[0001] Область техники
[0002] Настоящее изобретение относится к α- и β-ненасыщенному амидному соединению, представляющему собой производное бензотриазола, применяемому в качестве ингибитора TGF-βRI, и в частности относится к соединению, представленному формулой (I), или его фармацевтически приемлемой соли.
[0003] Уровень техники
[0004] Трансформирующий фактор роста бета (TGF-β) представляет собой многофункциональный цитокин, относящийся к суперсемейству трансформирующих факторов роста с широким спектром биологической активности, участвующих в раннем эмбриональном развитии, формировании хряща и кости, синтезе внеклеточного матрикса, воспалении, интерстициальном фиброзе, регуляции иммунной и эндокринной функций, образовании и развитии опухолей.
[0005] Суперсемейство TGF-β состоит из класса полипептидных факторов роста, структура и функция которых взаимосвязаны, и включает TGF-β (то есть узко определяемый TGF-β), активины, ингибины и костные морфогенетические белки (BMP), а именно мюллеров ингибирующий фактор, и т.д. TGF-β является одним из важных членов этого семейства. У млекопитающих TGF-β в основном существует в трех формах: TGF-β1, TGF-β2 и TGF-β3, - которые локализованы на разных хромосомах. Среди них в соматических клетках наибольшую долю составляет TGF-β1 (> 90%), он наиболее активен, наиболее универсален и наиболее широко распространен. Вновь синтезированный TGF-β находится в виде неактивного предшественника, состоящего из сигнального пептида, ассоциированного с латентностью полипептида (LAP) и зрелого TGF-β. После ферментативного гидролиза он образует активный TGF-β и затем связывается с рецептором для оказания биологического действия.
[0006] Передача сигналов с участием сигнальных молекул TGF-β осуществляется посредством трансмембранного рецепторного комплекса. Рецептор TGF-β представляет собой трансмембранный белок, присутствующий на поверхности клеток, и его разновидностями являются рецептор типа I (TGF-βRI), рецептор типа II (TGF-βRII) и рецептор типа III (TGF-βRIII), из которых TGF-PRI также известен как активин-рецептороподобная киназа 5 (ALK5). TGF-βRIII не обладает собственной активностью, и это отсутствие активности в основном связано с запасанием TGF-β. TGF-βRI и TGF-βRII относятся к семейству серин-треониновых киназ. Рецепторы типа II связываются с лигандами TGF-β с более высокой аффинностью и образуют гетерологичные рецепторные комплексы с рецепторами типа I. Фосфорилирование участка, богатого глицином и серином (GS-домена), в близких к мембране участках рецепторов типа I инициирует внутриклеточные каскадные реакции передачи сигнала.
[0007] Smads является важной молекулой для передачи и регуляции сигнала TGF-β в клетках, и она может непосредственно передавать сигнал TGF-β из клеточной мембраны в ядро. Таким образом, сигнальный путь TGF-β/Smads играет важную роль в возникновении и развитии опухолей. При передаче сигнала TGF-β/Smads активированный TGF-β сначала связывается с TGF-βRII на поверхности клеточной мембраны с образованием гетеродимерного комплекса, а TGF-βRI распознает бинарный комплекс и связывается с ним.
[0008] TGF-βRII фосфорилирует серин/треонин в GS-домене цитоплазматического домена TGF-βRI, что приводит к активации TGF-βRI. Затем активированный TGF-βRI в свою очередь фосфорилирует белок R-Smads (Smad2/Smad3), и последний связывается с Co-Smad (Smad4) с образованием гетеротримерного комплекса, который поступает в ядро и действует синергетически с другими коактиваторами и коингибиторами, обеспечивая регуляцию транскрипции генов-мишеней. Любое изменение в сигнальном пути TGF-бета/Smads может привести к аномалиям в пути передачи сигнала.
[0009] Современные исследования показывают, что в опухолевых клетках TGF-β может непосредственно влиять на рост опухоли (несобственные эффекты сигнала TGF-β) или косвенно влияет на рост опухоли (собственные эффекты TGF-β) путем индукции эпителиально-мезенхимальной трансформации, блокирования противоопухолевых иммунных ответов, усиления ассоциированного с опухолью фиброза и усиления ангиогенеза. В то же время TGF-β способен к сильной индукции фиброзных процессов, которая является активатором ассоциированных с опухолью фибробластов. Эти фибробласты являются основным источником коллагена типа I и других факторов фиброза. Продукты индукции фибробластов и других факторов фиброза могут продолжать развитие микроокружения, которое может ослаблять иммунные ответы, повышает устойчивость к лекарственным средствам и стимулирует ангиогенез в опухоли. Кроме того, TGF-β влияет на ангиогенез как во время онтогенеза, так и во время роста опухоли. Например, у дефицитных по TGF-βRI эмбрионов мыши наблюдаются тяжелые дефекты развития сосудов, свидетельствующие о том, что сигнальный путь TGF-β является ключевым регулятором в развитии эндотелия сосудов и клеток гладких мышц.
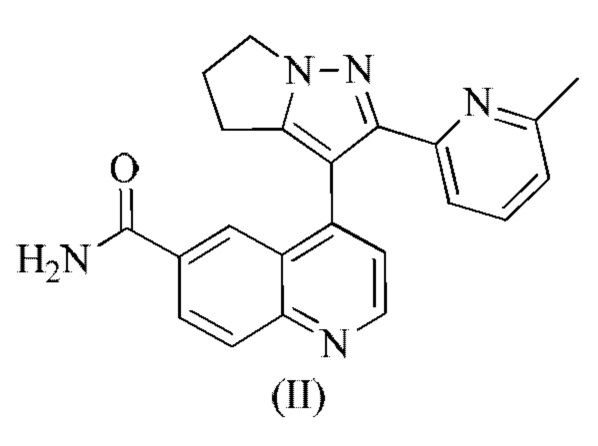
[00010] В 2013 году Федеральное управление по контролю качества пищевых продуктов и лекарственных средств США (FDA) отметило низкомолекулярный ингибитор TGF-βRI LY2157299 от компании Eli Lilly (WO 2002/094833) для лечения глиомы и рака печени. LY2157299 представляет собой малоизученное орфанное лекарственное средство, названное галунисертиб. Галунисертиб ингибирует инвазию и метастазирование опухолевых клеток, ингибируя при этом инфильтрацию опухолевых клеток в кровеносные сосуды. В фазе 2 клинических испытаний на пациентах с раком печени примерно у 23% пациентов, получавших галунисертиб, происходило снижение уровня альфа-фетопротеина (АФП) в сыворотке более чем на 20%. Для этих пациентов были характерны более медленное прогрессирование опухоли и более долгосрочная выживаемость, чем для пациентов без ответа АФП, и у этих пациентов также наблюдали повышенную экспрессию кадгерина в эпителиальных клетках, что позволяет предположить, что галунисертиб может регулировать эпителиально-мезенхимальную трансформацию (ЕМТ) посредством ингибирования пути передачи сигнала TGF-β, тем самым подавляя прогрессирование рака печени.
[00011] Структура галунисертиба (LY2157299) показана в виде формулы (II):
[00012] Ссылки на предшествующий уровень техники:
[00013] WO 2009/009059; WO 2007/076127; WO 2004/026306; WO 2004/072033; WO 2002/094833.
[00014] Раскрытие сущности изобретения
[00015] Согласно настоящему изобретению предложено соединение, представленное формулой (I), или его фармацевтически приемлемая соль,
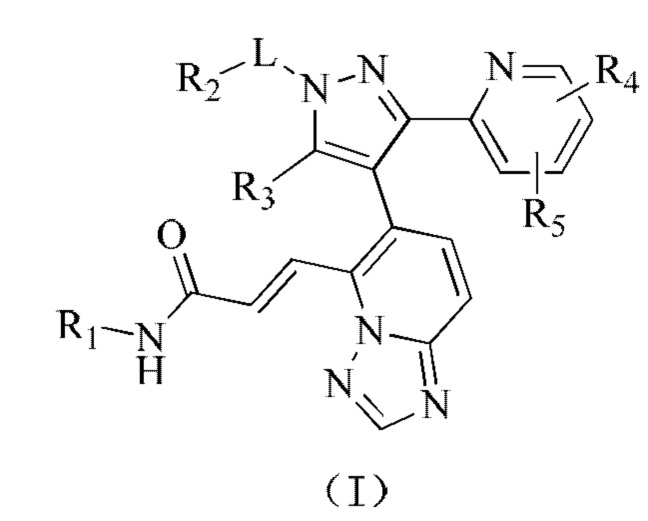
[00016] где
[00017] R1 выбран из водорода, гидроксила, амино или из группы, состоящей из C1-3 алкила и С3-6 циклоалкила, и указанная группа необязательно замещена 1, 2 или 3 R;
[00018] R2 выбран из группы, состоящей из C1-3 алкила, С3-6 циклоалкила и фенила, и указанная группа необязательно замещена 1, 2 или 3 R;
[00019] R3 выбран из водорода или из C1-3 алкила, который необязательно замещен 1, 2 или 3 R;
[00020] необязательно, R2 и R3 соединены с образованием 5-6-членного кольца, которое необязательно замещено 1, 2 или 3 R;
[00021] каждый из R4 и R5 независимо выбран из водорода, галогена или выбран из группы, состоящей из C1-3 алкила и C1-3 гетероалкила, и указанная группа необязательно замещена 1, 2 или 3 R;
[00022] L выбран из простой связи, -(CRR)1-3-;
[00023] R выбран из F, Cl, Br, I, CN, ОН, NH2, СООН или из группы, состоящей из C1-6 алкила, C1-6 гетероалкила, С3-6 циклоалкила, 3-6-членного гетероциклоалкила, фенила и 5-6-членного гетероарила, и указанная группа необязательно замещена 1, 2 или 3 R;
[00024] R' выбран из F, Cl, Br, I, ОН, CN, NH2, СООН, Me, Et, CF3, CHF2, CH2F, NHCH3, N(CH3)2;
[00025] «гетеро» относится к гетероатому или гетероатомной группе, выбранным из группы, состоящей из -C(=O)N(R)-, -N(R)-, -C(=NR)-, -S(=O)2N(R)-, -S(=O)N(R)-, -О-, -S-, =O, =S, -O-N=, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2-, -N(R)C(=O)N(R)-;
[00026] в любом из указанных выше случаев число гетероатомов или гетероатомных групп независимо выбрано из 1, 2, или 3.
[00027] В некоторых вариантах реализации настоящего изобретения R выбран из F, Cl, Br, I, CN, ОН или из группы, состоящей из C1-6 алкила, С3-6 циклоалкила и фенила, и указанная группа необязательно замещена 1, 2 или 3 R'.
[00028] В некоторых вариантах реализации настоящего изобретения R выбран из группы, состоящей из F, Cl, Br, I, CN, ОН, метила, CHF2, этила, пропила, циклопропила и фенила.
[00029] В некоторых вариантах реализации настоящего изобретения R1 выбран из водорода или из группы, состоящей из метила, этила, 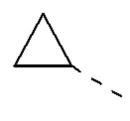 , и указанная группа необязательно замещена 1, 2 или 3 R.
, и указанная группа необязательно замещена 1, 2 или 3 R.
[00030] В некоторых вариантах реализации настоящего изобретения R1 выбран из водорода, метила, этила, 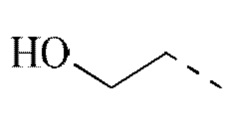 ,
, 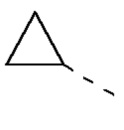 .
.
[00031] В некоторых вариантах реализации настоящего изобретения R2 выбран из группы, состоящей из метила, этила, изопропила, циклопентила и фенила, и указанная группа необязательно замещена 1, 2 или 3 R.
[00032] В некоторых вариантах реализации настоящего изобретения R2 выбран из группы, состоящей из метила, этила, изопропила, циклопентила 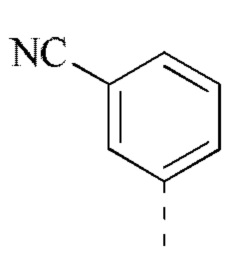 и
и 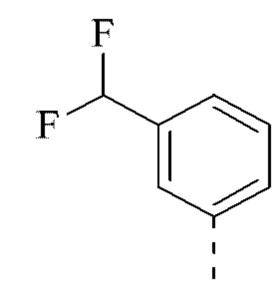 .
.
[00033] В некоторых вариантах реализации настоящего изобретения R2 и R3 соединены и фрагмент 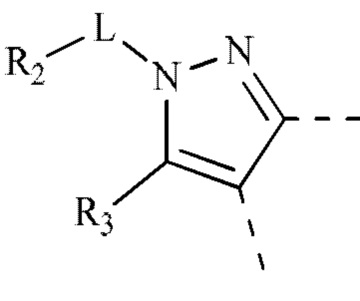 представляет собой
представляет собой 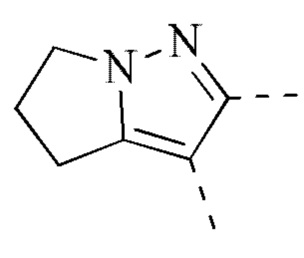 .
.
[00034] В некоторых вариантах реализации настоящего изобретения каждый из R4 и R5 независимо выбран из группы, состоящей из водорода, F, Cl, Br, метила и этила.
[00035] В некоторых вариантах реализации настоящего изобретения фрагмент 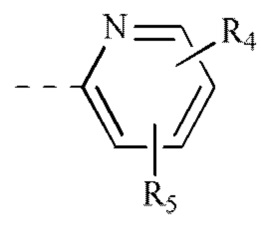 выбран из
выбран из 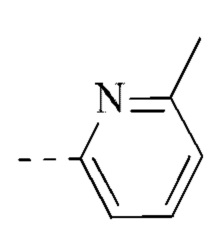 ,
, 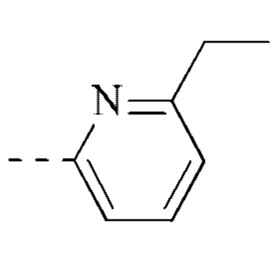 ,
, 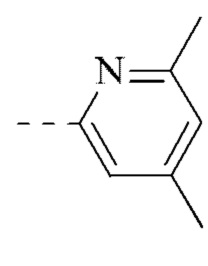 ,
, 
[00036] В некоторых вариантах реализации настоящего изобретения L выбран из простой связи, -(СН2)1-3-.
[00037] В некоторых вариантах реализации настоящего изобретения L выбран из простой связи, -СН2-, -СН2СН2-.
[00038] В некоторых вариантах реализации настоящего изобретения R выбран из F, Cl, Br, I, CN, ОН или из группы, состоящей из C1-6 алкила, С3-6 циклоалкила и фенила, и указанная группа необязательно замещена 1, 2 или 3 R', и другие переменные определены, как указано выше.
[00039] В некоторых вариантах реализации настоящего изобретения R выбран из F, Cl, Br, I, CN, ОН, метила, CHF2, этила, пропила, циклопропила и фенила, и другие переменные определены, как указано выше.
[00040] В некоторых вариантах реализации настоящего изобретения R1 выбран из водорода или из группы, состоящей из метила, этила, 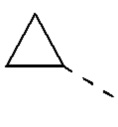 , и указанная группа необязательно замещена 1, 2 или 3 R, и другие переменные определены, как указано выше.
, и указанная группа необязательно замещена 1, 2 или 3 R, и другие переменные определены, как указано выше.
[00041] В некоторых вариантах реализации настоящего изобретения R1 выбран из водорода, метила, этила, 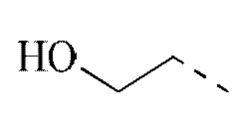 ,
, 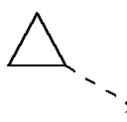 , и другие переменные определены, как указано выше.
, и другие переменные определены, как указано выше.
[00042] В некоторых вариантах реализации настоящего изобретения R2 выбран из группы, состоящей из метила, этила, изопропила, циклопентила и фенила, и указанная группа необязательно замещена 1, 2 или 3 R, и другие переменные определены, как указано выше.
[00043] В некоторых вариантах реализации настоящего изобретения R2 выбран из группы, состоящей из метила, этила, изопропила, циклопентила, 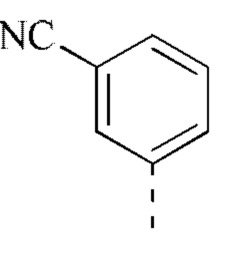 и
и 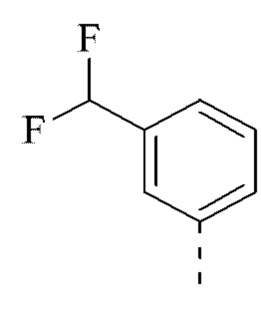 , и другие переменные определены, как указано выше.
, и другие переменные определены, как указано выше.
[00044] В некоторых вариантах реализации настоящего изобретения R2 и R3 соединены и фрагмент 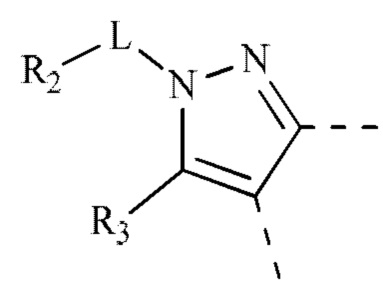 представляет собой
представляет собой 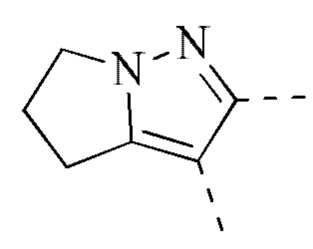 , и другие переменные определены, как указано выше.
, и другие переменные определены, как указано выше.
[00045] В некоторых вариантах реализации настоящего изобретения каждый из R4 и R5 независимо выбран из группы, состоящей из водорода, F, Cl, Br, метила и этила, и другие переменные определены, как указано выше.
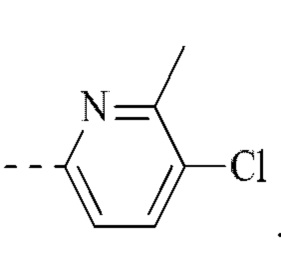
[000461 В некоторых вариантах реализации настоящего изобретения фрагмент 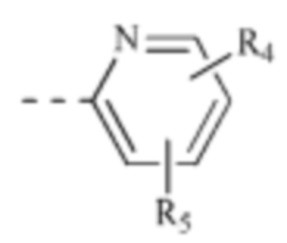 выбран из
выбран из  ,
,  ,
,  ,
,  , и другие переменные определены, как указано выше.
, и другие переменные определены, как указано выше.
[00047] В некоторых вариантах реализации настоящего изобретения L выбран из простой связи, -(СН2)1-3-, и другие переменные определены, как указано выше.
[00048] В некоторых вариантах реализации настоящего изобретения L выбран из простой связи, -СН2-, -СН2СН2-, и другие переменные определены, как указано выше.
[00049] В некоторых вариантах реализации настоящего изобретения соединение выбрано из
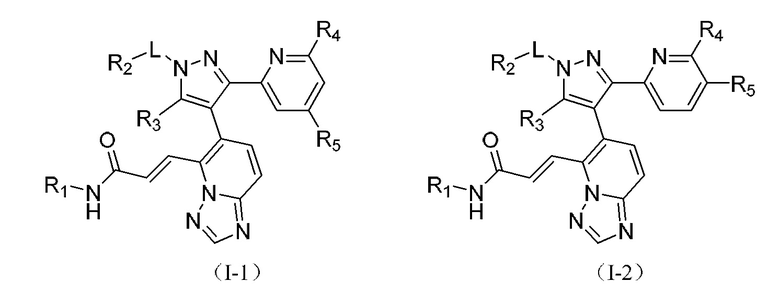
[00050] где R1, R2, R3, R4, R5 и L определены, как указано выше, и R4 и R5 одновременно оба не являются водородом.
[00051] В некоторых вариантах реализации настоящего изобретения соединение выбрано из
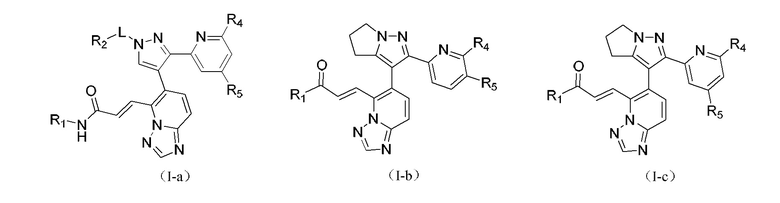
[00052] где R1, R2, R4, R5 и L определены, как указано выше, и R4 и R5 одновременно оба не являются водородом.
