Изобретение относится к аналитической химии, а именно к способам количественного и полуколичественного определения нитрит-ионов, и может использоваться при мониторинге загрязнений окружающей среды и контроле качества пищевых продуктов.
Известен способ определения нитрит-ионов (см. патент РФ №2578024 по кл. МПК G01N 33/18, опубл. 20.03.2016), включающий приготовление раствора нитритов, извлечение нитритов полиметакрилатной мембраной с иммобилизованным сафранином в качестве реагента, последующее ее отделение от раствора, измерение аналитического сигнала и оценку содержания нитрит-ионов, при этом в качестве аналитического сигнала используют светопоглощение при 530 нм или визуальную оценку интенсивности окраски оптической мембраны, оценку содержания нитритов проводят по градуировочному графику методом добавок или визуально-тестовым методом.
При взаимодействии сафранина с раствором нитрит-ионов в сильнокислой среде наблюдается уменьшение интенсивности малиновой окраски соответствующей матрицы (обесцвечивание) за счет протекания реакции диазотирования в твердой фазе пропорционально увеличению концентрации нитрит-ионов в растворе. Диапазон определяемых содержаний составил 1000-5000 нг/мл с пределом обнаружения 500 нг/мл.
Однако данный способ характеризуется недостаточно высокой чувствительностью, что не позволяет определять количество нитрит-ионов на уровне долей предельно-допустимых концентраций (ПДК).
Известен также способ определения нитрит-ионов в образцах воды и почвы (Cherian T., Narayana B. New system of spectrophotometric determination of trace amounts of nitrite in environmental samples // Journal of the Brazilian Chemical Society. 2006. V. 17. №. 3, p. 577-581), основанный на их реакции с п-нитроанилином в кислой среде с образованием иона диазония, который в сочетании со сложным эфиром (этоксиэтиленмалеиновый эфир или этилцианоацетат) в основной среде образует азокрасители, измерение поглощения которых позволяет определять нитрит-ионы в диапазонах 500-15000 нг/мл или 200-18000 нг/мл соответственно.
Недостатком известного способа является недостаточно высокая чувствительность определения и, как следствие, невозможность определения нитрит-ионов на уровне долей ПДК.
Известен способ фотометрического определения нитрит-ионов в жидкой среде (см. патент РФ №2265828 по кл. МПК G01N21/75, опубл. 10.12.2005), включающий взаимодействие нитритов с композицией хромогенных реагентов, а именно 3-гидрокси-7,8-бензо-1,2,3,4-тетрагидрохинолином в сочетании с 2-аминобензойной (4-аминобензойной), либо с сульфаниловой кислотой, либо с сульфаниламидом, закрепленных на бумажном носителе, который помещают в фотометрируемый раствор поперек светового потока. Способ позволяет определять концентрацию нитритов в диапазоне 100 - 20000 нг/мл.
К недостаткам способа можно отнести использование малодоступного реагента 3-гидрокси-7,8-бензо-1,2,3,4-тетрагидрохинолина, а также недостаточно высокая чувствительность определения, что не позволяет определять нитрит-ионы на уровне долей ПДК.
Наиболее близким аналогом к заявляемому изобретению является способ спектрофотометрического определения нитрит-ионов, основанный на их цветной реакции с п-нитроанилином в присутствии дифениламина в кислых средах и последующей мицеллярной экстракции в точке помутнения неионным ПАВ образующегося красителя (Afham A. et al. Micelle-mediated extraction for the spectrophotometric determination of nitrite in water and biological samples based on its reaction with p-nitroaniline in the presence of diphenylamine // Analytical biochemistry. 2005. V. 336. № 2, p. 295 - 299). Способ применен для обнаружения нитрит-ионов в сточных водах, водопроводной воде и образцах мочи человека. Диапазон определяемых содержаний составил 2-40 нг/мл с пределом обнаружения 0,87 нг/мл.
