Изобретение относится к способу получения бис(гидроксиметил) циклобутил пуринов или пиримидинов новых биологически активных соединений, проявляющих противовирусное действие, которые могут найти применение в ветеринарии.
Поставлена задача способ получения новых производных пурина и пиримидина малотоксичных и обладающих активностью против одного или нескольких видов вирусов, например, вирус герпеса 1 и 2, вирус ветряной оспы, вирус коровьей оспы, marine luhemia вирус и вирус человеческого иммунодефицита [1-2]
Поставленная задача решается описываемым способом получения бис/гидроксиметил/циклобутил пуринов или пиримидинов общей формулы I: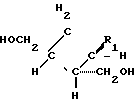





заключающийся в том, что соединения общей формулы: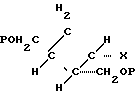 где Р бензоил
где Р бензоил
Х арилсульфонилокси группа, подвергают взаимодействию с соединениям формулы: R1-H, значения R1 указаны выше, причем это соединение является необязательно защищенным, и удаляют защитные группы.
Следующие примеры являются характерной чертой настоящего изобретения [3]
П р и м е р 1. (1α, 2β, 3α)-9-[2,3-бис(гидроксиметил)циклобутил]гуанин.
А) Кетен диэтил ацеталь.
Это соединение было получено, используя метод, описанный в "Organic Syntheses", Coll Vol III E.C. Horning, Ed, John Wiley and Sons, N.I стр. 506 (1955). К раствору трет-бутоксида калия (28,5 г, 0,254 моль) в сухом трет-бутаноле (150 мл, высушенном над 3А молекулярном сите) при 50о, добавили бромацетальдегид диэтил ацеталь (38,5 мл, 0,254 моль). Колонна, заполненная стеклянными спиральными насадками (20 х 1,4 см) с частично-съемной головкой для полного орошения была помещена на верх реакционной колбы. Температура масляной ванны была постепенно поднята до 100оС. После орошения в течение 35 мин отогнали трет-бутанол за 16 ч со скоростью 4,5 капли в 1 мин при соотношении орошения в головке 22:4:5. Масляную ванну охладили до 20о и колонну, заполненную спиральными насадками, заменили на дистиллятор короткого прохождения. Дистилляция при 20-50о и 4 мм дала 26,96 г смеси, содержащей 23,31 г кетен диэтил ацеталя и 3,66 г трет-бутанола, как было определено 1Н-ЯМР интеграцией.
Б) (транс)-3,3-диэтокси-1,2-циклобутандикарбоновая кислота, диэтиловый эфир.
Это соединение было получено модификацией способа K.C.Brannock et al. J. Org Chem 29,840 (1964). К раствору 25,27 г вышеупомянутой смеси, содержащей 21,63 г (0,186 моль) кетен диэтил ацеталя в сухом трет-бутаноле (60 мл), добавили диэтил фумарат (28,28 мл, 0,173 моль). Все это нагревали при 82о в течение 7 дней. Реакцию концентрировали в вакууме и остаток разделили на порции 3 г (А) и 39 г (В).
Порцию А хроматографировали на Мерк силикагель (1,5 х 30 см) в смеси гексан(этилацетат в соотношении 19:1. Фракции, содержащие продукт, объединили и концентрировали и получили 567 мг. Порцию В хроматографировали дважды на Мерк силикагель 60 (35 х 5 см) в том же растворителе. Фракции, содержащие продукт, объединили, концентрировали и получили 10,43 г. Общий выход желаемого вещества составил 10,99 г.
В) (транс)-3,3-диэтокси-1,2-циклобутандиметанол
К суспензии гидрида метилалюминия (2,38 г, 0,0627 моль) в сухом тетрагидрофуране (50 мл) медленно добавили (тран)-3,3-диэтокси-1,2-циклобутандикарбоновую кислоту, диэтиловый эфир (11,29 г, 0,0392 моль) в тетрагидрофуране (25 мл) так, чтобы поддерживать легкое орошение. Реакцию нагревали при 55о в течение 4 ч, а затем разбавили эфиром (100 мл) и вылили в насыщенный водный хлорид аммония. (100 мл). Понизили рН до 4 3М серной кислотой. Суспензию экстрагировали эфиром (4 х 100 мл), а затем хлороформом (3 х 100 мл). Эфирные экстракты объединили, высушили над сульфатом натрия, профильтровали, сконцентрировали и получили 5,613 г желаемого вещества. Объединили экстракты хлороформа, высушили над сульфатом натрия, профильтровали и сконцентрировали, получили 112 мг дополнительного желаемого вещества.
Г) (транс)-3,3-диэтокси-1,2-циклобутандиметанол, эфир дибензоата.
К раствору (транс)-3,3-диэтокси-1,2-циклобутандиметанола (5,7 г, 0,028 моль) в сухом пиридине (40 мл) под действием азота при 0о добавили за 5 мин хлорид бензоила (9,73 мл, 0,0838 моль). Все это нагрели до комнатной температуры, после чего образовался осадок. Через 2 ч добавили воду (20 мл) и реакционную смесь перемешивали в течение ночи. В вакууме удалили растворители. Остаток растворили в диэтилацетате (400 мл) и промыли водой (2 х 150 мл), 1N хлористоводородной кислотой (2 х 150 мл) и насыщенным бикарбонатом натрия (3 х 150 мл). Органический слой высушили над сульфатом натрия, профильтровали и сконцентрировали, что дало 10,97 г целевого вещества.
Д) (транс)-2,3-бис[(тензоилокси)метил]циклобутанон
К раствору (транс)-3,3-диэтокси-1,2-дициклобутандиметанол, эфира дибензоата (10,87 г, 0,0263 моль) в ацетоне (200 мл) добавили п-толуолсерную кислоту (250 мг, 0,00132 моль). Реакцию орошали в течение 3 ч. Раствор концентрировали в вакууме. Остаток растворили в этилацетате (200 мл) и промыли насыщенным бикарбонатом натрия (2 х 200 мл). Водный слой снова экстрагировали этилацетатом (50 мл). Соединили органические слои, высушили над сульфатом натрия, профильтровали и сконцентрировали в вакууме, что дало 8,7 г загрязненного вещества. Этот остаток очистили хроматографией в колонне на Мерк силикагель-60 (5 х 27 см) с использованием в качестве растворителя для элюирования гексан/этилацетата в соотношении 5:1.
Объединили и сконцентрировали фракции, содержащие вещество и получили 6,71 г целевого вещества.
Альтернативное получение (транс)-2,3-бис[(бензоилокси)метил]циклобутанона.
К раствору (транс)-3,3-диэтокси-1,2-циклобутандиметанола, эфира дибензоата (50 г, 0,12 моль) в 1,5 л ацетонитрила добавили 570 мл 0,5М серной кислоты в воде. Реакцию перемешивали под действием аргона в течение 16 ч при 25оС, а затем разбавили 5 л этилацетата. Смесь промыли водой (2 х 1 л) насыщенным бикарбонатом натрия (2 х 1 л), водой (2 х 1 л) и наконец 1 л рассола. Органическую фазу высушили над сульфатом натрия и концентрировали до масла. Растирание в порошок с гексаном дало 34 г сырого вещества. Растирание этого сырого твердого вещества в порошок с 300 мл диэтилового эфира дало 10 г целевого вещества, т.пл. 76-78оС. Охлаждение фильтрата при -30оС в течение 4 ч дало 12 г второй урожай, т.пл. 76-78оС, такой же чистоты.
Е) (1α 2β3β) -3-гидрокси-1,2-циклобутандиметанол, эфир 1,2-дибензоата и (1α 2β3β), 3-гидрокси-1,2-циклобутандиметанол, эфир 1,2-дибензоата
К раствору (транс)-2,3-бис)(бензоилокси)метил/циклобутанона (2,46 г, 7,28 моль) в безводном метаноле (40 мл) добавили цианоборгидрид натрия (1,01 г, 16 моль).