[00053] Согласно настоящему изобретению также предложено соединение или его фармацевтически приемлемая соль, которое выбрано из группы, состоящей из
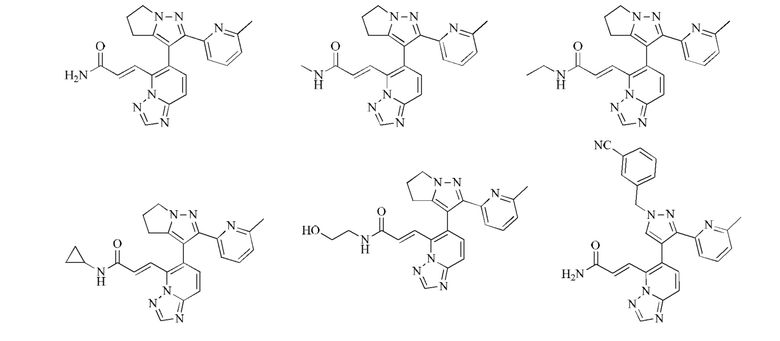
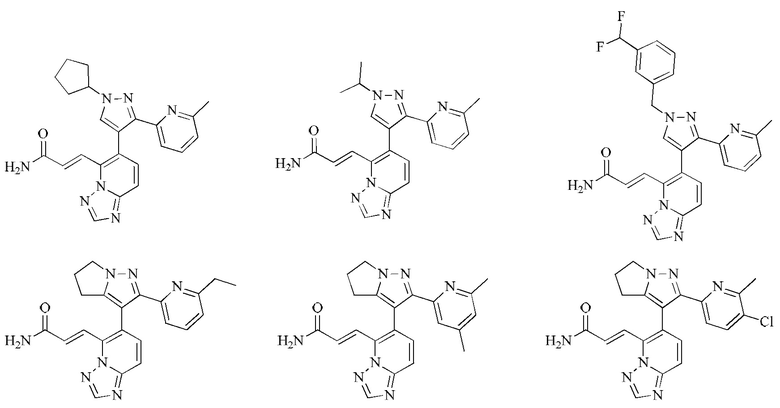
[00054] Согласно настоящему изобретению также предложена фармацевтическая композиция, содержащая терапевтически эффективную дозу соединения или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
[00055] Согласно настоящему изобретению также предложено применение соединения или его фармацевтически приемлемой соли или фармацевтической композиции для получения лекарственного средства для лечения рака.
[00056] В некоторых вариантах реализации настоящего изобретения рак относится к раку молочной железы.
[00057] Другие варианты реализации настоящего изобретения получены в результате случайного сочетания указанных выше переменных.
[00058] Технический результат
[00059] Соединение согласно настоящему изобретению главным образом применяют в качестве ингибитора TGF-бета R1, блокирующего нисходящий сигнальный путь TGF-бета путем ингибирования TGF-бета R1, обеспечивая тем самым желаемое фармакологическое действие. В отличие от решений, известных из уровня техники, бензотриазольная структура соединения согласно настоящему изобретению является важным фармакофором, который связывается с TGF-бета R1. Неожиданно было обнаружено, что комбинация химических структур соединений согласно настоящему изобретению обеспечивает большую биологическую активность по сравнению с предшествующим уровнем техники. При одной и той же дозе в сингенной модели на мышах с использованием клеток СТ-26 эффекты подавления опухоли для соединения согласно настоящему изобретению, применяемого как отдельно, так и в комбинации с PDL-1, превосходили эффекты препаратов, известных из уровня техники, а это позволяет сделать вывод о том, что соединение согласно настоящему изобретению обладает лучшей способностью активировать противоопухолевый иммунный ответ; в ортотопической трансплантационной модели на клетках рака молочной железы мышей линии 4Т1 соединение согласно настоящему изобретению обладает значительно лучшей способностью подавлять метастазирование по сравнению с решениями, известными из уровня техники. Соединение согласно настоящему изобретению оказывает очевидное подавляющее действие в отношении метастазирования и интенсивности метастазирования опухолей органов, содержащих различные ткани, что указывает на его большой потенциал в качестве терапевтического средства. Соединение согласно настоящему изобретению является очень перспективным в качестве средства подавления метастазирования рака молочной железы, играет важную роль в подавлении метастазирования при комбинированной терапии рака молочной железы и обеспечивает потенциально новую терапевтическую стратегию для лечения клинического рака молочной железы.
[00060] Определения и описание
[00061] Если не указано иное, следующие термины и фразы, используемые в настоящем описании, имеют следующие значения. Конкретный термин или фразу не следует считать неопределенными или неясными, если они не определены конкретно, их следует понимать в обычном смысле. В случае, когда в настоящем описании встречается товарный знак, он предназначен для указания соответствующего препарата или его активного ингредиента. Термин «фармацевтически приемлемый» в настоящем описании используется для обозначения тех соединений, материалов, композиций и/или лекарственных форм, которые в рамках обоснованного медицинского заключения подходят для применения в контакте с тканями людей и животных без чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений, соответствуя разумному соотношению польза/риск.
[00062] Термин «фармацевтически приемлемые соли» относится к солям соединений согласно настоящему изобретению, которые получают из соединений, содержащих конкретные заместители согласно настоящему изобретению, и относительно нетоксичных кислот или оснований. В случае, когда соединения согласно настоящему изобретению содержат относительно кислые функциональные группы, соли присоединения основания могут быть получены путем введения в контакт нейтральной формы таких соединений с достаточным количеством основания в чистом растворе или в подходящем инертном растворителе. Фармацевтически приемлемые соли присоединения основания включают соли натрия, калия, кальция, аммония, органических производных аммиака или магния или аналогичные соли. В случае, когда соединения согласно настоящему изобретению содержат относительно основные функциональные группы, соли присоединения кислоты могут быть получены путем введения в контакт нейтральной формы таких соединений с достаточным количеством кислоты в чистом растворе или в подходящем инертном растворителе. Примеры фармацевтически приемлемых солей присоединения кислоты включают соли неорганических кислот, включая, например, соляную кислоту, бромоводородную кислоту, азотную кислоту, угольную кислоту, бикарбонат, фосфорную кислоту, моногидрофосфат, дигидрофосфат, серную кислоту, бисульфат, иодоводородную кислоту, фосфористую кислоту и т.п.; и соли органических кислот, включая, например, уксусную кислоту, пропионовую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, фумаровую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфокислоту п-толуолсульфокислоту лимонную кислоту, винную кислоту, метанилульфокислоту и т.п.; также включают соли аминокислот (например, аргинина и т.д.), а также соли органических кислот, таких как глюкуроновая кислота (см. Berge et al., «Pharmaceutical Salts», Journal of Pharmaceutical Science 66: 1-19 (1977)). Некоторые конкретные соединения согласно настоящему изобретению содержат основные и кислотные функциональные группы, так что их можно превращать в любую соль присоединения основания или кислоты.
[00063] Предпочтительно, соль вводят в контакт с основанием или кислотой обычным способом, и выделяют исходное соединение, регенерируя таким образом нейтральную форму соединения. Исходная форма соединения отличается от его различных солевых форм по определенным физическим свойствам, таким как растворимость в полярных растворителях.
[00064] В настоящем описании «фармацевтически приемлемые соли» относятся к производным соединений согласно настоящему изобретению, где исходное соединение модифицировано путем образования соли с кислотой или путем образования соли с основанием. Примеры фармацевтически приемлемых солей включают, но не ограничиваются перечисленными: соли неорганических или органических кислот основных радикалов, таких как амины, неорганические или органические соли кислотных радикалов, таких как карбоновые кислоты, и т.п. Фармацевтически приемлемые соли включают обычные нетоксичные соли или четвертичные аммониевые соли исходного соединения, такие как соли, образованные нетоксичными неорганическими или органическими кислотами. Обычные нетоксичные соли включают, но не ограничиваются перечисленными, соли, полученные из неорганических и органических кислот, которые выбраны из группы, состоящей из 2-ацетоксибензойной кислоты, 2-гидроксиэтилсульфокислоты, уксусной кислоты, аскорбиновой кислоты, бензосульфокислоты, бензойной кислоты, бикарбоната, угольной кислоты, лимонной кислоты, этилендиаминтетрауксусной кислоты, этандисульфокислоты, этансульфокислоты, фумаровой кислоты, глюкогептонозы, глюконовой кислоты, глутаминовой кислоты, гликолевой кислоты, бромоводородной кислоты, соляной кислоты, гидроиодида, гидрокси, гидроксинафтила, изэтионовой кислоты, молочной кислоты, лактозы, додецилсульфокислоты, малеиновой кислоты, яблочной кислоты, миндальной кислоты, метансульфокислоты, азотной кислоты, щавелевой кислоты, памовой кислоты, пантотеновой кислоты, фенилуксусной кислоты, фосфорной кислоты, полигалактанальдегида, пропионовой кислоты, салициловой кислоты, стеариновой кислоты, уксусной кислоты, янтарной кислоты, сульфаминовой кислоты, сульфаниловой кислоты, серной кислоты, таннинов, винной кислоты и п-толуолсульфокислоты.
[00065] Фармацевтически приемлемые соли согласно настоящему изобретению могут быть синтезированы из исходного соединения, содержащего кислотные радикалы или основные радикалы, с помощью обычных химических способов. Обычно такие соли получают путем введения этих соединений в форме свободной кислоты или свободного основания в реакцию со стехиометрическим количеством подходящего основания или кислоты в воде или органическом растворителе или их смеси. Как правило, неводные среды, такие как эфир, этилацетат, этанол, изопропиловый спирт или ацетонитрил, являются предпочтительными.
[00066] Помимо солевых форм, соединения, предложенные согласно настоящему изобретению, также существуют в формах пролекарств. Пролекарства соединений согласно настоящему описанию легко изменяются химическим путем в физиологических условиях с превращением в соединения согласно настоящему изобретению. Кроме того, пролекарства могут быть превращены в соединения согласно настоящему изобретению с помощью химических или биохимических способов в среде in vivo.
[00067] Некоторые соединения согласно настоящему изобретению могут существовать в несольватированных или сольватированных формах, включая гидратированные формы. В общем, сольватированные формы эквивалентны несольватированным формам, и оба вида форм включены в объем настоящего изобретения.
[00068] Некоторые соединения согласно настоящему изобретению могут содержать асимметрические атомы углерода (оптические центры) или двойные связи. Рацематы, диастереомеры, геометрические изомеры и отдельные изомеры включены в объем настоящего изобретения.
[00069] Графическое представление рацемических, амбискалемических и скалемических или энантиомерных чистых соединений, приведенное в настоящем описании, взято из Maehr, J. Chem. Ed. 1985, 62: 114-120. Если не указано иное, абсолютная конфигурация стереоцентра представлена с помощью клиновидной связи и пунктирной связи. В случае, когда соединения согласно настоящему описанию содержат олефиновые двойные связи или другие центры геометрической асимметрии, они включают Е, Z геометрические изомеры, если не указано иное. Аналогично, все таутомерные формы включены в объем настоящего изобретения.
[00070] Соединения согласно настоящему изобретению могут существовать в конкретных геометрических или стереоизомерных формах. Настоящее изобретение включает все такие соединения, включая цис- и транс-изомеры, пары (-)- и (+)-энантиомеров, (R)- и (S)-энантиомеры, диастереомеры, (D)-изомер, (L)-изомер и рацемические смеси и другие их смеси, такие как энантиомерно или диастереомерно обогащенные смеси, все из которых включены в объем настоящего изобретения. Дополнительные асимметрические атомы углерода могут присутствовать в заместителях, таких как алкильные группы. Все эти изомеры и их смеси включены в объем настоящего изобретения.
[00071] Оптически активные (R)- и (S)-изомеры и D- и L-изомеры могут быть получены путем хирального синтеза или с применением хиральных реагентов или другими обычными способами. Если необходим энантиомер определенного соединения согласно настоящему изобретению, его можно получить путем асимметрического синтеза или дериватизации с хиральным вспомогательным веществом, при этом полученную смесь диастереомеров разделяют и вспомогательные группы отщепляют с получением чистого желаемого энантиомера. В качестве альтернативы, когда молекула содержит основную функциональную группу (такую как аминогруппа) или кислотную функциональную группу (такую как карбоксильная группа), образуется диастереомерная соль с подходящими оптически активными кислотой или основанием, затем проводят разделение диастереомеров обычными способами, известными в данной области техники, и далее выделяют чистый энантиомер. Кроме того, разделение энантиомеров и диастереомеров, как правило, выполняют посредством хроматографии с применением хиральной неподвижной фазы и, необязательно, в сочетании с химической дериватизацией (например, с образованием карбамината из аминов).
[00072] Соединения согласно настоящему изобретению могут содержать неприродные соотношения атомных изотопов при одном или более атомов, содержащихся в соединении. Например, соединения можно метить с помощью радиоактивных изотопов, таких как тритий (3Н), иод-125 (125I) или С-14 (14С). Все варианты изотопных композиций соединений согласно настоящему изобретению, являющихся или не являющихся радиоактивными, включены в объем настоящего изобретения.
[00073] Термин «фармацевтически приемлемый носитель» относится к любому агенту или переносящей среде, способным осуществлять доставку эффективного количества активного агента согласно настоящему изобретению без нарушения биологической активности активного агента и без проявления токсических побочных эффектов в отношении организма-хозяина или пациента. Типичные носители включают воду, масло, вещества растительного и минерального происхождения, основы для кремов, основы для лосьонов, основы для мазей и т.д. Эти основы содержат суспендирующие агенты, агенты, придающие клейкость, усилители проникновения через кожу и т.п. Их составы хорошо известны специалистам в области косметики или в области лекарственных средств для местного применения. Дополнительную информацию по носителям можно найти в публикации Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott, Williams & Wilkins (2005), содержание которой включено в настоящее описание посредством ссылки.
[00074] Термин «вспомогательное вещество» обычно относится к носителю, разбавителю и/или среде, необходимым для приготовления эффективной фармацевтической композиции.
[00075] Для лекарственного средства или фармакологически активного агента термин «эффективное количество» или «терапевтически эффективное количество» относится к достаточному количеству лекарственного средства или агента, которое не является токсичным, но может обеспечить достижение желаемого эффекта. В случае лекарственной формы для перорального введения согласно настоящему изобретению «эффективное количество» активного вещества в композиции относится к количеству, необходимому для достижения желаемого эффекта при применении в сочетании с другим активным веществом в композиции. Определение эффективного количества варьирует от человека к человеку в зависимости от возраста и общего состояния реципиента и также от конкретного активного вещества, и подходящее в отдельном случае эффективное количество может быть определено специалистом в данной области техники в соответствии с установленными экспериментами.
[00076] Термины «активный ингредиент», «терапевтический агент», «активное вещество» или «активный агент» относятся к химическому соединению, которое может обеспечить эффективное лечение целевого расстройства, заболевания или состояния.
[00077] «Необязательный» или «необязательно» означает, что событие или ситуация, описанные ниже, могут, но не обязательно, возникать, и настоящее описание включает возникновение упомянутых выше события или ситуации и отсутствие описанных в нем события или ситуации.
[00078] Термин «замещенный» означает, что любые один или более атомов водорода при конкретном атоме заменены на заместители, включая варианты дейтерия и водорода, при условии, что конкретный атом имеет нормальную валентность и замещенное соединение является стабильным. В случае, когда заместитель представляет собой кето (то есть =O), это означает, что два атома водорода являются замещенными. Замещение кетогруппой не встречается в ароматических группах. Термин «необязательно замещенный» означает, что он может быть или не быть замещенным. Если не указано иное, тип и количество заместителей могут быть произвольными в зависимости от достижимости с химической точки зрения.
[00079] В случае, когда какой-либо вариант (например, R) встречается более чем один раз в композиции или структуре соединения, его определение в каждом случае является независимым. Таким образом, например, если группа замещена 0-2 R, группа может необязательно быть замещена не более чем двумя R, и R в каждом случае является независимым вариантом. Кроме того, комбинации заместителей и/или их вариантов допустимы, только если такие комбинации приводят к стабильным соединениям.
[00080] В случае, когда индекс при соединяющей группе равен 0, например, -(CRR)0-, это означает, что соединяющая группа представляет собой простую связь.
[00081] В случае, когда один из вариантов выбран из простой связи, это означает, что две группы, которые она соединяет, связаны непосредственно. Например, когда L представляет собой простую связь в A-L-Z, структура фактически представляет собой A-Z.
[00082] В случае, когда заместитель является вакантным, это означает, что заместитель не существует. Например, когда X является вакантным в А-Х, это означает, что структура фактически представляет собой А. В случае, когда связь заместителя может быть перекрестно сшита с двумя атомами в кольце, заместитель может быть связан с любым атомом в кольце. В случае, когда для перечисленных заместителей не указано, посредством какого атома они присоединены к соединению, будучи включенными в общую формулу химической структуры, но конкретно не упомянутыми, такие заместители могут быть связаны посредством любого из их атомов. Комбинации заместителей и/или их вариантов допустимы, только если такие комбинации приводят к стабильным соединениям. Например, структурная единица 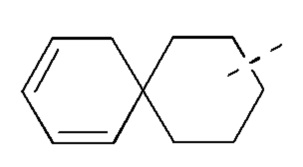 или
или 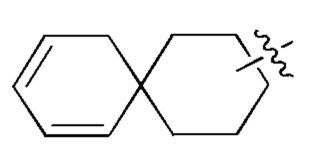 означает, что она может быть замещенной в любом положении циклогексила или циклогексадиена.
означает, что она может быть замещенной в любом положении циклогексила или циклогексадиена.
[00083] Если не указано иное, термин «гетеро» обозначает гетероатом или гетероатомную группу (то есть группу атомов, содержащую гетероатомы), содержащую атомы, отличные от углерода (С) и водорода (Н), и группы атомов, содержащие эти гетероатомы, включают, например, кислород (О), азот (N), серу (S), кремний (Si), германий (Ge), алюминий (Al), бор (В), -О-, -S-, =O, =S, -С(=O)O-, -С(=O)-, -C(=S)-, -S(=O), -S(=O)2- и необязательно замещенные -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O)2N(H)- или -S(=O)N(H)-.
[00084] Если не указано иное, «кольцо» относится к замещенному или незамещенному циклоалкилу гетероциклоалкилу, циклоалкенилу, гетероциклоалкенилу циклоалкинилу гетероциклоалкинилу, арилу или гетероарилу. Так называемое кольцо включает одиночное кольцо, бициклическое кольцо, спирокольцо, кольцевую систему, содержащую два кольца, имеющих одну общую связь, или мостиковое кольцо. Число атомов в кольце обычно определяется как число членов кольца. Например, «5-7-членное кольцо» относится к тому, что от 5 до 7 атомов замкнуты в круг. Если не указано иное, кольцо необязательно содержит от 1 до 3 гетероатомов. Таким образом, «5-7-членное кольцо» включает, например, фенил, пиридинил и пиперидинил; в другом аспекте термин «5-7-членное гетероциклоалкильное кольцо» включает пиридил и пиперидинил, но не включает фенил. Термин «кольцо» также включает кольцевые системы, содержащие по меньшей мере одно кольцо, при этом каждое «кольцо» независимо соответствует вышеуказанному определению.
[00085] Если не указано иное, термин «гетероцикл» или «гетероциклил» означает стабильные моноциклические, бициклические или трициклические кольца, содержащие гетероатомы или гетероатомные группы, которые могут быть насыщенными, частично ненасыщенными или ненасыщенными (ароматическими) и содержать атомы углерода и 1, 2, 3 или 4 гетероциклических атома, независимо выбранных из N, О и S, где любой из вышеуказанных гетероциклов может быть конденсирован с бензольным кольцом с образованием бициклического кольца. Гетероатомы азота и серы необязательно могут быть окисленными (то есть NO и S(O)p, р равен 1 или 2). Атом азота может быть замещенным или незамещенным (то есть N или NR, где R представляет собой Н или другие заместители, определенные в настоящем описании). Гетероциклы могут быть присоединены к боковым группам любого гетероатома или атома углерода с образованием стабильной структуры. Если полученное соединение является стабильным, гетероциклы согласно настоящему описанию могут быть замещены в положении атома углерода или азота. Атом азота в гетероцикле необязательно является четвертичным. В предпочтительном варианте реализации общее число атомов S и О в гетероцикле превышает 1, и эти гетероатомы не являются смежными. В другом предпочтительном варианте реализации общее число атомов S и О в гетероцикле не превышает 1. В настоящем описании термин «ароматическая гетероциклическая группа» или «гетероарил» означает стабильное 5-, 6- или 7-членное моноциклическое или бициклическое или 7- 8-, 9- или 10-членное бициклическое гетероциклильное ароматическое кольцо, которое содержит атомы углерода и 1, 2, 3 или 4 гетероциклических атома, независимо выбранных из N, О и S. Атом азота может быть замещенным или незамещенным (то есть N или NR, где R представляет собой Н или другие заместители, определенные в настоящем описании). Гетероатомы азота и серы необязательно могут быть окисленными (то есть NO и S(O)p, р равен 1 или 2). Следует отметить, что общее число атомов S и О в ароматическом гетероцикле не превышает 1. Мостиковые кольца также включены в определение гетероциклов. Мостиковое кольцо образуется, когда два несмежных атома углерода или азота соединены посредством одного или более атомов (то есть С, О, N или S). Предпочтительно, мостиковое кольцо содержит, не ограничиваясь перечисленными, один атом углерода, два атома углерода, один атом азота, два атома азота и одну группу углерод-азот. Следует отметить, что мостик всегда превращает одиночное кольцо в три кольца. В мостиковом кольце заместители в кольце также могут появляться на мостике.
