[0001] Настоящая заявка подана 20 февраля 2018 года как международная патентная заявка PCT и по ней испрашивается приоритет по временной патентной заявке США № 62/461672, поданной 21 февраля 2017 года, и временной патентной заявке США № 62/595190, поданной 6 декабря 2017 года, описание каждой из которых включено в настоящее описание в качестве ссылки в полном объеме.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
[0002] Настоящее изобретение относится к способам лечения рака легких, включающим введение нуждающемуся в этом индивидууму терапевтически эффективного количества антитела, специфически связывающегося с рецептором программируемой гибели клеток 1 (PD-1).
УРОВЕНЬ ТЕХНИКИ
[0003] Рак легких является одним из наиболее часто диагностируемых злокачественных новообразований и основной причиной смерти, связанной со злокачественными новообразованиями, во всем мире (Bray et al 2013, Int. J. Cancer 132:1133-45; Siegel et al 2016, CA Cancer J. Clin. 66: 7-30). На немелкоклеточный рак легких (NSCLC) приходится 80-85% всех случаев рака легких, и он представляет собой несколько гистологических подтипов, наиболее распространенные из которых включают аденокарциному (40-60%) и плоскоклеточную карциному (30%). Большинство пациентов с NSCLC на момент диагностики имеют злокачественное новообразование на поздней стадии (Leighl 2012, Curr. Oncol. 19:S52-8). При использовании химиотерапии у этих пациентов наблюдают медиану общей выживаемости (OS) до 12-18 месяцев и 5-летний коэффициент выживаемости приблизительно 18% (Leighl 2012, Curr. Oncol. 19:S52-8; Siegel et al 2016, CA Cancer J. Clin. 66: 7-30).
[0004] Системная терапия с использованием двухкомпонентых схем химиотерапии препаратами на основе платины с поддерживающей терапией или без нее до недавнего времени являлась стандартным лечением первой линии для всех пациентов с NSCLC на поздней стадии, опухоли у которых не содержат мутацию рецептора эпидермального фактора роста (EGFR), транслокацию киназы анапластической лимфомы (ALK) или мутацию онкогенной рецепторной тирозинкиназы C-ros (ROS1) (Besse et al 2014, Ann. Oncol. 25: 1475-84; Ettinger et al 2016, J. Natl. Compr. Canc. Netw. 14: 255-64; Reck et al 2014, Ann. Oncol. 25 Suppl 3: iii27-39). Несмотря на начальную терапию с использованием двухкомпонентных схем химиотерапии препаратами на основе платины, заболевание зачастую прогрессирует, а варианты дополнительного лечения ограничены. Таким образом, необходимы более новые терапевтические подходы, которые улучшат длительную выживаемость и качество жизни (QOL) у пациентов с NSCLC на поздней стадии.
[0005] В последние годы проводятся исследования иммунотерапии в качестве потенциального терапевтического подхода, который улучшит длительную выживаемость и QOL у пациентов с NSCLC на поздней стадии. Опухоли модулируют иммунный ответ организма-хозяина и избегают его с помощью ряда механизмов, включая образование в опухоли иммуносупрессорного окружения. Белок программируемой гибели клеток-1 (PD-1) является корецептором, экспрессирующимся на поверхности активированных T-клеток и опосредующим иммуносупрессию. Связывание PD-1 с одним из его лигандов, лигандом программируемой гибели клеток 1 (PD-L1) или лигандом программируемой гибели клеток 2 (PD-L2), приводит к ингибированию ответа цитотоксических T-клеток. Повышенная экспрессия PD-L1 в микроокружении опухоли облегчает ускользание от механизма иммунного надзора (индуцируемой T-клетками противоопухолевой активности). И наоборот, блокирование этого взаимодействия приводит к повышенному T-клеточному ответу с противоопухолевой активностью.
[0006] Показано, что блокирование пути T-клеточных контрольных точек PD-1/PD-L1 является эффективным и хорошо переносимым подходом для стимуляции иммунного ответа, и с его помощью достигают объективных ответов у пациентов с NSCLC (Topalian et al 2012, N. Engl. J. Med. 366: 2443-54; Borghaei et al 2015, N. Engl. J. Med. 373: 1627-39; Brahmer et al 2015, N. Engl. J. Med. 373: 123-35; Herbst et al 2016, Lancet 387: 1540-50; Fehrenbacher et al 2016, Lancet 387: 1837-46; Rittmeyer et al 2017, Lancet 389: 255-65; Reck et al 2016; N. Engl. J. Med. 375: 1823-33; Roach et al 2016, App.l Immunohistochem. Mol. Morphol. 24: 392-7; Socinski et al 2016, Ann. Oncol. 27 Suppl 6:LBA7_PR).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0007] В некоторых вариантах осуществления настоящее изобретение относится к способам лечения или улучшения по меньшей мере одного симптома или признака рака легких, ингибирования роста рака легких и/или повышения выживаемости у индивидуума. Способы по этому аспекту включают введение нуждающемуся в этом индивидууму одной или более доз терапевтически эффективного количества антитела или его антигенсвязывающего фрагмента, специфически связывающегося с белком программируемой гибели клеток 1 (PD-1). В некоторых вариантах осуществления рак легких является немелкоклеточным раком легких. В одном из вариантов осуществления индивидуум имеет рецидивирующий или метастазирующий рак легких на поздней стадии. В одном из вариантов осуществления индивидуум имеет плоскоклеточный немелкоклеточный рак легких. В одном из вариантов осуществления индивидуум имеет неплоскоклеточный немелкоклеточный рак легких. В некоторых вариантах осуществления индивидуум имеет рак легких, где опухоли экспрессируют лиганд программируемой гибели клеток 1 (PD-L1) в <50% опухолевых клеток. В некоторых вариантах осуществления индивидуум имеет рак легких (например, немелкоклеточный рак легких), где опухоли экспрессируют PD-L1 в <50%, ≤45%, ≤40%, ≤30%, ≤20%, ≤10%, ,5%, ≤2%, ≤1% или приблизительно 0% опухолевых клеток. В некоторых других вариантах осуществления индивидуум имеет рак легких, где опухоли экспрессируют PD-L1 в ≥50% опухолевых клеток. В некоторых вариантах осуществления индивидуум имеет рак легких (например, немелкоклеточный рак легких), где опухоли экспрессируют PD-L1 в ≥50%, ≥60%, ≥70%, ≥80% или ≥90% опухолевых клеток. В некоторых вариантах осуществления индивидуума подвергают лечению рака легких (противоопухолевой терапии, например, химиотерапии). В некоторых вариантах осуществления способы включают введение нуждающемуся в этом индивидууму одной или более доз антитела против PD-1, где каждая доза содержит от 20 мг до 1500 мг антитела против PD-1, и где каждую дозу вводят через 1 неделю, 2 недели, 3 недели или 4 недели после предыдущей дозы. В некоторых вариантах осуществления способы включают введение нуждающемуся в этом индивидууму терапевтически эффективного количества антитела против PD-1, необязательно, в комбинации с химиотерапией или ингибитором CTLA-4 (например, антитела против CTLA-4, такого как ипилимумаб). В одном из вариантов осуществления химиотерапия включает химиотерапевтическое средство на основе платины (например, пеметрексед, цисплатин, гемцитабин или их комбинацию). В одном из вариантов осуществления антитело против PD-1 является REGN2810.
[0008] В некоторых вариантах осуществления настоящее изобретение относится к способам лечения злокачественного новообразования, включающим выбор индивидуума с раком легких и введение одной или более доз антитела против PD-1, где введение приводит к ингибированию роста опухоли, повышению общей выживаемости и/или повышению выживаемости без прогрессирования у индивидуума.
[0009] В некоторых вариантах осуществления настоящее изобретение относится к способам лечения или улучшения по меньшей мере одного симптома или признака или ингибирования роста злокачественного новообразования у индивидуума. В некоторых вариантах осуществления настоящее изобретение относится к способам замедления роста опухоли или профилактики рецидивирования опухоли. В некоторых вариантах осуществления настоящее изобретение относится к способам повышения общей выживаемости или выживаемости без прогрессирования у пациента со злокачественным новообразованием. Способы по изобретению по этому аспекту включают последовательное введение одной или более доз терапевтически эффективного количества антитела или его антигенсвязывающего фрагмента, специфически связывающегося с PD-1. В одном из вариантов осуществления антитело против PD-1 вводят в комбинации с химиотерапией. В одном из вариантов осуществления антитело против PD-1 вводят в комбинации с ингибитором CTLA-4 (например, антителом против CTLA-4, таким как ипилимумаб). В некоторых вариантах осуществления злокачественное новообразование или опухоль является солидной опухолью или злокачественным новообразованием. В некоторых вариантах осуществления солидная опухоль выбрана из группы, состоящей из колоректального рака, рака яичников, рака предстательной железы, рака молочной железы, злокачественного новообразования головного мозга, рака шейки матки, рака мочевого пузыря, рака анального канала, рака матки, рака толстого кишечника, рака печени, рака поджелудочной железы, рака легких, рака эндометрия, злокачественного новообразования костной ткани, рака яичка, рака кожи, рака почки, рака желудка, рака пищевода, рака головы и шеи, рака слюнных желез и миеломы.
[0010] В некоторых вариантах осуществления способы включают введение одной или более доз антитела против PD-1 пациенту со злокачественным новообразованием на поздней стадии или метастазирующим злокачественным новообразованием (например, немелкоклеточным раком легких на поздней стадии), где опухолевая ткань пациента экспрессирует PD-L1 в ≤1%, ≤2%, ≤5%, ≤10%, ≤20%, ≤30%, ≤40%, ≤45% или <50% опухолевых клеток. В некоторых вариантах осуществления способы включают введение одной или более доз антитела против PD-1 пациенту со злокачественным новообразованием (например, раком легких на поздней стадии или метастазирующим раком легких), где опухолевая ткань пациента экспрессирует PD-L1 в ≥50%, ≥60%, ≥70%, ≥80% или ≥90% опухолевых клеток.
[0011] В некоторых вариантах осуществления антитело против PD-1 вводят пациенту со злокачественным новообразованием как лечение первой линии, где пациента ранее не подвергали системному лечению злокачественного новообразования. В некоторых вариантах осуществления антитело против PD-1 вводят пациенту со злокачественным новообразованием (например, метастазирующим злокачественным новообразованием) как лечение второй линии, где пациента ранее подвергали лечению, включающему, в качестве неограничивающих примеров, антитело против PD-1 (например, ниволумаб или пембролизумаб), ингибитор CTLA-4 (например, антитело против CTLA-4), химиотерапию, хирургическую операцию и/или лучевую терапию.
[0012] В некоторых вариантах осуществления каждая доза антитела против PD-1 содержит 0,1-20 мг/кг массы тела индивидуума. В некоторых вариантах осуществления каждая доза антитела против PD-1 содержит 0,3, 1, 3, 5 или 10 мг/кг массы тела индивидуума. В некоторых вариантах осуществления каждая доза антитела против PD-1 содержит 20-1500 мг. В одном из вариантов осуществления каждая доза антитела против PD-1 содержит приблизительно 200 мг. В одном из вариантов осуществления каждая доза антитела против PD-1 содержит приблизительно 250 мг. В одном из вариантов осуществления каждая доза антитела против PD-1 содержит приблизительно 350 мг. В одном из вариантов осуществления каждая доза антитела против PD-1 содержит приблизительно 1000 мг или приблизительно 1050 мг.
[0013] В некоторых вариантах осуществления способы по настоящему изобретению включают введение терапевтически эффективного количества антитела против PD-1 до, одновременно или после химиотерапии. В одном из вариантов осуществления способы по настоящему изобретению включают введение антитела против PD-1 до введения дозы химиотерапии.
[0014] В некоторых вариантах осуществления способы по настоящему изобретению включают введение одной или более терапевтических доз антитела против PD-1, где каждую дозу вводят через 0,5-12 недель после предыдущей дозы. В одном из вариантов осуществления каждую дозу вводят через 1 неделю после предыдущей дозы. В одном из вариантов осуществления каждую дозу вводят через 2 недели после предыдущей дозы. В одном из вариантов осуществления каждую дозу вводят через 3 недели после предыдущей дозы.
[0015] В некоторых вариантах осуществления одну или более доз антитела против PD-1 и, необязательно, второго терапевтического средства (например, химиотерапевтического средства) включают в цикл лечения. Способы по изобретению по этому аспекту включают подвергание нуждающегося в этом индивидуума по меньшей мере одному циклу лечения, где по меньшей мере один цикл лечения включает одну или более доз антитела против PD-1. В некоторых вариантах осуществления по меньшей мере один цикл лечения дополнительно включает одну или более доз химиотерапевтического средства (например, химиотерапевтического средства на основе платины, такого как гемцитабин, пеметрексед, цисплатин).
[0016] В некоторых вариантах осуществления антитело против PD-1 вводят в комбинации с дополнительным терапевтическим средством или способом терапии (например, антителом против CTLA-4 или любым средством или способом терапии, представленным в настоящем описании).
[0017] В некоторых вариантах осуществления лечение приводит к одному или более терапевтическим эффектам, выбранным из группы, состоящей из регрессирования опухоли, абскопального эффекта, ингибирования метастазирования опухоли, уменьшения метастазов с течением времени, снижения использования химиотерапевтических или цитотоксических средств, снижения опухолевой массы, повышения выживаемости без прогрессирования, повышения общей выживаемости, полного ответа, частичного ответа и стабильного заболевания.
[0018] В некоторых вариантах осуществления антитело против PD-1 или антигенсвязывающий белок содержит определяющие комплементарность области (HCDR) вариабельной области тяжелой цепи (HCVR), содержащей аминокислотную последовательность SEQ ID NO: 1, и CDR вариабельной области легкой цепи (LCVR), содержащей аминокислотную последовательность SEQ ID NO: 2. Одним из таких типов антигенсвязывающего белка, который можно использовать в способах по настоящему изобретению, является антитело против PD-1, такое как REGN2810 (также известное как цемиплимаб).
[0019] В некоторых вариантах осуществления настоящее изобретение относится к применению антитела против PD-1 или его антигенсвязывающего фрагмента в производстве лекарственного средства для лечения или ингибирования роста злокачественного новообразования у индивидуумов, включая людей. В некоторых вариантах осуществления злокачественное новообразование является раком легких. В некоторых вариантах осуществления рак легких является немелкоклеточным раком легких. В некоторых вариантах осуществления злокачественное новообразование является колоректальным раком, раком яичников, раком предстательной железы, раком молочной железы, злокачественным новообразованием головного мозга, раком шейки матки, раком мочевого пузыря, раком анального канала, раком матки, раком толстого кишечника, раком печени, раком поджелудочной железы, раком легких, раком эндометрия, злокачественным новообразованием костной ткани, раком яичка, раком кожи, раком почки, раком желудка, раком пищевода, раком головы и шеи, раком слюнных желез или миеломой.
[0020] Другие варианты осуществления настоящего изобретения будут очевидны из следующего подробного описания.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[00921] На фигуре 1 показан дизайн исследования, включающий введение доз антитела против PD-1 и лучевую терапию (XRT) у мышей, которым имплантировали опухоли MC38 (исследование, описанное в примере 1 в настоящем описании).
[0022] На фигуре 2 показан средний рост опухоли у мышей, которым вводили антитело изотипического контроля ( ), антитело против PD-1 (
), антитело против PD-1 ( ), изотипический контроль+лучевую терапию (XRT) (
), изотипический контроль+лучевую терапию (XRT) ( ) или антитело против PD-1+XRT (
) или антитело против PD-1+XRT ( ), в исследовании, описанном в примере 1 в настоящем описании.
), в исследовании, описанном в примере 1 в настоящем описании.
[0023] На фигуре 3 показана общая выживаемость мышей, которым вводили антитело изотипического контроля ( ), антитело против PD-1 (
), антитело против PD-1 ( ), изотипический контроль+лучевую терапию (XRT) (
), изотипический контроль+лучевую терапию (XRT) ( ) или антитело против PD-1+XRT (
) или антитело против PD-1+XRT ( ), в исследовании, описанном в примере 1 в настоящем описании.
), в исследовании, описанном в примере 1 в настоящем описании.
[0024] На фигуре 4 показан дизайн исследования, включающий введение доз антитела против PD-1 и лучевую терапию (XRT) у мышей, которым имплантировали опухоли B16F10.9 (исследование, описанное в примере 2 в настоящем описании).
[0025] На фигуре 5 показан средний рост опухоли у мышей, которым вводили антитело изотипического контроля ( ), антитело против PD-1 (
), антитело против PD-1 ( ), изотипический контроль+лучевую терапию (XRT) (
), изотипический контроль+лучевую терапию (XRT) ( ) или антитело против PD-1+XRT (О) в исследовании, описанном в примере 2 в настоящем описании.
) или антитело против PD-1+XRT (О) в исследовании, описанном в примере 2 в настоящем описании.
[0026] На фигуре 6 показана общая выживаемость мышей, которым вводили антитело изотипического контроля ( ), антитело против PD-1 (
), антитело против PD-1 ( ), изотипический контроль+лучевую терапию (XRT) (
), изотипический контроль+лучевую терапию (XRT) ( ) или антитело против PD-1+XRT (О), в исследовании, описанном в примере 2 в настоящем описании.
) или антитело против PD-1+XRT (О), в исследовании, описанном в примере 2 в настоящем описании.
[0027] На фигуре 7 показан дизайн исследования, включающий введение доз антитела против PD-1 и лучевую терапию (XRT) у мышей, которым имплантировали опухоли MC38 (исследование, описанное в примере 4 в настоящем описании).
[0028] На фигуре 8 показан средний рост первичной опухоли у мышей, которым вводили антитело изотипического контроля ( ), антитело против PD-1 (
), антитело против PD-1 ( ), изотипический контроль+лучевую терапию (XRT) (
), изотипический контроль+лучевую терапию (XRT) ( ) или антитело против PD-1+XRT (
) или антитело против PD-1+XRT ( ), в исследовании, описанном в примере 4 в настоящем описании.
), в исследовании, описанном в примере 4 в настоящем описании.
[0029] На фигуре 9 показана общая выживаемость мышей, которым вводили антитело изотипического контроля ( ), антитело против PD-1 (
), антитело против PD-1 ( ), изотипический контроль+лучевую терапию (XRT) (
), изотипический контроль+лучевую терапию (XRT) ( ) или антитело против PD-1+XRT (), в исследовании, описанном в примере 4 в настоящем описании.
) или антитело против PD-1+XRT (), в исследовании, описанном в примере 4 в настоящем описании.
[0030] На фигуре 10 показан рост вторичной опухоли у мышей, которым вводили антитело изотипического контроля ( ), антитело против PD-1 (
), антитело против PD-1 ( ), изотипический контроль+лучевую терапию (XRT) () или антитело против PD-1+XRT (
), изотипический контроль+лучевую терапию (XRT) () или антитело против PD-1+XRT ( ), в исследовании, описанном в примере 4 в настоящем описании.
), в исследовании, описанном в примере 4 в настоящем описании.
[0031] На фигуре 11 показан дизайн исследования, включающий введение доз антитела против PD-1, антитела против GITR и лучевую терапию (XRT) у мышей, которым имплантировали опухоли MC38 (исследование, описанное в примере 5 в настоящем описании).
[0032] На фигуре 12 показан средний рост опухоли у мышей, которым вводили антитело изотипического контроля ( ), антитело против PD-1 (
), антитело против PD-1 ( ), антитело против GITR (
), антитело против GITR ( ), комбинацию антитела против PD-1 и антитела против GITR (), изотипический контроль+лучевую терапию (XRT) (
), комбинацию антитела против PD-1 и антитела против GITR (), изотипический контроль+лучевую терапию (XRT) ( ),антитело против PD-1+XRT (О), антитело против GITR+XRT (
),антитело против PD-1+XRT (О), антитело против GITR+XRT ( ) или комбинацию антитела против PD-1, антитела против GITR+XRT (
) или комбинацию антитела против PD-1, антитела против GITR+XRT ( ), в исследовании, описанном в примере 5 в настоящем описании.
), в исследовании, описанном в примере 5 в настоящем описании.
[0033] На фигуре 13 показана общая выживаемость мышей, которым вводили антитело изотипического контроля ( ), антитело против PD-1 (
), антитело против PD-1 ( ), антитело против GITR (
), антитело против GITR ( ), комбинацию антитела против PD-1 и антитела против GITR (
), комбинацию антитела против PD-1 и антитела против GITR ( ), изотипический контроль+радиация (XRT) (
), изотипический контроль+радиация (XRT) ( ),антитело против PD-1+XRT (О), антитело против GITR+XRT () или комбинацию антитела против PD-1, антитела против GITR+XRT (), в исследовании, описанном в примере 5 в настоящем описании.
),антитело против PD-1+XRT (О), антитело против GITR+XRT () или комбинацию антитела против PD-1, антитела против GITR+XRT (), в исследовании, описанном в примере 5 в настоящем описании.
[0034] На фигуре 14A показано рентгеновское изображение метастазов в легких у пациента с базально-клеточной карциномой (BCC), указанных стрелками, на исходном уровне (слева) и на неделе 24 (справа).
[0035] На фигуре 14B показано рентгеновское изображение массы в шее у пациента с плоскоклеточной карциномой кожи (CSCC) на исходном уровне (слева) и на неделе 16 (справа).
ПОДРОБНОЕ ОПИСАНИЕ
[0036] Перед описанием настоящего изобретения необходимо отметить, что оно не ограничено конкретными описанными способами и экспериментальными условиями, т.к. такие способы и условия могут варьироваться. Также следует понимать, что терминология, используемая в настоящем описании, предназначена для описания исключительно конкретных вариантов осуществления, а не для ограничения, т.к. объем настоящего изобретения будет ограничен исключительно формулой изобретения.
[0037] Если не указано иначе, все технические и научные термины, используемые в настоящем описании, обладают значением, общепринято понимаемым специалистом в области, к которой принадлежит изобретение. В рамках изобретения термин "приблизительно" при использовании в отношении конкретного приведенного числового значения означает, что значение может варьироваться относительно приведенного значения не более чем на 1%. Например, в рамках изобретения выражение "приблизительно 100" включает 99 и 101 и все значения между ними (например, 99,1, 99,2, 99,3, 99,4 и т.д.).
[0038] Хотя в практическом осуществлении настоящего изобретения можно использовать любые способы и материалы, схожие или эквивалентные представленным в настоящем описании, далее описаны предпочтительные способы и материалы. Все публикации, упомянутые в настоящем описании, включены в него в качестве ссылки в полном объеме.
Способы лечения или ингибирования роста злокачественного новообразования
[0039] Настоящее изобретение относится к способам лечения, улучшения или снижения тяжести по меньшей мере одного симптома или признака или ингибирования роста злокачественного новообразования у индивидуума. Способы по изобретению по этому аспекту включают введение нуждающемуся в этом индивидууму терапевтически эффективного количества антитела или его антигенсвязывающего фрагмента, специфически связывающегося с PD-1. В некоторых вариантах осуществления антитело против PD-1 вводят в комбинации с противоопухолевой терапией (описываемой где-либо в настоящем описании). В рамках изобретения термины "лечить", "лечение" или т.п. означают улучшение симптомов, временное или постоянное устранение причины симптомов, замедление или ингибирование роста опухоли, снижение опухолевой нагрузки или опухолевой массы, стимуляцию регрессирования опухоли, индуцирование уменьшения опухоли, некроз и/или исчезновение, профилактику рецидивирования опухоли, профилактику или ингибирование метастазирования, ингибирование роста метастазирующей опухоли и/или увеличение длительности выживаемости индивидуума.
[0040] В рамках изобретения выражение "нуждающийся в этом индивидуум" означает человека или не являющегося человеком млекопитающего, демонстрирующего один или более симптомов или признаков злокачественного новообразования, и/или у которого диагностировали злокачественное новообразование, включая солидную опухоль, и нуждающегося в его лечении. Во многих вариантах осуществления термин "индивидуум" можно использовать взаимозаменяемо с термином "пациент". Например, у человека можно диагностировать первичную или метастазирующую опухоль и/или один или более симптомов или признаков, включая, в качестве неограничивающих примеров, необъяснимую потерю веса, общую слабость, постоянную утомляемость, потерю аппетита, повышение температуры, ночную потливость, боль в костях, одышку, отек области живота, боль/давление в груди, увеличение селезенки и повышение уровня биомаркеров злокачественных новообразований (например, CA125). Выражение включает индивидуумов с первичными или развившимися опухолями. В некоторых вариантах осуществления выражение включает людей, подвергаемых лечению и/или нуждающихся в лечении солидной опухоли, например, рака толстого кишечника, рака молочной железы, рака легких, рака предстательной железы, рака кожи, рака печени, злокачественного новообразования костной ткани, рака яичников, рака шейки матки, рака поджелудочной железы, рака головы и шеи и злокачественного новообразования головного мозга. В некоторых предпочтительных вариантах осуществления выражение включает людей, подвергаемых лечению и/или нуждающихся в лечении рака легких, включая немелкоклеточный рак легких. В одном из предпочтительных вариантов осуществления выражение включает пациентов, подвергаемых лечению и/или нуждающихся в лечении рецидивирующего или метастазирующего немелкоклеточного рака легких на поздней стадии. В другом предпочтительном варианте осуществления выражение включает пациентов, подвергаемых лечению и/или нуждающихся в лечении плоскоклеточного или неплоскоклеточного немелкоклеточного рака легких. Термин включает индивидуумов с первичными или метастазирующими опухолями (злокачественными новообразованиями на поздней стадии). В некоторых вариантах осуществления выражение "нуждающийся в этом индивидуум" включает пациентов с солидной опухолью, резистентных, или рефрактерных, или в недостаточной степени контролируемых с помощью предшествующей терапии (например, лечения противоопухолевым средством). Например, выражение включает индивидуумов, которых подвергали лечению с использованием одной или более линиям предшествующей терапии, такой как лечение с использованием химиотерапии (например, карбоплатина или доцетаксела). В некоторых вариантах осуществления выражение "нуждающийся в этом индивидуум" включает пациентов с солидной опухолью, которых подвергали лечению с использованием одной или более линий предшествующей терапии, но у которых затем наблюдали рецидив или метастазирование. Например, пациентов с солидной опухолью, которых могли подвергать лечению одним или более противоопухолевыми средствами, приводившему к регрессированию опухоли, которые, однако, затем перенесли рецидив злокачественного новообразования, резистентного к одному или более противоопухолевым средствам (например, злокачественного новообразования, резистентного к химиотерапии), лечат способами по настоящему изобретению. Выражение также включает индивидуумов с солидной опухолью, для которых общепринятая противоопухолевая терапия нецелесообразна, например, по причине токсических побочных эффектов. Например, выражение включает пациентов, которых подвергали одному или более циклам химиотерапии с токсическими побочными эффектами.
[0041] В некоторых вариантах осуществления способы по настоящему изобретению можно использовать для лечения пациентов, у которых наблюдают повышенные уровни одного или более биомаркеров злокачественных новообразований [например, лиганда программируемой гибели клеток 1 (PD-L1), CA125, CA19-9, простат-специфического антигена (PSA), лактатдегидрогеназы, KIT, карциноэмбрионального антигена, рецептора эпидермального фактора роста (EGFR), перестановки генов ALK]. Например, способы по настоящему изобретению включают введение терапевтически эффективного количества антитела против PD-1 пациенту с повышенным уровнем PD-L1 в опухолевой ткани. В некоторых вариантах осуществления способы по настоящему изобретению включают введение терапевтически эффективного количества антитела против PD-1 пациенту с раком легких, где опухолевая ткань пациента экспрессирует PD-L1 в <50%, ≤45%, ≤40%, ≤30%, ≤20%, ≤10%, ≤5%, ≤2% или ≤1% опухолевых клеток. В некоторых вариантах осуществления способы по настоящему изобретению включают введение терапевтически эффективного количества антитела против PD-1 пациенту с раком легких, где опухолевая ткань пациента экспрессирует PD-L1 в ≥50%, ≥60%, ≥70%, ≥80% или ≥90% опухолевых клеток.
[0042] В некоторых вариантах осуществления способы по настоящему изобретению используют для индивидуума с солидной опухолью. Термины "опухоль" и "злокачественное новообразование" используют в настоящем описании взаимозаменяемо.
[0043] В рамках изобретения термин "солидная опухоль" относится к аномальной массе ткани, как правило, не содержащей кисты или области жидкости. Солидные опухоли могут быть доброкачественными (незлокачественными новообразованиями) или злокачественными. В целях по настоящему изобретению термин "солидная опухоль" означает злокачественные солидные опухоли. Термин включает различные типы солидных опухолей, названных по типам образующих их клеток, а именно саркомы, карциномы и лимфомы. Однако термин не включает лейкозы. В различных вариантах осуществления термин "солидная опухоль" включает злокачественные новообразования, возникающие из соединительной или опорной ткани (например, костной или мышечной) (обозначаемые как саркомы), злокачественные новообразования, возникающие из железистых клеток и эпителиальных клеток, выстилающих ткани организма (обозначаемые как карциномы), и злокачественные новообразования лимфоидных органов, таких как лимфоузлы, селезенка и тимус (обозначаемые как лимфомы). Лимфоидные клетки встречаются почти во всех тканях организма, и, таким образом, лимфомы могут развиваться в различных органах. В некоторых вариантах осуществления термин "солидная опухоль" включает злокачественные новообразования, включающие, в качестве неограничивающих примеров, колоректальный рак, рак яичников, рак предстательной железы, рак молочной железы, злокачественное новообразование головного мозга, рак шейки матки, рак мочевого пузыря, рак анального канала, рак матки, рак толстого кишечника, рак печени, рак поджелудочной железы, рак легких, рак эндометрия, злокачественное новообразование костной ткани, рак яичка, рак кожи, рак почки, рак желудка, рак пищевода, рак головы и шеи, рак слюнных желез и миелому. В некоторых вариантах осуществления термин "солидная опухоль" включает злокачественные новообразования, включающие, в качестве неограничивающих примеров, печеночноклеточную карциному, немелкоклеточный рак легких, плоскоклеточный рак головы и шеи, базально-клеточную карциному, карциному молочной железы, плоскоклеточная карцинома кожи, хондросаркому, ангиосаркому, холангиокарциному, саркому мягких тканей, колоректальный рак, меланому, карциному Меркеля и мультиформную глиобластому. В некоторых вариантах осуществления термин "солидная опухоль" включает несколько очагов солидной опухоли, расположенных отдельно друг от друга, например, 2, более 2, более 5, более 10, более 15, более 20 или более 25 очагов у индивидуума, нуждающегося в лечении. В некоторых вариантах осуществления несколько очагов находятся дистально друг от друга в одном органе. В некоторых других вариантах осуществления очаги опухоли могут находиться в разных органах.
[0044] В некоторых вариантах осуществления настоящее изобретение относится к способам лечения или ингибирования роста злокачественного новообразования, включая, в качестве неограничивающих примеров, колоректальный рак, рак яичников, рак предстательной железы, рак молочной железы, злокачественное новообразование головного мозга, рак шейки матки, рак мочевого пузыря, рак анального канала, рак матки, рак толстого кишечника, рак печени, рак поджелудочной железы, рак легких, рак эндометрия, злокачественное новообразование костной ткани, рак яичка, рак кожи, рак почки, рак желудка, рак пищевода, рак головы и шеи, рак слюнных желез и миелому. В некоторых вариантах осуществления настоящее изобретение относится к способам лечения или ингибирования роста злокачественного новообразования, включая, в качестве неограничивающих примеров, печеночноклеточную карциному, немелкоклеточный рак легких, плоскоклеточный рак головы и шеи, базально-клеточную карциному, плоскоклеточную карциному кожи, хондросаркому, ангиосаркому, холангиокарциному, саркому мягких тканей, колоректальный рак, меланому, карциному Меркеля и мультиформную глиобластому. В некоторых вариантах осуществления настоящее изобретение относится к способам лечения солидных опухолей на поздней стадии, включая, в качестве неограничивающих примеров, метастазирующую плоскоклеточную карциному кожи (CSCC), неоперабельный местнораспространенный CSCC, метастазирующий колоректальный рак, печеночноклеточный рак на поздней стадии или метастазирующий печеночноклеточный рак, немелкоклеточный рак легких на поздней стадии, рецидивирующую мультиформную глиобластому, кастрационно-резистентный рак предстательной железы и любую солидную опухоль на поздней стадии, рефрактерную к терапии первой линии. Способы по этому аспекту, включают введение терапевтически эффективного количества антитела против PD-1, необязательно, в комбинации с противоопухолевой терапией. Противоопухолевая терапия включают, в качестве неограничивающих примеров, общепринятую противоопухолевую терапию, такую как химиотерапия, лучевая терапия, ингибитор CTLA-4 (например, антитело против CTLA-4) и хирургическая операция. Другую противоопухолевую терапию описывают где-либо в настоящем описании. В одном из вариантов осуществления противоопухолевая терапия включает химиотерапию препаратами платины. В некоторых вариантах осуществления нуждающемуся в этом индивидууму вводят одну или более доз антитела против PD-1, где каждую дозу вводят через 0,5, 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 недель после предыдущей дозы. В некоторых вариантах осуществления каждая доза содержит 0,1-10 мг/кг (например, 0,3 мг/кг, 1 мг/кг, 3 мг/кг или 10 мг/кг) массы тела индивидуума. В некоторых других вариантах осуществления каждая доза содержит 20-1500 мг антитела против PD-1, например, 50 мг, 100 мг, 150 мг, 200 мг, 250 мг, 300 мг, 350 мг, 400 мг, 500 мг, 550 мг, 600 мг, 700 мг, 750 мг, 800 мг, 900 мг, 1000 мг, 1050 мг, 1200 мг или 1500 мг антитела против PD-1.
[0045] В некоторых вариантах осуществления настоящее изобретение относится к способам лечения злокачественного новообразования или ингибирования роста злокачественного новообразования с микросателлитной нестабильностью (MSI). В рамках изобретения термин "микросателлитная нестабильность", также известный как "MSI", относится к изменениям в микросателлитных повторах в опухолевых клетках или генетической гипермутабельности, вызванной недостаточностью репарации ошибочно спаренных оснований в ДНК. Микросателлиты, также известные как простые повторяющиеся последовательности, являются повторяющимися последовательностями ДНК, содержащими повторяющиеся единицы 1-6 пар оснований в длину. Хотя длина микросателлитов широко варьируется от индивидуума к индивидууму и вносит свой вклад в ДНК-фингерпринт, каждый индивидуум имеет микросателлиты определенной длины. MSI является результатом нестабильности белков репарации ошибочно спаренных оснований (MMR) при коррекции ошибок репликации ДНК. MSI включает полиморфизмы ДНК, где ошибки репликации варьируются по своей длине, а не по последовательности. MSI включает мутации со сдвигом рамки считывания посредством инсерций или делеций или гиперметилирование, приводящие к сайленсингу генов. В этой области известно, что микросателлитная нестабильность может приводить к раку толстого кишечника, раку желудка, раку эндометрия, раку яичников, раку гепатобилиарной системы, раку мочевыводящих путей, злокачественному новообразованию головного мозга и раку кожи. Настоящее изобретение относится к способам лечения злокачественных новообразований с MSI, включающим введение нуждающемуся в этом пациенту терапевтически эффективного количества антитела против PD-1, необязательно, в комбинации со вторым противоопухолевым средством (например, химиотерапией, лучевой терапией).
[0046] В рамках изобретения термин "химиотерапия" относится к использованию химиотерапевтического средства (химического соединения, используемого для противоопухолевой терапии). Термин включает, в качестве неограничивающих примеров, алкилирующие средства, антиметаболиты, ингибиторы киназ, растительные алкалоиды, являющиеся веретенными ядами, цитотоксические/противоопухолевые антибиотики, ингибиторы топоизомераз, фотосенсибилизаторы, противоэстрогенные средства и селективные модуляторы рецепторов эстрогена (SERM), антипрогестероновые средства, супрессоры рецепторов эстрогена (ERD), антагонисты рецепторов эстрогена, агонисты рилизинг-фактора лютеинизирующего гормона, антиандрогенные средства, ингибиторы ароматазы, ингибиторы EGFR и ингибиторы VEGF. Примеры химиотерапевтических средств приведены где-либо в настоящем описании. В одном из вариантов осуществления термин относится к химиотерапевтическим средствам на основе платины (например, гемцитабину, цисплатину, карбоплатину, пеметрекседу или их комбинации). Химиотерапевтические средства вводят в соответствии со схемами введения, известными в этой области.
[0047] В рамках изобретения термин "лучевая терапия", также обозначаемый как "XRT", означает использование ионизирующей радиации для уничтожения злокачественных клеток, как правило, как часть противоопухолевой терапии. Для получения ионизирующей радиации используют рентгеновское излучение, гамма-излучение или заряженные частицы (например, протоны или электроны). Лучевую терапию можно осуществлять с помощью устройства, расположенного вне организма пациента (дистанционная лучевая терапия), или с помощью источника, расположенного внутри организма пациента (внутренняя лучевая терапия или брахитерапия), или с помощью системных радиоактивных изотопов, вводимых внутривенно или перорально (системная терапия радиоактивными изотопами). Лучевую терапию можно планировать и осуществлять в комбинации со способами визуализации, такими как компьютерная томография (КТ), магнитно-резонансная терапия (МРТ), для точного определения дозы и положения прилагаемой радиации. В различных вариантах осуществления лучевая терапия выбрана из группы, состоящей из тотальной лучевой терапии всего тела, общепринятой дистанционной лучевой терапии, стереотаксической радиохирургии, стереотаксической лучевой терапии тела, 3D-конформной лучевой терапии, лучевой терапии с модулированной интенсивностью, лучевой терапии под визуальным контролем, томотерапии, брахитерапии и системной лучевой терапии. В зависимости от целей, в некоторых вариантах осуществления лучевая терапия является излечивающей, адъювантной или паллиативной. В некоторых вариантах осуществления термин "лучевая терапия" относится к гипофракционированной лучевой терапии. Термин "гипофракционированная лучевая терапия" относится к лучевой терапии, при которой доза радиации содержится в 2 или более фракциях. В различных вариантах осуществления каждая фракция содержит 2-20 Гр. Например, дозу радиации 50 Гр можно разделять на 10 фракций, каждая из которых содержит 5 Гр. В некоторых вариантах осуществления 2 или более фракций вводят в последовательные дни. В некоторых других вариантах осуществления 2 или более фракций вводят раз в 2 дня, раз в 3 дня, раз в 4 дня, раз в 5 дней, раз в 6 дней, раз в 7 дней или в их комбинации.
[0048] В некоторых вариантах осуществления настоящее изобретение относится к способам лечения, или замедления, или ингибирования роста опухоли. В некоторых вариантах осуществления настоящее изобретение относится к способам стимуляции регрессирования опухоли. В некоторых вариантах осуществления настоящее изобретение относится к способам снижения опухолевой нагрузки или снижения опухолевой массы. В некоторых вариантах осуществления настоящее изобретение относится к способам профилактики рецидивирования опухоли. Способы по изобретению по этому аспекту включают последовательное введение нуждающемуся в этом индивидууму терапевтически эффективного количества антитела против PD-1 в комбинации со второй противоопухолевой терапией, где антитело вводят индивидууму во множестве доз, например, как часть конкретной терапевтической схемы дозирования. Например, терапевтическая схема дозирования может включать введение индивидууму одной или более доз антитела против PD-1 с частотой приблизительно раз в день, раз в два дня, раз в три дня, раз в четыре дня, раз в пять дней, раз в шесть дней, раз в неделю, раз в две недели, раз в три недели, раз в четыре недели, раз в месяц, раз в два месяца, раз в три месяца, раз в четыре месяца или реже. В некоторых вариантах осуществления одну или более доз антитела против PD-1 вводят в комбинации с одной или более дозами второй противоопухолевой терапии, где одну или более доз второй противоопухолевой терапии вводят индивидууму с частотой приблизительно раз в неделю, раз в две недели, раз в три недели, раз в четыре недели, раз в месяц, раз в два месяца, раз в три месяца, раз в четыре месяца или реже.
[0049] В некоторых вариантах осуществления одну или более доз включают в цикл лечения. Способы по этому аспекту включают подвергание нуждающегося в этом индивидуума по меньшей мере одному циклу лечения, где по меньшей мере один цикл лечения включает 1-10 доз антитела против PD-1 и, необязательно, одну или более доз химиотерапии. В некоторых вариантах осуществления нуждающегося в этом индивидуума подвергают 2-12 или более циклам лечения.