Бромкрезолевый зеленый (3 мг) добавили к рН индикатору. Когда индикатор стал синим, добавили 1N HCl в метаноле пока индикатор не стал желтым. Через 5 ч цвет уже не менялся и исходное вещество было израсходовано. В вакууме удалили растворитель, а остаток растворили в этилацетате (100 мл) и промыли насыщенным хлоридом натрия (50 мл). Водный слой еще раз экстрагировали с этилацетатом (50 мл). Соединили органические слои, высушили над сульфатом натрия, профильтровали и сконцентрировали в вакууме. Остаток очистили хроматографией в колонне на Мерк силикагель-60 (5 х 55 см).
Элюирование с гексаном/этилацетатом в соотношении 7:3 дало 521 мг (1α 2β 3β)-3-гидрокси-1,2-циклобутандиметанол, эфир 1,2-дибензоат.
Элюирование с гексаном/этилацетатом в соотношении 6:3 дало 1,78 г (1α 2β 2α )- 3-гидрокси-1,2-диклобутандиметанол, эфир 1,2-дибензоата.
Альтернативное получение (1α 2β3β)- 3-гидрокси-1,2-циклобутандиметанола, эфира 1,2-дибензоата и (1α 2β3α)-3-гидрокси-1,2-циклобутандиметанола, эфира 1,2-дибензоата.
К перемешенному раствору (транс)-2,3-бис-((бензоилокси/метил/циклобутанона (12,0 г, 0,0355 моль) в безводном тетрагидрофуране при -78оС в азоте добавили 35,5 мл (0,0355 моль) 1М литий три-фторбутил-боргидрид в тетрагидрофуране на 3 мин. Реакцию нагрели до комнатной температуры, а затем добавили насыщенный водный бикарбонат натрия (34 мл) с последующим добавлением по каплям 30% перекиси водорода (13,0 мл, 0,127 моль), поддерживая температуру в 30оС с помощью ледяной водной ванны. Реакцию нагрели до комнатной температуры, перемешали в течение 30 мин и разбавили этилацетатом (400 мл) и водой (120 мл). Разделили слои и органический слой экстрагировали водой (100 мл). Образовалась эмульсия, после чего добавили твердый хлорид натрия для разделения слоев. Два водных слоя объединили и экстрагировали с этилацетатом. Объединили все этилацетатовые слои, высушили над сульфатом натрия, профильтровали и концентрировали в вакууме до остатка (12,5 г). Часть этого остатка 7 г очистили препаративной с высоким давлением жидкой хроматографией на двух Waters Prep Pak 500 силикагель зарядах, используя в качестве растворителя для элюирования 30% этилацетат в гексане со скоростью 250 мл/мин. (1α 2β3β)-3-гидрокси-1,2-циклобутандиметанол, эфир 1,2-дибензоата элюировали 14-22 мин, а (1α 2β3β)- 3-гидрокси-1,2-циклобутандиметанол, эфир 1,2-дибензоата элюировали 23-34 мин. Аналогичная хроматография оставшейся части 12,5 г остатка (в две стадии, одна с использованием 25% этилацетата в гексане, а вторая с использованием 35% этилацетата в гексане) дала всего 8,80 г (1α 2β1β)-3-гидрокси-1,2-циклобутандиметанола, эфира 1,2-дибензоата и 2,6 г (1α 2β3α)-3-гидрокси-1,2-циклобутандиметанола, эфира дибензоата.
Альтернативное получение (1α 2β3β)- 3-гидрокси-1,2-циклобутандиметанола, эфира 1,2-дибензоата
К раствору (транс)2,3-бис((бензоилокси(метил) циклобутанона (13,55 г, 0,0389 моль) в тетрагидрофуране (180 мл) при -78оС в азоте добавили за 5 мин 38,9 мл (0,0389 моль) 1М литий тризиамилборгидрида в тетрагидрофуране. Реакцию перемешивали 10 минут, а затем нагрели до комнатной температуры. Добавили насыщенный бикарбонат натрия (36,9 мл), а затем медленно добавили 30% перекиси водорода (14,19 мл, 0,138 моль), поддерживая температуру 30о с помощью ледяной ванны. Смесь разбавили водой (120 мл) и экстрагировали с этилацетатом (400 мл). Органический слой промыли водой (100 мл), высушили над сульфатом натрия и концентрировали в вакууме, что дало 17,8 г сырого целевого вещества, как остаток, содержащий не выявленный (1α 2β3α )-3-гидрокси-1,2-циклобутандиметанол, эфир 1,2-дибензоата. Остаток очистили препаративной с высоким давлением жидкой хроматографией на двух Waters Prep 500 силикагель колоннах, используя в качестве растворителя для элюирования 30% этилацетат в гексане, что дало 9,17 г (1α 2β3β)-3-гидрокси-1,2-циклобутандиметанола, эфира 1,2-дибензоата.
Альтернативно сырой продукт (42 г), полученный восстановлением 40,5 г (0,12 моль) (транс)-2,3-бис((бензоилокси)метил)циклобутанона с 1М литий тризиамилборгидридом (как указано выше) был растворен в 100 мл смеси гексана и этилацетата (2) и нанесли на сухую подушку 1,2 л мерксиликагель-60. Подушку промыли 5 л того же растворителя 500 мл фракциями. Продукт, содержащий фракции, объединили и выпарили, что дало 39,8 г целевого вещества в виде бесцветной жидкости, достаточно чистой для применения в следующей стадии синтеза.
Ж) (1α 2β3β)-3-[4-метилфенил(сульфонил)-окси]-1,2-циклобутандиметанол, эфир дибензоата.
К раствору (1α 2β3β)-3-гидрокси-1,2-циклобутандиметанола, эфиру 1,2-дибензоата (7,31 г, 0,0215 моль), ранее высушенного концентрацией из безводного пиридина (2 х 20 мл) в 36 мл безводного пиридина добавили хлорид пара-толуолсульфонила (6,56 г, 0,344 моль). Реакцию перемешивали 16 ч при 60о в азоте и удалили пиридин в вакууме. Оставшийся пиридин удалили совместной перегонкой с толуолом (2 х 30 мл). Остаток растворили в этилацетате (480 мл) и промыли насыщенным карбонатом калия. Высушили над сульфатом натрия органический слой, профильтровали и концентрировали в вакууме до остатка, который был очищен хроматографией на Мерк силикагель-60 (1500 мл). Колонна элюировалась 3000 мл этилацетата и гексана в соотношении 1:5. Колонну затем элюировали смесью этилацетата и гексана в соотношении 1:3, собирая 50 мл фракции. Соответствующие фракции объединили и концентрировали, что дало 7,00 г (1α 2β 3β)-3-[(4-метилфенилсульфонил)окси]-1,2- циклобутандиметанола, эфира дибензоата.
Альтернативно, после нагревания (1α 2β3β)-3-гидрокси-1,2-циклобуандиметанола, эфира 1,2-дибензоата (39,8 г, 117 ммоль) с хлоридом пара-толуол-сульфонила (24,65 г, 128,5 ммоль) в 60 мл пиридина при 60оС в течение 22 ч, температуру понизили до 40оС и добавили 2 мл воды. Через 2 ч удалили летучие соединения и остаток разделили между этилацетатом и водой. Органический слой промыли 3% бикарбонатом натрия и высушили над сульфатом натрия. Полученный при концентрации растворителя сырой продукт растерли в порошок с пентаном и получили 38,4 г сырого вещества. Это вещество было растворено в 120 мл этилацетата при легком нагревании. Раствор охладили до комнатной температуры и разбавили 120 мл пентана. Отставание в течение нескольких часов при +5оС дало кристаллы, которые профильтровали, высушили и получили 32,6 г чистого целевого продукта.
