Настоящее изобретение относится к тимозину альфа-1 (Тα1) для применения в лечении муковисцидоза. Конкретней, настоящее изобретение относится к тимозину альфа-1 для применения в лечении муковисцидоза в качестве агента, корректирующего CFTR (регулятор трансмембранной проводимости при CF), агента, усиливающего CFTR, и противовоспалительного агента.
Муковисцидоз (CF, OMIM 219700) представляет собой опасное для жизни генетическое расстройство, которое прежде всего поражает легкие и пищеварительную систему. Оно представляет собой ограничивающее жизнь аутосомально-рецессивное расстройство, которое поражает 70000 человек по всему миру. Прогноз для пациентов, страдающих этим заболеванием, постоянно улучшается в течение многих лет, в основном в результате более раннего диагноза, более активной терапии и обеспечения ухода в специализированных центрах (1). У исследователей теперь есть более полное понимание молекулярно-биологического дефекта, который лежит в основе CF, что приводит к новым подходам в лечении.
CF вызван мутациями в гене, кодирующем регулятор трансмембранной проводимости при CF (CFTR), который регулирует секрецию жидкости через эпителиальную поверхность в респираторном и желудочно-кишечном трактах. Процессинг CFTR в клетке включает трансляцию, фолдинг в эндоплазматическом ретикулуме, транспорт через аппарат Гольджи, посттрансляционные модификации, нацеливание на апикальную плазматическую мембрану и эндосомальную рециркуляцию и возврат. CFTR плазматической мембраны интернализуется путем эндоцитоза и затем рециркулирует в плазматическую мембрану или направляется для деградации в лизосомы (2, 3).
В базе данных мутаций, приводящих к муковисцидозу, представлено почти 2000 вариантов (4). Эти мутации сгруппированы в шесть классов: мутанты класса I включают делеции, сдвиги рамки считывания и нонсенс-мутации, которые приводят в результате к преждевременно усеченным белковым продуктам CFTR, мутанты класса II являются дефектными по внутриклеточной миграции, мутанты класса III представляют собой полноразмерные белки, обладающие небольшой или отсутствием активности ионного канала, мутанты класса IV приводят в результате к CFTR с лишь слегка уменьшенной активностью канала, и белки мутантов класса V являются функциональными, но экспрессируются с пониженными уровнями, в то время как мутанты класса VI экспрессируются с уровнями, характерными для дикого типа, но демонстрируют уменьшенную стабильность на плазматической мембране (5). Несмотря на это, большое количество аллелей заболевания CFTR, у подавляющего большинства (более 90%) пациентов, страдающих CF, имеющих североевропейское происхождение, существует по меньшей мере одна копия единичной мутантной аллели ΔF508, которая кодирует молекулу CFTR, лишенную фенилаланина в положении 508. Делеция фенилаланина в положении 508 (ΔF508) в CFTR приводит в результате к чувствительному к температуре дефекту фолдинга, задержке белка в эндоплазматическом ретикулуме, и впоследствии направлению в протеасому для преждевременной деградации (6). Фолдинг ΔF508-CFTR является неэффективным, причем больше чем 99% ΔF508-CFTR направляются для протеасомальной деградации до того, как достигают аппарата Гольджи.
Изменение внутриклеточного поведения неправильно свернутого CFTR путем вмешательства в фолдинг, процессинг белка и протеолитические пути продемонстрировало перспективу для прерывания "последующей патологии". Идентификация специфических мишеней для корректирования дефектного фолдинга ΔF508-CFTR или процессинга в клетке (корректирующие агенты) и воротного механизма ионных каналов (усиливающие агенты) обеспечивает стратегию терапии CF, которая корректирует основной дефект. Корректирующие агенты могут действовать как "фармакологические шапероны" путем взаимодействия с самим F508del-CFTR, облегчая его фолдинг и клеточный процессинг, или как "регуляторы протеостаза" путем модулирования внутриклеточной структуры, контролирующей качество, для изменения распознавания и процессинга ΔF508-CFTR (7). В отличие от существующих в настоящее время способов терапии, таких как антибиотики, противовоспалительные агенты, муколитические агенты, распыляемый гипертонический физиологический раствор и замещение панкреатического фермента, которые лечат проявления болезни CF, корректирующие агенты и усиливающие агенты исправляют дефект анионного канала, в основе которого лежит CFTR.
К настоящему времени идентифицированы многочисленные агенты, усиливающие CFTR, и подтверждена их эффективность в исследованиях как in vitro, так и in vivo (8, 9), хотя эффективность большинства корректирующих агентов была несколько неудовлетворительной. В 2012 FDA (Управление по контролю за продуктами питания и лекарственными средствами США) одобрило ивакафтор (VX-770, Kalydeco, Vertex Pharmaceuticals), представляющий собой усиливающий агент, который может увеличить опосредованный CFTR хлоридный транспорт для лечения пациентов, страдающих CF, с мутацией G551D-CFTR, которая вызывает только дефект воротного механизма (10). Kalydeco также был протестирован на пациентах, являющихся гомозиготными по F508del-CFTR, продемонстрировав небольшую клиническую пользу, поскольку лишь небольшое количество F508del-CFTR направляется к клеточной плазматической мембране. После этого комбинированную терапию усиливающими агентами (циклопропан карбоксамид VX-809 (лумакафтор, от Vertex Pharmaceuticals)/ивакафтор (ORKAMBI) тестировали на гомозиготных пациентах F508del-CFTR, страдающих CF. Комбинация лекарственных средств улучшала функцию легкого на 3-4 процента и уменьшала осложнения на 35% (11). Хотя исследование подтвердило, что острая комбинированная терапия ивакафтор/лумакафтор может усилить восстанавливаемую лумакафтором активность ΔF508-CFTR, также сообщали об разочаровывающих результатах. В частности, некоторые усиливающие агенты, действующие на воротный механизм (включая ивакафтор), могли уменьшать корректирующую эффективность лумакафтора, вероятно из-за дестабилизации ивакафтором скорректированного ΔF508-CFTR (12).
Эффект отмены ивакафтора на корректирующую эффективность лумакафтора свидетельствует о необходимости дополнительной оптимизации усиливающих агентов для максимизации клинической пользы комбинированной терапии CF корректирующим агентом-усиливающим агентом.
Кроме того, принимая во внимание то, что ΔF508 следует рассматривать не как простой мутант класса II, но как смешанный мутант со свойствами классов II, III и V, предполагается рассмотрение или разработка более сложной терапевтической стратегии для корректирования множественных дефектов процесссинга белка, вызванных единичной мутацией ΔF508 (6).
Более того, отсутствует значительный прогресс в идентификации терапевтических стратегий возврата заболевания легких с хронических стадий. Таким образом, остается открытый вопрос, является ли восстановление функции хлоридный канал адекватным для обращения воспалительной патологии хронического заболевания легкого CF (13).
Действительно, дефект гена CF (или мутация), приводящий к нарушению функционирования белка CFTR, приводит к последствиям как в эпителиальных, так и воспалительных клетках легкого, что в результате приводит к уменьшенному выходу хлорида и увеличенному воспалительному ответу (14).
В отношении чрезмерного воспаления в дыхательных путях при CF, вопрос о том, является ли гипервоспалительный ответ результатом хронической инфекции или является первичным результатом дисфункции CFTR, все еще остается предметом спора (15).
Дефектный ионный транспорт и транспорт жидкостей, обусловленный мутацией CFTR, приводит в результате к ненадлежащему клиренсу слизи и захваченного ей материала в дыхательных путях пациентов, страдающих CF. Задержка материала приводит в результате к циклу обструкции дыхательных путей, воспалению и инфекции. Соответственно, легочный иммунный ответ при CF характеризуется ранней и неспецифической активацией системы врожденного иммунитета, которая разрегулирована на нескольких уровнях (16), не приводит в результате к усилению бактериального или грибкового клиренса (17) и играет основную роль в патогенезе легочного заболевания при CF (18). Нарушенное уничтожение бактерий на ранней стадии жизни вызывает преимущественно нейтрофильный воспалительный ответ, который повреждает легкое и способствует ремоделированию и обструкции дыхательных путей. Заметное присутствие хронических инфекций легких при CF, несмотря на интенсивную антибиотическую терапию (19), стимулировало некоторые инновационные подходы, в рамках которых выявление исходного воспалительного ответа в дыхательных путях при заболевании легкого CF стало приоритетной областью трансляционного исследования (20). Доказательства указывают на то, что нацеливание на специфические воспалительные/противовоспалительные пути может представлять собой допустимую терапевтическую стратегию при экспериментальном CF (21, 22).
Только одно противовоспалительное лекарственное средство (нестероидный агент ибупрофен) до настоящего времени продемонстрировало эффективность с улучшением уровня снижения FEV1 в течение 2-летнего периода и благоприятный профиль риска; поэтому его рекомендуют в качестве благоприятной терапии для пациентов в возрасте 7-18 лет (23).
Разработка средств противовоспалительной терапии CF является сложной, поскольку механизм их действия может представлять собой предупреждение длительного клинического ухудшения, а не влияние на острые изменения в чаще всего используемых показателях исхода заболевания, таких как легочная функция или частота легочных приступов. Несмотря на эти трудности, несколько терапевтических средств в настоящее время переведены на стадию фазы 2. Три примера включают N-ацетилцистеин, докозагексаеновую кислоту и силденафил. N-ацетилцистеин представляет собой антиоксидантное лекарственное средство для перорального введения, которое, как показано в одном из испытаний фазы 2, влияет на воспалительные показатели у пациентов, страдающих CF, и основное воспаление, причем, как предполагают, действие осуществляется за счет устранения окислительно-восстановительного дисбаланса в нейтрофилах (24). Недавнее клиническое испытание фазы 2b N-ацетилцистеина действительно демонстрировало действие на легочную функцию, но действие в отношении воспаления не было воспроизведено (24). Похожим образом, докозагексаеновая кислота, представляющая собой омега-3 жирную кислоту, продемонстрировала противовоспалительную активность в коротком клиническом испытании в 2003 году и, было завершено ее испытание в фазе 2 среди младенцев; результаты скоро будут получены. Фаза 2 испытания силденафила в настоящее время оценивает действие этого ингибитора фосфодиэстеразы для перорального введения в отношении показателей воспаления дыхательных путей (25).
В свете вышеприведенного, очевидна потребность в том, чтобы предложить новые способы терапии муковисцидоза (CF), способные преодолеть недостатки известных способов терапии, включающие сильные противовоспалительные лекарственные средства кортикостероиды, которые обладают существенными побочными действиями, в частности, подавлением роста и адренальной супрессией у детей (26).
Общий консенсус в терапевтических препаратах при CF состоит в том, что будущие способы терапии будут направлены скорее на предупреждение, нежели чем улучшение существующего поражения органа, включая патогенное воспаление, прежде чем у пациентов начнут проявляться симптомы.
