Область техники, к которой относится изобретение
Настоящее изобретение относится к новым соединениями, обладающим биологической активностью. В частности, настоящее изобретение относится к новым соединениям, способным ингибировать NMDA-рецепторы. Изобретение также относится к применению соединений по изобретению для лечения заболеваний, для которых характерна повышенная активация NMDA рецептора, таких как нейродегенеративные заболевания, невропатическая боль, депрессия и другие, к фармацевтическим композициям на основе этих соединений, и к соответствующим способам лечения.
Уровень техники
Возбуждающие аминокислоты (L-глутамат, L-аспартат) являются важнейшими нейромедиаторами в центральной нервной системе млекопитающих. Эти аминокислоты активируют в том числе семейство тетрамерных катионных каналов, расположенных преимущественно постсинаптически и называемых ионотропными глутаматными рецепторами. Фармакологически данное семейство рецепторов можно разделить на три вида по типу селективного активатора: рецепторы, чувствительные к N-метил-D-аспартату (NMDA), рецепторы, чувствительные к α-амино-5-метил-4-изоксазолилпропионовой кислоте (АМРА), и рецепторы, чувствительные к каиновой кислоте.
NMDA рецепторы играют особую роль в синаптической передаче: инициируют долговременную потенциацию, являющуюся базовым механизмом в процессах, обеспечивающих обучение и формирование памяти. NMDA рецепторы также вовлечены в другие процессы, такие, как передача сенсорной информации (MacDermott A.B., Dale N. Receptors, ion channels and synaptic potentials underlying the integrative actions of excitatory amino acids, Trends in Neuroscience, 1987, V. 10, pp. 280-284). При этом, аномально низкие уровни глутаминовой кислоты могут приводить, например, к ухудшению способностей к обучению и нарушить формирование памяти. Помимо этого, NMDA рецепторы способны пропускать ионы кальция в нервную клетку, поэтому гиперстимуляция NMDA рецепторов мозга, наблюдаемая в случаях аноксии, ишемии и гипогликемии, приводит к увеличению концентрации Ca2+ в активированных нейронах и, как следствие, к каскаду внутриклеточных событий (активации фосфолипаз PLA2, PLC, протеаз и эндонуклеаз), приводящих к смерти нервных клеток (Farooqui A.A., Horrocks L.A. Excitatory amino acid receptors, neural membrane phospholipid metabolism and neurological disorders, Brain Research Reviews, 1991, V. 16, pp. 171-191). Считается, что этот процесс имеет место при следующих патологиях: в случаях церебрального паралича, церебральной ишемии, эпилепсии, болезни Альцгеймера, при слабоумии, связанном со СПИДом, при травматических повреждениях мозга и других нейродегенеративных расстройствах (Olney J.W. Excitotoxic Amino Acids and Neuropsychiatric Disorders, Annual Review of Pharmacology and Toxicology, 1990, V. 30, pp. 47-71; и Rogawski M.A., Porter R.J. Antiepileptic drugs: pharmacological mechanisms and clinical efficacy with consideration of promising developmental stage compounds, Pharmacological Reviews, 1990, V. 42, pp. 223-286).
Для антагонистов NMDA рецептора было показано наличие быстро наступающего антидепрессантного эффекта. Следует отметить, что один из энантиомеров неселективного блокатора NMDA-рецептора кетамина, S-кетамин, был одобрен FDA в 2019 для лечения клинической депрессии в США. Один из предложенных механизмов объясняет антидепрессантное действие прямой блокировкой GluN2B-содержащих NMDA рецепторов кетамином, что приводит к ингибированию ГАМК-ергических ингибиторных интернейронов, индуцирующих повышение уровня глутамата в префронтальной коре (Moghaddam B., Adams B., Verma A., Daly D., Activation of Glutamatergic Neurotransmission by Ketamine: A Novel Step in the Pathway from NMDA Receptor Blockade to Dopaminergic and Cognitive Disruptions Associated with the Prefrontal Cortex, Journal of Neuroscience, 1997, V. 17, pp. 2921-2927), синтез BDNF и, как следствие, проявление антидепрессантного эффекта (Pochwat B., An update on NMDA antagonists in depression, Expert Review of Neurotherapeutics 2019, 19 (11), 1055-1067).
Ингибирование GluN2B-содержащих NMDA рецепторов также способно снизить или предотвратить избыточную активацию NMDA рецепторов при повышении уровня глутамата, наблюдаемую, в частности, в случае гибели окружающих клеток при повреждении тканей мозга (Peters A., Ketamine Alters Hippocampal Cell Proliferation and Improves Learning in Mice after Traumatic Brain Injury, 2018, Anesthesiology, 129 (2), 278–295) или при нейропатических болях (Aiyer R., A Systematic Review of NMDA Receptor Antagonists for Treatment of Neuropathic Pain in Clinical Practice, Clin. J. Pain., 2018, 34 (5), 450-467).
В уровне техники описаны некоторые соединения, способные ингибировать GluN2B-содержащие NMDA рецепторы. Одним из таких соединений является ифенпродил (4-[2-(4-бензилпиперидин-1-ил)-1-гидроксипропил]фенол), описанный в статье Reynolds I. J., Miller R. J., Ifenprodil is a novel type of N-methyl-D-aspartate receptor antagonist: interaction with polyamines, Mol. Pharmacol., 1989, 36 (5), 758-765 и являющийся ингибитором GluN1- и GluN2B-содержащих NMDA рецепторов.
Схожие с ифенпродилом антагонисты GluN2B-содержащих NMDA рецепторов описаны также в статьях Tamiz A. P. Structure-Activity Relationship of N-(Phenylalkyl)cinnamides as Novel NR2B Subtype-Selective NMDA Receptor Antagonists, J. Med. Chem., 1999, 42 (17), 3412-3420, Borza I, Benzimidazole-2-carboxamides as novel NR2B selective NMDA receptor antagonists, Chem. Lett., 2006, 16 (17), 4638-4640, McCauley J.A., NR2B-Selective N-Methyl-d-aspartate Antagonists: Synthesis and Evaluation of 5-Substituted Benzimidazoles, J. Med. Chem., 2004, 47 (8), 2089-2096, и Nagy J., Inducible expression and pharmacology of recombinant NMDA receptors, composed of rat NR1a/NR2B subunits, Neurochem. Intern., 2003, 43 (1), 19-29.
В статье Stroebel D., A Novel Binding Mode Reveals Two Distinct Classes of NMDA Receptor GluN2B-selective Antagonists, Mol. Pharmacol., 2016, 89 (5), 541-551 описано также соединение EVT-101 (5-(3-(дифторметил)-4-фторфенил)-3-((2-метил-1H-имидазол-1-ил)метил)пиридазин], являющееся антагонистом GluN2B-содержащих NMDA рецепторов, но отличающееся от ифенпродила в том, что касается механизма связывания с GluN1/GluN2B.
В настоящее время структура, особенности активации и ингибирования NMDA рецептора являются объектом пристального внимания. Особенный интерес вызывают ингибиторы, связывающиеся с аминоконцевыми доменами и уменьшающие вероятность открытия канала (ифенпродил, траксопродил и др.) вследствие наличия селективности к GluN2B-содержащим рецепторам и особенностей процессов ингибирования. Однако большинство известных ингибиторов аминоконцевых доменов, являются производными 2-фенилэтиламина и обладают существенным сродством к hERG, что при длительном употреблении может вызвать фатальную аритмию и смерть. Для антагонистов, связывающихся с аминоконцевыми доменами, характерно неполное подавление токов через рецептор при насыщающих концентрациях, что может вести к снижению побочных эффектов по сравнению с прямыми блокаторами ионного канала (Smith P. F., Therapeutic N-methyl-D-aspartate receptor antagonists: will reality meet expectation? Curr. Opin. Investig. Drugs, 2003 Jul; 4(7), 826-832).
Существует потребность в новых селективных антагонистах NMDA рецептора, проявляющих антидепрессантный, анальгетический и/или нейропротекторный эффекты.
Сущность изобретения
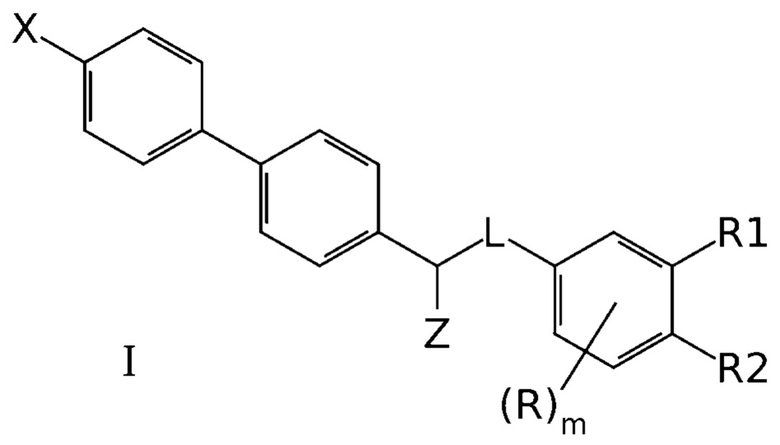
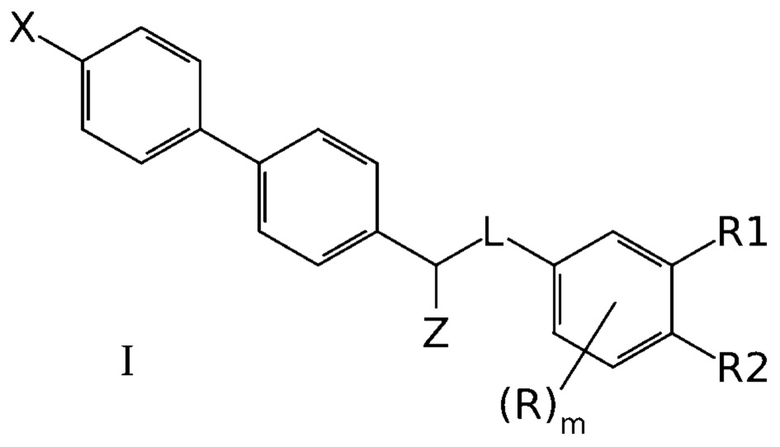
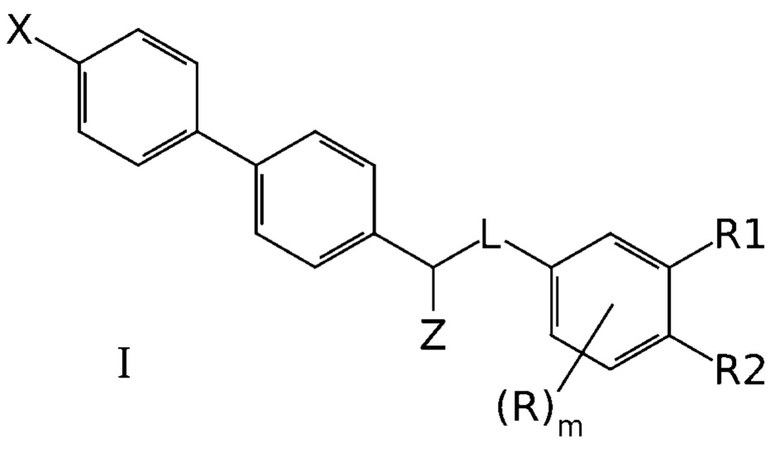
Задачей создателей данного изобретения является получение новых эффективных антагонистов NMDA рецептора. Эта задача была успешно решена и необходимый технический результат был успешно достигнут с использованием производных бифенила общей формулы I, эффективно ингибирующих NMDA рецепторы:
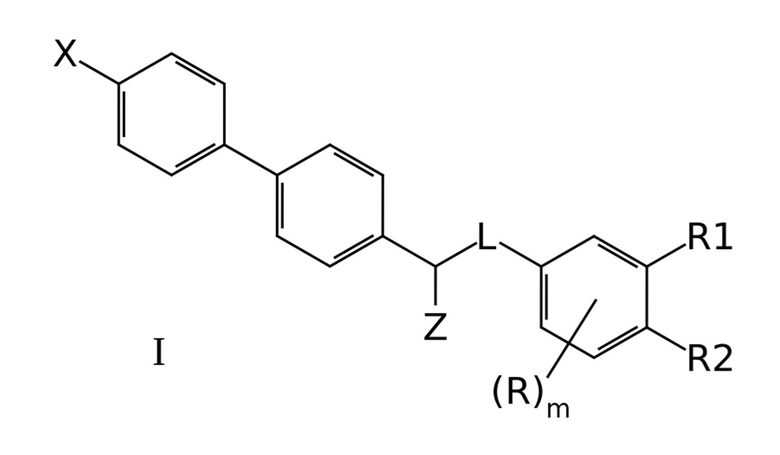
где X означает -H, атом галогена или C1-6-алкил, необязательно замещенный одним или несколькими атомами галогена;
Z означает -OR` или -NR`R``;
линкер L означает группу -(C0-2алкилен)-Y-(С0-2алкилен)-, где под “С0-алкиленом” понимается простая связь, и где Y означает группу -O- или -NR`-;
R1 означает -H, -OR`, -NR`R`` или -NR`-SO2-(C1-6-алкил);
R2 означает -H, -OR` или -NR`R``;
R в каждом случае независимо означает H, атом галогена, C1-6-алкил, необязательно замещенный одним или несколькими атомами галогена, -OR`, -NR`R``, -NR`-SO2-(C1-6-алкил);
R` и R`` в каждом случае независимо представляют собой группы, выбранные из H и C1-6-алкила; и
m представляет собой целое число от 0 до 3,
или их фармацевтических приемлемых солей.
