Область техники
Настоящее изобретение относится к фармацевтическому составу, обеспечивающему длительное высвобождение аримокломола и сопутствующую пониженную Cmax, пониженное ингибирование ОСТ2 и/или пониженный эффект в отношении уровней креатинина в сыворотке крови.
Уровень техники
Аримокломол представляет собой амплификатор белков теплового шока, в настоящее время оцениваемый в лечении детских лизосомных болезней накопления и бокового амиотрофического склероза (ALS).
Физические свойства аримокломола делают лекарственное средство более сложным в обращении. Лекарственное вещество является белым на вид, легким и рыхлым. Аримокломол является гигроскопичным, т.е. он абсорбирует влагу (молекулы воды) из его окружения. Аримокломол характеризуется относительно коротким периодом полувыведения из плазмы крови (2-4 часа), и в настоящее время требуется введение нескольких доз в сутки.
Аримокломол до настоящего времени вводят в виде заполненных порошком желатиновых капсул с покрытием (капсулы с аримокломолом). Капсулы с аримокломолом относятся к типу препаратов с немедленным высвобождением (IR).
Аримокломол исследовался на здоровых людях-добровольцах, и не была достигнута максимальная переносимая доза. Всего 261 субъектов подвергались воздействию пероральных отдельных возрастающих или повторных доз аримокломола в диапазоне от 50 до 800 мг в семи заключительных испытаниях фазы I и двух заключительных испытаниях фазы II, и оказалось, что он является безопасным и хорошо переносимым.
В исследованиях фазы I с отдельными и многократными дозами наблюдались незначительные и обратимые повышения уровней креатинина в сыворотке крови у ряда добровольцев, но они не считались клинически значимыми (см., например, Cudkowicz et al., Muscle & Nerve, July 2008, p. 837-844).
Сущность изобретения
Аспект настоящего изобретения заключается в обеспечении состава с длительным высвобождением аримокломола.
За счет замедления высвобождения аримокломола после приема внутрь пероральной дозы у состава с длительным высвобождением проявляется относительно низкая пиковая концентрация аримокломола в крови (Cmax) в сравнении с общим обеспечиваемым воздействием, что выражается площадью под фармакокинетической кривой (AUC). В течение повторяющегося перорального введения состав с длительным высвобождением, таким образом, снижает соотношение пиковой и самой низкой концентрацией аримокломола в крови. Это дает более преимуществ для клинического применения по сравнению с традиционным пероральным составом с немедленным высвобождением:
За счет поддержки менее частого введения доз состав с длительным высвобождением аримокломола будет поддерживать лучшее соблюдение режима лечения при лечении в домашних условиях, а также регулярность назначения в ситуациях регулируемого медицинского обслуживания.
За счет улучшения физических характеристик текучести состав с длительным высвобождением будет более пригодным для выпуска в форме саше или пакетиков, что может быть желательно для введения дозы посредством смешивания с напитками или пищевым продуктом, а также будет способствовать применению через зонды для искусственного кормления. Это будет представлять собой заметное улучшение для лечения пациентов с дисфагией или другими нервно-мышечными нарушениями.
В случае, когда некоторые пациенты испытывают неблагоприятные эффекты перорального дозирования с немедленным высвобождением, состав с длительным высвобождением может обеспечивать метод лечения, который может поддерживать относительно высокий уровень воздействия аримокломола при ограничении неблагоприятных эффектов по сравнению с составом с немедленным высвобождением.
ОСТ2 представляет собой почечный транспортер органических катионов, вовлеченный в секрецию креатинина (см., например, Lepist et al., Kidney International (2014) 86, 350-357). OCT2 (транспортер органических катионов 2) также известен как член 2 семейства 22 транспортеров растворенных веществ и экспрессируется с SLC22A2 (UniProt S22A2_HUMAN).
В данном документе показано, что аримокломол является ингибитором почечного поглощающего транспортера ОСТ2, полумаксимальное ингибирование (IC50) аримокломолом было определено при концентрации 10 мкМ для ОСТ2. Таким образом, вскоре после перорального приема человеком дозы, которая выше чем 400 мг, у него может присутствовать временная и обратимая ингибирующая активность в отношении этого транспортера при Cmax.
Для преодоления наблюдаемого ингибирования ОСТ2, а также незначительного и обратимого повышения уровня креатинина в сыворотке крови предполагается состав аримокломола с длительным высвобождением.
Состав также потенциально представляет ценность для пациентов, которые получают дополнительные лекарственные препараты, причем данные препараты сами по себе оказывают воздействие на уровни креатинина в сыворотке крови, и/или данные препараты зависят по меньшей мере частично от ОСТ2 в том, что касается клиренса и выведения.
Этот состав может представлять дополнительную ценность, например, для пациентов-детей и пациентов, у которых присутствуют повышенные базальные уровни креатинина в сыворотке крови; в том числе пациентов с заболеванием почек или пониженной функцией почек, а также пациентов с сахарным диабетом или артериальной гипертензией.
Данные составы характеризуются длительным высвобождением, обеспечивая возможность введения уменьшенного количества доз в сутки. Данные составы, кроме того, являются легкими для проглатывания и имеют приемлемые органолептические характеристики. Данный состав также обеспечивает более эффективное производство, получение партии с установленными техническими характеристиками и обеспечение возможности лучшей стандартизации.
Аспект настоящего изобретения заключается в обеспечении фармацевтического состава, содержащего активный фармацевтический ингредиент (API), выбранный из N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, причем указанный состав обеспечивает длительное высвобождение указанного активного фармацевтического ингредиента.
В соответствии с одним вариантом осуществления данные фармацевтические составы обеспечивают более низкую Cmax, более высокое Tmax, пониженное ингибирование транспортера ОСТ2 и/или пониженный эффект в отношении уровня креатинина в сыворотке крови по сравнению с этими параметрами после введения состава с немедленным высвобождением или болюсной IV инъекции того же активного фармацевтического ингредиента.
Краткое описание чертежей
Фиг. 1. Ингибирование аримокломолом OATP1B1-опосредованного транспорта маркерного субстрата в анализе ингибирования поглощающего транспортера.
Фиг. 2. Ингибирование аримокломолом ОАТР1В3-опосредованного транспорта маркерного субстрата в анализе ингибирования поглощающего транспортера.
Фиг. 3. Ингибирование аримокломолом OAT1-опосредованного транспорта маркерного субстрата в анализе ингибирования поглощающего транспортера.
Фиг. 4. Ингибирование аримокломолом ОАТ3-опосредованного транспорта маркерного субстрата в анализе ингибирования поглощающего транспортера.
Фиг. 5. Ингибирование аримокломолом ОСТ2-опосредованного транспорта маркерного субстрата в анализе ингибирование поглощающего транспортера.
Фиг. 6. Накопление аримокломола в экспрессирующих ОАТ1 и контрольных СНО клетках в анализе доступности субстрата поглощающему транспортеру.
Фиг. 7. Накопление аримокломола в экспрессирующих ОАТ3 и контрольных MDCKII клетках в анализе доступности субстрата поглощающему транспортеру.
Фиг. 8. Накопление аримокломола в экспрессирующих ОСТ2 и контрольных СНО клетках в анализе доступности субстрата поглощающему транспортеру.
Фиг. 9. Профиль растворения мини-таблеток (смесь 1) после
покрытия дисперсией этилцеллюлозы на водной основе (Surelease ТМ) до увеличения массы на 5%, 10% и 20%.
Фиг. 10. Профиль растворения мини-таблеток (смесь 1) после покрытия дисперсией ЕС на основе растворителя (Surelease ТМ) до увеличения массы на 5%, 10% и 20%.
Фиг. 11. Профиль растворения мини-таблеток после покрытия изолирующим покрытием из НРМС и дисперсией этилцеллюлозы на водной основе (Surelease ТМ) до увеличения массы на 5%, 10% и 20%.
Фиг. 12. Профиль растворения отдельных мини-таблеток после покрытия изолирующим покрытием из НРМС и дисперсией этилцеллюлозы на водной основе (Surelease ТМ) до увеличения массы на 20%.
Фиг. 13. Схематическое представление состава сферы.
Фиг. 14. Профиль растворения сахарных сфер, покрытых этилцеллюлозой до увеличения массы на 5% масс., 10% масс. и 20% масс., в буфере с рН 6,8.
Фиг. 15. Профиль растворения сахарных сфер, покрытых этилцеллюлозой до увеличения массы на 5% масс., 10% масс., в буфере с 0,1 М HCl.
Фиг. 16. Профиль растворения микрокристаллических сфер, покрытых этилцеллюлозой до увеличения массы на 5% масс., 10% масс.
Фиг. 17. Профиль растворения гранул аримокломола, полученных с помощью экструзии горячего расплава. Растворение образцов порошка в концентрации 33, 50 и 66 масс. % с различными размерами частиц в фосфатном буфере с рН 6,8. Голубые линии относятся к материалам в концентрации 33 масс. %, розовые линии относятся к материалам в концентрации 50 масс. %, а зеленые линии относятся к материалу в концентрации 66 масс. %.
Подробное описание изобретения
Настоящим изобретением предполагаются составы аримокломола с длительным высвобождением (ER), обладающие улучшенными фармакокинетическими характеристиками, характеризующиеся стандартизированным производством и лучшим соблюдением режима лечения пациентом. Термины "длительное высвобождение", "пролонгированное высвобождение", "замедленное высвобождение" и "контролируемое высвобождение" используются в данном документе взаимозаменяемо.
Настоящим изобретением в одном аспекте предполагается фармацевтический состав, содержащий активный фармацевтический ингредиент (API), выбранный из N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида (аримокломола), его стереоизомеров и их солей присоединения кислоты, причем указанный состав обеспечивает длительное высвобождение указанного активного фармацевтического ингредиента.
Термины "фармацевтический состав", "фармацевтически безопасный состав" и "фармацевтически приемлемый состав" используются в данном документе взаимозаменяемо.
В соответствии с одним вариантом осуществления указанный фармацевтический состав содержит указанный API в фармацевтически эффективном или фармацевтически активном количестве.
В соответствии с одним вариантом осуществления указанный состав содержит внутреннюю матрицу и по меньшей мере одно наружное покрытие.
В соответствии с другим вариантом осуществления указанный состав содержит гранулы с длительным высвобождением. В соответствии с одним вариантом осуществления указанные гранулы с длительным высвобождением получают с помощью экструзии горячего расплава (НМЕ) и необязательно измельчения.
В соответствии с одним вариантом осуществления данный фармацевтический состав снижает Cmax, повышает Tmax, снижает ингибирование ОСТ2 и/или снижает эффект в отношении уровней креатинина в сыворотке крови по сравнению с эквивалентным количеством N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида (аримокломола), его стереоизомеров и их солей присоединения кислоты, вводимых с помощью пероральной лекарственной формы с немедленным высвобождением и/или с помощью болюсной IV инъекции.
Технология длительного или контролируемого высвобождения представляет собой механизм, применяемый в составах для медленного растворения и высвобождения лекарственного средства с течением времени. Составы с длительным высвобождением можно принимать менее часто, нежели составы с немедленным высвобождением, и они поддерживают более устойчивые уровни лекарственного средства в кровотоке.
В соответствии с одним вариантом осуществления термин "обеспечение длительного высвобождения активного фармацевтического ингредиента согласно настоящему изобретению" означает, что API растворяется или высвобождается из фармацевтического состава с течением времени.
Лекарственные средства с длительным высвобождением можно составлять таким образом, чтобы активный ингредиент был погружен в матрицу из нерастворимого вещества(веществ), благодаря чему растворяющееся лекарственное средство должно проникать через отверстия в матрице. Некоторые лекарственные средства заключены в таблетки на основе полимера с прорезанным с помощью лазера отверстием с одной стороны и пористой мембраной с другой стороны. Желудочные кислоты пробиваются через пористую мембрану, тем самым выталкивая лекарственное средство наружу через прорезанное с помощью лазера отверстие. Со временем вся доза лекарственного средства высвобождается в систему, в то время как полимерный контейнер остается нетронутым, позднее выводясь из организма благодаря нормальному пищеварению. В некоторых составах лекарственное средство растворяется в матрице, и матрица физически набухает с образованием геля, обеспечивая возможность выхода лекарственного средства через наружную поверхность геля. Микроинкапсуляция также дает сложные профили растворения, благодаря нанесению активного фармацевтического ингредиента в виде покрытия вокруг инертного ядра и наслаиванию на него нерастворимых веществ с образованием микросферы получают более устойчивую и воспроизводимую скорость растворения при удобном формате, в котором можно обеспечить смешивание с другими фармацевтическими ингредиентами с быстрым высвобождением, например, в любой желатиновой капсуле из двух половин.
Лекарственная форма представляет собой смесь активных компонентов лекарственного средства и компонентов, не являющихся лекарственным средством. Фармацевтический состав в соответствии с настоящим изобретением в одном варианте осуществления представляет собой лекарственную форму, такую как пероральная лекарственная форма. В соответствии с одним вариантом осуществления указанная лекарственная форма представляет собой твердую лекарственную форму, такую как таблетка. В соответствии с одним вариантом осуществления указанная лекарственная форма представляет собой гранулярную лекарственную форму, как например, включающую гранулы с длительным высвобождением.
В соответствии с одним вариантом осуществления указанный фармацевтический состав является доступным перорально. В соответствии с одним вариантом осуществления указанный состав представляет собой твердую лекарственную форму. В соответствии с одним вариантом осуществления указанный состав представляет собой перорально доступную твердую лекарственную форму.
Таблетка представляет собой фармацевтическую лекарственную форму, содержащую смесь (какого-либо) активного вещества(активных веществ) и вспомогательного вещества(вспомогательных веществ), спрессованную или уплотненную в дозу в виде твердого вещества. Таблетки являются простыми и удобными для применения. Они обеспечивают точно отмеренную дозу активного ингредиента(активных ингредиентов) в удобной портативной упаковке. Процессы и методики производства могут обеспечивать таблеткам специальные свойства, например, составы с длительным высвобождением или быстрорастворимые составы. Таблетки легко отвешивать, и они характеризуются высокой физической целостностью.
Мини-таблетки представляют собой таблетки с диаметром ≤3 мм и представляют новую тенденцию в создании твердых лекарственных форм, причем ее главной целью является преодоление некоторых терапевтических препятствий, таких как нарушенное глотание и терапия с одновременным назначением нескольких лекарственных средств, а также они предоставляют некоторые терапевтические выходы, такие как широкий диапазон дозировки и сочетающийся характер высвобождения. В соответствии с одним вариантом осуществления мини-таблетка согласно настоящему изобретению представляет собой таблетку с диаметром, меньшим или равным (≤) 3 мм, как например, ≤2,5 мм, например, ≤2 мм, как например, ≤1,5 мм, например, приблизительно 1 мм. В соответствии с одним вариантом осуществления мини-таблетка согласно настоящему изобретению представляет собой таблетку с диаметром от 1 до 1,5 мм, как например, от 1,5 до 2 мм, например, от 2 до 2,5 мм, как например, от 2,5 до 3 мм.
В соответствии с одним вариантом осуществления микро-таблетка согласно настоящему изобретению представляет собой таблетку с диаметром, меньшим или равным (≤) 1 мм, как например, ≤0,9 мм, например, ≤0,8 мм, как например, ≤0,7 мм, например, ≤0,6 мм, как например, ≤0,5 мм, например, ≤0,4 мм, как например, ≤0,3 мм, например, ≤0,3 мм, как например, ≤0,1 мм. В соответствии с одним вариантом осуществления мини-таблетка согласно настоящему изобретению представляет собой таблетку с диаметром от 0,1 до 0,2 мм, как например, от 0,2 до 0,3 мм, например, от 0,3 до 0,4 мм, как например, от 0,4 до 0,5 мм, как например, от 0,5 до 0,6 мм, например, от 0,6 до 0,7 мм, как например, от 0,7 до 0,8 мм, например, от 0,8 до 0,9 мм, как например, от 0,9 до ≤1 мм.
В производстве фармацевтических средств термин "инкапсуляция" относится к ряду лекарственных форм в относительно стабильной оболочке, известной как капсула, которая позволяет, например, принимать их перорально или применять в качестве суппозиториев. Существует два основных типа капсул:
Капсулы с твердой оболочкой, выполненные из двух половинок: "корпуса" меньшего диаметра, который заполняется, а затем запечатывается с использованием "крышки" большего диаметра; и капсулы с мягкой оболочкой, преимущественно используемые для масел и для активных ингредиентов, которые растворяются или суспендируются в масле. Оба типа капсул выполнены из водных растворов гелеобразующих средств, включающих животный белок, главным образом желатин, и растительные полисахариды или их производные, такие как каррагенаны и модифицированные формы крахмала и целлюлозы. К раствору гелеобразующего средства могут быть добавлены другие ингредиенты, такие как пластификаторы, как например, глицерин и/или сорбит, для снижения твердости капсулы, красящие вещества, консерванты, разрыхлители, смазывающие средства и средства для поверхностной обработки.
Фармацевтический состав в соответствии с настоящим изобретением в одном варианте осуществления содержит активный фармацевтический ингредиент (API), который подробно описан в другом месте в данном документе, а также одно или более вспомогательных веществ.
Вспомогательное вещество представляет собой фармакологически неактивное (или химически неактивное) вещество, составленное с активным ингредиентом лекарственного препарата. Вспомогательные вещества обычно применяют для увеличения объема составов, которые содержат сильнодействующие активные ингредиенты (из-за этого часто называются "объемообразующими средствами", "наполнителями" или "разбавителями"), обеспечивая возможность удобного и точного распределения лекарственного вещества при получении лекарственной формы.
В соответствии с одним вариантом осуществления фармацевтический состав согласно настоящему изобретению содержит одно или более вспомогательных веществ. Указанные один или более вспомогательных веществ могут действовать как твердый носитель, разбавитель, ароматизатор, солюбилизатор, смазывающее средство, скользящее вещество, суспендирующее средство, связующее, наполнитель, консервант, антиадгезив, смачивающее средство, средство, обеспечивающее распадаемость таблетки, сорбент и/или инкапсулирующий/покровный материал.
Данный фармацевтический состав в соответствии с одним вариантом осуществления содержит по меньшей мере одно вспомогательное вещество для получения подходящего состава с желаемыми характеристиками длительного высвобождения.
В соответствии с одним вариантом осуществления фармацевтический состав согласно настоящему изобретению содержит одно или более контролирующих высвобождение вспомогательных веществ.
Состав с длительным высвобождением
Один аспект заключается в обеспечении фармацевтического состава, содержащего
- активный фармацевтический ингредиент (API), выбранный из N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, и
- контролирующее высвобождение вспомогательное вещество,
причем указанный состав обеспечивает длительное высвобождение указанного активного фармацевтического ингредиента.
В соответствии с одним вариантом осуществления указанный фармацевтический состав представляет собой лекарственную форму, такую как пероральная лекарственная форма (перорально доступная лекарственная форма).
В соответствии с одним вариантом осуществления фармацевтический состав представляет собой лекарственную форму с длительным высвобождением N-[2-гидрокси-3-(1 -пиперидинил)-пропокси |-пиридин-1-оксид-3-карбоксимидоил-хлорида. его стереоизомеров и их солей присоединения кислоты, содержащую
- фармацевтически эффективное количество N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты и
- контролирующее высвобождение вспомогательное вещество.
"Cmax" представляет собой термин, который применяют в фармакокинетике и который относится к максимальной (или пиковой) концентрации в сыворотке крови, которой лекарственное средство достигает в определенной части или исследуемой области организма после того, как лекарственное средство было введено, и перед введением второй дозы.
"Tmax" представляет собой термин, применяемый в фармакокинетике для описания времени, когда наблюдается Cmax.
В соответствии с одним вариантом осуществления фармацевтический состав способен к одному или более из следующего:
- снижение Cmax по сравнению с Cmax для эквивалентного количества N-[2-гидрокси-3-(1-пиперидинил)-пропокси] -пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, вводимых с помощью пероральной лекарственной формы с немедленным высвобождением и/или с помощью болюсной IV инъекции,
- снижение Cmax с 1 или 2 дозами в сутки по сравнению с Cmax для эквивалентного количества N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, вводимых с помощью пероральных лекарственных форм с немедленным высвобождением и/или с помощью болюсных IV инъекций три раза в сутки,
- проявление относительно низкой пиковой концентрации аримокломола в крови (Cmax) в сравнении с общим обеспечиваемым воздействием, что выражается площадью под фармакокинетической кривой (AUC),
- снижение соотношения пиковой и самой низкой концентрации аримокломола в крови (или сыворотке крови),
- сохранение воздействия аримокломола при снижении пикового уровня в плазме крови (Cmax)
- сохранение воздействия аримокломола и/или AUC с меньшим количеством введений (как например, один или два раза в сутки) при снижении пикового уровня в плазме крови (Cmax)
- повышение Tmax по сравнению с Tmax для эквивалентного количества N-[2-гидрокси-3-(1-пиперидинил)-пропокси|-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, вводимых с помощью пероральной лекарственной формы с немедленным высвобождением и/или с помощью болюсной IV инъекции,
- снижение ингибирования ОСТ2 по сравнению с ингибированием ОСТ2 для эквивалентного количества N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, вводимых с помощью пероральной лекарственной формы с немедленным высвобождением и/или с помощью болюсной IV инъекции,
- снижение эффекта в отношении уровней креатинина в сыворотке крови по сравнению с эффектом в отношении уровней креатинина в сыворотке крови для эквивалентного количества N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, вводимых с помощью пероральной лекарственной формы с немедленным высвобождением и/или с помощью болюсной IV инъекции,
- снижение эффекта в отношении почечного клиренса креатинина по сравнению с эффектом в отношении почечного клиренса креатинина для эквивалентного количества N-[-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, вводимых с помощью пероральной лекарственной формы с немедленным высвобождением и/или с помощью болюсной IV инъекции,
- достижение Cmax, которая является более низкой, чем полумаксимальная ингибирующая концентрация (IC50) активного фармацевтического ингредиента для ОСТ2.