К недостаткам этого способа можно отнести: 1) необходимость термостатирования системы на водяной бане в течение 10 минут при 70°С для завершения реакции азосочетания и достижения температуры помутнения неионного ПАВ; 2) центрифугирование системы в течение 5 минут при 3000 об/мин для достижения в ней разделения фаз; 3) применение метанола в качестве растворителя для разбавления мицеллярной фазы при ее фотометрировании.
Технической проблемой является разработка упрощенного способа определения нитрит-ионов при повышении его чувствительности.
Техническим результатом настоящего изобретения является снижение предела обнаружения нитрит-ионов до уровня сотых долей предельно-допустимых концентраций, расширение диапазона определяемых содержаний и понижение погрешности определения.
Для достижения заявляемого результата в способе определения нитрит-ионов, включающем обработку анализируемой пробы растворами органических реагентов, один из которых на основе п-нитроанилина, а другой - дифениламина, выделение из полученной реакционной смеси мицеллярной фазы в присутствии поверхностно-активного вещества и оценку содержания нитрит-ионов, согласно изобретению, в качестве поверхностно-активного вещества используют додецилсульфат натрия, а в качестве органических реагентов используют водные растворы 4-нитроанилина и дифениламина, полученные их диспергированием в растворе додецилсульфата натрия, причем диспергированный раствор 4-нитроанилина дополнительно подкисляют соляной кислотой до рН 1-3, при этом добавляют полученную смесь 4-нитроанилина и додецилсульфата натрия к анализируемой пробе в количестве 1⋅10-4 - 2⋅10-3 М и 2⋅10-3 - 1⋅10-2 М соответственно, а смесь дифениламина и додецилсульфата натрия в количестве 1⋅10-4 - 2⋅10-3 М и 2⋅10-2 - 1⋅10-1 М, выделение мицеллярной фазы осуществляют путем добавления к реакционной смеси соляной кислоты в количестве 2,7-4,1 М, причем оценку содержания нитрит-ионов проводят по градуировочному графику или визуально-тестовым методом путем сравнения окраски мицеллярных фаз с окраской цветовой шкалы.
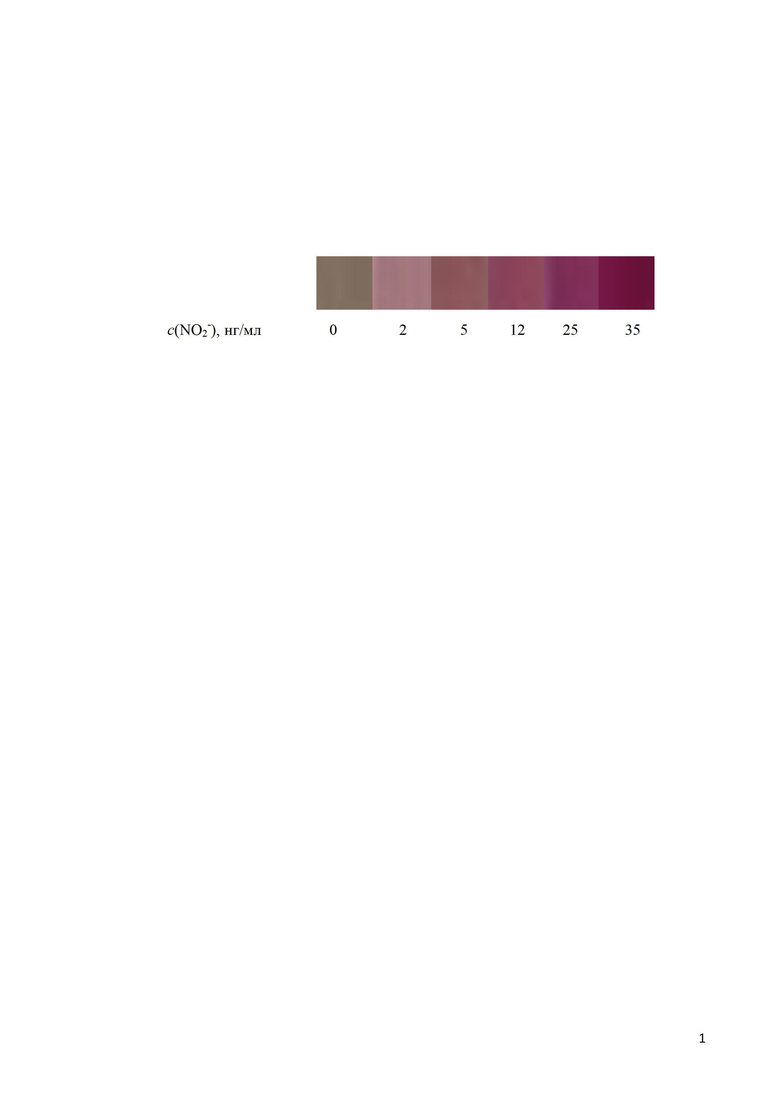
