Область техники, к которой относится изобретение
Настоящее изобретение относится к бициклическому октапептидному производному, которое может быть конъюгировано с соответствующей связывающейся с мишенью группой в специальной химической структуре. Эта структура является стабильной в плазме крови и разлагается на активный фармацевтический ингредиент в определенной биологической среде, максимизируя таким образом киллинг-эффекты на клетки-мишени и минимизируя токсичные побочные эффекты на не являющиеся мишенями клетки, которую можно использовать для лечения различных злокачественных опухолей.
Предпосылки создания изобретения
Аманитин является одним из пептидных аманитатоксинов, выделенных из смертельно ядовитых грибов, и представляет собой бициклический пептид, состоящий из восьми аминокислот. Существует девять природных аманитинов, которые были выделены и очищены, которые, соответственно, представляют собой α-аманитин, β-аманитин, γ-аманитин, ε-аманитин, аманин, аманинамид, амануллин, амануллиновую кислоту и проамануллин, при этом α-аманитин и β-аманитин являются основными токсинами, вызывающими смерть. Аманитин представляет собой класс медленно действующих токсинов и может ингибировать транскрипцию эукариотической РНК-полимеразы II и РНК-полимеразы III, приводя к дефициту белка и клеточной гибели. Этот класс токсинов обладает чрезвычайно высоким ингибиторным эффектом на РНК-полимеразу II, и его KD может достигать 3 нМ. Токсины периодически абсорбируются в организме в результате энтерогепатической циркуляции в желудочно-кишечном тракте и могут вызывать устойчивое серьезное повреждение органов, таких как печень, почки, сердце и легкие.
После конъюгации аманитина с большим биомолекулярным носителем (таким как молекула антитела) токсичность аманитина существенно снижается, и аманитин становится даже относительно нетоксичным; и цитотоксичность аманитина может наблюдаться только после удаления биомолекулярного носителя в специфических физиологических условиях.
В соответствии с проведенными группой Теодора Виланда исследованиями, когда сульфоксидную структуру в природном аманитине заменяют на элементарную серу, токсичность для клеток у неприродного аманитина, образованного таким образом, изменяется незначительно. Молекула природного аманитина была конъюгирована с моноклональным антителом компанией Heidelberg Pharmaceutical Co., Ltd., Germany, в результате чего была получена молекула лекарственного средства, обладающая противоопухолевыми активностями.
В настоящем изобретении неприродный аманитин, по токсичности подобный природному аманитину, конъюгируют с биомолекулой, способной связываться с мишенью через биофармацевтически приемлемую связывающую структуру, чтобы таким образом получить соединение, которое является стабильным в плазме и может эффективно убивать опухолевые клетки в клетках.
Определения
В соответствии со стандартами и практикой в данной области техники, символ 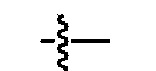 , используемый в формулах и таблицах, представляет собой связь, использующуюся как точка для присоединения фрагмента или заместителя к ядру структуры соединения.
, используемый в формулах и таблицах, представляет собой связь, использующуюся как точка для присоединения фрагмента или заместителя к ядру структуры соединения.
В соответствии со стандартами и практикой в данной области техники, гетеро (атом, алкил, арил, цикло) относится к соответствующей химической структуре, содержащей атом (или атомы), отличный от углерода.
Сущность изобретения
Настоящее изобретение предоставляет конъюгат бициклического октапептидного аманитинового производного и биомакромолекулы, который является стабильным в кровотоке и расщепляется после эндоцитоза клетками-мишенями, высвобождая при этом аманитиновое производное в качестве ингибитора РНК-полимеразы, и проявляет сильную токсичность в клетках посредством специфического ингибирования синтеза мРНК у эукариот, и конъюгат, в частности, представляет собой конъюгат токсина структурной формулы (I):
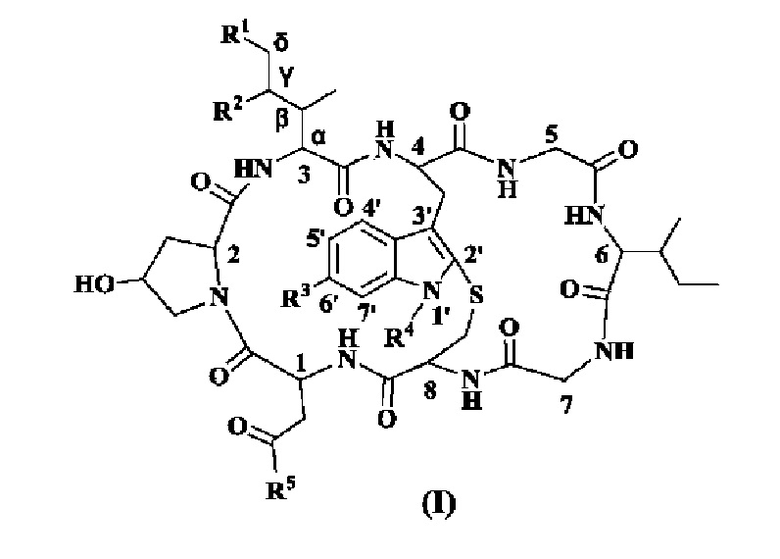
где:
R1 представляет собой H, -OH или -O-L-A;
R2 представляет собой H и -OH;
R3 представляет собой OC1-6 алкил;
R4 представляет собой H или -L-A;
R5 представляет собой -NH2, -OH, -NH-L-A или -O-L-A;
где O представляет собой атом кислорода, N представляет собой атом азота и H представляет собой атом водорода.
A представляет собой биомакромолекулярный фрагмент, связывающийся с сайтом-мишенью;
в качестве предпочтительного варианта осуществления, L включает следующую структуру:
-L1-[L2]m-[AA]n-L3-
где: L1 представляет собой линкер для связывания с биомакромолекулой A; L2 представляет собой спейсер, и L2 представляет собой фрагмент, связывающий L1 с AA; m представляет собой целое число 1-6; L3 связывается с токсином, показанным в структурной формуле (I); AA представляет собой фрагмент, состоящий из 1-6 аминокислот, и n имеет значение 0 или 1.
В качестве предпочтительного варианта осуществления, A представляет собой биомакромолекулярный фрагмент, связывающийся с сайтом-мишенью, и включает антитело или его антиген-связывающие фрагменты, антитело-подобный белок, нуклеиновокислотный аптамер и т.д.
В качестве предпочтительного варианта осуществления, биомакромолекула, связывающаяся с сайтом-мишенью, представляет собой антитело или его антиген-связывающий фрагмент, которое выбрано из химерного антитела, деиммунизованного антитела, гуманизированного антитела, человеческого антитела, диатела, триатела и нанотела.
Еще более предпочтительно, антиген-связывающий фрагмент выбран из группы, состоящей из Fab, F(ab’), Fd, Fv, одноцепочечного Fv и дисульфид-связанного Fv(dsFv).
В качестве предпочтительного варианта осуществления, один и только один из R1, R4 и R5 содержит структуру -L-A.
В качестве предпочтительного варианта осуществления, L1 представляет собой линкер для связывания с биомакромолекулой A, и выбран из:

и т.д.; где волнистая линия соединяет с биомакромолекулой A.
В качестве предпочтительного варианта осуществления, L2 представляет собой спейсер, выбранный из одной или нескольких одинаковых или отличных друг от друга комбинаций замещенного или незамещенного C1-C6 алкила, замещенного или незамещенного C3-C20 циклоалкила, замещенного или незамещенного C3-C20 гетероциклоалкила, замещенного или незамещенного C5-C20 арила, замещенного или незамещенного C5-C20 гетероарила и -(CH2CH2O)a- (где a представляет собой целое число от 1 до 20), и спейсеры связаны друг с другом через соответствующую химическую связь.
Еще более предпочтительно, заместитель выбран из одной или нескольких одинаковых или отличных друг от друга комбинаций гидроксильной группы, сульфгидрильной группы, галогена, карбоксильной группы, аминогруппы, фосфатной группы, нитрогруппы, цианогруппы, сульфогруппы, замещенного или незамещенного C1-C6 алкила и т.д.
Еще более предпочтительно, заместитель выбран из одной или нескольких одинаковых или отличных друг от друга комбинаций гидроксильной группы, сульфгидрильной группы, галогена, карбоксильной группы, аминогруппы, фосфатной группы, нитрогруппы, цианогруппы, сульфогруппы и т.д.
В качестве предпочтительного варианта осуществления, AA представляет собой фрагмент, состоящий из 1-6 аминокислот, которые представляют собой L-аминокислоты, выбранные из глицина, аланина, валина, лейцина, изолейцина, фенилаланина, пролина, триптофана, серина, тирозина, цистеина, метионина, аспарагина, глутамина, треонина, аспартата, глутамата, лизина, аргинина, гистидина и т.д.,
и еще более предпочтительно из фенилаланина, цитруллина, валина, лизина, серина, глутамата, аспартата и глицина.
В качестве предпочтительного варианта осуществления, L3 выбран из
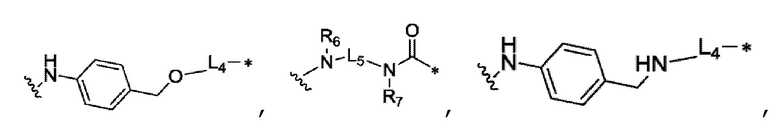
любой связывающей группы, которая связывает с токсином и т.д.,
где волнистая линия связывается с AA, а звездочка связывается с токсином, показанным в структурной формуле (I).
Еще более предпочтительно, L4 выбран из карбонильной группы или простой связи.
Еще более предпочтительно, R6 и R7 каждый независимо выбран из водорода, C1-C6 алкила и т.д.
Еще более предпочтительно, L5 представляет собой C2-C12 алкил.
