Настоящая заявка касается новых замещенных 4-арил-1,4-дигидро-1,6-нафтиридин-3-карбоксамидов, способа их получения, их применения для лечения и/или профилактики болезней, а также их применения для приготовления лекарственных средств для лечения и/или профилактики болезней, в частности кардиоваскулярных заболеваний.
Альдостерон играет ключевую роль в поддержании жидкостного и электролитного гомеостазов, поскольку он обеспечивает ретенцию натрия и секрецию калия в эпителии дистального нефрона, что способствует сохранению постоянства внеклеточного объема и вместе с тем регуляции кровяного давления. Кроме того, альдостерон оказывает непосредственное воздействие на структуру и функцию сердечно-сосудистой системы, причем лежащие в основе этого действия механизмы еще окончательно не выяснены [R.E.Booth, J.P.Johnson, J.D.Stockand, Adv. Physiol. Educ. 26(1), 8-20 (2002)].
Альдостерон - это стероидный гормон, который образуется в коре надпочечника. В основном его продукция косвенно регулируется в зависимости от почечного кровотока. Любое снижение почечного кровотока приводит в почках к вымыванию энзима ренин в систему кровообращения. Это активизирует образование ангиотензина II, который, с одной стороны, оказывает сужающее действие на артериальные кровеносные сосуды, а, с другой стороны, стимулирует образование альдостерона в коре надпочечника. Таким образом, почка действует как датчик кровяного давления и вместе с тем как косвенный датчик объема в кровообращении и через систему ренин-ангилтензин-альдостерон противодействует критической потере объема, так как, с одной стороны, повышается кровяное давление (действие ангиотензина II), а, с другой стороны, благодаря усиленной реабсорбции натрия и воды в почке снова уравновешивается состояние наполнения сосудистой системы (действие альдостерона).
Эта система регулирования может быть нарушена самыми разными способами. Например, хроническое пониженное кровоснабжение почек (например, в результате сердечной недостаточности и вызванная этим задержка оттока крови в венозной системе) приводит к хроническому повышенному вымыванию альдостерона. С другой стороны, это приводит к увеличению объема циркулирующей крови и вместе с этим усиливает сердечную слабость из-за лишнего объема циркулирующей крови к сердцу. Следствием могут быть застой крови в легких с одышкой и образованием отеков в конечностях, асцит и плевральные выпоты; почечный кровоток еще больше ослабевает. Вдобавок повышенное действие альдостерона приводит к уменьшению концентрации калия в крови и во внеклеточной жидкости. В уже поврежденных сердечных мышцах может произойти невыполнение критической минимальной величины нарушений сердечного ритма со смертельным исходом. В этом следует искать одну из главных причин внезапной сердечной смерти, часто наступающей у пациентов с сердечной недостаточностью.
Дополнительно альдостерон ответственен за ряд процессов превращений сердечной мышцы, обычно наблюдаемых при сердечной недостаточности. Таким образом, гиперальдостеронизм является решающим компонентом в патогенезе и прогнозе сердечной недостаточности, которая изначально может быть вызвана различными повреждениями, как, например, инфарктом миокарда, миокардитом или артериальной гипертонией. Это предположение подтверждается тем фактом, что в многочисленных клинических исследованиях в группах пациентов с хронической сердечной недостаточностью и после острого инфаркта миокарда значительно снижалась общая смертность путем применения антагонистов альдостерона [В.Pitt, F.Zannad, W.J.Remme et al., N.Engl. J.Med. 341. 709-717 (1999); В.Pitt, W.Remme, F.Zannad et al., N.Engl. J.Med. 348, 1309-1321 (2003)]. Это подтверждается, среди прочего, уменьшением частоты внезапной сердечной смерти.
В результате новых исследований у незначительной части пациентов, страдающих от эссенциальной гипертонии, обнаружен также так называемый нормокалиевый вариант первичного гиперальдостеронизма [преимущественно у 11% всех гипертоников: L.Seller und M.Reincke, Der Aldosteron-Renin-Quotient bei sekundärer Hypertonie, Herz 28, 686-691 (2003)]. Лучшим диагностическим методом при нормокалиевом гиперальдостеронизме служит показатель отношения альдостерон/ренин соответствующих концентраций плазмы, с тем, чтобы можно было диагностировать и, в конечном счете, лечить даже относительные повышения альдостерона по отношению к концентрациям ренина в плазме. Поэтому гиперальдостеронизм, диагностированный в связи с эссенциальной гипертонией, является отправной точкой для каузальной профилактики и для рациональной терапии.
Гораздо реже, чем приведенные выше формы гиперальдостеронизма, встречаются такие картины болезни, при которых нарушение либо следует искать в производящих гормоны клетках надпочечника, либо их количество или масса увеличивается за счет гиперплазии или разрастания. Аденомы или диффузные гиперплазии коры надпочечника - это наиболее частые причины первичного гиперальдостеронизма, называемого также как синдром Конна, ведущими симптомами которого являются гипертония и гипокалиемический алкалоз. При этом на переднем плане кроме хирургического удаления нездоровой ткани стоит медикаментозная терапия антагонистами альдостерона [Н.А. Kühn und J. Schirmeister (Hrsg.), Innere Medizin, 4. Aufl., Springer Verlag, Berlin, 1982].
Другой типичной картиной болезни, сопровождаемой повышением концентрации альдостерона в плазме, является цирроз печени в далеко зашедшей стадии. Здесь причина увеличения альдостерона лежит преимущественно в ограниченном распаде альдостерона из-за нарушения функции печени. Объемная перегрузка сердца, отеки и гипокалиемия представляют типичные последствия, которые в клинической практике могут успешно облегчаться антагонистами альдостерона.
Действия альдостерона сообщаются через минералокортикоидный рецептор, локализованный в клетках-мишенях. Имеющиеся в распоряжении до сего времени антагонисты альдостерона имеют, как и альдостерон, стероидную базовую структуру. Применимость такого рода стероидальных антагонистов ограничена из-за их взаимодействий с рецепторами других стероидных гормонов, что частично приводит к таким побочным действиям, как гинекомастия и импотенция, и к прерыванию лечения [М.А.Zaman, S.Opahl, D.A.Calhoun, Nature Rev. Drug Disc. 1, 621-636 (2002)].
Применение эффективных, нестероидальных и селективных к минералокортикоидному рецептору антагонистов делает возможным обойти этот профиль побочных действий и добиться существенного преимущества в лечении.
Задачей настоящего изобретения является получение новых соединений, которые могут использоваться в качестве селективных к минералокортикоидному рецептору антагонистов для лечения болезней, в частности кардиоваскулярных заболеваний.
В патентах ЕР 0133530-А, ЕР 0173933-А, ЕР 0189898-А и ЕР 0234516-А сообщается о 4-арил-замещенных 1,4-дигидро-1,6-нафтиридинах и -нафтиридинонах с антагонистическим действием кальция для лечения заболеваний кровеносных сосудов. О фармакологическом профиле этих соединений сообщается, среди прочего, в работе G. Werner et al., Naunyn-Schmiedeberg′s Arch. Pharmacol. 344(3), 337-344 (1991). Кроме того, в WO 02/10164 производные 1,4-дигидро-1,6-нафтиридина предназначаются как открыватели калиевых каналов для лечения различных заболеваний и, прежде всего, урологических. Производные 4-фторнонил- и 4-хромнонил-1,4-дигидропиридина в качестве антагонистов минералокортикоидного рецептора описаны в WO 2005/087740 и WO 2007/009670. В WO 2006/066011 приведены 4-арил-3-циано-1,4-дигидропиридин-5-карбоновой кислоты эфиры и амиды в качестве частично дуальных модуляторов рецепторов стероидных гормонов и кальциевого канала L-типа, а в WO 2005/097118 описаны соединения со структурой 4-арил-1,4-дигидропиридина в качестве антагонистов рецептора альдостерона.
Предметом настоящего изобретения являются соединения общей формулы (I)
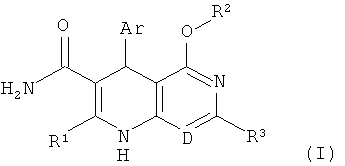
D означает N или C-R4, где
R4 означает водород, фтор, трифторметил или (С1-С4)-алкил,
Ar означает группу формул
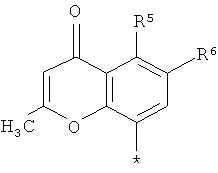 ,
, 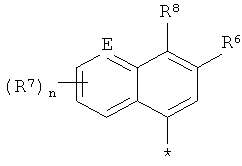 или
или 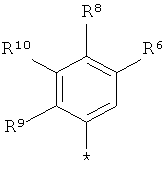
где
* означает место соединения,
R5 означает водород, фтор, хлор, циано, нитро, трифторметил или (С1-С4)-алкил,
R6 означает водород или фтор,
R7 означает галоген, (С1-С4)-алкил, трифторметил, (С1-С4)-алкокси или трифторметокси,
R8 означает циано или нитро,
R9 означает водород, галоген, (С1-С4)-алкил, (С1-С4)-алкокси, (С1-С4)-алкилтио или ди-(С1-С4)-алкиламино, причем алкильная группа в названных остатках (С1-С4)-алкил, (С1-С4)-алкокси и (С1-С4)-алкилтио может быть до трех раз замещена фтором,
или
фенил, который может быть замещен галогеном, (С1-С4)-алкилом или трифторметилом,
R10 означает водород, галоген или (С1-С4)-алкил,
Е означает CH, C-R7 или N
и
n означает число 0, 1 или 2,
причем в случае, если заместитель R7 появляется несколько раз, его значения могут быть одинаковыми или разными,
R1 - это (С1-С4)-алкил, который до трех раз может быть замещен фтором,
R2 - это (С1-С6)-алкил, который может быть замещен (С3-С7)-циклоалкилом
или до трех раз замещен фтором, или это группа формулы -SO2-R11, где
R11 означает (С1-С6)-алкил, трифторметил, (С3-С7)-циклоалкил, фенил или 5- или 6-членный гетероарил, имеющий до двух гетероатомов из ряда N, О и/или S,
причем фенил и гетероарил, в свою очередь, могут быть однократно или двукратно, одинаково или различно замещены галогеном, циано, нитро, (С1-С4)-алкилом, трифторметилом, (С1-С4)-алкокси и/или трифторметокси,
и
R3 - это водород, фтор, трифторметил или (С1-С4)-алкил,
а также их соли, сольваты и сольваты солей.
Соединениями согласно изобретению являются соединения формулы (I) и их соли, сольваты и сольваты солей, соединения приведенных далее формул, охватываемые формулой (I), и их соли, сольваты и сольваты солей, а также приведенные далее в качестве примеров исполнения соединения, охватываемые формулой (I), их соли, сольваты и сольваты солей, даже если для приведенных далее соединений, охватываемых формулой (I), речь не идет об их солях, сольватах и сольватах солей.
Соединения согласно изобретению в зависимости от их структуры могут быть в стереоизомерных формах (энантиомеры, диастереомеры). Поэтому настоящее изобретение охватывает энантиомеры и диастереомеры и их соответствующие смеси. Из таких смесей энантиомеров и/или диастереомеров известным способом могут быть выделены стереоизомерные однородные компоненты.
Если соединения согласно изобретению могут быть в таутомерных формах, то настоящее изобретение охватывает все таутомерные формы.
В качестве солей в рамках настоящего изобретения предпочтительны физиологически неопасные соли соединений согласно изобретению. Охватываются также соли, которые сами не пригодны для фармацевтических применений, но могут использоваться, например, для выделения или очистки соединений согласно изобретению.
Физиологически неопасные соли соединений согласно изобретению включают соли таких кислот, как минеральные кислоты, карбоновые и сульфоновые кислоты, например, соли соляной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, метансульфокислоты, этансульфокислоты, толуолсульфокислоты, бензолсульфокислоты, нафталиндисульфокислоты, уксусной кислоты, трифторуксусной кислоты, пропионовой кислоты, молочной кислоты, винной кислоты, яблочной кислоты, лимонной кислоты, малеиновой и бензойной кислот.
Физиологически неопасные соли соединений согласно изобретению включают соли обычных оснований, как например и преимущественно, соли щелочных металлов (например, соли натрия и калия), соли щелочноземельных металлов (например, соли кальция и магния) и соли аммония, полученные из аммиака или органических аминов с 1-16 атомами углерода, как например и преимущественно, этиламин, диэтиламин, триэтиламин, этилдиизопропиламин, моноэтаноламин, диэтаноламин, триэтаноламин, дициклогексиламин, диметиламиноэтанол, прокаин, дибензиламин, N-метилморфолин, аргинин, лизин, этилендиамин и N-метилпиперидин.
Сольватами в рамках настоящего изобретения называются такие формы соединений согласно изобретению, которые в твердом или жидком состоянии образуют комплекс путем координации с молекулами растворителя. Гидраты представляют особую форму сольватов, у которых координация осуществляется с водой. Предпочтительными сольватами в рамках настоящего изобретения являются гидраты.
Кроме того, настоящее изобретение охватывает также пролекарства соединений согласно изобретению. Понятие «пролекарства» означает соединения, которые сами могут быть биологически активными или неактивными, однако при нахождении их в организме преобразуются в соединения согласно изобретению (например, в результате метаболитических или гидролитических процессов).
В рамках настоящего изобретения заместители имеют следующие значения, если не указано другое определение:
(С1-С6)-Алкил и (С1-С4)-алкил означают в рамках изобретения алкильный остаток с разветвленной или неразветвленной цепью с 1-6 или 1-4 атомами углерода. Предпочтителен алкильный остаток с неразветвленной или разветвленной цепью с 1-4 атомами углерода. Например, и преимущественно могут быть названы следующие остатки: метил, этил, н-пропил, изопропил, н-бутил, изо-бутил, сек-бутил, трет-бутил, 1-этилпропил, н-пентил, изо-пентил и н-гексил.
(С3-С7)-Циклоалкил в рамках изобретения означает моноциклическую насыщенную циклоалкильную группу с 3-7 атомами углерода. Предпочтителен циклоалкильный остаток с 3-6 атомами углерода. Например, и преимущественно могут быть названы следующие остатки: циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
(С1-С4)-Алкокси в рамках изобретения означает остаток алкокси с неразветвленной или разветвленной цепью с 1-4 атомами углерода. Например, и преимущественно могут быть названы следующие остатки: метокси, этокси, н-пропокси, изопропокси, н-бутокси и трет-бутокси.
(С1-С4)-Алкилтио в рамках изобретения означает остаток алкилтио с неразветвленной или разветвленной цепью с 1-4 атомами углерода. Например, и преимущественно могут быть названы следующие остатки: метилтио, этилтио, н-пропилтио, изопропилтио, н-бутилтио и трет-бутилтио.
Ди-(С1-С4)-алкиламино означает в рамках изобретения аминогруппу с двумя одинаковыми или разными алкильными заместителями с неразветвленной или разветвленной цепью, имеющими по 1-4 атома углерода. Например, и преимущественно могут быть названы следующие остатки: N,N-диметиламино, N,N-диэтиламино, N-этил-N-метиламино, N-метил-N-н-пропиламино, N,N-диизопропиламино, N-изопропил-N-н-пропиламино, N-н-бутил-N-метиламино и N-трет-бутил-N-метиламино.
5- или 6-членный гетероарил означает в рамках изобретения ароматический гетероцикл (гетероароматические соединения) с 5 или 6 кольцевыми атомами, который содержит один или два кольцевых гетероатома и привязан через кольцевой атом углерода. Например, и преимущественно могут быть названы следующие остатки: фурил, пирролил, тиэнил, пиразолил, имидазолил, тиазолил, оксазолил, изоксазолил, изотиазолил, пиридил, пиримидинил, пиридазинил и пиразинил.
Галоген в рамках изобретения включает фтор, хлор, бром и йод. Предпочтителен фтор или хлор.
Если остатки в соединениях согласно изобретению являются замещенными, то они могут быть моно- или полизамещенными, если нет другого определения. В рамках настоящего изобретения действует правило, что все появляющиеся многократно остатки имеют независимые одно от другого значения. Предпочтительным является замещение одним, двумя или тремя одинаковыми или разными заместителями. Особенно предпочтительно замещение одним заместителем.
В рамках настоящего изобретения предпочтительны соединения формулы (I), в которой
D означает C-R4, где
R4 означает водород, метил или трифторметил,
Ar означает группу формул
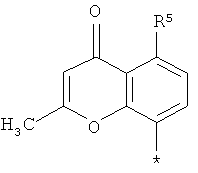 или
или 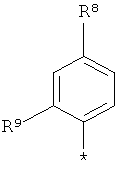 ,
,
где
* означает место соединения,
R5 означает водород, фтор, хлор или циано,
R8 означает циано или нитро
и
R9 означает хлор, бром, (С1-С4)-алкил, трифторметил, (С1-С4)-алкокси, трифторметокси, (С1-С4)-алкилтио или трифторметилтио,
R1 это метил или трифторметил,
R2 - это (С1-С4)-алкил, трифторметил или группа формулы -SO2-R11, в которой
R11 означает (С1-С4)-алкил или трифторметил,
и
R3 - это водород, метил или трифторметил,
а также их соли, сольваты и сольваты солей.
Особенно предпочтительными в рамках настоящего изобретения являются соединения формулы (I), в которых
D - это C-R4, где
R4 означает водород или метил,
Ar - это группа формулы
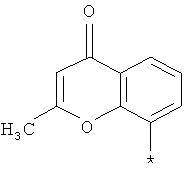 или
или 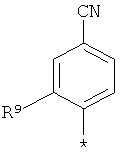 ,
,
или
в которой
* означает место соединения
и
R9 означает этил, метокси или трифторметокси,
R1 - это метил или трифторметил,
R2 - это метил, этил, н-пропил или изопропил
и
R3 - это водород или метил,
а также их соли, сольваты и сольваты солей.
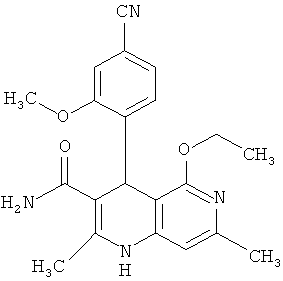
Наиболее предпочтительны соединения согласно формуле (I) со следующими структурами:
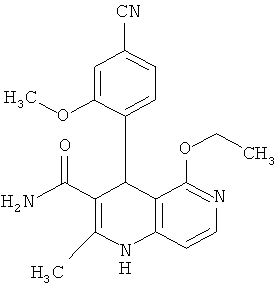 ,
, 
и 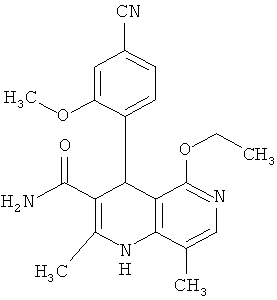
а также их соли, сольваты и сольваты солей.
При этом в особенности предпочтительны энантиомерные соединения со следующими структурами:
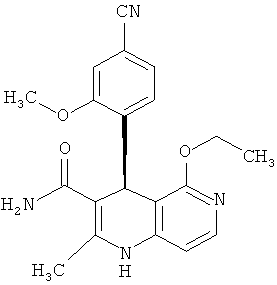 ,
, 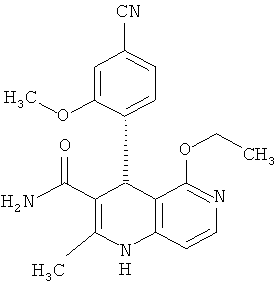 ,
,
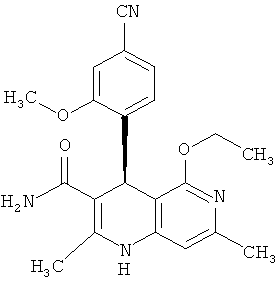 ,
, 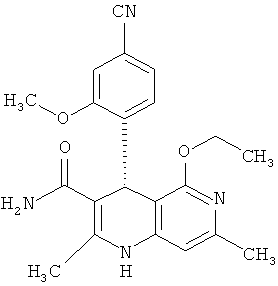 ,
,
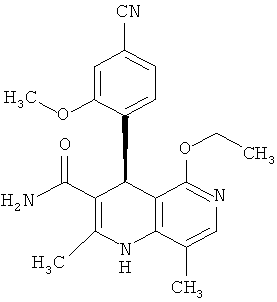 и
и 
а также их соли, сольваты и сольваты солей.
Указанные, в частности, определения остатков в имеющихся или предпочтительных их комбинациях произвольно заменяются определениями остатков других комбинаций независимо от имеющихся указанных комбинаций остатков.
Особенно предпочтительными являются комбинации двух или нескольких названных выше предпочтительных остатков.