[0050] В некоторых вариантах осуществления настоящее изобретение относится к способам повышения противоопухолевой эффективности или повышения ингибирования опухоли. Способы по изобретению по этому аспекту включают введение индивидууму с солидной опухолью терапевтически эффективного количества антитела против PD-1 перед введением дозы второй противоопухолевой терапии (например, химиотерапии или антитела против CTLA-4), где антитело против PD-1 можно вводить приблизительно за 1 день, более 1 дня, более 2 дней, более 3 дней, более 4 дней, более 5 дней, более 6 дней, более 7 дней или более 8 дней до второй противоопухолевой терапии. В некоторых вариантах осуществления способы обеспечивают ингибирование опухоли, повышенное, например, на приблизительно 20%, более 20%, более 30%, более 40%, более 50%, более 60%, более 70% или более 80% по сравнению с индивидуумом, которому вводят дозу противоопухолевой терапии (например, химиотерапии) до антитела против PD-1. В некоторых вариантах осуществления химиотерапия включает химиотерапию препаратами платины. В некоторых вариантах осуществления вторая противоопухолевая терапия включает ингибитор CTLA-4 (например, антитело против CTLA-4).
[0051] В некоторых вариантах осуществления настоящее изобретение относится к способам лечения злокачественного новообразования, включающим выбор индивидуума с первым очагом опухоли и по меньшей мере вторым очагом опухоли и введение одной или более доз антитела против PD-1 в комбинации с лучевой терапией таким образом, что лечат оба очага. В некоторых вариантах осуществления способы включают подвергание лучевой терапии первого очага опухоли, но не второго очага опухоли, где лучевая терапия приводит к регрессированию опухоли в обоих очагах опухоли (абскопальному эффекту). В некоторых вариантах осуществления способы включают выбор индивидуума с первым очагом опухоли и по меньшей мере вторым очагом опухоли и введение одной или более доз антитела против PD-1 в комбинации с гипофракционированной лучевой терапией, где гипофракционированной лучевой терапии подвергают первый очаг, но не второй очаг, и где оба очага лечат посредством такой лучевой терапии. В некоторых вариантах осуществления антитело против PD-1 вводят до лучевой терапии.
[0052] В некоторых вариантах осуществления настоящее изобретение относится к способам лечения злокачественного новообразования, включающим введение нуждающемуся в этом индивидууму одной или более субтерапевтических доз антитела против PD-1 в комбинации с одним или более способами противоопухолевой терапии, например, лучевой терапией. Как определено где-либо в настоящем описании, термин "субтерапевтическая доза" относится к дозе, меньшей, чем терапевтическая доза, и которую можно использовать для снижения токсичности проводимой терапии. В некоторых вариантах осуществления введение субтерапевтической дозы антитела против PD-1 в комбинации с лучевой терапией приводит к терапевтической противоопухолевой эффективности по сравнению с введением субтерапевтической дозы антитела против PD-1 в отдельности. В некоторых других вариантах осуществления способы по настоящему изобретению включают введение терапевтически эффективного количества антитела против PD-1 в комбинации с субтерапевтической дозой противоопухолевой терапии, такой как химиотерапия или лучевая терапия. Например, терапевтически эффективное количество антитела против PD-1 можно вводить в комбинации с субтерапевтической дозой циклофосфамида для повышения эффективности по сравнению с любой монотерапией.
[0053] В некоторых вариантах осуществления настоящее изобретение относится к способам ингибирования, задержки или прекращения метастазирования опухоли или инфильтрации опухолевыми клетками периферических органов. Способы по этому аспекту включают введение нуждающемуся в этом индивидууму терапевтически эффективного количества антитела против PD-1. В некоторых вариантах осуществления антитело против PD-1 вводят в комбинации с химиотерапией. В одном из вариантов осуществления химиотерапия является химиотерапией препаратами платины. В одном из вариантов осуществления химиотерапию проводят до, одновременно или после введения одной или более доз антитела против PD-1.
[0054] В некоторых вариантах осуществления способы по настоящему изобретению включают введение терапевтически эффективного количества антитела против PD-1 индивидууму с солидными опухолями на поздней стадии. В некоторых вариантах осуществления солидная опухоль на поздней стадии является метастазирующим раком легких, раком головы и шеи, печеночноклеточным раком или раком молочной железы. В некоторых других вариантах осуществления солидная опухоль на поздней стадии является плоскоклеточным раком кожи. В некоторых вариантах осуществления солидная опухоль на поздней стадии является невыраженной или агрессивной. В некоторых вариантах осуществления индивидуум не отвечает на предшествующую терапию или перенес рецидив после предшествующей терапии (например, с использованием карбоплатина). В некоторых вариантах осуществления индивидуум имеет солидную опухоль на поздней стадии, рефрактерную к химиотерапии первой линии. В некоторых дополнительных вариантах осуществления способы по настоящему изобретению дополнительно включают подвергание индивидуума с солидной опухолью на поздней стадии дополнительной противоопухолевой терапии (например, ингибитором CTLA-4).
[0055] В некоторых вариантах осуществления настоящее изобретение относится к способам лечения или ингибирования роста злокачественного новообразования, включая, в качестве неограничивающих примеров, колоректальный рак, рак яичников, рак предстательной железы, рак молочной железы, злокачественное новообразование головного мозга, рак шейки матки, рак мочевого пузыря, рак анального канала, рак матки, рак толстого кишечника, рак печени, рак поджелудочной железы, рак легких, рак эндометрия, злокачественное новообразование костной ткани, рак яичка, рак кожи, рак почки, рак желудка, рак пищевода, рак головы и шеи, рак слюнных желез и миелому. В некоторых вариантах осуществления настоящее изобретение относится к способам лечения или ингибирования роста злокачественного новообразования, включая, в качестве неограничивающих примеров, печеночноклеточную карциному, немелкоклеточный рак легких, плоскоклеточный рак головы и шеи, базально-клеточную карциному, плоскоклеточную карциному кожи, хондросаркому, ангиосаркому, холангиокарциному, саркому мягких тканей, колоректальный рак, меланому, карциному Меркеля и мультиформную глиобластому. В некоторых вариантах осуществления настоящее изобретение относится к способам лечения солидных опухолей на поздней стадии, включая, в качестве неограничивающих примеров, метастазирующую плоскоклеточную карциному кожи (CSCC), неоперабельную местнораспространенную CSCC, метастазирующий колоректальный рак, печеночноклеточный рак на поздней стадии или метастазирующий печеночноклеточный рак, немелкоклеточный рак легких на поздней стадии, рецидивирующую мультиформную глиобластому, впервые диагностированную мультиформную глиобластому, кастрационно-резистентный рак предстательной железы и любую солидную опухоль на поздней стадии, рефрактерную к терапии первой линии.
[0056] В одном из аспектов настоящее изобретение относится к способам лечения или ингибирования роста опухоли, включающим: (a) выбор пациента с плоскоклеточной карциномой кожи (CSCC), где пациента выбирают с учетом характеристики, выбранной из группы, состоящей из следующего: (i) пациент имеет местнораспространенную CSCC; (ii) пациент имеет метастазирующую CSCC; (iii) опухоль является неоперабельной; (iv) пациента ранее лечили с использованием по меньшей мере одной противоопухолевой терапии; (v) пациент имеет заболевание, считающееся неоперабельным; (vi) хирургическая операция и/или лучевая терапия противопоказаны; (vii) пациента ранее лечили лучевой терапией, и опухоль является резистентной к лучевой терапии или не отвечает на нее; и (viii) опухоль содержит УФ-индуцированное повреждение ДНК; и (b) введение терапевтически эффективного количества антитела против PD-1 нуждающемуся в этом пациенту. В некоторых вариантах осуществления одну или более доз антитела против PD-1 вводят через 1-12 недель после предыдущей дозы, например, через 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 или 12 недель после предыдущей дозы. В некоторых вариантах осуществления каждая доза антитела против PD-1 содержит 0,1, 1, 0,3, 3, 4, 5, 6, 7, 8, 9 или 10 мг/кг массы тела пациента. В некоторых вариантах осуществления каждая доза содержит 50-500 мг антитела против PD-1. В одном из вариантов осуществления антитело против PD-1 является REGN2810.
[0057] В одном из аспектов настоящее изобретение относится к способам лечения опухоли или повышения выживаемости пациента со злокачественным новообразованием, включающим: (a) выбор пациента, имеющего рак легких, где пациента выбирают с учетом характеристики, выбранной из группы, состоящей из следующего: (i) пациент имеет немелкоклеточный рак легких; (ii) опухолевая ткань пациента экспрессирует PD-L1 в <50% опухолевых клеток; (iii) пациент имеет плоскоклеточный или неплоскоклеточный рак легких стадии III или IV; (iv) пациента ранее не подвергали системному лечению рецидивирующего рака легких; и (v) пациента подвергали предыдущему лечению с использованием противоопухолевой терапии; и (b) введение пациенту одной или более доз терапевтически эффективного количества антитела или его антигенсвязывающего фрагмента, специфически связывающегося с PD-1. В одном из вариантов осуществления пациент имеет немелкоклеточный рак легких на поздней стадии или рецидивирующий немелкоклеточный рак легких, опухолевая ткань пациента экспрессирует PD-L1 в <50% опухолевых клеток, и пациента ранее не подвергали системному лечению рака легких. В одном из вариантов осуществления пациент имеет немелкоклеточный рак легких на поздней стадии или рецидивирующий немелкоклеточный рак легких, опухолевая ткань пациента экспрессирует PD-L1 в <50% опухолевых клеток, и пациента ранее подвергали системному лечению рака легких (например, химиотерапии). В некоторых вариантах осуществления пациент имеет немелкоклеточный рак легких на поздней стадии или рецидивирующий немелкоклеточный рак легких, и опухолевая ткань пациента экспрессирует PD-L1 в <50%, ≤45%, ≤40%, ≤30%, ≤20%, ≤10%, ≤5%, ≤2% или ≤1% опухолевых клеток.
[0058] В некоторых вариантах осуществления настоящее изобретение относится к способам лечения злокачественного новообразования или повышения выживаемости пациента со злокачественным новообразованием, включающим: (a) выбор пациента, имеющего рак легких, где опухолевая ткань пациента экспрессирует PD-L1 в ≥50% опухолевых клеток; и (b) введение пациенту одной или более доз терапевтически эффективного количества антитела или его антигенсвязывающего фрагмента, специфически связывающегося с PD-1. В одном из вариантов осуществления опухолевая ткань пациента экспрессирует PD-L1 в ≥60%, ≥70%, ≥80% или ≥90% опухолевых клеток. В одном из вариантов осуществления пациент имеет немелкоклеточный рак легких на поздней стадии или метастазирующий немелкоклеточный рак легких. В одном из вариантов осуществления пациент имеет плоскоклеточный или неплоскоклеточный немелкоклеточный рак легких стадии III или стадии IV. В одном из вариантов осуществления пациента ранее не подвергали системному лечению рака легких.
[0059] В одном из аспектов настоящее изобретение относится к способам лечения злокачественного новообразования или повышения выживаемости пациента со злокачественным новообразованием, включающим: (a) выбор пациента, имеющего рак легких, где пациент имеет по меньшей мере одну из следующих характеристик: (i) пациент имеет немелкоклеточный рак легких на поздней стадии или метастазирующий немелкоклеточный рак легких; (ii) пациент имеет плоскоклеточный или неплоскоклеточный рак легких стадии III или стадии IV; (iii) пациента ранее не подвергали системному лечению рака легких; и (iv) пациента ранее подвергали противоопухолевой терапии (например, химиотерапии препаратами платины, хирургической операции и/или лучевой терапии); (b) определение экспрессии PD-L1 в опухолевой ткани; и (c) введение пациенту одной или более доз терапевтически эффективного количества антитела или его антигенсвязывающего фрагмента, специфически связывающегося с PD-1, если опухолевая ткань экспрессирует PD-L1 в <50% опухолевых клеток. В одном из вариантов осуществления опухолевая ткань экспрессирует PD-L1 в <50%, ≤45%, ≤40%, ≤30%, ≤20%, ≤10%, ,5%, ≤2%, ≤1% или 0% опухолевых клеток.
[0060] В одном из аспектов настоящее изобретение относится к способам лечения злокачественного новообразования или повышения выживаемости пациента со злокачественным новообразованием, включающим: (a) выбор пациента, имеющего рак легких, где пациент имеет по меньшей мере одну из следующих характеристик: (i) пациент имеет немелкоклеточный рак легких на поздней стадии или метастазирующий немелкоклеточный рак легких; (ii) пациент имеет плоскоклеточный или неплоскоклеточный рак легких стадии III или стадии IV; (iii) пациента ранее не подвергали системному лечению рака легких; и (iv) пациента ранее подвергали противоопухолевой терапии (например, химиотерапии препаратами платины, хирургической операции и/или лучевой терапии); (b) определение экспрессии PD-L1 в опухолевой ткани; и (c) введение пациенту одной или более доз терапевтически эффективного количества антитела или его антигенсвязывающего фрагмента, специфически связывающегося с PD-1, если опухолевая ткань экспрессирует PD-L1 в ≥50% опухолевых клеток. В одном из вариантов осуществления опухолевая ткань экспрессирует PD-L1 в ≥60%, ≥70%, ≥80% или ≥90% опухолевых клеток.
[0061] В некоторых вариантах осуществления каждую дозу антитела против PD-1 вводят через 1 неделю, 2 недели, 3 недели или 4 недели после предыдущей дозы, где каждая доза содержит 20-1500 мг антитела против PD-1. В одном из вариантов осуществления каждая доза содержит 200, 250, 300, 350, 500, 600, 700, 800, 900, 1000 или 1050 мг антитела против PD-1. В одном из вариантов осуществления антитело против PD-1 является REGN2810 (цемиплимабом).
[0062] В некоторых вариантах осуществления способы включают введение одной или более доз терапевтически эффективного количества антитела против PD-1 пациенту со злокачественным новообразованием, где пациента выбирают с учетом экспрессии PD-L1 в менее чем 1% опухолевых клеток. В некоторых вариантах осуществления опухолевая ткань пациента экспрессирует PD-L1 в менее чем 2%, менее чем 5%, менее чем 10%, менее чем 20%, менее чем 30%, менее чем 40% или менее чем 50% опухолевых клеток. В некоторых вариантах осуществления способы включают выбор пациента со злокачественным новообразованием, где пациента выбирают с учетом экспрессии PD-L1 в ≥50% опухолевых клеток, и введение пациенту одной или более доз терапевтически эффективного количества антитела против PD-1. В некоторых вариантах осуществления экспрессию PD-L1 в опухолевой ткани определяют с помощью любого анализа, известного в этой области, например, анализа ELISA или иммуногистохимического (IHC) анализа, как описано в публикациях PCT №№ WO2016124558 или WO2016191751 или публикации патентной заявки США № 20160305947. В некоторых вариантах осуществления экспрессию PD-L1 определяют посредством количественного анализа экспрессии РНК, например, гибридизации in situ или RT-ПЦР. В некоторых вариантах осуществления экспрессию PD-L1 определяют посредством визуализации с использованием меченого антитела против PD-L1, например, иммунной позитронно-эмиссионной томографии или iPET [см., например, The Oncologist, 12: 1379 (2007); Journal of Nuclear Medicine, 52(8): 1171 (2011); временную патентную заявку США № 62/428672, поданную 1 декабря 2016 года].
[0063] В некоторых вариантах осуществления введение по меньшей мере одной дозы антитела против PD-1 приводит к повышению выживаемости без прогрессирования (PFS) или общей выживаемости (OS) пациента по сравнению с пациентом, которого подвергали химиотерапии препаратами платины в качестве монотерапии. В некоторых вариантах осуществления PFS повышают на по меньшей мере один месяц, приблизительно 2 месяца, приблизительно 3 месяца, приблизительно 4 месяца, приблизительно 5 месяцев, приблизительно 6 месяцев, приблизительно 7 месяцев, приблизительно 8 месяцев, приблизительно 9 месяцев, приблизительно 10 месяцев, приблизительно 11 месяцев, приблизительно 1 год, приблизительно 2 года, приблизительно 3 года или более по сравнению с пациентом, подвергаемым химиотерапии препаратами платины. В некоторых вариантах осуществления OS повышают на по меньшей мере один месяц, приблизительно 2 месяца, приблизительно 3 месяца, приблизительно 4 месяца, приблизительно 5 месяцев, приблизительно 6 месяцев, приблизительно 7 месяцев, приблизительно 8 месяцев, приблизительно 9 месяцев, приблизительно 10 месяцев, приблизительно 11 месяцев, приблизительно 1 год, приблизительно 2 года, приблизительно 3 года или более по сравнению с пациентом, подвергаемым химиотерапии препаратами платины.
[0064] В одном из аспектов настоящее изобретение относится к способам лечения или ингибирования роста опухоли, включающим выбор индивидуума со злокачественным новообразованием головного мозга и введение терапевтически эффективного количества антитела против PD-1 или его антигенсвязывающего фрагмента нуждающемуся в этом индивидууму. В некоторых вариантах осуществления злокачественное новообразование головного мозга является мультиформной глиобластомой. В одном из вариантов осуществления индивидуум имеет впервые диагностированную мультиформную глиобластому. В одном из вариантов осуществления возраст индивидуума составляет ≥65 лет. В одном из вариантов осуществления антитело против PD-1 вводят в одной или более дозах, где каждую дозу вводят через 0,5-4 недели после предыдущей дозы. В одном из вариантов осуществления каждая доза антитела против PD-1 содержит 1, 3 или 10 мг/кг массы тела индивидуума. В некоторых вариантах осуществления антитело против PD-1 вводят в комбинации с лучевой терапией. В одном из вариантов осуществления лучевая терапия является гипофракционированной лучевой терапией. В одном из вариантов осуществления индивидууму вводят 20-60 Гр в 2-20 фракциях. В некоторых вариантах осуществления одну или более доз антитела против PD-1 включают в один или более циклов лечения, где каждый цикл лечения включает 1-6 доз антитела против PD-1. В одном из вариантов осуществления по меньшей мере один цикл лечения дополнительно включает лучевую терапию. В дополнительном варианте осуществления лучевая терапия является гипофракционированной лучевой терапией. В некоторых вариантах осуществления индивидуума подвергают гипофракционированной лучевой терапии в первом цикле лечения, где гипофракционированная лучевая терапия включает 20-60 Гр в 2-20 фракциях. В одном из вариантов осуществления индивидуума подвергают гипофракционированной лучевой терапии через неделю после введения антитела против PD-1 в первом цикле лечения. В некоторых вариантах осуществления способы по настоящему изобретению дополнительно включают введение индивидууму антиангиогенного средства, если у индивидуума развивается отек головного мозга после введения антитела против PD-1. В одном из вариантов осуществления антиангиогенное средство является ингибитором фактора роста эндотелия сосудов (VEGF). В одном из вариантов осуществления антиангиогенное средство является ингибитором ангиопоэтина-2 (Ang-2) (например, антителом против Ang-2, таким как несвакумаб). В некоторых вариантах осуществления ингибитор VEGF выбран из группы, состоящей из VEGF-ингибирующего слитого белка (например, "VEGF-ловушки", такой как афлиберцепт или другой VEGF-ингибирующий слитый белок, как описано в патенте США № 7087411), антитела против VEGF (например, бевацизумаба) и низкомолекулярного ингибитора киназ рецептора VEGF (например, сунитиниба, сорафениба или пазопаниба).
[0065] В некоторых вариантах осуществления способы по настоящему изобретению включают введение индивидууму терапевтически эффективного количества антитела против PD-1 в комбинации с дополнительным терапевтическим средством, или схемой лечения, или способом. Дополнительное терапевтическое средство, или схему лечения, или способ можно использовать для повышения противоопухолевой эффективности, снижения токсических эффектов одного или более способов терапии и/или снижения дозы одного или более терапевтических средств. В различных вариантах осуществления дополнительное терапевтическое средство, или схема лечения, или способ выбраны из группы, состоящей, например, из химиотерапии, циклофосфамида, хирургической операции, лучевой терапии, противоопухолевой вакцины, ингибитора лиганда программируемой гибели клеток 1 (PD-L1) (например, антитела против PD-L1), ингибитора гена активации лимфоцитов 3 (LAG3) (например, антитела против LAG3), ингибитора антигена цитотоксических T-лимфоцитов 4 (CTLA-4) (например, антитела против CTLA-4, такого как ипилимумаб), ингибитора глюкокортикоид-индуцируемого рецептора фактора некроза опухоли (GITR) (например, антитела против GITR), ингибитора белка-3, содержащего T-клеточный иммуноглобулин и домен муцина (TIM3), ингибитора B- и T-лимфоцитарного аттенюатора (BTLA), ингибитора T-клеточного иммунорецептора с доменами Ig и ITIM (TIGIT), ингибитора CD47, ингибитора индоламин-2,3-диоксигеназы (IDO), антагониста фактора роста эндотелия сосудов (VEGF) [выбранного из группы, состоящей из VEGF-ингибирующего слитого белка (например, "VEGF-ловушки", такой как афлиберцепт или другой VEGF-ингибирующий слитый белок, как описано в патенте США № 7087411), антитела против VEGF (например, бевацизумаба) и низкомолекулярного ингибитора киназ рецептора VEGF (например, сунитиниба, сорафениба или пазопаниба)], ингибитора ангиопоэтина-2 (Ang2), ингибитора трансформирующего фактора роста бета (TGFβ), ингибитора рецептора эпидермального фактора роста (EGFR), антитела против опухолеспецифичного антигена [например, CA9, CA125, меланома-ассоциированного антигена 3 (MAGE3), карциноэмбрионального антигена (CEA), виментина, опухолевого M2-PK, простат-специфического антигена (PSA), муцина-1, MART-1, и CA19-9], биспецифического антитела против CD3/против CD20, вакцины (например, бациллы Кальметта-Герена), гранулоцитарно-макрофагального колониестимулирующего фактора, цитотоксина, химиотерапевтического средства, ингибитора ИЛ-6R, ингибитора ИЛ-4R, ингибитора ИЛ-10, цитокина, такого как ИЛ-2, ИЛ-7, ИЛ-21 и ИЛ-15, T-клеточной терапии, противовоспалительного лекарственного средства, такого как кортикостероиды и нестероидные противовоспалительные лекарственные средства, и пищевой добавки, такой как антиоксиданты. В некоторых вариантах осуществления антитело против PD-1 можно вводить в комбинации с терапией, включающей химиотерапевтическое средство и хирургическую операцию. В рамках изобретения фраза "в комбинации с" означает, что антитело против PD-1 вводят индивидууму одновременно, непосредственно перед или непосредственно после осуществления лучевой терапии и введения дополнительного терапевтического средства. В некоторых вариантах осуществления дополнительное терапевтическое средство вводят в виде совместного состава с антителом против PD-1.
[0066] В некоторых вариантах осуществления настоящее изобретение относится к способам лечения крупных опухолей или злокачественных новообразований на поздней стадии, включающим введение нуждающемуся в этом индивидууму антитела против PD-1 в комбинации с лучевой терапией и дополнительным терапевтическим средством, где дополнительное терапевтическое средство вводят для преодоления опосредованной регуляторными T-клетками (Treg) иммуносупрессии. В некоторых вариантах осуществления дополнительное терапевтическое средство выбрано из группы, состоящей из антитела против GITR, антитела против LAG3, циклофосфамида и ГМ-КСФ.
[0067] В рамках изобретения термин "крупная опухоль" относится к размеру опухоли. Как правило, он коррелирует с более высокой опухолевой массой или опухолевой нагрузкой. В некоторых вариантах осуществления он коррелирует со стадией заболевания, например, злокачественным новообразованием на поздней стадии. В некоторых вариантах осуществления он коррелирует с повышенной вероятностью метастазирования.
[0068] В некоторых вариантах осуществления настоящее изобретение относится к способам, включающим введение одной или более доз антитела против PD-1 в комбинации с лучевой терапией и субтерапевтической дозой циклофосфамида. В рамках изобретения термин "субтерапевтическая доза циклофосфамида" (также обозначаемый в настоящем описании как "циклофосфамид в низких дозах") означает количество циклофосфамида, которое само по себе не имеет терапевтического эффекта и, предпочтительно, токсичность. Примеры доз циклофосфамида, считающиеся "субтерапевтическими" в контексте настоящего изобретения включают 100 мг/м2, 90 мг/м2, 80 мг/м2 или менее.
[0069] В некоторых вариантах осуществления лучевую терапию используют для первого очага опухоли, но не второго очага опухоли, где ее осуществление в комбинации с антителом против PD-1 приводит к регрессированию опухоли в первом и втором очагах опухоли (абскопальному эффекту). В некоторых вариантах осуществления способы по настоящему изобретению включают введение антитела против PD-1 в комбинации с лучевой терапией для достижения длительного абскопального эффекта.
[0070] В некоторых вариантах осуществления способы по настоящему изобретению включают введение нуждающемуся в этом индивидууму терапевтически эффективного количества антитела против PD-1, необязательно, в комбинации со второй противоопухолевой терапией, где введение комбинации приводит к повышенному ингибированию роста опухоли. В некоторых вариантах осуществления рост опухоли ингибируют по меньшей мере на приблизительно 10%, приблизительно 20%, приблизительно 30%, приблизительно 40%, приблизительно 50%, приблизительно 60%, приблизительно 70% или приблизительно 80% по сравнению с индивидуумом, неподвергнутым лечению, или индивидуумом, которому вводили антитело или подвергали второй противоопухолевой терапии в качестве монотерапии. В некоторых вариантах осуществления введение антитела против PD-1 и/или осуществление второй противоопухолевой терапии приводит к повышенному регрессированию опухоли, уменьшению и/или исчезновению опухоли. В некоторых вариантах осуществления введение антитела против PD-1 и/или химиотерапевтического средства приводит к замедлению роста и развития опухоли, например, рост опухоли можно замедлять на приблизительно 3 дня, более 3 дней, приблизительно 7 дней, более 7 дней, более 15 дней, более 1 месяца, более 3 месяцев, более 6 месяцев, более 1 года, более 2 лет или более 3 лет по сравнению с индивидуумом, неподвергнутым лечению, или индивидуумом, которого лечили с помощью антитела или химиотерапии в качестве монотерапии. В некоторых вариантах осуществления введение антитела против PD-1 в комбинации со второй противоопухолевой терапией (например, химиотерапией) предотвращает рецидивирование опухоли и/или повышает длительность выживания индивидуума, например, повышает длительность выживания более чем на 15 дней, более чем на 1 месяц, более чем на 3 месяца, более чем на 6 месяцев, более чем на 12 месяца, более чем на 18 месяцев, более чем на 24 месяца, более чем на 36 месяцев или более чем на 48 месяцев по сравнению с индивидуумом, неподвергнутым лечению, или индивидуумом, которому вводят антитело или подвергают второй противоопухолевой терапии в качестве монотерапии. В некоторых вариантах осуществления введение антитела против PD-1 в комбинации с дополнительной противоопухолевой терапией повышает выживаемость без прогрессирования или общую выживаемость. В некоторых вариантах осуществления введение антитела против PD-1 в комбинации с химиотерапией повышает ответ и длительность ответа у индивидуума, например, более чем на 2%, более чем на 3%, более чем на 4%, более чем на 5%, более чем на 6%, более чем на 7%, более чем на 8%, более чем на 9%, более чем на 10%, более чем на 20%, более чем на 30%, более чем на 40% или более чем на 50% по сравнению с индивидуумом, неподвергнутым лечению, или индивидуумом, которому вводили антитело или химиотерапевтическое средство в качестве монотерапии. В некоторых вариантах осуществления введение индивидууму со злокачественным новообразованием антитела против PD-1 и/или осуществление второй противоопухолевой терапии приводит к полному исчезновению всех признаков опухолевых клеток ("полному ответу"). В некоторых вариантах осуществления введение индивидууму со злокачественным новообразованием антитела против PD-1 и/или осуществление второй противоопухолевой терапии приводит к по меньшей мере 30%-ному или большему снижению опухолевых клеток или размера опухоли ("частичному ответу"). В некоторых вариантах осуществления введение индивидууму со злокачественным новообразованием антитела против PD-1 и/или осуществление второй противоопухолевой терапии приводит к полному или частичному исчезновению опухолевых клеток/очагов, включая новые измеримые очаги. Уменьшение опухоли можно измерять любыми из известных в этой области способов, например, посредством рентгенографии, позитронно-эмиссионной томографии (PET), компьютерной томографии (КТ), магнитно-резонансной томографии (МРТ), цитологических, гистологических или молекулярно-генетических анализов.
[0071] В некоторых вариантах осуществления способы по настоящему изобретению включают введение нуждающемуся в этом индивидууму терапевтически эффективного количества антитела против PD-1, где введение антитела против PD-1 приводит к повышению общей выживаемости (OS) или выживаемости без прогрессирования (PFS) пациента по сравнению с пациентом, подвергаемым лечению в соответствии со "стандартом лечения" (например, химиотерапии, хирургической операции или лучевой терапии). В некоторых вариантах осуществления PFS повышают по меньшей мере на один месяц, приблизительно 2 месяца, приблизительно 3 месяца, приблизительно 4 месяца, приблизительно 5 месяцев, приблизительно 6 месяцев, приблизительно 7 месяцев, приблизительно 8 месяцев, приблизительно 9 месяцев, приблизительно 10 месяцев, приблизительно 11 месяцев, приблизительно 1 год, приблизительно 2 года, приблизительно 3 года или более по сравнению с пациентом, подвергаемым химиотерапии препаратами платины. В некоторых вариантах осуществления OS повышают по меньшей мере на один месяц, приблизительно 2 месяца, приблизительно 3 месяца, приблизительно 4 месяца, приблизительно 5 месяцев, приблизительно 6 месяцев, приблизительно 7 месяцев, приблизительно 8 месяцев, приблизительно 9 месяцев, приблизительно 10 месяцев, приблизительно 11 месяцев, приблизительно 1 год, приблизительно 2 года, приблизительно 3 года или более по сравнению с пациентом, подвергаемым химиотерапии препаратами платины.
Способы супрессии регуляторных T-клеток
[0072] В некоторых аспектах настоящее изобретение относится к способам супрессии или ингибирования активации и/или пролиферации регуляторных T-клеток (Treg). В некоторых вариантах осуществления настоящее изобретение относится к способам супрессии активности клеток Treg. В этих аспектах способы включают выбор индивидуума с солидной опухолью и введение индивидууму антитела против PD-1 или его антигенсвязывающего фрагмента в комбинации по меньшей мере с одним из (i) лучевой терапии и (ii) антагониста глюкокортикоид-индуцируемого рецептора фактора некроза опухоли (GITR). В некоторых вариантах осуществления способы включают введение нуждающемуся в этом индивидууму антитела против PD-1 или его антигенсвязывающего фрагмента в комбинации с лучевой терапией и антагонистом GITR.
[0073] В некоторых вариантах осуществления антагонист GITR является антителом против GITR или его антигенсвязывающим фрагментом. В некоторых иллюстративных вариантах осуществления настоящего изобретения антитело против GITR или его антигенсвязывающий фрагмент содержит вариабельную область тяжелой цепи (HCVR), вариабельную область легкой цепи (LCVR) и/или определяющие комплементарность области (CDR), содержащие аминокислотные последовательности любых из антител против GITR, описанных в патентной заявке США № 62/256922 (поданной 18 ноября 2015 года), содержание которой включено в настоящее описание в полном объеме. Другие антитела против GITR, которые можно использовать в контексте способов по настоящему изобретению, включают любые из антител против GITR, описанных, например, в патентах США №№ 9228016, 8709424, 8591886, 7812135 или патентной публикации США № 20150368349.
[0074] В некоторых вариантах осуществления настоящее изобретение относится к способам супрессии или устранения активности Treg, включающим введение нуждающемуся в этом индивидууму антитела против PD-1 или его антигенсвязывающего фрагмента в комбинации с одной или более дозами лучевой терапии и антагониста антигена-4 цитотоксических T-лимфоцитов (CTLA). В некоторых вариантах осуществления антагонист CTLA является антителом против CTLA (например, ипилимумабом).
[0075] В некоторых вариантах осуществления настоящее изобретение относится к способам супрессии или устранения активности Treg, включающим введение нуждающемуся в этом индивидууму антитела против PD-1 или его антигенсвязывающего фрагмента в комбинации с одной или более дозами лучевой терапии и антагониста гена активации лимфоцитов 3 (LAG-3). В некоторых вариантах осуществления антагонист LAG-3 является антителом против LAG-3. Антитела против LAG-3, которые можно использовать в контексте способов по настоящему изобретению, описаны в патентной заявке США № 62/239524 (поданной 9 октября 2015 года), содержание которой включено в настоящее описание в полном объеме.
[0076] В некоторых вариантах осуществления настоящее изобретение относится к способам супрессии или устранения активности Treg, включающим введение нуждающемуся в этом индивидууму антитела против PD-1 или его антигенсвязывающего фрагмента в комбинации с одной или более дозами лучевой терапии и циклофосфамида.
[0077] В одном из аспектов способы по настоящему изобретению включают введение индивидууму с солидной опухолью антитела против PD-1 в комбинации с лучевой терапией и дополнительным терапевтическим средством, выбранным из группы, состоящей из антагониста GITR, антитела против LAG-3 и циклофосфамида, где введение приводит к эффекту, выбранному из группы, состоящей из ингибирования роста опухоли, уменьшения размера опухоли, замедления роста опухоли, ингибирования метастазирования опухоли, уменьшения метастазов с течением времени, снижения использования химиотерапевтических или цитотоксических средств, повышения выживаемости, полного ответа, частичного ответа и стабильного заболевания. В некоторых вариантах осуществления введение приводит к уменьшению опухолевой массы у индивидуума. В некоторых вариантах осуществления индивидуум имеет крупную опухоль. Как определено где-либо в настоящем описании, термин "крупная опухоль" относится к размеру опухоли, и он коррелирует с повышенной опухолевой массой и повышенной вероятностью метастазирования. В некоторых вариантах осуществления термин относится к злокачественному новообразованию на поздней стадии.
Антитела против PD-1 и их антигенсвязывающие фрагменты
[0078] В некоторых иллюстративных вариантах осуществления настоящего изобретения способы включают введение терапевтически эффективного количества антитела против PD-1 или его антигенсвязывающего фрагмента. В рамках изобретения термин "антитело" включает молекулы иммуноглобулинов, содержащие четыре полипептидные цепи, две тяжелые (H) цепи и две легкие (L) цепи, соединенные друг с другом дисульфидными связями, а также их мультимеры (например, IgM). В типичном антителе каждая тяжелая цепь содержит вариабельную область тяжелой цепи (сокращенно обозначаемую в настоящем описании как HCVR или VH) и константную область тяжелой цепи. Константная область тяжелой цепи содержит три домена, CH1, CH2 и CH3. Каждая легкая цепь содержит вариабельную область легкой цепи (сокращенно обозначаемую в настоящем описании как LCVR или VL) и константную область легкой цепи. Константная область легкой цепи содержит один домен (CL1). Области VH и VL можно дополнительно разделять на гипервариабельные области, обозначаемые как определяющие комплементарность области (CDR), чередующиеся с областями, являющимися более консервативными, обозначаемыми как каркасные области (FR). Каждая VH и VL состоит из трех CDR и четырех FR, расположенных от амино-конца до карбокси-конца в следующем порядке: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. В различных вариантах осуществления изобретения FR антитела против ИЛ-4R (или его антигенсвязывающей части) может быть идентичной последовательностям зародышевой линии человека или природно или искусственно модифицированной. Консенсусную аминокислотную последовательность можно определять с учетом одновременного анализа двух или более CDR.
[0079] В рамках изобретения термин "антитело" также включает антигенсвязывающие фрагменты полных молекул антител. В рамках изобретения термины "антигенсвязывающая часть" антитела, "антигенсвязывающий фрагмент" антитела и т.п. включают любой природный, получаемый ферментативно, синтетический или генетически сконструированный полипептид или гликопротеин, специфически связывающийся с антигеном с образованием комплекса. Антигенсвязывающие фрагменты антитела можно получать, например, из полных молекул антител любыми подходящими стандартными способами, такими как протеолитическое расщепление или рекомбинантные способы генетической инженерии, включающими воздействие на ДНК, кодирующую вариабельные и, необязательно, константные домены антитела, и ее экспрессию. Такая ДНК известна и/или доступна, например, в коммерческих источниках, библиотеках ДНК (включая, например, фаговые библиотеки антител), или ее можно синтезировать. ДНК можно секвенировать и воздействовать на нее химически или способами молекулярной биологии, например, для расположения одного или более вариабельных и/или константных доменов в подходящей конфигурации или для встраивания кодонов, создания остатков цистеина, модификации, добавления или делеции аминокислот и т.д.
[0080] Неограничивающие примеры антигенсвязывающих фрагментов включают: (i) Fab-фрагменты; (ii) F(ab')2-фрагменты; (iii) Fd-фрагменты; (iv) Fv-фрагменты; (v) одноцепочечные молекулы Fv (scFv); (vi) dAb-фрагменты; и (vii) минимальные распознающие единицы, состоящие из аминокислотных остатков, имитирующих гипервариабельную область антитела (например, выделенной определяющей комплементарность области (CDR), такой как пептид CDR3), или пептид FR3-CDR3-FR4 с ограниченной конформационной свободой. Другие сконструированные молекулы, такие как домен-специфические антитела, однодоменные антитела, антитела с делетированным доменом, химерные антитела, антитела с пересаженными CDR, диатела, триатела, тетратела, миниантитела, нанотела (например, моновалентные нанотела, бивалентные нанотела и т.д.), малые модульные иммунофармацевтические средства (SMIP) и вариабельные домены IgNAR акулы, также включены в выражение "антигенсвязывающий фрагмент" в рамках изобретения.
[0081] Антигенсвязывающий фрагмент антитела, как правило, будет содержать по меньшей мере один вариабельный домен. Вариабельный домен может иметь любой размер или аминокислотную композицию и, как правило, будет содержать по меньшей мере одну CDR, смежную с одной или более каркасными последовательностями или находящуюся с ними в рамке считывания. В антигенсвязывающих фрагментах, содержащих домен VH, связанный с доменом VL, домены VH и VL можно располагать относительно друг друга в любой подходящей конфигурации. Например, вариабельная область может быть димерной и содержать димеры VH-VH, VH-VL или VL-VL. Альтернативно, антигенсвязывающий фрагмент антитела может содержать мономерный домен VH или VL.
[0082] В некоторых вариантах осуществления антигенсвязывающий фрагмент антитела может содержать по меньшей мере один вариабельный домен, ковалентно связанный с по меньшей мере одним константным доменом. Неограничивающие примеры конфигураций вариабельных и константных доменов, которые можно обнаружить в антигенсвязывающем фрагменте антитела по настоящему изобретению, включают: (i) VH-CH1; (ii) VH-CH2; (iii) VH-CH3; (iv) VH-CH1-CH2; (v) VH-CH1-CH2-CH3; (vi) VH-CH2-CH3; (vii) VH-CL; (viii) VL-CH1; (ix) VL-CH2; (x) VL-CH3; (xi) VL-CH1-CH2; (xii) VL-CH1-CH2-CH3; (xiii) VL-CH2-CH3; и (xiv) VL-CL. В любой конфигурации вариабельных и константных доменов, включая любой из указанных выше примеров конфигураций, вариабельные и константные домены можно соединять напрямую друг с другом или с помощью полной или частичной шарнирной или линкерной области. Шарнирная область может состоять из по меньшей мере 2 (например, 5, 10, 15, 20, 40, 60 или более) аминокислот, что приводит к получению гибкого или полугибкого соединения между смежными вариабельными и/или константными доменами в одной полипептидной молекуле. Кроме того, антигенсвязывающий фрагмент антитела по настоящему изобретению может содержать гомодимер или гетеродимер (или другой мультимер) из любой из конфигураций вариабельных и константных доменов, указанных выше, в нековалентном соединении друг с другом и/или с одним или более мономерными доменами VH или VL (например, с помощью дисульфидных связей).
[0083] В рамках изобретения термин "антитело" также включает мультиспецифические (например, биспецифические) антитела. Мультиспецифическое антитело или антигенсвязывающий фрагмент антитела, как правило, будет содержать по меньшей мере два разных вариабельных домена, где каждый вариабельный домен может специфически связываться с отдельным антигеном или с другим эпитопом на том же антигене. Любой формат мультиспецифического антитела можно адаптировать для использования в контексте антитела или антигенсвязывающего фрагмента антитела по настоящему изобретению с использованием общепринятых способов, доступных в этой области. Например, настоящее изобретение относится к способам, включающим использование биспецифических антител, где одно плечо иммуноглобулина является специфическим для PD-1 или его фрагмента, а другое плечо иммуноглобулина является специфическим для второй терапевтической мишени или конъюгировано с терапевтическим веществом. Неограничивающие примеры биспецифических форматов, которые можно использовать в контексте настоящего изобретения, включают, например, биспецифические форматы на основе scFv или диатела, слитые белки IgG-scFv, Ig с двойным вариабельным доменом (DVD), квадрому, белки со структурой "выступы во впадину", общую легкую цепь (например, общую легкую цепь с "выступами во впадину" и т.д.), CrossMab, CrossFab, антитело, содержащее домен, сконструированный с помощью замены цепей (SEED), лейциновую молнию, дуотело, IgG1/IgG2, IgG с Fab двойного действия (DAF) и биспецифические форматы Mab2 (см. обзор указанных выше форматов, например, в Klein et al. 2012, mAbs 4:6, 1-11 и цитируемых в ней ссылках). Биспецифические антитела также можно конструировать с использованием конъюгации пептида/нуклеиновой кислоты, например, где неприродные аминокислоты с независимой реакционной способностью используют для получения сайт-специфических конъюгатов антитело-олигонуклеотид, которые затем самостоятельно собираются в мультимерные комплексы с определенной композицией, валентностью и конфигурацией (см., например, Kazane et al., J. Am. Chem. Soc. [Epub: Dec. 4, 2012]).
