления. Отслеживали жизнеспособность клеток, и через 4-6 дней клетки седиментировали, и моноклональные антитела очищали от надосадочной жидкости при помощи системы Bio-Rad FPLC с использованием колонок с белком A (Tosoh Biosience). Агрегаты и эндотоксин удаляли при помощи хроматографической очистки с использованием колонок гель-фильтрации Superdex S-200 (GE Healthcare) и физиологического раствора с фосфатным буфером с рН 7,4. Антитела определяли при помощи SDS-PAGE (электрофорез белков в полиакриламидном геле), УФ-спектроскопии, гель-проникающей хроматографии (SEC-HPLC) и иммуноферментного анализа (ELISA) на эндотоксин. Типичные выходы очищенных антител составляли приблизительно 80-120 мг на литр питательной среды с агрегатами <1%.
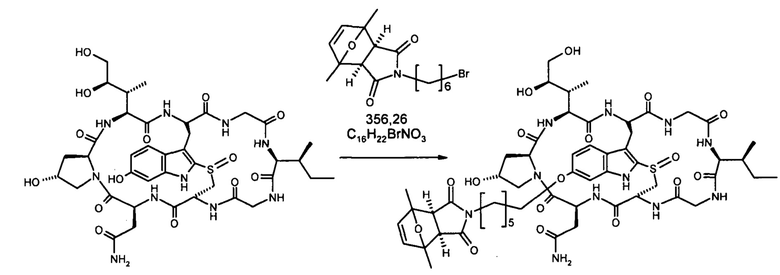
1.2 Связывание малеимид-аматоксин
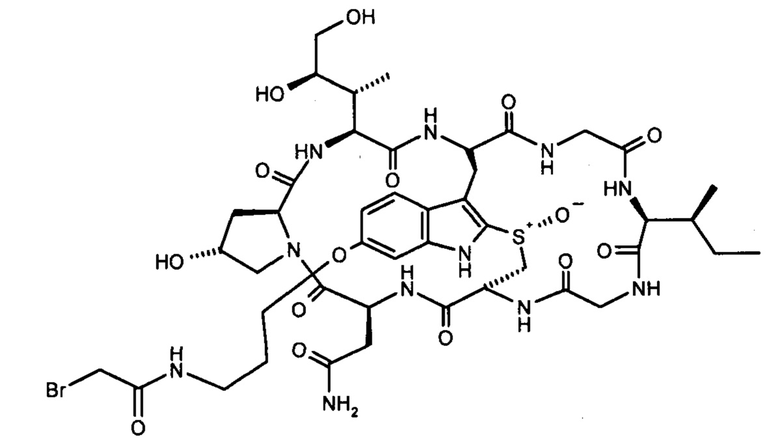
Для конъюгации производных малеимида-аматоксина, например, HDP 30.0880 и HDP 30.1699, антитела с замещениями цистеина довели до 5,0 мг/мл в 1 мМ EDTA в фосфатном буферном солевом растворе (PBS) с рН 7,4 и восстанавливали 40 эквивалентами трихлорэтилфосфата (ТСЕР) в течение 3 часов при 37°С. Восстановленные антитела очищали путем двух последовательных этапов диализа в 1 мМ EDTA в PBS, рН 7,4, и затем межцепочечные дисульфиды окисляли при помощи 20 эквивалентов дегидроаскорбиновой кислоты (dhAA) в течение 4 часов при комнатной температуре. Связывание токсина с замещенными цистеинами проводили посредством добавления 8-15 эквивалентов производных малеимида-аматоксина в течение 1 часа при комнатной температуре с последующей реакцией гашения при помощи 25 эквивалентов N-ацетил-L-цистеина. Конъюгаты аматоксин-ADCs очищали при помощи гельфильтрационной хроматографии с использованием колонок PD-10 или хроматографии G-25 Sephadex® (GE Healthcare). Соотношение лекарственных веществ к антителам (DAR) в ADCs определяли с помощью УФ-спектроскопии при длинах волн 280 нм и 310 нм с использованием коэффициентов экстинкции антител и α-аманитина. Кроме того, DAR определяли нативной LC/MS (фиг. 4(A)) и анализа LC/MS тяжелой/легкой цепи (фиг. 4(B), 4(C)). По данным жидкостной хроматографии с масс-спектрометрией DAR находится в диапазоне от 1,8 до 2,2 единиц аманитина на IgG, и лекарственное вещество расположено исключительно в тяжелой цепи. Качественное состояние ADCs проверяли с помощью SDS-PAGE, вестерн-блоттинга с использованием антисыворотки анти-аманитина, SEC-HPLC, хроматографии с гидрофобным взаимодействием (HIC-HPLC) и обращенно-фазной хроматографии (RP-HPLC). ADCs доводили до 3,0-5,0 мг/мл и хранили в PBS, рН 7,4 при 4°С до дальнейшего использования с клеточными культурами и в моделях in vivo.
Пример 2
6'(6-N-малеимидогексил)-α-аманитин (HDP 30.0880)
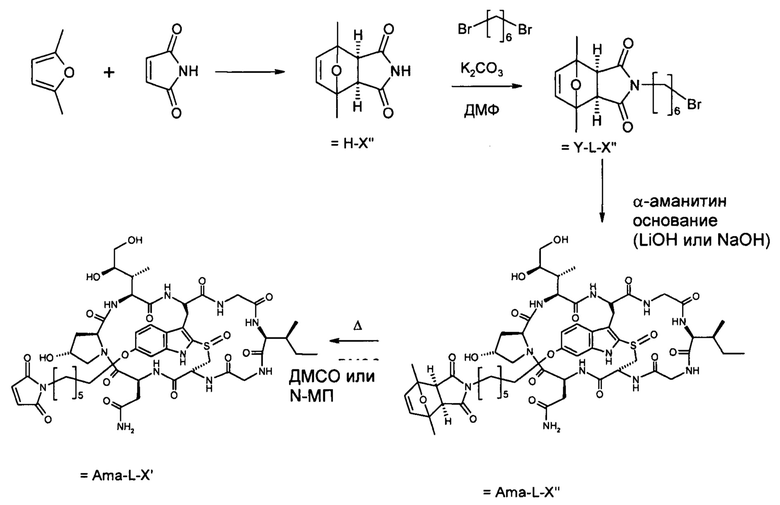
Этап 1: 1,7-диметил-10-окса-4-азатрицикло[5.2.1.02,6] дец-8-ен-3,5-дион, экзо-изомер (HDP 30.0891)
4,00 г (41,2 ммоль) 2,5-диметилфурана и 5,93 г (61,7 ммоль, 1,5 эквивалента) малеимида растворяли в 30 мл диэтилового эфира и нагревали до 90°С в реакторе Parr в течение 12 часов. Полученный осадок отфильтровывали и перекристаллизовывали из метанола:
6,62 г (83%) кристаллов, т.пл. 137°С.
1Н NMR (CDCl3, 500 МГц) δ(ppm): 8,68 (широкий синглет, 1Н), 6,31 (синглет, J, 2Н), 2,88 (синглет, 2Н), 1,73 (синглет, 6Н).
13С NMR (CDCl3, 100 МГц) δ(ppm): 175,04, 140,82, 87,68, 53,77, 15,76.
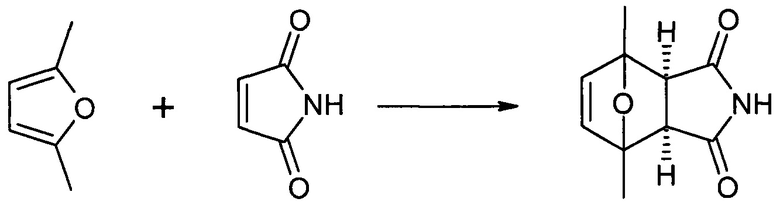
Этап 2: 4-(6-бромгексил)-1,7-диметил-10-окса-4-азатрицикло[5.2.1.02,6]дец-8-ен-3,5-дион, экзо-изомер (HDP 30.0916)
386 мг (2 ммоль) HDP 30.0891 и 1,952 г (8 ммоль) 1,6-дибромгексана растворяли в 20 мл ДМФ, добавляли 276 мг (2 ммоль) карбоната калия и нагревали суспензию до 50°С в течение 3 часов. Затем ДМФ выпаривали, остаток растворяли в 100 мл дихлорметана. Неорганические соли удаляли фильтрованием, к фильтрату добавляли кизельгур (3 г) и растворитель удаляли в вакууме. Остаток очищали посредством хроматографии на силикагеле с элюированием с градиентом от н-гексана до этилацетата, получая HDP 30.0916 (483 мг) в виде воскообразных кристаллов с выходом 68%.
1Н NMR (500 МГц, CDCl3) δ(ppm) 6,31 (s, 2Н), 3,48 (t, J=7,2 Гц, 2Н), 3,39 (t, J=6,8 Гц, 2Н), 2,81 (s, 1Н), 1,90-1,77 (m, 2Н), 1,70 (s, 5Н), 1,64-1,52 (m, 2Н), 1,44 (dddd, J=9,2, 7,4, 6,5, 5,4 Гц, 2Н), 1,35-1,23 (m, 2Н).
13С NMR (126 МГц, CDCl3) δ 174,81, 140,81, 87,52, 52,33, 38,42, 33,65, 32,50, 27,54, 27,33, 25,64, 15,87
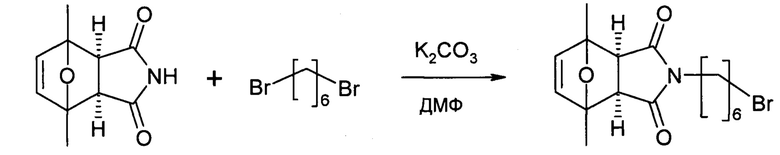
Этап 3: 6'-(6-(1,7-диметил-10-окса-4-азатрицикло[5.2.1.02,6]дец-8-ен-3,5-дион-4-ил-гексил)-α-аманитин (HDP 30.0903)
В атмосфере аргона и при комнатной температуре 34,5 мг (37,5 мкмоль) высушенного в вакууме α-аманитина растворяли в 1000 мкл сухого диметилсульфоксида (ДМСО). Добавляли HDP 30.0916 (106,8 мг, 8 эквивалентов) и 1 М гидроксид натрия (41,2 мкл, 1,1 эквивалента). Через 3 часа при комнатной температуре реакционную смесь подкисляли до рН=5 добавлением 41,2 мкл 1 М раствора уксусной кислоты в ДМСО. Растворитель удаляли в вакууме, и остаток получали препаративной ВЭЖХ на колонке С18 с градиентом от 5 до 100% метанола. Продукт, содержащий фракции, выпаривали до 27,2 мг (59%) HDP 30.0903 в виде бесцветного твердого вещества.
МС (ESI+) 1194,17 [М+Н]+, 1216,10 [M+Na]+
Этап 4: 6'-(6-N-малеимидогексил)-α-аманитин (HDP 30.0880)
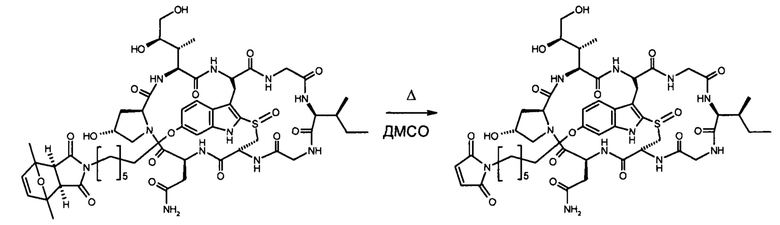
HDP 30.0903 (27,2 мг, 22,7 мкмоль) растворяли в 3000 мкл сухого диметилсульфоксида. Реакционную смесь нагревали до 100°С при перемешивании в течение 1,5 часов. После охлаждения до 40°С ДМСО удаляли в вакууме и остаток очищали препаративной ВЭЖХ указанным выше способом.
Фракцию со временем удерживания 17,3-18,1 минут собирали и растворители выпаривали. Остаток лиофилизировали из 3 мл трет-бутанола с получением 23,6 мг (94%) HDP 30.0880 в виде порошка беловатого цвета.
МС (ESI+) 1098,29 [М+Н]+, 1120,36 [M+Na]+
Используя способы примера 2 с изменениями, очевидными для специалиста, были получены следующие примеры:
Пример 15
6'-O-(6-(6-(N-малеимидо)-гексанамидо)гексил)-α-аманитин (HDP 30.1948)
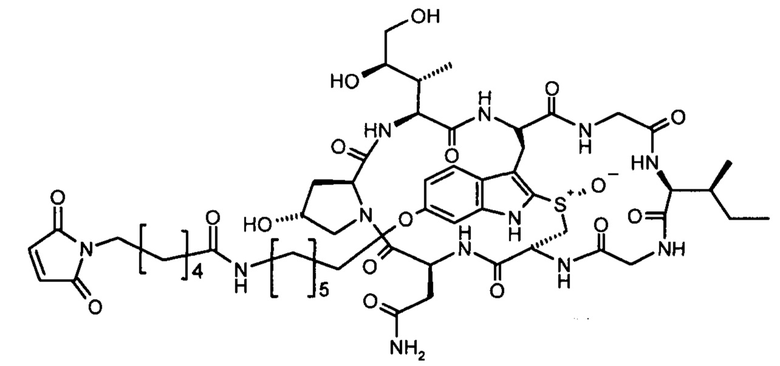
К 10,0 мг (8,83 мкмоль) 6'-O-(-6-аминогексил)-α-аманитина (HDP 30.0134, синтезированный как описано в ЕР 2621536), растворенного в 400 мкл сухого ДМФ, добавляли последовательно 663 мкл 20 мМ 6-(малеимидо)гексановой кислоты N-гидроксисукцинимида эфира (EMCS) в ДМФ и 17,7 мкл 1 М диизопропилэтиламина (DIPEA) в ДМФ. После выдерживания в течение 5 часов при комнатной температуре к реакционной смеси добавляли 100 мкл воды и выпаривали летучие вещества. Неочищенный продукт очищали с помощью RP18 ВЭЖХ с градиентом вода-метанол и чистые фракции лиофилизировали из трет-бутанола/воды: 9,02 мг (84%) HDP 30.1948 в виде бесцветного порошка.
МС (ESI+) получено: 1210,99; расчет: 1211,54 [МН]+ (C55H79N12O17S)
получено: 1233,32; расчет: 1233,52 [M+Na]+ (C55H78N12NaO17S)
Пример 16
6'-O-(6-(6-(N-α-малеимидо)-L-2,3-диаминопропанамидо)гексил)-α-аманитин (HDP 30.1958)
Этап 1: К 8,22 мг (17,66 мкмоль = 2 эквивалента) Mal-L-Dap(Boc)-OH × дициклогексиламин (DCHA) в 700 мкл ДМФ и 17,7 мкл 1 М DIPEA в ДМФ последовательно добавляли 9,19 мг (17,66 мкмоль = 2 эквивалента) РуВор (гексафторфосфат бензотриазол-1-илокси-трис(пирролидино)фосфония) и 6,01 мкл (35,33 мкмоль = 4 эквивалента) DIPEA. Через 1 мин к смеси добавляли 10,0 мг (8,83 мкмоль) 6'-O-(-6-аминогексил)-α-аманитина (HDP 30.0134), растворенного в 200 мкл сухого ДМФ. После выдерживания в течение 2 часов при комнатной температуре к реакционной смеси добавляли 100 мкл воды и выпаривали летучие вещества. Неочищенный продукт очищали посредством RP18 ВЭЖХ и чистые фракции выпаривали: 5,45 мг (48%) HDP 30.1954 в виде аморфного твердого вещества.
МС (ESI+) получено: 1306,58; расчет.: 1306,54 [M+Na]+ (C57H81N13NaO19S)
Этап 2: Полученный на этапе 1 продукт, защищенный Вос-группой, растворяли в 1 мл трифторуксусной кислоты. Через 2 минуты смесь выпаривали досуха при комнатной температуре. Остаток очищали посредством RP18 ВЭЖХ с градиентом от 0,05% трифторуксусной кислоты до метанола и чистые фракции выпаривали: 1,72 мг (31%) HDP 30.1958 в виде аморфного твердого вещества.