Следующим предметом изобретения является способ получения соединений формулы (I) согласно изобретению, отличающийся тем, что соединение формулы (II)
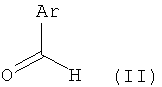 ,
,
в которой Ar имеет указанное выше значение,
в инертном растворителе, при необходимости, в присутствии кислоты, комбинации кислота/основание и/или дегидратирующего реагента с соединением формулы (III)
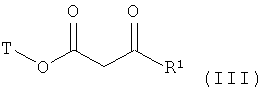 ,
,
в которой R1 имеет указанное выше значение и
Т означает аллил или 2-цианоэтил,
преобразуется в соединение формулы (IV)
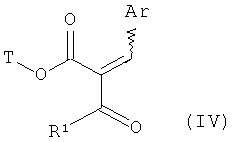 ,
,
в которой Ar, Т и R1 соответственно имеют указанные выше значения,
затем это соединение конденсируется в инертном растворителе с соединением формулы (V)
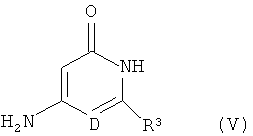 ,
,
в которой D и R3 имеют указанные выше значение,
в соединение формулы (VI)
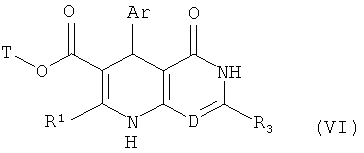 ,
,
в которой Ar, D, Т, R1 и R3 соответственно имеют указанные выше значения,
затем соединения формулы (VI) в инертном растворителе, при необходимости, в присутствии основания с соединением формулы (VII) или солью триалкилоксония формулы (VIII)
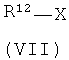
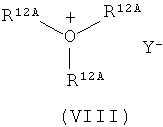
в которых
R12 означает (С1-С6)-алкил, который может быть замещен (С3-С7)-циклоалкилом или до трех раз замещен фтором,
R12A означает метил или этил,
Х означает исходную группу, как, например, галоген, мезилат, тозилат или трифлат
и
Y- означает не нуклеофильный анион, как, например, тетрафтороборат,
или в присутствии кислоты алкилируются триалкилортоформиатом формулы (IX)
 ,
,
в которой R12A имеет указанное выше значение,
в соединения формулы (Х-А)
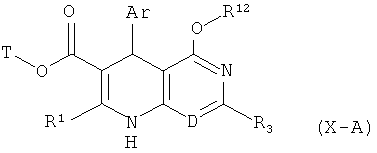 ,
,
в которой Ar, D, Т, R1, R3 и R12 соответственно имеют указанные выше значения,
или соединения формулы (VI) в инертном растворителе в присутствии основания с соединением формулы (XI)
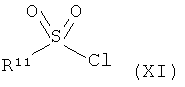 ,
,
в которой R11 имеет указанное выше значение,
преобразуются в соединения формулы (Х-В)
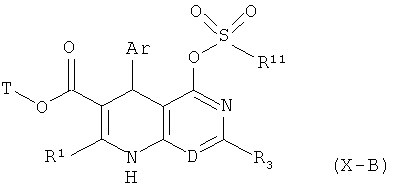 ,
,
в которой Ar, D, Т, R1, R3 и R11 соответственно имеют указанные выше значения,
после этого в соединениях формулы (Х-А) или (Х-В) известным специалисту способом отщепляется группа эфира Т в карбоновые кислоты формулы (XII)
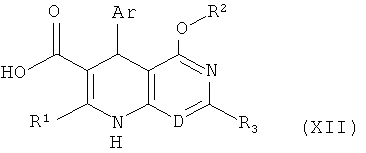 ,
,
в которой Ar, D, R1, R2 и R3 соответственно имеют указанные выше значения, затем 1,1'-карбонилдиимидазолом переводятся в имидозолиды формулы (XIII)
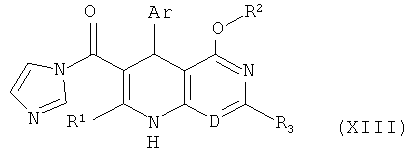 ,
,
в которой Ar, D, R1, R2 и R3 соответственно имеют указанные выше значения, и потом эти соединения в инертном растворителе, при необходимости, в присутствии основания преобразуются с аммиаком в амиды формулы (I) и, при необходимости, соединения формулы (I) известными специалисту методами разделяются на их энантиомеры и/или диастереомеры, и/или с помощью соответствующих растворителей (i) и/или оснований или кислот (ii) превращаются в их сольваты, соли и/или сольваты солей.
Последовательность процесса (II)+(III)→(IV) и (IV)+(V)→(VI) может также выполняться одним этапом как 3-компонентная реакция (II)+(III)+(V)→(VI), без выделения промежуточной стадии (IV).
Стадии процесса (II)+(III)→(IV) и (IV)+(V)→(VI) или (II)+(III)+(V)→(VI) в общем случае выполняются в инертном растворителе в интервале температур от +20°С до точки кипения растворителя при нормальном давлении.
В качестве инертного растворителя для этого пригодны, например, спирты, как метанол, этанол, н-пропанол, изопропанол, н-бутанол или трет-бутанол, галогенуглеводороды, как дихлорметан, трихлорметан, тетрахлорметан, трихлорэтан или 1,2-дихлорэтан, либо другие растворителе, как ацетонитрил, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, гексан, бензол, толуол, хлорбензол, пиридин или ледяная уксусная кислота. Предпочтительно проводить реакции обмена в дихлорметане, толуоле, этаноле или изопропаноле при температуре рефлюкса и при нормальном давлении.
Названные реакции могут, при необходимости, выполняться в присутствии кислоты, комбинации кислота/основание и/или обезвоживающего средства, как, например, молекулярное сито. В качестве кислот пригодны, например, уксусная кислота, трифторуксусная кислота, метансульфокислота или р-толуолсульфокислота; в качестве оснований особенно годится пиперидин или пиридин [по поводу синтеза 1,4-дигидропиридинов см. также D.M. Stout, A.I.Meyers, Chem. Rev. 1982, 82, 223-243; H.Meier et al., Liebigs Ann. Chem. 1977, 1888; H.Meier et al., ibid. 1977, 1895; H. Meier et al., ibid. 1976, 1762; F.Bossert et al., Angew. Chem. 1981, 93, 755].
Инертными растворителями для стадий процесса (VI)+(VII)→(Х-А), (VI)+(VIII)→(Х-А) и (VI)+(XI)→(Х-В) являются, например, простые эфиры, как диэтиловый эфир, метил-трет-бутиловый эфир, диоксан, тетрагидрофуран, гликольдиметиловый или диэтиленгликольдиметиловый эфир; углеводороды, как бензол, толуол, ксилол, гексан, циклогексан или нефтяные фракции; галогенуглеводороды, как дихлорметан, трихлорметан, тетрахлорметан, 1,2-дихлорэтан, трихлорэтан, тетрахлорэтан, трихлорэтилен, хлорбензол или хлортолуол; или другие растворители как N,N-диметилформамид (ДМФ), диметил-сульфоксид (ДМСО), N,N′-диметилпропиленкарбамид (ДМПК), N-метилпиролидон (NMP), пиридин или асетонитрил. Возможно также использование смесей названных растворителей. Предпочтительно в стадии процесса (VI)+(VII)→(Х-А) применяются тетрагидрофуран или диметилформамид, в стадии (VI)+(VIII)→(X-А) применяется дихлорметан и в стадии (VI)+(XI)→(Х-В) - пиридин.
Вариант процесса (VI)+(IX)→(Х-А) выполняется преимущественно со значительным превышением ортомуравьиного эфира в диметилформиаде или без добавления другого растворителя; в качестве реакционного катализатора пригодны, например, сильные неорганические кислоты, как серная кислота [см., например, I.I. Barabanov et al., Russ. Chem. Bl 47(11), 2256-2261 (1998)].
В качестве оснований для стадии процесса (VI)+(VII)→(Х-А) пригодны, в частности, карбонаты щелочных и щелочноземельных металлов, как карбонат лития, натрия, калия, кальция или цезия; гидриды щелочных металлов, как гидрид натрия или калия; амиды, как литий-, натрий- или калий-бис(триметилсилил)амид или литийдиизопропиламид; металлоорганические соединения, как бутиллитий или фениллитий, а также фосфазен-основания, как, например, P2-t-Bu или P4-t-Bu [так называемые "основания Швезингера", см. R. Schwesinger, H. Schlemper, Angew. Chem. Int. Ed. Engl. 26, 1167 (1987); Т. Pietzonka, D. Seebach, Chem. Ber. 124, 1837 (1991)]. Предпочтительно применение гидрида натрия или фосфазен-основания P4-t-Bu.
В качестве оснований для стадии процесса (VI)+(XI)→(Х-В) пригодны, в частности, карбонаты щелочных и щелочноземельных металлов, как карбонат лития, натрия, калия, кальция или цезия; гидриды щелочных металлов, как гидрид натрия или калия; металлоорганические соединения, как бутиллитий или фениллитий, или органические амины, как триэтиламин, N-метилморфолин, N-метилпиперидин, N,N-диизопропилэтиламин, пиридин, 1,5-диазабицикло[4.3.0]нон-5-ен (DBN), 1,8-диазабицикло[5.4.0]ундек-7-ен (DBU) или 1,4-диазабицикло[2.2.2]-октан (DABCO®). Предпочтительно применение пиридина, который одновременно служит растворителем.
Стадия процесса (VI)+(VIII)→(Х-А) выполняется в общем случае без добавления основания.
Реакции обмена (VI)+(VII)→(Х-А), (VI)+(VIII)→(Х-А) и (VI)+(XI)→(Х-В) осуществляются в общем случае в интервале температур от -20°С до +100°С, предпочтительно при температурах от 0°С до +60°С; вариант процесса (VI)+(IX)→(Х-А) выполняется, как правило, при температуре от +100°С до +150°С. Реакции могут проводиться при нормальном, повышенном или пониженном давлении (например, от 0,5 до 5 бар); в общем случае работают при нормальном давлении.
Отщепление аллильного или 2-цианоэтилового эфира в стадии процесса (X-А) или (Х-В)→(XII) происходит известными методами, опубликованными в литературе. В случае отщепления 2-цианоэтилового эфира предпочтительно использовать водный раствор гидроксида щелочного металла, как, например, раствор едкого натра или калия. Реакция проводится, в общем случае, с применением смешиваемого с водой инертного со-растворителя, как, например, тетрагидрофуран, диоксан или 1,2-диметоксиэтан, в интервале температур от 0°С до +40°С. В случае аллильного эфира отщепление осуществляется с помощью катализатора Уилкинсона [трис(трифенилфосфин)родий(I)хлорид] в смеси из воды, спирта и уксусной кислоты при температуре от +50°С до +100°С [см., например, Moseley, J.D., Tetrahedron Lett. 46, 3179-3181 (2005)].
В качестве инертного растворителя пригодны, например, простые эфиры, как простой диэтиловый эфир, метил-трет-бутиловый эфир, диоксан, тетрагидрофуран, гликольдиметиловый или диэтиленгликольдиметиловый эфир; галогенуглеводороды, как дихлорметан, трихлорметан, 1,2-дихлорэтан, трихлорэтан, тетрахлорэтан, хлорбензол или хлортолуол, или другие растворители, как N,N-диметилформамид (ДМФ), диметилсульфоксид (ДМСО), N,N′-диметилпропиленкарбамид (ДМПК), N-метилпиролидон (NMH), ацетон, ацетонитрил или этилацетат. Возможно также применение смесей названных растворителей. Предпочтительно использование тетрагидрофурана, диметилформамида или этилацетата. Как правило, реакция выполняется при температуре от 0°С до +40°С.
Для стадии процесса (XIII)→(I) в качестве инертных растворителей используются, например, спирты, как метанол, этанол, н-пропанол, изопропанол, н-бутанол или трет-бутанол; простые эфиры, как диэтиловый эфир, метил-трет-бутиловый эфир, диоксан, тетрагидрофуран, гликольдиметиловый или диэтиленгликольдиметиловый эфир, или другие растворители, как N,N-диметилформамид (ДМФ), диметилсульфоксид (ДМСО), N,N′-диметил-пропиленкарбамид (ДМПК), N-метилпиролидон (NMП), ацетонитрил или даже вода. Возможно также применение смесей этих растворителей. Предпочтительно использование тетрагидрофурана или диметилформамида.
Источниками аммиака для этих реакций обмена могут быть растворы газообразного аммиака в одном из названных выше растворителей, в частности в воде. Реакция преимущественно выполняется в присутствии третичного амина в качестве вспомогательного основания, как, например, триэтиламин, N-метилморфолин, N-метилпиперидин, N,N-диизопропилэтиламин или 4-N,N-диметиламинопиридин. Реакция обмена проходит, в общем случае, при температуре от +20°С до +120°С, предпочтительно при температуре от +50°С до +100°С.
Соединения формулы (II) имеются в продаже, известны из литературы и могут быть получены по аналогии с описанными в литературе способами (см. приведенные далее схемы реакций 1-7). Соединения формул (III), (VII), (VIII), (IX) и (XI) имеются в продаже, известны из литературы или могут быть получены по аналогии с описанными в литературе способами.
Соединения формулы (V) описаны в литературе или могут быть получены по аналогии с описанными в литературе способами [см., например, Т.Searls, L.W.McLaughlin, Tetrahedron 55, 11985-11996 (1999); D. McNamara, P.D.Cook, J. Med. Chem. 30, 340-347 (1987); S.Nesnow, С.Heidelberger, J.Heterocycl. Chem. 12,941-944 (1975); N.C.Hung, E.Bisagni, Synthesis 1984. 765-766; Z.Földi et al., Chem. Ber. 75(7), 755-763 (1942); G.W.Kenner et al., J. Chem. Soc., 388 (1943)].
При необходимости отделение энантиомеров и/или диастереомеров может осуществляться уже на промежуточном этапе (VI), (Х-А), (Х-В) или (XII), которые далее могут обособленно подвергаться следующим реакциям обмена.
Получение соединений согласно изобретению может быть наглядно показано следующими схемами синтеза:
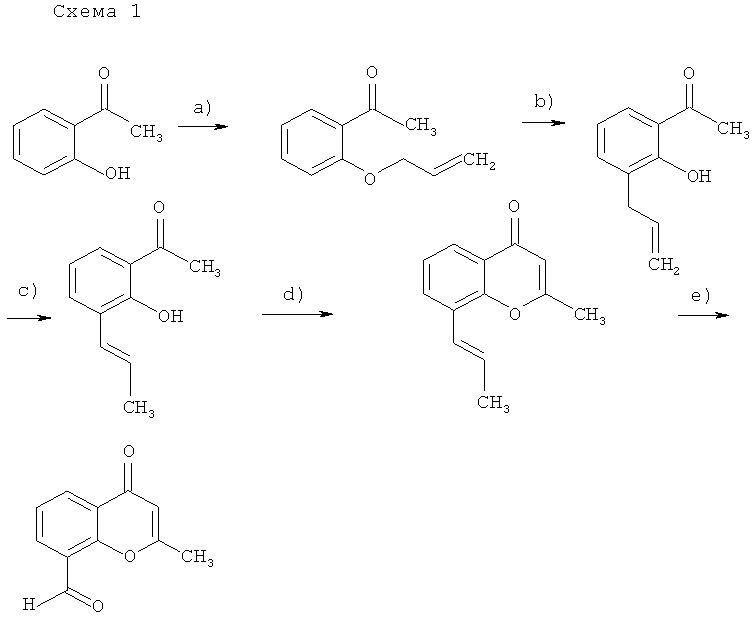
[а): аллилбромид, карбонат калия, кат. йодид калия, ацетон, рефлюкс; b): 230°С, 4 часа; с): бис(бензонитрил)дихлорпалладий(II), толуол, 120°С, 16 часов; d): ацетилхлорид, гидрид натрия, ТГФ, 10-25°С, 16 часов; е): 1. озон, дихлорметан, -60°С, 30 минут; 2. диметилсульфид].
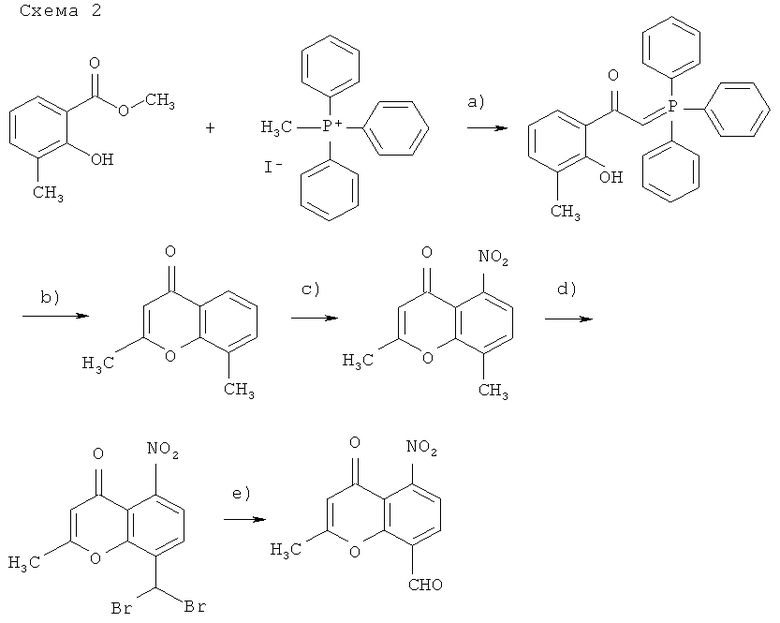
[а): n-бутиллитий, ТГФ, 60°С, 3 часа; b): уксусный ангидрид, пиридин, рефлюкс, 6 часов; с): конц. H2SO4, HNO3, 0°С, 1 час; d): N-бромсукцинимид, AIBN, четыреххлористый углерод, рефлюкс; е): N-метилморфолин-N-оксид, ацетонитрил, рефлюкс].
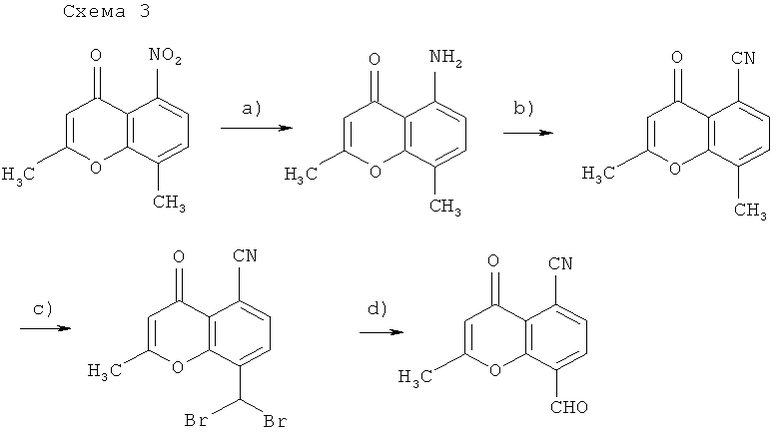
[а): цинк(II)хлорид-дигидрат, этилацетат, 70°С; b): 1. нитрит натрия, серная кислота, 0°С, 1.5 часа; 2. цианид меди(I), цианид натрия, вода/этилацетат, 0°С, 45 минут; с): N-бромсукцинимид, AIBN, четыреххлористый водород, рефлюкс; d): N-метилморфолин-N-оксид, ацетонитрил, рефлюкс].
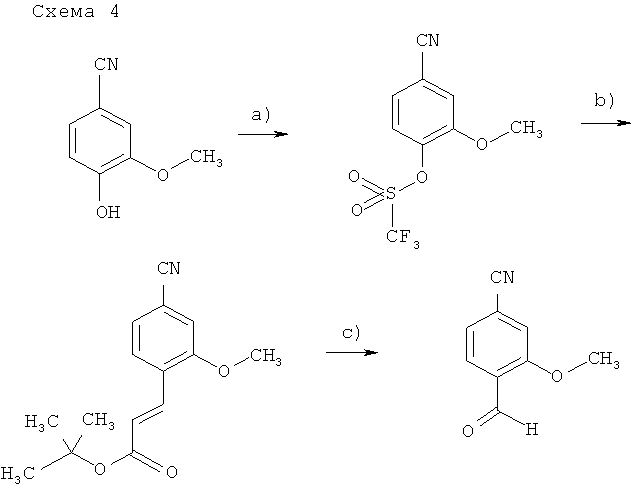
[а): ангидрид трифторметансульфокислоты, пиридин, 0°С → комн. темп., 30 мин; b): акриловой кислоты трет-бутиловый эфир, бис(трифенилфосфин)дихлорпалладий(II), ДМФ, 120°С, 24 часа; с): кат. четырехоксид осьмия, кат. бензилтриэтиламмония хлорид, периодат натрия, ТГФ/вода, 20-25°С, 2 часа].
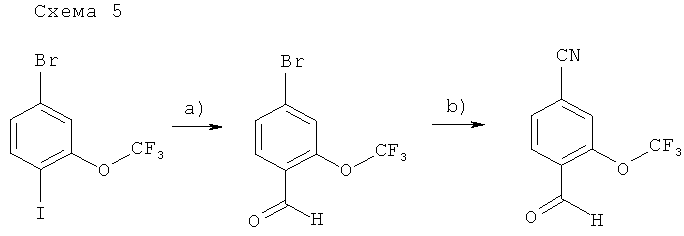
[а): н-бутиллитий, ТГФ, -78°С, затем N-формилморфолин; b): цианид цинка, тетракис(трифенилфосфин)палладий(0), ДМФ, СВЧ 250°С / 5 минут].

[а): N,N-диметилформамид-диметилацетал, ДМФ, 140-180°С; b): периодат натрия, ТГФ/вода].
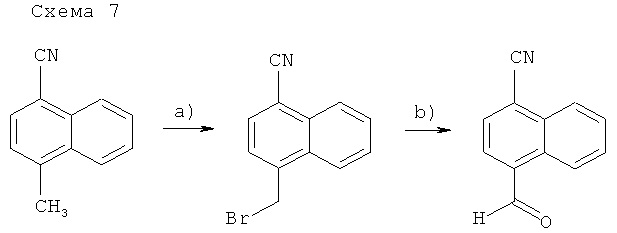
[а): N-бромсукцинимид, ABBN, тетрахлорметан, рефлюкс; b): N-метилморфолин-N-оксид, ацетонитрил, 3Å-молекулярное сито].