[0084] Антитела, используемые в способах по настоящему изобретению, могут являться антителами человека. В рамках изобретения термин "антитело человека" предназначен для включения антител, содержащих вариабельные и константные области, полученные из последовательностей иммуноглобулинов зародышевой линии человека. Тем не менее, антитела человека по изобретению могут включать аминокислотные остатки, не кодируемые последовательностями иммуноглобулинов зародышевой линии человека (например, мутации, встраиваемые при случайном или сайт-специфическом мутагенезе in vitro или посредством соматической мутации in vivo), например, в CDR и, в частности, CDR3. Однако, в рамках изобретения термин "антитело человека" не предназначен для включения антител, в которых последовательности CDR, полученные из зародышевой линии другого вида млекопитающих, такого как мышь, пересаживают на каркасные последовательности человека.
[0085] Антитела, используемые в способах по настоящему изобретению, могут являться рекомбинантными антителами человека. В рамках изобретения термин "рекомбинантное антитело человека" предназначен для включения всех антител человека, полученных, экспрессируемых, созданных или выделенных рекомбинантными способами, таких как антитела, экспрессируемые с использованием рекомбинантного экспрессирующего вектора, с помощью которого трансфицируют клетку-хозяина (описанную ниже), антитела, выделенные из рекомбинантной, комбинаторной библиотеки антител человека (описанной ниже), антитела, выделенные из животного (например, мыши), являющегося трансгенным по генам иммуноглобулинов человека [см. например, Taylor et al. (1992) Nucl. Acids Res. 20:6287-6295], или антитела, полученные, экспрессируемые, созданные или выделенные любыми другими способами, включающими сплайсинг последовательностей генов иммуноглобулинов человека с другими последовательностями ДНК. Такие рекомбинантные антитела человека содержат вариабельные и константные области, полученные из последовательностей иммуноглобулинов зародышевой линии человека. Однако в некоторых вариантах осуществления такие рекомбинантные антитела человека подвергают мутагенезу in vitro (или, если используют животное, трансгенное по последовательностям Ig человека, соматическому мутагенезу in vivo), и, таким образом, аминокислотные последовательности областей VH и VL рекомбинантных антител являются последовательностями, которые, хотя и получены из последовательностей VH и VL зародышевой линии человека и связаны с ними, могут не существовать в природе в репертуаре антител зародышевой линии человека in vivo.
[0086] В некоторых вариантах осуществления антитела, используемые в способах по настоящему изобретению, специфически связываются с PD-1. Термин "специфически связывается" или т.п. означает, что антитело или его антигенсвязывающий фрагмент образует комплекс с антигеном, являющийся относительно стабильным в физиологических условиях. Способы определения того, связывается ли антитело специфически с антигеном, хорошо известны в этой области и включают, например, равновесный диализ, поверхностный плазмонный резонанс и т.п. Например, антитело, "специфически связывающееся" с PD-1, в контексте настоящего изобретения включает антитела, связывающиеся с PD-1 или его частью с KD менее приблизительно 500 нМ, менее приблизительно 300 нМ, менее приблизительно 200 нМ, менее приблизительно 100 нМ, менее приблизительно 90 нМ, менее приблизительно 80 нМ, менее приблизительно 70 нМ, менее приблизительно 60 нМ, менее приблизительно 50 нМ, менее приблизительно 40 нМ, менее приблизительно 30 нМ, менее приблизительно 20 нМ, менее приблизительно 10 нМ, менее приблизительно 5 нМ, менее приблизительно 4 нМ, менее приблизительно 3 нМ, менее приблизительно 2 нМ, менее приблизительно 1 нМ или менее приблизительно 0,5 нМ, как измеряют с помощью анализа поверхностного плазмонного резонанса. Однако, выделенное антитело, специфически связывающееся с PD-1 человека, может иметь перекрестную реактивность в отношении других антигенов, таких как молекулы PD-1 другого вида (не принадлежащие человеку).
[0087] В некоторых иллюстративных вариантах осуществления настоящего изобретения антитело против PD-1 или его антигенсвязывающий фрагмент содержит вариабельную область тяжелой цепи (HCVR), вариабельную область легкой цепи (LCVR) и/или определяющие комплементарность области (CDR), содержащие аминокислотные последовательности любых из антител против PD-1, описанных в патентной публикации СЩА № 20150203579, включенной, таким образом, в настоящее описание в качестве ссылки в полном объеме. В некоторых иллюстративных вариантах осуществления антитело против PD-1 или его антигенсвязывающий фрагмент, который можно использовать в контексте способов по настоящему изобретению, содержит определяющие комплементарность области (HCDR) вариабельной области тяжелой цепи (HCVR), содержащей аминокислотную последовательность SEQ ID NO: 1, и определяющие комплементарность области (LCDR) вариабельной области легкой цепи (LCVR), содержащей аминокислотную последовательность SEQ ID NO: 2. В некоторых вариантах осуществления антитело против PD-1 или его антигенсвязывающий фрагмент содержит три HCDR (HCDR1, HCDR2 и HCDR3) и три LCDR (LCDR1, LCDR2 и LCDR3), где HCDR1 содержит аминокислотную последовательность SEQ ID NO: 3; HCDR2 содержит аминокислотную последовательность SEQ ID NO: 4; HCDR3 содержит аминокислотную последовательность SEQ ID NO: 5; LCDR1 содержит аминокислотную последовательность SEQ ID NO: 6; LCDR2 содержит аминокислотную последовательность SEQ ID NO: 7; и LCDR3 содержит аминокислотную последовательность SEQ ID NO: 8. В других вариантах осуществления антитело против PD-1 или его антигенсвязывающий фрагмент содержит HCVR, содержащую SEQ ID NO: 1, и LCVR, содержащую SEQ ID NO: 2. В некоторых вариантах осуществления способы по настоящему изобретению включают использование антитела против PD-1, где антитело содержит тяжелую цепь, содержащую аминокислотную последовательность SEQ ID NO: 9. В некоторых вариантах осуществления антитело против PD-1 содержит легкую цепь, содержащую аминокислотную последовательность SEQ ID NO: 10. Примером антитела, содержащего тяжелую цепь, содержащую аминокислотную последовательность SEQ ID NO: 9, и легкую цепь, содержащую аминокислотную последовательность SEQ ID NO: 10, является полностью человеческое антитело против PD-1, известное как REGN2810 (также известное как цемиплимаб). В некоторых иллюстративных вариантах осуществления способы по настоящему изобретению включают использование REGN2810 или его биоэквивалента. В рамках изобретения термин "биоэквивалент" относится к антителам против PD-1 или PD-1-связывающим белкам или его фрагментам, являющимся фармацевтическими эквивалентами или фармацевтическими альтернативами, скорость и/или степень абсорбции которых не отличаются значительно от скорости и/или степени абсорбции REGN2810 при введении в той же молярной дозе в схожих экспериментальных условиях в однократной дозе или многократных дозах. В контексте изобретения термин относится к антигенсвязывающим белкам, связывающимся с PD-1 и не имеющим клинически значимых отличий от REGN2810 в безопасности, чистоте и/или активности.
[0088] Другие антитела против PD-1, которые можно использовать в контексте способов по настоящему изобретению, включают, например, антитела, обозначенные и известные в этой области как ниволумаб (патент США № 8008449), пембролизумаб (патент США № 8354509), MEDI0608 (патент США № 8609089), пидилизумаб (патент США № 8686119) или любые из антител против PD-1, описанных в патентах США №№ 6808710, 7488802, 8168757, 8354509, 8779105 или 8900587.
[0089] Антитела против PD-1, используемые в контексте способов по настоящему изобретению, могут иметь pH-зависимые характеристики связывания. Например, антитело против PD-1 для использования в способах по настоящему изобретению может проявлять сниженное связывание с PD-1 при кислом pH по сравнению с нейтральным pH. Альтернативно, антитело против PD-1 по изобретению может проявлять повышенное связывание со своим антигеном при кислом pH по сравнению с нейтральным pH. Выражение "кислый pH" включает значения pH менее приблизительно 6,2, например, приблизительно 6,0, 5,95, 5,9, 5,85, 5,8, 5,75, 5,7, 5,65, 5,6, 5,55, 5,5, 5,45, 5,4, 5,35, 5,3, 5,25, 5,2, 5,15, 5,1, 5,05, 5,0 или менее. В рамках изобретения выражение "нейтральный pH" означает pH от приблизительно 7,0 до приблизительно 7,4. Выражение "нейтральный pH" включает значения pH приблизительно 7,0, 7,05, 7,1, 7,15, 7,2, 7,25, 7,3, 7,35 и 7,4.
[0090] В некоторых случаях "сниженное связывание с PD-1 при кислом pH по сравнению с нейтральным pH" выражают в терминах соотношения значения KD связывания антитела с PD-1 при кислом pH и значения KD связывания антитела с PD-1 при нейтральном pH (или наоборот). Например, антитело или его антигенсвязывающий фрагмент можно рассматривать как проявляющий "сниженное связывание с PD-1 при кислом pH по сравнению с нейтральным pH" в целях по настоящему изобретению, если антитело или его антигенсвязывающий фрагмент демонстрирует соотношение KD при кислом/нейтральном pH приблизительно 3,0 или более. В некоторых иллюстративных вариантах осуществления соотношение KD при кислом/нейтральном pH для антитела или антигенсвязывающего фрагмента по настоящему изобретению может составлять приблизительно 3,0, 3,5, 4,0, 4,5, 5,0, 5,5, 6,0, 6,5, 7,0, 7,5, 8,0, 8,5, 9,0, 9,5, 10,0, 10,5, 11,0, 11,5, 12,0, 12,5, 13,0, 13,5, 14,0, 14,5, 15,0, 20,0, 25,0, 30,0, 40,0, 50,0, 60,0, 70,0, 100,0 или более.
[0091] Антитела с pH-зависимыми характеристиками связывания можно получать, например, посредством скрининга популяции антител на сниженное (или повышенное) связывание с конкретным антигеном при кислом pH по сравнению с нейтральным pH. Кроме того, с помощью модификаций антигенсвязывающего домена на уровне аминокислот можно получать антитела с pH-зависимыми характеристиками. Например, заменяя одну или более аминокислот антигенсвязывающего домена (например, в CDR) остатком гистидина, можно получать антитело со сниженным связыванием антигена при кислом pH относительно нейтрального pH. В рамках изобретения выражение "кислый pH" означает pH 6,0 или менее.
Способы комбинированного лечения
[0092] В некоторых вариантах осуществления способы по настоящему изобретению включают подвергание индивидуума дополнительной противоопухолевой терапии в комбинации с антителом против PD-1. В некоторых вариантах осуществления способы по настоящему изобретению включают осуществление лучевой терапии или химиотерапии в комбинации с антителом против PD-1 для аддитивной или синергичной активности для лечения злокачественного новообразования. В рамках изобретения выражение "в комбинации с" означает, что дополнительную противоопухолевую терапию проводят до, после или одновременно с введением антитела против PD-1. Термин "в комбинации с" также включает последовательное или одновременное введение антитела против PD-1 и осуществление дополнительной противоопухолевой терапии. Например, при введении "до" дополнительной противоопухолевой терапии антитело против PD-1 можно вводить за более чем 150 часов, приблизительно 150 часов, приблизительно 100 часов, приблизительно 72 часа, приблизительно 60 часов, приблизительно 48 часов, приблизительно 36 часов, приблизительно 24 часа, приблизительно 12 часов, приблизительно 10 часов, приблизительно 8 часов, приблизительно 6 часов, приблизительно 4 часа, приблизительно 2 часа, приблизительно 1 час или приблизительно 30 минут, приблизительно 15 минут или приблизительно 10 минут до осуществления дополнительной терапии. При введении "после" дополнительной противоопухолевой терапии антитело против PD-1 можно вводить через приблизительно 10 минут, приблизительно 15 минут, приблизительно 30 минут, приблизительно 1 час, приблизительно 2 часа, приблизительно 4 часа, приблизительно 6 часов, приблизительно 8 часов, приблизительно 10 часов, приблизительно 12 часов, приблизительно 24 часа, приблизительно 36 часов, приблизительно 48 часов, приблизительно 60 часов, приблизительно 72 часа или более 72 часов после осуществления дополнительной противоопухолевой терапии. Введение "одновременно" с дополнительной противоопухолевой терапией означает, что антитело против PD-1 вводят индивидууму в пределах менее 10 минут (до, после или одновременно) относительно осуществления дополнительной противоопухолевой терапии.
[0093] В некоторых вариантах осуществления способы по настоящему изобретению включают введение дополнительного терапевтического средства, где дополнительное терапевтическое средство является противоопухолевым лекарственным средством. В рамках изобретения термин "противоопухолевое лекарственное средство" означает любое средство, которое можно использовать для лечения злокачественного новообразования, включая в качестве неограничивающих примеров, цитотоксины и такие средства, как антиметаболиты, алкилирующие средства, антрациклины, антибиотики, антимитотические средства, прокарбазин, гидроксимочевина, аспарагиназа, кортикостероиды, митотан (O,P′-(DDD)), биологические средства (например, антитела и интерфероны) и радиоактивные средства. В рамках изобретения термин "цитотоксин или цитотоксическое средство" также относится к химиотерапевтическому средству и означает любое средство, являющееся вредным для клеток. Неограничивающие примеры включают Таксол® (паклитаксел), темозоломид, цитохалазин B, грамицидин D, бромистый этидий, эметин, цисплатин, митомицин, этопозид, тенипозид, винкристин, винбластин, колхицин, доксорубицин, даунорубицин, дигидроксиантрациндион, митоксантрон, митрамицин, актиномицин D, 1-дегидротестостерон, глюкокортикоиды, прокаин, тетракаин, лидокаин, пропранолол и пуромицин и их аналоги или гомологи.
[0094] В некоторых вариантах осуществления способы по настоящему изобретению включают использование дополнительного терапевтического средства, или схемы лечения, или способа, выбранного из группы, состоящей из хирургической операции, лучевой терапии, ингибитора лиганда программируемой гибели клеток 1 (PD-L1) (например, антитела против PD-L1, как описано в патентной публикации США № 2015/0203580, или атезолизумаба), ингибитора гена активации лимфоцитов 3 (LAG-3) (например, антитела против LAG-3), ингибитора антигена-4 цитотоксических T-лимфоцитов (CTLA-4) (например, антитела против CTLA-4, такого как ипилимумаб), ингибитора глюкокортикоид-индуцируемого рецептора фактора некроза опухоли (GITR) (например, антитела против GITR), ингибитора белка-3, содержащего T-клеточный иммуноглобулин и домен муцина (TIM3), ингибитора B- и T-лимфоцитарного аттенюатора (BTLA), ингибитора T-клеточного иммунорецептора с доменами Ig и ITIM (TIGIT), ингибитора CD47, антагониста другого T-клеточного коингибитора или лиганда (например, антитела против CD28, 2B4, LY108, LAIR1, ICOS, CD160 или VISTA), ингибитора CD20 (например, антитела против CD20 или биспецифического антитела CD3/CD20), ингибитора индоламин-2,3-диоксигеназы (IDO), антагониста фактора роста эндотелия сосудов (VEGF) [например, "VEGF-ловушки", такой как афлиберцепт или другой VEGF-ингибирующий слитый белок, как описано в патенте США № 7087411, или антитело против VEGF или его антигенсвязывающий фрагмент (например, бевацизумаб или ранибизумаб) или низкомолекулярный ингибитор киназ рецептора VEGF (например, сунитиниб, сорафениб или пазопаниб)], ингибитора ангиопоэтина 2 (Ang2) (например, несвакумаба), ингибитора трансформирующего фактора роста бета (TGFβ), ингибитора рецептора эпидермального фактора роста (EGFR) (например, эрлотиниба, цетуксимаба), агониста костимуляторного рецептора (например, агониста глюкокортикоид-индуцируемого TNFR-родственного белка), антитела против опухолеспецифичного антигена [например, CA9, CA125, меланома-ассоциированного антигена 3 (MAGE3), карциноэмбрионального антигена (CEA), виментина, опухолевого M2-PK, простат-специфического антигена (PSA), муцина-1, MART-1 и CA19-9], вакцины (например, бациллы Кальметта-Герена, противоопухолевой вакцины), циклофосфамида, адъюванта для повышения презентации антигена (например, гранулоцитарно-макрофагального колониестимулирующего фактора), цитотоксина, химиотерапевтического средства (например, дакарбазина, темозоломида, доцетаксела, доксорубицина, даунорубицина, цисплатина, карбоплатина, гемцитабина, метотрексата, митоксантрона, оксалиплатина, паклитаксела и винкристина), ингибитора рецептора интерлейкина-6 (ИЛ-6R) (например, сарилумаба), ингибитора ИЛ-4R (например, дупилумаба), ингибитора ИЛ-10, цитокина, такого как ИЛ-2, ИЛ-7, ИЛ-21 и ИЛ-15, конъюгата антитело-лекарственное средство (ADC) (например, ADC против CD19-DM4 и ADC против DS6-DM4), T-клеток с химерным антигенным рецептором (например, T-клеток против CD19), противовоспалительного лекарственного средства (например, кортикостероидов и нестероидных противовоспалительных лекарственных средств) и пищевой добавки, такой как антиоксиданты.
[0095] В некоторых вариантах осуществления способы по изобретению включают введение антитела против PD-1 в комбинации с лучевой терапией и, необязательно, антителом против GITR для достижения длительных, устойчивых противоопухолевых ответов и/или повышения выживаемости пациентов со злокачественным новообразованием. В некоторых вариантах осуществления способы по изобретению включают осуществление лучевой терапии до, одновременно или после введения антитела против PD-1 и антитела против GITR пациенту со злокачественным новообразованием. Например, лучевую терапию можно осуществлять в одной или более дозах в отношении очагов опухоли после введения одной или более доз антитела. В некоторых вариантах осуществления можно местно подвергать очаг опухоли лучевой терапии для повышения местной иммуногенности в опухоли пациента (адъювантная лучевая терапия) и/или для уничтожения опухолевых клеток (абляционная лучевая терапия) после системного введения антитела против PD-1 и/или антитела против GITR. В некоторых вариантах осуществления лучевой терапии подвергают первый очаг опухоли, но не второй очаг опухоли, где введение в комбинации с антителом против PD-1 приводит к регрессированию опухоли в первом и втором очагах опухоли (абскопальному эффекту). В некоторых вариантах осуществления способы по настоящему изобретению включают введение антитела против PD-1 в комбинации с лучевой терапией и, необязательно, антитела против GITR для достижения длительного абскопального эффекта.
[0096] В некоторых вариантах осуществления антитело против PD-1 можно вводить в комбинации с лучевой терапией и химиотерапевтическим средством (например, темозоломидом или циклофосфамидом), антагонистом VEGF (например, афлиберцептом) или гранулоцитарно-макрофагальным колониестимулирующим фактором.
Фармацевтические композиции и введение
[0097] Настоящее изобретение относится к способам, включающим введение индивидууму антитела против PD-1 в комбинации с лучевой терапией, где антитело против PD-1 содержится в фармацевтической композиции. Фармацевтические композиции по изобретению можно составлять с подходящими носителями, эксципиентами и другими средствами, обеспечивающими подходящий перенос, доставку, переносимость и т.п. Множество соответствующих составов можно найти в справочниках, известных всем химикам-фармацевтам: Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA. Эти составы включают, например, порошки, пасты, мази, желе, воски, масла, липиды, везикулы, содержащие липид (катионный или анионный) (такие как LIPOFECTIN™), конъюгаты ДНК, безводные абсорбционные пасты, эмульсии "масло-в-воде" и "вода-в-масле", эмульсии карбовакса (полиэтиленгликолей различных молекулярных масс), полутвердые гели и полутвердые смеси, содержащие карбовакс. Также см. Powell et al. "Compendium of excipients for parenteral formulations" PDA (1998) J Pharm Sci Technol 52:238-311.
[0098] Известны различные системы доставки, и их можно использовать для введения фармацевтической композиции по изобретению, например, инкапсуляция в липосомах, микрочастицы, микрокапсулы, рекомбинантные клетки, которые могут экспрессировать мутантные вирусы, рецептор-опосредованный эндоцитоз (см., например, Wu et al., 1987, J. Biol. Chem. 262: 4429-4432). Способы введения включают, в качестве неограничивающих примеров, внутрикожный, внутримышечный, интраперитонеальный, внутривенный, подкожный, интраназальный, эпидуральный и пероральный пути. Композицию можно вводить любым удобным путем, например, посредством инфузии или болюсной инъекции, посредством абсорбции через эпителиальные или слизистые выстилки (например, слизистую оболочку ротовой полости, слизистую оболочку кишечника и т.д.), и ее можно вводить вместе с другими биологически активными средствами.
[0099] Фармацевтическую композицию по настоящему изобретению можно вводить подкожно или внутривенно с использованием стандартной иглы и шприца. Кроме того, что касается подкожного введения, для введения фармацевтической композиции по настоящему изобретению легко можно использовать шприц-ручку. Такой шприц-ручка может быть многоразовым или одноразовым. В многоразовом шприце-ручке, как правило, используют сменный картридж, содержащий фармацевтическую композицию. После введения всей фармацевтической композиции в картридже и опустошения картриджа, пустой картридж можно выбрасывать и заменять новым картриджем, содержащим фармацевтическую композицию. Затем шприц-ручку можно повторно использовать. В одноразовом шприце-ручке нет сменного картриджа. Вместо этого одноразовый шприц-ручку заранее наполняют фармацевтической композицией, содержащейся в резервуаре устройства. После опустошения резервуара все устройство выбрасывают.
[00100] В некоторых случаях фармацевтическую композицию можно вводить в системе с контролируемым высвобождением. В одном из вариантов осуществления можно использовать помпу. В другом варианте осуществления можно использовать полимерные материалы; см., Medical Applications of Controlled Release, Langer and Wise (eds.), 1974, CRC Pres., Boca Raton, Florida. В еще одном варианте осуществления систему с контролируемым высвобождением можно помещать вблизи целевого очага для композиции, таким образом, требуется лишь часть системной дозы (см., например, Goodson, 1984, в Medical Applications of Controlled Release, выше, vol. 2, pp. 115-138). Другие системы с контролируемым высвобождением описывают в обзоре Langer, 1990, Science 249:1527-1533.
[00101] Инъецируемые препараты могут включать лекарственные формы для внутривенных, подкожных, внутрикожных и внутримышечных инъекций, капельных инфузий и т.д. Эти инъецируемые препараты можно получать известными способами. Например, инъецируемые препараты можно получать, например, посредством растворения, суспендирования или эмульгирования антитела или его соли, описанных выше, в стерильной водной среде или масляной среде, общепринято используемой для инъекций. В качестве водной среды для инъекций можно упомянуть, например, физиологический раствор, изотонический раствор, содержащий глюкозу и другие вспомогательные средства и т.д., которые можно использовать в комбинации с подходящим солюбилизатором, таким как спирт (например, этанол), полиспирт (например, пропиленгликоль, полиэтиленгликоль), неионное поверхностно-активное вещество [например, полисорбат 80, HCO-50 (аддукт полиоксиэтилена (50 моль) и гидрогенизированного касторового масла)] и т.д. В качестве масляной среды используют, например, сезамовое масло, соевое масло и т.д., которые можно использовать в комбинации с солюбилизатором, таким как бензил бензоат, бензиловый спирт и т.д. Полученным таким образом инъекционным препаратом, предпочтительно, наполняют подходящую ампулу.
[00102] Предпочтительно, фармацевтические композиции для перорального или парентерального использования, описанные выше, получают в лекарственных формах в однократной дозе, соответствующей дозе активных ингредиентов. Такие лекарственные формы в однократной дозе включают, например, таблетки, пилюли, капсулы, инъекционные препараты (ампулы), суппозитории и т.д.
[00103] В некоторых вариантах осуществления настоящее изобретение относится к фармацевтическому составу, содержащему терапевтическое количество антитела против PD-1 и фармацевтический носитель. В некоторых вариантах осуществления настоящее изобретение относится к антителу против PD-1, составленному в фармацевтической композиции для внутривенного введения.
Схемы введения
[00104] Настоящее изобретение относится к способам, включающим введение индивидууму антитела против PD-1 с частотой приблизительно четыре раза в неделю, дважды в неделю, раз в неделю, раз в две недели, раз в три недели, раз в четыре недели, раз в пять недель, раз в шесть недель, раз в восемь недель, раз в двенадцать недель или реже при условии, что достигают терапевтического ответа. В некоторых вариантах осуществления способы включают введение антитела против PD-1 в комбинации со второй противоопухолевой терапией (например, химиотерапией) с частотой приблизительно дважды в неделю, раз в неделю, раз в две недели, раз в три недели, раз в четыре недели, раз в пять недель, раз в шесть недель, раз в восемь недель, раз в девять недель, раз в двенадцать недель или реже при условии, что достигают терапевтического ответа.
[00105] В некоторых вариантах осуществления способы по настоящему изобретению включают осуществление лучевой терапии, где лучевая терапия является гипофракционированной лучевой терапией. В некоторых вариантах осуществления гипофракционированная лучевая терапия включает 2-12 фракций. В некоторых вариантах осуществления 2-12 фракций вводят в последовательные дни. В некоторых вариантах осуществления лучевую терапию осуществляют после введения одной или более доз антитела против PD-1. В некоторых вариантах осуществления антитело против PD-1 вводят за 0,5-2 недели до использования одной или более фракций лучевой терапии.
[00106] В некоторых вариантах осуществления настоящего изобретения многократные дозы антитела против PD-1 в комбинации со второй противоопухолевой терапией (например, химиотерапией) можно вводить индивидууму в течение определенного времени. Способы по изобретению по этому аспекту включают последовательное введение индивидууму одной или более доз антитела против PD-1 в комбинации с одной или более дозами указанной второй противоопухолевой терапии. В рамках изобретения термин "последовательное введение" означает, что каждую дозу антитела вводят индивидууму в разные моменты времени, например, в разные дни с заранее определенными интервалами (например, часы, дни, недели или месяцы). В некоторых вариантах осуществления способы по настоящему изобретению включают последовательное введение одной или более доз антитела против PD-1, где каждую дозу вводят через 0,5-12 недель после предыдущей дозы. В некоторых дополнительных вариантах осуществления способы дополнительно включают осуществление второй противоопухолевой терапии (например, химиотерапии). В некоторых вариантах осуществления химиотерапия может являться химиотерапией препаратами платины. В некоторых вариантах осуществления способы дополнительно включают введение одной или более доз химиотерапевтического средства, где каждую дозу вводят через 1-6 недель после предыдущей дозы.
[00107] В некоторых вариантах осуществления настоящее изобретение относится к способам, включающим последовательное введение пациенту одной начальной дозы антитела против PD-1 с последующими одной или более вторичными дозами антитела против PD-1 и, необязательно, одной или более третичными дозами антитела против PD-1. В некоторых вариантах осуществления способы дополнительно включают последовательное введение пациенту одной начальной дозы химиотерапевтического средства с последующими одной или более вторичными дозами химиотерапевтического средства и, необязательно, одной или более третичными дозами химиотерапевтического средства.
[00108] Термины "начальная доза", "вторичные дозы" и "третичные дозы" относятся к временной последовательности введения. Таким образом, "начальная доза" является дозой, которую вводят в начале схемы лечения (также обозначаемой как "исходная доза"), "вторичные дозы" являются дозами, которые вводят после начальной дозы, и "третичные дозы" являются дозами, которые вводят после вторичных доз. Начальные, вторичные и третичные дозы могут содержать одинаковое количество антитела (антитела против PD-1). Однако, в некоторых вариантах осуществления количество, содержащееся в начальной, вторичных и/или третичных дозах варьируется друг относительно друга (например, их повышают или снижают, при необходимости) в течение лечения. В некоторых вариантах осуществления одну или более (например, 1, 2, 3, 4 или 5) доз вводят в начале схемы лечения как "ударные дозы" с последующими дозами, которые вводят реже (например, "поддерживающими дозами"). Например, антитело против PD-1 можно вводить пациенту со злокачественным новообразованием в ударной дозе приблизительно 1-3 мг/кг с последующей одной или более поддерживающими дозами от приблизительно 0,1 до приблизительно 20 мг/кг массы тела пациента.
[00109] В одном примере варианта осуществления настоящего изобретения каждую вторичную и/или третичную дозу вводят через ½-14 (например, ½, 1, 1½, 2, 2½, 3, 3½, 4, 4½, 5, 5½, 6, 6½, 7, 7½, 8, 8½, 9, 9½, 10, 10½, 11, 11½, 12, 12½, 13, 13½, 14, 14½ или более) недель после предыдущей дозы. В рамках изобретения фраза "предшествующая доза" в последовательности множественных введений означает дозу антитела против PD-1 (и, необязательно, второй противоопухолевой терапии), которую вводят пациенту перед введением непосредственно следующей дозы в последовательности без промежуточных доз.
[00110] Способы по изобретению по этому аспекту могут включать введение пациенту любого количества вторичных и/или третичных доз антитела против PD-1 (и/или второй противоопухолевой терапии). Например, в некоторых вариантах осуществления пациенту вводят только одну вторичную дозу. В других вариантах осуществления пациенту вводят две или более (например, 2, 3, 4, 5, 6, 7, 8 или более) вторичных доз. Аналогично, в некоторых вариантах осуществления пациенту вводят только одну третичную дозу. В других вариантах осуществления пациенту вводят две или более (например, 2, 3, 4, 5, 6, 7, 8 или более) третичных доз.
[00111] В вариантах осуществления, включающих множество вторичных доз, каждую вторичную дозу можно вводить с той же частотой, что и другие вторичные дозы. Например, каждую вторичную дозу можно вводить пациенту через 1-2 недели после предыдущей дозы. Аналогично, в вариантах осуществления, включающих множество третичных доз, каждую третичную дозу можно вводить с той же частотой, что и другие третичные дозы. Например, каждую третичную дозу можно вводить пациенту через 2-4 недели после предыдущей дозы. Альтернативно, частота, с которой пациенту вводят вторичные и/или третичные дозы, может варьироваться на протяжении схемы лечения. Лечащий врач также может корректировать частоту введения в течение лечения после клинического осмотра в зависимости от потребностей отдельного пациента.
[00112] В некоторых вариантах осуществления одну или более доз антитела против PD-1 и/или второй противоопухолевой терапии вводят в начале схемы лечения в качестве "индукционных доз" чаще (дважды в неделю, раз в неделю или раз в 2 недели) с последующими дозами ("консолидирующими дозами" или "поддерживающими дозами"), которые вводят реже (например, раз в 2-12 недель). В некоторых вариантах осуществления одну или более доз антитела против PD-1 и/или лучевой терапии вводят в начале схемы лечения в качестве "индукционных доз" чаще (дважды в неделю, раз в неделю или раз в 2 недели) с последующими дозами антитела против PD-1.
[00113] Настоящее изобретение относится к способам, включающим последовательное введение одной или более доз антитела против PD-1 в комбинации с одной или более дозами дополнительной противоопухолевой терапии, где одну или более доз включают в один или более циклов лечения.
[00114] В некоторых вариантах осуществления настоящего изобретения способы включают осуществление по меньшей мере одного цикла лечения, где по меньшей мере один цикл лечения включает введение одной или более доз антитела против PD-1 и, необязательно, одной или более доз второй противоопухолевой терапии (например, химиотерапии, лучевой терапии). В некоторых вариантах осуществления цикл лечения включает 1-10 доз антитела против PD-1, где каждую дозу антитела против PD-1 вводят через 0,5-8 недель после предыдущей дозы. В некоторых вариантах осуществления способы по настоящему изобретению включают осуществление до 6 или 8 циклов лечения. В некоторых других вариантах осуществления способы по настоящему изобретению включают осуществление до 100 или более циклов лечения, как это требуется для терапевтического эффекта. В некоторых вариантах осуществления по меньшей мере один цикл лечения дополнительно включает вторую противоопухолевую терапию (например, химиотерапию). В некоторых вариантах осуществления химиотерапия является химиотерапией препаратами платины. В некоторых вариантах осуществления дозы химиотерапевтического средства вводят раз в неделю, каждые 2 недели, каждые 3 недели, каждые 4 недели или более.
[00115] Настоящее изобретение относится к способам, включающим последовательное введение пациенту антитела против PD-1 в комбинации с химиотерапией для лечения злокачественного новообразования (например, рака легких), что приводит к повышенной противоопухолевой эффективности (например, большему ингибированию роста опухоли, усиленной профилактике рецидивирования опухоли по сравнению с индивидуумом, неподвергнутым лечению, или индивидуумом, которому вводят антитело или химиотерапевтическое средство в качестве монотерапии). В некоторых вариантах осуществления химиотерапевтическое средство вводят до, после или одновременно с антителом против PD-1.
Дозировка
[00116] Количество антитела против PD-1, вводимого индивидууму способами по настоящему изобретению, как правило, является терапевтически эффективным количеством. В рамках изобретения фраза "терапевтически эффективное количество" означает количество антитела (антитела против PD-1), приводящее к одному или более из: (a) снижения тяжести или длительности симптома или признака злокачественного новообразования, например, солидной опухоли; (b) ингибирования роста опухоли или повышения некроза опухоли, уменьшения опухоли и/или исчезновения опухоли; (c) замедления роста и развития опухоли; (d) ингибирования метастазирования опухоли; (e) профилактики рецидивирования роста опухоли; (f) повышения выживаемости индивидуума со злокачественным новообразованием и/или (g) снижения использования или потребности в общепринятой противоопухолевой терапии (например, снижения или прекращения использования химиотерапевтических или цитотоксических средств) по сравнению с индивидуумом, неподвергнутым лечению, или индивидуумом, которому вводили антитело в качестве монотерапии.
[00117] В случае антитела против PD-1 терапевтически эффективное количество может составлять от приблизительно 0,05 мг до приблизительно 1500 мг, от приблизительно 1 мг до приблизительно 1500 мг, от приблизительно 10 мг до приблизительно 1400 мг, от приблизительно 50 мг до приблизительно 1400 мг, от приблизительно 75 мг до приблизительно 1400 мг или от приблизительно 100 мг до приблизительно 1300 мг антитела. Например, в различных вариантах осуществления количество антитела против PD-1 составляет приблизительно 0,05 мг, приблизительно 0,1 мг, приблизительно 1,0 мг, приблизительно 2,0 мг, приблизительно 10 мг, приблизительно 20 мг, приблизительно 30 мг, приблизительно 40 мг, приблизительно 50 мг, приблизительно 60 мг, приблизительно 70 мг, приблизительно 80 мг, приблизительно 90 мг, приблизительно 100 мг, приблизительно 150 мг, приблизительно 200 мг, приблизительно 220 мг, приблизительно 240 мг, приблизительно 260 мг, приблизительно 280 мг, приблизительно 300 мг, приблизительно 350 мг, приблизительно 400 мг, приблизительно 450 мг, приблизительно 500 мг, приблизительно 550 мг, приблизительно 600 мг, приблизительно 650 мг, приблизительно 700 мг, приблизительно 750 мг, приблизительно 800 мг, приблизительно 850 мг, приблизительно 900 мг, приблизительно 950 мг, приблизительно 1000 мг, приблизительно 1050 мг, приблизительно 1100 мг, приблизительно 1150 мг, приблизительно 1200 мг, приблизительно 1250 мг, приблизительно 1300 мг, приблизительно 1400 мг или приблизительно 1500 мг антитела против PD-1. В одном из вариантов осуществления 250 мг антитела против PD-1 вводят способами по настоящему изобретению. В одном из вариантов осуществления 200 мг антитела против PD-1 вводят способами по настоящему изобретению. В одном из вариантов осуществления 350 мг антитела против PD-1 вводят способами по настоящему изобретению. В одном из вариантов осуществления 1050 мг антитела против PD-1 вводят способами по настоящему изобретению.
[00118] Количество антитела против PD-1, содержащееся в отдельных дозах, можно выражать в миллиграммах антитела на килограмм массы тела индивидуума (т.е. мг/кг). В некоторых вариантах осуществления антитело против PD-1, используемое в способах по настоящему изобретению, можно вводить индивидууму в дозе от приблизительно 0,0001 до приблизительно 100 мг/кг массы тела индивидуума. В некоторых вариантах осуществления антитело против PD-1 можно вводить в дозе от приблизительно 0,1 мг/кг до приблизительно 20 мг/кг массы тела пациента. В некоторых вариантах осуществления способы по настоящему изобретению включают введение антитела против PD-1 в дозе приблизительно 1 мг/кг, 3 мг/кг, 5 мг/кг или 10 мг/кг массы тела пациента.
[00119] В некоторых вариантах осуществления количество антитела против PD-1, вводимого пациенту, может быть меньше терапевтически эффективного количества, т.е. может являться субтерапевтической дозой. Например, если терапевтически эффективное количество антитела против PD-1 содержит 3 мг/кг, субтерапевтическая доза содержит количество менее 3 мг/кг, например, 2 мг/кг, 1,5 мг/кг, 1 мг/кг, 0,5 мг/кг или 0,3 мг/кг. Как определено в настоящем описании, термин "субтерапевтическая доза" относится к количеству антитела против PD-1, которое само по себе не приводит к терапевтическому эффекту. Однако в некоторых вариантах осуществления субтерапевтическую дозу антителу против PD-1 вводят со вторым и, необязательно, третьим терапевтическим средством для стимуляции терапевтического эффекта.
ПРИМЕРЫ
[00120] Следующие примеры представлены для обеспечения специалистов в этой области полным описанием того, как получать и использовать способы и композиции по изобретению, и они не предназначены для ограничения объема того, что авторы считают своим изобретением. Были предприняты попытки обеспечить точность в отношении используемых чисел (например, количеств, температуры и т.д.), но необходимо учитывать некоторые экспериментальные ошибки и отклонения. Если не указано иначе, части являются массовыми частями, молекулярная масса является средней молекулярной массой, температура указана в градусах Цельсия, и давление является атмосферным или близким к нему.
Пример 1: Эффективность антитела против PD-1 in vivo в комбинации с лучевой терапией в отношении опухолей MC38
[00121] В этом примере исследовали эффект блокирования PD-1 в комбинации с лучевой терапией в отношении развившихся опухолей MC38 у мышей.
[00122] 5×105 клеток карциномы толстого кишечника MC38 подкожно имплантировали в правый бок самок мышей C57BL/6 (Jackson Laboratory). Лечение начинали в день 9 после имплантации, когда средние объемы опухолей достигали приблизительно 100 мм3. Мышей случайным образом распределяли по группам, которым вводили изотипический контроль (2A3, BioXcell) или блокирующее антитело против PD-1 (RMP1-14, BioXCell) в дозе 5 мг/кг 2 раза в неделю всего в количестве 5 интраперитонеальных инъекций. Через день после начала введения антитела мышей распределяли по группам лучевой терапии, которые подвергали облучению опухолей в правом боку в дозе 12 Гр. Лучевую терапию осуществляли с использованием излучателя RS 2000 Biological Research Irradiator (Rad Source) в отношении мышей под анестезией (кетамин/ксилазин), экранированных с помощью устройств для частичного облучения тела (Precision X-ray) и свинцового листа (Images Scientific Instruments). Рост опухоли оценивали 3 раза в неделю до дней 70-80, когда всех мышей умерщвляли. На фигуре 1 показан дизайн исследования для эксперимента, включающего введение доз антитела против PD-1 и лучевую терапию.
[00123] На фигуре 2 и в таблице 1 показаны средние объемы опухолей у мышей, которым вводили антитело против PD-1 в отдельности или в комбинации с лучевой терапией.
