ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области медицины, и в частности относится к производным гинзенозида панаксдиолового типа, способам их получения и применения.
УРОВЕНЬ ТЕХНИКИ
В настоящее время атеросклероз является одним из заболеваний с высоким уровнем заболеваемости. Атеросклероз - это общая патологическая основа многих серьезных сердечнососудистых заболеваний (таких как ишемическая болезнь сердца, стенокардия, цереброваскулярная эмболия и т.д.), представляющих серьезную угрозу здоровью человека. В настоящее время лекарственные средства для лечения атеросклероза в основном регулируют содержание липидов крови, включая статины, фибраты и ниацины. Хотя при помощи указанных выше категорий лекарственных средств можно достигнуть хороших эффектов по снижению уровня липидов крови, уровень заболеваемости атеросклерозом остается высоким и из года в год увеличивается, что свидетельствует о том, что лекарственные средства, обладающие только функцией регуляции липидов крови, не могут отвечать требованиям по предотвращению и лечению атеросклероза. Исследования показали, что воспалительные иммунные факторы играют важную роль в патогенезе атеросклероза, а атеросклероз был признан хроническим воспалительным заболеванием кровеносных сосудов. Таким образом, важно разработать антиатеро склеротические лекарственные средства, которые одновременно оказывают регулирующее действие на липиды крови и обладают противовоспалительной активностью.
С древних времен женьшень был провозглашен "королем трав", который обладает эффектом восполнения жизненной энергии, восстановления пульса и предотвращения разрушения, активации работы селезенки и тонизирования легких, стимулирования выделения слюны или жидкости организма и обновления крови, а также успокаивает разум и способствует интеллектуальной деятельности. Гинзенозид является основным активным ингредиентом женьшеня и обладает основной фармакологической активностью, присущей женьшеню. Существует более 40 видов гинзенозидных мономеров с идентифицированными структурами. Исследования показали, что гинзенозиды панаксдиолового типа обладают определенным действием по предотвращению и лечению атеросклероза, и дополнительные исследования механизмов показали, что их фармакологическое действие включает регулирование липидов крови, уменьшение воспаления и уменьшение разрастания гладких мышц и т.д., однако они имеют низкую активность, обладают определенной цитотоксичностью и не имеют практической значимости.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Задачей настоящего изобретения является обеспечение производных гинзенозида панаксдиолового типа, способов их получения и применения. Изобретение предназначено для обеспечения производных гинзенозида панаксдиолового типа, обладающих относительно высокой активностью и низкой токсичностью, которые можно применять для получения лекарственных средств для предотвращения и лечения атеросклероза.
В настоящем изобретении предложено производное гинзенозида панаксдиолового типа, имеющее структуру, представленную в формуле I или формуле II:

в формуле I и формуле II R1 представляет собой 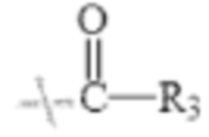 , при этом R3 представляет собой С1-С4 алкил;
, при этом R3 представляет собой С1-С4 алкил;
R2 имеет структуру, представленную в формуле III:

в формуле III n=0, 1 или 2, R4 представляет собой метил или этил, R5 представляет собой одно из атомов водорода, замещенного или незамещенного С1-С5 алкила, замещенного или незамещенного бензила и С4-С9 гетероциклоалкила.
Предпочтительно R5 представляет собой С1-С5 алкил, С1-С5 гидроксиалкил или С3-С5 сложноэфирный алкил.
Предпочтительно R5 представляет собой -СН2СН(СН3)2, -СН(СН3)2, -СН(ОН)СН3, -СН2ОН, -СН2СН2СООСН2СН3 или -СН2СООСН2СН3.
Предпочтительно R5 представляет собой 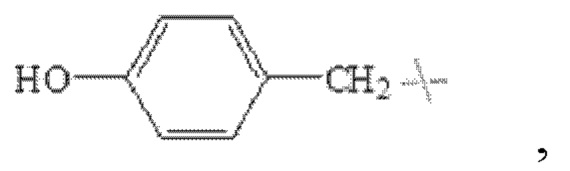
Предпочтительно R2 представляет собой 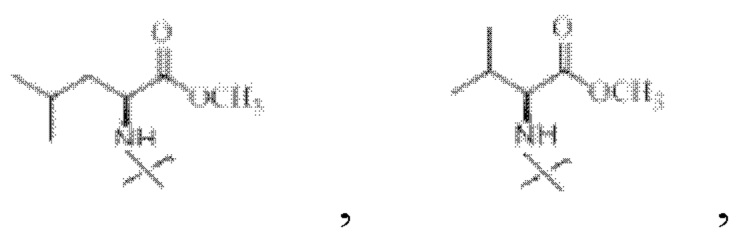
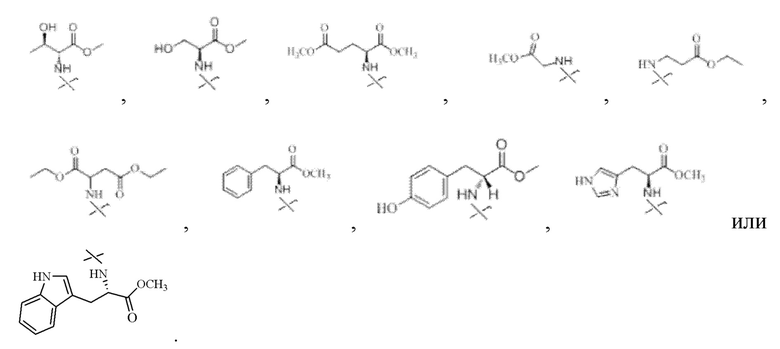
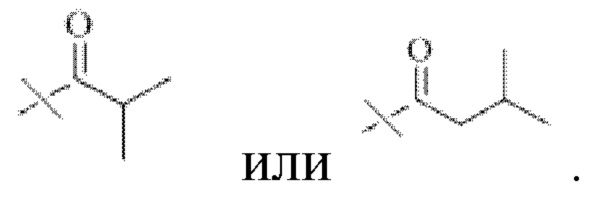
Предпочтительно R1 представляет собой 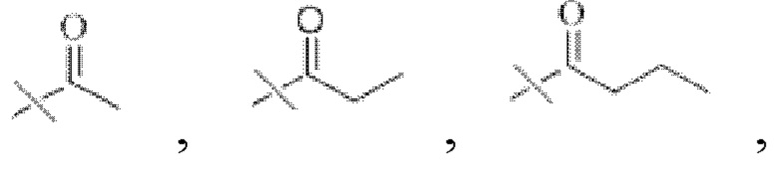
В настоящем изобретении предложен способ получения производного гинзенозида панаксдиолового типа, описанного в указанном выше техническом решении, включающий следующие стадии:
(1) введение исходного соединения в реакцию нуклеофильного замещения с ангидридом кислоты в присутствии щелочного реагента с получением первого промежуточного продукта;
(2) введение указанного первого промежуточного продукта стадии (1) в реакцию окисления в присутствии окисляющего агента и органического растворителя с получением второго промежуточного продукта;
(3) введение указанного второго промежуточного продукта стадии (2) в реакцию восстановительного аминирования с аминосоединением в присутствии органического растворителя и восстанавливающего агента с получением производного гинзенозида панаксдиолового типа, имеющего структуру, представленную в формуле I или формуле II;
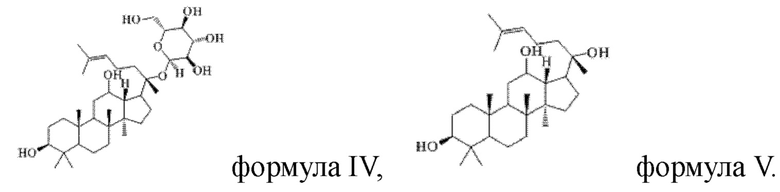
причем указанное исходное соединение в стадии (1) имеет структуру, представленную в формуле IV или формуле V:
Предпочтительно указанная реакция окисления в стадии (2) включает в частности:
(21) введение указанного промежуточного продукта стадии (1) в первичную реакцию окисления в присутствии первого окисляющего агента и органического растворителя с получением предшественника указанного второго промежуточного продукта;
(22) введение указанного предшественника второго промежуточного продукта стадии (21) во вторичную реакцию окисления в присутствии второго окисляющего агента и органического растворителя с получением указанного второго промежуточного продукта.
Предпочтительно первый окисляющий агент в стадии (21) представляет собой пероксид водорода, хлорноватистую кислоту, гипохлорит кальция, пероксид ацетона или мета-хлорпербензойную кислоту; второй окисляющий агент в стадии (22) представляет собой перманганат калия, диоксид марганца, йодную кислоту или реагент Саретта.
В настоящем изобретении предложено применение производного гинзенозида панаксдиолового типа, описанного в указанном выше техническом решении, или производного гинзенозида панаксдиолового типа, полученного согласно способу получения, описанному в указанном выше техническом решении, для получения лекарственного средства для предотвращения и лечения атеросклероза.
В настоящем изобретении предложены производные гинзенозида панаксдиолового типа, имеющее структуру, представленную в формуле I или формуле II, и предлагаемые в настоящем изобретении указанные производные гинзенозида панаксдиолового типа обладают низкой цитотоксичностью, могут значительно снижать процент площади атеросклеротических бляшек у мышей ароЕ-/-, они также могут эффективно снижать уровни холестерина липопротеинов низкой плотности и повышать уровни холестерина липопротеинов высокой плотности в сыворотке мышей и могут значительно снижать локальные уровни ФНО-α в артериях мышей ароЕ-/- и обладают хорошим противовоспалительным действием; при дозе 30 мкмоль/л производные гинзенозида панаксдиолового типа могут значительно уменьшать степень образования пенистых клеток, полученных из клеток RAW264.7, что свидетельствует о том, что предложенные в настоящем изобретении производные гинзенозида панаксдиолового типа можно применять в качестве активных ингредиентов для получения лекарственных средств для предотвращения и лечения атеросклероза.
Помимо этого, в настоящем изобретении предложены способы получения производных гинзенозида панаксдиолового типа, которые просты в обращении и которые можно получить с высоким выходом.
ВАРИАНТЫ РЕАЛИЗАЦИИ
В настоящем изобретении предложено производное гинзенозида панаксдиолового типа, имеющее структуру, представленную в формуле I или формуле II:
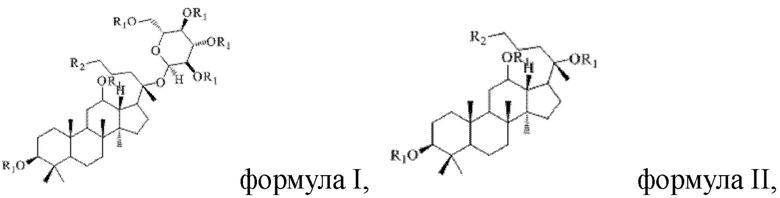
в формуле I и формуле II R1 представляет собой 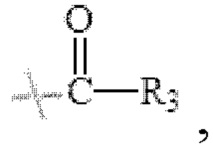 при этом R3 представляет собой С1-С4 алкил;
при этом R3 представляет собой С1-С4 алкил;
R2 имеет структуру, представленную в формуле III:
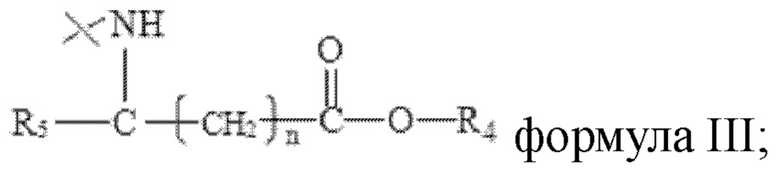
в формуле III n=0, 1 или 2, R4 представляет собой метил или этил, R5 представляет собой одно из атома водорода, замещенного или незамещенного С1-С5 алкила, замещенного или незамещенного бензила и С4-С9 гетероциклоалкила.
В настоящем изобретении R1 представляет собой  при этом R3 представляет собой С1-С4 алкил, предпочтительно метил, этил, н-пропил, изопропил или изобутил. Соответственно в настоящем изобретении R1 предпочтительно представляет собой
при этом R3 представляет собой С1-С4 алкил, предпочтительно метил, этил, н-пропил, изопропил или изобутил. Соответственно в настоящем изобретении R1 предпочтительно представляет собой 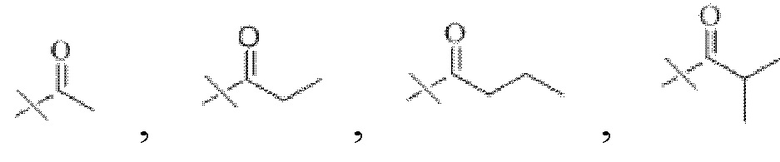 или
или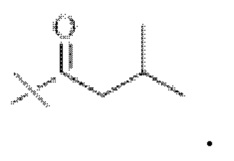
В настоящем изобретении R2 имеет структуру, представленную в формуле III:
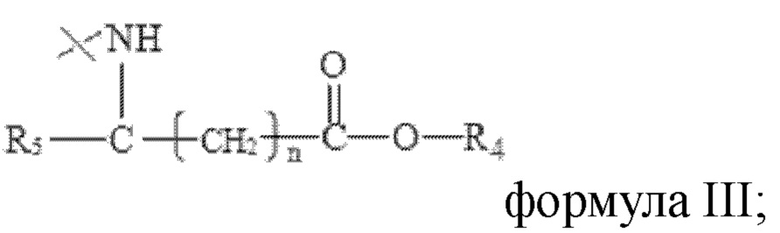
в формуле III n=0, 1 или 2, R4 представляет собой метил или этил, R5 представляет собой одно из атома водорода, замещенного или незамещенного С1-С5 алкила, замещенного или незамещенного бензила и С4-С9 гетероциклоалкила.
В настоящем изобретении указанный замещенный или незамещенный С1-С5 алкил предпочтительно представляет собой С1-С5 алкил, С1-С5 гидроксиалкил или С3-С5 сложноэфирный алкил. В настоящем изобретении указанный С1-С5 алкил предпочтительно представляет собой -CH2CH(СН3)2 или -СН(СН3)2; указанный С1-С5 гидроксиалкил предпочтительно представляет собой -СН(ОН)СН3 или -СН2ОН; указанный С3-С5 сложноэфирный алкил предпочтительно представляет собой - СН2СН2СООСН2СН3 ИЛИ -СН2СООСН2СН3.
В настоящем изобретении указанный замещенный бензил предпочтительно представляет собой гидроксибензил, в частности 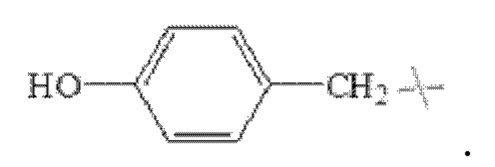
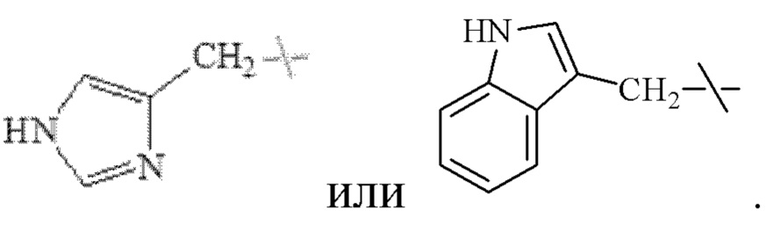
В настоящем изобретении указанный С4-С9 гетероциклоалкил предпочтительно представляет собой 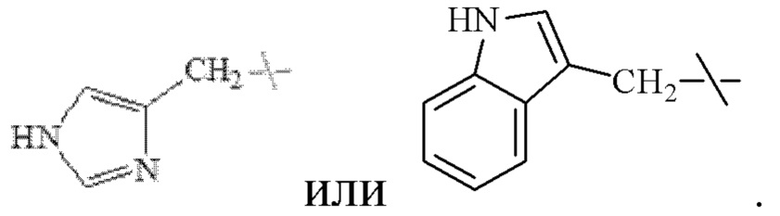
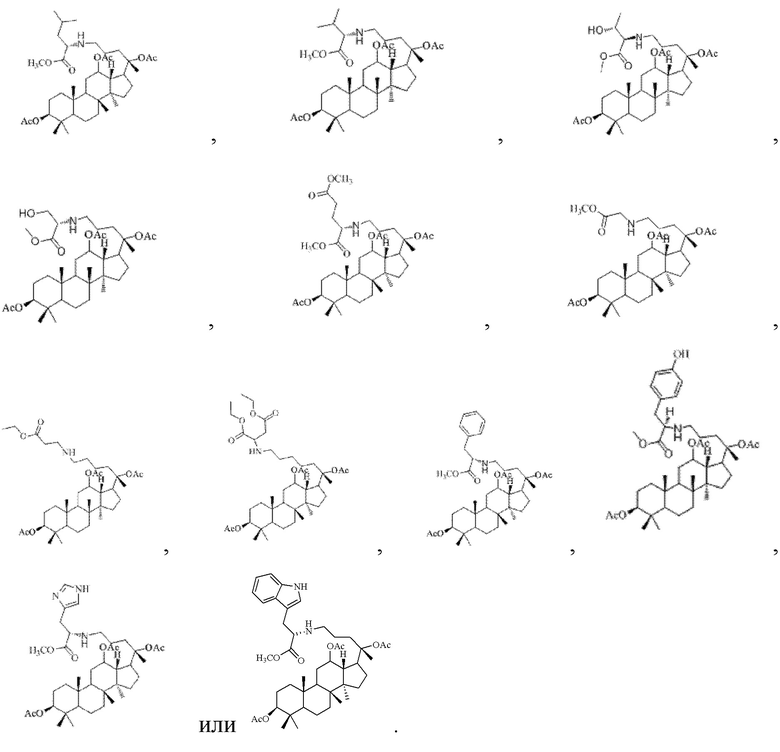
Соответственно в настоящем изобретении R2 предпочтительно представляет собой 

