Перекрестные ссылки на родственные заявки
Настоящее изобретение претендует на приоритет согласно 35 U.S.C. § 119(e), заявленный в предварительной заявке США №62/319,689, поданной 7 апреля 2016 г., содержание которой включено в настоящий текст посредством ссылки во всей своей полноте.
Область техники, к которой относится изобретение
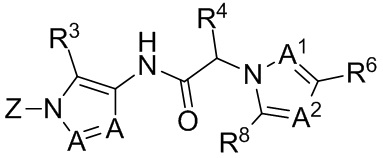
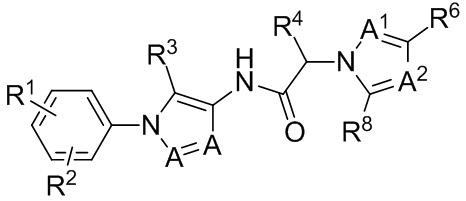
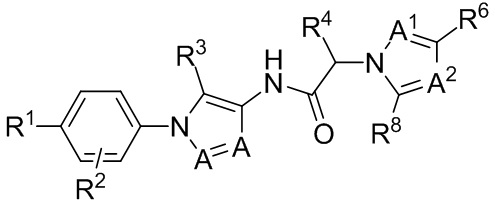
Настоящее изобретение касается способа уменьшения веса опухоли, роста опухоли, прогрессирования опухоли и/или образования метастазов у субъекта, страдающего раковой солидной опухолью, такой как трижды негативный рак груди. Способ включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества PD-L1 ингибитора или PD-1 ингибитора в комбинации с низкомолекулярным антагонистом хемокинового рецептора, который блокирует CCR1.
Уровень техники
Раковые опухоли используют многочисленные механизмы для уклонения от естественной цитотоксической иммунной реакции организма, поэтому иммунная система не справляется с опухолями. Эти механизмы включают нарушение передачи сигналов Т-клеткам, подавление регуляторных клеток и иммунных контрольных точек, которые в норме снижают интенсивность адаптивных иммунных реакций и защищают здоровые ткани от побочных эффектов. Например, у опухолей развивается иммунорезистентность, в частности к Т-клеткам, которые специфичны к опухолевым антигенам, благодаря притоку супрессивных клеток миелоидного происхождения (MDSC) в опухоли и их микроокружение.
MDSC экспрессируют хемокиновые рецепторы, такие как хемокиновый рецептор CCR1, и обладают функцией подавления иммунитета. MDSC играют ключевую роль в способности опухолей подавлять иммунный ответ. Другим ключевым компонентом этого подавления является активация иммунных контрольных точек, которые, в свою очередь, ограничивают активацию Т-клеток и их поступление в опухоли. Иммунные контрольные точки относятся к ингибирующим путям иммунной системы, которые необходимы для сохранения толерантности к «своему» и управления иммунными ответами в периферических тканях для минимизации сопутствующих повреждений в тканях.
PD-1 (Programmed Death-1) представляет собой один из многочисленных рецепторов иммунных контрольных точек, которые экспрессируются активированными Т-клетками и участвуют в подавлении иммунитета. Лиганды для PD-1 включают PD-L1 (Programmed Death Ligand-1) и PD-L2 (Programmed Death Ligand-2), которые экспрессируются на антиген-представляющих клетках, а также на многих человеческих раковых клетках. PD-L1 и PD-L2 могут снижать активацию Т-клеток и выработку цитокинов при связывании с PD-1.
Было показано, что ингибиторы взаимодействия PD-l/PD-Ll могут оказывать сильное противоопухолевое действие и эффективны для лечения некоторых видов рака. Ингибирование CCR1 ассоциируют с уменьшением размера опухоли в мышиной модели миеломы костей (Dairaghi et al., Blood, 2012, 12(7):1449-1457). Сохраняется потребность в эффективном лечении раковых заболеваний, таких как раковые солидные опухоли.
Раскрытие изобретения
В одном аспекте, в настоящем изобретении описан способ лечения субъекта с раковой солидной опухолью. Способ включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества антагониста хемокинового рецептора CCR1 и терапевтически эффективного количества PD-1 ингибитора или PD-L1 ингибитора.
В другом аспекте, в настоящем изобретении описана композиция для лечения субъекта с раковой солидной опухолью. Композиция содержит терапевтически эффективное количество антагониста хемокинового рецептора CCR1 и терапевтически эффективного количества PD-1 ингибитора или PD-L1 ингибитора, и фармацевтически приемлемый носитель или вспомогательное вещество.
В другом аспекте, в настоящем изобретении описан набор для лечения субъекта с раковой солидной опухолью. Набор содержит терапевтически эффективное количество антагониста хемокинового рецептора CCR1 и терапевтически эффективное количество PD-1 ингибитора или PD-L1 ингибитора, с инструкцией по эффективному применению.
Другие объекты, отличительные черты и преимущества настоящего изобретения будут понятны квалифицированному специалисту в данной области из приведенного далее описания и фигур.
Краткое описание фигур
На фиг. 1A-1E показан уровень экспрессии лигандов хемокинового рецептора CCR1 (CCL3, CCL5 и CCL7), PD-L1 и CCR1 в образцах от пациентов с раком груди. Значительно усиленная экспрессия CCL5 (RANTES; фиг. 1A), CCL7 (MCP-3; фиг. 1B), и PD-L1 (фиг. 1D) наблюдается у пациентов с трижды негативным раком груди. Уровень экспрессии CCL3 (MIP-1α) показан на фиг. 1C. На фиг. 1E показано, что экспрессия CCR1 и PD-L1 хорошо коррелирует в образцах от пациентов с раком груди.
На фиг. 2 изображена мышиная модель 4T1 карциномы молочной железы, которую можно применять в качестве мышиной модели трижды негативного рака груди.
Фиг. 3A-3E иллюстрируют, что комбинированная терапия CCR1 антагонистом с анти-PD-L1 антителами ослабляет развитие агрессивных метастатических опухолей (например, опухолей 4T1). Мышам с 4T1 карциномой молочной железы вводили носитель (фиг. 3A), CCR1 антагонист отдельно (фиг. 3B), PD-L1 антитело отдельно (фиг. 3C) или комбинацию CCR1 антагониста и PD-L1 антитела (фиг. 3D). Сравнение финального веса опухоли на фиг. 3E показывает, что комбинированная терапия эффективно минимизирует или уменьшает развитие опухоли.
Фиг. 4A и 4B показывают, что CCR1 антагонист уменьшил метастазы в легких у мышей с опухолью 4T1. Фиг. 4A иллюстрирует, что мыши, которым вводили CCR1 антагонист, имели меньше метастатических узлов на легкое, по сравнению с похожими мышами, которым вводили только носитель. На фиг. 4B даны изображения метастатических узлов в легких.
Фиг. 5A-5C показывает, что гранулоцитные супрессорные клетки миелоидного происхождения (G-MDSC) присутствуют у мышей с опухолью 4T1. Уровень G-MDSC сильно повышен в крови (фиг. 5A) и в селезенке (фиг. 5B) этих мышей. Также наблюдался небольшой рост концентрации моноцитных супрессорных клеток миелоидного происхождения (M-MDSC) в крови и селезенке. Большинство иммунных клеток, проникающих в 4T1 опухоли, представляет собой G-MDSC (фиг. 5C).
Фиг. 6A и 6B показывают, что большинство CCR1-экспрессирующих клеток в селезенке мышей с опухолью 4T1 представляют собой G-MDSC. Результаты анализа методом поточной цитометрии приведены на фиг. 6A. На фиг. 6B показано, что наибольшую долю CCR1-экспрессирующих клеток составляют G-MDSC.
Фиг. 7A и 7B иллюстрируют корреляцию между весом опухоли и соотношением G-MDSC и CD8-положительных T-клеток. На фиг. 7A сравнивается доля G-MDSC в иммунных клетках, проникающих в опухоль, и вес опухоли. На фиг. 7B сравнивается доля CD8+ T-клеток в иммунных клетках, проникающих в опухоль, и вес опухоли.
На фиг. 8A и 8B показано, что комбинированная терапия CCR1 антагонистом и PD-L1 mAb понижает уровень G-MDSC (фиг. 8A), но не уровень M-MDSC (фиг. 8B) в крови мышей с опухолью 4T1.
На фиг. 9A-9D показаны уровни G-MDSC, M-MDSC, CD8 T-клеток и B-клеток, проникающих в 4T1 опухоли после введения носителя, CCR1 антагониста, PD-L1 mAb и после комбинированной терапии. На фиг. 9A показано, что CCR1 антагонист уменьшает проникновение G-MDSC в опухоли 4T1. На фиг. 9B показан уровень проникновения M-MDSC в опухоли 4T1 у мышей, которым вводили препарат. На фиг. 9C показано увеличение проникновения CD8 T-клеток в опухоли 4T1 у мышей, которым вводили CCR1 антагонист в отдельности или в комбинации с PD-L1 mAb. На фиг. 9D показан уровень проникновения В-клеток в опухоли 4T1 у мышей, которым вводили препарат.
На фиг. 10 приведена схематическая диаграмма CCR1-опосредуемого поступления G-MDSC в микроокружение опухоли. На фиг. 10 также показано как данный путь может промотировать прогрессирование опухоли.
Раскрытие изобретения
Введение
В настоящем изобретении описаны способы, композиции и наборы для лечения ракового заболевания, такого как раковая солидная опухоль, у субъекта, нуждающегося в этом, путем применения комбинированной терапии CCR1 антагонистом и PD-1 ингибитором или PD-L1 ингибитором. Изобретение частично основано на синергетическом эффекте CCR1 антагониста в комбинации с PD-1 ингибитором или PD-L1 ингибитором в плане уменьшения веса опухоли, прогрессирования опухоли и/или формирования метастазов. В некоторых случаях, терапия с применением только CCR1 антагониста может уменьшить формирование метастазов, например метастазов в легких.
Определения
[0001] Термины, использующиеся в настоящем тексте в единственном числе, охватывают не только аспекты с одним указанным объектом, но также включают аспекты с более чем одним указанным объектом. Например, единственная форма существительного включает указание на возможность многих представителей, если контекст ясно не противоречит этому. Так, например, термин «клетка» включает допустимость множества клеток, а термин «средство» включает допустимость одного или больше средств, известных квалифицированным специалистам в данной области, и т.д.
Термины “около” и “примерно” в целом означают допустимую степень ошибки для измеряемого количества, с учетом природы и точности измерения. Обычно степень ошибки составляет не более 20 процентов (%), предпочтительно не более 10%, и более предпочтительно – не более 5% от указанного значения или интервала значений. Альтернативно, и особенно в биологических системах, термины “около” и “примерно” могут означать значения, которые находятся в пределах одного порядка, предпочтительно отличаются не более чем в 5 раз, и более предпочтительно – отличаются не более чем в 2 раза от приведенного значения. Числовые значения, приведенные в настоящем тексте, являются приблизительными, если явно не указано иное; и термины “около” и “примерно” могут применяться взаимозаменяемо, если явно не указано иное.
Термин "алкил", сам по себе и как часть другого заместителя, означает, если не указано иное, линейный или разветвленный углеводородный радикал, имеющий обозначенное число атомов углерода (например, C1-8 означает 1-8 атомов углерода). Примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, н-пентил, н-гексил, н-гептил, н-октил и т.п. Термин "алкенил" означает ненасыщенную алкильную группу, содержащую одну или больше двойных связей. Аналогично, термин «алкинил» означает ненасыщенную алкильную группу, содержащую одну или больше тройных связей. Примеры таких ненасыщенных алкильных групп включают винил, 2-пропенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и их высшие гомологи и изомеры. Термин "циклоалкил" относится к углеводородным циклам, имеющим указанное число атомов в цикле (например, C3-6циклоалкил) и являющимся полностью насыщенными или имеющими не более одной двойной связи между вершинами цикла. "Циклоалкил" относится также к бициклическим и полициклическим углеводородным кольцам, таким как, например, бицикло[2.2.1]гептан, бицикло[2.2.2]октан и т.д. Термин "гетероциклоалкан” или “гетероциклоалкил" относится к циклоалкильной группе, содержащей 1-5 гетероатомов, выбранных из N, O, и S, где атомы азота и серы необязательно окислены, и атом(ы) азота необязательно кватернизован(ы). Гетероциклоалкан может представлять собой моноциклическую, бициклическую или полициклическую кольцевую систему. Неограничивающие примеры гетероциклоалкановых групп включают пирролидин, имидазолидин, пиразолидин, бутиролактам, валеролактам, имидазолидинон, гидантоин, диоксолан, фталимид, пиперидин, 1,4-диоксан, морфолин, тиоморфолин, тиоморфолин-S-оксид, тиоморфолин-S,S-оксид, пиперазин, пиран, пиридон, 3-пирролин, тиопиран, пирон, тетрагидрофуран, тетрагидротиофен, хинуклидин и т.п. Гетероциклоалкановая группа может быть присоединена к остальной части молекулы через атом углерода в цикле или гетероатом в цикле.
Термин "алкилен" в отдельности или как часть другого заместителя означает двухвалентный радикал, являющийся производным алкана, в качестве примера можно привести -CH2CH2CH2CH2-. В типичном случае алкильная (или алкиленовая) группа содержит от 1 до 24 атомов углерода, предпочтительными по настоящему изобретению являются группы, содержащие 10 или меньше атомов углерода. «Низший алкил» или «низший алкилен» представляет собой короткоцепочечную алкильную или алкиленовую группу, обычно содержащую четыре или меньше атомов углерода. Аналогично, «алкенилен» или «алкинилен» означает ненасыщенные формы «алкилена», содержащие двойные или тройные связи, соответственно.
При использовании в настоящем тексте, волнистая линия "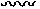 ", пересекающая простую, двойную или тройную связь в любой изображенной в настоящем тексте химической структуре, означает точку присоединения простой, двойной или тройной связи к остальной части молекулы.
", пересекающая простую, двойную или тройную связь в любой изображенной в настоящем тексте химической структуре, означает точку присоединения простой, двойной или тройной связи к остальной части молекулы.
Термины "алкокси," "алкиламино" и "алкилтио" (или тиоалкокси) применяются в их обычном смысле и относятся к алкильным группам, присоединенным к остальной части молекулы через атом кислорода, аминогруппу или атом серы, соответственно. Кроме того, для диалкиламино-групп, алкильные фрагменты могут быть одинаковыми или разными, а также могут объединяться с формированием 3-7-членного цикла с атомом азота, к которому они присоединены. Соответственно, группа, изображаемая как -NRaRb, включает пиперидинил, пирролидинил, морфолинил, азетидинил и т.п.
Термин "ди-(C1-4 алкил)амино-C1-4 алкил" относится к амино-группе, несущей две C1-4 алкильные группы, которые могут быть одинаковыми или разными (например, метил, этил, пропил, изопропил, н-бутил, втор-бутил, изобутил и трет-бутил), и которая присоединена к остальной части молекулы через C1-4 алкильную группу (1-4-углеродная алкиленовая связывающая группа). Примеры ди-(C1-4 алкил)амино-C1-4 алкильных групп включают диметиламинометил, 2-(этил(метил)амино)этил, 3-(диметиламино)бутил и т.п.
Термин "галоген" сам по себе или как часть другого заместителя означает, если не указано иное, атом фтора, хлора, брома или иода. Кроме того, такие термины как "галогеналкил" включают моногалогеналкил и полигалогеналкил. Например, термин "C1-4 галогеналкил" включает трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и т.п.
Термин "арил" означает, если не указано иное, полиненасыщенную, в типичном случае ароматическую, углеводородную группу, которая может представлять собой один цикл или несколько циклов (до трех циклов), сопряженные или связанные ковалентно. Термин «гетероарил» означает арильные группы (или циклы), содержащие от одного до пяти гетероатомов, выбранных из N, O и S, где атомы азота и серы необязательно окислены, и атом(ы) азота необязательно кватернизован(ы). Гетероарильная группа может быть присоединена к остальной части молекулы через гетероатом. Неограничивающие примеры арильных групп включают фенил, нафтил и бифенил, а неограничивающие примеры гетероарильных групп включают пиридил, пиридазинил, пиразинил, пиримидинил, триазинил, хинолинил, хиноксалинил, хиназолинил, циннолил, фталазинил, бензотриазинил, пуринил, бензоимидазолил, бензопиразолил, бензотриазолил, бензизоксазалил, изобензофурил, изоиндолил, индолизинил, бензотриазинил, тиенопиридинил, тиенопиримидинил, пиразолопиримидинил, имидазопиридины, бензотиаксолил, бензофуранил, бензотиенил, индолил, хинолил, изохинолил, изотиазолил, пиразолил, индазолил, птеридинил, имидазолил, триазолил, тетразолил, оксазолил, изоксазолил, тиадиазолил, пирролил, тиазолил, фурил, тиенил и т.п. Заместители в каждой из перечисленных выше арильных или гетероарильных циклических системах выбраны из группы приемлемых заместителей, описанных ниже.
Термин "арилалкил" включает радикалы, в которых арильная группа присоединена к алкильной группе (например, бензил, фенетил и т.п.). Аналогично, термин "гетероарил-алкил" включает радикалы, в которых гетероарильная группа присоединена к алкильной группе (например, пиридилметил, тиазолилэтил и т.п.).
Описанные выше термины (например, "алкил," "арил" и "гетероарил") в некоторых вариантах осуществления включают и замещенные, и незамещенные формы указанных радикалов. Предпочтительные заместители для каждого типа радикалов описаны ниже.
Заместителями в алкильных радикалах (включая группы, которые часто называют алкилен, алкенил, алкинил и циклоалкил) может быть широкий ряд групп, выбранных из следующих: -галоген, -OR’, -NR’R”, -SR’, -SiR’R”R”’, -OC(O)R’, -C(O)R’, -CO2R’, -CONR’R”, -OC(O)NR’R”, -NR”C(O)R’, -NR’-C(O)NR”R”’, -NR”C(O)2R’, -NH-C(NH2)=NH, -NR’C(NH2)=NH, -NH-C(NH2)=NR’, -S(O)R’, -S(O)2R’, -S(O)2NR’R”, -NR’S(O)2R”, -CN и -NO2, в количестве от нуля до (2m’+1), где m’ это общее число атомов углерода в таком радикале. R’, R” и R”’ каждый независимо означают атом водорода, незамещенный C1-8 алкил, незамещенный арил, арил, замещенный 1-3 галогенами, незамещенный C1-8 алкил, C1-8 алкокси- или C1-8 тиоалкокси-группы, или незамещенные арил-C1-4 алкильные группы. Когда R’ и R” присоединены к одному атому азота, они могут быть объединены с атомом азота с образованием 3-, 4-, 5-, 6- или 7-членного кольца. Например, -NR’R” включает 1-пирролидинил и 4-морфолинил.
Аналогично, заместители в арильных и гетероарильных группах варьируются и обычно выбраны из следующих: -галоген, -OR’, -OC(O)R’, -NR’R”, -SR’, -R’, -CN, -NO2, -CO2R’, -CONR’R”, -C(O)R’, -OC(O)NR’R”, -NR”C(O)R’, -NR”C(O)2R’, ,-NR’-C(O)NR”R”’, -NH-C(NH2)=NH, -NR’C(NH2)=NH, -NH-C(NH2)=NR’, -S(O)R’, -S(O)2R’, -S(O)2NR’R”, -NR’S(O)2R”, -N3, перфтор(C1-C4)алкокси и перфтор(C1-C4)алкил, в количестве от нуля до общего числа незанятых валентностей в ароматической системе; и где R’, R” и R”’ независимо выбраны из атома водорода, C1-8 алкила, C3-6 циклоалкила, C2-8 алкенила, C2-8 алкинила, незамещенного арила и гетероарила, (незамещенный арил)-C1-4 алкила и незамещенный арилокси-C1-4 алкила. Другие подходящие заместители включают каждый из перечисленных выше заместителей для арила, присоединенный к атому в цикле через алкиленовый мостик, содержащий 1-4 атомов углерода.
Два из заместителей на соседних атомах арильного или гетероарильного цикла необязательно могут быть заменены на заместитель, имеющий формулу -T-C(O)-(CH2)q-U-, где T и U независимо представляют собой -NH-, -O-, -CH2- или простую связь, и q это целое число от 0 до 2. Альтернативно, два из заместителей на соседних атомах арильного или гетероарильного цикла необязательно могут быть заменены на заместитель, имеющий формулу -A-(CH2)r-B-, где A и B независимо представляют собой -CH2-, -O-, -NH-, -S-, -S(O)-, -S(O)2-, -S(O)2NR’- или простую связь, и r это целое число от 1 до 3. Одна из простых связей в образованном таким образом новом цикле необязательно может быть заменена на двойную связь. Альтернативно, два из заместителей на соседних атомах арильного или гетероарильного цикла необязательно могут быть заменены на заместитель, имеющий формулу -(CH2)s-X-(CH2)t-, где s и t независимо представляют собой целые числа от 0 до 3, и X представляет собой -O-, -NR’-, -S-, -S(O)-, -S(O)2- или -S(O)2NR’-. Заместитель R’ в -NR’- и -S(O)2NR’- выбран из атома водорода или незамещенного C1-6 алкила.
При использовании в настоящем тексте, термин "гетероатом" включает в себя кислород (О), азот (N), серу (S) и кремний (Si).
Термин "фармацевтически приемлемые соли" включает соли веществ, полученные с относительно нетоксичными кислотами или основаниями, в зависимости от конкретных заместителей в описанных в настоящем тексте соединениях. Когда соединения по настоящему изобретению содержат относительно кислые функциональные группы, можно получить основно-аддитивные соли путем взаимодействия нейтральной формы таких соединений с достаточным количество желаемого основания, даже без растворителя или в подходящем инертном растворителе. Примеры солей, являющихся производными фармацевтически приемлемых неорганических оснований, включают соли алюминия, аммония, кальция, меди, железа(II), железа (III), лития, магния, марганца, калия, натрия, цинка и т.д. Соли, являющиеся производными фармацевтически приемлемых органических оснований, включают соли первичных, вторичных и третичных аминов, включая замещенные амины, циклические амины, природные амины и т.д., такие как аргинин, бетаин, кофеин, холин, N,N’-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п. Когда соединения по настоящему изобретению содержат относительно основные функциональные группы, можно получить кислотно-аддитивные соли путем взаимодействия нейтральной формы таких соединений с достаточным количеством желаемой кислоты, без растворителя или в подходящем инертном растворителе. Примеры фармацевтически приемлемых кислотно-аддитивных солей включают соли с неорганическими кислотами, такими как хлористоводородная, бромистоводородная, азотная, угольная, моногидроугольная, фосфорная, моногидрофосфорная, дигидрофосфорная, серная, моногидросерная, иодистоводородная или фосфористая кислота и т.п., а также соли с относительно нетоксичными органическими кислотами, такими как уксусная, пропионовая, изомасляная, малоновая, бензойная, янтарная, субериновая, фумаровая, миндальная, фталевая, бензолсульфоновая, пара-толуолсульфоновая, лимонная, винная, метансульфоновая и т.п. Также охватываются соли с аминокислотами, такие как аргинаты и т.п., и соли таких органических кислот, как глюкуроновая или галактуроновая кислоты и т.п. (см, например, Berge, S.M., et al, “Pharmaceutical Salts”, Journal of Pharmaceutical Science, 1977, 66, 1-19). Некоторые частные соединения по настоящему изобретению содержат и основные, и кислотные функциональные группы, что позволяет таким соединениям образовывать как основно-аддитивные, так и кислотно-аддитивные соли.
Нейтральные формы соединений можно регенерировать путем взаимодействия соли с основанием или кислотой и выделения материнского соединения обычным способом. Материнская форма соединения отличается от различных солевых форм определенными физическими характеристиками, такими как растворимость в полярных растворителях, но во всем остальном соли эквивалентны материнским соединениям, в терминах настоящего изобретения.
Помимо солевых форм, в изобретении описаны соединения в форме пролекарства. Пролекарства описанных в настоящем тексте соединений представляют собой соединения, которые легко претерпевают химические изменения в физиологических условиях, давая соединения по настоящему изобретению. Кроме того, пролекарства могут превращаться в соединения по настоящему изобретению химическими или биохимическими методами в условиях in vivo. Например, пролекарства могут медленно превращаться в соединения по настоящему изобретению при помещении в резервуар чрезкожного пластыря с подходящим ферментативным или химическим реагентом.
Некоторые соединения по изобретению могут существовать в несольватированных формах, а также в сольватированных формах, включая гидратированные формы. В целом, сольватированные формы эквивалентны несольватированным формам, и все они охватываются настоящим изобретением. Некоторые соединения по настоящему изобретению могут существовать в нескольких кристаллических или аморфных формах. В целом, все физические формы эквивалентны для областей применения, охватываемых настоящим изобретением, и входят в объем настоящего изобретения.
Некоторые соединения по изобретению имеют асимметрические атомы углерода (оптические центры) или двойные связи; все рацематы, диастереомеры, геометрические изомеры, региоизомеры и индивидуальные изомеры (например, отдельные энантиомеры) входят в объем настоящего изобретения. Когда показана стерохимическая конфигурация, это означает соединение, в котором присутствует один из изомеров, и оно практически не содержит другого изомера. “Практически не содержит” другого изомера означает соотношение этих двух изомеров по меньшей мере 80/20, более предпочтительно 90/10 или 95/5 или больше. В некоторых вариантах осуществления, один из изомеров присутствует в количестве по меньшей мере 99%.
Соединения по изобретению могут также иметь неприродные соотношения изотопов по одному или больше атомов, составляющих эти соединения. Неприродные соотношения изотопов можно определить как находящиеся в диапазоне от природного количества до количества рассматриваемого атома равного 100%. Например, соединения могут быть радиоактивно мечены радиоактивными изотопами, такими как, например, тритий (3H), иод-125 (125I) или углерод-14 (14C), или нерадиоактивными изотопами, такими как дейтерий (2H) или углерод-13 (13C). Такие вариации изотопов могут открыть дополнительные области применения к описанным в других разделах настоящего описания. Например, изотопные модификации соединений по настоящему изобретению могут найти дополнительное применение, включая (но не ограничиваясь только ими) применение в качестве диагностических и/или визуализирующих реагентов, или в качестве цитотоксических/радиотоксических терапевтических средств. Кроме того, изотопные варианты соединений по настоящему изобретению могут иметь измененные фармакокинетические и фармакодинамические характеристики, которые могут вносить свой вклад в улучшение характеристик безопасности, переносимости или эффективности при лечении. Все изотопные вариации соединений по настоящему изобретению, радиоактивные и нерадиоактивные, входят в объем настоящего изобретения.
Соединения по изобретению, имеющие формулу I-V, могут существовать в различных изомерных формах. В настоящем тексте, термины цис или транс используются в их обычном для химии значении, т.е. они означают взаимное расположение заместителей относительно определенной плоскости, например двойной связи или циклической системы, такой как система декалинового типа или гидрохинолонового типа: в цис-изомере заместители находятся по одну сторону от плоскости, в транс-изомере заместители находятся по разные стороны от плоскости. Кроме того, настоящее изобретение охватывает различные конформеры, а также различные ротамеры. Конформеры представляют собой конформационные изомеры, которые могут различаться вследствие вращения вокруг одной или больше σ-связей. Ротамеры представляют собой конформеры, которые различаются вращением вокруг только одной σ-связи.
Термин “рак” относится к заболеванию, характеризующемуся неконтролируемым ростом клеток, отклоняющихся от нормального типа. Раковые клетки могут распространяться локально или через кровеносную систему и лимфатическую систему в другие части организма. Примеры различных видов рака описаны в настоящем тексте и включают (но не ограничиваются только ими) рак груди, рак предстательной железы, рак яичника, рак шейки матки, рак кожи, рак поджелудочной железы, рак толстой и прямой кишки, рак почки, рак печени, рак мозга, лимфому, лейкоз, рак легких и т.п. Термины “опухоль” и “рак” применяются в настоящем тексте взаимозаменяемо, например, оба термина охватывают солидные опухоли и опухоли жидких тканей, диффузные или циркулирующие. При использовании в настоящем тексте, термин “рак” или “опухоль” включает предраковые, а также злокачественные опухоли и раковые заболевания.
Термин “PD-1” или “PD-1 рецептор” означает белок запрограммированной смерти 1 (programmed death-1), со-ингибитор T-клеток, также известный как CD279. Последовательность аминокислот человеческого полноразмерного белка PD-1 приведена, например, под номером GenBank Accession Number NP_005009.2. PD-1 представляет собой белок, построенный из 288 остатков аминокислот, имеющий внеклеточный N-терминальный конец, который представляет собой IgV домен, трансмембранный участок и внутриклеточный домен, содержащий иммунорецепторный тирозиновый ингибирующий мотив (ITIM) и иммунорецепторный тирозиновый свитч-мотив (ITSM) (Chattopadhyay et al., Immunol Rev, 2009, 229(1):356-386). Термин “PD-1” включает рекомбинантный PD-1 или его фрагмент или его варианты. PD-1 рецептор имеет два лиганда, PD-лиганд-1 (PD-L1) и PD-лиганд-2 (PD-L2).
