Данное изобретение относится к соединению формулы
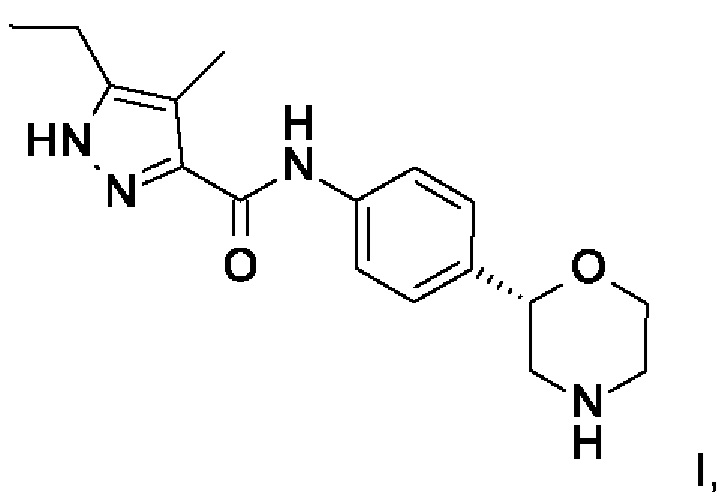
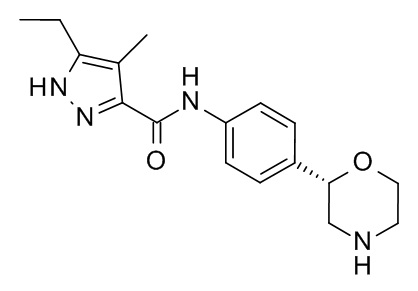 I,
I,
и к его фармацевтически приемлемым солям присоединения кислоты.
Соединение, описанное в данном документе и подпадающее под Формулу I, указанную выше, может демонстрировать таутомеризм. Изобретение охватывает любые таутомерные формы данного соединения или смеси таких форм и не ограничено какой-либо таутомерной формой, подпадающей под формулу, приведенную выше.
Обнаружили, что соединение формулы I (5-этил-4-метил-N-[4-[(2S) морфолин-2-ил]фенил]-1H-пиразол-3-карбоксамид) обладает хорошей аффинностью к рецепторам, ассоциированным со следовыми аминами (TAAR), в частности, к TAAR1, и вызывает меньше побочных эффектов по сравнению с соединениями, известными на уровне техники.
Аналогичные лиганды мышиного TAAR1 и крысиного TAAR1 описаны в WO2011/076678 и WO2012/16826.
Соединение формулы I и его фармацевтически приемлемые соли присоединения обладают ценными фармацевтическими свойствами. В частности, обнаружили, что соединение по данному изобретению является парциальным агонистом человеческого ассоциированного со следовыми аминами рецептора 1 (hTAAR1).
Соединение по данному изобретению обладает существенными преимуществами по сравнению с соединениями, известными на уровне техники, в частности
- выраженной агонистической активностью в отношении человеческого рецептора TAAR1,
- селективностью в отношении дофаминового переносчика (DAT),
- селективностью в отношении ионного канала hERG,
- низкой амфифильностью, и поэтому обусловливает низкий риск развития лекарственно-индуцированного фосфолипидоза (ЛИФ) (см. ниже).
Таким образом, соединение формулы I можно применять в качестве безопасного лекарственного средства для лечения депрессии, тревожных расстройств, биполярного расстройства, синдрома гиперактивности с дефицитом внимания (СГДВ), связанных со стрессом расстройств, психотических расстройств, таких как шизофрения, неврологических расстройств, таких как болезнь Паркинсона, нейродегенеративных расстройств, таких как болезнь Альцгеймера, эпилепсии, мигрени, гипертензии, аддикции, злоупотребления веществами и метаболических расстройств, таких как нарушения пищевого поведения, диабет, диабетические осложнения, ожирение, дислипидемия, нарушений поглощения энергии и ассимиляции, расстройств и нарушения гомеостаза температуры тела, расстройств сна и циркадного ритма и сердечно-сосудистых расстройств.
Классические биогенные амины (серотонин, норадреналин, адреналин, дофамин, гистамин) играют важную роль в качестве нейротрансмиттеров в центральной и периферической нервной системе[1]. Их синтез и хранение, а также их расщепление и обратный захват после высвобождения строго регулируются. Известно, что дисбаланс уровней биогенных аминов отвечает за нарушение функции головного мозга при многих патологических состояниях[2-5]. Второй класс эндогенных аминных соединений, так называемые следовые амины (ТА, от англ. trace amines) в значительной степени пересекается с классическими биогенными аминами по структуре, метаболизму и субклеточной локализации. ТА включают пара-тирамин, β-фенилендиамин, триптамин и октофамин и в целом они присутствуют в нервной системе млекопитающих в более низких количествах по сравнению с классическими биогенными аминами[6].
Нарушение регуляции ТА связывали с различными психическими заболеваниями, такими как шизофрения и депрессия[7] и с другими состояниями, такими как синдром гиперактивности с дефицитом внимания, головная боль по типу мигрени, болезнь Паркинсона, злоупотребление препаратами и нарушения пищевого поведения[8,9].
Долгое время существование ТА-специфических рецепторов предполагали лишь гипотетически на основании анатомически дискретных высокоаффинных ТА-связывающих сайтов в центральной нервной системе (ЦНС) человека и других млекопитающих[10,11]. Соответственно, считали, что фармакологические эффекты ТА опосредованы хорошо известным механизмом классических биогенных аминов, посредством либо запуска их высвобождения, либо ингибирования их обратного захвата, либо «перекрестной реакции» с их рецепторными системами[9,12,13]. Эта точка зрения существенно изменилась после обнаружения нескольких представителей нового семейства рецепторов, сопряженных с G-белком (GPCR, от англ. G-protein coupled receptor), представляющих собой рецепторы, ассоциированные со следовыми аминами (TAAR)[7,14]. Существует 9 генов TAAR (включая 3 псевдогена) и 16 генов у мышей (включая 1 псевдоген). Гены TAAR не содержат интронов (за одним исключением, TAAR2 содержит 1 интрон) и расположены друг за другом на одном и том же хромосомном сегменте. Филогенетические отношения генов рецепторов в соответствии с углубленным сравнением подобия фармакофоров GPCR и фармакологические данные позволяют предположить, что эти рецепторы образуют три отдельных подсемейства[7,14]. TAAR1 находится в первом подклассе из четырех генов (TAAR1-4), высоко консервативных между людьми и грызунами. TA активируют TAAR1 посредством Gαs. Показано, что нарушение регуляции ТА вносит вклад в этиологию различных заболеваний, таких как депрессия, тревожные расстройства, биполярное расстройство, синдром гиперактивности с дефицитом внимания (СГДВ), связанные со стрессом расстройства, психотические расстройства, такие как шизофрения, неврологические расстройства, такие как болезнь Паркинсона, нейродегенеративные расстройства, такие как болезнь Альцгеймера, эпилепсия, мигрень, гипертензия, аддикция, злоупотребление веществами и метаболические расстройства, такие как нарушения пищевого поведения, диабет, диабетические осложнения, ожирение, дислипидемия, нарушения поглощения энергии и ассимиляции, расстройства и нарушения гомеостаза температуры тела, расстройства сна и циркадного ритма и сердечно-сосудистые расстройства.
Таким образом, существует широкий интерес к расширению знаний о рецепторах, ассоциированных со следовыми аминами.
Список литературы:
[1] Deutch, A.Y. and Roth, R.H.; “Neurotransmitters.” In Fundamental Neuroscience (2nd Edn) (Zigmond, M.J., Bloom, F.E., Landis, S.C., Roberts, J.L, and Squire, L.R., Eds.), pp. 193-234, Academic Press (1999);
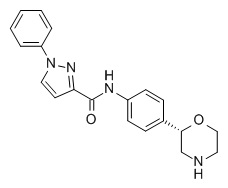
[2] Wong, M.L. and Licinio, J.; “Research and treatment approaches to depression.” Nat. Rev. Neurosci. 2001, 2, 343-351;
[3] Carlsson, A. et al.; “Interactions between monoamines, glutamate, and GABA in schizophrenia: new evidence.” Annu. Rev. Pharmacol. Toxicol. 2001, 41, 237-260;
[4] Tuite, P. and Riss, J.; “Recent developments in the pharmacological treatment of Parkinson's disease.” Expert Opin. Investig. Drugs 2003, 12, 1335-1352;
[5] Castellanos, F.X. and Tannock, R.; “Neuroscience of attention-deficit/hyperactivity disorder: the search for endophenotypes.” Nat. Rev. Neurosci. 2002, 3, 617-628;
[6] Usdin, Earl; Sandler, Merton; Editors; Psychopharmacology Series, Vol. 1: Trace Amines and the Brain. [Proceedings of a Study Group at the 14th Annual Meeting of the American College of Neuropsychoparmacology, San Juan, Puerto Rico] (1976);
[7] Lindemann, L. and Hoener, M.; “A renaissance in trace amines inspired by a novel GPCR family.” Trends in Pharmacol. Sci. 2005, 26, 274-281;
[8] Branchek, T.A. and Blackburn, T.P.; “Trace amine receptors as targets for novel therapeutics: legend, myth and fact.” Curr. Opin. Pharmacol. 2003, 3, 90-97;
[9] Premont, R.T. et al.; “Following the trace of elusive amines.” Proc. Natl. Acad. Sci. USA 2001, 98, 9474-9475;
[10] Mousseau, D.D. and Butterworth, R.F.; “A high-affinity [3H] tryptamine binding site in human brain.” Prog. Brain Res. 1995, 106, 285-291;
[11] McCormack, J.K. et al.; “Autoradiographic localization of tryptamine binding sites in the rat and dog central nervous system.” J. Neurosci. 1986, 6, 94-101;
[12] Dyck, L.E.; “Release of some endogenous trace amines from rat striatal slices in the presence and absence of a monoamine oxidase inhibitor.” Life Sci. 1989, 44, 1149-1156;
[13] Parker, E.M. and Cubeddu, L.X.; “Comparative effects of amphetamine, phenylethylamine and related drugs on dopamine efflux, dopamine uptake and mazindol binding.” J. Pharmacol. Exp. Ther. 1988, 245, 199-210;
[14] Lindemann, L. et al.; “Trace amine associated receptors form structurally and functionally distinct subfamilies of novel G protein-coupled receptors.” Genomics 2005, 85, 372-385.
Объектами данного изобретения являются новые соединения формулы I и их фармацевтически приемлемые соли, их применение в лечении заболеваний, связанных с биологической функцией рецепторов, ассоциированных со следовыми аминами, их получение и лекарственные средства на основе соединений по изобретению для контроля или предупреждения таких заболеваний, как депрессия, тревожные расстройства, биполярное расстройство, синдром гиперактивности с дефицитом внимания, связанные со стрессом расстройства, психотические расстройства, такие как шизофрения, неврологические расстройства, такие как болезнь Паркинсона, нейродегенеративные расстройства, такие как болезнь Альцгеймера, эпилепсия, мигрень, злоупотребление веществами, аддикция и метаболические расстройства, такие как нарушения пищевого поведения, диабет, диабетические осложнения, ожирение, дислипидемия, нарушения поглощения энергии и ассимиляции, расстройства и нарушения гомеостаза температуры тела, расстройства сна и циркадного ритма и сердечно-сосудистые расстройства.