MC(ESI+) получено: 1306,58; расчет: 1184,50 [M+Na]+ (C52H74N13O17S)
Пример 17
6'-O-(2-бромоацетамидо)гексил)-α-аманитин (HDP 30.1619)

К 5,03 мг (4,44 мкмоль) 6'-O-(-6-аминогексил)-α-аманитина (HDP 30.0134), растворенного в 400 мкл сухого ДМФ, добавляли последовательно 66,6 мкл 100 мМ N-гидроксисукцинимида эфира бромуксусной кислоты в ДМФ и 88,8 мкл 100 мМ DIPEA в ДМФ. После выдерживания в течение 3 часов при комнатной температуре добавляли 50 мкл воды и реакционную смесь по каплям вливали в 10 мл метил-трет-бутилового эфира (МТВЕ). Осадок выделяли центрифугированием и промывали 10 мл МТВЕ. Неочищенный продукт очищали с помощью RP18 ВЭЖХ с градиентом вода-метанол и чистые фракции лиофилизировали из трет-бутанола/воды: 3,70 мг (73%) HDP 30.1619 в виде бесцветного порошка.
MC(ESI+) получено: 1139,58; расчет: 1138,39 [М+Н]+ (C47H69BrN11O15S)
получено: 1160,42; расчет: 1160,37 [M+Na]+ (C47H68BrN11NaO15S))
Пример 18
6'-O-(2-бромоацетамидо)пропил)-α-аманитин (HDP 30.1618)
Способом из примера 16 для 6'-O-(-3-аминопропил)-α-аманитина, описанного в ЕР 2621536, синтезировали бромацетамид HDP 30.1618:
МС (ESI+) получено: 1096,22; расчет: 1096,34 [МН]+ (C44H63BrN11O15S)
получено: 1118,45; расчет: 1118,32 [M+Na]+ (C44H62BrN11NaO15S)
Пример 19
6'-[6-(6-(4-(5-(метилсульфонил)-1,2,4-оксадиазол-2-ил)-фенилокси)гексиламинокарбонил)-аминогексил)]-α-аманитин (HDP 30.1926)
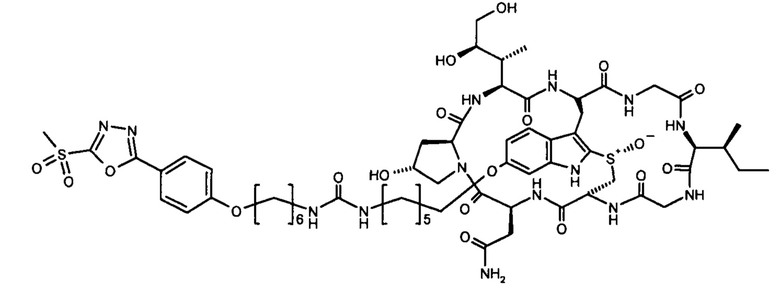
Линкеры метилсульфонил-1,2,4-оксадиазол синтезировали путем изменения способов, описанных у Toda et al. в Angew. Chem. Int. Ed. 2013, 52, 12592-12596.
Этап 1: 1-азидо-6-бромгексан
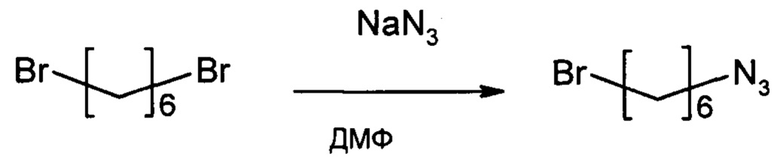
1,6-дибромгексан (7,32 г, 30 ммоль) перемешивали в течение ночи с азидом натрия (1,95 мг, 30 ммоль) в 60 мл ДМФ. Растворитель выпаривали и остаток перемешивали с 100 мл этилацетата в течение 5 минут. Неорганические соли отфильтровали и моноазид отделяли от дибромида и диазида колоночной хроматографией на силикагеле с градиентом от 0 до 20% дихлорметана в гексане с получением 2,26 г (37%) продукта в виде маслянистого вещества.
1Н NMR (500 МГц, CDCl3) δ 3,42 (t, J=6,7 Гц, 2Н), 3,28 (t, J=6,9 Гц, 2Н), 1,88 (dt, J=14,6, 6,8 Гц, 2Н), 1,62 (dt, J=14,3, 7,0 Гц, 2Н), 1,53-1,36 (m, 4Н).
13С NMR (126 МГц, CDCl3) δ 51,43, 33,80, 32,67, 28,82, 27,81, 26,03
Этап 2: Этил 4-[(6-азидогексил)окси]бензоат (HDP 30.1897)
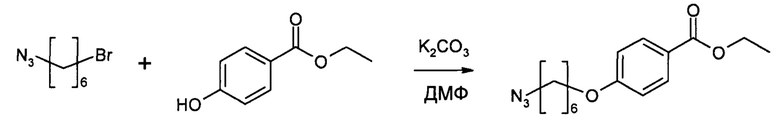
К раствору полученного на этапе 1 продукта (4,122 г, 20 ммоль) в ДМФ (40 мл) добавляли этил-4-гидроксибензоат (3,324 г, 20 ммоль) и K2CO3 (5,528 г, 40 ммоль) при комнатной температуре на 4 часа. Затем реакционную смесь разбавляли 200 мл МТВЕ и 200 мкл воды. Органический слой отделяли и промывали 3×100 мл воды, сушили (MgSO4) и выпаривали досуха. После очистки колоночной хроматографией на силикагеле (гексан/МТВЕ) получали указанное в заголовке соединение (5,199 г, 89%) в виде бесцветного маслянистого вещества.
МС (ESI+) получено: 314,33; расчет: 314,15 [M+Na]+ (C15H21N3NaO3)
Этап 3: 4-[(6-азидогексил)окси]бензоилгидразид (HDP 30.1899)
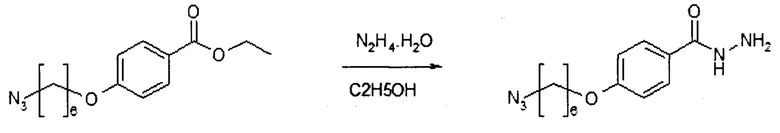
К раствору полученного на этапе 2 продукта (5,19 г, 17,8 ммоль) в этаноле (9,0 мл) добавляли гидразина моногидрат (1459 мкл, 30 ммоль) при комнатной температуре, затем смесь перемешивали с обратным холодильником в течение 22 часов. Реакционную смесь выпаривали под вакуумом. Остаток очищали колоночной хроматографией на силикагеле (градиент от дихлорметана до дихлорметан/этилацетат/метанол 6:3:1) с получением производного бензоилгидразида HDP 30.1899 (1,08 г, 22%) в виде бесцветного твердого вещества.
МС (ESI+) получено: 278,27; расчет: 278,16 [М+Н]+ (C13H20N5O2)
Этап 4: 5-[4-((6-азидогексил)окси)фенил]-1,3,4-оксадиазол-2-тиол (HDP 30.1903)
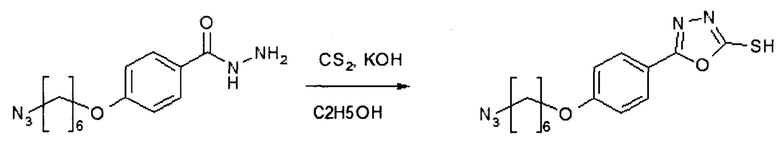
К раствору производного бензоилгидразида (1,08 г, 3,89 ммоль) в этаноле (10,0 мл) добавляли дисульфид углерода (1552 мкл, 25,68 ммоль) и порошковый КОН (218 мг, 3,89 ммоль) при комнатной температуре, а затем раствор перемешивали при 85°С в течение 3 часов. К раствору добавляли этилацетат и 1 М HCl. Органический слой промывали насыщенным бикарбонатом натрия и солевым раствором, сушили над MgSO4, отфильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат) с получением указанного в заголовке соединения HDP 30.1903 (1,18 г, выход 95%) в виде бесцветного твердого вещества.
Этап 5: 5-[4-((6-азидогексил)окси)фенил]-5-(метилсульфанил)-1,3,4-оксадиазол (HDP 30.1905)
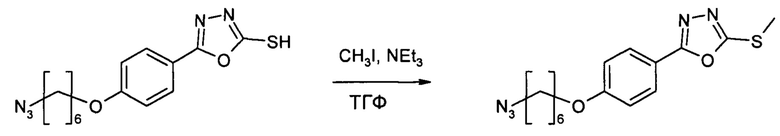
К раствору полученного на этапе 4 тиола (1,18 г, 3,69 ммоль) в ТГФ (15 мл) добавляли иодид метила (257 мкл, 4,13 ммоль) и триэтиламин (NEt3) (625 мкл, 4,51 ммоль) при 0°С. Затем смесь перемешивали в течение 1 часа при комнатной температуре. Затем добавляли воду (50 мл) и этилацетат (100 мл) и смесь обесцвечивали 10% раствором тиосульфата натрия (10 мл). Органический слой отделяли, промывали солевым раствором (50 мл) и сушили над MgSO4. После фильтрации органический растворитель удаляли в вакууме и остаток очищали на силикагеле гексаном / МТВЕ с получением указанного в заголовке соединения HDP 30.1905 (1,03 мг, выход 84%) в виде белого твердого вещества.
МС (ESI+) получено: 334,27; расчет: 334,13 [М+Н]+ (C15H20N5O2S)
получено: 356,29; расчет: 356,12 [M+Na]+ (C15H19N5NaO2S)
Этап 6: 5-[4-((6-азидогексил)окси)фенил]-5-(метилсульфонил)-1,3,4-оксадиазол (HDP 30.1910)
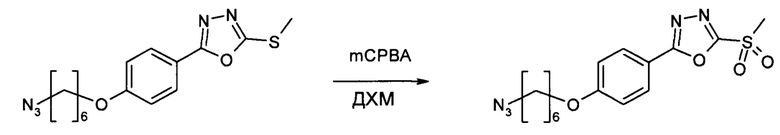
К раствору полученного на этапе 5 продукта (1,00 г, 3,00 ммоль) в дихлорметане (100 мл) добавляли м-хлорнадбензойную кислоту (mCPBA) (68% по весу, 2,79 мг, 3,66 ммоль) при 0°С, затем смесь перемешивали в течение 24 часов при комнатной температуре. Добавляли сульфат магния, нерастворимый материал удаляли фильтрованием и растворитель удаляли в вакууме. Остаток очищали колоночной хроматографией на силикагеле (гексан/ МТВЕ) с получением указанного в заголовке соединения HDP 30.1910 (858 мг, 78%).
МС (ESI+) получено: 366,00; расчет: 366,12 [М+Н]+ (C15H20N5O4S)
получено: 388,19; расчет: 388,11 [M+Na]+ (C15H19N5NaO4S)
Этап 7: 5-[4-(((6-сукцинимидилоксикарбониламино)гексил)окси)фенил]-5-(метилсульфонил)-1,3,4-оксадиазол (HDP 30.1916)
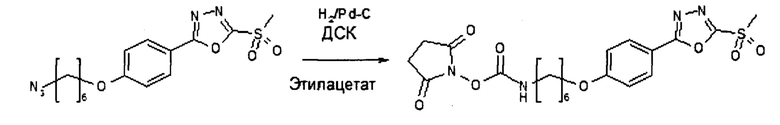
К раствору полученного на этапе 6 продукта (370 мг, 1,01 ммоль) в этилацетате (100 мл) добавляли N,N'-дисукцинимидилкарбонат (519 мг, 2,02 ммоль). Аппарат продували аргоном, добавляли палладий (10% на активированном угле) и затем перемешивали в течение ночи в атмосфере водорода. После фильтрации и выпаривания неочищенный продукт очищали на силикагеле с градиентом от гексана до этилацетата. Чистые фракции объединяли и лиофилизировали из 1,4-диоксана с получением сукцинимидилкарбамата HDP 30.1916 (280 мг, 58%) в виде бесцветного порошка.
МС (ESI+) получено: 481,10; расчет: 481,14 [М+Н]+ (C20H25N4O8S)
1Н NMR (500 МГц, CDCl3) d 8,08-8,01 (m, 2Н), 7,06-6,99 (m, 2Н), 5,57 (t, J=5,9 Гц, 1Н), 4,05 (t, J=6,4 Гц, 2H), 3,51 (s, 3Н), 3,28 (q, J=6,8 Гц, 2H), 2,83 (s, 4H), 1,88-1,77 (m, 2H), 1,67-1,39 (m, 6H).
13C NMR (126 МГц, CDCl3) d 170,05, 166,73, 163,11, 161,53, 151,44, 129,68, 115,28, 114,03, 68,14, 42,98, 41,90, 29,39, 28,85, 26,23, 25,55, 25,47.
Этап 8: 6'-[6-((6-((4-(5-(метилсульфонил)-1,2,4-оксадиазол-2-ил)фенил)окси)гексил)-аминокарбониламино)гексил]-α-аманитин (HDP 30.1926)
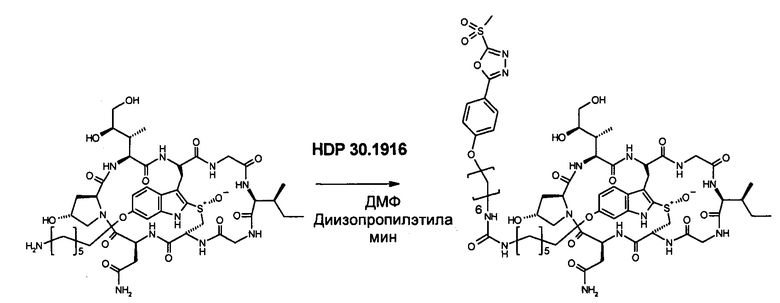
К 10,0 мг (8,83 мкмоль) 6'-O-(-6-аминогексил)-α-аманитина (HDP 30.0134, синтезированного как описано в ЕР 2621536), растворенного в 500 мкл сухого ДМФ, добавляли последовательно 8,49 мг (17,66 мкмоль) растворенного в 500 мкл ДМФ и 6,01 мкл (35,33 мкмоль) диизопропилэтиламина DIPEA. После выдерживания в течение 18 часов при комнатной температуре летучие вещества выпаривали. Неочищенный продукт очищали с помощью RP18 ВЭЖХ с градиентом от воды до метанола и чистые фракции лиофилизировали из трет-бутанола/воды: 7,04 мг (58%) HDP 30.1926 в виде бесцветного порошка.
МС (ESI+) получено: 1383,62; расчет: 1383,57 [МН]+ (C61H87N14O19S2)
получено: 1405,54; расчет: 1405,55 [M+Na]+ (C61H86N14NaO19S2)
Пример 20
6'[(3-малеидопропанамидо)-аминокапроновая кислота-Val-Cit-РАВ]-α-аманитин (HDP 30.1426)
Дипептид п-аминобензилбромиды синтезировали из соответствующих бензиловых спиртов путем адаптации способов, описанных у Jeffrey et al. в J. Med. Chem. 2005, 48, 1344-1358. Общую процедуру можно представить на примере следующей схемы:
Этап 1: Вос-аминокапроновая кислота-Val-Cit-PAB-OH
(HDP 30.1267)
Получение валин-цитруллин-n-аминобензилового спирта (H-Val-Cit-РАВ-ОН) и N-гидроксисукцинимид-сложного эфира N-Boc-аминокапроновой кислоты (Boc-Ahx-NHS) описано у Firestone et al. в US 6,214,345. При помощи данных методов получали неочищенный H-Val-Cit-РАВ-ОН из 4,79 г (7,96 ммоль) Fmoc-Val-Cit-РАВ-ОН и растворяли в 50 мл ДМФ. Добавляли Вос-аминокапроновая кислота-NHS (2,88 г, 8,76 ммоль) и 1489 мкл (8,76 ммоль) N-этилдиизопропиламина и перемешивали при комнатной температуре в течение ночи. После выпаривания ДМФ остаток перемешивали с 100 мл МТВЕ в течение ночи, и твердые вещества выделяли центрифугированием. Осадок ресуспендировали в МТВЕ и снова центрифугировали. Продукт сушили в вакууме до 4,44 г (выход 94%) порошка слегка коричневатого цвета.