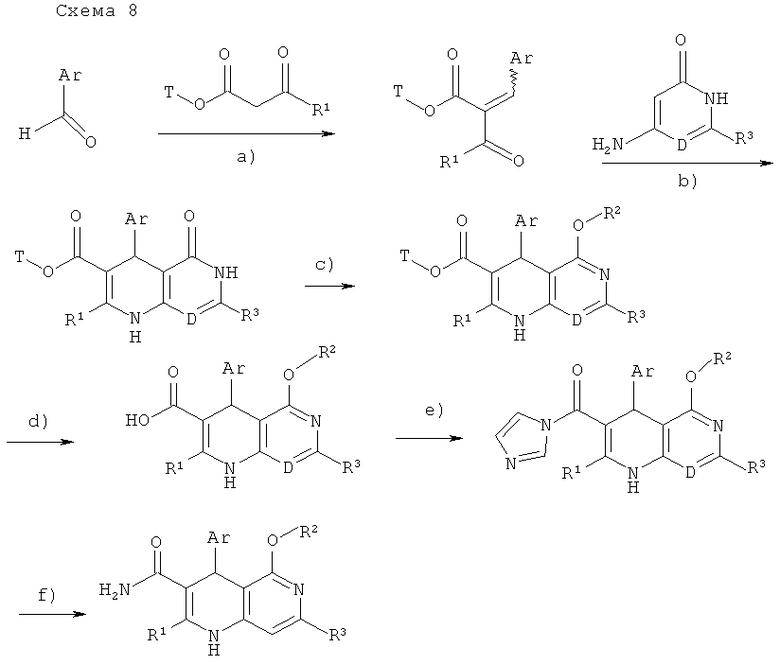
[а): кат. пиперидин/уксусная кислота, дихлорметан, рефлюкс, 24 часа; b): изопропанол, рефлюкс, 12-72 часа; с): алкилтрифлат или -йодид, основание, ТГФ или ДМФ, комн. темп.; или триалкилоксонийтетрафтороборат, дихлорметан, комн. темп.; или триалкилортоформиат, кат.серная кислота, 100-130°С; или R11-SO2-Cl, пиридин, комн. темп.; d): Т = 2-цианоэтил: водный раствор NaOH, ДМЭ/вода, комн. темп.; Т=аллил: (PPh3)3RhCl, вода/этанол/уксусная кислота, 75°С; е): 1,1′-карбонилдиимидазол, этиловый эфир уксусной кислоты, комн. темп., 12 часов; f): водный раствор аммиака, ДМФ, 50-100°С, 0.5-12 часов].
Соединения согласно изобретению действуют как антагонисты минералкортикоидного рецептора и демонстрируют не предполагаемый, широкий спектр фармакологического действия. Они пригодны для применения в качестве лекарственных средств для лечения и/или профилактики болезней у людей и животных.
Соединения согласно изобретению пригодны для профилактики и/или лечения различных заболеваний и осложнений, в частности болезней, вызванных либо повышением концентрации альдостерона в плазме, либо изменением соотношения концентраций в плазме альдостерона и ренина, или сопутствующих этим изменениям болезней. К примеру, можно назвать: идиопатический первичный гиперальдостеронизм, гиперальдостеронизм при гиперплазии надпочечника, аденомах и/или карциноме надпочечника, гиперальдостеронизм при сердечной недостаточности, а также (относительный) гиперальдостеронизм при эссенциальной гипертонии.
Кроме того, соединения согласно изобретению, благодаря их механизму действия, пригодны для профилактики внезапной сердечной смерти у пациентов, подверженных повышенному риску скончаться от разрыва сердца. В частности, это относится к пациентам, страдающим, например, одним из следующих заболеваний: первичная и вторичная гипертония; болезнь сердца, обостренная повышенным давлением, с застойной сердечной недостаточностью или без нее; не поддающаяся лечению гипертония; острая и хроническая сердечная недостаточность; коронарная болезнь сердца; стабильная и нестабильная стенокардия; ишемия миокарда, инфаркт миокарда; дилатационные кардиомиопатии; врожденные первичные кардиомиопатии, как, например, синдром Бругада, кардиомиопатии, вызванные болезнью Шагаса; шок, артериосклероз, предсердная и желудочковая аритмия, транзиторные и ишемические атаки, кровоизлияние в мозг, воспалительные кардиоваскулярные заболевания, заболевания периферийных и сердечных сосудов, нарушения периферийного кровоснабжения, облитерирующий эндартериит, как перемежающаяся хромота, бессимптомная дисфункция левого желудочка, миокардит, гипертрофические изменения сердца, легочная гипертония, спазмы коронарных и периферических артерий, тромбозы, тромбоэмболические заболевания и васкулит.
Далее, соединения согласно изобретению могут применяться для профилактики и/или лечения образования отеков, как, например, легочного отека, почечного отека или отека, обусловленного сердечной недостаточностью, для профилактики и/или лечения рестенозов, например, после тромболитической терапии, чрескожной транслюминальной ангиопластики (РТА) и коронарной ангиопластики (РТСА), трансплантации сердца, а также после шунтирования.
Кроме того, соединения согласно изобретению пригодны для использования в качестве сберегающего калий мочегонного средства и при нарушениях электролитного баланса, как, например, гиперкальциемия, гипернатриемия или гипокалиемия.
Соединения согласно изобретению пригодны также для лечения таких заболеваний почек, как острая и хроническая почечная недостаточность, болезни почек с гипертонией, артериосклеротического нефрита (хронического и интерстициального), нефросклероза, хронической почечной недостаточности и кистозных болезней почек, для предотвращения поражения почек, которые могут быть вызваны, например, блокаторами иммунного ответа, как циклоспорин А при трансплантации органов.
Кроме того, соединения согласно изобретению могут применяться для профилактики и/или лечения сахарного диабета и вызванных диабетом заболеваний, как, например, невропатия и нефропатия.
Далее, соединения согласно изобретению могут применяться для профилактики и/или лечения микроальбуминурии, вызванной, например, сахарным диабетом или артериальной гипертонией, а также для профилактики и/или лечения протеинурии.
Соединения согласно изобретению пригодны также для профилактики и/или лечения болезней, сопровождающихся либо повышением концентрации глюкокортикоидов в плазме либо локальным повышением концентрации глюкокортикоидов в ткани (например, сердца). Например, можно назвать: функциональные расстройства надпочечников, которые приводят к гиперпродукции глюкокортикоидов (синдром Кушинга), опухоли коры надпочечника с результирующей гиперпродукцией глюкокортикоидов и опухоли гипофиза, который автономно продуцирует АКТГ (адренокортикотропный гормон) и этим приводит к гиперплазии надпочечников с последующей болезнью Кушинга.
Кроме того, соединения согласно изобретению могут использоваться для профилактики и/или лечения общего ожирения, метаболического синдрома и обструктивной остановки дыхания во время сна.
Далее, соединения согласно изобретению могут применяться для профилактики и/или лечения воспалительных заболеваний, вызванных, например, вирусами, спирохетами, грибками, бактериями или микобактериями, а также воспалительных заболеваний неизвестной этиологии, как полиартрит, системная красная волчанка, пери- и полиартериит, дерматомикоз, склеродермия и саркоидоз.
Дальше, соединения согласно изобретению могут применяться для лечения заболеваний центральной нервной системы, как депрессии, приступы беспричинного страха и хронические боли, в частности мигрени, а также нейродегенеративные заболевания, как болезнь Альцгеймера и синдром Паркинсона.
Соединения согласно изобретению пригодны также для профилактики и/или лечения сосудистых повреждений, например, после таких вмешательств, как чрескожная транслюминальная коронарная ангиопластика (РТСА), имплантация стентов, коронарная ангиоскопия, закупорка или рестеноз после операции шунтирования и эндотелиальной дисфункции, при болезни Рейно, при облитерирующем тромбангиите (синдром Бюргера) и при синдроме ушного шума.
Следующим предметом настоящего изобретения является применение соединений согласно изобретению для лечения и/или предупреждения болезней, в частности названных выше болезней.
Следующим предметом настоящего изобретения является применение соединений согласно изобретению для приготовления лекарственных средств для лечения и/или предупреждения болезней, в частности названных выше болезней.
Следующим предметом настоящего изобретения является способ лечения и/или предупреждения болезней, в частности названных выше болезней, с применением действующего количества, по крайней мере, одного из соединений согласно изобретению.
Соединения согласно изобретению могут применяться самостоятельно или, при необходимости, в комбинации с другими биологически активными веществами. Следующим предметом настоящего изобретения являются лекарственные средства, содержащие, по крайней мере, одно из соединений согласно изобретению и одно или несколько других биологически активных веществ, в частности, для лечения и/или профилактики названных выше болезней. Подходящими для комбинирования биологически активными веществами могут быть названы, например и преимущественно, следующие:
- биологически активные вещества, понижающее кровяное давление, например и преимущественно, из группы антагонистов кальция, антагонистов ангиотензина AII, ингибиторов АСЕ, антагонистов эндотелина, ингибиторов ренина, блокаторов альфа-рецерторов и ингибиторов бета-рецепторов;
- мочегонные средства, в частности петлевые диуретики, а также тиазиды и подобные им диуретики;
- антитромботические средства, например и преимущественно, из группы ингибиторов агрегации тромбоцитов, антикоагулянтов или профибринолитических субстанций;
- биологически активные вещества, изменяющие жировой обмен, например и преимущественно, из группы агонистов тироидных рецепторов, ингибиторов синтеза холестерина, как, например и преимущественно, ингибиторов синтеза НМG-СоА-редуктазы или сквалена, ингибиторов АСАТ, ингибиторов СЕТР, ингибиторов МТР, агонистов PPAR-альфа, PPAR-гамма и/или PPAR-дельта рецепторов, ингибиторов абсорбции холестерина, ингибиторов липазы, полимерных адсорбентов желчной кислоты, ингибиторов реабсорбции желчной кислоты и антагонистов липопротеина(а);
- органические нитраты и NO-доноры, как, например, нитропруссид натрия, нитроглицерин, мононитрат изосорбида, динитрат изосорбида, молсидомин или SIN-1, а также ингалятивный NO;
- позитивно действующие инотропные соединения, как, например, сердечный гликозид (дигоксин), бета-адренергические и допаминергические агонисты, как изопротеренол, адреналин, норадреналин, допамин и добутамин;
- соединения, которые ингибируют расщепление циклического гуанозинмонофосфата (цГМФ) и/или циклического аденозинмонофосфата (цАМФ), как, например, ингибиторы фосфодиестеразы (ФДЕ) 1, 2, 3, 4 и/или 5, в частности ингибиторы ФДЕ-5, как силденафил, варденафил и тадалафил, а также ингибиторы ФДЕ-3, как амринон и милринон;
- натрийуретические пептиды, как, например, "атриальный натрийуретический пептид" (ANP, анаритиды), "натрийуретический пептид В-типа" или "натрийуретический пептид головного мозга" (BNP, несиритиды), "натрийуретический пептид с-типа" (CNP), а также уродилатин;
- синсебилизаторы кальция, как например и преимущественно, левосимендан;
- NO-независимые, но гем-зависимые стимуляторы гуанилатциклазы, как соединения, описанные, в частности, в патентах WO 00/06568, WO 00/06569, WO 02/42301 и WO 03/095451;
- NO- и гем-независимые активаторы гуанилатциклазы, как соединения, описанные, в частности, в патентах WO 01/19355, WO 01/19776, WO 01/19778, WO 01/19780, WO 02/070462 и WO 02/070510;
- ингибиторы человеческой нейтрофильной эластазы (ЧНЭ), как, например, сивелестат или DX-890 (релтран);
- соединения, ингибирующие каскады трансдукционных сигналов, как, например, ингибиторы тирозинкиназы, в частности, сорафениб, иматиниб, гефитиниб и эрлотиниб; и/или
- соединения, влияющие на энергию метаболизма сердца, как например и преимущественно, этомоксир, дихлорацетат, ранолазин или триметазидин.
В предпочтительной форме исполнения изобретения соединения согласно изобретению отпускаются в комбинации с каким-либо диуретиком, как например и преимущественно, фуроземид, буметанид, торземид, бендрофлуметиазид, хлортиазид, гидрохлортиазид, гидрофлумстиазид, метиклотиазид, политиазид, трихлорметиазид, хлорталидон, индапамид, метолазон, квинетазон, ацетазоламид, дихлорфенамид, метазоламид, глицерин, изосорбид, маннитол, амилорид или триамтерсн.
Под средствами, понижающими кровяное давление, понимаются преимущественно соединения из группы антагонистов кальция, антагонистов ангиотензина AII, ингибиторов энзима АСЕ, антагонистов эндотелина, ингибиторов ренина, блокаторов альфа-рецепторов, блокаторов бета-рецепторов, ингибиторов ро-киназы, а также мочегонные средства.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо антагонистом кальция, как например и преимущественно, нифедипин, амлодипин, верапамил или дилтиазем.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо антагонистом ангиотензина AII, как например и преимущественно, лосартан, кандесартан, валсартан, телмисартан или эмбусартан.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо ингибитором АСЕ, как например и преимущественно, эналаприл, каптоприл, лизиноприл, рамиприл, делаприл, фозиноприл, квиноприл, периндоприл или трандоприл.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо антагонистом эндотелина, как например и преимущественно, бозентан, дарузентан, ампризентан или ситаксзентан.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо ингибитором ренина, как например и преимущественно, алискирен, SPP-600, SPP-635, SPP-676, SPP-800 или SPP-1148.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо ингибитором альфа1-рецепторов, как например и преимущественно, празосин.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо блокатором бета-рецепторов, как например и преимущественно, пропранолол, атенолол, тимолол, пиндолол, альпренолол, окспренолол, пенбутолол, бупранолол, метипранолол, надолол, мепиндолол, каразалол, соталол, метопролол, бетаксолол, целипролол, бисопролол, картеолол, эсмолол, лабеталол, карведилол, адапролол, ландиолол, небиволол, эпанолол или бусиндолол.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо ингибитором ро-киназы, как например и преимущественно, фазудил, Y-27632, SLx-2119, BF-66851, BF-66852, BF-66853, KI-23095 или ВА-1049.
Под антитромботическими средствами понимаются преимуществественно соединения из группы ингибиторов агрегации тромбоцитов, антикоагулянтов или профибринолитических субстанций.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо ингибитором агрегации тромбоцитов, как например и преимущественно, аспирин, клопидогрель, тиклопидин или дипиридамол.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо ингибитором тромбина, как например и преимущественно, ксимелагатран, мелагатран, бивалирудин или клексан.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо GPIIb/IIIa-антагонистом, как например и преимущественно, тирофибан или абсиксимаб.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо ингибитором фактора Ха, как например и преимущественно, ривароксабан (BAY 59-7939), DU-176b, апиксабан, отамиксабан, фидексабан, разаксабан, фондапаринукс, идапаринукс, PMD-3112, YM-150, КFА-1982, EMD-503982, MCM-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR-126512 или SSR-128428.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с гепарином или производным гепарина с малым молекулярным весом (LMW).
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо антагонистом витамина К, как например и преимущественно, кумарин.
Под средствами, изменяющими жировой обмен, понимаются преимущественно соединения из группы ингибиторов СЕТР, агонистов тироидных рецепторов, ингибиторов синтеза холестерина, как ингибиторы HMG-CoA-редуктазы или синтеза сквалена, ингибиторов АСАТ, ингибиторов МТР, агонистов PPAR-альфа, PPAR-гамма и/или PPAR-дельта рецепторов, ингибиторов абсорбции холестерина, полимерных адсорбентов желчной кислоты, ингибиторов реабсорбции желчной кислоты, ингибиторов липазы, а также антагонистов липопротеина(а).
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо ингибитором СЕТР, как например и преимущественно, торсетрапиб (СР-529 414), JJT-705, BAY 60-5521, BAY 78-7499 или СЕТР-вакцина (авант).
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо агонистом тироидных рецепторов, как например и преимущественно, D-тироксин, 3,5,3'-трийодотиронин (Т3), CGS 23425 или акситиром (CGS 26214).
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо ингибитором HMG-CoA-редуктазы из класса статинов, как например и преимущественно, ловастатин, симвастатин, правастатин, флувастатин, аторвастатин, розувастатин, церивастатин или питавастатин.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо ингибитором синтеза сквалена, как например и преимущественно, BMS-188494 или ТАК-475.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо ингибитором АСАТ, как например и преимущественно, авазимиб, мелинамид, пактимиб, эфлусимиб или SMP-797.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо ингибитором МТР, как например и преимущественно, имплитапид, BMS-201038, R-103757 или JTT-130.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо агонистом PPAR-гамма рецепторов, как например и преимущественно, пиоглитазон или росиглитазон.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо агонистом PPAR-дельта рецепторов, как например и преимущественно, GW-501516 или BAY 68-5042.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо ингибитором абсорбции холестерина, как например и преимущественно, эзетимиб, тиквесид или памаквесид.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо ингибитором липазы, как например и преимущественно, орлистат.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо полимерным адсорбентом желчной кислоты, как например и преимущественно, холестирамин, колестипол, колесольвам, холеста-гель или колестимид.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо ингибитором реабсорбции желчной кислоты, как например и преимущественно, ингибиторы ASBT (=IBAT), как AZD-7806, S-8921, AK-105, BARI-1741, SC-435 или SC-635.
В предпочтительной форме исполнения изобретения соединения согласно изобретению применяются в комбинации с каким-либо антагонистом липопротеина (а), как например и преимущественно, гемкабен кальций (CI-1027) или никотиновая кислота.
Следующим предметом данного изобретения являются лекарственные средства, которые содержат, по крайней мере, одно соединение согласно изобретению, как правило, вместе с одним или несколькими инертными, нетоксичными, приемлемыми в фармацевтике вспомогательными веществами, а также их применение для названных выше целей.
Соединения согласно изобретению могут действовать систематически и/или локально. Для этой цели они могут приниматься подходящим способом, например, орально, парентерально, пульмонально, назально, сублингвально, лингвально, буккально, ректально, дермально, трансдермально, конъюнктивально, через уши или как имплантант или стент.
Для этих способов приема соединения согласно изобретению могут отпускаться в подходящих формах.
Для орального приема пригодны действующие формы применения, отдающие быстро и/или в модифицированном виде соединения согласно изобретению, которые соединения согласно изобретению содержат в кристаллическом и/или аморфном и/или растворенном виде, как например, таблетки (непокрытые или покрытые, например, с устойчивыми к действию желудочного сока или растворяющимися с задержкой или нерастворимыми оболочками, которые контролируют высвобождение соединения согласно изобретению), быстро распадающиеся в полости рта таблетки или облатки в пленке, лиофилизаты в пленке, капсулы (например, твердые или мягкие желатиновые капсулы), драже, гранулят, пеллеты, порошки, эмульсии, суспензии, аэрозоли или растворы.
Парентеральный прием может происходить без этапа всасывания (например, внутривенное, внутриартериальное, внутрисердечное, интраспинальное или интралюмбальное введение) или при включении процесса всасывания (например, внутримышечное, подкожное, внутрикожное, чрескожное или внутрибрюшинное введение). Для парентерального приема в качестве форм применения приемлемы препараты для инъекции и вливания в виде растворов, суспензий, эмульсий, лиофилизат или стерильных порошков.
Для прочих способов приема пригодны, например, лекарственные формы для ингаляции (в том числе порошковые ингаляторы, распылители), капли, растворы или спреи в нос, принимаемые на язык, под язык или трансбуккально таблетки, облатки или капсулы, свечи, ушные или глазные препараты, вагинальные капсулы, водные суспензии (лосьоны, болтушки), жирорастворимые суспензии, мази, кремы, трансдермальные терапевтические системы (например, пластырь), молоко, пасты, пенки, присыпки, имплантанты или стенты.
Предпочтителен оральный или парентеральный прием, в особенности оральный и внутривенный приемы.
Соединения согласно изобретению могут переводиться в приведенные формы применения. Это может осуществляться известным изготовителю способом путем смешивания с инертными, нетоксичными, приемлемыми в фармацевтике вспомогательными веществами. К таким вспомогательным веществам относятся, в том числе, наполнители (например, микрокристаллическая целлюлоза, лактоза, маннитол), растворители (например, жидкий полиэтиленгликоль), эмульгаторы и диспергаторы или смачивающие средства (например, додецилсульфат натрия, полиоксисорбитанолеат), связывающие средства (например, поливинилпирролидон), синтетические и природные полимеры (например, альбумин), стабилизаторы (например, антиоксиданты как аскорбиновая кислота), красители (например, неорганические пигменты как оксид железа) и вещества, улучшающие вкус и запах лекарства.
В общем случае при парентеральном применении для достижения эффективного результата оказалось полезным отпускать лекарство весом 0,001-1 мг/кг, преимущественно 0,01-0,5 мг/кг массы тела. При оральном применении доза составляет 0,01-100 мг/кг, преимущественно 0,01-20 мг/кг и особенно предпочтительно 0,1-10 мг/кг массы тела.
Но в некоторых случаях возможны отклонения от названных количеств, а именно дозировка может изменяться в зависимости от массы тела, пути приема, индивидуальной переносимости биологически активного вещества, способа приготовления и времени и интервалов приема. Так, в некоторых случаях может быть достаточна доза меньше названного минимального количества, в то время как в других случаях должна быть превышена названная верхняя граница. В случае приема большего количества рекомендуется разделить лекарство на несколько разовых доз для приема в течение дня.
Следующие примеры исполнения объясняют изобретение. Изобретение не ограничивается примерами.
