Предметом настоящего изобретения является способ получения {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил)фенил]карбамата и/или его фармацевтически приемлемых солей, имеющего высокую чистоту. Такой способ позволяет получить продукт, имеющий количество любой единичной неидентифицированной примеси, равное или ниже 0,10%, а также продукт, имеющий чистоту выше 99,5%, предпочтительно равную или выше 99,6%.
Еще одним предметом настоящего изобретения является способ ВЭЖХ для определения чистоты продукта и его возможных примесей.
Уровень техники
Гивиностат®, также известный под названием ITF2357, (по номенклатуре IUPAC {6-[(диэтиламино)метил]нафталин-2-ил}метил[4- (гидроксикарбамоил)фенил]карбамат представляет собой гидроксамовую кислоту, используемую в форме ее гидрохлорида, в частности, гидрохлорида моногидрата, который функционирует как ингибитор гистондеацетилазы (HDAC) и оказывает свое действие на одноименные ферменты I и II классов.
Гивиностат проявляет очень многообещающий профиль активности при множественной миеломе и остром миелогенном лейкозе как in vitro, так и in vivo, и также функционирует в качестве противовоспалительного средства и в качестве ингибитора фактора некроза опухоли альфа (TNF-α), IL-1, и секреции IL-6.
В настоящее время гивиностат проходит испытания в многочисленных исследованиях фазы III при воспалительных заболеваниях (мышечной дистрофии Дюшенна и Беккера, ювенильном артрите и истинной полицитемии) и в клинических испытаниях при раке крови (миеломах и лимфомах).
В патенте США № 6034096 описано получение гивиностата, в патенте США № 7329689 и международной заявке WO2004/65355 описано получение и характеристика одного его полиморфа моногидрата.
В патенте США № 8518988 описано получение и характеристика безводного полиморфа гивиностата.
В способах получения гивиностата, описанных в цитированных документах, сначала проводят синтез ацилхлорида (II) из промежуточного соединения (I) (стадия 1), который затем добавляют в виде смоченного ТГФ твердого вещества, к раствору гидроксиламина в воде и ТГФ с получением конечного продукта (стадия 2) в соответствии со следующей схемой 1:
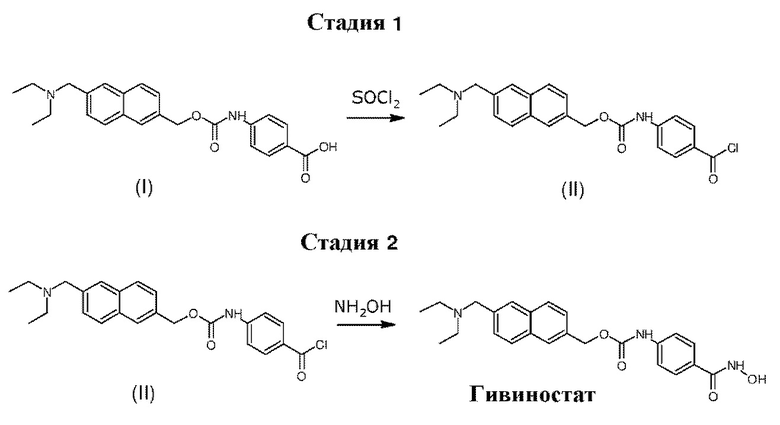
В вышеприведенных документах не описаны примеси, образующиеся в результате синтеза.
В литературе (A. Furlan, V. Monzani, L.L. Reznikov, F. Leoni, G. Fossati, D. Modena, P. Mascagni, C.A. Dinarello, Mol. Med. 2011, 17 (5-6) 353-362) описано данное промежуточное соединение (I) (ITF2375) и соответствующий амид (ITF2374), которые являются двумя основными метаболитами гивиностата in vivo (ITF2357), образующимися в результате биотрансформации гидроксамовой группы в карбоновую кислоту и амид соответственно.
Амид (ITF2374) имеет следующую формулу (Ia):

Следовательно, присутствие промежуточного соединения (I) или амида (Ia) в гивиностате в количествах выше 0,15% является обоснованным, поскольку они представляют собой квалифицированные примеси.
На основе современных руководств, действующих в области фармации, которые предусматривают подробное описание профиля примесей активных ингредиентов, предназначенных для применения у людей, разработка новых способов производства гивиностата в соответствии с современными стандартами качества и определением новых аналитических методов определения чистоты гивиностата, например, для идентификации присутствия любых примесей, имеет фундаментальное значение.
Определения
Если не указано иное, то все термины из области техники, обозначения и другая научная терминология, используемые здесь, имеют значения, обычно понимаемые специалистам в данной области, к которой относится настоящее изобретение. В некоторых случаях термины с общепринятыми значениями определены здесь для ясности и/или для справочных ссылок; таким образом, включение здесь таких определений не следует истолковывать в качестве существенного отличия от того, что обычно понимается в данной области техники.
В рамках настоящего изобретения, термин «физиологически приемлемый эксипиент» относится к веществу, не обладающему каким-либо собственным фармакологическим эффектом и которое не вызывает побочных реакций при введении млекопитающему, предпочтительно человеку. Физиологически приемлемые эксципиенты хорошо известны в данной области и раскрыты, например, в Handbook of Pharmaceutical Excipients, шестое издание 2009 г., включенном здесь в качестве ссылки.
Термин «фармацевтически приемлемые соли» относится к таким солям, которые обладают биологической эффективностью и свойствами солевого соединения и которые не вызывают побочных реакций при введении млекопитающему, предпочтительно человеку. Фармацевтически приемлемые соли могут представлять собой неорганические или органические соли; примеры фармацевтически приемлемых солей включают, не ограничиваясь этим, карбонат, гидрохлорид, гидробромид, сульфат, гидросульфат, цитрат, малеат, фумарат, трифторацетат, 2-нафталинсульфонат и пара-толуолсульфонат. Более подробную информацию о фармацевтически приемлемых солях можно найти в Handbook of pharmaceutical salts, P. Stahl, C. Wermuth, WILEY-VCH, 127-133, 2008, включенному здесь посредством ссылки.
Термины «содержащий», «имеющий», «включающий» и «состоящий» следует толковать как неограничивающие термины (т. е. означающие «включая, не ограничиваясь этим»), и их следует рассматривать как обеспечивающие поддержку также для таких терминов, как «состоят по существу из», «состоящий по существу из», «состоят из» или «состоящий из».
