ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Для этой заявки испрашивается приоритет в соответствии с заявкой США с серийным № 62/640865, поданной 9 марта 2018 года, и международной заявкой № PCT/CN 2017/085276, поданной 22 мая 2017 года, каждая из которых включена в данное описание посредством ссылки во всей своей полноте.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Изобретение относится к соединениям, которые являются ингибиторами янус-киназы, такой как JAK1, а также композициям, содержащим эти соединения, и способам применения, включая, но не ограничиваясь этим, диагностику или лечение пациентов, страдающих от состояния, отвечающего на ингибирование JAK-киназы.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Цитокиновые пути опосредуют большое разнообразие биологических функций, в том числе многие аспекты воспаления и иммунитета. Янус-киназы (JAK), включая JAK1, JAK2, JAK3 и TYK2, представляют собой цитоплазматические протеинкиназы, которые ассоциированы с цитокиновыми рецепторами I типа и II типа и регулируют передачу цитокинового сигнала. Взаимодействие цитокинов со своими рецепторами инициирует активацию ассоциированных с рецепторами JAK, и это приводит к JAK-опосредуемому фосфорилированию тирозиновых остатков в белках-передатчиках сигнала и активаторах транскрипции (STAT) и в конечном итоге к активации транскрипции конкретных наборов генов (Schindler et al., 2007, J. Biol. Chem., 282: 20059-63). JAK1, JAK2 и TYK2 демонстрируют многообразные картины генной экспрессии, в то время как экспрессия JAK3 ограничивается лейкоцитами. В типичном случае цитокиновые рецепторы функционируют в виде гетеродимеров и, как результат, с цитокин-рецепторными комплексами обычно ассоциирована JAK-киназа более чем одного типа. Во многих случаях с помощью генетических исследований были определены конкретные JAK, ассоциированные с разными цитокин-рецепторными комплексами, что подтверждено другими экспериментальными данными. Дискуссия на предмет типичной терапевтической пользы от ингибирования ферментов JAK приведена, например, в международной заявке № WO 2013/014567.
Первоначально JAK1 была идентифицирована при скрининге новых киназ (Wilks A.F., 1989, Proc. Natl. Acad. Sci. U.S.A., 86: 1603-1607). Генетические и биохимические исследования показали, что JAK1 функционально и физически ассоциирована с цитокин-рецепторными комплексами интерферонов I типа (например, IFN-альфа), интерферонов II типа (например, IFN-гамма) и интерлейкина-2 (IL-2) и IL-6 (Kisseleva et al., 2002, Gene, 285: 1-24; Levy et al., 2005, Nat. Rev. Mol. Cell Biol., 3: 651-662; O'Shea et al., 2002, Cell, 109 (suppl.): S121-S131). Мыши с нокаутом по JAK1 погибают в перинатальном периоде вследствие нарушений в передаче сигнала посредством рецептора LIF (фактор, ингибирующий лейкоз) (Kisseleva et al., 2002, Gene, 285: 1-24; O'Shea et al., 2002, Cell, 109 (suppl.): S121-S131). Характеристика тканей, извлеченных из мышей с нокаутом по JAK1, продемонстрировала критическую роль этой киназы в путях, опосредованных IFN, IL-10, IL-2/IL-4 и IL-6. Гуманизированное моноклональное антитело, направленное на IL-6-путь, (тоцилизумаб) было одобрено Комиссией Европейского союза для лечения ревматоидного артрита от умеренного до тяжелого (Scheinecker et al., 2009, Nat. Rev. Drug Discov., 8: 273-274).
CD4 Т-клетки играют важную роль в патогенезе астмы посредством продуцирования цитокинов ТН2-типа в легких, включая IL-4, IL-9 и IL-13 (Cohn et al., 2004, Annu. Rev. Immunol., 22: 789-815). IL-4 и IL-13 индуцируют усиленную выработку слизи, рекрутмент эозинофилов в легкие и усиленное продуцирование иммуноглобулина Е (IgE) (Kasaian et al., 2008, Biochem. Pharmacol., 76(2): 147-155). IL-9 вызывает активацию тучных клеток, что обостряет симптомы астмы (Kearley et al., 2011, Am. J. Resp. Crit. Care Med., 183(7): 865-875). α-Цепь рецептора IL-4 (IL-4R) активирует JAK1 и связывается либо с IL-4, либо с IL-13 при объединении с общей гамма-цепью или с α1-цепью IL-13R, соответственно (Pernis et al., 2002, J. Clin. Invest., 109(10): 1279-1283). Общая гамма-цепь также может объединяться с α-цепью IL-9R для связывания с IL-9, и α-цепь IL-9R также активирует JAK1 (Demoulin et al., 1996, Mol. Cell Biol., 16(9): 4710-4716). Несмотря на то, что общая гамма-цепь активирует JAK3, показано, что JAK1 играет доминирующую роль по сравнению с JAK3, и ингибирования JAK1 достаточно, чтобы инактивировать передачу сигнала через общую гамма-цепь, несмотря на наличие активности у JAK3 (Haan et al., 2011, Chem. Biol., 18(3): 314-323). Ингибирование опосредуемой IL-4, IL-13 и IL-9 передачи сигнала посредством блокирования JAK/STAT-сигнального пути может облегчать астматические симптомы в докпинических моделях воспаления легких (Mathew et al., 2001, J. Exp. Med., 193(9): 1087-1096; Kudlacz et. al., 2008, Eur. J. Pharmacol., 582(1-3): 154-161).
Биохимические и генетические исследования выявили взаимосвязь между JAK2 и семействами одноцепочечных рецепторов цитокинов (например, ЕРО (эритропоэтин)), рецепторов цитокинов IL-3 и интерферона-гамма (Kisseleva et al., 2002, Gene, 285: 1-24; Levy et al., 2005, Nat. Rev. Mol. Cell Biol., 3: 651-662; O'Shea et al., 2002, Cell, 109 (suppl.): S121-S131). В соответствии с этой взаимосвязью, мыши с нокаутом по JAK2 погибают от анемии (O'Shea et al., 2002, Cell, 109 (suppl.): S121-S131). Мутации, усиливающие киназную активность JAK2, (например, V617F в JAK2) связаны с миелопролиферативными расстройствами у людей.
JAK3 ассоциирована исключительно с общей гамма-цепью цитокиновых рецепторов, которая присутствует в цитокин-рецепторных комплексах IL-2, IL-4, IL-7, IL-9, IL-15 и IL-21. JAK3 критична для развития и пролиферации лимфоидных клеток, и мутации в JAK3 приводят к тяжелому комбинированному иммунодефициту (SCID) (O'Shea et al., 2002, Cell, 109 (suppl.): S121-S131). С учетом ее роли в регуляции лимфоцитов, JAK3 и JAK3-опосредуемые пути стали мишенью в случае иммуносупрессивных показаний (например, отторжения при трансплантации и ревматоидного артрита) (Baslund et al., 2005, Arthritis & Rheumatism, 52: 2686-2692; Changelian et al., 2003, Science, 302: 875-878).
TYK2 ассоциирована с цитокин-рецепторными комплексами интерферонов I типа (например, IFN-альфа), IL-6, IL-10, IL-12 и IL-23 (Kisseleva et al., 2002, Gene, 285: 1-24; Watford, W.T. & O'Shea, J.J., 2006, Immunity, 25: 695-697). В соответствии с этим, первичные клетки, полученные от человека с дефицитом по TYK2, являются дефектными в отношении передачи сигнала посредством интерферонов I типа, IL-6, IL-10, IL-12 и IL-23. Полностью человеческое моноклональное антитело, направленное на общую для цитокинов IL-12 и IL-23 субъединицу р40, (устекинумаб) было одобрено Комиссией Европейского союза для лечения бляшковидного псориаза от умеренного до тяжелого (Krueger et al., 2007, N. Engl. J. Med., 356: 580-92; Reich et al., 2009, Nat. Rev. Drug Discov. 8: 355-356). Кроме того, антитело, направленное на IL12- и IL23-пути, проходило клинические испытания в отношении лечения болезни Крона (Mannon et al., 2004, N. Engl. J. Med., 351: 2069-79).
В публикациях международных патентных заявок с номерами WO 2010/051549, WO 2011/003065, WO 2015/177326 и WO 2017/089390 обсуждаются некоторые соединения пиразолопиримидинов, которые, как сообщается, полезны в качестве ингибиторов одной или нескольких янус-киназ. В них представлены данные для некоторых конкретных соединений, демонстрирующих ингибирование JAK1, а также киназ JAK2, JAK3 и/или TYK2.
В настоящее время сохраняется необходимость в дополнительных соединениях, которые представляют собой ингибиторы янус-киназ. Например, существует потребность в соединениях, обладающих полезной эффективностью в качестве ингибиторов одной или нескольких янус-киназ (например, JAK1) в сочетании с другими фармакологическими свойствами, которые необходимы для достижения полезного терапевтического эффекта. Например, существует потребность в сильнодействующих соединениях, демонстрирующих селективность в отношении одной янус-киназы по сравнению с другими киназами в целом (например, селективность в отношении JAK1 по сравнению с другими киназами, такими как обогащенная лейциновыми повторами киназа 2 (LRRK2)). Также существует потребность в сильнодействующих соединениях, демонстрирующих селективность в отношении одной янус-киназы по сравнению с другими янус-киназами (например, селективность в отношении JAK1 по сравнению с другими янус-киназами). Киназы, демонстрирующие селективность в отношении JAK1, могут обеспечить терапевтическую пользу, с незначительным числом побочных эффектов, при состояниях, отвечающих на ингибирование JAK1. Кроме того, в настоящее время имеется потребность в сильнодействующих ингибиторах JAK1, которые обладают другими свойствами (например, точкой плавления, pK, растворимостью и т.д.), необходимыми для приготовления композиций и введения посредством ингаляции. Такие соединения будут особенно полезны для лечения таких состояний, как, например, астма.
В данной области техники существует потребность в дополнительных или альтернативных схемах лечения состояний, опосредуемых киназами JAK, например, таких, которые описаны выше.
Краткое описание сущности изобретения
Согласно данному изобретению предложены пиразолопиримидины, которые ингибируют JAK-киназу, как например, выбранные из соединения формулы (I), его стереоизомера или соли, такой как его фармацевтически приемлемая соль. JAK-киназа может представлять собой JAK1.
В одном из воплощений предложено соединение формулы (I):
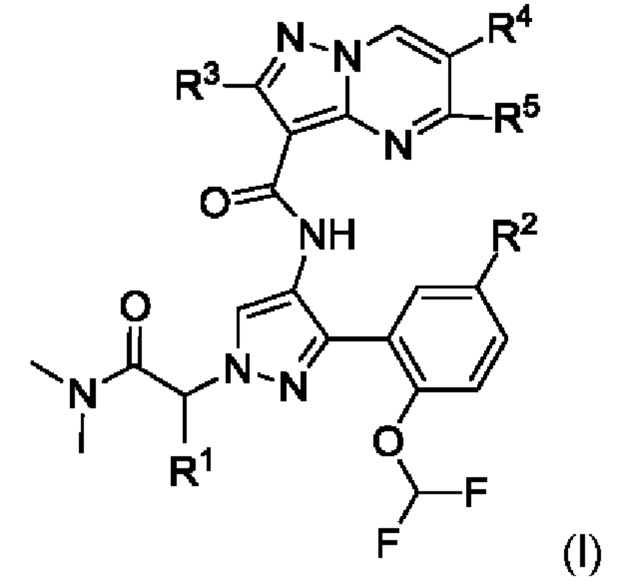
или его стереоизомер или соль (например, фармацевтически приемлемая соль), где:
R1 представляет собой атом водорода или СН3;
R2 представляет собой галоген, C1-С6алкил, С2-С6алкенил, С2-С6алкинил, С3-С6циклоалкил или -ORa, при этом R2 возможно замещен одной или несколькими группами, независимо выбранными из группы, состоящей из атома галогена, С1-С3алкила, циано, гидрокси и оксо;
Ra представляет собой C1-С6алкил, -фенил-CORbRc, -фенил-(3-6-членный гетероциклил) или 3-11-членный гетероциклил, при этом Ra возможно замещен одной или несколькими группами, независимо выбранными из группы, состоящей из атома галогена, C1-С3алкила, циано, гидрокси и оксо;
Rb и Rc каждый независимо представляет собой атом водорода или СН3;
R3 представляет собой атом водорода или NH2;
R4 представляет собой атом водорода или СН3; и
R5 представляет собой атом водорода или NH2.
Также предложена фармацевтическая композиция, содержащая ингибитор JAK, описанный в данной заявке, или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или эксципиент.
Также предложено применение ингибитора JAK, описанного в данной заявке, или его фармацевтически приемлемой соли в терапии, как например, в лечении воспалительного заболевания (например, астмы). Также предложено применение ингибитора JAK, описанного в данной заявке, или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения воспалительного заболевания. Также предложен способ предупреждения, лечения или ослабления тяжести заболевания или состояния, отвечающего на ингибирование активности янус-киназы у пациента, включающий введение пациенту терапевтически эффективного количества ингибитора JAK, описанного в данной заявке, или его фармацевтически приемлемой соли.
Некоторые соединения или их соли (например, их фармацевтически приемлемые соли), описанные в данной заявке, обладают существенной эффективностью в качестве ингибиторов одной или нескольких янус-киназ (например, JAK1). Некоторые соединения или их соли (например, их фармацевтически приемлемые соли) также обладают а) селективностью в отношении одной янус-киназы по сравнению с другими киназами, b) селективностью в отношении JAK1 по сравнению с другими янус-киназами, и/или c) обладают другими свойствами (например, точкой плавления, pK, растворимостью и т.д.), необходимыми для приготовления композиции и введения посредством ингаляции. Некоторые соединения или их соли (например, их фармацевтически приемлемые соли), описанные в данной заявке, могут быть особенно полезны для лечения таких состояний, как астма.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ОПРЕДЕЛЕНИЯ
"Галоген" или "атом галогена" относится к F, Cl, Br или I. Кроме того, подразумевается, что такие термины, как "галогеналкил", включают в себя моногалогеналкил и пол и галогеналкил, где атом(ы) водорода алкильной группы заменен(ы) на один или несколько атомов галогена.
Термин "алкил" относится к насыщенному одновалентному углеводородному радикалу с линейной или разветвленной цепью, при этом алкильный радикал возможно может быть замещен. В одном из примеров алкильный радикал содержит от одного до восемнадцати атомов углерода (C1-C18). В других примерах алкильный радикал представляет собой С0-С6, С0-С5, С0-С3, С1-C12, C1-C10, C1-C8, C1-C6, C1-C5, C1-C4 или C1-С3. С0алкил обозначает связь. Примеры алкильных групп включают метил (Me, -СН3), этил (Et, -СН2СН3), 1-пропил (н-Pr, н-пропил, -СН2СН2СН3), 2-пропил (i-Pr, изопропил, -СН(СН3)2), 1-бутил (н-Bu, н-бутил, -СН2СН2СН2СН3), 2-метил-1-пропил (i-Bu, изобутил, -СН2СН(СН3)2), 2-бутил (втор-Bu, втор-бутил, -СН(СН3)СН2СН3), 2-метил-2-пропил (t-B, трет-бутил, -С(СН3)3), 1-пентил (н-пентил, -СН2СН2СН2СН2СН3), 2-пентил (-СН(СН3)СН2СН2СН3), 3-пентил (-СН(СН2СН3)2), 2-метил-2-бутил (-С(СН3)2СН2СН3), 3-метил-2-бутил (-СН(СН3)СН(СН3)2), 3-метил-1-бутил (-СН2СН2СН(СН3)2), 2-метил-1-бутил (-СН2СН(СН3)СН2СН3), 1-гексил (-СН2СН2СН2СН2СН2СН3), 2-гексил (-СН(СН3)СН2СН2СН2СН3), 3-гексил (-СН(CH2CH3)(СН2СН2СН3)), 2-метил-2-пентил (-С(СН3)2СН2СН2СН3), 3-метил-2-пентил (-СН(СН3)СН(СН3)СН2СН3), 4-метил-2-пентил (-СН(СН3)СН2СН(СН3)2), 3-метил-3-пентил (-С(СН3)(СН2СН3)2), 2-метил-3-пентил (-СН(СН2СН3)СН(СН3)2), 2,3-диметил-2-бутил (-С(СН3)2СН(СН3)2), 3,3-диметил-2-бутил (-СН(СН3)С(СН3)3), 1-гептил и 1-октил. В некоторых воплощениях заместители для "возможно замещенных алкилов" включают от одного до четырех вариантов, выбранных из группы F, Cl, Br, I, ОН, SH, CN, NH2, NHCH3, N(СН3)2, NO2, N3, С(O)СН3, СООН, CO2CH3, метила, этила, пропила, изопропила, бутила, изобутила, циклопропила, метокси, этокси, пропокси, оксо, трифторметила, дифторметила, сульфониламино, метансульфониламино, SO, SO2, фенила, пиперидинила, пиперазинила и пиримидинила, при этом их алкильные, фенильные и гетероциклические части возможно могут быть замещены, например, одним-четырьмя вариантами заместителей, выбранных из этого же списка.
Термин "алкенил" относится к одновалентному углеводородному радикалу с линейной или разветвленной цепью, имеющему по меньшей мере один сайт ненасыщенности, т.е. углерод-углеродную двойную связь, при этом алкенильный радикал возможно может быть замещен и включает радикалы в "цис"- и "транс"- ориентациях или, альтернативно, "Е"- и "Z"-ориентациях. В одном из примеров алкенильный радикал содержит от двух до восемнадцати атомов углерода (C2-C18). В других примерах алкенильный радикал представляет собой C2-C12, С2-С10, С2-С8, C2-C6 или С2-С3. Примеры включают, но не ограничиваются этим, этенил или винил (-СН=СН2), проп-1-енил (-СН=СНСН3), проп-2-енил (-СН2СН=СН2), 2-метилпроп-1-енил, бут-1-енил, бут-2-енил, бут-3-енил, бута-1,3-диенил, 2-метилбута-1,3-диен, гекс-1-енил, гекс-2-енил, гекс-3-енил, гекс-4-енил и гекса-1,3-диенил. В некоторых воплощениях заместители для "возможно замещенных алкенилов" включают от одного до четырех вариантов, выбранных из группы F, Cl, Br, I, ОН, SH, CN, NH2, NHCH3, N(CH3)2, NO2, N3, С(O)СН3, СООН, CO2CH3, метила, этила, пропила, изопропила, бутила, изобутила, циклопропила, метокси, этокси, пропокси, оксо, трифторметила, дифторметила, сульфониламино, метансульфониламино, SO, SO2, фенила, пиперидинила, пиперазинила и пиримидинила, при этом их алкильные, фенильные и гетероциклические части возможно могут быть замещены, как например, одним-четырьмя вариантами заместителей, выбранных из этого же списка.
Термин "алкинил" относится к линейному или разветвленному одновалентному углеводородному радикалу, имеющему по меньшей мере один сайт ненасыщенности, т.е. углерод-углеродную тройную связь, при этом алкинильный радикал возможно может быть замещен. В одном из примеров алкинильный радикал содержит от двух до восемнадцати атомов углерода (C2-C18). В других примерах алкинильный радикал представляет собой С2-С12, С2-С10, C2-C8, С2-С6 или С2-С3. Примеры включают, но не ограничиваются этим, этинил (-С≡СН), проп-1-инил (-С≡ССН3), проп-2-инил (пропаргил, -СН2С≡СН), бут-1-инил, бут-2-инил и бут-3-инил. В некоторых воплощениях заместители для "возможно замещенных алкинилов" включают от одного до четырех вариантов, выбранных из группы F, Cl, Br, I, ОН, SH, CN, NH2, NHCH3, N(СН3)2, NO2, N3, С(O)СН3, СООН, CO2CH3, метила, этила, пропила, изопропила, бутила, изобутила, циклопропила, метокси, этокси, пропокси, оксо, трифторметила, дифторметила, сульфониламино, метансульфониламино, SO, SO2, фенила, пиперидинила, пиперазинила и пиримидинила, при этом их алкильные, фенильные и гетероциклические части возможно могут быть замещены, например, одним-четырьмя вариантами заместителей, выбранных из этого же списка.
Термин "алкилен" относится к насыщенной углеводородной группе с разветвленной или прямой цепью, имеющей два одновалентных радикальных центра, получающиеся в результате удаления двух атомов водорода от одного и того же или от двух разных атомов углерода исходного алкана. В одном из примеров двухвалентная алкиленовая группа содержит от одного до восемнадцати атомов углерода (C1-C18). В других примерах двухвалентная алкиленовая группа представляет собой C0-C6, C0-C5, С0-С3, C1-C12, C1-С10, C1-C8, C1-C6, C1-C5, C1-C4 или C1-С3. Группа C0алкилен обозначает связь. Примеры алкиленовых групп включают метилен (-СН2-), 1,1-этил (-СН(СН3)-), (1,2-этил (-СН2СН2-), 1,1-пропил (-СН(СН2СН3)-), 2,2-пропил (-С(СН3)2-), 1,2-пропил (-СН(СН3)СН2-), 1,3-пропил (-СН2СН2СН2-), 1,1-диметилэт-1,2-ил (-С(СН3)2СН2-), 1,4-бутил (-СН2СН2СН2СН2-) и тому подобное.
Термин "гетероалкил" относится к одновалентному углеводородному радикалу с прямой или разветвленной цепью, состоящему из указанного числа атомов углерода, или, если не указано ничего, то состоящему из атомов углерода числом до 18 включительно, и из одного-пяти гетероатомов, выбранных из группы, состоящей из О, N, Si и S, и при этом атомы азота и серы возможно могут быть окислены и гетероатом азота возможно может быть кватернизован. В некоторых воплощениях гетероатом выбран из О, N и S, при этом атомы азота и серы возможно могут быть окислены и гетероатом азота возможно может быть кватернизован. Гетероатом(ы) может/могут располагаться в любом внутреннем положении гетероалкильной группы, включая положение, в котором алкильная группа присоединена к остальной части молекулы (например, -O-CH2-CH3). Примеры включают -СН2-СН2-О-СН3, -СН2-СН2-NH-СН3, -СН2-СН2-N(СН3)-СН3, -СН2-S-СН2-СН3, -S(O)-СН3, -СН2-СН2-S(O)2-СН3, -Si(СН3)3 и -СН2-СН=N-ОСН3. Гетероатомы могут следовать друг за другом в количестве до двух включительно, как например, в случаях -СН2-NH-ОСН3 и -СН2-O-Si(СН3)3. Гетероалкильные группы возможно могут быть замещены. В некоторых воплощениях заместители для "возможно замещенных гетероалкилов" включают от одного до четырех вариантов, выбранных из группы F, Cl, Br, I, OH, SH, CN, NH2, NHCH3, N(СН3)2, NO2, N3, С(O)СН3, СООН, CO2CH3, метила, этила, пропила, изопропила, бутила, изобутила, циклопропила, метокси, этокси, пропокси, оксо, трифторметила, дифторметила, сульфониламино, метансульфониламино, SO, SO2, фенила, пиперидинила, пиперазинила и пиримидинила, при этом их алкильные, фенильные и гетероциклические части возможно могут быть замещены, например, одним-четырьмя вариантами заместителей, выбранных из этого же списка.
Термин "амино" относится к первичным (т.е. -NH2), вторичным (т.е. -NRH), третичным (т.е. -NRR) и четвертичным (т.е. -N(+)RRR) аминам, которые возможно замещены, в которых каждый R является таким же или другим и выбран из алкила, циклоалкила, арила и гетероциклила, при этом алкильные, циклоалкильные, арильные и гетерециклильные группы являются такими, как определено в данном описании. Конкретные вторичные и третичные амины представляют собой алкиламин, диалкиламин, ариламин, диариламин, аралкиламин и диаралкиламин, при этом алкильные и арильные части возможно могут быть замещены. Конкретными вторичными и третичными аминами являются метиламин, этиламин, пропиламин, изопропиламин, фениламин, бензиламин, диметиламин, диэтиламин, дипропиламин и диизопропиламин. В некоторых воплощениях каждая из групп R четвертичного амина независимо представляет собой возможно замещенную алкильную группу.
"Арил" означает карбоциклическую ароматическую группу независимо от того, конденсирована она или нет с одной или более группами, имеющую указанное количество атомов углерода или, если количество не указано, имеющую до 14 атомов углерода включительно. Один из примеров включает арильные группы, имеющие 6-14 атомов углерода. Другой пример включает арильные группы, имеющие 6-10 атомов углерода. Примеры арильных групп включают фенил, нафтил, бифенил, фенантренил, нафтаценил, 1,2,3,4-тетрагидронафталинил, 1Н-инденил, 2,3-дигидро-1Н-инденил и тому подобное (см., например, Lang's Handbook of Chemistry (Dean J.A., ed.), 13е изд., Таблица 7-2 [1985]). Конкретным арилом является фенил. Замещенный фенил или замещенный арил означает фенильную группу или арильную группу, замещенную одним, двумя, тремя, четырьмя или пятью заместителями, например, такими 1-2, 1-3 или 1-4 заместителями, как выбранные из группы, указанной в данном описании (см. определение для "возможно замещенный"), такими как F, Cl, Br, I, ОН, SH, CN, NH2, NHCH3, N(СН3)2, NO2, N3, С(O)СН3, СООН, CO2CH3, метил, этил, пропил, изопропил, бутил, изобутил, циклопропил, метокси, этокси, пропокси, оксо, трифторметил, дифторметил, сульфониламино, метансульфониламино, SO, SO2, фенил, пиперидинил, пиперазинил и пиримидинил, при этом их алкильные, фенильные и гетероциклические части возможно могут быть замещены, например, одним-четырьмя вариантами заместителей, выбранных из этого же списка. Примеры термина "замещенный фенил" включают моно- или ди(галоген)фенильную группу, такую как 2-хлорфенил, 2-бромфенил, 4-хлорфенил, 2,6-дихлорфенил, 2,5-дихлорфенил, 3,4-дихлорфенил, 3-хлорфенил, 3-бромфенил, 4-бромфенил, 3,4-дибромфенил, 3-хлор-4-фторфенил, 2-фторфенил, 2,4-дифторфенил и тому подобное; моно- или ди(гидрокси)фенильную группу, такую как 4-гидроксифенил, 3-гидроксифенил, 2,4-дигидроксифенил, их защищенные по группе гидрокси производные и тому подобное; нитрофенильную группу, такую как 3- или 4-нитрофенил; цианофенильную группу, например, 4-цианофенил; моно- или ди(алкил)фенильную группу, такую как 4-метилфенил, 2,4-диметилфенил, 2-метилфенил, 4-(изопропил)фенил, 4-этилфенил, 3-(н-пропил)фенил и тому подобное; моно- или ди(алкокси)фенильную группу, например, 3,4-диметоксифенил, 3-метокси-4-бензилоксифенил, 3-этоксифенил, 4-(изопропокси)фенил, 4-(трет-бутокси)фенил, 3-этокси-4-метоксифенил и тому подобное; 3- или 4-трифторметилфенил; моно- или дикарбоксифенильную или (защищенную по карбокси)фенильную группу, такую как 4-карбоксифенил, моно- или ди(гидроксиметил)фенил или (защищенный гидроксиметил)фенил, такой как 3-(защищенный гидроксиметил)фенил или 3,4-ди(гидроксиметил)фенил; моно- или ди(аминометил)фенил или (защищенный аминометил)фенил, такой как 2-(аминометил)фенил или 2,4-(защищенный аминометил)фенил; или моно- или ди(N-(метилсульфониламино))фенил, такой как 3-(N-метилсульфониламино)-фенил. Кроме того, термин "замещенный фенил" означает дизамещенные фенильные группы, где заместители будут разными, например, 3-метил-4-гидроксифенил, 3-хлор-4-гидроксифенил, 2-метокси-4-бромфенил, 4-этил-2-гидроксифенил, 3-гидрокси-4-нитрофенил, 2-гидрокси-4-хлорфенил, 2-хлор-5-дифторметокси и тому подобное, а также тризамещенные фенильные группы, где заместители будут разными, например, 3-метокси-4-бензилокси-6-метил-сульфониламино, 3-метокси-4-бензилокси-6-фенил-сульфониламино, и тетразамещенные фенильные группы, где заместители будут разными, такие как 3-метокси-4-бензилокси-5-метил-6-фенил-сульфониламино. В некоторых воплощениях заместитель у арила, такого как фенил, содержит амид. Например, заместителем у арила (например, фенила) может быть -(СН2)0-4CONR'R'', где R' и R'' каждый независимо относится к группам, включая, например, атом водорода; незамещенный C1-С6алкил; C1-С6алкил, замещенный атомом галогена, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''; незамещенный С1-С6гетероалкил; C1-С6гетероалкил, замещенный атомом галогена, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''; незамещенный С6-С10арил; С6-С10арил, замещенный атомом галогена, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси или NR'R''; незамещенный 3-11-членный гетероциклил (например, 5-6-членный гетероарил, содержащий 1-4 гетероатома, выбранных из О, N и S, или 4-11-членный гетероциклоалкил, содержащий 1-4 гетероатома, выбранных из О, N и S); и 3-11-членный гетероциклил (например, 5-6-членный гетероарил, содержащий 1-4 гетероатома, выбранных из О, N и S, или 4-11-членный гетероциклоалкил, содержащий 1-4 гетероатома, выбранных из О, N и S), замещенный галогеном, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''; или R' и R'' могут быть объединены с атомом азота с образованием 3-, 4-, 5-, 6- или 7-членного кольца, при этом какой-либо атом в кольце возможно замещен атомом N, О или S, и при этом кольцо возможно замещено галогеном, ОН, CN, незамещенным C1-С6алкилом, незамещенным С1-С6алкокси, оксо или NR'R''.
"Циклоалкил" относится к неароматической насыщенной или частично ненасыщенной углеводородной кольцевой группе, при этом указанная циклоалкильная группа возможно может быть независимо замещена одним или более заместителями, изложенными в данном описании. В одном из примеров циклоалкильная группа имеет 3-12 атомов углерода (С3-С12). В других примерах циклоалкил представляет собой группу С3-C8, С3-С10 или С5-С10. В других примерах циклоалкильная группа, в виде моноцикла, представляет собой группу С3-C8, С3-С6 или С5-С6. В другом примере циклоалкильная группа, в виде бицикла, представляет собой группу C7-C12. В другом примере циклоалкильная группа, в виде спиросистемы, представляет собой группу C5-C12. Примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, 1-циклопент-1-енил, 1-циклопент-2-енил, 1-циклопент-3-енил, циклогексил, полностью дейтерированный циклогексил, 1-циклогекс-1-енил, 1-цикпогекс-2-енил, 1-циклогекс-3-енил, циклогексадиенил, циклогептил, циклооктил, циклононил, циклодецил, циклоундецил и цикпододецил. Типичные конфигурации бициклических циклоалкилов, имеющих 7-12 атомов в кольце, включают, но не ограничиваются этим, [4,4]-, [4,5]-, [5,5]-, [5,6]- или [6,6]-кольцевые системы. Типичные мостиковые бициклические циклоалкилы включают, но не ограничиваются этим, бицикло[2.2.1]гептан, бицикло[2.2.2]октан и бицикло[3.2.2]нонан. Примеры спиро-циклоалкила включают спиро[2.2]пентан, спиро[2.3]гексан, спиро[2.4]гептан, спиро[2.5]октан и спиро[4.5]декан. В некоторых воплощениях заместители для "возможно замещенный циклоалкилов" включают от одного до четырех вариантов, выбранных из группы F, Cl, Br, I, ОН, SH, CN, NH2, NHCH3, N(СН3)2, NO2, N3, С(O)СН3, СООН, CO2CH3, метила, этила, пропила, изопропила, бутила, изобутила, циклопропила, метокси, этокси, пропокси, оксо, трифторметила, дифторметила, сульфониламино, метансульфониламино, SO, SO2, фенила, пиперидинила, пиперазинила и пиримидинила, при этом их алкильные, арильные и гетероциклические части возможно могут быть замещены, например, одним-четырьмя вариантами заместителей, выбранных из этого же списка. В некоторых воплощениях заместитель у циклоалкила содержит амид. Например, заместителем у циклоалкила может быть -(СН2)0-4CONR'R'', где R' и R'' каждый независимо относится к группам, включая, например, атом водорода; незамещенный C1-С6алкил; C1-С6алкил, замещенный атомом галогена, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''; незамещенный C1-С6гетероалкил; C1-С6гетероалкил, замещенный атомом галогена, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''; незамещенный С6-С10арил; С6-С10арил, замещенный атомом галогена, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси или NR'R''; незамещенный 3-11-членный гетероцикпил (например, 5-6-членный гетероарил, содержащий 1-4 гетероатома, выбранных из О, N и S, или 4-11-членный гетероциклоалкил, содержащий 1-4 гетероатома, выбранных из О, N и S); и 3-11-членный гетероциклил (например, 5-6-членный гетероарил, содержащий 1-4 гетероатома, выбранных из О, N и S, или 4-11-членный гетероциклоалкил, содержащий 1-4 гетероатома, выбранных из О, N и S), замещенный атомом галогена, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''; или R' и R'' могут быть объединены с атомом азота с образованием 3-, 4-, 5-, 6- или 7-членного кольца, при этом какой-либо атом в кольце возможно замещен атомом N, О или S, и при этом кольцо возможно замещено галогеном, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''.
Термины "гетероциклическая группа", "гетероциклический", "гетероцикл", "гетероциклил" или "гетероцикло" используются взаимозаменяемо и относятся к любой моно-, би-, трициклической или спиро, насыщенной или ненасыщенной, ароматической (гетероарильной) или неароматической (например, гетероциклоалкильной) кольцевой системе, имеющей 3-20 атомов в кольце (например, 3-10 атомов в кольце), при этом атомы в кольце представляют собой атомы углерода, и по меньшей мере один атом в кольце или кольцевой системе представляет собой гетероатом, выбранный из атомов азота, серы или кислорода. Если каким-либо находящимся в кольце атомом циклической системы является гетероатом, то такая система представляет собой гетероцикп, независимо от места присоединения циклической системы к остальной части молекулы. В одном из примеров гетероциклил содержит 3-11 атомов в кольце ("членов") и включает моноциклы, бициклы, трициклы и спиро-кольцевые системы, где атомы в кольце представляют собой атомы углерода, при этом по меньшей мере один атом в кольце или кольцевой системе представляет собой гетероатом, выбранный из атома азота, серы или кислорода. В одном из примеров гетероциклил содержит 1-4 гетероатома. В одном из примеров гетероциклил содержит 1-3 гетероатома. В другом примере гетероциклил включает 3-7-членные моноциклы, имеющие 1-2, 1-3 или 1-4 гетероатома, выбранных из атома азота, серы или кислорода. В другом примере гетероциклил включает 4-6-членные моноциклы, имеющие 1-2, 1-3 или 1-4 гетероатома, выбранных из атома азота, серы или кислорода. В другом примере гетероциклил включает 3-членные моноциклы. В другом примере гетероциклил включает 4-членные моноциклы. В другом примере гетероциклил включает 5-6-членные моноциклы, например, 5-6-членный гетероарил. В другом примере гетероциклил включает 3-11-членные гетероциклоалкилы, такие как 4-11-членные гетероциклоалкилы. В некоторых воплощениях гетероциклоалкил содержит по меньшей мере один атом азота. В одном из примеров гетероциклильная группа содержит 0-3 двойные связи. Любой гетероатом азота или серы возможно может быть окислен (например, NO, SO, SO2) и любой гетероатом азота возможно может быть кватернизован (например, [NR4]+Cl-, [NR4]+OH-). Примерами гетероциклов являются оксиранил, азиридинил, тииранил, азетидинил, оксетанил, тиетанил, 1,2-дитиетанил, 1,3-дитиетанил, пирролидинил, дигидро-1Н-пирролил, дигидрофуранил, тетрагидрофуранил, дигидротиенил, тетрагидротиенил, имидазолидинил, пиперидинил, пиперазинил, изохинолинил, тетрагидроизохинолинил, морфолинил, тиоморфолинил, 1,1-диоксо-тиоморфолинил, дигидропиранил, тетрагидропиранил, гексагидротиопиранил, гексагидропиримидинил, оксазинанил, тиазинанил, тиоксанил, гомопиперазинил, гомопиперидинил, азепанил, оксепанил, тиепанил, оксазепинил, оксазепанил, диазепанил, 1,4-диазепанил, диазепинил, тиазепинил, тиазепанил, тетрагидротиопиранил, оксазолидинил, тиазолидинил, изотиазолидинил, 1,1-диоксоизотиазолидинонил, оксазолидинонил, имидазолидинонил, 4,5,6,7-тетрагидро[2Н]индазолил, тетрагидробензоимидазолил, 4,5,6,7-тетрагидробензо[d]имидазолил, 1,6-дигидроимидазол[4,5-d]пирроло[2,3-b]пиридинил, тиазинил, оксазинил, тиадиазинил, оксадиазинил, дитиазинил, диоксазинил, оксатиазинил, тиатриазинил, оксатриазинил, дитиадиазинил, имидазолинил, дигидропиримидил, тетрагидропиримидил, 1-пирролинил, 2-пирролинил, 3-пирролинил, индолинил, тиапиранил, 2Н-пиранил, 4Н-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, пиразолидинил, дитианил, дитиоланил, пиримидинонил, пиримидиндионил, пиримидин-2,4-дионил, пиперазинонил, пиперазиндионил, пиразолидинилимидазолинил, 3-азабицикпо-[3.1.0]гексанил, 3,6-диазабицикло[3.1.1]гептанил, 6-азабицикло[3.1.1]гептанил, 3-азабицикло[3.1.1]гептанил, 3-азабицикло[4.1.0]гептанил, азабицикло[2.2.2]гексанил, 2-азабицикло[3.2.1]октанил, 8-азабицикло[3.2.1]октанил, 2-азабицикло[2.2.2]октанил, 8-азабицикло[2.2.2]октанил, 7-оксабицикло[2.2.1]-гептан, азаспиро[3.5]нонанил, азаспиро[2.5]октанил, азаспиро[4.5]деканил, 1-азаспиро[4.5]декан-2-онил, азаспиро[5.5]ундеканил, тетрагидроиндолил, октагидроиндолил, тетрагидроизоиндолил, тетрагидроиндазолил, 1,1-диоксогексагидротиопиранил. Примерами 5-членных гетероциклов, содержащих атом серы или кислорода и от одного до трех атомов азота, являются тиазолил, включая тиазол-2-ил и тиазол-2-ил-N-оксид, тиадиазолил, включая 1,3,4-тиадиазол-5-ил и 1,2,4-тиадиазол-5-ил, оксазолил, например оксазол-2-ил, и оксадиазолил, такой как 1,3,4-оксадиазол-5-ил и 1,2,4-оксадиазол-5-ил. Типичные 5-членные кольцевые гетероциклы, содержащие от 2 до 4 атомов азота, включают имидазолил, такой как имидазол-2-ил; триазолил, такой как 1,3,4-триазол-5-ил, 1,2,3-триазол-5-ил, 1,2,4-триазол-5-ил; и тетразолил, такой как 1Н-тетразол-5-ил. Типичными бензо-конденсированными 5-членными гетероциклами являются бензоксазол-2-ил, бензтиазол-2-ил и бензимидазол-2-ил. Типичные 6-членные гетероциклы содержат от одного до трех атомов азота и возможно атом серы или кислорода, например, пиридил, такой как пирид-2-ил, пирид-3-ил и пирид-4-ил; пиримидил, такой как пиримид-2-ил и пиримид-4-ил; триазинил, такой как 1,3,4-триазин-2-ил и 1,3,5-триазин-4-ил; пиридазинил, в частности, пиридазин-3-ил и пиразинил. Пиридин-N-оксиды и пиридазин-N-оксиды и пиридильная, пиримид-2-ильная, пиримид-4-ильная, пиридазинильная и 1,3,4-триазин-2-ильная группы, представляют собой другие типичные гетероциклические группы. Гетероциклы возможно могут быть замещены. Например, заместители для "возможно замещенных гетероцикпов" включают от одного до четырех вариантов, выбранных из группы F, Cl, Br, I, ОН, SH, CN, NH2, NHCH3, N(СН3)2, NO2, N3, С(O)СН3, СООН, СО2СН3, метила, этила, пропила, изопропила, бутила, изобутила, циклопропила, метокси, этокси, пропокси, оксо, трифторметила, дифторметила, сульфониламино, метансульфониламино, SO, SO2, фенила, пиперидинила, пиперазинила и пиримидинила, при этом их алкильные, арильные и гетероциклические части возможно могут быть замещены, например, одним-четырьмя вариантами заместителей, выбранных из этого же списка. В некоторых воплощениях заместитель у гетероциклической группы, такой как гетероарил или гетероциклоалкил, содержит амид. Например, заместителем у гетероциклической (например, гетероарильной или гетероциклоалкильной) группы может быть -(CH2)0-4CONR'R'', где R' и R'' каждый независимо относится к группам, включая, например, атом водорода; незамещенный C1-С6алкил; C1-С6алкил, замещенный атомом галогена, ОН, CN, незамещенным C1-С6алкилом, незамещенным С1-С6алкокси, оксо или NR'R''; незамещенный C1-С6гетероалкил; C1-С6гетероалкил, замещенный атомом галогена, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''; незамещенный С6-С10арил; С6-С10арил, замещенный атомом галогена, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси или NR'R''; незамещенный 3-11-членный гетероциклил (например, 5-6-членный гетероарил, содержащий 1-4 гетероатома, выбранных из О, N и S, или 4-11-членный гетероциклоалкил, содержащий 1-4 гетероатома, выбранных из О, N и S); и 3-11-членный гетероциклил (например, 5-6-членный гетероарил, содержащий 1-4 гетероатома, выбранных из О, N и S, или 4-11-членный гетероциклоалкил, содержащий 1-4 гетероатома, выбранных из О, N и S), замещенный галогеном, ОН, CN, незамещенным C1-С6алкилом, незамещенным С1-С6алкокси, оксо или NR'R''; или R' и R'' могут быть объединены с атомом азота с образованием 3-, 4-, 5-, 6- или 7-членного кольца, при этом какой-либо атом в кольце возможно замещен атомом N, О или S, и при этом кольцо возможно замещено галогеном, ОН, CN, незамещенным C1-С6алкилом, незамещенным С1-С6алкокси, оксо или NR'R''.
"Гетероарил" относится к любой моно-, би- или трициклической кольцевой системе, в которой по меньшей мере одно кольцо представляет собой 5- или 6-членное ароматическое кольцо, содержащее 1-4 гетероатома, выбранных из атомов азота, кислорода и серы, и в типичном воплощении по меньшей мере одним гетероатомом является атом азота. См., например, Lang's Handbook of Chemistry (изд. Dean J.A.) 13ое изд., Таблица 7-2 [1985]. В данное определение включены любые бициклические группы, где любое из вышеупомянутых гетероарильных колец конденсировано с арильным кольцом, при этом данное арильное кольцо или гетероарильное кольцо соединено с остальной частью молекулы. В одном из воплощений гетероарил включает 5-6 членные моноциклические ароматические группы, где одним или более чем одним атомом в кольце является атом азота, серы или кислорода. Типичные гетероарильные группы включают тиенил, фурил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, изоксазолил, триазолил, тиадиазолил, оксадиазолил, тетразолил, тиатриазолил, оксатриазолил, пиридил, пиримидил, пиразинил, пиридазинил, триазинил, тетразинил, тетразоло[1,5-b]пиридазинил, имидазол[1,2-a]пиримидинил и пуринил, а также бензо-конденсированные производные, например, бензоксазолил, бензофурил, бензотиазолил, бензотиадиазолил, бензотриазолил, бензоимидазолил и индолил. Гетероарильные группы возможно могут быть замещены. В некоторых воплощениях заместители для "возможно замещенных гетероарилов" включают от одного до четырех вариантов, выбранных из группы F, Cl, Br, I, ОН, SH, CN, NH2, NHCH3, N(CH3)2, NO2, N3, С(O)СН3, СООН, CO2CH3, метила, этила, пропила, изопропила, бутила, изобутила, циклопропила, метокси, этокси, пропокси, трифторметила, дифторметила, сульфониламино, метансульфониламино, SO, SO2, фенила, пиперидинила, пиперазинила и пиримидинила, при этом их алкильные, фенильные и гетероциклические части возможно могут быть замещены, например, одним-четырьмя вариантами заместителей, выбранных из этого же списка. В некоторых воплощениях заместитель у гетероарила содержит амид. Например, заместителем у гетероарила может быть -(CH2)0-4CONR'R'', где R' и R'' каждый независимо относится к группам, включая, например, атом водорода; незамещенный C1-С6алкил; C1-С6алкил, замещенный атомом галогена, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''; незамещенный C1-С6гетероалкил; C1-С6гетероалкил, замещенный атомом галогена, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''; незамещенный С6-С10арил; С6-С10арил, замещенный атомом галогена, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-Салкокси или NR'R''; незамещенный 3-11-членный гетероциклил (например, 5-6-членный гетероарил, содержащий 1-4 гетероатома, выбранных из О, N и S, или 4-11-членный гетероциклоалкил, содержащий 1-4 гетероатома, выбранных из О, N и S); и 3-11-членный гетероциклил (например, 5-6-членный гетероарил, содержащий 1-4 гетероатома, выбранных из О, N и S, или 4-11-членный гетероциклоалкил, содержащий 1-4 гетероатома, выбранных из О, N и S), замещенный галогеном, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-Салкокси, оксо или NR'R''; или R' и R'' могут быть объединены с атомом азота с образованием 3-, 4-, 5-, 6- или 7-членного кольца, при этом какой-либо атом в кольце возможно замещен атомом N, О или S, и при этом кольцо возможно замещено галогеном, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''.
В конкретных воплощениях гетероциклильная группа присоединена через атом углерода этой гетерециклильной группы. В качестве примера, присоединенные через атом углерода гетерециклильные группы включают конфигурации с размещением точек присоединения (bonding arrangements) no положению 2, 3, 4, 5 или 6 пиридинового кольца, положению 3, 4, 5 или 6 пиридазинового кольца, по положению 2, 4, 5 или 6 пиримидинового кольца, положению 2, 3, 5 или 6 пиразинового кольца, по положению 2, 3, 4 или 5 фуранового, тетрагидрофуранового, тиофуранового, тиофенового, пиррольного или тетрагидропиррольного кольца, по положению 2, 4 или 5 оксазольного, имидазольного или тиазольного кольца, по положению 3, 4 или 5 изоксазольного, пиразольного или изотиазольного кольца, по положению 2 или 3 азиридинового кольца, по положению 2, 3 или 4 азетидинового кольца, по положению 2, 3, 4, 5, 6, 7 или 8 хинолинового кольца или по положению 1, 3, 4, 5, 6, 7 или 8 изохинолинового кольца.
В некоторых воплощениях гетероциклильная группа является N-связанной. В качестве примера, присоединенные через атом азота гетерециклильные или гетероарильные группы включают конфигурации с размещением точек присоединения по положению 1 азиридина, азетидина, пиррола, пирролидина, 2-пирролина, 3-пирролина, имидазола, имидазолидина, 2-имидазолина, 3-имидазолина, пиразола, пиразолина, 2-пиразолина, 3-пиразолина, пиперидина, пиперазина, индола, индолина, 1Н-индазола, по положению 2 изоиндола или изоиндолина, по положению 4 морфолина и по положению 9 карбазола или β-карболина.
Термин "алкокси" относится к линейному или разветвленному одновалентному радикалу, представленному формулой -OR, в которой R представляет собой алкил, как он определен в данном описании. Группы алкокси включают метокси, этокси, пропокси, изопропокси, моно-, ди- и три-фторметокси и циклопропокси.
"Ацил" означает карбонилсодержащий заместитель, представленный формулой -C(O)-R, где R представляет собой атом водорода, алкил, цикпоалкил, арил или гетероциклил, при этом алкил, циклоалкил, арил и гетероциклил являются такими, как они определены в данном описании. Ацильные группы включают алканоил (например, ацетил), ароил (например, бензоил) и гетероароил (например, пиридиноил).
Термин "возможно замещенный", если не указано иное, означает, что группа может быть незамещенной или может быть замещена одним или несколькими заместителями (например, 0, 1, 2, 3, 4 или 5 либо более, или любым количеством заместителей, выбранным из этого диапазона) из заместителей, перечисленных для этой группы, при этом указанные заместители могут быть одинаковыми или разными. В одном из воплощений возможно замещенная группа имеет 1 заместитель. В другом воплощении возможно замещенная группа имеет 2 заместителя. В другом воплощении возможно замещенная группа имеет 3 заместителя. В другом воплощении возможно замещенная группа имеет 4 заместителя. В другом воплощении возможно замещенная группа имеет 5 заместителей.
Возможные заместители для алкильных радикалов, сами по себе или как часть другого заместителя (например, алкокси), а также для алкиленила, алкенила, алкинила, гетероалкила, гетероциклоалкила и цикпоалкила, также сами по себе или как часть другого заместителя, могут представлять собой ряд групп, таких как группы, изложенные в данном описании, а также могут быть выбраны из группы, состоящей из галогена; оксо; CN; NO; N3; -OR'; перфтор-С1-С4алкокси; незамещенного С3-С7циклоалкила; С3-С7циклоалкила, замещенного атомом галогена, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''; незамещенного С6-С10арила (например, фенила); С6-С10арила, замещенного атомом галогена, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси или NR'R''; незамещенного 3-11-членного гетероцикпила (например, 5-6-членного гетероарила, содержащего 1-4 гетероатома, выбранных из О, N и S, или 4-11-членного гетероциклоалкила, содержащего 1-4 гетероатома, выбранных из О, N и S); 3-11-членного гетероциклила (например, 5-6-членного гетероарила, содержащего 1-4 гетероатома, выбранных из О, N и S, или 4-11-членного гетероциклоалкила, содержащего 1-4 гетероатома, выбранных из О, N и S), замещенного галогеном, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''; -NR'R''; -SR'; -SiR'R''R'''; -OC(O)R'; -C(O)R'; -CO2R'; -CONR'R''; -OC(O)NR'R''; -NR''C(O)R'; -NR'''C(O)NR'R''; -NR''C(O)2R'; -S(O)2R'; -S(O)2NR'R''; -NR'S(O)2R''; -NR'''S(O)2NR'R''; амидинила; гуанидинила; -(CH2)1-4-OR'; -(CH2)1-4-NR'R''; -(CH2)1-4-SR'; -(CH2)1-4-SiR'R''R'''; -(CH2)1-4-OC(O)R'; -(CH2)1-4-C(O)R'; -(CH2)1-4-CO2R'; и -(CH2)1-4CONR'R'' или их комбинаций, в количестве, изменяющемся от нуля до (2m'+1), где m' означает общее число атомов углерода в таком радикале. Каждый R', R'' и R''' независимо обозначает группы, включая, например, атом водорода; незамещенный C1-С6алкил; С1-С6алкил, замещенный атомом галогена, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''; незамещенный C1-С6гетероалкил; С1-С6гетероалкил, замещенный атомом галогена, ОН, CN, незамещенным С1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''; незамещенный С6-С10арил; С6-С10арил, замещенный атомом галогена, ОН, CN, незамещенным С1-С6алкилом, незамещенным C1-С6алкокси или NR'R''; незамещенный 3-11-членный гетероциклил (например, 5-6-членный гетероарил, содержащий 1-4 гетероатома, выбранных из О, N и S, или 4-11-членный гетероциклоалкил, содержащий 1-4 гетероатома, выбранных из О, N и S); и 3-11-членный гетероциклил (например, 5-6-членный гетероарил, содержащий 1-4 гетероатома, выбранных из О, N и S, или 4-11-членный гетероцикпоалкил, содержащий 1-4 гетероатома, выбранных из О, N и S), замещенный галогеном, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''. Если R' и R'' присоединены к одному и тому же атому азота, они могут быть объединены с атомом азота с образованием 3-, 4-, 5-, 6- или 7-членного кольца, при этом какой-либо атом в кольце возможно замещен атомом N, О или S, и при этом кольцо возможно замещено галогеном, ОН, CN, незамещенным С1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''. Например, подразумевается, что -NR'R'' включает 1-пирролидинил и 4-морфолинил.
Аналогичным образом варьируются возможные заместители для арильных и гетероарильных групп. В некоторых воплощениях заместители для арильных и гетероарильных групп выбраны из группы, состоящей из галогена; CN; NO; N3; -OR'; перфтор-С1-С4алкокси; незамещенного С3-С7циклоалкила; С3-С7циклоалкила, замещенного галогеном, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''; незамещенного С6-С10арила (например, фенила); С6-С10арила, замещенного галогеном, ОН, CN, незамещенным C1-С6алкилом, незамещенным С1-С6алкокси или NR'R''; незамещенного 3-11-членного гетероциклила (например, 5-6-членного гетероарила, содержащего 1-4 гетероатома, выбранных из О, N и S, или 4-11-членного гетероциклоалкила, содержащего 1-4 гетероатома, выбранных из О, N и S); 3-11-членного гетероциклила (например, 5-6-членного гетероарила, содержащего 1-4 гетероатома, выбранных из О, N и S, или 4-11-членного гетероциклоалкила, содержащего 1-4 гетероатома, выбранных из О, N и S), замещенного галогеном, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''; -NR'R''; -SR'; -SiR'R''R'''; -OC(O)R'; -C(O)R'; -CO2R'; -CONR'R''; -OC(O)NR'R''; -NR''C(O)R'; -NR'''C(O)NR'R''; -NR''C(O)2R'; -S(O)2R'; -S(O)2NR'R''; -NR'S(O)2R''; -NR'''S(O)2NR'R''; амидинила; гуанидинила; -(CH2)1-4-OR'; -(CH2)1-4-NR'R''; -(CH2)1-4-SR'; -(CH2)1-4-SiR'R''R'''; -(CH2)1-4-OC(O)R'; -(CH2)1-4-C(O)R'; -(CH2)1-4-CO2R'; и -(CH2)1-4CONR'R'' или их комбинаций, в количестве, изменяющемся от нуля до (2m'+1), где m' означает общее число атомов углерода в таком радикале. Каждый R', R'' и R''' независимо обозначает группы, включая, например, атом водорода; незамещенный C1-С6алкил; С1-С6алкил, замещенный галогеном, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''; незамещенный C1-С6 гетероалкил; С1-C6гетероалкил, замещенный галогеном, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''; незамещенный С6-С10арил; C6-С10арил, замещенный галогеном, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси или NR'R''; незамещенный 3-11-членный гетероциклил (например, 5-6-членный гетероарил, содержащий 1-4 гетероатома, выбранных из О, N и S, или 4-11-членный гетероциклоалкил, содержащий 1-4 гетероатома, выбранных из О, N и S); и 3-11-членный гетероциклил (например, 5-6-членный гетероарил, содержащий 1-4 гетероатома, выбранных из О, N и S, или 4-11-членный гетероциклоалкил, содержащий 1-4 гетероатома, выбранных из О, N и S), замещенный галогеном, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''. Если R' и R'' присоединены к одному и тому же атому азота, они могут быть объединены с атомом азота с образованием 3-, 4-, 5-, 6- или 7-членного кольца, при этом какой-либо атом в кольце возможно замещен атомом N, О или S, и при этом кольцо возможно замещено галогеном, ОН, CN, незамещенным C1-С6алкилом, незамещенным C1-С6алкокси, оксо или NR'R''. Например, подразумевается, что -NR'R'' включает 1-пирролидинил и 4-морфолинил.
Термин ''оксо'' относится к =O или (=O)2.
Использованная в данном описании волнистая линия " ", которая пересекает связь в химической структуре, указывает место присоединения атома, к которому отмеченная волнистой линией связь присоединяется в этой химической структуре к остальной части молекулы или к остальной части фрагмента молекулы. В некоторых воплощениях для указания места присоединения используют стрелку вместе со звездочкой наподобие волнистой линии.
", которая пересекает связь в химической структуре, указывает место присоединения атома, к которому отмеченная волнистой линией связь присоединяется в этой химической структуре к остальной части молекулы или к остальной части фрагмента молекулы. В некоторых воплощениях для указания места присоединения используют стрелку вместе со звездочкой наподобие волнистой линии.
В некоторых воплощениях двухвалентные группы описываются в общем, без конкретизации конфигураций относительно точки присоединения. Очевидно, что такое общее описание предполагает включение обеих конфигураций относительно точки присоединения, если не указано иное. Например, если группа R2 в группе R1-R2-R3 описана как -СН2С(O)-, то понимают, что эта группа может быть связана как в виде R1-CH2C(O)-R3, так и в виде R1-C(O)CH2-R3, если не указано иное.
Термины "соединение(ия) по данному изобретению" и "соединение(ия) по настоящему изобретению" и тому подобные, если не указано иное, включают в себя приведенные в данном описании соединения формулы (I), такие как соединения 1-18, иногда упоминаемые как ингибиторы JAK, в том числе их стереоизомеры (включая атропоизомеры), геометрические изомеры, таутомеры, сольваты, метаболиты, изотопно меченные варианты, соли (например, фармацевтически приемлемые соли) и пролекарства. В некоторых воплощениях исключены сольваты, метаболиты, изотопно меченные варианты или пролекарства либо любая их комбинация.
Фраза "фармацевтически приемлемые" относится к молекулярным субстанциям и композициям, которые не вызывают неблагоприятную, аллергическую или другую нежелательную реакцию при введении, в случае необходимости, такому животному, как, например, человек.
Соединения по настоящему изобретению могут быть в форме соли, такой как фармацевтически приемлемая соль. Термин "фармацевтически приемлемые соли" включает в себя соли присоединения как кислоты, так и основания. Термин "фармацевтически приемлемая соль присоединения кислоты" относится к таким солям, которые сохраняют биологическую эффективность и свойства свободных оснований и которые не являются нежелательными ни биологически, ни в других отношениях, к солям, которые образуются с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, угольная кислота, фосфорная кислота и тому подобное, и с органическими кислотами, которые могут быть выбраны из классов алифатических, циклоалифатических, ароматических, аралифатических, гетероциклических карбоновых и сульфоновых органических кислот, таких как муравьиная кислота, уксусная кислота, пропионовая кислота, гликолевая кислота, глюконовая кислота, молочная кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, аспарагиновая кислота, аскорбиновая кислота, глутаминовая кислота, антраниловая кислота, бензойная кислота, коричная кислота, миндальная кислота, эмбоновая кислота, фенилуксусная кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, л-толуолсульфоновая кислота, салициловая кислота и тому подобное.
Термин "фармацевтически приемлемые соли присоединения основания" включает в себя таковые, которые происходят из неорганических оснований, как например, соли натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди, марганца, алюминия и тому подобные соли. В особенности, солями присоединения основания являются соли аммония, калия, натрия, кальция и магния. Соли, происходящие из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, в том числе природных замещенных аминов, циклических аминов и основных ионообменных смол, таких как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, 2-диэтиламиноэтанол, трометамин, дицикпогексиламин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, этилендиамин, глюкозамин, метилглюкамин, теобромин, пурины, пиперазин, пиперидин, N-этилпиперидин, полиаминные смолы и им подобные. В особенности, органические нетоксичные основания включают изопропиламин, диэтиламин, этаноламин, трометамин, дицикпогексиламин, холин и кофеин.
В некоторых воплощениях соль выбрана из гидрохлорида, гидробромида, трифторацетата, сульфата, фосфата, ацетата, фумарата, малеата, тартрата, лактата, цитрата, пирувата, сукцината, оксалата, метансульфоната, п-толуолсульфоната, бисульфата, бензолсульфоната, этансульфоната, малоната, ксинафоата, аскорбата, олеата, никотината, сахарината, адипата, формиата, гликолята, пальмитата, L-лактата, D-лактата, аспартата, малата, L-тартрата, D-тартрата, стеарата, фуроата (например, 2-фуроата или 3-фуроата), нападизилата (нафталин-1,5-дисульфоната или нафталин-1-(сульфоновая кислота)-5-сульфоната), эдизилата (этан-1,2-дисульфоната или этан-1-(сульфоновая кислота)-2-сульфоната), изетионата (2-гидроксиэтилсульфоната), 2-мезитиленсульфоната, 2-нафталинсульфоната, 2,5-дихлорбензолсульфоната, D-манделата, L-манделата, циннамата, бензоата, адипата, эзилата, малоната, мезитилата (2-мезитиленсульфоната), напсилата (2-нафталинсульфоната), камзилата (камфор-10-сульфоната, например соли (1S)-(+)-10-камфорсульфоновой кислоты), глутамата, глутарата, гиппурата (2-(бензоиламино)ацетата), оротата, ксилата (п-ксилол-2-сульфоната) и памоата (2,2'-дигидрокси-1,1'-динафтилметан-3,3'-дикарбоксилата).
"Стерильная" композиция является асептической или не содержит никаких живых микроорганизмов и их спор.
Термин "стереоизомеры" относится к соединениям, которые имеют идентичный химический состав, но различаются расположением атомов или групп в пространстве. Стереоизомеры включают диастереомеры, энантиомеры, конформеры и тому подобное.
Термин "хиральный" относится к молекулам, обладающим свойством не совпадать при наложении на зеркально отображаемого партнера, тогда как термин "ахиральный" относится к молекулам, совпадающим при наложении на своего зеркально отображаемого партнера.
Термин "диастереомер" относится к стереоизомеру с двумя или более центрами хиральности, и такие молекулы не являются зеркальными отображениями друг друга. Диастереомеры имеют разные физические свойства, например, точки плавления, точки кипения, спектральные свойства и биологические активности. Смеси диастереомеров можно разделить с помощью аналитических методик высокого разрешения, таких как электрофорез и хроматография, как например, HPLC (высокоэффективная жидкостная хроматография).
Термин "энантиомеры" относится к двум стереоизомерам соединения, которые не совпадают с зеркальными отображениями друг друга.
В данном описании в основном использованы стереохимические определения и правила, представленные в S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984), McGraw-Hill Book Company, New York; и Eliel E. and Wilen S., "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., New York, 1994. Многие органические соединения существуют в оптически активных формах, т.е. они обладают способностью вращать плоскость плоскополяризованного света. При описании оптически активного соединения для обозначения абсолютной конфигурации молекулы относительно ее хирального(ых) центра(ов) используют префиксы D и L или R и S. Для обозначения знака направления вращения плоскополяризованного света соединением применяют префиксы d и I или (+) и (-), при этом (-) или I означает, что соединение является левовращающим. Соединение с префиксом (+) или d является правовращающим. Для данной химической структуры эти стереоизомеры являются идентичными, за исключением того, что они являются зеркальными отображениями друг друга. Конкретный стереоизомер также может быть определен как энантиомер, и смесь таких изомеров часто называют энантиомерной смесью. Смесь с соотношением энантиомеров 50:50 называется рацемической смесью или рацематом, и это может иметь место, если не соблюдалось никакой стереоизбирательности или стереоспецифичности в химической(ом) реакции или процессе. Термины "рацемическая смесь" и "рацемат" относятся к эквимолярной смеси двух энантиомерных разновидностей, не обладающей оптической активностью.
Термин "таутомер" или "таутомерная форма" относится к структурным изомерам, обладающим разной энергией, взаимопревращение которых протекает через низкий энергетический барьер. Например, протонные таутомеры (также известные как прототропные таутомеры) характеризуются взаимопревращениями в результате миграции протона, как например, при кето-енольной и имин-енаминной изомериях. Валентные таутомеры включают взаимопревращения посредством реогранизации некоторых из связывающих электронов.
Некоторые соединения по настоящему изобретению могут существовать в несольватированных формах, а также сольватированных формах, в том числе гидратированных формах. Термин "сольват" относится к ассоциации или комплексу одной или более молекул растворителя и соединения по настоящему изобретению. Примеры растворителей, с которыми образуются сольваты, включают воду, изопропанол, этанол, метанол, DMSO (диметилсульфоксид), этилацетат, уксусную кислоту и этаноламин. Некоторые соединения по настоящему изобретению могут существовать во множественных кристаллических или аморфных формах. В общем случае подразумевается, что все физические формы включены в объем настоящего изобретения. Термин "гидрат" относится к комплексу, где молекулой растворителя является молекула воды.
Термин "метаболит" относится к продукту, образуемому в процессе метаболизма конкретного соединения или его соли в организме. Такие продукты могут получаться из вводимого соединения, например, в результате его окисления, восстановления, гидролиза, амидирования, дезамидирования, этерификации, дезэтерификации, ферментативного расщепления и тому подобного.
Продукты метаболизма обычно идентифицируют посредством получения меченного радиоактивным (например, 14С или 3H) изотопом соединения по изобретению, введения его в детектируемой дозе (например, более чем примерно 0,5 мг/кг) животному, такому как крыса, мышь, морская свинка, обезьяна, или человеку, с обеспечением достаточного для осуществления метаболизма времени (обычно примерно от 30 секунд до 30 часов) и выделения продуктов его превращения из мочи, крови или других биологических образцов. Выделение этих продуктов отличается простотой, поскольку они являются мечеными (другие продукты выделяют посредством применения антител, способных к связыванию с эпитопами, сохранившимися в метаболите). Структуры метаболитов определяют традиционным образом, например, анализом с использованием масс-спектрометрии (MS), жидкостной хроматографии в сочетании с MS (LC/MS) или ядерного магнитного резонанса (ЯМР). В общем случае анализ метаболитов осуществляют так же, как и традиционные исследования метаболизма лекарственных средств, хорошо известные специалистам в данной области техники. Продукты метаболизма, при условии, что в других случаях они не обнаруживаются in vivo, полезны в диагностических анализах для определения терапевтической дозы соединений по изобретению.
"Субъектом", "индивидом" или "пациентом" является позвоночное животное. В некоторых воплощениях позвоночным животным является млекопитающее. Млекопитающие включают, но не ограничиваются этим, сельскохозяйственных животных (таких как коровы), спортивных животных, домашних животных (таких как морские свинки, кошки, собаки, кролики и лошади), приматов, мышей и крыс. В некоторых воплощениях млекопитающим является человек. В воплощениях, включающих введение пациенту ингибитора JAK, описанного в данной заявке, или его фармацевтически приемлемой соли, пациент может нуждаться в этом.
Термин "янус-киназа" относится к протеинкиназам JAK1, JAK2, JAK3 и TYK2. В некоторых воплощениях янус-киназа также может быть определена как одна киназа из JAK1, JAK2, JAK3 или TYK2. В любом из воплощений какая-либо одна киназа из JAK1, JAK2, JAK3 и TYK2 может быть специально исключена как янус-киназа. В некоторых воплощениях янус-киназой является JAK1. В некоторых воплощениях янус-киназа представляет собой комбинацию JAK1 и JAK2.
Термины "ингибирование" и "снижение" или любой вариант этих терминов включают в себя любое измеряемое уменьшение или полное ингибирование с достижением желаемого результата. Например, уменьшением может быть снижение активности (например, активности JAK1) примерно, самое большее примерно или по меньшей мере примерно на 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 99% или больше либо на любую величину в процентах, выбранную из этого диапазона, по сравнению с обычным уровнем.
В некоторых воплощениях соединение или его соль (например, его фармацевтически приемлемая соль) описаны в данной заявке как селективные в отношении ингибирования JAK1 по сравнению с JAK3 и TYK2. В некоторых воплощениях соединение или его соль (например, его фармацевтически приемлемая соль) селективны в отношении ингибирования JAK1 по сравнению с JAK2, JAK3 или TYK2 либо с любой комбинацией JAK2, JAK3 или TYK2. В некоторых воплощениях соединение или его соль (например, его фармацевтически приемлемая соль) селективны в отношении ингибирования JAK1 и JAK2 по сравнению с JAK3 и TYK2. В некоторых воплощениях соединение или его соль (например, его фармацевтически приемлемая соль) селективны в отношении ингибирования JAK1 по сравнению с JAK3. Под "селективностью в отношении ингибирования" понимают, что соединение или его соль (например, его фармацевтически приемлемая соль) является более сильным, по меньшей мере на 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 99% или больше, либо на любую величину, выбранную из этого диапазона, ингибитором конкретной янус-киназной (например, JAK1) активности по сравнению с другой конкретной янус-киназной (например, JAK3) активностью, или является по меньшей мере в 2, 3, 4, 5, 10, 25, 50, 100, 250 или 500 раз более сильным ингибитором конкретной янус-киназной (например, JAK1) активности по сравнению с другой конкретной янус-киназной (например, JAK3) активностью.
Фраза "терапевтически эффективное количество" означает количество соединения или его соли (например, его фармацевтически приемлемой соли) по настоящему изобретению, которое (1) лечит или предотвращает конкретное заболевание, состояние или расстройство, либо (2) ослабляет, уменьшает интенсивность или устраняет один или более симптомов конкретного заболевания, состояния или расстройства, и возможно (3) предотвращает или задерживает начало развития одного или более симптомов конкретного заболевания, состояния или расстройства, описанного в данной заявке. В некоторых воплощениях терапевтически эффективным количеством является количество, достаточное для снижения или ослабления симптомов аутоиммунного или воспалительного заболевания (например, астмы). В некоторых воплощениях терапевтически эффективным количеством является количество химического соединения, описанного в данной заявке, достаточное для существенного уменьшения активности или количества В-клеток. В случае рака терапевтически эффективное количество лекарственного средства может уменьшать количество раковых клеток; уменьшать размер опухоли; ингибировать (т.е. замедлять до некоторой степени и предпочтительно останавливать) инфильтрацию раковых клеток в периферические органы; ингибировать (т.е. замедлять до некоторой степени и предпочтительно останавливать) опухолевый метастаз; ингибировать, до некоторой степени, опухолевый рост; или ослаблять до некоторой степени один или более симптомов, ассоциированных с раком. В зависимости от возможности лекарственного средства предотвращать рост или уничтожать существующие раковые клетки, оно может быть цитостатическим или цитотоксическим. В случае терапии рака эффективность можно измерить, например, оценивая время до прогрессирования заболевания (ТТР) или определяя коэффициент ответа (RR).
Термин "лечение" (и такие варианты, как "лечить" или "подвергать лечению") относится к клиническому вмешательству с попыткой изменения естественного течения заболевания у подвергаемых лечению индивида или клетки, которое может быть проведено либо для профилактики, либо в процессе развития клинической патологии. Желательные эффекты лечения включают предупреждение проявления или рецидива заболевания, облегчение симптомов, сокращение любых прямых или косвенных патологических последствий данного заболевания, стабильное (т.е. без ухудшения) состояние заболевания, уменьшение скорости прогрессирования заболевания, уменьшение интенсивности симптомов или временное облегчение болезненного состояния, продление выживаемости по сравнению с ожидаемой выживаемостью в отсутствие получения лечения и ремиссию или улучшенный прогноз. В некоторых воплощениях соединение по изобретению или его соль (например, его фармацевтически приемлемую соль) применяют для задержки развития заболевания или расстройства либо для замедления прогрессирования заболевания или расстройства. Нуждающиеся в таком лечении включают тех, кто уже имеет данное состояние или расстройство, а также тех, кто предрасположен к данному состоянию или расстройству (например, в результате генетической мутации), или тех, у которых данное состояние или расстройство должно быть предотвращено.
Термин "воспалительное расстройство" относится к любому заболеванию, расстройству или синдрому, при котором чрезмерный или нерегулируемый воспалительный ответ вызывает гипертрофированные воспалительные симптомы, повреждение ткани реципиента или утрату функции тканей. Термин "воспалительное расстройство" также относится к патологическому состоянию, опосредуемому притоком лейкоцитов или хемотаксисом нейтрофилов.
Термин "воспаление" относится к локализованному протективному ответу, вызываемому повреждением или разрушением тканей, который направлен на разрушение, ослабление или создание барьера (изолирование) как в отношении вредоносного агента, так и поврежденной ткани. Воспаление в значительной мере ассоциировано с притоком лейкоцитов или хемотаксисом нейтрофилов. Воспаление может возникнуть в результате инфекции патогенными микроорганизмами и вирусами и возникнуть неинфекционными путями, например, в результате травмы или реперфузии после инфаркта миокарда или инсульта, иммунного ответа на чужеродные антигены и аутоиммунных ответов. Соответственно, воспалительные расстройства, поддающиеся лечению соединением или его солью (например, его фармацевтически приемлемой солью) по настоящему изобретению охватывают расстройства, ассоциированные с ответами специфической защитной системы, а также с ответами неспецифической защитной системы.
Термин "специфическая защитная система" относится к компоненту иммунной системы, который реагирует на присутствие специфических антигенов. Примеры воспаления, являющегося результатом ответа специфической защитной системы, включают классический ответ на чужеродные антигены, аутоиммунные заболевания и реакцию гиперчувствительности замедленного типа, опосредуемую Т-клетками. Хронические воспалительные заболевания, отторжение твердых трансплантированных тканей и органов, например, трансплантатов почки и костного мозга, и реакция "трансплантат против хозяина" (GVHD), представляют собой другие примеры воспалительных ответов специфической защитной системы.
Термин "неспецифическая защитная система" относится к воспалительным расстройствам, которые опосредованы лейкоцитами, не обладающими иммунологической памятью (например, гранулоцитами и макрофагами). Примеры воспаления, являющегося результатом, по меньшей мере отчасти, ответа неспецифической защитной системы, включают воспаление, ассоциированное с такими состояниями, как (острый) респираторный дистресс-синдром взрослых (ARDS) или синдромы полиорганной недостаточности; реперфузионная травма; острый гломерулонефрит; реактивный артрит; дерматозы с острыми воспалительными компонентами; острый гнойный менингит или другие воспалительные расстройства центральной нервной системы, такие как инсульт; термическое повреждение; воспалительное заболевание кишечника; синдромы, ассоциированные с переливанием гранулоцитов, и цитокин-индуцируемая токсичность.
Термин "аутоиммунное заболевание" относится к любой группе расстройств, при которых повреждение ткани ассоциировано с гуморальными или клеточно-опосредуемыми ответами на собственные компоненты организма. Неограничивающие примеры аутоиммунных заболеваний включают ревматоидный артрит, волчанку и рассеянный склероз.
Термин "аллергическое заболевание", использованный в данном описании, относится к любым симптомам, поражению ткани или утрате функции ткани в результате аллергической реакции. Фраза "ассоциированное с артритом заболевание", использованная в данном описании, относится к любому заболеванию, характеризующемуся воспалительными поражениями суставов различной этиологии. Термин "дерматит", использованный в данном описании, относится к любому заболеванию из большого семейства заболеваний кожи, характеризующихся воспалением кожи различной этиологии. Термин "отторжение трансплантата", использованный в данном описании, относится к любой иммунной реакции, направленной против трансплантированной ткани, например, против органов, или клеток (например, костного мозга), характеризующейся утратой функции трансплантированной и окружающих ее тканей, болью, отеком, лейкоцитозом и тромбоцитопенией. Способы терапии по настоящему изобретению включают способы лечения расстройств, ассоциируемых с активацией воспалительных клеток.
Термин "активация воспалительных клеток" относится к индуцированию под действием стимулирующего вещества (в том числе, но не ограничиваясь этим, цитокинов, антигенов или аутоантител) пролиферативного клеточного ответа, к продуцированию растворимых медиаторов (в том числе, но не ограничиваясь этим, цитокинов, кислородных радикалов, ферментов, простаноидов или вазоактивных аминов) или к экспрессии на клеточной поверхности новых медиаторов или более высоких количеств медиаторов (в том числе, но не ограничиваясь этим, антигенов главного комплекса гистосовместимости или молекул клеточной адгезии) в воспалительных клетках (в том числе, но не ограничиваясь этим, моноцитах, макрофагах, Т-лимфоцитах, В-лимфоцитах, гранулоцитах (т.е. полиморфоядерных лейкоцитах, таких как нейтрофилы, базофилы и эозинофилы), тучных клетках, дендритных клетках, клетках Лангерганса и эндотелиальных клетках). Специалистам в данной области техники будет очевидно, что активация таких клеток одного фенотипа или комбинации этих фенотипов может способствовать инициации, сохранению или обострению воспалительного расстройства.
В некоторых воплощениях воспалительные расстройства, которые можно лечить согласно способам по данному изобретению, включают, но не ограничиваются этим, астму, ринит (например, аллергический ринит), аллергический синдром дыхательных путей, атопический дерматит, бронхит, ревматоидный артрит, псориаз, контактный дерматит, хроническую обструктивную болезнь легких (COPD) и реакции гиперчувствительности замедленного типа.
Термины "рак" и "раковый", "новообразование" и "опухоль", а также родственные термины относятся к физиологическим состояниям или описывают физиологические состояния у млекопитающих, которые обычно характеризуются нерегулируемым клеточным ростом. Термин "опухоль" подразумевает наличие одного или более типов раковых клеток. Примеры рака включают карциному, бластому, саркому, семиному, глиобластому, меланому, лейкоз и миелоидные или лимфоидные неоплазии. Более конкретные примеры таких видов рака включают плоскоклеточный рак (например, плоскоклеточный рак эпителиальной ткани), рак легкого, в том числе мелко клеточный рак легкого, немелкоклеточный рак легкого ("NSCLC"), аденокарциному легкого и плоско клеточный рак легкого. Другие виды рака включают рак кожи, кератоакантому, фолликулярную карциному, волосатоклеточный лейкоз, рак щеки, глотки, губ, языка, полости рта, слюнных желез, пищевода, гортани, гепатоклеточный рак, желудочный рак, рак желудка, желудочно-кишечного тракта, тонкого кишечника, толстого кишечника, поджелудочной железы, шейки матки, яичников, печени, мочевого пузыря, гепатому, рак молочной железы, толстой кишки, прямой кишки, колоректальный рак, рак органов мочеполовой системы, желчного протока, щитовидной железы, папиллярный рак, печеночный рак, рак эндометрия, матки, слюнных желез, почки или почечный рак, рак предстательной железы, яичка, вульвы, брюшины, анальный рак, рак пениса, кости, множественную миелому, В-клеточную лимфому, рак центральной нервной системы, головного мозга, головы и шеи, лимфому Ходжкина и ассоциированые метастазы. Примеры опухолевых заболеваний включают миелопролиферативные расстройства, такие как истинная полицитемия, эссенциальный тромбоцитоз, миелофиброз, такой как первичный миелофиброз, и хронический миелогенный лейкоз (CML).
"Химиотерапевтический агент" представляет собой агент, полезный в лечении заданного расстройства, например, рака или воспалительных расстройств. Примеры химиотерапевтических агентов хорошо известны в данной области техники и включают такие примеры, которые описаны в публикации заявки США №2010/0048557, включенной в данное описание посредством ссылки. Кроме того, химиотерапевтические агенты включают фармацевтически приемлемые соли, кислоты или производные любого из химиотерапевтических агентов, а также комбинации двух или более из них.
Термин "инструкция по применению препарата" обычно имеет отношение к инструкциям, традиционно включаемым в промышленные упаковки терапевтических продуктов, которые содержат информацию о показаниях, применении, дозировке, введении, противопоказаниях и/или предупреждениях касательно применения таких терапевтических продуктов.
Если не указано иное, то структуры, приведенные в данном описании, включают соединения, отличающиеся только наличием одного или более чем одного обогащенного изотопом атома. Типичные изотопы, которые могут быть инкорпорированы в соединения по настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, хлора и йода, такие как 2H, 3H, 11С, 13С, 14С, 13N, 15N, 15O, 17О, 18О, 32Р, 33Р, 35S, 18F, 36Cl, 123I и 125I, соответственно. Меченные изотопом соединения (например, соединения, меченные 3H и 14С) могут быть полезны в анализах распределения соединения или субстрата в тканях. Изотопы тритий (т.е. 3H) и углерод-14 (т.е. 14С) могут быть полезны ввиду простоты их получения и способности к детекции. Кроме того, замена на более тяжелые изотопы, такие как дейтерий (т.е. 2H), может давать некоторые терапевтические преимущества, обусловленные большей метаболической стабильностью (например, увеличенный in vivo период полувыведения или снижение дозировки). В некоторых воплощениях один или более атомов водорода заменены на 2H или 3H либо один или более атомов углерода заменены на 13С- или 14С-обогащенный атом углерода. Испускающие позитроны изотопы, такие как 15О, 13N, 11С и 18F, полезны в исследованиях с использованием позитронно-эмиссионной томографии (PET) для изучения степени занятости рецепторов субстратом. Меченные изотопом соединения обычно могут быть получены путем осуществления методик, аналогичных методикам, изложенным в данном описании на реакционных схемах или в разделе Примеры, в результате замены не меченного изотопом реагента на меченный изотопом реагент.
Специально оговаривается, что любое ограничение, рассматриваемое в отношении одного из воплощений изобретения, может применяться в отношении любого другого воплощения изобретения. Помимо этого, любое соединение или его соль (например, его фармацевтически приемлемую соль) либо любую композицию по изобретению можно использовать в любом способе по изобретению, а любой способ по изобретению можно использовать для получения или для применения любого соединения или его соли (например, его фармацевтически приемлемой соли) либо любой композиции по изобретению.
Применение термина "или" используется для обозначения "и/или", если явно не указано, что он относится только к альтернативам или что альтернатива является взаимоисключающей, хотя данное описание поддерживает определение, которое относится только к альтернативам и "и/или".
По всему объему этой заявки термин "примерно" используется для указания на то, что какое-либо значение включает в себя стандартное отклонение ошибки устройства или метода, используемых для определения этого значения.
Использованный в данном описании английский артикль "а" или "an" означает "один или более", если явно не указано иное. Использованный в данном описании термин "другой" означает по меньшей мере "второй" или более.
Заголовки, используемые в данном описании, предназначены только для организации представления материала.
Ингибиторы янус-киназ
В одном из воплощений предложено соединение формулы (I):

