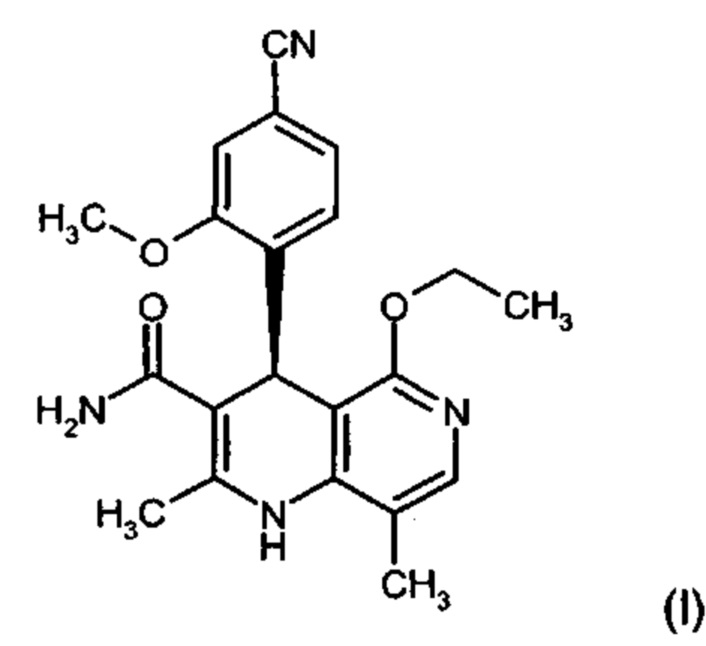
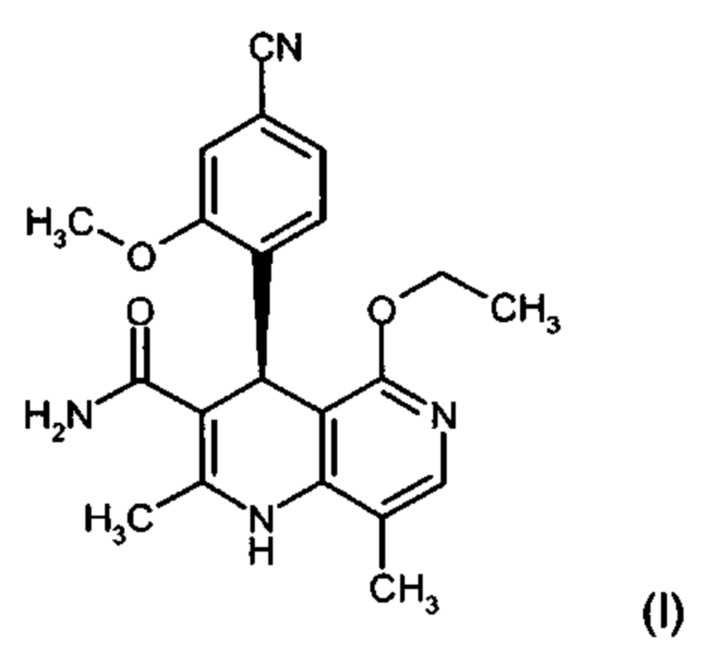
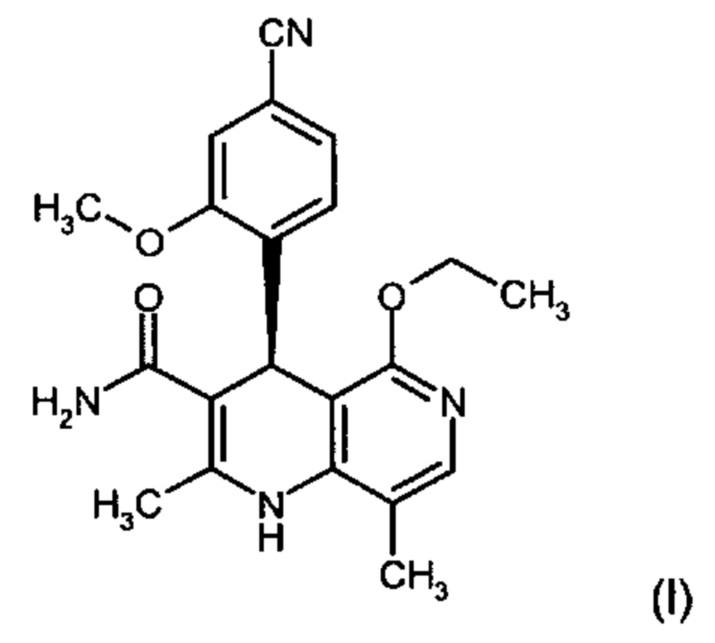
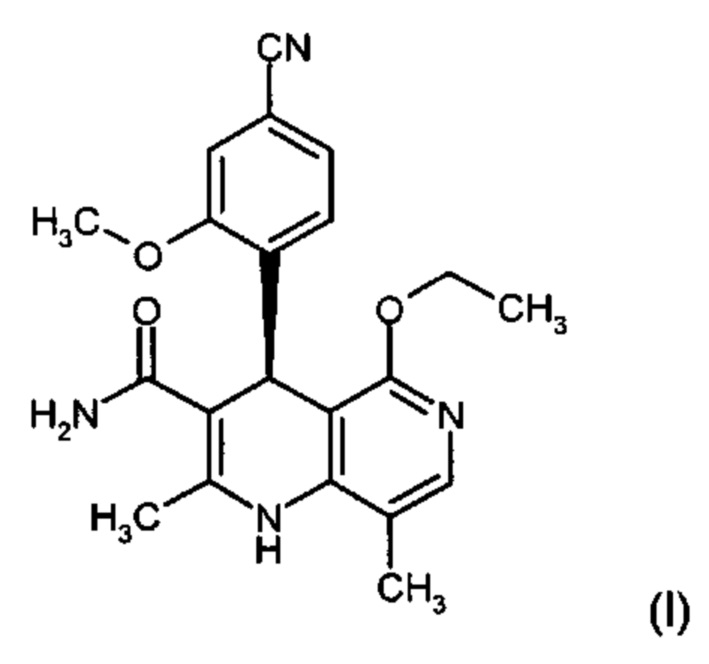
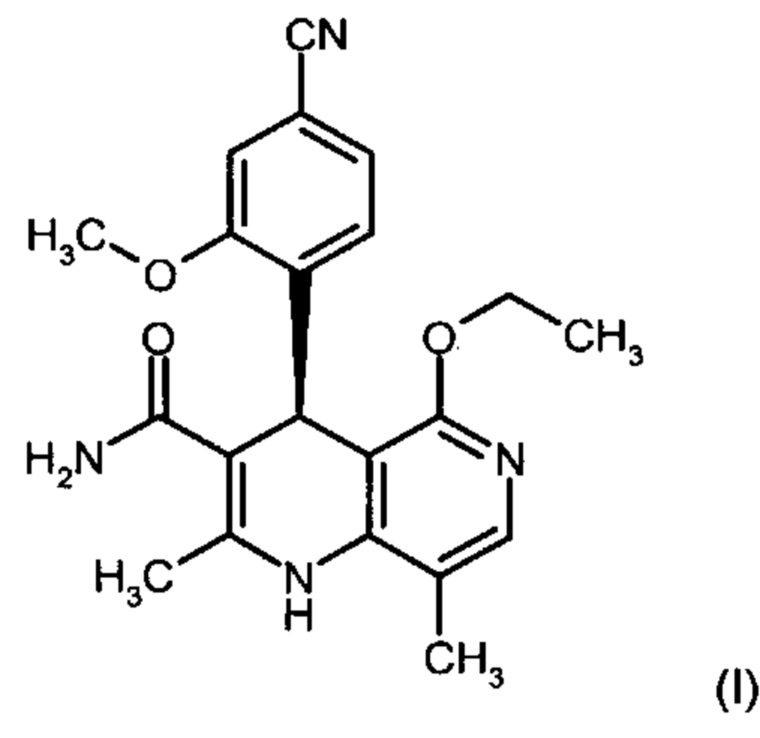
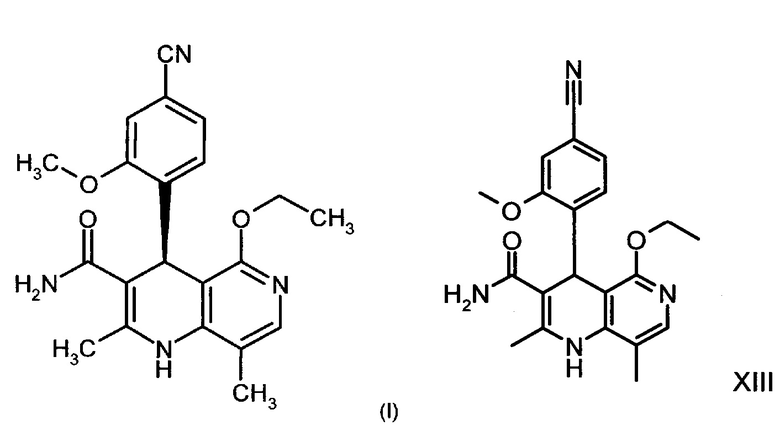
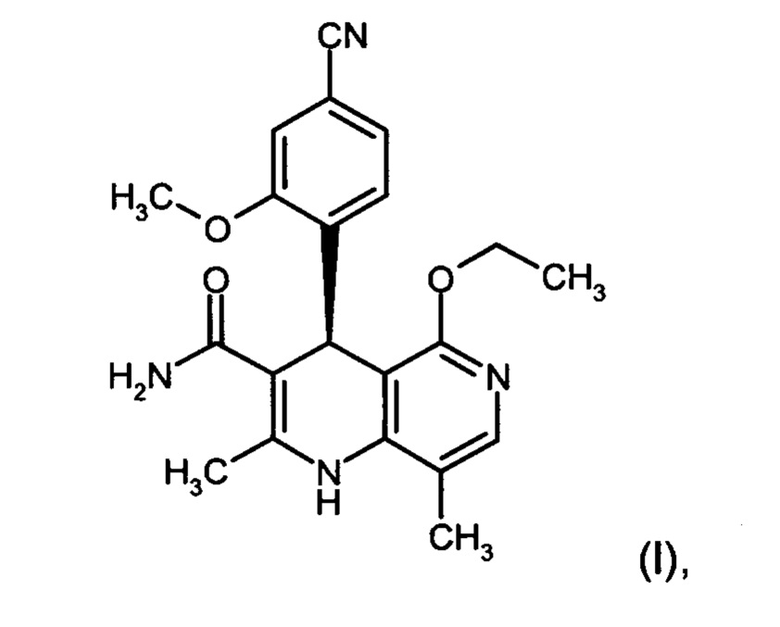
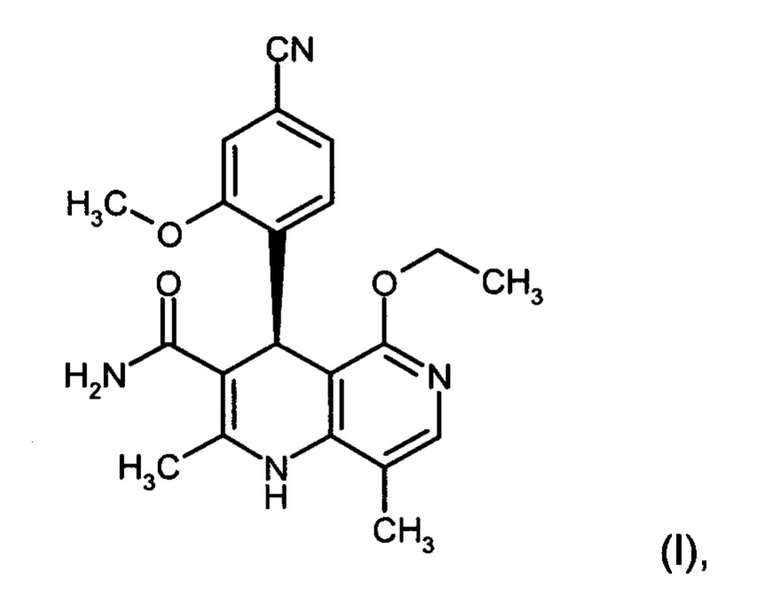
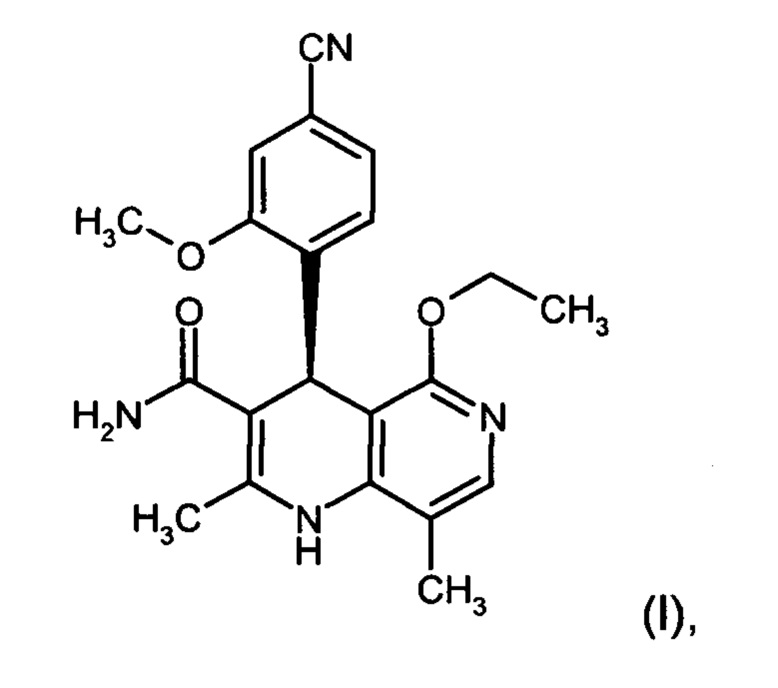
Изобретение относится к новому улучшенному способу получения (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида формулы (I):
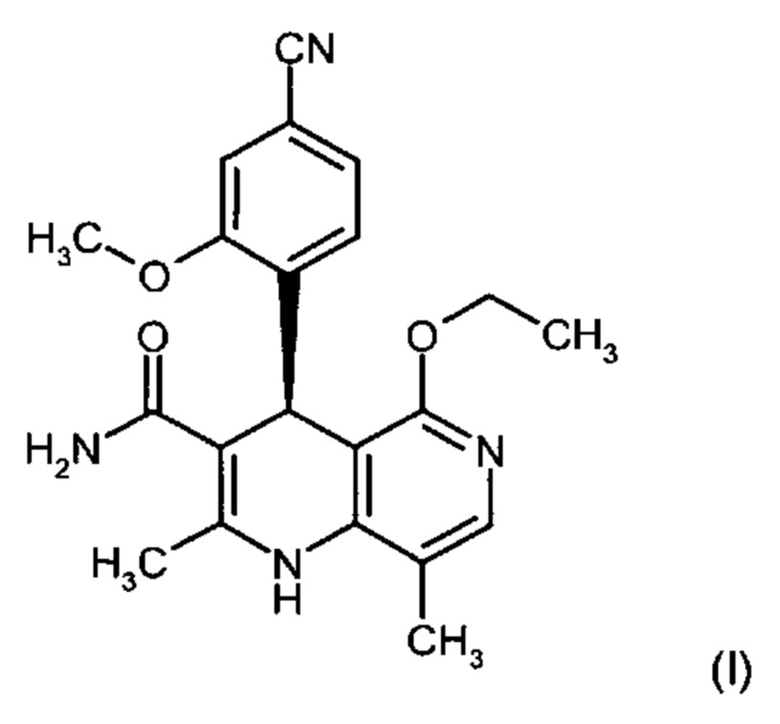 .
.
Соединение формулы (I) обладает действием нестероидного антагониста минералокортикоидного рецептора и может найти применение в качестве средства для профилактики и/или лечения сердечнососудистых и почечных заболеваний, например, сердечной недостаточности и диабетической нефропатии.
Соединение формулы (I) и процесс его получения описаны в международной заявке WO 2008/104306 и ChemMedChem 2012, 7, с. 1385, причем в обеих публикациях подробно обсуждаются результаты опытного синтеза указанного соединения. Недостаток подобного опытного синтеза соединения формулы (I) состоит в том, что он непригоден для реализации в промышленном масштабе, поскольку многие его стадии протекают при очень сильном разбавлении и очень большом избытке реагентов, а, следовательно, указанное соединение получают с относительно низким общим выходом. Кроме того, подобный синтез требует осуществления множества промежуточных стадий хроматографической очистки, как правило, отличающихся очень высокой технической трудоемкостью, необходимостью использования больших количеств растворителей и высокой затратностью, а следовательно, по возможности подлежащих исключению. Некоторые стадии не могут быть реализованы в промышленном масштабе в связи с проблемами безопасности и технологическими затруднениями.
Таким образом, существует потребность в реализуемом в промышленном масштабе синтезе, позволяющем с высоким общим выходом и низкими производственными расходами воспроизводимо получать высокочистое соединение формулы (I), удовлетворяющее всем требованиям, регламентируемым при клинических испытаниях биологически активных веществ и последующем ведомственном согласовании.
В соответствии с настоящим изобретением обнаружен чрезвычайно эффективный синтез, который удовлетворяет указанным выше требованиям.
Описанный в ChemMedChem 2012, 7, с. 1385 опытный синтез соединения формулы (I) из ванилина включает десять стадий, причем общий выход указанного соединения от теоретического составляет 3,76%. Соединение формулы (I) в виде аморфного твердого вещества получают посредством упаривания хроматографических фракций, причем на конечной стадии для установления полиморфии используют до последнего времени не описанный метод кристаллизации.
Известный метод получения соединения формулы (I) показан на схеме 1:
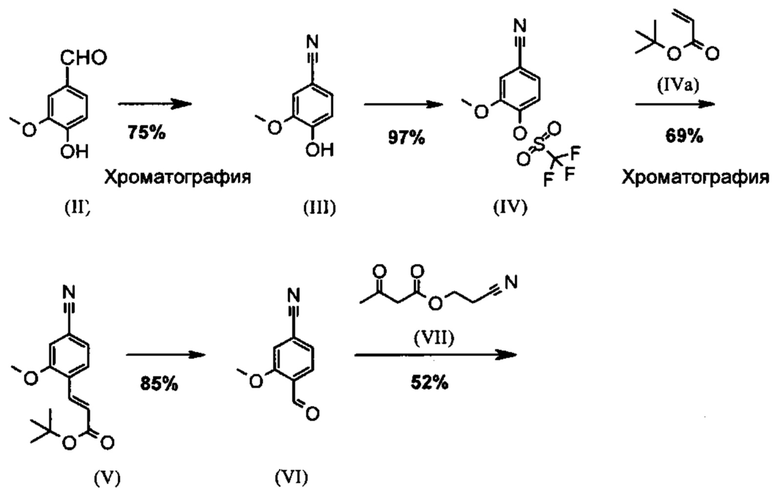
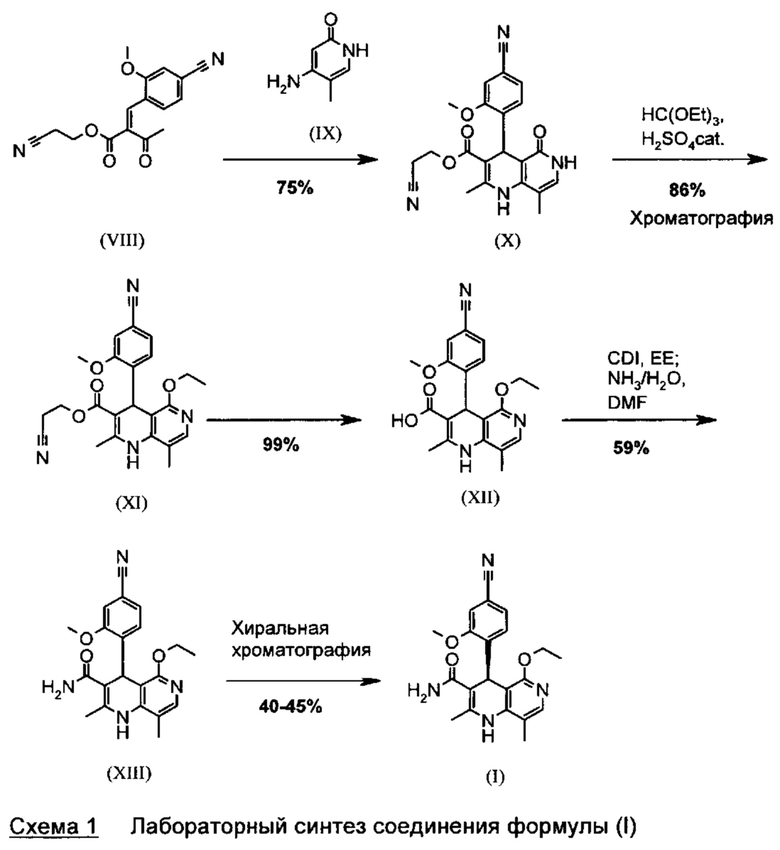
Опытный синтез включает три стадии хроматографической очистки, а также стадию хиральной хроматографии для разделения энантиомеров рацемата формулы (XIII). Часть стадий осуществляют при очень сильном разбавлении и использовании очень больших количеств реагентов.
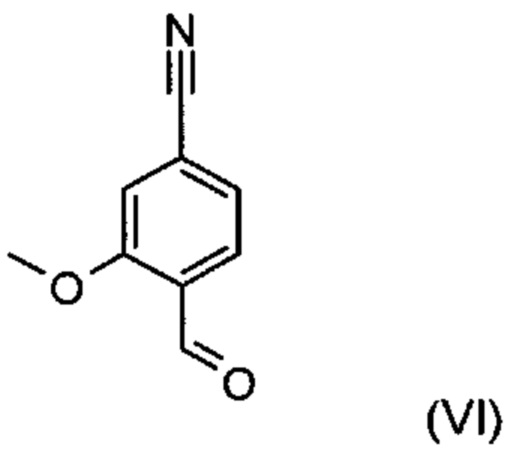
С точки зрения атомной эффективности неприемлемой, в частности, является также последовательность получения промежуточного нитрилальдегида (VI), играющая центральную роль при данном синтезе.
На схеме 2 показан предлагаемый в изобретении новый способ четырех-стадийного получения соединения формулы (I) из нитрилальдегида (VI) без промежуточных стадий хроматографической очистки с общим выходом 31,5% от теоретического (выход из альдегида (VI) в соответствии с указанным выше опытным синтезом составляет 8,8%).
