Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к новым способам и соединениям для синтеза производных аманитина. Настоящее изобретение, в частности, относится к способам синтеза производных (S)-6-гидрокситриптофана, которые можно использовать в качестве структурных элементов для синтеза производных аманитина или конъюгатов лекарственных средств и аматоксина. Настоящее изобретение также относится к промежуточным соединениям указанных путей синтеза для использования в синтезе производных аманитина и конъюгатов лекарственных средств и аматоксина и к использованию конкретных катализаторов, подходящих для содействия указанным путям синтеза.
Предшествующий уровень техники настоящего изобретения
Аматоксины представляют собой циклические пептиды, состоящие из 8 аминокислот, которые находятся в грибах Amanita phalloides (см. фиг. 1). Аматоксины специфически ингибируют ДНК-зависимую РНК-полимер азу II клеток млекопитающих и при этом также транскрипцию и биосинтез белков поврежденных клеток. Ингибирование транскрипции в клетке вызывает остановку роста и пролиферации. Хотя и не ковалентно связанный, комплекс между аманитином и РНК-полимеразой II очень крепкий (KD=3 нМ). Диссоциация аманитина из фермента является очень медленным процессом, таким образом делая извлечение поврежденной клетки маловероятным. Когда ингибирование транскрипции продолжается достаточно долго, клетка будет подвергаться запрограммированной гибели клетки (апоптозу).
Аматоксины можно выделять из собранных плодовых тел гриба Amanita phalloides или из чистых культур (Zhang Р, et al, FEMS Microbiol Lett. 2005 Nov 15;252(2):223-8. Epub 2005 Sep 15). Однако количества аматоксинов, которые можно получить, довольно небольшие (в диапазоне приблизительно 0,3-3 мг/г сухого вещества из природных плодовых тел и приблизительно их 10% из чистой культуры), и гибкость для дальнейшей модификации встречающихся в природе вариант аматоксина ограничена. Альтернативно, аматоксины можно получать ферментацией при помощи базидиомицета (Muraoka S, and Shinozawa Т., J Biosci Bioeng. 2000;89(l):73-6) или A. fissa (Guo XW, et al, 2006 Jun;46(3):373-8). Снова выходы низкие, и гибкость для дальнейшей модификации встречающихся в природе вариант аматоксина также ограничена. Наконец, аматоксины получали путем частичного или полного синтеза (например, Zanotti G, Mahringer С, and Wieland Т., lnt J Pept Protein Res. 1987 Oct;30(4): 450-9; Zanotti G, Wieland T, Benedetti E, Di Blasio 8, Pavone V, and Pedone C, lnt J Pept Protein Res. 1989 Sep; 34(3):222-8). Альтернативно, применение полностью синтетических путей для аматоксинов могут предлагать получение больших количеств аматоксинов, требуемых для терапевтических применений, и могут предлагать создание множества новых аматоксинов при помощи соответствующих исходных материалов в качестве структурных элементов.
Встречающиеся в природе аманитины, такие как α-аманитин, β-аманитин или γ-аманитин, содержат фенольную гидроксигруппу (-ОН) в 6'-положении триптофана, который представляет аминокислоту 4 в циклическом октапептиде-аманитине, что обеспечивает соединение линкера с аматоксином (см. фиг. 1). Мишень-связывающие макромолекулы, такие как антитела или аптамеры, могут затем присоединяться посредством указанного линкера для создания конъюгатов, например, конъюгатов антитела и лекарственного средства. Использование аматоксинов в качестве цитотоксических фрагментов для терапии опухолей уже было исследовано в 1981 г. путем соединения антитела к Thy 1.2 с α-аманитином, используя линкер, прикрепленный к индольному кольцу триптофана (Тгр, аминокислота 4; см. фиг. 1) посредством диазотирования (Davis & Preston, 1981, Science 213: 1385-1388).
При помощи исследованных в настоящее время полностью синтетических соединений аманитина включение функционализированного триптофана для присоединения линкерных элементов было достигнуто только совсем недавно. Перед этим, таким образом, линкеры соединяли с полностью синтетическими аманитинами главным образом в положениях аминокислоты 1 (аспарагиновой кислоты) или азота индола (N1) аминокислоты 4 (триптофана). Поскольку профили биологической активности аманитинов, присоединенных посредством различных якорных положений структуры аманитина, как было показано, являются очень различными, представляет большой интерес наличие возможности обеспечивать синтетический триптофан, гидроксилированный в его 6'-положении, т.е. соответствующем нативному α-аманитину, β-аманитину или γ-аманитину, в качестве структурного элемента, который можно включать в ходе синтеза аманитина.
Синтетическое введение триптофана в структуру аманитина можно проводить путем реакции Савига-Фонтаны (Savige & Fontana, 1980, lnt J Pept Protein Res. 15(3): 285-97). Согласно этой реакции триптофан превращается в смесь Вос-защищенного цис-2-карбокси-3а-гидрокси-1,2,3,3а,8,8а-гексагидропирроло[2,3-b]индола и транс-2-карбокси-3а-гидрокси-1,2,3,3а,8,8а-гексагидропирроло [2,3-b] индола, а затем включается в аминокислотную последовательность линейного предшественника аманитина.
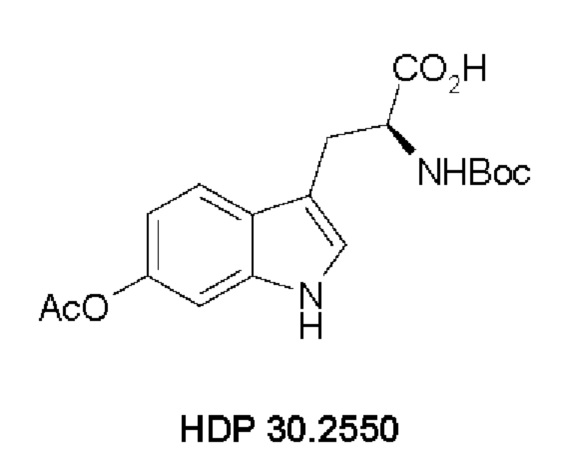
Для полностью синтетического получения аманитина синтез (S)-6-гидрокситриптофана и синтез соответствующих структурных элементов представляют особую важность. В частности, один из этих структурных элементов особой важности представляет собой (S)-6-ацетилокси-М-трет-бутоксикарбонил-триптофан (HDP 30.2550).
До сих пор удовлетворительный и эффективный путь синтеза (S)-6-гидрокситриптофана и его структурных элементов не был описан в уровне техники. В частности, не был доступен путь синтеза, дающий необходимую чистоту энантиомеров (L или S) этого аминокислотного производного. Различные базовые варианты синтеза можно в принципе рассматривать. Первый вариант может быть кристаллизацией рацемической формы с хиральными вспомогательными основаниями или кислотами. Однако этим способом можно было получить максимальный выход только 50%. Второй вариант представляет ферментативное получение; использование этого способа, однако, является времязатратным, результаты неустойчивые, и воспроизводимость слабая.
Таким образом, одной целью настоящего изобретения было обеспечение эффективного, простого и воспроизводимого способа синтеза (S)-6-гидрокситриптофана, его производных и его структурных элементов. Предпочтительно одной целью настоящего изобретения было обеспечение эффективного способа синтеза (S)-6-ацетилокси-М-трет-бутоксикарбонилтриптофана (HDP 30.2550) с достаточно высокой энантиомерной чистотой.
В качестве одной дополнительной цели настоящего изобретения следует обеспечивать применение указанного (S)-6-гидрокситриптофана и его производных, предпочтительно (S)-6-ацетилокси-К-трет-бутоксикарбонилтриптофана, в качестве структурных элементов для полностью синтетического производства аматоксинов.
Эти и другие цели удовлетворяются способами и средствами согласно независимым пунктам формулы настоящего изобретения. Зависимые пункты формулы связаны с конкретными вариантами осуществления.
Краткое раскрытие настоящего изобретения
Настоящее изобретение обеспечивает способ синтеза (S)-6-гидрокситриптофана, (S)-6-ацетилокси-N-трет-бутилоксикарбонилтриптофана, и их производных и структурных элементов для их синтеза. Настоящее изобретение также обеспечивает соединения и структурные элементы для применения в синтезе аманитина или производных аманитина или конъюгатов аматоксина и лекарственного средства.
Настоящее изобретение и общие преимущества его признаков будут обсуждаться более подробно ниже.
Краткое описание фигур
Фиг. 1 показывает структурные формулы различных аматоксинов. Числа, указанные жирным шрифтом (1-8), обозначают стандартную нумерацию восьми аминокислот, образующих аматоксин. Также показаны стандартные обозначения атомов в аминокислотах 1, 3 и 4 (греческие буквы α-γ, греческие буквы α-δ и числа 1'-7', соответственно).
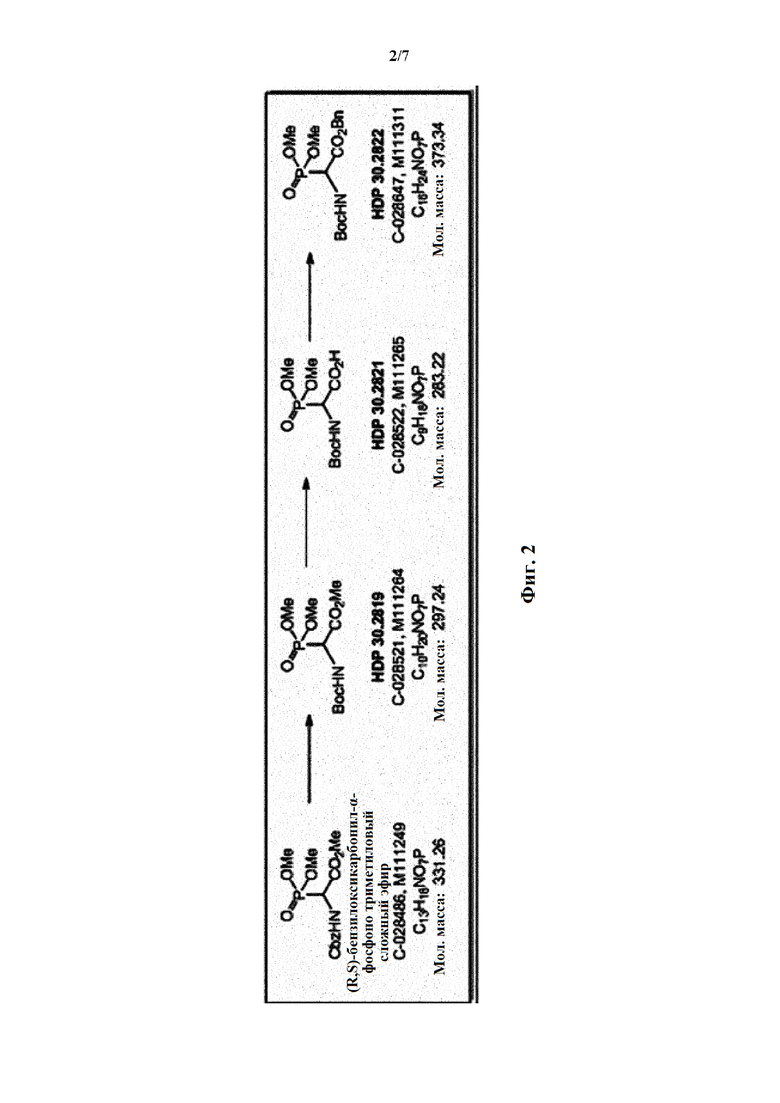
Фиг. 2 показывает синтез соединения HDP 30.2822.
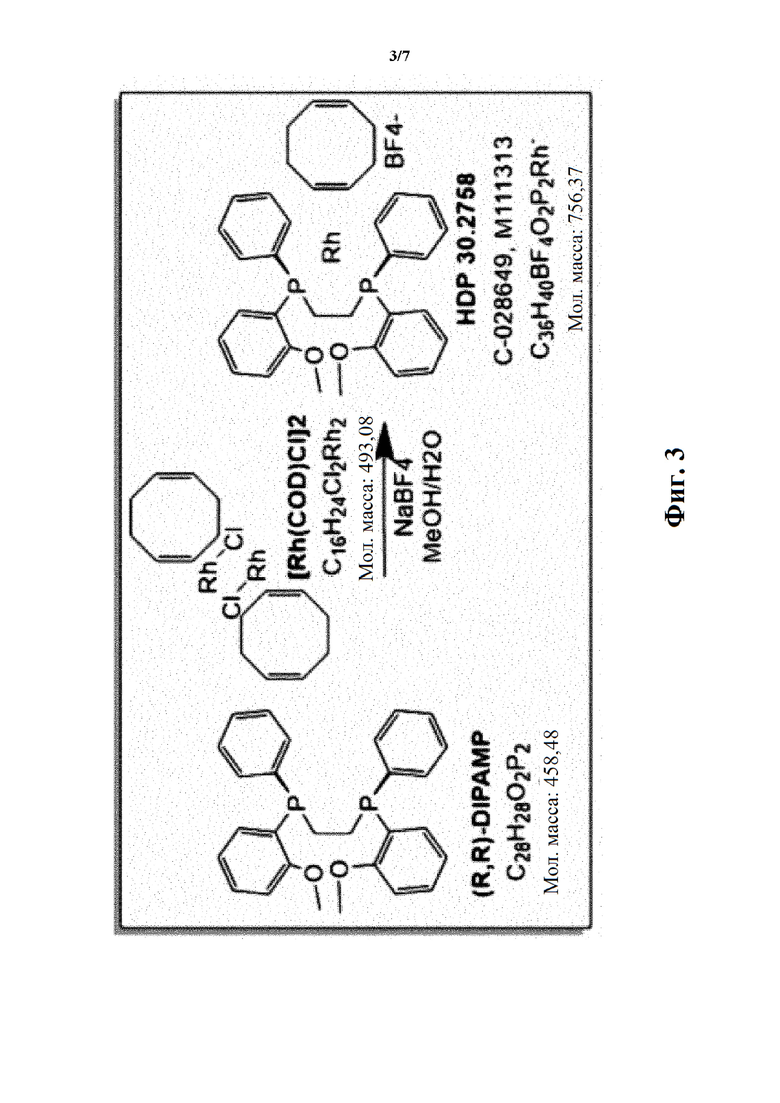
Фиг. 3 показывает синтез соединения HDP 30.2758.