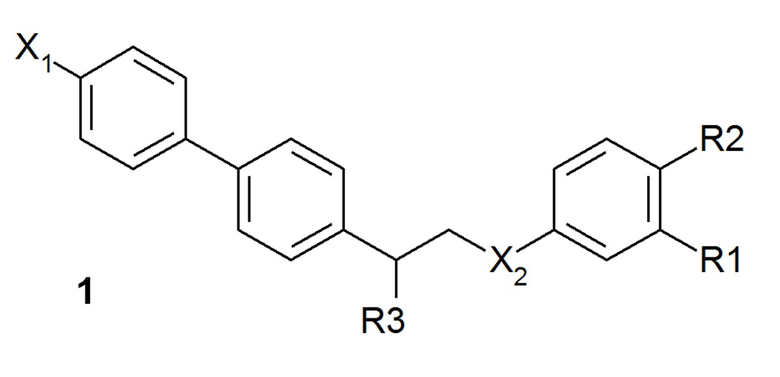
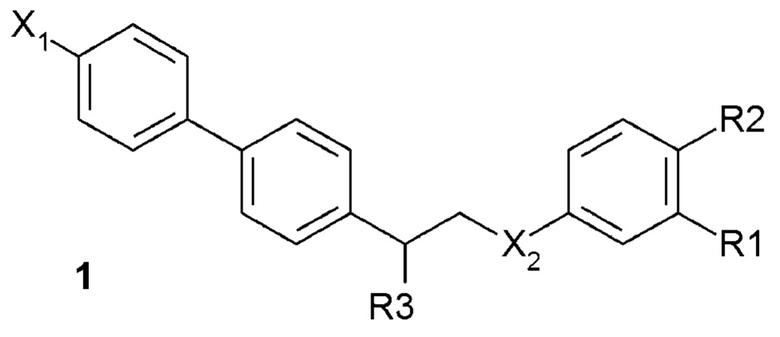
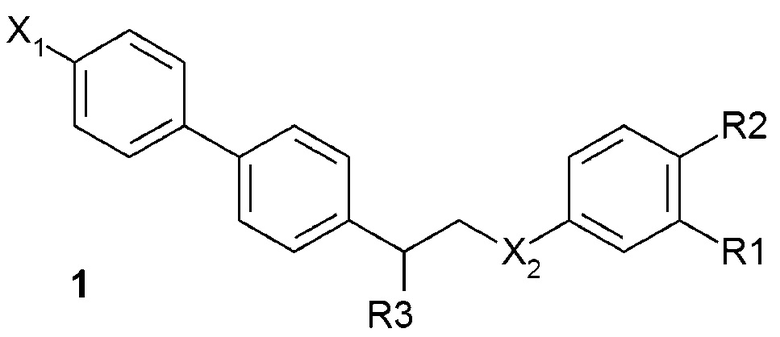
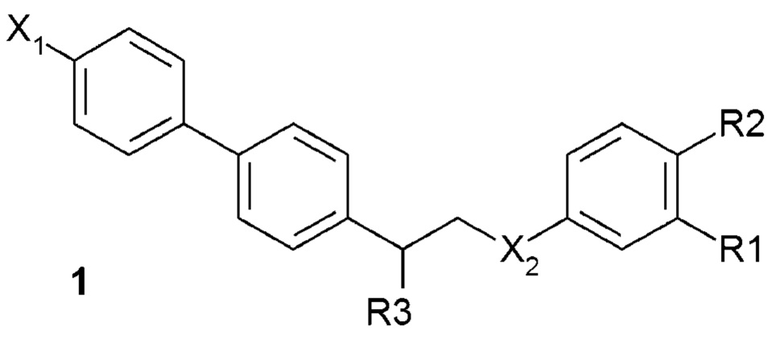
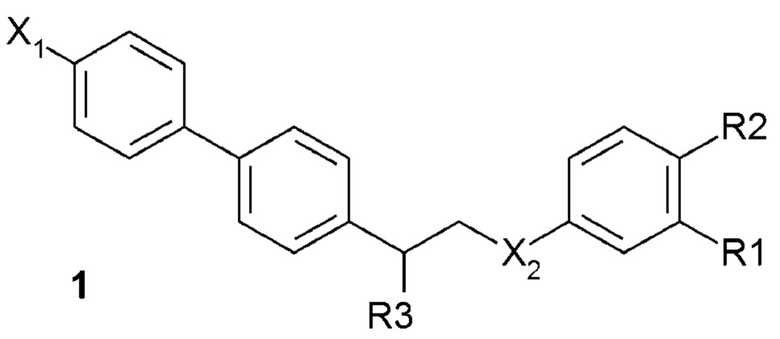
В одном из предпочтительных вариантов осуществления соединение по изобретению представляет собой соединение формулы 1:
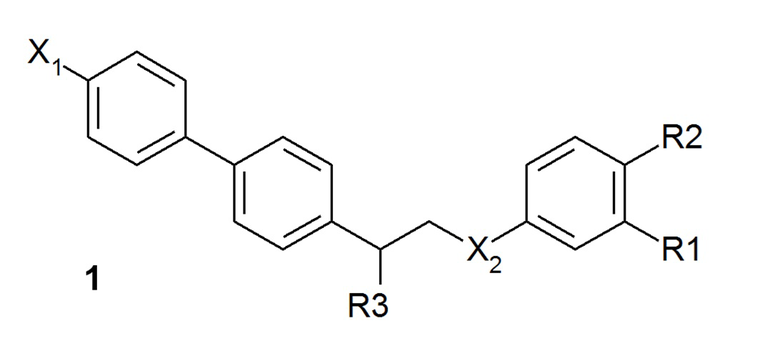
где X1 означает -H, C1-4-алкил (предпочтительно -CH3) или атом галоген (предпочтительно -F, -Cl или -Br);
X2 означает -O- или -NH-;
R1 означает -H, -OH, -NH2, -NH-(C1-6-алкил) или -NH-SO2-(C1-6-алкил);
R2 означает -H, -OH или -NH2;
R3 означает -NH2 или -OH,
или его фармацевтически приемлемую соль.
Соединения по изобретению обладают биологической активностью и могут быть использованы для лечения заболеваний, для которых характерна повышенная активация NMDA рецептора, таких как нейродегенеративные заболевания, инсульт и травмы мозга, нейропатическая боль, депрессия и другие.
В настоящем изобретении также описываются фармацевтические композиции, содержащие в качестве активного ингредиента одно или несколько соединений по изобретению или их фармацевтически приемлемых солей, а также способ лечения нарушений или заболеваний, сопровождающихся повышенной активацией NMDA-рецептора, включающий введение нуждающемуся в этом пациенту эффективного количества одного или нескольких соединений по изобретению или их фармацевтически приемлемых солей.
Краткое описание чертежей
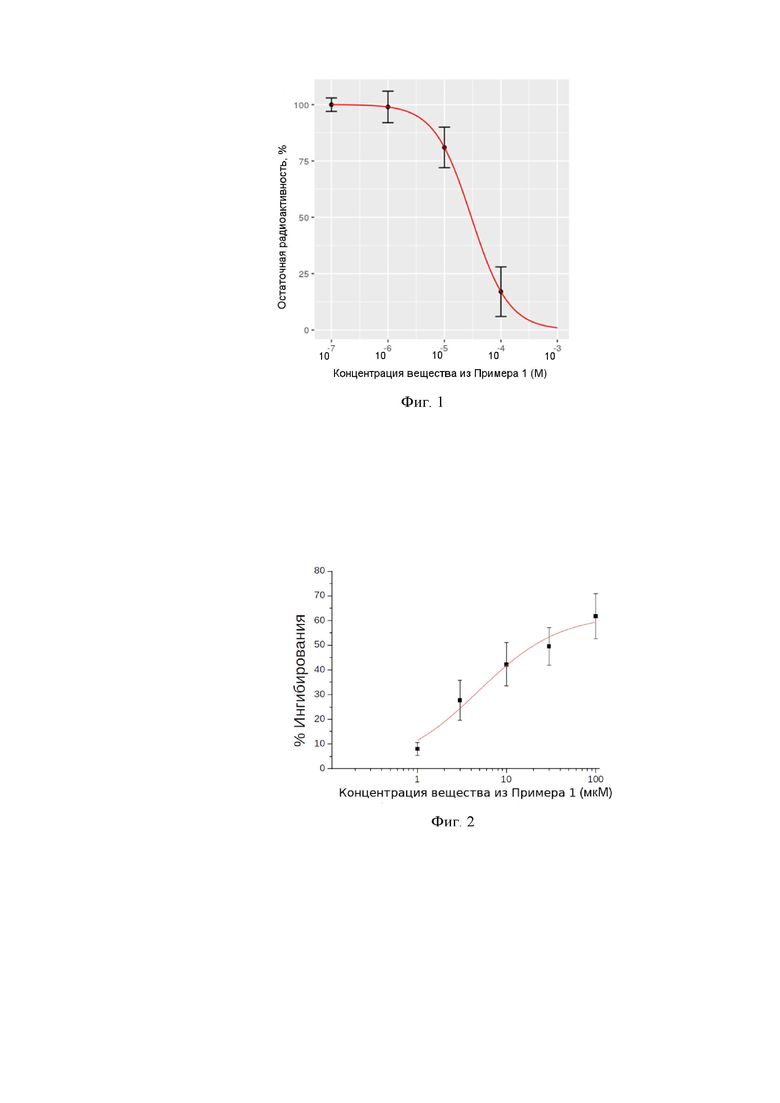
На фиг. 1 проиллюстрирована концентрационная зависимость вытеснения радиоактивно меченого [H3]-ифенпродила веществом 1k из Примера 1.
На фиг. 2 проиллюстрирована концентрационная зависимость ингибирования NMDA/Gly-индуцированных токов.
