Данное изобретение относится к соединениям, которые являются агонистами мускаринового рецептора M1 и/или M4 и пригодны для лечения заболеваний, опосредованных мускариновыми рецепторами M1/M4. Предложены также фармацевтические композиции, содержащие эти соединения, и терапевтическое применение этих соединений.
УРОВЕНЬ ТЕХНИКИ
Мускариновые ацетилхолиновые рецепторы (mAChR) являются представителями суперсемейства рецепторов, сопряженных с G белком, которые опосредуют работу нейромедиатора ацетилхолина, как в центральной, так и в периферической нервной системе. Было клонировано пять подтипов mAChR, от M1 до M5. Преимущественно mAChR M1 экспрессируются постсинаптически в коре головного мозга, гиппокампе, стриатуме и таламусе; mAChR M2 расположены преимущественно в стволе головного мозга и таламусе, а также и в коре головного мозга, гиппокампе и стриатуме, где они расположены на холинергических синаптических окончаниях (Langmead et al., 2008 Br J Pharmacol). Однако mAChR M2 также экспрессируются на мышечной ткани сердца (где они опосредуют вагальную иннервацию сердца), гладкой мускулатуре и экзокринных железах. В ЦНС mAChR M3 экспрессируются на относительно низком уровне, но повсеместно экспрессируются в гладкой мускулатуре и железистой ткани, такой как потовые и слюнные железы (Langmead et al., 2008 Br J Pharmacol).
Мускариновые рецепторы в центральной нервной системе, особенно mAChR M1, играют критически важную роль в опосредовании высшей когнитивной деятельности. Заболевания, связанные с когнитивными нарушениями, такие как болезнь Альцгеймера, сопровождаются потерей холинергических нейронов в базальном отделе переднего мозга (Whitehouse et al., 1982 Science). При шизофрении, которая также сопровождается когнитивным нарушением как важным компонентом клинической картины, плотность mAChR снижена в префронтальной коре, гиппокампе и дорсальном стриатуме больных шизофренией субъектов (Dean et al., 2002 Mol Psychiatry). Кроме того, на моделях животных показано, что блокада или повреждение центральных холинергических путей приводит к существенным расстройствам познавательных способностей, а неселективные антагонисты mAChR у пациентов с психическими заболеваниями индуцируют психотомиметические эффекты. Холинергическая заместительная терапия в основном основана на применении ингибиторов ацетилхолинэстеразы для предотвращения распада эндогенного ацетилхолина. Эти соединения показали эффективность против симптоматического снижения когнитивных способностей при клиническом применении, но дают повышение ограниченных дозой нежелательных явлений, возникающих при стимуляции периферических mAChR M2 и M3, включая нарушенную моторику желудочно-кишечного тракта, брадикардию, тошноту и рвоту (http://www.drugs.com/pro/donepezil.html; http://www.drugs.com/pro/rivastigmine.html).
Дополнительно усилия по изучению направлены на определение прямых агонистов mAChR M1 с помощью индуцирования селективных улучшений в отношении когнитивной функции с благоприятным профилем в отношении неблагоприятных эффектов. Такие усилия привели к идентификации ряда агонистов, представленных в качестве примера такими соединениями, как ксаномелин, AF267B, сабкормелин, миламелин и цевимелин. Показано, что многие из этих соединений являются очень эффективными в доклинических моделях познавательных способностей как на грызунах, так и/или на животных приматах. Для миламелина показана эффективность против индуцированных скополамином расстройств в отношении кратковременной и пространственной памяти у грызунов; для сабкомелина продемонстрирована эффективность в задании визуального выделения объекта у игрунковых мартышек, а ксаномелин обеспечивает реверсию опосредованных антагонистами mAChR расстройств в отношении когнитивного функционирования в парадигме пассивного избегания.
Болезнь Альцгеймера (БА) представляет собой наиболее распространенное нарушение (26,6 миллионов человек в мире в 2006 г.), которая поражает старшее поколение, приводя к существенной потере памяти и когнитивной дисфункции. Этиология данного заболевания сложна, но характеризуется двумя признаками патологий головного мозга: наличие скоплений амилоидных бляшек, в основном состоящих из пептида амилоида-β (Aβ), и наличие нейрофибриллярных клубков, образованных гиперфосфорилированными тау-белками. Считается, что накопление Aβ является главным признаком прогрессирования БА и, по существу, множество предполагаемых видов терапии для лечения БА в настоящее время нацелены на ингибирование продукции Aβ. Aβ образуется в результате протеолитического расщепления связанного с мембраной белка-предшественника амилоида (APP). APP процессируется двумя путями, неамилоидогенным и амилоидогенным. Расщепление APP γ-секретазой обычно происходит обоими путями, но в первом APP расщепляется α-секретазой с образованием растворимого APPα. Однако в амилоидогенном пути APP расщепляется β-секретазой с образованием растворимого APPβ, а также Aβ. Исследования in vitro показали, что агонисты mAChR могут способствовать процессингу APP в направлении растворимой формы по неамилоидогенному пути. Исследования in vivo показали, что агонист mAChR, AF267B, изменяет болезненную патологию у трансгенной мыши 3xTgAD, модели с различными компонентами болезни Альцгеймера (Caccamo et al., 2006 Neuron). Для агониста mAChR, цевимелина, показано обеспечение небольшого, но существенного снижения уровней Aβ в спинномозговой жидкости у пациентов с болезнью Альцгеймера, тем самым демонстрируется потенциальная эффективность модификации заболевания (Nitsch et al., 2000 Neurol).
Результаты доклинических исследований дают основание предполагать, что агонисты mAChR проявляют атипичный антипсихотический профиль в ряде доклинических подходов. Агонист mAChR, ксаномелин, обеспечивает реверсию ряда опосредованных дофамином поведенческих реакций, включая индуцированную амфетамином двигательную активность у крыс, индуцированное апоморфином лазание у мышей, стимулируемое агонистами дофамина переворачивание у крыс с односторонним повреждением 6-OH-DA и индуцированное амфетамином моторное беспокойство у обезьян (без отрицательного влияния по EPS). Показано также, что у крыс это соединение ингибирует A10, но не A9, возбуждение клеток дофамином и кондиционированное избегание, и индуцирует экспрессию c-fos в префронтальной коре и центре удовольствия, но не в стриатуме. Эти все данные являются предположительными для атипичного антипсихотически подобного профиля (Mirza et al., 1999 CNS Drug Rev). Мускариновые рецепторы также вовлечены в нейробиологию зависимости. Подкрепляющее влияние кокаина и других вызывающих привыкание веществ опосредовано мезолимбической дофаминовой системой, причем поведенческие и нейрохимические исследования показали, что подтипы холинергических мускариновых рецепторов играют важные роли в регуляции дофаминэргической нейропередачи. Например, для мышей M(4) (-/-) продемонстрировано существенно усиленное стимулированное подкреплением поведение в результате воздействия кокаина (Schmidt et al Psychopharmacology (2011) Aug;216(3):367-78). Кроме того, продемонстрировано, что ксаномелин блокирует воздействие кокаина в этих моделях.
Мускариновые рецепторы также участвуют в управлении движением и потенциально предоставляют новые виды лечения для таких расстройств движения, как болезнь Паркинсона, ADHD, болезнь Хантингтона, синдром Туретта и других синдромов, связанных с дофаминэргической дисфункцией, лежащей в основе патогенетического фактора, стимулирующего заболевание.
Ксаномелин, сабкомелин, миламелин и цевимелин все находятся на различных стадиях клинических исследований касательно лечения болезни Альцгеймера и/или шизофрении. В клинических исследованиях ксаномелина II фазы продемонстрирована его эффективность по сравнению с различными блоками когнитивных симптомов, включая поведенческие нарушения и галлюцинации, связанные с болезнью Альцгеймера (Bodick et al., 1997 Arch Neurol). Данное соединение также проходит оценку в небольшом исследовании II фазы больных шизофренией и оно обеспечивает значимое снижение положительных и отрицательных симптомов по сравнению с контрольным плацебо (Shekhar et al., 2008 Am J Psych). Однако во всех клинических исследованиях ксаномелин и другие родственные агонисты mAChR продемонстрировали неприемлемый диапазон безопасного использования применительно к нежелательным явлениям, включающим тошноту, боли в области желудочно-кишечного тракта, диарею, диафорез (избыточное потовыделение), гиперсаливацию (избыточное слюновыделение), обморок и брадикардию.
Мускариновые рецепторы участвуют в возникновении центральных и периферических болей. Боль можно разделять на три различных типа: острую, воспалительную и нейропатическую. Острая боль выполняет важную защитную функцию в обеспечении безопасности организма от воздействий, которые могут вызывать повреждение тканей; однако требуется коррекция боли после хирургических операций. Воспалительная боль может возникать вследствие множества причин, включая повреждение тканей, аутоиммунный ответ и проникновение патогенов, и запускается воздействием медиаторов воспаления, таких как нейропептиды и простагландины, которые приводят к возникновению нейронального воспаления и боли. Нейропатическая боль ассоциируется с аномальными болезненными ощущениями на неболевые воздействия. Нейропатическая боль связана с рядом различных заболеваний/травм, таких как повреждение спинного мозга, рассеянный склероз, диабет (диабетическая нейропатия), вирусная инфекция (такая как ВИЧ или герпес). Она также является обычной при раковом заболевании, как результат побочного эффекта химиотерапии данного заболевания. Показано, что мускариновые рецепторы оказывают анальгезирующее действие при нескольких болезненных состояниях посредством активации рецепторов в спинном мозге и высших центрах боли в головном мозге. Показано, что увеличение эндогенных уровней ацетилхолина посредством ингибиторов ацетилхолинэстеразы, прямая активация мускариновых рецепторов с участием агонистов или аллостерических модуляторов обладает анальгезирующей активностью. В противоположность этому, блокада мускариновых рецепторов с участием антагонистов или использование мышей с нокаутом увеличивает чувствительность к боли. Подтверждение роли рецептора M1 в возникновении боли обсуждается D. F. Fiorino and M. Garcia-Guzman, 2012.
Недавно был идентифицирован ряд соединений, которые демонстрируют улучшенную селективность к подтипу mAChR M1 по сравнению с периферически экспрессируемыми подтипами mAChR (Bridges et al., 2008 Bioorg Med Chem Lett; Johnson et al., 2010 Bioorg Med Chem Lett; Budzik et al., 2010 ACS Med Chem Lett). Несмотря на увеличенные уровни чувствительности по сравнению с подтипом mAChR M3, некоторые из этих соединений сохраняют значимую агонистическую активность как для этого подтипа, так и для подтипа mAChR M2. В данном документе авторы изобретения описывают группы соединений, которые показывают неожиданно высокие уровни селективности к mAChR M1 и/или M4 в отличие от подтипов рецепторов M2 и M3.
Описание изобретения
В данном изобретении предложены соединения, обладающие активностью в качестве агонистов мускариновых рецепторов M1 и/или M4. В частности, в изобретении предложены соединения, которые проявляют чувствительность к рецептору M1 и/или рецептору M4 относительно подтипов рецепторов M2 и M3.
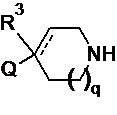
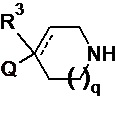
Соответственно, в одном варианте реализации изобретения (вариант реализации 1.1) в изобретении предложено соединение формулы (1) или формулы (1a):
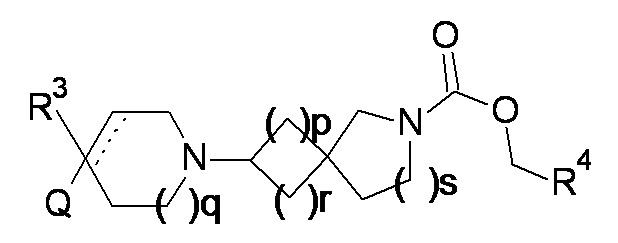 (1a)
(1a)
или соль указанного соединения, где
p равно 1 или 2;
q равно 0, 1 или 2;
r равно 1 или 2;
s равно 0 или 1, причем сумма r и s равна 1 или 2;
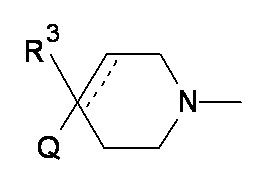
Q представляет собой CR1R2NR5R6, NR5R6, OR7, SR7;
R1 выбран из атома водорода или C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм;
R2 выбран из атома водорода или C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм;
R3 выбран из атома водорода; фтора; циано-; гидрокси-; аминогруппы и C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один, два или три, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм;
R4 представляет собой атом водорода или C1-6 неароматическую углеводородную группу, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм;
R5 выбран из гидроксигруппы; OR7; COR7; COOR7; CH2COR7; CH2COOR7; C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм; и группы W или CH2W, где W представляет собой необязательно замещенное 5- или 6-членное кольцо, содержащее 0, 1, 2 или 3 гетероатома, выбранных из O, N и S и их окисленных форм;
R6 выбран из гидроксигруппы; OR7; COR7; COOR7; CH2COR7; CH2COOR7; C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм; и группы W или CH2W, где W представляет собой необязательно замещенное 5- или 6-членное кольцо, содержащее 0, 1, 2 или 3 гетероатома, выбранных из O, N и S и их окисленных форм; и
R7 выбран из атома водорода, C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм; и группы W или C1-4 углеводородной группы W, где W представляет собой необязательно замещенное 5- или 6-членное кольцо, содержащее 0, 1, 2 или 3 гетероатома, выбранных из O, N и S и их окисленных форм;
а пунктирная линия показывает необязательную вторую углерод-углеродную связь, при условии, что когда присутствует углерод-углеродная связь, тогда отсутствует R3.
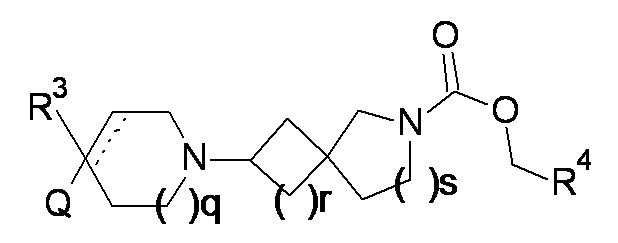 (1)
(1)
или соль указанного соединения, где
q равно 0, 1 или 2;
r равно 1 или 2;
s равно 0 или 1, причем сумма r и s равна 1 или 2;
Q представляет собой CR1R2NR5R6, NR5R6, OR7, SR7;
R1 выбран из атома водорода или C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм;
R2 выбран из атома водорода или C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм;
R3 выбран из атома водорода; фтора; циано-; гидрокси-; аминогруппы и C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один, два или три, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм;
R4 представляет собой атом водорода или C1-6 неароматическую углеводородную группу, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм;
R5 выбран из гидроксигруппы; OR7; COR7; COOR7; CH2COR7; CH2COOR7; C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм; и группы W или CH2W, где W представляет собой необязательно замещенное 5- или 6-членное кольцо, содержащее 0, 1, 2 или 3 гетероатома, выбранных из O, N и S и их окисленных форм;
R6 выбран из гидроксигруппы; OR7; COR7; COOR7; CH2COR7; CH2COOR7; C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм; и группы W или CH2W, где W представляет собой необязательно замещенное 5- или 6-членное кольцо, содержащее 0, 1, 2 или 3 гетероатома, выбранных из O, N и S и их окисленных форм; и
R7 выбран из атома водорода, C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм; и группы W или CH2W, где W представляет собой необязательно замещенное 5- или 6-членное кольцо, содержащее 0, 1, 2 или 3 гетероатома, выбранных из O, N и S и их окисленных форм;
а пунктирная линия показывает необязательную вторую углерод-углеродную связь, при условии, что когда присутствует углерод-углеродная связь, тогда отсутствует R3.
Конкретные соединения формулы (1) или формулы (1a) определены в вариантах реализации от 1.2 до 1.50, изложенных ниже.
1.2 Соединение в соответствии с вариантом реализации 1.1, в котором Q представляет собой NR5R6.
1.3 Соединение в соответствии с вариантом реализации 1.1, в котором Q представляет собой CR1R2NR5R6.
1.4 Соединение в соответствии с вариантами реализации от 1.1 до 1.3, в котором R1 выбран из атома водорода или C1-3 алкильной группы.
1.5 Соединение в соответствии с вариантом реализации 1.4, в котором R1 выбран из атома водорода, метила или этила.
1.6 Соединение в соответствии с вариантами реализации от 1.1 до 1.5, в котором R2 выбран из атома водорода или C1-3 алкильной группы.
1.7 Соединение в соответствии с вариантом реализации 1.6, в котором R2 выбран из атома водорода, метила или этила.
1.8 Соединение в соответствии с вариантом реализации 1.6, в котором R1 представляет собой H, а R2 выбран из атома водорода или метила.
1.9 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.8, в котором R5 выбран из C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм; и группы W или CH2W, где W представляет собой необязательно замещенное 5- или 6-членное кольцо, содержащее 0, 1, 2 или 3 гетероатома, выбранных из O, N и S и их окисленных форм.
1.10 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.8, в котором R5 выбран из C1-4 алкильной группы, которая необязательно замещена от одного до четырех атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм; и группы W или CH2W, где W представляет собой необязательно замещенное 5- или 6-членное ароматическое кольцо, содержащее 0, 1, 2 или 3 гетероатома, выбранных из O, N и S.
1.11 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.8, в котором R5 выбран из C1-4 алкильной группы, которая необязательно замещена от одного до четырех атомами фтора; и группы W или CH2W, где W представляет собой необязательно замещенное 5- или 6-членное ароматическое кольцо, содержащее 0, 1, 2 или 3 гетероатома, выбранных из O, N и S.
1.12 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.8, в котором R5 выбран из метила, этила, пропила, изопропила, циклопропила, фторэтила, дифторэтила, трифторэтила, бутила или циклобутила.
1.13 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.8, в котором R5 представляет собой группу W или CH2W, где W представляет собой необязательно замещенное фенильное, пиридильное или изоксазольное кольцо.
1.14 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.13, в котором R6 выбран из: COR7; COOR7; CH2COR7; CH2COOR7 или C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм.
1.15 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.13, в котором R6 выбран из метила, этила, пропила, трифторэтила, гидроксиэтила или метоксиэтила.
1.16 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.13, в котором R6 выбран из: COR7; COOR7; CH2COR7; CH2COOR7, где R7 выбран из H, метила, фторметила, дифторметила, трифторметила, этила, фторэтила, дифторэтила или трифторэтила.
1.17 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.16, в котором пунктирная линия показывает вторую углерод-углеродную связь, а R3 отсутствует.
1.18 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.16, в котором присутствует R3, а необязательная вторая углерод-углеродная связь отсутствует.
1.19 Соединение в соответствии с вариантом реализации 1.18, в котором R3 выбран из атома водорода; фтора; циано-; гидрокси-; аминогруппы и C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм.
1.20 Соединение в соответствии с вариантом реализации 1.19, в котором R3 выбран из атома водорода; фтора; циано-; гидрокси-; аминогруппы и C1-4 алкильной группы, которая необязательно замещена от одного до четырех атомами фтора.
1.21 Соединение в соответствии с вариантом реализации 1.20, в котором R3 выбран из атома водорода; фтора; циано-; гидрокси-; амино-; C1-4 алкил- и C1-4 алкоксигруппы, где C1-4 алкил- и C1-4 алкоксигруппа каждая необязательно замещена от одного до четырех атомами фтора.
1.22 Соединение в соответствии с вариантом реализации 1.21, в котором R3 выбран из атома водорода, фтора; гидрокси- и метоксигруппы.
1.23 Соединение в соответствии с вариантом реализации 1.22, в котором R3 представляет собой атом водорода.
1.24 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.23, в котором R4 представляет собой атом водорода или ациклическую C1-6 углеводородную группу.
1.25 Соединение в соответствии с вариантом реализации 1.24, в котором R4 представляет собой атом водорода или ациклическую C1-3 углеводородную группу.
1.26 Соединение в соответствии с вариантом реализации 1.25, в котором R4 представляет собой атом водорода или C1-3 алкильную группу или C2-3 алкинильную группу.
1.27 Соединение в соответствии с вариантом реализации 1.26, в котором R4 выбран из атома водорода, метила, этила, этинила и 1-пропинила.
1.28 Соединение в соответствии с вариантом реализации 1.27, в котором R4 выбран из атома водорода и метила.
1.29 Соединение в соответствии с вариантом реализации 1.28, в котором R4 представляет собой метил.
1.30 Соединение в соответствии с любым одним из предшествующих вариантов реализации, в котором R7, если присутствует, выбран из атома водорода, C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и их окисленных форм; и группы W или CH2W или C1-4 углеводородной группы W, где W представляет собой необязательно замещенное 5- или 6-членное кольцо, содержащее 0, 1, 2 или 3 гетероатома, выбранных из O, N и S и их окисленных форм.
1.31 Соединение в соответствии с вариантом реализации 1.30, в котором R7 представляет собой неароматическую C1-4 углеводородную группу, необязательно замещенную одним или более атомами фтора.
1.32 Соединение в соответствии с вариантом реализации 1.30, в котором R7 представляет собой C1-4 алкильную группу.
1.33 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.32, в котором q равно 0.
1.34 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.32, в котором q равно 1.
1.35 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.32, в котором q равно 2.
1.36 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.35, в котором r равно 1.
1.37 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.35, в котором s равно 0.
1.38 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.36, в котором r равно 1 и s равно 1.
1.39 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.37, в котором r равно 1, а s равно 0.
1.40 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.39, в котором p равно 1.
1.41 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.39, в котором p равно 2.
