Настоящее изобретение относится к области гетерогенного катализа. В частности, настоящее изобретение относится к способу получения катализаторов, преимущественно пригодных в процессах гидроочистки, например, в процессах гидродесульфурации, гидроденитрификации, гидродеароматизации углеводородов.
В частности, настоящее изобретение относится к способу получения указанных катализаторов, которые содержат смешанные оксиды никеля, алюминия, молибдена и вольфрама и, возможно, переходный металл Me, выбранный из группы, состоящей из Zn, Mn, Cd и их смеси, органический компонент С и, возможно, неорганическое связующее В.
Указанные смешанные оксиды содержат аморфную фазу и псевдокристаллическую фазу, изоструктурную вольфрамиту.
Настоящее изобретение также относится к указанным катализаторам гидроочистки и процессу гидроочистки, в котором используют указанные катализаторы.
Множество способов превращения тяжелых фракций сырой нефти в полезные топлива и химические соединения включают применение катализаторов. В частности, каталитические композиции на основе переходных металлов показали эффективность в способе очистки, в котором выполняют типовые операции гидроочистки.
Важные разработки в этом секторе были стимулированы экологическими нормативными требованиями, относящимися к содержанию в топливах загрязнителей на основе серы и азота, которые требуют специальных обработок фракций сырой нефти для уменьшения содержания таких загрязнителей. Обычно такие обработки выполняют путем гидрирования в присутствии катализаторов на основе молибдена или вольфрама на носителе, к которым добавляют Со или Ni в качестве промоторов активности этих катализаторов. Промотор действует синергетично с основным металлом и позволяет получать повышение активности катализатора в связи со способом получения, типом материала и другими факторами (С. Giavarini, "Hydrotreating", в Encyclopaedia of Hydrocarbons, Treccani, (2005), vol. 2, section 3.1, p. 115-135). Важно отметить, что для того, чтобы проявлять каталитическую активность, металлы в вышеуказанных композициях должны быть в форме сульфидов.
Ужесточение экологических нормативных требований и одновременно ухудшение топливного качества сырой нефти делают необходимым поиск новых катализаторов, подходящих для повышенной десульфуризации фракций сырой нефти, особенно по отношению к тем соединениям, которые трудно поддаются обработке, таким, например, как замещенные дибензотиофены (ДБТ), в которых воздействие на серу стерически затруднено. Более того, существует потребность в получении катализаторов гидродесульфурации, способных оказывать свое действие также в присутствии азотных загрязнителей, которые показывают, даже когда присутствуют в ограниченном количестве, сильную склонность к деактивации действия самих катализаторов с неблагоприятными воздействиями на качество конечного продукта.
Так называемые объемные катализаторы показали особенную эффективность для этой цели, причем указанные катализаторы описаны, например, M.V. Landau, D. Berger и М. Herskowitz в J. Catalysis (1996), vol. 159, p. 236-245.
«Объемные» катализаторы, используемые в способах гидроочистки, можно получать исключительно из одного или более неблагородных металлов Группы VIII и, возможно, одного или более металлов Группы VIB, и они не содержат «несущих» основ или подложек. Обычно объемные катализаторы не обеспечивают присутствия связующего в конечной каталитической композиции; когда необходимо использование связующего, последний повышает агрегацию частиц объемного катализатора, предпочтительно не изменяя их морфологию.
Этот тип катализаторов и их получение описаны, например, в патентах US 4596785, US 4820677, US 6299760, US 6635599 и в патентных заявках US 2007/0286781 и ЕР 1941944 А1.
В US 4596785 и US 4820677 описаны катализаторы гидроочистки и/или гидропереработки нефтяных погонов, полученные путем совместного осаждения металлов, составляющих указанные погоны, исходя из растворимых в воде солей, в присутствии сульфидов. Такие способы включают работу со сложными технологиями и в инертной атмосфере для того, чтобы избежать последующего превращения серных соединений металлов в соответствующие оксиды. В случае US 4596785 полученный катализатор имеет кристаллическую структуру, которая похожа на молибденит, в случае сульфида металла Группы VB3, и кристаллическую структуру, которая похожа на пирит, в случае сульфида металла Группы VIII. Вместо этого, катализатор, описанный в US 4820677, является аморфным.
В US 6299760 и US 6635599 описаны объемные катализаторы гидроочистки, полученные путем объединения в присутствии кислорода металлических компонентов в растворе и их реакции с получением стабильного осадка, который затем сульфидируют.
В частности, некоторые режимы, посредством которых можно получить указанный осадок, перечислены в US 6635599, например, путем изменения температуры и/или рН в течение или после объединения растворов металлических компонентов, или путем подходящего добавления комплексообразующих реагентов или нерастворителей, или путем уменьшения количества растворителя, или путем добавления избытка одного из металлических компонентов вплоть до осуществления его осаждения.
Также в заявке US 2007/0286781 описаны способы получения полиметаллических материалов на основе переходных металлов с использованием технологий совместного осаждения.
Наконец, в патентной заявке ЕР 1941944 А1 описаны катализаторы гидроочистки нефтяных фракций, полученные путем совместного осаждения металлов, исходя из раствора соответствующих аммониевых солей, с последующим нагреванием до высокой температуры в инертной атмосфере и сульфидирующей обработкой.
В композициях объемных катализаторов могут содержаться неорганические связующие, такие, например, как диоксид кремния, оксид алюминия, алюмосиликат, для поддержания целостности частиц катализатора.
Например, в предпочтительном аспекте ЕР 1171549 В1 предусмотрено добавление неорганического связующего в ходе получения катализатора или, в другом предпочтительном аспекте, вышеуказанное неорганическое связующее добавляют в каталитическую композицию перед стадией образования катализатора. В частности, в ЕР 1171549 В1 отмечено, что когда связующее добавляют в виде растворимого предшественника в ходе процесса получения, необходимо обеспечить, чтобы оно превращалось в твердое состояние как таковое, а именно без реакции с другими металлическими компонентами. В общем, такие связующие обладают более низкой каталитической активностью (или не имеют никакой каталитической активности) по отношению к объемному катализатору. Соответственно, добавление неорганического связующего в общем вызывает уменьшение активности каталитической композиции, которая его содержит.
В других случаях композиции объемных катализаторов могут содержать металлы в форме так называемых «тугоплавких» оксидов, таких, например, как диоксид кремния, оксид алюминия, оксид магния, диоксид титана, диоксид циркония, борный ангидрид и оксид цинка. Например, в заявке WO 2004/073859 описано получение объемного катализатора формулы (X)b(M)c(Z)d(O)e, в которой X является по меньшей мере одним неблагородным металлом Группы VIII, М является по меньшей мере одним неблагородным металлом группы VIB, Z является одним или более чем одним элементом, выбранным из алюминия, кремния, магния, титана, циркония, бора и цинка, О является кислородом, индекс, выбранный из b и с равен 1, в то время, как d, е и другие индексы, выбранные из b и с и отличные от 1, являются числом больше 0, так чтобы молярное отношение b/с находилось в интервале 0,5:1 - 5:1, молярное отношение d/c находилось в интервале 0,2:1-50:1 и молярное отношение е/с находилось в интервале 3,7:1-108:1.
Описанную в указанной заявке каталитическую композицию получают с помощью способа регулируемого совместного осаждения металлических соединений вместе с тугоплавким оксидом и щелочными соединениями (обычно аммиака) в протонсодержащей жидкости, обеспечивая таким образом комплекс металла и тугоплавкого оксида, который затем подвергают тепловой обработке. В предпочтительной форме изобретения вышеупомянутая каталитическая композиция может содержать небольшое количество цинка в форме тугоплавкого оксида (в интервале от 1 масс. % до 3 масс. % по отношению к общей массе каталитической композиции), к которому относят положительное воздействие на площадь поверхности каталитической композиции.
Катализатор WO 2004/073859 определяют как катализатор «низкой кристалличности», как показано «изображением» рентгеновской дифракции, которое не указывает никакого отражения, отличающегося полной шириной на половине высоты максимума (ПШПМ) меньше 2,5° 2θ.
В заявках WO 2009/058783 и WO 2010/126689 описано получение объемных катализаторов, содержащих дополнительные компоненты, выбранные из Групп IIA, IIB или IVB, среди которых Zn.
В WO2009/058783 описан каталитический предшественник, содержащий Zn/Mo/W/малеат, который после сульфидирования позволяет получать лучшие выходы гидрокрекинга по отношению к известным в уровне техники катализаторам на основе Ni/Mo/W, даже работая при более низких температурах, однако являющийся менее активным, чем эти последние в реакции гидродесульфуризации.
В WO 2009/126689 описан каталитический предшественник, характеризующийся «остаточной геометрической объемной усадкой» ниже 12% после сульфидирования при температуре по меньшей мере 100°С в течение по меньшей мере 30 минут и с мономодальным распределением пор в макроскопической области.
Дополнительное развитие достигнуто посредством так называемых полиметаллических «объемных слоистых» катализаторов, имеющих состав NixZnyMoW, в котором Ni и Zn присутствуют в переменном молярном отношении, как описано Y. Chen, L. Wang, Y. Zhang, T. Liu, X. Liu, Z. Jiang e C. Li в "A new multi-metallic bulk catalyst with high hydrodesulphurization activity of 4,6-DMDBT prepared using layered hydroxide salts as structural templates" (2014) Applied Catalysis A: General, vol. 474 p. 69-77 и в WO 2014/043993. Авторы относили высокую активность таких катализаторов к высокому содержанию активных металлов и к слоистой структуре, которая способна обеспечивать более высокое число активных центров и таким образом повышать взаимодействие сульфидов активных металлов, где совместное действие Ni/Zn и Mo/W играет важную роль.
В US 7648941 описан «объемный» катализатор, содержащий оксиды Ni и W в качестве основного компонента. В процесс синтеза можно добавлять второй металл Группы VIB (в количестве менее 10% по отношению к общему количеству молей металлов Группы VIB) и металл Группы V (в количестве менее 10% по отношению к общему количеству молей металлов Группы VIB).
В этом случае получение каталитической композиции включает тепловую обработку реакционной смеси в течение времени, достаточно продолжительного для обеспечения полного превращения исходного материала. Полученный катализатор определяют как «не аморфный», в котором присутствует по меньшей мере одна кристаллическая фаза, даже если в катализаторе также присутствуют одна или более неупорядоченных фаз. В этих условиях катализатор показывает метастабильную гексагональную структуру, которая отличается дифракционным спектром с одним отражением между примерно 60° и примерно 65° угла 2θ, и основными отражениями между примерно 32° и примерно 36° 2θ и между примерно 50° и примерно 55° 2θ, где полные ширины на половине высоты максимума (ПШПМ) ниже 2,5° 2θ. Авторы, не ограничивая себя какой-либо теорией, связывают высокую каталитическую активность с вышеупомянутой метастабильной гексагональной фазой. При более высоких температурах гексагональная фаза превращается в орторомбическую регулярную структуру, состоящую из NiWO4, изоструктурную вольфрамиту, которую можно определить путем сравнения в «стандартной порошковой дифракционной базе данных»: JCPDS-ICDD PDF card 15-0755 о 72-1189 о 72-480, где ее называют «оксид вольфрамата никеля». Поэтому в US 7648941 возможность получения изоструктурной вольфрамиту фазы связывают с композициями, в которых вольфрам составляет по меньшей мере 90% от общего количества молей металлов Группы VIB.
В WO 2012/130728 описаны конкретные смешанные оксиды, содержащие Ni, Mo, W, по меньшей мере один элемент, выбранный из Si, Al и их смесей, и возможно содержащие органический компонент, полученный из соединения, содержащего углерод и азот, характеризующиеся тем, что они содержат аморфную фазу и моноклинную кристаллическую фазу, изоструктурную вольфрамиту, со степенью кристалличности выше 0% и ниже 100%, предпочтительно выше или равной 3% и ниже 100%. Такие смешанные оксиды имеют композицию NiaYbZcOd⋅pC, где Y является смесью Мо и W и отношение Mo/W выше 0,1 и ниже 10. Образование кристаллической фазы происходит в течение тепловой обработки, и выбор температуры позволяет модулировать степень кристалличности смешанного оксида. Фактически, смешанные оксиды, содержащие аморфную фазу и кристаллическую фазу, изоструктурную вольфрамиту, где указанные оксиды имеют степень кристалличности выше 0% и ниже 70%, можно получить при определенных условиях, в которых тепловую обработку выполняют при температурах выше или равных 150°С и ниже или равных 500°С, предпочтительно при температуре выше или равной 170°С и ниже или равной 500°С. Тепловые обработки при температурах выше 500°С и ниже 900°С позволяют получать смешанный оксид, содержащий аморфную фазу и кристаллическую фазу, изоструктурную вольфрамиту, где указанный оксид имеет степень кристалличности выше 70% и ниже 100%.
В последнее время еще более ограничивающие нормативные требования по контролю выбросов отработанных газов сделали необходимым использование еще более активных катализаторов. Например, современные европейские нормативные требования обеспечивают максимальный уровень содержания серы в автомобильном дизельном топливе 10 мг/кг.
Более того, последние нормативные требования также требуют уменьшения содержания ароматических углеводородов, в особенности полициклических ароматических углеводородов (ПАУ). Эти последние являются известными канцерогенами и могут присутствовать в топливе и могут образовываться путем сжигания и затем поступать в состав отработанного газа. Сжигание ПАУ может, в свою очередь, вызывать образование бензола. Содержание ароматических углеводородов в дизельном топливе может обуславливать температуру пламени, влияя таким образом на выбросы NOx, и оно непосредственно связано с образованием частиц и выбросами СО2.