Способ поясняется иллюстрацией, где представлена цветовая шкала для визуально-тестового определения нитрит-ионов, представляющая собой цифровые изображения ПАВ-обогащенных фаз с различным содержанием азосоединения, образующегося по реакции взаимодействия реагентов (4-нитроанилина и дифениламина) с нитрит-ионами.
Способ осуществляется следующим образом. Исходный раствор 4-нитроанилина диспергируют в растворе одного из наиболее часто применяемых представителей анионных ПАВ - додецилсульфата натрия (ДДС) и подкисляют соляной кислотой (HCl) до рН (1-3). Водный раствор ДДС является альтернативой органическим растворителям, например, этанолу, увеличивает растворимость 4-нитроанилина и стабилизирует его раствор, в то время как низкие значения рН способствуют увеличению скорости образования диазотирующей частицы (хлорида нитрозила) и стабилизации малоустойчивой соли диазония.
К анализируемой пробе (вода или водная вытяжка), содержащей нитрит-ионы, добавляют диспергированный в ДДС 4-нитроанилин (4-НА) в кислой среде, в результате чего протекает реакция его диазотирования с образованием соли 4-нитрофенилдиазония. Выбор диазотируемого ариламина обусловлен его высокой реакционной способностью (электроноакцепторная группа в пара-положении повышает электрофильность катиона диазония), устойчивостью к окислению и выраженными гидрофобными свойствами. Кроме того, 4-нитроанилин - слабое основание и, поэтому, оно способно диазотироваться в сильнокислых средах без предварительного протонирования, что, с одной стороны, благоприятно для образования соли диазония, а с другой - необходимо для фазового разделения раствора анионного ПАВ.
После обработки анализируемой пробы раствором 4-НА, диспергированного в растворе ДДС, в количестве 1 ⋅ 10-4 ≤ С4-НА ≤ 2 ⋅ 10-3 М и 2 ⋅ 10-3 ≤ СДДС ≤ 1 ⋅ 10-2 М, к соли диазония добавляют дифениламин (ДФА), исходный раствор которого диспергируют в ДДС, в количестве 1 ⋅ 10-4 ≤ СДФА ≤ 2 ⋅ 10-3 М и 2 ⋅ 10-2 ≤ СДДС ≤ 1 ⋅ 10-1 М, в результате чего образуется окрашенное азосоединение. Выбор ДФА в качестве азосоставляющей обусловлен его выраженным гидрофобным характером, низкой растворимостью в воде и, следовательно, хорошей растворимостью в мицеллах ПАВ. Дифениламин (вторичный ариламин) с небольшим значением константы кислотности (рКа) не подвергается диазотированию, поэтому с его применением исключается ряд побочных реакций.