Приведенные процентные величины в следующих тестах и примерах являются массовыми процентами, части - массовыми долями, если не указано другое значение. Соотношения растворителей, разбавителей и данные концентрации растворов двух жидкостей соответственно относятся к объемам.
А. Примеры
LC-MS- и GC-MS-методы:
Метол 1 (LC-MS):
Тип прибора MS: Micromass ZQ; тип прибора HPLC: Waters Alliance 2795; колонка: Phenomcnex Synergi 2 мк Hydro-RP Mercury 20 мм×4 мм; элюент А: 1 л воды + 0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 90% А → 2.5 мин 30% А → 3.0 мин 5% А → 4.5 мин 5% А; поток: 0.0 мин 1 мл/мин → 2.5 мин/3.0 мин/4.5 мин 2 мл/мин; печь: 50°С; УФ-индикация: 210 нм.
Метод 2 (LC-MS):
Инструмент: Micromass Quattro LCZ с HPLC Agilent серии 1100; колонка:
Phenomenex Synergi 2µ Hydro-RP Mercury 20 мм × 4 мм; элюент А: 1 л воды + 0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 90% А → 2.5 мин 30% А → 3.0 мин 5% А→4.5 мин 5% А; поток: 0.0 мин 1 мл/мин → 2.5 мин/3.0 мин/4.5 мин 2 мл/мин; печь: 50°С; УФ-индикация: 208-400 нм.
Метод 3 (LC-MS):
Тип прибора MS: Micromass ZQ; тип прибора HPLC: HP 1100 Series; UV DAD; колонка: Phenomenex Gemini 3 мк 30 мм × 3.00 мм; элюент А: 1 л воды + 0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила +0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 90% А → 2.5 мин 30% А → 3.0 мин 5% А → 4.5 мин 5% А; поток: 0.0 мин 1 мл/мин → 2.5 мин/3.0 мин/4.5 мин 2 мл/мин; печь: 50°С; УФ-индикация: 210 нм.
Метод 4 (LC-MS):
Инструмент: Micromass Platform LCZ с HPLC Agilent серии 1100; колонка: Thermo Hypersil GOLD 3 мк 20 мм × 4 мм; элюент А: 1 л воды +0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 100% А → 0.2 мин 100% А → 2.9 мин 30% А → 3.1 мин 10% А → 5.5 мин 10% А; печь: 50°С; поток: 0.8 мл/мин; УФ-индикация: 210 нм.
Метод 5 (GC-MS):
Инструмент: Micromass GCT, GC 6890; колонка: Restek RTX-35MS, 30 м × 250 мкм × 0.25 мкм; постоянный поток с гелием: 0.88 мл/мин; печь: 60°С; впуск: 250°С; градиент: 60°С (0.30 минут выдержать), 50°С/мин → 120°С, 16°С/мин → 250°С, 30°С/мин → 300°С (1.7 минут выдержать).
Метод 6 (LC-MS):
Тип прибора MS: Waters ZQ; тип прибора HPLC: Waters Alliance 2795; колонка: Phenomenex Onyx Monolithic С18, 100 мм × 3 мм; элюент А: 1 л воды + 0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 90% А → 2 мин 65% А → 4.5 мин 5% А → 6 мин 5% А; поток: 2 мл/мин; печь: 40°С; УФ-индикация: 210 нм.
Метод 7 (LC-MS):
Инструмент: Micromass Quattro LCZ с HPLC Agilent серии 1100; колонка: Phenomenex Onyx Monolithic C18, 100 мм × 3 мм; элюент А: 1 л воды + 0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 90% А → 2 мин 65% А → 4.5 мин 5% А → 6 мин 5% А; поток: 2 мл/мин; печь: 40°С; УФ-индикация: 208-400 нм.
Метод 8 (LC-MS):
Тип прибора MS: Micromass ZQ; тип прибора HPLC: Waters Alliance 2795; колонка: Phenomenex Synergi 2.5 мк MAX-RP 100A Mercury 20 мм × 4 мм; элюент А: 1 л воды + 0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила +0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 90% А → 0.1 мин 90% А → 3.0 мин 5% А → 4.0 мин 5% А → 4.01 мин 90% А; поток: 2 мл/мин; печь: 50°С; УФ-индикация: 210 нм.
Метод 9 (LC-MS):
Инструмент: Micromass Quattro Premier с Waters UPLC Acquity; колонка: Thermo Hypersil GOLD 1.9 мк 50 мм × 1 мм; элюент А: 1 л воды +0.5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0.5 мл 50%-ной муравьиной кислоты; градиент: 0.0 мин 90% А → 0.1 мин 90% А → 1.5 мин 10% А → 2.2 мин 10% А; поток: 0.33 мл/мин; печь: 50°С; УФ-индикация: 210 нм.
Исходные и промежуточные соединения:
Пример 1А
1-[2-(Аллилокси)фенил]этанон
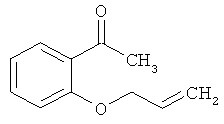
542 г (3.9 моль) 2-гидроксиацетофенона с 592 г (4.9 моль) аллилбромида, 1000 г (7.2 моль) карбоната калия и 13.2 г (79 ммоль) йодида калия в 2.4 литрах ацетона медленно нагреваются в течение 24 часов до рефлюкса. После охлаждения до комнатной температуры смесь фильтруется и растворитель удаляется в вакууме. Осадок растворяется в толуоле и промывается 10%-ным раствором едкого натра и водой. После сгущения получается 689 г (98% теор. возм.) выведенного в заголовке соединения.
1Н-ЯМР (300 МГц, CDCl3): δ=2.68 (s, 3Н), 4.68 (dd, 2H), 5.89 (dd, 2H), 6.09 (m, 1H), 6.99 (dd, 2H), 7.44 (m, 1H), 7.71 (d, 1H).
Пример 2А
1-(3-Аллил-2-гидроксифенил)этанон
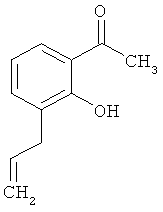
160 г 0.9 моль) 1-[2-(аллилокси)фенил]этанона перемешиваются в металлической бане при 230-240°С в течение 4 часов. После охлаждения до комнатной температуры продукт перегоняется в пленочном выпарном аппарате при 140°С и 0,4 барах. Получается 155 г (97% теор. возм.) выведенного в заголовке соединения.
1Н-ЯМР (300 MГц, CDCl3): δ=2.68 (s, 3Н), 3.44 (d, 2H), 5.09 (m, 2H), 6.01 (m, 1H), 6.85 (t, 1H), 7.38 (dd, 1H), 7.62 (dd, 1H), 12.61 (s, 1H).
Пример 3А
1-{2-Гидрокси-3-[(1Е)-проп-1-ен-1-ил]фенил}этанон
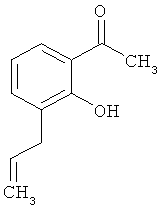
40 г (227 ммоль) 1-(3-аллил-2-гидроксифенил)этанона растворяются в 120 мл толуола и смешиваются с 2.17 г (5.6 ммоль) бис(бензонитрил)дихлорпалладия(II). Реакционная смесь в течение ночи нагревается до 120°С. После охлаждения до комнатной температуры смесь фильтруется через кизельгур и растворитель удаляется в вакууме. Получается 20.9 г (95% теор. возм.) выведенного в заголовке соединения, которое используется в следующей стадии без дополнительной очистки.
LC-MS (метод 1): Rt=2.36 мин; [М+Н]+=177
1Н-ЯМР (300 МГц, CDCl3): δ=1.91 (dd, 3Н), 2.63 (s, 3Н), 6.32 (m, 1H), 6.73 (dd, 1H), 6.85 (t, 1H), 7.59 (m, 2H), 12.74 (s, 1H).
Пример 4А
2-Метил-8-[(1Е)-проп-1-ен-1-ил]-4H-хромен-4-он
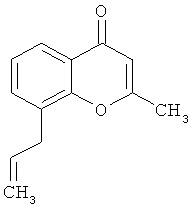
12.52 г (313.2 ммоль) 60%-ного гидрида натрия (суспензия в минеральном масле) в атмосфере аргона при 10°С разбавляются 300 мл абсолютного ТГФ. К суспензии каплями медленно добавляются 18.4 г (104.4 ммоль) 1-{2-гидрокси-3-[(1Е)-проп-1-ен-1-ил]фенил}этанона. Спустя 15 минут добавляются 9 г (114.9 ммоль) ацетилхлорида. Реакционная смесь перемешивается в течение ночи при комнатной температуре. Смесь подвергается гидролизу с 300 мл воды и несколько раз экстрагируется с помощью этилацетата. Органическая фаза после промывания насыщенным раствором хлорида натрия высушивается над сульфатом натрия. Затем растворитель удаляется в вакууме. Осадок разводится в 200 мл метанола и вместе с 50 мл 20%-ной соляной кислоты 30 минут нагревается до 80°С. После этого растворитель удаляется в вакууме и осадок разбавляется 400 мл воды. Несколько раз экстрагируется с помощью дихлорметана. После сушки органической фазы над сульфатом магния растворитель удаляется в вакууме и осадок очищается посредством колоночной хроматографии (растворитель: дихлорметан/метанол 98:2). Получается 10.5 г (50.2% теор. возм.) выведенного в заголовке соединения в виде желтого масла.
LC-MS (метод 2): Rt=2.07 мин; [М+Н]+=201
1Н-ЯМР (300 МГц, CDCl3): δ=1.98 (dd, 3Н), 2.43 (s, 3Н), 6.18 (s, 1H), 6.40 (m, 1H), 6.85 (dd, 1H), 7.31 (t, 1H), 7.72 (dd, 1H), 8.05 (dd, 1H).
Пример 5А
2-Метил-4-оксо-4H-хромен-8-карбалдегид
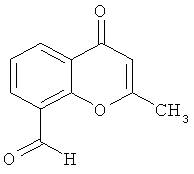
18.5 г (62.8 ммоль) 2-метил-8-[(1Е)-проп-1-ен-1-ил]-4H-хромен-4-она растворяются в 400 мл дихлорметана и охлаждаются до -60°С. В реакционную смесь в течение 30 минут вводится озон. Затем реакционная смесь разбавляется диметилсульфидом. После нагревания до комнатной температуры растворитель удаляется в вакууме и осадок взмучивается с небольшим количеством метанола. После фильтрации остающееся твердое вещество из диэтилового эфира перекристаллизовывается. Получается 9.1 г (77.4% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 1): Rt=1.31 мин; [М+Н]+=189
1Н-ЯМР (300 МГц, CDCl3): δ=2.48 (s, 3Н), 6.27 (s, 1H), 7.51 (m, 1H), 8.21 (dd, 1H), 8.46 (dd, 1H), 10.67 (s, 1H).
Пример 6А
4-Бром-2-(трифторметокси)бензалдегид
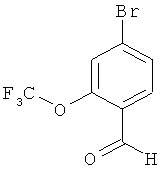
20.00 г (54.51 ммоль) 4-бром-2-(трифторметокси)йодбензола растворяются в 200 мл ТТФ и раствор охлаждается до -78°С. Затем каплями добавляются 26.16 мл (65.41 ммоль) 2.5 М раствора н-бутиллития в гексане. Смесь мешают в течение 30 минут и затем подмешивают 14.43 г (125.37 ммоль) N-формилморфолина. После того как зафиксирована полнота превращения (DC-контроль), изопрапонол при -78°С расщепляется ионами растворителя. После нагревания до комнатной температуры смесь разбавляется водой и дважды экстрагируется с помощью дихлорметана. Объединенные органические фазы промываются насыщенным раствором хлорида натрия, высушиваются сульфатом натрия и растворитель отгоняется при пониженном давлении. Осадок очищается с помощью колоночной хроматографии (силикагель, растворитель: циклогексан/этиловый эфир уксусной кислоты 5:1). Получается 11.43 г (78% теор. возм.) выведенного в заголовке соединения.
GC-MS (метод 5): Rt=4.24 мин; MS (EIpos): m/z=270 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=7.85-7.92 (m, 3H), 10.20 (s, 1H).
Пример 7А
4-Формил-3-(трифторметокси)бензонитрил
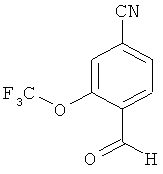
10.63 г (39.51 ммоль) 4-бром-2-(трифторметокси)бензалдегида, 3.43 г (29.24 ммоль) цианида цинка и 1.37 г (1.19 ммоль) тетракис(трифенилфосфин)палладия(0) растворяются в 80 мл диметилформамида. Затем реакционная смесь несколькими порциями преобразуется микроволнами в одномодовом режиме (Emrys Optimizer, 5 минут при 220°С). Объединенные осадки разбавляются водой и дважды экстрагируются с помощью толуола. Объединенные органические фазы промываются насыщенным раствором хлорида натрия, высушиваются сульфатом натрия, затем растворитель удаляется в ротационном выпарном аппарате. Осадок очищается с помощью колоночной хроматографии (силикагель, растворитель: циклогексан/этиловый эфир уксусной кислоты 10:1). Получается 3.32 г (78% теор. возм.) выведенного в заголовке соединения с чистотой 80% (согласно LC-MS).
MS (EIpos): m/z=215 [M]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=7.85-7.91 (m, 3H), 10.20 (s, 1H).
Пример 8А
4-Циано-2-метоксифенил-трифторметансульфонат
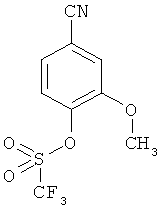
К раствору 20 г (134 ммоль) 4-гидрокси-3-метоксибензонитрила в пиридине (80 мл) медленно прикалываются 24 мл (141 ммоль) трифторметансульфокислоты ангидрида, причем температура реакции поддерживается с помощью ледяной ванны ниже 25°С. Затем суспензия перемешивается 1 час при комнатной температуре. Добавляется ледяная вода (400 мл) и перемешивание суспензии продолжается до достижения комнатной температуры. Потом фильтруется, твердое вещество растворяется в этилацетате и этот раствор промывается насыщенным раствором хлорида натрия. Органическая фаза высушивается над сульфатом магния и сгущается. Получается 37.13 г (92% теор. возм.) выведенного в заголовке соединения в виде твердого вещества белого цвета.
LC-MS (метод 3): Rt=2.54 мин; MS (EIpos): m/z=282 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=3.97 (s, 3Н), 7.60 (dd, 1H), 7.71 (d, 1H), 7.92 (d, 1H).
Пример 9А
трет-Бутил(2E)-3-(4-циано-2-метоксифенил)акрилат
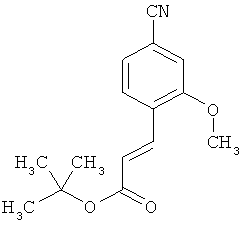
К дегазированному раствору из 37.13 г (132 ммоль) 4-циано-2-метоксифенил-трифторметансульфоната, 35 мл (245 ммоль) трет-бутилакрилата и 90 мл (645 ммоль) триэтиламина в диметилформамиде (250 мл) добавляются 4 г (5.7 ммоль) хлорида бис(трифенилфосфин)палладия(II). Раствор перемешивается 24 часа при 100°С в атмосфере защитного газа. Затем добавляется ледяная вода (1000 мл) и суспензия экстрагируется с помощью этилацетата (3 раза по 100 мл). Органическая фаза промывается насыщенным раствором хлорида натрия, высушивается над сульфатом магния и сгущается. Осадок очищается колоночной хроматографией (силикагель, растворитель: циклогексан/этиловый эфир уксусной кислоты 10:1). Получается 24.6 г (72% теор. возм.) выведенного в заголовке соединения в виде твердого вещества белого цвета.
LC-MS (метод 1): Rt=2.59 мин; MS (EIpos): m/z=260 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.48 (s, 9H), 3.93 (s, 3Н), 6.65 (d, 1H), 7.42 (d, 1H), 7.58 (s, 1H), 7.74 (d, 1H), 7.89 (d, 1H).
Пример 10А
4-Формил-3-метоксибензонитрил
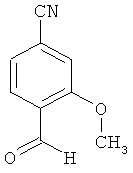
К интенсивно перемешанному раствору из 48 г (185 ммоль) трет-бутил(2Е)-3-(4-циано-2-метоксифенил)акрилата, 207 мг (0.81 ммоль) четырехоксида осмия и 1.4 г (6.14 ммоль) хлорида бензилтриэтиламмония в 750 мл воды/ТТФ (2:1) порциями добавляются 79 г (370 ммоль) метапериодата натрия, причем температура реакции поддерживается ниже 30°С. Раствор перемешивается еще 1 час при комнатной температуре. Добавляется вода (2000 мл), после чего смесь фильтруется. Остающееся твердое вещество растворяется в этилацетате и раствор промывается насыщенным раствором хлорида натрия. Органическая фаза высушивается над сульфатом магния и сгущается. Осадок смешивается с петролейным эфиром. Получается 21.18 г (71% теор. возм.) выведенного в заголовке соединения в виде твердого вещества белого цвета.
LC-MS (метод 3): Rt=1.87 мин; MS (EIpos): m/z=162 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=3.98 (s, 3Н), 7.53 (d, 1H), 7.80 (s, 1H), 7.81 (d, 1H), 10.37 (s, 1H).
Пример 11А
4-Формил-3-гидроксибензонитрил
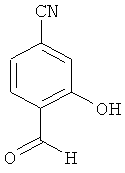
К раствору из 8 г (49.64 ммоль) 4-формил-3-метоксибензонитрила в 80 мл безводного дихлорметана в атмосфере аргона при -78°С прикапываются 100 мл раствора трибромида бора в дихлорметане (1 М, 100 ммоль). Реакционная смесь перемешивается при комнатной температуре до полного преобразования продукта (около 5 дней). Затем реакционный раствор нейтрализуется при 0°С насыщенным раствором гидрокарбонатом натрия. Фазы разделяются, и органическая фаза промывается насыщенным раствором хлорида натрия, высушивается над сульфатом магния и сгущается. Осадок очищается посредством колоночной хроматографии на силикагеле (растворитель: циклогексан/этилацетат 3:1). Получается 4.5 г (61% теор. возм.) выведенного в заголовке соединения в виде твердого вещества желтого цвета.
LC-MS (метод 1): Rt=1.38 мин; [М-Н]-=146
1Н-ЯМР (300 МГц, CDCl3): δ=7.38 (d, 1Н), 7.38 (s, 1H), 7.77 (d, 1H), 10.33 (s, 1H), 11.38 (s, 1H).
Пример 12А
5-Циано-2-формилфенил-трифторметансульфонат
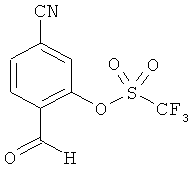
К раствору из 2 г (13.59 ммоль) 4-формил-3-гидроксибензонитрила и 2.5 мл (14.27 ммоль) N,N-диизопропилэтиламина в 37 мл безводного дихлорметана в атмосфере аргона при 0°С каплями добавляются 2.4 мл (14.27 ммоль) ангидрида трифторметансульфокислоты. Реакционная смесь перемешивается 1 час при комнатной температуре, затем разбавляется дихлорметаном (70 мл) и последовательно промывается 1 М соляной кислотой, насыщенным раствором гидрогенкарбоната натрия и насыщенным раствором хлорида натрия. Органический раствор высушивается над сульфатом магния и сгущается. Осадок очищается посредством колоночной хроматографии на силикагеле (растворитель: циклогексан/этилацетат 7:1). Получается 2.36 г (62% теор. возм.) выведенного в заголовке соединения в виде твердого вещества белого цвета.
LC-MS (метод 3):Rt=2.34 мин; [М+Н]+=280
1Н-ЯМР (300 МГц, CDCl3): δ=8.27 (m, 2H), 8.33 (s, 1H), 10.13 (s, 1H).
Пример 13А
4-Формил-3-винилбензонитрил
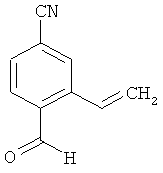
К раствору из 1 г (3.58 ммоль) 5-циано-2-формилфенил-трифторметансульфоната и 1.15 мл (3.94 ммоль) три-н-бутилвинилстаннана в 6 мл безводного и дегазированного диметилформамида в атмосфере аргона добавляются 125 мг (0.18 ммоль) хлорида бис(трифенилфосфин)палладия(II). Затем реакционная смесь перемешивается 90 минут при 80°С. После этого прибавляются 100 мл 10%-ного раствора фторида калия и смесь перемешивается 1 час при комнатной температуре. Суспензия трижды экстрагируется с помощью этилацетата (по 20 мл) и очищенные органические фазы последовательно промываются насыщенным раствором гидрогенкарбоната натрия и насыщенным раствором хлорида натрия. Органический раствор высушивается над сульфатом магния и сгущается. Остаток (0.6 г) без дополнительной очистки используется в следующей стадии.
GC-MS (метод 5):Rt=5.02 мин; [М]+=157
1Н-ЯМР (300 МГц, CDCl3): δ=5.62 (d, 1Н), 6.05 (d, 1H), 7.58 (dd, 1H), 7.95 (d, 1H), 8.00 (d, 1H), 8.24 (s, 1H), 10.32 (s, 1H).
Пример 14А
3-Этил-4-формилбензонитрил
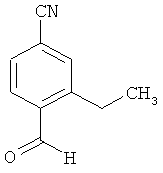
Раствор 1.3 г (8.27 ммоль) 4-формил-3-винилбензонитрила в 35 мл этанола смешивается с 880 мг 10%-ного палладия на угле и в течение 2 часов интенсивно перемешивается в атмосфере водорода. Суспензия фильтруется через слой силикагеля, осадок промывается этанолом и фильтрат сгущается. Осадок (890 мг) используется в следующей стадии без дополнительной очистки.
