ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к гетеробициклическим соединениям, которые ингибируют действие p38 митоген-активируемой протеинкиназы, протеинкиназы млекопитающих, которая участвует в клеточной пролиферации, ответной реакции клетки на стимулы и некрозе клеток. В частности, настоящее изобретение относится к гетеробициклическим соединениям, которые являются селективными и эффективными ингибиторами p38 митоген-активируемой протеинкиназы. Настоящее изобретение также относится к фармацевтическим композициям, содержащим данные гетеробициклические соединения, которые ингибируют p38 митоген-активируемую протеинкиназу.
УРОВЕНЬ ТЕХНИКИ
Митоген-активируемые протеинкиназы (MAP) представляет собой семейство пролин-направленных серин/треониновых киназ, которые активируются двойным фосфорилированием и, в свою очередь, фосфорилируют свои субстраты, или по участку треонин-пролин или по участку серин-пролин.
MAP киназы активируются в ответ на ряд сигналов, включая стресс, вызванный нарушением питания, и осмотический стресс, УФ-свет, факторы роста, эндотоксин и воспалительные цитокины. p38 подгруппа MAP киназ (p38, также известная как CSBP и RK) представляет собой семейство MAP киназ различных изоформ, которые являются ответственными за фосфорилирование большого набора субстратов, включая транскрипционные факторы (например, ATF2, CHOP и MEF2C), другие киназы (например, MAPKAP-2 и MAPKAP-3), онкосупрессоры (например, p53) и регуляторы трансляции (например, 3EBP, PRAK).
Установлено, что большой ряд хронических и острых заболеваний связан с нарушением воспалительной реакции. В данной реакции принимает участие большой ряд цитокинов, включая IL-I, IL-6, IL-8 и TNF. Выяснено, что экспрессия, секреция и активация данных цитокинов при регуляции воспаления зависит, по меньшей мере, отчасти, от активации p38. Данная киназа активируется двойным фосфорилированием после стимулирования физиохимическим стрессом, обработки липополисахаридами или провоспалительными цитокинами, такими как IL-I, и TNF.
TNF и интерлейкины, такие как IL-I и IL-8, влияют на большой ряд клеток и тканей и представляют собой важные воспалительные посредники большого ряда болезненных состояний и заболеваний. TNF-α представляет собой цитокин, синтезируемый преимущественно активированными моноцитами и макрофагами. Избыточный или нерегулируемый TNF синтез вовлечен в опосредование ряда заболеваний. Недавние исследования обнаружили, что TNF играет причинную роль в патогенезе ревматоидного артрита. Дополнительные исследования показывают, что ингибирование TNF широко применяется для лечения воспаления, воспалительного заболевания кишечника, множественного склероза и астмы. TNF также связан с вирусными инфекциями, такими как ВИЧ, вирус гриппа и вирус герпеса, включая вирус простого герпеса 1 типа (HSV-I), вирус простого герпеса 2 типа (HSV-2), цитомегаловирус (CMV), вирус ветряной оспы (VZV), вирус Эпштейна-Барр, человеческий вирус герпеса-6 (HHV-6), человеческий вирус герпеса-7 (HHV-7), человеческий вирус герпеса-8 (HHV-8), псевдобешенство и ринотрахеин, среди других. IL-8 представляет собой другой провоспалительный цитокин, который синтезируется мононуклеарами, фибробластами, эндотелиальными клетками и кератиноцитами и связан с патологическими состояниями, включая воспаление.
IL-1 синтезируется активированными моноцитами и макрофагами и вовлечен в воспалительную реакцию. IL-I участвует во многих патофизиологических реакциях, включая ревматоидный артрит, лихорадку и снижение резорбции кости.
TNF, IL-I и IL-8 воздействуют на большой ряд клеток и тканей и являются важными медиаторами воспаления большого ряда заболеваний и состояний. Ингибирование данных цитокинов ингибированием p38 киназы является полезным для контролирования, ослабления и облегчения многих из данных заболеваний.
В течение последних нескольких лет было показано, что p38 включает группу MAP I киназ, обозначенных p38δ, p38γ, p38β, p38α. Jiang, Y. et al. (A Biol Chem I (1996) 271: 17920-17926) сообщают о классификации p38β как белка, состоящего из 372 аминокислот, близко родственного p38-α. При сравнении активности p38α с активностью p38β, авторы утверждают, что тогда как оба из них активируются провоспалительными цитокинами и стрессом под влиянием окружающей среды, p38β преимущественно активируется MAP киназа-киназой-6 (MKK6) и преимущественно активирует транскрипционный фактор 2, тем самым указывая на то, что с данными формами могут быть связаны различные механизмы действия. Kumar, S. et al. (Biochem Biophys Res Comm (1997) 235:533-538) и Stein, B. et al. (JBiol Chem (1997) 272: 19509-19517) сообщали о второй изоформе p38β--p38β2, содержащей 364 аминокислот с 73% идентичностью p38α. Все из данных сообщений доказывают, что p38β активируется провоспалительными цитокинами и стрессом под влиянием окружающей среды, хотя вторые из них сообщали о p38β изоформе - p38β2, по-видимому, преимущественно экспрессируемой в ЦНС, сердце и скелетных мышцах, по сравнению с более повсеместной экспрессией в тканях p38α. Кроме того, наблюдалось, что активированный транскрипционный фактор-2 (ATF-2) является более хорошим субстратом для p38β2, чем для p38α, тем самым указывая на то, что с данными формами могут быть связаны различные механизмы действия. Физиологическая роль p38β1 подверглась сомнению в последних двух сообщениях, поскольку ее не могли обнаружить в человеческой ткани и она не проявляла заметную киназную активность с субстратами p38α.
Об обнаружении p38γ сообщалось Li, Z. et al. (Biochem Biophys Res Comm (1996)228:334-340), и об p38δ сообщалось Wang, X. et al. (JBiol Chem (1997) 272:23668-23674) и Kumar, S. et al. (Biochem Biophys Res Comm (1997) 235:533-538). Данные предполагают, что эти две p38 изоформы (γ и δ) представляют собой уникальный поднабор MAPK семейства, исходя из их характера распределения в тканях, применения субстрата, ответной реакции на прямые и непрямые стимулы и чувствительности к киназным ингибиторам. О различных результатах относительно различной ответной реакции на лекарственные средства, направленные на p38 семейство, как между p38α и предполагаемой p38β1 или p38β2, или обеими, сообщают Jiang, Kumar и Stein, цитируемые выше, так же как Eyers, P. A. et al. (Chem and Biol (1995)5:321-328). В дополнительной статье Wang, Y. et al. (JBiol Chem (1998)273:2161-2168) предполагается важность данных различных эффектов. Как отмечено Wang et al., ряд стимулов, таких как инфаркт миокарда, гипертензия, пороки клапанов, вирусный миокардит и дилатационная кардиомиопатия, приводят к увеличению нагрузки на сердце и повышенному механическому стрессу на кардиомиоциты.
Сообщается, что они приводят к адаптивной гипертрофированной ответной реакции, которая, если ее не контролировать, имеет несомненно негативные последствия. Wang et al. приводят предшествующие исследования, которые показали, что в сердце с ишемически-реперфузионным повреждением, p38 MAPK активности увеличиваются в связи с гипертрофией и запрограммированной клеточной смертью. Wang et al. показывают в приводимой статье, что активация p38β активности приводит в результате к гипертрофии, тогда как активация p38α активности приводит к апоптозу миоцитов.
Таким образом, селективное ингибирование p38α активности по сравнению с p38β активностью будет полезным для лечения состояний, связанных с сердечной недостаточностью. Данные состояния I включают застойную сердечную недостаточность, кардиомиопатию, миокардит, васкулит, сосудистый рестеноз, клапанный порок, состояния, связанные с сердечно-легочным шунтированием, аорто-коронарным шунтированием, трансплантатами и сосудистыми трансплантатами. Далее, поскольку α-изоформа является токсичной в других типах мышечных клеток, α-селективные ингибиторы будут полезны для состояний, связанных с кахексией, приписываемой TNF, или других состояний, таких как рак, инфекция или аутоиммунное заболевание.
PCT заявки WO 98/06715, WO 98/07425, WO 98/28292 и WO 96/40143 описывают влияние ингибиторов p38 киназы на различные заболевания. Как упоминалось в данных заявках, ингибиторы p38 киназы являются пригодными для лечения ряда заболеваний, связанных с хроническим воспалением. В данных заявках приводятся ревматоидный артрит, ревматоидный спондилит, остеоартрит, подагрический артрит и другие артритические заболевания, сепсис, септический шок, эндотоксический шок, грамотрицательный сепсис, синдром токсического шока, астма, синдром расстройства дыхания у взрослых, инсульт, реперфузионное повреждение, повреждения ЦНС, такие как повреждение нейронов и ишемия, псориаз, рестеноз, церебральная малярия I, хроническое легочное воспалительное заболевание, силикоз, патологическое разрастание мягких тканей легкого, заболевания, связанные с резорбцией кости, такие как остеопороз, реакция трансплантат против хозяина, болезнь Крона, язвенный колит, включая воспалительное заболевание кишечника (IBD), и лихорадка.
Сущность настоящего изобретения
Соединения, описанные химической формулой (A), или их фармацевтически приемлемые соли:
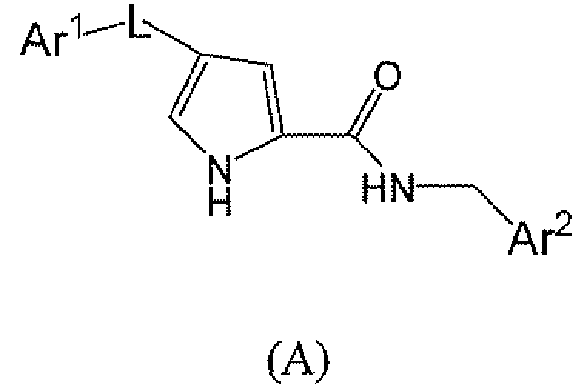
представляют собой ингибиторы p38 и являются пригодными для лечения воспаления, такого как лечение астмы, COPD, ARDS, ревматоидного артрита, ревматоидного спонделита, остеоартрита, подагрического артрита и других артритических заболеваний; воспаленных сосудов, экземы, псориаза или других воспалительных заболеваний кожи, таких как солнечные ожоги; воспалительных заболеваний глаз, включая конъюнктивит; лихорадки, боли, и других заболеваний, связанных с воспалением.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
В одном варианте осуществления настоящее изобретение относится к p38 ингибирующим соединениям химической формулы (A):
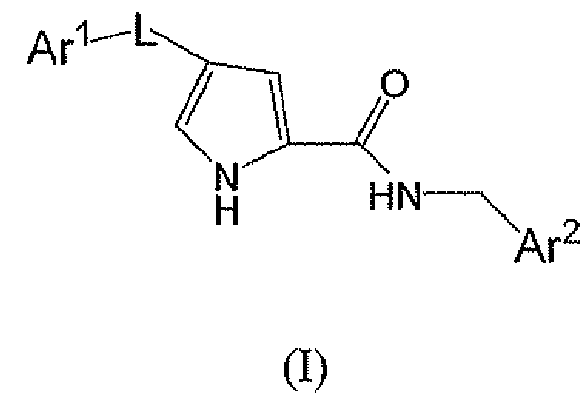
или их фармацевтически приемлемой соли, где:
L выбирают из группы, состоящей из:
(a) -C(O)-,
(b) -CH(OH)-,
(c) -CH(NR3R4)-,
(d) -C(=NOR3)-,
(e) -CH2- и
(f) -S(O)n-, где n равно 0, 1 или 2;
Ar1 представляет собой необязательно моно-, ди- или тризамещенное фенильное или гетероароматическое кольцо с 6 атомами, где гетероароматическое кольцо может содержать 1, 2 или 3 гетероатома, выбранные из N, S и O, где заместители независимо выбирают из группы, состоящей из:
(a) галогена,
(b) -C1-4алкила,
(c) -O-C1-4алкила,
(d) -CF3,
(e) -NH2,
(f) -NH-CH3,
(g) -CN,
(h) -C(О)NH2 и
(J) -S(O)n-CH3;
Ar2 представляет собой необязательно замещенное тиадиазольное или оксадиазольное кольцо, где заместитель представляет собой фенильное или 5- или 6-членное моноциклическое гетероароматическое или гетероциклическое кольцо, или бициклическое гетероароматическое или гетероциклическое кольцо с 9 или 10 атомами, причем упомянутое гетероароматическое или гетероциклическое кольцо содержит 1, 2 или 3 гетероатома, выбранные из группы, состоящей из S, O и N, где упомянутое фенильное, гетероароматическое или гетероциклическое кольцо является необязательно моно- или дизамещенным заместителями, независимо выбранными из группы, состоящей из:
(a) галогена,
(b) -C1-6алкила, необязательно замещенного 1-4 атомами фтора,
(с) -O-C1-6алкила,
(d) -CF3,
(e) -NH2 и
(f) -NH2-CH3,
(g) -NH2-CH2CF3,
(h) -C(O)-морфолинила,
(i) -C(O)-NR1R2,
(j) -C(O)OH,
(k) -CN,
(1) оксо и
(m) C3-6циклоалкила;
R1, R2, R3 и R4 независимо выбирают из группы, состоящей из
(a) водорода и
(b) C1-4алкила,
R1 и R2 или R3 и R4 могут соединяться, образуя 5- или 6-членное насыщенное кольцо, причем упомянутое кольцо необязательно содержит гетероатом, выбранный из S, N и O.
В данном варианте осуществления есть группа, где
L выбирают из группы, состоящей из:
(a) -C(O)- и
(b) -CH2-.
В данной группе есть подгруппа, где
L представляет собой -C(O)-.
В данном варианте осуществления есть группа, где
Ar1 представляет собой необязательно моно-, ди- или тризамещенное фенильное или гетероароматическое кольцо с 6 атомами, где гетероароматическое кольцо может содержать 1, 2 или 3 гетероатома, выбранные из N, S и O, где заместители независимо выбирают из группы, состоящей из:
(a) галогена,
(b) -C1-4алкила и
(c) -O-C1-4алкила.
В данной группе есть подгруппа, где
Ar1 представляет собой необязательно моно-, ди- или тризамещенный фенил или пиридил, где заместители независимо выбирают из группы, состоящей из
(a) фтора,
(b) хлора и
(C) -CH3.
В данной подгруппе есть класс, где
Ar1 представляет собой необязательно моно-, ди- или тризамещенный фенил, где заместители независимо выбирают из группы, состоящей из
(a) фтора,
(b) хлора и
(C) -CH3.
В данном варианте осуществления есть группа, где
Ar2 представляет собой необязательно замещенный тиадиазолил.
В данной группе есть подгруппа, где заместитель представляет собой фенил или 5- или 6-членное моноциклическое гетероароматическое или гетероциклическое кольцо или бициклическое гетероароматическое или гетероциклическое кольцо с 9 или 10 атомами, причем упомянутое гетероароматическое или гетероциклическое кольцо содержит 1, 2 или 3 гетероатома, выбранные из группы, состоящей из S, O и N, где упомянутое фенильное, гетероароматическое или гетероциклическое кольцо является необязательно моно- или дизамещенным заместителями, независимо выбранными из группы, состоящей из:
(a) галогена,
(b) -C1-6алкила, необязательно замещенного CF3,
(c) -O-C1-4алкила,
(d) -CF3 и
(e) C3-6циклоалкила.
В данной подгруппе есть класс, где заместитель представляет собой фенил или 5- или 6-членное моноциклическое гетероароматическое или гетероциклическое кольцо, упомянутое гетероароматическое или гетероциклическое кольцо содержит 1, 2 или 3 гетероатома, выбранные из группы, состоящей из S, O и N, где упомянутое фенильное, гетероароматическое или гетероциклическое кольцо является необязательно моно- или дизамещенным заместителями, независимо выбранными из группы, состоящей из:
(a) галогена,
(b) -C1-6алкила, необязательно замещенного CF3,
(c) -O-C1-4алкила,
(d) -CF3 и
(e) C3-6циклоалкила.
В данном варианте осуществления есть группа, где
R1, R2, R3 и R4 независимо выбирают из группы, состоящей из
(a) водорода и
(b) метила.
В данном варианте осуществления есть группа формулы I
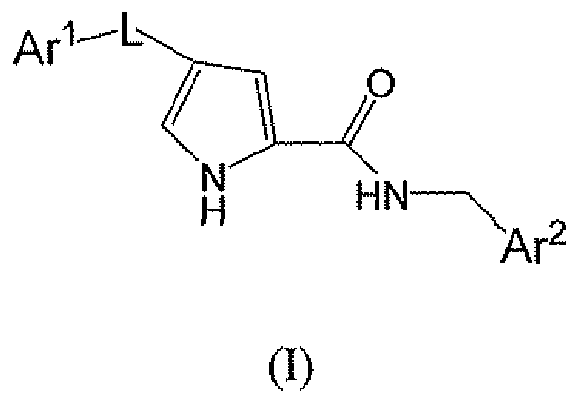
или его фармацевтически приемлемая соль, где
L представляет собой -C(O)-;
Ar1 представляет собой необязательно моно-, ди- или тризамещенный фенил, где заместители независимо выбирают из группы, состоящей из:
(a) F,
(b) Cl,
(c) -C1-4алкила и
(d) -O-C1-4алкила;
Ar2 является необязательно замещенным тиадиазолилом, и заместитель представляет собой фенил или 5- или 6-членное моноциклическое гетероароматическое или гетероциклическое кольцо, причем упомянутое гетероароматическое или гетероциклическое кольцо содержит 1, 2 или 3 гетероатома, выбранные из группы, состоящей из S, O и N, где упомянутое фенильное, гетероароматическое или гетероциклическое кольцо является необязательно моно- или дизамещенным заместителями, независимо выбранными из группы, состоящей из:
(a) галогена,
(b) -C1-4алкила,
(c) -O-C1-4алкила,
(d) -CF3,
(e) C3-6циклоалкила.
В данной группе есть подгруппа формулы II
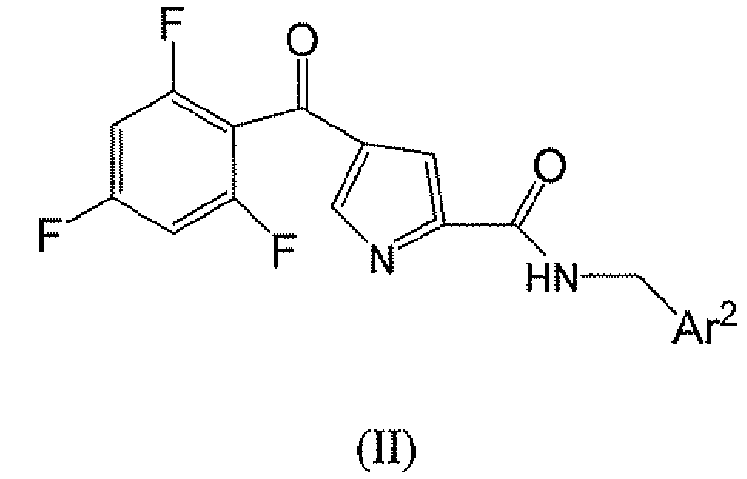
или его фармацевтически приемлемая соль, где
Ar2 является необязательно замещенным тиадиазолилом, где заместитель представляет собой фенильное или -5 или 6-членное моноциклическое гетероароматическое или гетероциклическое кольцо, причем упомянутое гетероароматическое или гетероциклическое кольцо содержит 1, 2 или 3 гетероатома, выбранные из группы, состоящей из S, O и N, где упомянутое фенильное, гетероароматическое или гетероциклическое кольцо является необязательно моно- или дизамещенным заместителями, независимо выбранными из группы, состоящей из:
(a) галогена,
(b) -C2-6алкила,
(c) -O-C1-4алкила и
(d) -CF3.
Как обсуждалось выше, p38 подгруппа MAP киназ представляет собой семейство MAP киназ различных изоформ (включая p38δ, p38γ, p38β, p38α), которые являются ответственными за фосфорилирование большого набора последующих в цепи субстратов. Данные указывают на то, что две p38 изоформы (γ и δ) представляют собой уникальную подгруппу MAPK семейства, исходя из их характера распределения в тканях, применения субстрата, ответной реакции на прямые и непрямые стимулы и чувствительности к киназным ингибиторам. О различных результатах относительно различной ответной реакции на лекарственные средства, направленные на p38 семейство, как между p38α и или предполагаемой p38β1, или p38β2, или обеими, сообщают Jiang, Kumar и Stein, цитируемые выше, так же как Eyers, P. A. et al. (Chem and Biol (1995)5:321-328). В дополнительной статье Wang, Y. et al. (J Biol Chem (1998)273:2161-2168) предполагается важность данных различных эффектов селективного ингибирования p38-α. Традиционные ингибиторы p38-α ингибируют фосфорилирование последующих в цепи субстратов, включая, но не ограничиваясь, MK2, MK3, ATF2, Mnk2a, MSK1, TAB1, CREB и HSP27. На основе этих данных p38-α ингибиторы, которые преимущественно ингибируют фосфорилирование одной подгруппы данных последующих в цепи субстратов, должны обладать повышенным терапевтическим индексом по сравнению с традиционными p38 ингибиторами.
Соответственно, в одном аспекте, настоящее изобретение относится к соединениям формулы I, которые селективно и предпочтительно ингибируют p38-α по сравнению с p38β, и/или p38δ, и/или p38γ. Данный аспект относится к соединениям формулы I, которые предпочтительно ингибируют p38-α по сравнению с p38β, и/или p38δ, и/или p38γ, как измерено in vitro киназным анализом.
В еще другом аспекте, настоящее изобретение относится к соединениям формулы I, которые представляют собой эффективные ингибиторы p38-α и селективно и предпочтительно ингибируют фосфорилирование одного или более из MK2, MK3, ATF2, Mnk2a, MSK1 и TAB1, по сравнению с оставшимися или другими последующими в цепи субстратами. Например, в одном аспекте, настоящее изобретение относится к соединениям формулы I, которые селективно и предпочтительно ингибируют фосфорилирование MK2 и MK3 по сравнению с MSK1, ATF2 или пептидным субстратом. Данный аспект включает соединения формулы 1, которые представляют собой эффективные ингибиторы p38-α и селективно и предпочтительно ингибируют фосфорилирование MK2 по сравнению с пептидным субстратом, как измерено in vitro киназным анализом.
Термин "ацеталь" обозначает функциональную группу или молекулу, содержащую CH группу, связанную с двумя -OR группами. Таким образом, "циклический ацеталь" обозначает циклическую или кольцевую структуру, содержащую ацетальную группу.
Термин "алкил" обозначает углеродную цепь, которая не содержит двойных или тройных связей и которая может быть линейной, разветвленной или их комбинацией. Таким образом, C1-C6 алкил определяют для обозначения группы как содержащей 1, 2, 3, 4, 5 или 6 атомов углерода в таком расположении, который является линейным, разветвленным или их комбинацией. Примеры алкильных групп включают метил, этил, пропил, н-пропил, изопропил, бутил, втор- и трет-бутил, пентил, гексил, гептил и подобные. Термин "C0-C4алкил" включает алкилы, содержащие 4, 3, 2, 1 или не содержащие атомов углерода. Алкил, не содержащий атомов углерода, представляет собой заместитель, являющийся атомом водорода, когда алкил представляет собой концевую группировку. Алкил, не содержащий атомов углерода, представляет собой прямую связь, когда алкил представляет собой мостиковую группировку. Термин "алкен" обозначает линейные или разветвленные структуры и их комбинации, с указанным количеством атомов углерода, содержащие, по меньшей мере, одну двойную связь углерод-углерод, где водород может быть замещен дополнительной двойной связью углерод-углерод. C2-C6 алкен, например, включает этилен, пропилен, 1-метилэтилен, бутилен и подобные.
Термин "алкинил" обозначает линейные или разветвленные структуры и их комбинации, с указанным количеством атомов углерода, содержащие, по меньшей мере, одну тройную связь углерод-углерод. Таким образом, C2-C6 алкинил определяют для обозначения группы как содержащей 2, 3, 4, 5 или 6 тома углерода с линейной или разветвленной структурой, так что C2-C6 алкинил более определенно включает 2-гексинил и 2-пентинил.
Термин "алкокси", как применяют в настоящем изобретении, отдельно или в комбинации, включает алкильную группу, соединенную с соединительным кислородным атомом. Термин "алкокси" также включает алкилэфирные группы, где термин "алкил" определен выше, и "эфир" обозначает две алкильные группы с атомом кислорода между ними. Примеры подходящих алкокси групп включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, метоксиметан (также называемый "диметиловый эфир") и метоксиэтан (также называемый "этилметиловый эфир").
Термин "амин", если специально не указано иначе, включает первичные, вторичные и третичные амины.
Подразумевается, что термин "арил", если специально не указано особо, обозначает любое стабильное моноциклическое или конденсированное бициклическое углеродное кольцо, содержащее вплоть до 7 членов в каждом кольце, где, по меньшей мере, одно кольцо является ароматическим. Примеры данных арильных элементов включают фенил, нафтил и толил.
Термин "арилокси", если специально не указано особо, включает большое количество кольцевых систем, а также системы с одним кольцом, такие как, например, фенил или нафтил, соединенные через соединяющий кислородный атом с местом присоединения.
Термин "циклоалкил" обозначает карбоциклы, не содержащие гетероатомов, и включает моно-, би- и трициклические насыщенные карбоциклы, а также конденсированные кольцевые системы. Данные конденсированные кольцевые системы могут содержать одно кольцо, которое является частично или полностью ненасыщенным, такое как бензольное кольцо, для образования конденсированных кольцевых систем, таких как карбоциклы, конденсированные с бензольным кольцом. Циклоалкил включает данные конденсированные кольцевые системы как спиро-конденсированные кольцевые системы. Примеры циклоалкилов включают циклопропил, циклобутил, циклопентил, циклогексил, декагидронафталенил, адамантанил, инданил, инденил, флуоренил, 1,2,3,4-тетрагидронафталенил и подобные. Аналогично, "циклоалкенил" обозначает карбоциклы, не содержащие гетероатомы и содержащие, по меньшей мере, одну двойную C-C связь, и включают моно-, би- и три-циклические частично насыщенные карбоциклы, а также циклоалкены, конденсированные с бензольным кольцом. Примеры циклоалкенила включают циклогексенил, инденил и подобные.
