ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] В соответствии с разделом 35 Кодекса законов США, 119 (e), данная заявка притязает на приоритет по предварительной заявке на патент США с порядковым номером 62/271172, зарегистрированной 12 декабря 2015 г., описание которой включено настоящим посредством ссылки в данный документ во всей ее полноте.
ВВЕДЕНИЕ
[0002] Фенфлурамин является амфетаминовым лекарственным препаратом, который широко предписывался в качестве препарата для подавления аппетита, чтобы решить проблему ожирения. Фенфлурамин лишен психомоторной стимулирующей активности и аддиктивного потенциала D-амфетамина и взаимодействует с 5-гидрокситриптаминовыми (серотониновыми, 5-HT) рецепторами, что высвобожает 5-HT из нейронов. Фенфлурамин был исследован как имеющий противосудорожную активность при лечении синдрома Драве или тяжелой миоклонической эпилепсии в младенчестве, редкого и злокачественного эпилептического синдрома. Этот тип эпилепсии имеет раннее проявление в первоначально здоровых детях.
[0003] Анорексигенное лечение фенфлурамином было связано с исследованием поражения клапанов сердца и легочной гипертензии, включая условия фиброза миокарда, что привело к удалению фенфлурамина с мировых рынков. Взаимосвязь основного метаболита фенфлурамина норфенфлурамина с 5-HT2B-рецептором приводит к гипертрофии сердечного клапана. При лечении эпилепсии, известные сердечно-сосудистые риски фенфлурамина оцениваются в сопоставлении с благоприятной противосудорожной активностью.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0004] Данное изобретение предоставляет способы получения фенфлураминового активного фармацевтического ингредиента. Аспекты способов, являющихся предметом данного изобретения, включают гидролиз 2-(3-(трифторметил)фенил)ацетонитрильной композиции, чтобы получить композицию 2-(3-(трифторметил)фенил)уксусной кислоты; реакционное взаимодействие композиции 2-(3-(трифторметил)фенил)уксусной кислоты с уксусным ангидридом и катализатором, чтобы получить композицию 1-(3-(трифторметил)фенил)пропан-2-она; и восстановительное аминирование композиции 1-(3-(трифторметил)фенил)пропан-2-она этиламином при применении борогидридного восстановителя, чтобы получить фенфлураминовую композицию.
Также предоставлены фенфлураминовые композиции и фармацевтические ингредиенты, полученные в соответствии со способами, являющимися предметом данного изобретения, которые включают уменьшенное количество одного или нескольких второстепенных компонентов, таких как примеси или продукты побочных реакций. В некоторых случаях композиции включают фармацевтически приемлемую соль фенфлурамина, имеющую менее чем 0,2 мас.% суммарно трифторметильных региоизомеров. Также предоставлены фармацевтические композиции, включающие фенфлураминовые композиции, являющиеся предметом данного изобретения.
[0005] Эти и другие предметы, преимущества и отличительные признаки данного изобретения станут очевидными специалистам в данной области из подробностей устойчивых к метаболизму фенфлураминовых аналогов и способов их применения, как более подробно описано ниже.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0006] Данное изобретение легче всего понять из представленного ниже подробного описания при его рассмотрении в сочетании с приложенными чертежами. Следует подчеркнуть, что, в соответствии с обычной практикой, различные элементы чертежей не являются масштабированными. Напротив, размеры различных элементов произвольным образом расширены или уменьшены для ясности. Включенными в чертежи являются указанные ниже фигуры.
[0007] Фиг. 1 иллюстрирует вклады различных предшествующих материалов в структуру фенфлурамина (1) в типичном ретросинтетическом анализе до кислоты (4).
[0008] Фиг. 2 иллюстрирует типичную хроматограмму высокоэффективной жидкостной хроматографии (HPLC) для полученного исходного неочищенного гидрохлорида фенфлурамина (спектральную поглощательную способность для УФ при 210 нм).
[0009] Фиг. 3 иллюстрирует типичную хроматограмму высокоэффективной жидкостной хроматографии (HPLC) для композиции кристаллизованного гидрохлорида фенфлурамина (спектральную поглощательную способность для УФ при 210 нм).
[0010] Фиг. 4 иллюстрирует различные пути синтеза для получения кетона (2). Типичным способом, который находит применение в способах, являющихся предметом данного изобретения, является получение кетона (2) из нитрила (5) посредством кислоты (4).
[0011] Фиг. 5 иллюстрирует маршрут получения кетона (2) из исходного арилнитро-материала посредством промежуточного диазония. Маршрут с диазонием имеет недостаток, обусловленный потенциальной возможностью формирования генотоксических промежуточных продуктов, показанных как заключенные в рамку соединения (например, N-гидроксиарил, N-нитрозамин и нитросоединение).
ОПРЕДЕЛЕНИЯ
[0012] Как использовано в данном документе, термин «субъект» относится к млекопитающему. Примеры млекопитающих включают, однако без ограничения ими, людей, домашних животных (например, собак, котов или т.п.), сельскохозяйственных животных (например, коров, овец, свиней, лошадей или т.п.) или лабораторных животных (например, обезьян, крыс, мышей, кроликов, морских свинок или т.п.). В определенных вариантах осуществления, субъектом является человек. «Пациент» относится к человеку и субъектам, не относящимся к человеку, главным образом к субъектам, являющимися млекопитающими.
[0013] Как использовано в данном документе, термины «обработка», «лечение» и т.п., относятся к получению желательного фармакологического и/или физиологического действия. Данное действие может быть профилактическим с точки зрения полного или частичного предотвращения болезни или ее симптома и/или может быть терапевтическим с точки зрения частичного или полного излечения болезни и/или устранения неблагоприятного действия, свойственного данной болезни. Как использовано в данном документе, термины «лечение», «обработка», «терапевтическое» или «терапия» не обязательно означают полное излечение или устранение болезни или состояния. Любое частичное снятие каких-либо нежелательных признаков или симптомов болезни или состояния, в той или иной мере может рассматриваться как лечение и/или терапия. Кроме того, лечение может включать действия, которые могут ухудшать общее самочувствие пациента или его внешний вид. «Лечение», как использовано в данном документе, относится к любой обработке или лечению болезни млекопитающего, в некоторых случаях человека, и включает: (a) предотвращение возникновения болезни или расстройства здоровья, такое как профилактическая обработка субъекта; (b) уменьшение интенсивности болезни или расстройства здоровья, такое как устранение или вызывание ремиссии болезни или расстройства здоровья пациента; (c) подавление болезни или расстройства здоровья, например, посредством замедления или задержки развития болезни или расстройства здоровья пациента; или (d) облегчение симптома болезни или расстройства здоровья пациента.
[0014] Как использовано в данном документе, термин «pKa» относится к отрицательному логарифму (p) константы кислотной диссоциации (Ka) кислоты, и он соответствует величине pH, при которой равные концентрации кислоты и сопряженного с ней основания присутствуют в растворе.
[0015] Термин «соль» относится к ионному соединению, которое получено в результате реакции нейтрализации кислоты и основания и состоит из по меньшей мере одного катиона (положительно заряженного иона) и по меньшей мере одного аниона (отрицательного иона). В некоторых вариантах осуществления соль является электрически нейтральной (без результирующего заряда). При необходимости, соль является фармацевтически приемлемой солью, хотя это не требуется для солей промежуточных соединений, которые не предназначены для применения к пациенту. В качестве примера, соли представленных соединений включают те, где основное соединение протонировано неорганической или органической кислотой, чтобы образовать сопряженный с кислотой катион, с сопряженной основой неорганической или органической кислоты в качестве анионного компонента соли. Соли, представляющие интерес, включают, однако без ограничения ими, гидрохлоридные соли. Понятно, что для любой из структур, представленных в данном документе, такие структуры могут также включать любые обычные формы соли.
[0016] Термин «фармацевтически приемлемый» относится к фармацевтическому препарату, одобренному органом государственного регулирования федерального правительства или правительства штата или перечисленному в Фармакопее США или к другому общепризнанному фармацевтическому препарату для применения на млекопитающих, таких как люди.
[0017] Термин «фармацевтически приемлемая соль» означает соль, которая является подходящей для применения к пациенту, такому как млекопитающее, (соли с противоионами, обладающие приемлемой безопасностью для млекопитающего при заданном режиме дозирования). Такие соли могут быть производными от фармацевтически приемлемых неорганических или органических оснований и от фармацевтически приемлемых неорганических или органических кислот. «Фармацевтически приемлемая соль» относится к фармацевтически приемлемым солям соединения, соли которого являются производными от различных органических и неорганических противоионов, хорошо известных в данной области, и включающих, в качестве лишь примера, натрий и т.п.; и, когда молекула обладает основной функциональностью, соли органических или неорганических кислот, такие как гидрохлорид и т.п. Фармацевтически приемлемые соли, представляющие интерес, включают, однако без ограничения ими, гидрохлоридные соли.
[0018] Термин «активный фармацевтический ингредиент» (API) относится к веществу или смеси веществ, предназначенных для применения при изготовлении лекарственного средства, и которые, при применении для изготовления лекарственного препарата, становятся активным ингредиентом в лекарственном средстве. Такие вещества предназначены, чтобы предоставлять фармакологическую активность или другой непосредственный эффект при диагностике, лечении, ослаблении симптомов, обработке или предотвращении болезни, или чтобы воздействовать на структуру и функцию тела.
[0019] «Сольват» относится к комплексу, образованному комбинацией молекул растворителя с молекулами или ионами растворенного вещества. Растворитель может являться органическим соединением, неорганическим соединением или смесью их обоих. Некоторые примеры растворителей включают, однако без ограничения ими, метанол, N, N-диметилформамид, тетрагидрофуран, диметилсульфоксид и воду. Когда растворителем является вода, сформированный сольват является гидратом.
[0020] «Стереоизомер» и «стереоизомеры» относятся к соединениям, которые имеют одну и ту же атомную связь, однако разное пространственное расположение атомов. Стереоизомеры включают цис-транс-изомеры, E- и Z-изомеры, энантиомеры и диастереомеры.
[0021] «Таутомер» относится к альтернативным формам молекулы, которые отличаются лишь в электронном связывании атомов и/или в расположении протонов, таким как энол-кето и имино-енамино таутомеры или таутомерические формы гетероарильных групп, содержащие кольцеобразное расположение атомов -N=C(H)-NH-, такие как пиразолы, имидазолы, бензимидазолы, триазолы и тетразолы. Среднему специалисту в данной области техники будет понятно, что другие таутомерические расположения групп, описанных в данном документе, являются возможными.
[0022] Следует принимать во внимание, что термин «или соль или сольват или стереоизомер» предполагает включение всех комбинаций солей, сольватов и стереоизомеров, таких как сольват фармацевтически приемлемой соли стереоизомера соединения, являющегося предметом данного изобретения. Понятно, что термин «или его соль» предполагает включение всех измененных форм солей. Понятно, что термин «или его фармацевтически приемлемую соль» предполагает включение всех измененных форм солей. Понятно, что термин «или его сольват» предполагает включение всех измененных форм сольватов. Понятно, что термин «или его соль» предполагает включение всех измененных форм солей. Понятно, что термин «или его таутомер» предполагает включение всех измененных форм таутомеров. Соответственно, например, из этого следует, что это предполагает включение сольвата фармацевтически приемлемую соль таутомера стереоизомера соединения, являющегося предметом данного изобретения.
[0023] «Фармацевтически эффективное количество» и «терапевтически эффективное количество» относятся к количеству соединения, достаточному для лечения определенного расстройства или болезни или одного или нескольких их симптомов, и/или чтобы предотвратить возникновение болезни или расстройства. В отношении канцерогенных пролиферативных расстройств, фармацевтически или терапевтически эффективное количество содержит количество, необходимое для того, чтобы, помимо прочего, вызывать уменьшение опухоли или уменьшать скорость роста опухоли.
[0024] Термин «среда» относится к разбавителю, адъюванту, вспомогательному веществу или носителю, посредством которого соединение по данному изобретению образовано в составе для применения к млекопитающему.
[0025] Когда приведен интервал величин, понятно, что каждая промежуточная величина в интервале, до десятой доли величины нижнего предела, если только из контекста ясно не следует иное, между верхним и нижним пределами этого интервала также указана конкретным образом. Каждый меньший интервал между любой установленной величиной или промежуточной величиной в установленном интервале и любая другая установленная или промежуточная величина в интервале в этом установленном интервале входит в объем данного изобретения. Верхний и нижний пределы этих меньших интервалов могут быть независимым образом включены в интервал или исключены из него, и каждый интервал, где один из пределов, ни один из пределов или оба предела, включенные в меньшие интервалы, также входят в объем данного изобретения, с учетом любого конкретного исключенного предела в заявленном интервале. Когда заявленный интервал включает один или оба предела, интервалы, исключающие один или оба из этих включенных пределов, также включены в данное изобретение.
[0026] Если не указано иное, все технические и научные термины, используемые в данном описании, имеют такое же значение, которое является общепринятым для специалистов в области, к которой относится данное изобретение. Хотя в практике или испытаниях настоящего изобретения могут быть использованы любые способы и материалы, подобные или эквивалентные раскрытым в данном описании, некоторые возможные и предпочтительные способы и материалы описаны ниже. Все публикации, указанные в данном документе, включены в данный документ посредством ссылки, чтобы раскрыть и описать способы и/или материалы, в связи с которыми противопоставлены данные публикации. Понятно, что данное изобретение замещает любое раскрытие включенной публикации до той степени, когда имеет место несоответствие.
[0027] Следует заметить, что как использовано в описании и в прилагаемой формуле изобретения, формы единственного числа подразумевают включение формы множественного числа, если только из контекста ясно не следует иное. Таким образом, например, ссылка на некоторое «соединение» включает несколько таких соединений, и ссылка на «данный способ» включает ссылку на один или несколько способов и их эквивалентов, известных специалистам в данной области, и так далее.
[0028] Публикации, рассматриваемые в данном документе, предоставлены только лишь в отношении описания их сведений перед датой подачи данной заявки. Ни один из аспектов данного описания не должен истолковываться как допущение того, что данное изобретение не обладает правом относить к более ранней дате такую публикацию в силу действия предшествующего изобретения. Кроме того, данные предоставленной публикации могут отличаться от данных фактической публикации, которые могут нуждаться в подтверждении независимым образом.
[0029] Перед тем как описаны представленные соединения и способы, следует понимать, что это изобретение не ограничено описанными конкретными соединениями и способами, которые могут, разумеется, варьироваться. Следует также понимать, что терминологию, используемую в данном документе, применяют лишь с целью описания конкретных вариантов осуществления и не предназначают для ограничения, поскольку объем данного изобретения будет ограничиваться лишь прилагаемой формулой изобретения.
ПОДРОБНОЕ ОПИСАНИЕ
[0030] Как представлено в обобщенном виде выше, данное изобретение предоставляет способы получения фенфлураминового активного фармацевтического ингредиента. Аспекты данного изобретения включают фенфлураминовые композиции и фармацевтические ингредиенты, полученные в соответствии со способами, являющимися предметом данного изобретения, где определенные нежелательные второстепенные компоненты, представляющие интерес, по существу удалены из композиции. Способы, являющиеся предметом данного изобретения, предоставляют комбинацию стадий, чтобы получить исходную неочищенную композицию, которая достигает желательного минимального порога в отношении нежелательных второстепенных компонентов, таких как трудноочищаемые региоизомеры, продукты побочных реакций и реагенты. Активные фармацевтические ингредиенты для фармацевтических составов получают посредством контролируемых и воспроизводимых способов, чтобы достигать композиций высокой чистоты активного агента, который предоставляет высокие уровни безопасности, эффективность и качество в результирующих фармацевтических составах. В некоторых случаях примеси или нежелательные второстепенные компоненты в фармацевтических композициях могут вызывать нестабильность лекарственного средства, потерю активности и токсичность. Существенное удаление таких второстепенных компонентов из фенфлураминовых композиций, являющихся предметом данного изобретения, предоставляет композицию, которая подходит для применения в фармацевтических композициях в в качестве активного фармацевтического ингредиента (API). Композиции, являющиеся предметом данного изобретения, могут быть получены эффективным образом при уменьшенной необходимости в очистке, устранении стадий очистки или стадий способа для улучшения выхода, таких, как те стадии, которые включают удаление трудных для удаления региоизомеров фенфлурамина.
[0031] Термин «композиция», при применении в связи со способами, являющимися предметом данного изобретения, характеризует материал, который является исходным материалом или продуктом одной или нескольких стадий способов, являющихся предметом данного изобретения, и который может содержать смесь компонентов. Композиция может относиться к ее доминирующему или целевому компоненту, например, фенфлураминовой композиции. В общих чертах, композиция может включать, в дополнение к доминирующему целевому компоненту, смесь других компонентов, таких как целевые изомеры (например, стереоизомер или региоизомер), примеси, продукты побочных реакций, исходные материалы, перенесенные компоненты от предшествующих стадий, реагенты, растворители и т.п. Как использовано в данном документе, термин «исходная неочищенная композиция» относится к материалу, полученному при выполнении процедуры химической реакции, который не был подвергнут дополнительным стадиям очистки, например, отдельным стадиям процедуры после реакции, таким как стадии хроматографии или рекристаллизации. При получении исходной неочищенной композиции материал может быть подвергнут воздействию простых стадий, например, таких как промывка водой, экстракция и/или отфильтровывание растворителя, которые считаются составной частью реакционной процедуры, поскольку такие стадии являются обычно применяемыми для завершения химической реакции и/или для «обработки» продукта реакции. Такие стадии реакционной обработки не рассматриваются как являющиеся дополнительными стадиями очистки, как описано выше, однако как являющиеся единственно частью получения исходной неочищенной композиции.
Способы получения фенфлураминовых композиций
[0032] Аспекты способов, являющихся предметом данного изобретения, включают получение фенфлураминовой композиции из композиции предшественника 1-(3-(трифторметил)фенил)-пропан-2-она посредством восстановительного аминирования (Схема 1).

Схема 1: Получение фенфлурамина (1) из 1-(3-(трифторметил)фенил)-пропан-2-она (2) посредством восстановительного аминирования.
[0033] Любые обычные способы восстановительного аминирования могут быть применены, что чтобы преобразовать кетон (2) в фенфлурамин (1) посредством промежуточного имина (1a), например, посредством основания Шиффа, образованного между этиламином (например, Et-NH2) и кетоном (2). Способы и реагенты, представляющие интерес, включают, однако без ограничения ими, способы и реагенты, описанные в статье «Abdel-Magid et al. ʺReductive Amination of Aldehydes and Ketones with Sodium Triacetoxyborohydride. Studies on Direct and Indirect Reductive Amination Proceduresʺ («Восстановительное аминирование альдегидов и кетонов посредством триацетоксиборогидрида натрия. Исследование прямой и косвенной процедур восстановительного аминирования»), J. Org. Chem., 1996, 61 (11), pp 3849-3862». В некоторых вариантах осуществления реакцию восстановительного аминирования выполняют при условиях, которые включают контактирование композиции 1-(3-(трифторметил)фенил)пропан-2-она с раствором 70 мас.% этиламин/вода и примерно 2,25 эквивалента или более триацетоксиборогидрида натрия, растворенного в метаноле в качестве растворителя. В некоторых случаях реакцию (например, по схеме 1) выполняют в промышленном масштабе (например, как описано в данном документе). В некоторых случаях выход реакции (например, по Схеме 1) составляет 80% или более, например, 85% или более, 90% или более, 95% или более, 98% или более или 99% или более.
[0034] Любые подходящие восстановители могут быть применены на стадии восстановительного аминирования способов, являющихся предметом данного изобретения, например, чтобы восстанавливать промежуточное основание Шиффа до вторичного аминового продукта, фенфлурамина. В некоторых случаях восстановителем является борогидридный восстановитель. Как использовано в данном документе, термин «борогидридный восстановитель» означает включение любого восстановителя, который включает группу BH-, такого как любой подходящий борогидрид, цианоборогидрид или триацетоксиборогидридный восстановитель, имеющий формулу MBR3H, где каждый R является независимым образом H, алкилом, цианогруппой или ацетоксигруппой, и M является металлом, таким как Na, Li или K. В некоторых случаях восстановителем является цианоборогидридный восстановитель. В некоторых случаях восстановителем является триацетоксиборогидридный восстановитель. В некоторых случаях восстановитель выбирают из борогидрид натрия, цианоборогидрид натрия, триацетоксиборогидрид натрия, триэтилборогидрид лития, борогидрид никеля, борогидрид калия и борогидрид кальция. В определенных случаях борогидридным восстановителем является триацетоксиборогидрид натрия (STAB; Na(CH3COO)3BH).