Данные соединения формулы (I) и их фармацевтически приемлемые соли могут быть получены способами, известными в области техники, например, описанными ниже способами, включающими
а) отщепление N-защитной группы (PG) у соединений формулы
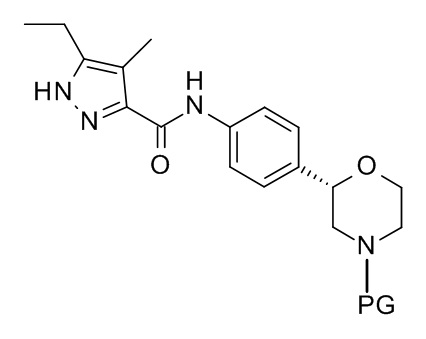 3'
3'
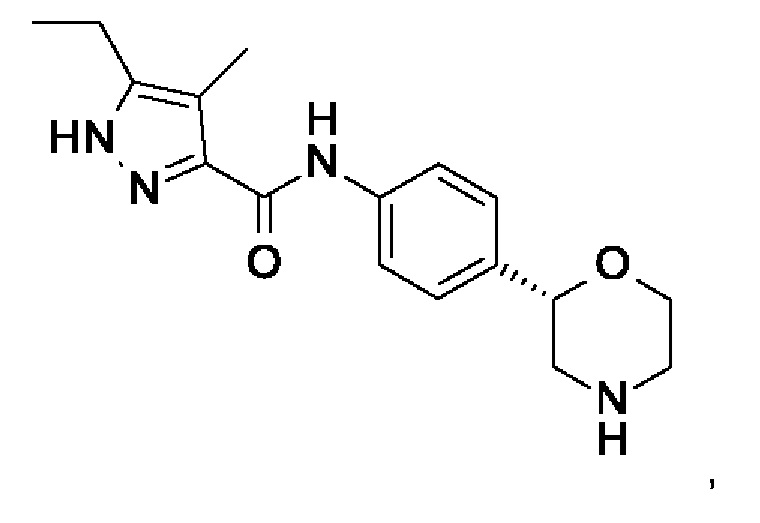
с образованием соединения формулы
I
где PG является N-защитной группой, выбранной из -C(O)O-трет-бутил (BOC) и,
при желании, превращение полученного соединения в фармацевтически приемлемые соли присоединения кислоты.
ОБЩАЯ МЕТОДИКА
Схема 1.
Образование амидной связи
Отщепление защитной группы
или 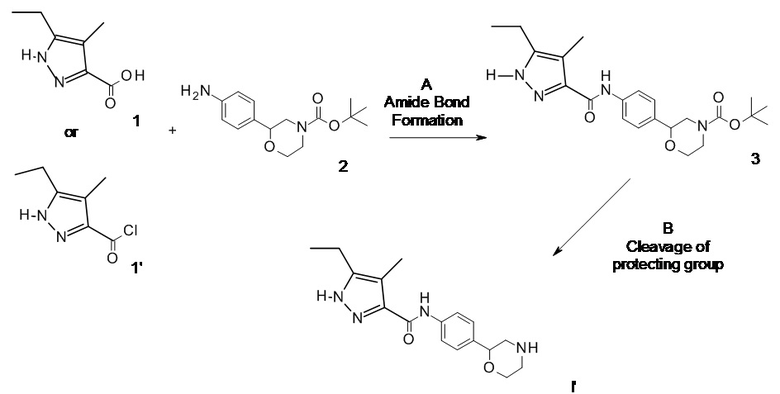
Исходные материалы 1, 1' и 2 доступны для приобретения или могут быть получены способами, хорошо известными в области техники. Рацемический трет-бутил 2-(4-аминофенил)морфолин-4-карбоксилат (CAS RN: 1002726-96-6) доступен для приобретения. Трет-бутил (2R)-2-(4-аминофенил)морфолин-4-карбоксилат (CAS RN: 1260220-42-5) доступен для приобретения. Трет-бутил (2S)-2-(4-аминофенил)морфолин-4-карбоксилат (CAS RN: 1260220-43-6) доступен для приобретения или может быть получен, как описано в литературе, например, Trussardi, R. & Iding, H, PCT Int. Appl. WO 2015/086495 A1.
Стадия А: Образование амидной связи может осуществляться посредством реакции сочетания между амином 2 и карбоновой кислотой 1 в присутствии реагента сочетания, такого как DCC, EDC, TBTU или HATU в присутствии органического основания, такого как триэтиламин, N,N-диизопропилэтиламин или N-метилморфолин, в галогенированных растворителях, таких как дихлорметан или 1,2-дихлорэтан или в эфирных растворителях, таких как диэтиловый эфир, диоксан, THF, DME или TBME.
Предпочтительными являются условия: N-метилморфолин в THF при 50-60°C в течение 12-48 часов.
В альтернативном варианте образование амидной связи может осуществляться посредством реакции сочетания между амином 2 и хлорангидридным соединением 1' в галогенированных растворителях, таких как дихлорметан или 1,2-дихлорэтан или в эфирных растворителях, таких как диэтиловый эфир, диоксан, THF, DME или TBME в присутствии органического основания, такого как триэтиламин или N,N-диизопропилэтиламин.
Предпочтительными являются условия: триэтиламин в THF при комнатной температуре в течение 18 ч.
При желании хлорангидридное соединение 1' может быть получено in situ из соответствующей карбоновой кислоты 1 путем обработки оксалилхлоридом в галогенированных растворителях, таких как дихлорметан или 1,2-дихлорэтан или в эфирных растворителях, таких как диэтиловый эфир, диоксан, THF, DME или TBME в присутствии катализатора, такого как DMF.
Предпочтительными являются условия: дихлорэтан при комнатной температуре в течение 1 ч.
В альтернативном варианте хлорангидридное соединение 1' может быть получено in situ из соответствующей карбоновой кислоты 1 путем обработки 1-хлор-N,N,2-триметилпропениламином [CAS 26189-59-3] в дихлорметане, с последующим удалением растворителя в вакууме согласно способу, предложенному Ghosez и соавт. (J. Chem. Soc., Chem. Commun. 1979, 1180; Org. Synth. 1980, 59, 26-34).
Стадия Б: Удаление N-защитной группы BOC можно выполнять с минеральными кислотами, такими как HCl, H2SO4 или H3PO4, или органическими кислотами, такими как CF3COOH, CHCl2COOH, HOAc или пара-толуолсульфоновая кислота в растворителях, таких как CH2Cl2, CHCl3, THF, MeOH, EtOH или H2O при температуре от 0 до 80°C.
Предпочтительными являются условия: CF3COOH в водном растворе ацетонитрила при 80°C в течение 3 часов или 4 N HCl в диоксане при комнатной температуре в течение 16 часов.
В случае применения рацемического исходного материала 2, полученную рацемическая смесь морфолинового соединения I’ можно разделять на составляющие ее энантиомеры, используя хиральную ВЭЖХ. В альтернативном варианте, соединение I может быть получено в энантиомерно чистой форме из энантиомерно чистого соединения 2.
Выделение и очистка соединений
Выделение и очистку соединений и промежуточных соединений, описанных в данном документе, можно осуществлять, при желании, любым подходящим способом разделения или очистки, например, таким как фильтрование, экстрагирование, кристаллизация, колоночная хроматография, тонкослойная хроматография, толстослойная хроматография, препаративная жидкостная хроматография низкого или высокого давления, или комбинацией указанных способов. Конкретные иллюстрации подходящих способов разделения или очистки можно получить путем ссылки на примеры получения и примеры, приведенные ниже. Тем не менее, конечно можно также применять и другие эквивалентные способы разделения или очистки. Рацемические смеси хиральных соединений формулы I можно разделять при помощи хиральной высокоэффективной жидкостной хроматографии (ВЭЖХ). Рацемические смеси хиральных синтетических промежуточных соединений также можно разделять при помощи хиральной ВЭЖХ.
Соли соединения формулы (I)
Соединение формулы I является основным и может быть преобразовано в соответствующую соль присоединения кислоты. Преобразование выполняют путем обработки по меньшей мере стехиометрическим количеством подходящей кислоты, такой как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., и органическими кислотами, такими как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, малоновая кислота, янтарная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, пара-толуолсульфоновая кислота, салициловая кислота и т.п. Обычно свободное основание растворяют в инертном органическом растворителе, таком как диэтиловый эфир, этилацетат, хлороформ, этанол или метанол и т.п. и добавляют кислоту в аналогичном растворителе. Поддерживают температуру между 0°C и 50°C. Полученная в результате соль выпадает в осадок спонтанно или может быть выделена из раствора менее полярным растворителем.
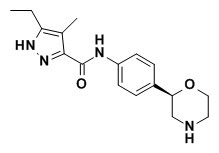
Пример 1
5-Этил-4-метил-N-[4-[(2S)-морфолин-2-ил]фенил]-1H-пиразол-3-карбоксамид
a) трет-бутил (2S)-2-[4-[(5-этил-4-метил-1H-пиразол-3-карбонил)амино]фенил]морфолин-4-карбоксилат
К перемешиваемому раствору трет-бутил (2S)-2-(4-аминофенил)морфолин-4-карбоксилата (CAS
RN: 1260220-43-6, 350 мг, 1,26 ммоль, 1,00 эквив.) и 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты (CAS RN: 957129-38-3, 245 мг, 1,51 ммоль, 1,20 эквив.) в THF (8 мл) добавляли TBTU (807 мг, 2,51 ммоль, 2,00 эквив.) и N-метилморфолин (509 мг, 553 мкл, 5,03 ммоль, 4,00 эквив.). Реакционную смесь перемешивали при температуре 50°C в течение 15 минут. TLC в момент времени t = 15 ч показала, что реакция прошла полностью. Реакционную смесь концентрировали в вакууме. Неочищенный материал очищали флэш-хроматографией (силикагель, элюент: от 0% до 100% EtOAc в гептане) с получением трет-бутил (2S)-2-[4-[(5-этил-4-метил-1H-пиразол-3-карбонил)амино]фенил]морфолин-4-карбоксилата в виде беловатого твердого вещества (501 мг, 96%). MS (ISP): 413,7 ([M-H]-).
b) 5-Этил-4-метил-N-[4-[(2S)-морфолин-2-ил]фенил]-1H-пиразол-3-карбоксамид
К перемешиваемому раствору трифторуксусной кислоты (1,37 г, 918 мкл, 12,0 ммоль, 10 эквив.) в воде (8 мл) добавляли суспензию трет-бутил (2S)-2-[4-[(5-этил-4-метил-1H-пиразол-3-карбонил)амино]фенил]морфолин-4-карбоксилата (497 мг, 1,2 ммоль, 1,00 эквив.) в ацетонитриле (4 мл). Реакционную смесь перемешивали при температуре 80°C в течение 3 часов. MS в момент времени t = 3 ч показала, что реакция прошла полностью. Реакционную смесь вливали в 1 M водный раствор NaOH и дважды экстрагировали EtOAc. Органические слои высушивали над Na2SO4 и концентрировали в вакууме. Неочищенный материал очищали колоночной флэш-хроматографией (SiliaSep™ картридж Amine, элюент: от 0% до 100% EtOAc в гептане, затем от 0% до 10% MeOH в EtOAc) с получением 5-этил-4-метил-N-[4-[(2S)-морфолин-2-ил]фенил]-1H-пиразол-3-карбоксамида в виде беловатого твердого вещества. MS (ISP): 315,7 ([M+H]+).
Пример 2 (приведенный для сравнения пример)
5-Этил-4-метил-N-[4-[(2R)-морфолин-2-ил]фенил]-1H-пиразол-3-карбоксамид
Указанное в заголовке соединение получали аналогично Примеру 1, используя (2R)-2-(4-аминофенил)морфолин-4-карбоксилат (CAS RN: 1260220-42-5) вместо (2S)-2-(4-аминофенил)морфолин-4-карбоксилата на стадии (a). Белое твердое вещество. MS (ISP): 315,6 ([M+H]+).
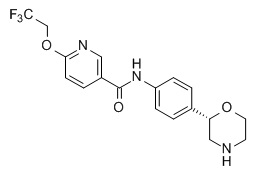
Пример 3 (приведенный для сравнения пример)
N-[4-[(2S)-Морфолин-2-ил]фенил]-6-(2,2,2-трифторэтокси)пиридин-3-карбоксамид
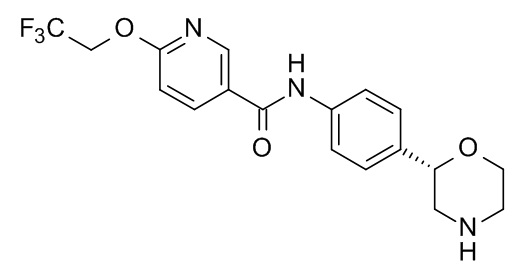
Указанное в заголовке соединение получали аналогично Примеру 1, используя 6-(2,2,2-трифторэтокси)никотиновую кислоту (CAS RN: 159783-29-6) вместо 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты на стадии (a). Белое твердое вещество. MS (ISP): 382,1 ([M+H]+).