В качестве предпочтительного варианта осуществления, лекарственное средство содержит конъюгат токсина структурной формулы (I), описанной выше, или его соль.
В качестве предпочтительного варианта осуществления, раскрыто применение конъюгата токсина структурной формулы (I), описанной выше, или его соли для получения противоопухолевого лекарственного средства или противоракового лекарственного средства.
В качестве предпочтительного варианта осуществления, противоопухолевое лекарственное средство или противораковое лекарственное средство представляет собой лекарственное средство от рака легкого, лекарственное средство от рака почек, лекарственное средство от рака уретры, лекарственное средство от колоректального рака, лекарственное средство от рака предстательной железы, лекарственное средство от глиобластомы, лекарственное средство от рака яичников, лекарственное средство от рака поджелудочной железы, лекарственное средство от рака молочной железы, лекарственное средство от меланомы, лекарственное средство от рака печени, лекарственное средство от рака мочевого пузыря, лекарственное средство от злокачественной лимфомы, лекарственное средство от лейкоза, лекарственные средства от рака желудка или лекарственное средство от рака пищевода.
Подробное описание вариантов осуществления
Настоящее изобретение далее проиллюстрировано путем комбинирования конкретных примеров, представленных ниже. Эти примеры используются только для описания настоящего изобретения, но не для ограничения объема настоящего изобретения. Если не указано иное, все используемые профессиональные и научные термины имеют такие же значения, которые обычно известны специалистам в данной области. Кроме того, любые методы и материалы, подобные или эквивалентные содержанию, описанному в настоящей заявке, все могут применяться в способе настоящего изобретения. Описанные предпочтительные варианты осуществления и материалы предназначены только для иллюстративных целей.
Пример 1 Синтез низкомолекулярной нагрузки ama-0301
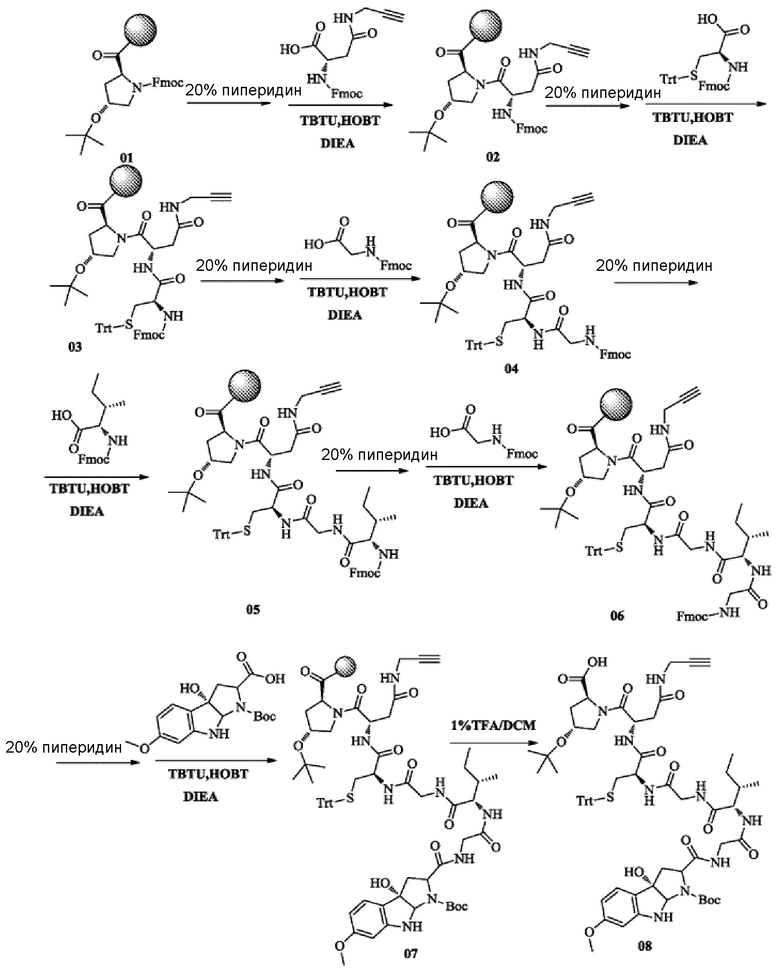
1) Твердофазный синтез промежуточного соединения 08
Предварительно нагруженную N-флуоренилметоксикарбонил-O-трет-бутил-L-гидроксипролином смолу, которую использовали в качестве исходного вещества, обрабатывали 20% раствором пиперидина (добавляя 20 мл 20% раствора пиперидина к 1 г смолы) для удаления защитной группы Fmoc и промывали при помощи DMF 5 раз до достижения нейтрального pH. Затем к DMF, который использовали в виде раствора (20 мл/г), последовательно добавляли Fmoc-N-пропинил-L-аспарагин (Fmoc-Asn(Trt)-OH) (3 экв.), TBTU (2,5 экв.), HOBT (1,8 экв.) и DIPEA (6 экв.), осуществляли взаимодействие смеси в течение 2 ч при комнатной температуре (28°C) и затем промывали 3 раза при помощи DMF (каждый раз добавляя 20 мл DMF к 1 г смолы), с последующим связыванием с аминокислотами в соответствии с предыдущей процедурой. После завершения конечного связывания полученное соединение отщепляли от смолы с использованием 1% раствора TFA в дихлорметане (каждый раз добавляя по 20 мл к 1 г смолы; 1% TFA в течение 5 мин; повторяли три раза); этот раствор удаляли на роторном испарителе; и осуществляли перемешивание с метил-трет-бутиловым эфиром для кристаллизации, с получением таким образом соединения 08 с общим выходом около 43% и высокой чистотой 81,3%. MS: [M+H]+ 1244,6521.
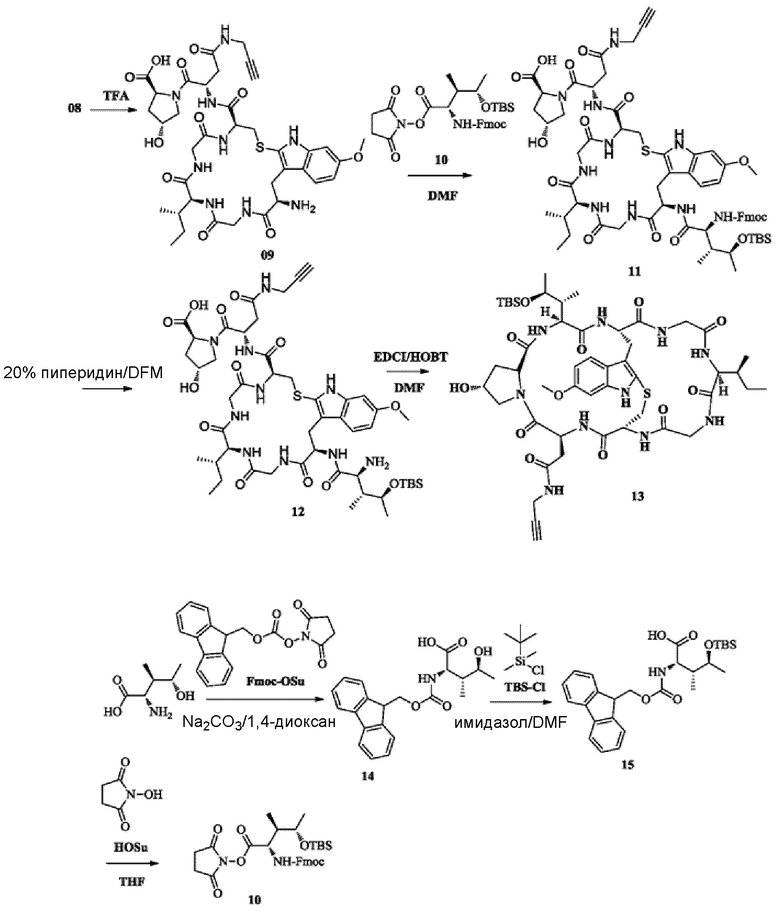
2) Синтез соединения 09
8 г соединения 08, неочищенного продукта, растворяли с использованием TFA (10 мл/г) и затем перемешивали и осуществляли взаимодействие в течение 5 ч при комнатной температуре; TFA удаляли при пониженном давлении при 50°C; и осуществляли очистку с использованием препаративной жидкостной хроматографии с получением около 3,8 г чистого продукта соединения 09 с выходом 71% и чистотой 96,4%. MS: [M+H]+ 828,3241.
3) Синтез соединения 14
2,94 г (4S)-гидроксилизолейцина, 40 мл 1,4-диоксана и 40 мл насыщенного раствора карбоната натрия добавляли в 250-мл одногорлую колбу и гомогенно перемешивали, с последующим добавлением по порциям Fmoc-OSu. Через 10 минут перемешивание продолжали при комнатной температуре в течение 12 ч до завершения взаимодействия исходных веществ. К реакционной жидкости добавляли 50 мл воды и использовали 5% раствор лимонной кислоты для доведения pH до около 4. Для экстракции использовали этилацетат, 3 раза (каждый раз по 50 мл). Органический слой собирали, промывали один раз 50 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия и концентрировали с получением бледно-желтого масла, которое непосредственно использовали на следующей стадии без очистки, с выходом > 100%.
4) Синтез соединения 15
Неочищенный продукт указанного выше соединения 13 растворяли с использованием 40 мл DMF и затем добавляли 2,68 г (2 экв.) имидазола с последующим добавлением по порциям TBS-Cl; после этого осуществляли перемешивание при комнатной температуре в течение 12 ч до завершения взаимодействия исходных веществ. Добавляли 50 мл воды и 50 мл этилацетата и перемешивали. Органический слой отделяли и водный слой экстрагировали два раза этилацетатом (каждый раз по 50 мл). Органический слой собирали, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением бледно-желтого масла, которое подвергали колоночной хроматографии на силикагеле (элюирование: PE:EA=5:1), с получением 4,5 г масла с выходом около 46,6% для двух стадий.
