Область техники
Данное изобретение относится к новому алкинил-замещенному гетероциклическому соединению, действующему как ингибитор FGFR, или его фармацевтически приемлемой соли; к фармацевтической композиции, содержащей указанное соединение или его фармацевтически приемлемую соль; к способу получения алкинил-замещенного гетероциклического соединения или его фармацевтически приемлемой соли; к применению алкинил-замещенного гетероциклического соединения или его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей алкинил-замещенное гетероциклическое соединение или его фармацевтически приемлемую соль, при получении лекарства для лечения и/или предупреждения заболеваний, связанных с FGFR, в частности, опухолей; и к способу лечения и/или предупреждения заболеваний, связанных с FGFR, в частности, опухолей, с применением указанного соединения или указанной композиции.
Уровень техники
Рецептор фактора роста фибробластов (FGFR) представляет собой тип рецепторной тирозинкиназы (RTK), структурно состоящей из внемембранного лиганд-связывающего домена, одного трансмембранного домена и внутримембранной тирозинкиназы. Он включает, главным образом, четыре подтипа, FGFR1, FGFR2, FGFR3 и FGFR4. Он и его лиганд, фактор роста фибробластов (FGF), играют важную регуляторную роль в сигнальной системе клетки. В качестве внеклеточного стимулирующего сигнала, FGF связывается с внеклеточным доменом FGFR, вызывая фосфорилирование его внутримембранной тирозинкиназы, что приводит к активации серии последующих сигнальных путей, которые регулируют клеточную пролиферацию, дифференцировку и метастазы.
С экспрессией и активацией FGF/FGFR тесно связаны различные опухоли, такие как немелкоклеточный рак легких, рак молочной железы, рак желудка, рак печени, рак мочевого пузыря, эндометриальный рак, рак предстательной железы, рак шейки матки, рак толстой кишки, рак пищевода, миелома и меланома, и т.д. (Clin. Cancer Res. 2012, 18, 1855). В исследованиях показано, что на амплификацию FGFR1 приходится 20% случаев немелкоклеточного рака легких, на амплификацию FGFR2 приходится около 5% рака желудка, на мутации FGFR3 приходится около 70% случаев неинвазивного рака мочевого пузыря, и FGFR4 амплифицируется в раке печени (PloS One 2012, 7, е36713). Таким образом, разработка ингибиторов, направленно в со действующих на FGFR, становится актуальным направлением разработки противоопухолевых лекарств (Drug Disc. Today 2014, 19, 51).
В настоящее время на рынке представлены некоторые неспецифические в отношении FGFR препараты, такие как сунитиниб компании Pfizer, ленватиниб компании Eisai и нинтеданиб компании Boehnnger Ingelheim, но в настоящее время отсутствуют FGFR-специфические ингибиторы. Специфические ингибиторы FGFR, для которых начаты клинические испытания, включают HMPL-4 53, BGJ-398, LY-2874455, AZ-4547, JKH 2756493, TAS-120, ARQ-087 и BLTJ-554.
Разработка ингибиторов FGFR привлекает внимание многих биофармацевтических компаний, но все еще существует потребность в разработке новых соединений с точки зрения их перспективности при лечении различных злокачественных опухолей. В ходе непрерывных поисков, осуществленных авторами данного изобретения, разработано соединение по данному изобретению, имеющее структуру, представленную общей формулой (I), и было обнаружено, что соединение, имеющее такую структуру, демонстрирует превосходное действие и эффект.
Сущность изобретения
В данном изобретении предложено соединение, представленное общей формулой (I), в качестве ингибитора FGFR, его пролекарство, его стабильное изотопное производное, его фармацевтически приемлемая соль, его изомер или их смесь:
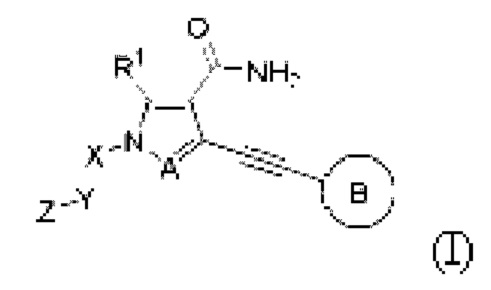
где:
А представляет собой N или CR2;
кольцо В представляет собой бензольное кольцо или 5-6-членное гетероарильное кольцо, где указанное бензольное кольцо и гетероарильное кольцо необязательно замещены одним или более G1;
R1 независимо выбран из Н, галогена, циано, С1-6 алкила или -NHR3;
R2 независимо выбран из Н, галогена, циано, С1-6 алкила, где указанный алкил необязательно замещен галогеном, циано, гидрокси или -OC1-6 алкилом;
R3 независимо выбран из Н, С1-6 алкила, С3-6 циклоалкила или 3-6-членного гетероциклила, где указанный алкил, циклоалкил и гетероциклил необязательно замещены галогеном, цианидом, -OR4, -NR5R6, С1-6 алкилом, С3-6 циклоалкилом или 3-6-членным гетероциклилом;
Х отсутствует или представляет собой С1-6 алкилен;
Y отсутствует или выбран из С3-8 циклоалкилена, 3-8-членного гетероциклилена, арилена или гетероарилена, где указанный циклоалкилен, гетероциклилен, арилен и гетероарилен необязательно замещены одним или более G2;
Z независимо выбран из циано, -NR7CN, 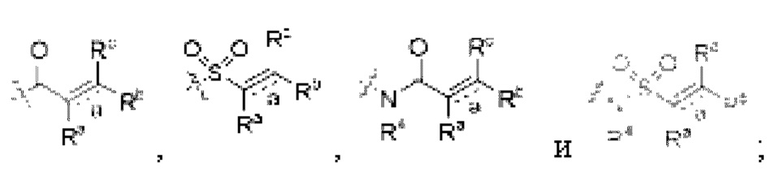
связь а представляет собой двойную связь ил и тройную связь;
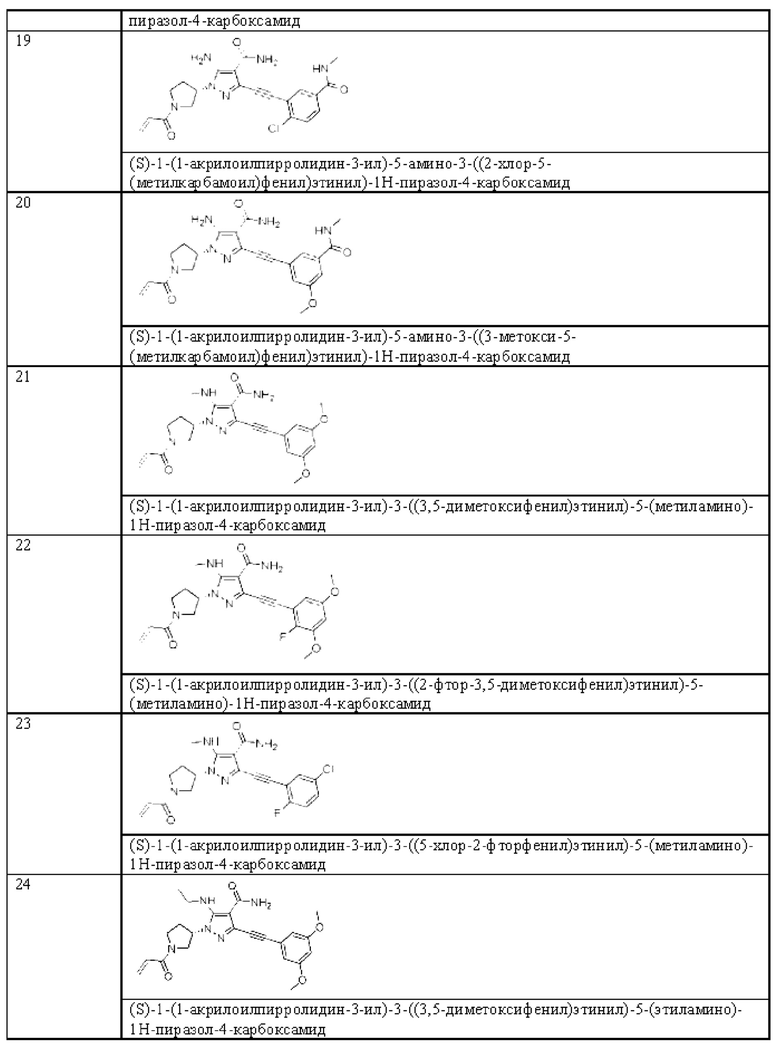
в том случае, если указанная связь а представляет собой двойную связь, каждый Ra, Rb и Rc независимо выбраны из Н, циано, галогена, С1-6 алкила, С3-6 циклоалкила и 3-6-членного гетероциклила, где указанный алкил, циклоалкил и гетероциклил необязательно замещены одним или более G3;
Ra и Rb или Rb и Rc необязательно вместе с атомом углерода, с которым они связаны, образуют необязательное 3-6-членное кольцо, содержащее гетероатом;
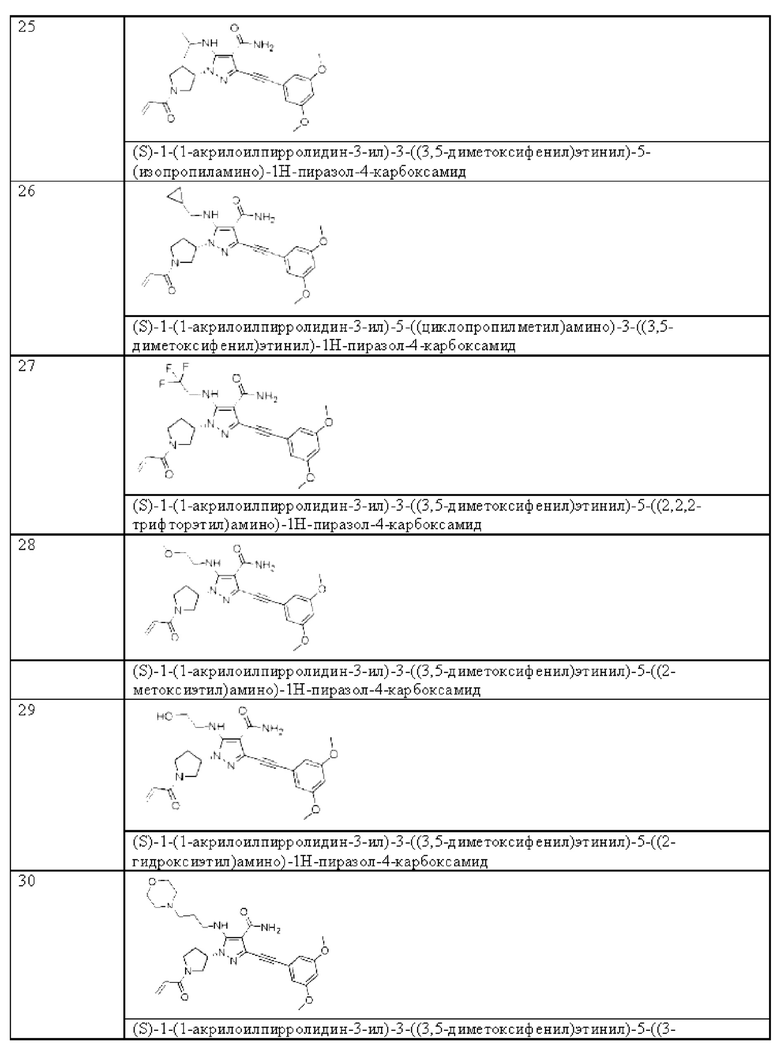
в том случае, если указанная связь а представляет собой тройную связь, Ra и Rc отсутствуют, Rb независимо выбран из Н, циано, галогена, С1-6 алкила, С3-6 циклоалкила и 3-6-членного гетероциклила, где указанный алкил, циклоалкил и гетероциклил необязательно замещены одним или более G4;
R4 независимо выбран из Н, С1-6 алкила, С3-6 циклоалкила или 3-6-членного гетероциклила, где указанный алкил, циклоалкил и гетероциклил необязательно замещены одним или более G5;
G1, G2, G3, G4 и G5, каждый независимо, выбраны из группы, состоящей из галогена, циано, С1-6 алкила, С3-6 алкенила, С2-6 алкинила, C3-8 циклоалкила, 3-8-членного гетероциклила, С6-10 арила, 5-10-членного гетероарила, -OR8, -ОС(O)NR8R9, -C(O)OR8, -C(O)NR8R9, -C(O)R8, -NR8R9, -NR8C(O)R9, -NR8C(O)NR9R10, -S(O)mR8 и -NR8SC(O)mR9, где указанный алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил и гетероарил необязательно замещены одной или более группами заместителей, выбранными из группы, состоящей из галогена, циано, С1-6 алкила, С3-8 циклоалкила, 3-8-членного гетероциклила, -OR11, -OC(O)NR11R12, -С(О)OR11, -C(O)NR11R12, -C(O)R11, -NR11R12, -NR11C(O)R12, -NR11C(O)NR12R13, -S(O)mR11 и -NR11S(О)mR12;
R4, R5, R6, R8, R9, R10, R11, R12 и R13, каждый независимо, выбраны из группы, состоящей из Н, С1-6 алкила, C3-8 циклоалкила, 3-8-членного моноциклического гетерециклила, моноциклического гетероарила и фенила; и
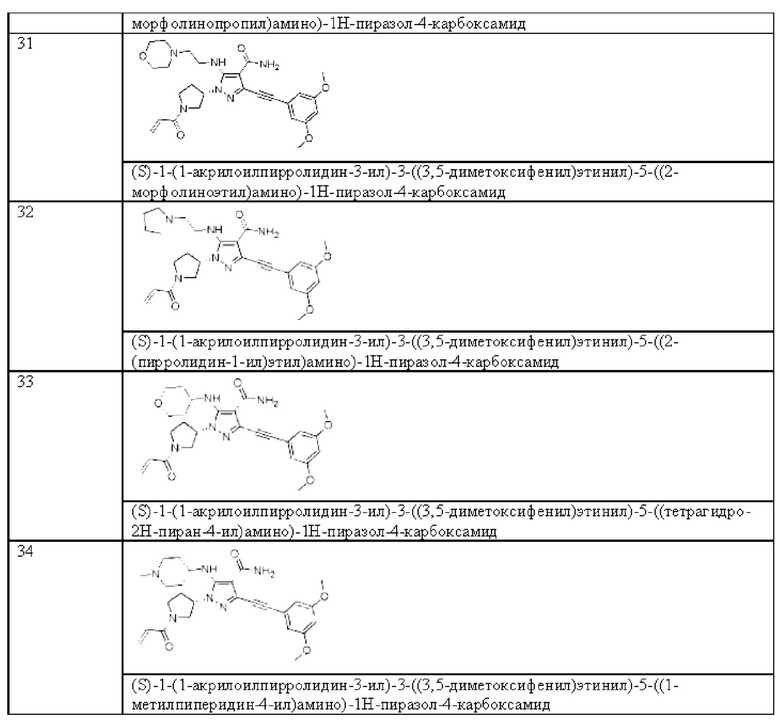
m равен 1 или 2.
Один вариант реализации данного изобретения относится к соединению, представленному общей формулой (I), как указано выше, его пролекарству, его стабильному изотопному производному, его фармацевтически приемлемой соли, его изомеру или их смеси, где А представляет собой N или CH, и предпочтительно представляет собой N.
Другой вариант реализации данного изобретения относится к соединению, представленному общей формулой (I), как указано выше, его пролекарству, его стабильному изотопному производному, его фармацевтически приемлемой соли, его изомеру или их смеси, где кольцо В представляет собой бензольное кольцо.
В одном аспекте данного изобретения предложено соединение, представленное общей формулой (II), его пролекарство, его стабильное изотопное производное, его фармацевтически приемлемая соль, его изомер или их смесь:
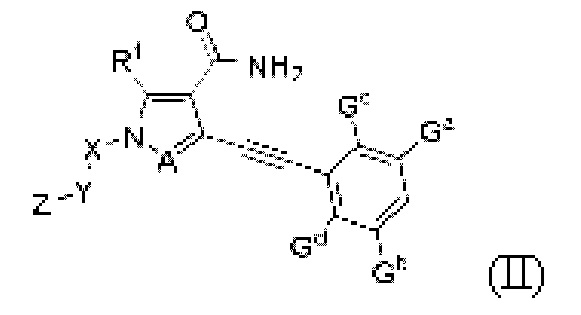
где:
Ga, Gb, Gc и Gd, каждый независимо, выбраны из группы, состоящей из Н, галогена, циано, С1-6 алкила, С3-8 циклоалкила, 3-8-членного гетероциклила, -OR8, -NR8R9 и -C(O)NR8R9, где указанный алкил, циклоалкил и гетероциклил необязательно замещены одной или более группами заместителей, выбранными из группы, состоящей из галогена, циано, С1-6 алкила, С3-8 циклоалкила, 3-8-членного гетероциклила, -OR11 и -NR11R12, где A, R1, R8, R9, R11, R12, X, Y, Z являются такими, как определено выше.
Другой вариант реализации данного изобретения относится к соединению, представленному общей формулой (I), как описано выше, его пролекарству, его стабильному изотопному производному, его фармацевтически приемлемой соли, его изомеру или их смеси, которые представляют собой соединение, представленное общей формулой (III), изображенной ниже, его пролекарство, его стабильное изотопное производное, его фармацевтически приемлемую соль, его изомер ил и их смесь:
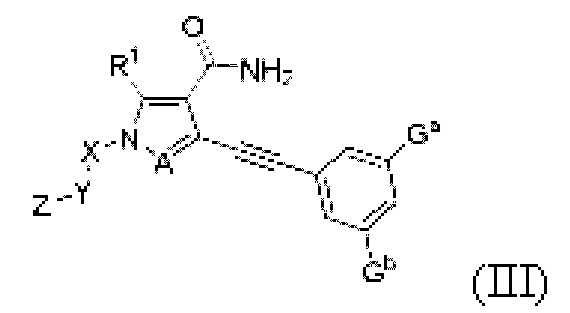
где:
Ga и Gb, каждый независимо, выбраны из группы, состоящей из Н, галогена, циано, С1-6 алкила, C3-8 циклоалкила, 3-8-членного гетероциклила, -OR8, -NR8R9 и -C(O)NR8R9, где указанный алкил, циклоалкил и гетероциклил необязательно замещены одной или более группами заместителей, выбранными из группы, состоящей из галогена, циано, С1-6 алкила, С3-8 циклоалкила, 3-8-членного гетероциклила, -OR11 и -NR11R12, где A, R1, R8, R9, R11, R12, X, Y, Z являются такими, как определено выше.
Другой вариант реализации данного изобретения относится к соединению, представленному общей формулой (I), как указано выше, его пролекарству, его стабильному изотопному производному, его фармацевтически приемлемой соли, его изомеру или их смеси, где R1 независимо выбран из Н, -NH2 и -NHC1-6 алкила.
В одном варианте реализации данного изобретения R1 может представлять собой Н или -NH2.
В одном варианте реализации данного изобретения Ga, Gb, Gc и Gd, каждый независимо, выбраны из -OC1-6 алкила и галогена.
В одном варианте реализации данного изобретения R1 независимо выбран из Н, -NH2 и -NHR3; и R3 независимо выбран из С1-6 алкила, С3-6 циклоалкила или 3-6-членного гетероциклила, где указанный алкил, циклоалкил и гетероциклил замещены галогеном, циано, -OR4, -NR5R6, С1-6 алкилом, С3-6 циклоалкилом или 3-6-членным гетероциклилом.
Другой вариант реализации данного изобретения относится е соединениям, представленным общей формулой (I), (II) и (III), их пролекарствам, их стабильным изотопным производным, их фармацевтически приемлемым солям, их изомерам или их смесям, где
Х отсутствует или представляет собой С1-6 алкилен;
Y отсутствует или представляет собой С3-8 циклоалкилен или 3-8-членный гетероциклилен,
Z независимо выбран из циано, -NR7CN, 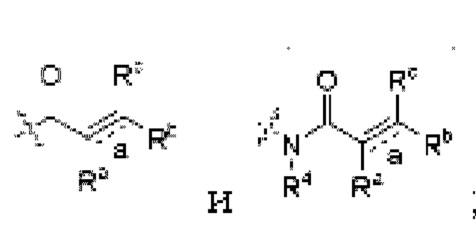
связь а представляет собой двойную связь или тройную связь;
в том случае, если указанная связь а представляет собой двойную связь, каждый Ra, Rb и Rc независимо выбраны из Н, циано, галогена, С1-6 алкила, С3-6 циклоалкила и 3-6-членного гетероциклила, где указанный алкил, циклоалкил и гетероциклил необязательно замещены одной или более группами заместителей, выбранными из группы, состоящей из галогена, циано, С1-6 алкила, С3-6 циклоалкила, 3-6-членного гетероциклила, -OR8 и -NR8R9;
в том случае, если указанная связь а представляет собой тройную связь, Ra н Rc отсутствуют, Rb независимо выбран из Н, циано, галогена, С1-6 алкила, С3-6 циклоалкила и 3-6-членного гетероциклила, где указанный алкил, циклоалкил и гетероциклил необязательно замещены одной или более группами заместителей, выбранными из группы, состоящей из галогена, циано, С1-6 алкила, С3-6 циклоалкила, 3-6-членного гетероциклила, -OR8 и -NR8R9;
R4, R8 и R9, каждый независимо, выбраны из Н и С1-6 алкила.
Другой вариант реализации данного изобретения относится к соединению, представленному общей формулой (I), приведенной выше, где указанное соединение выбрано из:
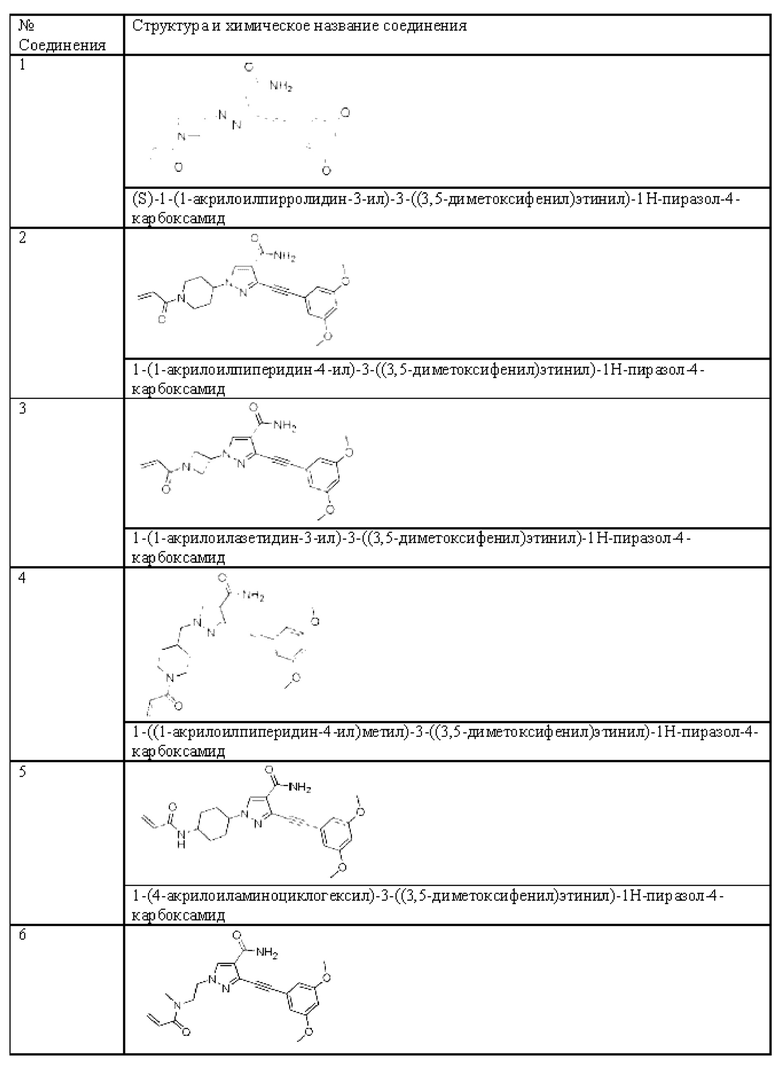
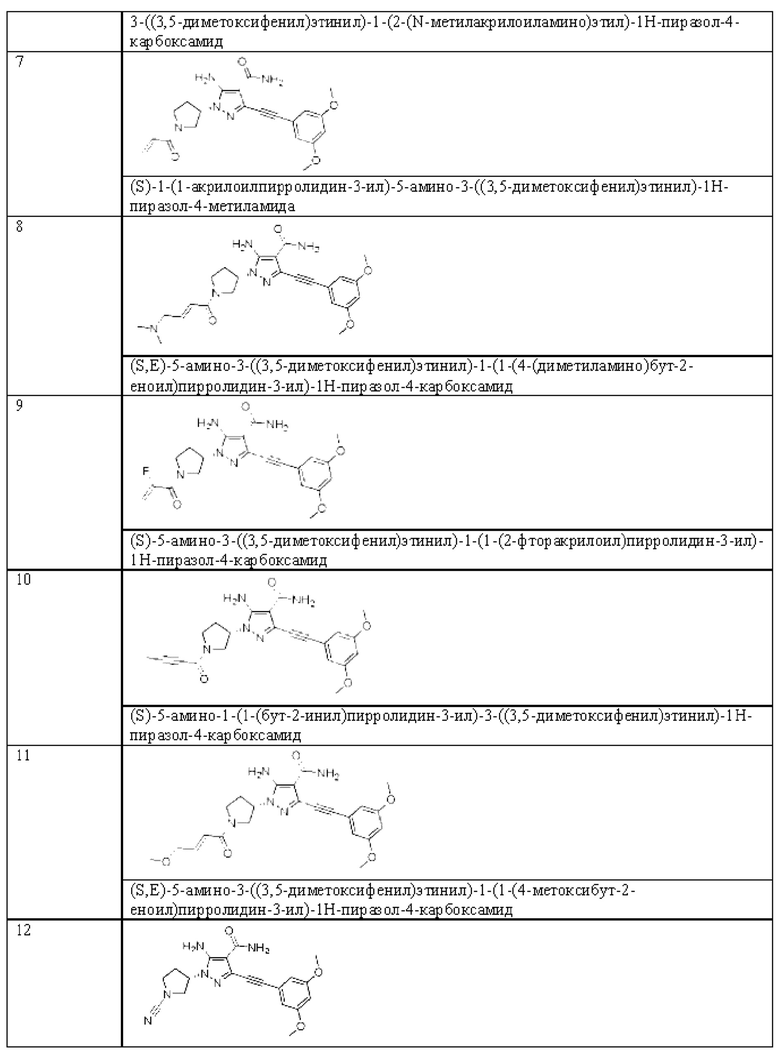
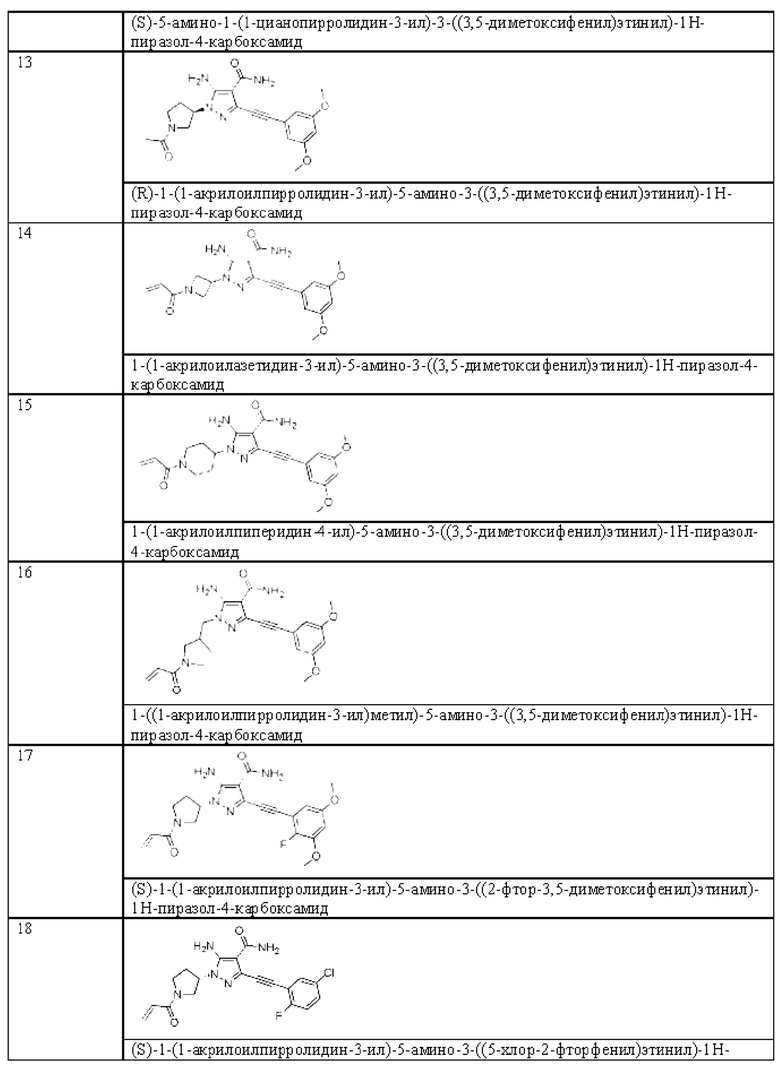
или его пролекарству, его стабильному изотопному производному, его фармацевтически приемлемой соли, его изомеру или их смеси.
