Область изобретения
Настоящее изобретение принадлежит к области медицины и относится к производному гетероарил[4,2-с]пиримидин-5-амина формулы (I), способу его получения, фармацевтической композиции, содержащей данное соединение, его применению в качестве терапевтического агента, в частности в качестве антагониста рецептора А2а, и его применению в получении лекарственного средства для лечения заболевания или состояния, улучшающегося в результате ингибирования рецептора А2а.
Предшествующий уровень техники
Аденозин представляет собой встречающийся в природе пуриновый нуклеозид и представляет собой эндогенный регулятор многих физиологических функций. Он играет важную роль в регуляции сердечно-сосудистой системы, центральной нервной системы, дыхательной системы, почки, жировой ткани и тромбоцитов.
Действие аденозина опосредовано семейством рецепторов, сопряженных с G-белком. В настоящее время известно, что существует по меньшей мере четыре подтипа аденозиновых рецепторов, которые подразделяются на A1, А2а, A2b и A3. Среди них, рецепторы A1 and A3 ингибируют активность фермента аденилатциклаза, тогда как рецепторы А2а и A2b стимулируют активность того же фермента, модулируя, таким образом, уровень циклического АМФ (циклический аденозинмонофосфат) в клетках. Аденозин регулирует широкий ряд физиологических функций посредством данных рецепторов.
Рецептор А2а (A2aR) широко распространен в организме и главным образом экспрессируются в полосатом теле в центральной нервной системе, и также экспрессируется в тканях, таких как периферическая нервная система, сердце, печень, легкое и почка. Несколько доклинических исследований показывают, что антагонисты рецептора А2а обладают неожиданной эффективностью в лечении нейродегенеративных заболеваний, главным образом, болезни Паркинсона, Болезни Гентингтона или болезни Альцгеймера (Trends in Neurosci. 2006, 29(11), 647-654; Expert Opinion on Therapeutic Patents, 2007, 17, 979-991 и тому подобное). Кроме того, антагонисты аденозинового рецептора А2а можно также использовать для лечения других заболеваний, связанных с центральной нервной системой (ЦНС), таких как депрессия, синдром беспокойства, нарушения сна и тревожный невроз (Clin. Neuropharmacol, 2010, 33, 55-60; J. Neurosci, 2010, 30 (48), 16284-16292; Parkinsonisn Relat. Disord. 2010, 16 (6), 423-426; и ссылки в них: Mov. Disorders, 2010, 25(2), S305). Кроме того, аденозиновый рецептор А2а также обладает терапевтическим потенциалом в качестве нейропротекторов (см. Jenner P. J Neuro I. 2000; 24 7Supp12: 1143-50).
Недавние исследования показывают, что активация аденозинового рецептора А2а может оказывать важное иммуномодулирующее действие во многих патологических процессах, таких как ишемия, гипоксия, воспаление, травма, трансплантация и тому подобное, которые могут быть связаны с более высоким уровнем экспрессии рецептора А2а в разных клетках иммунной системы, таких как Т-клетки, В-клетки, моноцит, макрофаги, нейтрофилы и тому подобное. Кроме того, активация рецептора А2а может способствовать генерации организмом иммунологической толерантности и активно участвовать в формировании «ускользания от иммунного ответа» или «иммунодепрессии» опухолевых клеток, создавая, таким образом, благоприятные условия для возникновения и развития опухолей. Lokshin и его коллеги (Cancer Res. 2006, Aug 1; 66(15):7758-65) демонстрируют, что активация A2aR в клетках естественных киллерах может ингибировать уничтожение опухолевых клеток клетками-естественными киллерами в результате повышения уровня цАМФ и активации протеинкиназы А (PKA - от англ. protein kinase А). Исследования также показывают, что активация рецептора А2а может стимулировать пролиферацию опухолевых клеток, таких как клетки меланомы А375, клетки фибробласты NIH3T3, клетки феохромоцитомы РС12 и тому подобное, что может быть связано с тем фактом, что активация рецептора А2а в Т-клетках может ингибировать Т-клеточную активацию, пролиферацию, адгезию к опухолевым клеткам и вызывать цитотоксическое действие на опухолевые клетки. Однако, у мышей, нокаутированных по рецептору А2а, противоопухолевый иммунитет CD8+ Т-клеток может быть усилен, и пролиферация опухолевых клеток значительно ингибируется. Таким образом, антагонисты рецептора А2а можно также использовать в лечении опухоли.
Несмотря на то, что соединения, обладающие значительной биологической активностью в отношении различных подтипов аденозиновых рецепторов, могут оказывать терапевтическое действие, они могут вызывать нежелательные побочные эффекты. Например, во время ишемии/гипоксии тканей, когда клетки центральной системы, кровеносной системы, пищеварительной системы и скелетной мускулатуры, находятся в среде аноксического и гипоксического стресса, внеклеточный агрегированный аденозин инициирует соответствующий защитный механизм посредством активирования аденозинового рецептора А1 на клеточной мембране, повышая, таким образом, устойчивость клеток к аноксии и гипоксии. Рецептор А1, расположенный на клетках иммунной системы, может стимулировать клеточные иммунные ответы в гипоксической среде. Кроме того, рецептор А1 может также снижать уровень свободных жирных кислот и триглицеридов, и вовлечен в регуляцию уровня глюкозы в крови. Таким образом, продолжительное блокирование рецептора А1 может вызывать разные вредные воздействия в тканях организма (Chinese Pharmacological Bulletin, 2008, 24(5), 573-576). Например, сообщается, что блокирование рецептора А1 будет вызывать вредные воздействия, такие как тревожность, пробуждение и тому подобное в животных моделях (Basic & Clinical Pharmacology & Toxicology, 2011, 109(3), 203-7). Аденозин, высвобождающийся посредством аденозинового рецептора А3 во время ишемии миокарда, вызывает сильное защитное действие в сердце (как описано Gessi S et al, Pharmacol, Ther. 117 (1), 2008, 123-140). Продолжительное блокирование рецептора А3 может повышать вероятность осложнений, вызываемых уже существующим или развивающимся ишемическим заболеванием сердца, таких как ангина или сердечная недостаточность.
В настоящее время, разработано много соединений в качестве антагонистов рецептора А2а для лечения разных заболеваний, как описано в WO 2007116106, WO 2009080197, WO 2011159302, WO 2011095625, WO 2014101373 и WO 2015031221. Однако, все еще существуют проблемы, такие как низкая растворимость, светочувствительность, низкая активность, низкая селективность и низкая биологическая доступность.
Согласно настоящему изобретению предложена новая структура гетероарил[4,3-с]пиримидин-5-амина в качестве антагониста адинозинового рецептора А2а, и обнаружено, что соединения, имеющие такую структуру, обладают сильной ингибирующей активностью, высокой селективностью, низкой концентрацией свободного лекарственного средство в мозге, слабой способностью проходить через гематоэнцефалитический барьер и меньшим количеством побочных эффектов, которые вероятно будут вызваны после поступления лекарственного средства в мозг.
Краткое изложение сущности изобретения
Цель настоящего изобретения заключается в предложении соединения формулы (I):

или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, где:
G представляет собой N или CR4;
кольцо А выбрано из группы, состоящей из циклоалкила, арила и гетероарила;
каждый R1 является идентичным или отличным, и каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, алкокси, галоалкила, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила;
R2 выбран из группы, состоящей из алкокси, гидрокси, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила, где каждый из алкокси, циклоалкила, гетеро цикл ила, арила и гетероарила независимо возможно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, алкила, алкокси, галоалкила, гидрокси, гидроксиалкила, оксо, циано, амино, нитро, ицклоалкила, гетероциклила, арила, гетероарила, C(O)OR5 и Rb;
R3 выбран из группы, состоящей из водорода, галогена, алкила, алкокси, галоалкила, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила;
R4 выбран из группы, состоящей из водорода, галогена, алкила, алкокси, галоалкила, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила;
R5 выбран из группы, состоящей из водорода, алкила, амино, галоалкила, циклоакила, гетероциклила, арила и гетероарила;
Rb представляет собой гетеро циклилалкил, где гетероциклил гетероциклилалкила возможно замещен одним или более заместителями, выбранными из группы, состоящей из алкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, циклоалкилокси, гетероциклила, арила, гетероарила и C(O)OR5; и
n представляет собой 1, 2, 3 или 4.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) R2 выбран из группы, состоящей из циано, циклоалкила, гетероциклила, арила и гетероарила, где каждый из циклоалкила, гетероциклила, арила и гетероарила независимо возможно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, алкила, галоалкила, алкокси, оксо, циклоалкила, гетероциклила и Rb; Rb представляет собой гетеро циклилалкил, где гетероциклил гетероциклилалкила возможно замещен одним или более алкилами.
В предпочтительном воплощении настоящего изобретения соединение формулы (I) представляет собой соединение формулы (II):

или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, или его фармацевтически приемлемую соль,
где:
кольцо В выбрано из группы, состоящей из циклоалкила, гетероциклила, арила и гетероарила;
каждый R6 является идентичным или отличным, и каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, алкокси, галоалкила, гидрокси, гидроксиалкила, оксо, циано, амино, нитро, циклоалкила, гетероциклила, арила, гетероарила, C(O)OR5 и Rb;
s представляет собой 0, 1, 2, 3 или 4; и
кольцо A, G, R1, R3, R5, Rb и n являются такими, как определено в формуле (I). В предпочтительном воплощении настоящего изобретения соединение формулы (I) представляет собой соединение формулы (III):

или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, или его фармацевтически приемлемую соль,
где
каждый R6 является идентичным или отличным, и каждый независимо выбран из группы, состоящей из галогена, алкила, галоалкила, алкокси, оксо, циклоалкила, гетероциклила, и Rb; Rb представляет собой гетероциклилалкил, где гетероциклил гетероциклилалкила возможно замещен одним или более алкилами;
кольцо А, кольцо В, R1, R3, n и s являются такими, как определено в формуле (II).
В предпочтительном воплощении настоящего изобретения в соединении формулы (II), кольцо В выбрано из группы, состоящей из фенила, 5-6-членного гетероциклила и 5-10-членного гетероарила и, предпочтительно, фенила, пиридила, пиразолила, пиридин-2-она, имидазолила, пирролила, фурила, тиенила, пиперидинила, 1,2,3,6-тетрагидропиридила, изохинолила, хинолила, хиноксалинила, индолила, индазолила, бензофуранила и бензотиенила.
В предпочтительном воплощении настоящего изобретения соединение формулы (I) представляет собой соединение формулы (III'):

или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, или его фармацевтически приемлемую соль,
где:
каждый R6 является идентичным или отличным, и каждый независимо выбран из группы, состоящей из атома водорода, атома галогена, алкила, галоалкила, алкокси, оксо, циклоалкила, гетероциклила и Rb; Rb представляет собой гетероциклилалкил, где гетероциклил гетероциклилалкила возможно замещен одним или более алкилами;
s равен 0, 1, 2, 3 или 4; кольцо A, R1, R3 и n являются такими, как определено в формуле (I).
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) кольцо А представляет собой арил или гетероарил, и предпочтительно фенил или фурил.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) R1 выбран из группы, состоящей из водорода, галогена и алкила.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) R3 выбран из группы, состоящей из водорода, галогена и алкила.
Типичные соединения по настоящему изобретению включают, но не ограничиваются следующими соединениями:



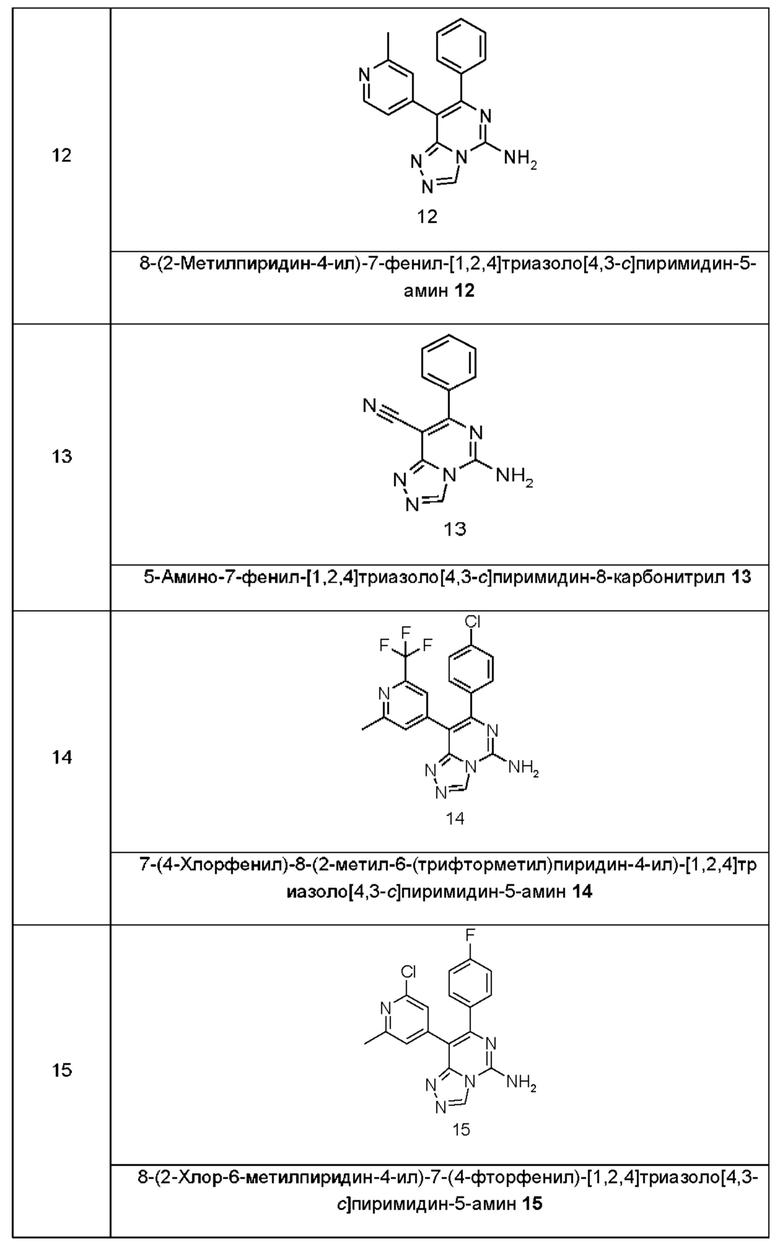


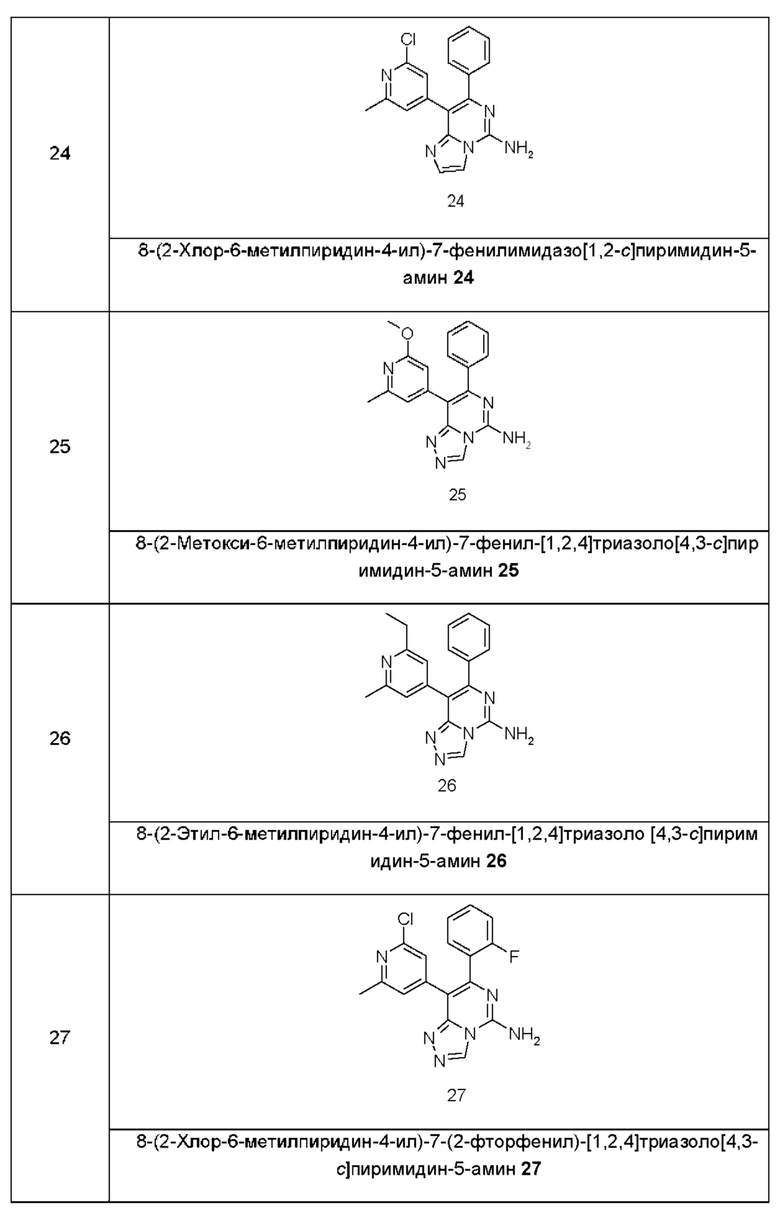

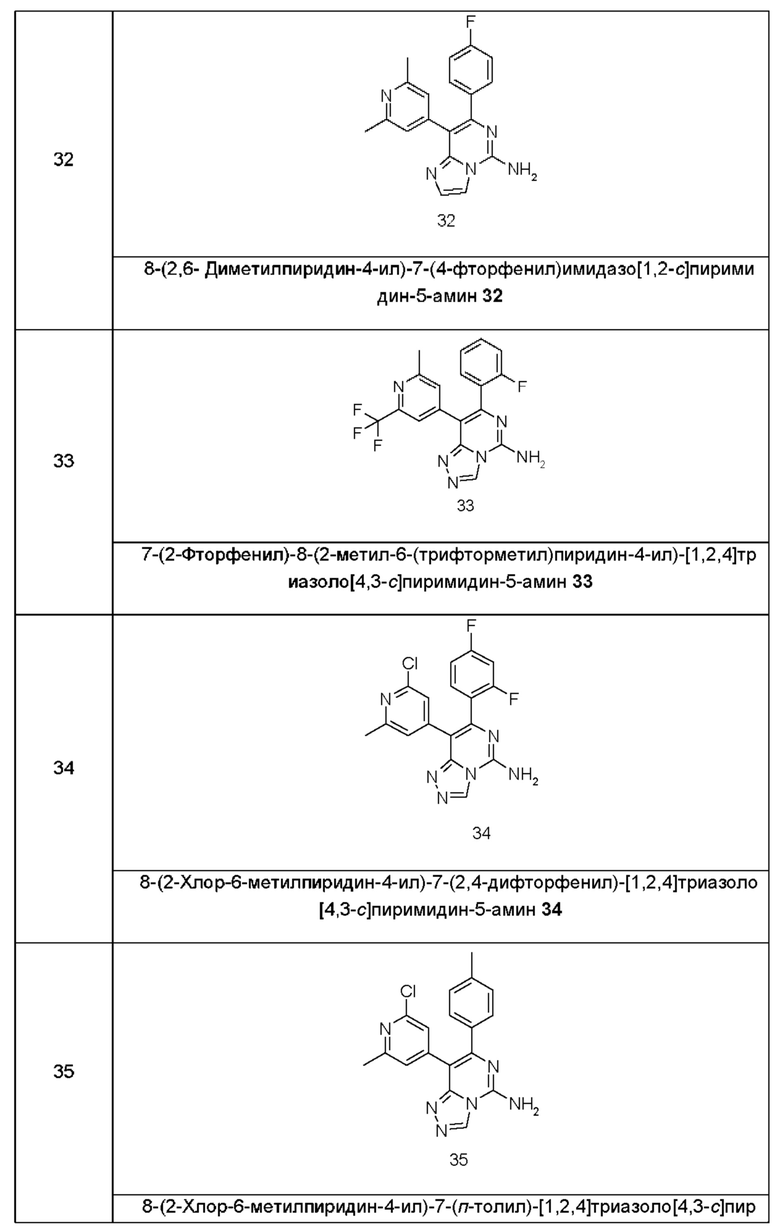

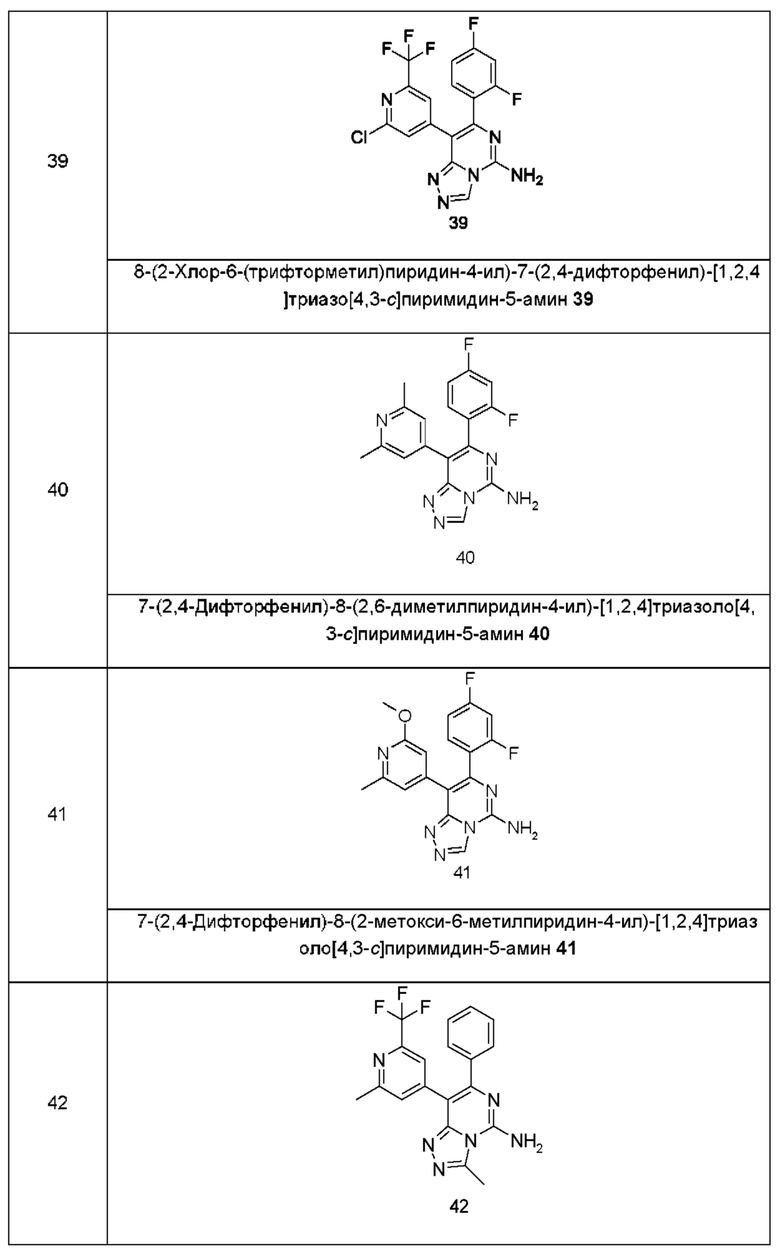


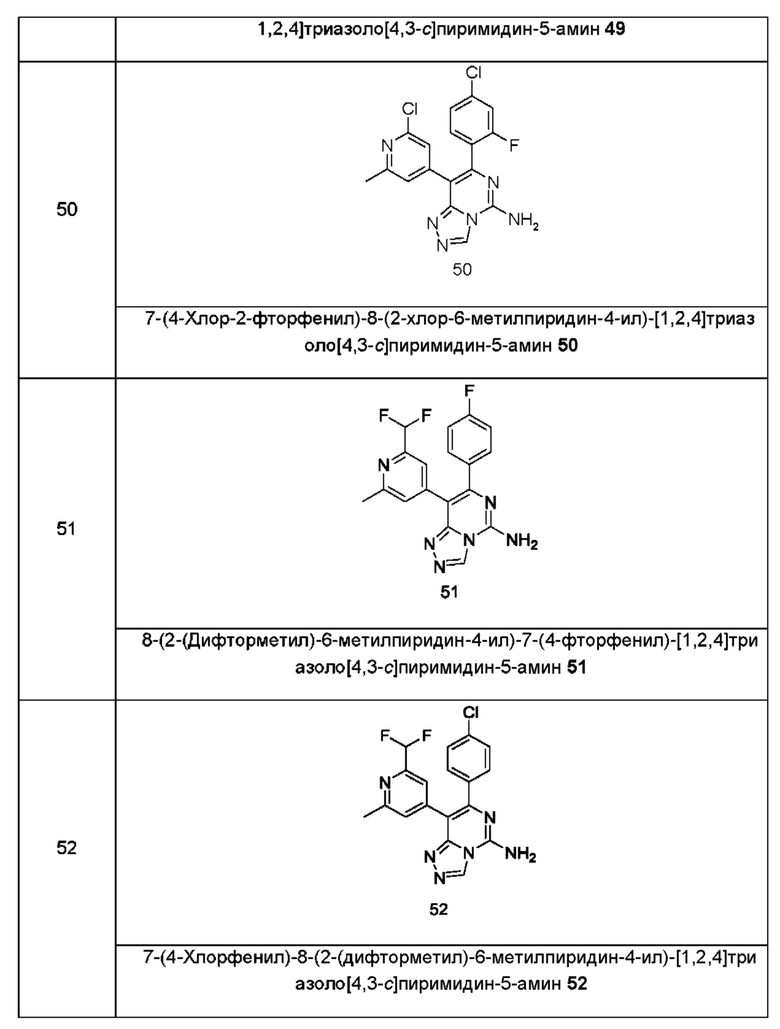
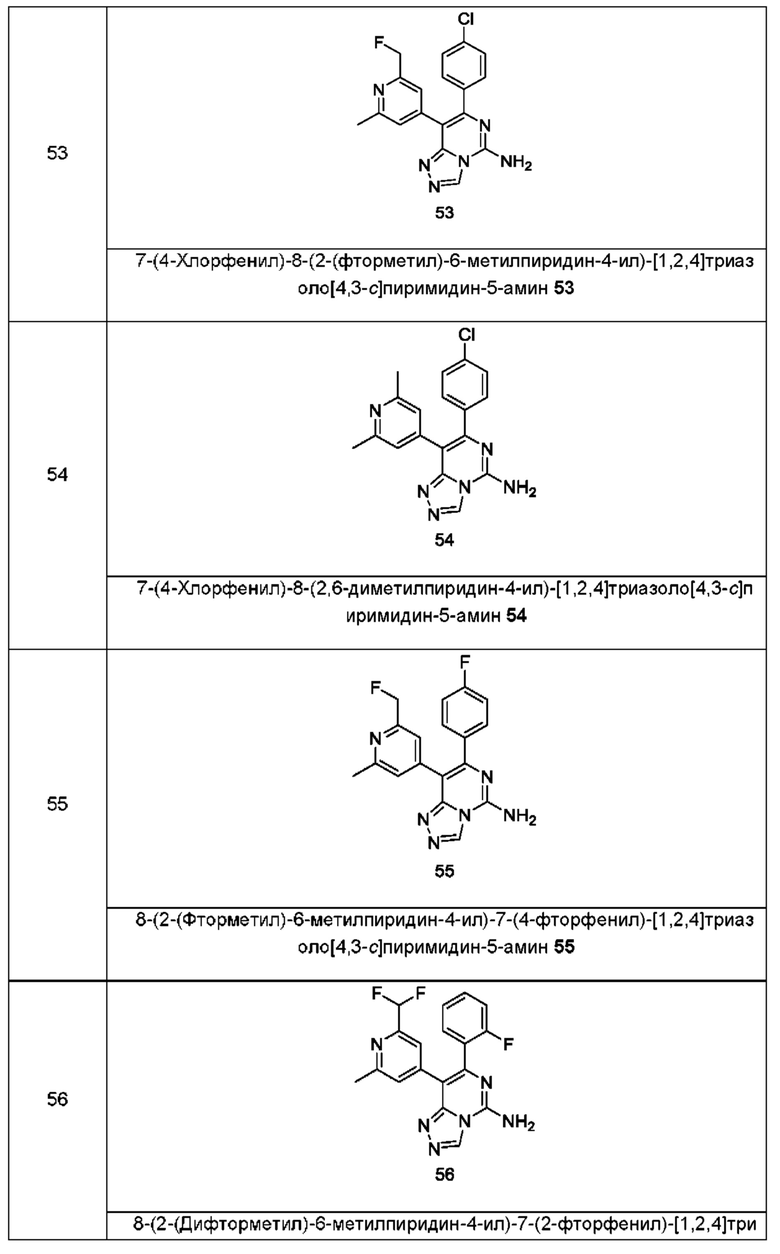

или их таутомером, мезомером, рацематом, энантиомером, диастереомером или их смесью, или их фармацевтически приемлемой солью.
В другом аспекте настоящее изобретение относится к соединению формулы (IV):

или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или его фармацевтически приемлемой соли,
где:
X представляет собой галоген;
кольцо A, G, R1, R3 и n являются такими, как определено в формуле (I). Типичные соединения по настоящему изобретению включают, но не ограничиваются следующими соединениями:


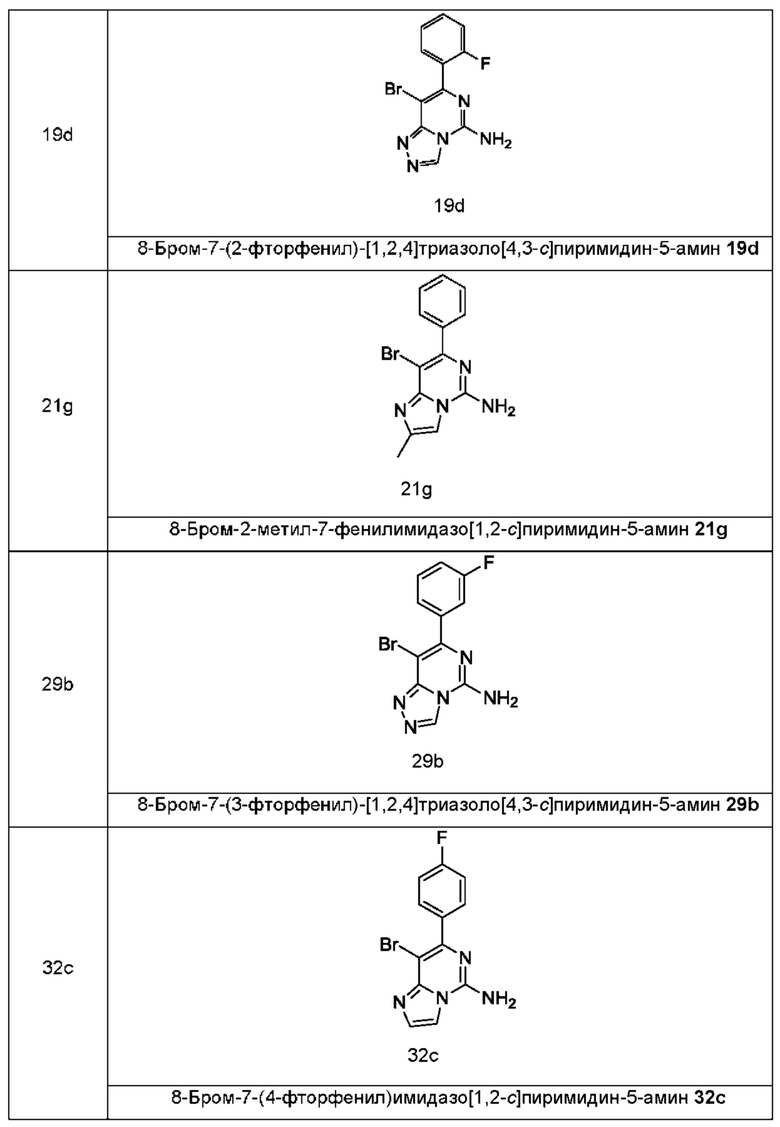
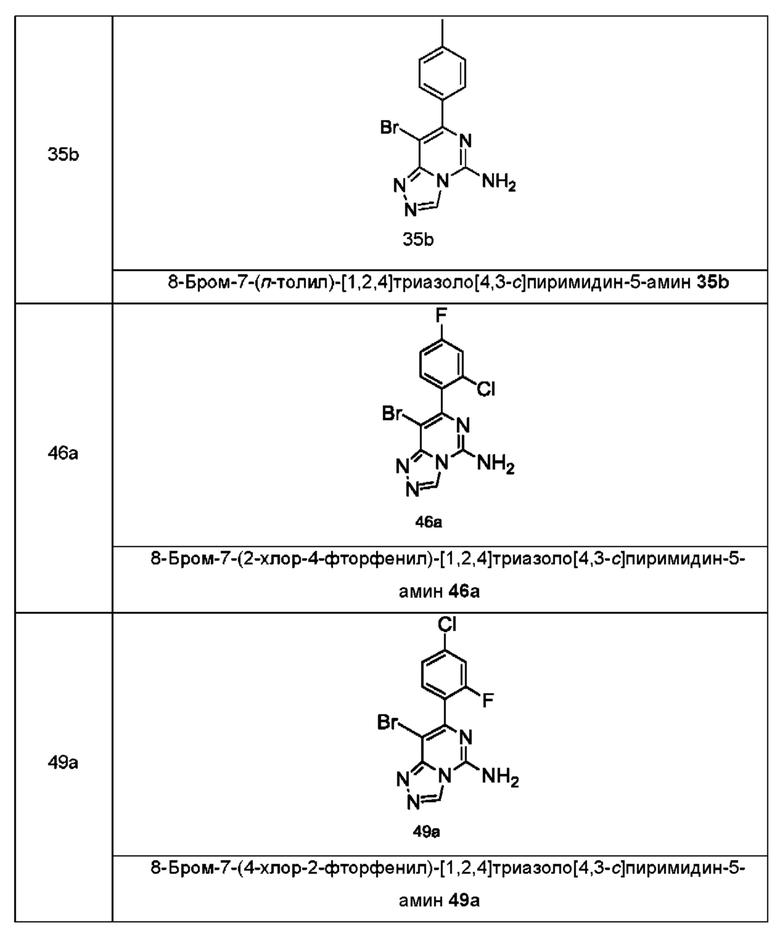
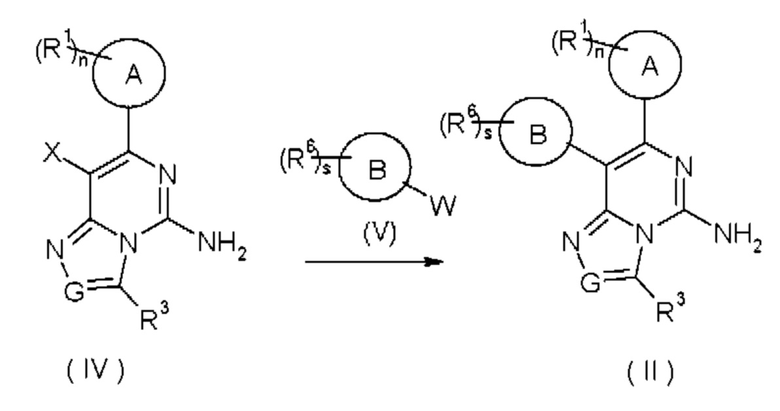
В другом аспекте настоящее изобретение относится к способу получения соединения формулы (II), включающему стадию:

вступления соединения формулы (IV) в реакцию с соединением формулы (V) с получением соединения формулы (II), где:
X представляет собой атом галогена;
W представляет собой 
кольцо А, кольцо В, G, R1, R3, R6, n и s представляют собой такие, как определено в формуле (I).
В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, и один или более фармацевтически приемлемых носителей, разбавителей или вспомогательных веществ.
Настоящее изобретение дополнительно относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей вышеуказанное, в получении лекарственного средства для ингибирования рецептора А2а.
Настоящее изобретение дополнительно относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей вышеуказанное, в получении лекарственного средства для лечения заболевания или состояния, улучшаемого посредством ингибирования рецептора А2а.
В настоящем изобретении заболевание или состояние, улучшаемое посредством ингибирования рецептора А2а, выбрано из группы, состоящей из опухоли, депрессии, нарушения когнитивной функции, нейродегенеративного расстройства (болезнь Паркинсона, болезнь Гентингтона, болезнь Альцгеймера или латеральный амиотрофический склероз и тому подобное), расстройства, связанного с вниманием, экстрапирамидального синдрома, аномального двигательного расстройства, цирроза, фиброза печени, жировой инфильтрации печени, кожного фиброза, нарушения сна, инсульта, повреждения головного мозга, нейровоспаления и зависимого поведения и предпочтительно опухоли.
Настоящее изобретение дополнительно относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей вышеуказанное, в получении лекарственного средства для лечения опухоли, депрессии, нарушения когнитивной функции, нейродегенеративного расстройства (болезнь Паркинсона, болезнь Гентингтона, болезнь Альцгеймера или латеральный амиотрофический склероз и тому подобное), расстройства, связанного с вниманием, экстрапирамидального синдрома, аномального двигательного расстройства, цирроза, фиброза печени, жировой инфильтрации печени, кожного фиброза, нарушения сна, инсульта, повреждения головного мозга, нейровоспаления и зависимого поведения, и предпочтительно опухоли.
Настоящее изобретение дополнительно относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли или фармацевтической композиции, содержащей вышеуказанное, в получении лекарственного средства для лечения опухоли.
Настоящее изобретение относится к способу ингибирования рецептора А2а, включающему стадию введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли или фармацевтической композиции, содержащей вышеуказанное.
Настоящее изобретение также относится к способу лечения заболевания или состояния, улучшаемого посредством ингибирования рецептора А2а, включающему стадию введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли или фармацевтической композиции, содержащей вышеуказанное.
Настоящее изобретении относится к способу лечения опухоли, депрессии, нарушения когнитивной функции, нейродегенеративного расстройства (болезнь Паркинсона, болезнь Гентингтона, болезнь Альцгеймера или латеральный амиотрофический склероз и тому подобное), расстройства, связанного с вниманием, экстрапирамидального синдрома, аномального двигательного расстройства, цирроза, фиброза печени, жировой инфильтрации печени, кожного фиброза, нарушения сна, инсульта, повреждения головного мозга, нейровоспаления и зависимого поведения, и предпочтительно опухоли, включающему стадию введения нуждающемуся в этом пациенту терапевтически эффективного количества соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли или фармацевтической композиции, содержащей вышеуказанное.
Настоящее изобретение дополнительно относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или его фармацевтически приемлемой соли или фармацевтической композиции, содержащей вышеуказанное, для применения в качестве лекарственного средства.
Настоящее изобретение также относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или его фармацевтически приемлемой соли или фармацевтической композиции, содержащей вышеуказанное, для применения в качестве антагониста рецептора Ага.
Настоящее изобретение также относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или его фармацевтически приемлемой соли или фармацевтической композиции, содержащей вышеуказанное, для применения в лечении заболевания или состояния, улучшаемого посредством ингибирования рецептора Ага.
Настоящее изобретении также относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или его фармацевтически приемлемой соли или фармацевтической композиции, содержащей вышеуказанное, для применения в лечении опухоли, депрессии, нарушения когнитивной функции, нейродегенеративного расстройства (болезнь Паркинсона, болезнь Гентингтона, болезнь Альцгеймера или латеральный амиотрофический склероз и тому подобное), расстройства, связанного с вниманием, экстрапирамидального синдрома, аномального двигательного расстройства, цирроза, фиброза печени, жировой инфильтрации печени, кожного фиброза, нарушения сна, инсульта, повреждения головного мозга, нейровоспаления и зависимого поведения, и предпочтительно опухали.
Настоящее изобретение дополнительно относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или его фармацевтически приемлемой соли или фармацевтической композиции, содержащей вышеуказанное, для применения в лечении опухоли.
Опухоль, описанная в настоящем изобретении, выбрана из группы, состоящей из меланомы, опухоли головного мозга, рака пищевода, рака желудка, рака печени, рака поджелудочной железы, колоректального рака, рака легкого, рака почки, рака молочной железы, рака яичника, рака предстательной железы, рака кожи, нейробластомы, саркомы, осгеохондромы, остеомы, остеосаркомы, семиномы, опухоли яичка, рака матки, рака головы и шеи, множественной миеломы, злокачественной лимфомы, истинной полицитемии, лейкоза, опухоли щитовидной железы, опухоли мочеточника, рака мочевого пузыря, рака желчного пузыря, холангиокарциномы, хорионэпителиомы и детской опухоли, и предпочтительно рака легкого.
Фармацевтическая композиция, содержащая действующее вещество, может быть представлена в форме, подходящей для перорального введения, например, таблетки, пастилки, леденца, водной или масляной суспензии, диспергируемого порошка или гранулы, эмульсии, жесткой или мягкой капсулы, сиропа или эликсира. Преоральная композиция может быть получена в соответствии с любым известным способом в данной области для получения фармацевтической композиции. Такая композиция может содержать один или более ингредиентов, выбранных из группы, состоящей из подсластителей, корригентов, красителей и консервантов, для предоставления приятной и привлекательной по вкусу фармацевтической композиции. Таблетка содержит действующее вещество в смеси с нетоксичными, фармацевтически приемлемыми вспомогательными веществами, подходящими для изготовления таблеток. Данные вспомогательные вещества могут представлять собой инертные вспомогательные вещества, гранулирующие агенты, дезинтегрирующие агенты, связующие вещества и смазывающие вещества. Таблетка может быть непокрыта оболочкой или покрыта оболочкой посредством известной методики для маскировки вкуса лекарственного средства или задержки дезинтеграции и поглощения действующего вещества в желудочно-кишечном тракте, обеспечивая, таким образом, замедленное высвобождение на протяжении длительного периода времени.
Пероральная композиция может быть также предложена в виде мягких желатиновых капсул, в которых действующее вещество смешано с инертным твердым разбавителем, или активное вещество смешано с водорастворимым носителем или масляной средой.
Водная суспензия содержит действующее вещество в смеси с вспомогательными веществами, подходящими для изготовления водной суспензии. Такие вспомогательные вещества представляют собой суспендирующие агенты, диспергирующие вещества или увлажнители. Водная суспензия может также содержать один или более консервантов, один или более красителей, один или более корригентов и один или более подсластителей.
Масляная суспензия может быть приготовлена посредством суспендирования действующего вещества в растительном масле или минеральном масле. Масляная суспензия может содержать загуститель. Упомянутые выше подсластители и корригенты могут быть добавлены для предоставления приятной на вкус композиции. Данные композиции можно хранить посредством добавления антиоксиданта.
Действующее вещество в смеси с диспергирующими веществами или увлажнителями, суспендирующим агентом или одним или более консервантами, можно получить в виде диспергируемых порошков или гранул, подходящих для получения водной суспензии в результате добавления воды. В качестве примера подходящих диспергирующих веществ или увлажнителей и суспендирующих агентов приведены диспергирующие вещества, увлажнители и суспендирующие агенты, уже упомянутые выше. Могут быть добавлены дополнительные вспомогательные вещества, такие как подсластители, корригенты и красители. Данные композиции можно хранить посредством добавления антиоксиданта, такого как аскорбиновая кислота.
Фармацевтическая композиция по настоящему изобретению может находиться в форме эмульсии типа «масло в воде». Масляная фаза может представлять собой растительное масло или минеральное масло или их смесь. Подходящие эмульгаторы могут представлять собой встречающиеся в природе фосфолипиды. Эмульсия может также содержать подсластитель, корригент, консервант и антиоксидант. Такая композиция может также содержать мягчитель, консервант, краситель и антиоксидант.
Фармацевтическая композиция по настоящему изобретению может находиться в форме стерильного инъецируемого водного раствора. Применимые носители или растворители, которые можно использовать, представляют собой воду, раствор Рингера или изотонический раствор хлорида натрия. Стерильная инъецируемая композиция может представлять собой стерильную, инъецируемую микроэмульсию типа «масло в воде», в которой действующее вещество растворено в масляной фазе. Инъецируемый раствор или микроэмульсию можно вводить в кровоток пациента посредством местной болюсной инъекции. В качестве альтернативы, раствор и микроэмульсию предпочтительно вводят таким образом, при котором сохраняется постоянная концентрация циркулирующего соединения по настоящему изобретению. Для поддержания данной постоянной концентрации может быть использовано устройство для непрерывной внутривенной доставки. Примером такого устройства является насос для осуществления внутривенных инъекций Deltec CADD-PLUS. ТМ. 5400.
Фармацевтическая композиция по настоящему изобретению может находиться в форме стерильной инъецируемой водной или масляной суспензии для внутримышечного и подкожного введения. Такая суспензия может быть приготовлена с помощью подходящих диспергирующих веществ или увлажнителей и суспендирующих агентов, как описано выше, в соответствии с известными методиками. Стерильная инъецируемая композиция может также представлять собой стерильный инъецируемый раствор или суспензию, приготовленную в нетоксичном парентерально приемлемом разбавителе или растворителе. Кроме того, стерильные жирные масла можно легко использовать в качестве растворителя или суспендирующей среды. С этой целью, можно использовать любое смешанное жирное масло. Кроме того, жирные кислоты можно также использовать для получения инъекций.
Соединение по настоящему изобретению можно вводить в форме суппозитория для ректального способа введения. Данную фармацевтическую композицию можно получать посредством смешивания лекарственного средства с подходящим нераздражающим вспомогательным веществом, которое является твердым при обычных температурах, но жидким в прямой кишке, плавясь, таким образом, в прямой кишке с высвобождением лекарственного средства.
Специалистам в данной области хорошо известно, что дозировка лекарственного средства зависит от множества факторов, включая следующие факторы: активность конкретного соединения, возраст пациента, масса пациента, общее состояние здоровья пациента, поведение пациента, рацион пациента, время введения, способ введения, скорость экскреции, сочетание лекарственных средств и тому подобное, но, не ограничиваясь ими. Кроме того, оптимальное лечение, такое как способ лечения, суточная доза соединения формулы (I) или тип его фармацевтически приемлемой соли, можно проверять посредством традиционных схем лечения.
Подробное описание изобретения
Если не указано иное, термины, используемые в описании и формуле изобретения, имеют значения, описанные ниже.
Термин «алкил» относится к насыщенной алифатической углеводородной группе, которая представляет собой группу с прямой или разветвленной цепью, содержащую от 1 до 20 атомов углерода, предпочтительно алкил, имеющий от 1 до 12 атомов углерода, и более предпочтительно алкил, имеющий от 1 до 6 атомов углерода. Неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, n-окстил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3- этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и их разные разветвленные изомеры. Более предпочтительно, алкильная группа представляет собой низший алкил, имеющий от 1 до 6 атомов углерода, и неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и т.п. Алкильная группа может быть замещенной или незамещенной. При замещении, замещающая(ие) группа(ы) может(гут) быть замещена(ы) в любой доступной точке соединения. Замещающая(ие) группа(ы) предпочтительно представляет (ют) собой одну или более групп, независимо выбранных из группы, состоящей из атома водорода, атома галогена, ал кила, алкокси, галоалкила, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила, гетероарила и -C(O)OR5.
Термин «алкокси» относится к -О-(алкильной) или -O-(незамещенной циклоалкильной) группе, где алкил представляет собой такой, как определено выше. Неограничивающие примеры алкокси включают метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси. Алкокси может быть, возможно, замещенным или незамещенным. При замещении замещающая(ие) группа(ы) предпочтительно представляет(ют) собой одну или более групп, независимо выбранных из группы, состоящей из атома водорода, атома галогена, ал кила, алкокси, галоалкила, гидрокси, гадроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила, гетероарила и -C(O)OR5.
Термин «циклоалкил» относится к насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной замещающей группе, имеющей от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода, предпочтительно от 3 до 10 атомов углерода и более предпочтительно от 3 до 6 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т.п. Полициклический циклоалкил включает циклоалкил, имеющий спирокольцо, конденсированное кольцо или мостиковое кольцо. Циклоалкил может быть замещенным или незамещенным. При замещении замещающая(ие) группа(ы) может(гут) быть замещена(ы) в любой доступной точке соединения. Замещающая(ие) группа(ы) предпочтительно представляет(ют) собой одну или более групп, независимо, возможно, выбранных из группы, состоящей из атома водорода, атома галогена, алкила, алкокси, галоалкила, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила, гетероарила и -C(O)OR5.
Термин «гетероциклил» относится к 3-20-членной насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной группе, где кольцевые один или более кольцевых атомов представляют собой гетероатомы, выбранные из группы, состоящей из N, О и S(O)m (где m представляет собой целое число от 0 до 2), но за исключением -О-О-, -O-S- или -S-S- в кольце, причем остальные кольцевые атомы представляют собой атомы углерода. Предпочтительно гетероциклил имеет от 3 до 12 кольцевых атомов, где 1-4 атома представляют собой гетероатомы, более предпочтительно от 3 до 10 кольцевых атомов, где 1-4 атома представляют собой гетероатомы, и более предпочтительно от 5 до 6 кольцевых атомов, где 1-3 атома представляют собой гетероатомы. Неограничивающие примеры моноциклического гетероциклила включают пирролидинил, тетрагидрофуранил, 1,2,3,6-тетрагидропиридил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил и т.п. Полициклический гетероциклил включает гетероциклил, имеющий спирокольцо, конденсированное кольцо или мостиковое кольцо.
Гетероциклильное кольцо может быть конденсировано с кольцом арила, гетероарила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой гетероциклил. Его неограничивающие примеры включают:

Гетероциклил может быть замещенным или незамещенным. При замещении замещающая(ие) группа(ы) может(гут) быть замещена(ы) в любой доступной точке соединения. Замещающая(ие) группа(ы) предпочтительно представляет(ют) собой одну или более групп, независимо возможно выбранных из группы, состоящей из атома водорода, атома галогена, алкила, алкокси, галоалкила, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила, гетероарила и -C(O)OR5.
Термин «арил» относится к 6-14-членному полностью углеродному моноциклическому кольцу или полициклическому конденсированному кольцу (т.е. каждое кольцо в системе имеет общую пару смежных атомов углерода с другим кольцом в системе), имеющему конъюгированную π-электронную систему, предпочтительно 6-10-членному арилу, например, фенилу и нафтилу. Кольцо арила может быть конденсировано с кольцом гетероарила, гетероциклила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой арильное кольцо. Неограничивающие примеры включают:

Арил может быть замещенным или незамещенным. При замещении замещающая(ие) группа(ы) может(гут) быть замещена(ы) в любой доступной точке соединения. Замещенная(ые) группа(ы) предпочтительно представляет(ют) собой одну или более групп, независимо возможно выбранных из группы, состоящей из атома водорода, атома галогена, алкила, алкокси, галоалкила, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила, гетероарила и -C(O)OR5.
Термин «гетероарил» относится к 5-14-членной гетероароматической системе, имеющей от 1 до 4 гетероатомов, выбранных из группы, состоящей из О, S и N. Гетероарил предпочтительно представляет собой 5-10-членный гетероарил, более предпочтительно 5- или 6-членный гетероарил, например, фуранил, тиенил, пиридил, пирролил, N-алкилпирролил, пиримидинил, пиразинил, пиридазинил, имидазолил, пиразолил, тетразолил и тому подобное. Кольцо гетероарила может быть конденсировано с кольцом арила, гетероциклила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой гетероарильное кольцо. Его неограничивающие примеры включают:


Гетероарил может быть замещенным или незамещенным. При замещении замещающая(ие) группа(ы) может(гут) быть замещена(ы) в любой доступной точке соединения. Замещающая(ие) группа(ы) представляет(ют) собой одну или более групп, независимо возможно выбранных из группы, состоящей из атома водорода, атома галогена, алкила, алкокси, галоалкоила, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила, гетероарила и -C(O)OR5.
Термин «оксо» относится к группе =O.
Термин «циклоалкилокси» относится к группе циклоалкил-O-.
Термин «гетероциклилалкил» относится к алкильной группе, замещенной одним или более гетероциклилами, где алкил и гетероциклил представляют собой такие, как определено выше. Термин «галоалкил» относится к алкильной группе, замещенной одним или более атомами галогена, где алкил представляет собой такой, как определено выше.
Термин «гидрокси» относится к группе -ОН.
Термин «гидроксиалкил» относится к алкильной группе, замещенной гидрокси, где алкил представляет собой такой, как определено выше.
Трермин «галоген» относится к атому фтора, хлора, брома или йода.
Термин «амино» относится к группе -NH2.
Термин «циано» относится к группе -CN.
Термин «нитро» относится к группе -NO2.
Термин «возможный» или «возможно» означает, что описанное впоследствии событие или обстоятельство может произойти, но необязательно произойдет, и такое описание включает ситуацию, в которой данное событие или обстоятельство происходит или не происходит. Например, фраза «гетероциклил, возможно замещенный алкилом» означает, что алкильная группа может присутствовать, но необязательно присутствует, и такое описание включает ситуацию, в которой гетероциклил замещен алкилом и в которой гетероциклил не замещен алкилом.
Термин «замещенный» относится к одному или более атомам водорода в группе, предпочтительно вплоть до 5 и более предпочтительно 1-3 атомам водорода, независимо замещенным соответствующим количеством заместителей. Совершенно очевидно, что заместители существуют только в их возможном химическом положении. Специалист в данной области способен определить, является ли замещение возможным или невозможным, с помощью экспериментов или теории, не прилагая слишком больших усилий. Например, комбинация амино- или гидрокси группы, имеющей свободный атом водорода, и атомов углерода, имеющих ненасыщенные связи (такие как олефиновые), может быть нестабильной.
Термин «фармацевтическая композиция» относится к смеси одного или более соединений по настоящему изобретению или их физиологически/фармацевтически приемлемых солей или пролекарств с другими химическими компонентами, и других компонентов, таких как физиологически/фармацевтически приемлемые носители и вспомогательные вещества. Цель фармацевтической композиции заключается в том, чтобы облегчать введение соединения в организм, которое способствует поглощению активного вещества, демонстрируя, таким образом, биологическую активность.
Фраза «фармацевтически приемлемая соль» относится к соли соединения по настоящему изобретению, которая является безопасной и эффективной у млекопитающих и обладает желаемой биологической активностью.
Где: R5 представляет собой такой, как определено в формуле (I).
Способ синтеза соединения по настоящему изобретению
Для достижения цели настоящего изобретения в настоящем изобретении применяют следующие технические решения:
Схема I
Способ получения соединения формулы (II) по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, включает следующую стадию:
соединение формулы (IV) и соединение формулы (V) подвергают реакции сочетания Сузуки в присутствии катализатора в щелочных условиях с получением соединения формулы (II).
Где:
Реагент, который обеспечивает щелочные условия, включает органические основания и неорганические основания. Органические основания включают триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, бис(триметилсилил)амин лития, ацетат калия, трет-бутоксид натрия, трет-бутоксид калия и н-бутоксид натрия, но не ограничиваются ими. Неорганические основания включают гидрид натрия, фосфат калия, карбонат натрия, карбонат калия, ацетат калия, карбонат цезия, гидроксид натрия и гидроксид лития, но не ограничиваются ими.
Катализатор включает Pd/C, Ni Ренея, тетра-трифенилфосфин палладия, дихолорид палладия, ацетат палладия, бис(дибензилиденацетон)палладий, хлор(2-дихлоргексилфисфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий, [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий, 1,1'-бис(дибензилфосфорил)ферроцен палладия дихлорид и трис(дибензилиденацетон)дипалладий, и предпочтительно [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий, но не ограничивается ими.
Приведенную выше реакцию предпочтительно проводят в растворителе. Используемый растворитель включает уксусную кислоту, метанол, этанол, н-бутанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, диметиловый эфир гликоля, воду, N,N-диметилформамид и их смеси, но не ограничивается ими.
Где:
X представляет собой галоген;
W представляет собой 
кольцо А, кольцо В, G, R1, R3, R6, n и s являются такими, как определено в формуле (II).
Схема II
Способ получения соединения формулы (III) по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли включает следующие стадии:

На стадии 1 соединение формулы (II1-1) вступает в реакцию с гидратом гидразина с получением соединения формулы (III-2);
На стадии 2 соединение формулы (III-2) вступает в реакцию с соединением формулы (III-3) с получением соединения формулы (IIIA);
На стадии 3 соединение формулы (IIIA) и соединение формулы (V) подвергают взаимодействию в реакции сочетания Сузуки в присутствии катализатора в щелочных условиях с получением соединения формулы (III).
Где:
Реагент, который обеспечивает щелочные условия, включает органические основания и неорганические основания. Органические основания включают триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, бис(триметилсилил)амин лития, ацетат калия, трет-бутоксид натрия, трет- бутоксид калия и н-бутоксид натрия, но не ограничиваются ими. Неорганические основания включают гидрид натрия, фосфат калия, карбонат натрия, карбонат калия, ацетат калия, карбонат цезия, гидроксид натрия и гидроксид лития, но не ограничиваются ими.
Катализатор включает Pd/C, Ni Ренея, тетра-трифенилфосфин палладия, дихолорид палладия, ацетат палладия, бис(дибензилиденацетон)палладий, хлор(2-дихлоргексилфосфино-2',4',6'-триизопропил-1, 1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий, [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий, 1,1'-бис(дибензилфосфорил)ферроцен палладия дихлорид и трис(дибензилиденацетон)дипалладий, и предпочтительно [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий, но не ограничивается ими.
Приведенную выше реакцию предпочтительно проводят в растворителе. Используемый растворитель включает уксусную кислоту, метанол, этанол, н-бутанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, диметиловый эфир гликоля, воду, N,N-диметилформамид и их смеси, но не ограничивается ими.
Где:
X представляет собой галоген;
W представляет собой  или
или  ;
;
кольцо А, кольцо В, G, R1, R3, R6, n и s представляют собой такие, как определено в формуле (II).
Схема III
Способ получения соединения формулы (III) по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли включает следующие стадии:

На стадии 1 соединение формулы (III-1) вступает в реакцию с гидратом гидразина с получением соединения формулы (III-2);
На стадии 2 соединение формулы (III-2) и соединение формулы (V) подвергают взаимодействию в реакции сочетания Сузуки в присутствии катализатора в щелочных условиях с получением соединения формулы (IIIB);
На стадии 3 соединение формулы (IIIB) вступает в реакцию с соединением формулы (III-3) с получением соединения формулы (III).
Где:
Реагент, который обеспечивает щелочные условия, включает органические основания и неорганические основания. Органические основания включают триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, бис(триметилсилил)амин лития, ацетат калия, трет-бутоксид натрия, трет- бутоксид калия и н-бутоксид натрия, но не ограничиваются ими. Неорганические основания включают гидрид натрия, фосфат калия, карбонат натрия, карбонат калия, ацетат калия, карбонат цезия, гидроксид натрия и гидроксид лития, но не ограничиваются ими.
Катализатор включает Pd/C, Ni Ренея, тетра-трифенилфосфин палладия, дихолорид палладия, ацетат палладия, бис(дибензилиденацетон)палладий, хлор(2-дихлоргексилфосфино-2',4',6'-триизопропил-1, 1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий, [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий, 1,1'-бис(дибензилфосфорил)ферроцен палладия дихлорид и трис(дибензилиденацетон)дипалладий, и предпочтительно [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий, но не ограничивается ими.
Приведенную выше реакцию предпочтительно проводят в растворителе. Используемый растворитель включает уксусную кислоту, метанол, этанол, н-бутанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, диметиловый эфир гликоля, воду, N,N-диметилформамид и их смеси, но не ограничивается ими.
Где:
X представляет собой галоген;
W представляет собой  или
или  ;
;
кольцо А, кольцо В, R1, R3, R6, n и s являются такими, как определено в формуле (III).
Схема IV
Способ получения соединения формулы (III') по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли включает следующие стадии:
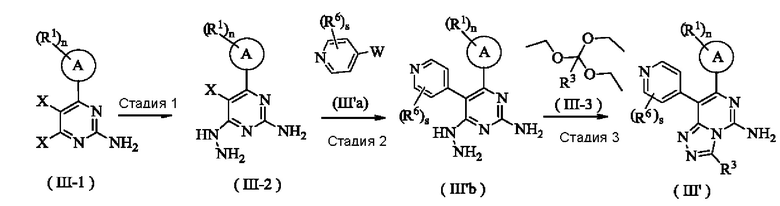
На стадии 1 соединение формулы (II1-1) вступает в реакцию с гидратом гидразина с получением соединения формулы (III-2);
На стадии 2 соединение формулы (III-2) и соединение формулы (III'а) подвергают взаимодействию в реакции сочетания Сузуки в присутствии катализатора в щелочных условиях с получением соединения формулы (III'b);
На стадии 3 соединение формулы (III'b) вступает в реакцию с соединением формулы (III-3) с получением соединения формулы (III').
Где:
Реагент, который обеспечивает щелочные условия, включает органические основания и неорганические основания. Органические основания включают триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, бис(триметилсилил)амин лития, ацетат калия, трет-бутоксид натрия, трет- бутоксид калия и н-бутоксид натрия, но не ограничиваются ими. Неорганические основания включают гидрид натрия, фосфат калия, карбонат натрия, карбонат калия, ацетат калия, карбонат цезия, гидроксид натрия и гидроксид лития, но не ограничиваются ими.
Катализатор включает Pd/C, Ni Ренея, тетра-трифенилфосфин палладия, дихолорид палладия, ацетат палладия, бис(дибензилиденацетон)палладий, хлор(2-дихлоргексилфисфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий, [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий, 1,1'-бис(дибензилфосфорил)ферроцен палладия дихлорид и трис(дибензилиденацетон)дипалладий, и предпочтительно [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий, но не ограничивается ими.
Приведенную выше реакцию предпочтительно проводят в растворителе. Используемый растворитель включает уксусную кислоту, метанол, этанол, н-бутанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, диметиловый эфир гликоля, воду, N,N-диметилформамид и их смеси, но не ограничивается ими.
Где:
X представляет собой галоген;
W представляет собой  или
или  ;
;
кольцо A, R1, R3, R6, n и s являются такими, как определено в формуле (III).
Предпочтительные воплощения
Настоящее изобретение будет дополнительно описано со ссылкой на следующие примеры, но данные примеры не следует рассматривать как ограничивающие объем настоящего изобретения.
Примеры
Структуры соединений идентифицировали посредством ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (МС). Сдвиги ЯМР (δ) приведены в 10-6 (млн-1). ЯМР определяли с помощью прибора Bruker AVANCE-400. Растворители для определения представляли собой дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD), и внутренний стандарт представлял собой тетраметилсилан (ТМС).
МС определяли с помощью масс-спектрометра FINNIGAN LCQAd (ионизация электрораспылением (ИЭР)) (производитель: Thermo, тип: Finnigan LCQ advantage MAX).
Высокоэффективную жидкостную хроматографию (ВЭЖХ) осуществляли на хроматографе для жидкостной хроматографии высокого давления Agilent 1200DAD (хроматографическая колонка Sunfire С18 150×4,6 мм) и на хроматографе для жидкостной хроматографии высокого давления Waters 2695-2996 (хроматографическая колонка Gimini С18 150×4,6 мм).
Анализ на основе хиральной ВЭЖХ осуществляли на LC-10A vp (Shimadzu) или SFC-analytical (Berger Instruments Inc.).
В качестве пластины для тонкослойной хроматографии на силикагеле (ТСХ) использовали пластину с силикагелем Yantai Huanghai HSGF254 или Qingdao GF254. Размеры пластины с силикагелем, используемой в ТСХ, составляли от 0,15 мм до 0,2 мм, а размеры пластины с силикагелем, используемой для очистки продукта, составляли от 0,4 мм до 0,5 мм.
В качестве носителя для колоночной хроматографии использовали силикагель Yantai Huanghai от 200 до 300 меш.
Для хиральной препаративной колоночной хроматографии использовали Prep Star SD-1 (Varian Instruments Inc.) или SFC-multigram (Berger Instruments Inc.).
Используемая система для флеш-хроматографии представляла собой Combiflash Rf200 (TELEDYNE ISCO).
Средние скорости ингибирования киназы и значения IC50 (концентрация полумаксимального ингибирования) определяли посредством NovoStar для твердофазного иммуноферментного анализа (ELISA - от англ. Enzyme-linked immunosorbent assay) (BMG Co., Германия).
Известные исходные вещества по настоящему изобретению могут быть получены способами, известными в данной области, или могут быть приобретены у ABCR GmbH & Co. KG, Acros Organnics, Aldrich Chemical Company, Accela ChemBio Inc. или Dari Chemical Company и т.д.
Если не указано иное, реакции проводили в атмосфере аргона или в атмосфере азота.
Термин «атмосфера аргона» или «атмосфера азота» означает, что реакционная колба оснащена баллоном аргона или азота (примерно 1 л).
Термин «атмосфера водорода» означает, что реакционная колба оснащена баллоном водорода (примерно 1 л).
Реакции гидрогенизации под давлением проводили на аппарате для гидрогенизации Parr 3916EKX и генераторе водорода QL-500 или аппарате для гидрогенизации HC2-SS.
В реакциях гидрогенизации в реакционной системе обычно создавали вакуум и заполняли ее водородом, причем описанную выше операцию повторяли три раза.
В микроволновых реакциях использовали микроволновый реактор типа СЕМ Discover-S 908860.
Если не указано иное, раствор относится к водному раствору.
Если не указано иное, температура реакции представляет собой комнатную температуру от 20°С до 30°С.
Реакционный процесс в примерах отслеживали посредством тонкослойной хроматографии (ТСХ). Используемый в реакциях проявляющий растворитель, система элюентов в колоночной хроматографии и система проявляющих растворителей в тонкослойной хроматографии для очистки соединений включали: А: систему дихлорметан/метанол, В: систему н-гексан/этил ацетат, С: систему петролейный эфир/этил ацетат, D: ацетон, Е: систему дихлорметан/ацетон, F: систему этилацетат/дихлорметан, G: этилацетат/дихлорметан/н-гексан и Н: этилацетат/дихлорметан/ацетон. Объемную долю растворителя регулировали в соответствии с полярностью соединений, и для регуляции также может быть добавлено небольшое количество щелочного реагента, такого как триэтиламин, или кислотного реагента, такого как уксусная кислота.
Пример 1
8-(2-Метил-6-(трифторметил)пиридин-4-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 1

Стадия 1
4-Хлор-5-йод-6-фенилпиримидин-2-амин 1b
4-Хлор-6-фенилпиримидин-2-амин 1a (2 г, 9,75 ммоль, полученный в соответствии с известным способом, раскрытым в «Bioorganic & Medicinal Chemistry Letters, 2011, 21(8), 2497-2501») и N-йодсукцинимид (2,6 г, 11,7 ммоль) растворяли в 30 мл уксусной кислоты. После завершения добавления реакционный раствор перемешивали в течение 16 часов. В реакционный раствор добавляли 200 мл насыщенного раствора бикарбоната натрия и проводили экстракцию этилацетатом (100 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл), сушили над безводным сульфатом натрия и фильтровали со сбором фильтрата. Фильтрат концентрировали при пониженном давлении, и остаток очищали посредством хроматографии на силикагеле с системой элюентов В с получением указанного в заголовке соединения 1b (2,88 г, выход: 89%).
МС m/z (ИЭР): 332,2 [М+1]
Стадия 2
4-Хлор-5-(2-метил-6-(трифторметил)пиридин-4-ил)-6-фенилпиримидин-2-амин 1d Соединение 1b (2,88 г, 8,7 ммоль), 2-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-6-(трифторметил)пиридин 1с (2,7 г, 9,17 ммоль, полученный в соответствии с известным способом, раскрытым в «Journal of Medicinal Chemistry, 2012, 55(5), 1898-1903»), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (0,64 г, 0,87 ммоль) и карбонат калия (3,6 г, 26,1 ммоль) последовательно растворяли в 66 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 10:1) в атмосфере азота. Реакционный раствор нагревали до 83°С и перемешивали в течение 3 часов. Реакцию останавливали, и реакционный раствор охлаждали до комнатной температуры и фильтровали. В фильтрат добавляли 200 мл этилацетата, промывали насыщенным раствором хлорида натрия (100 мл), сушили над безводным сульфатом натрия и фильтровали со сбором фильтрата. Фильтрат концентрировали при пониженном давлении, и остаток очищали посредством хроматографии на силикагеле с системой элюентов В с получением указанного в заголовке соединения 1d (2,4 г, выход: 76%).
МС m/z (ИЭР): 365,4 [М+1]
Стадия 3
4-Гидразинил-5-(2-метил-6-(трифторметил)пиридин-4-ил)-6-фенилпиримидин-2-амин 1е
Соединение 1d (100 мг, 0,275 ммоль) и 85% гидрат гидразина (62 мг, 2,747 ммоль) последовательно растворяли в 10 мл этанола. Реакционный раствор перемешивали с обратным холодильником в течение 17 часов. Реакцию останавливали, и реакционный раствор охлаждали до комнатной температуры. В реакционный раствор добавляли 30 мл воды и экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл × 2), сушили над безводным сульфатом натрия и фильтровали со сбором фильтрата. Фильтрат концентрировали при пониженном давлении с получением неочищенного, указанного в заголовке соединения 1е (85 мг), которое непосредственно использовали на следующей стадии без очистки.
МС m/z (ИЭР): 361,4 [М+1]
Стадия 4
8-(2-Метил-6-(трифторметил)пиридин-4-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 1
Неочищенный продукт 1е (85 мг, 0,275 ммоль) добавляли к 2 мл (три)этилортоформиата. Реакционный раствор перемешивали при 140°С в течение 0,5 часа. Реакцию останавливали, и реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный в результате остаток очищали посредством высокоэффективной жидкостной хроматографии с получением указанного в заголовке соединения 1 (6 мг, выход: 6,9%).
МС m/z (ИЭР): 371,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.38 (s, 1Н), 8.42 (brs, 2Н), 7.59 (s, 1Н), 7.42(s, 1Н), 7.35-7.31 (m, 5Н), 2.48 (s, 3Н).
Пример 2
7-Фенил-8-(1Н-пиразол-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 2

Стадия 1
5-Бром-4-хлор-6-фенилпиримидин-2-амин 2a
Соединение 1a (500 мг, 2,431 ммоль) и N-бромсукцинимид (519 мг, 2,918 ммоль) растворяли в 16 мл N,N-диметилформамида. После завершения добавления реакционный раствор перемешивали в течение 1 часа. В реакционный раствор добавляли 50 мл воды и экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, последовательно промывали водой (100 мл × 3) и насыщенным раствором хлорида натрия (200 мл), сушили над безводным сульфатом натрия и фильтровали со сбором фильтрата. Фильтрат концентрировали при пониженном давлении с получением неочищенного, указанного в заголовке соединения 2а (698 мг), которое использовали непосредственно на следующей стадии без очистки.
МС m/z (ИЭР): 284,2 [М+1]
Стадия 2
5-Бром-4-гидразинил-6-фенилпиримидин-2-амин 2b
Неочищенный продукт 2а (692 мг, 2,432 ммоль) и 85% гидрат гидразина (1,432 мг, 24,32 ммоль) последовательно растворяли в 20 мл этанола. Реакционный раствор перемешивали с обратным холодильником в течение 1 часа. Реакционный раствор охлаждали до комнатной температуры и перемешивали в течение 0,5 часа. Реакционный раствор фильтровали, и осадок на фильтре последовательно промывали этанолом (3 mL × 2) и простым эфиром (3 мл × 2). Осадок на фильтре собирали и сушили с получением неочищенного указанного в заголовке соединения 2b (480 мг), которое непосредственно использовали на следующей стадии без очистки.
МС m/z (ИЭР): 280,3 [М+1]
Стадия 3
8-Бром-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 2с
Неочищенный продукт 26 (480 мг, 1,713 ммоль) добавляли к 5 мл (три)этилортоформиата. Реакционный раствор перемешивали при 140°С в течение 15 минут. Реакцию останавливали, и реакционный раствор охлаждали до комнатной температуры и фильтровали. Осадок на фильтре последовательно промывали этанолом (3 мл × 3) и диэтиловым эфиром (5 мл × 3). Осадок на фильтре собирали и сушили с получением указанного в заголовке соединения 2с (348 мг, выход: 62,4%).
МС m/z (ИЭР): 290,3 [М+1]
Стадия 4
7-Фенил-8-(1Н-пиразол-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 2
Соединение 2с (120 мг, 0,414 ммоль), 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол 2 г (120 мг, 0,620 ммоль, полученный в соответствии с известным способом, раскрытым в «Journal of the American Chemical Society, 2014, 136(11), 4287-4299»), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (30 мг, 0,041 ммоль) и карбонат калия (171 мг, 1,241 ммоль) последовательно растворяли в 6 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 5:1) в атмосфере аргона. Реакционный раствор нагревали до 90°С и перемешивали в течение 3 часов. Реакцию останавливали, и в реакционный раствор добавляли 50 мл воды и экстрагировали этилацетатом (30 мл × 3). Органическую фазу концентрировали при пониженном давлении, и остаток очищали посредством высокоэффективной жидкостной хроматографии с получением указанного в заголовке соединения 2 (33 мг, выход: 28,7%).
МС m/z (ИЭР): 278,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 12,79 (brs, 1Н), 9.32 (s, 1Н), 7.98 (brs, 2Н), 7.41-7.37 (m, 7Н).
Пример 3
7-Фенил-8-(хинолин-6-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 3

Соединение 2c (100 мг, 0,345 ммоль), 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан -2-ил)хинолин 3а (106 мг, 0,414 ммоль, полученный в соответствии с известным способом, раскрытым в «Journal of the American Chemical Society, 2013, 135(50), 18730-18733»), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (25 мг, 0,034 ммоль) и карбонат калия (143 мг, 1,034 ммоль) последовательно растворяли в 6 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 5:1) в атмосфере аргона. Реакционный раствор нагревали до 90°С и перемешивали в течение 2 часов. Реакцию останавливали, и к реакционному раствору добавляли 50 мл воды и экстрагировали этилацетатом (50 мл × 3). Органическую фазу концентрировали при пониженном давлении, и остаток очищали посредством хроматографии на силикагеле с системой элюентов А с получением указанного в заголовке соединения 3 (49 мг, выход: 41,9%).
МС m/z (ИЭР): 339,2 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.38 (s, 1 Н), 8.88-8.87 (m, 1Н), 8.29-8.27 (m, 1Н), 8.20 (brs, 2Н), 8.05-8.04 (m, 1Н), 7.88-7.85 (m, 1Н), 7.54-7.50 (m, 2Н), 7.37-7.34 (m, 2Н), 7.24-7.20 (m, 3Н).
Пример 4
8-(2-Хлор-6-метилпиридин-4-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 4