1.42 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.41, в котором фрагмент:
выбран из групп от A до KKK, описанных ниже:
A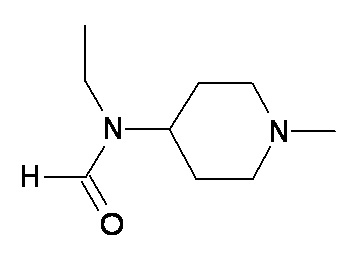
B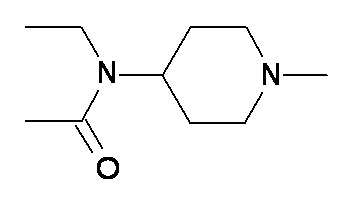
C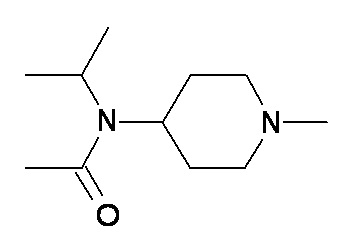
D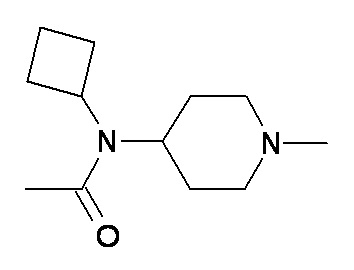
E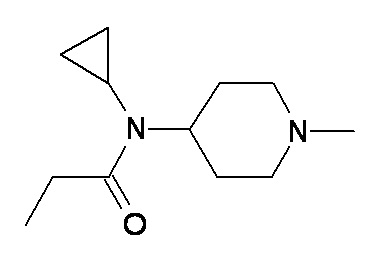
F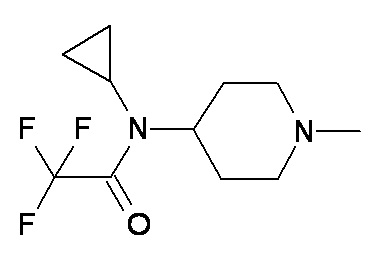 G
G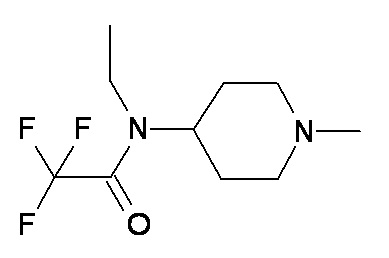
H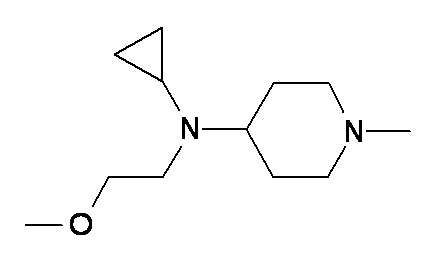
I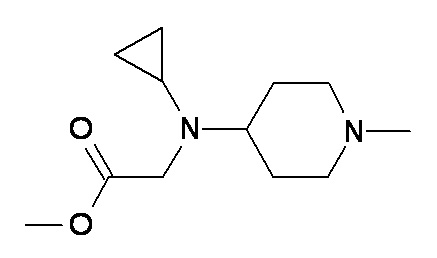 J
J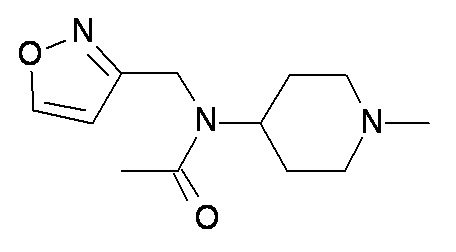
K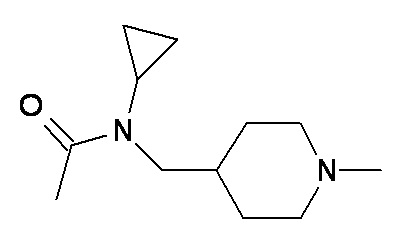 L
L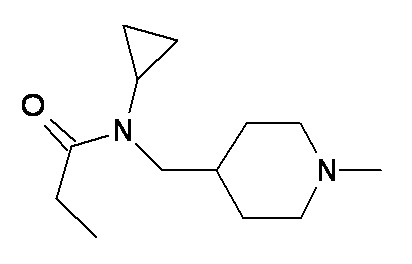 M
M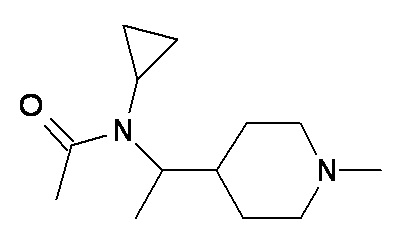
N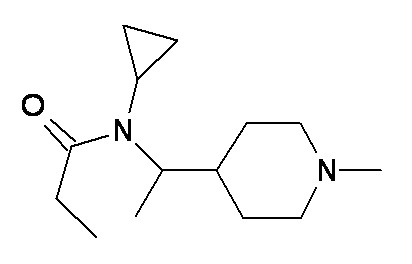 O
O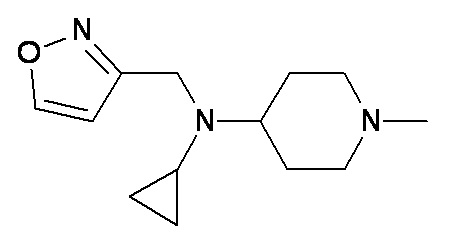 P
P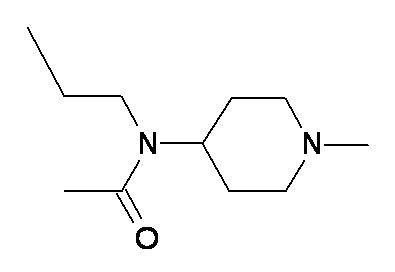
Q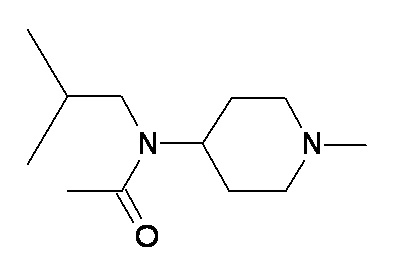
R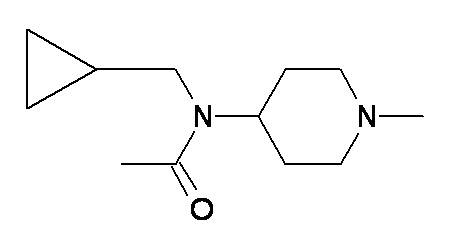
S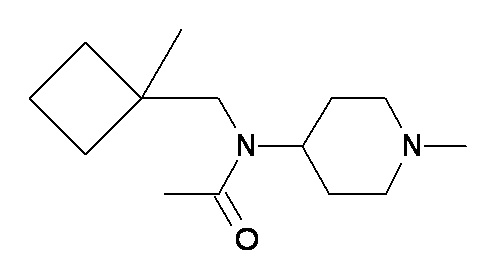
T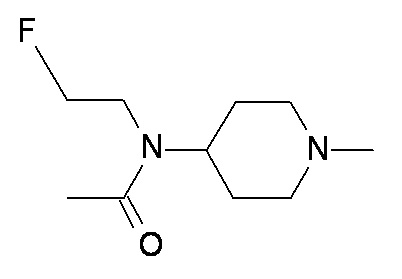
U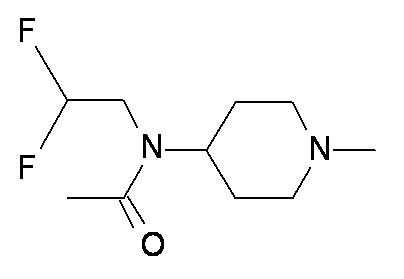 V
V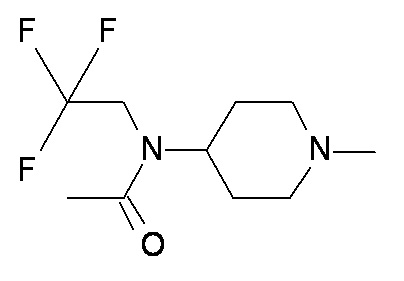
W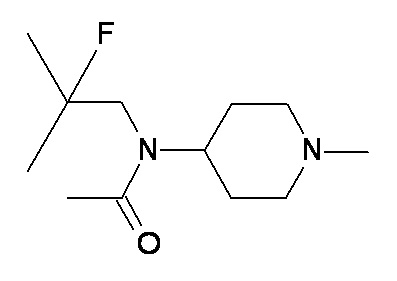
X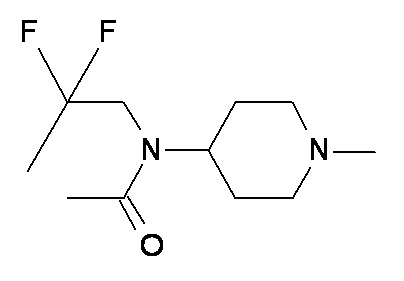
Y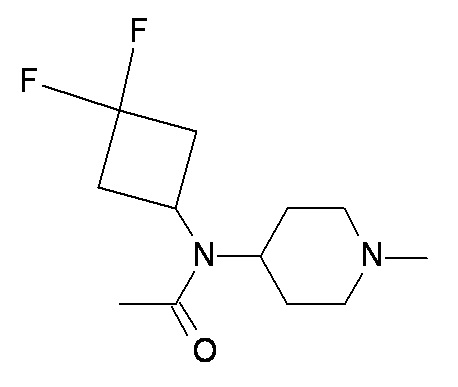
Z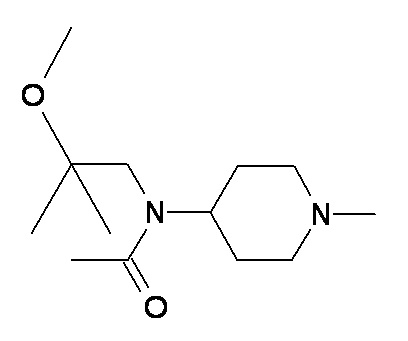
AA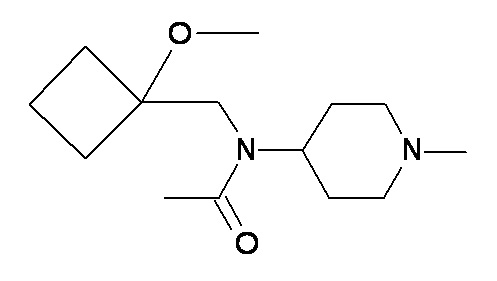
BB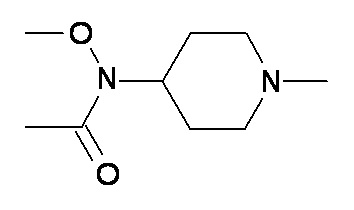
CC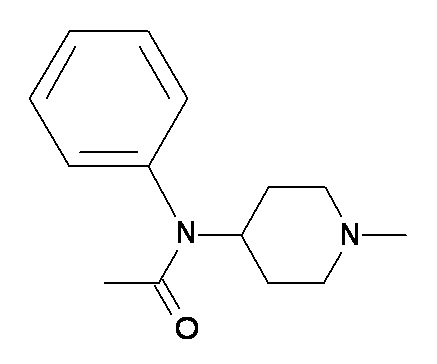 DD
DD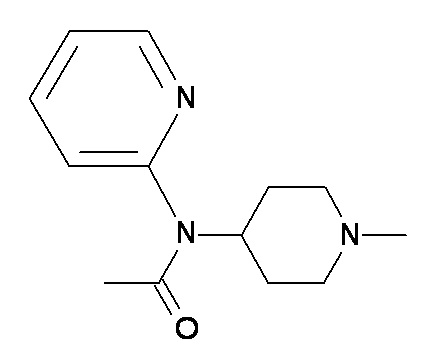
EE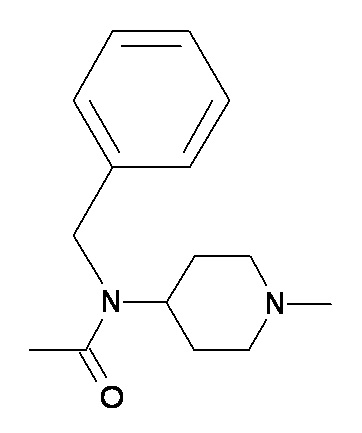
FF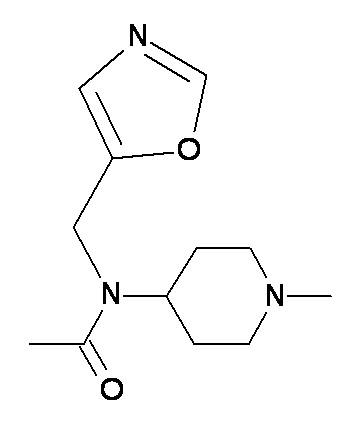
GG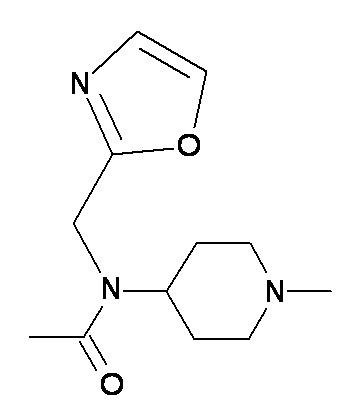
HH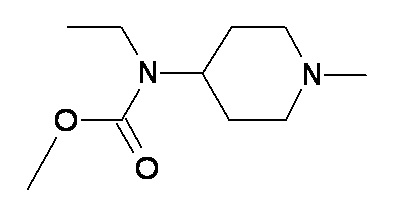
II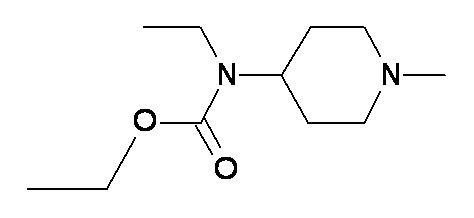
JJ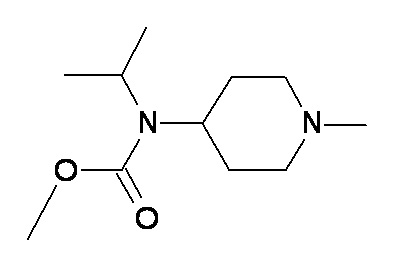
KK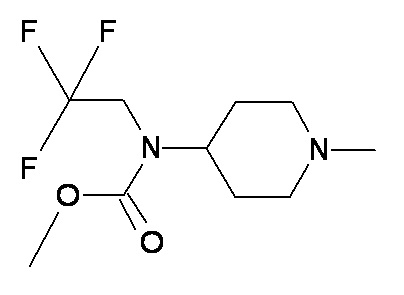
LL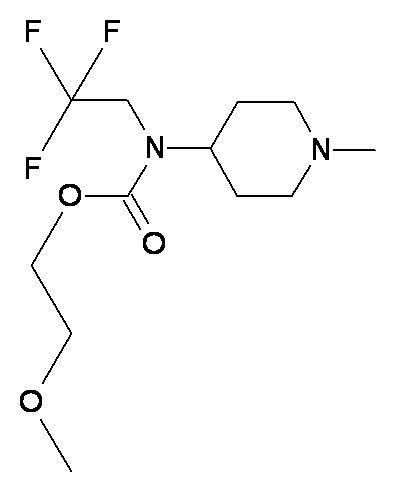
MM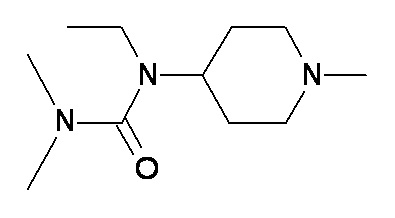 NN
NN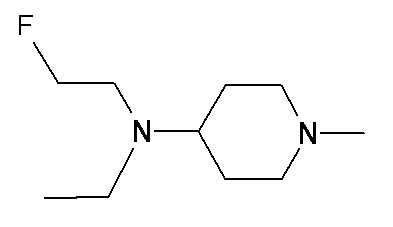
OO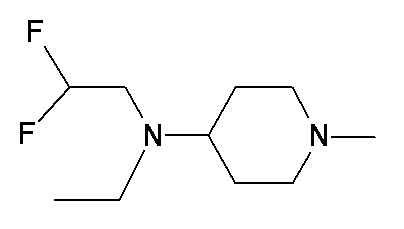
PP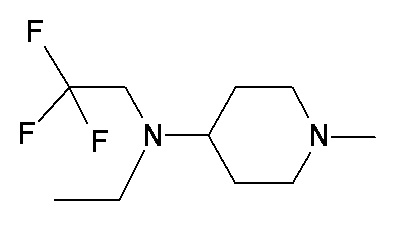
QQ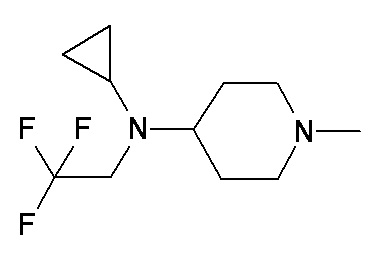
RR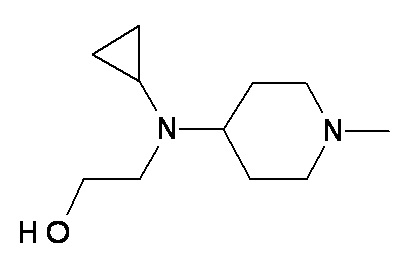
SS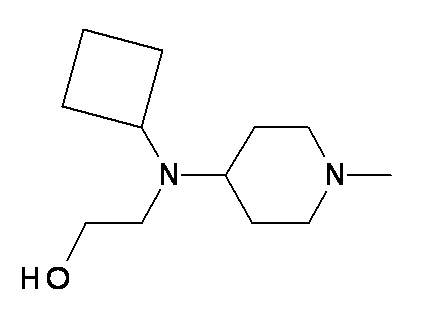 TT
TT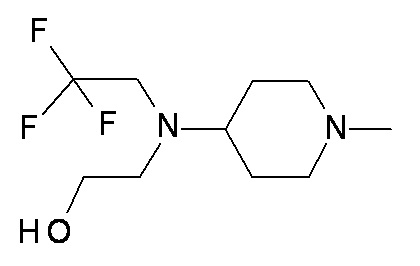 UU
UU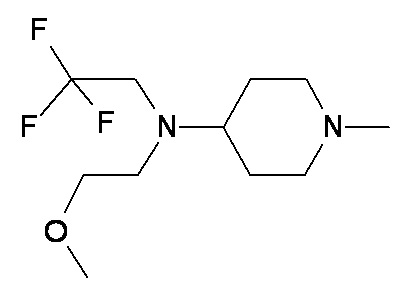 VV
VV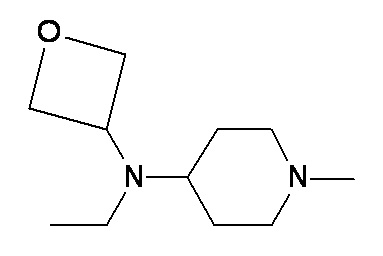
WW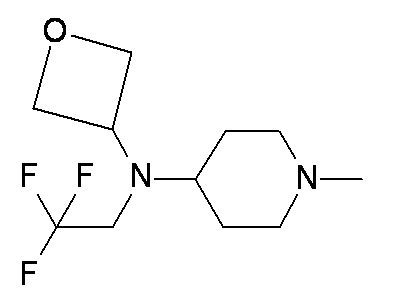
XX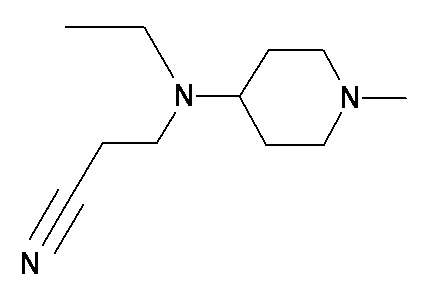
YY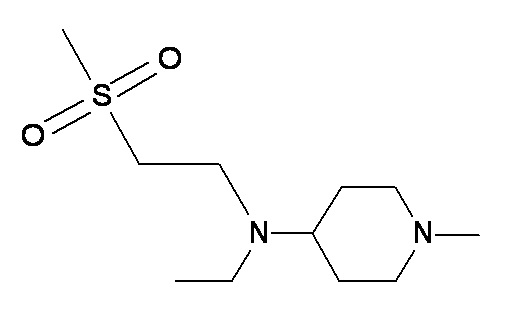 ZZ
ZZ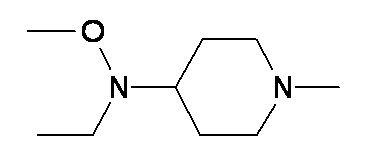
AAA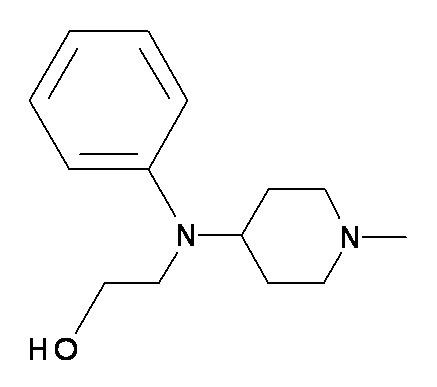 BBB
BBB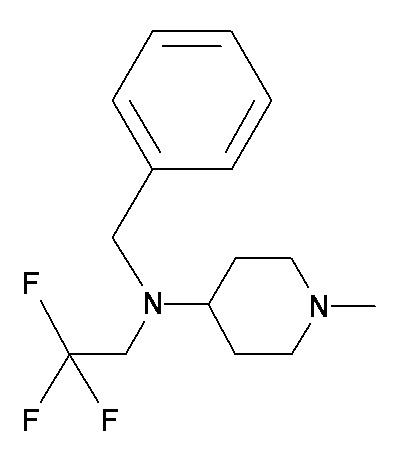 CCC
CCC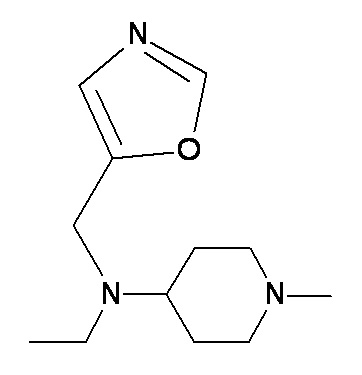
DDD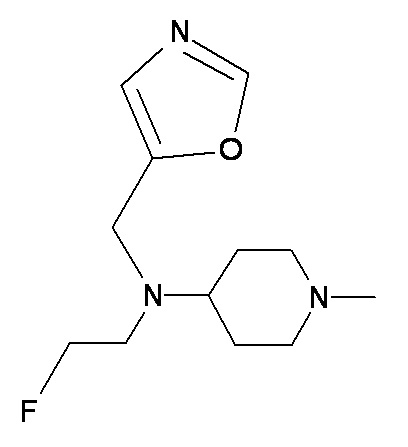
EEE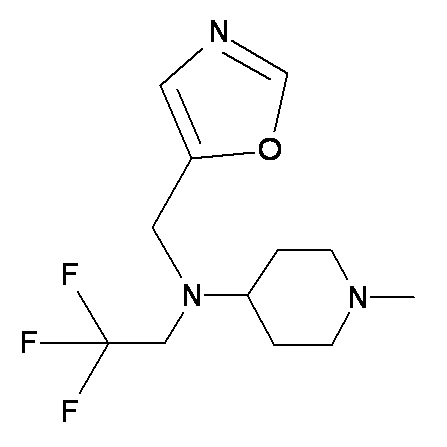
FFF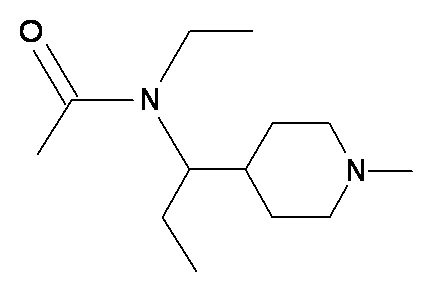
GGG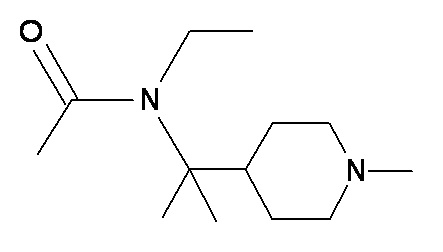
HHH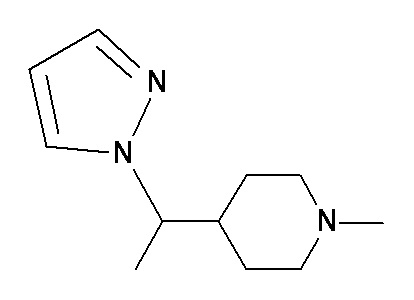
III
JJJ
KKK
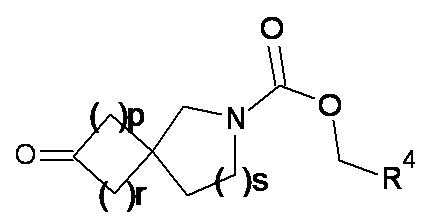
1.43 Соединение в соответствии с формулой (2) или формулой (2a):
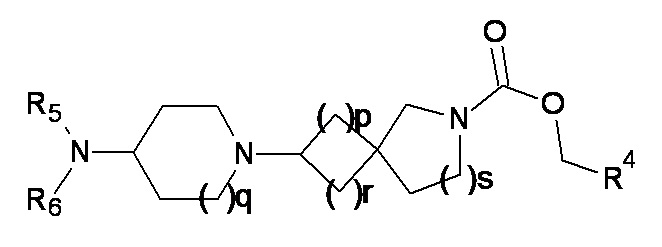 (2a)
(2a)
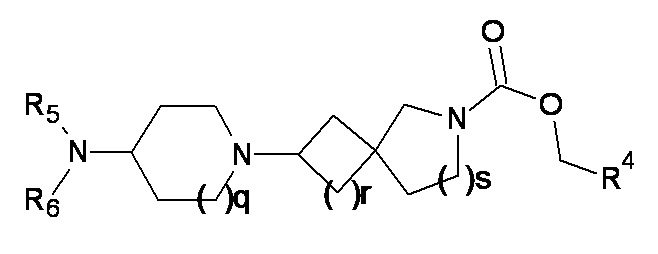 (2)
(2)
где p, q, r, s, R4, R5 и R6 являются такими, как определено в любом одном из вариантов реализации от 1.1 до 1.39.
1.44 Соединение в соответствии с формулой (3) или формулой (3a):
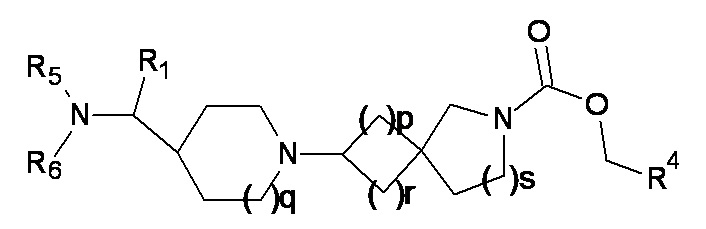 (3a)
(3a)
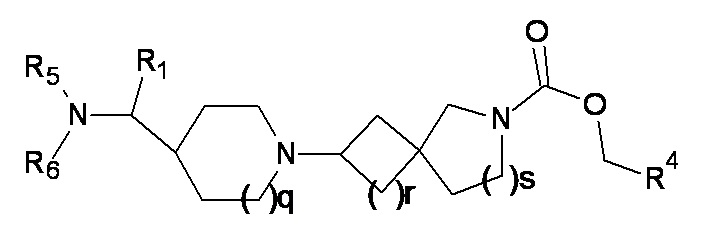 (3)
(3)
где R1 представляет собой H или метил, а p, q, r, s, R4, R5 и R6 являются такими, как определено в любом одном из вариантов реализации от 1.1 до 1.41.
1.45 Соединение в соответствии с формулой (4):
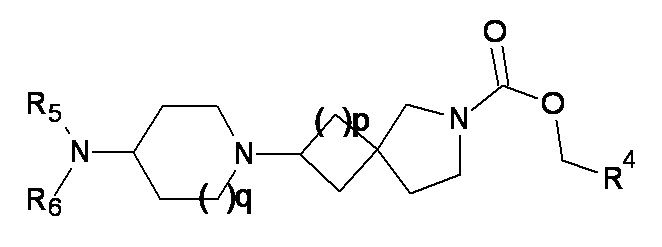 (4a)
(4a)
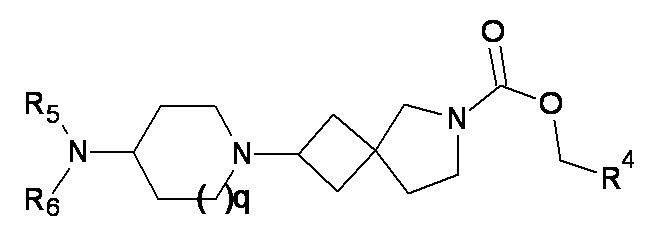 (4)
(4)
где q равно 1 или 2, а p, R4, R5 и R6 являются такими, как определено в любом одном из вариантов реализации от 1.1 до 1.32.
1.46 Соединение в соответствии с вариантом реализации 1.1, которое является таким, как определено в любом одном из примеров от 1-1 до 4-1.
1.47 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.46 с молекулярной массой менее 550.
1.48 Соединение в соответствии с вариантом реализации 1.47 с молекулярной массой менее 500.
1.49 Соединение в соответствии с вариантом реализации 1.48 с молекулярной массой менее 450.
1.50 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.49, которое представляет собой форму соли.
1.51 Соединение в соответствии с вариантом реализации 1.50, в котором соль представляет собой соль присоединения кислоты.
1.52 Соединение в соответствии с вариантом реализации 1.50 или вариантом реализации 1.51, в котором соль представляет собой фармацевтически приемлемую соль.
Определения
В данной заявке, если только не указано иное, применяются следующие определения.
Термин «лечение», в отношении применений соединений формулы (1) или формулы (1a), используется для описания любой формы воздействия, при котором соединение вводят субъекту, страдающему или имеющему риск заболеть, или потенциально имеющему риск заболеть обсуждаемой болезнью или расстройством. Таким образом, термин «лечение» охватывает как предупредительное (профилактическое) лечение, так и лечение, при котором проявляются измеряемые или выявляемые симптомы заболевания.
Термин «эффективное терапевтическое количество», как используется в данном документе (например, в отношении способа лечения заболевания или патологического состояния), относится к количеству соединения, которое является эффективным для получения требуемого терапевтического эффекта. Например, если патологическим состоянием является боль, то эффективное терапевтическое количество представляет собой количество, достаточное для обеспечения требуемого уровня облегчения боли. Требуемый уровень облегчения боли может быть, например, полным удалением боли или снижением тяжести боли.
Термин «неароматическая углеводородная группа», как во фразе «C1-10 неароматическая углеводородная группа» или «ациклическая C1-5 неароматическая углеводородная группа», относится к группе, состоящей из атомов углерода и водорода и не содержащей ароматических колец. Углеводородная группа может быть полностью насыщенной или может содержать одну или более углерод-углеродных двойных связей или углерод-углеродных тройных связей, или совокупностей двойных и тройных связей. Углеводородная группа может быть группой с прямой цепью или разветвленной цепью, или может состоять из или содержать циклическую группу. Таким образом, термин неароматический углеводород включает алкил, алкенил, алкинил, циклоалкил, циклоалкенил, циклоалкилалкил, циклоалкенилалкил и т. п.
Термины «алкил», «алкенил», «алкинил», «циклоалкил», «арил», «гетероарил» и «циклоалкенил» используются в своем традиционном смысле (например, как определено в Золотой Книге ИЮПАК), если только не указано иное.
Термин «насыщенная углеводородная группа», как во фразе «C1-4 насыщенная углеводородная группа» относится к углеводородной группе, не содержащей углерод-углеродные двойные связи или тройные связи. Поэтому насыщенная углеводородная группа может быть алкильной группой, циклоалкильной группой, циклоалкилалкильной группой, алкилциклоалкильной группой или алкилциклоалкильной группой. Примеры C1-4 насыщенных гидрокарбоновых групп включают C1-4 алкильные группы, циклопропил, циклобутил и циклопропилметил.
Термин «циклоалкил», как используется в данном документе, где допускается указанное число атомов углерода, включает как моноциклические циклоалкильные группы, такие как циклопропил, циклобутил, циклопентил, циклогексил, так и циклогептил и бициклические и трициклические группы. Бициклические циклоалкильные группы включают мостиковые системы колец, такие как бициклогептан, бициклооктан и адамантан.
В вышеприведенных определениях R1, R2, R3 и R4, где указано, один или два, но не все атомы углерода неароматической углеводородной группы необязательно могут быть замещены гетероатомом, выбранным из O, N и S и (в случае R1 и R4) их окисленных форм. Следует понимать, что когда атом углерода замещается гетероатомом, то низшие валентности гетероатомов, по сравнению с углеродом, означают, что с гетероатомами будет связано меньше атомов, чем могло быть бы связано с атомом углерода, который замещен. Таким образом, замещение атома углерода (валентность равна четырем) в группе CH2 кислородом (валентность равна двум) будет означать, что получаемая молекула будет содержать на два атома водорода меньше, а замещение атома углерода (валентность равна четырем) в группе CH2 азотом (валентность равна трем) будет означать, что получаемая молекула будет содержать на один атом водорода меньше.
Примеры замещений атомов углерода гетероатомами включают замещение атома углерода в цепи -CH2-CH2-CH2- атомом кислорода или серы с получением либо простого эфира -CH2-O-CH2-, либо тиоэфира -CH2-S-CH2-, замещение атома углерода в группе CH2-C≡C-H атомом азота с получением нитрильной (циано-) группы CH2-C≡N, замещение атома углерода в группе -CH2-CH2-CH2- на C=O с получением кетона -CH2-C(O)-CH2-, замещение атома углерода в группе -CH2-CH2-CH2- на S=O или SO2 с получением сульфоксида -CH2-S(O)-CH2- или сульфона -CH2-S(O)2-CH2-, замещение атома углерода в цепи -CH2-CH2-CH2- на C(O)NH с получением амида -CH2-CH2-C(O)-NH-, замещение атома углерода в цепи -CH2-CH2-CH2- атомом азота с получением амина -CH2-NH-CH2- и замещение атома углерода в цепи -CH2-CH2-CH2- на C(O)O с получением сложного эфира (или карбоксильной кислоты) -CH2-CH2-C(O)-O-. При каждом таком замещении должен оставаться по меньшей мере один атом углерода углеводородной группы.
Соли
Многие соединения формулы (1) или формулы (1a) могут существовать в форме солей, например в виде солей присоединения кислот, в некоторых случаях соли органических и неорганических оснований, такие как соли карбоксилаты, сульфонаты и фосфаты. Все такие соли входят в рамки объема данного изобретения, а ссылки на соединения формулы (1) или формулы (1a), включают формы солей соединений, определенных в вариантах реализации от 1.50 до 1.52.
Данные соли как правило являются солями присоединения кислот.
Соли по настоящему изобретению могут быть синтезированы из исходного соединения, которое содержит основной или кислотный фрагмент, традиционными химическими методами, такими как описаны в Pharmaceutical Salts: Properties, Selection, and Use, P. Heinrich Stahl (Editor), Camille G. Wermuth (Editor), ISBN: 3-90639-026-8, Hardcover, 388 pages, August 2002. Обычно, такие соли могут быть получены путем взаимодействия данных соединений в свободной кислотной или основной формах с соответствующим основанием или кислотой в воде или в органическом растворителе, или в их смеси; обычно используют неводную среду, такую как эфир, этилацетат, этанол, изопропанол или ацетонитрил.
Соли присоединения кислот (как определено в варианте реализации 1.120) могут образовываться из большого разнообразия кислот, как неорганических, так и органических. Примеры солей присоединения кислот, попадающие в вариант реализации 1.120 включает моно- или дисоли, образованные из кислоты, выбранной из группы, состоящей из уксусной, 2,2-дихлоруксусной, адипиновой, альгиновой, аскорбиновой (например L-аскорбиновой), L-аспарагиновой, бензолсульфоновой, бензойной, 4-ацетамидобензойной, бутановой, (+)-камфорной, камфорсульфоновой, (+)-(1S)-камфор-10-сульфоновой, каприновой, капроновой, каприловой, коричной, лимонной, цикламовой, додецилсульфоновой, этан-1,2-дисульфоновой, этансульфоновой, 2-гидроксиэтансульфоновой, муравьиной, фумаровой, галактаровой, гентизиновой, глюкогептоновой, D-глюконовой, глюкуроновой (например D-глюкуроновой), глютаминовой (например L-глютаминовой), α-оксоглютаровой, гликолиевой, гиппуровой, галогенводородных кислот (например бромистоводородной, хлористоводородной, йодистоводородной), изотионовой, молочной (например (+)-L-молочной, (±)-DL-молочной), лактобионовой, малеиновой, яблочной, (-)-L-яблочной, малоновой, (±)-DL-миндальной, метансульфоновой, нафталин-2-сульфоновой, нафталин-1,5-дисульфоновой, 1-гидрокси-2-нафтойной, никотиновой, азотной, олеиновой, оротовой, щавелевой, пальмитиновой, памоевой, фосфорной, пропионовой, пировиноградной, L-пироглютаминовой, салициловой, 4-аминосалициловой, себациновой, стеариновой, янтарной, серной, танниновой, (+)-L-виннокаменной, тиоциановой, п-толуолсульфоновой, ундециленовой и валериановых кислот, а также ацилированных аминокислот и катионообменных смол.
Если соединения формулы (1) и формулы (1a) содержат аминную функциональную группу, то они могут образовывать четвертичные аммонийные соли, например при взаимодействии с алкилирующим агентом, в соответствии со способами, хорошо известными специалисту в данной области техники. Такие четвертичные аммонийные соединения находятся в рамках формулы (1) или формулы (1a), соответственно.
Соединения по данному изобретению могут существовать в виде моно- и дисолей, в зависимости от pKa кислоты, из которой образована соль.
Соль, образованная из соединений по данному изобретению, как правило является фармацевтически приемлемой солью, и примеры фармацевтически приемлемых солей обсуждаются в Berge et al., 1977, "Pharmaceutically Acceptable Salts," J. Pharm. Sci., Vol. 66, pp. 1-19. Однако соли, которые не являются фармацевтически приемлемыми, могут также получаться в виде промежуточных форм, которые затем могут превращаться в фармацевтически приемлемые соли. Такие соли, не являющиеся фармацевтически приемлемыми, которые могут применяться, например, при очистке или разделении соединений по данному изобретению, также образуют часть изобретения.
Стереоизомеры
Стереоизомеры представляют собой изомерные молекулы, которые имеют одинаковую молекулярную формулу и последовательность связанных атомов, но они отличаются только по трехмерной ориентации атомов в пространстве. Стереоизомеры могут быть, например, геометрическими изомерами или оптическими изомерами.
Геометрические изомеры
В отношении геометрических изомеров изомеризация происходит из-за различных ориентаций атома или группы относительно двойной связи, как при цис и транс (Z и E) изомеризации относительно двойной углерод-углеродной связи, или цис- и транс-изомерах относительно амидной связи, или син- и анти-изомеризации относительно связи, когда имеется ограниченное вращение, цис- и транс изомеризации относительно кольца, такого как циклоалкановое кольцо.
Соответственно, в другом варианте реализации изобретения (вариант реализации 1.121) в изобретении предложен геометрический изомер соединения в соответствии с любым одним из вариантов реализации от 1.1 до 1.52.
Оптические изомеры
Если соединения формулы содержат один или более хиральных центров и могут существовать в форме двух или более оптических изомеров, то ссылки на эти соединения включают все их оптические изомерные формы (например, эпимеры и диастереоизомеры), либо в виде отдельных оптических изомеров, либо в виде смесей (например, рацемических смесей), или двух или более оптических изомеров, если в контексте не указано противоположное.
Соответственно, в другом варианте реализации изобретения (вариант реализации 1.132) в изобретении предложено соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.121, которое содержит хиральный центр.
Оптические изомеры могут быть охарактеризованы и идентифицированы по их оптической активности (т. е. как + и - изомеры или d и l изомеры) или они могут быть охарактеризованы в категориях их абсолютной стереохимии, используя номенклатуру «R и S», разработанную Cahn, Ingold and Prelog, см. Advanced Organic Chemistry by Jerry March, 4th Edition, John Wiley & Sons, New York, 1992, pages 109-114, а также см. Cahn, Ingold & Prelog, Angew. Chem. Int. Ed. Engl., 1966, 5, 385-415. Оптические изомеры могут быть разделены несколькими методиками, включающими хиральную хроматографию (хроматография на хиральной подложке) и такие методики хорошо известны специалисту в данной области техники. В качестве альтернативы хиральной хроматографии оптические изомеры могут быть разделены путем образования диастереоизомерных солей с хиральными кислотами, такими как (+)-виннокаменная кислота, (-)-пироглютаминовая кислота, (-)-дитолуол-L-виннокаменная кислота, (+)-миндальная кислота, (-)-яблочная кислота и (-)-камфорсульфоновая кислота, разделение стереоизомеров происходит путем предпочтительной кристаллизации и последующей диссоциацией солей с образованием отдельных энантиомеров свободного основания.
Если соединения по данному изобретению существуют в виде двух или более оптических изомерных форм, то один энантиомер в паре энантиомеров может проявлять преимущества над другим энантиомером, например, в категориях биологической активности. Таким образом, при некоторых обстоятельствах может требоваться использование в качестве лекарственного средства только одного из пары энантиомеров или только одного из множества диастереоизомеров.
Соответственно, в другом варианте реализации изобретения (варианте реализации 1.133) в изобретении предложена композиция, содержащая соединение в соответствии с вариантом реализации 1.132, имеющим один или более хиральных центров, причем по меньшей мере 55 % (например по меньшей мере 60 %, 65 %, 70 %, 75 %, 80 %, 85 %, 90 % или 95 %) соединения по варианту реализации 1.108 представлено в виде одного оптического изомера (например, энантиомера или диастереоизомера).
В одном варианте общем реализации (вариант реализации 1.134) 99 % или более (например, практически все) от общего количества соединения (или соединения для применения) по варианту реализации 1.132 представлено в виде одного оптического изомера.
Например, в одном варианте реализации (вариант реализации 1.135) соединение представлено в виде единственного энантиомера.
В другом варианте реализации (вариант реализации 1.136) соединение представлено в виде единственного диастереоизомера.
В изобретении также предложены смеси оптических изомеров, которые могут быть рацемическими или нерацемическими. Таким образом, в изобретении предложено:
1.137 Соединение в соответствии с вариантом реализации 1.132, которое представляет собой форму рацемической смеси оптических изомеров.
1.138 Соединение в соответствии с вариантом реализации 1.132, которое представляет собой форму нерацемической смеси оптических изомеров.
Изотопы
Соединения по данному изобретению, определенные в любом одном из вариантов реализации изобретения от 1.1 до 1.138, могут содержать одну или более изотопных замен, а ссылка на конкретный элемент включает перечень всех изотопов данного элемента. Например, ссылка на атом водорода включает перечень 1H, 2H (D) и 3H (T). Сходным образом, ссылка на атом углерода и кислорода включает перечень из, соответственно, 12C, 13C и 14C, и 16O и 18O.
Аналогичным образом, ссылка на конкретную функциональную группу также включает перечень изотопных вариаций, если только в контексте не указано противоположное. Например, ссылка на алкильную группу, такую как этильная группа, также охватывает вариации, в которых один или более атомов водорода в группе находятся в форме изотопа дейтерия или трития, например как в этильной группе, в которой пять атомов водорода находятся в изотопной форме дейтерия (пердейтероэтильная группа).
Изотопы могут быть радиоактивными или нерадиоактивными. В одном варианте реализации изобретения (вариант реализации 1.142) соединение по любому одному из вариантов реализации изобретения от 1.1 до 1.140 не содержит радиоактивные изотопы. Такие соединения являются предпочтительными для терапевтического применения. Однако в другом варианте реализации изобретения (вариант реализации 1.143) соединение по любому одному из вариантов реализации изобретения от 1.1 до 1.140 может содержать один или более радиоактивных изотопов. Соединения, содержащие такие радиоизотопы, могут использоваться в диагностическом контексте.
Сольваты
Соединения формулы (1) или формулы (1a), определенные в любом одном из вариантов реализации от 1.1 до 1.143, могут образовывать сольваты. Предпочтительные сольваты представляют собой сольваты образованные путем включения в структуру твердого тела (например кристаллическую структуру) соединений по данному изобретению из молекул нетоксического фармацевтически приемлемого растворителя (называемого ниже сольватирующий растворитель). Примеры таких растворителей включают воду, спирты (такие как этанол, изопропанол и бутанол) и диметилсульфоксид. Сольваты можно получать путем перекристаллизации соединений по данному изобретению с растворителем или смесью растворителей, содержащих сольватирующий растворитель. Образование сольвата в любом данном случае можно определять путем проведения анализа кристаллов соединения с использованием хорошо известных стандартных методик, таких как термогравиметрический анализ (ТГА), дифференциальная сканирующая калориметрия (ДСК) и рентгеновская кристаллография. Сольваты могут быть стехиометрическими или нестехиометрическими сольватами. В частности, предпочтительные сольваты представляют собой гидраты, а примеры гидратов включают гемигидраты, моногидраты и дигидраты.
Соответственно, в дополнительных вариантах реализации 1.153 и 1.154 в изобретение предложено:
1.153 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.143 в форме сольвата.
1.154 Соединение в соответствии с вариантом реализации 1.153, в котором сольват представляет собой гидрат.
Для получения более подробного обсуждения сольватов и способов, используемых для получения и определения их параметров, см. Bryn et al., Solid-State Chemistry of Drugs, Second Edition, published by SSCI, Inc of West Lafayette, IN, USA, 1999, ISBN 0-967-06710-3.
В альтернативном варианте вместо существования в виде гидрата соединение по данному изобретению может быть безводным. Поэтому в другом варианте реализации изобретения (вариант реализации 1.155) в изобретении предложено соединение, определенное в соответствии с любым одним из вариантов реализации от 1.1 до 1.143, в безводной форме (например в безводной кристаллической форме).