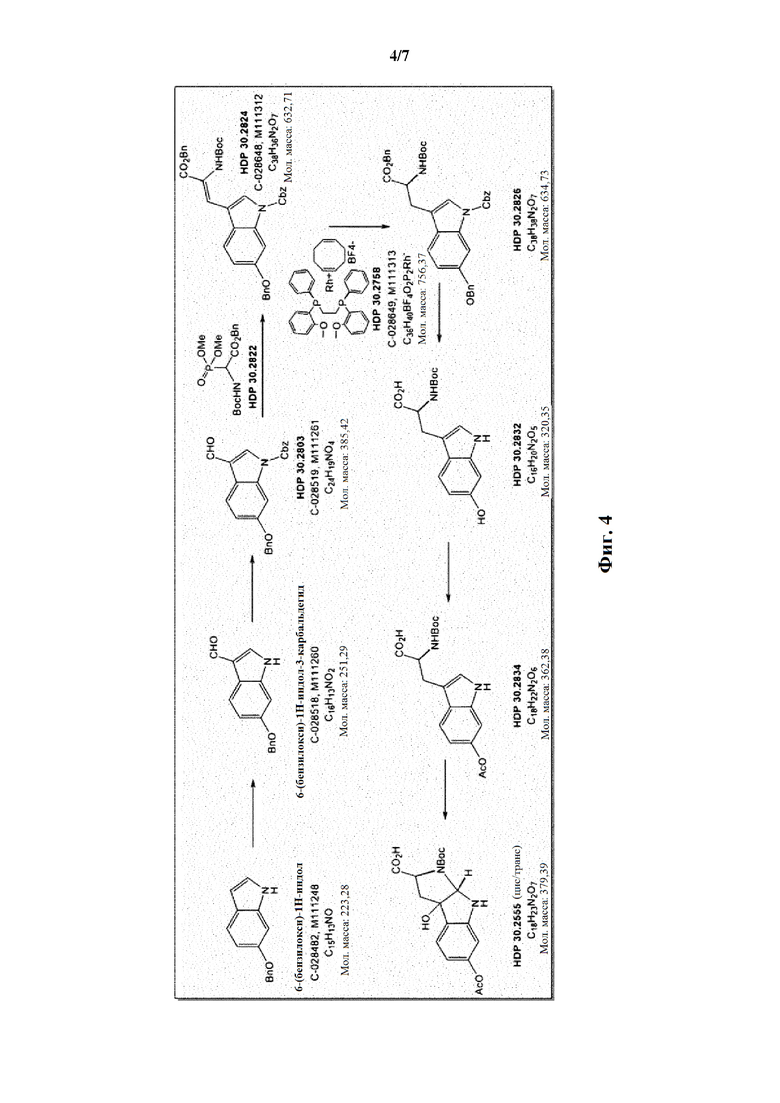
Фиг. 4 показывает синтез соединений HDP 30.2550 и HDP 30.2555.
Фиг. 5 показывает структурные композиции катализаторов, используемых для конверсии HDP 30.2824 в HDP 30.2826.
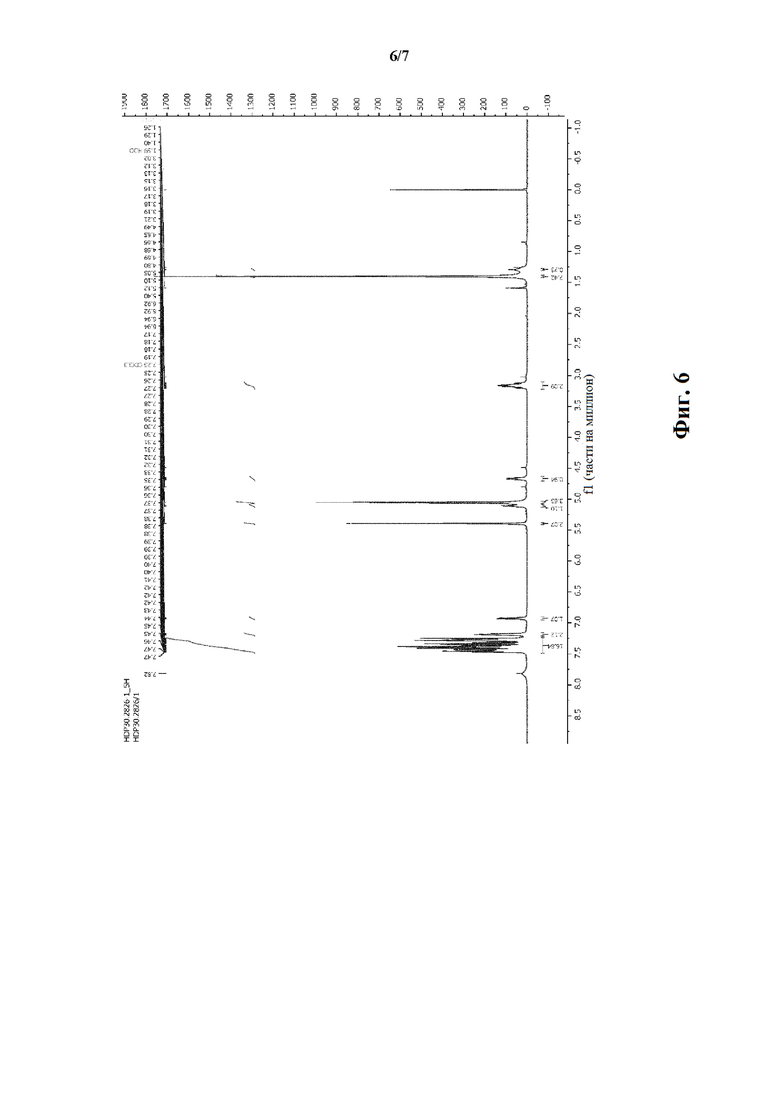
Фиг. 6 показывает результаты 1H-ЯМР спектроскопии соединения HDP 30.2826 (400 МГц, CDCl3, 5=части на миллион).
Фиг. 7 показывает результаты 13С-ЯМР спектроскопии соединения HDP 30.2826 (100 МГц, CDCl3, 5=части на миллион).
Подробное раскрытие настоящего изобретения
Перед подробным описанием настоящего изобретения следует понять, что данное изобретение не ограничено конкретными составляющими частями описанных средств или описанными технологическими стадиями способов, поскольку такие средства и способы могут меняться. Также следует понимать, что терминология, используемая в настоящем документе, представлена только с целью описания конкретных вариантов осуществления, а не предназначена для ограничения. Необходимо отметить, что при использовании в описании и приложенной формуле изобретения формы единственного числа включают ссылки на единственное и/или множественное, если контекст явно не указывает иное. Кроме того, следует понимать, что в случае, когда даны диапазоны параметров, которые ограничены численными значениями, диапазоны подразумевают включение этих ограничивающих значений.
Также следует понимать, что варианты осуществления, раскрытые в настоящем документе, не следует понимать как отдельные варианты осуществления, которые не связаны друг с другом. Признаки, обсуждаемые с одним вариантом осуществления, подразумеваются раскрытыми также в связи с другими вариантами осуществления, показанными в настоящем документе. Если в одном случае конкретный признак не раскрыт в связи с одним вариантом осуществления, а с другим, специалист будет понимать, что это не обязательно означает, что указанный признак не подразумевается как раскрытый с указанным другим вариантом осуществления. Специалист будет понимать, что сутью данной заявки является раскрытие указанного признака также для другого варианта осуществления, и что только с целью ясности и для сохранения удобного объема описания это не было сделано.
Кроме того, содержание документов уровня техники, на которые ссылаются в настоящем документе, включено ссылкой. Это относится, в частности, к документам уровня техники, которые раскрывают стандартные или обычные способы. В этом случае включение ссылкой имеет главной целью обеспечение достаточного для воспроизведения раскрытия и избегает растянутых повторений.
Крупномасштабное, промышленно осуществимое производство (S)-6-гидрокситриптофана, как он содержится во встречающемся в природе α-, β- и γ-аманитине, и/или его химически защищенные формы требует эффективного, простого и воспроизводимого способа синтеза. Впервые авторы настоящего изобретения смогли обеспечить такой простой и эффективный способ получения путем использования энантиомерно селективного гидрирования олефиновых аминокислотных предшественников. Используя специфичные хиральные катализаторы, они неожиданно обнаружили, что такой тип реакции может давать очень высокую энантиомерную чистоту.
Простой раскрытый путь синтеза дает (S)-6-гидрокситриптофан, который полностью идентичен соответствующей нативной структуре. Возможность использования указанного соединения и его защищенных форм для промышленного крупномасштабного производства аманитинов будет значительно снижать стоимость и повышать эффективность, например, изготовления синтетического α-, β- и γ-аманитина, а также конъюгатов антитела и лекарственного средства на основе аматоксина для терапевтических применений.
Согласно первому аспекту настоящее изобретение относится к способу синтеза (S)-6-ацетилокси-N-трет-бутилоксикарбонилтриптофана (HDP 30.2550) или (S)-6-гидрокситриптофана, причем указанный способ предусматривает по меньшей мере одну стадию энантиомерно селективного гидрирования олефинового соединения-предшественника путем использования по меньшей мере одного хирального катализатора.
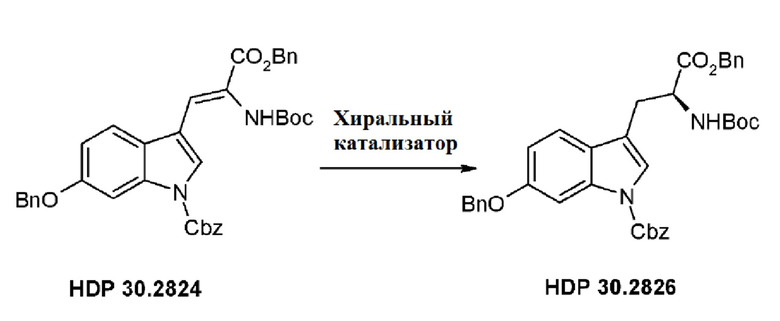
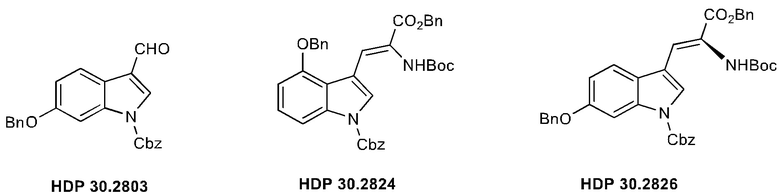
В одном варианте осуществления заявленного способа указанный олефиновый предшественник, используемый для асимметричного гидрирования, представляет олефиновый ненасыщенный аминокислотный предшественник. Предпочтительно это соединение HDP 30.2824.
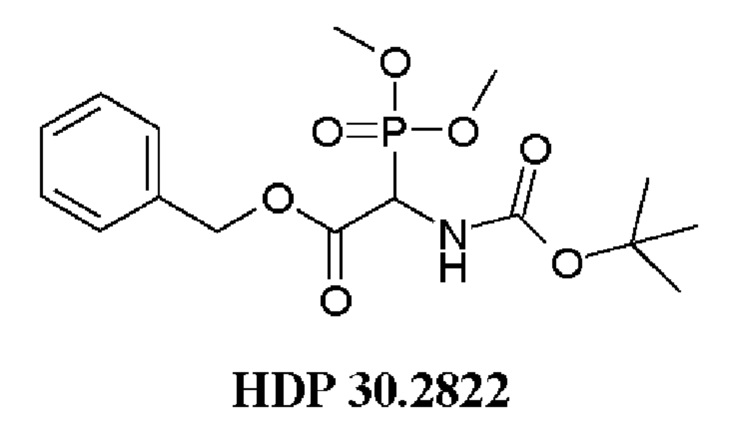
В одном варианте осуществления заявленного способа указанный олефиновый предшественник синтезируют путем использования следующего соединения:
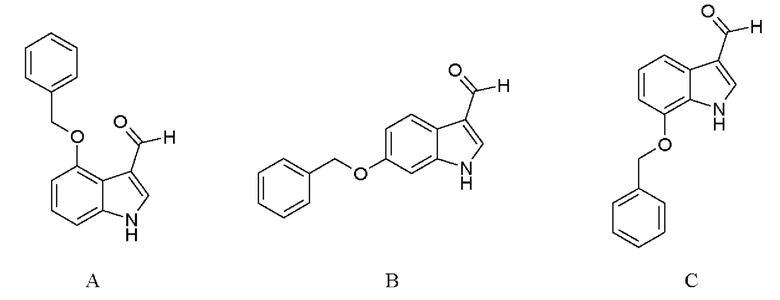
Указанный олефиновый предшественник можно также синтезировать при помощи любого из соединений А, В или С.
В дополнительном варианте осуществления заявленного способа указанный способ включает, по меньшей мере, следующие стадии:
В еще одном дополнительном варианте осуществления заявленного способа указанный способ включает, по меньшей мере, следующие стадии:
В контексте настоящего изобретения термин «аматоксин» включает все циклические пептиды, состоящие из 8 аминокислот, выделенные из рода Amanita и описанные у Wieland, Т. и Faulstich Н. (Wieland Т, Faulstich FL, CRC Crit Rev Biochem. 5 (1978) 185-260), также все их химические производные; также все их полусинтетические аналоги; также все их синтетические аналоги, состоящие из структурных элементов согласно основной структуре природных соединений (циклических, из 8 аминокислот), также все синтетические или полусинтетические аналоги, содержащие негидроксилированные аминокислоты вместо гидроксилированных аминокислот, также все синтетические или полусинтетические аналоги, в которых тиоэфирный сульфоксидный фрагмент заменен на сульфид, сульфон, тиоэфир или атомы, отличные от серы, например, атом углерода как в карбаналоге аманитина. Функционально аматоксины определены как пептиды или депсипептиды, которые ингибируют РНК-полимер азу II млекопитающих. Предпочтительные аматоксины представляют аматоксины с функциональной группой (например, карбоксильной группой, аминогруппой, гидроксигруппой, тиольной или захватывающей тиол группой), которые могут реагировать с молекулами-линкерами или мишень-связывающими фрагментами, как определено ниже.
В контексте настоящего изобретения термин «аманитины», в частности, относится к бициклической структуре, которая основана на аспарагиновой кислоте или аспарагиновом остатке в положении 1, пролиновом остатке, в частности, гидроксипролиновом остатке в положении 2, изолейцине, гидроксиизолейцине или дигидроксиизолейцине в положении 3, триптофановом или гидрокситриптофановом остатке в положении 4, глициновом остатке в положениях 5 и 7, изолейциновом остатке в положении 6 и цистеиновом остатке в положении 8, в частности, производном цистеина, которое окислено до сульфоксидного или сульфонового производного (касательно нумерации и типичных примеров аманитинов, см. фиг. 1), и, кроме того, включает все их химические производные; также все их полусинтетические аналоги; также все их синтетические аналоги, полученные из структурных элементов согласно основной структуре природных соединений (циклические, из 8 аминокислот), также все синтетические или полусинтетические аналоги, содержащие негидроксилированные аминокислоты вместо гидроксилированных аминокислот, также все синтетические или полусинтетические аналоги, причем в каждом случае любое такое производное или аналог является функционально активным посредством ингибирования РНК-полимеразы II млекопитающих.
Термин «мишень-связывающий фрагмент» при использовании в настоящем документе относится к любой молекуле или части молекулы, которая может специфически связываться с молекулой-мишенью или эпитопом-мишенью. Предпочтительными мишень-связывающими фрагментами в контексте настоящей заявки являются (i) антитела или их антигенсвязывающие фрагменты; (ii) подобные антителам белки и (iii) аптамеры нуклеиновых кислот.«Мишень-связывающие фрагменты», подходящие для использования в настоящем изобретении, обычно имеют молекулярную массу 40000 Да (40 кДа) или более.