Альтернативное приготовление (1α 2β3β)-3-[(4-метилфенил/сульфонил)окси-] -1,2-циклобутандиметанола, эфира дибензоата.
К раствору (1α 2β3α)-3-гидрокси-1,2-циклобутандиметанола, эфира дибензоата (3,096 г, 9,10 моль) в безводном толуоле (25 мл) добавили моногидрат пара-толуолсульфокислоты (2,08 г, 10,9 ммоль), триэтиламин (1,51 мл, 10,9 ммоль), трифенилфосфин (3,81 г, 14,6 ммоль) и диизопропил азодикарбоксилат (2,87 мл, 14,6 ммоль). Реакцию нагрели до 80о под действием азота. Через 1 ч, а затем еще через 3 ч добавили еще трифенилфосфин (1,90 г, 7,3 ммоль) и диизопропил азодикарбоксилат (1,43 мл, 7,3 ммоль). Еще через 3 ч нагревания добавили дополнительное количество трифенилфосфина (0,95 г, 3,65 ммоль) и диизопропила азодикарбоксилата (0,717 мл, 3,65 ммоль). Реакцию продолжали нагревать еще один час, охладили до комнатной температуры и профильтровали. Осадок промыли толуолом (20 мл), фильтрат и промытый осадок соединили и концентрировали в вакууме до остатка, который был растворен в этилацетате (100 мл). Раствор этилацетата промыли водой (2 х 30 мл), высушили над сульфатом натрия, профильтровали и концентрировали в вакууме. Остаток очистили хроматографией на Мерк силикагель-60 (300 мл) с использованием смеси гексана и этилацетата в соотношении 5:1 и соответствующие фракции объединили и сконцентрировали до 20 мл. Этот концентрат разбавили 30 мл гексана и оставили на ночь при комнатной температуре. Фильтрацией собрали кристаллы, промыли гексаном и высушили, что дало 2,18 г (порция 1) чистого целевого вещества.
Маточные растворы из порции 1 подвергли концентрации до 40 мл и оставили на ночь при комнатной температуре. Фильтрацией собрали кристаллы (порция 2), высушили в вакууме и подвергли хроматографии на Мерк силикагель-60 (300 мл) с использованием 2% этилацетата в толуоле, что дало 1,02 г (порция 3) целевого вещества с примесями. Маточные растворы из порции 2 подвергли хроматографии на Мерк силикагель-60 (300 мл) с использованием 2% этилацетата в толуоле, что дало 187 мг (порция 4) целевого вещества с примесями. Объединили порции 3 и 4 и перекристаллизовали из гексана-этилацетата, что дало дополнительные 770 мг чистого целевого вещества.
Общий выход чистого целевого вещества составил 2,95 г.
З) (1α 2β3α)-3-[2-амин-6-[(фенилметокси)-9Н-пирин-9-ил]-1,2-циклобутандиме- танол, эфир 1,2-дибензоата
К раствору (1α 2β3β)-3-[(4-метилфенил-(сульфонил)окси]-1,2-циклобутандиметанола, дибензоата (1,072 г, 2,17 ммоль) в диметилформамиде (20 мл) добавили 2-амино-6-/фенилметокси/-9H-пурин (784 мг, 3,25 ммоль), 18-краун-6 (573 мг, 2,17 ммоль) и карбонат калия (600 мг, 4,34 ммоль). Реакцию перемешивали в азоте при 110о в течение 24 часов. В вакууме удалили растворители в остаток хроматографировали на Мерк силикагель-60 (2,5 х 20 см) с использованием смеси этилацетата в соотношении 3:1, что дало 400 мг чистого целевого продукта. Другие фракции, содержащие целевой продукт с примесями, были объединены и еще раз хроматографированы на Мерк силикагель-60 (1,5 х 30 см) с использованием смеси этилацетата и гексана в соотношении 2:1, что дало дополнительные 52 мг целевого вещества, а общий выход целевого вещества составил 452 мг.
И) (1α 2β3α)-3-[2-амин-6-(фенилметокси)-9Н-пурин-9-ил]1,2-циклобутандиме-танол
К раствору (1α 2β3β)-3-[2-амин-6-(фенилметокси)-9Н-пурин-9-ил]-1,2-цикло-бутандиметанола, эфира 1,2-дибензоата (4252 мг, 0,803 ммоль) в безводном метаноле (12 мл) добавили 25%-ный раствор метоксида натрия в метаноле (109 мл, 0,482 мл). Реакцию перемешивали в азоте 1 ч при 40оС. В вакууме удалили растворитель и добавили воду (10 мл). С помощью 1N HCl понизили рН до 7. Удалили в вакууме растворители и остаток растерли в порошок с эфиром (2 х 20 мл) и высушили, что дало 358 мг сырого целевого продукта, который затем использовали в следующей стадии.
К) (1α 2β3α )-9-[2,3-бис(гидроксиметил)циклобутил]гуанин.
К суспензии (1α 2β3α)-3-[2-амин-6-(фенилметокси)-9Н-пурин-9-ил]-1,2-цикло-бутандиметанола (358 мг, 1,0 ммоль) в метаноле (5 мл) добавили 3N HCl (2,5 мл). Реакцию перемешивали 4 ч при 45оС. В вакууме удалили растворители и остаток растворили в воде (20 мл). С помощью 1N KOH подняли рН до 7. Взяли 10% аликвота и удалили растворители в вакууме. Остаток концентрировали из метанола (3 х 4 мл) и этилацетата (2 х 4 мл). Остаток растворили в воде (4 мл) при нагревании и поместили в колонку с СНР-20Р полимером (1,1 х 20 см, Митсубиси Кемикал Индустриз Лтд. (75-150 микрон). Элюирование с водой, 2% ацетонитрилом (водой и 4% ацетонитролом)водой дало 11 мг целевого вещества.
Оставшиеся 90% реакции концентрировали в вакууме, а затем концентрировали из метанола (3 х 20 мл) и этилацетат (2 х 2 мл). Остаток растворили в воде (30 мл) при нагревании и поместили в колонку с СНР-20Р полимером (2,5 х 15 см). Элюирование с водой, 2% ацетонитрилом/водой, 4% ацетонитрилом/водой и 10% ацетонитрилом/водой дало дополнительные Шмг (1α 2β 3α)-9-[2,3-бис(гидроксиметил/циклобу- тил]гуанин с температурой плавления >220оС.
Расчетный для C11H15N5O3 1,43 3H2O;
C 45,40; H 6,18; N 24,08; H2O 8,83
Экспериментальный: С 45,66; Н 5,95; N 23,82; H2O 8,83.
П р и м е р 2. (1α 2β3α)-3-(6-амин-9Н-пурин-9-ил)-1,2-циклобутандиметанола
А) (1α 2β 3α)-3-(6-амин-9Н-пурин-9-ил)-1,2-циклобутандиметанол, эфир дибензоата
К раствору (1α 2β3β)-3-{[(4-метилфенил)сульфонил]окси}-1,2-циклобутанди-метанола, эфир дибензоата (988 мг, 2 ммоль в безводном диметилформамиде (20 мл) в азоте добавили аденин (405 мг, 33 ммоль), 18-краун-6 (538 мг,2 ммоль) и карбонат калия (276 мг, 2 ммоль). Реакцию нагревали при 110оС 16 ч, а затем в вакууме удалили растворитель, что дало остаток, который очистили хроматографией в колонке на Мерк силикагель-60 (400 мл). Элюирование с 0,1, 0,5, 5 и 10% метаноле в этилацетате дало 522 мг целевого продукта с примесями. Хроматография в колонне этого вещества на Мерк силикагель-60 (400 мл) с использованием смеси дихлорметанметанола в соотношении 20:1 дала 400 мг чистого (1α 2β 3α)-3-(6-амин-9Н-пурин-9-ил)-1,2-циклобу- тандиметанола, эфира дибензоата.