Термины «состоят по существу из», «состоящий по существу из» следует толковать как термины полузакрытого типа, означающие, что не включаются никакие другие ингредиенты, которые существенно влияют на основные и новые характеристики изобретения (таким образом, могут быть включены необязательные эксципиенты).
Термины «состоит из», «состоящий из» следует толковать как термины закрытого типа.
Термин «имеющий высокую чистоту» относится к чистоте выше 99,5%, предпочтительно равной или выше 99,6%.
В рамках настоящего изобретения, термины гивиностат или ITF2357 предназначены для обозначения гидрохлоридной соли {6- [(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил) фенил]карбамата, в частности, гидрохлорида моногидрата (номер CAS 732302-99-7). Вместо этого гидрохлоридная соль имеет номер CAS 199657-29-9, и свободное основание имеет номер CAS 497833-27-9.
Термин «галоген» относится к атому фтора (F), хлора (Cl), брома (Br) или йода (I).
Термин «диполярный апротонный растворитель, не чувствительный к кислоте» относится к растворителю, не содержащему кислоточувствительных компонентов. ТГФ является примером кислоточувствительного растворителя. ДМСО, ацетонитрил, диметилацетамид или диметилформамид вместо этого являются примерами растворителей, не чувствительных к кислоте.
Термин «неизвестная примесь» относится к любой неидентифицированной примеси, находящейся в {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил)фенил]карбамате и/или его фармацевтически приемлемых солях.
Сущность изобретения
В отношении способов получения гивиностата, используемых на предшествующем уровне техники, авторы изобретения обнаружили, что данные способы обеспечивают гивиностат, имеющий содержание единичной неидентифицированной примеси выше 0,10%.
Таким образом, авторы настоящего изобретения столкнулись с необходимостью разработки нового способа получения гивиностата с содержанием любой единичной неидентифированной примеси, равным или ниже 0,10% (% площади на хроматограмме ВЭЖХ), что является уровнем, предполагаемым в руководстве ICH для неизвестных примесей, когда суточная доза действующего вещества составляет менее 2 г, как это имеет место в случае с гивиностатом.
Неожиданно было обнаружено, что на стадии 1 (схема 1) критически важно провести реакцию галогенирования промежуточного соединения (I) в растворителе, не содержащем кислоточувствительных компонентов, чтобы снизить количество эквивалентов галогенирующего агента, необходимых для завершения требуемой реакции, с получением тем самым промежуточного продукта (II), имеющего более низкое количество примесей, чем полученное с применением известного уровня техники.
В равной степени неожиданно авторы настоящего изобретения обнаружили, что на стадии 2 (схема 1) порядок добавления промежуточного соединения (II) и гидроксиламина является критическим для получения конечного продукта, имеющего единичную неидентифицированную примесь на уровне, равном или ниже 0,10% по площади на хроматограмме анализа относительной чистоты.
Следовательно, в первом аспекте настоящее изобретение относится к способу получения {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил)фенил]карбамата, и/или его фармацевтически приемлемых солей, имеющего количество любой единичной неидентифицированной примеси, равное или ниже 0,10%, и/или высокую чистоту.
В предпочтительном варианте способ по настоящему изобретению позволяет получить {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил)фенил]карбамат, и/или его фармацевтически приемлемые соли, имеющий чистоту выше 99,5%, предпочтительно 99,6% или выше.
Согласно второму аспекту настоящее изобретение относится к {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил) фенил]карбамату, и/или его фармацевтически приемлемым солям, предпочтительно гидрохлориду, более предпочтительно гидрохлориду моногидрату, имеющему количество любой единичной неидентифицированной примеси, равное или ниже 0,10%, или имеющему количество любой единичной примеси, кроме промежуточного соединения (I) или амида (Ia), равное или ниже 0,15%, предпочтительно равное или ниже 0,10%.
Согласно третьему аспекту настоящее изобретение относится к {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил) фенил]карбамату, и/или его фармацевтически приемлемым солям, предпочтительно гидрохлориду, более предпочтительно гидрохлориду моногидрату, имеющему чистоту выше 99,5%, предпочтительно равную или выше 99,6%.
Согласно четвертому аспекту настоящее изобретение относится к новому аналитическому способу ВЭЖХ для определения чистоты {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил) фенил]карбамата, и/или их фармацевтически приемлемых солей, и детектирования его примесей.
Согласно пятому аспекту настоящее изобретение относится к {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил) фенил]карбамату, и/или его фармацевтически приемлемым солям, предпочтительно гидрохлориду, более предпочтительно гидрохлориду моногидрату, имеющему количество неидентифицированной примеси, равное или ниже 0,10%, с RRT 0,93±0,02 и/или неидентифицированной примеси с RRT 1,21±0,02, и/или неидентифицированной примеси с RRT 1,51±0,02, и/или неидентифицированной примеси с RRT 1,75±0,02, где RRT измеряется с использованием способа ВЭЖХ по изобретению.
Согласно его шестому аспекту настоящее изобретение относится к способу получения {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил)фенил]карбамата и/или его фармацевтически приемлемых солей, включающему способ ВЭЖХ для определения чистоты по изобретению.
Краткое описание фигур
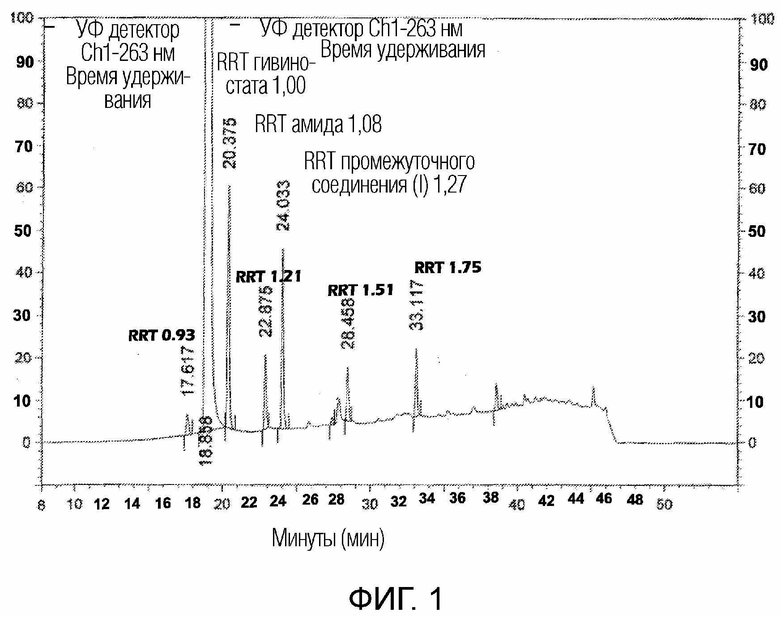
На фиг. 1 показана хроматограмма, полученная способом ВЭЖХ по изобретению на специально приготовленной смеси, содержащей все типичные примеси гивиностата.
