Настоящее изобретение относится к применению модуляторов CFTR (регулятор трансмембранной проводимости при муковисцидозе) для предотвращения и/или лечения цереброваскулярных состояний и к соответствующим способам лечения.
Среди пожилого населения число людей, живущих с заболеваниями сердца, быстро растет. Основными причинами являются более высокий уровень заболеваемости среди пожилых людей и достижения в медицине, позволившие значительно улучшить уровень выживаемости. За последнее десятилетие стало очевидно, что увеличение продолжительности жизни связано со значительным снижением когнитивных функций (1) и подпитывает начинающуюся "тихую эпидемию" когнитивных нарушений (2). Воздействие будет значительным: помимо прямых медицинских потребностей, когнитивные нарушения серьезно влияют на способность пациентов справляться со своим основным заболеванием (например, снижение соблюдения режима лечения) и имеют значительные социальные последствия для семей, друзей и общества в целом. Таким образом, истинное бремя этой начинающейся эпидемии невозможно количественно измерить простыми экономическими показателями. Несомненно то, что наши сети медицинского и психического здоровья структурно или финансово не оснащены для того, чтобы справиться с надвигающимся кризисом.
Снижение когнитивных функций у пациентов с сердечной недостаточностью (СН) обычно связывают с "сосудистым когнитивным нарушением", термином, описывающим множественные формы когнитивных нарушений (например, потеря памяти, потеря исполнительной функции, спутанность сознания), которые возникают по причине сосудистых нарушений. В качестве обобщающего термина этиология сосудистых когнитивных нарушений разнообразна и многофакторна; клинический диагноз сложен и неточен; и существует несколько различных и сложных подтипов травм (1). Большинство сердечно-сосудистых и цереброваскулярных патологий (включая гипертензию, ишемический и геморрагический инсульт, заболевания сердца и диабет) считаются основными и значительными причинами снижения когнитивных функций вследствие сосудистых нарушений (1). Хотя сосудистые когнитивные нарушения охватывают множество сложных случайных механизмов, объединяющим аспектом является недостаточная перфузия головного мозга: в этом отношении недавняя работа авторов настоящего изобретения была сосредоточена на улучшении перфузии головного мозга в экспериментальных моделях СН и субарахноидального кровоизлияния (САК) (3-6).
Проблема, лежащая в основе настоящего изобретения, состоит в обеспечении нового режима для улучшения цереброваскулярных состояний.
Решение указанной выше технической проблемы обеспечено вариантами осуществления настоящего изобретения, раскрытыми в формуле изобретения, настоящем описании и прилагаемых графических материалах.
Авторами настоящего изобретения обнаружены ключевые молекулярные механизмы, снижающие перфузию головного мозга при СН и САК (4-6). Примечательно, что обе патологии изменяют цереброваскулярную ауторегуляцию системы и, следовательно, церебральный кровоток (ЦК) по одному и тому же фундаментальному механизму. В частности, СН и САК индуцируют устойчивую экспрессию фактора некроза опухоли (TNF) в стенке сосуда мозговой артерии: TNF, локализованный в клетках гладких мышц, действует по аутокринному/паракринному механизму, стимулируя выработку сфингозин-1-фосфата (S1P), что затем вызывает сужение сосудов посредством рецептора подтипа S1P2 (3, 4, 6). Авторы настоящего изобретения воспользовались этим механистическим пониманием и успешно подтвердили два способа лечения: блокирование TNF (этанерцепт) или антагонизм передачи сигналов рецептора S1P2 в обеих моделях устраняет опосредованное СН и САК усиление цереброваскулярного сужения сосудов (т.е. измеряется как усиленный миогенный тонус) (3, 4, 6), нормализует перфузию головного мозга (3, 4, 6) и, следовательно, уменьшает повреждение нейронов (5, 6). Однако функциональность этих двух способов может быть ограничена: этанерцепт несет значительный риск, связанный с подавлением иммунитета (7), и может поставить под угрозу контроль артериального давления из-за воздействия на резистивные артерии скелетных мышц (8); в то время как антагонизм рецепторам S1P2, несомненно, вызовет непредсказуемые вторичные эффекты, поскольку эти рецепторы опосредуют важные и разнообразные функции в нескольких органах (9).
При поиске альтернативных терапевтических мишеней интересным кандидатом оказался регулятор трансмембранной проводимости при муковисцидозе (CFTR). Предыдущая работа авторов настоящего изобретения продемонстрировала, что при СН и САК подавляется экспрессия белка CFTR в мозговых артериях: поскольку CFTR является важным переносчиком, участвующим в деградации S1P клеток гладких мышц (4), подавление CFTR должно повышать биодоступность S1P и его про-констриктивные действия (3, 4). Действительно, сосудистая экспрессия TNF, экспрессия CFTR и миогенная реактивность тесно коррелированы (3, 4, 6), подпитывая механистическое предположение в соответствии с настоящим изобретением о том, что TNF-зависимое увеличение миогенного тонуса мозговой артерии опосредуется сниженной экспрессией белка CFTR (4). В этом контексте CFTR становится привлекательной и логичной терапевтической мишенью для коррекции миогенной реактивности в микроциркуляции головного мозга, поскольку известно, что CFTR терапевтические средства не нарушают иммунную функцию, и доступны одобренные управлением по контролю качества пищевых продуктов и лекарственных средств США (FDA) терапевтические средства, которые увеличивают экспрессию/активность CFTR (10).
Настоящее изобретение относится к применению соединений модуляторов CFTR, т.е. согласно настоящему изобретению, можно применять один или более модуляторов CFTR для лечения и/или предотвращения цереброваскулярного состояния.
Согласно настоящему изобретению "цереброваскулярное состояние" представляет собой состояние, которое снижает, нарушает или иным образом поражает цереброваскулярную систему индивидуума, предпочтительно пациента, представляющего собой человека. В частности, при цереброваскулярном состоянии снижается или нарушается цереброваскулярная перфузия. Термин "цереброваскулярная перфузия" также известен как и является синонимом, соответственно, "церебрального кровотока", "перфузии головного мозга" и "перфузия сосудов головного мозга".
Согласно настоящему изобретению модулятор CFTR оказывает свое действие на белки CFTR, присутствующие или экспрессируемые, соответственно, в клетках гладких мышц сосудов головного мозга. При этом модулятор CFTR нормализует цереброваскулярное фосфорилирование Erk1/2 и передачу сигналов сфингозин-1-фосфата и/или цереброваскулярную миогенную реактивность, и/или цереброваскулярный кровоток, и/или апоптоз нейронов и цереброваскулярное повреждение, и/или корректирует нарушения поведения и обучения у пациентов с цереброваскулярными состояниями, предпочтительно сниженной или нарушенной, соответственно, цереброваскулярной перфузией.
Обычно пациент, имеющий цереброваскулярное состояние или болезненное состояние, соответственно, имеет первичное болезненное состояние, обычно основное заболевание, которое, в свою очередь, вызывает указанное цереброваскулярное состояние, или по меньшей мере болезненное состояние, которое связано с указанным цереброваскулярным состоянием. Согласно настоящему изобретению предпочтительные цереброваскулярные состояния лечат или предотвращают, соответственно, с помощью указанного одного или более модуляторов CFTR, где состояние(я) вызвано(вызваны) заболеванием сердца, сердечной недостаточностью (СН), субарахноидальным кровоизлиянием (САК), внезапной нейросенсорной тугоухостью (SSHL), сосудистой деменцией, гипертензией, ишемическим инсультом, геморрагическим инсультом, заболеванием сердца, диабетом и болезнью Альцгеймера.
"Модулятор CFTR" по настоящему изобретению представляет собой соединение, модулирующее функцию белка CFTR, так что он может выполнять свою основную функцию, а именно, создавать канал для хлорида (компонента соли) для прохождения через клеточную поверхность.
Несколько пригодных модуляторов CFTR в контексте настоящего изобретения известны в данной области техники и предпочтительно включают усилители CFTR, корректоры CFTR и амплификаторы CFTR. Усилители CFTR увеличивают активность каналов CFTR. Корректоры CFTR увеличивают экспрессию CFTR на клеточной поверхности. Амплификаторы CFTR увеличивают количество экспрессированного CFTR за счет увеличения количества мРНК CFTR. Более предпочтительные модуляторы CFTR по настоящему изобретению представляют собой лекарственные средства, оказывающие протеостатическое действие на CFTR, в частности, корректоры CFTR и амплификаторы CFTR, особенно предпочтительно корректоры CFTR. Корректоры CFTR также очень предпочтительны для применения в настоящем изобретении, поскольку они проявляют очень хорошую активность даже при низком содержании CFTR.
Особенно пригодные соединения корректоры CFTR для применения в настоящем изобретении представляют собой производные циклопропанкарбоксамида, такие как раскрыты в WO-A-2005/075435, WO-A-2007/021982, WO-A-2008/127399, WO-A-2009/108657 и WO-A-2009/123896. Другие предпочтительные соединения корректоры CFTR для применения в настоящем изобретении представляют собой производные аминогетероциклила, такие как производные пиримидина, предпочтительно соединения, раскрытые в WO-A-2010/068863, WO-A-2010/151747 и WO-A-2011/008931, производные аминотиазола, предпочтительно соединения, раскрытые в WO-A-2006/101740, и производные хинолина/хиназолина, предпочтительно соединения, раскрытые в WO-A-2012/166654, производные кумарина, предпочтительно соединения, раскрытые в WO-A-2014/152213, и производные триметилангелицина, предпочтительно соединения, раскрытые в WO-A-2012/171954. Дополнительные пригодные соединения корректоры CFTR, которые можно применять в настоящем изобретении, раскрыты в WO-A-2014/081821, WO-A-2014/081820 и WO-A-2010/066912. Дополнительные соединения корректоры CFTR и их композиции для применения в настоящем изобретении включают ингибиторы транглутаминазы 2 (TG2) и ингибиторы казеинкиназы 2 (CK2). Предпочтительный ингибитор TG2 для применения в настоящем изобретении представляет собой цистеамин. Предпочтительный ингибитор СК2 для применения в настоящем изобретении представляет собой эпигаллокатехин галлат (ЭГКГ). Согласно предпочтительному варианту осуществления настоящего изобретения ингибитор TG2, предпочтительно цистеамин, и ингибитор CK2, предпочтительно ЭГКГ, применяют в комбинации. Такая комбинация может быть обеспечена в виде одновременного введения, предпочтительно в одной композиции, но также предполагается одновременное введение двух ингибиторов по-отдельности, а именно ингибитора TG2 и ингибитора CK2, каждого ингибитора как такового или в качестве компонента фармацевтической композиции. В альтернативном варианте осуществления ингибитор TG2 и ингибитор CK2 также можно вводить последовательно (так же, либо как таковые, либо в составе подходящей композиции). Комбинация ингибитора TG2 и ингибитора CK2, предпочтительно цистеамина и ЭГКГ, также раскрыта в US-A-2013/0310329.
Конкретные примеры модуляторов CFTR для применения в настоящем изобретении включают, но не ограничиваются ими, C18 (VRT-534), лумакафтор (VX-809), тезакафтор (VX-661), 4,4',6-триметилангелицин, VRT-768, VRT-422, VRT-325, CFpot-532, Copo-22, 002_NB_28 (DBM228), DBM_003_8Cl (DBM308), а также любую комбинацию таких соединений. Наиболее предпочтительные модуляторы CFTR по настоящему изобретению представляют собой C18 и лумакафтор. Следует понимать, что настоящее изобретение также относится к применению фармацевтически приемлемых солей, сольватов, сложных эфиров, солей таких сложных эфиров, а также любого другого аддукта или производного, которые при введении пациенту, нуждающемуся в лечении, могут обеспечить прямо или косвенно модулятор CFTR для применения в настоящем изобретении или его метаболит, или его остаток.
Настоящее изобретение также относится к модулятору(ам) CFTR, как описано выше, для применения для лечения и/или предотвращения описанных выше состояний. Настоящее изобретение также относится к применению модулятора(ов) CFTR, как описано выше, для получения лекарственного средства для лечения и/или предотвращения описанных выше состояний.
Кроме того, настоящее изобретение относится к способу предотвращения и/или лечения описанных выше состояний путем введения по меньшей мере одного (т.е. одного или более) модулятора CFTR пациенту, нуждающемуся в таком лечении, предпочтительно пациенту, представляющему собой человека, более предпочтительно пациенту, страдающему цереброваскулярным состоянием, связанным или вызванным, соответственно, одним или более состояниями, выбранными из заболевания сердца, сердечной недостаточности (СН), субарахноидального кровотечения (САК), внезапной нейросенсорной тугоухости (SSHL), сосудистой деменции, гипертензии, ишемического инсульта, геморрагического инсульта, заболевания сердца, диабета и болезни Альцгеймера.
Согласно настоящему изобретению один или более модулятор(ов) CFTR можно применять в его/их свободной форме. В других вариантах осуществления модулятор(ы) CFTR (т.е. по меньшей мере один модулятор CFTR), применяемый в настоящем изобретении, присутствует в фармацевтической композиции, содержащей указанный по меньшей мере один модулятор CFTR, как правило, в комбинации по меньшей мере с одним фармацевтически приемлемым эксципиентом, разбавителем, носителем и/или наполнителем.
Эффективное количество модулятора CFTR, необходимое для применения в способе и применениях по настоящему изобретения, т.е. конкретный уровень эффективной дозы для любого конкретного пациента или организма, будет зависеть от множества факторов, включая расстройство, подлежащее лечению, и тяжесть расстройства; активность конкретного применяемого соединения; конкретную применяемую композицию; возраст, массу тела, общее состояние здоровья, пол и диету пациента; время введения, способ введения и скорость выведения конкретного применяемого соединения; продолжительность лечения; лекарственные средства, применяемые в комбинации или одновременно с конкретным применяемым соединением, и подобные факторы, хорошо известные в области медицины.
Предпочтительные дозы в контексте настоящего изобретения, в частности, применительно к лумакафтору и C18, составляют от приблизительно 0,1 до приблизительно 10 мг на кг массы тела (далее обозначение "мг/кг"), предпочтительно от приблизительно 1 до приблизительно 8 мг/кг, более предпочтительно от приблизительно 3 до приблизительно 5 мг/кг. Дозу можно вводить один или более раз, например, два или три раза в день. Предпочтительно дозы вводят один или два раза в день.
Способ введения модулятора CFTR не имеет особого значения, и выбранный способ зависит от определенного применяемого соединения или соединений модулятора CFTR и субъекта, нуждающегося в лечении. Предпочтительно модулятор(ы) CFTR вводят системно, например, путем перорального или внутривенного введения, особенно предпочтительно путем перорального введения. Также можно рассмотреть возможность местного применения, где особенно предпочтительный местный способ введения представляет собой инъекцию в спинномозговую жидкость.
В контексте данного документа термин "пациент" означает животное, предпочтительно млекопитающее и наиболее предпочтительно человека.
По меньшей мере один модулятор CFTR, предпочтительно присутствующий в фармацевтической композиции, как описано выше, для применения в соответствии с настоящим изобретением можно вводить с применением любого количества и любого способа введения, эффективных для лечения цереброваскулярного состояния. В соответствии с настоящим изобретением следует понимать, что термин "обработка" или "лечение" означает, что тяжесть состояния по меньшей мере не прогрессирует по сравнению с состоянием, для которого лечение не проводили, предпочтительно тяжесть состояния не прогрессирует, более предпочтительно тяжесть состояния уменьшается, еще более предпочтительно существенно уменьшается и особенно предпочтительно состояние в значительной степени излечивается. Предпочтительно тяжесть состояния в соответствии с настоящим изобретением снижается по меньшей мере на 30%, более предпочтительно по меньшей мере на 50%, еще более предпочтительно по меньшей мере на 70%, особенно предпочтительно по меньшей мере на 90% до полного излечения состояния, что представляет собой наиболее предпочтительный результат лечения в соответствии с настоящим изобретением.
В отношении цереброваскулярного состояния, представляющего собой цель применения и способа по настоящему изобретению, соответственно, степень уменьшения тяжести или степень излечения, соответственно, цереброваскулярное состояние обычно оценивают посредством неврологической оценки субъекта, получающего лечение, в соответствии с медицинскими и диагностическими способами, известными специалисту в данной области техники, предпочтительно перед лечением в соответствии с настоящим изобретением, предпочтительно через обычные интервалы после начала лечения в соответствии с настоящим изобретением. Такие оценки включают, предпочтительно, измерение перфузии головного мозга с помощью МРТ (магнитно-резонансная томография), например, как описано в приведенном ниже примере (необязательно адаптировано для обследуемого пациента, например, когда пациент представляет собой человека).
Настоящее изобретение также относится к применению модуляторов CFTR, предпочтительно таких, как раскрыто в данном документе, для предотвращения и/или лечения легочных расстройств, предпочтительно легочной гипертензии, более предпочтительно вторичных легочных расстройств, особенно вторичной легочной гипертензии, связанной или вызванной, соответственно, определенными первичными расстройствами, такими как атеросклероз, заболевания сердечно-сосудистой системы (заболевания сердечно-сосудистой системы, такие как ишемическая болезнь сердца (ИБС) (стенокардия и инфаркт миокарда (обычно известный как сердечный приступ)), инсульт, сердечная недостаточность, гипертоническая болезнь сердца, ревматическая болезнь сердца, кардиомиопатия, нарушения сердечного ритма, врожденные заболевания сердца, порок клапана сердца, кардит, аневризмы аорты, заболевание периферических артерий, тромбоэмболическое заболевание и венозный тромбоз. Настоящее изобретение также относится к способу предотвращения и/или лечения легочных расстройств, предпочтительно легочной гипертензии, более предпочтительно вторичных легочных расстройств, таких как вторичная легочная гипертензия, связанная или вызванная, соответственно, определенными первичными расстройствами, такими как атеросклероз, заболевания сердечно-сосудистой системы (заболевания сердечно-сосудистой системы, такие как ишемическая болезнь сердца (ИБС) (стенокардия и инфаркт миокарда (обычно известный как сердечный приступ)), инсульт, сердечная недостаточность, гипертоническая болезнь сердца, ревматическая болезнь сердца, кардиомиопатия, нарушения сердечного ритма, врожденные заболевания сердца, порок клапана сердца, кардит, аневризмы аорты, заболевание периферических артерий, тромбоэмболическое заболевание и венозный тромбоз, включающему стадию введения эффективного количества по меньшей мере одного модулятора CFTR, предпочтительно модуляторов CFTR, как раскрыто данном документе, пациенту, нуждающемуся в этом.
ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фигура 1. Церебральный кровоток снижен у мышей CFTRΔF508.
(A) Миогенное сужение кровеносных сосудов сильнее в задних мозговых артериях (ЗМА), полученных от мутантных по регулятору трансмембранной проводимости при муковисцидозе (CFTR) ΔF508 мышей, по сравнению с контрольными однопометными мышами дикого типа (WT). (B) Задние мозговые артерии, полученные от мышей CFTRΔF508, демонстрируют сдвиг в сторону увеличения их зависимости реакции от дозы фенилэфрина. (C) Однако, как только реакции на фенилэфрин нормализованы по базовому тонусу (тонусактивный - тонуспокой, где тонусактивный представляет собой тонус при заданной концентрации фенилэфрина, а тонуспокой представляет собой тонус непосредственно перед стимуляцией), зависимость реакции от дозы фенилэфрина у WT и CFTRΔF508 практически идентичны (двухфакторный дисперсионный анализ P=N.S.). Средние максимальные диаметры сосудов при 45 мм рт. ст. (диаметрмакс) составляют: CFTRΔF508: 186 ± 2 мкм, n = 6 от 4 мышей и WT: 169 ± 8 мкм, n = 5 от 3 мышей; (критерий Манна-Уитни P=N.S. для диаметрмакс). (D) Магнитно-резонансную томографию применяли для измерения церебрального кровотока в заранее определенных кортикальных областях переднего мозга. Показаны типичные карты перфузии переднего мозга мышей WT и CFTRΔF508. Церебральный кровоток значимо ниже у мышей CFTRΔF508 (n = 10) по сравнению с однопометниками дикого типа (n = 11); однако ни (E) объемная скорость кровотока сердца (CO) (n = 5 для обеих групп), ни (F) общее периферическое сопротивление сосудов (TRP) (n = 5 для обеих групп) не различались между двумя генотипами. (G) У мышей WT экспрессия мРНК CFTR значимо выше в задних мозговых артериях (n = 5) по сравнению с артериями скелетных мышц кремастера (мышца, поддерживающая яичко) (Cre; n = 6). (H) Миогенный тонус резистивной артерии скелетных мышц кремастера не изменяется ингибированием CFTR in vitro (100 нмоль/л CFTR(ингибитор)-172 в течение 30 минут). (I) Делеция гена CFTR (CFTR KO), однако, вызывает умеренное, но значительное ослабление миогенного тонуса. Средние максимальные диаметры сосудов при 60 мм рт. ст. (диаметрмакс) составляют: WT (панель G): 72 ± 3 мкм, n = 5 от 4 мышей; CFTR KO (панель H): 88 ± 4 мкм, n = 6 от 4 мышей; и однопометники дикого типа (панель H): 78 ± 4 мкм, n = 5 от 2 мышей. Все данные представлены в виде среднего значения ± стандартная ошибка среднего. * обозначает P < 0,05 для непарного сравнения при помощи критерия Манна-Уитни. Сокращения: CFTR - регулятор трансмембранной проводимости при муковисцидозе; Cre - кремастер; KO - нокаут; ЗМА - задняя мозговая артерия; WT - дикий тип.
Фигура 2. C18 увеличивает экспрессию и функцию белка CFTR дикого типа за счет протеостатического механизма.
(A) Мозговые артерии, полученные от наивных мышей, получавших C18 (3 мг/кг в день в течение 2 дней; n = 5), имеют более высокую экспрессию белка регулятора трансмембранной проводимости при муковисцидозе (CFTR), по сравнению с артериями, полученными от контрольных носителей (n = 5). (B) C18 не влияет на экспрессию мРНК CFTR мозговой артерии (контроль n = 6, C18 n = 5). (C) C18 (6 мкмоль/л; 24 ч) увеличивает экспрессию белка CFTR в клетках фибробластов почек детенышей хомяка, стабильно экспрессирующих человеческий CFTR (n = 12 для обеих групп); (D) C18 не влияет на экспрессию мРНК CFTR в данной системе (n = 6 для обеих групп). (E) Фактор некроза опухоли (TNF; 10 нг/мл в течение 48 часов) подавляет экспрессию белка CFTR в первичных сосудистых гладкомышечных клетках брыжеечной артерии (контроль: n = 8; TNF: n = 8); совместная инкубация TNF с C18 (6 мкмоль/л; 24 ч) после 24 ч инкубации TNF (т.е. 48 ч TNF + 24 ч C18) полностью восстанавливает экспрессию белка CFTR (n = 7). Восстановление экспрессии белка CFTR в сосудистых гладкомышечных клетках коррелирует с нормализацией поглощения ослабленного (F) FITC-меченного сфингозин-1-фосфата (измеряется как увеличение интенсивности флуоресценции при 525 нм с помощью стандартного способа анализа сортировки клеток с активацией флуоресценции; контроль: n = 7; TNF: n = 10; TNF + C18: n = 6) и (G) стимулированный форсколином отток йодида (контроль: n = 7; TNF: n = 6; TNF + C18: n = 6). Все данные представлены в виде среднего значения ± стандартная ошибка среднего. На панелях от A до D * обозначает P < 0,05 для непарного сравнения при помощи критерия Манна-Уитни; на панелях от E до G * обозначает P < 0,05 для непарных сравнений с контролем при помощи критерия Краскела-Уоллиса и апостериорного критерия Даннетта. Сокращения: Конт - контроль; CFTR - регулятор трансмембранной проводимости при муковисцидозе; TNF - фактор некроза опухоли.
Фигура 3. C18 восстанавливает перфузию головного мозга при сердечной недостаточности.
(A) Мозговые артерии, полученные от мышей с сердечной недостаточностью (СН; 6 недель после перевязки левой передней нисходящей коронарной артерии), имеют сниженную экспрессию белка регулятора трансмембранной проводимости при муковисцидозе (CFTR) (n = 5) по сравнению с артериями, полученными от ложнооперированных контрольных животных (n = 6). Обработка C18 in vivo (3 мг/кг ежедневно в течение 2 дней) устраняет такое снижение экспрессии белка CFTR в мозговой артерии (n = 8). (B) Обработка C18 in vivo снижает миогенный тонус в задних мозговых артериях, полученных от мышей с СН, эффект (C), не наблюдаемый в задних мозговых артериях, выделенных от мышей с нокаутом CFTR (KO). Средние максимальные диаметры сосудов при 45 мм рт.ст. (диаметрмакс) составляют: ложнооперированные = 146 ± 9 мкм, n = 5 от 3 мышей; СН = 149 ± 8 мкм, n = 6 от 4 мышей; СН + C18 = 155 ± 8 мкм, n = 8 от 6 мышей (критерий Краскела-Уоллиса P=N.S. для диаметрмакс); и CFTR KO = 142 ± 5 мкм, n = 6 от 3 мышей; CFTR KO + C18 = 145 ± 5 мкм, n = 6 от 3 мышей (критерий Манна-Уитни P=N.S. для диаметрмакс). По сравнению с ложнооперированными контрольными животными, мыши с СН имеют (D) сниженную объемную скорость кровотока сердца (ложнооперированные: n = 6; СН: n = 8; СН + C18: n = 8), (E) повышенное общее периферическое сопротивление сосудов (n = 6 для всех групп) и (F) снижение среднего артериального давления (САД) (n = 6 для всех групп). (G) Показаны типичные карты перфузии магнитно-резонансной томографии, которые применяли для определения церебрального кровотока коры переднего мозга. СН провоцирует снижение церебральной перфузии; обработка C18 значимо улучшает перфузию головного мозга у мышей с СН (ложнооперированные: n = 6; СН: n = 8; СН + C18: n = 8). Все данные представлены в виде среднего значения ± стандартная ошибка среднего. Для панелей A и G * обозначает P < 0,05 для непарных сравнений с ложнооперированными животными при помощи критерия Краскела-Уоллиса и апостериорного критерия Даннетта; для панели B * обозначает P < 0,05 для непарных сравнений с ложнооперированными животными при помощи двухфакторного дисперсионного анализа и ретроспективного анализа с критерием Тьюки; на панели C группы статистически сравнивали при помощи двухфакторного дисперсионного анализа (P=N.S.); и на панелях от E до G * обозначает P < 0,05 для непарных сравнений с СН при помощи критерия Краскела-Уоллиса и апостериорного критерия Даннетта. Все образцы на типичном изображении вестерн-блоттинга, отображаемом на панели А, получены от одной и той же пленки, но образец СН + C18 не был расположен рядом на этой пленке, как показано на данной фигуре. Сокращения: Конт - контроль; CFTR - регулятор трансмембранной проводимости при муковисцидозе; диаметрмакс максимальный диаметр сосуда; СН - сердечная недостаточность; KO - нокаут.
Фигура 4. C18 не вызывает отек головного мозга.
Показаны типичные количественные карты T2, которые оценивают содержание воды в головном мозге как время релаксации T2 на изображениях магнитного резонанса. В общей сложности для каждой мыши оценивали девять карт Т2, которые покрывают переднюю, среднюю и заднюю области мозга. На типичных изображениях показаны срезы передней (слева), средней (в центре) и задней (справа) областей мозга ложнооперированных мышей (вверху), мышей с сердечной недостаточностью (СН; в центре) и мышей с СН, обработанных C18 (3 мг/кг в день в течение 2 дней) (внизу). Ни СН, ни обработка C18 у мышей с СН не вызывает изменения времени релаксации T2 в любой из интересующих областей в любом из оцениваемых срезов. Таким образом, данные объединяли для получения среднего времени релаксации T2 для корковой и подкорковой областей и представляли графически (ложнооперированные: n = 7; СН: n = 7; СН + C18: n = 6; критерий Манна-Уитни P=N.S. между группами в корковой и подкорковой областях соответственно). Все данные представлены в виде среднего значения ± стандартная ошибка среднего. Группы статистически сравнивали при помощи критерия Краскела-Уоллиса (P = N.S.). Сокращения: СН - сердечная недостаточность.
Фигура 5. C18 восстанавливает перфузию головного мозга при субарахноидальном кровоизлиянии.
(A) Мозговые артерии, полученные от мышей с субарахноидальным кровоизлиянием (САК; через 2 дня после индукции САК), имеют пониженную экспрессию белка регулятора трансмембранной проводимости при муковисцидозе (CFTR) (n = 6), по сравнению с артериями, полученными от ложнооперированных контрольных животных (n = 6). Обработка C18 in vivo (3 мг/кг в день в течение 2 дней) устраняет это снижение в экспрессии белка CFTR в артериях (n = 6). (B) Обработка C18 in vivo снижает миогенный тонус в обонятельных артериях, полученных от мышей с САК, эффект (C), не наблюдаемый в обонятельных артериях, полученных от мышей с нокаутом CFTR (KO). Средние максимальные диаметры сосудов при 45 мм рт.ст. (диаметрмакс) составляют: ложнооперированные = 113 ± 3 мкм, n = 5 от 3 мышей; САК = 109 ± 6 мкм, n = 6 от 6 мышей; САК + C18 = 104 ± 12 мкм, n = 5 от 4 мышей (критерий Краскела-Уоллиса P=N.S. для диаметрмакс); и CFTR WT = 98 ± 6 мкм, n = 8 от 4 мышей; CFTR KO = 110 ± 8 мкм, n = 5 от 4 мышей; CFTR KO + C18 = 96 ± 6 мкм, n = 6 от 3 мышей (критерий Краскела-Уоллиса P=N.S. для диаметрмакс). (D) Показаны типичные карты перфузии магнитно-резонансной томографии, которые применяли для определения церебрального кровотока коры переднего мозга. САК стимулирует снижение перфузии головного мозга; обработка C18 значительно улучшает перфузию головного мозга у мышей с САК (ложнооперированные: n = 10; САК: n = 5; САК + C18: n = 9). Все данные представлены в виде среднего значения ± стандартная ошибка среднего. На панелях A и D * обозначает P < 0,05 для непарных сравнений с САК при помощи критерия Краскела-Уоллиса и апостериорного критерия Даннетта; на панели B * обозначает P < 0,05 для непарных сравнений с САК при помощи двухфакторного дисперсионного анализа и ретроспективного анализа с критерием Тьюки; на панели C группы статистически сравнивали при помощи двухфакторного дисперсионного анализа (P = N.S.). Сокращения: CFTR - регулятор трансмембранной проводимости при муковисцидозе; диаметрмакс - максимальный диаметр сосуда; СН - сердечная недостаточность; KO - нокаут; САК - субарахноидальное кровоизлияние.
Фигура 6: Лумакафтор увеличивает экспрессию белка CFTR дикого типа с помощью протеостатического механизма и восстанавливает перфузию головного мозга при субарахноидальном кровоизлиянии.
(A) Мозговые артерии, полученные от наивных мышей, получавших лумакафтор (Лум; 3 мг/кг ежедневно в течение 2 дней; n = 6), имеют более высокую экспрессию белка регулятора трансмембранной проводимости при муковисцидозе (CFTR), по сравнению с артериями, полученными от контрольных носителей (n = 7). (B) лумакафтор не влияет на экспрессию мРНК CFTR в мозговой артерии (n = 5 для обеих групп). (C) лумакафтор (6 мкмоль/л; 24 ч) увеличивает экспрессию белка CFTR в клетках фибробластов почек детенышей хомяка, стабильно экспрессирующих человеческий CFTR (n = 13 для обеих групп); (D) лумакафтор не влияет на экспрессию мРНК CFTR в данной системе (n = 6 для обеих групп). (E) Мозговые артерии, полученные от мышей с субарахноидальным кровоизлиянием (САК; 2 дня после индукции САК), имеют пониженную экспрессию белка CFTR (n = 5) по сравнению с артериями, полученными от ложнооперированных контрольных животных (n = 6). Обработка лумакафтором in vivo (3 мг/кг ежедневно в течение 2 дней) устраняет такое снижение экспрессии белка CFTR в мозговой артерии (n = 6). (F) Обработка лумакафтором in vivo снижает миогенный тонус в обонятельных артериях, выделенных от мышей с САК. Средние максимальные диаметры сосудов при 45 мм рт. ст. (диаметрмакс) составляют: ложнооперированные = 91 ± 3 мкм, n = 7 от 4 мышей, САК = 86 ± 2 мкм, n = 6 от 3 мышей; САК + Лум = 87 ± 4 мкм, n = 7 из 4 мышей (критерий Краскела-Уоллиса P=N.S. для диаметрмакс). (G) Показаны типичные карты перфузии магнитно-резонансной томографии, которые применяли для определения церебрального кровотока. САК провоцирует снижение перфузии головного мозга; лечение лумакафтором значительно улучшает перфузию головного мозга у мышей с САК (ложнооперированные: n = 6; САК: n = 5; САК + Лум: n = 6). Все данные представлены в виде среднего значения ± стандартная ошибка среднего. На панелях от A до D * обозначает P < 0,05 для непарного сравнения при помощи критерия Манна-Уитни; на панелях от E до G * обозначает P < 0,05 для непарных сравнений с САК при помощи критерия Краскела-Уоллиса и апостериорного критерия Даннетта. На панели F * обозначает P < 0,05 для непарных сравнений с САК при помощи двухфакторного дисперсионного анализа и ретроспективного анализа с критерием Тьюки. Сокращения: CFTR - регулятор трансмембранной проводимости при муковисцидозе; диаметрмакс - максимальный диаметр сосуда; Лум - лумакафтор; САК - субарахноидальное кровоизлияние.
Фигура 7: Коррекция CFTR снижает повреждение нейронов при субарахноидальном кровоизлиянии.
Показаны типичные изображения кортикальных клеток, окрашенных по экспрессии расщепленной каспазы-3 (вверху) и фтор-нефритом (внизу) для когорт, включающих схемы лечения (A) C18 (3 мг/кг ежедневно в течение 2 дней) и (B) лумакафтор (Лум; 3 мг/кг ежедневно в течение 2 дней). Субарахноидальное кровоизлияние (САК; n = 13 мышей) увеличивает количество (C) каспазы-3 и (D) положительных по фтор-нефриту клеток по сравнению с ложнооперированными контрольными животными (n = 12); как C18 (n = 6), так и лумакафтор (n = 5) устраняют усиление окрашивания каспазы-3 и фтор-нефритом. (E) Мыши с САК (n = 11) имеют более низкий балл по модифицированной шкале Гарсия (максимальный балл = 18; слепые оценки, сделанные через 2 дня после САК), по сравнению с ложнооперированными мышами (n = 11); обработка C18 (n = 10) восстанавливает неврологическую оценку до уровня ложнооперированных животных. Все данные представлены в виде среднего значения ± стандартная ошибка среднего. * обозначает P < 0,05 для непарных сравнений с ложнооперированными животными при помощи критерия Краскела-Уоллиса и апостериорного критерия Даннетта. На панелях C и D знак + означает P < 0,05 по сравнению с САК без обработки при помощи критерия Краскела-Уоллиса и апостериорного критерия Даннетта. Сокращения: Лум - лумакафтор; САК - субарахноидальное кровоизлияние.
Фигура 8: Размещение интересующей области на изображениях FAIR-EPI и анализах.
Показаны типичные анатомические T2-взвешенные карты, T1-взвешенные карты потокозависимой последовательности инверсия-восстановление (FAIR), и карты FAIR церебрального кровотока (ЦК) для мышей, перенесших хирургическую процедуру по сердечной недостаточности (верхний ряд) или субарахноидальному кровоизлиянию (САК; нижний ряд). Интересующие области были вручную нарисованы на изображениях FAIR среза переднего мозга, полученных при времени инверсии (TI) 825 мс с применением программного обеспечения MIPAV. Как показано, интересующие области охватывают приблизительно 1 мм3 объема коры головного мозга в пределах одного полушария. Затем интересующие области скопировали непосредственно на карты ЦК, полученные из изображений FAIR в отдельные моменты времени инверсии. Мыши с САК и ложнооперированные контрольные животные демонстрировали различную степень искажения эхо-планарной визуализации (EPI) (лучше всего визуализируемое на изображении FAIR); искажение EPI было минимальным у мышей с сердечной недостаточностью.
Сокращения: ЦК - церебральный кровоток; EPI - эхо-планарная визуализация; FAIR - потокозависимая последовательность инверсия-восстановление; САК - субарахноидальное кровоизлияние.