Б) (1α 2β3α)-3-(6-амин-9Н-пурин-9-ил)-1,2-циклобутандиметанол
К суспензии (1α 2β3α)-3-(6-амин-9Н-пурин-9-ил)-1,2-циклобутандиметанола, эфира дибензоата (400 мг, 0,899 ммоль) в безводном метаноле (20 мл) добавили 25% -ный раствор метоксида натрия в метаноле (123 мл, 0,539 ммоль). Смесь перемешивали 45 мин при 40оС, а затем удалили в вакууме растворитель.
Остаток суспендировали в воде (20 мл) с помощью 1N HCl, pH довели до 7,0 и удалили летучие вещества. Остаток очистили хроматографией в колонне на СНР-20Р полимера. Элюирование с водой, градиент 0-20% метанола в воде, а затем 20 и 30% метанола в воде дало 128 мг (1α 2β3α)-3-(6-амин-9Н-пурин-9Н-пурин-9-ил)-1,2-циклобутандиметанола в качестве твердого вещества с температурой плавления 181-183оС.
Найдено, C 52,64; H 6,10; N 28,00
C11H15N2O5 ˙0,1 H2O
Рассчитано, C 52,63; H 6,10- N 27,90
П р и м е р 3. (1α 2β3α)-1-[2,3-бис(гидроксиметил)циклобутил]-5-метил-2,4-(1Н, 3Н)-пиримидиндион.
А) (1α 2β3α)-1-{2,3-бис[(бензоилокси)метил]циклобутил}-5-метил-2,4 (1Н, 3Н)-пиримидиндион
Смесь (1α 2β3α)-3-{[(4-метилфенил)сульфонил]окси}-1,2-циклобутандиме-танола, эфира дибензоата (1,245 г, 2,52 ммоль), тимина (625 мг, 4,96 ммоль), карбоната калия (1,39 г, 10,1 ммоль) в безводном диметилформамиде (12,5 мл) в аргоне, перемешивая, нагревали 15 ч при 105оС, а затем 1 ч при 125оС. Добавили еще тимин (310 мг, 2,2 ммоль) и карбонат калия (354 мг, 2,6 ммоль) и продолжили нагревание при 125оС еще 2 ч. Реагирующую смесь охладили и профильтровали и нерастворимые вещества промыли диметилформамидом. Фильтраты диметилформамида объединили и выпарили до остатка, который растерли в порошок с этилацетатом. Твердые вещества удалили фильтрацией и фильтрат выпарили до остатка. Этот остаток растворили в небольшом количестве этилацетата и гексана (1:1) и поместили в колонну с Мерк силикагель-60 (5 х 11,5 см) и гексане. Элюирование с этилацетатом и гексаном (4:1), а затем этилацетатом дало 219 мг частично очищенного целевого продукта. Хроматография этого вещества на колонне Мерк силикагель-60 в дихлорэтане элюированием с 10, 20, 30 и 50% этилацетатом в дихлорметане дало 166 мг чистого (1α 2β3α)-1-{2,2 бис[(бензоилокси)метил] -циклобутил} -5-метил-2,4-(1Н, 3Н)-пиримидиндиона в виде белого твердого вещества.
Б) (1α 2β 3α)-1-[2,3-бис(гидроксиметил)циклобутил] -5-метил-2,4-(1Н, 3Н)-пиримидиндион.
25% -ный раствор метоксида натрия в метаноле (44,7 мл, 0,196 ммоль) добавили к перемешанной суспензии (1α 2β3α)-1-{2,3-бис[(бензоилокси)метил]циклобутил}-5-метил-2,4 (1Н, 3Н)-пиримидиндиона (146 мг, 0,326 ммоль) в безводном метаноле (4,9 мл) в аргоне при 40оС. Через 4 ч прозрачный раствор охладили до комнатной температуры и концентрировали в вакууме до остатка, который поглотили в воде. рН довели до 7, используя разбавленную хлористоводородную кислоту, и раствор поместили в колонну (1,5 х 21 см) СНР-20Р полимера в воде. Элюирование водой 2, 4 и 10% ацетонитрилом в воде дало после выпаривания и последующей лиофилизации из воды 58 мг (1α 2β3α)-[2,3-бис(гидроксиметил)циклобутил] -5-ме-тил-2,4(1Н, 3Н)-пиримидиндион в виде расплывающего твердого вещества.
Протонный ЯМР (270 мГц, CD3- -CD3 тетраметилсилан II, II (широкий синглет, 1Н), 7,64 (дублет, J= 1,1 Гц, 1Н), 4,56 (мультиплет, 2Н), 4,47 (мультиплет, 2Н), 4,47 (мультиплет, 1Н), 3,44 (мультиплет, 4Н), 2,5 (мультиплет, CD3-
-CD3 тетраметилсилан II, II (широкий синглет, 1Н), 7,64 (дублет, J= 1,1 Гц, 1Н), 4,56 (мультиплет, 2Н), 4,47 (мультиплет, 2Н), 4,47 (мультиплет, 1Н), 3,44 (мультиплет, 4Н), 2,5 (мультиплет, CD3- -CD3 растворитель +1Н), 1,85 (мультиплет, 1Н), 1,84 (мультиплет, 1Н), 1,79 (дублет, J=1,1 Гц, 3Н).
-CD3 растворитель +1Н), 1,85 (мультиплет, 1Н), 1,84 (мультиплет, 1Н), 1,79 (дублет, J=1,1 Гц, 3Н).
П р и м е р 4. (1α 2β3α)-4-амин-1-[2,3-бис(гидроксиметил)-циклобутил]-2 (1Н)-пиримидинон
А) (1α 2β3α)-4-амин-1-{2,3-бис[(бензоилокси)-метил]циклобутил}-2(1H)-пири-мидинон
Смесь (1α 2β3β)-3-{[(4-метилфенил)сульфонил]окси}-1,2-циклобутандиме-танола, эфира дибензоата (1,51 г 3,05 ммоль), цитозина (6,78 мг, 6,10 ммоль), карбоната калия (1,69 г, 12,2 ммоль) и 18-краун-6 (804 мг, 3,04 ммоль) в 12,5 мл безводного диметилсульфоксида перемешивали в аргоне при 112о в течение 4,5 ч. Реакционную смесь охладили до комнатной температуры и нейтрализовали добавлением ледяной уксусной кислоты (0,7 мл, 12,2 ммоль). Растворитель удалили в вакууме и остаток был поглощен в этилацетате. Твердое вещество удалили фильтрацией и фильтровальный осадок промыли этилацетатом. Фильтрат этилацетата концентрировали до остатка, который растворили в толуоле и поместили в колонну с Мерк силикагель-60 в толуоле. Элюирование изопропанолом в толуоле дало 156 мг целевого вещества.
В) (1α 2β3α)-4-амин-1-[2,3-бис(гидроксиметил)циклобутил]-2(1H)-пиримиди-ном
25% -ный раствор метоксида натрия в метаноле (48 мл, 0,209 ммоль) добавили к раствору (1α 2β3α)-4-амин-1-[2,3-бис[(бензоилокси)метил/циклобутил]-2 (1Н)-пиримидинон (151,3 мг, 0,349 ммоль) в 5,25 мл безводного метанола. Реакцию перемешивали 75 мин при 40о и охладили до комнатной температуре. В вакууме удалили растворитель и остаток растворили в воде. С помощью 1N HCl довели рН до 7. Водный раствор загрузили в колонну с СНР-20Р полимером в воде и колонну вымыли 50 мл воды и элюировали с градиентом 0-50% ацетонитрила в воде. Комбинирование соответствующих фракций и удаление растворителя в вакууме дало целевой продукт в виде прозрачного стекла (52 мг).