Подробное описание изобретения
Предметом настоящего изобретения является способ получения {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил) фенил]карбамата, и/или его фармацевтически приемлемых солей, содержащих количество любой единичной неидентифицированной примеси, равное или ниже 0,10%, где указанный способ включает стадии:
i) приготовление раствора или суспензии соединения формулы (II) в органическом растворителе:
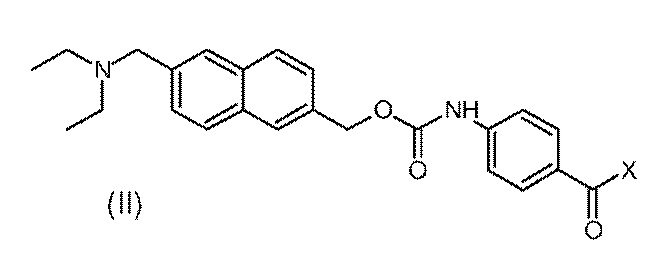
где X представляет собой атом галогена, предпочтительно атом хлора;
ii) добавление гидроксиламина к раствору или суспензии, полученным на стадии i).
В рамках настоящего изобретения, стадии i) и ii) будут называться стадией 2 в соответствии со способом изобретения.
В предпочтительном варианте осуществления органический растворитель на стадии i) на стадии 2 выбран из группы, включающей ТГФ, метил-ТГФ, диоксан, диметиловый эфир этиленгликоля и бис(2-метоксиэтиловый)эфир.
Предпочтительно указанный органический растворитель используется в количестве 1-100 частей по объему на 1 часть по массе соединения формулы (II).
Предпочтительно указанный органический растворитель имеет содержание воды ниже 0,5%.
Предпочтительно стадию ii) на стадии 2 способа по изобретению проводят при комнатной температуре.
В предпочтительном варианте осуществления способ по настоящему изобретения (стадия 2) отличается тем, что он дополнительно включает стадию iii) выделения {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил) фенил]карбамата в виде свободного основания или в виде фармацевтически приемлемой соли, предпочтительно гидрохлорида, более предпочтительно гидрохлорида моногидрата.
В еще одном предпочтительном варианте осуществления соединение формулы (II) получают из соответствующей кислоты формулы (I) взаимодействием с галогенирующим агентом, предпочтительно хлорирующим агентом (стадия а), в апротонном диполярном растворителе, нечувствительном к кислоте.
Предпочтительно соединение формулы (II) выделяют из реакционной смеси осаждением органическим растворителем, предпочтительно с последующей фильтрацией (стадия b).
В рамках настоящего изобретения, стадии а) и b) будут называться стадией 1 в соответствии со способом изобретения.
Нечувствительный к кислоте растворитель представляет собой растворитель, не содержащий кислоточувствительных компонентов (следовательно, например, без ТГФ). Такая характеристика растворителя позволяет использовать меньшие количества галогенирующего агента по сравнению с предшествующим уровнем техники.
Например, ТГФ представляет собой кислоточувствительный растворитель, поскольку в кислых условиях он может разлагаться и образовывать химически реакционноспособные молекулы, которые могут давать нежелательные побочные продукты в реакции галогенирования.
Предпочтительно на стадии а) стадии 1 указанный апротонный диполярный растворитель, нечувствительный к кислоте, выбран из ДМСО, ацетонитрила, диметилацетамида или диметилформамида, более предпочтительно диметилформамида.
Примерами хлорирующих агентов, которые можно использовать в способе по настоящему изобретению, являются тионилхлорид (SOCl2), трихлорид фосфора (PCl3), оксихлорид фосфора (POCl3) или пентахлорид фосфора (PCl5). В качестве альтернативы можно использовать соответствующие бромирующие агенты SOBr2, PBr3, POBr3 или PBr5.
В предпочтительном варианте осуществления стадии b) на стадии 1 органический растворитель, используемый для осаждения соединения (II), выбран из алифатических или ароматических углеводородов, простых эфиров, сложных эфиров или спиртов, более предпочтительно толуола или ТГФ.
Еще одним предметом настоящего изобретения является {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил) фенил]карбамат, и/или его фармацевтически приемлемые соли, имеющий количество любой единичной неидентифицированной примеси, равное или ниже 0,10%, полученным способом по изобретению только на стадии 2, относительно предшествующего уровня, как определено выше.
В еще одном предпочтительном варианте осуществления настоящее изобретение относится к {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил) фенил]карбамату, и/или его фармацевтически приемлемым солям, имеющему чистоту выше 99,5%, предпочтительно равное или выше 99,6%, полученным способом по изобретению как на стадии 1, так и стадии 2, относительно предшествующего уровня, как определено выше.
Еще одним предметом настоящего изобретения является {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил) фенил]карбамат, и/или его фармацевтически приемлемые соли, предпочтительно гидрохлорид, более предпочтительно гидрохлорид моногидрат, имеющий количество любой единичной неидентифицированной примеси, равное или ниже 0,10% или имеющий количество любой единичной примеси, кроме промежуточного соединения (I) или амида (Ia), равное или ниже 0,15%, предпочтительно равное или ниже 0,10%.
Еще одним предметом настоящего изобретения является {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил) фенил]карбамат, и/или его фармацевтически приемлемые соли, предпочтительно гидрохлорид, более предпочтительно гидрохлорид моногидрат, имеющий чистоту выше 99,5%, предпочтительно равную или выше 99,6%.
Еще одним предметом настоящего изобретения является {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил) фенил]карбамат, и/или его фармацевтически приемлемые соли, предпочтительно гидрохлорид, более предпочтительно гидрохлорид моногидрат, имеющий количество неидентифицированной примеси, равное или ниже 0,10% с RRT 0,93±0,02 и/или неидентифицированной примеси с RRT 1,21±0,02, и/или неидентифицированной примеси с RRT 1,51±0,02, и/или неидентифицированной примеси с RRT 1,75±0,02, RRT измеряется с использованием способа ВЭЖХ по изобретению.
В предпочтительном варианте RRT измеряется с использованием следующего способа ВЭЖХ:
- неподвижная фаза: носитель на основе частиц силикагеля, содержащего C18-алкильные цепи и имеющего содержание углерода менее 9% по массе;
подвижная фаза A: вода, забуференная с pH 3,7-3,8;
подвижная фаза B: забуференный метанол с pH 3,7-3,8.