5) Синтез соединения 10
Соединение 14, HOSu (1,23 г, 1,15 экв.), DCC (2,23 г, 1,15 экв.) и 50 мл THF последовательно добавляли в 250-мл одногорлую колбу и перемешивали при комнатной температуре в течение 6 ч в атмосфере азота. После завершения реакции добавляли 50 мл воды и 50 мл этилацетата и перемешивали в течение 10 минут и затем органический слой отделяли. Водный слой затем экстрагировали два раза этилацетатом (каждый раз по 50 мл) и органический слой объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением бледно-желтого масла, которое очищали препаративной жидкостной хроматографией с получением около 3,24 г белого пенистого твердого вещества с выходом 60%. 1H-ЯМР (400 МГц, DMSO-d6): 0,08 (с, 6H), 0,86 (с, 9H), 0,98 (д, 3H, J=8,0 Гц), 1,06 (д, 3H, J=5,6), 1,95 (т, J=10,8), 2,83 (с, 4H), 4,21 (дд, 1H, J=16,8 Гц, 8,0 Гц), 4,34 (дд, 1H, J=12 Гц, 4 Гц), 4,67-4,73 (м, 1H), 7,31 (д, 2H, J=8,0 Гц), 7,34-7,46 (м, 2H), 7,70-7,76 (м, 2H), 7,89 (т, 2H, J=12,0 Гц), 8,24 (д, 1H, J=8,8 Гц); MS: 581,34[M+H].
6) Синтез соединения 11
0,5 г соединения 09 растворяли с использованием 1,5 мл безводного DMF и затем добавляли соединение 10 (701 мг, 2 экв.). pH доводили до 8-9 при помощи DIPEA; реакцию осуществляли в атмосфере азота в течение 5 ч при комнатной температуре и отслеживали при помощи ВЭЖХ до тех пор, пока реакция исходного вещества 09 не была по существу завершена. Продукт непосредственно использовали на следующей стадии без последующей обработки.
7) Синтез соединения 12
0,3 мл (20%) пиперидина добавляли к вышеуказанной реакционной жидкости; реакционную смесь перемешивали в течение 2 часов при комнатной температуре и перемешивание прекращали, когда реакция исходных веществ завершилась (отслеживали при помощи ВЭЖХ). Для очистки использовали препаративную жидкостную хроматографию (нейтральные условия, ацетонитрил/чистая водная система) для сбора фракции, соответствующей целевому пику; после удаления ацетонитрила при пониженном давлении получали 277 мг белого порошкообразного твердого вещества путем лиофилизации с выходом около 42,8% для двух стадий. MS: [M+H]+ 1071,5120.
8) Синтез соединения 13
270 мг соединения 12 растворяли с использованием безводного DMF. Добавляли EDCI (96,6 мг, 2 экв.); HOBT (170 мг, 5 экв.) и DIPEA (0,22 мл, 5 экв.) и перемешивали в течение 4 ч при комнатной температуре до тех пор, пока реакция не завершилась (отслеживали при помощи ВЭЖХ). Для очистки использовали препаративную жидкостную хроматографию (нейтральные условия, ацетонитрил/чистая водная система) для сбора фракции, соответствующей целевому пику; после удаления ацетонитрила при пониженном давлении получали около 136,4 мг белого порошкообразного твердого вещества путем лиофилизации с выходом 51,4% и MS: [M+H]+ 1053,4908.

9) Синтез соединения 16
1 г N-Сукцинимидил-6-малеимидогексаноата растворяли с использованием 10 мл тетрагидрофурана и затем добавляли 0,678 г 2-[2-(2-азидоэтокси)этокси]этиламина (1,2 экв.); осуществляли перемешивание при комнатной температуре в течение 4 ч в атмосфере азота до тех пор, пока реакция не завершилась (отслеживали при помощи ТСХ); после этого добавляли воду и использовали EA для экстракции 3 раза (каждый раз по 10 мл). Органический слой объединяли, сушили, концентрировали и очищали колоночной хроматографией с получением около 0,8 г целевого продукта с выходом 84%.
10) Синтез соединения 17
80 мг соединения 16 растворяли с использованием 5 мл DMSO, добавляли соединение 13(114,7 мг, 0,5 экв.), 44,7 мг (1,5 экв.) бромида меди и 0,2 мл очищенной воды; осуществляли перемешивание в течение 3 ч при комнатной температуре в атмосфере азота до завершения реакции соединения 13 (отслеживали при помощи ВЭЖХ); очистку осуществляли с использованием препаративной жидкостной хроматографии для сбора фракции, соответствующей целевому пику, и органический растворитель удаляли на роторном испарителе; получали около 50,5 мг белого твердого вещества путем лиофилизации с выходом 32,6%. MS: [M+H]+ 1420,7031.
11) Синтез соединения ama-0301
К 45 мг соединения 17 добавляли 1 мл 5% TFA/MeOH для достижения осветления при растворении. Реакцию осуществляли при комнатной температуре в течение 1 часа в атмосфере азота. После завершения реакции исходного вещества 17 (отслеживали при помощи ВЭЖХ) и удаления растворителя путем продувки азотом осуществляли очистку с использованием препаративной жидкостной хроматографии и органический растворитель концентрировали и удаляли с получением 15,4 мг белого твердого вещества путем лиофилизации с выходом 37,2%. MS: [M+H]+ 1306,6013.
Пример 2 Синтез низкомолекулярной нагрузки ama-0302
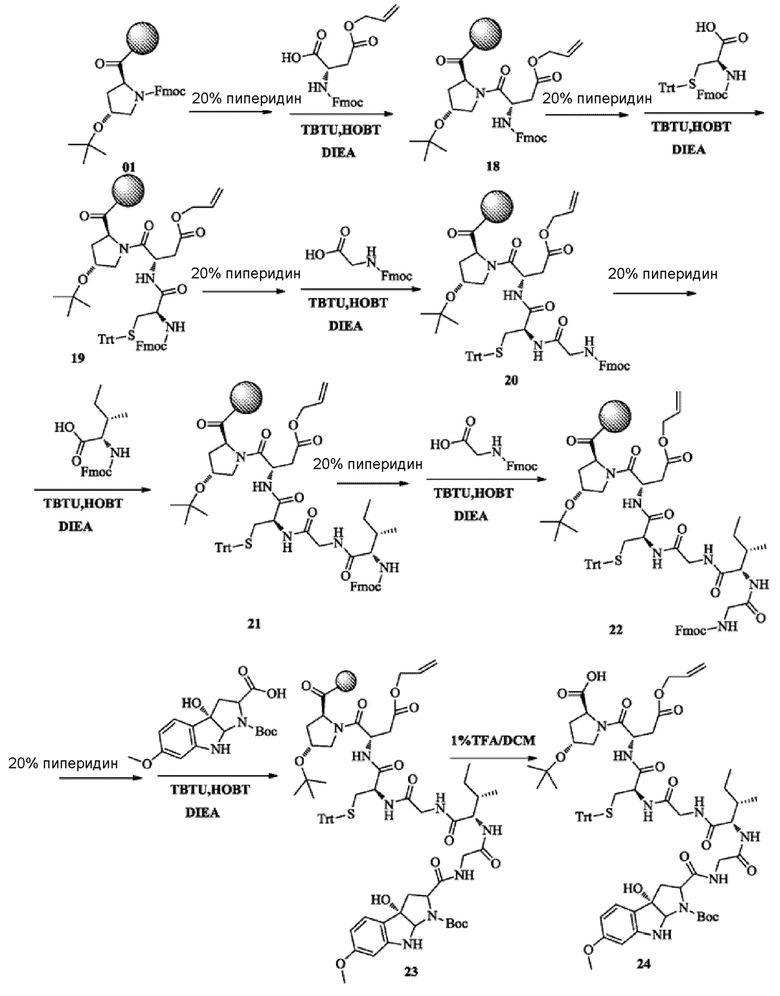
1) Твердофазный синтез ключевого промежуточного соединения 24
Получали со ссылкой на синтез соединения 08. После кристаллизаци метил-трет-бутиловым эфиром получали около 10,6 г желто-коричневого твердого вещества с высокой чистотой около 79,8%.
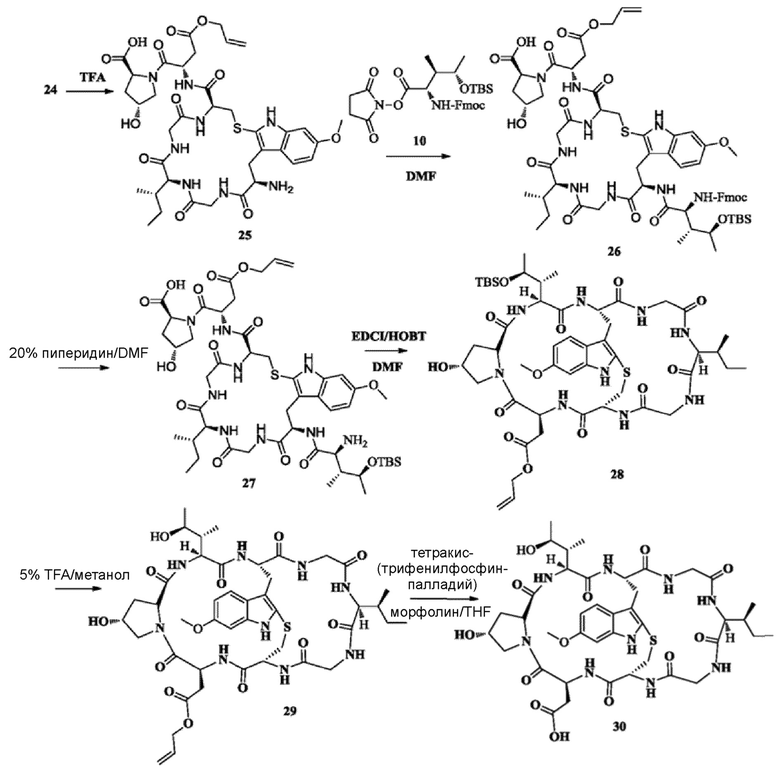
2) Синтез соединения 25
Синтез осуществляли со ссылкой на синтез соединения 09. Неочищенный продукт соединения 24 растворяли с использованием 50 мл TFA и перемешивали при комнатной температуре в течение 5 ч в атмосфере азота до тех пор, пока реакция исходных веществ по существу не завершилась, согласно данным ВЭЖХ; осуществляли очистку с использованием препаративной жидкостной хроматографии для сбора фракции, соответствующей целевому пику. После того, как органический растворитель удаляли при пониженном давлении, получали 2,38 г белого твердого вещества путем лиофилизации. MS: [M+H]+ 831,4251.