Соединение 2c (100 мг, 0,345 ммоль), 2-хлор-6-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин 4а (105 мг, 0,414 ммоль, полученный в соответствии с известным способом, раскрытым в «Organic Syntheses, 2005, 82, 126-133»), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (25 мг, 0,034 ммоль) и карбонат калия (143 мг, 1,034 ммоль) последовательно растворяли в 10 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 4:1) в атмосфере аргона. Реакционный раствор нагревали до 90°С и перемешивали в течение 3 часов. Реакцию останавливали, и в реакционный раствор добавляли 50 мл воды и экстрагировали этилацетатом (30 мл × 4). Органическую фазу концентрировали при пониженном давлении, и остаток очищали посредством высокоэффективной жидкостной хроматографии с получением указанного в заголовке соединения 4 (20,7 мг, выход: 17,8%).
МС m/z (ИЭР): 337,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.36 (s, 1Н), 8.38 (brs, 2Н), 7.36-7.32 (m, 5Н), 7.19-7.14 (m, 2Н), 2.35 (s, 3Н).
Пример 5
8-(8-Метилхинолин-6-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 5

Стадия 1
8-Метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)хинолин 5c
6-Бром-8-метилхинолин 5b (444 мг, 2,00 ммоль, полученный в соответствии с известным способом, раскрытым в «Journal of Organic Chemistry, 2014, 79(11), 5379-5385»), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) 5a (508 мг, 2,00 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (292 мг, 0,40 ммоль) и ацетат калия (588 мг, 6,00 ммоль) последовательно растворяли в 10 мл диметилового эфира глицерина в атмосфере аргона. Реакционный раствор нагревали до 80°С и перемешивали в течение 12 часов. Реакцию останавливали, и реакционный раствор охлаждали до комнатной температуры и фильтровали. В фильтрат добавляли 20 мл этилацетата, последовательно промывали водой (10 мл) и насыщенным раствором хлорида натрия (10 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали посредством тонкослойной хроматографии с системой проявляющих растворителей В с получением указанного в заголовке соединения 5в (320 мг, выход: 59,5%).
МС m/z (ИЭР): 270,1 [М+1]
Стадия 2
8-(8-Метилхинолин-6-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин
Соединение 2с (100 мг, 0,345 ммоль), 5с (130 мг, 0,482 ммоль), [1,Г-бис(дифенилфосфино)ферроцен]дихлорпалладий (25 мг, 0,034 ммоль) и карбонат калия (143 мг, 1,034 ммоль) последовательно растворяли в 5 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 4:1) в атмосфере аргона. Реакционный раствор нагревали до 90°С и перемешивали в течение 2 часов. Реакцию останавливали, и в реакционный раствор добавляли 50 мл воды и экстрагировали этилацетатом (30 мл ×3). Органическую фазу концентрировали при пониженном давлении, и остаток очищали посредством высокоэффективной жидкостной хроматографии с получением указанного в заголовке соединения 5 (32 мг, выход: 26,4%).
МС m/z (ИЭР): 353,2 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 19.37 (s, 1Н), 8.90-8.89 (m, 1Н), 8.21-8.19 (m, 3Н), 7.78 (s, 1 Н), 7.51-7.49 (m, 2Н), 7.37-7.36 (m, 2Н), 7.22-7.20 (m, 3Н), 2.60 (s, 3Н).
Пример 6
8-(7-Фтор-1Н-индазол-5-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 6


Стадия 1
7-Фтор-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индазол 6b
5-Бром-7-фтор-1Н-индазол 6а (1,27 г, 5,90 ммоль, полученный в соответствии со способом, раскрытым в заявке на патент «WO 2012037410»), соединение 5а (2,25 г, 8,86 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (432 мг, 0,56 ммоль) и ацетат калия (1,74 г, 17,7 ммоль) последовательно растворяли в 40 мл диметилового эфира гликоля в атмосфере аргона. Реакционный раствор нагревали до 80°С и перемешивали в течение 12 часов. Реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры, в него добавляли 10 мл этилацетата и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали посредством хроматографии на силикагеле с системой элюентов В с получением указанного в заголовке соединения 6b (1,178 г, выход: 76,0%).
МС m/z (ИЭР): 263,2 [М+1]
Стадия 2
8-(7-Фтор-1Н-индазол-5-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 6 Соединение 2с (117 мг, 0,348 ммоль), 6b (100 мг, 0,382 ммоль), [1,Г-бис(дифенилфосфино)ферроцен]дихлорпалладий (25 мг, 0,034 ммоль) и карбонат калия (192 мг, 1,387 ммоль) последовательно растворяли в 10 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 4:1) в атмосфере аргона. Реакционный раствор нагревали до 90°С и перемешивали в течение 2 часов. Реакцию останавливали, в реакционный раствор добавляли 50 мл воды и экстрагировали этилацетатом (30 мл × 3). Органическую фазу концентрировали при пониженном давлении, и остаток очищали посредством высокоэффективной жидкостной хроматографии с получением указанного в заголовке соединения 6 (13 мг, выход: 10,8%).
МС m/z (ИЭР): 346,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 13.65 (brs, 1Н), 9.34 (s, 1Н), 8.13-8.11 (m, 3Н), 7.55 (s, 1Н), 7.35-7.33 (m, 2Н), 7.23-7.22 (m, 3Н), 7.05-7.02 (m, 1 Н).
Пример 7
8-(8-Фторхинолин-6-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 7
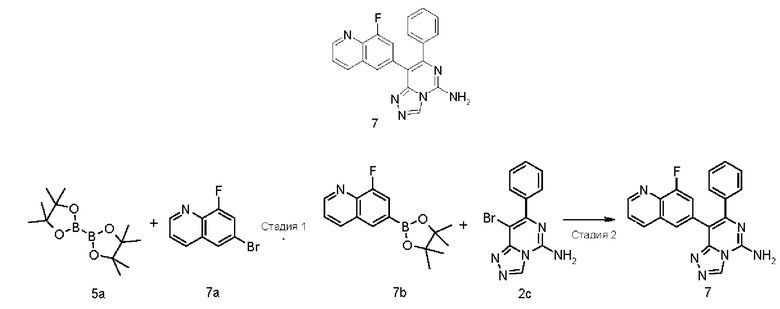
Стадия 1
8-Фтор-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)хинолин 7b
6-Бром-8-фторхинолин 7a (226 мг, 1,00 ммоль, полученный в соответствии с известным способом, раскрытым в «Journal of Medicinal Chemistry, 2010, 53(10), 4066-4084»), соединение 5a (305 мг, 1,20 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (146 мг, 0,20 ммоль) и ацетат калия (294 мг, 3,00 ммоль) последовательно растворяли в 10 мл диметилового эфира гликоля в атмосфере аргона. Реакционный раствор нагревали до 80°С и перемешивали в течение 12 часов. Реакцию останавливали, и реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали посредством системы для флеш-хроматографии CombiFlash с системой элюентов В с получением указанного в заголовке соединения 7b (220 мг, выход: 80,1%).
МС m/z (ИЭР): 274,1 [М+1]
Стадия 2
8-(8-Фторхинолин-6-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 7
Соединение 2с (100 мг, 0,345 ммоль), 7b (100 мг, 0,414 ммоль), [1,1'-бис(дифенилфосфино)феррецен]дихлорпалладий (25 мг, 0,034 ммоль) и карбонат калия (143 мг, 1,034 ммоль) последовательно растворяли в 6 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 5:1) в атмосфере аргона. Реакционный раствор нагревали до 90°С и перемешивали в течение 3 часов. Реакцию останавливали, и в реакционный раствор добавляли 50 мл воды и экстрагировали этилацетатом (40 мл × 4). Органическую фазу концентрировали при пониженном давлении, и остаток очищали посредством высокоэффективной жидкостной хроматографии с получением указанного в заголовке соединения 7 (20 мг, выход: 16,3%).
МС m/z (ИЭР): 357,2 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.38 (s, 1Н), 8.94-8.93 (m, 1Н), 8.35-8.33 (m, 1Н), 8.26 (brs, 2Н), 7.86 (s, 1Н), 7.62-7.59 (m, 1Н), 7.40-7.36 (m, 3Н), 7.26-7.24 (m, 3Н).
Пример 8
8-(2-Метил-6-(трифторметил)пиридин-4-ил)-7-(5-метилфуран-2-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 8
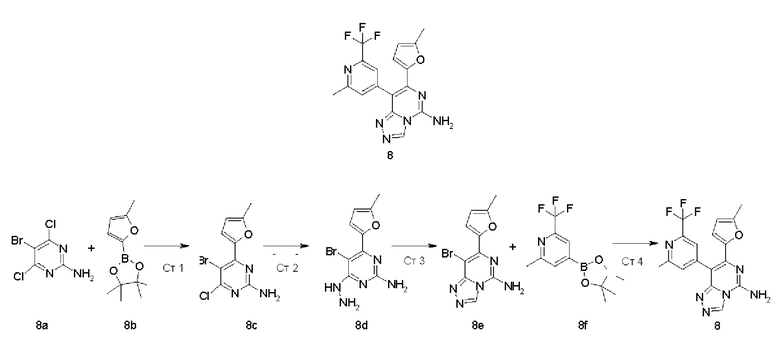
Стадия 1
5-Бром-4-хлор-6-(5-метилфуран-2-ил)пиримидин-2-амин 8c
5-Бром-4,6-дихлорпиримидин-2-амин 8a (5 г, 20,585 ммоль, полученный согласно способу, раскрытому в заявке на патент «US 20100331294»), 4,4,5,5-тетраметил-2-(5-метилфуран-2-ил)-1,3,2-диоксаборолан 8b (4,283 г, 20,585 ммоль, полученный в соответствии с известным способом, раскрытым в «Organometallics, 2015, 34(7), 1307-1320»), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (1,506 г, 2,058 ммоль) и карбонат калия (8,535 г, 61,756 ммоль) последовательно растворяли в 150 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 4:1) в атмосфере аргона и перемешивали в течение 2 часов. Реакцию останавливали, и в реакционный раствор добавляли 200 мл воды и экстрагировали этилацетатом (200 мл × 3). В органическую фазу добавляли диоксид кремния 100-200 меш и концентрировали при пониженном давлении. Остаток очищали посредством системы для флеш-хроматографии CombiFlash с системой элюентов В с получением указанного в заголовке соединения 8с (2,0 г, выход: 33,7%).
МС m/z (ИЭР): 288,2 [М+1]
Стадия 2
5-Бром-4-гидразинил-6-(5-метилфуран-2-ил)пиримидин-2-амин 8d
Соединение 8с (1,88 г, 6,516 ммоль) и 15 мл 85% гидрата гидразина последовательно растворяли в 120 мл этанола. Реакционный раствор перемешивали в течение 17 часов. Реакцию останавливали, и реакционный раствор фильтровали. Осадок на фильтре сушили с получением неочищенного, указанного в заголовке соединения 8d (1,5 г), которое непосредственно использовали на следующей стадии без очистки.
МС m/z (ИЭР): 284,3 [М+1]
Стадия 3
8-Бром-7-(5-метилфуран-2-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 8е
Неочищенный продукт 8d (1,5 г, 5,279 ммоль) добавляли к 20 мл (три)этилортоформиата. Реакционный раствор перемешивали при 140°С в течение 15 минут. Реакцию останавливали, и реакционный раствор охлаждали до комнатной температуры и фильтровали. Осадок на фильтре промывали н-гексаном (20 мл × 2) и сушили с получением неочищенного указанного в заголовке соединения 8е (1,13 г, выход: 72,8%).
МС m/z (ИЭР): 294,3 [М+1]
Стадия 4
8-(2-Метил-6-(трифторметил)пиридин-4-ил)-7-(5-метилфуран-2-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 8
Неочищенный продукт 8е (140 мг, 0,476 ммоль), 2-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-6-(трифторметил)пиридин 8f (205 мг, 0,714 ммоль, полученный согласно известному способу, раскрытому в «Journal of Medicinal Chemistry, 2012, 55(5), 1898-1903»), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (35 мг, 0,048 ммоль) и карбонат калия (197 мг, 1,428 ммоль) последовательно растворяли в 6 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 5:1) в атмосфере аргона. Реакционный раствор нагревали до 90°С и перемешивали в течение 2,5 часов. Реакцию останавливали, и в реакционный раствор добавляли 50 мл воды и экстрагировали этилацетатом (50 мл × 3). Органическую фазу концентрировали при пониженном давлении, и остаток очищали посредством высокоэффективной жидкостной хроматографии с получением указанного в заголовке соединения 8 (80 мг, выход: 44,9%).
МС m/z (ИЭР): 375,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.30 (s, 1Н), 8.26 (brs, 2Н), 7.68 (s, 1Н), 7.64 (s, 1Н), 6.70-6.69 (m, 1 Н), 6.20-6.19 (m, 1 Н), 2.60 (s, 3Н), 2.03 (s, 3Н).
Пример 9
7-Фенил-8-(7-(трифторметил)-1Н-индазол-5-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 9
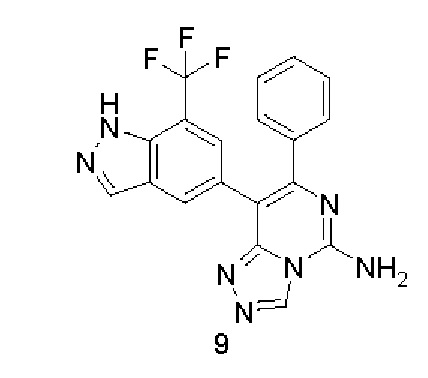
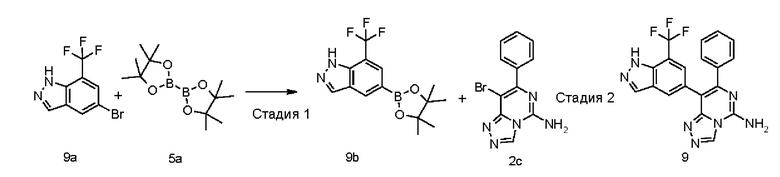
Стадия 1
5-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)-7-(трифторметил)-1Н-индазол 9b
5-Бром-7-(трифторметил)-1Н-индазол 9а (0,5 г, 1,88 ммоль, полученный в соответствии со способом, раскрытым в заявке на патент «WO 2012056372»), соединение 5а (575 мг, 2,26 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (275 мг, 0,38 ммоль) и ацетат калия (554 мг, 5,66 ммоль) последовательно растворяли в 10 мл диметилового эфира гликоля в атмосфере аргона. Реакционный раствор нагревали до 80°С и перемешивали в течение 2 часов. Реакцию останавливали, и реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали посредством системы для флеш-хроматографии CombiFlash с системой элюентов С с получением указанного в заголовке соединения 9b (270 мг, выход: 45,9%).
МС m/z (ИЭР): 313,2 [М+1]
Стадия 2
7-Фенил-8-(7-(трифторметил)-1Н-индазол-5-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 9
Соединение 2с (117 мг, 0,348 ммоль), 9b (35 мг, 0,11 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (25 мг, 0,035 ммоль) и карбонат калия (192 мг, 1,387 ммоль) последовательно растворяли в 5 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 4:1) в атмосфере аргона. Реакционный раствор нагревали до 90°С и перемешивали в течение 2 часов. Реакцию останавливали, и к реакционному раствору добавляли 10 мл воды и экстрагировали смешанным раствором дихлорметана и метанола (V/V составляет 8:1) (20 мл × 3). Органическую фазу концентрировали при пониженном давлении, и остаток очищали посредством высокоэффективной жидкостной хроматографии с получением указанного в заголовке соединения 6 (10 мг, выход: 22%).
МС m/z (ИЭР): 396,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 13.66 (s, 1Н), 9.36 (s, 1Н), 8.25-8.09 (m, 4Н), 7.53 (s, 1Н), 7.33-7.31 (m, 2Н), 7.25-7.23 (m, 3Н).
Пример 10
8-(1Н-индазол-5-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 10

Соединение 2с (140 мг, 0,482 ммоль), 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индазол 10а (141 мг, 0,579 ммоль, полученный в соответствии с известным способом, раскрытым в «Journal of Medicinal Chemistry, 2014, 57(9), 3856-3873»), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (35 мг, 0,048 ммоль) и карбонат калия (200 мг, 1,448 ммоль) последовательно растворяли в 12 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 5:1) в атмосфере аргона. Реакционный раствор нагревали до 90°С и перемешивали в течение 3 часов. Реакцию останавливали, и в реакционный раствор добавляли 50 мл воды и экстрагировали смешанным раствором дихлорметана и воды (V/V составляет 8:1) (30 мл × 3). Органическую фазу концентрировали при пониженном давлении, и остаток очищали посредством высокоэффективной жидкостной хроматографии с получением указанного в заголовке соединения 10 (8,6 мг, выход: 5,4%).
МС m/z (ИЭР): 328,1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 13.05 (brs, 1Н), 9.33 (s, 1Н), 8.05-8.01 (m, 3Н), 7.75 (s, 1Н), 7.53-7.41 (m, 1Н), 7.35-7.33 (m, 2Н), 7.20-7.07 (m, 4Н).
Пример 11
8-(2,6-Диметилпиридин-4-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 11

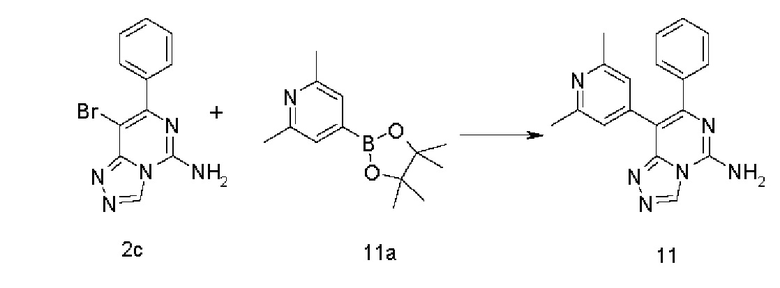
Соединение 2с (100 мг, 0,345 ммоль), 2,6-диметил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин 11а (91 мг, 0,414 ммоль, полученный в соответствии с известным способом, раскрытым в «Organic Letters, 2009, 11(16), 3586-3589»), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (25 мг, 0,034 ммоль) и карбонат калия (143 мг, 1,034 ммоль) последовательно растворяли в 6 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 5:1) в атмосфере аргона. Реакционный раствор нагревали до 90°С и перемешивали в течение 3 часов. Реакцию останавливали, и в реакционный раствор добавляли 50 мл воды и экстрагировали этилацетатом (30 мл × 3). Органическую фазу концентрировали при пониженном давлении, и остаток очищали посредством высокоэффективной жидкостной хроматографии с получением указанного в заголовке соединения 11 (27 мг, выход: 24,8%).
МС m/z (ИЭР): 317,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.35 (s, 1Н), 8.25 (brs, 2Н), 7.32-7.30 (m, 5Н), 6.97 (s, 2Н), 2.32 (s, 6Н).
Пример 12
8-(2-Метилпиридин-4-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 12
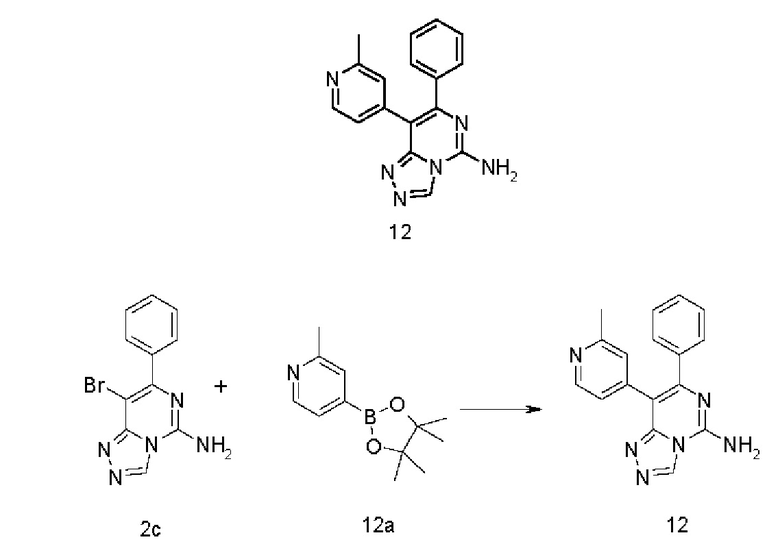
Соединение 2с (100 мг, 0,345 ммоль), 2-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин 12а (91 мг, 0,414 ммоль, полученный в соответствии с известным способом, раскрытым в «Journal of the American Chemical Society, 2014, 136(11), 4133-4136»), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (25 мг, 0,034 ммоль) и карбонат калия (143 мг, 1,034 ммоль) последовательно растворяли в 6 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 5:1) в атмосфере аргона. Реакционный раствор нагревали до 90°С и перемешивали в течение 3 часов. Реакцию останавливали, и в реакционный раствор добавляли 50 мл воды и экстрагировали этилацетатом (30 мл × 3). Органическую фазу концентрировали при пониженном давлении, и остаток очищали посредством высокоэффективной жидкостной хроматографии с получением указанного в заголовке соединения 12 (6,8 мг, выход: 6,5%).
МС m/z (ИЭР): 303,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.35 (s, 1Н), 8.29-8.26 (m, 3Н), 7.32-7.25 (m, 6Н), 6.99 (s, 1H),2.39(s,3H).
Пример 13
5-Амино-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-8-карбонитрил 13

Соединение 2с (1,0 г, 3,45 ммоль), цианид цинка (484 мг, 4,13 ммоль), цинк (22 мг, 0,34 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (124 мг, 0,17 ммоль) и трис(дибензилиденацетон)дипалладий (156 мг, 0,17 ммоль) растворяли в 30 мл N,N-диметилформамида в атмосфере аргона и перемешивали при 60°С в течение 12 часов. После завершения реакции реакционный раствор фильтровали через целит, и осадок на фильтре промывали метанолом. К фильтрату добавляли 30 мл воды, и экстрагировали смешанным раствором дихлорметана и метанола (V/V составляет 8:1) (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали посредством колоночной хроматографии на силикагеле с системой элюентов А с получением указанного в заголовке соединения 13 (650 мг, выход: 80,0%).
МС m/z (ИЭР): 237,0 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.24 (brs, 1Н), 8.83 (brs, 1Н), 8.64 (s, 1Н), 7.92-7.89 (m, 2Н), 7.59-7.57 (m, 3Н).
Пример 14
7-(4-Хлорфенил)-8-(2-метил-6-(трифторметил)пиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 14

Стадия 1
5-Бром-4-хлор-6-(4-хлорфенил)пиримидин-2-амин 14b
4-Хлор-6-(4-хлорфенил)пиримидин-2-амин 14а (11 г, 38,03 ммоль, полученный согласно способу, раскрытому в заявке на патент «DE 102006008880 A1») и N-бромсукцинимид (7,11 г, 39,93 ммоль) растворяли в 300 мл N,N-диметилформамида. После завершения добавления реакционный раствор перемешивали в течение 2 часов. В реакционный раствор добавляли 1 л воды и экстрагировали этилацетатом (300 мл × 4). Органические фазы объединяли, последовательно промывали водой (100 мл × 3) и насыщенным раствором хлорида натрия (200 мл × 2), сушили над безводным сульфатом натрия и фильтровали со сбором фильтрата. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 14b (12 г), которые непосредственно использовали на следующей стадии без очистки.
МС m/z (ИЭР): 318,2 [М+1]
Стадия 2
5-Бром-4-(4-хлорфенил)-6-гидразинилпиримидин-2-амин 14с
Неочищенное соединение 14b (12 г, 37,62 ммоль) и 40 мл 85% гидрата гидразина последовательно растворяли в 400 мл этанола. Реакционный раствор перемешивали в течение 17 часов. Реакционный раствор фильтровали, и осадок на фильтре последовательно промывали этанолом (50 мл) и н-гексаном (100 мл × 2). Осадок на фильтре собирали и сушили с получением неочищенного указанного в заголовке соединения 14с (10,28 г), которое использовали непосредственно на следующей стадии без очистки.
МС m/z (ИЭР): 314,3 [М+1]
Стадия 3
8-Бром-7-(4-хлорфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 14d
Неочищенное соединение 14с (2 г, 6,36 ммоль) и 1,16 мл (три)этилортоформиата (1,04 г, 6,99 ммоль) добавляли к 50 мл этанола. Реакционный раствор перемешивали с обратным холодильником в течение 4 часов. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. К остатку добавляли 51 мл смешанного раствора этанола и н-гексана (V/V составляет 1:50), перемешивали и фильтровали. Остаток на фильтре собирали с получением указанного в заголовке соединения 14d (2 г, выход: 96,9%).
МС m/z (ИЭР): 324,3 [М+1]
Стадия 4
7-(4-Хлорфенил)-8-(2-метил-6-(трифторметил)пиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 14
Соединение 14d (250 мг, 770,27 мкмоль), соединение 1с (265,36 мг, 924,32 мкмоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (56,36 мг, 77,03 мкмоль) и карбонат калия (319,36 мг, 2,31 ммоль) последовательно добавляли к 10 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 4:1) в атмосфере аргона. Реакционный раствор нагревали до 90°С и перемешивали в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали посредством системы для флеш-хроматографии CombiFlash с системой элюентов В. Полученный в результате неочищенный продукт очищали посредством высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, система элюирования: бикарбонат аммония, вода, ацетонитрил) с получением указанного в заголовке соединения 14 (64,4 мг, выход: 20,7%).
МС m/z (ИЭР): 405,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.39 (s, 1Н), 8.46 (brs, 2Н), 7.58 (s, 1Н), 7.49 (s, 1Н), 7.43-7.35 (m, 4Н), 2.50 (s, 3Н).
Пример 15
8-(2-Хлор-6-метилпиридин-4-ил)-7-(4-фторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 15