Фигура 9: Среднее артериальное давление у мышей с нокаутом CFTR
Среднее артериальное давление ниже у мышей с нокаутом регулятора трансмембранной проводимости при муковисцидозе (CFTR KO; CFTRtm1Unc; n = 6), по сравнению с однопометниками дикого типа (WT; n = 6). * обозначает P < 0,05 для непарного сравнения при помощи t-критерия.
Сокращения: CFTR - регулятор трансмембранной проводимости при муковисцидозе; KO - нокаут; WT - дикий тип
Фигура 10: Реакции на фенилэфрин в артериях кремастера мышей
Фенилэфрин стимулирует дозозависимое сужение кровеносных сосудов в артериях скелетных мышц кремастера, полученных от мышей дикого типа и с нокаутом регулятора трансмембранной проводимости при муковисцидозе (CFTR KO; CFTRtm1Unc). Зависимость реакции от дозы фенилэфрина не изменяется (А) ингибированием CFTR in vitro (100 нмоль/л CFTR(ингибитор)-172 в течение 30 минут) или (B) делецией гена CFTR. Средние максимальные диаметры сосудов при 60 мм рт. ст. (диаметрмакс) составляют: дикий тип (панель A): 72 ± 3 мкм, n = 5 от 4 мышей; CFTR KO (панель B): 88 ± 4 мкм, n = 6 от 4 мышей; и однопометники дикого типа (панель B): 79 ± 4 мкм, n = 5 от 2 мышей. Кривые "доза-реакция" на панели A сравниваются при помощи парного двухфакторного дисперсионного анализа (P=N.S.); кривые на панели B сравниваются при помощи непарного двухфакторного дисперсионного анализа (P=N.S.). Для панели B значения диаметрмакс KO и дикого типа не различаются (t-критерий P=N.S.).
Сокращения: CFTR - регулятор трансмембранной проводимости при муковисцидозе; диаметрмакс - максимальный диаметр сосуда; KO - нокаут; N.S. - незначимый.
Фигура 11: Реакции на фенилэфрин в задних мозговых артериях мыши после обработки С18 in vivo
(A) Фенилэфрин стимулирует дозозависимое сужение кровеносных сосудов в задних мозговых артериях, полученных от мышей с сердечной недостаточностью (СН) и мышей с сердечной недостаточностью, получавших C18 in vivo (3 мг/кг внутрибрюшинно ежедневно в течение 2 дней). Статистический анализ выявляет значимую разницу между двумя кривыми (т.е. значимо более высокий тонус артерий от мышей с СН по сравнению с мышами СН + C18). Однако (B), когда данные нормализованы по базовому тонусу (тонусактивный - тонуспокой, где тонусактивный представляет собой тонус при данной концентрации фенилэфрина, а тонуспокой представляет собой тон непосредственно перед стимуляцией), зависимости реакции от дозы существенно не различаются. Средние максимальные диаметры сосудов при 45 мм рт.ст. (диаметрмакс) составляют: СН: 137 ± 7 мкм, n = 5 от 3 мышей и СН + C18: 140 ± 8 мкм, n = 5 от 4 мышей (t-критерий P=N.S.). На обеих панелях * обозначает P < 0,05 при помощи двухфакторного дисперсионного анализа.
Сокращения: диаметрмакс - максимальный диаметр сосуда; СН - сердечная недостаточность; NS - незначимый.
Фигура 12: Обработка C18 in vivo не изменяет миогенный тонус или реакции на фенилэфрин в задних мозговых артериях, полученных от ложнооперированных мышей.
Обработка С18 in vivo (3 мг/кг внутрибрюшинно ежедневно в течение 2 дней) не изменяет (А) миогенный тонус или (В) реакции на фенилэфрин в задних мозговых артериях, полученных от ложнооперированных мышей. Средние максимальные диаметры сосудов при 45 мм рт.ст. (диаметрмакс) в панели A: ложнооперированные: 146 ± 9 мкм, n = 5 от 3 мышей и ложнооперированные + C18: 138 ± 9 мкм, n = 7 от 4 мышей (t-критерий P=N.S.); и на панели B: ложнооперированные: 148 ± 7 мкм, n = 6 от 3 мышей и ложнооперированные + C18: 130 ± 10 мкм, n = 5 от 3 мышей (t-критерий P=N.S.). Кривые на обеих панелях сравниваются при помощи двухфакторного дисперсионного анализа (P=N.S.).
Сокращения: диаметрмакс - максимальный диаметр сосуда; NS - незначимый.
Фигура 13: Реакции на фенилэфрин в задних мозговых артериях от мышей с нокаутом CFTR, получавших C18
Фенилэфрин провоцирует дозозависимое сужение кровеносных сосудов в задних мозговых артериях, полученных от мышей с нокаутом регулятора трансмембранной проводимости при муковисцидозе (CFTR KO; CFTRtm1Unc). Обработка C18 in vivo (3 мг/кг внутрибрюшинно ежедневно в течение 2 дней) не изменяет зависимость реакции от дозы фенилэфрина. Средние максимальные диаметры сосудов при 45 мм рт.ст. (диаметрмакс) составляют: CFTR KO: 142 ± 5 мкм, n = 6 от 3 мышей и CFTR KO + C18: 145 ± 5 мкм, n = 6 от 3 мышей (t-критерий P=N.S.). Кривые сравниваются при помощи двухфакторного дисперсионного анализа (P=N.S.).
Сокращения: CFTR - регулятор трансмембранной проводимости при муковисцидозе; диаметрмакс- максимальный диаметр сосуда; KO - нокаут; NS - незначимый.
Фигура 14: Анализ Шолла апикальных и базальных дендритов.
(A) Показаны гистограммы анализа Шолла, отображающие количество пересечений дендритов (т.е. ветвление дендритов) в зависимости от длины дендрита (т.е. расстояния от сомы нейрона) для апикальных дендритов. Не обнаружено различий морфологии ветвления между группами ложнооперированных мышей (n = 13 нейронов от N = 4 мышей), с сердечной недостаточностью (СН; n = 11; N = 4) и СН + C18 (3 мг/кг внутрибрюшинно ежедневно в течение 2 недель; n = 11; N = 4); однако длина апикальных дендритов короче у мышей с СН по сравнению с группами ложнооперированных контрольных животных и СН + C18. Соответственно, (B) средняя длина апикального дендрита (т.е. максимальный радиус) значимо уменьшена у мышей с СН по сравнению с ложнооперированными мышами; данный эффект нормализуется обработкой C18. Точно так же (C) гистограммы анализа Шолла для базальных дендритов не показывают различий в морфологии ветвления в группах ложнооперированных животных (n = 12; N = 4), с СН (n = 13; N = 4) и СН + C18 (n = 11; N = 4); однако длина базальных дендритов короче у мышей с СН по сравнению с ложнооперированными мышами и группой СН + C18. Соответственно, (D) средняя длина базального дендрита (т.е. максимальный радиус) значимо уменьшена у мышей с СН по сравнению с ложнооперированными мышами, эффект, который нормализуется обработкой C18. * обозначает P < 0,05 для непарных сравнений с ложнооперированными животными при помощи однофакторного дисперсионного анализа и апостериорного критерия Даннета.
Сокращения: СН - сердечная недостаточность.
Фигура 15: Анализ плотности шипиков апикальных дендритов
(A) У ложнооперированных (n = 8 нейронов от N = 4 мышей), с сердечной недостаточностью (СН; n = 6; N = 3) и мышей с СН + C18 (3 мг/кг внутрибрюшинно ежедневно в течение 2 недель; n = 10; N = 4), плотность шипиков апикальных дендритов (т.е. мера плотности синапсов) относительно постоянна по длине дендрита, давая наклоны, которые статистически не отличаются от нуля. (B) Хотя тенденции присутствуют, средняя плотность шипиков апикальных дендритов, измеренная на расстоянии от 60 до 100 мкм от сомы, не достигает статистической значимости. Группы статистически сравнивали с помощью однофакторного дисперсионного анализа (P=N.S.).
Сокращения: СН - сердечная недостаточность; NS - незначимый.
Фигура 16: Реакции на фенилэфрин в обонятельных мозговых артериях мыши после обработки С18 in vivo.
(A) Фенилэфрин стимулирует дозозависимое сужение кровеносных сосудов в обонятельных мозговых артериях, полученных от мышей с субарахноидальным кровоизлиянием (САК) и мышей с САК, получавших C18 in vivo (3 мг/кг внутрибрюшинно в течение 2 дней). Несмотря на явную тенденцию к разделению, две кривые статистически не различаются. (B) Когда данные нормализованы по базовому тонусу (тонусактивный - тонуспокой, где тонусактивный представляет собой тонус при данной концентрации фенилэфрина, а тонуспокой представляет собой тонус, непосредственно перед стимуляцией), разделение зависимостей реакции от дозы минимально. Средние максимальные диаметры сосудов при 45 мм рт.ст. (диаметрмакс) составляют: САК: 109 ± 6 мкм, n = 6 от 6 мышей и САК + C18: 105 ± 12 мкм, n = 5 от 4 мышей (t-тест P=N.S.). Кривые сравниваются при помощи двухфакторного дисперсионного анализа.
Сокращения: диаметрмакс - максимальный диаметр сосуда; NS - незначимый; САК - субарахноидальное кровоизлияние.
Фигура 17: C18 не влияет на миогенный тонус или реакции на фенилэфрин у ложнооперированных мышей.
Обработка C18 in vivo (3 мг/кг внутрибрюшинно ежедневно в течение 2 дней) не изменяет (A) миогенный тонус или (B) зависимость реакции от дозы фенилэфрина в обонятельных мозговых артериях, полученных от ложнооперированных мышей. Средние максимальные диаметры сосудов при 45 мм рт.ст. (диаметрмакс) составляют: ложнооперированные: 113 ± 3 мкм, n = 5 от 3 мышей; и ложнооперированные + C18: 110 ± 8 мкм, n = 6 от 4 мышей (t-критерий P=N.S.). Кривые сравниваются при помощи двухфакторного дисперсионного анализа.
Сокращения: диаметрмакс - максимальный диаметр сосуда; NS - незначимый.
Фигура 18: Реакции на фенилэфрин в обонятельных мозговых артериях, полученных от мышей с нокаутом CFTR
Фенилэфрин провоцирует дозозависимое сужение кровеносных сосудов в обонятельных мозговых артериях, полученных от мышей с нокаутом регулятора трансмембранной проводимости при муковисцидозе (CFTR KO; CFTRtm1Unc), что аналогично таковому у однопометников дикого типа (WT). Обработка C18 in vivo (3 мг/кг внутрибрюшинно ежедневно в течение 2 дней) не изменяет зависимостей реакции от дозы фенилэфрина. Средние максимальные диаметры сосудов при 45 мм рт.ст. (диаметрмакс) составляют: WT: 98 ± 6 мкм, n = 8 от 4 мышей; CFTR KO: 110 ± 8 мкм, n = 5 от 4 мышей и CFTR KO + C18: 96 ± 6 мкм, n = 6 от 3 мышей (однофакторный дисперсионный анализ P=N.S.). Кривые сравниваются при помощи двухфакторного дисперсионного анализа.
Сокращения: CFTR - регулятор трансмембранной проводимости при муковисцидозе; диаметрмакс - максимальный диаметр сосуда; KO - нокаут; NS - незначимый; WT - дикого типа.
Фигура 19. Реакции на фенилэфрин в обонятельных мозговых артериях мыши после обработки лумакафтором in vivo.
Обработка лумакафтором (Лум) in vivo (3 мг/кг внутрибрюшинно ежедневно в течение 2 дней) не изменяет зависимостей реакции от дозы фенилэфрина в обонятельных мозговых артериях, полученных от мышей с субарахноидальным кровоизлиянием (САК). Средние максимальные диаметры сосудов при 45 мм рт.ст. (диаметрмакс) составляют: САК: 86 ± 2 мкм, n = 6 от 3 мышей; и САК + Лум: 87 ± 4 мкм, n = 7 от 4 мышей (t-критерий P=N.S.). Кривые сравниваются при помощи двухфакторного дисперсионного анализа.
Сокращения: диаметрмакс - максимальный диаметр сосуда; Лум - лумакафтор; NS - незначимый; САК - субарахноидальное кровоизлияние.
Настоящее изобретение дополнительно проиллюстрировано следующим неограничивающим примером.
ПРИМЕР
Способы и реактивы
Реактивы
Антитело к регулятору трансмембранной проводимости при муковисцидозе (CFTR) человека, применяли для обнаружения CFTR в клетках фибробластов почек детенышей хомяка ("антитело 596"), терапевтический корректор CFTR "C18" (CF-106951; см. WO 2007/021982; 1-(бензо[d][1,3]диоксол-5-ил)-N-(5-((2-хлорфенил)(3-гидроксипирролидин-1-ил)метил)тиазол-2-ил)циклопропанкарбоксамид) приобретали в рамках программ по распространению реактивов и антител Фонда лечения муковисцидоза (http://www.cff.org/research). Д-р Джон Риордан (John Riordan) (Университет Северной Каролины - Чапел-Хилл) предоставил "антитело 596 CFTR", и д-р Роберт Бриджес (Robert Bridges) (Университет медицины и науки Розалинды Франклин, Чикаго, США) предоставил соединение C18. Антитело 596 CFTR нацелено на аминокислоты с 1204 по 1211 (т.е. расположенные в нуклеотид-связывающем домене 2) (1). Меченный флуоресцеином S1P (FITC-S1P) приобретали в Echelon Biosciences (через Cedarlane Laboratories; Берлингтон, Канада); лумакафтор приобретали в компании Selleck Chemicals (Cedarlane Laboratories); рекомбинантный фактор некроза опухоли приобретали у Sigma-Aldrich Canada (Оквилл, Canada; номер по каталогу T6674); и таблетки смеси ингибиторов протеаз (Complete®; применяют в лизирующих буферах для вестерн-блотов) приобретали у Roche (Миссиссога, Канада). Все остальные химические реактивы приобретали у Sigma-Aldrich. Сбалансированный солевой раствор MOPS содержал (ммоль/л): NaCl 145, KCl 4,7, CaCl2 3,0, MgSO4⋅7H2O 1,17, NaH2PO4⋅2H2O 1,2, пируват 2,0, этилендиаминтетрауксусная кислота (ЭДТА) 0,02, 3-морфолинопропансульфоновая кислота (MOPS) 3,0, и глюкоза 5,0.
Мутантные и нокаутные мыши CFTR
Самцы мышей, гомозиготные по мутации ΔF508 CFTR (CFTRtm1EUR; обозначены как "ΔF508" в настоящем документе) (2), делеции гена CFTR (CFTRtm1Unc; обозначены как CFTR-/-), и комплементарные контрольные однопометники дикого типа были получены из стабилизированной колонии в Госпитале для больных детей, Торонто (все CFTR-/-, CFTRΔF508 и однопометники дикого типа представляют собой смешанные линии). Авторами настоящего изобретения было обнаружено, что мыши CFTR-/- очень склонны к смерти под наркозом. Хотя эта смертность не была проблемой для экспериментов по выделению мозговой артерии (например, фиг. 3C и 5C), она значительно мешала церебральному кровотоку и системным измерениям гемодинамики. В отличие от мышей CFTR-/-, мутантные мыши с CFTRΔF508 были не так предрасположены к смерти во время анестезии. Поскольку мутация CFTRΔF508 значительно снижает доставку CFTR к плазматической мембране (3), активность CFTR очень низкая в модели мышей CFTRΔF508 (2, 4, 5). Таким образом, модель CFTRΔF508 обеспечила подходящую альтернативу мышам CFTR-/- для церебрального кровотока и системных гемодинамических измерений. Авторы настоящего изобретения не исследовали основную причину смертности CFTR-/-. Оба варианта CFTR-/-, и CFTRΔF508, как хорошо известно, неблагоприятно реагируют на стресс. Например, условия содержания и перемещения являются двумя заметными стрессогенными факторами, которые могут значительно увеличить смертность (6). Интересно, что мыши CFTRΔF508 демонстрируют менее тяжелый фенотип, чем мыши CFTR-/- (5, 6): предполагается, что это связано с небольшим количеством остаточной активности CFTR, которая присутствует у животных CFTRΔF508 (2). В этом контексте авторы настоящего изобретения предполагают, что мыши CFTR-/- могли быть более чувствительны к внешним стрессогенным факторам окружающей среды в экспериментальном помещении для животных (например, условиям содержания, шуму и уходу), что в конечном итоге проявилось в повышенной смертности под наркозом. В качестве экспериментальной модели мыши CFTRΔF508 обладают несколькими аномалиями, связанными с потерей функции CFTR. Кишечные осложнения являются наиболее выраженным патологическим действием мутации CFTR: и также является основной причиной послеродовой смертности (6, 7). Общие желудочно-кишечные проблемы включают задержку густой слизи, нарушение моторики и сильную склонность к непроходимости кишечника. Мыши CFTRΔF508 на 40-50 % меньше по массе/размеру по сравнению с аналогами дикого типа, хотя они хорошо развиваются во взрослой жизни (6, 7). Наблюдалось несколько других отличий от мышей дикого типа; однако они кажутся относительно небольшими (6, 7). Например, были отмечены измененные токи хлоридов и/или транспорт натрия в определенных тканях (например, почках, желчном пузыре, назальном эпителии), но патологическое значение этих различий неясно, так как ткани не кажутся гистологически дефектными (6, 7). Следует отметить, что мыши CFTRΔF508 (и мутантные мыши CFTR в целом) обладают довольно мягким фенотипом муковисцидоза по сравнению с муковисцидозом человека. Нижние дыхательные пути мышей CFTRΔF508 в основном нормальны: эпителиальных аномалий нижних дыхательных путей нет, и воспаление легких не развивается спонтанно без заражения (6, 7). Поджелудочная железа, желчный пузырь, печень, желчный проток, мужской половой тракт, слезная железа и подчелюстные железы не имеют явной гистопатологии (5-7). Отсутствие тяжелого фенотипа муковисцидоза частично объясняется экспрессией без CFTR, кальций-активированного хлоридного канала (CACC) в определенных тканях мыши, что компенсирует потерю CFTR (7, 8).