[0035] Композиция 1-(3-(трифторметил)фенил)-пропан-2-она (2) может быть получена из любой подходящей композиции предшественника. В некоторых случаях композицию 1-(3-(трифторметил)фенил)-пропан-2-она получают из 2-(3-(трифторметил)фенил)уксусной кислоты (4), например, в соответствии со Схемой 2 посредством реакции Дакина-Веста (Dakin-West). Как таковые, аспекты способов, являющихся предметом данного изобретения, включают реакционное взаимодействие композиции 2-(3-(трифторметил)фенил)уксусной кислоты с уксусным ангидридом и катализатором, чтобы получить композицию 1-(3-(трифторметил)фенил)пропан-2-она.
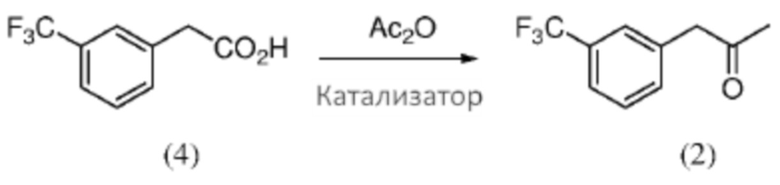
Схема 2: Получение кетона (2) из кислоты (4) посредством реакции Дакина-Веста (Dakin-West).
[0036] Реакция Дакина-Веста (Dakin-West) обеспечивает преобразование энолизируемой карбоновой кислоты в соответствующий метилкетон посредством реакционного взаимодействия с ацетилирующим агентом (например, уксусным ангидридом и катализатором). В некоторых случаях катализатор является нуклеофильным катализатором. Любой подходящий нуклеофильный катализатор может быть применен совместно с уксусным ангидридом при получении кетона (2) посредством Схемы 2. В некоторых вариантах осуществления катализатором является N-метилимидазол (т.е. 1-метилимидазол). Катализатор и уксусный ангидрид могут быть объединены, чтобы образовать ацетилирующий агент in situ. Понятно, что различные другие ацетилирующие агенты и реагенты, являющиеся предшественником для получения ацетилирующего агента in situ могут быть применены на реакционной стадии. В некоторых случаях данная стадия способа включает добавление предварительно образованного ацетилирующего агента непосредственно к кислоте (4). Способы и реагенты, представляющие интерес, которые находят применение при получении кетона (2), включают, однако без ограничения ими, те, что описаны Бьюкененом в «The Dakin-West reaction» («Реакция Дакина-Веста»), Chem. Soc. Rev., 1988, 17, 91-109. В некоторых вариантах осуществления, реакцию выполняют при условиях, которые включают приведение композиции 2-(3-(трифторметил)фенил)уксусной кислоты в контактирование с примерно 0,5 эквивалента 1-метилимидазола и примерно 5 эквивалентами или более уксусного ангидрида, необязательно в растворителе. В некоторых случаях выход реакции (например, по схеме 2) составляет 80% или более, например, 85% или более, 90% или более, 95% или более, 98% или более или 99% или более.
[0037] Кетон (2) может быть необязательно очищен перед применением на стадии, указанной на Схеме 1, при применении любого подходящего способа. В некоторых случаях кетон (2) очищают посредством формирования бисульфитного аддукта. Как использовано здесь, термины «бисульфитный аддукт» и «продукт присоединения бисульфита» применяют взаимозаменяемым образом в отношении продукта добавления бисульфитного иона к кетоновому соединению. Бисульфитный аддукт кетона (2) может быть твердотельным, что предоставляет более легко достижимое удаление примесей из аддукта композиции, чем это возможно из соответствующей исходной композиции кетона.
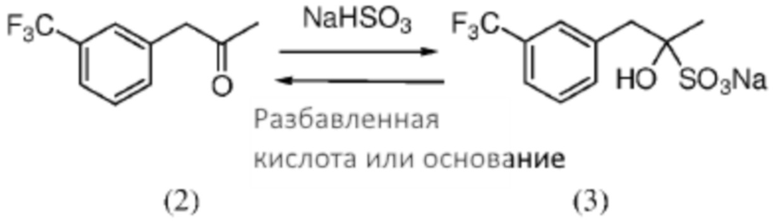
Схема 3: Очистка кетона (2) посредством формирования кетонового бисульфитного аддукта (3).
[0038] Аспекты способов, являющихся предметом данного изобретения, включают комбинацию отдельных стадий, описанных в данном документе, например, комбинацию стадий в качестве стадии далее на Схеме 4. Перед или после любой из описанных стадий, может быть выполнена необязательная стадия дополнительной очистки (например, стадия кристаллизации). В некоторых вариантах осуществления способ включает реакционное взаимодействие композиции 2-(3-(трифторметил)фенил)уксусной кислоты с уксусным ангидридом и катализатором, чтобы получить композицию 1-(3-(трифторметил)фенил)пропан-2-она; и восстановительное аминирование композиции 1-(3-(трифторметил)фенил)пропан-2-она этиламином при применении борогидридного восстановителя, чтобы получить фенфлураминовую композицию.

Схема 4: Получение фенфлурамина (1) из кислоты (4) посредством кетона (2)
[0039] Композиция 2-(3-(трифторметил)фенил)уксусной кислоты (4) может быть получена из любой подходящей композиции предшественника. В некоторых случаях композицию 2-(3-(трифторметил)фенил)уксусной кислоты получают из 2-(3-(трифторметил)фенил)ацетонитрильной композиции, например, в соответствии с реакционным взаимодействием по Схеме 5. Как таковые, аспекты способов, являющихся предметом данного изобретения, включают гидролиз композиции 2-(3-(трифторметил)фенил)ацетонитрила (5), чтобы получить композицию 2-(3-(трифторметил)фенил)уксусной кислоты (4).

Схема 5: Гидролиз нитрила (5) до кислоты (4).
[0040] Гидролиз нитрила (5) до кислоты (4) может быть достигнут при применении любых подходящих способов. В некоторых случаях гидролиз нитрила (5) достигают посредством гидролиза, катализированного кислотой. В некоторых случаях гидролиз нитрила (5) достигают посредством гидролиза, катализированного основанием. Гидролиз может быть выполнен посредством промежуточного амида (4a) при условиях водного раствора кислоты. В некоторых вариантах осуществления способа, гидролиз нитрила (5) до кислоты (4) выполняют при условиях водного раствора кислоты. В некоторых случаях выход реакции (например, по схеме 5) составляет 80% или более, например, 85% или более, 90% или более, 95% или более, 98% или более или 99% или более.
[0041] В некоторых случаях способ включает гидролиз композиции 2-(3-(трифторметил)фенил)ацетонитрила (5), чтобы получить композицию 2-(3-(трифторметил)фенил)уксусной кислоты (4); и реакционное взаимодействие композиции 2-(3-(трифторметил)фенил)уксусной кислоты (4) с уксусным ангидридом и катализатором, чтобы получить композицию 1-(3-(трифторметил)фенил)пропан-2-она (2) (см., например, Схему 6).
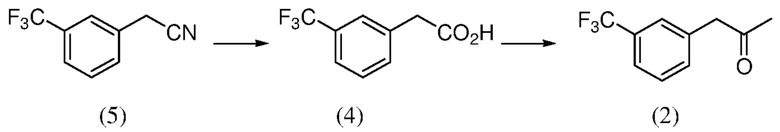
Схема 6: Получение кетона (2) из нитрила (5) посредством кислоты (4)
[0042] Аспекты способов, являющихся предметом данного изобретения, включают комбинацию стадий, как описано на Схеме 7. Перед или после любой из описанных стадий, может быть выполнена необязательная стадия дополнительной очистки (например, стадия кристаллизации). В некоторых вариантах осуществления способ включает гидролиз композиции 2-(3-(трифторметил)фенил)ацетонитрила (5), чтобы получить композицию 2-(3-(трифторметил)фенил)уксусной кислоты (4); реакционное взаимодействие композиции 2-(3-(трифторметил)фенил)уксусной кислоты (4) с уксусным ангидридом и катализатором, чтобы получить композицию 1-(3-(трифторметил)фенил)пропан-2-она (2); и восстановительное аминирование композиции 1-(3-(трифторметил)фенил)пропан-2-она (2) этиламином при применении борогидридного восстановителя, чтобы получить композицию фенфлурамина (1).
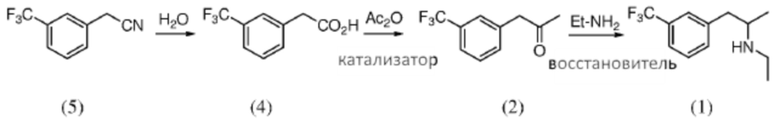
Схема 7: Получение фенфлурамина (1) из нитрила (5) посредством кислоты (4) и кетона (2)
[0043] В некоторых вариантах осуществления способа полученная фенфлураминовая композиция (например, исходная неочищенная фенфлураминовая композиция) имеет следующий состав: 80% или более по массе фенфлурамина или его соли, например, 90% или более, 95% или более, 96% или более, 97% или более, 98% или более, 99% или более, 99,5% или более или даже более по массе фенфлурамина или его соли; 1% или менее по массе региоизомера 2-фенфлурамина или его соли, например, 0,5% или менее, 0,2% или менее или 0,1% или менее, 0,05% или менее, 0,01% или менее или даже менее по массе региоизомера 2-фенфлурамина или его соли; 1% или менее по массе региоизомера 4-фенфлурамина или его соли, например, 0,5% или менее, 0,2% или менее или 0,1% или менее, 0,05% или менее, 0,01% или менее или даже менее по массе региоизомера 4-фенфлурамина или его соли; и 10% или менее по массе спиртового побочного продукта восстановления фенфлурамина, например, 5% или менее, 2% или менее или 1% или менее, 0,5% или менее, 0,1% или менее по массе спиртового побочного продукта восстановления фенфлурамина.
[0044] В некоторых вариантах осуществления способ является способом получения свободного основания фенфлурамина. Как таковая, фенфлураминовая композиция может включать свободное основание фенфлурамина. Свободное основание фенфлурамина, которое получено в соответствии со способами, являющимися предметом данного изобретения, может быть преобразовано в любую подходящую форму соли, например, соль сопряженной кислоты вторичной аминогруппы фенфлурамина (фенфлурамин.H+X-), при применении различных способов. Формирование соли фенфлурамина может быть выполнено в качестве части стадии восстановительного аминирования Схемы 1 (например, in situ), или формирование соли может быть выполнено на необязательной последующей стадии. В некоторых случаях сформированная соль является фармацевтически приемлемой солью фенфлурамина. Соли, представляющие интерес, включают, однако без ограничения ею, гидрохлоридную соль. В некоторых случаях фармацевтически приемлемая форма соли фенфлурамина является гидрохлоридной солью.

Схема 8: Получение соли фенфлурамина.
[0045] Способы, являющиеся предметом данного изобретения, предоставляют существенное удаление одного или нескольких нежелательных второстепенных компонентов из исходной неочищенной фенфлураминовой композиции или композиции фенфлураминовой соли, так что заключительные дополнительные стадии очистки могут быть выполнены простым образом при высокой эффективности и/или высоком выходе, чтобы получить высококачественную активную фармацевтическую композицию.
[0046] Одна или несколько дополнительных стадий очистки могут быть выполнены для исходной неочищенной фенфлураминовой композиции (например, которая включает свободное основание или фенфлурамин в форме соли), полученной в соответствии со способами, являющимися предметом данного изобретения, В некоторых случаях стадия очистки включает кристаллизацию фенфлурамина или фенфлурамина в форме соли из исходной неочищенной композиции. Кристаллическая форма соли фенфлурамина может иметь желательные полиморфизм, высокую кристалличность, растворимость в воде и/или стабильность. В некоторых случаях способы, являющиеся предметом данного изобретения, предоставляют кристаллическую соль гидрохлорида фенфлурамина, которая является единичным полиморфом, который является сыпучим, негигроскопичным и имеющим высокую температуру плавления.
[0047] В некоторых вариантах осуществления способа, полученная композиция содержит фармацевтически приемлемую соль фенфлурамина и имеет следующий профиль чистоты: 90% или более фармацевтически приемлемой соли фенфлурамина, например, 95% или более, 96% или более, 97% или более, 98% или более, 99% или более, 99,5% или более, 99,8% или более, 99,9% или более, или даже более по массе фармацевтически приемлемой соли фенфлурамина; 1% или менее по массе 2-фенфлурамина; 1% или менее по массе региоизомера 2-фенфлурамина или его соли, например, 0,5% или менее, 0,2% или менее или 0,1% или менее, 0,05% или менее, 0,01% или менее, или даже менее по массе региоизомера 2-фенфлурамина или его соли; 1% или менее по массе региоизомера 4-фенфлурамина или его соли, например, 0,5% или менее, 0,2% или менее или 0,1% или менее, 0,05% или менее, 0,01% или менее или даже менее по массе региоизомера 4-фенфлурамина или его соли; и 5% или менее по массе спиртового побочного продукта восстановления фенфлурамина, например, 3% или менее, 2% или менее или 1% или менее, 0,5% или менее, 0,1% или менее по массе спиртового побочного продукта восстановления фенфлурамина. В определенных вариантах осуществления композиция, полученная в соответствии со способами, являющимися предметом данного изобретения, является фенфлураминовым активным фармацевтическим ингредиентом, содержащим фармацевтически приемлемую соль фенфлурамина и содержащую 0,2% или менее по массе суммарно трифторметильных региоизомеров, например, 0,1% или менее, 0,05% или менее, 0,03% или менее, 0,01% или менее или даже менее по массе трифторметильных региоизомеров. В определенных вариантах осуществления фенфлураминовый активный фармацевтический ингредиент имеет профиль чистоты, включающий: по меньшей мере 90% (например, по меньшей мере 95%, по меньшей мере 96%, по меньшей мере 97%, по меньшей мере 98%, по меньшей мере 99%, по меньшей мере 99,5%, по меньшей мере 99,8%, по меньшей мере 99,9%, или более) по массе фармацевтически приемлемой соли фенфлурамина; менее чем 0,2 мас.% (например, менее чем 0,1%, менее чем 0,05%, менее чем 0,03%, менее чем 0,01 мас.%) 2-фенфлурамина; менее чем 0,2 мас.% (например, менее чем 0,1%, менее чем 0,05%, менее чем 0,03%, менее чем 0,01 мас.%) 4-фенфлурамина; и менее чем 1 мас.% (например, менее чем 0,5%, менее чем 0,3%, менее чем 0,1%, менее чем 0,05 мас.%) фенфлураминового спирта.
[0048] Способы, являющиеся предметом данного изобретения, предоставляют получение рацемической смеси энантиомеров фенфлурамина. Энантиомеры фенфлурамина могут рассматриваться как: дексфенфлурамин (т.е., (S)-N-этил-1-[3-(трифторметил)фенил]-пропан-2-амин, (+)-фенфлурамин или (S)-фенфлурамин); и левофенфлурамин (т.е., (2R)-N-этил-1-[3-(трифторметил)фенил]-2-пропанамин, (-)-фенфлурамин или (R)-фенфлурамин). Фенфлураминовые энантиомеры или их соли могут быть отделены одна от другой при применении любых подходящих способов. Способы, представляющие интерес, для отделения и очистки фенфлураминовых энантиомеров включают, однако без ограничения ими, хиральное разделение посредством кристаллизации и хиральной колоночной хроматографии. В некоторых вариантах осуществления, как таковых, способ дополнительно включает выполнение хирального разделения рацемической фенфлураминовой композиции, или ее соли, чтобы получить нерацемическую фенфлураминовую композицию, содержащую доминирующий стереоизомер фенфлурамина. Под нерацемической понимают композицию, имеющую энантиомерный избыток по меньшей мере 50%, например, по меньшей мере 60%, по меньшей мере 70%, по меньшей мере 80%, по меньшей мере 90%, по меньшей мере 95% или по меньшей мере 99% одного стереоизомера, например, доминирующего стереоизомера. Как использовано в данном документе, термин «доминирующий стереоизомер» подразумевает заключение в себе композиции, включающей лишь один стереоизомер, или композиции, которая включает смеси стереоизомеров.
[0049] В некоторых случаях композиция активного фармацевтического ингредиента является нерацемической композицией, включающей (S)-фенфлурамин или его фармацевтически приемлемую соль в качестве доминирующего стереоизомера. В некоторых случаях композиция активного фармацевтического ингредиента является нерацемической композицией, включающей (R)-фенфлурамин или его фармацевтически приемлемую соль в качестве доминирующего стереоизомера. В некоторых случаях полученная нерацемическая композиция включает лишь один стереоизомер.
Второстепенные компоненты
[0050] Как представлено в обобщенном виде выше, композиции в соответствии со способами, являющимися предметом данного изобретения, например, композиции исходного материала, промежуточные композиции и конечные фенфлураминовые композиции, могут предоставлять существенное удаление одного или нескольких второстепенных компонентов, что достигается способами, являющимися предметом данного изобретения, чтобы получить композиции, которые находят применение в качестве активного фармацевтического ингредиента (API) или его предшественника для фармацевтических композиций. Способы, являющиеся предметом данного изобретения, предоставляют существенное удаление нежелательных второстепенных компонентов различным образом. Как использовано в данном документе, «по существу удаленный » означает достижение желательного минимального порога для второстепенного компонента, представляющего интерес, таким образом, что второстепенный компонент, если он присутствует, присутствует при уровне или ниже порогового значения. Как использовано в данном документе, термин «по существу не имеет» относится к композиции, где второстепенный компонент, представляющий интерес, либо не присутствует, либо присутствует при уровне или ниже минимального порогового значения. Желательное минимальное пороговое значение для второстепенного компонента, представляющего интерес, может варьироваться в соответствии с природой компонента и любой композиции из промежуточной композиции или фенфлураминовой композиции, представляющей интерес. В некоторых случаях желательное минимальное пороговое значение второстепенного компонента, представляющего интерес, достигает 10 мас.% или менее, например, 5% или менее, 4% или менее, или 3% или менее, 2% или менее, 1% или менее 0,5% или менее, 0,4% или менее, 0,3% или менее, 0,2% или менее, 0,15% или менее, 0,1% или менее, 0,09% или менее, 0,08% или менее, 0,07% или менее, 0,06% или менее, 0,05% или менее, 0,03% или менее или 0,01% или менее. В некоторых случаях второстепенный компонент, представляющий интерес, полностью удален из композиции, представляющей интерес, т.е. композиция не содержит второстепенный компонент (например, при необнаруженном содержании компонента или его содержании ниже предела определения).
[0051] В некоторых случаях определенная комбинация стадий, примененных в способе, являющемся предметом данного изобретения, функционирует, чтобы устранять второстепенный компонент, представляющий интерес. В некоторых случаях очистка промежуточной композиции, например, посредством кристаллизации, выполняет существенное удаление второстепенного компонента, который являлся бы трудным для удаления, если бы второстепенный компонент, например, региоизомер, был перенесен к последующей стадии синтеза. В некоторых случаях выполнение конкретной стадии способа предоставляет селективность реакции, посредством чего второстепенный компонент, представляющий интерес, не преобразуется посредством условий реакции, как это имеет место для основного компонента, и соответственно может быть удален более простым образом, например, в качестве региоизомера исходного материала, а не продукта реакции или отдельной стадии способа. В некоторых случаях определенная комбинация стадий, использованных в способах, являющихся предметом данного изобретения, избегает применения одного или нескольких химических реагентов, растворителей и/или реагентов, которые требуются при обычных способах и которые приводят к нежелательным второстепенным компонентам в композициях продукта. Второстепенные компоненты, представляющие интерес, которые могут быть по существу удалены, включают, однако без ограничения ими, изомерные продукты, побочные продукты, альдегиды, кетоны, пероксиды, металлы (например, тяжелый металл и металлические катализаторы), нитрат/нитрит, следовые растворители и органические кислоты. Различные второстепенные компоненты и подробности их существенного удаления из композиций, являющихся предметом данного изобретения, описаны теперь более подробно ниже. Второстепенные компоненты, представляющие интерес, которые могут быть по существу удалены в соответствии со способами, являющимися предметом данного изобретения, включают любые примеси, побочные продукты, исходные материалы и второстепенные компоненты, описанные в данном документе, включающие, однако не ограниченные ими, примесь ацетата, примесь димера, примесь ацетамида, 1-((3-трифторметил)фенил)ацетон, региоизомеры фенфлурамина, фенфлураминовый спирт, N-(3-(трифторметил)-бензил)этанамин, норфенфлурамин и любую из примесей, представленных в Таблице 7.