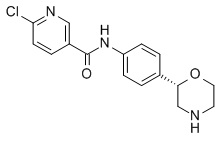
Пример 4 (приведенный для сравнения пример)
6-хлор-N-[4-[(2S)-морфолин-2-ил]фенил]пиридин-3-карбоксамид
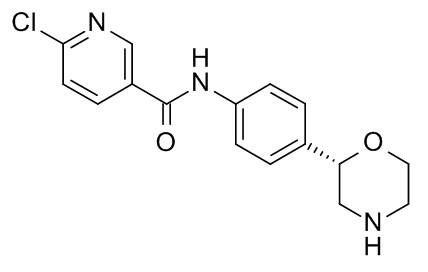
Указанное в заголовке соединение получали аналогично Примеру 1, используя 6-хлор-никотиновую кислоту (CAS RN: 5326-23-8) вместо 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты на стадии (a). Белое твердое вещество. MS (ISP): 320,1 ([{37Cl}M+H]+), 318,2 ([{35Cl}M+H]+).
Пример 5 (приведенный для сравнения пример)
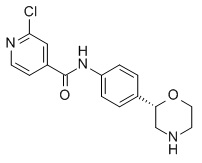
2-Хлор-N-[4-[(2S)-морфолин-2-ил]фенил]пиридин-4-карбоксамид
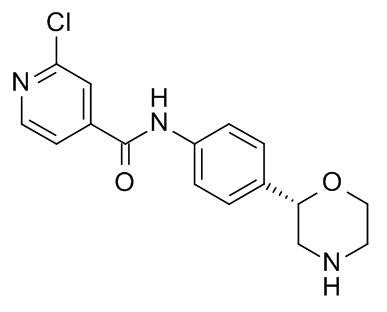
Указанное в заголовке соединение получали аналогично Примеру 1, используя 2-хлор-изоникотиновую кислоту (CAS RN: 6313-54-8) вместо 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты на стадии (a). Белое твердое вещество. MS (ISP): 320,1 ([{37Cl}M+H]+), 318,1 ([{35Cl}M+H]+).
Пример 6 (приведенный для сравнения пример)
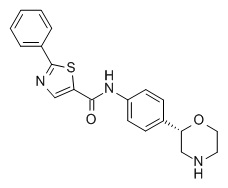
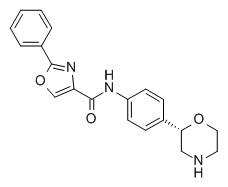
N-[4-[(2S)-Морфолин-2-ил]фенил]-2-фенил-1,3-тиазол-5-карбоксамид
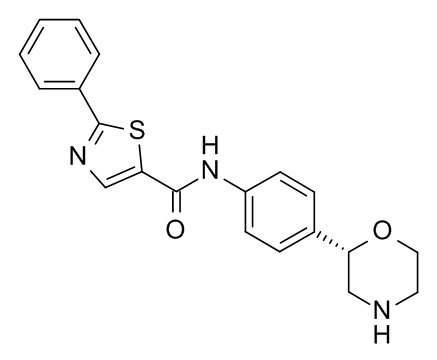
Указанное в заголовке соединение получали аналогично Примеру 1, используя 2-фенилтиазол-5-карбоновую кислоту (CAS RN: 10058-38-5) вместо 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты на стадии (a). Белое твердое вещество. MS (ISP): 366,1 ([M+H]+).
Пример 7 (приведенный для сравнения пример)
2,6-дихлор-N-[4-[(2S)-морфолин-2-ил]фенил]пиридин-4-карбоксамид
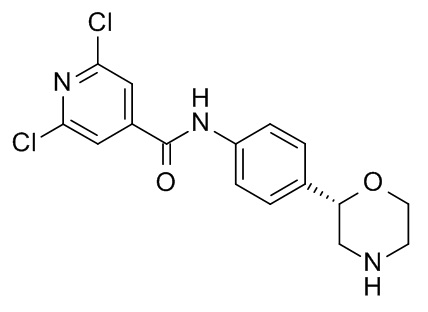
Указанное в заголовке соединение получали аналогично Примеру 1, используя 2,6-дихлор-изоникотиновую кислоту (CAS RN: 5398-44-7) вместо 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты на стадии (a). Белое твердое вещество. MS (ISP): 356,1 ([{37Cl}M+H]+), 354,1 ([{37Cl35Cl }M+H]+), 352,1 ([{35Cl}M+H]+).
Пример 8 (приведенный для сравнения пример)
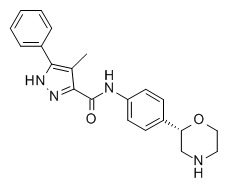
4-Метил-N-[4-[(2S)-морфолин-2-ил]фенил]-5-фенил-1H-пиразол-3-карбоксамид
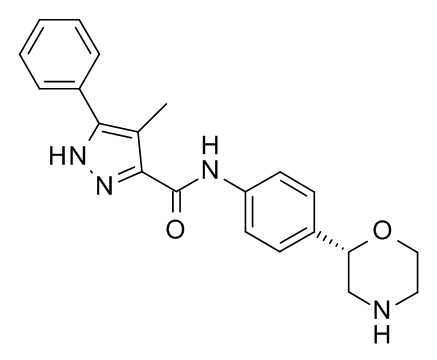
Указанное в заголовке соединение получали аналогично Примеру 1, используя 4-метил-5-фенил-1H-пиразол-3-карбоновую кислоту (CAS RN: 879770-33-9) вместо 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты на стадии (a). Белое твердое вещество. MS (ISP): 363,2 ([M+H]+).
Пример 9 (приведенный для сравнения пример)
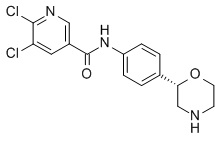
5,6-дихлор-N-[4-[(2S)-морфолин-2-ил]фенил]пиридин-3-карбоксамид
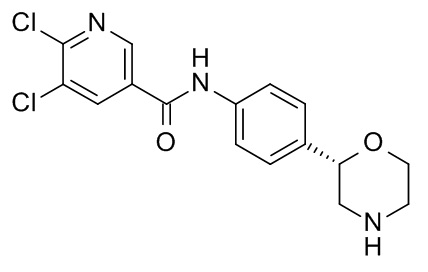
Указанное в заголовке соединение получали аналогично Примеру 1, используя 5,6-дихлор-никотиновую кислоту (CAS RN: 41667-95-2) вместо 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты на стадии (a). Белое твердое вещество. MS (ISP): 356,1 ([{37Cl35Cl }M+H]+), 354,1 ([{37Cl}M+H]+), 352,1 ([{35Cl}M+H]+).
Пример 10 (приведенный для сравнения пример).
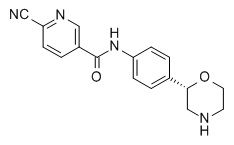
6-циано-N-[4-[(2S)-морфолин-2-ил]фенил]пиридин-3-карбоксамид
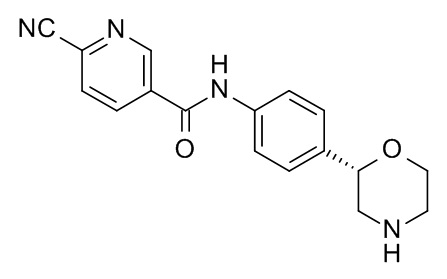
Указанное в заголовке соединение получали аналогично Примеру 1, используя 6-циано-никотиновую кислоту (CAS RN: 70165-31-0) вместо 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты на стадии (a). Белое твердое вещество. MS (ISP): 309,1 ([M+H]+).
Пример 11 (приведенный для сравнения пример).
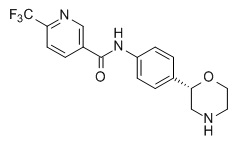
N-[4-[(2S)-Морфолин-2-ил]фенил]-6-(трифторметил)пиридин-3-карбоксамид

Указанное в заголовке соединение получали аналогично Примеру 1, используя 6-(трифторметил)-никотиновую кислоту (CAS RN: 158063-66-2) вместо 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты на стадии (a). Белое твердое вещество. MS (ISP): 352,2 ([M+H]+).
Пример 12 (приведенный для сравнения пример).
5-Хлор-N-[4-[(2S)-морфолин-2-ил]фенил]пиридин-2-карбоксамид
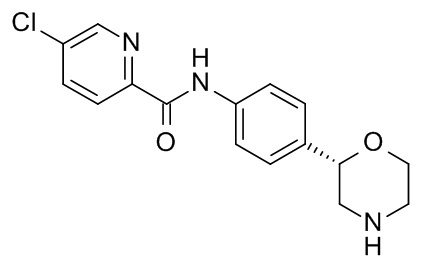
Указанное в заголовке соединение получали аналогично Примеру 1, используя 5-хлор-пиколиновую кислоту (CAS RN: 86873-60-1) вместо 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты на стадии (a). Белое твердое вещество. MS (ISP): 320,1 ([{37Cl}M+H]+), 318,2 ([{35Cl}M+H]+).
Пример 13 (приведенный для сравнения пример).
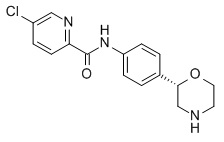
5-Хлор-N-[4-[(2S)-морфолин-2-ил]фенил]пиридин-3-карбоксамид
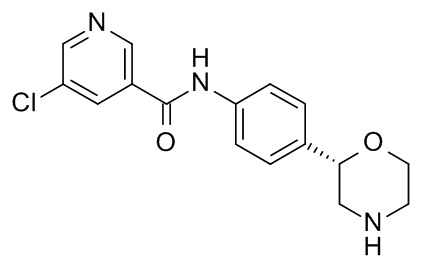
Указанное в заголовке соединение получали аналогично Примеру 1, используя 5-хлор-никотиновую кислоту (CAS RN: 22620-27-5) вместо 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты на стадии (a). Белое твердое вещество. MS (ISP): 320,1 ([{37Cl}M+H]+), 318,1 ([{35Cl}M+H]+).
Пример 14 (приведенный для сравнения пример).
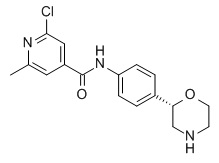
2-Хлор-6-метил-N-[4-[(2S)-морфолин-2-ил]фенил]пиридин-4-карбоксамид
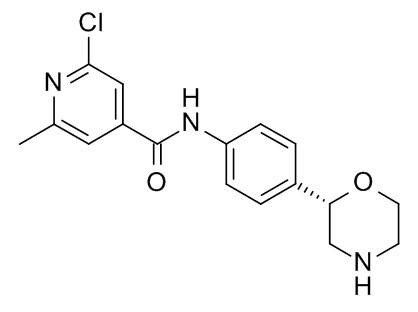
Указанное в заголовке соединение получали аналогично Примеру 1, используя 2-хлор-6-метилпиридин-4-карбоновую кислоту (CAS RN: 25462-85-5) вместо 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты на стадии (a). Белое твердое вещество. MS (ISP): 334,1 ([{37Cl}M+H]+), 332,1 ([{35Cl}M+H]+).
Пример 15 (приведенный для сравнения пример).
N-[4-[(2S)-Морфолин-2-ил]фенил]-2-фенил-1,3-оксазол-4-карбоксамид
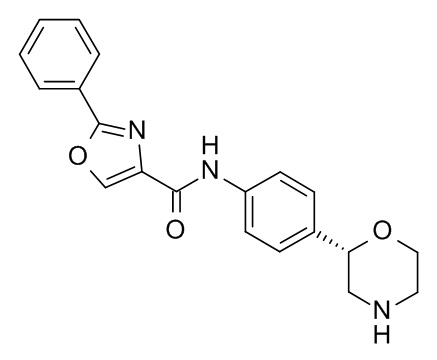
Указанное в заголовке соединение получали аналогично Примеру 1, используя 2-фенилоксазол-4-карбоновую кислоту (CAS RN: 23012-16-0) вместо 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты на стадии (a). Белое твердое вещество. MS (ISP): 350,2 ([M+H]+).