Осуществление изобретения
В настоящем документе термины и выражения имеют значения, обычно используемые специалистами в данной области техники. Однако в случаях, когда для какого-то термина или выражения здесь явно дается специальное определение, соответствующий термин следует понимать в соответствии с определением, данным в настоящем изобретении.
В настоящем изобретении под термином «алкил» понимается насыщенная линейная, разветвленная или циклическая углеводородная группа. Число атомов в алкильной группе может быть указано в виде Ci-k, что означает, что алкильная группа содержит как минимум i, но не более k атомов углерода. Например, выражение «C1-6алкил» означает алкильную группу, содержащую от 1 до 6 (в том числе, в некоторых вариантах от 1 до 5, от 1 до 4, от 1 до 3, от 1 до 2 или ровно 1) атомов углерода, включая (но не ограничиваясь этим) метильную, этильную, н-пропильную, изопропильную, н-бутильную, изобутильную, трет-бутильную, пентильную, гексильную, циклопентильную, циклогексильную группы и т.д. В настоящем изобретении алкильная группа в некоторых указанных случаях может быть замещена одним или несколькими атомами галогена. В таких случаях число атомов галогена в замещенной алкильной группе может составлять от 1 до максимального числа атомов галогена, которое может нести данная алкильная группа (пергалоалкил).
В настоящем изобретении под термином «алкилен» понимается описанная выше алкильная группа, связанная с двумя другими группами или фрагментами молекулы, т.е. являющаяся дизамещенной. Другие группы или фрагменты молекулы могут быть связаны с одним и тем же или с различными атомами алкиленовой группы. Число атомов в алкиленовой группы может быть указано в виде Ci-k, что означает, что алкиленовая группа содержит как минимум i, но не более k атомов углерода. Например, выражение «C1-2алкилен» означает алкиленовую группу, содержащую от 1 до 2 атомов углерода, т.е. это выражение охватывает группы -CH2-, -CH(CH3)- и -CH2-CH2-. Под выражением «С0-алкилен» понимается простая связь.
В настоящем изобретении под термином «галоген» понимается фтор, хлор, бром или йод. В наиболее предпочтительном варианте галоген означает фтор, хлор или бром.
В настоящем изобретении под выражением «фармацевтически приемлемая соль» понимаются соли соединений по изобретению, получаемые в присутствии фармацевтически приемлемых кислот или оснований. Например, фармацевтически приемлемая соль соединения по изобретению может представлять собой соль соединения по изобретению и фармацевтически приемлемой кислоты. Неограничивающими примерами таких солей могут быть соли, полученные в присутствии фармацевтически приемлемых неорганических кислот (хлороводород, бромоводород, фосфорная кислота и т.п.) или фармацевтически приемлемых органических кислот (уксусная кислота, лимонная кислота, яблочная кислота и т. п.). Более подробная информация о фармацевтически приемлемых кислотах и основаниях, которые могут быть использованы для получения фармацевтически приемлемых солей, может быть найдена в уровне техники.
В настоящем изобретении под выражением «пациент» понимаются млекопитающие, такие как приматы (предпочтительно человек), коровы, овцы, лошади, козы, собаки, кошки, кролики и т.д. В предпочтительном варианте пациентом является человек.
В настоящем изобретении под выражением «фармацевтически приемлемый вспомогательный агент» понимаются известные в данной области техники вспомогательные агенты, используемые для получения фармацевтических композиций. Неограничивающими примерами фармацевтически приемлемых агентов являются носители, наполнители, разбавители, связующие, разрыхляющие агенты, адсорбенты, ароматизирующие вещества, вкусовые агенты и т.п.
В настоящем изобретении под выражением «эффективное количество» или «фармакологически эффективное количество» понимается такое количество активного агента по изобретению, которое при его введении пациенту обеспечивает достижение желаемого фармацевтического/терапевтического эффекта. Это эффективное количество может быть выбрано специалистом в зависимости от таких известных факторов, как симптомы и тяжесть заболевания, которое предполагается лечить, возраст, пол, вес и состояние пациента, конкретное используемое соединение, способ его введения и форма готового препарата и т.п. Обычно, но не обязательно, фармакологически эффективное количество соединения по изобретению может находиться в диапазоне от 0,01 до 40 мг в день, и эффективное количество соединения в композиции может быть выбрано в соответствующим образом. В некоторых вариантах осуществления фармакологически эффективное количество соединения по изобретению может быть больше 0,1 мг, 0,5 мг или больше 1 мг в день, при этом в некоторых вариантах осуществления фармакологически эффективное количество может быть меньше 30 мг, меньше 20 мг или меньше 10 мг в день.
В настоящем изобретении под выражением «введение пациенту» подразумевается введение соединения или композиции по изобретению пациенту одним из известных в данной области техники способов, например (но не ограничиваясь указанным) пероральным, парентеральным, внутривенным, интраперитонеальным, интраназальным, подкожным путем или введение в виде суппозитория или местное нанесение.
В настоящем изобретении под выражением «лечение» подразумевается достижение какой-либо положительной динамики в том, что касается лечения, облегчения, замедления развития и/или профилактики какого-либо состояния, заболевания и/или расстройства, связанного с избыточной активацией NMDA-рецептора.
Настоящее изобретение имеет в основе соединения формулы I:
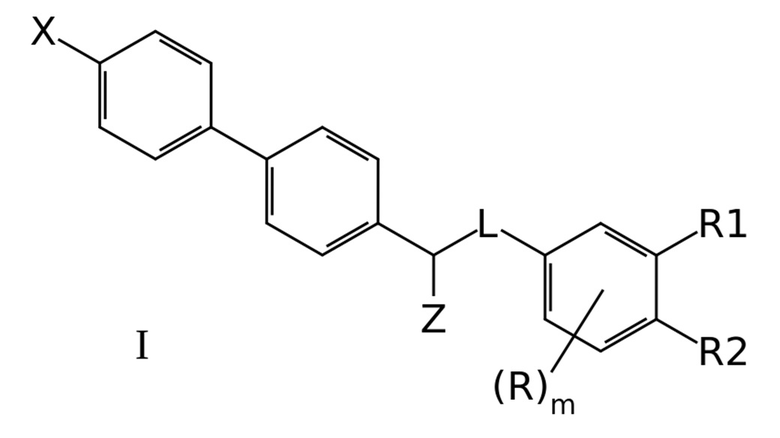
где X означает -H, атом галогена или C1-6-алкил, необязательно замещенный одним или несколькими атомами галогена;
Z означает –OR` или -NR`R``;
линкер L означает группу -(C0-2алкилен)-Y-(С0-2алкилен)-, где под “С0-алкиленом” понимается простая связь, и где Y означает группу -O- или -NR`-;
R1 означает -H, -OR`, -NR`R`` или -NR`-SO2-(C1-6-алкил);
R2 означает -H, -OR` или -NR`R``;
R в каждом случае независимо означает H, атом галогена, C1-6-алкил, необязательно замещенный одним или несколькими атомами галогена, -OR`, -NR`R``, -NR`-SO2-(C1-6-алкил);
R` и R`` в каждом случае независимо представляют собой группы, выбранные из H и C1-6-алкила; и
m представляет собой целое число от 0 до 3,
и их фармацевтически приемлемые соли.
В одном предпочтительном варианте осуществления X в формуле I выбран из группы, включающей -H, C1-4-алкил и атом галогена, более предпочтительно выбран из группы, включающей -H и атом галогена, такой как бром.
В еще одном предпочтительном варианте осуществления Z в формуле I выбран из -OH и -NH2.
В еще одном предпочтительном варианте осуществления линкер L в формуле I означает группу -(C0-1алкилен)-Y-(С0-1алкилен)-, наиболее предпочтительно группу -CH2-Y-.
В еще одном предпочтительном варианте осуществления R1 в формуле I выбран из группы, включающей -H, -OR`, -NHR` или -NR`-SO2-(C1-6-алкил).
Соединения формулы I по изобретению и их соли могут быть получены специалистами в области органической химии с использованием стандартных методик и реакций, известных в данной области техники. В качестве примера, один из общих методов синтеза соединений формулы I по настоящему изобретению показан на Схеме 1 ниже.
Схема. 1 Общая схема синтеза соединений общей формулы I
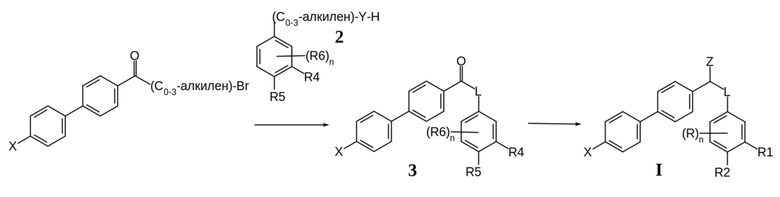
На Схеме 1 проиллюстрирована реакция исходного бромзамещенного бифенильного соединения с соединением 2, в котором группы R4, R5 и R6 соответствуют группам R1, R2 и R в формуле I, предшественникам этих групп или их защищенным формам. Затем промежуточное соединение 3 превращают в соединение I с использованием известных методик. Реакции, используемые для синтеза соединений формулы I по изобретению, хорошо известны специалистам.
В одном предпочтительном варианте осуществления соединение по изобретению представляет собой соединение формулы 1:
где X1 означает -H, C1-4-алкил (предпочтительно -CH3) или атом галоген (предпочтительно -F, -Cl или -Br);
X2 означает -O- или -NH-;
R1 означает -H, -OH, -NH2, -NH-(C1-6-алкил) или -NH-SO2-(C1-6-алкил);
R2 означает -H, -OH или -NH2;
R3 означает -NH2 или -OH,
или его фармацевтически приемлемую соль.
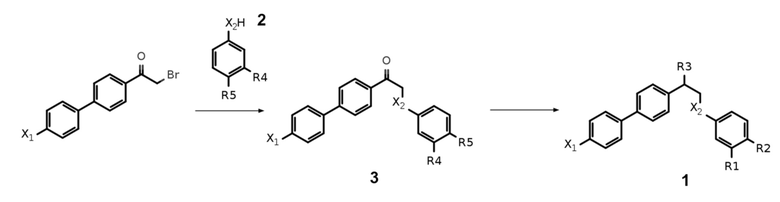
Общий метод синтеза предпочтительных соединений формулы 1 по настоящему изобретению показан на Схеме 2 ниже.
Схема 2. Общая схема синтеза соединений общей формулы 1
В одном варианте осуществления настоящего изобретения соединения формулы I согласно настоящему изобретению можно использовать в виде композиций, которые также являются объектом настоящего изобретения. Биологическая активность композиций по изобретению определяется активностью соединений формулы I, в частности, композиции по изобретению способы ингибировать NMDA-рецепторы.
Фармацевтические композиции согласно изобретению могут быть получены с использованием общепринятых в данной области техники приемов и включают фармакологически эффективное количество активного агента, представляющего собой соединение формулы I или его фармацевтически приемлемую соль (далее называются также "активное соединение"), и один или более фармацевтически приемлемых вспомогательных агентов.
В некоторых вариантах осуществления фармацевтическая композиция содержит от 0,01 до 50 вес. %, предпочтительно от 0,1 до 40 вес. %, более предпочтительно от 0,5 до 30 вес. %, например от 1 до 20 вес. %, например от 5 до 10 вес. % соединения по изобретению, причем в некоторых вариантах осуществления относительное количество соединения по изобретению может быть выше в дозированных формах, наносимых местно или вводимых перорально, и ниже в дозированных формах, предназначенных, например, для внутривенного или парентерального введения.
В соответствии с известным уровнем техники, фармацевтические композиции, в том числе такие, какие описаны в настоящем документе, могут быть представлены в виде различных жидких или твердых форм, которые могут быть получены методами, известными специалистам в данной области техники.