В настоящем изобретении производное гинзенозида панаксдиолового типа, имеющее структуру, представленную в формуле I, предпочтительно представляет собой
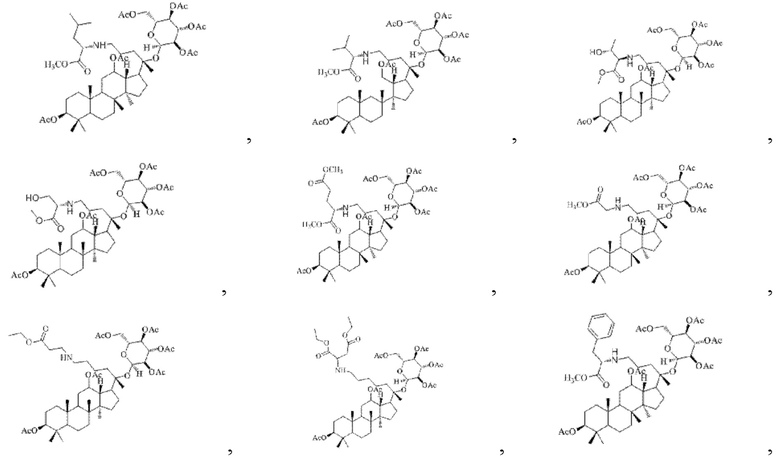
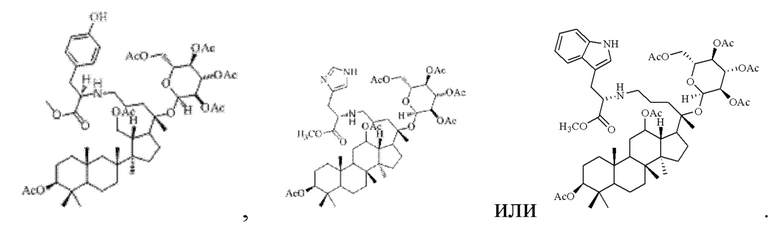
В настоящем изобретении производное гинзенозида панаксдиолового типа, имеющее структуру, представленную в формуле II, предпочтительно представляет собой
В настоящем изобретении предложен способ получения производного гинзенозида панаксдиолового типа, описанного в указанном выше техническом решении, включающий следующие стадии:
(1) введение исходного соединения в реакцию нуклеофильного замещения с ангидридом кислоты в присутствии щелочного реагента с получением первого промежуточного продукта;
(2) введение указанного первого промежуточного продукта стадии (1) в реакцию окисления в присутствии окисляющего агента и органического растворителя с получением второго промежуточного продукта;
(3) введение указанного второго промежуточного продукта стадии (2) в реакцию восстановительного аминирования с аминосоединением в присутствии органического растворителя и восстанавливающего агента с получением производного гинзенозида панаксдиолового типа, имеющего структуру, представленную в формуле I или формуле II;
причем указанное исходное соединение в стадии (1) имеет структуру, представленную в формуле IV или формуле V:

В настоящем изобретении исходное соединение, имеющее структуру, представленную в формуле IV или формуле V, вводят в реакцию нуклеофильного замещения с ангидридом кислоты в присутствии щелочного реагента с получением первого промежуточного продукта. В настоящем изобретении массовое соотношение исходное соединение: ангидрид кислоты: щелочной реагент предпочтительно составляет 1:(1-20):(1-20), более предпочтительно 1:(4-15):(4-15), наиболее предпочтительно 1:(8-12):(8-12).
В настоящем изобретении тип ангидрида кислоты конкретным образом не ограничен, и можно применять ангидрид кислоты, хорошо известный специалистам в данной области техники, который может иметь структуру R1. В настоящем изобретении указанный ангидрид кислоты предпочтительно представляет собой уксусный ангидрид, пропионовый ангидрид, масляный ангидрид, изомасляный ангидрид или изовалериановый ангидрид.
В настоящем изобретении тип щелочного агента конкретным образом не ограничен, и можно применять органическое щелочное соединение, неорганическое щелочное соединение и/или щелочной металл, которые хорошо известны в данной области техники.
В настоящем изобретении тип органического щелочного соединения конкретным образом не ограничен, и можно применять органическое щелочное соединение, хорошо известное специалистам в данной области техники. В настоящем изобретении указанное органическое щелочное соединение предпочтительно представляет собой триэтиламин, N,N-диизопропилэтиламин, 4-диметиламинопиридин, пиридин, N-метилморфолин, тетраметилэтилендиамин, калия трет-бутоксид, натрия метоксид, этилат калия или этилат натрия.
В настоящем изобретении тип неорганического щелочного соединения конкретным образом не ограничен, и можно применять неорганические щелочные соединения, хорошо известные специалистам в данной области техники. В настоящем изобретении указанное неорганическое щелочное соединение предпочтительно представляет собой гидроксид натрия, гидроксид калия, карбонат калия, карбонат натрий или гидрид натрия.
В настоящем изобретении тип щелочного металла конкретным образом не ограничен, и можно применять щелочной металл, хорошо известный специалистам в данной области техники, как, например, металлический натрий.
В настоящем изобретении температура реакции нуклеофильного замещения предпочтительно представляет собой 0-100°С, более предпочтительно 20-75°С, наиболее предпочтительно 35-60°С; и время реакции нуклеофильного замещения предпочтительно представляет собой 1-48 часов, более предпочтительно 5-38 часов, наиболее предпочтительно 15-25 часов. В настоящем изобретении реакцию нуклеофильного замещения предпочтительно проводят при перемешивании. В настоящем изобретении скорость перемешивания предпочтительно составляет 800-1200 об/мин, более предпочтительно 900-1100 об/мин. В настоящем изобретении способ перемешивания конкретным образом не ограничен, и можно применять способ перемешивания, хорошо известный специалистам в данной области техники. В настоящем изобретении предпочтительно применяют магнитное перемешивание.
В настоящем изобретении предпочтительно после завершения реакции нуклеофильного замещения, полученный продукт подвергают последующей обработке с получением первого промежуточного продукта. В настоящем изобретении указанная последующая обработка предпочтительно включает следующие стадии: после реакции нуклеофильного замещения осуществляют перегонку при пониженном давлении с получением неочищенного продукта; указанный неочищенный продукт очищают с помощью колоночной хроматографии на силикагеле с получением первого промежуточного продукта.
В настоящем изобретении определенные стадии операций и реагенты, применяемые в колоночной хроматографии на силикагеле, конкретным образом не ограничены, и можно применять методы колоночной хроматографии на силикагеле, хорошо известные специалистам в данной области техники. В настоящем изобретении неочищенный продукт предпочтительно смешивают с этилацетатом, и смесь применяют для проведения колоночной хроматографии после смешивания с силикагелем. В настоящем изобретении массовое отношение неочищенного продукта к этилацетату предпочтительно составляет 1:(1-10), более предпочтительно 1:(3-8) и наиболее предпочтительно 1:(4-6). В настоящем изобретении этилацетат и н-гексан предпочтительно применяют в качестве элюента, и объемное отношение этилацетата к н-гексану в указанном элюенте предпочтительно составляет 1:(1-10), более предпочтительно 1:(3-8), наиболее предпочтительно 1:(4-6).
В настоящем изобретении после получения первого промежуточного продукта первый промежуточный продукт подвергают реакции окисления в присутствии окисляющего агента и органического растворителя с получением второго промежуточного продукта. В настоящем изобретении реакция окисления конкретным образом включает:
введение первого промежуточного продукта в первичную реакцию окисления в присутствии первого окисляющего агента и органического растворителя с получением предшественника второго промежуточного продукта;
введение указанного предшественника второго промежуточного продукта во вторичную реакцию окисления в присутствии второго окисляющего агента и органического растворителя с получением указанного второго промежуточного продукта.
В настоящем изобретении первый промежуточный продукт подвергают первичной реакции окисления в присутствии первого окисляющего агента и органического растворителя с получением предшественника второго промежуточного продукта. В настоящем изобретении массовое отношение первый промежуточный продукт: первый окисляющий агент: органический растворитель предпочтительно составляет 1:(0,1-0,5):(5-10), более предпочтительно 1: (0,2-0,4): (6-8).
В настоящем изобретении первый окисляющий агент предпочтительно представляет собой пероксид водорода, гипохлорит кальция, пероксид ацетона или мета-хлорпербензойную кислоту.
В настоящем изобретении тип органического растворителя, необходимого для проведения первичной реакции окисления согласно настоящему изобретению, конкретным образом не ограничен, и можно применять полярный органический растворитель или неполярный органический растворитель, хорошо известный специалистам в данной области техники, который совместим с указанным первым промежуточным продуктом и указанным первым окисляющим агентом, такой как пиридин, N-метилпирролидон, хлороформ, дихлорметан, тетрахлорметан, тетрагидрофуран или 1,4-диоксан.
В настоящем изобретении температура первичной реакции окисления предпочтительно составляет 10-80°С, более предпочтительно 15-55°С и наиболее предпочтительно 20-35°С; в частности, в вариантах реализации согласно настоящему изобретению первичную реакцию окисления осуществляют при комнатной температуре без нагревания или охлаждения реакционной системы. В настоящем изобретении время первичной реакции окисления предпочтительно составляет 1-10 часов, более предпочтительно 3-8 часов и наиболее предпочтительно 4-6 часов.
В настоящем изобретении, предпочтительно после завершения первичной реакции окисления, полученный продукт подвергают последующей обработке с получением предшественника второго промежуточного продукта. В настоящем изобретении указанная последующая обработка предпочтительно включает следующие стадии: добавление воды к материалу, полученному после первичной реакции окисления, перемешивание, фильтрование, удаление растворителя, получение неочищенного продукта; очистку указанного неочищенного продукта с помощью метода колоночной хроматографии на силикагеле с получением второго промежуточного продукта.
В настоящем изобретении массовое отношение полученного после первичной реакции окисления материала к воде предпочтительно составляет 1:(1-100), более предпочтительно 1:(8-70) и наиболее предпочтительно 1:(20-40). В настоящем изобретении скорость перемешивания предпочтительно составляет 800-1200 об/мин, более предпочтительно 900-1100 об/мин; и время перемешивания предпочтительно составляет 1-10 часов, более предпочтительно 3-8 часов и наиболее предпочтительно 4-6 часов. В настоящем изобретении способ перемешивания конкретным образом не ограничен, и можно применять способ перемешивания, хорошо известный специалистам в данной области техники. В настоящем изобретении предпочтительно применяют магнитное перемешивание. В настоящем изобретении, способ фильтрования конкретным образом не ограничен, и может быть применено техническое решение для фильтрования, хорошо известное специалистам в данной области техники. В настоящем изобретении способ удаления растворителя конкретным образом не ограничен, и может быть применено техническое решение для удаления растворителя, хорошо известное специалистам в данной области техники. В настоящем изобретении для удаления растворителя предпочтительно применяют метод перегонки при пониженном давлении.
В настоящем изобретении определенные стадии операций и реагенты, применяемые в колоночной хроматографии на силикагеле, конкретным образом не ограничены, и можно применять методы колоночной хроматографии на силикагеле, хорошо известные специалистам в данной области техники. В настоящем изобретении неочищенный продукт предпочтительно смешивают с этилацетатом, и указанную смесь применяют для проведения колоночной хроматографии после смешивания с силикагелем. В настоящем изобретении массовое отношение неочищенного продукта к этилацетату предпочтительно составляет 1:(1-10), более предпочтительно 1:(3-8) и наиболее предпочтительно 1:(4-6). В настоящем изобретении этилацетат и н-гексан предпочтительно применяют в качестве элюента, и объемное отношение этилацетата к н-гексану в указанном элюенте предпочтительно составляет 1:(1-10), более предпочтительно 1:(3-8), наиболее предпочтительно 1:(4-6).
В настоящем изобретении, после получения предшественника второго промежуточного продукта, указанного предшественника второго промежуточного продукта вводят во вторичную реакцию окисления в присутствии второго окисляющего агента и органического растворителя с получением второго промежуточного продукта. В настоящем изобретении массовое отношение предшественник второго промежуточного продукта: второй окисляющий агент: органический растворитель предпочтительно составляет 1:(0,1-0,3):(5-10), более предпочтительно 1:(0,15-0,25):(6-8).
В настоящем изобретении второй окисляющий агент предпочтительно представляет собой представляет собой перманганат калия, диоксид марганца, йодную кислоту или реагент Саретта.
В настоящем изобретении тип органического растворителя, необходимого для проведения вторичной реакции окисления, конкретным образом не ограничен, и можно применять полярный органический растворитель или неполярный органический растворитель, хорошо известный специалистам в данной области техники, который совместим с предшественником второго промежуточного продукта и вторым окисляющим агентом, такой как хлороформ, дихлорметан, тетрахлорметан, ацетонитрил, тетрагидрофуран или 1,4-диоксан.
В настоящем изобретении температура вторичной реакции окисления предпочтительно составляет 10-100°С, более предпочтительно 15-65°С, наиболее предпочтительно 20-45°С; в частности, в вариантах реализации согласно настоящему изобретению вторичную реакцию окисления осуществляют при комнатной температуре без нагревания или охлаждения реакционной системы. В настоящем изобретении время вторичной реакции окисления предпочтительно составляет 1-10 часов, более предпочтительно 3-8 часов и наиболее предпочтительно 4-6 часов.
В настоящем изобретении предпочтительно после завершения вторичной реакции окисления, полученный продукт подвергают последующей обработке с получением второго промежуточного продукта. В настоящем изобретении указанная последующая обработка предпочтительно включает следующие стадии: экстракцию органическим растворителем материала, полученного после вторичной реакции окисления, промывку, удаление растворителя, получение неочищенного продукта; очистку указанного неочищенного продукта с помощью метода колоночной хроматографии на силикагеле с получением второго промежуточного продукта.
В настоящем изобретении массовое отношение полученного после вторичной реакции окисления материала к экстрагенту предпочтительно составляет 1:(10-30), более предпочтительно 1:(15-25) и наиболее предпочтительно 1:(18-22). В настоящем изобретении указанный экстрагент предпочтительно представляет собой этилацетат, хлороформ, дихлорметан или тетрахлорметан. В настоящем изобретении экстракцию предпочтительно проводят 2-4 раза. В настоящем изобретении, в частности, органическую фазу, полученную после экстракции, промывают.
В настоящем изобретении способ промывки конкретным образом не ограничен, и может быть применено техническое решение для промывки, хорошо известное специалистам в данной области техники. В настоящем изобретении предпочтительно материал, полученный после экстракции, последовательно промывают щелочным раствором и раствором хлорида натрия. В настоящем изобретении объемное отношение полученного после экстракции материала к щелочному раствору или раствору хлорида натрия предпочтительно составляет 1:(1-10), более предпочтительно 1:(3-8) и наиболее предпочтительно 1:(4-6). В настоящем изобретении тип щелочного раствора конкретным образом не ограничен, и можно применять щелочной раствор, хорошо известный специалистам в данной области техники, как, например, раствор бикарбоната натрия. В настоящем изобретении концентрации щелочного раствора и раствора хлорида натрия конкретным образом не ограничены, и можно применять подходящие для промывки концентрации растворов, хорошо известные специалистам в данной области техники. В вариантах реализации настоящего изобретения промывку проводят, в частности, с применением насыщенного щелочного раствора и насыщенного раствора хлорида натрия. В настоящем изобретении материал, полученный после экстракции, предпочтительно промывают щелочным раствором от 3 до 5 раз, и затем промывают раствором хлорида от 3 до 5 раз. В настоящем изобретении способ удаления растворителя конкретным образом не ограничен, и может быть применено техническое решение для удаления растворителя, хорошо известное специалистам в данной области техники. В настоящем изобретении предпочтительно применяют метод перегонки при пониженном давлении.
В настоящем изобретении определенные стадии операций и реагенты, применяемые в колоночной хроматографии на силикагеле, конкретным образом не ограничены, и можно применять методы колоночной хроматографии на силикагеле, хорошо известные специалистам данной области техники. В настоящем изобретении неочищенный продукт предпочтительно смешивают с этилацетатом, и указанную смесь применяют для проведения колоночной хроматографии после смешивания с силикагелем. В настоящем изобретении массовое отношение неочищенного продукта к этилацетату предпочтительно составляет 1:(1-10), более предпочтительно 1:(3-8) и наиболее предпочтительно 1:(4-6). В настоящем изобретении этилацетат и н-гексан предпочтительно применяют в качестве элюента, и объемное отношение этилацетата к н-гексану в указанном элюенте предпочтительно составляет 1:(1-10), более предпочтительно 1:(3-8), наиболее предпочтительно 1:(4-6).
В настоящем изобретении, после получения второго промежуточного продукта, второй промежуточный продукт вводят в реакцию восстановительного аминирования с аминосоединением в присутствии органического растворителя и восстанавливающего агента с получением производного гинзенозида панаксдиолового типа, имеющего структуру, представленную в формуле I или формуле II; в настоящем изобретении массовое отношение второго промежуточного продукта к указанному амино соединению, восстанавливающему агенту и органическому растворителю предпочтительно составляет 1:(1-100):(5-10), более предпочтительно 1:(8-70): (6-9), и еще более предпочтительно 1:(20-50):(7-8).
В настоящем изобретении аминосоединение предпочтительно представляет собой гидрохлорид метилового сложного эфира L-лейцина, гидрохлорид метилового сложного эфира L-валина, гидрохлорид метилового сложного эфира L-треонина, гидрохлорид метилового сложного эфира L-серина, гидрохлорид диметилового сложного эфира L-глутаминовой кислоты, гидрохлорид метилового сложного эфира глицина, гидрохлорид этилового сложного эфира β-аланина, гидрохлорид диэтиламиномалоната, гидрохлорид метилового сложного эфира L-фенилаланина, метиловый сложный эфир L-тирозина, гидрохлорид метилового сложного эфира L-гистидина или гидрохлорид метилового сложного эфира L-триптофана.
В настоящем изобретении восстанавливающий агент предпочтительно представляет собой борогидрид натрия, цианоборогидрид натрия или триацетилборогидрид натрия.
В настоящем изобретении тип органического растворителя, необходимого для проведения реакции восстановительного аминирования, конкретным образом не ограничен, и можно применять полярный органический растворитель или неполярный органический растворитель, хорошо известный специалистам в данной области техники, который совместим со вторым промежуточным продуктом, аминосоединением и восстанавливающим агентом, такой как метанол, этилацетат, хлороформ, дихлорметан и тетрахлорметан.
В настоящем изобретении восстанавливающий агент предпочтительно смешивают со вторым промежуточным продуктом, аминосоединением и органическим растворителем в диапазоне температур от -2 до +2°С. В настоящем изобретении способ регулирования температуры для смешивания восстанавливающего агента со вторым промежуточным продуктом, аминосоединением и органическим растворителем конкретным образом не ограничен, и можно применять способ регулирования температуры, хорошо известный специалистам в данной области техники. В вариантах реализации согласно настоящему изобретению, в частности, восстанавливающий агент смешивают со вторым промежуточным продуктом, аминосоединением и органическим растворителем в условиях ледяной водяной бани. В настоящем изобретении восстанавливающий агент предпочтительно смешивают со вторым промежуточным продуктом, аминосоединением и органическим растворителем при перемешивании. В настоящем изобретении скорость перемешивания предпочтительно составляет 800-1200 об/мин, более предпочтительно 900-1100 об/мин. В настоящем изобретении способ перемешивания конкретным образом не ограничен, и можно применять способ перемешивания, хорошо известный специалистам в данной области техники. В настоящем изобретении предпочтительно применяют магнитное перемешивание.
В настоящем изобретении температура реакции восстановительного аминирования предпочтительно составляет -10-80°С, более предпочтительно 0-55°С и наиболее предпочтительно 20-35°С; в частности, в вариантах реализации согласно настоящему изобретению реакцию восстановительного аминирования проводят при комнатной температуре без нагревания или охлаждения реакционной смеси. В настоящем изобретении время реакции восстановительного аминирования предпочтительно составляет 1-48 часов, более предпочтительно 8-32 часа и наиболее предпочтительно 13-20 часов.
В настоящем изобретении предпочтительно после завершения реакции восстановительного аминирования, полученный продукт подвергают последующей обработке с получением производного гинзенозида панаксдиолового типа, имеющего структуру, представленную в формуле I или формуле II. В настоящем изобретении указанная последующая обработка предпочтительно включает следующие стадии: экстракцию органическим растворителем материала, полученного после реакции восстановительного аминирования, промывку, удаление растворителя, получение неочищенного продукта; очистку указанного неочищенного продукта с помощью метода колоночной хроматографии на силикагеле с получением производного гинзенозида панаксдиолового типа, имеющего структуру, представленную в формуле I или формуле II.
В настоящем изобретении массовое отношение полученного после реакции восстановительного аминирования материала к экстрагенту предпочтительно составляет 1:(1-50), более предпочтительно 1:(8-40) и наиболее предпочтительно 1:(15-25). В настоящем изобретении указанный экстрагент предпочтительно представляет собой этилацетат, хлороформ, дихлорметан или тетрахлорметан. В настоящем изобретении экстракцию предпочтительно проводят 2-4 раза. В настоящем изобретении, в частности, органическую фазу, полученную после экстракции, промывают.
В настоящем изобретении способ промывки конкретным образом не ограничен, и может быть применено техническое решение для промывки, хорошо известное специалистам в данной области техники. В настоящем изобретении предпочтительно материал, полученный после экстракции, промывают раствором хлорида натрия. В настоящем изобретении концентрация раствора хлорида натрия конкретным образом не ограничена, и можно применять подходящие для промывки концентрации раствора, хорошо известные специалистам в данной области техники. В вариантах реализации согласно настоящему изобретению промывку осуществляют, в частности, с применением насыщенного раствора хлорида натрия. В настоящем изобретении объемное отношение материала, полученного после экстракции, к раствору хлорида натрия, предпочтительно составляет 1:(1-30), более предпочтительно 1:(5-23), наиболее предпочтительно 1:(10-15). В настоящем изобретении промывку предпочтительно проводят от 3 до 5 раз. В настоящем изобретении способ удаления растворителя конкретным образом не ограничен, и может быть применено техническое решение для удаления растворителя, хорошо известное специалистам в данной области техники. В настоящем изобретении предпочтительно применяют метод перегонки при пониженном давлении.
В настоящем изобретении определенные стадии операций и реагенты, применяемые в колоночной хроматографии на силикагеле, конкретным образом не ограничены, и можно применять методы колоночной хроматографии на силикагеле, хорошо известные специалистам в данной области техники. В настоящем изобретении неочищенный продукт предпочтительно смешивают с этилацетатом, и указанную смесь применяют для проведения колоночной хроматографии после смешивания с силикагелем. В настоящем изобретении массовое отношение неочищенного продукта к этилацетату предпочтительно составляет 1:(1-10), более предпочтительно 1:(3-8) и наиболее предпочтительно 1:(4-6). В настоящем изобретении этилацетат и н-гексан предпочтительно применяют в качестве элюента, и объемное отношение этилацетата к н-гексану в указанном элюенте предпочтительно составляет 1:(1-10), более предпочтительно 1:(3-8), наиболее предпочтительно 1:(4-6).
В настоящем изобретении предложено применение производного гинзенозида панаксдиолового типа из указанного выше технического решения или применение производного гинзенозида панаксдиолового типа, полученного при помощи способа получения согласно указанному выше техническому решению, для получения лекарственного средства для предотвращения и лечения атеросклероза. В настоящем изобретении указанное лекарственное средство для предотвращения и лечения атеросклероза предпочтительно содержит активный ингредиент и адъювант. В настоящем изобретении указанный активный ингредиент лекарственного средства для предотвращения и лечения атеросклероза представляет собой производное гинзенозида панаксдиолового типа из указанного выше технического решения согласно настоящему изобретению или производное гинзенозида панаксдиолового типа, полученное при помощи способа получения согласно указанному выше техническому решению. В настоящем изобретении тип адъюванта конкретным образом не ограничен, и можно применять адъювант, хорошо известный специалистам в данной области техники. В настоящем изобретении отношение активного ингредиента к адъюванту конкретным образом не ограничено, и указанное отношение активного ингредиента к адъюванту можно определить в соответствии с фактическими условиями. В настоящем изобретении лекарственная форма и дозировка лекарственного средства для предотвращения и лечения атеросклероза конкретным образом не ограничены, и лекарственную форму и дозировку лекарственного средства для предотвращения и лечения атеросклероза можно определить в соответствии с фактическими потребностями.
Технические решения в настоящем изобретении будут конкретно и полностью описаны ниже в сочетании с примерами настоящего изобретения. Очевидно, что описанные примеры являются только частью, но не всеми примерами настоящего изобретения. Все другие примеры, полученные специалистами в данной области техники на основе примеров настоящего изобретения без изобретательских усилий, входят в объем притязаний настоящего изобретения.
Пример 1
1 г (16 ммоль) соединения, имеющего структуру, представленную в формуле IV, смешивали с 10 мл уксусного ангидрида и 10 мл пиридина, и реакцию нуклеофильного замещения проводят в течение 3 часов в условиях перемешивания с магнитной мешалкой при 900 об/мин и 40°С; полученную систему подвергали перегонке при пониженном давлении, полученный неочищенный продукт смешивали с 6 мл этилацетата, и указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:5), получали 1,3 г соединения 1 в виде твердого вещества белого цвета, выход: 90%. Структура соединения 1 представляет собой:
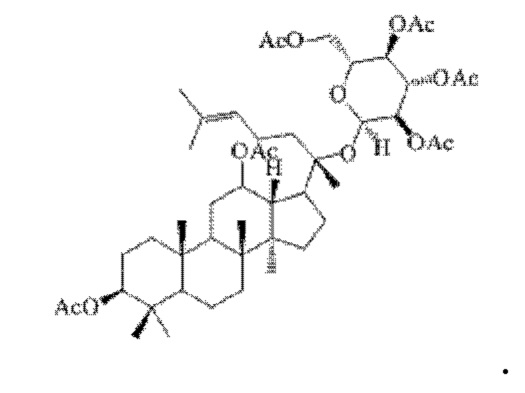
Пример 2
Смешивали 1 г (11 ммоль) соединения 1, полученного в примере 1, 5 мл дихлорметана и 0,2 г (11 ммоль) мета-хлорпероксибензойной кислоты, первичную реакцию окисления осуществляли при комнатной температуре в течение 1 часа; добавляли 25 мл воды к материалу, полученному после указанной первичной реакции окисления, проводили магнитное перемешивание при 1100 об/мин в течение 1 часа, осаждали твердое белое вещество, фильтровали с получением прозрачного раствора, указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 8 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:8), получали 0,87 г соединения 2 в виде твердого вещества белого цвета, выход: 90%. Структура соединения 2 представляет собой:
Пример 3
Смешивали 0,8 г (9 ммоль) соединения 2, полученного в примере 2, 5 мл тетрагидрофурана и 0,32 г (14 ммоль) йодной кислоты, вторичную реакцию окисления осуществляли при комнатной температуре в течение 2 часов; материал, полученный после указанной вторичной реакции окисления, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 40 мл), затем 4 раза последовательно промывали каждым из насыщенного раствора бикарбоната натрия и насыщенного раствора хлорида натрия с получением прозрачного раствора (объем насыщенного раствора бикарбоната натрия, необходимого для каждой промывки, составлял 50 мл, объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 60 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 4 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:4), получали 0,73 г соединения 3 в виде твердого вещества белого цвета, выход: 92%. Структура соединения 3 представляет собой:
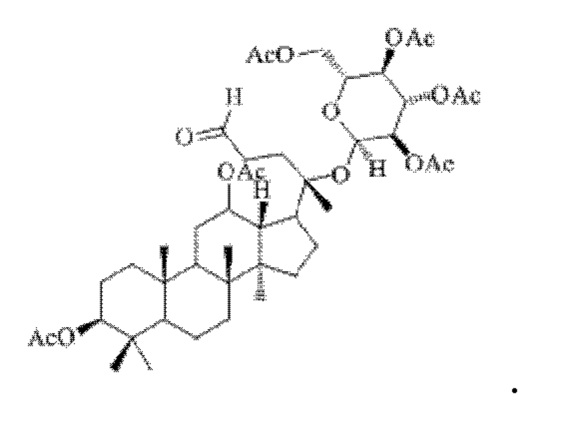
Пример 4
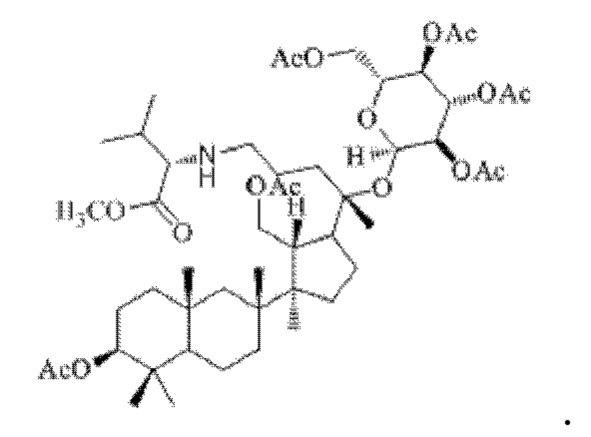
0,05 г (0,6 ммоль) соединения 3, полученного в примере 3, смешивали с 0,013 г (0,7 ммоль) гидрохлорида метилового сложного эфира L-лейцина и 2 мл метанола, добавляли 0,05 г (8 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 1000 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 1 часа; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 10 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 30 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 6 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:7), получали 0,03 г целевого соединения 4 в виде твердого вещества белого цвета, выход: 52%. Структура целевого соединения 4 представляет собой:

1H ЯМР (600 МГц, cdcl3) δ 5,90 (t, 1H), 5,70 (dd, 1H), 5,11(t, 1H), 4,87 (m, 2H), 4,55 (m, 1H), 4,4l(m, 1H), 4,29 (m, 1H), 4,19 (m, 1H), 3,62 (d, 2H), 3,05 (m, 1H), 2,55 (m, 2H), 2,283 (m, 1H), 2,062 (m, 2H), 2,00 (ddd, 18H), 1,92 (m, 2H), 1,85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 5
0,05 г (0,6 ммоль) соединения 3, полученного в примере 3, смешивали с 0,012 г (0,7 ммоль) гидрохлорида метилового сложного эфира L-валина и 2 мл этилацетата, добавляли 0,05 г (8 ммоль) борогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 800 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 3 часов; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 100 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 50 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 10 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:6), получали 0,03 г целевого соединения 5 в виде твердого вещества белого цвета, выход: 60%. Структура целевого соединения 5 представляет собой:
1H ЯМР (600 МГц, cdcl3) δ 6,98 (t, 1H), 5,22 (m, 3Н), 4,86 (m, 1H), 4,69 (dd, 1H), 4,50 (m, 1H), 4,42 (m, 2H), 4,27 (m, 1H), 4,01 (m, 1H), 3,66 (s, 3H), 2,67 (m, 1H), 2,58 (m, 2H), 2,33 (m, 1H), 2,283 (m, 1H), 2,062 (m, 2H), 2,00 (ddd, 18H), 1,92 (m, 2H), 1,85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,98 (s, 3H), 0,94 (s, 3H), 0,96 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 6
0,05 г (0,6 ммоль) соединения 3, полученного в примере 3, смешивали с 0,012 г (0,7 ммоль) гидрохлорида метилового сложного эфира L-треонина и 2 мл хлороформа, добавляли 0,05 г (8 ммоль) триацетилборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 900 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 6 часов; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 90 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 10 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 1 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:1), получали 0,02 г целевого соединения 6 в виде твердого вещества белого цвета, выход: 40%. Структура целевого соединения 6 представляет собой:

1Н ЯМР (600 МГц, cdcl3) δ 5,66 (t, 1H), 5,37 (m, 2H), 5,03 (m, 2H), 4,42 (s,2H), 4,28 (m, 1H), 4,09 (m, 1H), 3,77 (m, 1H), 3,66 (s, 3H), 2,74 (m, 1H), 2,57 (m, 2H), 2,283 (m, 1H), 2,062 (m, 2H), 12,00 (ddd, 18H), 1,92 (m, 2H), 1,85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,18 (s, 3H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 7
0,05 г (0,6 ммоль) соединения 3, полученного в примере 3, смешивали с 0,011 г (0,7 ммоль) гидрохлорида метилового сложного эфира L-серина и 2 мл дихлорметана, добавляли 0,05 г (8 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 1000 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 9 часов; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 20 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствор (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 90 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 2 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:2), получали 0,02 г целевого соединения 7 в виде твердого вещества белого цвета, выход: 40%. Структура целевого соединения 7 представляет собой:

1H ЯМР (600 МГц, cdcl3) δ 6,26 (t, 1H), 5,37 (t, 1H), 5,29 (m, 1H), 4,95 (m, 2H), 4,45 (m, 1H), 4,28 (m, 2H), 4,15 (m, 2H), 3,77 (m, 1H), 3,66 (s, 3H), 3,25 (t, 1H), 2,57 (m, 2H), 2,283 (m, 1H), 2,062 (m, 2H), 2,00 (ddd, 18H), 1,92 (m, 2H), 1,85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 8
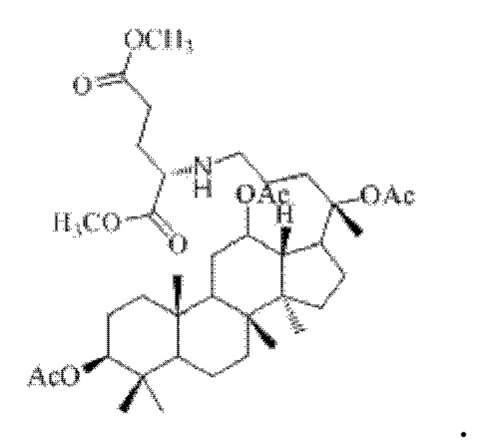
0,05 г (0,6 ммоль) соединения 3, полученного в примере 3, смешивали с 0,017 г (0,7 ммоль) гидрохлорида диметилового сложного эфира L-глутаминовой кислоты и 2 мл тетрахлорметана, добавляли 0,05 г (8 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 1100 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 1 часа; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 30 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 80 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 3 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:3), получали 0,03 г целевого соединения 8 в виде твердого вещества белого цвета, выход: 55%. Структура целевого соединения 8 представляет собой:
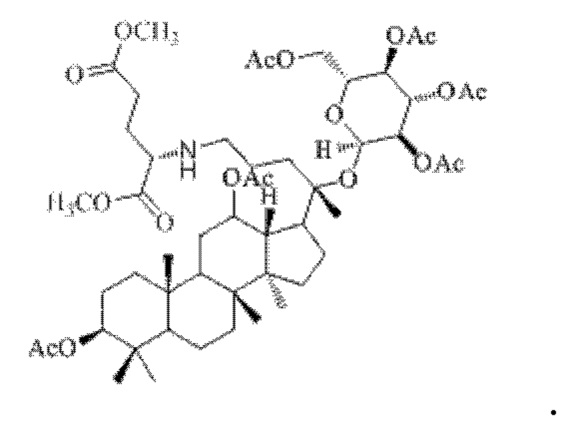
1H ЯМР (600 МГц, cdcl3) δ 6,68 (t, 1H), 5,63 (t, 1H), 5,06 (m, 1H), 4,95 (t, 2H), 4,82 (m, 1H), 4,55 (m, 2H), 4,26 (t, 1H), 3,99 (m, 1H), 3,66 (s, 3H), 3,60 (s, 3H), 3,55 (t, 1H), 2,57 (m, 2H), 2,35 (m, 2H), 2,28 (m, 1H), 2,22 (m, 1H), 2,09 (m, 1H), 2,062 (m, 2H), 2,00 (ddd, 18H), 1,92 (m, 2H), 1,85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 9
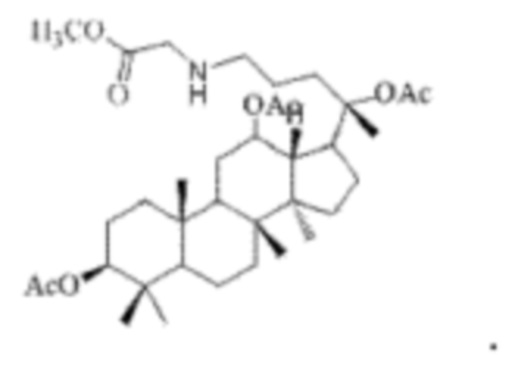
0,05 г (0,6 ммоль) соединения 3, полученного в примере 3, смешивали с 0,009 г (0,7 ммоль) гидрохлоридом метилового сложного эфира глицина и 2 мл метанола, добавляли 0,05 г (8 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 1200 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 1 часа; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 40 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 70 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 4 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:4), получали 0,03 г целевого соединения 9 в виде твердого вещества белого цвета, выход: 60%. Структура целевого соединения 9 представляет собой:

1H ЯМР (600 МГц, cdcl3) δ 6,66 (t, 1H), 4,95-4,82 (m, 5Н), 4,55 (m, 1H), 4,44 (m, 1H), 4,28 (t, 1H), 4,01 (m, 1H), 3,72 (s, 3H), 3,62 (d, 2H), 2,57 (m, 2H), 2,44 (m, 1H), 2,283 (m, 1H), 2,062 (m, 2H), 2,00 (ddd, 18H), 1,92 (m, 2H), 1.85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1.21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 10
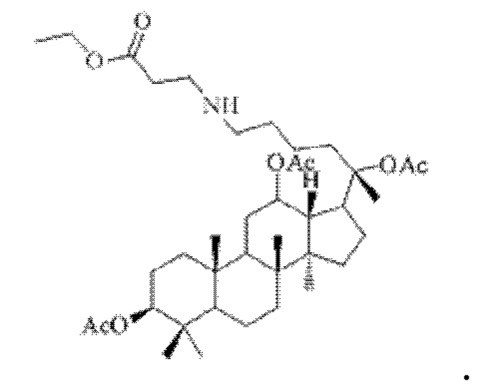
0,05 г (0,6 ммоль) соединения 3, полученного в примере 3, смешивали с 0,011 г (0,7 ммоль) гидрохлорида этилового сложного эфира β-аланина и 2 мл метанола, добавляли 0,05 г (8 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 1200 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 1 часа; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 50 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 60 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 5 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:5), получали 0,03 г целевого соединения 10 в виде твердого вещества белого цвета, выход: 60%. Структура целевого соединения 10 представляет собой:
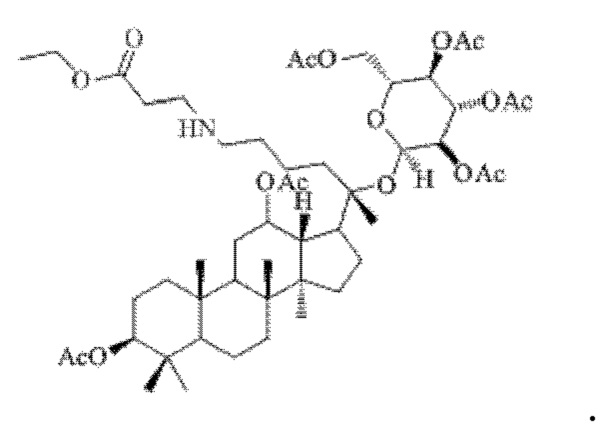
1H ЯМР (600 МГц, cdcl3) δ 5,16 (t, 1H), 4,97 (t, Гц, 1H), 4,89 (dd, 1H), 4,66 (d, 1H), 4,10-4,06 (m, 1H), 4,04 (dd, J=12,3, 5,3 Гц, 1H), 3,60 (ddd, 1H), 2,70 (ddd, 1H), 2,37-2,29 (m, 1H), 2,283 (m, 1H), 2,14-2,08 (m, 2Н), 2,062 (m, 2Н), 2,00 (ddd, 18Н), 1,92 (m, 2Н), 1,85 (m, 2Н), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2Н), 1,49 (m, 2Н), 1,39 (m,1Н), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2Н), 1,11 (s, 3H), 1,09 (m, 1H), 1,07 (m, 3H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3Н).
Пример 11
0,05 г (0,6 ммоль) соединения 3, полученного в примере 3, смешивали с 0,015 г (0,7 ммоль) гидрохлорида диэтиламиномалоната и 2 мл метанола, добавляли 0,05 г (8 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 1100 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 1 часа; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 40 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 70 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 4 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:4), получали 0,03 г целевого соединения 11 в виде твердого вещества белого цвета, выход: 50%. Структура целевого соединения 11 представляет собой:
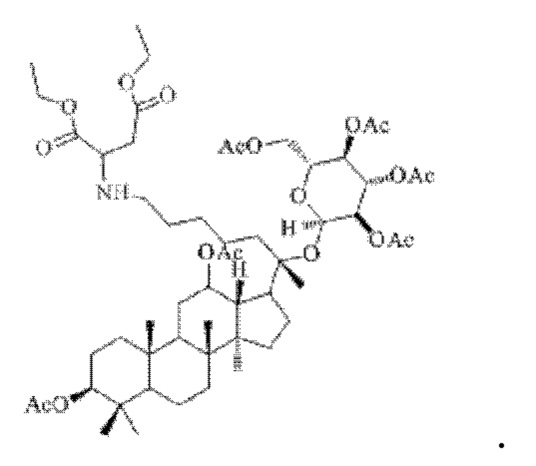
1H ЯМР (600 МГц, cdcl3) δ 5,16 (t, 1H), 4,97 (t, 1H), 4,89 (dd, 8,0 Гц, 1H), 4,78 (ddd, 1H), 4,66 (d, 1H), 4,46 (d, 1H), 4,11 (m, 5,3 Гц, 4H), 4,09 (m, 2H), 3,60 (ddd, 1H), 2,77 (ddd, 1H), 2,540 (m, 1H), 2,283 (m, 1H), 2,062 (m, 2H), 2,00 (m, 18H), 1,82 (dd, 1H), 1,81 (m, 1H), 1,74 (t, 1H), 1,67 (s, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,55 (m, 2H), 1,49 (m, 2H), 1,39 (m, 2H), 1,37 (m, 1H), 1,33 (s, 3H), 1,25 (m, 2H), 1,220 (m, 1H), 1,07 (m, 2H), 1,01 (s, 3H), 0,96 (s, 3H), 0,92 (s, 3H), 0,91 (s, 3H), 0,77 (s, 3H).
Пример 12
0,05 г (0,6 ммоль) соединения 3, полученного в примере 3, смешивали с 0,01 г (0,7 ммоль) гидрохлорида метилового сложного эфира L-фенилаланина и 2 мл метанола, добавляли 0,05 г (8 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 1100 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 1 часа; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 30 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 80 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 3 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:3), получали 0,03 г целевого соединения 12 в виде твердого вещества белого цвета, выход: 50%. Структура целевого соединения 12 представляет собой:
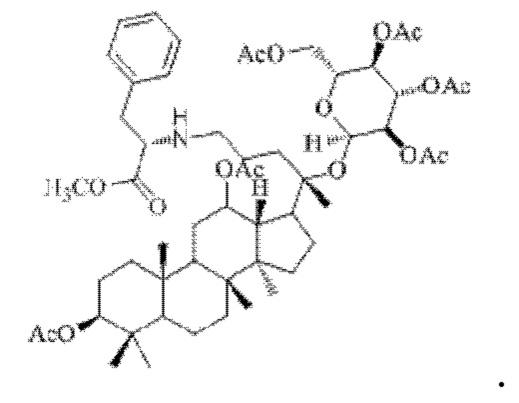
1H ЯМР (600 МГц, cdcl3) δ 7,31 (t, 1H), 7,11 (m, 5Н), 5,32 (m, 1H), 4,96 (m, 1H), 4,79 (m,2H), 4,61 (t, 1H), 4,42 (m, 1H), 4,31 (m, 1H), 4,16 (dd, 1H), 4,06 (t, 1H), 3,70 (s, 3H), 3,32 (m, 1H), 2,94 (m, 2H), 2,55 (m, 2H), 2,283 (m, 1H), 2,062 (m, 2H), 2,00 (ddd, 18H), 1,92 (m, 2H), 1,85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 13
0,05 г (0,6 ммоль) соединения 3, полученного в примере 3, смешивали с 0,014 г (0,7 ммоль) метилового сложного эфира L-тирозина и 2 мл метанола, добавляли 0,05 г (8 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 1100 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 1 часа; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 20 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 90 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 2 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:2), получали 0,02 г целевого соединения 13 в виде твердого вещества белого цвета, выход: 35%. Структура целевого соединения 13 представляет собой:
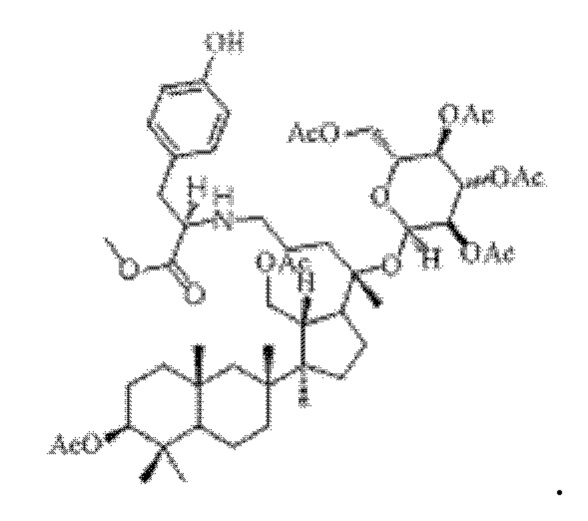
1H ЯМР (600 МГц, cdcl3) δ 7,01 (d, 2Н), 6,75 (d, 2Н), 6,04 (m, 1H), 5,49 (dd, 1H), 5,18 (m, 2Н), 4,56 (m, 1H), 4,50 (s, 2Н), 4,37 (m, 1H), 4,28 (m, 1H), 4,18 (m, 1H), 3,63 (m, 3H), 3,25 (s, 1H), 2,97 (m, 1H), 2,57 (t, 2H), 2,28 (m, 1H), 2,06 (m, 2H), 2,00 (ddd, 18H), 1,92 (m, 2H), 1,85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 14
0,05 г (0,6 ммоль) соединения 3, полученного в примере 3, смешивали с 0,017 г (0,7 ммоль) гидрохлорида метилового сложного эфира L-гистидина и 2 мл метанола, добавляли 0,05 г (8 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 900 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 1 часа; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 10 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 100 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 1 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:1), получали 0,03 г целевого соединения 14 в виде твердого вещества белого цвета, выход: 51%. Структура целевого соединения 14 представляет собой:
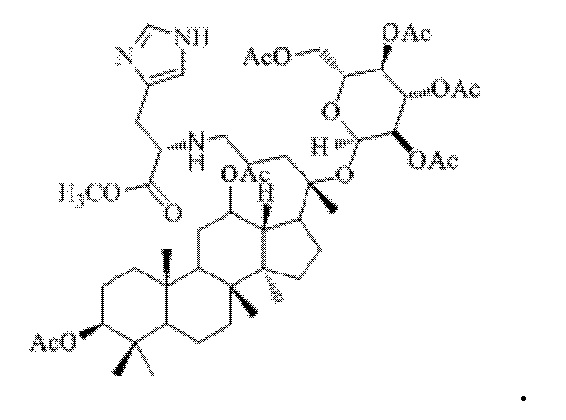
1H ЯМР (600 МГц, cdcl3) δ 8,67 (d, 1H), 7,63 (d, 1H), 6,02(t, 1H), 5,90 (dd, 1H), 4,99 (m, 1H), 4,75 (t, 1H), 4,55 (m, 1H), 4,42 (m, 2H), 4,26 (m, 1H), 4,16 (m, 1H), 3,66 (s, 3H), 3,48 (t, 1H), 3,14 (m, 1H), 2,77 (m, 1H), 2,55 (m, 2H), 2,283 (m, 1H), 2,062 (m, 2H), 2,00 (ddd, 18H), 1,92 (m, 2H), 1,85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 15
0,05 г (0,6 ммоль) соединения 3, полученного в примере 3 смешивали с 0,015 г (0,7 ммоль) гидрохлорида метилового сложного эфира L-триптофана и 2 мл метанола, добавляли 0,05 г (8 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 800 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 1 часа; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 50 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 50 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 5 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:5), получали 0,02 г целевого соединения 15 в виде твердого вещества белого цвета, выход: 33%. Структура целевого соединения 15 представляет собой:
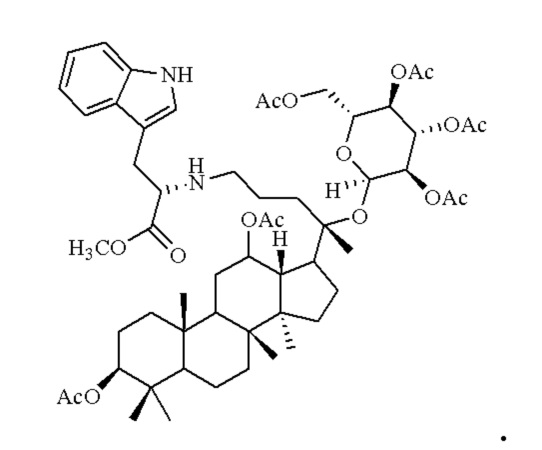
1Н ЯМР (600 МГц, cdcl3) δ 7,26 (dd, 1H), 6,85 (ddd, 1H), 6,76 (ddd, 1H), 6,67 (ddd, 1H), 6,26 (t, 1H), 5,15 (t, 1H), 4,92 (m, 1H), 4,55(dd, 1H), 4,44 (m, 1H), 4,28 (t, 1H), 4,11 (m, 2H), 3,68 (s, 3H), 3,52 (m, 2H), 3,45 (m, 1H), 3,37 (m, 1H), 2,54 (m, 2H), 2,48 (m, 1H), 2,20 (m, 1H), 2,283 (m, 1H), 2,062 (m, 2H), 2,00 (ddd, 18H), 1,92 (m, 2H), 1,85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 16
1 г (21 ммоль) соединения, имеющего структуру, представленную в формуле V, смешивали с 10 мл уксусного ангидрида и 10 мл пиридина, и реакцию нуклеофильного замещения осуществляли в условиях перемешивания с магнитной мешалкой при 1000 об/мин и 60°С в течение 3 часов; материал, полученный после указанной реакции нуклеофильного замещения, подвергали перегонке при пониженном давлении, полученный неочищенный продукт смешивали с 5 мл этилацетата, и указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:5), получали 1,1 г соединения 16 в виде твердого вещества белого цвета, выход: 90%. Структура соединения 16 представляет собой:
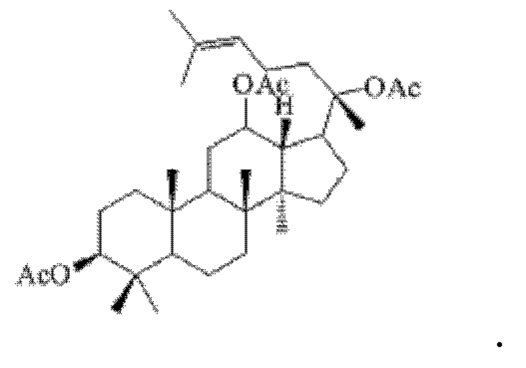
Пример 17
Смешивали 1 г (17 ммоль) соединения 16, полученного в примере 16, 5 мл дихлорметана и 0,31 г (11 ммоль) мета-хлорпербензойной кислоты, и первичную реакцию окисления осуществляли при комнатной температуре в течение 1 часа; добавляли 40 мл воды к материалу, полученному после указанной первичной реакции окисления, проводили магнитное перемешивание при 1000 об/мин в течение 2 часов, осаждали твердое белое вещество и фильтровали с получением прозрачного раствора, указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 5 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:5), получали 0,92 г соединения 17 в виде твердого вещества белого цвета, выход: 90%. Структура соединения 17 представляет собой:
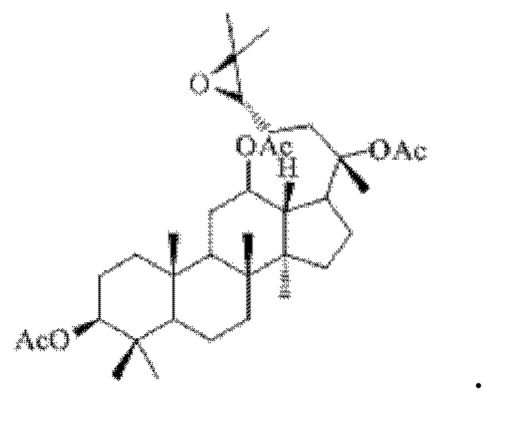
Пример 18
Смешивали 0,9 г (15 ммоль) соединения 17, полученного в примере 17, 5 мл тетрагидрофурана и 0,83 г (32 ммоль) йодной кислоты, и вторичную реакцию окисления осуществляли при комнатной температуре в течение 2 часов; материал, полученный после указанной вторичной реакции окисления, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 40 мл), затем четыре раза последовательно промывали каждым из насыщенного раствора бикарбоната натрия и насыщенного раствора хлорида натрия с получением прозрачного раствора (объем насыщенного раствора бикарбоната натрия, необходимого для каждой промывки, составлял 50 мл, объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 50 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 5 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:5), получали 0,71 г соединения 18 в виде твердого вещества белого цвета, выход: 85%. Структура соединения 18 представляет собой:
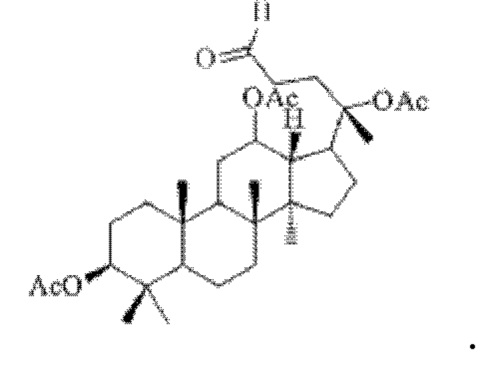
Пример 19
0,033 г (0,6 ммоль) соединения 18, полученного в примере 18, смешивали с 0,013 г (0,7 ммоль) гидрохлорида метилового сложного эфира L-лейцина и 2 мл метанола, добавляли 0,033 г (5,3 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 800 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 1 часа; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом в течение 3 раз (объем необходимого для каждой экстракции этилацетата составлял 10 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 100 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 10 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:10), получали 0,03 г целевого соединения 19 в виде твердого вещества белого цвета, выход: 72%. Структура целевого соединения 19 представляет собой:

1H ЯМР (600 МГц, cdcl3) δ 4,95 (m, 1H), 4,25 (t, 1H), 3,65 (s, 3H), 2,88 (m, 1H), 2,56 (t, 2Н), 2,00 (ddd, 9Н), 1,92 (m, 2Н), 1,85 (m, 2Н), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2Н), 1,49 (m, 2Н), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 20
0,033 г (0,6 ммоль) соединения 18, полученного в примере 18, смешивали с 0,012 г (0,7 ммоль) гидрохлорида метилового сложного эфира L-валина и 2 мл этилацетата, добавляли 0,033 г (5,3 ммоль) борогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 1000 об/мин, и реакция восстановительного аминирования осуществляли при комнатной температуре в течение 3 часов; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 20 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 90 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 9 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:9), получали 0,03 г целевого соединения 20 в виде твердого вещества белого цвета, выход: 75%. Структура целевого соединения 20 представляет собой:
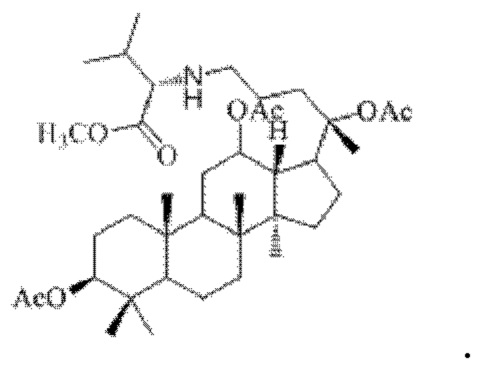
1H ЯМР (600 МГц, cdcl3) δ 4,95 (m, 1H), 4,25 (t, 1H), 3,65 (s, 3H), 2,88 (m, 1H), 2,56 (t, 2H), 2,00 (ddd, 9H), 1,92 (m, 2H), 1,85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,98 (s, 3H), 0,94 (s, 3H), 0,96 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 21
0,033 г (0,6 ммоль) соединения 18, полученного в примере 18, смешивали с 0,012 г (0,7 ммоль) гидрохлорида метилового сложного эфира L-треонина и 2 мл хлороформа, добавляли 0,033 г (5,3 ммоль) триацетоксиборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 1100 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 6 часов; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 30 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 80 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 8 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:8), получали 0,04 г целевого соединения 21 в виде твердого вещества белого цвета, выход: 50%. Структура целевого соединения 21 представляет собой:
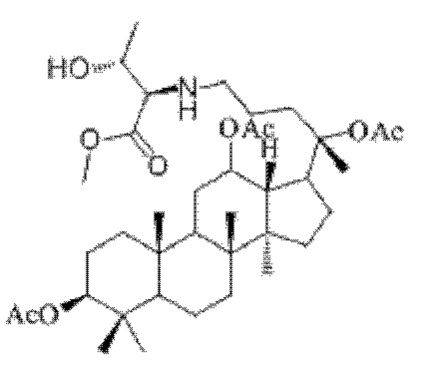
1H ЯМР (600 МГц, cdcl3) δ 4,95 (m, 1H), 4,25 (t, 1H), 3,65 (s, 3H), 2,88 (m, 1H), 2,56 (t, 2Н), 2,00 (ddd, 9Н), 1,92 (m, 2Н), 1,85 (m, 2Н), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2Н), 1,49 (m, 2Н), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2Н), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3Н).
Пример 22
0,033 г (0,6 ммоль) соединения 18, полученного в примере 18, смешивали с 0,011 г (0,7 ммоль) гидрохлорида метилового сложного эфира L-серина и 2 мл дихлорметана, добавляли 0,033 г (5,3 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 1200 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 9 часов; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 40 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 70 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 7 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:7), получали 0,028 г целевого соединения 22 в виде твердого вещества белого цвета, выход: 73%. Структура целевого соединения 22 представляет собой:
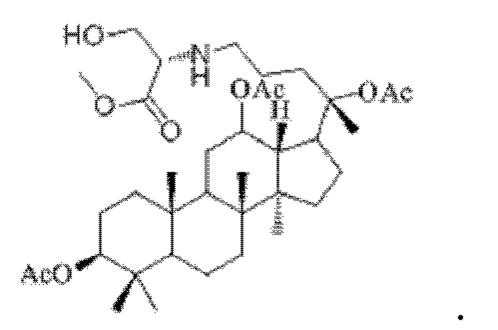
1H ЯМР (600 МГц, cdcl3) δ 4,95 (m, 1H), 4,25 (t, 1H), 3,65 (s, 3H), 2,88 (m, 1H), 2,56 (t, 2H), 2,00 (ddd, 9H), 1,92 (m, 2H), 1,85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 23
0,033 г (0,6 ммоль) соединения 18, полученного в примере 18, смешивали с 0,017 г (0,7 ммоль) гидрохлорида диметилового сложного эфира L-глутаминовой кислоты и 2 мл тетрахлорметана, добавляли 0,033 г (5,3 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 1200 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 1 часа; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 50 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 60 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 6 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:6), получали 0,028 г целевого соединения 23 в виде твердого вещества белого цвета, выход: 67%. Структура целевого соединения 23 представляет собой:
1H ЯМР (600 МГц, cdcl3) δ 4,95 (m, 1H), 4,25 (t, 1H), 3,65 (s, 3H), 2,88 (m, 1H), 2,56 (t, 2H), 2,00 (ddd, 9H), 1,92 (m, 2H), 1,85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 24
0,033 г (0,6 ммоль) соединения 18, полученного в примере 18, смешивали с 0,009 г (0,7 ммоль) гидрохлорида метилового сложного эфира глицина и 2 мл метанола, добавляли 0,033 г (5,3 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 1100 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 1 часа; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 60 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 50 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 5 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:5), получали 0,026 г целевого соединения 24 в виде твердого вещества белого цвета, выход: 70%. Структура целевого соединения 24 представляет собой:
1H ЯМР (600 МГц, cdcl3) δ 4,95 (m, 1H), 4,25 (t, 1H), 3,65 (s, 3H), 2,88 (m, 1H), 2,56 (t, 2H), 2,00 (ddd, 9H), 1,92 (m, 2H), 1,85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 25
0,033 г (0,6 ммоль) соединения 18, полученного в примере 18, смешивали с 0,011 г (0,7 ммоль) гидрохлорида этилового сложного эфира β-аланина и 2 мл метанола, добавляли 0,033 г (5,3 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 1000 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 1 часа; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 70 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 40 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 4 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:4), получали 0,025 г целевого соединения 25 в виде твердого вещества белого цвета, выход: 65%. Структура целевого соединения 25 представляет собой:
1H ЯМР (600 МГц, cdcl3) δ 4,95 (m, 1H), 4,25 (t, 1H), 3,92 (t, 2H), 2,93 (t, 2H), 2,56 (t, 2H), 2,46 (t, 2H), 2,00 (ddd, 9H), 1,92 (m, 2H), 1,85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 26
0,033 г (0,6 ммоль) соединения 18, полученного в примере 18, смешивали с 0,015 г (0,7 ммоль) гидрохлорида диэтилового аминомалоната и 2 мл метанола, добавляли 0,033 г (5,3 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 900 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 1 часа; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 80 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 30 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 3 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:3), получали 0,026 г целевого соединения 26 в виде твердого вещества белого цвета, выход: 60%. Структура целевого соединения 26 представляет собой:

1H ЯМР (600 МГц, cdcl3) δ 4,95 (m, 1H), 4,25 (t, 1H), 2,88 (m, 1H), 2,56 (t, 2Н), 2,00 (ddd, 9Н), 1,92 (m, 2Н), 1,85 (m, 2Н), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2Н), 1,49 (m, 2Н), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,07 (m, 3H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 27
0,033 г (0,6 ммоль) соединения 18, полученного в примере 18, смешивали с 0,01 г (0,7 ммоль) гидрохлорида метилового сложного эфира L-фенилаланина и 2 мл метанола, добавляли 0,033 г (5,3 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 800 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 1 часа; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 90 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 20 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 2 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:2), получали 0,029 г целевого соединения 27 в виде твердого вещества белого цвета, выход: 70%. Структура целевого соединения 27 представляет собой:

1H ЯМР (600 МГц, cdcl3) δ 7,31 (t, 1H), 7,11 (m, 5Н), 5 4,95 (m, 1H), 4,25 (t, 1H), 2,88 (m, 1H), 2,56 (t, 2H), 2,00 (ddd, 9H), 1,92 (m, 2H), 1,85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 28
0,033 г (0,6 ммоль) соединения 18, полученного в примере 18, смешивали с 0,014 г (0,7 ммоль) метилового сложного эфира L-тирозина и 2 мл метанола, добавляли 0,033 г (5,3 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 1000 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 1 часа; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 100 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 10 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 1 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:1), получали 0,024 г целевого соединения 28 в виде твердого вещества белого цвета, выход: 55%. Структура целевого соединения 28 представляет собой:
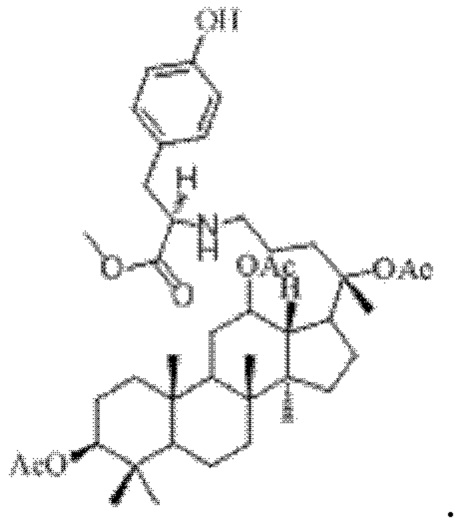
1H ЯМР (600 МГц, cdcl3) δ 7,01 (d, 2Н), 6,75 (d, 2Н), 5 4,95 (m, 1H), 4,25 (t, 1H), 3,66 (s, 3H), 3,50 (t, 1H), 3,25 (m, 1H), 3,05 (m, 1H), 2,56 (t, 2H), 2,00 (ddd, 9H), 1,92 (m, 2H), 1,85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 29
0,033 г (0,6 ммоль) соединения 18, полученного в примере 18, смешивали с 0,017 г (0,7 ммоль) гидрохлорида метилового сложного эфира L-гистидина и 2 мл метанола, добавляли 0,033 г (5,3 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 1000 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 1 часа; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 50 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 50 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 5 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:5), получали 0,025 г целевого соединения 29 в виде твердого вещества белого цвета, выход: 50%. Структура целевого соединения 29 представляет собой:
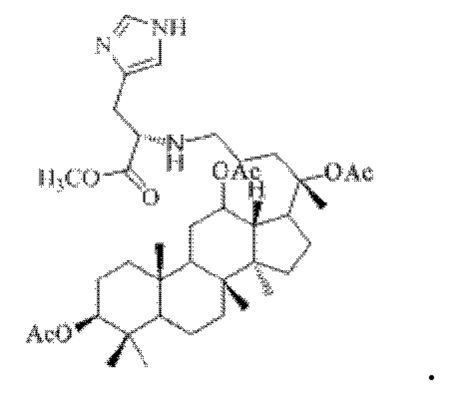
1H ЯМР (600 МГц, cdcl3) δ 8,73 (d, 1H), 7,65 (d, 1H), 4,95 (m, 1H), 4,25 (t, 1H), 3,45 (t, 2H), 3,98 (dd, 1H), 2,82 (dd, 1H), 2,56 (t, 2H), 2,00 (ddd, 9H), 1,92 (m, 2H), 1,85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 30
0,033 г (0,6 ммоль) соединения 18, полученного в примере 18, смешивали с 0,015 г (0,7 ммоль) гидрохлорида метилового сложного эфира L-триптофана и 2 мл метанола, добавляли 0,033 г (5,3 ммоль) цианоборогидрида натрия в условиях ледяной бани и перемешивания с магнитной мешалкой при 1000 об/мин, и реакцию восстановительного аминирования осуществляли при комнатной температуре в течение 1 часа; материал, полученный после указанной реакции восстановительного аминирования, три раза подвергали экстракции этилацетатом (объем необходимого для каждой экстракции этилацетата составлял 50 мл), затем четыре раза промывали насыщенным раствором хлорида натрия с получением прозрачного раствора (объем насыщенного раствора хлорида натрия, необходимого для каждой промывки, составлял 50 мл), указанный прозрачный раствор подвергали перегонке при пониженном давлении для удаления растворителя, полученный неочищенный продукт смешивали с 5 мл этилацетата, указанную смесь применяли для проведения колоночной хроматографии после смешивания с силикагелем. В качестве элюента применяли смесь этилацетата и н-гексана (объемное отношение этилацетата к н-гексану составляло 1:5), получали 0,02 г целевого соединения 30 в виде твердого вещества белого цвета, выход: 45%. Структура целевого соединения 30 представляет собой:

1H ЯМР (600 МГц, cdcl3) δ 7,26 (dd, 1H), 6,85 (ddd, 1H), 6,76 (ddd, 1H), 6,67 (ddd, 1H), 5 4,95 (t, 1H), 4,25 (m, 1H), 2,88 (m, 1H), 2,56 (t, 2H), 2,00 (ddd, 9H), 1,92 (m, 2H), 1,85 (m, 2H), 1,82 (m, 1H), 1,81 (m, 1H), 1,74 (m, 1H), 1,67 (m, 3H), 1,65 (m, 3H), 1,63 (m, 3H), 1,53 (m, 2H), 1,49 (m, 2H), 1,39 (m, 1H), 1,33 (m, 3H), 1,26 (m, 1H), 1,21 (m, 1H), 1,20 (m, 2H), 1,11 (s, 3H), 1,09 (m, 1H), 1,03 (m, 1H), 0,94 (s, 3H), 0,92 (s, 3H), 0,85 (m, 1H), 0,84 (s, 3H), 0,82 (s, 3H).
Пример 31
Исследовали токсичность соединений, полученных в примерах 1-30 согласно настоящему изобретению, и конкретные стадии были следующими:
186 мышей Kunming (18-22 г, самцы, были приобретены в Центре экспериментальных животных третьего военного медицинского университета) были случайным образом и в среднем разделены на 31 группу, и указанным мышам Kunming вводили соединения, полученные в примерах 1-30 согласно настоящему изобретению (внутриже луд очное введение 500 мг/кг однократно), а для мышей контрольной группы применяли солевой раствор (1 мл). За указанными мышами непрерывно наблюдали в течение 14 дней.
Результаты: среди мышей Kunming в каждой экспериментальной группе не было мертвых особей, и мыши находились в хорошем состоянии, они нормально принимали пищу, имели нормальный окрас шерсти, и значимых отличий в массах тела между мышами в экспериментальной группе и контрольной группе с солевым раствором не наблюдали.
Для дополнительной проверки токсичности соединений, полученных в примерах 1-30 согласно настоящему изобретению, культивировали линию клеток макрофагов мыши RAW264.7, и соединения, полученные в примерах 1-30 согласно настоящему изобретению, получали в концентрации 10 мкмоль/л с применением среды для культивирования, причем указанным растворителем был физиологический раствор, со-растворителем был диметилсульфоксид (ДМСО, конечная концентрация составляла 0,1%), и была представлена контрольная группа с 0,1% ДМСО в солевом растворе. Уровень выживаемости клеток определяли посредством способа CCK8. Данные были представлены в виде  . Однофакторный дисперсионный анализ между группами проводили при помощи пакета SPSS версии 10.0. Разница была достоверной в случае, когда р<0,05. Результат представлен в таблице 1. Как можно увидеть из таблицы 1, Соединения 1, 2 и 3 обладают значимой цитотоксичностью, и другие соединения при этой концентрации значимой токсичностью не обладают.
. Однофакторный дисперсионный анализ между группами проводили при помощи пакета SPSS версии 10.0. Разница была достоверной в случае, когда р<0,05. Результат представлен в таблице 1. Как можно увидеть из таблицы 1, Соединения 1, 2 и 3 обладают значимой цитотоксичностью, и другие соединения при этой концентрации значимой токсичностью не обладают.
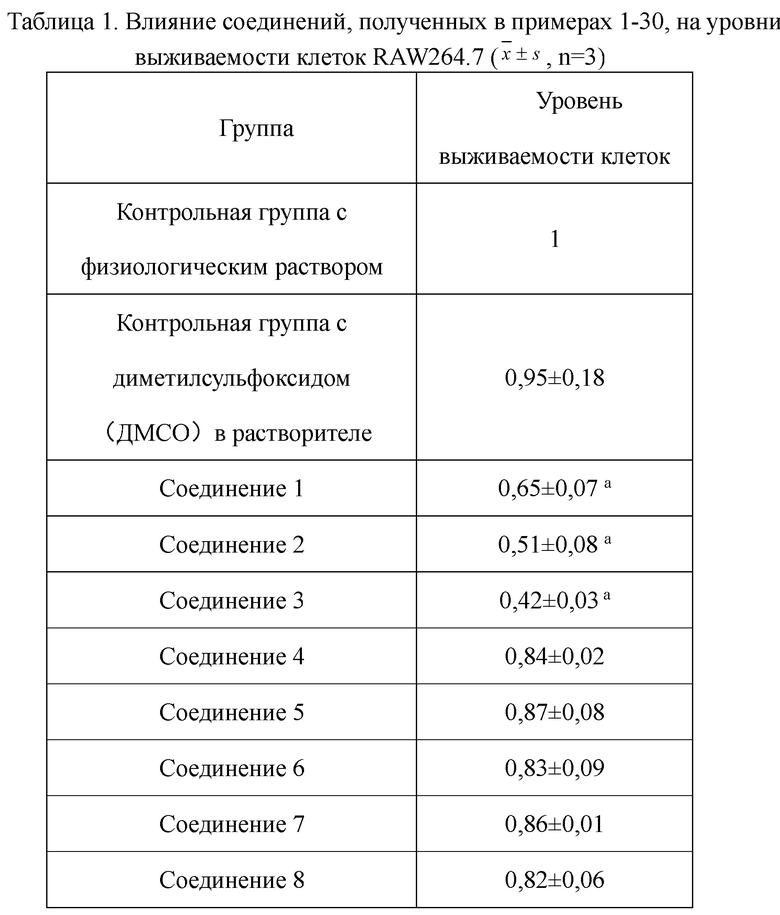


Пример 32
Исследовали влияние соединений, полученных в примерах 1-30 согласно настоящему изобретению, на процентное отношение площади атеросклеротических бляшек к площади всей артерии у мышей ароЕ-/-, получавших рацион с высоким содержанием жиров, и на уровни холестерина лпопротеинов низкой плотности и уровни холестерина липопротеинов высокой плотности, при этом конкретные стадии были следующими:
1. Животные: 160 здоровых и чистых мышей ароЕ-/- в возрасте 10 недель (были приобретены в Центре экспериментальных животных Пекинского университета), самцы, масса тела: 22-25 г, содержались в отдельных клетках в стерильном ламинарном помещении для кормления и имели свободный доступ к пище. Указанные мыши получали рацион с высоким содержанием жира (обычный корм + 0,15% холестерина + 21% сала) в течение 25 недель, комнатную температуру поддерживали на уровне 24°С, относительная влажность: 50%, продолжительность светового облучения: 7:30-19:30.
2. Группы, способ введения:
после 10 недель кормления животных были сбалансировано и случайным образом разделены на следующие 32 группы (n=5) по массе тела и умерщвлены через 15 недель после введения.
Контрольная группа с физиологическим раствором: физиологический раствор, внутрижелудочное введение.
Группа положительного контроля: симвастатин 10 мг/(кг⋅д), внутрижелудочное введение.
Группы, получавшие соединения, полученные в примерах 1-30 согласно настоящему изобретению (всего 30 групп): внутрижелудочное введение в каждой группе 30 мг/(кг⋅д).
3. Показатели и способы исследования:
 Анализ площади атеросклеротических бляшек и величина ее процентного отношения к общей площади аорты: после умерщвления животных отделяли аорты от их корней до конечных бифуркаций брюшных аорт. После фиксации формалином указанные аорты окрашивали при помощи Судана IV, окрашенные в красный цвет участки представляли собой участки атеросклеротических поражений. Процентное отношение площади области поражения к общей площади аорты рассчитывали с применением программного обеспечения Image Pro Plus 5.0.
Анализ площади атеросклеротических бляшек и величина ее процентного отношения к общей площади аорты: после умерщвления животных отделяли аорты от их корней до конечных бифуркаций брюшных аорт. После фиксации формалином указанные аорты окрашивали при помощи Судана IV, окрашенные в красный цвет участки представляли собой участки атеросклеротических поражений. Процентное отношение площади области поражения к общей площади аорты рассчитывали с применением программного обеспечения Image Pro Plus 5.0.
 Определение уровней общего холестерина (ОХ), холестерина липопротеинов низкой плотности (ХЛПНП) и холестерина липопротеинов высокой плотности (ХЛПВП): для определения применяли автоматический биохимический анализатор ROCHE 7060.
Определение уровней общего холестерина (ОХ), холестерина липопротеинов низкой плотности (ХЛПНП) и холестерина липопротеинов высокой плотности (ХЛПВП): для определения применяли автоматический биохимический анализатор ROCHE 7060.
 Определение уровня ФНО-α: для определения использовали способ с применением жидкофазных чипов Millipore (Mcytomag-70K-3, панель с магнитными микроносителями для определения цитокина/хемокина мыши).
Определение уровня ФНО-α: для определения использовали способ с применением жидкофазных чипов Millipore (Mcytomag-70K-3, панель с магнитными микроносителями для определения цитокина/хемокина мыши).
4. Способ статистического анализа:
Данные представляли в виде , и однофакторный дисперсионный анализ между группами проводили при помощи пакета SPSS версии 10.0, разница была достоверной в случае, когда р<0,05.
5. Результаты:
 Как показано в таблице 2, все соединения, полученные в примерах 1-30 согласно настоящему изобретению, в значительной степени уменьшают процент площади атеросклеротических бляшек у мышей ароЕ-/-, которое достоверно отличается от контрольной группы.
Как показано в таблице 2, все соединения, полученные в примерах 1-30 согласно настоящему изобретению, в значительной степени уменьшают процент площади атеросклеротических бляшек у мышей ароЕ-/-, которое достоверно отличается от контрольной группы.