Соединения по данному изобретению обладают существенным ингибирующим действием в отношении активности FGFR. Соединения по данному изобретению эффективны для ингибирования активности FG-FR1, FG-FR2, FG-FR3 или FG-FR4, предпочтительно имеют IC50 от 100 до 1000 нМ для ингибирования FG-FR1, FGFR2, FG-FR3 или FGFR4, более предпочтительно IC50 менее 100 нМ, наиболее предпочтительно IC50 менее 10 нМ. В частности, соединения по данному изобретению обладают существенным ингибирующим действием в отношении клеточной пролиферации опухолевых клеток (например, опухолевых клеток Нер3В, RT4 и SNU-16), предпочтительно имеют IC50 от 100 до 1000 нМ, более предпочтительно имеют IC50 менее 100 нМ, и наиболее предпочтительно имеют IC50 менее 10 нМ.
Таким образом, соединения по данному изобретению пригодны для лечения или предупреждения заболеваний, связанных с FGFR, включая, но не ограничиваясь этим, опухоли и воспалительные заболевания, такие как остеоартрит. Соединения по данному изобретению пригодны для лечения или предупреждения опухолей, связанных с FGFR, таких как немелкоклеточный рак легких, рак пищевода, меланома, рабдомиосаркома, почечно-клеточная карцинома, множественная миелома, рак молочной железы, рак яичника, эндометриальный рак, рак шейки матки, рак желудка, рак толстой кишки, рак мочевого пузыря, рак поджелудочной железы, рак легких, рак молочной железы, рак предстательной железы и рак печени (например, гепатоцеллюлярная карцинома), более конкретно, рак печени, рак желудка, немелкоклеточный рак легких и рак мочевого пузыря. Соответственно, в другом аспекте данного изобретения предложен способ лечения или предупреждения заболевания, опосредованного FGFR, такого как опухоль, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения по данному изобретению или его пролекарства, его стабильного изотопного производного, его фармацевтически приемлемой соли, его изомера или их смеси, или фармацевтической композиции, содержащей указанные соединения.
Другой аспект данного изобретения относится к применению соединения общей формулы (I) или его пролекарства, его стабильного изотопного производного, его фармацевтически приемлемой соли, его изомера или их смеси при получении лекарственного средства для лечения или предупреждения заболевания, опосредованного FGFR, такого как опухоль или воспалительное заболевание, включая, но не ограничиваясь этим, немелкоклеточный рак легких, рак пищевода, меланому, рабдомиосаркому, почечно-клеточный рак, множественную миелому, рак молочной железы, рак яичника, рак эндометрия, рак шейки матки, рак желудка, рак толстой кишки, рак мочевого пузыря, рак поджелудочной железы, рак легких, рак молочной железы, рак предстательной железы и рак печени.
Данное изобретение дополнительно относится к фармацевтической композиции, содержащей соединение по данному изобретению или его пролекарство, его стабильное изотопное производное, его фармацевтически приемлемую соль, его изомер или их смеси, и фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество.
Другой аспект данного изобретения относится к применению соединения общей формулы (I) или его пролекарства, стабильного изотопного производного, его фармацевтически приемлемой соли, его изомера или их смеси, или фармацевтической композиции для получения лекарственного средства, причем указанное лекарственное средство применяют для лечения или предупреждения заболевания, опосредованного FGFR, такого как опухоль и воспалительное заболевание.
В соответствии с данным изобретением, лекарственное средство может быть представлено в любой фармацевтической лекарственной форме, включая, но не ограничиваясь этим, таблетку, капсулу, раствор, лиофилизированный препарат и препарат для инъекции.
Фармацевтический препарат по данному изобретению можно вводить в форме единицы дозирования, содержащей определенное количество активного ингредиента на одну единицу дозирования. Такая единица может содержать, например, от 0,5 мг до 1 г, предпочтительно от 1 мг до 700 мг, особенно предпочтительно от 5 мг до 300 мг соединения по данному изобретению, в зависимости от заболевания, подлежащего лечению, способа введения, а также возраста, массы и состояния пациента, или фармацевтический препарат можно вводить в форме единиц дозирования, содержащих определенное количество активного ингредиента на одну единицу дозирования. Предпочтительные единичные лекарственные формы представляют собой формы, содержащие суточные или дробные дозы, указанные выше, или их соответствующие доли указанных доз активного ингредиента. Кроме того, фармацевтические препараты такого типа можно получать с применением способов, хорошо известных в области фармацевтики.
Фармацевтические препараты по данному изобретению можно адаптировать для введения любым пригодным способом, например, пероральным (включая буккальный или сублингвальный), ректальным, назальным, местным (включая буккальный, сублингвальный или трансдермальный), вагинальным или парентеральным (включая подкожный, внутримышечный, внутривенный или внутрикожный) способом введения. Лекарственные формы можно получать, например, посредством объединения активного ингредиента с одним или более вспомогательными веществами или одним или более адъювантами, с применением одного или более способов, известных в области фармацевтики.
Описание вариантов реализации изобретения
Если не указано иное, следующие термины, используемые в описании и формуле данной заявки, имеют следующие значения.
Выражение «Сх-у» в данном контексте означает диапазон количества атомов углерода, где оба х и у представляют собой целые числа, например, C3-8 циклоалкильная группа означает циклоалкильную группу, содержащую от 3 до 8 атомов углерода, то есть циклоалкильную группу, содержащую 3, 4, 5, 6, 7 или 8 атомов углерода. Следует понимать, что «С3-8» также включает любые поддиапазоны, входящие в него, такие как С3-7, С3-6, С4-7, С4-6, С5-6 и т.п.
«Алкил» относится к насыщенной неразветвленной линейной или разветвленной углеводородной группе, содержащей от 1 до 20 атомов углерода, например, от 1 до 18 атомов углерода, от 1 до 12 атомов углерода, от 1 до 8 атомов углерода, от 1 до 6 атомов углерода или от 1 до 4 атомов углерода. Неограничивающие примеры алкильных групп включают метил, этил, н-пропил, изо пропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил и 2-этилбутил. Алкильная группа может быть замещенной или незамещенной.
«Алкенил» относится к неразветвленной линейной или разветвленной углеводородной группе, содержащей по меньшей мере одну двойную углерод-углеродную связь и обычно от 2 до 20 атомов углерода, например, от 2 до 8 атомов углерод, от 2 до 6 атомов углерода или от 2 до 4 атомов углерода. Неограничивающие примеры алкенильных групп включают этенил, 1-пропенил, 2-пропенил, 1-бутенил, 2-бутенил, 3-бутенил, 2-метил-2-пропенил, 1,4-пентадиенил и 1,4-бутадиенил. Алкенильная группа может быть замещенной или незамещенной.
«Алкинил» относится к неразветвленной линейной или разветвленной углеводородной группе, содержащей по меньшей мере одну тройную углерод-углеродную связь и обычно от 2 до 20 атомов углерода, например, от 2 до 8 атомов углерод, от 2 до 6 атомов углерода или от 2 до 4 атомов углерода. Неограничивающие примеры алкинильных групп включают этинил, 2-пропинил, 2-пропинил, 1-бутинил, 2-бутинил и 3-бутинил. Алкинильная группа может быть замещенной или незамещенной.
«Циклоалкил» относится к насыщенной циклической углеводородной группе заместителя, содержащей от 3 до 14 циклических атомов углерода. Циклоалкильная группа может представлять собой одно углеродное кольцо и обычно содержит от 3 до 7 кольцевых атомов углерода. Неограничивающие примеры моноциклических циклоалкильных групп включают циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Альтернативно, циклоалкильная группа может содержать две или три кольцевые структуры, конденсированные друг с другом, например, декагидронафталин. Циклоалкильная группа может быть замещенной или незамещенной.
«Гетероциклическая или гетероциклильная группа» относится к насыщенной или частично ненасыщенной моноциклической или полициклической кольцевой группе, содержащей от 3 до 20 циклических атомов, например, от 3 до 16, от 3 до 14, от 3 до 12, от 3 до 10, от 3 до 8, от 3 до 6 или от 5 до 6 циклических атомов, причем один или более циклических атомов выбраны из группы, состоящей из азота, кислорода или S(O)m (где m представляет собой целое число от 0 до 2), но не содержащей кольцевой фрагмент -O-O-, -O-S- или -S-S-, и остальные циклические атомы представляют собой атомы углерода. Предпочтительно, она содержит от 3 до 12 циклических атомов, более предпочтительно от 3 до 10 циклических атомов, наиболее предпочтительно 5 или 6 циклических атомов, причем от 1 до 4 представляют собой гетероатомы, более предпочтительно от 1 до 3 представляют собой гетероатомы, наиболее предпочтительно от 1 до 2 представляют собой гетероатомы. Неограничивающие примеры моноциклических гетероциклических групп включают пирролидинил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил, оксеарил и азетидинил. Полициклические гетероциклические группы включают конденсированные, мостиковые или спирополициклические гетероциклические группы. Гетероциклическая или гетероциклильная группа может быть замещенной или незамещенной.
«Арил» относится к ароматической моноциклической или конденсированной полициклической группе, содержащей от 6 до 14 атомов углерода, предпочтительно от 6 до 10 членов, например, фенил и нафтил, наиболее предпочтительно фенил. Арильное кольцо может быть конденсировано с гетер о ар ильным, гетероциклильным или циклоалкильным кольцом, причем кольцо, к которому присоединена исходная структура, представляет собой арильное кольцо, неограничивающие примеры включают:
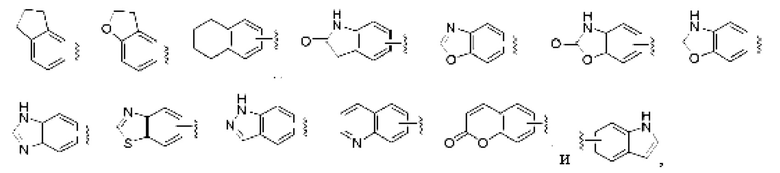
и арильная группа может быть замещенной или незамещенной.
«Гетероарил» или «гетероарильное кольцо» относится к гетероароматической системе, содержащей от 5 до 14 циклических атомов, причем от 1 до 4 циклических атомов выбраны из гетероатомов, включая кислород, серу и азот. Гетероарильная группа предпочтительно является 5-10-членной. Более предпочтительно, гетероарильная группа является 5- или 6-членной, такой как фурил, тиенил, пиридил, пирролил, N-алкилпрролил, пиримидинил, пиразинил, пиразолил, имидазолил, тетразолил, оксазолил, изоксазолил, тиазолил, изотиазолил и т.п. Гетероарильное кольцо может быть конденсировано с арильным, гетероциклическим или циклоалкильным кольцом, причем кольцо, к которому присоединена исходная структура, представляет собой гетероарильное кольцо, неограничивающие примеры включают:
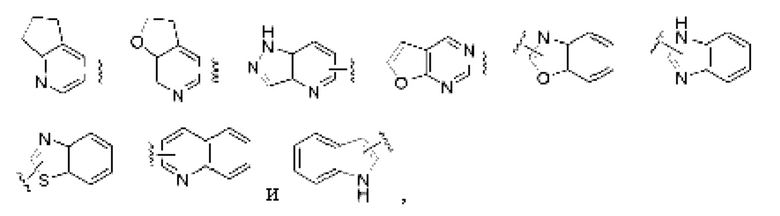
и гетероарильная группа может быть замещенной или незамещенной.
«Галоген» относится к фтору, хлору, брому или йоду.
«Циано» относится к -CN.
«Необязательный» или «необязательно» означает, что описанное далее явление или условие может иметь, но не обязательно имеет место, включая случаи, когда указанное явление или условие имеет или не имеет место. Например, «гетероциклическая группа, необязательно замещенная алкильная группой» означает, что указанная алкильная группа может быть, но не обязательно присутствует, и такое описание включает случай, в котором гетероциклическая группа замещена алкильной группой, и случай, в котором гетероциклическая группа не замещена алкильной группой.
«Замещенный» относится к одному или более атомам водорода в группе, предпочтительно к 5, более предпочтительно к 1-3 атомам водорода, которые независимо друг от друга замещены соответствующим количеством заместителей. Понятно, что группы заместителей находятся только в их возможных химических положениях, и специалисты в данной области техники могут без труда определить (экспериментально или теоретически) замещения, который могут быть или не быть возможными. Например, аминогруппа или гидроксильная группа, содержащая свободный атом водорода, может быть нестабильной при ассоциации с атомом углерода, имеющим ненасыщенную (например, олефиновую) связь. Такие группы заместителей включают, но не ограничиваются этим, гидроксил, амино, галоген, циано, С1-6 алкил, С1-6 алкокси, С2-6 алкенил, С2-6 алкинил, С3-8 циклоалкил и т.п.
«Фармацевтическая композиция» относится к композиции, содержащей одно или более соединений, описанных в данном документе, или его фармацевтически приемлемую соль, или пролекарство, а также другие компоненты, такие как фармацевтически приемлемые носители и вспомогательные вещества. Фармацевтическая композиция предназначена для облегчения введения в организм и для облегчения усвоения активного ингредиента, и, следовательно, проявления требуемой биологической активности.
«Изомер» относится к соединению, имеющему такую же молекулярную формулу, но другую природу или последовательность связывания атомов, или другое пространственное расположение атомов, которое называют «изомером». Изомер, имеющий другое пространственное расположение атомов, называют «стереоизомером». Стереоизомеры включают оптические изомеры, геометрические изомеры и конформационные изомеры.
Соединения по данному изобретению могут существовать в форме оптического изомера. Такие оптические изомеры находятся в «R» или «S» конфигурации в зависимости от расположения заместителей вокруг хирального атома углерода. Оптические изомеры включают энантиомеры и диастереомеры. Способы получения и выделения оптических изомеров известны в данной области техники.
Соединения по данному изобретению также могут существовать в форме геометрического изомера. Данное изобретение имеет различные геометрические изомеры и их смеси, образующиеся в результате различного распределения заместителей вокруг двойных углерод-углеродных связей, двойных углерод-азотных связей, циклоалкильных групп или гетероциклильных групп. Группы заместителей вокруг двойной углерод-углеродной связи или углерод-азотной связи обозначают как конфигурацию Z или Е, а группы заместителей вокруг циклоалкила или гетероцикла обозначают как цис- или трансконфигурацию.
Соединения по данному изобретению также могут проявлять таутомерию, такую как кето-енольная таутомерия.
Следует понимать, что данное изобретение включает любые таутомерные или стереомерные формы и их смеси, и оно не ограничено какой-либо одной из таутомерных или стереоизомерных форм, использованных в названии или химической структурной формуле соединения.
«Изотопы» представляют собой все изотопы атомов, встречающихся в соединениях по данному изобретению. Изотопы включают атомы, имеющие такой же атомный номер, но другое массовое число. Примеры изотопов, пригодных для внедрения в соединения по данному изобретению, представляют собой атомы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, например, но не ограничиваясь этим, 2Н, 3H, 13C, 14C, 15N, 18O, 17О, 31P, 32P, 35S, 18F и 36Cl. Соединения по данному изобретению с изотопной меткой, в целом, можно получать обычными техническими способами, известными специалистам в данной области техники, или способами, аналогичными тем, которые описаны в сопроводительных примерах, с применением соответствующих реагентов с изотопной меткой вместо реагентов без изотопной метки. Такие соединения имеют множество возможных областей применения, например, в качестве стандарта и реагента при определении биологической активности. В случае стабильных изотопов такие соединения могут преимущественно изменять биологические, фармакологические или фармакокинетические свойства.
«Пролекарство» относится к такому соединению по данному изобретению, которое можно вводить в форме пролекарства. Пролекарство представляет собой производное, которое может превращаться в биологически активное соединение по данному изобретению в физиологических условиях in vivo, например, в результате окисления, восстановления, гидролиза и т.д., каждый из которых протекает в присутствии фермента или без участия фермента. Примером пролекарства является соединение, в котором аминогруппа соединения по данному изобретению является ацилированной, алкилированной или фосфорилированной, например, эйкозиламино, аланиламино, пивалоилоксиметиламино, или в котором гидрокси-группа является ацилированной, алкилированной, фосфорилированной или превращенной в борат, например, ацетокси, пальмитоилокси, пивалоилокси, сукцинилокси, фумарилокси, аланилокси и т.п., или молекула-носитель, в которой карбоксильная группа эстерифицирована или амидирована, или соединение, в котором тиольная группа образует дисульфидный мостик с группой, обеспечивающей селективную доставку лекарства к мишени и/или в цитозоль клетки, например, пептид. Такие соединения можно получать из соединений по данному изобретению в соответствии с некоторыми известными способами.
«Фармацевтическая соль» или «фармацевтически приемлемая соль» относится к соли, полученной из фармацевтически приемлемого основания или кислоты, включая неорганические основания или кислоты и органические основания или кислоты; в том случае, если соединения по данному изобретению содержат одну или более кислотных или основных групп, данное изобретение также включает их соответствующие фармацевтически приемлемые соли. Таким образом, соединения по данному изобретению, содержащие кислотную группу, могут присутствовать в форме соли, и их можно использовать в соответствии с данным изобретением, например, в форме соли с щелочным металлом, в форме соли с щелочноземельным металлом или в форме аммониевой соли. Более конкретные примеры таких солей включают соли натрия, соли калия, соли кальция, соли магния или соли с аммиаком или органическими аминами, такими как этиламин, этаноламин, триэтаноламин или аминокислоты. Соединения по данному изобретению, содержащие основную группу, могут присутствовать в форме соли, и их можно использовать в соответствии с данным изобретением в форме их солей присоединения с неорганическими или органическими кислотами. Примеры пригодных кислот включают хлористоводородную кислоту, бромистоводородную кислоту, фосфорную кислоту, серную кислоту, азотную кислоту, метансульфоновую кислоту, п-толуолсульфоновую кислоту, нафталиндисульфоновую кислоту, щавелевую кислоту, уксусную кислоту, винную кислоту, молочную кислоту, салициловую кислоту, бензойную кислоту, муравьиную кислоту, пропионовую кислоту, пивалевую кислоту, малоновую кислоту, янтарную кислоту, пимелиновую кислоту, фумаровую кислоту, малеиновую кислоту, яблочную кислоту, сульфаминовую кислоту, фенилпропионовую кислоту, глюконовую кислоту, аскорбиновую кислоту, изоникотиновую кислоту, лимонную кислоту, адипиновую кислоту и другие кислоты, известные специалистам в данной области техники. Если соединение по данному изобретению содержит и кислотные, и основные группы в молекуле, то данное изобретение, помимо солевых форм, упомянутых выше, включает внутренние аммониевые соли. Каждую соль можно получать обычными способами, известными специалистам в данной области техники, например, посредством их приведения в контакт с органической или неорганической кислотой или основанием в растворителе или диспергаторе, или посредством анионного обмена или катионного обмена с другими солями.
Таким образом, при упоминании в данной заявке «соединения», «соединения по данному изобретению» или «соединений по данному изобретению» указанный термин включает также все такие формы соединения, такие как его пролекарства, его стабильные изотопные производные, его фармацевтически приемлемые соли, его изомеры, его мезомеры, его рацематы, его энантиомеры, его диастереомеры и их смеси.
В данном контексте термин «опухоль» включает доброкачественные опухоли и злокачественные опухоли (например, рак).
В данном контексте термин «рак» включает различные злокачественные опухоли, в которых участвует FGFR, включая, но не ограничиваясь этим, немелкоклеточный рак легких, рак пищевода, меланому, рабдомиосаркому, почечно-клеточную карциному, множественную миелому, рак молочной железы, рак яичника, эндометриальный рак, рак шейки матки, рак желудка, рак толстой кишки, рак мочевого пузыря, рак поджелудочной железы, рак легких, рак молочной железы, рак предстательной железы и рак печени (такой как гепатоцеллюлярная карцинома), более конкретно, рак печени, рак желудка, немелкоклеточный рак легких и рак мочевого пузыря.
В данном контексте термин «воспалительное заболевание» относится к любому воспалительному заболеванию, в котором FGFR участвует в возникновении воспаления, такому как остеоартрит.
В данном контексте термин «терапевтически эффективное количество» относится к количеству соединения по данному изобретению, которое является эффективным для подавления функции FGFR и/или для лечения или предупреждения заболевания.
Способы синтеза
В данном изобретении также предложены способы получения указанных соединений. Получение соединений общей формулы (I) по данному изобретению можно осуществлять в соответствии со следующими иллюстративными способами и вариантами реализации, но предложенные способы и варианты реализации никоим образом не следует толковать как ограничение данного изобретения. Соединения по данному изобретению также можно синтезировать другими методиками синтеза, известными специалистам в данной области техники, или можно использовать комбинацию способов, известных в данной области техники, и способов по данному изобретению. Продукт, получаемый на каждой стадии реакции, получают посредством технологий выделения, известных в данной области техники, включая, но не ограничиваясь этим, экстракцию, фильтрование, перегонку, кристаллизацию, хроматографическое разделение и т.п. Исходные вещества и химические реагенты, необходимые для синтеза, можно синтезировать обычным способом или приобрести в соответствии с литературными источниками (доступными при поиске из SciFinder).
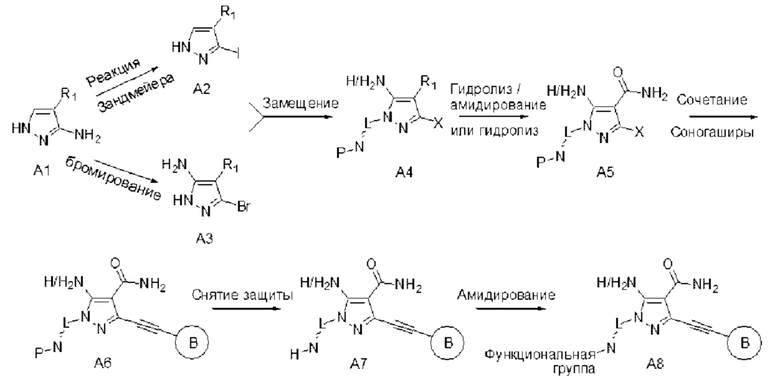
Пиразольное соединение общей формулы (I) по данному изобретению можно синтезировать по схеме, описанной в способе А: 1) исходное вещество А1 подвергают реакции Зандмейера с получением А2, или его можно бромировать с получением A3, где R1 может представлять собой -CN или сложный эфир (-COOR, где R представляет собой алкильную группу); 2) А2 или A3 и предшественник X-L~N-P (где X представляет собой уходящую группу, и L~N-P представляет собой функциональную группу, содержащую защищенную аминогруппу, Р представляет собой защитную группу для аминогруппы) подвергают реакции замещения, протекающей в условиях основного катализа, с получением А4, альтернативно, его и предшественника, содержащего гидроксильную группу (HO-L~N-P), можно подвергать несколько замедленной реакции (реакции Мицунобу) с получением А4; 3) если R1 в А4 представляет собой -CN, его подвергают гидролизу до амида А5 в условиях NaOH/Н2О2; если R1 в А4 представляет собой сложный эфир (-COOR, где R представляет собой алкильную группу), его сначала гидролизуют в основных условиях (таких как LiOH) до карбоновой кислоты, а затем подвергают амидированию с получением А5; 4) А5 и алкин связывают по реакции Соногаширы с получением А6; 5) с аминогруппы в А6 снимают защиту с получением А7; 6) аминогруппу в А7 подвергают дериватизацни с химическим реагентом (например, BrCN, акрилоилхлоридом и т.д.), содержащим функциональную группу, взаимодействующую с цистеиновым остатком в лиганд-связывающем домене киназы, с получением требуемого соединения А8.
Способ А:
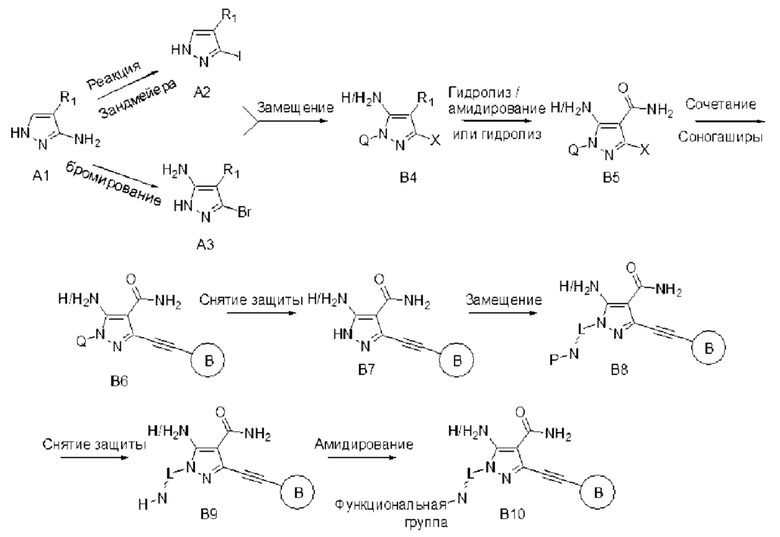
Альтернативно, его можно синтезировать по схеме, описанной в способе В. На второй стадии внедряют защитную группу Q для пиразольного NH, и на пятой стадии в результате снятия защиты получают общее промежуточное соединение В7, и затем пиразольный NH в В7 подвергают реакции с различными предшественниками с защищенной аминогруппой по реакции замещения, с которой затем снимают защиту и подвергают дериватизации с получением требуемого продукта А8.
Способ В:
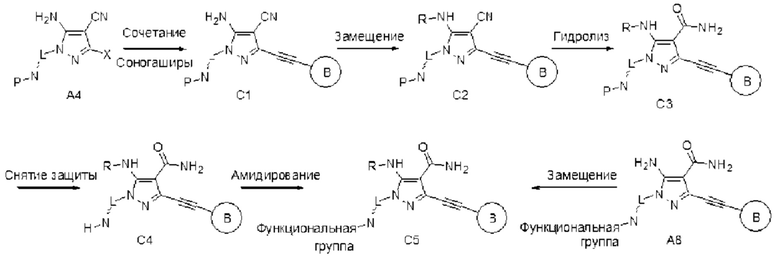
Пиразольное соединение общей формулы (I) по данному изобретению можно также синтезировать по схеме, описанной в способе С: 1) сначала А4 связывают с алкином по реакции Соногаширы с получением С1; 2) группу -NH2 в С1 замещают по реакции основного катализа или подвергают восстановительному аминированию с получением С2; 3) группу CN в С2 подвергают гидролизу до амида С3 в условиях NaOH/H2O2, в некоторых случаях ее сначала защищают группой Вос-NH-, а затем гидролизуют; наконец, снимают защиту и осуществляют дериватизацию с получением требуемого продукта С5; С5 также можно получать непосредственным замещением А8.