Стадия 1
4-Хлор-6-(4-фторфенил)пиримидин-2-амин 15с
4,6-Дихлорпиримидин-2-амин 15а (10 г, 60,98 ммоль, Adamas Reagent Ltd.), (4-фторфенил)бороновую кислоту 15b (8,53 г, 60,98 ммоль, Accela ChemBio (Shanghai) Inc.), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (4,46 г, 6,10 ммоль) и карбонат калия (16,83 г, 121,96 ммоль) последовательно добавляли к 250 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 4:1) в атмосфере аргона. Реакционный раствор нагревали до 60°С и перемешивали в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры, в него добавляли 300 мл воды и экстрагировали этилацетатом (150 мл × 3). Органические фазы объединяли и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле с системой элюентов В с получением указанного в заголовке соединения 15с (11,0 г, выход: 76,0%).
МС m/z (ИЭР): 224,3 [М+1]
Стадия 2
5-Бром-4-хлор-6-(4-фторфенил)пиримидин-2-амин 15d
Соединение 15с (11,0 г, 49,19 ммоль) растворяли в 100 мл N,N-диметилформамида. В реакционный раствор партиями добавляли N-бромсукцинимид (8,75 г, 49,19 ммоль) и перемешивали в течение 1 часа. В реакционный раствор добавляли 400 мл воды, и экстрагировали этилацетатом (100 мл × 3). Органические фазы объединяли, последовательно промывали водой (100 мл × 3) и насыщенным раствором хлорида натрия (200 мл), сушили над безводным сульфатом натрия и фильтровали со сбором фильтрата. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 15d (6,0 г), которые использовали непосредственно на следующей стадии без очистки.
МС m/z (ИЭР): 301,9 [М+1]
Стадия 3
5-Бром-4-(4-фторфенил)-6-гидразинилпиримид ин-2-амин 15е
Неочищенное соединение 15d (5,5 г, 18,18 ммоль) и 10 мл 85% гидрата гидразина последовательно растворяли в 20 мл этанола. Реакционный раствор перемешивали с обратным холодильником в течение 1 часа. Реакционный раствор охлаждали до комнатной температуры и перемешивали в течение 0,5 часа. Реакционный раствор фильтровали, и осадок на фильтре последовательно промывали этанолом (3 мл × 2) и диэтиловым эфиром (3 мл × 2). Осадок на фильтре собирали и сушили в вакууме с получением неочищенного указанного в заголовке соединения 15е (4,4 г), которое использовали непосредственно на следующей стадии без очистки.
МС m/z (ИЭР): 298,1 [М+1]
Стадия 4
8-Бром-7-(4-фторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 15f
Неочищенное соединение 15е (500 мг, 1,68 ммоль) и (три)этилортоформиат (497 мг, 3,35 ммоль) последовательно добавляли к 20 мл этанола. Реакционный раствор нагревали с обратным холодильником в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры и фильтровали. Осадок на фильтре один раз промывали 2 мл этанола, и затем собирали с получением неочищенного указанного в заголовке соединения 15f (460 мг), которое непосредственно использовали на следующей стадии без очистки.
МС m/z (ИЭР): 307,9 [М+1]
Стадия 5
8-(2-Хлор-6-метилпиридин-4-ил)-7-(4-фторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 15
Неочищенное соединение 15f (120 мг, 389,47 мкмоль), соединение 4а (138,24 мг, 545,26 мкмоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (28,50 мг, 38,95 мкмоль) и карбонат калия (161,48 мг, 1,17 ммоль) последовательно добавляли к 6,0 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 5:1) в атмосфере аргона. Реакционный раствор нагревали до 90°С и перемешивали в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры, в него добавляли 50 мл воды и экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли и концентрировали при пониженном давлении. Остаток очищали посредством высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, система элюирования: бикарбонат аммония, вода, ацетонитрил) с получением указанного в заголовке соединения 15 (22 мг, выход: 15,9%).
МС m/z (ИЭР): 355,1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.35 (s, 1Н), 8.38 (brs, 2Н), 7.40-7.37 (m, 2Н), 7.20-7.18 (m, 4H),2.37(s, 3Н).
Пример 16
7-(4-Фторфенил)-8-(2-метил-6-(трифторметил)пиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 16

Соединение 15f (100 мг, 324,56 мкмоль), соединение 1с (111,81 мг, 389,47 мкмоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (23,73 мг, 32,46 мкмоль) и карбонат калия (89,71 мг, 649,12 мкмоль) последовательно добавляли к 6,0 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 5:1) в атмосфере аргона. Реакционный раствор нагревали до 90°С и перемешивали в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры, добавляли в него 20 мл воды и экстрагировали этилацетатом (10 мл × 3). Органические фазы объединяли и концентрировали при пониженном давлении. Остаток очищали посредством высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, система элюирования: бикарбонат аммония, вода, ацетонитрил) с получением указанного в заголовке соединения 16 (34 мг, выход: 26,9%).
МС m/z (ИЭР): 389,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.35 (s, 1Н), 8.40 (brs, 2Н), 7.56 (s, 1Н), 7.43 (s, 1Н), 7.34-7.37 (m, 2Н), 7.13-7.17 (m, 2Н), 2.49 (s, 3Н).
Пример 17
8-(2-Метил-6-(трифторметил)пиридин-4-ил)-7-фенилимидазо[1,2-с]пиримидин-5-амин17
Стадия 1
5-Бром-2-(метилтио)-6-фенил пиримидин-4-амин 17b
2-(Метилтио)-6-фенилпиримидин-4-амин 17а (1,2 г, 5,5 ммоль, полученный в соответствии с известным способом, раскрытым в «Chemical & Pharmaceutical Bulletin, 1981, 29(4), 948-54») и N-бромсукцинимид (1,1 г, 6,1 ммоль) последовательно добавляли к 20 мл N,N-диметилформамида. Реакционный раствор перемешивали в течение 1 часа. К реакционному раствору добавляли воду и экстрагировали этилацетатом три раза. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 17b (550 мг), которое использовали непосредственно на следующей стадии без очистки.
МС m/z (ИЭР): 296,0 [М+1]
Стадия 2
8-Бром-5-(метилтио)-7-фенилимидазо[1,2-с]пиримидин 17с
Неочищенное соединение 17b (250 мг, 0,84 ммоль) растворяли в 10 мл 1,4-диоксана. К реакционному раствору по капли добавляли хлорацетальдегид (249 мг, 1,27 ммоль) и перемешивали при 90°С в течение 36 часов. Реакционный раствор охлаждали до комнатной температуры, добавляли в него насыщенный раствор бикарбоната натрия и три раза экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали посредством тонкослойной хроматографии с системой проявляющих растворителей А с получением указанного в заголовке соединения 17с (195 мг, выход: 72%).
МС m/z (ИЭР): 320,1 [М+1]
Стадия 3
8-Бром-5-(метилсульфонил)-7-фенилимидазо[1,2-с]пиримидин 17d
Соединение 17с (195 мг, 0,61 ммоль) растворяли в 10 мл дихлорметана. В реакционный раствор партиями добавляли м-хлорпероксибензойную кислоту (316 мг, 1,83 ммоль) и перемешивали в течение 3 часов. В реакционный раствор добавляли насыщенный раствор бикарбоната натрия и три раза экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 17d (210 мг), которое непосредственно использовали на следующей стадии без очистки.
МС m/z (ИЭР): 352,1 [М+1]
Стадия 4
8-Бром-7-фенилимидазо[1,2-с]пиримидин-5-амин 17е
Неочищенное соединение 17d (210 мг, 0,59 ммоль) растворяли в 10 мл 1,4-диоксана. В реакционный раствор по капли добавляли 1 мл 30% аммония и перемешивали в течение 1 часа. В реакционный раствор добавляли воду и три раза экстрагировали смешанным раствором дихлорметана и метанола (V/V составляет 10:1). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали посредством тонкослойной хроматографии с системой проявляющих растворителей А с получением указанного в заголовке соединения 17е (172 мг, выход: 46%).
МС m/z (ИЭР): 289,1 [М+1]
Стадия 5
8-(2-Метил-6-(трифторметил)пиридин-4-ил)-7-фенилимидазо[1,2-с]пиримидин-5-амин17
Соединение 17d (80 мг, 0,28 ммоль), соединение 1с (95 мг, 0,332 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпаладий (20 мг, 28 мкмоль) и карбонат калия (87 мг, 0,56 ммоль) последовательно добавляли к 10 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 4:1) в атмосфере аргона. Реакционный раствор нагревали до 90°С и перемешивали в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры, добавляли к нему воду и три раза экстрагировали смешанным раствором дихлорметана и метанола (V/V составляет 8:1). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали посредством тонкослойной хроматографии с системой проявляющих растворителей Ас получением указанного в заголовке соединения 17 (15 мг, выход: 14%).
МС m/z (ИЭР): 370,1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 8.02 (s, 1Н), 7.99 (brs, 2Н), 7.57 (s, 1Н), 7.55 (s, 1Н), 7.43 (s, 1 Н), 7.30 (brs, 5Н), 2.45 (s, 3Н).
Пример 18
7-(2,4-Дифторфенил)-8-(2-метил-6-(трифторметил)пиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 18

Стадия 1
4-Хлор-6-(2,4-дифторфенил)пиримидин-2-амин 18b
Соединение 15а (11 г, 63,72 ммоль), (2,4-дифторфенил)бороновую кислоту 18а (10,06 г, 63,72 ммоль, Shanghai Bepharm Ltd.), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (4,66 г, 6,37 ммоль) и карбонат калия (26,42 г, 191,17 ммоль) последовательно добавляли к 500 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 4:1) в атмосфере аргона. Реакционный раствор нагревали до 90°С и перемешивали в течение 2 часов. Реакционный раствор фильтровали, и фильтрат делили на две фазы. Водную фазу экстрагировали этилацетатом (200 мл × 2). Органические фазы объединяли и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле с системой элюентов В с получением указанного в заголовке соединения 18b (14,04 г, выход: 91,2%).
МС m/z (ИЭР): 242,3 [М+1]
Стадия 2
5-Бром-4-хлор-6-(2,4-дифторфенил)пиримидин-2-амин 18с
Соединение 18b (14,04 г, 58,11 ммоль) растворяли в 300 мл N,N-диметилформамида. В реакционный раствор добавляли N-бромсукцинимид (11,38 г, 63,92 ммоль) и перемешивали в течение 1 часа. Реакционный раствор наливали в 1 л воды, перемешивали в течение 30 минут и фильтровали. Осадок на фильтре собирали и сушили в вакууме с получением неочищенного указанного в заголовке соединения 18с (16 г), которое использовали непосредственно на следующей стадии без очистки.
МС m/z (ИЭР): 320,0 [М+1]
Стадия 3
5-Бром-4-(2,4-дифторфенил)-6-гидразинилпиримидин-2-амин 18d
Неочищенное соединение 18с (16 г, 49,92 ммоль) растворяли в 250 мл этанола. В реакционный раствор добавляли 50 мл 85% гидрата гидразина и перемешивали в течение 17 часов. Реакционный раствор фильтровали. Осадок на фильтре последовательно промывали этанолом (20 мл × 2) и н-гексаном (20 мл × 2), и сушили с получением указанного в заголовке соединения 18d (12 г, выход: 76,1%).
МС m/z (ИЭР): 316,0 [М+1]
Стадия 4
8-Бром-7-(2,4-дифторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 18е
Соединение 18d (4 г, 12,65 ммоль) и (три)этилортоформиат (2,25 г, 15,18 ммоль) растворяли в 50 мл этанола. Реакционный раствор перемешивали с обратным холодильником в течение 2 часов. Реакцию останавливали, и реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный в результате осадок превращали в мягкую массу в 5 мл этанола в течение 0,5 часа и фильтровали. Осадок на фильтре промывали безводным диэтиловым эфиром (10 мл × 2) и сушили с получением указанного в заголовке соединения 18е (3,85 г, выход: 93,4%).
МС m/z (ИЭР): 326,2 [М+1]
Стадия 5
7-(2,4-Дифторфенил)-8-(2-метил-6-(трифторметил)пиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 18
Соединение 18е (110 мг, 337,32 мкмоль), соединение 1с (145,26 мг, 505,98 мкмоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (24,7 мг, 33,73 мкмоль) и карбонат калия (93,1 мг, 674,64 мкмоль) последовательно добавляли к 12 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 5:1) в атмосфере аргона. Реакционный раствор нагревали до 90°С и перемешивали в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры, в него добавляли воду и экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали посредством высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, система элюирования: бикарбонат аммония, вода, ацетонитрил) с получением указанного в заголовке соединения 18 (31 мг, выход: 22,6%).
МС m/z (ИЭР): 407,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.41 (s, 1Н), 8.50 (brs, 2Н), 7.55 (brs, 2Н), 7.44 (s, 1Н), 7.20-7.18 (m, 2Н), 2.49 (s, 3Н).
Пример 19
8-(2,6-Диметилпиридин-4-ил)-7-(2-фторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 19

Стадия 1
5-Бром-4-хлор-6-(2,4-дифторфенил)пиримидин-2-амин 19b
4-Хлор-6-(2-фторфенил)пиримидин-2-амин 19а (1,5 г, 6,71 ммоль, полученный в соответствии со способом, раскрытым в заявке на патент «WO 2014162039 А1») растворяли в 20 мл N,N-диметилформамида. В реакционный раствор добавляли N-бромсукцинимид (1,31 г, 7,38 ммоль) и перемешивали в течение 1 часа. В реакционный раствор добавляли 120 мл воды, перемешивали и фильтровали. Осадок на фильтре собирали и сушили в вакууме с получением неочищенного указанного в заголовке соединения 19b (1,9 г), которое использовали непосредственно на следующей стадии без очистки.
МС m/z (ИЭР): 301,9 [М+1]
Стадия 2
5-Бром-4-(2-фторфенил)-6-гидразинилпиримидин-2-амин 19с
Неочищенное соединение 19b (1,9 г, 6,28 ммоль) растворяли в 30 мл этанола. В реакционный раствор добавляли 85% гидрат гидразина (125,61 ммоль, 7,18 мл) и перемешивали при 60°С в течение 1 часа. Реакционный раствор фильтровали. Осадок на фильтре промывали этанолом (20 мл × 2) и сушили с получением указанного в заголовке соединения 19с (1,7 г, выход: 90,8%).
МС m/z (ИЭР): 297,8 [М+1]
Стадия 3
8-Бром-7-(2-фторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 19d
Соединение 19с (1,7 г, 5,70 ммоль) добавляли к 10 мл (три)этилортоформиату. Реакционный раствор перемешивали при 130°С в течение 0,5 часа. Реакционный раствор охлаждали до комнатной температуры и фильтровали. К осадку на фильтре добавляли этилацетат, перемешивали и фильтровали. Осадок на фильтре сушили с получением указанного в заголовке соединения 19d (1,1 г, выход: 62,6%).
МС m/z (ИЭР): 307,9 [М+1]
Стадия 4
8-(2,6-Диметилпиридин-4-ил)-7-(2-фторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 19
Соединение 19d (200 мг, 649,12 мкмоль), соединение 11а (151,32 мг, 649,12 мкмоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (47,45 мг, 64,91 мкмоль) и карбонат калия (179,16 мг, 1,30 ммоль) последовательно добавляли к 12 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 5:1) в атмосфере аргона. Реакционный раствор нагревали до 80°С и перемешивали в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры, добавляли воду и экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали посредством высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, система элюирования: бикарбонат аммония, вода, ацетонитрил) с получением указанного в заголовке соединения 19 (26 мг, выход: 12%).
МС m/z (ИЭР): 335,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.38 (s, 1Н), 8.30 (s, 2Н), 7.41 (brs, 2Н), 7.19-7.22 (m, 1Н), 7.09-7.13 (m, 1 Н), 6.91 (s, 2Н), 2.29 (s, 6Н).
Пример 20
8-(2-(Дифторметил)-6-метилпиридин-4-ил)-7-(2,4-дифторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 20
2-(Дифторметил)-6-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин 20а (247,56 мг, 919,96 мкмоль, полученный в соответствии со способом, раскрытым в заявке на патент «WO 2011095625 A1»), соединение 18е (200 мг, 613,31 мкмоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (44,88 мг, 61,33 мкмоль) и карбонат калия (254,29 мг, 1,84 ммоль) последовательно добавляли к 12 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 5:1) в атмосфере аргона. Реакционный раствор нагревали до 90°С и перемешивали в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали посредством системы для флеш-хроматографии CombiFlash с системой элюентов В. Полученный в результате неочищенный продукт очищали посредством высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, система элюирования: бикарбонат аммония, вода, ацетонитрил) с получением указанного в заголовке соединения 20 (26 мг, выход: 12%).
МС m/z (ИЭР): 389,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.40 (s, 1Н), 8.44 (brs, 2Н), 7.54 (s, 1Н), 7.40-7.18 (m, 4Н), 6.96-6.69 (m, 1Н), 2.44 (s, 3Н).
Пример 21
8-(2,6-Диметилпиридин-4-ил)-2-метил-7-фенилимидазо[1,2-с]пиримидин-5-амин 21
Стадия 1
2-((2-(Метилтио)-6-фенилпиримидин-4-ил)амино)пропан-1-ол 21b
4-Хлор-2-метилтио-6-фенил-пиримидин 21а (1,5 г, 6,34 ммоль, полученный в соответствии с известным способом, раскрытым в «Tetrahedron, 1994, 50(34), 10299-308») растворяли в 40 мл ацетонитрила. В реакционный раствор добавляли 2-аминопропан-1-ол (713,91 мг, 9,50 ммоль) и N,N-диизопропилэтиламин (1,64 г, 12,67 ммоль) и перемешивали при 75°С в течение 72 часов. Реакционный раствор концентрировали при пониженном давлении, и остаток очищали посредством колоночной хроматографии на силикагеле с системой элюентов Ас получением указанного в заголовке соединения 21b (1,5 г, выход: 86%).
МС m/z (ИЭР): 276,2 [М+1]
Стадия 2
2-((5-Бром-2-(метилтио)-6-фенилпиримидин-4-ил)амино)пропан-1-ол 21с
Соединение 21b (1,5 г, 5,45 ммоль) растворяли в 30 мл N,N-диметилформамида. В реакционный раствор партиями добавляли N-бромсукцинимид (969,50 мг, 5,45 ммоль) и перемешивали в течение 1 часа. В реакционный раствор добавляли 150 мл воды и три раза экстрагировали смешанным раствором дихлорметана и метанола (V/V составляет 8:1). Органические фазы объединяли, последовательно промывали водой и насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и осадок очищали посредством колоночной хроматографии на силикагеле с системой элюентов А с получением указанного в заголовке соединения 21с (1,2 г, выход: 62.2%).
МС m/z (ИЭР): 354,1 [М+1]
Стадия 3
8-Бром-2-метил-5-(метилтио)-7-фенил-2,3-дигидроимидазо[1,2-с]пиримидин 21d
Соединение 21с (2,3 г, 6,49 ммоль) растворяли в 40 мл дихлорметана. В реакционный раствор добавляли триэтиламин (983,59 мг, 9,74 ммоль), по каплям добавляли метансульфонилхлорид (892,44 мг, 7,79 ммоль) и перемешивали в течение ночи. В реакционный раствор добавляли насыщенный раствор бикарбоната натрия и три раза экстрагировали дихлорметаном (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали посредством колоночной хроматографии на силикагеле с системой элюентов А с получением указанного в заголовке соединения 21d (1,2 г, выход: 55%).
МС m/z (ИЭР): 336,0 [М+1]
Стадия 4
8-Бром-2-метил-5-(метилтио)-7-фенилимидазо[1,2-с]пиримидин 21е
Соединение 21d (1,2 г, 3,57 ммоль) добавляли к 1,4-д и океану. К реакционному раствору добавляли диоксид марганца (3,14 г, 35,69 ммоль) и перемешивали при 90°С в течение 36 часов. Реакционный раствор охлаждали до комнатной температуры и фильтровали через целит. Осадок на фильтре промывали метанолом, и фильтрат концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле с системой элюентов А с получением указанного в заголовке соединения 21е (750 мг, выход: 62,9%).
МС m/z (ИЭР): 334,1 [М-1]
Стадия 5
8-Бром-2-метил-5-(метилсульфонил)-7-фенилимидазо[1,2-с]пиримидин 21f
Соединение 21е (320 мг, 957,41 мкмоль) растворяли в 5 мл трифторуксусной кислоты. В реакционный раствор по каплям добавляли пероксид водорода (0,5 мл, 957,41 мкмоль) и перемешивали в течение 1 часа. Реакционный раствор нейтрализовали насыщенным раствором карбоната натрия и экстрагировали дихлорметаном (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 21е (350 мг), которое непосредственно использовали на следующей стадии без очистки.
Стадия 6
8-Бром-2-метил-7-фенилимидазо[1,2-с]пиримидин-5-амин 21g
Неочищенное соединение 21f (350 мг, 955,68 мкмоль) растворяли в 10 мл 1,4-диоксана. В реакционный раствор добавляли моногидрат аммония (1,0 мл, 955,68 мкмоль) и перемешивали при 40°С в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении, добавляли в него воду и три раза экстрагировали смешанным раствором дихлорметана и метанола (V/V составляет 8:1).
Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над водным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали посредством тонкослойной хроматографии с системой проявляющих растворителей А с получением указанного в заголовке соединения 21g (215 мг, выход: 74,2%).
Стадия 7
8-(2,6-Диметилпиридин-4-ил)-2-метил-7-финилимидазо[1,2-с]пиримидин-5-амин 21
Соединение 21g (215 мг, 709,20 мкмоль), соединение 11а (247,99 мг, 1,06 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (51,84 мг, 70,92 мкмоль) и бикарбонат натрия (131,06 мг, 1,56 ммоль) последовательно добавляли к 12 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 5:1) в атмосфере аргона. Реакционный раствор нагревали до 80°С и перемешивали в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры, добавляли в него воду и экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлоридом натрия, сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали посредством высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, система элюирования: бикарбонат аммония, вода, ацетонитрил) с получением указанного в заголовке соединения 21 (45 мг, выход: 19,3%).
МС m/z (ИЭР): 330,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 7.68 (s, 1Н), 7.65 (s, 2Н), 7.28 (s, 2Н), 7.24 (s, 3Н), 6.89 (s, 2Н), 2.31 (s, 6Н), 2.27 (s, 3Н).
Пример 22
8-(2-Метил-6-((4-метилпиперазин-1-ил)метил)пиридин-4-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 22

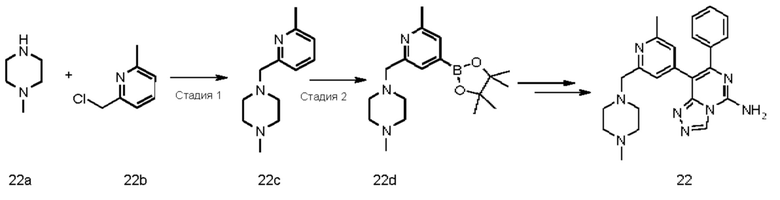
Стадия 1
1-Метил-4-((6-метилпиридин-2-ил)метил)пиперазин 22с
2-(Хлорметил)-6-метилпиридин 22b (1 г, 7,06 ммоль) и карбонат калия (1,46 г, 10,59 ммоль) растворяли в 40 мл ацетонитрила. В реакционный раствор добавляли 1-метилпиперазин 22а (848,57 мг, 8,47 ммоль, 939,72 мкл) и перемешивали при 80°С в течение 17 часов. Реакционный раствор очищали посредством колоночной хроматографии на силикагеле с системой элюентов А с получением указанного в заголовке соединения 22с (1 г, выход: 69%).
МС m/z (ИЭР): 206,4 [М+1]
Стадия 2
1-Метил-4-((6-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)метил)пиперазин 22d
Соединение 22с (1 г, 4,87 ммоль), соединение 5а (146,13 мкмоль) и димер (1,5-циклооктадиен)(метокси)иридия (I) (78,44 мг, 292,26 мкмоль) растворяли в 40 мл н-гексана в атмосфере аргона. Реакционный раствор перемешивали при 80°С в течение 17 часов. Реакционный раствор концентрировали при пониженном давлении и очищали посредством колоночной хроматографии на силикагеле с системой элюентов В с получением указанного в заголовке соединения 22d (217 мг, выход: 13,5%).
В соответствии с путем синтеза согласно Примеру 5 исходное соединение 5b на Стадии 1 заменяли 22d, соответственно, получали указанное в заголовке соединение 22 (27,6 мг).
МС m/z (ИЭР): 415,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.34 (s, 1Н), 8.26 (brs, 2Н), 7.32-7.28 (m, 6Н), 6.85 (s, 1Н), 3.38 (s, 2Н), 2.54 (s, 3Н), 2.43 (s, 3Н), 2.19-2.09 (m, 8Н).
Пример 23
8-(2-Хлор-6-(трифторметил)пиридин-4-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 23
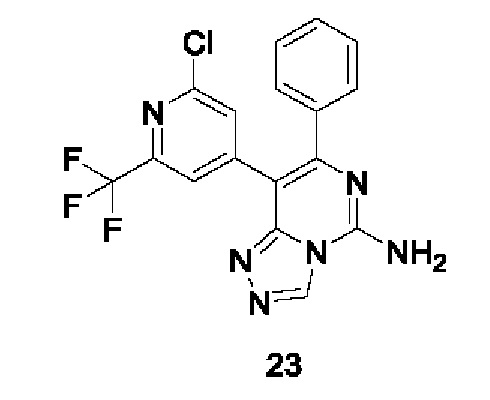
В соответствии с путем синтеза согласно Примеру 4 исходное соединение 4а на стадии 1 заменяли 2-хлор-4-(4,4,5,5-тетраметил-1,3,2-диоксоборолан-2-ил)-6-(трифторметил)пиридином 23а (полученным в соответствии с известным способом, раскрытым в «Research on Chemical Intermediates, 2013, 39(4), 1917-1926»), соответственно, получали указанное в заголовке соединение 23 (11,8 мг).
МС m/z (ИЭР): 391,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.40 (s, 1Н), 8.58 (brs, 2Н), 7.82 (s, 1Н), 7.65 (s, 1Н), 7.40-7.36 (m, 5Н).
Пример 24
8-(2-Хлор-6-метилпиридин-4-ил)-7-фенилимидазо[1,2-с]пиримидин-5-амин 24