Термин “PD-L1” или “programmed death ligand 1” означает лиганд PD-1 рецептора, также известный как CD274 и B7H 1. Последовательность аминокислот человеческого полноразмерного белка PD-L1 приведена, например, под номером GenBank Accession Number NP_054862.1. PD-L1 представляет собой белок, построенный из 290 остатков аминокислот, имеющий внеклеточный IgV домен, трансмембранный участок и высококонсервативный внутриклеточный домен, состоящий из примерно 30 аминокислот. PD-L1 конститутитивно экспрессируется на многих клетках, таких как антиген-представляющие клетки (например, дендритные клетки, макрофаги и В-клетки), и на гематопоетических и не-гематопоетических клетках (например, клетки сосудистого эндотелия, панкреатические островки и сайты иммунных привилегий). PD-L1 экспрессируется также на широком ряде опухолей, инфицированных вирусами клетках и аутоиммунных тканях.
Путь запрограммированной смерти 1 (PD-1/PD-L1) работает как контрольная точка для ограничения иммунных ответов, осуществляемых посредством Т-клеток. Оба PD-1 лиганда, PD-L1 и PD-L2, могут связываться с PD-1 рецептором и вызывать сигнал PD-1 и обратимое ингибирование активации и пролиферации Т-клеток. Когда PD-1 лиганды находятся на поверхности раковых клеток или соседних клеток, эти лиганды связываются с PD-1 рецептор позитивными иммунными эффекторными клетками и используют PD-1 механизм для уклонения от иммунного ответа.
Термин “ингибиторы иммунных контрольных точек” или “блокада иммунных контрольных точек” относится к любому средству, соединению, химикату, белку, полипептиду, макромолекуле и т.д., которые блокируют или ингибируют статистически, клинически или биологически значимым образом ингибирующие механизмы иммунной системы. Такие ингибиторы могут включать низкомолекулярные ингибиторы или антитела или их фрагменты, связывающиеся с антигеном, которые связываются и блокируют или ингибируют лиганды рецепторов иммунных контрольных точек. Иллюстративные молекулы иммунных контрольных точек, которые могут служить мишенями для блокирования или ингибирования, включают (но не ограничены только ими) CTLA-4, 4-1BB (CD137), 4-1BBL (CD137L), PDLl, PDL2, PD-l, B7-H3, B7-H4, BTLA, HVEM, TIM3, GAL9, LAG3, TIM3, B7H3, B7H4, VISTA, KIR, 2B4 (принадлежит к CD2 семейству молекул и экспрессируется на всех NK, γδ, и Т-клетках памяти CD8+ (αβ)), CD160 (также именуется BY55) и CGEN-15049. Иллюстративные ингибиторы иммунных контрольных точек включают дурвалумаб (анти-PD-L1 антитело; MEDI4736), пембролизумаб (анти-PD-1 моноклональное антитело), ниволумаб (анти-PD-l антитело), пидилизумаб (CT-011; гуманизированное анти-PD-l моноклональное антитело), AMP224 (рекомбинантный B7-DC-Fc химерный белок), BMS-936559 (анти-PD-Ll антитело), атезолизумаб (MPLDL3280A; человеческое Fc-оптимизированное анти-PD-Ll моноклональное антитело), авуелумаб (MSB0010718C; человеческое анти-PD-Ll антитело), ипилимумаб (ингибитор контрольной точки анти-CTLA-4), тремелимумаб (CTLA-4 блокирующее антитело) и анти-OX40.
Термины “CCR1 антагонист” и “антагонист хемокинового рецептора CCR1” используются взаимозаменяемо и относятся к малой молекуле, которая антагонизирует взаимодействие хемокинового рецептора CCR1 и любого из его лигандов. Такое соединение может ингибировать процессы, в норме запускаемые при взаимодействии лиганда с рецептором.
При использовании в настоящем тексте, “полный ответ” или “полная ремиссия” или “CR” означает исчезновение всех целевых поражений; “частичный ответ” или “частичная ремиссия” или “PR” означает по меньшей мере 30%-ное уменьшение суммы максимальных диаметров (SLD) целевых поражений, относительно исходного уровня SLD; и “стабильное заболевание” или “SD” означает отсутствие заметного уменьшения целевых поражений, характерного для PR, и отсутствие их заметного увеличения, характерного для PD, в сравнении с наименьшим значением SLD со времени начала лечения.
При использовании в настоящем тексте, “прогрессирование заболевания” или “PD” означает по меньшей мере 20%-ное увеличение SLD целевых поражений, в сравнении с наименьшим значением SLD со времени начала лечения, или появление одного или больше новых поражений.
При использовании в настоящем тексте, “выживаемость без прогрессирования заболевания” (PFS) означает длительность периода во время и после лечения, в течение которого заболевание, подвергающееся лечению (например, рак) не усиливается. Выживаемость без прогрессирования заболевания может включать интервал времени, в течение которого у пациентов наблюдался полный ответ или частичный ответ, а также интервал времени, в течение которого у пациентов наблюдалость стабильное заболевание.
При использовании в настоящем тексте, “общая частота ответа” (ORR) означает сумму частоты полного ответа (CR) и частичного ответа (PR).
При использовании в настоящем тексте, “общая выживаемость” означает процент пациентов в группе, для которых ожидается выживаемость в течение определенного периода времени.
Подробное описание вариантов осуществления
В одном аспекте, в настоящем изобретении описан способ лечения субъекта с раковой солидной опухолью. Способ включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества антагониста хемокинового рецептора CCR1 и терапевтически эффективного количества PD-1 ингибитора или PD-L1 ингибитора.
В некоторых вариантах осуществления, способ включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества антагониста хемокинового рецептора CCR1 и терапевтически эффективного количества PD-1 ингибитора.
В некоторых вариантах осуществления, способ включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества антагониста хемокинового рецептора CCR1 и терапевтически эффективного количества PD-L1 ингибитора.
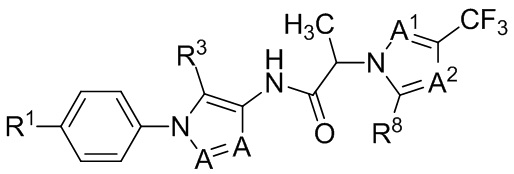
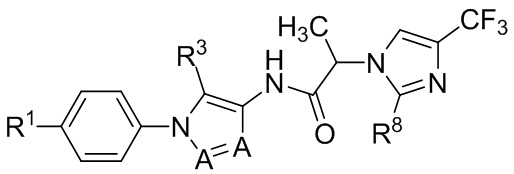
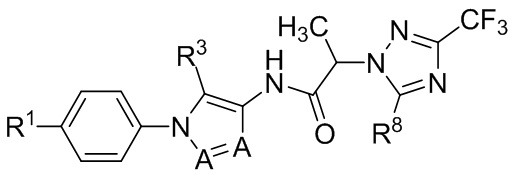
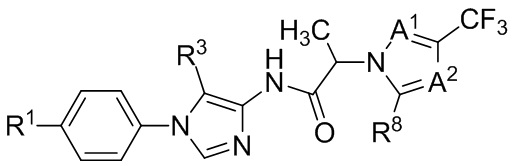
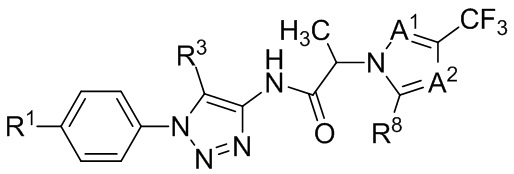
В некоторых вариантах осуществления, антагонист хемокинового рецептора CCR1 выбран из соединений, имеющих приведенные ниже формулы I-V, более конкретно – выбран из соединений 1.001, 3.002, 4.005, 5.005 и 3.001 или их фармацевтически приемлемых солей.
В некоторых вариантах осуществления, антагонист хемокинового рецептора CCR1 выбран из группы, состоящей из BL-5923, UCB-35625, BX-471, BI-638683, PS-031291, MLN-3701, AZD-4818, MLN-3897, CP-481715, F-18-CCR1, AOP-RANTES, PS-375179 и NSC-651016.
В некоторых вариантах осуществления, PD-1 ингибитор выбран из группы, состоящей из пембролизумаба, ниволумаба, IBI-308, mDX-400, BGB-108, MEDI-0680, SHR-1210, PF-06801591, PDR-001, GB-226, STI-1110, их биоаналогов, их биоулучшенных версий и их биоэквивалентов.
В некоторых вариантах осуществления, PD-1 ингибитор выбран из группы, состоящей из пембролизумаба, ниволумаба, IBI-308, mDX-400, BGB-108, MEDI-0680, SHR-1210, PF-06801591, PDR-001, GB-226 и STI-1110.
В некоторых вариантах осуществления, PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, авелумаба, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, STI-1014, KY-1003, их биоаналогов, их биоулучшенных версий и их биоэквивалентов.
В некоторых вариантах осуществления, PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, авелумаба, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, CA-327, STI-1014, KY-1003, их биоаналогов, их биоулучшенных версий и их биоэквивалентов.
В некоторых вариантах осуществления, PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, авелумаба, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-327, STI-1014, KY-1003, их биоаналогов, их биоулучшенных версий и их биоэквивалентов.
В некоторых вариантах осуществления, PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, авелумаба, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, STI-1014 и KY-1003.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из соединений, описанных в US2015291549, WO16039749, WO15034820 и US2014294898 (BRISTOL MYERS SQUIBB CO), которые включены в настоящий текст посредством ссылки.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из соединений, описанных в WO14151634, WO15160641, WO16039749, WO16077518, WO16100608, WO16149351, WO2016057624, WO2016100285, US2016194307, US2016222060 и US2014294898 (BRISTOL MYERS SQUIBB CO), которые включены в настоящий текст посредством ссылки.
В некоторых вариантах осуществления, PD-1 или PD-L1 ингибитор выбран из соединений, описанных в предварительной заявке на патент США № 62/355,119 или 62/440,100 которые включены в настоящий текст посредством ссылки.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из группы, состоящей из:
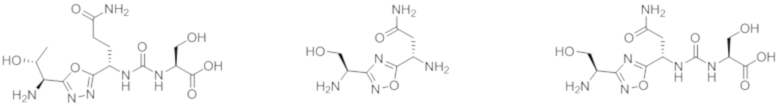
 .
.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из соединений, описанных в WO16142886, WO16142894, WO16142852, WO16142833, WO15033301, WO15033299, WO11161699, WO12168944, WO13132317, WO13144704, WO15033303, WO15036927, WO15044900, WO16142835, US2015073024, US8907053, US9044442, US9096642, US9233940 и US2016194295 (Aurigene discovery tech ltd), которые включены в настоящий текст посредством ссылки.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, пембролизумаба, ниволумаба, AP-106, AP-105, MSB-2311, CBT-501, авелумаба, AK-105, IO-102, IO-103, PDR-001, CX-072, SHR-1316, JTX-4014, GNS-1480, рекомбинантного гуманизированного анти-PD1 mAb (Shanghai Junshi Biosciences), REGN-2810, пелареорепа, SHR-1210, PD1/PDL1 ингибирующей вакцины (THERAVECTYS), BGB-A317, рекомбинантного гуманизированного анти-PD-1 mAb (Bio-Thera Solutions), Probody нацеленного на PD-1 (CytomX), XmAb-20717, FS-118, PSI-001, SN-PDL01, SN-PD07, PD-1 модифицированных TIL (Sangamo Therapeutics), PRS-332, FPT-155, jienuo mAb (Genor Biopharma), TSR-042, REGN-1979, REGN-2810, ресминостата, FAZ-053, PD-1/CTLA-4 биспецифического антитела (MacroGenics), MGA-012, MGD-013, M-7824, биспецифического антитела на основе PD-1 (Beijing Hanmi Pharmaceutical), AK-112, AK-106, AK-104, AK-103, BI-754091, ENUM-244C8, MCLA-145, MCLA-134, анти-PD-1 онколитического моноклонального антитела (Transgene SA), AGEN-2034, IBI-308, WBP-3155, JNJ-63723283, MEDI-0680, SSI-361, CBT-502, анти-PD-1 биспецифического антитела, нацеленного на две мишени анти-PD-1/LAG-3 mAbs (TESARO), нацеленного на две мишени анти-PD-1/TIM-3 mAbs (TESARO), PF-06801591, LY-3300054, BCD-100, STI-1110, биоаналога пембролизумаба, биоаналога ниволумаба, терапии PD-L1–ТФР-бета, KY-1003, STI-1014, GLS-010, AM-0001, GX-P2, KD-033,PD-L1/BCMA биспецифического антитела (Immune Pharmaceuticals), биспецифического антитела против PD-1/Ox40 (Immune Pharmaceuticals), BMS-936559, анти-PD-1/VEGF-A DARPins (Molecular Partners), mDX-400, ALN-PDL, PD-1 ингибирующего пептида (Aurigene), вакцины на основе дендритных клеток с миРНК (Alnylam Pharmaceuticals), GB-226, иммунотерапии PD-L1 на основе CAR-TNK (TNK Therapeutics/NantKwest), INSIX RA, INDUS-903, AMP-224, биспецифического гуманизированного антитела анти-CTLA-4/анти-PD-1 (Akeso Biopharma), вакцины B7-H1 (State Key Laboratory of Cancer Biology/Fourth Military Medical University) и GX-D1.
Антагонист хемокинового рецептора CCR1 и PD-1 ингибитор или PD-L1 ингибитор можно вводить параллельно. В некоторых случаях, антагонист хемокинового рецептора CCR1 и PD-1 ингибитор вводят одновременно. В некоторых случаях, антагонист хемокинового рецептора CCR1 и PD-L1 ингибитор вводят одновременно. В некоторых случаях, антагонист хемокинового рецептора CCR1 и PD-1 ингибитор или PD-L1 ингибитор вводят в виде комбинированного препарата. Антагонист хемокинового рецептора CCR1 и PD-1 ингибитор можно вводить в виде комбинированного препарата. Опционально, антагонист хемокинового рецептора CCR1 и PD-L1 ингибитор вводят в виде комбинированного препарата.
В других вариантах осуществления, антагонист хемокинового рецептора CCR1 и PD-1 ингибитор или PD-L1 ингибитор вводят последовательно. В некоторых случаях, антагонист хемокинового рецептора CCR1 и PD-1 ингибитор вводят последовательно. В некоторых случаях, антагонист хемокинового рецептора CCR1 и PD-L1 ингибитор вводят последовательно. Антагонист хемокинового рецептора CCR1 можно вводить до введения PD-1 ингибитора или PD-L1 ингибитора.
В некоторых вариантах осуществления, антагонист хемокинового рецептора CCR1 можно вводить до введения PD-L1 ингибитора. Антагонист хемокинового рецептора CCR1 можно вводить после введения PD-1 ингибитора или PD-L1 ингибитора.
В некоторых вариантах осуществления, антагонист хемокинового рецептора CCR1 можно вводить после введения PD-L1 ингибитора.
В некоторых вариантах осуществления, субъектом является человек.
В некоторых вариантах осуществления, раковая солидная опухоль может представлять собой рак мозга, рак груди, рак мочевого пузыря, рак кости, рак толстой и прямой кишки, рак легких, рак почки, рак печени, рак желудка, рак предстательной железы, саркому, меланому, карциному и лимфому.
В некоторых вариантах осуществления, раковая солидная опухоль представляет собой рак груди.
В некоторых вариантах осуществления, раковая солидная опухоль представляет собой трижды негативный рак груди.
Во втором аспекте, в настоящем изобретении описана композиция для лечения субъекта с раковой солидной опухолью. Композиция содержит терапевтически эффективное количество антагониста хемокинового рецептора CCR1 и терапевтически эффективное количество PD-1 ингибитора или PD-L1 ингибитора, и фармацевтически приемлемый носитель или вспомогательное вещество.
В некоторых вариантах осуществления, композиция содержит терапевтически эффективное количество антагониста хемокинового рецептора CCR1 и терапевтически эффективное количество PD-L1 ингибитора, и фармацевтически приемлемый носитель или вспомогательное вещество.
В некоторых вариантах осуществления, антагонист хемокинового рецептора CCR1 представляет собой соединение, выбранное из приведенных ниже формул I-V.
В некоторых вариантах осуществления, антагонист хемокинового рецептора CCR1 выбран из группы, состоящей из 1.001, 3.002, 4.005, 5.005 и 3.001 или их фармацевтически приемлемой соли.
В некоторых вариантах осуществления, антагонист хемокинового рецептора CCR1 выбран из группы, состоящей из BL-5923, UCB-35625, BX-471, BI-638683, PS-031291, MLN-3701, AZD-4818, MLN-3897, CP-481715, F-18-CCR1, AOP-RANTES, PS-375179 и NSC-651016.
В некоторых вариантах осуществления, PD-1 ингибитор выбран из группы, состоящей из пембролизумаба, ниволумаба, IBI-308, mDX-400, BGB-108, MEDI-0680, SHR-1210, PF-06801591, PDR-001, GB-226, STI-1110, их биоаналогов, их биоулучшенных версий и их биоэквивалентов.
В некоторых вариантах осуществления, PD-1 ингибитор выбран из группы, состоящей из пембролизумаба, ниволумаба, IBI-308, mDX-400, BGB-108, MEDI-0680, SHR-1210, PF-06801591, PDR-001, GB-226 и STI-1110.
В некоторых вариантах осуществления, PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, авелумаба, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, CA-327, STI-1014, KY-1003, их биоаналогов, их биоулучшенных версий и их биоэквивалентов.
В некоторых вариантах осуществления, PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, авелумаба, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, STI-1014, KY-1003, их биоаналогов, их биоулучшенных версий и их биоэквивалентов.
В некоторых вариантах осуществления, PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, авелумаба, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, STI-1014 и KY-1003.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из соединений, описанных в WO14151634, WO15160641, WO16039749, WO16077518, WO16100608, WO16149351, WO2016057624, WO2016100285, US2016194307, US2016222060, US2014294898, US2015291549 и US2016194307 (BRISTOL MYERS SQUIBB CO), которые включены в настоящий текст посредством ссылки.
В некоторых вариантах осуществления, PD-1 или PD-L1 ингибитор выбран из соединений, описанных в предварительной заявке на патент США № 62/355,119 или 62/440,100 которые включены в настоящий текст посредством ссылки.
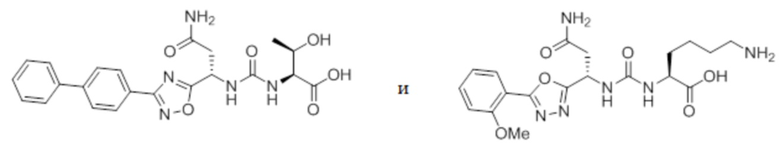
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из группы, состоящей из:
 .
.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из соединений, описанных в WO16142886, WO16142894, WO16142852, WO16142833, WO15033301, WO15033299, WO11161699, WO12168944, WO13132317, WO13144704, WO15033303, WO15036927, WO15044900, WO16142835, US2015073024, US8907053, US9044442, US9096642, US9233940 и US2016194295 (Aurigene discovery tech ltd), которые включены в настоящий текст посредством ссылки.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, пембролизумаба, ниволумаба, AP-106, AP-105, MSB-2311, CBT-501, авелумаба, AK-105, IO-102, IO-103, PDR-001, CX-072, SHR-1316, JTX-4014, GNS-1480, рекомбинантного гуманизированного анти-PD1 mAb (Shanghai Junshi Biosciences), REGN-2810, пелареорепа, SHR-1210, вакцины PD1/PDL1 ингибитора (THERAVECTYS), BGB-A317, рекомбинантного гуманизированного анти-PD-1 mAb (Bio-Thera Solutions), Probody нацеленного на PD-1 (CytomX), XmAb-20717, FS-118, PSI-001, SN-PDL01, SN-PD07, PD-1 модифицированных TIL (Sangamo Therapeutics), PRS-332, FPT-155, jienuo mAb (Genor Biopharma), TSR-042, REGN-1979, REGN-2810, ресминостата, FAZ-053, PD-1/CTLA-4 биспецифического антитела (MacroGenics), MGA-012, MGD-013, M-7824, биспецифического антитела на основе PD-1 (Beijing Hanmi Pharmaceutical), AK-112, AK-106, AK-104, AK-103, BI-754091, ENUM-244C8, MCLA-145, MCLA-134, анти-PD-1 онколитического моноклонального антитела (Transgene SA), AGEN-2034, IBI-308, WBP-3155, JNJ-63723283, MEDI-0680, SSI-361, CBT-502, анти-PD-1 биспецифического антитела, нацеленного на две мишени анти-PD-1/LAG-3 mAbs (TESARO), нацеленного на две мишени анти-PD-1/TIM-3 mAbs (TESARO), PF-06801591, LY-3300054, BCD-100, STI-1110, биоаналога пембролизумаба, биоаналога ниволумаба, терапии PD-L1–ТФР-бета, KY-1003, STI-1014, GLS-010, AM-0001, GX-P2, KD-033, PD-L1/BCMA биспецифического антитела (Immune Pharmaceuticals), биспецифического антитела против PD-1/Ox40 (Immune Pharmaceuticals), BMS-936559, анти-PD-1/VEGF-A DARPins (Molecular Partners), mDX-400, ALN-PDL, PD-1 ингибирующего пептида (Aurigene), вакцины на основе дендритных клеток с миРНК (Alnylam Pharmaceuticals), GB-226, иммунотерапии PD-L1 на основе CAR-TNK (TNK Therapeutics/NantKwest), INSIX RA, INDUS-903, AMP-224, биспецифического гуманизированного антитела анти-CTLA-4/анти-PD-1 (Akeso Biopharma), вакцины B7-H1 (State Key Laboratory of Cancer Biology/Fourth Military Medical University) и GX-D1.
В некоторых вариантах осуществления, антагонист хемокинового рецептора CCR1 и PD-1 ингибитор или PD-L1 ингибитор вводят в состав препаратов для параллельного введения.
В других вариантах осуществления, антагонист хемокинового рецептора CCR1 и PD-1 ингибитор или PD-L1 ингибитор вводят в состав препаратов для последовательного введения.
В другом аспекте, в настоящем изобретении описан набор для лечения субъекта с раковой солидной опухолью. Набор содержит терапевтически эффективное количество антагониста хемокинового рецептора CCR1 и терапевтически эффективное количество PD-1 ингибитора или PD-L1 ингибитора, с инструкцией по эффективному применению.
В некоторых вариантах осуществления, набор содержит терапевтически эффективное количество антагониста хемокинового рецептора CCR1 и терапевтически эффективное количество PD-L1 ингибитора, с инструкцией по эффективному применению.
В некоторых вариантах осуществления, антагонист хемокинового рецептора CCR1 выбран из группы, состоящей из 1.001, 3.002, 4.005, 5.005 и 3.001, или их фармацевтически приемлемой соли.
В некоторых вариантах осуществления, антагонист хемокинового рецептора CCR1 выбран из группы, состоящей из BL-5923, UCB-35625, BX-471, BI-638683, PS-031291, MLN-3701, AZD-4818, MLN-3897, CP-481715, F-18-CCR1, AOP-RANTES, PS-375179 и NSC-651016.
В некоторых вариантах осуществления, PD-1 ингибитор выбран из группы, состоящей из пембролизумаба, ниволумаба, IBI-308, mDX-400, BGB-108, MEDI-0680, SHR-1210, PF-06801591, PDR-001, GB-226, STI-1110, их биоаналогов, их биоулучшенных версий и их биоэквивалентов.
В некоторых вариантах осуществления, PD-1 ингибитор выбран из группы, состоящей из пембролизумаба, ниволумаба, IBI-308, mDX-400, BGB-108, MEDI-0680, SHR-1210, PF-06801591, PDR-001, GB-226 и STI-1110.
В некоторых вариантах осуществления, PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, авелумаба, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, CA-327, STI-1014, KY-1003, их биоаналогов, их биоулучшенных версий и их биоэквивалентов.
В некоторых вариантах осуществления, PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, авелумаба, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, STI-1014, KY-1003, их биоаналогов, их биоулучшенных версий и их биоэквивалентов.
В некоторых вариантах осуществления, PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, авелумаба, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, STI-1014 и KY-1003.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из соединений, описанных в WO14151634, WO15160641, WO16039749, WO16077518, WO16100608, WO16149351, WO2016057624, WO2016100285, US2016194307, US2016222060, US2015291549, US2016194307 и US2014294898 (BRISTOL MYERS SQUIBB CO), которые включены в настоящий текст посредством ссылки.
В некоторых вариантах осуществления, PD-1 или PD-L1 ингибитор выбран из соединений, описанных в предварительной заявке на патент США № 62/355,119 или 62/440,100 которые включены в настоящий текст посредством ссылки.
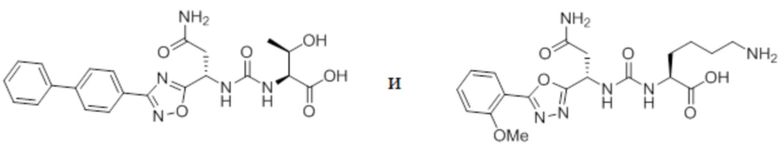
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из группы, состоящей из:
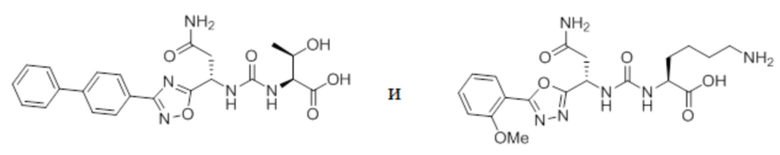 .
.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из соединений, описанных в WO16142886, WO16142894, WO16142852, WO16142833, WO15033301, WO15033299, WO11161699, WO12168944, WO13132317, WO13144704, WO15033303, WO15036927, WO15044900, WO16142835, US2015073024, US8907053, US9044442, US9096642, US9233940 и US2016194295 (Aurigene discovery tech ltd), которые включены в настоящий текст посредством ссылки.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, пембролизумаба, ниволумаба, AP-106, AP-105, MSB-2311, CBT-501, авелумаба, AK-105, IO-102, IO-103, PDR-001, CX-072, SHR-1316, JTX-4014, GNS-1480, рекомбинантного гуманизированного анти-PD1 mAb (Shanghai Junshi Biosciences), REGN-2810, пелареорепа, SHR-1210, PD1/PDL1 ингибирующей вакцины (THERAVECTYS), BGB-A317, рекомбинантного гуманизированного анти-PD-1 mAb (Bio-Thera Solutions), Probody нацеленного на PD-1 (CytomX), XmAb-20717, FS-118, PSI-001, SN-PDL01, SN-PD07, PD-1 модифицированных TIL (Sangamo Therapeutics), PRS-332, FPT-155, jienuo mAb (Genor Biopharma), TSR-042, REGN-1979, REGN-2810, ресминостата, FAZ-053, PD-1/CTLA-4 биспецифического антитела (MacroGenics), MGA-012, MGD-013, M-7824, биспецифического антитела на основе PD-1 (Beijing Hanmi Pharmaceutical), AK-112, AK-106, AK-104, AK-103, BI-754091, ENUM-244C8, MCLA-145, MCLA-134, анти-PD-1 онколитического моноклонального антитела (Transgene SA), AGEN-2034, IBI-308, WBP-3155, JNJ-63723283, MEDI-0680, SSI-361, CBT-502, анти-PD-1 биспецифического антитела, нацеленного на две мишени анти-PD-1/LAG-3 mAbs (TESARO), нацеленного на две мишени анти-PD-1/TIM-3 mAbs (TESARO), PF-06801591, LY-3300054, BCD-100, STI-1110, биоаналога пембролизумаба, биоаналога ниволумаба, терапии PD-L1–ТФР-бета, KY-1003, STI-1014, GLS-010, AM-0001, GX-P2, KD-033,PD-L1/BCMA биспецифического антитела (Immune Pharmaceuticals), биспецифического антитела против PD-1/Ox40 (Immune Pharmaceuticals), BMS-936559, анти-PD-1/VEGF-A DARPins (Molecular Partners), mDX-400, ALN-PDL, PD-1 ингибирующего пептида (Aurigene), вакцины на основе дендритных клеток с миРНК (Alnylam Pharmaceuticals), GB-226, иммунотерапии PD-L1 на основе CAR-TNK (TNK Therapeutics/NantKwest), INSIX RA, INDUS-903, AMP-224, биспецифического гуманизированного антитела анти-CTLA-4/анти-PD-1 (Akeso Biopharma), вакцины B7-H1 (State Key Laboratory of Cancer Biology/Fourth Military Medical University) и GX-D1.