Используя следующий метод градиентного элюирования:
(мин)
(об./об.)
(об./об.)
Предпочтительно использовать буфер на основе формиата аммония и муравьиной кислоты с pH 3,7-3,8.
Предпочтительно использовать УФ-детектор с длиной волны 263 нм.
Предпочтительно температура колонки составляет 25±1°C.
Предпочтительно объем инжектирования составляет 5 мкл.
Предпочтительно скорость потока составляет 0,25 мл/мин.
Предпочтительно образец продукта разбавляют ДМСО.
Еще одним предметом настоящего изобретения является фармацевтическая композиция, содержащая {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил) фенил]карбамат, и/или его фармацевтически приемлемые соли, как определено выше, и по меньшей мере, один фармацевтически приемлемый эксципиент.
В предпочтительном варианте осуществления фармацевтической композиции активное соединение находится в форме микронизированных частиц, имеющих средний размер менее 200 мкм, предпочтительно от 100 мкм до 1 мкм, более предпочтительно от 50 мкм до 5 мкм.
Еще одним предметом настоящего изобретения является также способ определения чистоты продукта {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил) фенил]карбамата, и/или его фармацевтически приемлемых солей, включающий элюирование продукта через колонку для ВЭЖХ, имеющую неподвижную фазу, содержащую алкильные цепочки, связанные с неорганическим носителем, например, силикагелем, и последующее детектирование продукта и его примесей с использованием детектора, подходящего для измерения количества аналита, элюированного с колонки, например, с детектором типа УФ, МС или RID.
В предпочтительном варианте осуществления способа по изобретению указанные алкильные цепочки относятся к октадецильным, октильным или бутильным (C18, C8 или C4) цепочкам, предпочтительно C18. В одном еще более предпочтительном варианте осуществления неподвижная фаза состоит из носителя силикагеля, дериватизированного C18-алкильными цепочками, с углеродной нагрузкой ниже 9% по массе.
Углеродная нагрузка означает содержание углерода, выраженное в мас.%, неподвижной фазы, связанной с силикагелем. Высокое содержание углерода (15-25%) делает поверхность неподвижной фазы более гидрофобной, и может удерживать наиболее гидрофобные примеси, что делает невозможным их правильную количественную оценку.
Несколько колонок такого типа коммерчески доступны, например ACE 5 C18-300, Halo C18 (твердое ядро), YMC-Pack OSD-A, BioBasic-18 PEEK. В предпочтительном варианте используется колонка Halo C18-90 Å (код 95812-902, производства Advanced Materials Technology).
Можно использовать различные элюенты, выбранные из воды, полярного органического растворителя или их смеси, с комбинациями неподвижной фазы и детектора, описанными выше. Указанный полярный органический растворитель представляет собой С1-С4 спирт, предпочтительно метанол или ацетонитрил.
Предпочтительно в способе ВЭЖХ по настоящему изобретению можно использовать смеси воды и метанола, воды и ацетонитрила, необязательно с или без градиентного элюирования, необязательно с буферами или без них. Предпочтительно вышеуказанные смеси можно использовать с добавлением буфера. Более предпочтительно использовать буфер на основе формиата аммония и муравьиной кислоты при pH 3,7-3,8.
В предпочтительном варианте осуществления в качестве метода элюирования используется хроматографический цикл по следующей схеме:
(мин)
%
(об./об.)
%
(об./об.)
где элюент A означает воду и буфер на основе формиата аммония и муравьиной кислоты с pH 3,7-3,8, и элюент B означает метанол и буфер на основе формиата аммония и муравьиной кислоты с pH 3,7-3,8.
Еще одним предметом настоящего изобретения также является способ получения {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил) фенил]карбамата, и/или его фармацевтически приемлемых солей, включающий способ определения чистоты по изобретению.
Примеры, приведенные в следующем экспериментальном разделе, следует рассматривать в качестве примеров предмета способа по настоящему изобретению и не ограничивают объем действительности самого изобретения.
Экспериментальный раздел
Пример 1 (сравнительный)
Синтез промежуточного соединения (II) в соответствии с предшествующим уровнем техники
73 г (0,1796 моль) промежуточного соединения (I) и 1,12 л сухого ТГФ загружают в реактор емкостью 2 л, предварительно высушенный и в инертной атмосфере. Полученную суспензию перемешивают при комнатной температуре в течение 30 мин. Внутреннюю температуру понижают до 3÷5°C и добавляют 64 г тионилхлорида (0,538 моль) в течение 5 мин, затем реакционную смесь нагревают до температуры кипения и выдерживают при перемешивании в течение примерно 1 ч. Добавляют 2 г сухого ДМФА и смесь перемешивают еще в течение 4 ч при той же температуре. Затем реакционную смесь концентрируют при пониженном давлении до остаточного объема примерно 0,3 л. Затем вносят 0,35 л толуола и смесь снова концентрируют. Данную операцию повторяют еще раз. После охлаждения смеси до комнатной температуры добавляют сухой ТГФ (0,7 л) и смесь перемешивают в течение примерно 30 мин. Затем белый осадок промежуточного соединения (II) отфильтровывают и промывают сухим ТГФ. Влажный продукт (107 г) хранят при 5°C и используют как таковой.
Пример 2 (сравнительный)
Синтез гивиностата согласно способу предшествующего уровня техники (эксперимент TT180)
125 мл сухого ТГФ и 60 мл воды загружают в реактор емкостью 1 л, предварительно высушенный и в инертной атмосфере. К этому раствору добавляют водный раствор гидроксиламина (21 г, 50% мас./мас.) и полученную смесь охлаждают до 5°C. 19 г влажного промежуточного продукта (II) из примера 1 (что соответствует 13 г исходного вещества, промежуточного продукта (I)) вносят одной порцией, и внутреннюю температуру постепенно повышают до 20±3°C. Наблюдают постепенное растворение, и полученную смесь выдерживают при перемешивании в течение 30-40 мин. Затем добавляют воду (55 мл) и затем медленно добавляют 6 Н водный раствор HCl (примерно 60 г) до достижения pH <2, поддерживая внутреннюю температуру на уровне 20±3°C. Затем ТГФ удаляют при пониженном давлении до тех пор, пока общий объем не уменьшится примерно наполовину, и не будет наблюдаться белый осадок. Смесь охлаждают до 10°C и выдерживают при перемешивании при этой температуре в течение 30 мин. Продукт выделяют вакуумным фильтрованием и промывают водой. Получают 16,4 г влажного неочищенного гивиностата.