В соответствии с путем синтеза согласно Примеру 17 исходное соединение 1с на Стадии 5 заменяли соединением 4а, соответственно, получали указанное в заголовке соединение 24 (32 мг).
МС m/z (ИЭР): 336,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 8.00 (s, 1Н), 7.93 (s, 2Н), 7.55 (s, 1Н), 7.31 (s, 5Н), 7.14 (d, 2Н), 2.33 (s, 3Н).
Пример 25
8-(2-Метокси-6-метилпиридин-4-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 25

В соответствии с путем синтеза согласно Примеру 4 исходное соединение 4а на Стадии 1 заменяли 2-метокси-6-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридином 25а (полученным в соответствии с известным способом, раскрытым в «Chemistry - A European Journal, 2017, 23(24), 5663-5667»), соответственно, получали указанное в заголовке соединение 25 (11,8 мг).
МС m/z (ИЭР): 333,2 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.33 (s, 1Н), 8.24 (brs, 2Н), 7.36-7.29 (m, 5Н), 6.76 (s, 1Н), 6.48 (s, 1 Н), 3.77 (s, 3Н), 2.29 (s, 3Н).
Пример 26
8-(2-Этил-6-метилпиридин-4-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 26

Этил-6-метилпиридин 26а (1 г, 8,25 ммоль, Accela ChemBio (Shanghai) Inc.), соединение 5a (2,31 г, 9,08 ммоль), (1Z,5Z)-циклоокта-1,5-диен; иридий; метилоксоний (164,10 мг, 247,57 мкмоль) и 4-трет-бутил-2-(4-трет-бутил-2-пиридил)пиридин (588 мг, 6,00 ммоль) последовательно добавляли к 40 мл н-гексана в атмосфере аргона. Реакционный раствор нагревали до 80°С и перемешивали в течение 17 часов. Реакционный раствор концентрировали при пониженном давлении, и остаток очищали посредством колоночной хроматографии на силикагеле с системой элюентов В с получением 2-этил-6-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина 26b (809 мг, выход: 39,7%).
МС m/z (ИЭР): 248,2 [М+1]
В соответствии с путем синтеза согласно Примеру 4 исходное соединение 4а на Стадии 1 заменяли 26b, соответственно, получали указанное в заголовке соединение 26 (136,6 мг).
МС m/z (ИЭР): 331,1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.35 (s, 1Н), 8.25 (brs, 2Н), 7.32-7.28 (m, 5Н), 7.12 (s, 1Н), 6.80 (s, 1Н), 2.57-2.55 (m, 2Н), 2.37 (s, 3Н), 1.03-1.00 (m, 3Н).
Пример 27
8-(2-Хлор-6-метилпиридин-4-ил)-7-(2-фторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 27
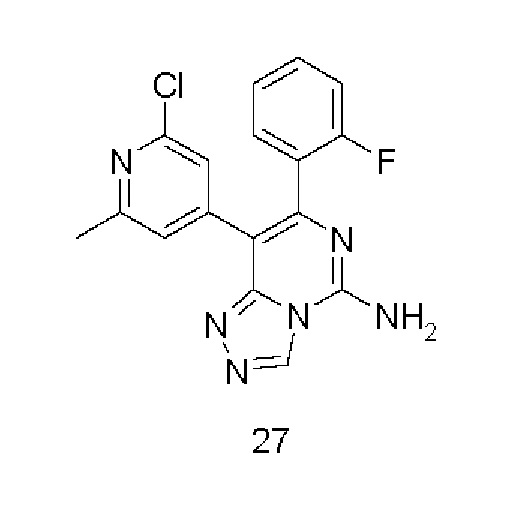
В соответствии с путем синтеза согласно Примеру 19 исходное соединение 11а на Стадии 4 заменяли соединением 4а, соответственно, получали указанное в заголовке соединение 27 (73 мг).
МС m/z (ИЭР): 355,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 8.39 (s, 1Н), 8.44 (s, 2Н), 7.49-7.45 (m, 2Н), 7.28-7.24 (m, 1Н), 7.17-7.14 (m, 2Н), 7.11 (s, 1Н), 2.33 (s, 3Н).
Пример 28
8-(2,6-Диметилпиридин-4-ил)-7-(4-фторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 28
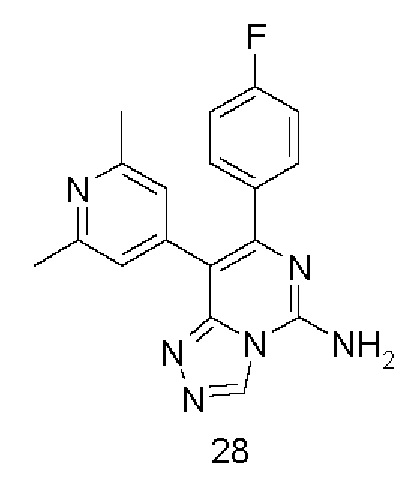
В соответствии с путем синтеза согласно Примеру 15 исходное соединение 4а на стадии 5 заменяли соединением 11а, соответственно, получали указанное в заголовке соединение 28 (38 мг).
МС m/z (ИЭР): 335,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.34 (s, 1Н), 8.24 (brs, 2Н), 7.39-7.35 (m, 2Н), 7.16-7.11 (m, 2Н), 6.95 (s, 2Н), 2.33 (s, 6Н).
Пример 29
7-(3-Фторфенил)-8-(2-метил-6-(трифторметил)пиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 29
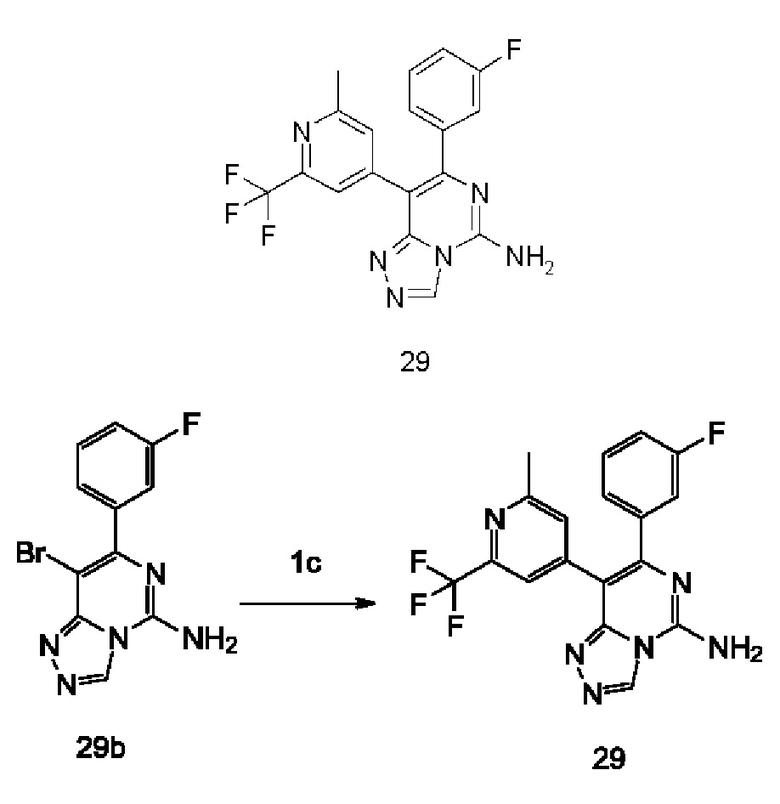
В соответствии с путем синтеза согласно Примеру 15 исходное соединение 15b на Стадии 1 заменяли (3-фторфенил)бороновой кислотой 29а, соответственно, получали соединение 8-бром-7-(3-фторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 29b;
Исходное соединение 4а на Стадии 5 согласно Примеру 15 заменяли соединением 1с, соответственно, получали указанное в заголовке соединение 29 (28 мг).
МС m/z (ИЭР): 389,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.39 (s, 1Н), 8.46 (brs, 2Н), 7.60 (s, 1Н), 7.48 (s, 1Н), 7.36-7.34 (m, 1Н), 7.23-7.19 (m, 2Н), 7.10-7.08 (m, 1Н), 2.49 (s, 3Н).
Пример 30
8-(2-Циклопропил-6-метилпиридин-4-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 30
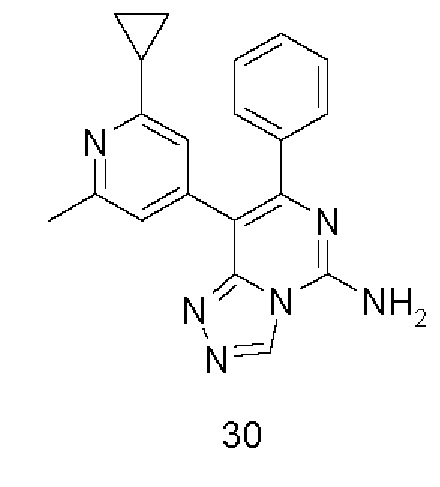
В соответствии с путем синтеза согласно Примеру 11 исходное соединение 11а на стадии 1 заменяли 2-циклопропил-6-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридином 30а (полученным в соответствии со способом, раскрытым в заявке на патент «WO 2011070131 А1»), соответственно, получали указанное в заголовке соединение.
МС m/z (ИЭР): 343,2 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.34 (s, 1Н), 8.23 (brs, 2Н), 7.47-7.31 (m, 5Н), 6.92-6.87 (m, 2Н), 2.28 (s, 3Н), 1.98-1.90 (m, 1Н), 0.84-0.72 (m, 4Н).
Пример 31
8-(2,6-Диметилпиридин-4-ил)-7-фенилимидазо[1,2-с]пиримидин-5-амин 31

В соответствии с путем синтеза согласно Примеру 17 исходное соединение 1 с на стадии 5 заменяли соединением 11а, соответственно, получали указанное в заголовке соединение 31 (40 мг).
МС m/z (ИЭР): 316,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 8.00 (s, 1Н), 7.99 (s, 2Н), 7.51 (d, 1Н), 7.31-7.29 (m, 2Н), 7.26-7.25 (m, 3Н), 6.91 (s, 2Н), 2.30 (s, 6Н).
Пример 32
8-(2,6-Диметилпиридин-4-ил)-7-(4-фторфенил)имидазо[1,2-с]пиримидин-5-амин 32
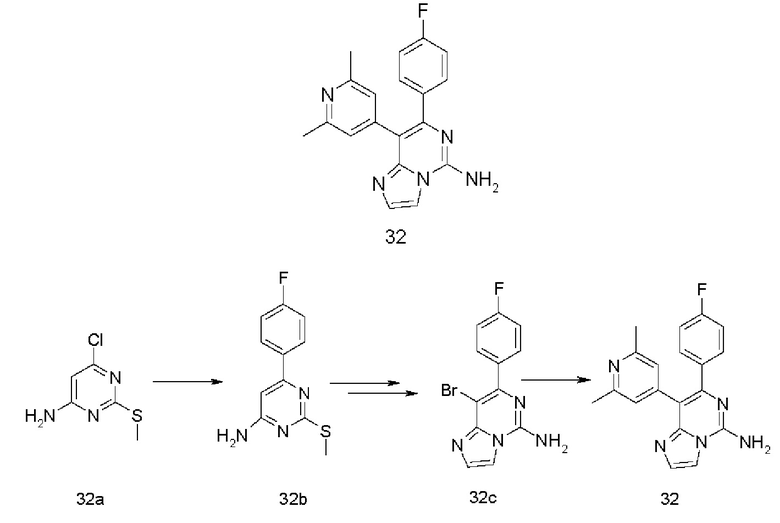
6-Хлор-2-(метилтио)пиримидин-4-амин 32а (2,0 г, 11,39 ммоль, Shanghai Bide Pharmatech Ltd.) и (4-фторфенил)бороновую кислоту (2,39 г, 17,08 ммоль) растворяли в 50 мл толуола в атмосфере аргона. Последовательно добавляли тетра(трифенилфосфин)палладий (657,60 мг, 569,35 мкмоль), карбонат натрия (2,41 г, 22,77 ммоль) и 10 мл воды. Реакционный раствор перемешивали при 90°С в течение 3 часов, затем в него добавляли воду и три раза экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и к остатку добавляли метанол, перемешивали и фильтровали. Осадок на фильтре собирали с получением 6-(4-фторфенил)-2-(метилтио)пиримидин-4-амина 32b (1,3 г, выход: 48,5%).
В соответствии с путем синтеза согласно Примеру 17 исходное соединение 17а на стадии 1 заменяли 32b, соответственно, получали 8-бром-7-(4-фторфенил)имидазо[1,2-с]пиримидин-5-амин 32с.
Исходное соединение 1с на стадии 5 согласно Примеру 17 заменяли соединением 11а, соответственно, получали указанное в заголовке соединение 32 (70 мг).
МС m/z (ИЭР): 334,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 7.98 (d, 1Н), 7.82 (s, 2Н), 7.52 (d, 1Н), 7.35-7.32 (m, 2Н), 7.28-7.08 (m, 2Н), 6.92 (s, 2Н), 2.32 (s, 6Н).
Пример 33
7-(2-Фторфенил)-8-(2-метил-6-(трифторметил)пиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 33

В соответствии с путем синтеза согласно Примеру 19 исходное соединение 11а на стадии 4 заменяли соединением 1с, соответственно, получали указанное в заголовке соединение 33 (25 мг).
МС m/z (ИЭР): 389,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.43 (s, 1Н), 8.48 (brs, 2Н), 7.55 (s, 1Н), 7.50-7.44 (m, 2Н), 7.42 (s, 1Н), 7.28-7.26 (m, 1Н), 7.15-7.12 (m, 1Н), 2.46 (s, 3Н)
Пример 34
8-(2-Хлор-6-метилпиридин-4-ил)-7-(2,4-дифторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 34

В соответствии с путем синтеза согласно Примеру 18 исходное соединение 1с на стадии 5 заменяли соединением 4а, соответственно, получали указанное в заголовке соединение 34 (30,4 мг).
МС m/z (ИЭР): 373,3 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.39 (s, 1Н), 8.46 (brs, 2Н), 7.55-7.54 (m, 1Н), 7.25-7.14 (m, 4H),2.36(s, 3Н).
Пример 35
8-(2-Хлор-6-метилпиридин-4-ил)-7-(п-толил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 35

В соответствии с путем синтеза согласно Примеру 15 исходное соединение 15b на стадии 1 заменяли п-толилбороновой кислотой 35а (Shanghai Dari chemical Ltd.), соответственно, получали 8-бром-7-(п-толил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 35b.
Исходное соединение 15f на стадии 5 Примера 15 заменяли соединением 35b, соответственно, получали указанное в заголовке соединение 35 (21,3 мг).
МС m/z (ИЭР): 351,1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.34 (s, 1Н), 8.33 (brs, 2Н), 7.26-7.13 (m, 6Н), 2.36 (s, 3Н), 2.30 (s, 3Н).
Пример 36
8-(2-Хлор-6-метилпиридин-4-ил)-7-(4-хлорфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 36

В соответствии с путем синтеза согласно Примеру 14 исходное соединение 1с на стадии 4 заменяли соединением 4а, соответственно, получали указанное в заголовке соединение 36 (45,9 мг).
MC m/z (ИЭР): 371,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.36 (s, 1Н), 8.40 (brs, 2Н), 7.43-7.35 (m, 4Н), 7.20-7.18 (m,2Н), 2.38 (s, 3Н).
Пример 37
7-Фенил-8-(пиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 37

В соответствии с путем синтеза согласно Примеру 2 исходное соединение 2d на стадии 4 заменяли 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридином 37а (Shanghai Bide Pharmatech Ltd.), соответственно, получали указанное в заголовке соединение 37 (20 мг).
MC m/z (ИЭР): 289,0 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.35 (s, 1Н), 8.47-8.46 (m, 2Н), 8.29 (brs, 2Н), 7.33-7.29 (m, 7Н).
Пример 38
8-(2-Хлорпиридин-4-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 38
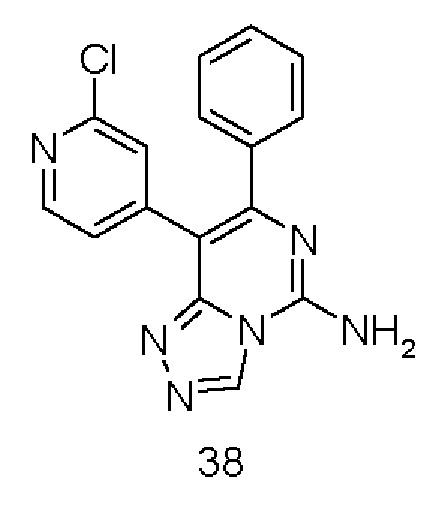
В соответствии с путем синтеза согласно Примеру 2 исходное соединение 2d на стадии 4 заменяли 2-хлор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридином 38а (Adamas Reagent Ltd.), соответственно, получали указанное в заголовке соединение 38 (20 мг).
MC m/z (ИЭР): 323,1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.36 (s, 1Н), 8.40 (brs, 2Н), 8.28-8.27 (m, 1Н), 7.47 (s, 1Н), 7.38-7.31 (m, 5Н), 7.23-7.22 (m, 1Н).
Пример 39
8-(2-Хлор-6-(трифторметил)пиридин-4-ил)-7-(2,4-дифторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 39

В соответствии с путем синтеза согласно Примеру 18 исходное соединение 1с на стадии 5 заменяли 2-хлор-4-(4,4,5,5-тетраметил-1,3,2-диоксоборолан-2-ил)-6-(трифторметил)пиридином (полученным в соответствии с известным способом, раскрытым «Research on Chemical Intermediates, 2013, 39(4), 1917-1926»), соответственно, получали указанное в заголовке соединение 39 (19,8 мг).
МС m/z (ИЭР): 427,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.66 (s, 1Н), 8.80 (brs, 2Н), 7.80 (s, 1Н), 7.70 (s, 1Н), 7.65-7.59 (m, 1Н), 7.29-7.22 (m, 2Н).
Пример 40
7-(2,4-Дифторфенил)-8-(2,6-диметилпиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 40

В соответствии с путем синтеза согласно Примеру 18 исходное соединение 1 с на стадии 5 замещали соединением 11а, соответственно, получали указанное в заголовке соединение 40 (40 мг).
МС m/z (ИЭР): 353,1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.54 (s, 1Н), 8.43 (brs, 2Н), 7.51-7.45 (m, 1Н), 7.21-7.10 (m, 2Н), 6.92 (s, 2Н), 2.31 (s, 6Н).
Пример 41
7-(2,4-Дифторфенил)-8-(2-метокси-6-метилпиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 41
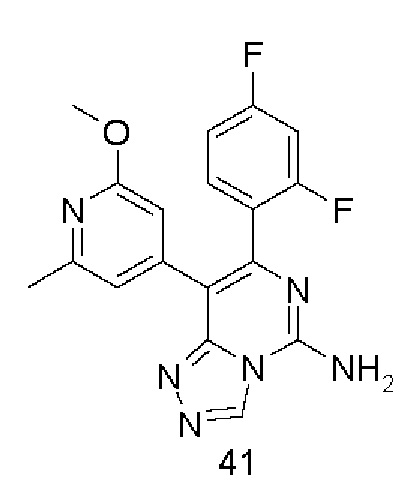
В соответствии с путем синтеза согласно Примеру 18 исходное соединение 1с на стадии 5 заменяли соединением 25а, соответственно, получали указанное в заголовке соединение 41 (10,6 мг).
МС m/z (ИЭР): 369,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.37 (s, 1Н), 8.33 (brs, 2Н), 7.50-7.48 (m, 1Н), 7.22-7.13 (m, 2Н), 6.76 (s, 1Н),6.45 (s, 1Н), 3.76 (s, 3Н), 2.30 (s, 3Н).
Пример 42
3-Метил-8-(2-метил-6-(трифторметил)пиридин-4-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 42
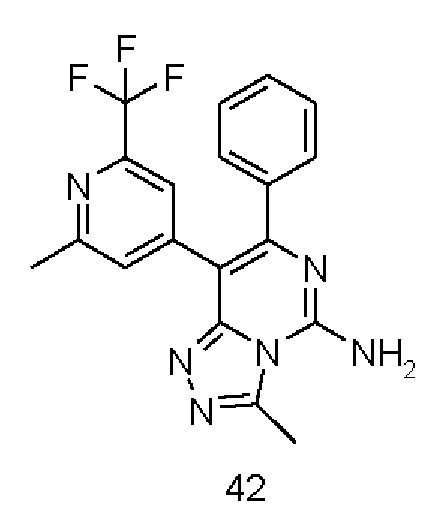
В соответствии с путем синтеза согласно Примеру 1 исходное соединение (три)этилортоформиат на стадии 4 заменяли триэтилортоацетатом, соответственно, получали указанное в заголовке соединение 42 (52 мг).
МС m/z (ИЭР): 385,1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 7.69 (brs, 2Н), 7.55 (s, 1Н), 7.41 (s, 1Н), 7.35-7.31 (m, 5Н), 2.96 (s, 3Н), 2.46 (s, 3Н).
Пример 43
5-(5-Амино-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-8-ил)-1-изопропилпиридин-2(1Н)-он 43
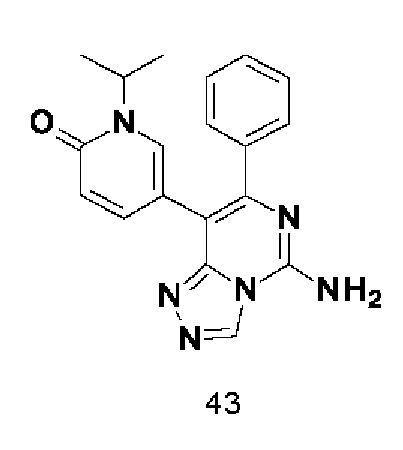
В соответствии с путем синтеза согласно Примеру 4 исходное соединение 4а на стадии 1 заменяли 1-изопропил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1H)-оном 43а (полученным в соответствии со способом, раскрытым в заявке на патент «WO 2011143426 A1»), соответственно, получали указанное в заголовке соединение 43 (31 мг).
МС m/z (ИЭР): 347,1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.33 (s, 1Н), 8.13 (brs, 2Н), 7.47-7.33 (m, 7Н), 6.38-6.35 (m, 1 Н), 4.97-4.90 (m, 1Н), 0.99-0.97 (m, 6Н).
Пример 44
8-(2-Циклопропил-6-метилпиридин-4-ил)-7-(2,4-дифторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 44

В соответствии с путем синтеза согласно Примеру 18 исходное соединение 1с на стадии 5 заменяли соединением 30а, соответственно, получали указанное в заголовке соединение 44 (79,4 мг).
МС m/z (ИЭР): 379,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.38 (s, 1Н), 8.33 (brs, 2Н), 7.52-7.46 (m, 1Н), 7.21-7.11 (m, 2Н), 6.90-6.85 (m, 2Н), 2.28 (s, 3Н), 1.95-1.85 (m, 1Н), 0.85-0.83 (m, 2Н), 0.72-0.70 (m, 2Н).
Пример 45
7-(2,4-Дифторфенил)-8-(2-метилпиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 45