Глобальные гемодинамические параметры
Как описано ранее (9-11), эхокардиографические измерения были получены с помощью механического секторного датчика 30 МГц (Vevo 770; Visual Sonics, Торонто, Канада) в сочетании с измерениями среднего артериального давления (MAP, САД) (манометрический катетер для мыши с микронаконечником Millar SPR-671; Inter V Medical Inc., Монреаль, Канада). Интеграл скорости кровотока в аорте (VTI) измеряли с помощью импульсно-волнового допплера чуть выше корня аорты. Площадь поперечного сечения аорты (CSA) рассчитывали из размера корня аорты (ARD): CSA = π(ARD/2)2. Ударный объем (SV), объемную скорость кровотока сердца (CO) и общее периферическое сопротивление сосудов (TPR) рассчитывали по формуле: SV = CSA × VTI; CO = SV × HR; TPR = MAP/CO.
Измерение перфузии головного мозга с помощью МРТ
Как описано ранее (10-12), авторы настоящего изобретения применяли способ магнитно-резонансной томографии (МРТ) с потокозависимой последовательностью инверсия-восстановление (FAIR) (13) для оценки церебрального кровотока (ЦК). Во время визуализации мышей иммобилизовали 1,8% изофлураном, доставляемым через назальный конус. Для поддержания температуры тела в МРТ встроены трубки, отводящие воду от внешнего нагревателя-насоса. Дыхание контролировали с помощью пневматической подушки (SA Instruments, Стоуни-Брук, США); уровни изофлурана регулировали для поддержания дыхания приблизительно 50 вдохов в минуту.
Изображения FAIR получали с применением системы микро-МРТ 7 Тесла (BioSpec 70/30 USR, Bruker BioSpin, Эттлинген, Германия), включая градиентную вставку B-GA12, линейный объемный резонатор с внутренним диаметром 72 мм для передачи радиочастот и расположенную спереди головную катушку для приема радиочастот. FAIR выделяет перфузию как ускоренную релаксацию сигнала T1 после срез-селективной по сравнению с неселективной инверсионной подготовкой согласно следующему уравнению: ЦК = l (1/T1, ss - 1/T1, ns) (мл/(100 г*мин), где "ss" и "ns" обозначают срез-селективные и неселективные измерения, и l представляет собой коэффициент распределения между кровью и мозгом, определяемый как соотношение между концентрацией воды на грамм ткани мозга и на мл крови. Этот коэффициент составляет приблизительно 90 мл/100 г у мышей (14). Оптимизация FAIR, примененная в настоящем изобретении, представляла собой способ одноимпульсной эхо-планарной визуализации (EPI) с предшествующей адиабатической инверсией. Параметры включали время эхо-сигнала 12,5 мс, время повторения 17 с, 18 значений времени инверсии от 25 до 6825 мс с шагом 400 мс, срез-селективная инверсионная пластина 3 мм, область сканирования 18×18 мм с матрицей 72×72 для разрешения в плоскости 250 мкм, толщина срезов 1 мм и время получения данных 10 мин 12 с. Получение данных повторяли в вертикальных срезах переднего, среднего и заднего мозга, соответственно передней, смешанной и задней зоне кровоснабжения. FAIR-изображения оценивали прописью (MIPAV, NIH, Bethesda, MD; http://mipav.cit.nih.gov) субполушарных областей интереса (ROI), называемых "глобальными", и локальных ROI, соответствующих кортикальной и субкортикальной паренхиме в отделах переднего мозга; кортикальной и паравентрикулярной паренхиме в средних отделах; и кортикальной и паравентрикулярной паренхиме среднего мозга в отделах заднего мозга. Области интереса нарисованы непосредственно на T1-взвешенных изображениях сигналов, чтобы обеспечить возможность ручной коррекции движения внутри сканирования. ROI регистрировали с помощью параметрических карт ЦК, чтобы проверить отсутствие смещения от сосудов с высокой перфузией и мозговых оболочек. Регрессии T1 и расчеты ЦК выполнялись с применением Matlab (Mathworks, Нейтик, Массачусетс). Все сообщенные значения ЦК попадают в опубликованный диапазон измерений ЦК у мышей для уровня изофлурана, в соответствии с настоящим изобретением (15). EPI особенно подвержен искажениям, связанным с магнитной восприимчивостью, в направлении фазового кодирования (16), которое соответствует вертикальному направлению в данном протоколе FAIR. Как видно на фиг. 8, коррекция неоднородности магнитного поля недостаточно ограничивает искажение EPI в хирургической модели субарахноидального кровоизлияния (САК) из-за границ воздух-ткань, непосредственно прилегающих к мозгу в моменты томографирования после операции. В некоторых случаях искажение было настолько серьезным, что области интереса приходилось размещать более латерально в коре головного мозга (фиг. 8). Однако, поскольку искажение одинаково во всех T1-взвешенных изображениях, это не влияет на измерение T1 при условии, что искаженная область не перекрывается с другой областью. Относительно согласованные значения ns-T1 в каждой когорте (т.е. стандартное отклонение 7% между когортами, отсутствие различий в группах лечения внутри когорт) обеспечивают подтверждающие доказательства того, что смещение искажения оказало незначительное влияние на измерения ЦК.
Культура клеток
Процедуры выделения и культивирования гладкомышечных клеток брыжеечной артерии выполняли, как описано в публикации (10). Кратко, сегменты брыжеечной артерии выделяли, разрезали на мелкие кусочки и подвергали воздействию трипсина, коллагеназы и эластазы. Полученную суспензию клеток промывали несколько раз фосфатно-солевым буфером и помещали в культуральную среду Игла, модифицированную по Дульбекко, (DMEM), содержащую 10% фетальную бычью сыворотку и 1% пенициллин и стрептомицин. Культуры клеток поддерживали при 37°C и 5% CO2 и разделяли при плотности посева 106 клеток. Клетки фибробластов почек детенышей хомяка, стабильно экспрессирующие человеческий CFTR дикого типа (10, 17), поддерживали в среде DMEM/F12, содержащей 5% фетальную бычью сыворотку и 250 мкмоль/л метотрексата (который активирует промотор трансгена CFTR). Клетки поддерживали в стандартных условиях культивирования клеток при 37°C и 5% CO2.
Измерение поглощения FITC-S1P на основе FACS
Как описано ранее (10), клеточные монослои (обработанные или необработанные) инкубировали с 1 мкмоль/л S1P-FITC в течение 60 минут; затем клетки отделяли трипсинизацией, дважды промывали ледяным фосфатно-солевым буфером, фильтровали через фильтр для клеток 35 мкм и анализировали, применяя Becton-Dickinson FACS Canto, при помощи программного обеспечения FACS DIVA версии 6.1. Монослои клеток, обработанные немеченым S1P, служили фоновым контролем. В ходе процедуры анализа определяли среднюю интенсивность флуоресценции (условные единицы) каждой клеточной популяции, которая является мерой поглощения.
Оценка оттока йодида
Измерения оттока йодида проводили, как описано ранее (18). Кратко, монослои конфлюэнтных клеток нагружали йодидом, инкубируя их в загрузочном буфере на основе HEPES (136 ммоль/л NaI, 3 ммоль/л KNO3, 2 ммоль/л Ca(NO3)2, 11 ммоль/л глюкозы и 20 ммоль/л HEPES; pH 7,4) в течение 1 часа (в стандартных условиях культивирования клеток при 37°C в 5% CO2). После нагрузки йодидом клетки промывали 4 раза буфером оттока, не содержащим йодида (где NaNO3 заменен на NaI). Уровни йодида в супернатанте количественно определяли с помощью йодид-селективного электрода (Lazar Research Laboratories; Лос-Анджелес, США), калиброванного стандартами NaI. Определяли уровни йодида перед стимуляцией, а затем клетки стимулировали смесью, содержащей 10 мкмоль/л форсколина, 1 ммоль/л изобутилметилксантина и 100 мкмоль/л cpt-цАМФ (в 1% об./об. ДМСО). Стимулирующая смесь сильно активирует протеинкиназу А и, следовательно, максимально фосфорилирует/активирует CFTR. Уровни йодида в супернатанте определяли через семь последовательных 1-минутных интервалов после стимуляции. В соответствии с предыдущими результатами (18), отток был очевидным в течение первой минуты и был достоверно максимальным между 60 и 120 секундами. Таким образом, максимальная скорость оттока была рассчитана с применением измеренного уровня оттока между 60 и 120 секундами после стимуляции (18).
Вестерн-блоттинг
Вестерн-блоттинг для CFTR проводили, как описано ранее (12, 18). Лизаты мозговых артерий получали измельчением образцов артерий в буфере для лизиса, содержащем 50 мМ Трис (pH 7,3), 150 мМ NaCl, 2 мМ ЭДТА, 0,1% тритон-X-100, 0,1% SDS и ингибиторы протеаз; тот же буфер для лизиса применяли для получения лизатов клеточных культур. После лизиса образцы центрифугировали (10 минут при 13500 g; при 4°C) для удаления нерастворимого материала. Непосредственно перед электрофорезом в полиакриламиде добавляли дополнительный SDS (до конечной концентрации 2%), глицерин (до конечной концентрации 2%), β-меркаптоэтанол (до конечной концентрации 2%) и дитиотреитол (конечная концентрация 2 мМ). Белки разделяли электрофоретически на 7% акриламидных гелях и переносили на мембраны из поливинилидендифторида (ПВДФ). Мембраны блокировали на время от 30 до 40 минут в 5% обезжиренном молоке (в фосфатно-солевом буфере, содержащем 1% Твин 20 (ФСБТ); 137 мМ NaCl, 2,7 мМ KCl, 10 мМ Na2HPO4, 1,76 мМ K2HPO4; pH 7,4). Обработка первичными антителами включала: (i) кроличьи поликлональные антитела к CFTR (1:1000 в 5% молоке/ФСБТ; Cell Signaling Technology от New England Biolabs Canada; Уитби, Канада; номер по каталогу 2269); лизаты мозговых артерий и сосудистых гладкомышечных клеток.
(ii) Мышиные моноклональные антитела к CFTR человека (разведение 1:20000 в 5% молоке/ФСБT; "антитело 596"); лизаты клеток фибробластов почек детенышей хомяков.
(iii) Мышиные моноклональные антитела к α-тубулину (1:5000 в 5% молоке/ФСБT; клон DM1A; Cell Signaling Technology от New England Biolabs Canada; номер по каталогу 3873).
Первичные антитела конъюгировали либо с вторичными меченными пероксидазой ослиными антителами к IgG кролика (GE Healthcare Amersham (Пискатауэй, США), номер по каталогу NA934), либо с вторичными меченными пероксидазой козьими антителами к IgG мыши (GE Healthcare Amersham, номер по каталогу NA931). Для экспонирования рентгеновской пленки или сбора цифровых изображений применяли стандартный способ хемилюминесценции (Westar ETA C; VPQ Scientific, Торонто, Канада) (ChemiDoc; Bio-Rad Laboratories; Миссиссога, Канада). Проявленные пленки оценивали при помощи денситометрии с применением программного обеспечения "Image J" (свободно доступного от NIH); цифровые изображения оценивали с помощью программного обеспечения Image Lab (Bio-Rad).
Выделение РНК и обратная транскрипция
РНК резистивной артерии выделяли при помощи спин-колонок Norgen Biotek (Торолд, Канада) "Total RNA Purification Micro", применяя способы расщепления протеиназой K и удаления ДНК, как указано в инструкциях производителя. Элюированную РНК количественно определяли с помощью набора Agilent Technologies RNA 6000 Pico и биоанализатора; анализом подтвердили получение высококачественной РНК из образцов ткани артерии (целостность РНК [RIN]: мозговая = 8,4 ± 0,1, n = 5; кремастер = 8,2 ± 0,4, n = 6). Выделение РНК из клеток фибробластов почки детеныша хомяка выполняли тем же способом, исключая стадию расщепления протеиназой К. Во всех случаях из РНК получали кДНК с применением набора для обратной транскрипции Superscript III (Invitrogen Life Technologies; Берлингтон, Канада) в соответствии с инструкциями производителя. Остаточную РНК удаляли путем инкубации полученной кДНК с РНКазой H (0,125 Ед/мкл; New England Biolabs Canada; Уитби, Канада).
Количественная ПЦР
Количественную ПЦР проводили с применением системы ПЦР в реальном времени Applied Biosystems ViiA™ 7 и мастер-микса Power SYBR® Green PCR (оба от Invitrogen Life Technologies). Каждый набор праймеров был тщательно проверен для гарантии специфичности и сопоставимой эффективности. Гены-мишени оценивали в трех повторах с применением 1 нг кДНК, полученной при обратной транскрипции; в качестве отрицательных контролей применяли воду. ПЦР-амплификация состояла из 10 минут денатурации при 95°C, с последующими 40 циклами амплификации (15 с при 95°C и 60 с при 60°C). После амплификации ампликоны плавили: полученная кривая диссоциации подтвердила получение одного продукта. Уровни экспрессии транскрипта в тканях мыши рассчитывали по значениям DCt относительно стандартного гена домашнего хозяйства гидроксиметилбилансинтазы (HMBS). Чтобы подтвердить надежность HMBS для нормализации, уровни экспрессии транскриптов также рассчитывали из значений DCt относительно гена глюкозо-6-фосфатдегидрогеназы (G6PD), с получением аналогичных результатов. В экспериментах с клетками фибробластов почек детенышей хомяка применяли праймеры, специфичные для CFTR человека и GAPDH хомяка, последний представляет собой ген домашнего хозяйства, подходящий для нормализации.
Фиксация мозга и подготовка срезов для иммуногистохимии и окрашивания фтор-нефритом
Развитие отсроченного сужения кровеносных сосудов и повреждения головного мозга происходит быстрее в экспериментальной модели мышей с САК (от 2 до 5 дней после САК) (12, 19-21) по сравнению с тем, что наблюдается клинически (от 4 до 12 дней после САК) (22, 23). Авторами изобретения была выбрана временная точка 2 дня после индукции САК для оценки маркеров повреждения на основе предыдущей работы, демонстрирующей значительное повреждение нейронов и поведенческий дефицит в этот момент времени (12). Через 2 дня после индукции САК животных анестезировали изофлураном; их мозг перфузировали фосфатно-солевым буфером (ФСБ), а затем перфузию фиксировали параформальдегидом в фосфатном буфере (4%; pH 7,4), оба раствора вводили через восходящую аорту. Мозг немедленно препарировали и разрезали на корональные срезы на 1 мм сзади от брегмы. Эти срезы головного мозга после фиксировали в 4% параформальдегиде (pH 7,4) в течение 48 часов при 4°C, а затем инкубировали в 10% сахарозе (3 часа), а затем 30% сахарозе в течение ночи (при 4°C) для цитопротекции. Затем срезы мозга помещали в реагент OCT (Sakura Finetek USA; Торранс, США) на изопентане и сухом льду. Криостатные срезы (корональные срезы толщиной 5 мкм) собирали на предметных стеклах "Tissue Path Superfrost Plus Gold" (Fisher Scientific; Уитби, Канада) и хранили при -80°C до применения.
Флуоресцентная иммуногистохимия
Все образцы предварительно обрабатывали 10 минут протеиназой (20 мкг/мл; Promega; Мэдисон, США) и затем в течение 60 минут блокировали стандартным способом 10% нормальной ослиной сывороткой (Jackson ImmunoResearch Laboratories; Уэст-Гроув, США) в ФСБ или 10% козьей сывороткой (Invitrogen Life Technologies; Берлингтон, Канада) в ФСБ, содержащей 1% бычий сывороточный альбумин. Срезы мозга инкубировали с моноклональными кроличьими антителами к активной расщепленной каспазе-3 человека (клон C92-605; разведение 1:1000; BD Biosciences Canada; Миссиссога, Канада) и конъюгировали с козьими антителами к IgG кролика Alexa Fluor 488 (Invitrogen). Оба вида антител разводили в растворе усилителя иммунной реакции Can Get Signal® и применяли время инкубации 1 час при комнатной температуре. Образцы промывали и закрепляли с помощью CC MountÔ.
Окрашивание фтор-нефритом
Срезы мозга последовательно инкубировали 1% NaOH/80% этанолом (5 минут), 70% этанолом (2 минуты), дистиллированной водой (2 минуты) и 0,06% перманганатом калия (10 минут). После промывания деионизированной водой срезы головного мозга окрашивали 0,0004% фтор-нефритом B (Histo-Chem Inc., Джефферсон, США) в 0,1% уксусной кислоте (15 минут). Затем образцы промывали деионизированной водой, сушили и очищали погружением в ксилол на 1 минуту. Слайды закрепляли с помощью среды для заключения срезов DPX (Sigma).
Цифровая визуализация и оценка повреждений мозга путем подсчета клеток
Цифровые иммунофлуоресцентные изображения получали с применением конфокального лазерно-сканирующего микроскопа Zeiss LSM 710 в сочетании с программным обеспечением Zeiss ZEN 2010 (изображения сосудов). Наложения создавались с помощью свободно доступного программного обеспечения ImageJ 1.44p (National Institutes of Health, США). Двумя независимыми экспертами были подсчитаны положительные по каспазе-3 и фтор-нефриту клетки в слепых условиях, как описано ранее (12). Для каждого животного подсчет проводили для кортикальной области на одном корональном срезе головного мозга 1 мм сзади от брегмы (увеличение 200×), которая включает левую и правую височные и теменные доли. Число положительных клеток, подсчитанное каждым экспертом, усредняли, получая среднее количество положительных клеток на животное, которое затем применяли для статистического анализа.