Термин "циклоалкилокси", если специально не указано особо, включает циклоалкильную группу, соединенную с соединительным атомом кислорода.
Термин "гетеро", если специально не указано особо, включает один или более O, S, или N атомов. Например, гетероциклоалкил и гетероарил включают кольцевые системы, которые содержат один или более O, S, или N атомов в кольце, включая смеси данных атомов. Гетероатомы замещают кольцевые атомы углерода.
Примеры гетероциклоалкилов включают азетидинил, пирролидинил, пиперидинил, пиперазинил, морфолинил, тетрагидрофуранил, имидазолинил, циклические ацетали, циклические кетали, пиролидин-2-он, пиперидин-2-он и тиоморфолинил. Как применяют в настоящем изобретении, "гетероциклоалкил" включает мостиковые гетероциклоалкилы, содержащие две или более гетероциклоалкильных групп, соединенных через соседние или не соседние атомы.
Подразумевается, что термин "гетероарил", как применяют в настоящем изобретении, за исключением случаев, где указано, обозначает стабильную 5- - 7-членную моноциклическую или стабильную 9- - 10-членную конденсированную бициклическую гетероциклическую кольцевую систему, которая содержит ароматическое кольцо, любое кольцо которого может быть насыщенным, таким как пиперидинил, частично насыщенным или ненасыщенным, таким как пиридинил, и которая состоит из атомов углерода и от одного до четырех гетероатомов, выбранных из группы, состоящей из N, O и S, и где гетероатомы азота и серы можно необязательно окислять и гетероатом азота можно необязательно кватернезировать, и содержащую любую бициклическую группу, в которой любое из определенных выше гетероциклических колец конденсируют с бензольным кольцом. Гетероциклическое кольцо может присоединяться по любому гетероатому или атому углерода, что приводит в результате к созданию стабильной структуры. Примеры данных гетероарильных групп включают, но не ограничиваются, пиридин, пиримидин, пиразин, тиофен, оксазол, тиазол, триазол, тиадиазол, оксадиазол, пиррол, 1,2,4-оксадиазол, 1,3,4-оксадиазол, 1,2,4-тиадиазол, 1,3,4-тиадиазол и 1,2,4-триазол.
Дополнительные примеры гетероарила включают хинолинил, пиримидинил, изохинолинил, пиридазинил, хиноксалинил, фурил, бензофурил, дибензофурил, тиенил, бензотиенил, индолил, индазолил, изоксазолил, изотиазолил, имидазолил, бензимидазолил, тиадиазолил, тетразолил.
Термин "гетероарилокси", если специально не указано особо, относится к гетероарильной группе, присоединенной через соединительный атом кислорода к месту присоединения.
Примеры гетероарил(C1-6)алкила включают, например, фурилметил, фурилэтил, тиенилметил, тиенилэтил, пиразолилметил, оксазолилметил, оксазолилэтил, изоксазолилметил, тиазолилметил, тиазолилэтил, имидазолилметил, имидазолилэтил, бензимидазолилметил, оксадиазолилметил, оксадиазолилэтил, тиадиазолилметил, тиадиазолилэтил, триазолилметил, триазолилэтил, тетразолилметил, тетразолилэтил, пиридинилметил, пиридинилэтил, пиридазинилметил, пиримидинилэтил, пиразинилметил, хинолинилметил, изохинолинилметил и хиноксалинилметил.
Если не указано особо, термин "карбамоил" применяют для включения -NHC(O)OC1-C4алкила и -OC(O)NHC1-C4алкила.
Термин "галоген" включает атом фтора, хлора, брома и йода.
Термин "кеталь" обозначает функциональную группу или молекулу, содержащую углерод, соединенный с двумя -OR группами. Таким образом, "циклический кеталь" обозначает циклическую или кольцевую структуру, содержащую кетальную группу.
Подразумевается, что термин "необязательно замещенный" включает и замещенный, и незамещенный. Таким образом, например, необязательно замещенный арил может представлять собой пентафторфенильное или фенильное кольцо. Затем, замещение можно осуществлять по любой из групп. Например, замещенный арил(C1-6)алкил включает замещение в арильной группе, а также замещение в алкильной группе.
Термин "оксид" гетероарильных групп применяют в стандартном хорошо известном химическом смысле, и он включает, например, N-оксиды гетероатомов азота.
Соединения, описанные в настоящем изобретении, содержат один или более двойных связей и таким образом могут приводить к цис/транс изомерам, а также другим конформационным изомерам. Настоящее изобретение включает все данные возможные изомеры, а также смеси данных изомеров.
Если специально не указано особо или не показано символом связи (пунктиром или двойным пунктиром), точка присоединения к описанной группе будет у крайней правой указанной группы. То есть, например, фенилалкильная группа соединена с основной структурой через алкил, и фенил представляет собой заместитель при алкиле.
Соединения настоящего изобретения являются пригодными в различных фармацевтически приемлемых солевых формах. Термин "фармацевтически приемлемая соль" относится к тем солевым формам, которые являются известными химикам, работающим в области фармацевтики, т.е. тем солевым формам, которые являются практически нетоксичными и которые обладают требуемыми фармакокинетическими свойствами, приятным вкусом, абсорбцией, распределением, метаболизмом или экскрецией. Другими факторами, более полезными по своему характеру, которые также являются важными при выборе, являются стоимость исходных соединений, простота кристаллизации, выход, стабильность, гигроскопичность и текучесть полученного в результате сыпучего лекарственного средства. Фармацевтические композиции можно удобно получать из активных ингредиентов в комбинации с фармацевтически приемлемыми носителями.
Соединения, описанные в настоящем изобретении, могут содержать один или более асимметрических центров и могут, таким образом, давать диастереомеры и оптические изомеры. Настоящее изобретение включает все данные возможные диастереомеры, а также их рацемические смеси, их практически чистые разделенные энантиомеры, все возможные геометрические изомеры и их фармацевтически приемлемые соли. Формула I выше показана без определенной стереохимии в определенных положениях. Настоящее изобретение включает все стереоизомеры формулы I и их фармацевтически приемлемые соли. Затем, смеси стереоизомеров, а также выделенные конкретные стереоизомеры являются также включенными. В процессе синтетических способов, применяемых для получения данных соединений, или при применении способов рацемизации или эпимеризации, известных специалистам в данной области техники, продукты данных способов могут представлять собой смеси стереоизомеров.
Термин "фармацевтически приемлемые соли" относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот. Когда соединение настоящего изобретения является кислым, его соответствующую соль можно удобно получить из фармацевтически приемлемых нетоксичных оснований, включая неорганические основания и органические основания. Соли, полученные из неорганических оснований, включают соли алюминия, аммония, кальция, меди (+1 и +2), железа (+3), железа (+2), лития, магния, марганца (+2 и +3), калия, натрия, цинка и подобные. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, а также циклических аминов и замещенных аминов, таких как природные и синтезированные замещенные амины. Другие фармацевтически приемлемые органические нетоксичные основания, из которых можно получить соли, включают ионообменные смолы, такие как, например, аргининовые, бетаиновые, кофеиновые, холиновые, N,N'-дибензилэтилендиаминовые, диэтиламиновые, 2-диэтиламиноэтаноловые, 2-диметиламиноэтаноловые, этаноламиновые, этилендиаминовые, N-этилморфолиновые, N-этилпиперидиновые, глюкаминовые, глюкозаминовые, гистидиновые, гидрабаминовые, изопропиламиновые, лизиновые, метилглюкаминовые, морфолиновые, пиперазиновые, пиперидиновые, полиаминовые смолы, прокаин, пурины, теобромин, тритиламин, триметиламин, трипропиламин, трометамин и подобные.
Когда соединение настоящего изобретения является основным, его соответствующую соль можно удобно получить из фармацевтически приемлемых нетоксичных кислот, включая неорганические и органические кислоты. Данные кислоты включают, например, уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глютаминовую, бромоводородную, хлороводородную, изэтиновую, молочную, малеиновую, оксиянтарную, миндальную, метансульфоновую, слизевую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, п-толуолсульфоновую кислоты и подобные. Примеры фармацевтически приемлемых солей включают, но не ограничиваются, минеральные или органические кислые соли основных остатков, таких как амины; щелочные или органические соли кислых остатков, таких как карбоновые кислоты; и подобные. Фармацевтически приемлемые соли включают общепринятые нетоксичные соли или четвертичные соли аммония родственных соединений, образованные, например, из нетоксичных неорганических или органических кислот. Например, данные общепринятые нетоксичные соли включают соли, полученные из неорганических кислот, таких как хлороводородная, бромоводородная, серная, сульфаминовая, фосфорная, азотная и подобные; и соли, полученные из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, оксиянтарная, винная, лимонная, аскорбиновая, памовая, малеиновая, гидроксималеиновая, фенилуксусная, глютаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изэтионовая кислоты и подобные.
Фармацевтически приемлемые соли настоящего изобретения можно получить общепринятыми химическими способами. Обычно, соли получают реакцией свободного основания или кислоты со стехиометрическими количествами или с избытком требуемой солеобразующей неорганической или органической кислотой или основанием, в подходящем растворителе или комбинации растворителей.
Соединения настоящего изобретения могут содержать асимметрические центры и находиться в виде рацематов, рацемических смесей и в виде отдельных диастереомеров. Все данные изомеры, включая оптические изомеры, включены в настоящее изобретение.
Настоящее изобретение, описанное в настоящем описании, также включает фармацевтическую композицию, которая состоит из соединения, описанного формулой (I), или его фармацевтически приемлемой соли, в комбинации с фармацевтически приемлемым носителем.
Настоящее изобретение, описанное в настоящем описании, также включает фармацевтическую композицию, которая состоит из соединения, описанного формулой (I), или его фармацевтически приемлемой соли, в комбинации с фармацевтически приемлемым носителем. Фармацевтические композиции настоящего изобретения содержат соединение, представленное формулой I (или его фармацевтически приемлемую соль), в качестве активного ингредиента, фармацевтически приемлемый носитель и необязательно другие терапевтические ингредиенты или вспомогательные вещества. Данные дополнительные терапевтические ингредиенты включают, например, i) антагонисты рецепторов лейкотриена, ii) ингибиторы биосинтеза лейкотриенов, iii) кортикостероиды, iv) антагонисты H1 рецептора, v) агонисты бета 2 адренорецептора, vi) COX-2 селективные ингибиторы, vii) статины, viii) нестероидные противовоспалительные лекарственные средства ("NSAID") и ix) M2/M3 антагонисты.
Настоящее изобретение, описанное в настоящем описании, также включает способ лечения артрита, который включает введение пациенту, являющемуся млекопитающим, нуждающемуся в данном лечении, соединения, описанного формулой (I), или его фармацевтически приемлемой соли, в количестве, которое является эффективным для лечения артрита. Настоящее изобретение, описанное в настоящем описании, также включает способ лечения артрита, который включает введение пациенту, являющемуся млекопитающим, нуждающемуся в данном лечении, соединения, описанного формулой (I), или его фармацевтически приемлемой соли, в количестве, которое является эффективным для лечения артрита. Настоящее изобретение включает способы лечения артрита введением пациенту, являющемуся млекопитающим, нуждающемуся в данном лечении, соединения, описанного формулой (I), или его фармацевтически приемлемой соли, в комбинации или одновременно вводимого с COX-2 ингибитором.
Настоящее изобретение, описанное в настоящем изобретении, также включает способ лечения заболевания, опосредованного цитокином, у млекопитающего, включающий введение пациенту, являющемуся млекопитающим, нуждающемуся в данном лечении, количества соединения, описанного формулой (I), или его фармацевтически приемлемой соли, в количестве, которое является эффективным для лечения упомянутого заболевания, опосредованного цитокином.
Особенно интересным является способ лечения воспаления у пациента, являющегося млекопитающим, нуждающегося в данном лечении, который состоит из введения упомянутому пациенту противовоспалительно эффективного количества соединения, описанного формулой (I), или его фармацевтически приемлемой соли.
Другой способ, который представляет особый интерес, представляет собой способ лечения заболевания, опосредованного цитокином, как описано в настоящем изобретении, где заболевание представляет собой остеопороз.
Другой способ, который представляет особый интерес, представляет собой способ лечения заболевания, опосредованного цитокином, как описано в настоящем изобретении, где заболевание представляет собой неостеопорозную резорбцию кости.
Еще другой способ, который представляет особый интерес, представляет собой способ лечения заболевания, опосредованного цитокином, как описано в настоящем изобретении, где заболевание представляет собой болезнь Крона.
Настоящее изобретение также относится к способу лечения артрита у нуждающегося в данном лечении млекопитающего, который включает введение упомянутому млекопитающему некоторого количества соединения формулы I, которое является эффективным для лечения артрита. Данный способ включает лечение ревматизма и остеоартрита.
При введении пациенту для лечения артрита, применяемая доза может изменяться в зависимости от типа артрита, возраста и общего состояния пациента, конкретного вводимого соединения, наличия или степени токсичности или неблагоприятных эффектов, проявляющихся с лекарственным средством, и других факторов. Типичным примером диапазона подходящих доз является диапазон от приблизительно 0,01 мг/кг до приблизительно 100 мг/кг. Однако вводимая доза остается на усмотрение врача.
Настоящее изобретение также относится к способу ингибирования действия p38 у нуждающегося в нем млекопитающего, который включает введение упомянутому млекопитающему эффективного количества соединения, описанного формулой (I), или его фармацевтически приемлемой соли, для ингибирования упомянутого действия p38, до нормального уровня или, в некоторых случаях, до почти нормального уровня, так чтобы улучшить, предотвратить или лечить болезненное состояние.
Соединения формулы I можно применять при профилактическом или терапевтическом лечении болезненных состояний у млекопитающих, которые обостряются или вызваны избыточным количеством цитокинов или нерегулируемыми цитокинами, более конкретно IL-I, IL-6, IL-8 или TNF.
Поскольку соединения формулы I ингибируют цитокины, такие как IL-I, IL-6, IL-8 и TNF, ингибированием действия p38, соединения являются пригодными для лечения заболеваний, при которых играет роль присутствие и активность цитокинов, таких как боль, ревматоидный артрит, ревматоидный спонделит, остеоартрит, подагрический артрит и другие артритические заболевания.
Соединения, описанные формулой I, или их фармацевтически приемлемая соль также являются полезными для лечения других болезненных состояний, опосредованных избыточными или нерегулируемыми TNF синтезом или активностью. Данные заболевания включают, но не ограничиваются, сепсис, септический шок, эндотоксический шок, грамотрицательный сепсис, синдром токсического шока, синдром расстройства дыхания у взрослых, церебральную малярию, хроническое легочное воспалительное заболевание, силикоз, легочный саркоидоз, заболевания, связанные с резорбцией кости, такие как остеопороз, реперфузионное повреждение, реакцию "трансплантат против хозяина", отторжение аллотрансплантата, лихорадку, миалгию в результате инфекции, кахексию, вторичную по отношению к инфекции или злокачественной опухоли, кахексию, вторичную к синдрому приобретенного иммунодефицита (СПИД), СПИД, ARC (СПИД-ассоциированный комплекс), келоидное образование, образование рубцовой ткани, болезнь Крона, язвенный колит, лихорадку, СПИД и другие вирусные инфекции, такие как цитомегаловирус (CMV), вирус гриппа и семейство вирусов герпеса, включая герпес зостер или простой I и II.
Соединения, описанные формулой (I), или их фармацевтически приемлемая соль также являются полезными для местного применения для лечения воспаления, такого как лечение ревматоидного артрита, ревматоидного спонделита, остеоартрита, подагрического артрита и других артритических заболеваний; воспаления суставов, экземы, псориаза или других воспалительных заболеваний кожи, таких как солнечные ожоги; воспалительных заболеваний глаз, включая конъюнктивит; лихорадки, боли и других заболеваний, связанных с воспалением.
Соединения, описанные формулой (I), или их фармацевтически приемлемая соль являются также пригодными для лечения заболеваний, таких как хроническое обструктивное заболевание легких и заболеваний, характеризующихся избыточной IL-8 активностью. Данные болезненные состояния включают псориаз, воспалительное заболевание кишечника, астму, сердечное или почечное реперфузионное повреждение, синдром расстройства дыхания у взрослых, тромбоз и гломерулонефрит.
Таким образом, настоящее изобретение включает способ лечения псориаза, воспалительного заболевания кишечника, астмы, сердечного или почечного реперфузионного повреждения, синдрома расстройства дыхания у взрослых, тромбоза и гломерулонефрита у млекопитающего, нуждающегося в данном лечении, который включает введение упомянутому млекопитающему соединения, описанного формулой (I), или его фармацевтически приемлемой соли, в количестве, которое является эффективным для лечения упомянутого заболевания или состояния.
Соединения, описанные формулой (I), или их фармацевтически приемлемая соль также являются пригодными для лечения болезни Альцгеймера. Таким образом, настоящее изобретение включает способ лечения болезни Альцгеймера у млекопитающего, нуждающегося в данном лечении, который включает введение упомянутому млекопитающему соединения формулы (I) или его фармацевтически приемлемой соли, в количестве, эффективном для лечения упомянутого заболевания или состояния.
При введении пациенту для лечения заболевания, при котором играет роль цитокин или цитокины, применяемая доза может изменяться в зависимости от типа заболевания, возраста и общего состояния пациента, конкретного вводимого соединения, наличия или степени токсичности или неблагоприятных эффектов, проявляющихся с лекарственным средством, и других факторов. Типичным примером подходящего диапазона доз является диапазон от приблизительно 0,01 мг/кг до приблизительно 100 мг/кг. Однако вводимая доза обычно остается на усмотрение врача.
Способы лечения можно осуществлять доставкой соединения формулы I парентерально. Термин "парентеральный", как применяют в настоящем изобретении, включает внутривенное, внутримышечное или внутрибрюшинное введение. Подкожная и внутримышечная формы парентерального введения являются обычно предпочтительными. Настоящее изобретение можно также осуществлять доставкой соединения формулы I подкожно, интраназально, внутриректально, трансдермально или внутривагинально.
Соединения формулы I можно также вводить ингаляцией. Под "ингаляцией" подразумевается интраназальное и пероральное ингаляционное введение. Подходящие лекарственные формы для данного введения, такие как аэрозольные составы или дозирующий ингалятор, можно получить общепринятыми способами.
Настоящее изобретение также относится к фармацевтической композиции, содержащей соединение формулы I и фармацевтически приемлемый носитель. Соединения формулы I можно также включать в фармацевтические композиции в комбинации со вторым терапевтически активным соединением.
Применяемый фармацевтический носитель может быть, например, твердый, жидкий или газообразный. Примеры твердых носителей включают лактозу, каолин, сахарозу, тальк, желатин, агар, пектин, камедь, стеарат магния, стеариновую кислоту и подобные. Примерами жидких носителей являются патока, арахисовое масло, оливковое масло, вода и подобные. Примеры газообразных носителей включают диоксид углерода и азот. Аналогично, носитель или разбавитель может содержать вещество, замедляющее высвобождение, хорошо известное в данной области техники, такое как моностеарат глицерина или дистеарат глицерина, отдельно или с воском.
Можно применять большой набор фармацевтических лекарственных форм. При применении твердой лекарственной формы для перорального введения, препарат может быть в форме таблетки, твердой желатиновой капсулы, пастилки или таблетки для рассасывания. Количество твердого носителя будет изменяться в широких пределах, но обычно будет находиться в диапазоне от приблизительно 0,025 мг до приблизительно 1 г. При необходимости в жидкой лекарственной форме для перорального введения, препарат обычно представляет собой сироп, эмульсию, мягкую желатиновую капсулу, суспензию или раствор. Когда предполагается применять парентеральную лекарственную форму, лекарственное средство может быть в твердой или жидкой форме и его можно формулировать для введения непосредственно или оно может быть подходящим для растворения.
Также включены лекарственные формы для местного применения. Примерами лекарственных форм для местного применения являются твердые, жидкие и полутвердые формы. Твердые формы могут включать присыпки, примочки и подобные. Жидкие включают растворы, суспензии и эмульсии. Полутвердые включают крема, мази, гели и подобные.
Количество применяемого соединения формулы I обычно будет, конечно, изменяться в зависимости от выбранного соединения, природы и тяжести заболевания, и оно может изменяться на усмотрение врача. Типичная доза для местного применения соединения формулы I составляет от приблизительно 0,01 мг до приблизительно 2,0 г, вводимая от одного до четырех или, предпочтительно, от одного до двух раз в день.
Активный ингредиент может составлять, для местного введения, от приблизительно 0,001% до приблизительно 10% в/в.
Капли согласно настоящему изобретению могут содержать стерильные или нестерильные водные или масляные растворы или суспензии, и их можно получить растворением активного ингредиента в подходящем водном растворе, необязательно содержащем бактерицидное и/или фунгицидное средство и/или любой другой подходящий консервант, и необязательно содержать поверхностно-активный агент. Затем, полученный в результате раствор можно процеживать фильтрацией, переносить в подходящую емкость, которую затем герметично закрывают и стерилизуют автоклавированием или выдерживанием при 98-100°C в течение 30 минут. Альтернативно, раствор можно стерилизовать фильтрацией и переносить в емкость в стерильных условиях. Примерами бактерицидных и фунгицидных агентов, подходящих для включения в капли, являются нитрат или ацетат фенилртути (0,002%), хлорид бензалкония (0,01%) и ацетат хлоргексидина (0,01%). Подходящие растворители для получения масляного раствора включают глицерин, разбавленный спирт и пропиленгликоль.
Лосьоны согласно настоящему изобретению включают лосьоны, подходящие для применения на коже или глазах. Лосьон для глаз может содержать стерильный водный раствор, необязательно содержащий бактерицидный агент, и его можно получить способами, аналогичными способам получения капель. Лосьоны или линименты для применения на коже могут также содержать агент для ускорения сушки и охлаждения кожи, такие как спирт или ацетон, и/или увлажняющий агент, такой как глицерин или масло, такое как касторовое масло или арахисовое масло.
Крема, мази или пасты согласно настоящему изобретению являются полутвердыми составами активного ингредиента для внешнего применения. Их можно получить смешением активного ингредиента в мелкоизмельченной или порошковой форме, отдельно или в растворе или суспензии в водной или неводной жидкости, с маслянистой или немаслянистой основой. Основа может содержать углеводороды, такие как твердый, мягкий или жидкий парафин, глицерин, пчелиный воск, металлосодержащее мыло; гуммиарабик; масло природного происхождения, такое как миндальное, кукурузное, арахисовое, касторовое или оливковое масло; шерстяной жир или его производные, или жирную кислоту, такую как стеариновая или олеиновая кислота, вместе со спиртом, таким как пропиленгликоль или макрогели. Состав может содержать любой подходящий поверхностно-активный агент, такой как анионное, катионное или неионное поверхностно-активное вещество, такое как эфиры сорбитана или его полиоксиэтиленовые производные. Можно также включать суспендирующие агенты, такие как натуральная камедь, производные целлюлозы или неорганические вещества, такие как диоксид кремния, и другие ингредиенты, такие как ланолин.
Что касается составов для ингаляции, количество на одно введение обычно является меньшим, чем количество для перорального состава, такого как таблетка или капсула. Например, дневная доза активного соединения, вводимая посредством состава для ингаляции, может быть в диапазоне от 0,010 мг до 10 мг, в частности от 0,010 мг до 2,5 мг. Можно применять в день единичную дозу или несколько доз для ингаляции, но единичная доза для ингаляции является предпочтительной.
Что касается введения ингаляцией, соли соединений формулы I настоящего изобретения удобно доставлять в форме аэрозоля, подходящего для доставки лекарственного средства в легкие. Данные аэрозольные лекарственные формы включают, но не ограничиваются, распыляемые растворы и суспензии, дозирующие ингаляторы или порошковые ингаляторы. Для распыления активный ингредиент (ингредиенты) обычно формулируют в водной среде и вводят струйным или электронным устройством, способным генерировать взвесь мелкодисперсного аэрозоля. В дозирующих ингаляторах (MDI) применяют пропелленты, такие как гидрофторуглероды для растворения или суспендирования активного ингредиента в емкости, находящейся под давлением, способной генерировать дисперсный аэрозоль. Что касается порошковых ингаляторов, соли соединений формулы I применяют отдельно или со вспомогательными веществами в сочетании с устройством для доставки, способным доставлять активное вещество в легкое.
В одном варианте осуществления лекарственный препарат приспособлен для применения с дозирующим ингалятором под давлением, который высвобождает отмеренную дозу лекарственного средства при каждом нажатии. Состав для pMDI может быть в форме растворов или суспензий в галогенированном углеводородном пропелленте. Тип пропеллента, который применяют в pMDI, заменяется на гидрофторалканы (HFA), также известные как гидрофторуглероды (HFC), поскольку отказались от применения хлорфторуглеродов (известных также как фреоны или CFC). В частности, 1,1,1,2-тетрафторэтан (HFA 134a) и 1,1,1,2,3,3,3-гептафторпропан (HFA 227) применяют в нескольких продаваемых в настоящее время на рынке фармацевтических продуктах для ингаляции. Композиция может содержать другие фармацевтически приемлемые вспомогательные вещества для ингаляционного применения, такие как этанол, олеиновая кислота, поливинилпирролидон и подобные.