Ясно, что идеальное лечение CF при помощи лекарственных средств должно быть способно не только восстановить мембранный белок CFTR, но также и облегчить гипервоспалительный ответ и потенциально уменьшить прогресс хронического заболевания легкого CF. Идентификация отдельных соединений, обладающих двойными корректирующими и усиливающими действиями, а также противовоспалительной активностью, была бы весьма желательна в области техники.
Тимозин альфа-1 (Тα1) представляет собой природный полипептид из 28 аминокислот, впервые описанный и охарактеризованный Goldstein et al. в 1972 году (27). Тα1 хорошо известен в медицинской области техники из-за своих иммунорегуляторных свойств в нескольких исследованиях in vitro и in vivo (27).
Предыдущее использование Тα1 уже известно. Пептид использовался во всем мире в качестве адъювантного или иммунотерапевтического агента для лечения разнородных заболеваний человека, включая вирусные инфекции, иммунодефициты и злокачественные образования (28, 29). Пептид может усиливать ответ Т-лимфоцитов, дендритных клеток и ответы в виде антител, модулировать продукцию цитокина и хемокина и блокирует вызванный стероидами апоптоз тимоцитов. Его центральная роль в модулировании функции дендритной клетки и активации множественных сигнальных путей, дифференцированно вносящих вклад в различные функции, может предложить вероятное объяснение его плейотропного действия. Кроме того, способность активировать фермент индолеамин-2,3-диокисгеназу, обеспечивающий иммунную толерантность во время трансплантации и ограничение неразрешимой проблемы, которая сохраняет хроническое воспаление, явилось поворотным моментом, свидетельствующим о потенциальной специфической функции в иммунитете (30). Соответственно, Тα1, как недавно показали, способствует иммуновостановительной терапии и улучшает выживание реципиентов трансплантатов от родственных индивидов стволовых клеток, истощенных по Т-клеткам, удовлетворяющих в соответствии с HLA, в фазе I/II клинического испытания (31).
В соответствии с настоящим изобретением обнаружено, что Тα1 обладает противовоспалительной эффективностью у пациентов, страдающих CF. Фактически Тα1 способен корректировать неправильный процессинг мутанта ΔF508-CFTR с дефектами миграции/стабильности; для восстановления активности хлоридного канала и для повторного воздействия на баланс воспалительной/противовоспалительной активности у мышей, страдающих CF, путем ингибирования продукции воспалительных цитокинов (IL1β и 1L1α), увеличения продукции блокирующего воспаление цитокина IL-1RA, стимуляции защитной толерогенной оси Th1/Treg, при повторном ограничении воспалительной оси Th17/Th2, вовлеченной в воспаление легкого при CF. Вместе эти результаты свидетельствуют о том, что коррекция Тα1 может увеличить уровни, которые влияют на функцию эпителия дыхательных путей и, таким образом, может быть клинически значимой. Последнее дополнительно к присущей Тα1 противовоспалительной активности делает его идеальным кандидатом для терапии CF.
Таким образом, настоящее изобретение относится к тимозину альфа-1 для применения в лечении муковисцидоза. Фактически, тимозин альфа-1 можно благоприятным образом использовать в качестве агента, корректирующего CFTR, агента, усиливающего CFTR, и противовоспалительного агента у пациентов, страдающих муковисцидозом.
Настоящее изобретение также относится к комбинации тимозина альфа-1 с по меньшей мере одним агентом, выбранным из группы, состоящей из антибиотика, противогрибкового агента, агента, корректирующего CFTR, агента, усиливающего CFTR, где указанный агент, корректирующий или усиливающий CFTR, не являются тимозином альфа-1, для раздельного или последовательного применения в лечении муковисцидоза.
В соответствии с комбинацией по настоящему изобретению антибиотический агент может быть выбран, например из группы, состоящей из тобрамицина, ципрофлоксацина, колистина; противогрибковый агент может быть выбран например из группы, состоящей из итраконазола, амфотерицина В; наконец, агент, корректирующий или усиливающий CFTR, который не является тимозином альфа-1, может быть выбран, например из группы, состоящей из ивафактора, лумакафтора.
Еще одной задачей настоящего изобретения является предложение фармацевтической композиции, содержащей или состоящей из тимозина альфа-1 в качестве действующего начала вместе с одним или более чем одним эксципиентом и/или вспомогательными веществами для применения в лечении муковисцидоза.
Фармацевтическая композиция в соответствии с настоящим изобретением дополнительно может включать по меньшей мере один агент, выбранный из группы, состоящей из антибиотика, противогрибкового агента, агента, корректирующего CFTR, агента, усиливающего CFTR, где указанный агент, корректирующий CFTR, или агент, усиливающий CFTR, не являются тимозином альфа-1.
Как упомянуто выше, антибиотический агент может быть выбран, например из группы, состоящей из тобрамицина, ципрофлоксацина, колистина; противогрибковый агент может быть выбран, например из группы, состоящей из итраконазола, амфотерицина В; наконец, агент, корректирующий CFTR, или агент, усиливающий CFTR, которые не являются тимозином альфа-1, могут быть выбраны, например из группы, состоящей из ивафактора, лумакафтора.
Как описано выше, тимозин альфа-1 демонстрирует противовоспалительную эффективность у пациентов, страдающих муковисцидозом.
Таким образом, еще одной задачей настоящего изобретения является предложение тимозина альфа-1 для применения в лечении и/или при предупреждении воспаления у пациентов, страдающих муковисцидозом.
Настоящее изобретение также относится к комбинации тимозина альфа-1 с по меньшей мере одним агентом, выбранным из группы, состоящей из антибиотика, противогрибкового агента, агента, корректирующего CFTR, агента, усиливающего CFTR, где указанный агент, корректирующий CFTR, или агент, усиливающий CFTR, не являются тимозином альфа-1, для раздельного или последовательного применения в лечении и/или предупреждении воспаления у пациентов, страдающих муковисцидозом.
В соответствии с комбинацией по настоящему изобретению антибиотический агент может быть выбран, например из группы, состоящей из тобрамицина, ципрофлоксацина, колистина; противогрибкового агента, где агент может быть выбран, например из группы, состоящей из итраконазола, амфотерицина В; наконец, агент, корректирующий CFTR, или агент, усиливающий CFTR, которые не являются тимозином альфа-1, могут быть выбраны, например из группы, состоящей из ивафактора, лумакафтора.
Еще одной задачей настоящего изобретения является предложение фармацевтической композиции, содержащей или состоящей из тимозина альфа-1 в качестве действующего начала, вместе с одним или более чем одним эксципиентом и/или вспомогательным веществом для применения в лечении и/или предупреждении воспаления у пациентов, страдающих муковисцидозом.
Фармацевтическая композиция в соответствии с настоящим изобретением дополнительно может содержать по меньшей мере один агент, выбранный из группы, состоящей из антибиотика, противогрибкового агента, агента, корректирующего CFTR, агента, усиливающего CFTR, где указанный агент, корректирующий CFTR, или агент, усиливающий CFTR, не являются тимозином альфа-1.
Как упомянуто выше, антибиотический агент может быть выбран, например из группы, состоящей из тобрамицина, ципрофлоксацина, колистина; противогрибковый агент может быть выбран, например из группы, состоящей из итраконазола, амфотерицина В; наконец, агент, корректирующий CFTR, или агент, усиливающий CFTR, которые не являются тимозином альфа-1, могут быть выбраны, например из группы, состоящей из ивафактора, лумакафтора.
При использовании в соответствии с изобретением термины "лечить" или "лечение" имеют свое обычное значение, которое включает предупреждение, подавление, ослабление, ингибирование, уменьшение интенсивности, остановку, сдерживание, замедление или обращение прогресса, активации или уменьшение тяжести заболевания, опосредованного воспалением.
В соответствии с настоящим изобретением под "раздельным применением" подразумевают одновременное введение двух или более соединений комбинации в соответствии с изобретением в отдельных фармацевтических формах. Под "последовательным применением" понимают последовательное введение двух или более соединений композиции в соответствии с изобретением, каждое из которых находится в отдельной фармацевтической форме.
Эффективное количество Тα1, которое вводят для лечения воспалительной реакции, представляет собой такое количество, которое требуется для предупреждения, препятствования, облегчения, уменьшения интенсивности, остановки, подавления, замедления или обращения прогресса, или уменьшения тяжести указанной воспалительной реакции, и вводимая суточная доза будет зависеть от решения основного лечащего врача, массы тела, возраста и общего состояния пациента.
Тα1 может быть введен в форме фармацевтической композиции в комбинации с фармацевтически приемлемыми носителями или эксципиентами, пропорцию и природу которых определяют в соответствии с растворимостью и химическими свойствами соединения в выбранных носителях и/или эксципиентах, выбранным путем введения и стандартной фармацевтической практикой.
Носитель или эксципиент может представлять собой твердый, полутвердый или жидкий материал, который может служить в качестве разбавителя или среды для активного ингредиента. Подходящие носители или эксципиенты хорошо известны в области техники. Фармацевтическая композиция может быть адаптирована для перорального введения, ингаляции, парентерального или местного применения и может быть введена пациенту в форме таблеток, капсул, аэрозолей, лекарственных форм для ингаляции, суппозиториев, раствора, суспензий, липосомы и т.п.