Таблица 1: Средние объемы опухолей у мышей, которым вводили антитело против PD-1 в отдельности или в комбинации с лучевой терапией
[00124] Блокирование PD-1 (RMP1-14) действовало синергически с местным облучением (XRT) и в значительной степени индуцировало регрессирование опухоли (4/6 мышей) у несущих опухоль MC38 мышей по сравнению с мышами, которых лечили XRT и изотипическим контролем (2/6 мышей). Рост опухоли ингибировали или замедляли у мышей, которым вводили антитело против PD-1 в комбинации с лучевой терапией. В случае мышей, которым вводили антитело против PD-1 и которых подвергали облучению, проходило более 40 дней до достижения объема опухоли 500 мм3 по сравнению с мышами, которых подвергали монотерапии, в случае которых проходило менее 20 дней до достижения объема опухоли 500 мм3. Регрессирование опухоли сохранялось до 4 недель в случае группы, которую лечили с помощью комбинации (XRT+антитело против PD-1) (в это время рецидивировала 1 из 4 отторженных опухолей), по сравнению с 1,5 неделями в случае группы, которую лечили XRT и изотипическим контролем (рецидивировала 1 из 2 отторженных опухолей). В этой модели опухоли блокирование PD-1 в качестве монотерапии не имело эффекта в отношении роста первичной опухоли.
Таблица 2: Процент выживания мышей, которым вводили антитело против PD-1 в отдельности или в комбинации с лучевой терапией
[00125] О терапевтической эффективности комбинированного лечения (XRT+антитело против PD-1) свидетельствовало статистически значимое повышение общей выживаемости в этой группе (50% оставались живыми через 70 дней после имплантации опухоли) по сравнению со всеми другими группами лечения: изотипический контроль (0% оставались живыми ко дню 70), антитело против PD-1 (0% оставались живыми ко дню 70) и XRT+изотипический контроль (17% оставались живыми ко дню 70) (фигура 3; таблица 2).
Пример 2: Эффективность антитела против PD-1 и лучевой терапии in vivo в отношении опухолей B16
[00126] В этом примере исследовали противоопухолевый эффект антитела против PD-1 мыши в комбинации с лучевой терапией в отношении развившихся опухолей B16 у мышей.
[00127] 2×105 клеток меланомы B16F10.9 подкожно имплантировали в правый бок самок мышей C57BL/6 (Jackson Laboratory). Лечение начинали, когда средние объемы опухолей достигали приблизительно 150 мм3. Мышей случайным образом распределяли по группам, которым вводили изотипический контроль (2A3, BioXcell) или блокирующее антитело против PD-1 (RMP1-14, BioXCell) в дозе 5 мг/кг 2 раза в неделю всего в количестве 5 интраперитонеальных инъекций. Через день после начала введения антитела мышей распределяли по группам лучевой терапии, которые подвергали облучению опухолей в правом боку в дозе 8 Гр. Лучевую терапию осуществляли с использованием излучателя RS 2000 Biological Research Irradiator (Rad Source) в отношении мышей под анестезией (кетамин/ксилазин), экранированных с помощью устройств для частичного облучения тела (Precision X-ray) и свинцового листа (Images Scientific Instruments). Рост опухоли оценивали 3 раза в неделю до дней 70-80, когда всех мышей умерщвляли. На фигуре 4 показан дизайн исследования для эксперимента, включающего введение доз антитела против PD-1 и лучевую терапию.
[00128] Введение блокирующего антитела против PD-1 (RMP1-14) в комбинации с местным облучением (XRT) замедляло рост первичной опухоли B16 по сравнению с монотерапии XRT или антителом против PD-1 (фигура 5; таблица 3).
Таблица 3: Средние объемы опухолей у мышей, которым вводили антитело против PD-1 в отдельности или в комбинации с лучевой терапией
Таблица 4: Процент выживания мышей, которым вводили антитело против PD-1 в отдельности или в комбинации с лучевой терапией
[00129] Лечение комбинацией XRT и антитела против PD-1 повышало общую выживаемость (50% оставались живыми ко дню 50 после имплантации) по сравнению с XRT в отдельности (0% оставались живыми ко дню 50), антитело против PD-1 в отдельности (0% оставались живыми ко дню 40) и изотипический контроль в отдельности (0% оставались живыми ко дню 30) (фигура 6; таблица 4).
Пример 3: Эффективность антитела против PD-1 в комбинации с лучевой терапией in vivo в отношении метастазирующих опухолей легких
[00130] В этом примере исследовали эффект блокирования PD-1 в комбинации с лучевой терапией в отношении развившихся и метастазирующих опухолей у мышей.
[00131] 1,5×105 клеток карциномы молочной железы 4T1 имплантировали подкожно в правый бок самок мышей Balb/c (Jackson Laboratory). Лечение начинали в день 12 после имплантации, когда средние объемы опухолей достигали приблизительно 100 мм3. Мышей случайным образом распределяли по группам, которым вводили изотипический контроль (2A3, BioXcell) или блокирующее антитело против PD-1 (RMP1-14, BioXCell) в дозе 5 мг/кг 2 раза в неделю всего в количестве 5 интраперитонеальных инъекций. Через день после начала введения антитела мышей распределяли по группам лучевой терапии, которые подвергали облучению опухолей в правом боку в дозе 8 Гр. Лучевую терапию осуществляли с использованием излучателя RS 2000 Biological Research Irradiator (Rad Source) в отношении мышей под анестезией (кетамин/ксилазин), экранированных с помощью устройств для частичного облучения тела (Precision X-ray) и свинцового листа (Images Scientific Instruments). Рост опухоли оценивали 3 раза в неделю до дня 28, когда всех мышей умерщвляли для оценки объема метастазов в легких с использованием клоногенного анализа. В кратком изложении, ткань легких подвергали диссоциации с использованием ДНКазы/либертазы TL (Roche) и культивировали в средах, дополненных 60 мкМ 6-тиогуанином. После двух недель культивирования планшеты докрашивали метиленовым синим и подсчитывали количество колоний (одна колония соответствует одной метастазирующей клетке 4T1).
[00132] Ожидают, что лечение с использованием антитела против PD-1 в комбинации с лучевой терапией способствует регрессированию опухоли, а также опосредует супрессию роста метастазов.
Пример 4: Эффективность антитела против человека PD-1 in vivo в комбинации с лучевой терапией способствует абскопальному эффекту в отношении дистальных опухолей
[00133] В этом примере исследовали эффект блокирования PD-1 в комбинации с лучевой терапией в отношении первичных и дистальных опухолей MC38 у мышей, гуманизированных по PD-1, с использованием антител против PD-1 человека.
[00134] Примером антитела против PD-1, используемого в этом примере, является REGN2810 (также известное как H4H7798N, как описано в US20150203579), полностью человеческое моноклональное антитело против PD-1, содержащее тяжелую цепь, содержащую аминокислотную последовательность SEQ ID NO: 9, и легкую цепь, содержащую аминокислотную последовательность SEQ ID NO: 10, пару аминокислотных последовательностей HCVR/LCVR, содержащих SEQ ID NO: 1/2, и последовательности CDR тяжелой и легкой цепи, содержащие SEQ ID NO: 3-8.
[00135] Мышей, гуманизированных по PD-1, получали с использованием технологии VelociGene® (Valenzuela et al 2003, Nat. Biotechnol. 21: 652-659; публикация патентной заявки США № 2015/0366174).
[00136] 5×105 клеток карциномы толстого кишечника MC38 имплантировали подкожно самкам гуманизированных мышей PD-1/C57BL/6 в день 0 (первичная опухоль в правом боку) и день 3 (опухоль в левом боку; дистальная опухоль). Лечение начинали, когда средние объемы первичных опухолей достигали приблизительно 150 мм3. Мышей случайным образом распределяли по группам, которым вводили изотипический контроль или блокирующее антитело против PD-1 (REGN2810) в дозе 5 мг/кг 2 раза в неделю всего в количестве 8 интраперитонеальных инъекций. Через день после начала введения антитела, мышей распределяли по группам лучевой терапии, которые подвергали облучению опухолей в правом боку в дозе 8 Гр. Лучевую терапию осуществляли с использованием излучателя RS 2000 Biological Research Irradiator (Rad Source) в отношении мышей под анестезией (кетамин/ксилазин), экранированных с помощью устройств для частичного облучения тела (Precision X-ray) и свинцового листа (Images Scientific Instruments). Рост первичных и вторичных опухолей оценивали 3 раза в неделю до дней 70-80, когда всех мышей умерщвляли. На фигуре 7 показан дизайн исследования для эксперимента, включающего введение доз антитела против PD-1 и лучевую терапию.
Результаты
[00137] Первичная опухоль: Лечение с блокированием PD-1 (REGN2810) действовало синергически с местным облучением (XRT) в отторжении первичных опухолей MC38 (4 из 6 мышей без опухолей) по сравнению с мышей, которых лечили XRT и изотипическим контролем (1/6 мышей без опухолей). Регрессирование опухоли сохранялось в группе, которую лечили с помощью комбинации, в течение 8 недель до конца эксперимента по сравнению с тремя неделями в группе, которую лечили XRT и изотипическим контролем (отторженная опухоль рецидивировала в этот момент времени) (фигура 8; таблица 5).
Таблица 5: Средние объемы первичных опухолей у мышей, которым вводили REGN2810 в отдельности или в комбинации с лучевой терапией
[00138] Блокирование PD-1 в качестве монотерапии опосредовало отторжение у 2 из 5 мышей; однако, 1 из мышей, отторгнувших первичную опухоль, погибла из-за вторичного роста опухоли, что привело к тому, что только 1 мышь выжила к концу эксперимента. О мощной терапевтической эффективности комбинированного лечения (XRT+REGN2810) свидетельствует статистически значимое повышение общей выживаемости (~67% оставались живыми через 70 дней после имплантации опухоли) по сравнению со всеми другими группами: изотипический контроль или XRT в отдельности (0% оставались живыми ко дню 70) и REGN2810 в качестве монотерапии (20% оставались живыми ко дню 70) (фигура 9; таблица 6).
Таблица 6: Процент выживания мышей, которым вводили REGN2810 в отдельности или в комбинации с лучевой терапией
[00139] Дистальная опухоль: REGN2810 в комбинации с XRT в значительной степени способствовало абскопальному эффекту (отторжению опухоли, имплантированной в дистальном участке) у 5 из 6 мышей без опухолей по сравнению с XRT в отдельности (2/6 без дистальных опухолей), REGN2810 в отдельности (1/6 без дистальных опухолей) и изотипический контроль (1/6 без дистальных опухолей) (фигура 10; таблица 7).
Таблица 7: Средние объемы дистальных опухолей у мышей, которым вводили REGN2810 в отдельности или в комбинации с лучевой терапией
Пример 5: Эффективность антитела против PD-1 in vivo в комбинации с лучевой терапией и антагонистом GITR в отношении опухолей MC38
[00140] В этом примере исследовали эффект блокирования PD-1 в комбинации с лучевой терапией и антагонистом глюкокортикоид-индуцируемого рецептора фактора некроза опухоли (GITR) (антитела против GITR) в отношении крупных развившихся опухолей MC38 у мышей.
[00141] 5×105 клеток карциномы толстого кишечника MC38 подкожно имплантировали в правый бок самок мышей C57BL/6 (Jackson Laboratory). Лечение начинали, когда средние объемы опухолей достигали приблизительно 150-200 мм3 (классифицируемых как "крупные опухоли"). Мышей случайным образом распределяли по группам, которым вводили антитело изотипического контроля (2A3 или LTF-2; BioXcell), антитело против PD-1 (RMP1-14; BioXcell), антитело против GITR (DTA-1; BioXcell) или комбинацию антитела против PD-1 и антитела против GITR в дозе 5 мг/кг 2 раза в неделю всего в количестве 5 интраперитонеальных инъекций. Через день после начала введения антитела мышей распределяли по группам лучевой терапии, которые подвергали облучению опухолей в правом боку в дозе 8 Гр. Лучевую терапию осуществляли с использованием излучателя RS 2000 Biological Research Irradiator (Rad Source) в отношении мышей под анестезией (кетамин/ксилазин), экранированных с помощью устройств для частичного облучения тела (Precision X-ray) и свинцового листа (Images Scientific Instruments). Рост опухоли оценивали 3 раза в неделю до дней 70-80, когда всех мышей умерщвляли. На фигуре 11 показан дизайн исследования для эксперимента, включающего введение антитела против PD-1, антитела против GITR и осуществление лучевой терапии.
[00142] Лечение антителом против PD-1 (RMP1-14) действовало синергически с местным облучением (XRT) и антителом против GITR в отторжении крупных опухолей MC38 (4 из 6 мышей без опухолей) по сравнению с мышами, которых лечили XRT и антителом против GITR (2/6 без опухолей), XRT и антителом против PD-1 (2/6 с отторжением) или XRT в отдельности (0/6 без опухолей). Монотерапия (антителом против PD-1 или антителом против GITR) или комбинированное лечение (антителом против PD-1 и антителом против GITR) имело минимальный эффект в отношении роста опухоли, при этом лечение антителом против PD-1 или антителом против GITR опосредовало отторжение у 1/5 мышей, а комбинация двух антител опосредовала отторжение у 2/5 мышей. Регрессирование опухоли сохранялось в течение до 6,5 недель после начала лечения в случае мышей, которых лечили с помощью тройной комбинации, по сравнению с 2 неделями в случае мышей, которых лечили XRT и антителом против GITR (фигура 12).
Таблица 8: Процент выживания мышей, которым вводили антитело против PD-1 в комбинации с лучевой терапией и антитело против GITR
[00143] В таблице 8 и на фигуре 13 показана выживаемость мышей, которым вводили антитело против PD-1 в комбинации с лучевой терапией и антителом против GITR. Кроме того, введение антитела против PD-1 и XRT приводили к регрессированию очень крупных опухолей (~300 мм3).
Пример 6: Эффективность антитела против PD-1 in vivo в комбинации с лучевой терапией и антагонистом GITR в отношении опухолей B16
[00144] В этом примере исследовали эффект блокирования PD-1 в комбинации с лучевой терапией и антагонистом GITR (антителом против GITR) в отношении развившихся опухолей B16 у мышей.
[00145] 2,5×105 клеток меланомы B16F10.9 подкожно имплантировали в правый бок самок мышей C57BL/6 (Jackson Laboratory). Лечение начинали, когда средние объемы опухолей достигали приблизительно 100 мм3. Мышей случайным образом распределяли по группам, которым вводили изотипический контроль (2A3, LTF-2; BioXcell), антитело против PD-1 (RMP1-14, BioXcell), антитело против GITR (DTA-1; BioXcell) или комбинацию антитела против PD-1 и антитела против GITR в дозе 5 мг/кг 2 раза в неделю всего в количестве 5 интраперитонеальных инъекций. Через день после начала введения антитела, мышей распределяли по группам лучевой терапии, которые подвергали облучению опухолей в правом боку в дозе 8 Гр. Лучевую терапию осуществляли с использованием излучателя RS 2000 Biological Research Irradiator (Rad Source) в отношении мышей под анестезией (кетамин/ксилазин), экранированных с помощью устройств для частичного облучения тела (Precision X-ray) и свинцового листа (Images Scientific Instruments). Рост опухоли оценивали 3 раза в неделю до дней 70-80, когда всех мышей умерщвляли.
[00146] Ожидают, что антитело против PD-1 в комбинации с антителом против GITR и лучевой терапией будет способствовать большему регрессированию опухоли и замедлению роста опухоли, чем монотерапия или антитело против PD-1 в комбинации с лучевой терапией.
Пример 7: Клиническое испытание антитела против PD-1 и лучевой терапии на пациентах с солидными опухолями на поздней стадии
[00147] Это исследование является открытым, многоцентровым исследованием с повышением доз с множеством групп с повышением доз и расширенных групп для изучения эффективности, безопасности и переносимости антитела против PD-1 в отдельности и в комбинации с другими способами противоопухолевой терапии (включая лучевую терапию) на взрослых пациентах с солидными опухолями на поздней стадии.
[00148] Примером антитела против PD-1, используемого в этом исследовании, является REGN2810 (также известное как H4H7798N, как описано в US20150203579), полностью человеческое моноклональное антитело против PD-1, содержащее тяжелую цепь, содержащую аминокислотную последовательность SEQ ID NO: 9, и легкую цепь, содержащую аминокислотную последовательность SEQ ID NO: 10, пару аминокислотных последовательностей HCVR/LCVR, содержащую SEQ ID NO: 1/2, и последовательности CDR тяжелой и легкой цепи, содержащие SEQ ID NO: 3-8.
Цели исследования
[00149] Первичной целью исследования является характеризация безопасности, переносимости, дозолимитирующей токсичности (DLT) REGN2810, вводимого внутривенно (IV) в качестве монотерапии или в комбинации с направленным облучением (предназначенным скорее для иммуностимулирующей, а не, главным образом, абляционной терапии), циклофосфамидом в низких дозах (терапией, которая, как показано, ингибирует ответы регуляторных T-клеток), гранулоцитарно-макрофагальным колониестимулирующим фактором, карбоплатином, доцетакселом или их комбинацией, на пациентах со злокачественными новообразованиями на поздней стадии.
[00150] Вторичными целями исследования являются: (1) определение рекомендуемой дозы REGN2810 для фазы 2 (RP2D) в качестве монотерапии и в комбинации с другими способами противоопухолевой терапии (направленным облучением, циклофосфамидом в низких дозах или и тем, и другим); (2) описание предварительной противоопухолевой активности REGN2810 в отдельности и в каждой комбинации; (3) характеризация PK REGN2810 в качестве монотерапии и в комбинации с другими способами противоопухолевой терапии (направленным облучением, циклофосфамидом в низких дозах или и тем, и другим); и (4) анализ иммуногенности REGN2810.
Обоснование дизайна исследования
[00151] Модель 3+3 для фазы повышения дозы в этом исследовании предназначена для осуществления оценки безопасности REGN2810 в качестве монотерапии с различными уровнями доз и в комбинации с иммуностимулирующим лечением: циклофосфамидом, ограниченным, направленным облучением, осуществляемым по 1 из 2 схем лечения, или комбинацией лучевой терапии и циклофосфамида.
[00152] После определения переносимости REGN2810 в отдельности и в комбинации с лучевой терапией и/или циклофосфамидом добавляют множество расширенных когорт, в которых используют различные комбинации или монотерапию по выбранным признакам [NSCLC, BC, HNSCC, CSCC, опухоли с MSI (колоректальные опухоли, опухоли эндометрия, опухоли предстательной железы или другие типы опухолей), HCC и другие солидные опухоли на поздней стадии] для дальнейшего подтверждения безопасности и оценки повышения противоопухолевой активности. В некоторые из этих комбинаций добавляют гранулоцитарно-макрофагальный колониестимулирующий фактор (ГМ-КСФ), карбоплатин и/или доцетаксел.
[00153] В таблице 9 приведены некоторые из когорт, в которых используют REGN2810 в качестве монотерапии и в комбинации с другими способами лечения.
Таблица 9: Список некоторых из расширенных когорт для монотерапии REGN2810 и способов комбинированного лечения
[00154] Исходное запланированное лечение REGN2810 осуществляют каждые 14 дней в течение до 48 недель с 24 неделями последующего наблюдения. Лучевую терапию осуществляют через неделю после введения первой дозы REGN2810. Циклофосфамид в низких дозах вводят пациентам, которым предписан циклофосфамид, за 1 день до каждой из первых 4 доз REGN2810.
Длительность исследования
[00155] Пациентов подвергали лечению в течение до 48 недель, после чего осуществляют последующее наблюдение в течение 24 недель. Пациента подвергают лечению до завершения 48-недельного периода лечения или до прогрессирования заболевания, возникновения неприемлемой токсичности, отзыва согласия на исследование или соответствия другому критерию исключения из исследования. После по меньшей мере 24 недель лечения пациенты с подтвержденными полными ответами (CR) могут решить прекратить участие в исследовании и продолжить проходить все соответствующие оценки в рамках исследования (например, оценки эффективности). После по меньшей мере 24 недель лечения пациенты с оценками опухолевой массы в виде стабильного заболевания (SD) или частичного ответа (PR), остающимися неизменными при 3 последовательных оценок опухоли, также могут решить прекратить участие в исследовании и продолжить проходить все соответствующие оценки в рамках исследования (например, оценки эффективности).
Исследуемая выборка
[00156] Целевая выборка для этого исследования включает пациентов со злокачественными новообразованиями на поздней стадии, неявляющихся кандидатами для стандартной терапии, нежелающих подвергаться стандартной терапии, или в случае которых нет доступной терапии, которая, как ожидают, принесла бы клиническую пользу; и пациентов со злокачественными новообразованиями, являющимися неизлечимыми, неотвечающими на стандартную терапию или демонстрирующими прогрессирование опухоли, несмотря на стандартную терапию.
[00157] Критерии включения: Пациент должен соответствовать следующим критериям включения в исследование: (1) прогрессирование солидной опухоли без доступных альтернативных вариантов терапии в соответствии со стандартами лечения; (2) по меньшей мере 1 очаг для оценки ответа. Пациенты, которым предписана лучевая терапия, должны иметь по меньшей мере один дополнительный очаг, который можно безопасно облучать, одновременно сберегая индексные очаги, и для которого лучевую терапию в предусмотренных ограниченных, паллиативных дозах будут считать целесообразной с медицинской точки зрения; (3) у пациентов должны наблюдать рецидив после терапии первой линии, или они должны быть рефрактерными к терапии первой линии (и до 2 предшествующим линиям терапии) в условиях рецидивирующего или метастазирующего заболевания и должны иметь заболевание, для которого показана паллиативная лучевая терапия; (4) пациенты с метастазирующим злокачественным новообразованием с микросателлитной нестабильностью (MSI), рефрактерным к до 2 предшествующим линиям терапии; (5) статус по шкале Восточной объединенной онкологической группы (ECOG) ≤1; (6) возраст более 18 лет; (7) функция печени: a. общий билирубин ≤1,5-кратного верхнего предела нормы (ULN; при метастазах в печени ≤3-кратного ULN), b. трансаминазы ≤3-кратного ULN (или ≤5,0-кратного ULN при метастазах в печени), c. щелочная фосфатаза (ALP) ≤2,5-кратного ULN (или 5,0-кратного ULN при метастазах в печени); (8) функция почек: креатинин сыворотки ≤1,5-кратного ULN; (9) количество нейтрофилов (ANC) ≥1,5×109/л, c. количество тромбоцитов ≥75×109/л; (10) возможность предоставления подписанного информированного согласия; и (11) возможность и желание следовать запланированным визитам, планам лечения, лабораторным анализам и другим предусмотренным исследованием процедурам.
Исследуемое лечение
[00158] REGN2810 поставляют в виде жидкости в стерильных одноразовых флаконах. Каждый флакон содержит объем, достаточный для забора 10 мл REGN2810 в концентрации 25 мг/мл. REGN2810 вводят в амбулаторных условиях в виде 30-минутной IV инфузии. Доза для каждого пациента зависит от массы тела индивидуума. Дозу REGN2810 корректируют для каждого цикла в случае изменений массы тела на ≥10%. REGN2810 вводят в отдельности или в комбинации с лучевой терапией и/или циклофосфамидом. Циклофосфамид вводят в дозе 200 мг/м2 или низкой дозе (100 мг/м2).
Монотерапия
[00159] REGN2810 вводят в амбулаторных условиях посредством IV инфузии в течение 30 минут каждые 14 дней в течение 48 недель (например, дни 1, 15±3, 29±3 и 43±3 56-дневного цикла). Запланированные схемы монотерапии, которые можно предписывать, могут включать: (i) IV инфузию 1 мг/кг в течение 30 минут каждые 14 дней в течение 48 недель; (ii) инфузию 3 мг/кг в течение 30 минут каждые 14 дней в течение 48 недель; (iii) инфузию 10 мг/кг в течение 30 минут каждые 14 дней в течение 48 недель; (iv) инфузию 0,3 мг/кг в течение 30 минут каждые 14 дней в течение 48 недель (если определяют, что MTD составляет менее 1 мг/кг); и (v) IV инфузию постоянной дозы 200 мг в течение 30 минут каждые 14 дней в течение 48 недель.
Комбинированное лечение
[00160] Сопутствующую лучевую терапию, введение циклофосфамида, ГМ-КСФ, карбоплатина и доцетаксела осуществляют по рецепту, и их использование, дозу, модификации доз, снижение или задержку введения, а также любые потенциальные AE, являющиеся результатом их использования, отслеживают вместе с такими же показателями для REGN2810.
[00161] Комбинация REGN2810 и лучевой терапии: REGN2810 вводят посредством IV инфузии в течение 30 минут каждые 14 дней в течение 48 недель в комбинации с лучевой терапией со дня 8 по день 12. Запланированные схемы комбинированного лечения с использованием REGN2810 и лучевой терапии могут включать:
инфузию 1 мг/кг REGN2810 в течение 30 минут каждые 14 дней в течение 48 недель и
лучевую терапию в дозе 30 Гр (6 Гр×5 раз/неделю; осуществляемую через 1 неделю после введения первой дозы REGN2810, предпочтительно, в последовательные дни)
инфузию 1 мг/кг REGN2810 в течение 30 минут каждые 14 дней в течение 48 недель и
лучевую терапию в дозе 27 Гр (9 Гр×3 раз/неделю; осуществляемую через 1 неделю после введения первой дозы REGN2810, предпочтительно, не в последовательные дни)
инфузию 3 мг/кг REGN2810 в течение 30 минут каждые 14 дней в течение 48 недель и
лучевую терапию в дозе 30 Гр (6 Гр×5 раз/неделю; осуществляемую через 1 неделю после введения первой дозы REGN2810, предпочтительно, в последовательные дни)
инфузию 3 мг/кг REGN2810 в течение 30 минут каждые 14 дней в течение 48 недель и
лучевую терапию в дозе 27 Гр (9 Гр×3 раз/неделю; осуществляемую через 1 неделю после введения первой дозы REGN2810, предпочтительно, не в последовательные дни)
[00162] Пациентам будут вводить 30 Гр в виде 5 фракций по 6 Гр, вводимые ежедневно, начиная с 1 недели после введения первой дозы REGN2810, или 27 Гр в виде 3 фракций по 9 Гр, вводимых через день, начиная с 1 недели после введения первой дозы REGN2810. Очаг, выбранный для облучения, должен являться очагом, который можно безопасно облучать с использованием фокального облучения, одновременно сберегая индексные очаги, и для которого лучевую терапию в предусмотренных ограниченных, паллиативных дозах будут считать целесообразной с медицинской точки зрения.
[00163] Комбинация REGN2810 и циклофосфамида: REGN2810 вводят посредством IV инфузии в течение 30 минут каждые 14 дней (2 недели) в течение 48 недель в комбинации с циклофосфамидом в низкой дозе 100 мг/м2 посредством IV инфузии каждые 14 дней в количестве 4 доз. Каждую из 4 доз циклофосфамида вводят за 1 до каждой из 4 первых доз REGN2810 (дни -1, 14, 28 и 42 первого 56-дневного цикла).
[00164] Запланированная схема комбинированного лечения REGN2810 и циклофосфамидом представляет собой:
Циклофосфамид в дозе 100 мг/м2 или 200 мг/м2 IV каждые 14 дней (дни -1, 14, 28 и 42 первого 56-дневного цикла) всего в количестве 4 доз; и
инфузия 3 мг/кг REGN2810 в течение 30 минут каждые 14 дней в течение 48 недель (обеспечивает монотерапевтическую дозу 3 мг/кг <MTD; если 3 мг/кг >MTD, доза будет составлять 1 мг/кг).
[00165] Комбинация REGN2810, лучевой терапии и циклофосфамида: Запланированная схема комбинированного лечения с использованием REGN2810, лучевой терапии и циклофосфамида включает:
циклофосфамид 100 мг/м2 (низкая доза) IV каждые 14 дней (дни -1, 14, 28 и 42 первого 56-дневного цикла) всего в количестве 4 доз; и
лучевую терапию в дозе 27 Гр (9 Гр×3 раз/неделю; осуществляемую через 7 или 8 дней после введения первой дозы REGN2810, предпочтительно, не в последовательные дни) или лучевую терапию в дозе 30 Гр (6 Гр×5 раз/неделю; осуществляемую через 7 или 8 дней после введения первой дозы REGN2810, предпочтительно, в последовательные дни); и
инфузию 3 мг/кг REGN2810 в течение 30 минут каждые 14 дней в течение 48 недель (обеспечивает монотерапевтическую дозу 3 мг/кг <MTD; если 3 мг/кг > MTD, доза будет составлять 1 мг/кг)
[00166] Комбинация REGN2810, лучевой терапии и ГМ-КСФ: Запланированная схема комбинированного лечения с использованием REGN2810, лучевой терапии и ГМ-КСФ включает:
250 мкг ГМ-КСФ SC ежедневно в течение 7 дней с четырьмя 7-дневными интервалами (дни 1-7, 15-21, 29-35 и 43-49 первого 56-дневного цикла); и
лучевую терапию в дозе 27 Гр (9 Гр×3 раз/неделю; осуществляемую через 1 неделю после введения первой дозы REGN2810, предпочтительно, не в последовательные дни); и
инфузию 3 мг/кг REGN2810 в течение 30 минут каждые 14 дней в течение 48 недель (обеспечивает монотерапевтическую дозу 3 мг/кг < MTD; если 3 мг/кг > MTD, доза будет составлять 1 мг/кг)
[00167] Комбинация REGN2810, лучевой терапии, ГМ-КСФ и циклофосфамида: Запланированная схема комбинированного лечения с использованием REGN2810, лучевой терапии, ГМ-КСФ и циклофосфамида включает:
250 мкг ГМ-КСФ SC ежедневно в течение 7 дней с четырьмя 7-дневными интервалами (дни 1-7, 15-21, 29-35 и 43-49 первого 56-дневного цикла); и
лучевую терапию в дозе 27 Гр (9 Гр×3 раз/неделю; осуществляемую через 1 неделю после введения первой дозы REGN2810, предпочтительно, не в последовательные дни); и
циклофосфамид в дозе 100 мг/м2 или 200 мг/м2 IV каждые 14 дней (дни -1, 14, 28 и 42 первого 56-дневного цикла) всего в количестве 4 доз; и
инфузию 3 мг/кг REGN2810 в течение 30 минут каждые 14 дней в течение 48 недель (обеспечивает монотерапевтическую дозу 3 мг/кг < MTD; если 3 мг/кг > MTD, доза будет составлять 1 мг/кг)
[00168] Комбинация REGN2810 и доцетаксела с карбоплатином или без него: Предполагаемая последовательность введения лекарственных средств представляет доцетаксел, затем карбоплатин (при включении в когорту с введением карбоплатина), затем REGN2810:
доцетаксел в дозе 30 мг/м2 IV в течение приблизительно 1 часа в дни 1, 8, 29 и 36 первого 56дневного цикла. Дексаметазон в дозе 8 мг IV будут вводить перед первой дозой доцетаксела. В случае последующего лечения доцетакселом, доза дексаметазона для премедикации может составлять 8 мг или 4 мг по решению исследователя
Карбоплатин AUC 2 IV в течение приблизительно 30 минут в дни 1, 8, 29 и 36 первого 56-дневного цикла. При дозировании карбоплатина необходимо использовать формулу Калверта на этикетке карбоплатина. Необходимо вычислять клиренс креатинина с использованием формулы Кокрофта-Голта.
инфузию 3 мг/кг REGN2810 в течение приблизительно 30 минут каждые 14 дней в течение 48 недель
Процедуры и оценки
[00169] Процедуры скрининга, которые необходимо осуществлять, включают определение бета-ХГЧ в сыворотке, МРТ головного мозга и рентгенографию грудной клетки.
[00170] Процедуры для оценки безопасности включают сбор анамнеза, физикальный осмотр, оценку показателей жизнедеятельности, электрокардиографию (ЭКГ), анализ свертывания, анализы иммунологической безопасности (для пациентов, которых лечат REGN2810), оценку симптомов B и оценку общего состояния, клинические лабораторные анализы, оценку AE и определение сопутствующих лекарственных средств.
[00171] Процедуры для оценки эффективности, которые необходимо осуществлять для оценки опухоли, включают КТ или МРТ, позитронно-эмиссионную томографию с 18F-тордезоксиглюкозуой (ФДГ-ПЭТ) и/или биопсию опухоли. КТ или МРТ для оценки опухоли осуществляют во время визита для скрининга (в пределах 28 дней перед инфузией) и в течение каждого цикла (приблизительно каждые 8 недель) в день 56±3, и когда подозревают прогрессирование заболевания. Кроме того, в случае пациентов, у которых не наблюдают прогрессирование в течение исследования, оценку опухоли осуществляют во время визитов для последующего наблюдения 3, 5 и 7. После выбора КТ или МРТ последующие оценки осуществляют тем же способом. Оценки ответов опухоли осуществляют в соответствии с Критериями оценки ответа солидных опухолей RECIST версии 1.1 (Eisenhauer et al 2009, Eur. J. Cancer 45: 228-247). Измеримые очаги, выбранные в качестве целевых очагов для измерения в соответствии с RECIST, также включали в качестве индексных очагов для иммуноопосредованных критериев ответа (irRC; Nishino et al 2013, Clin. Cancer Res. 19: 3936-3943). Ответу в соответствии с RECIST уделяют первостепенное внимание, как статистической оценке частоты ответа. В случае отдельного пациента irRC можно использовать при принятии решения о продолжении лечения по решению исследователя из-за вероятности необычных ответов.
[00172] Собирали образцы крови для анализа PK и антител против лекарственных средств (ADA).
Переменные в исследовании
[00173] Первичными переменными в исследовании являются частота DLT и частота и тяжесть TEAE и отклонений результатов лабораторных анализов в течение 48 недель исследования.
[00174] Вторичными переменными являются:
Противоопухолевая активность, оцениваемая с использованием соответствующих критериев для признака (как описано где-либо в настоящем описании):
√ Критерии оценки ответа солидных опухолей (RECIST; Eisenhauer et al 2009, Eur. J. Cancer 45: 228-247), определяемые посредством КТ или МРТ
√ Также используют другие критерии оценки для конкретных опухолей, для которых измерения RECIST не являются стандартом.
√ Иммуноопосредованные критерии ответа (irRC; Nishino et al 2013, Clin. Cancer Res. 19: 3936-3943), используемые в отношении измерений RECIST. Во всех случаях RECIST (или другие опухолеспецифические критерии) является основным инструментом для определения PD, SD, CR или PR. irRC определяют для принятия клинических решений и в информационных целях.
Частота возникновения антител против REGN2810
Противоопухолевая активность, измеряемая по PFS и общей выживаемости
[00175] В целях настоящего исследования пациентов повторно подвергают оценке ответа каждые 8 недель. Сканирования для подтверждения также осуществляют через 4 недели после исходного документирования объективного ответа или прогрессирования заболевания. Ответ и прогрессирование в этом исследовании оценивают с использованием международных критериев, предложенных в пересмотренном руководстве Критериев оценки ответа солидных опухолей (RECIST) (версии 1.1; Eisenhauer et al 2009, Eur. J. Cancer 45: 228-247). В критериях RECIST используют изменения наибольшего диаметра (одномерное измерение) очагов опухоли и наименьшего диаметра в случае злокачественных лимфоузлов.
Выбор очагов
[00176] Измеримое заболевание: Измеримые очаги определяют как очаги, которые можно точно измерить по меньшей мере в одном направлении (наибольший диаметр, подлежащий регистрации) как ≥20 мм (≥2 см) при рентгенографии грудной клетки или как ≥10 мм (≥1 см) при КТ, МРТ или с помощью калипера при клиническом осмотре. Все измерения опухолей необходимо регистрировать в миллиметрах (или десятичных дробях сантиметров). Примечание: см. ниже оценку облученных целевых очагов.
[00177] Злокачественные лимфоузлы: Чтобы считаться патологически увеличенным и измеримым, лимфоузел должен иметь размер ≥15 мм (≥1,5 см) по короткой оси при оценке посредством КТ (рекомендуемая толщина срезов на КТ должна составлять не более 5 мм [0,5 см]). На исходном уровне и при последующем наблюдении будут измерять и отслеживать только короткую ось.
[00178] Неизмеримое заболевание: Все другие очаги, включая небольшие очаги (наибольший диаметр <10 мм [<1 см] или патологические лимфоузлы с короткой осью от ≥10 до <15 мм [от ≥1 до <1,5 см]), считают неизмеримым заболеванием. Очаги в костях, лептоменингеальное заболевание, асцит, плевральный/перикардиальный экссудат, лимфангит кожи/пневмонит, воспалительное заболевание молочной железы и абдоминальные массы (неотслеживаемые посредством КТ или МРТ) считают неизмеримыми. Примечание: Кистозные очаги, соответствующие критериям для рентгенографически определяемых простых кист, следует считать злокачественными очагами (ни измеримыми, ни неизмеримыми), т.к. они по определению являются простыми кистами. "Кистозные очаги", как считают, представляющие собой кистозные метастазы, можно считать измеримыми очагами, если они соответствуют определению измеримости, описанному выше. Однако, если у того же пациента есть некистозные очаги, они являются предпочтительными для выбора в качестве целевых очагов.
[00179] Целевые очаги: Все измеримые очаги до максимум 2 очагов на орган и всего 5 очагов, представляющих все пораженные органы, необходимо идентифицировать как целевые очаги и регистрировать и измерять на исходном уровне. Целевые очаги выбирают с учетом их размера (очаги с наибольшим диаметром), они представляют все пораженные органы, но, кроме того, они включают очаги, которые поддаются воспроизводимым повторным измерениям. Может случиться так, что иногда наибольший очаг не поддается воспроизводимому измерению, в случае чего выбирают следующий наибольший очаг, который можно измерять. Вычисляют сумму диаметров (наибольших для неузловых очагов, короткую ось для узловых очагов) для всех целевых очагов и регистрируют как исходные суммарные диаметры. Если в сумму включают лимфоузлы, то в нее добавляют только короткую ось. Исходные суммарные диаметры используют в качестве референса для дальнейшей характеризации любого объективного регрессирования опухоли в измеримом направлении очага заболевания.
[00180] Нецелевые очаги: Все другие очаги, включая любые измеримые очаги, выходящие за пределы 5 целевых очагов, идентифицируют как нецелевые очаги и регистрируют на исходном уровне. Измерения этих очагов не требуются, но наличие, отсутствие или, в редких случаях, однозначное прогрессирование каждого из них регистрируют на всем протяжении последующего наблюдения.
Способы оценки измеримого заболевания
[00181] Все измерения осуществляют и регистрируют в метрических единицах с использованием линейки или калипера. Все исходные оценки осуществляют как можно ближе к началу лечения и никогда более чем за 4 недели до начала лечения. На исходном уровне и в течение последующего наблюдения необходимо использовать один и тот же способ оценки для характеризации каждого идентифицированного и зарегистрированного очага. Оценка посредством визуализации является предпочтительной для оценки при клиническом осмотре, но если отслеживаемые очаги нельзя визуализировать, их оценивают посредством клинического осмотра.
[00182] Клинические очаги: Клинические очаги считают измеримыми, только если они являются поверхностными (например, узелки в коже и пальпируемые лимфоузлы) и имеют диаметр ≥10 мм (≥1 см) при измерении с использованием калипера (например, узелки в коже). В случае кожных очагов, рекомендуют документировать их с помощью цветной фотографии с использованием линейки для оценки размера очага.
[00183] Рентгенография грудной клетки: Очаги при рентгенографии грудной клетки считают измеримыми очагами, если они четко определимы и окружены вентилируемым легким. Однако КТ является предпочтительной.
[00184] Общепринятые КТ и МРТ: В этом руководстве определена измеримость очагов при КТ с учетом предположения, что толщина срезов при КТ составляет 5 мм (0,5 см) или менее. Если при КТ используют толщину срезов более 5 мм (0,5 см), минимальный размер измеримого очага должен в два раза превышать толщину срезов. В некоторых случаях МРТ также является приемлемой.
[00185] ПЭТ-КТ: Если КТ, осуществляемая как часть ПЭТ-КТ, имеет диагностическое качество, идентичное диагностической КТ (с IV и пероральным контрастом), то КТ-часть ПЭТ-КТ можно использовать для измерений RECIST, а также ее можно использовать взаимозаменяемо с общепринятой КТ при точном измерении очагов злокачественного новообразования с течением времени.
[00186] Ультразвуковое исследование: Ультразвуковое исследование непригодно для оценки размера очага, и его нельзя использовать в качестве способа измерения. Если в течение исследования с помощью ультразвука идентифицируют новые очаги, рекомендуют подтверждать их с помощью КТ или МРТ. Если есть опасения, касающиеся радиационного воздействия при КТ, в некоторых случаях вместо КТ можно использовать МРТ.
[00187] Эндоскопия, лапароскопия: Эти способы не рекомендуют использовать для объективной оценки опухоли. Однако такие способы можно использовать для подтверждения патологического полного ответа при получении биоптатов или для определения рецидивирования в испытаниях, когда рецидивирование после полного ответа (CR) или хирургической резекции является конечной точкой.
[00188] Опухолевые маркеры: Опухолевые маркеры по отдельности нельзя использовать для оценки ответа. Если маркеры исходно превышают верхний предел нормы, их необходимо нормализовать для пациента, чтобы считать, что он имеет полный клинический ответ.