MDI под давлением обычно содержат два компонента. Во-первых, есть емкостной компонент, в котором частицы лекарственного средства хранятся под давлением в виде суспензии или раствора. Во-вторых, есть компонент, являющийся держателем, применяемый для удерживания и приведения в действие емкости. Обычно, емкость будет содержать большое количество доз состава, хотя также возможны емкости с одной дозой. Емкостной компонент обычно содержит выпускной клапан, из которого можно выпускать содержимое емкости. Аэрозольное лекарственное средство высвобождают из pMDI применением силы на емкостной компонент для того, чтобы задвинуть его в компонент, являющийся держателем, посредством чего открывается выпускной клапан, и это вызывает перекачивание частиц лекарственного средства из выпускного клапана через компонент, являющийся держателем, и высвобождение из выходного отверстия держателя. После высвобождения из емкости, частицы лекарственного средства распыляются, образуя аэрозоль. Предполагается, что пациент располагает поток аэрозольного лекарственного средства по отношению к его или ее лицу так, что частицы лекарственного средства попадают в дыхательный поток пациента и доставляются в легкие. Обычно, в pMDI применяют пропелленты для создания давления на содержимое емкости и для движения частиц лекарственного средства из выпускного отверстия компонента, являющегося держателем. В pMDI, состав предоставляют в виде жидкости или суспензии, и он находится в емкости вместе с пропеллентом. Пропеллент может принимать множество форм. Например, пропеллент может содержать сдавленный газ или сжиженный газ.
В другом варианте осуществления лекарственный препарат приспосабливают для применения с порошковым ингалятором. Композиция для ингаляции, подходящая для применения в DPI, обычно содержит частицы активного ингредиента и частицы фармацевтически приемлемого носителя. Размер частиц активного вещества может изменяться от приблизительно 0,1 мкм до приблизительно 10 мкм; однако для эффективной доставки в дистальное легкое, по меньшей мере, 95 процентов частиц активных агентов имеют размер 5 мкм или меньше. Каждый активный компонент может присутствовать в концентрации 0,01-99%. Однако обычно, каждый из активных агентов присутствует в концентрации приблизительно 0,05-50%, более обычно приблизительно 0,2-20% от суммарного веса композиции.
Как отмечалось выше, в добавление к активным ингредиентам порошок для ингаляции предпочтительно содержит фармацевтически приемлемый носитель, который может состоять из фармакологически инертного вещества или комбинации веществ, которые являются приемлемыми для ингаляции. Предпочтительно, чтобы частицы носителя состояли из одного или более кристаллических сахаров; частицы носителя могут состоять из одного или более сахарных спиртов или полиолов. Предпочтительно, чтобы частицы носителя представляли собой частицы декстрозы или лактозы, особенно лактозы. В вариантах осуществления настоящего изобретения, в которых применяют общепринятые порошковые ингаляторы, такие как Rotohaler, Diskhaler и Turbohaler, размер частицы носителя может находится в диапазоне от приблизительно 10 микрон до приблизительно 1000 микрон. В некоторых из данных вариантов осуществления размер частиц носителя может находиться в диапазоне от приблизительно 20 микрон до приблизительно 120 микрон. В некоторых других вариантах осуществления размер, по меньшей мере, 90% по весу частиц носителя является меньшим чем 1000 микрон и предпочтительно лежит в диапазоне от 60 микрон до 1000 микрон. Относительно большой размер данных частиц носителя дает хорошие характеристики течения и увлечения. При наличии, количество частицы носителя будет обычно составлять вплоть до 95%, например вплоть до 90%, предпочтительно вплоть до 80% и предпочтительно вплоть до 50% по весу относительно суммарного веса порошка. Количество любого мелкого материала наполнителя, при наличии, может составлять вплоть до 50% и предпочтительно вплоть до 30%, особенно вплоть до 20% по весу относительно суммарного веса порошка.
Настоящее изобретение в одном варианте осуществления обеспечивает композицией для применения в порошковом ингаляторе, который содержит монтелукастную кислоту и соединение формулы I и лактозу для ингаляции в качестве носителя, где упомянутую композицию приспосабливают для одновременного, последовательного или раздельного введения активных агентов. Весовое отношение лактозы к монтелукастной кислоте составляет от приблизительно 1:1 до приблизительно 30:1 и к соединению X составляет от приблизительно 20:1 до приблизительно 30:1. В одном примере весовое отношение лактозы к монтелукастной кислоте составляет приблизительно от 2:1 до приблизительно 25:1 и к соединению формулы I составляет от приблизительно 20:1 до приблизительно 25:1.
Настоящее изобретение в одном варианте осуществления обеспечивает композицией для применения в порошковом ингаляторе, которая содержит монтелукастную кислоту и кортикостероид для ингаляции и лактозу для ингаляции в качестве носителя, где упомянутую композицию приспосабливают для одновременного, последовательного или раздельного введения активных агентов. В данных композициях весовое отношение лактозы к монтелукастной кислоте обычно составляет от приблизительно 1:1 до приблизительно 30:1. В одной композиции, где кортикостероид для ингаляции представляет собой фуроат мометазона, весовое отношение лактозы к фуроату мометазона составляет от приблизительно 130:1 до приблизительно 4:1, и в одном варианте осуществления отношение составляет от приблизительно 124:1 до приблизительно 60:1. В композиции, где кортикостероидом для ингаляции является циклесонид, весовое отношение лактозы к циклесониду составляет от приблизительно 350:1 до приблизительно 100:1.
Порошок может также содержать мелкоизмельченные частицы вещества наполнителя, который может представлять собой, например, вещество, такое как одно из веществ, упомянутых выше в качестве подходящих для применения в качестве вещества носителя, особенно кристаллический сахар, такой как декстроза или лактоза. Мелкоизмельченное вещество наполнителя может представлять собой то же или отличное от частиц носителя вещество, при наличии обоих. Размер частиц мелкоизмельченного вещества наполнителя будет обычно не превышать 30 мкм, и предпочтительно не превышать 20 мкм. В некоторых обстоятельствах, например когда наличие частиц носителя и/или любого мелкоизмельченного вещества наполнителя само способно вызывать ощущение в орофарингеальной области, частицы носителя и/или мелкоизмельченного вещества наполнителя могут образовывать индикаторное вещество. Например, частицы носителя и/или любого мелкоизмельченного наполнителя в виде частиц могут содержать маннитол.
Составы, описанные в настоящем изобретении, могут также содержать одну или более добавок, в количестве от приблизительно 0,1% до приблизительно 10% по весу, и предпочтительно от приблизительно 0,15% до 5%, самое предпочтительное от приблизительно 0,5% до приблизительно 2%. Добавки могут включать, например, стеарат магния, лейцин, лецитин и стеарилфумарат натрия. Когда добавка представляет собой микронизированный лейцин или лецитин, предпочтительно обеспечивать ее в количестве от приблизительно 0,1% до приблизительно 10% по весу, предпочтительно от приблизительно 0,5% до приблизительно 5%, предпочтительно приблизительно 2% микронизированного лейцина. Предпочтительно, чтобы, по меньшей мере, 95% по весу микронизированного лейцина имело диаметр частиц меньше чем 150 микрон, предпочтительно меньше чем 100 микрон, и самое предпочтительное меньше чем 50 микрон. Предпочтительно, чтобы масс-медианный диаметр микронизированного лейцина был меньшим чем 10 микрон.
При применении в качестве добавки стеарата магния или стеарилфумарата натрия, предпочтительно обеспечивать их в количестве от приблизительно 0,05% до приблизительно 5%, предпочтительно от приблизительно 0,15% до приблизительно 2%, самое предпочтительное от приблизительно 0,25 до приблизительно 0,5%.
При ссылке на размер частиц порошка ясно, что если не указано обратное, размер частиц представляет собой средневзвешенный размер частиц. Размер частиц можно рассчитать способом лазерной дифракции. Когда частицы также содержат индикаторное вещество на поверхности частицы, предпочтительно, чтобы размер покрытых частиц также был в пределах предпочтительных диапазонов размеров, приведенных для непокрытых частиц.
Фармацевтические композиции в виде сухого порошка согласно настоящему изобретению можно получить, применяя стандартные способы. Фармацевтически активные агенты, частицы носителя и другие вспомогательные вещества, если они вообще есть, можно тщательно смешивать, применяя любой подходящий аппарат для смешивания, такой как барабанный смеситель. Отдельные компоненты состава можно смешивать в любом порядке. Можно обнаружить, что предварительное смешение конкретных компонентов является предпочтительным в определенных обстоятельствах. Затем, порошковую смесь применяют для заполнения капсул, блистеров, емкостей или других средств для хранения для применения в сочетании с порошковыми ингаляторами.
В порошковом ингаляторе дозу, которую будут вводить, хранят в форме сухого порошка, не находящегося под давлением, и, при нажатии на ингалятор, частицы порошка вдыхаются пациентом. DPI могут представлять собой устройства с одной дозой, в которых порошок содержится в отдельной капсуле, комбинированной дозой, в котором применяют большое количество капсул или блистеров, и устройство в виде емкости, в котором порошок отмеряется в момент дозирования из емкости для хранения. Порошковые ингаляторы могут представлять собой "пассивные" устройства, в которых дыхание пациента применяют для диспергирования порошка для доставки в легкие, или "активные" устройства, в которых для диспергирования порошка применяют механизм, отличный от приведения в действие с помощью дыхания. Примеры "пассивных" порошковых ингаляционных устройств включают Spinhaler, Handihaler, Rotahaler, Diskhaler, Diskus, Turbuhaler, Clickhaler и т.д. Примеры активных ингаляторов включают Nektar Pulmonary Inhaler (Nektar Therapeutics), Vectura Limited's Aspirair™ устройство, Microdose DPI (MicroDose) и Oriel DPI (Oriel). Однако должно быть ясно, что композиции настоящего изобретения можно вводить пассивными или активными ингаляционными устройствами.
Анализы
Экспрессия и очистка белка
Мышиный p38, содержащий FLAG эпитопную метку, экспрессировали в клетках Drosophila S2 при транскрипционном контроле индуцируемого медью металлотионеинового промотора. Экспрессию рекомбинантного p38 вызывали обработкой трансфецированных клеток 1 мМ CuSO4 в течение 4 часов. Для получения активного рекомбинантного мышиного p38, обработанные CuSO4 S2 клетки стимулировали за 10 минут до сбора 400 мМ NaCl, 2 мМ Na3VO4 и 100 мкг/л окадаиковой кислоты. Дебрис промывали фосфатно-солевым буферным раствором, 2 мМ Na3VO4, и лизировали в 20 мМ Tris HCl, pH 7,5, 120 мМ NaCl, 1% Triton X-100, 2 мМ EDTA, 20 мМ NaF, 4 мМ Na3VO4, 2 мМ Prefabloc SC (Boehringer Mannheim). Клеточные лизаты центрифугировали в течение 10 минут при 13000×g, и активированный рекомбинантный мышиный p38 очищали имунноафинной колоночной хроматографией из лизата с помощью анти-FLAG M2 смолы (Kodak), которая была сбалансирована с буфером для лизиса. После загрузки экстракта смолу промывали 10 объемами колонки буфера для лизиса, 10 объемами колонки буфера A (10 мМ Tris HCl, pH 7,5, 500 мМ NaCl, 20% глицерин) и 10 объемами колонки буфера B (10 мМ Tris HCl pH 7,5, 150 мМ NaCl, 20% глицерин). Химерный белок элюировали буфером B, содержащим 100 мкг/мл FLAG пептида (Kodak).
N-концевыве 115 аминокислот ATF-2 экспрессировали в E. coli в виде химерного белка с глютатион-S-трансферазой. Химерный белок очищали с помощью глютатионагарозы согласно стандартным способам (Pharmacia).
p38 киназный анализ
p38 киназный анализ проводили в реакционном объеме 100 мкл в 96-луночном планшете при 30° в течение 45-1200 минут при следующих условиях: 25 мМ Hepes, pH 7,4, 10 мМ MgCl2, 20 мМ β-глицеринфосфат, 2 мМ DTT, 5 мкм ATP, 10 мк Сi [γ-33P]-ATP и ~2 мкМ GST-ATF2. Последовательное разбавление соединений добавляли к каждой реакции в 2 мкл DMSO. 2 мкл DMSO добавляли к последнему ряду каждого планшета для реакции в качестве контроля, не содержащего ингибитора, для каждого титрования на ингибитор. Реакцию прекращали равным объемом стоп-раствора, содержащего 100 мМ EDTA и 15 мМ пирофосфата натрия. PVDF фильтровальные пластинки (MAIPNOB50, Millipore) предварительно смачивали метанолом и промывали стоп-раствором. 50 мкл аликвоты из одной реакционной смеси наносили на фильтр под вакуумом и фильтр промывали дважды 75 мМ фосфорной кислоты. Фильтровальные пластинки считали в сцинцилляционном счетчике (Top Count, Packard) и определяли ингибирование в процентах для каждой концентрации соединения.
Альтернативно, p38 киназные анализы проводили в реакционном объеме 70 мкл в 384-луночном планшете при 30° в течение 45-1220 минут в следующих условиях: 50 мМ Hepes, pH 7,4, 10 мМ MgCl2, 1 мг/мл FA Free BSA, 1 мМ DTT, 10 мкм ATP, 10 мкм p38 пептида [Caliper Life Sciences FL-пептид 8 (5-FAM-IPTSPITTTYFFFKKK-COOH)] и 5,7 нм p38-α (Millipore) или 14,3 нМ инактивированной MAPKAP киназы-2, 0,18 нМ p38-α (Millipore) и 2 мкМ RSK пептида [Caliper Life Sciences FL-пептид 11 (5-FAM-KKLNRTLSVA-COOH)]. Последовательное разбавление соединений добавляли к каждой реакции в 700 нл DMSO. 700 нл DMSO добавляли к контрольным лункам планшета для реакции в качестве контроля, не содержащего ингибитора, для каждого титрования на ингибитор. Реакцию прекращали добавлением 15 мкл 100 мМ EDTA. Образование продукта анализировали, применяя Caliper LabChip 3000. Буфер для разделения содержал 100 мМ HEPES pH 7,5, 0,015% Brij-35, 2,5% Coating Reagent #3 (Caliper Life Sciences) и 10 мМ EDTA. Расчеты отношений продукта к субстрату проводили, применяя программное обеспечение HTS Well Analyzer, предоставленное Caliper Life Sciences, и определяли ингибирование в процентах для каждой концентрации соединения.
Анализ высвобождения TNF-α
Кровь получали у здоровых добровольцев венепункцией, применяя гепарин натрия в качестве антикоагулянта. Мононуклеары периферической крови (PBMC) выделяли, применяя среду для фракционирования лимфоцитов (ICN) согласно инструкции производителя. Выделенные PBMC промывали 3 раза HBSS и разбавляли до плотности 2×106 клетки/мл в RPMI + 5% аутологичной человеческой плазмы. 50 мкл последовательного разбавления ингибитора добавляли к лункам 96-луночного планшета для выращивания тканей, с последующим добавлением 100 мкл PBMC, и затем 50 мкл RPMI полной среды, содержащей 400 нг/мл LPS. Контрольную лунку клеток без соединения, но с LPS (контроль с максимальным стимулированием) и лунку без соединения и без LPS (контроль фона) включали в каждое титрование. Клетки выдерживали в течение 16 часов во влажной камере при 37°C, 5% CO2. Затем кондиционированную среду собирали и TNF-α концентрацию определяли количественно имунноанализом, применяя реагенты, имеющиеся в продаже (R&D, Inc).
Соединения настоящего изобретения и, в частности, примеров показали эффективность (IC50) в вышеуказанных анализах с результатами меньше чем 10 мкм. Предпочтительные соединения имели результаты меньше чем 1 мкм. Даже более предпочтительные соединения имели результаты меньше чем 0,1 мкМ. Еще более предпочтительные соединения имели результаты в анализах меньше чем 0,01 мкм. Далее следует иллюстрация эффективности, продемонстрированная конкретными примерами:
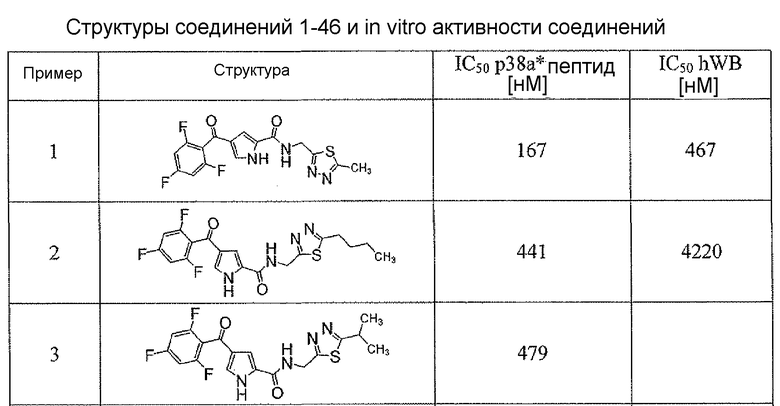
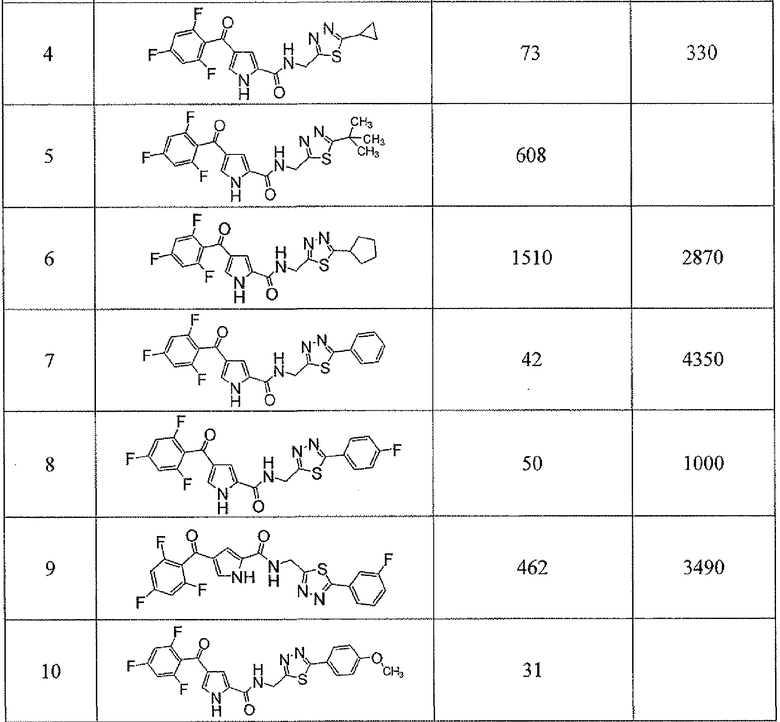
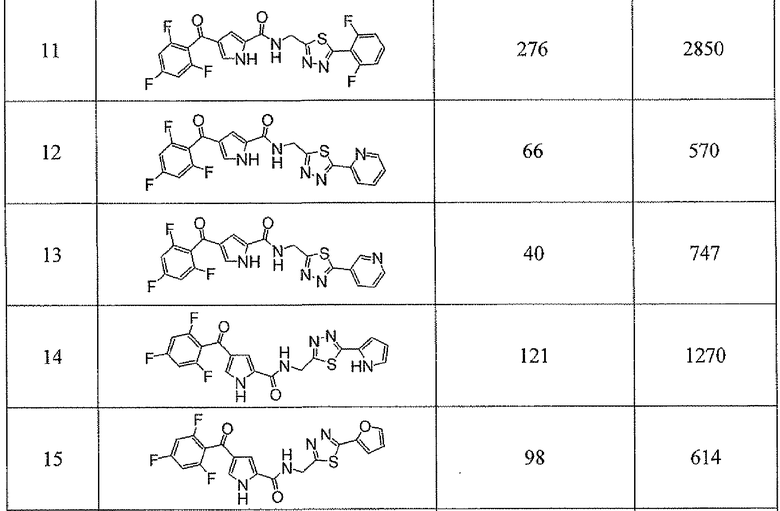
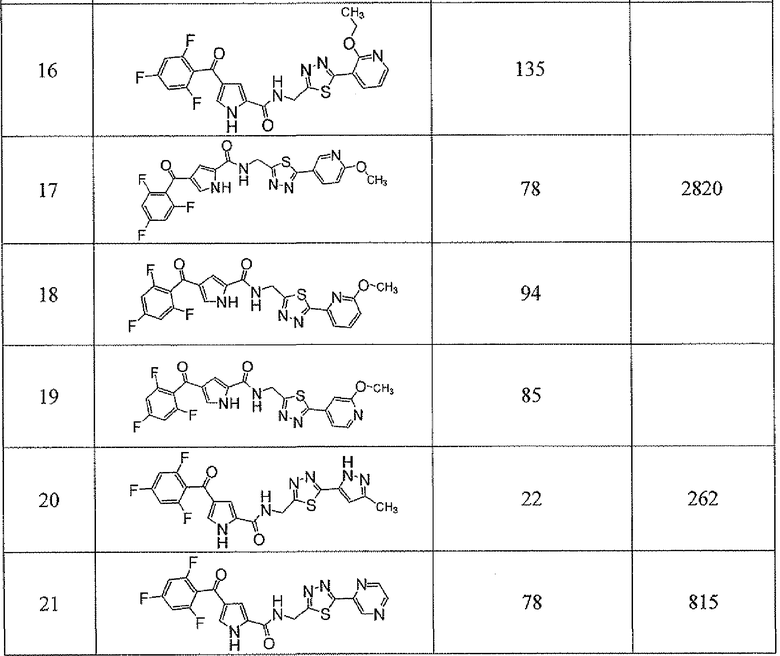
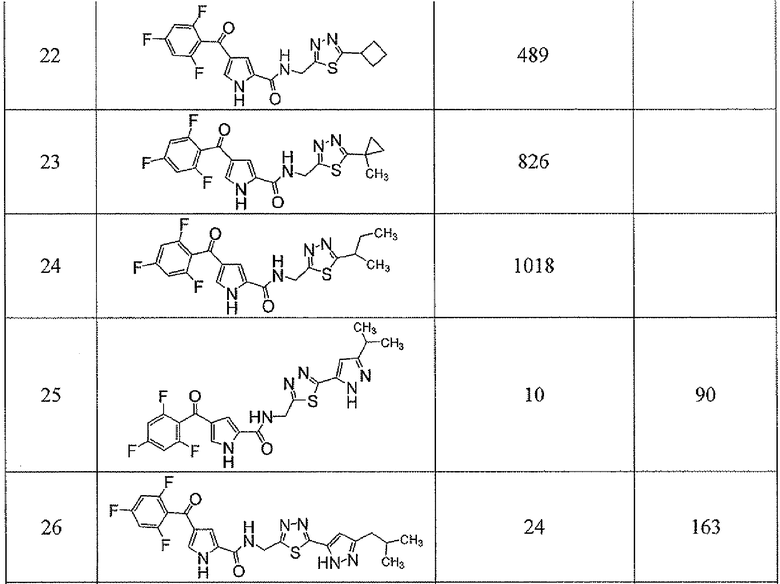
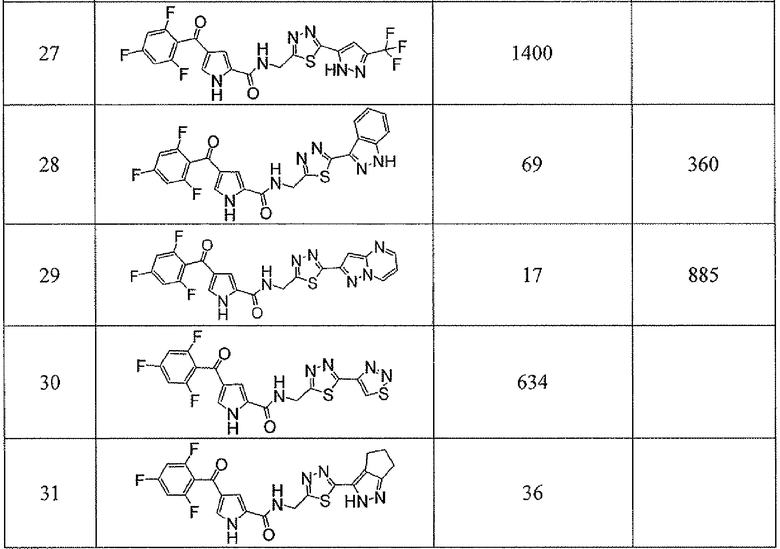
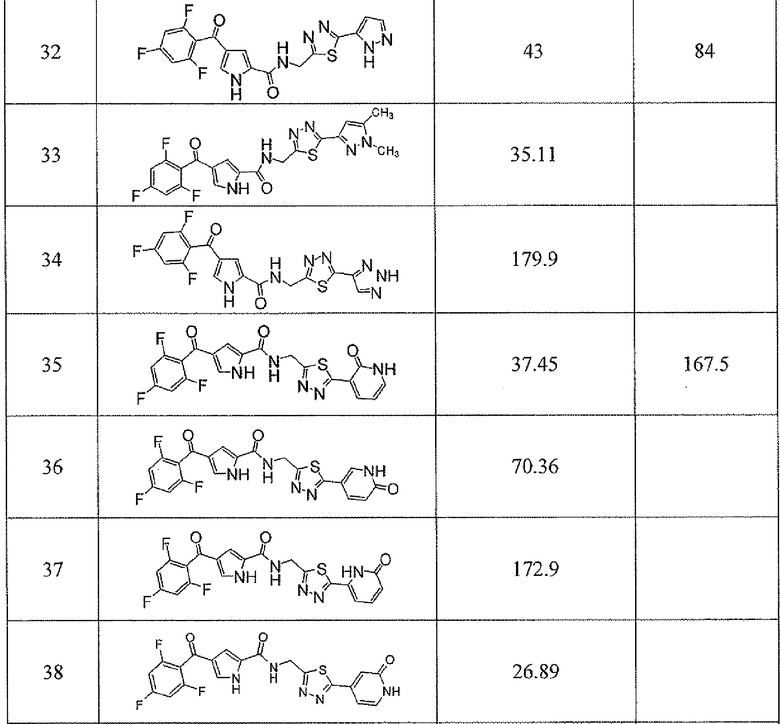
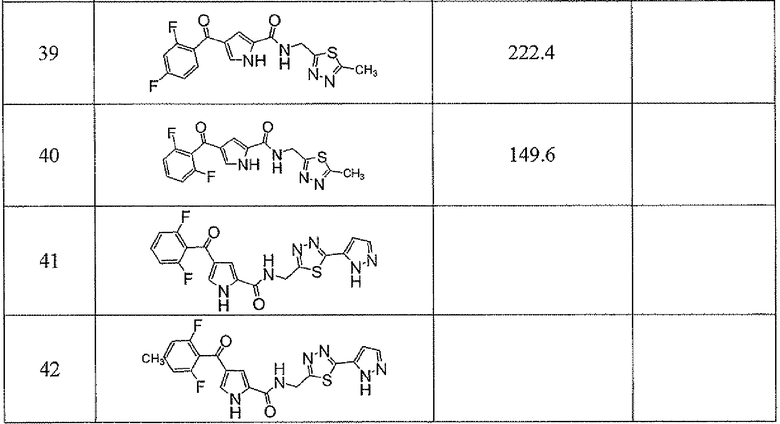
Сокращения, применяемые в настоящем изобретении, являются следующими, если не указано особо:

Настоящие соединения можно получить согласно общим схемам, показанным ниже, а также способам, представленным в промежуточных соединениях и примерах. Следующие схемы, примеры и промежуточные соединения дополнительно описывают, но не ограничивают, объем настоящего изобретения. Заместители являются такими же, как в вышеуказанных формулах, за исключением случаев, где определено обратное, или иначе ясно специалистам.