Настоящее изобретение далее будет описано иллюстративным, но не ограничивающего его объем образом, в соответствии с его предпочтительными воплощениями, с конкретной ссылкой на приложенные графические материалы, где:
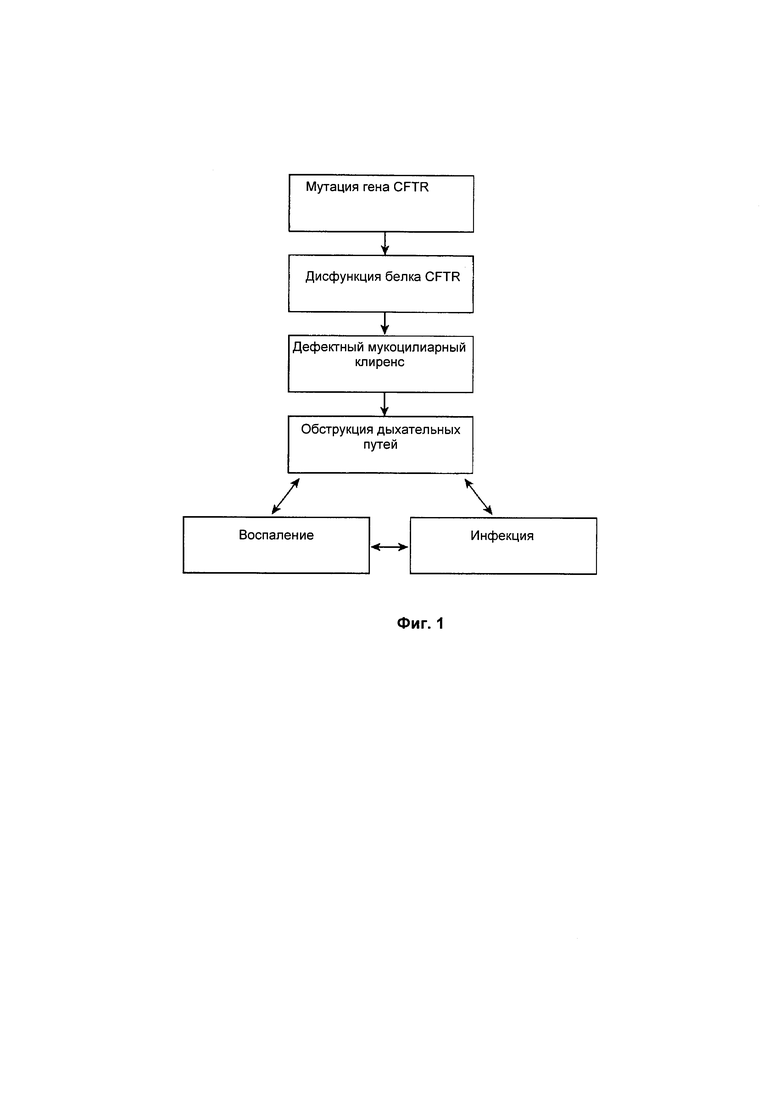
Фиг. 1 демонстрирует модель патофизиологии легочного заболевания муковисцидоза. В соответствии с этой моделью нарушенный ионный транспорт и транспорт жидкости вследствие мутации CFTR приводит в результате к ненадлежащему клиренсу слизи и захваченного ей материала в дыхательных путях пациентов, страдающих муковисцидозом. Задержка материала приводит в результате к циклу обструкции дыхательных путей, воспалению и инфекции. CFTR, регулятор трансмембранный проводимости при муковисцидозе.
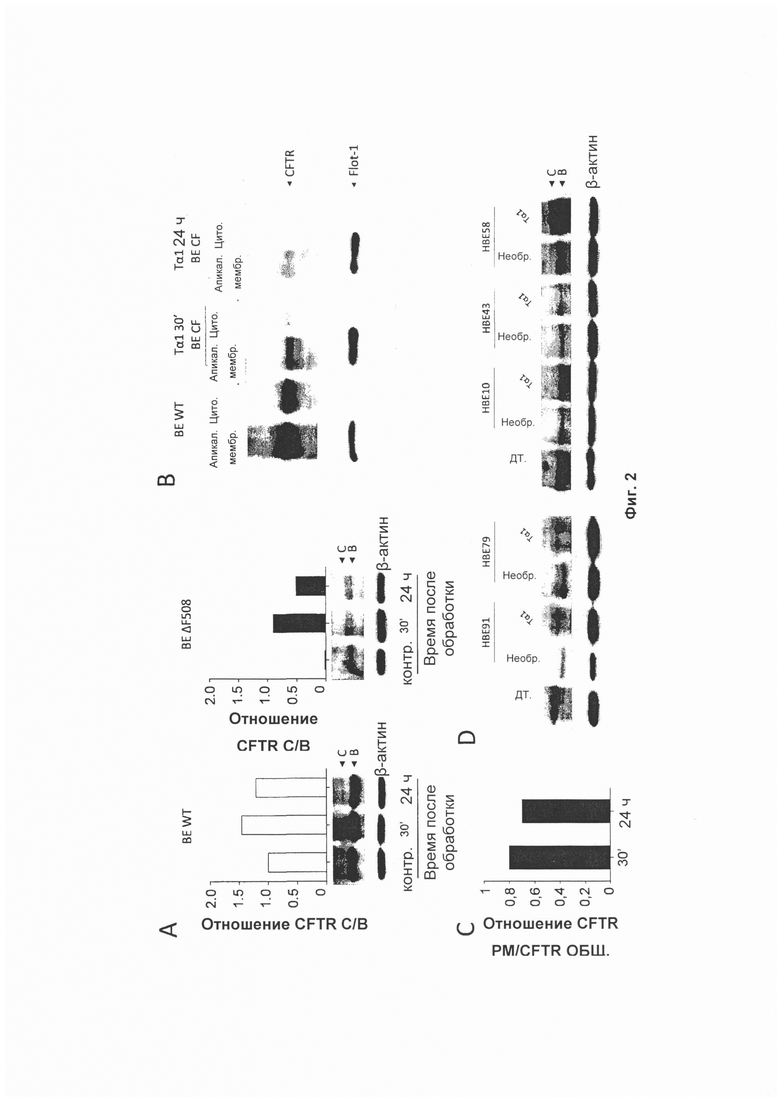
Фиг. 2 демонстрирует типичный иммуноблотинг общего клеточного белка контрольных клеток (НВЕ WT) и клеток ΔF508 (НВЕ ΔF508), обработанных Тα1 (А); Тα1 увеличивал экспрессию ΔF508 CFTR на РМ (плазматической мембране) (В); денситометрическое измерение CFTR (полоса С) на РМ выражается как общее отношение CFTR PM/CFTR (С); Тα1 увеличивал экспрессию CFTR (полоса С) у 3 (пациент №1, 2 и 5) из 5 пациентов (D).
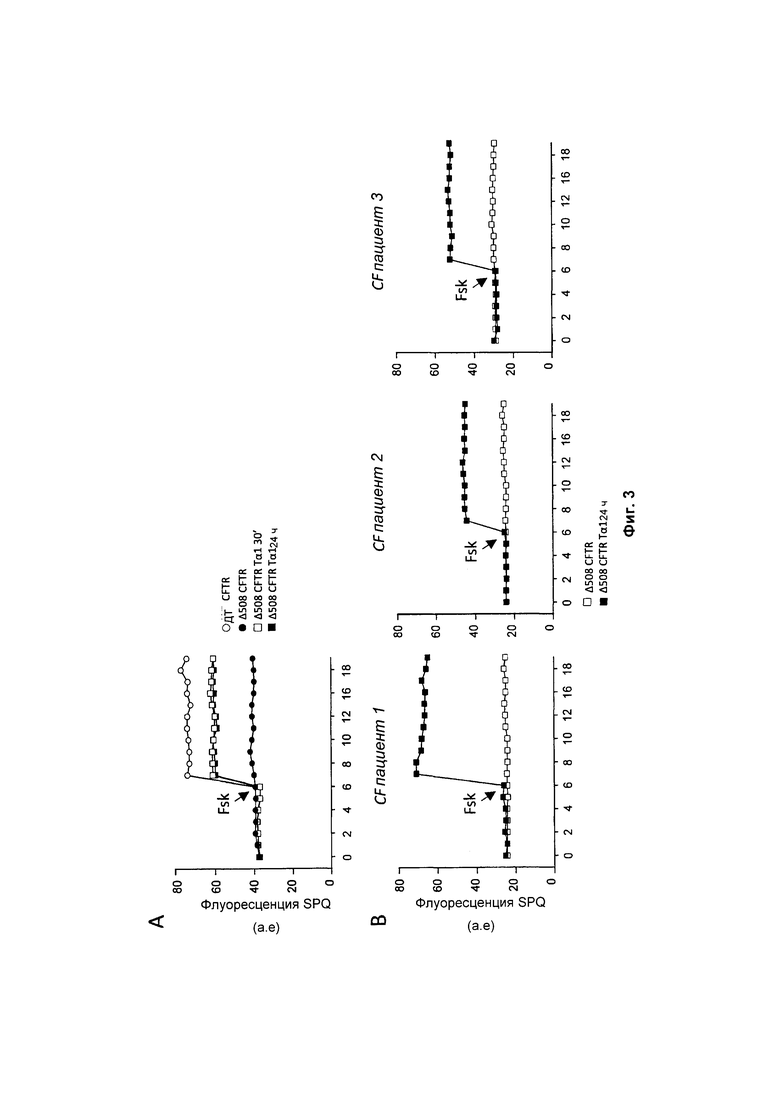
Фиг. 3 демонстрирует, что Тα1 увеличивал хлоридную проницаемость ΔF508 CFTR до приблизительно 70% относительно контроля (рассматривая контроль WT как 100% референсное значение) в клетках CFBE 41 о- (А) и клетках НВЕ пациентов, страдающих от CF (В).
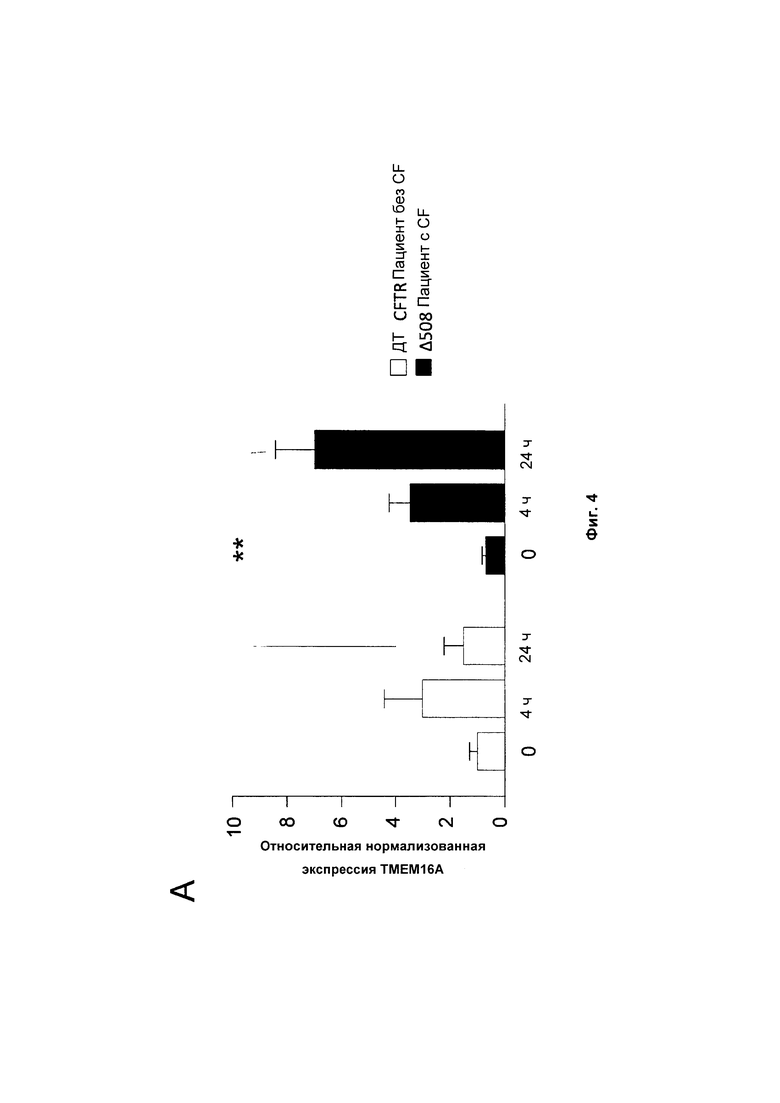
Фиг. 4 демонстрирует, что Тα1 значительно увеличивал экспрессию ТМЕМ16А в контрольных клетках и клетках CF после 4 часов воздействия. Тем не менее, хотя экспрессия ТМЕМ16А вернулась к базовым значениям в контрольных клетках через 24 часа, она оставалась увеличенной в клетках CF (А).