«Линкер» в контексте настоящей заявки относится к молекуле, которая увеличивает расстояние между двумя компонентами, например, для снижения стерического влияния между мишень-связывающим фрагментом и аматоксином, что может в ином случае снижать способность аматоксина к взаимодействию с РНК-полимеразой II. Линкер может служить другой цели, поскольку он может облегчать специфическое высвобождение аматоксина в клетку, на которую нацелен мишень-связывающий фрагмент. Предпочтительно, чтобы линкер и предпочтительно связь между линкером и аматоксином с одной стороны и связь между линкером и антителом с другой стороны была стабильна при физиологических условиях вне клетки, например, в крови, тогда как она может расщепляться внутри клетки, в частности, внутри клетки-мишени, например, раковой клетки или иммуноците. Для обеспечения этой селективной стабильности линкер может содержать функциональные группы, которые являются предпочтительно чувствительными к рН или чувствительными к протеазе. Альтернативно, связь, связывающая линкер с мишень-связывающим фрагментом, может обеспечивать селективную стабильность. Предпочтительно линкер имеет длину по меньшей мере 1, предпочтительно длину 1-30 атомов (например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30 атомов), причем одна сторона линкера реагировала с аматоксином, а другая сторона - с мишень-связывающим фрагментом. В контексте настоящего изобретения линкер предпочтительно представляет собой C1-30-алкильную, C1-30-гетероалкильную, С2-30-алкенильную, С2-30-гетероалкенильную, С2-30-алкинильную, С2-30-гетероалкинильную, циклоалкильную, гетероциклоалкильную, арильную, гетероарильную, аралкильную или гетер оар ал кил ьную группу, необязательно замещенную. Линкер может содержать один или несколько структурных элементов, таких как амидные, сложноэфирные, эфирные, тиоэфирные, дисульфидные, углеводородные фрагменты и подобное. Линкер может также содержать комбинацию двух или более из этих структурных элементов. Каждый из этих структурных элементов может быть представлен в линкере больше одного раза, например, два раза, три раза, четыре раза, пять раз или шесть раз. В некоторых вариантах осуществления линкер может содержать дисульфидную связь. Понятно, что линкер должен прикрепляться или за одну стадию, или за две или более последовательных стадий к аматоксину и мишень-связывающему фрагменту. Для этого линкер должен нести две группы, предпочтительно на проксимальном и дистальном конце, которые могут (i) образовывать ковалентную связь с группой, предпочтительно активированной группой на аматоксине или мишень-связывающиемпептиде, или (ii) который является или может быть активирован с образованием ковалентной связи с группой на аматоксине. Следовательно, если присутствует линкер, предпочтительно, чтобы химические группы были на дистальном и проксимальном конце линкера, что является результатом такой реакции сочетания, например, сложноэфирной, эфирной, уретановой, пептидной связи и пр. Присутствие «линкера» является необязательным, т.е. токсин может быть непосредственно связан с остатком мишень-связывающего фрагмента, в некоторых вариантах осуществления конъюгата мишень-связывающего фрагмента и токсина.
Согласно второму аспекту настоящее изобретение относится к (S)-6-ацетилокси-М-трет-бутоксикарбонилтриптофану (HDP 30.2550), (S)-6-гидрокситриптофану или любым соединениям-предшественникам согласно настоящему изобретению для использования в синтезе аманитина, или производных аманитина, или конъюгатов аматоксина и лекарственного средства.
В одном варианте осуществления настоящее изобретение относится к дегидроаминокислотному соединению, выбранному из группы, состоящей из соединений I, II, III, IV и V:
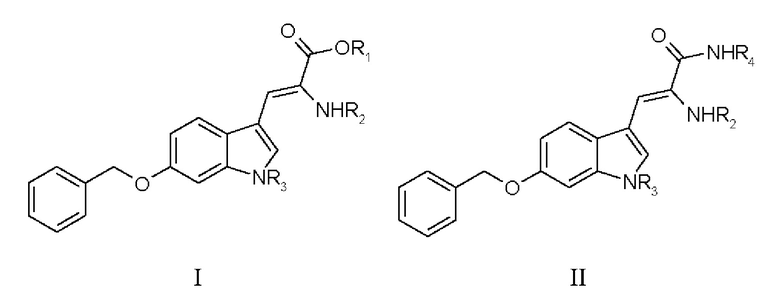
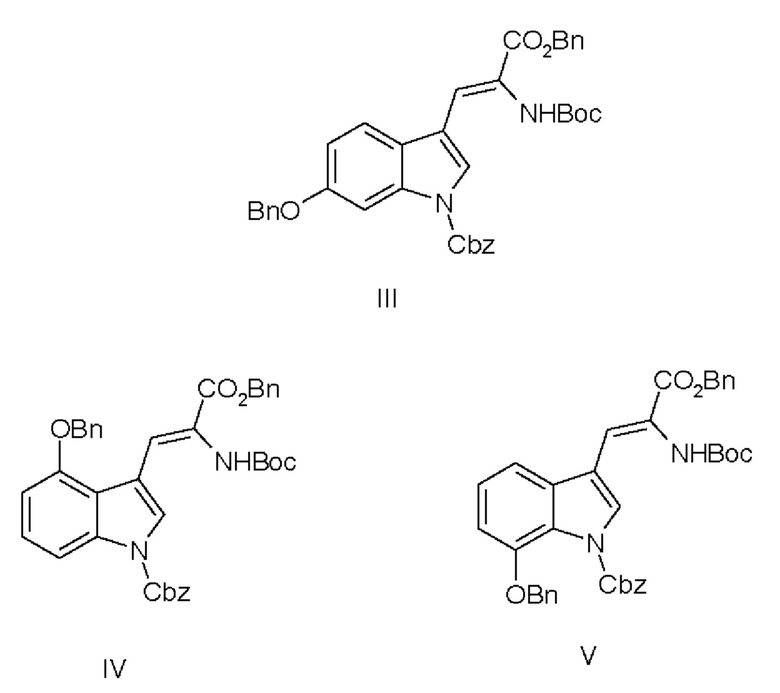
где
R1 выбран из: Н, алкила, алкенила, аралкила, необязательно замещенного,
R2 выбран из: Воc-, Cbz-, N-защитных групп,
R3 выбран из: Воc-, Cbz-, N-защитных групп,
R4 представляет собой аминокислотный остаток,
для использования в синтезе аманитина, или производных аманитина, или конъюгатов аматоксина и лекарственного средства.
Соединения III, IV и V являются предпочтительными.
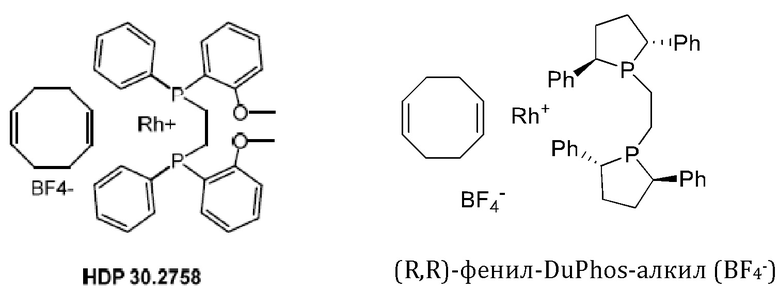
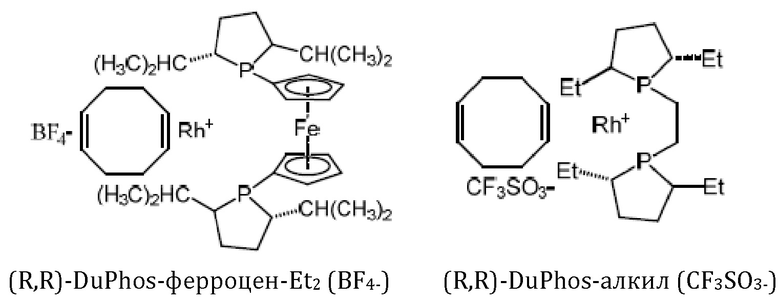
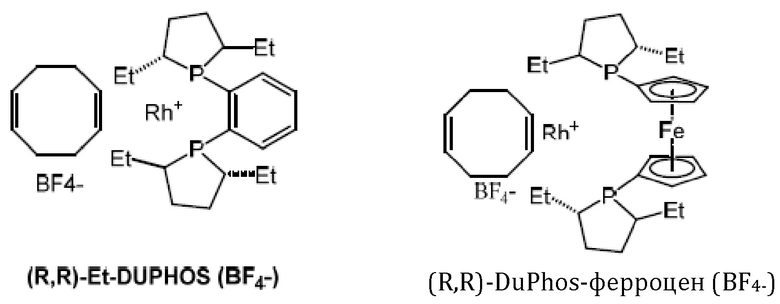
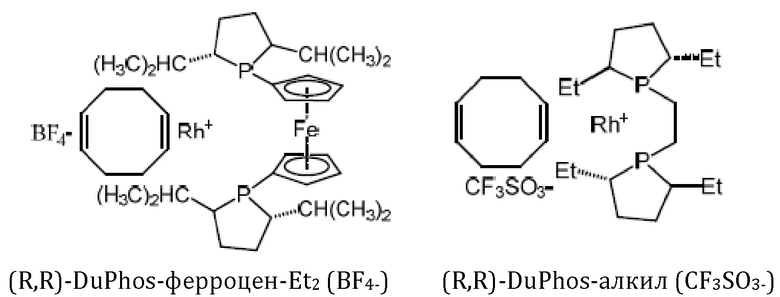
Кроме того, авторы настоящего изобретения неожиданно обнаружили, что из большего списка каталитических соединений, протестированных для асимметричного гидрирования (см. таблицу 1, фиг. 5), катализатор тетрафторборат циклооктадиен-1,5-[(R,R)-DIРАМР]родия, HDP 30.2758, давал наибольшую энантиомерную чистоту, которая составляла >98%).
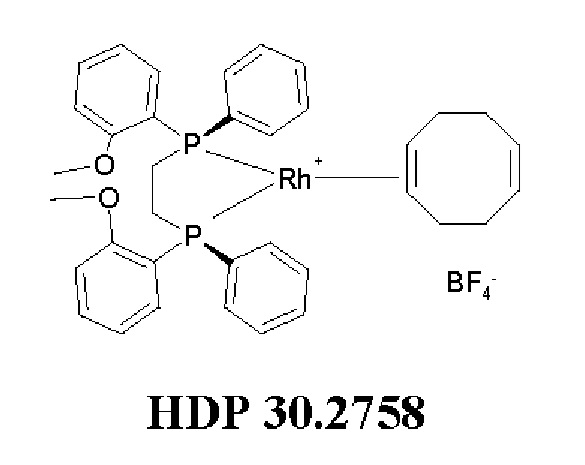
Обнаружили, что только катализатор HDP 30.2758 давал очень высокую чистоту более 98% (S)-энантиомеров. Все другие катализаторы, за исключением (R,R)-Et-DUPHOS (BF4-), давали значительно более низкие чистоты (S)-энантиомеров 50-70%. Кроме того, показатели общего абсолютного оборота и выхода соединения были намного хуже, чем с HDP 30.2758.
Катализаторы, протестированные для асимметричного гидрирования и соответствующие уровни чистоты (З)-энантиомеров подытожены в таблице 1.

Таким образом, согласно третьему аспекту настоящее изобретение относится к использованию соединения, выбранного из группы, состоящей из соединения HDP
30.2758, (R,R)-Et-DUPHOS (BF4-), (R,R)-DuPhos-ферроцeна (BF4-), (R,R)-DuPhos-ферроцен-Et2 (BF4-), (R,R)-DuPhos-алкила (СF3SО3-) и (R,R)-фенил-DuPhos-алкила (BF4-), в качестве катализатора дня гадрированияв следящей реакции:

Сошасно этому аспекту настоящее изобретение предпочтительно относится к использованию хиральных катализаторов - тетрафторборату циклооктадиен-1,5-[(R,R)-DIPAMP]родия (HDP 30.2753) или (R,R)-Et-DUPHOS (BF4-), наиболее предпочтительно хирального катализатора тетрафторбората циклооктадиен-1,5-[(R,R)-DIРАМР]родия(HDP 30.275S), в качестве катализатора дгшгадрирования в еле дующей реакции:
Включение производных триптофана в качестве структурных элементов в предшественники аманитина было описано ранее в заявке РСТ/ЕР 2018/071268, содержание которой включено в настоящий документ ссылкой. Например, HDP 30.2115 можно синтезировать путем включения структурного элемента незамещенного Hpi в молекулу пептидного предшественника аманитина для получения синтетического аманитина, 6-пщроксизамешенный структурный элемент HDP 30.2555 можно использовать для синтеза 6-гидроксизамещенных аманитинов согласно настоящему изобретению.
Примеры
Хотя настоящее изобретение было показано и описано подробно на фигурах и в вышеуказанном описании, такую иллюстрацию и описание следует рассматривать как иллюстративные или типичные, а не ограничительные; настоящее изобретение не ограничено раскрытыми вариантами осуществления. Другие варианты раскрытых вариантов осуществления могут пониматься и выполняться специалистами в данной области при осуществлении на практике заявленного изобретения, из изучения фигур, раскрытия и приложенной формулы изобретения. В формуле изобретения слово «содержащий» не исключает другие элементы или стадии, и формы единственного числа не исключают множественное. Сам факт того, что некоторые величины перечислены в различных зависимых пунктах формулы, не указывает на то, что комбинацию этих величин нельзя предпочтительно использовать. Любые номера позиций в формуле изобретения не следует рассматривать как ограничивающие объем.
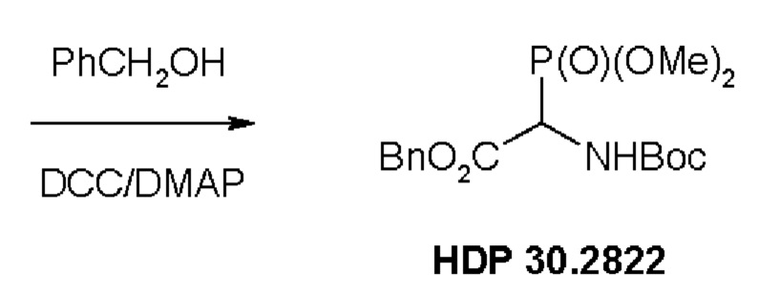
Пример 1. Синтез фосфониевого предшественника (структурного элемента) HDP 30.2822
Синтез фосфониевого предшественника (структурного элемента) HDP 30.2822 проводили, как описано (CHEMISTRY A European Journal, 2018, Vol. 24, Issue 7, pp 1544-1553):
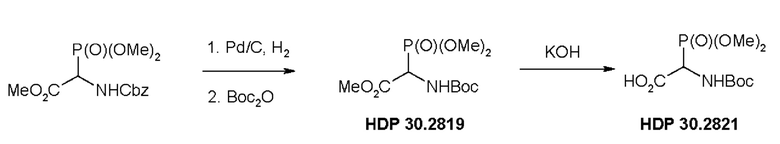
Пример 1.1. Получение триметилового сложного эфира (R,S)-Boc-α-фосфоноглицина HDP 30.2819
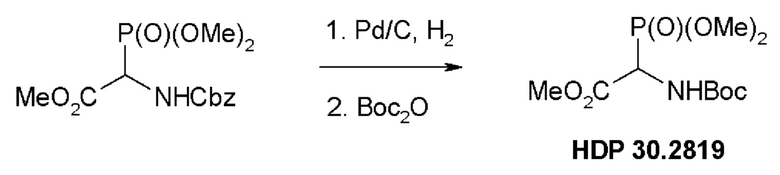
5,0 г (15,1 ммоль) триметилового сложного эфира (R,S)-М-Сbz-фосфоноглицина (CAS: 88568-95-0) гидрировали при помощи 1,4 г 10% Pd⋅C в 100 мл метанола при 1 атм., пока реакция не завершалась согласно TLC (хлороформ/метанол 15:1). Реакция завершалась за 3 часа. Катализатор отфильтровывали через набивку Celite® (диатомитовой земли) и метанольный раствор свободного амина концентрировали под вакуумом до бесцветного масла (2,9 г). Неочищенное масло использовали для следующей стадии без очистки.