Примеры твердых лекарственных форм включают, например, таблетки, пилюли, желатиновые капсулы и другие известные специалистам формы.
Примеры жидких лекарственных форм включают формы, предназначенные для инъекций и парентерального введения, в том числе растворы, эмульсии, суспензии и др.
Композиции по изобретению можно получить с использованием стандартных процедур и методик, например таких, которые предусматривают смешение активного соединения с жидким или тонко измельченным твердым носителем.
В предпочтительном варианте осуществления композиции по изобретению в форме таблеток содержат от 0,1 до 50 вес. %, предпочтительно от 0,5 до 40 вес. %, более предпочтительно от 1 до 30%, например от 2 до 20 %, например от 5 до 10 вес. % активного соединения, и один или более из наполнителей или носителей.
В качестве фармацевтически приемлемых вспомогательных агентов для таблеток могут быть использованы: а) разбавители (включая, без ограничения, свекловичный сахар, лактозу, глюкозу, натрия хлорид, сорбит, маннит, гликоль, фосфат кальция двузамещенный и т.п.); б) связующие вещества (включая, без ограничения, магниевый силикат алюминия, крахмальную пасту, желатин, трагакант, метилцеллюлозу, карбоксиметилцеллюлозу, поливинилпирролидон и т.п.); в) разрыхлители (включая, без ограничения, декстрозу, агар, альгиновую кислоту или ее соли, крахмал и т.п.) и другие известные специалистам вспомогательные вещества.
Для получения желатиновых капсул обычно дополнительно используют красители и стабилизаторы. В качестве красителей могут быть использованы, например, тартразин, индиго и т.п.; в качестве стабилизаторов могут быть использованы натрия метабисульфит, натрия бензоат и т.п.
В предпочтительном варианте осуществления желатиновые капсулы по изобретению могут содержать от 0,1 до 50 вес. %, предпочтительно от 0,5 до 20 %, например от 1 до 10 вес. % активного ингредиента.
Инъекционные формы композиций по изобретению обычно представляют собой изотонические растворы или суспензии. Вышеуказанные инъекционные формы могут содержать известные специалистам в данной области техники добавки, такие как консерванты (в частности, натрия метабисульфит, бензойная кислота, натрия бензоат, смеси метилпарабена и пропилпарабена или т.п.), стабилизаторы (в частности, абрикосовую или аравийскую камедь, декстрин, крахмальный клейстер, метилцеллюлозу или т.п.), соли, регулирующие осмотическое давление (например, хлорид натрия), и буферы. Кроме того, такие формы могут содержать и другие известные специалистам терапевтически полезные вещества. В предпочтительном варианту осуществления инъекционные формы композиций по изобретению являются стерильными.
Дополнительный вариант осуществления изобретения относится к способу лечения различных нарушений, сопровождающихся повышенной активацией NMDA-рецептора, где способ включает введение эффективного количества соединения общей формулы I, как оно описано в настоящем документе, или его фармацевтически приемлемой соли.
Назначаемая для приема доза активного компонента (соединения формулы I или его фармацевтически приемлемой(ых) соли(ей)) может зависеть от многих известных специалистам факторов, таких как возраст, пол, вес и состояние пациента, симптомы и тяжесть заболевания, конкретное используемое соединение, способ введения, форма препарата, в виде которой назначается активное соединение и т.п.
Обычно, совокупная доза, которую вводят пациенту, составляет от 0,01 до 40 мг в день, в некоторых вариантах осуществления от 1 до 40 мг в день. Совокупная доза может быть разделена на несколько доз, например, для приема от 1 до 4 раз в день. При пероральном введении диапазон совокупных доз активного вещества может составлять от 0,1 до 40 мг в день, предпочтительно от 0,1 до 10 мг. При парентеральном приеме диапазон совокупных доз может составлять от 0,5 до 20 мг в день, предпочтительно от 0,5 до 10 мг. При внутривенных инъекциях диапазон совокупных доз может составлять от 0,05 до 5,0 мг в день, предпочтительно от 0,05 до 2,5 мг. Точная доза может быть выбрана лечащим врачом с учетом специфики каждого отдельного случая.
Следует понимать, что признаки, раскрытые в настоящем документе для какого-то одного варианта осуществления изобретения, также могут быть использованы и в других вариантах осуществления, если это не противоречит здравому смыслу. Кроме того, в том случае, если для какой-то величины приведено несколько возможных диапазонов значений, все диапазоны, составленные путем комбинирования нижних и верхних граничных значений из указанных диапазонов, также включены в настоящее раскрытие, как если бы они были указаны в явном виде.
Приведенные ниже примеры иллюстрируют, но не ограничивают данное изобретение.
Примеры
Структуры соединений, получаемых в примерах, подтверждались данными химического, спектрального анализов и других физико-химических характеристик. Спектры ЯМР регистрировали на спектрометре Agilent 400-MR (400,0 МГц для 1H; 100,6 МГц для 13C) при комнатной температуре; химические сдвиги (δ) измеряли относительно растворителя CDCl3 для 1Н (δ = 7,26 ч/млн), 13C (δ = 77,16 ч/млн). Химические сдвиги (δ) приведены в миллионных долях; J значения приведены в Гц.
В примерах был получен ряд соединений общей формулы I согласно представленной ниже Таблице 1.
Таблица 1. Структуры заместителей в соединениях по изобретению
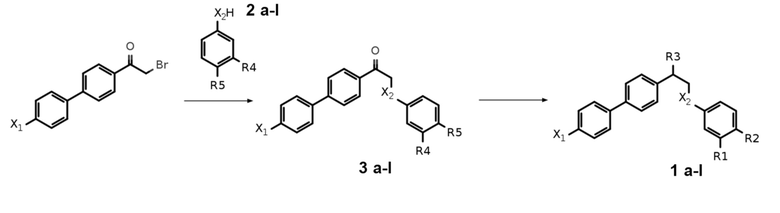
i** - 1i получали из 1h;
k*** - вещество 1k получали в 2 стадии из вещества 3l
Синтез соединений 3a, b, k, l
Смесь фенола 2 (1 ммоль) с подходящими заместителями и K2CO3 (0,28 г, 2 ммоль) в безводном MeCN (8 мл) перемешивали 30 минут при комнатной температуре. Затем добавляли 1-([1,1'-бифенил]-4-ил)-2-бромэтанон (0,28 г, 1 ммоль) в 4 мл MeCN. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов, затем выливали в ледяную воду и экстрагировали СН2Cl2 (3 порции по 15 мл). Объединенные органические фракции последовательно промывали водой (3 порции по 15 мл) и насыщенным раствором NaCl (2 порции по 15 мл). Органический слой сушили над MgSO4. Растворитель отгоняли при пониженном давлении, остаток очищали методом препаративной колоночной хроматографии на силикагеле (ПЭ:ЭА = 4:1) в случае соединений 3a, 3k и 3l или перекристаллизацией из ацетона в случае соединения 3b.
Синтез соединений 3c, 3f, 3h
Смесь подходящего анилина 2 (1 ммоль) и 1-([1,1'-бифенил]-4-ил)-2-бромэтанона (0,28 г, 1 ммоль) в 4 мл MeOH перемешивали при комнатной температуре в течение 2 часов, затем кипятили 30 минут. Реакционную смесь охлаждали до комнатной температуры, отфильтровывали осадок. Твёрдый остаток очищали методом препаративной колоночной хроматографии на силикагеле (ПЭ:ЭА = 4:1) в случае соединения 3c или перекристаллизацией из ацетона в случае соединений 3f и 3h.
Синтез соединений 3d, 3e
Смесь подходящего анилина 2 (1 ммоль) и 1-([1,1'-бифенил]-4-ил)-2-бромэтанона (0,28 г, 1 ммоль) в 4 мл MeOH перемешивали при комнатной температуре в течение 24 часов, затем нагревали при 50°C в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры, выпавший осадок отфильтровывали, затем очищали методом препаративной колоночной хроматографии на силикагеле (CH2Cl2/MeOH, 50/1).
Синтез соединения 3j
Соединение 3j получали аналогично соединениям 3d и 3e с использованием 1-([4'‐бромо‐1,1'-бифенил]-4-ил)-2-бромэтанона (0,36 г, 1 ммоль) вместо 1-([1,1'-бифенил]-4-ил)-2-бромэтанона.
Синтез соединения 1a
К суспензии LiAlH4 (0,04 г, 1,16 ммоль) в 9 мл абс. тетрагидрофурана медленно добавляли раствор кетона 3a (0,2 г, 0,58 ммоль) в 4 мл абс. тетрагидрофурана. Реакционную смесь кипятили в течение 3 часов, затем охлаждали до 0 ºС и по каплям последовательно добавляли 20 мл воды и 10 % раствор H2SO4 до pH 3. Эфирный раствор отделяли, высушивали над MgSO4, растворитель отгоняли при пониженном давлении. Продукт выделяли методом препаративной колоночной хроматографии на силикагеле (ПЭ:ЭА = 3:1).
Соединение 4-(2-([1,1'-бифенил]-4-ил)-2-гидроксиэтокси)фенол 1a: Выход 0,13 г (72%); бесцветное твердое вещество, т. пл. 123-124°С; Rf= 0,19 (гексан/этилацетат, 2/1);
Спектр 1H ЯМР (CD3OD, δ м.д.): 4,00 (d, 2H, CH2, J3= 5,8Hz), 5,02 (dd, J=5,8 Hz, 1H, СH), 6,68-6,74 (m, 2H, 2СH(Ar)), 6,76-7,82 (m, 2H, 2СH(Ar)), 7,27-7,33 (m, 1H, СH(Ar)), 7,36-7,43 (m, 2H, 2СH(Ar)), 7,47-7,52 (m, 2H, 2СH(Ar)), 7,56-7,61 (m, 4H, 4СH(Ar));
Спектр 13C ЯМР (101 MHz, CD3OD, δ м.д.): 73.3 (CH), 75.1 (CH2), 116.8 (2СH(Ar)), 116.9 (2СH(Ar)), 127.9 (4СH(Ar)), 128.0 (2СH(Ar)), 128.3(СH(Ar)), 129.8 (2СH(Ar)), 141.6(С(Ar)), 141.8 (С(Ar)), 142.0 (С(Ar)), 152.5 (С(Ar)), 153.5 (С(Ar));
HRMS-ESI [M + Na]+: рассчитано для C20H18O3Na 329,1148, найдено 329,1148.
Синтез соединений 1b, 1c, 1d, 1f, 1j
К раствору соответствующего кетона 3 (1 ммоль) в смеси ТГФ (8 мл) и воды (2 мл) порциями добавляли NaBH4 (0,04 г, 1 ммоль) в течение 3 минут и перемешивали 3 часа при комнатной температуре. Избыток NaBH4 разлагали насыщенным раствором NH4Cl (15 мл), затем водой (15 мл). Образовавшуюся смесь экстрагировали этилацетатом (3 порции по 15 мл). Объединенные органические фракции последовательно промывали водой и насыщенным раствором соли, сушили над MgSO4. Растворитель отгоняли при пониженном давлении, продукт выделяли методом препаративной колоночной хроматографии на силикагеле (гексан/этилацетат).