[00086] Примеры гетероциклических соединений включают, но не ограничиваются перечисленными, акридинил, азоцинил, бензимидазолил, бензофуранил, бензосульфидрилфуранил, бензосульфидрилфенил, бензоксазолил, бензоксазолинил, бензотиазолил, бензотриазолил, бензотетразолил, бензизоксазолил, бензизотиазолил, бензимидазолинил, карбазолил, 4аН-карбазолил, карболинил, хроманил, хромен, циннолинилдекагидрохинолинил, 2H,6H-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуранил, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1H-индолил, индолилалкенил, индолинил, индолизинил, индонил, 3H-индолил, изобензофуранил, изоиндолил, изоиндолинил, изохинолинил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, гидроксииндолил, пиримидинил, фенантридинил, фенантролинил, феназинил, фенотиазинил, бензоксантинил, феноксазинил, феназинил, пиперазинил, пиперидинил, пиперидинон, 4-пиперидинон, пиперонил, птеридил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридооксазол, пиридоимидазол, пиридотиазол, пиридил, пирролидинил, пирролинил, 2Н-пирролил, пирролил, хиназолинил, хинолинил, 4H-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, 6H-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантиенил, тиазолил, изотиазолилтиофенил, тиенооксазолил, тиенотиазолил, тиеноимидазолил, тиенил, триазинил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил и ксантен. Также включены соединения с конденсированными кольцами и спиросоединения.
[00087] Если не указано иное, термин «гидрокарбил» или его подчиненное понятие (такое как алкил, алкенил, алкинил, фенил и т.п.) как таковой или как часть другого заместителя означает неразветвленные, разветвленные или циклические углеводородные радикалы или их комбинации, которые могут быть полностью насыщенными (такими как алкил), содержащими одну двойную связь или полиненасыщенными (такими как алкенил, алкинил, фенил), могут быть монозамещенными, дизамещенными или полизамещенными и могут быть одновалентными (такими как метил), двухвалентными (такими как метилен) или поливалентными (такими как метин), могут включать двухвалентные или поливалентные радикалы и содержат указанное число атомов углерода (например, С1-С12 представляет от 1 до 12 атомов углерода, С1-С12 выбраны из группы, состоящей из C1, С2, С3, С4, С5, С6, С7, C8, С9, С10, С11 и С12; С3-12 выбраны из группы, состоящей из С3, С4, С5, С6, С7, C8, С9, С10, С11 и С12). «Гидрокарбил» включает, но не ограничивается перечисленными, алифатический и ароматический гидрокарбил, где алифатический гидрокарбил включает цепные и циклические структуры, включающие, но не ограничивающиеся перечисленными, алкил, алкенил, алкинил, и ароматический гидрокарбил включает, но не ограничивается перечисленными, 6-12-членный ароматический гидрокарбил, такой как бензол, нафталин и т.п. В некоторых вариантах реализации термин «гидрокарбил» относится к радикалам с неразветвленной или разветвленной цепью или их комбинациям, которые могут быть полностью насыщенными, содержащими одну двойную связь или полиненасыщенными и могут включать двухвалентные и поливалентные радикалы. Примеры насыщенных углеводородных радикалов включают, но не ограничиваются перечисленными, метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, изобутил, циклогексил, (циклогексил)метил, циклопропилметил и гомологи или изомеры н-пентила, н-гексила, н-гептила, н-октила и других групп атомов. Ненасыщенный алкил содержит одну или более двойных или тройных связей, и его примеры включают, но не ограничиваются перечисленными, винил, 2-пропенил, бутенил, кротил, 2-пренил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и высшие гомологи или изомеры.
[00088] Если не указано иное, термин «гетерогидрокарбил» или его подчиненное понятие (такое как гетероалкил, гетероалкенил, гетероалкинил, гетероарил и т.д.) как таковой или в сочетании с другим термином означает стабильные, неразветвленные, разветвленные или циклические углеводородные радикалы или их комбинации, состоящие из определенного числа атомов углерода и по меньшей мере одного гетероатома. В некоторых вариантах реализации термин «гетероалкил» как таковой или в сочетании с другим термином означает стабильные, неразветвленные, разветвленные углеводородные радикалы или их комбинации, состоящие из определенного числа атомов углерода и по меньшей мере одного гетероатома. В типичном варианте реализации гетероатом выбран из группы, состоящей из В, О, N, и S, где атомы азота и серы необязательно являются окисленными, а гетероатомы азота необязательно являются четвертичными. Гетероатом или гетероатомная группа может быть расположена в любом внутренней позиции гетерогидрокарбила (включая позицию, в которой гидрокарбил присоединен к остальному фрагменту молекулы). Примеры включают, но не ограничиваются перечисленными, -СН2-СН2-О-СН3, -СН2-СН2-NH-СН3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -СН2-СН2,- S(O)-CH3, -СН2-CH2-S(O)2-CH3, -СН=СН-O-СН3, -CH2-CH=N-OCH3 и -CH=CH-N(CH3)-CH3. До двух гетероатомов могут образовывать непрерывную последовательность, такую как -CH2-NH-OCH3.
[00089] Если не указано иное, термины «циклогидрокарбил», «гетероциклогидрокарбил» или их подчиненные понятия (такие как арил, гетероарил, циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, циклоалкинил, гетероциклоалкинил и т.д.) как таковые или в сочетании с другими терминами означают циклизованный «гидрокарбил», «гетерогидрокарбил», соответственно. Кроме того, для гетерогидрокарбила или гетероциклогидрокарбила (такого как гетероалкил, гетероциклоалкил) гетероатомы могут занимать позицию, в которой гетероцикл присоединен к остальному фрагменту молекулы. Примеры включают, но не ограничиваются перечисленными, циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил и т.п. Неограничивающие примеры гетероциклических групп включают 1-(1,2,5,6-тетрагидропиридинил), 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофураниндол-3-ил, тетрагидротиофен-2-ил, тетрагидротиофен-3-ил, 1-пиперазинил и 2-пиперазинил.
[00090] Если не указано иное, термин «алкил» означает неразветвленный или разветвленный насыщенный гидрокарбил, который может быть монозамещенным или полизамещенным и может быть одновалентным (таким как метил), двухвалентным (таким как метилен) или поливалентным (такой как метин). Примеры алкила включают метил (Me), этил (Et), пропил (такой как н-пропил и изопропил), бутил (такой как н-бутил, изобутил, втор-бутил, трет-бутил), пентил (такой как н-пентил, изопентил, неопентил) и т.д.
[00091] Если не указано иное, термин «алкенил» означает алкил, содержащий одну или более углерод-углеродных двойных связей в любом положении цепи, который может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или поливалентным. Примеры алкенила включают винил, пропенил, бутенил, пентенил, гексенил, бутадиенил, пиперилен, гексадиенил и т.д.
[00092] Если не указано иное, термин «алкинил» означает алкил, содержащий одну или более углерод-углеродных тройных связей в любом положении цепи, который может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или поливалентным. Примеры алкинила включают этинил, пропинил, бутинил, пентинил и т.д.
[00093] Если не указано иное, циклоалкил включает любую стабильную циклическую или полициклическую углеводородную группу, любой атом углерода которой является насыщенным, который может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или поливалентным. Примеры циклоалкила включают, но не ограничиваются перечисленными, циклопропил, норборнил, [2.2.2]бициклооктан, [4.4.0]бициклононан и т.д.
[00094] Если не указано иное, циклоалкенил включает любую стабильную циклическую или полициклическую углеводородную группу, содержащую одну или более ненасыщенных углерод-углеродных двойных связей в любом положении кольца, которая может быть монозамещенной или полизамещенной и может быть одновалентной, двухвалентной или поливалентной. Примеры циклоалкенила включают, но не ограничиваются перечисленными, циклопентенил, циклогексенил и т.д.
[00095] Если не указано иное, циклоалкинил включает любую стабильную циклическую или полициклическую углеводородную группу, содержащую одну или более ненасыщенных углерод-углеродных тройных связей в любом положении кольца, которая может быть монозамещенной или полизамещенной и может быть одновалентной, двухвалентной или поливалентной.
[00096] Если не указано иное, термин «гало» или «галоген» как таковой или как часть другого заместителя обозначает атом фтора, хлора, брома или иода. Кроме того, термин «галогеналкил» включает моногалогеналкил и полигалогеналкил. Например, термин «галоген(С1-С4)алкил» включает, но не ограничивается перечисленными, трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и т.п. Если не указано иное, примеры галогеналкила включают, но не ограничиваются перечисленными, трифторметил, трихлорметил, пентафторэтил и пентахлорэтил.
[00097] «Алкокси» представляет собой вышеуказанный алкил, содержащий указанное число атомов углерода, присоединенных посредством кислородного мостика, и, если не указано иное, C1-6 алкокси включает алкокси, содержащий C1, С2, С3, С4, С5 и С6. Примеры алкокси включают, но не ограничиваются этим, метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентилокси и S-пентилокси. Если не указано иное, термин «арил» относится к полиненасыщенному ароматическому углеводородному заместителю, который может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или поливалентным, и может представлять собой моноциклические или полициклические кольца (такие как от 1 до 3 колец; по меньшей мере одно из которых является ароматическим), конденсированные вместе или ковалентно связанные. Термин «гетероарил» относится к арильной группе (или кольцу), содержащей от одного до четырех гетероатомов. В одном иллюстративном примере гетероатом выбран из группы, состоящей из В, N, О и S, где атомы азота и серы необязательно являются окисленными и атом азота необязательно является четвертичным. Гетероарил может быть присоединен к остальному фрагменту молекулы посредством гетероатома. Неограничивающие примеры арильных или гетероарильных групп включают фенил, 1-нафтил, 2-нафтил, 4-бифенил, 1-пирролил, 2-пирролил, 3-пирролил, 3-пиразолил, 2-имидазолил, 4-имидазолил, пиразинил, 2-оксазолил, 4-оксазолил, 2-фенил-4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 2-фурил, 3-фурил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидил, 4-пиримидил, 5-бензотиазолил, пуринил, 2-бензимидазолил, 5-индолил, 1-изохинолинил, 5-изохинолинил, 2-хиноксалинил, 5-хиноксалинил, 3-хинолинил и 6-хинолинил. Заместители для любой из вышеуказанных арильных и гетероарильных кольцевых систем выбраны из приемлемых заместителей, описанных ниже.
[00098] Если не указано иное, арильные группы, при использовании в сочетании с другими терминами (например, арилокси, арилтио, арилалкил), включают арильные и гетероарильные кольца, определенные выше. Таким образом, термин «аралкил» включает те группы (например, бензил, фенэтил, пиридилметил и т.д.), в которых арильная группа присоединена к алкильной группе, и включает те алкильные группы, в которых атом углерода (например, метилен) был замещен атомом, таким как кислород, например, феноксиметил, 2-пиридилокеиметил, 3-(1-нафтилокси)пропил и т.п.
[00099] Термин «уходящая группа» относится к функциональной группе или атому, которые могут быть замещены другими функциональной группой или атомом в результате реакции замещения (например, реакции замещения по сродству). Например, типичные уходящие группы включают трифлат; хлор, бром, иод; сульфонатные группы, такие как мезилат, тозилат, 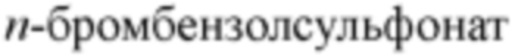 ,
, 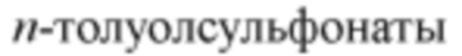 и т.п.; ацилокси, такой как ацетокси, трифторацетокси и т.п.
и т.п.; ацилокси, такой как ацетокси, трифторацетокси и т.п.
[000100] Термин «защитная группа» включает, но не ограничивается перечисленными, «аминозащищающую группу», «гидроксизащищающую группу» или «сульфгидрилзащищающую группу». Термин «аминозащищающая группа» относится к защитной группе, подходящей для блокирования побочной реакции по положению атома азота аминогруппы. Типичные аминозащищающие группы включают, но не ограничиваются перечисленными, формил; ацил, такой как алканоил (например, ацетил, трихлорацетил или трифторацетил); алкоксикарбонил, такой как трет-бутоксикарбонил (Boc); арилметоксикарбонил, такой как бензилоксикарбонил (Cbz) и 9-флуоренилметилоксикарбонил (Fmoc); арилметил, такой как бензил (Bn), тритил (Tr), 1,1-бис-(4'-метоксифенил)метил; силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS), и т.п. Термин «гидроксизащищающая группа» относится к защитной группе, которая подходит для блокирования побочной реакции гидроксильных групп. Типичные гидроксизащищающие группы включают, но не ограничиваются перечисленными, алкил, такой как метил, этил и трет-бутил; ацил, такой как алканоил (такой как ацетил); арилметил, такой как бензил (Bn), 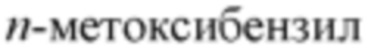 (РМВ), 9-флуоренилметил (Fm) и дифенилметил (бензгидрил, DPM); силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS), и т.п.
(РМВ), 9-флуоренилметил (Fm) и дифенилметил (бензгидрил, DPM); силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS), и т.п.
[000101] Соединения согласно настоящему изобретению могут быть получены различными синтетическими способами, хорошо известными специалистам в данной области техники, включая представленные ниже варианты реализации, их комбинации с другими способами химического синтеза и эквивалентные альтернативы, хорошо известные специалистам в данной области техники, предпочтительные варианты реализации включают, но не ограничиваются перечисленными, варианты реализации согласно настоящему изобретению.
[000102] Растворители, применяемые согласно настоящему изобретению, являются коммерчески доступными.
[000103] В настоящем описании использованы следующие сокращения: водн. для воды; HATU для гексафторфосфата O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония; EDC для гидрохлорида N-(3-диметиламинопропил)-N'-этилкарбодиимида; m-СРВА для 3-хлорпероксибензойной кислоты; экв. для эквивалентного, равного; CDI для карбонилдиимидазола; ДХМ для дихлорметана; ПЭ для петролейного эфира; DIAD для диизопропилазодикарбоксилата; ДМФ для N,N-диметилформамида; ДМСО для диметилсульфоксида; EtOAC для сложного эфира этилацетата; EtOH для этанола; МеОН для метанола; CBz для бензилоксикарбонила, аминозащищающей группы; ВОС для трет-бутоксикарбонила, аминозащищающей группы; НОАс для уксусной кислоты; NaCNBH3 для цианоборгидрида натрия; к.т для комнатной температуры; О/N для «в течение ночи»; ТГФ для тетрагидрофурана; Вос2О для ди-трет-бутилдикарбоната; ТФУ для трифторуксусной кислоты; DIPEA для диизопропилэтиламина; SOCl2 для тионилхлорида; CS2 для дисульфида углерода; TsOH для 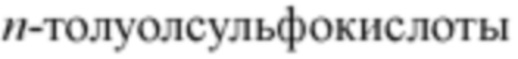 ; NFSI для N-фтор-N-(фенилсульфонил)фенилсульфониламида; NCS для 1-хлорпирролидин-2,5-диона;
; NFSI для N-фтор-N-(фенилсульфонил)фенилсульфониламида; NCS для 1-хлорпирролидин-2,5-диона;  для фторида тетрабутиламмония; iPrOH для 2-пропанола; т.п. для температуры плавления; LDA для литийдиизопропиламида, ФБС для фетальной бычьей сыворотки; ФСБД для фосфатно-солевого буфера Дульбекко; ЭДТА для этилендиаминтетрауксусной кислоты; DMEM для модифицированной по способу Дульбекко среды Игла; CellTiter-Glo (CTG) для способа определения флуоресцентной активности с помощью АТФ; ПО для введения через желудочно-кишечный тракт; ВБ для внутрибрюшинного введения.
для фторида тетрабутиламмония; iPrOH для 2-пропанола; т.п. для температуры плавления; LDA для литийдиизопропиламида, ФБС для фетальной бычьей сыворотки; ФСБД для фосфатно-солевого буфера Дульбекко; ЭДТА для этилендиаминтетрауксусной кислоты; DMEM для модифицированной по способу Дульбекко среды Игла; CellTiter-Glo (CTG) для способа определения флуоресцентной активности с помощью АТФ; ПО для введения через желудочно-кишечный тракт; ВБ для внутрибрюшинного введения.
[000104] Соединения названы вручную или с применением программного обеспечения ChemDraw®, и коммерчески доступные соединения названы по наименованиям в каталогах поставщиков.
[000105] Краткое описание чертежей
[000106] На фиг. 1 показано влияние соединения согласно варианту реализации 1, LY2157299 и BioXcell-mPD-L1 на массу тела самок мышей BALB/c в модели опухоли с подкожной трансплантацией клеток СТ-26.
[000107] Фиг. 2 представляет собой кривую роста опухоли у имеющих опухоль модельных мышей, которым трансплантировали клетки СТ-26, после введения соединения согласно варианту реализации 1, LY2157299 и BioXcell-mPD-L1.
[000108] На фиг. 3 представлены относительные изменения массы тела животных в исследовании ингибирования метастазирования опухолевых клеток у мышей BALB/c в ортотопической трансплантационной модели на клетках рака молочной железы мышей линии 4Т1.
[000109] Подробное описание предпочтительных вариантов реализации
[000110] Следующие примеры дополнительно иллюстрируют настоящее изобретение, но не ограничивают его. Несмотря на то, что настоящее изобретение было описано подробно и со ссылкой на его конкретные варианты реализации, специалистам в данной области техники будет очевидно, что различные изменения и модификации могут быть сделаны без выхода за рамки сущности и объема настоящего изобретения.
[000111] Соединение согласно варианту реализации 1
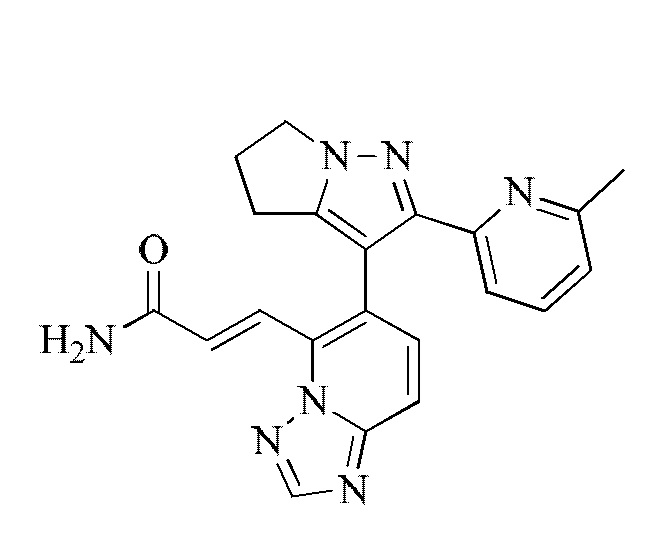
[000112] Получение промежуточного соединения 1-6
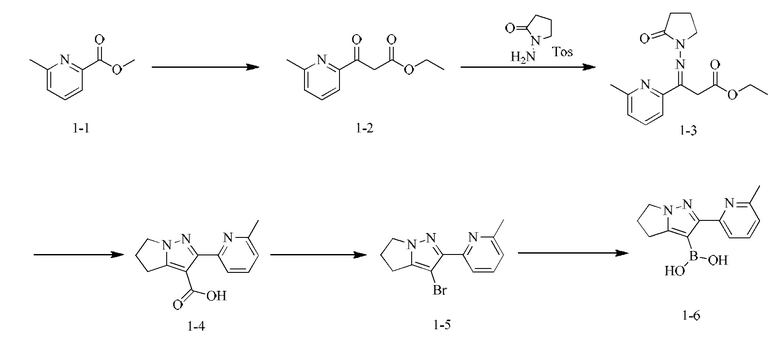
[000113] Стадия А: этилацетат (291,41 мл, 2,98 моль) растворяли в толуоле (750,00 мл), затем порциями при комнатной температуре добавляли этоксид натрия (135,06 г, 1,98 моль), и полученную смесь перемешивали при комнатной температуре в течение 1 ч. К вышеописанному реакционному раствору добавляли метил-6-метилпиридин-2-карбоксилат (150,00 г, 992,33 ммоль) при 25°С, затем нагревали до 95°С и перемешивали в течение 15 ч. Реакционную смесь охлаждали до 30°С, доводили рН до 7 с помощью уксусной кислоты, разбавляли водой (500 мл) и подвергали экстракции этилацетатом (500 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 50/1) с получением этил-3-(6-метилпиридин-2-ил)-3-оксопропаноата (120,00 г, выход: 58,35%).
[000114] Стадия В: этил-3-(6-метилпиридин-2-ил)-3-оксопропаноат (120,00 г, 579,07 ммоль) растворяли в пиридине (300 мл) и затем добавляли  1-аминопирролидин-2-она (172,01 г, 631,66 ммоль). Реакционную смесь перемешивали при 25°С в течение 16 ч и затем концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли водой (300 мл) и затем подвергали экстракции этилацетатом (300 мл × 2). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением этил-3 -(6-метилпиридин-2-ил)-3-((2-оксопирролидин-1-ил)имино)пропаноата (150 г). Выход: 90,28%.
1-аминопирролидин-2-она (172,01 г, 631,66 ммоль). Реакционную смесь перемешивали при 25°С в течение 16 ч и затем концентрировали при пониженном давлении для удаления растворителя. Остаток разбавляли водой (300 мл) и затем подвергали экстракции этилацетатом (300 мл × 2). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением этил-3 -(6-метилпиридин-2-ил)-3-((2-оксопирролидин-1-ил)имино)пропаноата (150 г). Выход: 90,28%.
[000115] Стадия С: этил-3-(6-метилпиридин-2-ил)-3-((2-оксопирролидин-1-ил)имино)пропаноат (155,00 г, 535,72 ммоль) растворяли в толуоле, затем добавляли этоксид натрия (72,91 г, 1,07 моль). Реакционную смесь нагревали до 100°С и перемешивали в течение 16 ч, затем охлаждали до комнатной температуры. Смесь медленно разбавляли водой (1,5 л), доводили рН до 4 с помощью концентрированной соляной кислоты и подвергали экстракции дихлорметаном/изопропиловым спиртом (10/1) (1 л × 7). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток растирали с петролейным эфиром/этилацетатом = 10/1 (200 мл), фильтровали и собирали твердое вещество. Затем твердое вещество сушили при пониженном давлении с получением 2-(6-метилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол-3-карбоновой кислоты (52,80 г, выход: 40,52%).