Способ С:
Примеры
Структуру соединения определяли ядерным магнитным резонансом (ЯМР) или масс-спектрометрией (МС). ЯМР записывали на ЯМР спектрометре Bruker AVANCE-400 или Varian Oxford-300, и в качестве растворителя использовали дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный хлороформ (CDCl3), дейтерированный метанол (CD3OD), и в качестве внутреннего стандарта использовали тетраметилсилан (TMS), химические сдвиги записывали в единицах 10-6 (м.д.).
МС записывали на масс-спектрометре Agilent SQD (ИЭР) (производитель: Agilent, модель: 6120).
Измерения ВЭЖХ проводили с помощью жидкостного хроматографа высокого давления Agilent 1200 DAD (колонка Sunfirc С18, 150×4,6 мм, 5 мкм) и жидкостного хроматографа высокого давления Waters 2695-2996 (колонка Gimini С18 150×4,6 мм, 5 мкм).
Для тонкослойной хроматографии на силикагелевых пластинах использовали силикагелевые пластины Qingdao Ocean GF254. Характеристики силикагелевой пластины, использованной для тонкослойной хроматографии (ТСХ): 0,15 мм - 0,2 мм. Характеристики для разделения и очистки методом тонкослойной хроматографии: силикагелевая пластина от 0,4 мм до 0,5 мм.
Для колоночной хроматографии в качестве носителя обычно использовали силикагель Qingdao Ocean с размером частиц 200-300 меш.
Известные исходные вещества для данного изобретения можно синтезировать способами, известными в данной области техники, или можно приобрести у ABCR GmbH & Co. KG, Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc., Beijing Coupling chemicals и других компаний.
В примерах по данному изобретению, если не указано иное, все реакции проводили в атмосфере аргона или в атмосфере азота.
Атмосфера аргона или атмосфера азота означает, что реакционную колбу подключали к баллону с аргоном или азотом, имеющему объем около 1 л.
Атмосфера водорода означает, что реакционную колбу подключали к баллону с водородом, имеющему объем около 1 л.
Реакции гидрирования под давлением проводили с применением генератора водорода высокой чистоты GCD-500G и гидрогенизатора среднего давления BLT-2000 производства компании Beijing Jiawei Kechuang Technology Co., Ltd.
Реакции гидрирования обычно проводили после вакуумирования, наполнения водородом и трехкратного повторения указанной последовательности.
Для реакций с микроволновым излучением использовали микроволновой реактор типа СЕМ Discover-SP.
В примерах по данному изобретению, если не указано иное, температура реакции была комнатной, и температурный диапазон составлял от 20°С до 30°С.
Ход реакции в приведенных примерах контролировали с помощью тонкослойной хроматографии (ТСХ), и система, использованная для реакции, представляла собой А: систему из дихлорметана и метанола; В: систему из петролейного эфира и этилацетата, объемное соотношение растворителей подбирали в зависимости от полярности соединения.
Система элюента для колоночной хроматографии, которую использовали в процессе очистки соединения, а также система проявляющего агента, которую использовали для проявления при проведении тонкослойной хроматографии, включала А: систему из дихлорметана и метанола; В: систему из петролейного эфира и этилацетата, объемное соотношение растворителей подбирали в зависимости от полярности соединения, и для коррекции можно добавлять небольшое количество триэтиламина и кислотного или основного реагента.
Пример 1
(S)-1-(1-Акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-карбоксамид
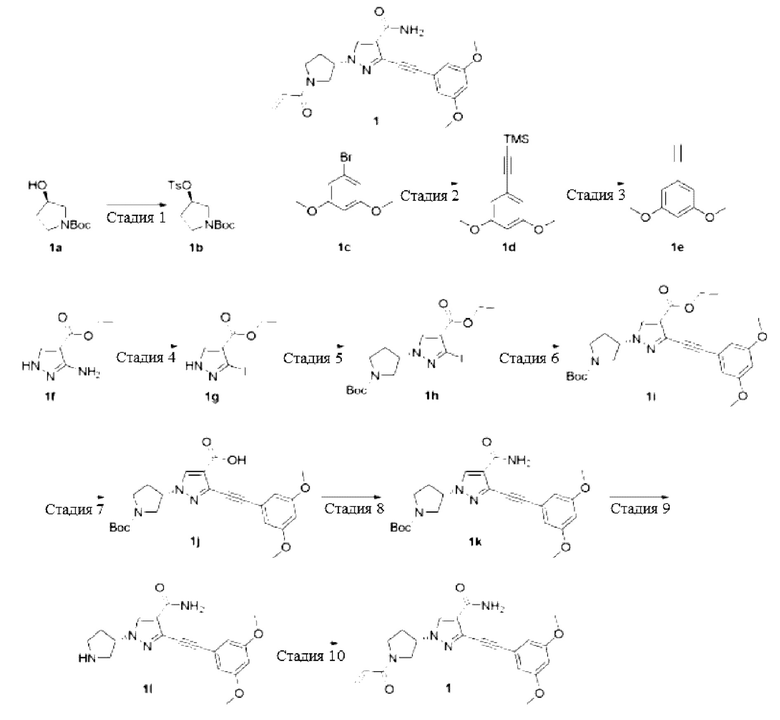
Стадия 1
Трет-бутиловый эфир (R)-3-(Толуолсульфонилокси)пирролидин-1-карбоновой кислоты
Соединение трет-бутиловый эфир (R)-3-гидроксипирролидин-1-карбоновой кислоты 1а (3,5 г, 18,7 ммоль), триэтиламин (5,25 мл, 37,9 ммоль), 4-диметиламинопиридин (0,35 г, 2,87 ммоль) растворяли в дихлорметане (50 мл), и затем добавляли п-толуолсульфонилхлорид (5,4 г, 28,1 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 12 часов, затем добавляли воду (50 мл) для разбавления и использовали этилацетат (100 мл × 3) для экстракции, полученные органические фазы объединяли и затем сушили безводным сульфатом натрия, затем удаляли осушающий агент посредством фильтрования и выпаривали растворитель при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (петролейный эфир / этилацетат = 2/1) с получением указанного в заголовке продукта, трет-бутилового эфира (R)-3-(толуолсульфонилокси)пирролидин-1-карбоновой кислоты 1b (6,0 г, маслянистое вещество желтого цвета), и выход составлял 94%.
МС m/z (ИЭР): 364 [М+23]
Стадия 2
((3,5-Диметоксифенил)этинил)триметилсилан
Смесь 1-бром-3,5-диметоксибензола 1с (6,51 г, 30 ммоль), триметилсилилацетилена (8,8 г, 90 ммоль), бис(трифенилфосфин)палладия хлорида (1,05 г, 1,5 ммоль), йодида меди (I) (0,56 г, 3,0 ммоль), триэтиламина (80 мл) и N,N-диметилформамида (150 мл) нагревали до 80°С и перемешивали в атмосфере азота в течение 12 часов; реакционную смесь охлаждали до комнатной температуры; концентрировали при пониженном давлении и очищали остаток колоночной хроматографией на силикагеле (петролейный эфир) с получением указанного в заголовке продукта, ((3,5-диметоксифенил)этинил)триметилсилана 1d (6,2 г, коричневое твердое вещество), и выход составлял 88%.
МС m/z (ИЭР): 235 [М+1]
Стадия 3
1-Этинил-3,5-диметоксибензол
((3,5-Диметоксифенил)этинил)триметилсилан 1d (3,0 г, 12,8 ммоль) растворяли в метаноле (100 мл) и добавляли карбонат калия (3,5 г, 25,6 ммоль), и перемешивали при комнатной температуре в течение 2 часов; фильтровали и концентрировали полученный фильтрат при пониженном давлении; остаток очищали колоночной хроматографией на силикагеле (петролейный эфир) с получением указанного в заголовке продукта, 1-этинил-3,5-диметоксибензола 1е (2 г, желтое твердое вещество), и выход составлял 96%.
Стадия 4
Этиловый эфир 3-Йод-1Н-пиразол-4-карбоновой кислоты
Этиловый эфир 3-Амино-1Н-пиразол-4-карбоновой кислоты 1f (4,7 г, 30,3 ммоль) растворяли в концентрированной хлористоводородной кислоте (12 М, 40 мл) и охлаждали до 0°С, добавляли раствор нитрита натрия (4,25 г, 60 ммоль) (7,5 мл) и затем перемешивали в течение 5 минут, затем медленно добавляли раствор йодида калия (12,5 г, 75 ммоль) (17,5 мл) и продолжали перемешивание в течение 30 минут, реакционную смесь выливали в насыщенный водный раствор тиосульфата натрия (200 мл) и экстрагировали этилацетатом (400 мл × 3), органические фазы объединяли и сушили безводным сульфатом натрия, затем удаляли осушающий агент посредством фильтрования и удаляли растворитель при пониженном давлении; остаток очищали колоночной хроматографией на силикагеле (петролейный эфир / этилацетат = 2/1) с получением указанного в заголовке продукта, этилового эфира 3-йод-1Н-пиразол-4-карбоновой кислоты 1g (6,4 г, бледно-желтое твердое вещество), и выход составлял 80%.
МС m/z (ИЭР): 267 [М+1]
Стадия 5
(S)-1-(1-(трет-Бутоксикарбонил)пирролидин-3-ил)-3-йод-1Н-пиразол-4-этилформиат Смесь этилового эфира 1-йод-1Н-пиразол-4-карбоновой кислоты 1 g (4,5 г, 17 ммоль), трет-бутилового эфира (R)-3-(толуолсульфонилокси)пирролидин-1-карбоновой кислоты 1b (6,1 г, 17,8 ммоль), карбоната цезия (7,5 г, 20,4 ммоль) и N,N-диметилформамида (50 мл) нагревали до 80°С и перемешивали в течение 3 часов, реакционную смесь охлаждали до комнатной температуры, и затем выливали в насыщенный раствор бикарбоната натрия (200 мл), который затем экстрагировали этилацетатом (300 мл × 3); органические фазы объединяли и сушили над безводным сульфатом натрия, затем отфильтровывали для удаления осушающего агента и удаляли растворитель при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (петролейный эфир / этил ацетат = от 5/1 до 2/1) с получением указанного в заголовке продукта, (S)-1-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-3-йод-1Н-пиразол-4-этилформиата 1Н (3,1 г, бледно-желтое твердое вещество), и выход составлял 42%. МС m/z (ИЭР): 458 [М+23]
Стадия 6
(S)-1-(1-(трет-Бутоксикарбонил)пирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-этилформиат
Смесь (S)-1-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-3-йод-1Н-пиразол-4-этилформиата 1h (1 г, 2,25 ммоль), 1-ацетилен-3,5-диметоксибензола 1е (0,75 г, 4,5 ммоль), бис(трифенилфосфин)палладия хлорида (175 мг, 0,25 ммоль), йодида меди (I) (95 мг, 0,5 ммоль), триэтиламина (12,5 мл) и N,N-диметилформамида (12,5 мл) нагревали до 80°С и перемешивали в течение 12 часов, реакционную смесь охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (петролейный эфир / этилацетат = 2/1) с получением указанного в заголовке продукта, (S)-1-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-этилформиата (0,95 г, желтое маслянистое вещество), и выход составлял 90%.
МС m/z (ИЭР): 414 [М+1-56]
Стадия 7
(S)-1-(1-(трет-Бутоксикарбонил)пирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-муравьиная кислота
(S)-1-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-этилформиат 1i (0,30 г, 0,64 ммоль) растворяли в тетрагидрофуране (3 мл) и добавляли раствор гидроксида натрия (4 М, 2 мл), и перемешивали при комнатной температуре в течение 1 часа, реакционную смесь концентрировали при пониженном давлении и подкисляли остаток хлористоводородной кислотой (6 М, 1 мл), и затем экстрагировали этилацетатом (10 мл × 3), органические фазы объединяли и сушили над безводным сульфатом натрия, и осушающий агент удаляли посредством фильтрования, растворитель удаляли при пониженном давлении с получением указанного в заголовке продукта, (S)-1-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-формиата 1j (200 мг, светло-желтое маслянистое вещество), и выход составлял 71%.
МС m/z (ИЭР): 386 [М+1-56]
Стадия 8
(S)-3-(4-Карбамоил-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир
Смесь (S)-1-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-формиата 1j (220 мг, 0,5 ммоль), хлорида аммония (270 мг, 5 ммоль), O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата (HATU) (228 мг, 0,6 ммоль), N,N-диизопропилэтиламина (129 мг, 1 ммоль) и N,N-диметилформамида (5 мл) перемешивали при комнатной температуре в течение ночи, и затем разбавляли водой и экстрагировали этилацетатом, органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования, растворитель удаляли при пониженном давлении, остаток очищали препаративной хроматографией на тонком слое силикагеля (дихлорметан / метанол = 20/1) с получением указанного в заголовке продукта, (S)-3-(4-карбамоил-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 1k (140 мг, белое твердое вещество), и выход составлял 64%.
МС m/z (ИЭР): 385 [М+1-56]
Стадия 9
(S)-3-((3,5-Диметоксифенил)этинил)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамид
Смесь (S)-3-(4-карбамоил-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 1k (50 мг, 0,11 ммоль), хлористоводородной кислоты (6 М, 5 мл) и диоксана (5 мл) перемешивали при комнатной температуре в течение 1 часа, и затем удаляли растворитель при пониженном давлении с получением указанного в заголовке продукта, (S)-3-((3,5-диметоксифенил)этинил)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамида 11 (42 мг, гидрохлоридная соль, неочищенный продукт), и выход составлял 100%.
МС m/z (ИЭР): 341 [М+1]
Стадия 10
(S)-1-(1-Акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-карбоксамид
К смеси (S)-3-((3,5-диметоксифенил)этинил)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамида гидрохлорида 11 (30 мг, 0,08 ммоль), N,N-диизопропилэтиламина (31 мг, 0,24 ммоль) и тетрагидрофурана (15 мл) по каплям добавляли раствор акрилоилхлорида (11 мг, 0,12 ммоль) в тетрагидрофуране (5 мл), реакционную смесь перемешивали при комнатной температуре в течение 30 минут, и затем гасили водой (30 мл), экстрагировали этилацетатом, органические фазы объединяли и сушили над безводным сульфатом натрия, и затем фильтровали для удаления осушающего агента, растворитель удаляли при пониженном давлении, остаток очищали препаративной хроматографией на тонком слое силикагеля (дихлорметан / метанол = 20/1) с получением указанного в заголовке продукта, (S)-1-(1-акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-карбоксамида 1 (15 мг, белое твердое вещество), и выход составлял 50%.
МС m/z (ИЭР): 395 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8,10 (д, J=9,8 Гц, 1H), 6,96 (шс, 1H), 6,71 (д, J=2,3 Гц, 2Н), 6,54-6,52 (м, 1H), 6,46-6,39 (м, 2Н), 5,80 (шс, 1H), 5,76-5,72 (м, 1H), 5,01-4,92 (м, 1Н), 4,13-4,00 (м, 2Н), 3,90-3,75 (м, 8Н), 2,62-2,44 (м, 2Н).
Пример 2
1-(1-Акрилоилпиперидин-4-ил)-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-карбоксамид
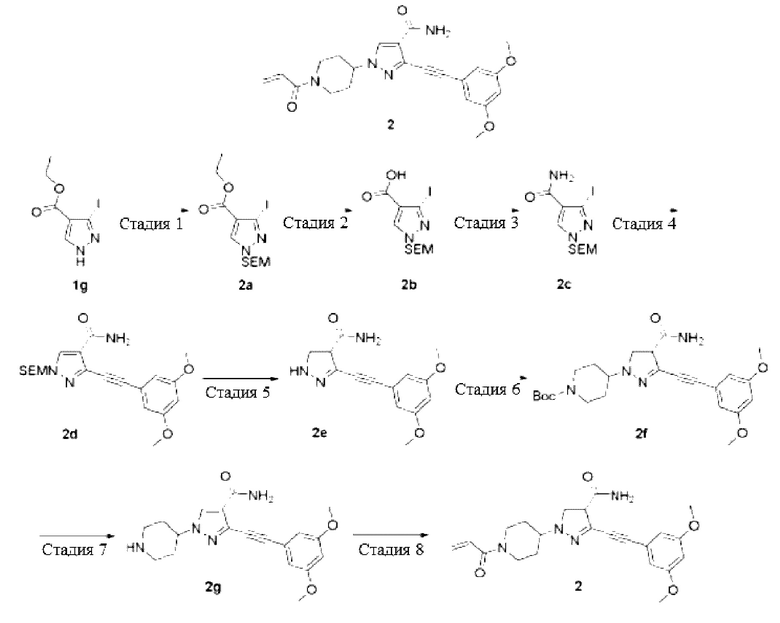
Стадия 1
3-Йод-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-этилформиат
Соединение 3-йод-1Н-пиразол-4-этилформиат 1g (2,01 г, 7,5 ммоль) растворяли в тетрагидрофуране (80 мл) и охлаждали до 0°С, добавляли гидрид натрия (60% дисперсия в минеральном масле, 0,42 г, 10,5 ммоль), перемешивали при комнатной температуре в течение 1 часа и добавляли к реакционной смеси 2-(триметилсилил)этоксиметилхлорид (1,76 г, 10,5 ммоль), продолжали перемешивание в течение 15 часов, и затем к реакционной смеси добавляли насыщенный солевой раствор (100 мл), затем смесь экстрагировали этилацетатом (150 мл × 2), органические фазы объединяли и промывали насыщенным солевым раствором (100 мл), растворитель удаляли при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (петролейный эфир / этилацетат = от 5/1 до 1/2) с получением указанного в заголовке продукта, 3-йод-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-этилформиата (2,6 г, бесцветное маслянистое вещество), и выход составлял 87%.
МС m/z (ИЭР): 397 [M+1]
Стадия 2
3-Йод-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-карбоновая кислота
Соединение 3-йод-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-этилформиат 2а (2,6 г, 6,5 ммоль) растворяли в тетрагидрофуране (40 мл), добавляли водный раствор гидроксида лития (1 М, 13 мл) и перемешивали при комнатной температуре в течение 15 часов, затем разбавляли водой (20 мл) и подкисляли до рН=4-5 хлористоводородной кислотой (1 М), затем экстрагировали этилацетатом (50 мл × 3), органические фазы объединяли и промывали насыщенным солевым раствором (100 мл), растворитель удаляли при пониженном давлении, и затем сушили с получением указанного в заголовке продукта, 3-йод-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-карбоновой кислоты 2b (2,03 г, белое твердое вещество), и выход составлял 85%.
МС m/z (ИЭР): 391[М+23]
Стадия 3
3-Йод-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-карбоксамид
Соединение 3-йод-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-карбоновую кислоту 2b (2,03 г, 5,5 ммоль), диизопропилэтиламин (2,13 г, 16,5 ммоль) и N,N-диметилформамид (20 мл) смешивали, затем последовательно добавляли O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (HATU) (2,5 г, 6,6 ммоль) и 1-гидроксибензотриазол (890 мг, 6,6 ммоль), после перемешивания при комнатной температуре в течение 1 часа добавляли твердый хлорид аммония (1,47 г, 27,5 ммоль), продолжали перемешивание в течение 15 часов и затем к реакционной смеси добавляли насыщенный солевой раствор (30 мл), экстрагировали этилацетатом (50 мл × 3), и органические фазы объединяли и промывали насыщенным солевым раствором (100 мл); после удаления растворителя при пониженном давлении остаток очищали колоночной хроматографией на силикагеле (дихлорметан / метанол = 20/1) с получением указанного в заголовке продукта, 3-йод-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-карбоксамида 2 с (2,3 г, желтое маслянистое вещество), и выход составлял 100%.
MC m/z (ИЭР): 368[М+1]
Стадия 4
3-((3,5-Диметоксифенил)этинил)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-карбоксамид
Соединения 3-йод-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-карбоксамид 2 с (2,7 г, 7,3 ммоль), 1-этинил-3,5-диметоксибензол (1,78 г, 11 ммоль), триэтиламин (2,2 г, 21,9 ммоль), бис(трифенилфосфин)палладия хлорид (512 мг, 0,73 ммоль) и безводный тетрагидрофуран (70 мл) смешивали и затем удаляли кислород, и перемешивали при комнатной температуре в течение 15 часов в атмосфере аргона, затем удаляли растворитель при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (этилацетат / петролейный эфир = от 10/1 до 2/1) с получением указанного в заголовке продукта, 3-((3,5-диметоксифенил)этинил)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-карбоксамида 2d (1,5 г, желтое твердое вещество), и выход составлял 51%.
MC m/z (ИЭР): 402[М+1]
Стадия 5
3-((3,5-Диметоксифенил)этинил)-1Н-пиразол-4-карбоксамид
3-((3,5-Диметоксифенил)этинил)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-карбоксамид 2d (1,4 г, 3,5 ммоль), этилендиамин (525 мг, 8,75 ммоль) и тетрагидрофуран (30 мл) смешивали и затем добавляли раствор фторида тетрабутиламмония в тетрагидрофуране (1 М, 17,5 мл, 17,5 ммоль), после нагревания до кипения с обратным холодильником в течение 15 часов смесь охлаждали до комнатной температуры и добавляли насыщенный солевой раствор (20 мл) и экстрагировали этилацетатом (100 мл × 3), полученные органические фазы объединяли и сушили над безводным сульфатом натрия, затем удаляли осушающий агент посредством фильтрования и удаляли растворитель при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (дихлорметан / метанол = 20/1) с получением указанного в заголовке продукта, 3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-карбоксамида 2е (600 мг, белое твердое вещество), и выход составлял 63%.
MC m/z (ИЭР): 272[М+1]
Стадия 6
Трет-бутиловый эфир 4-(4-карбамоил-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пиперидин-1-карбоновой кислоты
Соединения 3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-карбоксамид 2е (180 мг, 0,66 ммоль), 4-бромпиперидин-1-карбоновой кислоты трет-бутиловый эфир (264 мг, 0,99 ммоль), карбонат калия (182 мг, 1,32 ммоль) и N,N-диметилформамид (10 мл) смешивали и затем нагревали до 75°С, и перемешивали в течение 15 часов, затем добавляли воду, полученный раствор экстрагировали этилацетатом (50 мл × 3), органические фазы объединяли и промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования, и удаляли растворитель при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (дихлорметан/метанол = 20/1) с получением указанного в заголовке продукта, трет-бутилового эфира 4-(4-карбамоил-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пиперидин-1-карбоновой кислоты 2f (120 мг, желтое твердое вещество, содержащее региоизомер), и выход составлял 40%.
МС m/z (ИЭР): 477[М+23]
Стадия 7
3-((3,5-Диметоксифенил)этинил)-1-(пиперидин-4-ил)-1Н-пиразол-4-карбоксамид
Соединение 4-(4-карбамоил-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пиперидин-1-карбоновой кислоты трет-бутиловый эфир 2f (120 мг, 0,26 ммоль, смесь) растворяли в этаноле (20 мл) и затем добавляли хлороводород в этаноле (4 М, 1 мл, 4 ммоль), перемешивали при комнатной температуре в течение 15 часов, растворитель удаляли при пониженном давлении, после растворения остатка в метаноле (20 мл) доводили раствор до рН=8-9 с помощью насыщенного раствора бикарбоната натрия, затем растворитель удаляли при пониженном давлении и очищали остаток колоночной хроматографией на силикагеле (дихлорметан / метанол = 10/1) с получением указанного в заголовке продукта, 3-((3,5-диметоксифенил)этинил)-1-(пиперидин-4-ил)-1Н-пиразол-4-карбоксамида 2g (25 мг, белое твердое вещество), и выход составлял 27%.
МС m/z (ИЭР): 355[М+1]
Стадия 8
1-(1-Акрилоилпиперидин-4-ил)-3-((3,5-диметоксифенил)этинил)-1Н-пиразола-карбоксамид
Соединение 3-((3,5-диметоксифенил)этинил)-1-(пиперидин-4-ил)-1H-пиразол-4-карбоксамид 2g (25 мг, 0,07 ммоль), алкенпропионилхлорид (10 мг, 0,11 ммоль), твердый гидрокарбонат натрия (18 мг, 0,21 ммоль), воду (2 мл) и тетрагидрофуран (10 мл) смешивали при 0°С и перемешивали при указанной температуре в течение 10 часов, затем экстрагировали этилацетатом (20 мл × 3), органические фазы объединяли и сушили над безводным сульфатом натрия, затем удаляли осушающий агент посредством фильтрования и удаляли растворитель при пониженном давлении, затем остаток очищали колоночной хроматографией на силикагеле (дихлорметан / метанол = 10/1) с получением указанного в заголовке продукта, 1-(1-акрилоилпиперидин-4-ил)-3-((3,5-диметоксифенил)этинил)-1H-пиразол-4-карбоксамида 2 (17 мг, белое твердое вещество), и выход составлял 60%.
MCm/z (ИЭР): 409 [М+1]
1H ЯМР (400 МГц, CDCl3) δ 8,10 (с, 1H), 7,01 (шс, 1H), 6,72 (д, J=2,2 Гц, 2Н), 6,62 (дд, J=16,8, 10,6 Гц, 1Н), 6,55 (т, J=2,2 Гц, 1Н), 6,33 (дд, J=16,8, 1,5 Гц, 1Н), 5,80 (шс, 1Н), 5,76 (дд, J=10,6, 1,6 Гц, 1Н), 4,81 (шс, 1Н), 4,40 (т, J=11,4 Гц, 1Н), 4,18 (шс, 1Н), 3,82 (с, 6Н), 3,26 (шс, 1H), 2,89 (шс, 1Н), 2,42-2,25 (м, 2Н), 2,08-2,00 (м, 2Н).
Примеры 3-6 получали в соответствии со способом, предложенным в Примере 2:
Пример 3
1-(1-Акрилоилазетидин-3-ил)-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-карбоксамид
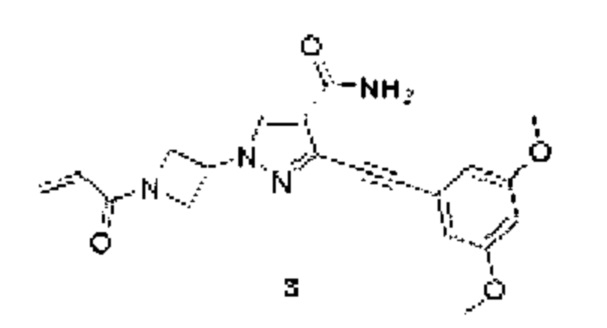
MCm/z (ИЭР): 381 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 8,43 (с, 1H), 7,30 (с, 2Н), 6,73 (д, J=2,2 Гц, 2Н), 6,60 (т, J=2,2 Гц, 1Н), 6,38 (дд, J=17,0, 10,3 Гц, 1H), 6,16 (дд, J=17,0, 2,1 Гц, 1H), 5,73 (дд, J=10,3, 2,1 Гц, 1H), 5,41-5,28 (м, 1H), 4,71 (т, J=8,6 Гц, 1H), 4,50 (дд, J=9,2, 4,9 Гц, 1H), 4,46-4,36 (м, 1Н), 4,20 (дд, J=10,7, 4,8 Гц, 1H), 3,78 (с, 6Н).
Пример 4
1-((1-Акрилоилпиперидин-4-ил)метил)-3-((3,5-диметоксифенил)этинил)-1H-пиразол-4-карбоксамид
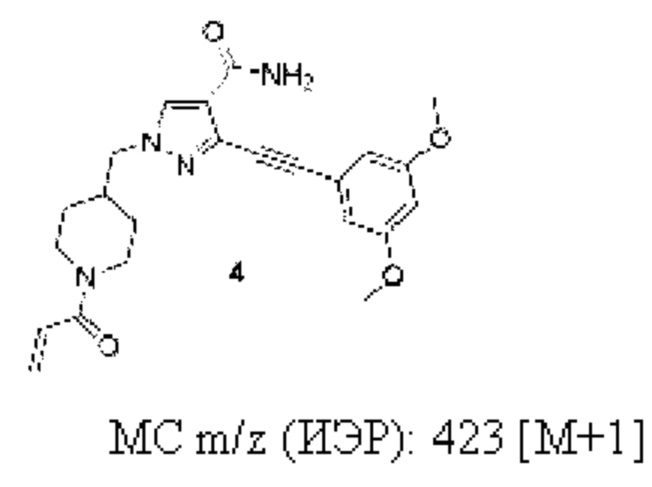
1Н ЯМР (400 МГц, CDCl3) δ 8,00 (с, 1H), 6,98 (шс, 1H), 6,72 (с, 2Н), 6,61-6,54 (м, 2Н), 6,28 (д, J=16,8 Гц, 1H), 5,87 (шс, 1Н), 5,70 (д, J=10,5 Гц, 1H), 4,72 (шс, 1H), 4,04 (шс, 3Н), 3,82 (с, 6Н), 3,05 (шс, 1H), 2,64 (шс, 1H), 2,27 (шс, 1H), 1,69 (шс, 2Н), 1,24 (шс, 2Н).