1Н-ЯМР (300 МГц, CDCl3,): δ=1.2 (t, 1H), 3.07 (q, 2H), 7.88 (d, 1H), 7.90 (s, 1H), 7.97 (d, 1H), 10.32 (s, 1H).
Пример 15А
Метил 4-циано-2-фторбензоат
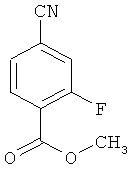
13.20 г (79.9 ммоль) 4-циано-2-фторбензойной кислоты растворяются в 300 мл ацетона. Затем последовательно добавляются 22.10 г (159.9 ммоль) карбоната калия и 9.08 мл (95.9 ммоль) диметилсульфата. Смесь перемешивается в течение 20 часов при температуре рефлюкса. После этого реакционная смесь разводится 300 мл воды и ацетон удаляется на ротационном выпарном аппарате. Несколько раз экстрагируется с помощью дихлорметана. Объединенные органические фазы промываются насыщенным раствором хлорида натрия и высушиваются над сульфатом натрия. Затем растворитель удаляется в вакууме. Оставшееся твердое вещество используется в дальнейшем без дополнительной очистки. Получается 16.1 г (84% теор. возм.) выведенного в заголовке соединения в виде бесцветного твердого вещества.
GC-MS (метод 5): Rt=6.23 мин; [М]+ (EIpos): m/z=179
1Н-ЯМР (300 МГц, DMSO-d6): δ=3.90 (s, 3Н), 7.83 (dd, 1H), 8.01-8.08 (m, 2H).
Пример 16А
3-Фтор-4-(гидроксиметил)бензонитрил
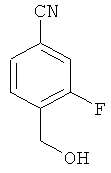
16.10 г (89.9 ммоль) метил-4-циано-2-фторбензоата растворяются в 150 мл метанола. Затем порциями добавляется 3.40 г (89.9 ммоль) боргидрида натрия. После завершения реакции обмена (DC-контроль) рН доводится разведенной соляной кислотой до 3 и смесь несколько раз экстрагируется с помощью дихлорметана. Объединенные органические фазы промываются насыщенным раствором хлорида натрия и высушиваются сульфатом магния. Затем растворитель удаляется в вакууме, а осадок очищается посредством колоночной хроматографии (силикагель, растворитель: циклогексан/этилацетат 15:1→3:7). Получается 3.70 г (27.2% теор. возм.) выведенного в заголовке соединения.
GC-MS (метод 5): Rt=6.51 мин; [М]+ (EIpos): m/z=151
1Н-ЯМР (300 МГц, DMSO-d6): δ=4.61 (s, 2H), 5.53 (s, 1H), 7.61-7.74 (m, 2H), 7.79 (dd, 1H).
Пример 17А
3-Фтор-4-формилбензонитрил
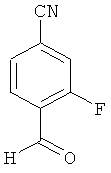
1.00 г (6.62 ммоль) 3-фтор-4-(гидроксиметил)бензонитрила растворяются в 50 мл дихлорметана и смешиваются с 9.20 г (105.9 ммоль) оксида марганца(IV). Смесь перемешивается в течение ночи при комнатной температуре и затем фильтруется через слой кизельгура. Растворитель отгоняется при пониженном давлении, а осадок очищается посредством колоночной хроматографии (силикагель, растворитель: дихлорметан). Получается 120 мг (12.1% теор. возм.) выведенного в заголовке соединения.
GC-MS (метод 5): Rt=5.11 мин; [М]+ (EIpos): m/z=149
1Н-ЯМР (300 МГц, DMSO-d6): δ=7.89 (d, 1H), 8.00 (t, 1H), 8.11 (d, 1H), 10.24 (d, 1H).
Пример 18А
3-Хлор-4-формилбензонитрил
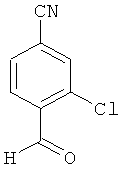
25.0 г (164.91 ммоль) 3-хлор-4-метилбензонитрила растворяются в 150 мл диметилформамида и смешиваются с 25.55 г (214.39 ммоль) N,N-диметилформамид-диметилацетала. Смесь мешается сначала 20 часов при 140°С, а затем 4 часа при температуре масляной бани 180°С. Летучие компоненты отделяются на ротационном выпарном аппарате, а остающийся осадок сразу подвергается реакции обмена.
Полученный таким образом сырой 3-хлор-4-[2-(диметиламино)винил]бензонитрил вводится в 500 мл раствора ТГФ/вода (1:1) и смешивается с 77.6 г (362.9 ммоль) периодата натрия. Смесь мешается 18 часов при комнатной температуре и затем выпавший осадок отделяется фильтрацией. Фильтрат разбавляется насыщенным раствором гидрогенкарбонатом натрия и трижды экстрагируется этилацетатом. Объединенные органические фазы высушиваются сульфатом натрия и растворитель удаляется в ротационном выпарном аппарате. Исходный продукт очищается посредством колоночной хроматографии (силикагель, растворитель: циклогексан/этилацетат 7:3). Получается 3.0 г (15% теор. возм.) выведенного в заголовке соединения.
GC-MS (метод 5): Rt=6.64 мин; [М]+(EIpos): m/z=165
1Н-ЯМР (300 МГц, DMSO-d6): δ=7.97-8.03 (m, 2H), 8.27 (s, 1H), 10.34 (d, 1H).
Пример 19А
4-Формил-1-нафтонитрил
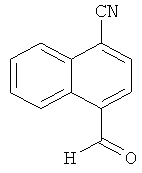
2.50 г (14.95 ммоль) 4-метил-1-нафтонитрил растворяются в 40 мл тетрахлорметана и смешиваются с 3.19 г (17.94 ммоль) N-бромсукцинимида и 245 мг (1.50 ммоль) 2,2′-азобис-2-метилпропаннитрила. Смесь мешается в течение ночи при температуре рефлюкса. После охлаждения продукт отфильтровывается. Получается 2.75 г (74.7% теор. возм.) 4-(бромметил)-1-нафтонитрила 90%-ной чистоты, который используется в дальнейшем без дополнительной очистки.
2.75 г (11.17 ммоль) полученного таким образом бромида растворяются в 60 мл ацетонитрила и смешиваются с 2 г молекулярного сита (3Å). Затем добавляются 1.44 г (12.29 ммоль) N-метилморфолин-N-оксида и смесь перемешивается в течение ночи при комнатной температуре. Отложение фильтруется через силикагель и фильтрат сгущается. Осадок очищается через Biotage-патрон (40 М) (элюент: изогексан/этилацетат 3:1). Фракции продукта очищаются, растворитель удаляется в ротационном выпарном аппарате и затем осадок смешивается с диэтиловым эфиром, причем наблюдается процесс кристаллизации. Продукт промывается небольшим количеством диэтилового эфира и высушивается в глубоком вакууме. Получается 254 мг (12.6% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 3): Rt=2.27 мин; [М+Н]+ (EIpos): m/z=182
1Н-ЯМР (300 МГц, CDCl3): δ=7.79-7.87 (m, 2H), 8.05 (d, 1H), 8.09 (d, 1H), 8.37 (m, 1H), 9.27 (m, 1H), 10.51 (s, 1H).
Пример 20А
2-Цианоэтил 4-(4-циано-2-метоксифенил)-2-метил-5-оксо-1,4,5,6-тетрагидро-1,6-нафтиридин-3-карбоксилат

14.63 г (90.81 ммоль) соединения из примера 10А, 10.00 г (90.81 ммоль) 4-аминопиридин-2(1H)-она [Searls, Т., McLaughlin, L.W., Tetrahedron 55. 11985-11996 (1999)] и 15.65 г (90.81 ммоль) 2-цианоэтил-3-оксобутаноата [Yamamoto, Т., et al., Bioorg. Med. Chem. Lett. 16, 798-802 (2006)] растворяются в 300 мл изопропанола и смесь мешается 3 дня в атмосфере аргона при температуре рефлюкса. Затем смесь сгущается и после этого очищается посредством колоночной хроматографии (силикагель; растворитель: сначала этиловый эфир уксусной кислоты, затем дихлорметан/метанол 10:1). Полученные фракции продукта концентрируются и впитываются в небольшое количество этилового эфира уксусной кислоты. Выпавший продукт отфильтровывается и высушивается в течение ночи в вакууме при 40°С. Получают 10.11 г (27% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 6): Rt=1.83 мин; MS (EIpos): m/z=391 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=2.27 (s, 3Н), 2.79 (m, 2H), 3.75 (s, 3Н), 3.96-4.14 (m, 2H), 5.19 (s, 1H), 5.87 (d, 1H), 7.10 (d, 1H), 7.23 (dd, 1H), 7.30-7.35 (m, 2H), 9.30 (s, 1H), 10.83 (s, 1H).
Пример 21А
2-Цианоэтил 4-(4-циано-2-метоксифенил)-5-этокси-2-метил-1,4-дигидро-1,6-нафтипиридин-3-карбоксилат
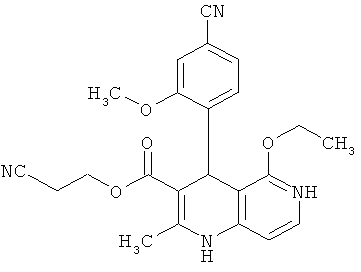
10.00 г (25.62 ммоль) соединения из примера 20А превращаются в суспензию в 250 мл триэтилового эфира ортомуравьиной кислоты и суспензия нагревается до 130°С. Затем в реакционную смесь каждый час в течение 8 часов добавляется по 15 капель концентрированной серной кислоты. Смесь перемешивается в течение ночи при равной температуре. После охлаждения избыточный ортоэфир удаляется в ротационном выпарном аппарате и исходный продукт очищается посредством колоночной хроматографии (силикагель; растворитель: циклогексан/этиловый эфир уксусной кислоты 1:1). Получают 7.20 г (65% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 6):Rt=2.82 мин; MS (EIpos): m/z=419 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.12 (t, 3H), 2.33 (s, 3H), 2.77 (m, 2H), 3.78 (s, 3H), 3.99-4.13 (m, 4H), 5.37 (s, 1H), 6.48 (d, 1H), 7.25 (dd, 1H), 7.29-7.35 (m, 2H), 7.73 (d, 1H), 9.53 (s, 1H).
Пример 22А
4-(4-Циано-2-метоксифенил)-5-этокси-2-метил-1,4-дигидро-1,6-нафтиридин-3-карбоновая кислота
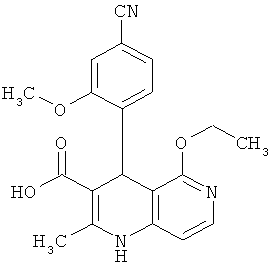
7.20 г (17.21 ммоль) соединения из примера 21А растворяются в 200 мл 1,2-диметоксиэтана и воды (3:1 отношение объемов), разбавляются 34.42 мл (34.42 ммоль) 1 N раствором едкого натра и смесь мешается в течение ночи при комнатной температуре. Затем отложение разводится 100 мл диэтилового эфира и 100 мл воды, органическая фаза отделяется, а водная фаза подкисляется 1 N соляной кислотой до рН 4-5. Образующаяся суспензия мешается 1 час и выпавший осадок отделяется фильтрацией. Осадок промывается водой и небольшим количеством диэтилового эфира. После сушки в вакууме при 40°С получают 3.57 г (57% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 7): Rt=2.32 мин; MS (EIpos): m/z=366 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.12 (t, 3H), 2.28 (s, 3H), 3.74 (s, 3H), 4.07 (m, 2H), 5.33 (s, 1H), 6.44 (d, 1H), 7.23-7.29 (m, 2H), 7.32 (s, 1H), 7.70 (d, 1H), 9.25 (s, 1H), 11.34 (s, 1H).
Пример 23А
4-[5-Этокси-3-(1H-имидазол-1-илкарбонил)-2-метил-1,4-дигидро-1,6-нафтиридин-4-ил]-3-метоксибензонитрил
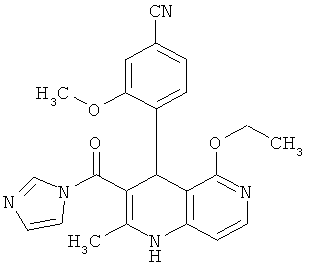
1.20 г (3.28 ммоль) соединения из примера 22А вводятся в 25 мл этилового эфира уксусной кислоты, разбавляются 0.666 г (4.11 ммоль) 1,1'-карбонилдиимидазола и перемешиваются в течение ночи при комнатной температуре. Реакционная смесь концентрируется в ротационном выпарном аппарате и полученный таким образом исходный продукт используется для дальнейших реакций без очистки.
MS (EIpos): m/z=416 [M+H]+.
Пример 24А
2-Цианоэтил 2-(4-циано-2-метоксибензилиден)-3-оксобутаноат
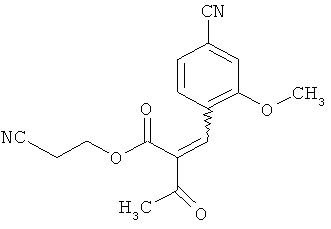
3.00 г (18.62 ммоль) соединения из примера 10А, 3.18 г (20.48 ммоль) 2-цианоэтил-3-оксобутаноата уксусной кислоты [Yamamoto, Т., et al., Bioorg. Med. Chem. Lett. 16. 798-802 (2006)], 213 мкл (3.72 ммоль) уксусной кислоты и 368 мкл (3.72 ммоль) пиперидина растворяются в 50 мл безводного дихлорметана и в течение ночи мешаются при рефлюксе в водоотделителе. После этого летучие компоненты отделяются в ротационном выпарном аппарате и осадок очищается посредством колоночной хроматографии (силикагель; растворитель: градиент циклогексан/этиловый эфир уксусной кислоты 7:3→1:1). Получается 2.77 г (48% теор. возм.) выведенного в заголовке соединения в виде смеси E/Z-изомеров.
LC-MS (метод 7): Rt=2.89 и 3.00 мин; MS (EIpos): m/z=299 [M+H]+.
Пример 25А
2-Цианоэтил 4-(4-циано-2-метоксифенил)-2,7-диметил-5-оксо-1,4,5,6-тетрагидро-1,6-нафтиридин-3-карбоксилат
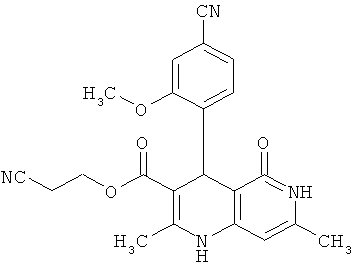
1.49 г (5.00 ммоль) соединения из примера 24А вводятся в 30 мл 2-пропанола, смешиваются с 620 мг (5.00 ммоль) 4-амино-6-метилпиридин-2(1H)-она [Bisagni, E., Hung, N.C., Synthesis, 765-766 (1984)] и перемешиваются в течение ночи при температуре рефлюкса. После охлаждения осадок отфильтровывается, промывается диэтиловым эфиром и высушивается в глубоком вакууме. Получают 1.53 г (76% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 3): Rt=1.73 мин; MS (EIpos): m/z=405 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=2.05 (s, 3H), 2.26 (s, 3H), 2.78 (m, 2H), 3.74 (s, 3H), 3.96-4.13 (m, 2H), 5.14 (s, 1H), 5.64 (d, 1H), 7.23 (dd, 1H), 7.28-7.33 (m, 2H), 9.22 (s, 1H), 10.82 (s, 1H).
Пример 26А
2-Цианоэтил 4-(4-циано-2-метоксифенил)-5-этокси-2,7-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксилат
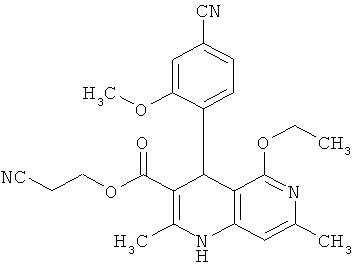
1.52 г (3.76 ммоль) соединения из примера 25А превращается в суспензию в 40 мл триэтилового эфира ортомуравьиной кислоты и суспензия нагревается до 130°С. После этого к реакционной смеси каждый чае в течение 8 часов добавляется по 10 капель концентрированной серной кислоты. Затем смесь перемешивается в течение ночи при этой же температуре. После охлаждения избыточный ортоэфир удаляется в ротационном выпарном аппарате, а исходный продукт очищается посредством колоночной хроматографии (силикагель; растворитель: циклогексан/этиловый эфир уксусной кислоты 1:1). Фракции продукта объединяются, растворитель удаляется, а к осадку добавляется небольшое количество метанола. Продукт кристаллизации отфильтровывается. После сушки в глубоком вакууме получают 1.09 г (67% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 3): Rt=2.23 мин; MS (EIpos): m/z=433 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.11 (t, 3H), 2.20 (s, 3H), 2.32 (s, 3H), 2.76 (m, 2H), 3.78 (s, 3H), 3.97-4.12 (m, 4H), 5.32 (s, 1H), 6.30 (s, 1H), 7.24 (d, 1H), 7.27-7.32 (m, 2H), 9.43 (s, 1H).
Пример 27А
4-(4-Циано-2-метоксифенил)-5-этокси-2,7-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоновая кислота
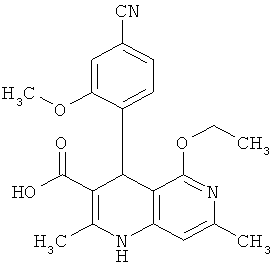
642 мг (2.52 ммоль) соединения из примера 26А растворяются в 40 мл смеси 1,2-диметоксиэтана и воды (3:1 отношение объемов), разбавляются 5.04 мл (5.04 ммоль) 1 N едкого натра и перемешиваются в течение ночи при комнатной температуре. После этого осадок разводится 30 мл диэтилового эфира и 30 мл воды, органическая фаза отделяется, а водная фаза подкисляется 1 N соляной кислотой до рН 4-5. Полученная суспензия мешается в течение 1 часа, и выпавшее твердое вещество отделяется фильтрацией. Осадок промывается водой и небольшим количеством диэтилового эфира. После сушки в вакууме при 40°С получают 642 мг (67% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 3): Rt=1.87 мин; MS (EIpos): m/z=380 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.11 (t, 3Н), 2.19 (s, 3H), 2.28 (s, 3H), 3.74 (s, 3H), 4.05 (m, 2H), 5.28 (s, 1H), 6.27 (s, 1H), 7.20-7.28 (m, 2H), 7.31 (s, 1H), 9.17 (s, 1H), 11.31 (s, 1H).
Пример 28А
2-Цианоэтил 4-(4-циано-2-метоксифенил)-2,8-диметил-5-оксо-1,4,5,6-тетрагидро-1,6-нафтиридин-3-карбоксилат
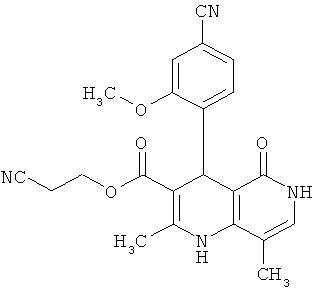
2.69 г (9.00 ммоль) соединения из примера 24А вводятся в 45 мл 2-пропанола, смешиваются с 1.17 г (9.00 ммоль) 4-амино-5-метилпиридин-2(1H)-она [Bisagni, Е., Hung, N.C., Synthesis, 765-766 (1984)] и мешаются в течение ночи при температуре рефлюкса. После охлаждения осадок отфильтровывается, промывается диэтиловым эфиром и высушивается в глубоком вакууме. Получают 2.22 г (61% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 3):Rt=1.75 мин; MS (EIpos): m/z=405 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=2.03 (s, 3H), 2.35 (s, 3H), 2.80 (m, 2H), 3.74 (s, 3H), 4.04 (m, 1H), 4.11 (m, 1H), 5.20 (s, 1H), 6.95 (s, 1H), 7.23 (dd, 1H), 7.28-7.33 (m, 2H), 8.18 (s, 1H), 10.76 (s, 1H).
Пример 29А
2-Цианоэтил 4-(4-циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксилат
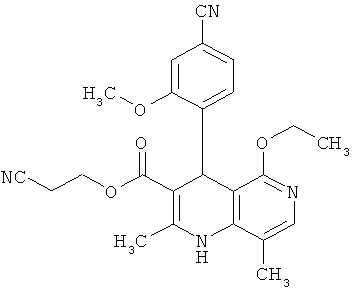
2.22 г (5.44 ммоль) соединения из примера 28А превращаются в суспензию в 100 мл триэтилового эфира ортомуравьиной кислоты и суспензия нагревается до 130°С. После этого к реакционной смеси в течение 8 часов каждый час добавляется по 10 капель концентрированной серной кислоты. Затем смесь мешается в течение ночи при этой же температуре. После охлаждения избыточный ортоэфир удаляется в ротационном выпарном аппарате и исходный продукт очищается посредством колоночной хроматографии (силикагель; растворитель: сначала дихлорметан, затем изогексан/этиловый эфир уксусной кислоты 1:1). Фракции продукта объединяются, растворитель удаляется, а осадок кристаллизуется из смеси этилового эфира уксусной кислоты и диэтилового эфира. Преципитат отфильтровывается и высушивается в глубоком вакууме. Получают 1.80 г (77% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 6): Rt=3.02 мин; MS (EIpos): m/z=433 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.11 (t, 3H), 2.16 (s, 3H), 2.42 (s, 3H), 2.78 (m, 2H), 3.77 (s, 3H), 4.01-4.13 (m, 4H), 5.37 (s, 1H), 7.25 (d, 1H), 7.28-7.33 (m, 2H), 7.60 (s, 1H), 8.35 (s, 1H).