или его соль (например, фармацевтически приемлемая соль), где:
R1 представляет собой атом водорода или СН3;
R2 представляет собой атом галогена, C1-С6алкил, С2-С6алкенил, C2-С6алкинил, С3-С6циклоалкил или -ORa, при этом R2 возможно замещен одной или несколькими группами, независимо выбранными из группы, состоящей из атома галогена, C1-С3алкила, циано, гидрокси и оксо;
Ra представляет собой C1-С6алкил, -фенил-CORbRc, -фенил-(3-6-членный гетероциклил) или 3-11-членный гетероциклил, при этом Ra возможно замещен одной или несколькими группами, независимо выбранными из группы, состоящей из атома галогена, C1-С3алкила, циано, гидрокси и оксо;
Rb и Rc каждый независимо представляет собой атом водорода или СН3;
R3 представляет собой атом водорода или NH2;
R4 представляет собой атом водорода или СН3; и
R5 представляет собой атом водорода или NH2.
В некоторых воплощениях R1 представляет собой атом водорода. В некоторых воплощениях R1 представляет собой СН3. В некоторых воплощениях R3 представляет собой атом водорода. В некоторых воплощениях R4 и R5 каждый представляет собой атом водорода. В некоторых воплощениях R1, R3, R4 и R5 каждый представляет собой атом водорода.
В некоторых воплощениях R2 выбран из группы, состоящей из атома галогена, C1-С6галогеналкила и C1-С6галогеналкилокси. В некоторых воплощениях R2 выбран из группы, состоящей из