2,9 г неочищенного продукта гидрирования растворяли в 20 мл дихлорметана и обрабатывали 3,23 мл (15,1 ммоль) ди-трет-бутилдикарбоната (Вoc2О). Через 17 часов перемешивания при температуре окружающей среды в атмосфере аргона реакционную смесь концентрировали досуха. Оставшееся бесцветное масло кристаллизовалось до белого твердого вещества (4,3 г). Неочищенный HDP 30.2819 использовали для следующей стадии без очистки.
Пример 1.2. Получение триметилового сложного эфира (R,S)-N-Boc-a-фосфоноглицина HDP 30.2821
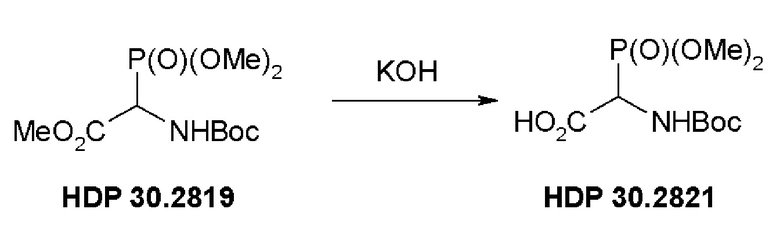
4,3 г (предполагалось 14,3 ммоль) неочищенного HDP 30.2819 растворяли в 10 мл 1,4-диоксана и быстро обрабатывали в атмосфере аргона и при комнатной температуре при помощи 14,5 мл 1 н KОН. Через 85 минут реакционную смесь разводили 36 мл воды и экстрагировали при помощи 35 мл этилацетата. Этилацетатный экстракт отбрасывали и водный раствор подкисляли до рН 3 добавлением по каплям 1 н НСl. Реакционную смесь экстрагировали при помощи 60 мл этилацетата (2х) и сушили над MgSO4. Полученное белое твердое вещество, 2,1 г HDP 30.2821, сушили под вакуумом и использовали непосредственно без очистки для следующей стадии реакции.
Мр: 148-150°С (Lit. JACS 111, 6244, 1989 mp: 154-155°С)
MS (ESI-) обнаружено: 282,00 [М-Н]-; рассчитано: 283,08 (C9H18NO7P)
MS (ESI-) обнаружено: 238,17 [М-СO2]-
Пример 1.3. Получение бензилового сложного эфира (R,S)-N-Boc-α-диметилфосфоно)-глицина HDP 30.2822
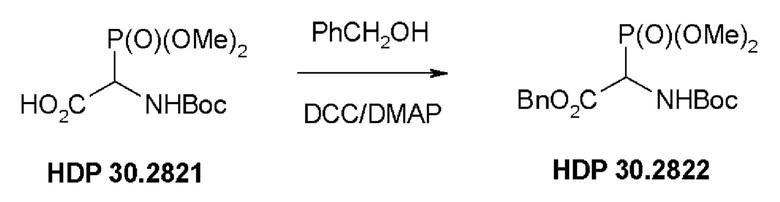
2,0 г (7,1 ммоль) HDP 30.2821 в 90 мл сухого дихлорметана обрабатывали при помощи 4,6 мл (44,1 ммоль) бензилового спирта, 230 мг DMAP и 2,2 г (10,6 ммоль) DCC, растворенного в 7 мл дихлорметана. Реакционную смесь перемешивали в атмосфере аргона при температуре окружающей среды в течение 24 часов. Затем мочевину отфильтровывали и органическую фазу промывали 5% лимонной кислотой и сушили над MgSO4. После испарения дихлорметана оставшееся полутвердое вещество отбирали в этилацетате и снова фильтровали для удаления дополнительной мочевины. Неочищенный продукт очищали флэш-хроматографией на 330 г силикагелевой колонке (длина волны обнаружения 254 нм) с градиентом н-гексана к н-гексану/этилацетату (1:2) и получали после испарения 1,94 г (73%) HDP 30.2822 в виде белого твердого вещества.
MS (ESI+) обнаружено: 373,92 [МН]+; рассчитано: 373,13 (С16Н242О7Р)
MS (ESI+) обнаружено: 396,17 [M+Na]+
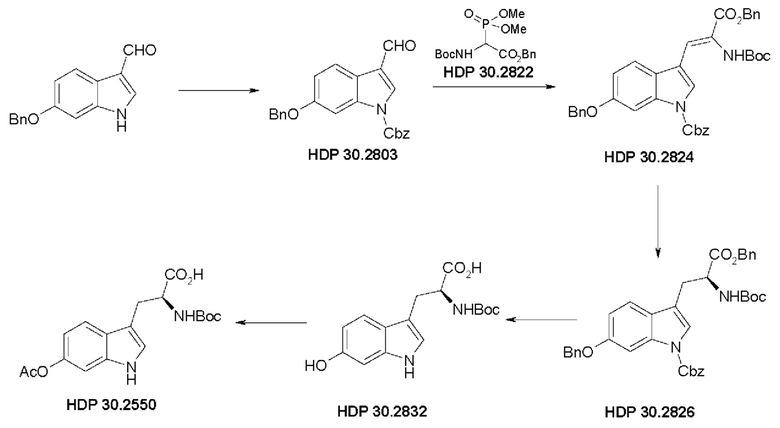
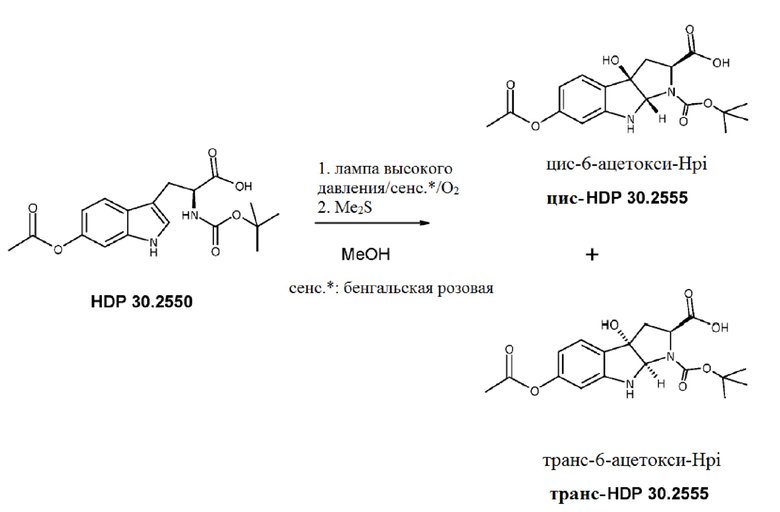
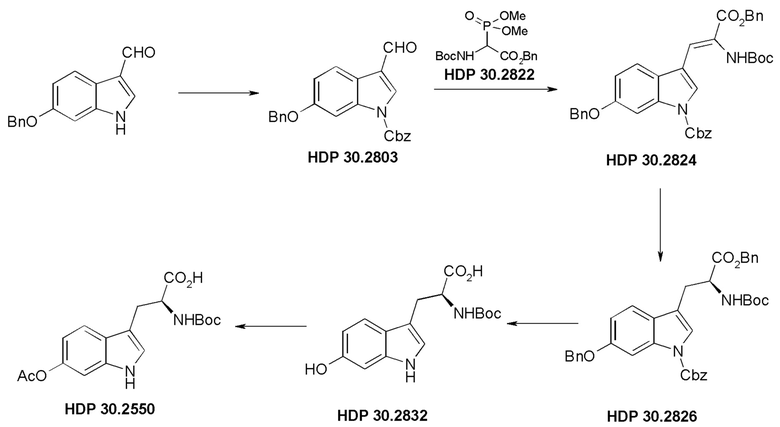
Пример 2. Синтез (S)-6-ацетилокси-N-трет-буталоксикарбонилтриптофана HDP 30.2550 в качестве предшественника HDP 30.2555 (гидрокси-Hpi)
Путь синтеза подытожен на следующей схеме синтеза.
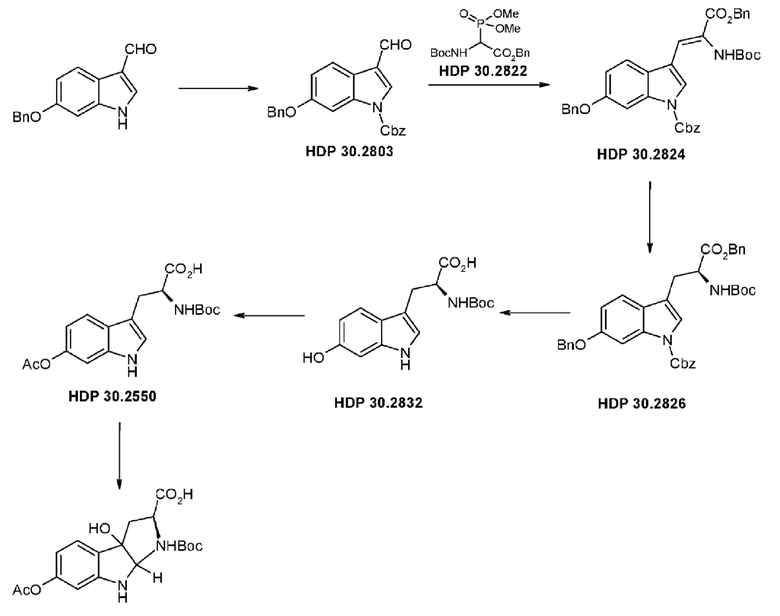
HDP 30.2555 (цис/транс)
Пример 2.1. Получение N-Cbz-6-бензилоксииндол-3-альдегида HDP 30.2803
Исходный материал 6-бензилоксииндол-3-альдегид для синтеза коммерчески доступен или может быть получен реакцией Вильсмейера с высокими выходами, начиная с 6-бензилоксииндола.
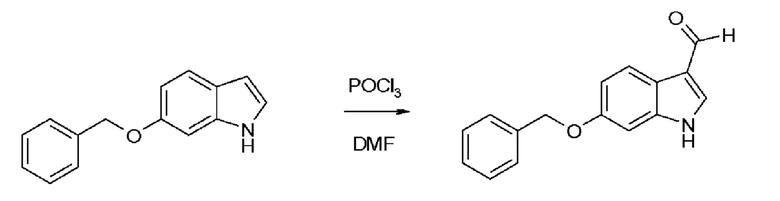
Не требуется хроматография для очистки.
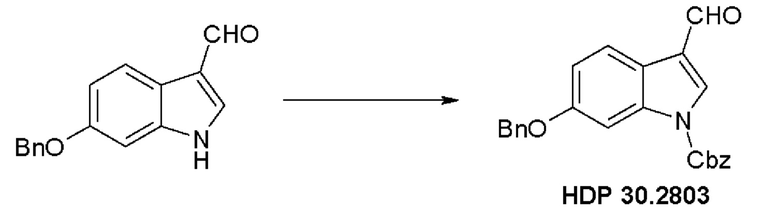
Триэтиламин (1,66 мл, 11,94 ммоль, 1,50 экв.) добавляли при помощи шприца в раствор 6-бензилокси-3-формилиндола (2,00 г, 7,96 ммоль, 1 экв.) и (DMAP) 4-диметиламинопиридина (97,23 мг, 796 мкмоль) в дихлорметане (20 мл) при 23°С. Бензилхлорформиат (1,45 мл, 10,35 ммоль, 1,30 экв.) добавляли по каплям в раствор при помощи шприца. Через 1 час другую часть бензилформиата (223 мкл, 1,59 ммоль, 0,20 экв.) добавляли при помощи шприца. Через 95 мин реакционную смесь разбавляли дихлорметаном (85 мл) и промывали насыщенным водным раствором бикарбоната натрия (85 мл). Водный слой дополнительно экстрагировали дихлорметаном (2 × 20 мл). Объединенные органические слои промывали водным хлороводородом (1 н, 85 мл) и полученный водный слой экстрагировали дихлорметаном (2 × 20 мл). Объединенные органические слои сушили над безводным MgSO4, фильтровали и концентрировали под пониженным давлением. Неочищенный продукт очищали флэш-хроматографией на 330 г силикагелевой колонке (длина волны обнаружения 254 нм) с градиентом н-гексана/этилацетата 4:1 к н-гексану/этилацетату (1:1) и получали после испарения 2,33 г (76%) HDP 30.2803 в виде белого твердого вещества.
1Н-ЯМР (400 МГц, CDCl3, 5=части на миллион)
δ=5,05 (s, 2Н, ОСН2); 5,47 (s, 2Н, СООСH2); 7,06-8,14 (m, Ar-H, 14Н); 10,01 (s, 1H, СНО)
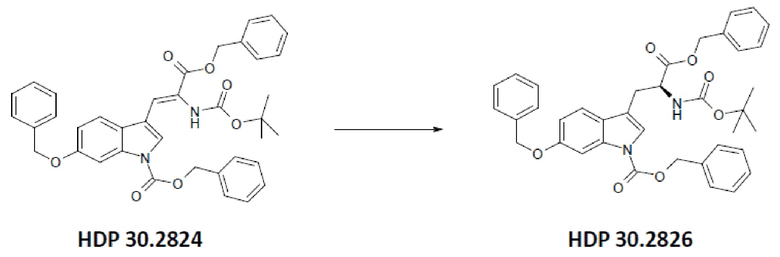
Пример 2.2. Получение бензилового сложного эфира [6-бензилокси-1Н-(бензилоксикарбонил)-3-индол]-2-(трет-бутилоксикарбониламино)акриловой кислоты HDP 30.2824
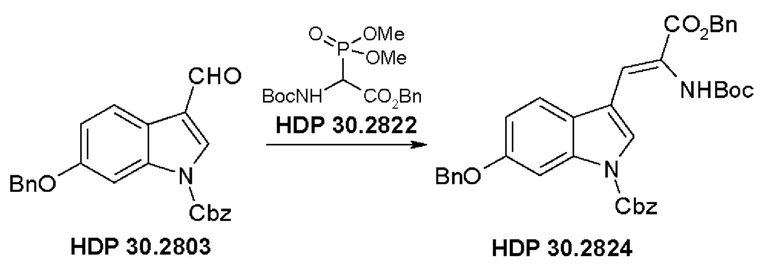
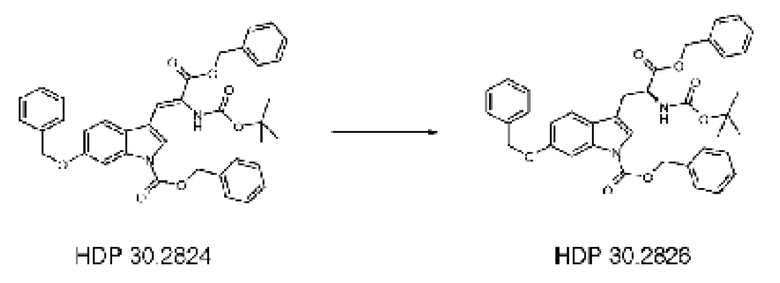
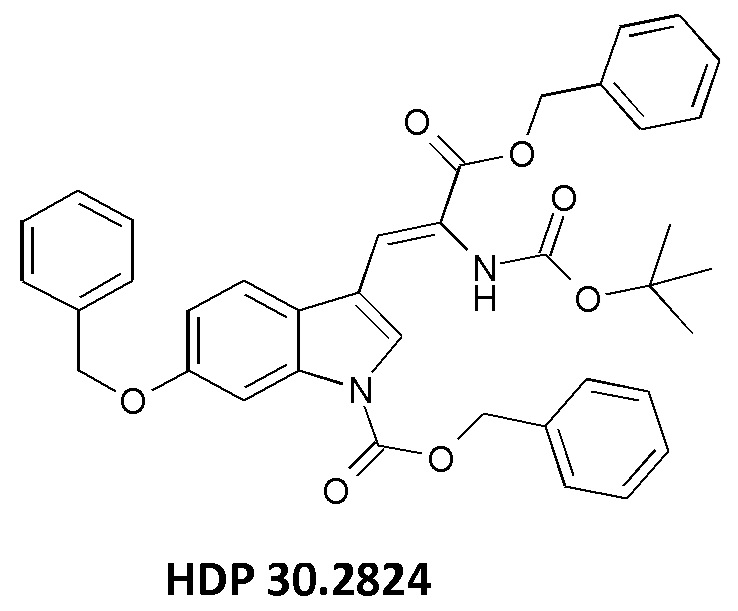
1,90 г (5,09 ммоль) бензилового сложного эфира (R,S)-N-Boc-α-диметилфосфоно)глицина HDP 30,2822 суспендировали в аргоне в 8 мл дихлорметана. Добавляли 0,705 мл (4,73 ммоль) DBU. Через 10 минут перемешивания медленно добавляли 1,66 г (4,31 ммоль) N-Cbz-6-бензилоксииндол-3-альдегида HDP 30.2803 в 4,7 мл дихлорметана. Реакционную смесь перемешивали в течение 5 часов и растворитель выпаривали под пониженным давлением. Остаток растворяли в 120 мл этилацетата и органический раствор промывали 2 раза при помощи 50 мл 1 н HCl и 50 мл рассола, сушили над MgSO4 и концентрировали под пониженным давлением с получением 2,70 г неочищенного материала. Неочищенный продукт очищали флэш-хроматографией на 330 г силикагелевой колонке (длина волны обнаружения 254 нм) с градиентом н-гексана к н-гексану/этилацетату (1:1) и получали после испарения 2,00 г (73%) HDP 30.2824 в виде белого твердого вещества.