Региоизомеры
[0052] В некоторых случаях региоизомер фенфлурамина, или его предшественник, может присутствовать в качестве второстепенного компонента любой из композиций, являющихся предметом данного изобретения, которые находят применение в способах, являющихся предметом данного изобретения. Фенфлурамин и предшественники его синтеза могут включать 3-трифторметил, замещенный фенильной группой. Как использовано здесь, термины, «трифторметильный региоизомер» и «трифторметил-фенильный региоизомер» применяют взаимозаменяемым образом в отношении изомера(ов) фенфлурамина или любого из предшественников синтеза, описанных в данном документе, где трифторметильный заместитель расположен в 2-позиции или 4-позиции замещенного фенильного кольца, а не в 3-позиции, соответствующей фенфлурамину. В качестве таковых, термины «2-трифторметильный региоизомер» и «4-трифторметильный региоизомер» могут быть применены в данном документе, чтобы описывать отдельные второстепенные компоненты любой промежуточной композиции или конечной композиции, которая находит применение в способах, являющихся предметом данного изобретения.
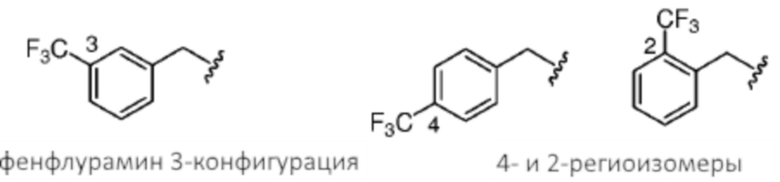
[0053] 2-(3-(трифторметил)фенил)ацетонитрильная композиция, являющаяся исходным материалом способов, являющихся предметом данного изобретения, может включать региоизомеры. В некоторых случаях региоизомеры являются производными при способе получения 2-(3-(трифторметил)фенил)ацетонитрила из трифторметилбензола. В некоторых случаях 2-(3-(трифторметил)фенил)ацетонитрильная композиция содержит по меньшей мере 0,2 мас.%, например, по меньшей мере 0,3%, по меньшей мере 0,4%, по меньшей мере 0,5%, по меньшей мере 1,0%, по меньшей мере 1,5%, по меньшей мере 2%, по меньшей мере 3%, по меньшей мере 4%, по меньшей мере 5%, по меньшей мере 10% или даже более по массе трифторметил-фенильных региоизомеров (например, суммарного содержания 2-(2-(трифторметил)фенил)ацетонитрила и 2-(4-(трифторметил)фенил)ацетонитрила). В некоторых случаях 2-(3-(трифторметил)фенил)ацетонитрильная композиция содержит по меньшей мере 0,2 мас.%, например, по меньшей мере 0,3%, по меньшей мере 0,4%, по меньшей мере 0,5%, по меньшей мере 1,0%, по меньшей мере 1,5%, по меньшей мере 2%, по меньшей мере 3%, по меньшей мере 4%, по меньшей мере 5%, по меньшей мере 10% или даже более по массе 2-(4-(трифторметил)фенил)ацетонитрила). В некоторых случаях 2-(3-(трифторметил)фенил)ацетонитрильная композиция содержит по меньшей мере 0,2 мас.%, например, по меньшей мере 0,3%, по меньшей мере 0,4%, по меньшей мере 0,5%, по меньшей мере 1,0%, по меньшей мере 1,5%, по меньшей мере 2%, по меньшей мере 3%, по меньшей мере 4%, по меньшей мере 5%, по меньшей мере 10% или даже более по массе 2-(2-(трифторметил)фенил)ацетонитрила). В некоторых случаях 2-(3-(трифторметил)фенил)ацетонитрильная композиция включает второстепенные региоизомерные компоненты, которые переносятся к следующей композиции, например, композиции 2-(3-(трифторметил)фенил)уксусной кислоты. Как таковая, композиция 2-(3-(трифторметил)фенил)уксусной кислоты, полученная в качестве промежуточного продукта в способе, являющемся предметом данного изобретения, может также включать региоизомеры (например, 2-(2-(трифторметил)фенил)уксусную кислоту и 2-(4-(трифторметил)фенил)уксусную кислоту при таких же уровнях, как и те, что описаны в данном документе для 2-(3-(трифторметил)фенил)ацетонитрильной композиции, являющейся исходным материалом.
[0054] Способы, являющиеся предметом данного изобретения, предоставляют удаление 2- и/или 4-региоизомеров в качестве второстепенных компонентов промежуточной композиции различным образом. В некоторых вариантах осуществления способ включает очистку композиции 2-(3-(трифторметил)фенил)уксусной кислоты, чтобы получить композицию, по существу не содержащую один или оба из трифторметил-фенильных региоизомеров. В некоторых случаях композиция также по существу не содержит бензальдегида, который присутствует в ацетонитрильном исходном материале. В некоторых случаях композиция также по существу не содержит трифторметил-бензальдегида, который присутствует в ацетонитрильном исходном материале. В некоторых случаях очистка композиции 2-(3-(трифторметил)фенил)уксусной кислоты для удаления части или всех второстепенных региоизомерных компонентов может быть достигнута посредством кристаллизации 2-(3-(трифторметил)фенил)уксусной кислоты. Как использовано в данном документе, термин «по существу не содержит трифторметил-фенильного региоизомера» означает менее чем 0,5 мас.%, например, менее чем 0,4%, менее чем 0,3%, менее чем 0,2%, менее чем 0,1%, менее чем 0,09%, менее чем 0,08%, менее чем 0,07%, менее чем 0,06%, менее чем 0,05%, менее чем 0,03% или даже менее. Любые подходящие способы кристаллизации или рекристаллизации могут быть применены в способах, являющихся предметом данного изобретения.
[0055] После очистки, например, кристаллизации, композиции 2-(3-(трифторметил)фенил)уксусной кислоты, композиция может включать менее чем 0,5 мас.%, например, менее чем 0,4%, менее чем 0,3%, менее чем 0,2%, менее чем 0,1%, менее чем 0,09%, менее чем 0,08%, менее чем 0,07%, менее чем 0,06%, менее чем 0,05%, менее чем 0,03% или даже менее 2-(2-(трифторметил)фенил)уксусной кислоты. После очистки, например, кристаллизации, композиции 2-(3-(трифторметил)фенил)уксусной кислоты, композиция может включать менее чем 0,5 мас.%, например, менее чем 0,4%, менее чем 0,3%, менее чем 0,2%, менее чем 0,1%, менее чем 0,09%, менее чем 0,08%, менее чем 0,07%, менее чем 0,06%, менее чем 0,05%, менее чем 0,03% или даже менее 2-(4-(трифторметил)фенил)уксусной кислоты. После очистки, например, кристаллизации, композиции 2-(3-(трифторметил)фенил)уксусной кислоты, композиция может включать менее чем 0,5 мас.%, например, менее чем 0,4%, менее чем 0,3%, менее чем 0,2%, менее чем 0,1%, менее чем 0,09%, менее чем 0,08%, менее чем 0,07%, менее чем 0,06%, менее чем 0,05%, менее чем 0,03% или даже менее бензальдегида.
[0056] В некоторых вариантах осуществления способ включает реакционное взаимодействие композиции 2-(3-(трифторметил)фенил)уксусной кислоты с уксусным ангидридом и катализатором, чтобы получить композицию 1-(3-(трифторметил)фенил)пропан-2-она; где 2-(3-(трифторметил)фенил)уксусную кислоту селективным образом преобразуют в кетон в присутствии непрореагировавшей 2-(2-(трифторметил)фенил)уксусной кислоты. Способ, являющийся предметом данного изобретения, предоставляет легко достижимое удаление 2-региоизомера, который присутствует, поскольку этот региоизомер не переносится на протяжении реакции в той же самой степени, что и целевое 3-трифторметильное соединение. В некоторых случаях способ дополнительно включает удаление непрореагировавшего региоизомера 2-(2-(трифторметил)фенил)уксусной кислоты из композиции 1-(3-(трифторметил)фенил)пропан-2-она. Как таковая, в некоторых случаях исходная неочищенная композиция 1-(3-(трифторметил)фенил)пропан-2-она по существу не содержит (например, включает менее чем 0,5 мас.%, например, менее чем 0,4%, менее чем 0,3%, менее чем 0,2%, менее чем 0,1%, менее чем 0,09%, менее чем 0,08%, менее чем 0,07%, менее чем 0,06%, менее чем 0,05%, менее чем 0,03%, или даже менее) 2-региоизомера кетонового продукта.
[0057] Удаление региоизомерных второстепенных компонентов, присутствующих в ацетонитрильном исходном материале, может быть достигнуто поэтапно во время выполнения данного способа синтеза. В некоторых вариантах осуществления первую часть региоизомерных второстепенных компонентов, присутствующих в исходном материале, удаляют из композиции 2-(3-(трифторметил)фенил)уксусной кислоты, например, посредством кристаллизации. В некоторых случаях вторую часть присутствующих региоизомерных второстепенных компонентов, которые переносятся посредством промежуточных композиций способа, являющегося предметом данного изобретения, удаляют посредством селективной реакции 2-(3-(трифторметил)фенил)уксусной кислоты, например, как описано в данном документе. В некоторых случаях третью часть присутствующих региоизомерных второстепенных компонентов, которые перенесены посредством промежуточных композиций способа, являющегося предметом данного изобретения, удаляют посредством очистки фенфлураминовой композиции.
Бензальдегид и трифторбензальдегид
[0058] В зависимости от способа получения 2-(3-(трифторметил)фенил)ацетонитрила, композиция исходного материала может включать бензальдегид или трифторбензальдегид в качестве второстепенного компонента. Нежелательно иметь присутствие такого второстепенного компонента в фармацевтическом активном ингредиенте. В некоторых случаях 2-(3-(трифторметил)фенил)ацетонитрильная композиция содержит по меньшей мере 0,2 мас.%, например, по меньшей мере 0,3%, по меньшей мере 0,4%, по меньшей мере 0,5%, по меньшей мере 1,0%, по меньшей мере 2%, по меньшей мере 5%, по меньшей мере 10% или даже более по массе бензальдегида или трифторбензальдегида в качестве второстепенного компонента. В некоторых случаях любой бензальдегид или трифторбензальдегид, который присутствует в качестве второстепенного компонента, в основном удаляют во время очистки, например, кристаллизацией, 2-(3-(трифторметил)фенил)уксусной кислоты из композиции, как описано в данном документе. В некоторых случаях бензальдегид не присутствует в исходной 2-(3-(трифторметил)фенил)-ацетонитрильной композиции вследствие способа ее получения.
Способ получения кетона (2)
[0059] Способы, являющиеся предметом данного изобретения, могут включать определенную комбинацию стадий получения кетона (2), которая предоставляет одно или несколько преимуществ по сравнению с другими возможными способами. Фиг. 4 иллюстрирует различные пути синтеза, которые могут быть применены для получения кетона (2). В некоторых случаях конкретным способом, который находит применение в способах, являющихся предметом данного изобретения, является получение кетона (2) из нитрила (5) посредством кислоты (4).
[0060] В способах, являющиеся предметом данного изобретения, второстепенные компоненты (например, примеси ацетата и димера), образованные во время реакция Дакина-Веста (например, как описано в Схеме 2), могут быть затем по существу удалены. В некоторых случаях эти второстепенные компоненты удаляют при применении процедуры дистилляции. В некоторых случаях эти второстепенные компоненты удаляют посредством процедуры, включающей отделение полученного кетона (2) в виде бисульфитной соли (например, как описано в данном документе). Примеси ацетата и димера представлены ниже.
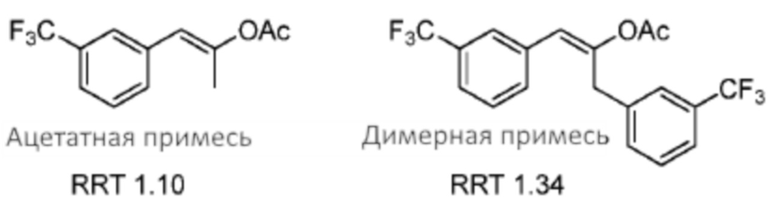
В некоторых случаях применение процедуры отделения в виде бисульфита улучшает чистоту кетона на по меньшей мере 30% (например, по меньшей мере 40%, по меньшей мере 50% или более) посредством удаления этих и других примесей. В некоторых вариантах осуществления способы, являющиеся предметом данного изобретения, предоставляют существенное удаление примеси ацетата из композиции кетона (2). В некоторых вариантах осуществления способы, являющиеся предметом данного изобретения, предоставляют существенное удаление примеси димера из композиции кетона (2).
[0061] Фиг. 5 иллюстрирует маршрут с диазонием для получения кетона (2) из исходного арилнитро-материала. Маршрут с диазонием имеет недостаток, обусловленный потенциальной возможностью формирования генотоксических промежуточных продуктов, показанных как заключенные в рамку соединения (например, N-гидроксиарил, N-нитрозамин и нитросоединение). В некоторых случаях удаление таких примесей и/или подтверждение их отсутствия является дорогостоящим и требующим много времени и иногда трудно достигается технически. Аспекты способов, являющихся предметом данного изобретения, включают маршрут синтеза, который по существу устраняет нежелательные второстепенные компоненты, который являются возможными посредством маршрута, показанного на Фиг. 5, устраняя тем самым потенциальную возможность присутствия таких токсичных и/или нежелательных соединений в композициях, являющихся предметом данного изобретения.
[0062] В некоторых случаях способы, являющиеся предметом данного изобретения, предоставляют удаление изомерных (например, региоизомерных) побочных продуктов исходного 3-трифторанилинового исходного материала, представленного на Фиг. 5. Такие побочные продукты могут присутствовать в 3-трифторанилиновой композиции, перенесенной на протяжении стадий синтеза, и являются затрудненными для существенного удаления из композиций ниже по потоку. В некоторых случаях способов, являющихся предметом данного изобретения, кристаллизация кислоты (4), результирующей от гидролиза нитрила (5), предоставляет кристаллическую кислоту (4) которая предоставляет легко достижимое удаление таких изомеров простым образом при синтезе. Удаление примесей и/или нежелательных изомеров в начале синтеза может быть предпочтительным, особенно, если такие примеси доводятся до конечных композиций продукта, поскольку очистка конечного продукта в конце синтеза является более дорогостоящей (например, при потерях ценного продукта) и оказывает большее влияние на стоимость товаров, чем удаление таких второстепенных компонентов в начале синтеза перед тем, как исходные материалы вводят во время процесса.
Удаленные токсичные реагенты
[0063] Способы, являющиеся предметом данного изобретения, включают конкретные путь синтеза и комбинацию химических реакций (например, как описано выше), которые предоставляют удаление определенных нежелательных реагентов и/или растворителей (например, растворителей Класса 1 или Класса 2, которые известны, или они в значительной степени подвергаются канцерогенной активности и/или являются вредо воздействующими на окружающую среду). Класс 1 и 2 растворителей, представляющих интерес, которые могут быть удалены из фенфлураминовой композиции посредством применяемых на практике способов, являющихся предметом данного изобретения, включают, однако без ограничения ими, любой растворитель, внесенный в реестр Международной конференции по гармонизации (ICH) Q3C и руководство для промышленности (Февраль 2012, Модификация 2, Департамент здравоохранения и социальных служб США), такой как ацетонитрил, бензол и замещенные бензолы, четыреххлористый углерод, хлороформ, циклогексан, 1,2-дихлорэтан, 1,1-дихлорэтан, 1,2-диметоксиэтан, диметилформамид (DMF), 1,4-диоксан, метанол, метилбутилкетон, N-метилпирролидинон, пиридин, толуол, 1,1,1-трихлорэтан, 1,1,2-трихлорэтен и ксилол. Способы, являющиеся предметом данного изобретения, также предоставляют удаление различных нежелательных и/или токсичны реагентов из фенфлураминовой композиции, которая получена посредством применяемых на практике способов, являющихся предметом данного изобретения. Например, посредством включения стадии восстановительного аминирования в соответствии со способом, описанным в Схеме 1, альтернативные пути синтеза, которые требуют применения потенциально токсичных металлических катализаторов, устраняются. Посредством исключения применения таких реагентов и/или растворителей из пути синтеза способов, являющихся предметом данного изобретения, потенциально токсичные второстепенные компоненты удаляют из фенфлураминовой композиции. В качестве таковой, фенфлураминовая композиция, являющаяся предметом данного изобретения, может относиться к композиции, которая по существу не содержит второстепенного компонента, представляющего интерес. В некоторых случаях один или более потенциальных компонентов тяжелых металлов, таких как Pb, As, Cd, Hg, Pb, Co, Mo, Se и V, по существу удалены. В некоторых случаях один или более растворителей Класса 1 по существу удалены (например, ниже допустимого порогового значения, как указано в реестре Международной конференции по гармонизации (ICH) Q3C). В некоторых случаях растворитель, являющийся бензолом, по существу удален, например, ниже предельной концентрации 2 млн-1. В некоторых случаях растворитель, являющийся четыреххлористым углеродом, по существу удален, например, ниже предельной концентрации 4 млн-1. В некоторых случаях растворитель, являющийся 1,2-дихлорэтаном, по существу удален, например, ниже предельной концентрации 5 млн-1. В некоторых случаях растворитель, являющийся 1,2-дихлорэтаном, по существу удален, например, ниже предельной концентрации 8 млн-1. В некоторых случаях растворитель, являющийся 1,1,1-трихлорэтаном, по существу удален, например, ниже предельной концентрации 1500 млн-1. Второстепенный компонент может считаться полностью удаленным из композиций, являющихся предметом данного изобретения, когда фенфлурамин получают способом, где второстепенный компонент не применяют на любой стадии синтеза или не присутствует в исходном материале.
Фенфлураминовый спирт
[0064] Как использовано в данном документе, термины «фенфлураминовый спирт» и «спиртовый побочный продукт восстановления» применяют взаимозаменяемым образом в отношении продукта восстановления кетона до спирта, которое может происходить на стадии восстановительного аминирования Схемы 1, как изображено ниже.
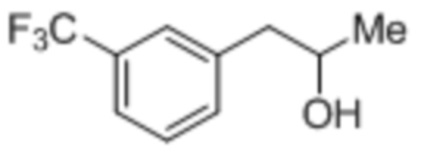
[0065] Способы, являющиеся предметом данного изобретения, предоставляют существенное удаление фенфлураминового спирта из композиций, являющихся предметом данного изобретения. В некоторых случаях исходная неочищенная фенфлураминовая композиция содержит менее чем 10 мас.% спиртового побочного продукта восстановления, например, менее чем 9%, менее чем 8%, менее чем 7%, менее чем 6%, менее чем 5%, менее чем 4%, менее чем 3%, менее чем 2%, менее чем 1%, менее чем 0,9%, менее чем 0,8%, менее чем 0,7%, менее чем 0,6%, менее чем 0,5%, менее чем 0,4%, менее чем 0,3%, менее чем 0,2%, менее чем 0,1%, менее чем 0,05% или даже менее. В некоторых случаях исходная неочищенная фенфлураминовая композиция имеет 10% или менее по массе спиртового побочного продукта восстановления, например, 9% или менее, 8% или менее, 7% или менее, 6% или менее, 5% или менее, 4% или менее, 3% или менее, 2% или менее, 1% или менее, 0,9% или менее, 0,8% или менее, 0,7% или менее, 0,6% или менее, 0,5% или менее, 0,2% или менее, 0,1% или менее, 0,05% или менее или даже менее.