В некоторых воплощениях предложено соединение, выбранное из представленных ниже соединений 1-18, или его соль (например, фармацевтически приемлемая соль) либо стереоизомер:


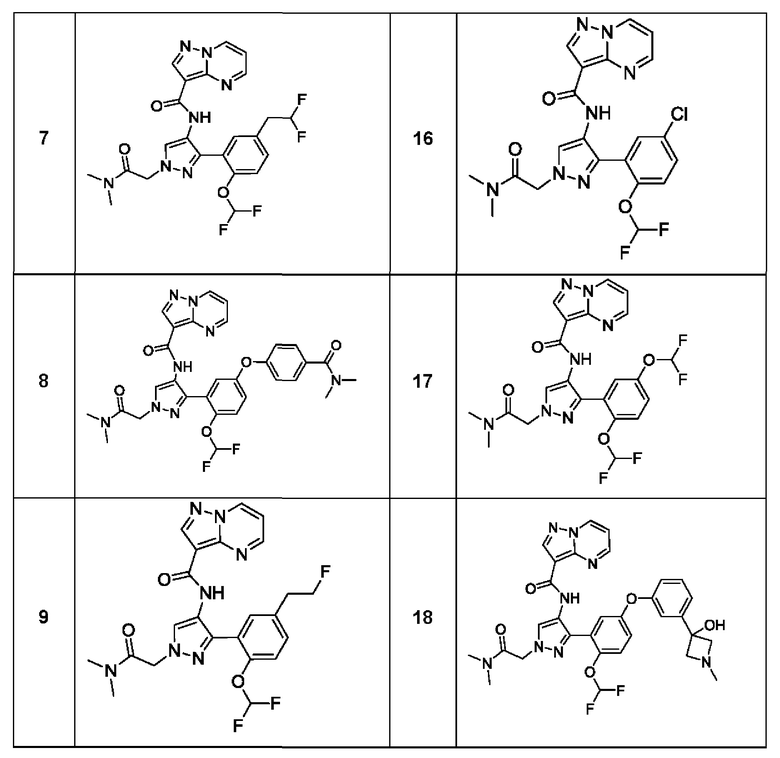
В некоторых воплощениях предложено следующее соединение:
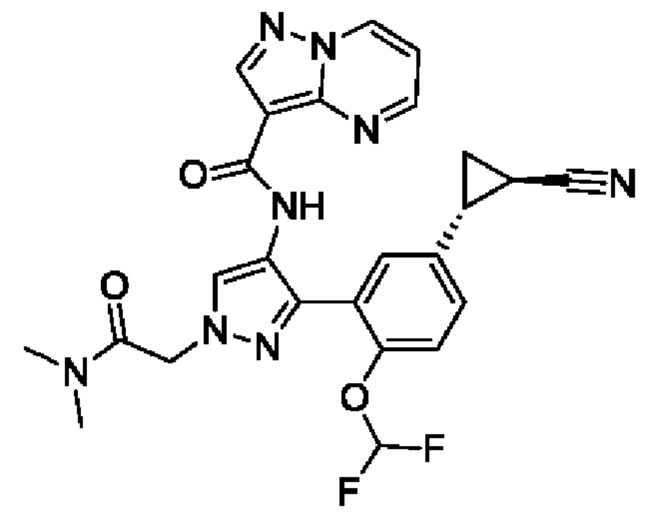
или его соль (например, фармацевтически приемлемая соль).
В некоторых воплощениях предложено следующее соединение:

или его соль (например, фармацевтически приемлемая соль).
В некоторых воплощениях предложено следующее соединение:

или его соль (например, фармацевтически приемлемая соль).
В некоторых воплощениях предложено следующее соединение:
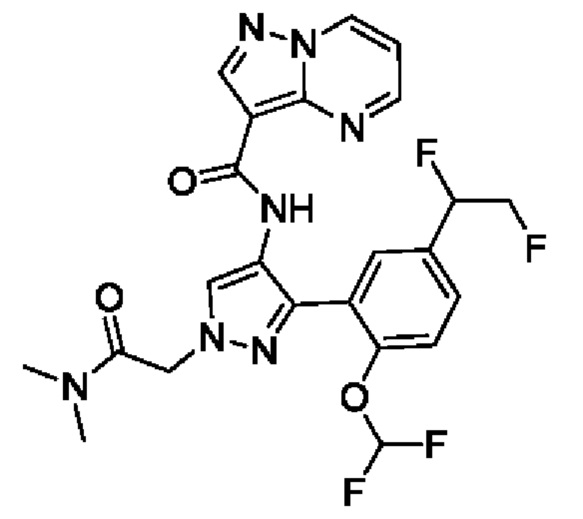
или его соль (например, фармацевтически приемлемая соль) или стереоизомер. В некоторых воплощениях предложено следующее соединение:

или его соль (например, фармацевтически приемлемая соль).
В некоторых воплощениях предложено следующее соединение:

или его соль (например, фармацевтически приемлемая соль) или стереоизомер. В некоторых воплощениях предложено следующее соединение:
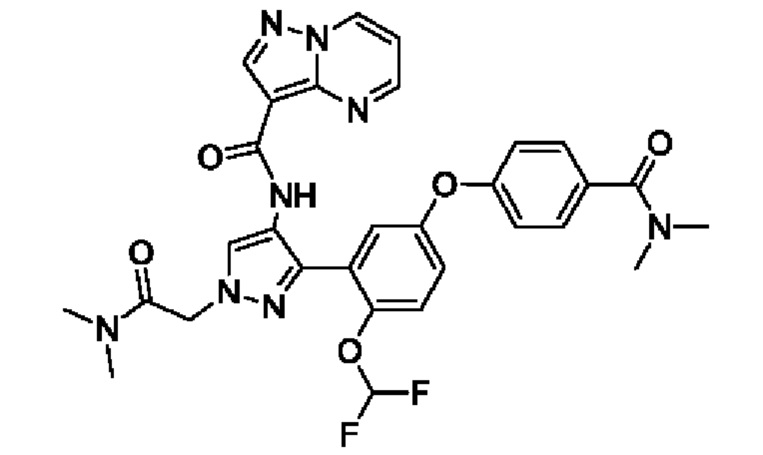
или его соль (например, фармацевтически приемлемая соль).
В некоторых воплощениях предложено следующее соединение:

или его соль (например, фармацевтически приемлемая соль).
В некоторых воплощениях предложено следующее соединение:
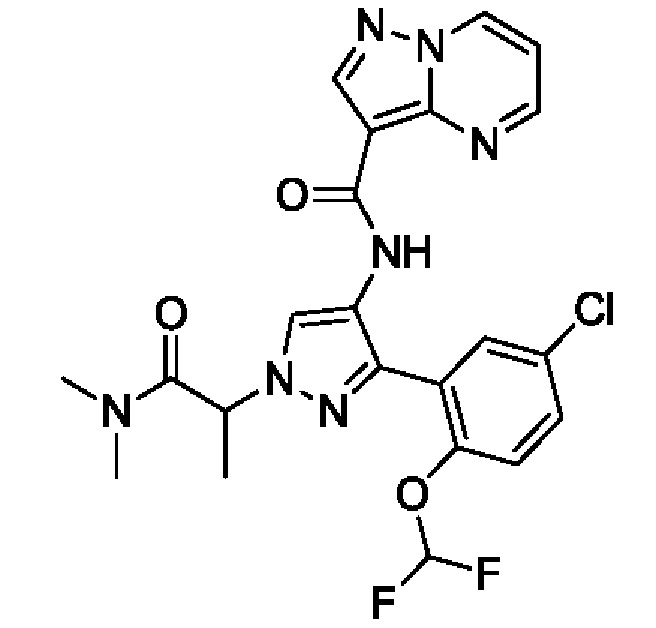
или его соль (например, фармацевтически приемлемая соль) или стереоизомер. В некоторых воплощениях предложено следующее соединение:

или его соль (например, фармацевтически приемлемая соль) или стереоизомер. В некоторых воплощениях предложено следующее соединение:

или его соль (например, фармацевтически приемлемая соль).
В некоторых воплощениях предложено следующее соединение:
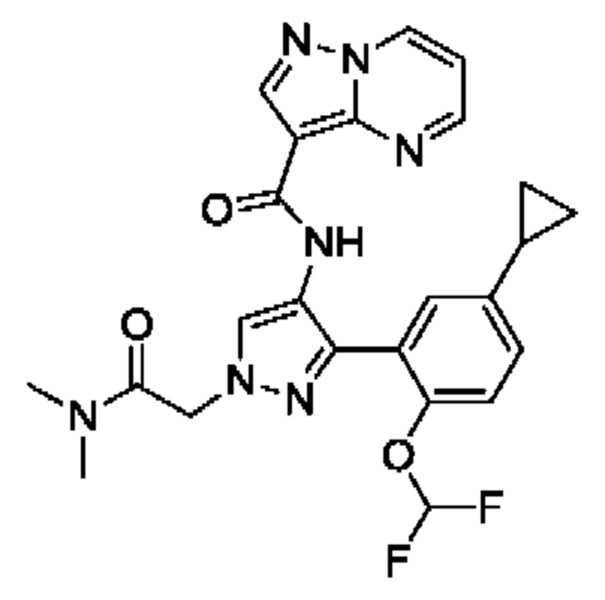
или его соль (например, фармацевтически приемлемая соль).
В некоторых воплощениях предложено следующее соединение:

или его соль (например, фармацевтически приемлемая соль).
В некоторых воплощениях предложено следующее соединение:
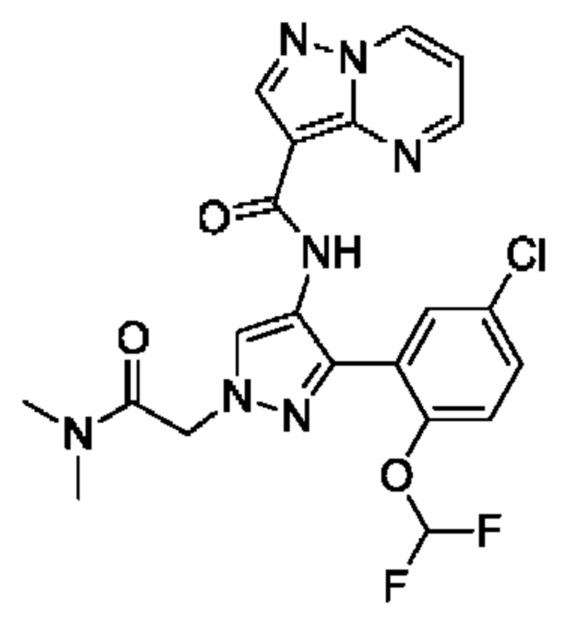
или его соль (например, фармацевтически приемлемая соль).
В некоторых воплощениях предложено следующее соединение:

или его соль (например, фармацевтически приемлемая соль).
В некоторых воплощениях предложено следующее соединение:

или его соль (например, фармацевтически приемлемая соль).
В некоторых воплощениях предложено соединение, выбранное из представленных ниже соединений (a)-(v), или его соль (например, фармацевтически приемлемая соль) либо стереоизомер:
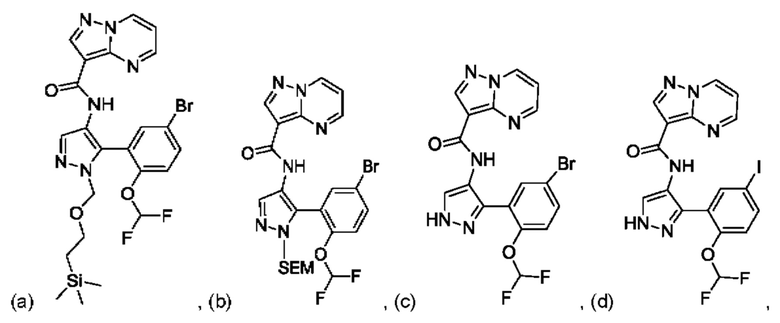
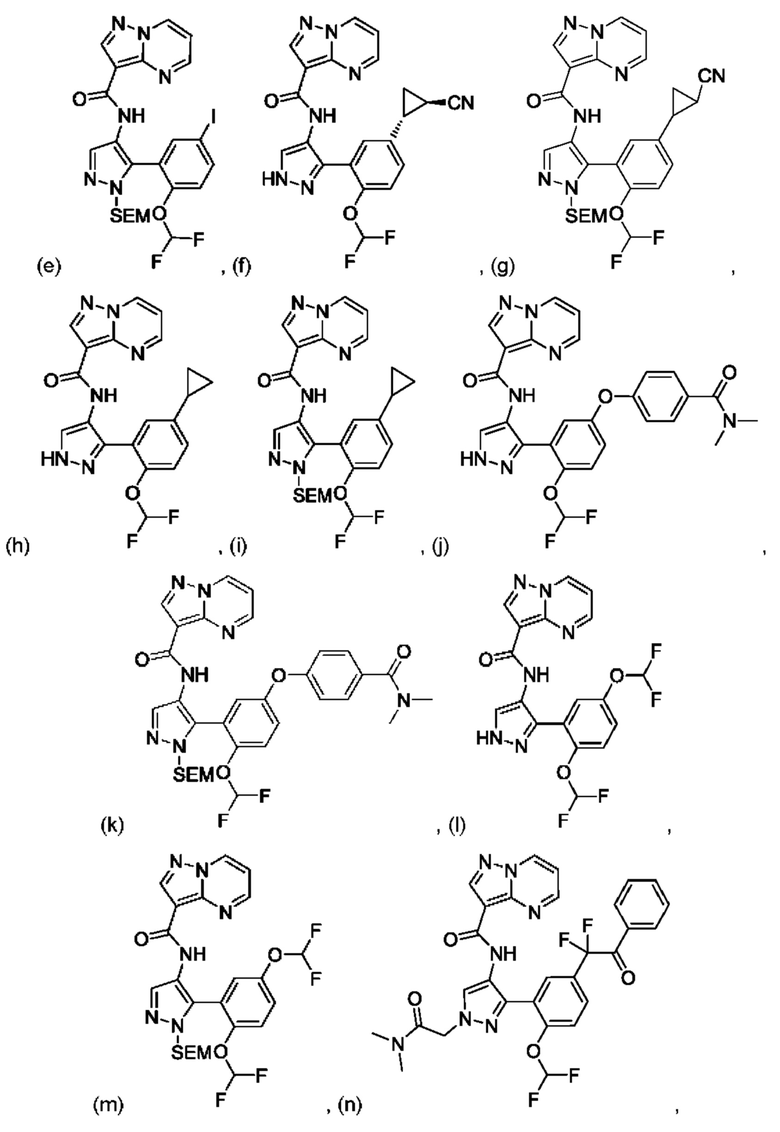
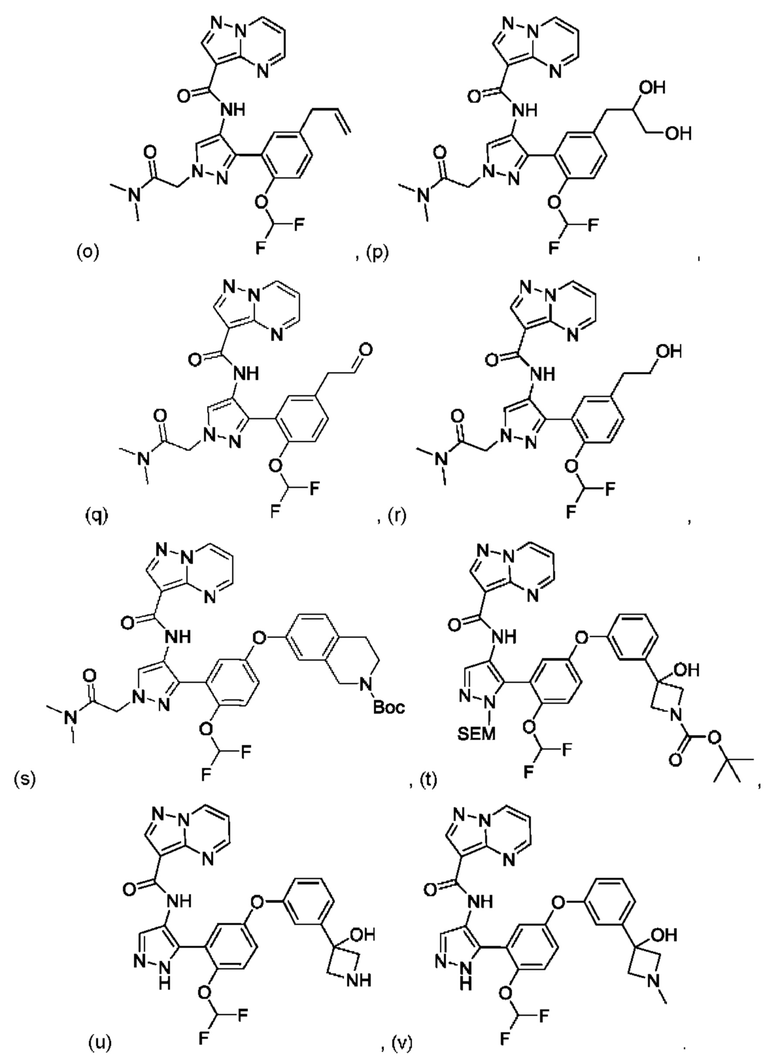
Также предложена фармацевтическая композиция, содержащая ингибитор JAK, описанный в данной заявке, или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или эксципиент.
Также предложено применение ингибитора JAK, описанного в данной заявке, или его фармацевтически приемлемой соли в терапии, как например, в лечении воспалительного заболевания (например, астмы). Также предложено применение ингибитора JAK, описанного в данной заявке, или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения воспалительного заболевания. Также предложен способ предупреждения, лечения или ослабления тяжести заболевания или состояния, отвечающего на ингибирование активности янус-киназы у пациента, включающий введение пациенту терапевтически эффективного количества ингибитора JAK, описанного в данной заявке, или его фармацевтически приемлемой соли.
В одном из воплощений заболеванием или состоянием, подлежащим терапии, является рак, истинная полицитемия, эссенциальный тромбоцитоз, миелофиброз, хронический миелогенный лейкоз (CML), ревматоидный артрит, синдром раздраженного кишечника, болезнь Крона, псориаз, контактный дерматит или реакции гиперчувствительности замедленного типа.
В одном из воплощений предложено применение ингибитора JAK, описанного в данной заявке, или его фармацевтически приемлемой соли, для лечения рака, истинной полицитемии, эссенциального тромбоцитоза, миелофиброза, хронического миелогенного лейкоза (CML), ревматоидного артрита, синдрома раздраженного кишечника, болезни Крона, псориаза, контактного дерматита или реакций гиперчувствительности замедленного.
В одном из воплощений предложена композиция, приготовленная для введения посредством ингаляции.
В одном из воплощений предложен дозирующий ингалятор, который содержит соединение по настоящему изобретению или его фармацевтически приемлемую соль.
В одном из воплощений ингибитор JAK, описанный в данной заявке, или его фармацевтически приемлемая соль является по меньшей мере в пять раз более сильным ингибитором JAK1, чем ингибитором LRRK2.
В одном из воплощений ингибитор JAK, описанный в данной заявке, или его фармацевтически приемлемая соль является по меньшей мере в десять раз более сильным ингибитором JAK1, чем ингибитором LRRK2.
В одном из воплощений ингибитор JAK, описанный в данной заявке, или его фармацевтически приемлемая соль является по меньшей мере в пять раз более сильным ингибитором JAK1, чем ингибитором JAK2.
В одном из воплощений ингибитор JAK, описанный в данной заявке, или его фармацевтически приемлемая соль является по меньшей мере в десять раз более сильным ингибитором JAK1, чем ингибитором JAK2.
В одном из воплощений ингибитор JAK, описанный в данной заявке, или его фармацевтически приемлемая соль обладает по меньшей мере является по меньшей мере в пять раз более сильным ингибитором JAK1, чем ингибитором JAK3.
В одном из воплощений ингибитор JAK, описанный в данной заявке, или его фармацевтически приемлемая соль является по меньшей мере в десять раз более сильным ингибитором JAK1, чем ингибитором JAK3.
В одном из воплощений ингибитор JAK, описанный в данной заявке, или его фармацевтически приемлемая соль является по меньшей мере в пять раз более сильным ингибитором JAK1, чем ингибитором TYK2.
В одном из воплощений ингибитор JAK, описанный в данной заявке, или его фармацевтически приемлемая соль является по меньшей мере в десять раз более сильным ингибитором JAK1, чем ингибитором TYK2.
В одном из воплощений предложен способ лечения выпадения волос у млекопитающего, включающий введение млекопитающему ингибитора JAK, описанного в данной заявке, или его фармацевтически приемлемой соли.
В одном из воплощений предложено применение ингибитора JAK, описанного в данной заявке, или его фармацевтически приемлемой соли для лечения выпадения волос.
В одном из воплощений предложено применение ингибитора JAK, описанного в данной заявке, или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения выпадения волос у млекопитающего.
Соединения по изобретению могут содержать один или более ассиметрических атомов углерода. Соответственно, соединения могут существовать в виде диастереомеров, энантиомеров или их смесей. В процедурах синтеза таких соединений могут быть использованы рацематы, диастереомеры или энантиомеры в качестве исходных веществ или в качестве промежуточных соединений. Смеси конкретных диастереомерных соединений могут быть разделены или обогащены по одному или более чем одному конкретному диастереомеру методами хроматографии или кристаллизации. Аналогичным образом могут быть разделены или энантиомерно обогащены энантиомерные смеси с использованием тех же или других методов, известных в данной области техники. Каждый из ассиметрических атомов углерода или азота может находиться в R- или S-конфигурации, и обе эти конфигурации включены в объем данного изобретения.
Если в структурах, приведенных в данном описании, стереохимическая конфигурация какого-либо конкретного хирального атома не указана, то в качестве соединений по изобретению охвачены и включены все стереоизомеры. Если стереохимическая конфигурация указана сплошным клином или пунктирной линией, изображающей конкретную конфигурацию, то указан и определен именно этот стереоизомер. Если не указано иное, то в случае использования сплошных клинов или пунктирных линий подразумевается относительная стереохимическая конфигурация.
Другой аспект включает пролекарства соединений, описанных в данной заявке, содержащие известные амино-защитные и карбокси-защитные группы, которые высвобождаются в физиологических условиях, например, в процессе гидролиза, с получением соединения по настоящему изобретению.
Термин "пролекарство" относится к форме предшественника или производного фармацевтически активного вещества, которая менее эффективна для пациента по сравнению с исходным лекарственным средством и способна к активации или превращению ферментативным или гидролитическим путем в более активную исходную форму. См., например, Wilman, "Prodrugs in Cancer Chemotherapy", Biochemical Society Transactions, 14, pp. 375-382, 615th Meeting Belfast (1986) и Stella et al., "Prodrugs: A Chemical Approach to Targeted Drug Delivery", Directed Drug Delivery, Borchardt et al., (ed.), pp. 247-267, Humana Press (1985). Пролекарства включают, но не ограничиваются этим, фосфатсодержащие пролекарства, тиофосфатсодержащие пролекарства, сульфатсодержащие пролекарства, пептидсодержащие пролекарства, модифицированные D-аминокислотами пролекарства, гликозилированные пролекарства, β-лактамсодержащие пролекарства, возможно замещенные феноксиацетамидсодержащие пролекарства или возможно замещенные фенилацетамидсодержащие пролекарства и пролекарства на основе 5-фторцитозина и 5-фторуридина.
Конкретным классом пролекарств являются соединения, у которых атом азота в группе амино, амидино, аминоалкиленамино, иминоалкиленамино или гуанидино замещен группой гидрокси, алкилкарбонильной (-CO-R) группой, алкоксикарбонильной (-COOR) или ацилоксиалкил-алкоксикарбонильной (-COO-R-O-COR) группой, где R представляет собой одновалентную или двухвалентную группу, например, алкил, алкилен или арил, либо группой, имеющей формулу -С(O)-O-СР1Р2-галогеналкил, где Р1 и Р2 являются одинаковыми или разными и представляют собой атом водорода, алкил, алкокси, циано, галоген, алкил или арил. В конкретном воплощении атом азота является одним из атомов азота группы амидино. Пролекарства могут быть получены в результате взаимодействия соединения с активированной группой, такой как ацильные группы, для образования связи, например, между атомом азота в данном соединении и типичным карбонилом активированной ацильной группы. Примерами активированных карбонильных соединений являются соединения, содержащие уходящую группу, связанную с карбонильной группой, и они включают, например, ацилгалогениды, ациламины, соли ацилпиридиния, ацилалкилаты, ацилфеноляты, такие как п-нитрофеноксиацил, динитрофеноксиацил, фторфеноксиацил и дифторфеноксиацил. Как правило, реакции осуществляют в инертных растворителях при пониженных температурах, например от -78°С до примерно 50°С. Данные реакции также могут быть проведены в присутствии неорганического основания, например, карбоната калия или бикарбоната натрия, либо органического основания, такого как амин, включая пиридин, триметиламин, триэтиламин, триэтаноламин или тому подобное.
Также включены другие типы пролекарств. Например, ингибитор JAK, описанный в данной заявке, может быть модифицирован по свободной карбоксильной группе с получением амида или сложного алкилового эфира. В качестве другого примера, соединения по настоящему изобретению, содержащие свободные гидроксигруппы, могут быть модифицированы с получением пролекарств в результате превращения гидроксигруппы в такую группу, как группа сложного фосфатного эфира, гемисукцината, диметиламиноацетата или фосфорилоксиметилоксикарбонильная группа, но не ограничиваясь этим, что приведено в Fleisher D. et al., (1996) Improved oral drug delivery: solubility limitations overcome by the use of prodrugs, Advanced Drug Delivery Reviews, 19: 115. Также включены карбаматные пролекарства на основе гидрокси- и аминогрупп в виде карбонатных пролекарств, сложных сульфонатных эфиров и сложных сульфатных эфиров на основе гидроксигрупп. Также включены производные по гидрокси группам в виде (ацилокси)метиловых и (ацилокси)этиловых простых эфиров, где ацильная группа может представлять собой группу сложного алкилового эфира, возможно замещенного группами, включая, но не ограничиваясь этим, функциональные группы простых эфиров, аминов и карбоновых кислот, или где ацильная группа представляет собой группу сложного эфира аминокислоты, как описано выше. Пролекарства этого типа описаны в J. Med. Chem. (1996), 39: 10. Более конкретные примеры включают замену атома водорода спиртовой группы на такую группу, как (С1-С6)алканоилоксиметил, 1-((С1-С6)алканоилокси)этил, 1-метил-1-((С1-С6)алканоил-окси)этил, (С1-С6)алкоксикарбонилоксиметил, N-(С1-С6)алкоксикарбониламинометил, сукциноил, (С1-С6)алканоил, альфа-амино(С1-С4)алканоил, арилацил и альфа-аминоацил или альфа-аминоацил-альфа-аминоацил, где каждая альфа-аминоацильная группа независимо выбрана из остатков природных L-аминокислот, Р(O)(ОН)2, -Р(O)(O(С1-С6)алкил)2 или гликозила (радикала, образующегося в результате удаления гидроксильной группы в полуацетальной форме углевода).
Термин "уходящая группа" относится к части первого реагента в химической реакции, которая заменяется в этом первом реагенте в данной химической реакции. Примеры уходящих групп включают, но не ограничиваются этим, атомы галогена, группы алкокси и сульфонилокси. Типичные группы сульфонилокси включают, но не ограничиваются этим, группы алкилсульфонилокси (например, метилсульфонилокси (мезилатную группу), и группы трифторметилсульфонилокси (трифлатную группу)) и арилсульфонилокси (например, п-толуолсульфонилокси (тозилатную группу), и п-нитросульфонилокси (нозилатную группу)).
СИНТЕЗ СОЕДИНЕНИЙ, ИНГИБИРУЮЩИХ ЯНУС-КИНАЗЫ
Соединения по настоящему изобретению могут быть синтезированы с использованием путей синтеза, изложенных в данном описании. В некоторых воплощениях можно использовать способы, хорошо известные в области химии, в дополнение к описанию или с учетом содержащегося в данной заявке описания. Обычно, исходные вещества приобретают в коммерческих источниках, таких как Aldrich Chemicals (Milwaukee, Wis.), или легко получают способами, хорошо известными специалистам в данной области техники (например, получают способами, в целом описанными в Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-19, Wiley, N.Y. (изд. 1967-1999 г.г.), Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, включая приложения (также доступные через онлайн-базу данных на основе справочника Бейльштейна), или Comprehensive Heterocyclic Chemistry, Editors Katrizky and Rees, Pergamon Press, 1984.
Соединения могут быть получены по отдельности или в виде библиотек соединений, содержащих по меньшей мере 2, например, от 5 до 1000 соединений или от 10 до 100 соединений. Библиотеки соединений можно получить, используя комбинаторный подход "разделения и смешивания" ("split and mix") или множественные параллельные процедуры синтеза с применением методик проведения химических реакций либо в растворе, либо в твердой фазе, известных специалистам в данной области техники. Таким образом, согласно другому аспекту изобретения предложена библиотека соединений, содержащая по меньшей мере 2 соединения по настоящему изобретению.
В иллюстративных целях на приведенных ниже реакционных схемах показаны пути синтеза соединений по настоящему изобретению, а также ключевых промежуточных соединений. Для более подробного описания отдельных реакционных стадий см. раздел Примеры, приведенный ниже. Специалистам в данной области техники будет очевидно, что можно использовать и другие пути синтеза. Хотя на реакционных схемах приведены и ниже рассмотрены конкретные исходные вещества и реагенты, они легко могут быть заменены на другие исходные вещества и реагенты для получения целого ряда производных и/или реакционных условий. Помимо этого, многие соединения, полученные описанными ниже способами, могут быть дополнительно модифицированы с учетом данного описания с использованием общепринятых методов химии, хорошо известных специалистам в данной области техники.
При получении соединений по настоящему изобретению может потребоваться защита функциональной группы (например, первичного или вторичного амина). Необходимость в такой защите будет варьировать в зависимости от природы такой функциональной группы и условий осуществления способов получения. Подходящие амино-защитные группы включают ацетил, трифторацетил, бензил, фенилсульфонил, трет-бутоксикарбонил (ВОС), бензилоксикарбонил (CBz) и 9-флуоренилметиленоксикарбонил (Fmoc). Необходимость в такой защите легко определяется специалистом в данной области техники. Общее описание защитных групп и их применение см. в Т. W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
Другие преобразования, обычно используемые в синтезе соединений по настоящему изобретению и которые можно осуществить с использованием ряда реагентов и условий, включают следующие далее.
(1) Взаимодействие карбоновой кислоты с амином с образованием амида. Такое преобразование можно осуществить с использованием различных реагентов, известных специалистам в данной области техники, а исчерпывающий обзор можно найти в Tetrahedron, 2005, 61, 10827-10852.
(2) Взаимодействие первичного или вторичного амина с арилгалогенидом или псевдогалогенидом, например, трифлатом, общеизвестное как "перекрестное сочетание Бухвальда-Хартвига", можно осуществить с использованием ряда катализаторов, лигандов и оснований. Обзор этих методов приведен в Comprehensive Organic Name Reactions and Reagents, 2010, 575-581.
(3) Палладий-опосредуемая реакция перекрестного сочетания между арилгалогенидом и винилбороновой кислотой или боронатным сложным эфиром. Такое преобразование представляет собой тип "перекрестного сочетания Сузуки-Мияура", класс реакции, подробно описанной в Chemical Reviews, 1995, 95(7), 2457-2483.
(4) Гидролиз сложного эфира с получением соответствующей карбоновой кислоты хорошо известен специалистам в данной области техники, и его условия включают: в случае метиловых и этиловых сложных эфиров, применение водного раствора сильного основания, такого как гидроксид лития, натрия или калия, или водного раствора сильной неорганической кислоты, такой как HCl; в случае трет-бутилового эфира гидролиз может быть осуществлен с использованием кислоты, например, HCl в диоксане или трифторуксусной кислоты (TFA) в дихлорметане (DCM).
Реакционная схема 1

Реакционная схема 1 иллюстрирует синтез показанных на ней соединений 6, 8 и 10. Соединение 1 можно подвергнуть арилированию с использованием 4 бром-1-(дифторметокси)-2-иодбензола в катализируемых палладием условиях с получением соединения 2. Нитрогруппу в соединении 2 можно восстановить в присутствии железа и хлорида аммония с получением аминоанилина 3. После осуществления реакции сочетания с образованием амидной связи с использованием имеющейся в продаже пиразоло[1,5-а]пиримидин-3-карбоновой кислоты в присутствии реагента сочетания, такого как, но не ограничиваясь этим, РуАОР (гексафторфосфат (7-азабензотриазол-1-илокси)трипирролидино-фосфония), и органического основания, такого как, но не ограничиваясь этим, DIPEA (диизопропилэтиламин) и DMAP (4-диметиламинопиридин), в органическом растворителе, таком как, но не ограничиваясь этим, DMF (N,N-диметилформамид), получают соединения 4. Соединение 4 может быть превращено в соответствующий иодид 5 с использованием таких условий, как наличие иодида натрия и CuI в присутствии основания, такого как N,N-диметилэтан-1,2-диамин, в растворителе, таком как tBuOH. Соединения формулы 7 могут образовываться в результате обработки соединения 4 замещенной бороновой кислотой (или сложным эфиром) или солью BF3K в катализируемых палладием условиях в присутствии основания, такого как, но не ограничиваясь этим, карбонат цезия, в растворителе, таком как, но не ограничиваясь этим, 1,4-диоксан. Кроме того, соединения формулы 9 могут быть синтезированы в результате обработки соединения 4 соответствующим образом замещенным фенолом в катализируемых Pd условиях сочетания в присутствии основания, такого как, но не ограничиваясь этим, карбонат цезия, в растворителе, таком как, но не ограничиваясь этим, толуол. Удаление защитной группы SEM ([β-(триметилсилил)этокси]метил) с соединений формул 5, 7 и 9 для получения соединений формул 6, 8 и 10 может быть осуществлено с использованием такой кислоты, как, но не ограничиваясь этим, HCl в растворителе, таком как, но не ограничиваясь этим, 1,4-диоксан.
Реакционная схема 2
53
Реакционная схема 2 иллюстрирует синтез показанных на ней соединений формулы I-III. Соединения формул 6, 8 и 10 можно обработать соответствующим образом замещенным 2-бром-N,N-диметилацетамидом в присутствии основания, такого как, но не ограничиваясь этим, карбонат цезия, в растворителе, таком как, но не ограничиваясь этим, DMF, с получением соединений формул I-III.
Реакционная схема 54
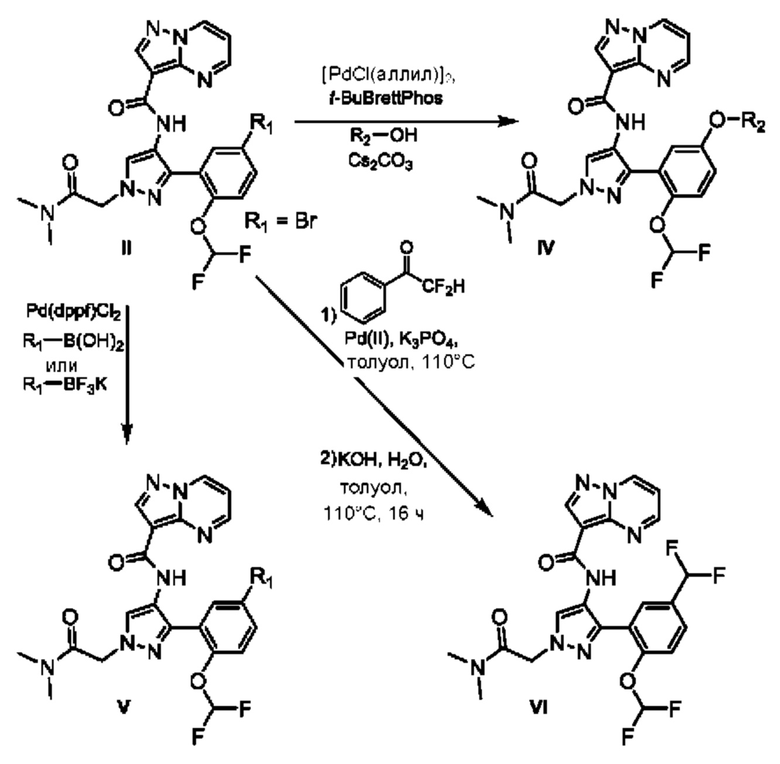
Реакционная схема 3 иллюстрирует синтез показанных на ней соединений формул IV-VI. Соединения формулы II (R1=Br) можно обработать соответствующим образом замещенным фенолом в катализируемых Pd условиях сочетания в присутствии основания, такого как, но не ограничиваясь этим, карбонат цезия, в растворителе, таком как, но не ограничиваясь этим, толуол, с получением соединений формулы IV. Соединения формулы V могут быть получены в результате обработки соединения 4 замещенной бороновой кислотой (сложным эфиром) или солью BF3K в катализируемых палладием условиях в присутствии основания, такого как, но не ограничиваясь этим, карбонат цезия, в растворителе, таком как, но не ограничиваясь этим, 1,4-диоксан. Содержащие дифторметильную группу соединения формулы VI могут быть получены с использованием методов, описанных в J. Am, Chem. Soc, 2014, 136, 4149-4152. Кроме того, когда R1 в соединениях формулы V представляет собой соответствующим образом замещенный олефин, с этим олефином можно производить дальнейшие действия, используя стандартные методы для получения фторированных алканов.
Реакционная схема 55