В соответствии с одним вариантом осуществления фармацевтический состав характеризуется длительным высвобождением для обеспечения возможности введения уменьшенного количества доз в сутки или пониженной частоты введения доз. В соответствии с предпочтительным вариантом осуществления состав вводят один раз или два раза в сутки по сравнению с традиционным IR составом, который вводят 3 раза в сутки.
В соответствии с одним вариантом осуществления фармацевтический состав вводят для достижения воздействия аримокломола или Cmax аримокломола, которая предотвращает i) ингибирование аримокломолом почечных транспортеров, ii) ингибирование ОСТ2 и/или iii) ингибирование клиренса креатинина.
Термин "контролирующее высвобождение вспомогательное вещество" предполагает присутствие по меньшей мере одного или одного или более контролирующих высвобождение вспомогательных веществ.
Контролирующее высвобождение вспомогательное вещество в соответствии с настоящим изобретением представляет собой вспомогательное вещество или средство, которое обеспечивает длительное высвобождение API, выбранного из N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты.
Контролирующее высвобождение вспомогательное вещество согласно настоящему изобретению в соответствии с одним вариантом осуществления контролирует скорость высвобождения API, выбранного из N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, из фармацевтического состава.
Контролирующее высвобождение вспомогательное вещество согласно настоящему изобретению в соответствии с одним вариантом осуществления контролирует скорость высвобождения API, выбранного из N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, для
- снижения Cmax,
- снижения ингибирования ОСТ2,
- снижения эффекта в отношении уровней креатинина в сыворотке крови и/или
- повышения Tmax
по сравнению с эквивалентным количеством N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, вводимых с помощью пероральной лекарственной формы с немедленным высвобождением и/или с помощью болюсной IV инъекции.
Также предполагается способ введения количества API, выбранного из N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, пациенту, нуждающемуся в этом, посредством длительного высвобождения таким образом, чтобы
- максимальная концентрация в сыворотке крови (Cmax) после введения N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты была понижена по сравнению с Cmax для их эквивалентного количества, вводимого с помощью пероральной лекарственной формы с немедленным высвобождением и/или с помощью болюсной IV инъекции,
- время, за которое концентрация в сыворотке крови достигнет своего максимума после введения N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, (Tmax) было повышено по сравнению с Tmax для их эквивалентного количества, вводимого с помощью пероральной лекарственной формы с немедленным высвобождением и/или с помощью болюсной IV инъекции,
- ингибирование ОСТ2 после введения N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты было понижено по сравнению с ингибированием ОСТ2 для их эквивалентного количества, вводимого с помощью пероральной лекарственной формы с немедленным высвобождением и/или с помощью болюсной IV инъекции,
- эффект в отношении уровней креатинина в сыворотке крови после введения N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорид, его стереоизомеров и их солей присоединения кислоты был понижен по сравнению с эффектом в отношении уровней креатинина в сыворотке крови для их эквивалентного количества, вводимого с помощью пероральной лекарственной формы с немедленным высвобождением и/или с помощью болюсной IV инъекции.
В соответствии с одним вариантом осуществления эффект N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида (аримокломола), его стереоизомеров и их солей присоединения кислоты в отношении уровней креатинина в сыворотке крови представляет собой незначительное повышение уровней креатинина в сыворотке крови.
В соответствии с одним вариантом осуществления фармацевтический состав обеспечивает более низкую Cmax API по сравнению с составом с немедленным высвобождением API.
В соответствии с одним вариантом осуществления фармацевтический состав обеспечивает более высокое Tmax у API по сравнению с составом с немедленным высвобождением API.
В соответствии с одним вариантом осуществления фармацевтический состав обеспечивает пониженное ингибирование транспортера ОСТ2 API (аримокломолом) по сравнению с составом с немедленным высвобождением API.
В соответствии с одним вариантом осуществления фармацевтический состав согласно настоящему изобретению снижает индуцированное аримокломолом ингибирование ОСТ2 или избегает его; причем пониженное ингибирование ОСТ2 в соответствии с одним вариантом осуществления снижает риск неблагоприятного воздействия на клиренс, выведение и/или период полувыведения из кровотока дополнительных лекарственных препаратов, в особенности, лекарственных препаратов, которые являются субстратами для транспортера ОСТ2.
В соответствии с одним вариантом осуществления фармацевтический состав обеспечивает пониженный эффект API в отношении креатинина в сыворотке крови по сравнению с составом с немедленным высвобождением API.
В соответствии с одним вариантом осуществления фармацевтический состав обеспечивает Cmax менее 15 мкМ, например, менее 10 мкМ, как например, менее 9 мкМ, например, менее 8 мкМ, как например, менее 7 мкМ, например, менее 6 мкМ, как например, менее 5 мкМ, например, менее 4 мкМ, как например, менее 3 мкМ, например, менее 2 мкМ, как например, менее 1 мкМ.
В соответствии с одним вариантом осуществления фармацевтический состав обеспечивает Cmax 1-2 мкМ, например, 2-3 мкМ, как например, 3-4 мкМ, например, 4-5 мкМ, как например, 5-6 мкМ, например, 6-7 мкМ, как например, 7-8 мкМ, например, 8-9 мкМ, как например, 9-10 мкМ, например, 10-11 мкМ, как например, 11-12 мкМ, например, 12-13 мкМ, как например, 13-14 мкМ, например, 14-15 мкМ.
В соответствии с предпочтительным вариантом осуществления фармацевтический состав обеспечивает Cmax, меньшую или равную 10 мкМ.
В соответствии с предпочтительным вариантом осуществления фармацевтический состав обеспечивает Cmax, меньшую или равную 10 мкМ, достигаемую при менее частом введении доз в течение суток, как например, достигаемую при введении дозы один раз в сутки или два раза в сутки.
В соответствии с одним вариантом осуществления Cmax снижается на величину по меньшей мере 10%, как например, на величину по меньшей мере 20%, как например, на величину по меньшей мере 30%, как например, на величину по меньшей мере 40%, как например, на величину по меньшей мере 50%, как например, на величину по меньшей мере 60%, как например, на величину по меньшей мере 70%, как например, на величину по меньшей мере 80%, как например, на величину по меньшей мере 90%, как например, на величину по меньшей мере 100%.
В соответствии с одним вариантом осуществления Cmax снижается на величину 10-20%, как например, на величину 20-30%, как например, на величину 30-40%, как например, на величину 40-50%, как например, на величину 50-60%, как например, на величину 60-70%, как например, на величину 70-80%, как например, на величину 80-90%, как например, на величину 90-100%.
В соответствии с одним вариантом осуществления Tmax повышается на величину по меньшей мере 10%, как например, на величину по меньшей мере 20%, как например, на величину по меньшей мере 30%, как например, на величину по меньшей мере 40%, как например, на величину по меньшей мере 50%, как например, на величину по меньшей мере 60%, как например, на величину по меньшей мере 70%, как например, на величину по меньшей мере 80%, как например, на величину по меньшей мере 90%, как например, на величину по меньшей мере 100%, как например, на величину по меньшей мере 125%, как например, на величину по меньшей мере 150%, как например, на величину по меньшей мере 175%, как например, на величину по меньшей мере 200%, как например, на величину по меньшей мере 250%.
В соответствии с одним вариантом осуществления Tmax повышается на величину 10-20%. как например, на величину 20-30%, как например, на величину 30-40%, как например, на величину 40-50%, как например, на величину 50-60%, как например, на величину 60-70%, как например, на величину 70-80%, как например, на величину 80-90%, как например, на величину 90-100%, как например, на величину 100-125%, как например, на величину 125-150%, как например, на величину 150-175%, как например, на величину 175-200%, как например, на величину 200-225%, как например, на величину 225-250%.
В соответствии с одним вариантом осуществления ингибирование ОСТ2 снижается на величину по меньшей мере 10%, как например, на величину по меньшей мере 20%, как например, на величину по меньшей мере 30%, как например, на величину по меньшей мере 40%, как например, на величину по меньшей мере 50%, как например, на величину по меньшей мере 60%, как например, на величину по меньшей мере 70%, как например, на величину по меньшей мере 80%, как например, на величину по меньшей мере 90%, как например, на величину по меньшей мере 100%.
В соответствии с одним вариантом осуществления ингибирование ОСТ2 снижается на величину 10-20%, как например, на величину 20-30%, как например, на величину 30-40%, как например, на величину 40-50%, как например, на величину 50-60%, как например, на величину 60-70%, как например, на величину 70-80%, как например, на величину 80-90%, как например, на величину 90-100%.
Фармацевтический состав в соответствии с одним вариантом осуществления характеризуется скоростью растворения, при которой 85% API высвобождается в течение 3-5 часов (средняя), и в соответствии с другим вариантом осуществления скоростью, при которой 85% API высвобождается спустя по меньшей мере (≥) 6 часов, как например, спустя по меньшей мере 7, 8, 9 или 10 часов (медленная).
Скоростью растворения описывается, как быстро соединение высвобождается из состава в раствор. Скорость растворения можно выразить с помощью уравнения Нойе-Уитни или уравнения Нернста и Брюннера.
В соответствии с одним вариантом осуществления фармацевтическим составом обеспечивается скорость растворения, при которой от 10 до 90% API высвобождается через 3-5 часов, как например, от 10 до 20%, от 20 до 30, от 30 до 40, от 40 до 50, от 50 до 60, от 60 до 70, от 70 до 75, от 75 до 80, от 80 до 85 или от 85 до 90% API высвобождается в течение 3-5 часов, как например, в течение 3 часов, в течение 4 часов или в течение 5 часов.
В соответствии с одним вариантом осуществления фармацевтическим составом обеспечивается скорость растворения, при которой от 10 до 90% API высвобождается в течение 6 часов, как например, от 10 до 20%, от 20 до 30, от 30 до 40, от 40 до 50, от 50 до 60, от 60 до 70, от 70 до 75, от 75 до 80, от 80 до 85 или от 85 до 90% API высвобождается в течение ≥6 часов, как например, в течение ≥7 часов, ≥8 часов, ≥9 часов, ≥10 часов, ≥11 часов, ≥12 часов, ≥13 часов, ≥14 часов, ≥15 часов, ≥16 часов, ≥17 часов, ≥18 часов.
Таблетки и сферы
Настоящим изобретением в одном аспекте предполагается фармацевтический состав, содержащий активный фармацевтический ингредиент, выбранный из N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида (аримокломола), его стереоизомеров и их солей присоединения кислоты, причем указанный состав содержит внутреннюю матрицу и по меньшей мере одно наружное покрытие, причем указанный состав обеспечивает длительное высвобождение указанного активного фармацевтического ингредиента.
В соответствии с одним вариантом осуществления указанный состав выбран из группы, состоящей из таблетки, мини-таблетки, микро-таблетки, таблетки с покрытием, мини-таблетки с покрытием, микро-таблетки с покрытием, сферы и сферы с покрытием.
В соответствии с одним вариантом осуществления фармацевтический состав согласно настоящему изобретению применяют в виде единичной пероральной лекарственной формы (также известной как неразделяемый состав). В соответствии с другим вариантом осуществления фармацевтический состав согласно настоящему изобретению применяют в виде пероральной лекарственной формы с множеством единиц (также известной как разделяемый состав). Пероральная лекарственная форма с множеством единиц представляет собой отдельные продукты на основе лекарственного средства, упакованные вместе, в данном контексте, например, мини-таблетки в капсуле.
В соответствии с одним вариантом осуществления внутренняя матрица состава содержит активный фармацевтический ингредиент. В соответствии с этими вариантами осуществления состав может быть выбран из таблетки с покрытием, мини-таблетки с покрытием и микро-таблетки с покрытием.
В соответствии с одним вариантом осуществления состав выбран из таблетки с покрытием, мини-таблетки с покрытием и микро-таблетки с покрытием, причем внутренняя матрица (или таблетка) содержит API, и при этом наружное покрытие не содержит API.
В соответствии с одним вариантом осуществления наружное покрытие состава содержит активный фармацевтический ингредиент. В соответствии с этими вариантами осуществления состав может представлять собой сферу с покрытием (сферу, нагруженную лекарственным средством).
В соответствии с одним вариантом осуществления указанный состав представляет собой сферу с покрытием, причем внутренняя матрица (основа сферы) не содержит API, а наружное покрытие содержит API.
В соответствии с одним вариантом осуществления наружное покрытие у сферы с покрытием содержит один или более отдельных слоев, причем самый внутренний слой, непосредственно окружающий сферу, содержит API.
В соответствии с одним вариантом осуществления фармацевтический состав согласно настоящему изобретению содержится в капсуле, как например, с обеспечением пероральной лекарственной формы с множеством единиц, такой как капсула, содержащая две или более единиц состава или таблеток/мини-таблеток/сфер согласно настоящему изобретению. В соответствии с одним вариантом осуществления капсула содержит или состоит из желатина. В соответствии с одним вариантом осуществления капсула представляет собой капсулу с твердой оболочкой, такую как твердая желатиновая капсула. В соответствии с дополнительным вариантом осуществления капсула дополнительно содержит наружное покрытие.
В соответствии с одним вариантом осуществления пероральная лекарственная форма с множеством единиц согласно настоящему изобретению представляет собой капсулу, содержащую две или более единиц состава в соответствии с настоящим изобретением, как например, содержащую 2-3, 3-4, 4-5, 5-6, 6-7, 7-8, 8-9, 9-10, 10-11, 11-12, 12-13, 13-14, 14-15, 15-16, 16-17, 17-18, 18-19, 19-20, 20-21, 21-22, 22-23, 23-24, 24-25, 25-26, 26-27, 27-28, 28-29, 29-30, 30-35, 35-40, 40-45, 45-50, 50-55, 55-60, 60-65, 65-70, 70-75, 75-80, 80-85, 85-90, 90-95, 95-100 единиц состава.
В соответствии с одним вариантом осуществления единица состава выбрана из группы, состоящей из мини-таблетки с покрытием, микро-таблетки с покрытием и сферы с покрытием.
Количество единиц состава в капсуле зависит от количества или концентрации API в каждой единице и отдельных характеристик пациента, которому следует вводить фармацевтический состав, что будет понятно специалисту в данной области техники.
Пероральная лекарственная форма - таблетка
В соответствии с одним вариантом осуществления фармацевтический состав согласно настоящему изобретению содержит составляющую-матрицу, такую как внутренняя матрица, содержащая API, и необязательно наружное покрытие. В соответствии с одним вариантом осуществления внутренняя матрица представляет собой таблетку, мини-таблетку или микро-таблетку, причем данная таблетка необязательно имеет покрытие.
В соответствии с одним вариантом осуществления фармацевтический состав согласно настоящему изобретению содержит по меньшей мере один тип гидроксипропилметилцеллюлозы (НРМС), также известный как гипромеллоза. НРМС применяют в качестве вспомогательного вещества в составах пероральной таблетки и капсулы, где в зависимости от марки она функционирует в качестве средства, обеспечивающего контролируемое высвобождение, или вспомогательного вещества, контролирующего высвобождение, для замедления высвобождения лекарственного соединения в пищеварительном тракте. Она также применяется в качестве связующего и в качестве компонента покрытий таблетки.
В соответствии с одним вариантом осуществления внутренняя матрица у данного состава содержит API и одно или более контролирующих высвобождение вспомогательных веществ. В соответствии с одним вариантом осуществления внутренняя матрица дополнительно содержит одно или более дополнительных вспомогательных веществ, таких как наполнители, связующие и смазывающие средства.
Как используется в данном документе, контролирующие высвобождение вспомогательные вещества могут представлять собой любое контролирующее высвобождение вспомогательное вещество, известное специалисту в данной области техники. Контролирующие высвобождение вспомогательные вещества в соответствии с одним вариантом осуществления представляют собой вспомогательное вещество, выбранное из группы, состоящей из гидроксипропилметилцеллюлозы (НРМС), этилцеллюлозы (ЕС), метилцеллюлозы, гидроксипропилцеллюлозы, гипромеллозы ацетата сукцинат, фталата гипромеллозы, ацетата целлюлозы, глицеринмоностеарата, глицерилмоноолеата, глицерилпальмитата, глицерилбегената, гидрирогенизированного растительного масла, гуаровой камеди, поливинилового спирта, альгинатов, ксантановой камеди, карнаубского воска, желтого воска, белого воска, зеина, каррагенана, карбомеров и агара.
В соответствии с одним вариантом осуществления внутренняя матрица дополнительно содержит наполнитель, такой как наполнитель, выбранный из группы, состоящей из карбоната кальция, фосфатов кальция, сульфата кальция, целлюлозы, ацетата целлюлозы, сжимаемого сахара, декстрана, декстрина, декстрозы, этилцеллюлозы, фруктозы, изомальта, лактита, лактозы, маннита, карбоната магния, оксида магния, мальтодекстрина, микрокристаллической целлюлозы (МСС), полидекстрозы, альгината натрия, сорбита, талька и ксилита.
В соответствии с одним вариантом осуществления внутренняя матрица дополнительно содержит связующее, такое как связующее, выбранное из группы, состоящей из аравийской камеди, альгиновой кислоты, карбомеров, карбоксиметилцеллюлозы натрия, каррагенана, ацетилфталата целлюлозы, хитозана, коповидона, декстрата, декстрина, декстрозы, этилцеллюлозы, желатина, гуаровой камеди, гидроксиэтилцеллюлозы, гидроксиэтилметилцеллюлозы, гидроксипропилцеллюлозы, гидроксипропилкрахмала, гипромеллозы, метилцеллюлозы, полоксамера, полидекстрозы, полиэтиленоксида, повидона, альгината натрия, сахарозы, крахмала, прежелатинизированного крахмала и мальтодекстрина.
В соответствии с одним вариантом осуществления внутренняя матрица дополнительно содержит смазывающее средство, такое как смазывающее средство, выбранное из группы, состоящей из стеарата кальция, глицеринмоностеарата, глицерилбегената, глицерилпальмитостеарата, гидрогенизированного касторового масла, гидрогенизированного растительного масла, лаурилсульфата магния, стеарата магния, триглицерида со средним размером цепи, пальмитиновой кислоты, полиэтиленгликоля, лаурилсульфата натрия, стеариновой кислоты, талька, диоксида кремния и стеарата цинка.
Любые другие вспомогательные вещества, подходящие для целей настоящего изобретения и известные специалисту в данной области техники, считаются охваченными настоящим изобретением.
Различные марки НРМС имеют разные характеристики в отношении, например, вязкости. Таким образом, разные НРМС будут оказывать разные воздействия на скорости высвобождения погруженного API. Также, количество НРМС в составе, твердость или степень прессования состава в таблетку, а также любые потенциально возможные покрытия будут потенциально оказывать воздействие на скорости высвобождения API. Скорости высвобождения можно определить посредством оценки профилей растворения у полученных партий. Данные растворения лекарственного средства in vitro, полученные из экспериментов по исследованию растворения, можно сопоставить с фармакокинетическими данными in vivo с использованием in vitro-in vivo корреляций (IVIVC).
В соответствии с одним вариантом осуществления внутренняя матрица содержит одно или более вспомогательных веществ, выбранных из группы, состоящей из гидроксипропилметилцеллюлозы (НРМС), крахмала, этилцеллюлозы (ЕС), микрокристаллической целлюлозы (МСС), диоксида кремния, стеарата магния и стеариновой кислоты. В соответствии с одним вариантом осуществления внутренняя матрица содержит по меньшей мере одну НРМС.
В химическом плане НРМС представляет собой смешанный простой алкил-гидроксиалкиловый эфир целлюлозы, содержащий метоксильные и гидроксипропильные группы. НРМС производится компанией Dow Chemical Company под торговым названием Methocel. В Methocel, используемой для применений в ER матрицах, используются два типа химических замещающих групп, обозначенных или 'Е', или 'K'. Полимеры Methocel также различаются по маркам на основании их вязкости (в сП) в водном растворе в концентрации 2% масса/объем при 20°С. Типичные марки НРМС, используемые для составов с ER, характеризуются вязкостью в диапазоне от 50 до 100000 сП при 20°С и включают Methocel Е50 Premium LV, K100 Premium LV CR, K4M Premium CR, K15M Premium CR, K100M Premium CR, E4M Premium CR и E10M Premium CR.
В соответствии с одним вариантом осуществления НРМС представляет собой НРМС марки, обеспечивающей вязкость от 50 до 100000 сП при 20°С. В соответствии с одним вариантом осуществления НРМС представляет собой НРМС марки с высокой вязкостью или НРМС марки со сверхвысокой вязкостью. В соответствии с одним вариантом осуществления НРМС представляет собой НРМС, позволяющую (или обеспечивающую) длительное высвобождение.
В соответствии с одним вариантом осуществления НРМС выбрана из группы, состоящей из Methocel Е50 Premium LV, K100 Premium LV CR, K4M Premium CR, K15M Premium CR, K100M Premium CR, E4M Premium CR, E10M Premium CR, K200M, E5 и E50.
Контролирующее высвобождение вспомогательное вещество, такое как НРМС, во внутренней матрице в соответствии с одним вариантом осуществления присутствует в количестве 20-50% масс., как например, 20-25% масс., например, 25-30% масс., как например, 30-35% масс., например, 35-40% масс., как например, 40-45% масс., например, 45-50% масс. В соответствии с конкретным вариантом осуществления контролирующее высвобождение вспомогательное вещество, такое как НРМС, присутствует в количестве приблизительно 30% масс., например, 35% масс., как например, приблизительно 40% масс.
В соответствии с одним вариантом осуществления матрица содержит два или более контролирующих высвобождение вспомогательных веществ, как например, три или более контролирующих высвобождение вспомогательных веществ.