Введение в предложенную систему ПАВ анионного типа приводит к: 1) стабилизации растворов 4-нитроанилина и его соли диазония; 2) увеличению растворимости образующегося азосоединения (1-(4-дифениламин)-4-нитробензола), малорастворимого в водных средах, вследствие его солюбилизации мицеллами ДДС, что делает возможным применять предложенную систему в фотометрическом анализе; 3) образованию мицеллярно-насыщенных ПАВ кислотно-индуцированных фаз, в которые экстрагируется аналитическая форма азокрасителя.
Затем добавляют соляную кислоту в количестве (2,7-4,1) М, которая способствует фазовому разделению системы и образованию двух изотропных фаз. Выбор HCl обусловлен отсутствием у нее окислительных свойств, тогда как применение кислот-окислителей (азотная или хлорная кислоты), может привести к нежелательным процессам окисления компонентов системы.
Полученные ПАВ-насыщенные фазы отбирали и фотометрировали в стеклянных кюветах (l = 1см) при λmax = 540 нм относительно дистиллированной воды.
В другом варианте оценивали содержание нитрит-ионов путем сравнения окраски мицеллярно-насыщенных фаз с цветовой шкалой.
Способ иллюстрируется следующими примерами.
Пример 1.
Приготовление исходных растворов реагентов:
1) Для приготовления раствора 4-нитроанилина (4-НА) концентрацией 1⋅10-1 М в 5⋅10-2 М в растворе анионного ПАВ додецилсульфата натрия (ДДС) навески 1,3800 г 4-НА и 1,4419 г ДДС помещают в мерную колбу вместимостью 100 мл, добавляют дистиллированную воду на 2/3 объема колбы, подкисляют соляной кислотой HCl (1 : 1) до рН (1-3), диспергируют компоненты смеси в ультразвуковой установке 5-10 минут. После осаждения пены ДДС доводят содержимое колбы дистиллированной водой до метки и тщательно перемешивают.
2) Для приготовления раствора дифениламина (ДФА) концентрацией 1⋅10-1 М в 2⋅10-1 М растворе ДДС навески 1,6920 г ДФА и 5,7676 г ДДС помещают в мерную колбу вместимостью 100 мл, добавляют дистиллированную воду на 2/3 объема колбы, диспергируют компоненты смеси в ультразвуковой установке 5-10 минут. После осаждения пены ДДС доводят содержимое колбы дистиллированной водой до метки и тщательно перемешивают.
Построение градуировочного графика.
Точную навеску свежеперекристаллизованного NaNO2 (х.ч.) 0,1725 г помещают в мерную колбу на 25 мл и растворяют в дистиллированной воде (раствор содержит 7 мг/мл NaNO2). Данный раствор разбавляют дистиллированной водой в 1000 раз для получения раствора нитрита натрия с концентрацией 7 мкг/мл (раствор А).
Для построения градуировочной характеристики в мерные колбы емкостью 25 мл помещают 0,01 мл (0,07мкг/мл), 0,05 мл (0,35 мкг/мл), 0,10 мл (0,7 мкг/мл), 0,15 мл (1,05 мкг/мл), 0,20 мл (1,4 мкг/мл), 0,25 мл (1,75 мкг/мл) раствора А, в каждую колбу добавляют по 2,5 мл 1⋅10-1 М раствора 4-НА в кислой среде (рН 1 - 3), приготовленного в 5⋅10-2 М растворе ДДС. Спустя 5-7 минут в каждую колбу добавляют по 2,5 мл 1⋅10-1 М раствора ДФА, приготовленного в 2⋅10-1 М растворе ДДС, тщательно перемешивают. Далее добавляют 7-8 мл HClконц., перемешивают и доводят дистиллированной водой объем каждой колбы до метки, после чего тщательно их перемешивают. Наблюдают помутнение системы и образование двухфазной системы с окрашенной в розовый - малиновый цвет, в зависимости от концентрации нитрит-иона, мицеллярно-насыщенной фазой, локализующейся в верхней части расслаивающейся системы. При фотометрическом детектировании полученные мицеллярные экстракты отделяют от водной фазы декантацией, разбавляют и измеряют оптическую плотность полученного окрашенного раствора с помощью спектрофотометра Shimadzu UV - 1800 в кювете с l = 1 см при длине волны λmax = 540 нм относительно дистиллированной воды. Уравнение градуировочной зависимости имеет вид: А540 = 17,52 ⋅ с(NO2-) - 0,02 (r = 0.991), где с(NO2) - концентрация нитрит-ионов, мкг/мл. Диапазон линейности полученной градуировочной зависимости составляет (0,07 - 7) ⋅ 10-2 мкг/мл или 0,7 - 70 нг/мл.