МС (ESI+) получено: 615,19; расчет: 615,35 [M+Na]+ (C29H48N6NaO7)
Этап 2: Вос-аминокапроновая кислота-Val-Cit-PAB-O-трет-бутилдиметилсилил (HDP 30.1362)
Полученный на этапе 1 продукт (4,44 г, 7,49 ммоль) растворяли в 20 мл ДМФ и добавляли 6,37 мкл (37,45 ммоль) N-этилдиизопропиламина и 3,39 г (22,47 ммоль) трет-бутилдиметилхлорсилана (TBDMSCl). Через 3 часа ДМФ выпаривали и остаток разделяли между 120 мл этилацетата/метанола 5:1 и 100 мл 0,2 М лимонной кислоты. Органический слой промывали водой и насыщенным бикарбонатом натрия, сушили (над MgSO4) и выпаривали при пониженном давлении. Неочищенный продукт элюировали из 120 г силикагеля с градиентом от 0 до 10% метанола в дихлорметане. Чистые фракции соединяли и выпаривали до 2,50 г (47%) продукта в виде твердого вещества.
МС (ESI+) получено: 729,29; расчет: 729,43 [M+Na]+ (C35H62N6NaO7Si)
Этап 3: Вос-аминокапроновая кислота-Val-Cit(SEM)-PAB-O-трет-бутилдиметилсилил (HDP 30.1368)
К раствору полученного на этапе 2 продукта (2,50 мг, 3,546 ммоль) в ТГФ (40 мл) добавляли NaH (142 мг 60% дисперсии в минеральном масле, 3,546 ммоль). Через 15 минут добавляли неразбавленный 2-(триметилсилил)-этоксиметилхлорид (SEMCl) (703 мкл, 3,546 ммоль), и реакционную смесь перемешивали в течение 8 часов. К реакционной смеси добавляли диатомовую землю (10 г) и летучие вещества удаляли при пониженном давлении. Оставшиеся твердые вещества наносили на верхнюю часть колонки с силикагелем и элюировали с градиентом от 0 до 10% метанола в дихлорметане. Чистые фракции соединяли и выпаривали до 637 мг (21%) аморфного указанного в заголовке соединения.
МС (ESI+) получено: 859,34; расчет: 859,52 [M+Na]+ (C41H76N6NaO8Si2)
Этап 4: Вос-аминокапроновая кислота-Val-Cit(SEM)-РАВ-ОН
(HDP 30.1370)
К раствору полученного на этапе 3 продукта (567 мг, 0,677 ммоль) в ТГФ (20 мл) добавляли фторид тетрабутиламмония (ТБАФ, TBAF)(833 мкл 1,0 М раствора, 0,833 ммоль, 1,2 эквивалента). Через 1 час к реакционной смеси добавляли диатомовую землю (1,5 г) и летучие вещества удаляли при пониженном давлении. Оставшиеся твердые вещества наносили на верхнюю часть колонки с силикагелем и элюировали с градиентом от 0 до 10% метанола в хлороформе. Чистые фракции соединяли и выпаривали до 279 мг (57%) продукта в виде белого твердого вещества.
МС (ESI+) получено: 745,28; расчет: 745,43 [M+Na]+ (C35H62N6NaO8Si)
Этап 5: Вос-аминокапроновая кислота-Cit(SEM)-РАВ-Br
(HDP 30.1381)
К раствору полученного на этапе 4 продукта (100 мг, 139 ммоль) в дихлорметане (ДХМ) (5 мл) добавляли трифенилфосфин (73 мг, 2 эквивалента), затем тетрабромид углерода (92 мг, 2 эквивалента). Через 1 час к реакционной смеси добавляли диатомовую землю (1 г) и летучие вещества удаляли при пониженном давлении. После элюирования из 12 г силикагеля с градиентом от 0 до 10% метанола в дихлорметане получали 61 мг (56%) указанного в заголовке продукта в виде маслянистого вещества.
MC(ESI+) получено: 807,15/809,17; расчет: 807,35/809,34 [M+Na]+ (C35H61BrN6NaO7Si)
Этап 6: 6'-[Вос-аминокапроновая кислота-Cit(SEM)-РАВ]-α-аманитин (HDP 30.1383)
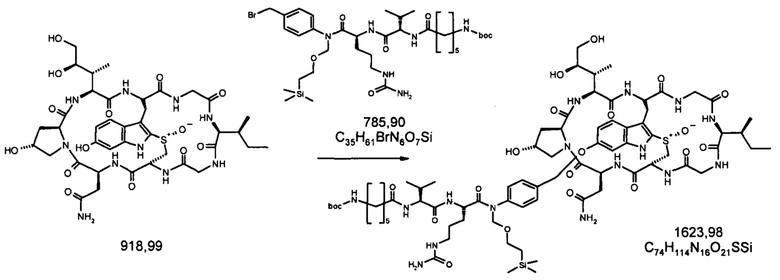
В атмосфере аргона и при комнатной температуре 44 мг (47,9 мкмоль) высушенного в вакууме α-аманитина растворяли в 1000 мкл сухого диметилсульфоксида (ДМСО). Добавляли полученный на этапе 5 продукт (61 мг, 78,1 мкмоль) и 1 М гидроксид натрия (52,7 мкл, 1,1 эквивалента). После выдерживания в течение 4 часов при комнатной температуре реакционную смесь подкисляли до рН=5 добавлением 52,7 мкл 1 М раствора уксусной кислоты в ДМСО. Растворитель удаляли в вакууме и остаток очищали препаративной ВЭЖХ на колонке С18 с градиентом от 5 до 100% метанола. Продукт, содержащий фракции, выпаривали до 25,1 мг (32%) HDP 30.1383 в виде бесцветного твердого вещества.
МС (ESI+) получено: 1646,40; расчет: 1645,77 [M+Na]+ (C74H114N16NaO21SSi)
Этап 7: 6'-[Н-аминокапроновая кислота-Cit-РАВ]-α-аманитин (HDP 30.1388)
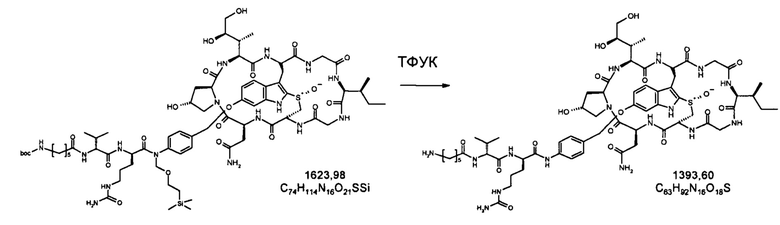
Полученный на этапе 6 продукт (18,4 мг, 11,3 мкмоль), защищенный Boc и SEM-группами, растворяли в 2 мл трифторуксусной кислоты. Через 2 минуты смесь выпаривали досуха при комнатной температуре, повторно растворяли в 2 мл воды и доводили до рН 10, добавляя по каплям 3,2% аммиака. Полученную суспензию сублимировали, применяли RP18 ВЭЖХ с градиентом от 5 до 100% метанола в 0,05% трифторуксусной кислоты, чистые фракции выпаривали и лиофилизировали из 2 мл воды: 12,1 мг (71%) HDP 30.1388 в виде бесцветного порошка.
МС (ESI+) получено: 1393,42; расчет: 1393,66 [М+Н]+ (C63H93N16O18S)
Этап 8:
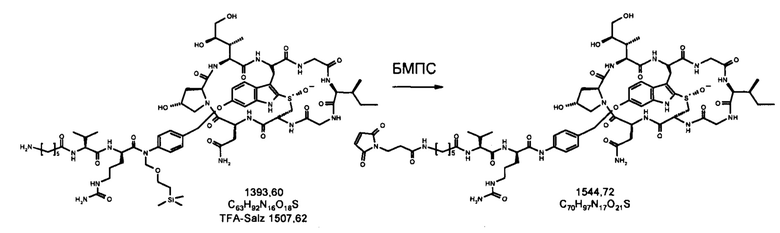
К 9,98 мг (6,62 мкмоль) полученного на этапе 7 продукта, растворенного в 500 мкл сухого ДМФ, последовательно добавляли 5,29 г (19,86 мкмоль) N-гидроксисукцинимидного эфира 3-(малеимидо) пропановой кислоты (BMPS) в 500 мкл ДМФ и 3,38 мкл (19,86 мкл) DIPEA. После выдерживания в течение 2 часов при комнатной температуре к реакционной смеси добавляли 100 мкл воды и выпаривали летучие вещества. Неочищенный продукт очищали с помощью RP18 ВЭЖХ с градиентом от воды до метанола и чистые фракции лиофилизировали из трет-бутанола/воды: 5,43 мг (53%) указанного в заголовке продукта 6'-[(3-малеидопропанамидо)-аминокапроновая кислота-Cit-РАВ]-α-аманитин в виде бесцветного порошка.
МС (ESI+) получено: 1544,25; расчет: 1544,68 [М+Н]+ (C70H98N17O21S)
получено: 1566,44; расчет: 1566,67 [M+Na]+ (C70H97N17NaO21S)
Пример 21
6'-[Н-Val-Ala-РАВ]-α-аманитин (HDP 30.1702)
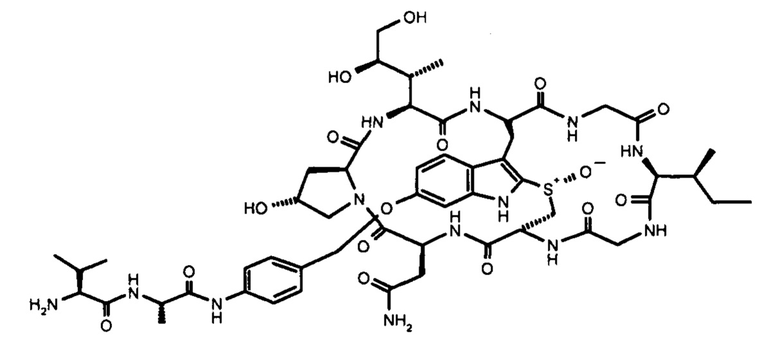
Повторением из способов примера 20 этапов 1-7 с H-Val-Ala-PAB-OH в качестве исходного материала получали указанное в заголовке вещество в виде бесцветного порошка:
МС (ESI+) получено: 1194,58; расчет: 1194,53 [М+Н]+ (C54H76N13O16S)
получено: 1216,75; расчет: 1216,51 [M+Na]+ (C54H75N13NaO16S)
Исходный материал H-Val-Ala-PAB-OH может быть получен общими способами, применяемыми в химии пептидов, и приведен в качестве примера у Howard et al. в US 2011/0256157.
Пример 22
6'-((3-малеидопропанамидо)-Val-Ala-РАВ]-α-аманитин
(HDP 30.1699)
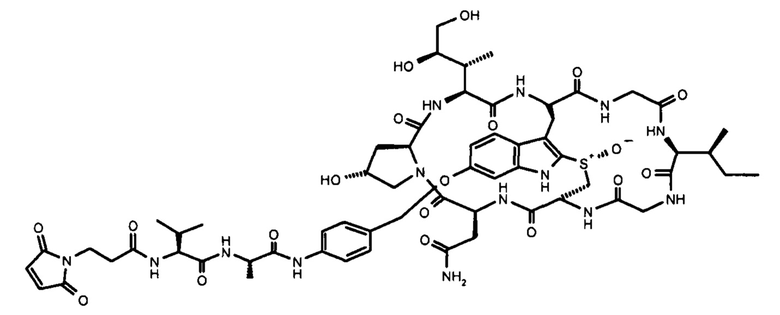
Применением способа примера 20, этапа 8 с 6'-[H-Val-Ala-PAB]-α-аманитином из примера 21 получали указанное в заголовке вещество с выходом 81% в виде бесцветного порошка:
МС (ESI+) получено: 1345,48; расчет: 1345,55 [МН]+ (C61H81N14O19S)
получено: 1368,17; расчет: 1367,53 [M+Na]+ (C61H80N14NaO19S)
Пример 23
6'-O-[((N-α-малеимидо)-L-2,3-Диаминопропанамидо)-Val-Ala-РАВ]-α-аманитин
(HDP 30.1957)
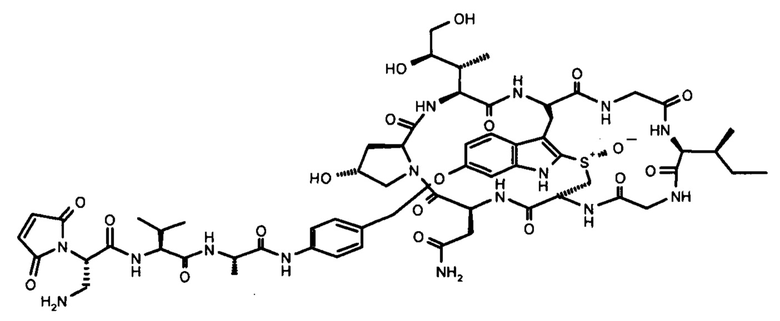
Применением способа примера 16 с 6'-[H-Val-Ala-РАВ]-α-аманитином из примера 21 получали указанное в заголовке вещество с выходом 39% в виде бесцветного порошка:
MC (ESI+) получено: 1345,48; расчет: 1360,56 (C61H82N15O19S)
Пример 24
6'-((2-бромацетамидо)-Val-Ala-РАВ]-α-аманитин (HDP 30.1704)

Применением способа из примера 17 с 6'-[H-Val-Ala-PAB]-α-аманитином из примера 21 получали указанное в заголовке вещество с выходом 26% в виде бесцветного порошка:
МС (ESI+) получено: 1314,28; расчет: 1314,45 [М+Н]+ (C56H77N13O17S)
получено: 1336,39; расчет: 1336,43 [M+Na]+ (C56H76N13NaO17S)
Пример 25
6'-((6-(4-(5-(метилсульфонил)-1,2,4-оксадиазол-2-ил)-фенилокси)гексиламинокарбонил)-Val-Ala-РАВ)-α-аманитин (HDP 30.1917)
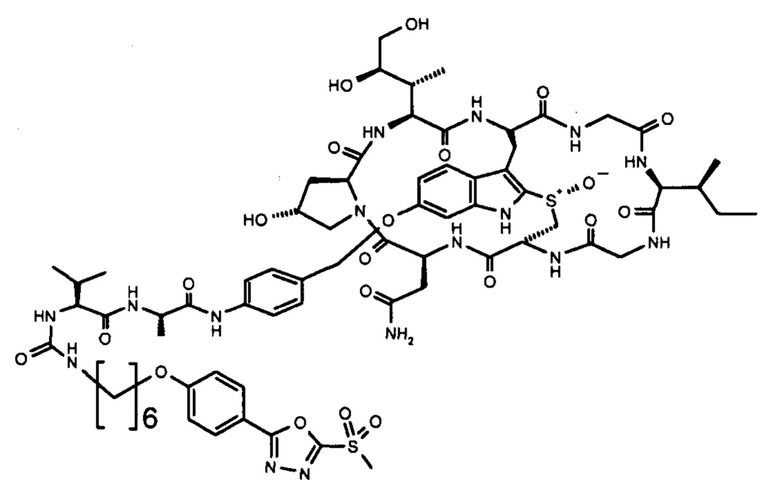
Применением способа из примера 19 с 6'-[H-Val-Ala-PAB]-α-аманитином из примера 21 получали указанное в заголовке вещество с выходом 44% в виде бесцветного порошка:
МС (ESI+) получено: 802,63 расчет: 802,30 [M+2Na]2+ (C70H94N16Na2O21S2)
Пример 26
6'-((6-(4-(5-(метилсульфонил)-1,2,4-оксадиазол-2-ил)-фенилокси)гексиламинокарбонил)-аминокапроновая кислота-Val-Ala-РАВ)-α-аманитин (HDP 30.1930)
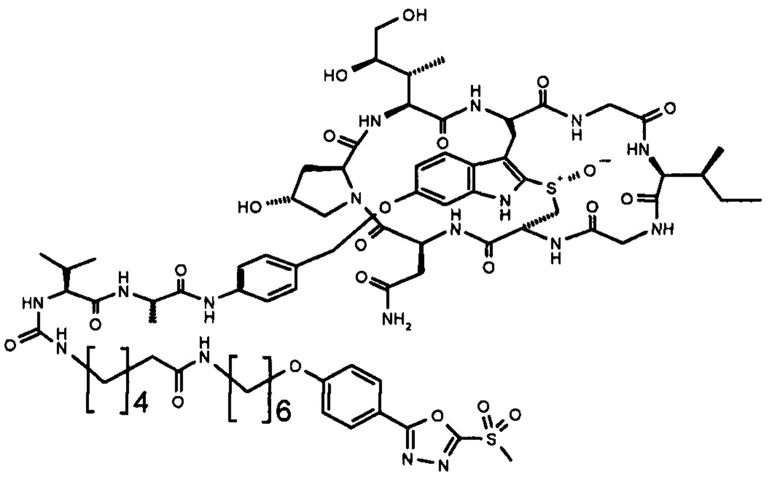
Применением способа из примера 19 с 6'-[Н-аминокапроновая кислота-Val-Ala-РАВ]-α-аманитином, полученным этапами 1-7 способа из примера 19 с заменой цитруллина аланином, получали указанное в заголовке вещество с выходом 37% в виде бесцветного порошка:
МС (ESI+) получено: 859,02; расчет: 858,85 [M+2Na]2+ (C76H105N17Na2O22S2)
Пример 27
6'-O-[3-(5-нитропиридин-2-илдисульфанил)пропил)-α-аманитин HDP 30.0951
Этап 1: 6'-O-(3-S-тритилсульфанил-пропил)-α-аманитин HDP 30.0517
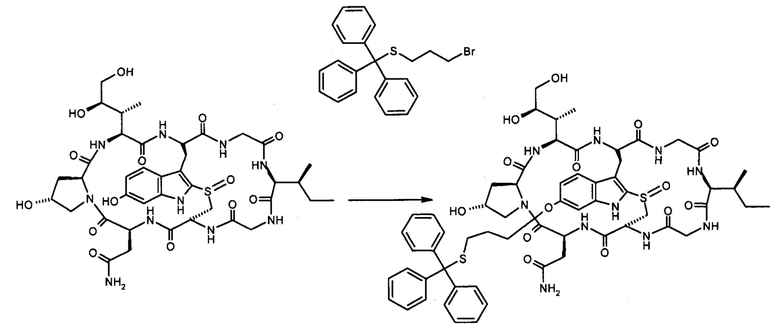
В атмосфере аргона 46 мг (50 мкмоль) высушенного в вакууме α-аманитина растворяли в 2500 мкл сухого диметилсульфоксида (ДМСО). Добавляли 3-(S-тритил)-меркаптопропил-1-бромид (159 мг, 8 эквивалентов), затем добавляли 60 мкл 1 М раствора гидроксида натрия. После выдерживания в течение 1,5 часов при комнатной температуре реакционную смесь подкисляли до рН=5 добавлением 50 мкл 1М уксусной кислоты в ДМСО и растворитель выпаривали. Остаток растворяли в 200 мкл метанола и добавляли по каплям в пробирку для центрифугирования, наполненную 10 мл трет-бутилметилового эфира (МТВЕ). Полученный осадок охлаждали до 0°С в течение 10 минут и выделяли центрифугированием (4000×g), затем промывали 10 мл МТВЕ. Надосадочные жидкости сливали и осадок растворяли в 750 мкл метанола и очищали в виде трех порций препаративной ВЭЖХ на колонке С18 (250×21,2 мм, Luna RP-18, 10 мкм, 100 А). Растворитель А: вода, растворитель В: метанол; Градиент: 0 мин. 5% В; 5 мин 5% В; 20 мин 100% В; 25 мин 100% В; 27 мин 5% В, 35 мин 5% В; Расход 30 мл/мин. Фракции со временем удерживания 21,1-21,8 минут собрали и растворители выпаривали до 36,5 мг (59%) HDP 30.0517 в виде бесцветного твердого вещества.