3) Синтез соединения 26
Синтез осуществляли со ссылкой на синтез соединения 11. Добавляли 1,0 г исходного вещества 25 и получали около 820 мг белого твердого вещества путем лиофилизации с выходом 52,5%. MS: [M+H]+ 1296,6431.
4) Синтез соединения 27
Синтез осуществляли со ссылкой на синтез соединения 12. Добавляли 800 мг исходного вещества 26 и получали около 308,8 мг белого твердого вещества путем лиофилизации с выходом около 46,6%. MS: [M+H]+ 1074,5184.
5) Синтез соединения 28
Синтез осуществляли со ссылкой на синтез соединения 13. Добавляли 300 мг исходного вещества 27 и получали около 208,4 мг белого твердого вещества путем лиофилизации с выходом около 70,6%. MS: [M+H]+ 1056,5243.
6) Синтез соединения 29
100 мг соединения 28 растворяли с использованием 1 мл 5% раствора TFA в метаноле; перемешивание осуществляли в течение 2 ч при комнатной температуре в атмосфере азота до тех пор, пока реакция исходных веществ не завершилась (отслеживали при помощи ВЭЖХ); осуществляли очистку с использованием препаративной жидкостной хроматографии для сбора фракции, соответствующей целевому пику, и получали около 69,5 мг бледно-желтого твердого вещества путем лиофилизации с выходом 77,9%. MS: [M+H]+ 942,4571.
7) Синтез соединения 30
50 мг соединения 29 добавляли в 10-мл реакционную колбу и в атмосфере азота добавляли 67,5 мг (1,1 экв.) тетракис(трифенилфосфинпалладий). Воздух заменяли азотом и в реакционную колбу добавляли 5 мл безводного тетрагидрофурана и 0,1 мл безводного морфолина через шприц. Исходные вещества растворяли и перемешивали в течение 12 ч при комнатной температуре до тех пор, пока реакция исходных веществ не завершилась (отслеживали при помощи ВЭЖХ). Осуществляли очистку с использованием препаративной жидкостной хроматографии для сбора фракции, соответствующей целевому пику, и получали около 35,2 мг бледно-желтого твердого вещества путем лиофилизации с выходом 73,5%. MS: [M+H]+ 902,4123.
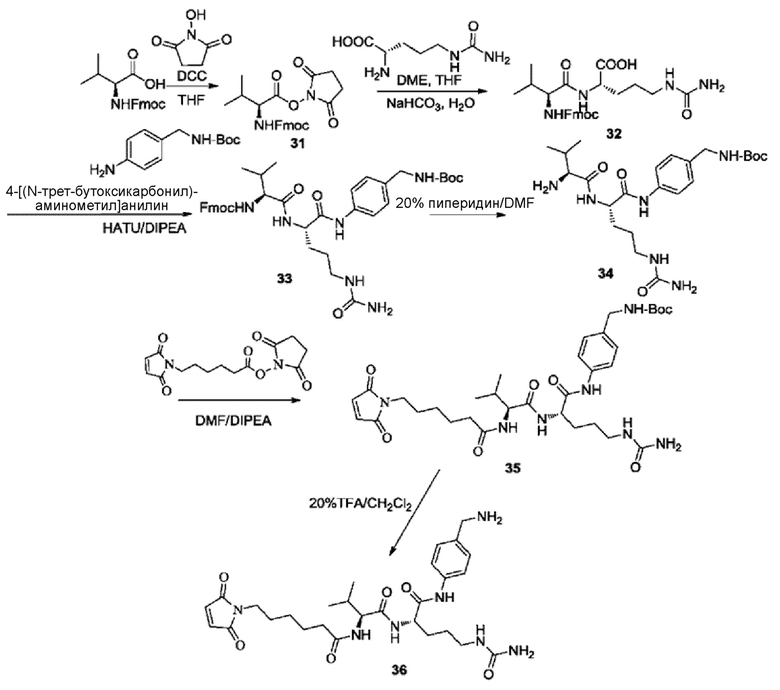
8) Синтез соединения 31
Fmoc-L-валин (20 г) и HoSu (7,46 г, 1,1 экв.) растворяли с использованием 200 мл THF; затем реакционную колбу помещали в баню с ледяной солью и охлаждали до 0°C. Медленно добавляли DCC конденсирующий агент (14,6 г, 1,1 экв.) и температуру реакции контролировали при 0-5°C, при этом добавление завершалось в течение 3 часов. Полученное вещество удаляли из ледяной бани и перемешивали и осуществляли взаимодействие в течение 12 ч. Реакцию не останавливали до тех пор, пока не завершалось взаимодействие Fmoc-L-валина (отслеживали при помощи ТСХ). Осуществляли фильтрование с отсасыванием при пониженном давлении и фильтровальную лепешку промывали 100 мл THF. Фильтрат подвергали сушке на роторном испарителе. К остатку добавляли 100 мл DCM, растворяли при перемешивании при 35°C, фильтровали для удаления незначительного количества нерастворимого вещества с использованием мембранного фильтра для органических соединений и затем помещали в масляную баню при 35°C. Добавляли 100 мл петролейного эфира при перемешивании; кристаллизацию осуществляли путем естественного охлаждения в течение 1 часа и затем охлаждения на бане с ледяной солью в течение 2 часов. Осуществляли фильтрование с отсасыванием и твердое вещество промывали петролейным эфиром и сушили в вакуумной печи при 40°C с получением 21,86 г белого порошкообразного твердого вещества с выходом около 85%.
9) Синтез соединения 32
20 г соединения 31 растворяли в 200 мл THF в качестве растворителя и добавляли 9,64 г (1,2 экв.) L-цитруллина; добавляли 1 M карбоната натрия для доведения pH до 8-9, и осветление растворяемой реакционной смеси не достигалось. Перемешивание осуществляли в течение 48 ч при комнатной температуре до тех пор, пока реакция не завершилась (отслеживали при помощи ТСХ). При перемешивании на ледяной бане реакционную жидкость доводили до pH 3-4 с использованием водного раствора лимонной кислоты и экстрагировали 3 раза смесью изопропанол:EA=1:5 (40 мл изопропанола+200 мл EA). Органическую фазу объединяли, сушили над безводным сульфатом натрия, фильтровали и подвергали сушке на роторном испарителе, с последующим добавлением 200 мл метил-трет-бутилового эфира и перемешиванием в течение 2 часов; затем осуществляли фильтрование с отсасыванием для сбора фильтровальной лепешки, которую сушили в вакуумной печи при 45°C, с получением 18,6 г белого твердого продукта с выходом около 81,7%.
10) Синтез соединения 33
18 г соединения 32 добавляли в 500-мл реакционную колбу и растворяли при помощи 200 мл DMF; затем добавляли последовательно 8 г (1,0 экв.) 4-[(N-трет-бутоксикарбонил)аминометил]анилина, 20 г (1,5 экв.) HATU и DIPEA (18 мл, 3 экв.) и перемешивали в течение 24 ч при комнатной температуре. После завершения реакции исходных веществ (отслеживали при помощи ТСХ) добавляли 200 мл воды и 200 мл дихлорметана и перемешивали в течение 10 минут. Затем органический слой отделяли и промывали два раза водой (каждый раз по 50 мл). Органический слой собирали, сушили над безводным сульфатом натрия и затем фильтровали и концентрировали с получением коричневого масла. К этому маслу добавляли 200 мл метил-трет-бутилового эфира и перемешивали в течение 30 мин; затем твердое вещество промывали и отфильтровывали и фильтровальную лепешку промывали два раза метил-трет-бутиловым эфиром (каждый раз по 50 мл). Фильтровальную лепешку собирали и сушили в вакуумной печи при 50°C с получением 18,9 г коричневого твердого вещества с выходом 74,4%.
11) Синтез соединения 34
15 г соединения 33 добавляли в 250-мл реакционную колбу, в которую добавляли 75 мл 20% раствора пиперидина в DMF; перемешивание осуществляли в течение 1 ч при комнатной температуре до тех пор, пока реакция исходных веществ не завершилась (отслеживали при помощи ТСХ). Растворитель отгоняли при пониженном давлении с использованием масляного насоса и затем добавляли 200 мл метил-трет-бутилового эфира и перемешивали в течение 2 ч при комнатной температуре. Осаждали коричневое твердое вещество и фильтровали. Фильтровальную лепешку промывали два раза метил-трет-бутиловым эфиром (каждый раз по 50 мл), собирали и сушили в вакуумной печи при 50°C с получением 6,8 г коричневого твердого вещества с выходом 66,4%.