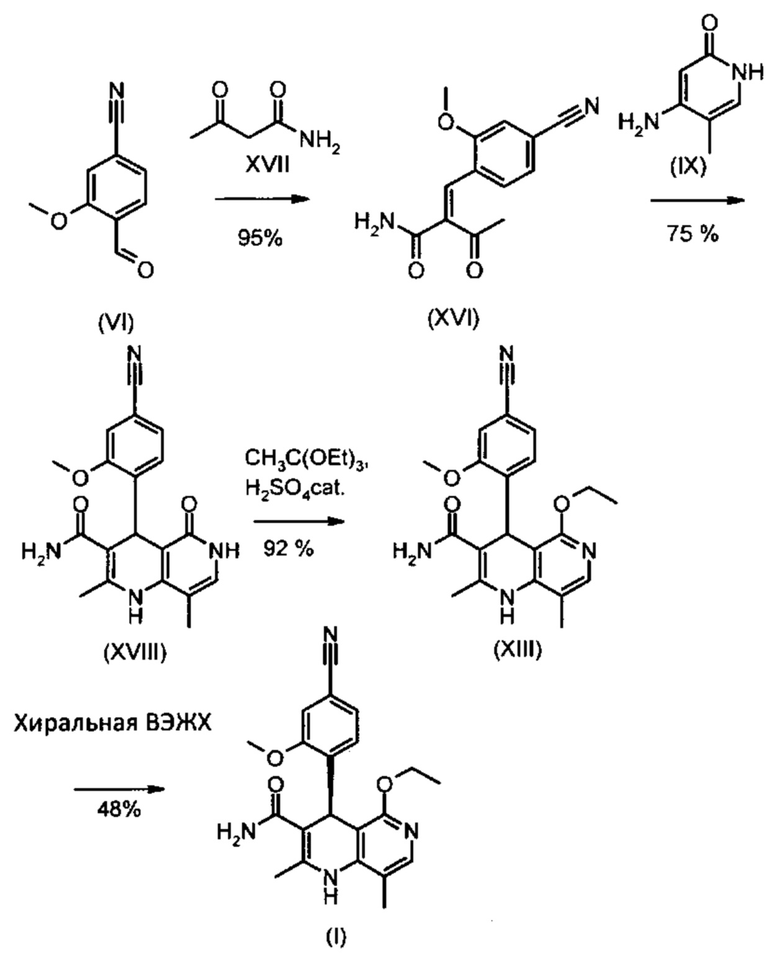
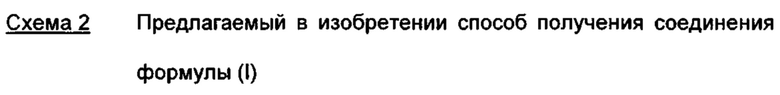
При этом для разделения энантиомеров используют метод препаративной хиральной высокоэффективной жидкостной хроматографии при высоком давлении (например, технологию хроматографического разделения на установке SMB, Varicol).
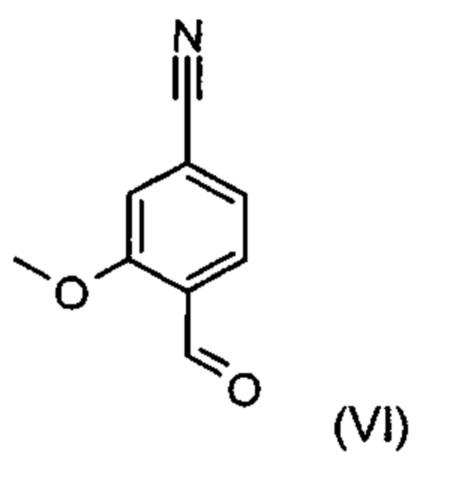
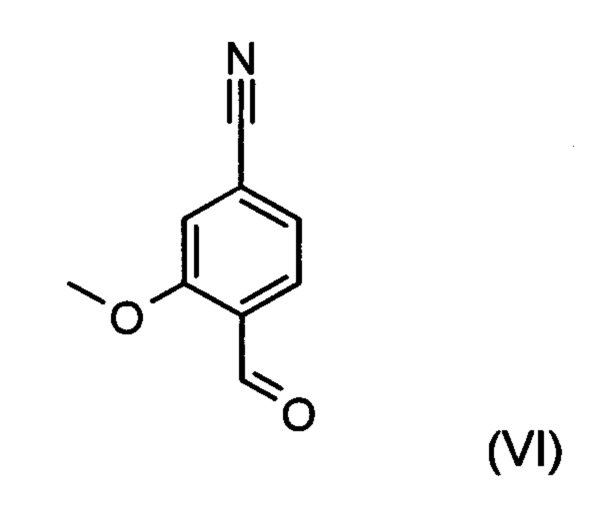
Альдегид формулы (VI) является известным из литературы соединением (J. Med. Chem., 2007, 50, сс. 2468-2485), получение которого представляет собой важную промежуточную стадию предлагаемого в изобретении ситеза. Кроме того, существует возможность комммерческого приобретения этого соединения.
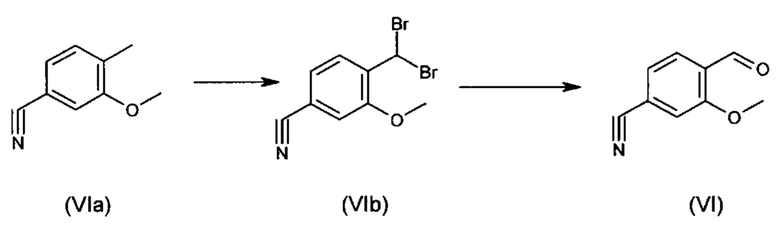
Из 4-циано-2-метокситолуола (VIa) с использованием N-бромсукцинамида получают дибромид (VIb), который в этаноле превращают с 2,46 эквивалентами нитрата серебра (в воде), получая целевой альдегид (VI). Подобный описанный в литературе синтез, а также указанный выше опытный синтез совершенно непригодны для перехода от опытного к крупнотоннажному промышленному производству, что обусловливает высокую потребность в новом более эффективном и экономичном синтезе.
Галогенбензойные кислоты (XIV) и (XIVa):
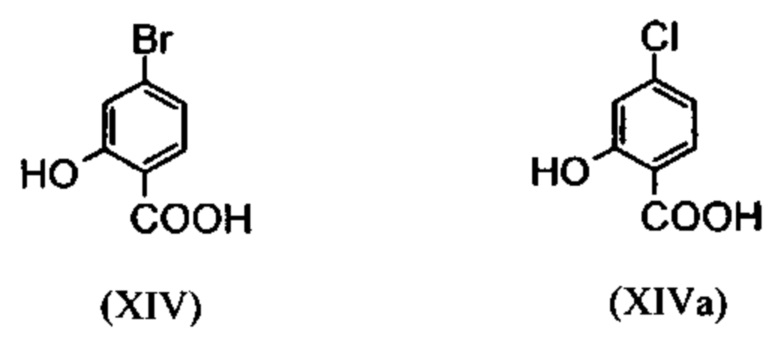
являются доступными в больших количествах торговыми продуктами. Был разработан весьма эффективный и недорогой процесс, в соответствии с которым промежуточные продукты (XV) и (XIX):
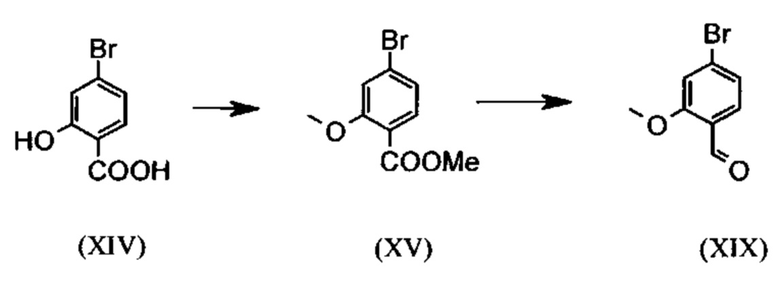
не выделяют, а подвергают дальнейшему превращению в растворе. Это оказывается возможным благодаря достижению при соответствующем превращении очень высокого выхода и степени чистоты (выход от теоретического более 95%). Простой/сложный метиловый эфир (XV) является известным из литературы соединением (Journal of Medicinal Chemistry, 1992, том 35, cc. 734-740), которое получают путем взаимодействия с чрезвычайно летучим, вредным для здоровья и дорогостоящим метил-йодидом.
Обнаружено, что для аналогичного превращения можно использовать труднолетучий и более дешевый диметилсульфат. Превращение кислоты (XIV) с диметилсульфатом в простой/сложный метиловый эфир (XV) осуществляют в растворителе, например, ацетоне, 2-бутаноне, тетрагидрофуране, 2-метилтетрагидрофуране, диметилформамиде, диметилацетамиде или N-метилпирролидоне, при температурах от 50 до 100°С и дополнительном использовании вспомогательного основания, например, карбоната калия, карбоната натрия, карбоната кальция, карбоната лития, N-метилимидазола, триэтиламина, пиридина или 2,6-лутидина. Речь при этом идет об известном специалистам методе этерификации кислот и получении простых эфиров фенолов (Tetrahedron, 2013, том 69, сс. 2807-2815, Journal of the American Chemical Society, 2013, том 135, cc. 5656-5668). Особенно предпочтительным является превращение в ацетоне при нагревании с обратным холодильником (56°С) и использовании диметилсульфата и карбоната калия. При этом диметилсульфат в течение четырех часов добавляют к кипящей реакционной смеси. Ацетон отгоняют и заменяют толуолом (редистилляция). Для переработки добавляют воду (деструкция избыточного диметилсульфата), толуольную фазу отделяют, промывают водой и насыщенным раствором поваренной соли, а затем до определенного объема отгоняют толуол (служит для азеотропной сушки, то есть для удаления воды на следующей стадии). Согласно результатам анализа раствора наблюдается почти полное превращение (более 96% от теоретического). Вместо бромсодержащего соединения можно использовать хлорсодержащее соединение, причем достигаемое превращение аналогично бромсодержащему соединению.
Получение альдегида (XIX) описано в литературе, например, в патенте США US 2008/312209 А1 (фирма Glaxo Group Limited, 2008), European Journal of Medicinal Chemistry, 1986, том 21, cc. 397-402, European Journal of Medicinal Chemistry, 1992, том 35, cc. 734-740, Journal of Medicinal Chemistry, 2011, том 21, cc. 9523-9531. Однако используемые для превращения исходные вещества очень дороги и недоступны в больших количествах, в связи с чем был разработан новый способ получения альдегида (XIX) из простого/сложного метилового эфира (XV). При этом превращение соединения (XV) в альдегид (XIX) осуществляют с продуктом REDAL (натрий-бис(2-метоксиэтокси)алюминий дигидридом) в толуоле при добавлении N-метилпиперазина. Данный метод описан в литературе (Synthesis, 2003, №6, 823-828, Tetrahedron, 57 (2001), cc. 2701-2710). В случае если реакцию осуществляют при стехиометрии, аналогичной указанной в литературе, помимо альдегида в реакционной смеси обнаруживают другое соединение. Речь при этом идет о соответствующем бензиловом спирте, образующемся в количестве до 10% вследствие перевосстановления. Важным обстоятельством при этом является установление точного соотношения между REDAL и N-метилпиперазином (1,21 эквивалента REDAL+1,28 эквивалента N-метилпиперазина), что позволяет уменьшить количество указанного выше побочного продукта, который мешает реализуемой на последующей стадии кристаллизации, до значений, составляющих менее 1%. Для этого при температуре от 0 до 5°С загружают 65-процентный раствор REDAL в толуоле (предпочтительно 1,21 эквивалента), к которому добавляют 1,28 эквивалента N-метилпиперазина. Полученный при этом раствор REDAL с N-метилпиперазином примерно в течение 30 минут добавляют к загруженному раствору сложного бромметилового эфира (XIV) в толуоле, после чего реагенты в течение часа перемешивают при 0°С. Реакционный раствор гасят водой/кислотой, предпочтительно водной серной кислотой, толуольную фазу отделяют, а затем промывают водой и насыщенным раствором поваренной соли. Толуол отгоняют и посредством редистилляции заменяют на диметилформамид (растворитель для следующей стадии). Достигаемый в результате указанного превращения выход от теоретического, как правило, составляет более 94%. Подобное превращение с аналогичными выходами протекает при использовании соответствующего хлорсодержащего соединения. Раствор диметилформамида непосредственно используют для осуществления последующей реакции.
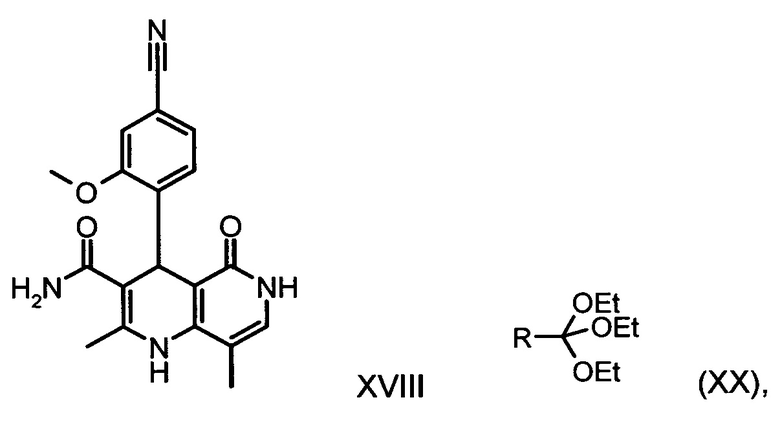
В ходе дальнейшего синтеза бромальдегид (XIX) известными специалистам методами (Synth. Commun. 1994, сс. 887-890, Angew. Chemie 2003, сс. 1700-1703, Tetrahedron Lett. 2007, сс. 2555-2557, Tetrahedron Lett. 2004, cc. 1441-1444, JACS 2003, 125, cc. 2890-2891, Journal of Organometallic Chemistry 689 (2004), cc. 4576-4583) превращают в нитрил, получая нитрилальдегид (VI). В случае бромсодержащего соединения особенно предпочтительным оказывается катализируемое палладием превращение с гексацианоферратом калия *3H2O в качестве источника цианида (Tetrahedron Lett. 48 (2007), сс. 1087-1090). При этом бромальдегид (XIX) вводят в 8-10-кратный избыток диметилформамида, добавляют 0,22 эквивалента гексацианоферрата калия *3H2O и 1,0 эквивалент карбоната натрия, а затем 0,005 эквивалента ацетата палладия. Реагенты в течение трех часов нагревают при 120°С. Раствор охлаждают до 20°С, а затем добавляют воду и этилацетат. Отделяют этилацетатную фазу, водную фазу вновь промывают этилацетатом, после чего этилацетат в объединенных этилацетатных фазах посредством редистилляции заменяют на изопропанол. Продукт при температуре кипения осаждают водой. Выделенный продукт сушат в вакууме. В некоторых случаях продукт осаждают из диметилформамида непосредственно путем добавления воды и после выделения и сушки непосредственно используют на следующей стадии. При описанном выше превращении выходы от теоретического, как правило, составляют более 85%. Использования ацетата палладия для превращения хлорсодержащего соединения недостаточно; в данном случае предпочтительными являются известные специалистам палладиевые катализаторы, описанные в Tetrahedron Lett. 48 (2007), сс. 1087-1090; при этом выходы от теоретического несколько ниже по сравнению с бромсодержащим соединением и, как правило, составляют от 80 до 85%.