Протонный ЯМР (270 мг, CD3- -CD3 тетраметилсилан 7,69 (дублет, y=7 Гц, 1Н), 6,98 (широкий синглет, 2Н), 5,71 (дублет, y=7,6 Гц, 1Н), 4,65 широкий мультиплет, 2Н), 4,37 (мультиплет, 1Н), 3,43 (мультиплет, 4Н), 2,43 (мультиплет, 1Н), 2,31 (мультиплет, 1Н), 2,20 (мультиплет, 1Н), 1,77 (мультиплет, 1Н).
-CD3 тетраметилсилан 7,69 (дублет, y=7 Гц, 1Н), 6,98 (широкий синглет, 2Н), 5,71 (дублет, y=7,6 Гц, 1Н), 4,65 широкий мультиплет, 2Н), 4,37 (мультиплет, 1Н), 3,43 (мультиплет, 4Н), 2,43 (мультиплет, 1Н), 2,31 (мультиплет, 1Н), 2,20 (мультиплет, 1Н), 1,77 (мультиплет, 1Н).
П р и м е р 5. (1α(E) 2β3α)-1-[2,3-бис(гидроксиметил)-циклобутил]-5-(2-бром- этенил)-2,4 (1Н, 3Н), пиримидиндион.
А) (1α 2β3α)-1-{2,3-бис[бензоилокси/метил]-циклобутил}-2,4(1Н0 3Н)-пиримидиндион
К раствору урацила (1,26 г, 11,23 ммоль), высушенного в течение 16 ч при 50оС и 18-краун-6 (1,98 г, 7,49 ммоль) в диметилсульфоксида (9 мл) при 50оС, добавили карбонат калия (2,07 г, 14,98 ммоль) и (1α 2β3β)-3-{[(4-метилфенил)сульфонил] окси} -1,2-цикло- бутандиметанол, эфир дибензоат (3,7 г, 7,49 ммоль). При нагревании при 100оС в азоте образовалась эмульсия. Добавили еще диметилсульфоксид (3 мл) и реакцию перемешивали при 100оС в течение 24 ч. В вакууме удалили растворители и получили остаток, который очистили хроматографией на Мерк силикагель-60 (700 мл), используя градиент толуола до 3% изопропилового спирта в толуоле. Соединили соответствующие фракции и получили 850 мг чистого целевого продукта. Фракции, содержащие целевой продукт с примесями были объединены и подвергли концентрации до остатка, который растворили в толуоле (1 мл). Собрали полученные кристаллы, высушили и получили 35 мг дополнительного чистого целевого продукта.
Б) (1α 2β3α)-1-[2,3-бис(гидроксиметил)-циклобутил]-2,4(1H, 3H)-пиримидиндион
К суспензии (1α 2β3α)-1-{2,3-бис [(бензоилокси)метил]циклобутил}-2,4(1H, 3H)-пиримидиндиона (885 мг, 2,04 ммоль) в безводном метаноле (25 мл) добавили 25%-ный раствор метоксида натрия в метаноле (264 мл, 1,22 ммоль). Реакцию нагревали 3 ч при 40оС в азоте. Растворители удалили в вакууме, а остаток растворили в воде (5 мл). Понизили рН до 7 с помощью 1N HCl и раствор оставили на ночь при 0о. Полученный осадок и всплывшее вещество очистили в колонне с единственным СНР-20Р полимером (200 мл), используя стадийный градиент воды, 2% ацетонитрила/воды и 4% ацетонитрила/воды и получили 423 мг целевого вещества.
В) (1α 2β3α)-1-[2,3-бис[гидроксиметил/циклобутил]-5-иод-2,4(1Н, 3Н)-пиримидиндион.
К суспензии (1α 2β3α)-1-[2,3-бис(гидроксиметил/циклобутил]-2,4(1H, 3H) пиримидиндиона 3423 мг, 1,87 ммоль/ в диоксане (38 мл, очищенном на основном оксиде алюминия) добавили иод (950 мг, 3,74 ммоль) и 0,8М азотной кислоты (2,5 мл, 2 ммоль). Этот раствор перемешивали 90 мин при 95оС, а затем охладили до комнатной температуры. Добавляли раствор насыщенного водного тиосульфата натрия до тех пор, пока не пропал красный цвет. Реакцию концентрировали в вакууме и получили слегка желтоватый остаток. Это вещество очистили хроматографией на СНР-20Р полимере (150 мл), используя градиент воды к 50% ацетонитрила в воде, что дало 557 мг целевого продукта.
Г) [1α(E) 2β3α-3-[1-[2,3-бис(гидроксиметил)-циклобутил] 1,2,3,4-тетрагидро-2,4-диоксо-5-пиримидинил]-2-пропеновая кислота, метиловый эфир
Суспензию палладий (П) ацетата (17,5 мг, 0,078 ммоль) трифенилфосфина (40,9 мг, 0,15 ммоль) и триэтиламина (290 мл, 2,08 ммоль) в диоксане 320 мл, очищенном на основном оксиде алюминия, нагревали 15 мин при 85о в азоте, а затем добавили раствор (1α 2β3α)-1-[2,3-бис(гидроксиметил)циклобутил]-5-иод-2,4-(1Н, 3Н)-пиримидиндионе (457 мг, 1,3 ммоль) и метилакрилат (468 мл, 5,2 ммоль) в диоксане 310 мл). Реакцию нагревали при 85оС в азоте. Через 4 ч добавили еще метилакрилат (234 мл, 2,7 ммоль). После дополнительного нагревания в течение 2 ч реакция еще не была завершена. Добавили целит (300 мг) и теплую реакцию профильтровали. В вакууме удалили растворители. Остаток высушили концентрацией из безводного диоксана (2 х 10 мл) и остаток подвергли следующим условиям реакции.
Реакцию повторили, но на этот раз удалили кислород из диоксана, пропуская пузырьки аргона через растворитель. После нагревания суспензии палладия (П) ацетата (17,5 мг, 0,078 ммоль), трифенилциофина (40,9 мг, 0,15 ммоль) и триэтиламина (290 мл, 2,08 ммоль) в диоксане (20 мл) в течение 15 мин при 85о в азоте, добавили раствор вышеупомянутого остатка и метилакрилата (468 мл, 5,2 ммоль) в диоксане (10 мл). Реакцию нагревали 3 ч при 85о. Добавили целит (300 мг) и теплую реакцию профильтровали, охладили до комнатной температуры и концентрировали в вакууме. Остаток поместили в колонну Мерк силикагель-60 (150 мл в хлороформе) и очистили, используя стадийный градиент из хлороформа к 5, 7,5, 10% метанол/хлороформа. Объединили соответствующие фракции и концентрировали, что дало 310 мг целевого вещества, загрязненного солями триэтиламмония. Эту смесь растворили в воде (5 мл) и этилацетате (50 мл). Разделили слои и слой воды экстрагировали этилацетатом (4 х 30 мл). Слои этилацетата соединили, высушили над сульфатом натрия, профильтровали и концентрировали, что дало 230 мг целевого вещества.