Гистологический анализ морфологии дендритов:
Мозг перфузировали гепаринизированным солевым раствором посредством транскардиальной перфузии, изолировали и обрабатывали/окрашивали с применением коммерческого, доступного в продаже набора Rapid GolgiStain (FD NeuroTechnologies Inc.; Колумбия, США). В наборе применяется модифицированный способ окрашивания по Гольджи-Коксу, первоначально описанный Глэзером и Ван дер Лоосом (24). Кратко, выделенные образцы ткани головного мозга погружали в раствор, входящий с состав набора, содержащий хлорид ртути, дихромат калия и хромат калия, в темноте в течение 8 дней; затем 6 дней инкубировали с раствором для защиты тканей. Затем образцы головного мозга промывали, заключали в 4% легкоплавкую агарозу, делали срезы в корональной плоскости (толщина 150 мкм; вибрационный микротом Campden Instruments 7000smz-2) и помещали на покрытые желатином стеклянные предметные стекла. Затем процедуру окрашивания завершали с применением реагентов для проявления/окрашивания, входящих в состав набора, в соответствии с инструкциями производителя. Нейроны и дендритные сегменты визуализировали с помощью стереологического программного обеспечения NIS Elements AR на микроскопе Nikon Eclipse Ti2 с моторизованным фокусом X, Y и Z для получения изображений с высоким разрешением (Nikon Instruments Europe; Амстердам, Нидерланды). Авторами изобретения проанализированы как базальные, так и апикальные дендритные сети пирамидных кортикальных нейронов лобной коры, с применением количественного подхода, первоначально описанного Шоллем (25). Применяли программное обеспечение Image J в сочетании с полуавтоматическим плагином Simple Neurite Tracer (оба находятся в свободном доступе от NIH) для идентификации и цифрового выделения дендритных сетей одного кортикального нейрона из изображения гиперпробы с увеличением 200×. Центр сомы отмечали стрелкой; затем морфологию дендритов характеризовали анализом Шолля (см. https://imagej.net/Sholl_Analysis, версия 3.7.4) с применением интервалов 5 мкм до максимального радиуса 300 мкм. Этот анализ характеризует морфологию дендритов с точки зрения пересечений дендритов (то есть ветвления) и длины дендрита. Для каждой группы обработки, от 2 до 4 нейронов на мышь от 4 мышей анализировали в слепых условиях. Для оценки плотности дендритных шипиков (то есть количество небольших выступов, обнаруженных на дендритах), визуализировали дендритные сегменты 3-го порядка ветви при 100× увеличении. Плотность шипиков измеряли как количество шипиков на сегмент для следующих сегментов: от 20 до 50 мкм от сомы; от 30 до 60 мкм от сомы; от 50 до 80 мкм от сомы; и от 60 до 100 мкм от сомы). Данный подход "скользящего измерения" позволяет оценить то, как плотность шипиков изменяется по длине дендритной ветви. Для каждой группы лечения плотность шипиков измеряли в пирамидных кортикальных нейронах от 2 до 4 (от 2 до 4 ветвей третьего порядка на нейрон) на мышь у от 3 до 4 мышей на группу в слепых условиях.
Как описано выше, корректор C18 CFTR и антитело к CFTR человека (обозначенное "596") приобретали в рамках программ по распространению реактивов и антител Фонда лечения муковисцидоза. Все остальные применяемые реагенты коммерчески доступны, как указано выше.
Животные: Институциональные комитеты по уходу за животными и их использованию в Университете Торонто и Университетской сети здравоохранения (UHN) одобрили все протоколы ухода за животными и экспериментов. Коммерчески доступных самцов мышей дикого типа (от 2 до 3 месяцев; C57BL/6N) приобретали в Charles River Laboratories (Монреаль, Канада). Самцов мышей, гомозиготных по мутации ΔF508 CFTR (CFTRtm1EUR; обозначены как "ΔF508" в настоящем документе) (11), делеции гена CFTR (CFTRtm1Unc; обозначены CFTR-/-), и комплементарных контрольных однопометников дикого типа получали из стабилизированной колонии в Госпитале для больных детей, Торонто (все CFTR-/-, CFTRΔF508 и однопометники представляют собой смешанные линии). Всех мышей содержали в соответствии со стандартным циклом 14 часов: 10 часов свет-темнота, кормили обычной пищей и обеспечивали неограниченный доступ к воде.
Инфаркт миокарда: сердечную недостаточность вызывали хирургической перевязкой левой передней нисходящей коронарной артерии (3). Кратко, мышей анестезировали изофлураном, интубировали ангиокатетером 20 размера и вентилировали комнатным воздухом. В стерильных условиях грудную клетку и перикард вскрывали, и левую переднюю нисходящую коронарную артерию необратимо перевязывали шелковой нитью 7-0 (Deknatel; Фолл-Ривер, США). В контрольной группе ложнооперированным животным вскрывали грудную клетку и перикард, но левую переднюю нисходящую коронарную артерию не перевязывали. После процедуры грудную клетку закрывали и мышам проводили экстубацию при самостоятельном дыхании. Задние мозговые артерии (ЗМА) выделяли спустя от 4 до 6 недель после инфаркта.
Индукция субарахноидального кровоизлияния: применяли хорошо изученную модель экспериментального САК (6). Кратко, каждую мышь анестезировали (изофлуран) и голову фиксировали в стереотаксической рамке; вдоль средней линии передней части волосистой части головы делали разрез 7 мм и просверливали отверстие 0,9 мм в черепе на 4,5 мм спереди от брегмы. Спинальную иглу вводили к хиазмальной цистерне: 80 мкл артериальной крови вводили во внутричерепное пространство в течение 10 секунд. Введенная кровь была получена от отдельной мыши-донора дикого типа (посредством пункции сердца) непосредственно перед инъекцией и не содержала антикоагулянты. После инъекции разрез скальпа зашивали. Бупренорфин (0,05 мг/кг; объем от 0,5 до 1,0 мл) вводили два раза в день (сразу после хирургической процедуры САК). Ложнооперированным животным проводили идентичную хирургическую процедуру с введением стерильного физиологического раствора вместо крови. Обонятельные мозговые артерии выделяли через 2 дня после индукции САК.
Выделение и функциональная оценка резистивных артерий: обонятельные артерии мыши (первая ветвь передней мозговой артерии) и ЗМА аккуратно рассекали, канюлировали на микропипетки, растянуты до их длины in vivo и подвергнуты давлению до 45 мм рт.ст., как описано ранее (3, 6); резистивные артерии скелетных мышц мышей отделяли от мышцы кремастера, канюлировали и создавали давление до 60 мм рт.ст. Все функциональные эксперименты проводили в солевом растворе с 3-морфолинопропансульфоновой кислотой (MOPS), при 37°C без перфузии. Вазомоторные реакции на фенилэфрин (5 мкмоль/л для задних мозговых артерий, 10 мкмоль/л для артерий скелетных мышц кремастера) обеспечивали оценку жизнеспособности сосудов в начале и в конце каждого эксперимента. Артерии, не показавшие реакции сужения на фенилэфрин более или равной 25 %, были исключены.
Миогенные реакции вызывали ступенчатым повышением трансмурального давления на 20 мм рт.ст. от 20 мм рт.ст. до 80 мм рт.ст. (обонятельные артерии) или 100 мм рт.ст. (задние мозговые артерии и резистивные артерии скелетных мышц кремастера). На каждой ступени давления измеряли диаметр сосуда (диаметрактивный) после достижения стационарного состояния (5 мин). Сосуды, требующие обработки (например, CFTR(ингибитор)-172), инкубировали с реагентом в MOPS в течение 30 минут, а затем оценивали миогенные реакции в присутствии данного реагента. После завершения всех измерений диаметраактивный, буфер MOPS заменяли версией, не содержащей Ca2+, и максимальный пассивный диаметр (диаметрмакс) регистрировали на каждой ступени давления.
Миогенный тонус рассчитывали в виде процента сужения по отношению к максимальному диаметру при каждом соответствующем трансмуральном давлении: тонус (%диаметрмакс) = [(диаметрмакс-диаметрактивный)/диаметрмакс] × 100, где диаметрактивный представляет собой диаметр сосуда в MOPS, содержащем Са2+, а диаметрмакс представляет собой диаметр в MOPS без Ca2+. При анализе вазомоторных реакций на фенилэфрин применяли то же уравнение, но в таком случае диаметрактивный представляет собой диаметр сосуда в стационарном состоянии после применения данного агента, а диаметрмакс представляет собой максимальный диаметр, измеренный в условиях отсутствия Ca2+.
Измерения церебрального кровотока на основе магнитно-резонансной томографии: для оценки церебральной перфузии применяли способ неинвазивной магнитно-резонансной томографии (МРТ) (способ FAIR), как описано ранее (6). Кратко, способ FAIR выделяет перфузию как ускоренную релаксацию сигнала T1. Сигналы МРТ (Bruker Corporation Biospec 70/30 USR; Эттлинген, Германия) получали на вертикальных срезах переднего, среднего и заднего мозга, которые соответствуют переднему, смешанному и заднему кровообращению. Изображения FAIR оценивали для обозначенных областей интереса (расположение областей показано на фиг. 8) с применением стандартизированных алгоритмов и способов обработки изображений (MIPAV; Национальные институты здравоохранения, Бетесда, США; http://mipav.cit.nih.gov/).
Измерение отека на основе магнитно-резонансной томографии: отек оценивали с помощью количественного картирования T2 (12) с применением системы микро-МРТ 7 Tesla (Bruker Corporation Biospec 70/30 USR; Эттлинген, Германия). Получение карт T2 позволило получить количественные карты T2 в 9 смежных двухмерных аксиальных срезах толщиной 1 мм, покрывающих объем от переднего мозга до заднего мозга. Карты T2 были сгенерированы из T2-взвешенных изображений с временем появления эхо-сигнала от 12 до 384 мс с применением встроенного программного обеспечения Bruker посредством линейной регрессии логарифмически преобразованных величин сигналов и времени появления эхо-сигналов для каждого вокселя. Значения T2 были извлечены с применением программного обеспечения MIPAV с применением вручную нарисованных областей интереса, помещенных в карты T2 переднего, среднего и заднего мозга.
Оценка неврологической функции: Неврологическую функцию оценивали с применением модифицированной шкалы Гарсиа, как описано ранее (6). Неврологическая оценка состоит из 6 доменов: спонтанная активность, спонтанное движение всех 4 конечностей, вытягивание передних конечностей, способность к лазанию, проприоцепция тела и реакция на прикосновение к вибриссам. Двое наблюдателей провели слепое неврологическое обследование через 2 дня после САК. Максимальный балл составляет 18, что свидетельствует о нормальной неврологической функции.
Статистика: все данные представлены в виде средних значений ± стандартная ошибка среднего, где n представляет собой количество независимых измерений (т.е. оценок образцов, сосудов или объектов эксперимента). Для статистических сравнений применяли непараметрический критерий Манна-Уитни с точным вычислением p-значения для сравнения двух независимых групп. Для сравнения нескольких независимых групп применяли непараметрический однофакторный дисперсионный анализ (критерий Краскела-Уоллиса), с последующим апостериорным критерием Даннетта. Для оценки миогенных ответов и зависимостей "доза-реакция" данные были проанализированы с помощью двухфакторного дисперсионного анализа с последующим ретроспективным анализом с критерием Тьюки. Различия считали достоверными при P < 0,05. Все данные, представленные на фиг. 6, были собраны в слепых условиях; все другие данные, представленные в настоящем документе, не требовали слепого анализа и не собирались в слепых условиях.
В приведенном выше разделе "Способы и реактивы" приведены следующие ссылки на источники:
1. Cui L, Aleksandrov L, Chang XB, et al. Domain interdependence in the biosynthetic assembly of CFTR. J Mol Biol 2007; 365:981-94.
2. van Doorninck JH, French PJ, Verbeek E, et al. A mouse model for the cystic fibrosis delta F508 mutation. EMBO J 1995; 14:4403-11.
3. Van Goor F, Straley KS, Cao D, et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol 2006; 290:L1117-30.
4. French PJ, van Doorninck JH, Peters RH, et al. A delta F508 mutation in mouse cystic fibrosis transmembrane conductance regulator results in a temperature-sensitive processing defect in vivo. J Clin Invest 1996; 98:1304-12.
5. Zeiher BG, Eichwald E, Zabner J, et al. A mouse model for the delta F508 allele of cystic fibrosis. J Clin Invest 1995; 96:2051-64.
6. Wilke M, Buijs-Offerman RM, Aarbiou J, et al. Mouse models of cystic fibrosis: phenotypic analysis and research applications. J Cyst Fibros 2011; 10 Suppl 2:S152-71.
7. Lavelle GM, White MM, Browne N, McElvaney NG, Reeves EP. Animal Models of Cystic Fibrosis Pathology: Phenotypic Parallels and Divergences. Biomed Res Int 2016; 2016:5258727.
8. Nilius B, Droogmans G. Amazing chloride channels: an overview. Acta Physiol Scand 2003; 177:119-47.
9. Hoefer J, Azam MA, Kroetsch JT, et al. Sphingosine-1-phosphate-dependent activation of p38 MAPK maintains elevated peripheral resistance in heart failure through increased myogenic vasoconstriction. Circ Res 2010; 107:923-33.
10. Meissner A, Yang J, Kroetsch JT, et al. Tumor Necrosis Factor-α-Mediated Downregulation of the Cystic Fibrosis Transmembrane Conductance Regulator Drives Pathological Sphingosine-1-hosphate Signaling in a Mouse Model of Heart Failure. Circulation 2012; 125:2739 50.
11. Yang J, Hossein Noyan-Ashraf M, Meissner A, et al. Proximal Cerebral Arteries Develop Myogenic Responsiveness in Heart Failure via Tumor Necrosis Factor-α-Dependent Activation of Sphingosine-1-Phosphate Signaling. Circulation 2012; 126:196-206.
12. Yagi K, Lidington D, Wan H, et al. Therapeutically Targeting Tumor Necrosis Factor-α/Sphingosine-1-Phosphate Signaling Corrects Myogenic Reactivity in Subarachnoid Hemorrhage. Stroke 2015; 46:2260-70.
13. Kim SG. Quantification of relative cerebral blood flow change by flow-sensitive alternating inversion recovery (FAIR) technique: application to functional mapping. Magn Reson Med 1995; 34:293-301.
14. Herscovitch P, Raichle ME. What is the correct value for the brain--blood partition coefficient for water? J Cereb Blood Flow Metab 1985; 5:65-9.
15. Foley LM, Hitchens TK, Kochanek PM, Melick JA, Jackson EK, Ho C. Murine orthostatic response during prolonged vertical studies: effect on cerebral blood flow measured by arterial spinlabeled MRI. Magn Reson Med 2005; 54:798-806.
16. Foltz WD, Porter DA, Simeonov A, et al. Readout-segmented echo-planar diffusion-weighted imaging improves geometric performance for image-guided radiation therapy of pelvic tumors. Radiother Oncol 2015; 117:525-31.
17. Sharma M, Benharouga M, Hu W, Lukacs GL. Conformational and temperature-sensitive stability defects of the delta F508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J Biol Chem 2001; 276:8942-50.
18. Malik FA, Meissner A, Semenkov I, et al. Sphingosine-1-Phosphate Is a Novel Regulator of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Activity. PLoS One 2015; 10:e0130313.
19. Lin CL, Calisaneller T, Ukita N, Dumont AS, Kassell NF, Lee KS. A murine model of subarachnoid hemorrhage-induced cerebral vasospasm. J Neurosci Methods 2003; 123:89-97.
20. Parra A, McGirt MJ, Sheng H, Laskowitz DT, Pearlstein RD, Warner DS. Mouse model of subarachnoid hemorrhage associated cerebral vasospasm: methodological analysis. Neurol Res 2002; 24:510-6.
21. Aum DJ, Vellimana AK, Singh I, et al. A novel fluorescent imaging technique for assessment of cerebral vasospasm after experimental subarachnoid hemorrhage. Sci Rep 2017; 7:9126.
22. Brilstra EH, Rinkel GJ, Algra A, van Gijn J. Rebleeding, secondary ischemia, and timing of operation in patients with subarachnoid hemorrhage. Neurology 2000; 55:1656-60.
23. Macdonald RL. Delayed neurological deterioration after subarachnoid haemorrhage. Nat Rev Neurol 2014; 10:44-58.
24. Glaser EM, Van der Loos H. Analysis of thick brain sections by obverse-reverse computer microscopy: application of a new, high clarity Golgi-Nissl stain. J Neurosci Methods 1981; 4:117-25.
25. Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat 1953; 87:387-406.
РЕЗУЛЬТАТЫ
Недостаточная функция CFTR приводит к снижению церебральной перфузии
Предыдущая работа продемонстрировала, что CFTR в значительной степени регулирует миогенный тонус мозговой артерии, и выявила сильную корреляцию между экспрессией белка CFTR в мозговой артерии и церебральной перфузией (4, 6). Таким образом, механистическая концепция предполагает, что коррекция недостаточной цереброваскулярной активности CFTR (то есть снижение транспорта S1P из-за снижения экспрессии CFTR на поверхности клетки) является жизнеспособной терапевтической стратегией для нормализации нарушенной церебральной миогенной реакции и, следовательно, церебрального кровотока (4).
Для установления причинной связи между активностью CFTR мозговой артерии и церебральной перфузией применяли мышей, мутантных по CFTR. Поскольку мыши CFTR-/- были чрезвычайно чувствительны к экспериментальным стрессогенным факторам, это не позволяло проводить точные и воспроизводимые измерения эхокардиографии/ЦК. Поэтому применяли более стабильных мышей CFTRΔF508, которые имеют минимальную экспрессию и активность CFTR на клеточной поверхности (11). Это подкрепило стратегическое решение авторов изобретения применять мышей CFTRΔF508 для установления связи миогенного тонуса мозговой артерии с гемодинамическими параметрами при фенотипе с потерей функции.
Оценивали вазомоторную реактивность в задних мозговых артериях (ЗМА), чтобы напрямую сравнить фенотип CFTRΔF508 с ранее опубликованной характеристикой ответов ЗМА от мышей с нокаутом CFTR (4). Как и ожидалось, ЗМА мышей CFTRΔF508 демонстрируют повышенный миогенный тонус по сравнению с однопометниками дикого типа (фиг. 8A); величина повышения качественно аналогична величине, обнаруженной для мышей CFTR-/- (4). Реакции на фенилэфрин демонстрируют сдвиг вверх из-за повышенного миогенного тонуса (фиг. 8B); однако значения EC50 не различаются (log EC50 WT: -5,83 ± 0,21, n = 5 от 3 мышей; log EC50 CFTRΔF508: -6,00 ± 0,04, n = 6 от 4 мышей; критерий Манна-Уитни P=N.S.) и различие между кривыми устраняется путем корректировки различия в базальном тонусе (т.е. миогенном тонусе при 45 мм рт.ст.; фиг. 8C). Таким образом, мутация ΔF508 увеличивает миогенную чувствительность, но не влияет на общую сократимость. ЦК значительно ниже у мышей CFTRΔF508 (фиг. 8D); однако ни CO (фиг. 8E), ни TPR (фиг. 8F) не подвержены мутации. На основании этих наблюдений очевидно, что дефицит церебральной перфузии имеет сосудистое происхождение.