Кристаллические и аморфные формы
Соединения по любому одному из вариантов реализации от 1.1 до 1.155 могут существовать в кристаллическом или некристаллическом (например аморфном) состоянии. Факт существования соединения в кристаллическом состоянии можно легко определять стандартными методиками, такими как порошковая рентгеновская дифракция (XRPD). Кристаллы и их кристаллические структуры можно охарактеризовать с использованием ряда методик, включающих рентгеновскую кристаллографию на одном кристалле, порошковую рентгеновскую дифракцию (XRPD), дифференциальную сканирующую калориметрию (ДСК) и инфракрасную спектроскопию, например инфракрасную спектроскопию с преобразованием Фурье (FTIR). Поведение кристаллов в условиях изменяющейся влажности можно анализировать путем гравиметрических исследований с сорбцией паров и также методом XRPD. Определение кристаллической структуры соединения можно выполнять рентгеновской кристаллографией, которая может проводиться в соответствии с традиционными методами, такими как описаны в данном документе и как описаны в Fundamentals of Crystallography, C. Giacovazzo, H. L. Monaco, D. Viterbo, F. Scordari, G. Gilli, G. Zanotti and M. Catti, (International Union of Crystallography/ Oxford University Press, 1992 ISBN 0-19-855578-4 (p/b), 0-19-85579-2 (h/b)). В этой методике используется анализ и интерпретация рентгеновской дифракции на одном кристалле. В аморфном твердом состоянии не существует трехмерная структура, которая обычно существует в кристаллической форме, и положения молекул относительно друг друга в аморфной форме являются по существу случайными, см. например Hancock et al. J. Pharm. Sci. (1997), 86, 1).
Соответственно, в дополнительных вариантах реализации в изобретение предложено:
1.160 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.155 в кристаллической форме.
1.161 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.155, которое является:
(a) от 50 % до 100 % кристаллическим, а более конкретно является по меньшей мере на 50 % кристаллическим или по меньшей мере на 60 % кристаллическим, или по меньшей мере на 70 % кристаллическим, или по меньшей мере на 80 % кристаллическим, или по меньшей мере на 90 % кристаллическим, или по меньшей мере на 95 % кристаллическим, или по меньшей мере на 98 % кристаллическим, или по меньшей мере на 99 % кристаллическим, или по меньшей мере на 99,5 % кристаллическим, или по меньшей мере на 99,9 % кристаллическим, например на 100 % кристаллическим.
1.162 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.155, которое находится в аморфной форме.
Пролекарства
Соединения формулы (1) или формулы (1a), определенные в любом одном из вариантов реализации от 1.1 до 1.162, могут быть представлены в форме пролекарства. «Пролекарствами» обозначают, например, любое соединение, которое превращается in vivo в биологически активное соединение формулы (1) или формулы (1a), соответственно, определенное в любом одном из вариантов реализации от 1.1 до 1.162.
Например, некоторые пролекарства представляют собой сложные эфиры активного соединения (например физиологически приемлемый метаболически нестабильный сложный эфир). Во время метаболизма сложноэфирная группа (-C(=O)OR) расщепляется с образованием активного лекарственного средства. Такие сложные эфиры могут получать путем этерификации, например, любой гидроксильной группы, присутствующей в исходном соединении с предварительной защитой, если необходимо, любой другой реакционноспособной группы, присутствующей в исходном соединении, с последующим снятием защиты, если требуется.
Некоторые пролекарства также активируются ферментативно с образованием активного соединения или соединения, которое при последующей химической реакции, дает активное соединение (например как в ADEPT, GDEPT, LIDEPT и т. п.). Например, пролекарство может быть производным сахаров или другого гликозидного конъюгата, или может быть производным сложного эфира аминокислоты.
Соответственно, в другом варианте реализации изобретения (вариант реализации 1.170) в изобретении предложено пролекарство, определенное в соответствии с любым одним из вариантов реализации от 1.1 до 1.170, причем соединение содержит функциональную группу, которая может превращаться в физиологических условиях с образованием гидроксильной группы или аминогруппы.
Комплексы и клатраты
Формулой (1) или формулой (1a) в вариантах реализации от 1.1 до 1.170 также охватываются комплексы (например комплексы включения или клатраты с такими соединениями, как циклодекстрины или комплексы с ионами металлов) соединений по вариантам реализации от 1.1 до 1.170.
Соответственно, в другом варианте реализации изобретения (вариант реализации 1.180) в изобретении предложено соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.170 в форме комплекса или клатрата.
Биологическая активность и виды терапевтического применения
Соединения по данному изобретению обладают активностью в качестве агонистов мускариновых рецепторов M1 и/или M4. Мускариновую активность соединений можно определять с использованием анализа фосфо-ERK1/2, описанного ниже в примере A.
Значительное преимущество соединений по данному изобретению заключается в том, что они являются высокоселективными к рецептору M1 и/или рецептору M4 относительно подтипов рецепторов M2 и M3. Соединения по данному изобретению не являются агонистами подтипов рецепторов M2 и M3. Например, когда соединения по данному изобретению обычно имеют значения pEC50 по меньшей мере 6 (предпочтительно по меньшей мере 6,5) и значения Emax более 80 (предпочтительно более 95) по отношению к рецептору M1 в функциональном анализе, описанном в примере A, то они могут иметь значения pEC50 менее 5 и значения Emax менее 20 %, при исследовании по сравнению с подтипами M2 и M3 в функциональном анализе примера A.
Соответственно, в вариантах реализации 2.1 и 2.9 в изобретение предложено:
2.1 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.180 для применения в медицине.
2.2 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.180 для применения в качестве агониста мускаринового рецептора M1 и/или M4.
2.3 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.180, которое является агонистом мускаринового рецептора M1, имеющим pEC50 в диапазоне от 6,0 до 8,0 и Emax по меньшей мере 90 по отношению к рецептору M1 в анализе примера A в данном документе или в практически аналогичном ему анализе.
2.4 Соединение в соответствии с вариантом реализации 2.3, которое является агонистом мускаринового рецептора M1, имеющим pEC50 в диапазоне от 6,5 до 7,5.
2.5 Соединение в соответствии с вариантом реализации 2.3 или вариантом реализации 2.4, имеющее Emax по меньшей мере 95 по отношению к рецептору M1.
2.6 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.180, которое является агонистом мускаринового рецептора M4, имеющим pEC50 в диапазоне от 6,0 до 8,5 и Emax по меньшей мере 90 по отношению к рецептору M4 в анализе примера A в данном документе или в практически аналогичном ему анализе.
2.7 Соединение в соответствии с вариантом реализации 2.6, которое является агонистом мускаринового рецептора M4, имеющим pEC50 в диапазоне от 6,5 до 8,5.
2.8 Соединение в соответствии с вариантом реализации 2.6 или вариантом реализации 2.7, имеющее Emax по меньшей мере 95 по отношению к рецептору M4.
2.9 Соединение в соответствии с любым одним из вариантов реализации от 2.3 до 2.8, которое является селективным для рецептора M1 и/или M4 по сравнению с мускариновыми рецепторами M2 и M3.
2.10 Соединение в соответствии с вариантом реализации 2.9, которое является селективным для рецептора M1 по сравнению с мускариновыми рецепторами M2 и M3.
2.11 Соединение в соответствии с вариантом реализации 2.9, которое является селективным для рецептора M4 по сравнению с мускариновыми рецепторами M2 и M3.
2.12 Соединение в соответствии с любым одним из вариантов реализации от 2.3 до 2.5, которое является селективным для рецептора M1 по сравнению с мускариновыми рецепторами M2, M3 и M4.
2.13 Соединение в соответствии с любым одним из вариантов реализации от 2.6 до 2.8, которое является селективным для рецептора M4 по сравнению с мускариновыми рецепторами M1, M2 и M3.
2.14 Соединение в соответствии с любым одним из вариантов реализации от 2.3 до 2.8, которое является селективным для рецепторов M1 и M4 по сравнению с мускариновыми рецепторами M2 и M3.
2.15 Соединение в соответствии с любым одним из вариантов реализации от 2.3 до 2.14, которое имеет pEC50 менее 5 и Emax менее 50 по отношению к подтипам мускариновых рецепторов M2 и M3.
2.16 Соединение в соответствии вариантом реализации 2.15, которое имеет pEC50 менее 4,5 и/или Emax менее 30 по отношению к подтипам мускариновых рецепторов M2 и M3.
2.17 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.180 и вариантами реализации от 2.3 до 2.16 для применения при лечении заболевания или патологического состояния, опосредованного мускариновым рецептором M1.
Вследствие своей агонистической активности для мускаринового рецептора M1 и/или M4, соединения по данному изобретению можно использовать для лечения болезни Альцгеймера, шизофрении и других психотических расстройств, когнитивных расстройств и других заболеваний, опосредованных мускариновым рецептором M1 и/или M4, а также их можно использовать для лечения различных типов боли.
Соответственно, в вариантах реализации от 2.18 до 2.34 в изобретение предложено:
2.18 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.180 для применения при лечении когнитивного расстройства или психотического расстройства.
2.19 Соединение в соответствии с вариантом реализации 2.18, в котором когнитивное расстройство или психотическое расстройство содержит, возникает из или связано с патологическим состоянием, выбранным из когнитивного нарушения, умеренного когнитивного нарушения, лобно-височной деменции, сосудистой деменции, деменции с тельцами Леви, пресенильной деменции, сенильной деменции, атаксии Фридрейха, синдрома Дауна, хореи Хатингтона, гиперкинезии, мании, синдрома Туретта, болезни Альцгеймера, прогрессирующего надъядерного паралича, нарушения когнитивных функций, включая нарушения внимания, ориентации, способности к обучению, памяти (т. е. нарушения памяти, амнезия, амнестические нарушения, синдром транзиторной глобальной амнезии и возрастное нарушение памяти) и функционирования языка; когнитивного нарушения в результате инсульта, болезни Хатингтона, болезни Пика, связанной с ВИЧ деменцией и других состояний деменции, таких как мультиинфарктная деменция, алкогольная деменция, связанная с гипотироидозом деменция и деменция, связанная с другими дегенеративными нарушениями, такими как церебеллярная атрофия и боковой амниотрофический склероз; других острых или подострых патологических состояний, которые могут вызывать снижение когнитивных способностей, такие как травма в результате делириума или депрессии (состояния псевдодеменции), травмы головы, возрастного снижения когнитивных способностей, инсульта, нейродегенерации, вызванных лекарственными средствами состояний, нейротоксических агентов, возрастного когнитивного нарушения, связанного с аутизмом когнитивного нарушения, синдрома Дауна, связанного с психозом когнитивного расстройства и пост-электросудорожной терапии, связанной с когнитивными расстройствами; когнитивных расстройств, возникших вследствие злоупотребления лекарственными средствами или их отмены, включая никотин, каннабис, амфетамин, кокаин, синдрома дефицита внимания и гиперактивности (ADHD) и дискинетических нарушений, таких как болезнь Паркинсона, вызванного нейролептиками паркинсонизма и поздней дискинезии, шизофрении, шизофрениформных заболеваний, психотической депрессии, мании, паранойи, галлюциногенных и бредовых расстройств, расстройств личности, обсессивно-компульсивных расстройств, шизотипичных расстройств, бредовых расстройств, психоза вследствие злоупотребления лекарственными средствами или их отмены, биполярных расстройств, эпилепсии и шизоаффективного расстройства.
2.20 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.180 для применения при лечении болезни Альцгеймера.
2.21 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.180 для применения при лечении шизофрении.
2.22 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.180 для применения при лечении болезни Альцгеймера и/или деменции с тельцами Леви.
2.23 Способ лечения когнитивного расстройства у субъекта (например пациента-млекопитающего, такого как человек, например нуждающийся в таком лечении человек), причем способ включает введение терапевтически эффективной дозы соединения в соответствии с любым одним из вариантов реализации от 1.1 до 1.180.
2.24 Способ в соответствии с вариантом реализации 2.20, в котором когнитивное расстройство содержит, возникает из или связано с патологическим состоянием, определенным в варианте реализации 2.19.
2.25 Способ в соответствии с вариантом реализации 2.24, в котором когнитивное расстройство возникает из или связано с болезнью Альцгеймера.
2.26 Способ в соответствии с вариантом реализации 2.24, в котором когнитивное расстройство представляет собой шизофрению.
2.27 Применение соединения в соответствии с любым одним из вариантов реализации от 1.1 до 1.180 для производства лекарственного средства для лечения когнитивного расстройства.
2.28 Применение в соответствии с вариантом реализации 2.27, в котором когнитивное расстройство содержит, возникает из или связано с патологическим состоянием, определенным в варианте реализации 2.11.
2.29 Применение в соответствии с вариантом реализации 2.28, в котором когнитивное расстройство возникает из или связано с болезнью Альцгеймера.
2.30 Применение в соответствии с вариантом реализации 2.28, в котором когнитивное расстройство представляет собой шизофрению.
2.31 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.180 для лечения или уменьшения тяжести острой, нейропатической или воспалительной боли, артрита, мигрени, кластерных головных болей, невралгии тройничного нерва, герпетической невралгии, генерализированных невралгий, висцеральной боли, остеоартритной боли, постгерпетической невралгии, диабетической нейропатии, радикулитной боли, невралгии седалищного нерва, боли спины, боли головы или шеи, тяжелой или не поддающейся лечению боли, ноцицептивной боли, прорывной боли, постхирургической боли или боли при раковом заболевании.
2.32 Способ лечения или уменьшения тяжести острой, хронической, нейропатической или воспалительной боли, артрита, мигрени, кластерных головных болей, невралгии тройничного нерва, герпетической невралгии, генерализированных невралгий, висцеральной боли, остеоартритной боли, постгерпетической невралгии, диабетической нейропатии, радикулитной боли, невралгии седалищного нерва, боли спины, боли головы или шеи, тяжелой или не поддающейся лечению боли, ноцицептивной боли, прорывной боли, постхирургической боли или боли при раковом заболевании, причем способ включает введение терапевтически эффективной дозы соединения в соответствии с любым одним из вариантов реализации от 1.1 до 1.180.
2.33 Соединение в соответствии с любым одним из вариантов реализации от 1.1 до 1.180 для лечения периферических нарушений, таких как снижение внутриглазного давления при глаукоме, и лечения симптомов сухости глаз и сухости рта, включая синдром Шегрена.
2.34 Способ лечения периферических нарушений, таких как снижение внутриглазного давления при глаукоме, и лечения симптомов сухости глаз и сухости рта, включая синдром Шегрена, причем способ включает введение терапевтически эффективной дозы соединения в соответствии с любым одним из вариантов реализации от 1.1 до 1.180.
2.35 Применение в соответствии с любым одним из вариантов реализации от 1.1 до 1.180 для производства лекарственного средства для лечения или уменьшения тяжести острой, хронической, нейропатической или воспалительной боли, артрита, мигрени, кластерных головных болей, невралгии тройничного нерва, герпетической невралгии, генерализированных невралгий, висцеральной боли, остеоартритной боли, постгерпетической невралгии, диабетической нейропатии, радикулитной боли, невралгии седалищного нерва, боли спины, боли головы или шеи, тяжелой или не поддающейся лечению боли, ноцицептивной боли, прорывной боли, постхирургической боли или боли при раковом заболевании, или для лечения периферических нарушений, таких как снижение внутриглазного давления при глаукоме, и лечения симптомов сухости глаз и сухости рта, включая синдром Шегрена.
2.36 Применение соединения в соответствии с любым одним из вариантов реализации от 1.1 до 1.180 для лечения зависимости.
2.37 Применение соединения в соответствии с любым одним из вариантов реализации от 1.1 до 1.180 для лечения таких расстройств движения, как болезнь Паркинсона, ADHD, болезнь Хантингтона, синдром Туретта и других синдромов, связанных с дофаминэргической дисфункцией, лежащей в основе патогенетического фактора, стимулирующего заболевание.
Способы получения соединений формулы (1) и формулы (1a)
Соединения формулы (1) и формулы (1a) можно получать в соответствии с методами синтеза, хорошо известными специалисту в данной области техники и описанными в данном документе.
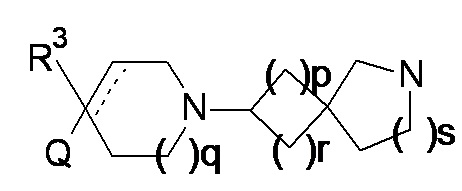
Соответственно, в другом варианте реализации изобретения (вариант реализации 3.1) в изобретении предложен процесс получения соединения, определенный в любом одном из вариантов реализации от 1.1 до 1.180, причем процесс включает:
(A) взаимодействие соединения формулы (10)
 (10)
(10)
с соединением формулы (11) или (11a):
 (11a) или
(11a) или 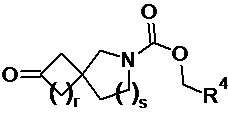 (11)
(11)
в условиях восстановительного аминирования; где p, q, r, s, R3, R4 и Q являются такими, как определено в любом одном из вариантов реализации от 1.1 до 1.180; или
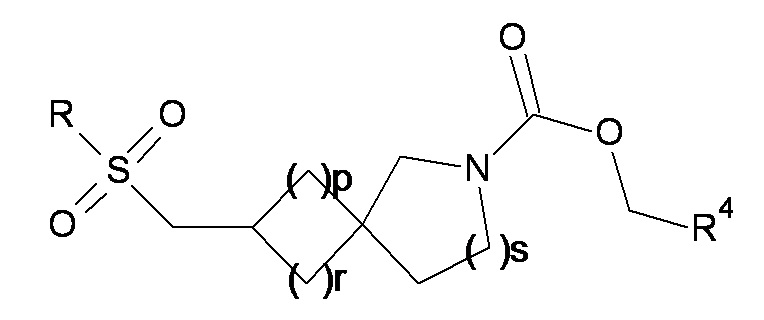
(B) взаимодействие соединения формулы (12) или (12a):
 (12a) или
(12a) или 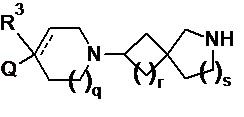 (12) с соединением формулы Cl-C(=O)-CH2-R4 в присутствии основания; или
(12) с соединением формулы Cl-C(=O)-CH2-R4 в присутствии основания; или
(C) взаимодействие соединения формулы (10)
 (10)
(10)
с соединением формулы (13) или (13a):
 (13a) или
(13a) или 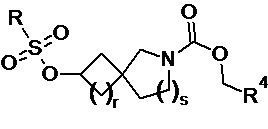 (13)
(13)
в условиях нуклеофильного замещения; где p, q, r, s, R3, R4 и Q являются такими, как определено в любом одном из вариантов реализации от 1.1 до 1.180; и необязательно:
(D) превращение одного соединения формулы (1) или формулы (1a) в другое соединение формулы (1) или формулы (1a), соответственно.
В варианте процесса (A) пиперидиновый гетероцикл (10) в условиях восстановительного аминирования взаимодействует с замещенным кетоном (11) или (11a). Реакция восстановительного аминирования, как правило, выполняется при комнатной температуре с использованием борогидридного восстанавливающего агента, такого как триацетоксиборогидрид натрия, в таком растворителе, как дихлорметан или дихлорэтан, содержащем уксусную кислоту.
В варианте процесса (C) пиперидиновый гетероцикл (10) реагирует с сульфоновым сложным эфиром (13 или 13a, R = метил, трифторметил или 4-метилфенил) в реакции нуклеофильного замещения, которая как правило выполняется при умеренном нагревании (например до температуры от около 40 °C до около 70° C) либо в чистом виде без растворителя, либо в подходящем растворителе, таком как тетрагидрофуран, ацетонитрил или диметилацетамид.
Промежуточные соединения формулы (12) и (12a) можно получать посредством ряда реакций, показанных ниже на схеме 1 и схеме 1a, соответственно.
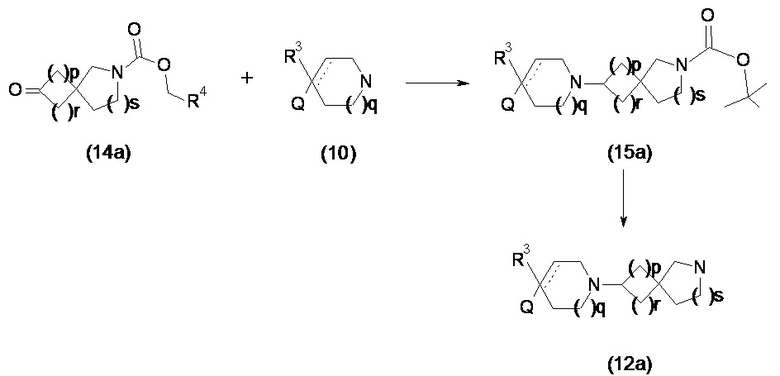
Схема 1a
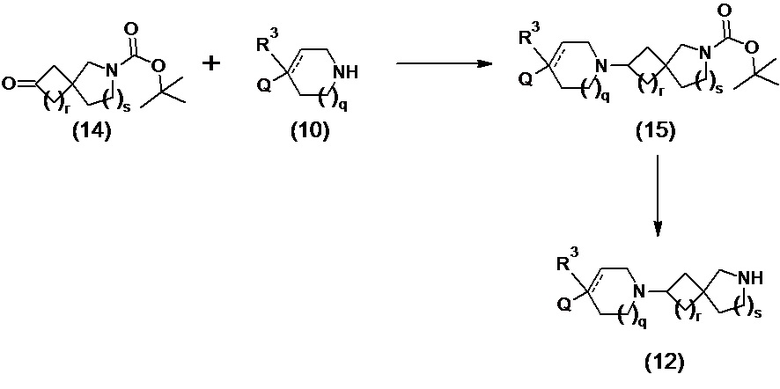
Схема 1
В реакции схемы 1 или схемы 1a пиперидиновый гетероцикл (10) взаимодействует в условиях восстановительного аминирования, соответственно, с Boc-защищенным спирокетоном (14) или (14a). Реакция восстановительного аминирования, как правило, выполняется при умеренном нагревании (например до температуры от около 40 °C до около 70 °C) в присутствии либо цианоборогидрида натрия в комбинации с хлоридом цинка, либо триацетоксиборогидрида натрия в комбинации с изопропоксидом титана, в таком растворителе как дихлорметан или дихлорэтан, содержащем уксусную кислоту, с получением промежуточного пиперидинового соединения (15) или (15a), с которого затем снимается защита путем удаления группы Boc обработкой кислотой (например трифторуксусной кислотой в дихлорметане) с получением, соответственно, соединения (12) или (12a).
Соединения формулы (12) и (12a) также можно получать путем последовательности реакций, показанных ниже на схеме 2 и схеме 2a, соответственно.
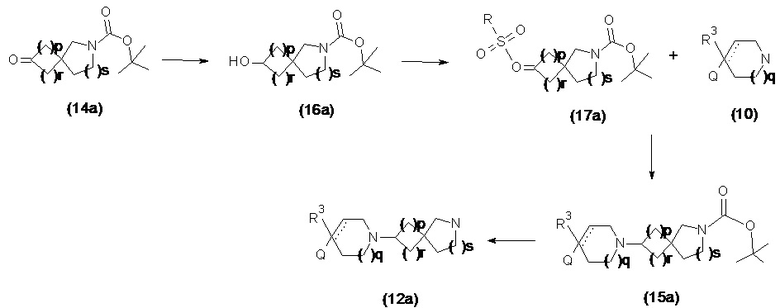
Схема 2a
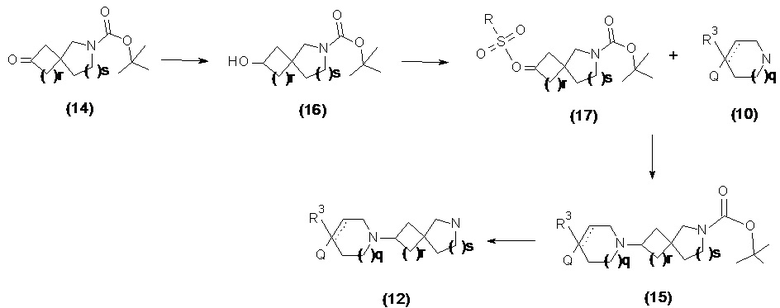
Схема 2
В реакции схемы 2 или схемы 2a Boc-защищенный спирокетон (14) или (14a) восстанавливается, соответственно, до спирта (16) или (16a) с помощью использования борогидрида натрия в метаноле. Затем спирт (16) или (16a) активируется в виде сульфонового сложного эфира (17 или 17a, R = метил, трифторметил или 4-метилфенил) с использованием соответствующего сульфонилхлорида в дихлорметане в присутствии третичного амина, такого как триэтиламин или N,N-диизопропилэтиламин. Сульфоновый сложный эфир (17) или (17a) взаимодействует с пиперидиновым гетероциклом (10) в реакции нуклеофильного замещения, которая как правило выполняется при умеренном нагревании (например до температуры от около 40 °C до около 70° C) либо в чистом виде без растворителя, либо в подходящем растворителе, таком как тетрагидрофуран, ацетонитрил или диметилацетамид с получением, соответственно, соединения (15) или (15a), с которого затем снимается защита путем удаления группы Boc путем обработки кислотой (например, трифторуксусной кислотой в дихлорметане) с получением, соответственно, соединения (12) или (12a).
После образования одно соединение формулы (1) или формулы (1a), или его защищенное производное, можно превращать в другое соединение формулы (1) или формулы (1a), соответственно, известными специалисту в данной области техники методами. Примеры процедур синтеза для превращения одной функциональной группы в другую функциональную группу изложены в стандартных текстах, таких как Advanced Organic Chemistry и Organic Syntheses (см. ссылки выше) или Fiesers’ Reagents for Organic Synthesis, Volumes 1-17, John Wiley, edited by Mary Fieser (ISBN: 0-471-58283-2). Примеры таких трансформаций включают образование амидной связи, образование мочевины, образование карбамата, реакции алкилирования, реакцию N-арилирования и реакции конденсации по C-C связям.
Во многих описанных выше реакциях может потребоваться защищать одну или более групп для предотвращения прохождения реакции в нежелательном положении молекулы. Примеры защитных групп и методы защиты и снятия защиты функциональных групп можно найти в Protective Groups in Organic Synthesis (T. Greene and P. Wuts; 3rd Edition; John Wiley and Sons, 1999).
Соединения, полученные представленными выше методами, можно выделять и очищать любым из разнообразных методов, хорошо известных специалистам в данной области техники, и примеры таких методов включают перекристаллизацию и хроматографические методики, такие как колоночная хроматография (например флэш-хроматография) и ВЭЖХ.
Фармацевтические составы
Если для активного соединения возможно самостоятельное введение, то предпочтительно предоставлять его в виде фармацевтической композиции (например препарата).
Соответственно, в другом варианте реализации (варианте реализации 4.1) изобретения предложена фармацевтическая композиция, содержащая по меньшей мере одно соединение формулы (1) или формулы (1a), определенное в любом одном из вариантов реализации от 1.1 до 1.180, вместе с по меньшей мере одним фармацевтически приемлемым вспомогательным веществом.
В одном варианте реализации (варианте реализации 4.2) композиция представляет собой таблетированную композицию.
В другом варианте реализации (варианте реализации 4.3) композиция представляет собой капсулированную композицию.
Фармацевтически приемлемое (-ые) вспомогательное (-ые) вещество (-а) можно выбирать из, например, носителей (например твердый, жидкий или пастообразный носитель), адъювантов, разбавителей (например твердых разбавителей, таких как наполнители или объемообразующие агенты; и жидких разбавителей, таких как растворители и сорастворители), способствующих гранулированию агентов, связывающих веществ, агентов для повышения сыпучести, формирующих покрытия агентов, контролирующих высвобождение агентов, разрыхлителей, буферизирующих агентов, корректирующих тоничность агентов, загустителей, ароматизаторов, подсластителей, пигментов, пластификаторов, маскирующих вкус агентов, стабилизаторов или других вспомогательных веществ, традиционно используемых в фармацевтических композициях.
Термин «фармацевтически приемлемый», как используется в данном документе, означает соединения, материалы, композиции и/или лекарственные формы, которые, в пределах здравого медицинского суждения, пригодны для использования в контакте с тканями субъекта (например субъекта-человека) без чрезмерной токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, соразмерного с разумным соотношением польза/риск. Каждое вспомогательное вещество также должно быть «приемлемым» в смысле совместимости с другими ингредиентами композиции.
Фармацевтические композиции, содержащие соединения формулы (1) или формулы (1a) могут быть приготовлены в соответствии с известными методиками, см. например Remington’s Pharmaceutical Sciences, Mack Publishing Company, Easton, PA, USA.
Фармацевтические композиции могут быть в любой форме, пригодной для перорального, парентерально, местного, интраназального, внутрибронхиального, сублингвального, офтальмологического, внутриушного, ректального, интравагинального или трансдермального введения.
Фармацевтические лекарственные формы, пригодные для перорального введения, включают таблетки (покрытые или непокрытые), капсулы (с твердой или мягкой оболочкой), каплеты, пилюли, леденцы, сиропы, растворы, порошки, гранулы, эликсиры и суспензии, сублингвальные таблетки, вафли или пластыри, такие как буккальные пластыри.
Таблетированные композиции могут содержать стандартную дозировку активного вещества вместе с инертным разбавителем или носителем, таким как сахар или сахарный спирт, например лактозу, сахарозу, сорбит или маннит; и/или разбавитель несахарного происхождения, такой как карбонат натрия, фосфат кальция, карбонат кальция или целлюлоза, или ее производное, такое как микрокристаллическая целлюлоза (МКЦ), метилцеллюлоза, этилцеллюлоза, гидроксипропилметилцеллюллоза и крахмалы, такие как кукурузный крахмал. Таблетки также могут содержать такие стандартные ингредиенты, как связующие агенты и способствующие гранулированию агенты, как поливинилпирролидон, разрыхлители (например набухающие перекрестно сшитые полимеры, такие как перекрестно сшитая карбоксиметилцеллюлоза), смазывающие агенты (например стеараты), консерванты (например парабены), антиоксиданты (например BHT), буферизирующие агенты (например фосфатные или цитратные буферы) и выделяющие газ агенты, такие как смеси цитратов/бикарбонатов. Такие вспомогательные вещества хорошо известны и не нуждаются в более детальном рассмотрении в данном документе.
Таблетки могут быть предназначены для высвобождения лекарственного средства при приведении в контакт с желудочным соком (таблетки немедленного высвобождения) или для высвобождения управляемым способом (таблетки с контролируемым высвобождением) в течение длительного периода времени или в конкретном отделе ЖК-тракта.
Фармацевтические композиции как правило содержат от приблизительно 1 % (мас./мас.) до приблизительно 95 %, предпочтительно % (мас./мас.) активного ингредиента и от 99 % (мас./мас.) до 5 % (мас./мас.) фармацевтически приемлемого вспомогательного вещества (например, как определено выше) или комбинации таких вспомогательных веществ. Предпочтительно композиции содержат от приблизительно 20 % (мас./мас.) до приблизительно 90 % (мас./мас.) активного ингредиента и от 80 % (мас./мас.) до 10 % фармацевтического вспомогательного вещества или комбинации вспомогательных веществ. Фармацевтические композиции содержат от приблизительно 1 % до приблизительно 95 %, предпочтительно от приблизительно 20 % до приблизительно 90 % активного ингредиента. Фармацевтические композиции в соответствии с изобретением могут быть, например, в стандартной дозированной форме, такой как форма ампул, флаконов, суппозиториев, предварительно наполненных шприцов, дражже, порошков, таблеток или капсул.
Таблетки и капсулы могут содержать, например, 0-20 % разрыхлителей, 0-5 % смазывающих веществ, 0-5 % агентов для повышения текучести и/или 0-99 % (мас./мас.) наполнителей или объемообразующих агентов (в зависимости от дозы лекарственного средства). Они также могут содержать 0-10 % (мас./мас.) полимерных связующих, 0-5 % (мас./мас.) антиоксидантов, 0-5 % (мас./мас.) пигментов. Таблетки с замедленным высвобождением могут обычно дополнительно содержать 0-99 % (мас./мас.) управляющих высвобождением (например вызывающих замедление) полимеров (в зависимости от дозы). Пленочные покрытия таблетки или капсулы, как правило, содержат 0-10 % (мас./мас.) полимеров, 0-3 % (мас./мас.) пигментов и/или 0-2 % (мас./мас.) пластификаторов.
Парентеральные композиции, как правило, содержат 0-20 % (мас./мас.) буферных растворов, 0-50 % (мас./мас.) сорастворителей и/или 0-99 % (мас./мас.) воды для инъекций (в зависимости от дозы и при лиофильном высушивании). Составы для внутримышечного введения могут также содержать 0-99 % (мас./мас.) масел.
Фармацевтические составы могут предлагаться пациенту в виде «упаковок для пациентов», содержащих полный курс лечения в одной упаковке, обычно в контурной ячейковой упаковке.
Соединения формулы (1) или формулы (1a) обычно будут представлены в стандартной лекарственной форме и, по существу, обычно будут содержать достаточное количество соединения для обеспечения требуемого уровня биологической активности. Например, состав может содержать от 1 нанограмма до 2 грамм активного ингредиента, например, от 1 нанограмма до 2 миллиграмм активного ингредиента. В пределах этих диапазонов конкретные поддиапазоны соединения составляют от 0,1 миллиграмм до 2 грамм активного ингредиента (чаще от 10 миллиграмм до 1 грамма, например от 50 миллиграмм до 500 миллиграмм) или от 1 микрограмма до 20 миллиграмм (например от 1 микрограмма до 10 миллиграмм, например, от 0,1 миллиграмм до 2 миллиграмм активного ингредиента).
Для пероральных композиций стандартная лекарственная форма содержит от 1 миллиграмма до 2 грамм, чаще от 10 миллиграмм до 1 грамма, например от 50 миллиграмм до 1 грамма, например от 100 миллиграм до 1 грамма активного соединения.
Активное соединение будет вводиться нуждающегося в этом пациенту (например, пациенту-человеку или животному) в количестве, достаточном для достижения требуемого терапевтического эффекта (эффективное количество). Точные количества вводимого соединения, может определять наблюдающий врач в соответствии со стандартными процедурами.
ПРИМЕРЫ
Далее изобретение будет проиллюстрировано, но не ограничиваться, ссылкой на конкретные варианты реализации изобретения, описанные в следующих примерах.
ПРИМЕРЫ ОТ 1-1 ДО 3-3
Были получены соединения примеров от 1-1 до 3-3, показанные ниже в табл. 1. Свойства по результатам анализа ЯМР и ЖХМС, а также методы, использованные для их получения, изложены в табл. 3.