Д) (1α(E) 2β3α)-3-{1-[2,3-бис(гидроксиметил)циклобутил]-1,2,3,4-тетрагидро-2, 4-диоксо-5-пиримидинил]-2-пропеновая кислота
Раствор [1α(E) 2β3β]-3-[1-[2,3-бис(гидроксиметил)циклобутил]-1,2,3,4-тет- рагидро-2, 4-диоксо-5-пиримидинил]-2-пропеновой кислоты, метиловый эфир (230 мл, 0,742 ммоль) в 2М гидроксиде натрия (3,7 мл, 7,42 ммоль) перемешивали 1,5 ч при комнатной температуре и реакцию охладили до 40оС. Понизили рН до 2 с 6 N HCl и оставили реакцию на 1 ч при 4оС. Фильтрацией собрали осадок, промыли водой и высушили над P2O5 в вакууме за 16 ч, что дало 120 мг целевого вещества. Маточные растворы и промывку концентрировали до 3 мл и оставили на 16 ч при 4оС. Собрали кристаллы, промыли водой, высушили над P2O5 в вакууме за 4 ч и получили 7 мг дополнительного целевого вещества.
Е) (1α(E) 2β3α)-3-[1-2,3-бис(гидроксиметил)-циклобутил]-5(2-бромэтенил)-2,4(1Н, 3Н)-пиримидиндион.
К раствору (1α(E) 2β 3α)-3-[1-[2,3-бис(гидроксиметил)циклобутил]-1,2,3,4-тет- рагидро-2, 4-диоксо-5-пиримидинил]-2-пропеновой кислоты (127 мг, 0,429 ммоль, высушенному выпариванием диметилформамида, 2 х 3 мл) в диметилформамиде (2 мл) добави- ли бикарбонат калия (129 мг, 1,29 ммоль). Добавили раствор N-бромсукцинимида (76 мг, 0,429 ммоль) в диметилформамиде (1 мл) и реакцию перемешивали 2,5 ч при комнатной температуре. Реакцию профильтровали и концентрировали в вакууме. Остаток концентрировали из воды (2 х 5 мл) и хроматографировали на СНР-20Р полимере (110 мл) с использованием градиента воды к 30% ацетонитрила в воде, что дало после концентрации в вакууме 99 мг [1α(E) 2β3α]-1-[2,3-бис(гидроксиметил)циклобутил]-5-(2- бромэтенил)-2,4 (1Н, 3Н)-пиримидиндиона с температурой плавления 155-157оС.
Рассчитано, C 42,79; H 4,68, N 8,32
C12H15N2O4Br˙0,31 H2O
Найдено, C 42,85; H 4,69; N 8,26
П р и м е р 6. (1α 2β3α)-2-амин-9-[2,3-бис(гидроксиметил)-циклобутил]-8- бром-1,9-дигидро- 6Н-пурин-6-ОН.
К перемешенной суспензии (1α 2β 3α)-9-[2,3-бис(гидроксиметил)циклобутил [гуанина (72 мг, 0,272 ммоль/в воде (9 мл) добавили 0,5 мл насыщенного раствора брома-воды. Через 25 мин добавили еще 0,5 мл раствора брома-воды. Через 15 мин дополнительного перемешивания профильтровали выпавшее в осадок вещество, промыли водой, суспендировали в воде (3 мл) и поместили в СНР-20Р колонну (24 мл) в воде. Элюирование водой, 4% ацетонитрилом/водой и 8% ацетонитрилом/водой дало 45 мг целевого вещества. Это вещество соединили с 49 мг целевого вещества из аналогичной реакции и соединенные вещества кристаллизировали из воды (7 мл) и получили 74 мг (1α 2β3α)2-амин-9-[2,3-бис(гидроксиметил)циклобутил] -8-бром-1,9-дигидро- 6Н-пурин-60Н с температурой плавления 130о.
ЯМР (270 мГц CD3- -CD3 тетраметилсилан δ: 10,67 (широкий синглет, 1Н), 6,43 (широкий синглет, 2Н), 4,59 (квартет, J=9 Гц, 1Н), 4,55 (триплет, J=5 Гц, 1Н), 4,48 (триплет, J= 5 Гц, 1Н), 3,59 (триплет, J=6 Гц, 2Н), 3,44 (триплет, J= 5 Гц, 2Н), 2,56 (мультиплет, 2Н), 2,26 (мультиплет, 1Н), 2,21 (мультиплет, 1Н).
-CD3 тетраметилсилан δ: 10,67 (широкий синглет, 1Н), 6,43 (широкий синглет, 2Н), 4,59 (квартет, J=9 Гц, 1Н), 4,55 (триплет, J=5 Гц, 1Н), 4,48 (триплет, J= 5 Гц, 1Н), 3,59 (триплет, J=6 Гц, 2Н), 3,44 (триплет, J= 5 Гц, 2Н), 2,56 (мультиплет, 2Н), 2,26 (мультиплет, 1Н), 2,21 (мультиплет, 1Н).
П р и м е р 7. (1α 2β3α)-1-[2,3-бис(гидроксиметил)циклобутил]-5-иод-2,4-(1Н, 3Н)-пиримидиндион.
А) (1α 2β3α)-1-[2,3-бис(бензоилокси)метил/-циклобутил]-2,4 (1H, 3H-пиримидиндион
Смесь (1α2β,3β)-3-{[4-метилфенил/сульфонил]окси}-1,2-циклобутандиметанола, эфира дибензоата (1,25 г, 5,07 ммоль), урацила (0,567 г, 5,07 ммоль), карбоната калия (1,40 г, 1012 ммоль) и 18-краун-6 (670 мг, 2,54 ммоль) в безводном диметилсульфоксиде (12,5 мл) нагревали 4,5 ч при 110оС. В вакууме удалили растворитель и полученное полутвердое вещество дважды растерли в порошок с этилацетатом. Соединенные образующие сверху вещества этилацетата концентрировали до небольшого объема, разбавили равным объемом гексана и поместили в Мерк силикагель-60 колонну (2,5 х 25 см) в гексане. Колонну элюировали смесью этилацетата и гексана в соотношении 1:4 и 1:1, а затем этилацетатом и получили частично очищенный целевой продукт (250 мг). Хроматография этого вещества на силикагелевой колонне (1,5 х 24,5 см) в метиленовом хлориде, используя в качестве элюента смесь этилацетата и метиленового хлорида в соотношении 1:4 и 1:1, а затем этилацетата не помогла растворить примеси. Последующая хроматография на силикагелевой колонне (1,5 х 25 см) в толуоле и элюирование смесью изопропанола и толуола в соотношении 4:96 дали целевой чистый продукт (56,5 мг), а также и целевой продукт с примесями. Перекристаллизация из толуола целевого продукта с примесями дало дополнительное целевое вещество (86,3 мг, общий выход 143 мг).
Б) (1α 2β3α)-1-[2,3-бис(гидроксиметил)циклобутил]-2,4-(1H, 3H)-пиримидиндион
Смесь (1α 2β3α)-1-[2,3-бис(бензоилокси)метилциклобутил]-2,4 (1H, 3H)-пиримидиндиона (142,9 мг, 0,329 ммоль), 45 мл 25%-ного раствора метоксида натрия в метаноле и 4,9 мл безводного метанола перемешивали при 40о в аргоне в течение 8,5 ч. Реакцию охладили до комнатной температуры и удалили в вакууме растворитель. Липкий остаток частично растворили в нескольких миллилитрах воды и довели рН до 7 разбавленной хлористоводородной кислотой и бикарбонатом натрия. Этот раствор (7-8 мл) поместили в колонну СНР-20Р полимера (1,5 х 23,5 см) в воде. После элюирования с водой ( ≈50 мл), колонну элюировали водным ацетонитрилом (2, 4 и 10%) и получили 55,8 мг целевого вещества в виде твердого вещества.