Норфенфлурамин
[0066] Норфенфлурамин является потенциальной присмесью композиций, котоые включают фенфлурамин. Способы, являющиеся предметом данного изобретения, предоставляют существенное удаление норфенфлурамина из композиций, являющихся предметом данного изобретения. В некоторых случаях исходная неочищенная фенфлураминовая композиция имеет менее чем 10 мас.% норфенфлурамина, например, менее чем 9%, менее чем 8%, менее чем 7%, менее чем 6%, менее чем 5%, менее чем 4%, менее чем 3%, менее чем 2%, менее чем 1%, менее чем 0,9%, менее чем 0,8%, менее чем 0,7%, менее чем 0,6%, менее чем 0,5%, менее чем 0,4%, менее чем 0,3%, менее чем 0,2%, менее чем 0,1%, менее чем 0,05% или даже менее. В некоторых случаях исходная неочищенная фенфлураминовая композиция включает 10% или менее по массе норфенфлурамина, например, 9% или менее, 8% или менее, 7% или менее, 6% или менее, 5% или менее, 4% или менее, 3% или менее, 2% или менее, 1% или менее, 0,9% или менее, 0,8% или менее, 0,7% или менее, 0,6% или менее, 0,5% или менее, 0,4% или менее, 0,3% или менее, 0,2% или менее, 0,1% или менее, 0,05% или менее, или даже менее.
Способы применения
[0067] Фенфлурамин и фенфлураминовые композиции, описанные в данном документе, могут быть применены в различных способах. Аспекты данного изобретения включают способ, который включает применение к субъекту, для которого это необходимо, терапевтически эффективного количества фенфлурамина в фармацевтической композиции (например, как описано в данном документе), чтобы лечить или предотвращать болезнь или нежелательное состояние. Посредством «терапевтически эффективного количества» означает концентрацию соединения, которая является достаточной, чтобы вызывать достаточное биологическое действие (например, обработку или предотвращение эпилепсии). Болезни и состояния, представляющие интерес, включают, однако без ограничения ими, эпилепсию, неврологические болезни, ожирение и болезни, связанные с ожирением.
[0068] В некоторых вариантах осуществления способ, являющийся предметом данного изобретения, включает применение к субъекту композиции, являющейся предметом данного изобретения, чтобы лечить неврологическое заболевание. Неврологические заболевания, представляющие интерес, включают, однако без ограничения ими, эпилепсию и синдром Драве. В определенных вариантах осуществления, субъектом является человек. В некоторых случаях субъект страдает от синдрома Драве. В определенных вариантах осуществления соединение применяют в качестве фармацевтического препарата.
[0069] Таким образом, в соответствии с еще одним аспектом данного изобретения, предоставлен способ стимулирования одного или нескольких 5-HT рецепторов в мозге пациента посредством применения эффективной дозы фенфлураминовой композиции указанному пациенту, указанные один или несколько 5-HT рецепторов выбраны из одного или нескольких из 5-HT1, 5-HT1A, 5-HT1B, 5-HT1C, 5-HT1D, 5-HT1E, 5-HT1F, 5-HT2, 5-HT2A, 5-HT2B, 5-HT2C, 5-HT3, 5-HT4, 5-HT5, 5-HT5A, 5-HT5B, 5-HT6 и 5-HT7, в частности. В некоторых случаях 5-HT рецептором является 5-HT2B. В определенных вариантах осуществления этого аспекта данного изобретения у пациента был диагностирован синдром Драве. В некоторых случаях способ является способом лечения синдрома Драве, который включает стимулирование одного или нескольких 5-HT рецепторов в мозге пациента посредством применения эффективной дозы фенфлураминовой композиции к указанному пациенту, указанные один или несколько 5-HT рецепторов выбраны из одного или нескольких из 5-HT1D, 5-HT2A и 5-HT2C, в частности.
[0070] Имеется ряд генетических мутаций, которые служат признаком синдрома Драве. Мутации в SCN1A (такие как частичные или полные делеционные мутации, усекающие мутации и/или миссенс-мутации, например, в потенциале или областях пор S4 по S6), SCN1B (например, области, кодирующей натриевый канал β1-субъединицы), SCN2A, SCN3A, SCN8A, SCN9A, GABRG2 (например, области, кодирующей γ2- субъединицу), GABRD (например, области, кодирующей δ субъединицу) и/или генах PCDH19, связанных с синдромом Драве.
[0071] Таким образом, в соответствии с другим аспектом данного изобретения, предоставлен способ лечения пациента, который проявляет мутацию в одной, некоторых или всех вышеуказанных генах, посредством применения к этому пациенту эффективной дозы фенфлураминовой композиции. В определенных вариантах осуществления этого аспекта данного изобретения у пациента был диагностирован синдром Драве.
[0072] В вариантах осуществления данного изобретения может быть применена любая эффективная доза фенфлураминовой композиции. Однако неожиданным образом низкие дозы фенфлураминовых композиций найдены авторами данного изобретения как являющиеся эффективными, конкретно для сдерживания или устранения острых приступов эпилепсии пациентов. Соответственно, в некоторых случаях в предпочтительном варианте осуществления данного изобретения, применяют суточную дозу менее чем примерно 10 мг/кг/день, например, менее чем примерно 9 мг/кг/день, менее чем примерно 8 мг/кг/день, менее чем примерно 7 мг/кг/день, менее чем примерно 6 мг/кг/день, менее чем примерно 5 мг/кг/день, менее чем примерно 4 мг/кг/день, менее чем примерно 3 мг/кг/день, менее чем примерно 2 мг/кг/день, менее чем примерно 1 мг/кг/день, например, примерно 1,0 мг/кг/день, примерно 0,9 мг/кг/день, примерно 0,8 мг/кг/день, примерно 0,7 мг/кг/день, примерно 0,6 мг/кг/день, примерно 0,5 мг/кг/день, примерно 0,45 мг/кг/день, примерно 0,4 мг/кг/день, примерно 0,3 мг/кг/день, примерно 0,25 мг/кг/день или примерно 0,2 мг/кг/день до примерно 0,1 мг/кг/день, примерно 0,05 мг/кг/день или примерно 0,01 мг/кг/день. Иными словами, предпочтительная доза составляет от менее чем примерно 10 мг/кг/день до примерно 0,01 мг/кг/день. В некоторых случаях доза составляет от менее чем примерно 5 мг/кг/день до примерно 0,1 мг/кг/день, например, от менее чем примерно 5 мг/кг/день до примерно 0,5, мг/кг/день, от менее чем примерно 4 мг/кг/день до примерно 0,5 мг/кг/день, от менее чем примерно 3 мг/кг/день до примерно 0,5 мг/кг/день, от менее чем примерно 2 мг/кг/день до примерно 0,5 мг/кг/день или от менее чем примерно 1,7 мг/кг/день до примерно 0,9 мг/кг/день.
[0073] Как указано выше, дозирование основано на весе пациента. Однако в целях удобства дозированные количества могут быть заранее установлены, например, при количестве 1 мг, 2.5 мг, 5 мг, 10 мг, 15 мг, 20 мг, 30 мг, 40 мг или 50 мг. В некоторых случаях дозированное количество может быть заранее установлено, например, при количестве от примерно 0,25 мг до примерно 5 мг, таком как примерно 0,5 мг, примерно 0,75 мг, примерно 1,0 мг, примерно 1,25 мг, примерно 1,5 мг, примерно 1,75 мг, примерно 2,0 мг, примерно 2,25 мг, примерно 2,5 мг, примерно 2,75 мг, примерно 3,0 мг, примерно 3,25 мг, примерно 3,5 мг, примерно 3,75 мг, примерно 4,0 мг, примерно 4,25 мг, примерно 4,5 мг, примерно 4,75 мг или примерно 5,0 мг. Дозированные количества, описанные в данном документе, могут быть применены один или несколько раз ежесуточно, чтобы предоставлять ежесуточное дозированное количество, например, один раз в сутки, два раза в сутки, три раза в сутки или четыре или более раз в сутки, и т.п. В определенных вариантах осуществления дозированное количество является суточной дозой 30 мг или менее, например, 30 мг, примерно 29 мг, примерно 28 мг, примерно 27 мг, примерно 26 мг, примерно 25 мг, примерно 24 мг, примерно 23 мг, примерно 22 мг, примерно 21 мг, примерно 20 мг, примерно 19 мг, примерно 18 мг, примерно 17 мг, примерно 16 мг, примерно 15 мг, примерно 14 мг, примерно 13 мг, примерно 12 мг, примерно 11 мг, примерно 10 мг, примерно 9 мг, примерно 8 мг, примерно 7 мг, примерно 6 мг, примерно 5 мг, примерно 4 мг, примерно 3 мг, примерно 2 мг или примерно 1 мг. Обычно наименьшая доза, которая является эффективной, должна быть применена для конкретного пациента. В некоторых случаях доза является обычно значительно ниже дозирования, применяемого при потере в весе.
[0074] Применение для субъекта фармацевтических композиций может быть системным или локальным. В определенных вариантах осуществления применение к млекопитающему будет приводить к системному высвобождению фенфлурамина (например, в кровоток). Способы применения могут включать энтеральные маршруты, такие как оральный, буккальный, сублингвальный и ректальный; топическое применение, такое как трансдермальное и интрадермальное; и парентеральное применение. Подходящие парентеральные маршруты включают инъекцию посредством иглы для подкожных инъекций или катетера, например, внутривенную, внутримышечную, подкожную, интрадермальную, интраперитонеальную, внутриартериальную, интравентрикулярную, интратекальную и интракамеральную инъекцию, и безынъекционные маршруты, такие как интравагинальное, ректальное или назальное применение. В определенных вариантах осуществления композиции данного изобретения применяют перорально. В определенных вариантах осуществления может являться желательным применение одного или нескольких компонентов по данному изобретению локальным образом для области, нуждающейся в лечении. Это может быть достигнуто, например, посредством локального введения во время наружного применения, посредством инъекции, посредством катетера, посредством суппозитория или посредством имплантата, указанный имплантат является пористым, непористым или желатинозным материалом, включающим мембраны, такие как сиаластиковые мембраны, или волокна.
[0075] Доза фенфлурамина, примененного в способах по данному изобретению, может быть образована в любой фармацевтически приемлемой лекарственной форме, включающей, однако не ограничиваемой ими, оральные лекарственные формы, такие как таблетки, включая перорально распадающиеся таблетки, капсулы, леденцы, оральные растворы или сиропы, оральные эмульсии, оральные гели, оральные пленки, трансбуккальные жидкости, порошок, например, для суспензии, и т.п.; инъецируемые лекарственные формы; трансдермальные лекарственные формы, такие как трансдермальные пластыри, мази, кремы; ингаляционные лекарственные формы; и/или назально, ректально, вагинально применяемые лекарственные формы. Такие лекарственные формы могут быть образованы в составе для применения один раз в сутки или для применения несколько раз в сутки (например, применения 2, 3 или 4 раза в сутки).
[0076] В некоторых вариантах осуществления способ, являющийся предметом данного изобретения, включает применение к субъекту аппетитоподавляющее количество соединения, являющегося предметом данного изобретения, чтобы лечить ожирение. Любой из способов применения и лекарственные формы композиций, являющихся предметом данного изобретения, могут быть применены для лечения ожирения.
[0077] Комбинированная терапия включает применение единственного фармацевтического лекарственного состава, который содержит композицию, являющуюся предметом данного изобретения, и один или более дополнительных агентов; а также применение композиции, являющейся предметом данного изобретения, и один или более дополнительных агентов в их собственном отдельном фармацевтическом лекарственном составе. Например, композиция, являющаяся предметом данного изобретения, и дополнительный агент, активный в отношении аппетитоподавляющего действия, (например, фентермин или топирамат) могут быть применены к пациенту в одной лекарственной композиции, такой как комбинированное лекарственное средством, или каждый агент может быть применен в отдельном лекарственном составе. Когда применяют отдельные лекарственные составы, композиция, являющаяся предметом данного изобретения, и один или более дополнительных агентов могут быть применены одновременно или в отдельные смещенные моменты времени, например, последовательно.
[0078] В некоторых вариантах осуществления способ, являющийся предметом данного изобретения, является способом in vitro, который включает контактирование пробы с композицией, являющейся предметом данного изобретения. Протоколы, которые могут быть применены в этих способах, являются многочисленными и включают, однако без ограничения ими, анализы выделения серотонина из нейронных клеток, бесклеточные анализы, анализы связующих проб (например, анализы связывания рецептора 5HT2B); клеточные анализы, в которых определяют клеточный фенотип, например, анализы экспрессии генов; и анализы, которые включают конкретную экспериментальную модель болезни на животном для состояния, представляющего интерес, (например, в отношении синдрома Драве).
Фармацевтические препараты
[0079] Также предоставлены фармацевтические препараты, которые включают композиции с фенфлураминовым активным фармацевтическим ингредиентом, полученные в соответствии со способами, являющимися предметом данного изобретения. Фармацевтические препараты являются композициями, которые включают соединение (или само по себе или при наличии одного или нескольких дополнительных активных агентов), присутствующее в фармацевтически приемлемой среде. В некоторых вариантах осуществления фармацевтическая композиция включает фенфлураминовую композицию (например, как описано в данном документе), образованную в составе фармацевтически приемлемого вспомогательного вещества.
[0080] Выбор вспомогательного вещества будет определяться отчасти конкретным соединением, а также конкретным способом, применяемым для введения композиции. Соответственно, имеет место широкий выбор подходящих составов фармацевтической композиции по данному изобретению.
[0081] Лекарственная форма фенфлурамина, применяемая в способах по данному изобретению может быть получена посредством комбинирования фенфлураминовой композиции с одним один или более фармацевтически приемлемыми разбавителями, носителями, адъювантами и т.п. таким образом, который известен специалистам в данной области фармацевтических составов.
[0082] Композиции, являющиеся предметом данного изобретения, могут быть образованы в виде составов для инъекции посредством растворения, суспендирования или эмульгирования их в водном или неводном растворителе, таком как растительные или другие подобные масла, синтетические глицериды алифатической кислоты, сложные эфиры высших алифатических кислот или пропиленгликоля; и, если желательно, с обычными добавками, такими как солюбилизаторы, изотонические агенты, суспендирующие агенты, эмульгирующие агенты, стабилизаторы и консерванты.
[0083] В некоторых вариантах осуществления составы, подходящие для орального применения, могут включать (a) жидкие растворы, такие как эффективное количество соединения, растворенного в разбавителях, например, воде или солевом растворе; (b) капсулы, саше или таблетки, содержащие заданные количества активного ингредиента (фенфлурамина), в виде твердых части или гранул; (c) суспензии в соответствующей жидкости; и (d) подходящие применимые эмульсии. Таблетированные формы могут включать один или более компонентов из лактозы, маннитола, кукурузного крахмала, картофельного крахмала, микрокристаллической целлюлозы, гуммиарабика, желатина, коллоидного диоксида кремния, кроскармеллозы натрия, талька, стеарата магния, стеариновой кислоты и других вспомогательных веществ, красителей, разбавителей, буферных агентов, увлажняющих средств, консервантов, ароматизирующих веществ и фармакологически совместимых вспомогательных веществ. Леденцовые формы могут включать активный вкусовой ингредиент, обычно сахарозу и гуммиарабик или трагакантовую камедь, а также пастилки, включающие активный ингредиент в инертной основе, такой как желатин и глицерин или сахарозу и гуммиарабик, эмульсии, гели и т.п., содержащие, в дополнение к активному ингредиенту, такие вспомогательные вещества как те, что описаны в данном документе.
[0084] Составы для субъекта могут быть изготовлены в виде аэрозольных составов для применения посредством ингаляции. Эти аэрозольные составы могут быть помещены в находящиеся под давлением приемлемые пропелленты, такие как дихлордифторметан, пропан, азот и т.п. Они могут также быть образованы в качестве лекарственных средств для препаратов, не находящихся под давлением, таких как для применения в ингаляторе или распылителе.
[0085] В некоторых вариантах осуществления составы, подходящие для парентерального применения, включают водные и неводные, изотонические стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостаты и растворенные вещества, которые предоставляют состав, изотоничный крови целевого реципиента, и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты, солюбилизаторы, загустители, стабилизаторы и консерванты. Составы могут быть представлены в однодозовых и многодозовых герметичных контейнерах, таких как ампулы и пузырьки, и могут храниться в высушенном сублимацией (лиофилизированном) состоянии, требующем лишь добавления стерильного жидкого вспомогательного вещества, например, воды, для инъекций, непосредственно перед применением. Экстемпоральные растворы для инъекций и суспензии могут быть получены из стерильных порошков, гранул и таблетки вида, описанного ранее.
[0086] Составы, подходящие для топического применения, могут быть представлены в качестве кремов, гелей, паст или пен, содержащих, в дополнение к активному ингредиенту, такие носители, которые являются подходящими. В некоторых вариантах осуществления топический состав содержит один или более компонентов, выбранных из структурирующего агента, загустителя или желатинирующего агента, и смягчающее вещество или смазывающее вещество. Часто применяемые структурирующие агенты включают спирты с длинной молекулярной цепью, такие как стеариловый спирт, и глицериловые простые или сложные эфиры и простые или сложные эфиры олигоэтиленоксида. Загустители и желатинирующие агенты включают, например, полимеры акриловой или метакриловой кислоты и ее сложные эфиры, полиакриламиды, и загустители природного происхождения, такие как агар, каррагенан, желатин и гуаровая смола. Примеры смягчающих веществ включают триглицеридные сложные эфиры, сложные эфиры и амиды жирной кислоты, воски, такие как пчелиный воск, спермацет, или карнаубский воск, фосфолипиды, такие как лецитин, и стероидные спирты и их сложные эфиры с жирной кислотой. Топические составы могут дополнительно включать другие компоненты, например, вяжущие средства, ароматизирующие вещества, пигменты, агенты для улучшения проникновения в роговой слой кожи, средства для защиты от солнечных ожогов (например, солнцезащитные агенты) и т.п.
[0087] Для оральных фармацевтических составов подходящие вспомогательные вещества включают фармацевтические категории носителей, такие как маннитол, лактоза, глюкоза, сахароза, крахмал, целлюлоза, желатин, стеарат магния, сахарин натрия и/или карбонат магния. Для применения в оральных жидких составах композиция может быть приготовлена в виде раствора, суспензии, эмульсии или сиропа и поставляться в твердотельной или жидкой форме, подходящей для гидратации в водном носителе, таком как, например, водный солевой раствор, водный раствор декстрозы, глицерин или этанол, предпочтительно воде или физиологическом растворе поваренной соли. Если желательно, композиция может также содержать небольшие количества нетоксичных вспомогательных веществ, таких как смачивающие агенты, эмульгирующие агенты или буферы.
[0088] В качестве иллюстрации, фенфлураминовая композиция может быть примешана к обычным фармацевтически приемлемым носителям и вспомогательным веществам (т.е. средам) и применена в форме водных растворов, таблеток, капсул, эликсиров, суспензий, сиропов, вафель и т.п. Такие фармацевтические композиции содержат, в определенных вариантах осуществления, от примерно 0,1% до примерно 90 мас.% активного соединения, и обычно от примерно 1% до примерно 30 мас.% активного соединения. Фармацевтические композиции могут содержать обычные носители и вспомогательные вещества, такие как кукурузный крахмал или желатин, лактоза, декстроза, сахароза, микрокристаллическая целлюлоза, каолин, маннитол, дикальцийфосфат, хлорид натрия и альгиновая кислота. Разрыхлители, обычно применяемые в составах по данному изобретению, включают кроскармеллозу, микрокристаллическую целлюлозу, кукурузный крахмал, натрия крахмалгликолят и альгиновую кислоту.