Пример 16 (приведенный для сравнения пример).
N-[4-[(2S)-Морфолин-2-ил]фенил]-2-фенил-1,3-тиазол-4-карбоксамид
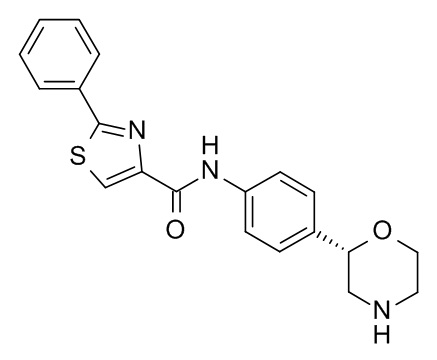
Указанное в заголовке соединение получали аналогично Примеру 1, используя 2-фенилтиазол-4-карбоновую кислоту (CAS RN: 7113-10-2) вместо 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты на стадии (a). Белое твердое вещество. MS (ISP): 366,1 ([M+H]+).
Пример 17 (приведенный для сравнения пример).
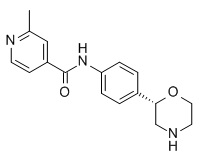
2-Метил-N-[4-[(2S)-морфолин-2-ил]фенил]пиридин-4-карбоксамид
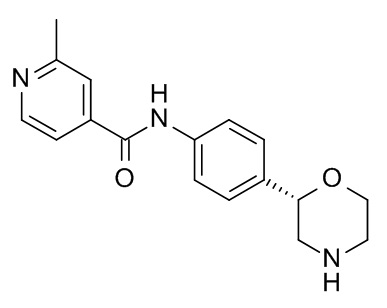
Указанное в заголовке соединение получали аналогично Примеру 1, используя 2-метил-изоникотиновую кислоту (CAS RN: 4021-11-8) вместо 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты на стадии (a). Белое твердое вещество. MS (ISP): 298,2 ([M+H]+).
Пример 18 (приведенный для сравнения пример).
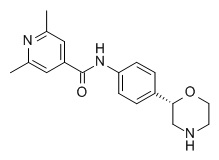
2,6-диметил-N-[4-[(2S)-морфолин-2-ил]фенил]пиридин-4-карбоксамид
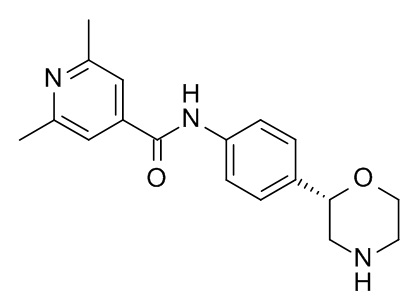
Указанное в заголовке соединение получали аналогично Примеру 1, используя 2,6-диметил-изоникотиновую кислоту (CAS RN: 54221-93-1) вместо 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты на стадии (a). Белое твердое вещество. MS (ISP): 312,2 ([M+H]+).
Пример 19 (приведенный для сравнения пример).
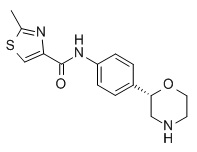
N-[4-[(2S)-Морфолин-2-ил]фенил]-2-метил-1,3-тиазол-4-карбоксамид
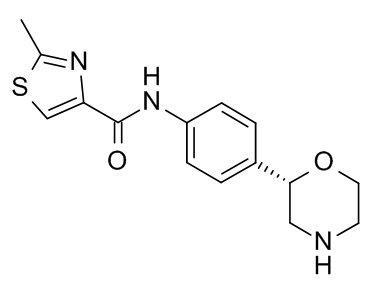
Указанное в заголовке соединение получали аналогично Примеру 1, используя 2-метилтиазол-4-карбоновую кислоту (CAS RN: 35272-15-2) вместо 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты на стадии (a). Белое твердое вещество. MS (ISP): 304,1 ([M+H]+).
Пример 20 (приведенный для сравнения пример)
N-[4-[(2S)-Морфолин-2-ил]фенил]-1-фенилпиразол-3-карбоксамид
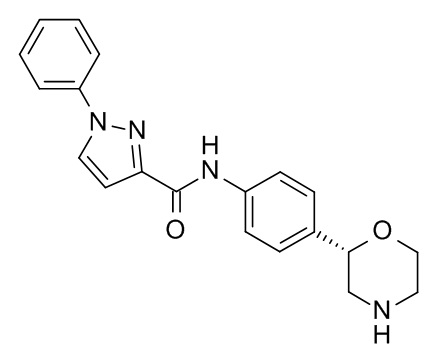
Указанное в заголовке соединение получали аналогично Примеру 1, используя 1-фенил-1H-пиразол-3-карбоновую кислоту (CAS RN: 4747-46-0) вместо 5-этил-4-метил-1H-пиразол-3-карбоновой кислоты на стадии (a). Белое твердое вещество. MS (ISP): 349,2 ([M+H]+).
Как упоминалось выше, соединение по данному изобретению обладает существенными преимуществами по сравнению с соединениями, известными на уровне техники, в частности, выраженной активностью агониста в отношении человеческого рецептора TAAR1, селективностью в отношении дофаминового переносчика (DAT), селективностью в отношении ионного канала hERG и низкой амфифильностью, и поэтому представляют низкий риск развития лекарственно-индуцированного фосфолипидоза (ЛИФ) (см. ниже).
Приведенные ниже сравнительные данные и комментарии позволяют продемонстрировать преимущества соединений формулы I по сравнению с соединениями, известными на уровне техники.
1. Фармакологические эффекты и терапевтический потенциал парциальных агонистов человеческого ассоциированного со следовыми аминами рецептора 1 (hTAAR1)
Существуют свидетельства существенных межвидовых различий во взаимодействиях лиганд-рецептор между TAAR1 человека и грызунов[1]. Таким образом, при выборе соединений для применения у человека в качестве лекарственных средств для лечения связанных с TAAR1 заболеваний важно отдавать предпочтение соединениям-кандидатам, основываясь на их функциональной активности в отношении человеческой формы рецептора TAAR1 (hTAAR1). hTAAR1 представляет собой связанный с G-белком трансмембранный рецептор (GPCR, от англ. G protein-coupled receptor), благодаря чему лиганды могут служить антагонистами, агонистами, парциальными агонистами или обратными агонистами рецептора. Соединение формулы I и приведенные для сравнения примеры исследовали in vitro по их функциональной активности в отношении hTAAR1, и обнаружили, что соединение формулы I является парциальным агонистом hTAAR1. Экспериментально установленные значения hTAAR1 EC50 для соединения формулы I и ряда приведенных для сравнения примеров показаны в Таблице 1 (см. ниже). Обнаружили, что соединение по Примеру 1, в частности, является мощным парциальным агонистом hTAAR1 in vitro.
Электрофизиологические эксперименты ex vivo с вентральной областью покрышки и дорсальным ядром шва показали, что парциальные агонисты TAAR1 усиливали частоту спайкового разряда DA и 5-HT нейронов у мышей дикого типа[2,3], тогда как полные агонисты, такие как пара-тирамин, снижали частоту спайкового разряда[3,4]. При этом продемонстрировали, что как полные, так и парциальные агонисты обладают защитными свойствами против подкрепляющего и усиливающего эффекта психостимулятора кокаина[5]. Тогда как полные агонисты, такие как амфетамин, индуцируют отрицательную обратную связь и купируют свое собственное влияние на DA и 5-HT системы[6,7], парциальные агонисты могут усиливать свое влияние на передачу сигнала нейронами, увеличивая частоту спайкового разряда, опосредовано через TAAR1. Эти данные, а также сведения о том, что фармакологические эффекты парциальных агонистов TAAR1 in vivo у грызунов более разнообразны по сравнению с полными агонистами[3,8], большое количество доклинических данных позволяет предположить, что парциальные агонисты TAAR1 имеют большой потенциал для применения у человека в качестве лекарственных средств для лечения заболеваний ЦНС, включая шизофрению, биполярное расстройство, депрессию, болезнь Паркинсона, а также для лечения пристрастия к алкоголю и лекарственной зависимости, но не ограничиваясь ими.
Например, предполагают, что парциальные агонисты TAAR1 превосходят существующие атипичные антипсихотические лекарственные средства, демонстрируя эффективность в качестве антипсихотических средств, при этом улучшая когнитивные функции и настроение, а также вызывая меньше побочных эффектов (например, не вызывают метаболический синдром, в отличие от существующих антипсихотических средств)[3,8]. В других публикациях возможные показания к применению включают биполярное расстройство,[8] лекарственную зависимость[5,9] и диабет[10].
Список литературы:
[1] Simmler, L. D.; Buchy, D.; Chaboz, S.; Hoener, M.C.; Liechti, M. E.; “In vitro characterization of psychoactive substances at rat, mouse and human trace amine-associated receptor 1”. J. Pharmacol. Exp. Ther. 2016, Fast Forward article DOI: 10.1124/jpet.115.229765;
[2] Bradaia, A. et al.; “The selective antagonist EPPTB reveals TAAR1-mediated regulatory mechanisms in dopaminergic neurons of the mesolimbic system”. Proc. Nat. Acad. Sci. USA 2009, 106, 20081-20086;
[3] Revel, F. G. et al.; “Trace amine-associated receptor 1 partial agonism reveals novel paradigm for neuropsychiatric therapeutics”. Biol. Psychiatry 2012, 72, 934-942;
[4] Revel, F. G. et al.; “TAAR1 activation modulates monoaminergic neurotransmission, preventing hyperdopaminergic and hypoglutamatergic activity”. Proc. Nat. Acad. Sci. USA 2011, 108, 8485-8490;
[5] Pei, Y.; Mortas, P.; Hoener, M. C.; Canales, J. J.; “Selective activation of the trace amine-associated receptor 1 decreases cocaine’s reinforcing efficacy and prevents cocaine-induced changes in brain reward thresholds”. Prog. Neuro-Psychopharmacol. & Biol. Psychiatry 2015, 63, 70-75;
[6] Lindemann, L. et al.; “Trace amine-associated receptor 1 modulates dopaminergic activity”. J. Pharmacol. Exp. Ther. 2008, 324, 948-956;
[7] Di Cara, B. et al.; “Genetic deletion of trace amine 1 receptors reveals their role in auto-inhibiting the actions of ecstasy (MDMA)”. J. Neuroscience 2011, 31, 16928-16940;
[8] Revel, F. G. et al.; “A new perspective for schizophrenia: TAAR1 agonists reveal antipsychotic- and antidepressant-like activity, improve cognition and control body weight”. Mol. Psychiatry 2013, 18, 543-556;
[9] Pei, Y.; Lee, J.; Leo, D.; Gainetdinov, R. R.; Hoener, M. C.; Canales, J. J.; “Activation of the trace amine-associated receptor 1 prevents relapse to cocaine seeking”. Neuropsychopharmacology 2014, 39, 2299-2308;
[10] Raab, S. et al.; “Incretin-like effects of small molecule trace amine-associated receptor 1 agonists”. Mol. Metabolism 2016, 5, 47-56.
1. Дофаминовый переносчик (DAT) и связанные с ним злоупотребление лекарственными средствами и/или аддиктивный потенциал
Психостимуляторные эффекты, аддиктивные механизмы и злоупотребление некоторыми психостимуляторными средствами, такими как кокаин и MDPV, обусловлено в том числе фармакологическим воздействием на дофаминовый переносчик (DAT, от англ. dopamine transporter).[1-3] Таким образом, для нового лекарственного средства, предназначенного для применения у человека, желательно избежать ингибирования или взаимодействия с дофаминовым переносчиком DAT для минимизации риска злоупотребления или возможной аддикции.