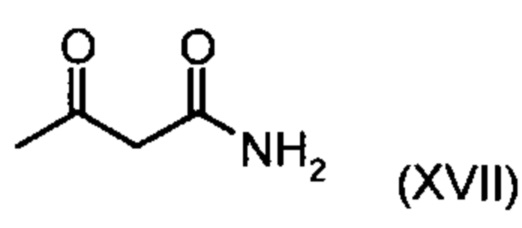
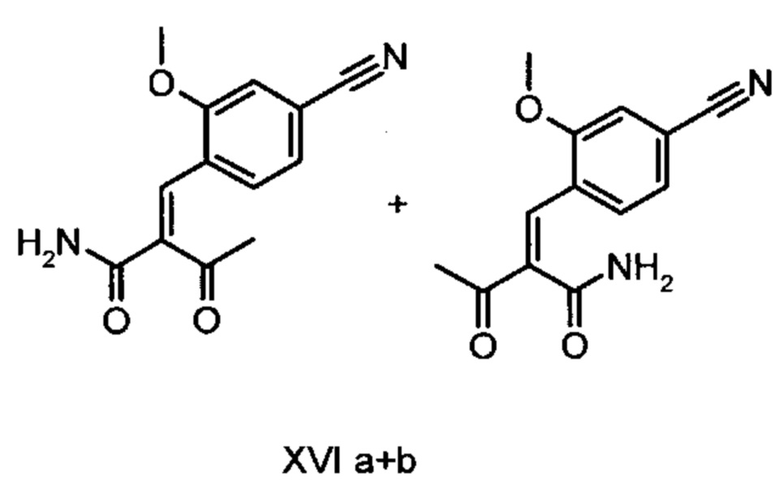
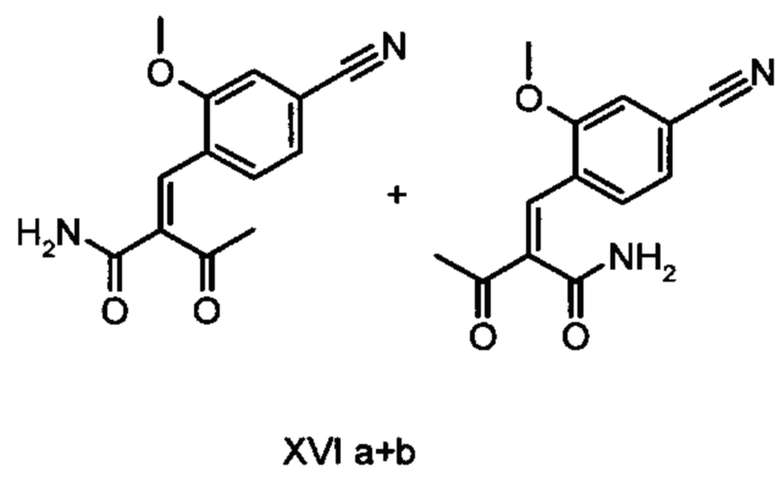
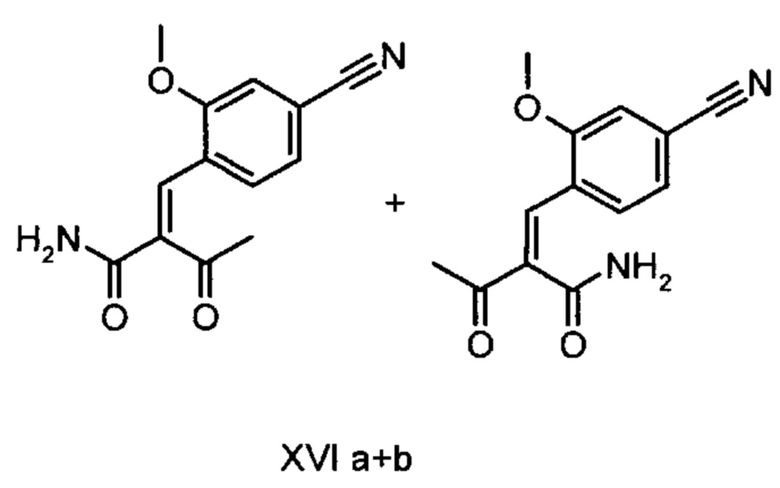
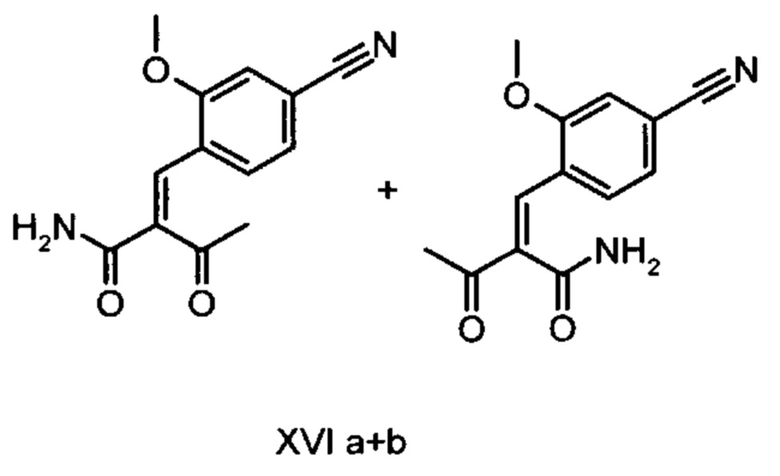
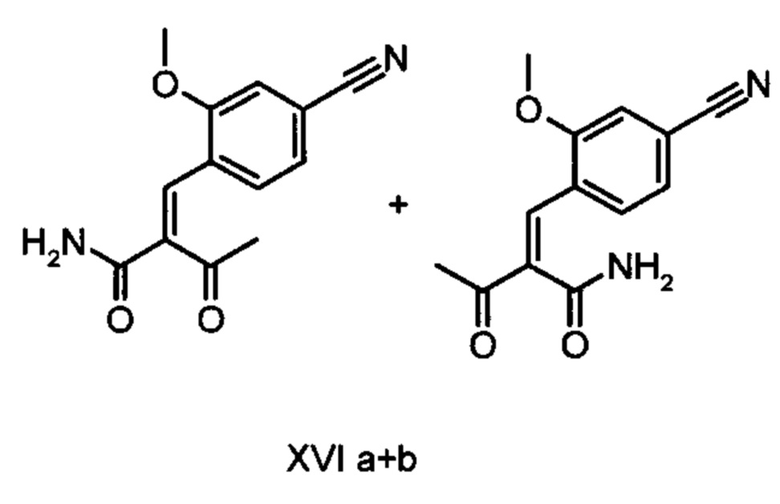
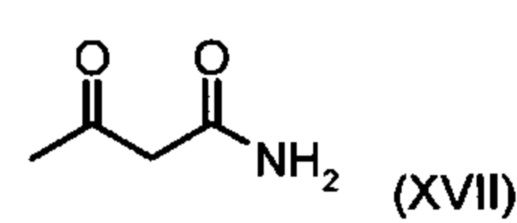
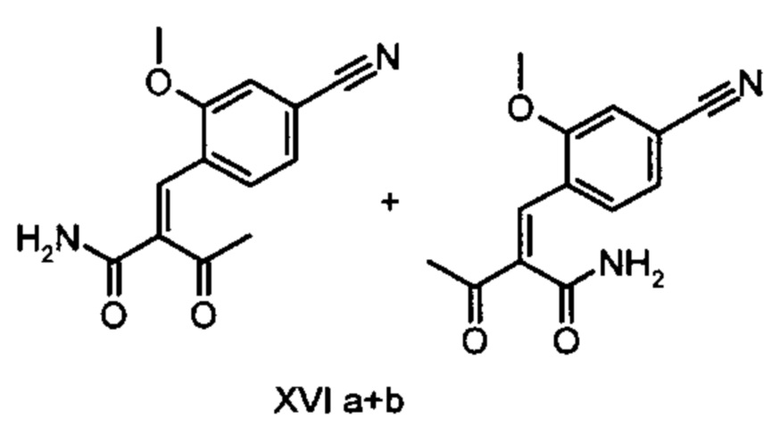
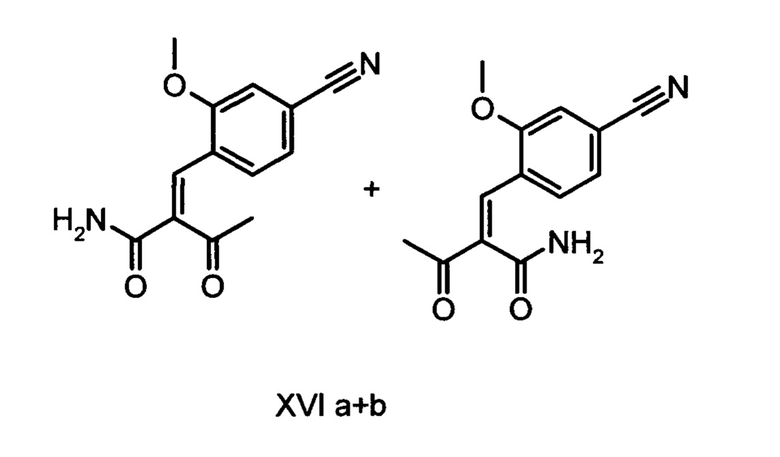
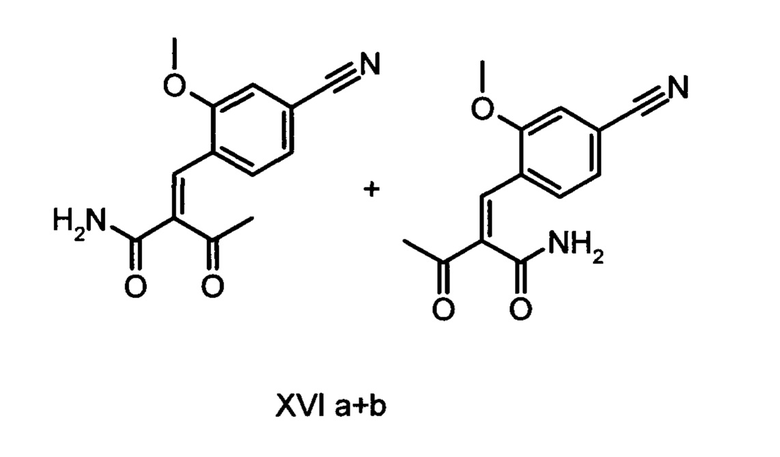
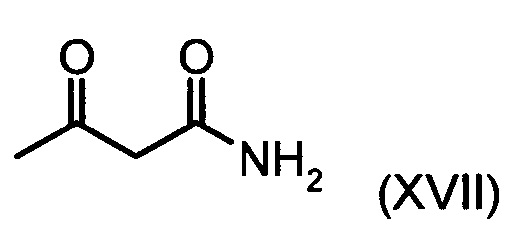
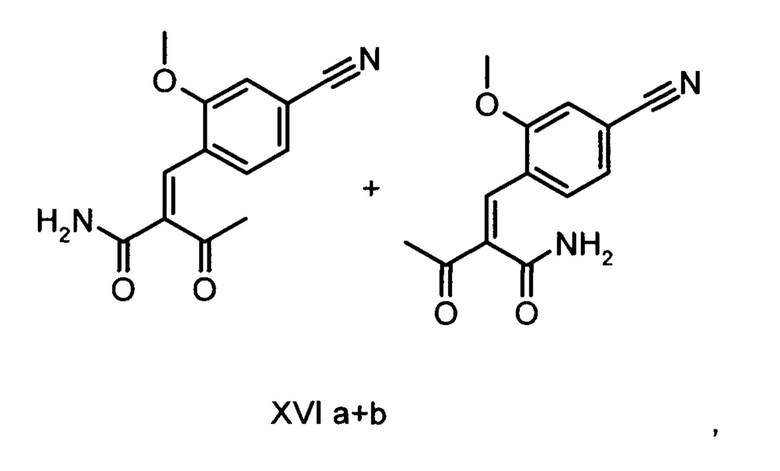
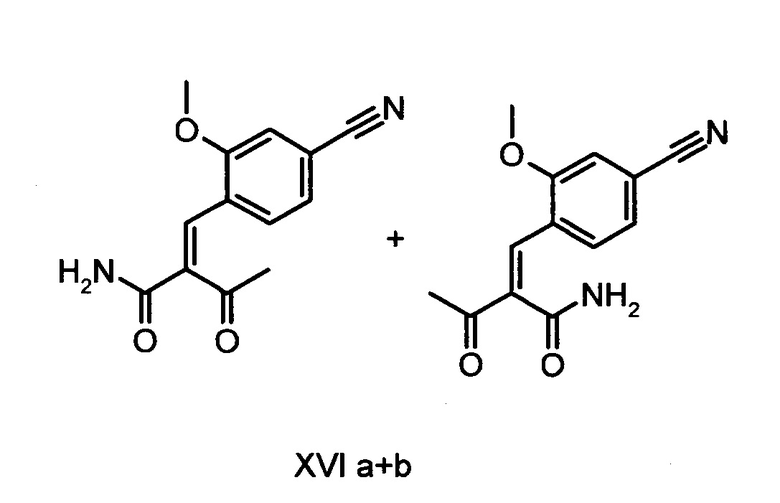
Коричный амид (XVI а, b) в виде смеси E/Z-изомеров получают из альдегида формулы (VI) по реакции Кневенагеля с кетоамидом (XVII):
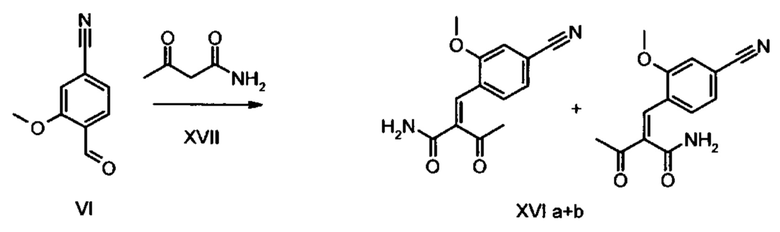
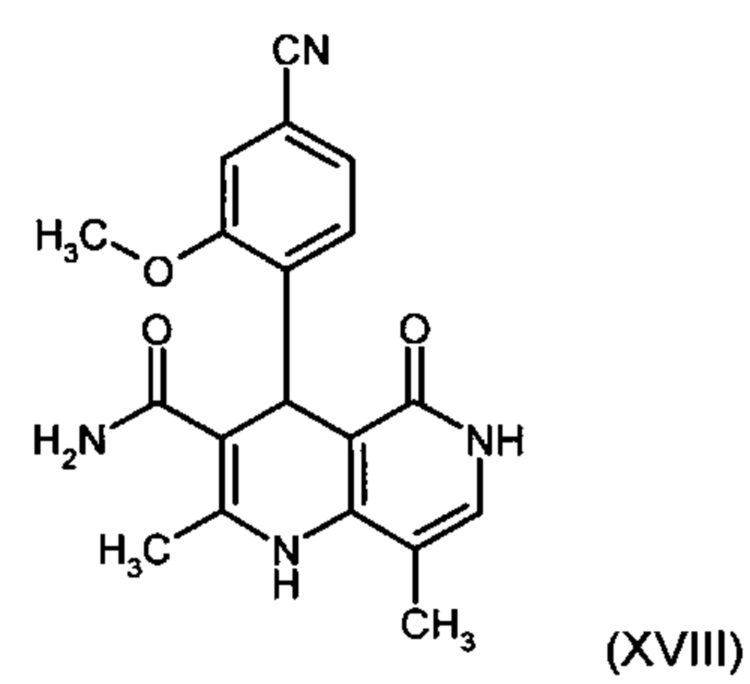
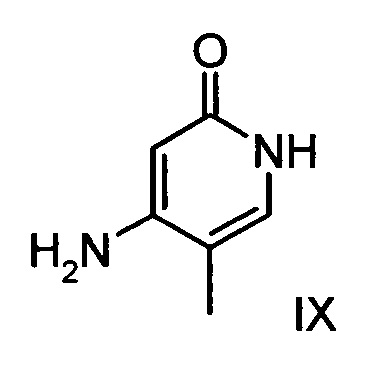
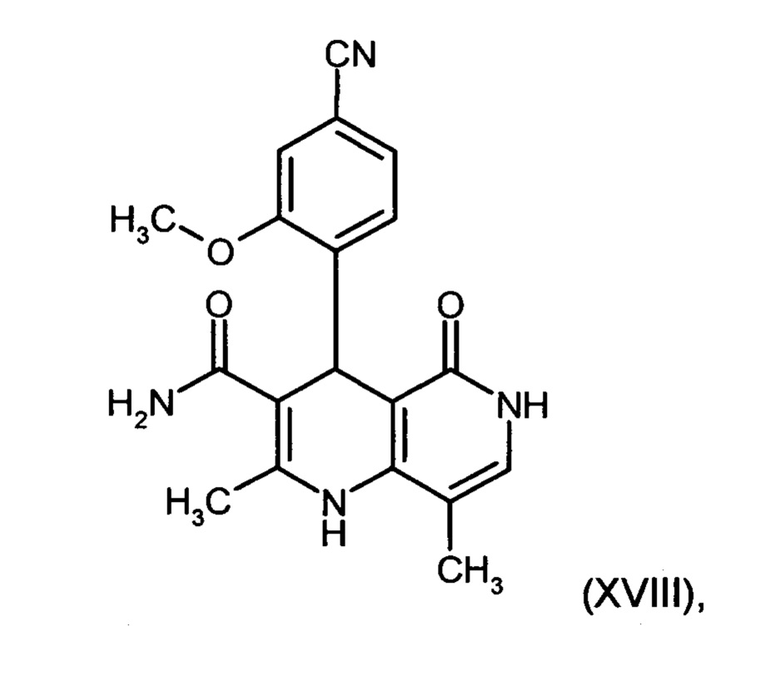
Данную реакцию предпочтительно осуществляют в кипящем дихлорметане (10-20-кратный избыток) при добавлении в водоотделитель от 5 до 20% мол., предпочтительно 10% мол. пиперидина, и от 5 до 20% мол., предпочтительно от 5 до 10% мол. ледяной уксусной кислоты. Время реакции составляет от 4 до 12 часов, предпочтительно от 5 до 6 часов, особенно предпочтительно 6 часов. Добавляют от 1,0 до 1,5 эквивалента, предпочтительно от 1,1 до 1,35 эквивалента, особенно предпочтительно 1,1 эквивалента кетоамида (XVII). Получение кетоамида (XVII) известно и описано в Tetrahedron Letters, 1993, том 34, сс. 6141-6142, однако речь может идти также о торговом продукте. По завершении реакции реакционную смесь охлаждают до 20°С, и органическую фазу дважды промывают водой. Растворитель в промытой органической фазе посредством ре-дистилляции заменяют на 2-бутанол, и коричный амид (XVI а+b) в виде E/Z-смеси без промежуточного выделения непосредственно используют для последующего получения дигидропиридина (XVIII) по реакции с гетероциклом (IX):
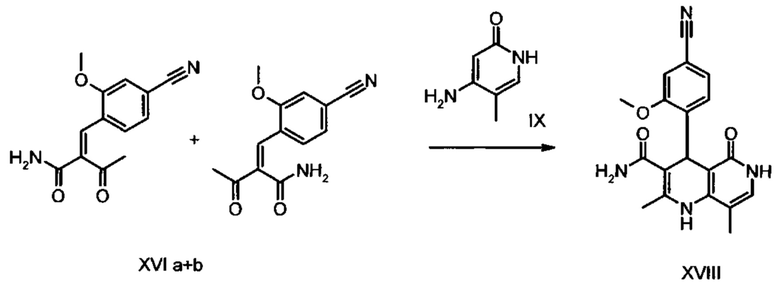
Обнаружено, что предпочтительным является осуществление реакции в спиртах, например, этаноле, изопропаноле, изобутаноле (2-бутаноле), 2-амиловом спирте или циклогексаноле, при температурах от 80 до 160°С и нормальном давлении, а также в автоклаве (от 2 до 10 бар), причем время реакции составляет от 8 до 40 часов, однако реакцию предпочтительно осуществляют в автоклаве в изопропаноле (от 100 до 130°С, от 2 до 10 бар, предпочтительно от 3 до 5 бар, от 8 до 24 часов), этаноле (от 90 до 130°С, от 3 до 10 бар, от 3 до 24 часов) или 2-бутаноле (от 100 до 130°С, от 2 до 10 бар, предпочтительно от 3 до 5 бар, от 8 до 24 часов). С целью переработки реакционную смесь охлаждают до температуры от 0 до 20°С, кристаллы отфильтровывают, промывают этанолом, а затем сушат в вакууме при 60°С.
В случае если по экологическим причинам необходимо отказаться от использования дихлорметана, коричный амид (XVI а, b) предпочтительно получают в изопропаноле, причем альдегид (VI) вводят в 3-9-кратный, предпочтительно 5-7-кратный избыток изопропанола и добавляют от 5 до 20% мол., предпочтительно от 5 до 10% мол. пиперидина и от 5 до 20% мол., предпочтительно от 5 до 10% мол. ледяной уксусной кислоты. При 30°С в течение трех часов добавляют от 1,0 до 1,5, предпочтительно от 1,1 до 1,35, особенно предпочтительно 1,1 эквивалента кетоамида (XVII), при необходимости растворенного в небольшом количестве изопропанола, и реагенты перемешивают в течение часа при 30°С. Во время реакции образуются кристаллы коричного амида (XVI а, b). Затем при необходимости после охлаждения продукт отфильтровывают предпочтительно при 0°С, промывают небольшим количеством охлажденного до 0°С изопропанола и используют во влажном состоянии для последующей реакции, как указано выше. Выход от теоретического составляет более 95%. Последующее превращение предпочтительно осуществляют в 10-15-кратном, предпочтительно 10-12-кратном (по отношению к альдегиду (VI)) избытке 2-бутанола или изопропанола в течение от 20 до 24 часов при 100°С под давлением. После завершения реакции и охлаждения продукт выделяют посредством фильтрования или центрифугирования. Затем продукт сушат в вакууме при температуре от 40 до 90°С.Поскольку превращение в коричный амид (XVI а, b) протекает почти количественно, последующий процесс может быть легко стандартизован без необходимости соответствующего адаптирования количества гетероцикла, что позволяет использовать содержащий изопропанол влажный продукт. Выходы от теоретического составляют более 75%. Гетероцикл (IX) может быть получен известными из литературы методами, например, методом, описанными в Synthesis 1984, сс. 765-766.
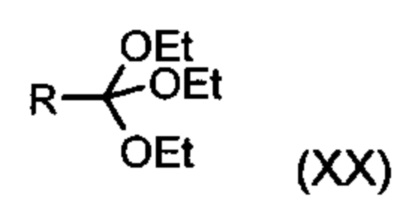
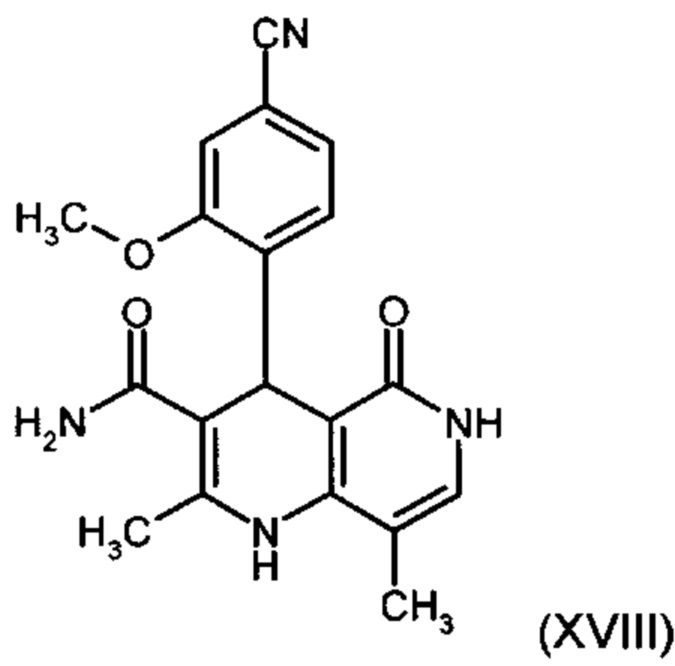
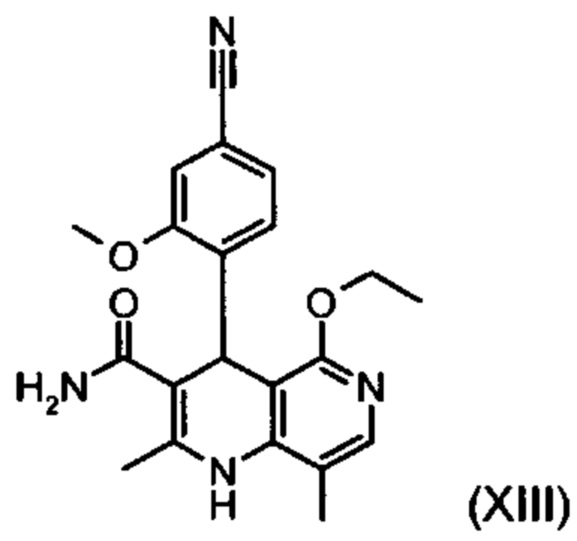
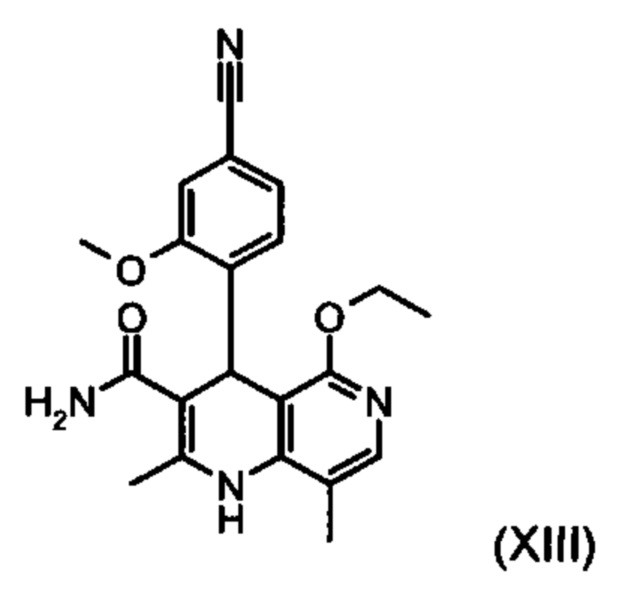
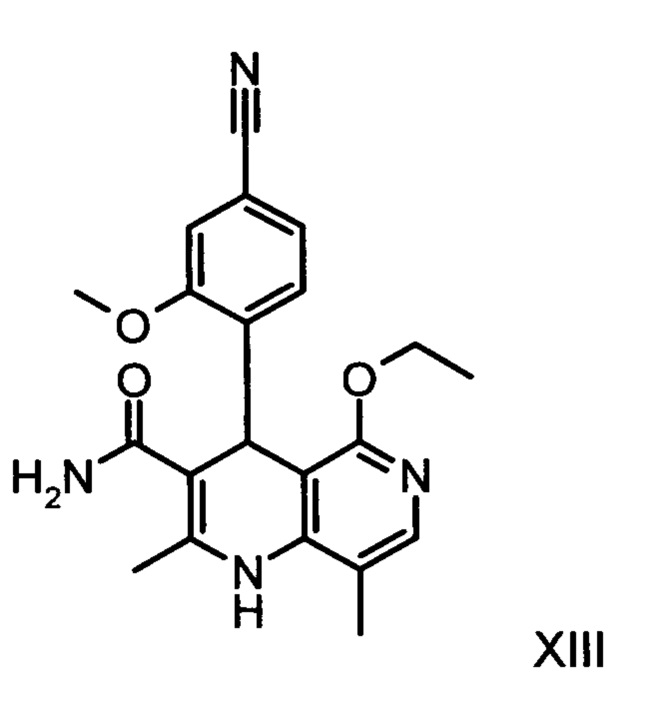
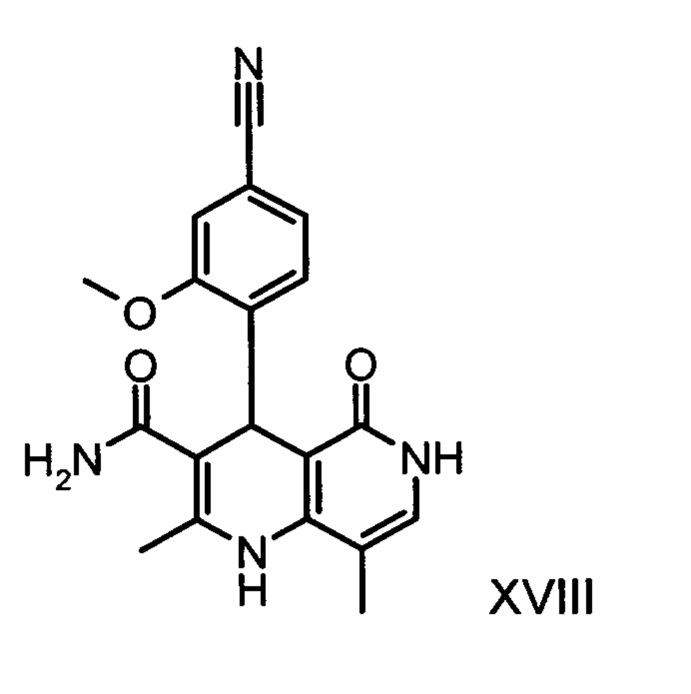
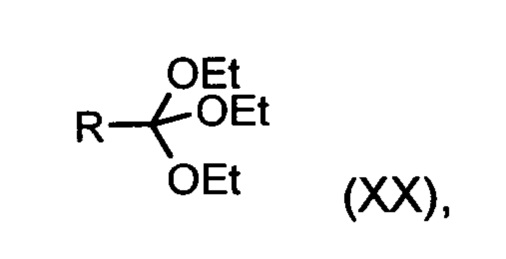
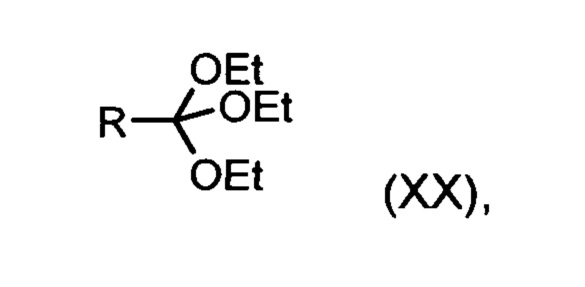
Из дигидропиридина (XVIII) посредством катализируемого кислотой превращения со сложным ортоэфиром формулы (XX), в которой R означает водород или метил, получают этиловый эфир (XIII):
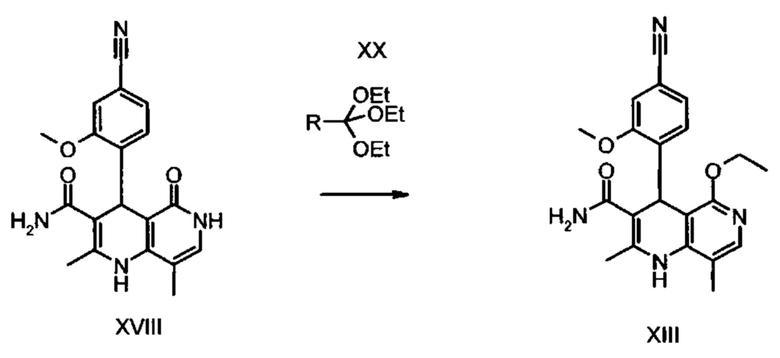
Неожиданно было обнаружено, что данную реакцию можно осуществлять при очень высокой концентрации эдукта в растворителях, например, диметилацетамиде, 1-метил-2-пирролидоне или диметилформамиде (до 1,5 г растворителя на 1 г эдукта) и добавлении от 4 до 10% масс, предпочтительно от 6 до 8% масс. концентрированной серной кислоты. При этом реакция неожиданно протекает уже с 2,5-5 эквивалентами ортоэфира (XX) (R означает водород или метил). Обнаружено, что для осуществления этой реакции гораздо более благоприятным является использование соответствующего триэтилортоэтилацетата, прежде всего поскольку он реагирует гораздо полнее и обладает гораздо более низкой воспламеняемостью, что предопределяет особенно высокую пригодность этого соединения для технического применения. Превращение предпочтительно осуществляют в диметилацетамиде или 1-метил-2-пирролидоне при температурах от 100 до 120°С, предпочтительно 115°С. Предпочтительной является отгонка части растворителя (диметилацетамида или 1-метил-2-пирролидона) при повышенной температуре (от 100 до 120°С, под вакуумом), выполняемая перед инициированием непосредственной реакции с целью удаления остатков изопропанола, в некоторых случаях присутствующих в исходном продукте, поскольку в противном случае возможно образование нежелательных побочных продуктов. Превращение осуществляют в течение промежутка времени от 1,5 до 3 часов, предпочтительно 2 часов, при перемешивании. Для выделения целевого продукта непосредственно к реакционной смеси добавляют воду, что сопровождается выпадением кристаллов целевого продукта. Для повышения стабильности и воспроизводимости добавляют лишь частичное количество воды (например, третью часть), после чего вносят кристаллическую затравку и добавляют остальное количество воды. Это позволяет всегда получать одинаковую кристаллическую модификацию, которая наилучшим образом пригодна для выделения. Продукт промывают водой и сушат. Выходы от теоретического составляют более 92%. Степень чистоты как правило превышает 99% (высокоэффективная жидкостная хроматография, 100-процентный метод).