[000116] Стадия D: 2-(6-метилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол-3-карбоновую кислоту (45,00 г, 184,99 ммоль) растворяли в N,N-диметилформамиде (650,00 мл) и затем добавляли NBS (49,09 г, 258,99 ммоль). Реакционную смесь перемешивали при 30-40°С в течение 60 ч, затем разбавляли водой (600 мл) и подвергали экстракции дихлорметаном/изопропиловым спиртом (10/1) (500 мл × 3). Объединенные органические фазы однократно промывали гидроксидом натрия (0,5 моль/л, 800 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Затем полученное твердое вещество растирали с петролейным эфиром/этилацетатом = 10/1 (200 мл), фильтровали и собирали твердое вещество. Твердое вещество сушили при пониженном давлении с получением 3-бром-2-(6-метилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразола (33,00 г, выход: 64,13%.).
[000117] Стадия Е: 3-бром-2-(6-метилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол (1,00 г, 3,60 ммоль) и триизопропилборат (1,79 г, 9,54 ммоль) растворяли в тетрагидрофуране (20,00 мл). Реакционную смесь охлаждали до минус 70°С, затем добавляли по каплям н-бутиллитий (2,5 М, 3,74 мл). После завершения добавления по каплям реакционную смесь перемешивали при 25°С в течение 1 ч, затем доводили рН до 7 с помощью водного раствора соляной кислоты (0,5 моль/л), далее концентрировали при пониженном давлении для удаления тетрагидрофурана и охлаждали до 15°С. Смесь фильтровали, и осадок на фильтре растирали с петролейным эфиром/этилацетатом = 10/1 (200 мл), фильтровали и собирали твердое вещество. Твердое вещество сушили при пониженном давлении с получением (2-(6-метилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол-3-ил)борной кислоты (750 мг, выход: 85,71%).
[000118] Получение соединения согласно варианту реализации 1
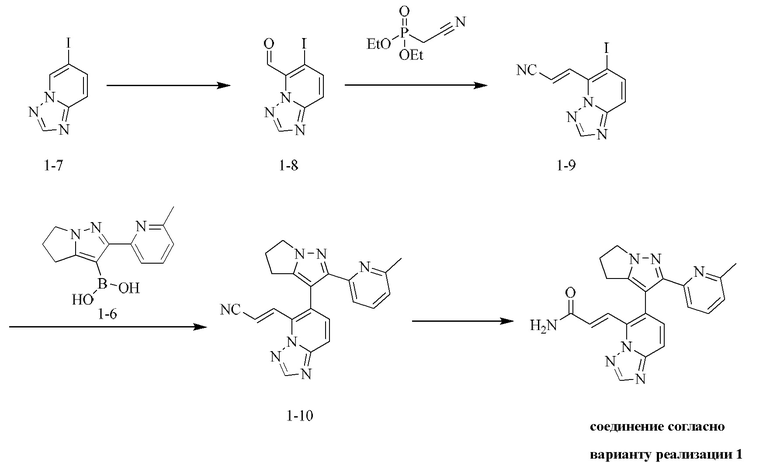
[000119] Стадия А: 6-иод-[1,2,4]триазоло[1,5-а]пиридин (16,00 г, 65,50 ммоль) растворяли в тетрагидрофуране (800,00 мл) и охлаждали до -60 - -70°С и затем добавляли по каплям гексаметилдисилазид лития (1 моль/л, 130,60 мл, 65,30 ммоль). Реакционную смесь перемешивали при -60 - -70°С в течение 15 мин и добавляли N,N-диметилформамид (14,32 г, 195,90 ммоль, 15,07 мл). Реакционную смесь дополнительно перемешивали при -60 - -70°С в течение 15 мин и затем в нее добавляли для гашения насыщенный водный раствор хлорида аммония (500 мл). Реакционную смесь нагревали до комнатной температуры и затем подвергали экстракции этилацетатом (500 мл × 2). Объединенные органические слои промывали солевым раствором (500 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (элюент: дихлорметан/этилацетат = 10/1) с получением 6-иод-[1,2,4]триазоло[1,5-а]пиридин-5-карбальдегида (6,40 г, выход: 35,90%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,46 (s, 1H), 8,62 (s, 1H), 8,16 (d, J=9,3 Гц, 1H), 7,88 (d, J=9,3 Гц, 1H).
[000120] Стадия В: 2-диэтоксифосфорилацетонитрил (3,83 г, 21,61 ммоль, 3,48 мл) и тетрагидрофуран (80 мл) добавляли в трехгорлую колбу объемом 500 мл, снабженную термометром и баллоном с азотом. Смесь охлаждали до 0°С. И затем добавляли порциями трет-бутоксид калия (2,42 г, 21,61 ммоль). Реакционную смесь перемешивали при 0°С в течение 15 мин и затем добавляли по каплям через капельную воронку к другой суспензии (6-иод-[1,2,4]триазоло[1,5-а]пиридин-5-карбальдегида, диспергированного в тетрагидрофуране (120 мл) и охлажденного до 0°С). Реакционную смесь перемешивали при 0°С в течение 15 мин, затем в нее добавляли для гашения воду (300 мл), подвергали экстракции этилацетатом (200 мл) и дихлорметаном (200 мл). Объединенную органическую фазу промывали солевым раствором (300 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (элюент: дихлорметан/этилацетат = от 200/1 до 10/1) с получением (E)-3-(6-иод-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилонитрила (4,2 г, выход: 65,66%). 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 8,42 (s, 1H), 8,03 (d, J=9,3 Гц, 1H), 7,98-7,91 (m, 1H), 7,85-7,78 (m, 1H), 7,60 (d, J=9,2 Гц, 1H).
[000121] Стадия С: (E)-3-(6-иод-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилонитрил (4,50 г, 15,20 ммоль), [2-(6-метил-2-пиридил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол-3-ил]борную кислоту (4,43 г, 18,24 ммоль), карбонат натрия (4,83 г, 45,60 ммоль), дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (556,07 мг, 759,96 мкмоль), 2-дициклогексилфосфин-2',6'-диметоксибифенил (311,98 мг, 759,96 мкмоль) и [2-(2-аминофенил)фенил]хлорпалладийциклогексил-[2-(2,6-диметокси)фенил)фенил]фосфин (547,64 мг, 759,96 мкмоль) добавляли к смеси растворителей, состоящей из диоксана (100 мл) и воды (20 мл). Смесь насыщали азотом 3 раза, затем нагревали до 90-100°С и перемешивали в течение 2 ч. В реакционную смесь добавляли для гашения воду (200 мл) и подвергали экстракции дихлорметаном (200 мл × 2). Объединенные органические слои промывали солевым раствором (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (элюент: дихлорметан/метанол = 30/1) с получением неочищенного продукта, и неочищенный продукт перемешивали в течение 12 ч в смеси растворителей, состоящей из петролейного эфира/этилацетата = 5/1, фильтровали, и твердое вещество собирали и концентрировали с получением (E)-3-(6-(2-(6-метилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол-3-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилонитрила (5,37 г, выход: 96,16%). 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 8,49 (s, 1H), 7,82-7,74 (m, 2Н), 7,59-7,46 (m, 4Н), 6,99(dd, J=2,6, 6,1 Гц, 1H), 4,39 (d, J=6,3 Гц, 2Н), 2,90-2,70 (m, 4Н), 2,20 (s, 3Н).
[000122] Стадия D: (E)-3-(6-(2-(6-метилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол-3-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилонитрил (5,37 г, 14,62 ммоль) растворяли в смеси растворителей, состоящей из дихлорметана (20 мл), диметилсульфоксида (70 мл) и воды (20 мл), и затем добавляли перекись водорода (8,29 г, 73,10 ммоль, 7,02 мл, 30%) и гидроксид натрия (2 моль/л, 14,62 мл). Смесь перемешивали при 15-20°С в течение 12 ч. Смесь гасили путем выливания в воду (200 мл) и подвергали экстракции смесью растворителей (200 мл × 1), состоящей из дихлорметана/изопропилового спирта (3/1). Органический слой промывали насыщенным водным раствором тиосульфата натрия (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной высокоэффективной жидкостной хроматографии (колонка: Phenomenex Gemini С18, 250 × 50 мм × 10 мкм; подвижная фаза: [вода (0,05% аммиак об/об) - ацетонитрил]; градиент: 5%-32%, 33; 80% мин) с получением соединения согласно варианту реализации 1 (3,6 г, выход: 63,82%). 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 8,45 (s, 1H), 8,09 (d, J=15,6 Гц, 1H), 7,85 (d, J=15,6 Гц, 1H), 7,69 (d, J=9,2 Гц, 1H), 7,55-7,45 (m, 2Н), 7,37 (d, J=7,8 Гц, 1H), 6,99 (d, J=7,7 Гц, 1H), 5,93-5,65 (m, 2Н), 4,35 (ушир. s, 2Н), 2,99-2,64 (m, 4Н), 2,33 (s, 3Н).
[000123] Соединение согласно варианту реализации 2
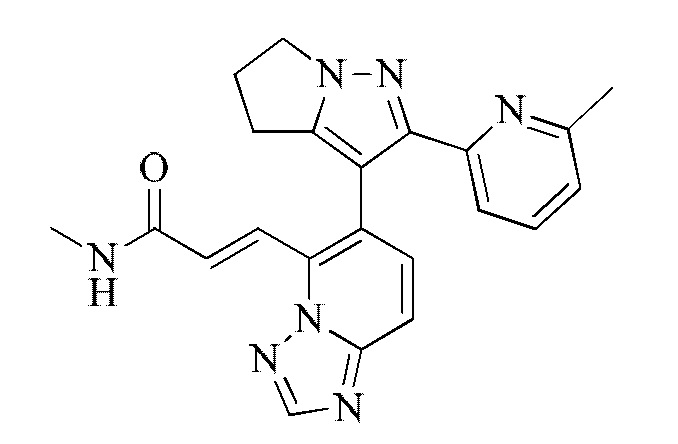
[000124] Получение соединения согласно варианту реализации 2
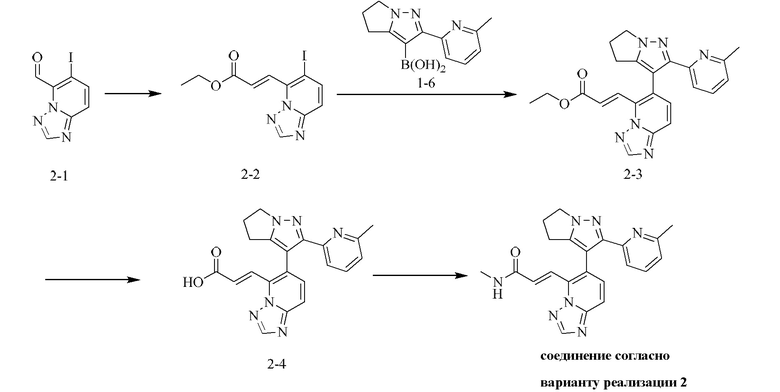
[000125] Стадия А: этил-2-диэтоксифосфорилацетат (295,93 мг, 1,32 ммоль, 261,88 мкл) растворяли в тетрагидрофуране (6 мл) и охлаждали до 0°С, и добавляли одной порцией гидрид натрия (52,80 мг, 1,32 ммоль). Реакционную смесь перемешивали при 0°С в течение 15 мин и затем добавляли по каплям к другой суспензии (6-иод-[1,2,4]триазоло[1,5-а]пиридин-5-карбальдегида (300 мг, 1,10 ммоль), диспергированного в тетрагидрофуране (6 мл) и охлажденного до -10 - -15°С). Реакционную смесь перемешивали при -10 - -15°С в течение 15 мин, гасили путем выливания в насыщенный водный раствор хлорида аммония (20 мл) и затем подвергали экстракции дихлорметаном (20 мл × 3). Объединенные органические слои промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (элюент: дихлорметан/этил ацетат = 10/1) с получением (E)-этил-3-(6-иод-[1,2,4]триазоло[1,5-а]пиридин)-5-ил)акрилата (330 мг, выход: 87,43%).
[000126] Стадия В: (E)-этил-3-(6-иод-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилат (330 мг, 961,76 мкмоль), [2-(6-метил-2-пиридил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол-3-ил]борную кислоту (268,84 мг, 1,11 ммоль), карбонат натрия (305,81 мг, 2,89 ммоль), дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия, дихлорметан (39,27 мг, 48,09 мкмоль), дициклогексилфосфин-2',6'-диметоксибифенил (19,74 мг, 48,09 мкмоль) и [2-(2-)аминофенил)фенил]хлорпалладийциклогексил-[2-(2,6-диметоксифенил)фенил]фосфин (34,65 мг, 48,09 мкмоль) добавляли к смеси растворителей, состоящей из диоксана (10 мл) и воды (2 мл). Реакционную смесь насыщали азотом три раза, затем нагревали до 90-100°С и перемешивали в течение 2 ч. Реакционную смесь гасили путем выливания в воду (20 мл) и подвергали экстракции дихлорметаном (200 мл × 3). Объединенные органические слои промывали солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1) с получением (E)-этил-3-(6-(2-(6-метилпиридин-2-ил)-5,6-дигидро)-4Н-пирроло[1,2-b]пиразол-3-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилата (359 мг, выход: 81,57%).
[000127] Стадия С: (E)-этил-3-(6-(2-(6-метилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол-3-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилат (359,00 мг, 866,19 мкмоль) растворяли в смеси растворителей, состоящей из тетрагидрофурана (6 мл) и воды (2 мл), затем добавляли одной порцией моногидрат гидроксида лития (109,04 мг, 2,6 ммоль). Реакционную смесь перемешивали при 15-20°С в течение 12 ч, затем разбавляли водой (15 мл) и доводили рН до 5-6 с помощью разбавленной соляной кислоты (1 моль/л) и затем подвергали экстракции дихлорметаном (20 мл × 1) Органическую фазу промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением (Е)-3-(6-(2-(6-метилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол-3-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акриловой кислоты (330 мг, выход: 98,59%).
[000128] Стадия D: (E)-3-(6-(2-(6-метилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол-3-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акриловую кислоту (65 мг, 168,22 мкмоль), гидрохлорид метиламина (22,72 мг, 336,44 мкмоль), HATU (127,92 мг, 336,44 мкмоль) и триэтиламин (68,09 мг, 672,88 мкмоль, 93,27 мкл) растворяли в N,N-диметилформамиде (2 мл). Реакционную смесь перемешивали при 15-20°С в течение 12 ч, разбавляли непосредственно метанолом (2 мл) и очищали посредством препаративной высокоэффективной жидкостной хроматографии (колонка: Phenomenex Gemini 150 × 25 мм × 10 мкм; подвижная фаза: [вода (0,05) % аммиачной воды (об/об)-ацетонитрил]; градиент: 21%-51%, 15 мин) с получением соединения согласно варианту реализации 2 (27,79 мг, выход: 41,36%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,67 (s, 1H), 8,43 (d, J=4,6 Гц, 1H), 7,93-7,80 (m, 2Н), 7,68-7,61 (m, 2Н), 7,60-7,49 (m, 2Н), 7,02 (dd, J=1,6, 6,8 Гц, 1H), 4,29 (d, J=9,0 Гц, 2Н), 2,84-2,72 (m, 2Н), 2,69-2,57 (m, 5Н), 1,99 (s, 3Н).
[000129] Соединения согласно вариантам реализации 3-5 могут быть получены в соответствии со способом получения соединения согласно варианту реализации 2.
[000130] Соединение согласно варианту реализации 3
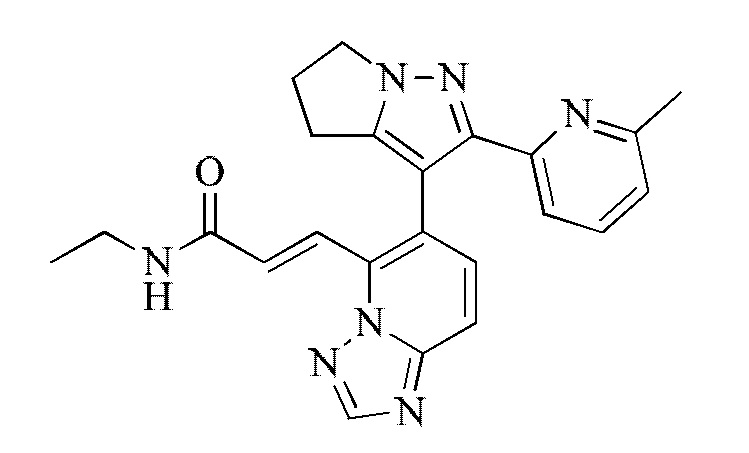
[000131] 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,67 (s, 1H), 8,48 (ушир. t, J=5,3 Гц, 1H), 7,95-7,78 (m, 2Н), 7,69-7,46 (m, 4Н), 7,02 (dd, J=1,6, 6,7 Гц, 1H), 4,29 (ушир. d, J=7,5 Гц, 2Н), 3,26-3,08 (m, 2Н), 2,81-2,58 (m, 4Н), 1,99 (s, 3Н), 1,04 (t, J=7,2 Гц, 3Н).
[000132] Соединение согласно варианту реализации 4
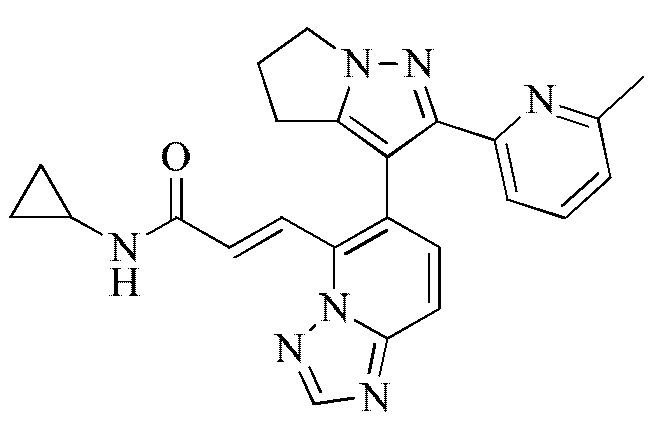
[000133] 1H ЯМР (400 МГц, ДМСО-d6) δ 8,66 (s, 1H), 8,53 (d, J=4,8 Гц, 1H), 8,14 (s, 1H), 7,89-7,81 (m, 2Н), 7,69-7,61 (m, 2Н), 7,60-7,48 (m, 2Н), 7,06-7,00 (m, 1H), 4,30 (d, J=8,9 Гц, 2Н), 2,83-2,73 (m, 3Н), 2,66-2,60 (m, 2Н), 1,99 (s, 3Н), 0,65 (d, J=5,6 Гц, 2Н), 0,46 (d, J=2,8 Гц, 2Н). [000134] Соединение согласно варианту реализации 5
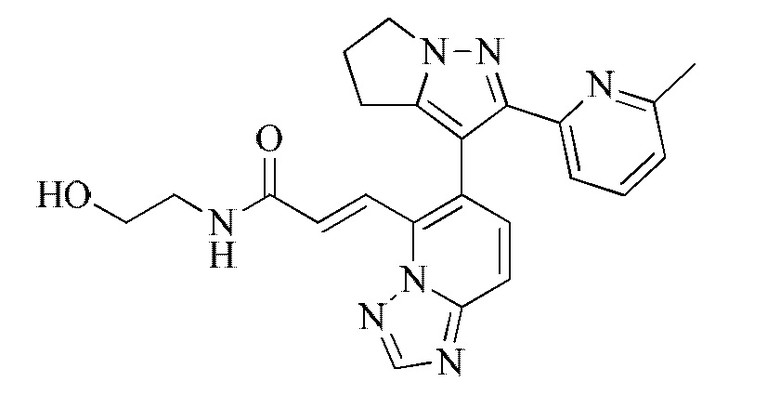
[000135] 1H ЯМР (400 МГц, ДМСО-d6) δ 8,67 (s, 1H), 8,50 (t, J=5,6 Гц, 1H), 7,93 (d, J=15,6 Гц, 1H), 7,83 (d, J=9,2 Гц, 1H), 7,69-7,61 (m, 2Н), 7,59-7,49 (m, 2Н), 7,02 (dd, J=1,8, 6,7 Гц, 1H), 4,69 (t, J=5,5 Гц, 1H), 4,34-4,24 (m, 2Н), 3,43 (q, J=5,9 Гц, 2Н), 3,21 (d, J=3,1 Гц, 2Н), 2,83-2,73 (m, 2Н), 2,62 (quin, J=7,2 Гц, 2Н), 1,99 (s, 3Н).