Пример 5
1-(4-Акрилоиламиноциклогексил)-3-((3,5-диметоксифенил)этинил)-1H-пиразол-4-карбоксамид
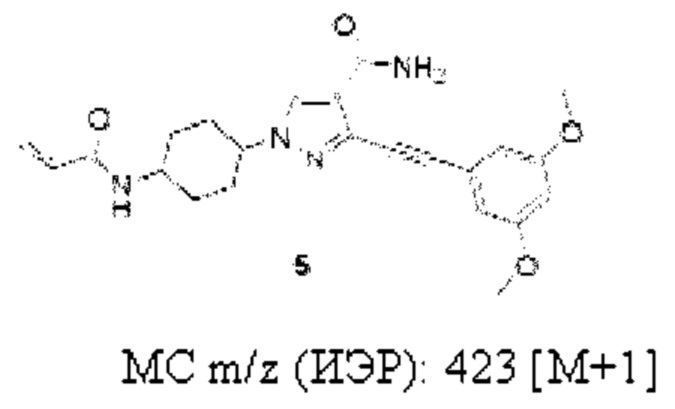
1H ЯМР (400 МГц, CD3OD) δ 8,25 (с, 1Н), 6,77 (д, J=2,3 Гц, 2Н), 6,58 (т, J=2,3 Гц, 1H), 6,38 (дд, J=17,1, 10,0 Гц, 1Н), 6,26 (дд, J=17,1, 2,0 Гц, 1H), 5,68 (дд, J=10,1, 2,0 Гц, 1Н), 4,38-4,33 (м, 1H), 4,13-4,11 (м, 1H), 3,82 (с, 6H), 2,28-2,18 (м, 2Н), 2,07-2,02 (м, 2Н), 1,96-1,80 (м, 4Н).
Пример 6
3-((3,5-Диметоксифенил)этинил)-1-(2-(N-метнлакрнлонламнно)этил)-1Н-пиразол-4-карбоксамид
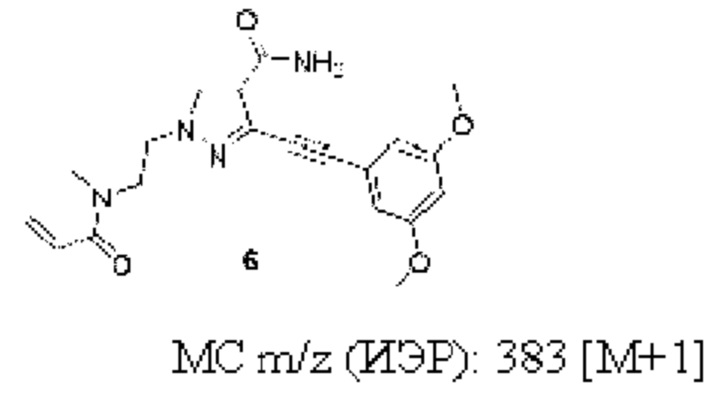
1Н ЯМР (300 МГц, ДМСО-d6) δ 8,24 (с, 1Н), 7,10-6,90 (м, 2Н), 6,76 (с, 2Н), 6,69-6,54 (м, 2Н), 6,07 (д, J=16,5 Гц, 1Н), 5,64 (д, J=9,8 Гц, 1Н), 4,37 (т, J=5,7 Гц, 2Н), 3,89-3,80 (м, 8Н), 2,94 (с, 3Н).
Пример 7
(S)-1-(1-Акрилоилпирролидин-3-ил)-5-амино-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-карбоксамид
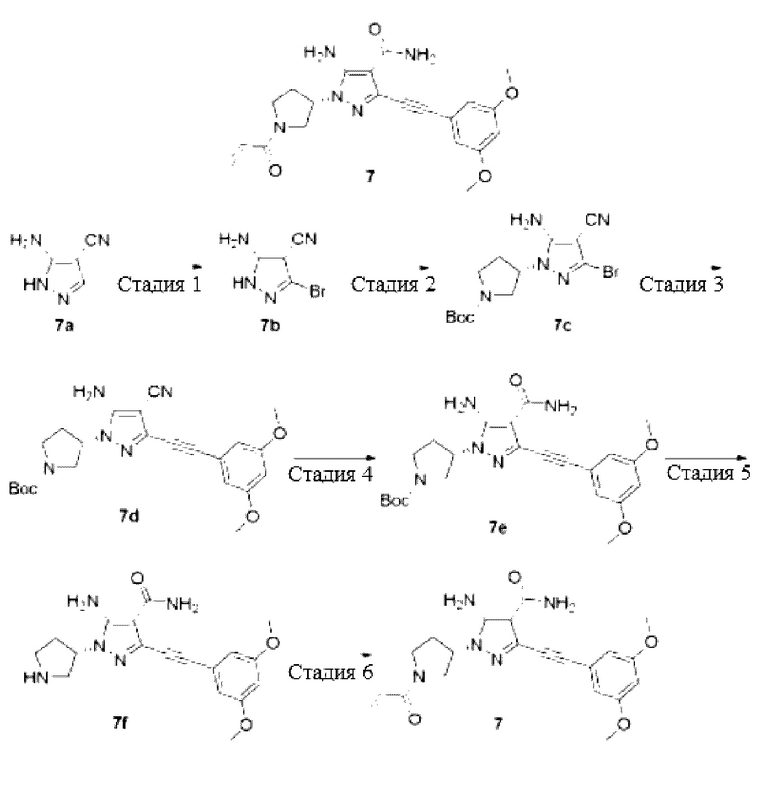
Стадия 1
5-Амино-3-бром-1H-пиразол-4-карбонитрил
Соединение 5-амино-1Н-пиразол-4-карбонитрил 7а (20 г, 185 ммоль) растворяли в N,N-диметилформамиде (200 мл) и охлаждали до 0°С, затем по частям добавляли N-бромсукцинимид (34 г, 190 ммоль), температуру повышали до комнатной температуры и перемешивали в течение 2 часов, затем реакционный раствор выливали в раствор сульфита натрия, экстрагировали этилацетатом (200 мл×3) и затем объединяли фазы и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования, и концентрировали реакционную систему при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (дихлорметан/метанол = 20/1) с получением указанного в заголовке продукта, 5-амино-3-бром-1Н-пиразол-4-карбонитрила 7b (32 г, желтое твердое вещество), и выход составлял 93%.
МС m/z (ИЭР): 187/189 [М+1]
Стадия 2
(S)-3-(5-Амино-3-бром-4-циано-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир
Смесь 5-амино-3-бром-1Н-пиразол-4-карбонитрила 7b (10 г, 53,8 ммоль), 3-(толуолсульфонилокси)пирролидин-1-карбоновой кислоты трет-бутилового эфира (22 г, 64,5 ммоль), карбоната цезия (58 г, 107,6 ммоль) и ацетонитрила (250 мл) нагревали до 90°C и проводили реакцию в течение 4 часов, и затем смесь охлаждали до комнатной температуры, фильтровали и промывали полученный осадок на фильтре дихлорметаном, фильтраты объединяли и концентрировали при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 5/1) с получением указанного в заголовке продукта, (S)-3-(5-амино-3-бром-4-циано-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 7 с (5 г, желтое маслянистое вещество), и выход составлял 26%.
МС m/z (ИЭР): 300/302 [М+1-56]
Стадия 3
(S)-3-(5-Амино-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир
Смесь (S)-3-(5-амино-3-бром-4-циано-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 7 с (5 г, 14,1 ммоль), йодида меди(I) (0,6 г, 2,8 ммоль), триэтиламина (9 мл), [1,1'-бис(дифенилфосфино)ферроцен]палладия дихлорида (2 г, 2,8 ммоль) и N,N-диметилформамида (150 мл) нагревали до 80°С в атмосфере аргона, и затем по частям добавляли 1-этинил-3,5-диметоксибензол (14 г, 84,5 ммоль), затем перемешивали в течение 2 часов, и затем охлаждали до комнатной температуры, выливали реакционный раствор в воду, экстрагировали этилацетатом (200 мл×3); затем органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования и концентрировали реакционную систему при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 5/1) с получением указанного в заголовке продукта, (S)-3-(5-амино-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 7d (5 г, коричневое маслянистое вещество), и выход составлял 81%.
MC m/z (ИЭР): 382 [M+1-56]
Стадия 4
(S)-3-(5-амино-4-карбамоил-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир
Смесь (S)-3-(5-амино-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 7d (5 г, 11,4 ммоль), гидроксида натрия (1,5 г, 37,5 ммоль, растворенного в 2 мл воды), этанола (50 мл) и диметилсульфоксида (10 мл) охлаждали до 0°C, добавляли пероксид водорода (20 мл), перемешивали при комнатной температуре в течение 2 часов, затем реакционный раствор выливали в раствор сульфита натрия, экстрагировали этилацетатом (100 мл×3), и органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования, и концентрировали реакционную систему при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 1/1) с получением указанного в заголовке продукта, (S)-3-(5-амино-4-карбамоил-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира, и выход составлял 96%.
MC m/z (ИЭР): 400 [М+1-56]
Стадия 5
(S)-5-Амино-3-((3,5-диметоксифенил)этинил)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамид
Соединение (S)-3-(5-амино-4-карбамоил-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир 7е (5 г, 11 ммоль) растворяли в дихлорметане (100 мл), затем добавляли трифторуксусную кислоту (15 мл), и затем перемешивали при комнатной температуре в течение 2 часов, и затем концентрировали при пониженном давлении с получением указанного в заголовке продукта, (S)-5-амино-3-((3,5-диметоксифенил)этинил)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамида 7f (7,1 г, коричневое маслянистое вещество, трифторацетат, неочищенный), и выход составлял >100%, продукт использовали для следующей реакции без очистки.
MC m/z (ИЭР): 356 [М+1]
Стадия 6
(S)-1-(1-акрилоилпирролидин-3-ил)-5-амино-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-карбоксамид
Соединение (S)-5-амино-3-((3,5-диметоксифенил)этинил)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамид 7f (7,1 г, 11 ммоль, трифторацетат, неочищенный) растворяли в тетрагидрофуране (50 мл) и охлаждали до 0°С, последовательно добавляли насыщенный раствор бикрабоната натрия (20 мл) и акрилоилхлорид (900 мг, 10 ммоль), перемешивали в течение 30 минут, затем реакционный раствор выливали в воду (100 мл) и экстрагировали дихлорметаном (100 мл×3), органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования и концентрировали реакционную систему при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 1/2) с получением указанного в заголовке продукта, (S)-1-(1-акрилоилпирролидин-3-ил)-5-амино-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-карбоксамида 7 (1,9 г, белое твердое вещество), и выход составлял 42%.
МС m/z(ИЭР): 410 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 7,18 (шс, 1H), 6,75 (д, J=2,3 Гц, 2Н), 6,69-6,55 (м, 3Н), 6,20-6,14 (м, 1Н), 5,72-5,67 (м, 1Н), 5,03-4,91 (м, 1H), 4,01-3,96 (м, 1Н), 3,84-3,70 (м, 7Н), 3,66-3,60 (м, 1Н), 3,55-3,48 (м, 1H), 2,36-2,21 (м, 2Н).
Пример 8
(S,E)-5-Амино-3-((3,5-диметоксифенил)этинил)-1-(1-(4-(диметиламино)бут-2-еноил)пирролидин-3-ил)-1Н-пиразол-4-карбоксамид
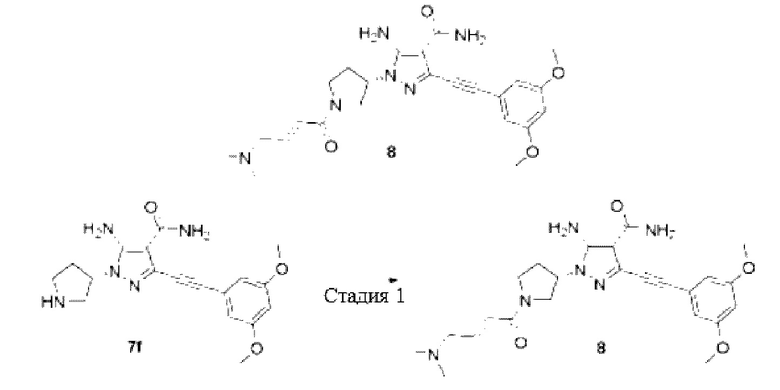
Стадия 1
(S,E)-5-Амино-3-((3,5-диметоксифенил)этинил)-1-(1-(4-(диметиламино)бут-2-енойл)пирролидин-3-ил)-1Н-пиразол-4-карбоксамид
Смесь (Е)-4-(диметиламино)бут-2-еновой кислоты (23 мг, 0,14 ммоль), O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилмочевины гексафторфосфата (HATU) (64 мг, 0,17 ммоль), (S)-5-амино-3-((3,5-диметоксифенил)этинил)-1-пирролидин-3-ил)-1Н-пиразол-4-карбоксамида 7f (50 мг, 0,14 ммоль), N,N-диизопропилэтиламина (2 мл) и дихлорметана (3 мл) перемешивали при комнатной температуре в течение 1 часа, реакционный раствор выливали в воду и экстрагировали дихлорметаном (20 мл×3), органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования и концентрировали реакционную систему при пониженном давлении, остаток очищали высокоэффективной жидкостной хроматографией с получением указанного в заголовке продукта, (S,E)-5-амино-3-((3,5-диметоксифенил)этинил)-1-(1-(4-(диметиламино)бут-2-еноил)пирролидин-3-ил)-1Н-пиразол-4-карбоксамида 8 (2,4 мг, белое твердое вещество, формиат), и выход составлял 4%.
МС m/z (ИЭР): 467 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 8,27 (шс, 1H), 7,20 (шс, 1H), 6,75 (д, J=2,3 Гц, 2Н), 6,70-6,61 (м, 3Н), 6,44-6,35 (м, 1Н), 5,01-4,93 (м, 1H), 4,01-3,93 (м, 1H), 3,77 (с, 6Н), 3,74-3,64 (м, 3Н), 3,06-3,03 (м, 2Н), 2,38-2,24 (м, 2Н), 2,17-2,15 (м, 6Н).
Пример 9
(S)-5-амино-3-((3,5-диметоксифенил)этинил)-1-(1-(2-фторакрилоил)пирролидин-3-ил)-1Н-пиразол-4-карбоксамид
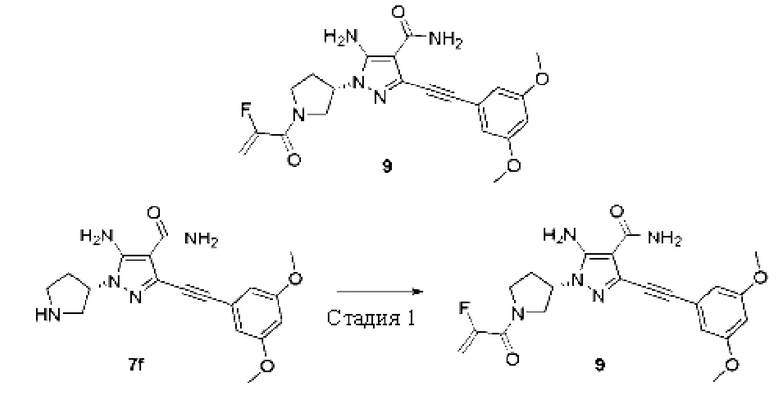
Стадия 1
(S)-5-Амино-3-((3,5-диметоксифенил)этинил)-1-(1-(2-фторакрилоил)пирролидин-3-ил)-1Н-пиразол-4-карбоксамид
Соединение (S)-5-амино-3-((3,5-диметоксифенил)этинил)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамид 7f (50 мг, 0,14 ммоль) и 2-фторакриловую кислоту (15 мг, 0,17 ммоль) растворяли в дихлорметане, затем добавляли N,N-диизопропилэтиламин (54 мг, 0,42 ммоль) и O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (НАТО) (69 мг, 0.18 ммоль), перемешивали при комнатной температуре в течение 2 часов, затем реакционную смесь разбавляли водой (10 мл), экстрагировали дихлорметаном (10 мл×3), и органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования, и концентрировали реакционную систему при пониженном давлении, остаток очищали тонкослойной колоночной хроматографией на силикагеле (дихлорметан/метанол = 20/1) с получением указанного в заголовке продукта, (S)-5-амино-3-((3,5-диметоксифенил)этинил)-1-(1-(2-фтсракрилоил)пирролидин-3-ил)-1Н-пиразолл-карбоксамида 9 (3,6 мг, белое твердое вещество), и выход составлял 6%.
МС m/z (ИЭР): 428 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 6,62 (т, J=2,5 Гц, 2Н), 6,47 (т, J=2,3 Гц, 1Н), 5,39 (дд, J=47,2, 3,5 Гц, 1H), 5,16 (ддд, J=16,6, 5,7, 3,5 Гц, 1Н), 4,86-4,81 (м, 1H), 4,02-3,91 (м, 2Н), 3,87-3,72 (м, 2Н), 3,71 (с, 6Н), 2,34-2,23 (м, 2Н).
Пример 10
(S)-5-Амино-1-(1-(бут-2-инил)пирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-карбоксамид
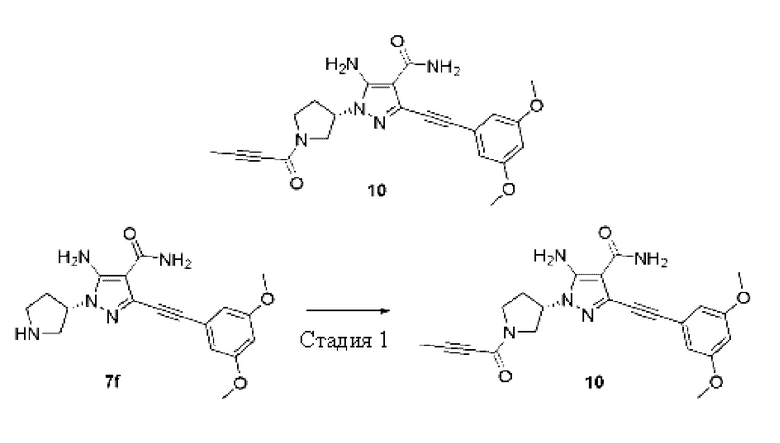
Стадия 1
(S)-5-Амино-1-(1-(6ут-2-инил)пирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-карбоксамид
(S)-5-Амино-3-((3,5-диметоксифенил)этинил)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамид 7f (50 мг, 0,14 ммоль) и 2-бутиновую кислоту (14 мг, 0,17 ммоль) растворяли в дихлорметане, затем добавляли N,N-диизопропилэтиламин (54 мг, 0,42 ммоль) и O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (HATU) (69 мг, 0,18 ммоль), перемешивали при комнатной температуре в течение 2 часов, затем реакционную смесь разбавляли водой (10 мл), экстрагировали дихлорметаном (10 мл×3), и органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования, и концентрировали реакционную систему при пониженном давлении, остаток очищали тонкослойной колоночной хроматографией на си лик are л е (цихлорметан/метанол = 20/1) с получением указанного в заголовке продукта, (S)-5-амино-1-(1-(бут-2-инил)пирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-карбоксамида 10 (5,1 мг, бледно-желтое твердое вещество), и выход составлял 9%.
MC m/z (ИЭР): 422 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 6,62 (т, J=2,1 Гц, 2Н), 6,47 (т, J=2,2 Гц, 1Н), 4,85-4,81 (м, 1H), 4,01-3,86 (м, 2Н), 3,77-3,62 (м, 7,5Н), 3,54-3,46 (м, 0,5Н), 2,32-2,27 (м, 2Н), 1,95-1,93 (м, 3Н).
Пример 11
(S,E)-5-амино-3-((3,5-диметоксифенил)этинил)-1-(1-(4-метоксибут-2-еноил)пирролидин-3-ил)-1Н-пиразол-4-карбоксамид
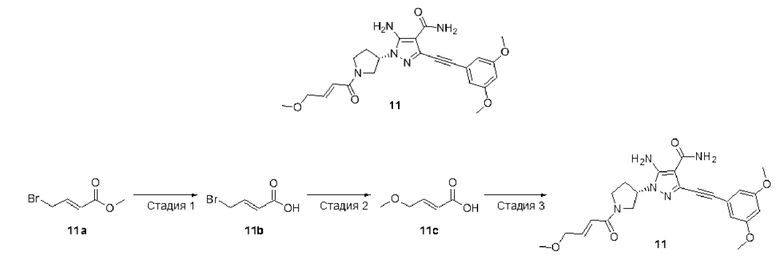
Стадия 1
(Е)-4-Бромбут-2-еновая кислота
Метил-(Е)-4-бромбут-2-еноат 11а (3 г, 16,8 ммоль), моногидрат гидроксида лития (1,1 г, 25,3 ммоль), тетрагидрофуран (50 мл) и воду (50 мл) смешивали при 0°С и перемешивали еще 2 часа; после завершения реакции тетрагидрофуран вымывали петролейным эфиром, а водную фазу доводили до рН=1 с помощью 2 М хлористоводородной кислоты, а затем экстрагировали этилацетатом (100 мл×2), и после объединения органических фаз выпаривали растворитель при пониженном давлении с получением указанного в заголовке продукта, (Е)-4-бромбут-2-еновой кислоты 11b (2,3 г, желтое маслянистое вещество), и выход составлял 83%.
MC m/z (ИЭР): 163 [М-1]
Стадия 2
(Е)-4-Метоксибут-2-еновая кислота
Соединение (Е)-4-бромбут-2-еновую кислоту 11b (100 мг, 0,61 ммоль) растворяли в метаноле (5 мл), добавляли метоксид натрия в метаноле (30%, 0,55 мл, 3,05 ммоль) и затем перемешивали в течение 15 часов, удаляли растворитель из реакционной смеси при пониженном давлении и затем растворяли остаток в воде, затем доводили до рН=1 с помощью разбавленной хлористоводородной кислоты, и затем экстрагировали дихлорметаном (10 мл×3), органические фазы объединяли и выпаривали растворитель при пониженном давлении с получением указанного в заголовке продукта, (Е)-4-метоксибут-2-еновой кислоты 11с (50 мг, желтое маслянистое вещество), и выход составлял 71%.
1Н ЯМР (400 МГц, CDCl3) δ 7,13-7,03 (м, 1H), 6,15-6,07 (м, 1H), 4,18-4,11 (м, 2Н), 3,48-3,38 (с, 3Н).
Стадия 3
(S,E)-5-Амино-3-((3,5-диметоксифенил)этинил)-1-(1-(4-метоксибут-2-еноил)пирролидин-3-ил)-1Н-пиразол-4-карбоксамид
Соединение (Е)-4-метоксибут-2-еновую кислоту 11с (22 мг, 0,19 ммоль), диизопропилэтиламин (67 мг, 0,52 ммоль), (S)-5-амино-3-((3,5-диметоксифенил)этинил)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамид 7f (50 мг, 0,13 ммоль), 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония гексафторфосфат (72 мг, 0,19 ммоль) и N,N-диметилформамид (10 мл) смешивали и перемешивали в течение 2 часов, растворитель удаляли при пониженном давлении и растворяли остаток в этилацетате (30 мл), и затем последовательно промывали водой и насыщенным солевым раствором, растворитель удаляли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (дихлорметан/метанол = 20/1) с получением указанного в заголовке продукта, (S,E)-5-амино-3-((3,5-диметоксифенил)этинил)-1-(1-(4-метоксибут-2-еноил)пирролидин-3-ил)-1Н-пиразол-4-карбоксамида (30 мг, белое твердое вещество), и выход составлял 51%.
МС m/z (ИЭР): 454 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 6,98 (д, 3=15,3 Гц, 1H), 6,86 (шс, 1H), 6,72 (д, J=2,1 Гц, 2Н), 6,54 (с, 1H), 6,39 (дд, J=27,7, 16,0 Гц, 1Н), 5,54 (шс, 1H), 4,73-4,70 (м, 1Н), 4,14-4,12 (м, 2Н), 4,05-4,00 (м, 2Н), 3,95-3,93 (м, 1Н), 3,82 (с, 6Н), 3,77-3,68 (м, 1Н), 3,43 (д, J=10,1 Гц, 3Н), 2,72 (шс, 0,5Н), 2,54 (шс, 0,5Н), 2,43-2,35 (м, 1H).
Пример 12
(S)-5-Амино-1-(1-цианопирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-1H-пиразол-4-карбоксамид
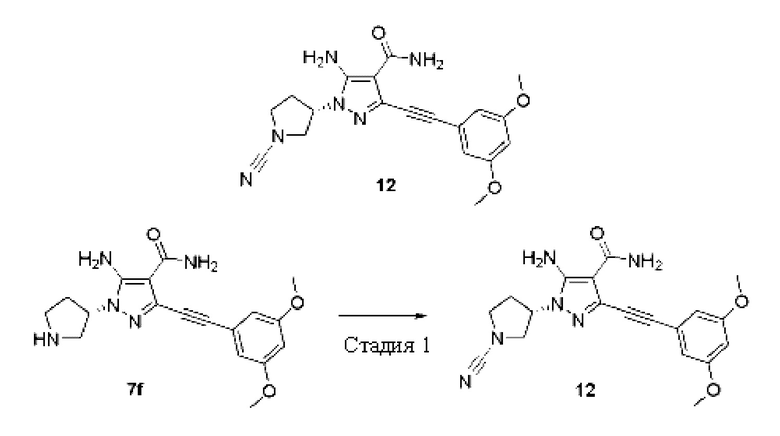
Стадия 1
(S)-5-Амино-1-(1-цианопирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-1H-пиразол-4-карбоксамид
Соединение (S)-5-амино-3-((3,5-диметоксифенил)этинил)-1-(пирролидин-3-ил)-1H-пиразол-4-карбоксамид 7f (50 мг, 0,14 ммоль) растворяли в тетрагидрофуране (2 мл), и затем добавляли триэтиламин (1 мл), охлаждали до 0°С, добавляли бромциан (17 мг, 0,15 ммоль), перемешивали при 0°С в течение 2 часов, затем температуру реакционной смеси повышали до комнатной температуры и продолжали перемешивание в течение 2 часов, реакционную смесь концентрировали при пониженном давлении и очищали остаток тонкослойной хроматографией на силикагеле (дихлорметан/метанол = 15/1) с получением указанного в заголовке продукта, (S)-5-амино-1-(1-цианопирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-1Н-пиразолл-карбоксамида 12 (18 мг, белое твердое вещество), и выход составлял 34%.
МС m/z (ИЭР): 381 [М+1]
1H ЯМР (400 МГц, CDCl3) δ 6,77 (шс, 1Н), 6,68 (д, J=1,9 Гц, 2Н), 6,50 (с, 1Н), 5,75 (с, 2Н), 5,67 (шс, 1Н), 4,79-4,73 (м, 1Н), 3,84-3,73 (м, 9Н), 3,61-3,53 (м, 1Н), 2,53-2,43 (м, 1Н), 2,37-2,26 (м, 1Н).