[00189] Цитологическое и гистологическое исследование: Эти способы можно использовать для дифференцирования частичных ответов (PR) и полных ответов (CR) в редких случаях (например, при остаточных очагах при таких типах опухоли, как опухоли половых клеток, когда могут оставаться известные остаточные доброкачественные опухоли). Цитологическое подтверждение неопластического происхождения любого экссудата, возникающего или ухудшающегося в течение лечения, если измеримая опухоль соответствует критериям ответа или стабильного заболевания, является обязательным для дифференцирования ответа или стабильного заболевания (экссудат может являться побочным эффектом лечения) и прогрессирующего заболевания.
[00190] ФДГ-ПЭТ: Хотя при оценке ответа посредством ФДГ-ПЭТ необходимо дополнительное исследование, иногда целесообразно включать использование сканирования ФДГ-ПЭТ, чтобы дополнить сканирование КТ при оценке прогрессирования (в частности, возможного "нового" заболевания). Новые очаги с учетом визуализации посредством ФДГ-ПЭТ можно идентифицировать с использованием следующего алгоритма: a. Отрицательные результаты ФДГ-ПЭТ на исходном уровне с положительными результатами ФДГ-ПЭТ при последующем наблюдении являются признаком PD с учетом нового очага. b. Отсутствие ФДГ-ПЭТ на исходном уровне и положительные результаты ФДГ-ПЭТ при последующем наблюдении: Если положительные результаты ФДГ-ПЭТ при последующем наблюдении соответствуют новому очагу заболевания, подтвержденному посредством КТ, это является PD. Если положительные результаты ФДГ-ПЭТ при последующем наблюдении не подтверждены как новый очаг заболевания при КТ, необходимо дополнительное сканирование КТ при последующем наблюдении для определения того, на самом ли деле имеет место прогрессирование в этом очаге (если это так, дата PD будет датой исходного сканирования ФДГ-ПЭТ с отклонениями). Если положительные результаты ФДГ-ПЭТ при последующем наблюдении соответствуют уже существующему очагу заболевания на КТ, непрогрессирующему с учетом анатомических изображений, это не является PD. c. ФДГ-ПЭТ можно использовать для повышения ответа до CR аналогично биопсии в случаях, когда считают, что остаточные рентгенографические отклонения представляют собой фиброз или рубцевание. Использование ФДГ-ПЭТ в этих случаях необходимо планово описать в протоколе и обосновать с помощью соответствующей заболеванию медицинской литературы по этому показанию. Однако следует отметить, что оба подхода могут приводить к ложноположительному CR по причине ограничений ФДГ-ПЭТ и разрешения/чувствительности биопсии. Примечание: Термин "положительные результаты" ФДГ-ПЭТ-сканирования очага, означает, что на изображении с поправкой на затухание захват ФДГ повышен более чем в два раза относительно окружающей ткани.
Критерии ответа для оценки целевых очагов
Полный ответ (CR): Исчезновение всех целевых очагов. Любые патологические лимфоузлы (целевые или нецелевые) должны уменьшиться по короткой оси до <10 мм (<1 см).
Частичный ответ (PR): по меньшей мере 30%н-ое снижение суммы диаметров целевых очагов при использовании исходных суммарных диаметров в качестве референса.
Прогрессирующее заболевание (PD): по меньшей мере 20%-ное увеличение суммы диаметров целевых очагов с использованием наименьшей суммы за исследование (она включает исходную сумму, если она является наименьшей за исследование) в качестве референса. В дополнение к относительному повышению на 20%, сумма также должна иметь абсолютное повышение по меньшей мере на 5 мм (0,5 см). (Примечание: появление одного или более новых очагов также считают прогрессированием).
Стабильное заболевание (SD): Отсутствие достаточного уменьшения опухоли для классификации в качестве PR и отсутствие достаточного увеличения для классификации в качестве PD при использовании наименьших суммарных диаметров за исследование в качестве референса.
Критерии ответа для оценки нецелевых очагов
Полный ответ (CR): Исчезновение всех нецелевых очагов и нормализация уровня опухолевых маркеров. Все лимфоузлы должны быть непатологическими по размеру (<10 мм [<1 см] по короткой оси). Примечание: Если опухолевые маркеры исходно превышают верхний предел нормы, они должны нормализоваться, чтобы считать, что пациент имеет клинический полный ответ.
Не-CR/Не-PD: Сохранение одного или более нецелевых очагов и/или поддержание уровня опухолевых маркеров выше пределов нормы.
Прогрессирующее заболевание (PD): Возникновение одного или более новых очагов и/или однозначное прогрессирование существующих нецелевых очагов. Однозначное прогрессирование, как правило, не должно перекрываться статусом целевых очагов. Оно должно соответствовать общему изменению статуса заболевания, а не увеличению одного очага.
Иммуноопосредованные критерии ответа
[00191] Иммуноопосредованные критерии ответа отличаются от RECIST (версии 1.1) тем, что сумму наибольших диаметров всех целевых очагов и новых очагов, если они есть, используют для определения ответа. Наличие новых очагов, по существу, не определяет прогрессирование, вместо этого учитывают общую опухолевую массу.
Оценка целевых очагов
Полный ответ (CR): Исчезновение всех целевых очагов. Любые патологические лимфоузлы (целевые или нецелевые) должны уменьшиться по короткой оси до <10 мм (<1 см).
Частичный ответ (PR): по меньшей мере 30%-ное снижение суммы диаметров целевых очагов, включая новые очаги, при использовании исходных суммарных диаметров в качестве референса.
Прогрессирующее заболевание (PD): по меньшей мере 20%-ное повышение суммы диаметров целевых очагов, включая новые очаги, при использовании наименьшей суммы за исследование (она включает исходную сумму, если она является наименьшей за исследование) в качестве референса. В дополнение к относительному повышению на 20%, сумма также должна иметь абсолютное повышение по меньшей мере на 5 мм (0,5 см).
Стабильное заболевание (SD): Отсутствие достаточного уменьшения опухоли для классификации в качестве PR и отсутствие достаточного увеличения для классификации в качестве PD при использовании наименьших суммарных диаметров за исследование в качестве референса и включении измерений новых очагов.
Оценка нецелевых очагов
Полный ответ (CR): Исчезновение всех нецелевых очагов и нормализация уровня опухолевых маркеров. Все лимфоузлы должны быть непатологическими по размеру (<10 мм [<1 см] по короткой оси). Примечание: Если опухолевые маркеры исходно превышают верхний предел нормы, они должны нормализоваться, чтобы считать, что пациент имеет клинический полный ответ.
Не-CR/не-PD: Сохранение одного или более нецелевых очагов и/или поддержание уровня опухолевых маркеров выше пределов нормы.
Прогрессирующее заболевание (PD): Однозначное прогрессирование существующих нецелевых очагов. Однозначное прогрессирование, как правило, не должно перекрываться статусом целевых очагов. Оно должно соответствовать общему изменению статуса заболевания, а не увеличение одного очага. Хотя четкое прогрессирование "нецелевых" очагов является необычным, в таких случаях мнение лечащего врача должно иметь приоритетное значение, и статус прогрессирования необходимо подтверждать позже.
Оценка общих критериев ответа
[00192] Лучшим общим ответом является общий ответ, регистрируемый с начала лечения до прогрессирования/рецидивирования заболевания (при использовании наименьших измерений, зарегистрированных с начала лечения, в качестве референса для прогрессирующего заболевания). Определение лучшего ответа пациента будет зависеть от достижения критериев измерения и подтверждения. Пересмотренные Критерии оценки ответа солидных опухолей (RECIST) версии 1.1 (Eisenhauer et al 2009, Eur. J. Cancer 45: 228-247) и иммуноопосредованные критерии ответа (irRC; Nishino et al 2013, Clin. Cancer Res. 19: 39363-943) приведены в таблицах 10 и 11 ниже.
Таблица 10: Ответ в соответствии с пересмотренными критериями RECIST (версии 1.1)
CR: полный ответ; PD: прогрессирующее заболевание; PR: частичный ответ; SD: стабильное заболевание
Таблица 11: оценка иммуноопосредованных критериев ответа
CR: полный ответ; PD: прогрессирующее заболевание; PR: частичный ответ; SD: стабильное заболевание
Оценка облученных целевых очагов
[00193] Облученные целевые очаги оценивают с использованием модифицированной версии международных критериев, предложенных комитетом по разработке Критериев оценки ответа солидных опухолей (RECIST) версии 1.1. Для определения локального контроля включены дополнительные определения, выходящие за пределы руководств RECIST 1.1 и характерные для этого протокола.
[00194] Критерии ответа для облученных очагов являются следующими:
[00195] Локальное увеличение (LE): По меньшей мере 20%-ное увеличение LD целевого очага при использовании наименьшего LD, зарегистрированного с начала лечения, в качестве референса. Теоретически, это определение будут осуществлять с учетом оценки изображений КТ.
[00196] Локальная неэффективность (LF): Термин относится к первичной опухоли, подвергнутой лечению, после терапии по протоколу и представляет собой соответствие двум критериям: (1) Увеличение размера опухоли на 20%, как определено выше для локального увеличения (LE); (2) Измеримая опухоль с критериями, соответствующими LE, должна демонстрировать повышенное накопление при позитронно-эмиссионной томографии (PET) с захватом с интенсивностью, схожей с ПЭТ для стадирования перед лечением, или измеримую опухоль необходимо подвергать биопсии, подтверждая возможную карциному.
[00197] Локальный контроль (LC): Отсутствие локальной неэффективности.
[00198] Наибольший диаметр (LD) облученного целевого очага, вычисленный из результатов сканирования КТ при планировании лечения с использованием соответствующего тканеспецифической регулировки окна, регистрируют как исходный LD. Исходный LD используют в качестве референса, с помощью которого характеризуют объективную опухоль. Для оценки при последующем наблюдении диагностическая КТ, осуществляемая с помощью алгоритма реконструкции с использованием прилегающих 5-миллиметровых срезов с использованием легочной регулировки окна, осуществляемой как часть запланированного последующего наблюдения по протоколу, является предпочтительной в качестве способа оценки ответа. Если КТ недоступна, допустимыми являются МРТ или рентгенография, при условии, что целевой очаг четко виден.
Результаты
[00199] REGN2810 в отдельности и в комбинации является безопасным и хорошо переносилось пациентами. Введение REGN2810 в отдельности или в комбинации с другими способами лечения ингибирует рост опухоли и/или способствует регрессированию опухоли у пациентов с солидными опухолями на поздней стадии. Общая частота ответа в случае комбинированного лечения с использованием лучевой терапии превосходит частоту ответа в случае монотерапии.
[00200] К настоящему времени лечению подвергнуты 60 пациентов со злокачественными солидными опухолями на поздней стадии (47% ранее подвергали четырем или более предшествующим терапиям). Злокачественные солидные опухоли на поздней стадии включают колоректальный рак, рак головы и шеи, рак молочной железы, саркому мягких тканей, рак надпочечников, рак анального канала, рак аппендикса, рак мочевого пузыря, рак шейки матки, рак эндометрия, рак пищевода, рак печени, немелкоклеточную аденокарциному легких, рак яичников, рак поджелудочной железы, рак предстательной железы, саркоматоидный рак почки, рак слюнных желез, немеланомный рак кожи, карциному Меркеля, плоскоклеточную карциному, базально-клеточную карциному, рак тонкого кишечника, рак щитовидной железы и рак матки.
[00201] У сорока двух пациентов (70%) наблюдали одно или более связанных с лечением нежелательных явлений (AE). Наиболее распространенными связанными с лечением AE являлись утомляемость (28,3%), артралгия (11,7%) и тошнота (11,7%). Из 60 пациентов, оцениваемых на ответы опухолей, у 11 (18,3%) наблюдали объективные ответы (PR/CR), в то время как у 31 пациента (51,7%) наблюдали контроль заболевания (CR/PR/SD). Из 36 пациентов, которых подвергали комбинированному лечению, включающему лучевую терапию, объективный ответ наблюдали у 6 пациентов (16,7%), а контроль заболевания - у 19 пациентов (52,8%). Из 24 пациентов, которых не подвергали лучевой терапии, объективный ответ наблюдали у пяти пациентов (20,8%), а контроль заболевания - у 12 пациентов (50%). В таблице 12 представлены обобщенные данные о пациентах, ответивших на лечение.
Таблица 12: Обобщенные данные о пациентах, ответивших на лечение
[00202] Среди пациентов, ответивших на лечение, медиана времени до достижения ответа в случае монотерапии составляла 113 дней (диапазон 52-226), а в случае пациентов, подвергнутых, в том числе, лучевой терапии - 59 дней (диапазон 56-113).
Пример 8: Описания случаев блокирования PD-1 с использованием моноклонального антитела REGN2810 с достижением длительных объективных ответов при метастазирующем немеланомном раке кожи - базально-клеточной карциноме и плоскоклеточной карциноме кожи
Введение
[00203] Базально-клеточная карцинома (BCC) и плоскоклеточная карцинома кожи (CSCC) имеют общий доминирующий фактор риска, УФ-излучение, и, таким образом, эти опухоли являются гипермутантными (Chalmers et al 2016, AACR Ann. Meeting, Abs 3576). При других злокачественных новообразованиях высокая мутационная нагрузка ассоциирована с клинической пользой терапии антителами против иммунных контрольных точек PD-1 [Le et al 2015, New Engl. J. Med. May 30 (Epub ahead of print)]. Более вероятно, что высокомутантные опухоли будут экспрессировать иммуногенные опухолевые неоантигены, привлекающие эффекторные T-клетки, которые могут активировать блокирование иммунных контрольных точек PD-1 (Mandal and Chan 2016, Cancer Discov. 6: 1-12). В этом примере описывают пациентку с метастазирующей BCC и пациента с метастазирующей CSCC, которых лечили REGN2810, полностью человеческим моноклональным антителом против PD-1 в продолжающемся испытании фазы 1 (NCT02383212; описанном в примере 7 в настоящем описании).
Описание случая 1
[00204] Пациентка являлся женщиной возрастом 66 лет, у которой диагностировали BCC стадии 1, возникшую на левой стороне подбородка и подвергнутую резекции с использованием операции по Моосу. Спустя 2 года определяли локализованный рецидив в том же месте, и при широком локальном иссечении выявляли инвазию в левую нижнюю челюсть и вовлечение одного из 18 лимфоузлов. Пациентку подвергали адъювантной лучевой терапии, и она оставалась в ремиссии в течение 4 лет, когда увеличенные узлы в легких, определяемые при визуализации грудной клетки при наблюдении, подвергали биопсии и подтверждали наличие метастазирующей BCC. Впоследствии пациентке вводили ингибитор пути Hedgehog (HHI) висмодегиб в течение 5 месяцев. Сначала у нее наблюдали ответ, но лечение прекратили из-за прогрессирования заболевания.
[00205] Через шесть месяцев после терапии висмодегибом и после продолжающегося медленного прогрессирования пациентку включали в исследование фазы 1 REGN2810 в когорту, которой вводили 10 мг/кг IV каждые 2 недель, и вводили ей первую дозу. Два метастаза в легких отслеживали в качестве целевых очагов. При оценке ответа в конце 8 недель (повышение на 3%) и 16 недель (повышение на 10%) наблюдали стабильное заболевание в соответствии с критериями RECIST. При оценке ответа в конце 24 недель наблюдали уменьшение размеров опухоли на 37% (фигура 14A) и подтверждали его через 32 недели. Пациентка хорошо переносила лечение и продолжает лечение REGN2810 в течение более чем 10 месяцев.
Описание случая 2
[00206] Пациент являлся мужчиной возрастом 52 лет, у которого диагностировали плоскоклеточную карциному кожи на левой щеке. Его подвергали операции по Моосу с четкими границами. Он перенес множество рецидивов и по меньшей мере 9 дополнительных операций по Моосу. Его подвергали широкому локальному иссечению на левой стороне нижней челюсти спустя 4 года и левой паротидэктомии через 20 месяцев. Кроме того, левую щеку, левую нижнюю челюсть, левую сторону шеи (с сопутствующим введением цетуксимаба) подвергали адъювантной лучевой терапии, а также осуществляли билатеральную адъювантную лучевую терапию шеи (с сопутствующим введением карбоплатина). Другая системная терапия представляла собой капецитабин и цисплатин+доцетаксел. Через десять лет после постановки диагноза его подвергали иссечению с четкими границами по причине рецидива опухоли в рубцовой ткани размером 2,2 см на левой стороне шеи. Впоследствии, инвазивная CSCC в телах позвонков C4-C5 повлекла за собой необходимость неотложной декомпрессии шейного отдела спинного мозга с использованием передней корпэктомии на уровне C4-C5 и задней ламинэктомии на уровне C4-C6. У него также развилась мышечная слабость в нижних конечностях, как считают, являющаяся результатом периневрального вовлечения, и в этом случае потребовалось использование ходунков для передвижения.
[00207] Его включали в исследование фазы 1 в первую когорту и вводили 1 мг/кг REGN2810 каждые две недели. В течение недель после начала лечения сила в его нижних конечностях постепенно восстанавливалась, и ему больше не требуются ходунки. Ответ на неделе 16 показан на фигуре 14B. Полного рентгенологического ответа в очаге на левой стороне шеи достигали на неделе 40. Пациент завершал запланированное 48-недельное лечение по протоколу с использованием REGN2810. Он продолжает находиться под тщательным, активным последующим наблюдением онколога без клинических или рентгенографических признаков рецидивирования заболевания.
Обсуждение
[00208] В этом примере описывают первый подтвержденный частичный ответ у пациентки с метастазирующей BCC, которую лечили ингибитором PD-1 (REGN2810), а также продолжающийся длительный полный ответ у пациента с метастазирующей CSCC. Глубокие и устойчивые ответы у этих пациентов, подвергнутых значительному предшествующему лечению, на монотерапию антителом против PD-1 в этом исследовании фазы 1 соответствует гипотезе о том, что высокая мутационная нагрузка при BCC и CSCC будет вызывать противоопухолевый клеточный иммунитет, который можно активировать посредством блокирования пути контрольных точек PD-1/PD-L1.
[00209] В этом примере подтверждают общий принцип, состоящий в том, что УФ-ассоциированные злокачественные новообразования кожи, помимо меланомы, являются чувствительными к блокированию PD-1. С помощью редукционистской модели прогнозировали, что у УФ-ассоциированных опухолей с более высокой нагрузкой несинонимичных мутаций будет более высокая отвечаемость на блокирование PD-1, чем у опухолей с более низкой мутационной нагрузкой.
Пример 9: Безопасность и эффективность антитела против PD-1 у пациентов с неоперабельной местнораспространенной или метастазирующей плоскоклеточной карциномой кожи (CSCC)
Предпосылки
[00210] Не существует установленного стандарта лечения неоперабельной местнораспространенной или метастазирующей CSCC. В результате УФ-индуцируемого повреждения ДНК большинство CSCC являются гипермутантными. Таким образом, эти опухоли могут отвечать на блокирование контрольных точек PD-1. В этом примере описывают пациентов с местнораспространенной или метастазирующей CSCC, которых лечили REGN2810, полностью человеческим моноклональным антителом против PD-1, в продолжающемся исследовании фазы 1 (NCT02383212; описанном в примере 7 в настоящем описании).
Способы
[00211] В расширенные когорты (EC) в исследовании фазы 1 REGN2810 включали пациентов с CSCC с отдаленными метастазами (EC 7) и местнораспространенной CSCC (EC8) (таблица 9). Всем пациентам вводили 3 мг/кг REGN2810 внутривенно каждые 2 недели в течение до 48 недель. Исследовательскую биопсию осуществляли на исходном уровне и в день 29 (и при прогрессировании, если возможно). Для определения общей частоты ответа измерение опухоли осуществляли каждые 8 недель в соответствии с RECIST 1.1.
Результаты
[00212] В исследование включали 25 пациентов (10 в EC 7 и 15 в EC 8): медиана возраста 72,5 лет (диапазон 56-88 лет); медиана PS 1 (диапазон 0-1); 20 M:5F; медиана количества схем предшествующей системной терапии 1 (диапазон 0-3). Медиана воздействия REGN2810 составляла 6 доз (диапазон 1-22). Наиболее распространенными связанными с лечением нежелательными явлениями любой степени являлись утомляемость (16,7%), тошнота, артралгия и сыпь (по 8,3% каждое). Каждое из следующих связанных с лечением нежелательных явлений (AE) степени ≥3 возникало однократно: повышение АСТ, повышение АЛТ, артралгия и сыпь.
[00213] Общая частота ответа (uPR+PR+CR) и частота контроля заболевания (ORR+SD) составляли 48% (11/23; 3uPR, 5 PR, 2 CR, 1 uCR) и 70% (16/23, включая 5 SD), соответственно. Два пациента еще не подвергнуты оценке. Вычисляют медиану PFS и медиану OS, и только у одного пациента наблюдали PD в течение лечения REGN2810 после исходного ответа. Продолжаются сравнительные научные исследования, включающие полноэкзомное секвенирование опухолевой ДНК.
Вывод
[00214] REGN2810 демонстрирует устойчивую противоопухолевую активность у пациентов с CSCC на поздней стадии.
Пример 10: Клиническое испытание антитела против PD-1 в комбинации с гипофракционированной лучевой терапии по сравнению со стандартом лечения на пациентах возрастом ≥65 лет с впервые диагностированной глиобластомой
Введение
[00215] Глиобластома является смертельным заболеванием с медианой выживаемости приблизительно 16 месяцев у пациентов с впервые диагностированной опухолью (nGBM) и приблизительно 9 месяцев в условиях рецидивирования (rGBM) (Friedman et al, 2009, J. Clin. Oncol. 27: 4733-4740). Существующим стандартом лечения для пациентов с впервые диагностированной глиобластомой является лучевая терапия (60 Гр за 6 недель) с сопутствующим введением темозоломида (TMZ) с последующим адъювантным лечением темозоломидом (Stupp et al, 2005, N. Engl. J. Med. 352: 987-996), хотя анализ в подгруппах позволяет предполагать, что добавление темозоломида может не улучшать эффективность у индивидуумов старшего возраста (Laperriere et al, 2013, Cancer Treat. Rev. 39: 350-357).
[00216] В этом примере описывают исследование фазы 3 для оценки эффективности антитела против PD-1 в комбинации с гипофракционированной лучевой терапией (hfRT) по сравнению со стандартом лечения (SoC) в терминах общей выживаемости у пациентов с nGBM возрастом ≥65 лет.
[00217] Примером антитела против PD-1, используемого в этом исследовании, является REGN2810 (также известное как H4H7798N, как описано в US20150203579), полностью человеческое моноклональное антитело против PD-1, содержащее тяжелую цепь, содержащую аминокислотную последовательность SEQ ID NO: 9, и легкую цепь, содержащую аминокислотную последовательность SEQ ID NO: 10, пару аминокислотных последовательностей HCVR/LCVR, содержащую SEQ ID NO: 1/2, и последовательности CDR тяжелой и легкой цепи, содержащие SEQ ID NO: 3-8.
Цели исследования
[00218] Первичной целью исследования является оценка эффективности в терминах общей выживаемости (OS) при введении REGN2810 в комбинации с hfRT по сравнению со стандартом лечения для пациентов с nGBM возрастом ≥65 лет.
[00219] Вторичной целью исследования является определение улучшения выживаемости без прогрессирования (PFS).
[00220] Другими целями исследования являются: (i) улучшение частоты объективных ответов (ORR), длительность ответа и длительность контроля заболевания; (ii) клиническая оценка с использованием шкалы неврологической оценки в нейроонкологии (NANO); (iii) оценка безопасности; (iv) улучшение качества жизни (QoL) и психического состояния; (v) изменения отека и использования стероидов; (vi) определение концентрации REGN2810 в сыворотке и антител против REGN2810; и (vii) исследование потенциальных фармакодинамических или прогностических биомаркеров.
Дизайн исследования
[00221] Это исследование являлось рандомизированным в соотношении 2:1 исследованием фазы 3 REGN2810, полностью человеческого антитела против PD-1, в комбинации с гипофракционированной лучевой терапией по сравнению со стандартом лечения у пациентов с впервые диагностированной глиобластомой возрастом ≥65 лет. Пациентов случайным образом распределяют по группам, которые подвергают лечению с использованием REGN2810 в комбинации с гипофракционированной лучевой терапией или стандартом лечения, в соотношении 2:1 с определением статуса метилирования (метилированный, или неметилированный, или неопределенный) и объема резекции (частичная или общая тотальная резекция) в качестве факторов стратификации. Эффективность оценивают по общей выживаемости.
[00222] Пациентов с nGBM, являющихся кандидатами для лучевой терапии, случайным образом распределяют в соотношении 2:1 в одну из следующих групп:
Исследовательская терапия: 3 мг/кг REGN2810 IV (каждые 2 недели) и гипофракционированная RT (6 Гр×5, только на второй неделе). Лучевую терапию осуществляют на неделе 2 цикла 1, но не последующих циклов.
Сравнительная терапия: стандарт лечения - TMZ (перорально, 75 мг/м2, ежедневно) в комбинации со стандартной RT (5 ежедневных фракций облучения/неделю по 2 Гр) в течение 6 недель, с последующей адъювантной терапией (перорально, от 150 мг/м2 до 200 мг/м2, 5 дней/28 дней) в течение 6 циклов. Лучевую терапию осуществляют только в течение первого 6-недельного цикла.
Длительность исследования
[00223] Исследование состоит из 28-дневного периода скрининга, после которого подходящих пациентов могли подвергать до двенадцати 56-дневным (8-недельным) циклам лечения всего в течение до 96 недель лечения. В течение периода скрининга (со дня -28 по день -1) все подходящие пациенты должны иметь доступные результаты резекции опухоли перед лечением (частичной или полной резекции) или биопсии для централизованного подтверждения патологии и определения и подтверждения метилирования MGMT.
[00224] После дня 1/исходного уровня пациенты возвращаются в клинику в течение цикла 1 в дни 8±3, 15±3, 29±3, 43±3 и 56±3. В каждый последующий 8-недельный цикл (циклы 2-12) пациенты возвращаются в клинику в дни 1, 15±3, 29±3, 43±3 и 56±3. Оценку опухоли (МРТ головного мозга, оценки iRANO и NANO, оценки по опросникам MMSE и EORTC QLQ-C30/BN20) осуществляют в день 1/на исходном уровне и в конце каждого цикла лечения. Обширную оценку безопасности осуществляют в день 1 каждого цикла; рутинную оценку безопасности будут осуществлять во время каждого визита. Также собирают образцы для оценки биомаркеров (клеточных и молекулярных маркеров, представленных в настоящем описании), связанных с воздействием REGN2810 при лечении, клинической активностью или основным заболеванием.
[00225] В течение 24-недельного периода последующего наблюдения пациенты возвращаются в клинику через 21-42 дней после последнего лечения в рамках исследования для первого визита для последующего наблюдения. Последующие визиты для последующего наблюдения (визиты для последующего наблюдения 2-7) осуществляют каждые 28 дней ± 7 дней. Оценку опухоли (МРТ головного мозга, оценки iRANO и NANO, оценки по опросникам MMSE и EORTC QLQ-C30/BN20) осуществляют во время визита для последующего наблюдения 3, визита для последующего наблюдения 5 и визита для последующего наблюдения 7. Обширную оценку безопасности осуществляют во время первого визита для последующего наблюдения, рутинную оценку безопасности будут осуществлять во время последующих визитов для последующего наблюдения. Собирают образцы для анализа биомаркеров (клеточных и молекулярных маркеров, представленных в настоящем описании), связанных с воздействием REGN2810 при лечении, клинической активностью или основным заболеванием.
Целевая выборка
[00226] Целевая выборка включает пациентов с nGBM возрастом ≥65 лет.
[00227] Критерии включения: Пациент должен соответствовать следующим критериям, чтобы подходить для включения в исследование: (1) впервые диагностированная первичная глиобластома с гистологическим подтверждением, имеющая максимальный диаметр ≤5 см и подвергнутая частичной или полной хирургической резекции; (2) общее состояние по шкале Восточной объединенной онкологической группы (ECOG) 0-2; (3) возраст ≥65 лет; (4) функция печени: (a) общий билирубин ≤1,5-кратного верхнего предела нормы; (b) АЛТ и АСТ ≤3-кратного ULN; (c) щелочная фосфатаза (ALP) ≤2,5-кратного ULN; (5) функция почек: креатинин сыворотки ≤1,5-кратного ULN; (6) функция костного мозга: гемоглобин ≥9,0 г/дл; абсолютное количество нейтрофилов (ANC) ≥1,5×109/л; количество тромбоцитов ≥75×109/л; (7) способность прочитать, понять и желание подписать ICF; и (8) способность и желание следовать запланированным визитам, планам лечения, лабораторным анализам и другим предусмотренным исследованием процедурам.
[00228] Критерии исключения: Пациента, соответствующего любым из следующих критериев, будут исключать из исследования: (1) любое предшествующее лечение GBM (иное, чем хирургическая операция); (2) имеет известное противопоказание к Gd-МРТ; (3) сохраняющиеся или недавние (в пределах 5 лет) признаки значительного аутоиммунного заболевания, для которого необходимо лечение посредством системной иммуносупрессии, что может вызывать риск иммуноопосредованных нежелательных явлений (irAE). Следующие заболевания не являются исключающими: витилиго, разрешенная астма детского возраста, остаточный гипотиреоз, в случае которого необходима лишь гормонозаместительная терапия, или псориаз, в случае которого не требуется системное лечение. (4) Продолжающееся лечение кортикостероидами, за исключением использования кортикостероидов по другим (неопухолевым и неиммуносупрессорным) показаниям максимум до 10 мг/день преднизона или его эквивалента. (5) Первичные опухоли, располагающиеся в стволе головного мозга, спинном мозге, или любая активная инфекция во вторичной опухоли головного мозга, для которой необходима терапия, включая известную инфекцию вируса иммунодефицита человека, или активная инфекция вируса гепатита B или гепатита C. (6) Пневмонит в анамнезе за последние 5 лет. (7) Осуществление любого исследовательского или противоопухолевого лечения в пределах 30 дней перед исходным введением REGN2810. (8) Задокументированные аллергические реакции или острая реакция гиперчувствительности в анамнезе, приписываемые лечению антителами в целом или средствам, конкретно используемым в этом исследовании. (9) Не контролируемая в достаточной степени гипертензия (определяемая как систолическое давление >150 мм рт.ст. и/или диастолическое давление >100 мм рт.ст.). (10) Известная аллергия на доксициклин или тетрациклин (предостережение по причине наличия их следовых количеств в REGN2810). (11) Гипертонический криз или гипертоническая энцефалопатия в анамнезе. (12) Инвазивное злокачественное новообразование в анамнезе за последние 5 лет, иное, чем злокачественное новообразование, подвергаемое лечению в рамках настоящего исследования, за исключением резецированной/аблированной базально-клеточной или плоскоклеточной карциномы кожи, или карциномы шейки матки in situ, или других локализованных опухолей, считающихся излечиваемыми посредством местного лечения. (13) Острые или хронические психиатрические расстройства по решению исследователя могут делать пациента непригодным для участия в исследовании. (14) Использование технологии Novocure Tumor Treating Fields (устройства Optune NovoTTF-100A) на момент скрининга. Планируемое или ожидаемое использование технологии Novocure Tumor Treating Fields в течение исследования. (15) Предшествующее лечение с использованием капсул-имплантатов с кармустином. (16) Сохраняющаяся половая активность у мужчин, нежелающих использовать соответствующую контрацепцию в течение исследования.
Исследуемое лечение
[00229] Для пациентов используют одну из следующих схем лечения:
[00230] Исследовательская терапия: 3 мг/кг REGN2810 (вводимого посредством IV инфузии в течение 30 минут каждые 2 недели в течение до 96 недель) и hfRT на неделе 2 цикла 1
[00231] Сравнительное лечение: стандарт лечения TMZ (перорально, 75 мг/м2, ежедневно) в комбинации со стандартной RT (5 ежедневных фракций облучения/неделю по 2 Гр) в течение 6 недель с последующей адъювантной терапией TMZ (перорально, от 150 мг/м2 до 200 мг/м2, 5 дней/28 дней) в течение 6 циклов. Лучевую терапию осуществляют только в течение первого цикла.
[00232] REGN2810 поставляют в виде жидкости в стерильном одноразовом флаконе. Каждый флакон содержит объем, достаточный для забора 10 мл REGN2810 в концентрации 25 мг/мл. REGN2810 вводят в виде 30-минутной IV инфузии. Доза для каждого пациента будет зависеть от массы тела индивидуума. Дозу REGN2810 необходимо корректировать во время каждого цикла при изменениях массы тела на ≥10%.
[00233] Лучевая терапия: Пациентов в контрольной группе подвергают стандартной лучевой терапии (60 Гр за 6 недель). Пациентов в группе экспериментального лечения подвергают hfRT (6 Гр×5 ежедневных фракций), осуществляемой через 1 неделю после введения первой дозы REGN2810.
[00234] REGN2810 и лучевая терапия (исследовательское лечение): REGN2810 вводят посредством IV инфузии в течение 30 минут каждые 14 дней в течение 96 недель в комбинации с hfRT со дня 8 по день 12.
[00235] Запланированная схема комбинированного лечения REGN2810 и hfRT: инфузия 3 мг/кг REGN2810 в течение 30 минут каждые 14 дней в течение 96 недель и лучевая терапия (hfRT в дозе 6 Гр×5 ежедневных фракций; осуществляемая через 1 неделю после введения первой дозы REGN2810, предпочтительно, в последовательные дни).
[00236] Характеристика лучевой терапии: Пациенты получают 30 Гр в виде 5 фракций по 6 Гр ежедневно, начиная через 1 неделю после введения первой дозы REGN2810.
[00237] Сравнительная группа: стандарт лечения: TMZ (перорально, 75 мг/м2, ежедневно) в комбинации со стандартной RT (5 ежедневных фракций облучения/неделю по 2 Гр) в течение 6 недель с последующей адъювантной терапией пероральным TMZ. Доза TMZ составляет 150 мг/м2 в течение первых 5 дней первого цикла адъювантной терапии, и ее повышают до 200 мг/м2 на 5 дней/28 дней, начиная со второго цикла, если нет неприемлемой гематологической токсичности во время первого цикла адъювантной терапии.
[00238] Если во время первого цикла адъювантной терапии все наблюдаемые случаи негематологической токсичности имеют степень ≤2 (за исключением алопеции, тошноты и рвоты), и количество тромбоцитов составляет ≥100×109/л, а ANC ≥1,5×109/л, то дозу TMZ необходимо повышать до уровня дозы 1 (200 мг/м2) и эту дозу необходимо использовать в качестве начальной дозы для последующих циклов. Если после цикла 1 введение TMZ необходимо прекратить по причине сохраняющейся негематологической токсичности степени ≥2, повышение дозы невозможно. Если дозу не повышали во время второго цикла, дозу не следует повышать в последующих циклах.
[00239] Лечение отека ЦНС: Введение REGN2810 и лучевую терапию прекращают в отношении любого пациента, у которого развивается симптоматический отек головного мозга во время исследования, до уменьшения отека.
[00240] Пациентам, у которых развивается отек головного мозга, вводят бевацизумаб IV по мере необходимости (PRN) в дозе, сниженной относительно стандартной дозы (предполагаемой дозе 5 мг/кг Q2W в количестве до 3 дозы, но не более 10 мг/кг Q2W на дозу), если нет противопоказаний (например, если пациента не подвергали хирургической операции за последние 28 дней).
[00241] Если введение бевацизумаба не приводит к разрешению отека головного мозга, в дополнение к бевацизумабу или вместо него можно вводить системные кортикостероиды в наименьшей дозе, считающейся подходящей для лечения симптома. В случае пациентов, непереносящих бевацизумаб, кортикостероиды используют в дозе, считающейся подходящей для лечения симптома.
Переменные в исследовании
[00242] Первичной конечной точкой при оценке эффективности является общая выживаемость (OS), определяемая как временной интервал между датой рандомизации и датой смерти по любой причине.
[00243] Ключевой вторичной конечной точкой является выживаемость без прогрессирования (PFS), определяемая как временной интервал между датой рандомизации до даты первого наблюдения прогрессирования заболевания или даты смерти (по любой причине). Прогрессирование заболевания определяют с помощью критериев iRANO.
[00244] Другими вторичными конечными точками при оценке эффективности являются:
[00245] Частота объективных ответов (ORR): определяемая как доля пациентов с подтвержденным полным ответом (CR) или подтвержденным частичным ответом (PR), определяемыми с помощью критериев оценки ответа на иммунотерапию в нейроонкологии (iRANO), относительно общего количества пациентов в анализируемой выборке.
[00246] Длительность ответа: определяют для пациентов с лучшим общим ответом CR или PR. Длительность ответа измеряют с моменты, когда критерии измерения впервые начинают соответствовать CR/PR (в зависимости от того, что регистрируют первым) до первой даты рецидивирования или прогрессирования заболевания (по данным рентгенографии) или смерти по любой причине.
[00247] Длительность контроля заболевания: определяют для пациентов с лучшим общим ответом SD, CR или PR. Длительность контроля заболевания измеряют с начала лечения до первой даты рецидивирования или прогрессирования заболевания (по данным рентгенографии) или смерти по любой причине.
[00248] Переменные качества жизни и контроля симптомов: Переменными качества жизни и контроля симптомов являются:
Пять функциональных шкал, три шкалы симптомов, одна шкала глобальной оценки состояния здоровья и шесть шкал из одного пункта для оценки симптомов с использованием опросников EORTC QLQ-C30 в течение исследования
Четыре шкалы и семь шкал из одного пункта с использованием опросников EORTC QLQ-BN20 в течение исследования
Клиническая оценка с использованием NANO;
Общие баллы MMSE в течение исследования
Использование кортикостероидов на исходном уровне, кумулятивное использование кортикостероидов в течение исследования и длительность отсутствия использования стероидов или использования стероидов в низких дозах в течение периода без прогрессирования в течение исследования
Использование бевацизумаба PRN на исходном уровне, кумулятивное использование бевацизумаба PRN в течение исследования и длительность отсутствия использования бевацизумаб в течение периода без прогрессирования в течение исследования
[00249] Исследовательские переменные биомаркеров: Другая конечная точка включает фармакодинамические и прогностические биомаркеры, связанные с клиническим ответом, механизмом действия и возможными AE, ассоциированными с REGN2810, после лечения. Переменные биомаркеров включают:
Уровни экспрессии рецепторов PD-L1, GITR и LAG3 иммунных контрольных точек, а также других потенциальных биомаркеров (например, EGFRvIII, Ki67 и т.д.) в образцах опухоли;
Количество и распределение TIL в образцах опухоли;
Мутационный статус IDH1, микросателлитная нестабильность (MSI) и мутационная нагрузка в образцах опухоли;
Циркулирующие биомаркеры, включая цитокины и ангиогенные факторы;
Субпопуляции клеток и уровни экспрессии интересующих биомаркеров в PBMC;
Статус метилирования промотора MGMT (также используемый для стратификации)
[00250] Другие переменные включают концентрацию REGN2810 в сыворотке (фармакокинетические переменные) и возникновение антител против REGN2810.
Процедуры и оценки
[00251] После периода скрининга в течение до 28 дней пациентов подвергают до двенадцати 56-дневным циклам лечения всего в течение до 96 недель лечения с последующим 24-недельным периодом последующего наблюдения. Осуществляют оценку эффективности, оценку безопасности, анализы PK, ADA и исследовательских биомаркеров.
Процедуры по оценке эффективности
[00252] МРТ: МРТ для оценки опухоли осуществляют через 72 часов после хирургической операции, во время визита для скрининга (в пределах 28 дней перед инфузией), в день 56±3 каждого цикла (приблизительно каждые 8 недель), и когда подозревают PD. Пациентов, у которых заболевание не прогрессирует, подвергают дополнительным оценкам опухоли, осуществляемым во время визитов для последующего наблюдения 3, 5 и 7. Примечание: если подтверждают PD, дополнительные сканирования во время визитов для последующего наблюдения не потребуются. Если МРТ до и после хирургической операции осуществляли перед включением в исследование, результаты этих сканирований также необходимо предоставлять для исследования, чтобы облегчить определение объема опухоли и прогрессирования опухоли.
[00253] Оценку ответа опухоли осуществляют в соответствии с iRANO; а клиническую неврологическую оценку будут осуществлять посредством NANO. Оценки в соответствии с RANO также осуществляют в качестве вспомогательного обследования; однако, первичное определение прогрессирования заболевания в случае отдельного пациента осуществляют в соответствии с iRANO.