[000136] Соединение согласно варианту реализации 6
[000137] Получение промежуточного соединения 6-4
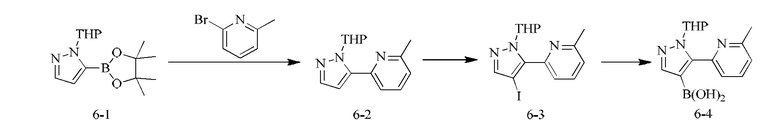
[000138] Стадия А: 1-тетрагидропиран-2-ил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиразол (10,96 г, 39,41 ммоль), 2-бром-6-метилпиридин (6,00 г, 34,88 ммоль, 3,97 мл), дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (1,28 г, 1,74 ммоль) и карбонат натрия (11,09 г, 104,64 ммоль) добавляли к смеси растворителей, состоящей из диоксана (200,00 мл) и воды (40,00 мл). Реакционную смесь насыщали азотом три раза, затем нагревали до 80-90°С и перемешивали в течение 3 ч, затем гасили путем выливания в воду (200 мл) и подвергали экстракции этилацетатом (180 мл × 2). Объединенную органическую фазу промывали солевым раствором (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 10/1-5/1) с получением 2-метил-6-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)пиридина (4,10 г, неочищенный). Продукт был идентифицирован как неочищенный методом ЯМР.
[000139] Стадия В: 2-метил-6-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)пиридин (2,10 г, неочищенный) растворяли в уксусной кислоте (20,00 мл), затем добавляли одной порцией NIS (2,04 г, 9,06 ммоль). Смесь нагревали до 70-80°С и перемешивали в течение 1 ч, затем гасили путем выливания в насыщенный раствор бикарбоната натрия (50 мл) и затем подвергали экстракции этилацетатом (50 мл × 3). Объединенную органическую фазу промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 10/1) с получением 2-(4-иод-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)-6-метилпиридина (1,60 г, выход: 50,22%).
[000140] Стадия С: 2-(4-иод-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)-6-метилпиридин (500,00 мг, 1,35 ммоль) и триизопропилборат (672,82 мг, 3,58 ммоль, 820,51 мкл) растворяли в тетрагидрофуране (10 мл). Реакционную смесь охлаждали до -78°С, добавляли по каплям н-бутиллитий (2,5 М, 1,40 мл) и перемешивали при -78 - -60°С в течение 30 мин. Реакционную смесь гасили путем выливания в насыщенный раствор хлорида аммония, перемешивали в течение 10 мин и подвергали экстракции этилацетатом (20 мл × 2). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1) с получением (5-(6-метилпиридин-2-ил)-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-4-ил) борной кислоты (200,00 мг, выход: 51,60%).
[000141] Получение соединения согласно варианту реализации 6
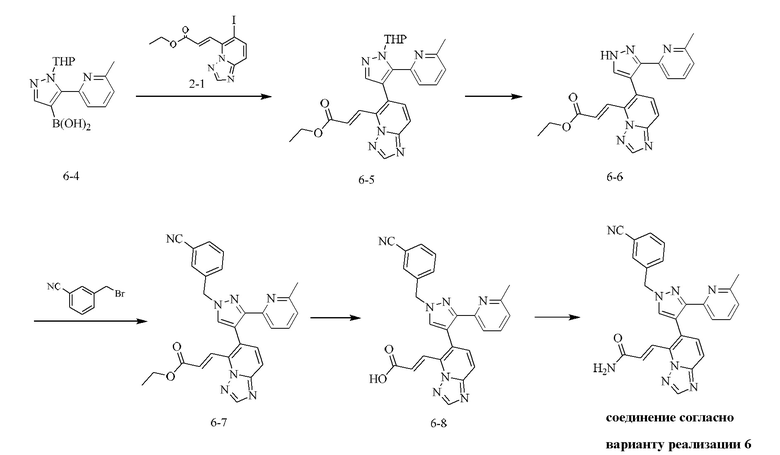
[000142] Стадия А: [5-(6-метил-2-пиридил)-1-тетрагидропиран-2-илпиразол-4-ил]борную кислоту (200,00 мг, 696,57 мкмоль), (E)-этил-3-(6-иод-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилат (239,01 мг, 696,57 мкмоль), карбонат натрия (221,49 мг, 2,09 ммоль), дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (50,97 мг, 69,66 мкмоль), дициклогексилфосфин-2',6'-диметоксибифенил (28,60 мг, 69,66 мкмоль) и [2-(2-аминофенил)фенил]хлорпалладийциклогексил-[2-(2,6-диметоксифенил)фенил]фосфин (50,20 мг, 69,66 мкмоль) добавляли к смеси растворителей, состоящей из диоксана (3 мл) и воды (1 мл). Смесь насыщали азотом 3 раза, затем нагревали до 80-90°С и перемешивали в течение 3 ч. Реакционную смесь гасили путем выливания в воду (30 мл) и подвергали экстракции этилацетатом (30 мл × 2). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной хроматографии на силикагеле (элюент: дихлорметан/метанол = 30/1) с получением (E)-этил-3-(6-(5-(6-метилпиридин-2-ил)-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилата (250,00 мг, выход: 78,27%).
[000143] Стадия В: (E)-этил-3-(6-(5-(6-метилпиридин-2-ил)-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилат (250,00 мг, 545,24 мкмоль) растворяли в этаноле (3 мл), затем добавляли гидрохлорид диоксана (4 М, 5,01 мл). Реакционную смесь перемешивали при 15-20°С в течение 12 ч, упаривали для удаления растворителя, затем доводили рН до 8-9 с помощью насыщенного раствора бикарбоната натрия (20 мл) и подвергали экстракции дихлорметаном (20 мл × 2). Объединенные органические фазы промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением (E)-этил-3-(6-(3-(6-метилпиридин-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилата (230,00 мг, неочищенный).
[000144] Стадия С: (E)-этил-3-(6-(3-(6-(6-метилпиридин-2-ил))-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилат растворяли в ТГФ (5 мл). Реакционную смесь охлаждали до -20°С, затем добавляли гидрид натрия (27,03 мг, 675,75 мкмоль) и перемешивали при -20°С в течение 30 мин. Затем добавляли 3-цианобензилбромид (132,47 мг, 675,75 мкмоль). Реакционную смесь нагревали до 15-20°С и дополнительно перемешивали в течение 4 ч, затем гасили путем выливания в воду (20 мл), рН доводили до 5-6 с помощью разбавленного водного раствора соляной кислоты (1 М) и подвергали экстракции этилацетатом (20 мл). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной хроматографии на силикагеле (элюент: дихлорметан/метанол = 30/1) с получением (E)-этил-3-(6-(1-(3-цианобензил)-3-(6-метилпиридин-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилата (150,00 мг, выход: 46,67%).
[000145] Стадия D: (E)-этил-3-(6-(1-(3-цианобензил)-3-(6-метилпиридин-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилат (150,00 мг, 306,42 мкмоль) растворяли в тетрагидрофуране (3 мл), затем добавляли одной порцией моногидрат гидроксида лития (38,57 мг, 919,26 мкмоль). Реакционную смесь перемешивали при 15-20°С в течение 12 ч, затем гасили путем выливания в воду (10 мл) и доводили рН до 5-6 с помощью разбавленного водного раствора соляной кислоты (1 М), затем подвергали экстракции дихлорметаном (20 мл × 2). Объединенные органические фазы промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением (E)-3-(6-(1-(3-цианобензил)-3-(6-метилпиридин-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акриловой кислоты (130,00 мг, выход: 91,94%).
[000146] Стадия Е: (E)-3-(6-(1-(3-цианобензил)-3-(6-метилпиридин-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акриловую кислоту (130,00 мг, 281,71 мкмоль), HATU (214,23 мг, 563,42 мкмоль) и триэтиламин (57,01 мг, 563,42 мкмоль, 78,10 мкл) растворяли в N,N-диметилформамиде (2 мл). После перемешивания реакционной смеси при 15-20°С в течение 1 ч добавляли 3 мл раствора аммиака в тетрагидрофуране (насыщенного при 0°С). Реакционную смесь дополнительно перемешивали при 15-20°С в течение 30 мин, концентрировали при пониженном давлении для удаления растворителя и затем разбавляли метанолом (2 мл). Остаток очищали посредством высокоэффективной жидкостной хроматографии (колонка: Phenomenex Synergi С18 150 × 30 мм × 4 мкм; подвижная фаза: [вода (0,225% муравьиная кислота)-ацетонитрил]; градиент: 15% - 45%, 12 мин) с получением соединения согласно варианту реализации 6 (53,00 мг, выход: 40,32%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,69 (s, 1H), 8,25 (s, 1H), 7,93-7,81 (m, 5Н), 7,67-7,52 (m, 6Н), 7,24 (ушир. s, 1H), 7,04 (dd, J=2,0, 6,2 Гц, 1H), 5,61 (s, 2Н), 1,98 (s, 3Н).
[000147] Соединение согласно варианту реализации 7 может быть получено в соответствии со способом получения соединения согласно варианту реализации 6.
[000148] Соединение согласно варианту реализации 7
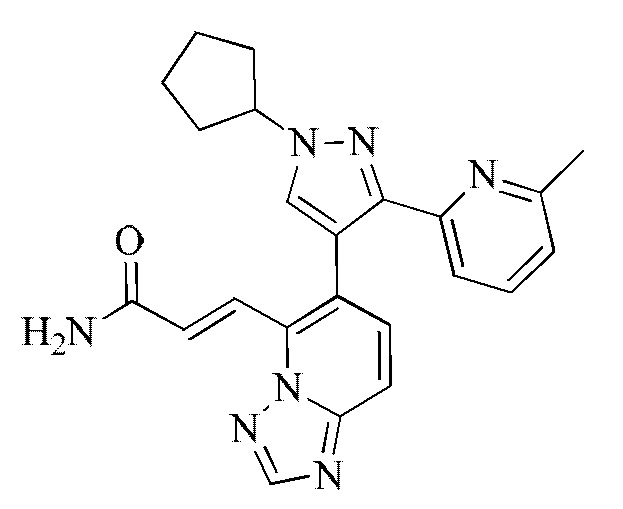
[000149] 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,67 (s, 1H), 8,41-8,36 (m, 1H), 8,38 (s, 1H), 8,09 (s, 1H), 7,89-7,78 (m, 3Н), 7,69-7,58 (m, 3Н), 7,51 (d, J=15,7 Гц, 1H), 7,16 (ушир. s, 1H), 7,02 (d, J=6,8 Гц, 1H), 4,86 (quin, J=6,9 Гц, 1H), 2,22-2,16 (m, 2Н), 2,14-2,04 (m, 2Н), 1,97 (s, 3Н), 1,92-1,83 (m, 2Н), 1,76-1,66 (m, 2Н).
[000150] Соединение согласно варианту реализации 8
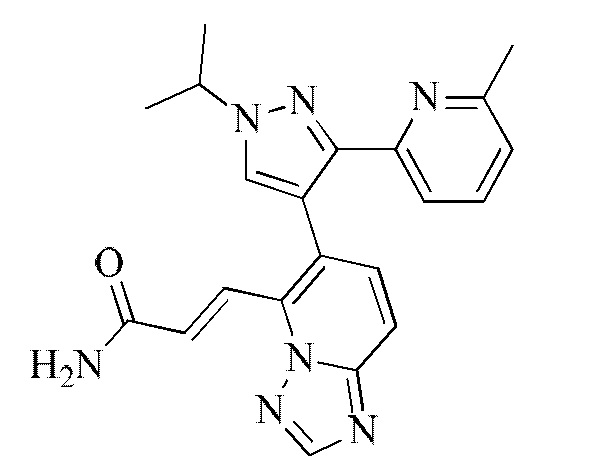
[000151] Получение промежуточного соединения 8-2
[000152] Стадия А: [5-(6-метил-2-пиридил)-1-тетрагидропиран-2-илпиразол-4-ил]борную кислоту (470,00 мг, 1,64 ммоль), (E)-3-(6-иод-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилонитрил (485,55 мг, 1,64 ммоль), карбонат натрия (521,47 мг, 4,92 ммоль), дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (36,00 мг, 49,20 мкмоль), бисциклогексилфосфино-2',6'-диметоксибифенил (6,73 мг, 16,40 мкмоль) и [2-(2-аминофенил)фенил]хлорпалладийциклогексил-[2-(2,6-диметоксифенил)фенил]фосфин (11,82 мг, 16,40 мкмоль) добавляли к смеси растворителей, состоящей из диоксана (20 мл) и воды (5 мл). Реакционную смесь насыщали азотом 3 раза, затем нагревали до 80-90°С и перемешивали в течение 12 ч, затем гасили путем выливания в воду (30 мл) и подвергали экстракции этилацетатом (30 мл × 2). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт перемешивали в течение 30 минут в смеси растворителей, состоящей из петролейного эфира (12 мл) и этилацетата (4 мл), и фильтровали. Твердое вещество собирали и концентрировали при пониженном давлении с получением (E)-3-(6-(5-(6-метилпиридин-2-ил)-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилонитрила (554,00 мг, выход: 82,32%).
[000153] Стадия В: (E)-3-(6-(5-(6-метилпиридин-2-ил)-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилонитрил (554,00 мг, 1,35 ммоль) растворяли в метаноле (5 мл), затем добавляли гидрохлорид диоксана (4 моль/л, 5 мл). Реакционную смесь перемешивали при 15-20°С в течение 12 ч, концентрировали при пониженном давлении для удаления растворителя и доводили рН до 8-9, затем подвергали экстракции дихлорметаном (20 мл × 2). Объединенные органические фазы промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением (E)-метил-3-(6-(3-(6-метилпиридин-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилата (500,00 мг, неочищенный).
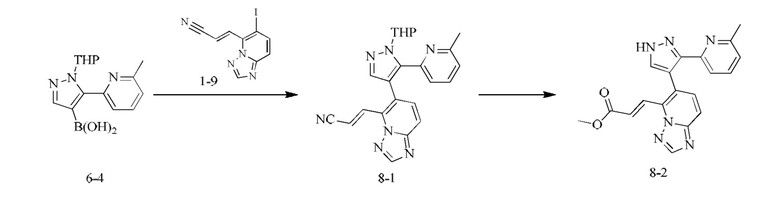
[000154] Получение соединения согласно варианту реализации 8
[000155] Стадия А: (E)-метил-3-(6-(3-(6-(6-метилпиридин-2-ил))-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилат (260,00 мг, неочищенный) растворяли в тетрагидрофуране (4 мл). Реакционную смесь охлаждали до 0°С, добавляли одной порцией гидрид натрия (31,75 мг, 793,63 мкмоль), затем перемешивали при 0°С в течение 30 мин, далее добавляли иодизопропан (134,91 мг, 793,63 мкмоль). Реакционную смесь перемешивали при 15-20°С в течение 12 ч. Контроль хода реакции методом ЖХ-МС показал, что (E)-метил-3-(6-(3-(6-метилпиридин-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилат был израсходован полностью, но целевой продукт не образовался (МС (ИЭР) m/z: 347 [М+Н+]). Смесь концентрировали при пониженном давлении для удаления тетрагидрофурана, и остаток растворяли в N,N-диметилформамиде (3 мл), затем добавляли 2-иодизопропан (613,22 мг, 3,61 ммоль, 360,72 мкл) и калий (498,58 мг, 3,61 ммоль). Реакционную смесь перемешивали в течение еще 12 ч при 15-20°С. Контроль хода реакции методом ЖХ-МС свидетельствовал о завершении реакции. Смесь гасили путем выливания в воду (20 мл) и затем подвергали экстракции этилацетатом (30 мл × 3). Объединенные органические фазы промывали солевым раствором (60 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной хроматографии на силикагеле (элюент: метиленхлорид/метанол = 30/1) с получением (E)-изопропил-3-(6-(1-изопропил-3-(6-метилпиридин-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилата (100,00 мг, неочищенный).
[000156] Стадия В: (E)-изопропил-3-(6-(1-изопропил-3-(6-метилпиридин-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилат (100,00 мг, неочищенный) растворяли в смеси растворителей, состоящей из из тетрагидрофурана (1 мл), метанола (1 мл) и воды (1 мл), затем добавляли одной порцией моногидрат гидроксида лития (29,24 мг, 696,87 мкмоль). Реакционную смесь перемешивали при 15-20°С в течение 3 ч, затем доводили рН до 5-6 с помощью разбавленной соляной кислоты (5%) и подвергали экстракции этилацетатом (20 мл × 3). Объединенные органические фазы промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением (E)-3-(6-(1-изопропил-3-(6-метилпиридин-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акриловой кислоты (100,00 мг, неочищенная).
[000157] Стадия С: (E)-3-(6-(1-изопропил-3-(6-метилпиридин-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акриловую кислоту (100,00 мг, неочищенная) растворяли в тетрагидрофуране (3 мл), затем добавляли HATU (195,78 мг, 514,90 мкмоль) и триэтиламин (52,10 мг, 514,90 мкмоль, 71,37 мкл) одной порцией, соответственно. После перемешивания реакционной смеси при 15-20°С в течение 1 ч добавляли 3 мл раствора аммиака в тетрагидрофуране (насыщенного при 0°С). Реакционную смесь дополнительно перемешивали при 15-20°С в течение 12 ч. Растворитель концентрировали при пониженном давлении для удаления растворителя и разбавляли метанолом (3 мл) и затем очищали посредством препаративной высокоэффективной жидкостной хроматографии (колонка: Phenomenex Synergi С18 150 × 30 мм × 4 мкм; подвижная фаза: [вода (0,225% муравьиная кислота)-ацетонитрил]; градиент: 10-40%, 12 мин) с получением соединения согласно варианту реализации 8 (30,50 мг, выход: 30,08%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,68 (s, 1H), 8,10 (s, 1H), 7,89-7,81 (m, 3Н), 7,68-7,59 (m, 3Н), 7,51 (d, J=15,7 Гц, 1H), 7,18 (ушир. s, 1H), 7,02 (dd, J=1,2, 7,1 Гц, 1H), 4,67 (quin, J=6,7 Гц, 1H), 1,97 (s, 3Н), 1,55 (d, J=6,7 Гц, 6Н).
[000158] Соединение согласно варианту реализации 9
[000159] Получение соединения согласно варианту реализации 9
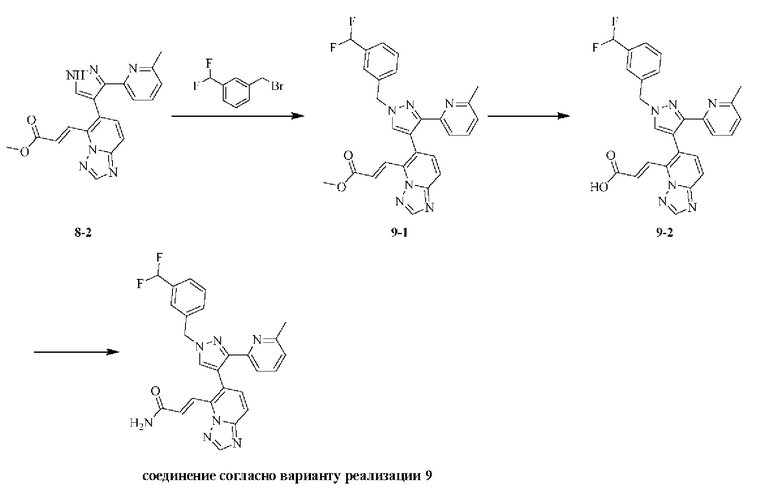
[000160] Стадия А: (E)-метил-3--(6-(3-(6-метилпиридин-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилат (200,00 мг, неочищенный) растворяли в тетрагидрофуране (5 мл). После охлаждения до 0°С добавляли одной порцией гидрид натрия (24,42 мг, 610,49 мкмоль). Реакционную смесь перемешивали при 0°С в течение 30 мин, затем добавляли 1-(бромметил)-3-(дифторметил)бензол (134,94 мг, 610,49 мкмоль), затем реакционную смесь нагревали до 15-20°С и далее перемешивали в течение 5 ч. Реакционную смесь гасили путем выливания в воду (20 мл) и подвергали экстракции этилацетатом (20 мл × 3). Объединенные органические фазы промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением (E)-метил-3-(6-(1-(3-(дифторметил)бензил)-3-(6-метилпиридин-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилата (150,00 мг, неочищенный).
[000161] Стадия В: (E)-метил-3-(6-(1-(3-(дифторметил)бензил)-3-(6-метилпиридин-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилат (150,00 мг, неочищенный) растворяли в смеси растворителей, состоящей из из тетрагидрофурана (1 мл), метанола (1 мл) и воды (1 мл), затем добавляли одной порцией моногидрат гидроксида лития (37,73 мг, 899,10 мкмоль). Реакционную смесь перемешивали при 15-20°С в течение 10 мин и доводили рН до 5-6 с помощью разбавленной соляной кислоты (0,5 М), при этом осаждались твердые вещества. Твердое вещество отфильтровывали и собирали с получением (Е)-3-(6-(1-(3-(дифторметил)бензил)-3-(6-метилпиридин-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акриловой кислоты (120,00 мг, выход: 60,34%).