12) Синтез соединения 35
1 г соединения 34 растворяли с использованием 10 мл DMF и затем добавляли 773 мг N-сукцинимидил-6-малеимидогексаноата (1,2 экв.); перемешивание осуществляли в течение 5 ч при комнатной температуре до тех пор, пока реакция исходных веществ не завершилась (отслеживали при помощи ТСХ). К реакционной жидкости добавляли 10 мл воды и 20 мл этилацетата и перемешивали в течение 10 минут. Органический слой затем отделяли, сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали колоночной хроматографией (петролейный эфир:этилацетат=50:1) с получением 956 мг бледно-желтого пенистого твердого вещества с выходом 68,3%.
13) Синтез соединения 36
30 мг соединения 35 растворяли с использованием 20% раствора TFA в дихлорметане и затем осуществляли перемешивание в течение 2 ч при комнатной температуре до тех пор, пока реакция не завершилась (отслеживали при помощи ТСХ). Растворитель удаляли на роторном испарителе при пониженном давлении с получением неочищенного продукта, соединения 36, который непосредственно использовали на следующей стадии без очистки.

14) Синтез ama-0302
Неочищенный продукт соединения 36 (1,2 экв.), полученный на предыдущей стадии, растворяли с использованием 1 мл DMF; затем добавляли 32,5 мг соединения 30 и 20,5 мг HATU (1,5 экв.) и pH доводили до 8-9 при помощи DIPEA; перемешивание осуществляли в течение 6 ч при комнатной температуре в атмосфере азота до завершения реакции исходного вещества 30 (отслеживали при помощи ВЭЖХ); осуществляли очистку с использованием препаративной жидкостной хроматографии для сбора фракции, соответствующей целевому пику. После лиофилизации получали 15,3 мг бледно-желтого твердого вещества с выходом 29,1%. MS: [M+H]+ 1455,7011.
Пример 3 Синтез низкомолекулярной нагрузки ama-0303
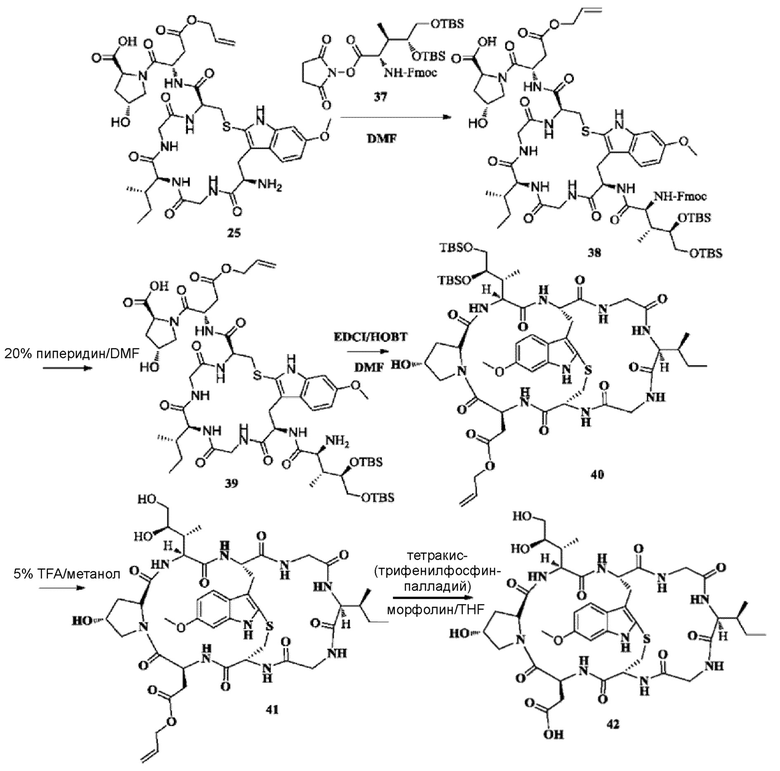
1) Синтез соединения 37
Синтез осуществляли со ссылкой на способ синтеза, описанный в J. Am. Chem. Soc. 2018, 140, 6513-6517.
2) Синтез соединения 38
Синтез осуществляли со ссылкой на синтез соединения 11. После лиофилизации получали около 751,5 мг целевого соединения с выходом 56,8%. MS: [M+H]+ 1426,6751.
3) Синтез соединения 39
Синтез осуществляли со ссылкой на синтез соединения 12. Добавляли 700 мг соединения 38 и получали около 482,3 мг белого твердого вещества путем лиофилизации с выходом 81,6%. MS: [M+H]+ 1205,6124.
4) Синтез соединения 40
Синтез осуществляли со ссылкой на синтез соединения 13. Добавляли 450 мг соединения 39. После очистки с использованием препаративной жидкостной хроматографии получали около 384,2 мг белого твердого вещества путем лиофилизации с выходом 86,7%. [M+H]+ 1186,6012.
5) Синтез соединения 41
Синтез осуществляли со ссылкой на синтез соединения 29. Добавляли 100 мг соединения 40; после очистки с использованием препаративной жидкостной хроматографии получали около 54,2 мг белого твердого вещества путем лиофилизации с выходом около 67,2%. [M+H]+ 958,4250.
6) Синтез соединения 42
Синтез осуществляли со ссылкой на синтез соединения 30. Добавляли 50 мг соединения 41; после очистки с использованием препаративной жидкостной хроматографии получали около 28,6 мг белого твердого вещества путем лиофилизации целевого пика, с выходом 59,7%. [M+H]+ 918,4413.
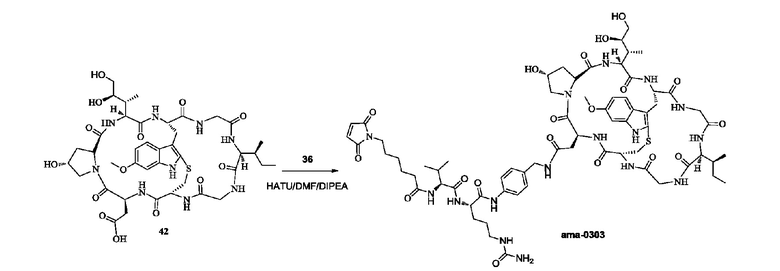
7) Синтез соединения ama-0303
Синтез осуществляли со ссылкой на синтез соединения ama-0302. Добавляли 25 мг соединения 42; после очистки с использованием препаративной жидкостной хроматографии получали около 15,2 мг бледно-желтого твердого вещества путем лиофилизации с выходом 38%. [M+H]+ 1471,6820.
Пример 4 Синтез низкомолекулярной нагрузки ama-0304
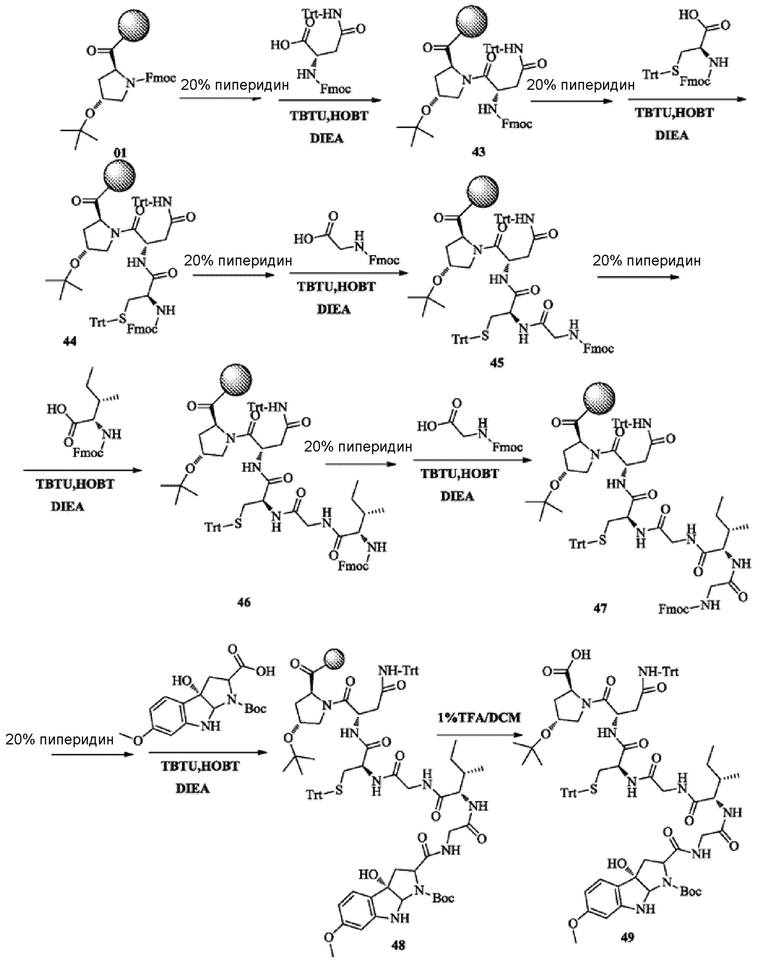
1) Синтез соединения 49
Что касается способа синтеза, его осуществляли со ссылкой на синтез соединения 08; получали около 2,4 г неочищенного продукта соединения 49 с чистотой около 86,2%, который непосредственно использовали на следующей стадии без очистки.
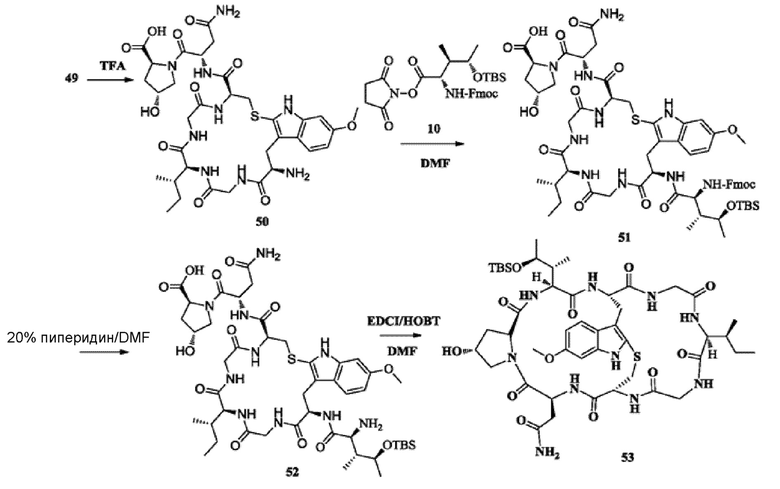
2) Синтез соединения 50
Синтез осуществляли со ссылкой на синтез соединения 09; после очистки с использованием препаративной жидкостной хроматографии получали около 1,1 г целевого соединения. [M+H]+ 790,4126.