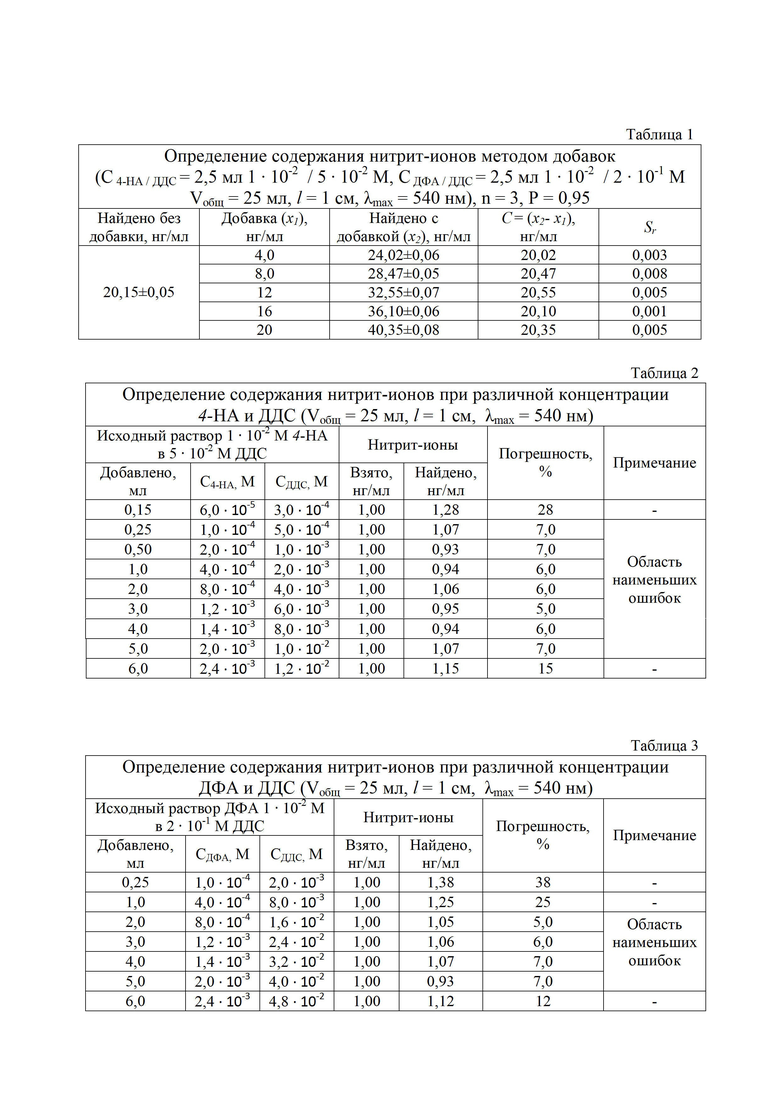
Пример 2. Определение содержания нитрит-ионов методом добавок
При определении содержания нитрит-ионов методом добавок готовят анализируемый раствор 25 мл с содержанием NO2- (0,0175-1,125 мкг), а также растворы с добавкой дополнительно 1-5 мл рабочего раствора нитрит-ионов с концентрацией 100 нг/мл.