Соединение 1-([1,1'-бифенил]-4-ил)-2-феноксиэтанол 1b: Выход 0,23 г (80 %); бесцветное твердое вещество, т. пл. 88-90 °С; Rf=0,85 (гексан/этилацетат, 1/1);
Спектр 1H ЯМР (400 MHz, CDCl3, δ м.д.): 2,98 (br.s, 1H, OH), 4,09 (dd, J=8,7 Hz, J= 9,6 Hz, 1H, CH2), 4,19 (dd, J=3,2 Hz, J=9,6 Hz, 1H, CH2),5,21 (dd, J=3,2 Hz, J=8,7 Hz, 1H, CH), 6,96-7,00 (m, 2H, 2СH(Ar)), 7,00-7,06 (m, 1H, СH(Ar)), 7,30-7,37 (m, 2H, 2СH(Ar)), 7,37-7,43 (m, 1H, 1СH(Ar)), 7,46-7,52 (m, 2H, 2СH(Ar)), 7,54-7,59 (m, 2H, 2СH(Ar)), 7,62-7,69 (m, 4H, 4СH(Ar));
Спектр 13C ЯМР (101 MHz, CDCl3, δ м.д.): 72,5 (CH), 73,3 (CH2), 114,8 (2СH(Ar)), 121,4 (СH(Ar)), 126,9 (2СH(Ar)), 127,2 (2СH(Ar)), 127,4 (2СH(Ar)), 127,5 (СH(Ar)), 128,9 (2СH(Ar)), 129,7 (2СН(Ar)), 138,8 (С(Ar)), 140,8 (С(Ar)), 141,2 (С(Ar)), 158,5 (С(Ar));
HRMS-ESI [M + Na]+: рассчитано для C20H18O2Na 313,1200, найдено 313,1199. Соединение 1-([1,1'-бифенил]-4-ил)-2-(фениламино)этанол 1c: Выход 0,24 г (82 %); бесцветное твердое вещество, т. пл. 116-117 °С; Rf=0,79 (гексан/этилацетат, 1/1);
Спектр 1H ЯМР (400 MHz, CDCl3, δ м.д.): 3,35 (dd, J=8,6 Hz, J=13,1 Hz, 1H, CH2), 3,48 (dd, J=3,9 Hz, J=13,1 Hz, 1H, CH2), 4,98 (dd, J=3,9 Hz, J=8,6 Hz, 1H, CH), 6,69-6,76 (m, 2H, 2СH(Ar)), 6,76-6,85 (m, 1H, СH(Ar)), 7,20-7,29 (m, 2H, 2СH(Ar)), 7,36-7,44 (m, 1H, 1СH(Ar)), 7,45-7,54 (m, 4H, 4СH(Ar)), 7,59-7,68 (m, 4H, 4СH(Ar));
Спектр 13C ЯМР (101 MHz, CDCl3, δ м.д.): 51,8 (CH2), 72,3 (CH), 113,6 (2СH(Ar)), 118,3 (СH(Ar)), 126,5 (2СH(Ar)), 127,2 (2СH(Ar)), 127,46 (2СH(Ar)), 127,52 (СH(Ar)), 128,9 (2СH(Ar)), 129,5 (2СН(Ar)), 140,8 (С(Ar)), 141,0 (С(Ar)), 141,1 (С(Ar)), 147,9 (С(Ar));
HRMS-ESI [M + H]+: рассчитано для C20H20NO 290,1540, найдено 290,1539.
Соединение N-(3-((2-([1,1'-бифенил]-4-ил)-2-гидроксиэтил)амино)фенил)метансульфонамид 1d: Выход 0,32 г (80 %); бесцветное твердое вещество, т. пл. 144-145 °С; Rf=0,32 (гексан/этилацетат, 1/1);
Спектр 1H ЯМР (400 MHz, CDCl3/CD3OD, δ м.д.): 2,61 (br.s, 2H, NH+OH), 2,94 (s, 3H, CH3), 3,28 (dd, J=8,4 Hz, J=13,1 Hz, 1H, CH2), 3,39 (dd, J=3,9 Hz, J= 13,1 Hz, 1H, CH2), 4,91 (dd, J=3,9 Hz, J=8,4 Hz, 1H, CH), 6,41-6,46 (m, 1H, 1СH(Ar)), 6,49-6,54 (m, 1H, 1СH(Ar)), 6,56-6,60 (m, 1H, СH(Ar)), 7,06-7,12 (m, 1H, 1СH(Ar)), 7,30-7,36 (m, 1H, 1СH(Ar)), 7,38-7,47 (m, 4H, 4СH(Ar)), 7,53-7,61 (m, 4H, 4СH(Ar));
Спектр 13C ЯМР (101 MHz, CDCl3/CD3OD, δ м.д.): 38,9 (CH3), 51,4 (CH2), 71,8 (CH), 105,1 (СH(Ar)), 109,5 (СH(Ar)), 110,0 (СH(Ar)), 126,4 (2СH(Ar)), 127,1 (2СH(Ar)), 127,3 (2СH(Ar)), 127,4 (СH(Ar)), 128,9 (2СН(Ar)), 130,4(СH(Ar)), 138,3(С(Ar)), 140,7 (С(Ar)), 140,8 (С(Ar)), 141,2 (С(Ar)), 149,2(С(Ar));
HRMS-ESI [M + H]+: рассчитано для C21H23N2O3S 383,1424, найдено 383,1426.
Соединение N-(3-((2-(4'-бром-[1,1'-бифенил]-4-ил)-2-гидроксиэтил)амино)фенил)метансульфонамид 1j: Выход 0,38 г (83 %); бесцветное твердое вещество, т. пл. 177-179°С; Rf=0,19 (гексан/этилацетат, 1/1);
Спектр 1H ЯМР (400 MHz, CDCl3/CD3OD, δ м.д.): 2,93 (s, 3H, CH3), 3,25 (dd, J=8,4 Hz, J=13,1 Hz, 1H, CH2), 3,36 (dd, J=4,1 Hz, J= 13,1 Hz, 1H, CH2), 4,88 (dd, J= 4,1 Hz, J=8,4 Hz, 1H, CH), 6,41-6,45 (m, 1H, 1СH(Ar)), 6,49-6,53 (m, 1H, 1СH(Ar)), 6,56-6,58 (m, 1H, СH(Ar)), 7,04-7,10 (m, 1H, 1СH(Ar)), 7,41-7,47 (m, 4H, 4СH(Ar)), 7,50-7,55 (m, 4H, 4СH(Ar));
Спектр 13C ЯМР (101 MHz, CDCl3/CD3OD, δ м.д.): 38,8 (CH3), 51,7 (CH2), 71,8 (CH), 105,2 (СH(Ar)), 109,6 (СH(Ar)), 110,1 (СH(Ar)), 121,7 (СH(Ar)), 126,8 (2СH(Ar)), 127,2 (2СH(Ar)), 128,9 (2СН(Ar)), 130,5 (СH(Ar)), 132,1 (2СH(Ar)), 138,9 (С(Ar)), 139,6 (С(Ar)), 140,0 (С(Ar)), 142,3(С(Ar)), 149,5 (С(Ar)).
HRMS-ESI [M + H]+: рассчитано для C21H22BrN2O3S 461,0529, найдено 461,0511.
Синтез соединений 1g и 1i.
Раствор трифторуксусной кислоты (1,6 ммоль) в дихлорметане (1 мл) медленно добавляли к раствору Вос-защищенного амина 1f (или спирта 1h) (1 ммоль) в дихлорметане (2 мл) при 0 °С. Реакционную смесь перемешивали при 0°С в течение 1 часа и затем при комнатной температуре - в течение 1 дня. Три раза добавляли бензол (порциями по 10 мл за раз), и растворители азеотропно удаляли при пониженном давлении. Незащищенные амины выделяли в виде соответствующих трифторуксусных солей. Остаток обрабатывали раствором NaHCO3 (25 мл). Водную часть экстрагировали этилацетатом (3 порции по 15 мл). Объединенные органические экстракты промывали водой и дважды солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией (SiO2, гексан/этилацетат, 1/3, соединение 1g) или перекристаллизацией из ацетона (соединение 1i).
Соединение 1-([1,1'-бифенил]-4-ил)-2-((4-аминофенил)амино)этанол 1g: Выход 0,18 г (60%); светло-коричневое твердое вещество, т. пл. 140-142°С; Rf=0,19 (гексан/этилацетат, 1/3);
Спектр 1H ЯМР (400 MHz, CDCl3, δ м.д.): 3,23 (dd, J=8,8 Hz, J=12,9 Hz, 1H, CH2), 3,37 (dd, J=3,7 Hz, J=12,9 Hz, 1H, CH2), 4,90 (dd, J=3,8 Hz, J=8,7 Hz, 1H, CH), 6,55-6,64 (m, 4H, 4СH(Ar)), 7,35-7,41 (m, 1H, СH(Ar)), 7,43-7,50 (m, 4H, 4СH(Ar)), 7,58-7,64 (m, 4H, 4СH(Ar));
Спектр 13C ЯМР (101 MHz, CDCl3, δ м.д.): 53,2 (CH2), 72,1 (CH), 115,5 (2СH(Ar)), 117,0 (СH(Ar)), 126,5 (2СH(Ar)), 127,2 (2СH(Ar)), 127,33 (2СH(Ar)), 127,43 (СH(Ar)), 128,9 (2СH(Ar)), 138,3 (2СН(Ar)), 140,75 (С(Ar)), 140,81 (С(Ar)), 140,90 (С(Ar)), 141,5 (С(Ar));
HRMS-ESI [M - H]+ рассчитано для C20H19N2O 303,1492, найдено 303,1489.
Соединение 3-((2-([1,1'-бифенил]-4-ил)-2-гидроксиэтил)амино)фенол 1i: Выход 0,21 г (63%); светло-желтое твердое вещество, т. пл. 153-155°С; Rf= 0,24 (гексан/этилацетат, 3/2);
Спектр 1H ЯМР (400 MHz, CD3OD, δ м.д.): 4,00 (d, 2H, CH2, J3= 5,8Hz), 5,02 (dd, J=5,8 Hz, 1H, СH), 6,68-6,74 (m, 2H, 2СH(Ar)), 6,76-7,82 (m, 2H, 2СH(Ar)), 7,27-7,33 (m, 1H, СH(Ar)), 7,36-7,43 (m, 2H, 2СH(Ar)), 7,47-7,52 (m, 2H, 2СH(Ar)), 7,56-7,61 (m, 4H, 4СH(Ar));
Спектр 13C ЯМР (101 MHz, CD3OD, δ м.д.): 52,8 (CH2), 72,8 (CH), 101,1 (СH(Ar)), 105,6 (СH(Ar)), 106,5(СH(Ar)), 127,6 (2СH(Ar)), 127,91 (2СH(Ar)), 127,94 (2СH(Ar)), 128,3 (СH(Ar)), 129,8 (2СH(Ar)), 130,9 (СH(Ar)), 141,7 (С(Ar)), 142,2 (С(Ar)), 143,5 (С(Ar)), 151,2 (С(Ar)), 159,3 (С(Ar));
HRMS-ESI [M + Na]+: рассчитано для C20H19NO2Na 328,1308, найдено 328,1309.
Синтез соединения 1e
К кетону 3е (0,2 г, 0,5807 ммоль) и NaBH4 (0,101 г, 2,671 ммоль) в сухом ТГФ (3 мл) добавляли йод (0,3 г, 1,161 ммоль) в сухом ТГФ (2,5 мл) в (несколько) порции под атмосферой аргона при 0 °С в течение 50 минут. Реакционную смесь кипятили с обратным холодильником (70 °С) в течение 3 часов; охлажденный до 0°С избыток NaBH4 разрушали осторожным добавлением 3М HCl (0,8 мл). После прекращения выделения газа его нейтрализовали с помощью 3М NaOH (1 мл). Органический слой отделяли, а водный слой экстрагировали этилацетатом (3 порции по 10 мл). Объединенные органические экстракты промывали водой, рассолом и сушили над MgSO4. Растворитель удаляли и продукт очищали колоночной хроматографией на силикагеле (гексан/этилацетат, 2/1).