По этим причинам Директива 2009/30/СЕ от 2009 требует уменьшения максимального содержания ПАУ в автомобильном дизельном топливе до 8 масс. %.
Поэтому очевидна необходимость создания новых катализаторов, еще более реакционноспособных и эффективных в удалении гетероатомов, в частности серы и азота, а также способных уменьшить уровень содержания ароматических углеводородов во фракциях сырой нефти, предназначенных для использования в качестве топлив.
Задачей заявителя является создание новых способов получения предшественников указанных выше объемных катализаторов, более дешевых и соответствующих современным направлениям защиты окружающей среды в сравнении с известными в уровне техники способами, используя меньшее количество органических добавок и/или металлов, которые являются дорогими и токсичными, использующих преимущество в показателях безопасности и затрат.
Задачей также является обнаружение объемного катализатора из предшественников, полученных посредством нового способа получения, отличающегося высокими характеристиками гидродесульфурации и гидроденитрификации, в котором также улучшена способность к ускорению гидродеароматизации углеводородов.
Заявитель обнаружил новый способ получения предшественников объемного катализатора, пригодного в способах гидроочистки фракций сырой нефти, содержащего полиметаллические смешанные оксиды, который обеспечивает решение вышеупомянутых задач и обладает многочисленными преимуществами по сравнению с известными в уровне техники способами.
Конкретно, первый аспект улучшения этого способа относится к тому, что в нем требуется меньшее количество благородных металлов Группы VIB и Группы VIII без влияния на каталитическую активность материалов, полученных посредством вышеупомянутого способа, и, соответственно, он отличается более низкими затратами по сравнению со способами получения известных объемных катализаторов.
Другой аспект улучшения относится к использованию пониженного количества органических добавок, с последующей меньшей потерей массы предшественников вышеупомянутых катализаторов и более высоким выходом процесса получения.
Другое преимущество состоит в том, что вышеупомянутый способ получения не включает каких-либо стадий разделения твердого вещества и жидкости и обеспечивает получение концентрированных суспензий металлов: это позволяет избежать образования вод отделения, загрязненных токсичными металлами, и уменьшает энергетические затраты и, таким образом, вносит вклад в увеличение выходов процесса получения.
Исходя из предшественников, содержащих смешанные оксиды переходных металлов, полученных посредством способа по настоящему изобретению, можно получить новые катализаторы, которые преимущественно показывают заметную селективность к реакциям гидродесульфуризации, гидроденитрификации и, в особенности, гидродеароматизации, таким образом минимизируя реакции гидрокрекинга.
Дополнительные признаки и преимущества станут понятны из следующего подробного описания.
Для лучшего понимания признаков способа, цели настоящего изобретения, ссылаются на приложенные чертежи, которые предназначены лишь для иллюстрации, а не для ограничения изобретения.
В частности, спектры 27Al-ЯМР-ВМУ (ЯМР при вращении под магическим углом) образцов смешанного оксида формулы (I) (спектр а) и формулы (I), связанной неорганическим связующим В на основе алюминия (спектр b), полученных согласно способу по настоящему изобретению, анализировали после обжига при температуре от 300 до 400°С, как представлено на Фиг. 1.
На Фиг. 2а представлены спектры рентгеновской дифракции смешанного оксида формулы (I), в котором отсутствует металл Me (индекс «а» равен 0), тогда как на Фиг. 2b представлены спектры рентгеновской дифракции смешанного оксида формулы (I), в котором присутствует металл Me и он является Zn. В обоих случаях спектры были получены на образцах, анализируемых после сушки (спектр а), после обжига при температуре от 300 до 400°С (спектр b) и после обжига при 600°С (спектр с) соответствующего смешанного оксида.
Спектры ИКСПФ (ИК спектроскопии с преобразованием Фурье), полученные после сушки (120°С), после обжига при температуре от 300 до 400°С (350°С) и после полного обжига (600°С) смешанного оксида формулы (I), содержащего металл Me и в котором Me=Zn, полученного способом по изобретению, представлены на Фиг 3а. Для сравнения, на Фиг. 3b представлены спектры ИКСПФ выбранных предшественников металлов, использованных в получении вышеупомянутого смешанного оксида.
Для целей настоящего описания и следующей формулы изобретения определения числовых интервалов всегда включают конечные значения, если не указано другое.
В описании воплощений настоящего изобретения использование терминов «включающий» или «содержащий» означает, что описанные варианты, например, относящиеся к стадиям способа, или компонентам продукта, или деталям устройства не обязательно являются исчерпывающими. Однако, важно отметить, что также объектами настоящего изобретения являются воплощения, в которых термин «включающий», относящийся к описанным вариантам, например, относящийся к стадиям способа или к компонентам продукта или деталям устройства, следует интерпретировать как «состоящий по существу из» или «который состоит из», даже если это явно не указано.
Для целей настоящего описания и следующей формулы изобретения для химических элементов и связанных с ними групп необходимо ссылаться на Периодическую таблицу элементов, представленную в CRC Handbook of Chemistry and Physics (Ed. 58, 1977-1978) и с использованием нумерации CAS. В частности, для целей настоящего изобретения выражения "Группа VIB" и «металл Группы VIB» включают хром, молибден, вольфрам и их смеси в элементной, ионной форме или в форме соединения, выражения «Группа VIII» и «неблагородные металлы Группы VIII» включают железо, кобальт, никель и их смеси в элементной, ионной форме или в форме соединения.
Для целей настоящего изобретения термин «объемный катализатор» означает катализатор без носителя, что означает таким образом, что каталитическая композиция не предусматривает предварительно образованного носителя, на который металлы загружают путем пропитки или осаждения. При этом не исключается возможность того, что композиции, описанные и приведенные в качестве примера в данном документе, могут содержать составляющие, отличные от тех, которые являются каталитически активными, такие как связующие, промоторы или добавки.
Для целей настоящего изобретения термин «предшественник кристаллической фазы, изоструктурной вольфрамиту» означает соединение, характеризующееся тем, что оно содержит аморфную фазу и псевдокристаллическую фазу, изоструктурную вольфрамиту, которое при обжиге в окислительной атмосфере при температуре равной или выше 600°С в течение периода времени выше или равного 3 часам развивается в изоструктурную вольфрамиту кристаллическую фазу, которая имеет изображение рентгеновской дифракции, содержащее сигналы, представленные в таблице 1.

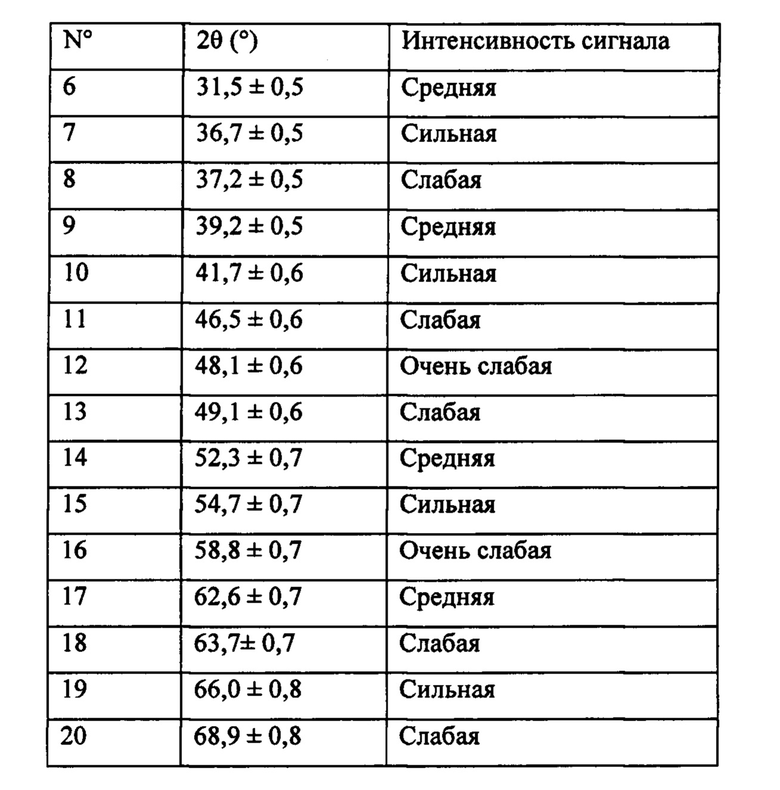
Термин «кристаллическая фаза, изоструктурная вольфрамиту» означает фазу, имеющую тот же тип кристаллической структуры натурального вольфрамита (минерала, состоящего из смешанного вольфрамата железа и марганца), например, класса призматической моноклинической симметрии, но отличающуюся химическим составом, в которой W, Fe, Mn могут быть частично или полностью замещены другими металлами. Вышеупомянутая фаза имеет изображение рентгеновской дифракции, содержащее сигналы, представленные в таблице 1.
Для целей настоящего изобретения термин «гидроочистка» означает ряд реакций, в которых углеводородная загрузка контактирует с водородом для изменения основных химических и физических свойств. Важно помнить, что в секторе очистки гидроочистка может иметь различные наименования в зависимости от цели (например, гидродесульфурация, гидроденитрификация, гидродеароматизация, гидродеметаллизация, гидродеоксигенация, гидродециклизация, гидроизомеризация, гидрокрекинг, гидродепарафинизация и т.п.). Обычно используют общий термин «гидрообработка», причем указанный термин может включать такие же вышеупомянутые способы гидроочистки и отличные от них, в общем, только для менее жестких, но не всегда, условий.
Гидроочистку по настоящему изобретению можно выполнять на многих нефтяных и нефтехимических производных, которые включают собственно сырую нефть или слабо крекированную нефть, остатки или продукты атмосферной или вакуумной перегонки, остатки процессов деасфальтизации пропаном, например, «клинкерные» фракции, тяжелые и легкие рецикловые газойли, остатки процесса каталитического крекинга с псевдоожиженным слоем катализатора, газойли атмосферной и вакуумной перегонки, газойли из коксовой печи, легкие и тяжелые дистилляты, включающие дистилляты «первичного сырья», продукты гидрокрекинга, продукты депарафинизации, петролатум, продукты процесса Фишера-Тропша, продукты нефтепереработки, лигроины, продукты способа EST (технологии глубокого гидрокрекинга) и их смеси.
В качестве продуктов способа EST необходимо иметь в виду углеводородные фракции, полученные, например, из способов, описанных в US 5932090, US 7255795, WO 2004/058922, WO 2004/056946, WO 2004/056947, WO 2005/047425, WO 2006/066911, WO 2006/066857, WO 2008/014947, WO 2008/014948, WO 2008/141830, WO 2008/141831, WO 2008/151792, WO 2009/003633, WO 2009/003634, WO 2009/149923.
Предпочтительно гидроочистка относится к углеводородным погонам, содержащим серные загрязнители и/или азотные загрязнители, в частности содержащим до 40000 частей на млн. серы, возможно содержащих до 2000 частей на млн. азота. В указанных погонах может присутствовать до 60 масс. % ароматических углеводородов и до 30 масс. % полициклических ароматических углеводородов.
В первом аспекте целью настоящего изобретения является способ получения смешанного оксида, содержащего Ni, Mo, W, Al, возможно по меньшей мере один металл Me и органический компонент С или остаток указанного органического компонента С, имеющего следующую формулу (I):
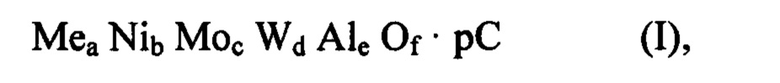
в которой
Me выбирают из группы, состоящей из Zn, Cd, Mn и их смеси,
С содержит полимерное органическое соединение,
а может быть больше или равно 0,
b, с, d, е и f больше 0,
f равно (2a+2b+6c+6d+3e)/2,
отношение (a+b)/(c+d) составляет от 0,9 до 1,1,
отношение а/b больше или равно 0 и меньше или равно 1,5,
отношение c/d составляет от 0,2 до 5,
отношение (a+b+c+d)/e составляет от 0,6 до 5, и
p является массовой процентной долей С по отношению к общей массе смешанного оксида формулы (I), и она больше 0% и меньше или равна 40%, причем указанный смешанный оксид формулы (I) содержит аморфную фазу и псевдокристаллическую фазу, изоструктурную вольфрамиту, где указанный способ включает следующие стадии:
1) смешивание по меньшей мере одного растворимого источника W и по меньшей мере одного растворимого источника Мо в подходящем объеме воды до тех пор, пока не получают прозрачный водный раствор,
2) возможно, добавление по меньшей мере одного источника по меньшей мере одного элемента Me в раствор, полученный на стадии 1,
3) добавление по меньшей мере одного источника Ni в смесь, полученную на предшествующей стадии,
4) подвергание смеси, полученной на стадии 3, первой тепловой обработке при температурах от 50°С до 80°С при перемешивании,
5) добавление к смеси, полученной на стадии 4, по меньшей мере одного растворимого, гидролизуемого или диспергируемого источника Al и по меньшей мере одного полимерного органического соединения,
6) подвергание смеси, полученной на предшествующей стадии, второй тепловой обработке при температурах от 80°С до 95°С при перемешивании с получением суспензии,
7) подвергание суспензии, полученной на стадии 6, сушке, получая таким образом твердую фазу,
8) обжиг указанной твердой фазы, полученной на предшествующей стадии, с получением смешанного оксида формулы (I).