Способы, описанные в настоящем изобретении для получения соединений, могут включать одну или более стадий манипуляции с защитными группами и очистки, такой как перекристаллизация, перегонка, колоночная хроматография, флэш-хроматография, тонкослойная хроматография (TLC), радиальная хроматография и хроматография высокого давления (ВЭЖХ). Продукты можно охарактеризовать, применяя различные способы, хорошо известные в химической области техники, включая протонный и углерод-13 ядерный магнитный резонанс (1H и 13C ЯМР), ИК- и УФ-спектроскопию (ИК и УФ), рентгеновскую кристаллографию, элементный анализ и ВЭЖХ и масс-спектрометрию (LC-MS). Способы манипуляций с защитными группами, очистки, определения структуры и определения величин являются известными специалистам в области химического синтеза.
Ясно, что можно дополнительно проводить манипуляции с функциональными группами, присутствующими в соединениях, описанных на схемах ниже, когда это целесообразно, применяя стандартные способы превращения функциональных групп, известные специалистам в данной области техники, для получения требуемых соединений, описанных в настоящем изобретении.
Другие варианты или модификации, которые будут ясны специалистам в данной области техники, включены в объем и идеи настоящего изобретения. Настоящее изобретение не должно ограничиваться, за исключением того, что изложено в формуле изобретения.
Настоящие соединения можно получить согласно общим схемам, приведенным ниже, а также способам, приведенным в промежуточных соединениях и примерах. Следующие схемы, примеры и промежуточные соединения дополнительно описывают, но не ограничивают, объем настоящего изобретения. Заместители являются такими же, как в вышеуказанных формулах, за исключением случаев, где определено обратное или иначе ясно специалистам.
Способы, описанные в настоящем изобретении для получения соединений, могут включать одну или более стадий с защитными группами и очистки, такой как перекристаллизация, перегонка, колоночная хроматография, флэш-хроматография, тонкослойная хроматография (TLC), радиальная хроматография и хроматография высокого давления (ВЭЖХ). Продукты можно охарактеризовать, применяя различные способы, хорошо известные в химической области техники, включая протонный и углерод-13 ядерный магнитный резонанс (1H и 13C ЯМР), ИК- и УФ-спектроскопию (ИК и УФ), рентгеновскую кристаллографию, элементный анализ и ВЭЖХ и масс-спектрометрию (LC-MS). Способы манипуляций с защитными группами, очистки, определения структуры и определения величин являются известными специалистам в области химического синтеза.
Ясно, что можно дополнительно проводить манипуляции с функциональными группами, присутствующими в соединениях, описанных на схемах ниже, когда это целесообразно, применяя стандартные способы превращения функциональных групп, известные специалистам в данной области техники, для получения требуемых соединений, описанных в настоящем изобретении.
Другие варианты или модификации, которые будут ясны специалистам в данной области техники, включены в объем и идеи настоящего изобретения. Настоящее изобретение не должно ограничиваться, за исключением того, что изложено в формуле изобретения.
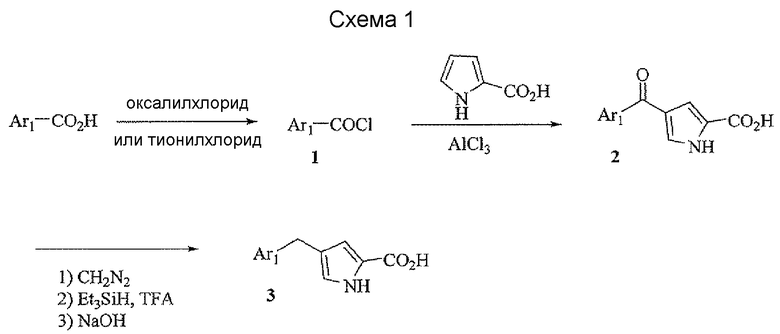
Соединения формулы I можно получить, как описано на схеме 1, 2 и 3. Подходящий хлорангидрид кислоты 1 можно получить способом, известным специалистам в данной области техники, из соответствующей кислоты или имеющегося в продаже соединения. Соединение 2 можно легко получить из соединения 1 любым из нескольких известных способов, таким как ацилирование по Фриделю-Крафтсу пиррол-2-кабоновой кислотой его эфирных производных. Соединение 3 можно получить восстановлением кетона триэтилсиланом в TFA.
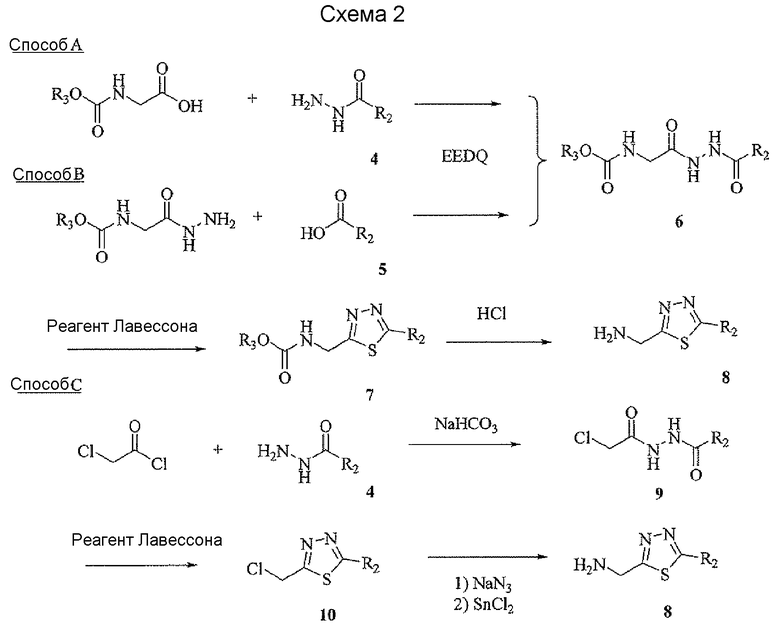
Соединение 6 можно получить из N-карбамоилглицина и подходящего карбогидразида 4 (способ A) или гидразида N-карбамоилглицина и карбоновой кислоты 5 (способ B), применяя реагент для образования амидной связи, такой как EEDQ. Обработка соединения 6 реагентом Лавессона дает тиадиазольное соединение 7. Деблокирование карбамоильной группы соединения 7 дает соединение 8. Альтернативно, соединение 8 можно получить, как описано в способе C. Таким образом, подходящим образом замещенный карбогидразид 4 ацилировали хлорацетилхлоридом, применяя основание, такое как бикарбонат натрия, для получения соединения 9. Обработка соединения 9 реагентом Лавессона дает соединение 10. Атом хлора соединения 10 замещают азидной группой, и последующее восстановление азидной группы дает соединение 8.
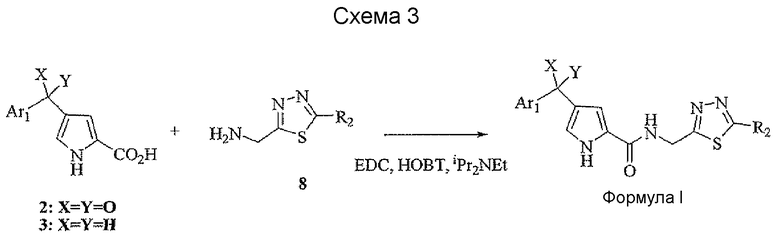
В стандартных условиях реакции пептидной конденсации кислоту 2 или 3 и амин 8 можно превратить в соединение формулы I. Стандартные условия реакции пептидной конденсации обозначают конденсацию карбоновой кислоты с амином, применяя реагент, активирующий кислоту, такой как EDC, DCC или BOP, в подходящем растворителе, таком как хлористый метилен или DMF, в присутствии HOBt.
Промежуточное соединение 1
4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоновая кислота
Стадия A: 2,4,6-трифторбензоилхлорид
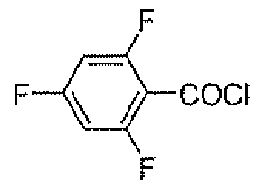
К DCM 200 мл раствору 2,4,6-трифторбензойной кислоты (20 г, 0,11 моль) и DMF (0,5 мл, 6,46 ммоль) добавляли по каплям оксалилхлорид (21,6 г, 0,17 моль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и растворитель удаляли при пониженном давлении, для того чтобы получить заявленное в заголовке соединение в виде неочищенного продукта (22 г).
Стадия B: 4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоновая кислота
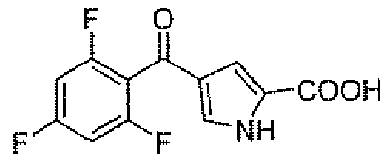
К 120 мл дихлорметановому раствору 2,4,6-трифторбензоилхлорида (4,3 г, 0,022 моль) добавляли AlCl3 (8,8 г, 0,066 моль) в атмосфере N2 при комнатной температуре. После перемешивания в течение 15 минут добавляли маленькими порциями 1H-пиррол-2-карбоновую кислоту (2,4 г, 0,022 моль) в течение 10-минутного периода.
Перемешивание продолжали при комнатной температуре в течение 1 часа, затем реакционную смесь обрабатывали добавлением по каплям ледяной воды (20 мл) и 1N HCl для доведения pΗ до 1. После перемешивания в течение дополнительных 30 минут, реакционную смесь экстрагировали AcOEt (3×30 мл). Объединенные органические слои промывали соляным раствором, сушили над безводным Na2SO4, фильтровали и концентрировали, для того чтобы получить заявленное в заголовке соединение (5,8 г, 97% выход). 1H-ЯМР (500 MГц, CDCl3): δ 12,48 (шир.с, 1Н), 7,48 (с, 1Н), 7,28-7,38 (м, 2H), 6,83 (с, 1Н).
Следующие промежуточные соединения получали, следуя способу для промежуточного соединения 1, стадии A и B, применяя подходящим образом замещенные карбоновые кислоты вместо 2,4,6-трифторбензойной кислоты.
Промежуточное соединение 2: 4-(2,6-дифторбензоил)-1H-пиррол-2-карбоновая кислота
Промежуточное соединение 3: 4-(2,4-дифторбензоил)-1H-пиррол-2-карбоновая кислота
Промежуточное соединение 4
4-(2,6-дифтор-4-метилбензоил)-1H-пиррол-2-карбоновая кислота
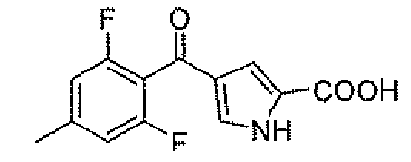
Стадия A: 1,3-дифтор-5-метилбензол
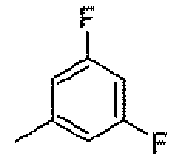
Смесь 1-(бромметил)-3,5-дифторбензола (50 г, 0,24 моль), 10% Pd/C (3 г) и ацетата натрия (140 г, 1,7 моль) в безводном эфире (250 мл) перемешивали в атмосфере водорода при атмосферном давлении в течение 24 часов. Смесь фильтровали и фильтрат сушили над безводным Na2SO4, фильтровали и затем применяли непосредственно в следующей стадии.
1H-ЯМР (500 MГц, CDCl3): δ 6,56 (д, 2H, J=6,0 Гц), 6,47 (т, 1Н, J=9,0 Гц), 2,22 (с, 3H).
Стадия B: 2,6-дифтор-4-метилбензальдегид
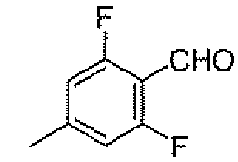
К раствору 1,3-дифтор-5-метилбензола (10,2 г, 80 ммоль) в безводном эфире (80 мл) добавляли н-BuLi (2,5 M раствор в гексане, 48 мл, 120 ммоль) в течение 20 минут, при этом поддерживали внутреннюю температуру в районе -50°C. После перемешивания при данной температуре в течение 1,5 часов добавляли DMF (14,6 г, 200 ммоль) в течение 20 минут. После перемешивания при той же самой температуре в течение дополнительных 1,5 часов, реакционную смесь медленно выливали в 1N водную серную кислоту (300 мл) и экстрагировали эфиром три раза. Объединенные органические слои промывали соляным раствором, сушили над безводным MgSO4, фильтровали и концентрировали, для того чтобы получить заявленное в заголовке соединение (11,2 г, 90%).
1H-ЯМР (500 MГц, CDCl3): δ 10,25 (с, 1H), 6,75 (д, 2H, J=9,9 Гц), 2,39 (с, 3H).
Стадия C: 2,6-дифтор-4-метилбензойная кислота
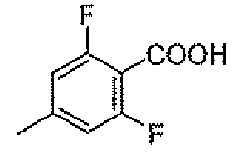
Оксид серебра (43,8 г, 0,189 моль) помещали в колбу вместе с водой (200 мл) и гидроксидом натрия (33,7 г, 0,842 моль). В нее добавляли маленькими порциями 2,6-дифтор-4-метилбензальдегид (29,23 г, 0,187 моль) в течение 30 минут. После энергичной экзотермической реакции цвет реакционной смеси изменялся с черного на серый. Полученную в результате густую суспензию перемешивали в течение 1 часа и затем фильтровали через воронку Бюхнера. Фильтрат подкисляли до pH 2 концентрированной HCl, для того чтобы получить суспензию. Осадок собирали фильтрованием с отсасыванием, растворяли в эфире, сушили над безводным Na2SO4, фильтровали и концентрировали, для того чтобы получить белый твердый остаток (17,0 г, 53%).
1H-ЯМР (500 MГц, d6-DMSO): δ 13,7 (шир.с, 1H), 7,02 (д, 2H, J=9,3 Гц), 2,32 (с, 3H).
Стадия D: 4-(2,6-дифтор-4-метилбензоил)-1H-пиррол-2-карбоновая кислота
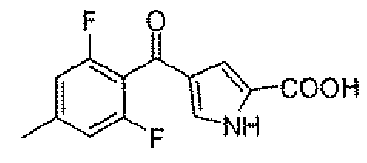
Заявленное в заголовке соединение получали, следуя способу для промежуточного соединения 1, стадии A и B, применяя 2,6-дифтор-4-метилбензойную кислоту вместо 2,4,6-трифторбензойной кислоты.
1Н-ЯМР (500 MГц, d6-DMSO): δ 12,9 (шир.с, 1Н), 12,6 (с, 1Н), 7,46 (с, 1H), 7,05 (д, 2H, J=8,8 Гц), 6,95 (с, 1H), 2,35 (с, 3H).
Промежуточное соединение 5
4-(2,4,6-трифторбензил)-1H-пиррол-2-карбоновая кислота
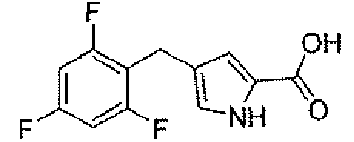
Стадия A: метил 4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксилат
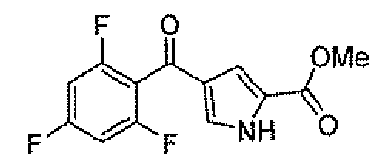
К дихлорметановой 70 мл суспензии 4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоновой кислоты (промежуточное соединение 1) (3 г, 11,2 ммоль) добавляли раствор диазометана в диэтиловом эфире при охлаждении в водяной бане со льдом. После перемешивания в течение 3 часов реакционную смесь концентрировали и хроматографировали на силикагеле, элюируя градиентной смесью растворителей AcOEt и гексана, для того чтобы получить заявленное в заголовке соединение (2,74 г).
1H-ЯМР (500 MГц, d6-DMSO): δ 12,9 (с, 1H), 7,65 (с, 1Н), 7,34 (т, 2H, J=9 Гц), 7,08 (с, 1H), 3,79 (с, 3H).
Стадия B: метил 4-(2,4,6-трифторбензил)-1H-пиррол-2-карбоксилат
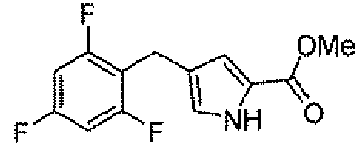
К TFA 30 мл раствору метил 4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксилата в герметичной пробирке добавляли триэтилсилан (2,55 г, 22 ммоль). Полученную в результате реакционную смесь грели в масляной бане при 70°C в течение ночи. Реакцию концентрировали и разбавляли изопропилацетатом и насыщенным водным раствором бикарбоната натрия. Органический слой отделяли. Водный слой экстрагировали изопропилацетатом дважды. Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали, концентрировали и хроматографировали на силикагеле, элюируя градиентной смесью растворителей AcOEt и гексана, для того чтобы получить заявленное в заголовке соединение (840 мг).
1H-ЯМР (500 MГц, d6-DMSO): δ 11,7 (с, 1Н), 7,16 (т, 2H, J=9 Гц), 6,79 (с, 1H), 6,52 (с, 1H), 3,72 (с,2H), 3,70 (с, 3H).
Стадия C: 4-(2,4,6-трифторбензил)-1H-пиррол-2-карбоновая кислота
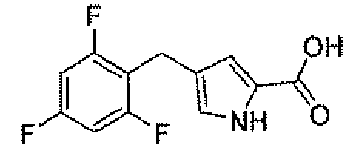
К метанольному 30 мл раствору метил 4-(2,4,6-трифторбензил)-1H-пиррол-2-карбоксилата (840 мг, 3,1 ммоль) добавляли 5N раствор гидроксида натрия (3,1 мл, 16 ммоль) и реакционную смесь перемешивали в масляной бане при 70°C в течение ночи. После охлаждения до комнатной температуры, pΗ реакционной смеси доводили до 1,5, для того чтобы получить серую суспензию. Осадок собирали фильтрованием с отсасыванием и сушили в вакууме, для того чтобы получить заявленное в заголовке соединение (796 мг).
1H-ЯМР (500 MГц, d6-DMSO): δ 12,2 (с, 1H), 11,5 (с, 1H), 7,15 (т, 2H, J=8 Гц), 6,73 (с, 1H), 6,47 (с, 1H), 3,73 (с, 2H).
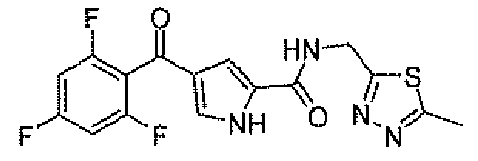
Пример 1
N-[(5-метил-1,3,4-тиадиазол-2-ил)метил]-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
Стадия A: трет-бутил[2-(2-ацетилгидразино)-2-оксоэтил]карбамат
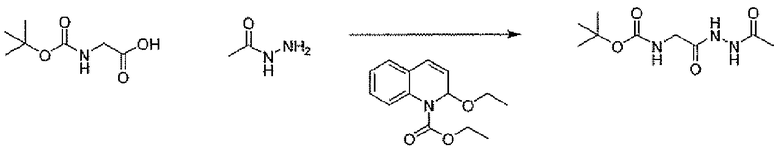
В атмосфере N2 к дихлорметановому 10 мл раствору N-трет-бутилоксикарбонилглицина (500 мг, 2,85 ммоль) добавляли EEDQ (706 мг, 2,85 ммоль). После перемешивания в течение 15 минут к ней добавляли ацетгидразид (260 мг, 3,51 ммоль) и перемешивание продолжали при комнатной температуре в течение ночи. Осадок собирали фильтрованием с отсасыванием, для того чтобы получить заявленное в заголовке соединение в виде белого твердого рыхлого остатка (496 мг).
1H-ЯМР (CDCl3, 400 MГц): δ 8,84 (шир.с, 1H), 8,27 (шир.с, 1H), 5,22 (шир.с, 1H), 3,87 (д, 2H), 2,02 (с, 3H), 1,41 (с, 9H).
Стадия B: трет-бутил[(5-метил-1,3,4-тиадиазол-2-ил)метил]карбамат
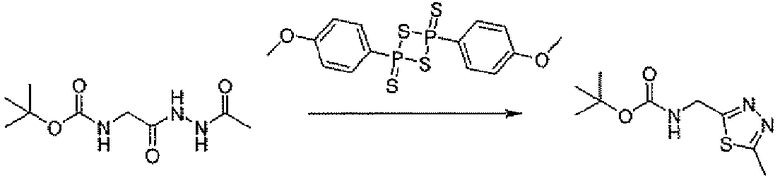
К 37,5 мл THF раствора трет-бутил[2-(2-ацетилгидразино)-2-оксоэтил]карбамата (496 мг, 2,15 ммоль) добавляли реагент Лавессона (900 мг, 2,23 ммоль). Полученную в результате реакционную смесь кипятили с обратным холодильником в течение 3 часов. Реакционную смесь концентрировали и хроматографировали на силикагеле, элюируя градиентной смесью растворителей AcOEt и дихлорметана, для того чтобы получить заявленное в заголовке соединение в виде белого кристаллического остатка (391 мг).
1H-ЯМР (CDCl3, 500 MГц): δ 5,31 (шир.с, 1H), 4,69 (д, 2H, J=6 Гц), 2,78 (с, 3H), 1,49 (с, 9H). LC/MS: m/z=230 (M+H), 252 (M+Na).
Стадия C: гидрохлорид 1-(5-метил-1,3,4-тиадиазол-2-ил)метанамина

После охлаждения в водяной бане со льдом к 6 мл 4M раствора хлороводорода в 1,4-диоксане добавляли трет-бутил[(5-метил-1,3,4-тиадиазол-2-ил)метил]карбамат (391 мг, 1,71 ммоль).
После перемешивания в течение 1 часа реакционную смесь концентрировали, для того чтобы получить заявленное в заголовке соединение в виде белого твердого остатка (351 мг).
1H-ЯМР (d6-DMSO, 500 MГц): δ 8,88 (шир.с, 2H), 4,49 (д, 2H, J=5,5 Гц), 2,74 (с, 3H). LC/MS: m/z=130 (M+H).
Стадия D: N-[(5-метил-1,3,4-тиадиазол-2-ил)метил]-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
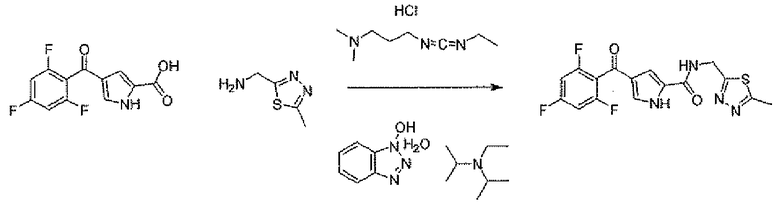
К DMF 3 мл раствору 4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоновой кислоты (промежуточное соединение 1) (30 мг, 0,11 ммоль), гидрата 1-гидроксибензотриазола (ΗOBT) (22 мг, 0,15 ммоль) добавляли гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида (EDC-гидрохлорид) (28 мг, 0,15 ммоль). Реакционную смесь перемешивали в течение 45 минут перед добавлением гидрохлорида 1-(5-метил-1,3,4-тиадиазол-2-ил)метанамина (19 мг, 0,17 ммоль) и диизопропилэтиламина (0,1 мл, 0,55 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, концентрировали и хроматографировали на силикагеле, элюируя AcOEt, для того чтобы получить заявленное в заголовке соединение 29 мг.
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (шир.с, 1H), 9,24 (т, 1H, J=6 Гц), 7,52 (с, 1H), 7,25 (с, 1H), 4,74 (д, 2H, J=6 Гц), 2,66 (с, 3H). LC/MS: m/z=381 (M+H), 403 (M+Na).
Пример 2-20: Заявленные в заголовке соединения получали, следуя способу, описанному для получения примера 1, применяя подходящим образом замещенные карбогидразид вместо ацетгидразида.
Пример 2
N-[(5-бутил-1,3,4-тиадиазол-2-ил)метил]-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,59 (шир.с, 1Н), 9,25 (т, 1Н, J=6 Гц), 7,51 (с, 1H), 7,35 (т, 2H, J=8 Гц), 7,25 (с, 1H), 4,75 (д, 2H, J=6 Гц), 2,99 (т, 2H, J=7,6 Гц), 1,65 (п, 2H, J=7,6 Гц), 1,32 (секст, 2H, J=7,6 Гц), 0,87 (т, 3H, J=7,6 Гц).
Пример 3
N-[(5-изопропил-1,3,4-тиадиазол-2-ил)метил]-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
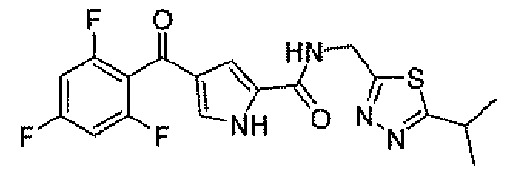
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (шир.с, 1H), 9,25 (т, 1H, J=6 Гц), 7,51 (с, 1Н), 7,35 (т, 2H, J=8 Гц), 7,25 (с, 1H), 4,75 (д, 2H, J=6 Гц), 3,35 (м, 1H, J=6,9 Гц), 1,31 (д, 6H, J=6,9 Гц). LC/MS: m/z=409 (M+H).