Соединение 1-([1,1'-бифенил]-4-ил)-2-((3-(этиламино)фенил)амино)этанол 1e: Выход 0,13 г (67 %); желто-коричневое твердое вещество, т. пл. 44°С; Rf=0,24 (гексан/этилацетат, 2/1);
Спектр 1H ЯМР (400 MHz, CDCl3, δ м.д.): 1,25 (t, J=7,2Hz, 3H, CH3), 3,14 (q, J=7,2Hz, 2H, CH2), 3,32 (dd, J=8,7Hz, J=13,1Hz, 1H, CH2), 3,46 (dd, J=4,0Hz, J=13,1Hz, 1H, CH2), 4,97 (dd, J=4,0Hz, J=8,7Hz, 1H, CH), 5,95-5,99 (m, 1H, 1СH(Ar)), 6,06-6,13 (m, 2H, 2СH(Ar)), 6,99-7,05 (m, 1H, СH(Ar)), 7,34-7,40 (m, 1H, 1СH(Ar)), 7,43-7,52 (m, 4H, 4СH(Ar)), 7,58-7,65 (m, 4H, 4СH(Ar));
HRMS-ESI [M + H]+: рассчитано для C22H25N2O 333,1961, найдено 333,1953.
Синтез соединения 1l
К суспензии LiAlH4 (0,03 г, 0,75 ммоль) в сухом диэтиловом эфире (6 мл) добавляли по каплям раствор 3-(2-([1,1'-бифенил]-4-ил)-2-оксоэтокси)фенилацетата 3l (0,13 г, 0,38 ммоль) в смеси диэтилового эфира (9 мл) и тетрагидрофурана (5 мл) при 0°C в атмосфере аргона. Полученную смесь перемешивали при кипячении с обратным холодильником в течение 2 ч, затем охлаждали до 0°С и гасили 20 мл воды и 10% H2SO4 (до рН 3). Органический слой сушили над безводным MgSO4 и концентрировали в вакууме. Продукт очищали колоночной хроматографией на силикагеле (CH2Cl2/CH3OH, 20/1).
Соединение 3-(2-([1,1'-бифенил]-4-ил)-2-аминоэтокси)фенол 1l: выход 0,1 г (65%); бесцветное твердое вещество, т. пл. 164-166°С; Rf=0,11 (гексан/этилацетат, 2/1);
Спектр 1H ЯМР (400 MHz, CD3OD, δ м.д.): 4,15 (dd, J=5,4 Hz, J=9,7 Hz, 1H, CH2), 4,19 (dd, J=9,7 Hz, J=7,4 Hz, 1H, CH2), 4,40 (dd, J= 5,4 Hz, J= 7,4 Hz, 1H, CH), 6,36-6,41 (m, 2H, 2СH(Ar)), 6,42-6,46 (m, 1H, СH(Ar)), 7,03-7,09 (m, 1H, СH(Ar)), 7,29-7,35 (m, 1H, 1СH(Ar)), 7,39-7,45 (m, 2H, 2СH(Ar)), 7,52-7,56 (m, 2H, 2СH(Ar)), 7,59-7,64 (m, 4H, 4СH(Ar)).
Двухстадийный синтез соединения 1k.
1. Гидрохлорид гидроксиламина (0,08 г, 1,155 ммоль) и пиридин (0,1 мл, 1,270 ммоль) добавляли при комнатной температуре к раствору 3-(2-([1,1-бифенил]-4-ил)-2-оксоэтокси)фенилацетата 3l (0,2 г, 0,5774) ммоль) в смеси MeOH (56 мл) и H2O (7 мл). Реакционную смесь перемешивали в течение 2 дней (контроль ТСХ). После завершения реакции смесь концентрировали при пониженном давлении, пока раствор не помутнел. Затем его разбавляли ледяной водой (30 мл) и перемешивали в течение несколько минут. Раствор оставляли в холодильнике на ночь. Осадок отфильтровывали и промывали водой. Очистка проводилась колоночной хроматографией на силикагеле (CH2Cl2/MeOH, 50/1), получали бесцветные кристаллы оксима (0,19 г, 91%).
2. К полученному на предыдущей стадии оксиму (0,19 г, 0,5258 ммоль) и NaBH4 (0,09 г, 2,419 ммоль) в сухом ТГФ (3 мл) добавляли йод (0,27 г, 1,052 ммоль) в сухом ТГФ (2,5 мл) в несколько порций в атмосфере аргона при 0°С в течение 50 минут. Реакционную смесь кипятили с обратным холодильником (70°С) в течение 3 часов; охлажденный до 0°С избыток NaBH4 разрушали осторожным добавлением 3М HCl (0,8 мл). После прекращения выделения газа его нейтрализовали с помощью 3М NaOH (1 мл). Органический слой отделяли, а водный слой экстрагировали этилацетатом (3 порции по 10 мл). Объединенные органические экстракты промывали водой, рассолом и сушили над MgSO4. Растворитель удаляли и продукт очищали колоночной хроматографией на силикагеле (гексан/этилацетат, 2/1).
Соединение 3-(2-([1,1'-бифенил]-4-ил)-2-гидроксиэтокси)фенол 1k: Выход 0,07 г (79 %); бесцветные кристаллы. т. пл. = 138-139ºС, Rf 0,19 (CH2Cl2:MeOH = 20:1).
Спектр 1H ЯМР (400 MHz, CDCl3+CD3OD, δ м.д.): 4,00 (дд, J3=8,3 Гц, J2=9,8 Гц, 1H, CH2), 4,05 (дд, J3=3,6 Гц, J2=9,8 Гц, 1H, CH2), 5,06 (дд, J3=3,6 Гц, J3=9,8 Гц, 1H, СH), 6,38-6,44 (м, 3H, 3СH(Ar)), 7,02-7,09 (м, 1H, СH(Ar)), 7,28-7,34 (м, 1H, СH(Ar)), 7,37-7,43 (м, 2H, 2СH(Ar)), 7,44-7,50 (м, 2H, 2СH(Ar)), 7,52-7,60 (м, 4H, 4СH(Ar));
Спектр 13C ЯМР (101 MHz, CD3OD, δ м.д.): 71,4 (CH), 72,3 (CH2), 101,4 (СH(Ar)), 105,1 (СH(Ar)), 107,4 (СH(Ar)), 126,16 (2СH(Ar)), 126,19 (2СH(Ar)), 126,22 (2СH(Ar)), 126,6 (СH(Ar)), 128,1 (2СН(Ar)), 129,2 (СН(Ar)), 139,8 (С(Ar)), 140,2 (С(Ar)), 140,3 (С(Ar)), 157,8 (С(Ar)), 159,7 (С(Ar));
HRMS-ESI (М+K)+: рассчитано для C20H18O3K: 345,0888, найдено 345,0894.
Пример 2. Оценка аффинности к сайту связывания ифенпродила, расположенному между аминоконцевыми доменами NMDA рецептора, по замещению [H3]-меченого ифенпродила.
Подготовку синаптических мембран головного мозга крысы осуществляли, как было описано в (Jones S.M., Snell L.D., Johnson K.M. Characterization of the binding of radioligands to the N-methyl-D-aspartate, phencyclidine, and glycine receptors in buffy coat membranes, Journal of Pharmacological Methods, 1989, V. 21, pp. 161-168). Ткань гиппокампа гомогенизировали с помощью гомогенизатора Поттера (тефлоновый пестик и стеклянный сосуд), используя измельчитель ткани при половине максимального значения, в 20 об. буфера № 1 (5 мМ HEPES / 4,5 мМ трис-буфера, рН 7,6, глюкоза 0,32 М). Гомогенат разбавляли буфером № 2 (5 мМ HEPES / 4,5 мМ Трис-буфер, рН 7,6) в соотношении 1:50, затем его центрифугировали при 1000 g в течение 10 мин. Супернатант собирали и затем центрифугировали при 25000g в течение 20 минут. Осадок гомогенизировали в буфере № 2 в соотношении 1:50 и центрифугировали в течение 20 минут при 8000 g. Осадок суспендировали в буфере № 3 (5 мМ HEPES / 4,5 мМ Трис-буфер, (рН 7,6), 5 мМ Na4EDTA) и суспензию снова центрифугировали. Эту процедуру промывки выполняли четыре раза, и при последней промывке Na4EDTA исключали из буферной композиции. Конечный осадок ресуспендировали в 5 объемах буфера № 2 и хранили в жидком азоте. В день использования мембранную фракцию размораживали. Рабочий раствор (конечный объем 0,5 мл) содержал 200 мкл буфера № 2, 50 мкл [H3]-ифенпродила (40 нМ) с удельной активностью 179 Ки/мМ и 250 мкл суспензии мембраны. Неспецифическое связывание определяют в присутствии 50 мкл немеченого ифенпродила (10 мкМ). Смесь инкубировали при комнатной температуре в течение 2 часов. В конце инкубации образцы фильтровали через фильтры из стекловолокна GF/B (Whatman), предварительно увлажненные в 0,3% полиэтиленамине в течение 2 часов при 4 оС. Каждую пробирку промывали один раз холодным буфером № 2, затем фильтры промывали три раза одинаковым объемом буфера. Фильтры сушили на воздухе до полного высыхания и переносили в сцинтилляционные флаконы, в которые добавляли 5 мл сцинтилляционной жидкости, содержащей 4 г дифенилоксазола (PPO), 0,2 г дифенилоксазолилбензола (POPOP) и 1 л толуола. Радиоактивность определяли на сцинтилляционном счетчике TriCarb2800 TR (PerkinElmer, Packard, США) с эффективностью счета около 65%. Исследование влияния исследуемых соединений на связывание [H3]-ифенпродила с мембранами гиппокампа крыс проводили с добавлением 50 мкл раствора исследуемых соединений к инкубационной среде в диапазоне концентраций 10-8-10-3 М. Каждую концентрацию измеряли 3 раза и определяли среднюю величину.
В таблице 2 представлены результаты измерения EC50 для веществ из примера 1.
Таблица 2. Результаты экспериментов по замещению радиоактивно-меченого [H3]-ифенпродила
В частности, как видно из Таблицы 2, в эксперименте было найдено, что вещество 1k из Примера 1 способно вытеснять радиоактивно меченый ифенпродил с EC50 ~ 30 мкМ (см. также Фиг. 1)
Пример 3. Оценка концентрационной зависимости ингибирования NMDA/Gly-индуцированных токов на нейронах, выделенных из гиппокампа крыс
Эксперименты проводились на нативных рецепторах нейронов, выделенных из переживающих срезов мозга крыс линии Вистар с помощью метода вибродиссоциации (Vorobjev V.S., Vibrodissociation of sliced mammalian nervous tissue, Journal of Neuroscience Methods, 1991, V. 38, pp. 145-150). Исследование действия соединений на NMDA рецепторы и проводилось на пирамидных нейронах гиппокампа. Активация NMDA рецепторов осуществлялась NMDA (100 мкМ) в присутствии глицина (10 мкМ). Для регистрации токов применялся метод фиксации потенциала в конфигурации «whole cell». Внеклеточный раствор содержал (в мМ): NaCl 143; KCl 5; CaCl2 2,5; D-глюкоза 10; HEPES 10 (pH доводился до 7,4 при помощи HCl). Пипеточный раствор содержал (в мМ): CsF 100; CsCl 40; NaCl 5; CaCl2 0,5; EGTA 5; HEPES 10 (pH доводился до 7,2 при помощи CsOH). Микропипетки изготавливались из боросиликатного стекла при помощи пуллера P-97 (Sutter Instruments, USA). Исследуемые вещества подавались при помощи перфузирующей системы RSC-200 (BioLogic Science Instruments, France), обеспечивающей смену раствора за не более чем 50мс. Для записи ответов использовался усилитель EPC 8 (HEKA Elektronik, Germany). Управление экспериментом и запись ответов осуществлялись при помощи персонального компьютера. Исследуемые соединения растворялись в DMSO для приготовления сток растворов. Экспериментальные растворы исследуемых веществ приготовлялись путем добавления необходимого количества сток раствора во внеклеточный раствор, содержащий также агонисты NMDA и глицин. Блокирующее действие исследуемых соединений определялось по их способности ингибировать поддерживающийся при аппликации агониста трансмембранный ток. Действие разных концентраций блокаторов использовалось для построения концентрационных зависимостей и определения величины IC50 при помощи уравнения Хилла. Статистическая обработка проводилась с помощью программы Microcal Origin 6.0.