Примеры 13-16 синтезировали со ссылкой на рабочие стадии Примера 7:
Пример 13
(R)-1-(1-Акрилоилпирролидин-3-ил)-5-амино-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-карбоксамид
МС m/z(H3P): 410 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 6,73 (д, J=1,9 Гц, 2Н), 6,71-6,60 (м, 1Н), 6,58 (шс, 1Н), 6,32 (дд, J=16,8, 1,7 Гц, 1Н), 5,84-5,74 (м, 1Н), 5,04-4,91 (м, 1Н), 4,09 (м, 0,5Н), 3,98 (тд, J=11,1, 4,0 Гц, 1Н), 3,91 (дд, J=7,8, 5,6 Гц, 1H), 3,86 (дд, J=9,9, 4,4 Гц, 1Н), 3,81 (с, 6Н), 3,73-3,63 (м, 0,5Н), 2,47 (дд, J=13,2, 6,7 Гц, 1Н), 2,38 (дд, J=13,6, 7,0 Гц, 1Н).
Пример 14
1-(1-Акрилоилазетидин-3-ил)-5-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразол-4-карбоксамид
МС m/z(ИЭР): 396 [M+1]
1Н ЯМР (400 МГц, CD3OD) δ 6,75 (д, J=2,3 Гц, 2Н), 6,60 (т, J=2,2 Гц, 1Н), 6,45-6,28 (м, 2Н), 5,80 (дд, J=10,1, 2,1 Гц, 1H), 5,29-5,21 (м, 1Н), 4,79-4,64 (м, 2Н), 4,54-4,47 (м, 1Н), 4,46-4,39 (м, 1H), 3,82 (с, 6Н).
Пример 15
1-(1-Акрилоилпиперидин-4-ил)-5-амино-3-((3,5-даметоксифенил)этинил)-1Н-пиразол-4-карбоксамид

МС m/z (ИЭР): 424 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 6,88-6,78 (м, 1Н), 6,73 (д, J=2,2 Гц, 2Н), 6,58 (т, J=2,2 Гц, 1Н), 6,24 (дд, J=16,8, 1,7 Гц, 1Н), 5,78 (дд, J=10,7, 1,7 Гц, 1Н), 4,73 (д, J=13,2 Гц, 1Н), 4,47-4,36 (м, 1H), 4,30 (д, J=13,3 Гц, 1Н), 3,81 (с, 6Н), 3,32-3,24 (м, 1Н), 2,91 (т, J=9,9 Гц, 1H), 2,02 (д, J=4,5 Гц, 4Н).
Пример 16
1-((1-Акрилоилпирролидин-3-ил)метил)-5-амино-3-((3,5-диметоксифенил)этинил)-1H-пиразол-4-карбоксамид
MC m/z (ИЭР): 424 [M+1]
1Н ЯМР (400 МГц, CD3OD) δ 6,73 (с, 2Н), 6,66-6,55 (м, 2Н), 6,28 (д, J=16,7 Гц, 1Н), 5,75 (д, J=10,4 Гц, 1Н), 4,13-3,99 (м, 2Н), 3,82 (с, 6Н), 3,78-3,61 (м, 2Н), 3,48 (дд, J=14,8, 7,4 Гц, 1Н), 3,39-3,34 (м, 1Н), 2,94-2,75 (м, 1Н), 2,20-2,02 (м, 1Н), 1,94-1,71 (м, 1Н).
Пример 17
(S)-1-(1-Акрилоилпирролидин-3-ил)-5-амино-3-((2-фтор-3,5-диметоксифенил)этинил)-1H-пиразол-4-карбоксамид
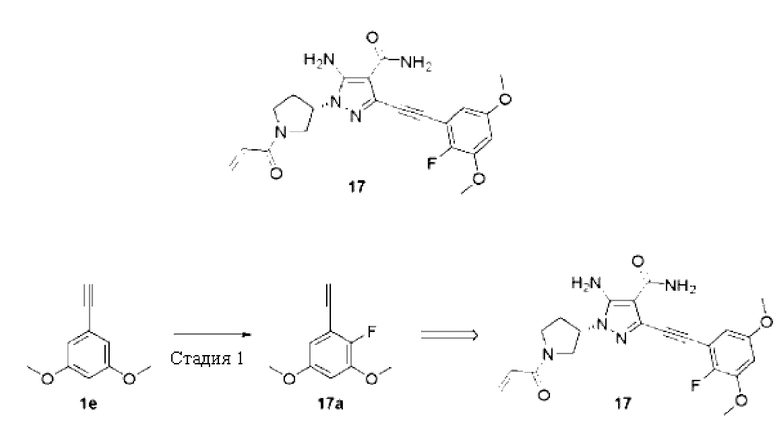
Стадия 1
1-Этинил-2-фтор-3,5-диметоксибензол
Смесь 1-этинил-3,5-диметокси бензол а 1е (2 г, 12,3 ммоль) растворяли в ацетонитриле (15 мл) и охлаждали до 0°С, затем по частям добавляли соль 1-хлорметил-4-фтор-1,4-диазония дицикло[2.2.2]октан-бис(тетрафторборат) (6,6 г, 18,5 ммоль), затем перемешивали при комнатной температуре в течение ночи, реакционный раствор выливали в воду (50 мл) и экстрагировали дихлорметаном (30 мл×3), и органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования, затем систему концентрировали при пониженном давлении и очищали остаток колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 30/1) с получением указанного в заголовке продукта, 1-этинил-2-фтор-3,5-диметоксибензола 17а (800 мг, желтое твердое вещество), и выход составлял 36%.
1Н ЯМР (400 МГц, CDCl3) δ 6,46 (дд, J=6,9, 2,9 Гц, 1H), 6,41 (дд, J=4,5, 3,0 Гц, 1Н), 3,78 (с, 3Н), 3,69 (с, 3Н), 3,22 (с, 1H).
Затем синтезировали Пример 17 со ссылкой на способ с первой по шестую стадии Примера 7, приведенного выше, но на третьей стадии использовали 1-этинил-2-фтор-3,5-диметоксибензол вместо 1-этинил-3,5-диметоксибензола.
МС m/z (ИЭР): 428 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7,00 (шс, 1Н), 6,59-6,57 (м, 2Н), 6,49-6,39 (м, 2Н), 5,74-5,70 (м, 1Н), 5,52 (д, J=8,5 Гц, 2Н), 5,35 (шс, 1H), 4,73-4,64 (м, 1H), 4,07-3,90 (м, 3Н), 3,88 (с, 3Н), 3,78 (д, J=5,3 Гц, 3Н), 3,75-3,67 (м, 1Н), 2,72-2,67 (м, 0,5Н), 2,54-2,31 (м, 1,5Н).
Пример 18
(S)-1-(1-Акрилоилпирролидин-3-ил)-5-амино-3-((5-хлор-2-фторфенил)этинил)-1Н-пиразол-4-карбоксамид
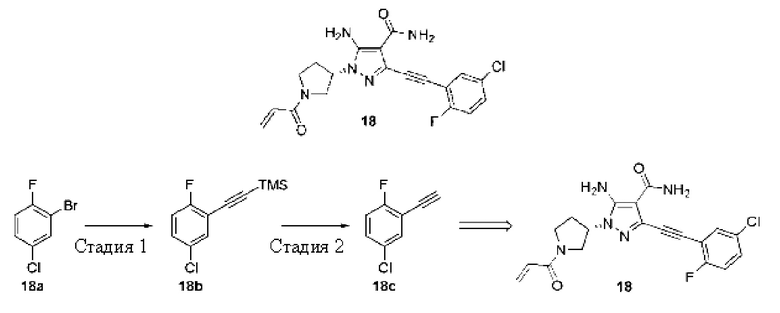
Стадия 1
((2-Фтор-5-хлорфенил)этинил)триметилсилан
2-фтор-5-хлорбромбензол 18а (11,0 г, 52,8 ммоль), этинилтриметилсилан (7,7 г, 79 ммоль) и триэтиламин (60 мл) смешивали и затем добавляли йодид меди (Г) (100 мг, 0,53 ммоль) и ди(трифенилфосфин)палладия хлорид (1,86 г, 2,65 ммоль), реакционную смесь нагревали до 80°C в атмосфере азота и продолжали перемешивание в течение 4 часов; после завершения реакции растворитель удаляли из раствора при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 100/1) с получением указанного в заголовке продукта, ((2-фтор-5-хлорфенил)этинил)триметилсилан а 18b (11,0 г, желтое маслянистое вещество), и выход составлял 90%.
1Н ЯМР (400 МГц, CDCl3) δ 7,45 (дд, J=6,0, 2,7 Гц, 1Н), 7,28-7,22 (м, 1H), 7,02 (т, J=8,8 Гц, 1Н), 0,29 (с, 9Н).
Стадия 2
4-Хлор-2-этинил-1-фторбензол
((2-Фтор-5-хлорфенил)этинил)триметилсилан 18b (11,0 г, 48 ммоль), карбонат калия (8,1 г, 58 ммоль), дихлорметан (80 мл) и метанол (40 мл) смешивали и перемешивали при комнатной температуре в течение 18 часов; после завершения реакции растворитель удаляли из раствора при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 100/1) с получением указанного в заголовке продукта, 4-хлор-2-этинил-1-фторбензола 18 с (5,5 г, желтое твердое вещество), и выход составлял 74%.
1Н ЯМР (400 МГц, CDCl3) δ 7,45 (дд, J=6,0, 2,7 Гц, 1H), 7,31-7,27 (м, 1H), 7,04 (т, J=8,0, 1Н), 3,35 (с, 1Н).
Затем синтезировали Пример 18 со ссылкой на способ с первой по шестую стадии Примера 7, приведенного выше, но на третьей стадии использовали 4-хлор-2-этинил-1-фторбензол вместо 1-этинил-3,5-диметоксибензола.
МС m/z (ИЭР): 402 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7,62-7,61 (м, 1H), 7,47-7,45 (м, 1H), 7,24 (т, J=9,0 Гц, 1H), 6,69-6,56 (м, 1H), 6,30 (д, J=16,8 Гц, 1H), 5,77 (т, J=9,2 Гц, 1H), 5,02-1,91 (м, 1Н), 4,09-3,95 (м, 2Н), 3,84-3,78 (м, 2Н), 2,46 (дд, J=13,1, 6,6 Гц, 1H), 2,37 (дд, J=13,6, 6,9 Гц, 1Н).
Пример 19
(S)-1-(1-акрилоилпирролидин-3-ил)-5-амино-3-((2-хлор-5-(метилкарбамоил)фенил)этинил)-1Н-пиразол-4-карбоксамид
Стадия 1
Метил-4-хлор-3-бромбензоат
4-Хлор-3-бромбензойную кислоту 19а (2 г, 8,5 ммоль) растворяли в метаноле (400 мл) и охлаждали до 0°С, затем по каплям добавляли ацетилхлорид (2,3 г, 30 ммоль) и продолжали перемешивание в течение 18 часов; после завершения реакции растворитель удаляли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 10/1) с получением указанного в заголовке продукта, метил 4-хлор-3-бромбензоата 19b (1,2 г, желтое твердое вещество), и выход составлял 57%.
1Н ЯМР (400 МГц, CDCl3) δ 8,31 (д, J=1,9 Гц, 1H), 7,93 (дд, J=8,3, 1,9 Гц, 1H), 7,55 (д, J=8,3 Гц, 1H), 3,95 (с, 3Н).
Стадия 2
Метил-4-хлор-3-((триметилсилил)этинил)бензоат
Соединение метил-4-хлор-3-бромбензоат 19b (1,2 г, 4,8 ммоль), триметилсилилацетилен (0,95 г, 9,7 ммоль), ацетат палладия (108 мг, 0,48 ммоль), трифенилфосфин (254 мг, 0,97 ммоль), йодид меди(I) (185 мг, 0,97 ммоль) и триэтиламин (25 мл) смешивали в закрытой пробирке и нагревали, и перемешивали при 100°C в течение 15 часов; после завершения реакции растворитель удаляли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 10/1) с получением указанного в заголовке продукта, метил-4-хлор-3-((триметилсилил)этинил)бензоата 19 с (1 г, желтое твердое вещество), и выход составлял 78%.
1Н ЯМР (400 МГц, CDCl3) δ 8,19 (д, J=2,0 Гц, 1H), 7,91 (дд, J=8,4, 2,1 Гц, 1H), 7,48 (д, J=8,4 Гц, 1H), 3,94 (с, 3Н), 0,30 (с, 9Н).
Стадия 3
Метил-4-хлор-3-этинилбензоат
Метил-4-хлор-3-((триметилсилил)этинил)бензоат 19 с (1 г, 3,76 ммоль) растворяли в метаноле (20 мл), затем добавляли карбонат калия (1,04 г, 7,52 ммоль), и после перемешивания при комнатной температуре в течение 1 часа удаляли растворитель при пониженном давлении, и остаток промывали водой, а затем отфильтровывали с получением указанного в заголовке продукта, метил-4-хлор-3-этинилбензоата 19d (380 мг, желтое твердое вещество), и выход составлял 52%.
1Н ЯМР (400 МГц, CDCl3) δ 8,23 (д, J=2,1 Гц, 1H), 7,96 (дд, J=8,4, 2,0 Гц, 1H), 7,51 (д, J=8,4 Гц, 1H), 3,95 (с, 3Н), 3,44 (с, 1H).
Стадия 4
(S)-3-(5-Амино-3-бром-4-карбамоил-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир
Соединение (S)-3-(5-амино-3-бром-4-циано-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир 7 с (2,20 г, 6,2 ммоль), водный раствор гидроксида натрия (0,5 М, 12,4 мл, 6,2 ммоль), водный раствор пероксида водорода (30%, 15 мл) и диметилсульфоксид (30 мл) смешивали и перемешивали при комнатной температуре в течение 2 часов, после чего реакционную смесь разбавляли насыщенным солевым раствором (50 мл) и экстрагировали этилацетатом (50 мл×3), затем органические фазы объединяли, растворитель удаляли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (дихлорметан/метанол = от 100/1 до 20/1) с получением указанного в заголовке продукта, (S)-3-(5-амино-3-бром-4-карбамоил-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 19е (1,92 г, светло-желтое твердое вещество), и выход составлял 83%.
МС m/z (ИЭР): 374 [М+1]
Стадия 5
(S)-3-(5-Амино-4-карбамоил-3-((2-хлор-5-(карбометокси<метосикарбонил>)фенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир
(S)-3-(5-Амино-3-бром-4-карбамоил-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир 19е (770 мг, 2,1 ммоль), триэтиламин (6 мл), 1,1'-бис(дифенилфосфиноферроцен)палладия дихлорид (307 мг, 0,42 ммоль), йодид меди(I) (80 мг, 0,42 ммоль) и N,N-диметилформамид (20 мл) смешивали, удаляли кислород, а затем нагревали до 90°C в атмосфере аргона, затем по каплям добавляли раствор метил-4-хлор-3-этинилбензоата 19d (3,20 г, 16,5 ммоль) в N,N-диметилформамиде (2 мл) и продолжали перемешивание в течение 12 часов, растворитель удаляли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (дихлорметан/метанол = 20/1) с получением указанного в заголовке продукта, (8)-3-(5-амино-4-карбамоил-3-((2-хлор-5-(карбометокси<метоксикарбонил>)фенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 19f (420 мг, желтое твердое вещество), и выход составлял 41%.
МС m/z (ИЭР): 488 [М+1]
Стадия 6
(S)-3-((5-Амино-1-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-4-карбамоил-1Н-пиразол-3-ил)этинил)-4-хлорбензойная кислота
(S)-3-(5-Амино-4-карбамоил-3-((2-хлор-5-(карбометокси<метоксикарбонил>)фенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир 19f (100 мг, 0,2 ммоль) растворяли в смешанном растворителе из метанола (4 мл) и воды (4 мл), затем добавляли гидроксид натрия (25 мг, 0,61 ммоль) и продолжали перемешивание в течение 2 часов; после завершения реакции органический растворитель удаляли при пониженном давлении, и остаток доводили до рН=4 5 с помощью хлористоводородной кислоты (1 М), и затем экстрагировали этилацетатом (30 мл×2), полученные органические фазы объединяли и промывали насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия и фильтровали, из фильтрата удаляли растворитель при пониженном давлении с получением указанного в заголовке продукта, (S)-3-((5-амино-1-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-4-карбамоил-1Н-пиразол-3-ил)этинил)-4-хлорбензойной кислоты 19g (80 мг, коричневое твердое вещество), и выход составлял 84%.
МС m/z (ИЭР): 418 [М+Н-56]
Стадия 7
(S)-3-(5-Амино-4-карбамоил-3-((2-хлор-5-(метилкарбамоил)фенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир
(S)-3-((5-Амино-1-(1-(трет-бутоксикарбонил)пирролидин-3-ил)-4-карбамоил-1Н-пиразол-3-ил)этинил)-4-хлорбензойную кислоту 19g (80 мг, 0,17 ммоль) растворяли в N,N-диметилформамиде (2,5 мл), затем последовательно добавляли гидрохлорид метиламина (34 мг, 0,50 ммоль), диизопропилэтиламин (129 мг, 1 ммоль) и 2-(7-бензотриазол)-N,N,N',N'-тетраметилмочевины гексафторфосфат (64 мг, 0,17 ммоль), реакционную смесь перемешивали при комнатной температуре в течение 2 часов, а затем гасили водой, затем экстрагировали этилацетатом (20 мл × 3), органические фазы объединяли, растворитель удаляли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = от 100/1 до 0/1) с получением указанного в заголовке продукта, (S)-3-(5-амино-4-карбамоил-3-((2-хлор-5-(метилкарбамоил)фенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 19h (41 мг, коричневое твердое вещество), и выход составлял 50%.
МС m/z (ИЭР): 387 [М+Н-Вос]
Стадия 8
(S)-5-Амино-3-((2-хлор-5-(метилкарбамоил)фенил)этинил)-1-(пирролидин-3-ил)-1Н-пиразол-4-формамида гидрохлорид
(S)-3-(5-Амино-4-карбамоил-3-((2-хлор-5-(метилкарбамоил)фенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир 19h (40 мг, 0,08 ммоль) растворяли в этилацетате (5 мл), затем добавляли раствор хлороводорода в этаноле (33%, 3 мл) и перемешивали при комнатной температуре в течение 1 часа; после завершения реакции растворитель удаляли при пониженном давлении с получением указанного в заголовке продукта, (S)-5-амино-3-((2-хлор-5-(метилкарбамоил)фенил)этинил)-1-(пирролидин-3-ил)-1Н-пиразол-4-формамида гидрохлорида 19i (40 мг, неочищенный, коричневое твердое вещество) и напрямую использовали продукт на следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 387 [М+Н]
Стадия 9
(S)-1-(1-Акрилоилпирролидин-3-ил)-5-амино-3-((2-хлор-5-(метилкарбамоил)фенил)этинил)-1Н-пиразол-4-карбоксамид
Соединение (S)-5-амино-3-((2-хлор-5-(метилкарбамоил)фенил)этинил)-1-(пирролидин-3-ил)-1Н-пиразол-4-формамида гидрохлорид 19i (40 мг, 0,08 ммоль, неочищенный), акрилоилхлорид (7,5 мг, 0,08 ммоль), водный раствор карбоната калия (0,4 М, 1,0 мл, 0,4 ммоль) и тетрагидрофуран (5 мл) смешивали при 0°С и перемешивали при указанной температуре в течение 0,5 часа, затем экстрагировали этилацетатом (20 мл × 2), органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования, растворитель удаляли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (дихлорметан/метанол = от 100/1 до 10/1) с получением указанного в заголовке продукта, (S)-1-(1-акрилоилпирролидин-3-ил)-5-амино-3-((2-хлор-5-(метилкарбамоил)фенил)этинил)-1Н-пиразол-4-карбоксамида 19 (18 мг, белое твердое вещество), и выход составлял 51% за две стадии.
МС m/z (ИЭР): 441 [М+Н]
1Н ЯМР (400 МГц, ДМСО-d6) δ 8,65 (с, 1Н), 8,18 (с, 1Н), 7,90 (д, J=8,0 Гц, 1H), 7,72 (д, J=8,3 Гц, 1Н),7,43 (с, 1Н), 6,70-6,62 (м, 4Н), 6,19-6,15 (м, 1H), 5,70 (т, J=10,2 Гц, 1H), 5,03-4,94 (м, 1H), 3,80-3,54 (м, 4Н), 2,78 (д, J=3,8 Гц, 3Н), 2,36-2,25 (м, 2Н).
Пример 20
(S)-1-(1-Акрилоилпирролидин-3-ил)-5-амино-3-((3-метокси-5-(метилкарбамоил)фенил)этинил)-1Н-пиразол-4-карбоксамид
Стадия 1
3-Бром-5-метокси-N-метилбензамид
3-Бром-5-метоксибензойную кислоту 20 а (500 мг, 2,17 ммоль) растворяли в N,N-диметилформамиде (15 мл), затем последовательно добавляли гидрохлорид метиламина (291 мг, 4,35 ммоль), диизопропилэтиламин (1,12 г, 8,68 ммоль) и 2-(7-бензотриазол)-N,N,N',N'-тетраметил моче вины гексафторфосфат (1,24 г, 3,26 ммоль), реакционную систему перемешивали при комнатной температуре в течение 2 часов и затем гасили водой, растворитель удаляли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этил ацетат = 1/1) с получением указанного в заголовке продукта, 3-бром-5-метокси-N-метилбензамида 20b (500 мг, белое твердое вещество), и выход составлял 95%.
MC m/z (ИЭР): 244 [М+Н]
Стадия 2
3-Метокси-N-метил-5-((триметилсилил)этинил)бензамид
Соединение 3-бром-5-метокси-N-метилбензамид 20b (500 мг, 2,1 ммоль), триметилсилилацетилен (302 мг, 3,1 ммоль), ацетат палладия (47 мг, 0,21 ммоль), трифенилфосфин (110 мг, 0,42 ммоль), йодид меди (I) (80 мг, 0,42 ммоль) и триэтиламин (20 мл) смешивали в закрытой пробирке и затем нагревали до 100°С, и перемешивали в течение 15 часов; после завершения реакции растворитель удаляли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 1/1) с получением указанного в заголовке продукта, 3-метокси-N-метил-5-((триметилсилил)этинил)бензамида 20 с (220 мг, желтое твердое вещество), и выход составлял 41%.
MC m/z (ИЭР): 262 [М+Н]
Стадия 3
3-Этинил-5-метокси-N-метилбензамид
3-Метокси-N-метил-5-((триметилсилил)этинил)бензамид 20 с (220 мг, 0,84 ммоль) растворяли в метаноле (8 мл), затем добавляли карбонат калия (233 мг, 1,68 ммоль), после перемешивания при комнатной температуре в течение 1 часа удаляли растворитель при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 1/1) с получением указанного в заголовке продукта, 3-этинил-5-метокси-N-метилбензамида 20d (140 мг, светло-желтое твердое вещество), и выход составлял 88%.
MC m/z (ИЭР): 190 [М+Н]
Стадия 4
(S)-3-(5-Амино-4-карбамоил-3-((3-метокси-5-(метилкарбамоил)фенил)этинил)-1Н-пиразол-1-карбоновой кислоты трет-бутиловый эфир
(S)-3-(5-Амино-3-бром-4-карбамоил-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир 19е (329 мг, 0,88 ммоль), триэтиламин (2 мл), 1,1'-бис(дифенилфосфиноферроцен)палладия дихлорид (129 мг, 0,2 ммоль), йодид меди (I) (34 мг, 0,18 ммоль) и N,N-диметилформамид (8 мл) смешивали и затем удаляли кислород, и нагревали до 90°С в атмосфере аргона, затем по каплям добавляли раствор 3-этинил-5-метокси-М-метилбензамида 20d (1,00 г, 5,3 ммоль) в N,N-диметилформамиде (2 мл), продолжали перемешивание в течение 12 часов, растворитель удаляли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (дихлорметан/метанол = 20/1) с получением указанного в заголовке продукта, (S)-3-(5-амино-4-карбамоил-3-((3-метокси-5-(метилкарбамоил)фенил)этинил)-1Н-пиразол-1-карбоновой кислоты трет-бутилового эфира 20е (400 мг, неочищенный, коричневое твердое вещество).
МС m/z (ИЭР): 383 [М+Н-100]
Стадия 5
(S)-5-Амино-3-((3-метокси-5-(метилкарбамоил)фенил)этинил)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамида гидрохлорид
(S)-3-(5-Амино-4-карбамоил-3-((3-метокси-5-(метилкарбамоил)фенил)этинил)-1Н-пиразол-1-карбоновой кислоты трет-бутиловый эфир 20е (400 мг, неочищенный) растворяли в дихлорметане (5 мл), затем добавляли раствор хлороводорода в этаноле (30%, 3 мл), перемешивали при комнатной температуре в течение 1 часа; после завершения реакции растворитель удаляли при пониженном давлении с получением указанного в заголовке продукта, (S)-5-амино-3-((3-метокси-5-(метилкарбамоил)фенил)этинил)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамида гидрохлорида 20f (300 мг, неочищенный, коричневое твердое вещество). Продукт напрямую использовали для следующей реакции без очистки.
МС m/z (ИЭР): 383 [М+Н]
Стадия 6
(S)-1-(1-Акрилоилпирролидин-3-ил)-5-амино-3-((3-метокси-5-(метилкарбамоил)фенил)этинил)-1Н-пиразол-4-карбоксамид
Соединение (S)-5-амино-3-((3-метокси-5-(метилкарбамоил)фенил)этинил)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамида гидрохлорид 20f (150 мг, 0,39 ммоль, неочищенный), акрилоилхлорид (42 мг, 0,47 ммоль), бикарбонат натрия (131 мг, 1,56 ммоль), воду (4 мл) и тетрагидрофуран (8 мл) смешивали при 0°С и перемешивали при указанной температуре в течение 0,5 часа, и экстрагировали этилацетатом (20 мл × 2), органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования, растворитель удаляли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (дихлорметан/метанол = 20/1) с получением указанного в заголовке продукта, (S)-1-(1-акрилоилпирролидин-3-ил)-5-амино-3-((3-метокси-5-(метилкарбамоил)фенил)этинил)-1H-пиразол-4-карбоксамида 20 (60 мг, белое твердое вещество), и выход составлял 35% за две стадии.
MCm/z (ИЭР): 437 [М+Н]
1Н ЯМР (400 МГц, CD3OD) δ 7,59 (с, 1Н), 7,45 (с, 1H), 7,28 (с, 1H), 6,73-6,58 (м, 1Н), 6,36-6,28 (м, 1Н), 5,83-5,75 (м, 1Н), 5,04-4,93 (м, 1Н), 4,12-3,91 (м, 2Н), 3,89 (с, 3Н), 3,86-3,66 (м, 2Н), 2,93 (с, 3Н), 2,51-2,44 (м, 1Н), 2,42-2,34 (м, 1Н).
Пример 21
(S)-1-(1-Акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-5-(метиламино)-1H-пиразол-4-карбоксамид
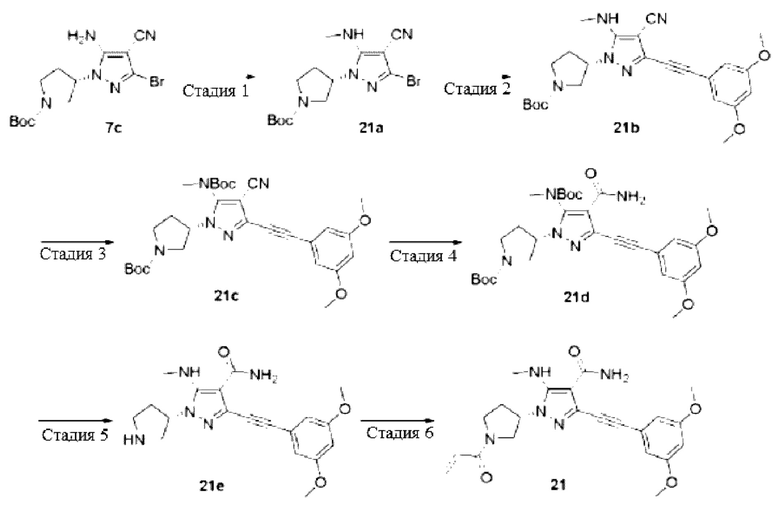
Стадия 1
(S)-3-(3-Бром-4-циано-5-(метиламино)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир
Соединение (S)-3-(5-амино-3-бром-4-циано-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир 7 с (178 мг, 0,5 ммоль) и моногидрат бензолсульфоновой кислоты (12 мг, 0,07 ммоль) растворяли в триэтил-ортоформиате (4 мл) и нагревали до кипения с обратным холодильником в течение 2 часов; после завершения реакции растворитель удаляли при пониженном давлении, и остаток диспергировали в воде, затем экстрагировали этилацетатом (30 мл × 2), органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования, из фильтрата удаляли растворитель при пониженном давлении, остаток растворяли в этаноле (10 мл) и охлаждали до 0°С, добавляли боргидрид натрия (89 мг, 2,35 ммоль) и затем перемешивали при комнатной температуре в течение 2 часов; после завершения реакции смесь гасили насыщенным солевым раствором и затем экстрагировали этилацетатом (30 мл × 2), органические фазы объединяли, растворитель удаляли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = от 100/1 до 1/1) с получением указанного в заголовке продукта, (S)-3-(3-бром-4-циано-5-(метиламино)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 21а (178 мг, белое твердое вещество), и выход составлял 100%.