[000162] Стадия С: (E)-3-(6-(1-(3-(дифторметил)бензил)-3-(6-метилпиридин-2-ил)-1H-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акриловую кислоту (120,00 мг, 180,83 мкмоль), HATU (187,59 мг, 493,36 мкмоль) и триэтиламин (49,92 мг, 493,36 мкмоль, 68,38 мкл) были растворяли в тетрагидрофуране (3 мл). Реакционную смесь перемешивали при 15-20°С в течение 1 ч, затем добавляли 3 мл раствора аммиака в тетрагидрофуране (насыщенного при 0°С). Реакционную смесь перемешивали при 15-20°С в течение 12 ч, затем гасили путем выливания в воду и подвергали экстракции этилацетатом (20 мл × 3). Объединенные органические фазы промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной высокоэффективной жидкостной хроматографии (колонка: Phenomenex Synergi С18 150 × 30 мм × 4 мкм; подвижная фаза: [вода (0,225% муравьиная кислота)-ацетонитрил]; градиент 15% - 45%, 12 мин) с получением соединения согласно варианту реализации 9 (47,85 мг, выход: 39,81%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,70 (s, 1H), 8,26 (s, 1H), 7,95-7,83 (m, 3Н), 7,67-7,51 (m, 8Н), 7,28 (d, J=11,2 Гц, 1H), 7,07-6,96 (m, 1H), 7,12 (s, 1H), 5,61 (s, 2Н), 1,97 (s, 3Н).
[000163] Соединение согласно варианту реализации 10
[000164] Получение промежуточного соединения 10-3
[000165] Стадия А: этил-6-хлорпиридин-2-карбоксилат (500,00 мг, 2,69 ммоль), винилтрибутилолово (887,12 мг, 2,80 ммоль, 813,87 мкл) и тетракис(трифенилфосфин)палладий (155,42 мг, 134,50 мкмоль) растворяли в толуоле (10 мл). Реакционную смесь насыщали азотом три раза, затем нагревали до 110-120°С и перемешивали в течение 3 ч. После охлаждения смесь выливали в насыщенный раствор фторида калия (30 мл) и перемешивали в течение 30 мин. Смесь фильтровали, и осадок на фильтре промывали этилацетатом (10 мл × 3). Фильтрат подвергали экстракции этилацетатом (30 мл × 2). Объединенные органические фазы промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 10/1) с получением этил-6-винилпиридин-2-карбоксилата (364,00 мг, выход: 76,21%). 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) 8,00 (dd, J=0,8, 7,8 Гц, 1H), 7,81 (t, J=7,8 Гц, 1H), 7,61 (dd, J=0,9, 7,9 Гц, 1H), 6,96 (dd, J=10,9, 17,6 Гц, 1H), 6,25 (dd, J=0,6, 17,6 Гц, 1H), 5,67-5,56 (m, 1H), 4,50 (q, J=7,2 Гц, 2Н), 1,46 (t, J=7,2 Гц, 3Н).
[000166] Стадия В: этил-6-винилпиридин-2-карбоксилат (364,00 мг, 2,05 ммоль) растворяли в этаноле (4 мл) и затем добавляли одной порцией палладий на угле (40,00 мг, 10%). Реакционную смесь насыщали водородом три раза и затем перемешивали при 15-20°С в течение 3 ч при давлении водорода 15 фунтов на квадратный дюйм. Далее палладий на угле удаляли путем фильтрования, и фильтрат концентрировали при пониженном давлении с получением этил-6-этилпиридин-2-карбоксилата (320,00 мг, выход 87,10%). 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) 7,95 (d, J=7,7 Гц, 1H), 7,75 (t, J=7,8 Гц, 1H), 7,37 (d, J=7,8 Гц, 1H), 4,49 (q, J=7,1 Гц, 2Н), 2,96 (q, J=7,7 Гц, 2Н), 1,44 (t, J=7,2 Гц, 3Н), 1,34 (t, J=7,7 Гц, 3Н).
[000167] Соединение согласно варианту реализации 10 может быть получено в соответствии со способом получения соединения согласно варианту реализации 1.
[000168] 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,66 (s, 1H), 7,81-7,91 (m, 3Н), 7,61-7,72 (m, 2Н), 7,57 (d, J=9,03 Гц, 1H), 7,49 (d, J=15,69 Гц, 1H), 7,21 (ушир. s, 1H), 6,96-7,03 (m, 1H), 4,21-4,37 (m, 2Н), 2,70-2,92 (m, 2Н), 2,62 (q, J=7,18 Гц, 2Н), 2,27 (q, J=7,53 Гц, 2Н), 0,48 (t, J=7,53 Гц, 3Н).
[000169] Соединение согласно варианту реализации 11
[000170] Получение промежуточного соединения 11-2
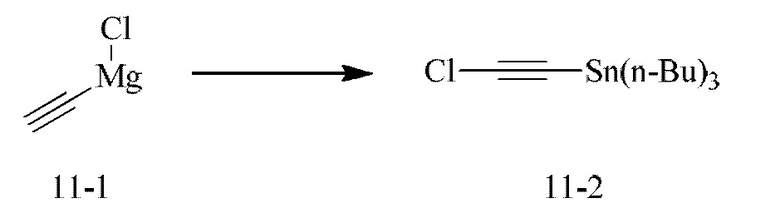
[000171] Стадия А: хлорид три-н-бутилолова (72,09 мл, 268,00 ммоль) добавляли по каплям (в течение не менее 30 мин) к раствору этинилмагнийхлорида в тетрагидрофуране (0,5 моль/л, 800,00 мл) при перемешивании при 0°С. Реакционную смесь перемешивали при 30°С в течение 0,5 ч, затем нагревали до 35°С и перемешивали в течение 1 ч. После этого смесь охлаждали до 0°С, затем в нее добавляли для гашения водный раствор хлорида аммония (800 мл) и затем подвергали экстракции петролейным эфиром (800 мл × 2). Объединенные органические фазы промывали солевым раствором (400 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением трибутил(хлорэтинил)станнана (84,00 г, выход: 60,08%). 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ: 1,54-1,67 (m, 6Н), 1,32-1,35 (m, 6Н), 0,88-1,04 (m, 15Н).
[000172] Получение промежуточного соединения 11-6
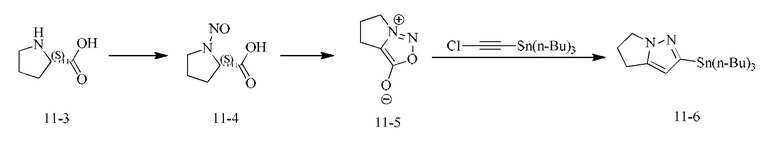
[000173] Стадия А: L-пролин (50 г, 434,29 ммоль) и нитрит натрия (41,95 г, 608,01 ммоль) растворяли в воде, затем добавляли концентрированную соляную кислоту (50 мл) при -10-0°С (с контролем температуры, чтобы она не превышала 10°С). После завершения добавления реакционную смесь перемешивали при 0°С в течение 0,5 ч, затем повышали температуру до 25°С и перемешивали в течение 16 ч. Смесь разбавляли водой (200 мл), затем подвергали экстракции метил-трет-бутиловым эфиром (300 мл × 5). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением N-нитро-L-пролина (57,00 г, неочищенный).
[000174] Стадия В: N-нитро-L-пролин (57,00 г, неочищенный) растворяли в толуоле (90 мл), охлаждали до 0°С и добавляли по каплям в течение 1 ч трифторуксусный ангидрид (82,51 мл, 593,21 ммоль). Реакционную смесь перемешивали при 25°С в течение 2 ч. Карбонат калия (87,45 г, 632,76 ммоль) диспергировали в воде (50 мл) и дихлорметане (100 мл), и к этому раствору добавляли по каплям предыдущий реакционный раствор при 0°С в течение 1 ч. После завершения добавления смесь перемешивали при 25°С в течение 1 ч. Смесь подвергали экстракции дихлорметаном (100 мл × 5) Объединенные органические фазы промывали солевым раствором (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 0/1) с получением 5,6-дигидро-4H-пирроло[1,2-с][1,2,3]оксадиазол-7-иум-3-олата (39,00 г, выход 78,20%).
[000175] Стадия С: 5,6-дигидро-4H-пирроло[1,2-с][1,2,3]оксадиазол-7-иум-3-олат (23,5 г, 186,35 ммоль) растворяли в толуоле (120 мл) в атмосфере азота, затем добавляли 2-хлорацетилен-три-н-бутилолово (84,67 г, 242,26 ммоль). Реакционную смесь перемешивали при 150°С в течение 40 ч и непосредственно очищали с помощью хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/0-20/1) с получением 2-(трибутилстаннил)-5,6-дигидро-4H-пирроло[1,2-b]пиразола (13,00 г, выход: 17,56%).
[000176] Получение соединения согласно варианту реализации 11
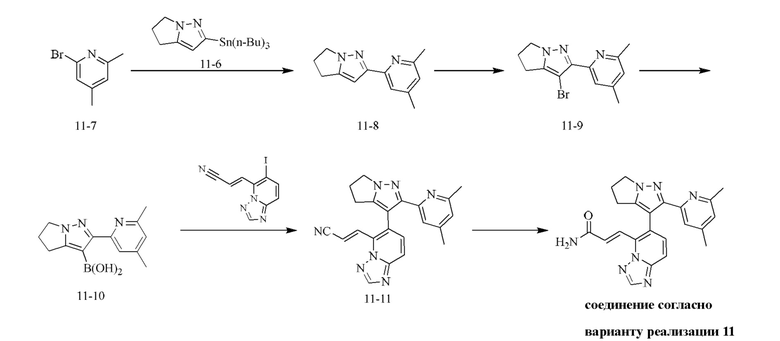
[000177] Стадия А: 2-(трибутилстаннил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол (1,49 г, 3,76 ммоль), 2-бром-4,6-диметилпиридин (700,00 мг, 3,76 ммоль), хлорид лития (318,98 мг, 7,52 ммоль) и тетракис(трифенилфосфин)палладий (434,77 мг, 376,24 мкмоль) добавляли к диоксану (20 мл). Реакционную смесь насыщали азотом три раза, затем нагревали до 100-110°С и перемешивали в течение 12 ч. Контроль хода реакции методом ЖХ-МС показал, что 2-(трибутилстаннил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол не был полностью израсходован. Реакционную смесь перемешивали еще в течение 12 ч при 100-110°С. Повторный контроль хода реакции методом ЖХ-МС показал, что 2-(трибутилстаннил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол не был полностью израсходован. Реакционную смесь дополнительно перемешивали в течение 12 ч при 100-110°С. Наконец, реакция, ход которой контролировали методом ЖХ-МС, была завершена. Затем реакционную смесь гасили путем выливания в воду (50 мл) и подвергали экстракции этилацетатом (50 мл × 3). Объединенные органические фазы промывали солевым раствором (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной хроматографии на силикагеле (элюент: дихлорметан/метанол = 30/1) с получением 2-(4,6-диметилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразола (649,00 мг, выход: 63,81%, чистота: 78,847%).
[000178] Стадия В: 2-(4,6-диметилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол (150,00 мг, 703,30 мкмоль) и NBS (137,69 мг, 773,63 мкмоль) растворяли в N,N-диметилформамиде (3 мл). Реакционную смесь перемешивали при 15-20°С в течение 2 ч, затем гасили путем выливания в воду (15 мл) и подвергали экстракции этилацетатом (20 мл × 3). Объединенные органические фазы промывали солевым раствором (15 мл), сушили с помощью безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной хроматографии на силикагеле (элюент: дихлорметан/метанол = 30/1) с получением 3-бром-2-(4,6-диметилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразола (150,00 мг, выход 73,00%).
[000179] Стадия С: 3-бром-2-(4,6-диметилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол (150,00 мг, 513,40 мкмоль) и триизопропилборат (255,87 мг, 1,36 ммоль, 312,04 мкл) растворяли в тетрагидрофуране (4 мл). Смесь охлаждали до -78 - -60°С, затем добавляли по каплям н-бутиллитий (2,5 М, 533,94 мкл). Реакционную смесь нагревали до 15-20°С и перемешивали в течение 30 мин, затем гасили путем выливания в насыщенный раствор хлорида аммония (20 мл) и подвергали экстракции этилацетатом (20 мл × 2). Объединенные органические фазы промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной хроматографии на силикагеле (элюент: дихлорметан/метанол = 30/1) с получением (2-(4,6-диметилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол-3-ил)бороновой кислоты (90,00 мг, выход: 68,18%).
[000180] Стадия D: (E)-3-(6-иод-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилонитрил (100,00 мг, 337,76 мкмоль), (2-(4,6-диметилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол-3-ил)борную кислоту (86,84 мг, 337,76 мкмоль), карбонат натрия (107,40 мг, 1,01 ммоль), дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (7,41 мг, 10,13 мкмоль), дициклогексилфосфин-2',6'-диметоксибифенил (1,39 мг, 3,38 мкмоль) и [2-(2-аминофенил)фенил]хлорпалладийциклогексил-[2-(2,6-диметоксифенил)фенил]фосфин (12,17 мг, 16,89 мкмоль) добавляли к смеси растворителей, состоящей из диоксана (20 мл) и воды (4 мл). Реакционную смесь насыщали азотом три раза, затем нагревали до 80-90°С и перемешивали в течение 12 ч. Затем смесь гасили путем выливания в воду (30 мл) и подвергали экстракции этилацетатом (30 мл × 2). Объединенные органические фазы промывали солевым раствором (40 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной хроматографии на силикагеле (элюент: дихлорметан/метанол = 30/1) с получением (E)-3-(6-(2-(4,6-диметилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол-3-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилонитрила (80,00 мг, неочищенный).
[000181] Стадия Е: (E)-3-(6-(2-(4,6-диметилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол-3-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилонитрил (80,00 мг, неочищенный) растворяли в смеси растворителей, состоящей из воды (1 мл) и диметилсульфоксида (2 мл), затем добавляли гидроксид натрия (10,49 мг, 262,18 мкмоль) и перекись водорода (74,31 мг, 655,45 мкмоль) одной порцией, соответственно. Реакционную смесь перемешивали при 15-20°С в течение 2 ч, затем гасили путем выливания в воду (20 мл) и подвергали экстракции дихлорметаном (20 мл × 3) Объединенные органические фазы промывали солевым раствором (50 мл), сушили с помощью безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной хроматографии на силикагеле (элюент: дихлорметан/метанол = 30/1), и контролировали загрязненность неочищенного продукта посредством ЖХ-МС. Неочищенный продукт повторно очищали посредством препаративной ВЭЖХ (колонка: Phenomenex Synergi С18 150 × 30 мм × 4 мкм; подвижная фаза: [вода (0,225% муравьиная кислота)-ацетонитрил]; градиент: 10%-40%, 12 мин) с получением соединения согласно варианту реализации 11 (12,00 мг, формиат, выход: 22,90%). 1Н ЯМР (400 МГц, МЕТАНОЛ-d4) δ 8,53 (s, 1H), 8,21 (ушир. s, 1H), 8,00 (d, J=15,8 Гц, 1H), 7,76 (d, J=9,2 Гц, 1H), 7,68-7,56 (m, 2Н), 7,38 (s, 1H), 6,99 (s, 1H), 4,34 (t, J=6,7 Гц, 2Н), 2,97-2,89 (m, 2Н), 2,80-2,69 (m, 2Н), 2,31 (s, 3Н), 2,17 (s, 3Н).
[000182] Соединение согласно варианту реализации 12
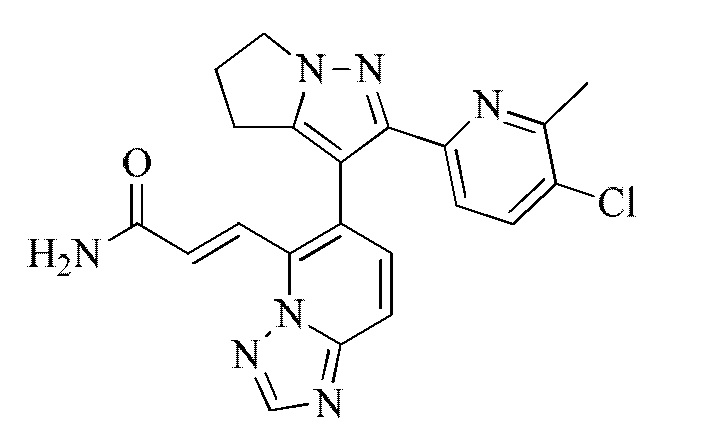
[000183] Получение промежуточного соединения 12-3
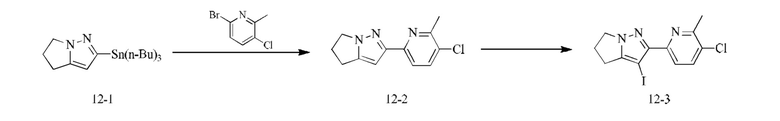
[000184] Стадия А: 2-(трибутилстаннил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол (2,69 г, 6,78 ммоль), 6-бром-3-хлор-2-метилпиридин (700,00 мг, 3,39 ммоль), хлорид лития (287,43 мг, 6,78 ммоль) и тетракис(трифенилфосфин)палладий (391,77 мг, 339,00 мкмоль) добавляли к диоксану (30 мл). Реакционную смесь насыщали азотом три раза, затем нагревали до 100-110°С и перемешивали в течение 12 ч. Смесь гасили путем выливания в воду (50 мл) и подвергали экстракции этилацетатом (50 мл × 3). Объединенные органические фазы промывали солевым раствором (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 10/1-5/1) с получением 2-(5-хлор-6-метилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразола (600,00 мг, выход 67,19%). 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) 7,65 (q, J=8,4 Гц, 2Н), 6,59 (s, 1H), 4,22 (t, J=7,2 Гц, 2Н), 2,95 (t, J=7,3 Гц, 2Н), 2,71-2,59 (m, 5Н).
[000185] Стадия В: 2-(5-хлор-6-метилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол (200,00 мг, 855,80 мкмоль) растворяли в N,N-диметилформамиде (3 мл) и затем добавляли одной порцией NIS (211,79 мг, 941,38 мкмоль). Реакционную смесь перемешивали при 15-20°С в течение 12 ч, затем фильтровали, и осадок на фильтре собирали, концентрировали и сушили с получением 2-(5-хлор-6-метилпиридин-2-ил)-3-иод-5,6-дигидро-4H-пирроло[1,2-b]пиразола (264,00 мг, выход: 85,79%).
[000186] Получение соединения согласно варианту реализации 12
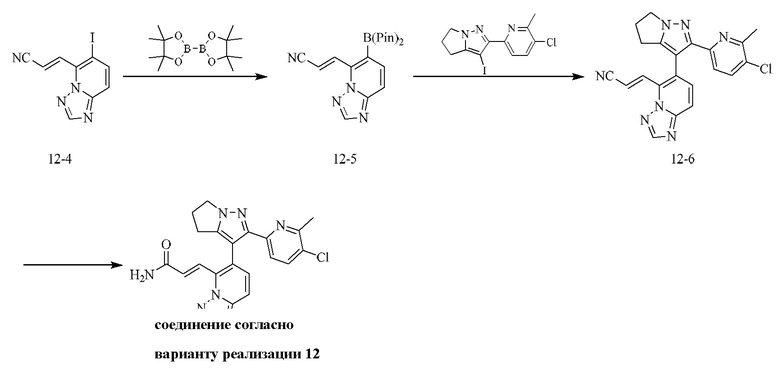
[000187] Стадия А: (E)-3-(6-иод-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилонитрил (200,00 мг, 675,52 мкмоль), бис(пинаколато)дибор (205,85 мг, 810,62 мкмоль), дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (49,43 мг, 67,55 мкмоль) и ацетат калия (132,59 мг, 1,35 ммоль) добавляли к диоксану (20 мл). Реакционную смесь насыщали азотом три раза, затем нагревали до 100-110°С и перемешивали в течение 12 ч. Реакцию оставляли необработанной, и раствор использовали непосредственно на следующей стадии.
[000188] Стадия В: 2-(5-хлор-6-метилпиридин-2-ил)-3-иод-5,6-дигидро-4H-пирроло[1,2-b]пиразол (121,43 мг, 337,69 мкмоль), карбонат натрия (107,38 мг, 1,01 мкмоль), дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (24,71 мг, 33,77 мкмоль), дициклогексилфосфин-2',6'-диметоксибифенил (13,86 мг, 33,77 мкмоль), 2-(2-аминофенил)фенил]хлорпалладий;циклогексил-[2-(2,6-диметоксифенил)фенил]фосфин (24,33 мг, 33,77 мкмоль), диоксан (4,00 мл) и воду (4,00 мл) добавляли к смеси, полученной на стадии А. Реакционную смесь насыщали азотом три раза, затем нагревали до 90-100°С и перемешивали в течение 2 ч. Смесь гасили путем выливания в воду (30 мл) и подвергали экстракции этилацетатом (30 мл × 2). Объединенные органические фазы промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной хроматографии на силикагеле (элюент: дихлорметан/метанол 30/1) с получением (E)-3-(6-(2-(5-хлор-6-метилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол-3-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилонитрила (120,00 мг, выход: 88,43%).