3) Синтез соединения 51
Синтез осуществляли со ссылкой на синтез соединения 11. Добавляли 500 мг соединения 50; после очистки с использованием препаративной жидкостной хроматографии получали 341,5 мг целевого соединения с выходом 43%. [M+H]+ 1255,6195.
4) Синтез соединения 52
Синтез осуществляли со ссылкой на синтез соединения 12. Добавляли 300 мг соединения 51; после очистки с использованием препаративной жидкостной хроматографии получали около 186,4 мг белого твердого вещества путем лиофилизации фракции, соответствующей целевому пику, с выходом 75,5%. [M+H]+ 1033,5013.
5) Синтез соединения 53
Синтез осуществляли со ссылкой на синтез соединения 13. Добавляли 150 мг соединения 52; после очистки с использованием препаративной жидкостной хроматографии получали около 86,5 мг белого твердого вещества путем лиофилизации фракции, соответствующей целевому пику, с выходом 58,7%. [M+H]+ 1015,5121.
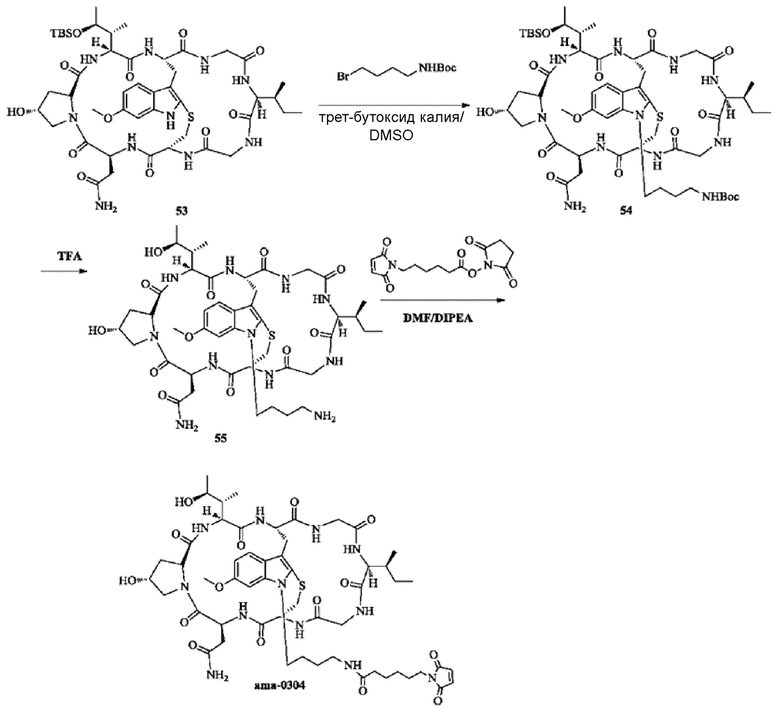
6) Синтез соединения 54
80 мг соединения 53 растворяли с использованием безводного DMSO; затем добавляли 159 мг трет-бутил-N-(4-бромбутил)карбаминовой кислоты (8 экв.) и 88 мг трет-бутоксида калия (10 экв.); реакционную смесь перемешивали в течение 12 ч при комнатной температуре и затем добавляли 159 мг трет-бутил-N-(4-бромбутил)карбаминовой кислоты (8 экв.) и 88 мг трет-бутоксида калия (10 экв.). Реакционную смесь перемешивали еще в течение 24 ч при комнатной температуре до исчезновения исходных веществ (отслеживали при помощи ВЭЖХ); осуществляли очистку с использованием препаративной жидкостной хроматографии для сбора фракции, соответствующей целевому пику; и получали 18,2 мг с выходом 19,5%. [M+H]+ 1186,6137.
7) Синтез соединения 55
18,2 мг соединения 54, полученного как описано выше, растворяли с использованием 0,1 мл трифторуксусной кислоты и перемешивали в течение 30 минут при комнатной температуре. Добавляли 2 мл дихлорметана; после перемешивания до гомогенности растворитель удаляли на роторном испарителе при пониженном давлении и полученное вещество сохраняли для последующего использования.
8) Синтез соединения ama-0304
Неочищенный продукт соединения 55, полученного как описано выше, растворяли с использованием 1 мл DMF и затем добавляли 9,5 мг N-сукцинимидил-6-малеимидогексаноата (2 экв.). pH доводили до 8-9 при помощи DIPEA; перемешивание осуществляли в атмосфере азота в течение 5 ч при комнатной температуре до тех пор, пока реакция исходных веществ не завершилась (отслеживали при помощи ВЭЖХ); осуществляли очистку с использованием препаративной жидкостной хроматографии для сбора фракции, соответствующей целевому пику. После удаления органического растворителя на роторном испарителе получали 10,4 мг не совсем белого твердого вещества путем лиофилизации с выходом 53%. [M+H]+ 1279,6537.
Пример 5 Синтез низкомолекулярной нагрузки ama-0305
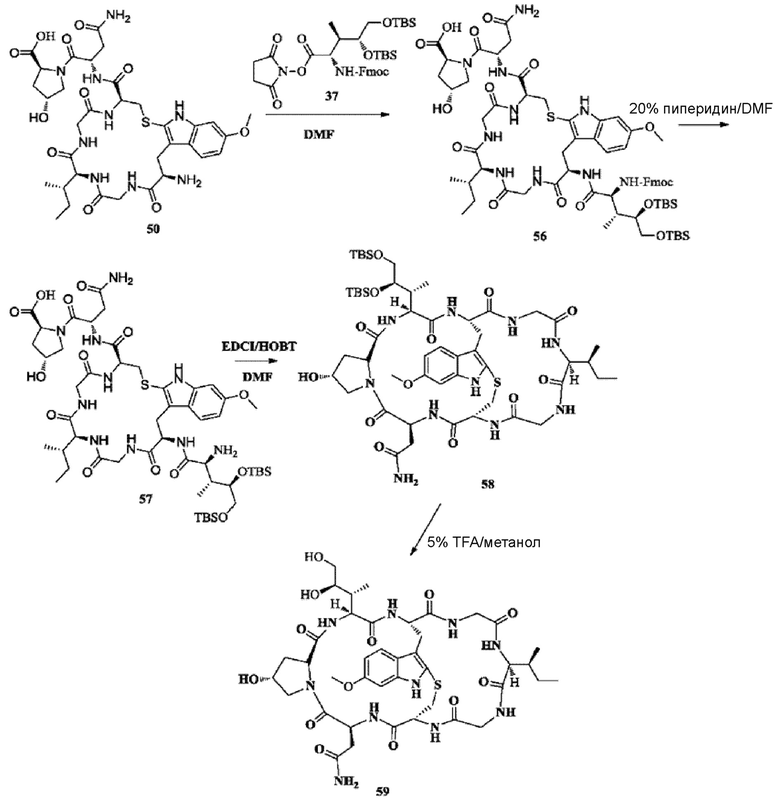
1) Синтез соединения 56
Синтез осуществляли со ссылкой на синтез соединения 11. Добавляли 200 мг исходного вещества 50 и получали около 211,3 мг белого твердого вещества путем лиофилизации с выходом 60,2%. [M+H]+ 1385,6713.
2) Синтез соединения 57
Синтез осуществляли со ссылкой на синтез соединения 12. Добавляли 200 мг исходного вещества 56 и получали около 114,8 мг белого твердого вещества путем лиофилизации с выходом 68,3%. [M+H]+ 1163,5793.
3) Синтез соединения 58
Синтез осуществляли со ссылкой на синтез соединения 13. Добавляли 110 мг исходного вещества 57 и получали около 72,5 мг не совсем белого твердого вещества путем лиофилизации с выходом 66,9%. [M+H]+ 1145,5901.
4) Синтез соединения 59
Около 70 мг соединения 58, полученного как описано выше, растворяли с использованием 0,5 мл раствора TFA в метаноле; затем осуществляли перемешивание в течение 1 ч при комнатной температуре до тех пор, пока реакция исходных веществ не завершилась (отслеживали при помощи ВЭЖХ); осуществляли очистку с использованием препаративной жидкостной хроматографии для сбора фракции, соответствующей целевому пику. После удаления органического растворителя на роторном испарителе получали 45,7 мг не совсем белого твердого вещества путем лиофилизации с выходом 81,6%. [M+H]+ 917,4013.