В каждую колбу добавляют по 2,5 мл 1 ⋅ 10-1 М раствора 4-НА в кислой среде (рН 1-3), приготовленного в 5⋅10-2 М растворе ДДС. Спустя 5-7 минут в каждую колбу добавляют по 2,5 мл 1⋅10-1 М раствора ДФА в 2⋅10-1 М растворе ДДС, тщательно перемешивают. Далее добавляют 7-8 мл HClконц., перемешивают, и доводят дистиллированной водой объем каждой колбы до метки, после чего тщательно их перемешивают.
Фотометрирование полученных мицеллярных экстрактов осуществляют аналогично примеру 1. Содержание нитрит-ионов определяют графическим способом, экстраполируя прямолинейную зависимость изменения поглощения ΔА540 от концентрации нитрит-ионов в добавке до значения А = 0, где ΔА540 = А0 - А1 (А0, А1 - проба без добавки и с добавкой NO2- соответственно).
Результаты определения приведены в таблице 1.
Пример 3. Визуально-тестовое определение содержания нитрит-ионов
Для визуально-тестового определения нитрит-ионов получена цветовая шкала путем регистрации окраски ПАВ-обогащенных фаз систем, полученных при построении градуировочной зависимости (пример 1), цифровым фотоаппаратом (разрешение ≥12 MegaPixeles).
Определение выполняется аналогично методике, прописанной в примере 1, с тем отличием, что полученные мицеллярные экстракты не отделяли от водной фазы и не фотометрировали, а проводили сравнение окраски с цветовой шкалой (см. ил.), что позволяет полуколичественно определять содержание нитрит-ионов с(NO2-) в интервале концентраций 2-35 нг/мл.
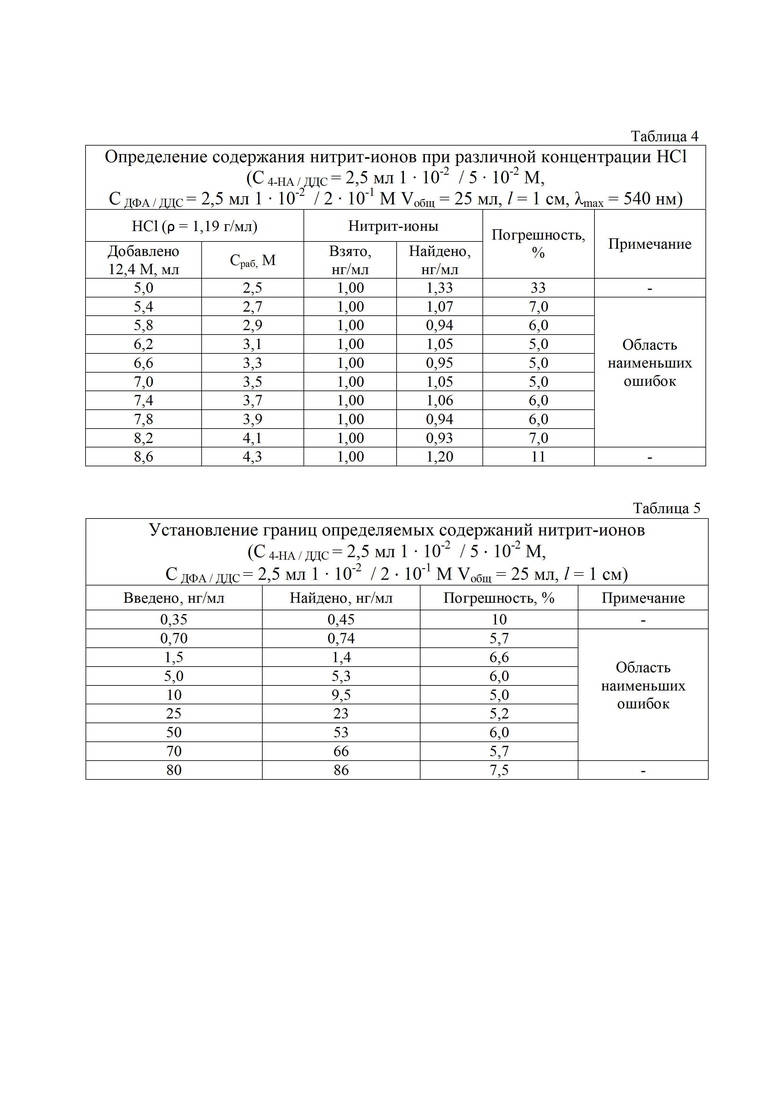
Установлены оптимальные концентрации реагентов в реакционной системе: 4-НА и ДДС (табл. 2), ДФА и ДДС (табл. 3), HCl (табл. 4). Последовательность проводимых операций аналогично примеру 1.