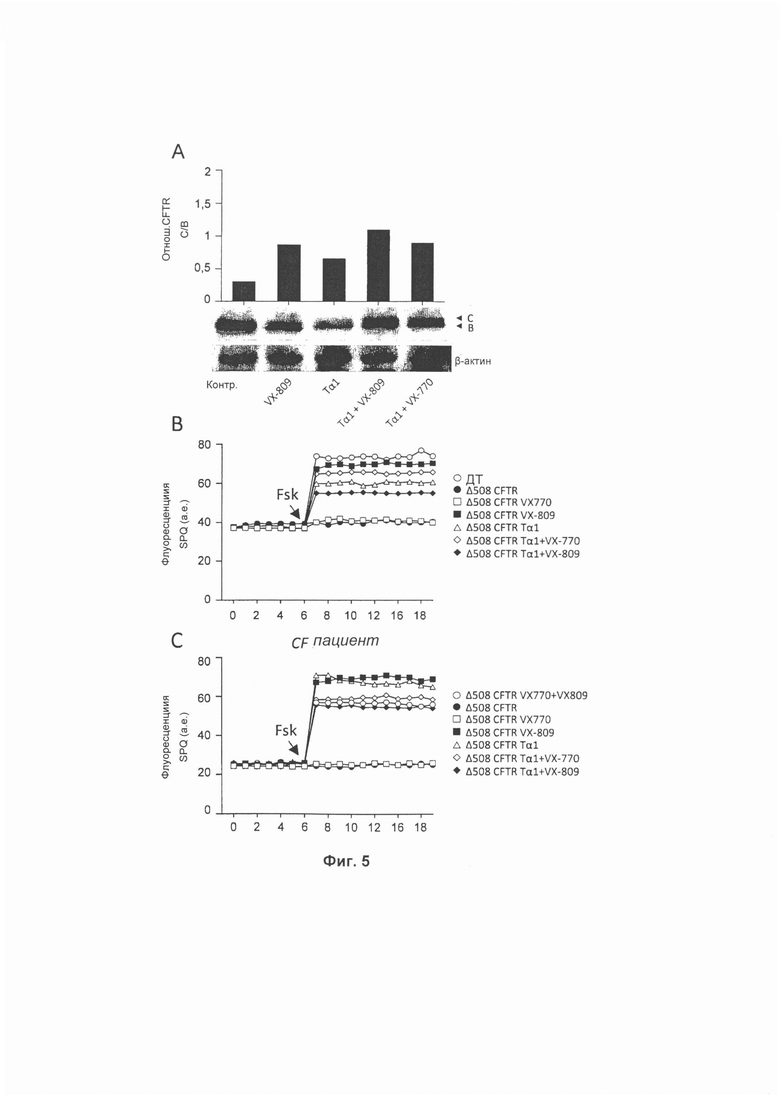
Фиг. 5 демонстрирует, что Tα1 увеличивал экспрессию зрелой формы (обозначенной С) по сравнению с незрелой формой (обозначенной В) CFTR в клетках, трансфицированных ΔF508 (А, типичный иммуноблотинг общего клеточного белка. Полосы CFTR количественно определяли путем денситометрии и выражали как отношение С/В). Стрелка указывает на положении форм В и С CFTR на основе относительной подвижности. Тα1 сам по себе или в комбинации с ивакафтором увеличивал хлоридную проницаемость ΔF508 CFTR в клетках до 60-70% относительно контроля (рассматривая контроль WT как 100% референсное значение)(В, определяли при помощи флуоресцентного анализа после стимулирования фосфорколином, Fsk). По сравнению с лумакафтором активность Тα1 была ниже в клетках CFBE41o, экспрессирующих ΔF508 CFTR (В), но была близка к клеткам НВЕ пациентов, страдающих CF (С). В комбинации с лумакафтором Тα1 не увеличивал активность лумакафтора (В и С).
Фиг. 6 демонстрирует, что мыши CF чувствительны к воспалительной патологии при инфекции. Длительный и устойчивый воспалительный ответ, характеризующийся рекрутингом нейтрофилов (PMN) и эозинофилов (EOS) в BAL (бронхоальвеолярном лаваже) (А) и легких (В и С), обнаружен у мышей Cftr-/-, у которых некоторая степень воспаления уже обнаруживалась до инфекции (С, гистология легкого (окрашивание Шифф-йодной кислотой и во вставке окрашивание по Гомори) в различные сутки после инфицирования (dpi)). Дифференцированный подсчет моноядерных клеток (MNC), клеток PMN и EOS осуществляли окрашиванием по Май-Грюнвальду-Гимзе в различные сутки после инфицирования (dpi). Величины представляют среднее значение ± SEM (стандартная ошибка) для трех мышей на группу и представляют собой типичные величины для 3 экспериментов. Фотографии получены при помощи микроскопии высокого разрешения на Olympus DP71 с использованием объектива ах20. Масштабная линейка 200 мкм. (С) Количество Gr1+положительных клеток оценивали путем проточной цитометрии на общих клетках легких в различные dpi. *Р меньше 0,05.
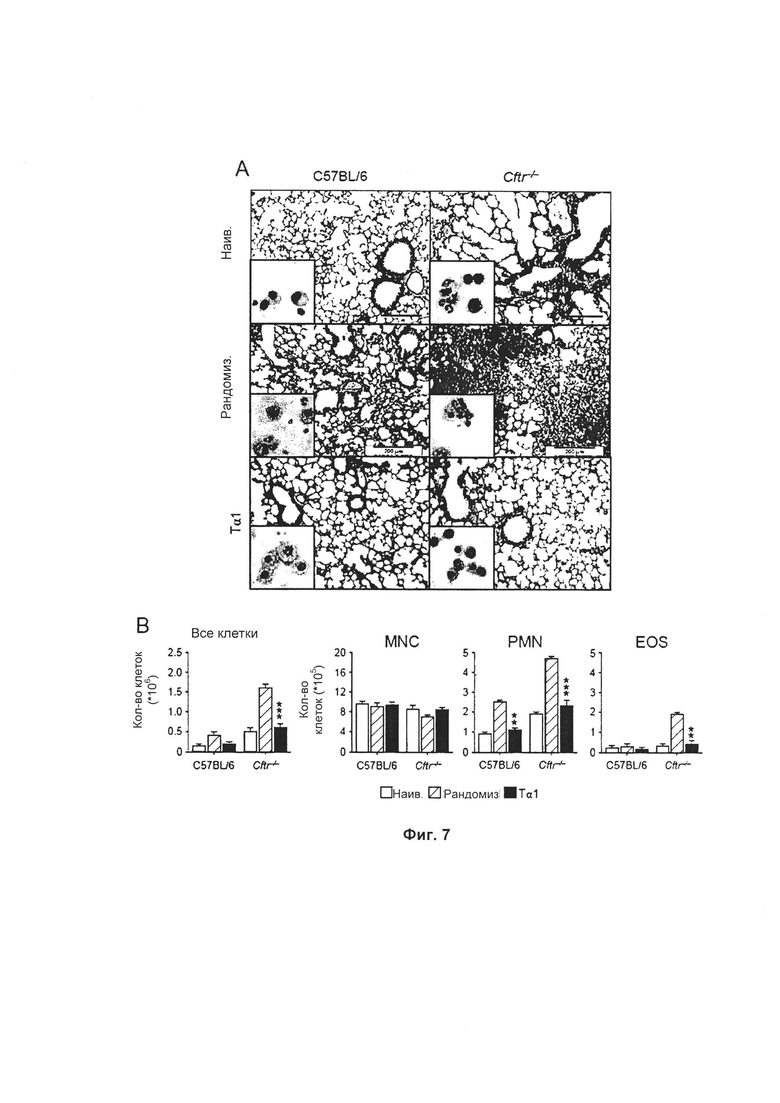
Фиг. 7 демонстрирует, что Тα1 защищает мышей, страдающих CF, от воспалительной патологии. Тα1, а не рандомизированный пептид, значительно снижал локальный рекрутинг воспалительных клеток и легочную патологию у мышей C57/BL6 и даже у мышей Cftr-/-, о чем свидетельствует уменьшение рекрутинга воспалительных клеток (в основном PMN, смотри вставки) в легком (А), а также в BAL (В). (А) Гистология легкого (окрашивание Шифф-йодной кислотой и клеточный рекрутинг во вставке). Фотографии получены при помощи микроскопии высокого разрешения на Olympus DP71 с использованием объектива ах20. Масштабная линейка 200 мкм. (В) Морфометрия BAL через 7 суток после инфицирования (dpi). Дифференцированный подсчет моноядерных клеток (MNC), полиморфоядерных (PMN) клеток и эозинофилов (EOS) осуществляли при помощи окрашивания по Май-Грюнвальду-Гимзе. Величины представляют среднее значение ± SEM для трех мышей на группу и являются типичными для 3 экспериментов. *Р меньше 0,05; **Р меньше 0,01; ***Р меньше 0,001.