Пример 4
N-[(5-циклопропил-1,3,4-тиадиазол-2-ил)метил]-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
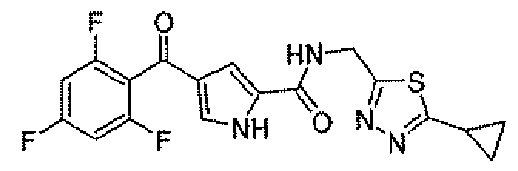
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,58 (шир.с, 1H), 9,225 (т, 1H, J=6 Гц), 7,51 (с, 1Н), 7,35 (т, 2H, J=8 Гц), 7,24 (с, 1H), 4,71 (д, 2H, J=6 Гц), 2,46 (м, 1H), 1,16 (м, 2H), 0,955 (м, 2H). LC/MS: m/z=407 (M+H).
Пример 5
N-[(5-трет-бутил-1,3,4-тиадиазол-2-ил)метил]-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
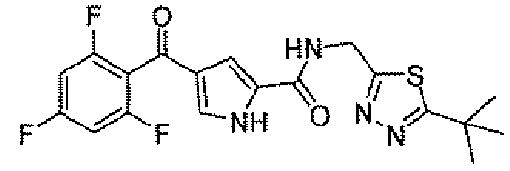
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,58 (шир.с, 1H), 9,257 (т, 1H, J=6 Гц), 7,52 (с, 1H), 7,34 (т, 2H, J=8 Гц), 7,25 (с, 1H), 4,75 (д, 2H, J=6 Гц), 1,37 (с, 9H). LC/MS: m/z=423 (M+H).
Пример 6
N-[(5-циклопентил-1,3,4-тиадиазол-2-ил)метил]-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
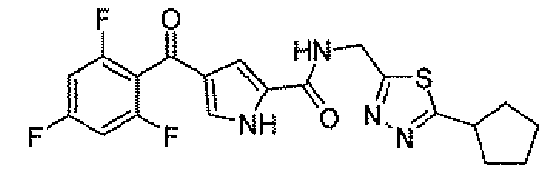
1H-ЯМР (CDCl3, 500 МГц): δ 10,4 (шир.с, 1H), 7,6 (шир.с, lH), 7,46 (с, 1H), 7,23 (с, 1H), 6,78 (т, 2H, J=8 Гц), 5,0 (д, 2H, J=6,2 Гц), 3,55 (п, 1H, J=7,8 Гц), 2,25 (м, 2H), 1,7-1,9 (м, 6H). LC/MS: m/z=435 (M+H).
Пример 7
N-[(5-фенил-1,3,4-тиадиазол-2-ил)метил]-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
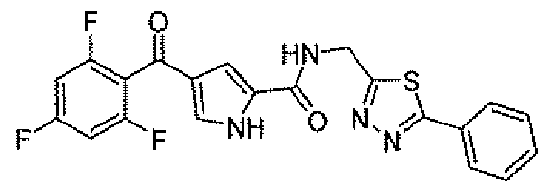
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (с, 1H), 9,35 (т, 1H, J=6 Гц), 7,96 (м, 2H), 7,54 (т, 2H, J=8 Гц), 4,85 (д, 2H, J=6 Гц). LC/MS: m/z=443 (M+H).
Пример 8
N-{[5-(4-фторфенил)-1,3,4-тиадиазолил-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
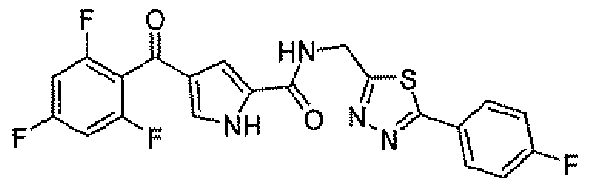
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,7 (с, 1H), 9,35 (т, 1Н, J=6 Гц), 8,02 (м, 2H), 7,53 (с, 1H), 7,28 (с, 1Н), 4,84 (д, 2H, J=6 Гц). LC/MS: m/z=461 (M+H).
Пример 9
N-{[5-(3-фторфенил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
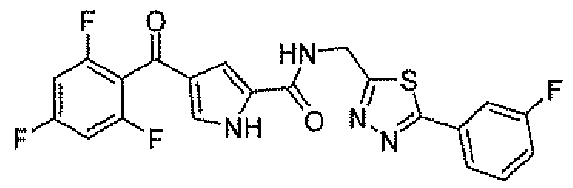
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (с, 1H), 9,36 (т, 1Н, J=6 Гц), 7,80 (д, 2H, J=7 Гц), 7,6-7,5 (м, 2H), 7,4-7,3 (м, 2H), 7,28 (с, 1H), 4,86 (д, 2H, J=6 Гц). LC/MS: m/z=461 (M+H).
Пример 10
N-{[5-(4-метоксифенил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
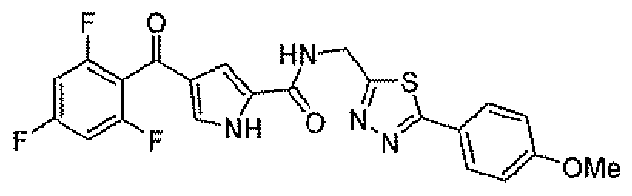
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (с, 1H), 9,32 (т, 1Н, J=6 Гц), 7,88 (д, 2H, J=7 Гц), 7,36 (т, 2H, J=8 Гц), 7,28 (с, 1H), 7,07 (д, 2H, J=6 Гц), 4,82 (д, 2H, J=6 Гц), 3,82 (с, 3H). LC/MS: m/z=473 (M+H).
Пример 11
N-{[5-(2,6-дифторфенил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
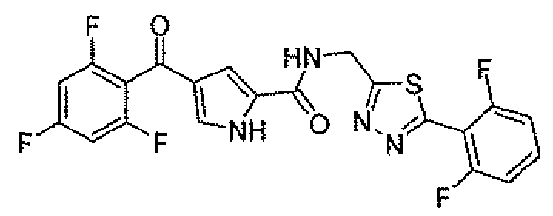
LC/MS: m/z=479 (M+Η).
Пример 12
N-[(5-пиридин-2-ил-1,3,4-тиадиазол-2-ил)метил]-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
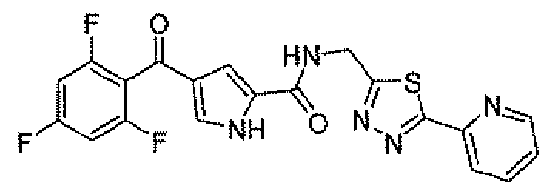
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (с, 1Н), 9,33 (т, 1H, J=6 Гц), 8,66 (д, 1H, J=5 Гц), 8,24 (д, 1Н, J=8 Гц), 8,02 (т, 1H, J=6 Гц), 7,55 (м, 1H), 7,53 (с, 1H), 7,36 (т, 2H, J=8 Гц), 7,28 (с, 1H), 4,85 (д, 2H, J=6 Гц). LC/MS: m/z=444 (M+H).
Пример 13
N-[(5-пиридин-3-ил-1,3,4-тиадиазол-2-ил)метил]-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
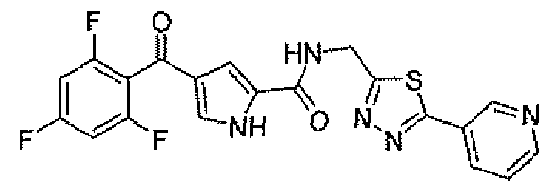
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (с, 1Н), 9,37 (т, 2H, J=6 Гц), 9,12 (д, 1H, J=2 Гц), 8,72 (м, 1Н), 8,34 (м, 1H), 7,55 (м, 1H), 7,53 (с, 1Н), 7,36 (т, 2H, J=8 Гц), 7,29 (с 1H), 4,87 (д, 2H, J=6 Гц). LC/MS: m/z=444 (M+H).
Пример 14
N-{[5-(1H-пиррол-2-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
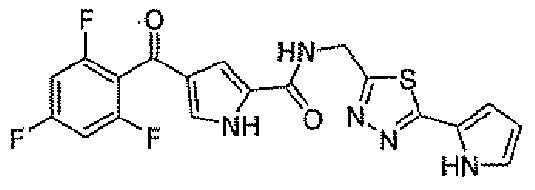
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (шир.с, 1Н), 12,0 (с, 1H), 9,3 (т, 1H, J=6 Гц), 7,53 (с, 1Н), 7,35 (т, 2H, J=8 Гц), 7,27 (с, 1Н), 6,98 (м, 1H), 6,71 (м, 1H), 6,19 (м, 1Н), 4,8 (д, 2H, J=6 Гц). LC/MS: m/z=432 (M+H).
Пример 15
N-{[5-(2-фурил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
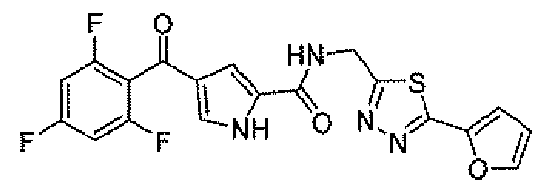
1H-ЯМР (CD3OD, 500 МГц): δ 7,77 (д, 1H, J=1,4 Гц), 7,5 (с, 1Н), 7,24 (д, 1H, J=1,4 Гц), 7,22 (д, 1H, J=3,4 Гц), 7,025 (т, 2H, J=8 Гц), 6,68 (дд, 1Н, J=1,4 Гц), 4,92 (с, 2H), 4,59 (шир.с, 1H). LC/MS: m/z=433 (M+H).
Пример 16
N-{[5-(2-этоксипиридин-3-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
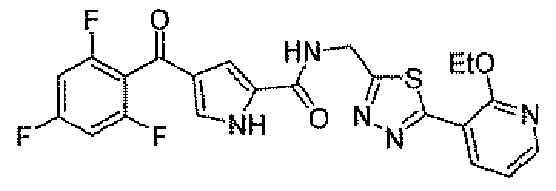
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (с, 1Н), 9,29 (т, 2H, J=6 Гц), 8,63 (дд, 1Н, J=2 Гц, 8 Гц), 8,36 (дд, 1Н, J=2 Гц, 5 Гц), 7,52 (д, 1H, J=2 Гц), 7,25 (м, 2H), 7,20 (с, 1H), 7,18 (м, 1H), 4,86 (д, 2H, J=6 Гц), 4,51 (кв, 2H, J=7 Гц), 1,39 (т, 3H, J=7 Гц). LC/MS: m/=488 (M+H).
Пример 17
N-{[5-(6-метоксипиридин-3-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
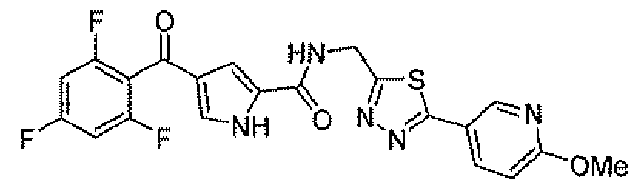
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (с, 1Н), 9,35 (т, 1H, J=6 Гц), 8,73 (д, 1Н, J=2 Гц), 8,25 (дд, 1H, J=2 Гц, 9 Гц), 7,53 (д, 1H, J=2 Гц), 7,36 (д, 2H, J=8 Гц), 7,28 (с, 1H), 6,97 (д, 1H, 9 Гц), 4,85 (д, 2H, J=6 Гц), 3,92 (с, 3H). LC/MS: m/z=474 (M+H).
Пример 18
N-{[5-(6-метоксипиридин-2-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
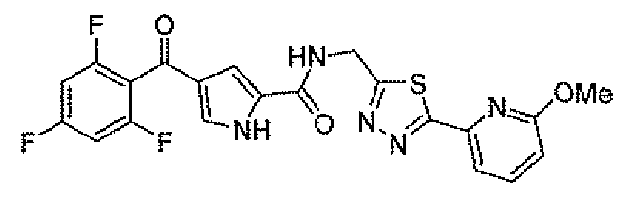
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (с, 1H), 9,34 (т, 1H, J=6 Гц), 7,91 (т, 1Н, J=8 Гц), 7,84 (д, 1H, J=7 Гц), 7,54 (с, 1Н), 7,36 (т, 2H, J=6 Гц), 7,28 (с, 1Н), 7,00 (д, 1H, J=7 Гц), 4,84 (д, 2H, J=6 Гц), 3,89 (с, 3H). LC/MS: m/z=474 (M+H).
Пример 19
N-{[5-(2-метоксипиридин-4-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
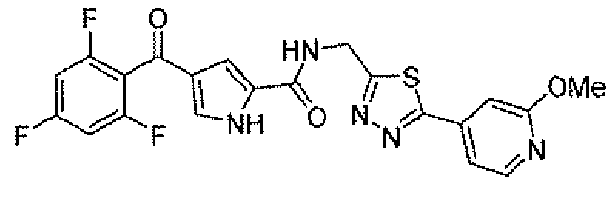
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (с, 1H), 9,38 (т, 1Н, J=6 Гц), 8,32 (т, 1H, J=6 Гц), 7,52 (м, 2H), 7,36 (т, 2H, J=8 Гц), 7,32 (с, 1H), 7,29 (с, 1H), 4,87 (д, 2H, J=6 Гц), 3,90 (с, 3H). LC/MS: m/z=474 (M+H).
Пример 20
N-{[5-(3-метил-1H-пиразол-5-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
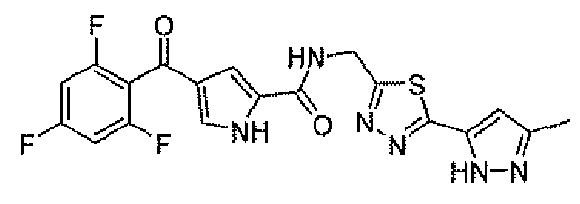
1H-ЯМР (d6-DMSO, 500 МГц): δ 13,1 (с, 1H), 12,6 (шир.с, 1Н), 9,3 (т, 1H, J=6 Гц), 7,53 (с, 1H), 7,33 (т, 2H, J=8 Гц), 7,27 (с, 1Н), 6,59 (с, 1H), 4,8 (д, 2H, J=6 Гц), 2,28 (с, 3H). LC/MS: m/z=447 (M+H).
Пример 21
N-[(5-пиразин-2-ил-1,3,4-тиадиазол-2-ил)метил]-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
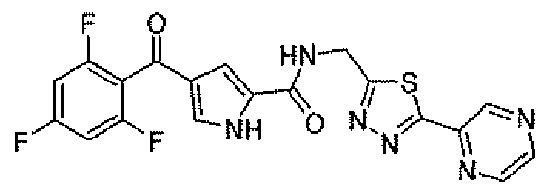
Стадия A: трет-бутил{2-оксо-2-[2-(пиразин-2-илуглеродил)гидразино]этил}карбамат
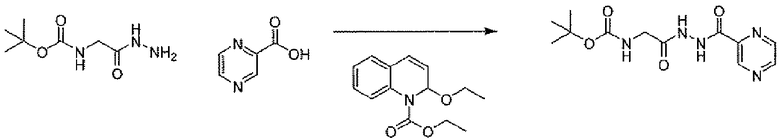
В атмосфере N2 к дихлорметановому 5 мл раствору пиразин-2-карбоновой кислоты (328 мг, 2,64 ммоль) добавляли EEDQ (653 мг, 2,64 ммоль). После перемешивания в течение 45 минут к нему добавляли N-(трет-бутилоксикарбонил)глицилгидразид (500 мг, 2,64 ммоль). Перемешивание продолжали при комнатной температуре в течение ночи. Реакционную смесь концентрировали и растирали из дихлорметана, для того чтобы получить заявленное в заголовке соединение (666 мг).
Стадия B-D: Следуя способу, описанному в примере 1, стадия B-D, получали заявленное в заголовке соединение.
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (с, 1H), 9,44 (с, 1H), 9,38 (т, 2H, J=6 Гц), 8,82 (с, 1Н), 8,78 (с, 1Н), 7,54 (с, 1H), 7,36 (т, 2H, J=8 Гц), 7,28 (с, 1Н), 4,88 (д, 2H, J=6 Гц). LC/MS: m/z =445 (M+H).
Заявленные в заголовке соединения в примерах 22-31 получали, следуя способу, описанному в примере 21, применяя подходящим образом замещенные карбоновые кислоты.
Пример 22
N-[(5-циклобутил-1,3,4-тиадиазол-2-ил)метил]-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
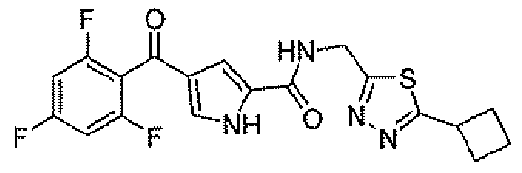
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,59 (с, 1H), 9,25 (т, 1Н, J=6 Гц), 7,516 (с, 1H), 7,35 (т, 2H, J=8 Гц), 7,25 (с, 1H), 4,75 (д, 2H, J=6 Гц), 3,93 (п, 1H, J=8,5 Гц), 2,4 (м, 2H), 2,234 (пд, 2H, J=9,2 Гц, J=2,4 Гц), 2,015 (секст, 1H, J=9 Гц), 1,89 (м, 1H). LC/MS: m/z=421 (M+H).
Пример 23
N-{[5-(1-метилциклопропил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
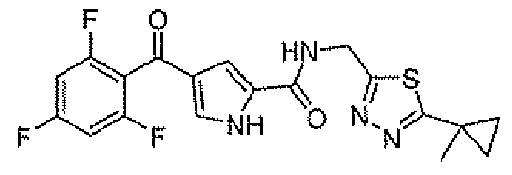
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,57 (шир.с, 1Н), 9,24 (т, 1Н, J=6 Гц), 7,51 (с, 1H), 7,34 (т, 2H, J=8 Гц), 7,24 (с, 1H), 4,72 (д, 2H, J=6 Гц), 1,49 (с, 3H), 1,15 (м, 2H), 1,04 (м, 2H). LC/MS: m/z=421 (M+H).
Пример 24
N-[(5-втор-бутил-1,3,4-тиадиазол-2-ил)метил]-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
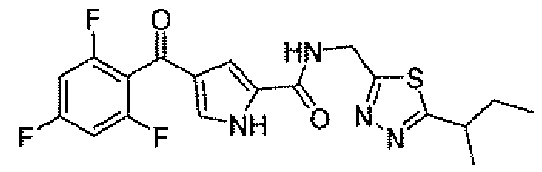
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,59 (с, 1H), 9,26 (т, 1H, J=6 Гц), 7,52 (с, 1H), 7,35 (т, 2H, J=8 Гц), 7,26 (с, 1Н), 4,76 (д, 2H, J=6 Гц), 3,195 (квт, 1H, J=6,9, 6,8 Гц), 1,66 (м, 2H, J=7,4, 6,8 Гц), 1,29 (д, 3H, J=6,9 Гц), 0,833 (т, 3H, J=7,4 Гц). LC/MS: m/z=423 (M+H).
Пример 25
N-{[5-(3-изопропил-1H-пиразол-5-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пирол-2-карбоксамид
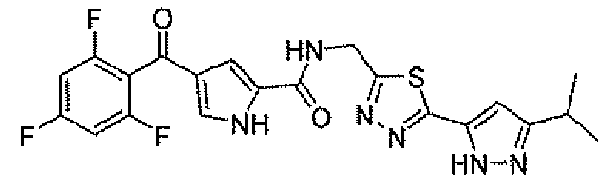
1H-ЯМР (d6-DMSO, 500 МГц): δ 13,16 (с, 1H), 12,61 (с, 1H), 9,29 (т, 1Н, J=6 Гц), 7,52 (с, 1H), 7,34 (т, 2H, J=8 Гц), 7,26 (с, 1H), 6,61 (с, 1H), 4,8 (д, 2H, J=6 Гц), 3,08 (септ, 1H, J=6,9 Гц), 1,24 (д, 6H, J=6,9 Гц). LC/MS: m/z=475 (M+H).
Пример 26
N-{[5-(3-изобутил-1H-пиразол-5-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
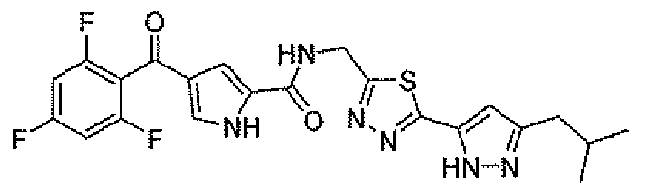
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (с, 1H), 9,29 (т, 1H, J=6 Гц), 7,52 (с, 1Н), 7,34 (т, 2H, J=8 Гц), 7,26 (с, 1Н), 6,6 (с, 1H), 4,8 (д, 2H, J=6 Гц), 2,51 (д, 2H, J=7,1 Гц), 1,91 (м, 1H, J=6,8 Гц, J=7,1 Гц), 0,88 (д, 6H, J=6,8 Гц). LC/MS: m/z=489 (M+H).
Пример 27
4-(2,4,6-трифторбензоил)-N-({5-[3-(трифторметил)-1H-пиразол-5-ил]-1,3,4-тиадиазол-2-ил}метил)-1H-пирол-2-карбоксамид
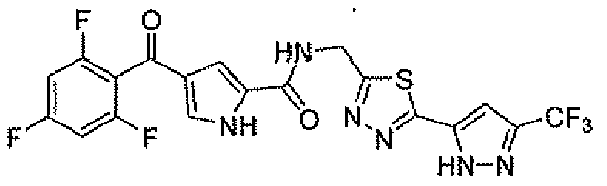
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,62 (с, 1H), 9,39 (т, 1H, J=5,8), 7,54 (с, 1Н), 7,47 (с, 1Н), 7,35 (т, 2H, J=8 Гц), 7,285 (с, 1H), 4,87 (д, 2H, J=5,8 Гц). LC/MS: m/z=501 (M+H).
Пример 28
N-{[5-(1H-индазол-3-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
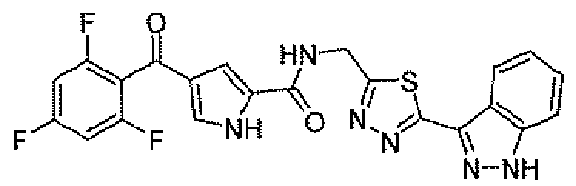
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,62 (с, 1H), 9,35 (т, 1H, J=6 Гц), 8,31 (д, 1Н, J=8,3 Гц); 7,66 (д, 1H, J=8,3 Гц), 7,54 (с, 1H), 7,49 (т, 1H, J=7,4 Гц), 7,35 (м, 3H), 7,29 (с, 1H), 4,87 (д, 2H, J=6 Гц). LC/MS: m/z=483 (M+H).
Пример 29
N-[(5-пиразолo[1,5-a]пиримидин-2-ил-1,3,4-тиадиазол-2-ил)метил]-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
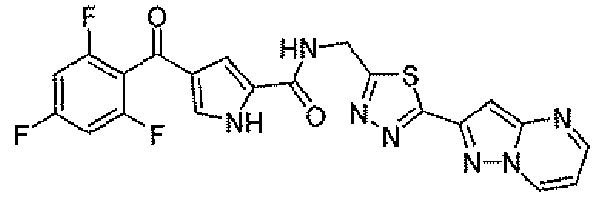
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,62 (с, 1H), 9,36 (т, 1Н, J=6 Гц), 9,17 (д, 1Н, J=7 Гц), 8,65 (д, 1H, J=4 Гц), 7,53 (с, 1Н), 7,342 (т, 2H, J=8 Гц), 7,32 (с, 1H), 7,28 (с, 1H), 7,176 (дд, 1H, J=7 Гц, J=4 Гц), 4,88 (д, 2H, J=6 Гц). LC/MS: m/z=484 (M+H).
Пример 30
N-{[5-(1,2,3-тиадиазол-4-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
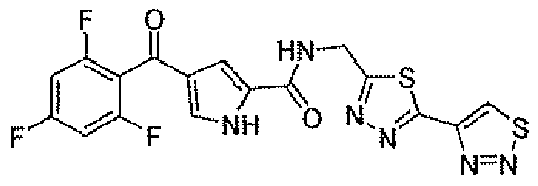
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,62 (с, 1Н), 9,93 (с, 1H), 9,4 (т, 1H, J=6 Гц), 7,53 (с, 1Н), 7,34 (т, 2H, J=8 Гц), 7,27 (с, 1H), 4,89 (д, 2H, J=6 Гц). LC/MS: m/z=451 (M+H).
Пример 31
N-{[5-(2,4,5,6-тетрагидроциклопента[c]пиразол-3-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,99 (шир.с, 1Н), 12,62 (с, 1Н), 9,29 (т, 1H, J=6 Гц), 7,53 (с, 1H), 7,343 (т, 2H, J=8 Гц), 7,27 (с, 1H), 4,8 (д, 2H, J=6 Гц), 2,73 (м, 4H), 2,53 (м, 2H). LC/MS: m/z=473 (M+H).
Пример 32
N-{[5-(1H-пиразол-5-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
Стадия A: N'-(2-хлорацетил)-1H-пиразол-5-карбогидразид
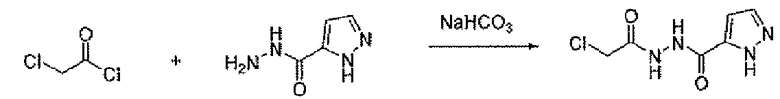
К суспензии 1H-пиразол-5-карбогидразида (102 мг, 0,81 ммоль) в 7 мл AcOEt добавляли 1,8 мл 1M раствора бикарбоната натрия и первоначальную белую суспензию перемешивали в течение нескольких минут, пока она не превратится в практически прозрачный двухфазный раствор. После охлаждения в водной бане со льдом к нему добавляли 0,7 мл AcOEt раствора хлорацетилхлорида (110 мг, 0,97 ммоль). После перемешивания в течение ночи реакционную смесь выливали в делительную воронку. Органический слой отделяли и водный слой экстрагировали дважды AcOEt. Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали, для того чтобы получить белый твердый остаток (110 мг).