В некоторых вариантах осуществления, антагонист хемокинового рецептора CCR1 и PD-1 ингибитор или PD-L1 ингибитор вводят в состав препаратов для параллельного введения.
В других вариантах осуществления, антагонист хемокинового рецептора CCR1 и PD-1 ингибитор или PD-L1 ингибитор вводят в состав препаратов для последовательного введения.
CCR1 Антагонисты
Способы, композиции и наборы, описанные в настоящем изобретении, включают CCR1 антагонист. В некоторых вариантах осуществления, описан CCR1 антагонист, выбранный из описанных ниже формул I, II, III, IV и V, а также их подструктур. В некоторых вариантах осуществления, CCR1 антагонист выбран из описанных ниже соединений 1.001, 3.002, 4.005, 5.005 или 3.001, или их фармацевтически приемлемых солей. В некоторых случаях, CCR1 антагонист может представлять собой любое соединение, которое может быть антагонистом или ингибитором хемокинового рецептора CCR1, включая (но не ограничиваясь только ими) описанные, например, в патентах США № 7,524,845; 7,576,106; 7,629,344; 8,343,975; 9,169,248; и в заявках на патент США № 2014/0171420 и 2014/0179733, которые включены в настоящий текст в полном объеме посредством ссылки.
В некоторых вариантах осуществления, соединение, которое подавляет одну или больше функций CCR1, можно вводить субъекту для лечения раковой солидной опухоли. В других вариантах осуществления, соединение, которое подавляет одну или больше функций CCR1, вводят для стимулирования (подавления или усиления) иммунного ответа, что приводит к благоприятному стимулированию противоракового ответа.
В некоторых вариантах осуществления, в настоящем изобретении описан способ уменьшения метастазов в легких путем применения монотерапии CCR1 антагонистом. CCR1 антагонист может быть любым из описанных в настоящем тексте.
В некоторых вариантах осуществления, CCR1 антагонисты имеют формулу:
 (I)
(I)
или представляют собой ее фармацевтически приемлемую соль, ротамер или оптический изомер.
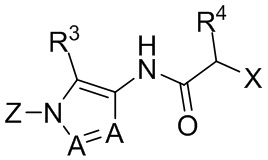
В формуле I, подстрочный индекс n представляет собой целое число от 0 до 3; каждый R1a и R1b независимо выбран из группы, состоящей из H, C1-8 алкила, C1-8 галогеналкила, C3-6 циклоалкила, -CORa, -CO2Ra, -CONRaRb, -NRaRb, -NRaCORb, -ORa, -X1CORa, -X1CO2Ra, -X1CONRaRb, -X1NRaCORb, -X1NRaRb и -X1ORa, где X1 выбран из группы, состоящей из C1-4 алкилена, и каждый Ra и Rb независимо выбран из группы, состоящей из атома водорода, C1-8 алкила, C1-8 галогеналкила и C3-6 циклоалкила, и опционально две R1a группы на соседних атомах углерода объединены с образованием 5-, 6- или 7-членного карбоциклического или гетероциклического кольца; каждый из R2a и R2b независимо выбран из группы, состоящей из H, гидроксила, C1-8 алкила, C1-8 галогеналкила, C1-8 алкокси-группы, C1-4 алкокси-C1-4 алкила, C1-8 гидроксиалкила, C1-4 алкокси-C1-4 алкокси-группы, C3-6 циклоалкила, C3-6 циклоалкил-C1-4 алкила, 3-7-членного гетероциклоалкила, 3-7-членного гетероциклоалкил-C1-4 алкила, -X1CO2Ra, -X1CONRaRb, -X1NRaCORb, -X1NRaRb, где X1, Ra и Rb имеют указанные выше значения.
Символ Ar1 представляет собой 6- или 10-членное моноциклическое или конденсированное бициклическое арильное кольцо, или 5-10-членное моноциклическое или конденсированное бициклическое гетероарильное кольцо; каждое из которых имеет от одного до пяти заместителей, R3, R3a, R3b, R4 и R4a, которые независимо выбраны из группы, состоящей из H, галогена, -ORc, -OC(O)Rc, -NRcRd, -SRc, -Re, -CN, -NO2, -CO2Rc, -CONRcRd, -C(O)Rc, -OC(O)NRcRd, -NRdC(O)Rc, -NRdC(O)2Re, -NRc-C(O)NRcRd, -NH-C(NH2)=NH, -NReC(NH2)=NH, -NH-C(NH2)=NRe, -NH-C(NHRe)=NH, -S(O)Re, -S(O)2Re, -NRcS(O)2Re, -S(O)2NRcRd, -N3, -X2ORc, -O-X2ORc, -X2OC(O)Rc, -X2NRcRd, -O-X2NRcRd, -X2SRc, -X2CN, -X2NO2, -X2CO2Rc, -O-X2CO2Rc, -X2CONRcRd, -O-X2CONRcRd, -X2C(O)Rc, -X2OC(O)NRcRd, -X2NRdC(O)Rc, -X2NRdC(O)2Re, -X2NRcC(O)NRcRd, -X2NH-C(NH2)=NH, -X2NReC(NH2)=NH, -X2NH-C(NH2)=NRe, -X2NH-C(NHRe)=NH, -X2S(O)Re, -X2S(O)2Re, -X2NRcS(O)2Re, -X2S(O)2NRcRd, -X2N3, -NRd-X2ORc, -NRd-X2NRcRd, -NRd-X2CO2Rc и -NRd-X2CONRcRd, где каждый X2 независимо выбран из группы, состоящей из C1-4 алкилена, и каждый Rc и Rd независимо выбран из атома водорода, C1-8 алкила, C1-8 гидроксиалкила, C1-8 галогеналкила и C3-6 циклоалкила, или опционально Rc и Rd, когда они присоединены к одному атому азота, могут быть объединены с атомом азота с образованием 5- или 6-членного кольца, содержащего от 0 до 2 дополнительных гетероатомов в качестве членов цикла; и каждый Re независимо выбран из группы, состоящей из C1-8 алкила, C1-8 гидроксиалкила, C1-8 галогеналкила и C3-6 циклоалкила.
Символ Ar2 представляет собой 6- или 10-членное моноциклическое или конденсированное бициклическое арильное кольцо, или 5-10-членное моноциклическое или конденсированное бициклическое гетероарильное кольцо; каждое из которых имеет от одного до пяти заместителей, R5, R6, R7, R8 и R9, независимо выбранных из группы, состоящей из H, галогена, -ORf, -OC(O)Rf, -NRfRg, -SRf, -Rh, -CN, -NO2, -CO2Rf, -CONRfRg, -C(O)Rf, -OC(O)NRfRg, -NRgC(O)Rf, -NRgC(O)2Rh, -NRf-C(O)NRfRg, -NH-C(NH2)=NH, -NRhC(NH2)=NH, -NH-C(NH2)=NRh, -NH-C(NHRh)=NH, -S(O)Rh, -S(O)2Rh, -NRfS(O)2Rh, -S(O)2NRfRg, -NRfS(O)2NRfRg, -N3, -X3ORf, -X3OC(O)Rf, -X3NRfRg, -X3SRf, -X3CN, -X3NO2, -X3CO2Rf, -X3CONRfRg, -X3C(O)Rf, -X3OC(O)NRfRg, -X3NRgC(O)Rf, -X3NRgC(O)2Rh, -X3NRf-C(O)NRfRg, -X3NH-C(NH2)=NH, -X3NRhC(NH2)=NH, -X3NH-C(NH2)=NRh, -X3NH-C(NHRh)=NH, -X3S(O)Rh, -X3S(O)2Rh, -X3NRfS(O)2Rh, -X3S(O)2NRfRg, -Y, -X3Y, -S(O)2Y, -C(O)Y, -X3N3, -O-X3ORf, -O-X3NRfRg, -O-X3CO2Rf, -O-X3CONRfRg, -NRg-X3ORf, -NRg-X3NRfRg, -NRg-X3CO2Rf и -NRg-X3CONRfRg, где Y представляет собой 5- или 6-членное арильное, гетероарильное или гетероциклическое кольцо, необязательно замещенное 1-3 заместителями, выбранными из группы, состоящей из галогена, -ORf, -OC(O)Rf, -NRfRg, -Rh, -SRf, -CN, -NO2, -CO2Rf, -CONRfRg, -C(O)Rf, -NRgC(O)Rf, -NRgC(O)2Rh, -S(O)Rh, -S(O)2Rh, -NRfS(O)2Rh, -S(O)2NRfRg, -X3ORf, X3SRf, -X3CN, -X3NO2, -X3CO2Rf, -X3CONRfRg, -X3C(O)Rf, -X3OC(O)NRfRg, -X3NRgC(O)Rf, -X3NRgC(O)2Rh, -X3NRf-C(O)NRfRg, -X3OC(O)Rf, -X3S(O)Rh, -X3S(O)2Rh, -X3NRfRg, -X3NRfS(O)2Rh, -X3S(O)2NRfRg, -O-X3ORf, -O-X3NRfRg, -O-X3CO2Rf, -O-X3CONRfRg, -NRg-X3ORf, -NRg-X3NRfRg, -NRg-X3CO2Rf и -NRg-X3CONRfRg, и где каждый X3 независимо выбран из группы, состоящей из C1-4 алкилена, и каждый Rf и Rg независимо выбран из атома водорода, C1-8 алкила, C1-8 гидроксиалкила, C1-8 галогеналкила и C3-6 циклоалкила, или, когда они присоединены к одному атому азота, могут быть объединены с атомом азота с образованием 5- или 6-членного кольца, содержащего от 0 до 2 дополнительных гетероатомов в качестве членов цикла, и каждый Rh независимо выбран из группы, состоящей из C1-8 алкила, C2-8 алкенила, C2-8 алкинила, C1-8 гидроксиалкила, C1-8 галогеналкила и C3-6 циклоалкила; или когда два из R5, R6, R7, R8 и R9 присоединены к двум соседним вершинам цикла Ar2, то они опционально объединены с образованием 5- или 6-членного кольца, содержащего 0, 1 или 2 гетероатома, выбранных из O и N, в качестве членов цикла.
В некоторых вариантах осуществления, соединения, имеющие формулу I, представляют собой соединения, в которых Ar1 выбран из группы, состоящей из фенила, нафтила, пиридила, пиразинила, пиридазинила, пиримидинила, триазинила, хинолинила, хиноксалинила и пуринила, каждый из которых необязательно имеет заместители R3, R3a, R3b, R4 и R4a.
В других вариантах осуществления, соединения, имеющие формулу I, представляют собой соединения, в которых Ar1 выбран из группы, состоящей из фенила, нафтила и пиридила, каждый из которых необязательно имеет заместители R3, R3a, R3b, R4 и R4a.
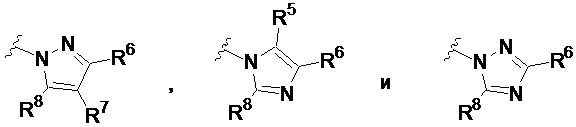
В других вариантах осуществления, соединения, имеющие формулу I, представляют собой соединения, в которых Ar2 выбран из группы, состоящей из фенила, пиразолила, имидазолила, триазолила, тетразолила, оксазолила, изоксазолила, оксадиазолила, оксатиадиазолила, пирролила, тиазолила, изотиазолила, бензимидазолила, бензоксазолила, бензопиразолила, бензотриазолила, пиразоло[3,4-b]пиридина, пиразоло[3,4-d]пиримидина, имидазо[4,5-b]пиридина, имидазо[1,5-a]пиридина и пирроло[2,3-b]пиридина, каждый из которых необязательно имеет заместитель R5, R6 и R7.
В других вариантах осуществления, соединения, имеющие формулу I, представляют собой соединения, в которых Ar2 выбран из группы, состоящей из пиразолила, имидазолила и триазолила, каждый из которых имеет заместитель R5, R6 и R7.
В некоторых вариантах осуществления, соединения, имеющие формулу I, представляют собой соединения, в которых Ar1 выбран из группы, состоящей из фенила, нафтила и пиридила, каждый из которых имеет от одного до пяти заместителей R3, R3a, R3b, R4 и R4a; и Ar2 выбран из группы, состоящей из пиразолила, имидазолила и триазолила, каждый из которых имеет заместитель R5, R6 и R7.
В частных вариантах осуществления, соединения, имеющие формулу I, представляют собой соединения, в которых Ar1 представляет собой фенил, который имеет от одного до пяти заместителей R3, R3a, R3b, R4 и R4a, и Ar2 выбран из группы, состоящей из пиразолила, имидазолила, бензимидазолила, бензопиразолила, пиразоло[3,4-b]пиридина, пиразоло[3,4-d]пиримидина, имидазо[4,5-b]пиридина, имидазо[1,5-a]пиридина и пирроло[2,3-b]пиридина, каждый из которых необязательно имеет заместитель R5, R6 и R7.
Другими вариантами осуществления настоящего изобретения являются соединения, имеющие формулы Ia, Ia1, Ia2, Ib, Ic, Id, II, IIa, IIb, IIb1, IIb2, IIb2a, IIb2b, IIb2c, IIc, IIb3, IIb2d, IIb2e, IIb2f, III, IIIa, IIIb, IIIc, IIb1, IIIb1a, IIIb1b, IIIb1c, IIIb2, IIIb3a, IIIb3b, IIIb3c, IV, IVa, IVb, IVc, IVd,V, Va и Vb.
Соответственно, в некоторых вариантах осуществления, указанные соединения представляют собой соединения, имеющие формулу Ia:
 (Ia)
(Ia)
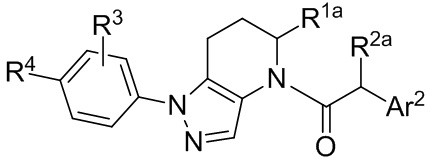
или их фармацевтически приемлемую соль, ротамер или оптический изомер, где R3 и R4 независимо выбраны из группы, состоящей из H, галогена, -Re, -CN и -SO2Re; и группы R1a, R2a и Ar2 имеют значения, указанные выше для формулы I или других вариантов осуществления.
В других вариантах формулы I или Ia, Ar2 представляет собой гетероарильную группу; в других вариантах осуществления, Ar2 представляет собой гетероарильную группу, необязательно замещенную и присоединенную к остальной части молекулы через входящий в состав цикла атом азота; и в других вариантах осуществления, Ar2 имеет формулу:
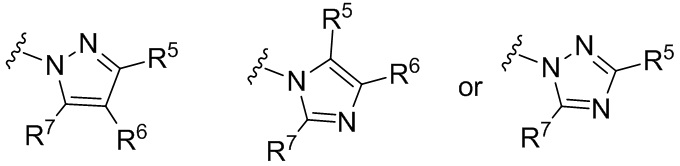
где R5, R6 и R7 независимо выбраны из группы, состоящей из H, галогена, -Rh, -CN, -SO2Rh, -CO2Rf, -CONRfRg и Y, где –Rh, Rf, Rg и Y имеют значения, указанные выше для формулы I.
В одной группе частных вариантов осуществления, соединения имеют формулу:
 (Ia1)
(Ia1)
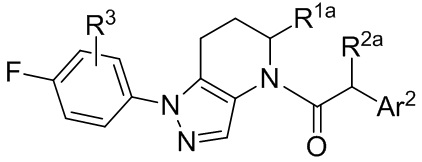
или представляют собой его фармацевтически приемлемую соль, ротамер или оптический изомер, где R4 выбран из группы, состоящей из F и Cl; и группы R1a, R2a, R3 и Ar2 имеют значения, указанные выше для формулы I или Ia или других вариантов осуществления.
В другой группе частных вариантов осуществления, соединения имеют формулу:
 (Ia2)
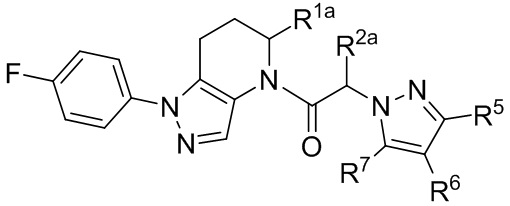
(Ia2)
или представляют собой его фармацевтически приемлемую соль, ротамер или оптический изомер, где R3 выбран из группы, состоящей из H, галогена, C1-8 алкила, C1-8 галогеналкила и C1-8 алкокси-группы; R1a и R2a независимо выбраны из группы, состоящей из H, C1-8 алкила, C1-8 галогеналкила, C1-8 алкокси-группы и C1-8 гидроксиалкила; и Ar2 имеет значение, указанное выше для формулы I или Ia или других вариантов осуществления.
В другой группе частных вариантов осуществления, соединения имеют формулу:
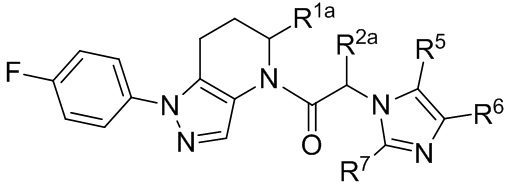
 (Ib)
(Ib)
или представляют собой его фармацевтически приемлемую соль, ротамер или оптический изомер, где R1a и R2a независимо выбраны из группы, состоящей из H, C1-8 алкила, C1-8 галогеналкила, C1-8 алкокси-группы и C1-8 гидроксиалкила; и R5, R6 и R7 независимо выбраны из группы, состоящей из H, галогена, -Rh, -CN, -SO2Rh, -CO2Rf, -CONRfRg и Y; где –Rh, Rf, Rg и Y имеют значения, указанные выше для формулы I.
В другой группе частных вариантов осуществления, соединения имеют формулу:
 (Ic)
(Ic)
или представляют собой его фармацевтически приемлемую соль, ротамер или оптический изомер, где R1a и R2a независимо выбраны из группы, состоящей из H, C1-8 алкила, C1-8 галогеналкила, C1-8 алкокси-группы и C1-8 гидроксиалкила; и R5, R6 и R7 независимо выбраны из группы, состоящей из H, галогена, -Rh, -CN, -SO2Rh, -CO2Rf, -CONRfRg и Y; где –Rh, Rf, Rg и Y имеют значения, указанные выше для формулы I.
В другой группе частных вариантов осуществления, соединения имеют формулу:
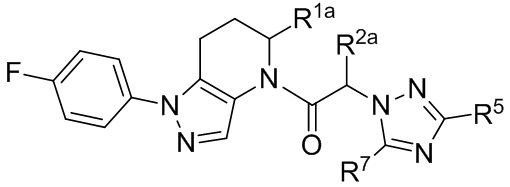 (Id)
(Id)
или представляют собой его фармацевтически приемлемую соль, ротамер или оптический изомер, где R1a и R2a каждый независимо выбраны из группы, состоящей из H, C1-8 алкила, C1-8 галогеналкила, C1-8 алкокси-группы и C1-8 гидроксиалкила; и R5 и R7 каждый независимо выбраны из группы, состоящей из H, галогена, -Rh, -CN, -SO2Rh, -CO2Rf, -CONRfRg и Y; где -Rh, Rf, Rg и Y имеют значения, указанные выше для формулы I.
Для любых из описанных выше вариантов осуществления, когда присутствует Y, частные варианты осуществления представляют собой соединения, в которых Y выбран из группы, состоящей из пиридила, пиримидинила, имидазолила, оксазолила, оксадиазолила, триазолила, тиазолила, имидазолинила и пиразолила.
В некоторых вариантах осуществления, CCR1 антагонист представляет собой соединение, выбранное из
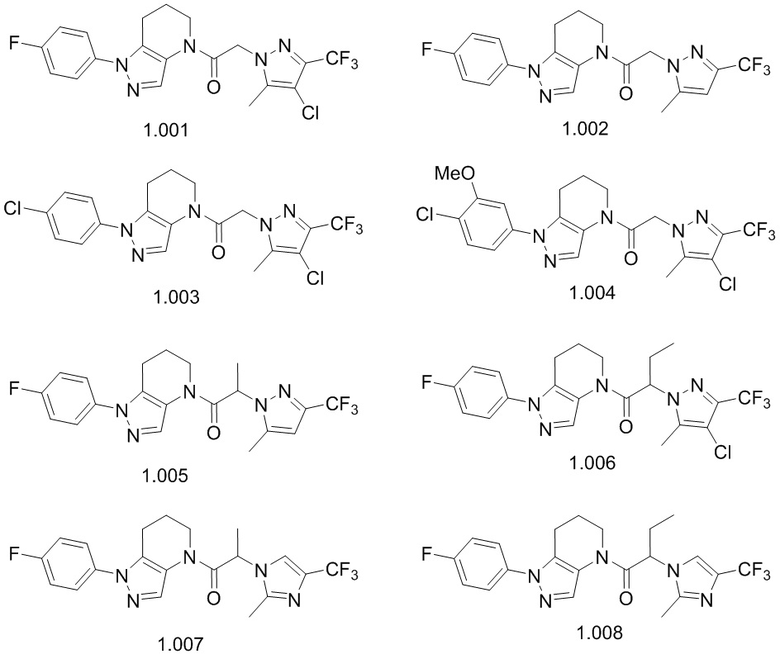 ,
,
или его фармацевтически приемлемую соль.
В частном варианте осуществления, CCR1 антагонист представляет собой соединение 1.001,
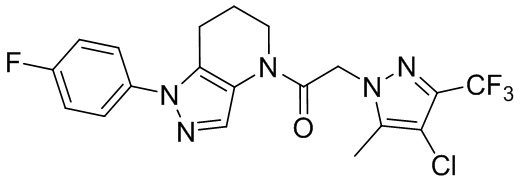 , или его фармацевтически приемлемую соль.
, или его фармацевтически приемлемую соль.
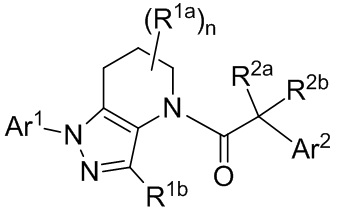
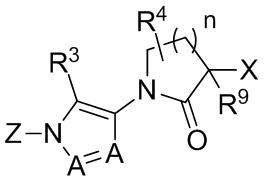
В других вариантах осуществления, CCR1 антагонист представляет собой соединение, имеющее Формулу II:
 (II)
(II)
или его фармацевтически приемлемую соль, гидрат, сольват, N-оксид или ротамер. В формуле II, каждый A независимо выбран из группы, состоящей из N и CH;
X и Z каждый независимо выбраны из группы, состоящей из
(i) моноциклического или конденсированного бициклического арила и гетероарила, где гетероарильная группа содержит в качестве членов цикла 1-4 гетероатомов, выбранных из N, O и S;
(ii) моноциклического 4-, 5-, 6- или 7-членного кольца, выбранного из группы, состоящей из циклоалкана и гетероциклоалкана, где гетероциклоалкановые кольца содержат в качестве членов цикла 1-3 гетероатомов, выбранных из N, O и S;
где каждое из колец в (i) и (ii) необязательно замещено 1-5 заместителями, выбранными из галогена, CN, C1-8 алкила, C3-8 циклоалкила, C2-8 алкенила, C2-8 алкинила, C1-8 галогеналкила, C1-8 гидроксиалкила, -ORa, -CO2Ra, -SO2Ra, -NRaRb, -CONRaRb, арила, 5- или 6-членного гетероарила, и 3-, 4-, 5- или 6-членного гетероциклоалкана, где гетероатомы, присутствующие в вершинах цикла гетероарильного и гетероциклоалканового колец, выбраны из N, O и S, и где алкильные, циклоалкильные, арильные, гетероарильные и гетероциклоалкановые фрагменты необязательно дополнительно замещены 1-3 группами Ra; и, необязательно, два заместителя на соседних вершинах цикла соединены с образованием дополнительного 5- или 6-членного кольца, являющегося насыщенным, ненасыщенным или ароматическим, с вершинами цикла, выбранными из C, O, N и S;
R3 выбран из группы, состоящей из H, галогена, CN, C1-8 алкила, C3-8 циклоалкила, C2-8 алкенила, C2-8 алкинила, C1-8 галогеналкила, C1-8 гидроксиалкила, -ORa, -CO2Ra, -NRaRb, -CONRaRb, арила, 5- или 6-членного гетероарила, и 3-, 4-, 5- или 6-членного гетероцикла, где гетероатомы, присутствующие в качестве вершин цикла в гетероарильных и гетероциклических кольцах, выбраны из N, O и S, и где алкильные, циклоалкильные, арильные, гетероарильные и гетероциклические фрагменты в R3 необязательно дополнительно замещены 1-3 группами Ra;
R4 выбран из группы, состоящей из H, -ORa и C1-8 алкила, необязательно имеющего заместитель -ORa или -NRaRb; или R4 объединен с X с образованием бициклической конденсированной системы; и
каждый Ra и Rb независимо выбран из группы, состоящей из атома водорода, гидроксила, галогена, циано-группы, C1-8 алкила, C1-8 алкокси-группы, C1-8 галогеналкила, C3-6 циклоалкила, C3-6 циклоалкилалкила, амино-группы, C1-8 алкиламино-группы, ди-C1-8 алкиламино-группы, карбоксамида, карбокси C1-4 алкилового эфира, карбоксильной группы и -SO2- C1-8 алкила.
Специалисту в данной области будет понятно, что перечисления заместителей относятся только к тем случаям, когда получающиеся в итоге соединения устойчивы (например, менее 20% разложения при хранении), так что группа -ORa не включает такие компоненты, где Ra представляет собой алкокси-группу (что дало бы перокси- или –OO-алкильную группу).
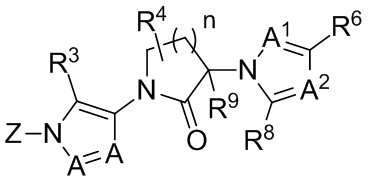
В некоторых частных вариантах осуществления, соединения, имеющие формулу II, представлены формулой IIa:
 (IIa)
(IIa)
где A1 представляет собой N или C(R5); A2 представляет собой N или C(R7); и R5, R6, R7 и R8 каждый независимо выбраны из H, галогена, CN, C1-8 алкила, C3-8 циклоалкила, C2-8 алкенила, C2-8 алкинила, C1-8 галогеналкила, C1-8 гидроксиалкила, -ORa, -CO2Ra, -NRaRb, -CONRaRb, арила, 5- или 6-членного гетероарила, и 3-, 4-, 5- или 6-членного гетероциклоалкана, где гетероатомы, присутствующие в вершинах цикла гетероарильного и гетероциклоалканового колец, выбраны из N, O и S, и где алкильные, циклоалкильные, арильные, гетероарильные и гетероциклоалкановые фрагменты в R5, R6, R7 и R8 необязательно дополнительно замещены 1-3 группами Ra; и опционально, R4 и R5, R4 и R8, или соседние представители R5, R6, R7 и R8 соединены с образованием дополнительного 5- или 6-членного кольца, являющегося насыщенным, ненасыщенным или ароматическим, с вершинами цикла, выбранными из C, O, N и S; или его фармацевтически приемлемая соль, гидрат, сольват, ротамер или N-оксид.
В других частных вариантах осуществления, соединения, имеющие формулу IIa, представляют собой соединения, где R8 отличается от H.
В других частных вариантах осуществления, соединения, имеющие формулу IIa, представлены формулой IIb:
 (IIb)
(IIb)
где R1 и R2 каждый независимо выбраны из H, галогена, CN, C1-8 алкила, C3-8 циклоалкила, C2-8 алкенила, C2-8 алкинила, C1-8 галогеналкила, C1-8 гидроксиалкила, -ORa, -CO2Ra, -SO2Ra, -NRaRb, -CONRaRb, и 3-, 4-, 5- или 6-членного гетероциклоалкана, где гетероатомы, присутствующие в вершинах цикла гетероциклоалканового кольца, выбраны из N, O и S, и где алкильные, циклоалкильные и гетероциклоалкановые фрагменты в R1 и R2 необязательно дополнительно замещены 1-3 группами Ra.
В частных вариантах формулы IIb, каждый R1 и R2 независимо выбран из H, галогена, CN, C1-8 алкила, C1-8 галогеналкила, -CO2Ra и -SO2Ra.
В других частных вариантах соединений, имеющих формулу IIb, соединения представлены структурой:
 (IIb1).
(IIb1).
В других частных вариантах соединений, имеющих формулу IIb и IIb1, кольцевой фрагмент, содержащий N, A1 и A2 в качестве вершин цикла, выбран из:
 .
.