Мутация CFTRΔF508 не изменяет TPR, указывая на то, что не все сосудистые русла подвержены CFTR-зависимой регуляции. Так как TPR в первую очередь генерируется и регулируется резистивными артериями скелетных мышц (14), было проанализировано, влияет ли CFTR на миогенный тонус в резистивных артериях скелетных мышц мышей. Экспрессия мРНК CFTR примерно в 10 раз ниже в резистивных артериях скелетных мышц мышей дикого типа по сравнению с мозговыми артериями (фиг. 1G). Ингибирование активности CFTR in vitro (100 нмоль/л CFTR(ингибитор)-172, 30 минут) не влияет на миогенный тонус артерии кремастера дикого типа (фиг. 1H), что противоречит недавним наблюдениям, согласно которым CFTR(ингибитор)-172 усиливает миогенный тонус обонятельной мозговой артерии (6). В отличие от ЗМА (4), делеция гена CFTR не увеличивает миогенный тонус в резистивных артериях скелетных мышц кремастера (фиг. 1I); на самом деле миогенный тонус слегка ослаблен и соответствует более низкому среднему артериальному давлению (САД) (фиг. 9). Зависимость реакции от дозы фенилэфрина не изменяется ни ингибированием CFTR in vitro, ни делецией гена CFTR (фиг. 10).
Таким образом, настоящие эксперименты с CFTRΔF508 устанавливают: (i) причинную связь между экспрессией CFTR, миогенной реактивностью ЗМА и ЦК; (ii) CFTR модулирует миогенный тонус в определенных сосудистых руслах; и (iii) CFTR не модулирует тонус резистивных артерий скелетных мышц, что примечательно, потому что указанные артерии вносят значительный вклад в TPR. Поскольку СН и САК подавляют экспрессию CFTR в мозговых артериях одновременно с микрососудистой дисфункцией (4, 6), эти данные обеспечивают прочную механистическую основу для применения лекарственных средств, модулирующих CFTR, для специфической коррекции цереброваскулярной дисфункции и дефицита ЦК при данной патологии.
C18 увеличивает активность CFTR после TNF-зависимого подавления регуляции
Лекарственные средства, применяемые в настоящее время, модулирующие CFTR, идентифицированные с помощью высокопроизводительных проверок лекарственных средств, экспериментально и клинически применяются для смягчения последствий муковисцидоза. В соответствии с настоящим изобретением, C18 был выбран для исследований, поскольку ранее было продемонстрировано, что он напрямую взаимодействует с CFTR дикого типа и стабилизирует его, тем самым увеличивая экспрессию CFTR на плазматической мембране (15).
В соответствии с настоящим изобретением сначала было подтверждено, что C18 способен увеличивать экспрессию CFTR как мыши, так и человека. Мозговые артерии, полученные от наивных мышей, которым вводили С18 (3 мг/кг ежедневно в течение 2 дней), демонстрируют более высокие уровни экспрессии белка CFTR по сравнению с контрольными животными, получавшими носитель (фиг. 2А); C18 не влияет на экспрессию мРНК CFTR (фиг. 2В). Чтобы подтвердить действие C18 на CFTR человека, применяли гетерологичную экспрессию системы клеток фибробластов почки детеныша хомяка, стабильно экспрессирующей конструкцию CFTR человека. Обработка C18 (6 мкмоль/л; 24 ч) более чем вдвое увеличивает экспрессию белка CFTR в клетках фибробластов почек детенышей хомяка (фиг. 2C); C18 не влияет на экспрессию мРНК CFTR в данной системе (фиг. 2D). В совокупности данные согласуются с предыдущими наблюдениями о том, что C18 оказывает прямое стабилизирующее действие на экспрессию белка CFTR (15) и демонстрирует его эффективность против мышиного и человеческого CFTR.
Затем оценивали, увеличивает ли обработка C18 содержание CFTR в условиях культивирования клеток, которые в достаточной мере имитируют соответствующие патофизиологические механизмы и клеточную среду. Как было ранее продемонстрировано (4), TNF (10 нг/мл) снижает экспрессию белка CFTR в первичных сосудистых гладкомышечных клетках и, таким образом, имитирует TNF-зависимую подавляющую регуляцию CFTR, наблюдаемую при СН (4) и САК (фиг. 2E) (6). Как и ожидалось, снижение экспрессии CFTR коррелирует с ослаблением поглощения FITC-S1P (т.е. CFTR-зависимым процессом (4); фиг. 2F) и оттоком йодида, стимулированным форсколином (т.е. классической оценкой активности CFTR (13); фиг. 2G). Обработка C18 (6 мкмоль/л; 24-часовая совместная инкубация с 10 нг/мл TNF после 24-часовой инкубации с 10 нг/мл TNF) обращает вспять снижение экспрессии CFTR (фиг. 2E) и полностью восстанавливает поглощение FITC-S1P (фиг. 2F) и отток йодида (фиг. 2G). Эти результаты полностью согласуются с предположением, сделанным в настоящем документе о том, что в условиях, когда CFTR снижается, протеостатический эффект C18 увеличивает общую экспрессию CFTR и восстанавливает его активность.
Обработка C18 устраняет дефицит церебрального кровотока при сердечной недостаточности
Как ранее было опубликовано Meissner et al. (4), индукция СН стимулирует значительное снижение экспрессии белка CFTR в мозговой артерии (фиг. 3A), что совпадает с заметным увеличением миогенного тонуса ЗМА (фиг. 3B). Как и было предсказано данными на фиг. 2, обработка C18 in vivo (3 мг/кг ежедневно в течение 2 дней) восстанавливает экспрессию CFTR в мозговых артериях, полученных от мышей с СН (фиг. 3A), и одновременно нормализует миогенный тонус ЗМА (фиг. 3B). Ослабление миогенного тонуса ассоциируется с ожидаемым сдвигом в реакции на фенилэфрин из-за снижения базального тонуса; однако значения EC50 не отличаются (log EC50 ложнооперированные: -6,30 ± 0,14, n = 6 от 4 мышей; log EC50 СН: -6,25 ± 0,07, n = 5 от 3 мышей; log EC50 СН + C18: -5,94 ± 0,21, n = 5 от 4 мышей; критерий Краскела-Уоллиса P=N.S.), и кривые перекрываются после поправки на разницу в базальном тонусе (фиг. 11). Обработка C18 не влияет на миогенный тонус ЗМА или реакции на фенилэфрин у ложнооперированных мышей (фиг. 12). Применяя ЗМА, полученные от мышей с нокаутом CFTR, авторами настоящего изобретения было подтверждено, что C18 опосредует его ослабляющее действие на миогенный тонус путем воздействия на CFTR. Как и ожидалось, ЗМА от мышей с нокаутом CFTR демонстрируют повышенный миогенный тонус, который не чувствителен к обработке C18 in vivo (фиг. 3C); обработка C18 не влияет на реакцию на фенилэфрин (фиг. 13).
На системном уровне индукция СН значительно снижает CO по сравнению с ложнооперированными контрольными животными (фиг. 3D; измерено через 6 недель после инфаркта); TPR увеличивается как компенсаторная реакция (фиг. 3E), чтобы предотвратить значительное снижение САД (фиг. 3F) (3, 4). Несмотря на относительно небольшое сокращение САД, ЦК заметно снижается (фиг. 3G). Обработка C18 in vivo восстанавливает ЦК у мышей с СН (фиг. 3G), точно коррелируя с нормализацией миогенного тонуса ЗМА (фиг. 3B). C18 не улучшает повреждение сердечно-сосудистой системы, вызванное процедурой перевязки левой передней нисходящей коронарной артерии: поэтому улучшение ЦК может быть однозначно отнесено к сосудистому механизму. Кроме того, поскольку C18 не влияет на TPR или САД (фиг. 3), C18-опосредованное восстановление ЦК должно представлять собой локализованный микрососудистый эффект, который не зависит от изменений системных гемодинамических параметров. Этот терапевтический профиль идеально согласуется с ранее описанным применением этанерцепта (3).
Помимо поддержания постоянной перфузии при колебаниях системного давления, миогенная реакция также защищает хрупкое капиллярное русло от повреждающего давления (16) и поддерживает капиллярное гидростатическое давление на уровнях, которые сводят к минимуму образование отеков (17). В этом контексте терапевтическое снижение миогенного тонуса церебральной артерии даже с повышенного уровня, который наблюдается при СН, потенциально может вызвать контрпродуктивный побочный эффект образования отека мозга. Таким образом, неинвазивную визуализацию отека применяли для подтверждения того, что ни лечение СН, ни C18 в контексте СН не вызывает явного отека в какой-либо области мозга (фиг. 4).
Обработка C18 устраняет дефицит церебрального кровотока при САК
Предыдущая работа демонстрирует, что САК имеет такой же цереброваскулярный фенотип, что и СН, в том, что (i) экспрессия белка CFTR в церебральной артерии снижена; (ii) повышен миогенный тонус мозговой артерии; и (iii) церебральная перфузия снижена (6). Как в СН (фиг. 3), обработка C18 in vivo (3 мг/кг ежедневно в течение 2 дней) восстанавливает экспрессию CFTR в мозговых артериях мышей с САК (фиг. 5A) и одновременно нормализует миогенный тонус обонятельной мозговой артерии (фиг. 5B). Ослабление миогенного тонуса связано с ожидаемым сдвигом базовой линии в ответной реакции на фенилэфрин из-за снижения базального тонуса; однако значения EC50 не различаются (log EC50 ложнооперированные: -5,52 ± 0,23, n = 5 от 3 мышей; log EC50 САК: -6,01 ± 0,16, n = 6 от 6 мышей; log EC50 САК + C18: -6,01 ± 0,31, n = 5 от 4 мышей; критерий Краскела-Уоллиса P=N.S.), и кривые перекрываются после поправки на разницу в базальном тонусе (фиг. 14). Обработка C18 не влияет на миогенный тонус обонятельных мозговых артерий или реакции на фенилэфрин у ложнооперированных мышей (фиг. 15). Применяя обонятельные мозговые артерии, полученные от мышей с нокаутом CFTR, авторами настоящего изобретения было подтверждено, что C18 опосредует его ослабляющее действие на миогенный тонус путем нацеливания на CFTR: как и ожидалось, обонятельные мозговые артерии от мышей с нокаутом CFTR проявляют повышенный миогенный тонус, который не поддается лечению C18 in vivo. (фиг. 5C); обработка C18 не влияет на чувствительность к фенилэфрину в данных артериях (фиг. 16). Важно отметить, что обработка C18 in vivo восстанавливает ЦК (фиг. 5D), что снова коррелирует с нормализацией миогенного тонуса обонятельной мозговой артерии (фиг. 5B).
Лечение лумакафтором устраняет дефицит церебрального кровотока при САК
Данные по C18 дополняются сообщениями о лечении с применением лумакафтора, корректора CFTR, клинически значимого аналога C18, одобренного Управлением по контролю качества пищевых продуктов и лекарственных средств США для лечения муковисцидоза, в сочетании с Ивакафтором (т.е. Orkambi®), усилителем CFTR. Впервые было подтверждено, что лумакафтор способен увеличивать экспрессию CFTR как мыши, так и человека: как наблюдали для C18 (фиг. 2), лумакафтор увеличивает экспрессию белка CFTR в мозговых артериях мышей (мыши, которым вводили 3 мг/кг ежедневно в течение 2 дней) и клетках фибробластов почек детенышей хомяка, стабильно экспрессирующих CFTR человека (фиг. 6). В обоих случаях экспрессия мРНК CFTR не изменилась, что указывает на нетранскрипционный механизм (фиг. 6). В соответствии с настоящими данными по C18 при САК (фиг. 5), лечение лумакафтором in vivo (3 мг/кг в день в течение 2 дней) восстанавливает экспрессию CFTR в мозговых артериях мышей с САК (фиг. 6) и одновременно нормализует миогенный тонус обонятельной мозговой артерии (фиг. 6) и церебральную перфузию (фиг. 6). Лумакафтор не влияет на чувствительность к фенилэфрину (фиг. 17), и, следовательно, значения EC50 не различаются (log EC50 ложнооперированные: -5,79 ± 0,09, n = 7 от 4 мышей; log EC50 САК: -5,93 ± 0,19, n = 6 от 3 мышей; log EC50 САК + Лум: -5,90 ± 0,18, n = 7 из 4 мышей; критерий Краскела-Уоллиса P=N.S.).
C18 и лумакафтор уменьшают повреждение нейронов при САК
Повреждение нейронов, вызванное САК, может быть легко охарактеризовано стандартными гистологическими способами (окрашивание фтор-нефритом и активированной каспазой-3) и простым неврологическим тестированием (модифицированная шкала Гарсиа) (6): эти способы применяли, чтобы определить, коррелирует ли восстановление нормальной миогенной реакции и ЦК с улучшением неврологического исхода. В самом деле, как C18, так и лумакафтор резко снижают повреждение нейронов при САК, что оценивали по окрашиванию активированной каспазой-3 и фтор-нефритом (фиг. 7); уменьшение повреждения нейронов у мышей, получавших C18, коррелирует с улучшением неврологической функции (фиг. 7E). В частности, мыши с САК набрали более низкие баллы по модифицированному тесту неврологической функции Гарсиа, чем ложнооперированные мыши; однако у мышей с САК, получавших C18, показатели неврологической функции были сопоставимы с ложнооперированными мышами.
Обсуждение
Предыдущая работа усовершенствовала первоначальное открытие о том, что ингибирование CFTR увеличивает миогенный тонус микрососудов, в новую механистическую концепцию (4, 6): по своей сути CFTR критически регулирует деградацию S1P и, таким образом, в значительной степени регулирует цереброваскулярный тонус в здоровом состоянии и при болезни (4). Настоящее изобретение применяет эти знания для лечения СН и САК. Настоящее изобретение обеспечивает новый класс терапевтических средств для лечения заболеваний сердечно-сосудистой системы. В настоящем изобретении обеспечена проверка in vivo того, что CFTR представляет собой цереброваскулярный регулятор, и подтверждается, что его можно применять для улучшения церебральной перфузии как при СН, так и при САК. Будучи первым новым лекарством-мишенью для микрососудов, появившимся за последние годы, терапевтические средства, которые усиливают функцию CFTR, могут представлять собой неиспользованный ресурс для управления широким спектром патологий, которые вызывают цереброваскулярную дисфункцию и дефицит церебральной перфузии.
Проксимальные ЗМА были стратегически выбраны для исследований СН, представленных в настоящем документе, и обонятельные мозговые артерии были выбраны для исследований САК в соответствии с настоящим примером, чтобы напрямую сравнить способы лечения, нацеленные на CFTR, с предыдущими исследованиями (3, 4, 6). Хотя ЗМА и обонятельные артерии происходят из разных областей церебральной микроциркуляции (то есть, задней и передней, соответственно) и демонстрируют различия в базовых кривых миогенного тонуса, тем не менее, в терминах патологических сигнальных механизмов, которые увеличивают миогенную чувствительность, они ведут себя одинаково (3, 4, 6). Сравнимый успех способов лечения, нацеленных на CFTR, для ЗМА и обонятельных артерий предполагает, что CFTR регулирует сосудистую реактивность в целом по всей церебральной микроциркуляции.
Очевидно, что CFTR не регулирует реактивность сосудов во всех сосудистых руслах, как показали сравнения (i) резистивных артерий мозговых и скелетных мышц, (ii) измерения TPR у мутантных мышей CFTR и мышей дикого типа и (iii) отсутствие эффекта обработки C18 на TPR. Различия в экспрессии CFTR, измеренные с помощью количественной ПЦР, объясняют, почему ингибирование CFTR регулирует миогенный тонус в мозговых артериях, но не играет очевидной роли в резистивных артериях скелетных мышц. В совокупности настоящие данные демонстрируют, что терапевтические средства CFTR будут иметь ограниченные эффекты, которые специфичны для дискретных микрососудистых руслах (например, церебральной микроциркуляции). Это свойство является предпосылкой для эффективного перераспределения кровотока и дает значительное терапевтическое преимущество перед общими сосудорасширяющими средствами, которые могут опасно снижать артериальное давление за счет неизбирательного расширения сосудов.
Интересно, что в то время как острое/химическое ингибирование CFTR не имело действия на резистивные артерии кремастера, делеция гена CFTR зародышевой линии немного снижала миогенный тонус в этом специфическом микрососудистом русле. Поскольку глобальная делеция CFTR приводит к сложному сердечно-сосудистому фенотипу (18), это небольшое несоответствие объясняется непрямым и/или адаптивным ответом. Авторы настоящего изобретения ранее продемонстрировали эффективность ингибитора CFTR как в клеточной (4), так и в изолированной артериальной (6) системах. Таким образом, можно сделать вывод, что CFTR играет минимальную роль в модуляции миогенного тонуса артерии кремастера.
Миогенный ответ является основой ауторегуляции ЦК, постоянного согласования сосудистого сопротивления с преобладающим трансмуральным давлением. Таким образом, вмешательства, направленные на увеличение патологически сниженного ЦК, должны стремиться к восстановлению нормальной миогенной реактивности, а не к неизбирательному подавлению сужения кровеносных сосудов. Ожидается, что посредством специфической нормализации миогенной реактивности при СН и САК, способы лечения по настоящему изобретению, нацеленные на CFTR, сохранят как ауторегуляцию ЦК, так и реакцию на местные вазоактивные стимулы (например, нейрососудистое сцепление) in vivo.
Миогенная реакция не только определяет перфузию головного мозга через свою ауторегуляторную функцию, но и поддерживает капиллярное давление на уровнях, которые минимизируют повреждение и образование отеков (16, 17). Следовательно, снижение миогенной реактивности может вызвать образование отека, позволяя дополнительному давлению проникать в капиллярные русла. В этой связи, поскольку S1P в значительной степени регулирует проницаемость гематоэнцефалического барьера (19), CFTR-зависимые изменения в передаче сигналов S1P могут вызывать отек из-за изменений барьерной функции. Таким образом, было крайне важно исключить отек как серьезный отрицательный эффект способа лечения по настоящему изобретению. Действительно, лечение корректором CFTR (C18) не вызывает отек у мышей с сердечной недостаточностью. Это подтверждает, что: (i) гидростатическое давление остается в допустимых пределах (этого следовало ожидать, поскольку измерения ЦК и САД сопоставимы с ложнооперированными животными); и (ii) функция гематоэнцефалического барьера сохраняется во время лечения.