Концентрационная зависимость ингибирования NMDA/Gly-индуцированных токов для соединения 1k из Примера 1 представлена на Фиг. 2. Аппроксимация концентрационной зависимости уравнением Хилла дает IC50 = 5 ± 2 мкМ. Значения IC50 для соединений 1k, 1d и 1i приведены в Таблице 3.
Таблица 3. Результаты измерения ингибирования NMDA/Gly-индуцированных токов (IC50) для некоторых соединений из Примера 1
Таким образом, исследуемые соединения обладают ингибирующим эффектом на NMDA рецептор.
Пример 4. Композиции по изобретению
Ниже приведен иллюстративный пример состава таблеток массой 100 мг, содержащих 5 мг соединения 1d.
Далее приведен иллюстративный пример состава капсул массой 500 мг, содержащих 10 мг соединения 1d
Далее приведен иллюстративный пример состава раствора для инъекций (2 мл), содержащего 1 мг соединения 1d.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ | 2018 |
|
RU2837261C2 |
| ДИАРИЛЬНЫЕ СОЕДИНЕНИЯ С МОСТИКОВОЙ СВЯЗЬЮ | 2002 |
|
RU2297409C2 |
| АНТИГИПЕРТЕНЗИВНЫЕ СОЕДИНЕНИЯ ДВОЙНОГО ДЕЙСТВИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2476427C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ВКЛЮЧАЮЩИЕ 2-АМИНОПИРИДИН-3-СУЛЬФОНОВЫЙ ФРАГМЕНТ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФОКУСИРОВАННАЯ БИБЛИОТЕКА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2004 |
|
RU2263667C1 |
| ИНГИБИТОРЫ НЕПРИЛИЗИНА | 2011 |
|
RU2622288C2 |
| 3,5-ДИЗАМЕЩЕННЫЕ 1,2,4-ТИАДИАЗОЛЬНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ СВЯЗЫВАНИЯ ТИОЛОВ | 1997 |
|
RU2173319C2 |
| СОЧЕТАННАЯ ТЕРАПИЯ С ИСПОЛЬЗОВАНИЕМ ПЕНТАФТОРБЕНЗОЛСУЛЬФОНАМИДОВ | 2000 |
|
RU2268054C2 |
| ИНГИБИТОРЫ НЕПРИЛИЗИНА | 2011 |
|
RU2605557C2 |
| ЗАМЕЩЕННЫЕ 3-ПИРИДИЛМЕТИЛАМИНЫ И ФОКУСИРОВАННАЯ БИБЛИОТЕКА | 2003 |
|
RU2228930C1 |
| СЕЛЕКТИВНЫЕ АНТАГОНИСТЫ АДЕНОЗИНОВЫХ A РЕЦЕПТОРОВ | 2007 |
|
RU2467009C2 |
Изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, которые обладают антагонистической активностью по отношению к NMDA-рецепторам. В формуле I X означает -Н, атом галогена или -СН3; Z означает -OR' или -NR'R''; линкер L означает группу -(С0-2алкилен)-Y-(С0-2алкилен)-, где под "С0-алкиленом" понимается простая связь и где Y означает группу -О- или -NR'-; R1 означает -Н, -OR', -NR'R'' или -NR'-SO2-(C1-6-алкил); R2 означает -Н, -OR' или -NR'R''; R в каждом случае означает Н; R' и R'' в каждом случае независимо представляют собой группы, выбранные из Н и C1-6-алкила; и m равно 3, при условии, что X, R1 и R2 не означают все одновременно -Н, и при условии, что когда одна группа из R1 и R2 означает -NH2, а другая означает -Н, X означает -Н, и L означает -СН2-O-, то Z не представляет собой -ОН. Изобретение относится также к фармацевтической композиции для ингибирования NMDA-рецепторов, содержащей фармакологически эффективное количество одного или нескольких соединений формулы I или их фармацевтически приемлемых солей и один или более фармацевтически приемлемых вспомогательных агентов. В формуле I X означает -Н, атом галогена или -СН3; Z означает -OR' или -NR'R''; линкер L означает группу -(С0-2алкилен)-Y-(С0-2алкилен)-, где под "С0-алкиленом" понимается простая связь и где Y означает группу -О- или -NR'-; R1 означает -H, -OR', -NR'R'' или -NR'-SO2-(С1-6-алкил); R2 означает -Н, -OR' или -NR'R''; R в каждом случае означает Н; R' и R'' в каждом случае независимо представляют собой группы, выбранные из Н и С1-6-алкила; и m равно 3. Описан также способ лечения нарушения или заболевания, сопровождающегося повышенной активацией NMDA-рецептора, включающий стадию, на которой нуждающемуся в этом пациенту вводят эффективное количество одного или нескольких соединений формулы I или их фармацевтически приемлемых солей или фармацевтической композиции. В формуле I X означает -Н, атом галогена или -СН3; Z означает -OR' или -NR'R''; линкер L означает группу -(С0-2алкилен)-Y-(С0-2алкилен)-, где под "С0-алкиленом" понимается простая связь и где Y означает группу -О- или -NR'-; R1 означает -H, -OR', -NR'R'' или -NR'-SO2-(С1-6-алкил); R2 означает -Н, -OR' или -NR'R''; R в каждом случае означает Н; R' и R'' в каждом случае независимо представляют собой группы, выбранные из Н и С1-6-алкила; и m равно 3. Также раскрыто применение соединений формулы I в качестве ингибиторов NMDA-рецепторов. В формуле I X означает -Н, атом галогена или -СН3; Z означает -OR' или -NR'R''; линкер L означает группу -(С0-2алкилен)-Y-(С0-2алкилен)-, где под "С0-алкиленом" понимается простая связь, и где Y означает группу -О- или -NR'-; R1 означает -H, -OR', -NR'R'' или -NR'-SO2-(С1-6-алкил); R2 означает -Н, -OR' или -NR'R''; R в каждом случае означает Н; R' и R'' в каждом случае независимо представляют собой группы, выбранные из Н и С1-6-алкила; и m равно 3. 4 н. и 28 з.п. ф-лы, 3 табл., 4 пр., 2 ил.
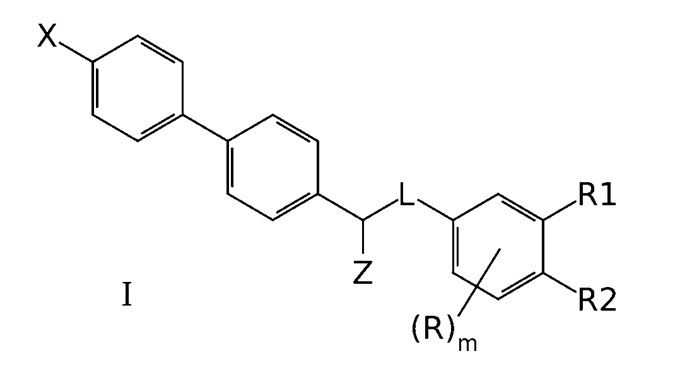
1. Соединение формулы I
 ,
,
где X означает -Н, атом галогена или -СН3;
Z означает -OR' или -NR'R'';
линкер L означает группу -(С0-2алкилен)-Y-(С0-2алкилен)-, где под "С0-алкиленом" понимается простая связь и где Y означает группу -О- или -NR'-;
R1 означает -Н, -OR', -NR'R'' или -NR'-SO2-(C1-6-алкил);
R2 означает -Н, -OR' или -NR'R'';
R в каждом случае означает Н;
R' и R'' в каждом случае независимо представляют собой группы, выбранные из Н и C1-6-алкила; и
m равно 3,
или его фармацевтически приемлемая соль,
при условии, что X, R1 и R2 не означают все одновременно -Н, и
при условии, что когда одна группа из R1 и R2 означает -NH2, а другая означает -Н, X означает -Н, и L означает -СН2-O-, то Z не представляет собой -ОН.
2. Соединение по п.1, в котором Z выбран из -ОН и -NH2.
3. Соединение по любому из пп. 1, 2, в котором L означает группу -(C0-1алкилен)-Y-(С0-1алкилен)-, более предпочтительно группу -СН2-Y-.
4. Соединение по любому из пп. 1-3, в котором R1 выбран из группы, включающей -Н, -OR', -NHR' или -NR'-SO2-(С1-6-алкил).
5. Соединение по любому из пп.1-4, представляющее собой соединение формулы 1
 ,
,
где X1 означает -Н, -СН3 или атом галогена (предпочтительно -F, -Cl или -Br);
Х2 означает -О- или -NH-;
R1 означает -Н, -ОН, -NH2, -NH-(С1-6-алкил) или -NH-SO2-(С1-6-алкил);
R2 означает -Н, -ОН или -NH2;
R3 означает -NH2 или -ОН,
или его фармацевтически приемлемую соль.
6. Соединение по любому из пп. 1-5, выбранное из следующих:
4-(2-([1,1'-бифенил]-4-ил)-2-гидроксиэтокси)фенол;
N-(3-((2-([1,1'-бифенил]-4-ил)-2-гидроксиэтил)амино)фенил)метансульфонамид;
N-(3-((2-(4'-бром-[1,1'-бифенил]-4-ил)-2-гидроксиэтил)амино)фенил)метансульфонамид;
1-([1,1'-бифенил]-4-ил)-2-((4-аминофенил)амино)этанол;
3-((2-([1,1'-бифенил]-4-ил)-2-гидроксиэтил)амино)фенол;
1-([1,1'-бифенил]-4-ил)-2-((3-(этиламино)фенил)амино)этанол;
3-(2-([1,1'-бифенил]-4-ил)-2-аминоэтокси)фенол;
3-(2-([1,1'-бифенил]-4-ил)-2-гидроксиэтокси)фенол.
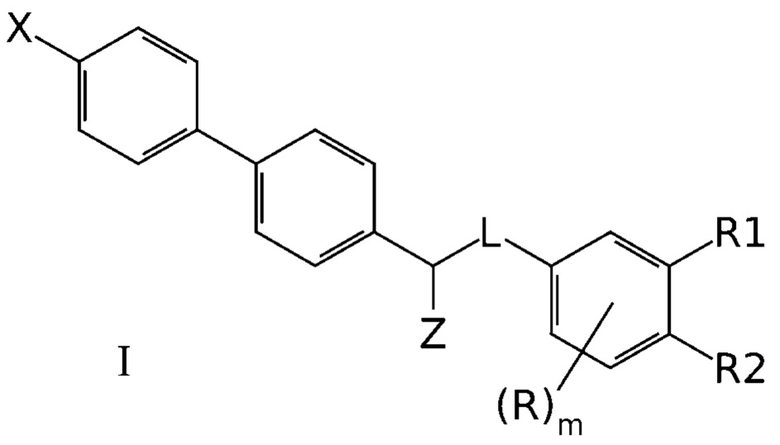
7. Фармацевтическая композиция для ингибирования NMDA-рецепторов, содержащая фармакологически эффективное количество одного или нескольких соединений формулы I
 ,
,
где X означает -Н, атом галогена или -СН3;
Z означает -OR' или -NR'R'';
линкер L означает группу -(С0-2алкилен)-Y-(С0-2алкилен)-, где под "С0-алкиленом" понимается простая связь и где Y означает группу -О- или -NR'-;
R1 означает -H, -OR', -NR'R'' или -NR'-SO2-(С1-6-алкил);
R2 означает -Н, -OR' или -NR'R'';
R в каждом случае означает Н;
R' и R'' в каждом случае независимо представляют собой группы, выбранные из Н и С1-6-алкила; и
m равно 3,
или их фармацевтически приемлемых солей,
и один или более фармацевтически приемлемых вспомогательных агентов.