MS (ESI+) обнаружено: 632,92 [МН]+; рассчитано: 632,25 (С38Н36N2О7)
MS (ESI+) обнаружено: 655,25 [M+Na]+
Пример 2.3. Получение бензилового сложного эфира (S)-6-бензилокси-N-трет-бутоксикарбонил-1-Сbz-L-триптофана HDP 30.2826
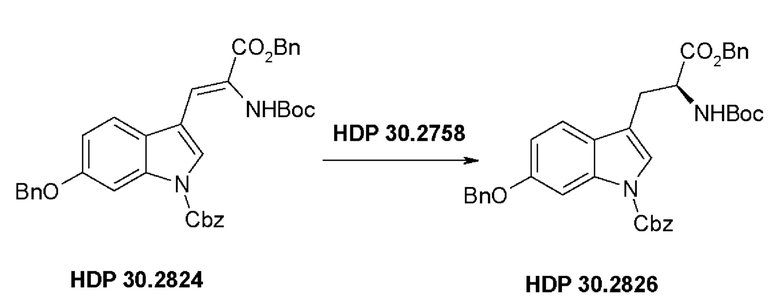
Пример 2.3.1. Синтез катализатора тетрафторбората циклооктадиен-1,5-[(R,R)-DIPAMP]родия HDP 30.2758
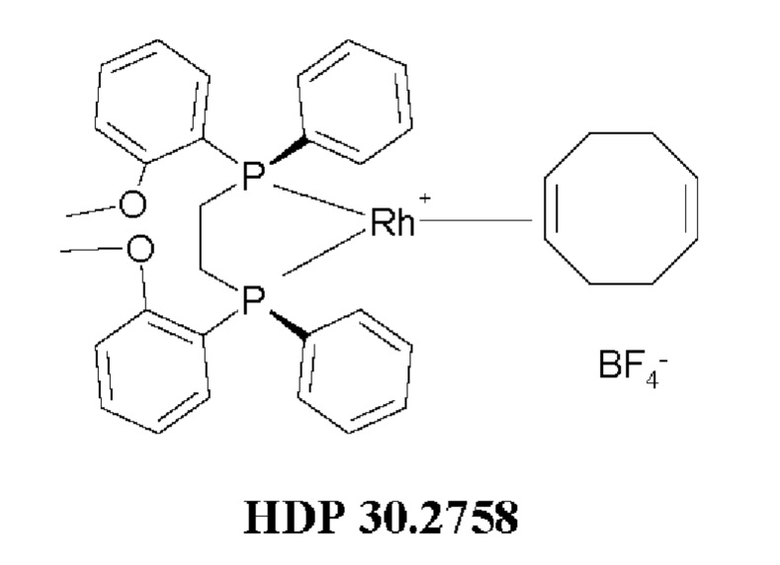
97,0 мг (0,20 ммоль) бис(циклооктадиен-1,5)-дихлордиродия [Rh(COD)Cl]2 (CAS: 12092-47-6. Alfa Aesar) добавляли в суспензию 180,0 мг (0,39 ммоль) (R,R)-DIPAMP (CAS:55739-58-7, Alfa Aesar) в 2,0 мл метанола/воды (1,5 мл /0,5 мл). Окрашенная оранжевым суспензия, перемешанная в течение 1 часа в атмосфере аргона, давала оранжевый раствор. Комплекс осаждался путем медленного добавления (в течение 30 минут) раствора 65,0 мг (0,6 ммоль) тетрафторбората натрия в 0,5 мл воды. Через 2,5 часа перемешивания при комнатной температуре оранжевые кристаллы отфильтровывали, промывали дважды небольшими порциями воды и сушили под высоким вакуумом. 240 мг (81%) катализатора тетрафторбората циклооктадиен-1,5-[(R,R)- DIPAMP]родия HDP 30.2758 получали в виде ярко-желтого порошка. Катализатор использовали без дополнительной очистки.
Пример 2.3.2. Синтез бензилового сложного эфира (S)-6-бензилокси-N-трет-бутоксикарбонил-1-Сbz-L-триптофана HDP 30.2826
В 250 мл автоклав из нержавеющей стали загружали 35,0 мг (0,08 ммоль) тетрафторбората циклооктадиен-1,5-[(R,R)- DIPAMP]родия HDP 30.2758, 1000 мг (1,8 ммоль) бензилового сложного эфира [6-бензилокси-1Н-(бензилоксикарбонил)-3-индол]-2-(трет-бутилоксикарбониламино)акриловой кислоты HDP 30.2824 в 40 мл сухого метанола/15 мл дихлорметана. После четырех циклов вакуума/Ar и H2 повышали давление реакционной смеси до исходного давления 12 бар. Реакции позволяли проходить в течение 4 дней при температуре окружающей среды. После испарения растворителя неочищенный продукт очищали флэш-хроматографией на 220 г силикагелевой колонке (длина волны обнаружения 254 нм) с н-гексаном/этилацетатом (3:1) и получали после испарения 0,79 г (79%) HDP 30.2826 в виде белого порошка.
MS (ESI+) рассчитано: 634,26 (C38H38N2O7)
MS (ESI+) обнаружено: 657,33 [M+Na]+
Пример 2.4. Получение N-трет-бутоксикарбонил-(S)-триптофана HDP 30.2832
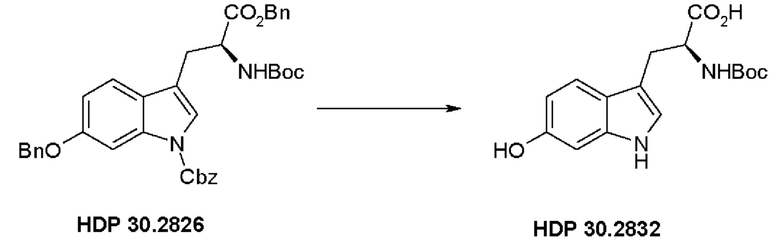
700 мг (1,10 ммоль) бензилового сложного эфира (S)-6-бензилокси-N-трет-бутоксикарбонил-1-Сbz-триптофана HDP 30.2826 гидрировали с 100 мг Pd-C 10% в смеси 7 мл этилацетата и 4 мл метанола. После 3 часов гидрирования (контроль TLC с хлороформом/метанолом 19:1+1% АсОН) при комнатной температуре и 1 атм. катализатор удаляли фильтрацией через набивку Celite®. Растворитель удаляли и оставшийся неочищенный остаток, 378 мг HDP 30.2832, использовали для следующей стадии без очистки.
Пример 2.5. Получение (S)-6-ацетилокси-N-трет-бутилоксикарбонилтриптофана HDP 30.2550
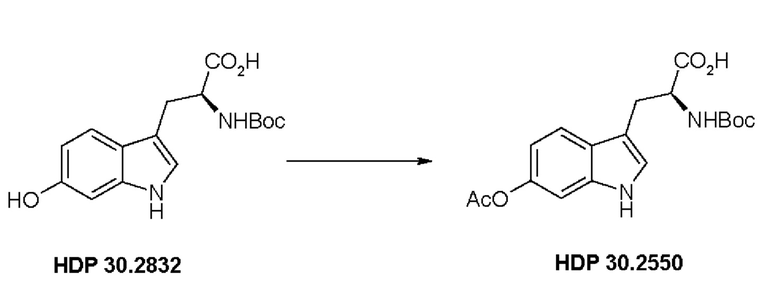
378 мг неочищенного HDP 30.2832 (предполагалось 1,10 ммоль) растворяли в 2,21 мл 1 н NaOH. В атмосфере аргона и при температуре окружающей среды 208,5 мкл (2,20 ммоль) уксусного ангидрида добавляли за один раз. Смесь перемешивали в течение 3,5 часов и подкисляли 5% лимонной кислотой. Реакционную смесь экстрагировали 3х этилацетатом, и объединенные органические фазы промывали 5% хлоридом натрия и сушили над MgSO4. Фильтрация и выпаривание досуха давали 380 мг неочищенного материала. Неочищенный продукт очищали флэш-хроматографией на 120 г силикагелевой колонке (длина волны обнаружения 254 нм) с градиентом дихлорметана +2% АсОН / дихлорметана / метанола (15:1)+2% АсОН и получали после испарения 270 мг (68%) HDP 30.2550 в виде белого твердого вещества.
MS (ESI-) обнаружено: 361,17 [М-Н]-; рассчитано: 362,15 (C18H22N2O6)
MS (ESI-) обнаружено: 723,08 [2М-Н]-
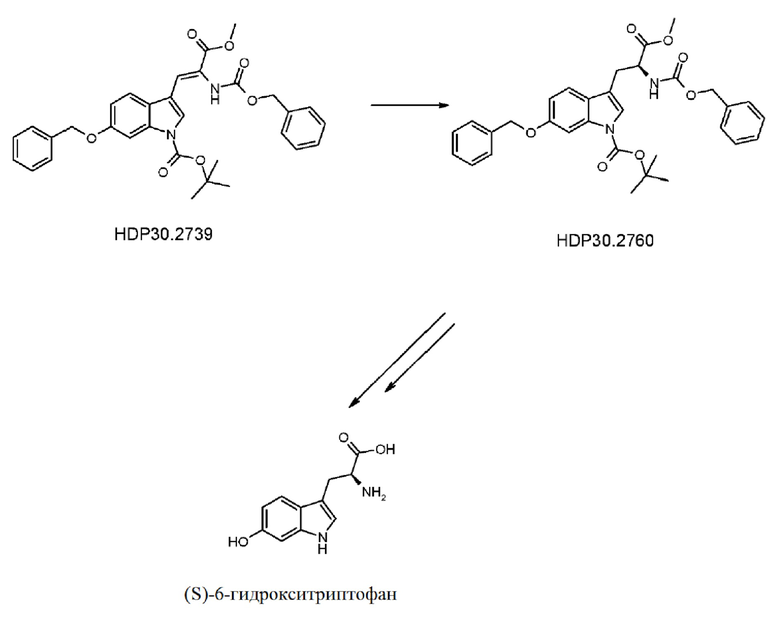
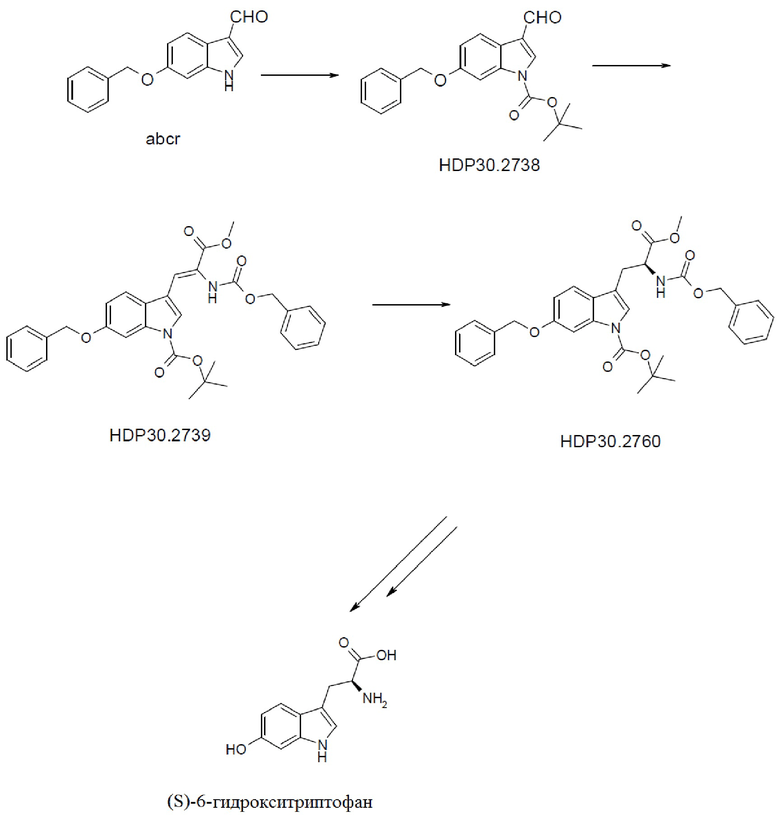
Пример 3. Получение (S)-6-гидрокситриптофана при помощи асимметричного гидрирования дегидроаминокислоты
Путь синтеза подытожен на следующей схеме синтеза.