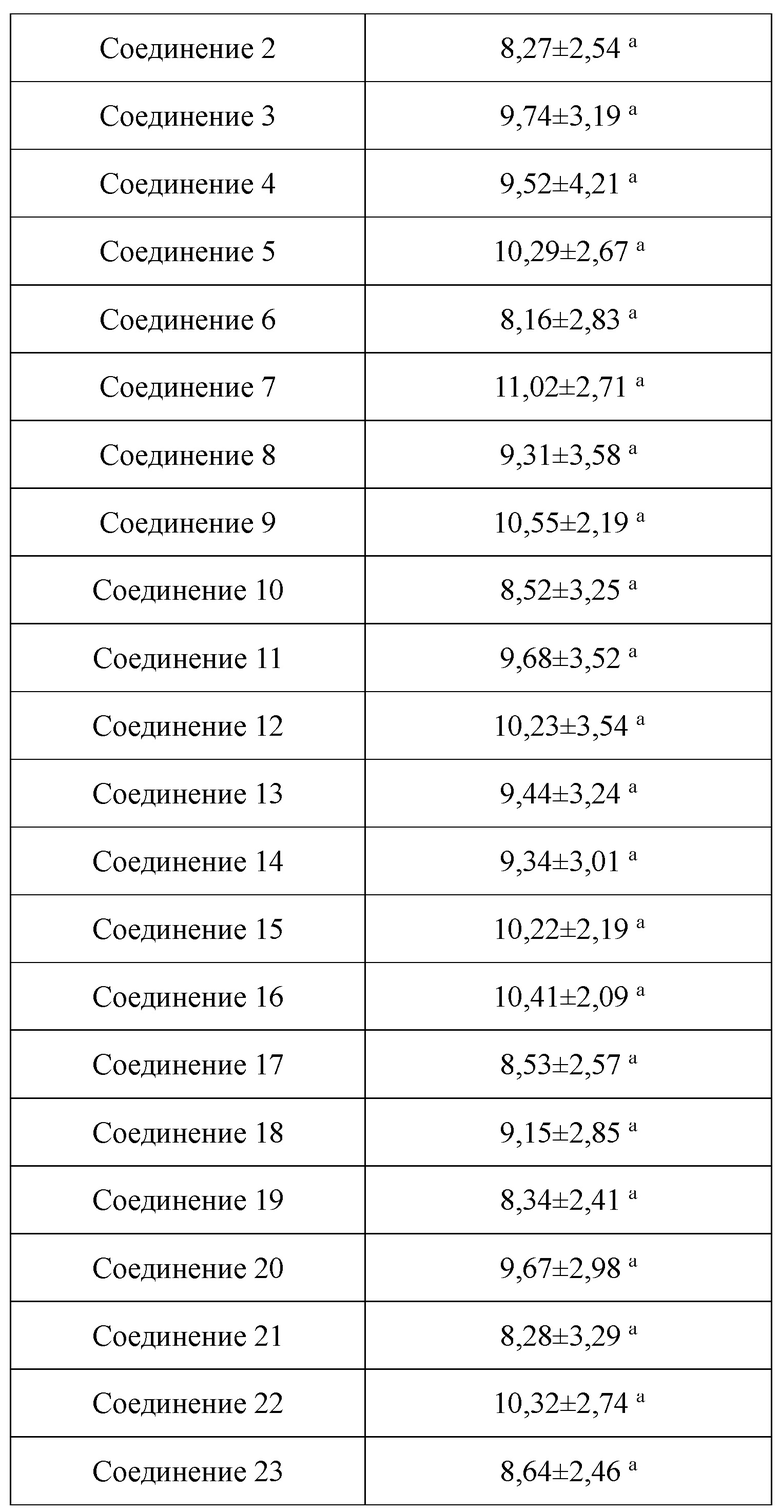

 Как показано в таблице 3, все соединения, полученные в примерах 1-30 согласно настоящему изобретению, могут в значительной степени повышать уровни ХЛПВП, а некоторые соединения могут в значительной степени понижать уровни ХЛПНП в сыворотке мышей, которые достоверно отличаются от уровней в контрольной группе.
Как показано в таблице 3, все соединения, полученные в примерах 1-30 согласно настоящему изобретению, могут в значительной степени повышать уровни ХЛПВП, а некоторые соединения могут в значительной степени понижать уровни ХЛПНП в сыворотке мышей, которые достоверно отличаются от уровней в контрольной группе.
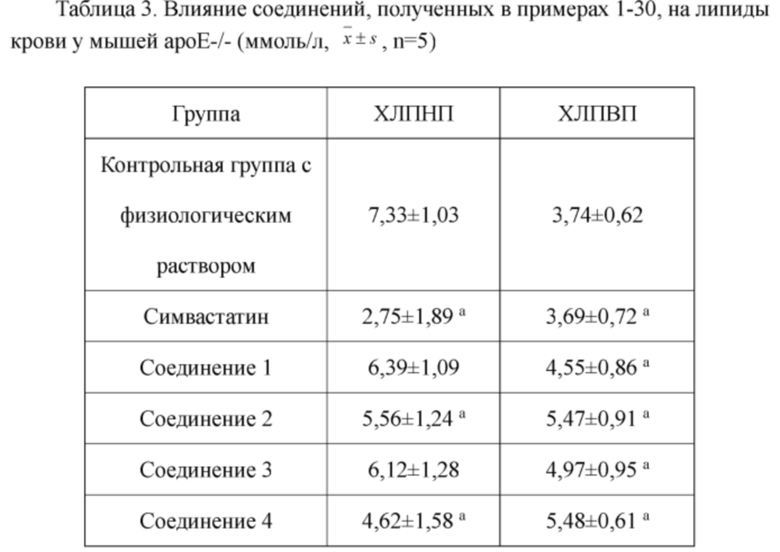
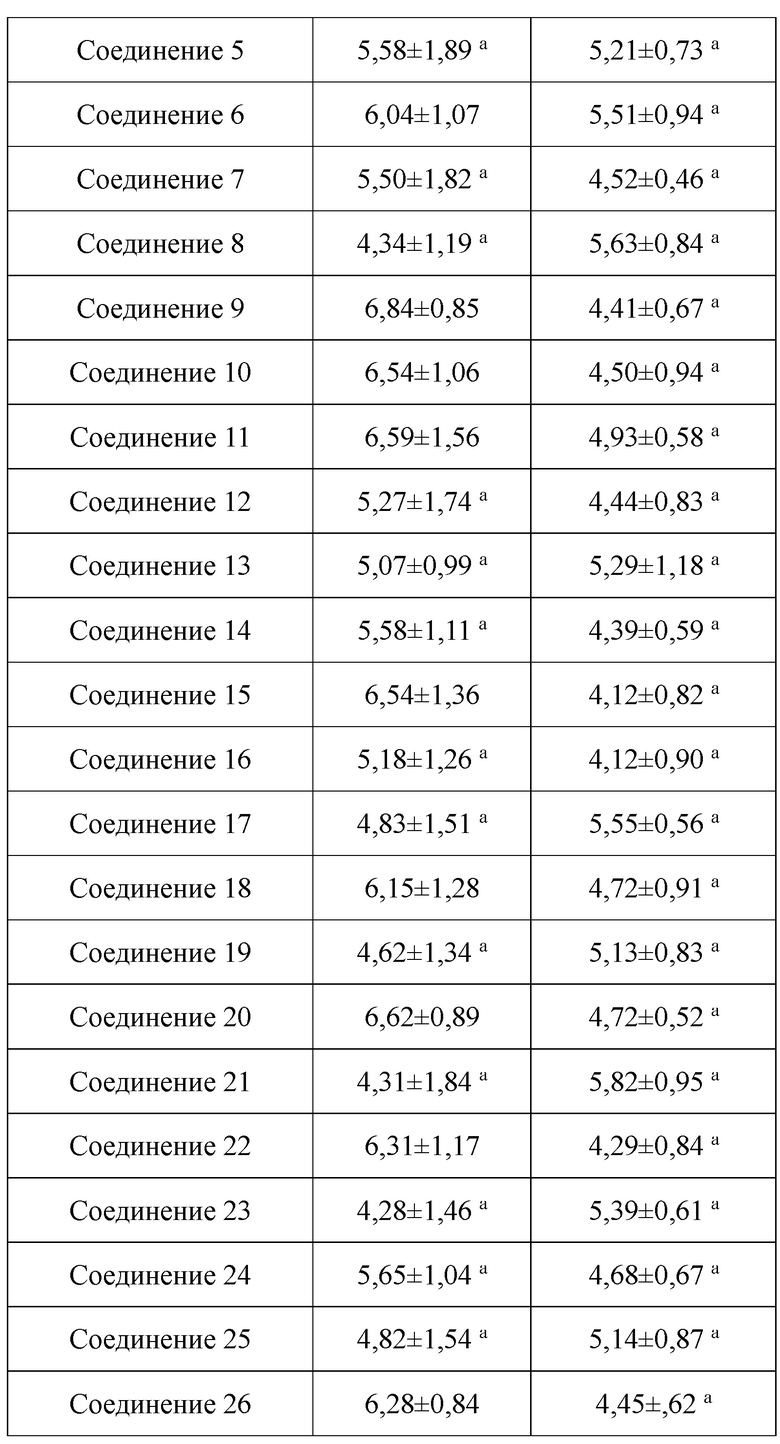
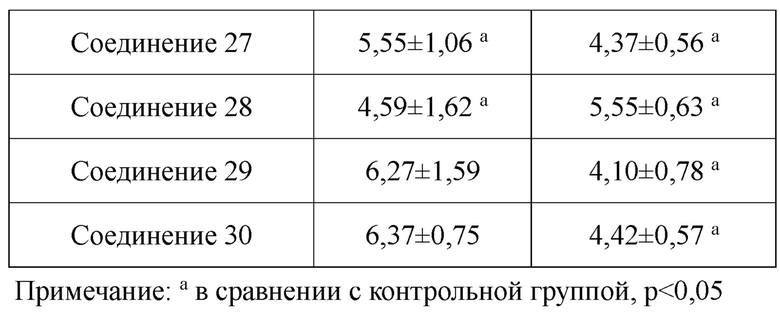
 Как показано в таблице 4, соединения, полученные в примерах 1-30 согласно настоящему изобретению, могут в значительной степени уменьшать локальные уровни ФНО-α в артериях мышей ароЕ-/-, которые достоверно отличаются от уровней в контрольной группе, что свидетельствует о том, что соединения, полученные в примерах 1-30 согласно настоящему изобретению, обладают хорошим противовоспалительным действием.
Как показано в таблице 4, соединения, полученные в примерах 1-30 согласно настоящему изобретению, могут в значительной степени уменьшать локальные уровни ФНО-α в артериях мышей ароЕ-/-, которые достоверно отличаются от уровней в контрольной группе, что свидетельствует о том, что соединения, полученные в примерах 1-30 согласно настоящему изобретению, обладают хорошим противовоспалительным действием.