Неочищенный гивиностат суспендируют в растворе NaHCO3 (6,5 г) в 285 мл воды при 20±3°C, затем добавляют ТГФ (265 мл) и наблюдают постепенное растворение. Полученный раствор перемешивают в течение 30 мин, затем добавляют этилацетат (132 мл). Через 15 мин перемешивание прекращают и дают возможность двум фазам разделиться. Водную фазу отбрасывают, и органическую фазу обрабатывают 37% HCl при интенсивном перемешивании до достижения pH <2. Полученную суспензию перемешивают в течение 30 мин, затем осадок отделяют вакуумным фильтрованием и промывают ТГФ. Получают 17 г влажного чистого гивиностата.
Продукт сушат в сушильном шкафу при 30°C при пониженном давлении в течение 16 ч. Получают 7,2 г сухого чистого гивиностата, чистота которого составляет 98,8%, и который показывает присутствие неидентифицированной примеси на уровне 0,23%.
Пример 3
Синтез гивиностата согласно способу предшествующего уровня техники для стадии 1 и способу по изобретению для стадии 2 (эксперимент TT177)
В реакторе емкостью 1 л, предварительно высушенном и в инертной атмосфере, 29,3 г влажного промежуточного продукта (II) из примера 1 (что соответствует 20 г исходного промежуточного продукта (I)) суспендируют в 192 мл сухого ТГФ и внутреннюю температуру доводят до 20±3°С. Одной порцией добавляют водный расвтор гидроксиламина (32 г, 50% мас./мас.) и смесь выдерживают при перемешивании в течение 30-40 мин. Затем добавляют воду (176 мл) и наблюдают постепенное растворение осадка. Медленно добавляют 6 Н водный раствор HCl (примерно 93 г) до достижения pH <2, поддерживая внутреннюю температуру на уровне 20±3°C. Затем ТГФ удаляют при пониженном давлении до тех пор, пока общий объем не уменьшится примерно наполовину, и не будет наблюдаться белый осадок. Смесь охлаждают до 10°C и выдерживают при перемешивании при этой температуре в течение 30 мин. Продукт выделяют вакуумным фильтрованием и промывают водой. Получают 29 г влажного неочищенного гивиностата.
Влажный неочищенный гивиностат суспендируют в растворе NaHCO3 (10 г) в 408 мл воды при 20±3°C, затем добавляют ТГФ (408 мл) и наблюдают постепенное растворение. Полученный раствор перемешивают в течение 30 мин, затем добавляют этилацетат (204 мл). Через 15 мин перемешивание прекращают и дают возможность двум фазам разделиться. Водную фазу отбрасывают, и органическую фазу обрабатывают 37% HCl при интенсивном перемешивании до достижения pH <2. Полученную суспензию перемешивают в течение 30 мин, затем осадок отделяют вакуумным фильтрованием и промывают ТГФ. Получают 31 г влажного чистого гивиностата.
Продукт сушат в сушильном шкафу при 30°C при пониженном давлении в течение 16 ч. Получают 15,6 г сухого чистого гивиностата. Анализ чистоты ВЭЖХ показывает, что продукт не содержит каких-либо неизвестных примесей в количестве выше 0,10%.
Пример 4
Синтез гивиностата согласно способу предшествующего уровня техники для стадии 1 и способу по изобретению для стадии 2 (эксперимент TT183)
В реакторе емкостью 1 л, предварительно высушенном и в инертной атмосфере, 29,3 г влажного промежуточного продукта (II) из примера 1 (что соответствует 20 г исходного промежуточного продукта (I)) суспендируют в 192 мл сухого ТГФ, и внутреннюю температуру доводят до 20±3°С. Добавляют водный раствор гидроксиламина (32 г, 50% мас./мас.), выливая его в течение 15 мин, и смесь оставляют при перемешивании на 30-40 мин. Затем добавляют воду (176 мл) и наблюдают постепенное растворение осадка. Медленно добавляют 6 Н водный раствор HCl (примерно 91 г) до достижения pH <2, поддерживая внутреннюю температуру на уровне 20±3°C. Затем ТГФ удаляют при пониженном давлении до тех пор, пока общий объем не уменьшится примерно наполовину, и не будет наблюдаться белый осадок. Смесь охлаждают до 10°C и выдерживают при перемешивании при этой температуре в течение 30 мин. Продукт выделяют вакуумным фильтрованием и промывают водой. Получают 36 г влажного неочищенного гивиностата.
Влажный неочищенный гивиностат суспендируют в растворе NaHCO3 (10 г) в 408 мл воды при 20±3°C, затем добавляют ТГФ (408 мл) и наблюдают постепенное растворение. Полученный раствор перемешивают в течение 30 мин, затем добавляют этилацетат (204 мл). Через 15 мин перемешивание прекращают и дают возможность двум фазам разделиться. Водную фазу отбрасывают, и органическую фазу обрабатывают 37% HCl при интенсивном перемешивании до достижения pH <2. Полученную суспензию перемешивают в течение 30 мин, затем осадок отделяют вакуумным фильтрованием и промывают ТГФ. Получают 27 г влажного чистого гивиностата.
Продукт сушат в сушильном шкафу при 30°C при пониженном давлении в течение 16 ч. Получают 15,4 г сухого чистого гивиностата. Анализ чистоты ВЭЖХ показывает, что продукт не содержит каких-либо неизвестных примесей в количестве выше 0,10%.
Пример 5
Синтез гивиностата согласно способу предшествующего уровня техники для стадии 1 и способу по изобретению для стадии 2. Промышленный масштаб
В реакторе емкостью 2000 л, предварительно высушенном и в инертной атмосфере, 50,0 кг влажного промежуточного продукта (II) (что соответствует 40 г исходного промежуточного продукта (I)) суспендируют в 400 л сухого ТГФ и внутреннюю температуру доводят до 20±3°C. Добавляют водный раствор гидроксиламина (67,2 кг, 50% мас./мас.), выливая его в течение 15 мин, и смесь оставляют при перемешивании на 30-40 мин. Затем добавляют воду (447 л) и наблюдают постепенное растворение осадка. Медленно добавляют 6 Н водный раствор HCl (примерно 170 кг) до достижения pH <2, поддерживая внутреннюю температуру на уровне 20±3°C. Затем ТГФ удаляют при пониженном давлении до тех пор, пока общий объем не уменьшится примерно наполовину, и не будет наблюдаться белый осадок. Смесь охлаждают до 10°C и выдерживают при перемешивании при этой температуре в течение 30 мин. Продукт выделяют вакуумным фильтрованием и промывают водой. Получают 60 кг влажного неочищенного гивиностата.