Для получения соединения формулы (I) рацемическую смесь амидов (XIII) необходимо разделять на соответствующие антиподы. При описанном выше опытном синтезе для этой цели используют специально синтезированную собственными силами хиральную фазу, которая в качестве хирального селектора содержит N-(дициклопропилметил)-N2-метакрилоил-D-лейцинамид. Данный селектор получают в несколько стадий, а затем полимеризуют на особом силикагеле. В качестве хроматографического растворителя используют смесь метанола с этилацетатом. Существенным недостатком этого метода является чрезвычайно низкая загрузка хроматографической колонки (30 мг при разделении на колонке 500×63 мм), в связи с чем существует настоятельная потребность в максимально эффективном методе разделения, позволяющем выполнять крупнотоннажное разделение антиподов. Неожиданно было обнаружено, что разделение можно осуществлять также на легко доступной торговой фазе. Речь при этом идет о фазе Chiralpak AS-V (20 мкм). В качестве хроматографического растворителя используют смесь метанола с ацетонитрила в соотношении 60:40. Важное преимущество данной смеси состоит в том, что ее можно регенерировать посредством дистилляционной переработки в виде хроматографического растворителя идентичного состава (60:40, что соответствует азеотропу). Это способствует существенному повышению эффективности процесса разделения (выход от теоретического составляет более 47% при теоретически возможном выходе 50%). Достигаемая оптическая чистота составляет более 93%, предпочтительно более 98,5%. Хроматографирование можно осуществлять на стандартной хроматографической колонке, однако предпочтительно используют известные специалистам технические средства хроматографического разделения, например, установку SMB или Varicol (Computers and Chemical Engineering 27, 2003, cc. 1883-1901). Так, например, на установке SMB разделяют около 500 кг рацемического амида (XIII), причем выход достигает 48%. Продукт, выгружаемый в виде раствора в смеси метанола с ацетонитрилом (60:40) концентрацией от 3 до 8%, предпочтительно от 5 до 7%, может быть использован для непосредственной «окончательной переработки». В качестве альтернативы возможны также другие соотношения между ацетонитрилом и метанолом, варьируемые в диапазоне от 90:10 до 10:90. В качестве альтернативы для разделения на установке SMB можно использовать также смеси других растворителей, например, смеси ацетонитрила с этанолом с соотношением компонентов в диапазоне от 10:90 до 90:10. Соотношение между соответствующими растворителями в некоторых случаях зависит от технических характеристик установки SMB и при необходимости должно быть приведено в соответствии с этими характеристиками (например, варьируемыми скоростями течения, рециклом растворителей в тонкослойном испарителе).
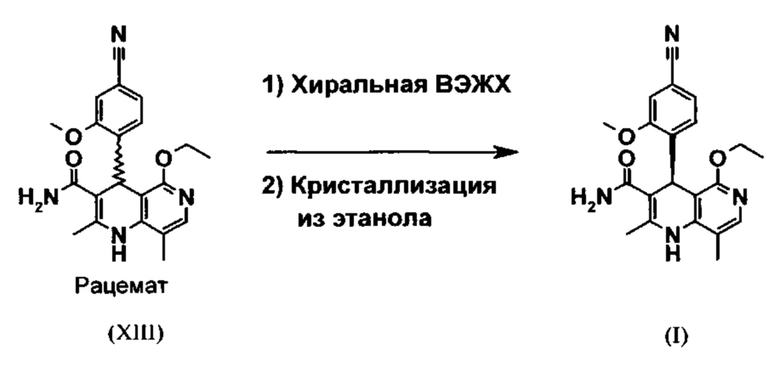
Поскольку согласно изобретению предусматривается формирование соединения формулы (I) в виде таблеток, существует настоятельная потребность в воспроизводимом выделении указанного соединения в определенной кристаллической форме, что позволяет обеспечить его воспроизводимую биологическую доступность. Неожиданно было обнаружено, что кристаллизацию соединения формулы (I) можно осуществлять из метанола, этанола, тетрагидрофурана, ацетонитрила, а также их смесей с водой, причем воспроизводимо образуется лишь полиморфная модификация I, которая характеризуется определенной точкой плавления (252°С). При этом предпочтительному использованию подлежит этанол, соответственно денатурированный этанол.
Технология заключительной кристаллизации. Руководствуясь правилами организации производства и контроля качества лекарственных средств, поступающий со стадии хроматографии раствор продукта в смеси метанола и ацетонитрила (60:40), примерная концентрация которого составляет от 5 до 7% (в случае раствора продукта в смеси этанола и ацетонитрила 50:50 она составляет от 3 до 4%), сначала подвергают фильтрованию с целью отделения частиц, после чего растворитель заменяют на этанол, причем предпочтительно используют этанол, денатурированный толуолом. С этой целью несколько раз выполняют повторную перегонку, концентрирование и соответственно добавление свежего этанола. По завершении замены растворителя добавляют этанол в количестве, необходимом для перехода к температуре испарения фазы растворителя, после чего при нормальном, соответственно несколько пониженном давлении осуществляют примерно трех-четырех кратное объемное концентрирование, сопровождаемое образованием кристаллов продукта. После охлаждения до 0°С кристаллы выделяют и сушат в вакууме при температуре от 40 до 50°С. Выходы от теоретического, как правило, составляют более 90%. Достигаемая степень химической чистоты, превышающая 99,8%, и содержание основного вещества около 100% соответствуют критериям, принятым для оценки торговых продуктов согласно рекомендациями ICH (Международной конференции по гармонизации технических требований к регистрации лекарственных средств для человека). Содержание остаточного растворителя (в случае этанола) составляет менее 0,02%. Оптическая чистота существенно превышает 99%.
Соединение формулы (I) в кристаллической модификации I:
отличается тем, что на его рентгеновской дифрактограмме обнаруживаются максимумы угла 2 тета при 8,5, 14,1, 17,2, 19,0, 20,5, 25,6, 26,5.
Соединение формулы (I) в кристаллической модификации I:
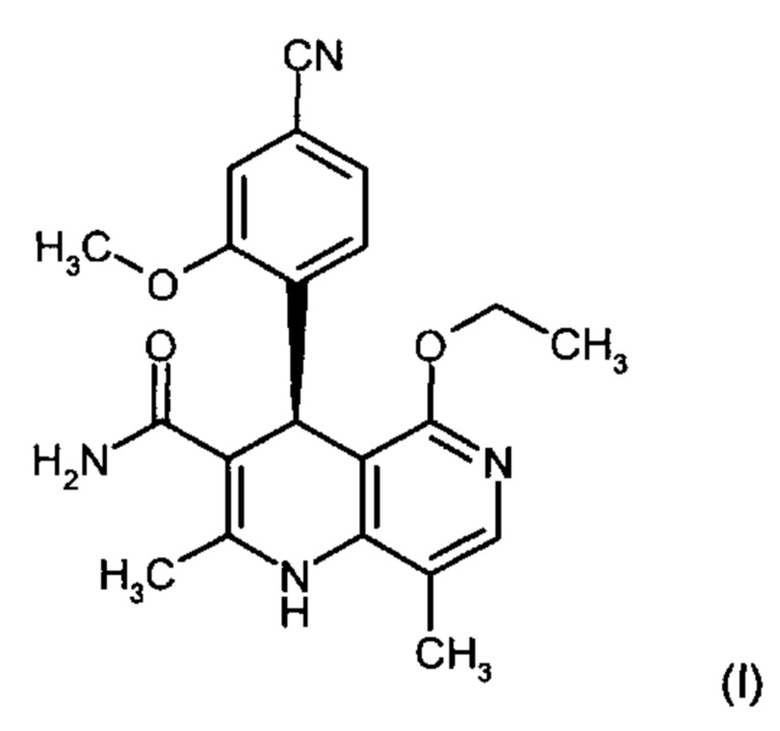
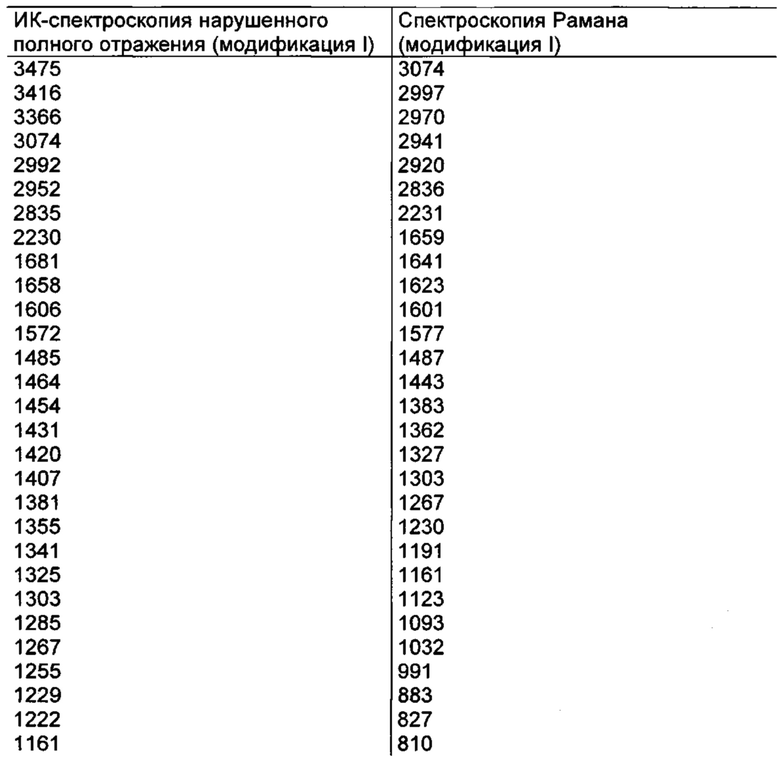
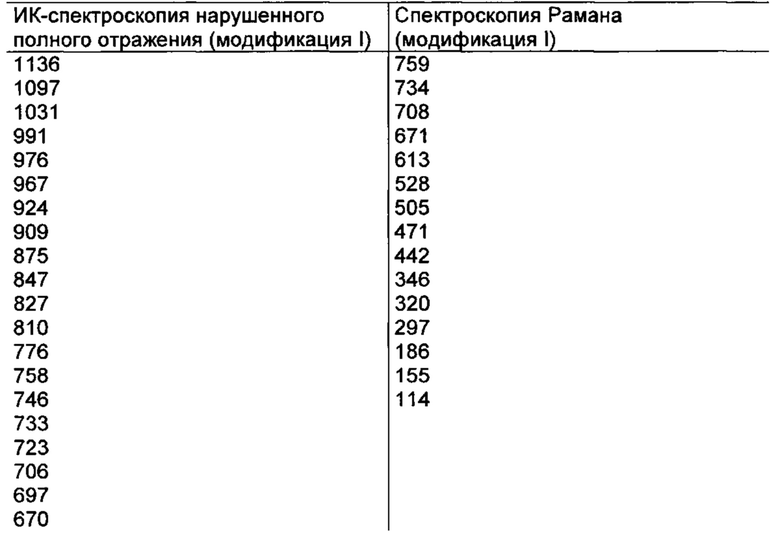
отличается тем, что на его инфракрасном спектре (ИК-спектре нарушенного полного отражения) обнаруживаются максимумы при 3475, 2230, 1681, 1658, 1606, 1572, 1485, 1255, 1136 и 1031 см-1.
Соединение формулы (I) в кристаллической модификации I:
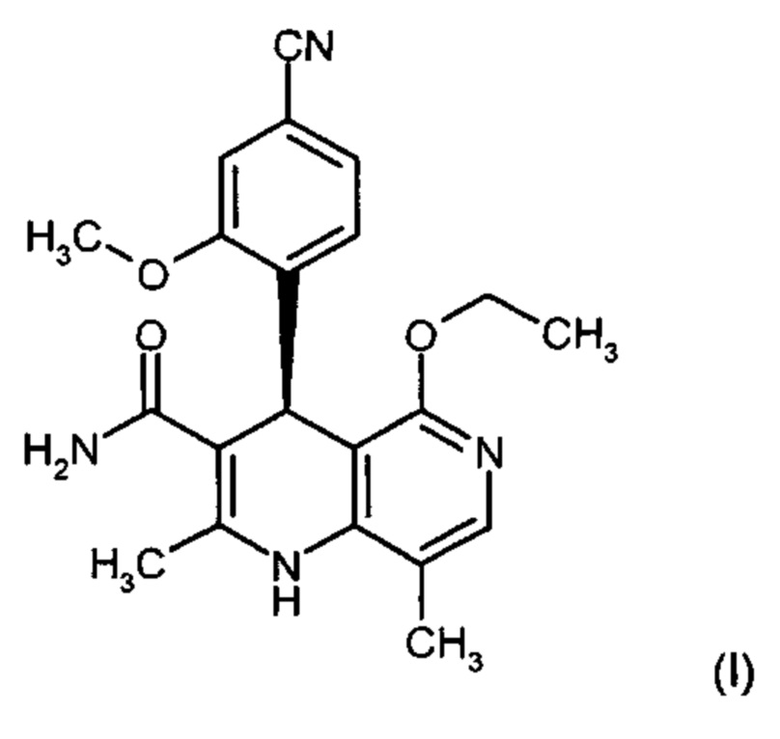
отличается тем, что на его спектре Рамана обнаруживаются максимумы при 3074, 2920, 2231,1601, 1577, 1443, 1327, 1267, 827 и 155 см-1.
Соединение формулы (I), как правило, микронизируют и компонуют в фармацевтические таблетки. Обнаружено, что соединение формулы (I) в кристаллической модификации I отличается чрезвычайно высокой стабильностью (в том числе и при повышенной влажности воздуха) и может без проблем храниться в течение более двух лет.
Предлагаемый в изобретении синтез позволяет чрезвычайно эффективным способом получать соединение формулы (I). По сравнению с уровнем техники способ обладает существенными преимуществами, касающимися возможности варьирования производственного масштаба и технического осуществления. Общий выход гораздо выше, чем в уровне техники, а биологически активное вещество характеризуется очень высокой степенью чистоты. Новый способ допускает возможность неизвестного из уровня техники, воспроизводимого и рентабельного получения соединения формулы (I) в кристаллической модификации I.
Объектом настоящего изобретения является способ получения соединения формулы (I):
 ,
,
отличающийся тем, что рацемическое соединение формулы (XIII) разделяют на соответствующие энантиомеры,
причем соединение формулы (XIII):
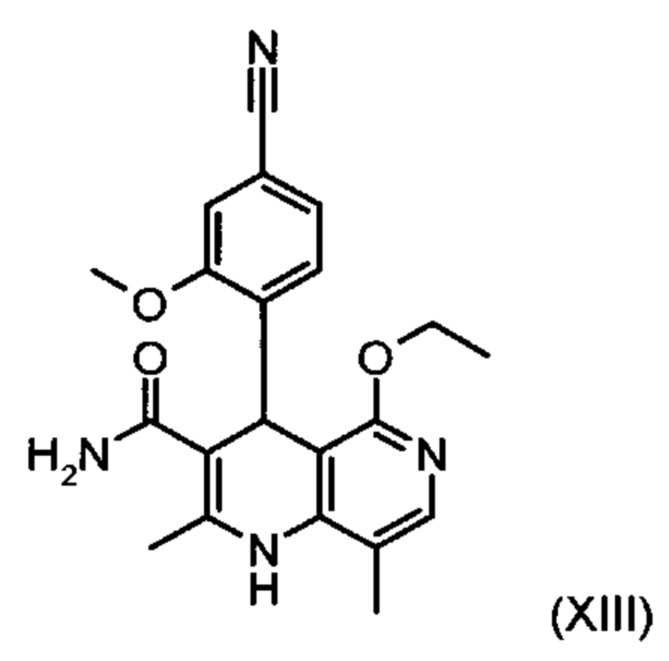
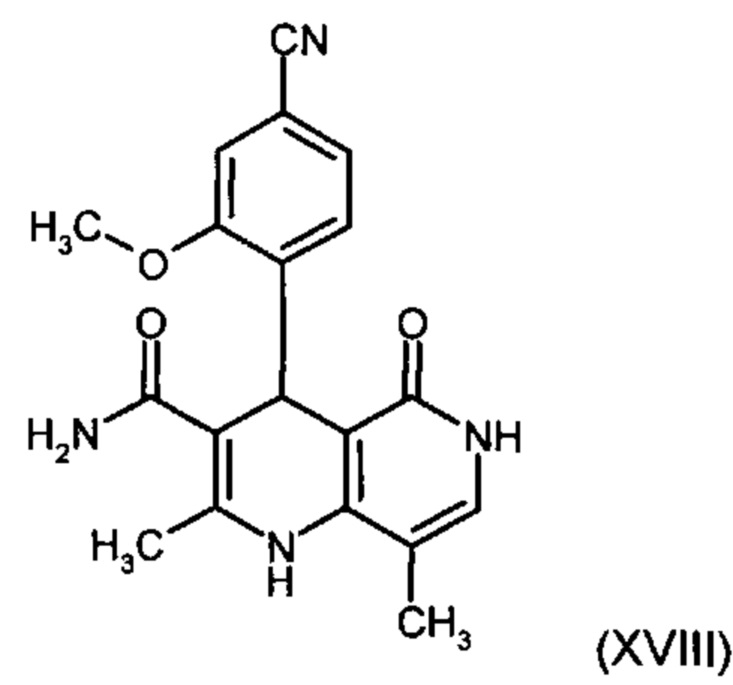
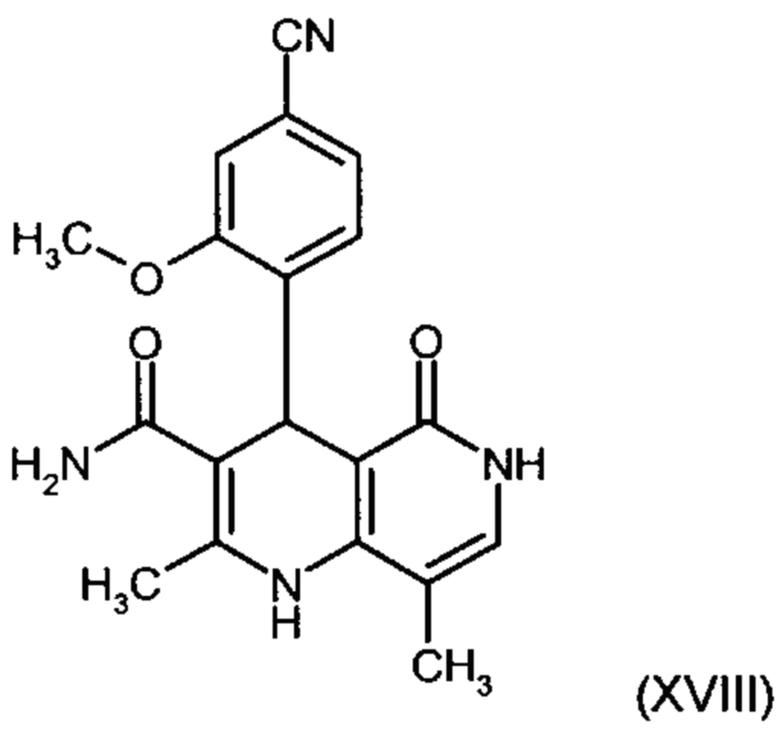
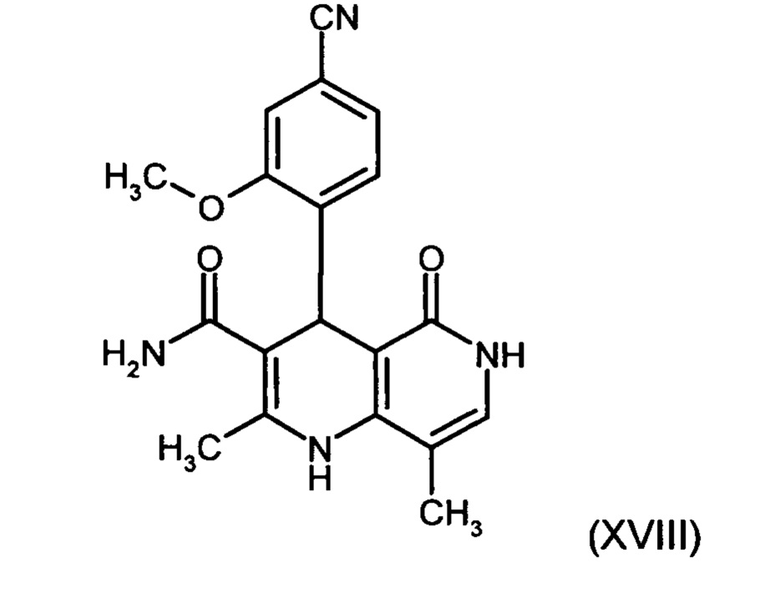
получают путем взаимодействия соединения формулы (XVIII):
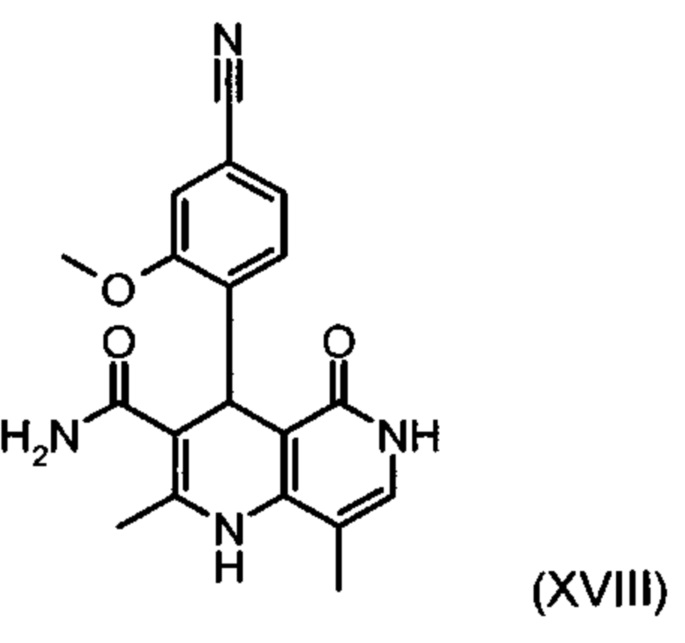
со сложным ортоэфиром формулы (XX):
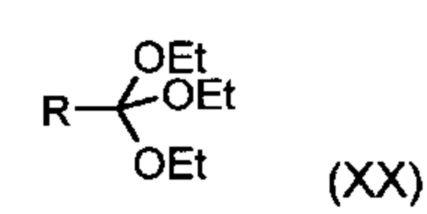 ,
,
в которой R означает водород или метил,
причем соединение формулы (XVIII):
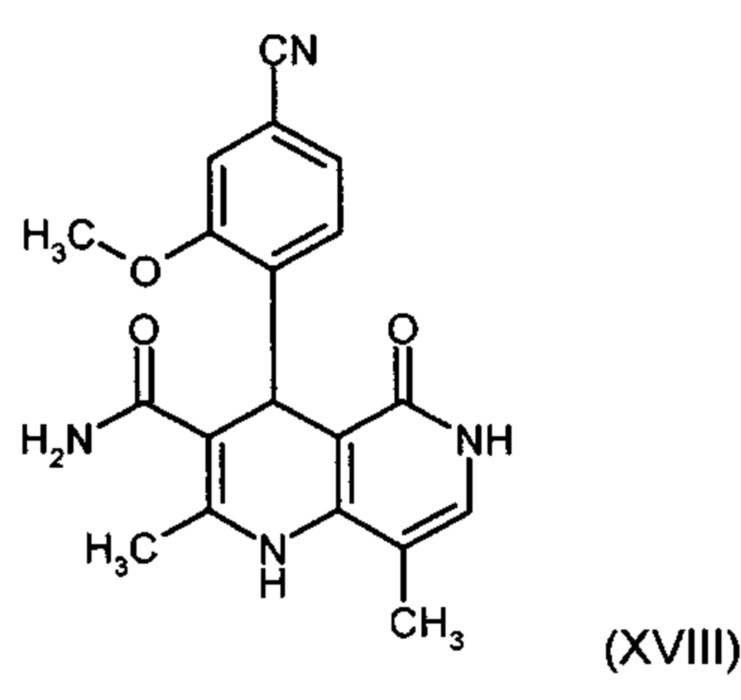 ,
,
получают путем взаимодействия соединений формулы (XVI а, b):
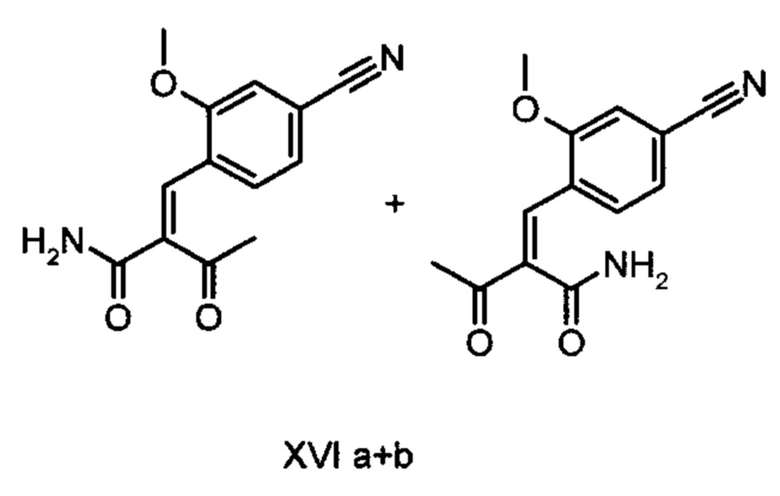
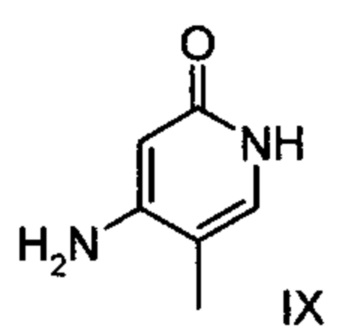
с соединением формулы (IX):
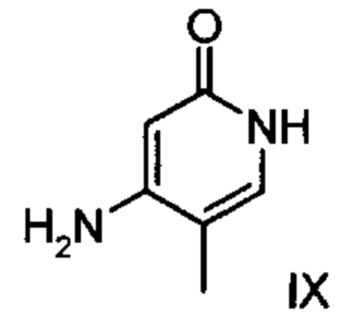
и причем соединение формулы (XVI а, b) получают путем взаимодействия соединения формулы (VI):
с соединением формулы (XVII):
 .