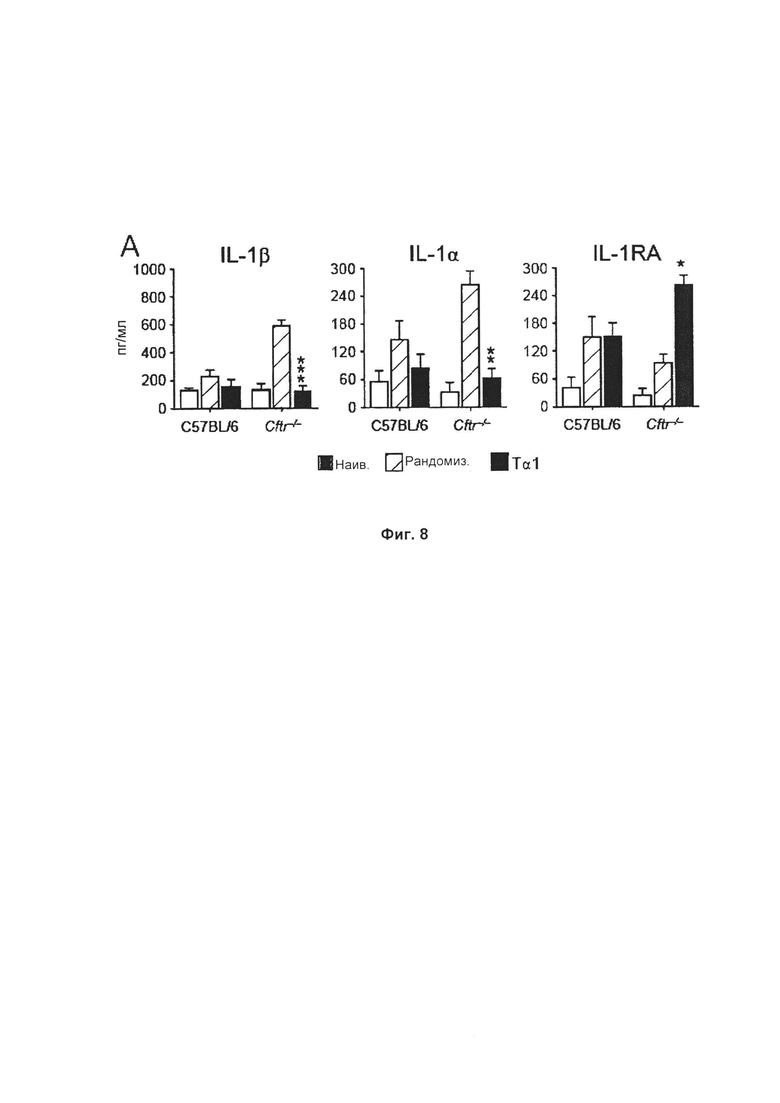
Фиг. 8 демонстрирует, что Тα1 осуществляет понижающую регуляцию воспалительных цитокинов у мышей, страдающих CF и аспергиллезом. Уровни IL-1β, 1L-1α и IL-1RA в BAL измеряли при помощи ELISA (иммуноферментный анализ) (пг/мл) на 7 сутки после инфицирования. Результаты демонстрировали типичные совокупные данные для двух экспериментов. *Р меньше 0,05; **Р меньше 0,01; ***Р меньше 0,001.
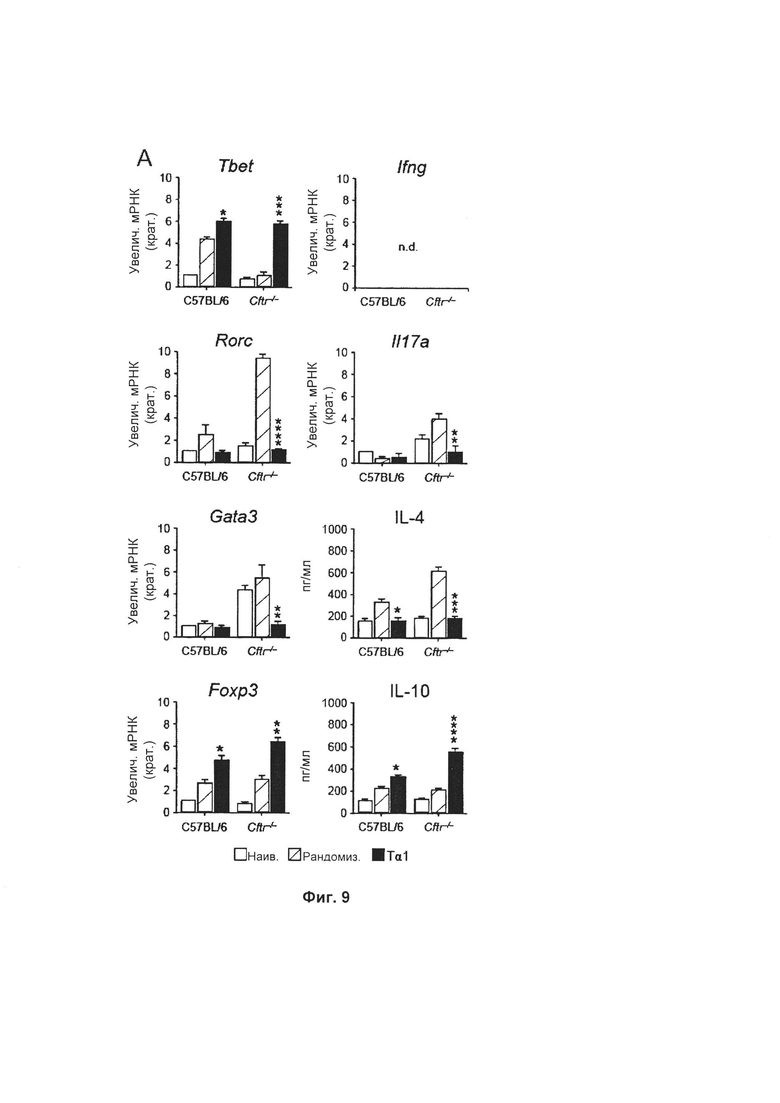
Фиг. 9 демонстрирует то, что Тα1 регулирует баланс клеток Th у мышей, страдающих CF и аспергиллезом. Тα1 уменьшал ТМ7 (уменьшенные уровни IL-17А и транскрипционного фактора Th17 Rorc), активация клеток Th2 (низкий уровень IL-4 и Gata3), в то же время способствуя активности клеток Th1 (увеличенные уровни IFN-γ и Tbet) и Treg (увеличенные уровни IL-10 и Foxp3). Эти результаты демонстрируют типичные совокупные данные двух экспериментов, n.d. не осуществляли. *Р меньше 0,05; **Р меньше 0,01; ***Р меньше 0,001.
ПРИМЕР 1: Исследования, относящиеся к действию тимозина α1 в качестве агента, корректирующего CFTR, защитного агента и противовоспалительного агента при муковисцидозе
Материалы и методы
Клетки. Клеточные линии и культура клеток - человеческие клетки бронхиального эпителия (НВЕ), гомозиготные по мутации δF508 и их изогенный дикий тип получали из легочных трансплантатов (пациенты, страдающие CF) или резекций легких (пациенты, не страдающие CF) (любезно предоставлен LJ Galietta в Italian Cystic Fibrosis Foundation (Итальянский фонд по муковисцидозу)). Клетки поддерживали при 37°С в увлажненном инкубаторе в атмосфере, содержащей 5% CO2, и эксперименты осуществляли через 5 суток после высевания (21, 22). Стабильную основанную на лентивирусе трансдукцию родительских клеток CFBE41o- (ΔF508/ΔF508), исходно иммортализированных и охарактеризованных Dr. D. Gruenert и сотрудниками (32) как WT-CFTR или ΔF508-CFTR, осуществляли в Tranzyme, Inc. (Birmingham, AL). Трансдуцированные клетки CFBE41o- поддерживали в минимальной среде Игла, дополненной 50 единиц/мл пенициллина, 50 мкг/мл стрептомицина, 2 мМ L-глутамином, 10% фетальной бычьей сывороткой и 1 мкг/мл бластицидина (WT-CFTR) или 2 мкг/мл пуромицина (ΔF508-CFTR) в инкубаторе с 5% CO2, 95% воздухом при 37°С. Родительские клетки CFBE41o- поддерживали в тех же самых культуральных условиях, но без бластицидина или пуромицина. Для получения поляризованных монослоев клетки CFBE41o- высевали на проницаемые подложки диаметром 24 мм Transwell (размер поры 0,4 мм; Corning Corp., Corning, NY) в количестве 2×106 и выращивали в виде культуры на границе воздух-жидкость при 37°С в течение 6-9 суток и затем при 27°С в течение 36 часов. Клетки инкубировали с каждым из 100 нг/мл Тα1 (CRIBI Biotechnology, Padova, смотри ниже), 3 мкМ VX-809 (Лумакафтор, Aurogene Rome, Italy), 1 мкМ VX-770 (Ивакафтор, Aurogene) или в комбинации в течение 24 часов, а затем оценивали экспрессию и функцию белка CFTR. В качестве контроля использовали сам разбавитель DMSO (диметилсульфоксид) (0,1%, об./об.) в течение 24 ч.
Мыши. Инбредных мышей дикого типа (WT) C57BL6 в возрасте от 8 до 12 недель приобретали в Charles River Breeding Laboratories (Calco, Italy). Генетически сконструированных гомозиготных мышей Cftr-/- (33) выращивали в виварии группы по исследованию муковисцидоза в San Raffaele Hospital, Milan, Italy. Эксперименты осуществляли в соответствии с протоколами, одобренными ведомственной комиссией по животным и в соответствии с директивой Европейского Экономического Сообщества, а также ведомственными руководствами по уходу за животными и их использованию.
Инфицирование и обработки. Мышей анестезировали путем i.p. (и.п.) (интраперитонеальной) инъекции 2,5% авертина (Sigma Chemical Со, St. Louis, МО) до интраназального закапывания 2×107 A. fumigatus (Af293) покоящихся конидий/20 мкл физиологического раствора. Для гистологических исследований заключенную в парафин ткань окрашивали Шифф-йодной кислотой (PAS), и сбор жидкости BAL осуществляли в соответствии с описанным (21, 22). Обработки были следующими: Тα1 и рандомизированный полипептид поставлялись в виде очищенного (уровни эндотоксинов меньше 0,03 пг/мл, в соответствии со стандартным анализом лизата амебоцитов мечехвоста), стерильного, лиофилизированного, ацетилированного полипептида. Последовательности были следующими: Ac-Ser-Asp-Ala-Ala-Val-Asp-Thr-Ser-Ser-Glu-Ile-Thr-Thr-Lys-Asp-Leu-Lys-Glu-Lys-Lys-Glu-Val-Val-Glu-Glu-Ala-Glu-Asn-О (Тα1) (SEQ ID NO: 1) и Ac-Ala-Lys-Ser-Asp-Val-Lys-Ala-Glu-Thr-Ser-Ser-Glu-Ile-Asp-Thr-Thr-Glu-Leu-Asp-Glu-Lys-Val-Glu-Val-Lys-Ala-Asn-Glu-OH (рандомизированный пептид) (SEQ ID NO: 2). Лиофилизированные порошки разбавляли в стерильной воде и 200 мкг/кг/и.п. давали ежесуточно в течение 6 последовательных суток, начиная со дня инфицирования.
Проточная цитометрия. Окрашивание на экспрессию клеточного антигена осуществляли в соответствии с описанным (21, 22). Клетки анализировали при помощи проточного цитофлуориметра FACScan (Becton Dickinson, Mountain View, CA), оборудованного программным обеспечением CELLQuestTM. Перед мечением осуществляли блокирование FcR. Контрольное окрашивание клеток нерелевантными антителами используют для получения величин фоновой флуоресценции. Данные выражены в виде процентной доли положительных клеток относительно всех проанализированных клеток.