MC m/z (ИЭР): 314 [М+Н-56]
Стадия 2
(S)-3-(4-Циано-3-((3,5-диметоксифенил)этинил)-5-(метиламино)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир
(S)-3-(3-Бром-4-циано-5-(метиламино)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир 21а (1,85 г, 5,0 ммоль), триэтиламин (20 мл), 1,1'-бис(дифенилфосфиноферроцен)палладия дихлорид (816 мг, 1 ммоль), йодид меди (I) (190 мг, 1 ммоль) и N,N-диметилформамид (20 мл) смешивали, удаляли кислород и затем нагревали до 90°С в атмосфере аргона, затем по каплям добавляли раствор 1-этинил-3,5-диметоксибензола (4,86 г, 30 ммоль) в N,N-диметилформамиде (10 мл), продолжали перемешивание в течение 12 часов, растворитель удаляли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = от 50/1 до 0/1) с получением указанного в заголовке продукта, (S)-3-(4-циано-3-((3,5-диметоксифенил)этинил)-5-(метиламино)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 21b (2,1 г, коричневое твердое вещество), и выход составлял 80%.
MC m/z (ИЭР): 496 [М+Н-56]
Стадия 3
(S)-3-(5-((трет-Бутоксикарбонил)(метил)амино)-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиридил-2-илиден-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир
(S)-3-(4-Циано-3-((3,5-диметоксифенил)этинил)-5-(метиламино)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир 21b (225 мг, 0,5 ммоль) растворяли в дихлорметане (10 мл), затем последовательно добавляли триэтиламин (150 мг, 1,5 ммоль), Вос-ангидрид (218 мг, 1 ммоль) и 4-диметиламинопиридин (6 мг, 0,05 ммоль), перемешивали при комнатной температуре в течение 2 часов, затем добавляли насыщенный солевой раствор (10 мл) и затем экстрагировали этилацетатом (20 мл × 2), органические фазы объединяли и удаляли растворитель при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = от 50/1 До 1/1) с получением указанного в заголовке продукта, (S)-3-(5-((трет-бутоксикарбонил)(метил)амино)-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиридил-2-илиден-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 21 с (200 мг, бледно-желтое твердое вещество), и выход составлял 72%.
МС m/z (ИЭР): 440 [М+Н-112]
Стадия 4
(S)-3-(5-((трет-Бутоксикарбонил)(метил)амино)-4-карбамоил-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир
(S)-3-(5-((трет-Бутоксикарбонил)(метил)амино)-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиридил-2-илиден-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир 21 с (55 мг, 0,1 ммоль), водный раствор гидроксида натрия (0,5 М, 0,1 мл, 0,05 ммоль), водный раствор пероксида водорода (30%, 0,5 мл) и диметилсульфоксид (1 мл) смешивали и перемешивали при комнатной температуре в течение 2 часов, реагенты разбавляли насыщенным солевым раствором (10 мл) и экстрагировали этилацетатом (20 мл × 2), органические фазы объединяли, растворитель удаляли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = от 100/1 до 1/100) с получением указанного в заголовке продукта, (S)-3-(5-((трет-бутоксикарбонил)(метил)амино)-4-карбамоил-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 21d (30 мг, коричневое твердое вещество), и выход составлял 50%.
МС m/z (ИЭР): 414 [М+Н-156]
Стадия 5
(S)-3-((3,5-Диметоксифенил)этинил)-5-(метиламино)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамида гидрохлорид
(S)-3-(5-((трет-Бутоксикарбонил)(метил)амино)-4-карбамоил-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир 21d (570 мг, 1 ммоль) растворяли в этилацетате (10 мл), затем добавляли раствор хлороводорода в этаноле (33%, 5 мл) и перемешивали при комнатной температуре в течение 1 часа; после завершения реакции растворитель удаляли при пониженном давлении с получением указанного в заголовке продукта, (S)-3-((3,5-диметоксифенил)этинил)-5-(метиламино)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамида гидрохлорида 21е (400 мг, неочищенный, коричневое твердое вещество). Полученный продукт напрямую использовали на следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 370 [М+Н]
Стадия 6
(S)-1-(1-Акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-5-(метиламино)-1Н-пиразол-4-карбоксамид
Соединение (S)-3-((3,5-диметоксифенил)этинил)-5-(метиламино)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамида гидрохлорид 21е, акрилоилхлорид (254 мг, 2,82 ммоль), водный раствор карбоната калия (2,5 М, 4,7 мл, 11,78 ммоль) и тетрагидрофуран (10 мл) смешивали и перемешивали при указанной температуре в течение, 5 часа, затем экстрагировали этилацетатом (50 мл × 2), органические фазы объединяли и сушили над безводным сульфатом натрия, растворитель удаляли при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (дихлорметан/метанол = от 100/1 до 10/1) с получением указанного в заголовке продукта, (S)-1-(1-акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-5-(метиламино)-1Н-пиразол-4-карбоксамида 21 (720 мг, белое твердое вещество), и выход составлял 76%.
МС m/z (ИЭР): 424 [М+Н]
1Н ЯМР (400 МГц, CDCl3) δ 6,88 (с, 1Н), 6,69 (д, J=2,3 Гц, 2Н), 6,51 (т, J=2,2 Гц, 1Н), 6,46-6,40 (м, 2Н), 5,74-5,72 (м, 1H), 5,52 5,48 (м, 1H), 5,06-5,01 (м, 1Н), 4,09-3,94 (м, 3Н), 3,80 (с, 6Н), 3,72-3,70 (м, 1H), 3,00 (с, 3Н), 2,71-2,56 (м, 1H), 2,45-2,35 (м, 1H).
Пример 22
(S)-1-(1-Акрилоилпирролидин-3-ил)-3-((2-фтор-3,5-диметоксифенил)этинил)-5-(метиламино)-1Н-пиразол-4-карбоксамид
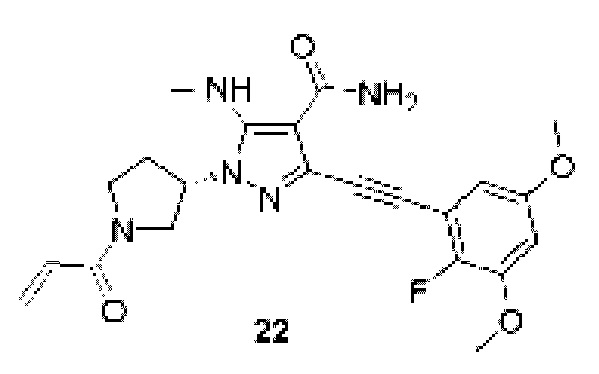
Пример 22 синтезировали в соответствии со способом Примера 21, но на второй стадии использовали 1-этинил-2-фтор-3,5-диметоксибензол вместо 1-этинил-3,5-диметоксибензола.
MC m/z (ИЭР): 442 [М+Н]
1Н ЯМР (400 МГц, CDCl3) δ 7,08 (с, 1Н), 6,68 (д, J=7,2 Гц, 1H), 6,60-6,57 (м, 2Н), 6,51-6,40 (м, 2Н), 5,74-5,69 (м, 1Н), 5,35 (с, 1H), 5,08-4,99 (м, 1H), 4,11-4,08 (м, 1H), 4,05-3,94 (м, 2Н), 3,88 (с, 3Н), 3,79 (с, 3Н), 3,75-3,65 (м, 1Н), 3,00 (т, J=5,2 Гц, 3Н), 2,72-2,58 (м, 1Н), 2,44-2,33 (м, 1Н).
Пример 23
(S)-1-(1-Акрилоилпирролидин-3-ил)-3-((5-хлор-2-фторфенил)этинил)-5-(метиламино)-1H-пиразол-4-карбоксамид
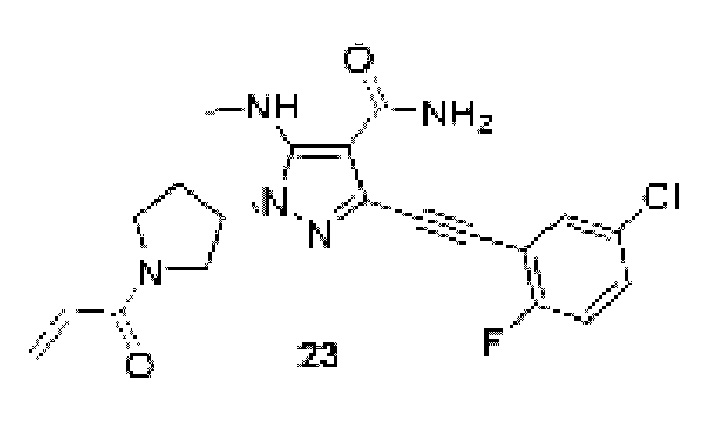
Пример 23 синтезировали в соответствии со способом Примера 21, но на второй стадии использовали 4-хлор-2-этинил-1-фторбензол вместо 1-этинил-3,5-диметоксибензола.
MCm/z (ИЭР): 416 [М+Н]
1Н ЯМР (400 МГц, CDCl3) δ 7,56-7,52 (м, 1H), 7,35-7,33 (м, 1H), 7,08 (т, J=8,8 Гц, 1Н), 7,02-6,92 (м, 1H), 6,51-6,39 (м, 2Н), 5,74 (д, J=9,3 Гц, 1Н), 5,55-5,44 (м, 1H), 5,09-4,98 (м, 1H), 4,14-3,90 (м, 3Н), 3,80-3,65 (м, 1Н), 3,01 (с, 3Н), 2,74-2,55 (м, 1Н), 2,49-2,34 (м, 1H).
Пример 24
(S)-1-(1-Акрилоилпирролидин-3-ил)-3-((3,5-диметоксифеннл)этинил)-5-(этиламино)-1Н-пиразол-4-карбоксамид
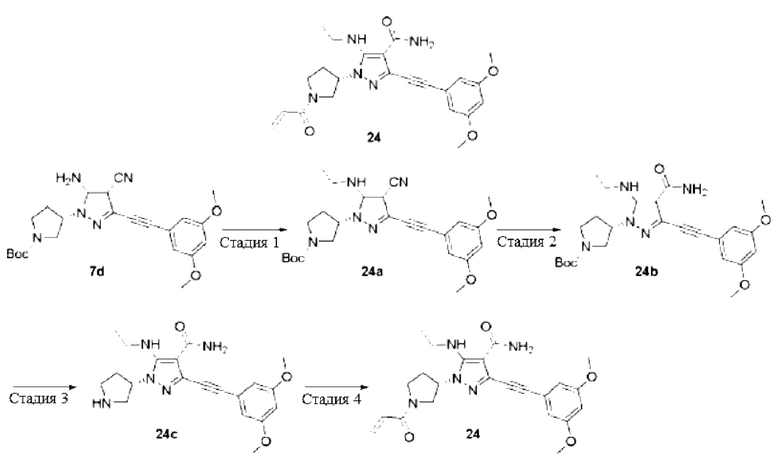
Стадия 1
(S)-3-(5-Этиламино-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир
Смесь (S)-3-(5-амино-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 7d (500 мг, 1,14 ммоль) и гидрида натрия (91 мг, 2,28 ммоль, 60%) добавляли к N,N-диэтилацетамиду (5 мл) и перемешивали в течение 10 минут, затем добавляли йодэтан (106 мг, 0,68 ммоль) и перемешивали в течение 0,5 часа, реакционный раствор выливали в воду и затем концентрировали при пониженном давлении, остаток очищали обращенно-фазовой препаративной высокоэффективной жидкостной хроматографией [ацетонитрил/вод а (содержащая 0,1% муравьиной кислоты): 50%-90%] с получением указанного в заголовке продукта, (S)-3-(5-этиламино-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 24а (70 мг, белое твердое вещество), и выход составлял 22%.
МС m/z (ИЭР): 410 [М+1-56]
Стадия 2
(S)-3-(5-Этиламино-4-карбамоил-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир
(S)-3-(5-Этиламино-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1 -карбоновой кислоты трет-бутиловый эфир 24а (55 мг, 0,12 ммоль) растворяли в диметилсульфоксиде (3 мл), затем добавляли пероксид водорода (2 мл) и гидроксид натрия (300 мг, 7,5 ммоль), перемешивали при комнатной температуре в течение 10 минут, затем нагревали до 40°С; после завершения реакции смесь разбавляли водой (20 мл), экстрагировали этилацетатом (30 мл) и промывали водой (20 мл × 3), органическую фазу концентрировали при пониженном давлении, остаток очищали обращенно-фазовой препаративной высокоэффективной жидкостной хроматографией [ацетонитрил/вода (содержащая 0,1% муравьиной кислоты): 50%-90%] с получением указанного в заголовке продукта, (S)-3-(5-этиламино-4-карбамоил-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 24b (15 mg), и выход составлял 26%.
МС m/z (ИЭР): 484 [М+1]
Стадия 3
(S)-3-((3,5-Диметоксифенил)этинил)-5-(этиламино)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамид
(S)-3-(5-Этиламино-4-карбамоил-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир 24b (15 мг, 0,031 ммоль) растворяли в дихлорметане (2 мл), затем добавляли трифторуксусную кислоту (0,5 мл), перемешивали в течение получаса; после завершения реакции реакционную систему концентрировали при пониженном давлении с получением указанного в заголовке продукта, (S)-3-((3,5-диметоксифенил)этинил)-5-(этиламино)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамида 24 с (20 мг, неочищенный, коричневое маслянистое вещество), и выход составлял >100%. Продукт использовали на следующей стадии без очистки.
МС m/z (ИЭР): 384 [М+1]
Стадия 4
(S)-1-(1-Акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-5-(этиламино)-1Н-пиразол-4-карбоксамид
Соединение (S)-3-((3,5-диметоксифенил)этинил)-5-(этиламино)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамид 24 с (20 мг, 0,031 ммоль, неочищенный) растворяли в тетрагидрофуране (5 мл), и затем добавляли насыщенный раствор бикарбоната натрия (2 мл), затем добавляли раствор акрилоилхлорида (2,7 мг, 0,03 ммоль) в тетрагидрофуране, перемешивали в течение 0,5 часа, реакционный раствор концентрировали при пониежнном давлении, остаток растворяли в этилацетате (30 мл) и затем промывали водой (20 мл × 3), затем органические фазы концентрировали при пониженном давлении и очищали остаток обращенно-фазовой препаративной высокоэффективной жидкостной хроматографией [ацетонитрил/вода (содержащая 0,1% муравьиной кислоты): 20%-70%] с получением указанного в заголовке продукта, (S)-1-(1-акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-5-(этиламино)-1Н-пиразол-4-карбоксамида 24 (4,7 мг, белое твердое вещество), и выход составлял 24%.
МС m/z (ИЭР): 438 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8,87 (шс, 1H), 6,74 (с, 2Н), 6,54 (с, 1Н), 6,52 (с, 1H), 6,48-6,40 (м, 2Н), 5,74-5,69 (м, 1H), 5,06-4,97 (м, 2Н), 4,13-3,93 (м, 3Н), 3,84 (с, 6Н), 3,80-3,67 (м, 1Н), 3,42 (шс, 2Н), 2,75-2,35 (м, 2Н), 1,31-1,25 (м, 3Н).
Пример 25
(S)-1-(1-Акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-5-(изопропиламино)-1Н-пиразол-4-карбоксамид
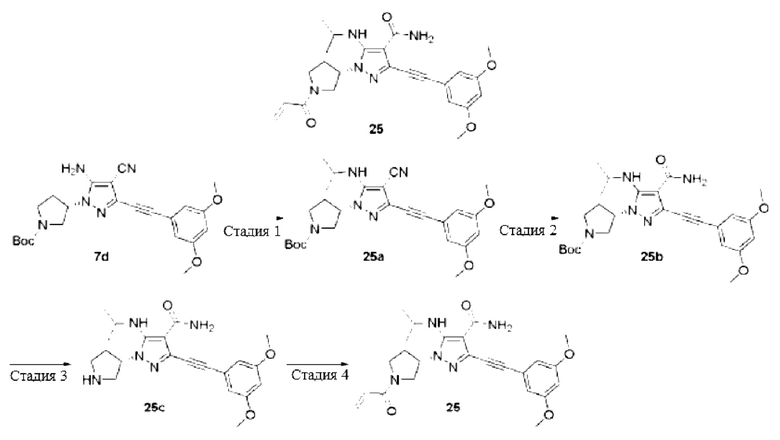
Стадия 1
(S)-3-(4-Циано-3-((3,5-диметоксифенил)этинил)-5-(изопропиламино)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир
Смесь (S)-3-(5-амино-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 7d (600 мг, 1,37 ммоль), карбоната цезия (893 мг, 2,74 ммоль) и ацетонитрила (25 мл) перемешивали в течение 10 минут, затем быстро добавляли 2-бромпропан (186 мг, 1,51 ммоль), нагревали до 72°С, перемешивали в течение 6 часов и затем охлаждали до комнатной температуры, и концентрировали при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этил ацетат = 2/1) с получением указанного в заголовке продукта, (S)-3-(4-циано-3-((3,5-диметоксифенил)этинил)-5-(изопропиламино)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 25а (600 мг, бледно-желтое твердое вещество), и выход составлял 91%.
МС m/z (ИЭР): 424 [М+1-56]
Пример 25 синтезировали со ссылкой на осуществление второй-четвертой стадий, выполненных в Примере 24.
МС m/z (ИЭР): 452 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 6,88 (шс, 1H), 6,70 (с, 2Н), 6,54 (с, 1H), 6,51-6,39 (м, 2Н), 6,03 (т, J=10,3 Гц, 1H), 5,74-5,69 (м, 1H), 5,49 (шс, 1H), 4,96-4,87 (м, 1H), 4,09-3,86 (м, 3Н), 3,80-3,66 (м, 7Н), 3,45-3,43 (м, 1H), 2,69-2,32 (м, 2Н), 1,27-1,15 (м, 6Н).
Пример 26
(S)-1-(1-Акрилоилпирролидин-3-ил)-5-((циклопропилметил)амино)-3-((3,5-диметоксифенил)этинил)-1H-пиразол-4-карбоксамид
Стадия 1
(S)-1-(1-Акрилоилпирролидин-3-ил)-5-((циклопропилметил)амино)-3-((3,5-диметоксифенил)этинил)-1H-пиразол-4-карбоксамид
Соединение (S)-1-(1-акрилоилпирролидин-3-ил)-5-амино-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-карбоксамид 7 (50 мг, 0,12 ммоль) растворяли в ацетонитриле (2 мл), и затем добавляли карбонат цезия (80 мг, 0,24 ммоль) и (бромметил)циклопропан (19 мг, 0,13 ммоль), нагревали до 70°С, перемешивали в течение 4 часов, затем реакционный раствор выливали в воду (30 мл) и экстрагировали этилацетатом (30 мл × 3)5 органические фазы объединяли и сушили над безводным сульфатом натрия, остаток очищали хроматографией на тонком слое силикагеля (дихлорметан/метанол = 12/1) с получением указанного в заголовке продукта, (S)-1-(1-акрилоилпирролидин-3-ил)-5-((циклопропилметил)амино)-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-карбоксамида 26 (14 мг, белое твердое вещество), и выход составлял 28%.
MC m/z (ИЭР): 464 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 6,89 (шс, 1H), 6,71 (с, 2Н), 6,54 (с, 1H), 6,51-6,37 (м, 2Н), 5,76-5,71 (м, 1H), 5,40 (шс, 1H), 5,03-4,95 (м, 1H), 4,06-3,89 (м, 3Н), 3,82 (с, 6Н), 3,78-3,67 (м, 1Н), 3,06-3,02 (м, 2Н), 2,69-2,52 (м, 1H), 2,46-2,35 (м, 1H), 1,15-0,98 (м, 1H), 0,63-0,60 (м, 2Н), 0,29-0,27 (м, 2Н).
Пример 27
(S)-1-(1-Акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-5-((2,2,2-трифторэтил)амино)-1Н-пиразол-4-карбоксамид
Стадия 1
(S)-3-(4-Циано-3-((3,5-диметоксифенил)этинил)-5-((2,2,2-трифторэтил)амино)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир
(S)-3-(5-Амино-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир 7d (430 мг, 0,98 ммоль), водный раствор трифторацетальдегида (75%) (304 мг, 1,96 ммоль) и тетраэтилтитанат (448 мг, 1,96 ммоль) добавляли к дихлорметану (15 мл), перемешивали в течение 2 часов; после завершения реакции к реакционной смеси добавляли боргидрид натрия (75 мг, 1,96 ммоль) и продолжали перемешивание при комнатной температуре в течение 1 часа, реакционную смесь выливали в воду и экстрагировали этилацетатом (20 мл × 3), органическую фазу объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования, и концентрировали реакционную систему при пониженном давлении, и остаток быстро очищали на колонке с получением указанного в заголовке продукта, (S)-3-(4-циано-3-((3,5-диметоксифенил)этинил)-5-((2,2,2-трифторэтил)амино)-1H-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 27а (120 мг, желтое маслянистое вещество), и выход составлял 26%.
МС m/z (ИЭР): 464 [М+1-56]
Пример 27 синтезировали со ссылкой на осуществление второй-четвертой стадий, выполненных в Примере 24.
МС m/z (ИЭР): 492 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 6,92 (шс, 1H), 6,70 (с, 2Н), 6,52 (с, 1H), 6,47-6,39 (м, 2Н), 6,31-6,25 (м, 1H), 5,75-5,65 (м, 1H), 5,65 (шс, 1H), 5,05-4,98 (м, 1Н), 4,10-3,88 (м, 3Н), 3,80 (с, 6Н), 3,75-3,61 (м, 3Н), 2,63-2,34 (м, 2Н).
Пример 28
(S)-1-(1-Акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-5-((2-метоксиэтил)амино)-1Н-пиразол-карбоксамид
Пример 28 синтезировали способом из Примера 25, но на первой стадии использовали 1-бром-2-метоксиэтан вместо 2-бромпропана.
MC m/z (ИЭР): 468 [M+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 7,32 (шс, 1H), 6,74 (д, J=2,2 Гц, 2Н), 6,64 (дд, J=16,8, 10,4 Гц, 1H), 6,60 (т, J=2,2 Гц, 1H), 6,50 (т, J=6,0 Гц, 1H), 6,16 (дд, J=16,8, 5,0 Гц, 1H), 5,68 (т, J=10,8 Гц, 1H), 5,15-5,05 (м, 1H), 4,05-4,01 (м, 0,5Н), 3,86-3,81 (м, 1,5Н), 3,77 (с, 6Н), 3,70-3,61 (м, 1H), 3,59-3,50 (м, 1H), 3,46 (т, J=5,1 Гц, 2Н), 3,39-3,34 (м, 2Н), 3,26 (с, 3Н), 2,42-2,23 (м, 2Н).
Пример 29
(S)-1-(1-Акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-5-((2-гидроксиэтил)амино)-1Н-пиразол-4-карбоксамид
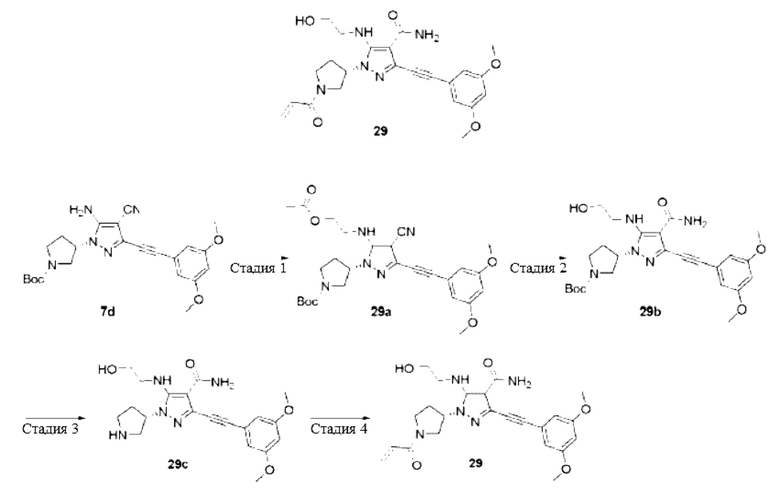
Стадия 1
(S)-3-(5-((2-Ацетоксиэтил)амино)-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир
Смесь (S)-3-(5-амино-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 7d (300 мг, 0,685 ммоль), 2-бромэтилацетата (126 мг, 0,753 ммоль), карбоната цезия (447 мг, 1,37 ммоль) и ацетонитрила (4 мл) нагревали до 90°С, перемешивали в течение 2 часов, реакционный раствор охлаждали до комнатной температуры и выливали в воду (50 мл), и затем экстрагировали этилацетатом (30 мл × 3), органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования, и концентрировали реакционную систему при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (дихлорметан/метанол = 15/1) с получением указанного в заголовке продукта, (S)-3-(5-((2-ацетоксиэтил)амино)-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 29а (148 мг, желтое твердое вещество), и выход составлял 41%.
MC m/z (ИЭР): 468 [М+1-56]
Стадия 2
(S)-3-(4-Карбамоил-3-((3,5-диметоксифенил)этинил)-5-((2-гидроксиэтил)амино)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир
К смеси (S)-3-(5-((2-ацетоксиэтил)амино)-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 29а (68 мг, 0,146 ммоль), этанола (5 мл) и диметилсульфоксида (1 мл) добавляли насыщенный раствор гидроксида натрия (3 мл) и пероксида водорода (4 мл), перемешивали при 30°С в течение 1 часа; после завершения реакции реакционный раствор выливали в насыщенный раствор сульфита натрия (30 мл) и экстрагировали этилацетатом (30 мл × 3), органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования, и концентрировали реакционную систему при пониженном давлении с получением указанного в заголовке продукта, (S)-3-(4-карбамоил-3-((3,5-диметоксифенил)этинил)-5-((2-гидроксиэтил)амино)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 29b (110 мг, неочищенный, желтое маслянистое вещество), и выход составлял >100%. Продукт использовали для следующей реакции без очистки.
MC m/z (ИЭР): 444 [М+1-56]
Стадия 3
(S)-3-((3,5-Диметоксифенил)этинил)-5-((2-гидроксиэтил)амино)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамид
Соединение (S)-3-(4-карбамоил-3-((3,5-диметоксифенил)этинил)-5-((2-гидроксиэтил)амино)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир 29b (110 мг, 0,146 ммоль, неочищенный) растворяли в растворе хлористоводородной кислоты в метаноле (5 мл), нагревали до 40°С и перемешивали в течение 1 часа; после завершения реакции реакционную систему концентрировали при пониженном давлении с получением указанного в заголовке продукта, (S)-3-((3,5-диметоксифенил)этинил)-5-((2-гидроксиэтил)амино)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамида 29 с (160 мг, неочищенный, белое твердое вещество), и выход составлял >100%. Продукт использовали для следующей реакции без очистки.