В других частных вариантах соединений, имеющих формулу IIb и IIb1, кольцевой фрагмент, содержащий N, A1 и A2 в качестве вершин цикла, выбран из:
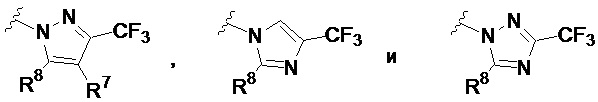
где R7 представляет собой H или Cl, и R8 представляет собой C1-8 алкил, необязательно замещенный 1 или 2 группами Ra.
В других частных вариантах Формулы IIb или IIb1, R4 представляет собой H или CH3.
Возвращаясь к формуле II, некоторые частные варианты осуществления представляют собой соединения, имеющие формулу IIb2:
 (IIb2)
(IIb2)
где R1 представляет собой Cl или F; R3 выбран из группы, состоящей из C1-8 алкила, C1-8 галогеналкила, C1-8 гидроксиалкила, где алкильный фрагмент в R3 необязательно дополнительно замещен 1-3 группами Ra; и где R7 и R8 не соединены с образованием кольца.
В других частных вариантах осуществления, соединения, имеющие формулу II, IIb, IIb1 и IIb2, представлены формулами IIb2a, IIb2b и IIb2c.
 (IIb2a)
(IIb2a)
 (IIb2b)
(IIb2b)
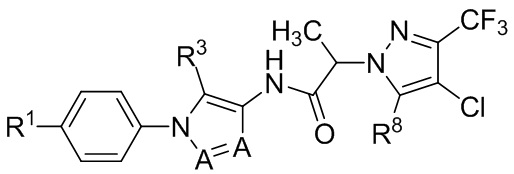 (IIb2c).
(IIb2c).
В некоторых частных вариантах осуществления, соединения представлены формулой IIc:
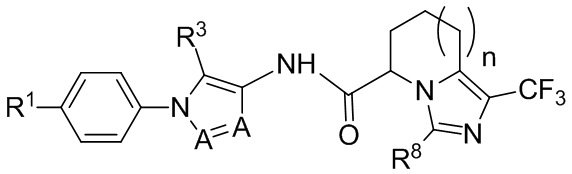 (IIc)
(IIc)
где подстрочный индекс n равен 0 или 1.
В некоторых частных вариантах осуществления, соединения представлены формулой IIb3:
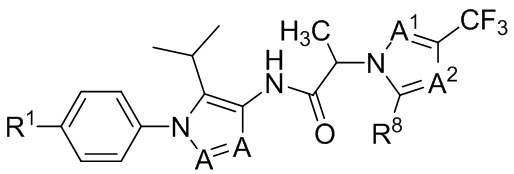 (IIb3).
(IIb3).
В некоторых частных вариантах формулы IIb, соединения представлены формулами IIb2d, IIb2e и IIb2f.
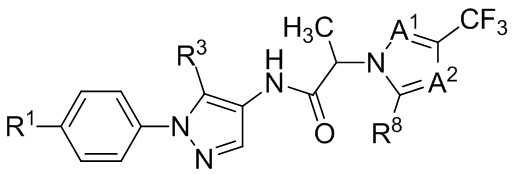 (IIb2d)
(IIb2d)
 (IIb2e)
(IIb2e)
 (IIb2f).
(IIb2f).
В частных вариантах любой из формул II, IIa, IIb, IIb1, IIb2, IIb2a, IIb2b, IIb2c, IIb2d, IIb2e, IIb2f, IIb3 и IIc, R3 представляет собой C1-8 алкил.
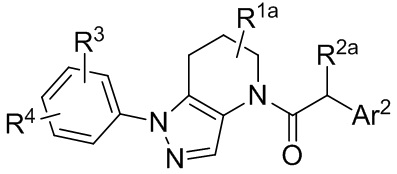
В других вариантах осуществления, CCR1 антагонисты, которые могут применяться в способах и композициях по настоящему изобретению, представлены формулой III:
 (III)
(III)
или представляют собой его фармацевтически приемлемую соль, гидрат, сольват, N-оксид или ротамер. В формуле III, индекс n представляет собой целое число от 0 до 3;
каждый A независимо выбран из группы, состоящей из N и CH;
X и Z каждый независимо выбраны из группы, состоящей из
(i) моноциклического или конденсированного бициклического арила и гетероарила, где гетероарильная группа содержит в качестве членов цикла 1-4 гетероатомов, выбранных из N, O и S;
(ii) моноциклического 4-, 5-, 6- или 7-членного кольца, выбранного из группы, состоящей из циклоалкана и гетероциклоалкана, где гетероциклоалкановые кольца содержат в качестве членов цикла 1-3 гетероатомов, выбранных из N, O и S;
где каждое из колец в (i) и (ii) необязательно замещены 1 - 5 заместителями, выбранными из галогена, CN, C1-8 алкила, C3-8 циклоалкила, C2-8 алкенила, C2-8 алкинила, C1-8 галогеналкила, C1-8 гидроксиалкила, -ORa, -CO2Ra, -SO2Ra, -NRaRb, -CONRaRb, арила, 5- или 6-членного гетероарила, и 3-, 4-, 5- или 6-членного гетероциклоалкана, где гетероатомы, присутствующие в вершинах цикла гетероарильного и гетероциклоалканового колец, выбраны из N, O и S, и где алкильные, циклоалкильные, арильные, гетероарильные и гетероциклоалкановые фрагменты необязательно дополнительно замещены 1-3 группами Ra; и необязательно, два заместителя на соседних вершинах цикла соединены с образованием дополнительного 5- или 6-членного кольца, являющегося насыщенным, ненасыщенным или ароматическим, с вершинами цикла, выбранными из C, O, N и S;
R3 выбран из группы, состоящей из H, галогена, CN, C1-8 алкила, C3-8 циклоалкила, C2-8 алкенила, C2-8 алкинила, C1-8 галогеналкила, C1-8 гидроксиалкила, -ORa, -CO2Ra, -NRaRb, -CONRaRb, арила, 5- или 6-членного гетероарила, и 3-, 4-, 5- или 6-членного гетероцикла, где гетероатомы, присутствующие в качестве вершин цикла в гетероарильных и гетероциклических кольцах, выбраны из N, O и S, и где алкильные, циклоалкильные, арильные, гетероарильные и гетероциклические фрагменты в R3 необязательно дополнительно замещены 1-3 группами Ra;
R4 выбран из группы, состоящей из H, -ORa и C1-8 алкила, необязательно имеющего заместитель -ORa;
R9 выбран из группы, состоящей из H и C1-8 алкила, необязательно имеющего заместитель -ORa;
каждый Ra и Rb независимо выбран из группы, состоящей из атома водорода, гидроксила, галогена, циано-группы, C1-8 алкила, C1-8 алкокси-группы, C1-8 галогеналкила, C3-6 циклоалкила, C3-6 циклоалкилалкила, амино-группы, C1-8 алкиламино-группы, ди-C1-8 алкиламино-группы, карбоксамида, карбокси C1-4 алкилового эфира, карбоксильной группы и -SO2- C1-8 алкила.
В некоторых частных вариантах осуществления, соединения, имеющие формулу III, представлены формулой IIIa:
 (IIIa)
(IIIa)
где A1 представляет собой N или C(R5); A2 представляет собой N или C(R7); и R5, R6, R7 и R8 каждый независимо выбраны из H, галогена, CN, C1-8 алкила, C3-8 циклоалкила, C2-8 алкенила, C2-8 алкинила, C1-8 галогеналкила, C1-8 гидроксиалкила, -ORa, -CO2Ra, -NRaRb, -CONRaRb, арила, 5- или 6-членного гетероарила, и 3-, 4-, 5- или 6-членного гетероциклоалкана, где гетероатомы, присутствующие в вершинах цикла гетероарильного и гетероциклоалканового колец, выбраны из N, O и S, и где алкильные, циклоалкильные, арильные, гетероарильные и гетероциклоалкановые фрагменты R5, R6, R7 и R8 необязательно дополнительно замещены 1-3 группами Ra; и, необязательно, соседние фрагменты R5, R6, R7 и R8 соединены с образованием дополнительного 5- или 6-членного кольца, являющегося насыщенным, ненасыщенным или ароматическим, с вершинами цикла, выбранными из C, O, N и S; или его фармацевтически приемлемая соль, гидрат, сольват, ротамер или N-оксид.
В других частных вариантах осуществления, соединения, имеющие формулу IIIa, представляют собой соединения, где R8 отличается от H.
В других частных вариантах осуществления, соединения, имеющие формулу IIIa, представлены формулой IIIb:
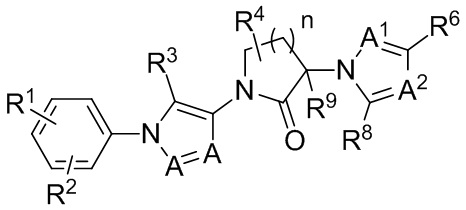 (IIIb)
(IIIb)
где R1 и R2 каждый независимо выбраны из H, галогена, CN, C1-8 алкила, C3-8 циклоалкила, C2-8 алкенила, C2-8 алкинила, C1-8 галогеналкила, C1-8 гидроксиалкила, -ORa, -CO2Ra, -SO2Ra, -NRaRb, -CONRaRb, и 3-, 4-, 5- или 6-членного гетероциклоалкана, где гетероатомы, присутствующие в вершинах цикла гетероциклоалканового кольца, выбраны из N, O и S, и где алкильные, циклоалкильные и гетероциклоалкановые фрагменты в R1 и R2 необязательно дополнительно замещены 1-3 группами Ra.
В частных вариантах формулы IIIb, каждый R1 и R2 независимо выбран из H, галогена, CN, C1-8 алкила, C1-8 галогеналкила, -CO2Ra и -SO2Ra.
В других частных вариантах соединений, имеющих формулу IIIb, кольцевой фрагмент, содержащий N, A1 и A2 в качестве вершин цикла, выбран из:
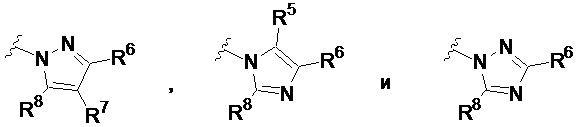 .
.
В других частных вариантах соединений, имеющих формулу IIIb, кольцевой фрагмент, содержащий N, A1 и A2 в качестве вершин цикла, выбран из:
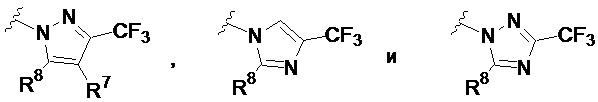
где R7 представляет собой H или Cl, и R8 представляет собой C1-8 алкил, необязательно замещенный 1 или 2 группами Ra.
В других частных вариантах формулы IIIb, R9 представляет собой H или CH3.
Возвращаясь к формуле III, некоторые частные варианты осуществления представляют собой соединения, имеющие формулу IIIc:
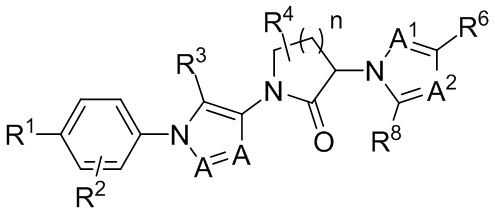 (IIIc)
(IIIc)
где индекс n равен 1, 2 или 3. Другие частные варианты осуществления представляют собой соединения, где n равен 1.
В других частных вариантах осуществления, соединения, имеющие формулу IIIb, представляют собой соединения, имеющие формулу IIIb1:
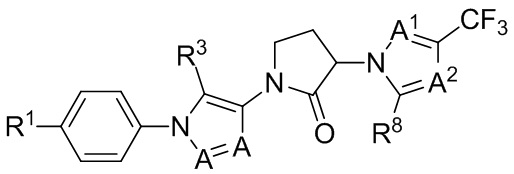 (IIIb1)
(IIIb1)
где R1 представляет собой Cl или F.
В других частных вариантах осуществления, соединения, имеющие формулу IIIb1, представлены формулами IIIb1a, IIIb1b и IIIb1c.
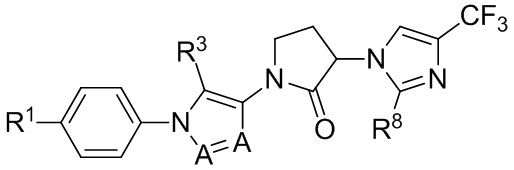 (IIIb1a)
(IIIb1a)
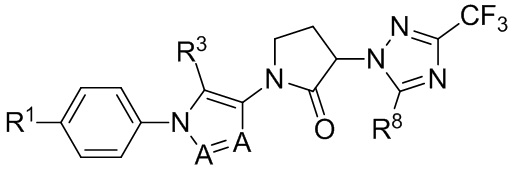 (IIIb1b)
(IIIb1b)
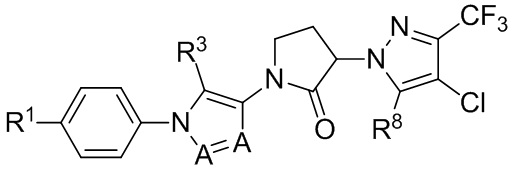 (IIIb1c).
(IIIb1c).
В некоторых частных вариантах формулы IIIb, соединения представлены формулой IIIb2:
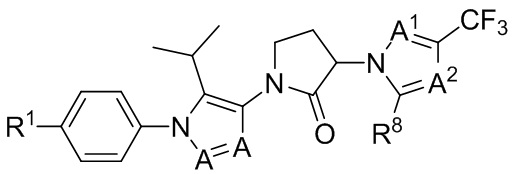 (IIIb2)
(IIIb2)
где R1 представляет собой Cl или F.
В некоторых частных вариантах формулы IIIb, соединения представлены формулами IIIb3a, IIIb3b и IIIb3c.
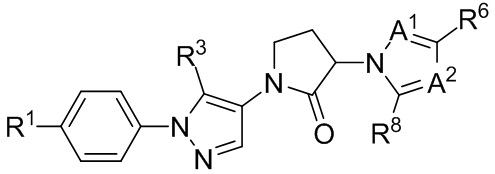 (IIIb3a)
(IIIb3a)
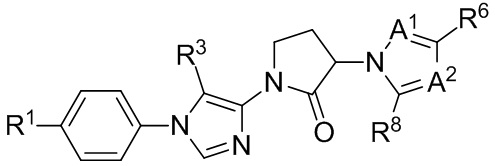 (IIIb3b)
(IIIb3b)
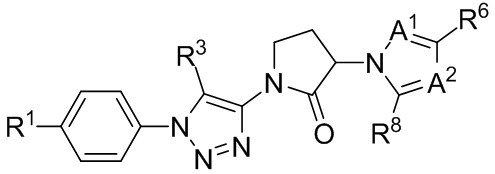 (IIIb3c).
(IIIb3c).
В частных вариантах любой из формул III, IIIa, IIIb, IIIc, IIIb1, IIIb1a, IIIb1b, IIIb1c, IIIb2, IIIb3a, IIIb3b и IIIb3c, R3 выбран из H, C1-8 алкила, C3-8 циклоалкила и C2-8 алкенила.
В некоторых частных вариантах осуществления, CCR1 антагонист выбран из:
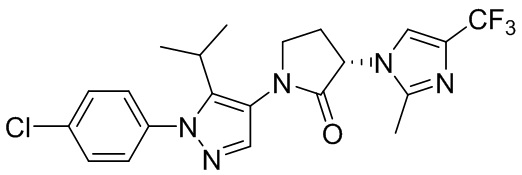 и
и 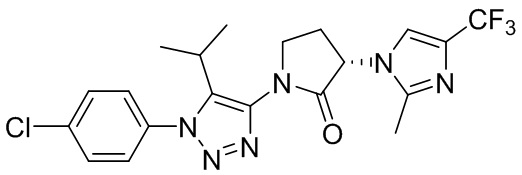
3.001 3.002
В некоторых частных вариантах осуществления, CCR1 антагонист представляет собой:
3.001.
В некоторых частных вариантах осуществления, CCR1 антагонист представляет собой:
3.002.
В других вариантах осуществления, CCR1 антагонисты, которые могут применяться в способах и композициях по настоящему изобретению, представлены формулой IV:
 (IV)
(IV)
или его фармацевтически приемлемой солью, гидратом или N-оксидом. В формуле IV, R1 представляет собой C1-4 алкил или C1-4 галогеналкил, и подстрочный индекс m представляет собой целое число от 0 до 1. В формуле IV, R2a, R2c, R2d каждый независимо выбраны из группы, состоящей из атома водорода, галогена, C1-4 алкокси-группы, C1-4 алкила, -O-C1-4 галогеналкила и C1-4 галогеналкила; R3 выбран из группы, состоящей из атома водорода и C1-4 алкила; и R4 представляет собой C1-4 алкил, и подстрочный индекс n представляет собой целое число от 0 до 2. В одном варианте осуществления, R3 представляет собой атом водорода.
В другом варианте осуществления, R3 представляет собой атом водорода, и подстрочный индекс n равен 0.
В другом варианте осуществления, R1 в формуле IV представляет собой метил, трифторметил или этил, и подстрочный индекс m равен 1.
В другом варианте осуществления подстрочный индекс m равен 0.
В другом варианте осуществления, R2a, R2c и R2d каждый независимо выбраны из группы, состоящей из фтора, хлора, брома, иода, метокси-группы, этокси-группы, трифторметокси-группы, метила, этила, трифторметила и 2-фторэтокси-группы.
В другом варианте осуществления, R2a представляет собой атом водорода, и R2c и R2d каждый независимо выбраны из группы, состоящей из фтора, хлора, брома, иода, метокси-группы, этокси-группы, трифторметокси-группы, метила, этила, трифторметила и 2-фторэтокси-группы.
В одном предпочтительном варианте осуществления, CCR1 антагонисты имеют формулу IVa или IVb.
IVa IVb.
В одном варианте осуществления, R2a и R2c в формуле IVa или IVb каждый независимо выбраны из группы, состоящей из фтора, хлора, брома и иода; и R2d выбран из группы, состоящей из метокси-группы, этокси-группы, трифторметокси-группы, метила, этила, трифторметила и 2-фторэтокси-группы.
В частном варианте осуществления, соединения, имеющие формулу IVa или IVb, выбраны из группы, состоящей из:
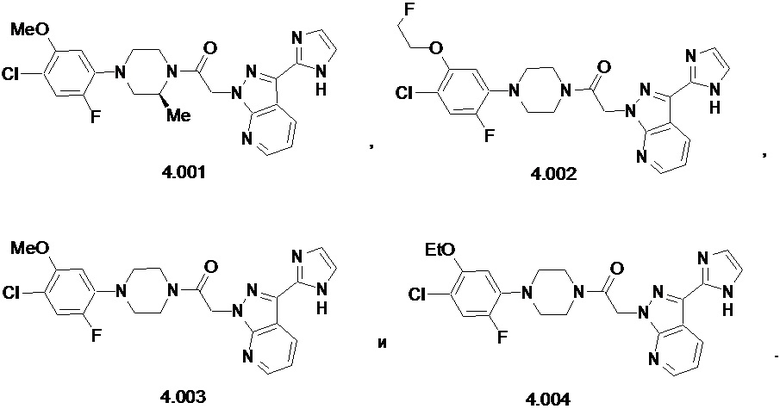 .
.
В другом частном варианте осуществления, соединения по настоящему изобретению имеют формулу IVc или IVd:
 ;
;
IVc IVd.
В формуле IVc и IVd, в некоторых вариантах осуществления, R2c выбран из группы, состоящей из фтора, хлора, брома и иода; и R2d выбран из группы, состоящей из метокси-группы, этокси-группы, трифторметокси-группы, метила, этила, трифторметила и 2-фторэтокси-группы.
В частном варианте осуществления, соединения, имеющие формулу IVc или IVd, выбраны из группы, состоящей из:
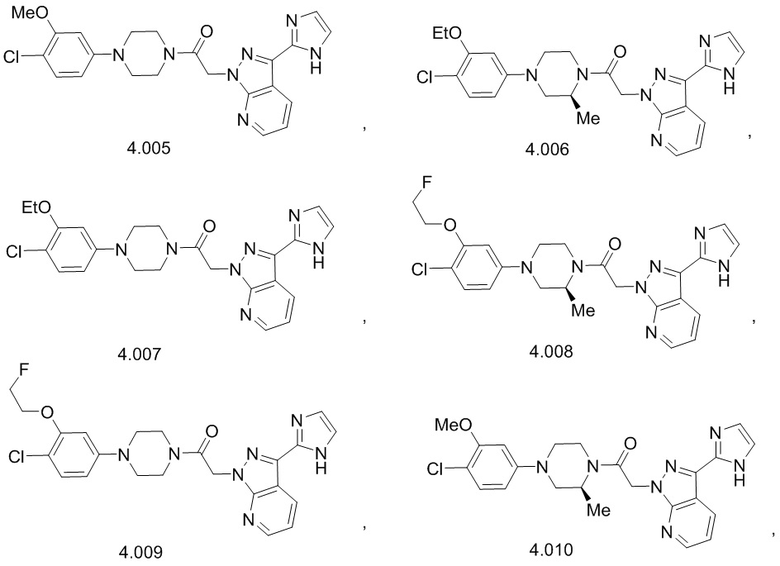
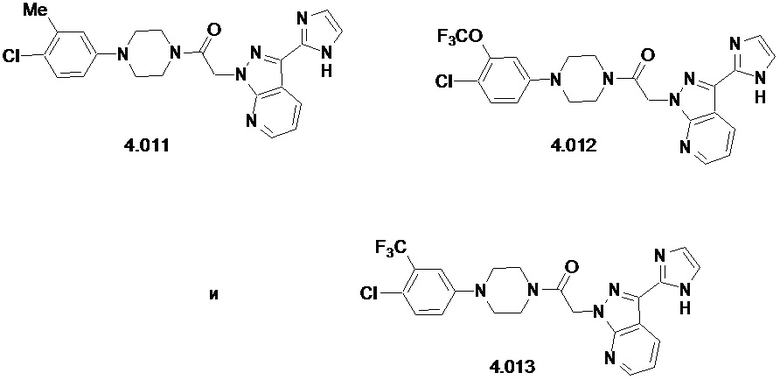 .
.
В другом варианте осуществления настоящего изобретения, соединение, имеющее формулу IVd, имеет следующую структуру:
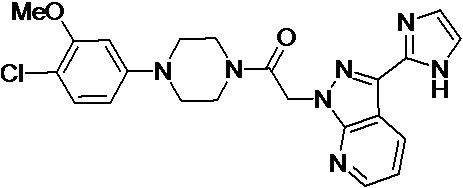
4.005.
В других вариантах осуществления, CCR1 антагонисты, которые могут применяться в способах и композициях по настоящему изобретению, представлены формулой V:
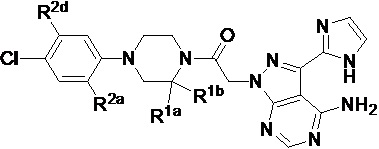 (V)
(V)
или его фармацевтически приемлемыми солями, гидратами или N-оксидами. В формуле V, R1a и R1b каждый независимо выбраны из группы, состоящей из H и CH3; R2a выбран из группы, состоящей из H и F; и R2d выбран из группы, состоящей из H, C1-4 алкокси-группы и C1-4 галогеналкокси-группы. В одном варианте осуществления, R2a представляет собой атом водорода.
В другом варианте осуществления, R1a и R1b каждый представляют собой H.
В другом варианте осуществления, R1b представляет собой метил, и R1a представляет собой H.
В другом варианте осуществления, R1a и R1b каждый представляют собой метил.
В другом варианте осуществления, R2a представляет собой атом водорода, и R2d выбран из группы, состоящей из метокси-группы, этокси-группы и трифторметокси-группы.
В одном предпочтительном варианте осуществления, соединения имеют формулу Va или Vb:
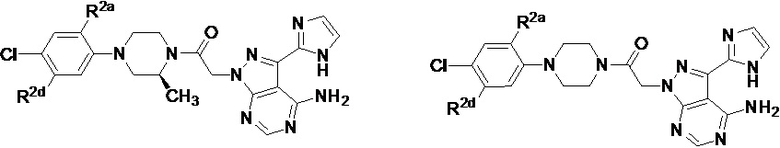
Va Vb
где R2a выбран из группы, состоящей из H и F; и R2d выбран из группы, состоящей из метокси-группы, этокси-группы и трифторметокси-группы.
В частном варианте осуществления, CCR1 антагонисты выбраны из группы, состоящей из:
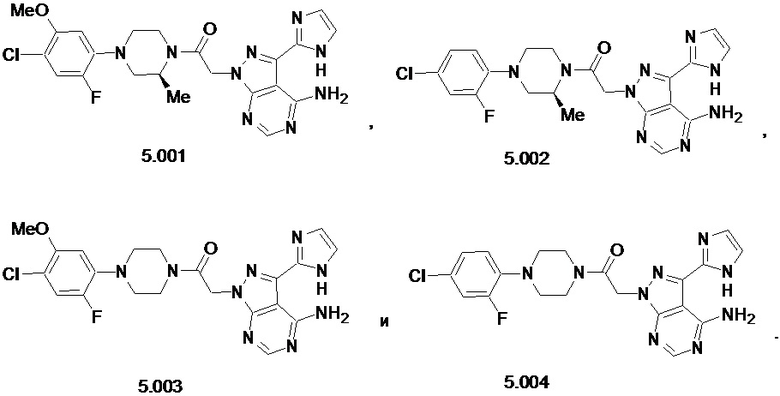 .
.
В другом частном варианте осуществления, CCR1 антагонисты выбраны из группы, состоящей из:
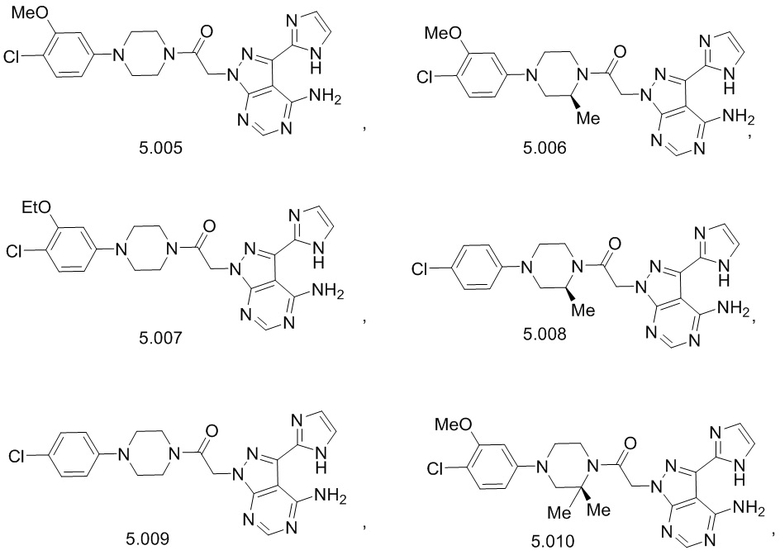
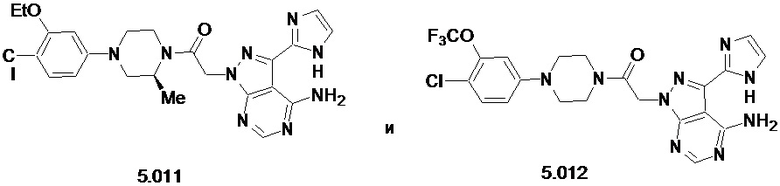 .
.
В другом варианте осуществления, CCR1 антагонист имеет структуру 5.005:
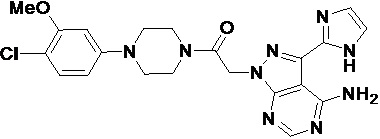
5.005
или представляет собой его фармацевтически приемлемая соль.
В некоторых вариантах осуществления, антагонист хемокинового рецептора CCR1 выбран из группы, состоящей из BL-5923, UCB-35625, BX-471, BI-638683, PS-031291, MLN-3701, AZD-4818, MLN-3897, CP-481715, F-18-CCR1, AOP-RANTES, PS-375179 и NSC-651016.
PD-1 ингибиторы и PD-L1 ингибиторы
Cпособы, композиции, и наборы, описанные в настоящем изобретении, включают ингибиторы иммунных контрольных точек, такие как ингибиторы PD-1/PD-L1 механизма.
В некоторых вариантах осуществления, ингибитор PD-1 механизма может представлять собой PD-1 антагонист, связывающийся с PD-1 антагонист, низкомолекулярный PD-1 антагонист, PD-1 ингибитор, анти-PD-1 биологический продукт (например, антитело или его фрагмент, специфично связывающийся с PD-1), PD-L1 антагонист, низкомолекулярный PD-L1 антагонист, связывающийся с PD-L1 антагонист, PD-L1 ингибитор, анти-PD-L1 биологический продукт (например, антитело или его фрагмент, специфично связывающийся с PD-L1) и т.п.