Например, существуют данные, что подкрепляющие эффекты кокаина зависят от его способности быстро блокировать дофаминовый переносчик (DAT). В экспериментах на животных также наблюдается самовведение ингибиторов обратного захвата дофамина отличных от кокаина, с относительной эффективностью, которая обычно положительно коррелирует с их эффективностью ингибирования DAT, но не переносчиков серотонина или норадреналина (SERT, NET)[4-8]. Животные, обученные самовведению кокаина, будут также осуществлять самовведение прямых агонистов дофаминовых рецепторов[9-11]. Кроме того, разрушение дофаминовых нервных окончаний может привести к угашению самовведения кокаина[12,13], и этот эффект наблюдается даже при сохранении поведенческой реакции на другие подкрепляющими агенты[14,15]. У человека индуцированный кокаином «подъем» коррелирует с загруженностью DAT в головном мозге[16].
Для дальнейшей проверки гипотезы о том, что DAT играет важную роль для подкрепляющих эффектов кокаина, создали функциональный, но «кокаин-нечувствительный» DAT и экспрессировали его у мышей[17,18]. Этот мутантный DAT демонстрировал аффинность к кокаину в 89 раз ниже по сравнению с DAT дикого типа, и кокаин не повышал внеклеточный дофамин в центре удовольствия и не усиливал локомоторную активность, стереотипию или предпочтение мест введения психоактивного вещества у мышей с нок-ином (DATki), экспрессирующих указанный мутантный DAT[18-20]. Кроме того, кокаин не оказывал положительного подкрепляющего эффекта у указанных мышей DATki, тогда как пища, дексамфетамин и прямой агонист дофаминовых рецепторов стабильно поддерживали оперантное поведение у этих мышей на уровне, сопоставимом с мышами дикого типа[21]. Повторное введение чувствительного к кокаину DAT дикого типа в участки головного мозга, включая в том числе вентральную область покрышки (VTA, от англ. ventral tegmental area), приводило к восстановлению «награждаемого» кокаином поведения у мышей DATki[22]. В заключение, способность кокаина блокировать DAT достаточна для устранения его подкрепляющих эффектов у мышей, что служит веским доказательством того, что блокада DAT имеет ключевое значение для подкрепляющих эффектов кокаина.
Таким образом, эти данные в совокупности позволяют предположить, что для новых лекарственных средств, предназначенных для применения у человека, весьма актуально избегать ингибирования или взаимодействия с дофаминовым переносчиком DAT для минимизации риска злоупотребления или возможной аддикции.
Значения DAT Ki, экспериментально установленные in vitro для ряда соединений TAAR1, приведены в Таблице 1 (см. ниже). Неожиданно обнаружили, что соединение по Примеру 1 является существенно более слабым лигандом DAT, чем другие соединения, при этом одновременно является мощным парциальным агонистом hTAAR1, следовательно, индекс селективности hTAAR1/DAT у соединения по Примеру 1 существенно выше, чем у других соединений.
Список литературы:
[1] Meil, W. M. and Boja, J. W.; “The dopamine transporter and addiction.” Chapter 1, pp. 1-21 in Neurochemistry of Abused Drugs (Karch, S. B., Ed.), CRC Press (2008);
[2] Simmler, L. D. et al.; “Pharmacological characterization of designer cathinones in vitro”. Brit. J. Pharmacol. 2013, 168, 458-470;
[3] Baumann, M. H.; Partilla, J. S.; Lehner, K. R.; “Psychoactive ‘bath salts’: not so soothing”. Eur. J. Pharmacol. 2013, 698, 1-5;
[4] Ritz., M. C.; Lamb, R. J.; Goldberg, S. R.; Kuhar, M. J.; “Cocaine receptors on dopamine transporters are related to self-administration of cocaine”. Science 1987, 237, 1219-1223;
[5] Bergmann., J.; Madras, B. K.; Johnson, S. E.; Spealman, R. D.; “Effects of cocaine and relared drugs in nonhuman primates. III. Self-administration by squirrel monkeys”. J. Pharmacol. Exp. Ther. 1989, 251, 150-155;
[6] Howell., L. L. & Byrd, L. D.; “Serotonergic modulation of the behavioural effects of cocaine in the squirrel monkey”. J. Pharmacol. Exp. Ther. 1995, 275, 1551-1559;
[7] Roberts, D. C. S. et al.; “Self-administration of cocaine analogs by rats”. Psychopharmacology 1999, 144, 389-397;
[8] Wee, S. et al.; “Relationship between serotonergic activity and reinforcing effects of a series of amphetamine analogs”. J. Pharmacol. Exp. Ther. 2005, 313, 848-854;
[9] Woolverton, W. L.; Goldberg, L. I.; Ginos, J. Z.; “Intravenous self-administration of dopamine receptor agonists by Rhesus monkeys”. J. Pharmacol. Exp. Ther. 1984, 230, 678-683;
[10] Wise, R. A.; Murray, A.; Bozarth, M. A.; “Bromocriptine self-administration and bromocriptine-reinstatement of cocaine-trained and heroin-trained lever pressing in rats”. Psychopharmacology 1990, 100, 355-360;
[11] Caine, S. B. & Koob, G. F.; “Modulation of cocaine self-administration in the rat through D-3 dopamine receptors”. Science 1993, 260, 1814-1816;
[12] Roberts, D. C. S.; Corcoran, M. E.; Fibiger, H. C.; “On the role of ascending catecholaminergic systems in intravenous self-administration of cocaine”. Pharmacol. Biochem. Behaviour 1977, 6, 615-620;
[13] Roberts, D. C. S.; Koob, G. F.; Klonoff, P.; Fibiger, H. C.; “Extinction and recovery of cocaine self-administration following 6-hydroxydopamine lesions of the nucleus accumbens”. Pharmacol. Biochem. Behaviour 1980, 12, 781-787;
[14] Pettit, H. O.; Ettenberg, A.; Bloom, F. E.; Koob, G. F.; “Destruction of dopamine in the nucleus accumbens selectively attenuates cocaine but not heroin self-administration in rats”. Psychopharmacology 1984, 84, 167-173;
[15] Caine, S. B. & Koob, G. F.; “Effects of mesolimbic dopamine depletion on responding maintained by cocaine and food”. J. Exp. Anal. Behavior 1994, 61, 213-221;
[16] Volkow, N. D. et al.; “Relationship between subjective effects of cocaine and dopamine transporter occupancy”. Nature 1997, 386, 827-830;
[17] Chen, R.; Han, D. D.; Gu, H. H.; “A triple mutation in the second transmembrane domain of mouse dopamine transporter markedly decreases sensitivity to cocaine and methylphenidate”. J. Neurochem. 2005, 94, 352-359;
[18] Chen, R. et al.; “Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter”. Proc. Nat. Acad. Sci. USA 2006, 103, 9333-9338;
[19] Tilley, M. R. & Gu, H. H.; “Dopamine transporter inhibition is required for cocaine-induced stereotypy”. Neuroreport 2008, 19, 1137-1140;
[20] Tilley, M. R.; O’Neill, B.; Han, D.D.; Gu, H. H.; “Cocaine does not produce reward in absence of dopamine transporter inhibition”. Neuroreport 2009, 20, 9-12;
[21] Thomsen, M.; Han, D.D.; Gu, H. H.; Caine, S. B.; “Lack of cocaine self-administration in mice expressing a cocaine-insensitive dopamine transporter”. J. Pharmacol. Exp. Ther. 2009, 331, 204-211;
[22] Wu, H. et al.; “Restoration of cocaine stimulation and reward by reintroducing wild type dopamine transporter in adult knock-in mice with a cocaine-insensitive dopamine transporter”. Neuropharmacology 2014, 86, 31-37.
3. Блокада hERG и связанное с ней удлинение интервала QT
Минимизация вероятности вызывать побочные эффекты со стороны сердца весьма актуальна для терапевтического агента, предназначенного для применения у человека, особенно для лекарства, предназначенного для долгосрочного применения. За последние годы регулирующие органы отложили одобрение или наложили ограничения на применение или даже запретили, или отозвали с рынка терапевтические агенты, удлиняющие интервал QT на кардиограмме. Интервал QT представляет собой время от начала комплекса QRS до конца волны T на электрокардиограмме (ЭКГ) и является мерой продолжительности деполяризации и реполяризации желудочков. Лекарства, удлиняющие интервал QT, связывают с полиморфной желудочковой тахикардией типа «пируэт» (Torsades de Pointes, TdP). Данная аритмия может вызывать серьезные кардиологические исходы и может прогрессировать в необратимую фибрилляцию желудочков с летальным исходом. Руководящий нормативный документ международного общества кардиологов ICH S7B[1] рекомендует общий комплекс мер для доклинической оценки сердечно-сосудистого риска новых химических соединений (NME, от англ. new molecular entities), включающую исследование IKr [калиевого тока каналов hERG] in vitro. Установили, что ингибирование hERG каналов является главным механизмом удлинения QT.[2] Таким образом, на доклиническом этапе минимальный рекомендуемый комплекс мер по снижению риска удлинения интервала QT заключается в исследовании влияния репрезентативных соединений из заданной группы химических соединений на активность hERG каналов in vitro.[3] Задачей является отбор соединений, ингибирующих hERG каналы не более чем на 20% в концентрациях, которые по меньшей мере в 30 раз ниже их эффективной концентрации in vitro (или in vivo, при наличии данных), необходимой для реализации их терапевтической активности. В случае агонистов TAAR1 соответствующей концентрацией in vitro, позволяющей предсказать терапевтическую активность, можно считать hTAAR1 EC50 (см. выше). Таким образом, желательно выбрать агонисты TAAR1, у которых соотношение hERG IC20/hTAAR1 EC50 составляет по меньшей мере 30.
Значения hERG IC20 и IC50 экспериментально установленные in vitro для ряда соединений TAAR1, приведены в Таблице 1 (см. ниже).
Основные соединения отличаются своей способностью вызывать ингибирование канала hERG.[4] Все соединения TAAR1 несут одинаковую морфолиновую концевую группу, поэтому полагают, что все соединения будут в одинаковой степени основными. Основная группировка необходима для агонистической активности в отношении hTAAR1. Неожиданно обнаружили, что соединение по Примеру 1 является существенно более слабым ингибитором hERG каналов, чем приведенные для сравнения соединения, следовательно, соотношение hERG IC20/hTAAR1 EC50 для соединения по Примеру 1 существенно выше, чем рекомендуемый 30-кратный минимум.
Список литературы:
[1] ICH Guideline. “The nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals (S7B)” issued as CPMP/ICH/423/02, adopted by CHMP in May 2005; http://www.ich.org/products/guidelines/safety/safety-single/article/the-non-clinical-evaluation-of-the-potential-for-delayed-ventricular-repolarization-qt-interval-pro.html
[2] Redfern, W. S.; Carlsson, L.; Davis, A. S.; Lynch, W. G.; MacKenzie, I.; Palethorpe, S.; Siegl, P.K.; Strang, I; Sullivan, A. T.; Wallis, R.; Camm, A.J.; Hammond, T. G.; “Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development”. Cardiovasc. Res. 2003, 58, 32-45;
[3] Helliwell, R. M.: “Potassium Channels”. Methods in Molecular Biology (Totowa, NJ, United States) 2008, 491, 279-295;
[4] Zolotoy, A. B.; Plouvier, B. P.; Beatch, G. B.; Hayes, E. S.; Wall, R. A.; Walker, M. J. A.; “Physicochemical determinants for drug-induced blockade of hERG potassium channels: Effect of charge and charge shielding”. Curr. Med. Chem. - Cardiovascular & Hematological Agents 2003, 1, 225-241.
4. Амфифильность и связанный с ней лекарственно-индуцированный фосфолипидоз (ЛИФ)
Фосфолипидоз (ФЛ) представляет собой лизосомальную болезнь накопления, характеризующуюся избыточным накоплением фосфолипидов в тканях[1][2][3]. Известно, что многие катионные амфифильные лекарственные средства, включая антидепрессанты, обезболивающие, противомалярийные и снижающие холестерин агенты вызывают лекарственно-индуцированный фосфолипидоз (ЛИФ) у животных и человека. Механизм ЛИФ включает захват или селективное поглощение ассоциированных с ЛИФ лекарственных средств в лизосомы и кислые везикулы пораженных клеток. Захват лекарств сопровождается постепенным накоплением комплексов лекарство-фосфолипид во внутренних мембранах лизосом. Нарастание нерасщепленных веществ приводит к аномальному накоплению мультиламелярных телец (миелоидных телец) в тканях. Несмотря на то, что фосфолипидоз преимущественно считают болезнью накопления, известно, что в случае некоторых соединений нарушение накопления ассоциировано с воспалением и некрозом, приводящими к функциональным нарушениям пораженных тканей. Таким образом, весьма актуально, чтобы лекарственное средство не создавало риск ЛИФ. Это особенно актуально для лекарственных средств, предназначенных для долгосрочного применения, например, лекарственных средств, предназначенных для лечения хронических психиатрических расстройств, таких как шизофрения, биполярное расстройство или депрессия, или лекарственных средств, предназначенных для лечения хронических метаболических расстройств, таких как диабет.