[000189] Стадия С: (E)-3-(6-(2-(5-хлор-6-метилпиридин-2-ил)-5,6-дигидро-4H-пирроло[1,2-b]пиразол-3-ил)-[1,2,4]триазоло[1,5-а]пиридин-5-ил)акрилонитрил (120,00 мг, 298,62 мкмоль) растворяли в смеси растворителей, состоящей из диметилсульфоксида (2 мл) и воды (1 мл), затем последовательно добавляли перекись водорода (338,54 мг, 2,99 ммоль) и гидроксид натрия (2 моль/л, 597,24 мкл). Реакционную смесь перемешивали при 15-20°С в течение 12 ч. Контроль хода реакции методом ЖХ-МС показал, что реакция не была завершена. Затем реакционную смесь нагревали до 40-50°С и перемешивали в течение 2 ч. Контроль хода реакции методом ЖХ-МС свидетельствовал о завершении реакции. Смесь гасили путем выливания в воду (10 мл) и подвергали экстракции дихлорметаном (30 мл × 2). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной высокоэффективной жидкостной хроматографии (колонка: Phenomenex Synergi С18 150 × 30 мм × 4 мкм; подвижная фаза: [вода (0,225% муравьиная кислота)-ACN]; градиент: 24% -54%, 12 мин) с получением соединения согласно варианту реализации 12 (21,46 мг, выход 15,72%). 1Н ЯМР (400 МГц, МЕТАНОЛ-d4) δ 8,56 (s, 1H), 8,06 (d, J=15,6 Гц, 1H), 7,79 (d, J=9,0 Гц, 1H), 7,73-7,61 (m, 4Н), 4,34 (ушир. d, J=6,0 Гц, 2Н), 2,94-2,85 (m, 2Н), 2,73 (ушир. s, 2Н), 2,17 (s, 3Н).
[000190] Эксперимент 1: тест на ингибирующую активность в отношении TGFβ-RI in vitro
[000191] Методика проведения эксперимента:
[000192] 1) Тестируемое соединение: IC50 определяли путем ступенчатого разведения с получением 10 точек с начальной концентрацией 5 мкМ и дальнейшим трехкратным ступенчатым разведением.
[000193] 2) IC50 соединения сравнения LDN193189 определяли путем ступенчастого разведения с получением 10 точек с начальной концентрацией 20 мкМ и дальнейшим трехкратным ступенчатым разведением.
[000194] 3) Реакционная система содержит 10 мкМ АТФ.
[000195] 4) Значение IC50 рассчитывали с помощью графика, когда процент ферментативной активности (по сравнению с группой растворителя) ниже 65% при наибольшей концентрации образца.
[000196] Результаты эксперимента: см. таблицу 1.
[000197] Заключение: соединение согласно настоящему изобретению обладает превосходной ингибирующей активностью in vitro.

[000198] [Примечание] Диапазон значений IC50 показан следующим образом: 50 нМ ≥ А ≥ 1 нМ; 500 нМ ≥ В ≥ 50 нМ; С > 500 нМ.
[000199] Эксперимент 2: тест на ингибирование пролиферации на эмбриональных клетках мышей NIH/3T3
[000200] Принцип эксперимента:
[000201] Люминесцентный анализ жизнеспособности клеток Promega (метод CellTiter-Glo®, например, определение флуоресцентной активности и анализ с помощью АТФ), соединение вносят в планшет для клеточных культур для инкубации. В день определения добавляли субстратный буфер для определения содержания внутриклеточной АТФ. Слегка встряхивали и центрифугировали при 1000 об/мин в течение 1 мин. Тест проводили после 10-минутного выстаивания пробы. Содержимое планшета анализировали, применяя мультифункциональный ферментный маркер Envision производства PerkinElmer, Inc., аналитическим методом для определения флуоресценции, данные получали при считывании хемилюминесцентного сигнала при 400-700 нм.
[000202] Стадии эксперимента
[000203] 1) Когда рост клеток на поверхности составлял примерно 70%, клеточный слой промывали 10 мл фосфатного буфера Дюшенна (D-PBS), не содержащего кальций и магний, затем добавляли 2 мл 0,25% гидролизующего раствора (digest) трипсин-ЭДТА. Колбу с клеточной культурой помещали в СО2-инкубатор при 37°С и инкубировали в течение 3-5 мин. Затем к 8 мл полной культуральной среды, содержащей 2% фетальной бычьей сыворотки, добавляли клетки в DMEM, клетки равномерно рассеивали по ячейке, подсчитывали с помощью Vi-клеточного питометра, и полученную суспензию клеток NIH/3T3 разбавляли до 0,375×105 клеток/мл.
[000204] 2) 50 мкл of 2% DMEM-содержащей среды вносили в 384-луночный планшет для культивирования клеток, затем в остальные лунки вносили 40 мкл клеточной суспензии до 1500 клеток на лунку. Распределение клеток наблюдали под микроскопом, и планшеты с клетками помещали в инкубатор для клеточных культур с 5% CO2 при 37°С.
[000205] 3) Разбавление соединения относится к получению соединения.
[000206] 4) Смесь 2% фетальной бычьей сыворотки, содержащей 1 нг/мл TGF-β1 в среде DMEM, добавляли вручную в промежуточный планшет с соединением, 20 мкл на лунку.
[000207] 5) Промежуточный планшет с соединением слегка встряхивали в течение 10 с при 1000 об/мин и центрифугировали в течение 10 с.
[000208] 6) 10 мкл смешанной жидкости из этапов 4 и 5 из каждой лунки переносили в планшет с клетками после инокуляции до конечного объема 50 мкл с использованием жидкой рабочей станции Bravo. Конечная концентрация TGF-β1 была снижена разбавлением до 0,2 нг/мл, далее проводили центрифугирование в течение 10 с при 1000 об/мин. Планшеты помещали в инкубатор с 5% диоксида углерода при 37°С, 5% диоксида углерода выдерживали 72 часа. Конечная концентрация соединения составляет: (единица: мкМ)

[000209] 7) Клеточный планшет, содержащий соединение, выдерживали в клеточном инкубаторе с 5% СО2 при 37°С в течение 3 суток.
[000210] 8) После этого в каждую лунку клеточного планшета вносили 25 мкл раствора для определения флуоресцентной активности с помощью АТФ, затем содержимое планшета аккуратно встряхивали примерно 1 минуту, центрифугировали при 500 об/мин примерно 30 с, выдерживали при комнатной температуре в течение 10 минут в темноте и проводили считывание на приборе Envision.
[000211] Результаты эксперимента: см. таблицу 2.

[000212] [Примечание] Диапазон значений IC50 показан следующим образом: 2 мкМ ≥ А ≥ 0,5 мкМ; 5 мкМ ≥ В ≥ 2 мкМ; С>5 мкМ.
[000213] Заключение: соединение согласно настоящему изобретению обладает превосходной ингибирующей активностью в отношении полиферации клеток NIH/3T3.
[000214] Эксперимент 3: эксперимент на мышах BALB/c по ингибированию пролиферации опухолевых клеток на модели опухоли мышиного ректального рака, в которой клетки СТ-26 трансплантируются подкожно в комбинации с BioXcell-mPD-L1
[000215] План эксперимента:
[000216] В следующей таблице перечислены группы животных и приведена схема введения соединения согласно варианту реализации 1, соединения LY2157299, используемого в качестве положительного контроля, и моноклонального антитела BioXcell PD-L1 (BioXcell-mPD-L1), которые применяются отдельно или в комбинации in vivo. См. таблицу 3.

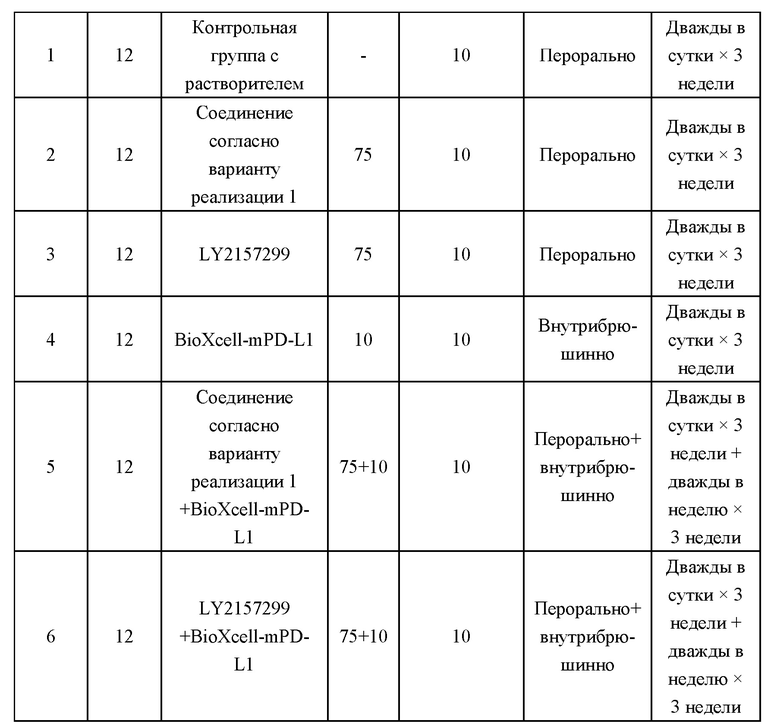
[000217] Методы и стадии проведения эксперимента:
[000218] 1) Клеточная культура
[000219] Клетки рака толстой кишки мыши СТ-26 культивировали in vitro в одном слое при следующих условиях: среда RPMI1640 (среда №1640, Roswell Parker Memorial Institute) с добавлением 10% фетальной бычьей сыворотки при 37°С, 5% СО2. Пассаж обычно гидролизовали трипсином-ЭДТА дважды в неделю. Когда насыщение клеток достигает 80-90%, клетки собирают, подсчитывают и инокулируют.
[000220] 2) Инокуляция клеток опухоли
[000221] 0,1 мл (1×105) клеток СТ-26 подкожно инокулировали в правый бок каждой BALB/c мыши. Мышей разделяли на группы по массе тела мыши на вторые сутки после инокуляции клеток.
[000222] 3) Измерение опухоли и индикаторы при проведении эксперимента
[000223] Индикатор при проведении эксперимента предназначен для исследования того, ингибирован ли рост опухоли, отсрочен или излечен. Диаметры опухолей измеряли с помощью штангенциркуля с нониусом дважды в неделю. Объем опухоли рассчитывали следующим образом: V=0,5а×b2, где а и b представляют собой длинный и короткий диаметром опухоли, соответственно.
[000224] Противоопухолевый эффект соединения оценивали по показателю TGI (%) или по уровню опухолевой пролиферации Т/С (%). TGI (%) отражает скорость ингибирования роста опухоли. Расчет TGI (%): TGI (%) = [(1-(средний объем опухоли на конец введения в группе с лечением - средний объем опухоли на начало введения в группе с лечением))/(средний объем опухоли на конец введения в контрольной группе с растворителем) - средний объем опухоли на начало введения в контрольной группе с растворителем)] × 100%.
[000225] Уровень опухолевой пролиферации Т/С (%): Использовали следующую формулу: Т/С % = Т/С × 100% (Т: группа с лечением; С: отрицательная контрольная группа).
[000226] Массу опухоли измеряли в конце эксперимента, а также подсчитывали процент Т/Смасса. Тмасса и Смасса представляют собой вес опухолей группы, которой вводили лекарство, и группы, получавшей носитель, соответственно.
[000227] 4) Отбор проб для определения фармакокинетических характеристик
[000228] На 20-е сутки после введения проводили введение согласно схеме.
[000229] Двенадцать мышей разделяли на 4 группы, кровь отбирали через 0,25, 1, 1,5, 4 и 8 часов после последнего введения; мышей умерщвляли через 0,25, 1, 4 и 8 часов для забора опухолей и печени. Всю кровь помещали в антикоагулянтные пробирки с 0,5 мл ЭДТА-2K, центрифугировали при 7000 об/мин, 4°С в течение 10 минут с получением плазмы. Опухолевую ткань помещали в 10 мл криопробирку. Плазму и опухолевые ткани быстро перемещали в холодильник с температурой -80°С на хранение.
[000230] 5) Статистический анализ
[000231] Статистический анализ, включая среднее и стандартную ошибку (SEM), объема опухоли в каждой временной точке для каждой группы (см. таблицу 4 для конкретных данных). Группа с лечением показала лучший терапевтический эффект на 20-е сутки после введения в конце испытания. Следовательно, статистический анализ проводили, основываясь на этих данных, для оценки различий между группами. Т-тест применяли для сравнения между двумя группами, и одномерный анализ различий применяли для сравнения между тремя и более групп. Если находили значимые отличия в значениях F, то для теста применяли метод Гасса-Хоуэлла. Если в значениях F не было значимых отличий, то для анализа применяли метод Даннета (2-сторонний). Весь анализ данных проводили при помощи SPSS 17.0. Отличия считали значимыми при р<0,05.
[000232] Результаты эксперимента:
[000233] 1) Смертность, заболеваемость и изменения массы
[000234] Массу опытных животных применяли как индикатор для сравнения при проведении непрямой оценки токсичности препарата. В этой модели никто из групп введения препарата не показал значительной потери массы тела, не было заболеваемости или смертей.
[000235] Эффекты от соединения согласно варианту реализации 1, LY2157299 и BioXcell-mPD-L1 на массу тела клеток СТ-26 на модели у самок мышей BALB/c при подкожном введении ксенотрансплантата опухоли показаны на фигуре 1. Точки данных представляют среднюю массу тела в группе, а столбики ошибок представляют стандартные ошибки (SEM).
[000236] 2) Объем опухоли
[000237] Объем опухоли меняется у самок мышей BALB/c при подкожном введении клеток СТ-26 после соединения согласно варианту реализации 1, LY2157299 и BioXcell-mPD-Ll лечения, что показано в таблице 4.
[000238] 3) Кривая роста опухоли
[000239] Кривые роста опухоли ксенотрансплантата СТ-26 на модели опухоли-носителя после введения соединения согласно варианту реализации 1 показаны на фигуре 2. Точки данных показывают средний объем опухоли в группе, а столбики ошибок отображают стандартные ошибки (SEM).
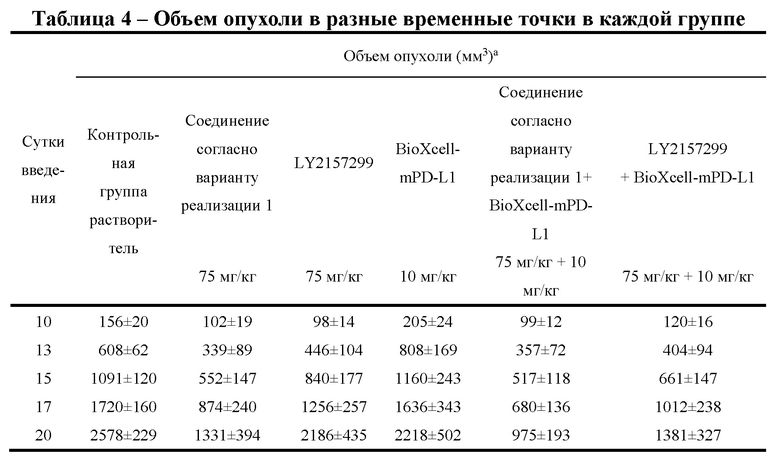
[000240] [Примечание]: а. Среднее ± SEM.
[000241] Заключение:
[000242] По этому эксперименту оценивали эффективность in vivo соединения согласно варианту реализации 1, положительного контроля LY2157299 и BioXcell-mPD-L1 на мышиной модели рака прямой кишки с ксенотрансплантатом СТ-26. Через двадцать суток после начала введения объем опухоли у мышей, имеющих опухоль (контрольная группа с растворителем), достигал 2578 мм3. Комбинация из соединения согласно варианту реализации 1 (75 мг/кг) и BioXcell-mPD-L1 (10 мг/кг) обладает значительным противоопухолевым действием по сравнению с контрольной группой (Т/С = 38%, TGI = 62,2%, р = 0,012), объем опухоли составлял 975 мм3; отдельно взятые соединение согласно варианту реализации 1 (75 мг/кг), LY2157299 (75 мг/кг), BioXcell-mPD-L1 (10 мг/кг) и комбинированные дозы LY2157299 (75 мг/кг) и BioXcell-mPD-L1 (10 мг/кг) не показали значимого противоопухолевого эффекта по сравнению с контрольной группой растворителя. Объемы опухолей составляли соответственно 1331, 2186, 2218 и 1381 мм3 (Т/С = 51%, 85%, 86% и 54%, р = 0,071, 0,906, 0,932 и 0,089).
[000243] Эксперимент 4: эксперимент по ингибированию метастазирования опухолевых клеток в ортотопической трансплантационной модели на клетках рака молочной железы мышей линии 4Т1 у мышей линии BALB/c
[000244] Метод проведения эксперимента:
[000245] 1) Создание опухолевой модели 4Т1 in situ:
[000246] Флуоресцентно меченные клетки рака молочной железы мыши 4Т1-Luc выращивали in vitro. Перед забором клеток, мышей анестезировали внутрибрюшинной инъекцией пентобарбитала натрия. После фиксации анестезированных мышей кожу на животе дезинфицировали 70%-ным спиртом. 100 мкл фосфатного буфера (содержащего 0,5×106 клеток 4Т1-luc2) инокулировали в левую сторону четвертой пары брюшных подушечек молочной железы у мышей и ушивали разрез для дезинфекции кожи. Животным поддерживали температуру под теплым одеялом, наблюдали, когда они проснуться, затем возвращали их в клетки. Для облегчения боли подкожно вводили 0,1 мг/кг бупренорфина за 30 минут до хирургической операции и через 6 часов после нее.
[000247] 2) План обработки групп:
[000248] На третьи сутки после получения модели животных подвергали биолюминесцентному исследованию с помощью формирования изображения в инфракрасном диапазоне, группы формировали случайным образом в соответствии со значениями флуоресценции, введения проводили в соответствии со следующим протоколом проведения эксперимента, см. таблицу 5.
[000249] 3) План по определению конечной точки эксперимента:
[000250] Для оценки ингибирующего эффекта соединения согласно варианту реализации 1 на рост и метастазирование опухоли, экспериментальная конечная точка была разработана так, чтобы она составляла 30-35 суток после введения, со ссылкой на прежде полученные данные по этой модели. В конце эксперимента опухоль in situ и различные ткани органов иссекали, опухоль взвешивали и определяли интенсивность флуоресценции каждого органа по флуоресценции IVIS. Ингибирование роста ортотопических опухолей можно сравнить с весом in situ опухоли в экспериментальной конечной точке. Кривую ингибирования формируют из данных измерения объема опухоли дважды в неделю в течение эксперимента. Ингибирование метастазирования опухоли определяли по наличию или отсутствию флуоресценции каждого органа и анализу относительной интенсивности флуоресценции.
[000251] В конце эксперимента определяют массу опухоли и рассчитывают относительную скорость роста опухоли Т/С (%). Объем опухоли рассчитывают по формуле: V=0.5а×b2, а и b представляют собой длинный и короткий диаметры опухоли соответственно. В то же время легкое, печень, селезенку, почку, кишечник и левую верхнюю конечность удаляли, и измеряли флуоресценцию для определения наличия метастазов и интенсивности метастазирования, а также их соотношение.
[000252] Противоопухолевый эффект препарата рассчитывали по показателю ингибирования роста опухоли (TGI (%)) или по относительной скорости роста опухоли Т/С (%).
[000253] Расчет TGI (%):
[000254] TGI (%) = [(1-(средний объем опухоли на конец введения в группе с лечением - средний объем опухоли на начало введения в группе с лечением))/(средний объем опухоли на конец введения в контрольной группе с растворителем - средний объем опухоли на начало введения в контрольной группе с растворителем)] × 100%.
[000255] Расчет относительной скорости роста опухоли Т/С (%):
[000256] Т/С (%) = Tt (группа с лечением)/а (контрольная группа) × 100%, Tt - это средний объем опухоли в конкретном измерении, значение Ct берут из данных за эти же сутки.
[000257] Результаты эксперимента статистически обрабатывали, применяя однофакторный метод ANOVA. Если были обнаружены значимые отличия в значениях F, то проводили множественные сравнения после анализа ANOVA. Все данные этого эксперимента анализировали, применяя SPSS 17.0. Отличия считали значимыми при р<0,05.

[000258] Результаты эксперимента:
[000259] 1) Изменения в массе животных:
[000260] Относительные изменения массы тела рассчитывали на основании массы тела животного в начале дозирования, как показано на фигуре 3. Данные в точках показывают процент изменения средней массы тела в группе, а столбики ошибок показывают стандартные ошибки (SEM).
[000261] 2) Ингибирующее действие соединения согласно варианту реализации 1 в отношении числа возникающих опухолевых метастазов:
[000262] В конце эксперимента ткань каждого органа иссекали, проводили флуоресцентную визуализацию и определяли значения интенсивности флуоресценции с помощью IVIS с экспонированием 40 секунд в течение 8 минут. Результаты флуоресцентной визуализации для исследованных тканей 10 животных, за исключением максимального и минимального значения, показаны в таблице 6.
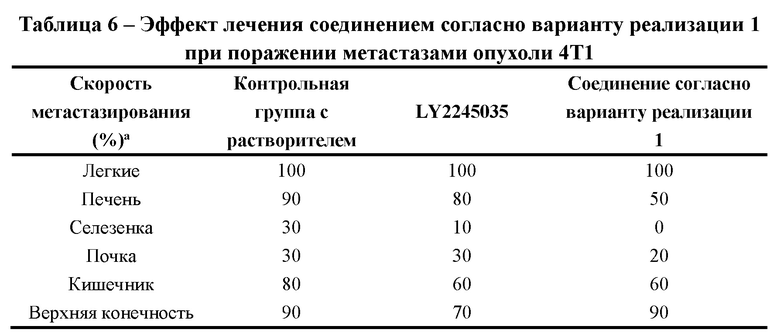
[000263] [Примечание]: а. Количество животных с метастазами в каждой группе / общее количество животных в группе.