В некоторых вариантах осуществления, PD-L1 ингибитор может представлять собой дурвалумаб, или атезолизумаб, или авелумаб, или BMS-936559 (MDX-1105), или ALN-PDL, или TSR-042, или KD-033, или CA-170, или CA-327, или STI-1014, или MEDI-0680, или KY-1003.
В некоторых вариантах осуществления, PD-L1 ингибитор может представлять собой дурвалумаб, или атезолизумаб, или авелумаб, или BMS-936559 (MDX-1105), или ALN-PDL, или TSR-042, или KD-033, или CA-170, или STI-1014, или MEDI-0680, или KY-1003. Дурвалумаб (MEDI4736) представляет собой человеческое моноклональное антитело против PD-L1. Атрексолизумаб (MPDL3280A) представляет собой полностью гуманизированное, сконструированное IgG1 моноклональное антитело против PD-L1. Авелумаб (MSB0010718C) представляет собой полностью гуманизированное, сконструированное IgG1 моноклональное антитело против PD-L1. BMS-936559 (MDX-1105) представляет собой полностью гуманизированное, сконструированное IgG4 моноклональное антитело против PD-L1. ALN-PDL представляет собой ингибирующую РНК (RNAi) против PD-L1. TSR-042 представляет собой сконструированное химерное антитело нацеленное против PD-1/PD-L1 механизма. KD-033 представляет собой бифункциональный анти-PD-L1/IL-15 химерный белок, в котором анти-PD-L1 антитело присоединено своим хвостом к цитокину IL-15 по sushi-домену IL-15 рецептора. CA-170 представляет собой низкомолекулярный антагонист PD-L1 и VISTA. STI-1014 представляет собой анти-PD-L1 антитело. KY-1003 представляет собой моноклональное антитело против PD-L1. CA-327 представляет собой низкомолекулярный антагонист PD-L1 и TIM3.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, пембролизумаба, ниволумаба, AP-106, AP-105, MSB-2311, CBT-501, авелумаба, AK-105, IO-102, IO-103, PDR-001, CX-072, SHR-1316, JTX-4014, GNS-1480, рекомбинантного гуманизированного анти-PD1 mAb (Shanghai Junshi Biosciences), REGN-2810, пелареорепа, SHR-1210, PD1/PDL1 ингибирующей вакцины (THERAVECTYS), BGB-A317, рекомбинантного гуманизированного анти-PD-1 mAb (Bio-Thera Solutions), Probody нацеленного на PD-1 (CytomX), XmAb-20717, FS-118, PSI-001, SN-PDL01, SN-PD07, PD-1 модифицированных TIL (Sangamo Therapeutics), PRS-332, FPT-155, jienuo mAb (Genor Biopharma), TSR-042, REGN-1979, REGN-2810, ресминостата, FAZ-053, PD-1/CTLA-4 биспецифического антитела (MacroGenics), MGA-012, MGD-013, M-7824, биспецифического антитела на основе PD-1 (Beijing Hanmi Pharmaceutical), AK-112, AK-106, AK-104, AK-103, BI-754091, ENUM-244C8, MCLA-145, MCLA-134, анти-PD-1 онколитического моноклонального антитела (Transgene SA), AGEN-2034, IBI-308, WBP-3155, JNJ-63723283, MEDI-0680, SSI-361, CBT-502, анти-PD-1 биспецифического антитела, нацеленного на две мишени анти-PD-1/LAG-3 mAbs (TESARO), нацеленного на две мишени анти-PD-1/TIM-3 mAbs (TESARO), PF-06801591, LY-3300054, BCD-100, STI-1110, биоаналога пембролизумаба, биоаналога ниволумаба, терапии PD-L1–ТФР-бета, KY-1003, STI-1014, GLS-010, AM-0001, GX-P2, KD-033,PD-L1/BCMA биспецифического антитела (Immune Pharmaceuticals), биспецифического антитела против PD-1/Ox40 (Immune Pharmaceuticals), BMS-936559, анти-PD-1/VEGF-A DARPins (Molecular Partners), mDX-400, ALN-PDL, PD-1 ингибирующего пептида (Aurigene), вакцины на основе дендритных клеток с миРНК (Alnylam Pharmaceuticals), GB-226, иммунотерапии PD-L1 на основе CAR-TNK (TNK Therapeutics/NantKwest), INSIX RA, INDUS-903, AMP-224, биспецифического гуманизированного антитела анти-CTLA-4/анти-PD-1 (Akeso Biopharma), вакцины B7-H1 (State Key Laboratory of Cancer Biology/Fourth Military Medical University) и GX-D1.
В некоторых вариантах осуществления, PD-1 ингибитор может представлять собой пембролизумаб, ниволумаб, IBI-308, mDX-400, BGB-108, MEDI-0680, SHR-1210, PF-06801591, PDR-001, GB-226 или STI-1110. Ниволумаб (также известен как OPDIVO™, MDX-1106, BMS-936558 и ONO-4538) представляет собой IgG4 моноклональное антитело против PD-1. Пембролизумаб (также известен как KEYTRUDA®, ламбролизумаб и MK-34) представляет собой гуманизированное IgG4 каппа изотипа моноклональное антитело против PD-1. IBI-308 представляет собой моноклональное антитело против PD-1. mDX-400 представляет собой мышиное антитело против PD-1. BGB-108 представляет собой гуманизированное моноклональное антитело против PD-1. MEDI-0680 (AMP-514) представляет собой гуманизированное IgG4 моноклональное антитело против PD-1. SHR-1210 представляет собой моноклональное антитело против PD-1. PF-06801591 представляет собой моноклональное антитело против PD-1. PDR-001 представляет собой моноклональное антитело против PD-1. GB-226 представляет собой моноклональное антитело против PD-1. STI-1110 представляет собой моноклональное антитело против PD-1.
Анти-PD-1 антитела и фрагменты антител, описанные в настоящем тексте, охватывают белки, имеющие последовательности аминокислот, варьирующиеся относительно описанных антител, но сохраняющих способность связываться с PD-1.
В некоторых вариантах осуществления, анти-PD-1 антитела включают биспецифичные антитела и антителоподобные терапевтические белки, включая DARTs®, DUOBODIES®, BITES®, XmAbs®, TandAbs®, Fab производные и т.п., которые связываются с PD-1.
Анти-PD-L1 антитела и фрагменты антител, описанные в настоящем тексте, охватывают белки, имеющие последовательности аминокислот, варьирующиеся относительно описанных антител, но сохраняющих способность связываться с PD-L1. Такие вариантные антитела и их фрагменты могут содержать одно или больше добавлений, делеций или замещений аминокислот по сравнению с материнской последовательностью, но обладают биологической активностью, по существу эквивалентной или практически биоэквивалентной активности описанных антител.
В некоторых вариантах осуществления, анти-PD-L1 антитела включают биспецифичные антитела и антителоподобные терапевтические белки, включая DARTs®, DUOBODIES®, BITES®, XmAbs®, TandAbs®, Fab производные и т.п., которые связываются с PD-L1.
Неограничивающие примеры дополнительных ингибиторов механизма PD-1/PD-L1 описаны, например, в работах Chen and Han, Jour Clin Invest, 2015, 125(9):3384-3391, патентах США № 8,168,757; 8,354,509; 8,552,154; 8,741,295 и 9,212,224; заявках на патент США № 2014/0341917; 2015/0203580 и 2015/0320859; международной заявке на патент № WO2015/026634.
Биологический продукт, например, антитело или его фрагмент, считается биоаналогом, если, например, биологический продукт в высокой степени сходен с уже одобренным FDA биологическим продуктом, считающимся референсным продуктом. Биоаналог не имеет значимых отличий по безопасности и эффективности от референсного продукта. Биоаналог может также иметь тот же механизм действия, способ введения, дозированную форму и силу, что и референсный продукт.
Два биологических продукта, например, антитела или их фрагменты, считаются биоэквивалентными, если, например, они являются фармацевтическими эквивалентами или фармацевтическими альтернативами, у которых скорость и степень абсорбции не показывают значительных отличий при введении в одинаковой молярной дозировке в сходных условиях эксперимента, в виде однократной дозы или нескольких доз. Некоторые антитела считают эквивалентами или фармацевтическими альтернативами, если они эквивалентны по степени абсорбции, но не по скорости абсорбции, и также могут считаться биоэквивалентами, поскольку такие различия в скорости абсорбции являются преднамеренными и отражаются на этикеточных надписях, не являются критичными для создания эффективных концентраций лекарственного средства в организме, например, при длительном применении, и считаются несущественными с медицинской точки зрения для конкретного изученного лекарственного продукта.
В некоторых вариантах осуществления, два биологических продукта (например, два антитела или их фрагменты) являются биоэквивалентными, если нет значимых отличий в их безопасности, чистоте или активности.
В других вариантах осуществления, два биологических продукта (например, два антитела или их фрагменты) являются биоэквивалентными, если пациент может переключаться один или больше раз между референсным продуктом и биологическим продуктом без увеличения ожидаемого риска побочных эффектов, включая клинически значимое изменение иммуногенности, или снижения эффективности, по сравнению с непрерывной терапией без такого переключения.
В других вариантах осуществления, два биологических продукта (например, два антитела или их фрагменты) являются биоэквивалентными, если они оба работают по одному механизму действия в условиях их применения, в той степени, в которой такие механизмы известны.
Биоэквивалентность может быть показана способами in vivo и/или in vitro. Критерии биоэквивалентности включают, например, (a) in vivo тест на людях или других млекопитающих, в котором концентрацию антител или их метаболитов замеряют в крови, плазме, сыворотке или других биологических жидкостях, как функцию от времени; (b) in vitro тест, который коррелирует и в разумной степени предсказывает данные биодоступности для людей in vivo; (c) in vivo тест на людях или других млекопитающих, в котором замеряют соответствующий острый фармакологический эффект антитела (или его мишени), как функцию от времени; и (d) надлежащим образом контролируемые клинические исследования, в которых определяют безопасность, эффективность или биодоступность или биоэквивалентность антител.
Биоулучшенные варианты антител, описанных в настоящем тексте, могут быть основаны на существующих референсных антителах, специфичных к таргетному антигену, например PD-1 или PD-L1, которые претерпели изменения, например, они приобрели более высокое сродство к своему таргетному антигену и/или они связываются с другим эпитопом, отличающимся от такового для референсного антитела, или имеют более желательные характеристики терапевтической эффективности, экспрессирования и/или биофизические свойства.
Фармацевтические композиции
Описанные в настоящем тексте фармацевтические композиции, такие как композиции, включающие соединения для модулирования CCR1 активности и средства для блокировки PD-1/PD-L1 пути, могут содержать фармацевтически приемлемый носитель или разбавитель.
Термин “композиция” при использовании в настоящем тексте охватывает продукт, содержащий указанные ингредиенты в указанных количествах, а также любой продукт, который получается, напрямую или опосредованно, при комбинировании указанных ингредиентов в указанных количествах. Под термином “фармацевтически приемлемый” понимается носитель, разбавитель или вспомогательное вещество, которое должно быть совместимым с другими ингредиентами препарата и не наносить вреда принимающему его пациенту.
Биологические продукты, такие как антитела по настоящему изобретению, могут присутствовать в фармацевтической композиции, содержащей антитела или их фрагменты и фармацевтически приемлемый носитель. При использовании в настоящем тексте, термин “фармацевтически приемлемый носитель” включает все и любые растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые средства, изотонические агенты и вещества, замедляющие всасывание и т.п., которые являются физиологически совместимыми. Предпочтительно, носитель подходит для внутривенного, внутримышечного, подкожного, парентерального, спинального введения или введения в эпидермис (например, посредством инъекции или вливания). Фармацевтическая композиция по настоящему изобретению может включать одну или больше фармацевтически приемлемых солей, антиоксидант, водные и неводные носители и/или вспомогательные вещества, такие как консерванты, увлажнители, эмульгаторы и диспергаторы.
Фармацевтические композиции для введения соединений и веществ по настоящему изобретению можно выпускать в виде дозированных лекарственных форм, и их можно готовить любым из способов, известных в фармакологии и введении лекарственных средств. Все способы включают стадию объединения действующего вещества с носителем, который содержит один или несколько вспомогательных ингредиентов. В целом, фармацевтические композиции получают путем однородного и тщательного смешивания действующего вещества с жидким носителем или тонко измельченным твердым носителем, или с обоими, и затем, при необходимости, придание продукту формы желаемого препарата. В фармацевтической композиции действующее вещество присутствует в количестве, достаточном для оказания целевого эффекта на болезнь или на патологическое состояние.
Фармацевтические композиции, содержащие действующее вещество, могут иметь форму, подходящую для перорального применения, например, они могут быть в виде таблеток, саше, пастилок, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий и самоэмульгирующихся препаратов, как описано в патенте США №6,451,339, 6,451,339, твердых или мягких капсул, сиропов, эликсиров, растворов, букаальных пластырей, пероральных гелей, жевательной резинки, жевательных таблеток, шипучих порошков и шипучих таблеток. Композиции, предназначенные для перорального приема, можно готовить любыми способами, известными в области производства фармацевтических композиций. Такие композиции могут содержать один или больше агентов, выбранных из группы, состоящей из подсластителей, ароматизаторов, красителей, антиоксидантов и консервантов, для создания фармацевтически привлекательных и приятных на вкус препаратов. Таблетки содержат действующее вещество в смеси с другими нетоксичными фармацевтически приемлемыми вспомогательными веществами, подходящими для производства таблеток. Такими вспомогательными веществами могут быть, например, инертные разбавители, такие как целлюлоза, диоксид углерода, оксид алюмини, карбонат кальция, карбонат натрия, глюкоза, маннит, сорбит, лактоза, фосфат кальция или фосфат нария; гранулирующие агенты и агенты, ускоряющие распад таблеток, например кукурузный крахмал или альгиновую кислоту; связующие агенты, например поливинилпирролидон, целлюлозу, крахмал, желатин или смолу акации, и лубриканты, например стеарат магния, стеариновую кислоту или тальк. Таблетки могут не иметь покрытия или они могут быть покрыты кишечнорастворимой оболочкой, или иметь покрытие, нанесенное каким-либо другим известным способом, с целью замедления распадения и всасывания в желудочнокишечном тракте, тем самым обеспечивая пролонгированное действие в течение более длительного времени. Например, можно применять такие замедляющие распадение материалы как глицерил моностеарат или глицерил дистеарат. На таблетки можно также наносить покрытие способами, описанными в патентах США № 4,256,108; 4,166,452 и 4,265,874, с образованием осмотических таблеток с замедленным высвобождением.
Препараты, предназначенные для перорального приема, могут также иметь вид твердых желатиновых капсул, где действующее вещество смешано с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, где действующее вещество смешано с водной или масляной средой, например арахисовым маслом, жидким парафином или оливковым маслом. Кроме того, можно готовить эмульсии с нерастворяющимися в воде ингредиентами, такими как масла, и стабилизировать их поверхностно-активными веществами, такими как моно-диглицериды, сложные эфиры ПЭГ и т.п.
Водные суспензии содержат действующие вещества в смеси со вспомогательынми веществами, подходящими для производства водных суспензий. Такие вспомогательные вещества представляют собой суспендирующие агенты, например натрия карбоксиметилцеллюлозу, метилцеллюлозу, гидроксипропилметилцеллюлозу, альгинат натрия, поливинилпирролидон, трагакантовую камедь, и смолу акации; диспергирующие или увлажняющие агенты могут представлять собой природные фосфатиды, например лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например полиоксиэтилен стеарат, или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с неполными сложными эфирами, полученными из жирных кислот и гекситола, такие как полиоксиэтилен сорбитмоноолеат, или продукты конденсации этиленоксида с неполными сложными эфирами, полученными из жирных кислот и ангидридами гекситола, например полиэтилен сорбитанмоноолеат. Водные суспензии могут также содержать один или больше консервантов, например этил или н-пропил пара-гидроксибензоат, один или больше красителей, один или больше ароматизаторов, и один или больше подсластителей, таких как сахароза или сахарин.
Масляные суспензии можно получать путем суспендирования действующего вещества в растительном масле, например в арахисовом масле, оливковом масле, сезамовом масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загустители, например пчелиный воск, твердый парафин или цетиловые спирты. Можно добавлять подсластители, такие как перечисленные выше, и красители, для получения приятной на вкус композиции для перорального приема. Такие композиции можно стабилизировать добавлением антиоксиданта, такого как аскорбиновая кислота.
Диспергируемые порошки и гранулы, подходящие для приготовления водной суспензии при добавлении воды, содержат действующее вещество в смеси с диспергирующим или увлажняющим агентом, суспендирующим агентом и одним или больше консервантами. Примеры диспергирующих или увлажняющих агентов, суспендирующих агентов приведены выше. Могут также присутствовать дополнительные вспомогательные вещества, например подсластители, ароматизаторы и красители.
Фармацевтические композиции по настоящему изобретению могут также иметь форму эмульсий типа масло-в-воде. Масляной фазой может служить растительное масло, например оливковое масло или арахисовое масло, или минеральное масло, например жидкий парафин, или их смесь. Подходящие эмульгаторы могут представлять собой природные смолы, например смолу акации или трагакантовую камедь, природные фосфатиды, например соевое масло, лецитин, и сложные эфиры или неполные сложные эфиры, полученные из жирных кислот и ангидридов гекситола, например сорбитан моноолеат, и продукты конденсации указанных неполных сложных эфиров с этиленоксидом, например полиоксиэтилен сорбитанмоноолеат. Эмульсия может также содержать подсластители и ароматизаторы.
В состав сиропов и эликсиров могут входить подсластители, например глицерин, пропиленгликоль, сорбит или сахароза. Такие препараты могут также содержать средства, уменьшающие раздражение, консервант, ароматизаторы и красители. Растворы для перорального приема можно готовить в комбинации с, например, циклодекстрином, ПЭГ и поверхностно-активными веществами.
Фармацевтические композиции могут иметь форму стерильных инъецируемых водных или масляных суспензий. Такую суспензию можно готовить по известным в данной области методикам, с применением подходящих диспергаторов или увлажняющих агентов и суспендирующих агентов, которые были указаны выше. Стерильные инъецируемые препараты могут также представлять собой стерильные инъецируемые растворы или суспензии в нетоксичном парентерально приемлемом разбавителе или растворителе, например, могут иметь форму раствора в 1,3-бутандиоле. Среди подходящих носителей и растворителей, которые можно использовать, находятся вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные масла часто применяют в качестве растворителя или суспендирующей среды. Для этой цели можно использовать любое безвкусное нелетучее масло, включая синтетические моно- и диглицериды. Кроме того, в приготовлении инъецируемых препаратов находят применение жирные кислоты, такие как олеиновая кислота.
Описанные в настоящем тексте соединения можно также вводить в форме суппозиториев для ректального введения лекарственного средства. Такие композиции можно готовить путем смешивания лекарственного средства с подходящим нераздражающим вспомогательным веществом, которое твердое при комнатной температуре, но переходит в жидкое состояние при температуре тела, и поэтому плавится в прямой кишке с высвобождением лекарственного средства. Такие материалы включают масло какао и полиэтиленгликоли. Кроме того, соединения можно вводить в виде глазных препаратов как капли или мази. Кроме того, можно осуществлять чрезкожное введение рассматриваемых соединений посредством ионофорезных пластырей и т.п. Для местного нанесения применяют кремы, мази, гели, растворы или супензии, содержащие соединения по настоящему изобретению. В контексте настоящего изобретения, местное нанесение включает также применение полосканий и растворов для рта.
Соединения по настоящему изобретению можно также соединять с носителем, представляющем собой подходящие полимеры, в качестве целенаправленных носителей лекарственного средства. Такие полимеры могут включать поливинилпирролидон, пирановый сополимер, полигидрокси-пропил-метакриламид-фенол, полигидроксиэтил-аспартамид-фенол или полиэтиленоксид-полилизин, замещенный пальмитоильными остатками. Кроме того, соединения по настоящему изобретению можно соединять с носителем, относящимся к классу биоразлагаемых полимеров, которые можно применять для достижения контролируемого высвобождения лекарственного средства, например: полимолочная кислота, полигликолевая кислота, сополимеры полимолочной и полигликолевой кислоты, поли-эпсилон-капролактон, полигидроксимасляная кислота, полиортоэфиры, полиацетали, полидигидропираны, полицианоакрилаты и сшитые или амфипатические блок-сополимеры гидрогелей. Полимеры и полупроницаемые полимерные матриксы можно формовать в изделия, такие как клапаны, стенты, трубки, протезы и т.п. В одном варианте осуществления настоящего изобретения, соединение по настоящему изобретению соединяют с полимером или полупроницаемым полимерным матриксом, сформованным в виде стента или стент-графта.
Соединения и средства по настоящему изобретению можно вводить в состав препаратов для депонирования в медицинское устройство, которое может включать любое из известного разнообразия имплантатов, шунтов, включая стент-графты, катетеры, баллоны, корзины или другое устройство, которое может быть размещено или на постоянной основе имплантировано в полости тела. Как частный пример, было бы желательно иметь устройства и способы, которые позволяют доставить соединения по настоящему изобретению в участок тела, который подвергся хирургическому вмешательству. Например, соединение и средство можно доставлять в опухоль или микроокружение опухоли.
В некоторых вариантах осуществления, соединения и средства можно депонировать на медицинское устройство, такое как стент, и доставлять в подвергающийся лечению участок для лечения части тела. Стенты применяют в качестве носителей для доставки терапевтических средств. Внутрисосудистые стенты обычно имплантируют на постоянной основе в коронарные или периферические сосуды. Конструкции стентов включают описанные в патентах США № 4,733,655; 4,800,882 и 4,886,062. Такие конструкции включают как металлические, так и полимерные стенты, а также саморасширяющиеся стенты и баллонорасширяемые стенты. Стенты можно также применять для доставки терапевтических средств в место контакта с сосудистой системой, как описано, например, в патенте США № 5,102,417, в международных заявках на патент WO 91/12779 и WO 90/13332, патентах США № 5,419,760 и 5,429,634.
Термин “депонированный” означает, что соединение и средство нанесены в виде покрытия, адсорбированы, помещены на поверхность или иным образом включены в устройство известными в данной области способами. Например, соединение и средство могут встраиваться и высвобождаться из внутренней части («матриксный тип») или быть окружены полимерными материалами, которые окружают медицинское устройство, и высвобождаться через них («резервуарный тип»). В последнем примере соединение и средство могут захватываться полимерными материалами или связываться с полимерными материалами посредством одной или больше методик создания таких материалов, известных в данной области. В других препаратах, соединение и средство может связываться с поверхностью медицинского устройства без необходимости нанесения покрытия, посредством связей, подверженных разрыву, и высвобождаться с течением времени, может удаляться активным механическим или химическим воздействием, или находятся в перманентно иммобилизованной форме, которая доставляет ингибирующее средство в сайт имплантирования.
В одном варианте осуществления, соединение и средство можно включать в полимерные композиции при формировании биосовместимых покрытий для медицинских устройств, таких как стенты. Покрытие, получаемое из таких компонентов, обычно гомогенное и может применяться для нанесения покрытий на различные устройства, предназначенные для имплантирования.
Полимер может быть биоустойчивым или биорассасывающимся полимером, в зависимости от желаемой скорости высвобождения или желаемой степени стабильности полимера, но биорассасывающийся полимер является предпочтительным в данном варианте осуществления, поскольку, в отличие от биоустойчивого полимера, он не будет присутствовать достаточно долгое время после имплантирования, чтобы вызвать какой-либо побочный хронический местный ответ. Биорассасывающиеся полимеры, которые подходят для применения, включают (но не ограничены только ими) следующие: поли(L-молочная кислота), поликапролактон, полигликолид (PGA), поли(лактид-со-гликолид) (PLLA/PGA), поли(гидроксибутират), поли(гидроксибутират-co-валерат), полидиоксанон, полиортоэфир, полиангидрид, поли(гликолевая кислота), поли(D-молочная кислота), поли(L-молочная кислота), поли(D,L-молочная кислота), поли(D,L-лактид) (PLA), поли(L-лактид) (PLLA), поли(гликолевая кислота-со-триметиленкарбонат) (PGA/PTMC), полиэтиленоксид (PEO), полидиоксанон (PDS), полифосфоэфир, полифосфоэфир уретан, поли(аминокислоты), цианоакрилаты, поли(триметиленкарбонат), поли(иминокарбонат), сополи(эфир-эстеры) (например, PEO/PLA), полиалкиленоксалаты, полифосфазены и биомолекулы, такие как фибрин, фибриноген, целлюлоза, крахмал, коллаген и гиалуроновая кислота, поли-эпсилон-капролактон, полигидроксимасляная кислота, полиортоэфиры, полиацетали, полидигидропираны, полицианоакрилаты, сшитые или амфипатические блок-сополимеры гидрогелей, и другие подходящие биорассасывающиеся полимеры, известные в данной области. Также могут применяться биоустойчивые полимеры с относительно низким хроническим тканевым ответом, такие как полиуретаны, силиконы и полиэфиры, и также могут применяться другие полимеры, если их можно растворить и отвердить или заполимеризовать на медицинском устройстве, такие как полиолефины, полиизобутилен и сополимеры этилен-альфаолефин; акриловые полимеры и сополимеры; полимеры и сополимеры винилгалогенидов, такие как поливинилхлорид; поливинилпирролидон; поливиниловые простые эфиры, такие как поливинилметиловый эфир; поливинилиден галогениды, такие как поливинилиден фторид и поливинилиден хлорид; полиакрилонитрил, поливиниловые кетоны; поливиниловые ароматические соединения, такие как полистирол, поливиниловые сложные эфиры, такие как поливинилацетат; сополимеры винильных мономеров друг с другом и с олефинами, такие как этилен-метилметакрилатные сополимеры, акрилонитрил-стирольные сополимеры, АБС полимеры и этилен-винилацетатные сополимеры; пирановые сополимеры; полигидрокси-пропил-метакриламид-фенол; полигидроксиэтил-аспартамид-фенол; полиэтиленоксид-полилизин, замещенный пальмитоильными остатками; полиамиды, такие как Nylon 66 и поликапролактам; алкидные смолы, поликарбонаты; полиоксиметилены; полиимиды; полиэфиры; эпоксидные смолы; полиуретаны; вискоза; вискоза-триацетат; целлюлоза, ацетат целлюлозы, бутират целлюлозы, ацетат-бутират целлюлозы; целлофан; нитрат целлюлозы; пропионат целлюлозы; простые эфиры целлюлозы; и карбоксиметилцеллюлоза.
В некоторых вариантах осуществления, соединение и средство высвобождаются из полимерного покрытия в среду, в которую помещено медицинское устройство. Предпочтительно, соединение и средство высвобождаются контролируемо в течение продолжительного периода времени (например, недели или месяцы), что достигается с применением одной из нескольких хорошо известных методик, включая полимерные носители или слои с контролируемым вымыванием. Некоторые из этих методик ранее были описаны в заявке на патент США № 20040243225.
Способы лечения раковых солидных опухолей
В некоторых аспектах, в настоящем изобретении описаны способы лечения раковой солидной опухоли у субъекта, нуждающегося в этом, путем введения пациенту, страдающему раковым заболеванием, терапевтически эффективного количества CCR1 антагониста и терапевтически эффективного количества ингибитора PD-1 или ингибитора PD-L1. Пациенту можно вводить CCR1 антагонист и PD-1 ингибитор. Альтернативно, пациенту можно вводить CCR1 антагонист и PD-L1 ингибитор.
В некоторых вариантах осуществления, CCR1 антагонист и PD-1 или PD-L1 ингибитор, описанные в настоящем тексте, можно применять или комбинировать с одним или больше дополнительными терапевтическими средствами или терапевтическими методами лечения (включая, но не ограничиваясь только ей, лучевую терапию).
Термин “пациент” в контексте настоящего изобретения включает животных, таких как млекопитающие, включая (но не ограничиваясь только ими) людей, других приматов, коров, овец, коз, лошадей, собак, кошек, кроликов, крыс, мышей и т.п. Соединения, средства и композиции, описанные в настоящем тексте, можно применять для лечения широкого ряда раковых заболеваний, включая раковые солидные опухоли.
Композиции, способы и наборы, описанные в настоящем тексте, можно применять для лечения пациента, у которого диагностирован рак или опухоль.
В некоторых вариантах осуществления, опухоль может быть злокачественным или потенциально злокачественным новообразованием или массой ткани любого размера, и включает первичные опухоли и вторичные новообразования. Солидная раковая опухоль может представлять собой аномальный рост или увеличение массы ткани, которая не содержит цист или областей жидкой ткани.