[00254] Опросник качества жизни Европейской организации по изучению и лечению рака (EORTC QLQ-C30) и Опросник модуля "Злокачественное новообразование головного мозга" EORTC (EORTC QLQ-BN20): EORTC QLQ-C30 является опросником из 30 пунктов, с помощью которого оценивают связанное со здоровьем качество жизни (HRQoL) у пациентов со злокачественными новообразованиями с использованием 15 шкал (из одного или множества пунктов), каждая из которых включает возможные баллы в диапазоне от 0 до 100. Из 30 пунктов 24 объединены в 9 шкал с множеством пунктов, представляющих собой различные показатели HRQoL: 5 шкал функциональности (физическая, ролевая, эмоциональная, когнитивная и социальная), 3 шкалы симптомов (утомляемость, боль и тошнота) и 1 глобальная шкала состояния здоровья. С помощью остальных 6 шкал из одного пункта оценивают симптомы: одышку, потерю аппетита, нарушения сна, запор и диарею и субъективные финансовые последствия лечения заболевания. Высокие баллы свидетельствуют о лучшем HRQoL в случае глобальной шкалы состояния здоровья и шкал функциональности и худшем HRQoL в случае шкал симптомов.
[00255] EORTC QLQ-BN20 является оценкой QoL из 20 пунктов, характерных для неоплазий головного мозга и предназначен для дополнения EORTC QLQ-C30 при оценке связанного со здоровьем качества жизни. С помощью опросника EORTC QLQ-BN20 оценивают симптомы заболевания, побочные эффекты лечения и некоторые конкретные психосоциальные вопросы, имеющие значение для пациентов со злокачественным новообразованием головного мозга, с использованием 4 шкал (оценка неуверенности в будущем, нарушений зрения, двигательной дисфункции и дефицита общения) и 7 отдельных пунктов (оценка других симптомов заболевания [например, головных болей и судорог] и токсических эффектов лечения [например, выпадения волос]). Диапазон возможных баллов составляет от 0 до 100, высокие баллы свидетельствуют о худшем HRQoL.
[00256] Краткая шкала оценки психического статуса: Краткая шкала оценки психического статуса (MMSE©) представляет собой короткое, количественное измерение когнитивного статуса у взрослых. Ее можно использовать для скрининга на когнитивные нарушения, оценки тяжести когнитивных нарушений в определенный момент времени и отслеживания динамики когнитивных изменений у индивидуума с течением времени. В настоящем исследовании баллы MMSE являются частью неврологического осмотра, осуществляемого в контексте оценки заболевания.
[00257] MMSE осуществляют в день 1/на исходном уровне, в конце каждого цикла лечения и каждые 8 недель в течение периода последующего наблюдения. Оценки MMSE совпадают со схемой оценок заболевания, но их необходимо завершать до информирования пациента о результатах рентгенографических исследований. MMSE необходимо завершать в начале следующего введения, запланированного по схеме лечения. В течение периода последующего наблюдения для оценки выживаемости необходимо продолжать оценивать MMSE на каждом втором визите для оценки выживаемости (каждые 8 недель), если у пациента еще не наблюдают прогрессирование.
[00258] Общие баллы MMSE имеют возможный диапазон от 0 (худшие) до 30 (лучшие).
Процедуры по оценке безопасности
[00259] В день 1 цикла 1 и во все последующие дни лечения будут оценивать показатели жизнедеятельности, включая температуру, артериальное давление в покое, пульс и частоту дыхательных движений, вместе с массой тела перед инфузией и через приблизительно 15 минут после завершения инфузии. Полный физикальный осмотр и ЭКГ в 12 отведениях осуществляют в начале каждого цикла.
Процедуры по оценке исследовательских биомаркеров опухоли
[00260] Интересующие биомаркеры, анализируемые посредством иммуногистохимии (IHC), включают, в качестве неограничивающих примеров, EGFRvIII и биомаркеры пролиферации клеток (например, Ki67). Для исследования потенциального эффекта REGN2810 в биоптатах опухоли анализируют уровни экспрессии (мРНК и/или белка) PD-L1, GITR и LAG-3, а также маркеры линий дифференцировки инфильтрирующих опухоль лимфоцитов (CD4, CD8, CD25, FoxP3).
[00261] Образцы опухолевой ткани можно использовать для выделения ДНК и РНК опухоли и последующих анализов предполагаемых генетических биомаркеров, относящихся к исследуемому лечению и глиобластоме. Образцы крови собирают для выделения ДНК зародышевой линии в день 1/на исходном уровне (перед введением дозы) или во время любого визита в рамках исследования, если забор в день 1/на исходном уровне невозможен. Анализы опухолевой ДНК включают (в качестве неограничивающих примеров) статус метилирования промотора MGMT, мутационный статус IDH1, микросателлитную нестабильность (MSI) и мутационную нагрузку опухоли (обе из которых могут быть прогностическими в отношении ответа на REGN2810 и другие иммунотерапевтические средства). Осуществляют анализ генетических вариантов в опухолевой (соматической) ДНК и ДНК зародышевой линии, которые могут влиять на прогрессирование заболевания, ответ на лекарственное средство и возможную токсичность. ДНК зародышевой линии также используют для сравнения с опухолевой ДНК для исследования потенциальных новых генетических вариантов, лежащих в основе злокачественных процессов.
Результаты
[00262] REGN2810 в комбинации с hfRT является безопасным и хорошо переносилось пациентами с nGBM. Введение REGN2810 в комбинации с hfRT ингибирует рост опухоли и/или способствует регрессированию опухоли у пациентов с nGBM по сравнению со стандартом лечения. У пациентов с nGBM, которых лечили REGN2810 и hfRT, наблюдали более длительную OS по сравнению со стандартом лечения.
Пример 11: Переносимость и противоопухолевая активность REGN2810 у пациентов с немелкоклеточным раком легких: промежуточные данные исследования фазы 1
[00263] В исследовании с повышением дозы (DE) фазы 1 (описанном в примере 7 в настоящем описании) оценивали монотерапию REGN2810 (цемиплимабом) в дозе 1 мг/кг, вводимой внутривенно (IV) в течение 30 минут каждые 2 недели (Q2W), в связи с немелкоклеточным раком легких (NSCLC). В расширенную когорту NSCLC (EC 1) включали пациентов, перенесших рецидив после терапии первой линии или рефрактерных к по меньшей мере терапии первой линии в условиях рецидивирующего или метастазирующего заболевания; пациентам вводили 200 мг цемиплимаба IV в течение 30 минут Q2W в течение до 48 недель. Исследовательскую биопсию осуществляли на исходном уровне и в день 29 (и при прогрессировании, если это возможно). Измерение опухоли осуществляли каждые 8 недель в соответствии с RECIST (Критериями оценки ответа солидных опухолей) 1.1.
[00264] Промежуточные результаты: в исследование включали 21 пациента с NSCLC (1 в DE; 20 в EC 1); медиана возраста составляла 65,0 лет (диапазон 50-82; 14 мужчин/7 женщин); 81,0% имели медиану статуса по шкале Восточной объединенной онкологической группы 1. Большинство (61,9%) имели гистологические признаки аденокарциномы на исходном уровне. В целом, наиболее распространенными связанными с лечениями нежелательными эффектами (TRAE) являлись астения, пневмонит и сыпь (каждое n=3, 14,3%). Каждое из следующих TRAE степени ≥3 возникало однократно: пневмонит, диабетический кетоацидоз и нефрит. У 6 из пациентов в EC 1 наблюдали частичный ответ (PR), а у 4 - стабильное заболевание (SD). Общая частота ответа (ORR=полный ответ [CR] + PR) по результатам централизованной независимой проверки (передача данных: 31 августа 2017 года) составляла 28,6% (n=6/21). Частота контроля заболевания (ORR+SD) составляла 57,1% (n=12/21; из которых 1 имел не-CR/не-прогрессирующее заболевание [PD]). В целом, у 9 пациентов (все из EC 1) наблюдали PD в течение лечения цемиплимабом.
[00265] Цемиплимаб, как правило, хорошо переносился, и наблюдали его противоопухолевую активность у пациентов NSCLC в этом исследовании.
Пример 12: Клиническое испытание REGN2810 при лечении первой линии пациентов с PD-L1+ немелкоклеточным раком легких на поздней стадии или метастазирующим PD-L1+ немелкоклеточным раком легких
[00266] Настоящее исследование является рандомизированным, международным, открытым исследованием фазы 3 монотерапии REGN2810 по сравнению со стандартом лечения, двухкомпонентной химиотерапии препаратами платины, на пациентах с плоскоклеточным или неплоскоклеточным NSCLC на поздней стадии или метастазирующим плоскоклеточным или неплоскоклеточным NSCLC, где опухоли экспрессируют PD-L1 в ≥50% опухолевых клеток, и где пациентов ранее не подвергали системному лечению их заболевания на поздней стадии.
Цели исследования
[00267] Основной целью исследования является определение того, улучшает ли REGN2810 выживаемость без прогрессирования (PFS) по сравнению со стандартом лечения, двухкомпонентной химиотерапией препаратами платины, у пациентов с плоскоклеточным или неплоскоклеточным NSCLC на поздней стадии или метастазирующим плоскоклеточным или неплоскоклеточным NSCLC, где опухоли экспрессируют PD-L1 в ≥50% опухолевых клеток. Ключевыми вторичными целями исследования являются сравнение REGN2810 с химиотерапией препаратами платины в отношении:
Общей выживаемости (OS)
Частоты объективных ответов (ORR)
[00268] Другими вторичными целями исследования являются следующие:
Сравнение длительности ответа (DOR) в случае REGN2810 по сравнению с химиотерапией препаратами платины
Оценка качества жизни (QOL) пациентов, подвергнутых лечению с использованием REGN2810, по сравнению с пациентами, подвергнутыми химиотерапии препаратами платины, осуществляемая с помощью Опросника качества жизни Core 30 Европейской организации по изучению и лечению рака (EORTC QLQ-C30) и Опросника качества жизни при раке легких 13 (EORTC QLQ-LC13).
Оценка безопасности и переносимости REGN2810 по сравнению с химиотерапией препаратами платины
Измерение концентраций REGN2810 в сыворотке и характеризация фармакокинетики (PK) REGN2810
[00269] Поисковые цели включают:
Оценку иммуногенности, измеряемой посредством оценки иммуногенности REGN2810
Анализ корреляции уровня экспрессии PD-L1 на исходном уровне и эффективности исследуемого лечения
Оценку времени до новой противоопухолевой терапии
Дизайн исследования
[00270] Это исследование является рандомизированным, многоцентровым, открытым, опорным исследованием фазы 3 монотерапии REGN2810 по сравнению с двухкомпонентной химиотерапией препаратами платины на пациентах с плоскоклеточным или неплоскоклеточным NSCLC стадии IIIB или стадии IV, где опухоли экспрессируют PD-L1 в ≥50% опухолевых клеток, и где пациентов ранее не подвергали системному лечению их заболевания на поздней стадии.
[00271] Исследование состоит из следующих 3 периодов: скрининга, лечения и последующего наблюдения. Пациентов подвергают скрининговой оценке для определения их пригодности в пределах 28 дней до рандомизации. Пригодных пациентов случайным образом распределяют в одну из следующих 2 групп лечения: монотерапии 350 мг REGN2810 или химиотерапии стандарта лечения. При рандомизации осуществляют стратификацию по гистологии (неплоскоклеточный и плоскоклеточный) и географической области (EU или ROW). Для пациентов с NSCLC, которым случайным образом предписывают химиотерапию, могут использовать одну из следующих схем лечения:
Паклитаксел+цисплатин или карбоплатин
Гемцитабин+цисплатин или карбоплатин
Пеметрексед+цисплатин или карбоплатин с последующим необязательным поддерживающим лечением пеметрекседом (в отношении пациентов с плоскоклеточным NSCLC рекомендуют не использовать схемы лечения, включающие пеметрексед)
[00272] Пациентам, приписанным к группе лечения REGN2810, вводят 350 мг REGN2810 в виде внутривенной (IV) инфузии в день 1 каждого цикла лечения (каждые 3 недели [Q3W]) в течение до 108 недель или до определяемого с помощью Критериев оценки ответа солидных опухолей (RECIST) 1.1 прогрессирования заболевания, неприемлемой токсичности, смерти или отзыва согласия на участие в исследовании. Пациенты в группе REGN2810, у которых наблюдают определяемое с помощью RECIST 1.1 прогрессирование заболевания во время терапии, могут продолжать лечение с использованием REGN2810, если определяют, что пациент получает клиническую пользу, и если пациент не завершил 108-недельный период лечения. Если подтверждают дальнейшее прогрессирование заболевания (определяемое как дополнительное 10%-ное повышение опухолевой массы с момента исходного прогрессирования заболевания), необходимо прекратить введение REGN2810 и, при необходимости, рассмотреть другую противоопухолевую терапию.
[00273] Для пациентов, приписанных в группу химиотерапии, выбирают один из вариантов двухкомпонентной химиотерапии препаратами платины по протоколу в течение до 4-6 циклов или до достижения определяемого с помощью RECIST 1.1 прогрессирования заболевания, неприемлемой токсичности, смерти или отзыва согласия на участие в исследовании. Пациентам, у которых наблюдают прогрессирование заболевания во время химиотерапии, предлагают вариант перехода на другое лечение с введением 350 мг REGN2810 Q3W в течение до 108 недель, при условии, что они соответствуют конкретным критериям. Пациенты осуществляют визиты для последующего наблюдения каждые 6 недель в течение 6 месяцев, а затем в 9 месяцев и 12 месяцев после введения последней дозы. Длительность исследования для каждого пациента составляет приблизительно 40 месяцев.
Исследуемая выборка
[00274] Пациенты, включенные в это исследования, являются мужчинами и женщинами возрастом ≥18 лет с диагностированным неплоскоклеточным или плоскоклеточным NSCLC стадии IIIB или стадии IV, у которых опухоли экспрессируют PD-L1 в ≥50% опухолевых клеток (что определяют с использованием диагностического анализа) и которых ранее не подвергали системному лечению их заболевания на поздней стадии.
[00275] Критерии включения: Пациент должен соответствовать следующим критериям, чтобы подходить для включения в исследование:
1. Мужчины и женщины возрастом ≥18 лет
2. Пациенты с гистологически или цитологически задокументированным плоскоклеточным или неплоскоклеточным NSCLC стадии IIIB или стадии IV, которых ранее не подвергали системному лечению рецидивирующего или метастазирующего NSCLC. Пригодными являются пациенты, подвергавшиеся адъювантной или неоадъювантной двухкомпонентной химиотерапии препаратами платины (после хирургической операции и/или лучевой терапии), и у которых развилось рецидивирующее или метастазирующее заболевание через более чем 6 месяцев после завершения терапии
3. Архивная или недавно полученная фиксированная в формалине опухолевая ткань из очага метастазирования/рецидивирования, который ранее не облучали
4. Опухолевые клетки, экспрессирующие PD-L1 в ≥50% опухолевых клеток по результатам IHC
5. По меньшей мере 1 рентгенографически измеримый очаг по результатам компьютерной томографии (КТ) или магнитно-резонансной томографии (МРТ) в соответствии с критериями RECIST 1.1. Целевые очаги могут локализоваться в ранее облученном поле, если в этом очаге задокументировано (рентгенографически) прогрессирование заболевания.
6. Статус по шкале ECOG ≤1
7. Ожидаемая продолжительность жизни по меньшей мере 3 месяца
8. Достаточная функция органов и костного мозга, как определено ниже: a. Гемоглобин ≥9,0 г/дл b. Абсолютное количество нейтрофилов ≥1,5×109/л c. Количество тромбоцитов ≥100000/мм3 d. Уровень клубочковой фильтрации (GFR) >30 мл/мин/1,73м2 e. Общий билирубин ≤1,5-кратного верхнего предела нормы (ULN) (при метастазах в печени ≤3-кратного ULN), за исключением пациентов, у которых диагностирован клинически подтвержденный синдром Жильбера f. Аспартатаминотрансфераза (АСТ) и аланинаминотрансфераза (АЛТ) ≤3-кратного ULN или ≤5-кратного ULN при метастазах в печени g. Щелочная фосфатаза ≤2,5-кратного ULN (или ≤5,0-кратного ULN при метастазах в печени или костях) h. Несоответствие критериям закона Хая (АЛТ >3-кратного ULN и билирубин >2-кратного ULN)
9. Желание и способность соблюдать визиты в клинику и предусмотренные исследованием процедуры
10. Предоставление подписанного информированного согласия
11. Способность понимать и заполнять связанные с исследованием опросники.
[00276] Критерии исключения: Пациента, соответствующего любым из следующих критериев, будут исключать из исследования: 1. Пациентов, которые никогда не курили, определяли как выкуривших ≤100 сигарет в течение жизни. 2. Активные или неподвергнутые лечению метастазы в головном мозге или компрессия спинного мозга. Пациенты пригодны, если метастазы в центральной нервной системе (ЦНС) подвергают достаточному лечению, и пациенты вернулись к неврологическому статусу исходного уровня (за исключением остаточных признаков или симптомов, связанных с лечением ЦНС) в течение по меньшей мере 2 недель перед включением в исследование. Пациенты должны прекратить терапию кортикостероидами (иммуносупрессорными дозами). 3. Пациенты с опухолями, положительными на мутации гена EGFR, транслокации гена ALK или слияния ROS1 по результатам тестирования. 4. Энцефалит, менингит или неконтролируемые судороги в течение года перед подписанием информированного согласия. 5. Интерстициальная болезнь легких в анамнезе (например, идиопатический легочный фиброз, организующаяся пневмония) или активный неинфекционный пневмонит, в случае которого необходимы иммуносупрессорные дозы глюкокортикоидов для облегчения лечения. Допустим радиационный пневмонит в облученном поле. 6. Пациенты с активным, известным или предполагаемым аутоиммунным заболеванием, для которого необходимо системное лечение, за последние 2 года. Допустимо включение пациентов с витилиго, сахарным диабетом I типа и гипотиреозом (включая гипотиреоз по причине аутоиммунного тиреоидита), которым необходима лишь гормонозаместительная терапия. 7. Пациенты с состоянием, при котором необходима терапия кортикостероидами (>10 мг преднизон/день или эквивалента) в пределах 14 дней перед рандомизацией. Допустимы физиологические заместительные дозы, даже если они составляют >10 мг преднизона/день или его эквивалента, при условии, что их не вводят в целях иммуносупрессии. Допустимы ингаляционные или местные стероиды, при условии, что они не предназначены для лечения аутоиммунного нарушения. 8. Другое злокачественное новообразование, прогрессирующее или требующее лечения, за исключением немеланомного рака кожи, подвергаемого потенциально излечивающей терапии, или карциномы шейки матки in situ или любой другой опухоли, подвергаемой лечению, и пациента считают находящимся в полной ремиссии в течение по меньшей мере 2 лет перед включением в исследование, и в течение исследования не требуется дополнительная терапия. 9. Известный активный гепатит B (положительный результат) или гепатит C (известный положительный результат) и известный количественный результат анализа на РНК HCV, превышающий нижний предел определения для анализа. 10. Вирус иммунодефицита человека (ВИЧ) в анамнезе или известный синдром приобретенного иммунодефицита с признаками неконтролируемой активной инфекции. Допустимо включение пациентов, подвергаемых высокоактивной антиретровирусной терапии, с неопределимыми уровнями РНК и количеством CD4 выше 350. 11. Активная инфекция, требующая системной терапии, в пределах 14 дней перед рандомизацией. 12. Предшествующая терапия антителами против PD-1 или против PD-L1. Допустимо предшествующее воздействие другой иммуномодулирующей терапии или вакцинотерапии, такой как антитела против антигена цитотоксических T-лимфоцитов 4 (CTLA-4), но последнюю дозу такого антитела необходимо вводить по меньшей мере за 3 месяца до введения первой дозы исследуемого лекарственного средства. 13. Связанные с лечением иммуноопосредованные нежелательные явления (AE) в ответ на иммуномодулирующие средства (включая, в качестве неограничивающих примеров, Mab против PD1/PD-L1, моноклональные антитела против CTLA4 и ингибиторы PI3K-δ), которые не разрешились к исходному уровню по меньшей мере за 3 месяца до начала лечения с использованием исследуемой терапии. Пациентов исключают из лечения REGN2810, если у них развиваются иммуноопосредованные AE, связанные с предшествующим лечением блокатором пути PD-1/PD-L1, имевшие степень 3 или 4 по своей тяжести и/или требовавшие прекращения введения средства, независимо от времени возникновения. 14. Предписание исследовательского лекарственного средства или устройства в пределах 30 дней перед скринингом или в пределах 5-кратного времени полужизни исследовательского лекарственного средства или терапии (в зависимости от того, что является более длительным). 15. Предписание живой вакцины в пределах 30 дней перед запланированным началом введения исследуемого лекарственного средства. 16. Обширная хирургическая операция или значительная травма в пределах 4 недель перед введением первой дозы 17. Задокументированная аллергическая реакция или острая реакция гиперчувствительности, приписываемая лечению антителами. 18. Известная аллергия на доксициклин или другие тетрациклиновые антибиотики. 19. Известное психиатрическое расстройство или наркотическая и алкогольная зависимость, которая будет мешать участию с соблюдением требований исследования, включая текущее использование каких-либо запрещенных лекарственных средств. 20. Беременные или кормящие грудью женщины. 21. Женщины детородного возраста, не желающие использовать высокоэффективную контрацепцию перед введением начальной дозы, в течение исследования и в течение по меньшей мере 6 месяцев после введения последней дозы. 22. Из исследования будут исключать пациентов, лишенных свободы на основании распоряжения судебных или административных органов.
Исследуемое лечение
[00277] Исследуемое лекарственное средство: REGN2810 вводят в дозе 350 мг в виде IV инфузии Q3W в течение до 108 недель.
[00278] Референсное лекарственное средство: Химиотерапию стандарта лечения (один из вариантов двухкомпонентной химиотерапии препаратами платины по протоколу, таблица 13) осуществляют в течение до 4-6 циклов или до определяемого с помощью RECIST 1.1 прогрессирования заболевания, неприемлемой токсичности, смерти или отзыва согласия на участие в исследовании.
Таблица 13: Руководство по схемам двухкомпонентной химиотерапии препаратами платины
Конечные точки исследования
[00279] Первичной конечной точкой является PFS, оцениваемая с использованием RECIST 1.1. Ключевыми вторичными конечными точками в исследовании являются OS и ORR. Другие вторичные конечные точки включают DOR и QOL, а также оценку безопасности и переносимости REGN2810.
Процедуры и оценка
[00280] Процедуры, осуществляемые при скрининге, включают получение информированного согласия; регистрацию медицинского и онкологического анамнеза и сопутствующих лекарственных средств; регистрацию демографических данных; получение опухолевой ткани для анализа PD-L1; тестирование опухолевой ткани на мутации рецептора эпидермального фактора роста (EGFR) и киназы анапластической лимфомы (ALK) и слияние человеческого гомолога трансформирующего гена v-ros вируса саркомы птиц UR2 (ROS1); рентгенографическую оценку опухоли; оценку опухолевой массы; рентгенографию грудной клетки; анализ сыворотки на беременность; электрокардиографию в 12 отведениях (ЭКГ); полный физикальный осмотр, включающий определение показателей жизнедеятельности, роста и массы тела; оценку статуса по шкале Восточной объединенной онкологической группы (ECOG) и лабораторные анализы. Также можно получать образцы для необязательного геномного подисследования. Во время лечения и последующего наблюдения осуществляют следующие процедуры для оценки безопасности: физикальный осмотр; оценку статуса по шкале ECOG; определение показателей жизнедеятельности; лабораторные анализы, включая тестирование женщин детородного возраста на беременность; ЭКГ и рентгенографию грудной клетки (по решению исследователя) и регистрацию нежелательных явлений (AE) и сопутствующих лекарственных средств. Компьютерную томографию (КТ) или магнитно-резонансную томографию (МРТ) для оценки опухоли осуществляют в различные моменты времени на всем протяжении исследования. Качество жизни измеряют с использованием валидированных опросников, самостоятельно заполняемых пациентами (EORTC QLQ-C30 и EORTC QLQ-LC13). Другие оценки включают получение образцов для анализа биомаркеров, образцов для измерения концентрации REGN2810 и образцов для анализа иммуногенности REGN2810.
Результаты
[00281] Ожидают, что лечение REGN2810 будет приводить к повышению выживаемости без прогрессирования и общей выживаемости по сравнению с лечением с использованием химиотерапии у пациентов с немелкоклеточным раком легких на поздней стадии, опухоли которых экспрессируют PD-L1 в ≥50% опухолевой ткани.
Пример 13: Комбинация стандартной и высокой дозы REGN2810 (цемиплимаба) и ипилимумаба (антитела против CTLA-4) при лечении второй линии у пациентов с метастазирующим немелкоклеточным раком легких с опухолями, экспрессирующими PD-L1 в <50%
[00282] В этом примере описывают клиническое исследование комбинаций стандартных и высоких доз REGN2810 (цемиплимаба, антитела против PD-1) и ипилимумаба (антитела против CTLA-4) при лечении второй линии у пациентов с метастазирующим немелкоклеточным раком легких с опухолями, экспрессирующими PD-L1 в <50%.
[00283] Основной целью исследования является сравнение частоты объективных ответов (ORR) для комбинированного лечения REGN2810 в высокой дозе ("HDREGN2810") и REGN2810 в стандартной дозе ("SDREGN2810/ipi") и ипилимумабом и для REGN2810 в стандартной дозе ("SDREGN2810") при лечении второй линии у пациентов с плоскоклеточным или неплоскоклеточным немелкоклеточным раком легких (NSCLC) на поздней стадии или метастазирующим плоскоклеточным или неплоскоклеточным немелкоклеточным раком легких (NSCLC) у пациентов, опухоли которых экспрессируют лиганд программируемой гибели клеток 1 (PD-L1) в <50% опухолевых клеток.
[00284] Вторичными целями исследования являются следующие: (1) Сравнение общей выживаемости (OS) для комбинированного лечения SDREGN2810, HDREGN2810 и SDREGN2810/ipi при лечении второй линии у пациентов с плоскоклеточным или неплоскоклеточным NSCLC на поздней стадии, опухоли которых экспрессируют PD-L1 в <50% опухолевых клеток. (2) Сравнение выживаемости без прогрессирования (PFS) HDREGN2810 и SDREGN2810/ipi с SDREGN2810 при лечении второй линии у пациентов с плоскоклеточным или неплоскоклеточным NSCLC на поздней стадии, опухоли которых экспрессируют PD-L1 в <50% опухолевых клеток. (3) Оценка безопасности и переносимости HDREGN2810 и SDREGN2810/ipi по сравнению с терапией SDREGN2810. (4) Оценка OS через 12 и 18 месяцев терапии HDREGN2810 и SDREGN2810/ipi по сравнению с терапией SDREGN2810 при лечении второй линии у пациентов с плоскоклеточным или неплоскоклеточным NSCLC на поздней стадии, опухоли которых экспрессируют PD-L1 в <50% опухолевых клеток. (5) Оценка качества жизни (QOL) у пациентов с плоскоклеточным или неплоскоклеточным NSCLC на поздней стадии, которым вводят HDREGN2810 и SDREGN2810/ipi, по сравнению с терапией SDREGN2810 (6) Оценка иммуногенности, измеряемой по антителам против лекарственного средства (ADA) против REGN2810. (7) Характеризация фармакокинетики (PK) REGN2810 при введении в комбинации с ипилимумабом или в виде HDREGN2810.
Дизайн исследования
[00285] Это исследование является клиническим исследованием HDREGN2810 и SDREGN2810/ipi по сравнению с терапией SDREGN2810 при лечении второй линии пациентов с плоскоклеточным или неплоскоклеточным NSCLC на поздней стадии или метастазирующим плоскоклеточным или неплоскоклеточным NSCLC с опухолями, экспрессирующими PD-L1 <50%. Исследование будет состоять из следующих 3 периодов: скрининга, лечения и последующего наблюдения. Пациентов будут подвергать скрининговой оценке для определения их пригодности для исследования в пределах 28 дней до рандомизации. Пригодных пациентов будут случайным образом распределять в соотношении 1:1:1 для использования одной из следующих схем лечения:
Группа лечения A: 350 мг REGN2810 каждые 3 недели (Q3W) в течение 108 недель (далее в настоящем описании обозначаемая как "SDREGN2810")
Группа лечения B: 350 мг REGN2810 Q3W в течение 108 недель и 50 мг ипилимумаба каждые 6 недель (Q6W) всего в количестве до 4 доз (далее в настоящем описании обозначаемая как "SDREGN2810/ipi")
Группа лечения C: 1050 мг REGN2810 Q3W в течение 108 недель (далее в настоящем описании обозначаемая как "HDREGN2810")
[00286] При рандомизации пациентов будут стратифицировать по гистологии (плоскоклеточный и неплоскоклеточный) и уровню экспрессии PD-L1 (<1% и 1%-<50%). Пациентов будут подвергать предписанному им лечению в течение периода лечения (как указано выше). Лечение можно прекращать досрочно по причине определяемого с помощью Критериев оценки ответа солидных опухолей версии 1.1 (RECIST 1.1) прогрессирования заболевания, неприемлемой токсичности, отзыва согласия на участие в исследовании, смерти, начала другой противоопухолевой терапии или в конкретных случаях подтвержденного полного ответа (CR), частичного ответа(PR) или стабильного заболевания (SD). Пациенты, у которых наблюдают определяемое с помощью RECIST 1.1 прогрессирующее заболевание, во время терапии могут продолжать исследуемое лечение, если исследователь считает, что пациент получит клиническую пользу, и если пациент не завершил 108-недельный период лечения. Если подтверждают дальнейшее прогрессирование заболевания (определяемое как дополнительное 10%-ное повышение опухолевой массы с момента исходного прогрессирования заболевания), необходимо прекратить введение REGN2810 (и ипилимумаба, при необходимости) и рассмотреть другую противоопухолевую терапию, при необходимости. После прекращения исследуемого лечения пациентов будут подвергать последующему наблюдению. Каждый пациент будет осуществлять первый визит для последующего наблюдения в течение от 14 до 30 дней (±7 дней) после введения последней дозы исследуемого средства, если лечение прекращают досрочно по причине прогрессирования заболевания, токсичности или по другой причине. В ином случае, каждый пациент будет осуществлять первый визит для последующего наблюдения в течение от 14 до 30 дней (±7 дней) после последнего визита цикла. Визиты для последующего наблюдения 2-7 будут осуществлять в пределах 28 дней (±7 дней) после предыдущего визита. Затем будут собирать данные о выживаемости посредством телефонного звонка или в кабинете врача во время визита каждые 3 месяца до смерти, неявки для последующего наблюдения или отзыва согласия на участие в исследовании.
Исследуемая выборка
[00287] Пациенты в этом исследовании будут включать мужчин и женщин возрастом ≥18 лет, у которых диагностирован неплоскоклеточный или плоскоклеточный NSCLC на поздней стадии или метастазирующий неплоскоклеточный или плоскоклеточный NSCLC, которых подвергали только 1 предшествующей линии лечения NSCLC на поздней стадии или метастазирующего NSCLC, и опухоли которых экспрессируют PD-L1 в <50%.
[00288] Критерии включения: 1. Мужчины и женщины возрастом ≥18 лет; 2. Пациенты с гистологически или цитологически задокументированным плоскоклеточным или неплоскоклеточным NSCLC стадии IIIb, не являющиеся кандидатами для лечения с использованием радикальной комбинированной химиолучевой терапии, или пациенты с заболеванием стадии IV, если их ранее подвергали системному лечению NSCLC на поздней стадии или метастазирующего NSCLC, и которых подвергали 1 предшествующей линии лечения NSCLC на поздней стадии; 3. Доступность архивного или полученного во время исследования фиксированного формалином, погруженного в парафин биоптата опухолевой ткани. Рекомендации по биопсии: a. Допустимы архивные или свежие биоптаты; b. Если используют архивный биоптат, он должен быть получен менее 5 месяцев назад; c. Биоптат необходимо получать из очага метастазирования или рецидивирования, который ранее не облучали. Исключение: первичная опухоль все еще находится на месте, а другие очаги метастазирования недоступны (головной мозг), или их нельзя использовать (кость), или биопсия будет нести риск для пациента. 4. Экспрессия PD-L1 в <50% опухолевых клеток, определяемая посредством анализа IHC PD-L1 22C3 pharmDx, осуществляемого в центральной лаборатории; 5. По меньшей мере 1 рентгенографически измеримый очаг по результатам компьютерной томографии (КТ) по критериям RECIST 1.1. Целевые очаги могут находиться в ранее облученном поле, если в этом очаге есть задокументированное (рентгенографически) прогрессирование заболевания; 6. Статус по шкале ECOG ≤1; 7. Ожидаемая продолжительность жизни по меньшей мере 3 месяца.
[00289] Критерии исключения: 1. Пациентов, которые никогда не курили, определяют как выкуривших ≤100 сигарет в течение жизни; 2. Активные или неподвергнутые лечению метастазы в головном мозге или компрессия спинного мозга. Пациенты пригодны, если метастазы в центральной нервной системе (ЦНС) подвергают достаточному лечению, и пациенты вернулись к неврологическому статусу исходного уровня (за исключением остаточных признаков или симптомов, связанных с лечением ЦНС) в течение по меньшей мере 2 недель перед включением в исследование. Пациенты должны прекратить терапию кортикостероидами (иммуносупрессорными дозами) (см. подробности временного режима прекращения введения стероидов в критерии исключения 7). 3. Пациенты с опухолями, положительными на мутации гена EGFR, транслокации гена ALK или слияния ROS1 по результатам тестирования. Всех пациентов будут подвергать анализу опухолей на мутации EGFR, перестановку в ALK и слияние ROS1. 4. Энцефалит, менингит или неконтролируемые судороги в течение года перед подписанием информированного согласия. 5. Интерстициальная болезнь легких в анамнезе (например, идиопатический легочный фиброз, организующаяся пневмония) или активный неинфекционный пневмонит, в случае которого необходимы иммуносупрессорные дозы глюкокортикоидов для облегчения лечения, или пневмонит за последние 5 лет. Допустим радиационный пневмонит в облученном поле в анамнезе при условии, что пневмонит разрешился за ≥6 месяцев перед включением в исследование. 6. Текущие или недавние признаки значительного аутоиммунного заболевания, для которого необходимо системное иммуносупрессорное лечение, в случае которого можно предположить риск иммуноопосредованных связанных с лечением нежелательных явлений (irTEAE). Следующие заболевания не являются исключающими: витилиго, разрешенная астма детского возраста, остаточный гипотиреоз, в случае которого необходима лишь гормонозаместительная терапия, или псориаз, в случае которого не требуется системное лечение. 7. Пациенты с состоянием, при котором необходима терапия кортикостероидами (>10 мг преднизона/день или его эквивалента) в пределах 14 дней перед рандомизацией. Допустимы физиологические заместительные дозы, даже если они составляют >10 мг преднизона/день или его эквивалента, при условии, что их не вводят в целях иммуносупрессии. Допустимы ингаляционные или местные стероиды, при условии, что они не предназначены для лечения аутоиммунного нарушения.
Исследуемое лечение
REGN2810, вводимое в дозе 350 мг в виде внутривенной (IV) инфузии Q3W в течение 108 недель ("SDREGN2810")
REGN2810, вводимое в дозе 350 мг в виде IV инфузии Q3W в течение 108 недель, в комбинации с ипилимумабом, вводимым IV в дозе 50 мг Q6W в количестве до 4 доз ("SDREGN2810/ipi")
REGN2810, вводимое в дозе 1050 мг в виде IV инфузии Q3W в течение 108 недель ("HDREGN2810")
Конечные точки исследования
[00290] Первичной конечной точкой является ORR, определяемая как доля пациентов, достигнувших CR или PR с учетом RECIST 1.1 по оценке маскированного независимого наблюдательного комитета (IRC).
[00291] Вторичными конечными точками в исследовании являются: (1) Общая выживаемость (OS), определяемая как время от рандомизации до даты смерти. Неумершего пациента будут оценивать по последней известной дате, когда этот пациент был жив. (2) Выживаемость без прогрессирования (PFS) определяют как время от рандомизации до даты первого задокументированного прогрессирования опухоли, определяемого с помощью RECIST 1.1 по оценке маскированного IRC, или до даты смерти по любой причине. (3) Общая выживаемость через 12 месяцев и 18 месяцев и к концу лечения. (4) Безопасность и переносимость комбинированного лечения SDREGN2810, HDREGN2810 и SDREGN2810/ipi, измеряемые по частоте возникших во время лечения нежелательных явлений, дозолимитирующей токсичности, серьезным нежелательным явлениям, смерти и отклонениям лабораторных показателей. (5) Качество жизни, измеряемое с помощью Опросника качества жизни Core 30 Европейской организации по изучению и лечению рака (EORTC QLQ-C30) и Опросника качества жизни при раке легких 13 (EORTC QLQ-LC13). (6) Характеризация PK REGN2810 при введении в комбинации с ипилимумабом или HDREGN2810. (7) Анализ иммуногенности, измеряемые по титрам ADA против REGN2810. (8) Оценка пигментации волос исследователем. (9) Мутационная нагрузка опухоли, оцениваемая с помощью панели Foundation Medicine "FoundationOne®". (10) Оценка объема опухоли. (11) Доля ICOS+ CD4 T-клеток и другие маркеры активации T-клеток.
Процедуры и оценка
[00292] Процедуры, которые следует осуществлять при скрининге, будут включать получение информированного согласия; оценку критериев включения/исключения; регистрацию медицинского и онкологического анамнеза и сопутствующих лекарственных средств; регистрацию демографических данных; получение и тестирование образцов опухолевой ткани для анализа PD-L1 и мутаций рецептора эпидермального фактора роста (EGFR) и киназы анапластической лимфомы (ALK) и слияния онкогена рецепторной тирозинкиназы C-ros (ROS1); рентгенографическую оценку опухоли; оценку опухолевой массы; рентгенографию грудной клетки; анализ сыворотки на беременность; электрокардиографию в 12 отведениях; регистрацию нежелательных явлений (AE); физикальный осмотр, включающий определение показателей жизнедеятельности, роста и массы тела; оценку статуса по шкале Восточной объединенной онкологической группы (ECOG) и лабораторные анализы. Также можно получать образцы для необязательного геномного подисследования. В период лечения будут осуществлять следующие процедуры для оценки эффективности и безопасности: определение QOL с использованием валидированных опросников, заполняемых пациентом; физикальный осмотр; оценку статуса по шкале ECOG; определение показателей жизнедеятельности; лабораторные анализы, включая тестирование женщин детородного возраста на беременность; регистрацию нежелательных явлений (AE) и сопутствующих лекарственных средств. Компьютерную томографию для рентгенографической оценки опухолевой массы и оценку опухолевой массы с помощью критериев RECIST 1.1 будут осуществлять в определенные моменты времени на всем протяжении исследования. Другие оценки будут включать оценку исследователем репигментации волос, измерение концентрации REGN2810, анализ ADA против REGN2810 и анализ биомаркеров. Анализ биомаркеров будет включать использование образцов опухолевой ткани для валидации дополнительных анализов PD-L1. Затем будут собирать данные о выживаемости посредством телефонного звонка или в кабинете врача во время визита каждые 3 месяца до смерти, неявки для последующего наблюдения или отзыва согласия на участие в исследовании.
Результаты
[00293] Ожидают, что SDREGN2810/ipi или HDREGN2810 будут приводить к более высокой частоте ответа, чем SDREGN2810, у пациентов, опухоли которых экспрессируют PD-L1 в <50% опухолевых клеток. Учитывая общую частоту ответа 10% у пациентов с экспрессией PD-L1 в 1-<50%, которых лечили SDREGN2810, ожидают, что с помощью HDREGN2810 или SDREGN2810/ipi можно достигать ORR 30%, абсолютного повышения на 20% по сравнению с SDREGN2810.
Пример 14: Клиническое исследование комбинаций REGN2810 (антитела против PD-1), ипилимумаба (антитела против CTLA4) и двухкомпонентной химиотерапии препаратами платины при лечении первой линии у пациентов с немелкоклеточным раком легких на поздней стадии или метастазирующим немелкоклеточным раком легких, опухоли которых экспрессируют PD-L1 в <50%
[00294] В этом примере описывают клиническое исследование комбинаций REGN2810 (антитела против PD-1), ипилимумаба (антитела против CTLA-4) и двухкомпонентной химиотерапии препаратами платины при лечении первой линии у пациентов с немелкоклеточным раком легких на поздней стадии или метастазирующим немелкоклеточным раком легких с опухолями, экспрессирующими PD-L1 в <50%, которых ранее не подвергали системному лечению их заболевания на поздней стадии.