В соответствии с одним вариантом осуществления внутренняя матрица содержит один или более разных типов (марок по вязкости) НРМС, как например, 1, 2, 3, 4 или 5 типов НРМС. В соответствии с одним вариантом осуществления внутренняя матрица содержит комбинацию полимеров НРМС.
В соответствии с одним вариантом осуществления внутренняя матрица содержит комбинацию НРМС с ионными, неионными и/или нерастворимыми в воде полимерами.
В соответствии с одним вариантом осуществления внутренняя матрица содержит комбинацию НРМС с одним или более ионными полимерами, выбранными из группы, состоящей из карбоксиметилцеллюлозы натрия (na CMC), альгината натрия, полимеров акриловой кислоты или карбомеров (карбопол 934, 940, 974Р NF), растворимых в кишечнике полимеров, таких как поливинилацетатфталат (PVAP), сополимеров метакриловой кислоты (Eudragit L100 L 30D 55, S и FS 30 D), гипромеллозы ацетата сукцинат (ацетилсукцината гипромеллозы) (AQOAT HPMCAS) и ксантановой камеди.
В соответствии с одним вариантом осуществления внутренняя матрица содержит комбинацию НРМС с одним или более неионными полимерами, выбранными из группы, состоящей из НРС (гидроксипропилцеллюлоза) и РЕО (POLYOX, Dow Chemical Company), имеющим марки, соответствующие различным молекулярным массам (от 100000 до 7000000 Да).
В соответствии с одним вариантом осуществления внутренняя матрица содержит комбинацию НРМС с одним или более нерастворимыми в воде полимерами, выбранными из группы, состоящей из этилцеллюлозы (например, ETHOCEL или Surrelease), ацетата целлюлозы, сополимеров метакриловой кислоты (например, Eudragit NE 30D), аммонио-метакрилатных сополимеров (например, Eudragit RL 100 или РО RS100) и поливинилацетата.
В соответствии с одним конкретным вариантом осуществления НРМС смешивают с микрокристаллической целлюлозой (МСС) с получением МСС/НРМС матрицы. Второе вспомогательное вещество, такое как МСС, в соответствии с одним вариантом осуществления присутствует в количестве 10-50% масс., как например, 10-15% масс., например, 15-20% масс., как например, 20-25% масс., например, 25-30% масс., как например, 30-35% масс., например, 35-40% масс., как например, 40-45% масс., например, 45-50% масс. МСС. В соответствии с конкретным вариантом осуществления МСС представляет собой Avicel РН 101 или Avicel РН 102.
Микрокристаллическая целлюлоза является коммерчески доступной под марками с разными размерами частиц и содержанием влаги, которые имеют различные свойства и применения. Avicel РН 101 имеет номинальный средний размер частиц 50 микрон, в то время как Avicel РН 102 имеет номинальный средний размер частиц 100 микрон. Обе они имеют содержание влаги <=5%
В соответствии с одним вариантом осуществления внутренняя матрица дополнительно содержит крахмал, как например, содержит крахмал в количестве 5-30% масс., как например, 5-10% масс., например, 10-15% масс., как например, 15-20% масс., например, 20-25% масс., как например, 25-30% масс. крахмала. В соответствии с конкретным вариантом осуществления крахмал присутствует в количестве приблизительно 5% масс., например, 10% масс., как например, приблизительно 15% масс., например, 20% масс. В соответствии с конкретным вариантом осуществления крахмал представляет собой StarCap 1500.
В соответствии с одним вариантом осуществления внутренняя матрица дополнительно содержит этилцеллюлозу (ЕС), как например, содержит ЕС в количестве 5-30% масс., как например, 5-10% масс., например, 10-15% масс., как например, 15-20% масс., например, 20-25% масс., как например, 25-30% масс. ЕС. В соответствии с конкретным вариантом осуществления ЕС присутствует в количестве приблизительно 5% масс., например, 10% масс., как например, приблизительно 15% масс., например, 20% масс. ЕС в соответствии с конкретным вариантом осуществления представляет собой Ethocel Standard 7 Premium.
В соответствии с одним вариантом осуществления внутренняя матрица дополнительно содержит диоксид кремния, такой как коллоидный диоксид кремния, причем диоксид кремния в соответствии с одним вариантом осуществления присутствует в количестве 0,05-1% масс., как например, 0,05-0,1, например, 0,1-0,2, как например, 0,2-0,3, например, 0,3-0,4, как например, 0,4-0,5, например, 0,5-0,6, как например, 0,6-0,7, например, 0,7-0,8, как например, 0,8-0,9, например, 0,9-1,0% масс. В соответствии с конкретным вариантом осуществления диоксид кремния присутствует в количестве приблизительно 0,2% масс.
В соответствии с одним вариантом осуществления внутренняя матрица дополнительно содержит стеарат магния, причем стеарат магния в соответствии с одним вариантом осуществления присутствует в количестве 0,1-5% масс., как например, 0,1-0,5, например, 0,5-1,0, как например, 1-2, например, 2-3, как например, 3-4, например, 4-5% масс. В соответствии с конкретным вариантом осуществления стеарат магния присутствует в количестве приблизительно 1% масс. В соответствии с конкретным вариантом осуществления стеарат магния представляет собой Ligamed MF-2-V.
В соответствии с одним вариантом осуществления внутренняя матрица дополнительно содержит стеариновую кислоту, причем стеариновая кислота в соответствии с одним вариантом осуществления присутствует в количестве 0,1-10% масс., как например, 0,1-0,5, например, 0,5-1,0, как например, 1-2, например, 2-3, как например, 3-4, например, 4-5, как например, 5-6, например, 6-7, как например, 7-8, например, 8-9, как например, 9-10% масс. В соответствии с конкретным вариантом осуществления стеариновая кислота присутствует в количестве приблизительно 2% масс.
В соответствии с одним вариантом осуществления внутреннюю матрицу подвергают прессованию с образованием таблетки с твердостью от 10 до 50 Кп (килопонд), как например, от 10 до 15 Кп, например, от 15 до 20 Кп, как например, от 20 до 25 Кп, например, от 25 до 30 Кп, как например, от 30 до 35 Кп, например, от 35 до 40 Кп, как например, от 40 до 50 Кп.
В соответствии с одним вариантом осуществления внутреннюю матрицу подвергают прессованию с образованием таблетки с твердостью от 15 до 80 Н (ньютон), как например, от 15 до 20 Н, например, от 20 до 25 Н, как например, от 25 до 30 Н, например, от 30 до 35 Н, как например, от 35 до 40 Н, например, от 40 до 45 Н, как например, от 45 до 50 Н, например, от 50 до 55, как например, от 55 до 60 Н, например, от 60 до 70 Н, как например, от 70 до 80 Н.
Таблетка с покрытием
В соответствии с одним вариантом осуществления фармацевтический состав согласно настоящему изобретению содержит составляющую-матрицу, такую как внутренняя матрица, содержащая API, и наружное покрытие. В соответствии с одним вариантом осуществления наружное покрытие не содержит активный фармацевтический ингредиент.
В соответствии с одним вариантом осуществления фармацевтический состав представляет собой таблетку с покрытием, мини-таблетку с покрытием или микротаблетку с покрытием.
Наружное покрытие предпочтительно способствует длительному высвобождению API, содержащегося во внутренней матрице. В соответствии с одним вариантом осуществления наружное покрытие представляет собой замедлитель высвобождения.
При упоминании "наружного покрытия" данный термин можно применить к одному или более отдельным слоям наружного покрытия. В соответствии с одним вариантом осуществления наружное покрытие содержит один или более отдельных слоев покрытия.
В соответствии с одним вариантом осуществления наружное покрытие содержит одно или более вспомогательных веществ. В соответствии с одним вариантом осуществления наружное покрытие содержит дисперсию этилцеллюлозы (ЕС) на водной основе, такую как Surrelease™. В соответствии с одним вариантом осуществления наружное покрытие содержит ЕС на основе растворителя. В соответствии с одним вариантом осуществления наружное покрытие содержит водную дисперсию на основе полиметакрилата, такую как Eudragit NE30D™. В соответствии с одним вариантом осуществления наружное покрытие содержит пленкообразующее вспомогательное вещество.
В соответствии с одним вариантом осуществления состав наносят в виде покрытия до тех пор, пока не достигается определенное увеличение массы (масса/масса). В соответствии с одним вариантом осуществления на состав наносят в виде покрытия до увеличения массы на 5% масс., как например, увеличения массы на 10% масс., например, увеличения массы на 15% масс., как например, увеличения массы на 20% масс., например, увеличения массы на 25% масс., как например, увеличения массы на 30% масс., например, увеличения массы на 35% масс., как например, увеличения массы на 40% масс. В соответствии с одним вариантом осуществления состав наносят в виде покрытия до увеличения массы на 5-40% масс., как например, на 10-15% масс., например, на 15-20% масс., как например, на 20-25% масс., например, на 25-30% масс., как например, на 30-35% масс., например, на 35-40% масс.
В соответствии с одним вариантом осуществления предполагается фармацевтический состав, содержащий
а. внутреннюю матрицу, содержащую активный фармацевтический ингредиент (API) выбранный из N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорид, его стереоизомеры и их соли присоединения кислоты,
причем указанная матрица содержит по меньшей мере одно контролирующее высвобождение вспомогательное вещество и необязательно одно или более дополнительных вспомогательных веществ, а также
b. необязательно наружное покрытие,
причем указанный состав обеспечивает длительное высвобождение указанного активного фармацевтического ингредиента.
В соответствии с одним вариантом осуществления внутренняя матрица содержит 5-40% масс. N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, как например, 5-10, например, 10-15, как например, 15-20, например, 20-25, как например, 25-30, например, 30-35, как например, 35-40% масс.
В соответствии с одним вариантом осуществления наружное покрытие дополнительно содержит наружное изолирующее покрытие. Наружное изолирующее покрытие наносят в качестве самого наружного слоя.
Пероральная лекарственная форма - сфера с покрытием
Фармацевтический состав согласно настоящему изобретению в соответствии с одним вариантом осуществления содержит составляющую-матрицу, такую как внутренняя матрица или основа сферы, и наружное покрытие, содержащее один или более отдельных слоев.
В соответствии с одним вариантом осуществления данный состав представляет собой сферу с покрытием, причем указанная сфера с покрытием содержит основу сферы и наружное покрытие, содержащее один или более отдельных слоев.
Наружное покрытие сферы с покрытием в соответствии с одним вариантом осуществления содержит один или более отдельных слоев, как например, два или более слоев, как например, три или более слоев, как например, четыре или более слоев, как например, пять или более слоев. В соответствии с одним вариантом осуществления наружное покрытие содержит 1-2, как например, 2-3, например, 3-4, как например, 4-5, например, 5-6 слоев.
В соответствии с одним вариантом осуществления внутренняя матрица или основа сферы не содержит активный фармацевтический ингредиент. В соответствии с одним вариантом осуществления наружное покрытие содержит активный фармацевтический ингредиент. В соответствии с одним вариантом осуществления самый внутренний слой наружного покрытия, содержащий два или более слоев, содержит активный фармацевтический ингредиент.
В соответствии с одним вариантом осуществления API осаждают или наносят в виде покрытия на поверхности указанной внутренней матрицы или основы сферы для обеспечения слоя лекарственного средства, и указанную сферу, покрытую API, дополнительно наносят один или более дополнительных слоев.
В соответствии с одним вариантом осуществления состав согласно настоящему изобретению содержит (изнутри-наружу): 1) основу сферы (или ядро из множества частиц), 2) слой лекарственного средства, содержащий активный фармацевтический ингредиент, 3) необязательно изолирующее покрытие, 4) покрытие для контролируемого высвобождения и 5) необязательно пленочное покрытие. Он проиллюстрирован на фиг. 13.
В соответствии с одним вариантом осуществления сфера с покрытием содержит: 1) основу сферы, 2) слой лекарственного средства до увеличения массы на 1-10% масс., 3) необязательно изолирующее покрытие до увеличения массы на 0,1-5% масс., 4) покрытие для контролируемого высвобождения до увеличения массы на 5-20% масс. и 5) необязательно пленочное покрытие до увеличения массы на 1-10% масс.
В соответствии с одним вариантом осуществления слой лекарственного средства наносят до увеличения массы на 1-10% масс., как например, на 1-2, например, на 2-3, как например, на 3-4, например, на 4-5, как например, на 5-6, например, на 6-7, как например, на 7-8, например, на 8-9, как например, до увеличения массы на 9-10% масс. В соответствии с одним вариантом осуществления слой лекарственного средства наносят до увеличения массы приблизительно на 1% масс., как например, на 2, например, на 3, как например, на 4, например, на 5, как например, на 6, например, на 7, как например, на 8, например, на 9, как например, до увеличения массы приблизительно на 10% масс.
В соответствии с одним вариантом осуществления изолирующее покрытие наносят до увеличения массы на 0,1-5% масс., как например, на 0,1-0,5, например, на 0,5-1, как например, на 1-2, например, на 2-3, как например, на 3-4, например, до увеличения массы на 4-5% масс. В соответствии с одним вариантом осуществления изолирующее покрытие наносят до увеличения массы приблизительно на 0,1% масс., как например, на 0,5, например, на 1, как например, на 2, например, на 3, как например, на 4, например, до увеличения массы на 5% масс.
В соответствии с одним вариантом осуществления покрытие для контролируемого высвобождения наносят до увеличения массы на 5-20% масс., как например, на 5-6, например, на 6-7, как например, на 7-8, например, на 8-9, как например, на 9-10, например, на 10-11, как например, на 11-12, например, на 12-13, как например, на 13-14, например, на 14-15, как например, на 15-16, например, на 16-17, как например, на 17-18, например, на 18-19, как например, до увеличения массы на 19-20% масс. В соответствии с одним вариантом осуществления покрытие для контролируемого высвобождения наносят до увеличения массы приблизительно на 5% масс., как например, на 6, например, на 7, как например, на 8, например, на 9, как например, на 10, например, на 11, как например, на 12, например, на 13, как например, приблизительно на 14, например, на 15, как например, на 16, например, на 17, как например, на 18, например, на 19, как например, до увеличения массы приблизительно на 20% масс.
В соответствии с одним вариантом осуществления пленочное покрытие наносят до увеличения массы на 1-10% масс., как например, на 1-2, например, на 2-3, как например, на 3-4, например, на 4-5, как например, на 5-6, например, на 6-7, как например, на 7-8, например, на 8-9, как например, до увеличения массы на 9-10% масс. В соответствии с одним вариантом осуществления пленочное покрытие наносят до увеличения массы приблизительно на 1% масс., как например, на 2, например, на 3, как например, на 4, например, на 5, как например, на 6, например, на 7, как например, на 8, например, на 9, как например, до увеличения массы приблизительно на 10% масс.
В соответствии с одним вариантом осуществления сфера с покрытием содержит: 1) основу сферы, 2) слой лекарственного средства до увеличения массы на 4% масс, 3) изолирующее покрытие до увеличения массы на 1% масс, 4) покрытие для контролируемого высвобождения до увеличения массы на 5-20% масс, и 5) пленочное покрытие до увеличения массы на 3-5% масс.
В соответствии с одним вариантом осуществления основа сферы содержит или состоит из сахара, как например, растворимая сахарная сфера, например, Suglets ТМ.
В соответствии с одним вариантом осуществления основа сферы содержит или состоит из сферы из МСС, как например, сфера из нерастворимой микрокристаллической целлюлозы, например, Vivapur ТМ.
В соответствии с одним вариантом осуществления сахарные сферы имеют размер 1000/1180 мкм.
В соответствии с одним вариантом осуществления сферы из МСС имеют размер 710-1000 мкм.
В соответствии с одним вариантом осуществления слой лекарственного средства содержит API и вспомогательное вещество, такое как НРМС. НРМС может представлять собой НРМС любой марки, которая является соответствующей, как например, марок, подробно описанных в других местах в данном документе. В соответствии с одним вариантом осуществления НРМС в слое лекарственного средства представляет собой Methocel Е6.
В соответствии с одним вариантом осуществления изолирующее покрытие и/или пленочное покрытие представляет собой пленочное покрытие на основе PVA, такое как Opadry 200 белый.
В соответствии с одним вариантом осуществления покрытие для контролируемого высвобождения содержит или состоит из этилцеллюлозы (ЕС) на водной или неводной основе, такой как Surrelease Е-7-19040™. В соответствии с другим вариантом осуществления покрытие для контролируемого высвобождения содержит или состоит из водной дисперсии на основе полиакрилата, такой как Eudragit E30D™.
В соответствии с одним вариантом осуществления покрытие для контролируемого высвобождения наносят до увеличения массы на 5-30% масс., как например, на 5-10, например, на 10-15, как например, на 15-20, например, на 20-25, как например, до увеличения массы на 25-30% масс. В соответствии с одним вариантом осуществления покрытие для контролируемого высвобождения наносят до увеличения массы приблизительно на 5% масс., как например, до увеличения массы приблизительно на 10% масс., например, до увеличения массы приблизительно на 15% масс., как например, до увеличения массы приблизительно на 20% масс., например, до увеличения массы приблизительно на 25% масс., как например, до увеличения массы приблизительно на 30% масс.
Гранулы с длительным высвобождением (гранулы, полученные с помощью экструзии горячего расплава)
В соответствии с одним вариантом осуществления предполагается фармацевтический состав, содержащий
- активный фармацевтический ингредиент (API), выбранный из N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, и
- контролирующее высвобождение вспомогательное вещество,
причем указанный состав обеспечивает длительное высвобождение указанного активного фармацевтического ингредиента,
при этом указанный состав присутствует в форме гранул с длительным высвобождением.
В соответствии с одним вариантом осуществления фармацевтический состав согласно настоящему изобретению представляет собой гранулы с длительным высвобождением, содержащие N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорид, его стереоизомеры и их соли присоединения кислоты, а также контролирующее высвобождение вспомогательное вещество.
В соответствии с одним вариантом осуществления указанные гранулы с длительным высвобождением получают с помощью экструзии горячего расплава (НМЕ).
В соответствии с одним вариантом осуществления предполагается фармацевтический состав, содержащий гранулы с длительным высвобождением, содержащие
- активный фармацевтический ингредиент (API), выбранный из N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, и
- контролирующее высвобождение вспомогательное вещество,
причем указанный состав можно получить с помощью экструзии горячего расплава.
В соответствии с одним вариантом осуществления контролирующее высвобождение вспомогательное вещество представляет собой вспомогательное вещество для НМЕ (или полимер для НМЕ).
В соответствии с одним вариантом осуществления гранулы с длительным высвобождением можно производить или получать с помощью экструзии горячего расплава, предусматривающей стадии
а. смешивания API, выбранного из N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, и вспомогательного вещества для НМЕ;
b. нагревания и экструзии указанных API и вспомогательного вещества для НМЕ с обеспечением экструдата, содержащего указанный API и вспомогательное вещество для НМЕ;
c. воздействия измельчением на указанный экструдат, как например, с помощью размола, и необязательно фракционирования по размеру, как например, с помощью просеивания через сито.
В соответствии с одним вариантом осуществления предполагается способ получения фармацевтического состава, содержащего гранулы с длительным высвобождением N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, причем указанный способ предусматривает стадии
i) обеспечения API, выбранного из N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, и вспомогательного вещества для НМЕ, причем указанное вспомогательное вещество для НМЕ характеризуется температурой плавления 65-75°С, как например, приблизительно 70°С,
ii) смешивания указанного API и вспомогательного вещества для НМЕ,
iii) воздействия температурой плавления 65-75°С, как например, 65-70°С, на API и вспомогательное вещество для НМЕ,
iv) экструзии указанных API и вспомогательного вещества для НМЕ при давлении расплава 0-10 бар, как например, 0-8 бар, с получением экструдата, содержащего указанный API и вспомогательное вещество для НМЕ,
v) предпочтительно измельчения, как например, посредством размола, указанного экструдата, содержащего API и вспомогательное вещество для НМЕ, и
vi) необязательно фракционирования по размеру, как например, с помощью просеивания указанного измельченного экструдата через сито.
Технология экструзии горячего расплава (НМЕ) становится все более значимой в области фармацевтической промышленности. Особый интерес представляет применение НМЕ для диспергирования активных фармацевтических ингредиентов в матрице на молекулярном уровне с образованием при этом твердых растворов. Саму технологию можно описать как процесс, при котором материал плавится или размягчается при повышенной температуре и давлении и продавливается через отверстие с помощью шнеков. Соответствующие термопластические свойства являются предпосылкой к применению любого полимера в экструзии горячего расплава. Число таких полимеров, одобренных для фармацевтического применения, в настоящее время ограничено.
Полимеры для НМЕ должны проявлять термопластические характеристики для обеспечения возможности процесса НМЕ, и они должны быть термически стабильными при температурах экструзии. Полимерные компоненты, используемые в процессе экструзии, могут функционировать в качестве вспомогательных веществ, контролирующих высвобождение лекарственного средства. В системах доставки экструдированного лекарственного средства полимер служит в качестве матрицы. Полимеры с высокой способностью к солюбилизации являются особенно подходящими, поскольку они могут растворять большие количества лекарственных средств.
В соответствии с одним вариантом осуществления вспомогательное вещество для НМЕ согласно настоящему изобретению выбрано из группы, состоящей из расплавленного липидного вспомогательного вещества, липидного вспомогательного вещества, липидной матрицы для длительного высвобождения и расплавленного покровного средства для состава с пролонгированным высвобождением лекарственного средства.
В соответствии с одним вариантом осуществления вспомогательное вещество для НМЕ представляет собой глицеринбегенат или глицериндибегенат.В соответствии с одним вариантом осуществления вспомогательное вещество для НМЕ представляет собой смесь разных сложных эфиров бегеновой кислоты с глицерином. В соответствии с одним вариантом осуществления вспомогательное вещество для НМЕ представляет собой Compritol®888 АТО. Compritol®888 и глицеринбегенат характеризуются температурой плавления приблизительно 70°С.