1H-ЯМР (d6-DMSO, 500 МГц): δ 13,4 (с, 1Н), 10,2 (с, 1H), 10,1 (с, 1Н), 7,86 (с, 1H), 6,71 (с, 1H), 4,17 (с, 2H).
Стадия B: 2-(хлорметил)-5-(1H-пиразол-5-ил)-1,3,4-тиадиазол

N-(2-хлорацетил)-1H-пиразол-5-карбогидразид (110 мг, 0,54 ммоль) и реагент Лавессона (220 мг, 0,54 ммоль) суспендировали в TΗF 5,5 мл и затем кипятили с обратным холодильником в течение 3 часов. Реакционную смесь концентрировали и хроматографировали на силикагеле, элюируя градиентной смесью растворителей AcOEt и гексана, для того чтобы получить заявленное в заголовке соединение в виде белого твердого остатка (60,5 мг).
1H-ЯМР (d6-DMSO, 500 МГц): δ 7,97 (с, 1H), 6,91 (с, 1H), 5,26 (с, 2H).
Стадия C: 2-(азидометил)-5-(1H-пиразол-5-ил)-1,3,4-тиадиазол
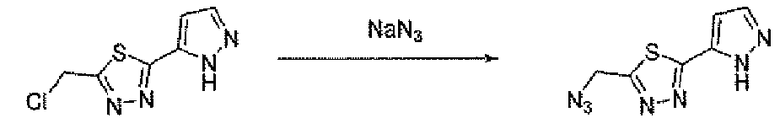
К 1 мл DMF раствора 2-(хлорметил)-5-(1H-пиразол-5-ил)-1,3,4-тиадиазола (58 мг, 0,29 ммоль) добавляли азид натрия (20 мг, 0,30 ммоль). Перемешивание продолжали в течение 3 часов. Реакционную смесь концентрировали и разбавляли AcOEt и водой. Органический слой отделяли. Водный слой экстрагировали AcOEt дважды и объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали, для того чтобы получить заявленное в заголовке соединение (60 мг).
1H-ЯМР (d6-DMSO, 500 МГц): δ 13,5 (с, 1H), 7,97 (с, 1H), 6,90 (с, 1H), 5,00 (с, 2H).
Стадия D: 1-[5-(1H-пиразол-5-ил)-1,3,4-тиадиазол-2-ил]метанамин
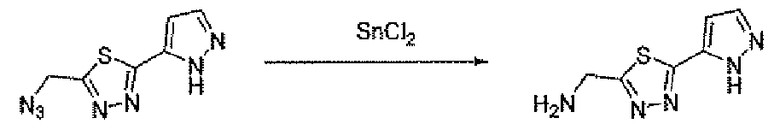
К 3 мл метанольному раствору 2-(азидометил)-5-(1H-пиразол-5-ил)-1,3,4-тиадиазола (50 мг, 0,24 ммоль) добавляли безводный хлорид олова (II) (82 мг, 0,43 ммоль) и полученный в результате желтый раствор перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали и очищали ВЭЖХ (элюент ацетонитрил-вода-гидроксид аммония), для того чтобы получить заявленное в заголовке соединение (37 мг).
1H-ЯМР (d6-DMSO, 500 МГц): δ 13,4 (с, 1Н), 7,92 (с, 1H), 6,84 (с, 1H), 4,12 (с, 2H).
Стадия E: N-{[5-(1H-пиразол-5-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
Следуя способу примера 1, стадия D, получали заявленное в заголовке соединение.
1H-ЯМР (d6-DMSO, 500 МГц): δ 13,48 (с, 1H), 12,64 (с, 1H), 9,35 (т, 1H, J=5,7 Гц), 7,93 (с, 1H), 7,53 (с, 1H), 7,35 (т, 2H, J=8 Гц), 7,28 (с, 1Н), 6,86 (с, 1Н), 4,81 (д, 2H, J=5,7 Гц). LC/MS: m/z=433 (M+H).
Заявленные в заголовке соединения в примерах 33-34 получали, следуя способу, описанному в примере 32, применяя подходящим образом замещенные карбогидразиды.
Пример 33
N-{[5-(1,5-диметил-1H-пиразол-3-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
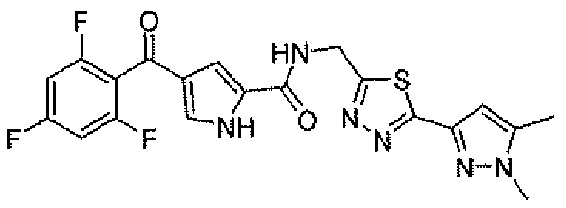
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (шир.с, 1H), 9,29 (т, 1H, J=6 Гц), 7,54 (с, 1H), 7,36 (т, 2H, J=8 Гц), 7,27 (с, 1Н), 6,65 (с, 1H), 4,8 (д, 2H, J=6 Гц), 3,78 (с, 3H), 2,31 (с, 3H). LC/MS: m/z=461 (M+H).
Пример 34
N-{[5-(2H-1,2,3-триазол-4-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
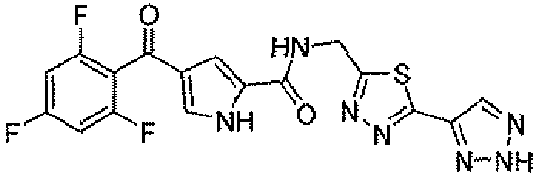
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,64 (с, 1Н), 9,34 (т, 1H, J=5,4 Гц), 8,87 (шир.с, 1H), 8,42 (шир.с, 1Н), 7,54 (с, 1Н), 7,36 (т, 2H, J=8 Гц), 7,28 (с, 1H), 4,85 (д, 2H, J=5,4 Гц). LC/MS: m/z=434 (M+H).
Пример 35
N-{[5-(2-оксо-1,2-дигидропиридин-3-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пирол-2-карбоксамид
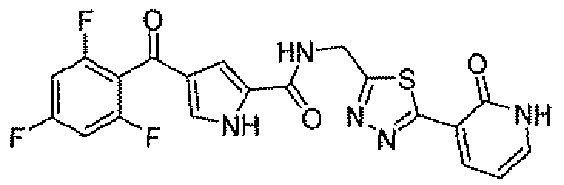
К N-{[5-(2-этоксипиридин-3-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамиду (пример 16) (102 мг, 021 ммоль) добавляли 3 мл 4M хлороводорода в 1,4-диоксане и полученную в результате суспензию перемешивали при комнатной температуре в течение 7 часов. Реакционную смесь концентрировали. Остаток растирали с дихлорметаном, для того чтобы получить заявленное в заголовке соединение (95 мг).
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (с, 1Н), 9,28 (т, 1H, J=6 Гц), 8,60 (дд, 1H, J=2 Гц, 8 Гц), 7,73 (с, 1H), 7,52 (с, 1H), 7,35 (т, 2H, J=2 Гц, 8 Гц), 7,27 (с, 1Н), 6,52 (т, 1H, J=7 Гц), 4,82 (д, 2H, J=6 Гц). LC/MS: m/z=460 (M+H).
Пример 36
N-{[5-(6-оксо-1,6-дигидропиридин-3-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
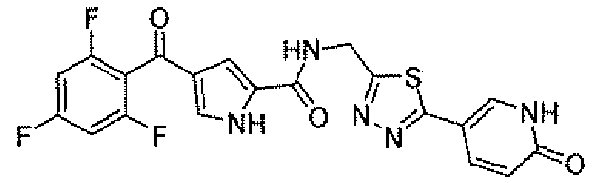
Следуя способу, описанному в примере 35, за исключением того, что реакционную смесь грели при 70°C в течение 6 часов, получали заявленное в заголовке соединение, применяя N-{[5-(6-метоксипиридин-3-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид (пример 17) в качестве исходного соединения.
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (с, 1H), 9,32 (т, 1H, J=6 Гц), 8,06 (д, 1H, J=3 Гц), 7,97 (дд, 1H, J=3 Гц: 10 Гц), 7,52 (с, 1H), 7,35 (т, 2H, J=8 Гц), 7,27 (с, 1H), 6,45 (д, 1Н, J=10 Гц), 4,79 (д, 2H, J=6 Гц). LC/MS: m/z=460(M+H).
Пример 37
N-{[5-(6-оксо-1,6-дигидропиридин-2-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
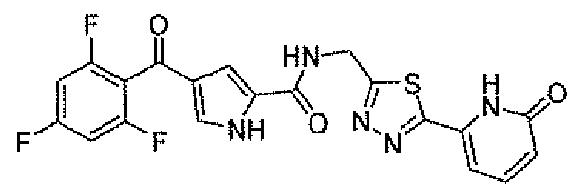
Следуя способу, описанному в примере 35, за исключением того, что реакционную смесь грели при 100°C в течение 6 часов, заявленное в заголовке соединение получали, применяя N-{[5-(6-метоксипиридин-2-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид (пример 18) в качестве исходного соединения.
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (с, 1Н), 11,2 (шир.с, 1Н), 9,35 (т, 1H, J=6 Гц), 7,81 (т, 1H, J=7 Гц), 7,69 (с, 1H), 7,54 (д, 1H, J=2 Гц), 7,36 (т, 2H, J=8 Гц), 7,28 (с, 1Н), 6,79 (д, 1Н, J=8 Гц), 4,82 (д, 2H, J=6 Гц). LC/MS: m/z=460 (M+H).
Пример 38
N-{[5-(2-оксо-1,2-дигидропиридин-4-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид
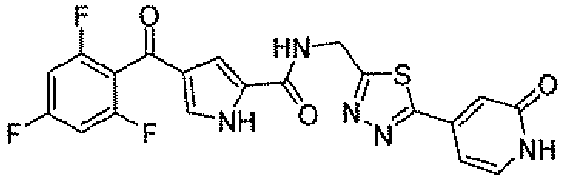
Следуя способу, описанному в примере 35, за исключением того, что реакционную смесь грели при 100°C в течение 6 часов, заявленное в заголовке соединение получали, применяя N-{[5-(2-метоксипиридин-4-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензоил)-1H-пиррол-2-карбоксамид (пример 19) в качестве исходного соединения.
LC/MS: m/z=460(M+H).
Пример 39
4-(2,4-дифторбензоил)-N-[(5-метил-1,3,4-тиадиазол-2-ил)метил]-1H-пиррол-2-карбоксамид
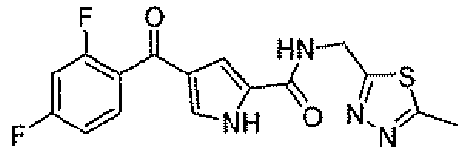
Заявленное в заголовке соединение получали, следуя способу, описанному в примере 1, применяя промежуточное соединение 3 вместо промежуточного соединения 1.
1H-ЯМР (CDCl3; 500 МГц): δ 7,93 (с, 1H), 7,73 (т, 1H, J=7 Гц), 6,86 (д, 1H, J=8 Гц), 5,33 (шир.с, 1H), 4,78 (д, 2H, J=6 Гц), 4,00 (с, 3H). LC/MS: m/z=363 (M+H).
Пример 40
4-(2,6-дифторбензоил)-N-[(5-метил-1,3,4-тиадиазол-2-ил)метил]-1H-пиррол-2-карбоксамид
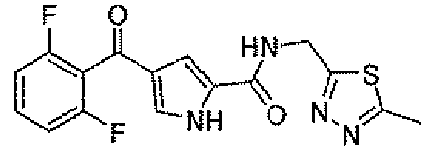
Заявленное в заголовке соединение получали, следуя способу, описанному в примере 1, применяя промежуточное соединение 2 вместо промежуточного соединения 1.
1H-ЯМР (CDCl3, 500 МГц): δ 10,7 (с, 1Н), 7,88 (т, 1Н, J=6 Гц), 7,01 (m, 2H), 5,00 (д, 2H, J=6 Гц), 2,77 (с, 3H). LC/MS: m/z=363 (M+H).
Пример 41
4-(2,6-дифторбензоил)-N-{[5-(1H-пиразол-5-ил)-1,3,4-тиадиазол-2-ил]метил}-1H-пиррол-2-карбоксамид
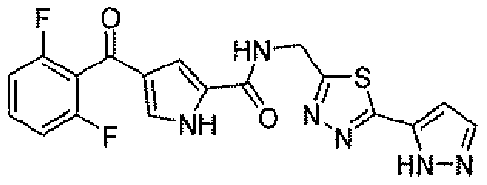
Заявленное в заголовке соединение получали, следуя способу, описанному в примере 32, применяя промежуточное соединение 2 вместо промежуточного соединения 1.
1H-ЯМР (d6-DMSO, 500 МГц): δ 13,47 (шир.с, 1Н), 12,60 (шир.с, 1H), 9,32 (м, 1H), 7,93 (м, 1H), 7,61 (м, 1H), 7,45 (с, 1H), 7,25 (м, 3H), 6,86 (с, 1H), 4,81 (с, 2H). LC/MS: m/z=415(M+H).
Пример 42
4-(2,6-дифтор-4-метилбензоил)-N-{[5-(1H-пиразол-5-ил)-1,3,4-тиадиазол-2-ил]метил}-1H-пиррол-2-карбоксамид
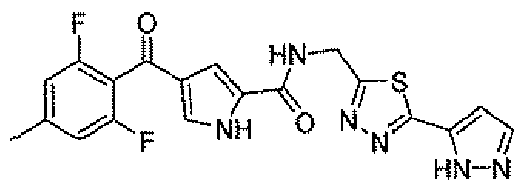
Заявленное в заголовке соединение получали, следуя способу, описанному в примере 32, применяя промежуточное соединение 4 вместо промежуточного соединения 1.
1H-ЯМР (d6-DMSO, 500 МГц): δ 13,45 (шир.с, 1H), 12,58 (шир.с, 1H), 9,30 (м, 1H), 7,94 (с, 1H), 7,43 (с, 1Н), 7,23 (с, 1H), 7,08 (м, 2H), 6,86 (с, 1Н), 4,81 (с, 2H), 2,37 (с, 3H). LC/MS: m/z=429(M+H).
Пример 43
N-{[5-(2-оксо-1,2-дигидропиридин-3-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензил)-1H-пиррол-2-карбоксамид
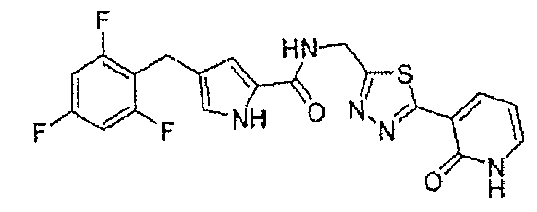
Заявленное в заголовке соединение получали, следуя способу, описанному в примере 35, применяя промежуточное соединение 5 вместо промежуточного соединения 1.
1H-ЯМР (d6-DMSO, 500 МГц): δ 12,6 (с, 1H), 11,4 (с, 1H), 8-90 (т, 1Н, J=6 Гц), 8,58 (дд, 1H, J=2 Гц, 7 Гц), 7,72 (м, 1H), 7,15 (т, 1H, J=8 Гц), 6,70 (с, 1Н), 6,62 (с, 1H), 6,50 (т, 1H, J=6 Гц), 4,74 (д, 2H, J=6 Гц), 3,71 (с, 2H).
Пример 44
N-[(5-пиридин-2-ил-1,3,4-тиадиазол-2-ил)метил]-4-(2,4,6-трифторбензил)-1H-пиррол-2-карбоксамид
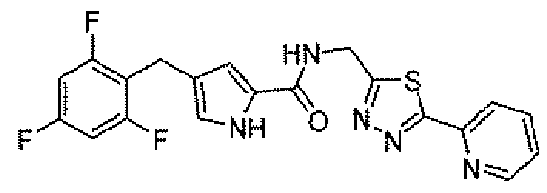
Заявленное в заголовке соединение получали, следуя способу, описанному в примере 12, применяя промежуточное соединение 5 вместо промежуточного соединения 1.
1H-ЯМР (CDCl3, 500 МГц): δ 9,22 (с, 1H), 8,64 (д, 1H, J=5 Гц), 8,34 (д, 1H, J=5 Гц), 7,87 (м, 1H), 7,41 (м, 1H), 6,84 (с, 1H), 6,67 (т, 2H, J=8 Гц), 6,51 (с, 1H), 5,04 (д, 2H, J=6 Гц), 3,82 (с, 2H).
Пример 45
N-{[5-(1H-пиразол-5-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензил)-1H-пиррол-2-карбоксамид
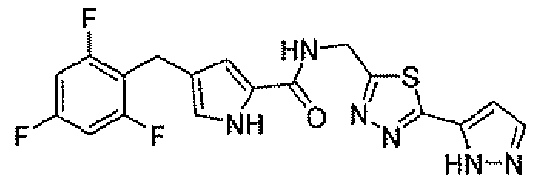
Заявленное в заголовке соединение получали, следуя способу, описанному в примере 32, применяя промежуточное соединение 5 вместо промежуточного соединения 1.
1H-ЯМР (d6-DMSO, 500 МГц): δ 13,4 (с, 1Н), 11,4 (с, 1H), 8,92 (т, 1H, J=6 Гц), 7,92 (с, 1H), 7,15 (т, 2H, J=8 Гц), 6,84 (с, 1H), 6,62 (с, 1H), 4,73 (д, 2H, J=6 Гц), 3,32 (с, 2H).
Пример 46
N-{[5-(3-метил-1H-пиразол-5-ил)-1,3,4-тиадиазол-2-ил]метил}-4-(2,4,6-трифторбензил)-1H-пиррол-2-карбоксамид
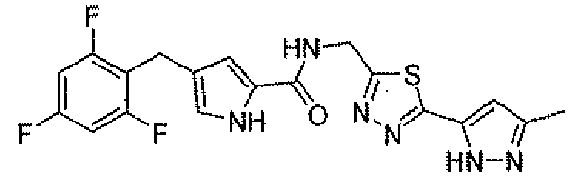
Заявленное в заголовке соединение получали, следуя способу, описанному в примере 20, применяя промежуточное соединение 5 вместо промежуточного соединения 1.
1H-ЯМР (d6-DMSO, 500 МГц): δ 13,1 (с, 1H), 11,4 (с, 1H), 8,9 (т, 1H, J=6 Гц), 7,15 (т, 2H, J=8 Гц), 6,71 (с, 1H), 6,62 (с, 1Н), 6,58 (с, 1Н), 4,72 (д, 2H, J=6 Гц), 3,69 (с, 2H), 2,28 (с, 3H). LC/MS: m/z=433 (M+H).
Альтернативный способ получения примера 32
Стадия 1. Фридель-Крафтс
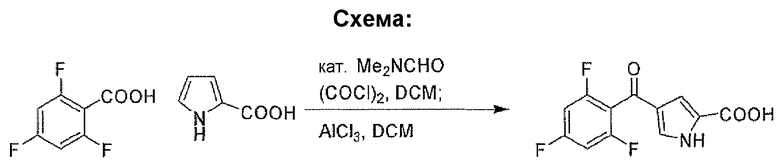
Способ: Данный способ проводили с двумя порциями и обе обрабатывали одинаково.
В 50 л многогорлую круглодонную колбу в паровой бане, снабженную верхнеприводной мешалкой, датчиком термопары и капельной воронкой с входным отверстием азота, сверху загружали 2,4,6-трифторбензойную кислоту, дихлорметан и DMF. Входное отверстие для азота соединяли с NaOH сепаратором. Оксалилхлорид загружали в капельную воронку и добавляли в течение 15 минут, причем в течение данного времени температура падала до 10°C с выделением газа. Реакционную смесь перемешивали в течение 1 часа, нагревая до 17°C. После еще трех часов при комнатной температуре образец проверяли с помощью ВЭЖХ на завершение реакции. Образец гасили в метаноле.
В 75 л многогорлую круглодонную колбу в паровой бане, снабженную верхнеприводной мешалкой, датчиком термопары и входным отверстием для азота, сверху загружали хлорид алюминия, суспендированный в дихлорметане. К данной суспензии добавляли раствор хлорангидрида кислоты в течение 5 минут с сопутствующим повышением температуры с 17°C до 22°C. Смесь выдерживали при комнатной температуре в течение 45 минут. Пиррол-2-карбоновую кислоту добавляли несколькими порциями в течение 40 минут с энергичным выделением газа после каждой загрузки. Температура повышалась до 23°C. После 30 минут при комнатной температуре отбирали образец и проверяли на завершенность реакции ВЭЖХ. Реакционную смесь перемешивали в течение дополнительных 2 часов перед помещением в лед и наносили сверху сухой лед для ее охлаждения в течение ночи. В 100 л многогорлую круглодонную колбу в паровой бане, снабженную верхнеприводной мешалкой, датчиком термопары и входным отверстием для азота сверху, загружали HCl и помещали в лед для охлаждения ее в течение ночи.
К охлажденному HCl раствору добавляли реакционную смесь в течение 1 3/4 часа с повышением температуры до 24°C. Полученную в результате суспензию выдерживали в течение 1 часа при комнатной температуре и затем фильтровали. Остаток на фильтре промывали большим количеством HCl, с последующей промывкой водой.
Остаток на фильтре сушили в емкости с азотом в течение ночи. Затем влажный остаток на фильтре сушили в вакуумной печи при 55°C с продувкой азотом в течение выходных. К полученному в результате твердому остатку в 100 л многогорлой круглодонной колбе в паровой бане, снабженной верхнеприводной мешалкой, датчиком термопары и капельной воронкой с входным отверстием азота, сверху загружали неочищенный продукт арилированной пирролкарбоновой кислоты и метанол. Данную суспензию грели при 48°C для растворения продукта, в процессе чего она охлаждалась. После достижения 30°C через капельную воронку добавляли воду в течение 1 часа.
Суспензию охлаждали до 5°C и выдерживали в течение 2 часов. Затем суспензию фильтровали при 5°C и остаток на фильтре промывали холодной смесью (5°C) метанол:вода. Влажный твердый продукт проверяли на соотношение региоизомеров ВЭЖХ. Остаток на фильтре сушили в емкости с азотом в течение ночи перед фасовкой.
ЯМР:
1Н (DMSO, 400 MГц) δ 12,92 (шир.с, 1H), 12,70 (шир.с, 1H), 7,58 (дд, 1Н, J=3,4, 1,5 Гц), 7,31 (дд, 2H, J=9,4, 7,9 Гц), 7,04 (т, 1H, J=2,0 Гц).
13C (DMSO, 100 MГц) δ 180,8, 162,8 (дт, JCF=249,41, 15,72 Гц), 161,4, 159,2 (ддд, J CF=248,60, 15,76, 11,15 Гц), 130,2, 125,9, 125,9, 114,7 (тд, JCF=18,75, 4,71 Гц), 114,3, 101,4 (тд, JCF=23,98, 3,01 Гц).
19F (DMSO, 376 MГц) δ -105,26, -105,28, -105,29, -111,11, -111,13.
HRMS: [M-H]- C12H5O3NF3 - рассчитанная 268,0222; найденная 268,0228; ошибка: 2,2 м.д.
2,4,6-Трифторбензойная кислота:
ЯМР:
1H (DMSO, 400 MГц) δ 13,89 (шир.с, 1H), 7,27 (м, 2H).
13C (DMSO, 100 MГц) δ 163,28 (дт, JCF=250,9, 16,0 Гц), 161,5, 160,3 (ддд, JCF=253,6, 15,8, 9,7 Гц), 109,0 (тд, JCF=19,5, 4,7 Гц), 101,5 (тд, JCF=26,8, 3,5 Гц).
19F (DMSO, 376 MГц) δ -103,6, -103,6, -103,7, -108,3, -108,3.
Пиррол-2-карбоновая кислота:
ЯМР:
1H (DMSO, 400 MГц) δ 12,16 (шир.с, 1H), 11,68 (шир.с, 1H), 5,70 (м, 1H), 6,71 (м, 1Н), 6,13 (м, 1H).
13C (DMSO, 100 MГц) δ 161,9, 123,4, 122,9, 114,7, 109,3.
Стадия 2. Образование ацилгидразида
Эксперимент:
В реакционный сосуд загружали метиловый эфир (140 г, 1110 ммоль), гидразингидрат (94 мл, 1554 ммоль) и метанол (700 мл), затем кипятили с обратным холодильником.
LC результаты: 45 мин: 72% превращение; 1,25 часов: 85% превращение.
Реакционную смесь выдерживали в течение дополнительных 2,5 часов, затем перемешивали при комнатной температуре в течение ночи, после чего LC анализ показал завершение реакции. Полученную в результате суспензию охлаждали до 3°C, твердый остаток фильтровали и промывали 200 мл воды, затем сушили в потоке азота, для того чтобы получить 120 г (86% выход) ацилгидразида.
Стадия 3. Хлорацетилирование
Эксперимент:
Реагент 1 (100 г, 793 ммоль) суспендировали в этилацетате (1000 мл) в 3 л 3-горлой RBF с верхнеприводным перемешиванием и обрабатывали бикарбонатом калия (600 мл, 1800 ммоль). Затем реакционную смесь охлаждали до 5°C, после чего добавляли в течение 7 минут хлорацетилхлорид (76 мл, 952 ммоль). Наблюдали нагревание до 16°C. LC анализ через 15 минут показал завершение реакции. Добавляли 100 мл 6N HCl, затем добавляли другие 15 мл 12N HCl для доведения pH до 4,6. Твердый остаток отфильтровывали и промывали 150 мл холодной воды и сушили в течение ночи в потоке азота, для того чтобы получить 143,7 г (89%) требуемого продукта.