[00295] Первичной целью исследования является сравнение выживаемости без прогрессирования (PFS) для комбинированного лечения REGN2810 и 4-6 циклами стандарта лечения, двухкомпонентной химиотерапии препаратами платины, (REGN2810/chemo-f) и комбинированного лечения REGN2810 и лишь 2 циклами стандарта лечения, двухкомпонентной химиотерапии препаратами платины, и ипилимумабом (REGN2810/chemo-l/ipi) со стандартом лечения, двухкомпонентной химиотерапией препаратами платины, при лечении первой линии у пациентов с плоскоклеточным или неплоскоклеточным немелкоклеточным раком легких (NSCLC) на поздней стадии в подгруппе пациентов, опухоли которых экспрессируют лиганд программируемой гибели клеток 1 (PD-L1) в 1-<50% опухолевых клеток, и в общей выборке исследуемых пациентов, опухоли которых экспрессируют PD-L1 в <50% опухолевых клеток.
[00296] Вторичные цели включают: (1) Сравнение общей выживаемости (OS) для REGN2810/chemo-f и REGN2810/chemo-l/ipi со стандартом лечения, двухкомпонентной химиотерапией препаратами платины, при лечении первой линии у пациентов с плоскоклеточным или неплоскоклеточным NSCLC на поздней стадии в подгруппе пациентов, опухоли которых экспрессируют PD-L1 в 1-<50% опухолевых клеток, и в общей популяции исследуемых пациентов, опухоли которых экспрессируют PD-L1 в <50% опухолевых клеток. (2) Сравнение частоты объективных ответов (ORR) для REGN2810/chemo-f и REGN2810/chemo-l/ipi по сравнению со стандартом лечения, двухкомпонентной химиотерапией препаратами платины, при лечении первой линии у пациентов с плоскоклеточным или неплоскоклеточным NSCLC на поздней стадии в подгруппе пациентов, опухоли которых экспрессируют PD-L1 в 1-<50% опухолевых клеток, и в общей выборке исследуемых пациентов, опухоли которых экспрессируют PD-L1 в <50% опухолевых клеток. (3) Оценка безопасности и переносимости REGN2810 и 4-6 циклов двухкомпонентной химиотерапии препаратами платины и REGN2810 и ипилимумаба. (4) Характеризация фармакокинетики REGN2810 и 4-6 циклов двухкомпонентной химиотерапии препаратами платины и REGN2810 и ипилимумаба. (5) Сравнение OS через 12 и 18 месяцев для REGN2810/chemo-f или REGN2810/chemo-l/ipi по сравнению со стандартом лечения, двухкомпонентной химиотерапией препаратами платины, при лечении первой линии у пациентов с плоскоклеточным или неплоскоклеточным NSCLC на поздней стадии в подгруппе пациентов, опухоли которых экспрессируют PD-L1 в 1-<50% опухолевых клеток, и в общей выборке исследуемых пациентов, опухоли которых экспрессируют PD-L1 в <50% опухолевых клеток. (6) Анализ иммуногенности посредством измерения антител против лекарственного средства против REGN2810.
Исследуемая выборка
[00297] Целевая выборка включает мужчин и женщин возрастом ≥18 лет с <50% PD-L1+ опухолевых клеток, плоскоклеточным или неплоскоклеточным NSCLC стадии IIIB или стадии IV, ранее не подвергаемых лечению их заболевания на поздней стадии.
[00298] Критерии включения:
1) Мужчины и женщины возрастом ≥18 лет
2) Пациенты с гистологически или цитологически задокументированным плоскоклеточным или неплоскоклеточным NSCLC стадии IIIB или стадии IV, которых ранее не подвергали системному лечению рецидивирующего или метастазирующего NSCLC
3) Пригодными являются пациенты, которых подвергали адъювантной или неоадъювантной двухкомпонентной химиотерапии препаратами платины (после хирургической операции и/или лучевой терапии), и у которых развивалось рецидивирующее или метастазирующее заболевание через более чем 6 месяцев после завершения терапии
4) Пригодными являются пациенты, которых подвергали адъювантному или неоадъювантному блокированию PD-1 или PD-L1, и у которых развивалось рецидивирующее или метастазирующее заболевание через более чем 12 месяцев после завершения терапии
5) Архивная или недавно полученная фиксированная формалином опухолевая ткань из очага метастазирования/рецидивирования, которую ранее не облучали
6) Опухолевые клетки, экспрессирующие PD-L1 в <50% опухолевых клеток по результатам IHC, осуществляемой центральной лабораторией
7) По меньшей мере 1 рентгенографически измеримый очаг по результатам компьютерной томографии (КТ) или магнитно-резонансной томографии (МРТ) в соответствии с критериями RECIST 1.1. Целевые очаги могут локализоваться в ранее облученном поле, если в этом очаге задокументировано (рентгенографически) прогрессирование заболевания.
8) Статус по шкале ECOG ≤1
9) Ожидаемая продолжительность жизни по меньшей мере 3 месяца
10) Достаточная функция органов и костного мозга, как определено ниже:
11) Гемоглобин ≥10,0 г/дл
12) Абсолютное количество нейтрофилов ≥1,5×109/л
13) Количество тромбоцитов ≥100000/мм3
14) Уровень клубочковой фильтрации (GFR) >30 мл/мин/1,73м2
15) Общий билирубин ≤1,5-кратного верхнего предела нормы (ULN) (при метастазах в печени ≤3-кратного ULN), за исключением пациентов, у которых диагностирован клинически подтвержденный синдром Жильбера
16) Аспартатаминотрансфераза (АСТ) и аланинаминотрансфераза (АЛТ) ≤3-кратного ULN или ≤5-кратного ULN при метастазах в печени
17) Щелочная фосфатаза ≤2,5-кратного ULN (или ≤5,0-кратного ULN при метастазах в печени или костях)
18) Несоответствие критериям закона Хая (АЛТ >3-кратного ULN и билирубин >2-кратного ULN)
19) Желание и способность соблюдать визиты в клинику и предусмотренные исследованием процедуры
20) Предоставление подписанного информированного согласия
21) Способность понимать и заполнять связанные с исследованием опросники
[00299] Критерии исключения: 1. Пациентов, которые никогда не курили, определяли как выкуривших ≤100 сигарет в течение жизни; 2. Активные или неподвергнутые лечению метастазы в головном мозге или компрессия спинного мозга. Пациенты пригодны, если метастазы в центральной нервной системе (ЦНС) подвергают достаточному лечению, и пациенты вернулись к неврологическому статусу исходного уровня (за исключением остаточных признаков или симптомов, связанных с лечением ЦНС) в течение по меньшей мере 2 недель перед включением в исследование. Пациенты должны прекратить терапию кортикостероидами (иммуносупрессорными дозами) (см. подробности временного режима прекращения введения стероидов в критерии исключения 7). 3. Пациенты с опухолями, положительными на мутации гена EGFR, транслокации гена ALK или слияния ROS1 по результатам тестирования. Всех пациентов будут подвергать анализу опухолей на мутации EGFR, перестановку в ALK и слияние ROS1. 4. Энцефалит, менингит или неконтролируемые судороги в течение года перед подписанием информированного согласия. 5. Интерстициальная болезнь легких в анамнезе (например, идиопатический легочный фиброз, организующаяся пневмония) или активный неинфекционный пневмонит, в случае которого необходимы иммуносупрессорные дозы глюкокортикоидов для облегчения лечения, или пневмонит за последние 5 лет. Допустим радиационный пневмонит в облученном поле в анамнезе при условии, что пневмонит разрешился за ≥6 месяцев перед включением в исследование. 6. Текущие или недавние признаки значительного аутоиммунного заболевания, для которого необходимо системное иммуносупрессорное лечение, в случае которого можно предположить риск иммуноопосредованных связанных с лечением нежелательных явлений (irTEAE). Следующие заболевания не являются исключающими: витилиго, разрешенная астма детского возраста, остаточный гипотиреоз, в случае которого необходима лишь гормонозаместительная терапия, или псориаз, в случае которого не требуется системное лечение. 7. Пациенты с состоянием, при котором необходима терапия кортикостероидами (>10 мг преднизона/день или его эквивалента) в пределах 14 дней перед рандомизацией. Допустимы физиологические заместительные дозы, даже если они составляют >10 мг преднизона/день или его эквивалента, при условии, что их не вводят в целях иммуносупрессии. Допустимы ингаляционные или местные стероиды, при условии, что они не предназначены для лечения аутоиммунного нарушения.
Дизайн исследования
[00300] Настоящее клиническое испытание является исследованием REGN2810/chemo-f по сравнению с REGN2810/chemo-l/ipi и стандартом лечения, двухкомпонентной химиотерапией препаратами платины, при лечении первой линии у пациентов с плоскоклеточным или неплоскоклеточным NSCLC стадии IIIB или стадии IV, опухоли которых экспрессируют PD-L1 в <50% опухолевых клеток, и которых ранее не подвергали системному лечению их заболевания на поздней стадии. Для анализа PD-L1 с использованием валидированного анализа PD-L1 будут получать опухолевую ткань (блок опухолевой ткани или по меньшей мере 12 неокрашенных срезов).
[00301] Пациентов с NSCLC на поздней стадии, ранее не подвергавшихся лечению, случайным образом распределяют в соотношении 1:1:1 в одну из следующих групп лечения:
[00302] Группа лечения A: Стандарт лечения, двухкомпонентная химиотерапия препаратами платины, каждые 3 недели (Q3W) в течение 4-6 циклов (с последующим необязательным поддерживающим лечением пеметрекседом в случае пациентов, которым исходно предписана включающая пеметрексед схема лечения)
[00303] Группа лечения B: 350 мг REGN2810 Q3W в течение 108 недель со стандартом лечения, двухкомпонентной химиотерапией препаратами платины, в течение 4-6 циклов (далее в настоящем описании обозначаемая как "REGN2810/chemo-f")
[00304] Группа лечения C: 350 мг REGN2810 Q3W в течение 108 недель со стандартом лечения, двухкомпонентной химиотерапией препаратами платины, в течение 2 циклов и 50 мг ипилимумаба каждые 6 недель (Q6W) в количестве до 4 доз (далее в настоящем описании обозначаемая как "REGN2810/chemo-l/ipi")
[00305] При рандомизации осуществляют стратификацию по гистологии (неплоскоклеточный и плоскоклеточный) и уровням экспрессии PD-L1 (<1% по сравнению с 1-24% и 25-<50%).
[00306] Пациентов будут подвергать предписанному лечению в течение периода лечения (как указано выше). Лечение можно прекращать досрочно по причине определяемого с помощью Критериев оценки ответа солидных опухолей версии 1.1 (RECIST 1.1) прогрессирования заболевания, неприемлемой токсичности, отзыва согласия на участие в исследовании, смерти, начала другого противоопухолевого лечения или, в случае пациентов в группах лечения B и C, в некоторых случаях подтвержденного полного ответа (CR) или частичного ответа (PR).
Исследуемое лечение
[00307] Группа лечения (A): Стандарт лечения, двухкомпонентная химиотерапия препаратами платины, осуществляемая IV Q3W в течение 4-6 циклов (с последующим необязательным поддерживающим лечением пеметрекседом в случае пациентов, которым исходно предписана включающая пеметрексед схема лечения)
[00308] Группа лечения (B): REGN2810, вводимое в дозе 350 мг в виде внутривенной (IV) инфузии Q3W в течение 108 недель, в комбинации со стандартом лечения, двухкомпонентной химиотерапией препаратами платины, осуществляемой Q3W IV в течение 4-6 циклов
[00309] Группа лечения (C): REGN2810, вводимое в дозе 350 мг в виде IV инфузии Q3W в течение 108 недель, в комбинации со стандартом лечения, двухкомпонентной химиотерапией препаратами платины, осуществляемой Q3W IV в течение 2 циклов, и ипилимумабом, вводимым IV в течение приблизительно 90 минут в дозе 50 мг Q6W в количестве до 4 дозы
[00310] 4-6 циклов стандарта лечения, двухкомпонентной химиотерапии препаратами платины, осуществляют в соответствии с одной из следующих схем лечения:
[00311] (i) Паклитаксел+Цисплатин: Участникам будут вводить 200 мг/м2 паклитаксела, вводимые IV, затем 75 мг/м2 цисплатина, вводимые IV в день 1 каждый 21 день в течение 4-6 циклов или до задокументированного прогрессирования заболевания. (ii) Паклитаксел+Карбоплатин: Участникам будут вводить 200 мг/м2 паклитаксела, вводимые IV, затем карбоплатин AUC в дозе 5 или 6 мг/мл/мин, вводимой IV в день 1 каждый 21 день в течение 4-6 циклов или до задокументированного прогрессирования заболевания. (iii) Гемцитабин+Цисплатин: Участникам будут вводить 1250 мг/м2 гемцитабина, вводимые IV в дни 1 и 8 каждого 21-дневного цикла, и 75 мг/м2 цисплатина, вводимые IV в день 1 каждый 21 день в течение 4-6 циклов или до прогрессирования заболевания. (iv) Гемцитабин+Карбоплатин: Участникам будут вводить 1250 мг/м2 гемцитабина, вводимые IV в дни 1 и 8 каждого 21-дневного цикла, и карбоплатин AUC в дозе 5 или 6 мг/мл/мин, вводимой IV в день 1 каждый 21 день в течение 4-6 циклов или до прогрессирования заболевания. (v) Пеметрексед+Цисплатин (только в случае неплоскоклеточного рака по результатам гистологического анализа): Участникам будут вводить 500 мг/м2 пеметрекседа IV, затем 75 мг/м2 цисплатина, вводимые IV в день 1 каждый 21 день в течение 4-6 циклов, затем необязательную поддерживающую дозу пеметрекседа 500 мг/м2 в течение остального времени исследования или до задокументированного прогрессирования заболевания. (vi) Пеметрексед+Карбоплатин (только в случае неплоскоклеточного рака по результатам гистологического анализа): Участникам будут вводить 500 мг/м2 пеметрекседа, вводимые IV, затем карбоплатин AUC в дозе 5 или 6 мг/мл/мин, вводимой IV в день 1 каждый 21 день в течение 4-6 циклов, затем необязательную поддерживающую дозу пеметрекседа 500 мг/м2 в течение остального времени исследования или до задокументированного прогрессирования заболевания.
Процедуры и оценки
[00312] Процедуры, которые следует осуществлять при скрининге, будут включать получение информированного согласия; оценку критериев включения/исключения; регистрацию медицинского и онкологического анамнеза и сопутствующих лекарственных средств; регистрацию демографических данных; получение и тестирование образцов опухолевой ткани для анализа PD-L1 и мутаций рецептора эпидермального фактора роста и киназы анапластической лимфомы и слияния онкогена рецепторной тирозинкиназы C-ros; рентгенографическую оценку опухоли; оценку опухолевой массы; рентгенографию грудной клетки; анализ сыворотки на беременность; электрокардиографию в 12 отведениях; регистрацию нежелательных явлений (AE); физикальный осмотр, включающий определение показателей жизнедеятельности, роста и массы тела; оценку статуса по шкале Восточной объединенной онкологической группы (ECOG) и лабораторные анализы. Также можно получать образцы для необязательного геномного подисследования.
[00313] В течение период лечения осуществляют следующие процедуры для оценки эффективности и безопасности: определение QOL с использованием валидированных опросников, заполняемых пациентом, физикальный осмотр, оценку статуса по шкале ECOG; определение показателей жизнедеятельности; лабораторные анализы, включая тестирование женщин детородного возраста на беременность; регистрацию нежелательных явлений (AE) и сопутствующих лекарственных средств. Компьютерную томографию или магнитно-резонансную томографию (или позитронно-эмиссионную томографию) для рентгенографической оценки опухолевой массы и оценку опухолевой массы с помощью критериев RECIST 1.1 осуществляют в определенные моменты времени на всем протяжении исследования.
[00314] Данные о выживаемости будут собирать посредством телефонного звонка или в кабинете врача во время визита каждые 3 месяца до смерти, неявки для последующего наблюдения или отзыва согласия на участие в исследовании.
Результаты
[00315] Ожидают, что схема комбинированного лечения с использованием R2810 будет повышать медиану PFS на 4 месяца (повышение с 6 месяцев для стандарта лечения, двухкомпонентной химиотерапии препаратами платины до 10 месяцев для любой из схем комбинированного лечения с использованием R2810) у пациентов с опухолями, экспрессирующими PD-L1 в 1-<50%, и в общей исследуемой выборке. REGN2810 с 4-6 циклами двухкомпонентной химиотерапии препаратами платины, или REGN2810, или REGN2810 с ипилимумабом будут пролонгировать OS и улучшать ORR по сравнению с двухкомпонентной химиотерапией препаратами платины.
Пример 15: Клиническое исследование комбинаций REGN2810 (антитела против PD-1), двухкомпонентной химиотерапии препаратами платины и ипилимумаба (антитела против CTLA-4) по сравнению с монотерапией пембролизумабом при лечении первой линии у пациентов с немелкоклеточным раком легких на поздней стадии или метастазирующим немелкоклеточным раком легких, опухоли которых экспрессируют PD-L1 в ≥50%
[00316] В примере описывают клиническое исследование комбинаций монотерапии REGN2810, двухкомпонентной химиотерапии препаратами платины и ипилимумаба (вводимого в количестве до 4 доз) по сравнению с монотерапией пембролизумабом у пациентов с плоскоклеточным или неплоскоклеточным NSCLC на поздней стадии или метастазирующим плоскоклеточным или неплоскоклеточным NSCLC, опухоли которых экспрессируют PD-L1 в ≥50% опухолевых клеток, и которых ранее не подвергали системному лечению их заболевания на поздней стадии.
[00317] Первичной целью исследования является сравнение выживаемости без прогрессирования (PFS) при комбинированном лечении REGN2810 (цемиплимабом) и ипилимумабом (далее в настоящем описании обозначаемом как REGN2810/ipi) и комбинированном лечении REGN2810 только с 2 циклами двухкомпонентной химиотерапии препаратами платины и ипилимумабом (далее в настоящем описании обозначаемом как "REGN2810/chemo/ipi") со стандартом лечения, монотерапией пембролизумабом, при лечении первой линии у пациентов с плоскоклеточным или неплоскоклеточным немелкоклеточным раком легких (NSCLC) на поздней стадии, опухоли которых экспрессируют лиганд программируемой гибели клеток 1 (PD-L1) в ≥50% опухолевых клеток. Дополнительные цели включают дальнейшую характеризацию общей выживаемости, ответов опухолей, сообщаемых пациентом исходов, безопасности и фармакокинетики (PK).
Исследуемая выборка
[00318] Целевая выборка включает мужчин и женщин возрастом ≥18 лет с ≥50% PD-L1+ опухолевых клеток, плоскоклеточным или неплоскоклеточным NSCLC стадии IIIB или стадии IV, которых ранее не подвергали лечению их заболевания на поздней стадии.
[00319] Критерии включения:
1) Мужчины и женщины возрастом ≥18 лет
2) Пациенты с гистологически или цитологически задокументированным плоскоклеточным или неплоскоклеточным NSCLC стадии IIIB или стадии IV, которых ранее не подвергали системному лечению рецидивирующего или метастазирующего NSCLC
3) Пригодными являются пациенты, которых подвергали адъювантной или неоадъювантной двухкомпонентной химиотерапии препаратами платины (после хирургической операции и/или лучевой терапии), и у которых развивалось рецидивирующее или метастазирующее заболевание через более чем 6 месяцев после завершения терапии
4) Пригодными являются пациенты, которых подвергали адъювантному или неоадъювантному блокированию PD-1 или PD-L1, и у которых развивалось рецидивирующее или метастазирующее заболевание через более чем 12 месяца после завершения терапии
5) Архивная или недавно полученная фиксированная формалином опухолевая ткань из очага метастазирования/рецидивирования, которую ранее не облучали
6) Опухолевые клетки, экспрессирующие PD-L1 в <50% опухолевых клеток по результатам IHC, осуществляемой центральной лабораторией
7) По меньшей мере 1 рентгенографически измеримый очаг по результатам компьютерной томографии (КТ) или магнитно-резонансной томографии (МРТ) в соответствии с критериями RECIST 1.1. Целевые очаги могут локализоваться в ранее облученном поле, если в этом очаге задокументировано (рентгенографически) прогрессирование заболевания.
8) Статус по шкале ECOG ≤1
9) Ожидаемая продолжительность жизни по меньшей мере 3 месяца
10) Достаточная функция органов и костного мозга, как определено ниже:
11) Гемоглобин ≥8,0 г/дл
12) Абсолютное количество нейтрофилов ≥1,0×109/л
13) Количество тромбоцитов ≥75000/мм3
14) Уровень клубочковой фильтрации (GFR) >30 мл/мин/1,73м2
15) Общий билирубин ≤1,5-кратного верхнего предела нормы (ULN) (при метастазах в печени ≤3-кратного ULN), за исключением пациентов, у которых диагностирован клинически подтвержденный синдром Жильбера
16) Аспартатаминотрансфераза (АСТ) и аланинаминотрансфераза (АЛТ) ≤3-кратного ULN или ≤5-кратного ULN при метастазах в печени
17) Щелочная фосфатаза ≤2,5-кратного ULN (или ≤5,0-кратного ULN при метастазах в печени или костях)
18) Несоответствие критериям закона Хая (АЛТ >3-кратного ULN и билирубин >2-кратного ULN)
19) Желание и способность соблюдать визиты в клинику и предусмотренные исследованием процедуры
20) Предоставление подписанного информированного согласия
21) Способность понимать и заполнять связанные с исследованием опросники
[00320] Критерии исключения: 1. Пациентов, которые никогда не курили, определяли как выкуривших ≤100 сигарет в течение жизни; 2. Активные или неподвергнутые лечению метастазы в головном мозге или компрессия спинного мозга. Пациенты пригодны, если метастазы в центральной нервной системе (ЦНС) подвергают достаточному лечению, и пациенты вернулись к неврологическому статусу исходного уровня (за исключением остаточных признаков или симптомов, связанных с лечением ЦНС) в течение по меньшей мере 2 недель перед включением в исследование. Пациенты должны прекратить терапию кортикостероидами (иммуносупрессорными дозами) (см. подробности временного режима прекращения введения стероидов в критерии исключения 7). 3. Пациенты с опухолями, положительными на мутации гена EGFR, транслокации гена ALK или слияния ROS1 по результатам тестирования. Всех пациентов будут подвергать анализу опухолей на мутации EGFR, перестановку в ALK и слияние ROS1. 4. Энцефалит, менингит или неконтролируемые судороги в течение года перед подписанием информированного согласия. 5. Интерстициальная болезнь легких в анамнезе (например, идиопатический легочный фиброз, организующаяся пневмония) или активный неинфекционный пневмонит, в случае которого необходимы иммуносупрессорные дозы глюкокортикоидов для облегчения лечения, или пневмонит за последние 5 лет. Допустим радиационный пневмонит в облученном поле в анамнезе при условии, что пневмонит разрешился за ≥6 месяцев перед включением в исследование. 6. Текущие или недавние признаки значительного аутоиммунного заболевания, для которого необходимо системное иммуносупрессорное лечение, в случае которого можно предположить риск иммуноопосредованных связанных с лечением нежелательных явлений (irTEAE). Следующие заболевания не являются исключающими: витилиго, разрешенная астма детского возраста, остаточный гипотиреоз, в случае которого необходима лишь гормонозаместительная терапия, или псориаз, в случае которого не требуется системное лечение. 7. Пациенты с состоянием, при котором необходима терапия кортикостероидами (>10 мг преднизона/день или его эквивалента) в пределах 14 дней перед рандомизацией. Допустимы физиологические заместительные дозы, даже если они составляют >10 мг преднизона/день или его эквивалента, при условии, что их не вводят в целях иммуносупрессии. Допустимы ингаляционные или местные стероиды, при условии, что они не предназначены для лечения аутоиммунного нарушения.
Дизайн исследования
[00321] Настоящее клиническое испытание является исследованием эффективности и безопасности REGN2810/ipi по сравнению с REGN2810/chemo/ipi и монотерапией пембролизумабом у пациентов с плоскоклеточным или неплоскоклеточным NSCLC стадии IIIB или стадии IV, опухоли которых экспрессируют PD-L1 в ≥50% опухолевых клеток, и которых ранее не подвергали системному лечению их заболевания на поздней стадии.
[00322] Исследование состоит из следующих 3 периодов: скрининга, лечения и последующего наблюдения. Пациентов подвергают скрининговой оценке для определения их пригодности в пределах 28 дней перед рандомизацией. Пригодных пациентов случайным образом распределяют в соотношении 1:1:1 в одну из следующих групп лечения:
Группа лечения A: монотерапия пембролизумабом в дозе 200 мг каждые 3 недели (Q3W) в течение 108 недель
Группа лечения B: 350 мг REGN2810 Q3W в течение 108 недель и 50 мг ипилимумаба каждые 6 недель (Q6W) в количестве до 4 доз
Группа лечения C: 350 мг REGN2810 Q3W в течение 108 недель, двухкомпонентная химиотерапия препаратами платины Q3W в течение 2 циклов и 50 мг ипилимумаба каждые 6 недель (Q6W) в количестве до 4 доз
[00323] Пациентов подвергают предписанному им лечению в течение 108-недельного периода лечения. Лечение можно прекращать досрочно по причине определяемого с помощью Критериев оценки ответа солидных опухолей версии 1.1 (RECIST 1.1) прогрессирования заболевания, неприемлемой токсичности, отзыва согласия на участие в исследовании, смерти, начала другого противоопухолевого лечения или, в случае пациентов в группах лечения B и C, в некоторых случаях подтвержденного полного ответа (CR) или частичного ответа (PR). Пациенты, у которых наблюдают определяемое с помощью RECIST 1.1 прогрессирование заболевания во время терапии, могут продолжать лечение, если исследователь считает, что пациент получит клиническую пользу, и если пациент не завершил 108-недельный период лечения. Если подтверждают дальнейшее прогрессирование заболевание (определяемое как дополнительное 10%-ное повышение опухолевой массы с момента исходного прогрессирования заболевания), необходимо прекратить лечение и, при необходимости, рассмотреть другую противоопухолевую терапию. Схожий подход лечения после первых признаков прогрессирования заболевания можно предлагать пациентам в группе лечения A, которым вводят пембролизумаб.
Исследуемое лечение
[00324] Группа лечения A: Пембролизумаб, вводимый в дозе 200 мг в виде IV инфузии Q3W в течение 108 недель
[00325] Группа лечения B: REGN2810, вводимое в дозе 350 мг в виде внутривенной (IV) инфузии Q3W в течение 108 недель, в комбинации с ипилимумабом, вводимым IV в течение приблизительно 90 минут в дозе 50 мг Q6W в количестве до 4 доз.
[00326] Группа лечения C: REGN2810, вводимое в дозе 350 мг в виде IV инфузии Q3W в течение 108 недель, в комбинации с двухкомпонентной химиотерапией препаратами платины, осуществляемой IV Q3W в течение 2 циклов, и ипилимумабом, вводимым IV в течение приблизительно 90 минут в дозе 50 мг Q6W в количестве до 4 доз
Процедуры и оценка
[00327] Процедуры, которые следует осуществлять при скрининге, будут включать получение информированного согласия; оценку критериев включения/исключения; регистрацию медицинского и онкологического анамнеза и сопутствующих лекарственных средств; регистрацию демографических данных; получение и тестирование образцов опухолевой ткани для анализа PD-L1 и мутаций рецептора эпидермального фактора роста и киназы анапластической лимфомы и слияния онкогена рецепторной тирозинкиназы C-ros; исходную рентгенографическую оценку опухоли и оценку опухолевой массы; рентгенографию грудной клетки; анализ сыворотки на беременность; электрокардиографию в 12 отведениях; полный физикальный осмотр, включающий определение показателей жизнедеятельности, роста и массы тела; оценку статуса по шкале Восточной объединенной онкологической группы (ECOG); регистрацию нежелательных явлений (AE) и лабораторные анализы. Также можно получать образцы для необязательного геномного подисследования.
[00328] В течение периода лечения осуществляют следующие процедуры для оценки эффективности и безопасности: определение QOL с использованием валидированных опросников, заполняемых пациентом, физикальный осмотр, оценку статуса по шкале ECOG; определение показателей жизнедеятельности; лабораторные анализы, включая тестирование женщин детородного возраста на беременность; регистрацию AE и сопутствующих лекарственных средств. Компьютерную томографию или магнитно-резонансную томографию (или позитронно-эмиссионную томографию) для рентгенографической оценки опухолевой массы и оценку опухолевой массы с помощью критериев RECIST 1.1 осуществляют в определенные моменты времени на всем протяжении исследования.
[00329] Данные о выживаемости собирают посредством телефонного звонка или в кабинете врача во время визита каждые 3 месяца до смерти, неявки для последующего наблюдения или отзыва согласия на участие в исследовании.
Результаты
[00330] Ожидают, что любая комбинация REGN2810 будет пролонгировать PFS на 1-5 месяцев по сравнению с монотерапией пембролизумабом. REGN2810 в комбинации с химиотерапией и/или антителом против CTLA-4 будет пролонгировать OS и улучшать ORR по сравнению с монотерапией пембролизумабом.
[00331] Объем настоящего изобретения не ограничен конкретными вариантами осуществления, представленными в настоящем описании. Фактически, различные модификации изобретения в дополнение к представленным в настоящем описании будут очевидны специалистам в этой области из изложенного выше описания и сопутствующих чертежей. Такие модификации предназначены для включения в объем формулы изобретения.
--->
СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Regeneron Pharmaceuticals, Inc.