До настоящего времени ни цереброваскулярная экспрессия CFTR в артериях головного мозга человека, ни перфузия мозга не определялась у пациентов с муковисцидозом. Клинически муковисцидоз не связан с когнитивными нарушениями или ишемическим повреждением; однако это не исключает снижения церебральной перфузии: как было продемонстрировано ранее (3, 20), мозг может переносить умеренное снижение ЦК без пересечения очевидного порога повреждения. Таким образом, не обязательно ожидать когнитивных нарушений у молодых и относительно здоровых пациентов с муковисцидозом. Хотя нет сомнений в том, что гипоперфузия значительно способствует снижению когнитивных функций (1) и церебральному повреждению (21), для преодоления клинически незаметным снижением ЦК порога повреждения, необходимы другие "удары по системе", включая старение и воспалительные реакции, вызванные заболеванием сердечно-сосудистой системы или САК.
Доступ к образцам мозговых артерий человека затруднен: таким образом, авторы настоящего изобретения полагались на перенос данных, включающий систематические сравнения экспрессии CFTR в брыжеечной артерии и артерии скелетных мышц у человека и мыши (22), чтобы установить, может ли CFTR экспрессироваться в мозговых артериях человека. В брыжеечных артериях человека присутствует белок CFTR, и его функциональное ингибирование приводит к ожидаемому увеличению миогенной реактивности (22): следовательно, подтверждается, что CFTR является модулятором миогенной реактивности резистивной артерии человека. В отличие от брыжеечных артерий человека, резистивные артерии скелетных мышц человека не содержат определяемого белка CFTR и, следовательно, нечувствительны к ингибированию CFTR (22). Поскольку функциональный профиль брыжеечных артерий и артерий скелетных мышц для человека и мышей перекрывается (то есть CFTR модулирует брыжеечные, но не резистивные артерии скелетных мышц; фиг. 1 (4, 22)), можно сделать вывод, что функциональный профиль также перекрывается для церебральной микроциркуляции. В отсутствие прямых оценок это предположение в настоящее время является наиболее разумным основанием для прогнозирования эффекта модуляторов CFTR на цереброваскулярную систему человека.
Соединения корректоров в настоящее время применяют для лечения муковисцидоза, вызванного мутацией CFTR ΔF508: эти соединения помогают в сопровождении неправильно свернутых мутантных белков CFTR к плазматической мембране. Ни патология недостаточного переноса CFTR, ни терапевтическое вмешательство в сопровождение не применимы к СН и САК, где происходит основанное на транскрипции снижение экспрессии CFTR. Таким образом, было крайне важно подтвердить, что модуляторы CFTR, предпочтительно корректоры CFTR, в частности C18 и лумакафтор, увеличивают количество CFTR дикого типа в экспериментальных условиях, где экспрессия белка CFTR снижается. Действительно, настоящие данные показывают, что модуляторы CFTR, предпочтительно корректоры CFTR, в частности C18 и лумакафтор, также увеличивают количество CFTR дикого типа посредством нетранскрипционного механизма. Этот протеостатический эффект стабилизирует локализованный на клеточной поверхности CFTR от интернализации и последующей деградации (15). Со временем стабилизация CFTR на клеточной поверхности повышает общие уровни экспрессии CFTR, поскольку меньшее количество CFTR на клеточной поверхности усваивается и обрабатывается механизмами деградации. Терапевтическое восстановление экспрессии CFTR в моделях заболевания, применяемых в настоящем изобретении, совпадает с ослаблением миогенного тонуса, нормализацией ЦК и САК, уменьшением повреждения нейронов.
Поскольку CFTR представляет собой критический регуляторный белок в нескольких органах, в первую очередь в легких, необходимо также учитывать влияние терапевтических средств CFTR на бессосудистые ткани. Примечательно, что СН подавляет экспрессию CFTR в эпителиальных клетках концевых бронхиол легких (4), и, таким образом, снижение CFTR не ограничивается микроциркуляцией в этих условиях (т.е. СН вызывает "фенотип муковисцидоза", который потенциально вызывает полиорганную недостаточность). Следовательно, терапевтические средства CFTR могут обладать широкими и существенными преимуществами для пациентов с сердечной недостаточностью, помимо улучшения церебральной перфузии. В этом контексте тот факт, что несколько серьезных сопутствующих заболеваний являются общими как для СН, так и для муковисцидоза (например, вторичная легочная гипертензия (24)), порождает провокационное, но все еще спорное утверждение о том, что определенные вторичные патологии при СН либо вызваны, либо усугублены недостаточной активностью CFTR и, следовательно, корректируются терапевтическими средствами CFTR.
Выводы
Настоящее изобретение обеспечивает первое доказательство того, что CFTR регулирует цереброваскулярную реактивность и, следовательно, перфузию головного мозга: это позиционирует CFTR как "главный переключатель" в контроле церебральной перфузии. Как СН, так и САК хронически снижают перфузию головного мозга, подавляя экспрессию белка CFTR в мозговой артерии, тем самым нарушая ауторегуляторный контроль. В настоящем изобретении раскрыто, что клинически доступные терапевтические средства CFTR могут восстанавливать экспрессию CFTR в мозговой артерии, реактивность сосудов и перфузию головного мозга. Примечательно, что данный терапевтический эффект локализован в церебральной микроциркуляции, поскольку CFTR действительно модулирует реактивность периферических артерий, участвующих в контроле артериального давления. Таким образом, терапия CFTR становится ценным клиническим инструментом для лечения цереброваскулярной дисфункции, нарушения перфузии головного мозга и повреждения нейронов. В настоящем изобретении раскрыта эффективность терапевтических средств CFTR для предотвращения и улучшения дефицита перфузии головного мозга, в частности, вызванного СН и САК.
В настоящем документе приведены следующие ссылки на источники, за исключением вышеприведенного раздела "Способы и реактивы", для которого приведен отдельный список ссылок на источники.
1. Román GC. Brain hypoperfusion: a critical factor in vascular dementia. Neurol Res 2004;26:454-8.