Анализ путем иммуноблотинга CFTR. Способы иммуноблотинга с использованием антитела против CFTR (клон CF3, Abcam) использовали для измерения созревания CFTR в клетках FRT, HEK-293 или НВЕ, экспрессирующих CFTR или F508del-CFTR (34). После инкубации клетки собирали в охлажденный на льду раствор D-PBS (забуференный фосфатом физиологический раствор Дульбекко) (без кальция и магния) и осаждали при 1000×g и 4°С. Клеточные осадки лизировали в 1% Nonidet Р-40, 0,5% дезоксихолата натрия, 200 мМ NaCl, 10 мМ Tris, рН 7,8 и 1 мМ EDTA плюс смесь протеазных ингибиторов (1:250; Roche) в течение 30 мин на льду. Лизаты центрифугировали в течение 10 мин при 10000×g при 4°С для осаждения ядер и нерастворимого материала. Приблизительно 12 мкг общего белка нагревали в буфере Лэмли с 5% β-меркаптоэтанолом при 37°С в течение 5 мин и наносили на 3%-8% Tris-ацетатный гель (Invitrogen). Гель переносили на нитроцеллюлозу и обрабатывали для вестерн-блотинга путем использования моноклонального антитела против CFTR или поликлонального против α-актина (Santa Cruz Biotechnology). Блоты визуализировали при помощи усиленной хемилюминесценции. LiteAblotPlus хемилюминисцентный субстрат (Euroclone S.p.A.), с использованием системы визуализации ChemiDocTM XRS+ (Bio-Rad Laboratories) и количественную оценку для блотинга осуществляли при помощи денситометрического анализа изображения с использованием программного обеспечения Image Lab 3.1.1 (Bio-Rad).
Активация CFTR. Поскольку канал CFTR является проницаемым для йода, то можно определить выход этого иона из ранее нагруженных клеток при помощи калориметрического анализа с использованием флуоресцентного зонда SPQ (6-метокси-N-(3-сульфопропил)хинолиний) (35).
Полимеразная цепная реакция с обратной транскрипцией (RT-PCR). Экстракцию общей РНК и синтез и PCR кДНК осуществляли на общих клетках легкого в соответствии с описанным (21, 22). Эффективность амплификации валидировали и нормализовали против Gapdh. Температурный профиль PCR в реальном времени с SYBR Green представлял собой 95°С в течение 3 мин, затем 40 циклов денатурации в течение 30 с при 95°С и стадия отжига/удлинения в течение 30 с при 60°С. Каждое значение данных проверяли в отношении надежности при помощи анализа графика амплификации. Нормализованные данные по мРНК выражали в виде отношения мРНК в обработанных клетках по сравнению с нестимулированными клетками.
Определение цитокинов при помощи ELISA (иммуноферментный анализ). Уровни цитокинов в легочных гомогенатах обработанных и необработанных мышей определяли при помощи цитокинспецифического ELISA (R&D Systems, Inc. Space Import-Export srl, Milan, Italy) в соответствии с описанным (21, 22)
Статистические анализы. Парный t-тест Стьюдента использовали для определения значимости величин в экспериментальных образцах (значимость определяли как Р меньше 0,05). Данные по выживанию анализировали с использованием U-теста Манна-Уитни. Группы in vivo состояли из 6 животных. Если не указано иное, то данные представляют собой среднее значение ± SE. Данные анализировали при помощи программы GraphPad Prism 4.03 (GraphPad Software).
Результаты
Tα1 действует в качестве корректирующего агента путем увеличения экспрессии ΔF508-CFTR на поверхности клеток CF
Использовали трансформированную SV40 клеточную линию эпителия дыхательных путей пациентов, страдающих CF (CFBE41o-), гомозиготную по мутации ΔF508-CFTR (32). Клетки CFBE41o-, стабильно экспрессирующие ΔF508 CFTR или WT CFTR, обрабатывали 100 нг/мл Тα1 в течение от 30 мин до 24 часов. Фиг. 2 демонстрирует типичный иммуноблотинг общего клеточного белка из контрольных клеток (НВЕ WT) и клеток ΔF508 (НВЕ ΔF508), обработанных Тα1 (панель А, Фиг. 2). Способы иммуноблотинга использовали для измерения выхода ΔF508-CFTR из эндоплазматического ретикулума и прохождения через аппарат Гольджи, который характеризуется увеличением молекулярной массы CFTR (от полосы 135-140 кДа до полосы 170-180 кДа) в результате гликозилирования. После процессинга CFTR в аппарате Гольджи зрелая подвергнутая комплексному гликозилированию форма CFTR доставляется на клеточную поверхность. Тα1 устойчиво повышал экспрессию в клетках ΔF508 зрелого CFTR (обозначенного как С) уже через 30 мин после воздействия и вплоть до 24 часов, в противоположность WT CFTR. Полосы CFTR количественно определяли при помощи денситометрии и выражали как отношение С/В. На Фиг. 2 стрелки указывают на положения С (зрелой) и В (незрелой) форм CFTR на основе относительной подвижности. Для подтверждения того, что увеличенная экспрессия ΔF508 CFTR коррелирует с увеличением его миграции и стабильности на плазматической мембране (РМ), белки РМ очищали из цитозольных компонентов клеток, обработанных Тα1, как описано выше. Фракции подвергали иммуноблоттингу с антителом против CFTR, и FLOT1 (клон С-2 Santa Cruz Biotechnology) использовали для подтверждения специфической локализации белка на клеточной поверхности. Эти результаты демонстрируют, что Тα1 увеличивал экспрессию ΔF508 CFTR в РМ (панель В, Фиг. 2). Денситометрическое измерение CFTR (полоса С) в РМ выражено как отношение CFTR РМ/общий CFTR (панель С, Фиг. 2). Для оценки того, увеличивает ли Тα1 также экспрессию CFTR у пациентов, страдающих CF, бронхоальвеолярные клетки (НВЕ, человеческий BE) от 5 пациентов, страдающих CF, обрабатывали 100 нг/мл Тα1 в течение 24 часов, и оценивали экспрессию белка CFTR. Эти результаты демонстрируют, что Тα1 увеличивал экспрессию CFTR (полоса С) у 3 (пациент №1, 2 и 5) из 5 пациентов (панель D, Фиг. 2). Эти результаты свидетельствуют о том, что Тα1 увеличивает созревание ΔF508 CFTR, приводящее в результате к увеличению плотности и стабильности клеточной поверхности, и качеству Тα1 в качестве корректирующего агента.
Тα1 действует в качестве усиливающего агента путем увеличения функциональной активности ΔF508-CFTR в клетках CF
Предполагается, что агенты восстанавливают активность мутантного CFTR в качестве сАМР-зависимого хлоридного канала на клеточной поверхности. Предполагается, что восстановление даже менее чем 30% функции CFTR in vivo (от 5 до 30%) подтверждает по меньшей мере частичную клиническую пользу для пациентов, страдающих CF, за счет улучшения функции легкого (36). Для оценки активности Тα1 в отношении канала Cl- клетки CFBE41o- и клетки НВЕ пациентов, страдающих CF, обрабатывали 100 нг/мл Тα1 в течение 24 часов и оценивали хлоридный транспорт путем использования галогенид-чувствительных флуоресцентных зондов (6-метокси-N-(3-сульфопропил)хинолиний (SPQ) при стимулировании форсколином для активации CFTR через путь сАМР/РКА (35). Результаты демонстрируют, что Тα1 увеличивал хлоридную проницаемость ΔF508 CFTR до приблизительно 70% относительно контроля (рассматривая контроль WT как 100% референсное значение) в клетках CFBE 41о- (панель А, Фиг. 3) и клетках НВЕ пациентов, страдающих CF (панель В, Фиг. 3). Таким образом, Тα1 значительно увеличивал опосредованную CFTR хлоридную проницаемость вместе с увеличенными уровнями ассоциированного с мембраной CFTR.
Тα1 индуцирует экспрессию альтернативного ионного канала ТМЕМ16А.
Фармакологическое корректирование дефектного ионного транспорта путем нацеливания на мутантный CFTR или альтернативные ионные каналы, которые могут компенсировать дисфункцию CFTR, длительное время рассматривалось как привлекательный подход для этиотропной терапии CF (37). ТМЕМ16А, представляющий собой активируемый Са2+ канал Cl-, ассоциирован с кальций-зависимым хлоридным потоком (38) и, хотя и отличается от CFTR, демонстрирует функциональное и молекулярное взаимодействие с CFTR (39). Активация ТМЕМ16А фармакологическими агентами может обойти первичный дефект в CF независимо от генотипа CFTR (40). Микроматричный анализ ДНК указывает на то, что экспрессия ТМЕМ16А значительно увеличивается у мышей, страдающих легочным аспергиллезом, после лечения Тα1 (неопубликованные результаты). На основе этих открытий оценивают, индуцируется ли экспрессия ТМЕМ16А Тα1 на клетках НВЕ контрольных индивидов или пациентов, страдающих CF. Для этой задачи клетки подвергают воздействию 100 нг/мл Тα1 в течение до 24 часов и определяют экспрессию мРНК ТМЕМ16А при помощи RT-PCR на общей РНК из клеток. Эти результаты демонстрируют, что Тα1 значительно увеличивал экспрессию ТМЕМ16А в контрольных клетках и клетках CF после 4 часов воздействия. Тем не менее, хотя экспрессия ТМЕМ16А возвращалась до базовых значений в контрольных клетках через 24 часа, она оставалась увеличенной в клетках CF (Фиг. 4А). Эти результаты свидетельствуют о том, что Tα1 путем индукции экспрессии альтернативного канала Cl- ТМЕМ 16А, может дополнительно улучшать активность ионного канала при CF.
Tα1, один или в комбинации с потенцирующим агентом ивакафтором, восстанавливает активность ΔF508 CFTR до той степени, которая близка к корректирующему агенту лумакафтору
Активность Tα1 самого по себе или в комбинации с ивакафтором сравнивали с активностью лумакафтора на клетках CFBE41o-, экспрессирующих ΔF508 CFTR, и на клетках НВЕ пациентов, страдающих CF. Для этой задачи клетки обрабатывали каждым из Tα1 (100 нг/мл), VX-770 (1 мкМ) или VX-809 (3 мкМ) или в комбинации в течение 24 часов и определяли экспрессию и функцию белка CFTR. Эти результаты демонстрируют то, что так же как лумакафтор Тα1 увеличивал экспрессию зрелой формы (обозначаемой как С) по сравнению с незрелой формой (обозначаемой как В) CFTR в клетках, трансфицированных ΔF508 (А, типичный иммуноблотинг общего клеточного белка (Фиг. 5). Полосы CFTR количественно определяли при помощи денситометрии и выражали как отношение С/В). Стрелки на Фиг. 5 указывают на положении форм В и С CFTR на основе относительной подвижности). Ивакафтор не увеличивал экспрессию CFTR, а комбинация Tα1/ивакафтор увеличивала. С точки зрения активности ионного канала Тα1 сам по себе или в комбинации с ивакафтором увеличивал хлоридную проницаемость клеток ΔF508 CFTR до 60-70% относительно контроля (рассматривая контроль WT как 100% референсное значение) (В, определяемое при помощи анализа флуоресценции после стимулирования фосфорколином, Fsk). По сравнению с лумакафтором активность Tα1 была ниже в клетках CFBE41o-, экспрессирующих ΔF508 CFTR (В), но была близка в клетках НВЕ пациентов, страдающих CF (С). Будучи комбинированным с лумакафтором Tα1 не увеличивал активность лумакафтора (В и С). Эти результаты указывают на то, что способность Tα1 увеличивать экспрессию CFTR сравнима с таковой у лумакафтора, и что его можно было бы использовать для комбинированной терапии с ивакафтором.