Традиционная температура при экструзии обычно составляет 100-200°С.Тем не менее API согласно настоящему изобретению, аримокломол, является нестабильным при этих температурах. Таким образом, вспомогательное вещество для НМЕ согласно настоящему изобретению предпочтительно характеризуется температурой плавления, которая обеспечивает возможность НМЕ при сохранении стабильности аримокломола.
В соответствии с одним вариантом осуществления вспомогательное вещество для НМЕ согласно настоящему изобретению характеризуется температурой плавления приблизительно 70°С. В соответствии с одним вариантом осуществления вспомогательное вещество для НМЕ согласно настоящему изобретению характеризуется температурой плавления приблизительно 65, 66, 67, 68, 69, 70, 71, 72, 73, 74 или 75°С. В соответствии с одним вариантом осуществления вспомогательное вещество для НМЕ согласно настоящему изобретению характеризуется температурой плавления 50-55°С, как например, 55-60°С, как например, 60-65°С, как например, 65-70°С, как например, 70-75°С. В соответствии с одним вариантом осуществления вспомогательное вещество для НМЕ согласно настоящему изобретению характеризуется температурой плавления менее 80°С, как например, менее 75°С, как например, равной или меньшей 70°С.
В соответствии с одним вариантом осуществления температура при экструзии или температура расплава в процессе экструзии горячего расплава составляет приблизительно 50-55°С, как например, 55-60°С, как например, 60-65°С, как например, 65-70°С, как например, 70-75°С. В соответствии с одним вариантом осуществления температура при экструзии составляет 60-61°С, как например, 61-62°С, как например, 62-63°С, как например, 63-64°С, как например, 64-65°С, как например, 65-66°С, как например, 66-67°С, как например, 67-68°С, как например, 68-69°С, как например, 69-70°С, как например, 70-71°С. В соответствии с одним вариантом осуществления температура при экструзии составляет 67-69°С. В соответствии с одним вариантом осуществления температура при экструзии составляет менее 80°С, как например, менее 75°С, как например, является равной или меньшей 70°С.
В процессе экструзии горячего расплава используется давление. В соответствии с одним вариантом осуществления давление экструзии или давление расплава составляет 0-10 бар. В соответствии с одним вариантом осуществления давление экструзии составляет 0-1 бар, как например, 1-2 бар, как например, 2-3 бар, как например, 3-4 бар, как например, 4-5 бар, как например, 5-6 бар, как например, 6-7 бар, как например, 7-8 бар, как например, 8-9 бар, как например, 9-10 бар.
В соответствии с одним вариантом осуществления крутящий момент инструмента составляет 5-20%, как например, 5-6%, 6-7%, 7-8%, 8-9%, 9-10%, 10-11%, 11-12%, 12-13%, 13-14%, 14-15%, 15-16%, 16-17%, 17-18%, 18-19%, как например, 19-20%.
Полоски или экструдаты, полученные посредством экструзии горячего расплава, можно подвергать размолу. В соответствии с одним вариантом осуществления экструдат горячего расплава, содержащий API и вспомогательное вещество для НМЕ дополнительно подвергают стадии измельчения, как например, посредством размола. В соответствии с одним вариантом осуществления экструдат горячего расплава охлаждают или позволяют ему остыть перед измельчением, как например, до комнатной температуры.
В соответствии с одним вариантом осуществления измельченный или размолотый экструдат горячего расплава, содержащий API и вспомогательное вещество для НМЕ, дополнительно подвергают стадии фракционирования по размеру, как например, с помощью просеивания через сито. Таким образом можно разделить порошки с разными размерами частиц.
Стадии измельчения, как например, посредством размола, и необязательно фракционирования по размеру с помощью просеивания через сито дают в результате гранулы (или микрогранулы) с длительным высвобождением.
Ситовые фракции можно собрать по отдельности с получением ситовых фракций с конкретными размерами частиц. Используемый в данном документе термин "размер частиц" может в равной степени относиться к "среднему размеру частиц".
В соответствии с одним вариантом осуществления размер частиц гранул с длительным высвобождением составляет 500-710 мкм, 710-1000 мкм или более 1000 мкм.
В соответствии с одним вариантом осуществления размер частиц гранул с длительным высвобождением составляет 500-750 мкм, как например, 750-1000 мкм, как например, более 1000 мкм, как например, 1000-1250 мкм, как например, 1250-1500 мкм, как например, 1500-1750 мкм, как например, 1750-2000 мкм, как например, 2000-2500 мкм, как например, 2500-3000 мкм.
В соответствии с одним вариантом осуществления гранулы с длительным высвобождением состоят из API и вспомогательного вещества для НМЕ. В соответствии с одним вариантом осуществления гранулы с длительным высвобождением состоят из 33 масс. % API и 67 масс. % вспомогательного вещества для НМЕ. В соответствии с одним вариантом осуществления гранулы с длительным высвобождением состоят из 50 масс. % API и 50 масс. % вспомогательного вещества для НМЕ. В соответствии с одним вариантом осуществления гранулы с длительным высвобождением состоят из 67 масс. % API и 33 масс. % вспомогательного вещества для НМЕ.
Гранулы с длительным высвобождением поддерживают высокое содержание лекарственного средства. В соответствии с одним вариантом осуществления гранулы с длительным высвобождением содержат примерно 33, 50 или 66 масс. % API, такого как аримокломол. В соответствии с одним вариантом осуществления гранулы с длительным высвобождением содержат 15-75 масс. % API, как например, 15-20, 20-25, 25-30, 30-35, 35-40, 40-45, 45-50, 50-55, 55-60, 60-65 или 65-70, 70-75 масс. % API, такого как аримокломол. В соответствии с конкретным вариантом осуществления гранулы с длительным высвобождением содержат 25-75 масс. % API, как например, 30-65 масс. % API, как например, 25-50 масс. % API, как например, 30-50 масс. % API.
В соответствии с одним вариантом осуществления гранулы с длительным высвобождением содержат 20-60 масс. % API, как например, 25-50 масс. % API, и характеризуются размером частиц более 710 мкм, как например, более 1000 мкм.
В соответствии с одним вариантом осуществления гранулы с длительным высвобождением содержат приблизительно 33 масс. % API, как например, 25-40 масс. % API, и характеризуются размером частиц более 1000 мкм. В соответствии с одним вариантом осуществления гранулы с длительным высвобождением содержат приблизительно 33 масс. % API, как например, 25-40 масс. % API, и характеризуются размером частиц более 710-1000 мкм. В соответствии с одним вариантом осуществления гранулы с длительным высвобождением содержат приблизительно 33 масс. % API, как например, 25-40 масс. % API, и характеризуются размером частиц более 500-710 мкм.
В соответствии с одним вариантом осуществления гранулы с длительным высвобождением содержат приблизительно 50 масс. % API, как например, 40-55 или 40-60 масс. % API, и характеризуются размером частиц более 1000 мкм. В соответствии с одним вариантом осуществления гранулы с длительным высвобождением содержат приблизительно 50 масс. % API, как например, 40-55 или 40-60 масс. % API, и характеризуются размером частиц 710-1000 мкм. В соответствии с одним вариантом осуществления гранулы с длительным высвобождением содержат приблизительно 50 масс. % API, как например, 40-55 или 40-60 масс. % API, и характеризуются размером частиц 500-710 мкм.
В соответствии с одним вариантом осуществления гранулы с длительным высвобождением содержат приблизительно 66 масс. % API, как например, 55-70 масс. % API, и характеризуются размером частиц более 1000 мкм.
В соответствии с одним вариантом осуществления гранулы с длительным высвобождением согласно настоящему изобретению содержатся в капсуле, как например, с обеспечением пероральной лекарственной формы с множеством единиц, такой как капсула, содержащая гранулы с длительным высвобождением согласно настоящему изобретению. В соответствии с одним вариантом осуществления капсула содержит или состоит из желатина. В соответствии с одним вариантом осуществления капсула представляет собой капсулу с твердой оболочкой, такую как твердая желатиновая капсула. В соответствии с дополнительным вариантом осуществления капсула дополнительно содержит наружное покрытие.
В соответствии с одним вариантом осуществления гранулы с длительным высвобождением согласно настоящему изобретению содержатся в пакетике или саше.
Гранулы с длительным высвобождением легко смешиваются в жидкостях или в пищевых продуктах для перорального приема внутрь или через зонд для искусственного кормления.
В соответствии с одним вариантом осуществления гранулы с длительным высвобождением согласно настоящему изобретению подвергают прессованию с образованием таблетки, мини-таблетки или микро-таблетки.
Активный фармацевтический ингредиент
Активный фармацевтический ингредиент (API), содержащийся в данных составах, выбран из N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида (аримокломола), его стереоизомеров и их солей присоединения кислоты. Аримокломол дополнительно описан, например, в патентном документе WO 00/50403.
Аспект настоящего изобретения заключается в обеспечении состава, содержащего N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорид (аримокломол), его оптически активный (+) или (-) энантиомер, смесь энантиомеров в любом соотношении и рацемическое соединение, кроме того, соли присоединения кислоты, образованные из любого из вышеуказанных соединений с минеральными или органическими кислотами, составляют объекты настоящего изобретения. Все возможные геометрические изомерные формы N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида относятся к объему настоящего изобретения. Термин "стереоизомеры N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида" относится ко всем возможным оптическим и геометрическим изомерам соединения.
Если необходимо, N-[2-гидрокси~3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорид или один из его оптически активных энантиомеров может быть превращен в соль присоединения кислоты с минеральной или органической кислотой с помощью известных способов.
В соответствии с одним вариантом осуществления активный фармацевтический ингредиент представляет собой рацемат N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида.
В соответствии с одним вариантом осуществления активный фармацевтический ингредиент представляет собой оптически активный стереоизомер N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида.
В соответствии с одним вариантом осуществления активный фармацевтический ингредиент представляет собой энантиомер N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида.
В соответствии с одним вариантом осуществления активный фармацевтический ингредиент выбран из группы, состоящей из (+)-R-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида и (-)-(S)-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида.
В соответствии с одним вариантом осуществления активный фармацевтический ингредиент представляет собой соль присоединения кислоты N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида.
В соответствии с одним вариантом осуществления активный фармацевтический ингредиент выбран из группы, состоящей из соли присоединения лимонной кислоты N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида (N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида цитрата) и соли присоединения малеиновой кислоты N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида (N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида малеата).
В соответствии с одним вариантом осуществления активный фармацевтический ингредиент выбран из группы, состоящей из соли присоединения лимонной кислоты (+)-R-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида ((+)-R-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида цитрата); соли присоединения лимонной кислоты (-)-S-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида ((-)-S-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида цитрата); соли присоединения малеиновой кислоты (+)-R-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида ((+)-R-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида малеата) и соли присоединения малеиновой кислоты (-)-S-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида ((-)-S-N-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида малеата).
Введение и дозировка
В соответствии с настоящим изобретением активный фармацевтический ингредиент (API) вводят индивидам, нуждающимся в лечении, в фармацевтически эффективных дозах. Терапевтически эффективное количество API в соответствии с настоящим изобретением представляет собой количество, достаточное для излечения, предупреждения, снижения риска, ослабления или частичной задержки клинических проявлений данного заболевания или состояния и его осложнений. Количество, которое является эффективным для конкретной терапевтической цели, будет зависеть от тяжести и типа заболевания, а также от массы тела и общего состояния субъекта.
Фармацевтический состав согласно настоящему изобретению в соответствии с одним вариантом осуществления вводят от 1 до 3 раз в сутки, как например, один раз в сутки, как например, два раза в сутки, например, 3 раза в сутки. Предпочтительно, фармацевтический состав вводят один раз в сутки или два раза в сутки.
В соответствии с одним вариантом осуществления введение происходит в течение ограниченного периода времени, как например, от 1 или 2 суток до 7 суток, например, от 7 суток до 14 суток, как например, от 14 суток до месяца, например, от месяца до нескольких месяцев (2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 месяцев); или введение в соответствии с одним вариантом осуществления является продолжительным, лечение может длиться с момента постановки диагноза, как например, в течение всего времени жизни индивида или до тех пор, пока индивид будет получать пользу от введения, т.е. когда присутствует заболевание, или в то время, когда существует повышенный риск развития заболевания.
Введение фармацевтического состава согласно настоящему изобретению в соответствии с одним вариантом осуществления вводят индивиду в различные моменты времени лечения. Лечение можно выполнять в течение одного непрерывного периода или с интервалами между периодами, причем введение прекращают, уменьшают или изменяют. Продолжительность таких периодов лечения или периодов без лечения может варьировать и в соответствии с одним вариантом осуществления составляет от 1 суток до 60 суток, как например, от 1 до 3 суток, от 3 до 6 суток, от 6 до 8 суток, от 8 до 14 суток, от 14 до 21 суток, от 21 до 30 суток, от 30 до 42 суток, от 42 до 49 суток или от 49 до 60 суток.
Фармацевтическая композиция согласно настоящему изобретению в соответствии с одним вариантом осуществления содержит API в количестве от 0,1 до 100 мг на дозу; как например, приблизительно 0,1, 0,5, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 50 или 100 мг API на дозу. Доза может относиться к лекарственной форме, таблетке или капсуле.
В соответствии с дополнительным вариантом осуществления API присутствует в составе в количестве от 0,1 до 0,5 мг на дозу, как например, от 0,5 до 1 мг, например, от 1 до 2 мг, как например, от 2 до 3 мг, например, от 3 до 4 мг, как например, от 4 до 5 мг, например, от 5 до 7,5 мг, как например, от 7,5 до 10 мг, например, от 10 до 15 мг, как например, от 15 до 20 мг, например, от 20 до 30 мг, как например, от 30 до 40 мг, например, от 40 до 50 мг, как например, от 50 до 60 мг на дозу, например, от 60 до 70 мг, как например, от 70 до 80 мг, например, от 80 до 90 мг, как например, от 90 до 100 мг API на дозу.
В соответствии с конкретным вариантом осуществления количество API на дозу составляет приблизительно 10 мг, как например, приблизительно 15 мг, как например, приблизительно 20 мг на дозу.
В соответствии с дополнительным вариантом осуществления API присутствует в одной лекарственной форме или единице состава, такой как отдельная таблетка и сфера, или в наборе таблеток или сфер, или композиции НМЕ гранул в общем количестве 5-1000 мг на дозу, как например, 5-10, 10-25, 25-50, 50-75, 75-100, 100-150, 150-200, 200-250, 250-300, 300-400, 400-500, 500-600, 600-700, 700-800, 800-900, 900-1000 мг API на дозу.
Целевая доза для API в соответствии с одним вариантом осуществления находится в пределах диапазона от 0,1 до 100 мг/кг массы тела, как например, от 0,1 до 0,5 мг/кг, например, от 0,5 до 1,0 мг/кг, как например, от 1 до 2 мг/кг, например, от 2 до 5 мг/кг, как например, от 5 до 10 мг/кг, например, от 10 до 15 мг/кг, как например, от 15 до 20 мг/кг, например, от 20 до 30 мг/кг, как например, от 30 до 40 мг/кг, например, от 40 до 50 мг/кг, как например, от 50 до 75 мг/кг, например, от 75 до 100 мг/кг массы тела.
В соответствии с конкретным вариантом осуществления диапазон доз составляет приблизительно от 15 до 50 мг, и целевая доза составляет приблизительно 1 мг/кг.
Целевая группа
Фармацевтический состав в соответствии с настоящим изобретением можно вводить любому индивиду, нуждающемуся в лечении. Индивид, нуждающийся в лечении, представляет собой любого индивида, на которого будет, или вероятно будет, оказывать благоприятное воздействие лечение активным фармацевтическим ингредиентом в соответствии с настоящим изобретением.
Аспект настоящего изобретения также заключается в обеспечении фармацевтического состава, содержащего активный фармацевтический ингредиент, выбранный из N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, причем указанный состав обеспечивает длительное высвобождение указанного активного фармацевтического ингредиента для введения индивиду, выбранному из группы, состоящей из пациентов-детей; пациентов, у которых присутствует повышенный уровень креатинина в сыворотке крови; и пациентов, подвергающихся лечению активным фармацевтическим ингредиентом, отличающимся от активного фармацевтического ингредиента в соответствии с настоящим изобретением
В соответствии с одним вариантом осуществления пациент-ребенок включает младенцев, детей и подростков возрастом в диапазоне от рождения до 18 лет. В соответствии с одним вариантом осуществления пациент-ребенок имеет возраст от 0 до 1 года, как например, от 1 до 2, например, от 2 до 3, как например, от 3 до 4, например, от 4 до 5, как например, от 5 до 6, например, от 6 до 7, как например, от 7 до 8, например, от 8 до 9, как например, от 9 до 10, например, от 9 до 10, как например, от 10 до 11, например, от 11 до 12, как например, от 12 до 13, например, от 13 до 14, как например, от 14 до 15, например, от 15 до 16, как например, от 16 до 17, например, от 17 до 18 лет. В соответствии с конкретным вариантом осуществления пациент-ребенок в соответствии с настоящим изобретением имеет возраст от 5 до 15 лет.
В соответствии с одним вариантом осуществления у пациента присутствует повышенный уровень креатинина в сыворотке крови в соответствии с настоящим изобретением представляет собой пациента, имеющего повышенные базальные уровни креатинина в сыворотке крови, как например, повышенные уровни по сравнению с уровнями, которые присутствовали бы в сыворотке крови у того же пациента, если бы этот пациент не страдал от состояния, которое вызывает повышение уровней креатинина. Таким образом, повышенный уровень креатинина у одного пациента может соответствовать концентрации креатинина в сыворотке крови, которая у другого индивида считается "нормальной", "не повышенной" или которая представляет собой базальный уровень для индивида.
Повышенный уровень креатинина в сыворотке крови может являться маркером заболевания, поскольку его уровни часто коррелируют с болезненными состояниями. Таким образом, пациент, имеющий одно или более медицинских состояний до получения лечения фармацевтическим составом согласно настоящему изобретению для заданного дополнительного состояния, может извлекать пользу от настоящего изобретения.
В соответствии с одним вариантом осуществления пациент, у которого присутствует повышенный уровень креатинина в сыворотке крови, представляет собой пациента с заболеванием почек (нефропатией), в том числе невоспалительной нефропатией (нефрозом) и воспалительной нефропатией (нефритом); и/или пациента с пониженной функцией почек; включая стадии почечной недостаточности, почечную недостаточность и уремию.
В соответствии с одним вариантом осуществления пациент с заболеванием почек представляет собой пациента, имеющего состояние, выбранные из группы, состоящей из IgA-нефропатии (включает отложение IgA-антител в клубочке), фокально-сегментального гломерулосклероза, вызванного лекарственным средством (например, анальгетиками, химиотерапевтическими средствами) и токсином хронического тубулоинтерстициального нефрита, недостаточности ксантиноксидазы, поликистозной болезни почек, острого повреждения почек (AKI), хронической болезни почек (CKD), гломерулонефрита, стеноза почечных артерий, ишемической нефропатии, гемолитико-уремического синдрома, васкулита, обструктивного заболевания почек (почечные камни и заболевание предстательной железы), длительного воздействия свинца или его солей; нефропатии, вызванной хроническими состояниями, в том числе системной красной волчанкой, сахарным диабетом и артериальной гипертензией, которые приводят к волчаночному нефриту, диабетической нефропатии и гипертонической нефропатии, соответственно; и хронического заболевания почек неизвестной этиологии (CKDu), такого как мезоамериканская нефропатия (MeN; также называемая, 'creatinina').
В соответствии с одним вариантом осуществления пациент, у которого присутствует повышенный уровень креатинина в сыворотке крови, представляет собой пациента с сахарным диабетом, в том числе с сахарным диабетом типа I и сахарным диабетом типа II.
В соответствии с одним вариантом осуществления пациент, у которого присутствует повышенный уровень креатинина в сыворотке крови, в соответствии с настоящим изобретением представляет собой пациента с артериальной гипертензией, как например, пациента с артериальной гипертензией, имеющего кровяное давление около 140/90 мм рт. ст.или выше.
В соответствии с одним вариантом осуществления пациент, получающий лечение активным фармацевтическим ингредиентом, отличным от активного фармацевтического ингредиента в соответствии с настоящим изобретением, представляет собой пациента, который получает один или более дополнительных активных фармацевтических ингредиентов для лечения или контроля течения заданного состояния. Указанное заданное состояние может представлять собой состояние, которое отличается от состояния, при котором эффективен фармацевтический состав согласно настоящему изобретению.
В соответствии с одним вариантом осуществления по меньшей мере два или более активных фармацевтических ингредиентов, которые пациент, получающий лечение активным фармацевтическим ингредиентом, отличающимся от активного фармацевтического ингредиента в соответствии с настоящим изобретением, содержит i) N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорид, его стереоизомеры и их соли присоединения кислоты, а также и) соединение, которое взаимодействует с N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлоридом, его стереоизомерами и его солями присоединения кислоты, или соединение, которое повышает уровень креатинина в сыворотке крови.
В соответствии с одним вариантом осуществления фармацевтический состав согласно настоящему изобретению избегает или снижает индуцированное аримокломолом повышение уровня креатинина в сыворотке крови и таким образом снижает риск противопоказаний у пациентов, которые получают дополнительные лекарственные препараты.
Медицинское применение
Аспект заключается в обеспечении фармацевтического состава в соответствии с настоящим изобретением для применения в качестве лекарственного препарата.
Аспект настоящего изобретения заключается в обеспечении фармацевтического состава в соответствии с настоящим изобретением для применения в способе лечения пациентов-детей, пациентов, у которых присутствует повышенный уровень креатинина в сыворотке крови; и пациентов, получающих лечение активным фармацевтическим ингредиентом, отличным от активного фармацевтического ингредиента в соответствии с настоящим изобретением.