Реакционная схема 4 иллюстрирует синтез показанных на ней соединений формулы VII. Имеющийся в продаже 4-(дифторметокси)фенол можно обработать бромирующим агентом, таким как, но не ограничиваясь этим, NBS (N-бромсукцинимид), в растворителе, таком как, но не ограничиваясь этим, уксусная кислота, с получением соединения 12. Дифторметилирование соединения 12 с образованием соединения 13 может быть осуществлено путем обработки соединения 12 диэтил-(бромдифторметил)фосфонатом в присутствии основания, такого как, но не ограничиваясь этим, водный раствор гидроксида калия, в растворителе, таком как, но не ограничиваясь этим, ацетонитрил. Соединение 13 можно обработать 4-нитро-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-бром-1-(дифторметокси)-2-иодбензолом в катализируемых палладием условиях в присутствии основания, такого как, но не ограничиваясь этим, карбонат калия, в растворителе, таком как, но не ограничиваясь этим, DMA (N,N-диметилацетамид), с получением соединения 14. Нитрогруппу в соединении 14 можно восстановить в присутствии железа и хлорида аммония с получением аминоанилина 15. После осуществления реакции сочетания с образованием амидной связи с использованием имеющейся в продаже пиразоло[1,5-а]пиримидин-3-карбоновой кислоты в присутствии реагента сочетания, такого как, но не ограничиваясь этим, РуАОР, и органического основания, такого как, но не ограничиваясь этим, DIPEA и DMAP, в органическом растворителе, таком как, но не ограничиваясь этим, DMF, получают соединения 16. Удаление защитной группы SEM с соединения 16 для получения соединения 16 может быть осуществлено с использованием такой кислоты, как, но не ограничиваясь этим, HCl в органическом растворителе, таком как, но не ограничиваясь этим, 1,4-диоксан, с получением соединений формулы VII.
Реакционная схема 5
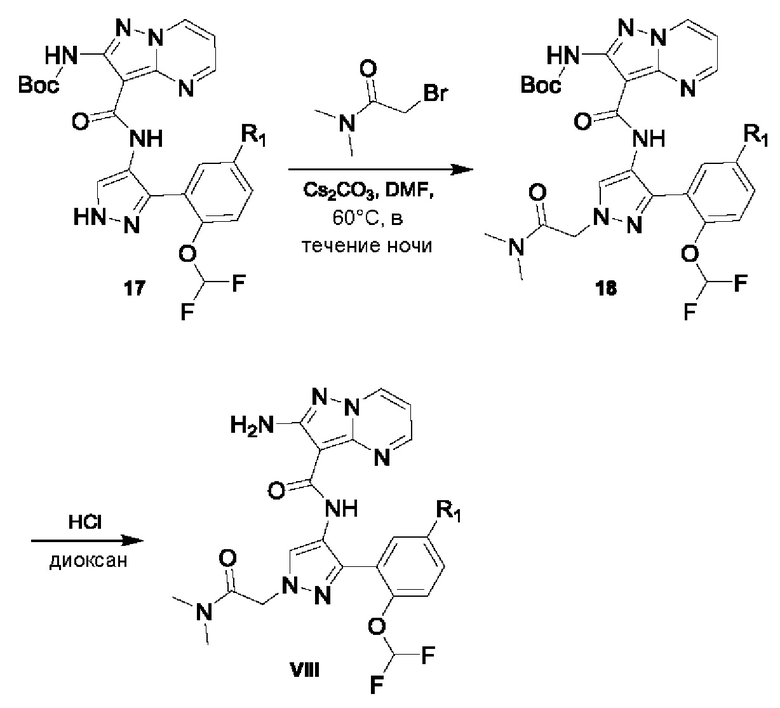
Реакционная схема 5 иллюстрирует синтез показанных на ней соединений формулы VIII. Соединение 17 можно обработать 2-бром-N,N-диметилацетамидом в присутствии основания, такого как, но не ограничиваясь этим, карбонат цезия, в растворителе, таком как, но не ограничиваясь этим, DMF, с получением соединения 18. Удаление защитной группы Boc с соединения 18 может быть осуществлено с использованием такой кислоты, как, но не ограничиваясь этим, HCl в органическом растворителе, таком как, но не ограничиваясь этим, 1,4-диоксан, с получением соединений формулы VIII.
Будет очевидно, что в тех случаях, когда имеются соответствующие функциональные группы, могут быть выполнены дополнительные модификации соединений различных формул или любых промежуточных соединений, используемых для их получения, с применением одного или нескольких стандартных методов синтеза, в которых используются реакции конденсации, замещения, окисления, восстановления или расщепления. Конкретные подходы с использованием реакций замещения включают традиционные процедуры алкилирования, арилирования, гетероарилирования, ацилирования, сульфонилирования, галогенирования, нитрования, формилирования и сочетания.
В другом примере первичные аминогруппы или вторичные аминогруппы могут быть превращены в амидные группы (-NHCOR' или -NRCOR') в результате ацилирования. Ацилирование может быть осуществлено путем взаимодействия с соответствующим хлорангидридом кислоты в присутствии основания, такого как триэтиламин, в подходящем растворителе, таком как дихлорметан, или путем взаимодействия с соответствующей карбоновой кислотой в присутствии подходящего агента сочетания, такого как HATU (O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат), в подходящем растворителе, таком как дихлорметан. Аналогично, аминогруппы могут быть превращены в сульфонамидные группы (-NHSO2R' или -NR''SO2R') путем взаимодействия с соответствующим сульфонилхлоридом в присутствии подходящего основания, такого как триэтиламин, в подходящем растворителе, таком как дихлорметан. Первичные или вторичные аминогруппы могут быть превращены в мочевинные группы (-NHCONR'R'' или -NRCONR'R'') в результате взаимодействия с соответствующим изоцианатом в присутствии или в отсутствие подходящего основания, такого как триэтиламин, в подходящем растворителе, таком как дихлорметан.
Аминогруппа (-NH2) может быть получена путем восстановления нитрогруппы (-NO2), например, посредством каталитического гидрирования с использованием, например, водорода в присутствии металлического катализатора, например, палладия на подложке, такой как уголь, в растворителе, таком как этилацетат или спирт, например, метанол. Альтернативно, такое превращение может быть осуществлено посредством химического восстановления с использованием, например, металла, например, олова или железа, в присутствии кислоты, такой как соляная кислота.
В другом примере аминогруппы (-CH2NH2) могут быть получены в результате восстановления нитрилов (-CN), например, посредством каталитического гидрирования с использованием, например, водорода в присутствии металлического катализатора, например, палладия на подложке, такой как уголь, или никеля Ренея, в растворителе, таком как простой эфир, например, циклический эфир, такой как тетрагидрофуран, при соответствующей температуре, например, от примерно -78°С до температуры дефлегмации растворителя.
В другом примере аминогруппы (-NH2) могут быть получены из карбоксильных групп (-CO2H) в результате превращения в соответствующий ацилазид (-CON3), перегруппировки Курциуса и гидролиза полученного изоцианата (-N=C=O).
Альдегидые группы (-СНО) могут быть превращены в аминогруппы (-CH2NR'R'') в результате восстановительного аминирования с применением амина и боргидрида, например, триацетоксиборгидрида натрия или цианоборгидрида натрия, в растворителе, таком как галогенированный углеводород, например, дихлорметан, или спирт, такой как этанол, если необходимо, то в присутствии кислоты, такой как уксусная кислота, при температуре, примерно равной температуре окружающей среды.
В другом примере альдегидные группы могут быть превращены в алкенильные группы (-CH=CHR') в результате применения реакции Виттига или Вудсворта-Эммонса с использованием соответствующего фосфорана или фосфоната в стандартных условиях, известных специалистам в данной области техники.
Альдегидные группы могут быть получены в результате восстановления сложноэфирных групп (таких как -CO2Et) или нитрилов (-CN) с использованием гидрида диизобутилалюминия в подходящем растворителе, таком как толуол. Альтернативно, альдегидные группы могут быть получены в результате окисления спиртовых групп с использованием любого подходящего окисляющего агента, известного специалистам в данной области техники.
Сложноэфирные группы (-CO2R') могут быть превращены в группу соответствующей кислоты (-CO2H) в результате катализируемого кислотами или основаниями, в зависимости от природы радикала R, гидролиза. Если R представляет собой трет-бутил, то катализируемый кислотами гидролиз может быть осуществлен, например, в результате обработки органической кислотой, такой как трифторуксусная кислота, в водном растворителе или в результате обработки неорганической кислотой, такой как соляная кислота, в водном растворителе.
Карбоксильные группы (-CO2H) могут быть превращены в амиды (CONHR' или -CONR'R'') в результате взаимодействия с соответствующим амином в присутствии подходящего агента сочетания, такого как HATU, в подходящем растворителе, таком как дихлорметан.
В другом примере карбоновые кислоты могут быть подвергнуты реакции одноуглеродной гомологизации (т.е. из -CO2H в -CH2CO2H) в результате превращения в соответствующий хлорангидрид кислоты (-COCl) с последующим проведением синтеза Арндта-Эйстерта.
В другом примере группы -ОН могут быть получены из соответствующего сложного эфира (например, -CO2R') или альдегида (-СНО) в результате восстановления с использованием, например, комплексного гидрида металла, такого как алюмогидрид лития, в диэтиловом эфире или тетрагидрофуране, или с использованием боргидрида натрия в таком растворителе, как метанол. Альтернативно, спирт может быть получен в результате восстановления соответствующей кислоты (-CO2H), используя, например, алюмогидрид лития в таком растворителе, как тетрагидрофуран, или используя боран в таком растворителе, как тетрагидрофуран.
Спиртовые группы могут быть превращены в уходящие группы, как например, атомы галогена или группы сульфонилокси, такие как алкилсульфонилокси, например, трифторметилсульфонилокси, или арилсульфонилокси, например, в группу п-толуолсульфонилокси, используя условия, известные специалистам в данной области техники. Например, спирт можно привести во взаимодействие с тионилхлоридом в галогенированном углеводороде (например, дихлорметане) с получением соответствующего хлорида. В данной реакции также можно использовать основание (например, триэтиламин).
В другом примере спиртовые, фенольные или амидные группы могут быть алкилированы в результате сочетания фенола или амида со спиртом в растворителе, таком как тетрагидрофуран, в присутствии фосфина, например, трифенилфосфина, и активатора, такого как диэтил-, диизопропил- или диметилазодикарбоксилат. Альтернативно, алкилирование может быть осуществлено посредством депротонирования с использованием подходящего основания, например, гидрида натрия, с последующим добавлением алкилирующего агента, такого как алкилгалогенид.
Ароматические галоген-содержащие заместители в соединениях можно подвергнуть реакции обмена галоген-металл путем обработки основанием, например, литиевым основанием, таким как н-бутил- или трет-бутил-литий, возможно при пониженной температуре, например, около -78°С, в растворителе, таком как тетрагидрофуран, с последующим гашением электрофилом для введения желаемого заместителя. Так, например, формильную группу можно ввести, используя N,N-диметилформамид в качестве электрофила. Альтернативно, ароматические галоген-содержащие заместители можно подвергнуть катализируемым металлами (например, палладием или медью) реакциям для введения, например, кислотных, сложноэфирных, циано-, амидных, арильных, гетероарильных, алкенильных, алкинильных, тио- или амино- заместителей. Подходящие методики, которые могут быть использованы, включают методики, описанные Хеком, Сузуки, Стилле, Бухвальдом или Хартвигом.
Ароматические галоген-содержащие заместители также можно подвергнуть нуклеофильному замещению посредством взаимодействия с соответствующим нуклеофилом, таким как амин или спирт. Предпочтительно, чтобы такое взаимодействие могло быть осуществлено при повышенной температуре в условиях облучения микроволнами.
МЕТОДЫ РАЗДЕЛЕНИЯ
Для каждой из типичных схем может иметь преимущество отделение продуктов реакции друг от друга или от исходных веществ. Желаемые продукты с каждой стадии или серии стадий разделяют или очищают (разделяют далее) до желаемой степени гомогенности методами, общеизвестными в данной области техники. Обычно такие методы разделения включают многофазную экстракцию, кристаллизацию или растирание из растворителя либо смеси растворителей, дистилляцию, сублимацию или хроматографию. Хроматография может включать любое количество методов, в том числе, например: обращенно-фазовую и нормально-фазовую; гель-проникающую; ионообменную; сверхкритическую жидкостную хроматографию; применение методов и аппаратуры для жидкостной хроматографии высокого, среднего и низкого давления; мелкомасштабную аналитическую хроматографию; хроматографию с псевдодвижущимся слоем (SMB) и препаративную тонкослойную и толстослойную хроматографию, а также методы мелкомасштабной тонкослойной и флэш-хроматографии.
Другой класс методов разделения включает обработку смеси реагентом, выбранным для связывания с желаемым продуктом, непрореагировавшим исходным веществом, побочным продуктом реакции или тому подобным либо оказывающим на все вышеперечисленное какое-либо иное действие, способствующее их разделению. Такие реагенты включают адсорбенты или абсорбенты, такие как активированный уголь, молекулярные сита, ионообменные среды или тому подобное. Альтернативно, такими реагентами могут быть кислоты в случае основного вещества, основания в случае кислотного вещества, связывающие реагенты, такие как антитела, связывающие белки, селективные хелаторы, такие как краун-эфиры, реагенты для жидкость/жидкостной ионной экстракции (LIX) или тому подобное.
Выбор соответствующих методов разделения зависит от природы используемых веществ. В типичных методах разделения учитывают: точку кипения и молекулярную массу при дистилляции и сублимации, наличие или отсутствие полярных функциональных групп при хроматографии, стабильность веществ в кислотных и щелочных средах при многофазной экстракции и тому подобное. Специалист в данной области техники будет применять методы, с наибольшей вероятностью приводящие к достижению желаемого разделения.
Диастереомерные смеси могут быть разделены на свои индивидуальные диастереомеры на основе различий в их физико-химических свойствах методами, хорошо известными специалистам в данной области техники, например, посредством хроматографии или фракционной кристаллизации. Энантиомеры можно разделить путем превращения энантиомерной смеси в диастереомерную смесь в результате взаимодействия с соответствующим оптически активным соединением (например, хиральным вспомогательным веществом, таким как хиральный спирт или хлорангидрид кислоты Мошера), разделения диастереомеров и превращения (например, посредством гидролиза) индивидуальных диастереоизомеров в соответствующие чистые энантиомеры. Кроме того, некоторые соединения по настоящему изобретению могут быть атропоизомерами (например, замещенные биарилы), и они рассматриваются как часть данного изобретения. Энантиомеры также могут быть разделены с применением колонки для хиральной HPLC или с использованием сверхкритической жидкостной хроматографии.
Индивидуальный стереоизомер, например, энантиомер, по существу не содержащий своего стереоизомера, можно получить путем разделения рацемической смеси, используя такой метод, как образование диастереомеров с применением оптически активных разделяющих агентов (Eliel Е. and Wilen S., "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., New York, 1994; Lochmuller C.H., J. Chromatogr., 113(3): 283-302 (1975)). Рацемические смеси хиральных соединений по изобретению могут быть разделены и соединения выделены любым подходящим методом, включая: (1) образование ионных диастереомерных солей с хиральными соединениями и разделение фракционной кристаллизацией или другими методами, (2) образование диастереомерных соединений с использованием хиральных дериватизирующих реагентов, разделение диастереомеров и превращение в чистые стереоизомеры и (3) разделение по существу чистых или обогащенных стереоизомеров непосредственно в хиральных условиях. См.: "Drug Stereochemistry, Analytical Methods and Pharmacology", Irving W. Wainer, Ed., Marcel Dekker, Inc., New York (1993).
Диастереомерные соли могут быть образованы в результате взаимодействия энантиомерно чистых хиральных оснований, таких как бруцин, хинин, эфедрин, стрихнин, α-метил-β-фенилэтиламин (амфетамин) и тому подобное, с ассиметрическими соединениями, несущими кислотную функциональную группу, такими как карбоновая кислота и сульфоновая кислота. Разделения диастереомерных солей можно достичь фракционной кристаллизацией или ионообменной хроматографией. Что касается разделения оптических изомеров аминосоединений, то к образованию диастереомерных солей может приводить добавление хиральных карбоновых или сульфоновых кислот, таких как камфорсульфоновая кислота, винная кислота, миндальная кислота или молочная кислота.
Альтернативно, подлежащий разделению субстрат приводят во взаимодействие с одним из энантиомеров хирального соединения с образованием диастереомерной пары (Eliel Е. and Wilen S., "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., New York, 1994, p. 322). Диастереомерные соединения могут образовываться в результате взаимодействия ассиметрических соединений с энантиомерно чистыми хиральными дериватизирующими реагентами, такими как ментильные производные, с последующим разделением диастереомеров и гидролизом с получением чистого или обогащенного энантиомера. Метод определения оптической чистоты включает в себя получение хиральных сложных эфиров, таких как ментиловый эфир, например (-)-ментил-хлорформиат, в присутствии основания, или эфир кислоты Мошера, α-метокси-α-(трифторметил)фенилацетат (Jacob, J. Org. Chem. 47: 4165 (1982)), в рацемической смеси и анализ ЯМР-спектра на наличие двух атропоизомерных энантиомеров или диастереомеров. Стабильные диастереомеры атропоизомерных соединений можно разделить и выделить нормально-фазовой и обращенно-фазовой хроматографией, следуя способам разделения атропоизомерных нафтил-изохинолинов (заявка WO 96/15111, включенная в данное описание посредством ссылки). Согласно методу (3) рацемическую смесь двух энантиомеров можно разделить хроматографией с использованием хиральной неподвижной фазы ("Chiral Liquid Chromatography", W. J. Lough, Ed., Chapman and Hall, New York (1989); Okamoto, J. Chromatogr. 513: 375-378 (1990)). Обогащенные или очищенные энантиомеры можно различить методами, используемыми для различения других хиральных молекул с ассиметрическими атомами углерода, такими как оптическое вращение и круговой дихроизм. Абсолютная стереохимическая конфигурация хиральных центров и энантиомеров может быть определена с использованием рентгеноструктурной кристаллографии.
Позиционные изомеры и промежуточные соединения для их синтеза могут быть охарактеризованы с применением таких методов, как ЯМР и аналитическая HPLC. Для некоторых соединений, где энергетический барьер для взаимопревращения является достаточно высоким, Е- и Z-изомеры могут быть разделены, например, с использованием препаративной HPLC.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ВВЕДЕНИЕ
Соединения, к которым относится данное изобретение, представляют собой ингибиторы киназы JAK, например, ингибиторы JAK1, и полезны для лечения некоторых заболеваний, например, воспалительных заболеваний, таких как астма.
Соответственно, в другом воплощении предложены фармацевтические композиции или лекарственные средства, содержащие соединение по изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или эксципиент, а также способы применения соединений по изобретению для изготовления таких композиций и лекарственных средств.
В одном из примеров могут быть изготовлены композиции на основе соединения по изобретению или его фармацевтически приемлемой соли желаемой степени чистоты путем смешивания при температуре окружающей среды и соответствующем значении рН с физиологически приемлемыми носителями, т.е. носителями, не являющимися токсичными для реципиентов в дозировках и концентрациях, применяемых в форме галенова препарата для введения. Значение рН композиции зависит главным образом от конкретного применения и концентрации соединения, но обычно изменяется везде в диапазоне от примерно 3 до примерно 8. В одном из примеров композицию на основе соединения по изобретению или его фармацевтически приемлемой соли изготавливают в ацетатном буфере при рН 5. В другом воплощении соединения по настоящему изобретению являются стерильными. Соединение можно хранить, например, в виде твердой или аморфной композиции, в виде лиофилизированного препарата или в виде водного раствора.
Изготовление, дозирование и введение композиций осуществляют способом, согласующимся с надлежащей медицинской практикой. Факторы, рассматриваемые в этом контексте, включают конкретное подлежащее лечению расстройство, конкретное подвергаемое лечению млекопитающее, клиническое состояние отдельного пациента, причину расстройства, место доставки агента, способ введения, схему введения и другие факторы, известные врачам.
Будет понятно, что специально подобранный уровень доз для любого конкретного пациента будет зависеть от ряда факторов, включая активность конкретного используемого соединения, возраст, массу тела, общее состояние здоровья, пол, режим питания, время введения, путь введения, скорость экскреции, лекарственную комбинацию и тяжесть конкретного заболевания, подлежащего лечению. Оптимальные уровни доз и частота введения будут определены по результатам клинического испытания, согласно требованиям, принятым в фармацевтической области. В общем случае, диапазон суточных доз для перорального введения будет лежать в диапазоне от примерно 0,001 мг до примерно 100 мг на кг массы тела человека, зачастую от 0,01 мг до примерно 50 мг на кг, например, от 0,1 до 10 мг на кг, при приеме в виде однократной дозы или разделенных доз. В общем случае диапазон суточных доз для введения посредством ингаляции будет лежать в диапазоне от примерно 0,1 мкг до примерно 1 мг на кг массы тела человека, предпочтительно от 0,1 мкг до 50 мкг на кг, при приеме в виде однократной дозы или разделенных доз. С другой стороны, в некоторых случаях может возникнуть необходимость в применении дозировок, лежащих за этими пределами.
Соединения по изобретению или их фармацевтически приемлемую соль можно вводить любым подходящим способом, включая пероральное, местное (в том числе трансбуккальное и сублингвальное), ректальное, вагинальное, трансдермальное, парентеральное, подкожное, внутрибрюшинное, внутрилегочное, интрадермальное, интратекальное, ингаляционное, эпидуральное и интраназальное введение, и, при необходимости локального лечения, введение в область поражения. Парентеральные инфузии включают внутримышечное, внутривенное, интраартериальное, внутрибрюшинное или подкожное введение. В некоторых воплощениях используют введение посредством ингаляции.
Соединения по настоящему изобретению или их фармацевтически приемлемую соль можно вводить в любой удобной для введения форме, например, в форме таблеток, порошков, капсул, пастилок, гранул, растворов, дисперсий, суспензий, сиропов, спреев, средств для ингаляции, суппозиториев, гелей, эмульсий, пластырей и так далее. Такие композиции могут содержать компоненты, традиционно используемые в фармацевтических препаратах, например, разбавители (например, глюкозу, лактозу или маннит), носители, модификаторы рН, буферы, подсластители, объемообразующие агенты, стабилизирующие агенты, поверхностно-активные вещества, увлажняющие агенты, смазывающие вещества, эмульгаторы, суспендирующие агенты, консерванты, антиоксиданты, придающие непрозрачность агенты, глиданты, технологические добавки, красители, отдушки, корригенты, другие известные вспомогательные вещества, а также дополнительные активные агенты.
Подходящие носители и эксципиенты хорошо известны специалистам в данной области техники и описаны подробно, например, в Ansel Howard С. et al., Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems. Philadelphia: Lippincott, Williams & Wilkins, 2004; Gennaro Alfonso R. et al. Remington: The Science and Practice of Pharmacy. Philadelphia: Lippincott, Williams & Wilkins, 2000; и Rowe, Raymond C. Handbook of Pharmaceutical Excipients. Chicago, Pharmaceutical Press, 2005. Например, носители включают растворители, дисперсионные среды, вещества покрытия, поверхностно-активные вещества, антиоксиданты, консерванты (например, антибактериальные агенты, противогрибковые агенты), изотонические агенты, замедляющие всасывание агенты, соли, консерванты, лекарственные средства, стабилизаторы лекарственных средств, гели, связующие вещества, эксципиенты, обеспечивающие распадаемость агенты, смазывающие вещества, подсластители, корригенты, красители, подобного рода вещества и их комбинации, которые будут известны специалисту средней квалификации в данной области техники (см., например, Remington's Pharmaceutical Sciences, pp. 1289-1329, 1990). За исключением случаев, когда любой традиционный носитель оказывается несовместимым с активным ингредиентом, его применение в терапевтических или фармацевтических композициях включено. Типичные эксципиенты включают дикальция фосфат, маннит, лактозу, крахмал, стеарат магния, натриевую соль сахарина, целлюлозу, карбонат магния или их комбинации. Фармацевтическая композиция может содержать носители или эксципиенты разных типов в зависимости от того, предполагается ли ее введение в твердой, жидкой или аэрозольной форме и имеется ли необходимость в ее стерильности для таких путей введения.
Например, таблетки и капсулы для перорального введения могут быть в форме, представляющей собой стандартную дозу, и могут содержать традиционные эксципиенты, такие как связующие вещества, например, сироп, аравийскую камедь, желатин, сорбит, трагакант или поливинил-пирролидон; наполнители, например, лактозу, сахар, кукурузный крахмал, фосфат кальция, сорбит или глицин; смазывающее вещество для таблеток, например, стеарат магния, тальк, полиэтиленгликоль или диоксид кремния; разрыхлители, например, картофельный крахмал, или приемлемые увлажняющие агенты, такие как лаурилсульфат натрия. Таблетки могут быть покрыты оболочкой в соответствии со способами, хорошо известными в обычной фармацевтический практике. Пероральные препараты в жидкой форме могут быть в форме, например, водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров или могут быть представлены в виде высушенного продукта для повторного разведения водой или другим подходящим разбавителем перед применением. Такие препараты в жидкой форме могут содержать традиционные вспомогательные вещества, такие как суспендирующие агенты, например, сорбит, сироп, метилцеллюлозу, глюкозный сироп, желатин, гидрогенизированные съедобные жиры; эмульгирующие агенты, например, лецитин, сорбитана моноолеат или аравийскую камедь; неводные разбавители (которые могут включать съедобные масла), например, миндальное масло, фракционированное кокосовое масло, сложные эфиры жирных кислот и таких соединений как глицерин, пропиленгликоль или этиловый спирт; консерванты, например, метил- или пропил-п-гидроксибензоат или сорбиновую кислоту и, при желании, традиционные корригенты или красители.
В случае местного применения на кожу соединение может быть представлено в составе крема, лосьона или мази. Композиции в форме крема или мази, которые могут быть использованы для приготовления лекарственного средства, являются традиционными композициями, хорошо известными в данной области техники, например, которые описаны в стандартных руководствах по фармацевтике, таких как Британская фармакопея.
Композиции на основе соединений по изобретению или их фармацевтически приемлемой соли также могут быть изготовлены для ингаляции, например, в виде назального спрея или ингаляторов сухого порошка либо аэрозольных ингаляторов. В случае доставки посредством ингаляции соединение обычно присутствует в форме микрочастиц, которые могут быть получены рядом методов, включая распылительную сушку, сублимационную сушку и микронизацию. Получение аэрозоля может быть осуществлено с использованием, например, струйных распылителей с приводом от давления или ультразвуковых распылителей, как например, с использованием дозированных аэрозолей, приводимых в действие пропеллентом, либо с использованием не содержащих пропеллентов систем введения микронизированных соединений, например, из ингаляционных капсул или других систем доставки "сухого порошка".
В качестве примера, композиция по изобретению может быть приготовлена в виде суспензии для доставки из небулайзера или в виде аэрозоля в жидком пропелленте, например, для применения в дозирующем ингаляторе под давлением (PMDI). Пропелленты, подходящие для применения в PMDI, известны специалисту и включают CFC-12, HFA-134a, HFA-227, HCFC-22 (CCl2F2) и HFA-152 (CH4F2 и изобутан).
В некоторых воплощениях композиция по изобретению находится в форме сухого порошка для доставки с использованием ингалятора сухого порошка (DPI). Известно много типов DPI.
Микрочастицы для доставки путем введения могут быть приготовлены содержащими эксципиенты, которые облегчают доставку и высвобождение. Например, в случае сухой порошковой композиции микрочастицы могут быть приготовлены содержащими большие частицы носителя, которые способствуют перенесению из DPI в легкое. Подходящие частицы носителя известны и включают частицы из лактозы; они могут иметь среднемассовый аэродинамический диаметр, например, больше 90 мкм.
В случае композиции на основе аэрозоля примером является следующая композиция:

Дозирование соединения по изобретению или его фармацевтически приемлемой соли может быть осуществлено, как описано, с учетом используемой системы ингалятора. Помимо данного соединения вводимые формы также могут содержать эксципиенты, которые описаны выше, или, например, пропелленты (например, хладон в случае дозированных аэрозолей), поверхностно-активные вещества, эмульгаторы, стабилизаторы, консерванты, корригенты, наполнители (например, лактозу в случае порошковых ингаляторов) или, если необходимо, другие активные соединения.
Для целей ингаляции доступно большое количество систем, с помощью которых могут быть образованы и введены аэрозоли частиц оптимальных размеров, при этом используется метод ингаляции, который подходит для пациента. Помимо применения адаптеров (насадок, расширителей) и контейнеров грушевидной формы (например, Nebulator®, Volumatic®) и, в случае дозированных аэрозолей, автоматических устройств, испускающих распыляемый аэрозоль (Autohaler®), в случае порошковых ингаляторов, в частности, доступен ряд технических решений (например, Diskhaler®, Rotadisk®, Turbohaler® или ингаляторы, например, которые описаны в патенте США №5263475, включенном в данное описание посредством ссылки). Кроме того, соединения по изобретению или их фармацевтически приемлемая соль могут быть доставлены с использованием многокамерных устройств, позволяющих таким образом доставлять комбинируемые агенты.
Соединение или его фармацевтически приемлемую соль также можно вводить парентерально в стерильной среде. В зависимости от используемого наполнителя и используемой концентрации соединение может быть либо суспендировано, либо растворено в наполнителе. Предпочтительно, чтобы в данном наполнителе могли быть растворены такие адъюванты, как анестетики местного действия, консерванты или буферные агенты.
НАПРАВЛЕННАЯ ДОСТАВКА ИНГАЛЯЦИОННЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ
Соединения по настоящему изобретению могут предусматривать направленную доставку ингаляцией. Не так давно был представлен обзор по оптимизации лекарственных средств для доставки в легкое путем местного (ингаляционного) введения (Cooper, А.Е. et al. Curr. Drug Metab., 2012, 13, 457-473).
Из-за ограничений, присущих устройству доставки, доза ингаляционного лекарственного средства по всей вероятности будет низкой (меньше приблизительно 1 мг/сутки) у людей, что обуславливает необходимость в сильнодействующих молекулах. Наличие высокой эффективности в отношении представляющей интерес мишени особенно важно в случае ингаляционного лекарственного средства вследствие таких факторов, как ограниченное количество лекарственного средства, которое может быть доставлено из ингалятора за одно нажатие, и проблемы безопасности, связанные с высокой нагрузкой аэрозолем легкого (например, кашель или раздражение). Например, в некоторых воплощениях в случае ингаляционного ингибитора JAK1 может быть желательно иметь значение Ki примерно 0,5 нМ или меньше в биохимическом анализе JAK1, например, описанном в данной заявке, и значение IC50 (концентрация, вызывающая 50%-ное ингибирование) примерно 20 нМ или меньше в анализе JAK1 с применением клеток, например, описанном в данной заявке. В других воплощениях планируемая доза соединения по настоящему изобретению или его фармацевтически приемлемой соли для человека по меньшей мере в два раза меньше планируемой дозы соединения, известного в данной области техники, для человека. Соответственно, в некоторых воплощениях соединения (или их фармацевтически приемлемая соль), описанные в данной заявке, демонстрируют такие значения эффективности.
IL13-опосредуемая передача сигнала вовлечена в высокой степени в патогенез астмы. IL13 представляет собой цитокин, для передачи сигнала с участием которого необходимо присутствие активной JAK1. Таким образом, в результате ингибирования JAK1 также ингибируется IL13-опосредуемая передача сигнала, что может принести пользу пациентам с астмой. Осуществление ингибирования IL13-опосредуемой передачи сигнала в животной модели (например, мышиной модели) может спрогнозировать будущий положительный эффект для страдающих астмой пациентов-людей. Таким образом, в случае ингаляционного ингибитора JAK1 может быть полезно проявление подавления IL13-опосредуемой передачи сигнала в животной модели. Способы измерения такого подавления известны в данной области техники. Например, как обсуждается в данном описании и как известно в данной области техники, JAK1-зависимое фосфорилирование STAT6 представляет собой известный процесс, протекающий после стимуляции под действием IL13. Соответственно, в некоторых воплощениях соединения (или их фармацевтически приемлемая соль), описанные в данной заявке, демонстрируют ингибирование индуцирования образования фосфорилированного STAT6 (pSTAT6) в легких. Чтобы исследовать фармакодинамические эффекты на уровни pSTAT6, соединения по изобретению вводили совместно с 1 мкг IL-13 интраназально самкам мышей Balb/c. Образцы соединений готовили в 0,2%-ном (об.:об.) твине 80 в физиологическом растворе и смешивали 1:1 (об.:об.) с IL-13 непосредственно перед введением. Интраназальные дозы вводили мышам со слабой анестезией (изофлураном) путем введения пипеткой фиксированного объема (50 мкл) непосредственно в ноздри для достижения целевого уровня доз (3 мг/кг, 1 мг/кг, 0,3 мг/кг, 0,1 мг/кг). Через 0,25 ч после введения дозы собирали образцы крови (приблизительно 0,5 мл) посредством пункции сердца и плазму крови получали центрифугированием (1500 g, 10 мин, +4°С). Легкие перфузировали охлажденным забуференным фосфатом физиологическим раствором (PBS), взвешивали и быстро замораживали в жидком азоте. Все образцы хранили при температуре приблизительно -80°С до анализа. Размороженные образцы легкого взвешивали и гомогенизировали после добавления 2 мл воды класса "для HPLC" из расчета на каждый грамм ткани, используя гомогенизатор Omni-Prep Bead Ruptor при 4°С. Экстрагирование образцов плазмы крови и легких проводили путем осаждения белка тремя объемами ацетонитрила, содержащего толбутамид (50 нг/мл) и лабеталол (25 нг/мл) в качестве аналитических внутренних стандартов. После перемешивания на вихревой мешалке и центрифугирования в течение 30 минут при 3200 g и 4°С супернатанты разбавляли соответствующим образом (например, 1:1 (об.:об.)) водой класса "для HPLC" в 96-луночном планшете. Репрезентативные аликвоты образцов плазмы крови и легких анализировали на предмет наличия исходного соединения посредством LC-MS/MS (жидкостная хроматография в сочетании с тандемной масс-спектрометрией), сравнивая с рядом согласованных с матрицей калибровочных стандартов и стандартов контроля качества. Стандарты готовили методом добавок аликвот плазмы крови или гомогената легкого контрольных мышей Balb/c (2:1 в воде класса "для HPLC") с тестируемым соединением и экстракцию проводили так, как описано для экспериментальных образцов. Соотношение легкое:
(плазма крови) определяли как отношение средней концентрации в легком (мкМ) к средней концентрации в плазме крови (мкМ) в момент отбора проб (0,25 ч). Теоретическое связывание с мишенью рассчитывали, используя приведенное далее уравнение, в предположении, что все лекарственное средство находилось в ткани легкого и несвязавшаяся доля была доступна для взаимодействия с мишенью:
(концентрация несвязавшегося средства в ткани/(концентрация несвязавшегося
средства в ткани + эффективность в клетках in vitro, т.е. IC50))*100.
Чтобы измерить уровни pSTAT6, легкие мышей хранили в замороженном состоянии при -80°С до анализа и гомогенизировали в 0,6 мл охлажденного во льду буфера для лизиса клеток (Cell Signalling Technologies, № по каталогу 9803S), дополненного 1 мМ PMSF (фенилметилсульфонилфторид) и смесью ингибиторов протеаз (Sigma Aldrich, № по каталогу Р8340) и фосфатаз (Sigma Aldrich, № по каталогу Р5726 и Р0044). Образцы центрифугировали при 16060 х g в течение 4 минут при 4°С для удаления тканевого дебриса и концентрацию белка в гомогенатах определяли, используя набор для анализа белков с применением бицинхониновой кислоты (ВСА) Pierce™ (№по каталогу 23225). Образцы разбавляли в охлажденной во льду дистиллированной воде до концентрации белка 5 мг/мл и уровни pSTAT6 определяли посредством электро-хемилюминесцентного иммуноанализа от MesoScale Discovery. Кратко, STAT6-захватывающее антитело (R&D Systems, № по каталогу МАВ 2169) в концентрации 150 мкг/мл вносили из расчета 5 мкл/лунка для создания покрытия в лунки 96-луночных планшетов с высокой связывающей способностью от MesoScale Discovery (№ по каталогу L15XB-3) и сушили на воздухе в течение 5 часов при комнатной температуре. Планшеты блокировали путем добавления блокатора А от MesoScale Discovery в концентрации 30 мг/мл из расчета 150 мкл/лунка (№ по каталогу R93BA-4) и инкубировали в течение 2 часов при комнатной температуре на микропланшетном шейкере. Блокированные планшеты промывали 4 раза буфером для промывки на основе ТРИСа от MesoScale Discovery (№ по каталогу R61TX-1), затем переносили гомогенат легких из расчета 50 мкл/лунка для достижения загрузки по белку 250 мкг/лунка. Используемые в анализе планшеты инкубировали в течение ночи при 4°С и промывали 4 раза буфером для промывки на основе ТРИСа, после чего добавляли из расчета 25 мкл/лунка sulfotag-меченное детектирующее антитело к pSTAT6 в концентрации 2,5 мкг/мл (BD Pharmingen, № по каталогу 558241) и инкубировали в течение 2 часов при комнатной температуре на микропланшетном шейкере. Планшеты промывали 4 раза буфером для промывки на основе ТРИСа и добавляли 1х буфер Т для считывания от MesoScale Discovery (№ по каталогу R92TC-1) из расчета 150 мкл/лунка. Количественное определение уровней pSTAT6 в гомогенате легких выполняли, детектируя сигналы электро-хемилюминесценции на приборе SECTOR S 600 от MesoScale Discovery.
Селективность в отношении JAK1 и JAK2 может играть важную роль в случае ингаляционного ингибитора JAK1. Например, GMCSF (гранулоцитарно-макрофагальный колониестимулирующий фактор) представляет собой цитокин, который передает сигналы исключительно через JAK2. Нейтрализация активности GMCSF ассоциируется с легочно-альвеолярным протеинозом (РАР) в легком. Однако, субмаксимальное подавление JAK2, по всей видимости, не ассоциировано с РАР. Таким образом, даже умеренная селективность в отношении JAK1 по сравнению с JAK2 может быть полезной для предотвращения полного подавления GMCSF-опосредуемого пути и предотвращения РАР. Например, в случае ингаляционного ингибитора JAK1 могут быть полезны соединения, демонстрирующие примерно в 2-5 раз более высокую селективность в отношении JAK1 по сравнению с JAK2. Соответственно, в некоторых воплощениях соединения (или их фармацевтически приемлемая соль), описанные в данной заявке, демонстрируют такую селективность. Методы измерения селективности в отношении JAK1 и JAK2 известны в данной области техники, и эту информацию также можно найти в данном описании в разделе Примеры.
Кроме того, может быть желательно наличие селективности у ингаляционного ингибитора JAK1 по сравнению с одной или несколькими другими киназами, чтобы снизить вероятность возможной токсичности благодаря подавлению нецелевого киназного пути. Таким образом, в случае ингаляционного ингибитора JAK1 также может быть полезно наличие селективности по сравнению с широкой панелью киназ, не являющихся JAK-киназами, таких как применяемые в протоколах, поставляемых Службой определения биохимического профиля ингибирования киназ SelectScreen™ компании ThermoFisher Scientific, в которых используются протокол скрининга и условия анализа Adapta™ (пересмотренные 29 июля 2016 г.), протокол скрининга и условия анализа LanthaScreen™ для исследования связывания киназ с применением Eu-мечения (пересмотренные 7 июня 2016 г.) и/или протокол скрининга и условия анализа Z'LYTE™ (пересмотренные 16 сентября 2016 г.). Например, соединение по настоящему изобретению или его фармацевтически приемлемая соль проявляет по меньшей мере в 50 раз более высокую селективность в отношении JAK1 по сравнению с панелью киназ, не являющихся JAK-киназами. Соответственно, в некоторых воплощениях соединения (или их фармацевтически приемлемая соль), описанные в данной заявке, демонстрируют такую селективность.
Токсичность в отношении гепатоцитов, общая цитотоксичность или цитотоксичность неизвестного механизма действия является нежелательным признаком для возможного лекарственного средства, в том числе для ингаляционных лекарственных средств. В случае ингаляционного ингибитора JAK1 может оказаться полезным, чтобы он обладал низкой собственной цитотоксичностью в отношении клеток различных типов. Обычные типы клеток, используемые для оценки цитотоксичности, включают как первичные клетки, такие как гепатоциты человека, так и пролиферирующие хорошо известные клеточные линии, такие как Jurkat, HEK-293 (клетки почки эмбриона человека) и Н23. Например, в случае ингаляционного ингибитора JAK1 может оказаться полезным, чтобы при измерении цитотоксичности в отношении клеток таких типов он имел IC50 выше 50 мкМ или выше 100 мкМ. Соответственно, в некоторых воплощениях соединения (или их фармацевтически приемлемая соль), описанные в данной заявке, демонстрируют такие значения. Методы измерения цитотоксичности известны в данной области техники. В некоторых воплощениях соединения, описанные в данной заявке, тестировали так, как приведено ниже:
(а) клетки Jurkat, Н23 и HEK293T поддерживали при плотности, соответствующей субконфлюэнтной культуре, во флаконах Т175. Клетки, из расчета 450 клеток/45 мкл среды, высевали в лунки 384-луночных, обработанных тканевой культурой черных/прозрачных планшетов Greiner (№ по каталогу Greiner 781091). После внесения клеток планшеты уравновешивали при комнатной температуре в течение 30 минут. Через 30 минут выдерживания при комнатной температуре клетки инкубировали в течение ночи при 37°С в инкубаторе с регулированием содержания CO2 и влажности. На следующие сутки клетки обрабатывали соединениями, разбавленными в 100%-ном DMSO (конечная концентрация DMSO в среде с клетками составляла 0,5%), с получением кривой зависимости доза-ответ по 10 точкам, при этом самая высокая концентрация составляла 50 мкМ. Затем клетки и соединения инкубировали в течение 72 часов при 37°С в инкубаторе с регулированием содержания CO2 и влажности. Через 72 часа инкубирования измеряли жизнеспособность, используя CellTiterGlo® (№ по каталогу Promega G7572) для всех лунок. После инкубирования при комнатной температуре в течение 20 минут планшеты прочитывали на EnVision™ (Perkin Elmer Life Sciences), используя метод на основе люминесценции;
(b) с использованием первичных гепатоцитов человека: готовили растворы тестируемого соединения в концентрации 10 мМ в DMSO. Кроме того, готовили раствор положительного контроля, такого как хлорпромазин, в концентрации 10 мМ в DMSO. Обычно оценку тестируемых соединений проводили, используя построенную по 7 точкам кривую зависимости доза-ответ с 2-кратными разведениями. Как правило, максимальная тестируемая концентрация составляла 50-100 мкМ. В типичном случае самая высокая концентрация определялась растворимостью тестируемого соединения. Хранящиеся при сверхнизких температурах первичные гепатоциты человека (BioreclamationIVT) (партия IZT) размораживали в среде для размораживания InVitroGro™ НТ (BioreclamationIVT) при 37°С, осаждали центрифугированием и ресуспендировали. Жизнеспособность гепатоцитов оценивали по исключению трипанового синего и клетки высевали в лунки 384-луночных планшетов с черными стенками, покрытые коллагеном BioCoat™ (Corning BD), при плотности 13000 клеток/лунка в среде для посева InVitroGro™ CP, дополненной 1% смеси антибиотиков Torpedo™ (BioreclamationIVT) и 5% фетальной телячьей сыворотки. Перед обработкой клетки инкубировали в течение 18 часов (37°С, 5% CO2). По прошествии 18 часов инкубирования среду для посева удаляли и гепатоциты обрабатывали соединениями, разбавленными в среде для инкубирования InVitroGro™ HI, содержащей 1% смеси антибиотиков Torpedo™ и 1% DMSO (бессывороточные условия). Гепатоциты обрабатывали тестируемыми соединениями в таких концентрациях, как 0,78; 1,56; 3,12; 6,25; 12,5; 25 и 50 мкМ, в конечном объеме 50 мкл. В данный анализ включали положительный контроль (например, хлорпромазин), обычно в тех же концентрациях, что и тестируемое соединение. Дополнительные клетки обрабатывали 1%-ным раствором DMSO в качестве разбавителя-контроля. Все процедуры обработки проводили в течение 48-часового периода времени (при 37°С, 5% CO2) и каждое условие обработки выполняли в трех повторах. По окончании 48 часов обработки соединением анализ жизнеспособности клеток CellTiter-Glo® (Promega) использовали в качестве окончательного анализа, измеряя содержание аденозинтрифосфата (АТФ) как меру жизнеспособности клеток. Анализ проводили в соответствии с инструкциями производителя. Сигнал люминесценции определяли на многопланшетном ридере EnVision™ (PerkinElmer, Waltham, MA, USA). Данные по люминесценции относили к контрольным лункам с разбавителем (1%-ным DMSO). Для получения кривых ингибирования и оценок IC50 методом нелинейной регрессии строили зависимость ответа с переменными коэффициентами Хилла от концентраций ингибитора (серийных разведений по 7 точкам, включая разбавитель) в логарифмически преобразованном виде, при этом в качестве верхней и нижней границ использовали постоянные значения 100 и 0, соответственно (GraphPad Prism™, программное обеспечение GraphPad, La Jolla, СА, USA).
Ингибирование калиевого hERG канала (специфического калиевого канала сердца человека (human ether-à-go-go-related gene)) может приводить к синдрому удлиненного интервала QT и сердечным аритмиям. Несмотря на то, что уровни ингаляционного ингибитора JAK1 в плазме крови будут ожидаемо уменьшаться, осажденное в легких соединение, выходящее из легкого посредством всасывания из легких в кровоток, будет циркулировать непосредственно в сердце. Таким образом, локальные концентрации ингаляционного ингибитора JAK1 в сердце могут быть временно более высокими, чем общие уровни в плазме, особенно непосредственно после введения. Таким образом, сведение к минимуму ингибирования hERG при применении ингаляционного ингибитора JAK1 может быть полезным. Например, в некоторых воплощениях предпочтительно, чтобы величина IC50 в случае hERG более чем в 30 раз превышала максимальную концентрацию (Cmax) свободного лекарственного средства в плазме крови. Соответственно, в некоторых воплощениях соединения по изобретению (или их фармацевтически приемлемая соль) демонстрируют сведенное к минимуму ингибирование hERG в таких условиях, как:
(а) использование условий 2pt-метода пэтч-клампа в автоматизированном режиме для hERG с целью исследования воздействия соединения in vitro на hERG, экспрессируемый в клетках млекопитающих, оцениваемого при комнатной температуре с использованием QPatch НТ® (Sophion Bioscience A/S, Denmark), автоматической параллельной системы пэтч-клампа. В некоторых случаях соединения тестировали только в одной или двух концентрациях, таких как 1 или 10 мкМ. В других случаях устанавливали более широкую зависимость между концентрацией и ответом, позволяющую оценить IC50. Например, концентрации тестируемых соединений выбирали такими, чтобы охватывать диапазон, соответствующий приблизительно 10-90% ингибирования, при этом использовали полулогарифмические приращения концентраций. Каждую концентрацию исследуемого изделия тестировали на двух или более клетках (n≥2). Продолжительность воздействия исследуемого изделия в каждой концентрации составляла минимум 3 минуты; и/или
(b) условия, описанные в WO 2014/074775 в разделе Примеры под заголовком "Воздействие на клонированные калиевые hERG каналы, экспрессированные в клетках млекопитающих", ChanTest™, Charles River Company; см. протокол со следующими изменениями: клетки, стабильно экспрессирующие hERG, выдерживали при -80 мВ. Начальную и стационарную стадии ингибирования соединением hERG-калиевого тока измеряли, используя последовательность импульсов с фиксированными амплитудами (подготовительная обработка предварительным импульсом: +20 мВ в течение 1 с; линейное изменение при тестировании реполяризации до -90 мВ (-0,5 В/с), повторяемое с интервалами продолжительностью 5 с). Каждое снятие показаний заканчивалось завершающим применением вещества сравнения, Е-4021 (500 нМ) (Charles River Company), в супрамаксимальной концентрации. Оставшийся неингибированным ток вычитали в режиме офлайн в цифровом формате из полученных данных для определения эффективности тестируемого вещества в отношении ингибирования hERG.
Ингибирование CYP (цитохрома Р450) не может быть желаемым свойством в случае ингаляционного ингибитора JAK1. Например, применение обратимого или время-зависимого ингибитора CYP может вызывать нежелательное повышение собственно его уровней в плазме крови или уровней других вводимых совместно лекарственных средств в плазме крови (эффект межлекарственных взаимодействий). Кроме того, причиной время-зависимого ингибирования CYP иногда является биопревращение исходного лекарственного средства в реакционноспособный метаболит. Такие реакционноспособные метаболиты могут ковалентно модифицировать белки, что может привести к токсичности. Таким образом, сведение к минимуму обратимого и время-зависимого ингибирования CYP может быть полезно в случае ингаляционного ингибитора JAK1. Соответственно, в некоторых воплощениях соединения по настоящему изобретению (или их фармацевтически приемлемая соль) демонстрируют минимальное ингибирование CYP или отсутствие обратимого и/или время-зависимого ингибирования CYP. Методы измерения ингибирования CYP известны в данной области техники. Оценку ингибирования CYP соединениями, описанными в данной заявке, проводили в диапазоне концентраций соединения 0,16-10 мкМ с использованием собранных в один пул (n=150) микросом из печени человека (Corning, Tewksbury, MA), применяя описанные ранее методы (Halladay et al., Drug Metab, Lett., 2011, 5, 220-230). Продолжительность инкубирования и концентрация белка зависели от изоформы CYP и оцениваемых маркерных субстратов/метаболитов. Для каждого CYP использовали следующие субстраты/метаболиты, времена инкубирования и концентрации белка: CYP1A2, фенацетин/ацетаминофен, 30 мин, 0,03 мг белка/мл; CYP2C9, варфарин/7-гидроксиварфарин, 30 мин, 0,2 мг белка/мл; CYP2C19, мефенитоин/4-гидроксимефенитоин, 40 мин, 0,2 мг белка/мл; CYP2D6, декстрометорфан/декстрорфан, 10 мин, 0,03 мг белка/мл; CYP3A4, мидазолам/1-гидроксимидазолам, 10 мин, 0,03 мг белка/мл и CYP3A4 тестостерон/6β-гидрокситестостерон, 10 мин, 0,06 мг белка/мл. Предварительно было определено, что эти условия отвечают линейному диапазону для скорости образования CYP-специфических метаболитов. Все реакции инициировали, используя 1 мМ NADPH (никотинамидадениндинуклеотидфосфат, восстановленная форма), и прекращали путем добавления 0,1%-ного раствора муравьиной кислоты в ацетонитриле, содержащего соответствующий стабильный меченный внутренний стандарт. Образцы анализировали с использованием LC-MS/MS.
Кроме того, в случае соединений, предполагаемых для доставки посредством ингаляции сухого порошка, выдвигается требование относительно их способности к образованию кристаллических форм соединения, которые можно микронизировать до размера 1-5 мкм. Размер частиц является важной детерминантой отложения в легких ингаляционного соединения. Обычно частицы с диаметром меньше 5 микрон (мкм) определяют как "пригодные для вдыхания". Частицы с диаметром более 5 мкм с большей долей вероятности оседают в ротоглотке и, соответственно, с меньшей долей вероятности осаждаются в легком. Кроме того, тонкодисперсные частицы с диаметром меньше 1 мкм скорее, чем большие по размерам частицы, будут оставаться в воздухе во взвешенном состоянии и, соответственно, скорее выдыхаться из легкого. Таким образом, в случае ингаляционного лекарственного средства, местом действия которого является легкое, полезным может быть диаметр частиц 1-5 мкм. Типичные методы, используемые для измерения размера частиц, включают метод с применением лазерной дифракции и метод каскадного импактора. Типичные величины, используемые для определения размера частиц, включают:
• D10, D50 и D90. Они представляют собой результаты измерения диаметра частиц, которые указывают, соответственно, что у 10%, 50% или 90% частиц в образце диаметр будет ниже этого значения. Например, значение D50, равное 3 мкм, указывает на то, что у 50% частиц в образце размер составляет ниже 3 мкм;
• среднемассовый аэродинамический диаметр (MMAD). MMAD представляет собой диаметр, относительно которого у 50% частиц по массе размер будет больше, а у 50% меньше. MMAD является параметром, характеризующим положение центра распределения;
• геометрическое стандартное отклонение (GSD). GSD является мерой величины дисперсности для MMAD или диапазона отклонений в аэродинамическом распределении частиц по размерам.
Общепринятой композицией в случае ингаляционных леакрственных средств является препарат в форме сухого порошка, включающий в себя активный фармацевтический ингредиент (API) в смеси с носителем, таким как лактоза, с добавлением или без добавления дополнительных вспомогательных веществ, таких как стеарат магния. В случае этой и других композиций может быть полезно, чтобы сам API обладал свойствами, позволяющими провести его измельчение до пригодного для вдыхания размера частиц 1-5 мкм. Следует избегать аггломерации частиц, которую можно измерить методами, известными в данной области техники, как например, исследование величины D90 в условиях разного давления. Соответственно, в некоторых воплощениях могут быть получены соединения (или их фармацевтически приемлемая соль) по настоящему изобретению, имеющие такой пригодный для вдыхания размер частиц при незначительной аггломерации или без нее.
Что касается кристалличности, то для некоторых композиций на основе ингаляционных лекарственных средств, включая смеси с лактозой, важно, что используется API конкретной кристаллической формы. Степень кристалличности и кристаллическая форма могут влиять на многие параметры, релевантные для ингаляционного лекарственного средства, включая, но не ограничиваясь этим: химическую и аэродинамическую стабильность во времени, совместимость с компонентами ингаляционной композиции, такими как лактоза, гигроскопичность, степень удержания в легких и степень раздражения легких. Таким образом, в случае ингаляционного лекарственного средства может быть полезна стабильная, воспроизводимая кристаллическая форма. Кроме того, методы, применяемые для измельчения соединений до частиц желаемого размера, зачастую являются энергоемкими и могут быть причиной превращения кристаллических форм с низкой точкой плавления в другие кристаллические формы либо становления их полностью или частично аморфными. Для кристаллической формы с точкой плавления ниже 150°С может оказаться невозможным выполнить измельчение, в то время как для кристаллической формы сточкой плавления ниже 100°С по всей вероятности процесс измельчения не возможен. Таким образом, в случае ингаляционного лекарственного средства может быть полезно иметь точку плавления по меньшей мере выше 100°С и в идеале выше 150°С. Соответственно, в некоторых воплощениях соединения (или их фармацевтически приемлемая соль), описанные в данной заявке, демонстрируют такие свойства.
Кроме того, уменьшение молекулярной массы до минимально возможной может помочь снизить эффективную дозу ингаляционного ингибитора JAK1. Более низкая молекулярная масса приводит к соответственно более высокому числу молекул на единицу массы активного фармацевтического ингредиента (API). Таким образом, может быть полезно обнаружить ингаляционный ингибитор JAK1 с самой низкой молекулярной массой, который сохраняет все другие желаемые свойства ингаляционного лекарственного средства.
И наконец, необходимо поддерживать достаточную концентрацию соединения в легком в течение определенного периода времени, чтобы иметь возможность оказывать фармакологическое действие желаемой продолжительности, а для фармакологических мишеней в тех случаях, когда системное ингибирование указанной мишени является нежелательным, иметь слабое системное воздействие. Легкое по своей природе характеризуется высокой проницаемостью как для крупных молекул (белков, пептидов), так и для малых молекул, имеющих при этом короткие периоды полувыведения из легкого, поэтому необходимо уменьшить скорость всасывания в легком посредством изменения одного или нескольких свойств соединений: сводя к минимуму проницаемость мембраны, увеличивая pKa, увеличивая cLogP, снижая растворимость, уменьшая скорость растворения или увеличивая уровень основности (например, путем введения аминогрупп) соединения с целью усиления связывания с богатой фосфолипидами тканью легкого или путем захвата в субклеточные компартменты с кислой средой, такие как лизосомы (рН 5). Методы измерения таких свойств известны в данной области техники.
Соответственно, в некоторых воплощениях соединение по настоящему изобретению (или его фармацевтически приемлемая соль) проявляет одно или более чем одно из вышеупомянутых свойств. Кроме того, в некоторых воплощениях соединение по настоящему изобретению благоприятно проявляет одно или более чем одно из этих свойств по сравнению с соединением, известным в данной области техники: это может быть особенно справедливо для соединений данной области техники, предназначенных в качестве пероральных лекарственных средств по сравнению с ингаляционными. Например, соединения, характеризующиеся быстрым всасыванием в ротовой полости, обычно плохо удерживаются в легком при ингаляции.
СПОСОБЫ ЛЕЧЕНИЯ ИНГИБИТОРАМИ ЯНУС-КИНАЗ И ИХ ПРИМЕНЕНИЯ
Соединения по настоящему изобретению или их фармацевтически приемлемая соль ингибируют активность янус-киназы, такой как киназа JAK1. Например, соединение или его фармацевтически приемлемая соль ингибирует фосфорилирование белков-передатчиков сигнала и активаторов транскрипции (STAT), осуществляемое под действием киназы JAK1, а также STAT-опосредуемое продуцирование цитокинов. Соединения по настоящему изобретению полезны для ингибирования активности киназы JAK1 в клетках через цитокиновые пути, как например пути, опосредованные IL-6, IL-15, IL-7, IL-2, IL-4, IL-9, IL-10, IL-13, IL-21, G-CSF (гранулоцитарный колониестимулирующий фактор), IFN-альфа, IFN-бета или IFN-гамма. Соответственно, в одном из воплощений предложен способ приведения в контакт клетки с соединением по настоящему изобретению или его фармацевтически приемлемой солью для ингибирования активности янус-киназы в данной клетке (например, активности JAK1).
Данные соединения могут быть использованы для лечения иммунологических расстройств, инициируемых аберрантной передачей сигнала через цитокины IL-6, IL-15, IL-7, IL-2, IL-4, IL9, IL-10, IL-13, IL-21, G-CSF, IFN-альфа, IFN-бета или IFN-гамма.
Соответственно, одно из воплощений включает соединение по настоящему изобретению или его фармацевтически приемлемую соль для применения в терапии.
В некоторых воплощениях предложено применение соединения по настоящему изобретению или его фармацевтически приемлемой соли в лечении воспалительного заболевания. Дополнительно предложено применение соединения по настоящему изобретению или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения воспалительного заболевания, такого как астма. Также предложены соединение по настоящему изобретению или его фармацевтически приемлемая соль для применения в лечении воспалительного заболевания, такого как астма.
Другое воплощение включает способ предупреждения, лечения или ослабления тяжести заболевания или состояния, такого как астма, отвечающего на ингибирование активности янус-киназы, такой как активность киназы JAK1, у пациента. Способ может включать стадию введения пациенту терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли. В одном из воплощений таким заболеванием или состоянием, отвечающим на ингибирование янус-киназы, такой как киназа JAK1, является астма.
В одном из воплощений заболевание или состояние представляет собой рак, инсульт, диабет, гепатомегалию, сердечно-сосудистое заболевание, рассеянный склероз, болезнь Альцгеймера, кистозный фиброз, вирусное заболевание, аутоиммунные заболевания, атеросклероз, рестеноз, псориаз, ревматоидный артрит, воспалительное заболевание кишечника, астму, аллергические расстройства, воспаление, неврологические расстройства, гормонозависимое заболевание, состояния, ассоциированные с трансплантацией органов (например, отторжение трансплантата), иммунодефицитные расстройства, деструктивные поражения костей, пролиферативные расстройства, инфекционные заболевания, состояния, ассоциированные с гибелью клеток, индуцированную тромбином агрегацию тромбоцитов, заболевание печени, патологические состояния иммунной системы, вовлекающие Т-клеточную активацию, расстройства центральной нервной системы (ЦНС) или миелопролиферативное расстройство.
В одном из воплощений воспалительным заболеванием является ревматоидный артрит, псориаз, астма, воспалительное заболевание кишечника, контактный дерматит или реакции гиперчувствительности замедленного типа. В одном из воплощений аутоиммунным заболеванием является ревматоидный артрит, волчанка или рассеянный склероз.
В другом воплощении соединение по настоящему изобретению или его фармацевтически приемлемая соль могут быть использованы для лечения заболеваний легких, таких как фибротическое заболевание легких или интерстициальное заболевание легких (например, интерстициальная пневмония). В некоторых воплощениях соединение по настоящему изобретению или его фармацевтически приемлемая соль могут быть использованы для лечения идиопатического легочного фиброза (IPF), ассоциированного с системным склерозом интерстициального заболевания легких (SSc-ILD), неспецифической интерстициальной пневмонии (NSIP), ассоциированного с ревматоидным артритом интерстициального заболевания легких (RA-ILD), саркоидоза, гиперчувствительного пневмонита или ILD, обусловленного заболеванием соединительной ткани без склеродермии (например, полимиозита, дерматомиозита, ревматоидного артрита, системной красной волчанки (SLE) или смешанного заболевания соединительной ткани).
В одном из воплощений рак представляет собой рак молочной железы, яичника, шейки матки, предстательной железы, яичка, пениса, мочеполового тракта, семиному, рак пищевода, гортани, области желудка, желудка, желудочно-кишечного тракта, кожи, кератоакантому, фолликулярную карциному, меланому, рак легкого, мелкоклеточный рак легкого, немелкоклеточный рак легкого (NSCLC), легочную аденокарциному, плоскоклеточную карциному легкого, рак толстой кишки, поджелудочной железы, щитовидной железы, папиллярный рак, рак мочевого пузыря, печени, желчных протоков, почки, кости, миелоидные нарушения, лимфоидные нарушения, волосатоклеточный лейкоз, рак ротоглотки (полости рта), губ, языка, ротовой полости, слюнных желез, глотки, тонкого кишечника, толстого кишечника, прямой кишки, анального отверстия, почек, предстательной железы, вульвы, щитовидной железы, толстой кишки, рак эндометрия, матки, головного мозга, центральной нервной системы, рак брюшины, гепатоклеточный рак, рак головы, рак шеи, болезнь Ходжкина или лейкоз.
В одном из воплощений таким заболеванием является миелопролиферативное расстройство. В одном из воплощений миелопролиферативное расстройство представляет собой истинную полицитемию, эссенциальный тромбоцитоз, миелофиброз или хронический миелогенный лейкоз (CML).
Другое воплощение включает применение соединения по настоящему изобретению или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения заболевания, описанного в данной заявке (например, воспалительного расстройства, иммунологического расстройства или рака). В одном из воплощений изобретения предложен способ лечения заболевания или состояния, описанного в данной заявке (например, воспалительного расстройства, иммунологического расстройства или рака), путем направленного ингибирования киназы JAK, такой как JAK1.
КОМБИНИРОВАННАЯ ТЕРАПИЯ
Данные соединения могут быть применены по отдельности или в комбинации с другими агентами для лечения. Второе или следующее (например, третье) соединение в фармацевтической композиции или режиме введения обычно обладает взаимодополняющими активностями по отношению к соединению по данному изобретению, такими, которые не оказывают неблагоприятного влияния друг на друга. Такие соединения соответственно присутствуют в комбинации в количествах, эффективных для решения предполагаемой задачи. Соединения могут быть введены вместе в единой фармацевтической композиции или по отдельности, и в случае раздельного введения оно может осуществляться одновременно или последовательно. Такое последовательное введение может осуществляться близко или отдаленно во времени.
Например, для предупреждения или лечения воспалительных заболеваний, таких как астма, с соединением по настоящему изобретению или его фармацевтически приемлемой солью могут быть скомбинированы другие соединения. Терапевтические агенты, подходящие для комбинированной терапии, включают, но не ограничиваются этим: антагонист аденозинового А2А-рецептора; противоинфекционное средство; нестероидный агонист глюкокортикоидного рецептора (GR рецептора); антиоксидант; агонист β2-адренорецептора; антагонист хемокинового рецептора 1 семейства С-С (CCR1); антагонист хемокинового рецептора (не CCR1); кортикостероид; антагонист CRTh2; антагонист DP1; антагонист рецептора формилпептидов; активатор гистон-деацетилазы; блокатор кальций-активируемого хлоридного канала человека (hCLCA1); блокатор эпителиального натриевого канала (блокатор ENAC); блокатор молекулы 1 межклеточной адгезии (блокатор ICAM); ингибитор IKK2; ингибитор c-Jun-N-концевой киназы (JNK); ингибитор канала с транзиторным рецепторным потенциалом, подсемейства анкирина 1 (TRPA1); ингибитор тирозинкиназы Брутона (BTK) (например, фенебрутиниб; ингибитор тирозинкиназы селезенки (SYK); антитело к бета-триптазе; антитело к ST2-рецептору (например, AMG 282); ингибитор циклооксигеназы (ингибитор СОХ); ингибитор липоксигеназы; антагонист рецептора лейкотриенов; обладающее двойным действием соединение - агонист β2-адренорецептора/антагонист мускаринового (М3)-рецептора (соединение МАВА); ингибитор MEK-1; ингибитор миелопероксидазы (ингибитор МРО); антагонист мускариновых рецепторов; ингибитор митоген-активируемой протеинкиназы (MAPK) р38; ингибитор фосфодиэстеразы (PDE) 4; ингибитор фосфатидилинозитол-3-киназы δ (ингибитор PI3-киназы δ); ингибитор фосфатидилинозитол-3-киназы γ (ингибитор PI3-киназа γ); агонист рецептора, активируемого пролифератором пероксисом (агонист PPARγ); ингибитор протеаз; модулятор рецептора ретиноевой кислоты (модулятор RARγ); статин; антагонист тромбоксанов; агонист toll-подобного рецептора (TLR) 7; или вазодилататор.
Помимо этого, соединение по настоящему изобретению или его фармацевтически приемлемая соль могут быть скомбинированы с: (1) кортикостероидами, такими как алклометазона дипропионат, амелометазон, беклометазона дипропионат, будесонид, бутиксокорта пропионат, биклесонид, клобетазола пропионат, дезизобутирилциклесонид, дексаметазон, этипреднола диклоацетат, флуоцинолона ацетонид, флутиказона фуроат, флутиказона пропионат, лотепреднола этабонат (местного действия) или мометазона фуроат; (2) агонистами β2-адренорецепторов, такими как сальбутамол, альбутерол, тербуталин, фенотерол, битолтерол, карбутерол, кленбутерол, пирбутерол, римотерол, тербуталин, третохинол, тулобутерол и длительно действующие агонисты β2-адренорецепторов, такие как метапротеренол, изопротеренол, изопреналин, сальметерол, индакатерол, формотерол (включая формотерола фумарат), арформотерол, кармотерол, абедитерол, вилантерола трифенат, олодатерол; (3) комбинированными продуктами, составленными из кортикостероидов/длительно действующих β2-агонистов, такими как сальметерол/флутиказона пропионат (Advair®, также продаваемый как Seretide®), формотерол/будесонид (Symbicort®), фор моте рол/флутиказона пропионат (Flutiform®), формотерол/циклесонид, формотерол/мометазона фуроат, индакатерол/мометазона фуроат, вилантерола трифенат/флутиказона фуроат (БРЕО ЭЛЛИПТА) или арформотерол/циклесонид; (4) антихолинергическими агентами, например, антагонистами мускариновых-3 (М3) рецепторов, такими как ипратропия бромид, тиотропия бромид, аклидиния бромид (LAS-34273), гликопиррония бромид, умеклидиния бромид; (5) комбинированными продуктами, составленными из М3-антихолинергических агентов/агонистов β2-адренорецепторов, такими как вилантерол/умеклидиний (Anoro® Ellipta®), олодатерол/тиотропия бромид, гликопиррония бромид/индакатерол (Ultibro®, также продаваемый как Xoterna®), фенотерола гидробромид/ипратропия бромид (Berodual®), альбутерола сульфат/ипратропия бромид (Combivent®), формотерола фумарат/гликопирролат или аклидиния бромид/формотерол; (6) обладающими двойным фармакологическим действием средствами, составленными из М3-антихолинергических агентов/агонистов β2-адренорецепторов, такими как батефентерола сукцинат, AZD-2115 или LAS-190792; (7) модуляторами лейкотриенов, например, антагонистами лейкотриенов, такими как монтелукаст, зафирлукаст или пранлукаст, или ингибиторами биосинтеза лейкотриенов, такими как зилейтон, или антагонистами лейкотриена В4 (LTB4), такими как амелубант, или ингибиторами 5-липоксигеназа-активирующего белка (FLAP), такими как фибофлапон, GSK-2190915; (8) ингибиторами фосфодиэстеразы-IV (PDE-IV) (для перорального или ингаляционного введения), такими как рофлумиласт, циломиласт, оглемиласт, ролипрам, тетомиласт, AVE-8112, ревамиласт, CHF 6001; (9) антигистаминными препаратами, например, селективными антагонистами гистаминовых-1 (Н1) рецепторов, такими как фексофенадин, цетиризин, лоратадин или астемизол, или обладающими двойным действием антагонистами Н1/Н3 рецепторов, такими как GSK 835726 или GSK 1004723; (10) противокашлевыми агентами, такими как кодеин или декстраморфан; (11) муколитическим средством, например, N-ацетилцистеином или фудостеином; (12) отхаркивающим средством/мукокинетическим модулятором, таким как амброксол, гипертонические растворы (например, физиологический раствор или раствор маннита) или поверхностно-активное вещество; (13) пептидным муколитическим средством, например, рекомбинантной человеческой дезоксирибонуклеазой I (дорназой-альфа и рчДНКазой) или гелицидином; (14) антибиотиками, например азитромицином, тобрамицином или азтреонамом; (15) неселективными ингибиторами СОХ-1/СОХ-2, такими как ибупрофен или кетопрофен; (16) ингибиторами СОХ-2, такими как целекоксиб и рофекоксиб; (17) антагонистами VLA-4 (очень поздний антиген-4), такими как те, которые описаны в заявках WO 97/03094 и WO 97/02289, каждая из которых включена в данное описание посредством ссылки; (18) ингибиторами ТАСЕ (TNFα-превращающий фермент) и ингибиторами TNF-α, например, моноклональными антителами к TNF, такими как Remicade® и CDP-870, и молекулами иммуноглобулинов к рецептору TNF, такими как Enbrel®; (19) ингибиторами матриксной металлопротеазы (ММР), например, ММР-12; (20) ингибиторами эластазы нейтрофилов человека, такими как BAY-85-8501 или как те, которые описаны в заявках WO 2005/026124, WO 2003/053930 и WO 06/082412, каждая из которых включена в данное описание посредством ссылки; (21) A2b-антагонистами, такими как те, которые описаны в заявке WO 2002/42298, включенной в данное описание посредством ссылки; (22) модуляторами функции хемокиновых рецепторов, например, антагонистами CCR3 и CCR8; (23) соединениями, которые модулируют действие других простаноидных рецепторов, например, антагонистом тромбоксана А2, антагонистами DP1, такими как ларопипрант или азапипрант, антагонистами CRTH2, такими как ОС000459, февипипрант, ADC 3680 или ARRY 502; (24) агонистами PPAR, включая агонисты PPAR-альфа (такие как фенофибрат), агонисты PPAR-дельта, агонисты PPAR-гамма, такие как пиоглитазон, розиглитазон и балаглитазон; (25) метилксантинами, такими как теофиллин или аминофиллин, и комбинациями метилксантинов/кортикостероидов, такими как теофиллин/будесонид, теофиллин/флутиказона пропионат, теофиллин/циклесонид, теофиллин/мометазона фуроат и теофиллин/беклометазона дипропионат; (26) А2а-агонистами, такими как те, которые описаны в ЕР 1052264 и ЕР 1241176; (27) антагонистами CXCR2 (хемокиновые рецепторы семейства С-Х-С) или IL-8, такими как AZD-5069, AZD-4721, данириксин; (28) модуляторами IL-R-опосредованной передачи сигнала, такими как кинерет и ACZ 885; (29) антагонистами МСР-1 (моноцитарный хемоаттрактантный белок 1), такими как ABN-912; (30) ингибитором MAPK р38, таким как ВСТ197, JNJ49095397, лосмапимод или РН-797804; (31) агонистами рецептора TLR7, такими как AZD 8848; (32) ингибиторами PI3-киназ, такими как RV1729 или GSK2269557 (немиралисиб); (33) комбинированными продуктами тройного действия, такими как ТРЕЛЕДЖИ ЭЛЛИПТА (комбинация флутиказона фуроата, умеклидиния бромида и вилантерола); или (34) низкомолекулярными ингибиторами TRPA1, BTK или SYK.
В некоторых воплощениях соединение по настоящему изобретению или его фармацевтически приемлемую соль можно использовать в комбинации с одним или более чем одним из дополнительных лекарственных средств, например, с антигиперпролиферативными, противораковыми, цитостатическими, цитотоксичными, противовоспалительными или химиотерапевтическими агентами, такими как агенты, описанные в публикации заявки США №2010/0048557, включенной в данное описание посредством ссылки. Соединение по настоящему изобретению или его фармацевтически приемлемую соль также можно использовать в сочетании с лучевой терапией или хирургическим вмешательством, что является известным фактом в данной области техники.
Особенно включены комбинации любого из перечисленных выше агентов с соединением по настоящему изобретению или его фармацевтически приемлемой солью.
ИЗДЕЛИЯ ПРОИЗВОДСТВА
Другое воплощение включает изделие производства (например, набор) для лечения заболевания или расстройства, отвечающего на ингибирование янус-киназы, такой как киназа JAK1. Набор может содержать:
(a) первую фармацевтическую композицию, содержащую соединение по настоящему изобретению или его фармацевтически приемлемую соль; и
(b) инструкции по применению.
В другом воплощении набор дополнительно содержит:
(c) вторую фармацевтическую композицию, такую как фармацевтическая композиция, содержащая агент для лечения, описанный выше, такой как агент для лечения воспалительного расстройства, или химиотерапевтический агент.
В одном из воплощений в инструкциях описывается одновременное, последовательное или раздельное введение указанных первой и второй фармацевтических композиций пациенту, нуждающемуся в этом.
В одном из воплощений первая и вторая композиции содержатся в разных контейнерах. В другом воплощении первая и вторая композиции содержатся в одном и том же контейнере.
Используемые контейнеры включают, например, бутылки, флаконы, шприцы, блистерную упаковку и так далее. Контейнеры могут быть изготовлены из разнообразных материалов, таких как стекло или пластик. Контейнер включает в себя соединение по настоящему изобретению или его фармацевтически приемлемую соль, которые эффективны для лечения указанного состояния, и может иметь стерильное входное отверстие (например, контейнер может представлять собой мешочек или флакон для внутривенных растворов, имеющий пробку, поддающуюся прокалыванию иглой для подкожных инъекций). На этикетке или в инструкции по применению указывается, что соединение применяют для лечения выбранного состояния, такого как астма или рак. В одном из воплощений на этикетке или в инструкциях по применению указывается, что соединение можно использовать для лечения какого-либо расстройства. Помимо этого, на этикетке или в инструкциях по применению может быть указано, что подлежащим лечению пациентом является пациент с расстройством, характеризующимся сверхвысокой или нерегулируемой активностью янус-киназы, такой как сверхвысокая или нерегулируемая активность JAK1. На этикетке или в инструкции по применению также может быть указано, что соединение или композицию можно использовать для лечения других расстройств.
Альтернативно или помимо этого, такой набор может дополнительно содержать второй (или третий) контейнер, содержащий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекций (BWFI), забуференный фосфатом физиологический раствор, раствор Рингера или раствор декстрозы. Кроме того, он может включать в себя другие материалы, подходящие с коммерческой точки зрения и точки зрения потребителя, в том числе другие буферы, разбавители, фильтры, иглы и шприцы.
Для иллюстрации данного изобретения включены приведенные ниже примеры. Однако следует понимать, что эти примеры не ограничивают изобретение, а предназначены только для рекомендации способа практического осуществления изобретения. Специалистам в данной области техники будет понятно, что описанные химические реакции можно легко адаптировать для получения других соединений по настоящему изобретению, и альтернативные способы получения этих соединений включены в объем данного изобретения. Например, синтез не описанных в качестве примеров соединений по изобретению с успехом можно провести, используя модификации, очевидные для специалистов в данной области техники, например, путем введения соответствующей защиты на мешающие проведению реакции группы, путем использования других подходящих реагентов, известных в данной области техники, отличающихся от описанных, и/или путем выполнения общепринятых модификаций реакционных условий. Альтернативно, другие реакции, описанные в данной заявке или известные в данной области техники, будут признаны как находящие применение для получения других соединений по изобретению.
ПРИМЕРЫ
Общая подробная информация касательно условий экспериментов
Все растворители и имеющиеся в продаже реагенты использовали по получении, если не указано иное. В тех случаях, когда продукты очищали хроматографией на диоксиде кремния, этот процесс выполняли с использованием либо стеклянной колонки, упакованной вручную силикагелем (кизельгелем 60, 220-440 меш, 35-75 мкм), либо Si II картриджа для твердофазной экстракции (SPE) Isolute®. "Si картридж для SPE Isolute®" означает предварительно упакованную колонку из полипропилена, содержащую немодифицированный активированный диоксид кремния с нерегулярными частицами, имеющими средний размер 50 мкм и номинальную пористость 60Å. В тех случаях, когда использовали SCX-2 картридж Isolute®, "SCX-2 картридж Isolute®" означает предварительно упакованную колонку из полипропилена, содержащую сильный катионообменный сорбент на основе диоксида кремния, функционализированного не блокированными группами пропилсульфоновой кислоты.
Условия LCMS
Метод А
Эксперименты проводили на приборе SHIMADZU LCMS-2020 с С18-обращенно-фазовой колонкой (50×3 мм, Shim-Pack XR-ODS, размер частиц 2,2 мкм), с элюированием растворителем А: вода + 0,05% трифторуксусной кислоты; растворителем В: ацетонитрил + 0,05% трифторуксусной кислоты. Градиент:

Детектирование по поглощению в ультрафиолетовом диапазоне (УФ) (220 и 254 нм) и светорассеянию испаренного образца (ELSD).
Метод В
Эксперименты проводили на приборе SHIMADZU LCMS-2020 с С18-обращенно-фазовой колонкой (50×3 мм, Shim-Pack XR-ODS, размер частиц 2,2 мкм), с элюированием растворителем А: вода + 0,05% трифторуксусной кислоты; растворителем В: ацетонитрил + 0,05% трифторуксусной кислоты. Градиент:

Детектирование - УФ (220 и 254 нм) и ELSD.
Метод С
Эксперименты проводили на приборе SHIMADZU LCMS-2020 с С18-обращенно-фазовой колонкой (50×3 мм, Shim-Pack XR-ODS, размер частиц 2,2 мкм), с элюированием растворителем А: вода + 0,05% трифторуксусной кислоты; растворителем В: ацетонитрил + 0,05% трифторуксусной кислоты. Градиент:


Детектирование - УФ (220 и 254 нм) и ELSD.
Метод D
Эксперименты проводили на приборе SHIMADZU LCMS-2020 с С18-обращенно-фазовой колонкой (50×3 мм, Shim-Pack XR-ODS, размер частиц 2,2 мкм), с элюированием растворителем А: вода + 0,05% трифторуксусной кислоты; растворителем В: ацетонитрил + 0,05% трифторуксусной кислоты. Градиент:

Детектирование - УФ (220 и 254 нм) и ELSD.
Метод Е
Эксперименты проводили на приборе SHIMADZU LCMS-2020 с С18-обращенно-фазовой колонкой (50×3 мм, Shim-Pack XR-ODS, размер частиц 2,2 мкм), с элюированием растворителем А: вода + 0,05% трифторуксусной кислоты; растворителем В: ацетонитрил + 0,05% трифторуксусной кислоты. Градиент:

Детектирование - УФ (220 и 254 нм) и ELSD.
Метод F
Эксперименты проводили на приборе SHIMADZU LCMS-2020 с С18-обращенно-фазовой колонкой (50×3 мм, Shim-Pack XR-ODS, размер частиц 2,2 мкм), с элюированием растворителем А: вода + 0,05% трифторуксусной кислоты; растворителем В: ацетонитрил + 0,05% трифторуксусной кислоты. Градиент:

Детектирование - УФ (220 и 254 нм) и ELSD.
Метод G
Эксперименты проводили на приборе для HPLC SHIMADZU 20А с С18-обращенно-фазовой колонкой (50×2,1 мм, Ascentis Express, С18, размер частиц 2,7 мкм), с элюированием растворителем А: вода + 0,05% трифторуксусной кислоты; растворителем В: ацетонитрил + 0,05% трифторуксусной кислоты. Градиент:

Детектирование - УФ (220 и 254 нм) и ELSD.
Метод Н
Эксперименты проводили на приборе SHIMADZU LCMS-2020 с С18-обращенно-фазовой колонкой (50×3 мм, Shim-Pack XR-ODS, размер частиц 2,2 мкм), с элюированием растворителем А: вода + 0,05% трифторуксусной кислоты; растворителем В: ацетонитрил + 0,05% трифторуксусной кислоты. Градиент:

Детектирование - УФ (220 и 254 нм) и ELSD.
Метод I
Эксперименты проводили на приборе для HPLC SHIMADZU 20А с колонкой Poroshell НРН-С18 (50×3 мм, размер частиц 2,7 мкм), с элюированием растворителем А: вода/5 мМ NH4HCO3; растворителем В: ацетонитрил. Градиент:

Детектирование - УФ (220 и 254 нм) и ELSD.
Метод J
Эксперименты проводили на приборе SHIMADZU LCMS-2020 с С18-обращенно-фазовой колонкой (50×3 мм, Kinetex ХВ-С18, размер частиц 2,6 мкм), с элюированием растворителем А: вода + 0,05% трифторуксусной кислоты; растворителем В: ацетонитрил + 0,05% трифторуксусной кислоты. Градиент:

Детектирование - УФ (220 и 254 нм) и ELSD.
Метод K
Эксперименты проводили на приборе SHIMADZU LCMS-2020 с С18-обращенно-фазовой колонкой (50×3 мм, Shim-Pack XR-ODS, размер частиц 2,2 мкм), с элюированием растворителем А: вода + 0,05% трифторуксусной кислоты; растворителем В: ацетонитрил + 0,05% трифторуксусной кислоты. Градиент:

Детектирование - УФ (220 и 254 нм) и ELSD.
Метод L
Эксперименты проводили на приборе SHIMADZU LCMS-2020 с С18-обращенно-фазовой колонкой (50×2,1 мм, Kinetex ХВ-С18, 100А, размер частиц 2,6 мкм), с элюированием растворителем А: вода + 0,05% трифторуксусной кислоты; растворителем В: ацетонитрил + 0,05% трифторуксусной кислоты. Градиент:

Детектирование - УФ (220 и 254 нм) и ELSD.
Список общих сокращений


Промежуточное соединение 1
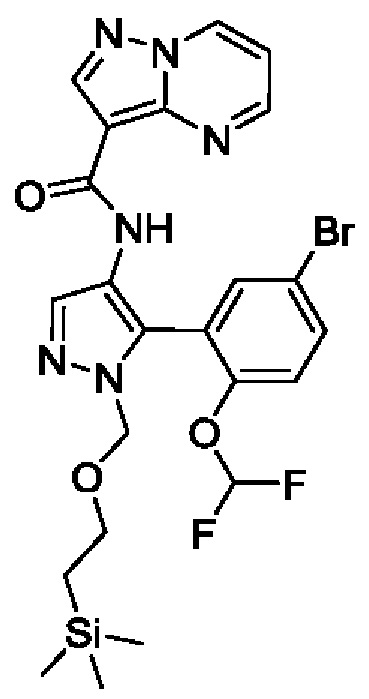
N-(5-(5-Бром-2-(дифторметокси)фенил)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид
Стадия 1: синтез 4-бром-1-(дифторметокси)-2-иодбензола