[0089] Конкретные составы данного изобретения находятся в жидкой форме. Жидкость может являться раствором или суспензией и может быть оральным раствором или сиропом, который включают во флакон посредством пипетки, которая градуирована, исходя из количеств в миллиграммах, которые будут получены в данном объеме раствора. Жидкий раствор делает возможным регулирование раствора для маленьких детей, который может быть применен в пределах от 0,5 мг до 15 мг и при любом количестве между ними при приращениях в половину миллиграмма и, соответственно, применен при 0,5, 1,0, 1,5, 2,0 мг и т.п.
[0090] Жидкая композиция будет обычно состоять из суспензии или раствора соединения или фармацевтически приемлемой соли в одном или нескольких подходящих жидких носителях, например, в этаноле, глицерине, сорбитоле, неводном растворителе, таком как полиэтиленгликоль, маслах или воде, вместе с суспендирующим агентом, консервантом, поверхностно-активным веществом, смачивающим агентом, ароматизирующим веществом или окрашивающей добавкой. В качестве альтернативы, жидкий состав может быть получен из ресуспендируемого или повторно растворяемого порошка.
ПРИМЕРЫ
[0091] Представленные ниже примеры приведены для того, чтобы предоставить средним специалистам в данной области полное раскрытие и описание того, каким образом реализовать и применить данное изобретение, и не предназначены для ограничения объема того, что авторы изобретения рассматривают в качестве своего изобретения, и они не предназначены представлять то, что эксперименты, приведенные ниже, являются всеми или единственными проведенными экспериментами. Были приняты меры, чтобы обеспечить точность по отношению к примененным числам (например, количествам, температуре и т.д.), однако некоторые экспериментальные ошибки и отклонения должны приниматься во внимание. Если не указано иное, части являются частыми по массе, молекулярная масса является среднемассовой молекулярной массой, температура выражена в градусах по Цельсию, и давление находится при атмосферном давлении или вблизи него. Под «средней величиной» понимают среднее арифметическое значение. Стандартные аббревиатуры могут быть применены, например, с или сек, секунда(ы); мин., минута(ы); ч, час(ы); ак, аминокислота(ы); i.m., внутримышечная(но); i.p., интраперитонеальная(но); s.c., подкожная(но); и т.п.
Общие процедуры синтеза
[0092] Многие общие ссылки, предоставляющие общеизвестные схемы химического синтеза и условия, применимые для синтеза раскрытых соединений являются доступными (см., например, Smith and March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure («Органическая химия: Реакции, механизмы и структура»), Fifth Edition, Wiley-Interscience, 2001; или Vogel, A Textbook of Practical Organic Chemistry, Including Qualitative Organic Analysis («Учебник практической органической химии, включающий качественный органический анализ»), Fourth Edition, New York: Longman, 1978).
[0093] Соединения как описано в данном документе, могут быть очищены посредством любого протокола очистки, известного в данной области, включая хроматографию, такую как высокоэффективная жидкостная хроматография (HPLC), препаративная тонкослойная хроматография, колоночная флэш-хроматография и ионообменная хроматография. Любая подходящая неподвижная фаза может быть применена, включая нормальную и обращенную фазы, а также ионные смолы. В определенных вариантах осуществления раскрытые соединения очищают посредством хроматографии на силикагеле и/или на глиноземе. См., например, Introduction to Modern Liquid Chromatography («Введение в современную жидкостную хроматографию»), 2nd Edition, ed. L. R. Snyder and J. J. Kirkland, John Wiley and Sons, 1979; и Thin Layer Chromatography («Тонкослойная хроматография»), ed E. Stahl, Springer-Verlag, New York, 1969.
[0094] Во время любого из способов получения соединений, являющихся предметом данного изобретения, может быть необходима и/или желательна защита чувствительных или реакционноспособных групп в любой из участвующих молекул. Это может быть достигнуто посредством обычных защитных групп, таких как группы, описанные в стандартных работах, например, в J. F. W. McOmie, ʺProtective Groups in Organic Chemistryʺ («Защитные группы в органической химии»), Plenum Press, London and New York 1973, в T. W. Greene and P. G. M. Wuts, ʺProtective Groups in Organic Synthesisʺ («Защитные группы в органическом синтезе»), Third edition, Wiley, New York 1999, в ʺThe Peptides (Пептиды)ʺ; Volume 3 (editors: E. Gross and J. Meienhofer), Academic Press, London and New York 1981, в ʺMethoden der organischen Chemie» («Методы органической химии»), Houben-Weyl, 4th edition, Vol. 15/1, Georg Thieme Verlag, Stuttgart 1974, в H.-D. Jakubke and H. Jescheit, ʺAminosauren, Peptide, Proteineʺ («Аминокислоты, пептиды, протеины»), Verlag Chemie, Weinheim, Deerfield Beach, and Basel 1982, и/или в Jochen Lehmann, ʺChemie der Kohlenhydrate: Monosaccharide and Derivateʺ («Химия углеводов: Моносахариды и производные»), Georg Thieme Verlag, Stuttgart 1974. Эти защитные группы могут быть удалены на подходящей последующей стадии при применении способов, известных в данной области.
[0095] Соединения, являющиеся предметом данного изобретения, могут быть синтезированы посредством ряда различных маршрутов синтеза при применении коммерчески доступных исходных материалов и/или исходных материалов, полученных обычными способами синтеза. Ряд примеров маршрутов синтеза, которые могут быть применены, чтобы синтезировать соединения, раскрытые в данном документе, описаны в схемах ниже.
ПРИМЕР 1
1. Номенклатура и структура фенфлурамина
[0096] Регистрационный номер (RN) Химической реферативной службы (CAS): 404-82-0 (Соль HCl), 458-24-2 (Исходное свободное основание)
[0097] Химическое наименование: N-этил-α-метил-3-(трифторметил)-бензолэтанамин (в виде гидрохлорида) (1:1). Другие наименования: Фенфлурамин HCl, DL-Фенфлурамин, (±)-Фенфлурамин
[0098] Структура хлористоводородной соли:

[0099] Стереохимия: Фенфлурамин HCl имеет лишь один хиральный центр и разработан в качестве рацемата и содержит дексфенфлурамин и левофенфлурамин
[00100] Молекулярная формула хлористоводородной соли: C12H16F3N • HCl
[00101] Молекулярная масса/Молекулярный вес: 267,72 г/моль
2. Общие свойства
[00102] Таблица 1 суммирует химические и физические свойства фенфлурамина HCl.
Таблица 1: Общие свойства фармацевтической субстанции фенфлурамина HCl
0,03% при 150°C
91% при 220°C (испарение)
(Динамическая сорбция паров (DVS))
При относительной влажности (RH) 60%: ~0,07%
При относительной влажности (RH) 90%: ~0,20%a)
3. Синтез фармацевтической субстанции фенфлурамина
[00103] Схема 3.1 показывает двухстадийный маршрут синтеза, применяемый для получения первичных клинических поставок фенфлурамина HCl из кетона (2). Партию в размере 4 кг производили в лабораторной стеклянной посуде (в масштабе полупромышленного производства). Хроматография не требуется, и стадии процесса могут быть увеличены в масштабе. В процессе 1 имеется одно отделенное промежуточное свободное основание фенфлурамина (1), производное от коммерчески доступного 1-(3-(трифторметил)фенил)ацетона (Кетона 2). Все стадии выполняют при условиях текущей надлежащей производственной практики (cGMPs), начиная от Кетона (2).
[00104] Схема 3.1 Схема протекания синтеза фенфлурамина HCl (Маршрут 1)

MTBE=Метил-трет-бутиловый эфир, EtOAc=Этилацетат.
[00105] Схема 3.2 показывает четырехстадийный маршрут синтеза до фенфлурамина HCl, который может быть применен для коммерческих поставок. Маршрут 2 использует тот же самый двухстадийный процесс, что использован посредством Маршрута 1, чтобы преобразовывать Кетон (2) до фенфлурамина HCl, за исключением того, что Кетон (2) синтезируют при условиях текущей надлежащей производственной практики (cGMP), начиная от 3-(трифторметил)-фенилуксусной кислоты (Кислота 4). Бисульфатный комплекс (3) является отделяемым твердотельным веществом и может быть очищен перед декомплексацией до Кетона (2). Промежуточные продукты in-situ, которые являются маслами, показаны в квадратных скобках. Производили партии в размере 10 кг. Коммерческие партии в размере 20 кг производили в стационарной опытно-промышленной установке. Стадии 1-2 Схемы 3.2 для производства Кетона (2) продемонстрировали в масштабе 100 г предоставление кетона (2) высокой чистоты >99,8% (газовая хроматография (GC) и высокоэффективная жидкостная хроматография (HPLC)). Преобразование Кетона (2) в Фенфлурамин при применении Маршрута 1 или 2 предоставляло аналогичные профили чистоты.
[00106] Схема 3,2 Схема протекания синтеза фенфлурамина HCl (Маршрут 2)
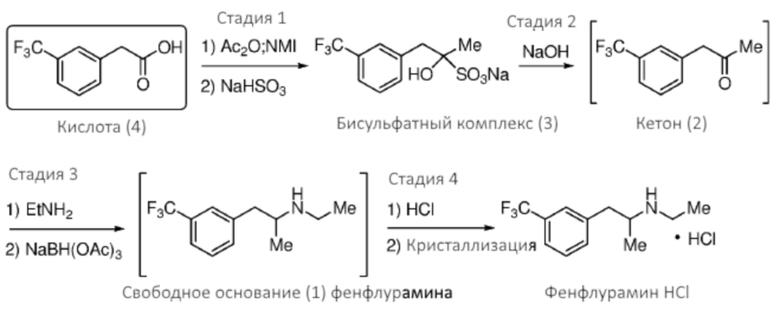
Исходные материалы обозначены прямоугольными контурами. Соединения, заключенные в скобки, и без скобок соответственно означают предполагаемые промежуточные продукты in-situ и отделенные. NMI=N-Метилимидазол.
4.1. Пояснительное описание (Маршрут 1)
[00107] Стадия 1: Восстановительное аминирование (Получение свободного основания фенфлурамина 1)
[00108] Раствор этиламина, воды, метанола и 1-(3-(трифторметил)фенил)ацетон (Кетона 2) обрабатывали триацетоксиборогидридом натрия и перемешивали в течение 16 ч при 25 °C, при данном времени анализ высокоэффективной жидкостной хроматографией (HPLC) (IPC-1; Контроль в процессе № 1) показал завершение реакционного взаимодействия, и раствор гидроксида натрия добавляли до достижения pH >10. Добавляли толуол и разделяли фазы, и водную фазу (IPC-2) и органическую фазу (IPC-3) проверяли на остаточный фенфлурамин и фенфлураминовый спирт, и органическую фазу уменьшали. Добавляли очищенную воду и pH регулировали до <2 при применении концентрированной HCl, и фазы разделяли. Водную фазу промывали толуолом, и фазу толуола (IPC-4) и the водную фазу (IPC-5) проверяли на содержание фенфлурамина и фенфлураминового спирта. Величину pH водной фазы, содержащей продукт, регулировали до >10 при применении раствора гидроксида натрия. Щелочную водную фазу извлекали посредством метил-трет-бутилового эфира (MTBE) до тех пор, пока удаление фенфлурамина из водной фазы не было определено высокоэффективной жидкостной хроматографией (HPLC) (<0,5 мг/мл) (IPC-6). Органическую фазу сушили поверх сульфата натрия и фильтровали. Фильтрат концентрировали в вакууме, чтобы предоставить промежуточный продукт Свободное основание фенфлурамина 1 в виде бледно-желтого масла, подвергнутого испытаниям в соответствии со спецификациями, описанными в данном документе, которые показали посредством ЯМР, что материал содержит 2,93% толуола, предоставляя активный выход 88,3%, с чистотой 98,23% при определении посредством высокоэффективной жидкостной хроматографии (HPLC) (0,67% фенфлураминового спирта).
[00109] Стадия 2: Формирование соли (Получение фенфлурамина HCl)
[00110] В колбу загружали этанол и ацетилхлорид. Раствор медленно перемешивали на протяжении ночи перед тем, как добавляли этилацетат. HCl в сформированном растворе этилацетата отфильтровывали фильтром тонкой очистки в чистую бутыль и оставляли для последующего применения. В сосуд добавляли Свободное основание фенфлурамина 1 и метил-трет-бутиловый эфир (MTBE). Раствор фенфлурамина раствор в метил-трет-бутиловом эфире (MTBE) собирали в две бутыли перед тем, как сосуд был очищен и проверен на остаток частиц. Раствор фенфлурамина отфильтровывали фильтром тонкой очистки в сосуд и охлаждали и добавляли HCl в раствор этилацетата, предоставляя конечную величину pH 6-7. Партию перемешивали в течение 1 ч и фильтровали. Продукт сушили в вакууме при 40°C. Продукт (выход 96,52%), подвергнутый тестированию в соответствии с IPC-7, имел чистоту 99,75% при применении высокоэффективной жидкостной хроматографии (HPLC), и анализ парофазной газовой хроматографией (GC-HS) показал, что присутствуют метил-трет-бутиловый эфир (MTBE) (800 млн-1) и этилацетат (EtOAc) (150 млн-1). Продукт затем тестировали в соответствии со спецификациями, представленными в данном документе.
4.2. Пояснительное описание (Маршрут 2)
[00111] Стадия 1: Получение кетонового бисульфитного аддукта
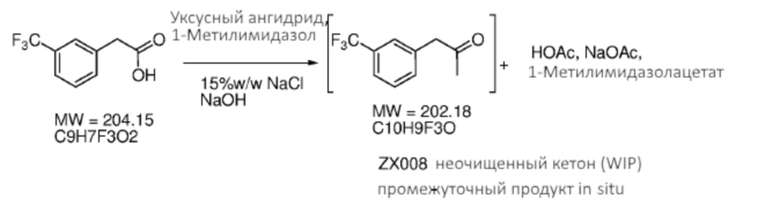

[00112] Процедура: Загружают уксусный ангидрид (2,8 по объему, 3,0 по массе, 5,0 экв.) в сосуд и начинают перемешивание. Охлаждают раствор до температуры от -5 до 5°C, целевой температуры -4°C. Загружают 1-метилимидазол, (0,2 по объему, 0,21 по массе, 0,5 экв.) в смесь при от -5 до 5°C. Предупреждение: очень экзотермическая реакция. При необходимости, температуру регулируют до 0-5°C. Загружают кислоту ZX008 (1,00 по массе, 1,0 экв.) в смесь при при 0-5°C. Предупреждение: экзотермическая реакция. Перемешивают смесь при 0-5°C до ≤2,1% площади кислоты ZX008 при анализе высокоэффективной жидкостной хроматографией (HPLC), типично от 7 до 9 часов. Загружают 15 мас.%-ный раствор хлорида натрия раствор (2,0 по объему) в смесь при 0-5°C, в течение от 60 до 90 минут. Предупреждение: очень экзотермическая реакция, которая будет немного замедляться. Нагревают смесь до 18-23°C на протяжении 45-60 минут и продолжают перемешивание в течение дополнительных 30-45 минут при 18-23°C. Загружают трет-бутилметиловый эфир (TBME), (5,0 по объему, 3,7 по массе) в смесь и перемешивают в течение 10-15 минут при 18-23°C. Отделяют водный слой и оставляют органический слой. После этого экстрагируют водный слой трет-бутилметиловым эфиром (TBME) (2×3,0 по объему, 2×2,2 по массе) при 18-23°C, оставляя каждый органический слой. Регулируют pH комбинированного органического слоя до pH 6,5-9,0, при целевом значении 7,0 посредством загрузки 20 мас.%-ного раствора гидроксида натрия (от 5,3 до 8,3 по объему) при 18-23°C. Предупреждение: экзотермическая реакция. Отделяют водный слой и оставляют органический слой. Промывают органический слой 4 мас.%-ным раствором гидрокарбоната натрия (2×3,0 по объему) при 18-23°C. Определяют остаточное содержание кислоты ZX008 в органическом слое посредством анализа высокоэффективной жидкостной хроматографией (HPLC), критерий прохождения ≤0,10% площади для кислоты ZX008. Промывают органический слой очищенной водой (2×3,0 по объему) при 18-23°C. Концентрируют органический слой при уменьшенном давлении до коэффициента приблизительно 2 по объему при 40-45°C, целевой температуре 43°C.
[00113] Определяют массовое содержание кетона ZX008 (полупродукта (WIP)) в смеси посредством анализа 1H-ЯМР только для информации и рассчитывают имеющий место выход кетона ZX008 (полупродукта (WIP)) в смеси. Примечание: Эта стадия может быть удалена из процесса, поскольку процесс является устойчивым и постоянно обеспечивает выход от 80 до 90%. Достигнутый выход учитывали в качестве коэффициента при загрузках последующих стадий.
[00114] Загружают н-гептан, (4,0 по объему, 2,7 по массе) в смесь при 40-45°C, целевой температуре 43°C. Концентрируют смесь до коэффициента приблизительно 2 по объему при 40-45°C, целевой температуре 43°C. Определяют содержание трет-бутилметилового эфира (TBME) в смеси посредством анализа 1H-ЯМР, (критерий прохождения ≤5,0 мас.% трет-бутилметилового эфира (TBME) по сравнению с кетоном ZX008). Загружают н-гептан, (2,4 по объему, 1,6 по массе) при 40-45°C, целевой температуре 43°C, в сосуд A. В сосуд B загружают метабисульфит натрия, (0,82 по массе, 0,88 экв.) при 18-23°C. В сосуд B загружают раствор гидрокарбоната натрия, (0,16 по массе, 0,4 экв.) в очищенной воде, код RM0120, (2,0 по объему) при 18-23°C, с последующей промывкой очищенной водой, код RM0120, (0,4 по объему) при 18-23°C. Предупреждение: газовыделение. Нагревают содержимое сосуда B до 40-45°C, целевая температура 43°C. Загружают содержимое из сосуда A в сосуд B с последующей промывкой н-гептаном (0,8 по объему, 0,5 по массе) при 40-45°C, целевой температуре 43°C. Перемешивают смесь в течение от 1 до 1,5 часа при 40-45°C, целевой температуре 43°C. Загружают н-гептан, код RM0174, (3,2 по объему, 2,2 по массе) в смесь при предоставлении возможности охлаждения до 18-45°C в конце добавления. Охлаждают смесь до 18-23°C при приблизительно постоянной скорости на протяжении от 45 до 60 минут. Перемешивают смесь при 18-23°C в течение 1,5-2 часов.
[00115] Образец смеси подвергают анализу 1H-ЯМР для определения содержания остаточного кетона ZX008 (критерий прохождения ≤10,0 мол.%, целевое содержание 5,0 мол.% кетона ZX008 по сравнению с кетоном ZX008 бисульфитного аддукта). Фильтруют смесь, и промывают осадок на фильтре н-гептаном (2×2,0 по объему, 2×1,4 по массе) при 18-23°C. Твердотельное вещество сушат при температуре вплоть до 23°C до тех пор, пока содержание воды не составит ≤10,0 мас.% при определении анализом фильтром Калмана в соответствии с AKX реагентом. По меньшей мере 16 часов. Определяют массовое содержание отделенного бисульфитного аддукта кетона ZX008 посредством анализа 1H-ЯМР и рассчитывают имеющий место выход бисульфитного аддукта кетона ZX008.
[00116] Выходы и профили: Выход для стадии 1 демонстрационной партии представлен в обобщенном виде в Таблице ниже. Ввод: 17000 г, нескорректировано, кислоты, 99,50% площади (по контролю качества высокоэффективной жидкостной хроматографией (HPLC)), 2-изомер не обнаружен, 4-isomer 0,02% площади, RRT 1.58 (первоначально не наблюдалось) 0,48% площади согласно способу получения. Аналитические данные представлены в обобщенном виде в Таблице 1A ниже.