.
Другим объектом настоящего изобретения является способ получения соединения формулы (I):
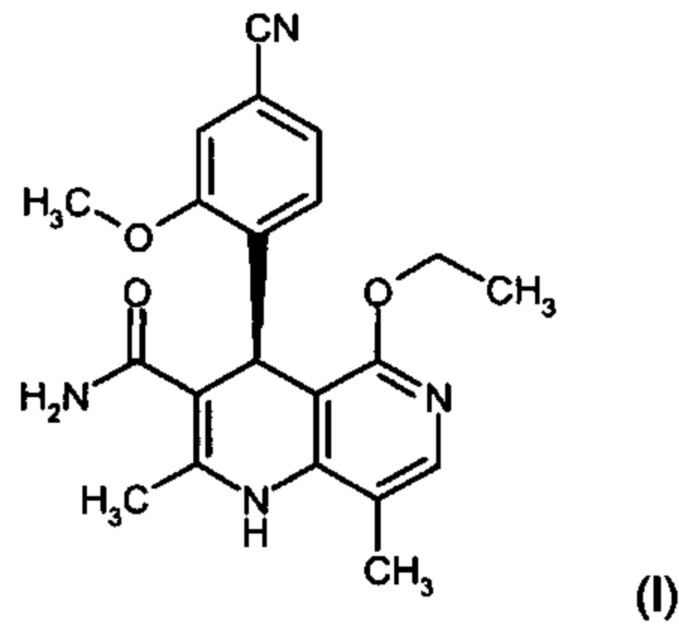 ,
,
отличающийся тем, что рацемическое соединение формулы (XIII) разделяют на соответствующие энантиомеры,
причем соединение формулы (XIII):
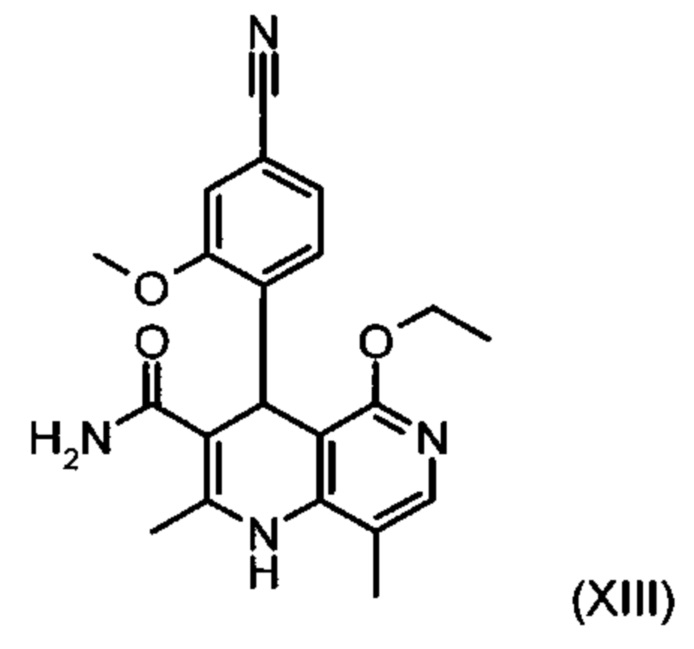
получают путем взаимодействия соединения формулы (XVIII):
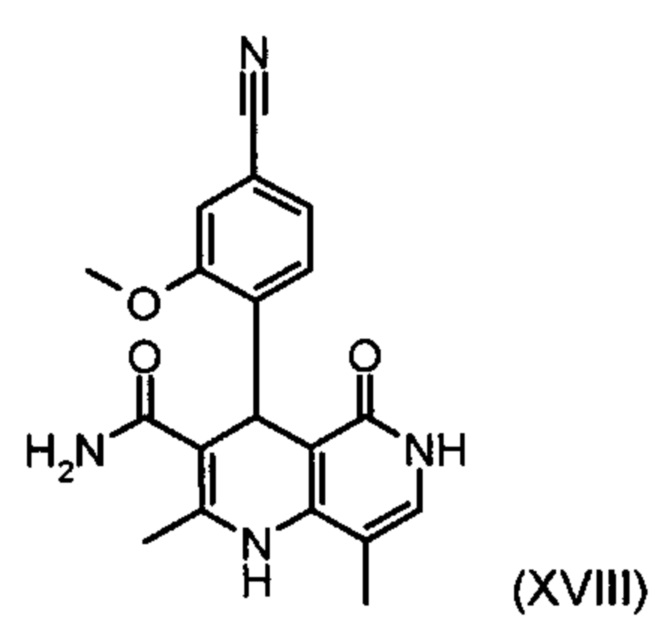
с ортоэфиром формулы (XX):
,
в которой R означает водород или метил,
и причем соединение формулы (XVIII):
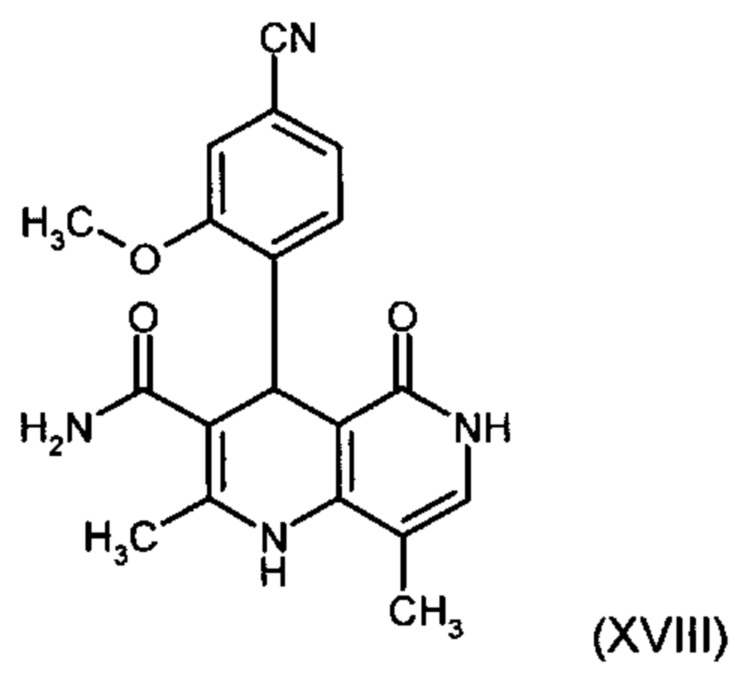
получают путем взаимодействия соединений формулы (XVI а, b):
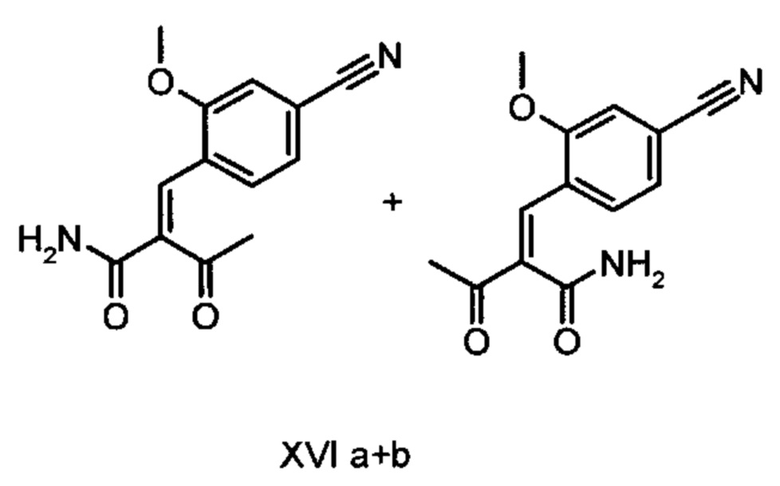
с соединением формулы (IX):
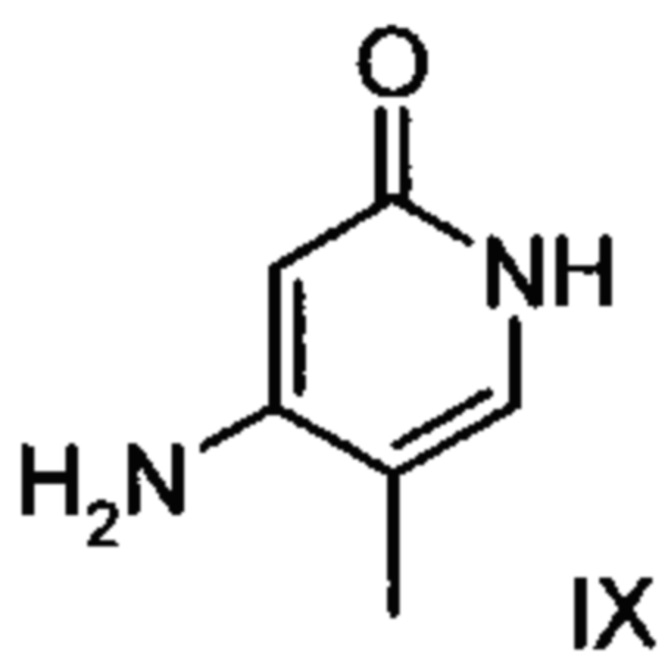 .
.
Другим объектом настоящего изобретения является способ получения соединения формулы (I):
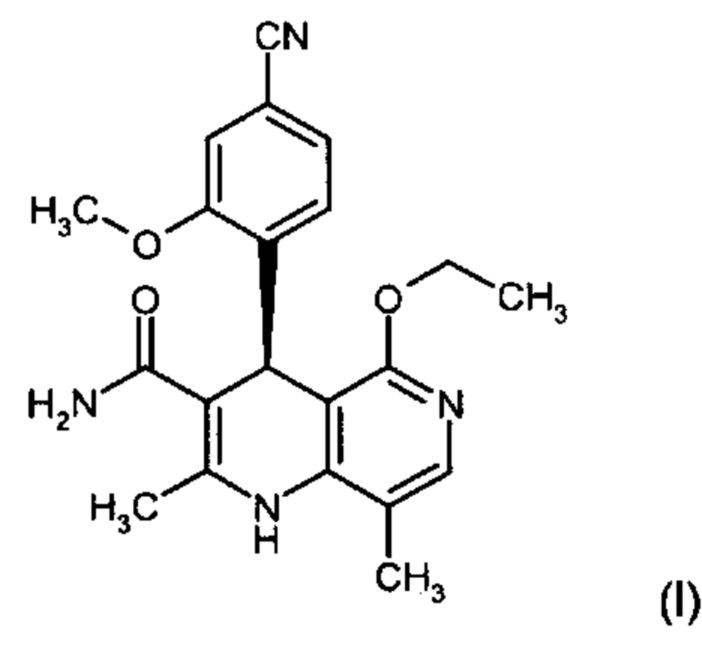 ,
,
отличающийся тем, что рацемическое соединение формулы (XIII) разделяют на соответствующие энантиомеры,
причем соединение формулы (XIII):
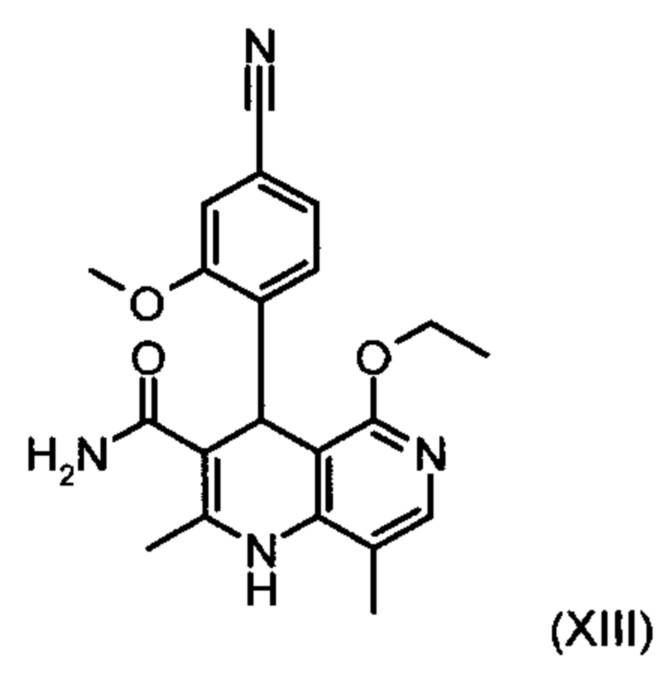
получают путем взаимодействия соединения формулы (XVIII):
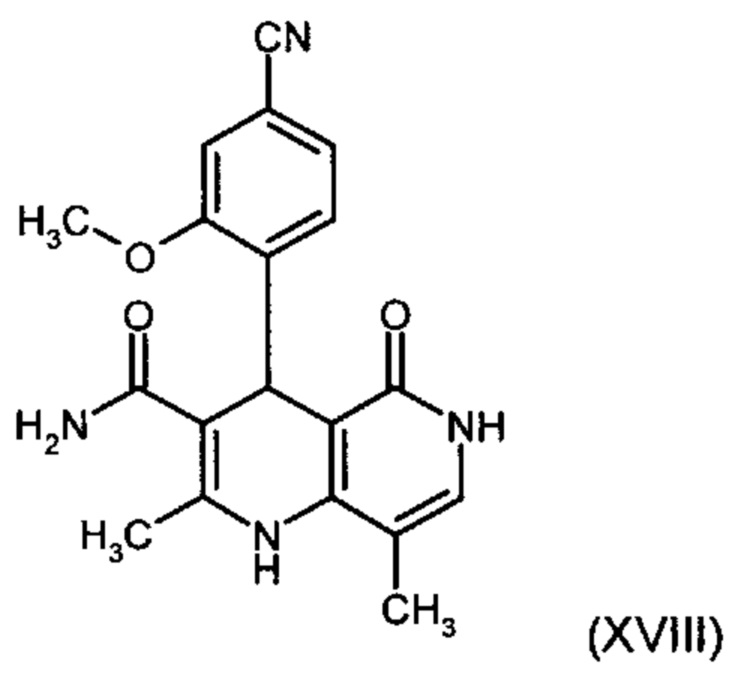
со сложным ортоэфиром формулы (XX):
 ,
,
в которой R означает водород или метил.
Другим объектом настоящего изобретения является способ получения соединения формулы (I):
с использованием соединения формулы (XVIII):
 .
.
Другим объектом настоящего изобретения является способ получения соединения формулы (I):
с использованием соединений формулы (XVI а, b):
 .
.
Другим объектом настоящего изобретения является способ получения соединения формулы (I):
с использованием соединения формулы (XVIII):
и соединений формулы (XVI а, b):
 .
.
Другим объектом настоящего изобретения является соединение формулы:
 ,
,
а также его соли, сольваты и сольваты солей.
Другим объектом настоящего изобретения является соединение формулы (XVI а, b) в виде E/Z- смеси:
 ,
,
а также его соли, сольваты и сольваты солей.
Другим объектом настоящего изобретения является способ получения соединения формулы (XVIII):
 ,
,
отличающийся тем, что соединения формулы (XVI а, b):
подвергают превращению с соединением формулы (IX):
 .
.
Другим объектом настоящего изобретения является способ получения соединений формулы (XVI а, b):
 ,
,
отличающийся тем, что соединение формулы (VI):
подвергают превращению с соединением формулы (XVII):
 .
.
Другим объектом настоящего изобретения является способ получения соединения (XIII):
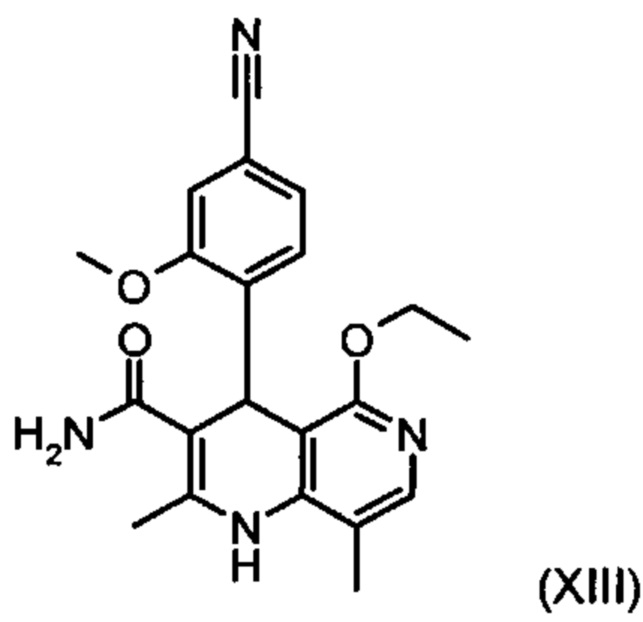 ,
,
отличающийся тем, что соединение формулы (XVIII):
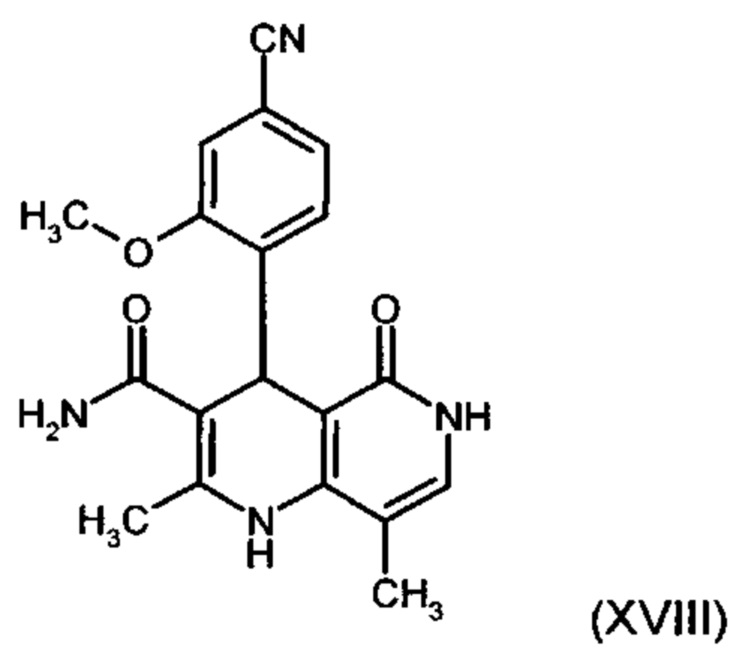
подвергают превращению со сложным ортоэфиром формулы (XX):
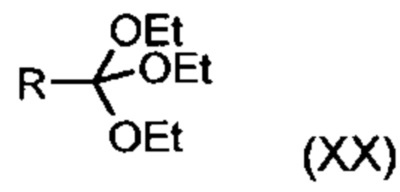 ,
,
в которой R означает водород или метил.