Мыши, страдающие CF, подвержены воспалительной патологии при инфекции
Для определения терапевтической активности Tα1 при CF оценивали подверженность мышей C57BL/6 и Cftr-/- воспалительной патологии, ассоциированной с инфицированием Aspergillus fumigatus, представляющим собой известный инвазивный микроорганизм в дыхательных путях пациентов, страдающих CF (41). Мышей C57BL/6 и Cftr-/- интраназально инфицировали живыми конидиями A. fumigatus и определяли параметры воспаления. Длительный и устойчивый воспалительный ответ, характеризующийся рекрутингом нейтрофилов (PMN) и эозинофилов (EOS) в BAL (панель А, Фиг. 6) и легком (панели В и С, Фиг. 6), обнаружен у мышей Cftr-/-, у которых степень воспаления уже обнаруживалась до инфицирования (С, гистология легкого (окрашивание Шифф-йодной кислотой и во вставке окрашивание по Гомори) в различные dpi). Дифференцированный подсчет моноядерных клеток (MNC), клеток PMN и EOS осуществляли при помощи окрашивания по Май-Грюнвальду-Гимзе в различные сутки после инфицирования (dpi). Величины представляют среднее значение ± SEM для трех мышей на группу и представляют собой типичные величины для 3 экспериментов. Фотографии получены при помощи микроскопии высокого разрешения на Olympus DP71 с использованием объектива ах20. Масштабная линейка 200 мкм. (С) Количество GM+положительных клеток оценивали путем проточной цитометрии на общих клетках легких в различные dpi. *Р меньше 0,05.
Тα1 защищает мышей, страдающих CF, от воспалительной патологии
Мыши, страдающие CF, подвержены воспалительному ответу, ассоциирующемуся с инфицированием Aspergillus, и аллергией, и таким образом представляют собой подходящую модель для оценки действий Tα1.
Мышей, инфицированных Aspergillus, ежесуточно обрабатывали 200 мкг/кг/и.п. Tα1 в течение 6 последовательных суток, начиная со дня инфицирования. У мышей контролировали воспалительную патологию легкого и клеточный рекрутинг на 7 сутки после инфицирования. Tα1, но не рандомизированный пептид, значительно уменьшал локальный рекрутинг воспалительных клеток и легочную патологию у мышей C57/BL6 и даже мышей Cftr-/-, о чем свидетельствует уменьшение рекрутинга воспалительных клеток (в основном PMN, смотри вставки) в легком (панель А, Фиг. 7), а также в BAL (панель В, Фиг. 7). Эти данные указывают на то, что Tα1 является эффективным для ограничения рекрутинга воспалительных клеток в легком мышей, страдающих CF, во время инфекции. (А) Гистология легкого (окрашивание Шифф-йодной кислотой и на вставке клеточный рекрутинг). Фотографии получены при помощи микроскопии высокого разрешения на Olympus DP71 с использованием объектива а х 20. Масштабная линейка 200 мкм. (С) Количество Gr1+положительных клеток оценивали путем проточной цитометрии на общих клетках легких в различные dpi. *Р меньше 0,05. (В) морфометрия BAL на 7 сутки после инфицирования (dpi). Дифференцированный подсчет моноядерных клеток (MNC), полиморфоядерных клеток (PMN) и эозинофилов (EOS) осуществляли при помощи окрашивания по Май-Грюнвальду-Гимзе. Величины представляют среднее значение ± SEM для трех мышей на группу и представляют собой типичные величины для 3 экспериментов. *Р меньше 0,05; **Р меньше 0,01; ***Р меньше 0,001.
Тα1 оказывает понижающую регуляцию в отношении воспалительных цитокинов у мышей CF. страдающих аспергиллезом.
Мышей C57BL/6 и Cftr-/- интраназально инфицировали живыми конидиями A. fumigatus и ежесуточно обрабатывали 200 мкг/кг/и.п. Тα1 в течение 6 последовательных суток, начиная со дня инфицирования. Результаты представлены на Фиг. 8. Они демонстрируют, что повышенный и неспецифический воспалительный ответ у мышей Cftr-/- ассоциирован с более высокими уровнями IL-1β и IL-1α по сравнению с C57/BL6 в BAL во время инфекции. После обработки Tα1 уровни IL-1β и IL-1α резко уменьшались у любого типа мышей, и самое интересное в том, что уровни блокирующего воспаление цитокина (IL-1RA) увеличивались. Принимая во внимание то, что высокие уровни IL-1β и IL-1α обнаружены у пациентов, страдающих CF (42), эти данные свидетельствуют о том, что Тα1 может вносить вклад в локальный баланс воспалительных/противовоспалительных цитокинов у пациентов, страдающих CF. Уровни IL-1β, IL-1α и IL-1RA в BAL измеряли при помощи ELISA (пг/мл) через 7 суток после инфицирования. Приведенные результаты представляют совокупные данные двух экспериментов. *Р меньше 0,05; **Р меньше 0,01; ***Р меньше 0,001.
Тα1 регулирует баланс клеток Th у мышей, страдающих CF и аспергиллезом
Воспалительный путь Th17 вовлечен в воспаление легкого при CF (21, 22, 43), тогда как путь Th2 ассоциирован с аллергией на грибы при CF (44). Для определения действий Тα1 на активацию клеток Th уровни цитокинов Th в легких и соответствующих транскрипционных факторов в дренирующих торакальных лимфатических узлах измеряли у мышей C57BL/6 и Cftr-/-, инфицированных и обработанных Тα1, как указано выше. Тα1 уменьшал активацию клеток Th17 (уменьшенные уровни IL-17A и транскрипционного фактора Th17 Rorc), Th2 (низкие IL-4 и Gata3), в то же самое время способствуя активности клеток Th1 (увеличенные IFN-γ и Tbet) и Treg (увеличенные IL-10 и Foxp3). Эти результаты свидетельствуют о том, что Тα1 представляет собой сильный активатор противовоспалительной оси Th1/Treg при CF, в то же самое время ограничивающий активацию воспалительных клеток Th17/Th2 (Фиг. 9). Уровни цитокинов определяли в легочных гомогенатах (при помощи ELISA или RT-PCR), и экспрессию соответствующих транскрипционных факторов Th (при помощи RT-PCR) в дренирующих торакальных лимфатических узлах. Приведенные результаты демонстрируют совокупные данные двух экспериментов, n.d. не осуществляли. *Р меньше 0,05;**Р меньше 0,01;***Р меньше 0,001.
Ссылки
1.  BP, Freedman SD. Cystic fibrosis. Lancet 2009; 373: 1891-1904.
BP, Freedman SD. Cystic fibrosis. Lancet 2009; 373: 1891-1904.
2. Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251-1254.
3. Guggino WB, Stanton BA. New insights into cystic fibrosis: molecular switches that regulate CFTR. Nature Rev. Mol. Cell. Biol. 2006;7:426-436.
4. Cystic Fibrosis Foundation. Cystic Fibrosis Foundation Patient Registry Annual Data Report 2011. Cystic Fibrosis Foundation; 2012.
5. The Cystic Fibrosis Genotype-Phenotype Consortium Correlation between genotype and phenotype in patients with cystic fibrosis. N. Engl. J. Med. 1993;329:1308-1313.
6. Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application Nat Rev Genet. 2015; 16: 45-56.
7. Mall MA, Galietta LJ. Targeting ion channels in cystic fibrosis. J Cyst Fibros. 2015. pii: S1569-1993(15)00150-2.
8. Pettit RS, Fellner C. CFTR modultators for the treatment of Cystic fibrosis. P&T 2014;39:500-511.
9. Yang H, Ma T. F508del-cystic fibrosis transmembrane regulator correctors for treatment of cystic fibrosis: a patent review. Expert Opin. Ther. Patents 2015.
10. Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC,  P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, Rowe SM, Dong Q, Rodriguez S, Yen K,
P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, Rowe SM, Dong Q, Rodriguez S, Yen K,  C, Elborn JS.
C, Elborn JS.
11. Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, Colombo C, Davies JC, De Boeck K, Flume PA, Konstan MW, McColley SA, McCoy K, McKone EF, Munck A, Ratjen F, Rowe SM, Waltz D, Boyle MP; TRAFFIC Study Group; TRANSPORT Study Group. Lumacaftor-lvacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373:220-31.
12. Cholon DM, Quinney NL, Fulcher ML, Esther CR Jr, Das J, Dokholyan NV, Randell SH, Boucher RC, Gentzsch M. Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Sci Transl Med. 2014;6:246ra96.
13. Corvol H, Thompson KE, Tabary O, le Rouzic P, Guillot L. Translating the genetics of cystic fibrosis to personalized medicine. Transl Res. 2015 Apr 15. pii: S1931-5244 (15)00131-0.
14. Belcher CN, Vij N. Protein processing and inflammatory signaling in Cystic Fibrosis: challenges and therapeutic strategies. Curr Mol Med. 2010;10:82-94.
15. Cantin AM, Hartl D, Konstan MW, Chmiel JF. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. J Cyst Fibros. 2015 Jul; 14:419-30).
16. Hartl D, Gaggar A, Bruscia E, et al. Innate immunity in cystic fibrosis lung disease. J Cyst Fibros 2012; 11: 363-382.
17. Mizgerd JP, Lupa MM, Kogan MS, et al. Nuclear factor-kappaB p50 limits inflammation and prevents lung injury during Escherichia coli pneumonia. Am J Respir Crit Care Med 2003; 168: 810-817.
18. Cohen TS, Prince A. Cystic fibrosis: a mucosal immunodeficiency syndrome. Nat Med 2012; 18: 509-519.
19. Hoffman LR, Ramsey BW. Cystic fibrosis therapeutics: the road ahead. Chest 2013; 143:207-213.
20. Ramsey B, Banks-Schlegel S, Accurso F, et al. Future Directions in Early Cystic Fibrosis Lung Disease Research. Am J Respir Crit Care Med 2012; 185: 887-892.