В предпочтительном аспекте изобретения b, с, d и е больше 0,1. В предпочтительном аспекте изобретения а больше 0 и более предпочтительно больше 0,1.
Описанный выше способ можно выполнять последовательно от стадии 1 до стадии 8. Однако в некоторых предпочтительных воплощениях можно пропустить по меньшей мере одну необязательную стадию или к указанному способу можно добавить по меньшей мере одну дополнительную стадию без изменения предмета изобретения, как лучше описано далее.
Первая стадия способа заключается в растворении источника вольфрама и источника молибдена в некотором количестве воды, предпочтительно деионизированной или дистиллированной воды, достаточном по меньшей мере для получения прозрачного раствора.
Подходящими источниками Мо и W являются, например, оксиды, в особенности кислые оксиды, соответствующих кислот и их солей аммония. Предпочтительно в качестве источника вольфрама используют метавольфрамат, и гептамолибдат аммония используют в качестве источника молибдена.
В вышеупомянутых источниках вольфрама и молибдена соответствующие металлы имеют валентность 6.
Полученную смесь поддерживают при перемешивании до тех пор, пока не получат прозрачный раствор, что указывает на полное растворение источников Мо и W.
Предпочтительно стадию 1 указанного способа можно выполнять при температуре от 25°С до 50°С для содействия вышеуказанному растворению.
Согласно необязательной стадии 2, в полученный прозрачный раствор можно добавлять растворимый или нерастворимый или частично растворимый источник элемента Me, предпочтительно растворимый источник.
Не желая связывать себя какой-либо теорией, авторы полагают, что вышеупомянутый элемент Me, когда он присутствует, может способствовать образованию псевдокристаллической фазы, изоструктурной вольфрамиту.
Когда способ включает уже упомянутое добавление элемента Me, указанный элемент можно выбрать из группы, состоящий из Zn, Cd и Mn и их смеси.
В предпочтительном аспекте изобретения элемент Me, возможно добавляемый на стадии 2 указанного способа, может быть Zn.
В дополнительном предпочтительном аспекте изобретения элемент Me, возможно добавляемый на стадии 2 указанного способа, может быть Mn.
Подходящим источниками элемента Me могут быть, например, соответствующие нитраты, ацетаты, карбонаты, гидроксикарбонаты.
Когда Me является Zn, в качестве источников элемента Me можно использовать ацетат цинка или гидроксикарбонат цинка, предпочтительно ацетат цинка.
Предпочтительно, когда Me является Mn, в качестве источника элемента Me можно использовать ацетат марганца.
В вышеуказанном источнике элемента Me все соответствующие металлы (Zn, Cd или Mn) имеют валентность 2.
В предпочтительном аспекте изобретения необязательную стадию 2 можно пропустить. В этом случае в смешанном оксиде формулы (I), получаемом в конце вышеупомянутого способа, элемент Me не присутствует, то есть индекс «а» указанного элемента Me в вышеупомянутой формуле (I) равен 0.
Далее, согласно стадии 3 данного способа добавляют растворимый, нерастворимый или частично растворимый источник, предпочтительно частично растворимый источник, никеля. Подходящими источниками Ni могут быть соответствующие нитраты, ацетаты, гидроксикарбонаты, карбонаты, ацетилацетонаты. Предпочтительно можно использовать гидроксикарбонат никеля.
Предпочтительно стадию 3 указанного способа можно выполнять при температуре от 50°С до 70°С для повышения растворения указанного источника Ni в смеси, полученной на предшествующей стадии.
Полученную смесь подвергают на стадии 4 первой тепловой обработке при температурах от 50°С до 80°С при перемешивании.
Предпочтительно указанную первую тепловую обработку стадии 4 можно выполнять в течение времени от 10 минут до 1 часа при температурах от 50°С до 80°С при перемешивании.
В предпочтительном аспекте указанную первую тепловую обработку стадии 4 данного способа получения можно выполнять при температурах от 55°С до 70°С, постоянно поддерживая смесь при перемешивании.
В предпочтительном аспекте первую тепловую обработку стадии 4 указанного способа можно выполнять при постоянном перемешивании в течение времени от 20 до 40 минут.
В особенно предпочтительном аспекте настоящего изобретения первую тепловую обработку стадии 4 указанного способа можно выполнять при температуре от 55°С до 70°С в течение времени от 20 до 40 минут при постоянном перемешивании.
В конце этой стадии тепловой обработки в способе предусматривают, что на стадии 5 в полученную таким образом смесь можно добавлять по меньшей мере один растворимый источник алюминия, гидролизуемый или диспергируемый, и по меньшей мере одно полимерное органическое соединение.
Можно использовать источники алюминия, например, лактат алюминия, или диспергируемый оксид алюминия, или моногидрат оксида алюминия, или тригидрат оксида алюминия, или триалкоксиды алюминия, в которых алкил является линейным или разветвленным и может содержать от 2 до 5 атомов углерода, и предпочтительно используют диспергируемый оксид алюминия.
Предпочтительно диспергируемый оксид алюминия, добавляемый в качестве источника алюминия на стадии 5 указанного способа, может быть бемитом или псевдобемитом, характеризующимся частицами, имеющими средний диаметр менее 100 мкм. Подходящими диспергируемыми оксидами алюминия могут быть, например, имеющиеся в продаже бемиты серий Versal®, Pural®, Catapal®, Dequadis®, Disperal® и Dispal®.
Среди источников диспергируемого алюминия могут быть особенно предпочтительными диспергируемые в воде оксиды алюминия или оксиды алюминия, диспергируемые в водных растворах, содержащих одновалентную кислоту, при комнатной температуре при перемешивании: эти оксиды алюминия имеют наноразмеры в дисперсной фазе и отличаются размерами частиц в дисперсии от 10 до 500 нм. Диспергируемыми оксидами алюминия этого типа, которые преимущественно пригодны, являются, например, имеющиеся в продаже бемиты серий Disperal Р3® (Al2O3 68 масс. %) и Dequadis HP®.
Предпочтительно источниками гидролизуемого алюминия, которые, исходя из мономерных предшественников алюминия, обеспечивают его хорошую дисперсию, могут быть триалкилалюминаты, в которых алкильная группа содержит от 3 до 4 атомов углерода.
Важно отметить, что γ-оксиды алюминия не содержатся среди источников алюминия, подходящих на этой стадии.
В предпочтительном аспекте изобретения источник Al можно добавлять на стадии 5 в виде дисперсии, полученной ранее следующим образом: растворимый источник Al, гидролизуемый или диспергируемый, в количестве от 5 до 25 масс. %, предпочтительно от 8 до 18 масс. %, добавляют в водный раствор, содержащий от 0,2 до 2 масс. % уксусной кислоты. Полученные дисперсии перемешивают в течение времени от 1 до 48 часов, предпочтительно от 3 до 24 часов, при температуре от 25 до 90°С, предпочтительно от 30 до 70°С.
Затем, для повышения однородности композиции и улучшения структурных свойств конечного смешанного оксида, к полученной дисперсии после добавления источника Al добавляют полимерное органическое соединение.
Вышеупомянутое полимерное органическое соединение, добавляемое на стадии 5 указанного способа, предпочтительно содержит один или более гетероатомов, выбранных из N и О, и его можно выбрать, например, из группы, состоящей из альгината аммония, метилцеллюлозы, пропилметилцеллюлозы, сополимеров этиленгликоля и пропиленгликоля, этоксилированного октилфенола, полиоксиэтиленцетилэфира.
В предпочтительном аспекте полимерное органическое соединение, добавляемое на стадии 5 указанного способа, является метилцеллюлозой. В этом случае можно использовать имеющуюся в продаже метилцеллюлозу, такую, например, как метилцеллюлоза Fluka® (вязкость суспензии 2% в воде = 1200-1800 сПз), метилцеллюлоза Acros® (вязкость суспензии 2% в воде = 15 сПз), пропилметилцеллюлоза Dow® Methocel® 311 (вязкость суспензии 2% в воде > 1000 сПз).
В другом предпочтительном аспекте изобретения полимерное органическое соединение, добавляемое на стадии 5 указанного способа, может быть альгинатом аммония.
В других предпочтительных формах настоящего изобретения в качестве полимерного органического соединения можно использовать блок-сополимер полиэтиленгликоль-полипропиленгликоль-полиэтиленгликоль (например, имеющийся в продаже полимер Pluronic® P123 с вязкостью при 60°С = 350 сПз), или этоксилированный октилфенол (имеющийся в продаже как Triton® Х-305, со свойствами неионного поверхностно-активного вещества, характеризующийся вязкостью при 25°С = 470 сПз), или полиоксиэтилен-цетиловый эфир (имеющийся в продаже как Brij® 58, со свойствами неионного поверхностно-активного вещества, характеризующийся относительной вязкостью (Н2О = 1) 5% в воде при 25°С = 1,2-1,5).
Указанные полимерные органические соединения можно добавлять в процентной доле, изменяющейся от 1 масс. % до 10 масс. % по отношению к массе металлов, присутствующих в смеси.
В предпочтительном аспекте указанное полимерное органическое соединение можно добавлять в процентной доле от 1 масс. % до 5 масс. % по отношению к массе металлов.
Отсюда видно, что количество полимерного органического соединения в способе получения катализатора по изобретению особенно понижено: это обеспечивает получение, в течение последующего обжига смешанного оксида, меньшего количества летучих органических соединений, в частности азотных соединений, так что не обязательно требуется обработка паров в камерах дожигания.
Согласно альтернативному воплощению настоящего изобретения на стадии 5 вышеупомянутого способа вместо отдельного добавления по меньшей мере одного растворимого, гидролизуемого или диспергируемого источника Al и затем по меньшей мере одного полимерного органического соединения, можно добавлять дисперсию, приготовленную отдельно и содержащую как указанный по меньшей мере один растворимый, гидролизуемый или диспергируемый источник Al, так и указанное по меньшей мере одно полимерное органическое соединение.
Процедура получения вышеупомянутой дисперсии, содержащей как Al, так и полимерное органическое соединение, является следующей: растворимый, гидролизуемый или диспергируемый источник Al добавляют в водный раствор, содержащий уксусную кислоту, и полученную дисперсию смешивают с водной суспензией, содержащей полимерное органическое соединение и ранее поддерживаемой при перемешивании в течение периода от 20 минут до 1 часа при температуре от 25 до 80°С.
Также в этом случае полимерные органические соединения можно добавлять в процентной доле, составляющей от 1 масс. % до 10 масс. % по отношению к массе металлов, присутствующих в смеси.
В предпочтительном аспекте указанное полимерное органическое соединение можно добавлять в процентной доле, составляющей от 1 масс. % до 5 масс. % по отношению к массе металлов.
После добавления полимерного органического соединения дисперсию, содержащую как растворимый, гидролизуемый или диспергируемый источник Al, так и полимерное органическое соединение, перемешивают в течение времени от 1 до 48 часов, предпочтительно от 3 до 24 часов, и при температуре от 25 до 90°С, предпочтительно от 30 до 70°С, перед добавлением в металлосодержащую смесь, полученную на стадии 3 способа по изобретению.
Как путем добавления источника Al отдельно от полимерного органического соединения, так и путем добавления этих двух компонентов совместно, как описано выше, полученную смесь, содержащую Мо, W, Ni, Al, полимерное органическое соединение и, возможно, Me, дополнительно подвергают на следующей стадии 6 второй тепловой обработке при температурах от 80°С до 95°С при перемешивании.
Предпочтительно указанную вторую тепловую обработку выполняют при температурах от 80°С до 95°С в течение времени от 5 до 30 часов при перемешивании.
Вторая тепловая обработка позволяет получить гомогенную суспензию, в которой металлические компоненты, возможно полученные из нерастворимых или частично растворимых источников, могут взаимодействовать и устанавливать близкий контакт друг с другом. Предпочтительно суспензия, полученная на стадии 6, может иметь теоретическое содержание оксидов металлов от 15 до 40 масс. %, предпочтительно от 18 до 30 масс. % по отношению к общей массе суспензии.
В предпочтительном аспекте изобретения указанную вторую тепловую обработку можно выполнять при температурах от 85°С до 92°С.
В предпочтительном аспекте изобретения указанную вторую тепловую обработку можно выполнять в течение времени от 15 до 25 часов.
В особенно предпочтительном аспекте изобретения указанную вторую тепловую обработку стадии 6 можно выполнять при температурах от 85°С до 92°С в течение времени от 15 до 25 часов.
Полученную таким образом суспензию подвергают стадии 7 сушки. Основной функцией сушки на стадии 7 является удаление присутствующей в суспензии воды, что позволяет таким образом получить твердую фазу.
Сушку на стадии 7 можно выполнять, используя любую из известных специалисту технологий, например, с помощью стационарной печи, или путем «конвейерной сушки», или «термической сушки», или «сушки распылением» и предпочтительно ее выполняют путем «сушки распылением».
В предпочтительном аспекте сушку можно выполнять при температуре от 100°С до 250°С.
Время, необходимое для получения твердой фазы, связано со способом, используемым для выполнения сушки стадии 7. Определение наиболее подходящего времени для каждого способа для получения требуемой твердой фазы находится в компетенции специалиста.
Важно отметить, что стадия 6, на которой выполняют вторую тепловую обработку, отлична от стадии 7 сушки, даже если эти две стадии можно выполнять с помощью одинакового принципа, а именно путем подачи тепла в течение подходящего времени, учитывая, что в отличие от стадии 6, на которой можно получить суспензию, даже чрезвычайно плотную, на стадии 7 сушки важным элементом является получение твердой фазы, по существу не содержащей жидкой фазы.