В соответствии с путем синтеза согласно Примеру 18 исходное соединение 1 с на стадии 5 замещали соединением 12а, соответственно, получали указанное в заголовке соединение 45 (79,4 мг).
MC m/z (ИЭР): 339,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.46 (s, 1Н), 8.40 (brs, 2Н), 8.31-8.30 (m, 1Н), 7.53-7.47 (m, 1Н), 7.22-7.11 (m, 3Н), 6.98-6.97 (m, 1Н), 2.39 (s, 3Н).
Пример 46
7-(2-Хлор-4-фторфенил)-8-(2-метил-6-(трифторметил)пиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 46


В соответствии с путем синтеза согласно Примеру 15 исходное соединение 15b на Стадии 1 заменили (2-хлор-4-фторфенил)бороновой кислотой (Accela ChemBio (Shanghai) Inc.), соответственно, получали 8-бром-7-(2-хлор-4-фторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 46а.
Исходное соединение 4а на стадии 5 Примера 15 заменяли соединением 1 с, соответственно, получали указанное в заголовке соединение 46 (32 мг).
МС m/z (ИЭР): 423,3 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.43 (s, 1Н), 8.54 (brs, 2Н), 7.54 (s, 1Н), 7.53-7.49 (m, 2Н), 7.47 (s, 1Н), 7.40-7.28 (m, 1Н), 2.47 (s, 3Н).
Пример 47
8-(2-Хлор-6-(морфолинометил)пиридин-4-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 47
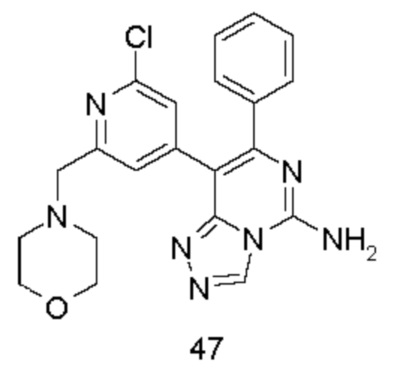
В соответствии с путем синтеза согласно Примеру 22 исходное соединение 22 с заменяли 4-((6-хлорпиридин-2-ил)метил)морфолином (полученным в соответствии со способом, раскрытым в заявке на патент «US 20150361100 А1»), соответственно, получали указанное в заголовке соединение 47 (34,8%).
МС m/z (ИЭР): 422,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.37 (s, 1Н), 7.58 (s, 1Н), 7.48-7.33 (m, 5Н), 7.05 (s, 1Н), 3.35-3.28 (m, 6Н), 2.11 (s, 2Н), 1.90-1.87 (m, 4Н).
Пример 48
8-(2-Метил-6-(морфолинометил)пиридин-4-ил)-7-фенил-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 48

В соответствии с путем синтеза согласно Примеру 43 исходное соединение 43а на стадии 1 заменяли морфолином 48а, соответственно, получали указанное в заголовке соединение 48 (30,8 мг).
МС m/z (ИЭР): 402,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.34 (s, 1Н), 8.23 (brs, 2Н), 7.35-7.28 (m, 6Н), 6.85 (s, 1Н), 3.42-3.35 (m, 6Н), 2.44 (s, 3Н), 2.13-2.05 (m, 4Н).
Пример 49
7-(4-Хлор-2-фторфенил)-8-(2-метил-6-(трифторметил)пиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин
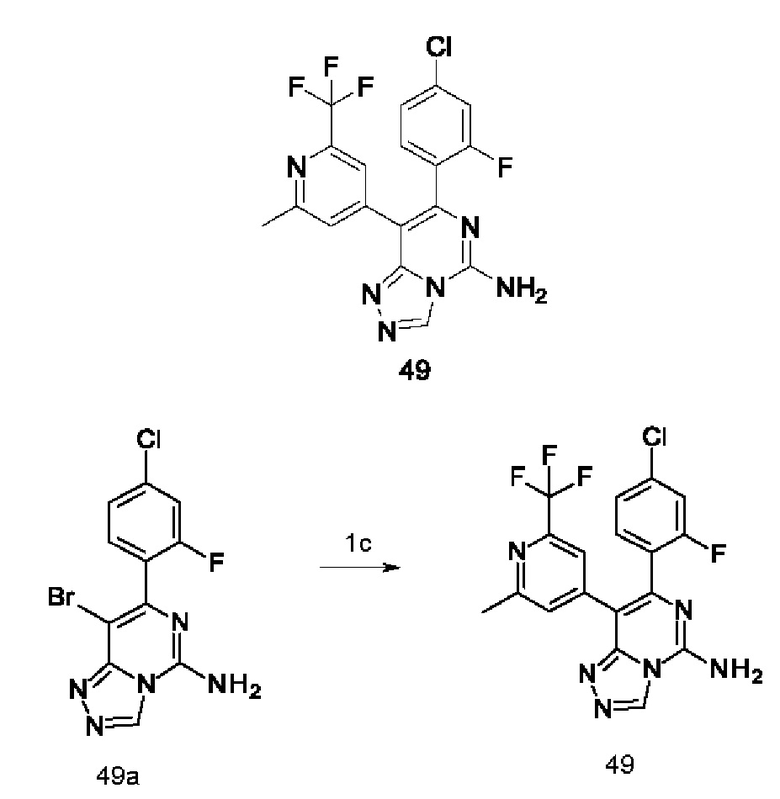
В соответствии с путем синтеза согласно Примеру 15 исходное соединение 15b на стадии 1 заменяли (4-хлор-2-фторфенил)бороновой кислотой (Accela ChemBio (Shanghai) Inc.), соответственно, получали 8-бром-7-(4-хлор-2-фторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 49а.
Исходное соединение 4а на стадии 5 Примера 15 заменяли соединением 1с, соответственно, получали указанное в заголовке соединение 49 (20 мг).
МС m/z (ИЭР): 422,7 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.43 (s, 1Н), 8.54 (brs, 2Н), 7.56-7.52 (m, 2Н), 7.45 (s, 1Н), 7.42-7.38 (m, 2Н), 2.48 (s, 3Н).
Пример 50
7-(4-Хлор-2-фторфенил)-8-(2-хлор-6-метилпиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 50

В соответствии с путем синтеза согласно Примеру 15 исходное соединение 15b на стадии 1 заменяли (4-хлор-2-фторфенил)бороновой кислотой (Accela ChemBio (Shanghai) Inc.), соответственно, получали указанное в заголовке соединение 50 (30 мг).
MC m/z (ИЭР): 389,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.41 (s, 1Н), 8.47 (brs, 2Н), 7.54-7.50 (m, 1Н), 7.44-7.39 (m, 2Н), 7.18 (s, 1H),7.14(s, 1Н), 2.36 (s, 3Н).
Пример 51
8-(2-(Дифторметил)-6-метилпиридин-4-ил)-7-(4-фторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 51

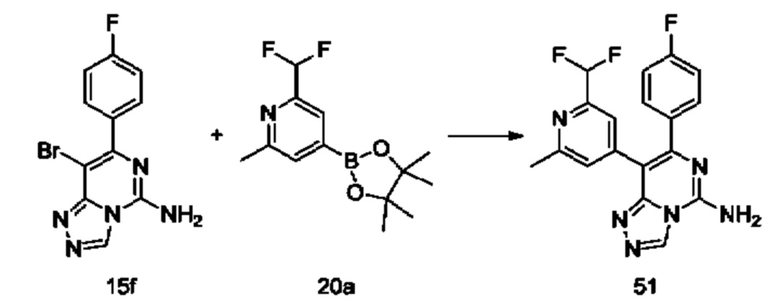
Соединение 15f (1,00 г, 3,25 ммоль), соединение 20а (1,05 г, 3,90 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (238 мг, 324,6 мкмоль) и бикарбонат натрия (682 мг, 8,11 ммоль) последовательно добавляли к 55 мл смешанного раствора 1,4-диоксана и воды (V/V составляет 9:2) в атмосфере аргона. Реакционный раствор нагревали до 95°С и перемешивали в течение 17 часов. Реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали посредством высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, система элюирования: бикарбонат аммония, вода, ацетонитрил) с получением указанного в заголовке соединения 51 (50 мг).
МС m/z (ИЭР): 371,2 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.37 (s, 1Н), 8.38 (brs, 2Н), 7.44 (s, 1Н), 7.39-7.36 (m, 2Н), 7.32 (s, 1Н), 7.19-7.14 (m, 2Н), 6.98-6.70 (m, 1Н), 2.46 (s, 3Н).
Пример 52
7-(4-Хлорфенил)-8-(2-(дифторметил)-6-метилпиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 52
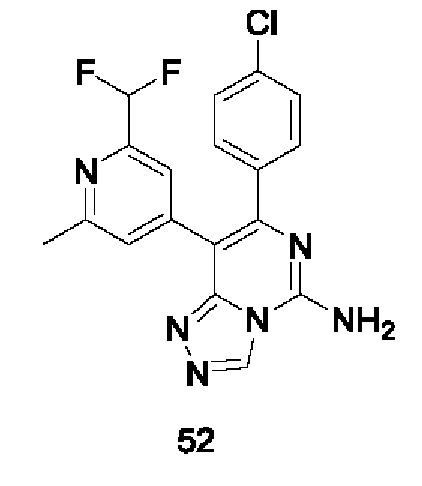
В соответствии с путем синтеза согласно Примеру 14 исходное соединение 1с заменяли соединением 20а, соответственно, получали указанное в заголовке соединение 52 (32 мг).
МС m/z (ИЭР): 387,0 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.37 (s, 1Н), 8.39 (brs, 2Н), 7.40-7.43 (m, 2Н), 7.33-7.38 (m, 4Н), 6.84 (t, 1Н), 2.46 (s, 3Н).
Пример 53
7-(4-Хлорфенил)-8-(2-(фторметил)-6-метилпиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 53
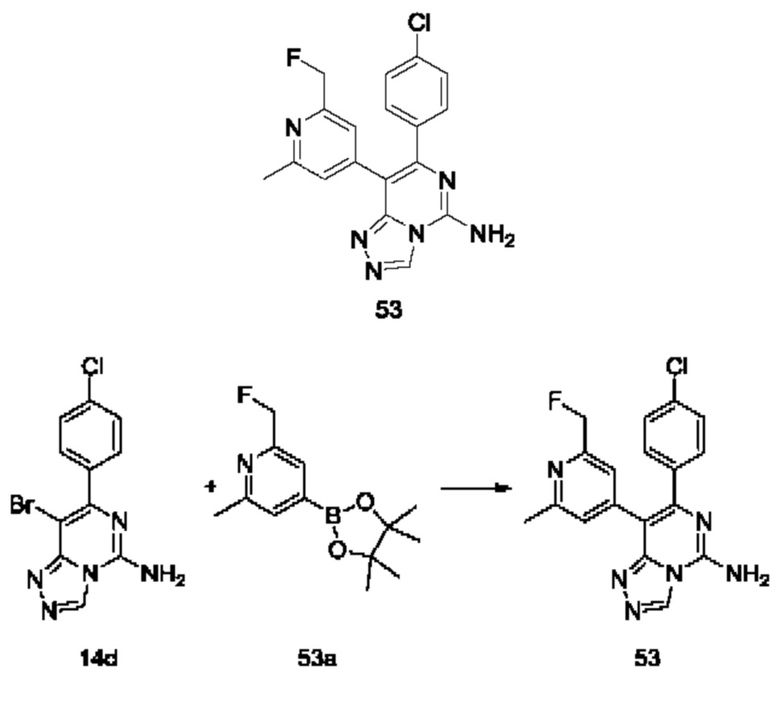
В соответствии с путем синтеза согласно Примеру 14 исходное соединение 1 с заменяли соединением 2-(фторметил)-6-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин 53а (полученным в соответствии со способом, раскрытым в заявке на патент «WO 201195625 A1»), соответственно, получали указанное в заголовке соединение 53 (4 мг).
МС m/z (ИЭР): 369,1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.36 (s, 1Н), 8.36 (brs, 2Н), 7.42-7.32 (m, 3Н), 7.19 (s, 1Н), 5.42-5.30 (m, 2Н), 7.19-7.14 (m, 2Н), 2.40 (s, 3Н).
Пример 54
7-(4-Хлорфенил)-8-(2,6-диметилпиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 54

В соответствии с путем синтеза согласно Примеру 14 исходное соединение 1с заменяли соединением 11а, соответственно, получали указанное в заголовке соединение 54 (47,5 мг).
МС m/z (ИЭР): 351,1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.34 (s, 1Н), 8.27 (brs, 2Н), 7.39-7.33 (m, 4Н), 6.96 (s, 2Н), 2.34 (s, 6Н).
Пример 55
8-(2-(Фторметил)-6-метилпиридин-4-ил)-7-(4-фторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 55
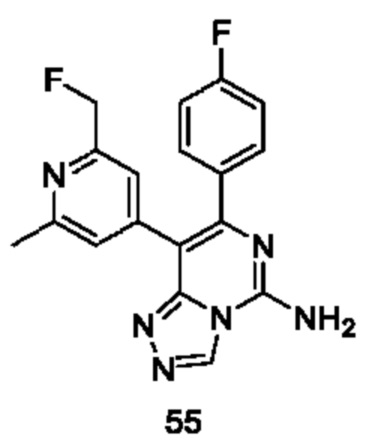
В соответствии с путем синтеза согласно Примеру 15 исходное соединение 4а заменяли соединением 53а, соответственно, получали указанное в заголовке соединение 55 (20 мг).
MC m/z (ИЭР): 353,2 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.36 (s, 1Н), 8.31 (brs, 2Н), 7.40-7.36 (m, 2Н), 7.20-7.13 (m, 4Н), 5.42-5.31 (m, 2Н), 2.41 (s, 3Н).
Пример 56
8-(2-(Дифторметил)-6-метилпиридин-4-ил)-7-(2-фторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 56

В соответствии с путем синтеза согласно Примеру 19 исходное соединение 11а заменяли соединением 20а, соответственно, получали указанное в заголовке соединение 5b (105,6 мг).
MC m/z (ИЭР): 371,1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.53 (s, 1Н), 8.50 (brs, 2Н), 7.47-7.42 (m, 2Н), 7.37 (s, 1Н), 7.31 (s, 1Н), 7.26-7.24 (m, 1Н), 7.13-7.11 (m, 1Н), 6.94-6.66 (m, 1Н), 2.41 (s, 3Н).
Пример 57
8-(2-(фторметил)-6-метилпиридин-4-ил)-7-(2-фторфенил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 57

В соответствии с путем синтеза согласно Примеру 19, исходное соединение 11а заменяли соединением 53а, соответственно, получали указанное в заголовке соединение 57 (58 мг).
МС m/z (ИЭР): 353,2 [М+1]
1Н ЯМР (400 МГц, МЕТАНОЛ-d6) δ 9.29 (s, 1Н), 7.51-7.48 (m, 1Н), 7.43-7.41 (m, 1Н), 7.26-7.21 (m, 3Н), 7.04-6.99 (m, 1Н), 5.37-5.25 (m, 2Н), 2.45 (s, 3Н).
Пример 58
7-(2,4-Дифторфенил)-8-(2-(фторметил)-6-метилпиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 58
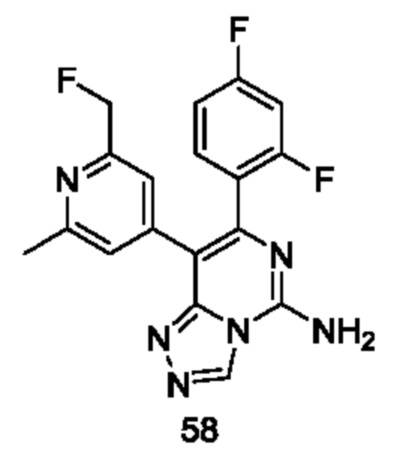
В соответствии с путем синтеза согласно Примеру 18 исходное соединение 1 с заменяли соединением 53а, соответственно, получали указанное в заголовке соединение 58 (30 мг).
МС m/z (ИЭР): 371,1 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 9.29 (s, 1Н), 7.59-7.55 (m, 1Н), 7.27-7.25 (m, 2Н), 7.06-7.02 (m, 1Н), 6.91-6.86 (m, 1Н), 5.40-5.28 (m, 2Н), 2.48 (s, 3Н).
Пример 59
7-(4-Хлор-2-фторфенил)-8-(2-(дифторметил)-6-метилпиридин-4-ил)-[1,2,4]триазоло[4,3-с]пиримидин-5-амин 59

В соответствии с путем синтеза согласно Примеру 49 исходное соединение 1с заменяли соединением 20а, соответственно, получали указанное в заголовке соединение 59 (49,1 мг).
МС m/z (ИЭР): 405,1 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 9.42 (s, 1 Н), 7.54-7.50 (m, 1Н), 7.40-7.30 (m, 4Н), 6.97-6.69 (m, 1 Н), 3.43 (brs, 2Н), 2.44 (s, 3Н).
Примеры анализов:
Биологический анализ
Пример анализа 1. Определение ингибирующей активности соединений по настоящему изобретению в отношении цАМФ-зависимого сигнального пути аденозинового А2а рецептора (A2aR), цАМФ-зависимого сигнального пути аденозинового A2b рецептора (A2bR), цАМФ-зависимого сигнального пути аденозинового A1 рецептора (A1R) и цАМФ-зависимого сигнального пути аденозинового А3 рецептора (A3R).
Ингибирующую активность соединений по настоящему изобретению в отношении цАМФ-зависимого сигнального пути аденозинового А2а рецептора, цАМФ-зависимого сигнального пути аденозинового A2b рецептора, цАМФ-зависимого сигнального пути аденозинового рецептора и цАМФ-зависимого сигнального пути аденозинового А3 рецептора определяли посредством следующего способа. Экспериментальный способ кратко описан следующим образом:
I. Экспериментальные материалы и оборудование
1. Клетки СНО (от англ. chinese hamster ovary cell - клетка яичника китайского хомяка)-K1/A2aR (NM_000675.5) или клетки CHO-K1/A2bR (NM_000676.2) или клетки СНО-K1/A1R (NM_000674.2) или клетки CHO-K1/A3R (NM_000677.3)
2. Фетальная телячья сыворотка (Gibco, 10099-141)
3. Блеомицин (Thermo, R25001) или G418 (ENZO, ALX-380-013-G005) или пуромицин (Thermo, 10687-010)
4. Среда DMEM (от англ. Dulbecco modified Eagle's medium - среда Иглу, модифицированная по Дульбекко)/F12 (GE, SH30023.01)
5. Буфер, разделяющий клетки (Thermo Fisher, 13151014)
6. HEPES (от англ. 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid - 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота) (Gibco, 42360-099)
7. Бычий сывороточный альбумин (MP Biomedicals, 219989725)
8. Ролипрам (sigma, R6520-10MG)
9. Аденозиндезаминаза (sigma, 10102105001)
10. Форсколин (sigma, F6886)
11. 2CI-IB-MECA(Tocrics, 1104/10)
12. N6- циклопентиладенозин (Tocris, 1702/50)
13. Сбалансированный солевой буфер (Thermo, 14025-092)
14. Набор для выявления динамики цАМФ 2 (Cisbio, 62АМ4РЕВ)
15. 384-Луночный планшет (Corning, 4514) или (Nunc, 267462#)
16. Этилкарбазол (Torcis, 1691/10)
17. Многофункциональный микропланшет-ридер PHERAstar (Cisbio, 62АМ4РЕВ)
II. Процедуры экспериментов
2.1 Аденозиновый А2а рецептор
Клетки CHO-K1/A2aR культивировали в среде DMEM/F12, содержащей 10% фетальную телячью сыворотку и 800 мкг/мл блеомицина. Клетки обрабатывали буфером, разделяющим клетки, на протяжении эксперимента. Клетки ресуспендировали в сбалансированном солевом буфере, содержащем 20 мМ НЕ PES и 0,1% бычий сывороточный альбумин, и подсчитывали, плотность клеток доводили до 106 клеток/мл. В 384-луночном планшете в каждую лунку добавляли 5 мкл клеточной суспензии и 2,5 мкл тестируемого соединения (4× концентрация), приготовленного с сбалансированным солевым буфером, содержащим 20 мМ HEPES, 0,1% бычий сывороточный альбумин, 54 мкМ ролипрам и 2,7 Ед/мл аденозиндезаминазы, и планшет инкубировали при комнатной температуре в течение 30 минут. Затем в каждую лунку добавляли 2,5 мкл этилкарбазола (4× концентрация), приготовленного с сбалансированным солевым буфером, содержащим 20 мМ HEPES, 0,1% бычий сывороточный альбумин, 54 мкМ ролипрам и 2,7 Ед/мл аденозиндезаминазы, и планшет инкубировали при комнатной температуре в течение 30 минут. Конечные концентрации соединений составляли: 10000, 2000, 400, 80, 16, 3,2, 0,64, 0,128, 0,0256, 0,00512 и 0,001024 нМ. Конечная концентрация этилкарбазола составляла 20 нМ. Внутриклеточную концентрацию цАМФ выявляли с помощью набора для выявления динамики цАМФ 2. цАМФ-d2 и антитело против цАМФ-Eu-Криптат разводили соответственно цАМФ лизирующим буфером в соотношении 1:4. В каждую лунку добавляли 5 мкл разведенного цАМФ-d2 с последующим добавлением 5 мкп разведенного антитела против цАМФ-Eu-Криптат, и планшет инкубировали при комнатной температуре в темноте в течение 1 часа. Значения сигналов HTRF (от англ. homogenous time-resolved fluorescence -гомогенная флуоресценция с временным разрешением) считывали посредством многофункционального микропланшета-ридера PHERAstar. Значения IC50 (от англ. half maximal inhibitory concentration - концентрация полумаксимального ингибирования) ингибирующей активности соединений рассчитывали посредством программного обеспечения Graphpad Prism, и они показаны в Таблице 1.
2.2 Аденозиновый А1 рецептор
Клетки CHO-K1/A1R культивировали в среде DMEM/F12, содержащей 10% фетальную телячью сыворотку и 1 мкг/мл G418. Клетки обрабатывали буфером, разделяющим клетки, на протяжении эксперимента. Затем клетки ресуспендировали в сбалансированном солевом буфере, содержащем 20 мМ НЕ PES и 0,1% бычий сывороточный альбумин, и подсчитывали, и плотность клеток доводили до 5×105 клеток/мл. В 384-луночном планшете в каждую лунку добавляли 12,5 мкл клеточной суспензии и 6,25 мкл тестируемого соединения (4х концентрация), приготовленного с сбалансированным солевым буфером, содержащим 20 мМ HEPES, 0,1% бычий сывороточный альбумин, 54 мкМ ролипрам и 2,7 Ед/мл аденозиндезаминазы, и планшет инкубировали при комнатной температуре в течение 30 минут. Затем в каждую лунку добавляли 6,25 мкл форсколина и М6-циклопентиладенозина (4× концентрация), приготовленного с сбалансированным солевым буфером, содержащим 20 мМ HEPES, 0,1% бычий сывороточный альбумин, 54 мкМ ролипрам и 2,7 Ед/мл аденозиндезаминазы, и планшет инкубировали при комнатной температуре в течение 30 минут. Конечные концентрации соединений составляли: 10000, 1000, 100, 10, 1, 0,1 и 0 нМ. Конечная концентрация форсколина составляла 10 мкМ. Конечная концентрация CPA составляла 10 нМ. Внутриклеточную концентрацию цАМФ выявляли с помощью набора для выявления динамики цАМФ 2. цАМФ-d2 и антитело против цАМФ-Eu-Криптат разводили соответственно цАМФ лизирующим буфером в соотношении 1:4. В каждую лунку добавляли 12,5 мкл разведенного цАМФ-d2 с последующим добавлением 12,5 мкл разведенного антитела против цАМФ-Eu-Криптат, и планшет инкубировали при комнатной температуре в темноте в течение 1 часа. Значения сигналов HTRF считывали посредством многофункционального микропланшета-ридера PHERAstar. Значения IC50 ингибирующей активности соединений рассчитывали посредством программного обеспечения Graphpad Prism, и они показаны в Таблице 2.
2.3 Аденозиновый А3 рецептор
Клетки CHO-K1/A3R культивировали в среде DMEM/F12, содержащей 10% фетальную телячью сыворотку и 10 мкг/мл пуромицина. Клетки обрабатывали буфером, разделяющим клетки, на протяжении эксперимента. Затем клетки ресуспендировали в сбалансированном солевом буфере, содержащем 20 мМ HEPES и 0,1% бычий сывороточный альбумин, и подсчитывали, и плотность клеток доводили до 5×105 клеток/мл. В 384-луночном планшете в каждую лунку добавляли 12,5 мкл клеточной суспензии и 6,25 мкл тестируемого соединения (4× концентрация), приготовленного с сбалансированным солевым буфером, содержащим 20 мМ HEPES, 0,1% бычий сывороточный альбумин, 54 мкМ ролипрам и 2,7 Ед/мл аденозиндезаминазы, и планшет инкубировали при комнатной температуре в течение 30 минут. Затем в каждую лунку добавляли 6,25 мкл форсколина и 2CI-IB-MECA (4× концентрация), приготовленного с сбалансированным солевым буфером, содержащим 20 мМ HEPES, 0,1% бычий сывороточный альбумин, 54 мкМ ролипрам и 2,7 Ед/мл аденозиндезаминазы, и планшет инкубировали при комнатной температуре в течение 30 минут. Конечные концентрации соединений составляли: 100000, 10000, 1000, 100, 10, 1, 0,1 и 0 нМ. Конечная концентрация форсколина составляла 10 мкМ. Конечная концентрация 2CI-IB-MECA составляла 5 нМ. Внутриклеточную концентрацию цАМФ выявляли с помощью набора для выявления динамики цАМФ 2. цАМФ-d2 и антитело против цАМФ-Eu-Криптат разводили соответственно цАМФ лизирующим буфером в соотношении 1:4. В каждую лунку добавляли 12,5 мкл разведенного цАМФ-d2 с последующим добавлением 12,5 мкл разведенного антитела против цАМФ-Eu-Криптат, и планшет инкубировали при комнатной температуре в темноте в течение 1 часа. Значения сигналов HTRF считывали посредством многофункционального микропланшета-ридера PHERAstar. Значения IC50 ингибирующей активности соединений рассчитывали посредством программного обеспечения Graphpad Prism, и они показаны в Таблице 2.
2.4 Аденозиновый A2b рецептор (A2bR)
Клетки CHO-K1/A2bR культивировали в среде DMEM/F12, содержащей 10% фетальную телячью сыворотку и 1 мкг/мл G418. Клетки обрабатывали буфером, разделяющим клетки, на протяжении эксперимента. Затем клетки ресуспендировали в сбалансированном солевом буфере, содержащем 20 мМ НЕ PES и 0,1% бычий сывороточный альбумин, и подсчитывали, и плотность клеток доводили до 106 клеток/мл. В 384-луночном планшете в каждую лунку добавляли 5 мкл клеточной суспензии и 2,5 мкл тестируемого соединения (4× концентрация), приготовленного с сбалансированным солевым буфером, содержащим 20 мМ HEPES, 0,1% бычий сывороточный альбумин, 54 мкМ ролипрам и 2,7 Ед/мл аденозиндезаминазы, и планшет инкубировали при комнатной температуре в течение 30 минут. Затем в каждую лунку добавляли 2,5 мкл этилкарбазола (4× концентрация) (Torcis, 1691/10), приготовленного с сбалансированным солевым буфером, содержащим 20 мМ HEPES, 0,1% бычий сывороточный альбумин, 54 мкМ ролипрам и 2,7 Ед/мл аденозиндезаминазы, и планшет инкубировали при комнатной температуре в течение 30 минут. Конечные концентрации соединений составляли: 100000, 10000, 1000, 100, 10, 1, 0,1 и 0 нМ. Конечная концентрация этилкарбазола составляла 1 мкМ. Внутриклеточную концентрацию цАМФ выявляли с помощью набора для выявления динамики цАМФ 2. цАМФ-d2 и антитело против цАМФ-Eu-Криптат разводили соответственно цАМФ лизирующим буфером в соотношении 1:4. В каждую лунку добавляли 5 мкл разведенного цАМФ-d2 с последующим добавлением 5 мкл разведенного антитела против цАМФ-Eu-Криптат, и планшет инкубировали при комнатной температуре в темноте в течение 1 часа. Значения сигналов HTRF считывали посредством многофункционального микропланшета-ридера PHERAstar. Значения IC50 ингибирующей активности соединений рассчитывали посредством программного обеспечения Graphpad Prism, и они показаны в Таблице 3.