К раствору 4-бром-2-иодфенола (282 г; 943 ммоль) в N,N-диметилформамиде (2000 мл) и воде (500 мл) добавляли 2-хлор-2,2-дифторацетат натрия (216 г; 1,42 моль) и Cs2CO3 (617 г; 1,89 моль). Реакционный сосуд был оборудован выходным отверстием для газа для высвобождения CO2. Полученную смесь перемешивали в течение ночи при 120°С, оставляли охлаждаться до комнатной температуры и выливали в ледяную воду (3000 мл). Полученный раствор экстрагировали этилацетатом (3×1500 мл) и органические слои объединяли. Этилацетатные экстракты промывали рассолом (1000 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с элюированием смесью этилацетат/петролейный эфир (1/10), получая 300 г (91%) 4-бром-1-(дифторметокси)-2-иодбензола в виде желтого масла. 1Н ЯМР (300 МГц, CDCl3) δ 7.96 (dd, J=5,7 Гц; 2,4 Гц, 1Н), 7.45 (dd, J=8,7 Гц; 2,4 Гц, 1Н), 7.03 (d, J=8,7 Гц, 1Н), 6.39 (t, J=72,9 Гц, 1Н).
Стадия 2: синтез 5-[5-бром-2-(дифторметокси)фенил]-4-нитро-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразола
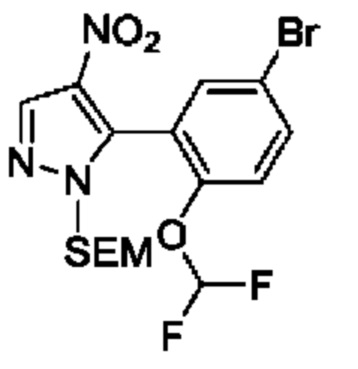
К раствору 4-нитро-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразола (100 г; 411 ммоль) в безводном THF (1000 мл) по каплям добавляли раствор LiHMDS (490 мл; 1,0 моль/л в THF) с перемешиванием при -70°С в атмосфере азота. Полученный раствор перемешивали в течение 1 ч при -50°С и затем охлаждали до -70°С. По каплям добавляли ZnCl2 (500 мл; 0,7 моль/л в THF) при -70°С. Полученный раствор оставляли нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение 1 ч. К смеси добавляли 4-бром-1-(дифторметокси)-2-иодбензол (150 г; 860 ммоль), Pd(PPh3)4 (24,0 г; 20,8 ммоль). Полученный раствор нагревали при температуре дефлегмации в течение ночи, оставляли охлаждаться до комнатной температуры и концентрировали при пониженном давлении. Данную реакцию в этом масштабе повторяли еще раз и неочищенные продукты с двух повторов объединяли для очистки. Остаток очищали флэш-хроматографией на силикагеле, элюируя смесью этилацетат/петролейный эфир (1/20). Соответствующие фракции объединяли и концентрировали при пониженном давлении. В результате этого в общей сложности получали 300 г (79%) 5-[5-бром-2-(дифторметокси)фенил]-4-нитро-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразола в виде светло-желтого твердого вещества. 1Н ЯМР (300 МГц, CDCl3) δ 8.27 (s, 1Н), 7.68 (dd, J=8,7; 2,4 Гц, 1Н), 7.62 (d, J=2,4 Гц, 1Н), 7.19 (d, J=8,4 Гц, 1Н), 6.39 (t, J=72,5 Гц, 1Н), 5.44-5.19 (m, 2Н), 3.72-3.54 (m, 2Н), 0.94-0.89 (m, 2Н), 0.02 (s, 9Н).
Стадия 3: синтез 5-(5-бром-2-(дифторметокси)фенил)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-амина
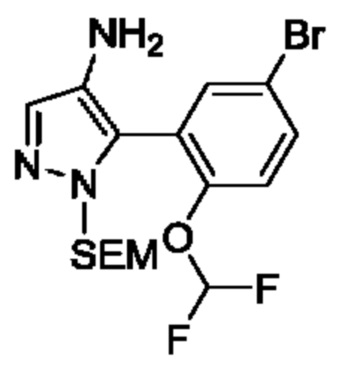
К раствору 5-(5-бром-2-(дифторметокси)фенил)-4-нитро-1-((2-(триметилсилил)этокси)метил)-1Н-пиразола (50,1 г; 108 ммоль) в этаноле (2000 мл) и воде (200 мл) добавляли порошок железа (60,1 г; 1,07 моль) и NH4Cl (28,0 г; 0,523 моль). Реакционную смесь перемешивали при температуре дефлегмации в течение 3 ч в атмосфере азота. Твердые вещества отфильтровывали и промывали этанолом (100 мл). Фильтрат концентрировали при пониженном давлении. Остаток растворяли в 3000 мл этилацетата. Этилацетатный раствор промывали 1×500 мл рассола, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении, получая 50,1 г неочищенного 5-(5-бром-2-(дифторметокси)фенил)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-амина в виде желтого масла. Неочищенный продукт использовали для следующуй стадии без дополнительной очистки. LC/MS (метод G; ESI (электрораспылительная ионизация)): [М+Н]+=434,2; RT=0,93 мин.
Стадия 4: синтез N-(5-(5-бром-2-(дифторметокси)фенил)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида

К раствору 5-(5-бром-2-(дифторметокси)фенил)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-амина (50,1 г; 115 ммоль) в DMA (1500 мл) добавляли пиразоло[1,5-а]пиримидин-3-карбоновую кислоту (32,1 г; 196,0 ммоль), РуАОР(102 г; 196 ммоль), DMAP (1,41 г; 11,0 ммоль) и DIPEA (44,1 г; 0,341 моль). Полученный раствор перемешивали в течение 3 ч при 60°С в масляной бане и затем оставляли охлаждаться до комнатной температуры. Затем реакционную смесь распределяли между ледяной водой (2000 мл) и этилацетатом (2000 мл). Водную фазу экстрагировали этилацетатом (2×). Органические слои объединяли, промывали рассолом (1000 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле, элюируя смесью этилацетат/петролейный эфир (4:1). Соответствующие фракции объединяли и концентрировали при пониженном давлении. К остатку добавляли воду (150 мл) и смесь перемешивали в воде в течение 1 ч при комнатной температуре. Твердое вещество собирали фильтрованием и сушили на воздухе, получая 60,1 г (91%) N-(5-(5-бром-2-(дифторметокси)фенил)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида в виде светло-желтого твердого вещества. LC/MS (метод G; ESI): [М+Н]+=579,1 и 581,1; RT=1,10 мин. 1Н ЯМР (300 МГц, CDCl3) δ 9.62 (s, 1Н), 8.80 (dd, J=6,9; 1,7 Гц, 1Н), 8.73 (s, 1Н), 8.53 (dd, J=4,2; 1,7 Гц, 1Н), 8.38 (s, 1Н), 7.79 (d, J=2,4 Гц, 1Н), 7.67 (dd, J=8,8; 2,5 Гц, 1Н), 7.29 (d, J=1,4 Гц, 1Н), 7.00 (dd, J=6,9; 4,2 Гц, 1Н), 6.43 (t, J=72,6 Гц, 1Н), 5.53-5.27 (m, 2Н), 3.73-3.50 (m, 2Н), 0.88 (ddd, J=9,5; 6,4; 4,4 Гц, 2Н), 0.00 (s, 9Н).
Промежуточное соединение 2

N-[3-[5-Бром-2-(дифторметокси)фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид
N-[5-[5-Бром-2-(дифторметокси)фенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид (промежуточное соединение 1; 5,00 г; 8,63 ммоль) обрабатывали смесью HCl/диоксан (150 мл; 4 М) в течение ночи при комнатной температуре. Полученную смесь концентрировали при пониженном давлении. В результате этого получали 3,80 г N-[3-[5-бром-2-(дифторметокси)фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде желтого твердого вещества. Чистота промежуточного соединения была достаточной для применения на следующей стадии без дополнительной очистки. LC/MS (метод I, ESI): [М+Н]+=449,0; RT=1,02 мин. 1Н ЯМР (400 МГц, CD3OD) δ 9.11 (dd, J=6,8; 1,6 Гц, 1Н), 8.67-8.64 (m, 2Н), 8.32 (s, 1Н), 7.80 (d, J=2,4 Гц, 1Н), 7.72 (dd, J=8,8; 2,4 Гц, 1Н), 7.37 (d, J=8,8 Гц, 1Н), 7.23 (dd, J=7,0; 4,2 Гц, 1Н), 6.81 (t, J=73,2 Гц, 1Н).
Промежуточное соединение 3
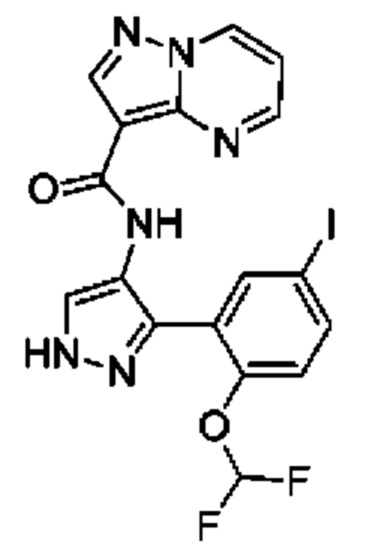
N-[3-[2-(Дифторметокси)-5-иодфенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид
Стадия 1: синтез N-[5-[2-(дифторметокси)-5-иодфенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида

К раствору N-[5-[5-бром-2-(дифторметокси)фенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (100 мг; 0,173 ммоль) в t-BuOH (2 мл) добавляли N,N-диметилэтан-1,2-диамин (2,28 мг; 0,0259 ммоль), NaI (155 мг; 1,04 ммоль), CuI (4,93 мг; 0,026 ммоль) в атмосфере азота. Полученный раствор перемешивали в течение 14 ч при 120°С в масляной бане в атмосфере азота, после чего охлаждали до комнатной температуры. Полученную смесь концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с элюированием смесью этилацетат/петролейный эфир (1/1), получая 80 мг (74%) N-[5-[2-(дифторметокси)-5-иодфенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде желтого твердого вещества. LC/MS (метод J, ESI): [М+Н]+=627,1; RT=1,31 мин.
Стадия 2: синтез N-[3-[2-(дифторметокси)-5-иодфенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида

N-[5-[2-(Дифторметокси)-5-иодфенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид (80,0 мг; 0,128 ммоль) обрабатывали CF3CO2H (3,0 мл) в течение 30 мин при комнатной температуре. Полученную смесь концентрировали при пониженном давлении. Остаток растворяли в воде. Медленно добавляли насыщенный раствор бикарбоната натрия до достижения рН раствора примерно 8. Твердое вещество собирали фильтрованием. Твердое вещество очищали флэш-хроматографией на силикагеле с элюированием смесью этилацетат/петролейный эфир (2/1), получая 23,0 мг (36%) N-[3-[2-(дифторметокси)-5-иодфенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде светло-желтого твердого вещества. LC/MS (метод K, ESI): [М+Н]+=497,1; RT=1,74 мин. 1Н ЯМР (300 МГц, CD3OD) δ 9.08 (dd, J=6,9; 1,5 Гц, 1Н), 8.65-8.61 (m, 2Н), 8.27 (s, 1Н), 7.94 (s, 1Н), 7.87 (d, J=8,7 Гц, 1Н), 7.21-7.18 (m, 2Н), 6.78 (t, J=73,2 Гц, 1Н).
Промежуточное соединение 4

N-(3-(5-((1R,2R)-2-Цианоциклопропил)-2-(дифторметокси)фенил)-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид
Стадия 1: синтез N-[5-[5-(2-цианоциклопропил)-2-(дифторметокси)фенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида

К раствору N-[5-[5-бром-2-(дифторметокси)фенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (промежуточного соединения 1; 1,00 г; 1,73 ммоль) в диоксане (15 мл) и воде (3,0 мл) добавляли (2-цианоциклопропил)трифторборат калия (449 мг; 2,60 ммоль), Pd(dppf)Cl2⋅CH2Cl2 (141 мг; 0,173 ммоль; 0,10 экв.) и Cs2CO3 (1,13 г; 3,47 ммоль; 2,01 экв.) в атмосфере азота. Полученный раствор перемешивали в течение ночи при 80°С в масляной бане. Полученную смесь концентрировали при пониженном давлении. Реакцию в этом масштабе повторяли пять раз и неочищенные продукты с 5 процедур объединяли вместе для очистки. Остаток пропускали через короткую набивку силикагеля, элюируя смесью этилацетат/петролейный эфир (6/4). Соответствующие фракции объединяли и концентрировали при пониженном давлении, получая 2,5 г (с чистотой 85% по данным LCMS при 254 нм) N-[5-[5-(2-цианоциклопропил)-2-(дифторметокси)фенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде желтого твердого вещества. Качество промежуточного соединения было достаточным для проведения следующей стадии. Никакой дополнительной очистки не требовалось. LC/MS (метод G; ESI): [М+Н]+=566,2; RT=1,07 мин.
Стадия 2: N-(3-[5-[(1R,2R)-2-цианоциклопропил]-2-(дифторметокси)фенил]-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид

N-[5-[5-(2-Цианоциклопропил)-2-(дифторметокси)фенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид (2,90 г; 5,13 ммоль) обрабатывали трифторуксусной кислотой (10 мл) в дихлорметане (20 мл) в течение ночи при комнатной температуре. Полученную смесь концентрировали под вакуумом. рН остатка подводили до значения больше 7, используя DIEA. Полученную смесь концентрировали под вакуумом. Добавляли воду и смесь перемешивали в течение 1 ч. Твердые вещества собирали фильтрованием. Неочищенный продукт (2,30 г) очищали препаративной HPLC в следующих условиях: колонка: XBridge Shield RP18 OBD, 19×150 мм, 5 мкм; 13 нм; подвижная фаза: вода с 0,05% NH4OH и MeCN (от 30,0% до 45,0% MeCN за 9 мин); детектор: УФ 254 нм. Рацемический продукт разделяли, используя препаративную сверхкритическую жидкостную хроматографию (SFC) в следующих условиях: колонка: CHIRALPAK-IC-SFC-02, 5 см × 25 см (5 мкм); подвижная фаза А: CO2:50, подвижная фаза В: EtOH:50; скорость потока: 180 мл/мин; 220 нм; RT1=13,97 мин (первый пик); RT2=17,84 мин (второй пик), получая две фракции.
Фракция 1 (R,R-изомер). Первый пик представлял собой желаемую фракцию, и ее далее очищали посредством перекристаллизации из изопропанола. В результате этого получали 340 мг (15%) N-(3-[5-[(1R,2R)-2-цианоциклопропил]-2-(дифторметокси)фенил]-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида в виде беловатого твердого вещества. 1Н ЯМР (300 МГц, CD3OD) δ 9.09 (dd, J=7,1; 1,7 Гц, 1Н), 8.67-8.62 (m, 2Н), 8.27 (s, 1Н), 7.43-7.35 (m, 3Н), 7.21 (dd, J=6,9; 4,2 Гц, 1Н), 6.76 (t, J=73,8 Гц, 1Н), 2.79-2.72 (m, 1Н), 1.94-1.88 (m, 1Н), 1.68-1.63 (m, 1Н), 1.62-1.56 (m, 1Н).
Фракция 2 (S,S-изомер). Из второго пика получали 474 мг (21%) N-(3-[5-[(1S,2S)-2-цианоцикпопропил]-2-(дифторметокси)фенил]-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида в виде белого твердого вещества. 1Н ЯМР (300 МГц, CD3OD) δ 9.09 (dd, J=7,1; 1,7 Гц, 1Н), 8.67-8.62 (m, 2Н), 8.27 (s, 1Н), 7.43-7.35 (m, 3Н), 7.21 (dd, J=6,9; 4,2 Гц, 1Н), 6.76 (t, J=73,8 Гц, 1Н), 2.79-2.72 (m, 1Н), 1.94-1.88 (m, 1Н), 1.68-1.63 (m, 1Н), 1.62-1.56 (m, 1Н).
Промежуточное соединение 5

N-[3-[5-Циклопропил-2-(дифторметокси)фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид
Стадия 1: синтез N-[3-[5-циклопропил-2-(дифторметокси)фенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида

К раствору N-(5-(5-бром-2-(дифторметокси)фенил)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (промежуточного соединения 1; 1,40 г; 2,41 ммоль) в диоксане (15 мл) и воде (3,0 мл) добавляли циклопропилбороновую кислоту (314 мг; 3,66 ммоль), Pd(dppf)Cl2-CH2Cl2 (200 мг; 0,245 ммоль) и Cs2CO3 (1,56 г; 4,79 ммоль) в атмосфере азота. Реакционную смесь перемешивали в течение ночи при 80°С в атмосфере азота. Полученную смесь концентрировали при пониженном давлении. Остаток пропускали через короткую набивку силикагеля с элюированием смесью дихлорметан/метанол (94/6). Соответствующие фракции объединяли и концентрировали при пониженном давлении, получая 1,40 г (чистота примерно 85% при 254 нм) N-[3-[5-циклопропил-2-(дифторметокси)фенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде темно-красного твердого вещества. LC/MS (метод G; ESI): [М+Н]+=541,2; RT=1,12 мин. Промежуточное соединение использовали без дополнительной очистки.
Стадия 2: синтез N-[3-[5-циклопропил-2-(дифторметокси)фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида

N-[3-[5-Циклопропил-2-(дифторметокси)фенил]-1-[[2-(триметилсилил)-этокси]метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид (145 мг) с предыдущей стадии обрабатывали смесью HCl/диоксан (5,0 мл; 4 М) в течение 2 ч при 25°С. Раствор концентрировали при пониженном давлении. Остаток очищали препаративной HPLC в следующих условиях: колонка: XBridge С18, 19×150 мм, 5 мкм; подвижная фаза А: вода/0,05% NH4HCO3, подвижная фаза В: ACN; скорость потока: 30 мл/мин; градиент: от 20% В до 85% В за 10 мин; 254 нм, получая 44,9 мг (41%) N-[3-[5-циклопропил-2-(дифторметокси)фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде желтого твердого вещества. LC/MS (метод Н, ESI): [М+Н]+=411,2; RT=1,14 мин. 1Н ЯМР (300 МГц, CD3OD) δ 9.09 (dd, J=6,9; 1,5 Гц, 1Н), 8.63-8.61 (m, 2Н), 8.27 (s, 1Н), 7.28-7.25 (m, 3Н), 7.20 (dd, J=7,2; 4,2 Гц, 1Н), 6.68 (t, J=73,8 Гц, 1Н), 2.04-1.95 (m, 1Н), 1.03-0.97 (m, 2Н), 0.79-0.71 (m, 2Н).
Промежуточное соединение 6

N-[3-[2-(Дифторметокси)-5-[4-(диметилкарбамоил)фенокси]фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид
Стадия 1: синтез N-[5-[2-(дифторметокси)-5-[4-(диметилкарбамоил)фенокси]фенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида

К раствору N-5-[5-бром-2-(дифторметокси)фенил]-1-[2-(триметилсилил)этокси]метил-1Н-пиразол-4-илпиразоло[1,5-а]пиримидин-3-карбоксамида (промежуточного соединения 1; 1,17 г; 2,02 ммоль) в толуоле (10 мл) добавляли 4-гидрокси-N,N-диметилбензамид (0,400 г; 2,42 ммоль), Cs2CO3 (0,790 г; 2,43 ммоль), [PdCl(аллил)]2 (37,0 мг; 0,101 ммоль) и t-BuBrettPhos (98,0 мг; 0,202 ммоль) в атмосфере азота. Реакционную смесь перемешивали в течение ночи при 90°С. Полученную смесь концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (95/5), получая 1,3 г (81%) N-[5-[2-(дифторметокси)-5-[4-(диметилкарбамоил)фенокси]фенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде желтого масла. LC/MS (метод Н, ESI): [М+Н]+=664,4; RT=1,36 мин.
Стадия 2: синтез N-[3-[2-(дифторметокси)-5-[4-(диметилкарбамоил)фенокси]фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида
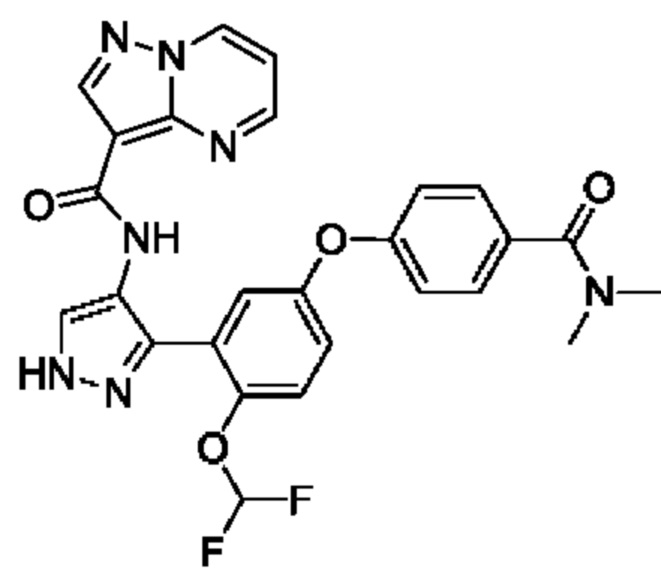
К раствору N-[5-[2-(дифторметокси)-5-[4-(диметилкарбамоил)фенокси]фенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (139 мг; 0,209 ммоль) в МеОН (3,0 мл) добавляли концентрированную соляную кислоту (1,5 мл; 12 М). Реакционную смесь перемешивали в течение 2 ч при 25°С и концентрировали при пониженном давлении. Остаток нейтрализовали, используя DIPEA. Полученную смесь концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (95/5), получая 28 мг (25%) N-[3-[2-(дифторметокси)-5-[4-(диметилкарбамоил)фенокси]фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде беловатого твердого вещества. LC/MS (метод Н, ESI): [М+Н]+=534,2; RT=1,09 мин. 1Н ЯМР (300 МГц, DMSO-d6) δ 13.05 (s, 1Н), 9.73 (s, 1Н), 9.35 (dd, J=7,1; 1,7 Гц, 1Н), 8.70 (dd, J=4,2; 1,5 Гц, 1Н), 8.69 (s, 1Н), 8.22 (s, 1Н), 7.47 (d, J=9,3 Гц, 1Н), 7.39 (d, J=8,4 Гц, 2Н), 7.32-7.25 (m, 3Н), 7.06 (d, J=8,7 Гц, 2Н), 7.18 (t, J=73,8 Гц, 1H), 2.94 (s, 6H).
Промежуточное соединение 7
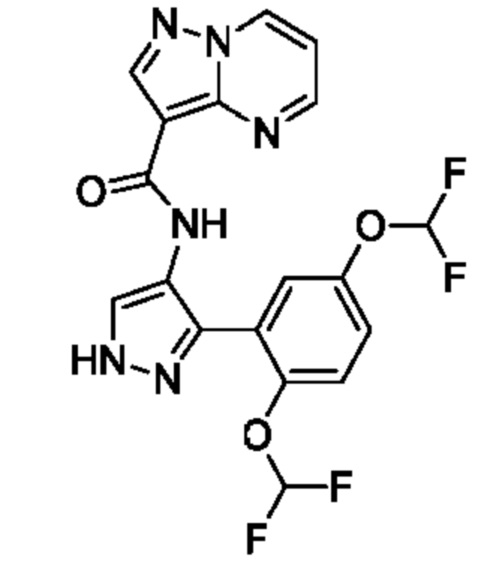
N-[3-[2,5-бис(Дифторметокси)фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид
Стадия 1: синтез 1-(бензилокси)-4-(дифторметокси)бензола
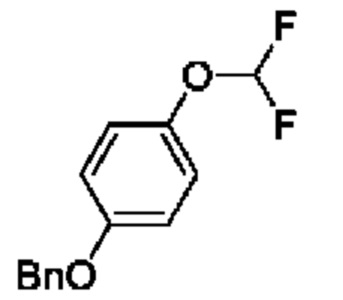
В круглодонную колбу емкостью 3000 мл в условиях продувания азотом и поддержания инертной атмосферы азота помещали N,N-диметилформамид (1500 мл), 4-(бензилокси)фенол (200 г; 999 ммоль) и Cs2CO3 (651 г; 1,99 моль). Реакционный сосуд был оборудован выходным отверстием для высвобождения CO2. После этого добавляли 2-хлор-2,2-дифторацетат натрия (228 г; 1,50 моль; 1,50 экв.) в виде нескольких порций при 120°С. По завершении добавления 2-хлор-2,2-дифторацетата натрия реакционную смесь перемешивали при 120°С в масляной бане до прекращения выделения газа (примерно 1 ч) и затем оставляли охлаждаться до комнатной температуры. Реакционную смесь медленно с перемешиванием добавляли к 3000 мл ледяной воды. Полученную смесь экстрагировали этилацетатом (3×4000 мл). Органические слои объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с элюированием смесью этилацетат/петролейный эфир (1/19). Соответствующие фракции объединяли и концентрировали при пониженном давлении. Эту реакцию повторяли четыре раза. В результате этого получали в общей сложности 450 г (36%) 1-(бензилокси)-4-(дифторметокси)бензола в виде белого твердого вещества.
Стадия 2: синтез 4-(дифторметокси)фенола
В круглодонную колбу емкостью 3000 мл помещали метанол (1500 мл), 1-(бензилокси)-4-(дифторметокси)бензол (140 г; 559 ммоль) и 10%-ный палладированный уголь (15 г; 141 ммоль). Полученную смесь перемешивали в атмосфере водорода (примерно 45 ф/кв. дюйм (310 кПа)) в течение ночи при комнатной температуре. Катализаторы отфильтровывали. Фильтрат концентрировали при пониженном давлении. Эту реакцию повторяли три раза. В результате этого получали 300 г (78%) 4-(дифторметокси)фенола в виде желтого масла.
Стадия 3: синтез 2-бром-4-(дифторметокси)фенола
В круглодонную колбу емкостью 1000 мл помещали уксусную кислоту (500 мл), 4-(дифторметокси)фенол (50 г; 312 ммоль) и NBS (55,6 г; 312 ммоль). Реакционную смесь перемешивали в течение 1 ч при 15°С. Затем полученную смесь медленно с перемешиванием добавляли к 1000 мл ледяной воды. Полученный раствор экстрагировали этилацетатом (3×1000 мл). Органические слои объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле, элюируя смесью дихлорметан/петролейный эфир (30/70). Соответствующие фракции собирали и концентрировали при пониженном давлении. В результате этого получали 50 г (67%) 2-бром-4-(дифторметокси)фенола в виде светло-желтого масла.
Стадия 4: синтез 2-бром-1,4-бис(дифторметокси)бензола
В круглодонную колбу емкостью 2000 мл помещали CH3CN (500 мл), воду (500 мл), 2-бром-4-(дифторметокси)фенол (54 г; 226 ммоль) и гидроксид калия (94 г; 1,68 моль). Эту колбу помещали в ледяную баню и реакционную смесь перемешивали в течение 30 мин в ледяной бане. Затем к реакционной смеси по каплям добавляли диэтил(бромдифторметил)фосфонат (120 г; 449 ммоль) при 0°С. По завершении добавления диэтил(бромдифторметил)фосфоната реакционную смесь перемешивали в течение 1 ч в бане с ледяной водой. Полученный раствор экстрагировали этилацетатом (3×300 мл). Органические слои объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле, элюируя смесью этилацетат/петролейный эфир (1/19), соответствующие фракции собирали и концентрировали при пониженном давлении. В результате этого получали 54 г (83%) 2-бром-1,4-бис(дифторметокси)бензола в виде светло-желтого масла.
Стадия 5: синтез 5-[2,5-бис(дифторметокси)фенил]-4-нитро-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразола

В круглодонную колбу емкостью 1000 мл в условиях продувания азотом и поддержания инертной атмосферы азота помещали DMA (500 мл), карбонат калия (112 г; 810 ммоль), 4-нитро-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол (66 г; 271 ммоль), 2-бром-1,4-бис(дифторметокси)бензол (79 г; 273 ммоль), 2,2-диметилпропановую кислоту (8,3 г; 81,3 ммоль), Pd(OAc)2 (6,0 г; 26,7 ммоль) и бис(адамантан-1-ил)(бутил)фосфин (19 г; 52,9 ммоль, 0,195 экв.). Реакционную смесь перемешивали при 120°С в течение ночи в масляной бане и оставляли охлаждаться до комнатной температуры. Затем реакционную смесь добавляли с перемешиванием к 1000 мл ледяной воды. Полученный раствор экстрагировали этилацетатом (3×1000 мл). Органические слои объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле, элюируя смесью этилацетат/петролейный эфир (1/1). Соответствующие фракции собирали и концентрировали при пониженном давлении. В результате этого получали 100 г (82%) 5-[2,5-бис(дифторметокси)фенил]-4-нитро-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразола в виде твердого вещества.
Стадия 6: синтез 5-[2,5-бис(дифторметокси)фенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-амина

В круглодонную колбу емкостью 3000 мл помещали этанол (1500 мл), воду (150 мл), 5-[2,5-бис(дифторметокси)фенил]-4-нитро-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол (100,00 г; 221 ммоль), порошок железа (124 г; 2,22 моль) и NH4Cl (59,2 г; 1,11 моль). Полученную смесь перемешивали при температуре дефлегмации в масляной бане в течение 2 ч, после чего охлаждали до комнатной температуры. Твердые вещества отфильтровывали и промывали этанолом. Фильтрат концентрировали при пониженном давлении. Остаток растворяли в 3000 мл этилацетата. Этилацетатный раствор промывали 1×1000 мл рассола, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. В результате этого получали 100 г неочищенного 5-[2,5-бис(дифторметокси)фенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-амина в виде светло-желтого масла, который использовали непосредственно без очистки.
Стадия 7: синтез N-[5-[2,5-бис(дифторметокси)фенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида
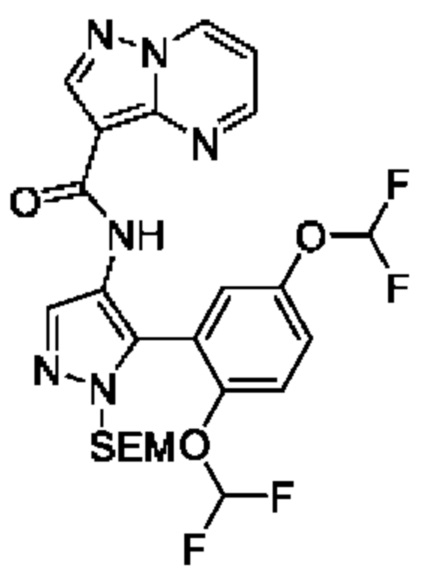
В круглодонную колбу емкостью 2000 мл помещали DMA (1000 мл), 5-[2,5-бис(дифторметокси)фенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-амин с предыдущей стадии, пиразоло[1,5-а]пиримидин-3-карбоновую кислоту (58,06 г; 355,9 ммоль), 7-азабензотриазол-1-илокси)трипирролидинофосфония гексафторфосфат (РуАОР) (185,56 г; 355,9 ммоль), 4-диметиламинопиридин (2,90 г; 23,7 ммоль) и DIPEA (92,0 г; 712 ммоль). Полученный раствор перемешивали в течение ночи при 65°С в масляной бане. Затем реакционную смесь медленно с перемешиванием добавляли к 2000 мл воды. Полученный раствор экстрагировали этилацетатом (3×2000 мл). Объединенные органические фазы промывали 1000 мл рассола, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле, элюируя смесью этилацетат/петролейный эфир (40/60). Соответствующие фракции объединяли и концентрировали при пониженном давлении, получая 120 г N-[5-[2,5-бис(дифторметокси)фенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде белого твердого вещества.
Стадия 8: синтез N-[3-[2,5-бис(дифторметокси)фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида

В круглодонную колбу емкостью 2000 мл помещали метанол (800 мл), концентрированную соляную кислоту (400 мл; 12 н.) и N-[5-[2,5-бис(дифторметокси)фенил]-1-[[2-(триметилсилил)этокси]метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид (80 г; 141 ммоль). Полученный раствор перемешивали в течение 4 ч при 25°С. Твердые вещества собирали фильтрованием. Твердые вещества помещали в колбу емкостью 1 л и добавляли H2O (200 мл). По каплям с перемешиванием добавляли насыщенный водный раствор NaHCO3 до тех пор, пока значение рН раствора не достигало примерно 8. Твердые вещества собирали фильтрованием, промывали водой и сушили, получая 55 г (89%) N-[3-[2,5-бис(дифторметокси)фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде светло-желтого твердого вещества. 1Н ЯМР (300 МГц, CD3OD) δ 9.08 (dd, J=7,2; 1,5 Гц, 1Н), 8.65-8.61 (m, 2Н), 8.28 (s, 1Н), 7.46 (d, J=9,0 Гц, 1Н), 7.40 (d, J=3,0 Гц, 1Н), 7.34 (dd, J=8,9; 2,9 Гц, 1Н), 7.19 (dd, J=6,7; 4,4 Гц, 1Н), 6.87 (t, J=73,7 Гц, 1Н), 6.73 (t, J=73,7 Гц, 1Н).
Пример 1
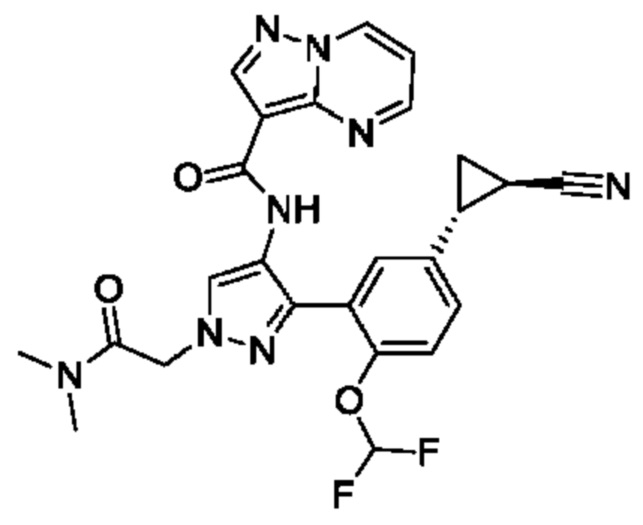
N-(3-(5-((1R,2R)-2-Цианоциклопропил)-2-(дифторметокси)фенил)-1-(2-(диметиламино)-2-оксоэтил)-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид
К раствору N-(3-(5-((1R,2R)-2-цианоциклопропил)-2-(дифторметокси)фенил)-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (промежуточного соединения 4; 100 мг; 0,230 ммоль) в DMF (10 мл) добавляли Cs2CO3 (160 мг; 0,491 ммоль). После этого добавляли 2-бром-N,N-диметилацетамид (80 мг; 0,482 ммоль). Полученную смесь перемешивали в течение 30 мин при 60°С в масляной бане. Полученную смесь концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (8% МеОН), получая 20,9 мг (17%) N-(3-(5-((1R,2R)-2-цианоциклопропил)-2-(дифторметокси)фенил)-1-(2-(диметиламино)-2-оксоэтил)-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида в виде белого твердого вещества. LC/MS (метод A, ESI): [М+Н]+=521,3; RT=1,50 мин; 1Н ЯМР (400 МГц, DMSO-d6) δ 9.76 (s, 1Н), 9.35 (dd, J=6,8; 1,6 Гц, 1H), 8.68 (s, 1Н), 8.65 (dd, J=4,0; 1,6 Гц, 1H), 8.28 (s, 1Н), 7.42-7.38 (m, 3Н), 7.29 (dd, J=7,0; 4,2 Гц, 1H), 7.23 (t, J=73,2 Гц, 1H), 5.20 (s, 2Н), 3.06 (s, 3Н), 2.89 (s, 3Н), 2.84-2.80 (m, 1Н), 2.12-2.09 (m, 1Н), 1.66-1.61 (m, 1Н), 1.54-1.50 (m, 1Н).
Пример 2

N-[3-[2-(Дифторметокси)-5-иодфенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид
К раствору N-[3-[2-(дифторметокси)-5-иодфенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (промежуточного соединения 3; 160 мг; 0,322 ммоль) в DMF (4,0 мл) добавляли Cs2CO3 (210 мг; 0,645 ммоль). К этой смеси добавляли 2-бром-N,N-диметилацетамид (107 мг; 0,645 ммоль). Полученный раствор перемешивали в течение 4 ч при 60°С в масляной бане. Реакционную смесь распределяли между водой и этилацетатом. Водную фазу экстрагировали этилацетатом (2×). Органические слои объединяли, последовательно промывали водой и рассолом, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток пропускали через короткую набивку силикагеля, элюируя смесью дихлорметан/метанол (90/10). Соответствующие фракции объединяли и концентрировали при пониженном давлении. Неочищенный продукт далее очищали препаративной HPLC в следующих условиях: препаративная колонка: SunFire С18 OBD, 19×150 мм, 5 мкм, 10 нм; подвижная фаза: вода (с 0,1% FA) и ACN (от 25,0% до 44,0% ACN за 12 мин); детектор: УФ 254/220 нм, получая 24,1 мг (13%) N-[3-[2-(дифторметокси)-5-иодфенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде белого твердого вещества. LC/MS (метод В, ESI): [М+Н]+=582,2, RT=2,92; 1Н ЯМР (400 МГц, CD3OD) δ 9.11 (dd, J=6,8; 1,6 Гц, 1Н), 8.67 (dd, J=4,4; 1,6 Гц, 1Н), 8.65 (s, 1Н), 8.36 (s, 1Н), 8.02 (d, J=2,4 Гц, 1Н), 7.90 (dd, J=8,8; 2,4 Гц, 1Н), 7.24-7.21 (m, 2Н), 6.82 (t, J=73,6 Гц, 1Н), 5.24 (s, 2Н), 3.18 (s, 3Н), 3.03 (s, 3Н).
Пример 3
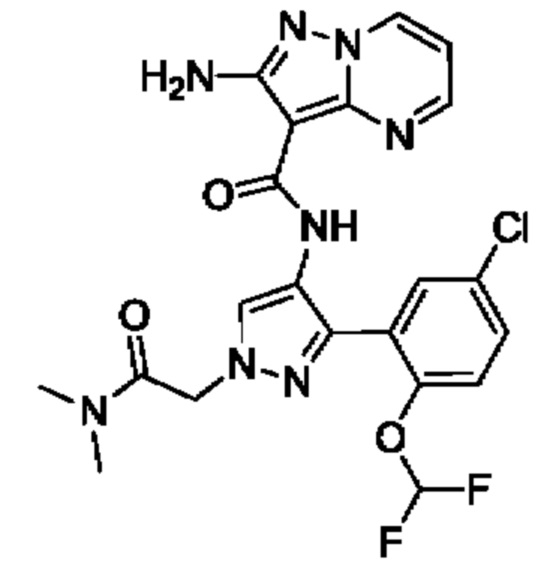
2-Амино-N-(3-(5-хлор-2-(дифторметокси)фенил)-1-(2-(диметиламино)-2-оксоэтил)-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид
Стадия 1: синтез трет-бутил-N-[3-([3-[5-хлор-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]карбамоил)пиразоло[1,5-а]пиримидин-2-ил]карбамата

К раствору трет-бутил-N-[3-([3-[5-хлор-2-(дифторметокси)фенил]-1Н-пиразол-4-ил]карбамоил)пиразоло[1,5-а]пиримидин-2-ил]карбамата (200 мг; 0,385 ммоль) в DMF (5,0 мл) добавляли Cs2CO3 (251 мг; 0,770 ммоль) и 2-бром-N,N-диметилацетамид (63,0 мг; 0,379 ммоль). Реакционную смесь перемешивали в течение 16 ч при 60°С в масляной бане. Полученную смесь концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (95/5). Соответствующие фракции объединяли и концентрировали при пониженном давлении, получая 200 мг (86%) трет-бутил-N-[3-([3-[5-хлор-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]карбамоил)пиразоло[1,5-а]пиримидин-2-ил]карбамата в виде желтого твердого вещества. LC/MS (метод Н, ESI): [М+Н]+=605,3; RT=1,32 мин.
Стадия 2: синтез 2-амино-N-[3-[5-хлор-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида
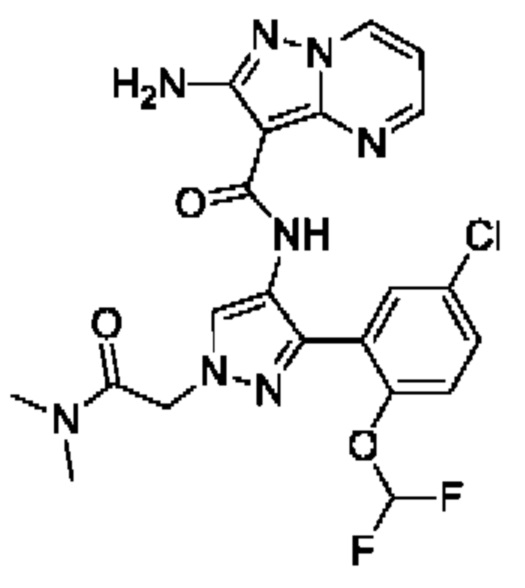
трет-Бутил-N-[3-([3-[5-хлор-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]карбамоил)пиразоло[1,5-а]пиримидин-2-ил]карбамат (100 мг; 0,165 ммоль) обрабатывали раствором HCl в диоксане (2,0 мл; 4 М) в течение 2 ч при комнатной температуре. Полученную смесь концентрировали при пониженном давлении. Неочищенный продукт очищали препаративной HPLC в следующих условиях: колонка: XBridge Shield RP18 OBD, 5 мкм, 19×150 мм; подвижная фаза: вода (с 0,05% NH3H2O) и ACN (от 10% до 45% ACN за 12 мин); детектор: УФ 220 нм, получая 27,4 мг (33%) 2-амино-N-[3-[5-хлор-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде белого твердого вещества. LC/MS (метод A, ESI): [М+Н]+=505,2; RT=1,58 мин; 1Н ЯМР (300 МГц, DMSO-d6) δ 9.55 (s, 1Н), 8.93 (dd, J=6,8; 1,7 Гц, 1Н), 8.36 (dd, J=4,5; 1,5 Гц, 1Н), 8.26 (s, 1Н), 7.62 (dd, J=8,7; 2,7 Гц, 1Н), 7.54 (d, J=2,7 Гц, 1Н), 7.45 (J=8,7 Гц, 1Н), 7.26 (t, J=73,2 Гц, 1Н), 7.00 (dd, J=6,6; 4,5 Гц, 1Н), 6.57 (s, 2Н), 5.18 (s, 2Н), 3.04 (s, 3Н), 2.87 (s, 3Н).
Примеры 4 и 5