Таблица 1
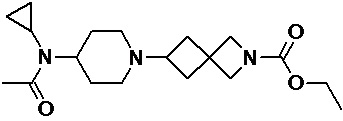
Пример 1-1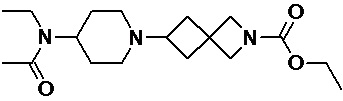
Пример 1-2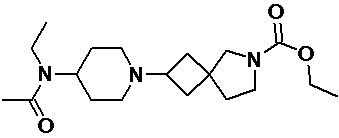
Пример 2-1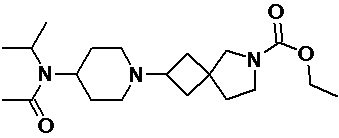
Пример 2-2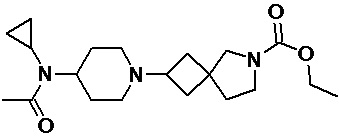
Пример 2-3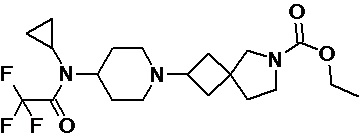
Пример 2-4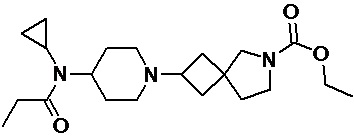
Пример 2-5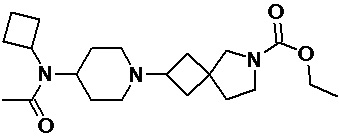
Пример 2-6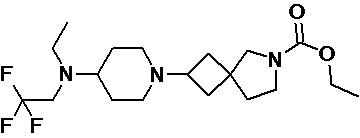
Пример 2-7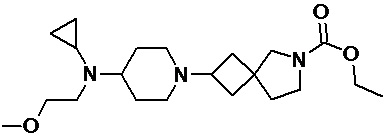
Пример 2-8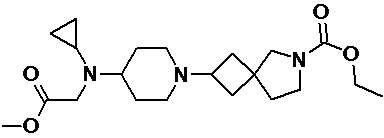
Пример 2-9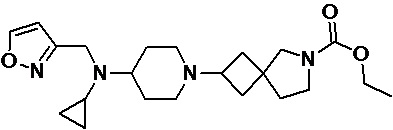
Пример 2-10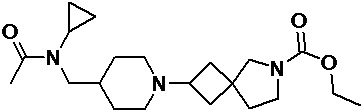
Пример 2-11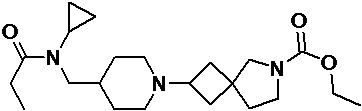
Пример 2-12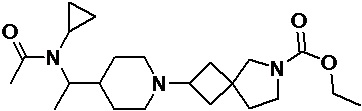
Пример 2-13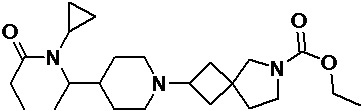
Пример 2-14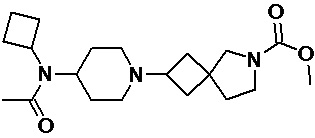
Пример 2-15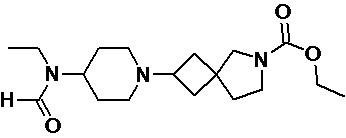
Пример 2-16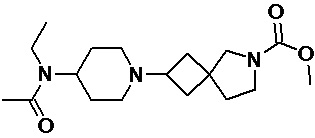
Пример 2-17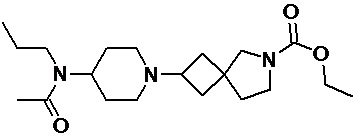
Пример 2-18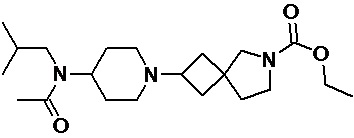
Пример 2-19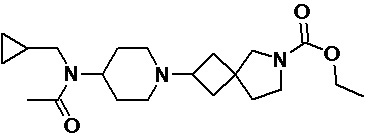
Пример 2-20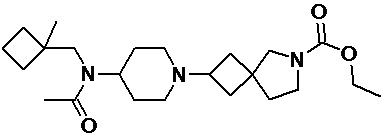
Пример 2-21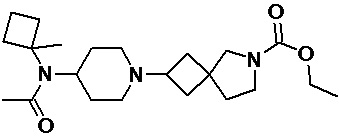
Пример 2-22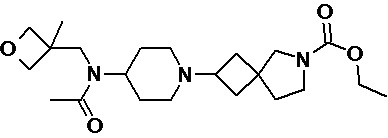
Пример 2-23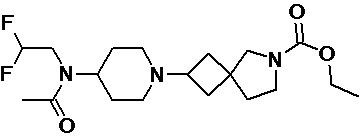
Пример 2-24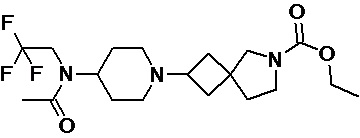
Пример 2-25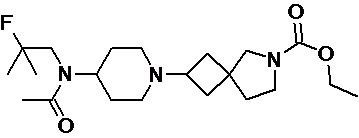
Пример 2-26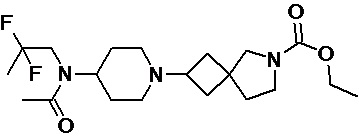
Пример 2-27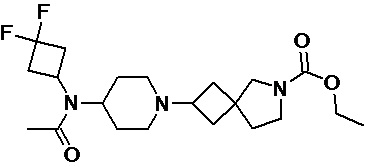
Пример 2-28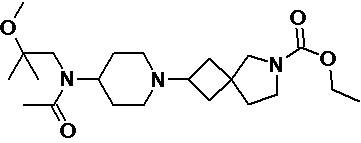
Пример 2-29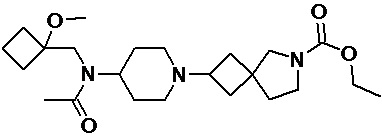
Пример 2-30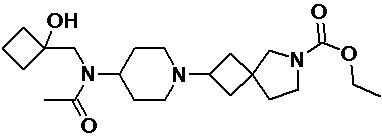
Пример 2-31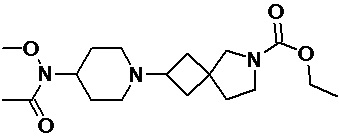
Пример 2-32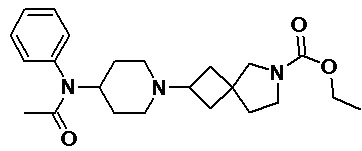
Пример 2-33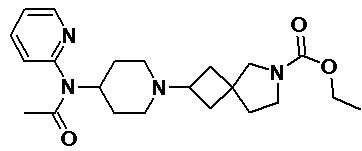
Пример 2-34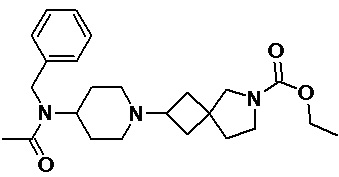
Пример 2-35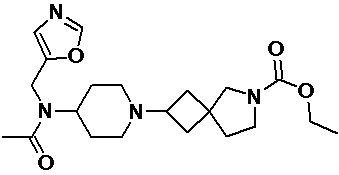
Пример 2-36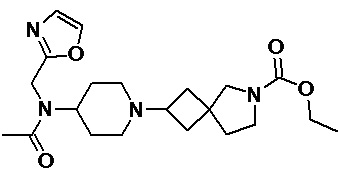
Пример 2-37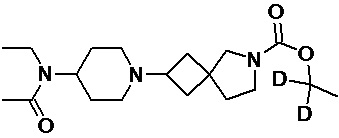
Пример 2-38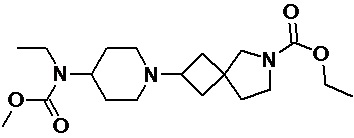
Пример 2-39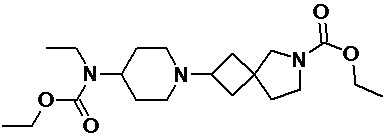
Пример 2-40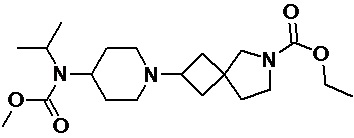
Пример 2-41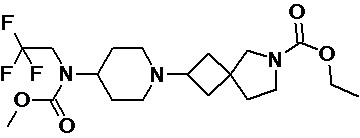
Пример 2-42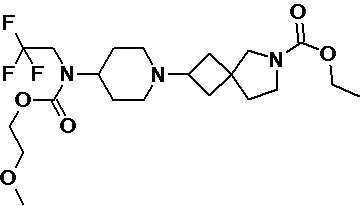
Пример 2-43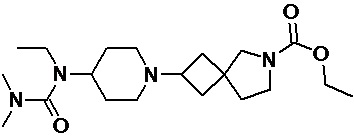
Пример 2-44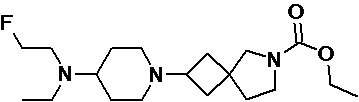
Пример 2-45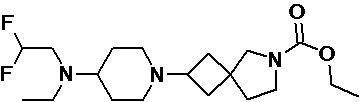
Пример 2-46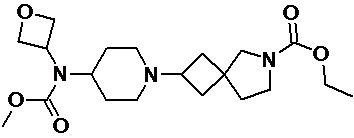
Пример 2-47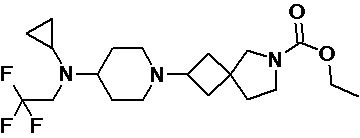
Пример 2-48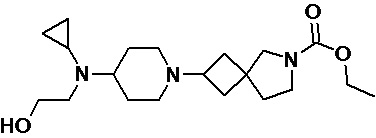
Пример 2-49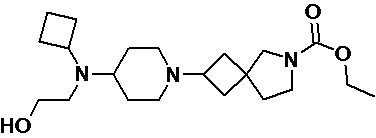
Пример 2-50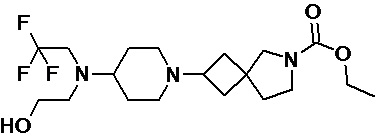
Пример 2-51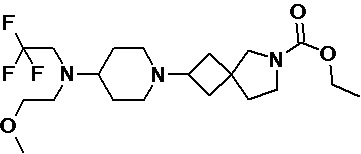
Пример 2-52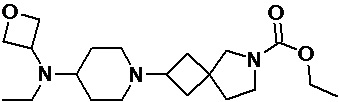
Пример 2-53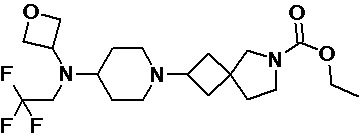
Пример 2-54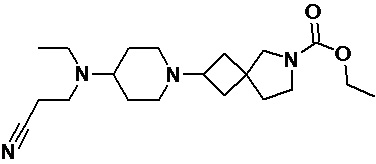
Пример 2-55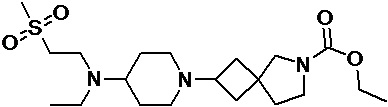
Пример 2-56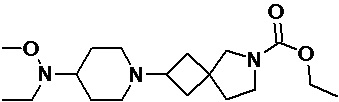
Пример 2-57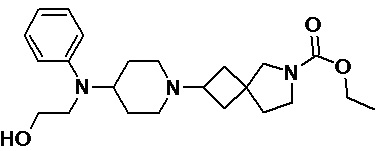
Пример 2-58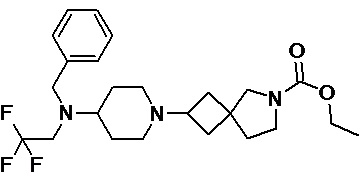
Пример 2-59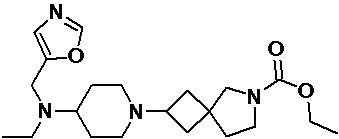
Пример 2-60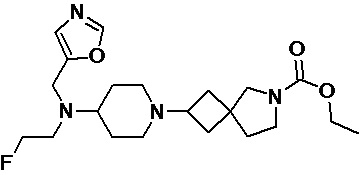
Пример 2-61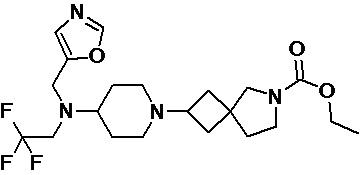
Пример 2-62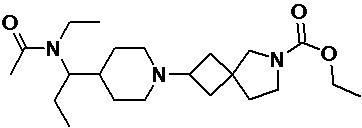
Пример 2-63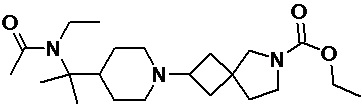
Пример 2-64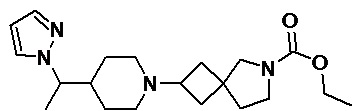
Пример 2-65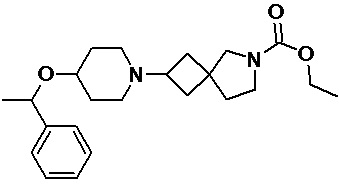
Пример 2-66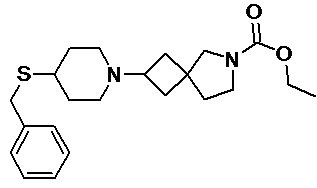
Пример 2-67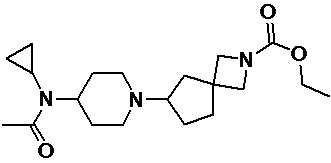
Пример 3-1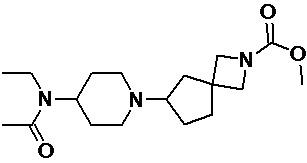
Пример 3-2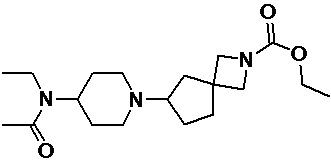
Пример 3-3
Общие процедуры
Если не описаны пути препаративного получения, то соответствующее промежуточное соединение имеется в продаже. Коммерческие реагенты использовались без дополнительной очистки. Комнатная температура (кт) относится к приблизительно 20-27 °C. 1H-ЯМР спектры записывали при 400 МГц либо на приборе Bruker, либо Jeol. Значения химических сдвигов выражаются в долях на миллион (м.д.), т. е. (δ:)-значениях. Следующие сокращения использованы для обозначения мультиплетности сигналов ЯМР: s=синглет, br=широкий, d=дублет, t=триплет, q=квартет, quint=квинтет, td=триплет из дублетов, tt= триплет из триплетов, qd=квартет из дублетов, ddd=дублет из дублетов, ddt=дублет из триплетов, m=мультиплет. Константы взаимодействия перечислены как значения J, измеренные в Гц. Результаты ЯМР и масс-спектроскопии корректировали для учета фоновых пиков. Хроматография относится к колоночной хроматографии, выполненной с использованием силикагеля с размером 60-120 меш и проведенной под давлением азота (флэш-хроматография). ТСХ для отслеживания прохождения реакций относится к анализу ТСХ с использованием указанной подвижной фазы и силикагеля F254 (Merck), как неподвижной фазы. Реакции под воздействием микроволнового излучения выполняли в микроволновых реакторах Biotage Initiator или CEM Discover.
Эксперименты ЖХМС, как правило, выполняли с использованием условий электрораспыления, как указано для каждого компонента в следующих условиях:
Метод C ЖХМС
Приборы: ЖХ Agilent 1260 Infinity с диодно-матричным детектором, МС Agilent 6120B с одним квадруполем с источником API-ES; колонка: Phenomenex Gemini-NX C-18, 3 микрона, 2,0 x 30 мм; градиент [время (мин)/растворитель B в A ( %)]: Метод: 0,00/5, 2,00/95, 2,50/95, 2,60/5, 3,00/5; растворители: растворитель A = 2,5 л H2O + 2,5 мл (28 % NH3 в H2O); растворитель B = 2,5 л MeCN + 129 мл H2O + 2,7 мл (28 % NH3 в H2O); инжектируемый объем 0,5 мкл; УФ-детекция от 190 до 400 нм; температура колонки 40 °C; скорость потока 1,5 мл/мин.
Методы D и Е ЖХМС
Приборы: HP 1100 с ДМД G1315A, Micromass ZQ; колонка: Waters X-Bridge C-18, 2,5 микрон, 2,1 x 20 мм или Phenomenex Gemini-NX C-18, 3 микрона, 2,0 x 30 мм; градиент [время (мин)/растворитель D в C ( %)]: Метод D: 0,00/2, 0,10/2, 2,50/95, 3,50/95 или Метод E: 0,00/2, 0,10/2, 8,40/95, 10,00/95; растворители: растворитель C = 2,5 л H2O + 2,5 мл 28 % раствор аммиака в H2O; растворитель D = 2,5 л MeCN + 135 мл H2O + 2,5 мл 28 % раствор аммиака в H2O); инжектируемый объем 1 мкл; УФ-детекция от 230 до 400 нм; масс-детекция от 130 до 800 а.е.м. (электрораспыление +ve и –ve); температура колонки 45 °C; скорость потока 1,5 мл/мин.
Метод F ЖХМС
Приборы: Waters Acquity H Class, детектор на фотодиодной матрице SQ; колонка: BEH C18, 1,7 микрон, 2,1 x 50 мм; градиент [время (мин)/растворитель B в A ( %)]: 0,00/5, 0,40/5, 0,8/35, 1,20/55, 2,50/100, 3,30/100 4,00/5; растворители: растворитель A = 5 мM ацетат аммония и 0,1 % муравьиная кислота в H2O; растворитель B = 0,1 % муравьиная кислота в MeCN; инжектируемый объем 2 мкл; УФ-детекция от 200 до 400 нм; масс-детекция от 100 до 1200 а.е.м. (электрораспыление +ve); колонка при комнатной температуре; скорость потока 0,5 мл/мин.
Метод H ЖХМС
Приборы: Waters 2695, детектор на фотодиодной матрице ZQ-2000; колонка: X-Bridge C18, 5 микрон, 150 x 4,6 мм; градиент [время (мин)/растворитель B в A ( %)]: 0,00/100, 7,00/50, 9,00/0, 11,00/0, 11,01/100, 12,00/100; растворители: растворитель A = 0,1 % аммиак в H2O; растворитель B = 0,1 % аммиак в MeCN; инжектируемый объем 10 мкл; УФ-детекция от 200 до 400 нм; масс-детекция от 60 до 1000 а.е.м. (электрораспыление +ve); колонка при комнатной температуре; скорость потока 1,0 мл/мин.
Метод I ЖХМС
Приборы: Waters 2695, детектор на фотодиодной матрице ZQ-2000; колонка: X-Bridge C18, 3,5 микрон, 150 x 4,6 мм; градиент [время (мин)/растворитель B в A ( %)]: 0,00/5, 5,00/90, 5,80/95, 10/95; растворители: растворитель A = 0,1 % аммиак в H2O; растворитель B = 0,1 % аммиак в MeCN; инжектируемый объем 10 мкл; УФ-детекция от 200 до 400 нм; масс-детекция от 60 до 1000 а.е.м. (электрораспыление +ve); колонка при комнатной температуре; скорость потока 1,0 мл/мин.
Метод K ЖХМС
Приборы: Waters 2695, детектор на фотодиодной матрице ZQ-2000; колонка: X-Bridge C18, 3,5 микрон, 50 x 4,6 мм; градиент [время (мин)/растворитель B в A ( %)]: 0,01/0, 0,20/0; 5,00/90, 5,80/95, 7,20/95, 7,21/100, 10,00/100; растворители: растворитель A = 0,1 % аммиак в H2O; растворитель B = 0,1 % аммиак в MeCN; инжектируемый объем 10 мкл; УФ-детекция от 200 до 400 нм; масс-детекция от 60 до 1000 а.е.м. (электрораспыление +ve); колонка при комнатной температуре; скорость потока 1,0 мл/мин.
Данные ЖХМС в экспериментальном разделе даны в формате: масса иона, время удерживания, УФ-активность.
Сокращения
AcOH = уксусная кислота
CDI = 1,1'-карбонилдиимидазол
д = дней
DAST = трифторид диэтиламиносеры
DCE = дихлорэтан
DCM = дихлорметан
DIPEA = диизопропилэтиламин
DIAD = диизопропилазодикарбоксилат
DMF = диметилформамид
DMP = периодинан Десса-Мартина
DMSO = диметилсульфоксид
ES = электрораспылительная ионизация
EtOAc = этилацетат
ч = часов
HATU = 1-[бис(диметиламино)метилен]-1H-1,2,3-триазол[4,5-b]
пиридиний-3-оксид гексафторфосфат
ВЭЖХ = высокоэффективная жидкостная хроматография
ЖХ = жидкостная хроматография
LiAlH4 / LAH = алюмогидрид лития
MeCN = ацетонитрил
MeOH = метанол
мин = минут
МС = масс-спектрометрия
Et3N = триэтиламин
ЯМР = ядерный магнитный резонанс
кт = комнатная температура
насыщ. = насыщенный
р-р = раствор
STAB = триацетоксиборогидрид натрия
THF = тетрагидрофуран
ТСХ = тонкослойная хроматография
Приставки н-, втор-, и-, т- и трет- имеют свои обычные значения: нормальный, вторичный, изо и третичный.
Готовые соединения названы с использованием пакета программного обеспечения ACD/ChemSketch, версия 12. Промежуточные продукты и реактивы названы либо с использованием пакета программного обеспечения ACD/ChemSketch, версия 12, либо обозначаются с использованием их обычного названия, как правило, найденного в каталогах поставщиков и т. п.
Общие процедуры синтеза для промежуточных соединений
Путь 1
Процедура получения промежуточного соединения 4, этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилата
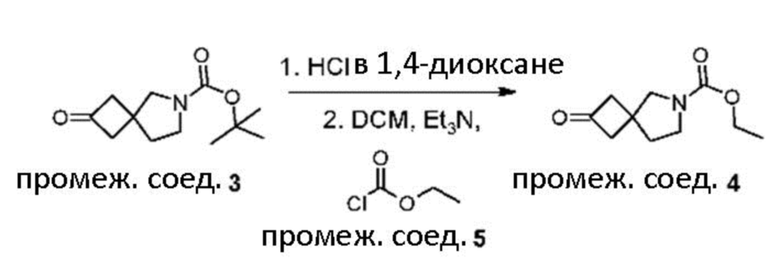
Промежуточное соединение 3, трет-бутил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (3,37 г, 15 ммоль) добавляли порциями к хлороводороду (4 M раствор в диоксане, 50 мл, 200 ммоль). Осторожно: вспенивание. Через 24 ч реакционную смесь концентрировали под вакуумом и твердый остаток растворяли в смеси Et3N (4,18 мл, 30 ммоль) и DCM (66 мл). После завершения растворения раствор немедленно охлаждали до 0 °C, затем по каплям добавляли промежуточное соединение 5, этилкарбонохлоридат (1,57 мл, 16 ммоль). Через 18 ч смесь выливали в DCM (100 мл) и NaHCO3 (водн. р-р) (100 мл) и экстрагировали DCM (2 × 100 мл). Собирали органические слои, промывали солевым раствором (20 мл), высушивали над MgSO4, затем после испарения остаток очищали колоночной хроматографией (нормальная фаза, [картридж Biotage SNAP KP-sil 100 г, 40-63 мкм, 60 Å, 50 мл в мин, градиент от 0 % до 4 % MeOH в DCM]) с получением промежуточного соединения 4, этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилата в виде масла (2,47 г, 83 %).
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 2
Типовая процедура получения аминов на примере получения промежуточного соединения 2, гидрохлорида N-этил-N-(пиперидин-4-ил)ацетамида
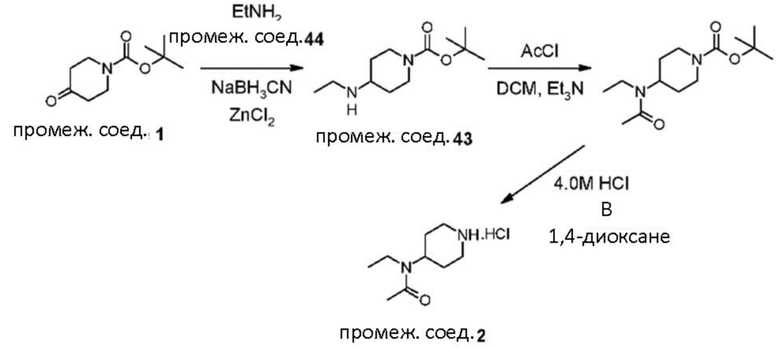
К промежуточному соединению 1, трет-бутил-4-оксопиперидин-1-карбоксилату (3,0 г, 15,1 ммоль), в MeOH (40 мл) добавляли промежуточное соединение 44, этанамин (12,6 мл, 25,1 ммоль, 2 M в THF), Et3N (4,2 мл, 30,3 ммоль) и ZnCl2 (0,1 г, 0,7 ммоль) и реакционную смесь перемешивали при 60 °C в течение 7 ч. Добавляли порциями NaBH3CN (1,2 г, 19,6 ммоль) и полученную реакционную смесь перемешивали при 25 °C в течение 17 ч. Растворители удаляли под вакуумом, а остаток распределяли между H2O (250 мл) и EtOAc (200 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 × 200 мл) и высушивали объединенные органические фазы (Na2SO4), а растворитель удаляли под вакуумом. Остаток очищали колоночной хроматографией (нормальный основной активированный оксид алюминия, от 10 до 30 % EtOAc в гексане) с получением промежуточного соединения 43, трет-бутил-4-(этиламино)пиперидин-1-карбоксилата (3,0 г, 88 %) в виде смолы.
Данные для промежуточного соединения 43 приведены в табл. 2.
К промежуточному соединению 43, трет-бутил-4-(этиламино)пиперидин-1-карбоксилату (0,20 г, 0,9 ммоль), в DCM (10 мл) по каплям добавляли триэтиламин (0,15 мл, 1,1 ммоль) и смесь перемешивали при 0 °C в течение 30 мин. По каплям добавляли ацетилхлорид (0,09 г, 1,1 ммоль) при 0 °C и перед удалением растворителей под вакуумом полученную реакционную смесь перемешивали при 25 °C в течение 8 ч. Остаток распределяли между H2O (120 мл) и EtOAc (100 мл) и проводили дополнительную экстракцию водного слоя EtOAc (2 x 100 мл). Высушивали объединенные органические фазы (Na2SO4), а растворитель удаляли под вакуумом. Остаток очищали колоночной хроматографией (нормальный основной активированный оксид алюминия, от 0,5 до 1,0 % MeOH в DCM) с получением трет-бутил-4-[ацетил(этил)амино]пиперидин-1-карбоксилата (0,15 г, 63 %) в виде жидкости.
ЖХМС (метод I): m/z 271 [M+H]+ (ES+), на 3,79 мин, УФ-активный.
К трет-бутил-4-[ацетил(этил)амино]пиперидин-1-карбоксилату (0,20 г, 0,7 ммоль) в 1,4-диоксане (5 мл) по каплям добавляли 4,0 M HCl в 1,4-диоксане (5 мл) и полученную реакционную смесь перемешивали при 25 °C в течение 16 ч. Растворитель удаляли под вакуумом, а остаток очищали растиранием с диэтиловым эфиром (3 x 5 мл) с получением промежуточного соединения 2, гидрохлоридной соли N-этил-N-(пиперидин-4-ил)ацетамида (0,15 г, 100 %) в виде твердого вещества.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 3
Типовая процедура получения аминов на примере получения промежуточного соединения 8, N-циклопропил-N-(пиперидин-4-ил)ацетамида
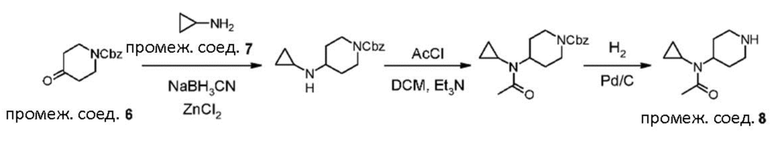
Промежуточное соединение 7, циклопропанамин (1,2 г, 21,5 ммоль), промежуточное соединение 6, бензил-4-оксопиперидин-1-карбоксилат (5,0 г, 21,5 ммоль) и хлорид цинка (0,15 г, 1,1 ммоль) растворяли в MeOH (15 мл) и подогревали до 50 - 60 °C в течение 3 ч в атмосфере N2. Затем смесь охлаждали до 0 - 10 °C перед порционным добавлением NaCNBH3 (1,8 г, 27,9 ммоль) и дополнительным перемешиванием при кт в течение 2 ч. Реакционную смесь распределяли между H2O (15 мл) и EtOAc (25 мл) и проводили дополнительную экстракцию водного слоя EtOAc (2 x 25 мл). Высушивали объединенные органические фазы (Na2SO4), а растворитель удаляли под вакуумом с получением бензил-4-(циклопропиламино)пиперидин-1-карбоксилата (4,0 г, 68 %) в виде смолы.
ЖХМС (метод F): m/z 275 [M+H]+ (ES+), на 1,60 мин, УФ-активный.
К бензил-4-(циклопропиламино)пиперидин-1-карбоксилату (2,5 г, 9,1 ммоль) в DCM (10 мл), охлажденном до 0 - 5 °C, по каплям добавляли Et3N (1,8 г, 18,2 ммоль) и ацетилхлорид (0,9 г, 11,9 ммоль) и реакционную смесь перемешивали при 25 °C в течение 16 ч. Реакционную смесь распределяли между H2O (15 мл) и DCM (25 мл) и проводили дополнительную экстракцию водного слоя DCM (2 x 25 мл). Высушивали объединенные органические фазы (Na2SO4), а растворитель удаляли под вакуумом с получением бензил-4-[ацетил(циклопропил)амино]пиперидин-1-карбоксилата (2,0 г, 70 %) в виде смолы, который использовали на следующей стадии без дополнительной очистки.
ЖХМС (метод I): m/z 317 [M+H]+ (ES+), на 4,17 мин, УФ-активный.
К бензил-4-[ацетил(циклопропил)амино]пиперидин-1-карбоксилату (2,0 г, 6,3 ммоль) в MeOH (15 мл) при кт добавляли 10 % Pd/C (0,2 г) и реакционную смесь перемешивали в атмосфере газообразного H2 (давление 10 кг) в течение 16 ч при кт. Затем реакционную смесь фильтровали через целит и из фильтрата под вакуумом удаляли растворитель с получением промежуточного соединения 8, N-циклопропил-N-(пиперидин-4-ил)ацетамида (1,0 г, 86 %) в виде смолы, которое использовали на следующей стадии без дополнительной очистки.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 4
Типовая процедура получения аминов, замещенных боковым сложным эфиром, на примере получения промежуточного соединения 13, гидрохлорида метил[циклопропил(пиперидин-4-ил)ацетата
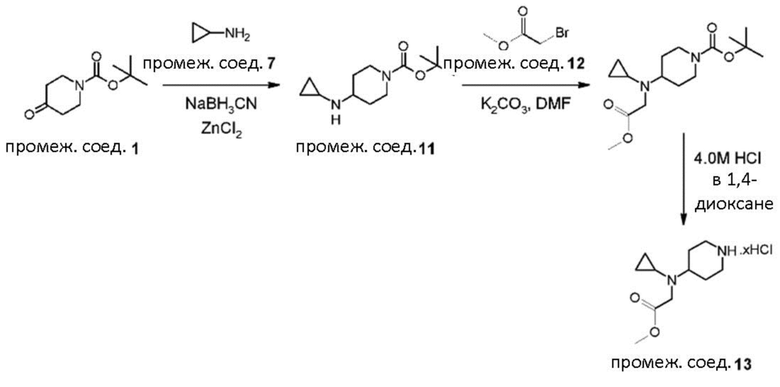
К промежуточному соединению 1, трет-бутил-4-оксопиперидин-1-карбоксилату (5,0 г, 25,1 ммоль), в MeOH (40 мл) добавляли промежуточное соединение 7, циклопропанамин (1,4 г, 25,1 ммоль), Et3N (10,0 мл, 75,3 ммоль) и ZnCl2 (0,3 г, 2,5 ммоль). Реакционную смесь перемешивали при 60 °C в течение 7 ч, затем порциями добавляли NaBH3CN (4,8 г, 75,3 ммоль). Полученную реакционную смесь перемешивали при 25 °C в течение 17 ч. Растворители удаляли под вакуумом, а остаток распределяли между H2O (250 мл) и EtOAc (200 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 × 200 мл), высушивали объединенные органические слои (Na2SO4), а растворитель удаляли под вакуумом. Остаток очищали колоночной хроматографией (нормальный основной активированный оксид алюминия, от 10 до 30 % EtOAc в гексане) с получением промежуточного соединения 11, трет-бутил-4-(циклопропиламино)пиперидин-1-карбоксилата (5,3 г, 88 %) в виде смолы.
Данные для промежуточного соединения 11 приведены в табл. 2.
Промежуточное соединение 11, трет-бутил-4-(циклопропиламино)пиперидин-1-карбоксилат (300 мг, 1,25 ммоль) растворяли в DMF (10 мл) и добавляли K2CO3 (517 мг, 3,75 ммоль). Реакционную смесь перемешивали при 70 °C в течение 3 ч, затем по каплям добавляли при 20 °C промежуточное соединение 12, метилбромацетат (229 мг, 1,50 ммоль). Полученную реакционную смесь перемешивали при 60 °C в течение 8 ч. Растворители удаляли под вакуумом, а остаток распределяли между H2O (150 мл) и EtOAc (100 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 x 100 мл) и высушивали объединенные органические слои (Na2SO4), а растворители удаляли под вакуумом. Остаток очищали колоночной хроматографией (нормальный основной активированный оксид алюминия, от 0,5 % до 1,0 % MeOH в DCM) с получением трет-бутил-4-[циклопропил(2-метокси-2-оксоэтил)амино]пиперидин-1-карбоксилата (310 мг, 80 %) в виде смолы.
ЖХМС (метод I): m/z 313 [M+H]+ (ES+), на 4,85 мин, УФ-активный.