В некоторых вариантах осуществления, введение соединений, средств и композиций по настоящему изобретению может уменьшить вес опухоли, опухолевую массу, размер опухоли и/или количество опухолей у пациента. В некоторых случаях, соединения, средства и композиции могут предотвратить или минимизировать метастазы опухоли. В других случаях, соединения, средства и композиции могут стимулировать или усиливать некроз опухоли.
В некоторых вариантах осуществления, введение соединений, средств и композиций по настоящему изобретению может приводить к частичной ремиссии или полной ремиссии (выживаемость без прогрессирования заболевания), отсрочиванию прогрессирования заболевания и/или улучшению общей выживаемости. В некоторых случаях, соединения, средства и композиции могут увеличивать продолжительность суммарного ответа на лечение, стимулировать регресс опухоли, регресс рака или стабилизацию заболевания и/или обеспечивать преимущество с клинической точки зрения. В других случаях, соединения, средства и композиции могут уменьшать степень тяжести по меньшей мере одного симптома заболевания, увеличивать частоту и длительность бессимптомных периодов заболевания или предотвращать ухудшения или потерю трудоспособности вследствие ракового заболевания. В некоторых случаях, можно уменьшить развитие рака или рецидивы рака.
Пациент, нуждающийся в лечении, может иметь раковое заболевание или раковую солидную опухоль, включая (но не ограничиваясь только ими) следующие: аденокарцинома, рак мочевого пузыря, рак груди, трижды негативный рак груди, рак толстой и прямой кишки, рак толстой кишки, рак печени, рак легких, немелкоклеточный рак легких, рак тонкого кишечника, рак почки, почечно-клеточный рак, рак пищевода, саркома, меланома, множественная меланома, рак кости, рак поджелудочной железы, рак предстательной железы, рак желудка, рак кожи, рак головы и шеи, накожная или внутриглазная злокачественная меланома, рак желудка, рак мочевого пузыря, уротелиальный рак мочевого пузыря, рак матки, рак яичника, рак прямой кишки, рак анальной области, рак яичка, рак матки, карцинома фаллопиевых труб, рак эндометрия, карцинома шейки матки, карцинома вагины, карцинома вульвы, метастатическая карцинома Меркеля, ходжкинская лимфома, неходжкинская лимфома, рак эндокринной системы, рак щитовидной железы, рак паращитовидной железы, рак надпочечника, саркома мягкой ткани, рак уретры, рак пениса, солидные опухоли детского возраста, карцинома почечной лоханки, новообразования в центральной нервной системе (ЦНС), первичная лимфома ЦНС, глиобластома, глиома, ангиогенез опухоли, опухоль оси позвоночника, глиома ствола мозга, аденома гипофиза, саркома Капоши, плоскоклеточный рак, Т-клеточная лимфома, рак, вызванный влиянием факторов внешней среды, любые комбинации перечисленных видов рака, метастатическое поражение от любых видов рака, распространенная раковая опухоль или злокачественная опухоль и распространенная солидная опухоль.
Дополнительные примеры раковых солидных опухолей включают (но не ограничены только ими) фибросаркому, миксосаркому, липосаркому, хондросаркому, остеогенную саркому, хордому, ангиосаркому, эндотелиосаркому, лимфангиосаркому, лимфангиоэндотелиосаркому, синовиому, мезотелиому, опухоль Юинга, лейомиосаркому, рабдомиосаркому, карциному толстой кишки, плоскоклеточную карциному, базальноклеточную карциному, карциному потовых желез, карциному сальной железы, папиллярную карциному, папиллярные аденокарциномы, цистаденокарциному, медуллярный рак, бронхогенную карциному, почечно-клеточный рак, гепатому, карциному желчного протока, хориокарциному, семиному, эмбриональный рак, опухоль Вильмса, опухоль яичка, карциному легкого, мелкоклеточный рак легкого, рак мочевого пузыря, эпителиальную карциному, глиому, астроцитому, медуллобластому, краниофарингиому, эпендимому, пинеалому, гемангиобастому, нейрому слухового нерва, олигодендроглиому, менингиому, меланому, нейробластому и ретинобластому.
В некоторых вариантах осуществления, CCR1 антагонист и PD-1 или PD-L1 ингибитор можно применять или комбинировать с одним или больше дополнительными терапевтическими средствами или методами терапевтического лечения (включая, но не ограничиваясь только ими: химиотерапия, лучевая терапия и другие методы лечения рака).
В некоторых вариантах осуществления, CCR1 антагонист и PD-1 или PD-L1 ингибитор, описанные в настоящем тексте, можно применять или комбинировать с одним или больше химиотерапевтическими средствами, противораковыми средствами, антиангиогенными средствами, антипролиферативными/антимитотическими средствами, ингибиторами поли(АДФ-рибоза)полимеразы, средствами, повреждающими ДНК, включая платинасодержащие средства, противофиброзными средствами, противогормональными средствами, иммунотерапевтическими средствами, терапевтическими антителами, биспецифическими антителами, “antibody-like” терапевтическими белками (такими как DARTs®, Duobodies®, Bites®, XmAbs®, TandAbs ®, Fab производные), конъюгатами антитело-лекарственное средство (ADC), средствами лучевой терапии, антинеопластическими средствами, антипролиферативными средствами, онколитическими вирусами, модификаторами или редакторами генов, такими как CRISPR (включая CRISPR/Cas9), нуклеазами с цинковыми пальцами или синтетическими нуклеазами (TALEN), с иммунотерапией с использованием Т-клеток с химерными антигенными рецепторами (CAR) или их комбинацией.
В некоторых вариантах осуществления, перечисленные терапевтические средства имеют форму соединений, антител, полипептидов или полинуклеотидов.
В некоторых вариантах осуществления, CCR1 антагонист и PD-1 или PD-L1 ингибитор, описанные в настоящем тексте, комбинируют с одним или больше из: ингибитора, агониста, антагониста, лиганда, модулятора, стимулятора, блокатора, активатора или супрессора: белка хеджхог, FGF рецептор, фактора роста плаценты, тирозинкиназы (включая, но не ограничиваясь только ими: Axl, MET, Abl, Bcr белок, EPH семейство, Fyn, Kit, Ltk, Lck, Src, Yes, Flt3, RET, Raf, Erbb, Erbb4, Erbb2, Jak, Jak1, Jak2, Btk, EGFR), DHFR, эпидермальный фактор роста, циклин-зависимая киназа, интерферон-бета, VEGF лиганд, PDGF-B лиганд, VEGF, CSF-1, тубулин, mTOR, HDAC, PARP, поли-АДФрибоза-полимераза, протеасома, циклооксигеназа 2, GNRH, эстрогеновый рецептор, соматостатиновый рецептор, ароматаза, тимидилат-синтаза, трансфераза, белок-антиген цитотоксическогоТ-лимфоцита 4, макрофагальный маннозный рецептор 1, MEK-1 протеинкиназа, MEK-2 протеинкиназа, тимидилат-синтаза, GCSF рецептор, CSF2 ген, ретиноид-X-рецептор, ДНК-полимераза, топоизомераза II, дигидропиримидин дегидрогеназа, оротат фосфорибозилтрансфераза, ДНК-гираза, цитозин-ДНК-метилтрансфераза, топоизомераза I, хорионический гонадотропин, рецептор лютеинизирующего гормона, интерлейкин-2 лиганд, интерлейкин-2, эстрадиол 17 бета дегидрогеназа, глюкокортикоид, рецептор прогестерона, допаминовый рецептор D2, ген IL17, интерлейкин 17E, нейрокининовый рецептор, ретиноид-X-рецептор, орнитин декарбоксилаза, интерферон альфа 2, интерлейкин-2 лиганд, интерлейкин-2, альбумин, микротрубочки, циклин G1 тимидилат-синтаза, ингибитор контрольной точки, CTLA-4, 4-1BB (CD137), 4-1BBL (CD137L), B7-H3, B7-H4, BTLA, HVEM, TIM3, GAL9, LAG3, TIM3, B7H3, B7H4, VISTA, KIR, 2B4 CD160 (именуемый также BY55) CGEN-15049, OX40, или любая их комбинация.
В некоторых вариантах осуществления, CCR1 антагонист и PD-1 или PD-L1 ингибитор, описанные в настоящем тексте, можно применять или комбинировать с одним или больше из следующих: сонидегиб, нинтеданиб, афлиберцепт, сорафениб, эрибулин мезилат, пралатрексат, цетуксимаб, палбоциклиб, бевацизумаб, рекомбинантный интерферон бета-1a, дасатиниб, рамуцирумаб, пазопаниб, трастузумаб эмтансин, эверолимус, сунитиниб, перфлубутан, золедроновая кислота, руксолитиниб, ибрутиниб, панобиностат, лапатиниб, олапариб, бортезомиб, целекоксиб, гефитиниб, госерелин ацетат, ралоксифен, октреотид ацетат, октреотид, анастрозол, гемцитабин, летрозол, пеметрексел динатрия, кабозатиниб, ипилимумаб, технеций Tc 99m тилманоцепт, кобиметиниб, паклитаксел, пертузумаб, капецитабин, эрлотиниб, филграстим, талимоген лагерпапервек, трастузумаб, базедоксифен, нимотузумаб, трабектедин, бексаротен, доксорубицин, доцетаксел, CS-055, абиратерон, вориностат, афатиниб, левпролид ацетат, вандетаниб, трипторелин памоат, тегафур, гимерацил, отерацил калия, пиксантрон, карбоплатин, экземестан, митоксантрон, технеций Tc 99m тетрофосмин, ганиреликс ацетат, децитабин, иринотекан, босутиниб, эпирубицин, апатиниб, темсиролимус, хориогонадотропин альфа, винорелбин, тамоксифен, трипторелин ацетат, трипторелин, алдеслейкин, мифепристон, винфлунин, гадобутрол, октафторпропан, талапорфин, милтефозин, бромокриптин, DaunoXome, цетрореликс, памидронат, вирулизин, DLBS-1425, 99mTc-сестамибин, арцитумомаб, нафарелин, алитретиноин, форместан, трилостан, пирарубицин, эфлорнитин, гистрелин, 111In-сатумомаб пендетид, интерферон альфа-2b, интерлейкин-2, рекомбинантный человеческий интерлейкин-2, гадофосвесет, глютоксим, Рексин-G, доксифлуридин, просорба, фадрозол, ипилимумаб, тремелимумаб, AGEN-1884, ADC-1015, PSI-001, JHL-1155 и лобаплатин, или любая их комбинация.
В некоторых вариантах осуществления, CCR1 антагонист и PD-1 ингибитор или PD-L1 ингибитор по настоящему изобретению можно вводить в комбинации с другими подходящими противораковыми терапевтическими средствами, включая, например, химиотерапевтические средства, облучение, биологические средства, иммунотерапию и т.д. Выбор подходящих средств для применения в составе комбинированной терапии может сделать квалифицированный специалист в данной области, в соответствии с общепринятыми фармацевтическими принципами. Комбинация терапевтических средств может работать синергетически, осуществляя лечение или профилактику различных нарушений, таких как, например, рак. Используя этот подход, можно достичь терапевтической эффективности с более низкими дозировками каждого средства, тем самым снижая вероятность нежелательных побочных эффектов.
В некоторых вариантах осуществления, CCR1 антагонист и PD-1 или PD-L1 ингибитор, описанные в настоящем тексте, и одно или больше дополнительных терапевтических средств вводят одновременно, по отдельности или последовательно.
Способы введения
В зависимости от вида ракового заболевания и состояния пациента или степени тяжести заболевания, соединения, средства и композиции по настоящему изобретению можно вводить перорально, парентерально (например, посредством внутримышечной, интраперитонеальной, внутривенной, интрацеребровентрикулярной, интрацистернальной инъекции или инфузии, подкожной инъекции или с помощью импланта), посредством имплантирования (например, когда соединение объединяют с шунтом), посредством ингаляционного спрея, назально, вагинально, ректально, сублингвально или местно; и их можно вводить, в отдельности или в комбинации, в составе подходящих однократных дозированных форм, содержащих общеизвестные нетоксичные фармацевтически приемлемые носители, адъюванты и вспомогательные вещества, подходящие для каждого способа введения.
В лечении раковых заболеваний, например солидных опухолей, которые требуют модулирования хемокинового рецептора, подходящий диапазон дозировок CCR1 антагониста обычно составляет примерно от 0,01 до 100 мг на кг веса тела пациента, которые можно вводить в виде одной или нескольких доз. Предпочтительно, диапазон дозировок составляет примерно от 0,01 до 25 мг/кг в сутки; более предпочтительно примерно от 0,05 до 10 мг/кг в сутки. Подходящий диапазон дозировок может составлять примерно от 0,01 до 25 мг/кг в сутки, примерно от 0,05 до 10 мг/кг в сутки, или примерно от 0,1 до 5 мг/кг в сутки. Внутри указанного диапазона, дозировка может составлять от 0,005 до 0,05, от 0,05 до 0,5, или от 0,5 до 5,0 мг/кг в сутки. При пероральном введении, композиции предпочтительно вводят в виде таблеток, содержащих от 1,0 до 1000 миллиграммов действующего вещества, в частности 1,0, 5,0, 10,0, 15,0, 20,0, 25,0, 50,0, 75,0, 100,0, 150,0, 200,0, 250,0, 300,0, 400,0, 500,0, 600,0, 750,0, 800,0, 900,0 и 1000,0 миллиграммов действующего вещества, с симптоматическим регулированием дозировки для пациента, подвергающегося лечению. Соединения можно вводить в режиме от 1 до 4 раз в сутки, предпочтительно один или два раза в сутки.
Подходящая дозировка PD-1 ингибитора или PD-L1 ингибитора обычно составляет от примерно 0,0001 до примерно 100 мг/кг, обычно от примерно 0,001 до примерно 20 мг/кг, и чаще от примерно 0,01 до примерно 10 мг/кг веса тела пациента. Предпочтительно, дозировка находится в диапазоне 0,1-10 мг/кг веса тела. Например, дозировка может составлять 0,1, 0,3, 1, 3, 5 или 10 мг/кг веса тела, и более предпочтительно – 0,3, 1, 3 или 10 мг/кг веса тела. Режим введения обычно составляют таким образом, чтобы достичь воздействия, которое обеспечивает устойчивую оккупацию рецепторов (ОР), с учетом типичных фармакокинетических свойств антитела. Пример режима введения предусматривает введение один раз в неделю, один раз в две недели, один раз в три недели, один раз в четыре недели, один раз в месяц, один раз в три месяца или один раз каждые 3-6 месяцев. Дозировку и режим можно изменять во время курса лечения. Например, режим дозирования может включать введение антитела: (i) каждые две недели в течение 6-недельного цикла; (ii) каждые четыре недели, шесть доз, затем каждые три месяца; (iii) каждые три недели; (iv) 3-10 мг/кг веса тела, затем 1 мг/кг веса тела каждые 2-3 недели. Учитывая, что IgG4 антитело обычно имеет время полужизни 2-3 недели, предпочтительный режим введения анти-PD-1 или анти-PD-L1 антитела предусматривает 0.3-10 мг/кг веса тела, предпочтительно 3-10 мг/кг веса тела, более предпочтительно 3 мг/кг веса тела внутривенно, при этом антитело вводят каждые 14 дней в ходе 6-недельных или 12-недельных циклов до полного ответа или подтвержденного прогрессирования заболевания.
В некоторых вариантах осуществления, два или более антител с различной специфичностью связывания вводят одновременно, в этом случае дозировка каждого вводимого антитела попадает в указанный диапазон дозировок. Антитело можно вводить в несколько приемов. Интервалы между отдельными введениями могут составлять, например, 1 раз в неделю, каждые 2 недели, каждые 3 недели, один раз в месяц, каждые три месяца и один раз в год. Интервалы могут также быть нерегулярными, в соответствии с измеренным у пациента уровнем в крови антител к таргетному антигену. В некоторых способах дозировку регулируют таким образом, чтобы достичь концентрации антител в плазме крови примерно 1-1000 мг/мл, и в некоторых способах – примерно 25-300 мг/мл.
Альтернативно, антитела можно вводить в виде препарата с замедленным высвобождением, в этом случае требуется меньшая частота введения. Дозировка и частота введения может варьироваться в зависимости от времени полужизни антител у пациента. Обычно человеческие антитела имеют самые большие времена полужизни, затем идут гуманизированные антитела, химерные антитела и нечеловеческие антитела. Дозировка и частота введения может варьироваться в зависимости от того, является лечение профилактическим или терапевтическим. При профилактическом применении вводят относительно низкую дозировку со сравнительно нечастыми интервалами в течение длительного периода времени. Некоторые пациенты продолжают получать такое лечение на протяжении всей жизни. При терапевтическом применении иногда требуется введение относительно высоких доз со сравнительно короткими интервалами, до остановки или замедления прогрессирования заболевания, и предпочтительно до тех пор, пока у пациента проявится частичное или полное ослабление симптомов заболевания. После этого пациента можно переводить на профилактический режим.
Уровень дозировки действующих веществ в фармацевтических композициях можно варьировать таким образом, чтобы достичь содержания действующего вещества, которое эффективно для достижения целевого терапевтического ответа для конкретного пациента, композиции и способа введения, при этом без проявления токсичности для пациента. Выбранный уровень дозировки будет зависеть от различных фармакокинетических факторов, включая активность конкретной композиции по настоящему изобретению, способ введения, время введения, скорость экскреции конкретного используемого соединения, длительность лечения, других лекарственных средств, соединений и/или материалов, применяемых в комбинации с конкретной композицией, возраст, пол, вес, состояние, общее состояние здоровья и история болезни пациента, подвергающегося лечению, и подобные факторы, известные в данной области. Композицию можно вводить в применением одного или больше способов введения, применяя один или больше из разнообразия способов, известных в данной области. Как будет понятно квалифицированному специалисту, способ или путь введения варьируется в зависимости от желаемых результатов лечения.
Терапевтическое соединение и средство в комбинированной терапии, описанные в настоящем тексте, можно вводить по отдельности или в составе фармацевтической композиции, которая содержит терапевтическое соединение и средство и один или больше фармацевтически приемлемых носителей, вспомогательных веществ и разбавителей.
В некоторых вариантах осуществления, каждое терапевтическое соединение и средство применяются в количестве, которое было бы субтерапевтическим при применении в отдельности или без другого. Специалисту будет понятно, что «комбинации» могут означать комбинирование в лечении (т.е. два или больше лекарственных средств можно вводить в виде смеси или по меньшей мере параллельно, или по меньшей мере вводить пациенту в разное время, но так, чтобы оба действовали в одно и то же время).
Аналогично, соединения, средства и композиции по настоящему изобретению можно применять в комбинации с другими лекарственными средствами, которые используются в лечении, профилактике, подавлении или ослаблении рака. Такие другие лекарственные средства можно вводить способом и в количестве, обычно применяемом для них, одновременно или последовательно с соединением, средством или композицией по настоящему изобретению. Когда соединение, средство или композиция по настоящему изобретению применяется одновременно с одним или больше другими лекарственными средствами, предпочтительна фармацевтическая композиция, содержащая такие другие лекарственные средства в дополнение к соединению, средству или композиции по настоящему изобретению. Соответственно, фармацевтические композиции могут включать такие, которые содержат также одно или больше других действующих веществ или терапевтических агентов, в дополнение к соединению, средству или композиции по настоящему изобретению.
Соединения, средства и композиции, описанные в настоящем тексте, можно вводить с уровнем дозировки и частотой введения, которые обеспечивают синергетический эффект CCR1 антагониста в комбинации с PD-1 ингибитором или PD-L1 ингибитором.
Однако следует понимать, что конкретная дозировка и частота введения для каждого конкретного пациента может варьироваться и зависит от разных факторов, включая активность конкретного применяемого соединения, метаболическую устойчивость и длительность действия этого соединения, возраст, вес тела, наследственность, общее состояние здоровья, пол и диету субъекта, а также способ и время введения, скорость экскреции, применяемые в комбинации лекарства и тяжесть конкретного патологического состояния у конкретного пациента, подвергающегося терапии.
Комбинированная терапия включает совместное введение CCR1 антагониста и PD-1 или PD-L1 ингибитора, последовательное введение CCR1 антагониста и PD-1 или PD-L1 ингибитора, введение композиции, содержащей CCR1 антагонист и PD-1 или PD-L1 ингибитор, или одновременное введение отдельных композиций, так что одна композиция содержит CCR1 антагонист, а другая композиция содержит PD-1 или PD-L1 ингибитор.
Наборы
В некоторых вариантах осуществления, в настоящем изобретении описаны наборы, содержащие антагонист хемокинового рецептора CCR1, описанный в настоящем тексте, и либо PD-1 ингибитор, либо PD-L1 ингибитор, описанный в настоящем тексте. Такие наборы можно применять для лечения пациента, страдающего раковым заболеванием, таким как раковая солидная опухоль. Набор может содержать фармацевтическую композицию, содержащую антагонист хемокинового рецептора CCR1 и PD-1 ингибитор или PD-L1 ингибитор. Набор может содержать фармацевтическую композицию, содержащую антагонист хемокинового рецептора CCR1 и PD-1 ингибитор. Набор может содержать фармацевтическую композицию, содержащую антагонист хемокинового рецептора CCR1 и PD-L1 ингибитор. Антагонист хемокинового рецептора CCR1 может представлять собой 1.001, 3.002, 4.005, 5.005 или 3.001, его аналог или его производное.
В некоторых вариантах осуществления, антагонист хемокинового рецептора CCR1 выбран из группы, состоящей из BL-5923, UCB-35625, BX-471, BI-638683, PS-031291, MLN-3701, AZD-4818, MLN-3897, CP-481715, F-18-CCR1, AOP-RANTES, PS-375179 и NSC-651016. PD-1 ингибитор может представлять собой пембролизумаб, ниволумаб, IBI-308, mDX-400, BGB-108, MEDI-0680, SHR-1210, PF-06801591, PDR-001, GB-226, STI-1110, его биоаналог, его биоулучшенный вариант или его биоэквивалент. PD-1 ингибитор может представлять собой пембролизумаб, ниволумаб, IBI-308, mDX-400, BGB-108, MEDI-0680, SHR-1210, PF-06801591, PDR-001, GB-226 или STI-1110. PD-L1 ингибитор может представлять собой дурвалумаб, атезолизумаб, авелумаб, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, STI-1014, KY-1003, его биоаналог, его биоулучшенный вариант или его биоэквивалент. PD-L1 ингибитор может представлять собой дурвалумаб, атезолизумаб, авелумаб, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, STI-1014 или KY-1003. PD-L1 ингибитор может представлять собой дурвалумаб, атезолизумаб, авелумаб, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, CA-327, STI-1014, KY-1003, его биоаналог, его биоулучшенный вариант или его биоэквивалент. PD-L1 ингибитор может представлять собой дурвалумаб, атезолизумаб, авелумаб, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, CA-327, STI-1014 или KY-1003.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из соединений, описанных в WO14151634, WO15160641, WO16039749, WO16077518, WO16100608, WO16149351, WO2016057624, WO2016100285, US2016194307, US2016222060, US2015291549, US2016194307 и US2014294898 (BRISTOL MYERS SQUIBB CO), которые включены в настоящий текст посредством ссылки.
В некоторых вариантах осуществления, PD-1 или PD-L1 ингибитор выбран из соединений, описанных в предварительной заявке на патент США № 62/355,119 или 62/440,100, которые включены в настоящий текст посредством ссылки.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из группы, состоящей из:
 .
.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из соединений, описанных в WO16142886, WO16142894, WO16142852, WO16142833, WO15033301, WO15033299, WO11161699, WO12168944, WO13132317, WO13144704, WO15033303, WO15036927, WO15044900, WO16142835, US2015073024, US8907053, US9044442, US9096642, US9233940, и US2016194295 (Aurigene discovery tech ltd), которые включены в настоящий текст посредством ссылки.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, пембролизумаба, ниволумаба, AP-106, AP-105, MSB-2311, CBT-501, авелумаба, AK-105, IO-102, IO-103, PDR-001, CX-072, SHR-1316, JTX-4014, GNS-1480, рекомбинантного гуманизированного анти-PD1 mAb (Shanghai Junshi Biosciences), REGN-2810, пелареорепа, SHR-1210, PD1/PDL1 ингибирующей вакцины (THERAVECTYS), BGB-A317, рекомбинантного гуманизированного анти-PD-1 mAb (Bio-Thera Solutions), Probody нацеленного на PD-1 (CytomX), XmAb-20717, FS-118, PSI-001, SN-PDL01, SN-PD07, PD-1 модифицированных TIL (Sangamo Therapeutics), PRS-332, FPT-155, jienuo mAb (Genor Biopharma), TSR-042, REGN-1979, REGN-2810, ресминостата, FAZ-053, PD-1/CTLA-4 биспецифического антитела (MacroGenics), MGA-012, MGD-013, M-7824, биспецифического антитела на основе PD-1 (Beijing Hanmi Pharmaceutical), AK-112, AK-106, AK-104, AK-103, BI-754091, ENUM-244C8, MCLA-145, MCLA-134, анти-PD-1 онколитического моноклонального антитела (Transgene SA), AGEN-2034, IBI-308, WBP-3155, JNJ-63723283, MEDI-0680, SSI-361, CBT-502, анти-PD-1 биспецифического антитела, нацеленного на две мишени анти-PD-1/LAG-3 mAbs (TESARO), нацеленного на две мишени анти-PD-1/TIM-3 mAbs (TESARO), PF-06801591, LY-3300054, BCD-100, STI-1110, биоаналога пембролизумаба, биоаналога ниволумаба, терапии PD-L1–ТФР-бета, KY-1003, STI-1014, GLS-010, AM-0001, GX-P2, KD-033,PD-L1/BCMA биспецифического антитела (Immune Pharmaceuticals), биспецифического антитела против PD-1/Ox40 (Immune Pharmaceuticals), BMS-936559, анти-PD-1/VEGF-A DARPins (Molecular Partners), mDX-400, ALN-PDL, PD-1 ингибирующего пептида (Aurigene), вакцины на основе дендритных клеток с миРНК (Alnylam Pharmaceuticals), GB-226, иммунотерапии PD-L1 на основе CAR-TNK (TNK Therapeutics/NantKwest), INSIX RA, INDUS-903, AMP-224, биспецифического гуманизированного антитела анти-CTLA-4/анти-PD-1 (Akeso Biopharma), вакцины B7-H1 (State Key Laboratory of Cancer Biology/Fourth Military Medical University) и GX-D1.
В некоторых случаях, набор включает печатные материалы, например, инструкции по применению соединения, средства, антитела или фармацевтических композиций с ними. Не ограничиваясь перечисленным, набор может включать буферы, разбавители, фильтры, иглы, шприцы и листовки-вкладыши с инструкциями по практической реализации описанных в настоящем изобретении способов.
Метастазы
В некоторых вариантах осуществления, описан способ уменьшения или предотвращения образования метастазов у субъекта с раковой солидной опухолью, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества антагониста хемокинового рецептора CCR1.
В некоторых вариантах осуществления, метастазы представляют собой метастазы в легких.
В некоторых вариантах осуществления, антагонист хемокинового рецептора CCR1 имеет формулу (IIIb1a):
(IIIb1a)
где каждый A представляет собой N или CH, и по меньшей мере один A представляет собой N; R1 представляет собой галоген; R3 выбран из группы, состоящей из C1-8 алкила, C3-8 циклоалкила и C2-8 алкенила; и R8 представляет собой C1-8 алкил; или его фармацевтически приемлемая соль.
В некоторых вариантах осуществления, антагонист хемокинового рецептора CCR1 выбран из группы, состоящей из:
и ,
или их фармацевтически приемлемой соли.
В некоторых вариантах осуществления, антагонист хемокинового рецептора CCR1 выбран из группы, состоящей из:
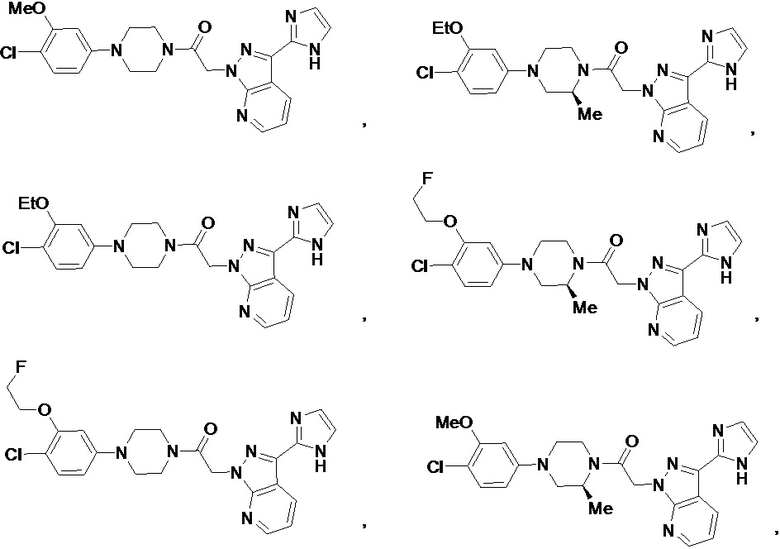
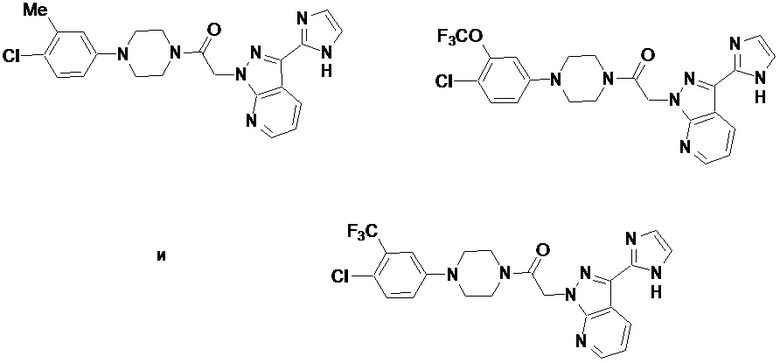 ,
,
или их фармацевтически приемлемой соли.