Стадия 4. Циклизация
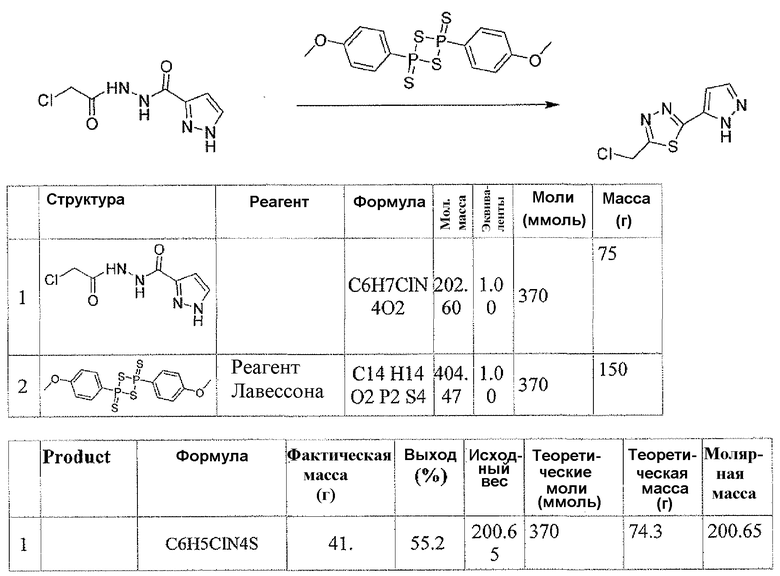
Эксперимент:
Суспендировали реагент 1 (75 г, 370 ммоль) и реагент Лавессона (150 г, 370 ммоль) в THF (1500 мл), затем кипятили с обратным холодильником в атмосфере азота в течение 2,5 часов. Затем реакцию охлаждали до комнатной температуры и концентрировали. Полученный в результате остаток растворяли в 250 мл EtOAc, обрабатывали 300 г силикагеля, затем фильтровали. Остаток на фильтре промывали 3×500 мл EtOAc. Первую фракцию пропускали через слой силикагеля снова и фракции с продуктом концентрировали и повторно растворяли в DCM; хроматографировали на силикагеле, 1,5 кг колонка; элюировали EtOAc/гептан 50-60%. Отбирали фракции с веществом, для того чтобы получить 41 г твердого остатка (55,2 %).
Стадия 5. Замещение азидом натрия
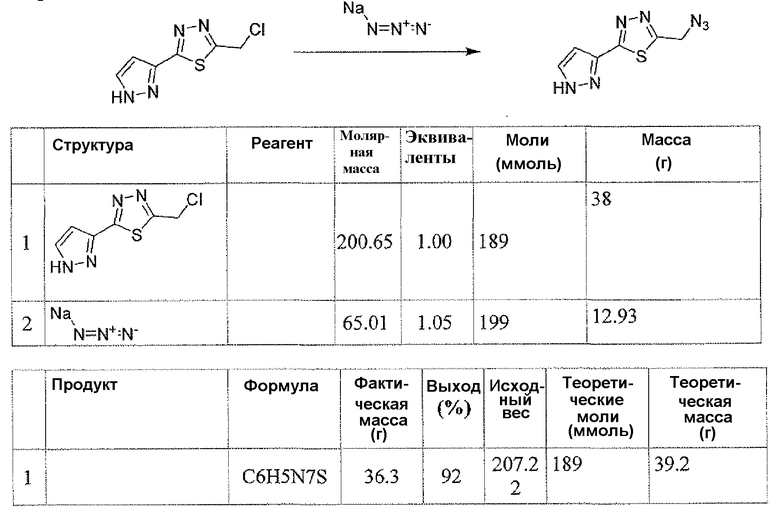
Эксперимент:
Растворяли реагент 1 (38 г, 189 ммоль) в DMF (160 мл) при комнатной температуре. Добавляли азид натрия (12,93 г, 199 ммоль); перемешивали при температуре окружающей среды. Смесь становилась оранжевой, через несколько минут начинал выпадать осадок.
После перемешивания реакционной смеси в течение выходных, LC анализ показал завершение реакции. Добавляли 250 мл воды (слегка экзотермическая), что приводило в результате к гомогенному раствору, затем начинал кристаллизоваться осадок. Суспензию охлаждали до 5°C, фильтровали, промывали 2×100 мл холодной водой и сушили, для того чтобы получить 36,6 г (92%) продукта.
Стадия 6. Восстановление азида
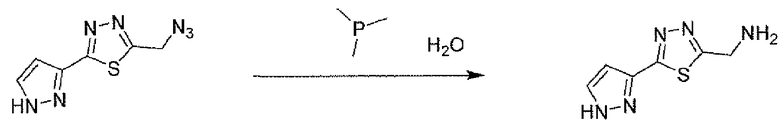
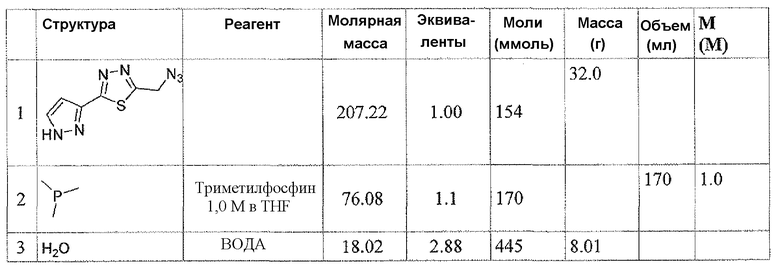
Эксперимент:
Суспендировали реагент 1 (32,0 г, 154 ммоль) в THF (160 мл); добавляли воду (8,01 г, 445 ммоль). Добавляли по каплям в течение часа триметилфосфин 1,0 M в THF (170 мл, 170 ммоль). Анализ через 2 часа показал отсутствие исходного вещества. Раствор концентрировали и полученный в результате остаток обрабатывали 160 мл 2N HCl и перемешивали при комнатной температуре в течение ночи.
Затем раствор подщелачивали 60 мл 5N NaOH. Добавляли 160 мл 1N NaHCO3 и применяли непосредственно в следующей конечной реакции конденсации.
Стадия 7a. Активация
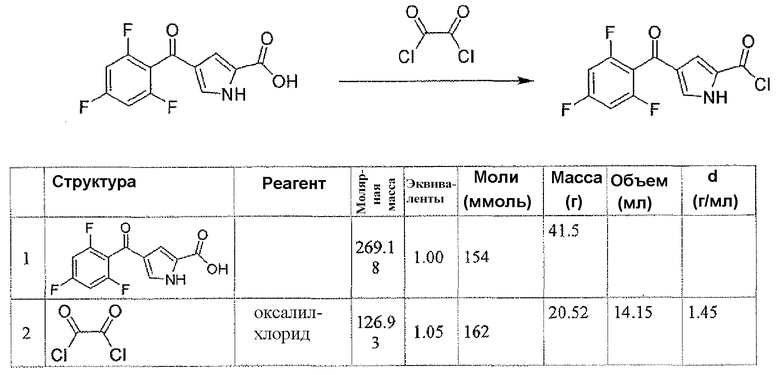
Эксперимент:
Растворяли реагент 1 (41,5 г, 154 ммоль) в THF (415 мл); добавляли 0,25 мл DMF, затем добавляли оксалилхлорид (14,15 мл, 162 ммоль). Раствор перемешивали при комнатной температуре в течение 1 часа, затем концентрировали. Полученный в результате остаток разбавляли 200 мл 2-MeTHF и применяли непосредственно в конечной стадии конденсации.
Стадия 7b. Конечная конденсация
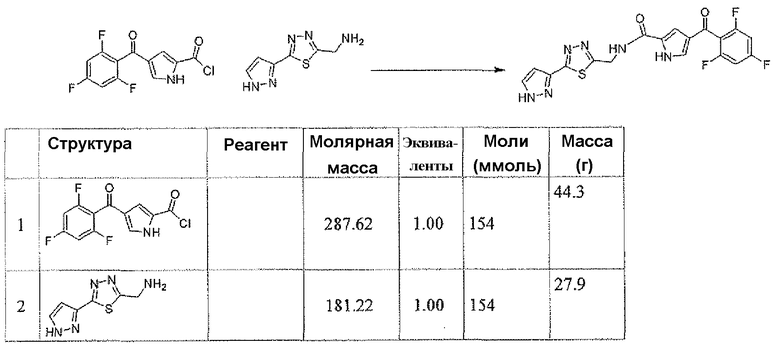

Эксперимент:
Растворы неочищенных хлорангидрида кислоты и амина смешивали и перемешивали при комнатной температуре в течение 2 часов. После завершения реакции (LC анализом), раствор концентрировали для удаления органических растворителей и полученную в результате водную суспензию фильтровали, твердый остаток промывали водой (100 мл, 150 мл), затем 150 мл ацетонитрила и сушили с отсасыванием, для того чтобы получить 72 г бледно-зеленого твердого остатка.
Перекристаллизация
Загружали 71,8 г твердого остатка в 22 л круглодонную колбу и добавляли 7,0 л ацетонитрила и 3,5 л воды, затем нагревали до 77°C. Фильтровали горячую смесь через воронку Бюхнера для удаления нерастворимых соединений. (Фильтрат был слегка мутным.) Перекачивали фильтрат (61°C) в чистую 22 л круглодонную колбу через 5 мк линейный фильтр, для того чтобы получить прозрачный желтый раствор. Медленно охлаждали до 30°C и затем охлаждали до 5°C. Отфильтровывали твердый остаток и промывали его 250 мл 2/1 смесь ацетонитрил/вода, сушили в вакууме в течение ночи, для того чтобы получить 44,65 г (67% выход на 2 стадии и перекристаллизацию) конечного продукта.
| название | год | авторы | номер документа |
|---|---|---|---|
| Анелированные 9-гидрокси-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-a]пиразин-7-карбоксамиды - ингибиторы интегразы ВИЧ, способы их получения и применения | 2019 |
|
RU2717101C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PRMT5 | 2018 |
|
RU2797822C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОАРИЛОМ ПИРИДИНЫ И СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2756743C2 |
| ПРОИЗВОДНЫЕ 2-БЕНЗОИЛ-ИМИДАЗО[1,2-a]ПИРИДИНА, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2008 |
|
RU2442785C1 |
| ТРИЦИКЛИЧЕСКИЕ ПИРРОЛО ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2012 |
|
RU2591191C2 |
| СПОСОБ ЛЕЧЕНИЯ | 2012 |
|
RU2621148C2 |
| ПРОИЗВОДНЫЕ 2-ПИРАЗИНОНА ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ ИЛИ СОСТОЯНИЯ, ПРИ КОТОРЫХ ПОЛЕЗНО ИНГИБИРОВАНИЕ АКТИВНОСТИ НЕЙТРОФИЛЬНОЙ ЭЛАСТАЗЫ | 2007 |
|
RU2448098C2 |
| ЗАМЕЩЕННЫЕ ПИРРОЛЫ, АКТИВНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2013 |
|
RU2666538C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| ПИРАЗОЛО[1,5-A]ПИРИМИДИНИЛКАРБОКСАМИДЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ПАТОЛОГИЧЕСКИХ СОСТОЯНИЙ | 2017 |
|
RU2799332C2 |
Изобретение относится к соединениям общей формулы (I), обладающим активностью в отношении цитокинов, вариантам фармацевтических композиций на их основе и их применению. Соединения формулы (I) могут найти применение для лечения или предотвращения астмы, COPD, ARDS, ревматоидного артрита, ревматоидного спонделита, остеоартрита или подагрического артрита. В общей формуле (I) L выбирают из группы, состоящей из
-С(O)-, -CH2-, Ar1 представляет собой моно-, ди- или тризамещенное фенильное кольцо, где заместители независимо выбраны из группы, состоящей из галогена и -C1-4алкила; Ar2 представляет собой необязательно замещенное тиадиазольное кольцо, где заместитель представляет собой -C1-4алкил, -C3-5циклоалкил, -метилциклопропил, фенильное или 5- или 6-членное моноциклическое гетероароматическое кольцо или бициклическое гетероароматическое кольцо с 9 или 10 атомами, причем упомянутое гетероароматическое кольцо содержит 1, 2 или 3 гетероатома, выбранные из группы, состоящей из S, О и N, где упомянутое фенильное или гетероароматическое кольцо является необязательно моно- или дизамещенным заместителями, независимо выбранными из группы, состоящей из галогена, -С1-6алкила, необязательно замещенного 1-4 атомами фтора, -O-C1-6алкила, -CF3 и оксо. 6 н. и 10 з.п. ф-лы, 1 табл., 46 пр.
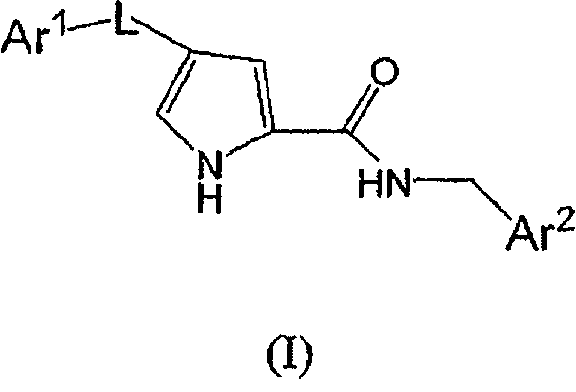
1. Соединение, представленное химической формулой (I)
где:
L выбирают из группы, состоящей из:
(а) -С(O)-,
(е) -CH2-;
Ar1 необязательно представляет собой моно-, ди- или тризамещенное фенильное кольцо, где заместители независимо выбраны из группы, состоящей из:
(a) галогена и
(b) -C1-4алкила;
Ar2 представляет собой необязательно замещенное тиадиазольное кольцо, где заместитель представляет собой -C1-4алкил, -C3-5циклоалкил, -метилциклопропил, фенильное или 5- или 6-членное моноциклическое гетероароматическое кольцо или бициклическое гетероароматическое кольцо с 9 или 10 атомами, причем упомянутое гетероароматическое кольцо содержит 1, 2 или 3 гетероатома, выбранные из группы, состоящей из S, О и N, где упомянутое фенильное или гетероароматическое кольцо является необязательно моно- или дизамещенным заместителями, независимо выбранными из группы, состоящей из:
(а) галогена,
(b) -С1-6алкила, необязательно замещенного 1-4 атомами фтора,
(c) -O-C1-6алкила,
(d) -CF3 и
(1) оксо.
2. Соединение по п.1, где L выбирают из группы, состоящей из:
(a) -С(O)- и
(b) -CH2-.
3. Соединение по п.2, где L представляет собой -С(O)-.
4. Соединение по п.1, где
Ar1 представляет собой необязательно моно-, ди- или тризамещенное фенильное кольцо, где заместители независимо выбраны из группы, состоящей из:
(a) галогена,
(b) -C1-4алкила и
(c) -O-C1-4алкила.
5. Соединение по п.4, где
Ar1 представляет собой необязательно моно-, ди- или тризамещенный фенил, где заместители независимо выбраны из группы, состоящей из
(a) фтора,
(b) хлора и
(С) -CH3.
6. Соединение по п.1, где
Ar2 представляет собой необязательно замещенный тиадиазолил.
7. Соединение по п.6, где заместитель представляет собой - C1-4алкил, -C3-5циклоалкил, -метилциклопропил, фенильное или 5- или 6-членное моноциклическое гетероароматическое кольцо или бициклическое гетероароматическое кольцо с 9 или 10 атомами, причем упомянутое гетероароматическое кольцо содержит 1, 2 или 3 гетероатома, выбранные из группы, состоящей из S, О и N, где упомянутое фенильное или гетероароматическое кольцо является необязательно моно- или дизамещенным заместителями, независимо выбранными из группы, состоящей из:
(a) галогена,
(b) -C1-6алкила, необязательно замещенного CF3,
(c) -O-C1-4алкила,
(d) -CF3 и
(e) C3-6циклоалкила.
8. Соединение по п.7, где заместитель представляет собой фенильное или 5- или 6-членное моноциклическое гетероароматическое кольцо, причем упомянутое гетероароматическое кольцо содержит 1, 2 или 3 гетероатома, выбранные из группы, состоящей из S, О и N, где упомянутое фенильное или гетероароматическое кольцо является необязательно моно- или дизамещенным заместителями, независимо выбранными из группы, состоящей из:
(a) галогена,
(b) -C1-6алкила, необязательно замещенного CF3,
(c) -O-C1-4алкила;
(d) -CF3 и
(e) C3-6циклоалкила.
9. Соединение по п.1 формулы I
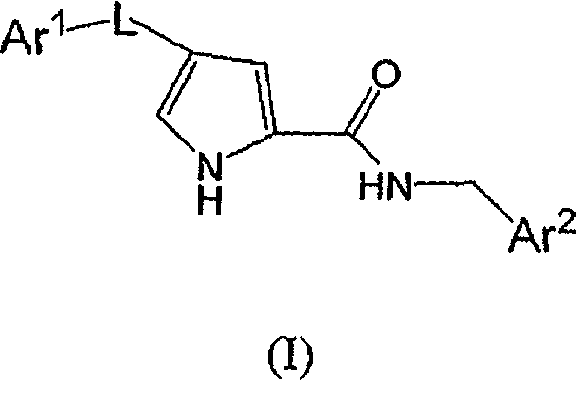
где
L представляет собой -С(O)-;
Ar1 представляет собой необязательно моно-, ди- или тризамещенный фенил, где заместители независимо выбраны из группы, состоящей из:
(a) F,
(b) Cl и
(c) -C1-4алкила;
Ar2 представляет собой необязательно замещенный тиадиазолил, и заместитель представляет собой фенильное или 5- или 6-членное моноциклическое гетероароматическое кольцо, причем упомянутое гетероароматическое кольцо содержит 1, 2 или 3 гетероатома, выбранные из группы, состоящей из S, О и N, где упомянутое фенильное или гетероароматическое кольцо является необязательно моно- или дизамещенным заместителями, независимо выбранными из группы, состоящей из:
(a) галогена,
(b) -C1-4алкила,
(c) -О-C1-4алкила,
(d) -CF3,
(e) C3-6циклоалкила.
10. Соединение по п.1 формулы (II)
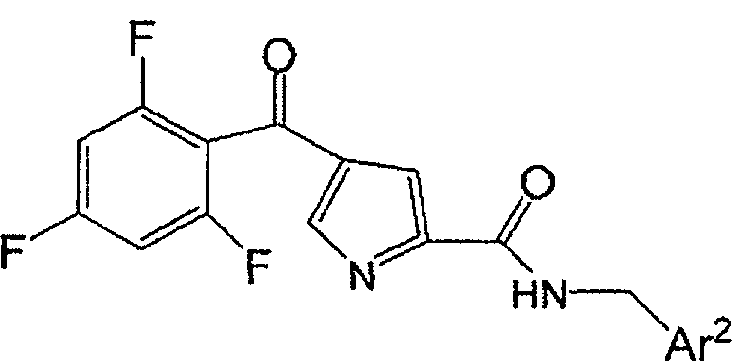
(II)
где
Ar2 представляет собой необязательно замещенный тиадиазолил, где заместитель представляет собой фенильное или 5- или 6-членное моноциклическое гетероароматическое кольцо, причем упомянутое гетероароматическое кольцо содержит 1, 2 или 3 гетероатома, выбранные из группы, состоящей из S, О и N, где упомянутое фенильное или гетероароматическое кольцо является необязательно моно- или дизамещенным заместителями, независимо выбранными из группы, состоящей из:
(a) галогена,
(b) -C2-6алкила,
(c) -O-C1-4алкила и
(d) -CF3.