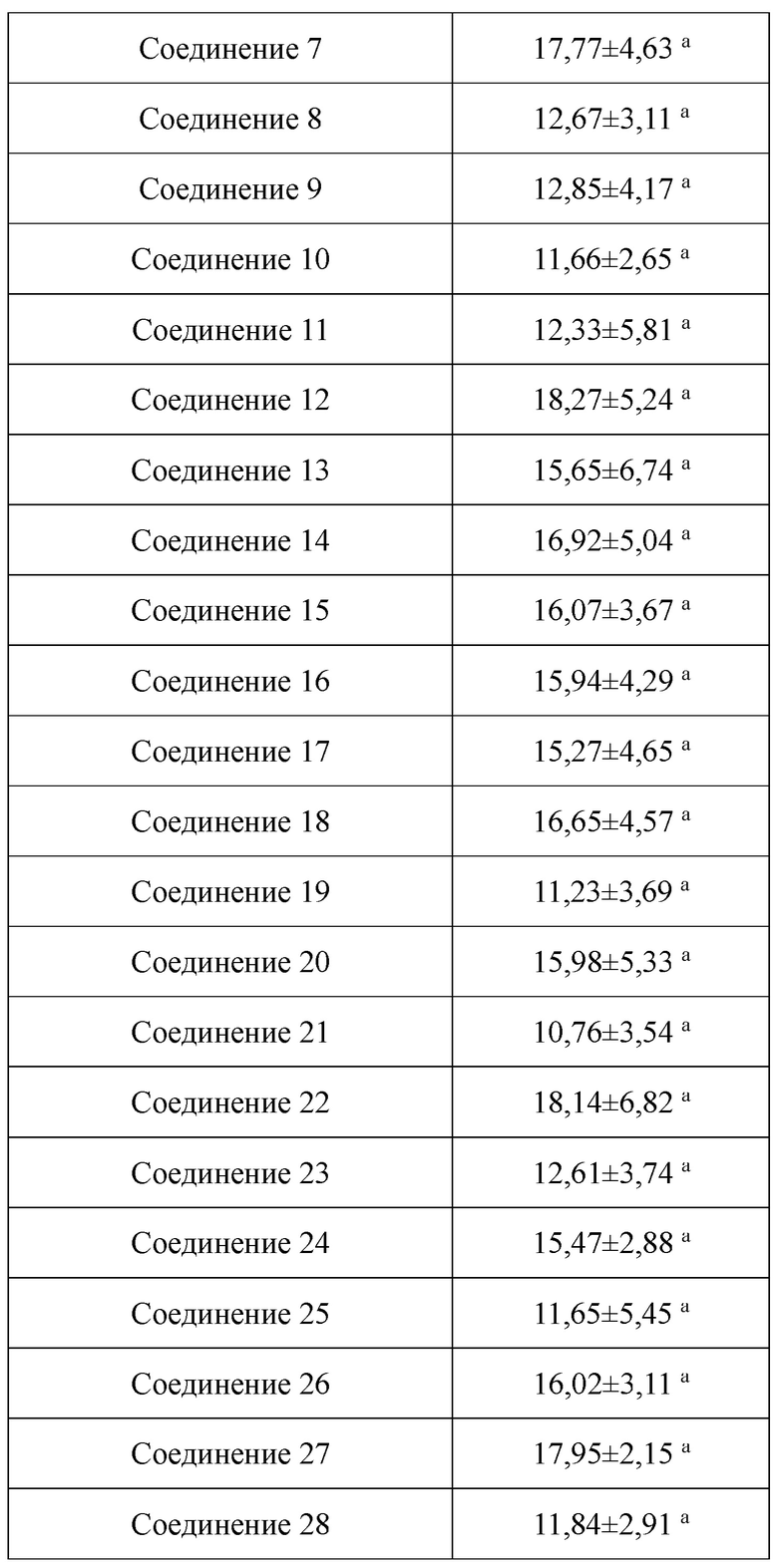

Пример 33
Исследовали влияние соединений, полученных в примерах 1-30 согласно настоящему изобретению, на образование пенистых клеток, полученных из клеток RAW264.7, при этом конкретные стадии были следующими:
1. Создание культуры клеток и модели пенистых клеток:
Клетки RAW264.7 культивировали в среде для культивирования DMEM с высоким уровнем глюкозы (GIBCO) до получения концентрации клеток 1×106, высаживали в 6-луночный планшет и добавляли 60 мг/л ох-ЛПНП (окисленный модифицированный липопротеин низкой плотности, Пекинский объединенный медицинский колледж) и пенистые клетки получали после 48 часов инкубации.
2. Определение групп и показателей:
Полученные пенистые клетки поделили на 61 группу, в каждой группе было по 3 луночных планшета. После обработки путем добавления лекарственных средств (100 мкл на лунку) в течение 48 часов среду отбрасывали, и после окрашивания масляным красным О излишки указанного красителя вымывали. Клетки растворяли в изопропаноле и значения поглощения измеряли при 490 нм. Значение ОП в модельной группе составляло 100%, и значение ОП для каждой группы, откорректированное по числу клеток, сравнивали с модельной группой для получения соотношения содержания липидов внутри клетки. Чем больше указанное значение, тем больше содержание липидов в клетке, тем выше степень образования пенистых клеток.
Контрольная группа с физиологическим раствором: физиологический раствор
Группы с низкой дозой соединений, полученных в примерах 1-30 согласно настоящему изобретению: 10 мкмоль/л (растворители представляли собой физиологический раствор).
Группы с высокой дозой соединений, полученных в примерах 1-30 согласно настоящему изобретению: 30 мкмоль/л (растворители представляли собой физиологический раствор).
3. Способ статистического анализа:
Данные представляли в виде 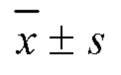 , и одно факторный дисперсионный анализ между группами проводили при помощи пакета SPSS версии 10.0, разница была достоверной в случае, когда р<0,05.
, и одно факторный дисперсионный анализ между группами проводили при помощи пакета SPSS версии 10.0, разница была достоверной в случае, когда р<0,05.
4. Результаты: соединения, полученные в примерах 1-30 согласно настоящему изобретению, могут в значительной степени уменьшать степень образования пенистых клеток при дозе 30 мкмоль/л, а некоторые соединения могут в значительной степени уменьшать степень образования пенистых клеток при дозе 10 мкмоль/л.
 )
)
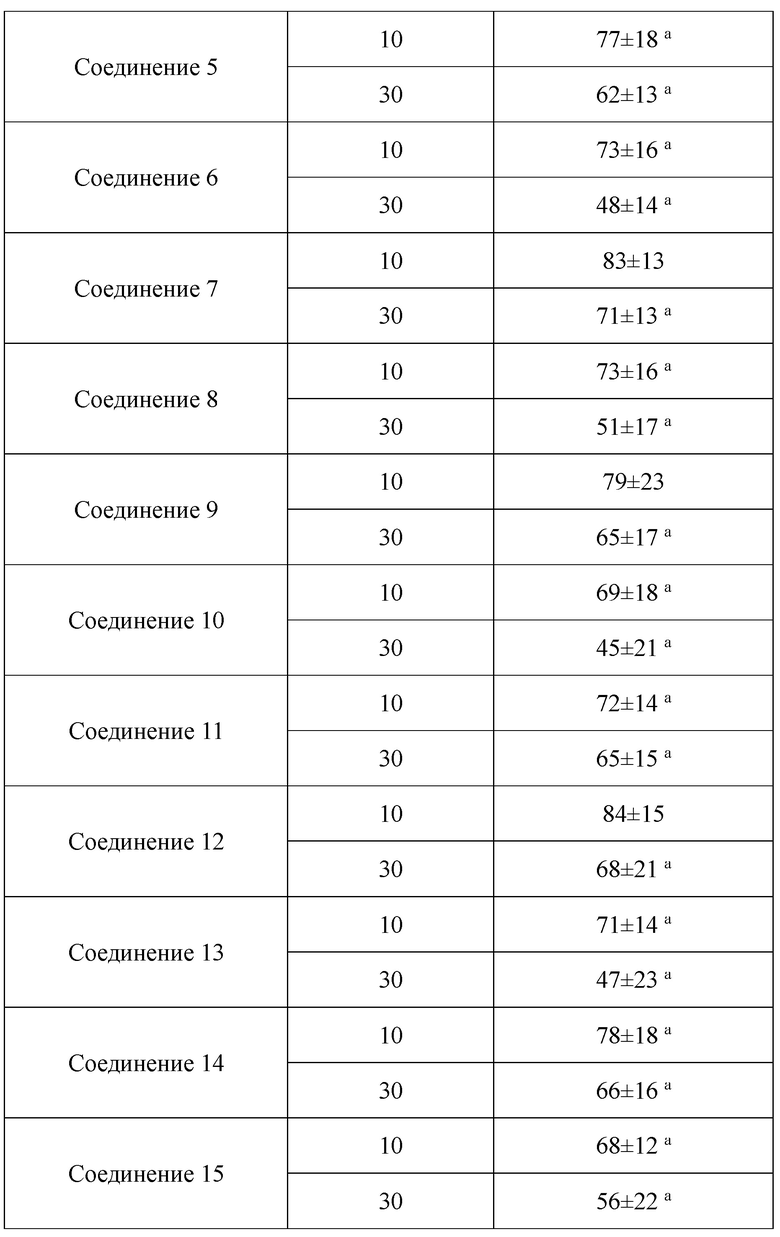
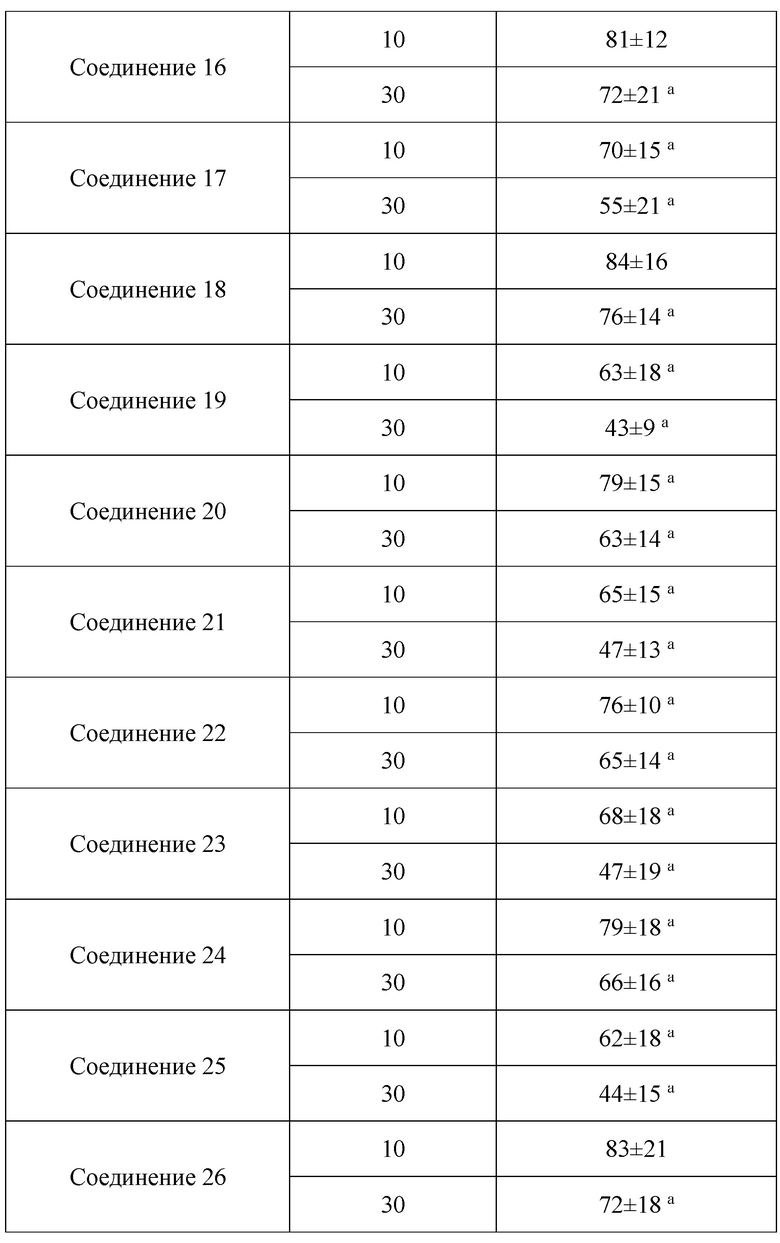
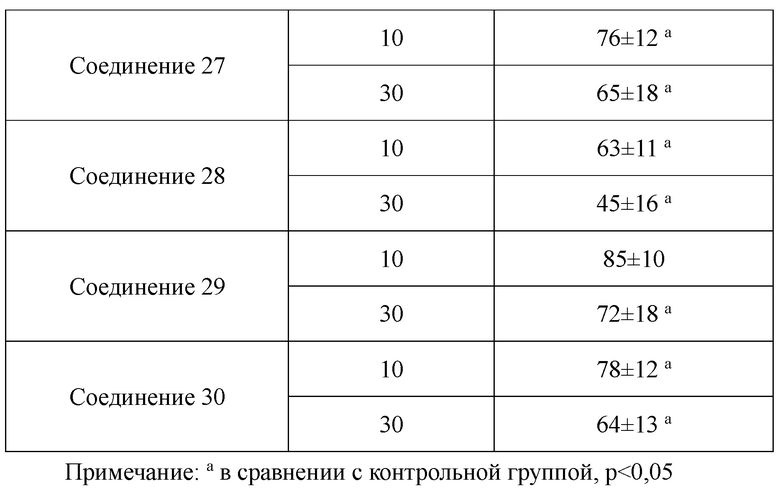
Как можно увидеть из указанных выше примеров, производные гинзенозида панаксдиолового типа, предложенные в настоящем изобретении, обладают низкой цитотоксичностью, могут значительно снижать процент площади атеросклеротических бляшек у мышей ароЕ-/-, они также могут эффективно снижать уровни холестерина липопротеинов низкой плотности и повышать уровни холестерина липопротеинов высокой плотности в сыворотке мышей, могут значительно снижать локальные уровни ФНО-α в артериях мышей ароЕ-/- и обладают хорошим противовоспалительным действием; при дозе 30 мкмоль/л производные гинзенозида панаксдиолового типа могут значительно уменьшать степень образования пенистых клеток, полученных из клеток RAW264.7. Это свидетельствует о том, что предложенные в настоящем изобретении производные гинзенозида панаксдиолового типа можно применять в качестве активных ингредиентов для получения лекарственных средств для предотвращения и лечения атеросклероза.
Указанные выше примеры являются только лишь предпочтительными вариантами реализации согласно настоящему изобретению. Необходимо отметить, что специалисты в данной области техники также могут внести некоторые улучшения и модификации без отступления от принципов настоящего изобретения, и указанные улучшения и модификации следует рассматривать в рамках объема притязаний настоящего изобретения.
Изобретение относится к производному гинзенозида панаксдиолового типа, имеющему структуру, представленную в формуле I или формуле II. В формуле I и формуле II R1 представляет собой 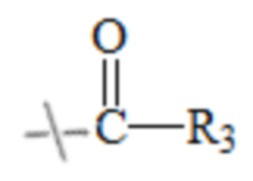 , где R3 представляет собой метил; R2 имеет структуру, представленную в формуле III:
, где R3 представляет собой метил; R2 имеет структуру, представленную в формуле III: 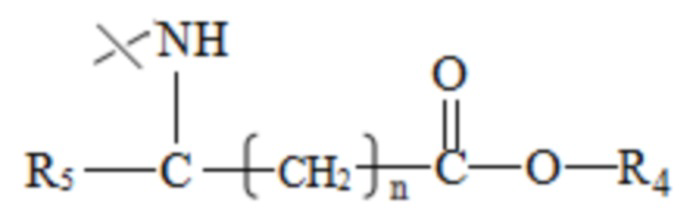 формула III; в формуле III n=0, 1 или 2, R4 представляет собой метил или этил, R5 представляет собой одно из атома водорода, незамещенного С3-С4 алкила, незамещенного бензила, C1-C2 гидроксиалкила, -CH2CH2COOCH3, -CH2COOCH2CH3, гидроксибензила, -CH2-(1H-имидазол-4-ил) и -CH2-(1H-индол-3-ил). Изобретение также относится к способу получения и к применению производного гинзенозида панаксдиолового типа. Технический результат: получены новые производные гинзенозида панаксдиолового типа, имеющие структуру формулы I или формулы II, которые можно применять для получения лекарственного средства для предотвращения и лечения атеросклероза. 3 н. и 5 з.п. ф-лы, 5 табл., 33 пр.
формула III; в формуле III n=0, 1 или 2, R4 представляет собой метил или этил, R5 представляет собой одно из атома водорода, незамещенного С3-С4 алкила, незамещенного бензила, C1-C2 гидроксиалкила, -CH2CH2COOCH3, -CH2COOCH2CH3, гидроксибензила, -CH2-(1H-имидазол-4-ил) и -CH2-(1H-индол-3-ил). Изобретение также относится к способу получения и к применению производного гинзенозида панаксдиолового типа. Технический результат: получены новые производные гинзенозида панаксдиолового типа, имеющие структуру формулы I или формулы II, которые можно применять для получения лекарственного средства для предотвращения и лечения атеросклероза. 3 н. и 5 з.п. ф-лы, 5 табл., 33 пр.
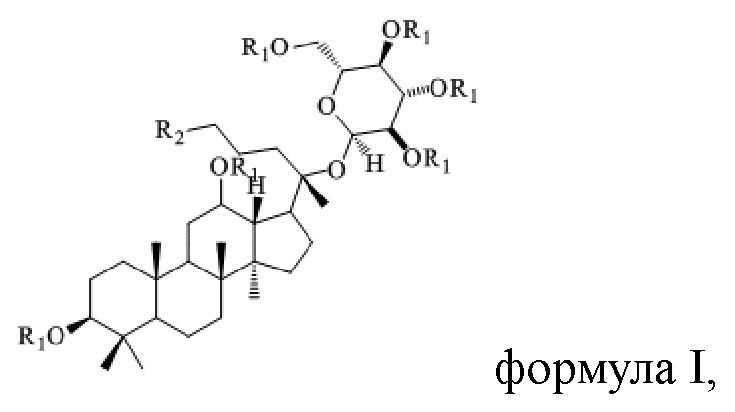
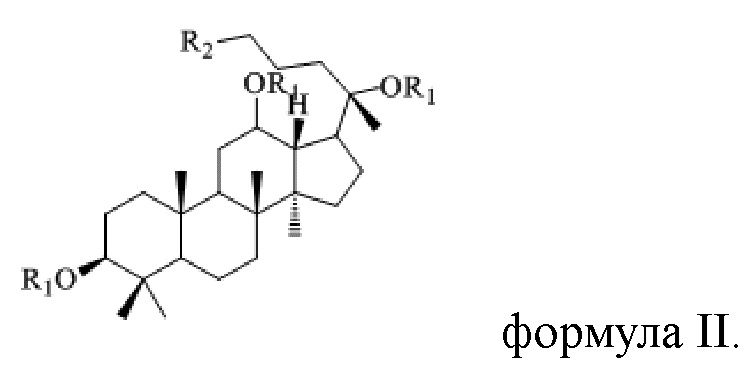
1. Производное гинзенозида панаксдиолового типа, имеющее структуру, представленную в формуле I или формуле II:
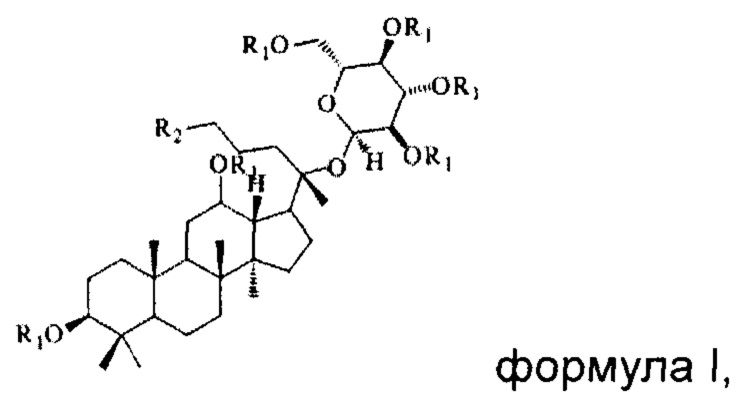

в формуле I и формуле II представляет собой  где R3 представляет собой метил;
где R3 представляет собой метил;
R2 имеет структуру, представленную в формуле III:

в формуле III n=0, 1 или 2, R4 представляет собой метил или этил, R5 представляет собой одно из атома водорода, незамещенного С3-С4 алкила, незамещенного бензила, С1-С2 гидроксиалкила, -СН2СН2СООСН3, -СН2СООСН2СН3, гидроксибензила, -СН2-(1Н-имидазол-4-ил) и -СН2-(1Н-индол-3-ил).
2. Производное гинзенозида панаксдиолового типа по п. 1, в котором R5 представляет собой -СН2СН(СН3)2, -СН(СН3)2, -СН(ОН)СН3, -СН2ОН, -СН2СН2СООСН3 или -СН2СООСН2СН3.
3. Производное гинзенозида панаксдиолового типа по п. 1, в котором
R5 представляет собой 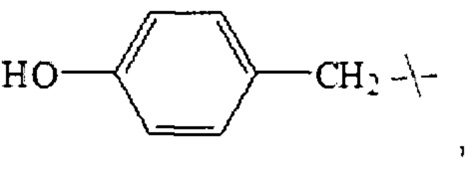

4. Производное гинзенозида панаксдиолового типа по любому из пп. 1-3, в котором
R2 представляет собой  ,
,  ,
,  ,
, 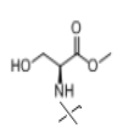 ,
, 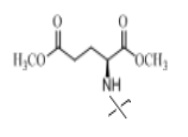 ,
, 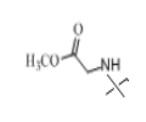 ,
,  ,
, 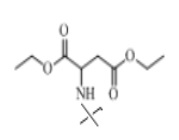 ,
,  ,
, 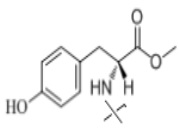 ,
, 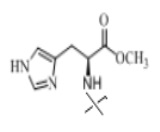 или
или 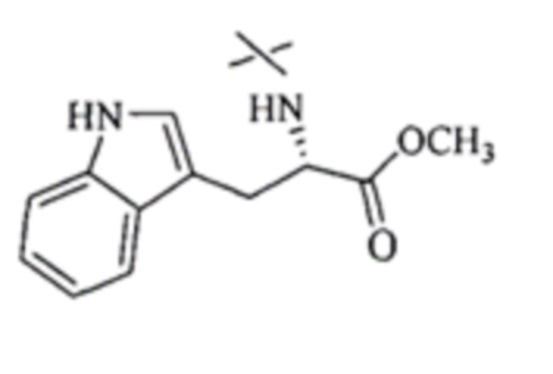 .
.
5. Производное гинзенозида панаксдиолового типа по п. 1, в котором R1 представляет собой 
6. Способ получения производного гинзенозида панаксдиолового типа в соответствии с любым из пп. 1-5, включающий следующие стадии:
(1) введение исходного соединения в реакцию нуклеофильного замещения с ангидридом кислоты в присутствии щелочного реагента, представляющего собой пиридин, при температуре от 35-60°С и при перемешивании с получением первого промежуточного продукта;
(2) введение указанного первого промежуточного продукта стадии (1) в первичную реакцию окисления в присутствии первого окисляющего агента и органического растворителя при комнатной температуре с получением предшественника указанного второго промежуточного продукта;
введение указанного предшественника второго промежуточного продукта во вторичную реакцию окисления в присутствии второго окисляющего агента и органического растворителя при комнатной температуре с получением указанного второго промежуточного продукта;
(3) введение указанного второго промежуточного продукта стадии (2) в реакцию восстановительного аминирования с аминосоединением, выбранным из гидрохлорида метилового сложного эфира L-лейцина, гидрохлорида метилового сложного эфира L-валина, гидрохлорида метилового сложного эфира L-треонина, гидрохлорида метилового сложного эфира L-серина, гидрохлорида диметилового сложного эфира L-глутаминовой кислоты, гидрохлорида метилового сложного эфира глицина, гидрохлорида этилового сложного эфира β-аланина, гидрохлорида диэтиламиномалоната, гидрохлорида метилового сложного эфира L-фенилаланина, метилового сложного эфира L-тирозина, гидрохлорида метилового сложного эфира L-гистидина или гидрохлорида метилового сложного эфира L-триптофана в присутствии органического растворителя и восстанавливающего агента, причем восстанавливающий агент смешивают со вторым промежуточным продуктом, аминосоединением и органическим растворителем при перемешивании в диапазоне температур от -2 до +2°С, и реакцию восстановительного аминирования проводят при комнатной температуре, с получением производного гинзенозида панаксдиолового типа, имеющего структуру, представленную в формуле I или формуле II;
причем указанное исходное соединение в стадии (1) имеет структуру, представленную в формуле IV или формуле V:

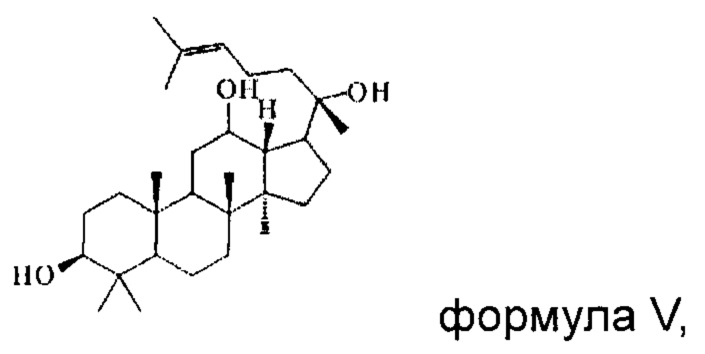
указанный первый промежуточный продукт имеет структуру, представленную формулой:
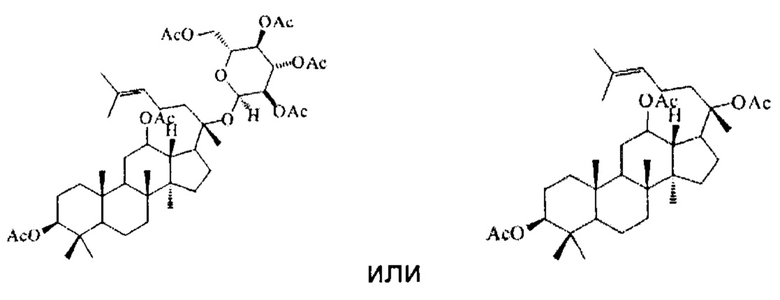 соответственно для исходных
соответственно для исходных
соединений формулы IV или формулы V;
предшественник второго промежуточного продукта имеет структуру, представленную формулой:

второй промежуточный продукт имеет структуру, представленную формулой:

7. Способ получения по п. 6, в котором указанный первый окисляющий агент для стадии (2) представляет собой пероксид водорода, хлорноватистую кислоту, гипохлорит кальция, пероксид ацетона или мета-хлорпербензойную кислоту; указанный второй окисляющий агент для стадии (2) представляет собой перманганат калия, диоксид марганца, йодную кислоту или реагент Саретта.
8. Применение производного гинзенозида панаксдиолового типа по любому из пп. 1-5 или производного гинзенозида панаксдиолового типа, полученного способом получения по любому из пп. 6, 7, для получения лекарственного средства для предотвращения и лечения атеросклероза.
| WO 2004058796 A1, 15.07.2004 | |||
| US 20110212909 A1, 01.09.2011 | |||
| KR 20090115280 A, 05.11.2009 | |||
| CN 101653447 A, 24.02.2010 | |||
| CN 102766187 A, 07.11.2012 | |||
| WO 2013189229 A1, 27.12.2013 | |||
| А | |||
| М | |||
| Попов "Механизмы биологической активности гликозидов женьшеня: сравнение с гликозидами голотурий" Вестник ДВО РАН, N6, 2006, 92-104. |