Пример 30А
4-(4-Циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоновая кислота
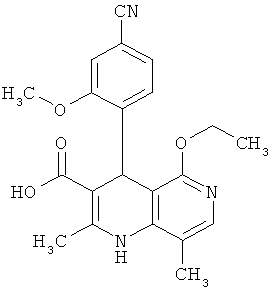
1.75 г (4.05 ммоль) соединения из примера 29А растворяется в 60 мл смеси из 1,2-диметоксиэтана и воды (2:1 отношение объемов), разбавляется 8.09 мл (8.09 ммоль) 1 N едкого натра и один час мешается при комнатной температуре. Затем исходная смесь смешивается с 30 мл диэтилового эфира и водная фаза подкисляется 6 N соляной кислотой. Образующийся осадок отфильтровывается и промывается водой и небольшим количеством диэтилового эфира. После сушки при 40°С в вакуумном сушильном шкафу получают 1.47 г (96% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 7): Rt=2.50 мин; MS (EIpos): m/z=380 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.14 (t, 3H), 2.14 (s, 3H), 2.37 (s, 3H), 3.73 (s, 3H), 4.04 (m, 2H), 5.33 (s, 1H), 7.26 (m, 2H), 7.32 (s, 1H), 7.57 (s, 1H), 8.16 (s, 1H), 11.43 (br. s, 1H).
Пример 31А
2-Цианоэтил 2-метил-4-(2-метил-4-оксо-4H-хромен-8-ил)-5-оксо-1,4,5,6-тетрагидро-1,6-нафтиридин-3-карбоксилат
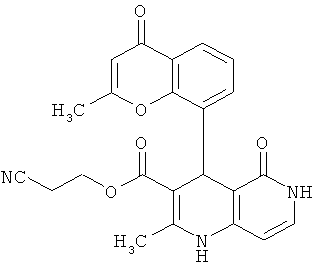
3.00 г (15.94 ммоль) соединения из примера 5А, 1.75 г (15.94 ммоль) 4-аминопиридин-2(1H)-она [Searls, Т., McLaughlin, L.W., Tetrahedron 55, 11985-11996 (1999)] и 2.47 г (15.94 ммоль) 2-цианоэтил-3-оксобутаноата [Yamamoto, Т., et al., Bioorg. Med. Chem. Lett. 16. 798-802 (2006)] растворяются в 60 мл этанола и мешаются в течение ночи в атмосфере аргона при температуре рефлюкса. После этого выпавший в осадок продукт отфильтровывается, промывается этанолом и диэтиловым эфиром и высушивается в глубоком вакууме. Получают 2.30 г (35% теор. возм.) выведенного в заголовке соединения в виде кристаллов бежевого цвета.
LC-MS (метод 7): Rt=1.59 мин; MS (EIpos): m/z=418 [M+H]+.
Пример 32А
2-Цианоэтил 5-этокси-2-метил-4-(2-метил-4-оксо-4Н-хромен-8-ил)-1,4-дигидро-1,6-нафтиридин-3-карбоксилат
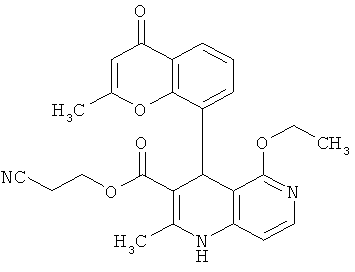
2.20 г (5.27 ммоль) соединения из примера 31А превращаются в суспензию в 80 мл триэтилового эфира ортомуравьиной кислоты и нагреваются до 130°С. После этого к реакционной смеси в течение 8 часов каждый час добавляется по 5 капель концентрированной серной кислоты. Затем смесь мешается в течение ночи при этой же температуре. После охлаждения избыточный ортоэфир удаляется в ротационном выпарном аппарате и исходный продукт очищается посредством колоночной хроматографии (силикагель; растворитель: сначала дихлорметан, затем этиловый эфир уксусной кислоты, в конце этиловый эфир уксусной кислоты/метанол 20:1). Фракции продукта сгущаются до объема порядка 5 мл. Выпавший в осадок продукт отфильтровывается и после промывания этиловым эфиром уксусной кислоты и диэтиловым эфиром высушивается в глубоком вакууме. Получают 282 мг (12% теор. возм.) выведенного в заголовке соединения в виде коричневых кристаллов.
LC-MS (метод 3):Rt=1.96 мин; MS (EIpos): m/z=446 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.01 (t, 3H), 2.38 (s, 6H), 2.74 (m, 2H), 3.96-4.13 (m, 4H), 5.54 (s, 1H), 6.19 (s, 1H), 6.53 (d, 1H), 7.31 (t, 1H), 7.67 (dd, 1H), 7.76 (d, 1H), 7.80 (dd, 1H), 9.69 (s, 1H).
Пример 33А
5-Этокси-2-метил-4-(2-метил-4-оксо-4H-хромен-8-ил)-1,4-дигидро-1,6-нафтиридин-3-карбоновая кислота
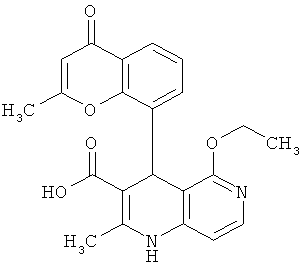
270 мг (0.61 ммоль) соединения из примера 32А растворяются в 15 мл смеси из 1,2-диметоксиэтана и воды (2:1 объемное отношение), смешиваются с 1.21 мл (1.21 ммоль) 1 N раствора едкого натра и мешаются 1 час при комнатной температуре. После этого реакционная смесь разбавляется 30 мл диэтилового эфира. Водная фаза отделяется и подкисляется 1 N соляной кислотой. Выпавший осадок отфильтровывается и промывается водой и небольшим количеством диэтилового эфира. После сушки при 40°С в вакууме получают 167 мг (70% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 7):Rt=2.01 мин; MS (EIpos): m/z=393 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.02 (t, 3H), 2.33 (s, 3H), 2.35 (s, 3H), 3.97 (m, 1H), 4.08 (m, 1H), 5.52 (s, 1H), 6.18 (s, 1H), 6.50 (d, 1H), 7.31 (t, 1H), 7.60 (dd, 1H), 7.74 (d, 1H), 7.79 (dd, 1H), 9.42 (s, 1H), 11.45 (br. s, 1H).
Пример 34А
2-Цианоэтил 4-[4-бром-2-(трифторметокси)фенил]-2-метил-5-оксо-1,4,5,6-тетрагидро-1,6-нафтиридин-3-карбоксилат
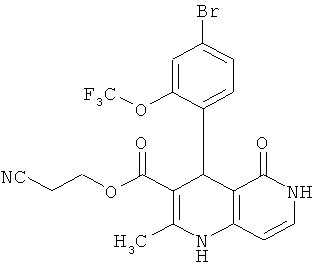
10.00 г (31.17 ммоль) соединения из примера 6А и 6.41 г (31.17 ммоль) 2-цианоэтил-3-оксобутаноата [Yamamoto, Т., et al., Bioorg. Med. Chem. Lett. 16, 798-802 (2006)] вводятся в 100 мл 2-пропанола, смешиваются с 4.09 г (37.17 ммоль) 4-аминопиридин-2(1H)-она [Searls, Т., McLaughlin, L.W., Tetrahedron 55, 11985-11996 (1999)] и смесь мешается три дня при температуре рефлюкса. После охлаждения растворитель удаляется при пониженном давлении, а исходный продукт очищается посредством колоночной хроматографии (силикагель; растворитель: дихлорметан/метанол 10:1). Получают 7.38 г (36% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 6): Rt=2.84 мин; MS (EIpos): m/z=499 [M+H]+.
Пример 35А
2-Цианоэтил 4-[4-бром-2-(трифторметокси)фенил]-5-этокси-2-метил-1,4-дигидро-1,6-нафтиридин-3-карбоксилат
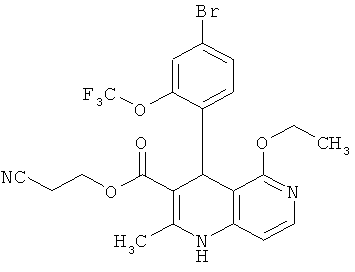
7.30 г (13.19 ммоль) соединения из примера 34А превращаются в суспензию в 150 мл триэтилового эфира ортомуравьиной кислоты и нагреваются до 130°С. После этого к реакционной смеси в течение 7 часов каждый час добавляется по 15 капель концентрированной серной кислоты. Затем смесь мешается в течение ночи при этой же температуре. После охлаждения избыточный ортоэфир удаляется в ротационном выпарном аппарате и исходный продукт очищается посредством колоночной хроматографии (силикагель; растворитель: циклогексан/этиловый эфир уксусной кислоты 1:1). Получают 4.59 г (64% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 3): Rt=2.74 мин; MS (EIpos): m/z=527 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.17 (t, 3H), 2.33 (s, 3H), 2.81 (m, 2H), 3.99-4.21 (m, 4H), 5.29 (s, 1H), 6.50 (d, 1H), 7.31 (t, 1H), 7.37 (d, 1H), 7.44 (dd, 1H), 7.78 (d, 1H), 9.63 (s, 1H).
Пример 36А
2-Цианоэтил-4-[4-циано-2-(трифторметокси)фенил]-5-этокси-2-метил-1,4-дигидро-1,6-нафтиридин-3-карбоксилат

4.59 г (8.72 ммоль) соединения из примера 35А, 758 мг (6.45 ммоль) цианида цинка и 504 мг (0.436 ммоль) тетракис(трифенилфосфин)палладия(0) растворяются в 40 мл диметилформамида и затем, разделенные на три смеси, нагреваются микроволнами в одномодовом режиме (Emrys Optimizer) 5 минут до 220°С. Отдельные смеси снова объединяются и растворитель удаляется в ротационном выпарном аппарате. Исходный продукт впитывается этиловым эфиром уксусной кислоты и фильтруется через кизельгур. Органическая фаза промывается водой (2 раза) и насыщенным раствором хлорида натрия. После отгонки растворителя исходный продукт очищается посредством колоночной хроматографии (силикагель; растворитель: циклогексан/этиловый эфир уксусной кислоты 7:3→1:1). Получают 1.40 г (31% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 3): Rt=2.48 мин; MS (EIpos): m/z=473 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.10 (t, 3H), 2.34 (s, 3H), 2.80 (m, 2H), 4.00-4.21 (m, 4H), 5.36 (s, 1H), 6.51 (d, 1H), 7.60 (d, 1H), 7.66-7.74 (2H), 7.79 (d, 1H), 9.70 (s, 1H).
Пример 37А
4-[4-Циано-2-(трифторметокси)фенил]-5-этокси-2-метил-1,4-дигидро-1,6-нафтиридин-3-карбоновая кислота
1400 мг (2.96 ммоль) соединения из примера 36А растворяются в смеси из 35 мл 1,2-диметоксиэтана и воды (2.5:1 отношение объемов), разбавляются 5.93 мл (5.93 ммоль) 1 N раствора едкого натра и смесь мешается 2 часа при комнатной температуре. Затем реакционная смесь смешивается с 50 мл диэтилового эфира и 50 мл воды. Водная фаза отделяется и подкисляется 1 N соляной кислотой до рН 4-5. Образующаяся суспензия перемешивается в течение 1 часа. Выпавший осадок отфильтровывается и промывается водой и небольшим количеством диэтилового эфира. После сушки в вакууме получают 850 мг (68% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 3): Rt=2.19 мин; MS (EIpos): m/z=420 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.11 (t, 3Н), 2.31 (s, 3Н), 4.06 (m, 1H), 4.13 (m, 1H), 5.37 (s, 1H), 6.49 (d, 1H), 7.51 (d, 1H), 7.65-7.72 (m, 2H), 7.76 (d, 1H), 9.42 (s, 1H), 11.62 (s, 1H).
Пример 38А
4-(4-Циано-2-метоксифенил)-5-этокси-2-(трифторметил)-1,4-дигидро-1,6-нафтиридин-3-карбоновая кислота
Выведенное в заголовке соединение может быть получено исходя из стехиометрических количеств 4-формил-3-метоксибензонитрила (пример 10А), 4-аминопиридин-2(1H)-она [Searls, Т., McLaughlin, L.W., Tetrahedron 55, 11985-11996 (1999)] и аллил 4,4,4-трифтор-3-оксобутаноата [Moseley, J.D., Tetrahedron Lett. 46. 3179-3181 (2005)]. При этом сначала в течение ночи при температуре рефлюкса проводится синтез дигидропиридина в этаноле без добавок. Получающийся вначале промежуточный продукт аллил 4-(4-циано-2-метоксифенил)-2-гидрокси-5-оксо-2-(трифторметил)-1,2,3,4,5,6-гексагидро-1,6-нафтиридин-3-карбоксилат может быть затем дегидратирован уксусной кислотой, как описано в литературе [см. Moseley, J.D., Tetrahedron Lett. 46, 3179-3181 (2005)]. Потом по аналогии с синтезом примера 29А получается аллил 4-(4-циано-2-метоксифенил)-5-этокси-2-(трифторметил)-1,4-дигидро-1,6-нафтиридин-3-карбоксилат путем реакции обмена с триэтил-ортоформиатом. Заключительное расщепление аллилового эфира посредством катализатора Уилкинсона [трис(трифенилфосфин)родия(1) хлорид] в уксусной кислоте дает соединение, выведенное в заголовке [см. Moseley, J.D., Tetrahedron Lett. 46, 3179-3181 (2005)].
LC-MS (метод 7): Rt=2.98 мин; MS (EIpos): m/z=420 [M+H+]
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.09 (t, 3Н), 3.77 (s, 3Н), 3.98-4.16 (m, 2H), 5.37 (s, 1H), 6.73 (d, 1H), 7.19 (d, 1H), 7.34 (dd, 1H), 7.42 (d, 1H), 7.78 (d, 1H), 9.62 (s, 1H).
Пример 39А
2-Цианоэтил 2,8-диметил-4-(2-метил-4-оксо-4Н-хромен-8-ил)-5-оксо-1,4,5,6-тетрагидро-1,6-нафтиридин-3-карбоксилат
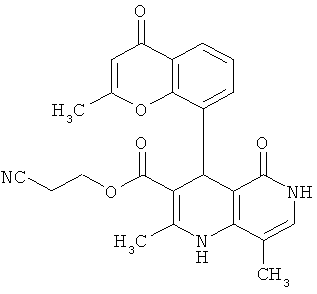
1.50 г (7.97 ммоль) соединения из примера 5А, 1.86 г (9.57 ммоль) 2-цианоэтил-3-оксобутаноата [Yamamoto, Т., et al., Bioorg. Med. Chem. Lett. 16, 798-802 (2006)], 91 мкл (1.59 ммоль) уксусной кислоты и 158 мкл (1.59 ммоль) пиперидина растворяются в 30 мл безводного дихлорметана и мешаются в водоотделителе в течение ночи в условиях рефлюкса. После этого смесь промывается водой, органическая фаза высушивается сульфатом магния и летучие компоненты удаляются в ротационном выпарном аппарате. 2.91 г (около 7.33 ммоль) полученного таким образом исходного продукта 2-цианоэтил-(2Z)-2-[(2-метил-4-оксо-4H-хромен-8-ил)метилиден]-3-оксобутаноата разбавляются 0.990 г (9.00 ммоль) 4-амино-5-метилпиридин-2(1H)-она [Bisagni, E., Hung, N.C., Synthesis, 765-766 (1984)], соединяются с 40 мл 2-пропанола и смесь мешается в течение ночи при температуре рефлюкса. После охлаждения выпавший осадок отфильтровывается и промывается небольшим количеством диэтилового эфира.
После сушки в глубоком вакууме получается 1.20 г (38% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 8): Rt=1.00 мин; MS (EIpos): m/z=432 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=2.06 (s, 3Н), 2.34 (s, 3H), 2.44 (s, 3H), 2.77 (m, 2H), 3.98-4.14 (m, 2H), 5.76 (s, 1H), 6.15 (s, 1H), 6.98 (s, 1H), 7.28 (t, 1H), 7.71 (dd, 1H), 7.78 (dd, 1H), 8.29 (s, 1H), 10.78 (br. s, 1H).
Пример 40А
2-Цианоэтил 5-этокси-2,8-диметил-4-(2-метил-4-оксо-4-хромен-8-ил)-1,4-дигидро-1,6-нафтиридин-3-карбоксилат
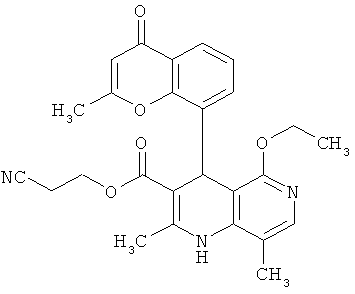
1.20 г (2.78 ммоль) соединения из примера 39А и 9.25 мл (55.6 ммоль) диэтилового эфира ортомуравьиной кислоты вводятся в 30 мл сухого диметилформамида, смесь нагревается до 130°С и разбавляется 5-ю каплями концентрированной серной кислоты. Спустя 2 часа HPLC-контроль показывает полное превращение компонентов. После охлаждения летучие компоненты удаляются в ротационном выпарном аппарате и исходный продукт очищается посредством жидкостной хроматографии среднего давления (Biotage-патрон 40 М, элюент: изогексан/ этилацетат 1:2). Получают 640 мг (50% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 9): Rt=0.99 мин; MS (EIpos): m/z=460 [M+H]+
1Н-ЯМР (300 MГц, DMSO-d6): δ=1.01 (t, 3Н), 2.19 (s, 3H), 2.38 (s, 3H), 2.48 (s,
3Н), 2.75 (m, 2H), 3.93-4.14 (m, 4H), 5.55 (s, 1H), 6.18 (s, 1H), 7.30 (t, 1H), 7.63 (s, 1H), 7.67 (dd, 1H), 7.79 (dd, 1H), 8.49 (s, 1H).
Пример 41 A
5-Этокси-2,8-диметил-4-(2-метил-4-оксо-4Н-хромен-8-ил)-1,4-дигидро-1,6-нафтиридин-3-карбоновая кислота
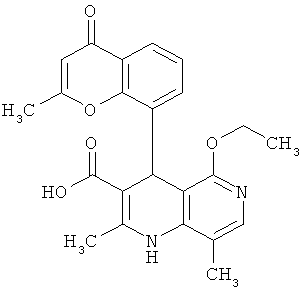
640 мг (1.39 ммоль) соединения из примера 40А растворяются в смеси из 30 мл 1,2-диметоксиэтана и воды (2:1 отношение объемов), разбавляются 2.76 мл (2.76 ммоль) 1 N раствора едкого натра и мешаются 30 минут при комнатной температуре. Затем реакционная смесь разбавляется 20 мл диэтиловым эфиром. Водная фаза отделяется, подкисляется 1 N соляной кислотой до рН 4-5 и трижды экстрагируется с помощью этилового эфира уксусной кислоты. Органические фазы объединяются и высушиваются сульфатом магния. Летучие компоненты отгоняются в ротационном выпарном аппарате. После сушки в вакууме получают 335 мг (56% теор. возм.) выведенного в заголовке соединения с чистотой 94% (LC-MS).
LC-MS (метод 8): Rt=1.21 мин; MS (EIpos): m/z=407 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.01 (t, 3H), 2.18 (s, 3H), 2.35 (s, 3H), 2.42 (s, 3H), 3.90-4.10 (m, 2H), 5.54 (s, 1H), 6.18 (s, 1H), 7.31 (t, 1H), 7.58 (dd, 1H), 7.60 (s, 1H), 7.78 (dd, 1H), 8.25 (s, 1H), 11.52 (br. s, 1H).
Примеры исполнения:
Пример 1
4-(4-Циано-2-метоксифенил)-5-этокси-2-метил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид
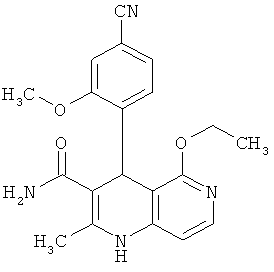
100 мг (около 0.24 ммоль) соединения из примера 23А вводятся в 3 мл диметилформамида. Затем смешиваются с 2.94 мг (0.024 ммоль) 4-N,N-диметиламинопиридина и 340 мкл аммиака (28 мас.%-ный раствор в воде, 2.41 ммоль) и смесь выдерживается 3 часа при 100°С. После охлаждения исходный продукт очищается непосредственно препаративной жидкостной хроматографией высокого давления (элюент: ацетонитрил/вода с 0.1% муравьиной кислоты, градиент 20:80 → 95:5). Получается 32 мг (37% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 3): Rt=1.57 мин; MS (EIpos): m/z=365 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.07 (t, 3H), 2.13 (s, 3H), 3.83 (s, 3H), 4.04 (m, 2H), 5.36 (s, 1H), 6.42 (d, 1H), 6.66 (br. s, 2H), 7.18 (d, 1H), 7.29 (dd, 1H), 7.38 (d, 1H), 7.67 (d, 1H), 8.80 (s, 1H).