Таблица 1A: Таблица для фактических выходов для стадии 1 Демонстрационной партии
[00117] Стадия 2: Получение кетона
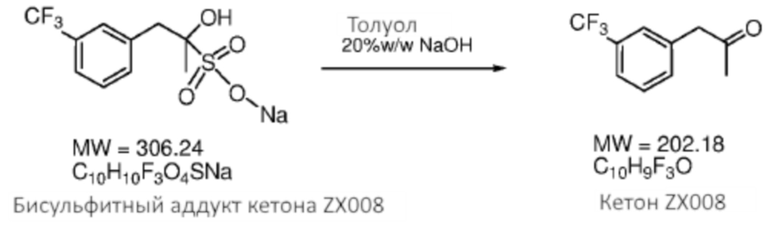
[00118] Процедура: Загружают толуол, (5,0 по объему, 4,3 по массе), и очищенную воду, (5,0 по объему) в сосуд и начинают перемешивание. При необходимости, температуру регулируют до 18-23°C и загружают бисульфитный аддукт кетона ZX008, (1,00 по массе, скорректировано для содержания в мас.%) в смесь при 18-23°C. Загружают 20 мас.%-ный раствор гидроксида натрия в смесь при 18-23°C, регулируя pH смеси до pH от 8,0 до 12,0, при целевом значении 9,0 (от 0,5 до 1,0 по объему).
Отделяют нижний водный слой и оставляют верхний органический слой. Промывают органический слой очищенной водой (3,0 по объему) при 18-23°C. Концентрируют органический слой при уменьшенном давлении до коэффициента приблизительно 2 по объему при 45-50°C, целевой температуре 48°C. Загружают метанол (5,0 по объему, 4,0 по массе) в смесь при 45-50°C, целевой температуре 48°C. Повторно концентрируют смесь при уменьшенном давлении до коэффициента приблизительно 2 по объему при 45-50°C, целевой температуре 48°C. Повторяют стадии 7 и 8 один раз перед переходом к стадии 9. Охлаждают смесь до 18-23°C. Очищают смесь в тарированном барабане подходящего размера с последующей промывкой метанолом (1,0 по объему, 0,8 по массе) при 18-23°C. Определяют массовое содержание кетона ZX008 (полупродукта (WIP)) в смеси посредством анализа 1H-ЯМР и рассчитывают имеющий место выход кетона ZX008 (полупродукта (WIP)) в смеси. Определяют содержание толуола в смеси посредством анализа 1H-ЯМР.
[00119] Выходы и профили: Выход для стадии 2 демонстрационной партии представлен в обобщенном виде в Таблице 1B ниже. Ввод: 1200,0 г с учетом поправки кетонового бисульфитного аддукта, 76,0 мас.% содержания (при анализе ЯМР, при применении диметилбутана (DMB) в качестве внутреннего эталона в дейтерированном диметилсульфоксиде (d6-DMSO)), (1,00 экв., 1,00 по массе с учетом поправки для массового содержания) для расчета ввода.
Таблица 1B: Таблица для фактических выходов для стадии 2 Демонстрационной партии
[00120] Стадия 3: Получение исходного неочищенного фенфлурамина.HCl
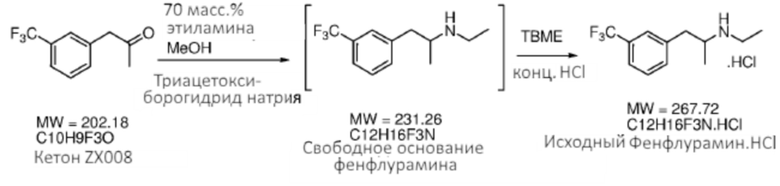
[00121] Процедура: Загружают кетон ZX008 (с учетом коррекции содержания, 1,00 по массе, 1,00 экв., отделенный в качестве раствора в MeOH на стадии 2) в сосуд. Загружают метанол, код RM0036 (5,0 по объему, 4,0 по массе) в смесь при 18-23°C. Охлаждают раствор до 0-5°C. Загружают 70%-ный по массе водный раствор этиламина (1,3 по объему, 1,6 по массе, 4.0 экв.) в смесь при 0-10°C, на протяжении 15-30 минут, с последующей промывкой метанолом (1,0 по объему, 0,8 по массе). Нагревают смесь до 15-20°C и перемешивают смесь в течение дополнительных 60-70 минут при 15-20°C. Регулируют смесь до 15-18°C, при необходимости, при целевом значении 15°C. Загружают триацетоксиборогидрид натрия (2,4 по массе, 2,25 экв.) в смесь при примерно 10 порциях, поддерживая смесь при 15-20°C, целевой температуре 17°C. Время добавления от 1,5 до 2 часов. Предупреждение: Экзотермическая реакция. Перемешивают смесь при 15-20°C до завершения при определении посредством анализа высокоэффективной жидкостной хроматографией (HPLC), критерий прохождения ≤3,0% площади кетона ZX008, типично от 2 до 3 часов. Регулируют pH смеси до pH>12 посредством загрузки 20 мас.%-ного водного раствора гидроксида натрия (от 5,0 до 6,0 по объему) в смесь при 15-40°C. Время добавления от 10 до 30 минут. Предупреждение: Экзотермическая реакция. При необходимости, температуру регулируют до 18-23°C. Экстрагируют смесь толуолом (3×3,0 по объему, 3×2,6 по массе) при 18-23°C, оставляя и объединяя верхний органический слой после каждой экстракции. Промывают комбинированный органический слой очищенной водой (1,0 по объему) при 18-23°C. Нагревают смесь до температуры от 40 до 50°C, целевой температуры 48°C. Концентрируют смесь при уменьшенном давлении при постоянном объеме, поддерживаемом при коэффициенте примерно 5 по объему посредством загрузки органического слоя при примерно таком же расходе, что и расход от дистилляции при 40-50°C, целевой температуре 48°C. Охлаждают смесь до 18-23°C. Загружают очищенную воду (10,0 по объему) в смесь при 18-23°C. Регулируют pH смеси до 0,1≤pH≤1,5 при 18-23°C посредством загрузки концентрированной хлористоводородной кислоты, 0,5 по объему. Не задерживаются на этой стадии, как только будет выполнена нейтрализация.
[00122] Отделяют слои при 18-23°C, оставляя нижний водный слой. Промывают водный слой толуолом (3,0 по объему, 2,6 по массе) при 18-23°C, оставляя водный слой. Регулируют pH водного слоя до pH>12 посредством загрузки 20 мас.%-ного раствора гидроксида натрия при 18-23°C (от 0,8 до 0,9 по объему). Предупреждение: Экзотермическая реакция. Загружают трет-бутилметиловый эфир (TBME), код RM0002 (2,0 по объему, 1,5 по массе) в основный водный слой. Отделяют слои при 18-23°C, оставляя верхний органический слой. После этого экстрагируют водный слой трет-бутилметиловым эфиром (TBME) (2×2,0 по объему, 2×1,5 по массе) при 18-23°C, оставляя органические слои. Промывают комбинированный органический слой очищенной водой (2×1,0 по объему) при 18-23°C. Концентрируют комбинированные органические слои при уменьшенном давлении при 40-50°C, целевой температуре 48°C, до коэффициента приблизительно 3 по объему. Определяют содержание остаточного толуола в смеси посредством анализа 1H-ЯМР. Образцом для определения остаточного содержания воды посредством анализа фильтром Калмана является AKX реагент. Загружают трет-бутилметиловый эфир (TBME) (8,7 по объему, 6,4 по массе) в смесь при 40-50°C. Охлаждают раствор до температуры от 0 до 5°C, целевой температуры 2°C. Загружают концентрированную хлористоводородную кислоту (0,54 по объему, 0,46 по массе), поддерживая температуру <15°C. Предупреждение: Экзотермическая реакция. Затем промывают трет-бутилметиловым эфиром (TBME) (1,0 по объему, 0,7 по массе). При необходимости, температуру регулируют до 0-10°C и перемешивают смесь при 0-10°C в течение дополнительных 2-3 часов. Фильтруют смесь и промывают осадок на фильтре трет-бутилметиловым эфиром (TBME) (2×4,4 по объему, 2×3,3 по массе) при 0-10°C. Твердотельное вещество сушат при температуре вплоть до 40°C до тех пор, пока содержание трет-бутилметилового эфира (TBME) не составит ≤0,5 мас.% при анализе 1H-ЯМР, от 4 до 8 часов.
[00123] Выходы и профили: Выход для стадии 3 демонстрационной партии представлен в обобщенном виде в Таблице 1C ниже. Ввод: 856,8 г с учетом поправки кетона, 44,2 мас.% содержания (при анализе ЯМР, при применении тетрацианбензола (TCNB) в качестве внутреннего эталона в CDCl3), (1,00 экв., 1,00 по массе с учетом поправки для массового содержания) для расчета ввода. Фиг. 2 и Таблица 1D показывают типичную хроматограмму высокоэффективной жидкостной хроматографии (HPLC) для полученного исходного неочищенного гидрохлорида фенфлурамина (спектральную поглощательную способность для УФ при 210 нм).
Таблица 1C: Таблица фактических выходов для стадии 3 Демонстрационной партии
Таблица 1D: Чистота исходного неочищенного гидрохлорида фенфлурамина при определении высокоэффективной жидкостной хроматографией (HPLC) (см. Фиг. 2)
Описание технологического канала Хроматография DAD AU Ch 1 Образец 210, Bw 4
Результаты измерения пиковых значений
[00124] Стадия 4,2: Кристаллизация гидрохлорида фенфлурамина
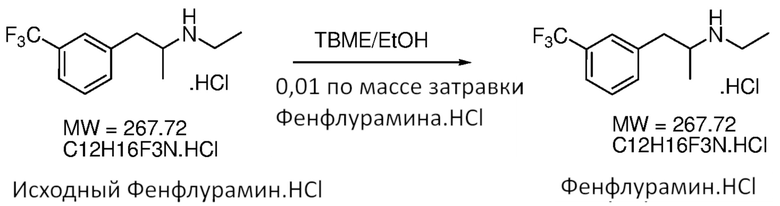
[00125] Процедура: Загружают фенфлурамин.HCl (исходный неочищенный) (1,00 по массе, 1,0 экв.) и трет-бутилметиловый эфир (TBME) (10,0 по объему, 7,4 по массе) в сосуд и начинают перемешивание. Нагревают суспензию для дефлегмирования (50-58°C). Загружают этанол (5,0 по объему, 3,9 по массе), поддерживая температуру при 50-58°C. Время добавления 20 минут. Перемешивают при 50-58°C в течение 5-10 минут и проверяют растворение. Перемешивают раствор при 50-58°C в течение 5-10 минут, при целевом значении 54-58°C. Очищают реакционную смесь посредством проходного фильтра 0,1 мкм при 54-58°C, с последующей промывкой трет-бутилметиловым эфиром (TBME) (1 по объему, 0,7 по массе). Охлаждают раствор до 48-50°C. Загружают фенфлурамин.HCl, код FP0188 (0,01 по массе). Проверяют кристаллизацию. Суспензии предоставляют возможность охлаждения до 15-20°C, целевой температуры 17°C, на протяжении 5-5,5 часов при приблизительно постоянной скорости. Перемешивают смесь при 15-20°C, целевой температуре 17°C, в течение 2-3 часов. Фильтруют смесь и промывают осадок на фильтре очищенным трет-бутилметиловым эфиром (TBME) (2×3,0 по объему, 2×2,2 по массе) при 5-15°C. Твердотельное вещество сушат при температуре вплоть до 40°C до тех пор, пока содержание трет-бутилметилового эфира (TBME) не составит ≤0,5 мас.%, и содержание этанола (EtOH) не составит <0,5 мас.%, при анализе 1H-ЯМР, от 4 до 8 часов. Определяют массовое содержание отделенного фенфлурамина.HCl посредством анализа 1H-ЯМР.
[00126] Выходы и профили: Выход для стадии 4 демонстрационной партии представлен в обобщенном виде в Таблице 1E ниже. Ввод: 750,0 г, нескорректировано, исходного неочищенного фенфлурамина.HCl (1,00 экв., 1,00 по массе, нескорректировано) для расчета ввода. Фиг. 3 показывает типичную хроматограмму высокоэффективной жидкостной хроматографии (HPLC) для примера кристаллизованного гидрохлорида фенфлурамина (спектральную поглощательную способность для УФ при 210 нм).
Таблица 1E: Таблица фактических выходов для стадии 4 Демонстрационной партии
5. Средства контроля в процессе производства
[00127] Таблица 2 суммирует средства контроля (IPCs) в процессе производства посредством номера IPC как указано пояснительных процедурах выше, применяемых для Процесса 1.
Таблица 2: Средства контроля в процессе производства, выполняемого во время Процесса 1
Менее 1,0% фенфлураминового спирта
Фенфлураминовый спирт
Этанол не более 0,50 мас.%
Этилацетат не более 0,50 мас.%
Метанол не более 0,50 мас.%
Толуол не более 0,50 мас.%
Метил-трет-бутиловый эфир (MTBE) не более 0,50 мас.%
6. Исходные материалы
[00128] Этот раздел предоставляет информацию и технические условия контроля для применяемых исходных материалов, чтобы создать клинические поставки фенфлурамина посредством маршрутов, представленных в данном документе.
Таблица 3: Исходные материалы при Маршруте 1
[Номер CAS]
Таблица 4: Исходные материалы при Маршруте 2
[Номер CAS]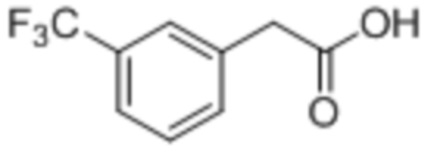

[00129] Таблица 5 предоставляет список промежуточных продуктов Маршрута 2 синтеза. Оба маршрута используют то же самое промежуточное Свободное основание фенфлурамина (1). Свободное основание фенфлурамина (1) обрабатывали в качестве отделенного промежуточного продукта в процессе Маршрута 1, однако процесс Маршрута 2 применяет стационарное оборудование, где как Кетон (2), так и Свободное основание фенфлурамина 1, оба неотделяемые масла, соединяют в качестве раствора и контролируют в качестве промежуточных продуктов in-situ. Бисульфатный комплекс (3) отделяют в качестве твердотельного вещества таким образом, что оно может быть подвергнуто обработке в качестве отделенного промежуточного вещества и высвобожденного как таковое. Исходный неочищенный фенфлурамин HCl может быть отделен в качестве промежуточного вещества перед рекристаллизацией.
[00130] Применяют спецификацию и стратегию тестирования для промежуточных продуктов. Дополнительные испытания и критерии приемки добавляют на основании обзора данных от исследований первичной стабильности партий и критических параметров оценки процесса. Аналитические стандарты применяли при полной характеризации каждого промежуточного вещества. Методы высокоэффективной жидкостной хроматографии (HPLC) для определения содержания и примесей являются такими же, что и способ высвобождения фармацевтической субстанции, и оцениваются в отношении точности, разброса, воспроизводимости, внутрилабораторной точности, селективности/специфичности, предела чувствительности, нижнего предела количественного определения, линейности, интервала и устойчивости.
Таблица 5: Промежуточные продукты in-situ и отделенные
[Номер CAS]

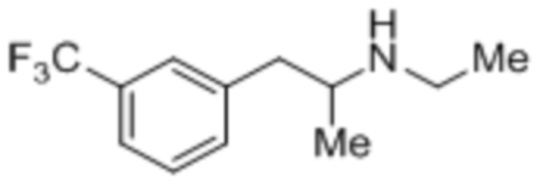
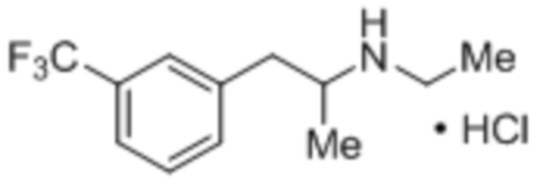
7. Характеризация
[00131] Физико-химические характеристики фармацевтической субстанции.
[00132] Фенфлурамин HCl получают в качестве одиночной полиморфной Формы 1. Выполняли исследование полиморфизм и предварительного состава. При широком интервале растворителей и условий кристаллический материал производит ту же самую полиморфную Форму 1 на основании четко определенной картины рентгеновской порошковой дифрактометрии (XRPD) и согласованно воспроизводимого эндотерма посредством анализа дифференциальной сканирующей калориметрией (ДСК). Сводка химико-физических свойств фенфлурамина HCl от этого исследования предоставлена ниже. Сведенные в таблицу данные включают типичные дифрактограммы, картины дифференциальной сканирующей калориметрии (ДСК) и микрофотографии.
[00133] Ввод фенфлурамина HCl (от отделения осаждением) характеризовали, чтобы предоставить ссылочные данные и также, чтобы определить, имела ли соль ту же самую форму, что и та, что идентифицирована от предыдущих формирований соли. Картина рентгеновской порошковой дифрактометрии (XRPD) соли показывает кристаллическое твердотельное вещество, которое визуально соответствует картинам отражений, полученных от соответствующей правилам кристаллизации фенфлурамина HCl, и она достаточным образом выражала Форму 1. Сравнение данных микро-ИК-спектроскопии нарушенного полного внутреннего отражения с преобразованием Фурье (μATR-FTIR) для соли от различных партий предоставляло профили, которые имели совпадение на 99,95%.
[00134] Данные термического анализа соответствовали предыдущим данным, полученным посредством лишь одной основной эндотермы на термограмме дифференциальной сканирующей калориметрии (ДСК), имеющей пик при 172,3°C, который соответствует началу потенциального разложения, показанного термографом для термогравиметрического анализа (ТГА). Это также соответствует описанной температуре плавления, указанной для эталонного образца.
[00135] Было показано, что отделение аморфной формы затруднено, при попытках применения трех обычных способах (быстрое испарение растворителя, осаждение антирастворителем и лиофилизация) все получаемые высококристаллические твердотельные вещества имели очень близкую долю по отношению к той же самой картине рентгеновской порошковой дифрактометрии (XRPD) вводимой Формы 1.
[00136] Анализ стабильности соли через одну неделю при 40°C/0% относительной влажности (RH), три недели при 40°C/75% относительной влажности (RH) и при условиях светоустойчивости показал, что ввод Формы 1 поддерживался при отсутствии новых примесей, обнаруживаемых при пороговом значении 0,1%.
[00137] Результаты анализа дифференциальной сканирующей калориметрией (ДСК) при циклическом воздействии температуры фенфлурамина HCl сравнимы с результатами, полученными, когда материал поддерживали при 170°C. Кристаллизация не наблюдалась, и аморфное вещество не образовывалось, а имел место возврат к Форме 1.
[00138] Поддерживание фенфлурамина HCl при приблизительно 170°C в течение нескольких часов вызывает то, что имеет место плавление и испарение, при рекомбинации и охлаждении для предоставления белого твердотельного вещества. Анализ белого твердотельного вещества рентгеновской порошковой дифрактометрии (XRPD), дифференциальной сканирующей калориметрией (ДСК) и 1H ЯМР указывает на отсутствие изменений в химической или физической форме, чистоте или разложении.
[00139] Выполненные исследования форсированной деградации доказали, что фенфлурамин HCl является стабильным при интервале условий. Термическая модуляция фенфлурамина HCl многократно увеличивало ввод Формы 1.
8. Примеси
[00140] Примеси в фармацевтической субстанции могут быть органическими примесями (технологическими примесями или продуктами разложения фармацевтической субстанции), неорганическими примесями (солевыми остатками или металлами) и остаточными растворителями; некоторые из этих примесей должны быть удалены, являются они или нет генотоксическими агентами. Эти примеси учитывают и регулируют в приготовлении фенфлурамина HCl посредством применения кратких руководств или ратифицированных аналитических методов согласно спецификациям или посредством отдельного тестирования «только для информации». Последующие разделы рассматривают фактические и потенциальные примеси в фенфлурамине HCl.
[00141] Фактические примеси и аттестация партии синтеза
[00142] Ни одна из указанных примесей в партиях фармацевтической субстанции, предназначенной для применения для людей, при условиях текущей надлежащей производственной практики (cGMP) не превышала пороги квалификации примеси ICH Q3A 0,15% (Таблица 8). Все примеси при >0,1% идентифицировали и обрабатывали, как описано в ICH Q3A, если они не являются генотоксическими примесями.
[00143] Технологические примеси
[00144] Таблица 6 перечисляет известные потенциальные примеси, являющиеся результатом маршрута синтеза. Все эти примеси регулируют ниже порога квалификации примеси ICH Q3A 0,15% посредством изменений процесса и/или регулирования степеней чистоты вводимого исходного материала.