Растворяли трет-бутил-4-[циклопропил(2-метокси-2-оксоэтил)амино]пиперидин-1-карбоксилат (300 мг, 0,96 ммоль) в 1,4-диоксане (5 мл) и по каплям добавляли 4 M HCl в 1,4-диоксане (3 мл). Полученную реакционную смесь перемешивали при 25 °C в течение 16 ч. Растворители удаляли под вакуумом, а остаток очищали растиранием с диэтиловым эфиром (3 x 5 мл) с получением промежуточного соединения 13, гидрохлоридной соли метил[циклопропил(пиперидин-4-ил)амино]ацетата (210 мг, 85 %) в виде твердого вещества.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 5
Типовая процедура получения аминов, замещенных арилметильной группой, на примере получения промежуточного соединения 15, гидрохлорида N-циклопропил-N-(1,2-оксазол-3-илметил)пиперидин-4-амина
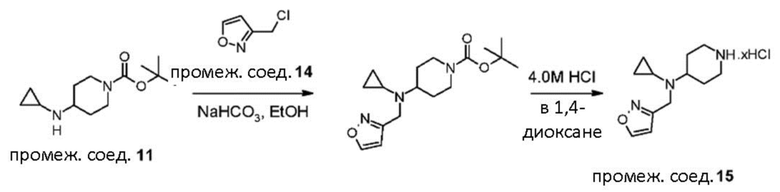
Промежуточное соединение 11, трет-бутил-4-(циклопропиламино)пиперидин-1-карбоксилат (200 мг, 0,83 ммоль), растворяли в этаноле (10 мл) и добавляли бикарбонат натрия (200 мг, 2,38 ммоль). Реакционную смесь перемешивали при 0 °C в течение 30 мин, затем по каплям при кт добавляли промежуточное соединение 14, 3-(хлорметил)-1,2-оказол (97 мг, 0,83 ммоль). Полученную реакционную смесь перемешивали при 60 °C в течение 16 ч. Растворители удаляли под вакуумом, а остаток распределяли между H2O (120 мл) и EtOAc (100 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 x 100 мл) и высушивали объединенные органические слои (Na2SO4), а растворители удаляли под вакуумом. Остаток очищали колоночной хроматографией (нормальный основной активированный оксид алюминия, от 0,5 % до 1,0 % MeOH в DCM) с получением трет-бутил-4-[циклопропил(1,2-оксазол-3-илметил)амино]пиперидин-1-карбоксилата (150 мг, 58 %) в виде жидкости.
ЖХМС (метод I): m/z 322 [M+H]+ (ES+), на 4,92 мин, УФ-активный.
Растворяли трет-бутил-4-[циклопропил(1,2-оксазол-3-илметил)амино]пиперидин-1-карбоксилат (150 мг, 0,46 ммоль) в 1,4-диоксане (5 мл) и по каплям добавляли 4 M HCl в 1,4-диоксане (5 мл). Полученную реакционную смесь перемешивали при 25 °C в течение 16 ч. Растворители удаляли под вакуумом, а остаток очищали растиранием с диэтиловым эфиром (3 x 5 мл) с получением промежуточного соединения 15, гидрохлоридной соли N-циклопропил-N-(1,2-оксазол-3-илметил)пиперидин-4-амина (120 мг, 100 %) в виде твердого вещества.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 6
Типовая процедура получения аминов, замещенных двумя алкильными группами, на примере получения промежуточного соединения 18, гидрохлорида N-этил-N-(2,2,2-трифторэтил)пиперидин-4-амина
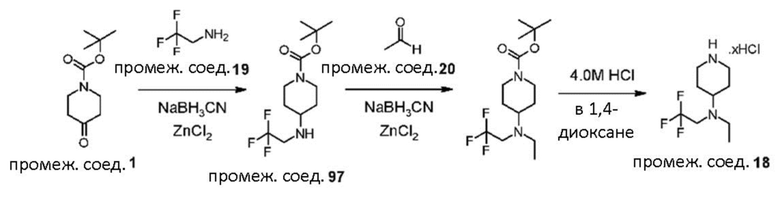
К промежуточному соединению 1, трет-бутил-4-оксопиперидин-1-карбоксилату (500 мг, 2,5 ммоль) в виде раствора в MeOH (10 мл), добавляли промежуточное соединение 19, 2,2,2-трифторэтанамин (273 мг, 2,8 ммоль), триэтиламин (1,0 мл, 7,5 ммоль) и ZnCl2 (34 мг, 0,3 ммоль) и реакционную смесь перемешивали при 60 °C в течение 7 ч. Добавляли порциями NaBH3CN (475 мг, 7,5 ммоль) и полученную реакционную смесь перемешивали при 25 °C в течение 17 ч. Растворитель удаляли под вакуумом, а остаток распределяли между H2O (150 мл) и EtOAc (120 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 × 120 мл) и высушивали объединенные органические фазы (Na2SO4), а растворитель удаляли под вакуумом. Остаток очищали колоночной хроматографией (нормальный основной активированный оксид алюминия, от 0,5 % до 1,0 % MeOH в DCM) с получением промежуточного соединения 97, трет-бутил-4-[(2,2,2-трифторэтил)амино]пиперидин-1-карбоксилата (350 мг, 49 %) в виде смолы.
Данные для промежуточного соединения 97 приведены в табл. 2.
К промежуточному соединению 97, трет-бутил-4-[(2,2,2-трифторэтил)амино]пиперидин-1-карбоксилату (300 мг, 1,1 ммоль) в виде раствора в MeOH (10 мл), добавляли промежуточное соединение 20, ацетальдегид (69 мг, 1,6 ммоль), триэтиламин (0,4 мл, 3,2 ммоль) и ZnCl2 (14 мг, 0,1 ммоль) и реакционную смесь перемешивали при 50 °C в течение 7 ч. Перед порционным добавлением NaBH3CN (201 мг, 3,2 ммоль) смеси давали остыть до кт. Смесь перемешивали при 25 °C в течение 17 ч, затем под вакуумом удаляли растворитель. Остаток распределяли между H2O (150 мл) и EtOAc (120 мл) и проводили дополнительную экстракцию водного слоя EtOAc (2 × 120 мл). Высушивали объединенные органические фазы (Na2SO4), а растворитель удаляли под вакуумом. Остаток очищали колоночной хроматографией (нормальный основной активированный оксид алюминия, от 0,5 % до 3 % MeOH в DCM) с получением трет-бутил-4-[этил(2,2,2-трифторэтил)амино]пиперидин-1-карбоксилата (280 мг, 85 %) в виде смолы.
ЖХМС (метод I): m/z 311 [M+H]+ (ES+), на 5,65 мин, УФ-активный.
К трет-бутил-4-[этил(2,2,2-трифторэтил)амино]пиперидин-1-карбоксилату (220 мг, 0,7 ммоль) в 1,4-диоксане (5 мл) по каплям добавляли 4,0 M HCl в 1,4-диоксане (5 мл) и полученную реакционную смесь перемешивали при 25 °C в течение 16 ч. Растворители удаляли под вакуумом, а остаток очищали растиранием с эфиром (3 x 5 мл) с получением промежуточного соединения 18, гидрохлоридной соли N-этил-N-(2,2,2-трифторэтил)пиперидин-4-амина (164 мг, 94 %) в виде твердого вещества.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 7
Типовая процедура получения аминов на примере получения промежуточного соединения 21, гидрохлорида N-циклопропил-N-(2-метоксиэтил)пиперидин-4-амина
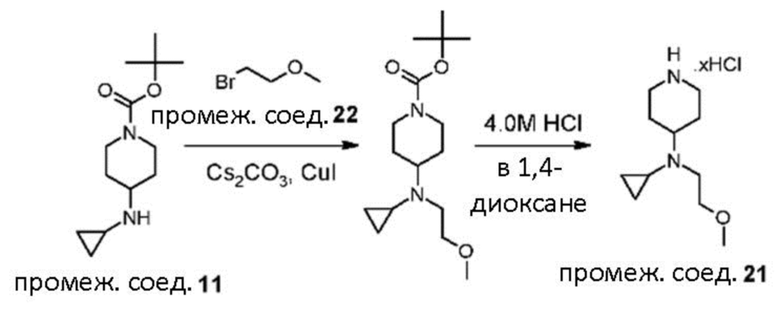
К промежуточному соединению 11, трет-бутил-4-(циклопропиламино)пиперидин-1-карбоксилату (0,50 г, 2,1 ммоль) в ацетонитриле (10 мл), добавляли Cs2CO3 (2,03 г, 6,2 ммоль) и CuI (20 мг) и реакционную смесь перемешивали при 70 °C в течение 1 ч. Затем при 25 °C по каплям добавляли промежуточное соединение 22, 1-бром-2-метоксиэтан (0,43 г, 3,1 ммоль) и полученную реакционную смесь перемешивали при 75 °C в течение 70 ч. Растворитель удаляли под вакуумом, а остаток распределяли между H2O (150 мл) и EtOAc (120 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 x 120 мл) и высушивали объединенные органические фазы (Na2SO4), а растворитель удаляли под вакуумом. Остаток очищали колоночной хроматографией (нормальный основной активированный оксид алюминия, 0,5 % MeOH в DCM) с получением трет-бутил-4-[хлорпропил(2-метоксиэтил)амино]пиперидин-1-карбоксилата (0,27 г, 44 %) в виде смолы.
ЖХМС (метод I): m/z 299 [M+H]+ (ES+), на 4,81 мин, УФ-активный.
К раствору трет-бутил-4-[циклопропил(2-метоксиэтил)амино]пиперидин-1-карбоксилату (0,27 г, 0,9 ммоль) в 1,4-диоксане (5 мл) по каплям добавляли 4,0 M HCl в 1,4-диоксане (5 мл) и полученную реакционную смесь перемешивали при 25 °C в течение 16 ч. Растворитель удаляли под вакуумом, а остаток очищали растиранием с эфиром (3 x 5 мл) с получением промежуточного соединения 21, гидрохлоридной соли N-циклопропил-N-(2-метоксиэтил)пиперидин-4-амина (0,17 г, 81 %) в виде твердого вещества. Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 8
Типовая процедура получения пиперидинов на примере получения промежуточного соединения 35, гидрохлорида N-циклопропил-N-(пиперидин-4-илметил)ацетамида
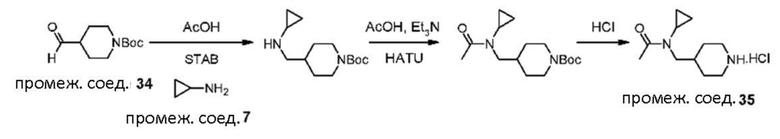
К промежуточному соединению 34, трет-бутил-4-формилпиперидин-1-карбоксилату (427 мг, 2,0 ммоль), и промежуточному соединению 7, циклопропанамину (114 мг, 2,0 ммоль) в виде раствора в DCM (10 мл), добавляли AcOH (0,23 мл, 4,0 ммоль). Смесь перемешивали в течение 3 ч, затем добавляли STAB (1,06 г, 5,0 ммоль) и перемешивали смесь при кт в течение ночи. Реакционную смесь гасили добавлением водн. р-ра NaHCO3 (20 мл). Добавляли твердый Na2CO3 для обеспечения основности водного слоя, затем проводили экстракцию реакционной смеси DCM (4 x 20 мл). Органические слои объединяли, высушивали (MgSO4), фильтровали и удаляли растворители под вакуумом с получением неочищенного трет-бутил-4-[(циклопропиламино)метил]пиперидин-1-карбоксилата (предположительно 100 %), который непосредственно использовали без дополнительной очистки.
ЖХМС (метод C): m/z 255 (M+H)+ (ES+), на 1,38 мин, УФ-активный.
К раствору трет-бутил-4-[(циклопропиламино)метил]пиперидин-1-карбоксилата (предположительно 2,0 ммоль) в DCM (10 мл) добавляли Et3N (1,12 мл, 8,0 ммоль), HATU (914 мг, 2,4 ммоль) и AcOH (0,23 г, 4,0 ммоль) и реакционную смесь перемешивали в течение ночи. Реакционную смесь гасили добавлением водн. р-ра NaHCO3 (20 мл) и экстрагировали DCM (4 x 20 мл). Органические слои объединяли, высушивали (MgSO4), фильтровали и удаляли растворители под вакуумом с получением неочищенного трет-бутил-4-{[ацетил(циклопропил)амино]метил}пиперидин-1-карбоксилата (предположительно 100 %), который непосредственно использовали без дополнительной очистки.
ЖХМС (метод C): m/z 319 (M+Na)+ (ES+), на 1,26 мин, УФ-активный.
К суспензии трет-бутил-4-{[ацетил(циклопропил)амино]метил}пиперидин-1-карбоксилата (предположительно 2,0 ммоль) в DCM (10 мл) добавляли 4,0 M HCl в 1,4-диоксане (2,5 мг, 10,0 ммоль) и смесь перемешивали при кт в течение ночи. Удаляли растворители под вакуумом с получением промежуточного соединения 35, гидрохлоридной соли N-циклопропил-N-(пиперидин-4-илметил)ацетамида (предположительно 2,0 ммоль) в виде твердого вещества, которое использовали без дополнительной очистки.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 9
Типовая процедура получения пиперидинов на примере получения промежуточного соединения 36, гидрохлорида N-циклопропил-N-(пиперидин-4-илметил)пропанамида
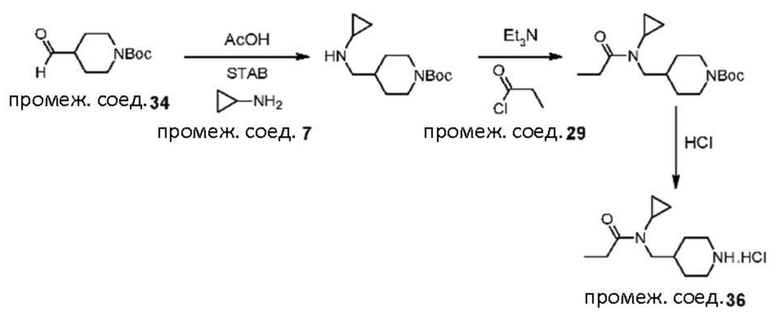
К раствору промежуточного соединения 34, трет-бутил-4-формилпиперидин-1-карбоксилата (0,43 г, 2,0 ммоль), и промежуточного соединения 7, циклопропанамина (0,11 г, 2,0 ммоль), в DCM (10 мл) при кт добавляли AcOH (0,23 мл, 4,0 ммоль) и полученную смесь перемешивали в течение 3 ч. Добавляли STAB (1,06 г, 5,0 ммоль) и перемешивали смесь при кт в течение ночи. Реакционную смесь гасили добавлением водн. р-ра Na2CO3 (20 мл), затем добавляли твердый Na2CO3 для обеспечения основности водного слоя. Проводили экстракцию смеси DCM (4 x 20 мл) и высушивали объединенные органические слои (MgSO4), фильтровали и удаляли растворители под вакуумом с получением неочищенного трет-бутил-4-[(циклопропиламино)метил]пиперидин-1-карбоксилата (предположительно 100 %), который непосредственно использовали без дополнительной очистки.
ЖХМС (метод C): m/z 255 (M+H)+ (ES+), на 1,38 мин, УФ-активный.
К раствору трет-бутил-4-[(циклопропиламино)метил]пиперидин-1-карбоксилата (предположительно 2,0 ммоль) в DCM (10 мл), Et3N (1,12 мл, 8,0 ммоль) и промежуточного соединения 29 добавляли пропаноилхлорид (0,26 мл, 3,0 ммоль) и реакционную смесь перемешивали в течение ночи. Реакционную смесь гасили добавлением водн. р-ра NaHCO3 (20 мл) и экстрагировали DCM (4 x 20 мл). Органические слои объединяли, высушивали (MgSO4), фильтровали и удаляли растворители под вакуумом с получением неочищенного трет-бутил-4-{[циклопропил(пропаноил)амино]метил}пиперидин-1-карбоксилата (предположительно 100 %), который непосредственно использовали без дополнительной очистки.
ЖХМС (метод C): m/z 333 (M+Na)+ (ES+), на 1,39 мин, УФ-активный.
К суспензии трет-бутил-4-{[циклопропил(пропаноил)амино]метил}пиперидин-1-карбоксилата (предположительно 2,0 ммоль) в DCM (10 мл) добавляли 4,0 M HCl в 1,4-диоксане (2,5 мл, 10,0 ммоль) и смесь перемешивали при кт в течение ночи. Удаляли растворители под вакуумом с получением промежуточного соединения 36, гидрохлоридной соли N-циклопропил-N-(пиперидин-4-илметил)пропанамида (предположительно 100 %) в виде твердого вещества, которое использовали без дополнительной очистки.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 10
Типовая процедура получения пиперидинов на примере получения промежуточного соединения 42, N-этил-N-(пиперидин-4-ил)формамида
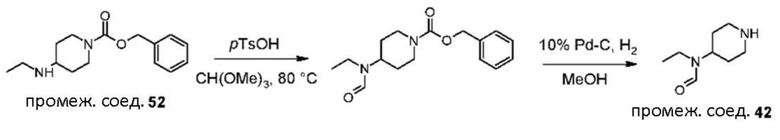
Растворяли промежуточное соединение 52, бензил-4-(этиламино)пиперидин-1-карбоксилат (500 мг, 1,91 ммоль) и п-толуолсульфоновую кислоту (10 мг, 0,06 ммоль) в триэтилортоформиате (3,3 мл, 19,84 ммоль) при кт и реакционную смесь перемешивали при 80 °C в течение 16 ч. Реакционную смесь гасили 0,1 N HCl (30 мл) и проводили экстракцию 10 % MeOH в DCM (2 x 30 мл). Объединяли органические слои, промывали насыщ. водным р-ром NaHCO3 (30 мл) и высушивали (Na2SO4). Удаляли растворители под вакуумом с получением неочищенного бензил-4-[этил(формил)амино)]пиперидин-1-карбоксилата (400 мг, 100 %) в виде смолы, который использовали без дополнительной очистки.
ЖХМС (метод F): m/z 291 [M+H]+ (ES+), на 1,86 мин, УФ-активный.
Растворяли бензил-4-[этил(формил)амино)]пиперидин-1-карбоксилат (180 мг, 0,62 ммоль) в MeOH (15 мл) и добавляли 10 % Pd/C (влажность 50 %) (100 мг, 0,09 ммоль) при кт в атмосфере азота. Систему продували азотом и помещали под газообразный водород с перемешиванием при кт в течение 16 ч. Катализатор удаляли фильтрацией и фильтрат концентрировали под вакуумом с получением неочищенного промежуточного соединения 42, N-этил-N-пиперидин-4-илформамида (100 мг, 100 %) в виде смолы.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 11
Типовая процедура получения пиперидинов на примере получения промежуточного соединения 53, трифторацетата N-этил-N-(пиперидин-4-ил)ацетамида
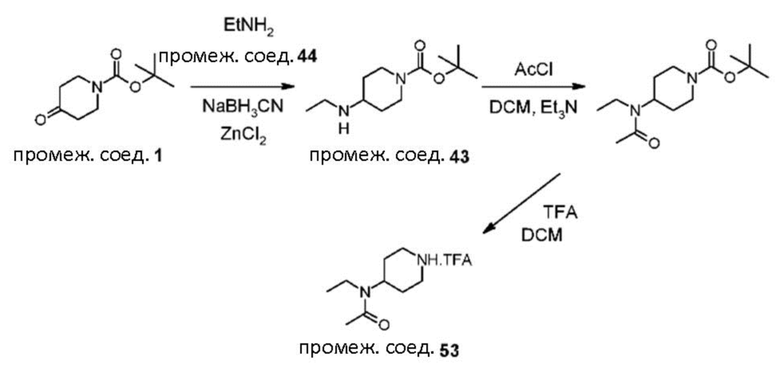
К промежуточному соединению 1, трет-бутил-4-оксопиперидин-1-карбоксилату (3,0 г, 15,1 ммоль), в MeOH (40 мл) добавляли промежуточное соединение 44, этанамин (2 M в THF, 12,6 мл, 25,1 ммоль), Et3N (4,2 мл, 30,3 ммоль) и ZnCl2 (0,1 г, 0,7 ммоль) и реакционную смесь перемешивали при 60 °C в течение 7 ч. Добавляли порциями NaBH3CN (1,2 г, 19,6 ммоль) и полученную реакционную смесь перемешивали при кт в течение 17 ч. Удаляли растворители под вакуумом, а остаток распределяли между H2O (250 мл) и EtOAc (200 мл) и проводили дополнительную экстракцию водного слоя EtOAc (2 x 200 мл). Объединенные органические фазы высушивали (Na2SO4), растворитель удаляли под вакуумом, а остаток очищали колоночной хроматографией (нормальный основной активированный оксид алюминия, от 10 до 30 % EtOAc в гексане) с получением промежуточного соединения 43, трет-бутил-4-(этиламино)пиперидин-1-карбоксилата (3,0 г, 88 %) в виде смолы.
Данные для промежуточного соединения 43 приведены в табл. 2.
К промежуточному соединению 43, трет-бутил-4-(этиламино)пиперидин-1-карбоксилату (0,20 г, 0,9 ммоль) в DCM (10 мл) по каплям добавляли триэтиламин (0,15 мл, 1,1 ммоль) и полученную смесь перемешивали при 0 °C в течение 30 мин. Затем по каплям добавляли ацетилхлорид (0,09 г, 1,1 ммоль) при 0 °C и полученную реакционную смесь перемешивали при кт в течение 8 ч. Растворители удаляли под вакуумом, а остаток распределяли между H2O (120 мл) и EtOAc (100 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 x 100 мл) и высушивали объединенные органические фазы (Na2SO4), а растворитель удаляли под вакуумом. Неочищенный остаток очищали колоночной хроматографией (нормальный основной активированный оксид алюминия, от 0,5 до 1,0 % MeOH в DCM) с получением трет-бутил-4-[ацетил(этил)амино]пиперидин-1-карбоксилата (0,15 г, 63 %) в виде жидкости.
ЖХМС (метод I): m/z 271 [M+H]+ (ES+), на 3,79 мин, УФ-активный.
К трет-бутил-4-[ацетил(этил)амино]пиперидин-1-карбоксилату (450 мг, 1,66 ммоль) в DCM (15 мл) при 0 °C по каплям добавляли трифторуксусную кислоту (1,3 мл, 16,66 ммоль). Полученную реакционную смесь перемешивали при кт в течение 16 ч. Растворитель удаляли под вакуумом, а остаток очищали растиранием с диэтиловым эфиром (3 x 5 мл) с получением промежуточного соединения 53, трифторуксусной соли N-этил-N-(пиперидин-4-ил)ацетамида (450 мг, 100 %) в виде смолы.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 12
Типовая процедура получения пиперидинов на примере получения промежуточного соединения 76, трифторацетата N-метокси-N-(пиперидин-4-ил)ацетамида
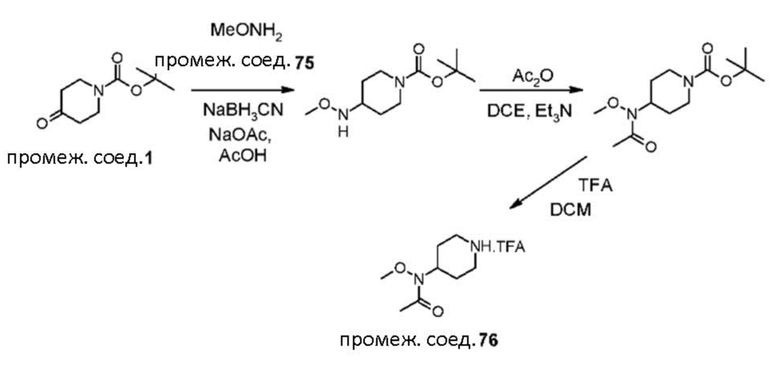
К промежуточному соединению 75, O-метилгидроксиламину (0,5 г, 6,0 ммоль), в MeOH (25 мл) добавляли ацетат натрия (0,51 г, 6,2 ммоль) и реакционную смесь перемешивали при кт в течение 5 минут. Добавляли промежуточное соединение 1, трет-бутил-4-оксопиперидин-1-карбоксилат (1,0 г, 5,0 ммоль), AcOH (0,5 г, 8,8 ммоль) и NaBH3CN (0,3 г, 5,0 ммоль). Полученную реакционную смесь перемешивали при кт в течение 24 ч, затем распределяли между H2O (30 мл) и EtOAc (50 мл). Органическую фазу промывали насыщенным водным р-ром NaHCO3 (20 мл) и насыщ. водным р-ром NaCl (20 мл). Органическую фазу высушивали (Na2SO4), растворитель удаляли под вакуумом, а остаток очищали колоночной хроматографией (силикагель для нормальной фазы, EtOAc и гексаны) с получением трет-бутил-4-(метоксиамино)пиперидин-1-карбоксилата (1 г, 90 %) в виде твердого вещества.
ЖХМС (метод F): m/z 231 [M+H]+ (ES)+, на 2,07 мин, УФ-активный.
К перемешиваемому раствору трет-бутил-4-(метоксиамино)пиперидин-1-карбоксилата (120 мг, 0,52 ммоль) в DCE (3 мл) при 0 oC добавляли Ac2O (79 мг, 0,78 ммоль) и Et3N (0,1 мл, 0,78 ммоль). Реакционную смесь нагревали до 50 – 60 oC в течение 3 ч, а затем распределяли между H2O (5 мл) и DCM (10 мл). Водный слой дополнительно экстрагировали DCM (2 x 10 мл) и объединенные органические слои промывали насыщ. водным р-ром NaHCO3 (30 мл) и насыщ. водным р-ром NaCl (30 мл), затем высушивали (Na2SO4), а растворитель удаляли под вакуумом с получением неочищенного трет-бутил-4-[ацетил(метокси)амино]пиперидин-1-карбоксилата (110 г, 77 %), который использовали на следующей стадии без дополнительной очистки.
ЖХМС (метод F): m/z 273 [M+H]+ (ES)+, на 2,02 мин, УФ-активный.
К перемешиваемому раствору трет-бутил-4-[ацетил(метокси)амино]пиперидин-1-карбоксилата (110 мг, 0,40 ммоль) в DCM (5 мл) при 0 oC по каплям добавляли TFA (2 мл) и полученную смесь перемешивали при кт в течение 3 ч. Удаляли растворитель под вакуумом, а остаток высушивали совместным испарением с толуолом (x 3) с получением промежуточного соединения 76, трифторуксусной соли N-метокси-N-(пиперидин-4-ил)ацетамида (120 мг, 100 %) в виде смолы, которую использовали без дополнительной очистки.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 13
Типовая процедура получения аминов на примере получения промежуточного соединения 89, гидрохлорида 1-(1,3-оксазол-5-ил)метанамина
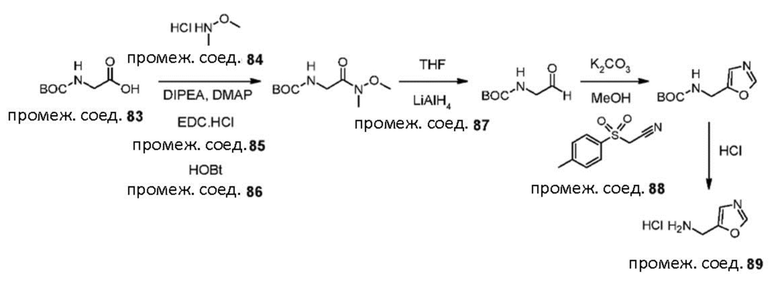
Промежуточное соединение 83, [(трет-бутоксикарбонил)амино]уксусная кислота (5,00 г, 28,5 ммоль), DIPEA (14,75 г, 104 ммоль) и промежуточное соединение 84, гидрохлорид N-метоксиметанамина (5,60 г, 57,0 ммоль), растворяли в DCM (100 мл), DMF (100 мл) и добавляли промежуточное соединение 85, гидрохлорид EDC (6,56 г, 34,0 ммоль). Реакционную смесь перемешивали в атмосфере азота при 0 oC в течение 1 ч, затем порциями добавляли промежуточное соединение 86, HOBt (4,63 г, 34,0 ммоль) и DMAP (100 мг) и полученную смесь перемешивали в течение 16 ч при комнатной температуре. Реакционную смесь распределяли между H2O (250 мл) и DCM (100 мл) и проводили дополнительную экстракцию водного слоя EtOAc (2 x 100 мл). Высушивали объединенные органические слои (Na2SO4), фильтровали и растворитель удаляли под вакуумом. Остаток очищали колоночной хроматографией (силикагель для нормальный фазы, от 0 до 3 % метанола в DCM) с получением трет-бутил-{2-[метокси(метил)амино]-2-оксоэтил}карбамата (4,50 г, 72 %) в виде твердого вещества.
ЖХМС (метод F): m/z 219 (M+H)+ (ES+), на 1,77 мин, УФ-активный.
Растворяли трет-бутил-{2-[метокси(метил)амино]-2-оксоэтил}карбамат (4,50 г, 20,6 ммоль) в THF (50,0 мл) и при -30 °C по каплям добавляли промежуточное соединение 87, алюмогидрид лития (1,0 M в THF, 20,6 мл, 20,6 ммоль). Смесь перемешивали в течение 20 мин при -30 °C, затем порциями добавляли избыток сульфата натрия декагидрата. Смесь перемешивали в течение 30 мин, затем фильтровали через целитную пластину и фильтрат концентрировали под вакуумом с получением неочищенного трет-бутил-(2-оксоэтил)карбамата (390 мг, 85 %) в виде смолы, который использовали без дополнительной очистки.
1H ЯМР: (400 МГц, DMSO-d6) δ: 1,39 - 1,40 (m, 12 H), 2,46 - 2,47 (m, 3 H), 2,89 - 2,95 (m, 1 H), 3,05 - 3,12 (m, 1 H), 4,11 - 4,19 (m, 1 H).
Смешивали трет-бутил-(2-оксоэтил)карбамат (3,00 г, 18,8 ммоль), промежуточное соединение 88, п-толуолсульфометилизоцианид (5,52 г, 28,2 ммоль) и K2CO3 (7,78 г, 56,4 ммоль) в метаноле (50 мл) и перемешивали при 0 °C в течение 70 ч. Реакционную смесь распределяли между H2O (30 мл) и EtOAc (20 мл) и проводили дополнительную экстракцию водного слоя EtOAc (2 x 20 мл). Высушивали объединенные органические слои (Na2SO4), фильтровали и концентрировали под вакуумом. Остаток очищали колоночной хроматографией (силикагель для нормальный фазы, от 0 до 3 % MeOH в DCM) с получением трет-бутил-(1,3-оксазол-5-илметил)карбамата (900 мг, 24 %) в виде смолы.
ЖХМС (метод F): m/z 199 (M+H)+ (ES+), на 1,72 мин, УФ-активный.
Растворяли трет-бутил-(1,3-оксазол-5-илметил)карбамат (900 мг, 0,45 ммоль) в 1,4-диоксане (10 мл) и по каплям добавляли 4 M HCl в 1,4-диоксане (10 мл) и полученную смесь перемешивали в течение 3 ч при комнатной температуре. Реакционную смесь концентрировали, а остаток высушивали совместным испарением с диэтиловым эфиром (5 мл) с получением промежуточного соединения 89, хлористоводородной соли 1-(1,3-оксазол-5-ил)метанамина (400 мг, 90 %) в виде смолы, которую использовали без дополнительной очистки.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 14
Типовая процедура получения аминов на примере получения промежуточного соединения 98, гидрохлорида метилпиперидин-4-ил(2,2,2-трифторэтил)карбамата
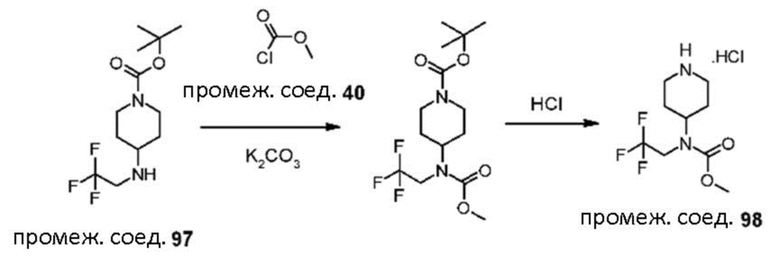
Промежуточное соединение 97, трет-бутил-4-[(2,2,2-трифторэтил)амино]пиперидин-1-карбоксилат (300 мг, 1,06 ммоль), растворяли в ацетонитриле (10 мл) и добавляли K2CO3 (450 мг, 3,19 ммоль). Смесь перемешивали при 70 °C в течение 2 ч, затем охлаждали до 0 °C. По каплям добавляли промежуточное соединение 40, метилкарбонохлоридат (0,12 мл, 1,59 ммоль) и полученную реакционную смесь перемешивали при 25 °C в течение 8 ч. Растворители удаляли под вакуумом, а остаток распределяли между H2O (120 мл) и EtOAc (100 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 x 100 мл) и высушивали объединенные органические слои (Na2SO4), фильтровали и концентрировали под вакуумом. Остаток очищали колоночной хроматографией (нормальный нейтральный активированный оксид алюминия, от 10 % до 15 % EtOAc в гексане) с получением трет-бутил-4-[(метоксикарбонил)(2,2,2-трифторэтил)амино]пиперидин-1-карбоксилата (330 мг, 92 %) в виде смолы.
ЖХМС (метод I): m/z 284 (M+H-56)+ (ES+), на 5,01 мин, УФ-активный.