В) (1α 2β3α)-1-[2,3-бис(гидроксиметил)циклобутил]-5-иод-2,4-1Н,3Н)-пирими-диндион
Раствор (1α 2β3α)[1-[2,3-бис(гидроксиметил)циклобутил]2,4(1H,3H)-пирими- диндиона (54,7 мг, 0,242 ммоль), иода (123 мг, 0,484 ммоль) и водной азотной кислоты (0,8 N, 0,256 ммоль) в 5 мл диоксана перемешивали при 105оС 85 мин. После охлаждения до комнатной температуры смесь обесцветили водным тиосульфатом натрия и концентрировали в вакууме до твердого вещества. Твердое вещество поглотили в воде и концентрировали в вакууме (3 раза). Полученное твердое вещество частично растворили в воде и поместили в колонну СНР-20Р полимера (1,5 х 20 см) в воде. После элюирования водой (50 мл) колонну элюировали непрерывным градиентом из воды -50% ацетонитрила в воде и получили 67,3 мг (1α 2β3α)-1-[2,3-бис(гидроксиметил)циклобутил] 5-иод-2,4(1Н,3Н)-пиримидиндиона в виде твердого вещества с температурой плавления 170-171о.
Рассчитано, C 33,65; H 3,82; N 7,85
C10H13/N2O4 ˙0,27 H2O
Найдено,C 33,68; H 3,77; N 7,82
П р и м е р 8. (1α 2β3α)-5-амин-3-[2,3-бис(гидроксиметил)циклобутил]-3,6-ди- гидро-7Н- 1,2,3-триазол[4,5-d] пиримидин-7-ОН.
А) Хлорид 4-хлорбензолдиазонил.
К суспензии 4-хлоранилина (21,14 г, 0,166 мл) в воде 156 мл и 12 NHCl (46 мл) при 0оС добавили нитрит натрия (12,69 г, 0,182 ммоль) в воде (156 мл) в течение 20 мин, поддерживая температуру реакции ниже 10оС. Раствор хлорида 4-хлорбензолдиазония профильтровали, выдержали 30 мин при 0о, а затем использовали в следующей стадии.
Б) 6-хлор-5-[(4-хлорфенил)азо]-2,4-пиримидиндиамин.
К суспензии 4-хлор-2,6-диаминпиримидина (21,68 г, 0,150 мл) в воде (750 мл) в уксусной кислоте (750 мл) добавили ацетат натрия (300 г). Раствор получился после 20 мин перемешивания и затем добавили хлорид 4-хлорбензолдиазония при охлаждении в течение 30 мин со скоростью, которая поддерживала реакцию при 18оС. Реакцию перемешивали в течение ночи при комнатной температуре, после чего профильтровали оранжевые кристаллы, промыли водой (4 х 400 мл) и высушили в вакууме, что дало 17,6 г 6-хлор-5-[(4-хлорфенил)азо]-2,4-пиримидиндиамина. Охладили до 5ов течение 20 ч маточные растворы, собрали кристаллы, высушили в вакууме и получили 6,94 г дополнительного 6-хлор-5[(4-хлорфенил)-азо]-2, 4-пиримидиндиамина.
В) 6-хлор-2,4,5-пиримидинтриамин.
Суспензию 6-хлор-5-[(4-хлорфенил)азо] -2,4-пиримидиндиамина (24,55 г, 0,0906 моль) в этаноле (640 мл) нагрели до 70оС в азоте. Медленно в течение 1 ч добавили цинковую пыль (75 г), а затем реакцию перемешивали еще 1 ч при 70оС. Затем реакцию охладили до комнатной температуры и профильтровали в азоте. Фильтраты охладили до 0о и подняли рН до 10 с помощью гидроокиси натрия (400 мл).
Выпавшую в осадок гидроокись цинка удалили фильтрацией через броунмиллерит, а темно-красный фильтрат нейтрализовали до рН 7 ледяной уксусной кислотой и концентрировали до 300 мл. Добавили воду (50 мл), охладили реакцию до 0о и рН подняли до 9 с помощью 10% NaOH. Раствор оставили на 3 дня при 5о. Собрали кристаллы, промыли водой (50 мл) и эфиром (50 мл), в течение 16 ч сушили при 35оС в вакууме, что дало 10,94 г целевого вещества.
Г) 7-хлор-1Н-1,2,3-триазол[4,5-d] пиримидин-5-амин
Раствор 6-хлор-2,4,5-пиримидинтриамина (10,94 г, 0,0686 моль) и нитрита изоамила (9,20 мл, 0,0686 моль) в диоксане (500 мл, только что очищенного прохождения через основный оксид алюминия) нагревали под действием азота 2 ч, перемешивая при 90оС. Реагирующую смесь охладили, обработали активированным углем, профильтровали и концентрировали до 150 мл. Добавили петролейный эфир (250 мл, температура кипения 35-60о). Профильтровали осадок, промыли петролейный эфир (50 мл) и высушили в вакууме над P2O5 при 40оС за 16 ч, что дало 9,23 г сырого целевого вещества, которое потом использовали в следующей стадии.
Д) 7-(фенилметокси)-1Н-1,2,3-триазол[4,5-d] пиримидин-5-амин.
Натриевый металл (3,7 г, 0,162 моль) добавили частями к бензиловому спирту (117 мл, 1,13 моль) под действием азота за 20 мин. Затем реакцию нагревали 90 мин при 80оС. Весь натриевый металл растворился и реакцию оставили на 16 ч при комнатной температуре. Затем добавили 7-хлор-1Н-1,2,3-триазол [4,5-d] пиримидин-5-амин (9,23 г, 0,0541 моль) и реакцию нагревали 5 ч при 60оС. Реакцию охладили и оставили на 16 ч при 5оС. Добавили воду (500 мл) для растворения остатка и затем смесь экстрагировали с эфиром (3 х 200 мл). Понизили рН водного слоя до 7 с помощью концентрированной HCl, а затем до 5,5 с 1N HCl. Осадок профильтровали и высушили при комнатной температуре над P2O5 в вакууме, что дало 8,05 г целевого вещества.
Е) (1α 2β3α)-3-[5-амин-7-(фенилметокси)-3Н-1,2,3-триазол [4,5-d]-пиримидин-3-ил]-1,2-циклобутандиметанол, эфир дибензоата
К суспензии 60% NaН (78 мг, 1,96 ммоль) в диметилформамиде (4 мл) под действием азота добавили 7-(фенилметокси)-1Н-1,2,3-триазол [4,5-d] пиримидин-5-амин (474 мг, 1,96 ммоль). Через 10 мин добавили (1α 2β3β)-3-{[(4-метилфенил)сульфонил] окси} -1,2-циклобутандиметанол, эфир дибензоата (880 мг, 1,78 ммоль) и реакцию нагревали 24 ч при 85оС. Удалили в вакууме растворители и остаток растерли в порошок с этилацетатом (3 х 30 мл) и профильтровали. Объединили экстракты этилацетата и концентрировали до остатка, который очистили на Мерк силикагель-60 (100 мл), элюируя с поэтапным градиентом в 10% этилацетате в гексане до 100% этилацетата. Целевой продукт, элюированный с 30% этилацетата в гексане, дал 205 мг (1α 2β 3α)-3-[5-амин-7-(фенилметокси)-3Н-1,2,3- триазол [4,5-d]-пиримидин-3-ил]-1,2-циклобутандиметанол, эфир дибензоата.
Ж) (1α 2β3α)-3-[5-амин-7-(фенилметокси)-3Н-1,2,3-триазол [4,5-d]-пиримидин-3-ил]-1,2-циклобутандиметанол.
К раствору (1α 2β3α)-3-[5-амин-7-(фенилметокси)-3Н-1,2,3-триазол [4,5-d] -пиримидин-3-ил] -1,2-циклобутандиметанола, эфира дибензоата (205 мг, 0,363 ммоль) в безводном метаноле (6 мл) добавили 25%-ный раствор метоксида натрия в метаноле (50 мл, 0,218 ммоль). Все это нагревали 1 ч до 40о под действием азота, затем добавили воду (2 мл), а рН довели до 7 с помощью 1М HCl. Реакцию концентрировали в вакууме и получили сырой целевой продукт.