Другим объектом настоящего изобретения является способ получения соединения формулы (XIII):
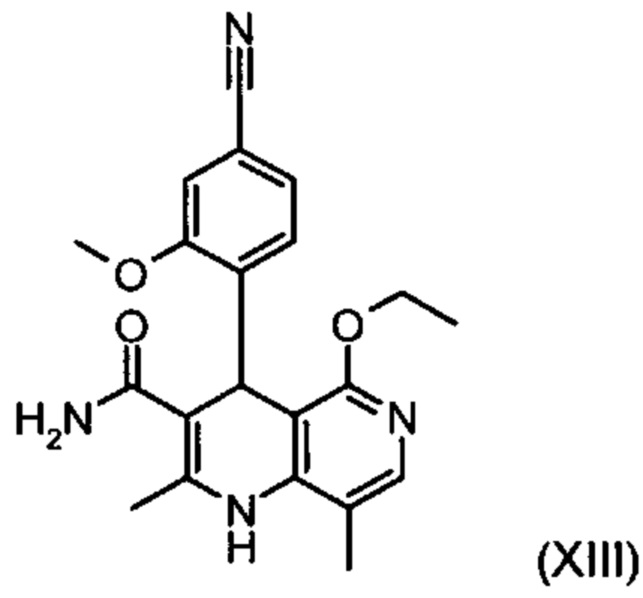 ,
,
отличающийся тем, что соединение формулы (XVIII):
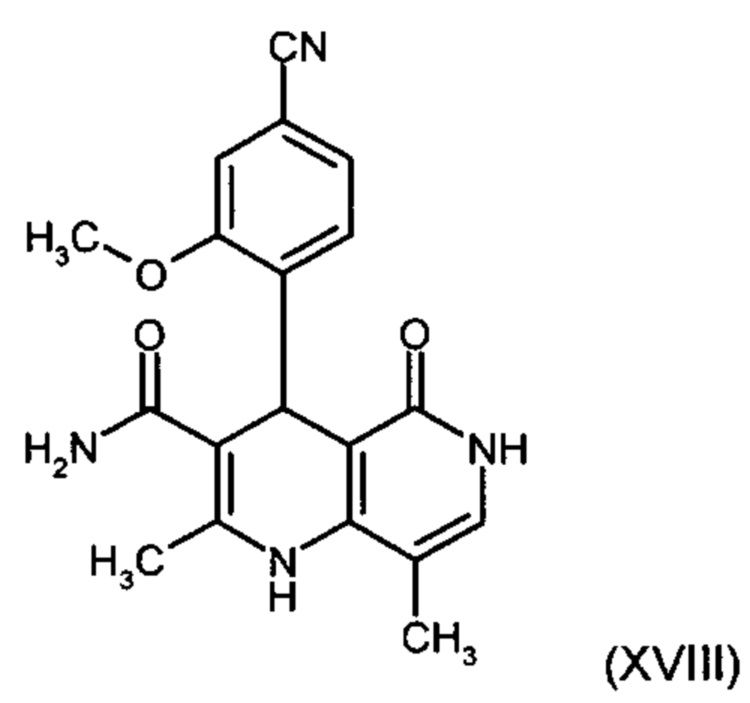
подвергают превращению со сложным ортоэфиром формулы (XX):
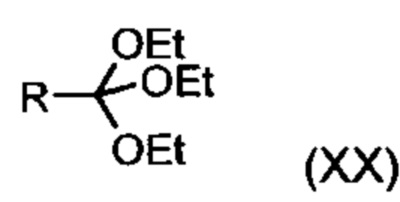 ,
,
в которой R означает водород или метил,
причем соединение формулы (XVIII) получают путем взаимодействия соединений формулы (XVI а, b):
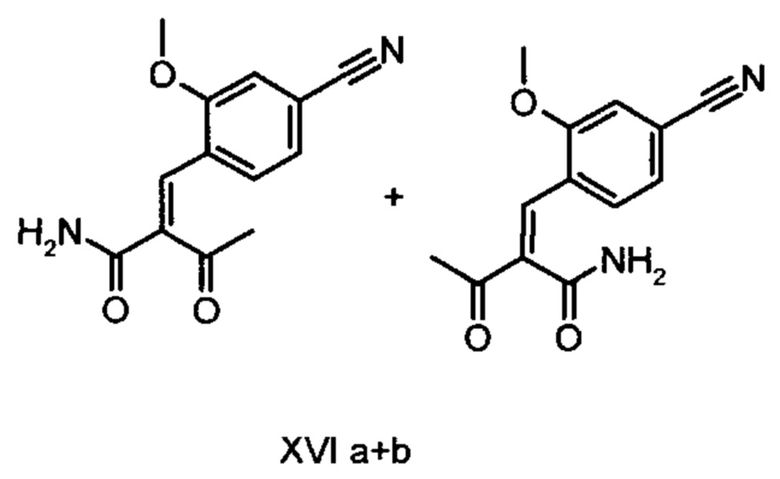
с соединением формулы (IX):
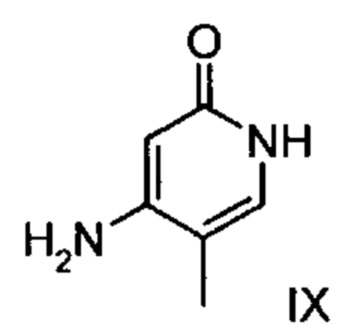 .
.
Другим объектом настоящего изобретения является реализуемый, как описано выше, способ получения соединения формулы (I) в кристаллической модификации I:
 ,
,
отличающийся тем, что соединение формулы (I), находящееся в одной или нескольких модификациях или в виде сольвата, перемешивают в инертном растворителе при температуре от 20 до 120°С и выделяют соединение формулы (I) в кристаллической модификации I.
Предлагаемые в изобретении соединения, то есть соединение формулы (I) и соединение формулы (I) в кристаллической модификации I, которые в данном случае и дальнейшем описании называют предлагаемыми в изобретении соединениями, действуют в качестве антагонистов минералокортикоидного рецептора и обладают непредсказуемым ценным спектром фармакологического действия. В связи с этим они пригодны для использования в качестве лекарственных средств для лечения и/или профилактики болезней людей и животных.
Предлагаемые в изобретении соединения пригодны для профилактики и/или лечения различных заболеваний и обусловленных ими состояний, в особенности заболеваний, которые либо характеризуются повышением концентрации альдостерона в плазме или изменением концентрации альдостерона в плазме относительно концентрации ренина в плазме, либо сопровождаются указанными изменениями. Примерами подобных заболеваний являются идиоматический первичный гиперальдостеронизм, гиперальдостеронизм при гиперплазии надпочечников, аденомы и/или карциномы надпочечников, гиперальдостеронизм при циррозе печени, гиперальдостеронизм при сердечной недостаточности, а также (относительный) гиперальдостеронизм при эссенциальной гипертонии.
Кроме того, предлагаемые в изобретении соединения в связи со специфическим механизмом их действия пригодны для предупреждения смерти пациентов, подверженных повышенному риску внезапной остановки сердца. Речь при этом прежде всего идет о пациентах, страдающих, например, следующими заболеваниями: первичная и вторичная гипертония, гипертензивная болезнь сердца с конгитивной сердечной недостаточностью или без конгетивной сердечной недостаточности, не поддающаяся лечению гипертония, острая или хроническая сердечная недостаточность, коронарное заболевание сердца, стабильная или нестабильная стенокардия, миокардиальная ишемия, инфаркт миокарда, дилатационные кардиомиопатии, врожденные первичные кардиомиопатии, например, синдром Бругада, обусловленные американским трипаносомозом кардиомиопатии, шок, артериосклероз, предсердная или вентрикулярная аритмия, преходящие или ишемические приступы, кровоизлияние в мозг, воспалительные сердечнососудистые заболевания, периферийные или кардиальные заболевания сосудов, периферийные нарушения кровоснабжения, артериальные облитерирующие заболевания, в частности, перемежающаяся хромота, бессимптомная дисфункция левого желудочка, миокардит, идеопатические изменения сердца, пульмональная гипертония, спазмы венечных и периферийных артерий, тромбозы, тромбоэмболические заболевания, а также васкулит.
Предлагаемые в изобретении соединения можно использовать также для профилактики и/или лечения образования отеков, например, пульмонального отека, почечного отека или отека, обусловленного сердечной недостаточностью, и рестенозами, в частности, после тромболитической терапии, чрескожной транслюминальной ангиопластики и чрескожной транслюминальной коронарной ангиопластики, трансплантации сердца, а также после операций шунтирования.
Кроме того, предлагаемые в изобретении соединения пригодны для применения в качестве калийсберегающих диуретинов, а также при электролитных нарушениях, например, гиперкальциемии, гипернатриемии или гипокалиемии.
Предлагаемые в изобретении соединения пригодны также для лечения заболеваний почек, например, острого или хронического нарушения функции почек, гипертензивной болезни почек, артериосклеротического нефрита (хронического и интерстициального), нефросклероза, хронической почечной недостаточности и кистозных заболеваний почек, для предотвращения повреждения почек, которые могут быть обусловлены, например, применяемыми при трансплантации органов иммунодепрессивными средствами, такими как циклоспорин А, а также при раке почек.
Кроме того, предлагаемые в изобретении соединения можно использовать для профилактики и/или лечения диабета и сопутствующих заболеваний, например, таких как невропатия и нефропатия.
Наряду с этим предлагаемые в изобретении соединения можно использовать для профилактики и/или лечения микроальбуминурии, например, обусловленной диабетом или артериальной гипертонией, а также протеинурии.
Предлагаемые в изобретении соединения пригодны также для профилактики и/или лечения заболеваний, которые сопровождаются повышением концентрации глюкокортикоидов в плазме или локальным повышением концентрации глюкокортикоидов в ткани (например, сердце). Речь при этом идет, например, о дисфункции надпочечников, обусловливающей гиперпродукцию глюкокортикоидов (синдром Кушинга), опухоли надпочечников, которая сопровождается гиперпродукцией глюкокортикоидов, а также опухоли гипофиза, которая сопровождается выработкой автономного адренокортикотропного гормона, а следовательно, приводит к гиперплазии надпочечников, вызывающей болезнь Кушинга.
Кроме того, предлагаемые в изобретении соединения можно использовать для профилактики и/или лечения общего ожирения, метаболического синдрома и обструктивной временной остановки дыхания во время сна.
Помимо этого предлагаемые в изобретении соединения можно использовать для профилактики и/или лечения воспалительных заболеваний, вызываемых, например, вирусами, спирохетами, грибками, бактериями или микобактериями, а также воспалительных заболеваний неизвестной этиологии, например, полиартрита, эритематозной волчанки, периартериита, полиартериита, дерматомиозита, склеродермии и доброкачественного гранулематоза.
Кроме того, предлагаемые в изобретении соединения можно использовать для лечения заболеваний центральной нервной системы, например, депрессий, тревожных состояний и хронических болей, в частности, мигрени, а также при нейродегенеративных заболеваниях, например, болезни Альцгеймера и синдроме Паркинсона.
Предлагаемые в изобретении соединения пригодны также для профилактики и/или лечения повреждения сосудов, например, после вмешательств, в частности, после чрескожной транслюминальной коронарной ангиопластики, имплантации стентов, коронарной ангиоскопии, реокклюзии или рестенозе после операций шунтирования, а также при эндотелиальной дисфункции, виброболезни, облитерирующем тромбангиите (синдроме Бюргера) и синдроме ушного шума.
Другим объектом настоящего изобретения является применение предлагаемых в изобретении соединений для лечения и/или профилактики заболеваний, в частности, указанных выше заболеваний.
Другим объектом настоящего изобретения является применение предлагаемых в изобретении соединений для изготовления лекарственного средства, предназначенного для лечения и/или профилактики заболеваний, в частности, указанных выше заболеваний.
Другим объектом настоящего изобретения является способ лечения и/или профилактики заболеваний, в частности, указанных выше заболеваний, предусматривающий использование эффективного количества по меньшей мере одного предлагаемого в изобретении соединения.
Предлагаемые в изобретении соединения можно использовать индивидуально или при необходимости в комбинации с другими биологически активными веществами. Другим объектом настоящего изобретения являются лекарственные средства, которые содержат по меньшей мере одно из предлагаемых в изобретении соединений и одно или несколько других биологически активных веществ и, в частности, предназначены для лечения и/или профилактики указанных выше заболеваний. Ниже приведены примеры предпочтительных пригодных комбинаций биологически активных веществ:
- снижающие кровяное давление биологически активные вещества, предпочтительно выбранные из группы, включающей, например, антагонисты кальция, антагонисты ангиотензина AII, ингибиторы превращающих ангиотензин ферментов, антагонисты эндотелина, ингибиторы ренина, блокаторы альфа-рецепторов, блокаторы бета-рецепторов и ингибиторы Rho-киназы,
- диуретики, в частности, петлевые диуретики, а также тиазиды и подобные тиазиду диуретики,
- средства, обладающие антитромботическим действием, предпочтительно выбранные из группы, например, включающей ингибиторы агрегации тромбоцитов, антикоагулянты и профибринолитические вещества,
- изменяющие жировой обмен биологически активные вещества, предпочтительно выбранные из группы, например, включающей агонисты рецепторов щитовидной железы, ингибиторы синтеза холестерина, например, предпочтительно ингибиторы HMG-CoA-редуктазы или синтеза сквалена, ингибиторы ацил-СоА-холестерол-ацилтрансферазы (АСАТ), ингибиторы транспортного белка холе-стерин-эстеразы (СЕТР), ингибиторы переносящего триглицериды микросомального белка (МТР), PPAR-альфа-, PPAR-гамма- и/или PPAR-дельта-агонисты, ингибиторы абсорбции холестерина, ингибиторы липазы, полимерные адсорберы желчной кислоты, реабсорбирующие ингибиторы желчной кислоты и антагонисты липопротеина(-ов),
- органические нитраты и доноры NO, например, нитропруссид натрия, нитроглицерин, изосорбид мононитрат, изосорбид динитрат, молсидомин или SIN-1, а также ингаляционный NO,
- соединения с положительным инотропным действием, например, сердечные гликозиды (дигоксин), бета-адренергические и допамин-эргические агонисты, в частности, изопротеренол, адреналин, норадреналин, допамин и добутамин,
- соединения, ингибирующие деструкцию циклического гуанозинмонофосфата (cGMP) и/или циклического аденозинмонофосфата (сАМР), например, ингибиторы фосфодиэстеразы 1, 2, 3, 4 и/или 5, в частности, ингибиторы фосфодиэстеразы 5 (например, силденафил, варденафил и тадалафил), а также ингибиторы фосфодиэстеразы 3 (например, амринон и милринон),
- натрийуретические пептиды, например, предсердный натрийуретический пептид (ANP, анаритид), натрийуретический пептид В-типа, мозговой натрийуретический пептид (BNP, незиритид) или натрийуретический пептид С-типа (CNP), а также уродилатин,
- сенсибилизаторы кальция, например, предпочтительно левосимендан,
- NO-независимые, однако гемзависимые стимуляторы гуаниатциклазы, в частности, соединения, описанные в международных заявках W0 00/06568, WO 00/06569, WO 02/42301 и WO 03/095451,
- NO-зависимые и гемнезависимые активаторы гуаниатциклазы, в частности, соединения, описанные в международных заявках WO 01/19355, WO 01/19776, WO 01/19778, WO 01/19780, WO 02/070462 и WO 02/070510,
- ингибиторы человеческой нейтрофильной эластазы (HNE), например, сивелестат или DX-890 (релтран),
- соединения, ингибирующие каскад сигнальной трансдукции, например, ингибиторы терозинкиназы, в частности, сорафениб, иматиниб, гефитиниб и эрлотиниб, и/или
- соединения, оказывающие влияние на энергетический обмен сердца, примерами которых предпочтительно являются этомоксир, дихлор-ацетат, ранолазин или триметазидин.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с диуретиками, примерами которых предпочтительно являются фуросемид, буметанид, торземид, бендрофлуметиазид, хлортиазид, гидрохлортиазид, гидрофлу-метиазид, метиклотиазид, политиазид, трихлорметиазид, хлортралогени-дон, индапамид, метолазон, хинэтазон, ацетозоламид, дихлорфенамид, метазоламид, глицерин, изосорбид, маннит, амилорид или триамтерен.
Снижающими кровяное давление средствами предпочтительно являются соединения, выбранные из группы, включающей антагонисты кальция, антагонисты ангиотензина AII, ингибиторы АСЕ, антагонисты эндотелина, ингибиторы ренина, блокаторы альфа-рецепторов, блокаторы бета-рецепторов и ингибиторы Rho-киназы, а также диуретики.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с антагонистом кальция, например, предпочтительно нифедипином, амлодипином, вера-памилом или дилтиаземом.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с антагонистом ангиотензина AII, например, предпочтительно лозартаном, кандесартаном, валсартаном, телмизартаном или эмбусартаном.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с ингибитором АСЕ, например, предпочтительно эналаприлом, каптоприлом, лисиноприлом, рамиприлом, делаприлом, фосиноприлом, хиноприлом, периндоприлом или трандоприлом.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с антагонистом эндотелина, например, предпочтительно босентаном, дарусентаном, амбрисентаном или ситакссентаном.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с ингибитором ренина, например, предпочтительно алискиреном, SPP-600, SPP-635, SPP-676, SPP-800 или SPP-1148.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с блокатором альфа-1-рецепторов, например, предпочтительно празосином.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с блокатором бета-рецепторов, например, предпочтительно пропранололом, атенонололом, тимололом, пиндилолом, алпренололом, окспренололом, пенбутололом, бупранололом, метипроналолом, надололом, мепиндололом, каразололом, солатолом, метопрололом, бетаксололом, целипрололом, бисопрололом, картеололом, эсмололом, лабетололом, карведилолом, адапрололом, ландиололом, небивололом, эпанололом или буциндололом.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с ингибитором Rho-киназы, например, предпочтительно фасудилом, Y-27632, SLX-2119, BF-66851, BF-66852, BF-66853, KI-23095 или ВА-1049.
Под средствами, обладающими антитромботическим действием, предпочтительно подразумевают соединения, выбранные из группы, включающей ингибиторы агрегации тромбоцитов, противосвертывающие вещества и профибринолитические вещества.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с ингибитором агрегации тромбоцитов, например, предпочтительно аспирином, клопидо-грелом, тиклопидином или дипиридамолом.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с ингибитором тромбина, например, предпочтительно ксимелагатраном, мелагатраном, бивалирудином или клексаном.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с антагонистом GPIIb/IIIa, например, предпочтительно тирофибаном или абциксимабом.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с ингибитором фактора Ха, например, предпочтительно ривароксабаном (BAY 59-7939), DU-176b, апиксабаном, отамиксабаном, фидексабаном, разаксабаном, фондапаринуксом, идрапаринуксом, PMD-3112, YM-150, KFA-1982, EMD-503982, МСМ-17, MLN-1021, DX 9065а, DPC 906, JTV 803, SSR-126512 или SSR-128428.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с гепарином или низкомолекулярным производным гепарина.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с антагонистами витамина K, например, предпочтительно кумарином.
Под изменяющими жировой обмен средствами предпочтительно подразумевают соединения, выбранные из группы, включающей ингибиторы СЕТР, агонисты рецепторов щитовидной железы, ингибиторы синтеза холестерина, в частности, ингибиторы HMG-CoA-редуктазы или синтеза сквалена, ингибиторы АСАТ, ингибиторы МТР, PPAR-альфа-, PPAR-гамма- и/или PPAR-дельта-агонисты, ингибиторы, абсорбирующие холестерин, полимерные адсорберы желчной кислоты, реабсорбирующие ингибиторы желчной кислоты, ингибиторы липазы, а также антагонисты липо-протеина(-ов).
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с ингибитором СЕТР, например, предпочтительно торцетрапибом (СР-529 414), JJT-705, BAY 60-5521, BAY 78-7499 или СЕТР-вакциной (Avant).
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с агонистом рецепторов щитовидной железы, например, предпочтительно D-тироксином, 3,5,3'-трийодтиронином (Т3), CGS 23425 или акситиромом (CGS 26214).
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с ингибитором HMG-СоА-редуктазы из класса статинов, например, предпочтительно ловастатином, симвастатином, правастатином, флувастатином, аторвастатином, розувастатином, церивастатином или питавастатином.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с ингибитором синтеза сквалена, например, предпочтительно BMS-188494 или ТАК-475.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с ингибитором АСАТ, например, предпочтительно авасимибом, мелинамидом, пактимибом, эфлуцимибом или SMP-797.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с ингибитором МТР, например, предпочтительно имплитапидом, BMS-201038, R-103757 или JTT-130.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с агонистом PPAR-гамма, например, предпочтительно пиоглитазоном или розиглитазоном.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с агонистом PPAR-дельта, например, предпочтительно GW-501516 или BAY 68-5042,
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с абсорбирующим холестерин ингибитором, например, предпочтительно эзетимибом, тиквузидом или памаквузидом.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с ингибитором липазы, например, предпочтительно орлистатом.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с полимерным адсорбером желчной кислоты, например, предпочтительно холестирамином, колестиполом, колесольвамом, холестагелем или колестимидом.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с реабсорбирующим ингибитором желчной кислоты, например, предпочтительно с ингибитором ASBT (=IBAT), например, таким как AZD-7806, S-8921, AK-105, BARI-1741, SC-435 или SC-635.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения назначают в комбинации с антагонистом липопротеина(-ов), например, предпочтительно препаратом Gemcabene calcium (CI-1027) или никотиновой кислотой.
Другим объектом настоящего изобретения являются лекарственные средства, содержащие по меньшей мере одно предлагаемое в изобретении соединение обычно совместно с одним или несколькими инертными, нетоксичными, фармацевтически приемлемыми вспомогательными веществами, а также применение этих лекарственных средств для указанных выше целей.
Предлагаемые в изобретении соединения могут обладать системным и/или локальным действием. Для реализации указанного действия их можно применять пригодным образом, например, орально, парентерально, пульмонально, назально, сублингвально, лингвально, буккально, ректально, дермально, трансдермально, коньюнктивально, введением в уши или в виде имплантата, соответственно стент.
Предлагаемые в изобретении соединения можно назначать в соответствующих указанным выше методам формах применения.
Для орального применения пригодны функционирующие в соответствии с уровнем техники формы применения, быстро и/или модифицированно высвобождающие предлагаемые в изобретении соединения, которые находятся в них в кристаллической, аморфизованной и/или растворенной форме, например, таблетки (непокрытые или покрытые таблетки, например, снабженные устойчивыми к действию желудочного сока, замедленно растворяющимися или нерастворимыми покрытиями, которые обеспечивают контролируемое высвобождение предлагаемого в изобретении соединения), быстро распадающиеся в ротовой полости таблетки или пленки/облатки, пленки/лиофилизаты, капсулы, например, из жесткого или мягкого желатина, драже, грануляты, пеллеты, порошки, эмульсии, суспензии, аэрозоли или растворы.
Парентеральное применение может происходить без стадии резорбции (например, внутривенно, внутриартериально, внутрикардиально, внутрипозвоночно или внутрипояснично) или с включением стадии резорбции (например, внутримышечно, подкожно, внутрикожно, чрескожно или внутрибрюшинно). В качестве форм, предназначенных для парентерального применения, пригодны, в частности, инъекционные и инфузионные препараты в виде растворов, суспензий, эмульсий, лиофилизатов или стерильных порошков.
Для прочих методов применения пригодны, например, ингаляционные лекарственные формы (в частности, порошковые ингаляторы, распылители), назальные капли, растворы или спреи, таблетки, подлежащие лингвальному, сублингвальному или буккальному применению, пленки/облатки или капсулы, суппозитории, ушные или глазные препараты, вагинальные капсулы, водные суспензии (лосьоны, микстуры типа «болтушка»), липофильные суспензии, мази, кремы, трансдермальные терапевтические системы (например, пластыри), препараты в виде молочка, паст, пен, присыпок, имплантатов или стентов.