МС (ESI+) 1234,8 [М+Н]+, 1257,3 [M+Na]+
Этап 2: 6'-O-[3-(5-нитропиридин-2-илдисульфанил)пропил)-α-аманитин HDP 30.0951
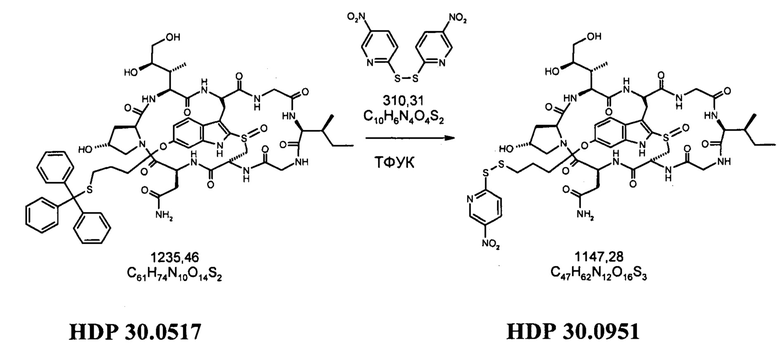
К полученному на этапе 1 продукту (5,00 мг, 4,05 мкмоль) добавляли 2,2'-дитиобис(5-нитропиридин), DTNP (6,28 мг, 5 эквивалентов), растворенный в 200 мкл трифторуксусной кислоты ТФУК (TFA). Через 4 минуты летучие вещества отгоняли и остаток выпаривали вместе с 1000 мкл метанола. Неочищенный продукт очищали ВЭЖХ, аналогично способу этапа 1. Фракции со временем удерживания 18,46-19,28 минут собирали, растворители выпаривали, и остаток лиофилизировали из 2 мл трет-бутанола до 2,99 мг (64%) HDP 30.0951 в виде твердого вещества светло-желтого цвета.
МС (ESI+) 1146,97 [М+Н]+, 1169,17 [M+Na]+
Пример 28
Условия проведения эксперимента для фиг. 5-14
Испытания жизнеспособности клеток, основанные на включении BrdU (фиг. 11, 12, 13)
Для оценки влияния соединений на линии опухолевых клеток, экспрессирующих антиген, высевали материал в объеме 2500 клеток/лунку в 90 мкл среды. На следующий день добавляли 10 мкл среды, содержащей различные концентрации конъюгатов антитело-лекарственное средство. Через 72 или 96 часов воздействия лекарственного средства проводили испытание на включение BrdU (иммуноферментный анализ на пролиферацию клеток, BrdU, Roche). Хемолюминесценцию измеряли с использованием хемилюминомера FLUOstar Optima (BMG LABtech). Значения EC50 для каждого соединения определяли анализом сигмоидальной кривой зависимости «доза-эффект» с использованием программного обеспечения Graphpad Prism 4.0.
Испытания жизнеспособности клеток на основании WST-I (фиг. 8)
Для оценки влияния соединений на клетки ТНР-1 с экспрессией Fcg рецептора высеивали материал в объеме 2500 клеток/лунку в 90 мкл среды. На следующий день добавляли 10 мкл среды, содержащей различные концентрации конъюгатов антитело- лекарственное средство. Через 96 часов после применения лекарственного средства в каждую лунку добавляли 10 мкл агента пролиферации клеток WST-1 (Roche). После дополнительной инкубации в течение 4-24 часов определяли абсорбцию при 440 нм/660 нм с использованием хемилюминомера FLUOstar Optima (BMG LABtech). Значения EC50 для каждого соединения определяли анализом сигмоидальной кривой зависимости «доза-эффект» с использованием программного обеспечения Graphpad Prism 4.0.
Испытание связывания с поверхностью клеток (фиг. 6)
100 мкл суспензии клеток в PBS, содержащей 1×106 антиген-экспрессирующих опухолевых клеток, инкубировали в течение 30 минут при 4°С с использованием или без использования 0,1-50 мкг/мл антитела или конъюгата антитело-лекарственное средство. Для детекции использовали фрагмент козьего антитела F(ab')2 к человеческому IgG (H+L)-Alexa Fluor 488 (Dianova). Фиксированные окрашенные клетки анализировали FACS (флуоресцентно-активированной сортировкой клеток) на устройстве BD FACScan. Среднюю интенсивность флуоресценции образца рассчитывали при помощи программного обеспечения BD CellQuest Pro и строили график ее зависимости от концентрации IgG после вычитания значения контрольного образца для получения кривых связывания.
SDS-PAGE с окрашиванием синим Кумасси и вестерн-блоттингом (фиг. 7)
Антитела и конъюгаты антитело-лекарственное средство анализировали с помощью восстановительного и невосстановительного SDS-PAGE с последующим окрашиванием синим Кумасси или обнаружением при помощи вестерн-блоттинг детекции полезной нагрузки аманитина в соответствии со стандартными методами. Для детекции использовали поликлональную кроличью сыворотку, что позволяет обнаруживать аманитин и соединения аманитин-линкер, конъюгированные с белками.
Эксперименты по переносимости и эффективности на мышах (рис. 9, 14)
Максимальную переносимую дозу (МПД), определяемую как доза, которая приводит к индивидуальной потере веса тела, не превышающей 10%, оценивали для однократных внутривенных введений конъюгатов антитело-лекарственное средство. Для моделей ксенотрансплантата 5×106 опухолевых клеток (номер раннего пассажа), суспендированных в 240 мкл среды, подкожно вводили в правый бок самок голых мышей линии NMRI (Janvier) в возрасте 7-8 недель. После достижения опухолями среднего объема от 100 до 200 мм3 животных случайным образом распределяли в терапевтические группы по 8 животных на группу. Всем животным выполняли процедуру одноразового внутривенного введения. Концентрации эндотоксинов <1 EU (единица эндотоксина) на мг белка были продемонстрированы для всех партий, используемых для экспериментов in vivo, при помощи системы Endosafe-PTS (Charles River). Вводимый объем составлял 10 мл/кг массы тела, в качестве контроля растворителем PBS. Рост опухоли измеряли с помощью штангенциркуля, объем опухоли рассчитывали по формуле
Тобъем = (больший диаметр × (меньший диаметр)2 × 0,5). Мышей умерщвляли в момент агонии или в указанные моменты времени ингаляцией CO2. Все исследования на животных выполнялись в соответствии с немецкими стандартами защиты прав животных (GV-SOLAS) и были одобрены ответственным советом (Региональный совет Карлсруэ, отдел 35). Статистический анализ выполняли с использованием программного обеспечения Prism (GraphPad Software, Inc.) и однофакторного дисперсионного анализа.