В некоторых вариантах осуществления, антагонист хемокинового рецептора CCR1 выбран из группы, состоящей из:
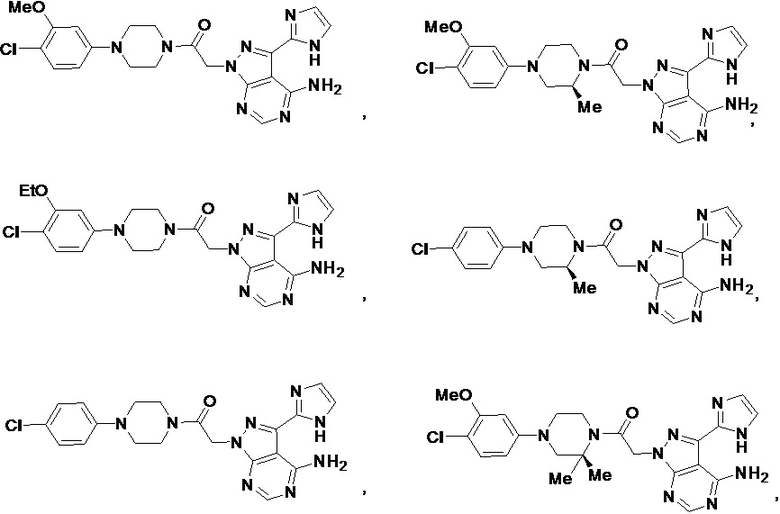
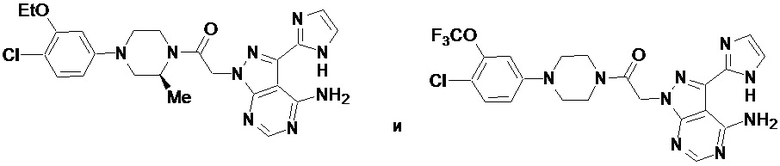 ,
,
или их фармацевтически приемлемой соли.
В некоторых вариантах осуществления, антагонист хемокинового рецептора CCR1 выбран из группы, состоящей из BL-5923, UCB-35625, BX-471, BI-638683, PS-031291, MLN-3701, AZD-4818, MLN-3897, CP-481715, F-18-CCR1, AOP-RANTES, PS-375179 и NSC-651016.
В некоторых вариантах осуществления, субъектом является человек.
В некоторых вариантах осуществления, раковая солидная опухоль выбрана из группы, состоящей из рака мозга, рака груди, трижды негативного рака груди, рака мочевого пузыря, рака кости, рака толстой и прямой кишки, рака легких, рака почки, рака печени, рака желудка, рака предстательной железы, саркомы, меланомы, карциномы и лимфомы.
В некоторых вариантах осуществления, раковая солидная опухоль представляет собой трижды негативный рак груди/молочной железы.
В некоторых вариантах осуществления, CCR1 антагонист вводят с одним или больше дополнительными терапевтическими средствами.
В некоторых вариантах осуществления, одно или больше дополнительных терапевтических средств представляют собой PD-1 ингибитор или PD-L1 ингибитор.
В некоторых вариантах осуществления, PD-1 ингибитор выбран из группы, состоящей из пембролизумаба, ниволумаба, IBI-308, mDX-400, BGB-108, MEDI-0680, SHR-1210, PF-06801591, PDR-001, GB-226, STI-1110, их биоаналогов, их биоулучшенных версий и их биоэквивалентов.
В некоторых вариантах осуществления, PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, авелумаба, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, CA-327, STI-1014, KY-1003, их биоаналогов, их биоулучшенных версий и их биоэквивалентов.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из соединений, описанных в WO14151634, WO15160641, WO16039749, WO16077518, WO16100608, WO16149351, WO2016057624, WO2016100285, US2016194307, US2016222060, и US2014294898 (BRISTOL MYERS SQUIBB CO), которые включены в настоящий текст посредством ссылки.
В некоторых вариантах осуществления, PD-1 или PD-L1 ингибитор выбран из соединений, описанных в предварительной заявке на патент США №. 62/355,119 или 62/440,100, которые включены в настоящий текст посредством ссылки.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из группы, состоящей из:
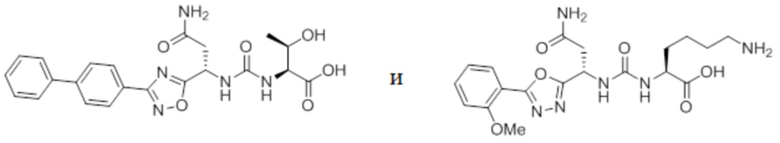 .
.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из соединений, описанных в WO16142886, WO16142894, WO16142852, WO16142833, WO15033301, WO15033299, WO11161699, WO12168944, WO13132317, WO13144704, WO15033303, WO15036927, WO15044900, WO16142835, US2015073024, US8907053, US9044442, US9096642, US9233940 и US2016194295 (Aurigene discovery tech ltd), которые включены в настоящий текст посредством ссылки.
В некоторых вариантах осуществления, PD-1 и/или PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, пембролизумаба, ниволумаба, AP-106, AP-105, MSB-2311, CBT-501, авелумаба, AK-105, IO-102, IO-103, PDR-001, CX-072, SHR-1316, JTX-4014, GNS-1480, рекомбинантного гуманизированного анти-PD1 mAb (Shanghai Junshi Biosciences), REGN-2810, пелареорепа, SHR-1210, PD1/PDL1 ингибирующей вакцины (THERAVECTYS), BGB-A317, рекомбинантного гуманизированного анти-PD-1 mAb (Bio-Thera Solutions), Probody нацеленного на PD-1 (CytomX), XmAb-20717, FS-118, PSI-001, SN-PDL01, SN-PD07, PD-1 модифицированных TIL (Sangamo Therapeutics), PRS-332, FPT-155, jienuo mAb (Genor Biopharma), TSR-042, REGN-1979, REGN-2810, ресминостата, FAZ-053, PD-1/CTLA-4 биспецифического антитела (MacroGenics), MGA-012, MGD-013, M-7824, биспецифического антитела на основе PD-1 (Beijing Hanmi Pharmaceutical), AK-112, AK-106, AK-104, AK-103, BI-754091, ENUM-244C8, MCLA-145, MCLA-134, анти-PD-1 онколитического моноклонального антитела (Transgene SA), AGEN-2034, IBI-308, WBP-3155, JNJ-63723283, MEDI-0680, SSI-361, CBT-502, анти-PD-1 биспецифического антитела, нацеленного на две мишени анти-PD-1/LAG-3 mAbs (TESARO), нацеленного на две мишени анти-PD-1/TIM-3 mAbs (TESARO), PF-06801591, LY-3300054, BCD-100, STI-1110, биоаналога пембролизумаба, биоаналога ниволумаба, терапии PD-L1–ТФР-бета, KY-1003, STI-1014, GLS-010, AM-0001, GX-P2, KD-033,PD-L1/BCMA биспецифического антитела (Immune Pharmaceuticals), биспецифического антитела против PD-1/Ox40 (Immune Pharmaceuticals), BMS-936559, анти-PD-1/VEGF-A DARPins (Molecular Partners), mDX-400, ALN-PDL, PD-1 ингибирующего пептида (Aurigene), вакцины на основе дендритных клеток с миРНК (Alnylam Pharmaceuticals), GB-226, иммунотерапии PD-L1 на основе CAR-TNK (TNK Therapeutics/NantKwest), INSIX RA, INDUS-903, AMP-224, биспецифического гуманизированного антитела анти-CTLA-4/анти-PD-1 (Akeso Biopharma), вакцины B7-H1 (State Key Laboratory of Cancer Biology/Fourth Military Medical University) и GX-D1.
Примеры
Описанные ниже примеры приведены для иллюстрации, а не для ограничения настоящего изобретения.
Применяемым низкомолекулярным CCR1 антагонистом является Соединение 3.002, а анти-PD-L1 моноклональное антитело представляет собой BioLegend® 10F.9G2.
Пример 1. Комбинированная терапия, состоящая в ингибировании хемокинового рецептора плюс блокада pd-l1, усиливает противоопухолевое действие в мышиной модели рака груди
Траффик и экспансия супрессорных клеток миелоидного происхождения (MDSC) играет большую роль в иммунном подавлении опухоли. MDSC экспрессируют хемокиновые рецепторы, которые обсулавливают их поступление в микроокружение опухоли. Подавление Т-клеток также осуществляется вследствие взаимодействия PD-1 (programmed death-1) и его лигандов, которые избыточно экспрессируются в раковых клетках и иммунных инфильтратах, включая MDSC.
Рак груди является одной из главных причин смертей, связанных с раковыми заболеваниями. По оценкам, число новых случаев в 2015 году составило 231840. Нацеленная терапия успешно работает для некоторых подтипов рака груди, например, трастузумаб (Herceptin®) против HER2+ подтипа, и гормональная терапия для подтипов эстрогенового рецептора (ЭР)+ или прогестеронового рецептора (ПР)+. К сожалению, нет эффективной нацеленной терапии против трижды негативного рака груди (TNBC; HER2-ER-PR-). TNBC более иммуногенный, чем другие типы. Трижды негативный рак груди в высшей степени мутагенный и продуцирует неоантигены, которые вызывают иммунный ответ. Иммуномодулирующая терапия может быть эффективной для лечения этого и схожих видов рака.
Этот пример показывает, что задействование обоих механизмов посредством введения низкомолекулярного антагониста хемокинового рецептора (Соединение 3.002, которое блокирует CCR1) и анти-PD-L1 моноклонального антитела (BioLegend® 10F.9G2) значительно уменьшает вес опухоли в мышиной модели ортотопического рака груди.
Анализ образцов от пациентов с человеческим раком груди из базы данных “Атлас ракового генома” (The Cancer Genome Atlas, TCGA) показал, что CCR1 лиганды, MCP-7 (CCL7) и RANTES (CCL5), присутствуют в значительно повышенных количествах при раке груди, по сравнению с нормальной тканью груди. Кроме того, CCR1 и его лиганды, а также PD-L1, присутствуют в значительно повышенных количествах в образцах при трижды негативном раке груди, по сравнению с другими подтипами рака груди.
Повышенные уровни экспрессии лигандов хемокинового рецептора CCR1 - CCL5 (RANTES; фиг. 1A) и CCL7 (MCP-3; фиг. 1B), а также лиганда PD-1 (PD-L1; фиг. 1D) были обнаружены в образцах ткани от людей с трижды негативным раком груди, в сравнении с образцами нормальной ткани и образцами ткани от всех других типов рака груди. Увеличение экспрессии было статистически значимым. Экспрессия мРНК CCR1 лиганда CCL3 (MIP-1α; фиг. 1C) не была повышенной в образцах рака груди, например в образцах трижды негативного рака груди.
Экспрессия CCR1 и PD-L1 хорошо коррелировала в образцах от пациентов с человеческим раком груди (фиг. 1E). Каждая точка на диаграмме представляет собой образец опухоли груди от отдельного пациента с раком груди. На фиг. 1E, “r” представляет собой коэффициент корреляции Пирсона.
В этом исследовании использовали мышиную модель трижды негативного рака груди (мышиная модель 4T1 карциномы молочной железы) для исследования терапевтического эффекта CCR1 блокады в комбинации с PD-L1 моноклональным антителом.
5x104 4T1 клеток рака груди ортотопически инъецировали в молочную железу 6-8-недельных самок мышей BALB/C. Через 13 дней после имплантирования мышей рандомно распределяли по группам по объему опухоли, так что каждая группа имела одинаковый средний объем опухоли около 150 мм3. Объемы опухолей измеряли кавернометром и вычисляли по формуле ширина x (длина)2/2. В день 13 после имплантирования, начинали введение CCR1 антагониста 3.002 в течение 10 последующих дней в дозировке 15 мг/кг через оральный зонд, два раза в сутки. Анти-PD-L1 моноклональное антитело (BioLegend, 10F.9G2) вводили в день 13, 17 и 20 интраперитонеально, 200 мкг на мышь. Такой же объем носителя (ПЭГ400) и такой же объем изотипа контрольного антитела (Rat IgG, BioLegend) вводили мышам в контрольной группе. Объем опухолей измеряли три раза в неделю до дня 23 после имплантирования (фиг. 3A-3D). Мыши, получавшие комбинированное лечение, продемонстрировали пониженное прогрессирование опухоли (фиг. 3D) по сравнению с мышами, получавшими только CCR1 антагонист (фиг. 3B), мышами, получавшими только PD-L1 mAb (фиг. 3C), и мышами из контрольной группы (фиг. 3A).
В день 23 после имплантирования мышей умерщвляли и отбирали опухоли и легкие. Измеряли влажный вес опухолей. На фиг. 3E показано, что мыши, терапия которых включала CCR1 антагонист в связке с анти-PD-L1 моноклональным антителом, имели наименьший вес опухолей.
Рост первичных опухолей не менялся заметно при введении какого-либо средства в отдельности (фиг. 3B и 3C), но комбинация CCR1 ингибитора и анти-PD-L1 антитела (фиг. 3D) приводила к значительному уменьшению веса опухоли. Вес опухолей значительно снижался у мышей, получавших комбинированную терапию (фиг. 3E).
Легкие фиксировали в растворе Боуина в течение ночи и подсчитывали число поверхностных метастатических узлов невооруженным глазом (фиг. 4A). Статистический анализ проводили с применением t-критерия Стьюдента. *;p<0.05, **; p<0.005, ****; p<0.0001. Мыши, которым вводили только CCR1 антагонист, имели меньше метастатических узлов на легкое, по сравнению с мышами из контрольной группы, которым вводили только носитель (фиг. 4B).
Метастазы в легких статистически значимо уменьшались при введении только CCR1 антагониста в мышиной модели 4T1. На фиг. 4A показано, что число метастатических узлов на легкое было меньше у мышей, получавших CCR1 антагонист, по сравнению с контрольной группой.
Число супрессорных клеток миелоидного происхождения (MDSC) быстро увеличивается во время воспаления, инфекции и ракового заболевания, и ингибирует иммунный ответ. MDSC активно участвуют в подавлении вызванного опухолью иммунного ответа. Существуют два основных типа MDSC, показанные в таблице 1.
Таблица 1
(G-MDSC)
(M-MDSC)