МС m/z (ИЭР): 400 [М+1]
Стадия 4
(S)-1-(1-акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-5-((2-гидроксиэтил)амино)-1Н-пиразол-4-карбоксамид
Соединение (S)-3-((3,5-диметоксифенил)этинил)-5-((2-гидроксиэтил)амино)-1-(пирролидин-3-ил)-1Н-пиразол-4-карбоксамид 29 с (160 мг, 0,146 ммоль, неочищенный) растворяли в тетрагидрофуране (5 мл) и затем добавляли насыщенный раствор бикарбоната натрия (10 мл), и затем добавляли акрилоилхлорид (12 мг, 0,13 ммоль), перемешивали при комнатной температуре в течение 10 минут. После завершения реакции реакционный раствор выливали в воду (50 мл) и экстрагировали этилацетатом (30 мл × 3), органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования и концентрировали реакционную систему при пониженном давлении, остаток очищали обращенно-фазовой высокоэффективной препаративной хроматографией [ацетонитрил/вода (с 0,2% муравьиной кислоты): от 20% до 60%] с получением указанного в заголовке продукта, (S)-1-(1-акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-5-((2-гидроксиэтил)амино)-1Н-пиразол-4-карбоксамида 29 (6 мг, белое твердое вещество), и выход составлял 9%.
МС m/z (ИЭР): 454 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 7,36 (шс, 1H), 6,76 (шс, 1Н), 6,74 (с, 2Н), 6,70-6,60 (м, 2Н), 6,55-6,52 (м, 1Н), 6,17 (д, J=16,9 Гц, 1H), 5,69 (т, J=10,9 Гц, 1H), 5,16-5,10 (м, 1Н), 4,87 (с, 1Н), 4,06-4,0 (м, 0,5Н), 3,83-3,81 (м, 1,5Н), 3,77 (с, 6Н), 3,68-3,63 (м, 2Н), 3,55-3,53 (м, 2Н), 3,28-3,26 (м, 2Н), 2,38 - 2,27 (м, 2Н).
Пример 30
(S)-1-(1-акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-5-((3-морфолинопропил)амино)-1Н-пиразол-4-карбоксамид
Стадия 1
3-Морфолинопрогаш-4-метилбензолсульфонат
Соединение 3-морфолинопропан-1-ол 30а (500 мг, 3,45 ммоль) растворяли в дихлорметане (100 мл) и затем добавляли 4-диметиламинопиридин (42 мг, 0,34 ммоль), триэтиламин (1,04 г, 10,3 ммоль) и п-толуолсульфонилхлорид (988 мг, 5,17 ммоль), перемешивали при комнатной температуре в течение ночи; после завершения реакции реакционный раствор выливали в воду (50 мл) и затем экстрагировали дихлорметаном (50 мл × 3), органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования и концентрировали реакционную систему при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 2/1) с получением указанного в заголовке продукта, 3-морфолинопропил 4-метилбензолеульфоната 30b (660 мг, желтое маслянистое вещество), и выход составлял 64%.
MC m/z(ИЭР): 300 [М+1]
Пример 30 синтезировали со ссылкой на способ из Примера 25, но на первой стадии использовали 3-морфолинопропил-4-метоксибензолсульфонат вместо 2-бромпропана.
МС m/z (ИЭР): 537 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8,23 (шс, 1H), 7,12 (шс, 1H), 6,94 (шс, 1H), 6,69 (с, 2Н), 6,52 (с, 1H), 6,49-6,40 (м, 2Н), 5,92 (шс, 1Н), 5,74-5,70 (м, 1H), 5,03-4,96 (м, 1H), 4,09-3,90 (м, 3Н), 3,80-3,68 (м, 11Н), 3,28 (шс, 2Н), 2,89 (шс, 6Н), 2,69-2,33 (м, 2Н), 1,93 (шс, 2Н).
Пример 31
(S)-1-(1-Акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-5-((2-морфолиноэтил)амино)-1Н-пиразол-карбоксамид
Пример 31 синтезировали со ссылкой на способ из Примера 25, но на первой стадии вместо 2-бромпропана использовали 4-(2-хлорэтил)морфолин.
МС m/z (ИЭР): 523 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8,14 (с, 1H), 6,95 (шс, 1H), 6,69 (с, 2Н), 6,63 (шс, 1H), 6,52 (с, 1H), 6,49-6,39 (м, 2Н), 6,14 (шс, 1H), 5,75-5,70 (м, 1H), 5,06-4,98 (м, 1H), 4,11-3,85 (м, 3Н), 3,80-3,72 (м, 11Н), 3,37-3,33 (м, 2Н), 2,80-2,73 (м, 2Н), 2,65 (шс, 4Н), 2,45-2,32 (м, 2Н).
Пример 32
(S)-1-(1-Акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-5-((2-(пирролидин-1-ил)этил)амино)-1Н-пиразол-4-карбоксамид
2 -Бромэтил-4-метилбензолсульфонат
Соединение 2-бромэтанол 32а (500 мг, 4,0 ммоль), 4-диметиламинопиридин (246 мг, 2,02 ммоль) и триэтиламин (1,22 г, 12,1 ммоль) растворяли в дихлорметане (50 мл), охлаждали до 0°С, затем по частям добавляли п-толуолсульфонилхлорид (1,15 г, 6,05 ммоль), после завершения добавления перемешивали при комнатной температуре в течение ночи; после завершения реакции реакционную смесь выливали в воду (50 мл) и экстрагировали дихлорметаном (50 мл × 3), органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования и концентрировали реакционную систему при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этил ацетат = 10/1) с получением указанного в заголовке продукта, 2-бромэтил-4-метилбензолсульфоната 32b (600 мг, желтое маслянистое вещество), и выход составлял 53%.
МС m/z (ИЭР): 277 [М+1]
Стадия 2
(S)-3-(5-((2-Бромэтал)амино)-4-циано-3-((3,5-диметоксифенил)этинил)-1H-пиразол-1-карбоновой кислоты трет-бутиловый эфир
Смесь (S)-3-(5-амино-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 7d (400 мг, 0,92 ммоль), 2-бромэтил-4-метилбензолсульфоната (380 мг, 1,37 ммоль), карбоната цезия (600 мг, 1,84 ммоль) и ацетонитрила (10 мл) нагревали до 70°С, перемешивали в течение 2 часов, реакционный раствор выливали в воду (50 мл) и экстрагировали этилацетатом (50 мл × 3), органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования и концентрировали реакционную систему при пониженном давлении, остаток быстро очищали на колонке с получением указанного в заголовке продукта, (S)-3-(5-((2-бромэтил)амино)-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-карбоновой кислоты трет-бутилового эфира 32 с (240 мг, коричневое маслянистое вещество), и выход составлял 48%.
МС m/z (ИЭР): 408 [М+1-56-80]
Стадия 3
(S)-3-(4-Циано-3-((3,5-диметоксифенил)этинил)-5-((2-(пирролидин-1-ил)этил)амино)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутиловый эфир
Смесь (S)-3-(5-((2-бромэтил)амино)-4-циано-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-1-карбоновой кислоты трет-бутилового эфира 32 с (240 мг, 0,44 ммоль), пирролидина (47 мг, 0,66 ммоль), карбоната цезия (288 мг, 0,88 ммоль) и ацетонитрила (5 мл) нагревали до 70°С, перемешивали в течение 1,5 часа и выливали реакционный раствор в воду (30 мл), и затем экстрагировали этилацетатом (30 мл × 3), органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования и концентрировали реакционную систему при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (дихлорметан/метанол = 10/1) с получением указанного в заголовке продукта, (S)-3-(4-циано-3-((3,5-диметоксифенил)этинил)-5-((2-(пирролидин-1-ил)этил)амино)-1Н-пиразол-1-ил)пирролидин-1-карбоновой кислоты трет-бутилового эфира 32d (200 мг, желтое маслянистое вещество), и выход составлял 85%.
МС m/z (ИЭР): 479 [М+1-56]
Пример 32 синтезировали со ссылкой на осуществление второй-четвертой рабочих стадий, выполненных в Примере 24.
МС m/z (ИЭР): 507 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 8,38 (с, 1H), 6,99 (шс, 1H), 6,69 (с, 2Н), 6,51 (с, 1H), 6,47-6,36 (м, 2Н), 5,72-5,67 m, 2Н), 5,16-5,08 (м, 1H), 4,12-3,86 (м, 3Н), 3,80-3,62 (м, 9Н), 3,33-3,29 (м, 6Н), 2,62-2,34 (м, 2Н), 2,07 (шс, 4Н).
Пример 33
(S)-1-(1-Акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-5-((тетрагидро-2Н-пиран-4-ил)амино)-1Н-пиразол-4-карбоксамид
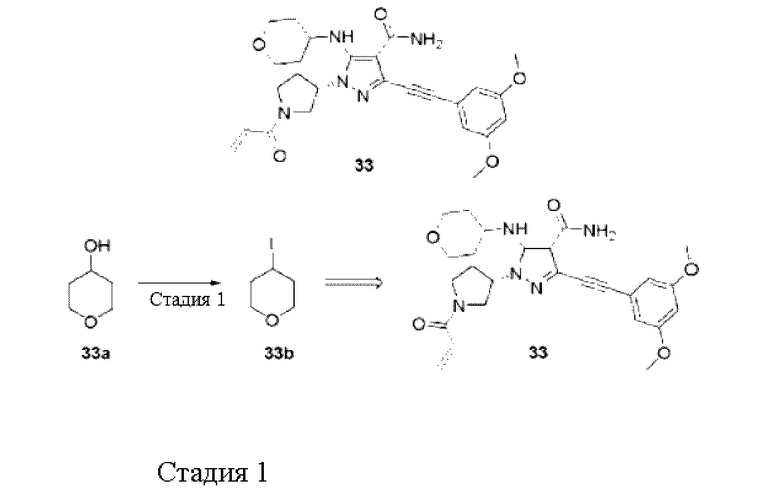
4-Йодтетрагидро-2Н-пиран
4-Гидрокситетрагидро-2Н-пиран 33а (2,04 г, 20 ммоль), трифенилфосфин (6,81 г, 26) и имидазол (2,04 г, 30 ммоль) растворяли в дихлорметане (100 мл) и охлаждали до 0°С, затем добавляли йод (6,09 г, 24 ммоль) и перемешивали при 45°С в течение 14 часов, реакцию гасили водой и затем экстрагировали этилацетатом (50 мл × 2), органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования и концентрировали реакционную систему при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 1/1) с получением указанного в заголовке продукта, 4-йодтетрагидро-2Н-пирана ЗЗЬ (2,12 г, белое твердое вещество), и выход составлял 50%.
1Н ЯМР (400 МГц, ДМСО-d6) δ 4,62 (дт, J=13,9, 4,5 Гц, 1H), 3,68-3,64 (м, 2Н), 3,47-3,42 (м, 2Н), 2,13-1,97 (м, 4Н).
Пример 33 синтезировали способом из Примера 24, но на первой стадии использовали 4-йодтетрагидро-2Н-пиран вместо этилйодида.
МС m/z (ИЭР): 494 [М+Н]
1Н ЯМР (400 МГц, CD3OD) δ 6,74 (т, J=2,2 Гц, 2Н), 6,71-6,62 (м, 1H), 6,60-6,58 (м, 1Н), 6,36-6,30 (м, 1H), 5,82-5,77 (м, 1H), 5,18-5,12 (м, 1H), 4,04-3,94 (м, 6Н), 3,81 (с, 6Н), 3,52-3,46 (м, 3Н), 2,55-2,39 (м, 2Н), 1,94-1,92 (м, 2Н), 1,60-1,55 (м, 2Н).
Пример 34
(S)-1-(1-Акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-5-((1-метилпиперидин-4-ил)амино)-1Н-пиразол-4-карбоксамид
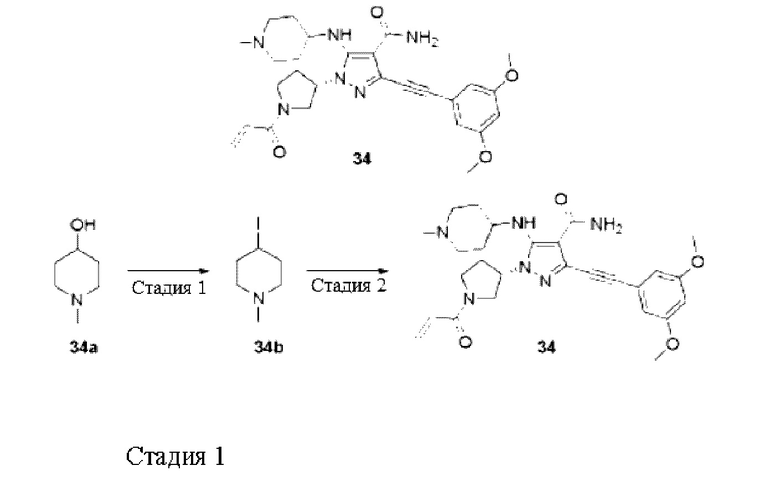
4-Йод-1-метилпиперидин
4-Гидрокси-1-метилпиперидин 34а (2,3 г, 20 ммоль), трифенилфосфин (6,81 г, 26 ммоль), имидазол (2,04 г, 30 ммоль) и дихлорметан (100 мл) смешивали и охлаждали до 0°С и затем добавляли йод (6,09 г, 24 ммоль), и продолжали перемешивание в течение 18 часов; после завершения реакции смесь гасили водой и затем экстрагировали дихлорметаном (50 мл × 2), органические фазы объединяли и сушили над безводным сульфатом натрия, осушающий агент удаляли посредством фильтрования и концентрировали реакционную систему при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (дихлорметан/метанол = 10/1) с получением указанного в заголовке продукта, 4-йод-1-метилпиперидина 34b (2,25 г, белое твердое вещество), и выход составлял 50%.
МС m/z (ИЭР): 226 [М+Н]
Стадия 2
(S)-1-(1-Акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-5-((1-метилпиперидин-4-ил)амино)-1Н-пиразол-4-карбоксамид
Соединение (S)-1-(1-акрилоилпирролидин-3-ил)-5-амино-3-((3,5-диметоксифенил)этинил)-1Н-пиразол-4-карбоксамид 7 (210 мг, 0,5 ммоль), 4-йод-1-метилпиперидин 34b (450 мг, 2 ммоль), карбонат калия (207 мг, 1,5 ммоль) и ацетонитрил (10 мл) смешивали и нагревали, и перемешивали при 90°С в течение 13 часов, растворитель удаляли из реакционной смеси при пониженном давлении и затем растворяли реакционную смесь, и затем экстрагировали этилацетатом (50 мл × 2), органическую фазу объединяли и удаляли растворитель при пониженном давлении, остаток очищали обращенно-фазовой препаративной жидкостной хроматографией с получением указанного в заголовке продукта, (S)-1-(1-акрилоилпирролидин-3-ил)-3-((3,5-диметоксифенил)этинил)-5-((1-метилпиперидин-4-ил)амино)-1Н-пиразол-4-карбоксамида 34 (8,1 мг, белое твердое вещество), и выход составлял 3,2%.
МС m/z (ИЭР): 507 [М+Н]
1Н ЯМР (400 МГц, CD3OD) δ 6,77 (с, 2Н), 6,68-6,65 (м, 1H), 6,59 (с, 1H), 6,34-6,30 (м, 1H), 5,81-5,78 (м, 1H), 5,69-5,67 (м, 1Н), 5,03-5,00 (м, 1H), 4,98-5,95(м, 1H), 4,92-4,90 (м, 1H), 4,36 (с, 2Н), 4,12-4,07 (м, 1H), 4,00-3,98 (м, 1H), 3,93-3,90 (м, 1H), 3,86-3,84 (м, 1H), 3,81 (с, 6Н), 3,72-3,68 (м, 1H), 2,53-2,47 (м, 3Н), 2,40-2,38 (м, 1H), 2,32 (с, 3Н), 2,27-2,21 (м, 1H).
Биологические эксперименты
Испытание ингибирования активности FGFR
Оценивали влияние соединений по данному изобретению на in vitro активность FGFR, измеряя степень фосфорилирования субстрата в киназной реакции с использованием набора для анализа киназ HTRF (таблица 1).
Испытание ингибирования активности FGFR1
Ниже представлено обобщенное описание экспериментальных способов:
Реакционный буфер содержал следующие компоненты: 5-кратно разбавленный раствор ферментативного буфера/киназы 5Х (Cisbio, кат. №62EZBFDD) (основной компонент 50 мМ HEPES, рН 7,0), 5 мМ MgCl2 и 1 мМ DTT; каталитический домен белка человеческого рекомбинантного FGFR1 (аминокислоты 308-731), очищенный указанной компанией, 0,6 нг/мкл раствор киназы, разбавленный реакционным буфером; реакционный раствор субстрата, содержащий 400 нМ биотинилированного тирозинкиназного субстрата, разбавленный реакционным буфером (Cisbio, кат. №62ТК0РЕС), и 40 мкМ АТФ; испытуемый раствор содержал 0,125 нг/мкл антитела Cage с меткой Eu3+ (Cisbio, кат. №61T66KLB), разбавленного экспериментальным буфером (Cisbio, кат. №62SDBRDF), и 25 нМ XL665 со стрептавидиновой меткой (Cisbio, кат. №610SAXLB).
Соединения разбавляли до 1 мМ в ДМСО, затем получали серийные 4-кратные разбавления с ДМСО до минимальной концентрации 0,061 мкМ, и каждую концентрацию дополнительно разбавляли в 40 раз реакционным буфером. Если значение IC50 соединения является слишком низким, то исходную концентрацию соединения можно уменьшить.
Добавляли 4 мкл раствора соединения и 2 мкл раствора киназы FGFR1 в 384-луночный аналитический планшет (Thermo, кат. №264706), тщательно смешивали и инкубировали в течение 15 минут при комнатной температуре; затем добавляли 4 мкл реакционного раствора субстрата и инкубировали реакционную смесь в течение 60 минут при комнатной температуре; и затем реакцию останавливали посредством добавления равного объема 10 мкл исследуемого раствора и тщательно перемешивали смесь, и оставляли стоять при комнатной температуре. Через 60 минут фосфорилированный продукт одновременно распознавали по антителу Cage с меткой Eu3+ (донор) и по антителу XL665 со стрептавидиновой меткой (акцептор); после лазерного возбуждения между донором и акцептором, находящимися вблизи друг друга, возникал резонансный перенос энергии, и энергия переходила от донора (620 нм) к акцептору (665 нм), что можно было обнаружить с помощью микропланшет-ридера EnVision (Perkin Elmer). Соотношение 665/620 положительно коррелирует со степенью фосфорилирования субстрата; таким образом, определяли активность киназы FGFR1.
В данном эксперименте группу без фермента использовали в качестве группы со 100% ингибированием, группу с ферментом, но без соединения использовали в качестве группы с 0% ингибированием. Таким образом, процент ингибирования активности FGFR1 под действием соединения рассчитывали по следующей формуле:
Процент ингибирования = 100-100* (соотношение с соединением - соотношение при 100% ингибировании) / (соотношение при 0% ингибировании - соотношение при 100% ингибировании)
Значение IC50 соединения рассчитывали для 10 точек концентрации с помощью программного обеспечения XLfit в Excel по следующей формуле:
Y = Нижнее + (верхнее-нижнее)/(1+10^((logIC50-Х)*коэффициент наклона))
Где Y представляет собой процент ингибирования, Нижнее представляет собой значение нижнего плато S-образной кривой, Верхнее представляет собой значение верхнего плато S-образной кривой, X представляет собой логарифм концентрации испытуемого соединения, и коэффициент наклона представляет собой коэффициент наклона кривой.
Испытание ингибирования активности FGFR2
Ниже представлено обобщенное описание экспериментальных способов:
Реакционный буфер содержал следующие компоненты: 5-кратно разбавленный раствор ферментативного буфера/киназы 5Х (Cisbio, кат. №62EZBFDD) (основной компонент 50 мМ HEPES, рН 7,0), 5 мМ MgCl2 и 1 мМ DTT; каталитический домен белка человеческого рекомбинантного FGFR2 (аминокислоты 400-821), который приобретали у компании Yiqiao Shenzhou Biotech Co., 0,45 нг/мкл раствор киназы, разбавленный реакционным буфером; реакционный раствор субстрата, содержащий 800 нМ биотинилированного тирозинкиназного субстрата, разбавленный реакционным буфером (Cisbio, кат. №62ТК0РЕС), и 50 мкМ АТФ; испытуемый раствор содержал 0,125 нг/мкл антитела Cage с меткой Eu3+ (Cisbio, кат. №61T66KLB), разбавленного экспериментальным буфером (Cisbio, кат. №62SDBRDF), и 50 нМ XL665 со стрептавидиновой меткой (Cisbio, кат. №610SAXLB).
Соединения разбавляли до 1 мМ в ДМСО, затем получали серийные 4-кратные разбавления с ДМСО до минимальной концентрации 0,061 мкМ, и каждую концентрацию дополнительно разбавляли в 40 раз реакционным буфером. Если значение IC50 соединения является слишком низким, то исходную концентрацию соединения можно уменьшить.
Добавляли 4 мкл раствора соединения и 2 мкл раствора киназы FGFR2 в 384-луночный аналитический планшет (Thermo, кат. №264706), тщательно смешивали и инкубировали в течение 15 минут при комнатной температуре; затем добавляли 4 мкл реакционного раствора субстрата и инкубировали реакционную смесь в течение 60 минут при комнатной температуре; и затем реакцию останавливали посредством добавления равного объема 10 мкл исследуемого раствора и тщательно перемешивали смесь, и оставляли стоять при комнатной температуре. Через 60 минут фосфорилированный продукт одновременно распознавали по антителу Cage с меткой Eu3+ донор) и по антителу XL665 со стрептавидиновой меткой (акцептор); после лазерного возбуждения между донором и акцептором, находящимися вблизи друг друга, возникал резонансный перенос энергии, и энергия переходила от донора (620 нм) к акцептору (665 нм), что можно было обнаружить с помощью микропланшет-ридера EnVision (Perkin Elmer). Соотношение 665/620 положительно коррелирует со степенью фосфорилирования субстрата; таким образом, определяли активность киназы FGFR2.
В данном эксперименте группу без фермента использовали в качестве группы со 100% ингибированием, группу с ферментом, но без соединения использовали в качестве группы с 0% ингибированием. Таким образом, процент ингибирования активности FGFR2 под действием соединения рассчитывали по следующей формуле:
Процент ингибирования=100-100* (соотношение с соединением - соотношение при 100% ингибировании) / (соотношение при 0% ингибировании - соотношение при 100% ингибировании)
Значение IC50 соединения рассчитывали для 10 точек концентрации с помощью программного обеспечения XLfit в Excel по следующей формуле:
Y=Нижнее+(верхнее-нижнее)/(1+10^((logIC50-X)*коэффициент наклона))
Где Y представляет собой процент ингибирования, Нижнее представляет собой значение нижнего плато S-образной кривой, Верхнее представляет собой значение верхнего плато S-образной кривой, X представляет собой логарифм концентрации испытуемого соединения, и коэффициент наклона представляет собой коэффициент наклона кривой.
Испытание ингибирования активности FGFR3
Ниже представлено обобщенное описание экспериментальных способов:
Реакционный буфер содержал следующие компоненты: 5-кратно разбавленный раствор ферментативного буфера/киназы 5Х (Cisbio, кат. №62EZBFDD) (основной компонент 50 мМ HEPES, рН 7,0), 5 мМ MgCl2 и 1 мМ DTT; каталитический домен белка человеческого рекомбинантного FGFR3 (аминокислоты 399-806), который приобретали у компании Yiqiao Shenzhou Biotech Co., 0,3 нг/мкл раствор киназы, разбавленный реакционным буфером; реакционный раствор субстрата, содержащий 1000 нМ биотинилированного тирозинкиназного субстрата, разбавленный реакционным буфером (Cisbio, кат. №62ТК0РЕС), и 90 мкМ АТФ; испытуемый раствор содержал 0,125 нг/мкл антитела Cage с меткой Eu3+ (Cisbio, кат. №61T66KLB), разбавленного экспериментальным буфером (Cisbio, кат. №62SDBRDF), и 62,5 нМ XL665 со стрептавидиновой меткой (Cisbio, кат. №610SAXLB).
Соединения разбавляли до 1 мМ в ДМСО, затем получали серийные 4-кратные разбавления с ДМСО до минимальной концентрации 0,061 мкМ, и каждую концентрацию дополнительно разбавляли в 40 раз реакционным буфером. Если значение IC50 соединения является слишком низким, то исходную концентрацию соединения можно уменьшить.
Добавляли 4 мкл раствора соединения и 2 мкл раствора киназы FGFR3 в 384-луночный аналитический планшет (Thermo, кат. №264706), тщательно смешивали и инкубировали в течение 15 минут при комнатной температуре; затем добавляли 4 мкл реакционного раствора субстрата и инкубировали реакционную смесь в течение 60 минут при комнатной температуре; и затем реакцию останавливали посредством добавления равного объема 10 мкл исследуемого раствора и тщательно перемешивали смесь, и оставляли стоять при комнатной температуре. Через 60 минут фосфорилированный продукт одновременно распознавали по антителу Cage с меткой Eu3+ донор) и по антителу XL665 со стрептавидиновой меткой (акцептор); после лазерного возбуждения между донором и акцептором, находящимися вблизи друг друга, возникал резонансный перенос энергии, и энергия переходила от донора (620 нм) к акцептору (665 нм), что можно было обнаружить с помощью микропланшет-ридера EnVision (Perkin Elmer). Соотношение 665/620 положительно коррелирует со степенью фосфорилирования субстрата; таким образом, определяли активность киназы FGFR3.
В данном эксперименте группу без фермента использовали в качестве группы со 100% ингибированием, группу с ферментом, но без соединения использовали в качестве группы с 0% ингибированием. Таким образом, процент ингибирования активности FGFR3 под действием соединения рассчитывали по следующей формуле:
Процент ингибирования=100-100* (соотношение с соединением - соотношение при 100% ингибировании) / (соотношение при 0% ингибировании - соотношение при 100% ингибировании)
Значение IC50 соединения рассчитывали для 10 точек концентрации с помощью программного обеспечения XLfit в Excel по следующей формуле:
Y=Нижнее+(верхнее-нижнее)/(1+10^((logIC50-Х)*коэффициент наклона))
Где Y представляет собой процент ингибирования, Нижнее представляет собой значение нижнего плато S-образной кривой, Верхнее представляет собой значение верхнего плато S-образной кривой, X представляет собой логарифм концентрации испытуемого соединения, и коэффициент наклона представляет собой коэффициент наклона кривой.
Испытание ингибирования активности FGFR4
Ниже представлено обобщенное описание экспериментальных способов:
Реакционный буфер содержал следующие компоненты: 5-кратно разбавленный раствор ферментативного буфера/киназы 5Х (Cisbio, кат. № 62EZBFDD) (основной компонент 50 мМ HEPES, рН 7,0), 5 мМ MgCl2 и 1 мМ DTT; каталитический домен белка человеческого рекомбинантного FGFR4 (аминокислоты 460-802), который приобретали у компании Tsinghua University Protein Research Technology Center, 0,5 нг/мкл раствор киназы, разбавленный реакционным буфером; реакционный раствор субстрата, содержащий 500 нМ биотинилированного тирозинкиназного субстрата, разбавленный реакционным буфером (Cisbio, кат. №62ТК0РЕС), и 90 мкМ АТФ; испытуемый раствор содержал 0,125 нг/мкл антитела Cage с меткой Eu3+ (Cisbio, кат.№61T66KLB), разбавленного экспериментальным буфером (Cisbio, кат. №62SDBRDF), и 31,25 нМ XL665 со стрептавидиновой меткой (Cisbio, кат. №610SAXLB).
Соединения разбавляли до 1 мМ в ДМСО, затем получали серийные 4-кратные разбавления с ДМСО до минимальной концентрации 0,061 мкМ, и каждую концентрацию дополнительно разбавляли в 40 раз реакционным буфером. Если значение IC50 соединения является слишком низким, то исходную концентрацию соединения можно уменьшить.