Пример 2
4-(4-Циано-2-метоксифенил)-5-этокси-2,7-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид
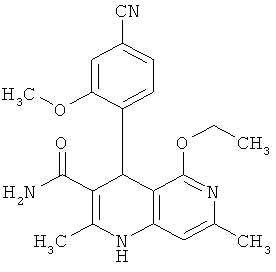
640 мг (1.69 ммоль) соединения из примера 27А вводятся в 30 мл этилового эфира уксусной кислоты, смешиваются с 342 мг (2.11 ммоль) 1,1′-карбонилдиимидазола, затем смесь мешается в течение ночи при комнатной температуре. Контроль тонкослойной хроматографией (силикагель; растворитель: циклогексан/этиловый эфир уксусной кислоты 1:1 или дихлорметан/метанол 9:1) показывает полное превращение компонентов. Летучие компоненты отгоняются в ротационном выпарном аппарате, а осадок пропитывается 20 мл диметилформамида. Затем добавляются 2.36 мл аммиака (28 мас.%-ный раствор в воде, 16.87 ммоль) и реакционная смесь выдерживается 8 часов при 50°С. Растворитель отгоняется при пониженном давлении, а осадок очищается посредством препаративной жидкостной хроматографии высокого давления (элюент: ацетонитрил/вода с 0.1% муравьиной кислоты, градиент 20:80→95:5). Получают 368 мг (58% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 7): Rt=1.91 мин; MS (EIpos): m/z=379 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.05 (t, 3H), 2.13 (s, 3H), 2.19 (s, 3H), 3.84 (s, 3H), 4.02 (q, 2H), 5.32 (s, 1H), 6.25 (s, 1H), 6.62 (br. s, 2H), 7.16 (d, 1H), 7.28 (dd, 1H), 7.37 (d, 1H), 8.71 (s, 1H).
Пример 3
энт-4-(4-Циано-2-метоксифенил)-5-этокси-2,7-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид [(-)-энантиомер и (+)-энантиомер]
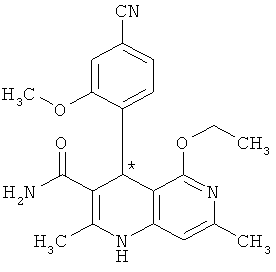
Рацемическое соединение из примера 2 может быть разделено в препаративном масштабе с помощью HPLC в хиральной фазе на его энантиомеры [колонка: Chiralpak IA, 250 мм × 20 мм; элюент: метил-трет-бутиловый эфир/метанол 85:15 (отношение объемов); поток: 15 мл/мин; температура: 30°С; УФ-индикация: 220 нм].
(-)-энантиомер:
HPLC: Rt=5.28 мин, ее>98% [колонка: Chiratpak IA, 250 мм × 4.6 мм; элюент: метил-трет-бутиловый эфир/метанол 80:20 (отношение объемов); поток: 1 мл/мин; температура: 25°С; УФ-индикация: 220 нм];
значение удельного вращения (хлороформ, 589 нм, 19.8°С, с=0.50500 г/100 мл): - 239.3°.
Рентгеноструктурный анализ на монокристалле дает для этого энантиомера S-конфигурацию на С*-атоме.
(+)-энантиомер:
HPLC: Rt=4.50 мин, ее>99% [колонка: Chiralpak IA, 250 мм × 4.6 мм; элюент: метил-трет-бутиловый эфир/метанол 80:20 (отношение объемов); поток: 1 мл/мин; температура: 25°С; УФ-индикация: 220 нм];
значение удельного вращения (хлороформ, 589 нм, 20°С, с=0.51000 г/100 мл):+222.7°.
Пример 4
4-(4-Циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид
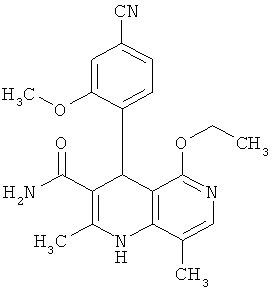
1.46 г (3.84 ммоль) соединения из примера 30А вводится в 50 мл этилового эфира уксусной кислоты, смешиваются с 777 мг (4.79 ммоль) 1,1'-карбонилдиимидазола и смесь перемешивается в течение ночи при комнатной температуре. DC-контроль (силикагель; растворитель: этиловый эфир уксусной кислоты) показывает полное превращение компонентов. Летучие компоненты отгоняются в ротационном выпарном аппарате, а осадок пропитывается 20 мл диметилформамида. Затем добавляются 10.74 мл аммиака (28 мас.%-ный раствор в воде, 76.8 ммоль) и реакционная смесь выдерживается 30 минут при 100°С. Растворитель отгоняется при пониженном давлении, а осадок очищается посредством препаративной HPLC (элюент: ацетонитрил/вода с 0.1% муравьиной кислоты, градиент 20:80→95:5). После сгущения фракций продукта осадок растворяется в 40 мл смеси из дихлорметана и метанола (1:1 отношение объемов) и разбавляется 100 мл этилового эфира уксусной кислоты. Растворитель концентрируется до объема порядка 20 мл, причем продукт кристаллизуется. Преципитат отфильтровывается и промывается небольшим количеством диэтилового эфира. После сушки при 40°С в вакуумном сушильном шкафу получают 1.40 г (96% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 3): Rt=1.64 мин; MS (EIpos): m/z=379 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.05 (t, 3H), 2.12 (s, 3H), 2.18 (s, 3H), 3.82 (s, 3H), 3.99-4.07 (m, 2H), 5.37 (s, 1H), 6.60-6.84 (m, 2H), 7.14 (d, 1H), 7.28 (dd, 1H), 7.37 (d, 1H), 7.55 (s, 1H), 7.69 (s, 1H).
Пример 5
энт-4-(4-Циано-2-метоксифенил)-5-этокси-2,8-диметил-1,4-дигидро-1,6-нафтиридин-3-карбоксамид [(-)-энантиомер и (+)-энантиомер]
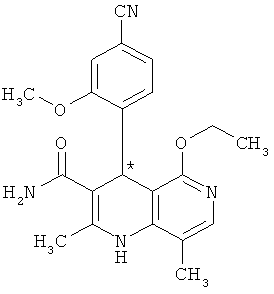
Рацемическое соединение из примера 4 может быть разделено в препаративном масштабе с помощью HPLC в хиральной фазе на его энантиомеры [колонка: 680 мм × 40 мм; фаза с силикагелем, базирующаяся на хиральном селекторе поли(N-метакрилоил-D-лейцин-дициклопропилметиламид; элюент: этиловый эфир уксусной кислоты; температура: 24°С; поток: 80 мл/мин; УФ-индикация: 260 нм].
(-)-энантиомер:
HPLC: Rt=2.48 мин, ee=99.6% [колонка: 250 мм × 4.6 мм; фаза с силикагелем, базирующаяся на хиральном селекторе поли(N-метакрилоил-D-лейцин-дициклопропилметиламид; элюент: этиловый эфир уксусной кислоты; температура: 24°С; поток: 2 мл/мин; УФ-индикация: 260 нм];
значение удельного вращения (хлороформ, 589 нм, 19.7°С, c=0.38600 г/100 мл): - 148.8°.
Рентгеноструктурный анализ на монокристалле дает для этого энантиомера S-конфигурацию на С*-атоме.
(+)-энантиомер:
HPLC: Rt=4.04 мин, ee=99.3% [колонка: 250 мм × 4.6 мм; фаза с силикагелем, базирующаяся на хиральном селекторе поли(N-метакрилоил-D-лейцин-дициклопропилметиламид; элюент: этиловый эфир уксусной кислоты; температура: 24°С; поток: 2 мл/мин; УФ-индикация: 260 нм];
значение удельного вращения (хлороформ, 589 нм, 19.8°С, с=0.36300 г/100 мл): +153.0°.
Пример 6
5-Этокси-2-метил-4-(2-метил-4-оксо-4H-хромен-8-ил)-1,4-дигидро-1,6-нафтиридин-3-карбоксамид
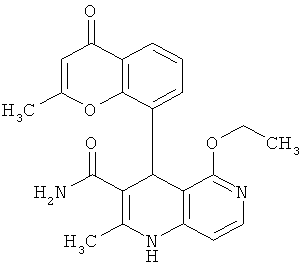
155 мг (0.395 ммоль) соединения из примера 31А вводятся в 10 мл ТГФ, смешиваются с 80.1 мг (0.494 ммоль) 1,1'-карбонилдиимидазола и смесь мешается в течение ночи при комнатной температуре. DC-контроль (силикагель; растворитель: этиловый эфир уксусной кислоты или дихлорметан/метанол 9:1) показывает полное превращение компонентов. Летучие компоненты отгоняются в ротационном выпарном аппарате, а осадок пропитывается 3 мл диметилформамидом. Затем добавляются 553 мг аммиака (28 мас.%-ный раствор в воде, 3.95 ммоль) и реакционная смесь выдерживается 10 минут при 100°С. Растворитель отгоняется при пониженном давлении, а осадок очищается посредством препаративной HPLC (элюент: ацетонитрил/вода с 0.1% муравьиной кислоты, градиент 20:80→95:5). Получают 30 мг (19% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 6): Rt=1.17 мин; MS (EIpos): m/z=392 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=0.96 (t, 3H), 2.09 (s, 3H), 2.36 (s, 3H), 3.94 (m, 1H), 4.03 (m, 1H), 5.59 (s, 1H), 6.19 (s, 1H), 6.42 (d, 1H), 6.66 (br. s, 1H), 7.00 (br. s, 1H), 7.31 (t, 1H), 7.53 (dd, 1H), 7.68 (d, 1H), 7.79 (dd, 1H), 8.83 (s, 1H).
Пример 7
4-[4-Циано-2-(трифторметокси)фенил]-5-этокси-2-метил-1,4-дигидро-1,6-нафтиридин-3 -карбоксамид
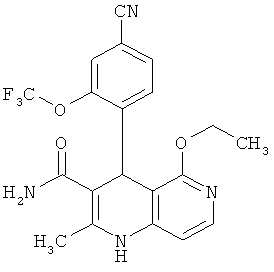
200 мг (0.477 ммоль) соединения из примера 37А вводятся в 5 мл этилового эфира уксусной кислоты, смешиваются с 96.7 мг (0.596 ммоль) 1,1′-кар6онилдиимидазола и затем смесь перемешивается в течение ночи при комнатной температуре (DC-контроль: недостаточное превращение). После этого добавляется 1 мл диметилформамида и смесь мешается еще одну ночь при комнатной температуре (DC-контроль: полное превращение). Летучие компоненты отгоняются в ротационном выпарном аппарате, а осадок пропитывается 4 мл диметилформамида. Затем добавляются 663 мкл аммиака (28 мас.%-ный раствор в воде, 4.77 ммоль) и реакционная смесь выдерживается в закрытом сосуде при 100°С в течение ночи. После охлаждения растворитель удаляется при пониженном давлении, а осадок очищается посредством препаративной HPLC (элюент: ацетонитрил/вода с 0.1% муравьиной кислоты, градиент 20:80→95:5). Получают 140 мг (70% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 6): Rt=2.26 мин; MS (EIpos): m/z=419 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.04 (t, 3Н), 2.04 (s, 3H), 4.06 (m, 2H), 5.42 (s, 1H), 6.41 (d, 1H), 6.80 (br. s, 1H), 6.97 (br. s, 1H), 7.45 (d, 1H), 7.68-7.74 (m, 3H), 8.82 (d, 1H).
Пример 8
4-(4-Циано-2-метоксифенил)-5-этокси-2-(трифторметил)-1,4-дигидро-1,6-нафтиридин-3-карбоксамид
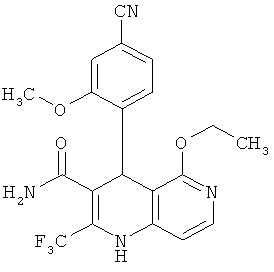
180 мг (0.429 ммоль) соединения из примера 38А вводятся в 5 мл этилового эфира уксусной кислоты, смешиваются с 87.0 мг (0.537 ммоль) 1,1′-карбонилдиимидазола и затем два часа мешаются при комнатной температуре. DC-контролем устанавливается полное превращение. Летучие компоненты отгоняются в ротационном выпарном аппарате, а осадок пропитывается 4 мл диметилформамида. Потом добавляются 597 мкл аммиака (28 мас.%-ный раствор в воде, 4.29 ммоль) и реакционная смесь три часа выдерживается в закрытом сосуде при 100°С. После охлаждения растворитель удаляется при пониженном давлении, а осадок очищается посредством препаративной HPLC (элюент: ацетонитрил/вода с 0.1% муравьиной кислоты, градиент 20:80→95:5). Получают 10 мг (5% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 3): Rt=1.85 мин; MS (EIpos): m/z=419 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=1.03 (t, 3H), 3.79 (s, 3H), 3.96-4.11 (m, 2H), 5.37 (s, 1H), 6.62 (d, 1H), 7.08-7.14 (m, 2H), 7.32 (dd, 1H), 7.37-7.46 (m, 2H), 7.73 (d, 1H), 9.18 (s, 1H).
Пример 9
5-Этокси-2,8-диметил-4-(2-метил-4-оксо-4H-хромен-8-ил)-1,4-дигидро-1,6-нафтиридин-3-карбоксамид
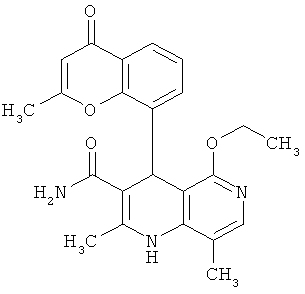
335.0 мг (0.824 ммоль) соединения из примера 41А вводятся в 10 мл этилового эфира уксусной кислоты и смешиваются с 167.1 мг (0.537 ммоль) 1,1′-карбонилдиимидазола. Затем суспензия перемешивается в течение ночи при комнатной температуре. Поскольку нет четкого образования раствора, добавляются 2 мл диметилформамида и смесь перемешивается еще два часа при комнатной температуре. После этого DC-контролем устанавливается полное превращение компонентов. Летучие компоненты отгоняются в ротационном выпарном аппарате, а осадок пропитывается 4 мл диметилформамида. Затем добавляются 2.293 мл аммиака (28 мас.%-ный раствор в воде, 16.5 ммоль) и 10.1 мг 4-N,N-диметиламинопиридина. Реакционная смесь выдерживается тридцать минут в закрытом сосуде при 100°С. После охлаждения растворитель удаляется при пониженном давлении, а осадок очищается посредством препаративной HPLC (элюент: ацетонитрил/вода с 0.1% муравьиной кислоты, градиент 20:80→95:5). Фракции продукта сгущаются, осадок пропитывается небольшим количеством дихлорметана и разбавляется диизопропиловым эфиром до помутнения. Оседающее твердое вещество выделяется и высушивается в вакууме. Получают 207 мг (59% теор. возм.) выведенного в заголовке соединения.
LC-MS (метод 9): Rt=0.67 мин; MS (EIpos): m/z=406 [M+H]+
1Н-ЯМР (300 МГц, DMSO-d6): δ=0.94 (t, 3H), 2.13 (s, 3H), 2.14 (s, 3H), 2.34 (s, 3H), 3.90 (m, 1H), 4.00 (m, 1H), 5.58 (s, 1H), 6.18 (s, 1H), 6.70 (br. s, 1H), 7.06 (br. s, 1H), 7.30 (t, 1H), 7.50 (dd, 1H), 7.56 (s, 1H), 7.71 (s, 1H), 7.78 (dd, 1H).
В. Оценка фармакологической эффективности
Преимущественные фармакологические свойства соединений согласно изобретению могут быть показаны следующими испытаниями:
1. Клеточное испытание in vitro для определения ингибиторной МР-активности и МР-селективности по сравнению с другими рецепторами стероидных гормонов
Идентификация антагонистов человеческого минералокортикоидного рецептора (МР) и количественное выражение эффективности описанных здесь соединений осуществляется с помощью рекомбинантной клеточной линии. Клетка изначально происходит от яичниковой эпителиальной клетки хомяка (Chinese Hamster Ovary, СНО K1, ATCC: American Type Culture Collection, VA 20108, USA).
В этой клеточной линии применяется химерная система клеточной линии СНО K1, в которой лиганд-связывающие домены человеческих рецепторов стероидных гормонов объединены с ДНК-связывающими доменами фактора транскрипции дрожжей GAL4. Образующиеся таким образом GAL4 химеры рецепторов стероидных гормонов перемещаются в СНО-клетки с репортерной структурой и стабильно копируются.
Клонирование:
Для генерирования GAL4 химер рецептора стероидных гормонов клонируется CAL4-ДНК-связующий домен (аминокислоты 1-147) из вектора pFC2-dbd (фирма Stratagene) с PCR-расширенными лиганд-связующими доменами минералокортикоидного рецептора (МР, аминокислоты 734-985), рецептора глюкокортикоида (ГР, аминокислоты 443-777), рецептора прогестерона (ПР, аминокислоты 680-933) и рецептора андрогена (АР, аминокислоты 667-919) в вектор pIRES2 (фирма Ctontech). Репортерная структура, которая содержит пять копий мест привязки GAL4, предвключенных перед промотором тимидинкиназы, приводит к экспрессии люциферазы светляков (Photinus pyralis) после активирования и сцепления GAL4 химер рецептора стероидных гормонов соответствующими специфическими агонистами альдостерона (МР), дексаметазона (ГР), прогестерона (ПР) и дигидротестостерона (АР).
Ход испытания:
Клетки МР, ГР, ПР и АР в день перед тестированием помещаются в среду (Optimem, 2,5% FGS, 2 мМ глутамина, 10 мМ HEFES) в микротитры с 96 (или 384 или 1536) отверстиями и выдерживаются в клеточном инкубаторе (влажность воздуха 96%, объемное отношение CO2 5%, 37°С). В день тестирования исследуемые субстанции вводятся в описанную выше среду и добавляются к клеткам. Через 10-30 минут после добавления исследуемых субстанций прибавляются соответствующие специфические агонисты рецепторов стероидных гормонов. После инкубационного периода в 5-6 часов с помощью видеокамеры измеряется люциферазная активность. Измеренные относительные световые единицы дают сигмоидную кривую стимуляции. Расчет IC50-значений осуществляется с помощью компьютерной программы GraphPad PRISM (версия 3.02).
В таблице А показаны IC50-значения (МР) представленных в примерах соединений:
2. Испытание in vitro для определения возможной активации связывания в кальциевом канале L-типа
Срезы церебральной коры головного мозга крыс Уистара служат в качестве исходного материала для радиоактивного тестирования связывания, которое подробно описано в литературе как стандартный метод испытания [Ehlert, F.J., Roeske, W.R., Itoga E., Yamamura, H.I., Life Sci. 30, 2191-2202 (1982); Gould, R.J., Murphy, K.M.M., Snyder, S.H., Proc. Natl. Acad. Sci. U.S.А 79, 3656-3660] и применяется коммерческими провайдерами (например, фирмой MDS Pharma Services) в рамках договорных исследований. В этом тестировании связывания ряд разведений исследуемых соединений в диметилсульфоксиде выдерживаются обычно 90 минут при 25°С в 50 мМ буферном растворе трис HCl, рН 7,7, со срезами и маркированными тритием лигандами нитрендипира (0,1 нМ) и специфическое связывание исследуемых соединений определяется количественной оценкой специфически вытесненных радиоактивно маркированных лиганд. Значения IC50 получаются с помощью нелинейного регрессионного анализа.
В этом тестировании связывания в кальциевых каналах L-типа для классических антагонистов кальция типа дигидропиридин, как, например, нитрендипин, определяется значение IC50, равное 0,3 нМ, в то время как для исследованных примеров описанных здесь соединений согласно изобретению получены значения IC50 больше 1 µМ, что, по крайней мере, показывает уменьшенное в 3000 раз сродство к кальциевому каналу L-типа. Соединения с таким меньшим остаточным сродством связывания в кальциевом канале L-типа не демонстрируют in vivo никаких выраженных гемодинамических эффектов, которые передаются через кальциевый канал L-типа.
3. Испытание in vivo для доказательства кардиоваскулярного действия: исследования диуреза на бодрствующих крысах в обменных клетках
Крысы Уистара (вес тела 250-350 г) содержатся со свободным доступом к пище (альтромин) и питьевой воде. За 72 часа до начала испытания животные вместо нормальной пищи получают пищу с пониженным содержанием поваренной соли, только 0,02% хлорида натрия, (ssniff R/M-H, 10 мм с 0,02% Na, S0602-E081, фирма. ssniff Spezialdiäten GmbH, D-59494 Soest). Во время испытания животные на 24 часа помещаются в обменные клетки, специально приспособленные для крыс этой весовой категории (фирма Tecniplast Deutschland GmbH, D-82383 Hohenpeiβenberg) со свободным доступом к пище с пониженным содержанием соли и питьевой воде. К началу испытания животным через желудочный зонд в желудок вводится исследуемое вещество объемом 0,5 мл/кг веса тела с подходящим растворителем. Контрольные животные получают только растворитель. Контроль и проверка вещества выполняются параллельно в один и тот же день. Контрольные группы и группы, получившие дозу вещества, состоят, соответственно, из 3-6 животных. Во время испытания выделяемая животными моча собирается в приемник на дне клетки. Для каждого животного определяется объем мочи в единицу времени и концентрация выделенных в моче ионов натрия и калия с помощью стандартных методов пламенной фотометрии. По измеренным величинам рассчитывается показатель натрий/калий как мера действия исследуемого вещества. Интервалы измерений составляют обычно до 8 часов после начала испытания (дневной интервал) и временной промежуток от 8 до 24 часов после начала испытания (ночной интервал). При дневном интервале моча собирается с промежутком в два часа. Чтобы при этом получить достаточное количество мочи, животным к началу испытания и впоследствии с промежутком в два часа через желудочный зонд вводится определенное количество воды.