ЛИФ представляет собой побочный эффект, в частности связанный с катионными амфифильными лекарственными средствами (CAD, от англ. cationic amphiphilic drugs).[4] Во избежание ЛИФ должна быть снижена либо pKa основания (pKa основания < 6,3), либо амфифильность (ΔΔGam > - 6 кДж моль-1) соединения (т.е. ΔΔGam должна быть увеличена).[5] Соединение считают DIPL-негативным, либо если значение pKa основания ниже 6,3, либо если амфифильность выше ΔΔGam = -6 кДж моль-1. Амфифильность заданного соединения можно рассчитать при помощи компьютерного моделирования непосредственно по молекулярной структурной формуле[6], следовательно предсказанный риск ЛИФ для этого соединения также можно рассчитать при помощи компьютерного моделирования[5], при этом в алгоритме предсказания используют классификацию риска ЛИФ, который определяют согласно следующим критериям на основе параметров, полученных на обучающей выборке, включающей экспериментально полученные результаты по фосфолипидозу для большой выборки соединений:
Амфифильность > -5,0 кДж/моль и BPKA1 <= 5,6 предсказывают отсутствие риска ЛИФ;
-7,0 кДж/моль < Амфифильность < -5,0 кДж/моль и/или 7,0 > BPKA1 > 5,60 предсказывают пограничный риск ЛИФ;
Амфифильность < -7,0 кДж/моль и BPKA1 >= 7,00 предсказывают риск возникновения ЛИФ.
Рассчитанные значения амфифильности (ΔΔGam в кДж моль-1), а также предсказание риска ЛИФ методами компьютерного моделирования (отрицательный/пограничный/положительный) для ряда соединений TAAR1 приведены в Таблице 1 (см. ниже).
Все соединения TAAR1 несут одинаковую морфолиновую концевую группу, следовательно, pKa основания у всех соединений очень схожи и явно превышают 6,3. Основная группировка необходима для агонистической активности в отношении hTAAR1. Таким образом, единственным способом избежать ЛИФ является снижение липофильности остова молекул. Неожиданно обнаружили, что у соединения по Примеру 1 липофильность снижена существенно больше, чем это можно было ожидать на основании результатов, полученных для аналогичных соединений и, таким образом, амфифильность соединения по Примеру 1 отчетливо снижена и, следовательно, согласно прогнозу, данное соединение не будет вызывать ЛИФ.
Список литературы:
[1] Anderson, N.; Borlak, J.; “Drug-induced phospholipidosis”. FEBS Lett. 2006, 580, 5533-540;
[2] Reasor, M. J.; Hastings, K. L.; Ulrich, R. G.; “Drug-induced phospholipidosis: issues and future directions”. Expert Opin. Drug Safety 2006, 5, 567-83;
[3] Nonoyama, T.; Fukuda, R.; “Drug induced phospholipidosis pathological aspects and its prediction”. J. Toxicol. Pathol. 2008, 21, 9-24;
[4] Lullmann, H.; Lullmann-Rauch, R.; Wassermann, O.; “Lipidosis induced by amphiphilic cationic drugs.” Biochem. Pharmacol. 1978, 27, 1103-8;
[5] Fischer, H.; Atzpodien, E. A.; Csato, M; Doessegger, L.; Lenz, B.; Schmitt, G.; Singer, T.; “In silico assay for assessing phospholipidosis potential of small drug like molecules: training, validation and refinement using several datasets.” J. Med. Chem. 2012, 55, 126-139;
[6] Fischer, H.; Kansy, M.; Bur, D.; “CAFCA: a novel tool for the calculation of amphiphilic properties of charged drug molecules”. Chimia 2000, 54, 640-645.
Соединения исследовали в тестах, приведенных ниже.
Материалы и методы
Человеческий TAAR1
Для конструирования экспрессионных плазмид кодирующие последовательности человеческого TAAR 1 амплифицировали из геномной ДНК, по существу, как описано Lindemann et al. [14]. Использовали систему высокоточной полимеразной цепной реакции (ПЦР) с корректирующей экзонуклеазной активностью (Roche Diagnostics) при концентрации ионов Mg2+ 1,5 мМ и клонировали очищенные ПЦР-продукты в клонирующий вектор pCR2.1-TOPO (Invitrogen), следуя инструкциям производителя. Продукты ПЦР субклонировали в вектор pIRESneo2 (BD Clontech, Palo Alto, California) и верифицировали последовательности экспрессирующих векторов перед внедрением в клеточные линии.
Клетки HEK293 (ATCC # CRL-1573) культивировали по существу, как описано Lindemann et al. (2005). Для получения стабильно трансфицированных клеточных линий клетки HEK293 трансфицировали экспрессирующими плазмидами pIRESneo2, содержащими кодирующие последовательности TAAR (описанные выше) с применением липофектамина 2000 (Invitrogen) согласно инструкциям производителя и через 24 ч после трансфекции в культуральную среду добавляли 1 мг/мл G418 (Sigma, Buchs, Switzerland). После культивирования в течение приблизительно 10 суток выделяли клоны, размножали и тестировали по их способности реагировать на следовые амины (все соединения приобретали в компании Sigma) с помощью системы иммуноферментного анализа (ИФА) cAMP Biotrak (Amersham), используя методику ИФА без ацетилирования, предложенную производителем. Моноклональные клеточные линии, демонстрирующие стабильную EC50 для культивирования в течение 15 пассажей, использовали для всех последующих исследований.
Определение цАМФ проводили, как описано ранее (Revel et al., Proc. Natl. Acad. Sci. USA 2011, 108, 8485-8490). Вкратце, клетки, экспрессирующие человеческий TAAR1, высаживали в 96-луночные планшеты (BIOCOAT 6640; Becton Dickinson, Allschwil, Switzerland) и инкубировали в течение 20 ч при 37°C. Перед стимуляцией клеток агонистами в широком диапазоне концентраций в течение 30 мин при 37°C клетки промывали ФСБ и предварительно инкубировали с ФСБ, содержащим 1 мМ 3-изобутил-1-метилксантина, в течение 10 мин при 37°C и 5% CO2. Стимуляцию 0,2% DMSO принимали за исходный уровень, а влияние 30 мкМ β-PEA принимали за максимальный ответ. Затем клетки лизировали и проводили определение цАМФ согласно инструкциям производителя (набор cAMP; Upstate/Millipore, Schaffhausen, Switzerland). Наконец, сигнал в планшетах считывали при помощи люминометра (1420 Multilabel counter; PerkinElmer, Schwerzenbach, Switzerland) и рассчитывали количество цАМФ. Получали результаты по меньшей мере в трех независимых экспериментах. Эксперименты проводили в дупликатах или в трипликатах. Значения EC50 представлены как среднее ± стандартное отклонение (в мкМ). В данных по функциональной активности в отношении TAAR1 значение Emax характеризует степень функциональной активности относительно 100% для эндогенного лиганда и полного агониста β-PEA.
Человеческий DAT
Связывание с дофаминовым переносчиком (DAT) in vitro. Культивировали эмбриональные клетки почки человека (HEK, от англ. Human embryonic kidney) 293 (Invitrogen, Zug, Switzerland), стабильно трансфицированные DAT. Клетки собирали и промывали три раза фосфатно-солевым буфером (ФСБ). Осадок замораживали при -80°C. Затем осадок ресуспендировали в 400 мл 20 мМ HEPES-NaOH, pH 7,4, содержавшем 10 мМ EDTA при 4°C. После гомогенизации с применением Polytron (Kinematica, Lucerne, Switzerland) при 10000 оборотов в минуту (об/мин) в течение 15 с гомогенаты центрифугировали при 48000 x g в течение 30 мин при 4°C. Аликвоты препаратов мембран замораживали при -80°C. Исследуемые соединения разводили в 20 мл буфера для связывания (252 мМ NaCl, 5,4 мМ KCl, 20 мМ Na2HPO4, 3,52 мМ KH2PO4, pH 7,4), делали разведения для построения кривых разведений по 10 точкам и переносили в 96-луночные белые планшеты из полистирола (Sigma-Aldrich, Buchs, Switzerland). [3H]-WIN35,428 (~86 Ки/ммоль; Perkin-Elmer) служил в качестве радиоактивного лиганда для определения DAT и имел Kd 12 нМ. В каждую лунку планшета для определения DAT добавляли 50 мкл [3H]-WIN35,428 (концентрация ~40 нМ) для получения конечной концентрации [3H]-WIN35,428 10 нМ. Для определения общего связывания в аналитическом планшете использовали 20 мкл связывающего буфера, а неспецифическое связывание определяли в присутствии 10 мкМ индатралина. Замороженные препараты мембран, несущих DAT, размораживали и ресуспендировали до концентрации приблизительно 0,04 мг белка/мл связывающего буфера (разведение 1:1 в H2O) с использованием гомогенизатора тканей polytron. Затем гомогенаты мембран (40 μг/мл) осторожно перемешивали в течение 5-30 мин с поливинилтолуоловыми частицами (PCT) для сцинтилляционного анализа сближения, покрытыми агглютинином из проростков пшеницы (WGASPA; Amersham Biosciences), в соотношении 7,7 мг частиц/мл гомогената. В каждую лунку аналитического планшета, содержавшую радиоактивный лиганд и исследуемые соединения, добавляли 130 мкл смеси мембраны/частицы (итоговый объем в каждой лунке составлял 200 мкл) и инкубировали приблизительно 2 ч при комнатной температуре при встряхивании. Затем регистрировали импульсы в аналитических планшетах при помощи счетчиков Packard Topcount в режиме измерений PVT SPA. Для определения общего количества импульсов радиоактивного лиганда, добавленного в соответствующих тестах, регистрировали радиоактивность 50 мкл матричного раствора [3H]-WIN35428 в 5 мл сцинтилляционного коктейля ReadySafe (Beckman Industries) при помощи жидкостного сцинтилляционного счетчика Packard 1900CA. Для аппроксимации данных сигмоидными кривыми и определения значений IC50 связывания и захвата использовали нелинейную регрессию. Значения Ki связывания и захвата рассчитывали с использованием уравнения Ченга-Прусоффа: Ki = IC50/(1+ [S]/Km).