Как видно из табл. 2, введение в реакционную смесь 0,25 - 5,0 мл 1⋅10-2 М раствора п-НА, диспергированного в 5⋅10-2 М растворе ДДС, позволяет достигать наименьших ошибок определения нитрит-ионов, не превышающих 5-7 %. Таким образом, оптимальные концентрации в реакционной смеси п-НА и ДДС составляют соответственно 1 ⋅ 10-4 ≤ С ≤ 2 ⋅ 10-3 М и 2 ⋅ 10-3 ≤ С ≤ 1 ⋅ 10-2 М.
Как видно из табл. 3, введение в реакционную смесь 0,25 - 5,0 мл 1 ⋅ 10-2 М раствора п-НА диспергированного в 2⋅10-1 М растворе ДДС, позволяет достигать наименьших ошибок определения нитрит-ионов не превышающих 5-7 %. Таким образом, оптимальные концентрации в реакционной смеси ДФА и ДДС составляют соответственно 1 ⋅ 10-4 ≤ С ≤ 2 ⋅ 10-3 М и 2 ⋅ 10-2 ≤ С ≤ 1 ⋅ 10-1 М.
Данные табл. 4 свидетельствуют о том, что в интервале концентраций соляной кислоты 2,7-4,1 М достигается наименьшая ошибка определения нитрит-ионов.
Установлены границы определяемых содержаний нитрит-ионов (l = 1 см), которые представлены в табл. 5, в интервале концентраций 0,7-70 нг/мл с погрешностью, не превышающей 7 %, в отличие от прототипа, в котором диапазон определяемых содержаний составляет 2-40 нг/мл.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ НОВОКАИНА | 2019 |
|
RU2715997C1 |
| 2,6-ДИФЕНИЛ-4- (4-ДИМЕТИЛАМИНОСТИРИЛ)ПИРИЛИЯ-ХЛОРИД В КАЧЕСТВЕ АНАЛИТИЧЕСКОГО РЕАГЕНТА ДЛЯ КОЛИЧЕСТВЕННОГО ФОТОМЕТРИЧЕСКОГО ОПРЕДЕЛЕНИЯ АНИОННЫХ ПОВЕРХНОСТНО-АКТИВНЫХ ВЕЩЕСТВ | 1992 |
|
RU2030414C1 |
| Способ определения свинца | 1990 |
|
SU1755185A1 |
| Способ количественного определения нитрит-иона | 1974 |
|
SU517846A1 |
| Способ определения меди в воде | 1989 |
|
SU1682866A1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ АНИЛИНА И ЕГО МОНОНИТРОПРОИЗВОДНЫХ | 1991 |
|
RU2011968C1 |
| ИОНОСЕЛЕКТИВНАЯ МЕМБРАНА ДЛЯ ОПРЕДЕЛЕНИЯ ИОННЫХ ПОВЕРХНОСТНО-АКТИВНЫХ ВЕЩЕСТВ | 2013 |
|
RU2546045C1 |
| МЕМБРАНА ИОНОСЕЛЕКТИВНОГО ЭЛЕКТРОДА ДЛЯ ОПРЕДЕЛЕНИЯ ИОННЫХ ПОВЕРХНОСТНО-АКТИВНЫХ ВЕЩЕСТВ В СТОЧНЫХ ВОДАХ И СИНТЕТИЧЕСКИХ МОЮЩИХ СРЕДСТВАХ | 2013 |
|
RU2531130C1 |
| СПОСОБ ФЛУОРИМЕТРИЧЕСКОГО ОПРЕДЕЛЕНИЯ ФЛУНИКСИНА | 2014 |
|
RU2582960C1 |
| Способ определения алкилсульфатов в водных растворах | 1989 |
|
SU1675746A1 |