Аспект настоящего изобретения заключается в обеспечении применения фармацевтического состава в соответствии с настоящим изобретением для производства лекарственного препарата для лечения пациентов-детей, пациентов, у которых присутствует повышенный уровень креатинина в сыворотке крови; и пациентов, получающих лечение активным фармацевтическим ингредиентом, отличным от активного фармацевтического ингредиента в соответствии с настоящим изобретением.
Аспект настоящего изобретения заключается в обеспечении способа лечения пациентов-детей, пациентов, у которых присутствует повышенный уровень креатинина в сыворотке крови; и пациентов, получающих лечение активным фармацевтическим ингредиентом, отличающимся от активного фармацевтического ингредиента в соответствии с настоящим изобретением, причем указанный способ предусматривает стадию введения фармацевтического состава в соответствии с настоящим изобретением.
Аспект настоящего изобретения заключается в обеспечении фармацевтического состава в соответствии с настоящим изобретением для применения в способе лечения детского заболевания.
Аспект настоящего изобретения заключается в обеспечении фармацевтического состава в соответствии с настоящим изобретением для применения в способе лечения лизосомной болезни накопления (LSD).
Аспект настоящего изобретения заключается в обеспечении применения фармацевтического состава в соответствии с настоящим изобретением для производства лекарственного препарата для лечения лизосомной болезни накопления (LSD).
Аспект настоящего изобретения заключается в обеспечении способа лечения лизосомной болезни накопления (LSD), предусматривающего введение пациенту, нуждающемуся в этом, фармацевтического состава в соответствии с настоящим изобретением.
Лизосомные болезни накопления представляют собой группу примерно из 40 редких наследственных метаболических нарушений, которые являются результатом дефектов в функционировании лизосом вследствие недостаточности одного фермента, необходимого для метаболизма липидов, гликопротеинов или мукополисахаридов. Хотя каждое нарушение является результатом мутаций в разных генах, которые обуславливают недостаточность активности фермента, у всех них имеется общая биохимическая характеристика - все лизосомные нарушения возникают в результате ненормального накопления веществ внутри лизосомы.
Аспект настоящего изобретения заключается в обеспечении фармацевтического состава в соответствии с настоящим изобретением для применения в способе лечения лизосомной болезни накопления, выбранной из группы, состоящей из нарушений накопления липидов (или липидоза), в том числе сфинголипидозов, ганглиозидозов и лейкодистрофий; мукополисахаридозов, нарушений накопления гликопротеинов (или гликопротеиноза) и муколипидозов.
В соответствии с одним вариантом осуществления лизосомная болезнь накопления выбрана из группы, состоящей из болезни Ниманна-Пика, болезни Фарбера, болезни Краббе, болезни Фабри, болезни Гоше, сиалидоза (муколипидоза типа I), метахроматической лейкодистрофий (поздняя младенческая, ювенильная и взрослая формы) и недостаточности сапозина.
В соответствии с одним вариантом осуществления болезнь Ниманна-Пика выбрана из группы, состоящей из болезни Ниманна-Пика типа А, болезни Ниманна-Пика типа В, болезни Ниманна-Пика типа С и болезни Ниманна-Пика типа D.
В соответствии с одним вариантом осуществления болезнь Гоше выбрана из группы, состоящей из болезни Гоше типа I (неневропатический тип), типа II (острая младенческая невропатическая болезнь Гоше) и типа III (хроническая невропатическая форма).
Аспект настоящего изобретения заключается в обеспечении фармацевтического состава в соответствии с настоящим изобретением для применения в способе лечения бокового амиотрофического склероза (ALS).
Аспект настоящего изобретения заключается в обеспечении применения фармацевтического состава в соответствии с настоящим изобретением для производства лекарственного препарата для лечения бокового амиотрофического склероза (ALS).
Аспект настоящего изобретения заключается в обеспечении способа лечения бокового амиотрофического склероза (ALS), предусматривающего введение пациенту, нуждающемуся в этом, фармацевтического состава в соответствии с настоящим изобретением.
Пункты настоящего изобретения:
1. Фармацевтический состав, содержащий активный фармацевтический ингредиент, выбранный из N-(2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты,
причем указанный состав содержит внутреннюю матрицу и по меньшей мере одно наружное покрытие,
при этом указанный состав обеспечивает длительное высвобождение указанного активного фармацевтического ингредиента.
2. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав представляет собой твердую лекарственную форму, такую как перорально доступная твердая лекарственная форма.
3. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная внутренняя матрица содержит активный фармацевтический ингредиент.
4. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная внутренняя матрица выбрана из группы, состоящей из таблетки, мини-таблетки и микро-таблетки.
5. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав выбран из группы, состоящей из таблетки с покрытием, мини-таблетки с покрытием и микро-таблетки с покрытием.
6. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанное наружное покрытие не содержит активный фармацевтический ингредиент.
7. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем каждое из указанной внутренней матрицы и/или указанного наружного покрытия содержит одно или более вспомогательных веществ, таких как одно или более контролирующих высвобождение вспомогательных веществ.
8. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанное контролирующее высвобождение вспомогательное вещество выбрано из группы, состоящей из гидроксипропилметилцеллюлозы (НРМС), этилцеллюлозы (ЕС), метилцеллюлозы, гидроксипропилцеллюлозы, ацетилсукцината гипромеллозы, фталата гипромеллозы, ацетата целлюлозы, глицеринмоностеарата, глицерилмоноолеата, глицерилпальмитата, глицерилбегената, гидрирогенизированного растительного масла, гуаровой камеди, поливинилового спирта, альгинатов, ксантановой камеди, карнаубского воска, желтого воска, белого воска, зеина, каррагенана, карбомеров и агара.
9. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанное покрытие содержит пленкообразующее вспомогательное вещество.
10. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная внутренняя матрица содержит по меньшей мере одну гидроксипропилметилцеллюлозу (НРМС).
11. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная НРМС представляет собой НРМС, имеющую марку, обеспечивающую вязкость от 50 до 100000 сП, НРМС марки с высокой вязкостью или НРМС марки со сверхвысокой вязкостью.
12. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная НРМС выбрана из группы, состоящей из Methocel Е50 Premium LV, K100 Premium LV CR, K4M Premium CR, K15M Premium CR, K100M Premium CR, E4M Premium CR, E10M Premium CR, K200M, E5 и E50.
13. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанное контролирующее высвобождение вспомогательное вещество присутствует в количестве 20-50% масс., как например, 20-25% масс., например, 25-30% масс., как например, 30-35% масс., например, 35-40% масс., как например, 40-45% масс., например, 45-50% масс.
14. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная внутренняя матрица содержит один или более ионных, неионных и/или нерастворимых в воде полимеров.
15. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный ионный полимер выбран из группы, состоящей из карбоксиметилцеллюлозы натрия (na CMC), альгината натрия, полимеров акриловой кислоты или карбомеров (карбопол 934, 940, 974Р NF), растворимых в кишечнике полимеров, таких как поливинилацетатфталат (PVAP), сополимеров метакриловой кислоты (Eudragit L100 L 30D 55, S и FS 30 D), ацетилсукцината гипромеллозы (AQOAT HPMCAS) и ксантановой камеди.
16. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный неионный полимер выбран из группы, состоящей из НРС (гидроксипропилцеллюлоза) и РЕО (POLYOX, Dow Chemical Company) марок с различными молекулярными массами (от 100000 до 7000000 Да).
17. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный нерастворимый в воде полимер выбран из группы, состоящей из этилцеллюлозы (например, ETHOCEL или Surrelease), ацетата целлюлозы, сополимеров метакриловой кислоты (например, Eudragit NE 30D), аммонио-метакрилатных сополимеров (например, Eudragit RL 100 или РО RS100) и поливинилацетата.
18. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная внутренняя матрица содержит одно или более дополнительных вспомогательных веществ, таких как один или более наполнителей, связующих и/или смазывающих средств.
19. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная внутренняя матрица содержит один или более наполнителей, таких как один или более наполнителей, выбранные из группы, состоящей из карбоната кальция, фосфатов кальция, сульфата кальция, целлюлозы, ацетата целлюлозы, сжимаемого сахара, декстрана, декстрина, декстрозы, этилцеллюлозы, фруктозы, изомальта, лактита, лактозы, маннита, карбоната магния, оксида магния, мальтодекстрина, микрокристаллической целлюлозы (МСС), полидекстрозы, альгината натрия, сорбита, талька и ксилита.
20. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная внутренняя матрица содержит одно или более связующих, таких как одно или более связующих, выбранные из группы, состоящей из аравийской камеди, альгиновой кислоты, карбомеров, карбоксиметилцеллюлозы натрия, каррагенана, ацетилфталата целлюлозы, хитозана, коповидона, декстрата, декстрина, декстрозы, этилцеллюлозы, желатина, гуаровой камеди, гидроксиэтилцеллюлозы, гидроксиэтилметилцеллюлозы, гидроксипропилцеллюлозы, гидроксипропилкрахмала, гипромеллозы, метилцеллюлозы, полоксамера, полидекстрозы, полиэтиленоксида, повидона, альгината натрия, сахарозы, крахмала, прежелатинизированного крахмала и мальтодекстрина.
21. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная внутренняя матрица содержит одно или более смазывающих средств, таких как одно или более смазывающих средств, выбранных из группы, состоящей из стеарата кальция, глицеринмоностеарата, глицерилбегената, глицерилпальмитостеарата, гидрогенизированного касторового масла, гидрогенизированного растительного масла, лаурилсульфата магния, стеарата магния, триглицерида со средним размером цепи, пальмитиновой кислоты, полиэтиленгликоля, лаурилсульфата натрия, стеариновой кислоты, талька, диоксида кремния и стеарата цинка.
22. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная внутренняя матрица содержит одно или более из вспомогательных веществ на основе гидроксипропилметилцеллюлозы (НРМС), крахмала, этилцеллюлозы (ЕС), микрокристаллической целлюлозы (МСС), диоксида кремния, стеарата магния и стеариновой кислоты.
23. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная внутренняя матрица дополнительно содержит микрокристаллическую целлюлозу (МСС), такую как Avicel РН 101 или Avicel РН 102.
24. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанное второе вспомогательное вещество, такое как МСС, присутствует в количестве 10-50% масс., как например, 10-15% масс., например, 15-20% масс., как например, 20-25% масс., например, 25-30% масс., как например, 30-35% масс., например, 35-40% масс., как например, 40-45% масс., например, 45-50% масс.
25. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная внутренняя матрица дополнительно содержит крахмал, как например, в количестве 5-30% масс., как например, 5-10% масс., например, 10-15% масс., как например, 15-20% масс., например, 20-25% масс., как например, 25-30% масс. крахмала.
26. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная внутренняя матрица дополнительно содержит этилцеллюлозу (ЕС), как например, в количестве 5-30% масс., как например, 5-10% масс., например, 10-15% масс., как например, 15-20% масс., например, 20-25% масс., как например, 25-30% масс.
27. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная внутренняя матрица дополнительно содержит диоксид кремния, такой как коллоидный диоксид кремния, как например, в количестве 0,05-1% масс, как например, 0,05-0,1, например, 0,1-0,2, как например, 0,2-0,3, например, 0,3-0,4, как например, 0,4-0,5, например, 0,5-0,6, как например, 0,6-0,7, например, 0,7-0,8, как например, 0,8-0,9, например, 0,9-1,0% масс.
28. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная внутренняя матрица дополнительно содержит стеарат магния, как например, в количестве 0,1-5% масс., как например, 0,1-0,5, например, 0,5-1,0, как например, 1-2, например, 2-3, как например, 3-4, например, 4-5% масс.
29. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная внутренняя матрица дополнительно содержит стеариновую кислоту, как например, в количестве 0,1-10% масс., как например, 0,1-0,5, например, 0,5-1,0, как например, 1-2, например, 2-3, как например, 3-4, например, 4-5, как например, 4-5, например, 5-6, как например, 5-6, например, 6-7, как например, 7-8, например, 8-9, как например, 9-10% масс.
30. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанную внутреннюю матрицу подвергают прессованию с образованием таблетки, такой как таблетка с твердостью от 10 до 50 Кп (килопонд), как например, от 10 до 15 Кп, например, от 15 до 20 Кп, как например, от 20 до 25 Кп, например, от 25 до 30 Кп, как например, от 30 до 35 Кп, например, от 35 до 40 Кп, как например, от 40 до 50 Кп.
31. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанное наружное покрытие содержит один или более слоев покрытия.
32. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанное наружное покрытие содержит одно или более вспомогательных веществ.
33. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанное наружное покрытие содержит или состоит из этилцеллюлозы (ЕС) на водной основе, ЕС на основе растворителя или полиметакрилата на водной основе.
34. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав наносят в виде покрытия до увеличения массы на 5% масс., как например, увеличения массы на 10% масс., например, увеличения массы на 15% масс., как например, увеличения массы на 20% масс., например, увеличения массы на 25% масс., как например, увеличения массы на 30% масс., например, увеличения массы на 35% масс., как например, увеличения массы на 40% масс.
35. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав наносят в виде покрытия до увеличения массы на 5-40% масс., как например, на 10-15% масс., например, на 15-20% масс., как например, на 20-25% масс., например, на 25-30% масс., как например, на 30-35% масс., например, на 35-40% масс.
36. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанное наружное покрытие дополнительно содержит наружное изолирующее покрытие.
37. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав содержит или состоит из:
i) внутренней матрицы, содержащей или состоящей из
a. 5-40% N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида (аримокломола), его стереоизомеров и их солей присоединения кислоты, как например, 5-10, например, 10-15, как например, 15-20, например, 20-25, как например, 25-30, например, 30-35, как например, 35-40% масс.;
b. 20-50% НРМС, как например, 20-25% масс., например, 25-30% масс., как например, 30-35% масс., например, 35-40% масс., как например, 40-45% масс., например, 45-50% масс. НРМС;
c. 10-50% МСС, например, 10-50% масс., как например, 10-15% масс., например, 15-20% масс., как например, 20-25% масс., например, 25-30% масс., как например, 30-35% масс., например, 35-40% масс., как например, 40-45% масс., например, 45-50% масс. МСС;
d. 5-30% крахмала, как например, 5-10% масс., например, 10-15% масс., как например, 15-20% масс., например, 20-25% масс., как например, 25-30% масс. крахмала;
e. 0,05-1% диоксида кремния, как например, 0,05-0,1, например, 0,1-0,2, как например, 0,2-0,3, например, 0,3-0,4, как например, 0,4-0,5, например, 0,5-0,6, как например, 0,6-0,7, например, 0,7-0,8, как например, 0,8-0,9, например, 0,9-1,0% масс. диоксида кремния;
f. 0,1-5% стеарата магния, как например, 0,1-0,5, например, 0,5-1,0, как например, 1-2, например, 2-3, как например, 3-4, например, 4-5% масс. стеарата магния;
g. 0,1-10% стеариновой кислоты, как например, 0,1-0,5, например, 0,5-1,0, как например, 1-2, например, 2-3, как например, 3-4, например, 4-5, как например, 5-6, например, 6-7, как например, 7-8, например, 8-9, как например, 9-10% масс. стеариновой кислоты; и
h. необязательно 5-30% (ЕС), как например, 5-10% масс., например, 10-15% масс., как например, 15-20% масс., например, 20-25% масс., как например, 25-30% масс. ЕС; и
ii) необязательно наружное покрытие, содержащее или состоящее из
a. этилцеллюлозы (ЕС) на водной основе, ЕС на основе растворителя и/или полиметакрилата на водной основе, наносимых до увеличения массы на 5-40% масс., как например, на 10-15% масс., например, на 15-20% масс., как например, на 20-25% масс., например, на 25-30% масс., как например, на 30-35% масс., например, на 35-40% масс.; и
b. необязательно наружного изолирующего покрытия.
38. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав содержит или состоит из:
i) внутренней матрицы, содержащей или состоящей из
a. 5-40% N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида (аримокломола), его стереоизомеров и их солей присоединения кислоты,
b. 20-50% НРМС, с. 10-50% МСС,
d. 5-30% крахмала,
e. 0,05-1% диоксида кремния,
f. 0,1 -5% стеарата магния и
g. 0,1-10% стеариновой кислоты, и
ii) наружного покрытия, содержащего или состоящего из
a. этилцеллюлозы (ЕС) на водной основе, ЕС на основе растворителя и/или полиметакрилата на водной основе, наносимых до увеличения массы на 5-25% масс.; и
b. необязательно наружного изолирующего покрытия.
39. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанное наружное покрытие содержит активный фармацевтический ингредиент.
40. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав содержит внутреннюю матрицу или основу сферы и наружное покрытие, содержащее один или более отдельных слоев.
41. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанное наружное покрытие содержит два или более слоев, как например, три или более слоев, как например, четыре или более слоев, как например, пять или более слоев.
42. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав представляет собой сферу с покрытием, такую как сфера с покрытием, содержащая основу сферы и наружное покрытие, содержащее один или более отдельных слоев.
43. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем первый или самый внутренний слой наружного покрытия содержит активный фармацевтический ингредиент.
44. Фармацевтический состав в соответствии с любым из предыдущих пунктов, содержащий 1) основу сферы, 2) слой лекарственного средства, содержащий активный фармацевтический ингредиент, 3) необязательно изолирующее покрытие, 4) покрытие для контролируемого высвобождения и 5) необязательно пленочное покрытие.
45. Фармацевтический состав в соответствии с любым из предыдущих пунктов, содержащий 1) основу сферы, 2) слой лекарственного средства до увеличения массы на 1-10% масс., 3) необязательно изолирующее покрытие до увеличения массы на 0,1-5% масс., 4) покрытие для контролируемого высвобождения до увеличения массы на 5-20% масс. и 5) необязательно пленочное покрытие до увеличения массы на 1-10% масс.
46. Фармацевтический состав в соответствии с любым из предыдущих пунктов, содержащий 1) основу сферы, 2) слой лекарственного средства до увеличения массы на 4% масс., 3) изолирующее покрытие до увеличения массы на 1% масс., 4) покрытие для контролируемого высвобождения до увеличения массы на 5-20% масс., и 5) пленочное покрытие до увеличения массы на 3-5% масс.
47. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная внутренняя матрица или основа сферы содержит или состоит из сахара, как например, растворимая сахарная сфера.
48. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная внутренняя матрица или основа сферы содержит или состоит из микрокристаллической целлюлозы (МСС), как например, нерастворимая сфера из микрокристаллической целлюлозы.
49. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный слой лекарственного средства содержит активный фармацевтический ингредиент и вспомогательное вещество, такое как НРМС.
50. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный слой лекарственного средства наносят до увеличения массы на 1-10% масс., как например, на 1-2, например, на 2-3, как например, на 3-4, например, на 4-5, как например, на 5-6, например, на 6-7, как например, на 7-8, например, на 8-9, как например, до увеличения массы на 9-10% масс.
51. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанное покрытие для контролируемого высвобождения содержит или состоит из этилцеллюлозы (ЕС) на водной или неводной основе или водной дисперсии на основе полиакрилата.
52. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанное покрытие для контролируемого высвобождения наносят до увеличения массы на 5-30% масс., как например, 5-10, например, 10-15, как например 15-20, например, 20-25, как например, увеличения массы на 25-30% масс.
53. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанное пленочное покрытие наносят до увеличения массы на 1-10% масс., как например, на 1-2, например, на 2-3, как например, на 3-4, например, на 4-5, как например, на 5-6, например, на 6-7, как например, на 7-8, например, на 8-9, как например, до увеличения массы на 9-10% масс. В соответствии с одним вариантом осуществления пленочное покрытие наносят до увеличения массы приблизительно на 1% масс., как например, на 2, например, на 3, как например, на 4, например, на 5, как например, на 6, например, на 7, как например, на 8, например, на 9, как например, до увеличения массы приблизительно на 10% масс.
54. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанное изолирующее покрытие наносят до увеличения массы на 0,1-5% масс., как например, на 0,1-0,5, например, на 0,5-1, как например, на 1-2, например, на 2-3, как например, на 3-4, например, до увеличения массы на 4-5% масс.
55. Фармацевтический состав в соответствии с любым из предыдущих пунктов, содержащий или состоящий из:
а. основы сферы, содержащей 50-90% масс. сахара или МСС, как например, 50-55% масс., например, 55-60% масс., как например, 60-65% масс., например, 65-70% масс., как например, 70-75% масс., например, 75-80% масс., как например, 80-85% масс., например, 85-90% масс., сахара или МСС;
b. слоя лекарственного средства, содержащего 1-10% масс. N-[-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида (аримокломола), его стереоизомеров и их солей присоединения кислоты, как например, 1-2, например, 2-3, как например, 3-4, например, 4-5, как например, 5-6, например, 6-7, как например, 7-8, например, 8-9, как например, 9-10% масс.; и содержащего 1-10% масс. НРМС, как например, Methocel Е6, как например, 1-2, например, 2-3, как например, 3-4, например, 4-5, как например, 5-6, например, 6-7, как например, 7-8, например, 8-9, как например, 9-10% масс. НРМС;
c. изолирующего покрытия, содержащего 0,1-5% масс. пленочного покрытия на основе PVA, как например, 0,1-0,5, например, 0,5-1, как например, 1-2, например, 2-3, как например, 3-4, например, 4-5% масс.;
d. покрытия для контролируемого высвобождения, содержащего 5-30% масс. ЕС или полиакрилата, как например, 5-10, например, 10-15, как например, 15-20, например, 20-25, как например, 25-30% масс. ЕС или полиакрилата; и
e. необязательно пленочного покрытия, содержащего 1-10% масс., пленочного покрытия на основе PVA, как например, 1-2, например, 2-3, как например, 3-4, например, 4-5, как например, 5-6, например, 6-7, как например, 7-8, например, 8-9, как например, 9-10% масс. пленочного покрытия на основе PVA.