Человеческий ERG (hERG)
Для исследования влияния тестируемых соединений на человеческие калиевые каналы, кодируемые геном hERG (от англ. ether-a-go-go related gene), использовали метод локальной фиксации потенциала в конфигурации «целая клетка» на стабильно трансфицированных клетках CHO при температурах близких к физиологическим (36 ± 1°C). Влияние соединений на показатели тока ионов K+ в hERG каналах оценивали в 4 концентрациях (0,3-3-30-300 мкМ) по меньшей мере на 3 клетках CHO, стабильно экспрессирующих hERG каналы. Для электрофизиологических экспериментов клетки высаживали в 35 мм стерильные культуральные чашки, содержащие 2 мл культуральной среды без гигромицина B. Клетки культивировали в плотности, позволяющей проводить измерения на одиночных клетках (без видимых контактов с соседними клетками). Клетки инкубировали во влажной атмосфере при 37°C с 5% CO2 (относительная влажность приблизительно 95%). Клетки непрерывно поддерживали и делали пассажи в стерильных культуральных флаконах, содержащих питательную среду F-12 (DMEM/F-12 с L-глутамином) с добавлением 10% эмбриональной телячьей сыворотки и 10% раствора пенициллина/стрептомицина. Ежедневно по меньшей мере три клетки обрабатывали селективным блокатором калиевого тока задержанного выпрямления IKr (E-4031, референтное вещество) для обеспечения точных результатов. 35 мм культуральные чашки, в которые клетки были посажены в плотности, обеспечивающей возможность регистрации потенциала на одиночных клетках, помещали на предметный стол микроскопа и осуществляли непрерывную перфузию (со скоростью приблизительно 1 мл/мин) омывающей жидкостью (хлорид натрия 150 мМ, хлорид калия 4 мМ, хлорид кальция 1,2 мМ, хлорид магния 1 мМ, HEPES 10 мМ, pH (NaOH) 7,4) при температуре, близкой к физиологической (36 ± 1°C)). После формирования между подведенными электродами и отдельными клетками CHO, стабильно трансфицированными hERG, контакта, имеющего гигаомное сопротивление (диапазон сопротивления пипетки: 2,0 МОм - 7,0 МОм; диапазон сопротивления в месте соединения пипетки с мембраной: > 1 ГОм), клеточную мембрану вокруг кончика пипетки разрушали для обеспечения доступа во внутриклеточное пространство (конфигурация «целая клетка»). Если качество контакта было неудовлетворительным, процесс формирования контакта повторяли с другой клеткой и новой пипеткой. После формирования стабильного контакта измеряли выходящие следовые токи, проводящиеся hERG каналами, при деполяризации клеточной мембраны до -40 мВ в течение 50 мс и затем до +20 мВ в течение 500 мс (активация каналов) от поддерживаемого потенциала -80 мВ и при последующей реполяризации до -40 мВ в течение 500 мс. Подачу потенциалов в данном режиме осуществляли по меньшей мере 10 раз с интервалами 10 с. Если считали, что плотность тока недостаточна для измерений, для регистрации использовали другую клетку. После завершения контрольных измерений осуществляли непрерывную перфузию клеток омывающей жидкостью, содержащей исследуемые соединения. Во время закачивания исследуемых соединений продолжали подавать потенциалы в указанном выше режиме с интервалами 10 с до достижения устойчивого состояния блокирования. 4 исследуемые концентрации соединения добавляли к 3 клеткам последовательно. Исследуемые соединения ингибировали следовые токи через hERG канал, строили кривую концентрация-эффект и рассчитывали значение IC50. На основе значений IC50 рассчитывали IC20. Каждую концентрацию исследуемого соединения анализировали в трех экспериментах (n=3).
Расчет амфифильности (ΔΔ Gam) и предсказание риска ЛИФ методом компьютерного моделирования
Определение амфифильности (ΔΔ Gam) и предсказание риска ЛИФ методом компьютерного моделирования проводили по молекулярной структурной формуле соединения формулы I и приведенных для сравнения соединений согласно опубликованным алгоритмам (Fischer, H.; Kansy, M.; Bur, D.; “CAFCA: a novel tool for the calculation of amphiphilic properties of charged drug molecules”. Chimia 2000, 54, 640-645; Fischer, H.; Atzpodien, E. A.; Csato, M; Doessegger, L.; Lenz, B.; Schmitt, G.; Singer, T.; “In silico assay for assessing phospholipidosis potential of small drug like molecules: training, validation and refinement using several datasets.” J. Med. Chem. 2012, 55, 126-139).
Как показано в Таблице 1, соединение формулы I обладает активностью парциального агониста в отношении hTAAR1 (EC50 в мкМ), аффинностью связывания с hDAT (Ki в мкМ) и активностью блокатора канала hERG (IC20 и IC50 в мкМ). В Таблице 1 также приведены рассчитанная амфифильность (ΔΔGam в кДж моль-1) и оценка риска фосфолипидоза методом компьютерного моделирования (прогноз возникновения ЛИФ in vitro и ЛИФ in vivo отрицательный/положительный/пограничный) для соединения формулы I и соединений, приведенных для сравнения, рассчитанные с помощью процедуры, описанной выше.
Таблица 1
(мкМ)
(%)
IC50 (мкМ)
IC20 (мкМ)
(кДж/моль)
Ki (мкМ)



* активность агониста hTAAR1 в % оценена по шкале, где эффект эндогенного лиганда β-фенилэтиламина соответствует 100% агонизму.
Неожиданно обнаружили, что соединение формулы I (Пример 1) в целом демонстрирует наилучшее сочетание свойств, как следует из мощной агонистической активности в отношении hTAAR1, высокой селективности в отношении hDAT, высокой селективности в отношении hERG, низкой амфифильности и, следовательно, низкого риска развития фосфолипидоза по сравнению с другими соединениями TAAR1, известными на уровне техники. Данные в Таблице 1 свидетельствуют, что соединение по Примеру 1 обладает выраженной активностью парциального агониста в отношении hTAAR1 (EC50 = 0,059 мкМ), является высоко селективным в отношении hDAT (Ki = 27,5 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 471), является высоко селективным в отношении hERG (IC20 = 36,2 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 619) и имеет низкую амфифильность (ΔΔGam = - 3,47 кДж моль-1), которая существенно ниже порогового уровня, вызывающего опасения по поводу развития фосфолипидоза (методом компьютерного моделирования предсказывается отсутствие риска ЛИФ).
Данные в Таблице 1 свидетельствуют, что близкие аналоги соединения по Примеру 1 уступают по свойствам соединению по Примеру 1 с одной или нескольких точек зрения.
Например, приведенное для сравнения соединение по Примеру 2, являющееся R энантиомером соединения по Примеру 1, менее эффективно в отношении hTAAR1 (EC50 = 0,2632 мкМ), из чего следует, что абсолютная стереохимическая конфигурация S, как в Примере 1, является предпочтительной для достижения большей эффективности в отношении hTAAR1.
Приведенное для сравнения соединение по Примеру 3 существенно более эффективно в отношении hERG (IC20 = 1,97 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 52), а также обладает существенно более высокой амфифильностью (ΔΔGam = - 6,3 кДж моль-1), следовательно, предсказывается риск возникновения ЛИФ.
Приведенное для сравнения соединение по Примеру 4 существенно более эффективно в отношении DAT (Ki = 2,5 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 40), а также обладает более высокой амфифильностью (ΔΔGam = - 5,3 кДж моль-1), следовательно, риск возникновения ЛИФ пограничный.
Приведенное для сравнения соединение по Примеру 5 существенно более эффективно в отношении DAT (Ki = 1,5 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 23), а также обладает более высокой амфифильностью (ΔΔGam = - 5,9 кДж моль-1), следовательно, риск возникновения ЛИФ пограничный.
Приведенное для сравнения соединение по Примеру 6 существенно более эффективно в отношении hERG (IC20 = 0,38 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 5), более эффективно в отношении DAT (Ki = 5,9 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 74), а также обладает существенно более высокой амфифильностью (ΔΔGam = - 8,46 кДж моль-1), следовательно, предсказывается риск возникновения ЛИФ.
Приведенное для сравнения соединение по Примеру 7 существенно более эффективно в отношении hERG (IC20 = 3,57 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 43), существенно более эффективно в отношении DAT (Ki = 0,79 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 9), а также обладает существенно более высокой амфифильностью (ΔΔGam = - 7,41 кДж моль-1), следовательно, предсказывается риск возникновения ЛИФ.
Приведенное для сравнения соединение по Примеру 8 существенно более эффективно в отношении hERG (IC20 = 3,01 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 28).
Приведенное для сравнения соединение по Примеру 9 менее эффективно в отношении hTAAR1 (EC50 = 0,144 мкМ), существенно более эффективно в отношении hERG (IC20 = 1,14 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 8), существенно более эффективно в отношении DAT (Ki = 0,48 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 3), а также обладает существенно более высокой амфифильностью (ΔΔGam = - 8,83 кДж моль-1), следовательно, предсказывается риск возникновения ЛИФ.
Приведенное для сравнения соединение по Примеру 10 менее эффективно в отношении hTAAR1 (EC50 = 0,184 мкМ) и более эффективно в отношении hERG (IC20 = 5,78 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 31).
Приведенное для сравнения соединение по Примеру 11 менее эффективно в отношении hTAAR1 (EC50 = 0,203 мкМ), существенно более эффективно в отношении hERG (IC20 = 2,59 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 13), существенно более эффективно в отношении DAT (Ki = 2,33 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 12), а также обладает существенно более высокой амфифильностью (ΔΔGam = - 6,18 кДж моль-1), следовательно, предсказывается риск возникновения ЛИФ.
Приведенное для сравнения соединение по Примеру 12 менее эффективно в отношении hTAAR1 (EC50 = 0,212 мкМ), существенно более эффективно в отношении hERG (IC20 = 3,21 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 15), существенно более эффективно в отношении DAT (Ki = 1,78 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 8), а также обладает существенно более высокой амфифильностью (ΔΔGam = - 6,59 кДж моль-1), следовательно, предсказывается риск возникновения ЛИФ.
Приведенное для сравнения соединение по Примеру 13 существенно менее эффективно в отношении hTAAR1 (EC50 = 0,405 мкМ), более эффективно в отношении hERG (IC20 = 9,14 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 23), существенно более эффективно в отношении DAT (Ki = 1,31 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 3), а также обладает существенно более высокой амфифильностью (ΔΔGam = - 6,23 кДж моль-1), следовательно, предсказывается риск возникновения ЛИФ.
Приведенное для сравнения соединение по Примеру 14 менее эффективно в отношении hTAAR1 (EC50 = 0,447 мкМ), существенно более эффективно в отношении DAT (Ki = 7,32 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 16), а также обладает существенно более высокой амфифильностью (ΔΔGam = - 6,96 кДж моль-1), следовательно, предсказывается риск возникновения ЛИФ.
Приведенное для сравнения соединение по Примеру 15 менее эффективно в отношении hTAAR1 (EC50 = 0,663 мкМ), существенно более эффективно в отношении hERG (IC20 = 2,40 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 4), а также обладает существенно более высокой амфифильностью (ΔΔGam = - 7,4 кДж моль-1), следовательно, предсказывается риск возникновения ЛИФ.
Приведенное для сравнения соединение по Примеру 16 менее эффективно в отношении hTAAR1 (EC50 = 0,666 мкМ), существенно более эффективно в отношении hERG (IC20 = 3,35 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 5), а также обладает существенно более высокой амфифильностью (ΔΔGam = - 8,48 кДж моль-1), следовательно, предсказывается риск возникновения ЛИФ.
Приведенное для сравнения соединение по Примеру 17 существенно менее эффективно в отношении hTAAR1 (EC50 = 0,673 мкМ) и более эффективно в отношении DAT (Ki = 10,32 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 15).
Приведенное для сравнения соединение по Примеру 18 существенно менее эффективно в отношении hTAAR1 (EC50 = 0,827 мкМ), а также обладает более высокой амфифильностью (ΔΔGam = - 5,82 кДж моль-1), следовательно, предсказывается пограничный риск ЛИФ.
Приведенное для сравнения соединение по Примеру 19 менее эффективно в отношении hTAAR1 (EC50 = 1,025 мкМ) и более эффективно в отношении hERG (IC20 = 8,32 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 8).
Наконец, приведенное для сравнения соединение по Примеру 20 существенно менее эффективно в отношении hTAAR1 (EC50 = 2,48 мкМ), существенно более эффективно в отношении hERG (IC20 = 2,49 мкМ; индекс селективности по отношению к hTAAR1 EC50 = 1), а также обладает существенно более высокой амфифильностью (ΔΔGam = - 5,37 кДж моль-1), следовательно, предсказывается пограничный риск ЛИФ.
Таким образом, принимая во внимание все данные, приведенные в Таблице 1, соединение формулы I (Пример 1) в целом является наиболее предпочтительным соединением для предполагаемого применения в качестве безопасного и эффективного терапевтического агента для лечения TAAR1-ассоциированных расстройств у человека, особенно для лечения хронических расстройств ЦНС, таких как депрессия, тревожные расстройства, биполярное расстройство, синдром гиперактивности с дефицитом внимания (СГДВ), связанные со стрессом расстройства, психотические расстройства, такие как шизофрения, неврологические расстройства, такие как болезнь Паркинсона, нейродегенеративные расстройства, такие как болезнь Альцгеймера, эпилепсия, мигрень, гипертензия, злоупотребление веществами, аддикция, и метаболических расстройств, таких как нарушения пищевого поведения, диабет, диабетические осложнения, ожирение, дислипидемия, нарушения поглощения энергии и ассимиляции, расстройств и нарушений гомеостаза температуры тела, расстройств сна и циркадного ритма и сердечно-сосудистых расстройств. Наиболее предпочтительными расстройствами являются шизофрения, биполярное расстройство или депрессия.