Заключение: соединения по настоящему изобретению обладают значимой ингибирующей активностью в отношении цАМФ-зависимого сигнального пути аденозинового А2а рецептора.


Заключение: соединения по настоящему изобретению обладают слабой ингибирующей активностью в отношении аденозинового А1 рецептора и аденозинового А3 рецептора, что указывает на то, что соединения по настоящему изобретению являются высокоселективными в отношении аденозинового А2а рецептора.

Заключение: соединения по настоящему изобретению обладают слабой ингибирующей активностью в отношении аденозинового A2b рецептора, что указывает на то, что соединения по настоящему изобретению являются высокоселективными в отношении аденозинового А2а рецептора.
Пример анализа 2. Определение проницаемости головного мозга в отношении соединений по настоящему изобретению у мышей
Проницаемость головного мозга в отношении соединений по настоящему изобретению у мышей определяли следующим экспериментальным способом:
I. Экспериментальные материалы и оборудование
1. Вставки RED Device Inserts (Thermo Scientific, QL21291110)
2. Масс-спектрометр с линейной ионной ловушкой API 4000 Q-trap (Applied Bio systems)
3. Система для жидкостной хроматографии сверхвысокого давления LC-30A (Shimadzu)
4. рН 7,4 PBS (100 мМ, хранящийся при 4°С в холодильнике)
5. Мыши С57, предоставленные Jiesijie Laboratory Animal Co., LTD, с сертификационным №: SCXK (Shanghai) 2013-0006.
II. Обработка подопытных животных
Четыре самки мышей С57 содержали при цикле 12 часов света/12 часов темноты, при постоянной температуре 24±3°С и влажности 50-60%, и они имели свободный доступ к пище и воде. Соединения внутрижелудочно вводили мышам после ночи воздержания от принятия пищи. Вводимая дозировка составляла 20 мг/кг. Группу введения лекарственного средства умерщвляли после забора крови в момент времени 0,5 ч после введения лекарственного средства (объем отобранной крови: 0,5 мл). Образец крови хранили в пробирках с гепарином и центрифугировали в течение 10 минут при 3500 об/мин для отделения плазмы, которую обозначали плазмой 1 и хранили при -80°С. Физиологический раствор впрыскивали в сердце умерщвленного животного для удаления избыточной крови из ткани головного мозга. Брали ткань головного мозга, и остаточную кровь в ткани головного мозга промакивали фильтровальной бумагой. Ткань головного мозга обозначали как ткань головного мозга 1 и хранили при -80°С. Еще три животных отбирали для холостой плазмы и ткани головного мозга 2, и способ обработки был таким же, как и способ обработки группы введения лекарственного средства.
III. Метод определения связывания с белком плазмы на основе равновесного диализа
3.1 Получение образцов
Лекарственные соединения растворяли в ДМСО до 20 мМ с получением стокового раствора I. Соответствующее количество стокового раствора I отбирали и разбавляли метанолом с получением 200 мкМ разбавленного стокового раствора II. 10 мкл стокового раствора II обирали в 1,5 мл пробирку Eppendorf, добавляли 990 мкл холостой плазмы и хорошо перемешивали с получением 2 мкМ образца плазмы 2 (концентрация DMSO меньше или равна 0,2%), который использовали для определения скорости связывания с белком плазмы при данной концентрации. Отбирали 50 мкл указанного выше приготовленного образца плазмы, обозначали как Т0 и хранили при -4°С в холодильнике для тестирования.
3.2 Процедуры эксперимента
Пробирку для равновесного диализа RED Device Inserts вставляли в 96-луночный планшет. 300 мкл указанного выше приготовленного образца плазмы 2, содержащего тестируемое соединение, и соответствующего образца холостой плазмы добавляли в лунку, обозначенную красным (камера для плазмы). 500 мкл фосфатного буферного раствора (рН 7,4) добавляли в другую лунку (камера для буфера) рядом с лункой, обозначенной красным. В соответствии с указанными выше процедурами, каждая концентрация каждого соединения имела по 2 образца. Затем, 96-луночный планшет покрывали запечатывающей лентой, и полный планшет помещали на шейкер с нагревом и уравновешивали при 37°С при 400 об/мин в течение 4 ч. Устройство 96-луночный планшет снимали с шейкера с нагревом после инкубации с достижением равновесного диализа. К 25 мкл уравновешенного образца плазмы или образца-диализата добавляли 25 мкл соответствующего неуравновешенного холостого фосфатного буферного раствора, не содержащего лекарственное средство, или холостой плазмы, не содержащей лекарственное средство, и затем добавляли 200 мкл внутреннего стандарта (приготовленного с ацетонитрилом), перемешивали на вортексе в течение 5 мин и центрифугировали в течение 10 минут (4000 об/мин). Супернатант отбирали для анализа ЖХ (жидкостная хроматография)/МС (масс-спектрометрия)/МС. Образец Т0 не подвергали инкубированию. Отношения площадей хроматографических пиков общего количества лекарственного средства (камера для плазмы) и свободного лекарственного средства (камера для буфера) к внутреннему стандарту прямо определяли посредством способа ЖХ/МС/МС, определенного выше, соответственно, и рассчитывали свободный процент (fu плазма %).
IV. Метод определения связывания с белком ткани головного мозга на основе равновесного диализа
Метод определения связывания с белком ткани головного мозга на основе равновесного диализа: холостую ткань головного мозга 2 превращали в гомогенат холостой ткани головного мозга с помощью PBS (рН 7,4) в соответствии с фактором разведения 11, и в него добавляли соединение с приготовлением 2 мкМ гомогената головного мозга. Другие процедуры были такими же, как процедуры определения связывания с белком плазмы. Отношения площадей хроматографических пиков общего количества лекарственного средства (камера для гомогената головного мозга) и свободного лекарственного средства (камера для буфера) к внутреннему стандарту определяли установленным способом ЖХ/МС/МС, соответственно, и рассчитывали свободный процент (fu голог головного мозга %).
V. Способ расчета данных анализа проницаемости головного мозга
5.1 Концентрации лекарственного средства в плазме 1 и ткани головного мозга 1 мышей, спустя 0,5 ч после введения лекарственного средства, определяли установленным способом ЖХ/МС/МС, соответственно, которые составляли общую концентрацию (Собщ.п и Собщ.г.м);
5.2 Скорости связывания соединения с белком в плазме и ткани головного мозга мышей определяли соответственно методом равновесного диализа с устройством RED Device Inserts, таким образом, чтобы рассчитать свободный процент (fu плазма %, fu головной мозг %);
Свободный процент плазмы (fu плазма %) = Сбуффер/Сплазма × 100%;
Свободный процент гомогената головного мозга (fu гомог головного мозга %) = Сбуфер / Сгом головного мозга × 100%,
Свободный процент ткани головного мозга (fu головной мозг %) = fu гомог головного мозга / (Df-(Df-1) × fu гомог головного мозга) × 100%, с Df=11.
5.3 Коэффициент проницаемости через гематоэнцефалическмй барьер Кр-несвяз. рассчитывали, используя следующую формулу:

VI. Результаты и обсуждение анализа
Коэффициенты проницаемости головного мозга в отношении соединений по настоящему изобретению показаны ниже:

Заключение: соединения по настоящему изобретению имеют низкую концентрацию свободного лекарственного средства в головном мозге, способность проходить через гематоэнцефалический барьер является слабой, и меньшее количество лекарственного средства поступает в головной мозг, что может иметь слабые побочные эффекты.
Настоящее изобретение относится к области медицины, а именно к соединению формулы (I) или его фармацевтически приемлемой соли
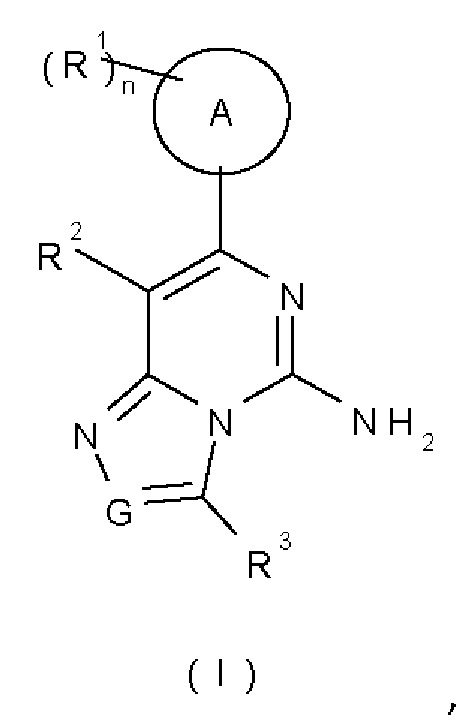
где G представляет собой N или CR4; кольцо А представляет собой фенил или фурил; каждый R1 является идентичным или отличным и каждый независимо выбран из группы, состоящей из водорода, галогена и C1-6алкила; R2 выбран из группы, состоящей из циано, 6-10-членного арила и 5-10-членного гетероарила, имеющего от 1 до 2 гетероатомов N, где каждый из 6-10-членного арила и 5-10-членного гетероарила независимо возможно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, C1-6алкила, C1-6алкокси, C1-6галоалкила, оксо, 3-6-членного циклоалкила и Rb; R3 выбран из группы, состоящей из водорода, галогена и C1-6алкила; R4 представляет собой водород или C1-6алкил; Rb представляет собой 6-членный гетероциклилС1-6алкил, где 1-2 атома гетероциклила представляют собой гетероатомы, выбранные из группы, состоящей из N и О, при этом 6-членный гетероциклил 6-членного гетероциклилС1-6алкила возможно замещен одним или более С1-6алкилами; и n представляет собой 1, 2, 3 или 4. Также предложены промежуточное соединение формулы (IV), способ получения соединения формулы (II), фармацевтическая композиция, применение соединения формулы (I) для получения лекарственного средства. Технический результат изобретения заключается в получении новых производных гетероарил[4,2-с]пиримидин-5-аминов, которые применимы в качестве антагонистов рецептора А2а. 6 н. и 9 з.п. ф-лы, 3 табл., 61 пр.
1. Соединение формулы (I) или его фармацевтически приемлемая соль

где G представляет собой N или CR4;
кольцо А представляет собой фенил или фурил;
каждый R1 является идентичным или отличным и каждый независимо выбран из группы, состоящей из водорода, галогена и C1-6алкила;
R2 выбран из группы, состоящей из циано, 6-10-членного арила и 5-10-членного гетероарила, имеющего от 1 до 2 гетероатомов N, где каждый из 6-10-членного арила и 5-10-членного гетероарила независимо возможно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, C1-6алкила, C1-6алкокси, C1-6галоалкила, оксо, 3-6-членного циклоалкила и Rb;
R3 выбран из группы, состоящей из водорода, галогена и C1-6алкила;
R4 представляет собой водород или C1-6алкил;
Rb представляет собой 6-членный гетероциклилС1-6алкил, где 1-2 атома гетероциклила представляют собой гетероатомы, выбранные из группы, состоящей из N и О, при этом 6-членный гетероциклил 6-членного гетероциклилС1-6алкила возможно замещен одним или более С1-6алкилами; и
n представляет собой 1, 2, 3 или 4.
2. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, представляющее собой соединение формулы (II) или его фармацевтически приемлемую соль

где кольцо В выбрано из группы, состоящей из 6-10-членного арила и 5-10-членного гетероарила, имеющего от 1 до 2 гетероатомов N;
каждый R6 является идентичным или отличным и каждый независимо выбран из группы, состоящей из водорода, галогена, С1-6алкила, С1-6алкокси, С1-6галоалкила, оксо, 3-6-членного циклоалкила и Rb;
s представляет собой 0, 1, 2, 3 или 4; и
кольцо А, G, R1, R3, Rb и n являются такими, как определено в п. 1.
3. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1 или 2, представляющее собой соединение формулы (III) или его фармацевтически приемлемую соль

где кольцо В выбрано из группы, состоящей из 6-10-членного арила и 5-10-членного гетероарила, имеющего от 1 до 2 гетероатомов N;
каждый R6 является идентичным или отличным и каждый независимо выбран из группы, состоящей из водорода, галогена, С1-6алкила, С1-6галоалкила, С1-6алкокси, оксо, 3-6-членного циклоалкила и Rb;
Rb представляет собой 6-членный гетероциклилС1-6алкил, где 1-2 атома гетероциклила представляют собой гетероатомы, выбранные из группы, состоящей из N и О, при этом 6-членный гетероциклил 6-членного гетероциклилС1-6алкила возможно замещен одним или более С1-6алкилами;
s представляет собой 0, 1, 2, 3 или 4; и
кольцо А, R1, R3 и n являются такими, как определено в п. 1.
4. Соединение формулы (II) или его фармацевтически приемлемая соль по любому из пп. 2 или 3, в котором кольцо В выбрано из группы, состоящей из фенила, пиридила, пиразолила, пиридин-2-она, имидазолила, пирролила, фурила, тиенила, изохинолила, хинолила, хиноксалинила, индолила, индазолила, бензофуранила и бензотиенила.
5. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-4, представляющее собой соединение формулы (III') или его фармацевтически приемлемую соль

где каждый R6 является идентичным или отличным и каждый независимо выбран из группы, состоящей из водорода, галогена, С1-6алкила, С1-6галоалкила, С1-6алкокси, оксо, 3-6-членного циклоалкила и Rb;
Rb представляет собой 6-членный гетероциклилС1-6алкил, где 1-2 атома гетероциклила представляют собой гетероатомы, выбранные из группы, состоящей из N и О, при этом 6-членный гетероциклил 6-членного гетероциклилС1-6алкила возможно замещен одним или более С1-6алкилами;
s представляет собой 0, 1, 2, 3 или 4;
кольцо А, R1, R3 и n являются такими, как определено в п. 1.
6. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-5, выбранное из группы, состоящей из:


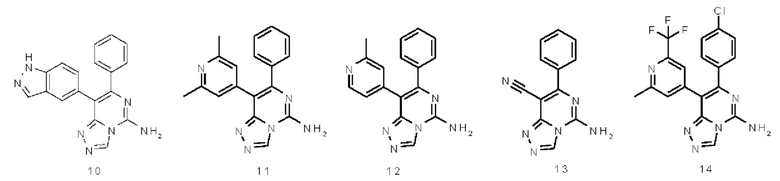

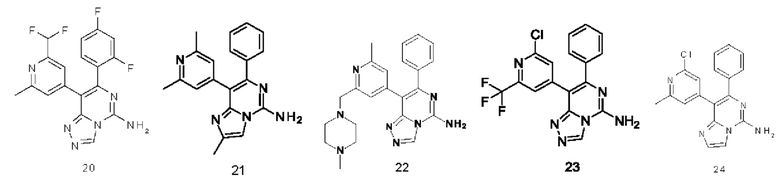
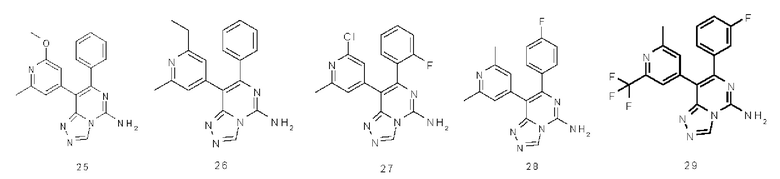
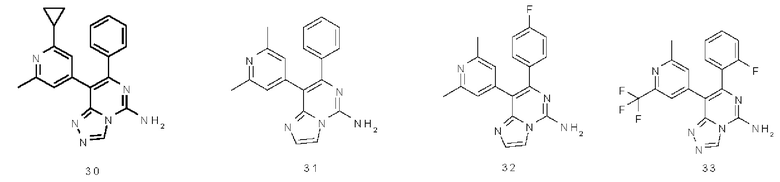


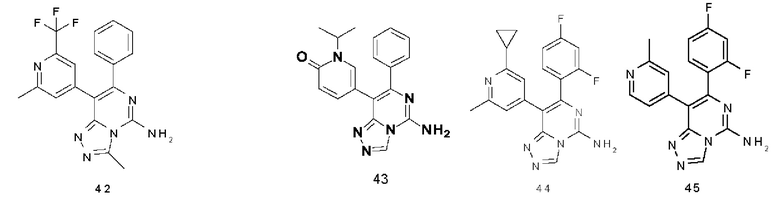


 и
и 
7. Соединение формулы (IV) или его фармацевтически приемлемая соль

где X представляет собой галоген;
кольцо А, G, R1, R3 и n являются такими, как определено в п. 1.
8. Соединение формулы (IV) или его фармацевтически приемлемая соль по п. 7, выбранное из группы, состоящей из:

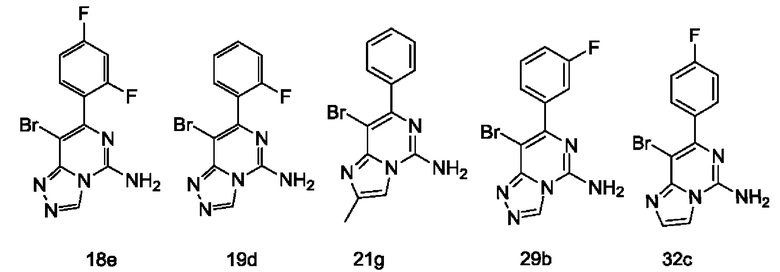
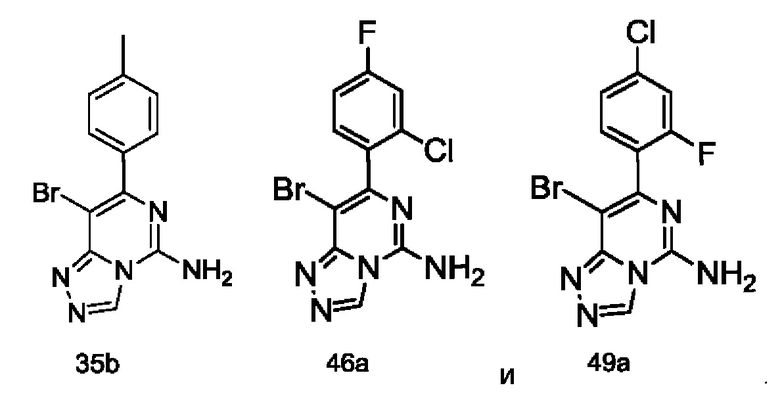
9. Способ получения соединения формулы (II) или его фармацевтически приемлемой соли по п. 2, включающий стадию:
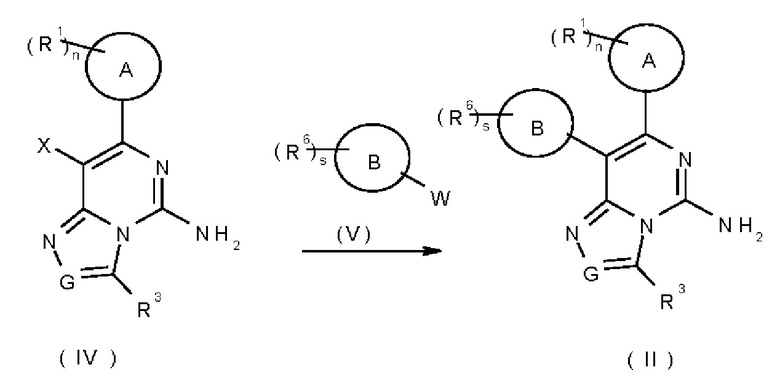
вступления соединения формулы (IV) в реакцию с соединением формулы (V) с получением соединения формулы (II),
где X представляет собой галоген;
W представляет собой  или
или 
кольцо А, кольцо В, G, R1, R3, R6, n и s являются такими, как определено в п. 2.
10. Фармацевтическая композиция для ингибирования рецептора А2а, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-6 и один или более фармацевтически приемлемых носителей, разбавителей или вспомогательных веществ.
11. Применение соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-6 или фармацевтической композиции по п. 10 в получении лекарственного средства для ингибирования рецептора А2а.
12. Применение соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-6 или фармацевтической композиции по п. 10 в получении лекарственного средства для лечения заболевания или состояния, улучшаемого посредством ингибирования рецептора А2а, при этом заболевание или состояние, улучшаемое посредством ингибирования рецептора А2а, выбрано из группы, состоящей из опухоли, депрессии, нарушения когнитивной функции, нейродегенеративного расстройства, расстройства, связанного с вниманием, экстрапирамидального синдрома, аномального двигательного расстройства, цирроза, фиброза печени, жировой инфильтрации печени, кожного фиброза, нарушения сна, инсульта, повреждения головного мозга, нейровоспаления и зависимого поведения.
13. Применение по п. 12, где заболевание или состояние, улучшаемое посредством ингибирования рецептора А2а, представляет собой опухоль.
14. Применение по п. 13, где опухоль выбрана из группы, состоящей из меланомы, опухоли головного мозга, рака пищевода, рака желудка, рака печени, рака поджелудочной железы, колоректального рака, рака легкого, рака почки, рака молочной железы, рака яичника, рака предстательной железы, рака кожи, нейробластомы, саркомы, остеохондромы, остеомы, остеосаркомы, семиномы, опухоли яичка, рака матки, рака головы и шеи, множественной миеломы, злокачественной лимфомы, истинной полицитемии, лейкоза, опухоли щитовидной железы, опухоли мочеточника, рака мочевого пузыря, рака желчного пузыря, холангиокарциномы, хорионэпителиомы и детской опухоли.
| WO 2009111449 A1, 11.09.2009 | |||
| Уплотнение ротора паровой турбины | 1986 |
|
SU1328560A1 |
| WO 03068776 A1, 21.08.2003 | |||
| JP 2002037787 A, 06.02.2002 | |||
| WO 2006138734 A1, 28.12.2006 | |||
| EP 0976753 A1, 02.02.2000 | |||
| EP 1116722 A1, 18.07.2001 | |||
| RU 2007134899 A, 27.04.2009. | |||