[000264] 3) Ингибирование соединением согласно варианту реализации 1 интенсивности метастазирования опухоли в каждом органе:
[000265] Согласно конечной точке эксперимента, интенсивность флуоресценции в каждой группе органов нормализовали по контрольной группе и определяли отношение относительной интенсивности флуоресценции для каждой группы органов. Данное отношение отражает уровень метастатической интенсивности в соответствующих органах. Результаты показаны в таблице 7.
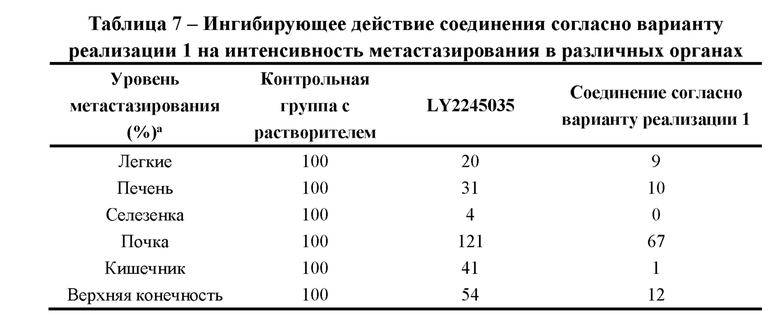
[000266] [Примечание]: а. Среднее значение флуоресценции определяли по отношению флуоресценции в группе с введением лекарства к среднему значению флуоресценции, определенному в органе для контрольной группы.
[000267] Заключение:
Оценка значений, приведенных в таблице сравнения, позволяет сделать вывод о том, что соединение согласно варианту реализации 1 значительно ингибировало метастазирование 4Т1 в печени, селезенке, почках и кишечнике, его ингибирующий эффект был значительно выше, чем у лекарства, использованного в качестве положительного контроля. Соединение согласно варианту реализации 1 демонстрирует значительное ингибирование возникновения и интенсивности метастазирования опухоли во множестве тканей органов и его эффект значительно превосходит эффект от лекарства, использованного в данном эксперименте в качестве положительного контроля.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ ПИРИДАЗИНАМИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ СЕЛЕЗЕНКИ (SYK) | 2013 |
|
RU2627661C2 |
| БЕНЗОКСЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2557658C2 |
| Применение 1H-пиразоло[4,3-b]пиридинов в качестве ингибиторов PDE1 | 2017 |
|
RU2759380C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| ПРОИЗВОДНЫЕ ИНДОЛИН-2-ОНА ИЛИ ПИРРОЛОПИРИДИН/ПИРИМИДИН-2-ОНА | 2014 |
|
RU2666532C2 |
| ПРОИЗВОДНОЕ ГЕТЕРОАРИЛ[4,3-с]ПИРИМИДИН-5-АМИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКИЕ ПРИМЕНЕНИЯ | 2018 |
|
RU2764655C2 |
| ИМИДАЗОПИРИДИНИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ РАССТРОЙСТВ | 2020 |
|
RU2833052C2 |
| Соединения триазоло-пиримидина и их применение | 2019 |
|
RU2802866C2 |
| МОДУЛЯТОРЫ ФОСФОДИЭСТЕРАЗЫ 10 | 2012 |
|
RU2567396C1 |
| АМИНОТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2009 |
|
RU2552642C2 |
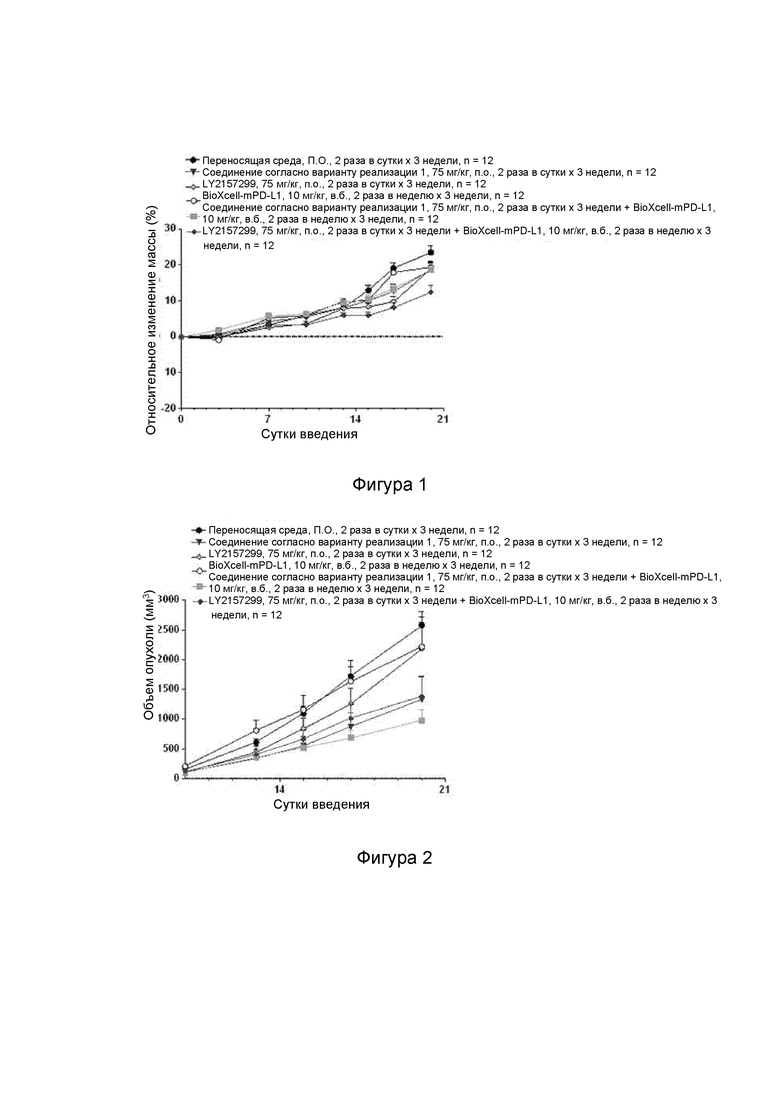
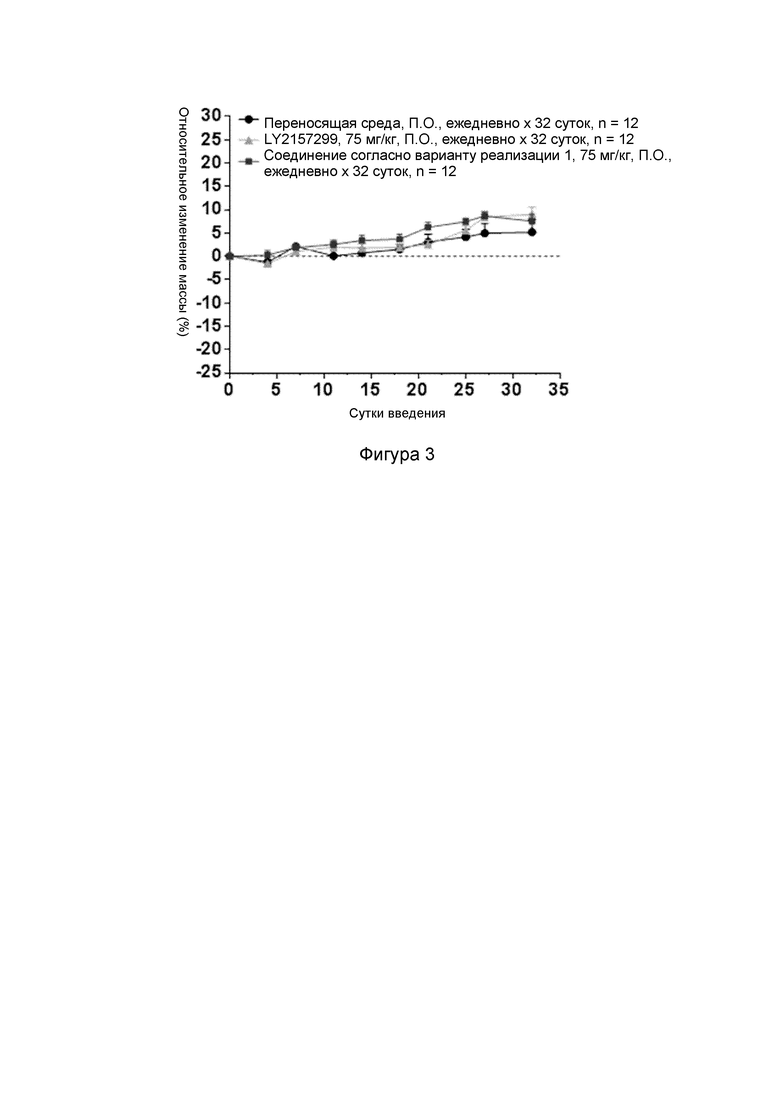
Изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, где в формуле (I) R1 выбран из водорода, амино или из группы, состоящей из C1-3 алкила и C3-6 циклоалкила, и указанная группа необязательно замещена 1 R; R2 выбран из группы, состоящей из C1-3 алкила, C3-6 циклоалкила и фенила, и указанная группа необязательно замещена 1 R; R3 представляет собой водород; необязательно, R2 и R3 соединены с образованием 5-членного кольца; каждый из R4 и R5 независимо выбран из водорода, галогена или представляет собой C1-3 алкил; L выбран из простой связи, -CH2-; R выбран из CN, OH, или C1-6 алкила, необязательно замещенного 1, 2 или 3 R'; R' представляет собой F. Изобретение также относится к индивидуальным соединениям, к фармацевтическим композициям, применениям соединения, к применениям фармацевтической композиции, к фармацевтической комбинации. Технический результат: получены новые соединения формулы (I), обладающие свойствами ингибитора TGF-βRI. 9 н. и 10 з.п. ф-лы, 7 табл., 3 ил.
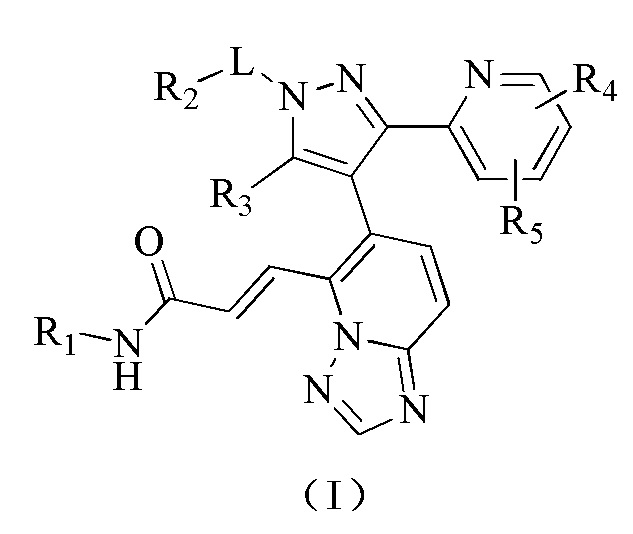
1. Соединение формулы (I) или его фармацевтически приемлемая соль,
где
R1 выбран из водорода, амино или из группы, состоящей из C1-3 алкила и C3-6 циклоалкила, и указанная группа необязательно замещена 1 R;
R2 выбран из группы, состоящей из C1-3 алкила, C3-6 циклоалкила и фенила, и указанная группа необязательно замещена 1 R;
R3 представляет собой водород;
необязательно, R2 и R3 соединены с образованием 5-членного кольца;
каждый из R4 и R5 независимо выбран из водорода, галогена или представляет собой C1-3 алкил;
L выбран из простой связи, -CH2-;
R выбран из CN, OH, или C1-6 алкила, необязательно замещенного 1, 2 или 3 R';
R' представляет собой F.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где R выбран из группы, состоящей из CN, OH, метила, CHF2, этила и пропила.
3. Соединение или его фармацевтически приемлемая соль по п. 1, где R1 выбран из водорода или из группы, состоящей из метила, этила,  , и указанная группа необязательно замещена 1 R.
, и указанная группа необязательно замещена 1 R.
4. Соединение или его фармацевтически приемлемая соль по п. 3, где R1 выбран из водорода, метила, этила,  , .
, .
5. Соединение или его фармацевтически приемлемая соль по п. 1, где R2 выбран из группы, состоящей из метила, этила, изопропила, циклопентила и фенила, и указанная группа необязательно замещена 1 R.
6. Соединение или его фармацевтически приемлемая соль по п. 5, где R2 выбран из группы, состоящей из метила, этила, изопропила, циклопентила  и
и 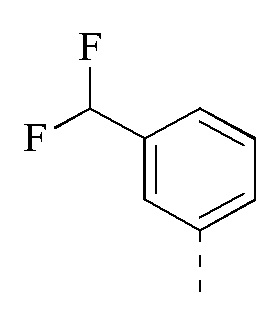 .
.
7. Соединение или его фармацевтически приемлемая соль по п. 1, где R2 и R3 соединены и фрагмент 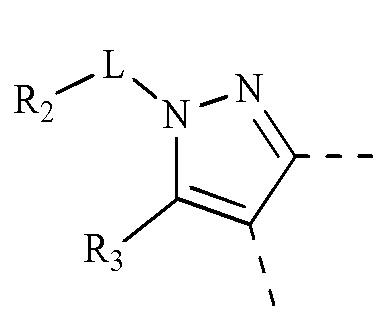 представляет собой
представляет собой 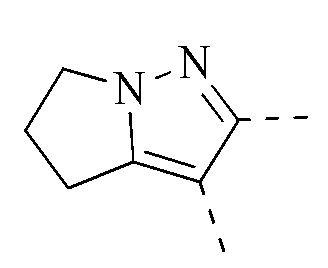 .
.
8. Соединение или его фармацевтически приемлемая соль по п. 1, где каждый из R4 и R5 независимо выбран из группы, состоящей из водорода, F, Cl, Br, метила и этила.
9. Соединение или его фармацевтически приемлемая соль по п. 1, где фрагмент
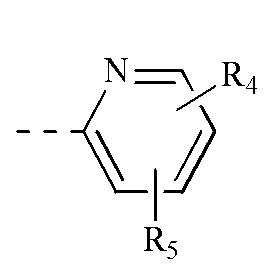 выбран из
выбран из 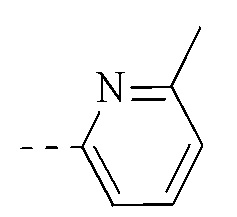 ,
, 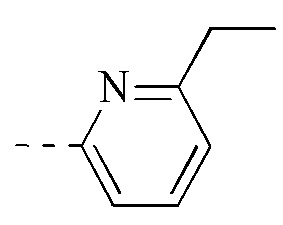 ,
, 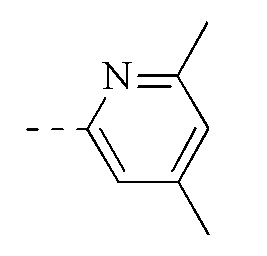 ,
, 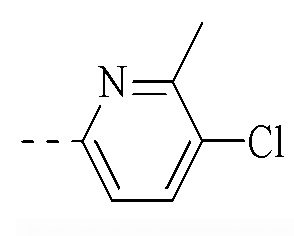 .
.
10. Соединение или его фармацевтически приемлемая соль по п. 1, выбранное из
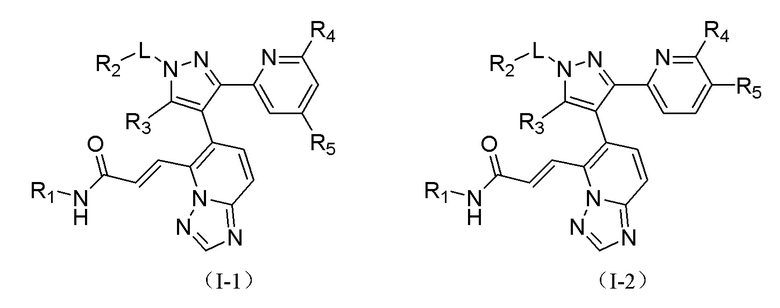
где R1, R2, R3, R4, R5 и L определены, как указано в п. 1, и R4 и R5 одновременно оба не являются водородом.
11. Соединение или его фармацевтически приемлемая соль по п. 10, выбранное из
где R1, R2, R4, R5 и L определены, как указано в п. 10, и R4 и R5 одновременно оба не являются водородом.
12. Соединение или его фармацевтически приемлемая соль, выбранное из группы, состоящей из
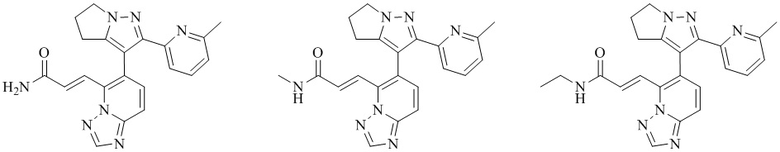
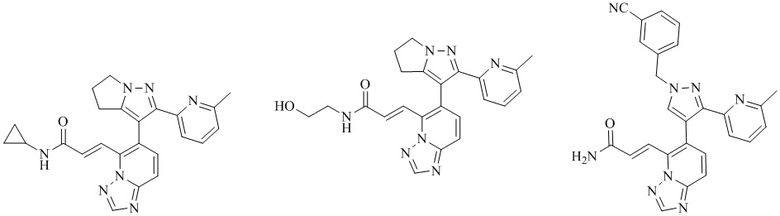
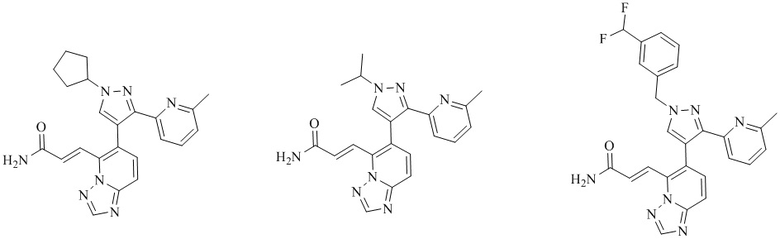
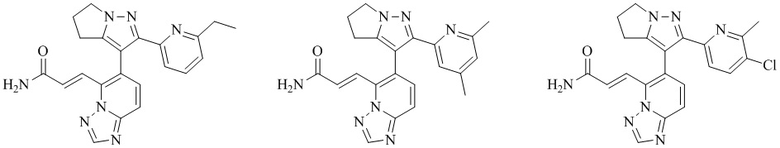 .
.
13. Фармацевтическая композиция, обладающая свойствами ингибитора TGF-βRI, содержащая терапевтически эффективную дозу соединения или его фармацевтически приемлемой соли по п. 1 и фармацевтически приемлемый носитель.
14. Фармацевтическая композиция, обладающая свойствами ингибитора TGF-βRI, содержащая терапевтически эффективную дозу соединения или его фармацевтически приемлемой соли по п. 12 и фармацевтически приемлемый носитель.
15. Применение соединения или его фармацевтически приемлемой соли по п. 1 для получения лекарственного средства для лечения рака, выбранного из рака молочной железы, рака печени, рака селезенки, рака почек и рака толстой кишки, путем ингибирования TGF-βRI.
16. Применение соединения или его фармацевтически приемлемой соли по п. 12 для получения лекарственного средства для лечения рака, выбранного из рака молочной железы, рака печени, рака селезенки, рака почек и рака толстой кишки, путем ингибирования TGF-βRI.
17. Применение фармацевтической композиции по п. 13 для получения лекарственного средства для лечения рака, выбранного из рака молочной железы, рака печени, рака селезенки, рака почек и рака толстой кишки, путем ингибирования TGF-βRI.
18. Применение фармацевтической композиции по п.14 для получения лекарственного средства для лечения рака, выбранного из рака молочной железы, рака печени, рака селезенки, рака почек и рака толстой кишки, путем ингибирования TGF-βRI.
19. Фармацевтическая комбинация для лечения рака толстой кишки, содержащая
(i) 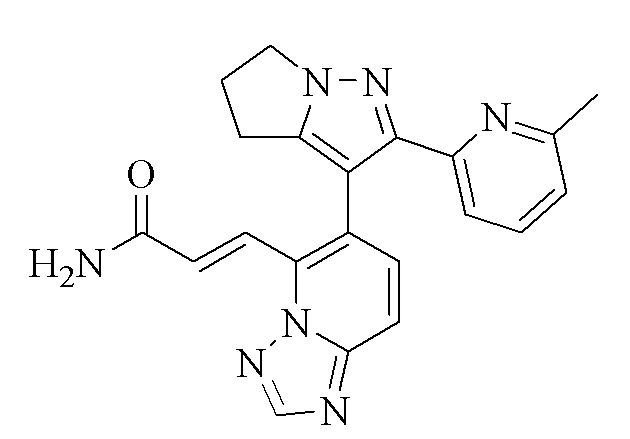 ; и
; и
(ii) антитело к PD-L1.
| EA 200500377 A1, 25.08.2005 | |||
| WO 2009009059 A1, 15.01.2009 | |||
| УКАЗАТЕЛЬНОЕ ПРИСПОСОБЛЕНИЕ ДЛЯ ИЗМЕРИТЕЛЬНЫХ ПРИБОРОВ | 1927 |
|
SU7782A1 |
| 2-ПИРИДИЛЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ В КАЧЕСИВЕ ТЕРАПЕВТИЧЕСКИХ ИНГИБИТОРОВ ALK5 И/ИЛИ ALK4 | 2011 |
|
RU2518069C1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |