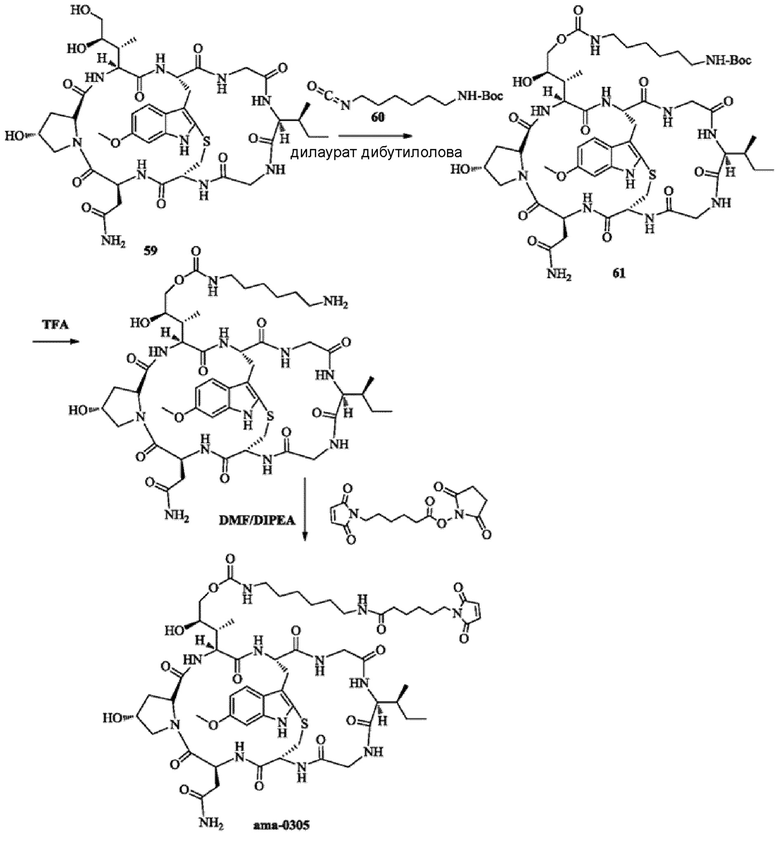
5) Синтез соединения 60
Синтез осуществляли со ссылкой на способ, описанный в патенте WO2012041504
6) Синтез соединения 61
Около 42 мг исходного вещества 59 растворяли с использованием 1 мл безводного DMF; затем добавляли 22 мг соединения 60 (2 экв.) и 58 мг дилаурата дибутилолова; после этого осуществляли перемешивание в течение 24 ч при комнатной температуре в атмосфере азота. Снова добавляли 22 мг соединения 60 (2 экв.) и перемешивание продолжали в течение 52 ч при комнатной температуре до исчезновения исходного вещества 59 (отслеживали при помощи ВЭЖХ). К вышеуказанной реакционной жидкости добавляли 0,2 мл метанола для остановки реакции; осуществляли очистку с использованием препаративной жидкостной хроматографии для сбора фракции, соответствующей целевому пику. Органический растворитель удаляли на роторном испарителе и получали около 9,6 мг бледно-желтого твердого вещества путем лиофилизации с выходом 18,1%. [M+H]+ 1159,5703.
7) Синтез соединения 62
9,6 мг соединения 61 полученного как описано выше, растворяли с использованием 0,1 мл трифторуксусной кислоты и перемешивали в течение 30 минут при комнатной температуре. Добавляли 2 мл дихлорметана; после перемешивания до гомогенности растворитель удаляли на роторном испарителе при пониженном давлении и полученное вещество использовали непосредственно на следующей стадии без очистки.
8) Синтез соединения ama-0305
Соединение 62, полученное как описано выше, растворяли с использованием 1 мл безводного DMF и затем добавляли 5,1 мг N-сукцинимидил-6-малеимидогексаноата (2 экв.); осуществляли перемешивание в атмосфере азота в течение 5 ч при комнатной температуре до тех пор, пока реакция исходных веществ не завершилась (отслеживали при помощи ВЭЖХ); осуществляли очистку с использованием препаративной жидкостной хроматографии для сбора фракции, соответствующей целевому пику; после сушки газообразным азотом получали 4,6 мг не совсем белого твердого вещества с выходом 44,4%. [M+H]+ 1252,6091.
Пример 6 Получение конъюгата антитело-лекарственное средство
1) Общий способ конъюгации: После предварительной очистки молекулы антител с выходом мономеров больше чем 95% подвергали замене среды и добавляли к фосфатно-буферному раствору, содержащему EDTA, через ультрафильтрационную центрифужную пробирку при концентрации 10 мг/мл. Добавляли TCEP в 10-кратном количестве относительно мольного количества молекул антител и осуществляли взаимодействие при комнатной температуре в течение 2 ч. Полученное вещество подвергали процедуре замены среды и добавляли к фосфатно-буферному раствору при pH 6,5 через ультрафильтрационную центрифужную пробирку; затем добавляли DHAA в 10-кратном количестве относительно мольного количества молекул антител и осуществляли взаимодействие при комнатной температуре в течение 2 ч. Затем добавляли полезную нагрузку в 3-кратном количестве относительно мольного количества молекул антител и осуществляли взаимодействие при комнатной температуре в течение 4 ч. После завершения реакции полученное вещество подвергали процедуре замены среды и добавляли к PBS через ультрафильтрационную центрифужную пробирку, имеющую отсечку по молекулярной массе 30 кДа, и несвязанную нагрузку удаляли.
2) Определение DAR антитела-лекарственного средства
Условия детекции для выхода мономера:
Образцы центрифугировали при 14000 об/мин в течение 5 минут и супернатант использовали для анализа вводимой пробы.
Устройство: Waters e2695 (2489 УФ/видимая область спектра)
Хроматографическая колонка: TSKgel G3000SWXL (7,8 × 300 мм, 5 мкм)
Подвижная фаза: A: 50 мМ PB, 300 мМ NaCl, 200 мМ Arg, 5% IPA, pH 6,5
изократическое элюирование с подвижной фазой A в течение 30 мин; скорость потока: 0,714 мл/мин; температура колонки: 25°C; и длина волны детекции: 280 нм.
Условия определения DAR:
Образцы центрифугировали при 14000 об/мин в течение 5 минут и супернатант использовали для анализа вводимой пробы.
Устройство: Waters H-class (TUV)
Хроматографическая колонка: Proteomix HIC Бутил-NP5 (4,6 × 35 мм, 5 мкм)
Подвижная фаза: A: 1,5 M сульфата аммония, 0,025 M безводного фосфата натрия, pH 7,0
B: 0,025 M безводного фосфата натрия, 25% IPA, pH 7,0
уравновешивание хроматографической колонки подвижной фазой A; градиентное элюирование с подвижной фазой A и B; скорость потока: 0,8 мл/мин; температура колонки: 25°C; и длина волны детекции: 214 нм.
3) Результаты
4) Заключение
После связывания ama-0301/ama-0302/ama-0303/ama-0304/ama-0305 с Трастузумабом эффективность связывания была выше, и выход мономера был выше.
Пример 7 Стабильность в плазме
1) Процедуры
Определенное количество образца ADC добавляли к человеческой плазме, из которой был удален человеческий IgG. Каждый ADC повторяли три раза в трех параллельных анализах и помещали в 37°C водяную баню для инкубации. После инкубации в течение 72 ч и 144 ч, соответственно, образец ADC извлекали и 100 мкл ProteinA смолы (MabSelect SuReTM LX Lot: # 10221479 GE, промытая PBS) добавляли в каждую пробирку, которую подвергали встряхиванию с использованием вертикального смесителя для адсорбции в течение 2 ч; после стадий промывки и элюирования получали ADC после инкубации и осуществляли детекцию образцов ADC, инкубированных в течение определенного времени, методом ОФ-ВЭЖХ.
2) Результаты
3) Заключение
В человеческой плазме каждый конъюгат антитело-лекарственное средство не показал почти никакого разложения через 3 и 6 дней и имел хорошую стабильность.
Пример 8 испытание на активность in vitro
1) Материалы, используемые в эксперименте
Клетки: от Cell Bank of Chinese Academy of Sciences
Среда для культивирования опухолевых клеток: Gibco
FBS: BIOWEST
2) Получение культуральной среды
Питательная среда (с 10% FBS, Пенициллин/стрептомицин (100 Ед/мл))
Среда для детекции (с 1% FBS, Пенициллин/стрептомицин (100 Ед/мл))
3) Процедуры
Ультрафиолетовую лампу бокса биологической безопасности включали заранее за 30 минут для облучения, а затем осуществляли вентиляцию в течение 3 минут. Питательную среду, среду для детекции, D-PBS и трипсин предварительно нагревали на водяной бане с термостатом 37°C, дезинфицировали поверхность спиртом и помещали в бокс биологической безопасности. Отбирали клетки с конфлюэнтностью около 80% и помещали в бокс биологической безопасности; после того, как старую среду удаляли, клетки промывали D-PBS, который затем аспирировали; и промытые клетки расщепляли трипсином в течение 2-3 минут, добавляли питательную среду для нейтрализации и центрифугировали при 1200 об/мин в течение 3 минут. Полученный после центрифугирования супернатант удаляли, и использовали 4 мл среды для детекции для гомогенного смешивания. 100 мкл использовали для подсчета, при этом отбирали 50 мкл клеточной жидкости и добавляли 50 мкл красителя трипанового синего для гомогенного смешивания и затем подсчета. В соответствии с определенным ранее количеством, клетки высевали при плотности 80 мкл/лунка в 96-луночный планшет, где только в лунку E11, F11 и G11 добавляли только 80 мкл среды для детекции, а крайние лунки заполняли 150 мкл DPBS. Разбавление раствора антител включает: использование среды для детекции для получения 300 мкл раствора испытываемого образца с начальной концентрацией 5 мкМ в первой колонке 96-луночного планшета (V-типа); добавление 210 мкл среды для детекции во вторую-десятую колонку с конца; добавление 30 мкл гомогенно смешанного раствора из первой колонки во вторую колонку; смешивание этого до гомогенности 10 раз пипеткой вверх и вниз; и удаление наконечника пипетки и повторение процедуры последовательно для следующих 7 концентраций; через 24 ч после посева добавление разбавленных антител при 20 мкл на лунку и установление контролей путем добавления только 20 мкл среды для детекции в колонку 11; повторение каждой концентрации в 2 лунках; и после добавления осуществление гомогенного смешивания с использованием вихревого встряхивателя для клеток при 550 об/мин в течение 3 мин.