Влажный неочищенный гивиностат суспендируют в растворе, состоящем из NaHCO3 (21 кг) в 860 л воды и 860 л ТГФ при 20±3°C, наблюдая постепенное растворение. Полученный раствор перемешивают в течение 30 мин, затем добавляют этилацетат (440 л). Через 15 мин перемешивание прекращают и дают возможность двум фазам разделиться. Водную фазу отбрасывают, и органическую фазу обрабатывают 37% HCl при интенсивном перемешивании до достижения pH <2. Полученную суспензию перемешивают в течение 30 мин, затем осадок отделяют вакуумным фильтрованием и промывают ТГФ. Получают 58 кг влажного чистого гивиностата.
Продукт сушат в сушильном шкафу при 30°C при пониженном давлении в течение 16 ч. Получают 35 кг сухого чистого гивиностата. Анализ чистоты ВЭЖХ показывает, что продукт не содержит каких-либо неизвестных примесей в количестве выше 0,10% и имеет общую чистоту выше 99,5%.
Пример 6 (эксперимент ТТ259)
Синтез гивиностата согласно способу по изобретению (стадия 1+стадия 2)
Стадия 1: синтез промежуточного соединения (II)
100 г (0,2460 моль) промежуточного соединения (I) и 300 мл диметилформамида загружают в реактор емкостью 3 л, предварительно высушенный и в инертной атмосфере. Полученную таким образом суспензию перемешивают при 20-25°C в течение 30 мин. Добавляют 40 г (0,3362 моль) тионилхлорида, наблюдая небольшой экзотермический эффект, который сохраняется для поддержания температуры реакционной смеси 20-25°C. Полученную суспензию выдерживают при 20-25°C еще в течение 2 ч, затем проводят два цикла вакуумирования и восстанавливают давление азотом для удаления газов, образующихся в результате реакции. Затем добавляют 2000 мл ТГФ, выдерживая смесь при 20-25°C в течение 1 ч. Затем суспензию фильтруют через воронку Бюхнера и промывают 300 мл ТГФ. Влажный продукт (133 г) хранят при 5°C и используют как таковой.
Стадия 2: синтез гивиностата
133 г промежуточного соединения (II) со стадии 1 и 960 мл ТГФ загружают в реактор емкостью 3 л, предварительно высушенный и в инертной атмосфере. Полученную суспензию перемешивают и термостатируют при 12-18°C. К суспензии добавляют 160 г (2,424 моль) 50% водного раствора гидроксиламина (реакция является экзотермической, и температура смеси возрастает от 15°C до 28°C). Затем смесь термостатируют при 17-23°C и выдерживают в этих условиях в течение 40 мин.
Затем, поддерживая ту же температуру, добавляют 1100 мл деионизированной воды, наблюдая постепенное растворение осадка. В конце добавляют 220 г 15% водного раствора HCl до достижения pH смеси 1,2-1,8. Смесь перемешивают в течение 30 мин при 17-23°C, затем концентрируют при пониженном давлении, поддерживая внутреннюю температуру 25°C, до остаточного объема примерно 1400 мл. Затем давление восстанавливают и загружают 1000 мл деионизированной воды. Затем смесь охлаждают до 7-13°C и выдерживают при перемешивании в течение 1 ч. Суспензию фильтруют, промывая 400 мл деионизированной воды, подкисленной 1,2 г 37% HCl.
Влажный фильтрат загружают обратно в реактор, в который добавляют 5 кг бикарбоната натрия, 1000 мл деионизированной воды и 1000 мл ТГФ. Смесь перемешивают и нагревают до 47-53°С, выдерживая в этих условиях в течение 3 ч. Затем смесь охлаждают до 17-23°C и дают фазам разделиться. Отделенную водную фазу повторно экстрагируют 500 мл этилацетата. Затем органические экстракты объединяют и к ним добавляют 200 мл 37% HCl при интенсивном перемешивании, наблюдая за осаждением продукта. Смесь перемешивают в течение 30 мин, затем фильтруют и панель промывают 400 мл ТГФ.
Влажный фильтрат загружают обратно в реактор, в который добавляют 5 кг бикарбоната натрия, 1000 мл деионизированной воды и 1000 мл ТГФ. Смесь перемешивают при 17-23°C, выдерживая в этих условиях в течение 30 мин. Затем к смеси добавляют 500 мл этилацетата при 17-23°C, перемешивая в течение 15 мин. Фазам дают разделиться, и отделенную органическую фазу фильтруют на микрофильтре 10 микрон. Затем реактор и линии промывают смесью 120 мл ТГФ и 60 мл этилацетата, и к объединенным органическим фазам добавляют 200 мл 37% HCl, наблюдая осаждение продукта. Смесь выдерживают при перемешивании в течение 30 мин, затем фильтруют и панель промывают 400 мл ТГФ. Продукт выгружают (157 г) и сушат в вакууме (<50 мбар) при 25-35°C в течение 15 ч. Получают 107 г конечного продукта.
Пример 7 (эксперимент ТТ267)
Синтез гивиностата согласно способу по изобретению (стадия 1+стадия 2)
Повторяют приготовление промежуточного соединения (II), которое затем превращают в гивиностат, как описано в примере 6. В конце способа получают 105 г продукта.
Пример 8 (эксперимент ТТ287)
Синтез гивиностата согласно способу по изобретению (стадия 1+стадия 2)
100 г (0,2460 моль) промежуточного соединения (I) и 300 мл диметилформамида загружают в реактор емкостью 3 л, предварительно высушенный и в инертной атмосфере. Полученную суспензию перемешивают при 20-25°C в течение 30 мин. Затем добавляют 40 г (0,3362 моль) тионилхлорида, наблюдая небольшой экзотермический эффект, который сохраняется для поддержания температуры реакционной смеси при 20-25°C. Полученную суспензию выдерживают при 20-25°C еще 2 ч, затем проводят два цикла вакуумирования и восстанавливают давление азотом для удаления газов, образующихся в результате реакции. Затем добавляют 2000 мл толуола, выдерживая смесь при 20-25°C в течение 1 ч. Затем суспензию фильтруют через воронку Бюхнера и промывают 300 мл толуола. Влажный продукт (133 г) хранят при 5°C и используют как таковой.