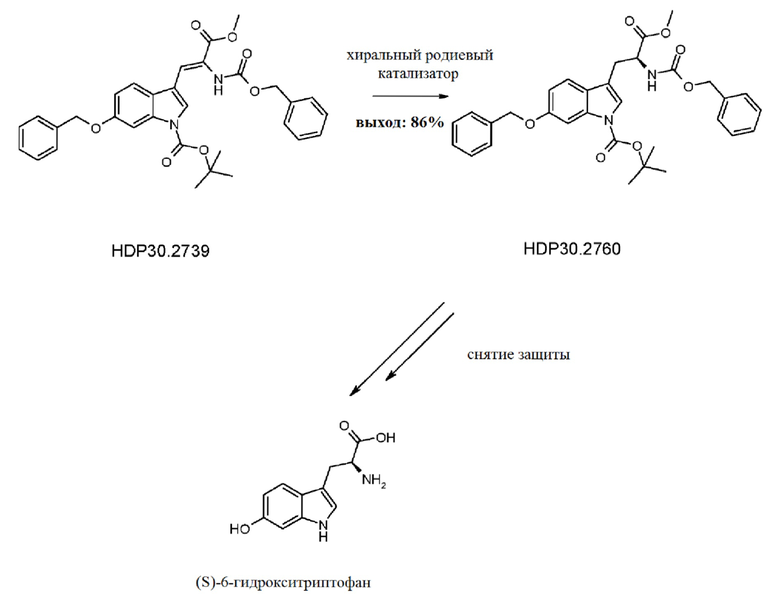
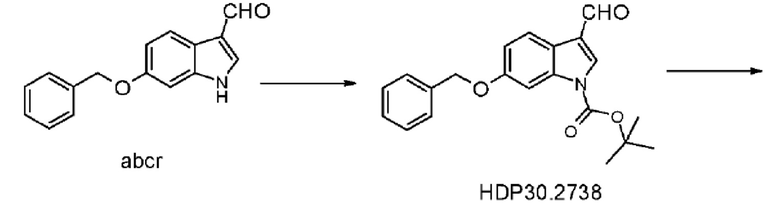
Пример 3.1. Получение 6-бензилокси-Ш-индол-3-карбальдегида
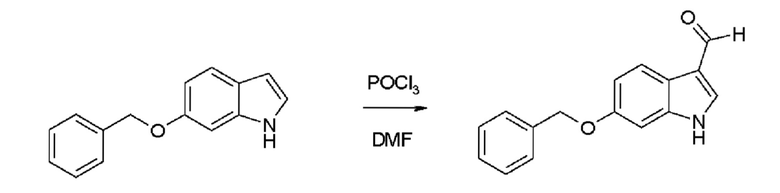
В перемешанный раствор оксихлорида фосфора (10,0 мл, 107,0 ммоль) в DMF (35 мл) добавляли раствор 6-бензилоксииндола (22,3 г, 100,0 ммоль) в DMF (25 мл) при комнатной температуре. Через 45 минут реакционную смесь выливали в ледяную воду (200 мл). В эту смесь добавляли твердый NaOH (19,0 г, 475,0 ммоль) и воду (100 мл). Через 30 минут дополнительное количество воды (200 мл) добавляли и всю смесь нагревали с обратным холодильником в течение 3 минут. Осадок собирали, промывали 5 порциями 50 мл холодной воды и сушили с получением 24,8 г (98,8%) 6-бензилокси-1H-индол-3-карбальдегида в виде белого порошка. Соединение было идентичным эталонному материалу и достаточно чистым для следующей реакции.
Пример 3.2. Получение 6-бензилокси-1Н-1-трет-бутоксикарбонилиндолкарбальдегида HDP 30.2738
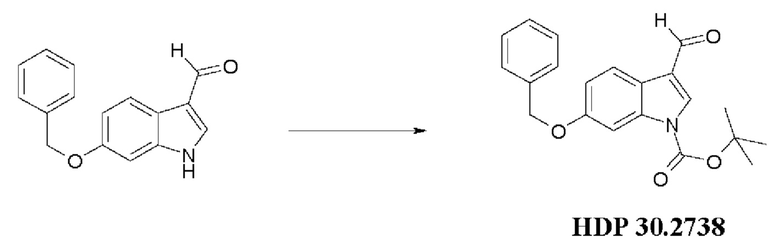
10,0 г (39,8 ммоль) 6-бензилокси-1Н-индол-3-карбальдегида суспендировали в 100 мл дихлорметана и обрабатывали при помощи 0,56 г (4,5 ммоль) 4-диметиламинопиридина DM АР и 10,5 г (47,3 ммоль) ди-трет-бутилдикарбоната Вoc2О, растворенного в 10 мл дихлорметана. После перемешивания в течение 2 часов добавляли 100 мл 1 н KHSO4 и дихлорметан выпаривали. Водный слой экстрагировали несколькими порциями диэтилового эфира (2 × 200 мл) и объединенные органические экстракты промывали 250 мл 1 н KHSO4, 250 мл 1 н NaHCO3 и 250 мл рассола. Органический слой сушили над MgSO4 и концентрировали под пониженным давлением с получением 12,0 г (86%) красно-коричневатого порошка. Соединение было достаточно чистым для следующей стадии реакции.
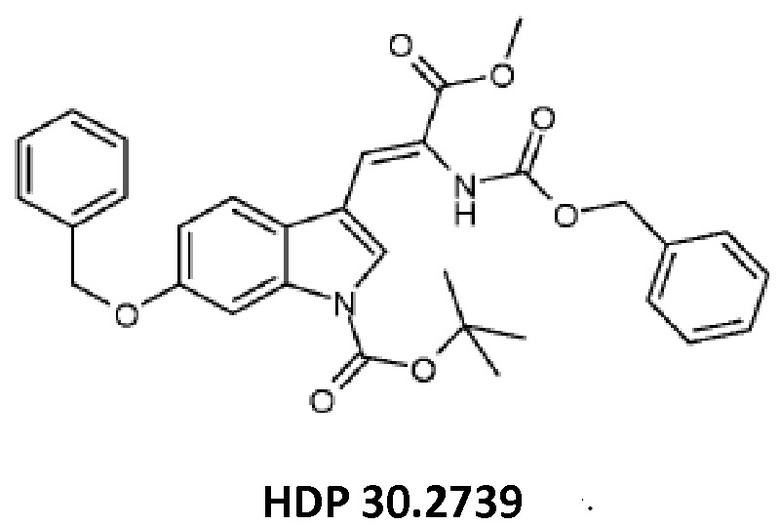
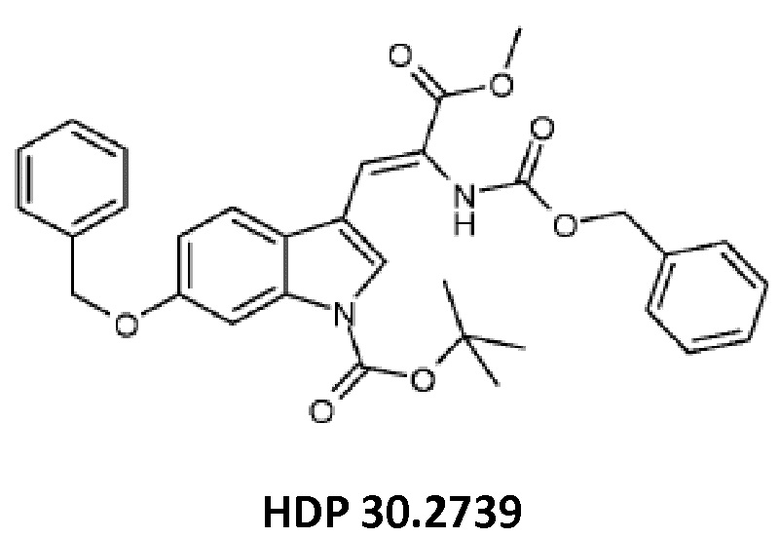
Пример 3.3. Получение метилового сложного эфира 3-[6-бензилокси-1Н-(1-трет-бутоксикарбонил)-3-индол]-2-(бензилоксикарбониламино)акриловой кислоты HDP 30.2739
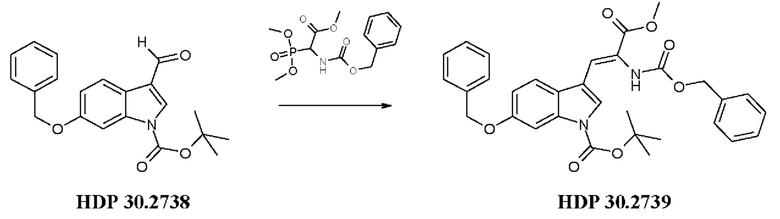
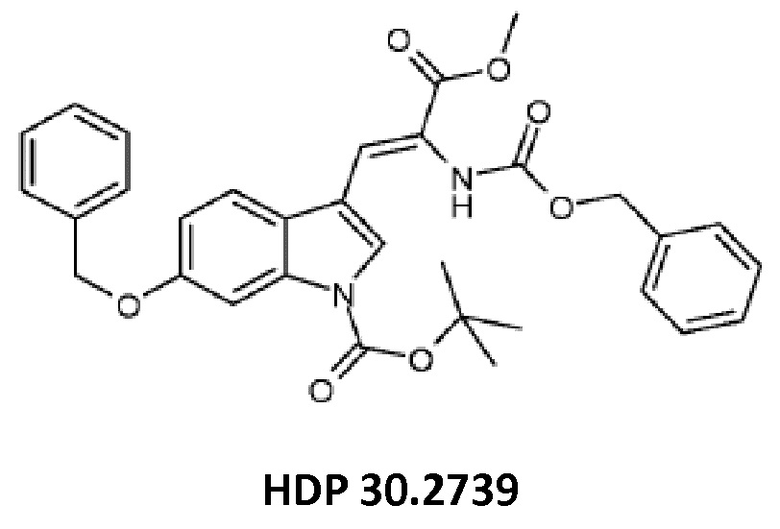
5,12 г (15,44 ммоль) триметилового сложного эфира (R,S)-бензилоксикарбонил-5-фосфоноглицина (CAS: 88568-95-0, Alfa Aesar) растворяли в атмосфере аргона в 18 мл дихлорметана. Добавляли 2,14 мл (14,31 ммоль) DBU. После 10 минут перемешивания медленно добавляли 4,60 г (13,07 ммоль) 6-бензилокси-1Н-1-трет-бутилоксикарбонилиндол-3-карбальдегида HDP 30.2738 в 14 мл дихлорметана. Реакционную смесь перемешивали в течение 6 часов, и растворитель выпаривали под пониженным давлением. Остаток растворяли в 300 мл этилацетата, затем органический раствор промывали 2 раза при помощи 120 мл 1 н HCl и 120 мл рассола, сушили над MgSO4 и концентрировали под пониженным давлением с получением 7,43 г неочищенного материала. Неочищенный продукт очищали флэш-хроматографией на 330 г силикагелевой колонке (длина волны обнаружения 254 нм) с градиентом н-гексана к н-гексану/этилацетату (2:1) и получали после испарения 5,23 г (72%) HDP 30.2739 в виде белого твердого вещества.
MS (ESI+) обнаружено: 557,17 [MH]+ рассчитано: 557,22 (C32H32N2O7)
MS (ESI+) обнаружено: 579,25 [M+Na]+
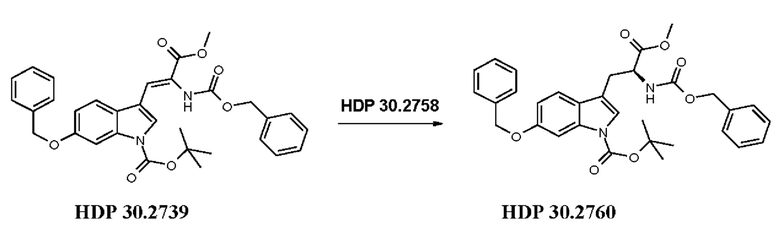
Пример 3.4. Получение метилового сложного эфира 6-бензилокси-N-карбобензилокси-1-трет-бутоксикарбонил-L-триптофана HDP 30.2760
Пример 3.4.1. Синтез тетрафторбората циклооктадиен-1,5-[(R,R)-DIPAMP]родия HDP 30.2758
Синтезировали катализатор тетрафторборат циклооктадиен-1,5-[(R,R)-DIPAMP]родия HDP 30.2758, как описано в примере 2.3.1.
Пример 3.4.2. Синтез метилового сложного эфира (S)-6-бензилокси-N-карбобензилокси-1-трет-бутоксикарбонилтриптофана HDP 30.2760
В 250 мл автоклав из нержавеющей стали загружали 60,0 мг (0,08 ммоль) тетрафторбората циклооктадиен-1,5-[(R,R)-DIPAMP]родия HDP 30.2758 и 1000 мг (1,8 ммоль) метилового сложного эфира [6-бензилокси-1Н-(1-трет-бутоксикарбонил)-3-индол]-2-(бензилоксикарбониламино)акриловой кислоты HDP 30.2739 в 40 мл сухого метанола. После четырех циклов вакуума/Ar и H2 повышали давление реакционной смеси до исходного давления 30 бар. Реакции позволяли проходить в течение 4 дней при температуре окружающей среды. После испарения растворителя неочищенный продукт очищали флэш-хроматографией на 120 г силикагелевой колонке (длина волны обнаружения 254 нм) с градиентом н-гексана к н-гексану/этилацетату (2:1) и получали после испарения 0,85 г (86%) HDP 30.2760 в виде белого твердого вещества.
MS (ESI+) рассчитано: 558,23 (C33H34N2O7)
MS (ESI+) обнаружено: 581,17 [M+Na]+; 1138,83 [2M+Na]+
Пример 3.5. Получение метилового сложного эфира 6-бензилокси-N-карбобензилокси-L-триптофана HDP 30.2790

100,0 мг (0,18 ммоль) метилового сложного эфира (S)-6-бензилокси-М-карбобензилокси-1-трет-бутоксикарбонилтриптофана HDP 30.2760 растворяли в 5,0 мл муравьиной кислоты и перемешивали 1 час при 40°С. Реакционную смесь выпаривали досуха и остаток растворяли в этилацетате. Этилацетатный раствор промывали водой, насыщенным NaHCO3 и рассолом и сушили над MgSO4. После испарения растворителя неочищенный продукт очищали флэш-хроматографией на 24 г силикагелевой колонке (длина волны обнаружения 254 нм) с градиентом н-гексана к н-гексану/этилацетату (1:1) и получали после испарения 29 мг (35%) HDP 30.2790 в виде белого твердого вещества.
MS (ESI+) рассчитано: 458,52 (C27H26N2O5)
MS (ESI+) обнаружено: 459,25 [М+Н]+
Пример 3.6. Получение (S)-6-бензилокси-N-карбобензилокситриптофана HDP 30.2782
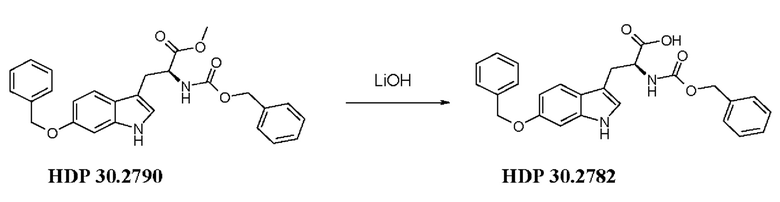
2н водный раствор LiOH (84,7 мкл) добавляли в раствор HDP 30.2790 25,9 мг (0,056 ммоль) в 1000 мкл тетрагидрофурана/воды (10:1) при температуре окружающей среды. Реакционную смесь перемешивали в течение 2,5 часов и разделяли между этилацетатом и 5% лимонной кислотой. Водный слой экстрагировали этилацетатом и органические слои объединяли, сушили (MgSO4) и концентрировали. Полученную карбоновую кислоту HDP 30.2782 очищали на силикагеле при помощи дихлорметана/метанола (+1% уксусной кислоты) в качестве подвижной фазы. 13,7 мг (55%) белого твердого вещества.