Приживление клеток ортотопического рака груди вызывало сильную экспансию CD11bposLy6GhiLy6Chi MDSC или G-MDSC, субпопуляция которого экспрессирует CCR1. На фиг. 5A показано значительное увеличение концентрации G-MDSC в крови (фиг. 5A) и в селезенке (фиг. 5B) у мышей с 4T1 опухолью. Основную часть иммунных клеток, которые проникают в 4T1 опухоли, составляют G-MDSC (фиг. 5C). Также, основную часть CCR1-экспрессирующих клеток в селезенке у мышей с 4T1 опухолью составляли G-MDSC. FACS анализ, изображенный на фиг. 6A, показывает популяцию клеток CD45pos и CCR1pos из селезенки. Эту популяцию дополнительно анализировали для определения процента G-MDSC (CD11bposLy6GhighLy6Chigh), M-MDSC (CD11bposLy6GnegLy6Chigh) и макрофагов (CD11bposLy6GnegLy6CnegF4/80pos) в этих клетках. Анализ также показал, что опухоли с большим проникновением G-MDSC и меньшим проникновением Т-клеток были больше по размеру. Например, фиг. 7A показыват, что вес опухоли коррелировал с увеличением доли G-MDSC в иммунных клетках, проникающих в опухоль. Фиг. 7B показывает, что вес опухоли коррелировал также с уменьшением процентной доли CD8+ T-клеток в иммунных клетках, проникающих в опухоль.
Количество или процентная доля G-MDSC в иммунных клетках, проникающих в опухоль, в крови мышей с опухолью 4T1 сильно снижались после применения к мышам комбинированной терапии низкомолекулярным CCR1 антагонистом (3.002) и PD-L1 mAb (фиг. 8A). Такая комбинированная терапия не влияла на количество или процентное содержание M-MDSC в крови (фиг. 8B).
Анализ клеток, проникающих в опухоль, показал, что 3.002 отдельно или в комбинации с PD-L1 mAb значительно уменьшал количество G-MDSC в первичных опухолях (фиг. 9A). Результаты показывают, что CCR1 блокада вследствие притока G-MDSC отражается в уменьшении веса опухоли. Режимы введения не изменяли количество или процентное содержание M-MDSC в первичных опухолях (фиг. 9B). Введение CCR1 антагониста отдельно или в комбинации с PD-L1 mAb значительно уменьшало процент CD8+ T-клеток (фиг. 9C). В некоторых 4T1 опухолях, 3.002 отдельно или в комбинации с PD-L1 mAb также уменьшал проникновение B-клеток.
Суммируя вышесказанное, полученные данные показывают, что CCR1 обуславливает поступление G-MDSC в опухоль или микроокружение опухоли и способствует разрастанию опухоли (фиг. 10). Экспрессия лигандов CCR1 рецептора и PD-L1 значительно повышена в опухолях при раке груди у человека, и в частности – при трижды негативном раке груди. Уровень G-MDSC повышен у мышей с 4T1 трижды негативными опухолями груди и положительно коррелируют с увеличением размера опухолей. Введение низкомолекулярного CCR1 антагониста, 3.002, в комбинации с PD-L1 mAb значительно уменьшает прогрессирование первичной опухоли. Введение низкомолекулярного CCR1 антагониста, 3.002, значительно уменьшает образование метастазов в легких. Также, CCR1 антагонист и PD-L1 mAb понижают уровень G-MDSC в кровеносной системе и их проникновение в опухоль, и повышают проникновение CD8 T-клеток и В-клеток в 4T1 трижды негативные опухоли. Такая комбинированная терапия усиливает противоопухолевый иммунный ответ. Так, комбинированная терапия, включающая CCR1 антагонист и PD-L1 mAb, может обладать терапевтической эффективностью при лечении людей с трижды негативным раком груди.
Хотя описанное выше изобретение было описано в некоторых подробностях посредством иллюстраций и примеров в целях ясности понимания, квалифицированному специалисту в данной области будет понятно, что определенные изменения и модификации могут быть осуществлены в рамках объема прилагаемой формулы изобретения. Кроме того, каждый литературный источник, приведенный в настоящем изобретении, включен в настоящий текст посредством сылки во всей своей полноте в той же степени, как если бы каждый литературный источник был отдельно включен в настоящий текст посредством ссылки.
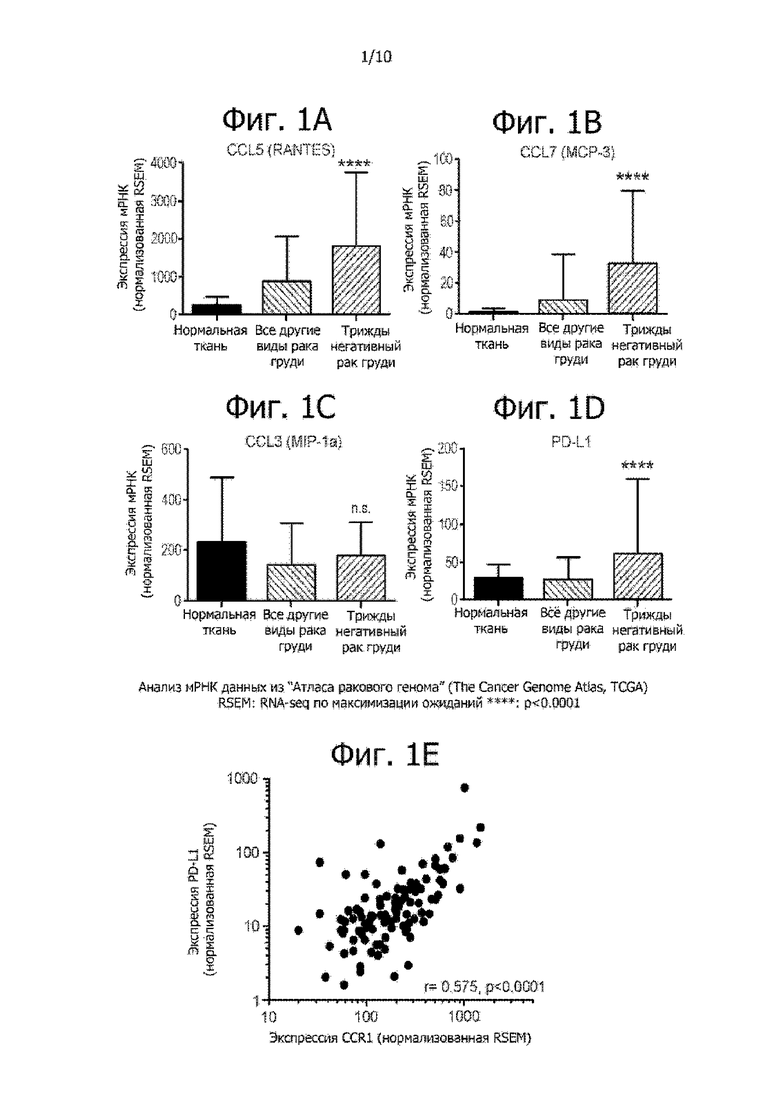
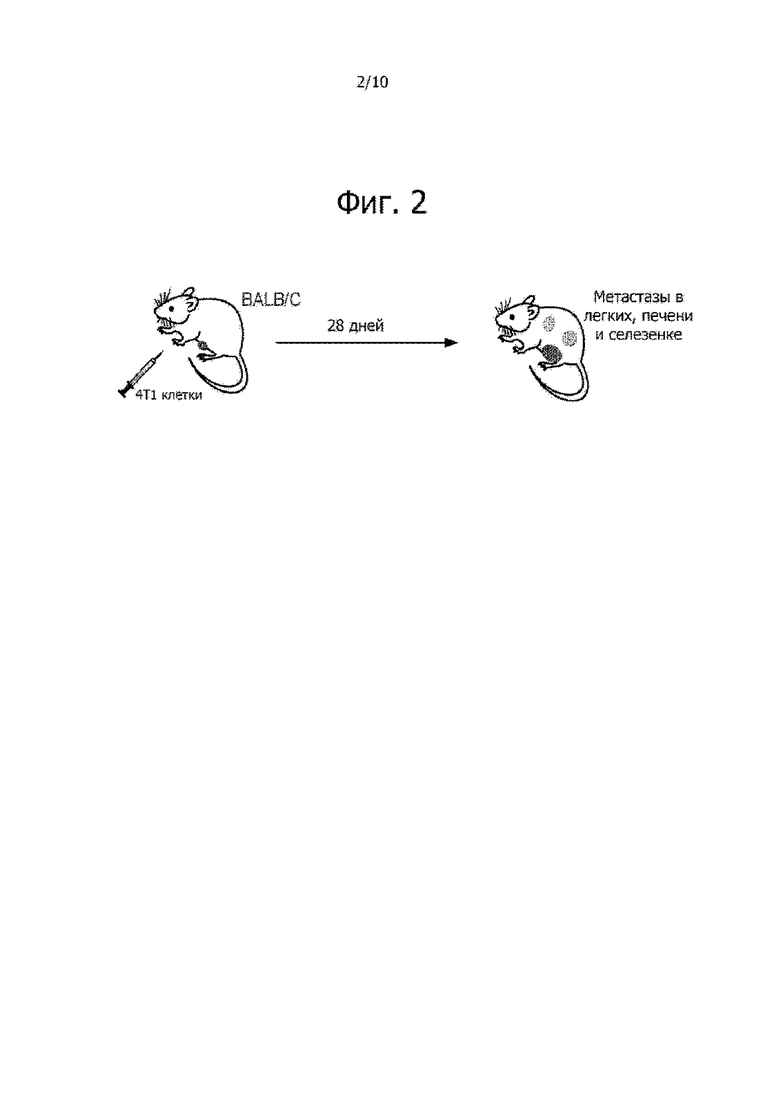
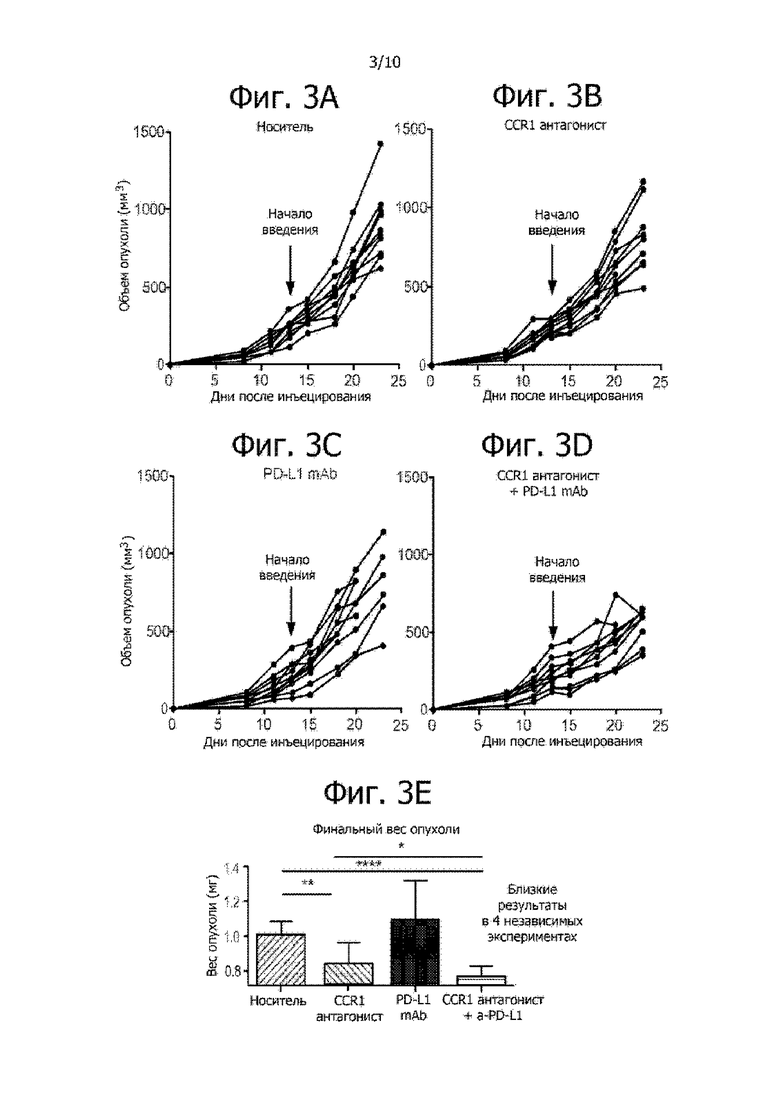
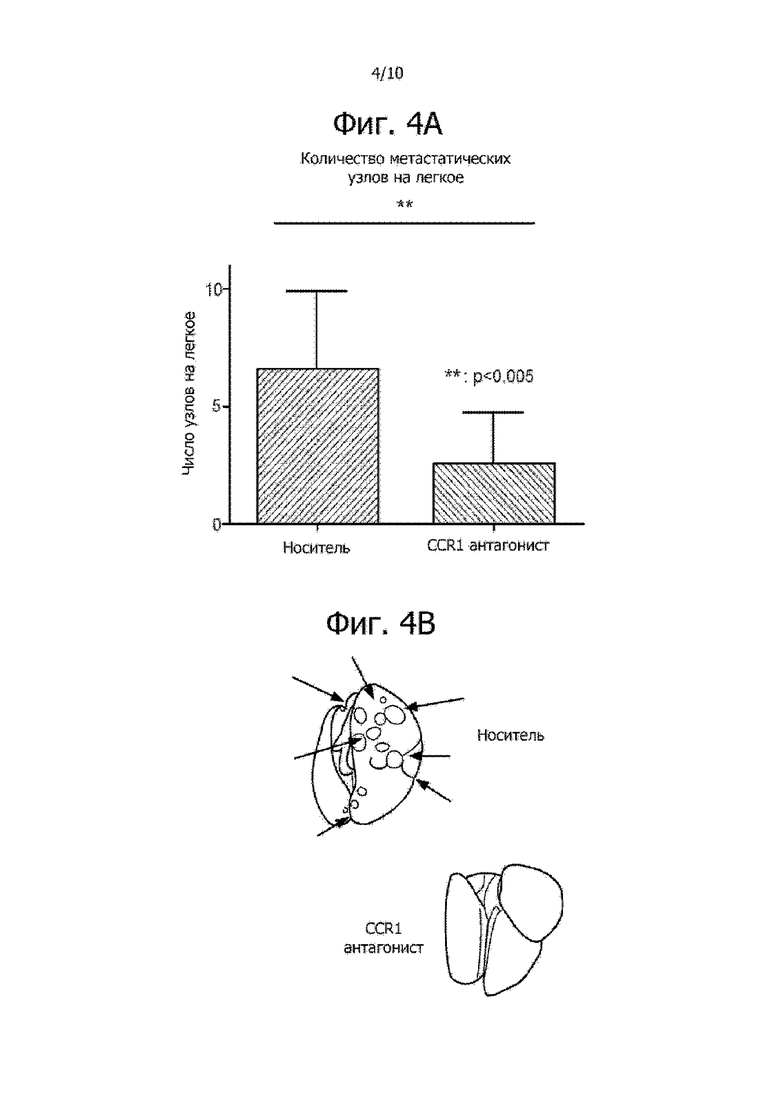
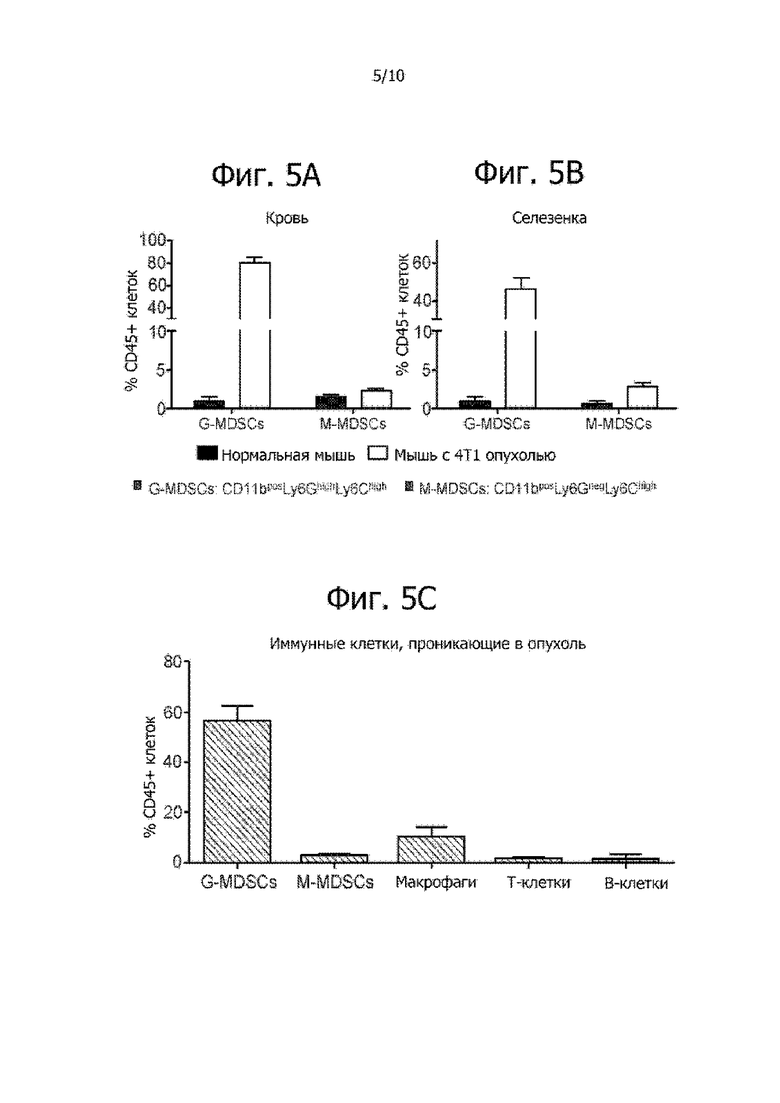
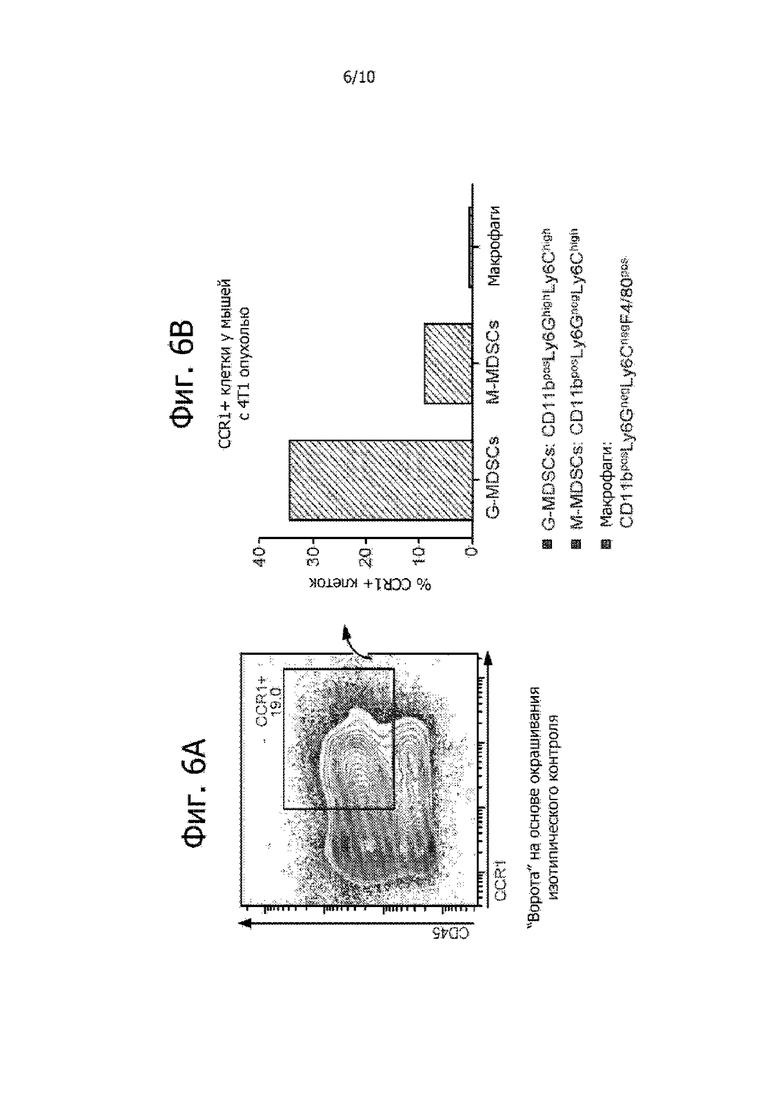
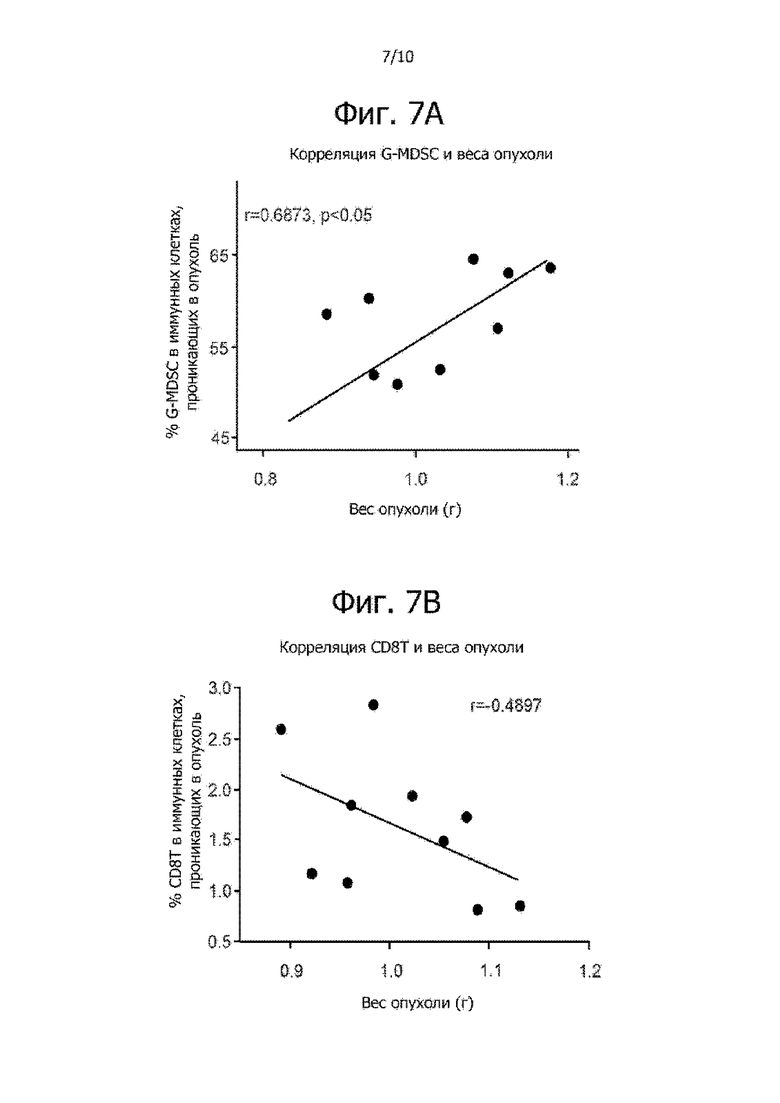
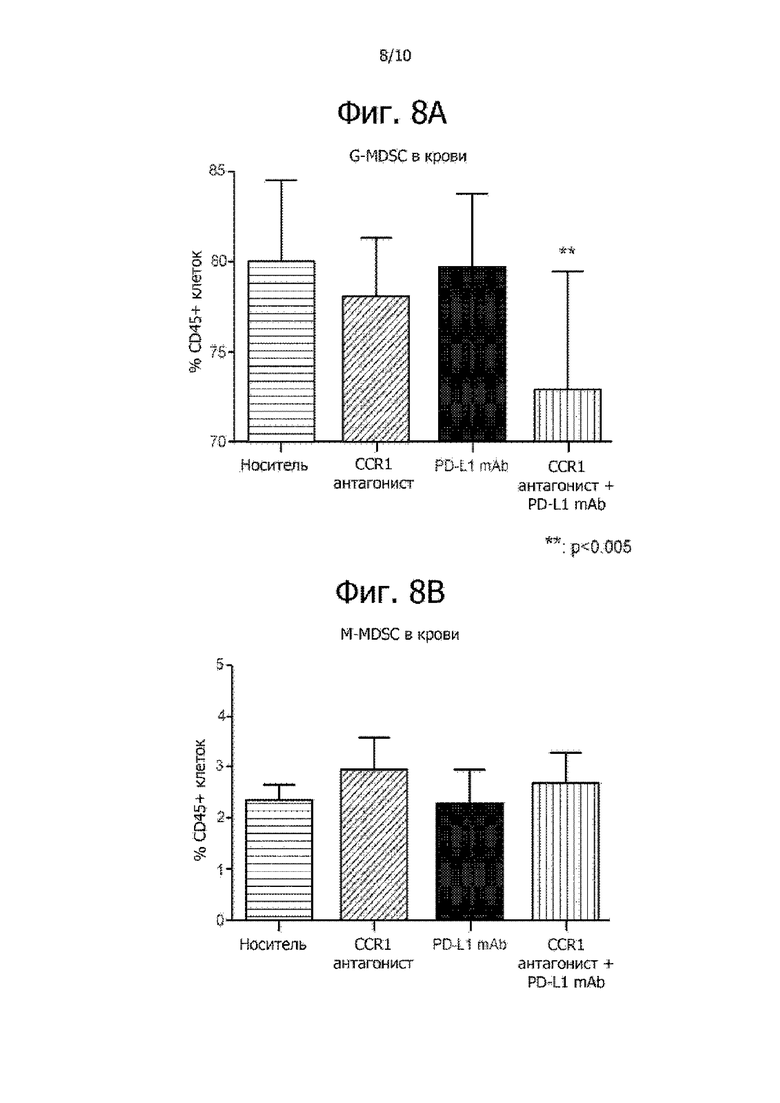
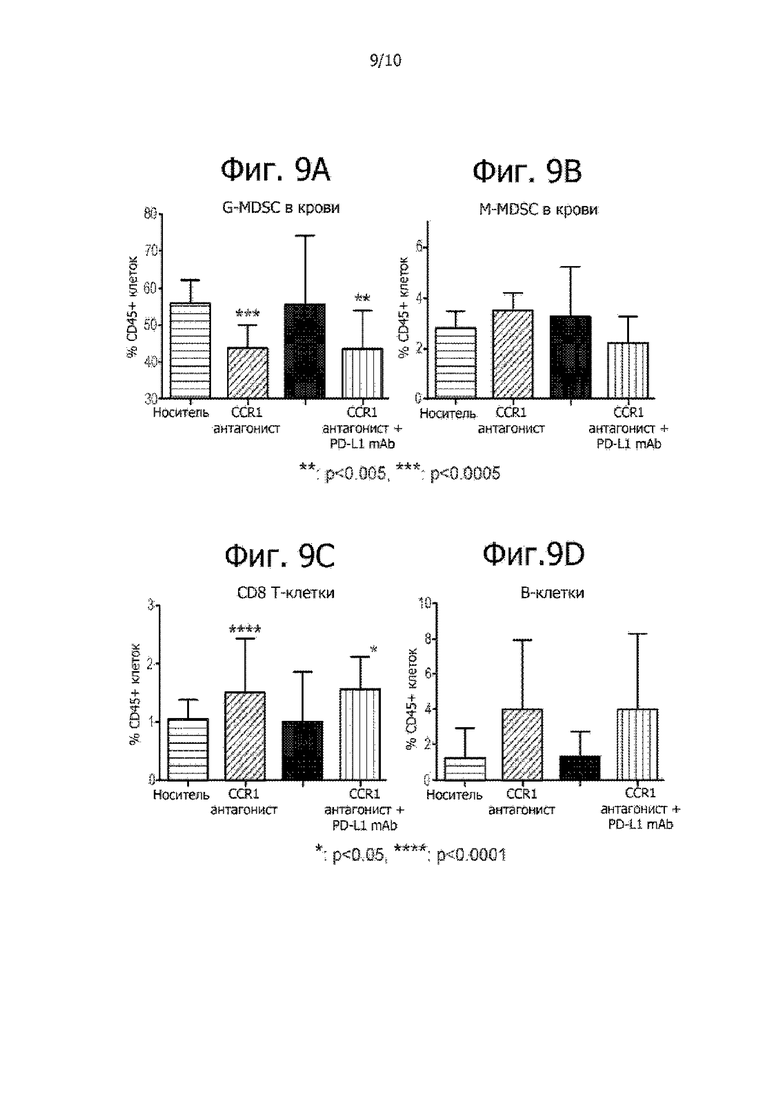
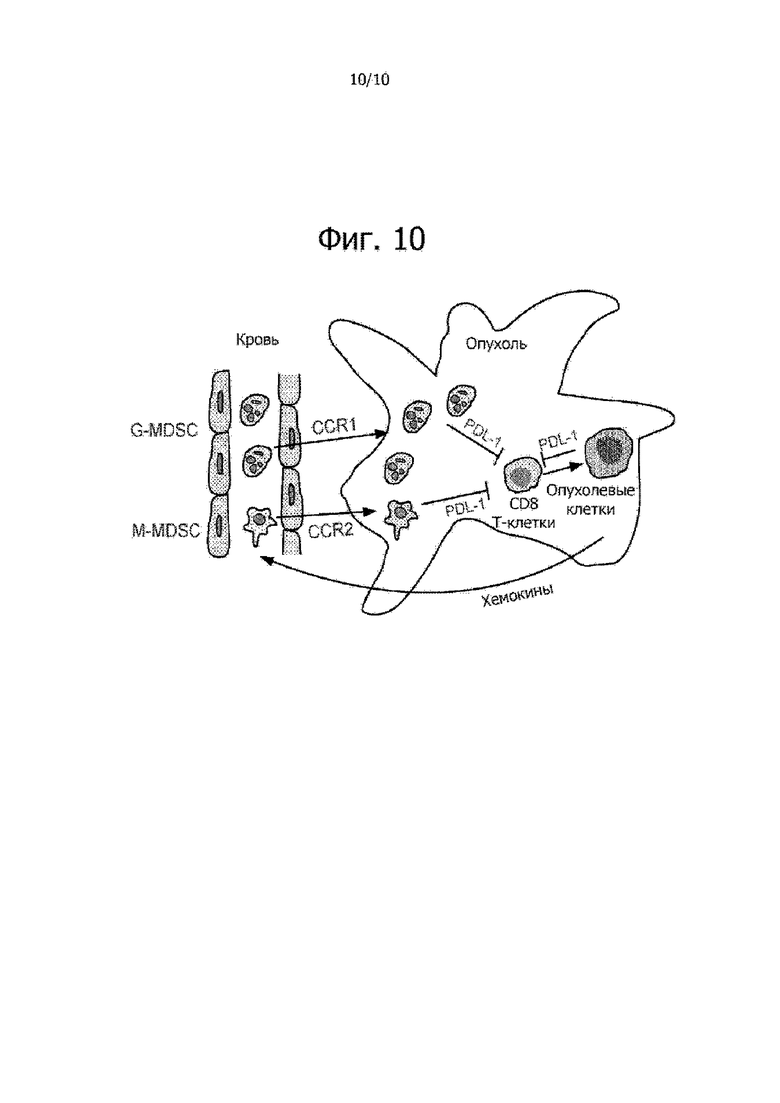
Группа изобретений относится к области медицины и фармацевтики. Первый объект представляет собой способ лечения субъекта с трижды негативным раком груди, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества антагониста хемокинового рецептора CCR1 и терапевтически эффективного количества PD-1 ингибитора или PD-L1 ингибитора, где антагонист хемокинового рецептора CCR1 представляет собой соединение формулы (IIIb1a):
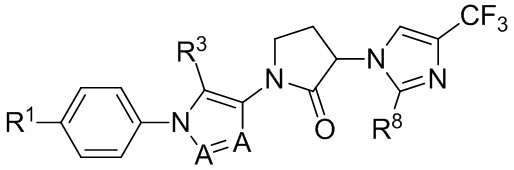 (IIIb1a),
(IIIb1a),
где каждый A представляет собой N или CH и по меньшей мере один A представляет собой N; R1 представляет собой галоген; R3 выбран из группы, состоящей из C1-8 алкила, C3-8 циклоалкила и C2-8 алкенила; и R8 представляет собой C1-8 алкил; или его фармацевтически приемлемую соль. Второй и третий объекты представляют собой соответствующие композицию и набор. Технический результат заключается в синергетическом эффекте комбинаций по изобретению при лечении трижды негативного рака груди. 3 н. и 24 з.п. ф-лы, 10 ил., 1 табл., 1 пр.
1. Способ лечения субъекта с трижды негативным раком груди, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества антагониста хемокинового рецептора CCR1 и терапевтически эффективного количества PD-1 ингибитора или PD-L1 ингибитора, где антагонист хемокинового рецептора CCR1 имеет формулу (IIIb1a):
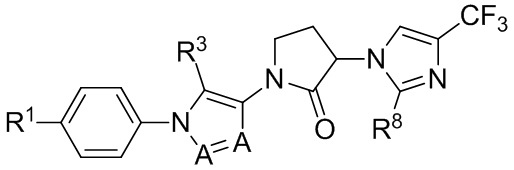 (IIIb1a)
(IIIb1a)
где каждый A представляет собой N или CH и по меньшей мере один A представляет собой N; R1 представляет собой галоген; R3 выбран из группы, состоящей из C1-8 алкила, C3-8 циклоалкила и C2-8 алкенила; и R8 представляет собой C1-8 алкил, или его фармацевтически приемлемой соли.
2. Способ по п. 1, где вводят антагонист хемокинового рецептора CCR1 и PD-1 ингибитор в виде комбинации.
3. Способ по п. 1, где вводят антагонист хемокинового рецептора CCR1 и PD-L1 ингибитор в виде комбинации.
4. Способ по п. 1, где антагонист хемокинового рецептора CCR1 выбран из группы, состоящей из:
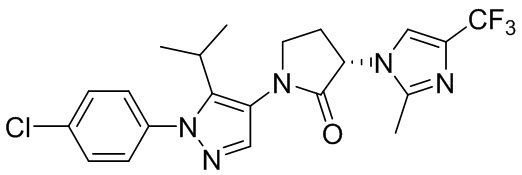 и
и 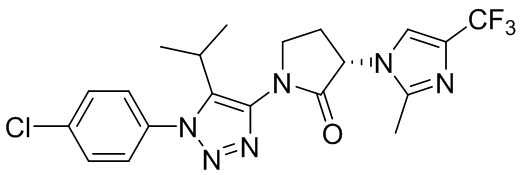
или их фармацевтически приемлемой соли.
5. Способ по любому из пп. 1, 2 или 4, где PD-1 ингибитор выбран из группы, состоящей из пембролизумаба, ниволумаба, IBI-308, mDX-400, BGB-108, MEDI-0680, SHR-1210, PF-06801591, PDR-001, GB-226 и STI-1110.
6. Способ по любому из пп. 1, 3 или 4, где PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, авелумаба, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, CA-327, STI-1014 и KY-1003.
7. Способ по любому из пп. 1-6, где антагонист хемокинового рецептора CCR1 и PD-1 ингибитор или PD-L1 ингибитор вводят одновременно.
8. Способ по п. 7, где антагонист хемокинового рецептора CCR1 и PD-1 ингибитор или PD-L1 ингибитор вводят в виде комбинированного препарата.
9. Способ по любому из пп. 1-6, где антагонист хемокинового рецептора CCR1 и PD-1 ингибитор или PD-L1 ингибитор вводят последовательно.
10. Способ по п. 9, где антагонист хемокинового рецептора CCR1 вводят до введения PD-1 ингибитора или PD-L1 ингибитора.
11. Способ по п. 9, где антагонист хемокинового рецептора CCR1 вводят после введения PD-1 ингибитора или PD-L1 ингибитора.
12. Способ по любому из пп. 1-11, где антагонист хемокинового рецептора CCR1 вводят перорально и PD-1 ингибитор или PD-L1 ингибитор вводят внутривенно.
13. Способ по любому из пп. 1-12, где субъект представляет собой человека.
14. Композиция для лечения субъекта с трижды негативным раком груди, содержащая терапевтически эффективное количество антагониста хемокинового рецептора CCR1, терапевтически эффективное количество PD-1 ингибитора или PD-L1 ингибитора и фармацевтически приемлемый носитель или вспомогательное вещество, где антагонист хемокинового рецептора CCR1 имеет формулу (IIIb1a):
(IIIb1a)
где каждый A представляет собой N или CH и по меньшей мере один A представляет собой N; R1 представляет собой галоген; R3 выбран из группы, состоящей из C1-8 алкила, C3-8 циклоалкила и C2-8 алкенила; и R8 представляет собой C1-8 алкил, или его фармацевтически приемлемой соли.
15. Композиция по п. 14, содержащая антагонист хемокинового рецептора CCR1, PD-1 ингибитор и фармацевтически приемлемый носитель или вспомогательное вещество.
16. Композиция по п. 14, содержащая антагонист хемокинового рецептора CCR1, PD-L1 ингибитор и фармацевтически приемлемый носитель или вспомогательное вещество.
17. Композиция по п. 14, где антагонист хемокинового рецептора CCR1 выбран из группы, состоящей из
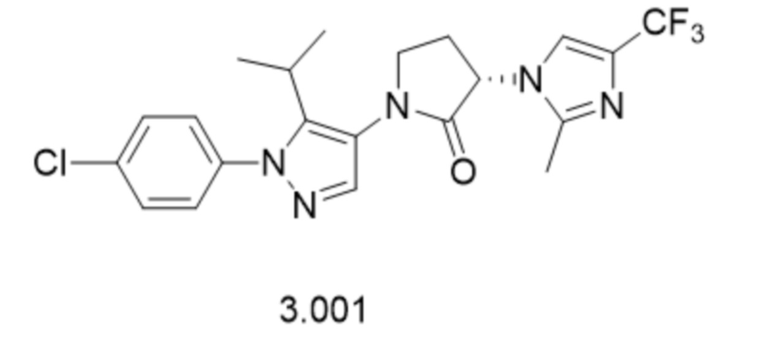
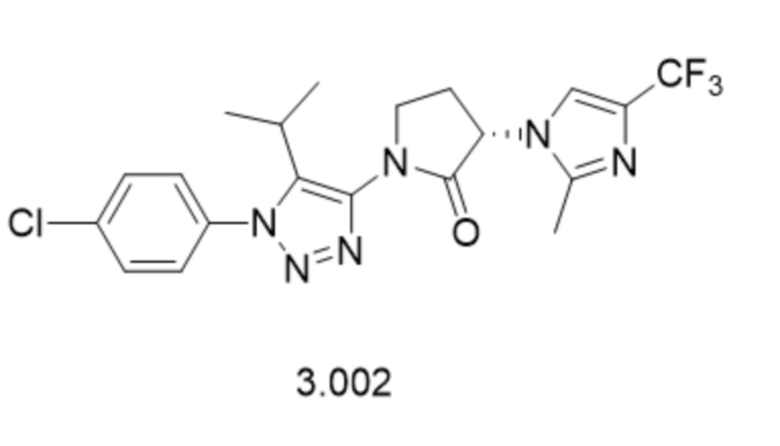
или их фармацевтически приемлемой соли.
18. Композиция по любому из пп. 14, 15 или 17, где PD-1 ингибитор выбран из группы, состоящей из пембролизумаба, ниволумаба, IBI-308, mDX-400, BGB-108, MEDI-0680, SHR-1210, PF-06801591, PDR-001, GB-226 и STI-1110.
19. Композиция по любому из пп. 14 или 17, где PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, авелумаба, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, CA-327, STI-1014 и KY-1003.
20. Набор для лечения субъекта с трижды негативным раком груди, содержащий терапевтически эффективное количество антагониста хемокинового рецептора CCR1 и терапевтически эффективное количество PD-1 ингибитора или PD-L1 ингибитора, с инструкцией по эффективному применению, где антагонист хемокинового рецептора CCR1 имеет формулу (IIIb1a):
(IIIb1a)
где каждый A представляет собой N или CH и по меньшей мере один A представляет собой N; R1 представляет собой галоген; R3 выбран из группы, состоящей из C1-8 алкила, C3-8 циклоалкила и C2-8 алкенила; и R8 представляет собой C1-8 алкил, или его фармацевтически приемлемой соли.
21. Набор по п. 20, содержащий антагонист хемокинового рецептора CCR1 и PD-1 ингибитор с инструкцией по эффективному применению.
22. Набор по п. 20, содержащий антагонист хемокинового рецептора CCR1 и PD-L1 ингибитор с инструкцией по эффективному применению.
23. Набор по п. 20, где антагонист хемокинового рецептора CCR1 выбран из группы, состоящей из
или их фармацевтически приемлемой соли.
24. Набор по любому из пп. 20, 21 или 23, где PD-1 ингибитор выбран из группы, состоящей из пембролизумаба, ниволумаба, IBI-308, mDX-400, BGB-108, MEDI-0680, SHR-1210, PF-06801591, PDR-001, GB-226 и STI-1110.
25. Набор по одному из пп. 20, 22 или 23, где PD-L1 ингибитор выбран из группы, состоящей из дурвалумаба, атезолизумаба, авелумаба, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, CA-327, STI-1014 и KY-1003.
26. Набор по любому из пп. 20-25, где антагонист хемокинового рецептора CCR1 и PD-1 ингибитор или PD-L1 ингибитор вводят в состав препаратов для одновременного введения.
27. Набор по любому из пп. 20-25, где антагонист хемокинового рецептора CCR1 и PD-1 ингибитор и/или PD-L1 ингибитор вводят в состав препаратов для последовательного введения.
| JUNG H | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| - Vol | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| - No | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| - P | |||
| Ротационный колун | 1919 |
|
SU227A1 |
| WO 2014089495 A1, 12.06.2014 | |||
| US 2008300257 A1, 04.12.2008 | |||
| WO 2010030815 A1, 18.03.2010 | |||
| WO 2014035967 A2, | |||