4) Детекция
Через 4 дня реагент MTS извлекали, размораживали при комнатной температуре, защищая от света, и гомогенно смешивали в вихревой мешалке. В боксе биологической безопасности 20 мкл CellTiter 96® One Solution Reagen MTS реагента добавляли вдоль боковой стенки лунок на каждые 100 мкл объема клеточной культуры. Раствор MTS гомогенно смешивали, осторожно похлопывая по поверхности планшета, и помещали в инкубатор для клеток с защитой от света на 2 часа для постоянной инкубации. После завершения реакции 96-луночный планшет извлекали и измеряли значение оптической плотности при OD 490 нм с использованием считывающего устройства для микропланшетов; и осуществляли регистрацию, сортировку и хранение данных.
5) Результаты
IC50 (нМ)
IC50 (нМ)
IC50 (нМ)
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТ АНТИТЕЛА С АМАТОКСИНОМ НЕПРИРОДНОГО ТИПА | 2018 |
|
RU2840926C2 |
| КОНЪЮГАТ АМАНТИНА С АНТИТЕЛОМ | 2018 |
|
RU2809116C2 |
| КОНЪЮГАТЫ АМАТОКСИН - АНТИТЕЛО | 2016 |
|
RU2724328C2 |
| НОВЫЙ СПОСОБ СИНТЕЗА АМАНИТИНОВ | 2018 |
|
RU2792210C2 |
| КОНЪЮГАТЫ АМАТОКСИНОВ С УЛУЧШЕННЫМИ ЛИНКЕРАМИ | 2011 |
|
RU2601411C2 |
| СИНТЕЗ (S)-6-ГИДРОКСИТРИПТОФАНА И ЕГО ПРОИЗВОДНЫХ | 2019 |
|
RU2810786C2 |
| ПРОИЗВОДНЫЕ АМАТОКСИНА | 2014 |
|
RU2695370C2 |
| КОНЪЮГАТЫ АМАТОКСИНА С УЛУЧШЕННЫМИ СВЯЗЯМИ | 2012 |
|
RU2575854C2 |
| ЛИНКЕР ДЛЯ КОНЪЮГАТОВ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2792201C2 |
| СПОСОБЫ СИНТЕЗА АМАТОКСИНОВОГО СТРУКТУРНОГО БЛОКА И АМАТОКСИНОВ | 2013 |
|
RU2637924C2 |
Изобретение относится к области биотехнологии. Описана группа изобретений, включающая конъюгат неприродного аматоксина с антителом, обладающий противоопухолевой или противораковой активностью, и применение конъюгата для получения противоопухолевого лекарственного средства или противоракового лекарственного средства. В одном из вариантов реализации конъюгат имеет структурную формулу (I)
 .
.
Изобретение расширяет арсенал средств, обладающих противоопухолевой или противораковой активностью. 2 н. и 11 з.п. ф-лы, 4 табл., 8 пр.
1. Конъюгат неприродного аматоксина с антителом, обладающий противоопухолевой или противораковой активностью, содержащий фрагмент токсина и биомакромолекулярный фрагмент A, где конъюгат имеет структурную формулу (I), или его фармацевтически приемлемая соль,

где:
R1 представляет собой H, -OH или -O-L-A;
R2 представляет собой H или -OH;
R3 представляет собой OC1-6 алкил;
R4 представляет собой H или -L-A;
R5 представляет собой -NH2, -OH, -NH-L-A или -O-L-A;
A представляет собой биомакромолекулярный фрагмент, связывающийся с сайтом-мишенью;
где O представляет собой атом кислорода, N представляет собой атом азота, и H представляет собой атом водорода;
где L включает следующую структуру:
-L1-[L2]m-[AA]n-L3-,
где:
L1 или L3 каждый независимо выбран из одной или нескольких групп из сукцинимида и его формы с раскрытием цикла, -C(=O)-, -C(=O)NH-, C1-C6 алкила, C1-C6 алкокси, триазола, аминобензилового спирта и их производных;
L2 представляет собой спейсер, выбранный из одного или более из замещенного или незамещенного C1-C6 алкила, замещенного или незамещенного C3-C20 циклоалкила, замещенного или незамещенного C3-C20 гетероциклоалкила, замещенного или незамещенного C5-C20 арила, замещенного или незамещенного C5-C20 гетероарила и -(CH2CH2O)a-, где a представляет собой целое число, выбранное из 1-20, и где заместитель выбран из одной или более из гидроксильной группы, сульфгидрильной группы, галогена, карбоксильной группы, аминогруппы, фосфатной группы, нитрогруппы, цианогруппы, сульфогруппы или C1-C6 алкила, который не замещен или замещен одной или более из гидроксильной группы, сульфгидрильной группы, галогена, карбоксильной группы, аминогруппы, фосфатной группы, нитрогруппы, цианогруппы или сульфогруппы;
L1 связывается с биомакромолекулярным фрагментом A;
L2 связывает L1 с AA;
L3 связывается с фрагментом токсина;
m представляет собой целое число, выбранное из 1-6;
AA представляет собой структурный фрагмент, состоящий из 1-6 аминокислот, и n имеет значение 0 или 1.
2. Конъюгат неприродного аматоксина с антителом по п. 1 или его фармацевтически приемлемая соль, где A представляет собой биомакромолекулярный фрагмент, связывающийся с сайтом-мишенью, и включает антитело, его антиген-связывающий фрагмент, антителоподобный белок.
3. Конъюгат неприродного аматоксина с антителом по п. 2 или его фармацевтически приемлемая соль, где биомакромолекула, связывающаяся с сайтом-мишенью, представляет собой антитело или его антиген-связывающий фрагмент, которое выбрано из химерного антитела, деиммунизованного антитела, гуманизированного антитела, человеческого антитела, диатела, триатела, тетратела или нанотела.
4. Конъюгат неприродного аматоксина с антителом по п. 2 или 3 или его фармацевтически приемлемая соль, где антиген-связывающий фрагмент выбран из группы, состоящей из Fab, F(ab’), Fd, Fv, одноцепочечного Fv или дисульфид-связанного Fv(dsFv).
5. Конъюгат неприродного аматоксина с антителом по п. 1 или его фармацевтически приемлемая соль, где один и только один из R1, R4 и R5 содержит структуру -L-A.
6. Конъюгат неприродного аматоксина с антителом по п. 1 или его фармацевтически приемлемая соль, где L1 представляет собой линкер для связывания с биомакромолекулой A и выбран из:
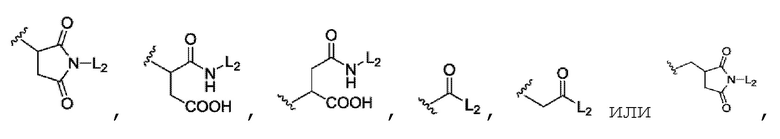
где волнистая линия связывает с биомакромолекулой A.
7. Конъюгат неприродного аматоксина с антителом по п. 1 или его фармацевтически приемлемая соль, где AA представляет собой фрагмент, состоящий из 1-6 аминокислот, которые выбраны из глицина, аланина, валина, лейцина, изолейцина, фенилаланина, пролина, триптофана, серина, тирозина, цистеина, метионина, аспарагина, глутамина, треонина, аспартата, глутамата, лизина, аргинина или гистидина.
8. Конъюгат неприродного аматоксина с антителом по п. 7 или его фармацевтически приемлемая соль, где аминокислоты представляют собой L-аминокислоты, выбранные из фенилаланина, цитруллина, валина, лизина, серина, глутамата, аспартата или глицина.
9. Конъюгат неприродного аматоксина с антителом по п. 1 или его фармацевтически приемлемая соль, где L3 выбран из
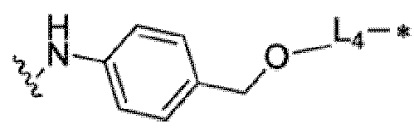 ,
, 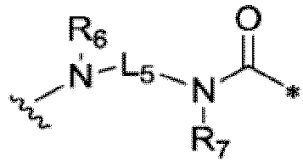 или
или 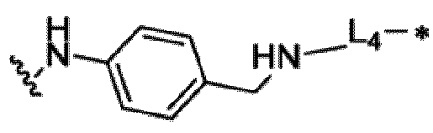
или любой связывающей группы, которая связывается с токсином;
где:
L4 или L5 каждый независимо выбран из одной или нескольких групп из сукцинимида и его формы с раскрытием цикла, -C(=O)-, -C(=O)NH-, C1-C6 алкила, C1-C6 алкокси, триазола, аминобензилового спирта и их производных;
R6 и R7 каждый независимо выбран из атома водорода, атома дейтерия, C1-C6 алкильной группы, замещенной алкильной группы, арильной группы, замещенной арильной группы или гетероарильной группы;
где волнистая линия связывает с AA, а звездочка связывает с токсином, как показано в структурной формуле формуле (I).
10. Конъюгат неприродного аматоксина с антителом по п. 9 или его фармацевтически приемлемая соль, где L4 представляет собой карбонильную группу или простую связь.
11. Конъюгат неприродного аматоксина с антителом по п. 9 или его фармацевтически приемлемая соль, где R6 и R7 каждый независимо выбран из водорода или C1-C6 алкила.
12. Конъюгат неприродного аматоксина с антителом по п. 9 или его фармацевтически приемлемая соль, где L5 представляет собой C2-C12 алкил.
13. Применение конъюгата неприродного аматоксина с антителом, обладающего противоопухолевой или противораковой активностью, по любому из пп. 1-12 или его фармацевтически приемлемой соли и фармацевтически приемлемого ингредиента для получения противоопухолевого лекарственного средства или противоракового лекарственного средства.
| КОНЪЮГАТЫ ЛИГАНДА С НЕСКОЛЬКИМИ ЛЕКАРСТВЕННЫМИ СРЕДСТВАМИ | 2006 |
|
RU2470668C2 |
| US 10245326 B2, 02.04.2019 | |||
| WO 2015073721 A1, 21.05.2015. | |||