В таблице 1 ниже показан терапевтический индекс различных Her2-аманитин-ADCs:
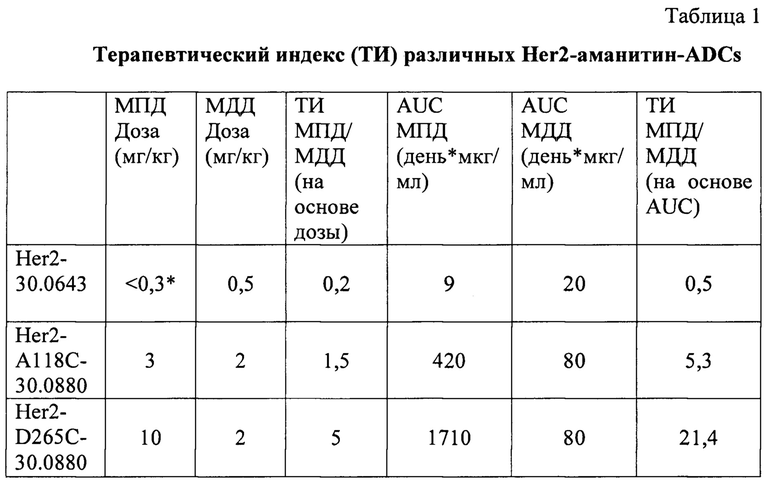
МПД - максимальная переносимая доза; МДД - минимальная допустимая доза; AUC - площадь под кривой
*: принято равным 0,1 для расчетов
Исследование переносимости на яванских макаках (фиг. 10)
Исследования токсичности на не человекообразных приматах (НЧП) проводились на самках яванских макак. Все протоколы исследований были одобрены испытательным органом «Комитет по институциональному контролю и использованию животных». Исследования однократной токсичности без соблюдения требований Надлежащей лабораторной практики выполняли с использованием внутривенного (в/в) введения повышающихся доз в течение трех недель. В каждом исследовании оценивали комплексные показатели токсикологии, в том числе физические обследования, вес тела, потребление пищи, поведение, клиническую химию, анализ мочи и гематологию. В различные моменты времени производили забор образцов крови. В качестве примера переносимости динамика во времени параметра ЛДГ в крови показана для (А) Her2-30.0643; (В) Her2-30.1465; (С) Her2-A118C-30.0880; (D) Her2-D265C-30.0880. Дозы: (i) 0,3 мг/кг; (ii) 1 мг/кг; (iii) 3 мг/кг; (iv) 10 мг/кг;
Масс-спектрометрия (LC-MS) (фиг. 4, 5)
Перед началом анализа МС антитела и конъюгаты дегликозилировали и восстанавливали согласно стандартным протоколам. Таким образом, после дегликозилирования с помощью PNGase F и диализа растворы антител и конъюгатов антитело-лекарственное средство осаждали посредством добавления ацетонитрила, центрифугировали, а осадок ресуспендировали в 6 М растворе гидрохлорида гуанидиния. Для анализа легких и тяжелых цепей ресуспендированные растворы восстанавливали добавлением 20 мМ дитиотреитола в течение 30 минут при 56°С. Дегликозилированные, а также восстановленные антитела и конъюгаты антитело-лекарственное средство затем анализировали с помощью нано- LC-MS с использованием масс-спектрометрической ионной ловушки Orbitrap Elite (Thermo Fisher Scientific). Способы оптимизировали для обеспечения наличия неповрежденных и восстановленных антител перед анализом. Полученные масс-спектры окончательно развернули при помощи соответствующего программного обеспечения (ProMass, Thermo Fisher Scientific) для интерпретации данных.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЙ СПОСОБ СИНТЕЗА АМАНИТИНОВ | 2018 |