56. Фармацевтический состав в соответствии с любым из предыдущих пунктов, содержащий или состоящий из:
a. основы сферы, содержащей 67% масс. сахара или МСС,
b. слоя лекарственного средства, содержащего 4% масс. N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида (аримокломола), его стереоизомеров и их солей присоединения кислоты и содержащего 5% масс. НРМС, такой как Methocel Е6,
c. изолирующего покрытия, содержащего 1% масс. пленочного покрытия на основе PVA,
d. покрытия для контролируемого высвобождения, содержащего 5-20% масс. ЕС или полиакрилата, и
e. необязательно пленочного покрытия, содержащего 3% масс. пленочного покрытия на основе PVA.
57. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав представляет собой единичную пероральную лекарственную форму.
58. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав представляет собой пероральную лекарственную форму с множеством единиц.
59. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав содержится в капсуле, такой как капсула с твердой оболочкой, как например, капсула, содержащая желатин, как например, капсула с твердой оболочкой, дополнительно содержащая наружное покрытие.
60. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав представляет собой пероральную лекарственную форму с множеством единиц, причем указанная лекарственная форма содержит капсулу, содержащую две или более единиц состава по любому из предыдущих пунктов.
61. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанная единица состава выбрана из группы, состоящей из мини-таблетки с покрытием, микро-таблетки с покрытием и сферы с покрытием.
62. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный активный фармацевтический ингредиент представляет собой рацемат N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида.
63. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный активный фармацевтический ингредиент представляет собой оптически активный стереоизомер N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида.
64. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный активный фармацевтический ингредиент представляет собой энантиомер N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида.
65. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный активный фармацевтический ингредиент выбран из группы, состоящей из
(+)-R-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида и (-)-(S)-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин--оксид-3-карбоксимидоил-хлорида.
66. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный активный фармацевтический ингредиент представляет собой соль присоединения кислоты N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида.
67. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный активный фармацевтический ингредиент выбран из группы, состоящей из
соли присоединения лимонной кислоты N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида и соли присоединения малеиновой кислоты N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридип-1-оксид-3-карбоксимидоил-хлорида.
68. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный активный фармацевтический ингредиент выбран из группы, состоящей из
соли присоединения лимонной кислоты (+)-R-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида; соли присоединения лимонной кислоты (-)-S-N-[2-гидрокси-3-(1-пииеридинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида; соли присоединения малеиновой кислоты (+)-R-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида и соли присоединения малеиновой кислоты (-)-S-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида.
69. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав содержит указанный активный фармацевтический ингредиент в количестве от 0,1 мг до 100 мг, например, от 0,1 до 0,5 мг, как например, от 0,5 до 1 мг, например, от 1 до 2 мг, как например, от 2 до 3 мг, например, от 3 до 4 мг, как например, от 4 до 5 мг, например, от 5 до 7,5 мг, как например, от 7,5 до 10 мг, например, от 10 до 15 мг, как например, от 15 до 20 мг, например, от 20 до 30 мг, как например, от 30 до 40 мг, например, от 40 до 50 мг, как например, от 50 до 60 мг на дозу, например, от 60 до 70 мг, как например, от 70 до 80 мг, например, от 80 до 90 мг, как например, от 90 до 100 мг.
70. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав содержит один или более дополнительных активных фармацевтических ингредиентов.
71. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный активный фармацевтический ингредиент следует вводить в дозе от 0,1 до 100 мг/кг массы тела, как например, от 0,1 до 0,5 мг/кг, например, от 0,5 до 1,0 мг/кг, как например, от 1 до 2 мг/кг, например, от 2 до 5 мг/кг, как например, от 5 до 10 мг/кг, например, от 10 до 15 мг/кг, как например, от 15 до 20 мг/кг, например, от 20 до 30 мг/кг, как например, от 30 до 40 мг/кг, например, от 40 до 50 мг/кг, как например, от 50 до 75 мг/кг, например, от 75 до 100 мг/кг массы тела.
72. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав характеризуется скоростью растворения, при которой 85% активного фармацевтического ингредиента высвобождается в течение 3-5 часов (средняя).
73. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав характеризуется скоростью растворения, при которой 85% активного фармацевтического ингредиента высвобождается спустя ≥6 часов (медленная).
74. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав обеспечивает скорость растворения, при которой от 10 до 90% API высвобождается через 3-5 часов, как например, от 10 до 20, от 20 до 30, от 30 до 40, от 40 до 50, от 50 до 60, от 60 до 70, от 70 до 75, от 75 до 80, от 80 до 85 или от 85 до 90% активного фармацевтического ингредиента высвобождается в течение 3-5 часов, как например, в течение 3 часов, в течение 4 часов или в течение 5 часов.
75. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав обеспечивает скорость растворения, при которой от 10 до 90% API высвобождается в течение 6 часов, как например, от 10 до 20, например, от 20 до 30, как например, от 30 до 40, например, от 40 до 50, как например, от 50 до 60, например, от 60 до 70, как например, от 70 до 75, например, от 75 до 80, как например, от 80 до 85, например, от 85 до 90% активного фармацевтического ингредиента высвобождается в течение ≥6 часов, как например, в течение ≥7 часов, ≥8 часов, ≥9 часов, ≥10 часов, ≥11 часов, ≥12 часов, ≥13 часов, ≥14 часов, ≥15 часов, ≥16 часов, ≥17 часов, ≥18 часов.
76. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав обеспечивает более низкую Стах активного фармацевтического ингредиента по сравнению с составом с немедленным высвобождением активного фармацевтического ингредиента.
77. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав обеспечивает пониженное ингибирование транспортера ОСТ2 активным фармацевтическим ингредиентом по сравнению с составом с немедленным высвобождением активного фармацевтического ингредиента.
78. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав обеспечивает пониженный эффект активного фармацевтического ингредиента в отношении уровня креатинина в сыворотке крови по сравнению с составом с немедленным высвобождением активного фармацевтического ингредиента.
79. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав обеспечивает Cmax менее 20 мкМ, как например, менее 15 мкМ, например, менее 10 мкМ, как например, менее 9 мкМ, например, менее 8 мкМ, как например, менее 7 мкМ, например, менее 6 мкМ, как например, менее 5 мкМ, например, менее 4 мкМ, как например, менее 3 мкМ, например, менее 2 мкМ, как например, менее 1 мкМ.
80. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав обеспечивает Cmax 1-2 мкМ, например, 2-3 мкМ, как например, 3-4 мкМ, например, 4-5 мкМ, как например, 5-6 мкМ, например, 6-7 мкМ, как например, 7-8 мкМ, например, 8-9 мкМ, как например, 9-10 мкМ, например, 10-11 мкМ, как например, 11-12 мкМ, например, 12-13 мкМ, как например, 13-14 мкМ, например, 14-15 мкМ, как например, 15-16 мкМ, например, 16-17 мкМ, как например, 17-18 мкМ, например, 18-19 мкМ, как например, 19-20 мкМ.
81. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав следует вводить один раз в сутки.
82. Фармацевтический состав в соответствии с любым из предыдущих пунктов, причем указанный состав следует вводить в комбинации с одним или несколькими дополнительными активными фармацевтическими ингредиентами раздельно, последовательно или одновременно.
83. Фармацевтический состав в соответствии с любым из предыдущих пунктов для введения индивиду, выбранному из группы, состоящей из пациентов-детей; пациентов, у которых присутствует повышенный уровень креатинина в сыворотке крови; и пациентов, получающих лечение активным фармацевтическим ингредиентом, отличным от активного фармацевтического ингредиента в соответствии с настоящим изобретением.
84. Фармацевтический состав в соответствии с любым из предыдущих пунктов для введения пациенту, имеющему заболевание, выбранное из заболевания почек (нефропатии), в том числе невоспалительной нефропатии (нефроза) и воспалительной нефропатии (нефрита); сахарного диабета типа I и сахарного диабета типа II и артериальной гипертензии.
85. Фармацевтический состав в соответствии с любым из предыдущих пунктов для применения в качестве лекарственного препарата.
86. Фармацевтический состав в соответствии с любым из предыдущих пунктов для применения в способе лечения пациентов-детей, пациентов, у которых присутствует повышенный уровень креатинина в сыворотке крови; и пациентов, получающих лечение активным фармацевтическим ингредиентом, отличным от активного фармацевтического ингредиента
87. Фармацевтический состав в соответствии с любым из предыдущих пунктов для применения в способе лечения заболевания, выбранного из группы, состоящей из детского заболевания; лизосомной болезни накопления (LSD); LSD, выбранной из группы, состоящей из нарушений накопления липидов (или липидоза), в том числе сфинголипидозов, ганглиозидозов и лейкодистрофий; мукополисахаридозов, нарушений накопления гликопротеинов (или гликопротеиноза) и муколипидозов; и бокового амиотрофического склероза (ALS).
ПРИМЕРЫ
Пример 1. In vitro исследования взаимодействия аримокломола с поглощающими транспортерами ОАТР1В1, ОАТР1В3, ОСТ2, ОАТ1 и OAT3 человека
Цель данного изобретения заключалась в получении данных о взаимодействии аримокломола с человеческими SLC (поглощающими) транспортерами: ОАТР1В1 (ОАТР2, ОАТР-С), ОАТР1В3 (ОАТР8), ОАТ1, ОАТ3 и ОСТ2 (Таблица 1).

Краткое изложение результатов
Аримокломол был растворимым до концентрации 300 мкМ в буфере для анализа, используемом для анализов.
Анализы ингибирования поглощающего транспортера
Аримокломол ингибировал ОСТ2-опосредованное накопление маркерного субстрата в применяемых концентрациях (дозозависимо) с максимальным ингибированием при 98,36 (фиг. 5). Расчетная IC50 составляла 9,72 мкМ. Аримокломол не оказывал влияния на ОАТР1В1-, ОАТР1В3-, ОАТ1- и ОАТ3-опосредованное накопление маркерного субстрата при применяемых концентрациях (фиг. 1, фиг. 2, фиг. 3 и фиг. 4).
Анализы субстратов поглощающих транспортеров
Не наблюдали соответствующего кратного уровня накопления аримокломола (кратность изменения накопления составляла <2) в клетках при применяемых концентрациях (1 и 10 мкМ) и в моменты времени (2 и 20 мин.) в экспериментах по изучению доступности субстрата ОАТ1 (фиг. 6), ОАТ3 (фиг. 7) и ОСТ2 (фиг. 8)). Наиболее высокие кратные уровни накопления аримокломола составляли 0,77 для ОАТ1 (1 мкМ и 2 мин.), 0,86 для ОАТ3 (1 мкМ, как спустя 2 мин., так и спустя 20 мин.) и 1,28 для ОСТ2 (1 мкМ и 20 мин.; таблица 6Таблица). Положительные контрольные эксперименты подтверждали функционирование транспортера в применяемых клетках.
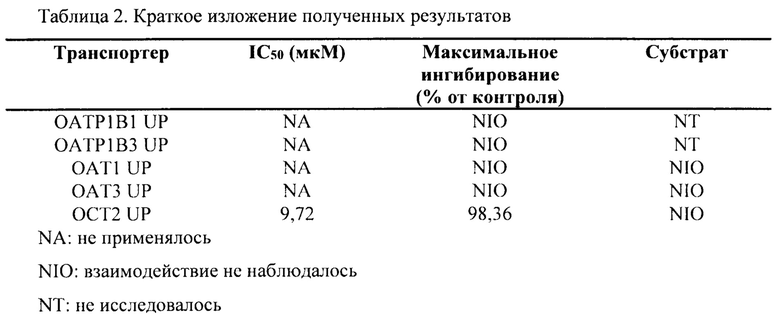
В соответствии с данными аримокломол является ингибитором транспортера ОСТ2.
В соответствии с данными аримокломол является ингибитором транспортеров ОАТР1В1, ОАТР1В3, ОАТ1 и ОАТ3.
В соответствии с данными аримокломол не является субстратом транспортеров ОАТР1В1, ОАТР1В3, ОАТ1, ОАТ3 и ОСТ2.
Материалы и способы
Исследуемые препараты, маточные растворы, химические продукты и инструменты
Исследуемый препарат аримокломола в концентрации 313,7799 г/моль хранили при RT. Растворимость составляет 14 г/100 мл при 25°С (вода) и 0,4 г/100 мл при 25°С (метанол). Маточные растворы (1, 10 и 30 мМ) готовили в воде. Последовательные разведения (7-этапные, специальные) готовили в DMSO, и использовали в качестве исследуемых растворов в разных анализах (100-кратное разведение в анализе поглощения). Кратность разведения в экспериментах с субстратами составляла 1000 раз. Концентрация растворителя в буфере для анализа не превышала 1,1% (об./об.) в остальных анализах.
Инструменты, используемые для выявления, включают хроматограф Thermo Scientific Dionex UltiMate 3000 series UHPLC (Thermo Scientific, Сан-Хосе, Калифорния) с тройным квадрупольным масс-спектрометрическим детектором Thermo Scientific TSQ Quantum Access Max triple quadrupole MS; жидкостный сцинтилляционный счетчик MicroBeta2 (Perkin Elmer, Уолтем, Массачусетс) и многофункциональный микропланшет-ридер BMG Labtech FluoStar Omega (BMG Labtech, Оффенбург, Германия).
Оценка кинетических характеристики растворения в буферах для анализа
Растворимость ТА в воде определяли с помощью спектрофотометрических измерений в сочетании с оценкой с помощью оптической микроскопии (5 и 10 × увеличение). Бесцветные соединения не поглощают свет в видимом диапазоне (400-700 нм), следовательно, когда растворы ТА измеряют с использованием спектрофотометра, скорректированные к фону значения оптического поглощения, которые выше, чем значения оптического поглощения у пустой пробы в этом диапазоне длины волны, указывают на наличие рассеяния света, возможно, вызванного выпавшими в осадок частицами. Временные рамки оценки растворимости охватывали время инкубирования в соответствующем эксперименте in vitro.
Экспериментальный метод для исследования растворимости
Маточный раствор и серии разведений (5-этапная серия разведений в 2 раза) ТА готовили в воде. Маточные растворы смешивали с соответствующими буферами для анализа в 96-луночном планшете и инкубировали в течение 10 минут при 37°С, перед тем как растворы оценивали при длине волны 500 нм. Измеренные значения оптического поглощения для буферных растворов, как правило, составляют от 0,010 до 0,030. Следовательно, чтобы считаться растворимым в заданной концентрации, скорректированное к фону оптическое поглощение раствора ТА должно составлять менее 0,030 единицы оптического поглощения (Δ = Aраствора - Aпустой пробы < 0,030). Определяли скорректированное к фону значение оптического поглощения для каждого раствора.
Анализы ингибирования поглощающих транспортеров и их субстратов
Эксперименты с поглощением проводили с использованием СНО, MDCKII или HEK293 клеток, стабильно экспрессирующих соответствующие поглощающие транспортеры. Параметры анализов поглощающих транспортеров представлены в таблице 3. Информация о контрольных клеточных линиях, культивировании клеток, а также о посеве, кратко представлена в таблице 4.
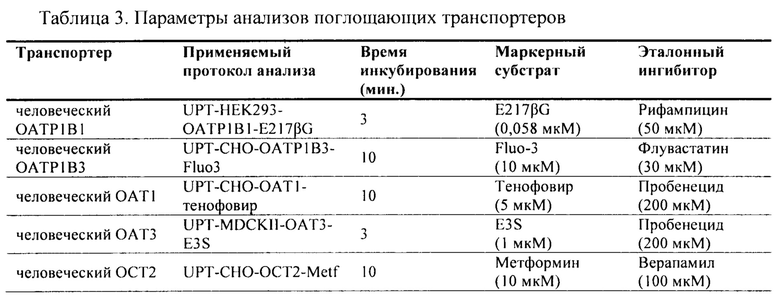
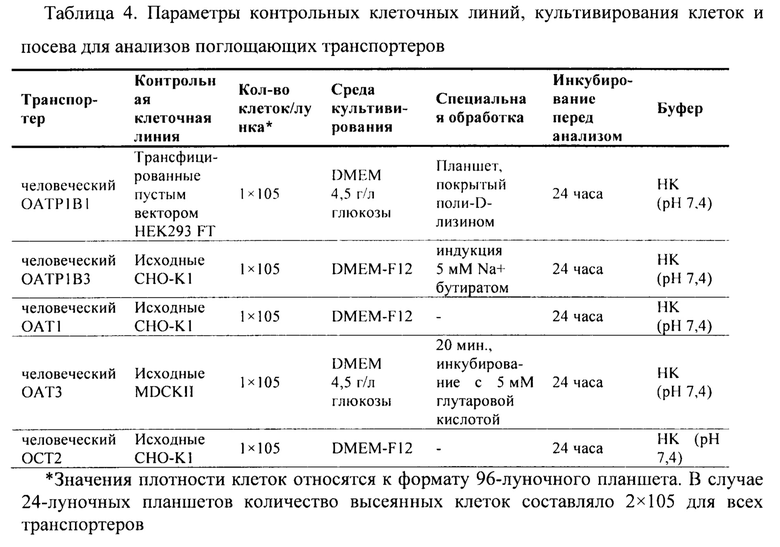
Экспериментальный метод для экспериментов по ингибированию поглощающих транспортеров
Клетки культивировали при 37±1°С в атмосфере воздух : СО2 в соотношении 95:5 и высевали на стандартные 96-луночные планшеты для тканевых культур в количестве клеток, описанном в таблице 4.
Перед экспериментом среду удаляли, и клетки промывали дважды 100 мкл HK буфера с рН 7,4 (приготовлен из химических продуктов Sigma, Sigma-Aldrich, Сент-Луис, Миссури). Эксперименты с поглощением проводили при 37±1°С в 50 мкл HK буфера (рН 7,4), содержащего маркерный субстрат и ТА или контроль с растворителем (воду).
После эксперимента клетки промывали дважды 100 мкл ледяного HK буфера и лизировали с использованием 50 мкл 0,1 М NaOH (1 мм CaCl2 в 5% SDS в случае ОАТР1В3). Транспорт Fluo-3 (ОАТР1В3) определяли посредством измерения флуоресценции с использованием длин волны возбуждения и излучения 485 нм и 520 нм, соответственно. Транспорт меченого радиоактивным изотопом маркерного субстрата определяли путем измерения на аликвоте (35 мкл) из каждой лунки с использованием жидкостно-сцинтилляционного подсчета импульсов.
Поглощение маркерного субстрата в контрольных клетках обеспечивало значения фоновой активности для всех измеряемых параметров. Инкубирование без ТА или эталонного ингибитора (только растворитель) обеспечивало значения 100% активности. Эталонный ингибитор служил в качестве положительного контроля для ингибирования.
Экспериментальный метод для экспериментов с субстратами поглощающих транспортеров
Клетки культивировали при 37±1°С в атмосфере воздух : СО2 в соотношении 95:5, как описано в таблице 4, и высевали на стандартные 24-луночные планшеты для тканевых культур в количестве 2×105 клеток/лунку. Поглощение ТА определяли с использованием клеток, сверхэкспрессирующих соответствующий поглощающий транспортер, и контрольных клеток в два момента времени инкубирования (2 и 20 мин.) и в двух концентрациях (1 и 10 мкМ) ТА для определения, активно ли поглощался ТА в клетки, или нет. Для подтверждения взаимодействия специфическое по отношению к транспортеру поглощение ТА определяли в присутствии известного ингибитора соответствующего транспортера.
Перед экспериментом среду среду удаляли и клетки промывали дважды 300 мкл HK буфера с рН 7,4 (приготовлен из химических продуктов Sigma). Поглощение клетками ТА внутрь клеток измеряли посредством добавления 300 мкл HK буфера, содержащего ТА, и инкубирования их при 37±1°С. Реакции гасили посредством удаления HK буфера, содержащего ТА, и клетки промывали дважды 300 мкл HK буфера. Клетки лизировали путем добавления 300 мкл смеси MeOH : H2O (2:1) и инкубировали в течение 20 минут при 4±1°С. Количество ТА в клеточном лизате определяли с помощью LC-MS/MS (жидкостная хроматография с тандемной масс-спектрометрией). Количество белка в каждой лунке количественно определяли с использованием набора ВСА для определения белка (Sigma-Aldrich, Сент-Луис, Миссури, США). Поглощение маркерного субстрата в контрольных клетках обеспечивало значения фоновой активности для всех измеряемых параметров. Инкубирование без эталонного ингибитора (только растворитель) обеспечивало значения 100% активности. Эталонный ингибитор служил в качестве положительного контроля для ингибирования.
Расчет относительных активностей
Количество перемещенного маркерного субстрата определяли для каждой лунки в cpm (количество импульсов счета в минуту) или RFU (условные единицы флуоресценции). Относительные активности рассчитывали на основании уравнения:

А: количество перемещенного субстрата в присутствии ТА в трансфицированных клетках
В: количество перемещенного субстрата в присутствии ТА в контрольных клетках
С: количество перемещенного субстрата в присутствии растворителя в трансфицированных клетках
D: количество перемещенного субстрата в присутствии растворителя в контрольных клетках
Расчет значений кратности изменения накопления
Значение кратности изменения накопления определяли как соотношение поглощения ТА или маркерного субстрата в трансфицированные и контрольные клетки:
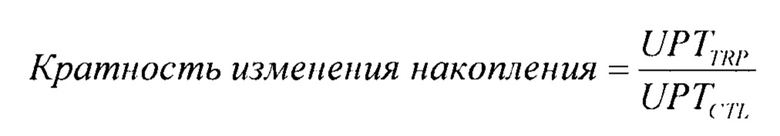
UPTTRP: накопленное количество ТА или маркерного субстрата в трансфицированных клетках, нормализованное к содержанию белка [пмоль/мг белка]
UPTCTL: накопленное количество ТА или маркерного субстрата в контрольных клетках, нормализованное к содержанию белка [пмоль/мг белка]
Если кратность изменения накопления составляет > 2, и оно может ингибироваться известным ингибитором транспортера, ТА может считаться субстратом соответствующего транспортера.