11. Соединение по п.1, выбранное из группы, состоящей из:
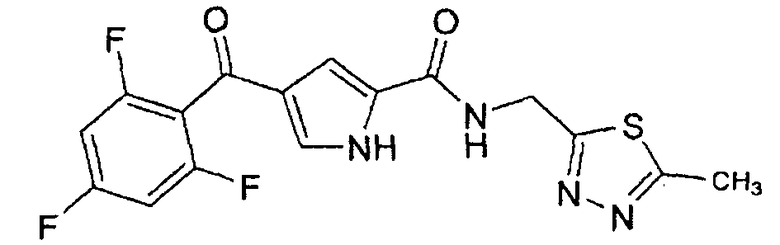
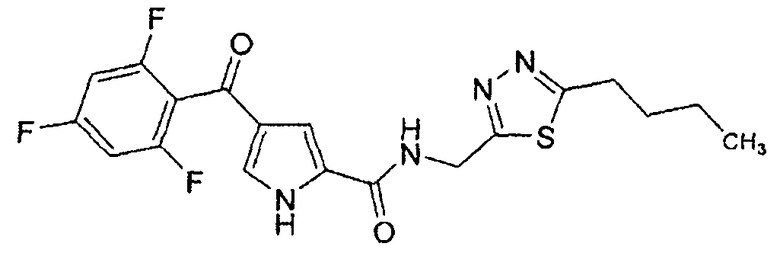
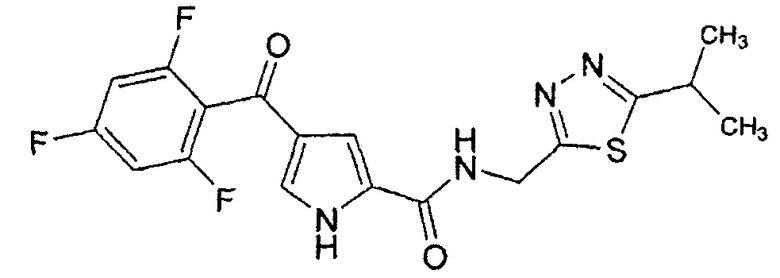
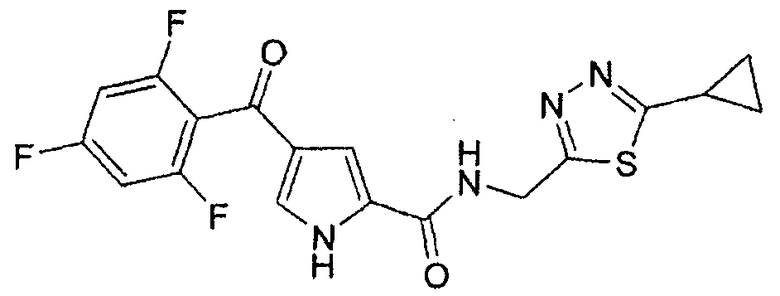
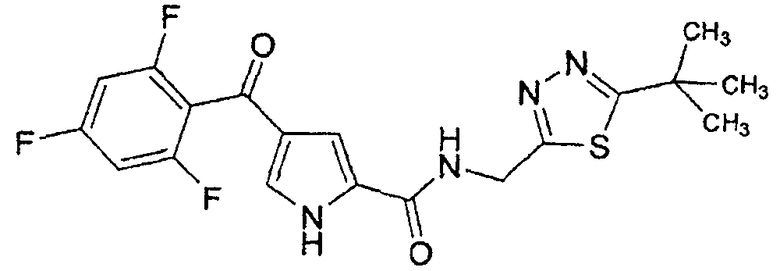
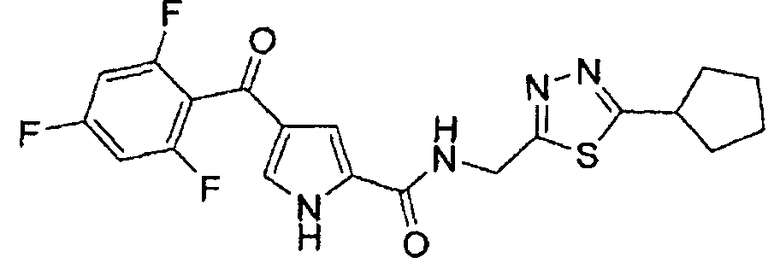
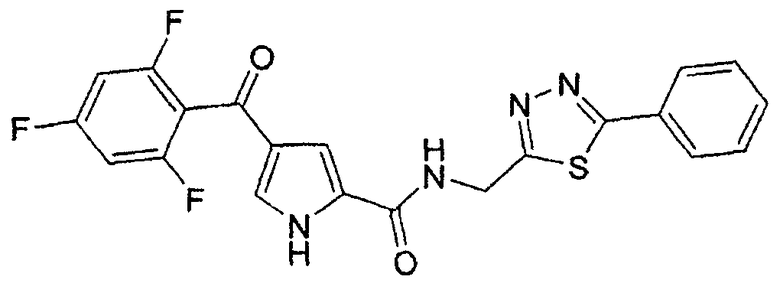
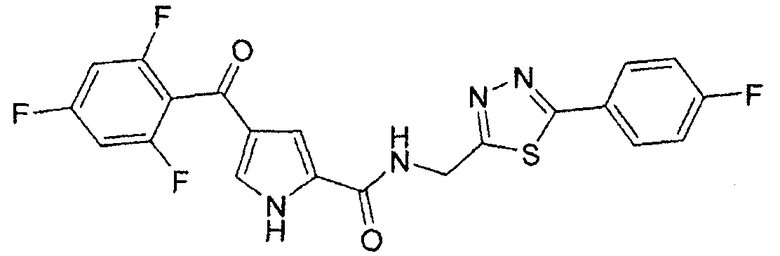
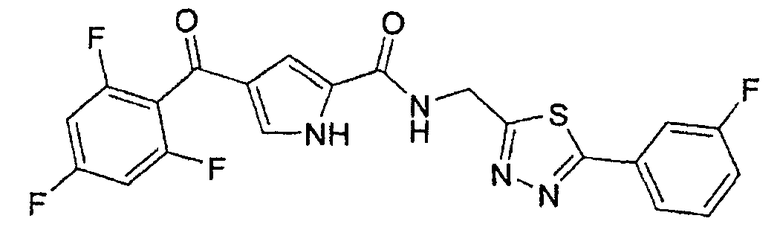
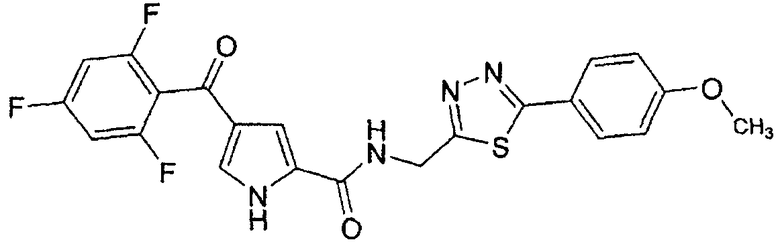
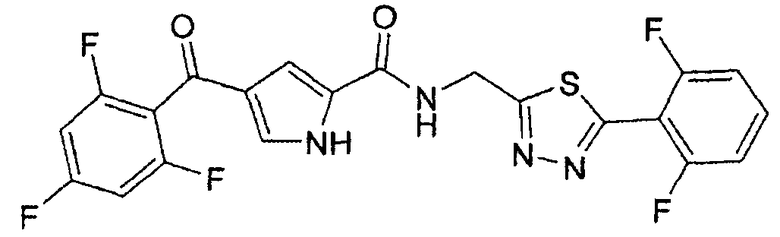
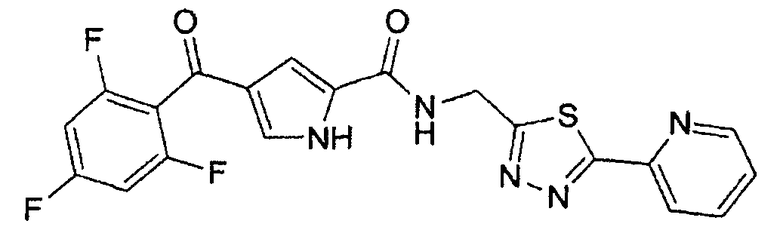
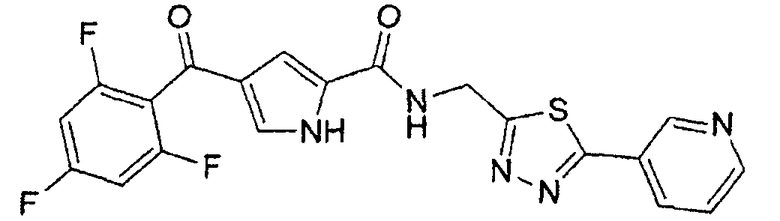

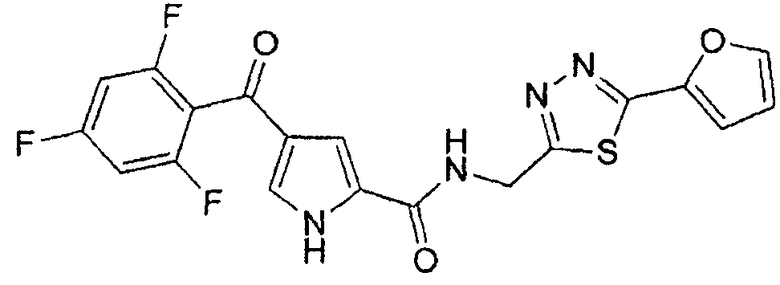
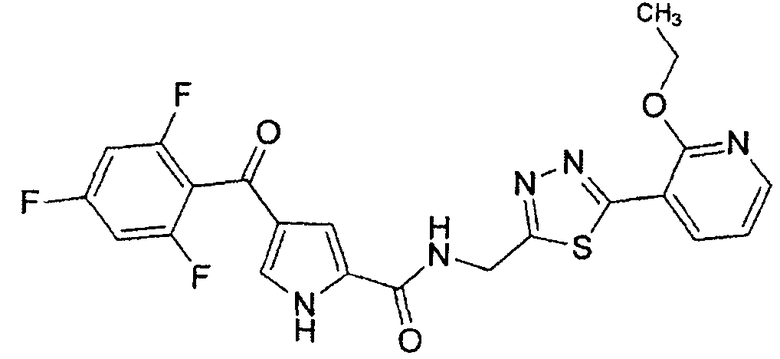
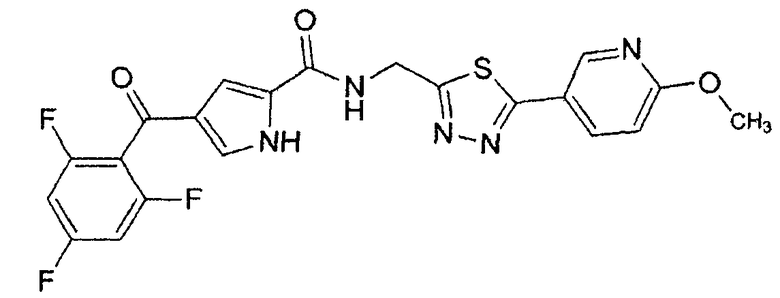
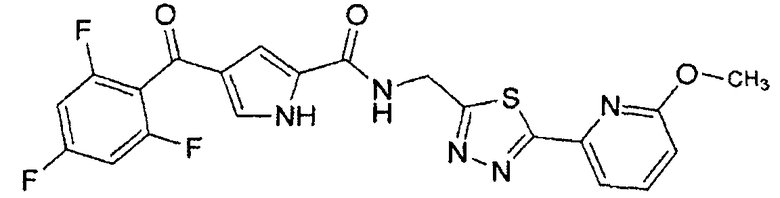
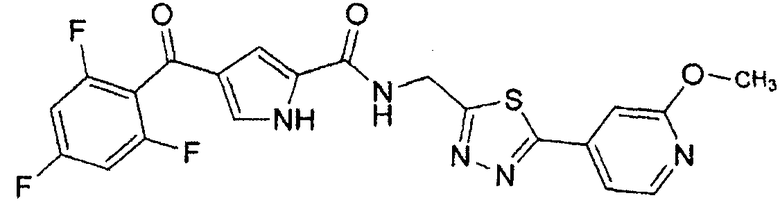
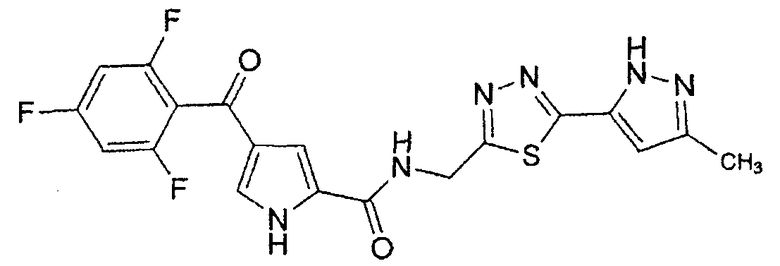
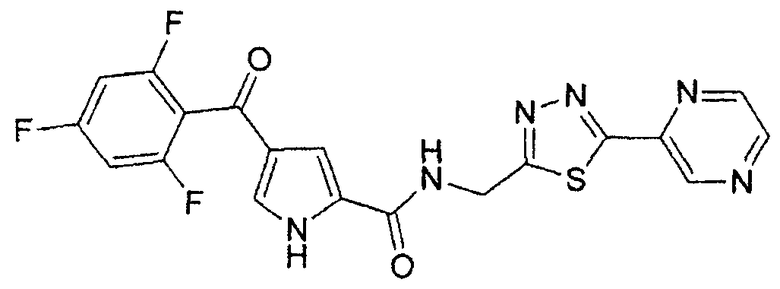
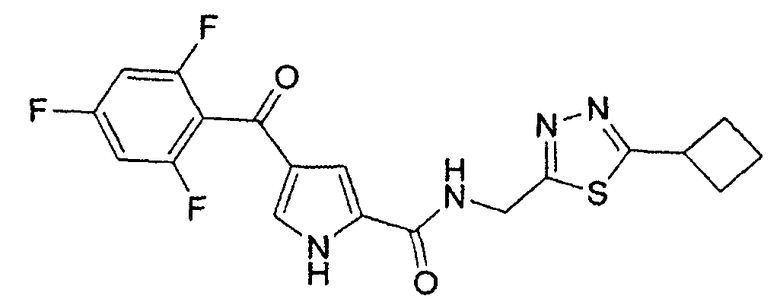
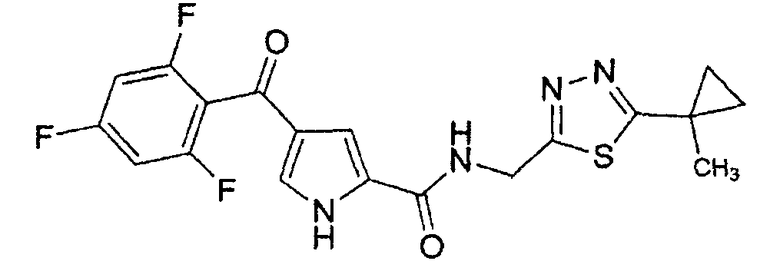
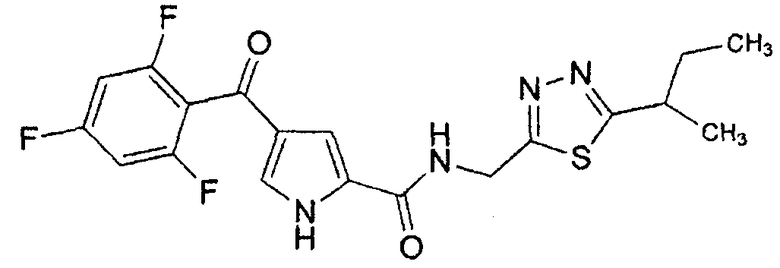
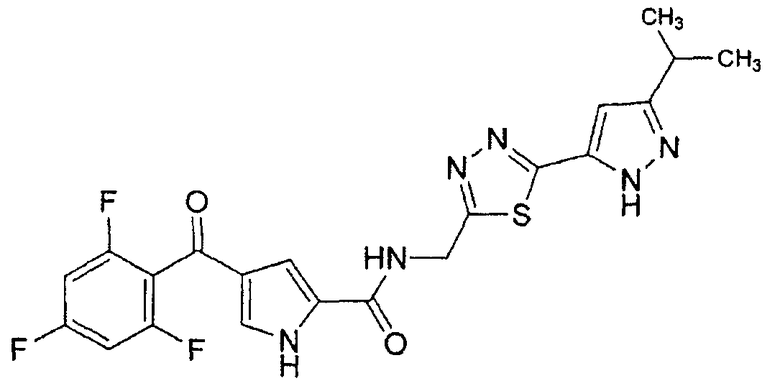
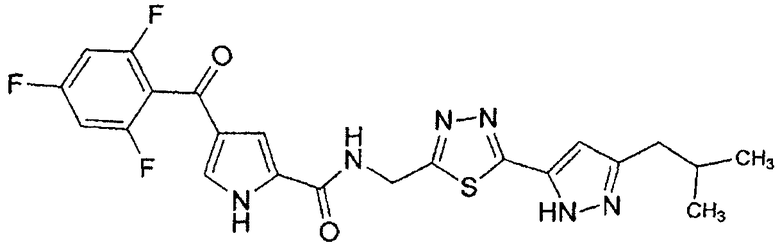
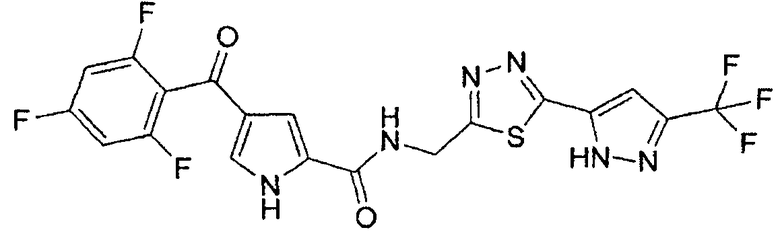
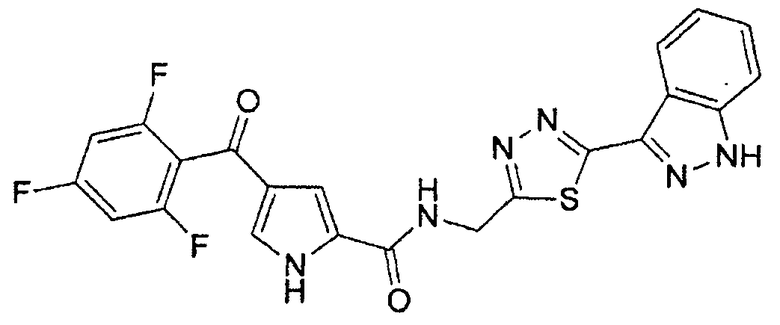
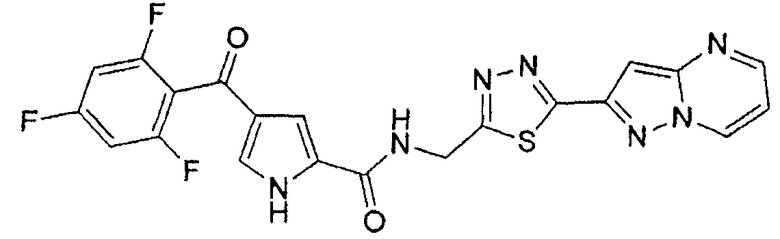
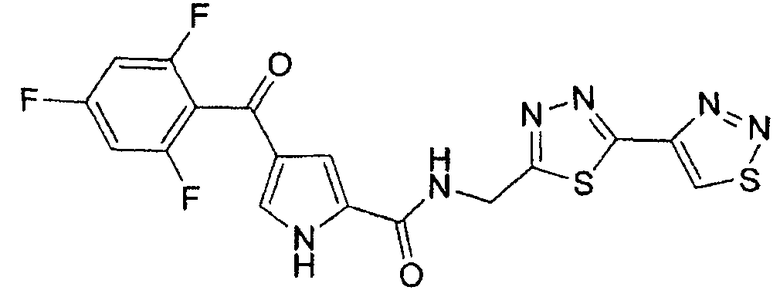
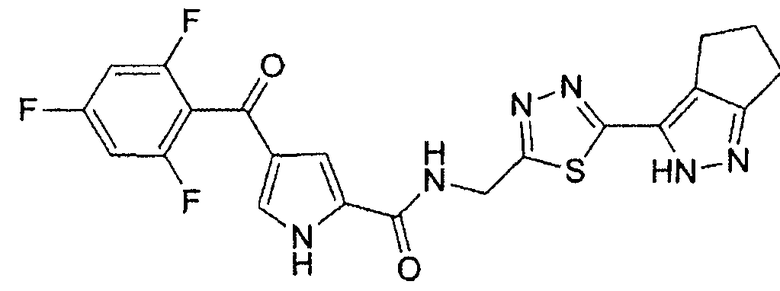
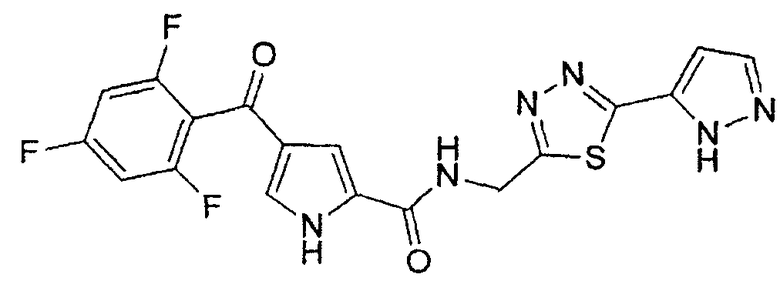
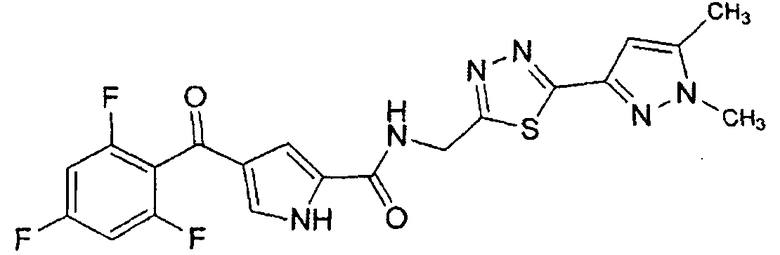
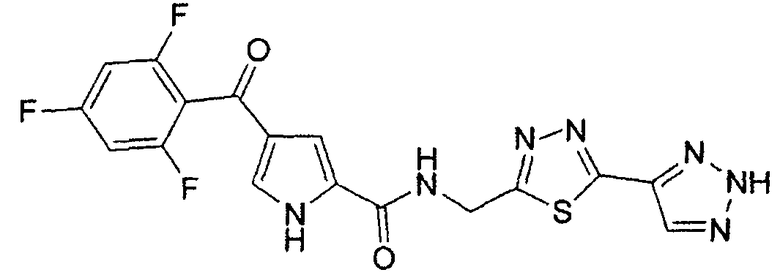
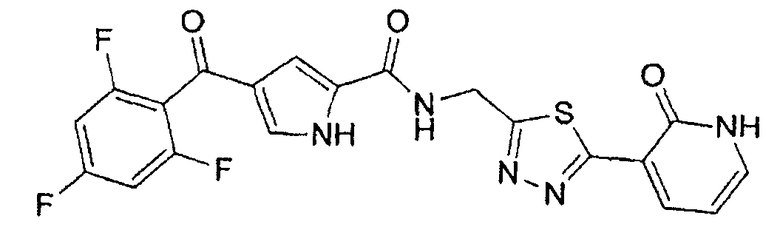
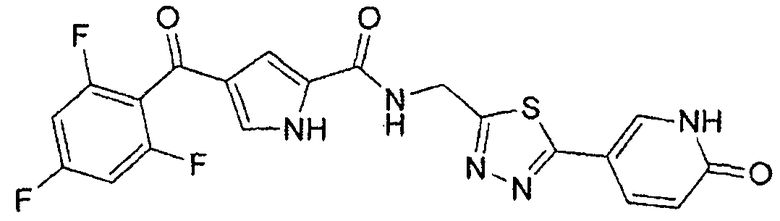
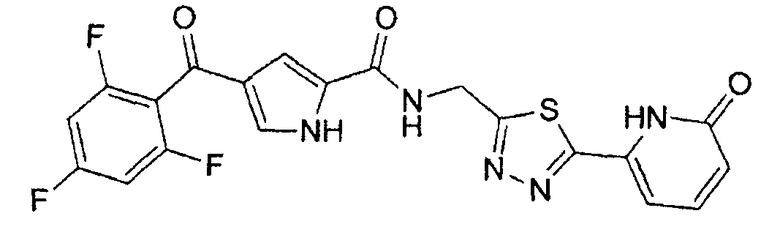
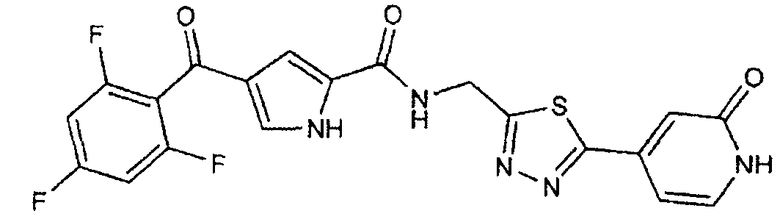
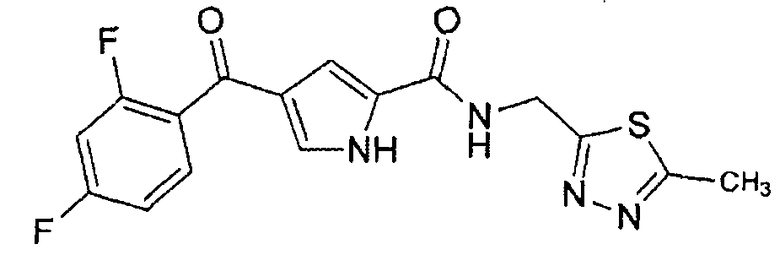
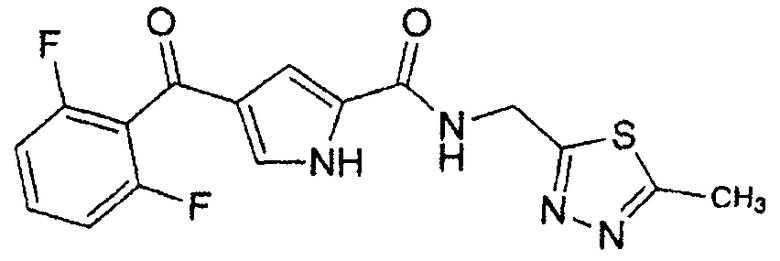
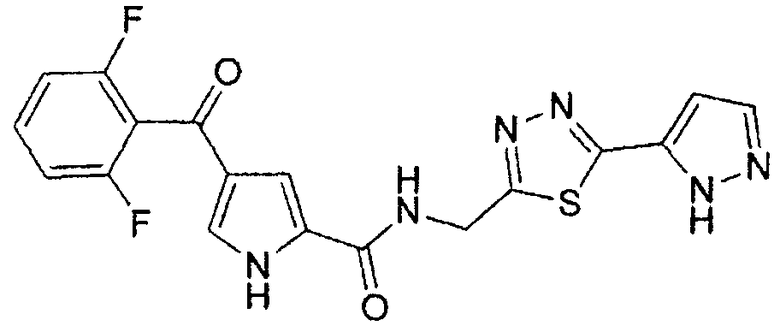
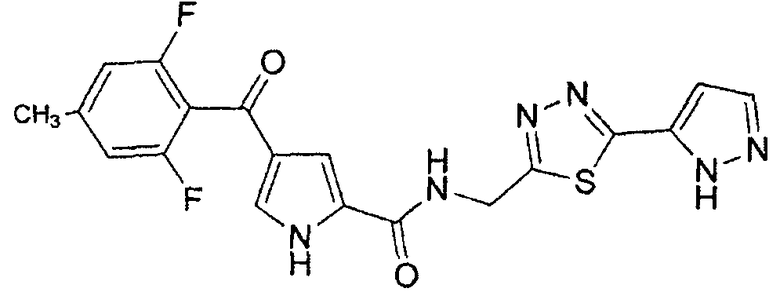
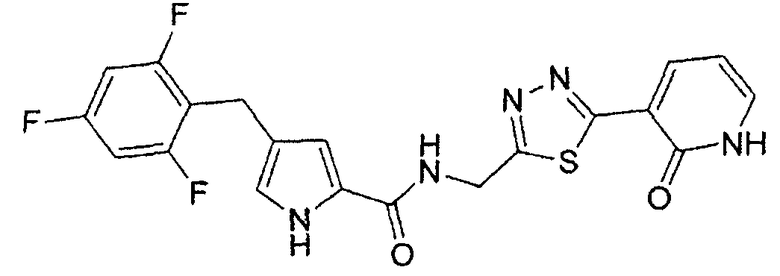
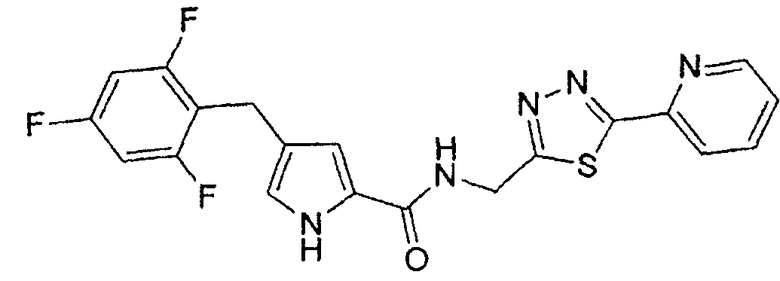
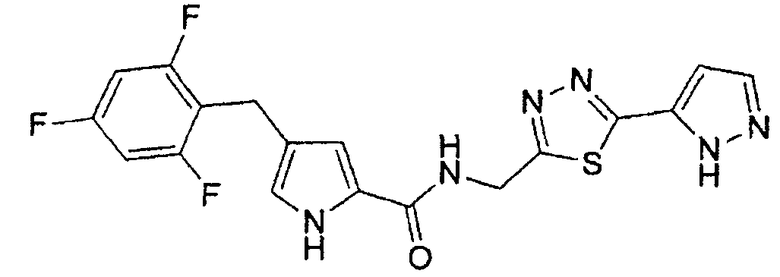
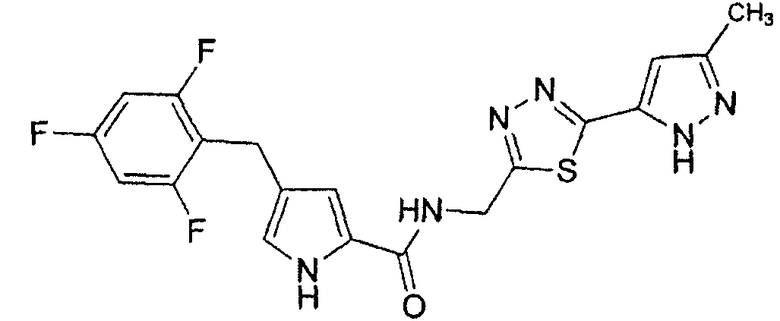
12. Фармацевтическая композиция для профилактики или лечения заболевания, расстройства или состояния, опосредованного цитокином, содержащая инертный носитель и эффективное количество соединения по п.1.
13. Применение соединения по п.1 для получения лекарственного средства для лечения или предотвращения астмы, COPD, ARDS, ревматоидного артрита, ревматоидного спонделита, остеоартрита или подагрического артрита.
14. Соединение, имеющее следующую структуру:
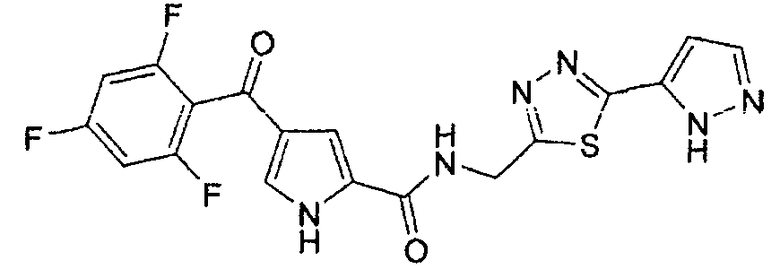
15. Фармацевтическая композиция для профилактики или лечения заболевания, расстройства или состояния, опосредованного цитокином, содержащая инертный носитель и эффективное количество соединения по п.14.
16. Применение соединения по п.14 для получения лекарственного средства для лечения или предотвращения астмы, COPD, ARDS, ревматоидного артрита, ревматоидного спонделита, остеоартрита или подагрического артрита.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| US 20040077707 A1, 22.04.2004 | |||
| RU 2007107388 A, 10.09.2008 | |||