Изобретение относится к аналитической химии, а именно к способу определения нитрит-ионов. Способ включает обработку анализируемой пробы растворами органических реагентов, один из которых на основе п-нитроанилина, а другой дифениламина, выделение из полученной реакционной смеси мицеллярной фазы в присутствии поверхностно-активного вещества и оценку содержания нитрит-ионов. В качестве поверхностно-активного вещества используют додецилсульфат натрия, а в качестве органических реагентов используют водные растворы 4-нитроанилина и дифениламина, полученные их диспергированием в растворе додецилсульфата натрия. Диспергированный раствор 4-нитроанилина дополнительно подкисляют соляной кислотой до рН 1-3. Добавляют полученную смесь 4-нитроанилина и додецилсульфата натрия к анализируемой пробе в количестве 1⋅10-4 - 2⋅10-3 М и 2⋅10-3 - 1⋅10-2 М соответственно, а смесь дифениламина и додецилсульфата натрия в количестве 8⋅10-4 - 2⋅10-3 М и 2⋅10-2 - 4-10-2 М. Выделение мицеллярной фазы осуществляют путем добавления к реакционной смеси соляной кислоты в количестве 2,7-4,1 М. Оценку содержания нитрит-ионов проводят по градуировочному графику или визуально-тестовым методом путем сравнения окраски мицеллярных фаз с окраской цветовой шкалы. 3 пр., 6 ил.
Способ определения нитрит-ионов, включающий обработку анализируемой пробы растворами органических реагентов, один из которых на основе п-нитроанилина, а другой дифениламина, выделение из полученной реакционной смеси мицеллярной фазы в присутствии поверхностно-активного вещества и оценку содержания нитрит-ионов, отличающийся тем, что в качестве поверхностно-активного вещества используют додецилсульфат натрия, а в качестве органических реагентов используют водные растворы 4-нитроанилина и дифениламина, полученные их диспергированием в растворе додецилсульфата натрия, причем диспергированный раствор 4-нитроанилина дополнительно подкисляют соляной кислотой до рН 1-3, при этом добавляют полученную смесь 4-нитроанилина и додецилсульфата натрия к анализируемой пробе в количестве 1⋅10-4 - 2⋅10-3 М и 2⋅10-3 - 1⋅10-2 М соответственно, а смесь дифениламина и додецилсульфата натрия в количестве 8⋅10-4 - 2⋅10-3 М и 2⋅10-2 - 4-10-2 М, выделение мицеллярной фазы осуществляют путем добавления к реакционной смеси соляной кислоты в количестве 2,7-4,1 М, причем оценку содержания нитрит-ионов проводят по градуировочному графику или визуально-тестовым методом путем сравнения окраски мицеллярных фаз с окраской цветовой шкалы.
| AFKHAMI A | |||
| et al | |||
| Micelle-mediated Extraction for the Spectrophotometric determination of Nitrite in Water and Biological Samples Based on its Reaction with p-Nitroaniline in the Presence of Diphenylamine, Analytical biochemistry, 2005, v | |||
| 336, p | |||
| УСТРОЙСТВО ПАРОПЕРЕГРЕВАТЕЛЯ | 1920 |
|
SU295A1 |
| ДОРОНИН С.Ю | |||
| и др., Мицелярный катализ в системах ариламин-дифениламин-NO2-, Журнал общей | |||