Обработка данных и статистические методы
Microsoft Excel 2010 (Microsoft Corporation, Редмонд, Вашингтон) использовали для обработки исходных данных, a GraphPad Prism 5.0 (GraphPad Software Inc., Сан-Диего, Калифорния) использовали для подбора кривой и определения параметров реакции.
В анализах ингибирования поглощающих транспортеров рассчитывали IC50 (мкМ) при необходимости. IC50 определяли как концентрацию ТА, необходимую для ингибирования максимальной активности на 50%.
Значения IC50 получали из четырехпараметрического логистического уравнения (log(ингибитор) в сравнении с откликом - переменный уклон кривой); подбирали кривую для графика относительной активности в сравнении с концентрацией ТА с использованием нелинейной регрессии. Самое высокое (максимальный отклик) и самое низкое (отклик с максимальным ингибированием) значения не ограничивали постоянными значениями 100 и 0, соответственно, если не указано иное.
Результаты
Результаты анализов поглощающих транспортеров
1) Результаты анализов ингибирования поглощающих транспортеров - см. фиг. 1 (ОАТР1В1), фиг. 2 (ОАТР1В3), фиг. 3 (ОАТ1), фиг. 4 (ОАТ3), фиг. 5 (ОСТ2).
2) Результаты анализов субстратов поглощающих транспортеров - см. фиг. 6 (ОАТ1), фиг. 7 (ОАТ3), фиг. 8 (ОСТ2).
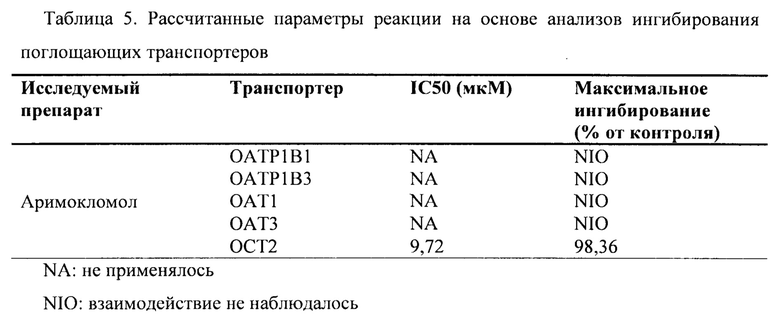
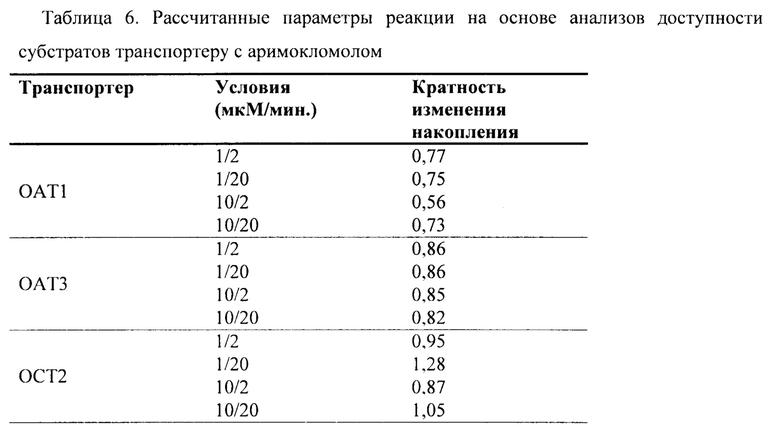
Пример 2. Разработка, производство, изучение высвобождения и исследования стабильности состава с контролируемым высвобождением, содержащего аримокломол
Целью этого исследования была разработка пероральной лекарственной формы для аримокломола, которую легко проглатывать с приемлемыми органолептическими характеристиками и которая характеризуется длительным высвобождением.
Оценивались альтернативные подходы для обеспечения контролируемого высвобождения лекарственного вещества в формате множества частиц, включающем мини-таблетки, содержащие замедлители растворения с покрытием и без него, и нагруженные лекарственным средством бусины или сферы, покрытые слоем для контролируемого высвобождения.
Лекарственное вещество (аримокломол) является белым на вид и является легким и рыхлым. У него проявляются свойства слабой текучести, и он является "липким". Определенная насыпная плотность до уплотнения, составляющая 0,214 г/см3, и значение индекса сжимаемости Карра, составляющее 44,4%, свидетельствовали об очень слабой текучести. Насыпная плотность после уплотнения, составляющая 0,386 г/см3, была определена после 250 встряхиваний.
Мини-таблетки
Исследовалось в общей сложности девять смесей для составов, кратко изложенных ниже:
Мини-таблетки с НРМС: Смеси 1, 2 и 3 содержат изменяющиеся соотношения НРМС и крахмала в матрице
Мини-таблетки с НРМС/ЕС: Смеси 4 и 5 содержат изменяющиеся соотношения НРМС, крахмала и ЕС в матрице
Воскообразная матрица: Получали в общей сложности 4 смеси, содержащие воскообразную основу (глицериндибегенат или глицериндистеарат в концентрации 30% масс. или 40% масс.). При использовании воскообразной основы получение мини-таблеток не было успешным.
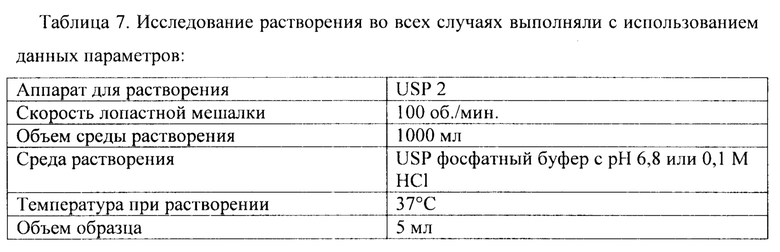
Мини-таблетки с НРМС и НРМС/ЕС
НРМС включали в матрицу мини-таблетки для исследования ее способности задерживать высвобождение лекарственного вещества из мини-таблеток, и, таким образом, она сама по себе действовала в качестве матрицы для контролируемого высвобождения. Все смеси легко засыпались в засыпную воронку для таблетирования, и их успешно подвергали прессованию в 2 мм мини-таблетки, что говорит о подходящей текучести. Не наблюдали видимых признаков налипания или расслоения.
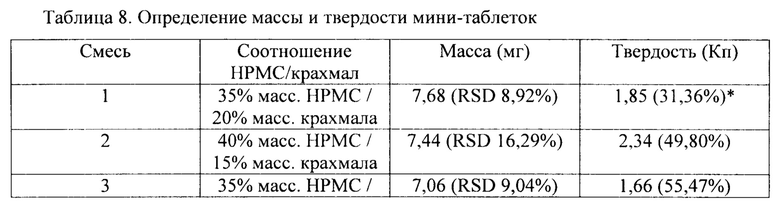

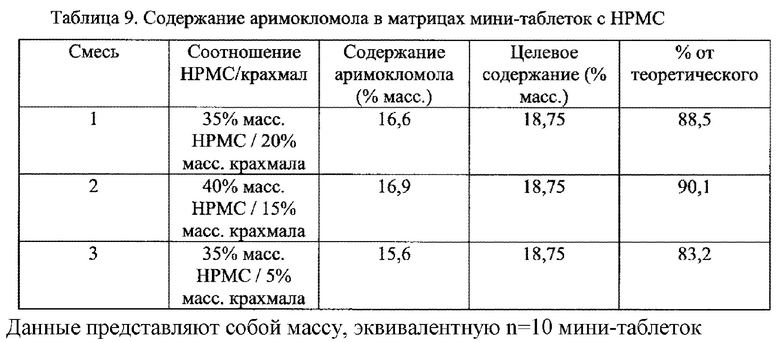
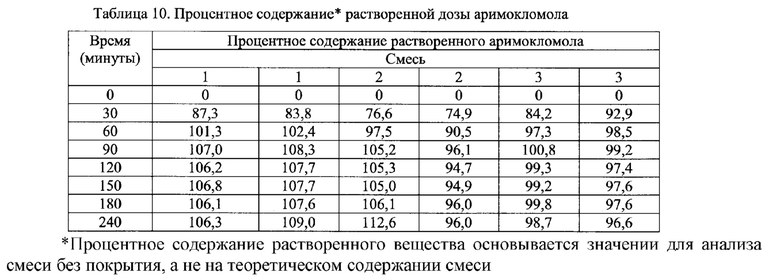
Не присутствует заметного различия в профилях растворения, наблюдаемых у смесей для мини-таблеток с НРМС, и матрица с НРМС сама по себе не работает как матрица для контролируемого высвобождения, поскольку не наблюдалось признаков замедленного высвобождения.
Получали две смеси, содержащие 5% масс. или 10% масс. этилцеллюлозы в матрице мини-таблетки, регулируя концентрацию крахмала в матрице. Обе смеси легко засыпались в засыпную воронку для таблетирования, и их успешно подвергали прессованию в 2 мм мини-таблетки и не наблюдали видимых признаков налипания или расслоения.
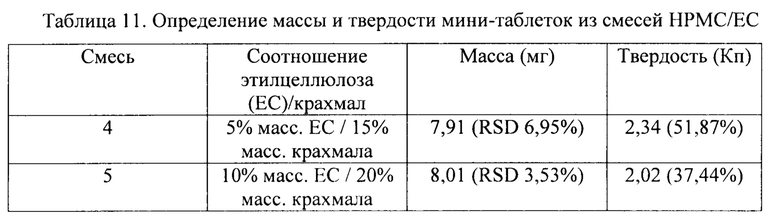
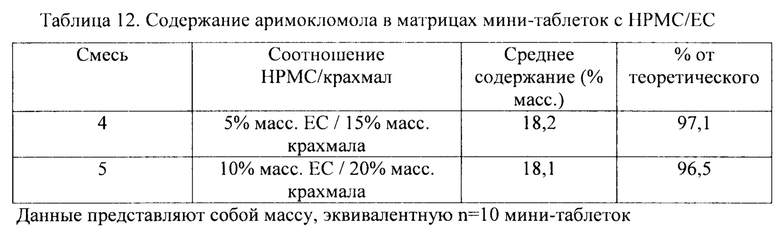

Не присутствует заметного различия в профилях растворения, наблюдаемых у смесей для мини-таблеток с НРМС/ЕС, при отсутствии заметного изменения в профиле высвобождения после включения ЕС независимо от концентрации.
Мини-таблетки с покрытием из НРМС и НРМС/ЕС
Профили растворения от смеси 1 к смеси 4, как было показано, являются сравнимыми; таким образом, испытания с покрытием изначально выполняли на смеси
1. Использовали следующие подходы к нанесению покрытия:
применение дисперсии ЕС на водной основе (SureleaseTM) до увеличения массы на 5%, 10% и 20%;
применение покрытия из ЕС на основе растворителя до увеличения массы на 5%, 10% и 20%;
применение дисперсии полиметакрилата на водной основе (Eudragit NE30D ТМ) до увеличения массы на 5%, 10% и 20%.
На мини-таблетки наносили вручную соответствующее покрытие для контролируемого высвобождения до необходимого увеличения массы. Использовали двухстадийный подход, в связи с этим, исходя из полученных данных о растворении, смеси НРМС/ЕС (смеси 1-4) собирали и наносили в виде покрытия с использованием дисперсии ЕС на водной основе (Surelease ТМ) после изолирующего покрытия до увеличения массы на 5%, 10% и 20%. После нанесения покрытия мини-таблетки подвергали анализу растворения.
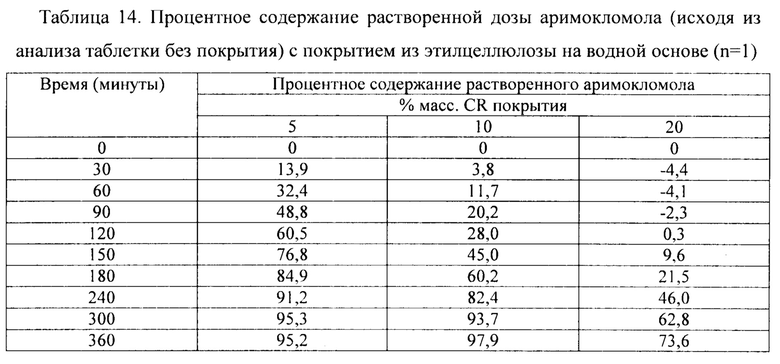
Профили растворения проиллюстрированы на фиг. 9.
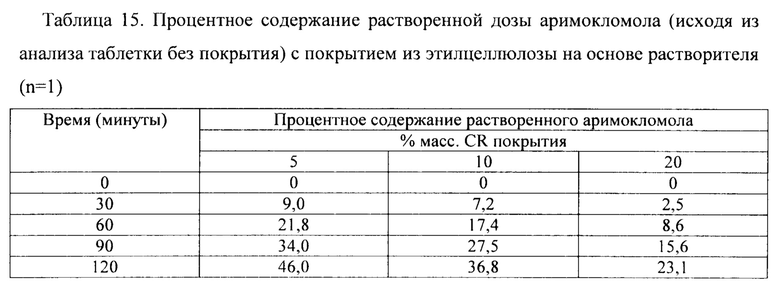

Профили растворения проиллюстрированы на фиг. 10.
Покрытие мини-таблеток с НРМС с использованием ЕС приводило в результате к контролируемому высвобождению лекарственного средства из матрицы, причем большую задержку наблюдали при повышенном увеличении массы. Это не зависит от состава покрывающего раствора/дисперсии, т.е. на водной основе или на основе растворителя. Слой покрытия из ЕС до увеличения массы на 5% масс. приводил в результате к высвобождению 13,9% лекарственного средства спустя 30 минут для покрытия на водной основе по сравнению с высвобождением 9% лекарственного средства для покрытия на основе растворителя. Примерно 85% лекарственного средства высвобождалось через 3 часа после нанесения покрытия до увеличения массы на 5% масс., высвобождение 84,9% лекарственного средства наблюдалось для покрытия ЕС на водной основе по сравнению с 82,2% для покрытия ЕС на основе растворителя.
Большую задержку высвобождения наблюдали после нанесения покрытия до увеличения массы на 10% масс. Через 3 часа высвобождение 60,2% и 69,5% лекарственного средства наблюдали в случае увеличения массы на 10% масс. покрытия ЕС на водной основе и на основе растворителя, соответственно. Нанесение покрытия до увеличения массы на 20% масс. приводило в результате к варьирующим данным между покрытием на водной основе и покрытием на основе растворителя. 73,6% лекарственного средства высвобождалось спустя 6 часов в случае покрытия ЕС на водной основе, значение 96,9% наблюдали в случае покрытия на основе растворителя. Изменения профилей растворения могут объясняться изменениями целостности слоя покрытия: любые трещины или сколы в слое будут обеспечивать возможность высвобождения лекарственного средства из матрицы мини-таблетки.
Покрытие мини-таблеток дисперсией на основе полиметакрилата (Eudragit NE30D) не было успешным, полученные мини-таблетки были мягкими после нанесения покрытия и впоследствии распадались.
Покрытие мини-таблеток с помощью автоматизированного процесса Glatt. На основании данных с нанесением покрытия вручную и данных растворения мини-таблеток без покрытия из НРМС/ЕС смесь 4 и смесь 5 собирали для обеспечения достаточного количества мини-таблеток для обеспечения возможности нанесения покрытия с использованием установки Mini-Glatt, оснащенной набором для работы в микро-формате. Перед покрытием дисперсией ЕС на водной основе (Surelease ТМ) наносили изолирующее покрытие. Это делалось для предотвращения набухания матрицы мини-таблетки во время процесса нанесения покрытия. Изолирующее покрытие (20% масс. НРМС) наносили до увеличения массы на 5%. После изолирующего покрытия наносили покрытие для контролируемого высвобождения (Surelease ТМ) до увеличения массы на 5%, 10% и 20%. Полученные в результате мини-таблетки были объектом исследования растворения в соответствии с параметрами в таблице 7, где иллюстрируется профиль растворения мини-таблеток после нанесения изолирующего покрытия и покрытия для контролируемого высвобождения.
Результаты растворения мини-таблеток с покрытием. Покрытие мини-таблеток из НРМС/ЕС с использованием ЕС приводило в результате к контролируемому высвобождению лекарственного средства из матрицы, причем большую задержку наблюдали при повышенном увеличении массы. Через 60 минут 46,0% лекарственного средства высвобождалось после нанесения покрытия до увеличения массы на 5% масс, в сравнении с 27,1% и 1,1% после нанесения покрытия при увеличении массы с 10% до 20%. Данные представляют собой средние значения из анализов с n=2.
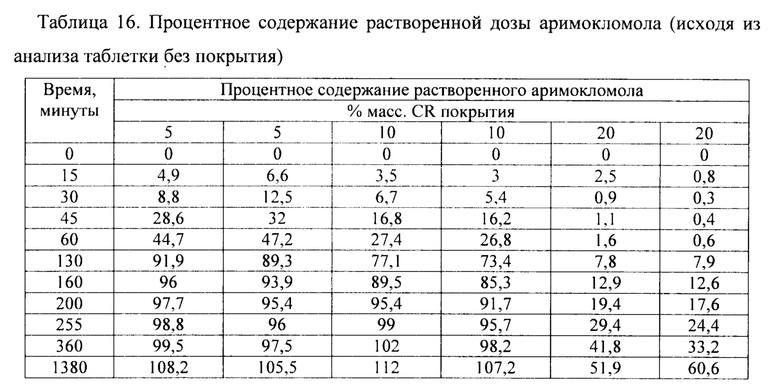
После нанесения покрытия до увеличения массы на 5% 95% лекарственного средства высвобождалось через 3 часа, что было сравнимо с данными, полученными после нанесения покрытия вручную. Сравнимые данные получают после нанесения покрытия до увеличения массы на 10%, причем 93,6% лекарственного средства высвобождаются через 200 минут. Нанесение покрытия до увеличения массы на 20% не приводило в результате к полному высвобождению лекарственного средства за 23 часа, причем высвобождалось 56,3% лекарственного средства. Несмотря на то что нанесение покрытия до увеличения массы на 20%, как представляется, задерживает замедление высвобождения лекарственного средства, у отдельных мини-таблеток в растворяемой партии проявляются отличающиеся свойства. Анализировали в общей сложности 10 мини-таблеток, для 5 из мини-таблеток через слой покрытия проникала среда растворения, приводя в результате к набуханию матрицы из НРМС, образованию трещин в покрытии из ЕС и высвобождению лекарственного средства. В отличие от этого, проникновения через покрытие у остальных мини-таблеток не происходило, а также не происходило набухания ядра таблетки и разрушения покрытия, и не наблюдалось высвобождение лекарственного средства. Исходя из данных, модифицированное исследование растворения выполняли в 250 мл среды растворения, анализируя n=1 мини-таблетку на сосуд для растворения, в общей сложности n=6 образцов оставалось для всех других параметров растворения, как определено в таблице 7.
Краткое изложение результатов - мини-таблетки с покрытием. На основании полученных данных следующие выводы можно сформулировать для мини-таблеток с покрытием: Покрытие мини-таблеток с использованием дисперсии ЕС на водной основе (Surelease ТМ) приводило в результате к замедлению высвобождения лекарственного средства, увеличивающемуся с ростом увеличения массы покрытия до увеличения массы на 10%. Покрытие мини-таблеток с использованием дисперсии полиметакрилата на водной основе (Eudragit NE30D ТМ) не было успешным после нанесения покрытия вручную. Покрытие мини-таблеток с использованием изолирующего покрытия перед дисперсией ЕС на водной основе (SureleaseTM) приводило в результате к замедлению высвобождения лекарственного средства, возрастающему с повышением увеличения массы покрытия. Не наблюдали видимых признаков набухания матрицы мини-таблетки после нанесения на мини-таблетки изолирующего покрытия перед покрытием дисперсией ЕС на водной основе (Surelease ТМ). Данные среднего значения для растворения показывают однородное растворение лекарственного средства, и профиль указывает на целостность покрытия.
Сферы с покрытием
Цель заключалась в получении сфер с покрытием при использовании двух разных ядер (из сахара и микрокристаллической целлюлозы) и разных материалов покрытия (водная дисперсия этилцеллюлозы, дисперсия этилцеллюлозы на основе растворителя и водная дисперсия полиакрилата) с рядом профилей растворения. Два типа сфер рассматривали в качестве основы для нанесения слоя лекарственного средства; растворимые сахарные сферы (Suglets ТМ) и нерастворимые сферы из микрокристаллической целлюлозы (Vivapur ТМ).
Сахарные сферы размером 1000/1180 мкм использовали для выполнения работы по разработке основной массы составов для сфер, причем выполняли следующие исследования:
Покрытие сахарных сфер с использованием покрытия для контролируемого высвобождения из этилцеллюлозы на неводной основе и на водной основе (эксперимент шесть);
Нанесение покрытия на сахарные сферы с использованием покрытия из водной дисперсии на основе полиакрилата (Eudragit E30D ТМ) (эксперимент восемь).
Данные для сахарных сфер оценивали и, исходя из данных, на сферы из микрокристаллической целлюлозы (размером 710-1000 мкм) наносили покрытие с использованием подхода, который предусматривает контролируемое высвобождение. Их использовали по той причине, что сфера является основой для обеспечения возможности нанесения слоя лекарственного средства, а также потому, что она не оказывает воздействия на характеристики контролируемого высвобождения лекарственного средств. Таким образом, на сферы из микрокристаллической целлюлозы наносили покрытие с использованием только покрытия для контролируемого высвобождения из этилцеллюлозы на водной основе.