MS (ESI+) рассчитано: 444,17.23 (C26H24N2O5)
MS (ESI+) обнаружено: 445,25 [М+Н]+; 467,17 [M+Na]+
Пример 3.7. Получение (S)-6-гидрокси-N-(трет-бутоксикарбонил)-триптофана HDP 30.2832
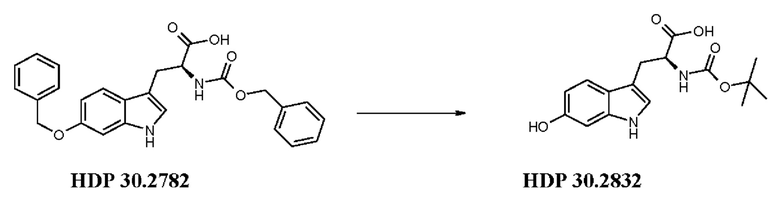
Палладий на угле 10 мг (10 масс. %) добавляли в раствор 50 мг (0,11 ммоль) HDP 30.2782 в 800 мкл метанола. Реакционную смесь продували три раза водородом и перемешивали в течение 2,5 ч при комнатной температуре. Суспензию фильтровали через набивку Celite®, промывали метанолом и концентрировали досуха. Твердый остаток (22,2 мг) (S)-6-гидрокситриптофана растворяли в 1000 мкл 1,4-диоксана/воды (1:1) и обрабатывали 101 мкл (0,101 ммоль) 1н NaOH и 21,57 мкл (0,10 ммоль) ди-трет-бутилдикарбоната. Реакционную смесь перемешивали в течение 2,5 часов и доводили до рН 2 при помощи 1н соляной кислоты. Водный раствор экстрагировали трижды этилацетатом и объединенные органические фазы промывали рассолом, сушили и выпаривали досуха. Неочищенный HDP 30.2832 очищали на силикагеле, используя дихлорметан/метанол (+1% уксусной кислоты) в качестве подвижной фазы. 9,9 мг (31%) белого твердого вещества. Материал был идентичен эталонному образцу.
MS (ES-) рассчитано: 320,14 (C16H20N2O5)
MS (ES-) обнаружено: 319,08 [М-Н]-
Пример 4. Получение цис,транс-1-(трет-бутоксикарбнил)-2-карбокси-3а-гилрокси-6-ацетокси-1,2,3,3а,8,8а-гексагидропирроло[2,3-b]индола цис-HDP 30.2555 и транс HDP 30.2555 (цис,транс-6-ацетокси-Hpi)
Пример 4.1. Получение (S)-М-(трет-бутоксикарбонил)-6-ацетокситриптофана HDP 30.2550
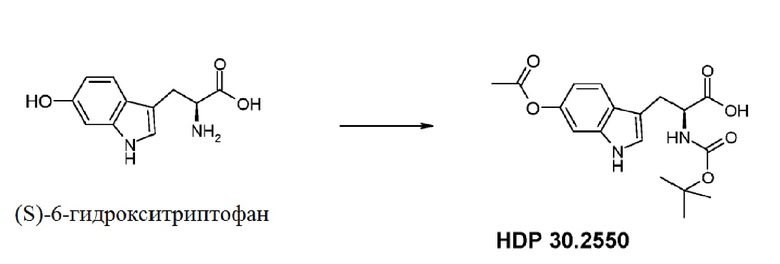
590,0 мг (2,68 ммоль) (S)-6-гидрокситриптофана со стадии гидрирования в примере 3.7 суспендировали в смеси 30 мл 1,4-диоксана/воды 1:1 (об.:об.). В атмосфере аргона 2,68 мл (2,68 ммоль) 1н NaOH добавляли за один раз при температуре окружающей среды. Полученный желтый раствор затем обрабатывали 574,6 мл (2,68 ммоль) Вос-ангидрида (Вос2О) и перемешивали в течение 24 часов при комнатной температуре. Раствор подкисляли при помощи 1 н соляной кислоты до рН 2,4 и экстрагировали 3 раза при помощи 25 мл этилацетата. Объединенные этилацетатные экстракты промывали насыщенным раствором NaCl и сушили над MgSO4. Фильтрация и выпаривание досуха давали 785,0 мг неочищенного материала. Неочищенный М-Вос-6-гидрокси-L-триптофан растворяли в 4,91 мл (4,91 ммоль) 1н NaOH и обрабатывали при помощи 463,2 мл (500,3 мг, 4,90 ммоль) ацетангидрида. Реакционную смесь перемешивали в течение 3 часов и подкисляли 5% лимонной кислотой. Водную фазу экстрагировали трижды при помощи 25 мл этилацетата, промывали насыщенным NaCl и сушили над MgSO4. Фильтрация и выпаривание давали 635 мг неочищенного твердого вещества. Неочищенный продукт очищали флэш-хроматографией на 330 г силикагелевой колонке (длина волны обнаружения 254 нм) с градиентом CH2Cl2+1% уксусной кислоты к CH2Cl2/МеОН (15:1)+1% уксусной кислоты и получали после совместного выпаривания с толуолом 564,4 мг (56% выход) белого порошка.
MS (EST) обнаружено: 361,08[М-Н]-; рассчитано: 362,15 (C18H22N2O6)
Пример 4.2. Получение цис,транс-6-ацетокси-Hpi
Фотооксигенацию проводили при помощи 400 Вт натриевой лампы высокого давления (лампа Sirius Х400 230 В, 400 Вт; 55000 люмен на расстоянии 1,3 м). Бенгальскую розовую использовали в качестве сенсибилизирующего красителя. Реакцию проводили в 500 мл цилиндрической реакционной емкости с теплообменной рубашкой, изготовленной из боросиликатного стекла, с плоским дном и плоским лабораторным фланцем (DN) с двумя коннекторами с резьбой GL 18. Расстояние от лампы до реакционной емкости составляло 15 см, а температура реакции была в диапазоне 3-4°С.
Готовый продукт очищали на системе флэш-хроматографии Teledyne ISCO с 330 г флэш-колонкой Silica Redi Sept (по каталогу Teledyne ISCO: 69-2203-330). Растворители CH2Cl2, СН3ОН, СН3СООН были стандартными для HPLC или BP. Сухой кислород (99,5% чистоты) продували через реакционную смесь со скоростью 2-4 л в минуту.
943,0 мг (2,60 ммоль) N-(трет-бутоксикарбонил)-L-6-ацетокситриптофана HDP 30.2550 и 100 мг бенгальской розовой растворяли в 500 мл метанола и охлаждали до 3°С при помощи криостата Huber с гликолем/водой в качестве охлаждающей среды. Реакционный раствор облучали 400 Вт натриевой лампой высокого давления. При облучении медленный поток кислорода продували через реакционный раствор. Через 5 часов облучения оксигенацию и охлаждение прекращали и реакционную среду обрабатывали 10 мл диметилсульфида. Смесь перемешивали в течение 2 часов и выпаривали досуха при помощи роторного испарителя с температурой водяной бани 35°С. Темно-красный остаток сушили дополнительно под высоким вакуумом до кристаллического твердого вещества 1,20 г. Неочищенный продукт очищали на 330 г силикагелевой колонке (длина волны обнаружения 254 нм) с градиентом CH2Cl2+5% уксусной кислоты к CH2Cl2/МеОН (30:1)+5% уксусной кислоты. 380 мг цис-HDP 30.2555 и 290 мг транс-HDP 30.2555 элюировали и совместно выпаривали с толуолом. После лиофилизации в трет-бутаноле оба изомера получали в виде не совсем белых порошков.
цис-1-(трет-бутоксикарбонил)-2-карбокси-3а-гидрокси-6-ацетокси-1,2,3,3а,8,8а-гексагидропирроло[2.3-b]индол (цис-HDP 30.2555)
380 мг цис-HDP 30.2555 выход: 39%
1H-ЯМР (400 МГц, CD3OD, δ=части на миллион)
δ=1,22, 1,44, 1,54 [s, 9Н, С(СН3)3]; 2,23 (s, 3Н, ОСОСН3); 2,46-2,63 (m, 2Н, СН2); 4,14-4,29 (m, 1H, 2-Н); 5,35 (s, 1H, 8а-Н); 6,39-6,46 (m, 2Н, 7-Н, 5-Н); 7,20-7,24 (m, 1H, 4-Н)
13С-ЯМР (100 МГц, CD3OD, δ=части на миллион)
δ=20,93, 28,45, 31,12, 42,80, 61,12, 69,44, 82,21, 85,82, 87,93, 104,97, 112,98, 124,84, 129,42, 151,51, 154,04, 155,97, 171,34, 175,79
MS (ESI+) обнаружено: 378,92 [МН]+; рассчитано: 378,14 (C18H22N2O7)
MS (ESI+) обнаружено: 401,17 [М+Ка]+;рассчитано: 401,14 (C18H22N2NaO7)
УФ/видимый (СН3ОН): λмакс=296 нм, 239 нм, 215 нм
λмин=266 нм, 227 нм транс-1-(трет-бутоксикарбонил)-2-карбокси-3а-гидрокси-6-ацетокси-1,2,3,3а,8,8а-гексагидропирроло[2,3-b]индол (транс-НРР 30.2555)
290 мг транс-HDP 30.2555 выход: 30%
1Н-ЯМР (400 МГц, CD3OD, δ=части на миллион)
δ=1,22,1,45, 1,54 [s, 9Н, С(СН3)3]; 2,22 (s, 3Н, ОСОСН3); 2,55-2,73 (m, 2Н, СН2); 4,51-4,57 (m, 1H, 2-Н); 5,21-5,24 (s, 1H, 8а-Н); 6,36-6,41 (m, 2Н, 7-Н, 5-Н); 7,17-7,18 (m, 1H, 4-Н)
13С-ЯМР (100 МГц, CD3OD, δ=части на миллион)
δ=20,95, 28,50, 31,12, 42,47, 60,97, 69,44, 82,06, 84,84, 87,54, 104,74, 112,67, 125,03, 128,70, 152,31, 154,22, 156,00, 171,23, 174,67
MS (ESI+) обнаружено: 379,00 [МН]+; рассчитано: 378,14 (C18H22N2O7)
MS (ESI+) обнаружено: 401,17 [M+Na]+; рассчитано: 401,14 (C18H22N2NaO7)
MS (ESI+) обнаружено: 779,00 [2M+Na]+; рассчитано: 779,28 (C36H44N4Na2O14)
УФ/видимый (СН3ОН): λмaкс=299 нм, 241 нм, 215 нм
λмин=268 нм, 228 нм
Пример 4.3. Введение цис,транс-6-ацетокси-Hpi в предшественник аманитина
Проводили синтез аманитина при помощи цис,транс-6-ацетокси-Hpi, как описано в заявке РСТ/ЕР 2018/071268, содержание которой включено в настоящий документ ссылкой, главным образом с целью передачи опыта.
Ссылки
Muraoka S, and Shinozawa Т., J Biosci Bioeng. 2000;89(l):73-6
Wieland Т., Faulstich H. 1978. CRC Crit Rev Biochem. Vol. 5: 185-260.
Zanotti G, Mahringer C, and Wieland Т., lnt J Pept Protein Res. 1987 Oct;30(4):450-9;
Zanotti G, Wieland T, Benedetti E, Di Blasio 8, Pavone V, and Pedone C, lnt J Pept Protein Res. 1989 Sep; 34(3):222-8
Zhang P, et al, FEMS Microbiol Lett. 2005 Nov 15; 252(2):223-8. Epub 2005 Sep 15
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЙ СПОСОБ СИНТЕЗА АМАНИТИНОВ | 2018 |
|
RU2792210C2 |
| ПРОИЗВОДНЫЕ АМАТОКСИНА | 2014 |
|
RU2695370C2 |
| КОНЪЮГАТЫ АМАТОКСИН - АНТИТЕЛО | 2016 |
|
RU2724328C2 |
| КОНЪЮГАТЫ АМАТОКСИНОВ С УЛУЧШЕННЫМИ ЛИНКЕРАМИ | 2011 |
|
RU2601411C2 |
| СПОСОБЫ СИНТЕЗА АМАТОКСИНОВОГО СТРУКТУРНОГО БЛОКА И АМАТОКСИНОВ | 2013 |
|
RU2637924C2 |
| КОНЪЮГАТЫ АМАТОКСИНА С УЛУЧШЕННЫМИ СВЯЗЯМИ | 2012 |
|
RU2575854C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПРОПИОНОВОЙ КИСЛОТЫ | 2011 |
|
RU2575345C2 |
| C-СИММЕТРИЧНЫЕ БИСФОСФИНОВЫЕ ЛИГАНДЫ И ИХ ПРИМЕНЕНИЕ В АСИММЕТРИЧЕСКОМ СИНТЕЗЕ ПРЕГАБАЛИНА | 2005 |
|
RU2335342C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПРОПИОНОВОЙ КИСЛОТЫ | 2010 |
|
RU2544989C2 |
| СПОСОБ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 5-БИФЕНИЛ-4-ИЛ-2-МЕТИЛПЕНТАНОВОЙ КИСЛОТЫ | 2009 |
|
RU2513521C2 |



Изобретение относится к способам синтеза производных (S)-6-гидрокситриптофана, к соединениям для синтеза производных аманитина или конъюгатов лекарственных средств, к промежуточным соединениям для использования в синтезе производных аманитина и конъюгатов лекарственных средств и аматоксина и к использованию конкретных катализаторов, подходящих для содействия указанным путям синтеза. Способ синтеза (S)-6-ацетилокси-N-трет-бутоксикарбонилтриптофана включает по меньшей мере одну стадию энантиоселективного гидрирования олефинового соединения-предшественника путем использования по меньшей мере одного хирального катализатора, причем указанное олефиновое соединение-предшественник выбрано из бензилового сложного эфира [6-бензилокси-1Н-(бензилоксикарбонил)-3-индол]-2-(трет-бутоксикарбониламино)акриловой кислоты (соединение HDP 30.2824)
и метилового сложного эфира 3-[6-бензилокси-1Н-(1-трет-бутоксикарбонил)-3-индол]-2-(бензилоксикарбониламино)акриловой кислоты (соединение HDP 30.2739)
Хиральный катализатор представляет собой соединение, выбранное из группы, состоящей из тетрафторборат циклооктадиен-1,5-[(R,R)-DIPAMP]родия (HDP 30.2758), (COD)Rh(I)(R,R)-Et-DUPHOS (BF4-), (COD)Rh(I)(R,R)-DuPhos-ферроцена (BF4-), (COD)Rh(I)(R,R)-DuPhos-ферроцена-Et2 (BF4-), (COD)Rh(I)(R,R)-DuPhos-алкила(CF3SO3-) и (COD)Rh(I)(R,R)-фенил-DuPhos-алкила(BF4-). Техническим результатом изобретения является обеспечение эффективного, простого и воспроизводимого способа синтеза (S)-6-гидрокситриптофана, его производных и его структурных элементов, а также обеспечение эффективного способа синтеза (S)-6-ацетилокси-N-трет-бутоксикарбонилтриптофана с достаточно высокой энантиомерной чистотой. 8 н. и 6 з.п. ф-лы, 7 ил., 1 табл., 22 пр.