В предпочтительном аспекте изобретения после сушки полученную твердую фазу можно подвергнуть формованию.
В этом случае способ по настоящему изобретению может включать дополнительную стадию 7', непосредственно следующую за стадией 7, на которой высушенную твердую фазу, полученную на указанной стадии 7, подвергают формованию.
Для этой цели можно использовать все технологии формования. Формование можно выполнять путем таблетирования, экструзии, грануляции, образования шариков или распыления путем сушки распылением.
Для облегчения формования в смешанный оксид можно добавить одну или более органических добавок для улучшения реологических свойств вышеупомянутого смешанного оксида. Эти добавки предпочтительно могут содержать крахмал, целлюлозу, стеарат, поверхностно-активные вещества или их смесь.
На последней стадии способа обеспечивают частичный обжиг полученного смешанного оксида. Обжиг на стадии 8 можно выполнять в воздухе или в инертной атмосфере при температуре выше или равной 200°С и ниже или равной 450°С. В предпочтительном аспекте обжиг можно выполнять при температуре от 300°С до 400°С.
Частичный обжиг можно выполнять при постоянной температуре или при температурном градиенте (или «программируемо»). Время обжига может изменяться предпочтительно от минимума, составляющего 3 часа, до 20 часов. На основе температуры обжига и времени обжига полученный смешанный оксид может содержать различные процентные доли, выше 0 и ниже или равные 40%, органического компонента С и/или остатка указанного органического компонента С.
Для целей настоящего изобретения термин «остаток органического компонента С» означает ряд соединений, не полностью изученных, возможно присутствующих и полученных из указанного органического компонента С путем термического разложения или термической деструкции в течение процесса обжига.
Вышеупомянутый органический компонент С может содержать, помимо ранее описанного полимерного органического соединения, органическую часть, получающуюся из металлических источников (например, ион ацетата, ион ацетилацетоната, ион лактата, алкоксид) и уксусной кислоты, возможно использованных в способе получения смешанного оксида по изобретению.
Органический компонент С (и, возможно, вышеупомянутый остаток органического компонента С) можно количественно измерить с помощью термогравиметрии и дифференциального термического анализа (ТГ-ДТА) на образце смешанного оксида с использованием термических весов Mettler (модель TG50). Испытание выполняют путем подвергания образца линейному нагреву со скоростью 10°С/мин в потоке воздуха (200 см3/мин) от 30°С до 600°С. Содержание компонента С (и, возможно, вышеупомянутого остатка органического компонента С) выражают в виде массовой процентной доли (процентная доля «p») по отношению к общей массе смешанного оксида, и ее вычисляют в зависимости от потери массы образца, анализируемого посредством ТГ-ДТА в интервале 150-600°С.
В предпочтительном аспекте после обжига процентная доля «p» указанного органического компонента С и, возможно, остатка указанного органического компонента С в композиции смешанного оксида, получаемого способом по настоящему изобретению, может быть выше или равна 0.01% и ниже или равна 30%.
В предпочтительном аспекте «p» может быть выше или равна 0,2% и ниже или равна 10%.
В предпочтительном воплощении изобретения в способе по изобретению обеспечивают введение элемента Me на стадии 2.
Поэтому согласно предпочтительному воплощению настоящего изобретения способ получения смешанного оксида, содержащего Ni, Мо, W, Al, по меньшей мере один металл Me и органический компонент С или остаток указанного органического компонента С, имеющего следующую формулу (I):
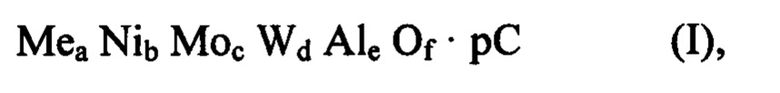
в которой
Me выбирают из группы, состоящей из Zn, Cd, Mn и их смеси,
С содержит полимерное органическое соединение,
а, b, с, d, е и f больше 0,
f равно (2a+2b+6c+6d+3e)/2,
отношение (a+b)/(c+d) может составлять от 0,9 до 1,1,
отношение а/b может быть больше или равно 0 и меньше или равно 1,5,
отношение c/d может составлять от 0,2 до 5,
отношение (a+b+c+d)/e может составлять от 0,6 до 5, и
p является массовой процентной долей С по отношению к общей массе смешанного оксида формулы (I), и она может быть больше 0% и меньше или равна 40%,
причем указанный смешанный оксид формулы (I) содержит аморфную фазу и псевдокристаллическую фазу, изоструктурную вольфрамиту, включает следующие стадии:
1) смешивание по меньшей мере одного растворимого источника W и по меньшей мере одного растворимого источника Мо в подходящем объеме воды до тех пор, пока не получают чистый водный раствор,
2) добавление по меньшей мере одного источника по меньшей мере одного элемента Me в раствор, полученный на стадии 1,
3) добавление по меньшей мере одного источника Ni, предпочтительно частично растворимого, в смесь, полученную на предшествующей стадии,
4) подвергание смеси, полученной на стадии 3, первой тепловой обработке при температурах от 50°С до 80°С при перемешивании,
5) добавление к смеси, полученной на стадии 4, по меньшей мере одного растворимого, гидролизуемого или диспергируемого источника Al и по меньшей мере одного полимерного органического соединения,
6) подвергание смеси, полученной на предшествующей стадии, второй тепловой обработке при температурах от 80°С до 95°С при перемешивании с получением суспензии,
7) подвергание суспензии, полученной на стадии 6, сушке с получением таким образом твердой фазы,
8) обжиг указанной твердой фазы, полученной на предшествующей стадии, с получением смешанного оксида формулы (I).
В предпочтительном аспекте а, b, с, d и е больше 0,1.
Предпочтительно первую тепловую обработку стадии 4 можно выполнять в течение времени от 10 минут до 1 часа при температурах от 50°С до 80°С при перемешивании.
Предпочтительно вторую тепловую обработку стадии 6 можно выполнять в течение времени от 5 до 30 часов при температурах от 80°С до 95°С при перемешивании.
Наоборот, согласно альтернативному воплощению в способе по изобретению можно исключить введение элемента Me на стадии 2.
В этом случае стадию 2 способа можно пропустить и индекс «а» в формуле (I) равен 0.
Преимущество данного способа состоит в том, что он не включает операций отделения и промывки полученной твердой фазы. Отсутствие этих стадий позволяет поддерживать атомные отношения присутствующих в смесях металлов, реагирующих в течение всего способа, в конечном твердом продукте: соответственно, не требуется аналитическое регулирование для определения конечного состава смешанных оксидов металлов, соответствующих атомным отношениям металлов, используемым в процессе, даже после обжига.
Более того, таким образом избегают получения вод, загрязненных смешанными оксидами металлов. Этот аспект имеет особую промышленную и экологическую важность, так как эти металлы в общем являются токсичными и некоторые из них относятся к канцерогенным соединениям.
В другом воплощении настоящего изобретения описанный выше способ можно модифицировать так, чтобы он обеспечивал добавление по меньшей мере одного неорганического связующего В.
Способ получения смешанных оксидов формулы (I), связанных по меньшей мере одним неорганическим связующим В, включает все стадии уже описанного способа и включает дополнительную стадию 5', которая следует сразу после стадии 5 указанного способа, а именно после добавления полимерного органического соединения или в случае, когда это последнее добавляют в смесь с источником алюминия, после добавления указанной смеси в смесь, полученную на стадии 4, и на которой добавляют неорганическое связующее В.
Вышеупомянутое неорганическое связующее В может состоять из материала, обычно используемого в качестве неорганического связующего для катализаторов. Неограничивающие примеры неорганического связующего могут включать, например, диоксид кремния, оксид алюминия, алюмосиликаты, покрытый оксидом алюминия диоксид кремния и покрытый диоксидом кремния оксид алюминия, гиббсит, диоксид титана, диоксид циркония, анионные и катионные глины, сапонит, бентонит, каолин, сепиолит или гидроталькит или их смеси. Предпочтительными связующими материалами являются диоксид кремния, оксид алюминия, алюмосиликат или их смеси. В частности, в качестве источника оксида алюминия можно использовать γ-оксид алюминия, возможно в форме водной дисперсии.
Когда смешанный оксид по настоящему изобретению связывают по меньшей мере одним неорганическим связующим В, где В содержит Al, в вышеупомянутом смешанном оксиде обнаруживают две структурно различные формы указанного металла, с которыми связаны различные функции (компонент каталитической композиции и функция неорганического связующего, соответственно).
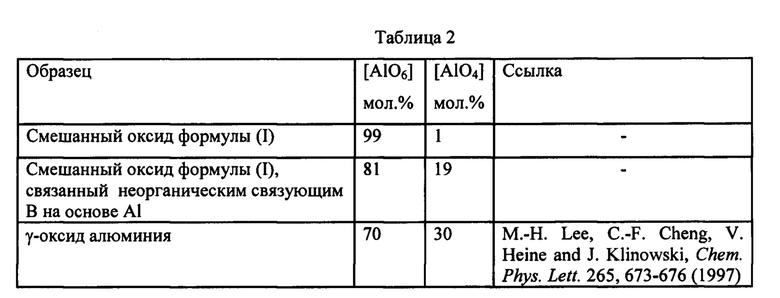
Присутствие двух структурно различных форм алюминия в описанном выше смешанном оксиде было подтверждено путем сравнения твердотельной спектроскопии 27Al ЯМР-ВМУ смешанного оксида формулы (I) и соответствующего смешанного оксида, связанного неорганическим связующим В на основе Al. Для испытания использовали прибор Varian V-500, время импульса 0,2 мкс (10° импульс) и задержка релаксации 1 с. 4 мм ротор, содержащий порошок, вращался при 14 кГц. Спектры получали при 130 МГц, химические сдвиги относили к трихлориду алюминия в растворе (0 м.д.). Спектры 27Al ЯМР-ВМУ образцов смешанного оксида формулы (I) без неорганического связующего (спектр а) и связанного неорганическим связующим В на основе Al (спектр b), полученные согласно способу по настоящему изобретению и проанализированные после обжига при температуре от 300 до 400°С, представлены на Фиг. 1. Не учитывая «боковые полосы вращения» (указанные звездочками на Фиг. 1), можно отметить присутствие двух различных сигналов с химическим сдвигом, равным 6±5 и 64±5 частей на млн., относимых к атомам Al в октаэдрической [AlO6] и тетраэдрической координации [AlO4], соответственно. Относительное содержание двух соединений, выраженное в мол. %, получали посредством способа обращения свертки сигнала и вычисления лежащей ниже площади, и оно представлено в таблице 2.
Очевидно, что образец смешанного оксида формулы (I) содержит почти исключительно Al в октаэдрической координации, в то время как образец, соответствующий смешанному оксиду, связанному неорганическим связующим В на основе Al, а также алюминиевый октаэдрический компонент, содержат второй Al компонент с октаэдрической и тетраэдрической координацией, сопоставимой с присутствием γ-оксида алюминия в качестве неорганического связующего. Алюминий, присутствующий в виде γ-оксида алюминия, составляет только часть всего алюминия, присутствующего в образце смешанного оксида, связанного по меньшей мере одним неорганическим связующим В, согласно более низкому содержанию [AlO4] в мол. % по отношению к образцу γ-оксида алюминия.
В предпочтительном аспекте изобретения в способ получения смешанного оксида формулы (I), связанного по меньшей мере одним неорганическим связующим В, также можно включить стадию формования после сушки полученной твердой фазы.
Аналогично описанному выше, для этого способ по настоящему изобретению может включать стадию 7', следующую сразу за стадией 7 сушки, на которой высушенную твердую фазу, полученную на указанной стадии 7, подвергают формованию.
Также смешанный оксид формулы (I), образованный в присутствии по меньшей мере одного неорганического связующего В, содержит аморфную фазу и псевдокристаллическую фазу, изоструктурную вольфрамиту.
Смешанный оксид формулы (I), связанный по меньшей мере одним неорганическим связующим В по настоящему изобретению, можно перед применением подвергнуть обжигу в воздухе или в инертной атмосфере при температуре выше 250°С и ниже или равной 450°С и предпочтительно можно подвергнуть обжигу при температурах от 300°С до 400°С.
Обжиг можно выполнять при постоянной температуре или при температурном градиенте (или «программируемо»). Время обжига можно изменять от минимум 3 часов до 20 часов. В зависимости от температуры обжига и времени обжига обожженный смешанный оксид может содержать различные процентные доли, выше 0 и ниже или равные 40%, органического компонента С и/или остатка указанного органического компонента С.
Второй задачей настоящего изобретения является смешанный оксид, содержащий Ni, Мо, W, Al, возможно по меньшей мере один металл Me и органический компонент С или остаток указанного органического компонента С, имеющий следующую формулу (I):
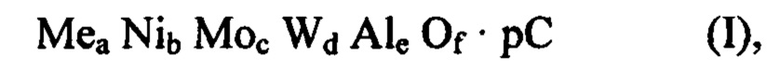
в которой
Me выбирают из группы, состоящей из Zn, Cd, Mn и их смеси,
С содержит полимерное органическое соединение,
а может быть больше или равно 0,
b, с, d, е и f больше 0,
f равно (2a+2b+6c+6d+3e)/2,
отношение (a+b)/(c+d) составляет от 0,9 до 1,1,
отношение а/b больше или равно 0 и меньше или равно 1,5,
отношение c/d составляет от 0,2 до 5,
отношение (a+b+c+d)/e составляет от 0,6 до 5, и
p является массовой процентной долей С по отношению к общей массе смешанного оксида формулы (I), и она больше или равна 0% и меньше или равна 40%,
причем указанный смешанный оксид формулы (I) содержит аморфную фазу и псевдокристаллическую фазу, изоструктурную вольфрамиту.