(R)-N-(3-(5-(1,2-Дифторэтил)-2-(дифторметокси)фенил)-1-(2-(диметиламино)-2-оксоэтил)-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид и (S)-N-(3-(5-(1,2-дифторэтил)-2-(дифторметокси)фенил)-1-(2-(диметиламино)-2-оксоэтил)-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид
Стадия 1: синтез N-[3-[2-(дифторметокси)-5-(1,2-дигидроксиэтил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида
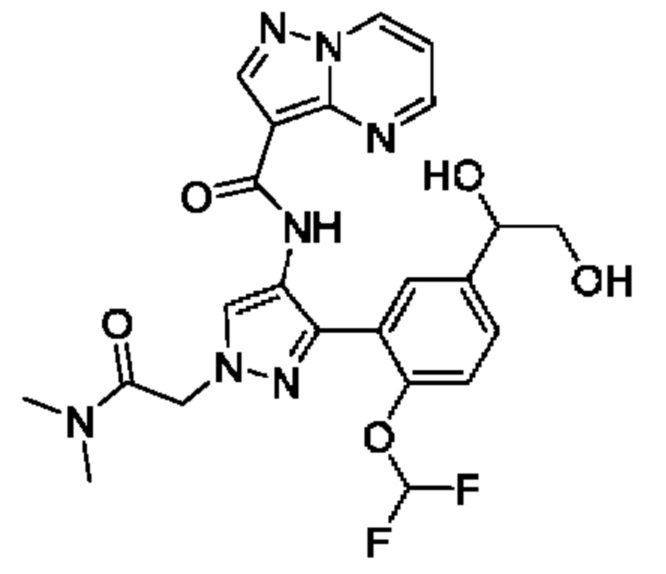
К раствору N-[3-[2-(дифторметокси)-5-этенилфенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (1,70 г; 3,53 ммоль) в тетрагидрофуране (20 мл) и воде (10 мл) добавляли OsO4 (1,82 г; 7,16 ммоль) и NMO (831 мг; 7,09 ммоль). Полученный раствор перемешивали в течение 2 ч при комнатной температуре. Полученный раствор разбавляли, используя 30 мл H2O. Полученный раствор экстрагировали, используя 3×50 мл дихлорметана. Органические слои объединяли, промывали рассолом, сушили и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле, элюируя смесью дихлорметан/метанол (9/1). Соответствующие фракции объединяли и концентрировали при пониженном давлении, получая 700 мг (38%) N-[3-[2-(дифторметокси)-5-(1,2-дигидроксиэтил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде коричневого твердого вещества.
Стадия 2: синтез N-[3-[5-(1,2-дифторэтил)-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида
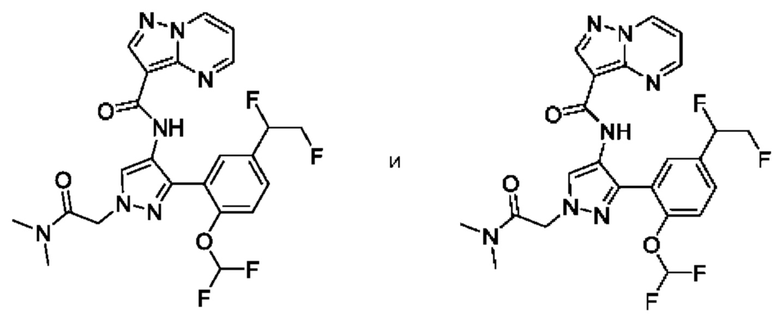
К раствору N-[3-[2-(дифторметокси)-5-(1,2-дигидроксиэтил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (300 мг; 0,582 ммоль) в дихлорметане добавляли трифторид (диэтиламино)серы (DAST; 374 мг; 2,32 ммоль). Реакционную смесь перемешивали в течение 2 ч при комнатной температуре в атмосфере азота. Затем реакцию гасили, добавляя 10 мл воды. Значение рН раствора подводили до 7, используя водный раствор (10%-ный) бикарбоната натрия. Полученную смесь экстрагировали, используя 3×10 мл дихлорметана, органические слои объединяли и концентрировали при пониженном давлении. Неочищенный продукт очищали препаративной HPLC в следующих условиях: препаративная колонка: XBridge ВЕН130 C18 OBD, 19×150 мм; 5 мкм; подвижная фаза: вода (с 0,05% NH3H2O) и ACN (от 30% до 34% ACN за 7 мин); детектор: УФ 254/220 нм, получая 100 мг рацемической смеси. Рацемическую смесь разделяли хиральной препаративной HPLC в следующих условиях: колонка: CHIRALPAK IA, 2,12×15 см, 5 мкм; подвижная фаза: гексан/DCM=5/1 и этанол (выдерживание при 50,0% этанола в течение 13 мин); скорость потока 16 мл/мин; детектор: УФ 220/254 нм, получая две фракции.
Изомер 1: элюировали на 7,15 мин; 12,4 мг (4%) N-[3-[5-(1,2-дифторэтил)-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде белого твердого вещества. LC/MS (метод A, ESI): [М+Н]+=520,2; RT=1,70 мин; 1Н ЯМР (400 МГц, DMSO-d6) δ 9.72 (s, 1Н), 9.34 (dd, J=7,0; 1,4 Гц, 1Н), 8.68 (s, 1Н), 8.66 (dd, J=4,0; 1,6 Гц, 1Н), 8.29 (s, 1Н), 7.65-7.61 (m, 2H), 7.51 (d, J=8,0 Гц, 1H), 7.32 (t, J=73,2 Гц, 1H), 7.29 (dd, J=7,0; 4,2 Гц, 1H), 6.11-5.84 (m, 1H), 5.22 (s, 2H), 4.88-4.68 (m, 2H), 3.06 (s, 3H), 2.89 (s, 3H).
Изомер 2: элюировали на 9,66 мин; 13,7 мг (5%) N-[3-[5-(1,2-дифторэтил)-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде белого твердого вещества. LC/MS (метод A, ESI): [М+Н]+=520,2; RT=1,70 мин; 1Н ЯМР (400 МГц, DMSO-d6) δ 9.72 (s, 1Н), 9.34 (dd, J=7,0; 1,4 Гц, 1Н), 8.68 (s, 1Н), 8.66 (dd, J=4,0; 1,6 Гц, 1Н), 8.29 (s, 1Н), 7.65-7.61 (m, 2Н), 7.51 (d, J=8,0 Гц, 1Н), 7.32 (t, J=73,2 Гц, 1Н), 7.29 (dd, J=7,0; 4,2 Гц, 1Н), 6.11-5.84 (m, 1Н), 5.22 (s, 2Н), 4.88-4.68 (m, 2Н), 3.06 (s, 3Н), 2.89 (s, 3Н).
Пример 6

Стадия 1: синтез N-[3-[5-(1,1-дифтор-2-оксо-2-фенилэтил)-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида

К раствору N-[3-[5-бром-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (50,0 мг; 0,0936 ммоль) в толуоле (5,0 мл) добавляли 2,2-дифтор-1-фенилэтан-1-он (29,0 мг; 0,186 ммоль), [2-(2-аминофенил)фенил](хлор)палладия трициклогексилфосфин (12,0 мг; 0,0203 ммоль) и K3РO4 (80,0 мг; 0,377 ммоль) в атмосфере азота. Полученный раствор перемешивали в течение 16 ч при 100°С в масляной бане. Реакционную смесь охлаждали до комнатной температуры. Полученную смесь концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (92/8), получая 23 мг (40%) N-[3-[5-(1,1-дифтор-2-оксо-2-фенилэтил)-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде светло-желтого твердого вещества. LC/MS (метод I, ESI): [М+Н]+=610,3; RT=1,11 мин.
Стадия 2: синтез N-[3-[2-(дифторметокси)-5-(дифторметил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида
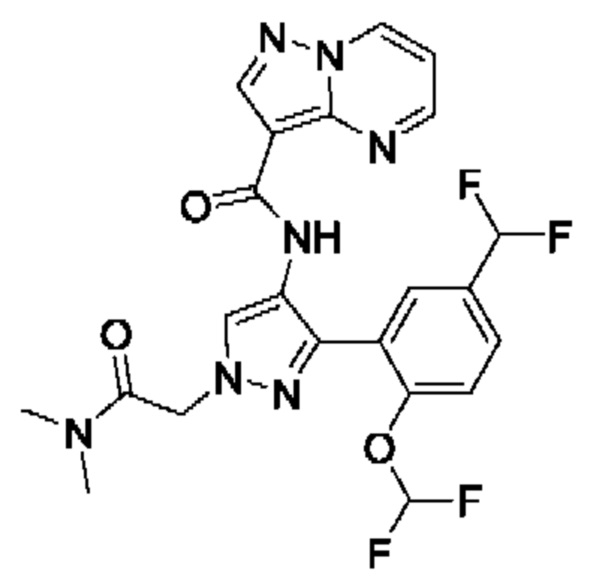
К раствору N-[3-[5-(1,1-дифтор-2-оксо-2-фенилэтил)-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (230 мг; 0,377 ммоль) в толуоле (30 мл) и воде (5,0 мл) добавляли гидроксид калия (43,0 мг; 0,766 ммоль). Полученный раствор перемешивали в течение 16 ч при 100°С в масляной бане. Реакционную смесь охлаждали до комнатной температуры. Полученный раствор разбавляли, используя 50 мл Н2O. Полученный раствор экстрагировали, используя 2×30 мл дихлорметана, органические слои объединяли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток пропускали через короткую набивку силикагеля, элюируя смесью дихлорметан/метанол (95/5). Соответствующие фракции объединяли и концентрировали при пониженном давлении. Остаток далее очищали препаративной HPLC в следующих условиях: препаративная колонка: XBridge ВЕН130 С18 OBD, 19×150 мм, 5 мкм; подвижная фаза: вода (с 0,05% NH4OH) и ACN (от 29,0% до 42,0% ACN за 7 мин); детектор: УФ 254/220 нм, получая 23,7 мг (12%) N-[3-[2-(дифторметокси)-5-(дифторметил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде белого твердого вещества. LC/MS (метод A, ESI): [М+Н]+=506,3; RT=1,52 мин. 1H ЯМР (400 МГц, DMSO-d6) δ 9.74 (s, 1Н), 9.35 (dd, J=6,8; 1,6 Гц, 1Н), 8.68 (s, 1Н), 8.66 (dd, J=4,4; 1,6 Гц, 1Н), 8.32 (s, 1Н), 7.79 (d, J=8,8 Гц, 1Н), 7.74 (s, 1Н), 7.59 (d, J=8,8 Гц, 1Н), 7.30 (dd, J=7,2; 4,4 Гц, 1Н), 7.39 (t, J=73,0 Гц, 1Н), 7.14 (t, J=55,8 Гц, 1Н), 5.24 (s, 2Н), 3.06 (s, 3Н), 2.89 (s, 3Н).
Пример 7

К раствору N-[3-[2-(дифторметокси)-5-(2-оксоэтил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (90,0 мг; 0,181 ммоль) в дихлорметане (2,0 мл) при 0°С добавляли DAST (84 мг; 0,521 ммоль) в атмосфере азота. Полученный раствор перемешивали в течение 2 ч при комнатной температуре. Полученную смесь концентрировали при пониженном давлении. Неочищенный продукт очищали препаративной HPLC в следующих условиях: колонка: XBridge Shield RP18 OBD, 5 мкм, 19×150 мм; подвижная фаза: вода (с 0,05% NH3H2O) и ACN (от 10% до 45% ACN за 12 мин); детектор: УФ 220 нм. Получали 27,4 мг продукта и концентрировали при пониженном давлении, получая 36,2 мг (39%) N-[3-[5-(2,2-дифторэтил)-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде белого твердого вещества. LC/MS (метод С, ESI): [М+Н]+=520,2; RT=1,89 мин; 1Н ЯМР (400 МГц, DMSO-d6) δ 9.72 (s, 1Н), 9.34 (dd, J=6,8; 1,6 Гц, 1Н), 8.67 (s, 1Н), 8.64 (dd, J=4,0; 1,6 Гц, 1Н), 8.28 (s, 1Н), 7.49-7.47 (m, 2Н), 7.41 (d, J=8,8 Гц, 1Н), 7.28 (dd, J=7,0; 4,2 Гц, 1Н), 7.25 (t, J=73,6 Гц, 1Н), 6.46-6.09 (m, 1Н), 5.20 (s, 2Н), 3.26-3.19 (m, 2Н), 3.06 (s, 3Н), 2.88 (s, 3Н).
Пример 8
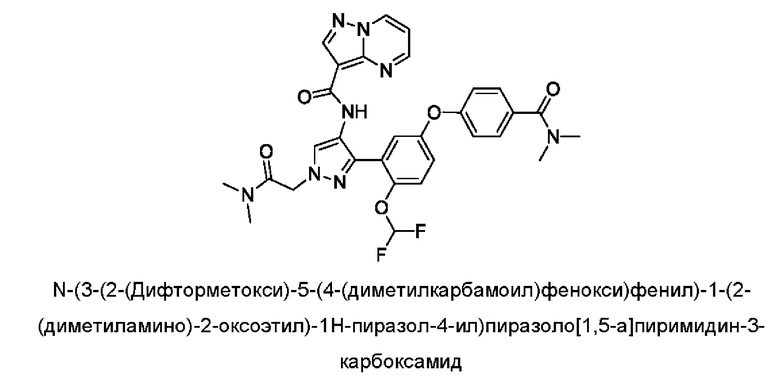
К раствору N-[3-[2-(дифторметокси)-5-[4-(диметилкарбамоил)фенокси]фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (промежуточного соединения 6; 150 мг; 0,281 ммоль) в DMF (4,0 мл) добавляли Cs2CO3 (183 мг; 0,562 ммоль) и 2-бром-N,N-диметилацетамид (69,0 мг; 0,416 ммоль). Реакционную смесь перемешивали в течение 2 ч при 25°С. Полученную смесь концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (93/7), получая 22,0 мг (13%) N-[3-[2-(дифторметокси)-5-[4-(диметилкарбамоил)фенокси]фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде белого твердого вещества. LC/MS (метод A, ESI): [М+Н]+=619,3; RT=1,50 мин; 1Н ЯМР (300 МГц, DMSO-d6) δ 9.78 (s, 1Н), 9.35 (dd, J=7,1; 1,7 Гц, 1Н), 8.70 (dd, J=4,4; 1,7 Гц, 1Н), 8.67 (s, 1Н), 8.26 (s, 1Н), 7.49 (d, J=9,0 Гц, 1Н), 7.42 (d, J=8,7 Гц, 2Н), 7.32-7.27 (m, 2Н), 7.19 (t, J=73,8 Гц, 1Н), 7.18 (d, J=3,0 Гц, 1Н), 7.07 (d, J=8,7 Гц, 2Н), 5.18 (s, 2Н), 3.03 (s, 3Н), 2.94 (s, 6Н), 2.86 (s, 3Н).
Пример 9
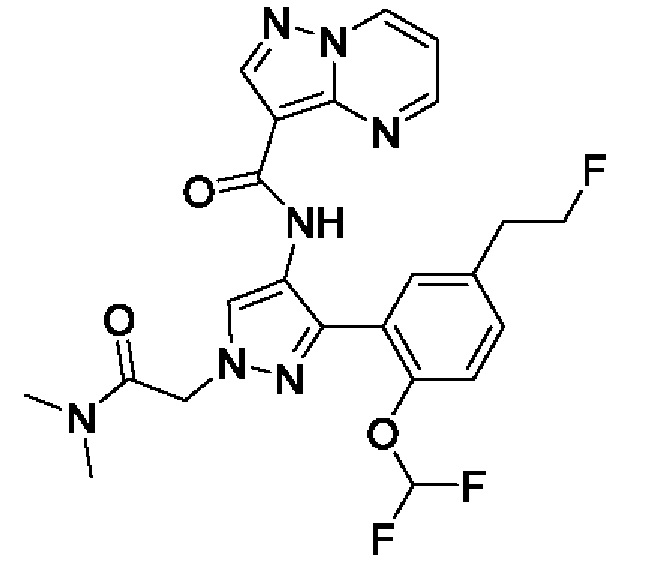

Стадия 1: синтез N-[3-[2-(дифторметокси)-5-(проп-2-ен-1-ил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида

К раствору N-[3-[5-бром-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (6,00 г; 11,23 ммоль) в 1,4-диоксане (100 мл) добавляли 4,4,5,5-тетраметил-2-(проп-2-ен-1-ил)-1,3,2-диоксаборолан (3,02 г; 18,0 ммоль; 1,600 экв.), комплекс Рd(dppf)Сl2-дихлорметан (459 мг; 0,562 ммоль) и раствор Cs2CO3 (6,60 г; 20,3 ммоль) в воде (20 мл) в атмосфере азота. Полученный раствор перемешивали в течение 3 ч при 85°С. Полученную смесь концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (32/1), получая 4,10 г (74%) N-[3-[2-(дифторметокси)-5-(проп-2-ен-1-ил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде коричневого твердого вещества. LC/MS (метод L, ESI): [М+Н]+=496,1; RT=0,83 мин.
Стадия 2: синтез N-[3-[2-(дифторметокси)-5-(2,3-дигидроксипропил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида

К раствору N-[3-[2-(дифторметокси)-5-(проп-2-ен-1-ил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (7,06 г; 14,2 ммоль) в тетрагидрофуране (140 мл) добавляли NMO (3,33 г; 28,4 ммоль) и воду (70 мл). После этого добавляли OsO4 (7,23 г; 28,4 ммоль) при комнатной температуре. Полученный раствор перемешивали в течение 2 ч при комнатной температуре. Затем реакцию гасили, добавляя 50 мл раствора Na2S2O3 (насыщ.). Полученную смесь экстрагировали этилацетатом (3×). Объединенные органические фазы промывали последовательно водой и рассолом, сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (10/1), получая 4,02 г (53%) N-[3-[2-(дифторметокси)-5-(2,3-дигидроксипропил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде светло-желтого твердого вещества. LC/MS (метод Н, ESI): [М+Н]+=530,2; RT=0,95 мин.
Стадия 3: синтез N-[3-[2-(дифторметокси)-5-(2-оксоэтил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида

К раствору N-[3-[2-(дифторметокси)-5-(2,3-дигидроксипропил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (670 мг; 1,27 ммоль) в CH3CN (15 мл) и воде (3,0 мл) при 0°С добавляли NaIO4 (325 мг; 1,52 ммоль). Полученный раствор перемешивали в течение ночи при комнатной температуре. Полученную смесь концентрировали при пониженном давлении. Остаток распределяли между водой и этилацетатом. Водную фазу экстрагировали этилацетатом (2×). Объединенные органические фазы промывали рассолом, сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле, элюируя смесью дихлорметан/метанол (92/8). Соответствующие фракции объединяли и концентрировали при пониженном давлении, получая 482 мг (77%) N-[3-[2-(дифторметокси)-5-(2-оксоэтил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде желтого твердого вещества. LC/MS (метод Н, ESI): [М+Н]+=498,2; RT=1,03 мин.
Стадия 4: синтез N-[3-[2-(дифторметокси)-5-(2-гидроксиэтил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида
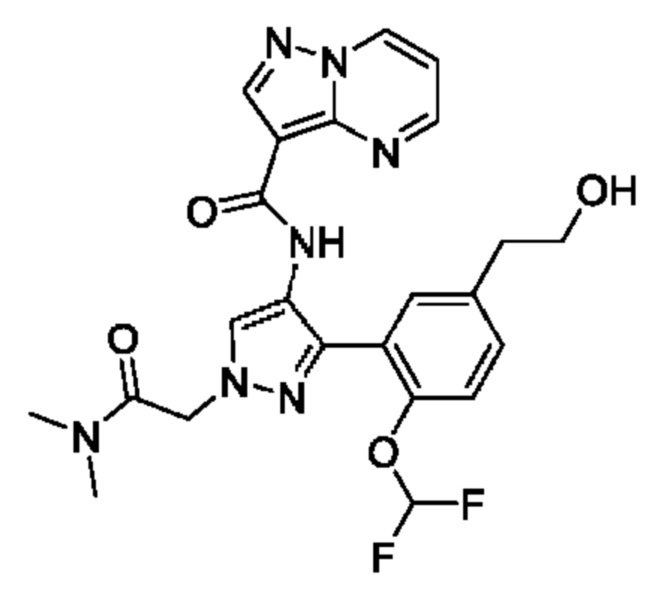
К раствору N-[3-[2-(дифторметокси)-5-(2-оксоэтил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (487 мг; 0,979 ммоль) в дихлорметане (20 мл) при 0°С добавляли NaBH(OAc)3 (300 мг; 1,42 ммоль). Полученный раствор перемешивали в течение 3 ч при комнатной температуре. Полученную смесь концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле, элюируя смесью дихлорметан/метанол (93/7). Собранные фракции объединяли и концентрировали при пониженном давлении, получая 430 мг (88%) N-[3-[2-(дифторметокси)-5-(2-гидроксиэтил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде светло-желтого твердого вещества. LC/MS (метод Н, ESI): [М+Н]+=500,3; RT=1,00 мин.
Стадия 5: синтез N-[3-[2-(дифторметокси)-5-(2-фторэтил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида
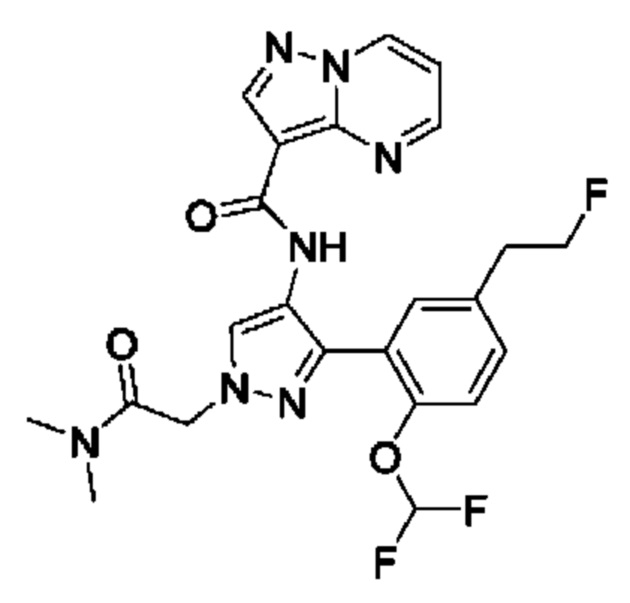
К раствору N-[3-[2-(дифторметокси)-5-(2-гидроксиэтил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (200 мг; 0,400 ммоль) в дихлорметане (10 мл) добавляли диэтил(трифтор-4-сульфанил)амин (96,6 мг; 0,599 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Полученную смесь концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (95/5). Соответствующие фракции объединяли и концентрировали при пониженном давлении. Неочищенный продукт далее очищали препаративной HPLC в следующих условиях: колонка: силикагель; подвижная фаза: [Н2O (с NH4HCO3)]/CH3CN=90/10 с изменением до [Н2O (с NH4HCO3)]/CH3CN=50/50 за 10 мин; детектор: УФ 254 нм, получая 9,3 мг (5%) N-[3-[2-(дифторметокси)-5-(2-фторэтил)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде беловатого твердого вещества. LC/MS (метод A, ESI): [М+Н]+=502,3; RT=1,48 мин; 1Н ЯМР (400 МГц, DMSO-d6) δ 9.74 (s, 1Н), 9.34 (dd, J=7,2; 1,6 Гц, 1Н), 8.68 (s, 1Н), 8.65 (dd, J=4,2; 1,4 Гц, 1Н), 8.28 (s, 1Н), 7.45-7.40 (m, 2Н), 7.39 (d, J=8,8 Гц, 1Н), 7.28 (dd, J=7,0; 4,2 Гц, 1Н), 7.23 (t, J=73,6 Гц, 1Н), 5.21 (s, 2Н), 4.74-4.72 (m, 1Н), 4.61-4.59 (m, 1Н), 3.08-3.07 (m, 1Н), 3.06 (s, 3Н), 3.02-3.00 (m, 1Н), 2.89 (s, 3Н).
Примеры 10 и 11

К раствору N-[3-[5-хлор-2-(дифторметокси)фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (200 мг; 0,494 ммоль) в N,N-диметилформамиде (5,0 мл) добавляли 2-бром-N,N-диметилпропанамид (133 мг; 0,739 ммоль) и Cs2CO3 (323 мг; 0,991 ммоль). Полученный раствор перемешивали в течение 16 ч при 60°С в масляной бане. Полученную смесь концентрировали при пониженном давлении. Остаток распределяли между этилацетатом и водой. Водную фазу экстрагировали этилацетатом (2×). Объединенные органические фазы промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали препаративной HPLC в следующих условиях: колонка: XBridge Shield RP18 OBD, 5 мкм, 19×150 мм; подвижная фаза: вода (с NH4HCO3, 10 ммоль/л) и ACN (от 25,0% до 65,0% ACN за 7 мин); детектор: УФ 220 нм, получая 120 мг рацемической смеси. Рацемическую смесь разделяли хиральной препаративной HPLC в следующих условиях: колонка: CHIRALPAK IF, 2×25 см, 5 мкм; подвижная фаза: гексан и этанол (выдерживание при 50,0% этанола в течение 27 мин); детектор: УФ 220/254 нм, получая две фракции.
Изомер 1 (1-ый пик): элюировали на 18,08 мин; 40,0 мг (16%) N-[3-[5-хлор-2-(дифторметокси)фенил]-1-[1-(диметилкарбамоил)этил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде белого твердого вещества, LC/MS (метод A, ESI): [М+Н]+=504,2; RT=1,65 мин; 1Н ЯМР (400 МГц, DMSO-d6) δ 9.76 (s, 1Н), 9.35 (dd, J=7,2; 1,6 Гц, 1Н), 8.68 (dd, J=4,0; 1,6 Гц, 1Н), 8.67 (s, 1Н), 8.36 (s, 1Н), 7.64 (dd, J=8,8; 2,8 Гц, 1Н), 7.61 (d, J=2,8 Гц, 1Н), 7.46 (d, J=8,8 Гц, 1Н), 7.30 (dd, J=7,0; 4,2 Гц, 1Н), 7.24 (t, J=73,4 Гц, 1Н), 5.65 (q, J=6,8 Гц, 1Н), 3.05 (s, 3Н), 2.88 (s, 3Н), 1.60 (d, J=6,8 Гц, 3Н).
Изомер 2 (2-ой пик): элюировали на 22,84 мин; 38,3 мг (15%) N-[3-[5-хлор-2-(дифторметокси)фенил]-1-[1-(диметилкарбамоил)этил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде белого твердого вещества. LC/MS (метод A, ESI): [М+Н]+=504,2; RT=1,65 мин; 1Н ЯМР (400 МГц, DMSO-d6) δ 9.76 (s, 1Н), 9.35 (dd, J=7,2; 1,6 Гц, 1Н), 8.68 (dd, J=4,0; 1,6 Гц, 1Н), 8.67 (s, 1Н), 8.36 (s, 1Н), 7.64 (dd, J=8,8; 2,8 Гц, 1Н), 7.61 (d, J=2,8 Гц, 1Н), 7.46 (d, J=2,8 Гц, 1Н), 7.30 (dd, J=7,0; 4,2 Гц, 1Н), 7.24 (t, J=73,4 Гц, 1Н), 5.65 (q, J=6,8 Гц, 1Н), 3.05 (s, 3Н), 2.88 (s, 3Н), 1.60 (d, J=6,8 Гц, 3Н).
Пример 12
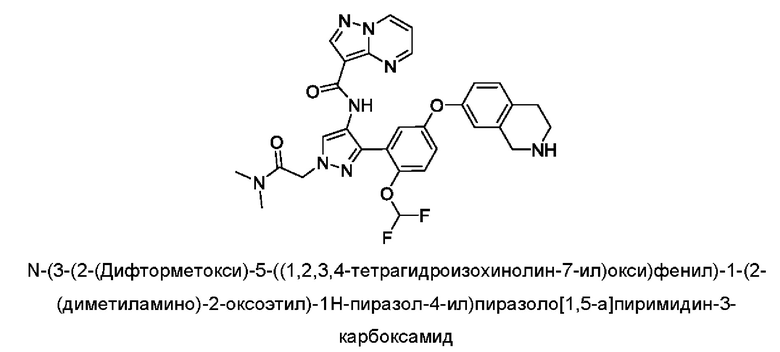
Стадия 1: синтез трет-бутил-7-[4-(дифторметокси)-3-[1-[(диметилкарбамоил)метил]-4-[пиразоло[1,5-а]пиримидин-3-амидо]-1Н-пиразол-3-ил]фенокси]-1,2,3,4-тетрагидроизохинолин-2-карбоксилата
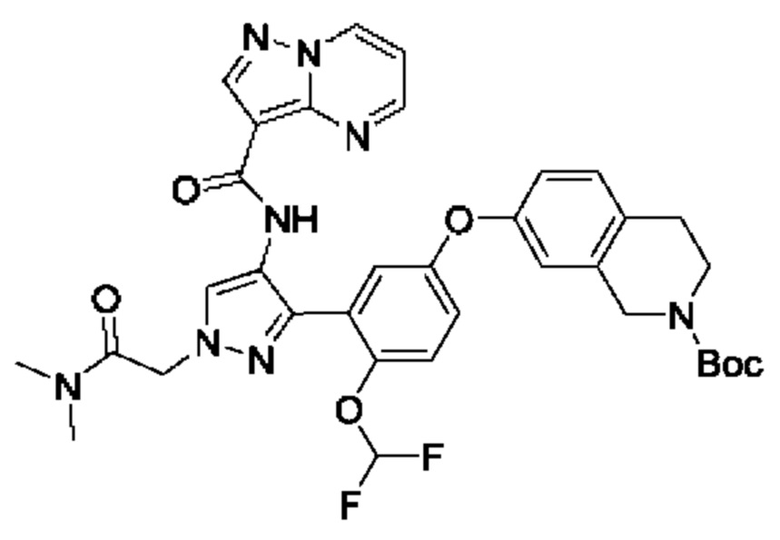
К раствору N-[3-[5-бром-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (157 мг; 0,294 ммоль) в толуоле (10 мл) добавляли Cs2CO3 (115 мг; 0,353 ммоль), f-BuBrettPhos (14,3 мг; 0,0295 ммоль), Рd2(аллил)2Cl2 (5,38 мг; 0,0148 ммоль) и трет-бутил-гидрокси-1,2,3,4-тетрагидроизохинолин-2-карбоксилат (87,5 мг; 0,351 ммоль) в атмосфере азота. Полученный раствор перемешивали в течение ночи при 100°С в масляной бане. Полученную смесь концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (10/1), получая 150 мг (73%) трет-бутил-7-[4-(дифторметокси)-3-[1-[(диметилкарбамоил)метил]-4-[пиразоло[1,5-а]пиримидин-3-амидо]-1Н-пиразол-3-ил]фенокси]-1,2,3,4-тетрагидроизохинолин-2-карбоксилата в виде желтого твердого вещества. TLC (тонкослойная хроматография): Rf (фактор удерживания)=0,4; этилацетат/гексан=4/1.
Стадия 2: синтез N-[3-[2-(дифторметокси)-5-(1,2,3,4-тетрагидроизохинолин-7-илокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида
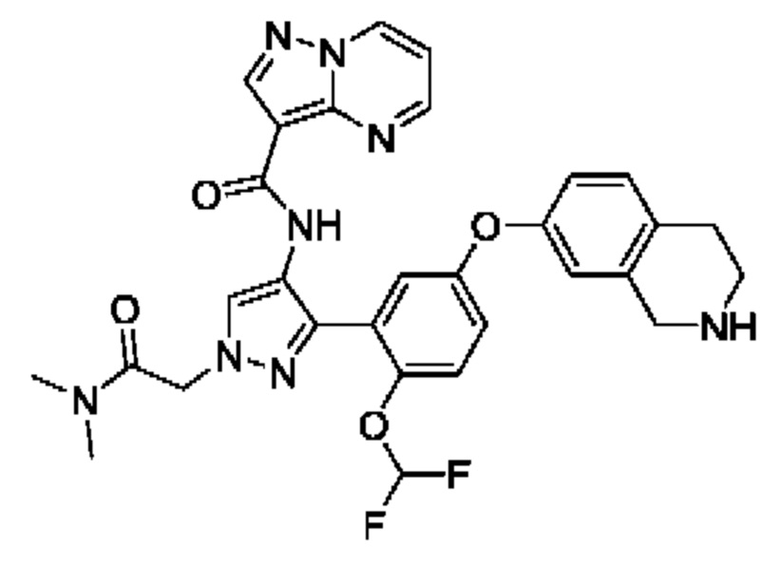
трет-Бутил-7-[4-(дифторметокси)-3-[1-[(диметилкарбамоил)метил]-4-[пиразоло[1,5-а]пиримидин-3-амидо]-1Н-пиразол-3-ил]фенокси]-1,2,3,4-тетрагидроизохинолин-2-карбоксилат (150 мг; 0,213 ммоль) обрабатывали раствором HCl в 1,4-диоксане (5,0 мл; 4 М) в течение 1 ч при комнатной температуре. Полученную смесь концентрировали при пониженном давлении. Неочищенный продукт очищали препаративной HPLC в следующих условиях: колонка: XBridge Shield RP18 OBD, 5 мкм, 19×150 мм; подвижная фаза: вода (с 0,05% NH3H2O) и ACN (от 15,0% до 55,0% ACN за 10 мин); детектор: УФ 254/220 нм, получая 120 мг (93%) N-[3-[2-(дифторметокси)-5-(1,2,3,4-тетрагидроизохинолин-7-илокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде белого твердого вещества. LC/MS (метод Е, ESI): [М+Н]+=603,3; RT=2,27 мин; 1Н ЯМР (300 МГц, DMSO-d6) δ 9.74 (s, 1Н), 9.35 (dd, J=6,9; 1,5 Гц, 1Н),8.68 (dd, J=4,4; 1,7 Гц, 1H), 8.67 (s, 1Н), 8.25 (s, 1Н), 7.43 (d, J=9,0 Гц, 1H), 7.33 (dd, J=7,2; 4,2 Гц, 1H), 7.18 (dd, J=9,0; 3,0 Гц, 1H), 7.13 (t, J=73,8 Гц, 1H), 7.06 (d, J=8,4 Гц, 1H), 7.02 (d, J=3,0 Гц, 1H), 6.82 (dd, J=8,4; 2,4 Гц, 1H), 6.75 (d, J=2,4 Гц, 1H), 5.17 (s, 2Н), 3.77 (s, 2Н), 3.03 (s, 3Н), 2.93 (t, J=5,7 Гц, 2H), 2.86 (s, 3Н), 2.65 (t, J=5,7 Гц, 2H).
Пример 13
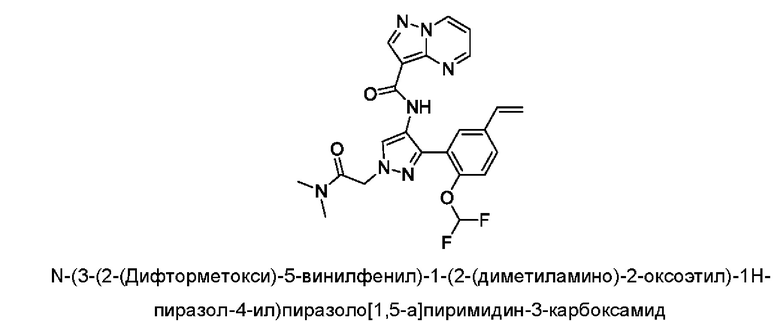
К раствору N-[3-[5-хлор-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (1,30 г; 2,65 ммоль) в диоксане (30 мл) добавляли винилтрифторборат калия (389 мг; 2,90 ммоль), K2СО3 (612 мг; 4,43 ммоль), Pd(dppf)Cl2.CH2Cl2 (197 мг; 0,241 ммоль) и воду (4,0 мл) в атмосфере азота. Полученный раствор перемешивали в течение 4 ч при 80°С в масляной бане. Полученную смесь концентрировали при пониженном давлении. Остаток распределяли между водой и этилацетатом. Водную фазу экстрагировали этилацетатом (2×). Объединенные органические фазы промывали рассолом, сушили и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с элюированием этилацетатом, получая 574 мг (45%) N-[3-[2-(дифторметокси)-5-этенилфенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде белого твердого вещества. LC/MS (метод F, ESI): [М+Н]+=482,3; RT=1,39 мин; 1Н ЯМР (400 МГц, DMSO-d6) δ 9.77 (s, 1Н), 9.35 (dd, J=7,2; 1,6 Гц, 1Н), 8.68 (s, 1Н), 8.62 (dd, J=4,4; 1,6 Гц, 1Н), 8.30 (s, 1 Н), 7.69 (dd, J=8,6; 2,2 Гц, 1Н), 7.62 (d, J=2,0 Гц, 1 Н), 7.41 (d, J=8,4 Гц, 1Н), 7.28 (dd, J=6,7; 4,4 Гц, 1Н), 7.27 (t, J=73,6 Гц, 1Н), 6.82 (dd, J=17,6; 11,2 Гц, 1Н), 5.87 (d, J=17,6 Гц, 1Н), 5.31 (d, J=11,2 Гц, 1Н), 5.22 (s, 2Н), 3.06 (s, 3Н), 2.89 (s, 3Н).
Пример 14

К раствору N-[3-[5-циклопропил-2-(дифторметокси)фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (промежуточного соединения 5; 100 мг; 0,244 ммоль) в DMF (10 мл) добавляли Cs2CO3 (160 мг; 0,491 ммоль). К этой смеси добавляли 2-бром-N,N-диметилацетамид (83 мг; 0,500 ммоль). Полученный раствор перемешивали в течение 30 мин при 60°С в масляной бане. Полученную смесь концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле с элюированием смесью дихлорметан/метанол (8% МеОН), получая 63,8 мг (53%) N-[3-[5-цикпопропил-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде белого твердого вещества. LC/MS (метод A, ESI): [М+Н]+=496,2; RT=1,64 мин; 1Н ЯМР (400 МГц, DMSO-d6) δ 9.74 (s, 1Н), 9.35 (dd, J=7,2; 4,0 Гц, 1Н), 8.68 (s, 1Н), 8.65 (dd, J=4,0; 1,6 Гц, 1Н), 8.27 (s, 1Н), 7.32-7.23 (m, 4Н), 7.16 (t, J=74,0 Гц, 1Н), 5.20 (s, 2Н), 3.06 (s, 3Н), 2.89 (s, 3Н), 2.03-1.97 (m, 1Н), 0.99-0.94 (m, 2Н), 0.73-0.68 (m, 2Н).
Пример 15
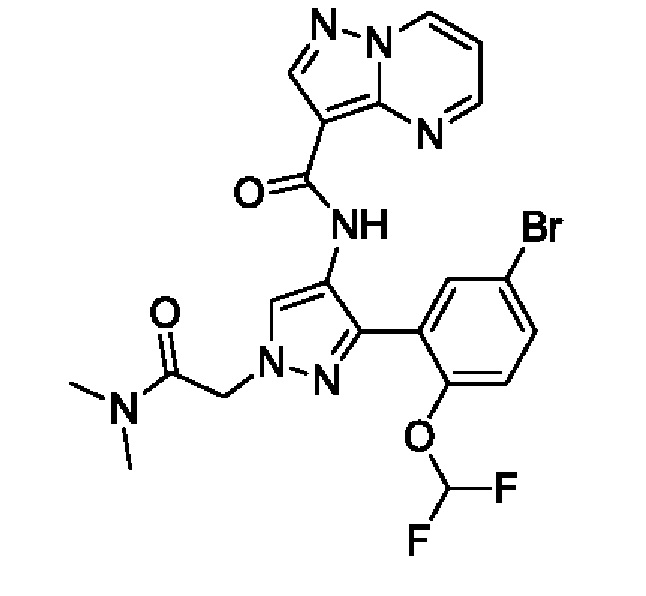