21. lannitti RG, Carvalho A, Cunha C, et al. Th17/Treg imbalance in murine cystic fibrosis is linked to indoleamine 2,3-dioxygenase deficiency but corrected by kynurenines. Am J Respir Crit Care Med 2013; 187: 609-620.
22. lannitti RG, Casagrande A, De Luca A, et al. Hypoxia promotes danger-mediated inflammation via receptor for advanced glycation end products in cystic fibrosis. Am J Respir Crit Care Med 2013; 188: 1338-1350.
23. Lands LC, Stanojevic S. Oral non-steroidal anti-inflammatory drug therapy for lung disease in cystic fibrosis. Cochrane Database Syst Rev. 2013 Jun 13;6:CD001505.
24. Conrad C, Lymp J, Thompson V, Dunn C, Davies Z, Chatfield B, Nichols D, Clancy J, Vender R, Egan ME, Quittell L, Michelson P, Antony V, Spahr J, Rubenstein RC, Moss RB, Herzenberg LA, Goss CH, Tirouvanziam R. Long-term treatment with oral N-acetylcysteine: affects lung function but not sputum inflammation in cystic fibrosis subjects. A phase II randomized placebo-controlled trial. J Cyst Fibros. 2015;14:219-27.
25. P.J. Mogayzel Jr., E.T. Naureckas, K.A. Robinson, G. Mueller, D. Hadjiliadis, J.B. Hoag, et al. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health Am J Respir Crit Care Med. 2013 187. 680-689.
26. De Benedictis FM, Bush A. Corticosteroids in respiratory diseases in children. Am J Respir Crit Care Med. 2012;185:12-23.
27. Goldstein AL, Guha A, Zatz MM, et al. Purification and biological activity of thymosin, a hormone of the thymus gland. Proc Natl Acad Sci U S A 1972; 69: 1800-1803.
28. Garaci E., Pica F., Rasi G., et al Combination therapy with BRM in cancer and infections disease. Mech. Ageing Dev, 1997,96,103-116.
29. Tuthill C, Rios I, McBeath R. Thymosin alpha 1: past clinical experience and future promise. Ann N Y Acad Sci 2010; 1194: 130-135.
30. Romani L, Bistoni F, Perruccio K, et al. Thymosin alpha1 activates dendritic cell tryptophan catabolism and establishes a regulatory environment for balance of inflammation and tolerance. Blood 2006; 108: 2265-2274.
31. Perruccio K, Bonifazi P, Topini F, et al. Thymosin alpha1 to harness immunity to pathogens after haploidentical hematopoietic transplantation. Ann N Y Acad Sci 2010; 1194: 153-161.
32. Bruscia E, Sangiuolo F, Sinibaldi P, Goncz KK, Novelli G, Gruenert DC. Isolation of CF cell lines corrected at DeltaF508-CFTR locus by SFHR-mediated targeting. Gene Ther. 2002;9:683-5.
33. Zhou L, Dey CR, Wert SE, et al. Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR. Science 1994; 266: 1705-1708.
34. Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu PA. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A. 2011;108:18843-8.
35. Munkonge F1, Alton EW, Andersson C, Davidson H, Dragomir A, Edelman A, Farley R, Hjelte L, McLachlan G, Stern M, Roomans GM. Measurement of halide efflux from cultured and primary airway epithelial cells using fluorescence indicators. J Cyst Fibros. 2004;3 Suppl 2:171-6.
36. Ramalho AS1, Beck S, Meyer M, Penque D, Cutting GR, Amaral MD. Five percent of normal cystic fibrosis transmembrane conductance regulator mRNA ameliorates the severity of pulmonary disease in cystic fibrosis. Am J Respir Cell Mol Biol. 2002;27:619-27).
37. Mall MA, Galietta LJ. Targeting ion channels in cystic fibrosis. J Cyst Fibros. 2015 23. pii: S1569-1993(15)00150-2.
38. Caputo A1, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590-4.
39. Ousingsawat J, Kongsuphol P, Schreiber R, Kunzelmann K. CFTR and TMEM16A are separate but functionally related Cl-channels. Cell Physiol Biochem. 2011;28(4):715-24).
40. Sondo E, Caci E, Galietta LJ. The TMEM16A chloride channel as an alternative therapeutic target in cystic fibrosis. Int J Biochem Cell Biol. 2014;52:73-6.
41. Felton IC, Simmonds NJ. Aspergillus and cystic fibrosis: old disease -new classifications. Curr Opin Pulm Med 2014; 20: 632-638.
42. Tang A, Sharma A, Jen R, et al. Inflammasome-mediated IL-1beta production in humans with cystic fibrosis. PLoS One 2012; 7: e37689.
43. Dubin PJ, McAllister F, Kolls JK. Is cystic fibrosis a TH17 disease? Inflamm Res 2007; 56: 221-227.
44. Kreindler JL, Steele C, Nguyen N, et al. Vitamin D3 attenuates Th2 responses to Aspergillus fumigatus mounted by CD4+ T cells from cystic fibrosis patients with allergic bronchopulmonary aspergillosis. J Clin Invest 2010; 120: 3242-3254.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Romani, Luigina
Garaci, Enrico
<120> Тимозин альфа 1 для применения в лечении муковисцидоза
<130> PCT30251
<150> RM2015A000056
<151> 2015-02-09
<150> 102015000053089
<151> 2015-09-18
<160> 2
<170> PatentIn версия 3.5
<210> 1
<211> 28
<212> ПРТ
<213> Homo sapiens
<400> 1
Ser Asp Ala Ala Val Asp Thr Ser Ser Glu Ile Thr Thr Lys Asp Leu
1 5 10 15
Lys Glu Lys Lys Glu Val Val Glu Glu Ala Glu Asn
20 25
<210> 2
<211> 28
<212> ПРТ
<213> Искусственная
<220>
<223> рандомизированный пептид в соответствии с SEQ ID NO:1
<400> 2
Ala Lys Ser Asp Val Lys Ala Glu Thr Ser Ser Glu Ile Asp Thr Thr
1 5 10 15
Glu Leu Asp Glu Lys Val Glu Val Lys Ala Asn Glu
20 25
<---

Настоящая группа изобретений относится к медицине, а именно к терапии и пульмонологии, и касается лечения муковисцидоза. Для этого вводят композицию, содержащую эффективное количество тимозина альфа-1 (Тα1), в качестве монотерапии или в комбинации с другими противовоспалительыми средствами. Это обеспечивает эффективное лечение муковисцидоза за счет противовоспалительной активности Тα1 и его способности увеличивать опосредованный CFTR хлоридный транспорт в легких. 7 н. и 11 з.п. ф-лы, 9 ил., 1 пр.
1. Способ лечения муковисцидоза у пациента, нуждающегося в этом,включающий
введение указанному пациенту фармацевтической композиции,содержащей или состоящей из тимозина альфа-1.
2. Способ увеличения экспрессии мутантного регулятора трансмембранной проводимости при муковисцидозе (CFTR) на поверхности клеток у пациента с муковисцидозом, включающий
введение указанному пациенту фармацевтической композиции, содержащей или состоящей из тимозина альфа-1 в качестве корректирующего агента.
3. Способ увеличения функциональной активности мутантного регулятора трансмембранной проводимости при муковисцидозе (CFTR) у пациента с муковисцидозом, включающий
введение указанному пациенту фармацевтической композиции, содержащей или состоящей из тимозина альфа-1 в качестве усиливающего агента.
4. Способ увеличения экспрессии альтернативного ионного канала ТМЕМ16А у пациента с муковисцидозом, включающий
введение указанному пациенту фармацевтической композиции, содержащей или состоящей из тимозина альфа-1.
5. Способ восстановления активности мутантного регулятора трансмембранной проводимости при муковисцидозе (CFTR) у пациента с муковисцидозом, включающий
введение указанному пациенту фармацевтической композиции, содержащей или состоящей из тимозина альфа-1.
6. Способ понижающей регуляции воспалительных цитокинов или регуляции баланса клеток Th у пациента с муковисцидозом, включающий
введение указанному пациенту фармацевтической композиции, содержащей или состоящей из тимозина альфа-1.
7. Способ по любому из пп. 1-6, включающий
введение указанному пациенту комбинации тимозина альфа-1 с по меньшей мере одним агентом, выбранным из группы, состоящей из
антибиотика, противогрибкового агента, агента, усиливающего CFTR, где указанный агент, усиливающий CFTR, не является тимозином альфа-1.
8. Способ по п. 7, где указанный по меньшей мере один антибиотик выбран из группы, состоящей из тобрамицина, ципрофлоксацина и колистина.
9. Способ по п. 7, где указанный по меньшей мере один противогрибковый аген выбран из группы, состоящей из итраконазола и амфотерицина B.
10. Способ по п. 7, где указанный по меньшей мере один агент, усиливающий CFTR, который не является тимозином альфа-1, выбран из группы, состоящей из ивафактора и лумакафтора.
11. Способ по п. 7, где тимозин альфа-1 и по меньшей мере один агент, выбранный из группы, состоящей из антибиотика, противогрибкового агента, агента, усиливающего CFTR, вводят пациенту раздельно или последовательно.
12. Способ по любому из пп. 1-10, где указанная фармацевтическая композиция содержит
один или более чем один эксципиент и/или вспомогательное вещество.
13. Способ лечения и/или предупреждения хронического воспаления у пациента,
страдающего муковисцидозом, включающий
введение указанному пациенту фармацевтической композиции, содержащей или состоящей из тимозина альфа-1.
14. Способ по п. 13, включающий введение указанному пациенту комбинации тимозина альфа-1 с по меньшей мере одним агентом, выбранным из группы, состоящей из антибиотика, противогрибкового агента, агента, усиливающего CFTR, где указанный агент, усиливающий CFTR, не является тимозином альфа-1.
15. Способ по п. 13, где антибиотик выбран из группы, состоящей из тобрамицина, ципрофлоксацина и колистина.
16. Способ по п. 13, где противогрибковый агент выбран из группы, состоящей из итраконазола и амфотерицина B.
17. Способ по п. 13, где агент, усиливающий CFTR, который не является тимозином альфа-1, выбран из группы, состоящей из
ивафактора, лумакафтора.
18. Способ по п. 14, где тимозин альфа-1 и по меньшей мере один агент, выбранный из группы, состоящей из антибиотика, противогрибкового агента, агента, усиливающего CFTR, вводят пациенту раздельно или последовательно.
| WO 2004087067 A2, 14.10.2004 | |||
| WO 2006101740 A2, 28.09.2006 | |||
| ОЖЕГОВ А.М | |||
| и др | |||
| "Применение иммуномодуляторов у детей с муковисцедозом" | |||
| Лечащий врач | |||
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| GOLDSTEIN AL | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Expert Opin | |||