З) (1α 2β3α)-5-амин-3-[2,3-бис(гидроксиметил)циклобутил-3,6-дигидро-7Н- 1,2,3-триазол [4,5-d] пиримидин-7-ОН]
Сырой (1α 2β3α)-3-[5-амин-7-(фенилметокси)-3Н-1,2,3-триазол [4,5-d]-пиримидин-3-ил]-1,2-циклобутандиметанол суспендировали в 10,5 мл метанола, а затем добавили 3N HCl (600 мл). Реакцию нагревали до 45оС в течение 4 ч и оставили при комнатной температуре на 16 ч. Подняли рН до 7 с 1N KOH и раствор концентрировали в вакууме до остатка. Хроматография этого остатка на ССР-20Р полимере (34 мл), используя градиент воды до 70% ацетонитрила в воде, дало 64 мг (1α 2β 3α)-5-амин-3-[2,3-бис(гидроксиметил)цик- лобутил]-3,6-дигидро-7Н-1,2,3-триазол [4,5-d] пиримидин-7-он с температурой плавления больше 200о.
Рассчитано, C 38,57; H 6,15; N 26,99
C10H14N6O3 ˙2,5H2O
Найдено, C 39,17; H 4,98; N 26,51.
П р и м е р 9. Лечение вирусных инфекций в культуре клеток.
Проведены анализы в системах культуры клеток для определения концентраций, которые эффективны для предотвращения нескольких видов вирусных инфекций. Анализы описываются ниже, а результаты приводятся в таблице.
Сокращения:
HSV-1 (вирус герпеса, тип 1)
HSV-2 (вирус герпеса, тип 2)
VZV (вирус ветряной оспы)
HCMV (чел. вирус цитомегалии)
MuLV (murine вирус лейкоза)
Анализы уменьшения бляшек:
Вирус (герпеса 1, герпеса 2, цитомегалии, ветряной оспы) был адсорбирован в WI-38 однослойных клетках культуры в чашках с культурой с 6 ячейками (Costar, Cambridge, MA) в течение 1 ч перед тем как добавили поддерживающую среду, содержащую сдвоенные разбавления опытного соединения. Торможение развития бляшек было определено на неподвижных и окрашенных монослоях через 4 дня инкубирования при 37оС для HSV-1 и HSV-2 и через 6-7 дней инкубирования при 37оС для HCMV и VZV.
LD50 величины были определены из концентрации лекарства, которые подтвердили по крайней мере 50% уменьшение бляшек по сравнению с контрольными вирусами.
Противовирусные анализы с MuLV были проведены с некоторыми модификациями, как описано Rowe et al. and Shanuon et al, SC-1. Клетки были посажены приблизительно 2 х 105 клеток на ячейку в чашки с 6 ячейками. После инкубирования в течение ночи при 37оС культуру клеток сенсибилизировали в течение 1 ч при 37оС с помощью DEAE DEXTRAN, промыли и инкубировали с MuLV. В культуру вновь добавили среду роста, содержащую различные концентрации опытного соединения. Через три дня при 37оС в культуру вновь добавили свежую среду и опытные соединения и инкубировали при 37оС еще 3 дня. Культуру затем промыли для удаления среды, облучили ультрафиолетовым светом и поместили в среду для роста клеток, содержащую соответствующую концентрацию опытного соединения, приблизительно 5 х 105 ХС клеток на ячейку. Затем культуру инкубировали еще 4 дня, с добавлением на второй день среды роста, содержащей опытное соединение, с последующим присоединением ХС клетки. Затем культуру ополоснули, окрасили и подсчитали соклеточные бляшки.
Сравнительные испытания.
Антивирусная активность и активность по ингибированию роста клеток определялись следующим образом:
Антивирусные опыты в культуре клеток. Вирус Herpes Siviplex тип-1 (HSV-1) штамм Shooler и штамм HSV-2 186 приготавливались в виде экстрактов из культур клеток, инфицированных Vero Штамм AD 169 циматогаловируса человека (HCMVI) и штамм Е11ЕП вируса варицелле зостер (VZV) приготавливались в виде суспензии клеток, инфицированных WI-38. Клетки WI-38 выращивались в минимально неотъемлемой среде Eagles с солью Eoples (EMEM) с добавкой 2 мкМ 2-глютамина, 100 единиц/мл пенициллина, 11 мкг/мл стрептомицина и 10% зародышевой бычьей сыворотки (Джибко Лабораториз, Гранд Айланд, НИ).
Вирусы анализировались на монослоях клеток WI-38. Вирусы абсорбировались на клеточные монослои на 6-ячеистых пластинах для культуры (Костар, Кембридж, МА) в течение 1-2 ч перед добавлением среды для поддержания жизнедеятельности (ЕМЕМ плюс добавки, 1% карбоксиметилцеллюлоза, 2,5% зародышевая (внутриутробная) бычья сыворотка + лекарство), содержащая двукратные разбавления испытываемого соединения. Спустя 4-6 дней после инкубации при 37оС оценивалось ингибирование развития бляшек (тромбоцитов). Величины ID50 оценивались по концентрации лекарства, которая обеспечивает 50% снижение количества бляшек, в сравнении с вирусными контролями. Все титрования производились в двух повторениях и выражались в виде интервала в повторных анализах.
Ингибирование роста клеток Клетки WI-38 наносились в количестве 1 х 105 клеток/мл на 12-ячеистые платины для культуры клеток Костар. Через 24 ч в культуры клеток повторно добавлялась среда для роста, содержащая антивирусное соединение с рядом разбавлений. С интервалами 24 ч культуры клеток в каждой концентрации с четырехкратным повторением повторно суспендировались с помощью трипсинизации, производился подсчет жизнеспособных и мертвых клеток с использованием критерия исключения (эксклюзии) трипана синего. Контрольные культуры оценивались аналогичным образом, на протяжении 96-часового периода увеличивались в 3-5 раз. Показатель ID50 для каждого соединения вычислялся в виде концентрации, которая ингибировала рост на 50% по отношению к контрольным культурам клеток.


Использование: в химической промышленности при получении соединений, обладающих антигерпесной активностью. Сущность изобретения: способ получения бис (гидроксиметил) циклобутил пуринов или пиридинов ф лы  , где R1 гуанин, пурин, пиримидин-2,6-дион, 5-метил-пиримидин-2,6 дион или 6-амино пиримидин 2 он. Реагент 1: [1 Альфа, 2 Бета, 3 Бета [3 -[арилсульфонилокси] 1,2 циклобутандиметанол, эфир дибензоата. Реагент 2: R1-H. Значения R1 указаны выше, причем это соединения необязательно защищено, полученный после конденсации продукт деблокируют. 1 табл.
, где R1 гуанин, пурин, пиримидин-2,6-дион, 5-метил-пиримидин-2,6 дион или 6-амино пиримидин 2 он. Реагент 1: [1 Альфа, 2 Бета, 3 Бета [3 -[арилсульфонилокси] 1,2 циклобутандиметанол, эфир дибензоата. Реагент 2: R1-H. Значения R1 указаны выше, причем это соединения необязательно защищено, полученный после конденсации продукт деблокируют. 1 табл.
СПОСОБ ПОЛУЧЕНИЯ БИС(ГИДРОКСИМЕТИЛ)ЦИКЛОБУТИЛ ПУРИНОВ ИЛИ ПИРИМИДИНОВ общей формулы





отличающийся тем, что соединение общей формулы
где P бензоил;
X арилсульфонилоксигруппа,
подвергают взаимодействию с соединением общей формулы
R1 H,
где R1 имеет указанные значения,
причем это соединение является не обязательно защищенным, и удаляют защитные группы.
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| H.Loibner и др., Helv | |||
| Chem | |||
| Acta, 59, с.2100 (1976). | |||