В предпочтительном аспекте а больше 0 и более предпочтительно больше 0,1. В предпочтительном аспекте изобретения b, с, d и е больше 0,1. В предпочтительном аспекте изобретения, когда а больше 0, элемент Me может быть Zn.
В другом аспекте изобретения, когда а больше 0, элемент Me может быть Mn.
В другом аспекте изобретения отношение (a+b)/(c+d) может составлять от 0,8 до 2. В другом предпочтительном аспекте указанное отношение (a+b)/(c+d) может быть равно 1.
В предпочтительном аспекте отношение а/b может быть больше или равно 0 и меньше или равно 1. В особенно предпочтительном аспекте отношение а/b может составлять от 0,1 до 0,4.
Предпочтительно отношение c/d может составлять от 0,4 до 3. В особенно предпочтительном аспекте отношение c/d может составлять от 1 до 2,5.
Предпочтительно p может быть больше или равно 0,01% и меньше или равно 30%. В другом предпочтительном аспекте p может быть больше или равно 0,2% и меньше или равно 10%.
В предпочтительном аспекте смешанный оксид формулы (I) можно получить посредством описанного выше способа по настоящему изобретению.
В предпочтительном аспекте смешанный оксид можно связать по меньшей мере одним неорганическим связующим В.
В предпочтительном аспекте указанный смешанный оксид формулы (I), связанный по меньшей мере одним неорганическим связующим В, можно получить посредством способа по настоящему изобретению в воплощении, которое предусматривает дополнительную стадию 5', на которой добавляют указанное неорганическое связующее В. Как уже ранее упоминалось, вышеупомянутую стадию 5' выполняют после стадии 5 указанного способа, а именно после добавления полимерного органического соединения, или в случае, когда последнее добавляют в смесь с источником алюминия, после добавления указанной смеси.
Смешанные оксиды формулы (I), полученные способом по настоящему изобретению, характеризовались, перед обжигом и после обжига, путем рентгеновской дифрактометрии порошков с помощью дифрактометра Philips модель  с вертикальным гониометром, оборудованного электронной системой счета импульсов с использованием Kα излучения Cu (длина волны λ=0,154 нм) с применением способов, известных специалисту.
с вертикальным гониометром, оборудованного электронной системой счета импульсов с использованием Kα излучения Cu (длина волны λ=0,154 нм) с применением способов, известных специалисту.
Например, на Фиг. 2а представлены спектры рентгеновской дифракции смешанного оксида формулы (I), в котором отсутствует металл Me (индекс «а» равен 0), в то время как на Фиг. 2b представлены спектры рентгеновской дифракции смешанного оксида формулы (I), в котором присутствует металл Me и он является Zn. В обоих случаях спектры получали на образцах, анализированных после сушки (спектр а), после обжига при температуре от 300 до 400°С (спектр b) и после обжига при 600°С соответствующего смешанного оксида.
После обжига при температуре от 300 до 400°С (Фиг. 2а и 2b, спектр b) вышеупомянутые смешанные оксиды формулы (I) характеризуются присутствием аморфной фазы и псевдокристаллической фазы.
Посредством тепловой обработки при 600°С псевдокристаллическая фаза изменяется по направлению к образованию кристаллической фазы, изоструктурной вольфрамиту. Псевдокристаллическая фаза, присутствующая в образцах после обжига при температуре от 300 до 400°С, поэтому является предшественником кристаллической фазы, изоструктурной вольфрамиту. В спектре рентгеновской дифракции обожженных при 600°С образцов (Фиг. 2а и 2b, спектры с) различают приведенные выше в таблице 1 сигналы. В частности, содержащий металл Me смешанный оксид (Фиг. 2b) дополнительно характеризуется по меньшей мере двумя дополнительными сигналами в интервале от 25° до 28° угла 2θ (при 26,2° очень слабой интенсивности и при 26,6° слабой интенсивности).
На Фиг. 2а и 2b (спектры а) спектры рентгеновской дифракции образцов указывают на сигналы, относимые к присутствующим смешанным аммониевым солям. Таким образом, обработка обжигом способствует образованию смешанных оксидов формулы (I), характеризующихся кристаллической фазой, изоструктурной вольфрамиту, которая является хорошо кристаллизованной после обработки при 600°С.
Важно отметить, что в смешанном оксиде формулы (I), содержащим металл Me, в спектре рентгеновской дифракции при 120°С, 350-400°С и 600°С (Фиг. 2b) всегда присутствуют по меньшей мере три сигнала в интервале от 25° до 28° угла 2θ.
Смешанный оксид формулы (I), полученный посредством способа по настоящему изобретению, дополнительно характеризовали путем инфракрасной спектроскопии с преобразованием Фурье (ИКСПФ) с помощью спектрометра Perkin-Elmer модель Spectrum ВХ с использованием таблеток порошков, разбавленных в KBr (2 масс. % в KBr) путем применения способов, известных специалисту.
Спектры ИКСПФ, полученные после сушки (120°С), после обжига при температуре от 300 and 400°С (350°С) и после полного обжига (600°С) смешанного оксида формулы (I), содержащего металл Me, в котором Me = Zn, полученного посредством способа по изобретению, представлены на Фиг. 3а.
Для сравнения на Фиг. 3b представлены спектры ИКСПФ выбранных предшественников металлов, использованных при получении вышеупомянутого смешанного оксида.
Основные сигналы ИКСПФ, присутствующие в твердом веществе после сушки, относят к предшественникам металлов, используемых в синтезе: в частности, сигнал при примерно 1400 см-1 является обычным для предшественников Мо, W, Ni, и сигнал при примерно 1070 см-1 является обычным для предшественника Al (псевдобемит).
В обожженных материалах эти сигналы больше не присутствуют, что указывает на реакцию в твердом состоянии между металлическими компонентами, связанную с образованием смешанного оксида. В частности, на Фиг. За (600°С) более не регистрируют обычный сигнал оксида алюминия при примерно 1070 см-1, что указывает, что он не присутствует как таковой в смеси со смешанным оксидом Zn, Ni, Мо и W. Другими словами, в смешанных оксидах формулы (I), полученных посредством способа по изобретению, алюминий, добавленный в виде растворимого, гидролизуемого или диспергируемого источника алюминия, не обладает функцией носителя или связующего и затем разбавления для других металлических компонентов, однако он находится в композиции смешанного оксида равным образом по отношению к другим металлам. В частности, смешанный оксид формулы (I), независимо от того, содержит он или не содержит металл Me, производит спектр ИКСПФ, в котором обнаруживают сигналы при 805±10 см-1, 605±10 см-1 и 445±10 с-1, которые обычно принадлежат фазе вольфрамита и определение которых повышается с температурой обжига.
Площадь поверхности и пористость смешанного оксида формулы (I), полученного посредством способа по настоящему изобретению, определяли из изотермы адсорбции/десорбции N2 при температуре -196°С, используя анализатор площади поверхности и пористости Micrometrics TriStar®. Перед получением изотерм образцы (примерно 0,3 г на испытание) подвергали предварительной обработке в вакууме в течение 16 часов при 150°С.
Удельную площадь поверхности (УПП) определяли с помощью известного специалисту способа БЭТ (Брунауэра - Эммета - Теллера), выполняя анализ в интервале относительного давления Р/Р0 от 0,05 до 0,3.
Вышеупомянутый смешанный оксид отличается удельной площадью поверхности, определенной после тепловой обработки при 350°С, больше или равной 80 м2/г.
Предпочтительно удельная площадь поверхности находится в интервале 90-230 м2/г и более предпочтительно она находится в интервале 90-190 м2/г.
Смешанный оксид формулы (I), полученный способом по настоящему изобретению, является мезопористым, а именно, в соответствии с терминологией IUPAC (Pure & Appl. Chem. Vol. 66, No. 8, pp. 1739-1758, 1994), характеризуется диаметром пор от 2 до 50 нм. Предпочтительно средний диаметр пор составляет от 4 до 10 нм.
Удельный общий объем пор вычисляли, используя метод Гурвича при 0,99 Р/Р0 и распределение пор определяли путем применения метода BJH (Barret-Joyner-Hallender) на кривой десорбции. Все указанные способы известны специалистам.
Смешанный оксид формулы (I), полученный способом по настоящему изобретению, характеризуется объемом пор больше или равным 0,15 мл/г. Предпочтительно указанный объем пор составляет от 0,15 до 0,35 мл/г.
Смешанный оксид формулы (I), возможно связанный по меньшей мере одним неорганическим связующим В, полученный способом по настоящему изобретению, можно превратить в соответствующий сульфид, и указанный сульфид можно преимущественно использовать в качестве катализатора гидроочистки.
Поэтому другой задачей настоящего изобретения является катализатор на основе сульфидов металлов, полученный путем сульфидирования смешанного оксида формулы (I), возможно связанного по меньшей мере одним неорганическим связующим В.
В предпочтительном аспекте смешанный оксид формулы (I), подвергаемый сульфидированию для предоставления вышеупомянутого катализатора на основе сульфидов металлов, получают с помощью способа получения указанного смешанного оксида формулы (I) по изобретению.
Согласно другому воплощению изобретения металлический сульфидированный катализатор можно получить путем сульфидирования смешанного оксида формулы (I), связанного по меньшей мере одним неорганическим связующим В.
В предпочтительном аспекте смешанный оксид формулы (I), связанный по меньшей мере одним неорганическим связующим В, подвергнутый сульфидированию для создания вышеупомянутого катализатора на основе сульфидов металлов, получают согласно изобретению с помощью способа получения указанного смешанного оксида формулы (I), связанного по меньшей мере одним неорганическим связующим В.
Для получения соответствующей сульфидированной композиции, активной в качестве катализатора гидроочистки, сульфидирование смешанного оксида по настоящему изобретению можно выполнять с помощью любой из известных специалисту технологий, используя любой сульфидирующий реагент, как описано, например, J.H. Gary и G.E. Handwerk в "Petroleum Refining - Technology and Economics" (2001, M. Dekker), p. 177.
Сульфидирование можно выполнять in situ, а именно в том же реакторе, в котором затем будут выполнять гидроочистку, или ex situ. Способ сульфидирования можно выполнять в восстанавливающей атмосфере, например, состоящей из H2S и Н2 или CS2 и Н2, при высокой температуре от 300°С до 500°С в течение периода, достаточного для сульфидирования исходного смешанного оксида. Например, сульфидирование можно выполнять в течение времени от 1 до 100 часов, и предпочтительно его выполняют в течение времени от 15 до 50 часов.
Альтернативно, сульфидирование смешанных оксидов можно выполнять, используя диметилдисульфид (ДМДС), растворенный в углеводородной загрузке, такой, например, как лигроин или газойль, при температурах от 300°С до 500°С.
Наконец, в другом предпочтительном аспекте сульфидирование можно выполнять путем контактирования смешанного оксида по настоящему изобретению непосредственно с обрабатываемой богатой серой углеводородной загрузкой, предпочтительно при температурах от 300°С до 500°С.
Как упоминалось выше, катализатор по настоящему изобретению, полученный путем сульфидирования смешанного оксида формулы (I) или смешанного оксида формулы (I), связанного по меньшей мере одним неорганическим связующим В, является катализатором, который очень активен и стабилен и обеспечивает особенно высокие каталитические характеристики широкого спектра в способе гидроочистки, включающем помимо гидродесульфурации и гидроденитрификации также гидродеароматизацию и уменьшение содержания полициклических ароматических углеводородов в обработанных углеводородных смесях.
Наоборот, вышеупомянутый катализатор показывает слабую склонность к катализу реакций гидрокрекинга.
Поэтому другой целью настоящего изобретения является способ гидроочистки сырья, содержащего один или более углеводородов, включающий контактирование указанного сырья с водородом в присутствии катализатора, полученного путем сульфидирования смешанного оксида формулы (I) или смешанного оксида формулы (I), связанного по меньшей мере одним неорганическим связующим В.
Любое сырье из углеводородной смеси, содержащей примеси, содержащие серу и/или азот, можно обработать с помощью катализатора по настоящему изобретению, например, продукты перегонки сырой нефти, остатки сырой нефти, лигроин и т.п.можно подвергнуть обработке, и предпочтительно гидроочистка относится к углеводородным погонам, содержащим содержащие серу загрязнители и/или содержащие азот загрязнители.
В частности, катализатор на основе сульфидов металлов по настоящему изобретению можно преимущественно использовать для гидроочистки сырья, включающего один или более углеводородов, содержащих до 4 масс. % S, до 0,2 масс. % N и до 50 масс. % полициклических ароматических углеводородов.
Предпочтительно обработка происходит при температуре от 100 до 450°С, более предпочтительно от 300 до 370°С, при давлении от 5,0 до 10,0 МПа, более предпочтительно от 5,0 до 7,0 МПа. ЧОСЖ (часовая объемная скорость жидкости) может составлять от 0,5 до 5 ч-1, предпочтительно от 0,8 до 2 ч-1. Количество водорода может составлять от 100 до 800 раз от количества углеводорода, выраженное как норм, л Н2/л углеводородной смеси.