|
RU2792210C2 |
| ПРОИЗВОДНЫЕ АМАТОКСИНА | 2014 |
|
RU2695370C2 |
| КОНЪЮГАТЫ АМАТОКСИНОВ С УЛУЧШЕННЫМИ ЛИНКЕРАМИ | 2011 |
|
RU2601411C2 |
| КОНЪЮГАТЫ АМАТОКСИНА С УЛУЧШЕННЫМИ СВЯЗЯМИ | 2012 |
|
RU2575854C2 |
| СИНТЕЗ (S)-6-ГИДРОКСИТРИПТОФАНА И ЕГО ПРОИЗВОДНЫХ | 2019 |
|
RU2810786C2 |
| КОНЪЮГАТ АНТИТЕЛА С АМАТОКСИНОМ НЕПРИРОДНОГО ТИПА | 2018 |
|
RU2840926C2 |
| МАТЕРИАЛЫ И СПОСОБЫ, СВЯЗАННЫЕ С ЛИНКЕРАМИ ДЛЯ ПРИМЕНЕНИЯ В КОНЪЮГАТАХ ЛЕКАРСТВЕННОГО СРЕДСТВА И БЕЛКА | 2015 |
|
RU2737553C2 |
| КОНЪЮГАТ АНТИТЕЛА С АМАТОКСИНОМ НЕПРИРОДНОГО ТИПА | 2018 |
|
RU2798981C2 |
| НЕЛИНЕЙНЫЕ САМОРАСЩЕПЛЯЮЩИЕСЯ ЛИНКЕРЫ И ИХ КОНЪЮГАТЫ | 2017 |
|
RU2755899C2 |
| КОНЪЮГАТ, ВКЛЮЧАЮЩИЙ ЛИГАНД, СПЕЙСЕР, ПЕПТИДНЫЙ ЛИНКЕР И БИОМОЛЕКУЛУ | 2020 |
|
RU2807081C2 |
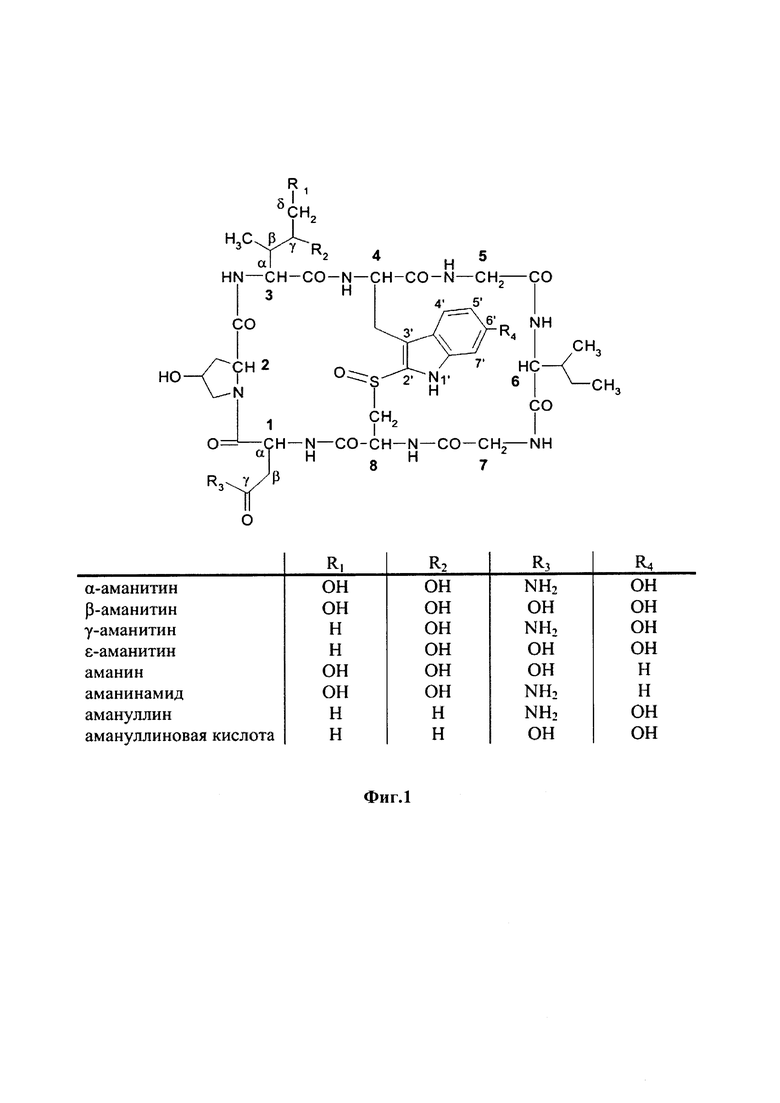
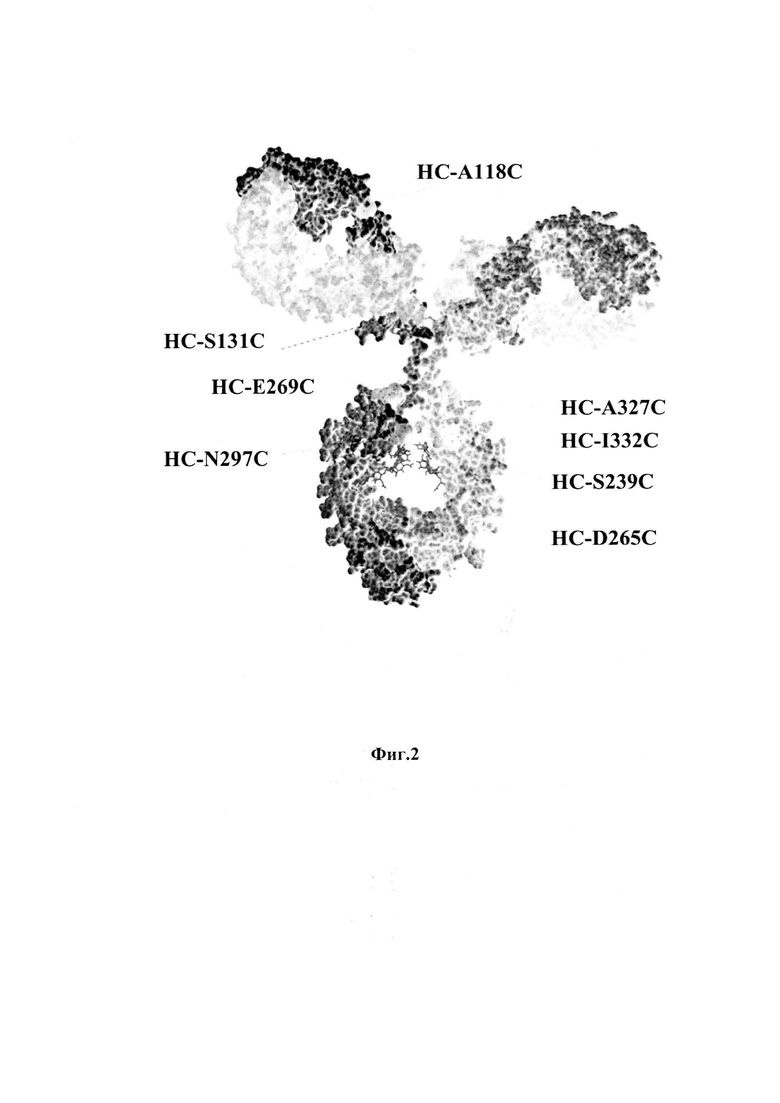
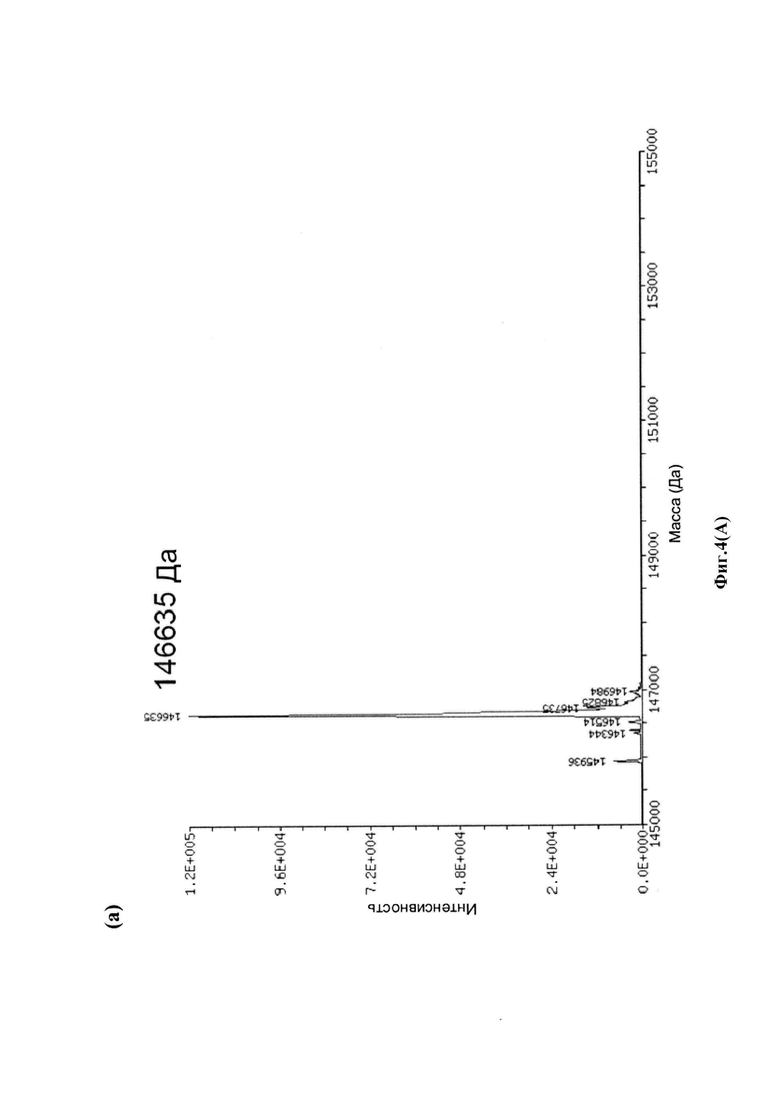

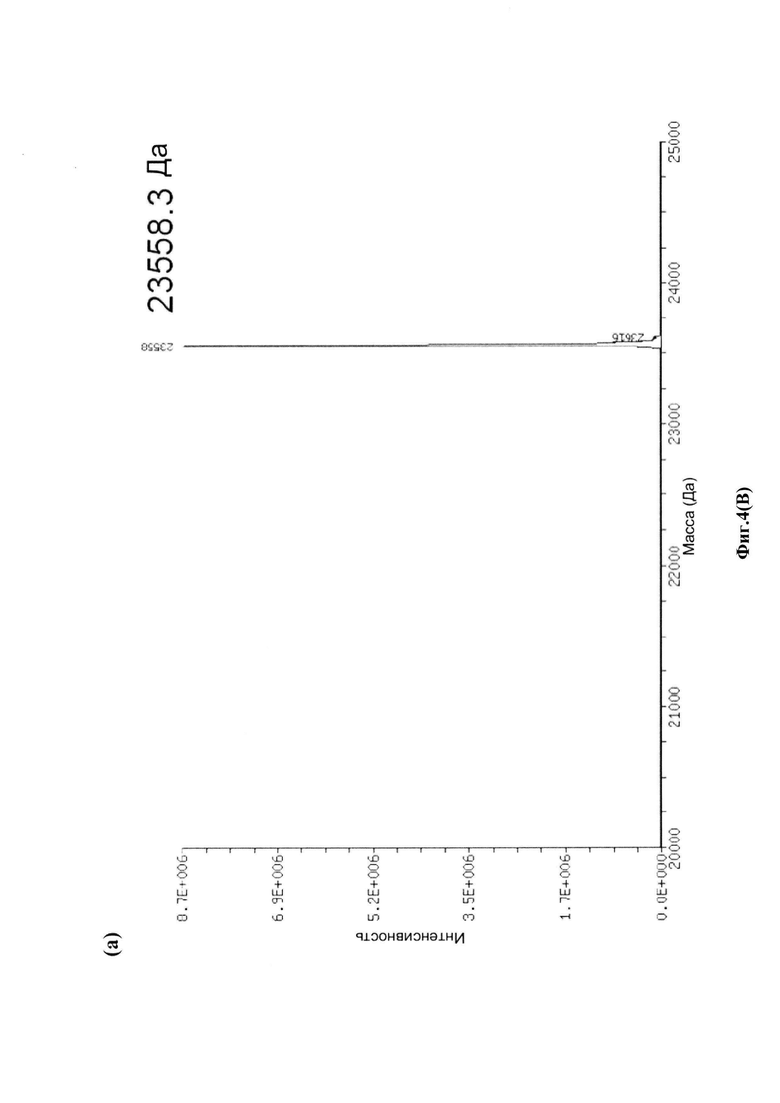


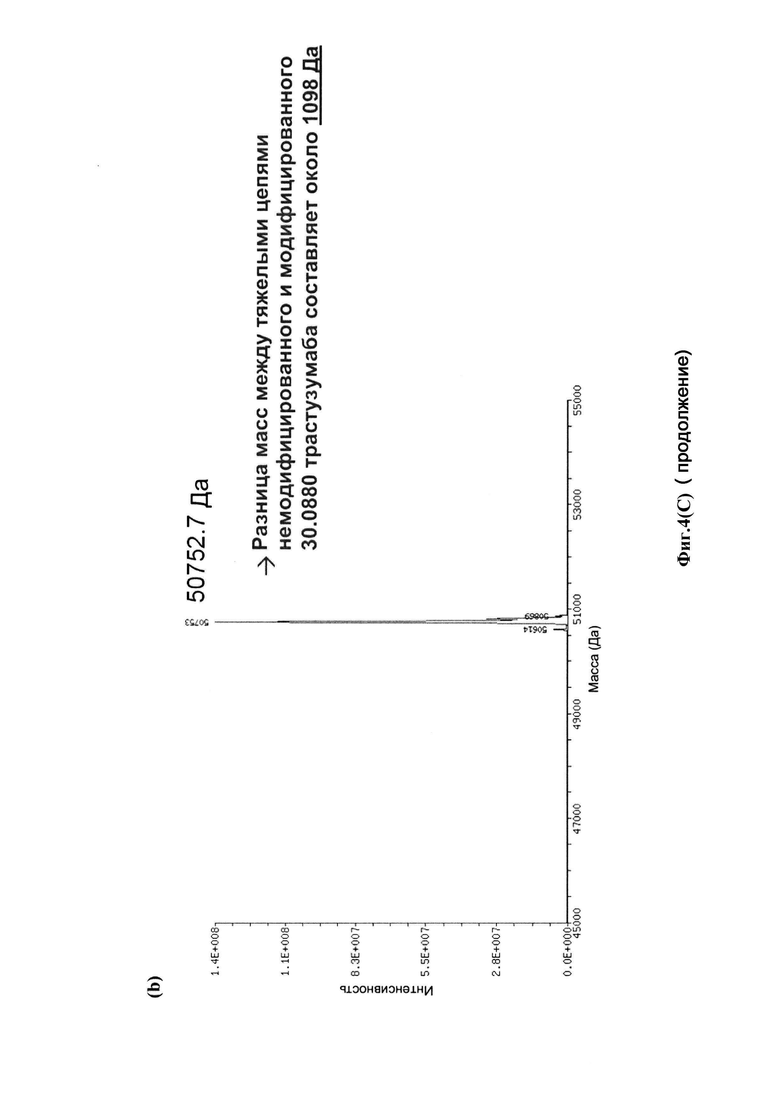
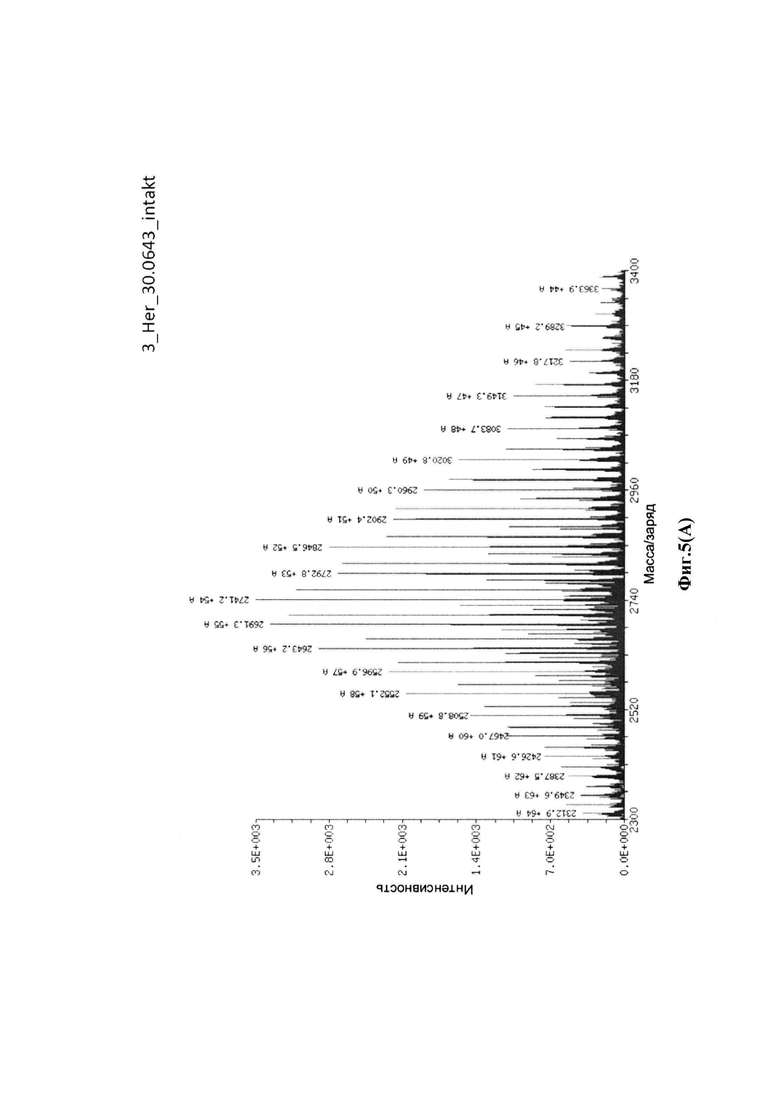

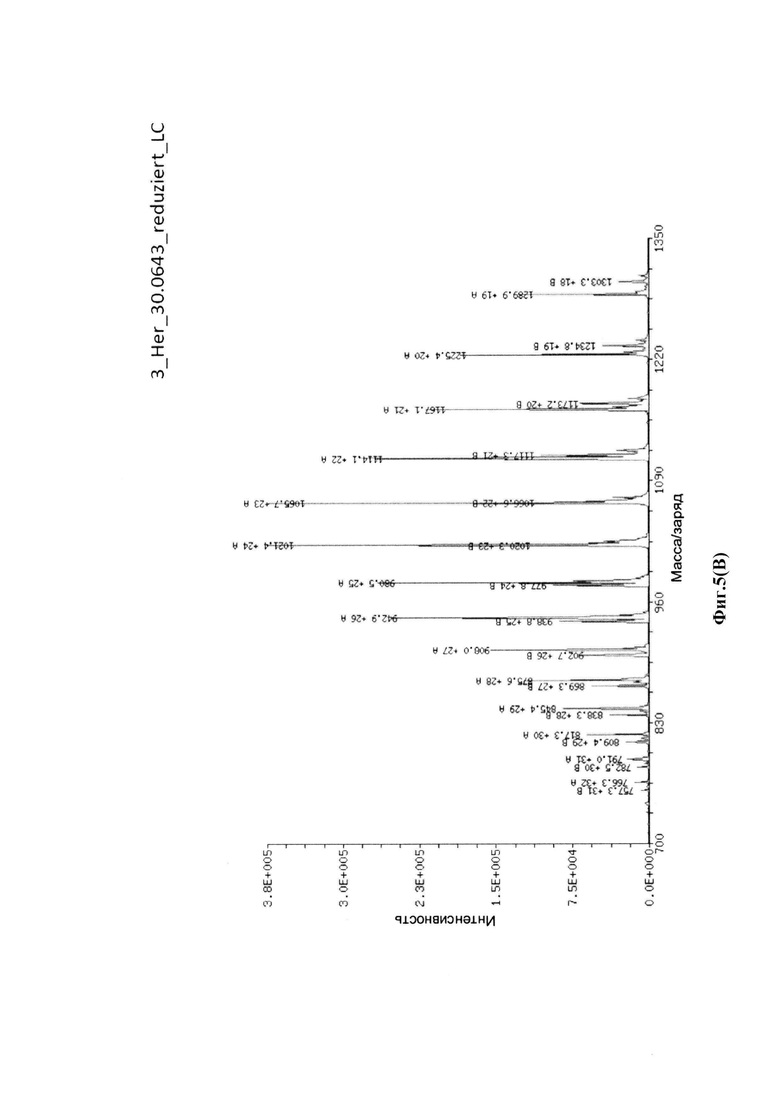

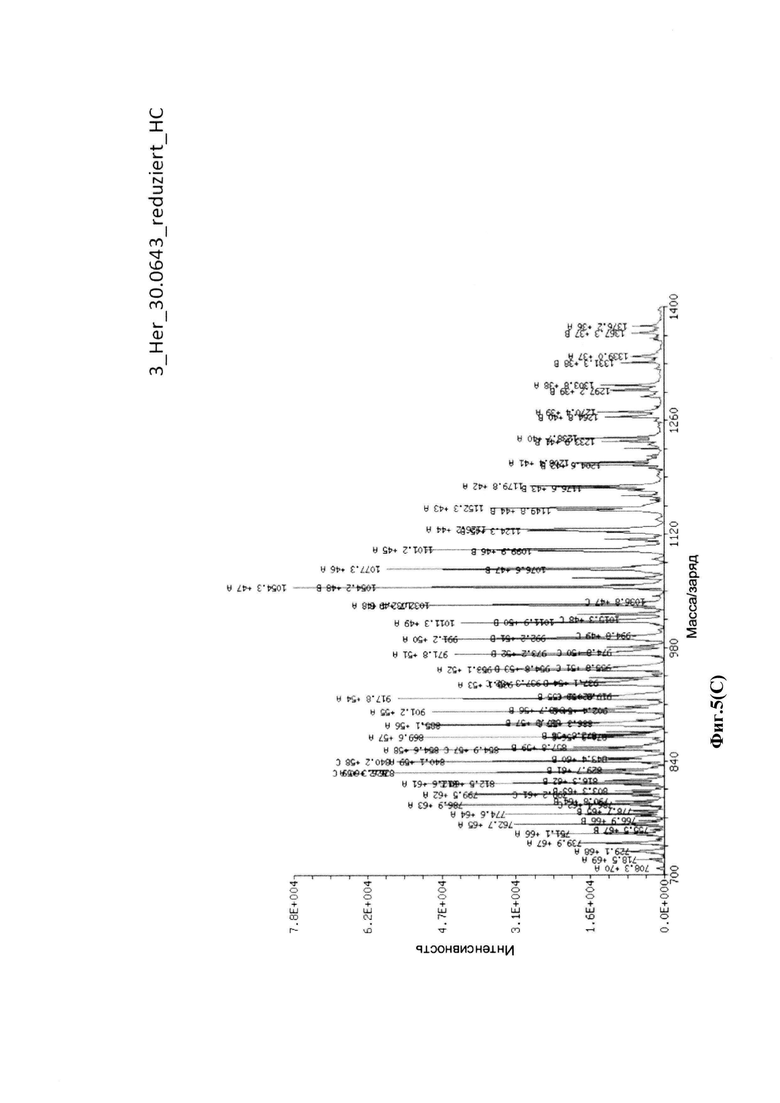

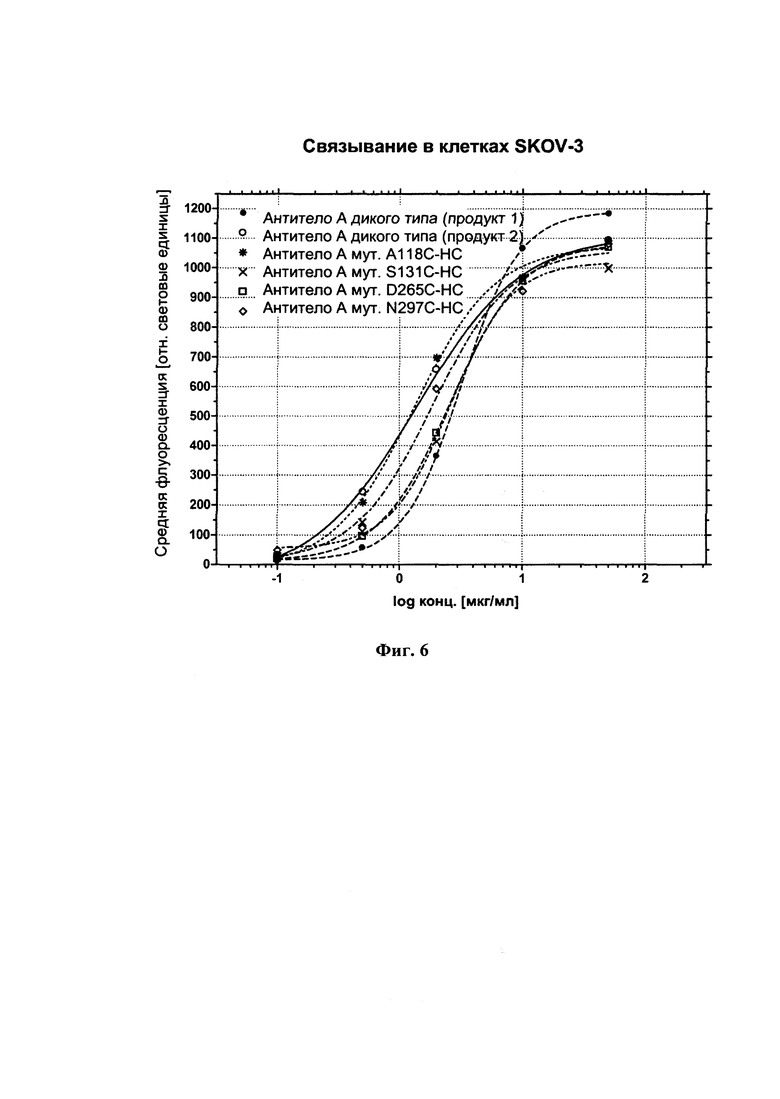
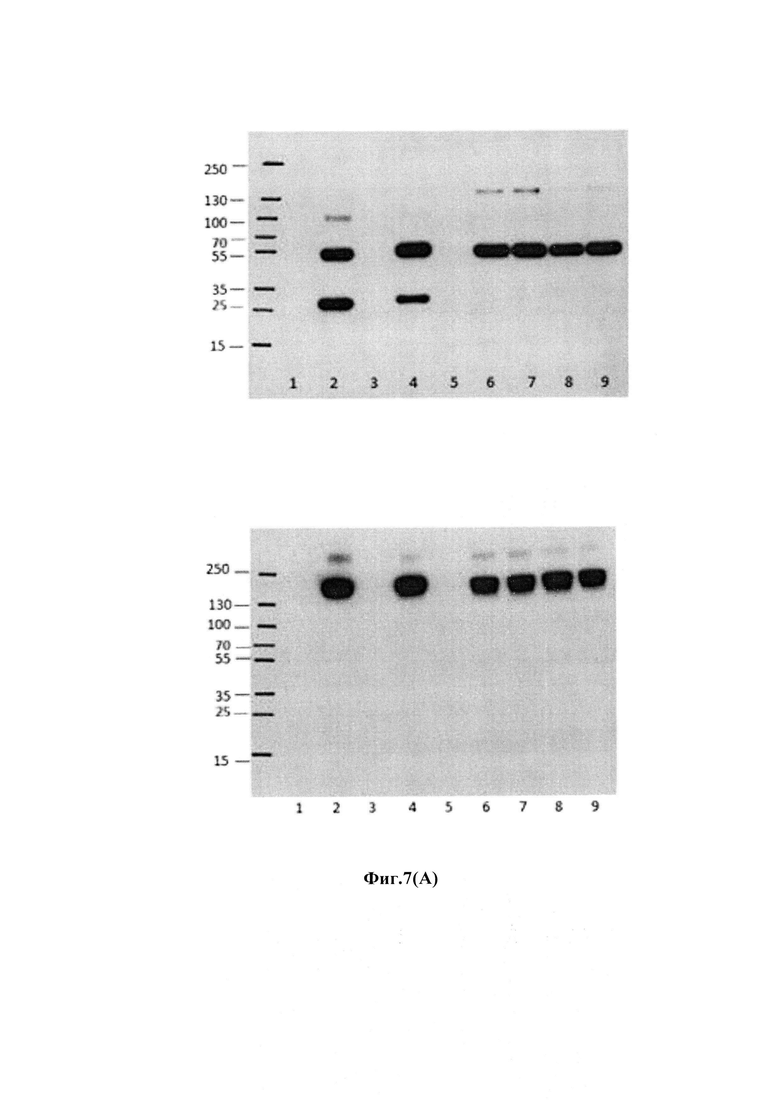
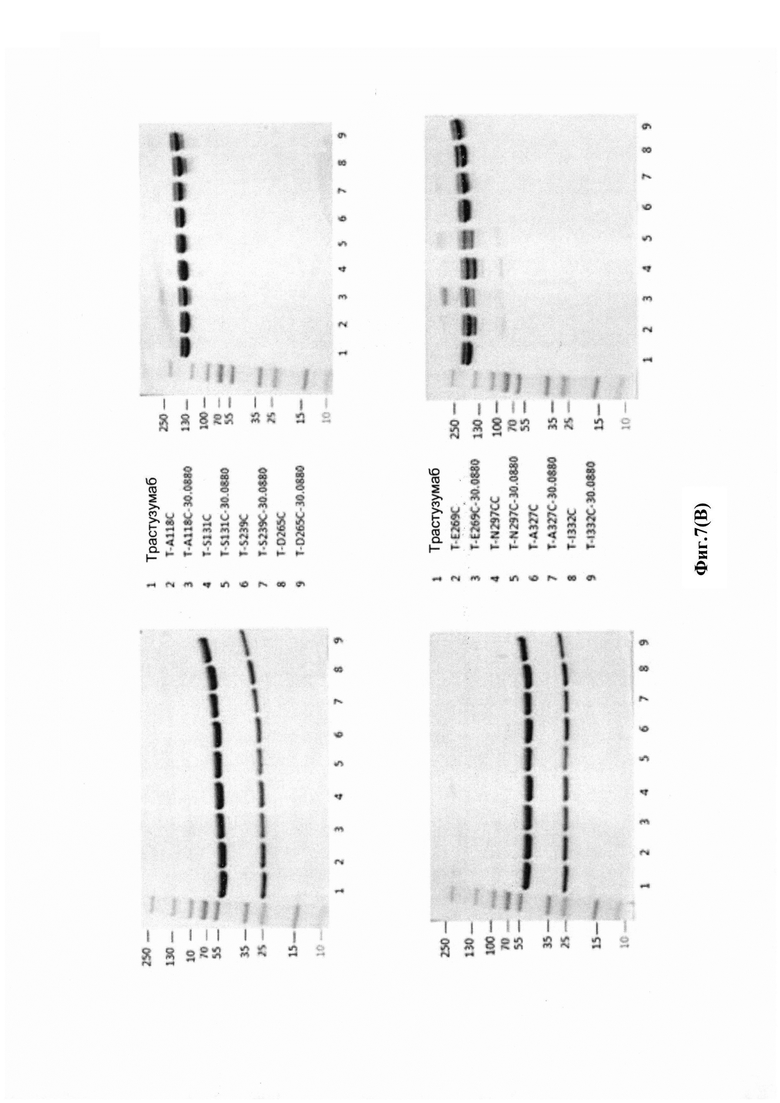
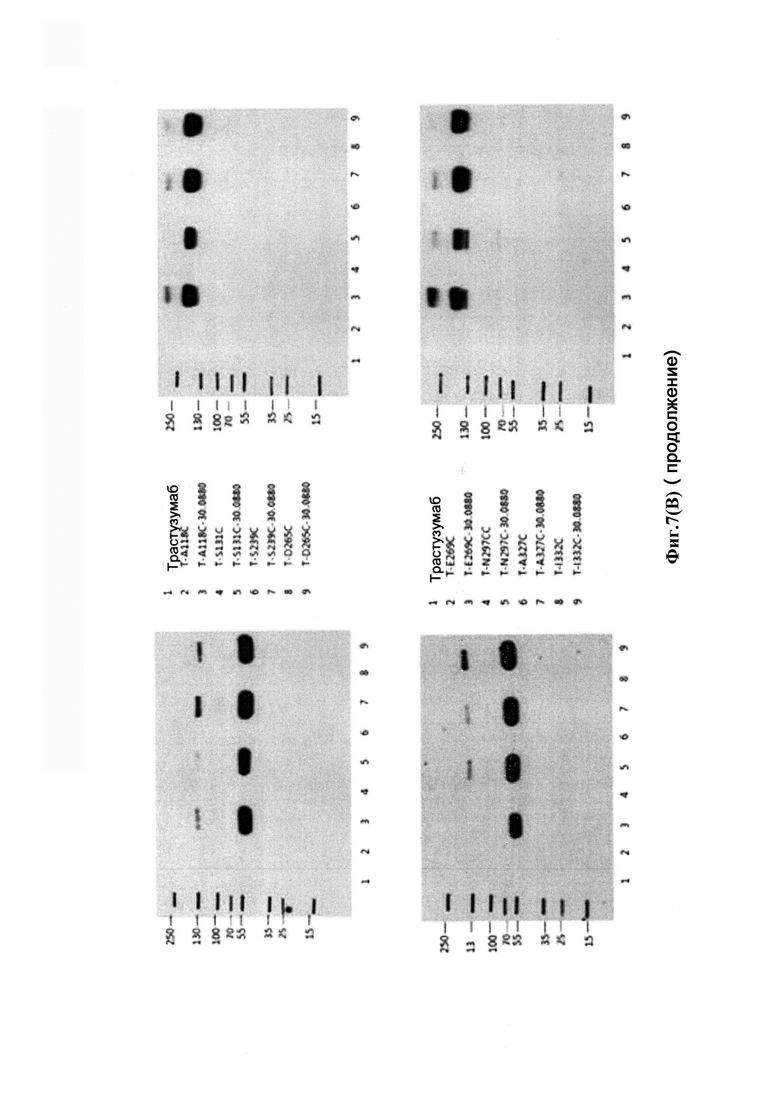
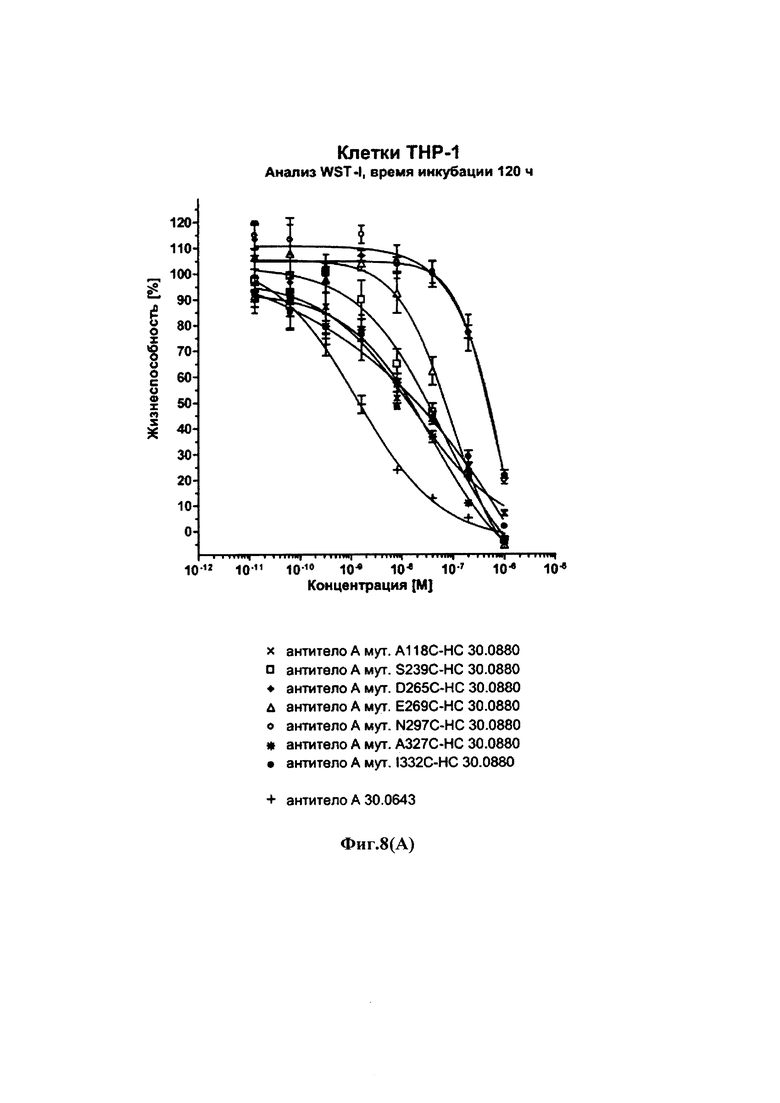
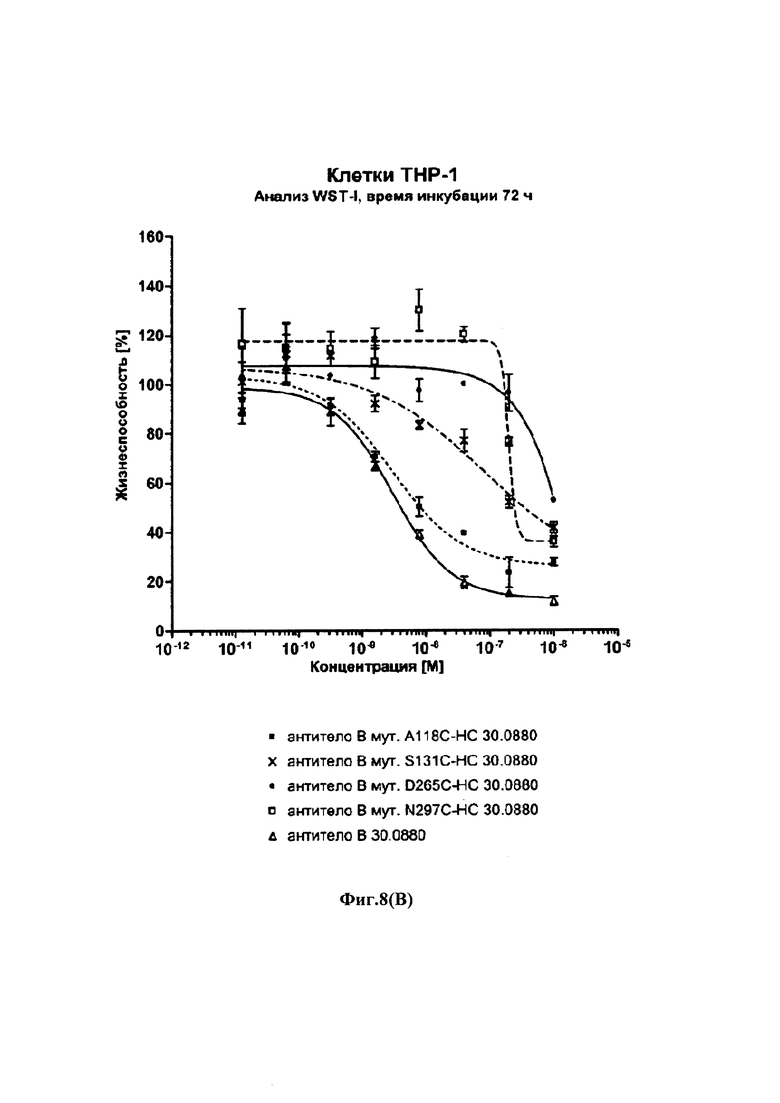
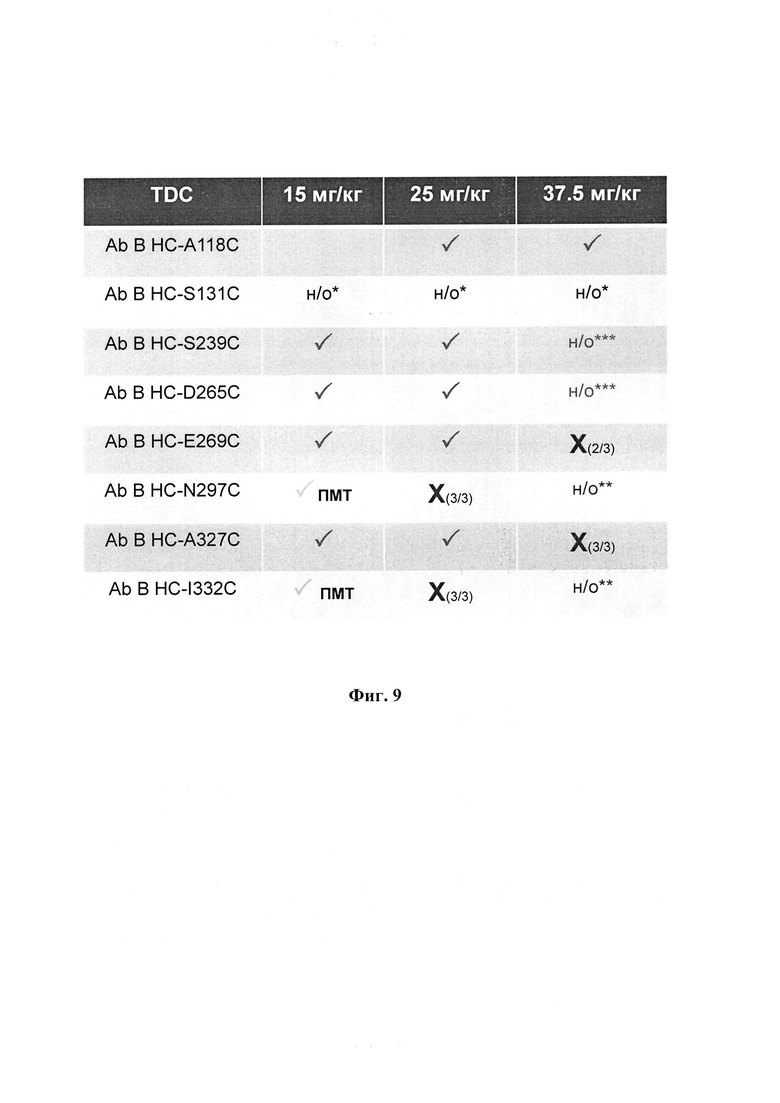

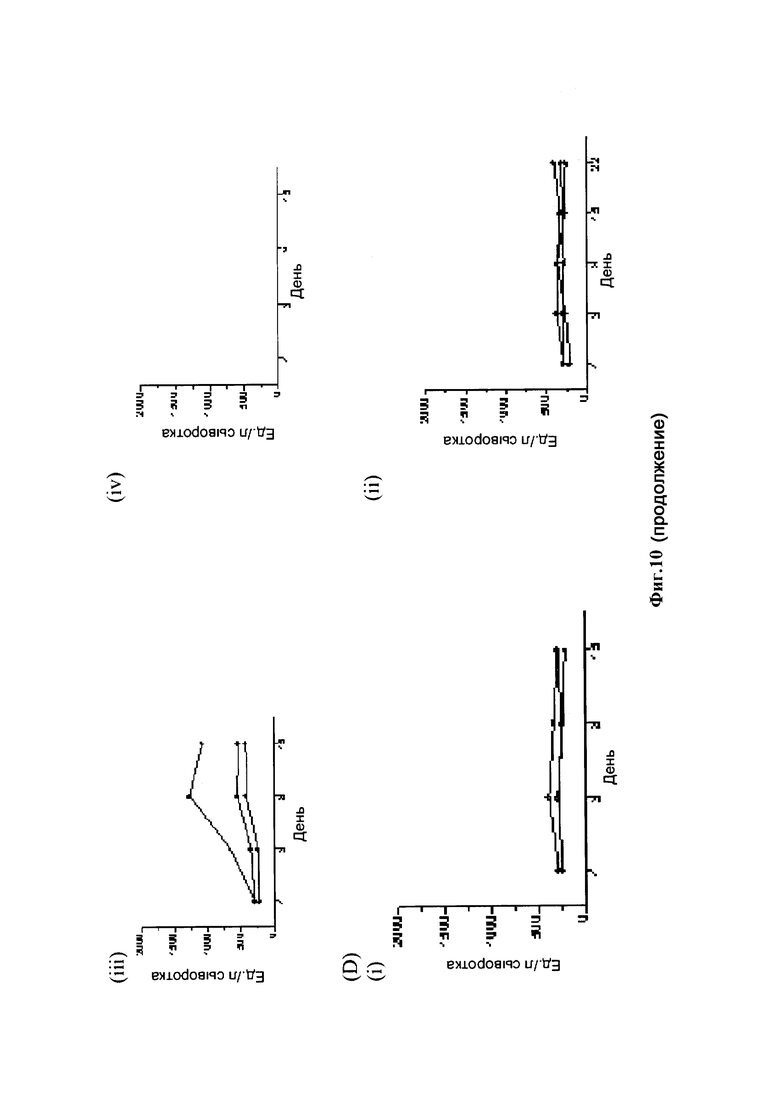
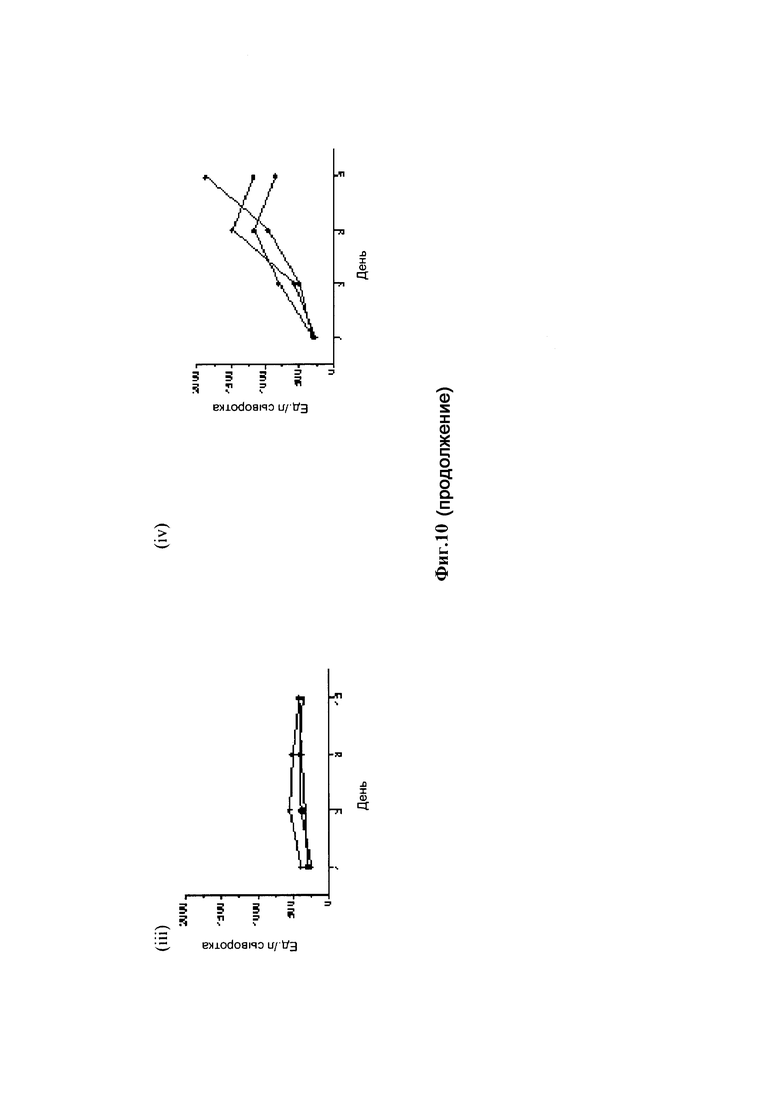
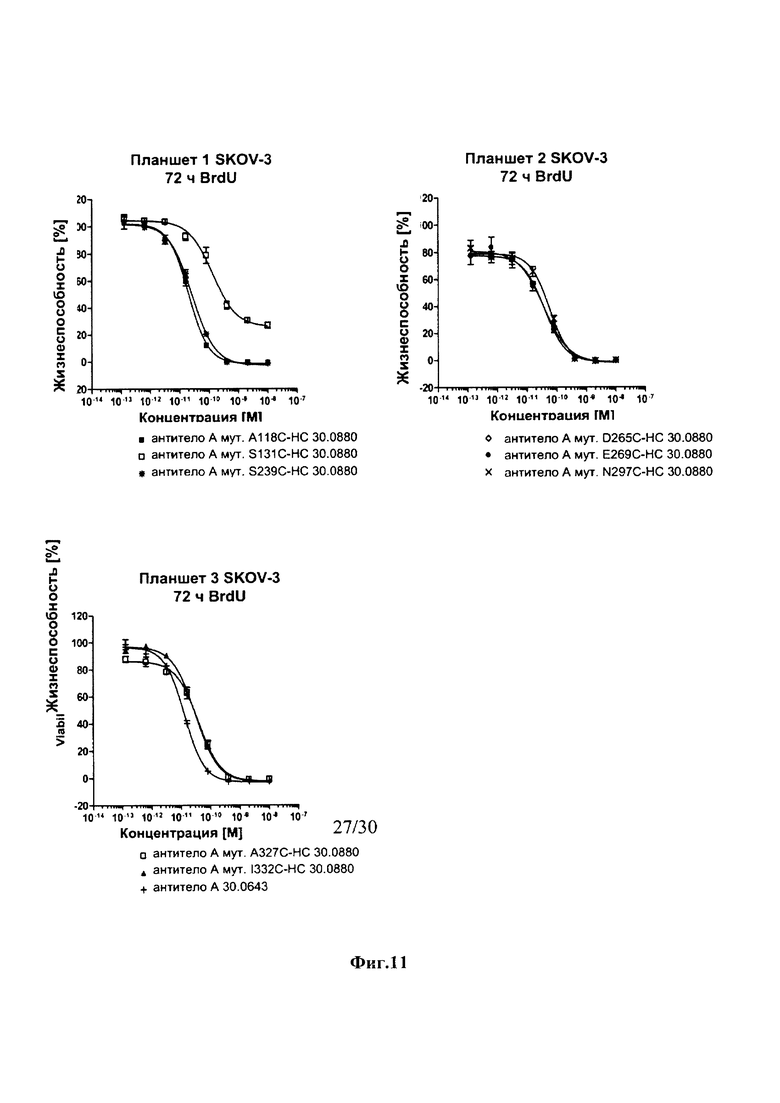
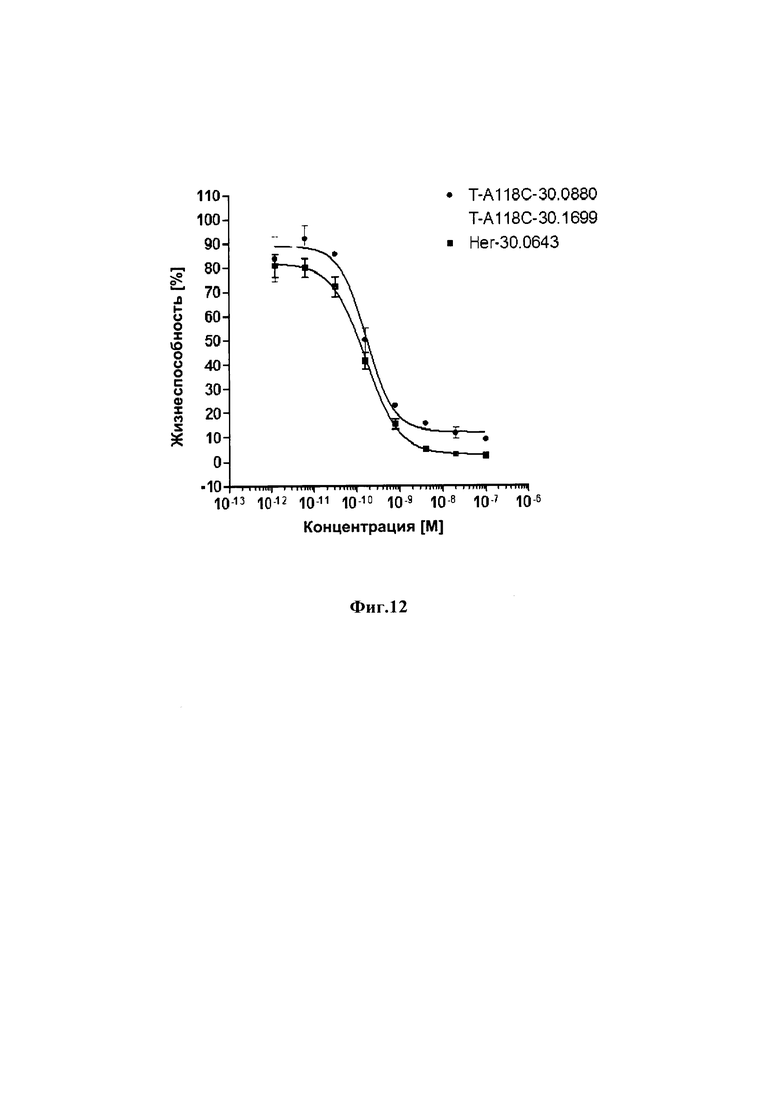
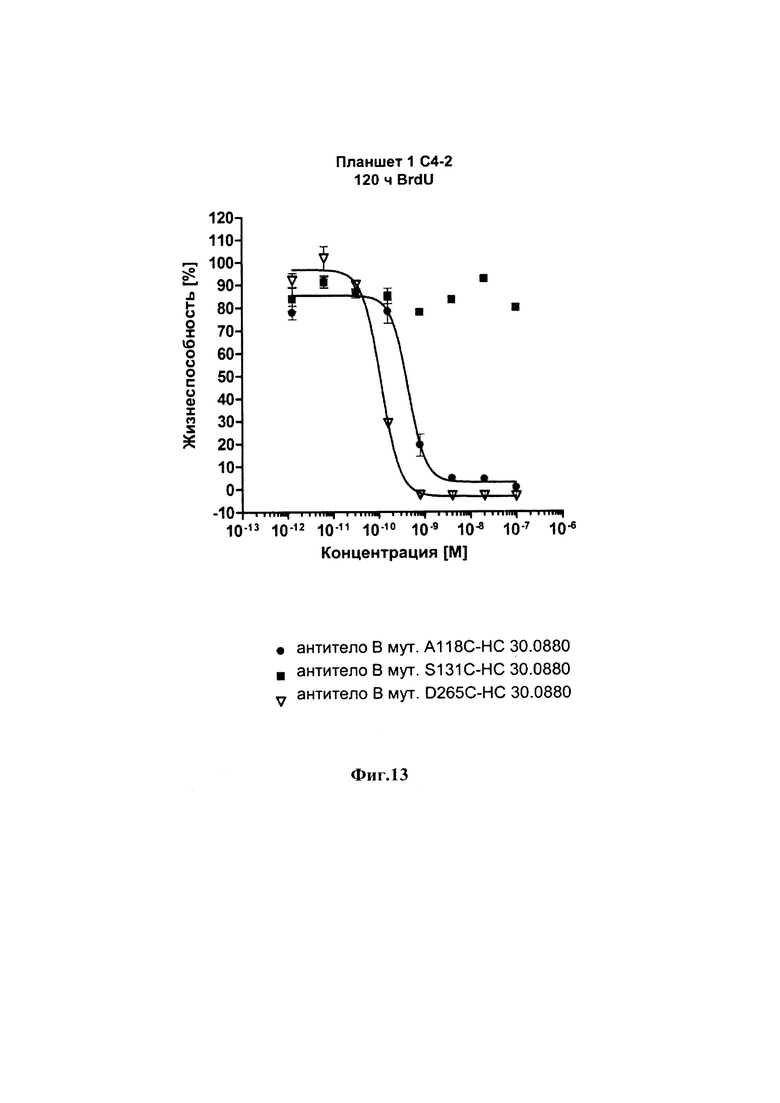
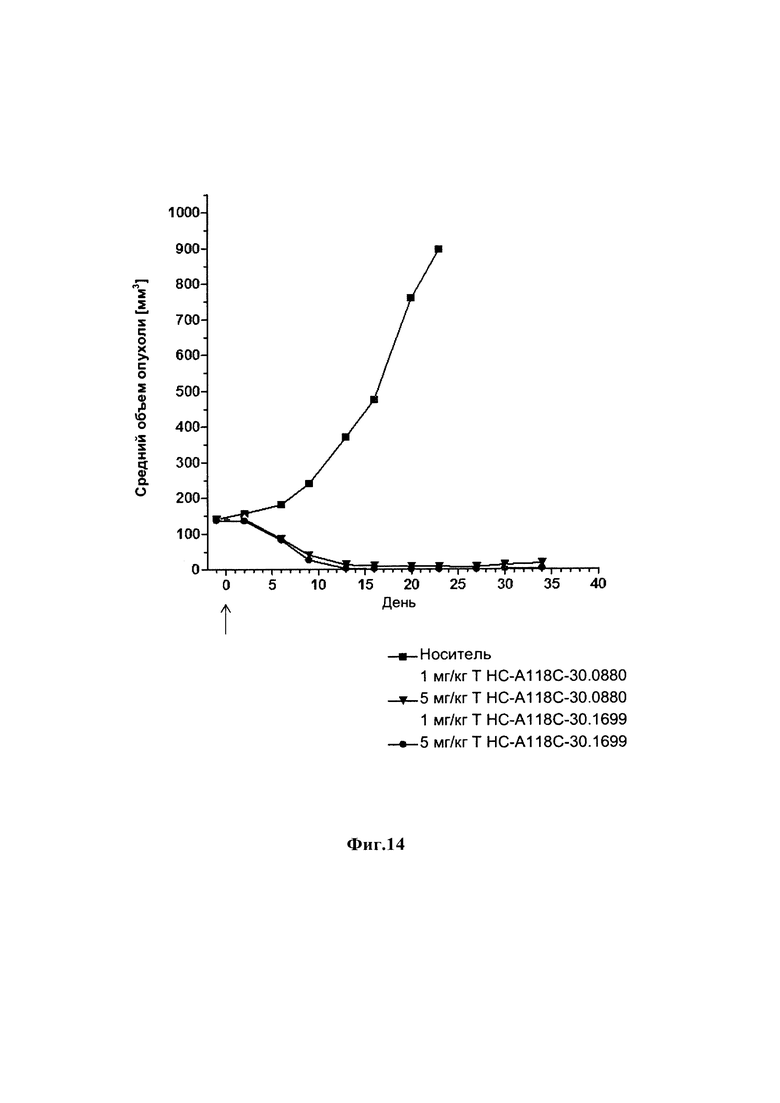
Изобретение относится к конъюгату общей формулы: Ama-L-X-S-Ab, в котором Ama - аматоксин, L - линкер, X - фрагмент, полученный в результате связывания тиольной группы с тиол-реагирующей группой, S - атом серы аминокислотного остатка цистеина и Ab - последовательность антитела или функциональный фрагмент антитела, содержащая(ий) указанный остаток цистеина, причем этим остатком