4. Модель ДОКА/соль
Введение дезоксикортикостеронацетата (ДОКА) в комбинации с диетой с повышенным содержанием соли и односторонним удалением почки вызывает у крыс гипертонус, который характеризуется относительно низким уровнем ренина. Эта эндокринная гипертония (ДОКА представляет непосредственную предстадию альдостерона) приводит в зависимости от выбранной концентрации ДОКА к гипертрофии сердца и последующим повреждениям конечных органов, например почек, которые характеризуются протеинурией и гломерулосклерозом. Такая модель с крысами позволяет исследовать тестируемые вещества на наличие антигипертрофического и защищающего конечные органы действия.
Мужские особи крыс Sprague Dawley (SD) в возрасте 8 недель (вес тела между 250 и 350 граммами) подвергаются унинефрэктомии. Для этого крысы анестезируются 1,5-2%-ным изофлураном в смеси из 66% N2O и 33% O2 и почка удаляется через боковой разрез. Контрольными животными служат так называемые фиктивно прооперированные особи, у которых почка не удаляется.
SD-крысы, подвергшиеся унинефрэктомии, получают 1% хлорида натрия в питьевой воде и один раз в неделю им делается подкожная инъекция дезоксикортикостеронацетата (растворенного в кунжутном масле; фирма Sigma) между лопатками (большая доза: 100 мг/кг в неделю s.c.; нормальная доза 30 мг/кг в неделю s.c.). За день до испытания животные в произвольном порядке разделяются на группы, как правило, по 10 особей. Во время всего испытания животные получают корм и питьевую воду по желанию (ad libitum). Вещества даются один раз в день на протяжении 4-8 недель как добавка к корму или путем насильственного кормления. К контрольной группе относятся те животные, которые содержатся в тех же условиях, но получают либо только растворитель, либо корм без исследуемого вещества.
Действие исследуемых веществ определяется путем измерения гемодинамических параметров [кровяное давление, частота сердечных сокращений, инотропия (dp/dt), время релаксации (тау), максимальное давление левого желудочка, давление в конце диастолы левого желудочка (ЛЖКДД)], определения веса сердца, почки и легких, измерения выделений протеина, а также путем измерения экспрессии генов биомаркеров (например, ANP, Atrial Natriuretic Peptide, и BNP, Brain Natriuretic Peptide) посредством RT/TaqMan-PCR после выделения РНК из сердечной ткани. Статистическая оценка осуществляется тестированием распределения Стьюдента (t-распределения) после предварительной проверки отклонений на однородность.
5. Испытание in vivo для доказательства антиминералокортикоидного действия на наркотизированных собаках
Собакам мужского или женского рода (Mongrels, Marshall BioResources, США) весом от 20 до 30 килограмм дается наркоз (30 мг/кг внутривенно, Narcoren®, Merial, Германия). При этом хлорид алькурониума (3 мг/животное внутривенно, Alloferin®, ICN Pharmaceuticals, Германия) предназначен для дополнительной миорелаксации. Собаки интубируются и вводится смесь кислорода и окружающего воздуха (40/60 об.%, приблизительно 5-6 литров в минуту). Вентиляция легких осуществляется аппаратом для искусственного дыхания фирмы Draeger (Sulla 808) и контролируется анализатором CO2 (фирмы Engström). Наркоз поддерживается постоянной инфузией пентобарбитала (50 мкг/кг/мин) или изофлурана (1-2 об.%). Аналгетиком служит фентанил (10 мкг/кг/час).
Первичной целью опыта является исследование влияния тестируемых соединений, действующих против минералокортикоидных рецепторов, на ретенцию натрия, индуцированную альдостероном. При этом действуют по аналогии с опубликованным методом [Н.Р.Ramjoe, U.M.Bucher, J.Richter и M. De Gasparo, Anti-mineralocorticoid activity of three novel aldosterone antagonists in the conscious dog and in man, в: Diuretics II: Chemistry, Pharmacology, and Clinical Applications, J.B.Puschett und A.Greenberg (Ed.), Elsevier Science Publishing Co., Inc., 1987]. Постоянная инфузия альдостерона через 3 часа приводит к уменьшению отношения натрия и калия в моче (определение натрия и калия осуществляется пламенным фотометром). При продолжающейся инфузии альдостерона вводится тестируемое вещество внутривенно, интрадуоденально или орально. Позитивным контролем служит спиронолактон, который в зависимости от дозы повышает отношение натрия и калия в моче.
Для обеспечения постоянной гемодинамики и для учета функциональных параметров сердечно-сосудистой системы контролируется гемодинамика собаки, и она обрабатывается следующим образом:
- ввод катетера в мочевой пузырь для измерения потока мочи и ее состава;
- прикрепление ЭКГ-проводов к конечностям для ЭКГ-измерения;
- ввод мягкой трубки Fluidmedic PE 300, наполненной раствором поваренной соли, в A.femoralis, которая подсоединена к датчику давления (фирма Braun Mclsungen, Германия) для измерения систематического кровяного давления;
- ввод катетера типа Millar (тип 350 PC, Millar Instruments, Хьюстон, США) через левое предсердие или через затвор, вделанный в предсердие (A.carotis) для измерения гемодинамики сердца;
- ввод катетера Swan-Ganz (CCOmbo 7.5F, Edwards, Irvine, США) через яремную вену (V.jugularis) в легочную артерию (A.pulmonalis) для измерения минутного объема сердца, насыщения кислородом, давления в легочной артерии и давления в центральной вене;
- прикрепление ультразвукового зонда флюксметра (Transsonic Systems, Ithaka, США) к A.descendens для измерения потока через аорту;
- прикрепление ультразвукового зонда флюксметра (Transsonic Systems, Ithaka, США) к левой коронарной артерии сердца (A.coronaria) для измерения коронарного потока;
- ввод бранулы в мозг (V.cephalicae) для инфузии пентобарбитала, замещения жидкости и для забора крови (определение уровня вещества в плазме или других клинических показателей крови);
- ввод бранулы в подкожную вену бедра (V.saphenae) для инфузии фентанила, альдостерона и для введения вещества. Первичные сигналы, при необходимости, усиливаются (усилитель Gould, Gould Instrument Systems, Valley View, США и монитор Edwards Vigilance, Edwards, Irvine, США) и затем заносятся в память в систему Ponemah (DataSciences Inc., Minneapolis, США). Сигналы постоянно в течение всего времени испытания регистрируются, обрабатываются программой в цифровом формате и усредняются через 30 секунд.
6. Модель хронического инфаркта миокарда бодрствующей крысы
Мужские особи крыс Уистар (вес тела 280-300 г, Harlan-Winkelmann) вводятся в наркоз 5%-ным изофураном в клетках для наркоза, интубируются, подсоединяются к насосу для ингаляции (ugo basile 7025 rodent, 50 тактов/мин, 7 мл) и вводится 2%-ный изофуран/N2O/O2. Температура тела поддерживается подогреваемыми матами на уровне 37-38°С. В качестве аналгетика подкожно вводится темгезик (0,05 мг/кг). Грудная клетка вскрывается сбоку между третьим и четвертым ребром и обнажается сердце. Венечная артерия левого желудочка сердца (LAD) с помощью окклюзионной нити (Prolene 1 metric 5-0 Ethicon1H) помещается ниже его начала (ниже левого предсердия) и перевязывается. Наступление инфаркта миокарда контролируется измерением ЭКГ (Cardioline, Remco, Италия). Грудная клетка снова закрывается и мышечный слой зашивается нитью Ethibond excel 1 metric 5/0 6951H, а верхний слой кожи нитью Ethibond excel 3/0 6558Н. Операционный шов увлажняется аэрозольным пластырем (например, Nebacetin®N Sprühverband, биологически активное вещество: неомицинсульфат) и наркоз завершается.
Через одну неделю после закупорки LAD оценивается размер инфаркта миокарда эхокардиографией (Sequoia 512, Acuson). Животные в произвольном порядке разбиваются на лечебные группы и выделяется контрольная группа, не получающая лечения веществом. В качестве дополнительного контроля ведется группа «симулянтов», которым была только сделана операция, но не была выполнена закупорка LAD.
Лечение веществом осуществляется в течение 8 недель насильственным кормлением или добавкой вещества в корм или в питьевую воду. Животные еженедельно взвешиваются, и каждые 14 дней определяется потребление воды и корма.
После 8-недельного лечения животным снова дается наркоз (2% изофлуран/N2O/воздух) и вводится катетер (Millar SPR-320 2F) через сонную артерию (A. carotis) в левый желудочек сердца. Регистрируются и оцениваются частота сердечных сокращений, давление в левом желудочке (ДЛЖ), давление в конце диастолы левого желудочка (ДКДЛЖ), сократимость (dp/dt) и скорость релаксации (τ) с помощью системы Powerlab (AD Instruments, ADI-PWLB-4SP) и программного обеспечения Chart-5 (SN 425-0586). Затем берется проба крови для определения уровня вещества в плазме и биомаркер плазмы, и животные умертвляются. Сердце (желудочки сердца, левый желудочек с перегородкой, правый желудочек), печень, легкие и почка извлекаются и взвешиваются.
7. Модель крысы, предрасположенной к удару при спонтанном повышении давления
Введение поваренной соли у так называемой предрасположенной к удару при спонтанном повышении давления крысы (СУ-СПДК) парадоксальным образом через несколько дней приводит у этой модели к прекращению физиологического подавления высвобождения ренина и ангиотензина, вызванного солью. Таким образом, гипертония у СУ-СПДК-животных отличается относительно высоким уровнем ренина. Результатом развивающейся гипертонии являются явно выраженные нарушения конечных органов сердца и почки, которые среди прочего характеризуются протеинурией и гломерулосклерозом, а также общим сосудистым изменением. Так, прежде всего из-за нарушения мозгового кровообращения в последующем течении болезни может развиваться апоплексия («апоплексические удары»), которая приводит к высокой смертности не охваченных лечением животных. На такой модели крысы может быть исследовано действие тестируемого вещества на понижение кровяного давления и защиту конечных органов.
Мужские особи СУ-СПДК-крыс в возрасте 10 недель (вес тела от 190 до 220 г) в день перед испытанием произвольным образом делятся на группы с одинаковым числом животных, как правило, по 10-12 крыс. На протяжении всего испытания животным по желанию предоставляется подсоленная питьевая вода (2% NaCl) и пища. Вещества вводятся ежедневно в течение 6-8 недель насильственным кормлением или путем добавки к корму (Ssniff, Германия). В контрольной группе животные содержатся в таких же условиях, но получают либо только растворитель, либо корм без тестируемого вещества. В рамках исследования смертности опыт прекращается, когда умрут приблизительно 50% животных из контрольной группы. Действие тестируемых веществ отслеживается путем измерений систолического кровяного давления (через манжету на хвосте) и выделения протеина в моче. После смерти определяется вес сердца, почки и легких, а также анализируется гистологическая патология сердца, почки и мозга полуколичественной оценкой гистологических изменений. Различные биомаркеры (например, ANP, Atrial Natriuretic Peptide, и BNP, Brain Natriuretic Peptide, KIM-1, kidney induced molecule 1, Osteopontin-1) определяются с помощью RT/TaqMan-PCR по выделению РНК из сердца и ткани почки или по сыворотке или плазме. Статистическая оценка осуществляется тестированием распределения Стьюдента (t-распределения) после предварительной проверки отклонений на однородность
С. Примеры исполнения фармацевтических составов
Соединения согласно изобретению могут быть переведены в фармацевтические препараты следующим образом:
Таблетки:
Состав:
100 мг соединения согласно изобретению, 50 мг лактозы (моногидрат), 50 мг кукурузного крахмала (природного), 10 мг поливинилпиролидона (PVP 25) (фирма BASF, Людвигсгафен, Германия) и 2 мг стеарата магния. Вес таблетки 212 мг, диаметр 8 мм, радиус выпуклости 12 мм.
Приготовление:
Смесь из соединения согласно изобретению, лактозы и крахмала вместе с 5%-ным раствором PVP в воде (отношение масс) превращается в гранулы. После сушки гранулят 5 минут перемешивается со стеаратом магния. Эта смесь прессуется стандартным прессом для таблетирования (формат таблетки см. выше). Ориентировочное значение усилия прессования составляет 15 кН.
Суспензия для орального применения:
Состав:
1000 мг соединения согласно изобретению, 1000 мг этанола (96%), 400 мг Родигеля® (ксантановая смола фирмы. FMC, Пенсильвания, США) и 99 г воды. Разовой дозе в 100 мг соединения согласно изобретению соответствуют 10 мл оральной суспензии.
Приготовление:
Родигель превращается в этаноле в суспензию, к ней добавляется соединение согласно изобретению. При помешивании добавляется вода. Смесь мешается приблизительно 6 часов до окончательного набухания Родигеля.
Раствор для орального применения:
Состав:
500 мг соединения согласно изобретению, 2.5 г полисорбата и 97 г попиэтиленгликоля 400. Разовой дозе в 100 мг соединения согласно изобретению соответствуют 20 г орального раствора.
Приготовление:
Соединение согласно изобретению при помешивании превращается в суспензию в смеси из полиэтиленгликоля и полисорбата. Процесс помешивания продолжается до полного растворения соединения согласно изобретению.
Раствор для внутривенного вливания:
Соединение согласно изобретению в концентрации, меньшей для растворимости до насыщения, растворяется в физиологически премлемом растворе (например, изотонический раствор поваренной соли, раствор глюкозы 5% и/или раствор PEG-400 30%). Раствор фильтруется в стерильных условиях и разливается в стерильные и апирогенные ампулы для инъекций.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ДИГИДРОПИРАЗОЛОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ HIF-ПРОЛИЛ-4-ГИДРОКСИЛАЗЫ | 2009 |
|
RU2509080C9 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГИДАНТОИНА В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2005 |
|
RU2376301C2 |
| ПРОИЗВОДНЫЕ ПРОПИОНОВЫХ КИСЛОТ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АКТИВАТОРОВ hPPARs. | 2003 |
|
RU2316539C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГИДАНТОИНА В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2005 |
|
RU2378269C2 |
| ПИПЕРИДИНИЛПИРАЗОЛОПИРИМИДИНОНЫ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2713937C2 |
| АНТАГОНИСТЫ TLR7/8 И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2833035C2 |
| НОВЫЕ АЗАХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ | 2018 |
|
RU2773290C2 |
| Бициклические конденсированные гетероарильные или арильные соединения в качестве модуляторов IRAK4 | 2016 |
|
RU2684324C1 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ АМИНОВ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА ЕР4 | 2011 |
|
RU2565596C2 |
| СОЕДИНЕНИЯ, ИНГИБИРУЮЩИЕ МЕТАЛЛОФЕРМЕНТЫ | 2017 |
|
RU2764666C2 |
Настоящее изобретение касается новых замещенных 4-арил-1,4-дигидро-1,6-нафтиридин-3-карбоксамидов, способа их получения, их применения для изготовления лекарственного средства для изготовления лекарственного средства, ингибирующего МР-активность. 4 н. и 7 з.п. ф-лы, 9 пр., 1 табл.
1. Соединение формулы (I)
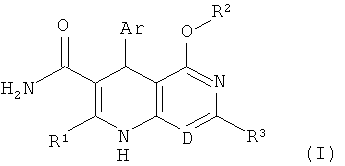
в которой
D означает C-R4, где
R4 означает водород или (С1-С4)-алкил,
Ar означает группу формулы
или
где
* означает место соединения,
R5 означает водород,
R6 означает водород,
R8 означает циано,
R9 означает (С1-С4)-алкокси, который может быть до трех раз замещен фтором,
R10 означает водород,
R1 означает (С1-С4)-алкил, который до трех раз может быть замещен фтором,
R2 означает (С1-С6)-алкил,
R3 означает водород или (С1-С4)-алкил,
а также их фармацевтически приемлемые соли.
2. Соединение формулы (I) по п.1, в которой
D означает C-R4, где
R4 означает водород или метил,
Ar означает группу формулы
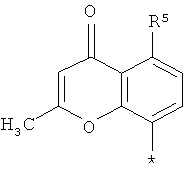 или
или
где
* означает место соединения,
R5 означает водород,
R8 означает циано и
R9 означает (С1-С4)-алкокси или трифторметокси,
R1 означает метил или трифторметил,
R2 означает (С1-С4)-алкил, и
R3 означает водород или метил,
а также их фармацевтически приемлемые соли.
3. Соединение формулы (I) по п.1, в которой
D означает C-R4, где
R4 означает водород или метил,
Ar означает группу формулы
или
где
* означает место соединения и
R9 означает метокси или трифторметокси,
R1 означает метил или трифторметил,
R2 означает метил, этил, н-пропил или изопропил и
R3 означает водород или метил,
а также их фармацевтически приемлемые соли.
4. Соединение по п.1, имеющее одну из следующих структур
и
а также их фармацевтически приемлемые соли.
5. Соединение по одному из пп.1-4, имеющее одну из следующих структур
и
а также их фармацевтически приемлемые соли.
6. Способ получения соединений формулы (I), определенной по пп.1-5,
отличающийся тем, что соединение формулы (II)
в которой Ar имеет значение, указанное по пп.1-5,
в инертном растворителе с соединением формулы (III)
в которой R1 имеет значение, указанное по пп.1-5, и
Т означает аллил или 2-цианоэтил,
превращают в соединение формулы (IV)
в которой Ar, Т и R1 соответственно имеют указанные выше значения, которое затем в инертном растворителе с соединением формулы (V)
в которой D и R3 имеют значения, указанные по пп.1-5, конденсируют в соединение формулы (VI)
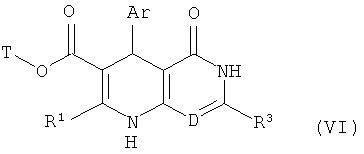
в которой Ar, D, T, R1 и R3 соответственно имеют указанные выше значения,
затем соединения формулы (VI) в инертном растворителе с соединением формулы (VII) или с солью триалкилоксония формулы (VIII)
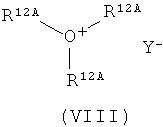
в которых
R12 означает (С1-С6)-алкил, который может быть замещен (С3-С7)-циклоалкилом или до трех раз замещен фтором,
R12A означает метил или этил,
X означает уходящую группу, как например галоген, мезилат, тозилат или
трифлат, и
Y- означает ненуклеофильный анион, как например тетрафтороборат, или в присутствии кислоты с триалкилортоформиатом формулы (IX)
в которой R12A имеет указанное выше значение, алкилируют в соединения формулы (Х-А)
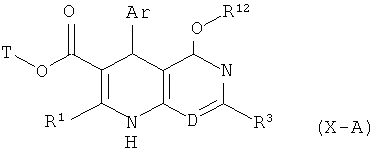
в которой Ar, D, T, R1, R3 и R12 соответственно имеют указанные выше значения,
или соединения формулы (VI) в инертном растворителе в присутствии основания преобразуют с соединением формулы (XI)
в которой R11 имеет указанное по п.1 или 2 значение, в соединение формулы (Х-В)
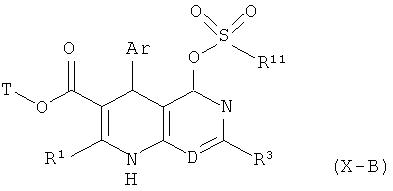
в которой Ar, D, T, R1, R3 и R11 соответственно имеют указанные выше значения,
после этого в соединениях формулы (Х-А) или (Х-В) сложно эфирную группу Т расщепляют на карбоновые кислоты формулы (XII)
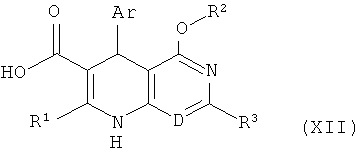
в которой Ar, D, R1, R2 и R3 соответственно имеют значения, указанные по пп.1-5,
затем 1,1′-карбонилдиимидазолом переводят в имидазолиды формулы (XIII)
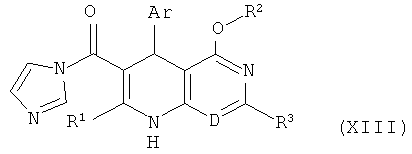
в которой Ar, D, R1, R2 и R3 соответственно имеют указанные выше значения,
и потом имидазолиды в инертном растворителе преобразуют аммиаком в амиды формулы (I) при последующем отделении конечного продукта в свободном виде как соли или в виде энантиомеров и/или диастереомеров.
7. Способ по п.6, отличающийся тем, что реакцию обмена соединения формулы (II) с соединением формулы (III) проводят в присутствии кислоты, комбинации из кислоты и основания и/или дегидратирующего реагента.
8. Способ по п.6, отличающийся тем, что реакцию обмена соединения формулы (VI) с соединением формулы (VII) проводят в присутствии основания.
9. Способ по одному из пп.6-8, отличающийся тем, что реакцию обмена соединения формулы (XIII) с аммиаком проводят в присутствии вспомогательного основания.
10. Применение соединения формулы (I), определенного по одному из пп.1-5, для изготовления лекарственного средства, ингибирующего МР-активность.
11. Фармацевтическая композиция, имеющая свойства селективных к минералокортикоидному рецептору антагонистов, содержащая соединение формулы (I), определенное по одному из пп.1-5, в комбинации с инертным, нетоксичным, фармацевтически приемлемым вспомогательным веществом.
| ЭЛЕКТРОЛЮМИНЕСЦЕНТНЫЙ ЦИФРОВОЙ ИНДИКАТОР | 0 |
|
SU234516A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| ПРОИЗВОДНЫЕ НАФТИРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2002 |
|
RU2296764C2 |