Вследствие способности одновременно осуществлять высокую активность гидродесульфурации, гидроденитрификации, гидродеароматизации и уменьшения полициклических ароматических соединений, катализатор по изобретению можно преимущественно использовать также в качестве гидрирующего компонента, связанного с кислыми компонентами, в способе гидрокрекинга. Сырьем, подходящим для гидрокрекинга, являются, например, тяжелая и сверхтяжелая сырая нефть, вакуумный газойль (ВГО), вакуумные остатки (ВО).
Для применения на практике и лучшей иллюстрации настоящего изобретения ниже представлены некоторые неограничивающие примеры способов получения смешанных оксидов формулы (I), которые являются предшественниками катализаторов, и каталитических испытаний.
Во всех примерах молярные формулы составов смешанных оксидов нормализованы по отношению к сумме (моли Ni + моли Zn) = 1,00.
Пример 1 по изобретению. Получение смешанного оксида, имеющего состав
Me0,00Ni1,00Mo0,60W0,40Al1,04O5.56·6,0%C
Процедура получения смешанного оксида включает последовательное добавление при подходящих температурных условиях и при перемешивании растворов или дисперсий, содержащих элементы композиции.
Сначала 49,4 г гидрата метавольфрамата аммония (NH4)6H2W12O40⋅xH2O добавляли в 154 г воды, и растворение соли ускоряли, используя ряд валов-мешалок при 180 об/мин (периферическая скорость=40 м/мин). Спустя примерно 15 минут в раствор добавляли 51,9 г гептамолибдата аммония (NH4)6Mo7O24⋅4H2O, и смесь нагревали при 50°С при перемешивании для облегчения растворения молибденовой соли. Спустя 30 минут добавляли 62,7 г гидроксикарбоната никеля NiCO3⋅Ni(ОН)2⋅4Н2О (58 масс. % NiO), доводя температуру смеси до 70°С, и эту смесь поддерживали при этой температуре все время при постоянном перемешивании в течение по меньшей мере 30 минут.
В это время 36,7 г Disperal® Р3 Sasol (псевдобемит, содержащий 70 масс. % Al2O3) смешивали с 188,1 г водного раствора 0,6 масс. % уксусной кислоты. Смесь поддерживали при перемешивании в течение примерно 2 часов для получения однородной дисперсии оксида алюминия.
Одновременно 4,7 г метилцеллюлозы Methocel® МС Fluka добавляли в 35,0 г воды при температуре 50°С.Смесь поддерживали при 50°С при перемешивании в течение примерно 10 минут для получения однородной дисперсии полимерного органического соединения.
Эту последнюю медленно добавляли в дисперсию оксида алюминия при перемешивании и поддерживая температуру при 50°С до получения однородной дисперсии, содержащей как оксид алюминия, так и полимерное органическое соединение.
Указанную однородную дисперсию добавляли к описанной выше суспензии, содержащей Ni, Мо и W, поддерживаемой, в свою очередь, при 70°С при перемешивании. Добавление выполняют очень медленно и с использованием капельной воронки. В конце температуру смеси доводили до 90°С и полученную суспензию поддерживали при этой температуре при перемешивании в течение примерно 18 часов. рН суспензии, измеренный в конце вышеупомянутой тепловой обработки, был равен 5,6 и содержание Methocel® МС было равно 3,1 масс. % по отношению к теоретическому содержанию смешанного оксида.
В конце этой обработки полученную таким образом суспензию, которая имеет теоретическое содержание оксидов, равное 26 масс. %, охлаждали и подвергали сушке в течение примерно 20 минут в предварительно нагретой печи при 200°С. Часть полученного твердого вещества (50 г) обжигали в статичном воздухе согласно следующей температурной программе: от комнатной температуры до 200°С за 30 минут с линейным изменением 6°С/мин, изотерма при 200°С в течение 10 минут, нагрев до 350°С за 2 часа 35 минут с линейным изменением примерно 1°С/мин, изотерма при 350°С в течение 5 часов.
Спектр 27Al ЯМР-ВМУ представлен на Фиг. 1а, из которого можно вычислить, что 99 мол. % алюминия занимает октаэдрическую координацию.
Часть обожженного при 350°С образца дополнительно обжигали при 600°С в течение 5 часов. Спектры рентгеновской дифракции образцов, подвергнутых нескольким тепловым обработкам, представлены на Фиг. 2а.
Остальную часть высушенного при 200°С образца затем помещали в механический смеситель и перемешивали при температуре примерно 50°С в течение примерно 2 часов до получения однородной пасты с подходящей для экструзии густотой. Экструдат выдерживали при комнатной температуре в течение 15 часов, затем обжигали с описанным выше линейным изменением.
Конечный экструдированный смешанный оксид имеет следующий молярный состав: Ni1,00Mo0,60W0,40Al1,04O5,56. Смешанный оксид содержит 6,0 масс. % остатка органического компонента по отношению к общей массе твердого вещества.
Удельная площадь поверхности (УПП) составляет 142 м2/г, общий объем пор (Vp) составляет 0,18 см3/г, средний размер пор, вычисленный из изотермы десорбции, составляет 4,0 нм.
Пример 2 по изобретению. Получение смешанного оксида, имеющего состав
Me0,00Ni1,00Mo0,50W0,50Al1,04O5,56⋅6,3%С
Повторяли процедуру примера 1, изменяя количество гидрата метавольфрамата аммония (NH4)6H2W12O40⋅xH2O (61,7 г) и гептамолибдата аммония (NH4)6Mo7O24⋅4Н2О (43,2 г).
Полученный смешанный оксид имеет следующий молярный состав: Ni1,00Mo0,50W0,50Al1,04O5,56. Смешанный оксид содержит 6,3 масс. % остатка органического компонента по отношению к общей массе твердого вещества. Удельная площадь поверхности (УПП) составляет 149 м2/г, общий объем пор (Vp) составляет 0,19 см3/г, средний размер пор, вычисленный из изотермы десорбции, составляет 4,4 нм.
Пример 3 по изобретению. Получение смешанного оксида, имеющего композицию
Me0,50Ni0,50Mo0,50W0,50Al1,02O5,53⋅13,1%С, где Me = Zn
61,7 г гидрата метавольфрамата аммония (NH4)6H2W12O40⋅xH2O добавляли в 154 г воды и растворение соли ускоряли, используя ряд валов-мешалок при 180 об/мин (периферическая скорость = 40 м/мин). Спустя примерно 15 минут в раствор добавляли 43,2 г гептамолибдата аммония (NH4)6Mo7O24⋅4Н2О и смесь нагревали при 50°С при перемешивании для облегчения растворения молибденовой соли. Спустя 30 минут добавляли 53,9 г ацетата цинка Zn(CH3COO)2⋅2H2O, затем смесь поддерживали при 50°С при перемешивании в течение еще 30 минут. Наконец, добавляли 31,7 г гидроксикарбоната никеля NiCO3⋅Ni(ОН)2⋅4Н2О (58 масс. % NiO), температуру смеси доводили до 70°С, и эту смесь поддерживали при этой температуре все время при постоянном перемешивании в течение по меньшей мере 30 минут.
В это время 36,7 г Disperal® Р3 Sasol (псевдобемит, содержащий 70 масс. % Al2O3) смешивали с 188,1 г водного раствора 0,6 масс. % уксусной кислоты. Смесь поддерживали при перемешивании в течение примерно 2 часов для получения однородной дисперсии оксида алюминия.
Одновременно 4,2 г метилцеллюлозы Methocel® МС Fluka добавляли в 35,0 г воды при температуре 50°С. Смесь поддерживали при 50°С при перемешивании в течение примерно 10 минут для получения однородной дисперсии полимерного органического соединения.
Эту последнюю медленно добавляли в дисперсию оксида алюминия при перемешивании и поддерживая температуру при 50°С до получения однородной дисперсии, содержащей как оксид алюминия, так и полимерное органическое соединение.
Указанную однородную дисперсию добавляли к описанной выше суспензии, содержащей Zn, Ni, Мо и W, поддерживаемой, в свою очередь, при 70°С при перемешивании. Добавление выполняют очень медленно и с использованием капельной воронки. В конце температуру смеси доводили до 90°С и полученную суспензию поддерживали при этой температуре при перемешивании в течение примерно 18 часов. рН суспензии, измеренный в конце вышеупомянутой тепловой обработки, был равен 5,6 и содержание Methocel® МС было равно 3,1 масс. % по отношению к теоретическому содержанию смешанного оксида.
В конце этой обработки полученную таким образом суспензию, которая имеет теоретическое содержание оксидов, равное 22 масс. %, охлаждали и подвергали сушке в течение примерно 20 минут в предварительно нагретой печи при 200°С.
Извлеченную твердую фазу затем обжигали в статичном воздухе согласно следующей температурной программе: от комнатной температуры до 200°С за 30 минут с линейным изменением 6°С/мин, изотерма при 200°С в течение 10 минут, нагрев до 350°С за 2 часа 35 минут с линейным изменением примерно 1°С/мин, изотерма при 350°С в течение 5 часов.
Конечный смешанный оксид имеет следующий молярный состав: Zn0,50Ni0,50Mo0,50W0,50Al1,02O5,53. Смешанный оксид содержит 13,1 масс. % остатка органического компонента по отношению к общей массе твердого вещества.
Удельная площадь поверхности (УПП) составляет 142 м2/г, общий объем пор (Vp) составляет 0,21 см3/г, средний размер пор, вычисленный из изотермы десорбции, составляет 9 нм.
Пример 4 по изобретению. Получение смешанного оксида, имеющего состав
Me0,25Ni0,75Mo0,60W0,40Al1,03O5,55⋅14,9%С, где Me = Zn
49,4 г гидрата метавольфрамата аммония (NH4)6H2W12O40⋅xH2O) добавляли в 154 г воды, и растворение соли ускоряли, используя ряд валов-мешалок при 180 об/мин (периферическая скорость = 40 м/мин). Спустя примерно 15 минут в раствор добавляли 51,9 г гептамолибдата аммония (NH4)6Mo7O24⋅4H2O, и смесь нагревали при 50°С при перемешивании для облегчения растворения молибденовой соли. Спустя 30 минут добавляли 27,2 г ацетата цинка Zn(СН3СОО)2⋅2Н2О, затем смесь поддерживали при 50°С при перемешивании в течение еще 30 минут. Наконец, добавляли 47,0 г гидроксикарбоната никеля NiCO3⋅Ni(ОН)2⋅4Н2О (58 масс. % NiO), температуру смеси доводили до 70°С, и смесь поддерживали при этой температуре все время при постоянном перемешивании в течение по меньшей мере 30 минут.
В это время 36,7 г Disperal® Р3 Sasol (псевдобемит, содержащий 70 масс. % Al2O3) смешивали с 188,1 г водного раствора 0,6 масс. % уксусной кислоты. Смесь поддерживали при перемешивании в течение примерно 2 часов для получения однородной дисперсии оксида алюминия.
Одновременно 4,4 г метилцеллюлозы Methocel® МС Fluka добавляли в 35,0 г воды при температуре 50°С. Смесь поддерживали при 50°С при перемешивании в течение примерно 10 минут для получения однородной дисперсии полимерного органического соединения.
Эту последнюю медленно добавляли в дисперсию оксида алюминия при перемешивании и поддерживая температуру при 50°С до получения однородной дисперсии, содержащей как оксид алюминия, так и полимерное органическое соединение.
Указанную однородную дисперсию добавляли к описанной выше суспензии, содержащей Zn, Ni, Мо и W, поддерживаемой, в свою очередь, при 70°С при перемешивании. Добавление выполняют очень медленно и с использованием капельной воронки. В конце температуру смеси доводили до 90°С и полученную суспензию поддерживали при этой температуре при перемешивании в течение примерно 18 часов. рН суспензии, измеренный в конце вышеупомянутой тепловой обработки, был равен 5,6 и содержание Methocel® МС было равно 3,1 масс. % по отношению к теоретическому содержанию смешанного оксида.
В конце этой обработки полученную таким образом суспензию, которая имеет теоретическое содержание оксидов, равное 24 масс. %, охлаждали и подвергали сушке в течение примерно 20 минут в предварительно нагретой печи при 200°С.
Извлеченную твердую фазу затем обжигали в статичном воздухе согласно следующей температурной программе: от комнатной температуры до 200°С за 30 минут с линейным изменением 6°С/мин, изотерма при 200°С в течение 10 минут, нагрев до 350°С за 2 часа 35 минут с линейным изменением примерно 1°С/мин, изотерма при 350°С в течение 5 часов.
Конечный смешанный оксид имеет следующий молярный состав: Me0,25Ni0,75Mo0,60W0,40Al1,03O5,55. Смешанный оксид содержит 14,9 масс. % остатка органического компонента по отношению к общей массе твердого вещества.
Удельная площадь поверхности (УПП) составляет 175 м2/г, общий объем пор (Vp) составляет 0,21 см3/г, средний размер пор, вычисленный из изотермы десорбции, составляет 4,1 нм.
Часть образца дополнительно обжигали при 600°С в течение 5 часов. Спектры рентгеновской дифракции образцов, подвергнутых нескольким тепловым обработкам, представлены на Фиг. 2b. Из спектров рентгеновской дифракции при 120°С, 350-400°С и 600°С наблюдали, что по меньшей мере три сигнала всегда присутствуют в интервале от 25° до 28° угла 2θ.
Спектры ИКСПФ образцов, подвергнутых нескольким тепловым обработкам, представлены на Фиг. 3а.
Пример 5 по изобретению. Получение смешанного оксида, имеющего состав Me0,25Ni0,75Mo0,60W0,40Al1,03O5,55⋅13,8%С⋅10% γ-Al2O3, где Me = Zn, и связанного 10%
неорганического связующего В В этом примере описано получение смешанного оксида, содержащего Zn и связанного неорганическим связующим В, в котором используемое полимерное органическое соединение представляет собой Methocel® МС.