<120> АНТИТЕЛА ПРОТИВ PD-1 ДЛЯ ЛЕЧЕНИЯ РАКА ЛЕГКИХ
<130> 10411WO01
<160> 10
<170> FastSEQ для Windows версии 4.0
<210> 1
<211> 117
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> HCVR R2810
<400> 1
Glu Val Gln Leu Leu Glu Ser Gly Gly Val Leu Val Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe Ser Asn Phe
20 25 30
Gly Met Thr Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ser Gly Ile Ser Gly Gly Gly Arg Asp Thr Tyr Phe Ala Asp Ser Val
50 55 60
Lys Gly Arg Phe Thr Ile Ser Arg Asp Asn Ser Lys Asn Thr Leu Tyr
65 70 75 80
Leu Gln Met Asn Ser Leu Lys Gly Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Val Lys Trp Gly Asn Ile Tyr Phe Asp Tyr Trp Gly Gln Gly Thr Leu
100 105 110
Val Thr Val Ser Ser
115
<210> 2
<211> 107
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> LCVR R2810
<400> 2
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Ser Ile Thr Ile Thr Cys Arg Ala Ser Leu Ser Ile Asn Thr Phe
20 25 30
Leu Asn Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Asn Leu Leu Ile
35 40 45
Tyr Ala Ala Ser Ser Leu His Gly Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Arg Thr Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Gln Gln Ser Ser Asn Thr Pro Phe
85 90 95
Thr Phe Gly Pro Gly Thr Val Val Asp Phe Arg
100 105
<210> 3
<211> 8
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> HCDR1 R2810
<400> 3
Gly Phe Thr Phe Ser Asn Phe Gly
1 5
<210> 4
<211> 8
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> HCDR2 R2810
<400> 4
Ile Ser Gly Gly Gly Arg Asp Thr
1 5
<210> 5
<211> 10
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> HCDR3 R2810
<400> 5
Val Lys Trp Gly Asn Ile Tyr Phe Asp Tyr
1 5 10
<210> 6
<211> 6
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> LCDR1 R2810
<400> 6
Leu Ser Ile Asn Thr Phe
1 5
<210> 7
<211> 3
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> LCDR2 R2810
<400> 7
Ala Ala Ser
1
<210> 8
<211> 9
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> LCDR3 R2810
<400> 8
Gln Gln Ser Ser Asn Thr Pro Phe Thr
1 5
<210> 9
<211> 444
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> HC R2810
<400> 9
Glu Val Gln Leu Leu Glu Ser Gly Gly Val Leu Val Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe Ser Asn Phe
20 25 30
Gly Met Thr Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ser Gly Ile Ser Gly Gly Gly Arg Asp Thr Tyr Phe Ala Asp Ser Val
50 55 60
Lys Gly Arg Phe Thr Ile Ser Arg Asp Asn Ser Lys Asn Thr Leu Tyr
65 70 75 80
Leu Gln Met Asn Ser Leu Lys Gly Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Val Lys Trp Gly Asn Ile Tyr Phe Asp Tyr Trp Gly Gln Gly Thr Leu
100 105 110
Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser Val Phe Pro Leu
115 120 125
Ala Pro Cys Ser Arg Ser Thr Ser Glu Ser Thr Ala Ala Leu Gly Cys
130 135 140
Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser Trp Asn Ser
145 150 155 160
Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val Leu Gln Ser
165 170 175
Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro Ser Ser Ser
180 185 190
Leu Gly Thr Lys Thr Tyr Thr Cys Asn Val Asp His Lys Pro Ser Asn
195 200 205
Thr Lys Val Asp Lys Arg Val Glu Ser Lys Tyr Gly Pro Pro Cys Pro
210 215 220
Pro Cys Pro Ala Pro Glu Phe Leu Gly Gly Pro Ser Val Phe Leu Phe
225 230 235 240
Pro Pro Lys Pro Lys Asp Thr Leu Met Ile Ser Arg Thr Pro Glu Val
245 250 255
Thr Cys Val Val Val Asp Val Ser Gln Glu Asp Pro Glu Val Gln Phe
260 265 270
Asn Trp Tyr Val Asp Gly Val Glu Val His Asn Ala Lys Thr Lys Pro
275 280 285
Arg Glu Glu Gln Phe Asn Ser Thr Tyr Arg Val Val Ser Val Leu Thr
290 295 300
Val Leu His Gln Asp Trp Leu Asn Gly Lys Glu Tyr Lys Cys Lys Val
305 310 315 320
Ser Asn Lys Gly Leu Pro Ser Ser Ile Glu Lys Thr Ile Ser Lys Ala
325 330 335
Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr Leu Pro Pro Ser Gln
340 345 350
Glu Glu Met Thr Lys Asn Gln Val Ser Leu Thr Cys Leu Val Lys Gly
355 360 365
Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu Ser Asn Gly Gln Pro
370 375 380
Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu Asp Ser Asp Gly Ser
385 390 395 400
Phe Phe Leu Tyr Ser Arg Leu Thr Val Asp Lys Ser Arg Trp Gln Glu
405 410 415
Gly Asn Val Phe Ser Cys Ser Val Met His Glu Ala Leu His Asn His
420 425 430
Tyr Thr Gln Lys Ser Leu Ser Leu Ser Leu Gly Lys
435 440
<210> 10
<211> 214
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> LC R2810
<400> 10
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Ser Ile Thr Ile Thr Cys Arg Ala Ser Leu Ser Ile Asn Thr Phe
20 25 30
Leu Asn Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Asn Leu Leu Ile
35 40 45
Tyr Ala Ala Ser Ser Leu His Gly Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Arg Thr Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Gln Gln Ser Ser Asn Thr Pro Phe
85 90 95
Thr Phe Gly Pro Gly Thr Val Val Asp Phe Arg Arg Thr Val Ala Ala
100 105 110
Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly
115 120 125
Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala
130 135 140
Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln
145 150 155 160
Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser
165 170 175
Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr
180 185 190
Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser
195 200 205
Phe Asn Arg Gly Glu Cys
210
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ПУТИ IL-4/IL-13 ДЛЯ ПОВЫШЕННОЙ ЭФФЕКТИВНОСТИ ПРИ ЛЕЧЕНИИ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ | 2020 |
|
RU2836467C2 |
| КОМБИНАЦИЯ АНТИ-PD-L1 АНТИТЕЛА И ИНГИБИТОРА ДНК-ПК ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННОГО НОВООБРАЗОВАНИЯ | 2018 |
|
RU2765997C2 |
| СПОСОБЫ, ТЕРАПИЯ И ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННОГО НОВООБРАЗОВАНИЯ | 2021 |
|
RU2831425C1 |
| СПОСОБЫ ЛЕЧЕНИЯ РАКА ШЕЙКИ МАТКИ ПУТЕМ ВВЕДЕНИЯ ИНИБИТОРА PD-1 | 2021 |
|
RU2833461C1 |
| СПОСОБЫ МОДУЛИРОВАНИЯ CART-КЛЕТОК | 2017 |
|
RU2774232C2 |
| СПОСОБ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННОГО НОВООБРАЗОВАНИЯ С ИСПОЛЬЗОВАНИЕМ КОМБИНАЦИИ АНТИТЕЛА К PD-1 И ХИМИОТЕРАПЕВТИЧЕСКОГО АГЕНТА | 2021 |
|
RU2787457C2 |
| ИНГИБИТОРЫ PD-1 / PD-L1 ДЛЯ ЛЕЧЕНИЯ РАКА | 2016 |
|
RU2714233C2 |
| СПОСОБЫ И КОМБИНИРОВАННОЕ ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ РАКА ЖЕЛЧНЫХ ПРОТОКОВ | 2019 |
|
RU2793543C2 |
| ПРИМЕНЕНИЕ АНТИТЕЛА ПРОТИВ PD-1 В ИЗГОТОВЛЕНИИ ЛЕКАРСТВЕННОГО СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ СОЛИДНЫХ ОПУХОЛЕЙ | 2020 |
|
RU2825845C2 |
| Способы лечения рака с помощью анти-PD-1-антител и анти-CTLA4-антител | 2019 |
|
RU2825835C2 |
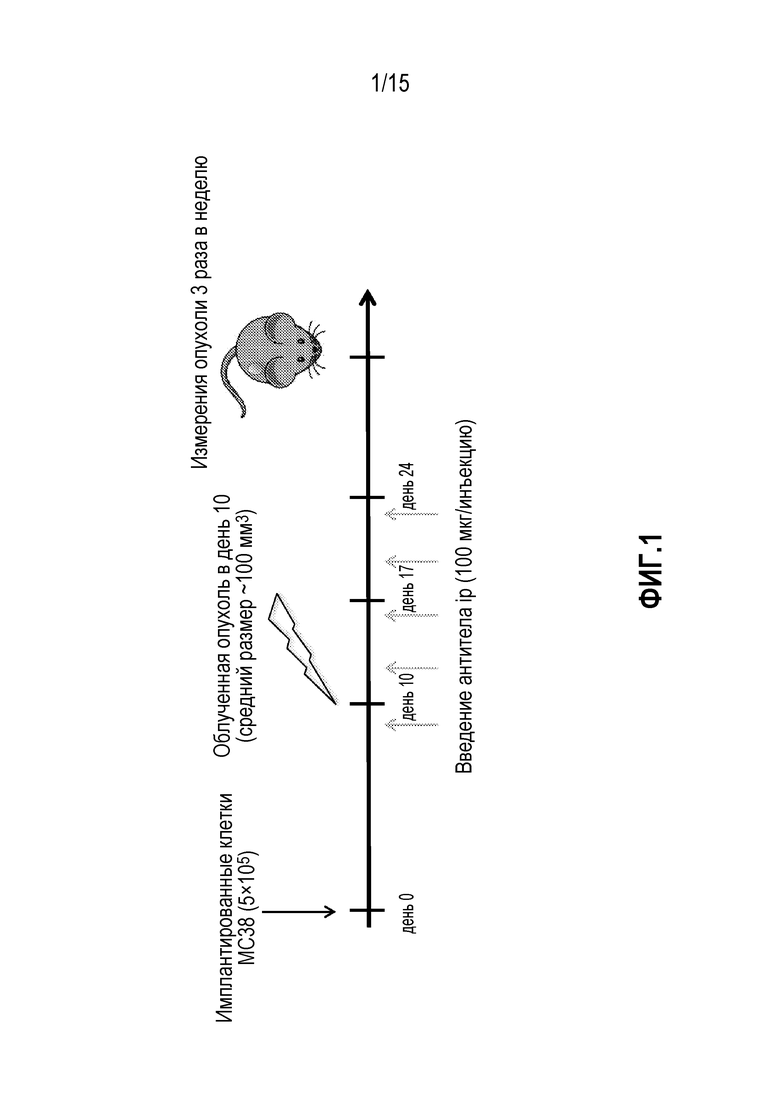
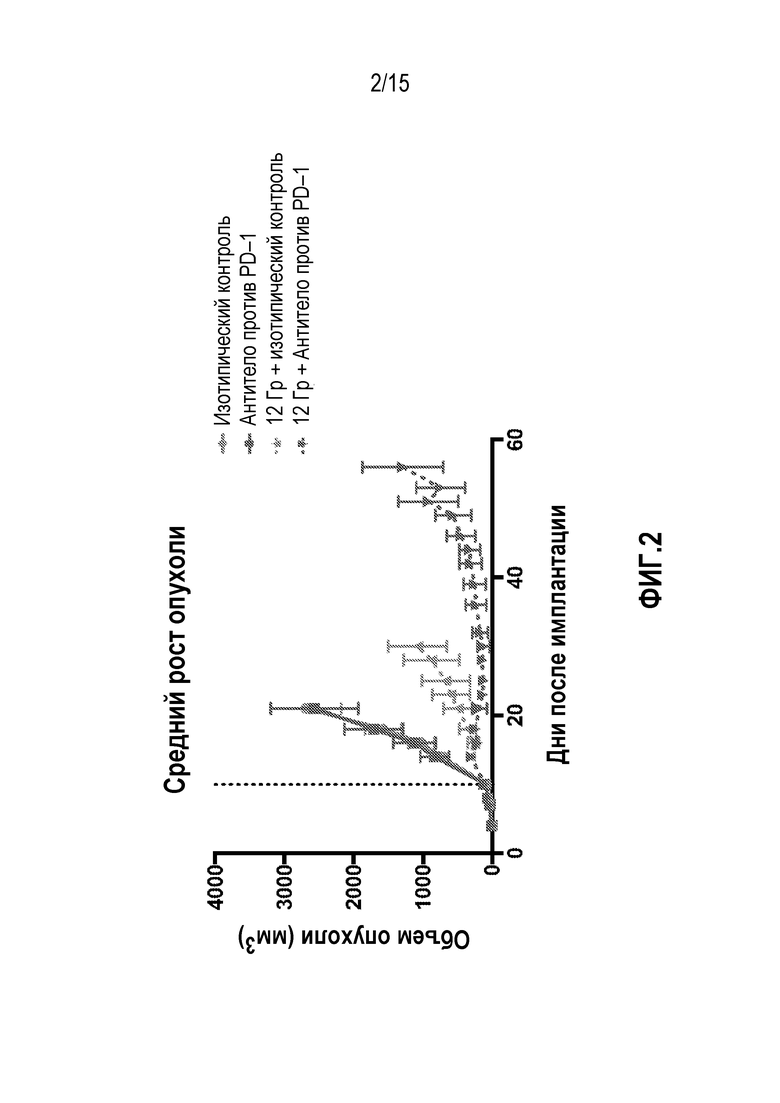
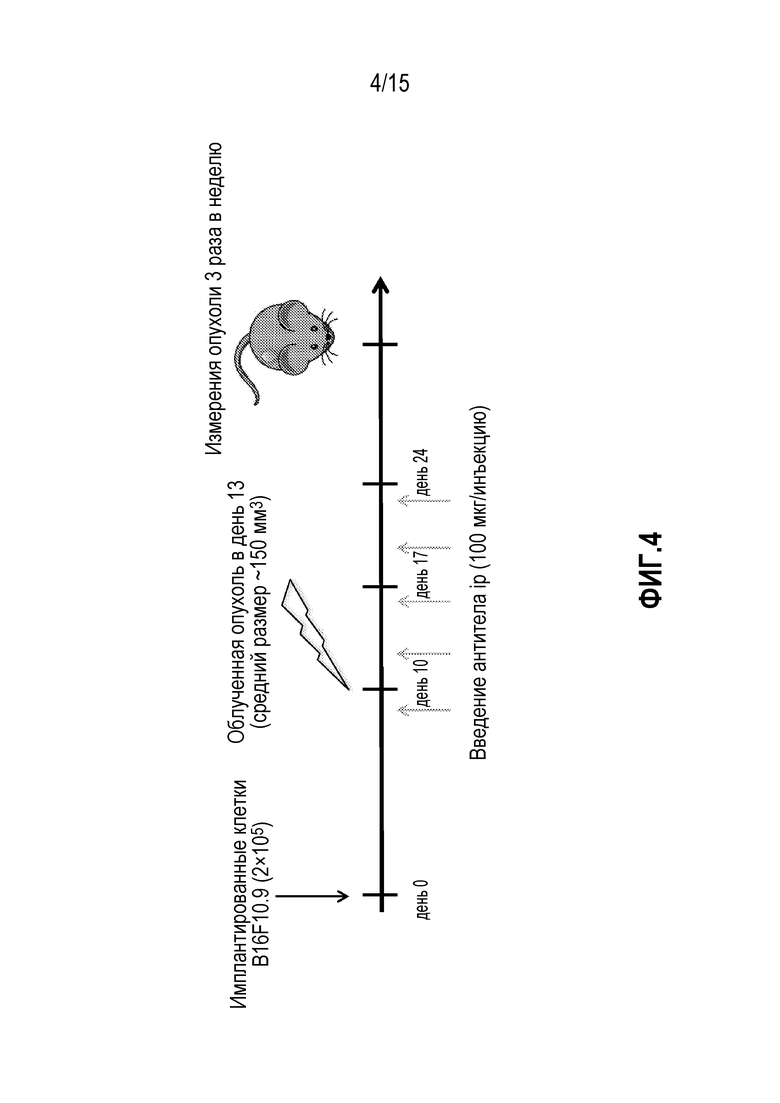
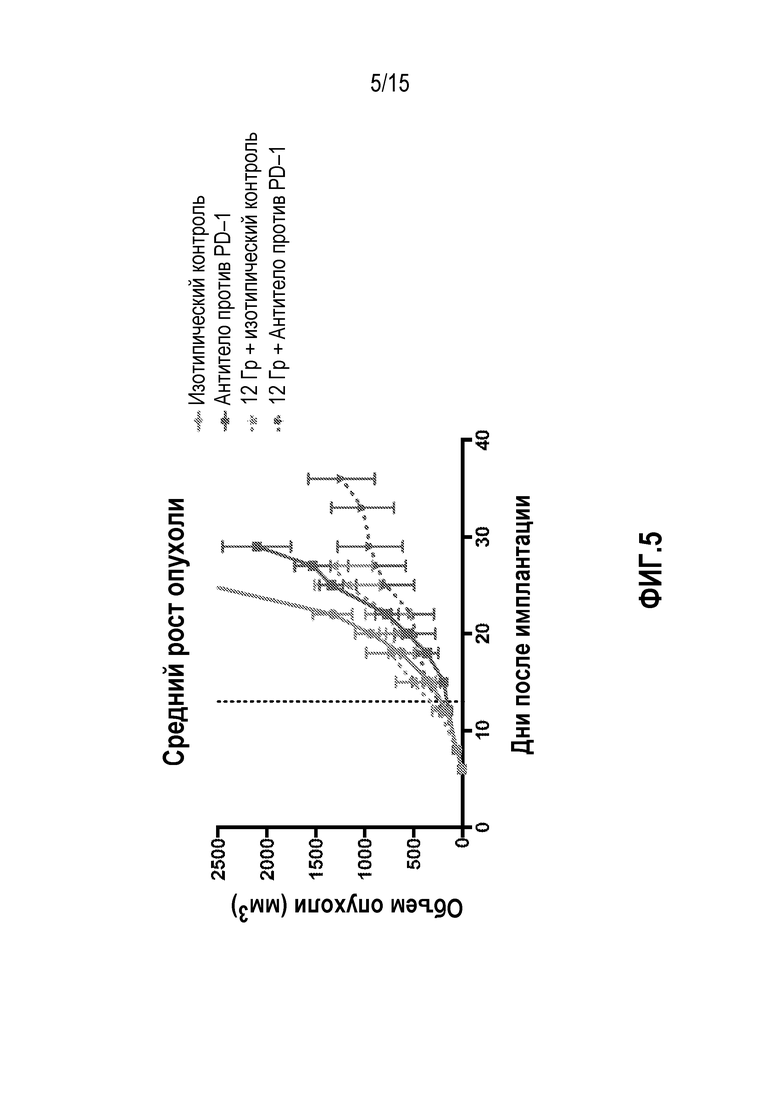
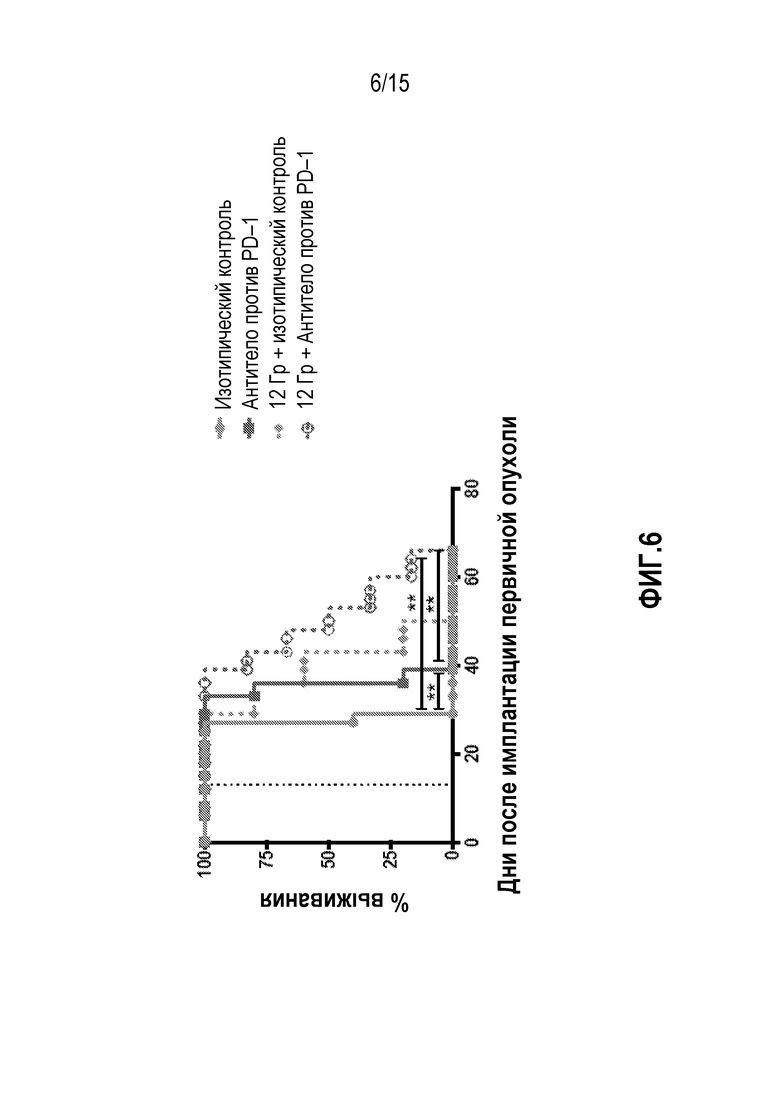
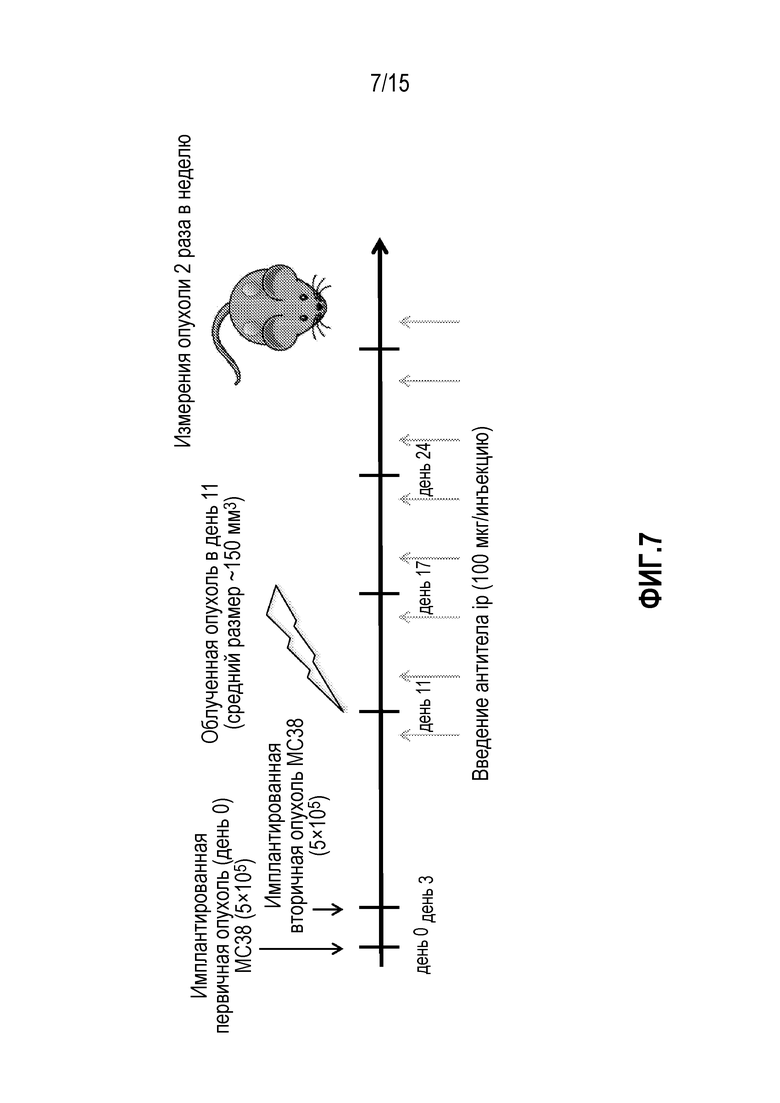
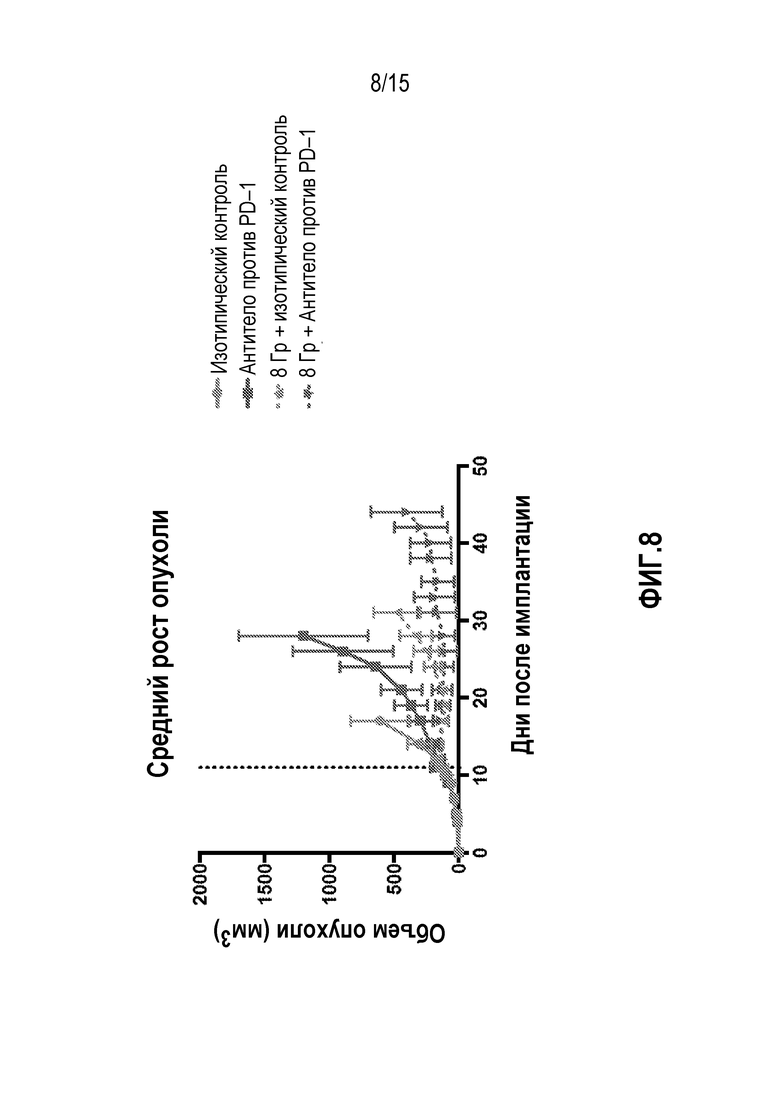
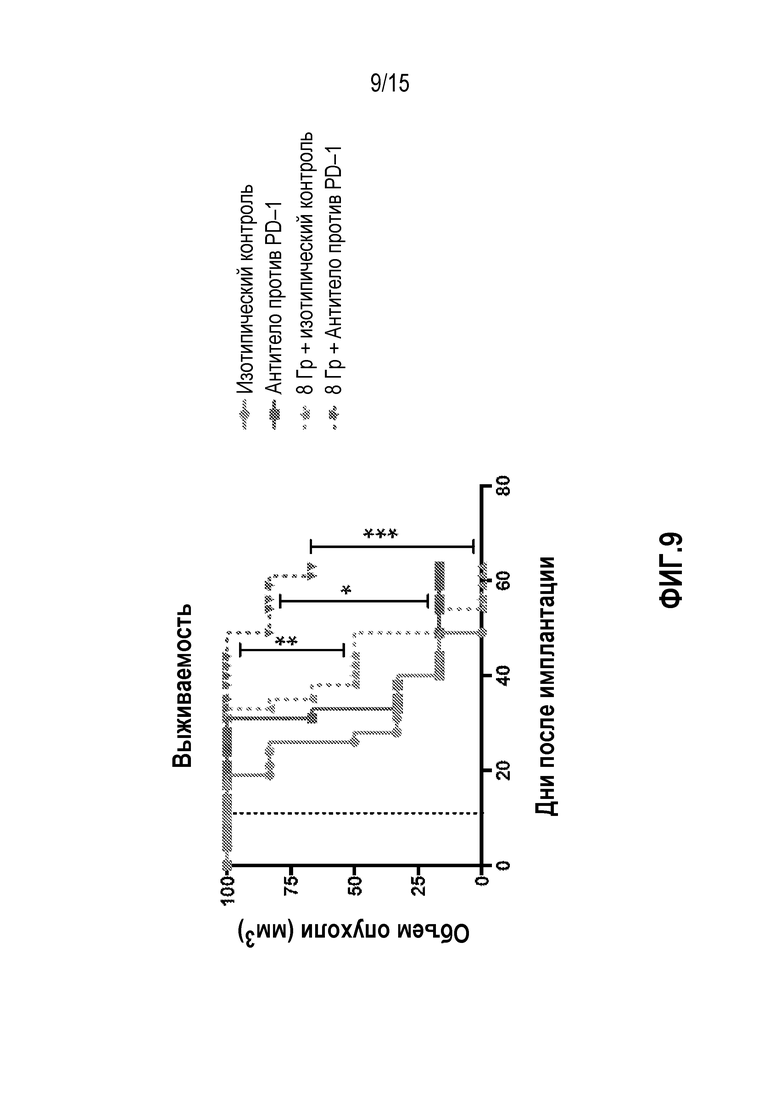
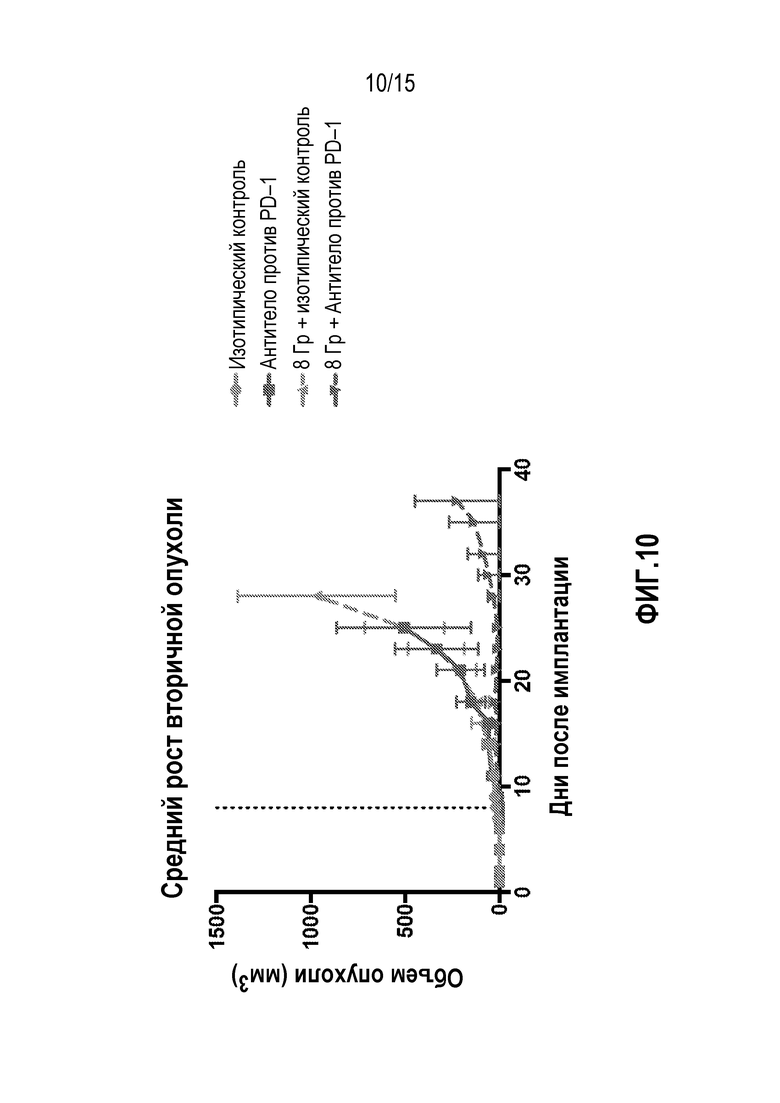
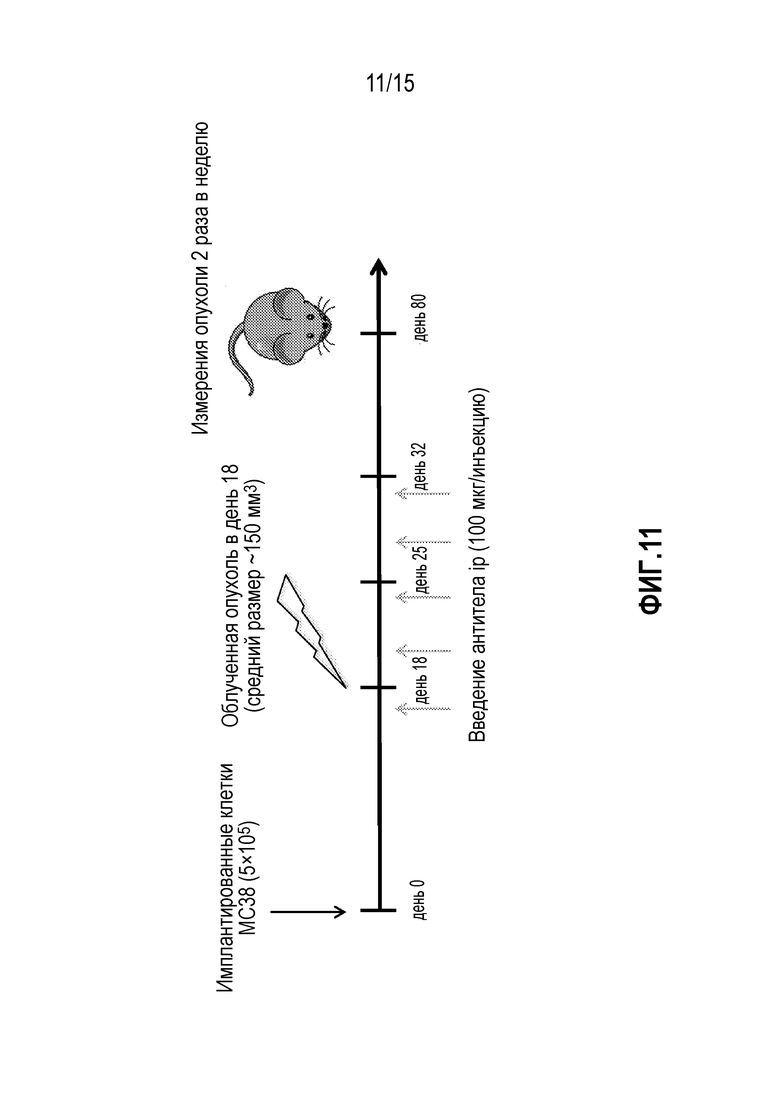
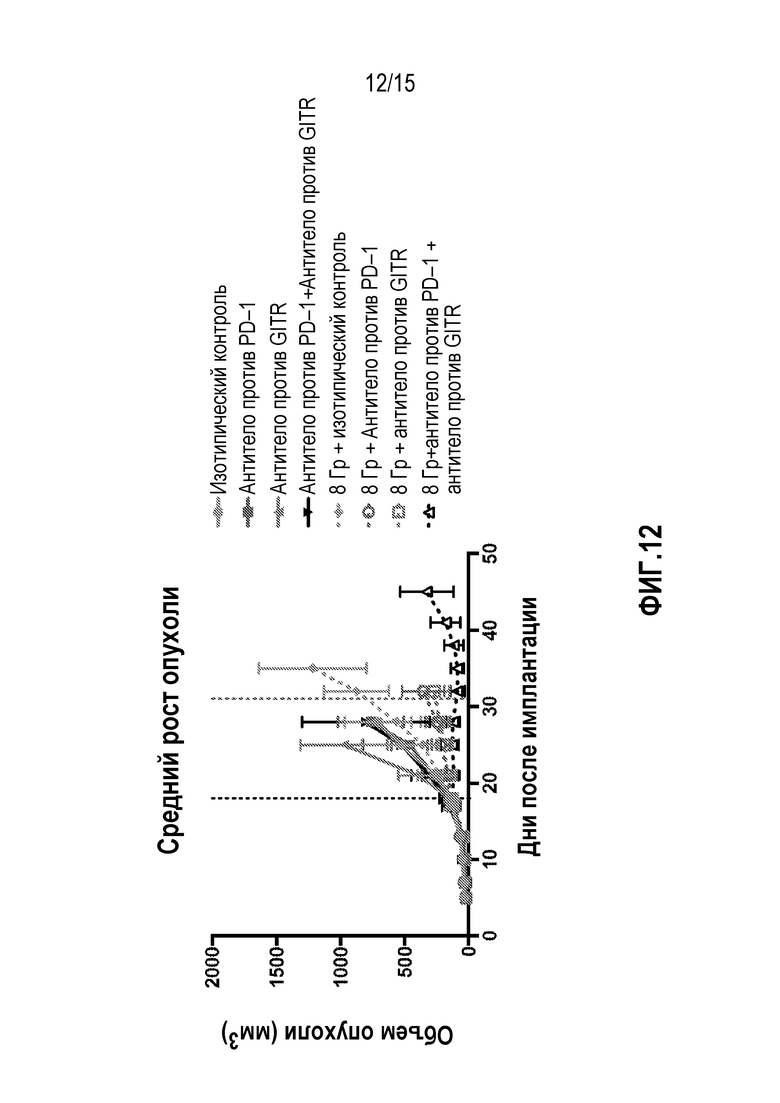
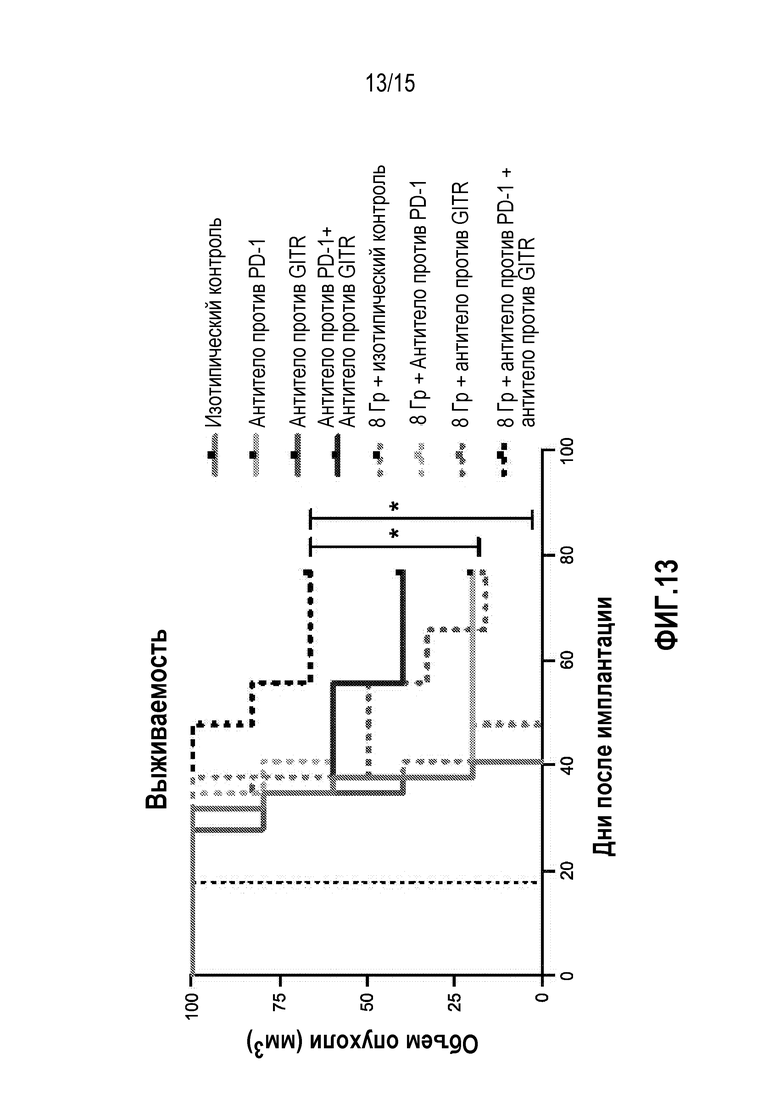
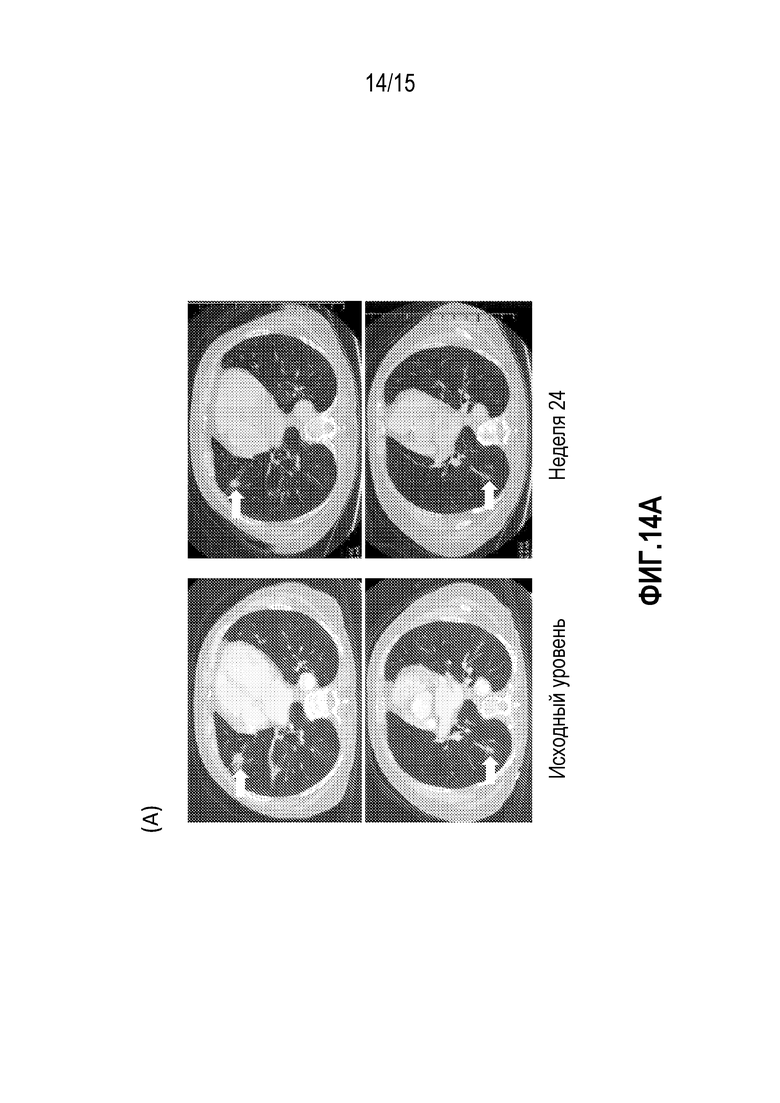

Изобретение относится к области биотехнологии, в частности к способам лечения рака легких. Способы предусматривают выбор пациента, имеющего немелкоклеточный рак легких, где опухолевая ткань пациента экспрессирует PD–L1 в ≥50% опухолевых клеток, и пациент не тестирован как положительный в отношении мутаций рецептора эпидермального фактора роста (EGFR), транслокаций киназы анапластической лимфомы (ALK) и/или слияний ROS1, а также введение пациенту терапевтически эффективного количества антитела, специфически связывающегося с PD–1. Изобретение эффективно для лечения немелкоклеточного рака легких. 2 н. и 26 з.п. ф-лы, 14 ил., 13 табл., 15 пр.
1. Способ лечения рака легких, включающий:
(a) выбор пациента, имеющего немелкоклеточный рак легких, где опухолевая ткань пациента экспрессирует PD–L1 в ≥50% опухолевых клеток, и пациент не тестирован как положительный в отношении мутаций рецептора эпидермального фактора роста (EGFR), транслокаций киназы анапластической лимфомы (ALK) и/или слияний ROS1; и
(b) введение пациенту одной или более доз терапевтически эффективного количества антитела или его антигенсвязывающего фрагмента, специфически связывающихся с белком программируемой гибели клеток 1 (PD–1), и, таким образом, лечение рака легких у пациента,
где антитело или его антигенсвязывающий фрагмент содержит три определяющих комплементарность области тяжелой цепи (HCDR1, HCDR2 и HCDR3) вариабельной области тяжелой цепи (HCVR) и три определяющих комплементарность области легкой цепи (LCDR1, LCDR2 и LCDR3) вариабельной области легкой цепи (LCVR), где HCDR1 содержит аминокислотную последовательность SEQ ID NO: 3; HCDR2 содержит аминокислотную последовательность SEQ ID NO: 4; HCDR3 содержит аминокислотную последовательность SEQ ID NO: 5; LCDR1 содержит аминокислотную последовательность SEQ ID NO: 6; LCDR2 содержит аминокислотную последовательность SEQ ID NO: 7; и LCDR3 содержит аминокислотную последовательность SEQ ID NO: 8.
2. Способ по п.1, где пациента ранее не подвергали системному лечению рака легких.
3. Способ по п.1, где пациента ранее подвергали химиотерапии.
4. Способ по любому из пп.1-3, где немелкоклеточный рак легких находится местно на поздней стадии и пациент не является кандидатом для лечения с использованием радикальной химиолучевой терапии, или немелкоклеточный рак легких представляет собой метастазирующий немелкоклеточный рак легких.
5. Способ по п.3 или 4, где противоопухолевая терапия включает химиотерапию на основе платины.
6. Способ по любому из пп.1-5, где пациент имеет плоскоклеточный или неплоскоклеточный немелкоклеточный рак легких стадии III или стадии IV.
7. Способ по любому из пп.1-5, где пациент имеет рецидивирующий немелкоклеточный рак легких или немелкоклеточный рак легких на поздней стадии.
8. Способ по любому из пп.1–7, где каждую дозу антитела вводят один раз в неделю, один раз в 2 недели, один раз в 3 недели или один раз в 4 недели.
9. Способ по любому из пп.1–8, где каждая доза содержит 20–1500 мг антитела.
10. Способ по любому из пп.1-7, где каждая доза содержит 200, 250, 300, 350, 450, 600, 750, 800, 1000 или 1050 мг антитела.
11. Способ по любому из пп.1-7, где каждая доза содержит 350 мг антитела, и ее вводят каждые 3 недели.
12. Способ по любому из пп.1–7, где каждая доза антитела содержит 0,1–10 мг/кг массы тела пациента.
13. Способ по п.12, где каждая доза антитела содержит 1, 3, 4, 5, 6 или 10 мг/кг массы тела пациента.
14. Способ по п.13, где каждую дозу вводят через один раз в неделю, один раз в 2 недели, один раз в 3 недели или один раз в 4 недели.
15. Способ по любому из пп.1–14, где антитело вводят пациенту в виде внутривенной инфузии.
16. Способ по любому из пп.1–15, где антитело вводят в комбинации со вторым терапевтическим средством или схемой лечения, выбранными из группы, состоящей из химиотерапии, ингибитора антигена цитотоксических T–лимфоцитов 4 (CTLA–4), лучевой терапии, хирургической операции, ингибитора гена активации лимфоцитов 3 (LAG–3), ингибитора рецептора эпидермального фактора роста (EGFR), ингибитора PD–L1, циклофосфамида, вакцины, антитела к глюкокортикоид-индуцируемому рецептору фактора некроза опухоли(GITR), слитого белка, ингибирующего фактор роста эндотелия сосудов (VEGF), антитела к VEGF, сунитиниба, сорафениба, пазопаниба, антитела к опухолеспецифическому антигену, гранулоцитарно-макрофагального колониестимулирующего фактора, цитотоксина, цитокина, Т-клеточной терапии, противовоспалительного препарата, пищевой добавки.
17. Способ по п.16, где антитело против PD–1 вводят в комбинации с химиотерапией препаратами платины.
18. Способ по п.16 или 17, где антитело против PD–1 вводят в комбинации с антителом против CTLA–4.
19. Способ по любому из пп.1–15, где введение по меньшей мере одной дозы антитела против PD–1 приводит к повышению выживаемости без прогрессирования (PFS) или общей выживаемости (OS) пациента по сравнению с пациентом, которого подвергали химиотерапии препаратами платины в качестве монотерапии.
20. Способ по п.19, где PFS повышают по меньшей мере на один месяц по сравнению с пациентом, которого подвергали химиотерапии препаратами платины.
21. Способ по п.19 или 20, где OS повышают по меньшей мере на один месяц по сравнению с пациентом, которого подвергали химиотерапии препаратами платины.
22. Способ по любому из пп.1-21, где HCVR содержит аминокислотную последовательность SEQ ID NO: 1 и LCVR содержит аминокислотную последовательность SEQ ID NO: 2.
23. Способ по любому из пп.1–22, где антитело против PD–1 содержит тяжелую цепь, содержащую аминокислотную последовательность SEQ ID NO: 9, и легкую цепь, содержащую аминокислотную последовательность SEQ ID NO: 10.
24. Способ по любому из пп.1–22, где антитело против PD–1 является REGN2810 (цемиплимабом).
25. Способ лечения рака легких, включающий:
(a) выбор пациента, имеющего немелкоклеточный рак легких, где: (i) опухолевая ткань пациента экспрессирует лиганд программируемой гибели клеток 1 (PD–L1) в ≥50% опухолевых клеток; (ii) пациент не тестирован как положительный в отношении мутаций рецептора эпидермального фактора роста (EGFR), транслокаций киназы анапластической лимфомы (ALK) и/или слияний ROS1; (iii) пациент не является кандидатом для лечения с использованием радикальной химиолучевой терапии или немелкоклеточный рак легких представляет собой метастазирующий немелкоклеточный рак легких; (iv) пациента ранее не подвергали системному лечению рака легких; и
(b) введение пациенту терапевтически эффективного количества антитела, специфически связывающегося с белком программируемой гибели клеток 1 (PD–1), и, таким образом, лечение рака легких у пациента,
где антитело или его антигенсвязывающий фрагмент содержит три определяющих комплементарность области тяжелой цепи (HCDR1, HCDR2 и HCDR3) вариабельной области тяжелой цепи (HCVR) и три определяющих комплементарность области легкой цепи (LCDR1, LCDR2 и LCDR3) вариабельной области легкой цепи (LCVR), где HCDR1 содержит аминокислотную последовательность SEQ ID NO: 3; HCDR2 содержит аминокислотную последовательность SEQ ID NO: 4; HCDR3 содержит аминокислотную последовательность SEQ ID NO: 5; LCDR1 содержит аминокислотную последовательность SEQ ID NO: 6; LCDR2 содержит аминокислотную последовательность SEQ ID NO: 7; и LCDR3 содержит аминокислотную последовательность SEQ ID NO: 8;
и указанное антитело вводят внутривенно каждые три недели в дозе 350 мг.
26. Способ по п.25, где HCVR содержит аминокислотную последовательность SEQ ID NO: 1 и LCVR содержит аминокислотную последовательность SEQ ID NO: 2.
27. Способ по п.25 или 26, где антитело содержит тяжелую цепь, содержащую аминокислотную последовательность SEQ ID NO: 9, и легкую цепь, содержащую аминокислотную последовательность SEQ ID NO: 10.
28. Способ по любому из пп. 25-27, где антитело к PD-1 представляет собой цемиплимаб.
| SUZANNE L | |||
| TOPALIAN et al., Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer, N Engl J Med., 2012, 366(26), pp | |||
| Полый вставной колосник для подведения воздуха к топливу в русской печи | 1924 |
|
SU2443A1 |
| HOSSEIN BORGHAEI et al., Nivolumab versus Docetaxel in Advanced Non-squamous Non-small Cell Lung Cancer, N Engl J Med, 2015, 373(17), pp | |||
| Электрический насос | 1923 |
|
SU1627A1 |
| EDWARD B | |||
| GARON et al., Pembrolizumab for the | |||