Растворяли трет-бутил-4-[(метоксикарбонил)(2,2,2-трифторэтил)амино]пиперидин-1-карбоксилат (330 мг, 0,97 ммоль) в 1,4-диоксане (5 мл) по каплям добавляли 4,0 M HCl в 1,4-диоксане (10 мл) и полученную реакционную смесь перемешивали при 25 °C в течение 8 ч. Растворители удаляли под вакуумом, а остаток очищали растиранием с диэтиловым эфиром (3 x 3 мл) с получением промежуточного соединения 98, гидрохлоридной соли метилпиперидин-4-ил(2,2,2-трифторэтил)карбамата (210 мг, 90 %) в виде твердого вещества, которую использовали без дополнительной очистки.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 15
Типовая процедура получения аминов на примере получения промежуточного соединения 99, гидрохлорида 2-метоксиэтилпиперидин-4-ил(2,2,2-трифторэтил)карбамата
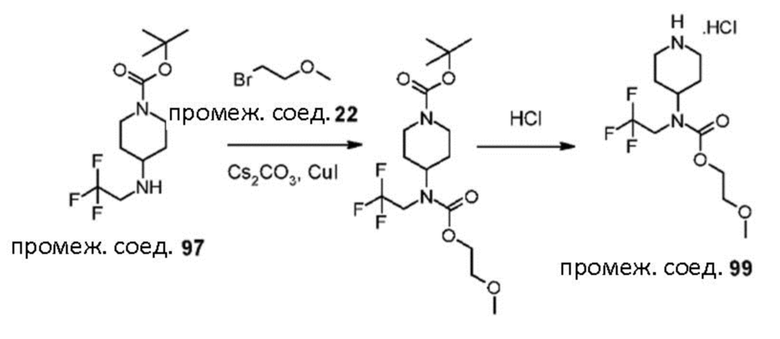
Растворяли промежуточное соединение 97, трет-бутил-4-((трифторэтил)амино)пиперидин-1-карбоксилат (1,0 г, 3,50 ммоль), в DMF (15 мл), добавляли Cs2CO3 (3,46 г, 10,6 ммоль) и CuI (336 мг, 1,77 ммоль) и реакционную смесь перемешивали при 70 °C в течение 5 ч, затем охлаждали до 25 °C. Добавляли промежуточное соединение 22, 1-бром-2-метоксиэтан (986 мг, 7,09 ммоль) и реакционную смесь перемешивали при 90 oC в течение 7 дней. Растворители удаляли под вакуумом, а остаток распределяли между H2O (180 мл) и EtOAc (120 мл). Проводили дополнительную экстракцию водного слоя EtOAc (3 × 120 мл) и высушивали объединенные органические слои (Na2SO4), фильтровали и концентрировали под вакуумом. Остаток очищали колоночной хроматографией (нормальный нейтральный активированный оксид алюминия, в 8 % EtOAc в гексане) с получением трет-бутил-4-{[(2-(метоксиэтокси)карбонил](2,2,2-трифторэтил)амино}пиперидин-1-карбоксилата (280 мг, 21 %) в виде смолы.
ЖХМС (метод I): m/z 329 (M+H-56)+ (ES+), на 4,90 мин, УФ-активный.
Растворяли трет-бутил-4-{[(2-метоксиэтокси)карбонил](2,2,2-трифторэтил)амино}пиперидин-1-карбоксилат (240 мг, 0,71 ммоль) в 1,4-диоксане (5 мл) по каплям добавляли 4,0 M HCl в 1,4-диоксане (10 мл) и полученную реакционную смесь перемешивали при 25 °C в течение 8 ч. Растворители удаляли под вакуумом, а остаток очищали растиранием с диэтиловым эфиром (3 x 3 мл) с получением промежуточного соединения 99, гидрохлоридной соли 2-метоксиэтилпиперидин-4-ил(2,2,2-трифторэтил)карбамата (170 мг, 96 %) в виде твердого вещества.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 16
Типовая процедура получения аминов на примере получения промежуточного соединения 109, гидрохлорида N-циклопропил-N-(22,2-трифторэтил)пиперидин-4-амина
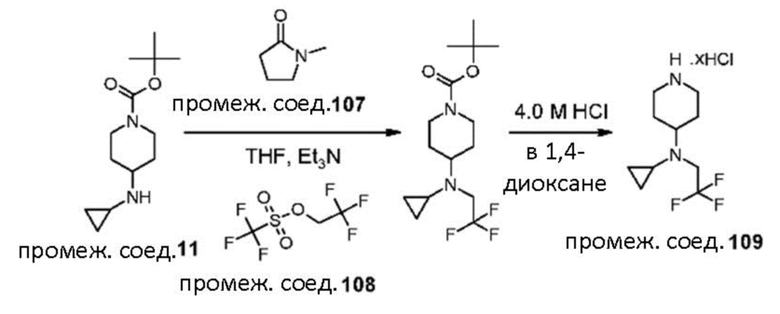
Растворяли промежуточное соединение 11, трет-бутил-4-(циклопропиламино)пиперидин-1-карбоксилат (200 мг, 0,83 ммоль), в THF (10 мл), добавляли промежуточное соединение 107, N-метил-2-пирролидинон (0,6 мл) и триэтиламин (0,5 мл, 3,30 ммоль) и реакционную смесь перемешивали при 70 oC в течение 1 ч, затем охлаждали до комнатной температуры. Добавляли промежуточное соединение 108, 2,2,2-трифторэтилтрифторметансульфонат (385 мг, 1,66 ммоль) и полученную реакционную смесь перемешивали при 80 °C в течение 70 ч. Удаляли растворители под вакуумом, а остаток распределяли между H2O (120 мл) и EtOAc (100 мл) и проводили дополнительную экстракцию водного слоя EtOAc (2 x 100 мл). Объединенные органические слои высушивали (Na2SO4), фильтровали и концентрировали под вакуумом, а остаток очищали колоночной хроматографией (нормальный нейтральный активированный оксид алюминия, 0,5 % MeOH в CH2Cl2) с получением трет-бутил-4-[циклопропил(2,2,2-трифторэтил)амино]пиперидин-1-карбоксилата (220 мг, 82 %) в виде смолы.
ЖХМС (метод I): m/z 267 (M+H-56)+ (ES+), на 5,90 мин, УФ-активный.
Растворяли трет-бутил-4-[циклопропил(2,2,2-трифторэтил)амино]пиперидин-1-карбоксилат (200 мг, 0,62 ммоль) в 1,4-диоксане (5 мл) по каплям добавляли 4,0 M HCl в 1,4-диоксане (5 мл) и полученную реакционную смесь перемешивали при 25 °C в течение 16 ч. Растворители удаляли под вакуумом, а остаток очищали растиранием с диэтиловым эфиром (3 x 5 мл) с получением промежуточного соединения 109, гидрохлоридной соли N-циклопропил-N-(2,2,2-трифторэтил)пиперидин-4-амина (160 мг, 100 %) в виде твердого вещества.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 17
Типовая процедура получения аминов на примере получения промежуточного соединения 111, трифторацетата 2-[циклопропил-(пиперидин-4-ил)амино)]этанола
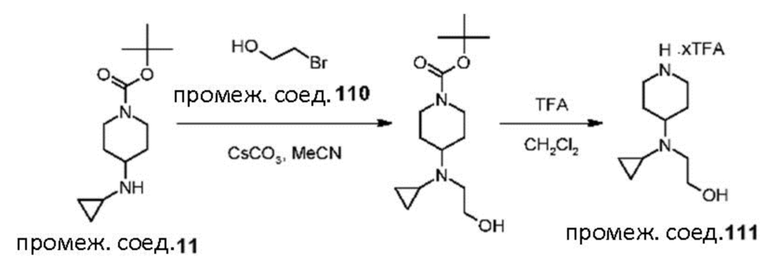
Добавляли промежуточное соединение 11, трет-бутил-4-(циклопропиламино)пиперидин-1-карбоксилат (200 мг, 0,833 ммоль), и карбонат цезия (0,812 г, 2,5 ммоль) к MeCN (10 мл) и перемешивали при 25 °C в течение 15 мин. Добавляли промежуточное соединение 110, 2-бромэтанол (134 мл, 1,08 ммоль) и реакционную смесь перемешивали при 60 oC в течение 16 ч. Реакционную смесь распределяли между водой (30 мл) и 10 % метанолом в CH2Cl2 (30 мл) и проводили дополнительную экстракцию водного слоя 10 % метанолом в CH2Cl2 (2 x 30 мл). Затем объединяли органические слои, высушивали (Na2SO4), фильтровали и концентрировали с получением трет-бутил-4-[циклопропил(2-гидроксиэтил)амино]пиперидин-1-карбоксилата (180 мг, 76 %) в виде смолы, который использовали без дополнительной очистки.
ЖХМС (метод F): m/z 285 (M+H)+ (ES+), на 1,46 мин, УФ-активный.
Растворяли трет-бутил-4-[циклопропил(2-гидроксиэтил)амино]пиперидин-1-карбоксилат (365 мг, 1,29 ммоль) в CH2Cl2 (10 мл), добавляли трифторуксусную кислоту (1,1 мл, 12,9 ммоль) и полученную реакционную смесь перемешивали при 25 °C в течение 16 ч. Растворители удаляли под вакуумом, а остаток очищали растиранием с диэтиловым эфиром (3 x 10 мл) с получением промежуточного соединения 111, трифторуксусной соли 2-[циклопропил(пиперидин-4-ил)амино]этанола (380 мг, 100 %) в виде смолы.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 18
Типовая процедура получения аминов на примере получения промежуточного соединения 112, трет-бутил-4-(циклобутиламино)пиперидин-1-карбоксилата
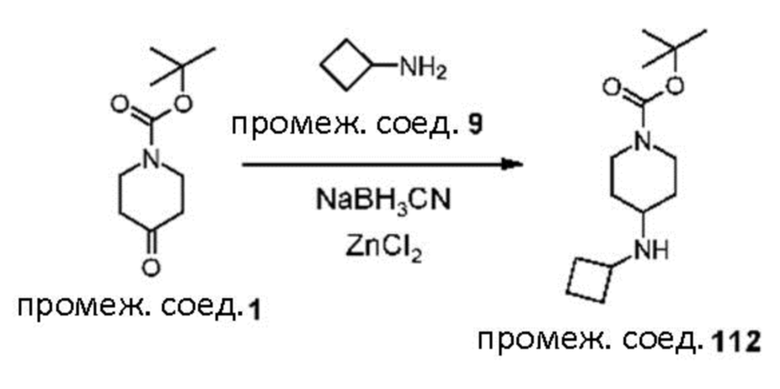
Растворяли промежуточное соединение 1, трет-бутил-4-оксопиперидин-1-карбоксилат (500 мг, 2,51 ммоль) в метаноле (10 мл), добавляли при комнатной температуре промежуточное соединение 9, циклобутанамин (178 мг, 2,51 ммоль), триэтиламин (1,0 мл, 7,53 ммоль) и ZnCl2 (34 мг, 0,25 ммоль) и реакционную смесь перемешивали при 60 °C в течение 4 ч. Добавляли порциями NaBH3CN (475 мг, 7,53 ммоль) и полученную реакционную смесь перемешивали при 25 °C в течение 12 ч, а затем удаляли растворители под вакуумом. Остаток распределяли между H2O (150 мл) и EtOAc (120 мл) и проводили дополнительную экстракцию водного слоя EtOAc (2 × 120 мл). Объединяли органические слои, высушивали (Na2SO4), фильтровали и концентрировали под вакуумом. Остаток очищали колоночной хроматографией (нормальный основной активированный оксид алюминия, от 10 до 30 % EtOAc в гексане) с получением промежуточного соединения 112, трет-бутил-4-(циклобутиламино)пиперидин-1-карбоксилата (560 мг, 88 %) в виде смолы.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 19
Типовая процедура получения пиперидинов на примере получения промежуточного соединения 119, трифторацетата N-(2-метоксиэтил)-N-(2,2,2-трифторэтил)пиперидин-4-амина
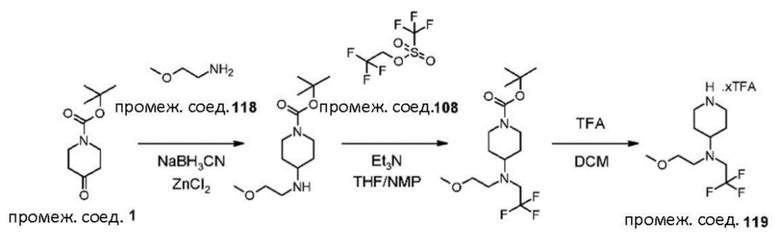
Растворяли промежуточное соединение 1, трет-бутил-4-оксопиперидин-1-карбоксилат (1,0 г, 5,02 ммоль) в метаноле (15 мл) и обрабатывали его промежуточным соединением 118, 2-метоксиэтиламином (490 мг, 6,53 ммоль), триэтиламином (2,1 мл, 15,1 ммоль) и ZnCl2 (68 мг, 0,50 ммоль). Реакционную смесь перемешивали при 65 °C в течение 7 ч, затем порциями добавляли NaBH3CN (949 мг, 15,1 ммоль). Полученную реакционную смесь перемешивали при 25 °C в течение 17 ч. Растворители удаляли под вакуумом, а остаток распределяли между H2O (150 мл) и EtOAc (120 мл). Проводили экстракцию водного слоя EtOAc (2 × 120 мл) и объединяли органические слои, высушивали (Na2SO4), а растворитель удаляли под вакуумом. Остаток очищали колоночной хроматографией (нормальный основной активированный оксид алюминия, от 40 до 50 % EtOAc в гексане) с получением трет-бутил-4-[(2-метоксиэтил)амино]пиперидин-1-карбоксилата (480 мг, 37 %) в виде жидкости.
ЖХМС (метод I): m/z 203 (M+H-56)+ (ES+), на 3,60 мин, УФ-активный.
Растворяли трет-бутил-4-[(2-метоксиэтил)амино]пиперидин-1-карбоксилат (300 мг, 1,16 ммоль) в THF (10 мл) и обрабатывали его N-метил-2-пирролидиноном (344 мг, 3,48 ммоль) и триэтиламином (0,7 мл, 4,65 ммоль). Реакционную смесь перемешивали при 70 oC в течение 1 ч, затем при 25 oC по каплям добавляли промежуточное соединение 108, 2,2,2-трифторэтилтрифторметансульфонат (297 мг, 1,28 ммоль). Полученную реакционную смесь перемешивали при 70 °C в течение 16 ч. Растворители удаляли под вакуумом, а реакционную смесь распределяли между H2O (120 мл) и EtOAc (100 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 x 100 мл) и высушивали объединенные органические слои (Na2SO4). Растворитель удаляли под вакуумом, а остаток очищали колоночной хроматографией (нормальный нейтральный активированный оксид алюминия, от 10 % до 20 % EtOAc в гексане) с получением трет-бутил-4-[(2-метоксиэтил)(2,2,2-трифторэтил)амино]пиперидин-1-карбоксилата (180 мг, 46 %) в виде смолы.
ЖХМС (метод I): m/z 341 (M+H)+ (ES+), на 5,31 мин, УФ-активный.
Растворяли трет-бутил-4-[(2-метоксиэтил)(2,2,2-трифторэтил)амино]пиперидин-1-карбоксилат (150 мг, 0,44 ммоль) в DCM (3 мл) и охлаждали до 0 °C. По каплям добавляли трифторуксусную кислоту (0,8 мл) и полученную реакционную смесь перемешивали при 25 °C в течение 8 ч. Растворители удаляли под вакуумом, а остаток очищали растиранием с диэтиловым эфиром (3 x 2 мл) с получением промежуточного соединения 119, трифторуксусной соли N-(2-метоксиэтил)-N-(2,2,2-трифторэтил(пиперидин-4-амина (105 мг, 67 %) в виде смолы.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 20
Типовая процедура получения пиперидинов на примере получения промежуточного соединения 120, трифторацетата N-этил-N-(оксетан-3-ил)пиперидин-4-амина
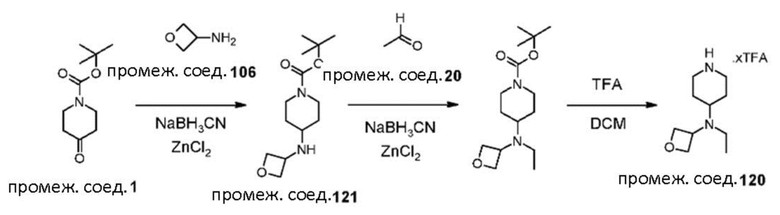
Растворяли промежуточное соединение 1, трет-бутил-4-оксопиперидин-1-карбоксилат (546 мг, 4,10 ммоль) в метаноле (20 мл). Добавляли промежуточное соединение 106, оксетан-3-амин (300 мг, 4,10 ммоль), триэтиламин (1,7 мл, 12,3 ммоль) и ZnCl2 (56 мг, 0,41 ммоль) и затем реакционную смесь перемешивали при 65 °C в течение 8 ч. Добавляли порциями NaBH3CN (776 мг, 1,23 ммоль) и полученную реакционную смесь перемешивали при 25 °C в течение 17 ч. Растворители удаляли под вакуумом, а остаток распределяли между H2O (120 мл) и EtOAc (100 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 × 100 мл) и высушивали объединенные органические слои (Na2SO4). Растворитель удаляли под вакуумом, а остаток очищали растиранием с пентаном и декантировали растворители с получением трет-бутил-4-(оксетан-3-иламино)пиперидин-1-карбоксилата (680 мг, 97 %) в виде смолы.
ЖХМС (метод I): m/z 257 (M+H)+ (ES+), на 2,92 мин, УФ-активный.
Растворяли трет-бутил-4-(оксетан-3-иламино)пиперидин-1-карбоксилат (200 мг, 0,78 ммоль) в метаноле (10 мл) и добавляли промежуточное соединение 20, ацетальдегид (103 мг, 2,34 ммоль), триэтиламин (0,3 мл, 2,34 ммоль) и ZnCl2 (11 мг, 0,08 ммоль). Реакционную смесь перемешивали при 50 °C в течение 7 ч, затем порциями добавляли NaBH3CN (148 мг, 2,34 ммоль). Полученную реакционную смесь перемешивали при 25 °C в течение 17 ч. Растворители удаляли под вакуумом, а остаток распределяли между H2O (100 мл) и EtOAc (80 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 × 80 мл) и высушивали объединенные органические слои (Na2SO4). Растворитель удаляли под вакуумом, а остаток очищали колоночной хроматографией (нормальный основной активированный оксид алюминия, от 0,5 % до 3 % MeOH в DCM) с получением трет-бутил-4-[этил(оксетан-3-ил)амино]пиперидин-1-карбоксилата (180 мг, 81 %) в виде смолы.
ЖХМС (метод I): m/z 285 (M+H)+ (ES+), на 3,84 мин, УФ-активный.
Растворяли трет-бутил-4-[этил(оксетан-3-ил)амино]пиперидин-1-карбоксилат (180 мг, 0,63 ммоль) в DCM (8 мл) и охлаждали до 0 °C. По каплям добавляли трифторуксусную кислоту (2 мл) и полученную реакционную смесь перемешивали при 25 °C в течение 6 ч. Растворители удаляли под вакуумом, а остаток очищали растиранием с диэтиловым эфиром (3 x 1 мл) с получением промежуточного соединения 120, трифторуксусной соли N-этил-N-(оксетан-3-ил)пиперидин-4-амина (110 мг, 95 %) в виде смолы.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 21
Типовая процедура получения пиперидинов на примере получения промежуточного соединения 128, трифторацетата N-этил-N-метоксипиперидин-4-амина
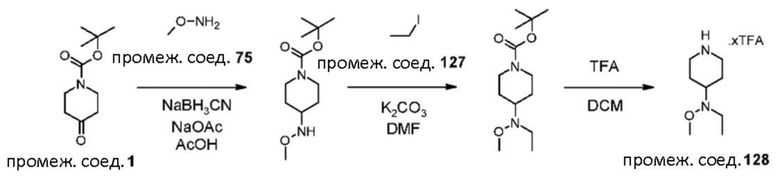
К перемешиваемому раствору промежуточного соединения 75, O-метилгидроксиламину (0,5 г, 6,0 ммоль), в MeOH (25 мл) добавляли NaOAc (0,51 г, 6,2 ммоль) и реакционную смесь перемешивали при кт в течение пяти минут. Добавляли промежуточное соединение 1, трет-бутил-4-оксопиперидин-1-карбоксилат (0,5 г, 8,8 ммоль) и NaBH3CN (0,3 г, 5,0 ммоль) и реакционную смесь перемешивали при кт в течение 24 часов. Реакционную смесь распределяли между EtOAc и водой, отделяли фазу EtOAc и промывали водным NaHCO3 и солевым раствором. Органическую фазу высушивали с помощью Na2SO4 и концентрировали под вакуумом с получением неочищенного продукта, который очищали колоночной хроматографией (силикагель для нормальной фазы, EtOAc и гексаны в качестве элюирующих растворителей) с получением трет-бутил-4-(метоксиамино)пиперидин-1-карбоксилата (1 г, 90 %) в виде твердого вещества.
ЖХМС (метод F): m/z 231 (M+H)+ (ES+), на 2,07 мин, УФ-активный.
К перемешиваемому раствору трет-бутил-4-(метоксиамино)пиперидин-1-карбоксилата (300 мг, 1,30 ммоль) в DMF (5 мл) добавляли K2CO3 (540 мл, 3,91 ммоль) и смесь перемешивали при 80 °C в течение 1 часа. Затем добавляли промежуточное соединение 127, йодэтан (305 мг, 1,96 ммоль) и смесь перемешивали при 80 °C в течение 16 ч. Реакционную смесь охлаждали до кт, разбавляли холодной водой (10 мл) и экстрагировали соединение EtOAc (20 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 × 20 мл) и промывали объединенные органические слои солевым раствором, затем высушивали с помощью Na2SO4. Растворитель удаляли под вакуумом с получением неочищенного продукта, который очищали колоночной хроматографией (силикагель для нормальной фазы, EtOAc и гексаны в качестве элюирующих растворителей) с получением трет-бутил-4-[этил(метокси)амино]пиперидин-1-карбоксилата (160 мг, 47 %).
ЖХМС (метод F): m/z 259 (M+H)+ (ES+), на 2,02 мин, УФ-активный.
К перемешиваемому раствору трет-бутил-4-[этил(метокси)амино]пиперидин-1-карбоксилата (160 мг, 0,62 ммоль) в DCM (8 мл) при 0 oC по каплям при добавляли TFA (3 мл) и полученную смесь перемешивали при комнатной температуре в течение 3 ч. Затем испаряли растворитель под вакуумом, а остаток высушивали совместным испарением с толуолом (x 3) с получением промежуточного соединения 128, трифторуксусной соли N-этил-N-метоксипиперидин-4-амина (150 мг, 63 %) в виде смолы.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 22
Типовая процедура получения пиперидинов на примере получения промежуточного соединения 130, гидрохлорида 2-[фенил(пиперидин-4-ил)амино]этанола
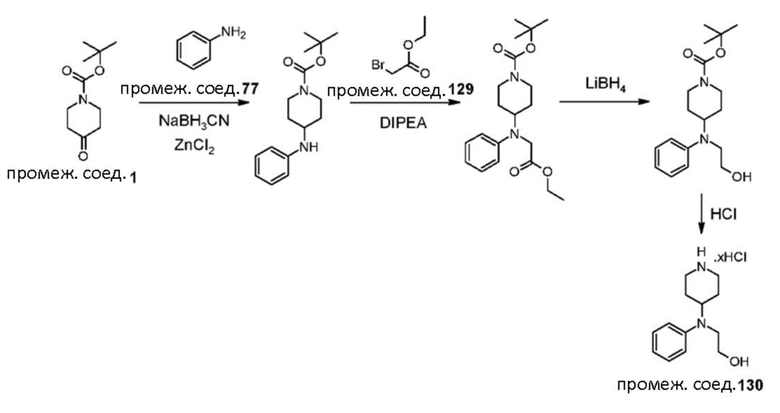
Растворяли промежуточное соединение 1, трет-бутил-4-оксопиперидин-1-карбоксилат (3,00 г, 15,0 ммоль), промежуточное соединение 77, анилин (1,40 г, 15,0 ммоль), триэтиламин (6,35 мл, 45,0 ммоль) и хлорид цинка (0,75 мл, 0,75 ммоль) в метаноле (25,0 мл) в атмосфере азота и перемешивали при 50 – 60 °C в течение 16 ч. При 0 – 10 °C добавляли порциями NaBH3CN (2,84 г, 45,0 ммоль) и полученную реакционную смесь перемешивали при 50 – 60 °C в течение 16 ч. Реакционную смесь распределяли между H2O (150 мл) и EtOAc (50 мл) и проводили дополнительную экстракцию водного слоя EtOAc (2 x 50 мл). Высушивали объединенные органические слои (Na2SO4) и удаляли растворитель под вакуумом с получением неочищенного продукта, который очищали колоночной хроматографией (силикагель для нормальный фазы, от 0 до 3 % метанола в DCM) с получением трет-бутил-4-(фениламино)пиперидин-1-карбоксилата (1,30 г, 31 %) в виде твердого вещества.
ЖХМС (метод F): m/z 277 (M+H)+ (ES+), на 2,33 мин, УФ-активный.
Растворяли трет-бутил-4-(фениламино)пиперидин-1-карбоксилат (350 мг, 1,26 ммоль) и промежуточное соединение 129, этилбромацетат (274 мг, 1,64 ммоль) в DIPEA (3,0 мл) и перемешивали в течение 16 ч при 90 °C. Реакционную смесь распределяли между H2O (30 мл) и EtOAc (20 мл) и проводили дополнительную экстракцию водного слоя EtOAc (2 x 20 мл). Высушивали объединенные органические слои (Na2SO4) и удаляли растворитель под вакуумом с получением неочищенного продукта, который очищали колоночной хроматографией (силикагель для нормальный фазы, от 0 до 22 % EtOAc в гексанах) с получением трет-бутил-4-[(2-этокси-2 оксоэтил)(фенил)амино]пиперидин-1-карбоксилата (390 мг, 85 %) в виде смолы.
ЖХМС (метод F): m/z 363 (M+H)+ (ES+), на 2,72 мин, УФ-активный.
Растворяли трет-бутил-4-[(2-этокси-2-оксоэтил)(фенил)амино]пиперидин-1-карбоксилат (350 мг, 0,96 ммоль) в THF (10,0 мл) и обрабатывали его при 0 OC раствором борогидрида лития в THF (3,0 M, 1,30 мл, 3,86 ммоль) и перемешивали при комнатной температуре в течение 48 ч. Реакционную смесь разделяли между холодным водным раствором NH4Cl (30 мл) и EtOAc (15 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 x 15 мл) и высушивали объединенные органические слои (Na2SO4) и удаляли растворитель под вакуумом с получением неочищенного продукта, который очищали колоночной хроматографией (силикагель для нормальный фазы, от 0 до 35 % EtOAc в гексанах) с получением трет-бутил-4-[(2-гидроксиэтил)(фенил)амино]пиперидин-1-карбоксилата (270 мг, 87 %) в виде смолы.
ЖХМС (метод F): m/z 321 (M+H)+ (ES+), на 1,94 мин, УФ-активный.
Растворяли трет-бутил-4-[(2-гидроксиэтил)(фенил)амино]пиперидин-1-карбоксилат (265 мг, 0,82 ммоль) при 0 OC в 4 M HCl в 1,4-диоксане (5,0 мл) и перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь концентрировали, а затем растирали с диэтиловым эфиром (3 x 10 мл) с получением промежуточного соединения 130, хлористоводородной соли 2-[фенил(пиперидин-4-ил)амино]этанола (200 мг, 94 %) в виде твердого вещества.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 23
Типовая процедура получения пиперидинов на примере получения промежуточного соединения 138, трифторацетата N-этил-N-[1-(пиперидин-4-ил)пропил]ацетамида
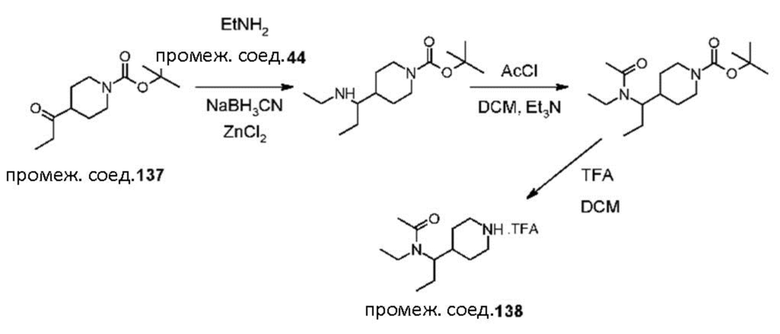
Во флакон помещали промежуточное соединение 137, трет-бутил-4-пропаноилпиперидин-1-карбоксилат (450 мг, 1,86 ммоль), промежуточное соединение 44, этанамин (2,0 M раствор в THF, 2,33 мл, 4,66 ммоль), Et3N (0,780 мл, 5,60 ммоль), ZnCl2 (0,2 мл) и MeOH (10 мл). Реакционную смесь нагревали при 60 °C в течение 4 ч, затем реакционную смесь охлаждали до 0 °C и добавляли NaBH3CN (351 мг, 5,60 ммоль). Реакционную смесь давали нагреться до комнатной температуры и перемешивали в течение 16 ч. Реакционную смесь концентрировали под вакуумом, а остаток распределяли между H2O (100 мл) и EtOAc (100 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 × 50 мл), высушивали объединенные органические слои (Na2SO4) и удаляли растворитель под вакуумом с получением неочищенного продукта, который очищали комбинированной флэш-колоночной хроматографией (нейтральный силикагель, нормальная фаза, 60-120 меш, от 0 до 1 % MeOH в DCM) с получением трет-бутил-4-[1-(этиламино)пропил]пиперидин-1-карбоксилата (440 мг, 87 %) в виде смолы.
ЖХМС (метод F): m/z 271 (M+H)+ (ES+), на 5,34 мин, УФ-активный.
Растворяли трет-бутил-4-[1-(этиламино)пропил]пиперидин-1-карбоксилат (435 мг, 1,61 ммоль) в DCM (10 мл) и при 0 – 5 °C по каплям добавляли триэтиламин (0,67 мл, 4,83 ммоль). Реакционную смесь перемешивали при 0 – 5 °C в течение 10 мин, затем при 0 – 5 °C по каплям добавляли ацетилхлорид (0,17 мл, 2,41 ммоль). Полученную реакционную смесь перемешивали при 25 °C в течение 8 ч, затем под вакуумом удаляли растворители. Остаток распределяли между H2O (50 мл) и DCM (50 мл) и проводили дополнительную экстракцию водного слоя DCM (2 x 30 мл). Высушивали объединенные органические слои (Na2SO4) и удаляли растворители под вакуумом. Остаток очищали колоночной хроматографией (нормальный основной активированный оксид алюминия, от 0,5 % до 1,0 % MeOH в DCM) с получением трет-бутил-4-{1-[ацетил(этил)амино]пропил}пиперидин-1-карбоксилата (415 мг, 63 %) в виде смолы.
ЖХМС (метод I): m/z 313 (M+H)+ (ES+), на 4,53 мин, УФ-активный.
Растворяли при 0 °C трет-бутил-4-{1-[ацетил(этил)амино]пропил}пиперидин-1-карбоксилат (415 мг, 1,33 ммоль) в DCM (5,0 мл) и к раствору при 0 °C добавляли TFA (2,5 мл). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 6 ч. Затем реакционную смесь концентрировали и высушивали совместным испарением с диэтиловым эфиром (3 x 5 мл) с получением промежуточного соединения 138, трифторуксусной соли N-этил-N-[1-(пиперидин-4-ил)пропил]ацетамида (250 мг, 80 %) в виде смолы.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 24
Типовая процедура получения пиперидинов на примере получения промежуточного соединения 140, гидрохлорида N-этил-N-[2-(пиперидин-4-ил)пропан-2-ил]ацетамида
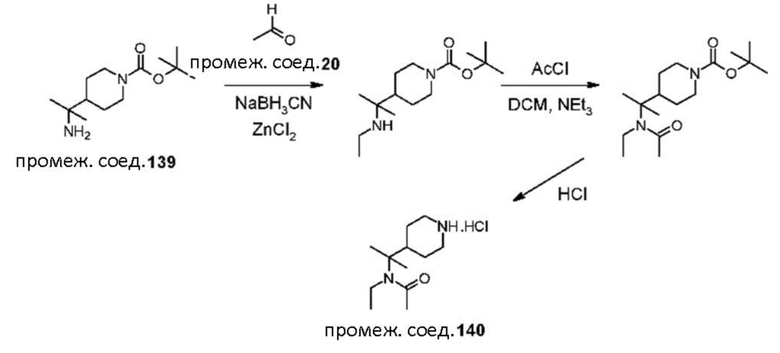
Растворяли промежуточное соединение 139, трет-бутил-4-(2-аминопропан-2-ил)пиперидин-1-карбоксилат (300 мг, 1,24 ммоль), промежуточное соединение 20, ацетальдегид (163 мг, 3,71 ммоль), триэтиламин (0,52 мл, 3,71 ммоль) и хлорид цинка (0,06 мл, 0,06 ммоль) в метаноле (10 мл) в атмосфере азота и перемешивали в течение 16 ч при 50 – 60 °C. Через 16 ч при 0 – 10 °C порциями добавляли NaCNBH3 (233 мг, 3,74 ммоль) и реакционную смесь перемешивали 6 ч при 50 – 60 °C. Реакционную смесь распределяли между H2O (40 мл) и EtOAc (25 мл) и проводили дополнительную экстракцию водного слоя EtOAc (2 x 25 мл). Высушивали объединенные органические слои (Na2SO4) и удаляли растворитель под вакуумом с получением неочищенного продукта, который очищали колоночной хроматографией (силикагель для нормальный фазы, от 0 до 4 % метанола в DCM) с получением трет-бутил-4-[2-(этиламино)пропан-2-ил]пиперидин-1-карбоксилата (180 мг, 54 %) в виде смолы.
ЖХМС (метод F): m/z 271 (M+H)+ (ES+), на 1,71 мин, УФ-активный.
Растворяли трет-бутил-4-[(2-(этиламино)пропан-2-ил]пиперидин-1-карбоксилат (180 мг, 0,66 ммоль) и триэтиламин (0,27 мл, 19,9 ммоль) в сухом DCM (5,0 мл) в атмосфере азота. При 0 °C добавляли ацетилхлорид (78,0 мг, 0,99 ммоль) и реакционную смесь перемешивали в течение 30 мин при комнатной температуре. Затем реакционную смесь разделяли между насыщ. водным раствором NaHCO3 (20 мл) и EtOAc (15 мл) и проводили дополнительную экстракцию водного слоя EtOAc (2 x 25 мл). Высушивали объединенные органические слои (Na2SO4) и удаляли растворитель под вакуумом с получением неочищенного продукта, который очищали колоночной хроматографией (силикагель для нормальный фазы, от 0 до 65 % EtOAc в гексанах) с получением трет-бутил-4-{2-[ацетил(этил)амино]пропан-2-ил}пиперидин-1-карбоксилата (160 мг, 77 %) в виде смолы.
ЖХМС (метод I): m/z 257 (M+H-56)+ (ES+), на 4,75 мин, УФ-активный.