цистеина является 265Cys с тяжелой цепью в соответствии с системой нумерации ЕС, и в котором одна или более одной молекулы аматоксина связаны с одним антителом или с одним функциональным фрагментом антитела. Также предложены его фармацевтическая композиция и способ его получения. Конъюгат предназначен для лечения опухолевых заболеваний. 5 н. и 7 з.п. ф-лы, 14 ил., 1 табл., 28 пр.
1. Конъюгат общей формулы:
Ama-L-X-S-Ab,
в котором Ama - аматоксин, L - линкер, X - фрагмент, полученный в результате связывания тиольной группы с тиол-реагирующей группой, S - атом серы аминокислотного остатка цистеина и Ab - последовательность антитела или функциональный фрагмент антитела, содержащая(ий) указанный остаток цистеина, причем этим остатком цистеина является 265Cys с тяжелой цепью в соответствии с системой нумерации ЕС, и в котором одна или более одной молекулы аматоксина связаны с одним антителом или с одним функциональным фрагментом антитела.
2. Конъюгат по п. 1, в котором линкер выбран из стабильного линкера и расщепляемого линкера.
3. Конъюгат по п. 1 или 2, в котором тиол-реагирующая группа выбрана из бромацетамида, йодацетамида, 4,6-дихлор-1,3,5-триазин-2-иламино, 4-[5-(метилсульфонил)-1Н-тетразол-1-ил]фенил, 2-(метилсульфонил)-бензо[d]тиазол-5-ил, 4-[5-(метилсульфонил)-1,3,4-оксадиазол-2-ил]-фенил, 2-пиридилдитио-, 2-(5-нитропиридил)дитио-, метилтиосульфонил и малеимид, особенно малеимид.
4. Конъюгат по любому из пп. 1-3, в котором две молекулы аматоксина связаны с одним антителом или с одним функциональным фрагментом антитела.
5. Конъюгат по любому из пп. 1-3, в котором 1-4 молекулы аматоксина связаны с одним антителом или с одним функциональным фрагментом антитела.
6. Способ синтеза конъюгата общей формулы:
Ama-L-X-S-Ab
по п. 1 реакцией соединения Ama-L-X', в котором Ama - аматоксин, L - линкер, X - фрагмент, полученный в результате связывания тиольной группы с тиол-реагирующей группой X', с антителом или функциональным фрагментом антитела, Ab-SH, в котором группа -SH - тиол аминокислотного остатка цистеина и Ab - последовательность антитела или последовательность функционального фрагмента антитела, содержащая указанный остаток цистеина, причем этим остатком цистеина является 265Cys с тяжелой цепью в соответствии с системой нумерации ЕС, и в котором одна или более одной молекулы аматоксина связаны с одним антителом или с одним функциональным фрагментом антитела.