К раствору неочищенного N-[3-[5-бром-2-(дифторметокси)фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (промежуточного соединения 2; 3,80 г; 8,46 ммоль) в N,N-диметилформамиде (150 мл) добавляли Cs2CO3 (8,30 г; 25,5 ммоль) и 2-бром-N,N-диметилацетамид (2,20 г; 13,3 ммоль). Реакционную смесь перемешивали в течение 2 ч при комнатной температуре и выливали при перемешивании в 1,0 л воды. Твердые вещества собирали фильтрованием. Неочищенный продукт очищали однократной перекристаллизацией из этилацетата, получая 3,30 г (72% за две стадии) N-[3-[5-бром-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде желтого твердого вещества. LC/MS (метод D, ESI): [М+Н]+=536,1; RT=2,72 мин. 1Н ЯМР (400 МГц, DMSO-d6): δ (млн-1) 9.75 (s, 1Н), 9.36 (dd, J=7,2; 1,6 Гц, 1Н), 8.71-8.69 (m, 2Н), 8.30 (s, 1Н), 7.76 (dd, J=8,8; 2,4 Гц, 1Н), 7.68 (d, J=2,8 Гц, 1Н), 7.41 (d, J=8,8 Гц, 1Н), 7.31 (dd, J=7,0; 4,2 Гц, 1Н), 7.28 (t, J=73,2 Гц, 1Н), 5.22 (s, 2Н), 3.06 (s, 3Н), 2.89 (s, 3Н).
Пример 16
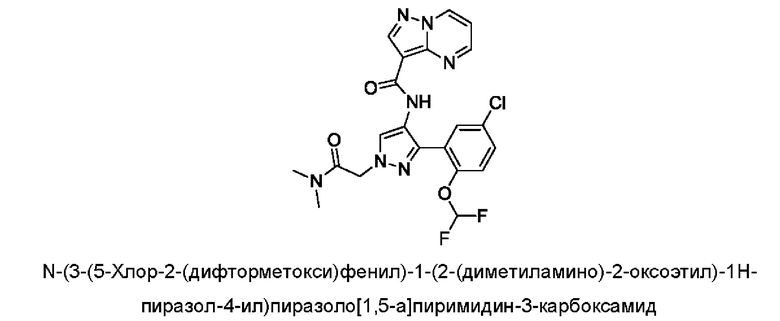
Перемешиваемую смесь Cs2CO3 (31 г; 95 ммоль), N,N-диметилформамида (150 мл) и N-[3-[5-хлор-2-(дифторметокси)фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (25 г; 62 ммоль) охлаждали до 0°С, используя баню с ледяной водой. По каплям добавляли 2-бром-N,N-диметилацетамид (12,5 г; 75,3 ммоль). По окончании добавления 2-бром-N,N-диметилацетамида ледяную баню удаляли, реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 1,5 ч при комнатной температуре. Реакционную смесь постепенно при перемешивании добавляли к 2 л Н2O. Осадок собирали фильтрованием и сушили при пониженном давлении. К неочищенному продукту добавляли МеОН (500 мл) и смесь нагревали до температуры дефлегмации в течение 0,5 ч с перемешиванием, после чего охлаждали до комнатной температуры. Твердые вещества собирали фильтрованием, затем добавляли еще 500 мл МеОН и смесь нагревали до температуры дефлегмации в течение 30 мин, после чего охлаждали до комнатной температуры. Твердые вещества собирали фильтрованием, получая указанное в заголовке соединение с чистотой 99,1% по данным HPLC. Эту же реакцию (с 25 г N-[3-[5-xлop-2-(дифторметокси)фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида) повторяли 4 раза и образцы N-[3-[5-хлор-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида с 4-х процедур (в общей сложности 81 г, все с чистотой более 99% по данным HPLC) объединяли для перевода в кристаллическую форму.
N-[3-[5-Хлор-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамид (81 г; 165 ммоль) с предыдущей стадии помещали в круглодонную колбу емкостью 1 л и добавляли метанол (400 мл). Смесь перемешивали в течение 3 суток при комнатной температуре. Твердые вещества собирали фильтрованием и сушили при пониженном давлении. В результате этого получали 79,46 г (98%) кристаллического соединения N-[3-[5-хлор-2-(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде желтого твердого вещества. LC/MS (метод D, ESI): [М+Н]+=490,2; RT=2,63 мин. 1Н ЯМР (400 МГц, DMSO-d6) δ 9.77 (s, 1Н), 9.36 (dd, J=7,0; 1,4 Гц, 1Н), 8.70-8.69 (m, 2Н), 8.31 (s, 1Н), 7.64 (dd, J=8,8; 2,8 Гц, 1Н), 7.57 (d, J=2,4 Гц, 1Н), 7.47 (d, J=8,8 Гц, 1 Н), 7.30 (dd, J=7,0; 4,2 Гц, 1Н), 7.27 (t, J=73,2 Гц, 1Н), 5.22 (s, 2Н), 3.06 (s, 3Н), 2.89 (s, 3Н).
Пример 17

Перемешиваемую смесь карбоната калия (30 г; 217 ммоль) и N-[3-[2,5-бис(дифторметокси)фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (промежуточного соединения 7; 60 г; 137 ммоль) в N,N-диметилформамиде (400 мл) в круглодонной колбе емкостью 1000 мл охлаждали до 0°С, используя баню с ледяной водой. По каплям добавляли 2-бром-N,N-диметилацетамид (28 г; 169 ммоль; 1,2 экв.), затем реакционную смесь оставляли нагреваться до комнатной температуры и перемешивание продолжали в течение ночи при комнатной температуре. Реакционную смесь постепенно при перемешивании добавляли к 4 л воды. Твердые вещества собирали фильтрованием, затем добавляли к 2 л этилацетата, смесь нагревали до температуры дефлегмации и перемешивали при этой температуре в течение 30 мин. Смесь оставляли охлаждаться до комнатной температуры и перемешивали при комнатной температуре в течение двух суток. Твердые вещества собирали фильтрованием. Твердые вещества разделяли на две равные части и помещали в две круглодонные колбы емкостью по 5 л. В эти две колбы добавляли этилацетат (по 3 л в каждую) и смеси нагревали до температуры дефлегмации. Каждую смесь перемешивали в течение 30 мин при температуре дефлегмации, после чего оставляли охлаждаться до комнатной температуры и далее перемешивали в течение 48 ч при комнатной температуре. Твердые вещества собирали фильтрованием и сушили под вакуумом, получая 40 г (56%) N-[3-[2,5-бис(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида в виде белого твердого вещества (с чистотой 99,0% по данным HPLC).
Перевод в кристаллическую форму. В колбе емкостью 500 мл к 68 г N-[3-[2,5-бис(дифторметокси)фенил]-1-[(диметилкарбамоил)метил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (с чистотой по данным HPLC 99,0%) добавляли ЕtOАс (200 мл) и смесь перемешивали при комнатной температуре в течение 2 суток. Твердое вещество отфильтровывали и сушили под вакуумом при комнатной температуре в течение 6 ч, получая кристаллическое соединение (64,8 г; с чистотой по данным HPLC 99,1%). LC/MS (метод D, ESI): [М+Н]+=522,2; RT=2,62 мин; 1Н ЯМР (400 МГц, DMSO-d6) δ 9.77 (s, 1Н), 9.36 (dd, J=6,8; 1,6 Гц, 1Н), 8.69 (s, 1Н), 8.67 (dd, J=4,4; 1,6 Гц, 1Н), 8.30 (s, 1Н), 7.51 (d, J=8,8 Гц, 1Н), 7.39 (dd, J=8,8; 2,8 Гц, 1Н), 7.33-7.31 (m, 2Н), 7.30 (t, J=73,6 Гц, 1Н), 7.24 (t, J=73,2 Гц, 1Н), 5.23 (s, 2Н), 3.05 (s, 3Н), 2.89 (s, 3Н).
Пример 18
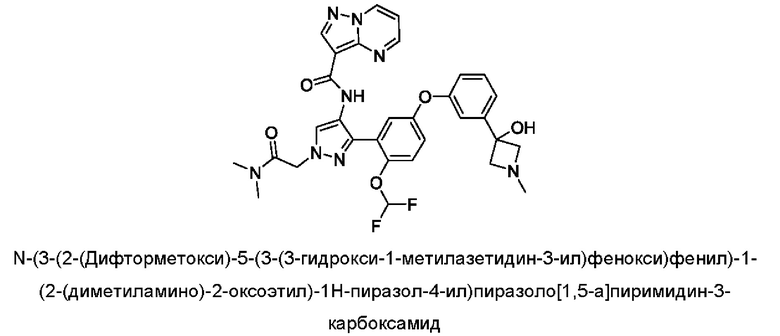

Стадия 1: синтез трет-бутил-3-[3-[4-(дифторметокси)-3-[4-(пиразоло[1,5-а]пиримидин-3-карбоксамидо)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-5-ил]фенокси]фенил]-3-гидроксиазетидин-1-карбоксилата stab

Раствор N-[5-[5-бром-2-(дифторметокси)фенил]-1-(2-триметилсилилэтоксиметил)пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (300 мг; 0,518 ммоль), трет-бутил-3-гидрокси-3-(3-гидроксифенил)азетидин-1-карбоксилата (275 мг; 1,04 ммоль), [РdСl(аллил)]2 (7,58 мг; 0,0207 ммоль), RockPhos (24,3 мг; 0,0518 ммоль), Cs2CO3 (337 мг; 1,04 ммоль) и толуола (4480 мг; 5,18 мл; 48,6 ммоль) перемешивали в течение 16 ч при 100°С в атмосфере азота. Полученную смесь концентрировали под вакуумом и очищали флэш-хроматографией на силикагеле, элюируя смесью метанол/дихлорметан (0-10%). Соответствующие фракции объединяли и концентрировали при пониженном давлении, получая трет-бутил-3-[3-[4-(дифторметокси)-3-[4-(пиразоло[1,5-а]пиримидин-3-карбоксамидо)-1-((2-(триметилсилил)этокси)метил)-1Н-пиразол-5-ил]фенокси]фенил]-3-гидроксиазетидин-1-карбоксилат (229 мг; 59%) в виде твердого вещества. LC/MS (метод L, ESI): [М+Н]+=764,2.
Стадия 2: синтез N-(3-(2-(дифторметокси)-5-(3-(3-гидроксиазетидин-3-ил)фенокси)фенил)-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида
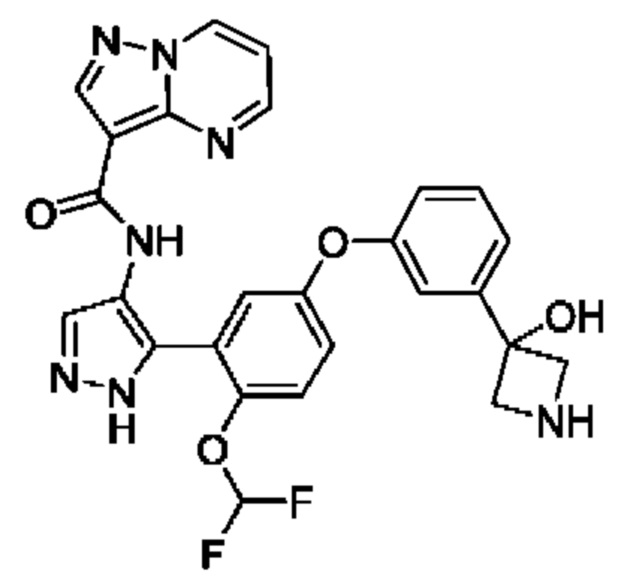
Раствор трет-бутил-3-[3-[4-(дифторметокси)-3-[4-(пиразоло[1,5-а]пиримидин-3-карбониламино)-2-(2-триметилсилилэтоксиметил)пиразол-3-ил]фенокси]фенил]-3-гидрокси-азетидин-1-карбоксилата (220 мг; 0,288 ммоль), дихлорметана (3830 мг; 2,88 мл; 45,1 ммоль) и трифторуксусной кислоты (536 мг; 0,360 мл; 4,70 ммоль) перемешивали при температуре окружающей среды в течение 16 ч. Раствор концентрировали под вакуумом и использовали без дополнительной очистки, получая N-(3-(2-(дифторметокси)-5-(3-(3-гидроксиазетидин-3-ил)фенокси)фенил)-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид (220 мг; 113%). LC/MS (метод X, ESI): [М+Н]+=534,1.
Стадия 3: синтез N-(3-(2-(дифторметокси)-5-(3-(3-гидрокси-1-метилазетидин-3-ил)фенокси)фенил)-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида

Раствор N-[3-[2-(дифторметокси)-5-[3-(3-гидроксиазетидин-3-ил)фенокси]фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (220 мг; 0,326 ммоль), 1,3,5-триоксана (39,1 мг; 0,434 ммоль), дихлорметана (4320 мг; 3,26 мл; 50,9 ммоль), метанола (516 мг; 0,652 мл; 15,8 ммоль) и триацетоксиборгидрида натрия (STAB; 207 мг; 0,979 ммоль) перемешивали при температуре окружающей среды в течение 3 ч. Полученную суспензию экстрагировали водой и использовали без дополнительной очистки, получая N-(3-(2-(дифторметокси)-5-(3-(3-гидрокси-1-метилазетидин-3-ил)фенокси)фенил)-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид (190 мг; 100%). LC/MS (метод X, ESI): [М+Н]+=548,2.
Стадия 4: синтез N-(3-(2-(дифторметокси)-5-(3-(3-гидрокси-1-метилазетидин-3-ил)фенокси)фенил)-1-(2-(диметиламино)-2-оксоэтил)-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида
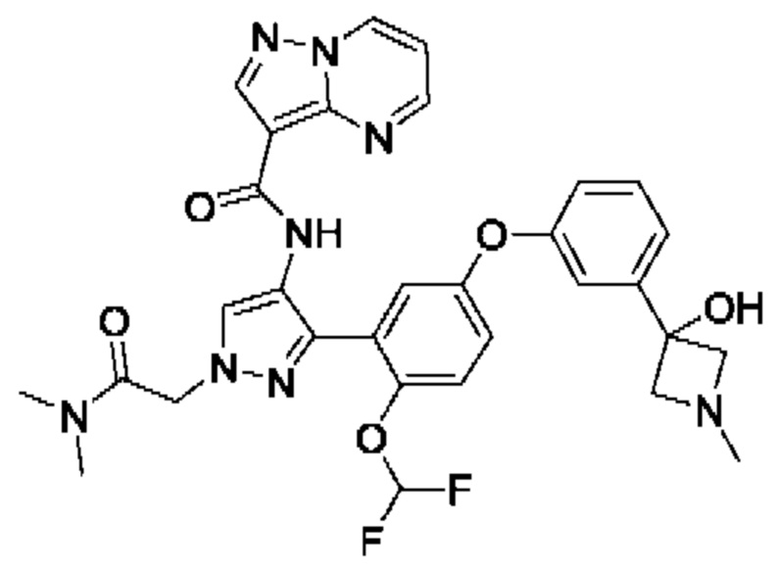
Раствор N-[3-[2-(дифторметокси)-5-[3-(3-гидрокси-1-метил-азетидин-3-ил)фенокси]фенил]-1Н-пиразол-4-ил]пиразоло[1,5-а]пиримидин-3-карбоксамида (190 мг; 0,330 ммоль), N,N-диметилформамида (3110 мг; 3,30 мл; 42,6 ммоль), 2-бром-N,N-диметил-ацетамида (65,7 мг; 0,0469 мл; 0,396 ммоль) и N,N-диизопропилэтиламина (174 мг; 0,230 мл; 1,32 ммоль) перемешивали при 50°С в течение 5 суток. Остаток концентрировали при пониженном давлении и очищали препаративной HPLC в следующих условиях: колонка: Gemini NX С18 110А, 30×100 мм, 10 мкм; подвижная фаза: вода (с 0,1% NH4OH) и ACN (от 20% до 60% ACN за 15 мин); детектор: УФ 254/220 нм. Соответствующие фракции объединяли и концентрировали при пониженном давлении, получая N-(3-(2-(дифторметокси)-5-(3-(3-гидрокси-1-метилазетидин-3-ил)фенокси)фенил)-1-(2-(диметиламино)-2-оксоэтил)-1Н-пиразол-4-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид (2,4 мг; 1%). LC/MS (метод Y, ESI): [М+Н]+=633,2; RT=3,42 мин. 1Н ЯМР (400 МГц, DMSO-d6) δ 13.01 (s, 1Н), 9.75 (s, 1Н), 9.36 (dd, J=7,0; 1,7 Гц, 1Н), 8.69 (dd, J=4,2; 1,5 Гц, 1Н), 8.65 (s, 1Н), 8.25 (s, 1Н), 7.44 (d, J=8,9 Гц, 1Н), 7.30 (dd, 1Н), 7.38-6.89 (m, 7Н), 3.32 (s, 2Н), 3.20 (d, J=13,7 Гц, 1Н), 3.04 (d, J=14,2 Гц, 1Н), 2.72-2.62 (m, 7Н), 2.47-2.39 (m, 1Н), 2.33 (р, J=1,8 Гц, 1Н), 2.16 (s, 3Н).
Приведенные далее в Таблице 1 репрезентативные соединения получали, используя методики, аналогичные методикам, описанным на схемах и в примерах данной заявки. Абсолютная стереохимическая конфигурация каждого приведенного ниже соединения может быть не указана, поэтому структуры могут появляться более одного раза, каждый раз представляя индивидуальный стереоизомер.


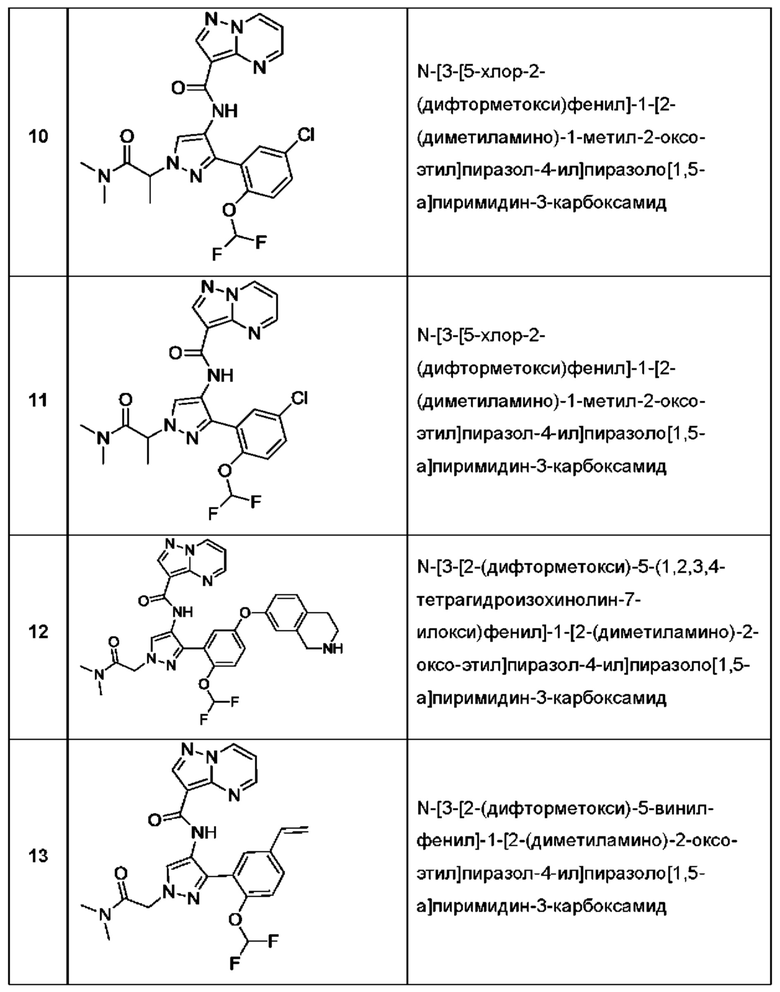

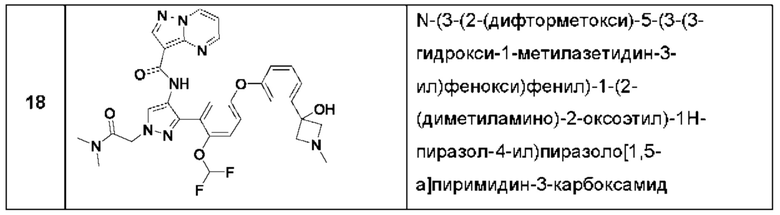
Ферментативные анализы
Анализы фермента JAK выполняли так, как приведено ниже
Активность выделенного рекомбинантного киназного домена JAK1 и JAK2 измеряли путем мониторинга фосфорилирования пептида, происходящего из JAK3 (Val-Ala-Leu-Val-Asp-Gly-Tyr-Phe-Arg-Leu-Thr-Thr, флуоресцентно меченного по N-концу 5-карбоксифлуоресцеином), с использованием технологии Caliper LabChip® (Caliper Life Sciences, Hopkinton, MA). Для определения констант ингибирования (Ki) готовили серийные разведения соединений в DMSO и добавляли к 50 мкл киназных реакционных смесей, содержащих очищенный фермент (в концентрации 1,5 нМ для JAK1 или 0,2 нМ для JAK2), 100 мМ буфер на основе 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновой кислоты (HEPES) (рН 7,2), 0,015% Brij-35, 1,5 мкМ пептидный субстрат, АТФ (25 мкМ), 10 мМ MgCl2, 4 мМ дитиотреит (DTT) в конечной концентрации DMSO, равной 2%. Реакционные смеси инкубировали при 22°С в 384-луночных полипропиленовых планшетах для микротитрования в течение 30 минут и затем реакции останавливали путем добавления 25 мкл раствора, содержащего этилендиаминтетрауксусную кислоту (EDTA) (100 мМ HEPES-буфер (рН 7,2), 0,015% Brij-35, 150 мМ EDTA), в результате чего конечная концентрация EDTA составляла 50 мМ. После остановки киназной реакции долю фосфорилированного продукта определяли как долю от общего пептидного субстрата, используя Caliper LabChip® 3000 в соответствии с указаниями производителя. Затем определяли значения Ки используя модель прочного связывания Моррисона (Morrison, J.F., Biochim. Biophys. Acta, 185: 269-296 (1969); William, J.W. and Morrison, J.F., Meth. Enzymol., 10 63: 437-467 (1979)), модифицированную для АТФ-конкурентного ингибирования [Ki=Ki,app/(1+[АТФ]/Km,арр)]. Данные для репрезентативных соединений показаны в Таблице 2.
Анализ пути JAK1 в клеточных линиях выполняли так, как приведено ниже
Эффективность ингибирования (ЕС50) определяли в анализах с применением клеток, разработанных для измерения JAK1-зависимого фосфорилирования STAT. Как отмечалось выше, ингибирование пути передачи сигнала с участием IL-4, IL-13 и IL-9 посредством блокирования Jak/Stat-опосредуемого сигнального пути может облегчить симптомы астмы в доклинических моделях воспаления легких (Mathew et al., 2001, J. Exp. Med., 193(9): 1087-1096; Kudlacz et. al., 2008, Eur. J. Pharmacol., 582(1-3): 154-161).
В одном из подходов к анализу клетки эритролейкоза человека линии TF-1, полученные из Американской коллекции типовых культур (АТСС; Manassas, VA), использовали для измерения JAK1-зависимого фосфорилирования STAT6 после стимуляции под действием IL-13. Перед применением в этих анализах клетки TF-1 лишали GM-CSF в течение ночи в среде OptiMEM (Life Technologies, Grand Island, NY), дополненной 0,5% фетальной телячьей сыворотки (FBS), очищенной активированным углем/декстраном, 0,1 мМ несущественными аминокислотами (NEAA) и 1 мМ пируватом натрия. Анализы проводили в 384-луночных планшетах в бессывороточной среде OptiMEM, используя 300000 клеток на одну лунку. Во втором подходе к анализу клетки эпителия бронхов человека линии BEAS-2B, полученные из АТСС, высевали в количестве 100000 клеток на одну лунку в 96-луночном планшете за одни сутки до проведения эксперимента. Анализ с использованием BEAS-2B проводили в полной ростовой среде (основной питательной среде для клеток эпителия бронхов с добавлением BulletKit™; Lonza; Базель, Швейцария).
Готовили растворы тестируемых соединений в серийном разведении в соотношении 1:2 в DMSO, а затем их разбавляли в соотношении 1:50 в среде непосредственно перед применением. Разбавленные растворы соединений добавляли к клеткам до конечной концентрации DMSO, равной 0,2%, и инкубировали в течение 30 мин (в анализе с TF-1) или 1 ч (в анализе с BEAS-2B) при 37°С. Затем клетки стимулировали рекомбинантным цитокином человека в концентрациях, соответствующих их ЕС90 (эффективные концентрации, вызывающие эффект величиной 90%), определенных ранее для каждой отдельной партии. Для стимулирования клеток использовали IL-13 (R&D Systems, Minneapolis, MN) в течение 15 мин при 37°С. Реакции с участием клеток TF-1 останавливали путем прямого добавления 10-кратного лизирующего буфера (Cell Signaling Technologies, Danvers, MA), тогда как инкубирование с клетками BEAS-2B прекращали посредством удаления среды и добавления 1-кратного лизирующего буфера. Полученные образцы замораживали в планшетах при -80°С. Опосредуемое соединениями ингибирование фосфорилирования STAT6 измеряли в клеточных лизатах, используя технологию MesoScale Discovery (MSD) (Gaithersburg, MD). Значения EC50 определяли как концентрацию соединения, необходимую для 50%-ного ингибирования фосфорилирования STAT, относительно случая такого измерения для контроля с использованием DMSO. Данные для репрезентативных соединений показаны в Таблице 2.

| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2015 |
|
RU2733400C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2700004C1 |
| ПИРИДОПИРИМИДИНОНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА N-МЕТИЛ-D-АСПАРТАТА | 2016 |
|
RU2717665C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2840815C2 |
| ПРОИЗВОДНЫЕ 1-ФЕНИЛ-2-ПИРИДИНИЛ-АЛКИЛОВЫХ СПИРТОВ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ | 2013 |
|
RU2637945C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛОВ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2689777C1 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| ПРОИЗВОДНЫЕ 2-ПИРАЗИНОНА ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ ИЛИ СОСТОЯНИЯ, ПРИ КОТОРЫХ ПОЛЕЗНО ИНГИБИРОВАНИЕ АКТИВНОСТИ НЕЙТРОФИЛЬНОЙ ЭЛАСТАЗЫ | 2007 |
|
RU2448098C2 |
| Соединения 6, 7-дигидро-5H-пиразоло[5,1-b][1,3]оксазин-2-карбоксамида | 2017 |
|
RU2719599C2 |
Изобретение относится к производным пиразолопиримидинов или к их стереоизомеру, выбранному из группы, состоящей из приведенных ниже соединений 1-18:
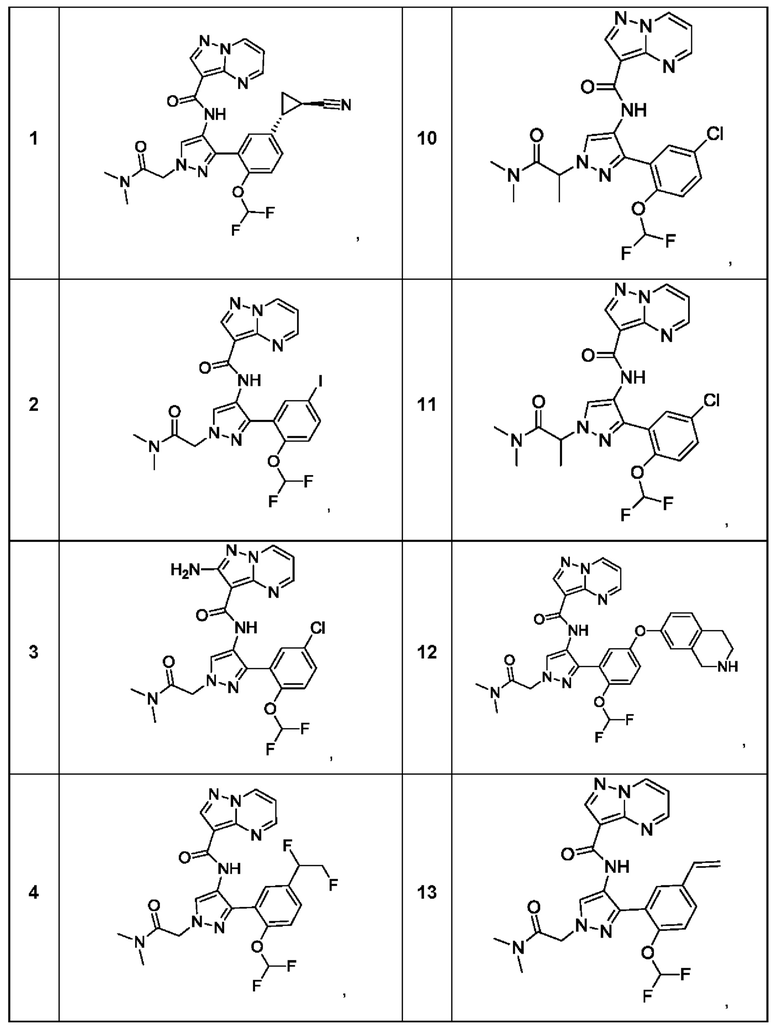

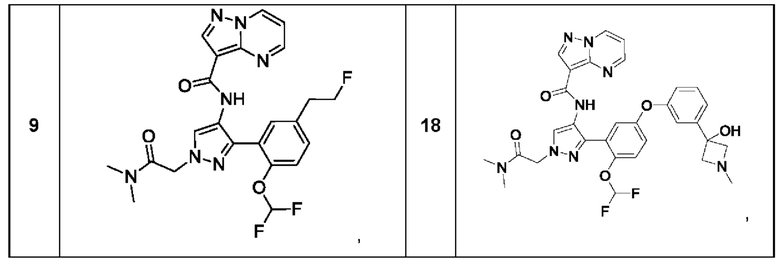
Изобретение также относится к фармацевтической композиции, обладающей ингибирующей активностью в отношении янус-киназы 1 (JAK1), на основе указанных соединений. Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине для лечения заболевания или состояния, отвечающего на ингибирование активности янус-киназы. 21 н. и 2 з.п. ф-лы, 2 табл., 18 пр.
1. Соединение, выбранное из группы, состоящей из приведенных ниже соединений 1-18:
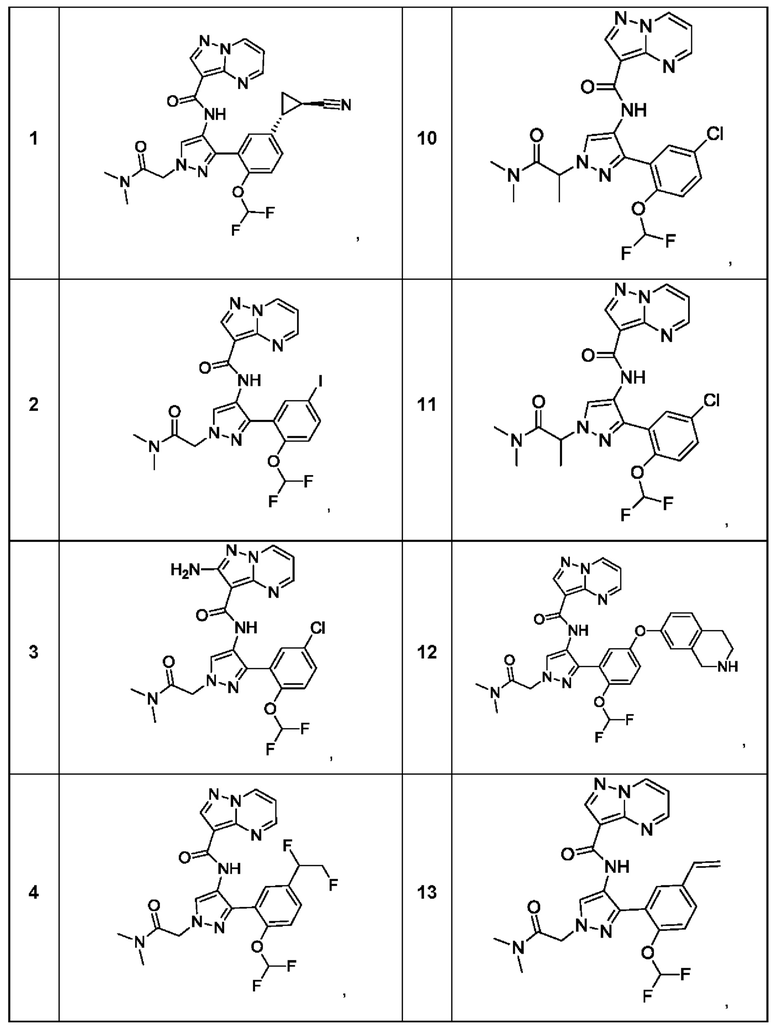
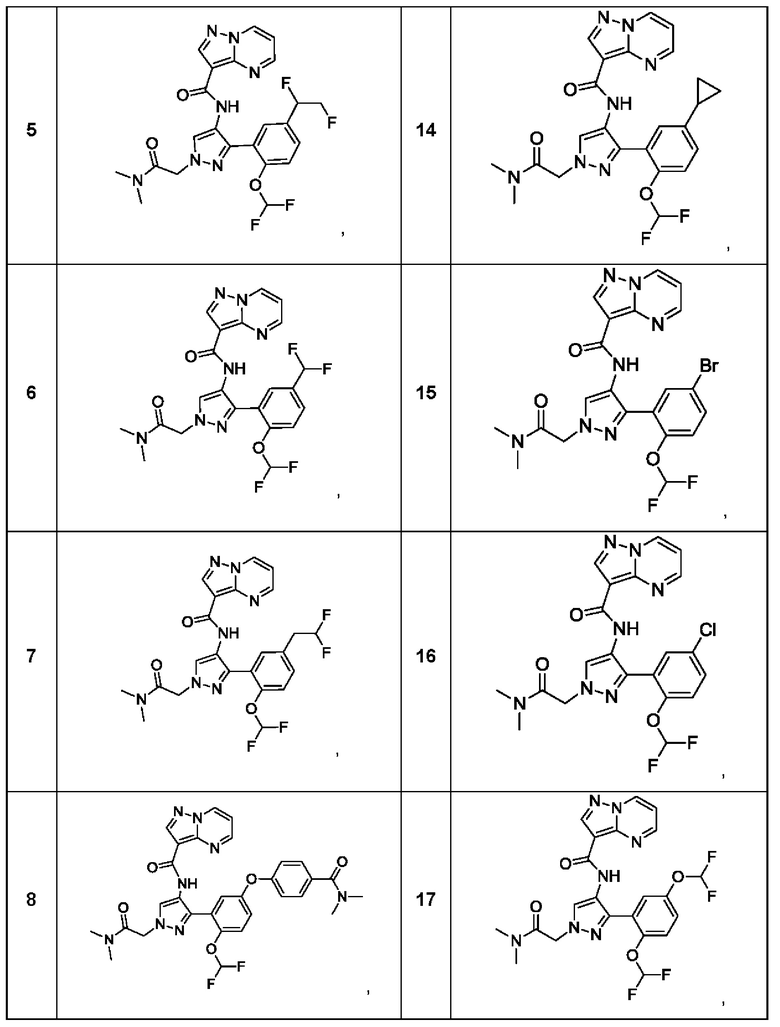

или его стереоизомер.
2. Соединение по п. 1, выбранное из группы, состоящей из:

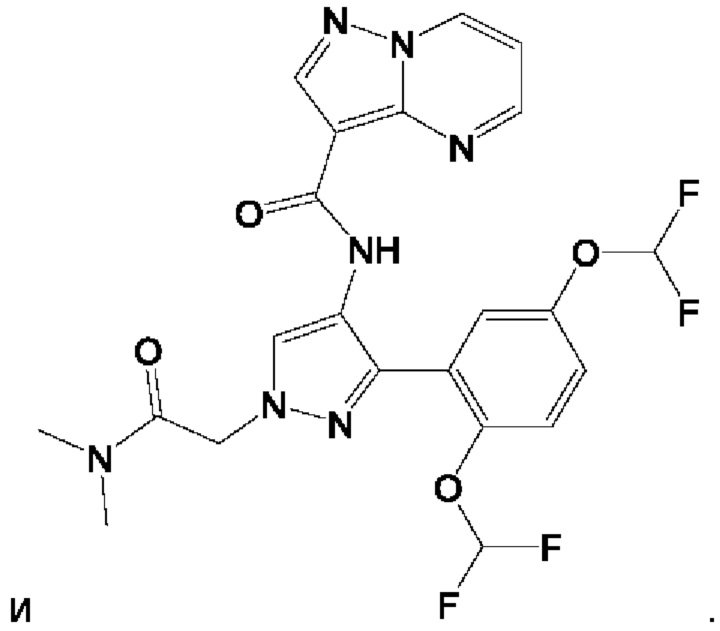
3. Соединение, представляющее собой:

4. Соединение, представляющее собой:

5. Соединение, представляющее собой:
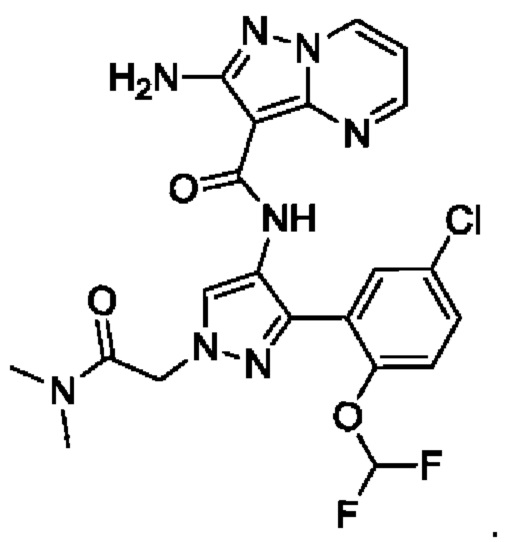
6. Соединение, представляющее собой:
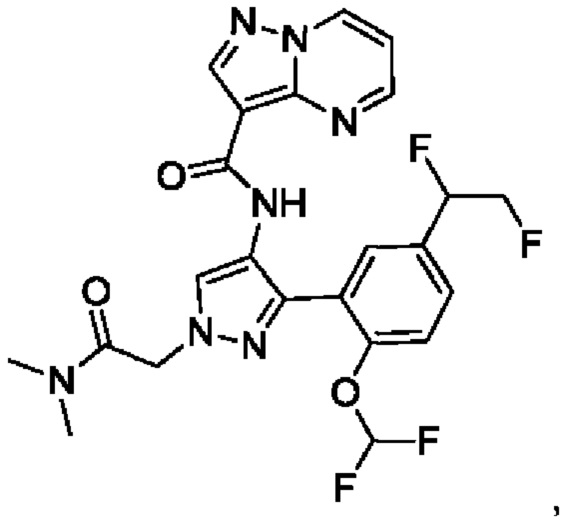
или его стереоизомер.
7. Соединение, представляющее собой:
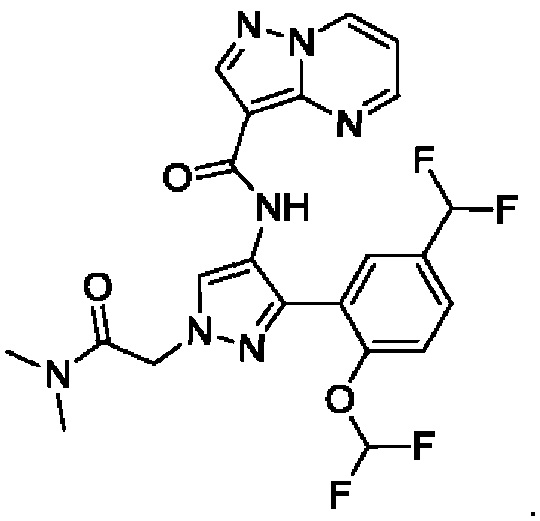
8. Соединение, представляющее собой:

или его стереоизомер.
9. Соединение, представляющее собой:

10. Соединение, представляющее собой:

11. Соединение, представляющее собой:

или его стереоизомер.
12. Соединение, представляющее собой:

или его стереоизомер.
13. Соединение, представляющее собой:

14. Соединение, представляющее собой:

15. Соединение, представляющее собой:

16. Соединение, представляющее собой:
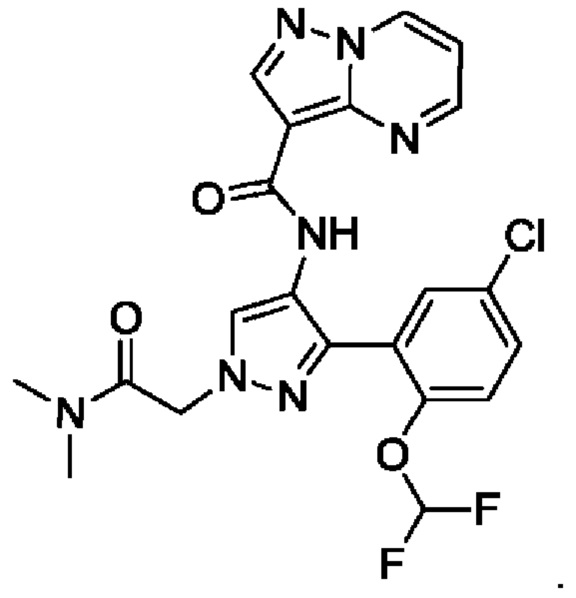
17. Соединение, представляющее собой:

18. Соединение, представляющее собой:
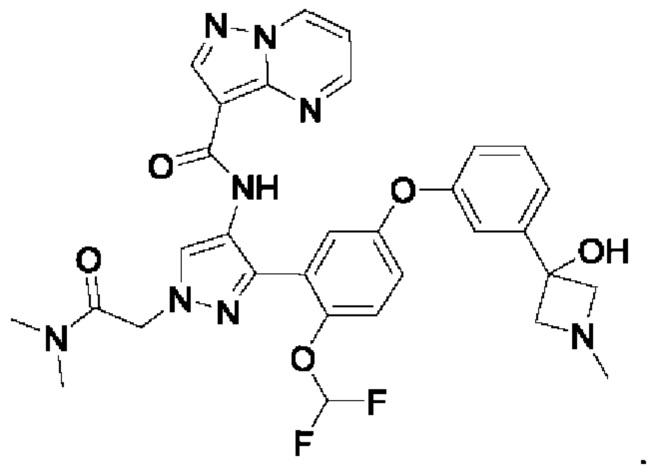
19. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении янус-киназы 1 (JAK1), содержащая терапевтически эффективное количество соединения по любому из пп. 1-18 или его стереоизомера и фармацевтически приемлемый носитель, разбавитель или эксципиент.
20. Применение соединения по любому из пп. 1-18 или его стереоизомера для ингибирования JAK1.
21. Применение соединения по любому из пп. 1-18 или его стереоизомера для приготовления лекарственного средства для ингибирования JAK1.
22. Соединение по любому из пп. 1-18 или его стереоизомер для применения в ингибировании JAK1.
23 Способ ингибирования JAK1 у пациента, включающий введение пациенту терапевтически эффективного количества соединения по любому из пп. 1-18 или его стереоизомера.
| WO 2011003065 A2, 06.01.2011 | |||
| WO 2015177326 A1, 26.11.2015 | |||
| WO 2012129258 A1, 27.09.2012 | |||
| US 20070083044 A1, 12.04.2007 | |||
| ПИРАЗОЛОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ-ИНГИБИТОРЫ JAK И СПОСОБЫ | 2009 |
|
RU2539568C2 |