Растворяли трет-бутил-4-{2-[ацетил(этил)амино]пропан-2-ил}пиперидин-1-карбоксилат (160 мг, 0,51 ммоль) и 4 M HCl в 1,4-диоксане (5 мл) в атмосфере азота и перемешивали вместе в течение 3 ч при комнатной температуре. Реакционную смесь выливали в толуол, а затем растирали с диэтиловым эфиром (2 x 5 мл) и концентрировали под вакуумом с получением промежуточного соединения 140, хлористоводородной соли гидрохлоридной соли N-этил-N-[2-(пиперидин-4-ил)пропан-2-ил]ацетамида (110 мг, 95 %) в виде твердого вещества.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 25
Типовая процедура получения пиперидинов на примере получения промежуточного соединения 144, трифторацетата 4-[1-(1H-пиразол-1-ил)этил]пиперидина
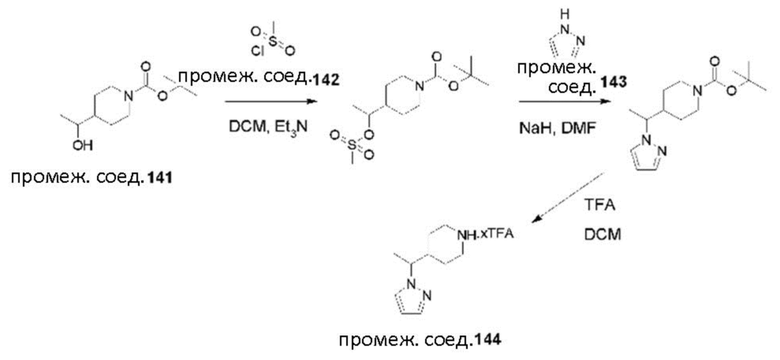
Растворяли промежуточное соединение 141, трет-бутил-4-(1-гидроксиэтил)пиперидин-1-карбоксилат (2,0 г, 8,73 ммоль) и Et3N (3,64 мл, 26,3 ммоль) в дихлорметане (20,0 мл) и охлаждали до 0 °C. По каплям добавляли промежуточное соединение 142, метансульфонилхлорид (0,82 мл, 10,4 ммоль) и реакционную смесь оставляли перемешиваться при 0 °C в течение 2 ч. Реакционную смесь разбавляли водой (100 мл) и экстрагировали DCM (2 x 30 мл). Высушивали объединенные органические слои (Na2SO4) и концентрировали под вакуумом с получением трет-бутил-4-{1-[(метилсульфонил)окси]этил}пиперидин-1-карбоксилата (2,0 г, 75 %) в виде масла. Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
ЖХМС (метод I): m/z 252 (M+H-56)+ (ES+), на 4,51 мин, УФ-активный.
Растворяли промежуточное соединение 143, 1H-пиразол (887 мг, 13,03 ммоль) в DMF (15,0 мл) и охлаждали до 0 °C. Добавляли суспензию 60 % гидрида натрия в минеральном масле (281 мг, 7,0 ммоль) и реакционную смесь оставляли перемешиваться при 0 °C в течение 1 ч. По прошествии 1 ч при 0 oC по каплям при добавляли трет-бутил-4-{1-[(метилсульфонил)окси]этил}пиперидин-1-карбоксилат (2,0 г, 6,51 ммоль) в DMF (1,0 мл) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Смесь разбавляли водой (100 мл) и экстрагировали DCM (2 x 30 мл). Высушивали объединенные органические слои (Na2SO4) и концентрировали под вакуумом с получением неочищенного продукта, который очищали колоночной хроматографией (нейтральный силикагель, нормальная фаза, 60-120 меш, от 0 до 30 % EtOAc в гексане) с получением трет-бутил-4-[1-(1H-пиразол-1-ил)этил]пиперидин-1-карбоксилата (430 мг, 24 %) в виде смолы.
ЖХМС (метод I): m/z 280 (M+H)+ (ES+), на 4,51 мин, УФ-активный.
Растворяли трет-бутил-4-[1-(1H-пиразол-1-ил)этил]пиперидин-1-карбоксилат (430 мг, 1,54 ммоль) в дихлорметане (8,0 мл) и охлаждали до 0 °C. Добавляли TFA (4,0 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Затем концентрировали реакционную смесь под вакуумом с получением неочищенного промежуточного соединения 144, трифторацетата 4-[1-(1H-пиразол-1-ил)этил]пиперидина (450 мг, 100 %) в виде смолы, которое использовали без дополнительной очистки.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 26
Типовая процедура получения пиперидинов на примере получения промежуточного соединения 147, гидрохлорида 4-(1-феноксиэтил)пиперидина
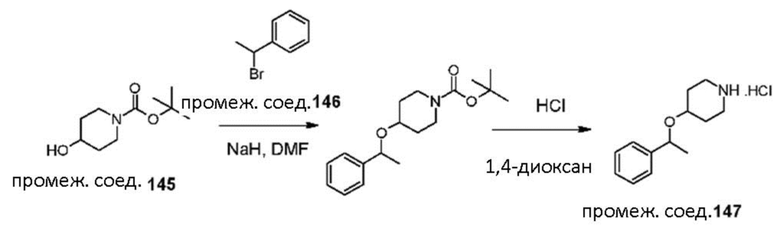
Растворяли промежуточное соединение 145, трет-бутил-4-гидроксипиперидина-1-карбоксилат (543 мг, 2,69 ммоль), в DMF (10 мл), при 0 °C в атмосфере азота порциями добавляли суспензию 60 % гидрида натрия в минеральном масле (183 мг, 4,58 ммоль) и смесь перемешивали при комнатной температуре в течение 1 ч. Через 1 ч по каплям добавляли промежуточное соединение 146, (1-бромэтил)бензол (500 мг, 2,69 ммоль) и полученную реакционную смесь перемешивали в течение 16 ч при 90 °C. Реакционную смесь распределяли между H2O (50 мл) и EtOAc (25 мл) и проводили дополнительную экстракцию водного слоя EtOAc (2 x 25 мл). Объединяли органические слои, высушивали (Na2SO4) и удаляли растворитель под вакуумом с получением неочищенного продукта, который очищали колоночной хроматографией (силикагель для нормальный фазы, от 0 до 15 % EtOAc в гексанах) с получением трет-бутил-4-(1-фенилэтокси)пиперидин-1-карбоксилата (161 мг, 20 %) в виде смолы.
ЖХМС (метод F): m/z 306 (M+H)+ (ES+), на 2,79 мин, УФ-активный.
Растворяли трет-бутил-4-(1-фенилэтокси)пиперидин-1-карбоксилат (160 мг, 5,27 ммоль) при 0 OC в 4 M HCl в 1,4-диоксане (5 мл) и перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь концентрировали, а остаток растирали с диэтиловым эфиром (3 x 10 мл) с получением промежуточного соединения 147, хлористоводородной соли 4-(1-фенилэтокси)пиперидина (100 мг, 89 %) в виде твердого вещества.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 27
Типовая процедура получения пиперидинов на примере получения промежуточного соединения 150, гидрохлорида 4-(бензилсульфанил)пиперидина
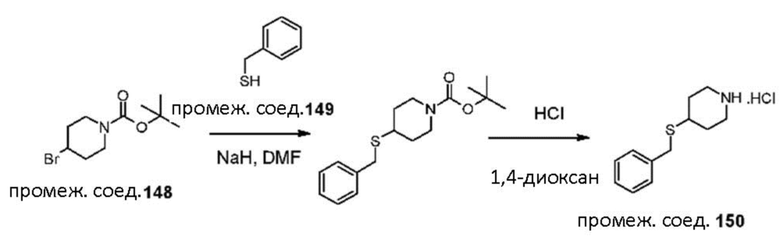
Растворяли промежуточное соединение 149, фенилметантиол (9,6 мг, 81,8 ммоль) в DMF (80 мл), при 0 °C в атмосфере азота порциями добавляли суспензию 60 % гидрида натрия в минеральном масле (3,27 г, 81,8 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 30 мин. Через 30 мин по каплям добавляли промежуточное соединение 148, трет-бутил-4-бромпиперидин-1-карбоксилат (5,4 мг, 20,4 ммоль) и полученную смесь перемешивали в течение 16 ч при комнатной температуре. Реакционную смесь распределяли между H2O (150 мл) и EtOAc (50 мл) и проводили дополнительную экстракцию водного слоя EtOAc (2 x 50 мл). Высушивали объединенные органические слои (Na2SO4) и удаляли растворитель под вакуумом с получением неочищенного продукта, который очищали колоночной хроматографией (силикагель для нормальный фазы, от 0 до 12 % EtOAc в гексанах) с получением трет-бутил-4-(бензилсульфанил)пиперидин-1-карбоксилата (1,59 г, 25 %) в виде смолы.
ЖХМС (метод F): m/z 252 (M+H-56)+ (ES+), на 2,73 мин, УФ-активный.
Растворяли трет-бутил-4-(бензилсульфанил)пиперидин-1-карбоксилат (1,00 г, 3,25 ммоль) при 0 OC в 4 M HCl в 1,4-диоксане (10 мл) и перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь концентрировали, а остаток растирали с диэтиловым эфиром (3 x 10 мл) с получением промежуточного соединения 150, хлористоводородной соли 4-(бензилсульфанил)пиперидина (750 мг, 95 %) в виде смолы.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Путь 28
Процедура получения промежуточного соединения 152, метил-6-оксо-2-азаспиро[3.4]октан-2-карбоксилата
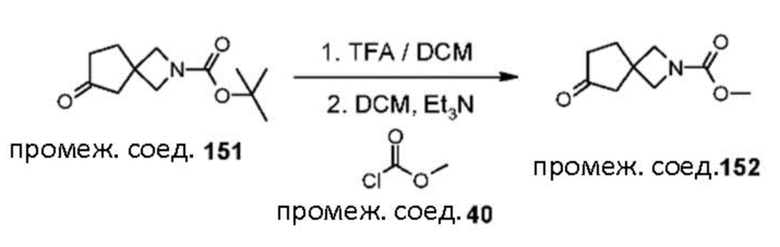
Растворяли промежуточное соединение 151, трет-бутил-6-оксо-2-азаспиро[3.4]октан-2-карбоксилат (120 мг, 0,533 ммоль) в DCM (2,0 мл) при 0 °C и добавляли TFA (1,0 мл). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 2 ч, затем концентрировали под вакуумом. Остаток сушили совместным испарением с диэтиловым эфиром (3 x 10 мл) с получением трифторуксусной соли 2-азаспиро[3.4]октан-6-она (120 мг, 100 %) в виде смолы.
ЖХМС (метод I): m/z 125 (M+H)+ (ES+), на 0,60 мин, УФ-активный.
Растворяли трифторуксусную соль 2-азаспиро[3.4]октан-6-она (60 мг, 0,251 ммоль) в DCM (5 мл) и при 0 °C добавляли триэтиламин (0,2 мл, 1,25 ммоль). При 0 °C добавляли промежуточное соединение 40, метилкарбонохлоридат (94,4 мг, 0,37 ммоль), реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 2 ч. Смесь концентрировали под вакуумом, а остаток распределяли между H2O (25 мл) и EtOAc (25 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 × 10 мл) и высушивали объединенные органические фазы (Na2SO4), а растворитель удаляли под вакуумом с получением промежуточного соединения 152, метил-6-оксо-2-азаспиро[3.4]октан-2-карбоксилата (30 мг, 34 %) в виде масла.
Данные для приведенного в заголовке соединения приведены в табл. 2.
Общие процедуры синтеза в качестве примеров
Путь a
Типовая процедура получения пиперидинов посредством восстановительного аминирования на примере получения примера 2-2, этил-2-{4-[ацетил(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
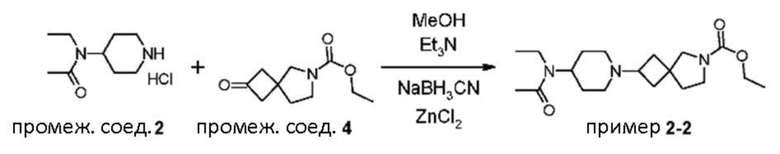
Растворяли промежуточное соединение 2, гидрохлорид N-этил-N-(пиперидин-4-ил)ацетамида (150 мг, 0,9 ммоль), промежуточное соединение 4, этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (170 мг, 0,9 ммоль), Et3N (0,24 мл, 1,9 ммоль) и ZnCl2 (6 мг) в MeOH (10 мл) и реакционную смесь перемешивали при 60 °C в течение 8 ч. Затем смесь охлаждали до 0 °C и добавляли порциями NaBH3CN (72 мг, 1,2 ммоль), после чего смесь перемешивали при 25 °C в течение 17 ч. Растворитель удаляли под вакуумом, а остаток распределяли между H2O (100 мл) и EtOAc (80 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 × 80 мл) и высушивали объединенные органические фазы (Na2SO4), а растворитель удаляли под вакуумом. Остаток очищали препаративной ВЭЖХ [обращенная фаза (X-BRIDGE C-18, 150 × 19 мм, 5 мкм, 15 мл в мин, градиент от 25 % до 100 % (в течение 20 мин), 100 % (в течение 3 мин), затем 30 % (в течение 2 мин), 0,1 % NH3 в MeCN/воде] с получением изомера 1 примера 2-2, этил-2-{4-[ацетил(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (30 мг, 9 %) в виде смолы, и изомера 2 примера 2-2, этил-2-{4-[ацетил(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (25 мг, 7 %) в виде смолы.
Данные для изомера 2 примера 2-12 приведены в табл. 3.
Путь b
Типовая процедура получения пиперидинов посредством восстановительного аминирования с помощью триацетоксиборогидрида натрия на примере получения примера 2-12, этил-2-(4-{[ацетил(циклопропил)амино]метил}пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
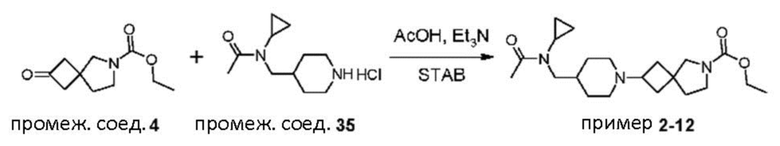
Растворяли промежуточное соединение 4, этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (99 мг, 0,5 ммоль), и промежуточное соединение 35, гидрохлорид N-циклопропил-N-(пиперидин-4-илметил)ацетамида (116 мг, 0,5 ммоль) в DCM (10 мл) при кт и добавляли Et3N (0,35 мл, 2,5 ммоль). Смесь перемешивали в течение 30 мин перед добавлением AcOH (0,29 мл, 5,0 ммоль). Смесь перемешивали в течение 3 ч, затем добавляли STAB (265 мг, 1,3 ммоль) и перемешивали смесь при кт в течение ночи. Реакционную смесь гасили добавлением водн. р-ра Na2CO3 (20 мл) и добавляли твердый Na2CO3 для обеспечения основности водного слоя. Проводили экстракцию полученной смеси DCM (4 x 20 мл) и объединяли органические слои, высушивали (MgSO4), фильтровали и удаляли растворители под вакуумом. Остаток очищали колоночной хроматографией (нормальная фаза, [картридж Biotage SNAP KP-sil 10 г, 40-63 мкм, 60 Å, 20 мл в мин, градиент от 0 % до 10 % MeOH в DCM]) с получением неразделяемой смеси диастереоизомеров. Очищали эту смесь препаративной обращенно-фазовой ВЭЖХ (колонка Phenomenex Gemini-NX 5 мкм C18 110A Axia, 100 x 30 мм, элюированием от 30 до 60 % MeCN/растворитель B в течение 14,4 мин при 30 мл/мин [где растворитель B состоит из 0,2 % (28 % NH3/H2O) в H2O] и собиранием фракций с отслеживанием при 205 нм) с получением изомера 1 примера 2-12, этил-2-(4-{[ацетил(циклопропил)амино]метил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата (42 мг, 22 %) в виде твердого вещества, и изомера 2 примера 2-12 этил-2-(4-{[ацетил(циклопропил)амино]метил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата (36 мг, 19 %) в виде твердого вещества.
Данные для изомера 2 примера 2-12 приведены в табл. 3.
Путь c
Типовая процедура получения пиперидинов посредством использования защищенного кетона на примере получения примера 2-23, этил-2-(4-{ацетил[(3-метоксиэтан-3-ил)метил]амино}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата
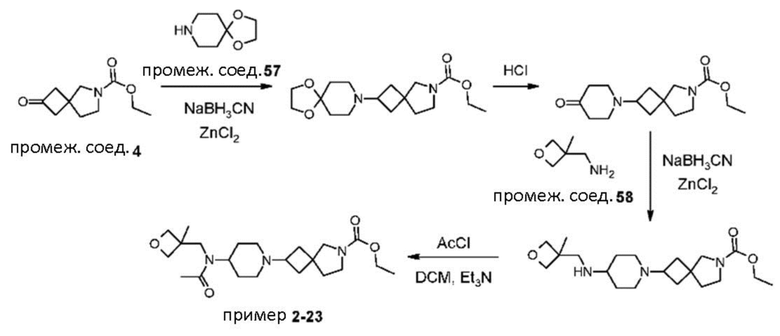
Растворяли промежуточное соединение 57, 1,4-диокса-8-азаспиро[4.5]декан (1,0 г, 6,99 ммоль), в метаноле (20 мл) и добавляли промежуточное соединение 4, 2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (1,38 г, 6,99 ммоль), триэтиламин (2,9 мл, 20,9 ммоль) и ZnCl2 (95 мг, 0,70 ммоль), а затем реакционную смесь перемешивали при 65 °C в течение 8 ч. Добавляли порциями NaBH3CN (1,32 г, 20,9 ммоль) и полученную реакционную смесь перемешивали при 25 °C в течение 17 ч. Растворители удаляли под вакуумом, а остаток распределяли между H2O (120 мл) и EtOAc (100 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 × 100 мл), объединяли органические слои, высушивали (Na2SO4), а растворитель удаляли под вакуумом. Остаток очищали растиранием с пентаном и декантировали растворители с получением этил-2-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-6-азаспиро[3.4]октан-6-карбоксилата (1,80 г, 79 %) в виде смолы.
ЖХМС (метод I): m/z 325 (M+H)+ (ES+), на 3,54 мин и 3,69 мин, УФ-активный.
Растворяли этил-2-(1,4-диокса-8-азаспиро[4.5]дец-8-ил)-6-азаспиро[3.4]октан-6-карбоксилат (1,80 г, 5,55 ммоль) в этаноле (20 мл) и добавляли 4,0 M HCl в 1,4-диоксане (30 мл). Полученную реакционную смесь перемешивали при 70 °C в течение 18 ч, а затем защелачивали насыщ. раствором бикарбоната натрия. Растворители удаляли под вакуумом, а остаток распределяли между H2O (100 мл) и EtOAc (80 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 × 80 мл) и высушивали объединенные органические слои (Na2SO4). Растворитель удаляли под вакуумом, а остаток очищали растиранием с пентаном с получением этил-2-(4-оксопиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата (1,20 г, 77 %) в виде смолы.
ЖХМС (метод I): m/z 281 (M+H)+ (ES+), на 3,30 мин и 3,41 мин, УФ-активный.
Растворяли промежуточное соединение 58, 1-(3-метилоксетан-3-ил)метанамин (72 мг, 0,72 ммоль), этил-2-(4-оксопиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат (200 мг, 0,71 ммоль), триэтиламин (0,4 мл, 2,85 ммоль) и ZnCl2 (9 мг, 0,07 ммоль) в MeOH (10 мл) и реакционную смесь перемешивали при 65 °C в течение 8 ч. Смесь охлаждали до 0 °C и порциями добавляли NaBH3CN (134 мг, 2,14 ммоль). Полученную реакционную смесь перемешивали при 25 °C в течение 17 ч. Растворители удаляли под вакуумом, а остаток распределяли между H2O (80 мл) и EtOAc (60 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 × 60 мл) и высушивали объединенные органические слои (Na2SO4), а растворитель удаляли под вакуумом. Остаток очищали растиранием с пентаном (3 x 1 мл) с получением этил-2-(4-{[(3-метилоксетан-3-ил)метил]амино}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата (210 мг, 81 %) в виде смолы.
ЖХМС (метод I): m/z 366 (M+H)+ (ES+), на 3,63 мин и 3,81 мин, УФ-активный.
Растворяли этил-2-(4-{[(3-метилоксетан-3-ил)метил]амино}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат (200 мг, 0,55 ммоль) в DCM (10 мл), добавляли триэтиламин (0,2 мл, 1,64 ммоль) и реакционную смесь перемешивали при 0 °C в течение 20 мин. Порциями добавляли ацетилхлорид (0,06 мл, 0,82 ммоль) и полученную смесь перемешивали при 25 °C в течение 2 ч. Удаляли растворители под вакуумом, а остаток распределяли между H2O (80 мл) и EtOAc (60 мл) и проводили дополнительную экстракцию водного слоя EtOAc (2 × 60 мл). Высушивали объединенные органические слои (Na2SO4), удаляли растворитель под вакуумом, а остаток очищали препаративной ВЭЖХ [обращенная фаза (X-BRIDGE C-18, 250 × 19 мм, 5 мкм, 15 мл в мин, градиент от 5 % до 30 % (в течение 36 мин), 30 % (в течение 9 мин), 100 % (в течение 5 мин), затем 5 % (в течение 5 мин), подвижная фаза (A) 5 мМ бикарбоната аммония + 0,1 % аммиак в воде и (B) 100 % ацетонитрил] с получением изомера 1 примера 2-23, этил-2-(4-{ацетил[(3-метилоксетан-3-ил)метил]амино}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата (47 мг, 21 %) в виде жидкости, и изомера 2 примера 2-23, этил-2-(4-{ацетил[(3-метилоксетан-3-ил)метил]амино}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата (45 мг, 20 %) в виде смолы.
Данные для изомера 2 примера 2 -23 приведены в табл. 3.
Путь d
Типовая процедура получения пиперидинов посредством нуклеофильного замещения на 4-нитрофенилкарбамате на примере получения примера 2-38, (1,1-2H2)-этил-2-{4-[ацетил(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
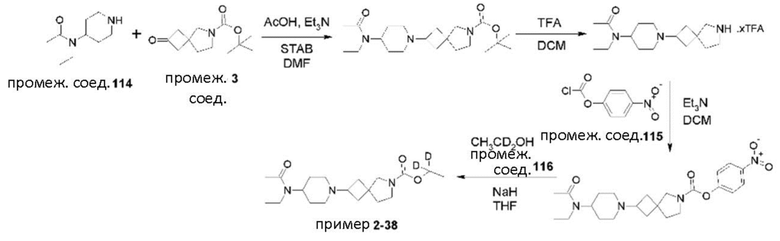
Смешивали промежуточное соединение 114, N-этил-N-(пиперидин-4-ил)ацетамид (1,70 г, 10 ммоль), и промежуточное соединение 3, трет-бутил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (2,25 г, 10 ммоль) в DMF (40 мл) в атмосфере азота. Добавляли AcOH (0,86 мл, 15 ммоль) и STAB (4,24 г, 20 ммоль) и полученную смесь перемешивали при кт в течение 6 дней. Смесь концентрировали для удаления DMF, а остаток обрабатывали толуолом и концентрировали для удаления AcOH. Остаток растворяли в MeOH и концентрировали на флэш-силикагеле (15 мл). Полученный порошок очищали колоночной хроматографией (нормальная фаза, [картридж Biotage SNAP KP-sil 100 г, 40-63 мкм, 60 Å, 40 мл в мин, градиент от 0 % до 10 % растворителя A в DCM в течение 15 CV, где растворитель A состоит из 10 % {7 M NH3 в MeOH} в MeOH]) с получением трет-бутил-2-{4-[ацетил(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата в виде смолы (2,92 г, 77 %).
ЖХМС (метод D): m/z 380 (M+H)+ (ES+), на 2,11 мин, УФ-активный.
Растворяли трет-бутил-2-{4-[ацетил(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат (2,80 г, 7,38 ммоль) в смеси DCM (50 мл) и TFA (50 мл) в атмосфере азота и перемешивали при кт в течение 3,5 ч. Смесь разбавляли толуолом и концентрировали. Разбавляли маслянистый остаток толуолом и концентрировали с получением трифторуксусной соли N-[1-(6-азаспиро[3.4]окт-2-ил)пиперидин-4-ил]-N-этилацетамида в виде смолы (5,73 г, предположительно 100 %).
ЖХМС (метод D): m/z 280 (M+H)+ (ES+), на 1,67 мин и 1,79 мин, слабо УФ-активный.
Растворяли трифторуксусную соль N-[1-(6-азаспиро[3.4]окт-2-ил)пиперидин-4-ил]-N-этилацетамида (5,73 г, предположительно 7,38 ммоль) в DCM (140 мл) в атмосфере азота. Добавляли Et3N (5,1 мл, 36,6 ммоль) и промежуточное соединение 115, 4-нитрофенилкарбонохлоридат (1,78 г, 8,83 ммоль), и полученную смесь перемешивали при кт в течение ночи. Добавляли больше Et3N (2 мл, 14,3 ммоль) и промежуточного соединения 115, 4-нитрофенилкарбонохлоридата (0,74 г, 3,67 ммоль), и смесь перемешивали при кт в течение 3 дней. Реакционную смесь концентрировали на флэш-силикагеле (15 мл) и полученный порошок очищали колоночной хроматографией (нормальная фаза, [картридж Biotage SNAP KP-sil 100 г, 40-63 мкм, 60 Å, 40 мл в мин, градиент от 0 % до 5 % растворителя A в DCM в течение 10 CV, где растворитель A состоит из 10 % {7 M NH3 в MeOH} в MeOH]) с получением масла, которое дополнительно очищали колоночной хроматографией (нормальная фаза, [картридж Biotage SNAP KP-sil 100 г, 40-63 мкм, 60 Å, 40 мл в мин, изократический 5 % EtOAc A в DCM в течение 5 CV, затем изократический 5 % растворитель A в DCM в течение 5 CV, где растворитель A состоит из 10 % {7 M NH3 в MeOH} в MeOH]) с получением масла (6,85 г). Масло растворяли в DCM, промывали H2O (x 2), высушивали пропусканием через картридж для разделения фаз и концентрировали с получением 4-нитрофенил-2-{4-[ацетил(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата в виде пены (2,41 г, 73 %).
ЖХМС (метод C): m/z 445 (M+H)+ (ES+), на 1,32 мин, УФ-активный.
Растворяли промежуточное соединение 116, (1,1-2H2)-этанол (0,42 мл, 7,19 ммоль) в THF (18 мл) в атмосфере азота и обрабатывали 60 % суспензией гидрида натрия в минеральном масле (0,29 г, 7,25 ммоль). Смесь перемешивали при кт в течение 1 ч, затем добавляли 4-нитрофенил-2-{4-[ацетил(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат (0,800 г, 1,80 ммоль) и полученную смесь перемешивали при кт в течение 4 дней. Реакционную смесь концентрировали для удаления THF, затем остаток разбавляли H2O и экстрагировали EtOAc (x 2). Объединенные органические фазы пропускали через картридж для разделения фаз и концентрировали на флэш-силикагеле (10 мл). Полученный порошок очищали колоночной хроматографией (нормальная фаза, [картридж Biotage SNAP KP-sil 50 г, 40-63 мкм, 60 Å, 40 мл в мин, изократический 20 % EtOAc в DCM в течение 5 CV, градиент 20 % EtOAc в DCM до 10 % растворителя A в DCM в течение 1 CV, изократический 10 % растворитель A в DCM в течение 10 CV, где растворитель A состоит из 10 % {7 M NH3 в MeOH} в MeOH]) с получением неразделяемой смеси диастереоизомеров (0,359 г, 56 %). Очищали эту смесь препаративной обращенно-фазовой ВЭЖХ (колонка Phenomenex Gemini-NX 5 мкм C18 110A Axia, 100 x 30 мм, элюированием от 20 до 50 % MeCN/растворитель B в течение 14,4 мин при 30 мл/мин [где растворитель B состоит из 0,2 % (28 % NH3/H2O) в H2O] и собиранием фракций с отслеживанием при 205 нм) с получением изомера 1 примера 2-38, (1,1-2H2)-этил-2-{4-[ацетил(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (140 мг, 22 %) в виде смолы, и изомера 2 примера 2-38 (1,1-2H2)-этил-2-{4-[ацетил(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (129 мг, 20 %) в виде смолы.
Данные для изомера 2 примера 2-38 приведены в табл. 3.
Путь e
Типовая процедура получения пиперидинов, в которой смесь более двух изомеров разделяется использованием обращенно-фазовой хроматографии с последующей хиральной хроматографией на примере получения примера 2-63, этил-2-(4-{1-[ацетил(этил)амино]пропил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата
Во флакон помещали промежуточное соединение 138, трифторацетат N-этил-N-[1-(пиперидин-4-ил)пропил]ацетамид (250 мг, 1,18 ммоль), промежуточное соединение 4, этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (232 мг, 1,18 ммоль), Et3N (0,821 мл, 5,89 ммоль), ZnCl2 (0,3 мл) и MeOH (5 мл). Полученную смесь нагревали при 60 °C в течение 4 ч, затем охлаждали до 0 °C. Добавляли при 0 °C NaBH3CN (222 мг, 3,53 ммоль) и смеси давали нагреться до комнатной температуры и перемешивали в течение 16 ч. Смесь концентрировали под вакуумом, а остаток распределяли между H2O (50 мл) и EtOAc (50 мл). Проводили дополнительную экстракцию водного слоя EtOAc (2 × 50 мл), высушивали объединенные органические слои (Na2SO4) и удаляли растворитель под вакуумом с получением неочищенного продукта, который очищали препаративной ВЭЖХ [обращенно-фазовая ВЭЖХ (X-BRIDGE, 250 × 19 мм, 5 мкм, 15 мл в мин, градиент 48 % (в течение 60 мин), 100 % (в течение 2 мин), затем 48 % (в течение 3 мин), (A) 10 мМ бикарбонат аммония в воде + 0,1 % аммиак в воде, (B) 50:50 (MeCN:MeOH)] с получением двух изомеров – изомера 1 и изомера 2.
В свою очередь эти два изомера дополнительно очищали хиральной препаративной ВЭЖХ [CHIRALCEL OX-H 250 x 4,6 мм, 5 мкм {0,3 % DEA в IPA:MeOH (50:50)} с получением изомера 1a примера 2-63, этил-2-{4-[(2-гидроксиэтил)(фенил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат (13 мг, 3 %) в виде смолы, изомера 1b примера 2-63, этил-2-{4-[(2-гидроксиэтил)(фенил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат (10 мг, 2 %) в виде смолы, изомера 2a примера 2-63, этил-2-{4-[(2-гидроксиэтил)(фенил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат (11 мг, 2 %) в виде смолы и изомера 2b примера 2-63, этил-2-{4-[(2-гидроксиэтил)(фенил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат (10 мг, 2 %) в виде смолы.
Данные для изомера 2b примера 2-63 приведены в табл. 3.
Путь f
Типовая процедура получения пиперидинов, в которой смесь более двух изомеров разделяется использованием обращенно-фазовой хроматографии с последующей хиральной хроматографией на примере получения примера 2-65, этил-2-{4-[1-1H-пиразол-1-ил)этил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
Растворяли промежуточное соединение 144, трифторуксусную соль 4-[1-(1H-пиразол-1-ил)этил]пиперидина (430 мг, 2,40 ммоль), и Et3N (1,6 мл, 12,0 ммоль) в метаноле (10 мл). Добавляли промежуточное соединение 4, этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (473 мг, 2,40 ммоль), добавляли ZnCl2 (0,12 мл, 0,12 ммоль) и реакционную смесь перемешивали при 70 °C в течение 5 ч. Реакционную смесь охлаждали до 0 °C, добавляли NaBH3CN (452 мг, 7,21 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь концентрировали под вакуумом, разбавляли водой (30 мл) и экстрагировали этилацетатом (2 x 10 мл). Объединенные органические фазы промывали солевым раствором, высушивали над Na2SO4 и концентрировали под вакуумом. Неочищенный продукт очищали препаративной ВЭЖХ [X-BRIDGE, (250 x 19 мм), 5 мкм, 12 мл в мин, градиент от 30 % до 100 % (в течение 120 мин), затем 100 % (5 мин), [5 мМ бикарбонат аммония в воде / MeCN:MeOH (50:50)] с получением двух изомеров – изомера 1 и изомера 2.
Изомер 1 дополнительно очищали хиральной СФХ [Chiral CEL OX-H (250 x 4,6 мм) 5 мкм, сорастворитель: 15 % из 0,3 % DEA в IPA:MeOH (50 : 50) с получением изомера 1a пример 2-65, этил-2-{4-[1-(1H-пиразол-1-ил)этил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат (17 мг, 28 %), и изомера 1b пример 2-65, этил-2-{4-[1-(1H-пиразол-1-ил)этил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат (25 мг, 42 %).
Данные для изомера 1a примера 2-65 приведены в табл. 3.
Изомер 2 дополнительно очищали хиральной СФХ [Chiral PAK ADH (250 x 4,6 мм) 5 мкм, сорастворитель: 35 % из 0,3 % DEA в MeOH с получением изомера 2a примера 2-65, этил-2-{4-[1-(1H-пиразол-1-ил)этил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат (19 мг, 32 %), и изомера 2b примера 2-65, этил-2-{4-[1-(1H-пиразол-1-ил)этил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат (21 мг, 35 %).
Данные для изомера 2a примера 2-65 приведены в табл. 3.