Предпочтительным является оральное или парентеральное применение, в частности, оральное или внутривенное применение.
Предлагаемые в изобретении соединения могут быть преобразованы в указанные выше формы применения. Подобное преобразование можно осуществлять известными методами, предусматривающими смешивание с инертными, нетоксичными, фармацевтически приемлемыми вспомогательными веществами. К подобным вспомогательным веществам относятся, в частности, вещества-переносчики (например микрокристаллическая целлюлоза, лактоза, маннит), растворители (например, жидкие полиэтиленгликоли), эмульгаторы, диспергирующие или смачивающие средства (например, натрийдодецилсульфат, полиоксисорбитанолеат), связующие вещества (например, поливинилпирролидон), синтетические и природные полимеры (например, альбумин), стабилизаторы (например, антиоксиданты, в частности, аскорбиновая кислота), красители (например, неорганические пигменты, в частности, оксиды железа), а также средства для корректирования вкуса и/или запаха.
В общем случае необходимое для обеспечения должной эффективности количество при парентеральном применении предпочтительно составляет примерно от 0,001 до 1 мг, предпочтительно примерно от 0,01 до 0,5 мг соответственно на килограмм массы тела. При оральном применении дозировка находится в примерном интервале от 0,01 до 100 мг, предпочтительно от 0,01 до 20 мг, еще более предпочтительно от 0,1 до 10 мг соответственно на килограмм массы тела.
Тем не менее в некоторых случаях может возникнуть необходимость в отклонении от указанных выше количеств, зависящая от массы тела, метода применения, индивидуального отношения к биологически активному веществу, типа препарата и момента времени применения, соответственно временного интервала, в течение которого реализуют применение. Так, например, в некоторых случаях может оказаться достаточным количество, меньшее указанного выше нижнего предела, тогда как в других случаях применяемое количество должно превышать соответствующий верхний предел. В случае применения больших количеств рекомендуется распределять их на несколько принимаемых в течение дня разовых доз.
Приведенные ниже примеры служат для пояснения настоящего изобретения и не ограничивают его объем.
В отсутствие особых указаний количественные данные в нижеследующих тестах и примерах приведены в массовых процентах или массовых частях. Соотношения растворителей, степени разбавления и концентрации растворов «жидкость/жидкость» соответственно указаны в пересчете на объем.
Экспериментальная часть
Сокращения и аббревиатуры

Примеры
Пример 1. Метил-4-бром-2-метоксибензоат (XV)
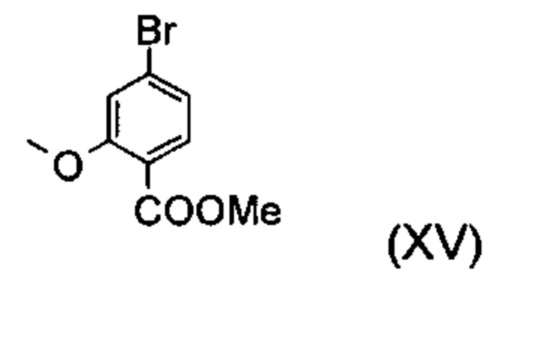
3,06 кг (22,12 моль) карбоната калия добавляют к 3,6 л ацетона и нагревают до образования флегмы. К суспензии карбоната калия добавляют 1,2 кг 4-бром-2-гидроксибензойной кислоты (5,53 моль), суспендированного в 7,8 л ацетона и промытого ацетоном (0,6 л). Реагенты в течение часа нагревают с обратным холодильником (происходит сильное газовыделение). Затем в течение 4 часов при кипении добавляют 2,65 кг (21,01 моль) диметилсульфата. В течение последующих 2,5 часов реакционную смесь перемешивают при нагревании с обратным холодильником. Отгоняют большую часть растворителя (пока сохраняется способность реакционной смеси к перемешиванию), добавляют 12 л толуола и при 110°С отгоняют остаточный ацетон. Отгоняют около 3 л дистиллята и это количество дополняют посредством добавления к реакционной смеси еще 3 л толуола. После охлаждения до 20°С добавляют 10,8 л воды, и смесь энергично перемешивают. Отделяют органическую фазу, водную фазу экстрагируют толуолом (6,1 л). Объединенные органические фазы промывают насыщенным раствором поваренной соли (3 л), толуольную фазу концентрируют до объема около 4 л. Выход продукта, определенный по его содержанию после упаривания частичного количества раствора, составляет 1,306 кг (96,4% от теоретического). Раствор непосредственно используют на следующей стадии.


Пример 2. 4-Бром-2-метоксибензальдегид (XIX)
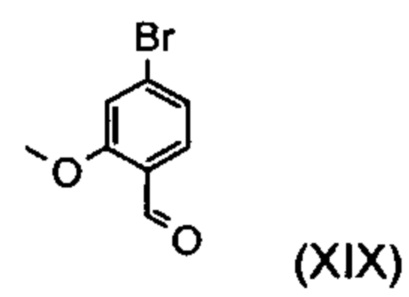
1,936 кг (6,22 моль) 65-процентного раствора Red-AI в толуоле при -5°С добавляют к 1,25 л толуола. К полученному раствору добавляют 0,66 кг (6,59 моль) 1-метилпиперазина и выполняют промывку толуолом (150 мл), причем температуру поддерживают в интервале от -7 до -5°С.Реагенты в течение 30 минут перемешивают при 0°С.Полученный раствор добавляют к раствору 1,261 кг (5,147 моль) метил-4-бром-2-метоксибензоата (XV) в 4 л толуола, поддерживая температуру в интервале от -8 до 0°С.Раствор дважды промывают толуолом (0,7 л) и в течение 1,5 часов перемешивают при 0°С.С целью переработки добавляют охлажденную до 0°С водную серную кислоту (12,5 л воды+1,4 кг концентрированной серной кислоты). При этом температура должна повышаться максимум до 10°С (медленное дозирование). Посредством дополнительного добавления серной кислоты показатель рН при необходимости устанавливают на уровне 1. Отделяют органическую фазу, водную фазу экстрагируют толуолом (7,6 л). Объединенные органические фазы промывают водой (5,1 л), сильно концентрируют и концентрированный остаток вводят в 10 л диметилформамида. Вновь осуществляют концентрирование до объема около 5 л. Выход продукта, определенный по его содержанию после упаривания частичного количества раствора, составляет 1,041 кг (94,1% от теоретического). Раствор непосредственно используют на следующей стадии.

Пример 3. 4-Формил-3-метоксибензонитрил (VI)
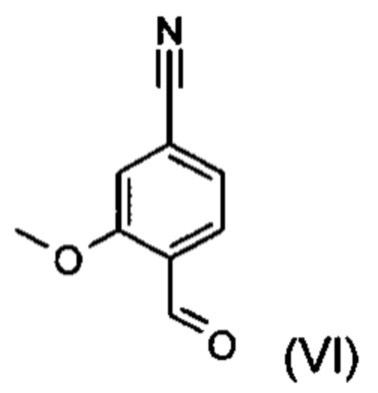
К раствору 719 г (3,34 моль) 4-бром-2-метоксибензальдегида (XVI) в 4,5 л диметилформамида добавляют 313 г (0,74 моль) гексацианоферрата калия (K4[Fe(CN)6]), 354 г (3,34 моль) карбоната натрия, дополнительно 1,2 л диметилформамида и 3,8 г (0,017 моль) ацетата палладия. Реакционную смесь в течение 3 часов перемешивают при 120°С.После охлаждения до 20°С добавляют 5,7 л воды. Затем смесь экстрагируют этилацетатом (17 л), водную фазу еще раз промывают этилацетатом (17 л). Органические фазы объединяют, сильно концентрируют, смешивают с 5 л изопропанола и вновь концентрируют до объема около 2 л. После нагревания до температуры кипения по каплям добавляют 2 л воды. После остывания смеси до 50°С вновь добавляют 2 л воды. Смесь охлаждают до 3°С и в течение одного часа перемешивают при этой температуре. Продукт отфильтровывают и дважды промывают водой (порциями соответственно по 1,2 л). Затем продукт сушат при 40°С в вакууме.

Пример 4. (2Е/2Z)-2-(4-циан-2-метоксибензилиден)-3-оксобутанамид (XVI а, b)
1000 г (6204,95 ммоль) 4-формил-3-метоксибензонитрила (VI), 721,5 г (7135,7 ммоль) 3-оксобутанамида (XVII), 53 г (620 ммоль) пиперидина и 37,3 г (620 ммоль) ледяной уксусной кислоты в 15 л дихлорметане в течение четырех часов нагревают с обратным холодильником и водоотделителем. Затем отгоняют около 10 л дихлорметана и реакционную смесь охлаждают до комнатной температуры. После охлаждения до 0°С реакционную смесь в течение 4 часов перемешивают при этой температуре, продукт отфильтровывают и дважды промывают холодным дихлорметаном (порциями соответственно по 1000 мл). Затем продукт сушат при 40°С в вакууме и атмосфере газа-носителя.

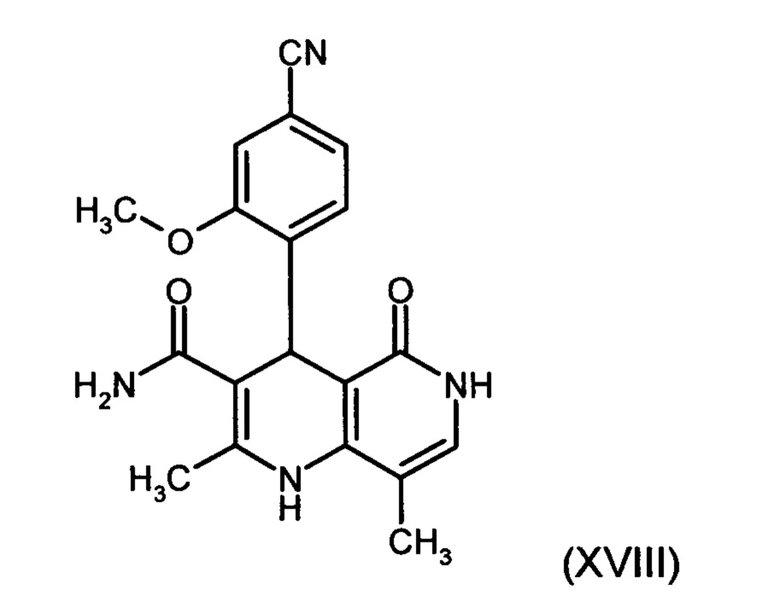
Пример 5. 4-(4-Циан-2-метоксифенил)-2,8-диметил-5-оксо-1,4,5,6-тетрагидро-1,6-нафтиридин-3-карбоксамид (XVIII)
2,128 кг (8,712 моль) (2Е/2Z)-2-(4-циан-2-метоксибензилиден)-3-оксобутан-амида (XVI а, b) смешивают с 29 л 2-бутанола, добавляют 1,277 кг (7,92 моль) 4-амино-5-метилпиридона и в течение 12 часов нагревают в закрытом резервуаре при внутренней температуре 120°С и избыточном давлении. Реакционную смесь в течение 5 часов плавно охлажадают до 0°С и в течение последующих 3 часов перемешивают при этой температуре. Продукт отфильтровывают и промывают холодным изопропанолом (2,1 л). Затем продукт сушат в вакууме при 60°С.

Пример 5. 4-(4-Циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (XIII)
1,857 кг (5,3 моль) 4-(4-циан-2-метоксифенил)-2,8-диметил-5-оксо-1,4,5,6-тетрагидро-1,6-нафтиридин-3-карбоксамида (XVIII) и 4,70 кг (29 моль) сложного триэтилового эфира ортоуксусной кислоты растворяют в 12,15 л диметилацетамида и добавляют 157,5 г концентрированной серной кислоты. Реагенты в течение 1,5 часов нагревают при 115°С, а затем охлаждают до 50°С. При 50°С в течение 30 минут по каплям добавляют 12,15 л воды. По завершении добавления воды вносят 10 г кристаллической затравки в виде титульного соединения (XI) и в течение 30 минут при 50°С дополнительно по каплям добавляют 12,15 л воды. Реакционную смесь охлаждают до 0°С (плавно, в течение двух часов) и в течение двух часов перемешивают при 0°С. Продукт отфильтровывавают, дважды промывают водой (порциями соответственно по 7,7 л) и сушат в вакууме при 50°С.

Пример 6. (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (I) в виде раствора в ацетонитриле/метаноле 40:60
Разделение энантиомеров на установке SMB
Питающим раствором является раствор соответствующей концентрации, состоящий из 50 г рацемата 4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида (XIII), растворенного в 1 литре смеси метанола с ацетонитрилом (60:40).
Хроматографирование осуществляют на установке SMB с использованием стационарной фазы Chiralpak AS-V (20 мкм). Давление составляет 30 бар, в качестве элюента используют смесь метанол с ацетонитрилом (60:40).
9,00 кг 4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида (XIII) растворяют в 180 л смеси метанола с ацетонитрилом (60:40) и хроматографируют на установке SMB. После концентрирования содержащих продукт фракций получают 69,68 литров раствора концентрацией 6,2%, что соответствует 4,32 кг (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида (I) в виде раствора в ацетонитриле/метаноле (40:60).
Выход: 4,32 кг продукта, растворенного в 69,68 литрах смеси ацетонитрила с метанолом (40:60), в виде бесцветной фракции (48% от теоретического).
Степень чистоты энантиомеров: более 98,5% (ВЭЖХ, метод D).
MS (Elpos) концентрированного в вакууме образца: m/z=379 [М+Н]+.
1Н-ЯМР (300 МГц, диметилсульфоксид-d6): δ=1,05 (t, 3Н), 2,12 (s, 3Н), 2,18 (s, 3Н), 3,82 (s, 3Н), 3,99-4,07 (m, 2Н), 5,37 (s, 1Н), 6,60-6,84 (m, 2Н), 7,14 (d, 1Н), 7,28 (dd, 1Н), 7,37 (d, 1Н), 7,55 (s, 1Н), 7,69 (s, 1Н).
Пример 7. (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид (I)
Кристаллизация и установление полиморфии
64,52 литра 6,2-процентного раствора продукта из примера 6 в смеси ацетонитрила с метанолом 40:60 (соответствует 4,00 кг соединения формулы I) фильтруют через фильтровальный патрон (1,2 um), а затем концентрируют дистилляцией при 250 мбар до такой степени, чтобы сохранялась возможность перемешивания раствора. Добавляют 48 л денатурированного толуолом этанола и вновь осуществляют дистилляцию при 250 мбар до предельной концентрации, при которой сохраняется способность к перемешиванию (редистилляция с заменой на этанол). Добавляют 48 л денатурированного толуолом этанола, а затем осуществляют дистилляцию при нормальном давлении до общего объема около 14 л (температура рубашки 98°С). Раствор в течение 4 часов плавно охлаждают до 0°С, в течение последующих 2 часов перемешивают при 0°С и отфильтровывают продукт. Продукт дважды промывают холодным этанолом (порциями соответственно по 4 л), а затем сушат в вакууме при 50°С.
Выход продукта в виде бесцветного кристаллического порошка составляет 3,64 кг (91% от теоретического).
Степень чистоты энантиомеров гораздо выше 99% (ВЭЖХ, метод D); время удерживания/RRT: (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида (I) около 11 минут (RRT: 1,00), (4R)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида (I) около 9 минут (RRT: 0,82).
Степень чистоты: более 99,8% (ВЭЖХ, метод В), время удерживания около 6,7 минут.
Содержание: 99,9% (относительно внешнего стандарта).
Значение удельного вращения (хлороформ, 589 нм, 19,7°С, концентрация 0,38600 г/100 мл): -148,8°.
MS (Elpos): m/z=379 [M+H]+
1Н-ЯМР (300 МГц, диметилсульфоксид-d6): δ=1,05 (t, 3Н), 2,12 (s, 3Н), 2,18 (s, 3Н), 3,82 (s, 3Н), 3,99-4,07 (m, 2Н), 5,37 (s, 1Н), 6,60-6,84 (m, 2Н), 7,14 (d, 1Н), 7,28 (dd, 1Н), 7,37 (d, 1Н), 7,55 (s, 1Н), 7,69 (s, 1Н).
Точка плавления: 252°С (соединение формулы (I) в кристаллической форме модификации I).
Физико-химические характеристики соединения формулы (I) в кристаллической форме модификации I
Соединение формулы (I) в кристаллической форме модификации I плавится при 252°С, ΔН от 95 до 113 Дж/г (скорость нагревания 20 K/мин).
В зависимости от скорости нагревания наблюдается депрессия точки плавления.
При уменьшении скорости нагревания (например, до 2 K/мин) точка плавления снижается, поскольку происходит деструкция.
Наблюдается отсутствие каких-либо других фазовых переходов. Весовая потеря при нагревании до температуры 175°С составляет около 0,1%.
Стабильность и влажность при хранении
Образцы соединения формулы (I) в кристаллической форме модификации I хранят при относительной влажности воздуха (25°С) 85%, соответственно 97%. Через 12 месяцев образцы оценивают методами дифференциальной сканирующей калориметрии, термогравиметрического анализа и порошкового рентгеноструктурного анализа. Изменение массы после 12-месячного хранения в обоих случаях составляет менее 0,1%. Таким образом, при хранении соединения формулы (I) в кристаллической форме модификации I в указанных выше условиях сколько-нибудь существенное водопоглощение отсутствует. Согласно результатам дифференциальной сканирующей калориметрии, термогравиметрического анализа и порошкового рентгеноструктурного анализа никакого изменения соединения формулы (I) в кристаллической форме модификации I не происходит.
Условия/методы высокоэффективной жидкостной хроматографии при высоком давлении
Метод А
YMC Hydrosphere С18
Колонка 150×4,6 мм (3,0 мкм)
25°С, 1 мл/мин, 270 нм, 4 нм
0 минут: 70% трифторуксусной кислоты (0,1% в воде), 30% ацетонитрила
17 минут: 20% трифторуксусной кислоты (0,1% в воде), 80% ацетонитрила
18 минут: 70% трифторуксусной кислоты (0,1% в воде), 30% ацетонитрила
Метод В
YMC Hydrosphere С18
Колонка 150×4,6 мм (3,0 мкм)
25°С, 1 мл/мин, 255 нм, 6 нм
0 минут: 90% трифторуксусной кислоты (0,1% в воде), 10% ацетонитрила
20 минут: 10% трифторуксусной кислоты (0,1% в воде); 90% ацетонитрила
18 минут: 10% трифторуксусной кислоты (0,1% в воде), 90% ацетонитрила
Метод С
Nucleodur Gravity С18
Колонка 150×2 мм (3,0 мкм)
35°С, 0,22 мл/мин, 255 нм, 6 нм
Раствор А: 0,58 г гидрофосфата аммония и 0,66 г дигидрофосфата аммония в 1 литре воды (рН аммонийфосфатного буфера 7,2)
Раствор В: ацетонитрил
0 минут: 30% В, 70% А
15 минут: 80% В, 20% А
25 минут: 80% В, 20% А
Метод D
Длина колонки: 25 см
Внутренний диаметр колонки: 4,6 мм
Насадка: Chiralpak IA (5 мкм)
Реагенты: 1. ацетонитрил (для ВЭЖХ), 2. метил-трет-бутиловый эфир (чда)
Испытуемый раствор: образец растворяют в ацетонитриле в концентрации 1,0 мг/мл (например, около 25 мг точно взвешенного образца растворяют в ацетонитриле и доводят объем до 25,0 мл)
Элюент: А - ацетонитрил, В - метил-трет-бутиловый эфир (чда)
Скорость течения: 0,8 мл/мин
Температура печи колонки: 25°С
Детектирование: длина волны 255 нм
Ширина полосы: 6 нм
Инжектируемый объем: 5 мкл
Элюенты А и В смешивают в объемном отношении 90:10
Длительность цикла хроматографии: 30 минут
Время удерживания/RRT: (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида (1) около 11 минут (RRT: 1,00), (4R)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамида (1) около 9 минут (RRT: 0,82)
Параметры рентгендифрактометрических измерений для соединения формулы (I) в кристаллической форме модификации I




Параметры измерения методами ИК-спектроскопии и спектроскопии Рамана для соединения формулы (I) в кристаллической форме модификации I
ИК-спектроскопия:

Спектроспопия Рамана:

Максимумы [см-1]
Максимумы [см-1]
Изобретение относится к области органической химии, а именно к способу получения соединения (4S)-4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-кар-боксамида формулы (I), отличающийся тем, что рацемическое соединение формулы (XIII) разделяют на соответствующие энантиомеры, причем соединение формулы (XIII) получают путем взаимодействия соединения формулы (XVIII) со сложным ортоэфиром формулы (XX), в которой R означает водород или метил, при кислотном катализе и температуре 100-120°С. Также изобретение относится к соединениям формул (XVIII) и (XVI a,b), их применению в способе получения соединения формулы (I), способам получения соединений формул (XVIII), (XVI a,b) и (XIII). Технический результат: разработан новый способ получения соединения формулы (I), полезного в качестве нестероидного антагониста минералокортикоидного рецептора, отличающийся высоким общим выходом и чистотой целевого соединения и низкими производственными расходами. 10 н. и 2 з.п. ф-лы, 8 пр.
1. Способ получения соединения формулы (I):
отличающийся тем, что рацемическое соединение формулы (XIII) разделяют на соответствующие энантиомеры,
причем соединение формулы (XIII):
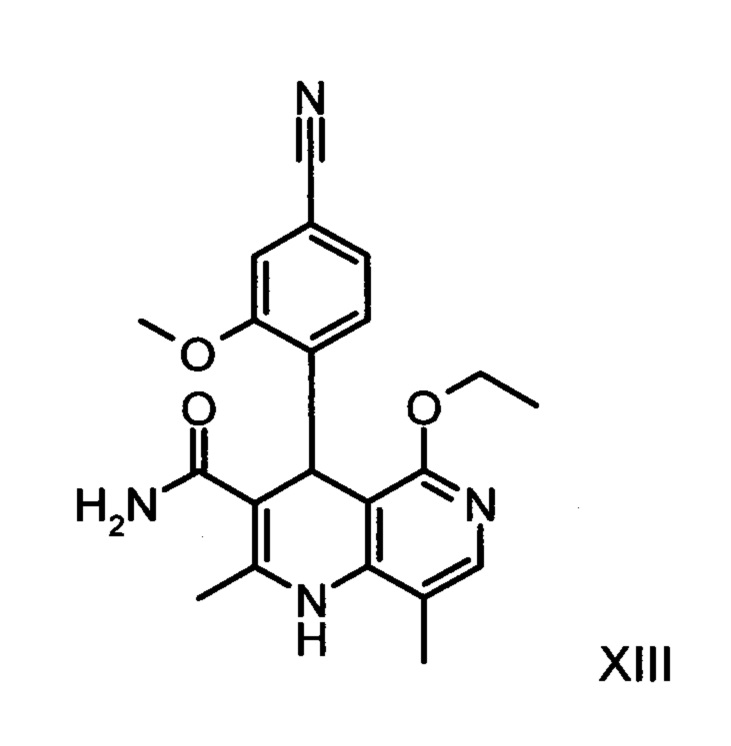
получают путем взаимодействия соединения формулы (XVIII):
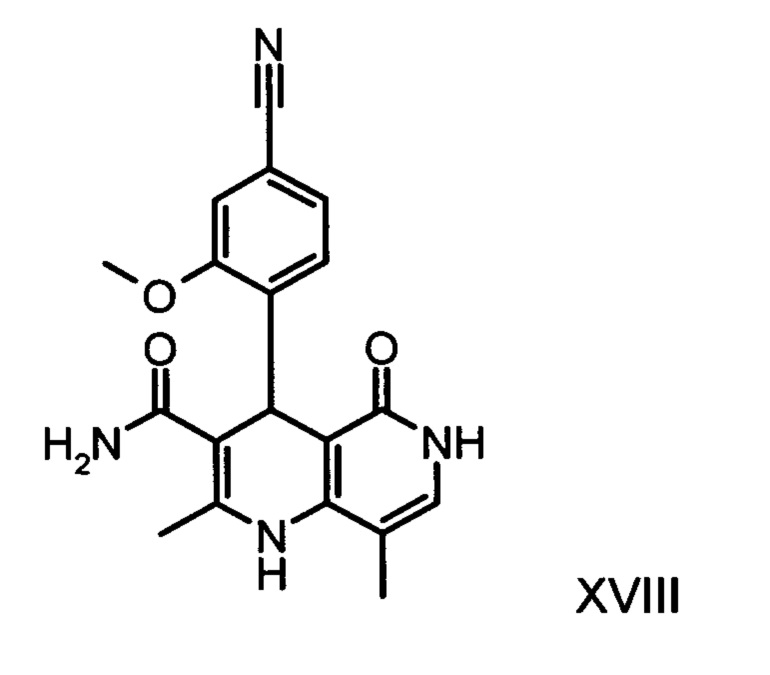
со сложным ортоэфиром формулы (XX):
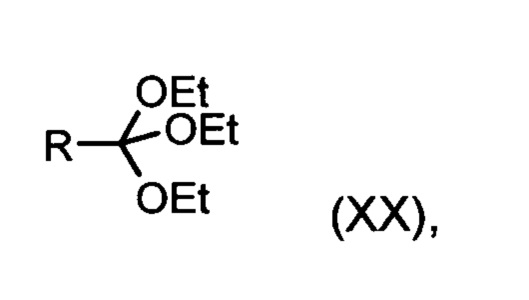
в которой R означает водород или метил,
при кислотном катализе и температуре 100-120°С.