8. Композиция по п. 7, в которой Z выбран из -ОН и -NH2.
9. Композиция по п. 7 или 8, в которой L означает группу -(С0-1алкилен)-Y-(С0-1алкилен)-, более предпочтительно группу -СН2-Y-.
10. Композиция по любому из пп. 7-9, в которой R1 выбран из группы, включающей -Н, -OR', -NHR' или -NR'-SO2-(С1-6-алкил).
11. Композиция по любому из пп. 7-10, в которой соединение формулы I представляет собой соединение формулы 1
 ,
,
где X1 означает -Н, -СН3 или атом галогена (предпочтительно -F, -Cl или -Br);
Х2 означает -О- или -NH-;
R1 означает -Н, -ОН, -NH2, -NH-(С1-6-алкил) или -NH-SO2-(С1-6-алкил);
R2 означает -Н, -ОН или -NH2;
R3 означает -NH2 или -ОН,
или его фармацевтически приемлемую соль.
12. Композиция по любому из пп. 7-11, в которой соединение формулы I выбрано из следующих:
4-(2-([1,1'-бифенил]-4-ил)-2-гидроксиэтокси)фенол;
1-([1,1'-бифенил]-4-ил)-2-феноксиэтанол;
1-([1,1'-бифенил]-4-ил)-2-(фениламино)этанол;
N-(3-((2-([1,1'-бифенил]-4-ил)-2-гидроксиэтил)амино)фенил)метансульфонамид;
N-(3-((2-(4'-бром-[1,1'-бифенил]-4-ил)-2-гидроксиэтил)амино)фенил)метансульфонамид;
1-([1,1'-бифенил]-4-ил)-2-((4-аминофенил)амино)этанол;
3-((2-([1,1'-бифенил]-4-ил)-2-гидроксиэтил)амино)фенол;
1-([1,1'-бифенил]-4-ил)-2-((3-(этиламино)фенил)амино)этанол;
3-(2-([1,1'-бифенил]-4-ил)-2-аминоэтокси)фенол;
3-(2-([1,1'-бифенил]-4-ил)-2-гидроксиэтокси)фенол.
13. Композиция по любому из пп. 7-12, в которой X, R1 и R2 не означают все одновременно -Н и/или в которой, когда одна группа из R1 и R2 означает -NH2, а другая означает -Н, X означает -Н и L означает -СН2-O-, то Z не представляет собой -ОН.
14. Композиция по любому из пп. 7-13, содержащая от 0,01 до 50 вес.%, предпочтительно от 0,1 до 40 вес.%, более предпочтительно от 0,5 до 30 вес.%, например от 1 до 20 вес.%, например от 5 до 10 вес.% одного или нескольких соединений формулы I.
15. Композиция по любому из пп. 7-14, предназначенная для введения путем, выбранным из перорального, парентерального, внутривенного, интраперитонеального, интраназального, подкожного введения, введения в виде суппозитория или местного нанесения.
16. Композиция по любому из пп. 7-15, в которой один или более фармацевтически приемлемых вспомогательных агентов выбраны из группы, включающей разбавители (например, свекловичный сахар, лактозу, глюкозу, натрия хлорид, сорбит, маннит, гликоль, фосфат кальция двузамещенный), связующие вещества (например, магниевый силикат алюминия, крахмальную пасту, желатин, трагакант, метилцеллюлозу, карбоксиметилцеллюлозу, поливинилпирролидон), разрыхлители (например, декстрозу, агар, альгиновую кислоту или ее соли, крахмал), красители (например, тартразин, индиго), стабилизаторы (например, натрия метабисульфит, натрия бензоат, абрикосовую или аравийскую камедь, декстрин, крахмальный клейстер, метилцеллюлозу), консерванты (например, натрия метабисульфит, бензойная кислота, натрия бензоат, смеси метилпарабена и пропилпарабена), соли, регулирующие осмотическое давление (например, хлорид натрия), и буферы.
17. Способ лечения нарушения или заболевания, сопровождающегося повышенной активацией NMDA-рецептора, включающий стадию, на которой нуждающемуся в этом пациенту вводят эффективное количество одного или нескольких соединений формулы I
 ,
,
где X означает -Н, атом галогена или -СН3;
Z означает -OR' или -NR'R'';
линкер L означает группу -(С0-2алкилен)-Y-(С0-2алкилен)-, где под "С0-алкиленом" понимается простая связь и где Y означает группу -О- или -NR'-;
R1 означает -Н, -OR', -NR'R'' или -NR'-SO2-(С1-6-алкил);
R2 означает -Н, -OR' или -NR'R'';
R в каждом случае означает Н;
R' и R'' в каждом случае независимо представляют собой группы, выбранные из Н и С1-6-алкила; и
m равно 3,
или их фармацевтически приемлемых солей,
или фармацевтической композиции по любому из пп. 7-16.
18. Способ по п. 17, в котором Z выбран из -ОН и -NH2.
19. Способ по любому из пп. 17 или 18, в котором L означает группу -(С0-1алкилен)-Y-(С0-1алкилен)-, более предпочтительно группу -СН2-Y-.
20. Способ по любому из пп. 17-19, в котором R1 выбран из группы, включающей -Н, -OR', -NHR' или -NR'-SO2-(С1-6-алкил).
21. Способ по любому из пп. 17-20, в котором соединение формулы I представляет собой соединение формулы 1
 ,
,
где X1 означает -Н, -СН3 или атом галогена (предпочтительно -F, -Cl или -Br);
Х2 означает -О- или -NH-;
R1 означает -Н, -ОН, -NH2, -NH-(С1-6-алкил) или -NH-SO2-(С1-6-алкил);
R2 означает -Н, -ОН или -NH2;
R3 означает -NH2 или -ОН,
или его фармацевтически приемлемую соль.
22. Способ по любому из пп. 17-21, в котором соединение формулы I выбрано из следующих:
4-(2-([1,1'-бифенил]-4-ил)-2-гидроксиэтокси)фенол;
1-([1,1'-бифенил]-4-ил)-2-феноксиэтанол;
1-([1,1'-бифенил]-4-ил)-2-(фениламино)этанол;
N-(3-((2-([1,1'-бифенил]-4-ил)-2-гидроксиэтил)амино)фенил)метансульфонамид;
N-(3-((2-(4'-бром-[1,1'-бифенил]-4-ил)-2-гидроксиэтил)амино)фенил)метансульфонамид;
1-([1,1'-бифенил]-4-ил)-2-((4-аминофенил)амино)этанол;
3-((2-([1,1'-бифенил]-4-ил)-2-гидроксиэтил)амино)фенол;
1-([1,1'-бифенил]-4-ил)-2-((3-(этиламино)фенил)амино)этанол;
3-(2-([1,1'-бифенил]-4-ил)-2-аминоэтокси)фенол;
3-(2-([1,1'-бифенил]-4-ил)-2-гидроксиэтокси)фенол.
23. Способ по любому из пп. 17-22, в котором X, R1 и R2 не означают все одновременно -Н и/или в котором, когда одна группа из R1 и R2 означает -NH2, а другая означает -Н, X означает -Н и L означает -СН2-O-, то Z не представляет собой -ОН.
24. Способ по любому из пп. 17-23, в котором нарушением или заболеванием, сопровождающимся повышенной активацией NMDA-рецептора, является нейродегенеративное заболевание, инсульт, травма мозга, нейропатическая боль или депрессия.
25. Способ по любому из пп. 17-24, в котором соединение или композицию вводят путем, выбранным из перорального, парентерального, внутривенного, интраперитонеального, интраназального, подкожного введения, введения в виде суппозитория или местного нанесения.
26. Применение соединения формулы I
 ,
,
где X означает -Н, атом галогена или -СН3;
Z означает -OR' или -NR'R'';
линкер L означает группу -(С0-2алкилен)-Y-(С0-2алкилен)-, где под "С0-алкиленом" понимается простая связь и где Y означает группу -О- или -NR'-;
R1 означает -Н, -OR', -NR'R'' или -NR'-SO2-(С1-6-алкил);
R2 означает -Н, -OR' или -NR'R'';
R в каждом случае означает Н;
R' и R'' в каждом случае независимо представляют собой группы, выбранные из Н и С1-6-алкила; и
m равно 3,
или его фармацевтически приемлемой соли,
в качестве ингибитора NMDA-рецепторов.
27. Применение по п. 26, в котором Z выбран из -ОН и -NH2.
28. Применение по п. 26 или 27, в котором L означает группу -(С0-1алкилен)-Y-(С0-1алкилен)-, более предпочтительно группу -СН2-Y-.
29. Применение по любому из пп. 26-28, в котором R1 выбран из группы, включающей -Н, -OR', -NHR' или -NR'-SO2-(С1-6-алкил).
30. Применение по любому из пп. 26-29, в котором соединение формулы I представляет собой соединение формулы 1
 ,
,
где X1 означает -Н, -СН3 или атом галоген (предпочтительно -F, -Cl или -Br);
Х2 означает -О- или -NH-;
R1 означает -Н, -ОН, -NH2, -NH-(С1-6-алкил) или -NH-SO2-(С1-6-алкил);
R2 означает -Н, -ОН или -NH2;
R3 означает -NH2 или -ОН,
или его фармацевтически приемлемую соль.
31. Применение по любому из пп. 26-30, в котором соединение формулы I выбрано из следующих:
4-(2-([1,1'-бифенил]-4-ил)-2-гидроксиэтокси)фенол;
1-([1,1'-бифенил]-4-ил)-2-феноксиэтанол;
1-([1,1'-бифенил]-4-ил)-2-(фениламино)этанол;
N-(3-((2-([1,1'-бифенил]-4-ил)-2-гидроксиэтил)амино)фенил)метансульфонамид;
N-(3-((2-(4'-бром-[1,1'-бифенил]-4-ил)-2-гидроксиэтил)амино)фенил)метансульфонамид;
1-([1,1'-бифенил]-4-ил)-2-((4-аминофенил)амино)этанол;
3-((2-([1,1'-бифенил]-4-ил)-2-гидроксиэтил)амино)фенол;
1-([1,1'-бифенил]-4-ил)-2-((3-(этиламино)фенил)амино)этанол;
3-(2-([1,1'-бифенил]-4-ил)-2-аминоэтокси)фенол;
3-(2-([1,1'-бифенил]-4-ил)-2-гидроксиэтокси)фенол.
32. Применение по любому из пп. 26-31, в котором X, R1 и R2 не означают все одновременно -Н и/или в котором, когда одна группа из R1 и R2 означает -NH2, а другая означает -Н, X означает -Н и L означает -СН2-O-, то Z не представляет собой -ОН.
| N.MANN et al., Potentielle Analgetika, 7 | |||
| Mitt | |||
| Uber die analgetische Wirksamkeit einiger reduzierter Biphenyl-Mannich-Basen, ARCHIV DER PHARMAZIE, 1976, V.309, No.4, p.320-325 | |||
| R.M.SHAFIK et al., New biphenyl derivatives II: 1-(4-biphenylyl)-1-hydroxy-2-aminoethanes and 1-(4-biphenylyl)-1-chloro-2-aminoethanes as potential β-adrenoceptor |