Используемый способ идентичен тому, который описан в предшествующем примере 4, с тем отличием, что 58,0 г диспергируемого у-оксида алюминия (AERODISP® W925, 25 масс. % γ-Al2O3) добавляли в смесь перед конечной тепловой обработкой, составляющей 18 часов.
Полученный смешанный оксид имеет следующий молярный состав:
Zn0,25Ni0,75Mo0,06W0,4Al1,03O5,55⋅10% γ-Al2O3. Смешанный оксид содержит 13,8 масс. % остатка органического компонента по отношению к общей массе твердого вещества.
Удельная площадь поверхности (УПП) составляет 145 м2/г, общий объем пор (Vp) составляет 0,23 см3/г, средний размер пор, вычисленный из изотермы десорбции, составляет 4,0 нм.
Спектр рентгеновской дифракции полученного смешанного оксида позволяет обнаружить по меньшей мере три сигнала в интервале угла 2θ от 21° до 28°.
Спектр 27Al ЯМР-ВМУ представлен на Фиг. 1b, из которого вычислили, что 81 мол. % алюминия занимает октаэдрическую координацию.
Сравнительный пример 6. Получение смешанного оксида, имеющего состав
Ni1,0Mo0,5W0,54Al0,57O4,98⋅2,0%C, не по изобретению
7,56 г октиламина растворяли в 40 г абсолютного этанола. Раствор, содержащий 14,89 г гексагидрата нитрата никеля Ni(NO3)2⋅6Н2О, 4,52 г гептамолибдата аммония (NH4)6Mo7O24⋅4Н2О и 6,98 г гидрата метавольфрамата аммония (NH4)6H2W12O40⋅H2O, растворенных в 50 мл водного раствора, содержащего 14,90 г 10 масс. % водной дисперсии бемита (Disperal® Р2 от Sasol™), добавляли при перемешивании в этот раствор. Молярное отношение октиламин/(Ni+Мо+W) равно 0,6. Образовывался светло-зеленый гель, который оставляли при перемешивании в течение 3 часов, нагревая при 70°С. Его оставляли в покое на 48 часов. Полученный гель не имел надосадочной жидкости, и его сушили в печи при 90°С в течение 48 часов. Высушенный материал подвергали тепловой обработке при 400°С в течение 5 часов в воздухе.
Твердое вещество имело следующий молярный состав: Ni1,00Mo0,50W0,54Al0,57O4,98.Смешанный оксид содержит 2,0 масс. % остатка органического компонента по отношению к общей массе твердого вещества.
Удельная площадь поверхности составляет 151 м2/г, общий объем пор составляет 0,381 см3/г, средний размер пор, вычисленный из изотермы десорбции, составляет 6,3 нм.
Пример 7. Каталитическое испытание Смешанные оксиды, полученные согласно примерам 1, 2, 4, 5, 6 использовали в экспериментах, которые подтверждали их эффективность в качестве катализаторов гидроочистки.
В каждом эксперименте 20 см3 смешанного оксида, предварительного спрессованного, гранулированного и просеянного (10-16 меш), разбавляли 20 см3 инертного материала (карборунда) и загружали в реактор с неподвижным слоем с объемом, равным 40 мл. Затем смешанный оксид обрабатывали сульфидирующей смесью, содержащей «прямогонный газойль» с добавленным диметилдисульфидом (ДМДС), так чтобы концентрация S была равна 2,5 масс. % по отношению к общей массе сульфидной смеси. Использованные условия сульфидирования были следующими:
ЧОСЖ = 3 ч-1
Р = 3,0 МПа
Т = 340°С
Н2/сульфидная смесь = 200 норм, л/л
Время сульфидирования = 26 ч
За стадией сульфидирования, на которой смешанный оксид приобретает каталитические свойства, следует стадия стабилизации при Т=330°С в течение 60-100 ч, с поддержанием каталитического слоя в контакте с «прямогонным» газойлем.
Реакцию гидроочистки выполняли путем подачи смеси, содержащей прямогонный газойль и газойль легкого крекинга нефти в массовой пропорции 75:25, и поэтому характеризующейся высоким количеством серы (2 масс. %) и полициклических ароматических углеводородов (17 масс. %).
Условия реакции были следующими:
Р (Н2)=4,9 МПа
Отношение Н2/сырье = 230 норм, л/л
ЧОСЖ=0,8 ч-1
Температура реакции = 340°С.
Активность каждого катализатора оценивали в процентах по отношению к каталитической активности сравнительного примера 6 (принятой за 100%). Данные приведены в следующей таблице 4.
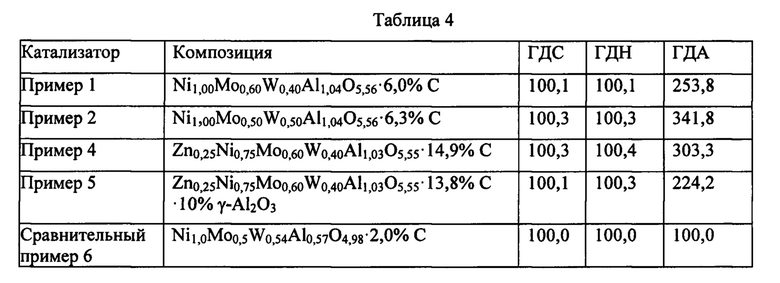
Помимо сравнимых характеристик гидродесульфурации (ГДС) и гидроденитрификации (ГДН), все образцы показали значительное увеличение превращения при гидродеароматизации (ГДА).
Важно отметить, что также катализатор примера 5, отличающийся более низким содержанием металлов, так как он содержит 10 масс. % неорганического связующего В, является более активным, в основном для целей реакции гидродеароматизации, по сравнению с катализатором сравнительного примера.
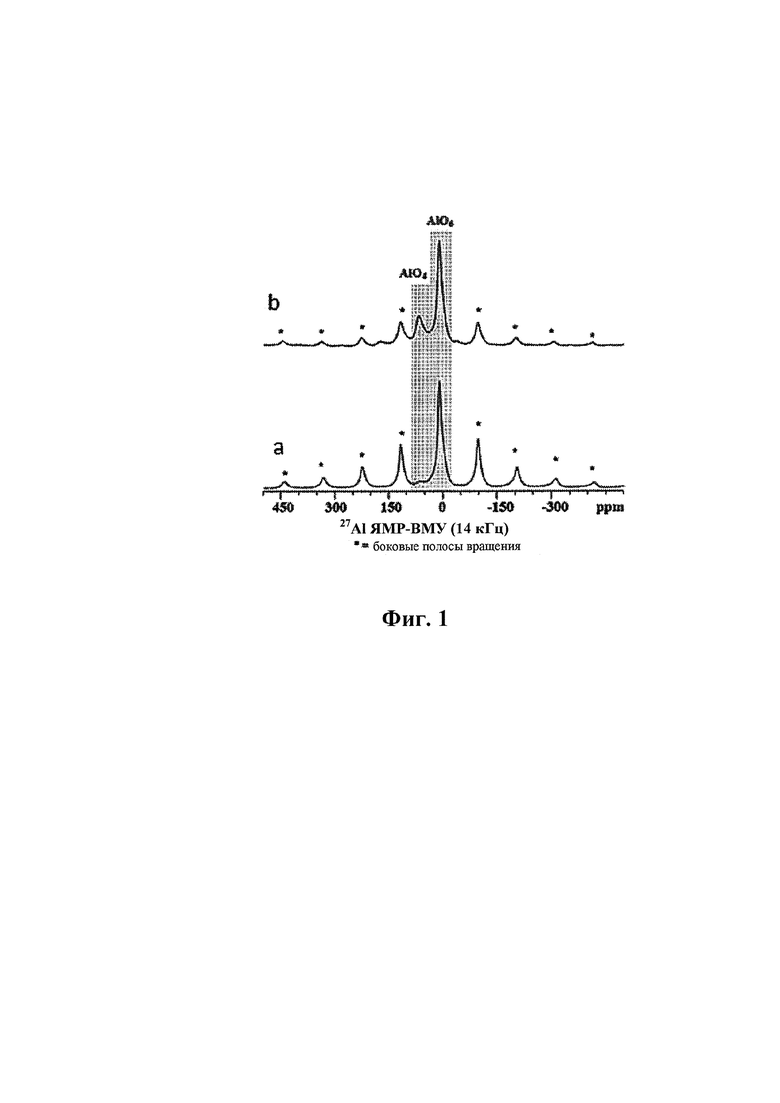
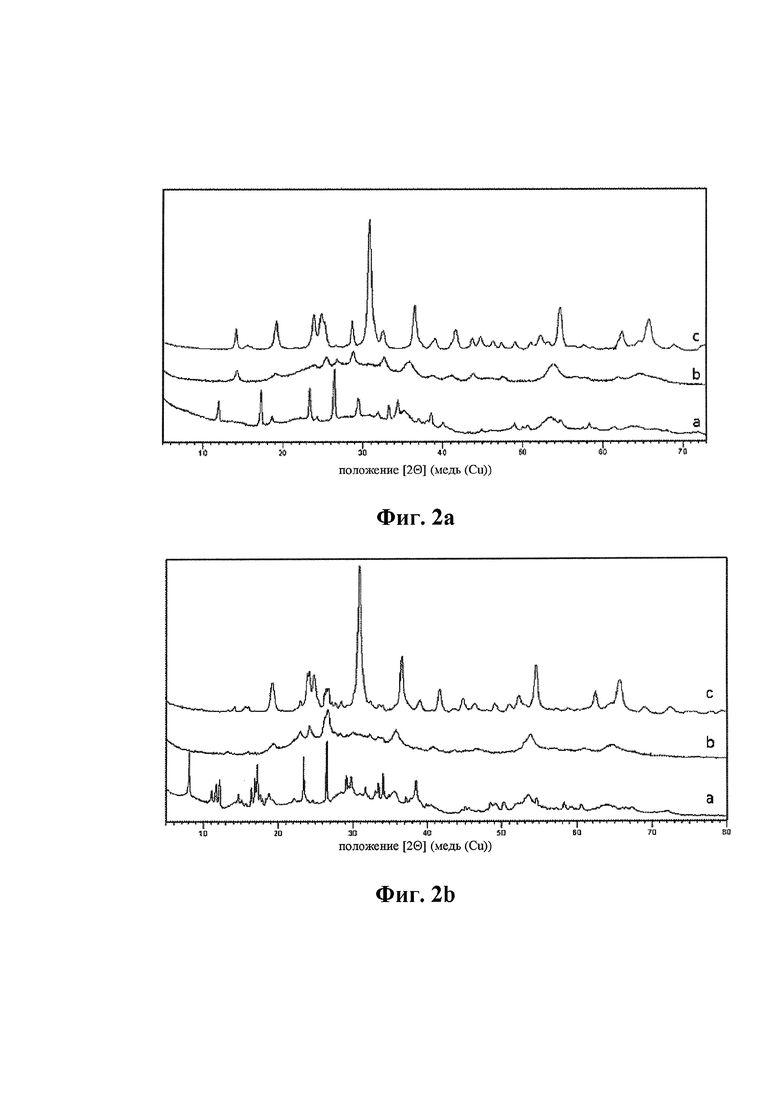
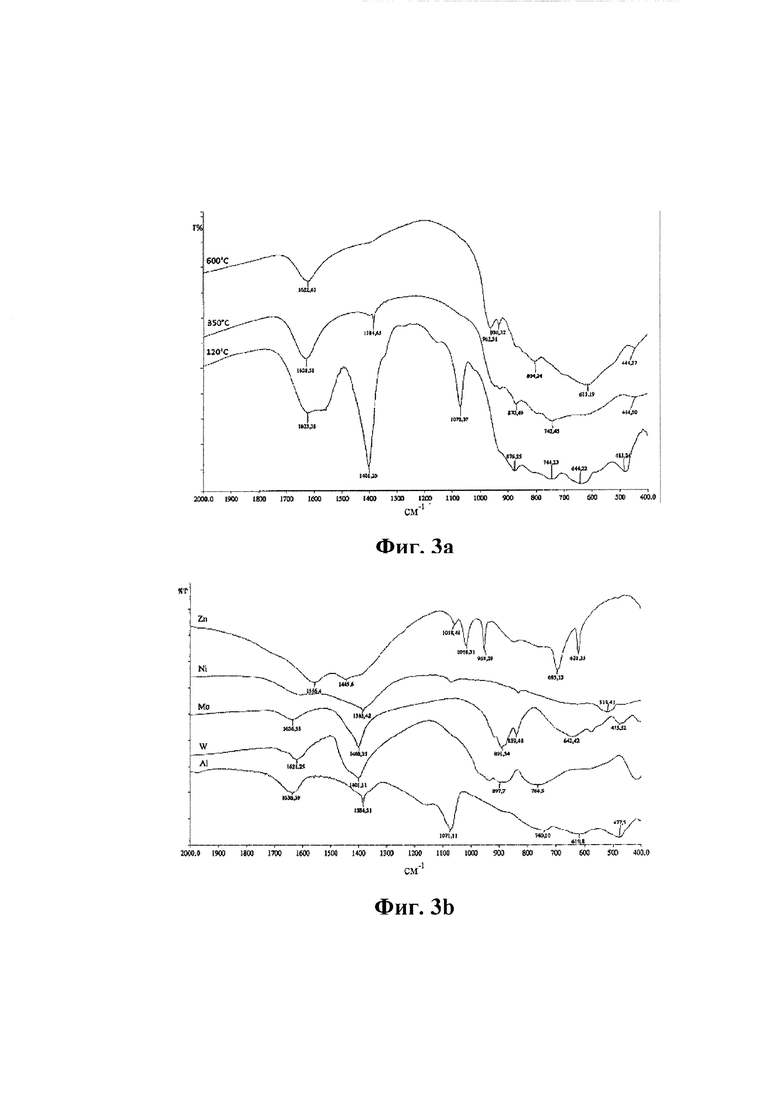
Изобретение относится к области гетерогенного катализа, в частности к способу получения смешанного оксида, содержащего Ni, Mo, W, Al, возможно, по меньшей мере один металл Ме и органический компонент С или остаток указанного органического компонента С, имеющего следующую формулу (I): 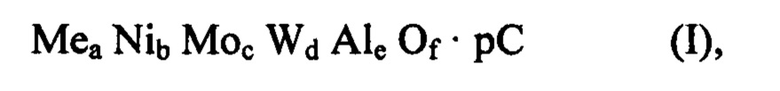 в которой Ме выбирают из группы, состоящей из Zn, Cd и их смеси, С содержит полимерное органическое соединение, а больше или равно 0, b, c, d, e и f больше 0, f равно (2a+2b+6c+6d+3e)/2, отношение (a+b)/(c+d) составляет от 0,9 до 1,1, отношение a/b больше или равно 0 и меньше или равно 1,5, отношение c/d составляет от 0,2 до 5, отношение (a+b+c+d)/e составляет от 0,6 до 5, и р является массовой процентной долей С по отношению к общей массе смешанного оксида формулы (I) и она больше 0% и меньше или равна 40%. Причем указанный смешанный оксид формулы (I) содержит аморфную фазу и псевдокристаллическую фазу, изоструктурную вольфрамиту, где указанный способ включает следующие стадии: 1) смешивание по меньшей мере одного растворимого источника W и по меньшей мере одного растворимого источника Мо в подходящем объеме воды до тех пор, пока не получают прозрачный водный раствор, 2) возможно, добавление по меньшей мере одного источника по меньшей мере одного элемента Ме в раствор, полученный на стадии 1, 3) добавление по меньшей мере одного источника Ni в смесь, полученную на предшествующей стадии, 4) подвергание смеси, полученной на стадии 3, первой тепловой обработке при температурах от 50 °С до 80 °С при перемешивании, 5) добавление к смеси, полученной на стадии 4, по меньшей мере одного растворимого, гидролизуемого или диспергируемого источника Al и по меньшей мере одного полимерного органического соединения, 6) подвергание смеси, полученной на предшествующей стадии, второй тепловой обработке при температурах от 80 °С до 95 °С при перемешивании с получением суспензии, 7) подвергание суспензии, полученной на стадии 6, сушке с получением таким образом твердой фазы, 8) обжиг указанной твердой фазы, полученной на предшествующей стадии, с получением смешанного оксида формулы (I). Изобретение также относится к смешанному оксиду в качестве предшественника катализаторов гидрообработки на основе сульфидов металлов, к катализатору на основе сульфидов металлов для гидробработки, полученному путем сульфидирования смешанного оксида формулы (I) или смешанного оксида формулы (I), связанного по меньшей мере одним неорганическим связующим В, и к способу гидроочистки сырья, содержащего один или более углеводородов. Технический результат заключается в получении катализатора, отличающегося высокими характеристиками гидродесульфурации и гидроденитрификации. 4 н. и 16 з.п. ф-лы, 5 ил., 3 табл., 7 пр.