Таблица 6: Известные потенциальные технологические примеси фенфлурамина HCl (Маршрут 1)
[Номер CAS]
[351-35-9]
[621-45-4]
[1886-26-6]
[172953-70-7]
[1683-15-4]
[90754-95-3]
1) ICH Q3A Идентификация порогового значения. Предел регистрации примеси (предел количественного определения (LOQ)) для метода высокоэффективной жидкостной хроматографии (HPLC) составляет 0,05%.
[00145] Примеси, являющиеся продуктами деградации
[00146] Не обнаружено изменений в составе примесей на протяжении длительного хранения на основании исследований форсированной деградации при условиях ICH Q1A(R2) воздействия нагревания (твердотельного вещества, раствора), кислоты, основания, окисления, и при условиях светоустойчивости ICH Q1B (твердотельное вещество, раствор). Фенфлурамин HCl является стабильным в течение 7 дней в качестве твердотельного вещества при 150°C (99,90% исходной площади), в качестве раствора в воде-ацетонитриле при 70°C (99,73% исходной площади), в качестве раствора в кислоте, основании, или при условиях светоустойчивости в окружающей среде. Только условия окисления (условия воздействия пероксида) создавали деградацию фенфлурамина HCl до 94,42% после 1 дня, создавая несколько новых родственных примесей при ~1% каждой в соответствии с жидкостной хроматографией/масс-спектрометрией (LC-MS) с +16 побочными продуктами окисления.
[00147] Органические летучие/остаточные растворители
[00148] Таблица 8 в разделе Анализ партии суммирует растворители, применяемые в процессе, и результирующие количества, найденные в фармацевтической субстанции. Все растворители, примененные на стадиях надлежащей производственной практики (GMP), контролировали при пределах ICH Q3A при применении подходящим образом квалифицированного метода газовой хроматографии в свободном пространстве над продуктом (HSGC).
[00149] Неорганические примеси
[00150] Тяжелые металлы соответствуют USP <231> или ICP способу USP <233>, а также ICH Q3D.
[00151] Генотоксические примеси
[00152] Рекомендации ICH Q3A и Q3B не являются достаточными для предоставления руководства по примесям, которые являются реакционноспособными в отношении ДНК. Рекомендацию Европейского агентства по лекарственным средствам (EMA) (2006) «Рекомендация по пределам генотоксических примесей» (EMA 2006) и Рекомендацию ICH M7 (2014) «Оценка и контроль примесей, реакционноспособных в отношении ДНК (мутагенных) в фармацевтических препаратах, чтобы ограничивать потенциальный онкогенный риск» (Рекомендация ICH M7) учитывают при контроле потенциальных генотоксических примесей. Маршрут с диазонием для получения кетона (2), представленный на Фиг. 5, имеет недостаток, обусловленный потенциальной возможностью формирования генотоксических промежуточных продуктов, показанных как заключенные в рамку соединения (например, N-гидроксиарил, N-нитрозамин и нитросоединение). Muller et al. (Regulatory Toxicology and Pharmacology 44 (2006) 198-211) составили список потенциально опасных функциональных групп, которые могут быть генотоксическими. Руководства нормативные документы по безопасности указывают, что анализ процесса и идентификация потенциальных генотоксических агентов, и регулирование таких примесей на уровнях менее 10 частей на миллион является критическим для безопасности. Часто удаление таких примесей и/или подтверждение их отсутствия является дорогостоящим и требующим много времени и иногда трудно достигается технически. По этим причинам, выбор маршрутов синтеза, которые устраняет потенциал для таких токсичных промежуточных продуктов, является важным. По причине потенциальных проблем с диазо-маршрутом, как рассмотрено выше, а также потенциальных проблем с безопасностью при применении промежуточных диазопродуктов (чувствительность к ударному воздействию), а также более низких профилей чистоты при этом маршруте, этот маршрут является менее предпочтительным, чем предпочтительный маршрут до Кетона (2), начиная от Нитрила (5). Этот маршрут не создает потенциальных генотоксических агентов и приводит к высокой чистоте Кетон (2) после отделения посредством дистилляции или посредством последовательности аддукт в виде бисульфитной соли - гидролиз.
[00153] Кроме того, попытки удаления изомерных побочных продуктов, присутствующих в коммерческих поставках анилина, были неудачными, тогда как кристаллизация Кислоты (4), получаемой от гидролиза Нитрила (5) предоставляет кристаллическую Кислоту (4), которая может быть очищена для удаления изомеров в начале синтеза. Удаление примесей и/или изомеров в начале синтеза является предпочтительным, если известно, что такие примеси доводятся до конечного продукта, поскольку необходимость в кристаллизации конечного продукта в конце синтеза является более дорогостоящей в отношении его потерь и оказывает большее влияние на стоимость товаров, чем удаление в начале синтеза перед тем, как исходные материалы вводят во время процесса.
Таблица 7: Потенциальные примеси в синтезе фенфлурамина

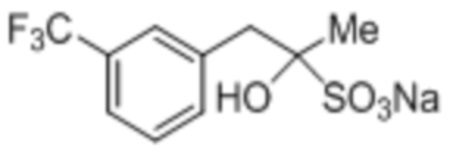



Таблица 8: Общие свойства фармацевтической субстанции фенфлурамина HCl
Безводное основание (высокоэффективная жидкостная хроматография (HPLC))
(RRT 0,53-0,57)
(Карл Фишер)
USP <61>
USP <62>
a) Эти испытания были добавлены к спецификациям в последнее время, соответственно последние партии протестированы при применении этого теста.
b) Эти испытания были исключены из спецификаций, соответственно лишь предыдущие партии были испытаны при применении этого теста.
ПРИМЕР 3
Способ гидролиза нитрила (5) до кислоты.
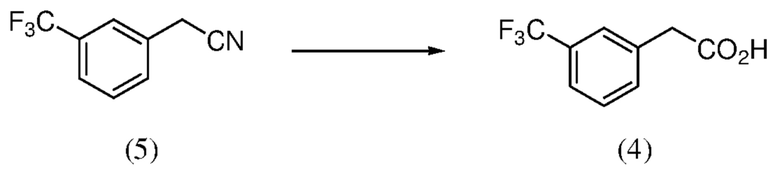
Таблица 9
Ожидаемый выход: 60-90% без учета поправки, 68-103 мас.%
Ожидаемая чистота: 93,00 до 99% площади при определении высокоэффективной жидкостной хроматографией (HPLC)
ПРИМЕР 4
Оценка второстепенных компонентов, образованных во время реакции Дакина-Веста при получении кетона (2)
[00154] Описаны примеси, образованные во время реакции Дакина-Веста, и их последующее удаление при применении дистилляции или посредством отделения продукта кетона в качестве бисульфитной соли. Найденные две основные примеси представлены ниже.
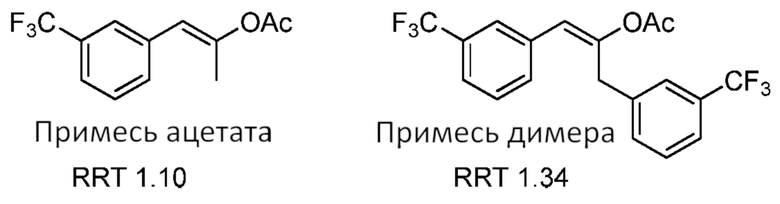
[00155] Таблица 10 представляет аналитические данные для исходного неочищенного Кетона (2), отделенного от реакции Дакина-Веста перед и после бисульфитной очистки. При введении 1 исходный неочищенный кетон, отделенный непосредственно от стадии Дакина-Веста (обработки предварительного бисульфита), имеет чистоту 61,66% (например, примерно 62%) и содержит 1,98% (например, примерно 2%) и 4,64% (например, примерно 5%), соответственно, примесей, имеющих относительное время удерживания (RRT) 1,20 и 1,34, которые предположительно являются примесями ацетата и димера (например, представленными выше), соответственно. При введении 2, после бисульфитной обработки, эти другие примеси удаляют, что приводит к общей чистоте 95,55% (например, примерно 96%). Другие введения, представленные в Таблице 10, предоставляют другие примеры этого улучшения в отношении загрязнений посредством бисульфитной обработки исходного неочищенного кетона Дакина-Веста. Последние два введения применяют чистый кетон Fluorochem в качестве ввода на стадию формирования соли стадия и повторное отделение кетона, иллюстрируя тем самым, что формирование соли и повторное отделение не создают каких-либо примесей сами по себе. Кроме того, применение процедуры экстрагирования бикарбоната во время реакционной обработки предоставляет улучшение в чистоте результирующей композиции, когда это служит для удаления любой непрореагировавшей кислоты. Исходный неочищенный Кетон (2), полученный посредством диазо-маршрута, показал подобные улучшения в чистоте, когда был обработан бисульфитом и отделен.
[00156] Таблица 10: Аналитические данные в отношении чистоты для для исходного неочищенного Кетона (2), отделенного от реакции Дакина-Веста перед и после бисульфитной очистки. Относительное время удерживания (RRT) является относительным временем удерживания (мин) в хроматограмме.
Анилин
Кетон
Нитрил
Кислота
Введение 1 (Исходный неочищенный кетон от Маршрута 1); Введение 2 (Кетон от Маршрута 1 после высвобождения бисульфита); Введение 3 (Исходный неочищенный кетон с применением исходной неочищенной кислоты); Введение 4 (Кетон с применением исходной неочищенной кислоты после бисульфита); Введение 5 (Исходный неочищенный кетон с применением кристаллической кислоты); Введение 6 (Исходный неочищенный кетон с применением кристаллической кислоты после бисульфита); Введение 7 (Исходный неочищенный кетон с применением кристаллической кислоты); Введение 8 (Кетон Fluorochem); Введение 9 (Кетон Fluorochem после бисульфита).
ПРИМЕР 5
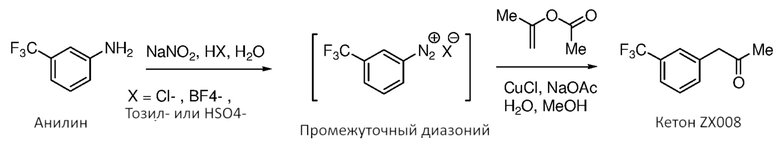
Дополнительный способ получения 1-(3-Трифторметил)фенил-пропан-2-она
[00157] 35 мл воды и 45 г 37 мас.%-ной водной хлористоводородной кислоты помещают в колбу, снабженную мешалкой и капельной воронкой. 24,25 г (0,151 моля) м-трифторметиланилина добавляют после охлаждения до 10°C посредством ледяной ванны и затем, при 5°C, медленной добавляют водный раствор, содержащий 12,43 г (0,180 моля) нитрита натрия в 150 мл воды. Реакционную смесь перемешивают в течение 30 минут и затем заливают на протяжении 30 минут в смесь, изготовленную из 90 мл воды, 1,35 г (0,014 моля) монохлорида меди, 2,30 г (0,013 моля) дигидрата дихлорида меди, 50 мл ацетона, 40,8 г (0,300 моля) тригидрата ацетата натрия и 23 г (0,230 моля) изопропенилацетата, при поддержании температуры реакции при 30°C. После перемешивания в в течение дополнительных 30 минут, реакционную смесь доводят до 20°C и добавляют 50 мл метиленхлорида и разделяют два слоя.
[00158] Водный слой удаляют, в то время как органический слой концентрируют в вакууме до тех пор, пока не получают масло, которое обрабатывают с применением 35 г метабисульфита натрия, 70 мл воды и 150 мл гептана при перемешивании при комнатной температуре в течение 12 часов. Суспензию фильтруют, бисульфитный комплекс промывают на фильтре с применением 50 мл гептана и затем суспендируют в двухфазной смеси, образованной из 100 мл метиленхлорида и 150 мл 10% (масса/объем) водного раствора гидроксида натрия. Слои отделяют после одного часа перемешивания при комнатной температуре, водную фазу удаляют, в то время как органический слой промывают водой и обезвоживают в вакууме, чтобы получить чистый кетон.
[00159] Несмотря на прилагаемую формулу изобретения, описание, изложенное в данном документе, также определено представленными ниже пунктами:
Пункт 1. Способ получения фенфлураминового активного фармацевтического ингредиента, данный способ включает:
(a) гидролиз 2-(3-(трифторметил)фенил)ацетонитрильной композиции, чтобы получить композицию 2-(3-(трифторметил)фенил)уксусной кислоты;
(b) реакционное взаимодействие композиции 2-(3-(трифторметил)фенил)уксусной кислоты с уксусным ангидридом и катализатором, чтобы получить композицию 1-(3-(трифторметил)фенил)пропан-2-она; и
(c) восстановительное аминирование композиции 1-(3-(трифторметил)фенил)пропан-2-она этиламином при применении борогидридного восстановителя, чтобы получить фенфлураминовую композицию.
[00160] Пункт 2. Способ по п. 1, где 2-(3-(трифторметил)фенил)ацетонитрильная композиция содержит по меньшей мере 0,2 мас.% трифторметил-фенильных региоизомеров.
[00161] Пункт 3. Способ по любому из пп. 1 и 2, где 2-(3-(трифторметил)фенил)ацетонитрильную композицию получают из трифторметилбензола.
[00162] Пункт 4. Способ по любому из пп. 1-3, дополнительно включающий, перед стадией (b), очистку композиции 2-(3-(трифторметил)фенил)уксусной кислоты, чтобы получить композицию, по существу не содержащую один или несколько трифторметил-фенильных региоизомеров и по существу не содержащую трифторметилбензальдегид и бензальдегид.
[00163] Пункт 5. Способ по п. 4, где очистка включает кристаллизацию 2-(3-(трифторметил)фенил)уксусной кислоты из композиции.
[00164] Пункт 6. Способ по любому из пп. 1-5, где стадия (b) включает очистку композиции 1-(3-(трифторметил)фенил)пропан-2-она кетоновым бисульфитным аддуктом.
[00165] Пункт 7. Способ по любому из пп. 1-6, где стадия (b) включает селективное реакционное взаимодействие 2-(3-(трифторметил)фенил)уксусной кислоты в присутствии непрореагировавшей 2-(2-(трифторметил)фенил)уксусной кислоты.
[00166] Пункт 8. Способ по любому из пунктов 1-7, где стадия (b) дополнительно включает удаление непрореагировавшего региоизомера 2-(2-(трифторметил)фенил)уксусной кислоты из композиции 1-(3-(трифторметил)фенил)пропан-2-она.
[00167] Пункт 9. Способ по любому из пп. 1-8, где фенфлураминовая композиция является исходной неочищенной и по существу не включает: трифторметил-фенильные региоизомеры фенфлурамина или их соли; металлические катализаторы; растворители Класса I или Класса II (ICH Q3C) (например, бензол, четыреххлористый углерод, 1,2-дихлорэтан, 1,1-дихлорэтен и/или 1,1,1-трихлорэтан); и спиртовый побочный продукт восстановления.
[00168] Пункт 10. Способ по любому из пп. 1-9, где фенфлураминовая композиция является исходной неочищенной и содержит менее чем 1 мас.% суммарно трифторметил-фенильных региоизомеров фенфлурамина или их солей.
[00169] Пункт 11. Способ по любому из пп. 1-10, где борогидридный восстановитель является триацетоксиборогидридом натрия.
[00170] Пункт 12. Способ по любому из пп. 1-11, где фенфлураминовая композиция является исходной неочищенной и имеет менее чем 10 мас.% спиртового побочного продукта восстановления.
[00171] Пункт 13. Способ по любому из пп. 1-12, дополнительно включающий кристаллизацию фенфлурамина или его соли из фенфлураминовой композиции.
[00172] Пункт 14. Способ по п. 1, где стадию (a) выполняют при условиях водного раствора кислоты.
[00173] Пункт 15. Способ по п. 14, где выход стадии (a) составляет 80% или более.
[00174] Пункт 16. Способ по п. 1, где стадию (b) выполняют при условиях, которые включают приведение композиции 2-(3-(трифторметил)фенил)уксусной кислоты в контактирование с примерно 0,5 эквивалента 1-метилимидазола и примерно 5 эквивалентами или более уксусного ангидрида, в необязательном растворителе.
[00175] Пункт 17. Способ по п. 16, где выход стадии (b) составляет 80% или более.
[00176] Пункт 18. Способ по п. 1, где стадию (c) выполняют при условиях, которые включают контактирование композиции 1-(3-(трифторметил)фенил)пропан-2-она с раствором 70 мас.% этиламина в воде и примерно 2,25 эквивалента или более триацетоксиборогидрида в метаноле в качестве растворителя.
[00177] Пункт 19. Способ по п. 18, где выход стадии (c) составляет 80% или более.
[00178] Пункт 20. Способ по п. 1, где фенфлураминовая композиция имеет следующий профиль: по меньшей мере 80 мас.% фенфлурамина или его соли; менее чем 1 мас.% 2-фенфлурамина или его соли; менее чем 1 мас.% 4-фенфлурамина или его соли; и менее чем 10 мас.% спиртового побочного продукта восстановления фенфлурамина.
[00179] Пункт 21. Способ по любому из пп. 1-20, дополнительно включающий преобразование фенфлурамина в фенфлураминовой композиции в фармацевтически приемлемую соль фенфлурамина.
[00180] Пункт 22. Способ по п. 21, дополнительно включающий кристаллизацию фармацевтически приемлемой соли фенфлурамина из фенфлураминовой композиции.
[00181] Пункт 23. Способ по п. 22, где фармацевтически приемлемая соль фенфлурамина имеет следующий профиль чистоты: по меньшей мере 90% или более фармацевтически приемлемой соли фенфлурамина; менее чем 1 мас.% 2-фенфлурамина; менее чем 5 мас.% 4-фенфлурамина; и менее чем 5 мас.% спиртового побочного продукта восстановления фенфлурамина.
[00182] Пункт 24. Способ по любому из пп. 20-22, где фармацевтически приемлемая соль фенфлурамина является гидрохлоридом фенфлурамина.
[00183] Пункт 25. Способ по любому из пп. 1-20, дополнительно включающий очистку свободного основания фенфлурамина из фенфлураминовой композиции.
[00184] Пункт 26. Способ по любому из пп. 1-25, дополнительно включающий выполнение хирального разделения рацемической фенфлураминовой композиции, чтобы получить нерацемическую фенфлураминовую композицию, содержащую доминирующий стереоизомер фенфлурамина.
[00185] Пункт 27. Способ по п. 26, где доминирующим стереоизомером фенфлурамина является (S)-N-этил-1-[3-(трифторметил)фенил]-пропан-2-амин.
[00186] Пункт 28. Способ по п. 26, где доминирующим стереоизомером фенфлурамина является (R)-N-этил-1-[3-(трифторметил)фенил]-пропан-2-амин.
[00187] Пункт 29. Фенфлураминовая композиция, полученная в соответствии со способом по любому из пунктов 1-28.
[00188] Пункт 30. Фенфлураминовый активный фармацевтический ингредиент, содержащий фармацевтически приемлемую соль фенфлурамина и имеющий менее чем 0,2 мас.% суммарно трифторметильных региоизомеров.
[00189] Пункт 31. Фенфлураминовый активный фармацевтический ингредиент по п. 30, имеющий следующий профиль: по меньшей мере 90% или по массе фармацевтически приемлемой соли фенфлурамина; менее чем 0,2 мас.% 2-фенфлурамина; менее чем 0,2 мас.% 4-фенфлурамина; и менее чем 1 мас.% фенфлураминового спирта.
[00190] Пункт 32. Фенфлураминовый активный фармацевтический ингредиент по любому из пп. 30-31, где компоненты тяжелых металлов по существу или полностью удалены из композиции.
[00191] Пункт 33. Фенфлураминовый активный фармацевтический ингредиент по любому из пп. 30-31, где растворители Класса 1 и/или Класса 2 по существу или полностью удалены из композиции (например, фенфлураминовый активный фармацевтический ингредиент по любому из пунктов 30-31 по существу не включает растворители Класса 1 и/или Класса 2).
[00192] Пункт 34. Фенфлураминовый активный фармацевтический ингредиент по любому из пп. 30-33, где фенфлураминовый спирт полностью удален из композиции.