133 г промежуточного соединения (II) из описанного способа получения и 960 мл ТГФ загружают в реактор емкостью 3 л, предварительно высушенный и в инертной атмосфере. Полученную суспензию перемешивают и термостатируют при 12-18°C. К суспензии добавляют 160 г (2,424 моль) 50% водного раствора гидроксиламина (реакция является экзотермической, и температура смеси повышается от 15°C до 28°C). Затем смесь термостатируют при 17-23°C и выдерживают в этих условиях в течение 40 мин.
Затем, поддерживая ту же температуру, добавляют 1100 мл деионизированной воды, поддерживая температуру 17-23°C и наблюдая постепенное растворение осадка. В конце добавляют 220 г 15% водного раствора HCl до достижения pH смеси 1,2-1,8. Реакционную смесь выдерживают при перемешивании в течение 30 мин при 17-23°C, затем концентрируют при пониженном давлении, поддерживая внутреннюю температуру на уровне 25°C, до достижения остаточного объема примерно 1400 мл. Затем восстанавливают давление и загружают 1000 мл деионизированной воды. Затем смесь охлаждают до 7-13°C и выдерживают при перемешивании в течение 1 ч. Суспензию фильтруют, промывая 400 мл деионизированной воды, подкисленной 1,2 г 37% HCl.
Влажный фильтрат загружают обратно в реактор, в который добавляют 5 кг бикарбоната натрия, 1000 мл деионизированной воды и 1000 мл ТГФ. Смесь перемешивают и нагревают до 47-53°C, поддерживая эти условия в течение 3 ч. Затем смесь охлаждают до 17-23°C и фазам дают разделиться. Отделенную водную фазу повторно экстрагируют 500 мл этилацетата. Затем органические экстракты объединяют и к ним добавляют 200 мл 37% HCl при интенсивном перемешивании, наблюдая за осаждением продукта. Смесь перемешивают 30 мин, затем фильтруют и панель промывают 400 мл ТГФ.
Влажный фильтрат загружают обратно в реактор, в который добавляют 5 кг бикарбоната натрия, 1000 мл деионизированной воды и 1000 мл ТГФ. Смесь перемешивают при 17-23°C, поддерживая эти условия в течение 30 мин. Затем к смеси добавляют 500 мл этилацетата при 17-23°C, перемешивая в течение 15 мин. Фазам дают разделиться, и отделенную органическую фазу фильтруют на микрофильтре 10 микрон. Затем реактор и линии промывают смесью 120 мл ТГФ и 60 мл этилацетата, и к объединенным органическим фазам добавляют 200 мл 37% HCl, наблюдая осаждение продукта. Смесь выдерживают при перемешивании в течение 30 мин, затем панель фильтруют и промывают 400 мл ТГФ. Продукт выгружают (157 г) и сушат в вакууме (<50 мбар) при 25-35°C в течение 15 ч. Получают 107 г конечного продукта.
Пример 9
Способ ВЭЖХ для определения чистоты гивиностата и его примесей
- фаза B: 1000 мл метанола+1,23 г формиата аммония+0,6 мл муравьиной кислоты
Фазы обрабатывают ультразвуком в течение 10-15 мин
Смотри градиент (таблица ниже)
(мин)
%
(об./об.)
%
(об./об.)
Подготовка образца
Взвешивают точную навеску примерно 20 мг образца ITF2357, переносят его в колбу емкостью 100 мл и доволят до объема ДМСО (концентрация 0,20 мг/мл).
Типичная хроматограмма, полученная способом ВЭЖХ в соответствии с изобретением, на специально приготовленной смеси, содержащей все типичные примеси продукта, приведена на фиг. 1.
Пример 11
Анализ чистоты гивиностата, полученного с использованием способа предшествующего уровня (пример 2), с использованием способа, известного в данной области для стадии 1 и способа по изобретению для стадии 2 (примеры 3 и 4), и с использованием способа по изобретению для стадии 1 и стадии 2 (примеры 6, 7 и 8)
(17,6 мин)
[гивиностат]
(18,9 мин)
[амид (Ia)]
(20,4 мин)
(22,9 мин)
[промежуточное соединение(I)]
(24,0 мин)
(28,5 мин)
(33,1 мин)
пример 2
(предшествующий уровень)
пример 3 (по изобретению, только стадия 2)
пример 4
(по изобретению, только стадия 2)
пример 6
(по изобретению, стадия 1+стадия 2)
пример 7
(по изобретению, стадия 1+стадия 2)
пример 8
(по изобретению, стадия 1+стадия 2)
В таблице 1 приведены, в частности, значения относительного времени удерживания гивиностата (RRT 1), метаболита амида (Ia) (RRT 1,08), промежуточного соединения (I) (RRT 1,27) и неизвестных примесей, определенные с использованием анализа ВЭЖХ, описанного в примере 9.
Эксперимент TT 180 (пример 2) относится к повторению способа, используемого на предшествующем уровне, в то время как эксперименты TT 177 (пример 3) и TT 183 (пример 4) относятся к экспериментам, в которых способ по настоящему изобретению применяется для стадии 2, где 50% водный раствор гидроксиламина добавляют к раствору промежуточного соединения (II), полученного в соответствии с предшествующим уровнем, растворенного в ТГФ.
Из анализа данных по чистоте, приведенных в таблице 1, очевидно, что обратный порядок добавления (гидроксиламина к промежуточному соединению (II)) по сравнению со стандартным порядком (промежуточное соединение (II) к гидроксиламину) позволяет получить снижение содержания всех неизвестных примесей ниже 0,10% в расчете на конечный продукт реакции. И наоборот, при использовании стандартного способа концентрация одной примеси значительно превышает предел 0,10%.
Еще одним критическим аспектом для ограничения образования примесей является количество воды, содержащейся в смеси, содержащей промежуточное соединение (II), к которой добавляется гидроксиламин, которое должно быть ограничено в пределах 0,5% от массы смеси. С другой стороны, время добавления гидроксиламина не является ограничением, в то время как, в отличие от способа предшествующего уровня, способ по настоящему изобретению позволяет работать при комнатной температуре, а не при 5°C.