2. Способ получения соединения формулы (I):
по п.1, отличающийся тем, что рацемическое соединение формулы (XIII) разделяют на соответствующие энантиомеры, причем соединение формулы (XIII):
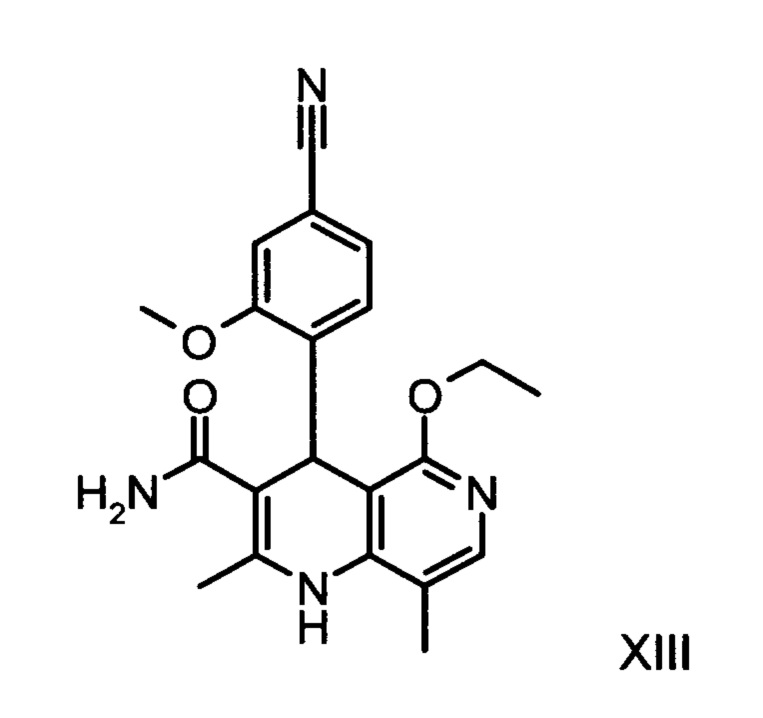
получают путем взаимодействия соединения формулы (XVIII)
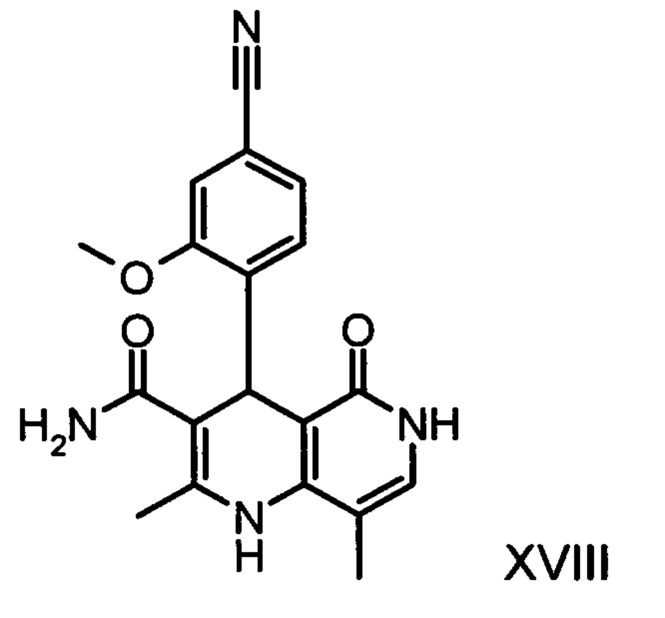
со сложным ортоэфиром формулы (XX)
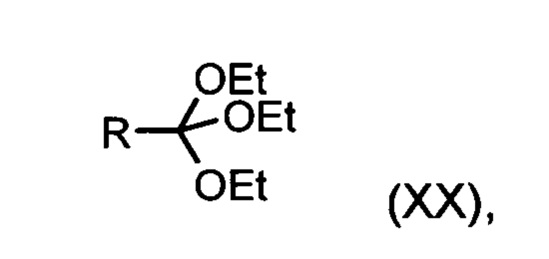
в которой R означает Н или метил,
при кислотном катализе и температуре 100-120°С,
и соединение формулы (XVIII)
получают путем взаимодействия соединений формулы (XVI а, b):
с соединением формулы (IX):
при температуре 80-160°С.
3. Способ получения соединения формулы (I):
по п.2, отличающийся тем, что рацемическое соединение формулы (XIII) разделяют на соответствующие энантиомеры, причем соединение формулы (XIII):
получают путем взаимодействия соединения формулы (XVIII):
со сложным ортоэфиром формулы (XX):
в которой R означает Н или метил,
при кислотном катализе и температуре 100-120°С,
и соединение формулы (XVIII)
получают путем взаимодействия соединений формулы (XVI а, b):
с соединением формулы (IX):
при температуре 80-160°С,
и соединение формулы (XVI а, b) получают путем взаимодействия соединения формулы (VI):
с соединением формулы (XVII)
в дихлорметане при добавлении от 5 до 20 % мол. пиперидина и от 5 до 20 % мол. ледяной уксусной кислоты.
4. Применение соединения формулы (XVIII):
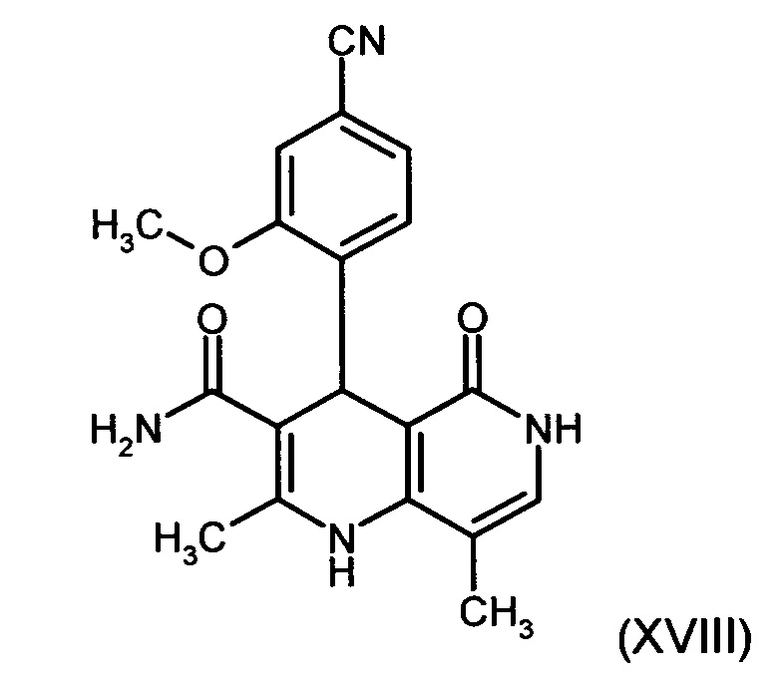
в способе получения соединения формулы (I).
5. Применение соединений формулы (XVI а, b):
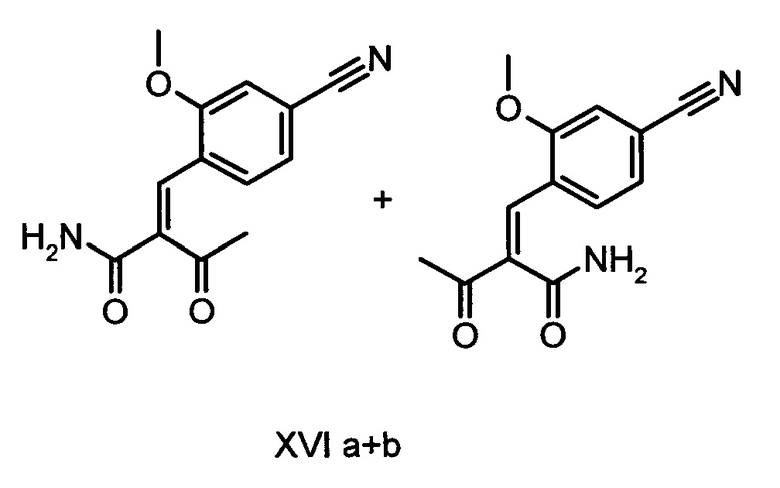
в способе получения соединения формулы (I).
6. Применение соединения формулы (XVIII):
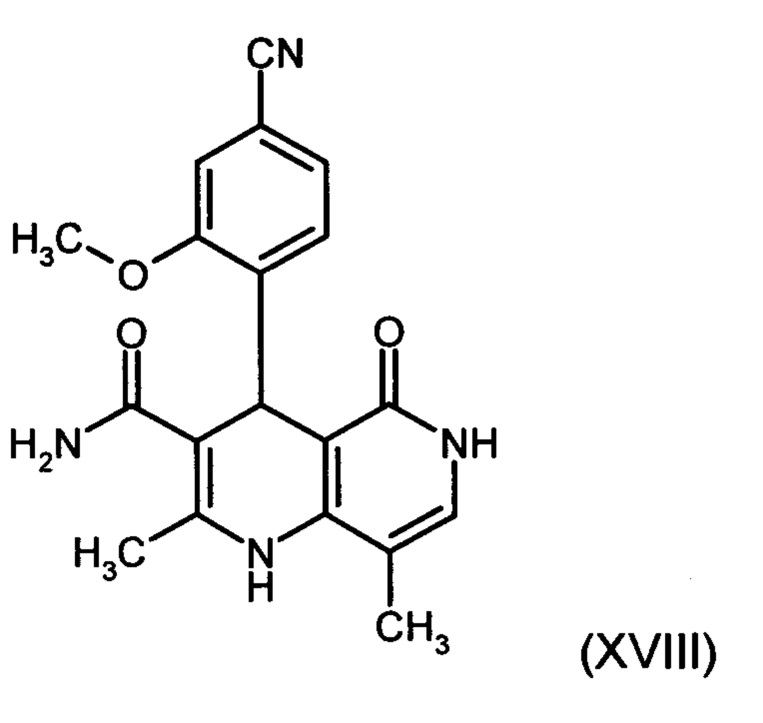
и соединений формулы (XVI а, b):
в способе получения соединения формулы (I).
7. Соединение формулы:
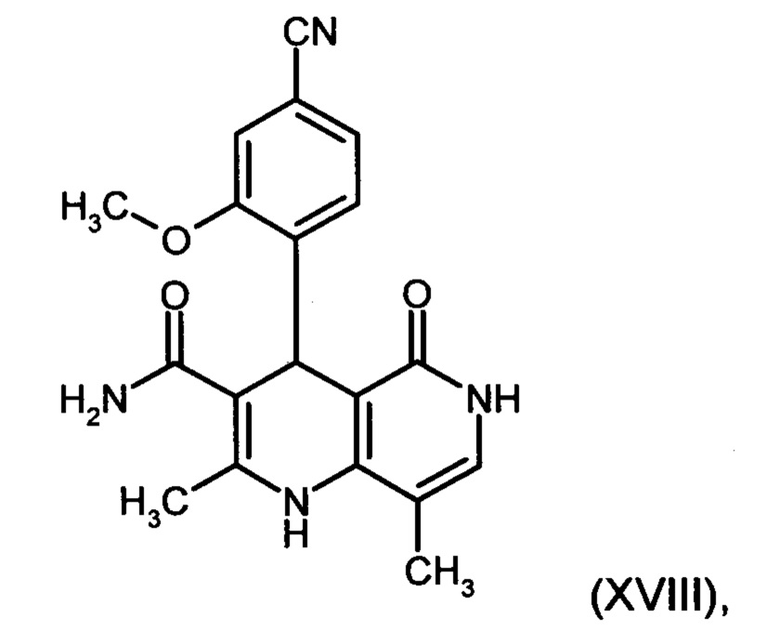
а также его соли, сольваты и сольваты солей.
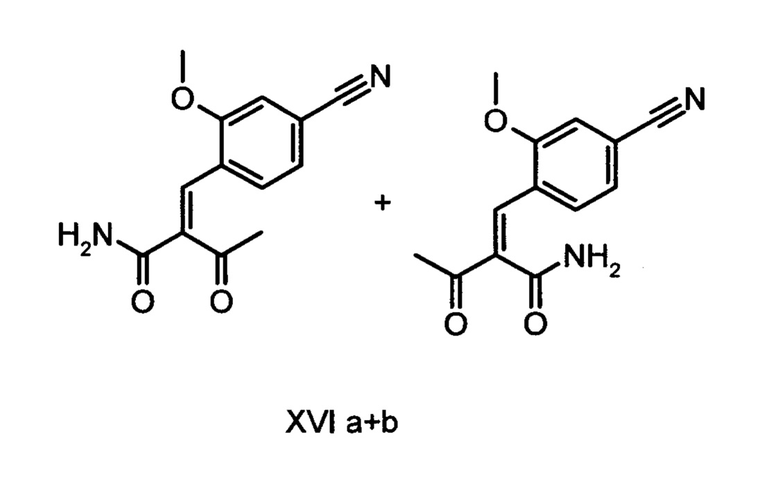
8. Соединение формулы (XVI а+b) в виде E/Z-смеси:
а также его соли, сольваты и сольваты солей.
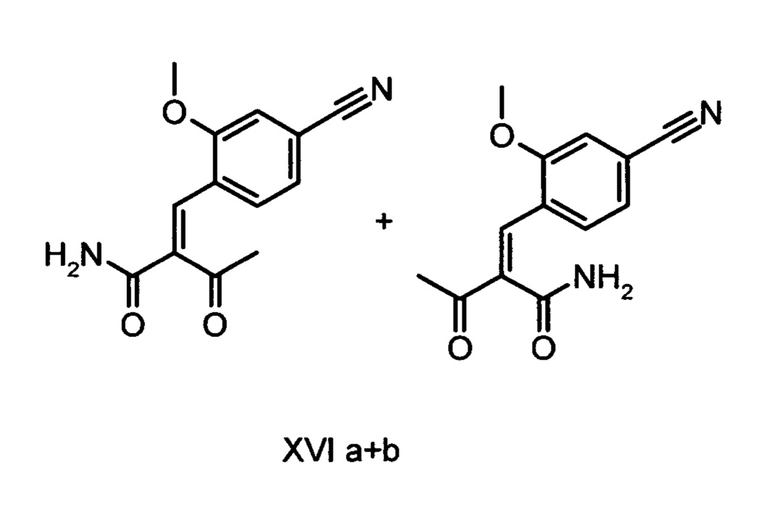
9. Способ получения соединения формулы (XVIII):
отличающийся тем, что соединения формулы (XVI а, b):
подвергают взаимодействию с соединением формулы (IX):
при кислотном катализе и температуре 100-120°С.
10. Способ получения соединений формулы (XVI а, b):
отличающийся тем, что соединение формулы (VI):
подвергают взаимодействию с соединением формулы (XVII):
в дихлорметане при добавлении от 5 до 20 % мол. пиперидина и от 5 до 20 % мол. ледяной уксусной кислоты.
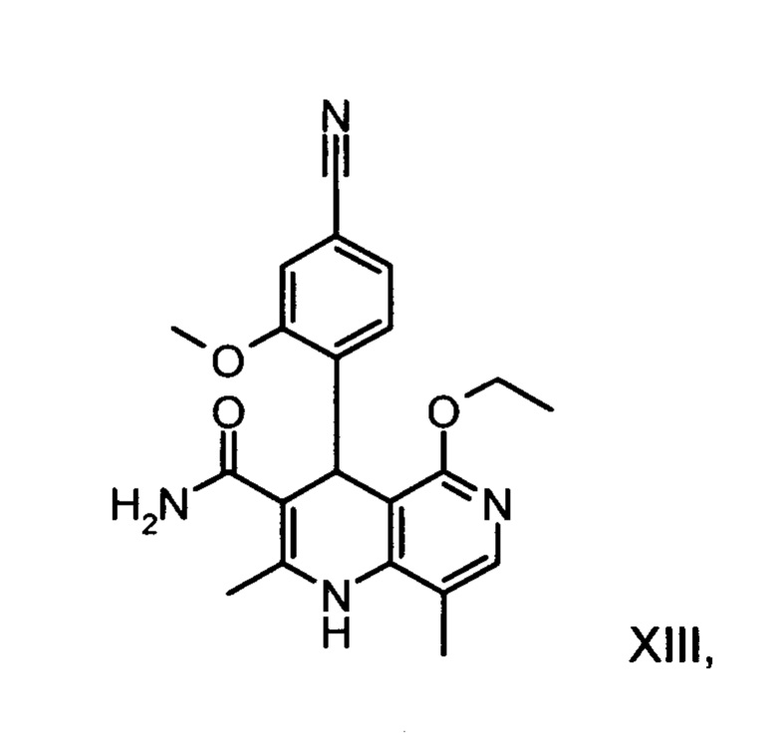
11. Способ получения соединения (XIII):
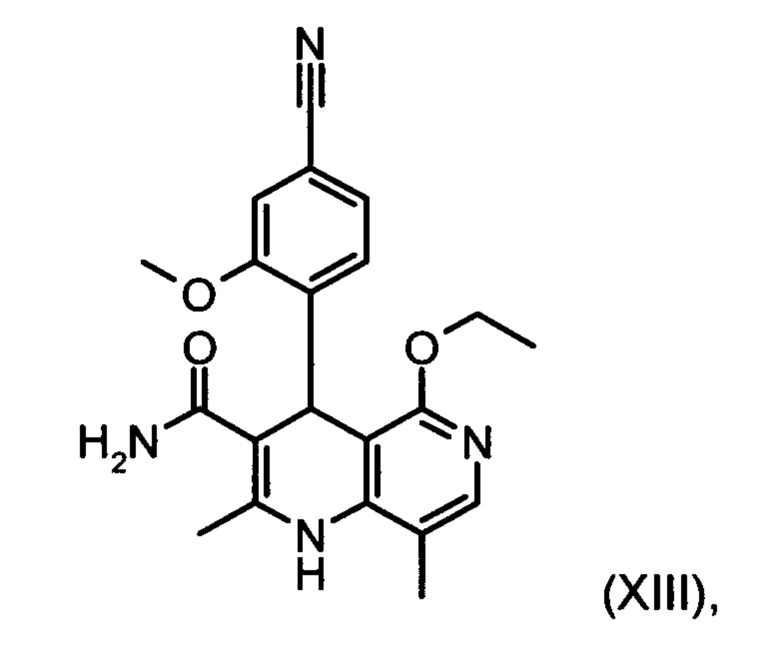
отличающийся тем, что соединение формулы (XVIII):
подвергают взаимодействию со сложным ортоэфиром формулы (XX):
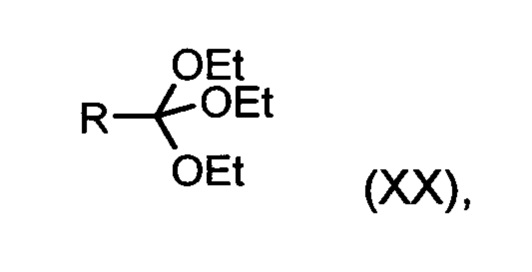
в которой R означает Н или метил,
при кислотном катализе и температуре 100-120°С.
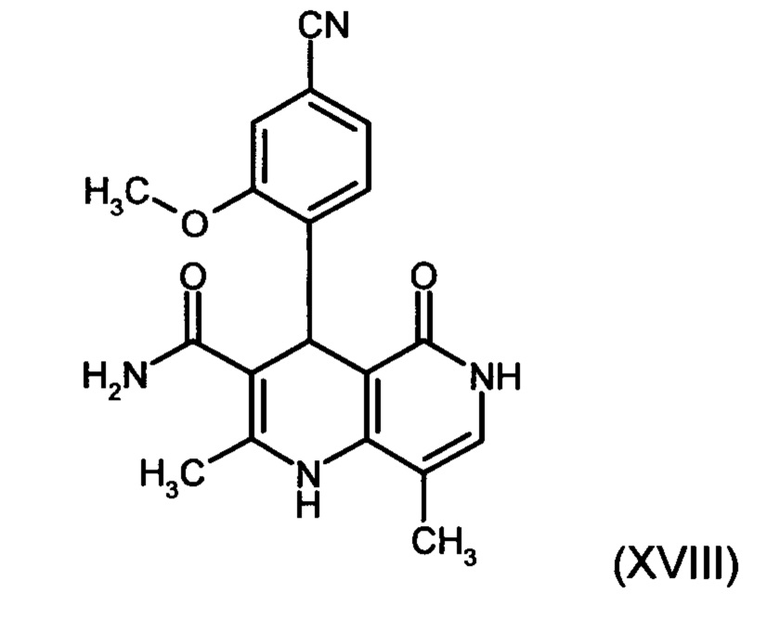
12. Способ получения соединения формулы (XIII):
отличающийся тем, что соединение формулы (XVIII):
подвергают взаимодействию со сложным ортоэфиром формулы (XX):
в которой R означает Н или метил,
при кислотном катализе и температуре 100-120°С, и
отличающийся тем, что соединение формулы (XVIII) получают посредством превращения соединений формулы (XVI а, b):
с соединением формулы (IX):
при температуре 80-160°С.
| Токарный резец | 1924 |
|
SU2016A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Baerfacker, L., Kuhl, A., Hillisch, A., Grosser, R., Figueroa-Perez, S., Heckroth, H | |||
| Kolkhof, P | |||
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Экономайзер | 0 |
|
SU94A1 |
| ChemMedChem, 7(8), | |||