2. Román GC. Stroke, cognitive decline and vascular dementia: the silent epidemic of the 21st century. Neuroepidemiology 2003;22:161-4.
3. Yang J, Hossein Noyan-Ashraf M, Meissner A, et al. Proximal Cerebral Arteries Develop Myogenic Responsiveness in Heart Failure via Tumor Necrosis Factor-α-Dependent Activation of Sphingosine-1-Phosphate Signaling. Circulation 2012;126:196-206.
4. Meissner A, Yang J, Kroetsch JT, et al. Tumor Necrosis Factor-α-Mediated Downregulation of the Cystic Fibrosis Transmembrane Conductance Regulator Drives Pathological Sphingosine-1-Phosphate Signaling in a Mouse Model of Heart Failure. Circulation 2012;125:2739-50.
5. Meissner A, Visanji NP, Momen MA, et al. Tumor Necrosis Factor-α Underlies Loss of Cortical Dendritic Spine Density in a Mouse Model of Congestive Heart Failure. J Am Heart Assoc 2015;4:e001920.
6. Yagi K, Lidington D, Wan H, et al. Therapeutically Targeting Tumor Necrosis Factor-α/Sphingosine-1-Phosphate Signaling Corrects Myogenic Reactivity in Subarachnoid Hemorrhage. Stroke 2015;46:2260-70.
7. Murdaca G, Spanò F, Contatore M, et al. Infection risk associated with anti-TNF-α agents: a review. Expert Opin Drug Saf 2015;14:571-82.
8. Kroetsch JT, Levy AS, Zhang H, et al. Constitutive smooth muscle tumour necrosis factor regulates microvascular myogenic responsiveness and systemic blood pressure. Nat Commun 2017;8:14805.
9. Blankenbach KV, Schwalm S, Pfeilschifter J, Meyer Zu Heringdorf D. Sphingosine-1-Phosphate Receptor-2 Antagonists: Therapeutic Potential and Potential Risks. Front Pharmacol 2016;7:167.
10. Boyle MP, Bell SC, Konstan MW, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med 2014;2:527-38.
11. van Doorninck JH, French PJ, Verbeek E, et al. A mouse model for the cystic fibrosis delta F508 mutation. EMBO J 1995;14:4403-11.
12. Loubinoux I, Volk A, Borredon J, et al. Spreading of vasogenic edema and cytotoxic edema assessed by quantitative diffusion and T2 magnetic resonance imaging. Stroke 1997;28:419-26.
13. Malik FA, Meissner A, Semenkov I, et al. Sphingosine-1-Phosphate Is a Novel Regulator of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Activity. PLoS One 2015;10:e0130313.
14. Henrion D. Pressure and flow-dependent tone in resistance arteries. Role of myogenic tone. Arch Mal Coeur Vaiss 2005;98:913-21.
15. Okiyoneda T, Veit G, Dekkers JF, et al. Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat Chem Biol 2013;9:444-54.
16. Bidani AK, Griffin KA, Williamson G, Wang X, Loutzenhiser R. Protective importance of the myogenic response in the renal circulation. Hypertension 2009;54:393-8.
17. Moien-Afshari F, Skarsgard PL, McManus BM, Laher I. Cardiac transplantation and resistance artery myogenic tone. Can J Physiol Pharmacol 2004;82:840-8.
18. Van Iterson EH, Karpen SR, Baker SE, Wheatley CM, Morgan WJ, Snyder EM. Impaired cardiac and peripheral hemodynamic responses to inhaled β2-agonist in cystic fibrosis. Respir Res 2015;16:103.
19. Prager B, Spampinato SF, Ransohoff RM. Sphingosine 1-phosphate signaling at the blood-brain barrier. Trends Mol Med 2015;21:354-63.
20. Baron JC. Perfusion thresholds in human cerebral ischemia: historical perspective and therapeutic implications. Cerebrovasc Dis 2001;11 Suppl 1:2-8.
21. Jefferson AL, Tate DF, Poppas A, et al. Lower cardiac output is associated with greater white matter hyperintensities in older adults with cardiovascular disease. J Am Geriatr Soc 2007;55:1044-8.
22. Hui S, Levy AS, Slack DL, et al. Sphingosine-1-Phosphate Signaling Regulates Myogenic Responsiveness in Human Resistance Arteries. PLoS One 2015;10:e0138142.
23. Balch WE, Roth DM, Hutt DM. Emergent properties of proteostasis in managing cystic fibrosis. Cold Spring Harb Perspect Biol 2011;3:a004499.
24. Roy R, Couriel JM. Secondary pulmonary hypertension. Paediatr Respir Rev 2006;7:36-44.







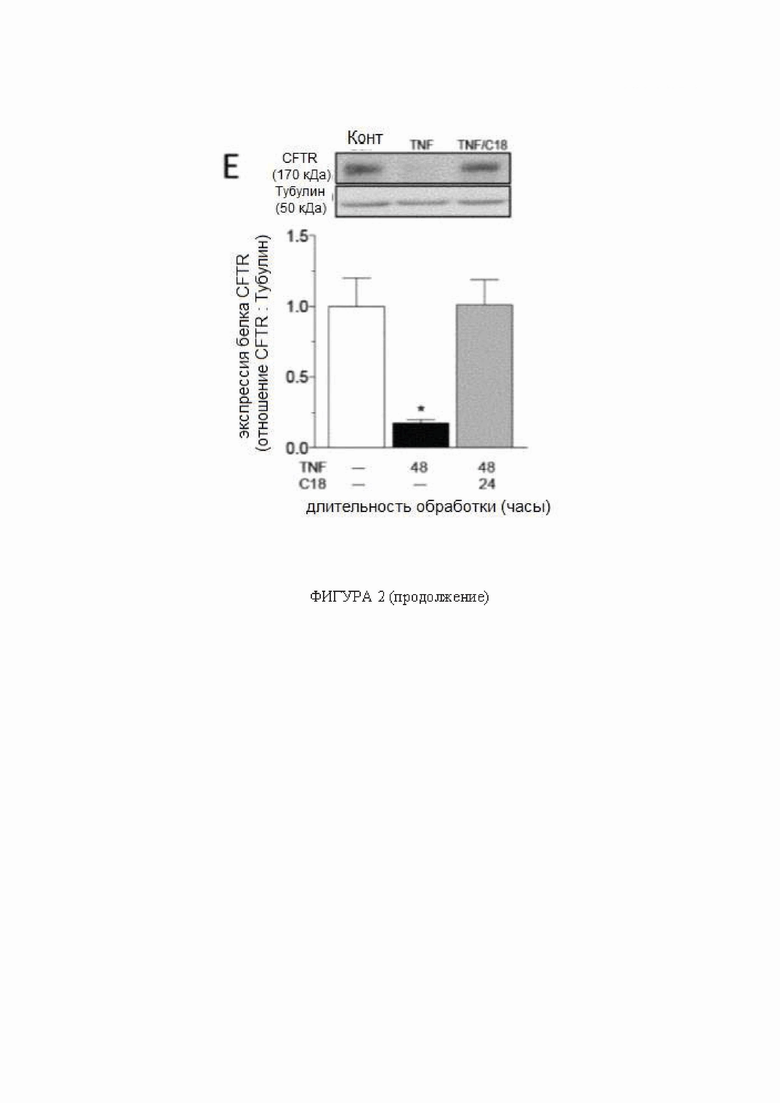

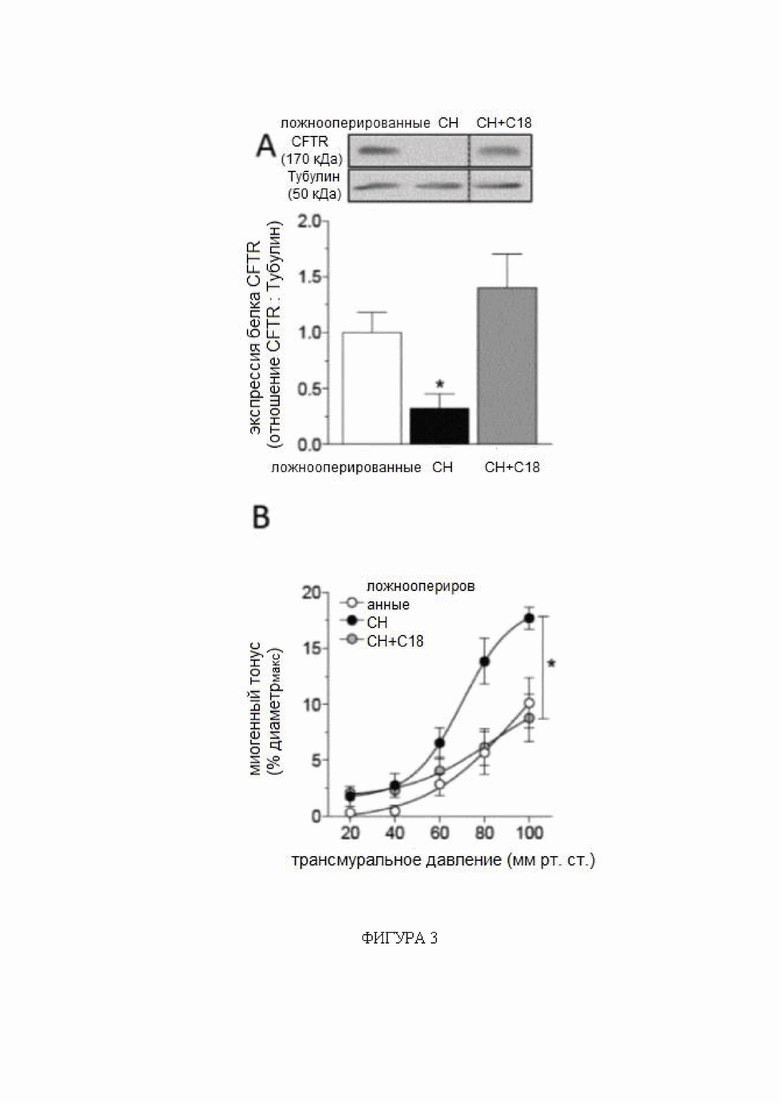
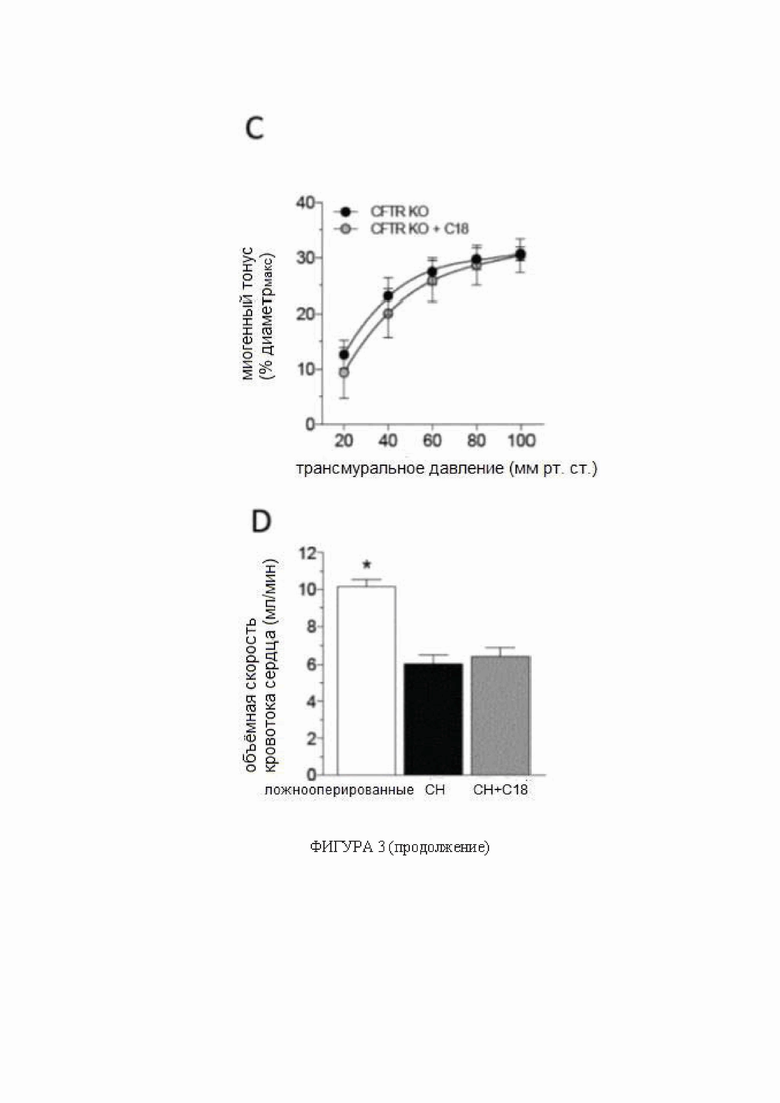
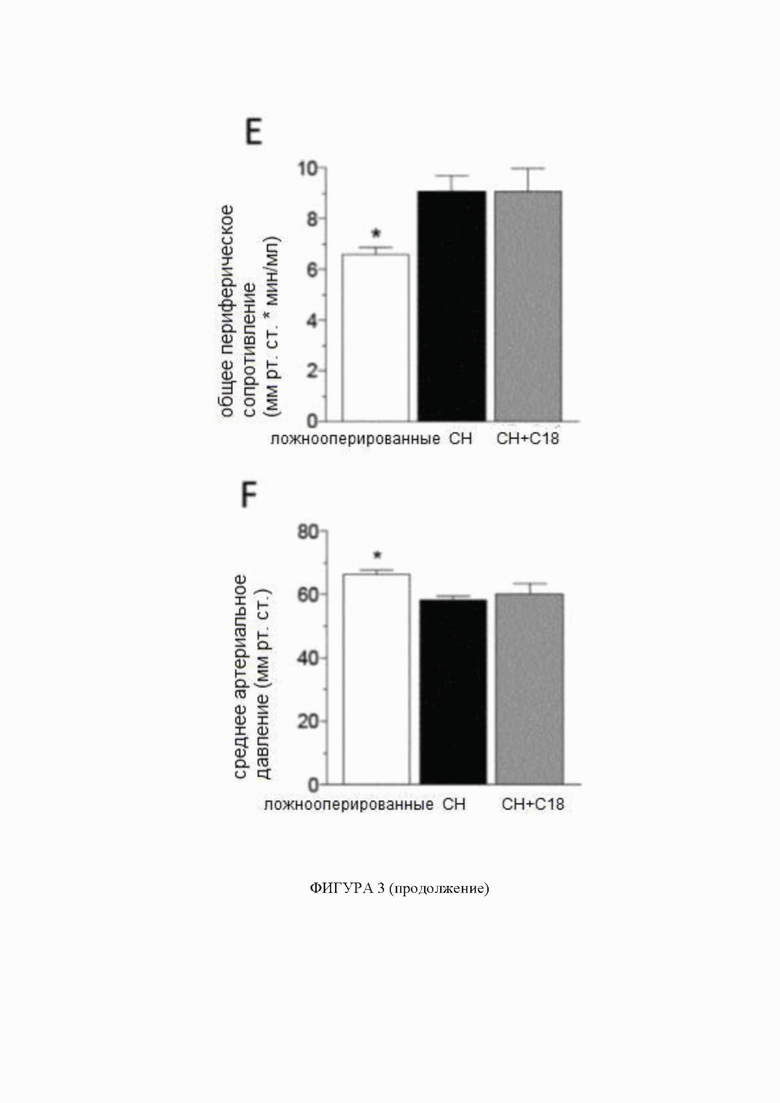
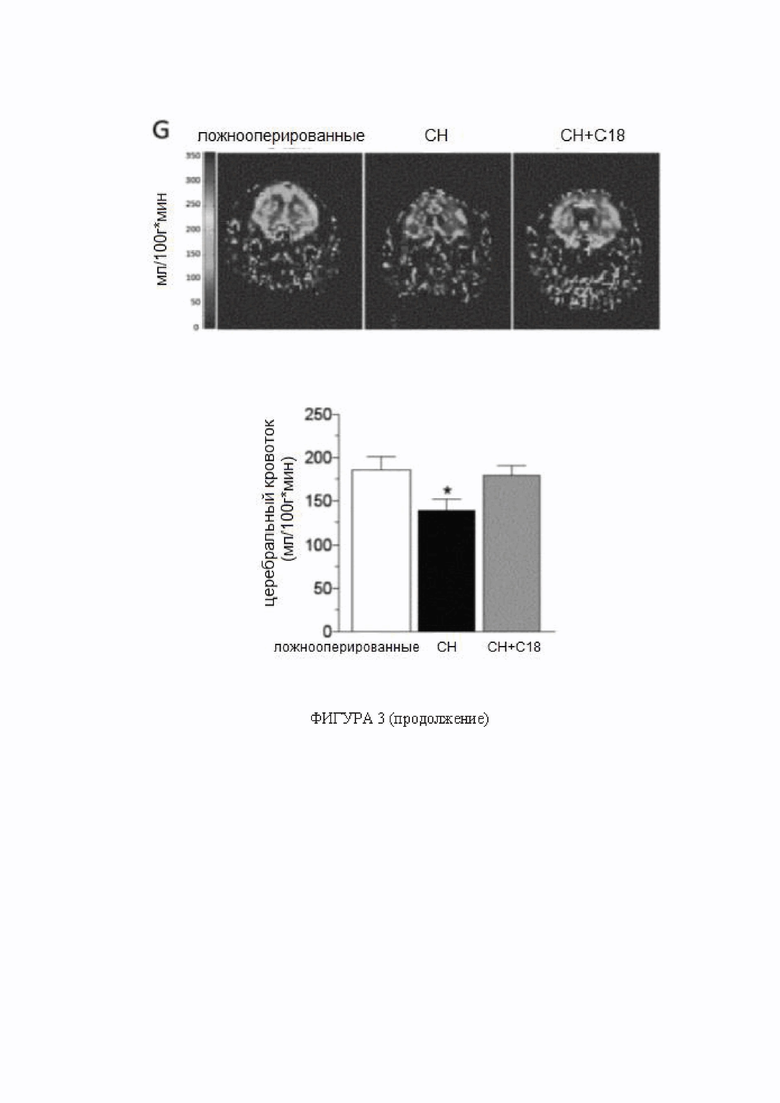

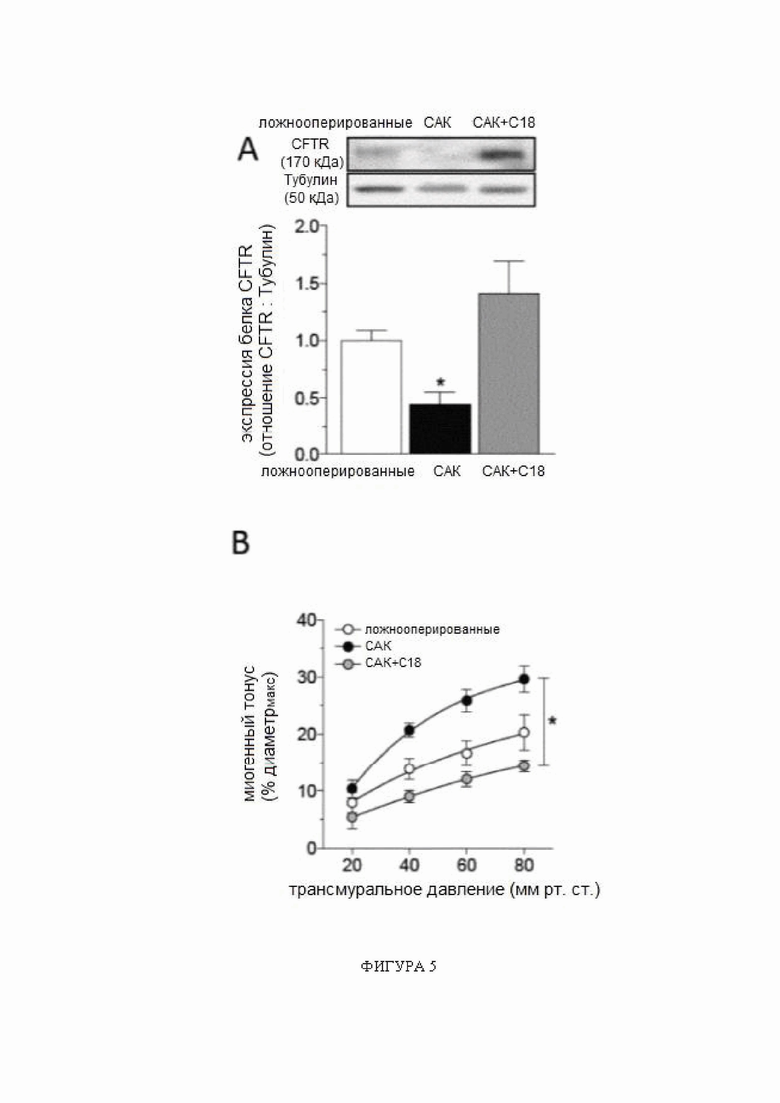
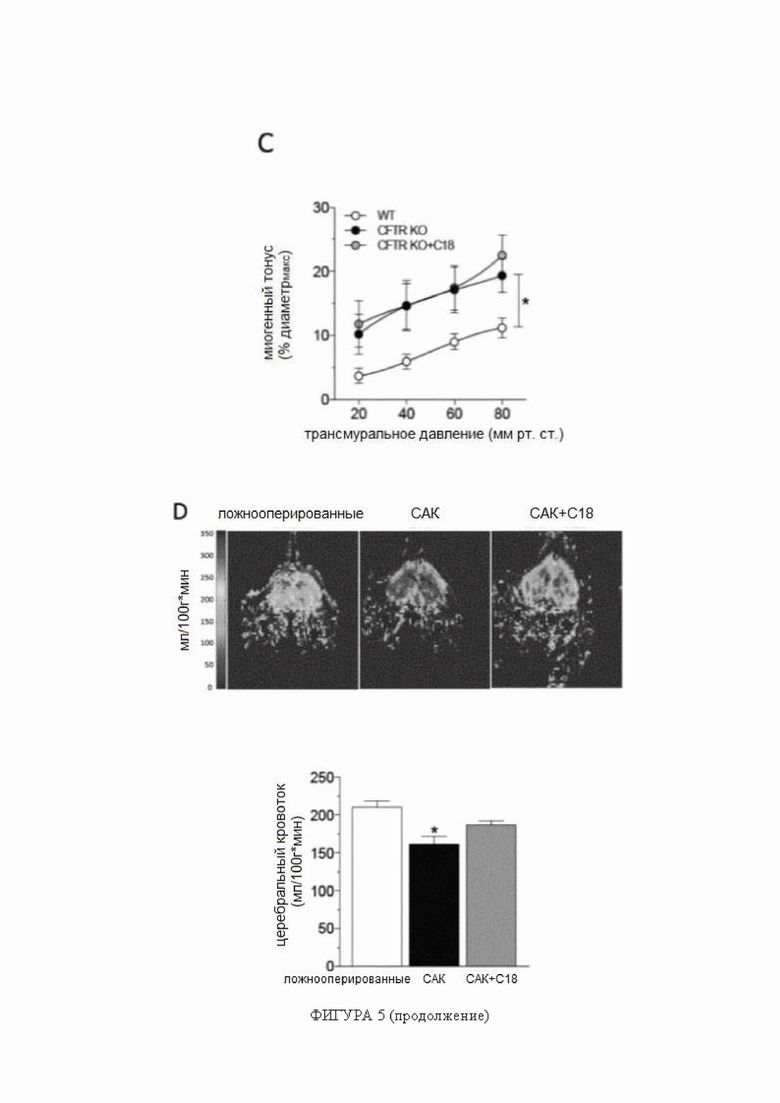








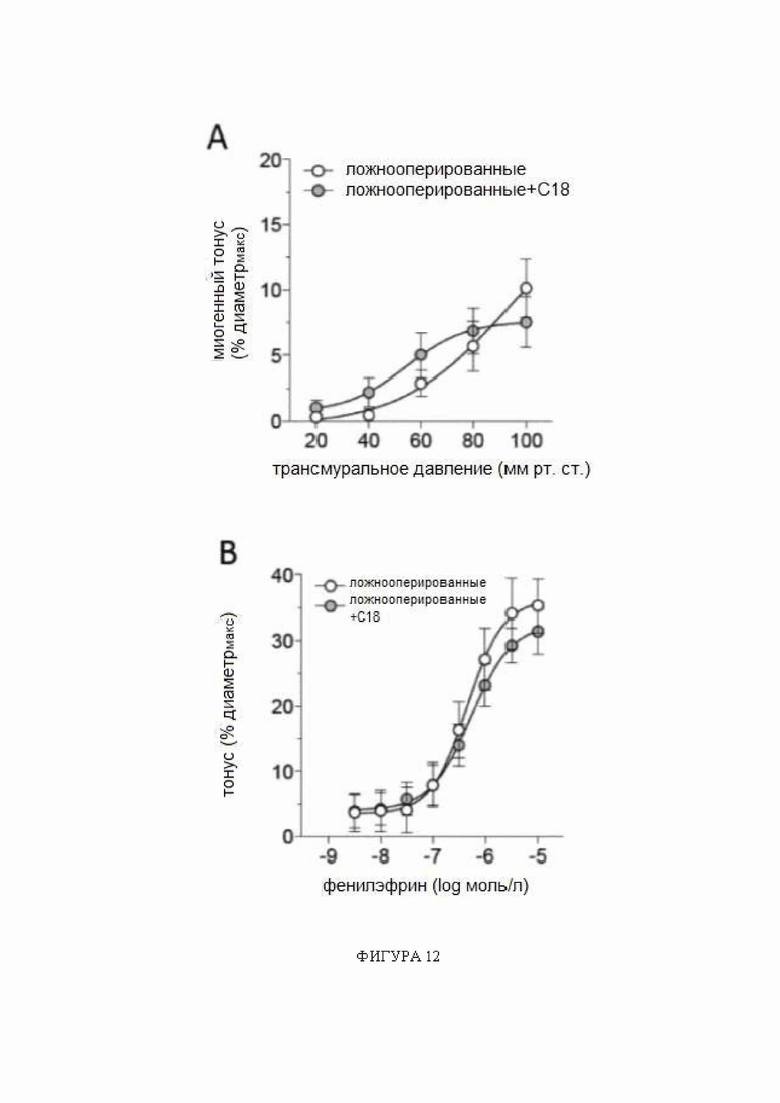





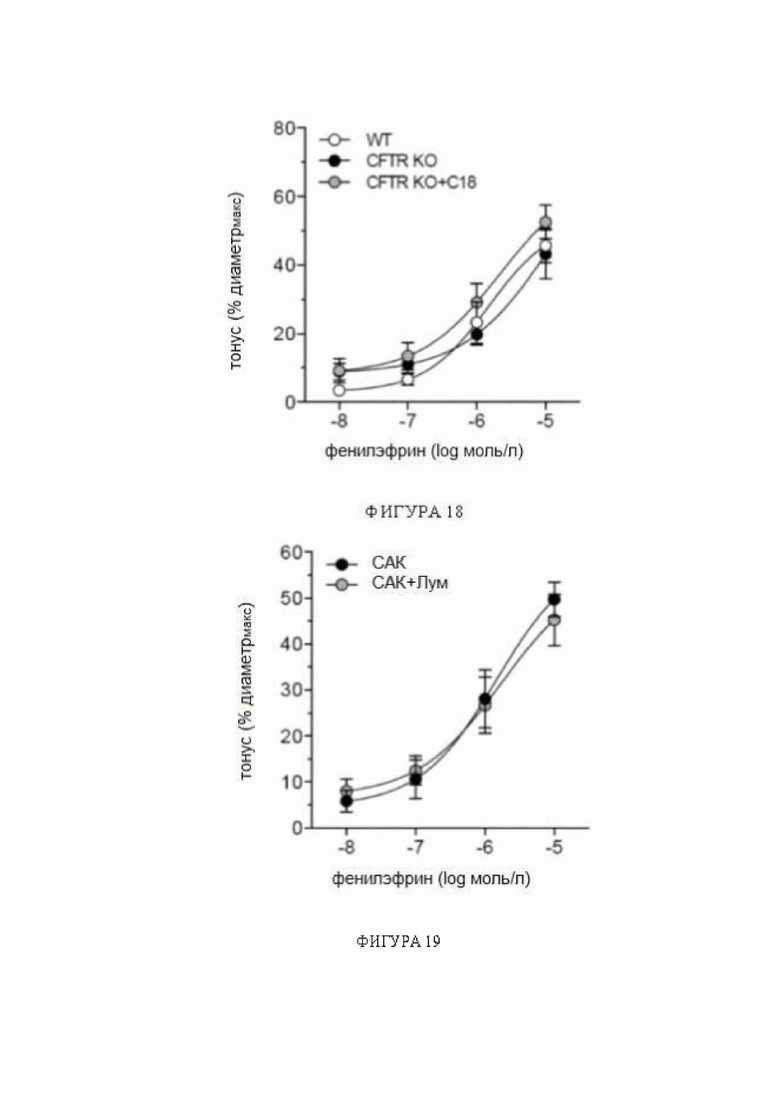
Изобретение относится к области медицины и фармацевтики и применения корректора CFTR (регулятор трансмембранной проводимости при муковисцидозе) для предотвращения и/или лечения снижения или нарушения перфузии головного мозга, где корректор CFTR представляет собой производное циклопропанкарбоксамида, выбранное из группы, состоящей из лумакафтора, тезакафтора, C18 и их фармацевтически приемлемых солей и сольватов. Использование изобретения позволяет эффективно предотвращать и/или лечить снижение или нарушение перфузии головного мозга. 2 з.п. ф-лы, 19 ил., 1 пр.
1. Применение корректора CFTR (регулятор трансмембранной проводимости при муковисцидозе) для предотвращения и/или лечения снижения или нарушения перфузии головного мозга, где корректор CFTR представляет собой производное циклопропанкарбоксамида, выбранное из группы, состоящей из лумакафтора, тезакафтора, C18 и их фармацевтически приемлемых солей и сольватов.
2. Применение корректора CFTR по п. 1, где корректор CFTR представляет собой лумакафтор.
3. Применение корректора CFTR по п. 1 или 2, где снижение перфузии головного мозга связано с состоянием, выбранным из группы, состоящей из заболевания сердца, сердечной недостаточности, субарахноидального кровоизлияния, внезапной нейросенсорной тугоухости, сосудистой деменции, артериальной гипертензии, ишемического инсульта, геморрагического инсульта, диабета и болезни Альцгеймера.
| WO 2016105484 A1, 30.06.2016 | |||
| US 20050245433 A1, 03.11.2005 | |||
| WO 2010068863 A2, 17.06.2010 | |||
| Lidington D, Kroetsch JT, Bolz S-S | |||
| Cerebral artery myogenic reactivity: The next frontier in developing effective interventions for subarachnoid hemorrhage | |||
| Journal of Cerebral Blood Flow & Metabolism | |||
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |
| RU 2011142297 A, 27.04.2013. | |||