В примерах 6, 7 и 8 были применены обе инновации по настоящему изобретению в отношении стадии 1 и стадии 2. Из значений, представленных в таблице, будет ясно, что гивиностат получают с чистотой более 99,5%, предпочтительно равной или превышающей 99,6%, и что неизвестные примеси не обнаруживаются (предел обнаружения метода 0,02%).
| название | год | авторы | номер документа |
|---|---|---|---|
| СЕЛЕКТИВНЫЕ ИНГИБИТОРЫ HDAC6 | 2018 |
|
RU2764718C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-ЗАМЕЩЕННОГО БЕНЗО[B]ТИОФЕНА (ВАРИАНТЫ), СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 2-ФЕНИЛ-3-АРОИЛБЕНЗО[B]ТИОФЕНА (ВАРИАНТЫ), И ПРОИЗВОДНЫЕ БЕНЗО[B]ТИОФЕНА | 1996 |
|
RU2147582C1 |
| БЕНЗО-N-ГИДРОКСИАМИДНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 2017 |
|
RU2737433C2 |
| СПОСОБ ПОЛУЧЕНИЯ АНТАГОНИСТОВ NMDA (N-МЕТИЛ-D-АСПАРТАТА) | 1997 |
|
RU2190597C2 |
| N-ГИДРОКСИ-БЕНЗАМИДЫ ДЛЯ ЛЕЧЕНИЯ РАКА | 2011 |
|
RU2577861C2 |
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ АНТАГОНИСТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ И АГОНИСТОВ БЕТА-2-АДРЕНЕРГИЧЕСКИХ РЕЦЕПТОРОВ | 2013 |
|
RU2661877C2 |
| АЛКИЛЗАМЕЩЕННЫЕ ЦИКЛИЧЕСКИЕ АМИНЫ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ЛИГАНДОВ D3-ДОПАМИНА | 1997 |
|
RU2185372C2 |
| СИНТЕЗ ТРАНС-8-ХЛОР-5-МЕТИЛ-1-[4-(ПИРИДИН-2-ИЛОКСИ)-ЦИКЛОГЕКСИЛ]-5,6-ДИГИДРО-4Н-2,3,5,10В-ТЕТРААЗАБЕНЗО[E]АЗУЛЕНА И ЕГО КРИСТАЛЛИЧЕСКИЕ ФОРМЫ | 2014 |
|
RU2775690C2 |
| ВОДОРАСТВОРИМЫЕ ПРОИЗВОДНЫЕ ФЕНИЛГЛИЦИНА | 2003 |
|
RU2283305C2 |
| ОКСАДИАЗОЛИДИНДИОНОВОЕ СОЕДИНЕНИЕ | 2007 |
|
RU2440994C2 |

Настоящее изобретение относится к области органической химии и фармацевтики, конкретно к методу синтеза гивиностата ({6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил)фенил]карбамата), который проявляет активность при множественной миеломе и остром миелогенном лейкозе, а также функционирует в качестве противовоспалительного средства. Способ получения гивиностата и/или его фармацевтически приемлемых солей, имеющего количество любой единичной неидентифицированной примеси, равное или ниже 0,10%, включает две стадии. Первая стадия характеризуется взаимодействием галогенирующего агента с соединением формулы (I) в апротонном диполярном растворителе, нечувствительном к кислоте, с получением соединения формулы (II), где X представляет собой атом галогена, предпочтительно атом хлора. Стадия 2 заключается в приготовлении раствора или суспензии соединения формулы (II) в органическом растворителе и добавлении к нему гидроксиламина. Техническим результатом изобретения является обеспечение способом получения высокочистого целевого продукта. 8 з.п. ф-лы, 1 ил., 1 табл., 11 пр.
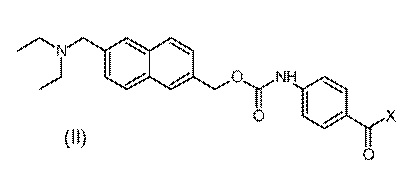
1. Способ получения {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил)фенил]карбамата и/или его фармацевтически приемлемых солей, имеющего количество любой единичной неидентифицированной примеси, равное или ниже 0,10%, где указанный способ включает следующие стадии:
стадия 1
взаимодействие галогенирующего агента с соединением формулы (I)

в апротонном диполярном растворителе, нечувствительном к кислоте, с получением соединения формулы (II)

где X представляет собой атом галогена, предпочтительно атом хлора;
стадия 2
i) приготовление раствора или суспензии соединения формулы (II) в органическом растворителе;
ii) добавление гидроксиламина к раствору или суспензии, полученным на стадии i).
2. Способ по п.1, отличающийся тем, что указанный органический растворитель выбран из группы, включающей ТГФ, метил-ТГФ, диоксан, диметиловый эфир этиленгликоля и бис(2-метоксиэтиловый)эфир.
3. Способ по п.1 или 2, отличающийся тем, что указанный органический растворитель используют в количестве 1-100 частей по объему на 1 часть по массе соединения формулы (II).
4. Способ по любому из предшествующих пунктов, отличающийся тем, что указанный органический растворитель имеет содержание воды ниже 0,5%.
5. Способ по любому из предшествующих пунктов, отличающийся тем, что стадию ii) проводят при комнатной температуре.
6. Способ по любому из предшествующих пунктов, отличающийся тем, что он дополнительно включает стадию iii) выделения {6-[(диэтиламино)метил]нафталин-2-ил}метил[4-(гидроксикарбамоил) фенил]карбамата в виде свободного основания или в виде фармацевтически приемлемой соли, предпочтительно гидрохлорида, более предпочтительно гидрохлорида моногидрата.
7. Способ по любому из предшествующих пунктов, отличающийся тем, что указанный апротонный диполярный растворитель выбран из ДМСО, ацетонитрила, диметилацетамида или диметилформамида.
8. Способ по любому из предшествующих пунктов, отличающийся тем, что указанный галогенирующий агент представляет собой хлорирующий агент, предпочтительно выбранный из тионилхлорида (SOCl2), трихлорида фосфора (PCl3), оксихлорида фосфора (POCl3) или пентахлорида фосфора (PCl5).
9. Способ по любому из предшествующих пунктов, отличающийся тем, что соединение формулы (II) выделяют осаждением органическим растворителем, предпочтительно выбранным из алифатических или ароматических углеводородов, простых эфиров, сложных эфиров или спиртов, более предпочтительно толуола или ТГФ.
| ITMI 20030063 A1, 18.07.2004 | |||
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ ПРОТИВОВОСПАЛИТЕЛЬНОЙ И ИММУНОДЕПРЕССИВНОЙ АКТИВНОСТЬЮ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1997 |
|
RU2177473C2 |
| ПОЛИМОРФ ГИДРОХЛОРИДА (6-ДИЭТИЛАМИНОМЕТИЛ-2-НАФТАЛЕНИЛ)МЕТИЛОВОГО ЭФИРА (4-ГИДРОКСИКАРБАМОИЛФЕНИЛ)-КАРБАМИНОВОЙ КИСЛОТЫ | 2010 |
|
RU2552354C2 |