Путь g
Типовая процедура получения пиперидинов, содержащих систему колец 2-азаспиро[3.4]октана, при которой смесь двух изомеров разделяется использованием обращенно-фазовой хроматографии с последующей хиральной хроматографией на примере получения примера 3-2, метил-6-{4-[ацетил(этил)амино]пиперидин-1-ил}-2-азаспиро[3.4]октан-2-карбоксилата
Растворяли промежуточное соединение 2, гидрохлорид N-этил-N-(пиперидин-4-ил)ацетамида (150 мг, 0,818 ммоль), промежуточное соединение 152, метил-6-оксо-2-азаспиро[3.4]октан-6-карбоксилат (139 мг, 0,818 ммоль), триэтиламин (0,342 мл, 2,45 ммоль) и ZnCl2 (1,0 M раствор в диэтиловом эфире, 0,2 мл, 0,2 ммоль) в MeOH (100 мл) и смесь перемешивали при 60 °C в течение 8 ч. Затем смесь охлаждали до 0 – 5 °C и порциями добавляли NaBH3CN (154 мг, 2,45 ммоль). Полученную реакционную смесь перемешивали при 25 °C в течение 17 ч, затем растворители удаляли под вакуумом. Остаток распределяли между H2O (100 мл) и EtOAc (100 мл) и проводили дополнительную экстракцию водного слоя EtOAc (2 × 50 мл). Высушивали объединенные органические слои (Na2SO4) и растворитель удаляли под вакуумом. Остаток очищали препаративной ВЭЖХ [обращенная фаза (PHENYL HEXYL, 250 × 19 мм, 5 мкм, 14 мл в мин, градиент от 35 % (в течение 9 мин), 100 % (в течение 2 мин), затем 35 % (в течение 2 мин), A: 0,1 % аммиак в воде, B: 100 % MeCN] с получением метил-6-{4-[ацетил(этил)амино]пиперидин-1-ил}-2-азаспиро[3.4]октан-2-карбоксилата (65 мг, 22 %) в виде смолы. Ее дополнительно очищали хиральной препаративной ВЭЖХ (CHIRALPAK AD-H 250 x 4,6 мм, 5 мкм, сорастворитель 0,3 % диэтиламин в MeOH с получением изомера 1 примера 3-2, метил-6-{4-[ацетил(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (27 мг, 9 %) в виде смолы, и изомера 2 примера 3-2, метил-6-{4-[ацетил(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (31 мг, 11 %) в виде смолы.
Данные для изомера 2 примера 3-2 приведены в табл. 3.
Таблица 2
Данные характеристик и коммерческие источники для исходных материалов и промежуточных соединений
CAS: 79099-07-3
CAS: 203661-71-6
CAS: 541-41-3
CAS: 19099-93-5
CAS: 765-30-0
CAS: 2516-34-9
CAS: 96-32-2
CAS: 57684-71-6
CAS: 1181816-12-5
CAS: 753-90-2
CAS: 75-07-0
CAS: 6482-24-2
CAS: 74-89-5
CAS: 75-31-0
CAS:79-03-8
CAS: 1363382-39-1
CAS: 407-25-0
CAS: 137076-22-3
CAS: 206989-61-9
CAS: 79-22-1
и промежуточное соединение 52
CAS: 75-04-7
CAS:107-10-8
CAS: 78-81-9
CAS: 2516-47-4
CAS: 1245647-53-3
CAS: 159874-38-1
CAS: 174886-05-6
CAS: 177-11-7
CAS: 153209-97-3
CAS: 430-67-1
CAS: 753-90-2
CAS: 879121-42-3
CAS: 421-00-1
CAS: 791061-00-2
CAS: 89282-70-2
CAS: 1443980-50-4
CAS: 180205-28-1
CAS: 67-62-9
CAS: 62-53-3
CAS: 504-29-0
CAS: 100-46-9
CAS: 4530-20-5
CAS: 6638-79-5
CAS: 25952-53-8
CAS: 123333-53-9
CAS: 16853-85-3
CAS: 36635-61-7
CAS: 87120-72-7
CAS: 65373-52-6
CAS: 79-44-7
CAS: 460-08-2
CAS: 430-67-1
CAS: 21635-88-1
CAS: 872-50-4
CAS: 6226-25-1
CAS: 540-51-2
CAS: 139062-99-0
CAS: 7693-46-1
CAS: 1859-09-2
CAS: 109-85-3
CAS: 151-18-8
CAS: 49773-20-8
CAS: 75-03-6
CAS: 105-36-2
CAS: 95353-04-1
CAS: 419571-73-6
CAS: 530116-33-7
CAS: 183170-69-6
CAS: 124-63-0
CAS: 288-13-1
CAS: 109384-19-2
CAS: 585-71-7
CAS: 180695-79-8
CAS: 100-53-8
CAS: 1363382-39-1
БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ
ПРИМЕР А
Анализы на фосфо-ERK1/2
Функциональные анализы выполняли с использованием анализа на фосфо-ERK1/2 Alphascreen Surefire (Crouch & Osmond, Comb. Chem. High Throughput Screen, 2008). Фосфорилирование ERK1/2 представляет собой следующую последовательность активации рецепторов, связанных с белками Gq/11 и Gi/o, что делает ее более подходящей для оценки рецепторов M1, M3 (связанных с Gq/11) и M2, M4 (связанных с Gi/o), чем использование различных форматов анализов для других подтипов рецепторов. Клетки CHO, стабильно экспрессирующие мускариновые рецепторы M1, M2, M3 или M4 человека, высевали на 96-луночные планшеты для культивирования клеток (25K / лунку) в MEM-альфа + 10 % диализированной FBS. После прикрепления клетки голодали без сыворотки в течение ночи. Стимуляцию агонистами выполняли добавлением к клеткам 5 мкл агониста в течение 5 мин (37 °C). Среду удаляли и добавляли 50 мкл буферного раствора для лизиса. Через 15 мин переносили 4 мкл образца в 384-луночный планшет и добавляли 7 мкл смеси для выявления. Планшеты инкубировали в течение 2 ч с осторожным перемешиванием в темноте, а затем считывали на устройство считывания планшетов PHERAstar. По полученным данным рассчитывали значения pEC50 и Emax для каждого подтипа рецепторов.
Результаты представлены ниже в таблице 4.
Для каждого примера, содержащего систему колец 6-азаспиро[3.4]октана, существует два диастереоизомера, которые, если не указано иное, разделяли и на основании их времени удерживания при анализе ЖХМС обозначали (изомер 1, изомер 2). В большинстве примеров, изомер 1 не активен. Если существуют дополнительные (хиральные) изомеры, иногда их разделяли и обозначали (изомер 1a, изомер 1b) на основании их времени удерживания при хиральном разделении.
Для каждого примера, содержащего систему колец 2-азаспиро[3.4]октана, существует два диастереоизомера, которые, если не указано иное, разделяли и на основании их времени удерживания при хиральном разделении обозначали (изомер 1, изомер 2).
Данные анализа для активных изомеров представлены в табл. 3. Данные для нескольких слабо активных соединений включены в табл. 4 для выделения преимущества абсолютной стереохимической формы.
(% Emax cf. ACh)
(% Emax cf. ACh)
(% Emax cf. ACh)
(% Emax cf. ACh)
ПРИМЕР B
Влияние нового исследуемого соединения и ксаномелина на индуцированную d-амфетамином гиперактивность у крыс
Цель исследования заключается в проверке влияния нового исследуемого соединения на индуцированную d-амфетамином гиперактивность у крыс. Шизофрения представляет собой сложное многофакторное заболевание, которое не может быть полностью представленным одной экспериментальной процедурой. У крыс оценивали антипсихотически подобное поведение путем ингибирования гиперактивности (или гиперлокомоции), вызванной d-амфетамином. Эта процедура чувствительна к клинически значимым антагонистам рецепторов дофамина и поэтому считается подходящей для сравнения мускариновых антагонистов, которые воздействуют на передачу дофаминэргических сигналов. Дозу ксаномелина, для которой раннее наблюдали значимое уменьшение индуцированной d-амфетамином гиперактивности, применяли в качестве положительного контроля. Статистический анализ, как правило, включал трехфакторный ковариационный анализ или устойчивую регрессию, связанную с лечением, сутками и стойкой, как факторами, и активностью в течение 30 минут перед лечением в качестве ковариаты с последующими соответствующими тестами множественного сравнения. Статистически значимым считали P-значение <0,05 и обозначали его соответственно на всех последующих фигурах.
Данные для изомера 2 примера 2-1 показаны на фигуре 1.
ПРИМЕР C
ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ
(i) Состав таблетки
Таблетированную композицию, содержащую соединение формулы (1) или формулы (1a), готовили путем смешения 50 мг соединения с 197 мг лактозы (BP), в качестве разбавителя, и 3 мг стеарата магния, в качестве смазывающего вещества, и прессования в форму таблетки известным путем.
(ii) Состав капсулы
Капсульный состав готовили смешиванием 100 мг соединения формулы (1) или формулы (1a) со 100 мг лактозы и необязательно 1 % по массе стеарата магния и наполняли полученной смесью стандартные непрозрачные твердые желатиновые капсулы.
Эквиваленты
Предыдущие примеры представлены с целью иллюстрации данного изобретения и не предназначены для какого-либо ограничения объема данного изобретения. Будет совершенно очевидно, что могут быть сделаны многочисленные модификации и изменения конкретных вариантов реализации изобретения, описанных выше и проиллюстрированных в примерах, без отступления от принципов, лежащих в основе данного изобретения. Предполагается, что все такие модификации и изменения охватываются этой заявкой.
| название | год | авторы | номер документа |
|---|---|---|---|
| Бициклические азотсодержащие соединения как агонисты М1 мускариновых рецепторов | 2019 |
|
RU2811601C1 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2767857C2 |
| Бициклические азотсодержащие соединения как агонисты М1 мускариновых рецепторов | 2015 |
|
RU2685230C2 |
| ОКСАДИАЗОЛЫ В КАЧЕСТВЕ АГОНИСТОВ МУСКАРИНОВОГО РЕЦЕПТОРА M1 И/ИЛИ M4 | 2019 |
|
RU2817018C2 |
| ПРОИЗВОДНЫЕ 5-ОКСА-2-АЗАСПИРО[3.4]ОКТАНА В КАЧЕСТВЕ АГОНИСТОВ M4 | 2020 |
|
RU2820477C1 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ АМИНОВ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА ЕР4 | 2011 |
|
RU2565596C2 |
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2017 |
|
RU2799820C2 |
| ИНГИБИТОРЫ ПРОТЕИНТИРОЗИНФОСФАТАЗЫ | 2020 |
|
RU2799449C2 |
| НОВЫЕ СУЛЬФОНАМИДКАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ | 2018 |
|
RU2808572C2 |
| СПИРО-КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ПИПЕРИДИНА ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КАЛИЕВОГО КАНАЛА НАРУЖНОГО МЕДУЛЛЯРНОГО СЛОЯ | 2013 |
|
RU2642066C2 |
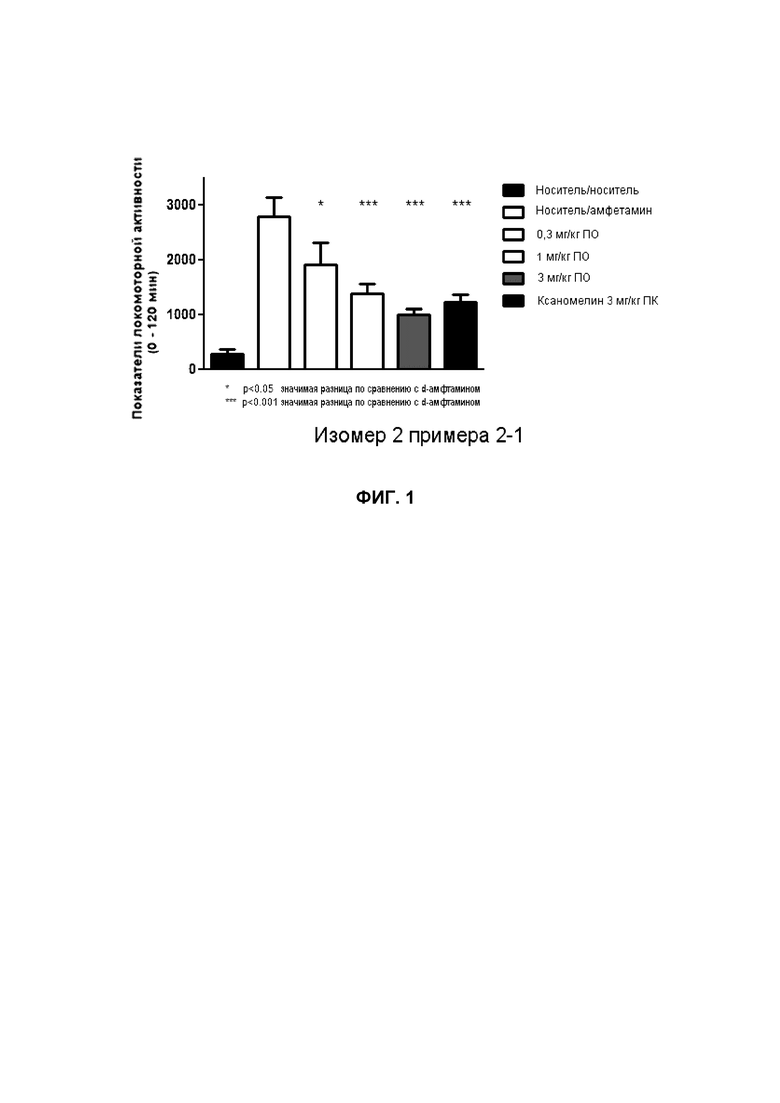
Данное изобретение относится к соединениям формулы (1a) или его фармацевтически приемлемой соли, где q равно 1; r равно 1 или 2; s равно 0 или 1, причем сумма r и s равна 1 или 2; Q представляет собой CR1R2NR5R6, NR5R6, OR7, SR7; R1 выбран из водорода или C1-6 алкильной группы; R2 выбран из водорода или C1-6 алкильной группы; R3 представляет собой водород; R4 представляет собой водород или C1-6 алкильную группу; R5 выбран из OR7; C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть заменены гетероатомом, выбранным из O, N и S и их окисленных форм; и группы W или CH2W, где W представляет собой 5- или 6-членное ароматическое кольцо, содержащее 0, 1, 2 или 3 гетероатома, выбранных из O и N; R6 выбран из COR7; COOR7; CH2COR7; CH2COOR7; C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть заменены гетероатомом O; или C3-6 циклоалкильной группы; и R7 выбран из водорода, C1-6 алкильной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один атом углерода необязательно может быть заменен гетероатомом N; и в случае, когда Q представляет собой группу OR7 или SR7, R7 выбран из группы W или (C1-4 алкил)W, где W представляет собой фенил; а пунктирная линия, которая показывает необязательную вторую углерод-углеродную связь, отсутствует. Кроме того, предложены конкретные соединения, фармацевтические композиции, содержащие соединения формулы (1а) или конкретные указанные соединения. Технический результат: получены и описаны новые соединения, которые являются агонистами мускаринового рецептора и/или M4 и пригодны для лечения заболеваний, опосредованных мускариновыми рецепторами M1/M4. 3 н. и 19 з.п. ф-лы, 1 ил., 4 табл.
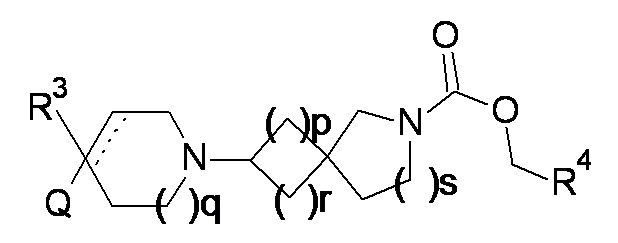 (1а)
(1а)
1. Соединение формулы (1):
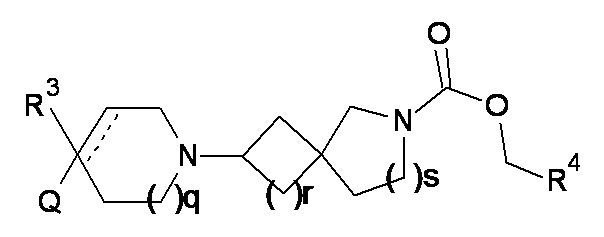 (1)
(1)
или его фармацевтически приемлемая соль, где
q равно 1;
r равно 1 или 2;
s равно 0 или 1, причем сумма r и s равна 1 или 2;
Q представляет собой CR1R2NR5R6, NR5R6, OR7, SR7;
R1 выбран из водорода или C1-6 алкильной группы;
R2 выбран из водорода или C1-6 алкильной группы;
R3 представляет собой водород;
R4 представляет собой водород или C1-6 алкильную группу;
R5 выбран из OR7; C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть заменены гетероатомом, выбранным из O, N и S и их окисленных форм; и группы W или CH2W, где W представляет собой 5- или 6-членное ароматическое кольцо, содержащее 0, 1, 2 или 3 гетероатома, выбранных из O и N;
R6 выбран из COR7; COOR7; CH2COR7; CH2COOR7; C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все атомы углерода углеводородной группы необязательно могут быть заменены гетероатомом O; или C3-6 циклоалкильной группы; и
R7 выбран из водорода, C1-6 алкильной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один атом углерода необязательно может быть заменен гетероатомом N; и в случае, когда Q представляет собой группу OR7 или SR7, R7 выбран из группы W или (C1-4 алкил)W, где W представляет собой фенил;
а пунктирная линия, которая показывает необязательную вторую углерод-углеродную связь, отсутствует.
2. Соединение по п. 1, отличающееся тем, что Q представляет собой NR5R6.
3. Соединение по п. 1, отличающееся тем, что Q представляет собой CR1R2NR5R6.
4. Соединение по любому одному из пп. 1-3, отличающееся тем, что R1 выбран из водорода, метила или этила.
5. Соединение по любому одному из пп. 1-4, отличающееся тем, что R2 выбран из водорода, метила или этила.
6. Соединение по любому одному из пп. 1-5, отличающееся тем, что R5 выбран из метила, этила, пропила, изопропила, циклопропила, фторэтила, дифторэтила, бутила или циклобутила.
7. Соединение по любому одному из пп. 1-5, отличающееся тем, что R5 представляет собой группу W или CH2W, где W представляет собой фенильное, пиридильное или изоксазольное кольцо.
8. Соединение по любому одному из пп. 1-7, отличающееся тем, что R6 выбран из метила, этила, трифторэтила, гидроксиэтила или метоксиэтила.
9. Соединение по любому одному из пп. 1-7, отличающееся тем, что R6 выбран из COR7; COOR7; CH2COR7; CH2COOR7, где R7 выбран из H, метила, фторметила, дифторметила, трифторметила, этила, фторэтила, дифторэтила или трифторэтила.
10. Соединение по п. 1, отличающееся тем, что фрагмент:
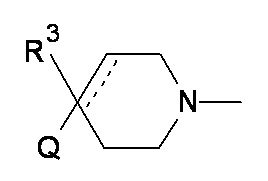
выбран из групп от A до KKK:
A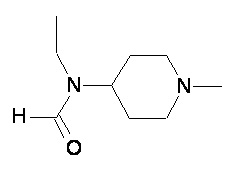
B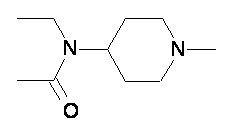
C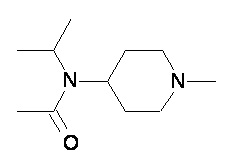
D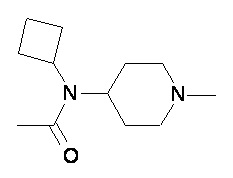
E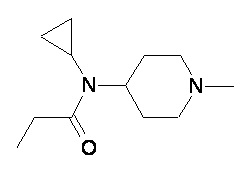
F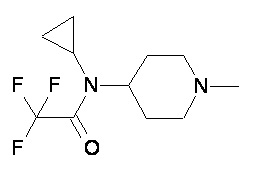 G
G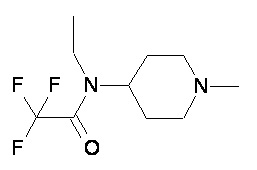
H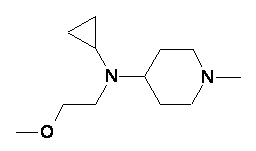
I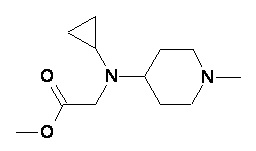 J
J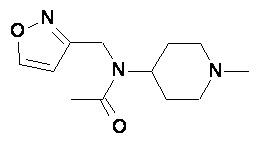
K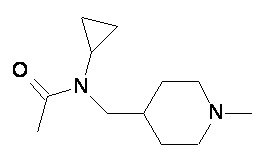 L
L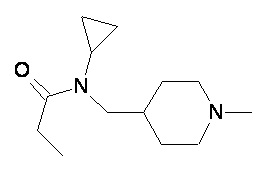 M
M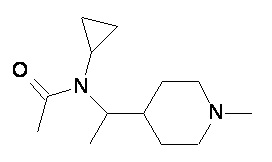
N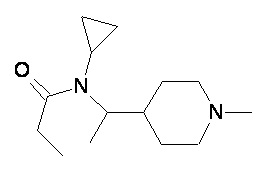 O
O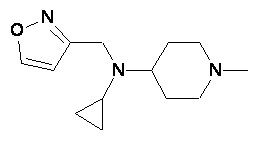 P
P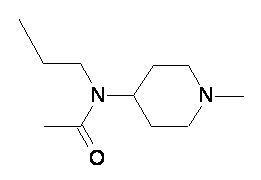
Q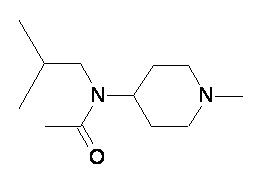
R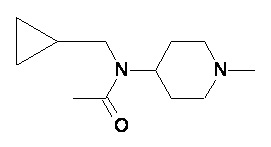
S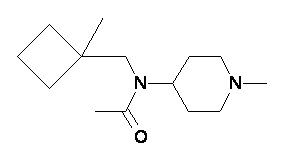
T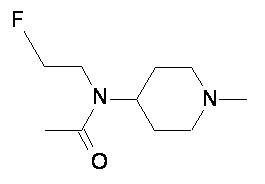
U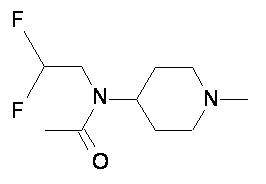 V
V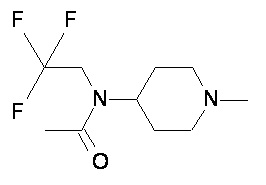
W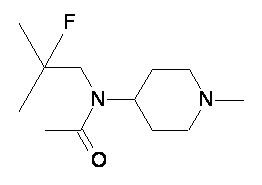
X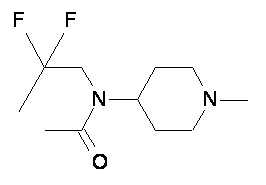 Y
Y
Z AA
AA
CC DD
DD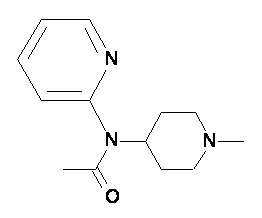 EE
EE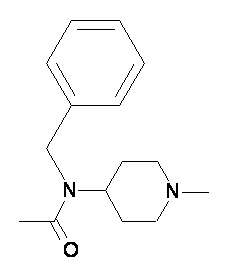
FF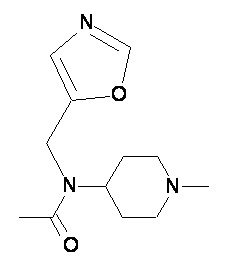
GG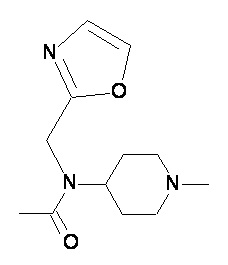
HH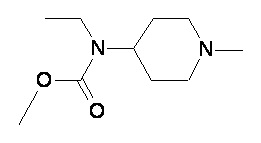
II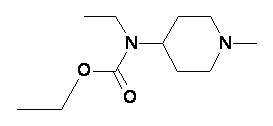 JJ
JJ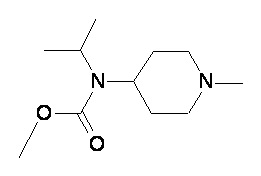 KK
KK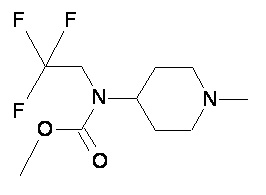
LL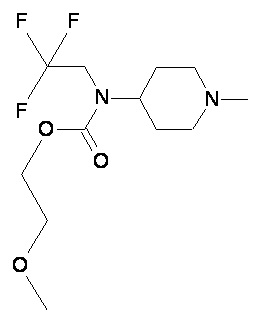
MM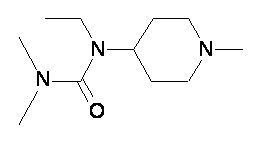 NN
NN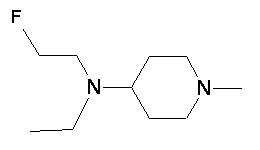
OO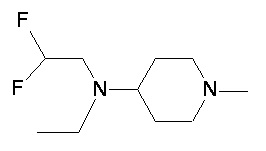
PP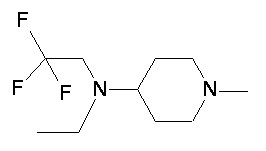
QQ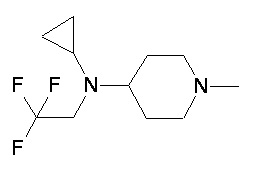
RR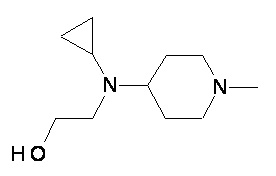
SS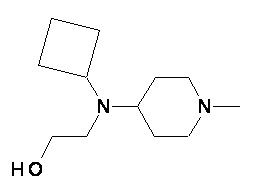 TT
TT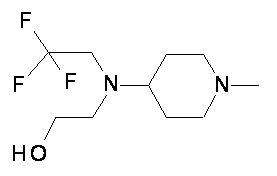 UU
UU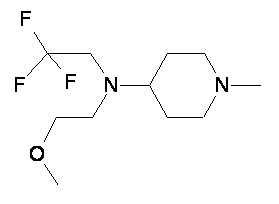 VV
VV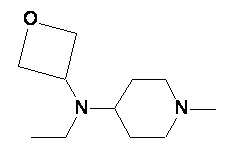
WW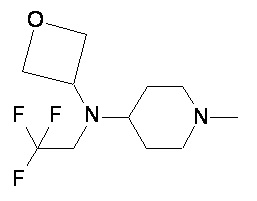
XX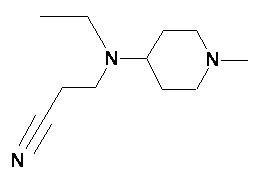
YY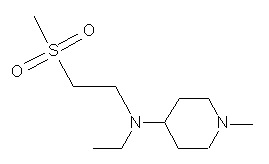 ZZ
ZZ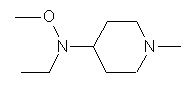
AAA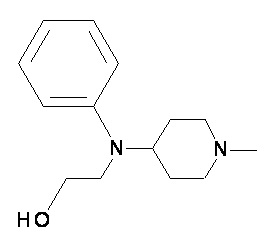 BBB
BBB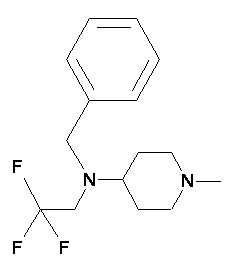 CCC
CCC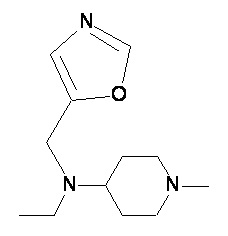
DDD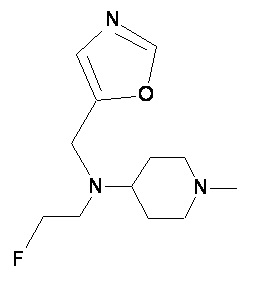
EEE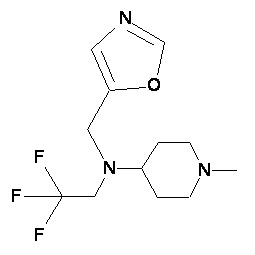 FFF
FFF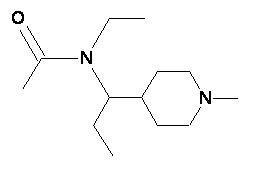
GGG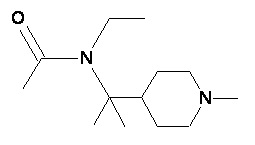
HHH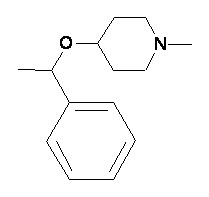
JJJ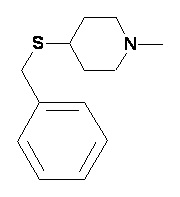
KKK
.
11. Соединение по любому одному из пп. 1-10, отличающееся тем, что R4 выбран из водорода и метила.
12. Соединение, которое представляет собой
Этил-6-{4-[ацетил(циклопропил)амино]пиперидин-1-ил}-2-азаспиро[3.3]гептан-2-карбоксилат;
Этил-6-{4-[ацетил(этил)амино]пиперидин-1-ил}-2-азаспиро[3.3]гептан-2-карбоксилат;
Этил-2-{4-[ацетил(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(пропан-2-ил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(циклопропил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[циклопропил(трифторацетил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[циклопропил(пропаноил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(циклобутил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[этил(2,2,2-трифторэтил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[циклопропил(2-метоксиэтил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[циклопропил(2-метокси-2-оксоэтил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[циклопропил(1,2-оксазол-3-илметил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-(4-{[ацетил(циклопропил)амино]метил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-(4-{[циклопропил(пропаноил)амино]метил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-(4-{1-[ацетил(циклопропил)амино]этил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-(4-{1-[циклопропил(пропаноил)амино]этил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат;
Метил-2-{4-[ацетил(циклобутил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[этил(формил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Метил-2-{4-[ацетил(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(пропил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(2-метилпропил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(циклопропилметил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-(4-{ацетил[(1-метилциклобутил)метил]амино}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(1-метилциклобутил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-(4-{ацетил[(3-метилоксетан-3-ил)метил]амино}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(2,2-дифторэтил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(2,2,2-трифторэтил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(2-фтор-2-метилпропил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(2,2-дифторпропил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(3,3-дифторциклобутил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(2-метокси-2-метилпропил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-(4-{ацетил[(1-метоксициклобутил)метил]амино}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-(4-{ацетил[(1-гидроксициклобутил)метил]амино}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(метокси)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(фенил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(пиридин-2-ил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(бензил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(1,3-оксазол-5-илметил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[ацетил(1,3-оксазол-2-илметил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
(1,1-2H2)-Этил-2-{4-[ацетил(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[этил(метоксикарбонил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[(этоксикарбонил)(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[(метоксикарбонил)(пропан-2-ил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[(метоксикарбонил)(2,2,2-трифторэтил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-(4-{[(2-метоксиэтокси)карбонил](2,2,2-трифторэтил)амино}пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[(диметилкарбамоил)(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[этил(2-фторэтил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[(2,2-дифторэтил)(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[(метоксикарбонил)(оксетан-3-ил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[циклопропил(2,2,2-трифторэтил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[циклопропил(2-гидроксиэтил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[циклобутил(2-гидроксиэтил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[(2-гидроксиэтил)(2,2,2-трифторэтил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[(2-метоксиэтил(2,2,2-трифторэтил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[этил(оксетан-3-ил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[оксетан-3-ил(2,2,2-трифторэтил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[(2-цианоэтил)(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-(4-{этил[2-(метилсульфонил)этил]амино}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[этил(метокси)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[(2-гидроксиэтил)(фенил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[бензил(2,2,2-трифторэтил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[этил(1,3-оксазол-5-илметил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[(2-фторэтил)(1,3-оксазол-5-илметил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[(1,3-оксазол-5-илметил)(2,2,2-трифторэтил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-(4-{1-[ацетил(этил)амино]пропил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-(4-{2-[ацетил(этил)амино]пропан-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-{4-[1-(1H-пиразол-1-ил)этил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-[4-(1-фенилэтокси)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-2-[4-(бензилсульфанил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат;
Этил-6-{4-[ацетил(циклопропил)амино]пиперидин-1-ил}-2-азаспиро[3.4]октан-2-карбоксилат;
Метил-6-{4-[ацетил(этил)амино]пиперидин-1-ил}-2-азаспиро[3.4]октан-2-карбоксилат;
Этил-6-{4-[ацетил(этил)амино]пиперидин-1-ил}-2-азаспиро[3.4]октан-2-карбоксилат;
или его фармацевтически приемлемую соль.
13. Соединение по п. 1, представляющее собой этил-6-{4-[ацетил(этил)амино]пиперидин-1-ил}-2-азаспиро[3.3]гептан-2-карбоксилат или его фармацевтически приемлемую соль.
14. Соединение по п. 1, представляющее собой этил-2-{4-[ацетил(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат или его фармацевтически приемлемую соль.
15. Соединение по п. 1, представляющее собой этил-2-{4-[ацетил(пропан-2-ил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат или его фармацевтически приемлемую соль.
16. Соединение по п. 1, представляющее собой этил-2-{4-[этил(2,2,2-трифторэтил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат или его фармацевтически приемлемую соль.
17. Соединение по п. 1, представляющее собой метил-2-{4-[ацетил(этил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат или его фармацевтически приемлемую соль.
18. Соединение по п. 1, представляющее собой этил-2-{4-[ацетил(2,2-дифторэтил)амино]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат или его фармацевтически приемлемую соль.
19. Соединение по любому из пп. 1-18 для применения в лечении заболеваний, опосредованных мускариновым рецептором M1 и/или M4.
20. Фармацевтическая композиция, содержащая соединение, определенное по любому из пп. 1-18, и фармацевтически приемлемое вспомогательное вещество, для применения в лечении заболеваний, опосредованных мускариновым рецептором M1 и/или M4.
21. Соединение по любому из пп. 1-18 для применения при лечении когнитивного расстройства или психотического расстройства, или для лечения или уменьшения тяжести острой, хронической, нейропатической или воспалительной боли, или для лечения зависимости, или для лечения расстройств движения.
22. Соединение по п. 21 для применения при лечении болезни Альцгеймера, деменции с тельцами Леви или шизофрении.
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| RU 2010135253 А, 10.04.2012. | |||