в которой Ме выбирают из группы, состоящей из Zn, Cd и их смеси, С содержит полимерное органическое соединение, а больше или равно 0, b, c, d, e и f больше 0, f равно (2a+2b+6c+6d+3e)/2, отношение (a+b)/(c+d) составляет от 0,9 до 1,1, отношение a/b больше или равно 0 и меньше или равно 1,5, отношение c/d составляет от 0,2 до 5, отношение (a+b+c+d)/e составляет от 0,6 до 5, и р является массовой процентной долей С по отношению к общей массе смешанного оксида формулы (I) и она больше 0% и меньше или равна 40%. Причем указанный смешанный оксид формулы (I) содержит аморфную фазу и псевдокристаллическую фазу, изоструктурную вольфрамиту, где указанный способ включает следующие стадии: 1) смешивание по меньшей мере одного растворимого источника W и по меньшей мере одного растворимого источника Мо в подходящем объеме воды до тех пор, пока не получают прозрачный водный раствор, 2) возможно, добавление по меньшей мере одного источника по меньшей мере одного элемента Ме в раствор, полученный на стадии 1, 3) добавление по меньшей мере одного источника Ni в смесь, полученную на предшествующей стадии, 4) подвергание смеси, полученной на стадии 3, первой тепловой обработке при температурах от 50 °С до 80 °С при перемешивании, 5) добавление к смеси, полученной на стадии 4, по меньшей мере одного растворимого, гидролизуемого или диспергируемого источника Al и по меньшей мере одного полимерного органического соединения, 6) подвергание смеси, полученной на предшествующей стадии, второй тепловой обработке при температурах от 80 °С до 95 °С при перемешивании с получением суспензии, 7) подвергание суспензии, полученной на стадии 6, сушке с получением таким образом твердой фазы, 8) обжиг указанной твердой фазы, полученной на предшествующей стадии, с получением смешанного оксида формулы (I). Изобретение также относится к смешанному оксиду в качестве предшественника катализаторов гидрообработки на основе сульфидов металлов, к катализатору на основе сульфидов металлов для гидробработки, полученному путем сульфидирования смешанного оксида формулы (I) или смешанного оксида формулы (I), связанного по меньшей мере одним неорганическим связующим В, и к способу гидроочистки сырья, содержащего один или более углеводородов. Технический результат заключается в получении катализатора, отличающегося высокими характеристиками гидродесульфурации и гидроденитрификации. 4 н. и 16 з.п. ф-лы, 5 ил., 3 табл., 7 пр.
1. Способ получения смешанного оксида, содержащего Ni, Mo, W, Al, возможно, по меньшей мере один металл Ме и органический компонент С или остаток указанного органического компонента С, имеющего следующую формулу (I):
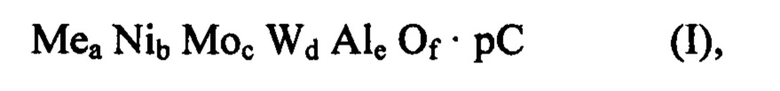
в которой
Ме выбирают из группы, состоящей из Zn, Cd и их смеси,
С содержит полимерное органическое соединение,
а больше или равно 0,
b, c, d, e и f больше 0,
f равно (2a+2b+6c+6d+3e)/2,
отношение (a+b)/(c+d) составляет от 0,9 до 1,1,
отношение a/b больше или равно 0 и меньше или равно 1,5,
отношение c/d составляет от 0,2 до 5,
отношение (a+b+c+d)/e составляет от 0,6 до 5, и
р является массовой процентной долей С по отношению к общей массе смешанного оксида формулы (I) и она больше 0% и меньше или равна 40%,
причем указанный смешанный оксид формулы (I) содержит аморфную фазу и псевдокристаллическую фазу, изоструктурную вольфрамиту,
где указанный способ включает следующие стадии:
1) смешивание по меньшей мере одного растворимого источника W и по меньшей мере одного растворимого источника Мо в подходящем объеме воды до тех пор, пока не получают прозрачный водный раствор,
2) возможно, добавление по меньшей мере одного источника по меньшей мере одного элемента Ме в раствор, полученный на стадии 1,
3) добавление по меньшей мере одного источника Ni в смесь, полученную на предшествующей стадии,
4) подвергание смеси, полученной на стадии 3, первой тепловой обработке при температурах от 50 °С до 80 °С при перемешивании,
5) добавление к смеси, полученной на стадии 4, по меньшей мере одного растворимого, гидролизуемого или диспергируемого источника Al и по меньшей мере одного полимерного органического соединения,
6) подвергание смеси, полученной на предшествующей стадии, второй тепловой обработке при температурах от 80 °С до 95 °С при перемешивании с получением суспензии,
7) подвергание суспензии, полученной на стадии 6, сушке с получением таким образом твердой фазы,
8) обжиг указанной твердой фазы, полученной на предшествующей стадии, с получением смешанного оксида формулы (I).
2. Способ по п.1, в котором b, c, d, e и f больше 0,1.
3. Способ по п.1 или 2, в котором элемент Ме, возможно добавленный на стадии 2 указанного способа, является Zn.
4. Способ по любому из пп. 1–3, в котором возможную стадию 2 пропускают.
5. Способ по любому из пп. 1–4, в котором первую тепловую обработку на стадии 4 выполняют в течение времени от 10 минут до 1 часа.
6. Способ по любому из пп. 1–5, в котором в качестве источника алюминия на стадии 5 указанного способа используют диспергируемый оксид алюминия и предпочтительно указанный диспергируемый оксид алюминия является бёмитом или псевдобёмитом, характеризующимся частицами со средним диаметром менее 100 мкм.
7. Способ по любому из пп. 1–6, в котором полимерное органическое соединение, добавляемое на стадии 5 указанного способа, является метилцеллюлозой.
8. Способ по любому из пп. 1–7, в котором на стадии 5 указанного способа добавляют дисперсию, получаемую отдельно и содержащую как указанный по меньшей мере один растворимый, гидролизуемый или диспергируемый источник Al, так и указанное по меньшей мере одно полимерное органическое соединение.
9. Способ по любому из пп. 1–8, в котором вторую тепловую обработку на стадии 6 выполняют в течение времени от 5 до 30 часов.
10. Способ по любому из пп. 1–9, включающий, сразу после стадии 7, дополнительную стадию 7’, на которой высушенную твердую фазу, полученную на указанной стадии 7, подвергают формованию.
11. Способ по любому из пп. 1–10, в котором обжиг на стадии 8 выполняют в инертной атмосфере или на воздухе при температурах, больше или равных 200 °С и меньше или равных 450 °С.
12. Способ по любому из пп. 1–11, в котором процентная доля «р» указанного органического компонента С и, возможно, остатка указанного органического компонента С в композиции смешанного оксида, получаемого указанным способом, больше или равна 0,01% и меньше или равна 30%.
13. Способ по любому из пп. 1–12, в котором указанный способ включает, сразу после стадии 5 указанного способа, дополнительную стадию 5’, на которой добавляют неорганическое связующее В.
14. Смешанный оксид в качестве предшественника катализаторов гидрообработки на основе сульфидов металлов, содержащий Ni, Mo, W, Al, возможно по меньшей мере один металл Ме и органический компонент С или остаток указанного органического компонента С, имеющий следующую формулу (I):
в которой
Ме выбран из группы, состоящей из Zn, Cd и их смеси,
С содержит полимерное органическое соединение, содержащее один или более гетероатомов, выбранных из N и О,
а больше или равно 0,
b, c, d, e и f больше 0,
f равно (2a+2b+6c+6d+3e)/2,
отношение (a+b)/(c+d) составляет от 0,9 до 1,1,
отношение a/b больше или равно 0 и меньше или равно 1,5,
отношение c/d составляет от 0,2 до 5,
отношение (a+b+c+d)/e составляет от 0,6 до 5, и
р является массовой процентной долей указанного органического соединения С или остатка указанного соединения С по отношению к общей массе смешанного оксида формулы (I) и она больше или равна 0,2% и меньше или равна 30%,
причем указанный смешанный оксид содержит аморфную фазу и псевдокристаллическую фазу, изоструктурную вольфрамиту.
15. Смешанный оксид по п. 14, в котором, когда а больше 0, элемент Ме является Zn.
16. Смешанный оксид по п.14 или 15, связанный по меньшей мере одним неорганическим связующим В.
17. Смешанный оксид по п.14 или 15, получаемый способом по любому из пп. 1–12.
18. Смешанный оксид по п. 16, получаемый способом по п. 13.
19. Катализатор на основе сульфидов металлов для гидробработки, полученный путем сульфидирования смешанного оксида формулы (I) или смешанного оксида формулы (I), связанного по меньшей мере одним неорганическим связующим В, по любому из пп. 14–18, где неорганическое связующее В выбрано из группы, состоящей из диоксида кремния, оксида алюминия, алюмосиликата, покрытого оксидом алюминия диоксида кремния и покрытого диоксидом кремния оксида алюминия, гиббсита, диоксида титана, диоксида циркония, анионных и катионных глин, сапонита, бентонита, каолина, сепиолита или гидроталькита или их смесей.
20. Способ гидроочистки сырья, содержащего один или более углеводородов, который включает контактирование указанного сырья с водородом в присутствии катализатора по п. 19.
| WO 2012130728 A1, 04.10.2012 | |||
| YANDIE CHEN et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| СМЕШАННЫЕ ОКСИДЫ ПЕРЕХОДНЫХ МЕТАЛЛОВ, ПОЛУЧАЕМЫЕ ИЗ НИХ КАТАЛИЗАТОРЫ ГИДРООЧИСТКИ И СПОСОБ ПОЛУЧЕНИЯ, ВКЛЮЧАЮЩИЙ ЗОЛЬ-ГЕЛЬ СПОСОБЫ | 2010 |
|
RU2554941C2 |
| МЕЗОПОРИСТЫЕ МАТЕРИАЛЫ С АКТИВНЫМИ МЕТАЛЛАМИ | 2003 |
|
RU2334554C2 |