[00193] Пункт 35. Фенфлураминовый активный фармацевтический ингредиент по любому из пп. 30-34, где бензальдегид и трифторбензальдегид по существу или полностью удалены из композиции.
[00194] Пункт 36. Фенфлураминовый активный фармацевтический ингредиент по любому из пп. 30-35, где композиция является неочищенной.
[00195] Пункт 37. Фармацевтическая композиция, содержащая фенфлураминовый активный фармацевтический ингредиент по любому из пп. 30-36 и фармацевтически приемлемое вспомогательное вещество.
[00196] Предшествующее изложение лишь иллюстрирует принципы данного изобретения. Следует принимать во внимание, что специалисты в данной области смогут разработать различные варианты, которые, хотя не описаны ясным образом и не представлены в данном документе, содержат в себе принципы данного изобретения и включены в его сущность и объем. Кроме того, все примеры и профессиональные термины, изложенные в данном документе, преимущественно предназначены, чтобы помочь читателю в понимании принципов данного изобретения и концепций, вносимых авторами данного изобретения для развития данной области, и должны истолковываться как являющиеся, без ограничения, такими конкретно изложенными примерами и состояниями. Более того, все формулировки изложенных в данном документе принципов, аспектов и вариантов осуществления данного изобретения, а также их конкретные примеры, предназначены, чтобы охватывать как структурные, так и функциональные их эквиваленты. Кроме того, предполагается, что такие эквиваленты включают как известные в настоящее время эквиваленты, так и эквиваленты, разработанные в будущем, т.е. любые разработанные элементы, которые выполняют такую же функцию, независимо от структуры. Объем данного изобретения, поэтому, не предполагает ограничения примерами вариантов осуществления, представленных и описанных в данном документе. Напротив, объем и сущность данного изобретения представлены прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 3-АМИНОПИПЕРИДИНА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2309147C9 |
| НОВЫЕ ИНГИБИТОРЫ S-НИТРОЗОГЛУТАТИОНРЕДУКТАЗЫ | 2011 |
|
RU2585763C2 |
| АЗОЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ МАЛОНИЛ-СОА-ДЕКАРБОКСИЛАЗЫ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ МЕТАБОЛИЗМА | 2002 |
|
RU2258706C2 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛА КАК ИНГИБИТОРЫ 11-БЕТА-ГИДРОКСИСТЕРОИДДЕГИДРОГЕНАЗЫ-1 | 2003 |
|
RU2360910C2 |
| СПОСОБ ПОЛУЧЕНИЯ { 6-[(ДИЭТИЛАМИНО)МЕТИЛ]НАФТАЛИН-2-ИЛ} МЕТИЛ[4-(ГИДРОКСИКАРБАМОИЛ)ФЕНИЛ]КАРБАМАТА, ИМЕЮЩЕГО ВЫСОКУЮ ЧИСТОТУ | 2020 |
|
RU2826216C2 |
| СИНТЕЗ ДИАРИЛПИРАЗОЛОВ | 2003 |
|
RU2319695C2 |
| ЗАМЕЩЕННЫЕ ИЗОКСАЗОЛЫ В КАЧЕСТВЕ ФУНГИЦИДОВ | 2005 |
|
RU2392273C2 |
| СПОСОБ СИНТЕЗА АНАЛОГОВ ГОРМОНОВ ЩИТОВИДНОЙ ЖЕЛЕЗЫ И ИХ ПОЛИМОРФОВ | 2013 |
|
RU2668960C2 |
| ИНГИБИТОРЫ MAGL | 2017 |
|
RU2754536C1 |
| ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ ИНГИБИТОР НАТРИЙЗАВИСИМОГО ПЕРЕНОСЧИКА ФОСФАТА | 2015 |
|
RU2740008C2 |
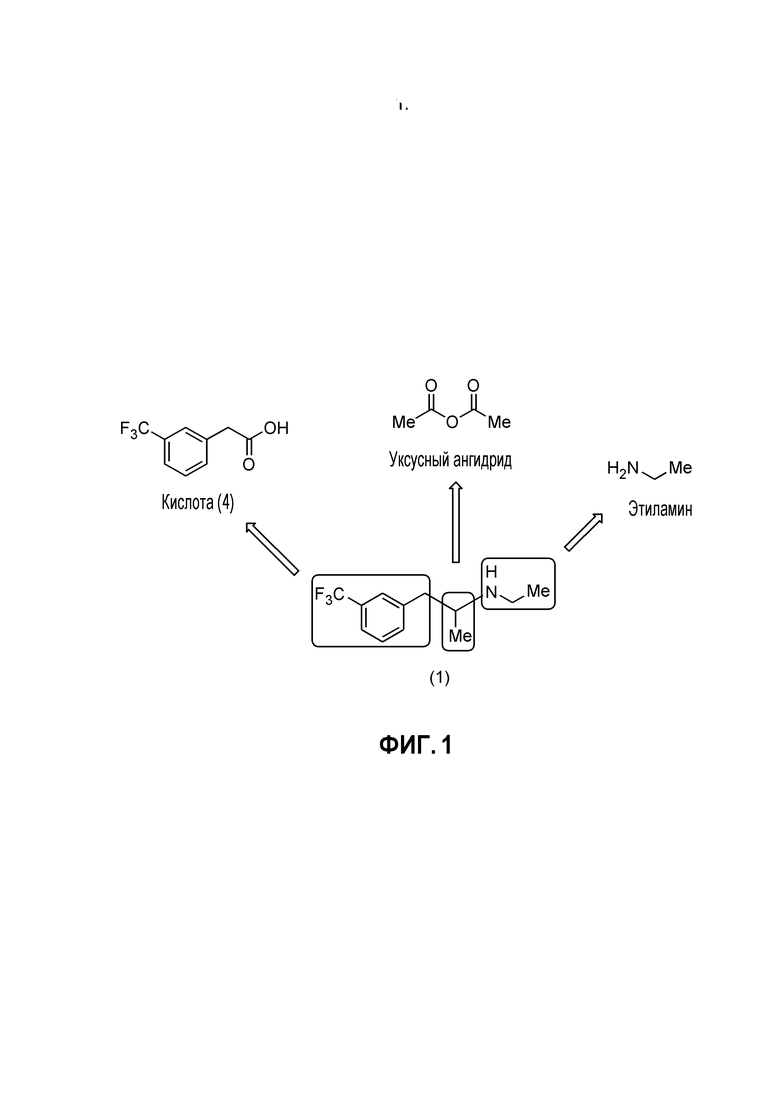


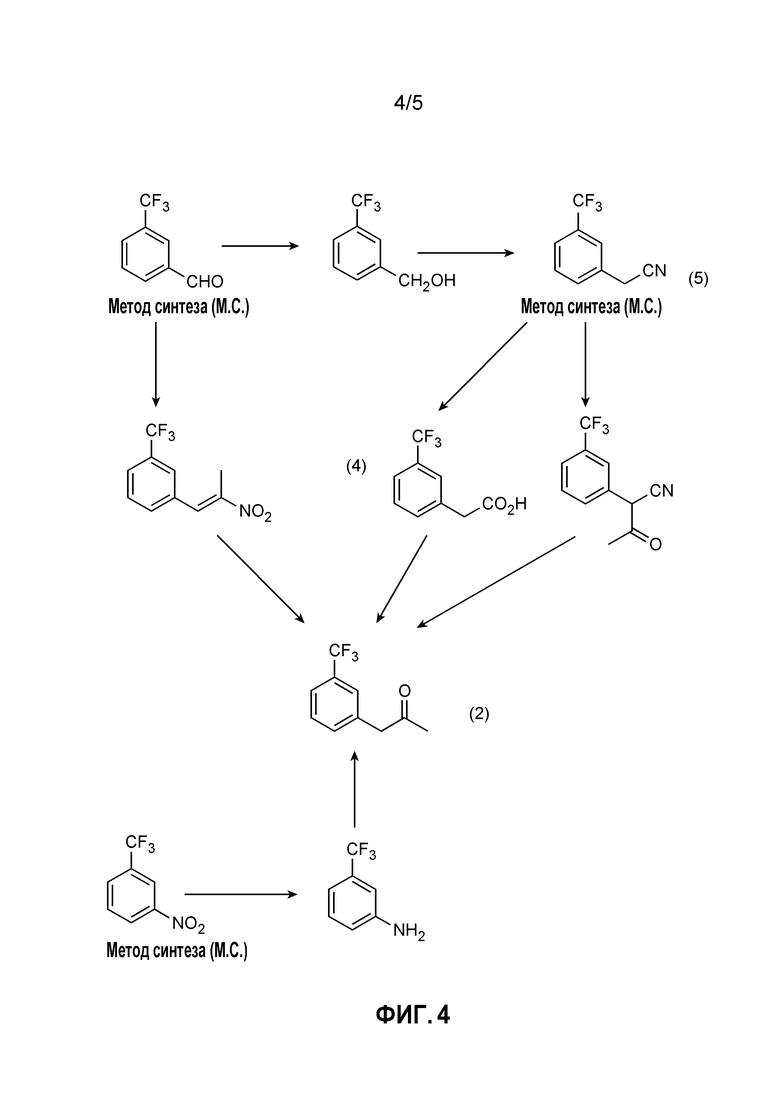
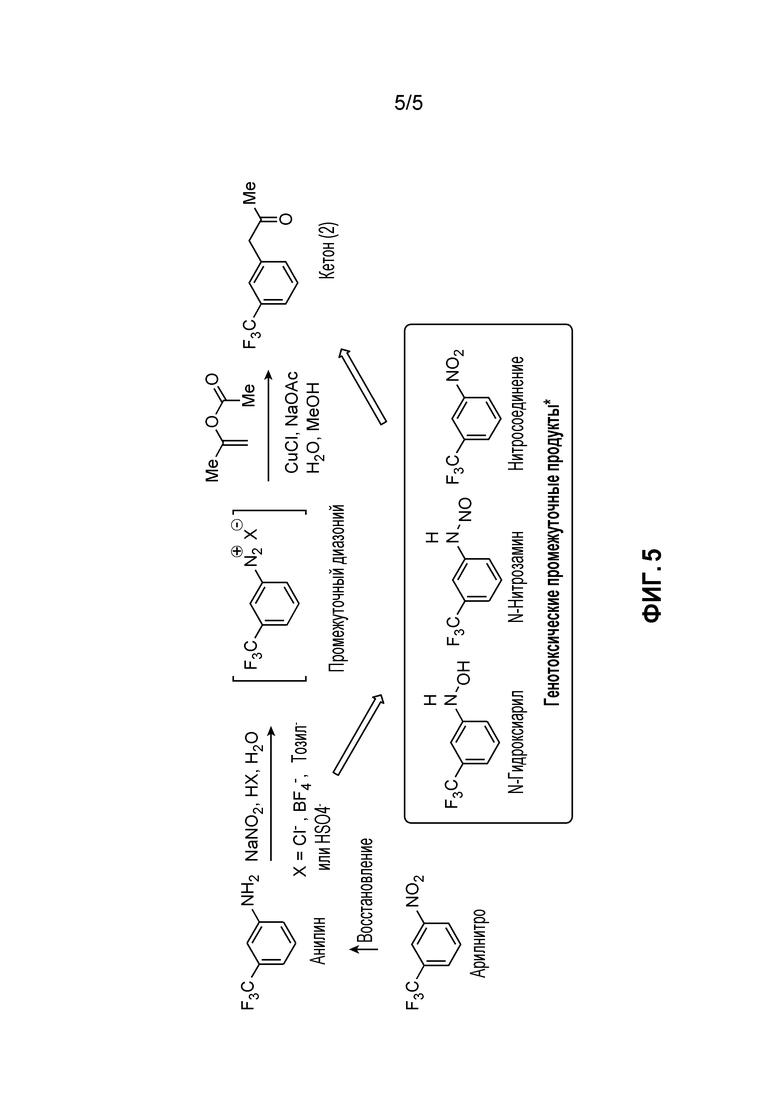
Изобретение относится к способу получения фенфлураминового активного фармацевтического ингредиента. Предлагаемый способ включает стадии (a)-(e). На стадии (a) осуществляют гидролиз 2-(3-(трифторметил)фенил)ацетонитрильной композиции с получением композиции 2-(3-(трифторметил)фенил)уксусной кислоты. На стадии (b) производят очистку композиции 2-(3-(трифторметил)фенил)уксусной кислоты с получением очищенной композиции 2-(3-(трифторметил)фенил)уксусной кислоты. На стадии (c) очищенная композиция 2-(3-(трифторметил)фенил)уксусной кислоты взаимодействует с уксусным ангидридом и катализатором с получением композиции 1-(3-(трифторметил)фенил)пропан-2-она. На стадии (d) осуществляют очистку композиции 1-(3-(трифторметил)фенил)пропан-2-она с использованием кетонового бисульфитного аддукта с получением очищенной композиции 1-(3-(трифторметил)фенил)пропан-2-она. На стадии (e) производят восстановительное аминирование композиции 1-(3-(трифторметил)фенил)пропан-2-она этиламином с использованием боргидридного восстановителя с получением неочищенной композиции фенфлурамина, содержащей трифторметил-фенильных региоизомеров фенфлурамина или их солей в общем количестве менее 0,2 мас.%. Предлагаемый способ позволяет получать фенфлураминовый активный фармацевтический ингредиент, из которого удалены определенные нежелательные второстепенные компоненты. 20 з.п. ф-лы, 10 табл., 5 пр., 5 ил.
1. Способ получения фенфлураминового активного фармацевтического ингредиента, где способ включает:
(a) гидролиз 2-(3-(трифторметил)фенил)ацетонитрильной композиции с получением композиции 2-(3-(трифторметил)фенил)уксусной кислоты;
(b) очистку композиции 2-(3-(трифторметил)фенил)уксусной кислоты с получением очищенной композиции 2-(3-(трифторметил)фенил)уксусной кислоты;
(c) взаимодействие очищенной композиции 2-(3-(трифторметил)фенил)уксусной кислоты с уксусным ангидридом и катализатором с получением композиции 1-(3-(трифторметил)фенил)пропан-2-она;
(d) очистку композиции 1-(3-(трифторметил)фенил)пропан-2-она с использованием кетонового бисульфитного аддукта с получением очищенной композиции 1-(3-(трифторметил)фенил)пропан-2-она; и
(e) восстановительное аминирование композиции 1-(3-(трифторметил)фенил)пропан-2-она этиламином с использованием боргидридного восстановителя с получением неочищенной композиции фенфлурамина, содержащей трифторметил-фенильных региоизомеров фенфлурамина или их солей в общем количестве менее 0,2 мас.%.
2. Способ по п. 1, где 2-(3-(трифторметил)фенил)ацетонитрильная композиция содержит по меньшей мере 1 мас.% трифторметил-фенильных региоизомеров.
3. Способ по п. 1, где композицию 2-(3-(трифторметил)фенил)ацетонитрила получают из трифторметилбензола.
4. Способ по п. 1, где очищенная композиция 2-(3-(трифторметил)фенил)уксусной кислоты на стадии (b) содержит общее количество трифторметил-фенильных региоизомеров менее 0,2 мас.% и общее количество трифторметилбензальдегида и бензальдегида 0,5 мас.% или менее.
5. Способ по п. 1, где очистка на стадии (b) включает кристаллизацию 2-(3-(трифторметил)фенил)уксусной кислоты.
6. Способ по п. 1, где очищенная композиция 1-(3-(трифторметил)фенил)пропан-2-она содержит примесь ацетата в количестве менее 0,2 мас.% и примесь димера в количестве менее 0,5 мас.%.
7. Способ по п. 1, где неочищенная композиции фенфлурамина:
содержит трифторметил-фенильных региоизомеров фенфлурамина или их соли в количестве менее 0,2 мас.%;
не содержит металлических катализаторов;
не содержит растворителей, выбранных из ацетонитрила, бензола и замещенных бензолов, четыреххлористого углерода, хлороформа, циклогексана, 1,2-дихлорэтана, 1,1-дихлорэтана, 1,2-диметоксиэтана, диметилформамида (DMF), 1,4-диоксана, метанола, метилбутилкетона, N-метилпирролидинона, пиридина, толуола, 1,1,1-трихлорэтана, 1,1,2-трихлорэтена и ксилола; и
содержит спиртовый побочный продукт восстановления в количестве менее 5 мас.%.
8. Способ по п. 1, где неочищенный фенфлурамин содержит 4-фенфлурамин или его соли в количестве менее 0,2 мас.%.
9. Способ по п. 8, где неочищенная композиция фенфлурамина содержит 4-фенфлурамин или его соли в количестве менее 0,1 мас.%.
10. Способ по п. 9, где неочищенная композиция фенфлурамина содержит 2-фенфлурамин или его соли в количестве менее 0,1 мас.%.
11. Способ по п. 1, где неочищенная композиция фенфлурамина содержит спиртовый побочный продукт восстановления в количестве менее 10 мас.%.
12. Способ по п. 9, дополнительно включающий кристаллизацию фенфлурамина или его соли из неочищенной композиции фенфлурамина.
13. Способ по п. 1, где стадию (с) выполняют при условиях, которые включают контактирование очищенной композиции 2-(3-(трифторметил)фенил)уксусной кислоты с 1-метилимидазолом в количестве примерно 0,5 эквивалента и уксусным ангидридом в количестве примерно 5 эквивалентов или более в необязательном растворителе.
14. Способ по п. 1, где стадию (е) выполняют при условиях, которые включают контактирование композиции 1-(3-(трифторметил)фенил)пропан-2-она с раствором этиламина в воде с концентрацией 70 мас.% и триацетоксиборгидридом в количестве приблизительно 2,25 эквивалента или более в метанольном растворителе.
15. Способ по п. 1, где композиция фенфлурамина имеет следующий профиль:
по меньшей мере 80 мас.% фенфлурамина или его соли;
менее 0,2 мас.% 2-фенфлурамина или его соли;
менее 0,2 мас.% 4-фенфлурамина или его соли; и
менее 10 мас.% спиртового побочного продукта восстановления фенфлурамина.
16. Способ по п. 1, дополнительно включающий:
преобразование фенфлурамина в фенфлураминовой композиции в фармацевтически приемлемую соль фенфлурамина; и
кристаллизацию фармацевтически приемлемой соли фенфлурамина из фенфлураминовой композиции, где фармацевтически приемлемая соль фенфлурамина имеет следующий профиль чистоты:
по меньшей мере 95 мас.% или более фармацевтически приемлемой соли фенфлурамина;
менее 0,2 мас.% 2-фенфлурамина;
менее 0,2 мас.% 4-фенфлурамина; и
менее 1 мас.% спиртового побочного продукта восстановления фенфлурамина.
17. Способ по п. 1, дополнительно включающий очистку свободного основания фенфлурамина из неочищенной фенфлураминовой композиции.
18. Способ по п. 1, дополнительно включающий выполнение хирального разделения рацемической композиции с получением нерацемической фенфлураминовой композиции, содержащей доминирующий стереоизомер фенфлурамина.
19. Способ по п. 1, где неочищенную фенфлураминовую композицию получают в масштабе килограммов.
20. Способ по п. 1, где очищенная композиция 2-(3-(трифторметил)фенил)уксусной кислоты на стадии (b) содержит 4-трифторметил-фенильный региоизомер в количестве менее 0,2 мас.%.
21. Способ по п. 20, где очищенная композиция 2-(3-(трифторметил)фенил)уксусной кислоты на стадии (b) содержит 2-трифторметил-фенильный региоизомер в количестве менее 0,2 мас.%.
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Устройство для поддержания постоянного уровня латекса в расходной ванне | 1972 |
|
SU441160A1 |
| US 5811586 A, 22.09.1998 | |||
| СПОСОБ МОДИФИКАЦИИ ПИЩЕВОГО ПОВЕДЕНИЯ И ПРИМЕНЕНИЕ PYY ИЛИ ЕГО АГОНИСТА В КАЧЕСТВЕ СРЕДСТВА ДЛЯ МОДИФИКАЦИИ ПИЩЕВОГО ПОВЕДЕНИЯ | 2002 |
|
RU2317104C2 |