1. Способ синтеза (S)-6-ацетилокси-N-трет-бутоксикарбонилтриптофана (HDP 30.2550), причем указанный способ включает по меньшей мере одну стадию энантиомерно селективного гидрирования олефинового соединения-предшественника путем использования по меньшей мере одного хирального катализатора,
причем указанное олефиновое соединение-предшественник выбрано из соединения HDP 30.2824 (бензилового сложного эфира [6-бензилокси-1Н-(бензилоксикарбонил)-3-индол]-2-(трет-бутоксикарбониламино)акриловой кислоты)
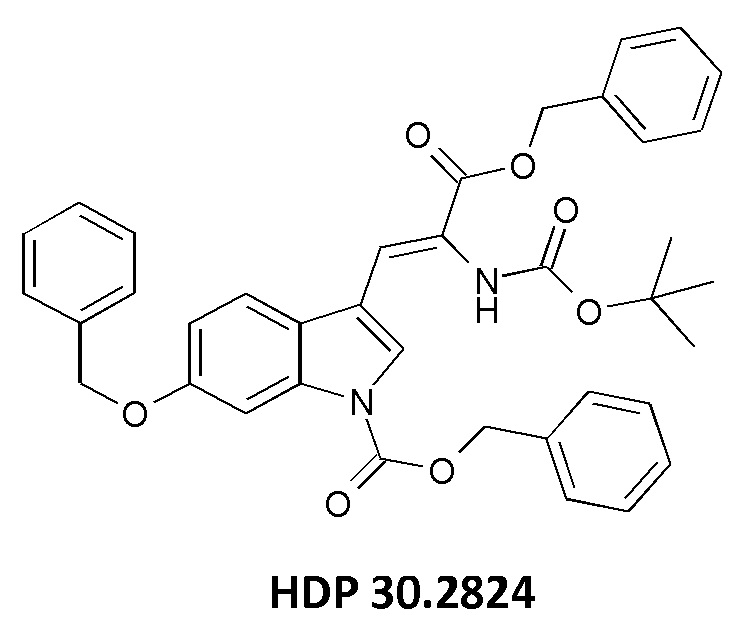
и соединения HDP 30.2739 (метилового сложного эфира 3-[6-бензилокси-1Н-(1-трет-бутоксикарбонил)-3-индол]-2-(бензилоксикарбониламино)акриловой кислоты)
и причем указанный хиральный катализатор представляет собой соединение, выбранное из группы, состоящей из тетрафторборат циклооктадиен-1,5-[(R,R)-DIPAMP]родия (HDP 30.2758), (COD)Rh(I)(R,R)-Et-DUPHOS (BF4-), (COD)Rh(I)(R,R)-DuPhos-ферроцена (BF4-), (COD)Rh(I)(R,R)-DuPhos-ферроцена-Et2 (BF4-), (COD)Rh(I)(R,R)-DuPhos-алкила(CF3SO3-) и (COD)Rh(I)(R,R)-фенил-DuPhos-алкила(BF4-)

причем указанный (S)-6-ацетилокси-N-трет-бутоксикарбонилтриптофан (HDP 30.2550) может быть использован в синтезе аманитинов, производных аманитина и конъюгатов аматоксина.
2. Способ синтеза (S)-6-гидрокситриптофана, причем указанный способ включает по меньшей мере одну стадию энантиомерно селективного гидрирования олефинового соединения-предшественника путем использования по меньшей мере одного хирального катализатора,
причем указанный олефиновый предшественник выбран из соединения HDP 30.2824 (бензилового сложного эфира [6-бензилокси-1Н-(бензилоксикарбонил)-3-индол]-2-(трет-бутоксикарбониламино)акриловой кислоты)
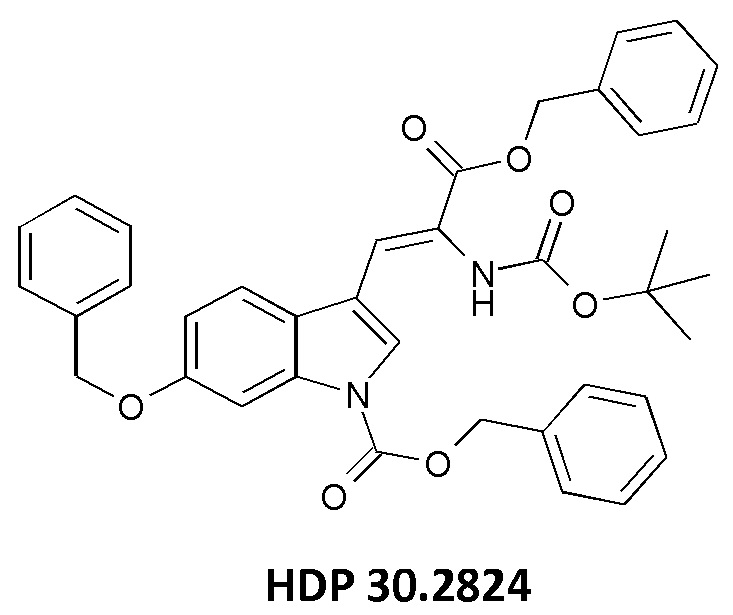
и соединения HDP 30.2739 (метилового сложного эфира 3-[6-бензилокси-1Н-(1-трет-бутоксикарбонил)-3-индол]-2-(бензилоксикарбониламино)акриловой кислоты)
и причем указанный хиральный катализатор представляет собой соединение, выбранное из группы, состоящей из тетрафторборат циклооктадиен-1,5-[(R,R)-DIPAMP]родия (HDP 30.2758), (COD)Rh(I)(R,R)-Et-DUPHOS (BF4-), (COD)Rh(I)(R,R)-DuPhos-ферроцена (BF4-), (COD)Rh(I)(R,R)-DuPhos-ферроцена-Et2 (BF4-), (COD)Rh(I)(R,R)-DuPhos-алкила(CF3SO3-) и (COD)Rh(I)(R,R)-фенил-DuPhos-алкила(BF4-)

причем указанный (S)-6-гидрокситриптофан может быть использован в синтезе аманитинов, производных аманитина и конъюгатов аматоксина.
3. Способ по любому из пп. 1 и 2, в котором указанный хиральный катализатор представляет собой соединение HDP 30.2758 или (R,R)-Et-DUPHOS (BF4-).
4. Способ по любому из пп. 1-3, в котором указанный олефиновый предшественник синтезируют путем использования соединения HDP 30.2822.
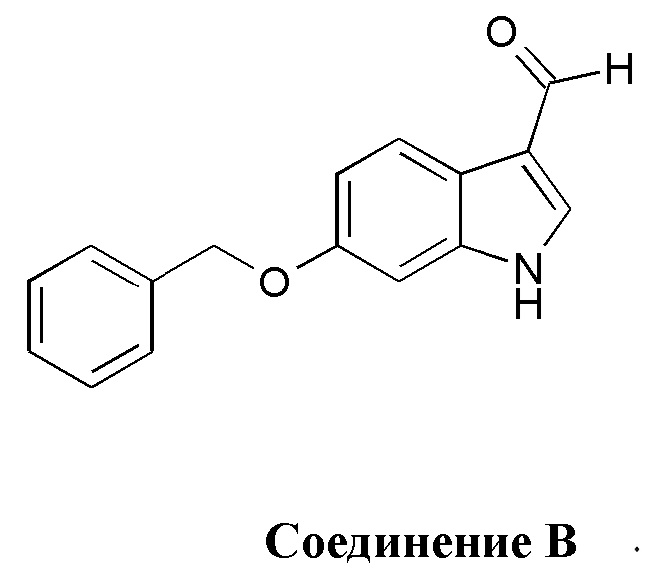
5. Способ по п. 4, в котором указанный олефиновый предшественник дополнительно синтезируют путем использования соединения В
6. Способ по любому из пп. 1-5, причем указанный способ включает использование по меньшей мере одного исходного или промежуточного соединения, выбранного из группы, состоящей из

7. Способ синтеза (S)-6-ацетилокси-N-трет-бутоксикарбонилтриптофана (HDP 30.2550) по п. 1, причем указанный способ включает, по меньшей мере, следующие стадии:
8. Способ синтеза (S)-6-гидрокситриптофана по п. 2, причем указанный способ включает, по меньшей мере, следующие стадии:
9. (S)-6-ацетилокси-N-трет-бутоксикарбонилтриптофан (HDP 30.2550) или любые из соединений-предшественников или производных, выбранных из группы, состоящей из:
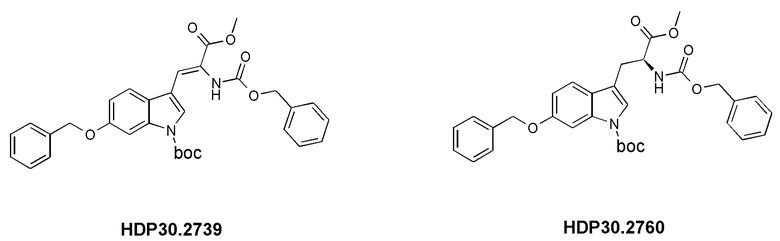
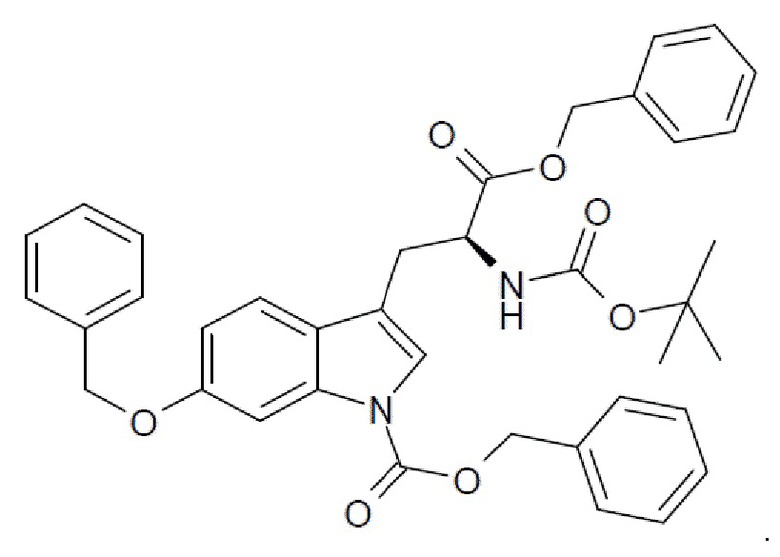
10. Применение (S)-6-ацетилокси-N-трет-бутоксикарбонилтриптофана (HDP 30.2550), (S)-6-гидрокситриптофана или любых из соединений-предшественников по пп. 6, 7 и 8, соответственно, в синтезе аманитина, или производных аманитина, или конъюгатов аматоксина и лекарственного средства, причем указанный конъюгат аматоксина и лекарственного средства необязательно содержит линкер.
11. Дегидроаминокислотное соединение, выбранное из группы, состоящей из соединений I и III
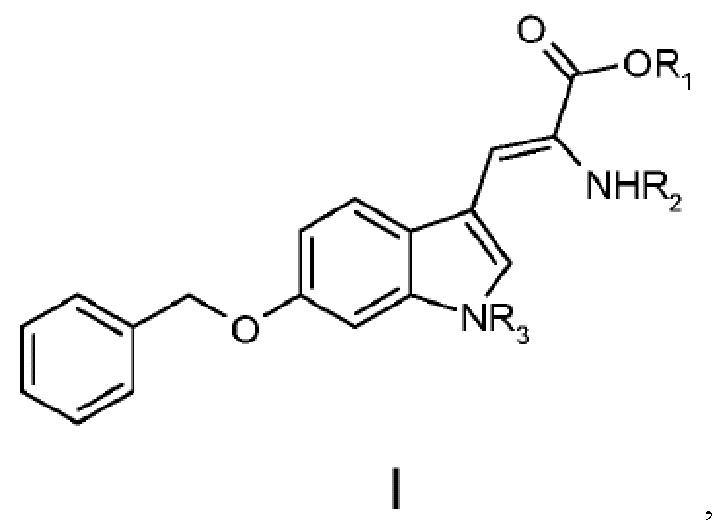
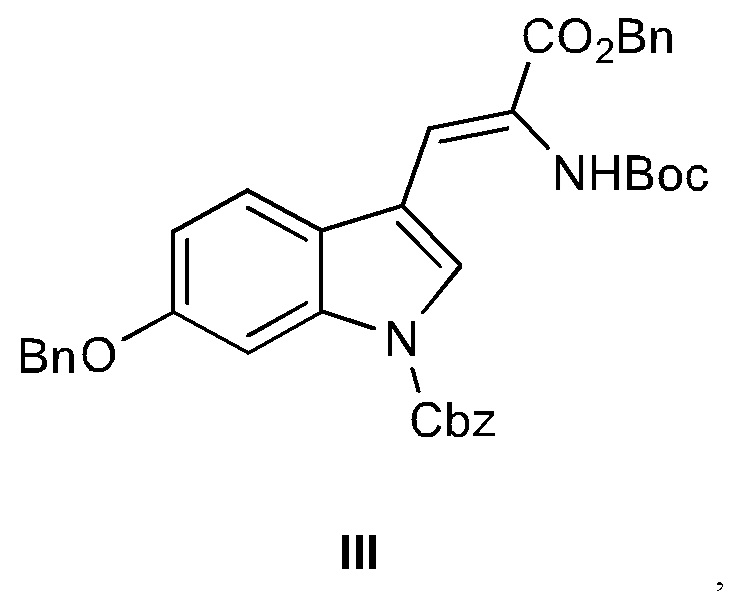
где R1 выбран из Н, алкила, аралкила,
R2 выбран из Boc-, Cbz-,
R3 выбран из Boc-, Cbz-,
для использования в синтезе аманитина, или производных аманитина, или конъюгатов аматоксина и лекарственного средства.
12. Применение (S)-6-ацетилокси-N-трет-бутоксикарбонилтриптофана (HDP 30.2550) или (S)-6-гидрокситриптофана для синтеза аманитина, или производных аманитина, или конъюгатов аматоксина и лекарственного средства, причем предпочтительно указанный конъюгат аматоксина и лекарственного средства содержит линкер.
13. Применение соединения, выбранного из группы, состоящей из тетрафторбората циклооктадиен-1,5-[(R,R)-DIPAMP]родия (HDP 30.2758), (COD)Rh(I)(R,R)-Et-DUPHOS (BF4-), (COD)Rh(I)(R,R)-DuPhos-ферроцена (BF4-), (COD)Rh(I)(R,R)-DuPhos-ферроцена-Et2 (BF4-), (COD)Rh(I)(R,R)-DuPhos-алкила(CF3SO3-) и (COD)Rh(I)(R,R)-фенил-DuPhos-алкила(BF4-) в качестве катализатора гидрирования в следующей реакции:
 .
.
14. Применение тетрафторбората циклооктадиен-1,5-[(R,R)-DIPAMP]родия (HDP 30.2758) в качестве катализатора гидрирования в следующей реакции:
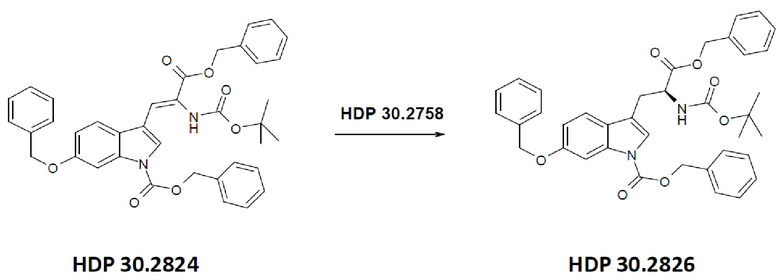 .
.
| CHIOTELLIS A | |||
| ET AL, Synthesis and Biological Evaluation of 18F-Labeled Fluoroethoxy Tryptophan Analogues as Potential PET Tumor Imaging Agents, MOLECULAR PHARMACEUTICS, 2014, 11(11), pp | |||
| СПОСОБ И АППАРАТ ДЛЯ ПОЛУЧЕНИЯ ЧИСТОЙ ПОВАРЕННОЙ СОЛИ ИЗ ЗАГРЯЗНЕННЫХ ХИМИЧЕСКИМИ И МЕХАНИЧЕСКИМИ ПРИМЕСЯМИ СОЛЕЙ | 1925 |
|
SU3839A1 |
| WO 2011073173 A1, 23.06.2011 | |||
| ПРОИЗВОДНЫЕ ТЕТРАЦИКЛИНА | 1994 |
|
RU2138507C1 |
| Li Y | |||
| ET AL, Synthesis of potent BCRP inhibitor—Ko143, TETRAHEDRON LETTERS, 2008, 49(9), pp | |||