Добавляли 4 мкл раствора соединения и 2 мкл раствора киназы FGFR4 в 384-луночный аналитический планшет (Thermo, кат. №264706), тщательно смешивали и инкубировали в течение 15 минут при комнатной температуре; затем добавляли 4 мкл реакционного раствора субстрата и инкубировали реакционную смесь в течение 60 минут при комнатной температуре; и затем реакцию останавливали посредством добавления равного объема 10 мкл исследуемого раствора и тщательно перемешивали смесь, и оставляли стоять при комнатной температуре. Через 60 минут фосфорилированный продукт одновременно распознавали по антителу Cage с меткой Eu3+ (донор) и по антителу XL665 со стрептавидиновой меткой (акцептор); после лазерного возбуждения между донором и акцептором, находящимися вблизи друг друга, возникал резонансный перенос энергии, и энергия переходила от донора (620 нм) к акцептору (665 нм), что можно было обнаружить с помощью микропланшет-ридера EnVision (Perkin Elmer). Соотношение 665/620 положительно коррелирует со степенью фосфорилирования субстрата; таким образом, определяли активность киназы FGFR4.
В данном эксперименте группу без фермента использовали в качестве группы со 100% ингибированием, группу с ферментом, но без соединения использовали в качестве группы с 0% ингибированием. Таким образом, процент ингибирования активности FGFR4 под действием соединения рассчитывали по следующей формуле:
Процент ингибирования=100-100* (соотношение с соединением - соотношение при 100% ингибировании) / (соотношение при 0% ингибировании - соотношение при 100% ингибировании)
Значение IC50 соединения рассчитывали для 10 точек концентрации с помощью программного обеспечения XLfit в Excel по следующей формуле:
Y=Нижнее+(верхнее-нижнее)/(1+10^((logIC50-Х)*коэффициент наклона))
Где Y представляет собой процент ингибирования, Нижнее представляет собой значение нижнего плато S-образной кривой, Верхнее представляет собой значение верхнего плато S-образной кривой, X представляет собой логарифм концентрации испытуемого соединения, и коэффициент наклона представляет собой коэффициент наклона кривой.
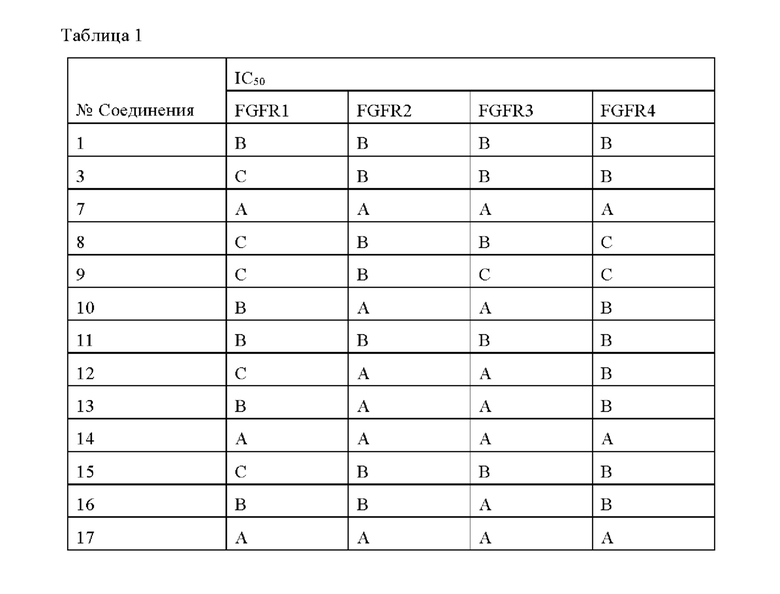
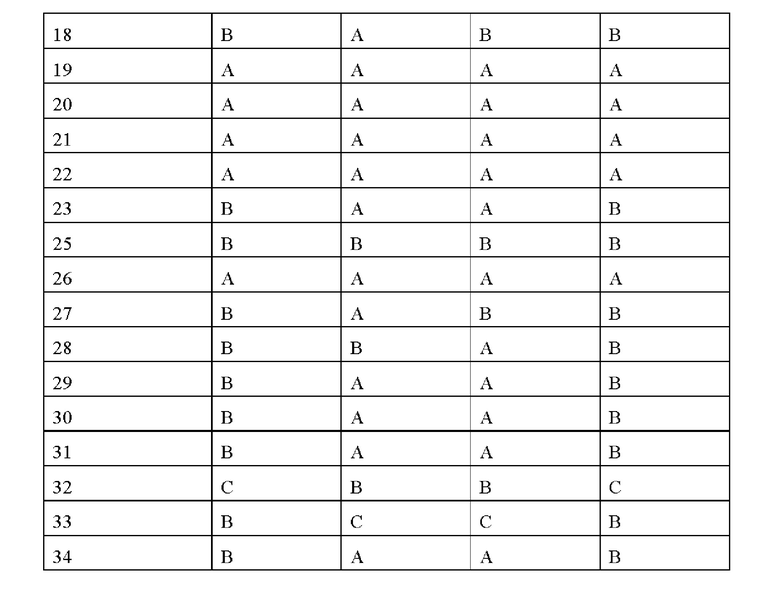
А<10 нМ; 10 нМ ≤В < 100 нМ; 100 нМ ≤ С < 1000 нМ
Соединения в примерах по данному изобретению оказывают существенное ингибирующее действие на активность FGFR, предпочтительно с IC50 от 100 до 1000 нМ, более предпочтительно IC50 менее 100 нМ, и наиболее предпочтительно IC50 менее 10 нМ.
Определение ингибирования пролиферации клеток Нер3В
Оценивали влияние соединений по данному изобретению на пролиферацию клеток в клеточной линии гепатомы Нер3В, используя люминесцентный анализ жизнеспособности клеток (таблица 2).
Ниже представлено обобщенное описание экспериментальных способов:
Реагент CellTilter-Glo (Promega, кат. №G7572) состоял из лиофилизированного порошка CTG и буфера CTG. Перед применением лиофилизированный порошок необходимо растворить в указанном буфере.
Соединение разбавляли в ДМСО (Sigma, кат. №D5879) до 5 мМ, затем серийно разбавляли в 4 раза с помощью ДМСО до минимальной концентрации 0,31 мкМ, и каждую точку концентрации дополнительно разбавляли в 50 раз, используя среду DMEM, не содержащую FBS (ThermoFisher, кат. №11995073). Если значение IC50 соединения является слишком низким, то исходную концентрацию соединения можно уменьшить.
Клетки Нер3В (приобретенные у компании Cell Resource Center Шанхайского института биологических наук Китайской Академии наук) выращивали в полной культуральной среде DMEM, содержащей смесь 10% раствора FBS (GBICO, кат. №10099-141) и 100 ед./мл стрептомицина (ThermoFisher, кат. №15140122). Когда клетки в культуральном контейнере достигали 80-90% слияния, клетки обрабатывали 0,25% трипсином (содержащим ЭДТК) (ThermoFisher, кат. 25200056), диспергировали и затем помещали на белый 384-луночный культуральный планшет (ThermoFisher, кат. №164610), и каждая лунка содержала около 1000 клеток (27 мкл полной культуральной среды DMEM), затем инкубировали 384-луночные планшеты в течение ночи (от 18 до 20 часов) в инкубаторе при 37°С с 5% CO2.
После инкубации в течение ночи в каждую лунку добавляли 3 мкл раствора соединения, разбавленного DMEM, и затем осторожно центрифугировали для тщательного перемешивания, и затем помещали 384-луночный планшет в инкубатор при 37°С с 5% CO2 для продолжения выращивания, и через 72 часа планшет извлекали из оставляли стоять при комнатной температуре в течение 30 минут, затем добавляли 15 мкл реагента CTG на лунку, нагревали до комнатной температуры, планшет осторожно встряхивали на шейкере в течение 3 минут для обеспечения достаточного лизиса клеток, оставляли стоять на 10 минут для стабилизации сигнала люминесценции, а затем считывали сигнал люминесценции на приборе EnVision (Perkin Elmer).
Люминесцентный сигнал в группе BLU9931 (Cancer Discovery 2015, 5, 424) с 10 мкМ Blueprint использовали в качестве сигнала при 100% ингибировании, а люминесцентный сигнал в группе с 0,2% ДМСО использовали в качестве сигнала при 0% ингибировании.
Процент ингибирования пролиферации клеток НерЗВ под действием соединения можно рассчитать по следующей формуле:
Процент ингибирования=100-100* (соотношение с соединением - соотношение при 100% ингибировании) / (соотношение при 0% ингибировании - соотношение при 100% ингибировании)
Значение IC50 соединения рассчитывали по 8 точкам концентрации, используя программное обеспечение XLfit (ID Business Solutions Ltd., Великобритания), по следующей формуле:
Y=Нижнее+(верхнее-нижнее)/(1+10^((logIC50-Х)*коэффициент наклона))
Где Y представляет собой процент ингибирования, Нижнее представляет собой значение нижнего плато S-образной кривой, Верхнее представляет собой значение верхнего плато S-образной кривой, X представляет собой логарифм концентрации испытуемого соединения, и коэффициент наклона представляет собой коэффициент наклона кривой.
Определение ингибирования пролиферации клеток RT4
Оценивали влияние соединений по данному изобретению на пролиферацию клеток в клеточной линии рака мочевого пузыря RT4, используя люминесцентный анализ жизнеспособности клеток (таблица 2).
Использовали такой же экспериментальный способ, как способ, описанный для определения ингибирования пролиферации клеток Нер3В. Клетки RT4 приобретали у компании Cell Resource Center Шанхайского Института биологических наук Китайской Академии наук, и в качестве положительного контроля использовали ((S)-1-(3-(4-амино-3-((3,5-диметоксифенил)этинил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)пирролидин-1-ил)проп-2-ен-1-он), описанный в примере 1 заявки на патент Taiho WO 2015008844 A1.
Определение ингибирования пролиферации клеток SNU-16
Оценивали влияние соединений по данному изобретению на пролиферацию клеток в клеточной линии рака желудка SNU-16, используя люминесцентный анализ жизнеспособности клеток (таблица 2).
Использовали такой же экспериментальный способ, как способ, описанный для определения ингибирования пролиферации клеток Нер3В. Клетки SNU-16 представляют собой продукт НВ-8064 Американской коллекции типовых культур (АТСС), в качестве положительного контроля использовали BJG398 производства компании Novartis.
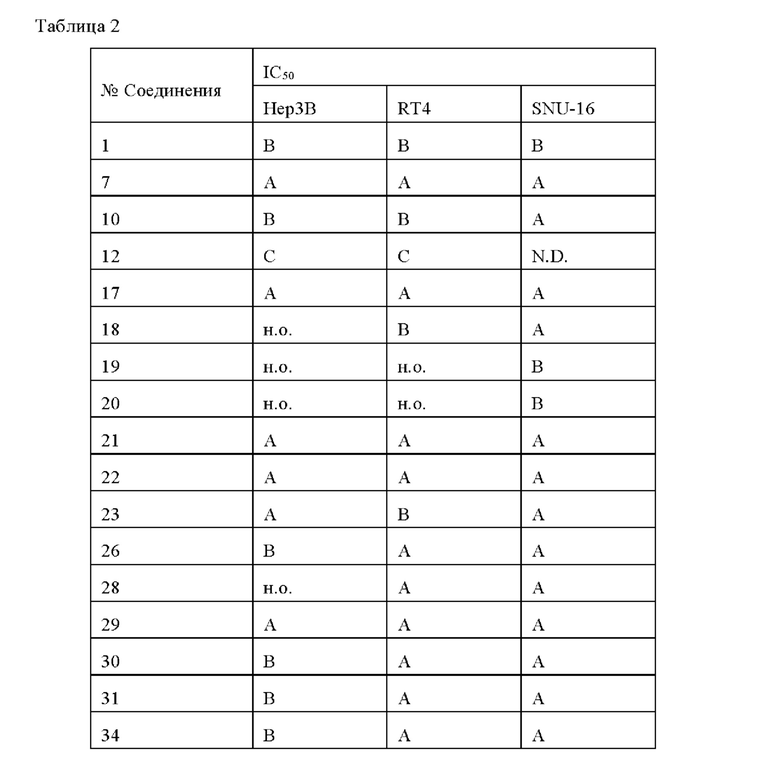
Примечание: А<10 нМ; 10 нМ ≤ В < 100 нМ; 100 нМ ≤ С < 1000 нМ и.о.: не обнаружено
Соединения, описанные в примерах данного изобретения, оказывают существенное ингибирующее действие на пролиферацию клеток Нер3В, RT4 и SNU-16, соответственно, предпочтительно с IC50 от 100 до 1000 нМ, и более предпочтительно IC50 менее 100 нМ.
| название | год | авторы | номер документа |
|---|---|---|---|
| 3,5-ДИЗАМЕЩЕННОЕ АЛКИНИЛБЕНЗОЛЬНОЕ СОЕДИНЕНИЕ И ЕГО СОЛЬ | 2013 |
|
RU2576384C1 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2011 |
|
RU2652638C2 |
| НОВОЕ КОНДЕНСИРОВАННОЕ ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2014 |
|
RU2666349C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛОПИРИМИДИНЫ И ЗАМЕЩЕННЫЕ ПУРИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ УБИКВИТИН-СПЕЦИФИЧЕСКОЙ ПРОЦЕССИРУЮЩЕЙ ПРОТЕАЗЫ 1 (USP1) | 2019 |
|
RU2833222C2 |
| ПРОИЗВОДНОЕ ПИПЕРИДИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2730508C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| КЛАСС БИФУНКЦИОНАЛЬНЫХ ХИМЕРНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ДЛЯ НАПРАВЛЕННОГО РАЗРУШЕНИЯ АНДРОГЕННЫХ РЕЦЕПТОРОВ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2825000C2 |
| ЗАМЕЩЕННЫЕ НИКОТИНИМИДНЫЕ ИНГИБИТОРЫ ВТК, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ В ТЕРАПИИ РАКОВЫХ, ВОСПАЛИТЕЛЬНЫХ И АУТОИММУННЫХ ЗАБОЛЕВАНИЙ | 2014 |
|
RU2677884C2 |
| ПРОТИВООПУХОЛЕВОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО ДЛЯ ПРЕРЫВИСТОГО ВВЕДЕНИЯ ИНГИБИТОРА FGFR | 2014 |
|
RU2664118C2 |
| Новое соединение 2,3,5-замещенного тиофена в качестве ингибитора протеинкиназы | 2017 |
|
RU2724957C2 |
Изобретение относится к области органической химии, а именно к соединению формулы (II), или его изомеру, или его фармацевтически приемлемой соли:
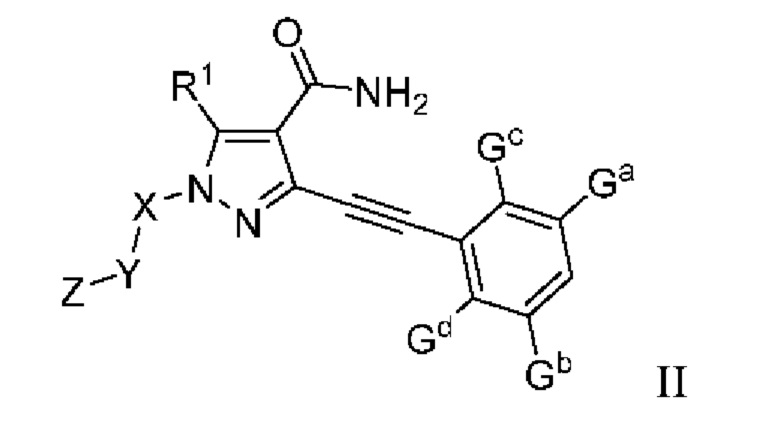
В формуле II: R1 представляет собой Н или -NHR3; R3 представляет собой Н, C1-6 алкил или 4-6-членный гетероциклил, где алкил и гетероциклил необязательно замещены галогеном, -OR5, C1-6алкилом, С3-6циклоалкилом или 4-6-членным гетероциклилом; X отсутствует или представляет собой C1-6алкилен; Y отсутствует или представляет собой С3-6циклоалкилен или 4-6-членный гетероциклилен; Z представляет собой циано,
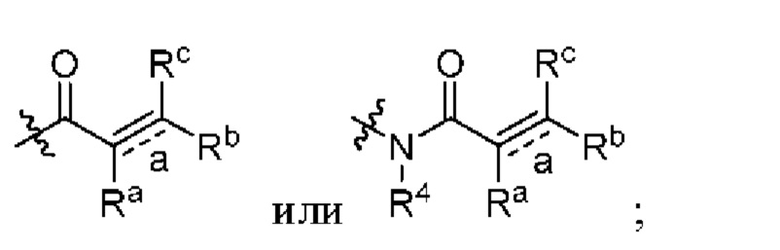
связь а представляет собой двойную связь или тройную связь; в том случае, если указанная связь а представляет собой двойную связь, каждый Ra, Rb и Rc независимо представляют собой Н, галоген или C1-6 алкил, где алкил необязательно замещен -ОС1-2алкилом или -N(C1-2 алкилом)2; в том случае, если указанная связь а представляет собой тройную связь, Ra и Rc отсутствуют и Rb представляет собой Н или C1-6 алкил; Ga, Gb, Gc и Gd, каждый независимо, выбраны из группы, состоящей из Н, галогена, -OR5 и -C(O)NR6R7; R4, R5, R6 и R7, каждый независимо, представляют собой Н или C1-6алкил, где Н в формуле II необязательно замещен 2Н. Также предложена фармацевтическая композиция и способ лечения. Технический результат: предложены новые органические соединения, действующие в качестве ингибитора рецептора фактора роста фибробластов (FGFR). 3 н. и 8 з.п. ф-лы, 2 табл., 34 пр.
1. Соединение формулы (II), или его изомер, или его фармацевтически приемлемая соль:
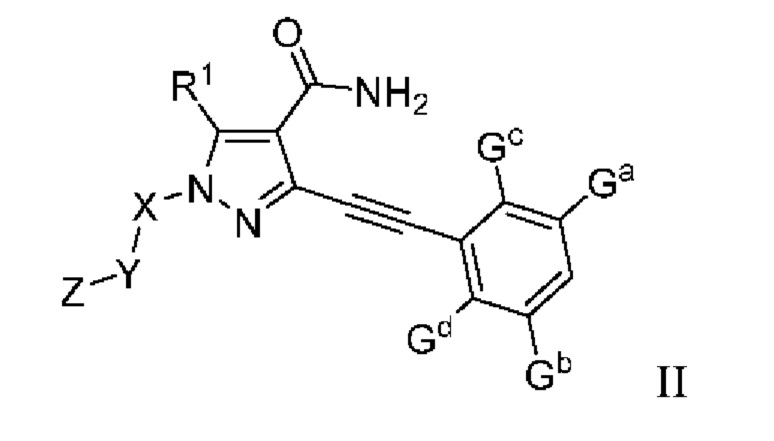
где R1 представляет собой Н или -NHR3;
R3 представляет собой Н, C1-6 алкил или 4-6-членный гетероциклил, где алкил и гетероциклил необязательно замещены галогеном, -OR5, C1-6алкилом, С3-6циклоалкилом или 4-6-членным гетероциклилом;
X отсутствует или представляет собой C1-6алкилен;
Y отсутствует или представляет собой С3-6циклоалкилен или 4-6-членный гетероциклилен;
Z представляет собой циано, 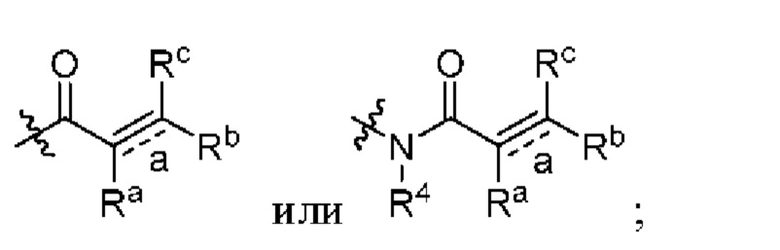
связь а представляет собой двойную связь или тройную связь;
в том случае, если указанная связь а представляет собой двойную связь, каждый Ra, Rb и Rc независимо представляют собой Н, галоген или C1-6 алкил, где алкил необязательно замещен -ОС1-2 алкилом или -N(C1-2 алкилом)2,
в том случае, если указанная связь а представляет собой тройную связь, Ra и Rc отсутствуют и Rb представляет собой Н или C1-6 алкил;
Ga, Gb, Gc и Gd, каждый независимо, выбраны из группы, состоящей из Н, галогена, -OR5 и -C(O)NR6R7;
R4, R5, R6 и R7, каждый независимо, представляют собой Н или C1-6 алкил,
где Н в формуле II необязательно замещен 2Н.
2. Соединение по п. 1, отличающееся тем, что X отсутствует;
Y представляет собой где
где  означает присоединение к Z и → означает присоединение к пиразолу;
означает присоединение к Z и → означает присоединение к пиразолу;
Z представляет собой CN или 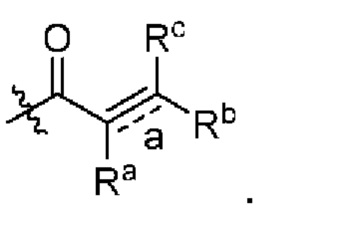
3. Соединение по п. 1, отличающееся тем, что
Ga представляет собой -OC1-2алкил, галоген или -C(O)NH(C1-2 алкил);
Gb представляет собой Н или -OC1-2алкил ;
Gd представляет собой Н или галоген;
Gc представляет собой Н.
4. Соединение по п. 1, отличающееся тем, что
R1 представляет собой Н или NHR3;
R3 представляет собой Н, C1-6 алкил или 4-6-членный гетероциклил, где алкил и гетероциклил необязательно замещены F, -ОН, С1-2 алкилом, С3-6 циклоалкилом или 4-6-членным гетероциклилом.
5. Соединение по п. 1, отличающееся тем, что
в том случае, если указанная связь а представляет собой двойную связь, Ra представляет собой Н или F; Rb и Rc представляют собой Н или C1-6 алкил, где алкил необязательно замещен -ОС1-2 алкилом или -N(C1-2 алкилом)2;
в том случае, если указанная связь а представляет собой тройную связь, Ra и Rc отсутствуют, Rb представляет собой Н или C1-6 алкил, где алкил необязательно замещен -ОС1-2 алкилом или -N(C1-2 алкилом)2.
6. Соединение по п. 1, выбранное из следующих:
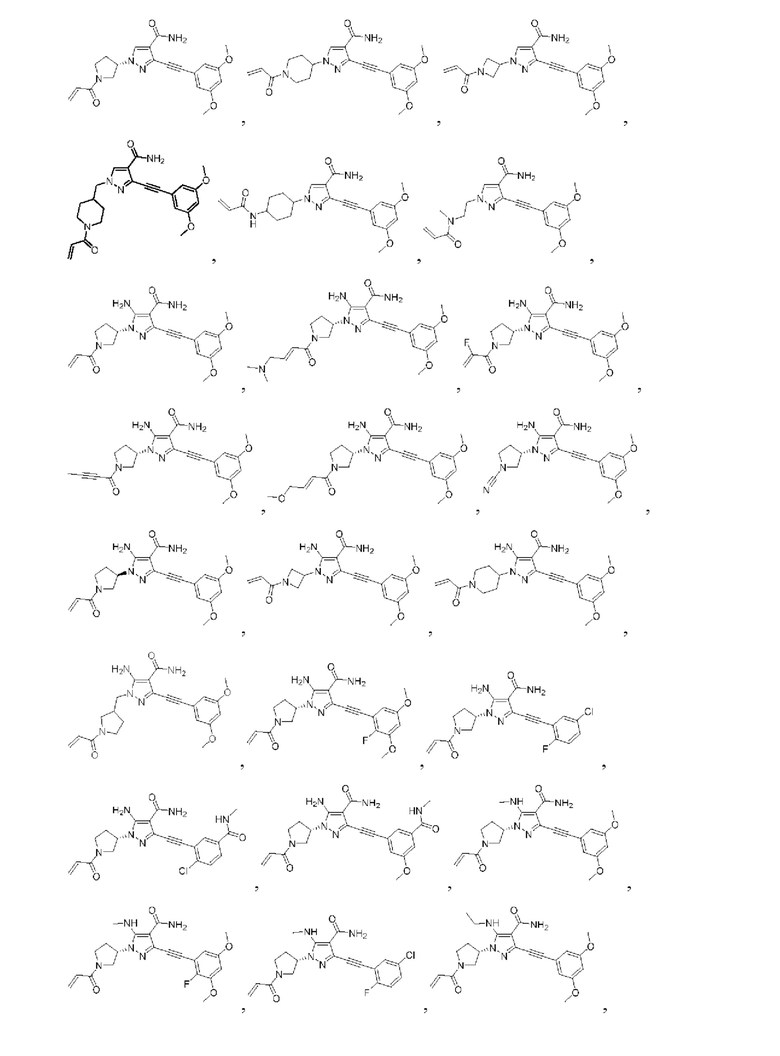

или его фармацевтически приемлемая соль.
7. Соединение по п. 6, представлющее собой
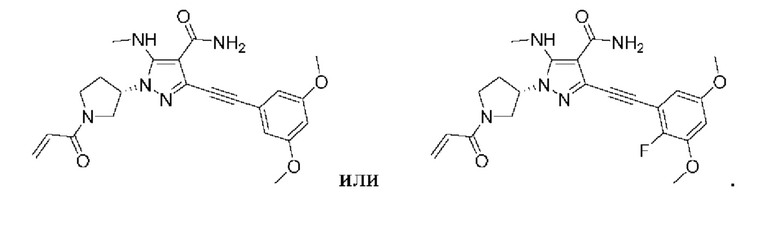
8. Соединение по п. 6, представляющее собой
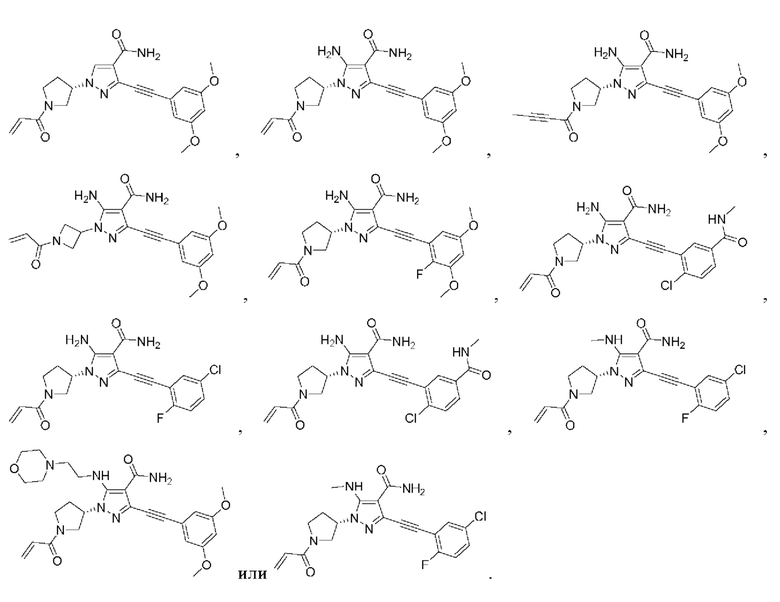
9. Фармацевтическая композиция, имеющая FGFR-ингибирующую активность, содержащая терапевтически эффективное количество соединения по п. 1 и фармацевтически приемлемый носитель и вспомогательное вещество.
10. Способ лечения заболевания, связанного с FGFR, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения по п. 1, причем указанное заболевание, связанное с FGFR, представляет собой рак печени, рак желудка, немелкоклеточный рак легких, рак мочевого пузыря, рак пищевода, меланому, рабдомиосаркому, почечно-клеточную карциному, множественную миелому, рак молочной железы, рак яичника, эндометриальный рак, рак шейки матки, рака толстой кишки, рак поджелудочной железы, рак легких или рак предстательной железы.
11. Способ по п. 10, в котором указанное заболевание, связанное с FGFR, представляет собой рак печени, рак желудка, немелкоклеточный рак легких или рак мочевого пузыря.
| WO 2013014039 A1, 31.01.2013 | |||
| WO 2015008844 A1, 22.01.2015 | |||
| Устройство для контроля времени работы тормозного компрессора | 1987 |
|
SU1472931A1 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ | 2002 |
|
RU2294326C2 |