7. Способ по п. 6, в котором две молекулы аматоксина связаны с одним антителом или с одним функциональным фрагментом антитела.
8. Способ по п. 6, в котором 1-4 молекулы аматоксина связаны с одним антителом или с одним функциональным фрагментом антитела.
9. Способ по п. 6, который дополнительно включает этап синтеза соединения Ama-L-X', в котором Ama - аматоксин, L - линкер, X' - малеимидная группа, включающего этап (а), на котором осуществляют реакцию аматоксина, содержащего нуклеофильную группу, с соединением Y-L-X'', в котором
Y - уходящая группа и
X'' - защищенная малеимидная группа.
10. Набор, содержащий (i) соединение Ama-L-X', где Ama - аматоксин, L - линкер, X' - тиол-реагирующая группа, и (ii) антитело, или функциональный фрагмент антитела, Ab-SH, в котором группа -SH - тиол аминокислотного остатка цистеина, и Ab - последовательность антитела или последовательность функционального фрагмента антитела, содержащая указанный остаток цистеина, причем этим остатком цистеина является 265Cys в соответствии с системой нумерации ЕС для применения в способе по п. 6.
11. Фармацевтическая композиция для лечения заболевания, ассоциированного с опухолевыми клетками, содержащая конъюгат по любому из пп. 1-5 в терапевтически эффективном количестве.
12. Способ лечения заболевания, ассоциированного с опухолевыми клетками, включающий этап, на котором осуществляют контакт указанных клеток с конъюгатом по любому из пп. 1-5, в котором указанное антитело или указанный функциональный фрагмент антитела, является специфичным для мишени, представленной указанными опухолевыми клетками.
| WO 2014043403 A1, 20.03.2014 | |||
| WO 2008070593 A2, 12.06.2008 | |||
| RU 2015142093 A, 07.04.2017 | |||
| Ben-Quan Shen et al | |||
| "Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates", Nature Biotechnology, 2012, Vol.30, No 2, Page(s) 184 - 189 | |||
| May S Kung; Sutherland et al | |||
| Способ сопряжения брусьев в срубах | 1921 |
|
SU33A1 |