Соединение формулы I и фармацевтически приемлемые соли соединения формулы I можно применять в качестве лекарственных средств, например, в форме фармацевтических препаратов. Фармацевтические препараты можно вводить перорально, например, в форме таблеток, таблеток с оболочкой, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий. Однако введение можно осуществлять и ректально, например, в форме суппозиториев, или парентерально, например, в форме инъекционных растворов.
Для получения фармацевтических препаратов соединение формулы I можно обрабатывать с фармацевтически инертными неорганическими или органическими носителями. В качестве таких носителей для таблеток, таблеток с оболочкой, драже и твердых желатиновых капсул можно применять, например, лактозу, кукурузный крахмал или его производные, тальк, стеариновую кислоту или ее соли и т.п. Подходящими носителями для мягких желатиновых капсул являются, например, растительные масла, воска, жиры, полутвердые и жидкие полиолы и т.п. Тем не менее, в зависимости от природы действующего вещества, в случае мягких желатиновых капсул носители обычно не требуются. Подходящими носителями для получения растворов и сиропов являются, например, вода, полиолы, глицерин, растительное масло и т.п. Подходящими носителями для суппозиториев являются, например, естественные или отвержденные масла, воска, жиры, полужидкие или жидкие полиолы и т.п.
Кроме того, фармацевтические препараты могут содержать консерванты, солюбилизаторы, стабилизаторы, увлажняющие агенты, эмульгаторы, подсластители, красители, корригенты, соли для изменения осмотического давления, буферы, маскирующие агенты или антиоксиданты. Они могут также содержать другие вещества, обладающие терапевтической ценностью.
Лекарственные средства, содержащие соединение формулы I или его фармацевтически приемлемую соль и терапевтически инертный носитель, также являются объектом данного изобретения, как и способ их получения, включающий приведение соединения формулы I и/или его фармацевтически приемлемой соли присоединения кислоты и, при желании, одного или более других веществ, обладающих терапевтической ценностью, в форму галенова препарата вместе с одним или более терапевтически инертными носителями.
Наиболее предпочтительными показаниями к применению в данном изобретении являются показания, включающие расстройства центральной нервной системы, например, лечение или предупреждение депрессии, шизофрении и биполярных расстройств.
Дозировка может варьировать в широких диапазонах и, конечно, должна быть отрегулирована по индивидуальным потребностям в каждом конкретном случае. В случае перорального введения доза для взрослых может варьировать от приблизительно 0,01 мг до приблизительно 1000 мг соединения общей формулы I или соответствующего количества его фармацевтически приемлемой соли в сутки. Суточную дозу можно вводить в виде однократной дозы или в нескольких дозах и, кроме того, верхний предел также может быть повышен, когда это считают показанным.


Способ изготовления
1. Смешивают ингредиенты 1, 2, 3 и 4 и гранулируют с очищенной водой.
2. Высушивают гранулы при 50°C.
3. Пропускают гранулы через подходящее оборудование для измельчения.
4. Добавляют ингредиент 5 и смешивают в течение трех минут; прессуют на подходящем прессе.

Способ изготовления
1. Смешивают ингредиенты 1, 2 и 3 в подходящем смесителе в течение 30 минут.
2. Добавляют ингредиенты 4 и 5 и смешивают в течение 3 минут.
3. Заполняют в подходящую капсулу.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ МОРФОЛИН-ПИРИДИНА | 2015 |
|
RU2690154C2 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛКАРБОКСАМИДА | 2013 |
|
RU2637938C2 |
| 2-ОКСА-5-АЗАБИЦИКЛО[2.2.1]ГЕПТАН-3-ИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2015 |
|
RU2697651C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ РЕЦЕПТОРОВ, АССОЦИИРОВАННЫХ СО СЛЕДОВЫМИ АМИНАМИ (TAARS) | 2013 |
|
RU2621050C2 |
| ПРОИЗВОДНЫЕ 1-[4-[БЕНЗОИЛ(МЕТИЛ)АМИНО]-3-(ФЕНИЛ)БУТИЛ]АЗЕТИДИНА ДЛЯ ЛЕЧЕНИЯ ЖЕЛУДОЧНО-КИШЕЧНЫХ РАССТРОЙСТВ | 2007 |
|
RU2439067C2 |
| ЗАМЕЩЕННЫЕ БЕНЗАМИДНЫЕ ПРОИЗВОДНЫЕ | 2010 |
|
RU2595902C2 |
| ФЕНИЛПИРАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ | 2008 |
|
RU2480456C2 |
| ХИНОЛИНКАРБОКСАМИДНЫЕ И ХИНОЛИНКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ mGLuR2-НЕГАТИВНЫХ АЛЛОСТЕРИЧЕСКИХ МОДУЛЯТОРОВ, КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2610262C2 |
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2769696C2 |
| 4-ИМИДАЗОЛИНЫ В КАЧЕСТВЕ ЛИГАНДОВ TAAR | 2007 |
|
RU2465269C2 |
Изобретение относится к соединению формулы (I), которое представляет собой 5-этил-4-метил-N-[4-[(2S)морфолин-2-ил]фенил]-1Н-пиразол-3-карбоксамид, или к его фармацевтически приемлемой соли присоединения кислоты. Изобретение также относится к способу получения соединения формулы (I), к его применению, к фармацевтической композиции, к способу активации TAAR. Технический результат: получено новое соединение формулы (I), обладающее агонистической активностью в отношении TAAR. 11 н. и 1 з.п. ф-лы, 20 пр., 1 табл.
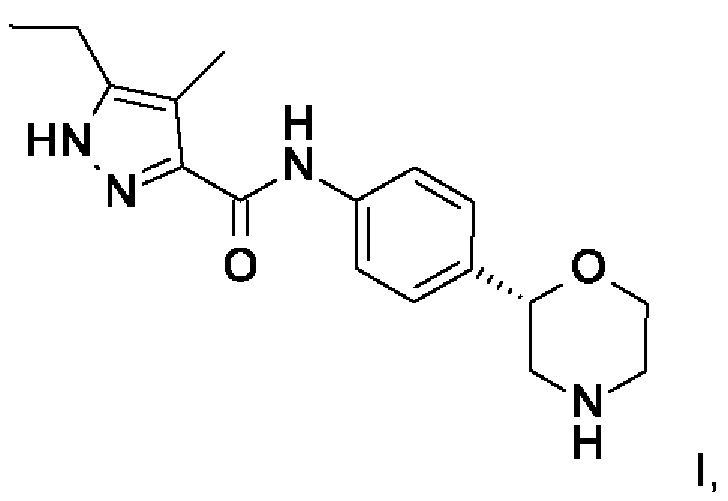
1. Соединение формулы I
которое представляет собой 5-этил-4-метил-N-[4-[(2S)морфолин-2-ил]фенил]-1Н-пиразол-3-карбоксамид, или его фармацевтически приемлемая соль присоединения кислоты.
2. Соединение формулы I
которое представляет собой 5-этил-4-метил-N-[4-[(2S)морфолин-2-ил]фенил]-1Н-пиразол-3-карбоксамид.
3. Способ получения соединения формулы I согласно п. 1 или 2, включающий
отщепление N-защитной группы (PG) у соединений формулы 3 в растворителе в присутствии кислоты при нагревании
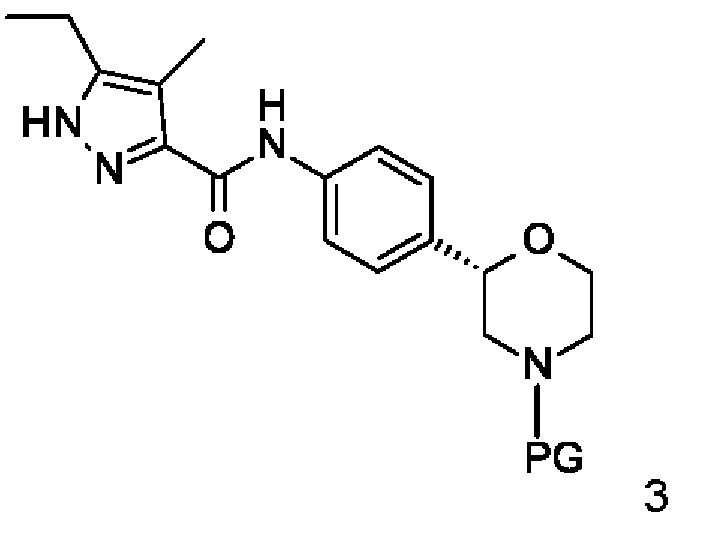
с образованием соединения формулы I
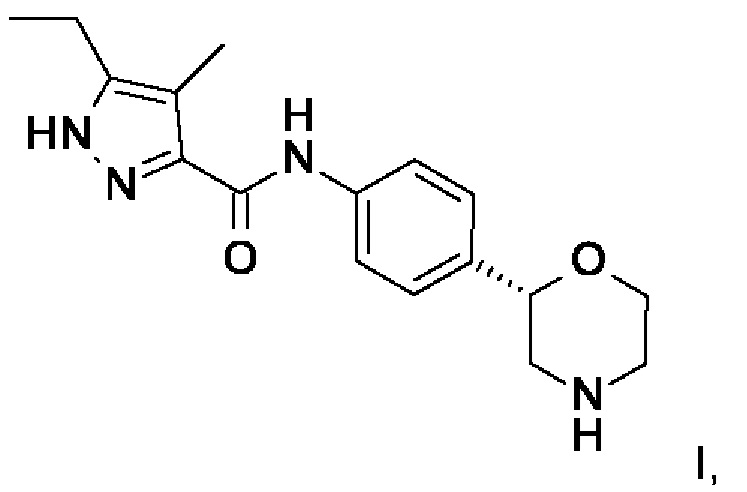
где PG представляет собой -C(O)O-трет-бутил (ВОС), и,
при желании, превращение полученного соединения формулы I в его фармацевтически приемлемую соль присоединения кислоты.
4. Применение соединения формулы I согласно п. 1 в качестве терапевтически активного вещества, обладающего агонистической активностью в отношении TAAR.
5. Применение соединения формулы I согласно п. 2 в качестве терапевтически активного вещества, обладающего агонистической активностью в отношении TAAR.
6. Применение соединения формулы I по п. 1 для лечения заболеваний, связанных с биологической функцией рецепторов, ассоциированных со следовыми аминами, при которых полезна активация TAAR.
7. Применение соединения формулы I по п. 2 для лечения заболеваний, связанных с биологической функцией рецепторов, ассоциированных со следовыми аминами, при которых полезна активация TAAR.
8. Фармацевтическая композиция, обладающая агонистической активностью в отношении TAAR, содержащая эффективное количество соединения формулы I согласно п. 1 или 2 и фармацевтически приемлемые эксципиенты.
9. Фармацевтическая композиция по п. 8, представляющая собой пероральный препарат, выбранный из группы, включающей таблетки, таблетки с оболочкой, драже, твердые и мягкие желатиновые капсулы, растворы, эмульсии и суспензии.
10. Применение соединения формулы I согласно п. 1 для изготовления лекарственного средства, обладающего агонистической активностью в отношении TAAR.
11. Применение соединения формулы I согласно п. 2 для изготовления лекарственного средства, обладающего агонистической активностью в отношении TAAR.
12. Способ активации TAAR, включающий введение эффективного количества соединения формулы I согласно п. 1 или 2.
| WO 2015165835 A1, 05.11.2015 | |||
| WO 2016016292 A1, 04.02.2016 | |||
| RU 2012129737 A, 27.01.2014 | |||
| EA 201391787 A1, 30.04.2014 | |||
| EA 201590454 A1, 30.09.2015 | |||
| EA 022376 B1, 30.12.2015 | |||
| WO 2014041106 A1, 20.03.2014. |