Используемый процесс нанесения покрытия на сферу являлся одинаковым для обоих вариантов сфер и всех растворов/дисперсий для покрытий для контролируемого высвобождения. Он представляет собой многостадийный процесс (см. также фиг. 13). Состав сферы, используемый для всех составов с целью достижения нагрузки лекарственным средством, составляющей 4% масс. (10 мг на дозу), описан в таблице ниже. Покрытие для контролируемого высвобождения вначале наносили слоем до 5% масс., после чего образец удаляли и нанесение покрытия до увеличения массы на 20% масс. осуществляли до окончательного состава продукта 100% масс. Конечная концентрация, выраженная в мг/единицу, будет более ниже, так как пленочное покрытие не наносили.
Следующие растворы/дисперсии покрытия готовили перед процессом нанесения покрытия:
Раствор для слоя с лекарственным средством: Готовили 100 г раствора, содержащего 4% масс. лекарственного средства и 5% масс. НРМС.
Изолирующее покрытие: Готовили 100 г дисперсии, содержащей от 3% масс. до 5% масс. твердых веществ покрытия, в соответствии с инструкциями производителя.
Раствор для контролируемого высвобождения: Готовили 100 г раствора, содержащего 15% масс. твердых веществ покрытия (Surelease Е-7-19040ТМ), путем добавления 60 г SureleaseTM к 100 г воды.
Пленочное покрытие: получали также, как и изолирующее покрытие.
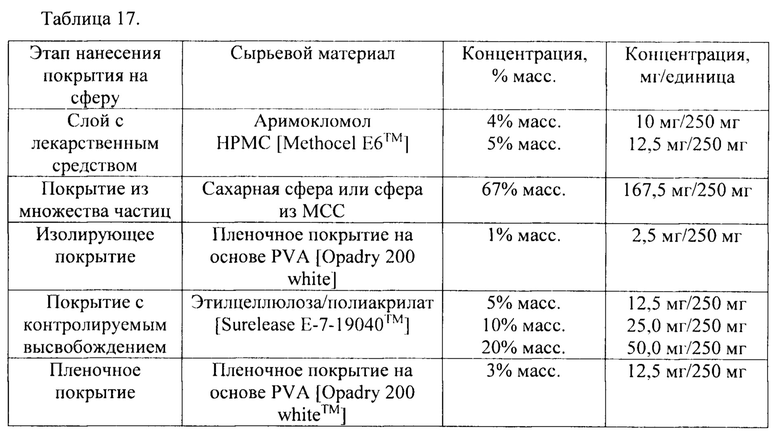
На сферы наносили покрытие путем распыления снизу с использованием установки Mini-Glatt, оснащенной набором для работы в микро-формате и колонной Вурстера, с использованием 0,5 мкм распылительной форсунки. Сферы подогревали в течение 10 минут перед добавлением раствора для получения слоя с лекарственным средством до достижения увеличения массы на 14% масс. с температурой воздуха на входе 50°С - 60°С, давлением воздуха на входе 0,55 бар, давлением воздуха при распылении 0,72 бар и скоростью доставки текучей среды для покрытия, возрастающей от 0,42 г/мин. вначале до 0,94 г/мин после нанесения достаточного покрытия. После чего наносили изолирующее покрытие до достижения увеличения массы на 1% масс., применяя вышеупомянутые параметры в сравнимом соотношении. Покрытие для контролируемого высвобождения наносили до увеличения массы на 5% масс., применяя те же параметры нанесения покрытия, образец сфер удаляли и дополнительный раствор для получения покрытия наносили до достижения увеличения массы на 10% масс. и 20% масс. Заключительное пленочное покрытие наносили на сферы с покрытием для контролируемого высвобождения до увеличения массы на 20% масс., применяя лишь такие же описанные параметры.
Оценка бусин с покрытием: подход с использованием этилцеллюлозы
Для исследований использовали коммерчески доступную водную дисперсию ЕС (Surelease™ Е-7-19040), а не подход на основе растворителя. ЕС наносили до достижения увеличения массы в диапазоне 5% масс., 10% масс. и 20% масс., применяя описанный способ. Этот подход к нанесению покрытия применяли для сахарных сфер и сфер из МСС, тем не менее, нанесение покрытия до увеличения массы на 20% масс. не выполняли при использовании сфер из МСС вследствие ограниченной доступности.
Исследование растворения сахарных сфер показывает замедленное высвобождение после нанесения покрытия для контролируемого высвобождения из ЕС, причем большую задержку наблюдали с повышенной концентрацией покрытия (фиг. 14). Это не зависело от используемой среды растворения. После нанесения покрытия до увеличения массы на 5% масс. 53,7% лекарственного средства высвобождались спустя 4 часа при применении буфера с рН 6,8 по сравнению с высвобождением 46,9% при использовании 0,1 М HCl (фиг. 14 и фиг. 15).
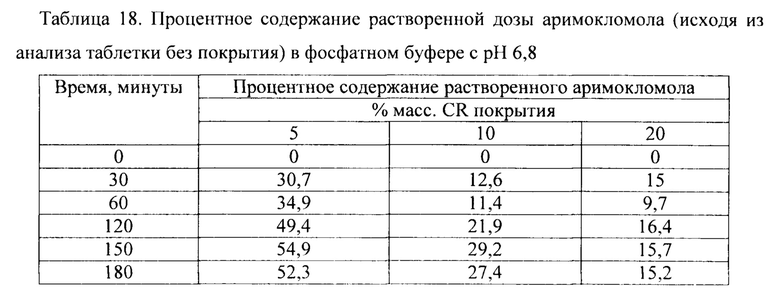
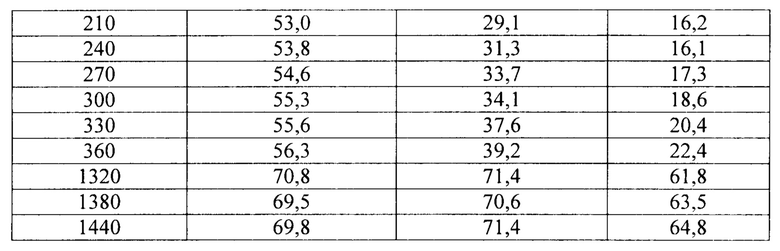
Сравнимые данные получали после нанесения покрытия до увеличения массы на 10% масс. Не было признаков, указывающих на сброс дозы при использовании 0,1 М HCl, и на основании полученных данных, среду растворения с рН 6,8 следует использовать для всех дальнейших исследований. См. фиг. 15.
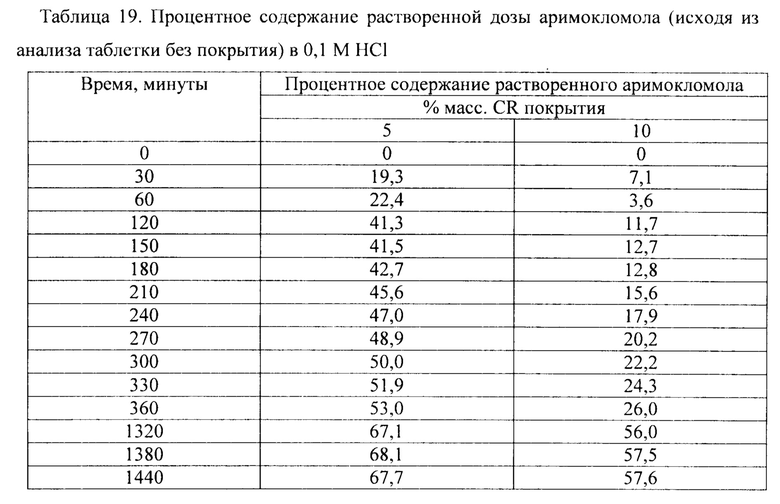
Также наблюдали замедленное высвобождение лекарственного средства из сфер из МСС (фиг. 16), причем большая задержка наблюдалась с увеличением толщины покрытия для контролируемого высвобождения, наносимого в количестве от 5% масс. до 10% масс. После нанесения покрытия до увеличения массы на 5% масс. 56,4% лекарственного средства высвобождалось спустя 4  часа, причем 100,4% лекарственного средства высвобождалось после 23 часов. В отличие от этого, 33,5% лекарственного средства высвобождалось спустя 4
часа, причем 100,4% лекарственного средства высвобождалось после 23 часов. В отличие от этого, 33,5% лекарственного средства высвобождалось спустя 4  часа, причем высвобождение 82,5% лекарственного средства наблюдали после 23 часов (фиг. 15).
часа, причем высвобождение 82,5% лекарственного средства наблюдали после 23 часов (фиг. 15).
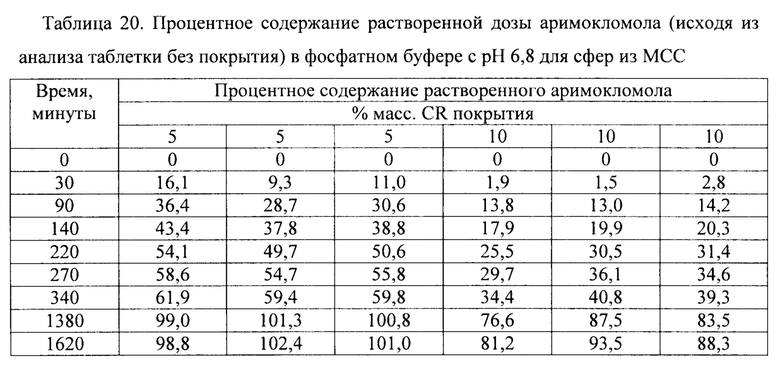
См. фиг. 16.
Исходя из полученных данных, оказывается, что существует различие в скоростях высвобождения, наблюдаемое при сравнении сахарных сфер со сферами из МСС, причем у сфер из МСС проявляется более медленное высвобождение. Сферы из МСС проявляют меньший размер, 710-1000 мкм, по сравнению с сахарными сферами, 1000-1100 мкм, следовательно, у них большая площадь поверхности, а значит, ожидается более быстрое высвобождение.
На основании данных следующие выводы можно сделать для покрытия для контролируемого высвобождения из ЕС: Сахарные сферы и сферы из микрокристаллической целлюлозы успешно нагружали лекарственным средством. Покрытие сфер слоем с 5% этилцеллюлозы приводило в результате к высвобождению 53,8% лекарственного средства спустя 4 часа для сахарных сфер и к высвобождению 56,4% лекарственного средства спустя 4  часа для сфер из МСС. Покрытие сфер слоем с 10% этилцеллюлозы приводило в результате к высвобождению 31,3% лекарственного средства спустя 4 часа для сахарных сфер и к высвобождению 33,5% лекарственного средства спустя 4
часа для сфер из МСС. Покрытие сфер слоем с 10% этилцеллюлозы приводило в результате к высвобождению 31,3% лекарственного средства спустя 4 часа для сахарных сфер и к высвобождению 33,5% лекарственного средства спустя 4  часа для сфер из МСС. Данные говорят о том, что сфера с покрытием обеспечивает подходящий подход для контролируемого высвобождения.
часа для сфер из МСС. Данные говорят о том, что сфера с покрытием обеспечивает подходящий подход для контролируемого высвобождения.
Оценка бусин с покрытием: подход с использованием ЕС/НРМС и подход с использованием поли(мет)акрилата. Оценку можно выполнить, как изложено выше для подхода с использованием ЕС.
Пример 3. НМЕ-гранулы с длительным высвобождением аримокломола
Оценка экструзии горячего расплава (НМЕ) для варьирования скоростей растворения и высвобождения для составов с длительным высвобождением аримокломола.
Цель: разработка состава с длительным высвобождением аримокломола в Compritol® 888, а также оценка практических аспектов включения больших объемов аримокломола в матрицу Compritol® 888 и получения порошков с разными размерами частиц, которые можно применять для наполнения капсул, или саше. В процессе изучения будет оцениваться воздействие нагрузки лекарственным средством и размера частиц на профили растворения лекарственного средства.
Составы аримокломол/Compritol® 888 с 33, 50 и 66 масс. % аримокломола успешно получали с помощью экструзии горячего расплава и подвергали измельчению с получением дискретных распределений размера посредством просеивания через сито. Частицы, полученные таким образом, не демонстрировали наблюдаемого разрушения.
Гранулы с размерами частиц в диапазонах 710-500, 1000-710 и ≥1000 мкм подвергали исследованию растворения в фосфатном буферном растворе с рН 6,8, и они демонстрировали различные профили длительного высвобождения аримокломола. Скорости высвобождения, как оказалось, зависят как от размера частиц, так и от концентрации аримокломола.
Последующий анализ подвергнутых растворению частиц с помощью SEM (сканирующая электронная микроскопия) продемонстрировал образование пористой структуры, что согласуется с механизмом высвобождения за счет образования извилистого пути, образующегося при растворении аримокломола, оставляющего после себя нерастворимую в воде матрицу Compritol® 888.
Получение составов с помощью экструзии горячего расплава
Составы с аримокломолом в количестве 33, 50 и 66 масс. %, активные в Compritol® 888 (С888), были намечены для получения с помощью экструзии горячего расплава с использованием экструдера Thermo Scientific Pharma 11 с двумя шнеками, вращающимися в одном направлении, для получения твердых продуктов из смесей порошков аримокломол/С888.
Композиции для составов для НМЕ подробно в таблице 21 (композиции для составов в виде партий из 50 г материала, подлежащего экструзии)
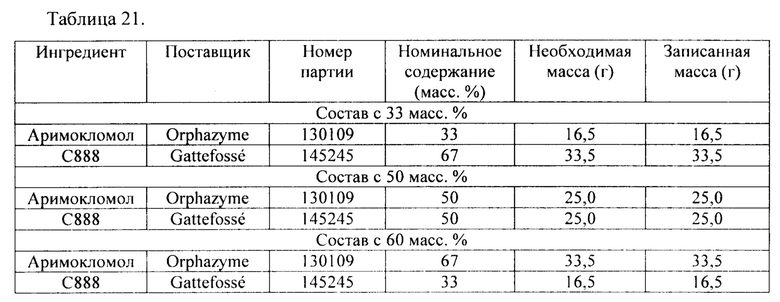
Выбирали номинальную скорость подачи сырьевого материала в экструдер, составляющую 1,4-1,6 г/мин. Скорость экструдирующего шнека, составляющую 100 об./мин., выбирали на основании предшествующих данных и литературы. Профиль нагревания экструдера устанавливали в конфигурацию, подробно описанную в таблице 22 (Конфигурация нагревания экструдера горячего расплава для получения всех экструдированных смесей).

Следующие выходные параметры при обработке наблюдали для процесса экструзии у всех трех смесей:
- давление расплава составляло 0-8 бар
- температура расплава составляла 67-69°С
- крутящий момент инструмента составлял 11-12%
Экструдированные лекарственные формы во всех концентрациях были мягкими/пластичными непосредственно на выходе из головки экструдера горячего расплава, а при охлаждении до комнатной температура представляли собой белые, воскообразные и крошащиеся твердые вещества. Из экструдера собирали полоски различной длины, варьирующей от 5 до 20 см.
Полоски, полученные посредством экструзии горячего расплава, помещали в маленький ручной блендер и подвергали воздействию 5 х 1 -секундных размалывающих импульсов, разделенных 5-секундными периодами охлаждения. Измельченный материал затем высыпали на колонну сит, состоящую из следующих сит: 1000 мкм, 710 мкм и 500 мкм. Они были ориентированы в вертикальном направлении, причем сита располагались от самого крупного в верхней части колонны к самому мелкому внизу колонны. Металлическую пластину для сбора помещали под нижнее сито для сбора частиц размером менее 500 мкм. Ориентируя сита таким образом, получали следующие ситовые фракции:<500 мкм, 710-500 мкм, 1000-710 мкм и ≥1000 мкм. Значения выхода этих ситовых фракций представлены ниже в таблице 23.
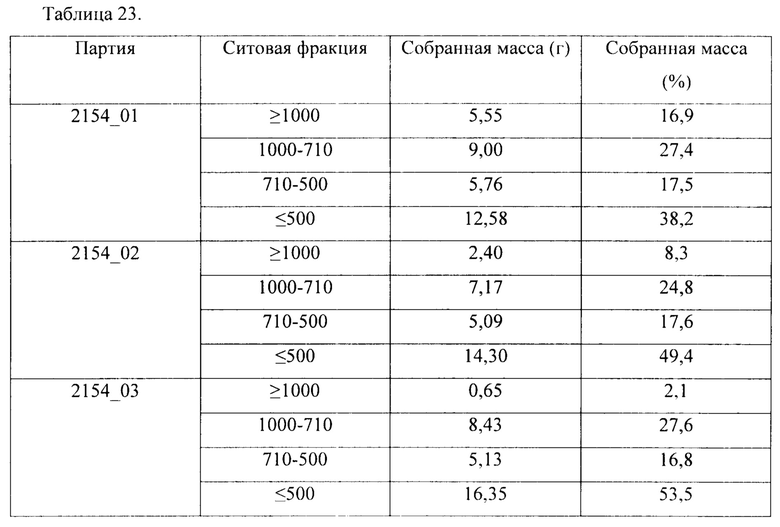
По мере того как возрастало содержание аримокломола, так же возрастала фракция тонкоизмельченного материала (ниже 500 мкм), что сопровождалось снижением количества более крупного материала (выше 1000 мкм).
Анализ сопутствующих веществ проводили на частицах среднего размера (1000-710 мкм) для оценки любого разрушения аримокломола, которое произошло в ходе обработки с получением гранул, его результаты включены ниже в таблице 24.
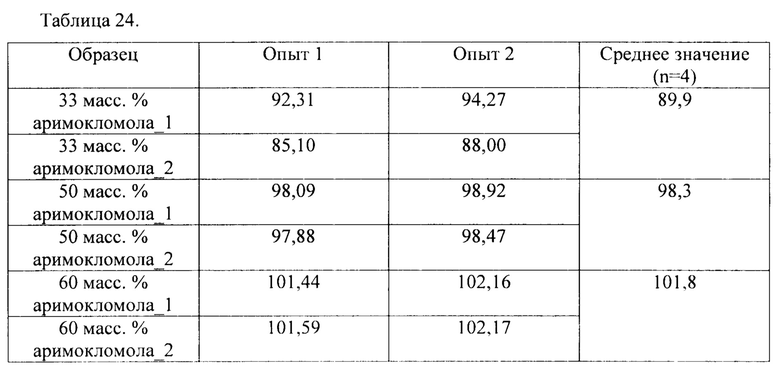
Не наблюдали сопутствующих веществ в количестве, большем чем 0,05% площади образца, в любом из анализируемых образцов.
IR микроскопия гранул. Инфракрасную (IR) микроскопию использовали для того, чтобы охарактеризовать поверхность гранул. Усредненные спектры записывали у 5 гранул для гранул с 33 масс. % (710-500 мкм), гранул с 50 масс. % (710-500 мкм) и гранул с 66 масс. % (710-500 мкм). IR спектроскопия продемонстрировала, что во всех случаях поверхность гранул представляла собой преимущественно С888, что говорило о том, что в процессе экструзии горячего расплава было достигнуто хорошее покрытие аримокломола.
По мере того как возрастало количество аримокломола в составе, так же возрастало количество аримокломола, наблюдаемого на поверхности состава, что демонстрировалось повышением интенсивности соответствующего пика (примерно 1590 см-1). Это согласуется с уменьшенным покрытием аримокломола С888 по мере того, как содержание аримокломола повышается, последствиями чего, вероятно, является повышение наблюдаемых скоростей растворения из нерастворимой в воде матрицы.
SEM-анализ размолотых образцов до и после растворения. Сканирующую электронную микроскопию (SEM) осуществляли на превращенных в порошок материалах до и после растворения для составов во всех трех концентрациях и для всех диапазонов размеров частиц 710-500 мкм, 1000-710 мкм и ≥1000 мкм, за исключением фракции<500 мкм.
Во всех случаях эти данные демонстрировали, что размолотый экструдат присутствовал преимущественно в виде непрерывной волнистой поверхности с отдельными угловатыми частицами, которые были как погруженными, так и располагались на поверхности, как правило, эти частицы связаны с присутствием кристаллического аримокломола.
Сканирующую электронную микроскопию (SEM) проводили на гранулах после того, как их подвергали экспериментам с растворением. Во всех случаях угловатые частицы (кристаллический аримокломол) растворялся с поверхности гранулы, оставляя пористую нерастворимую в воде матрицу.
Оказалось, что гранулы становятся более пористыми с повышением содержания аримокломола. Это говорит о том, что аримокломол способен растворяться из матрицы С888 с образованием извилистого пути, что приводило к длительному высвобождению аримокломола.
Результаты растворения. Все гранулы подвергали экспериментам с растворением. Наблюдали, что начальная скорость растворения аримокломола возрастала с повышением нагрузки аримокломолом в масс. %, причем 66 масс. % показывают наибольшую начальную скорость растворения, а 33 масс. % показывают самую медленную начальную скорость растворения. Также можно увидеть, что скорость растворения зависит от фракции частиц по размеру, причем фракция с размером частиц 500-710 мкм показывает наибольшую скорость растворения, а фракция с размером частиц ≥1000 мкм показывает самую медленную скорость растворения. См. фиг. 17.
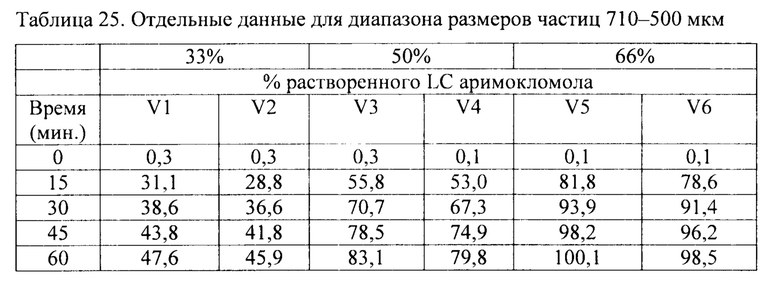

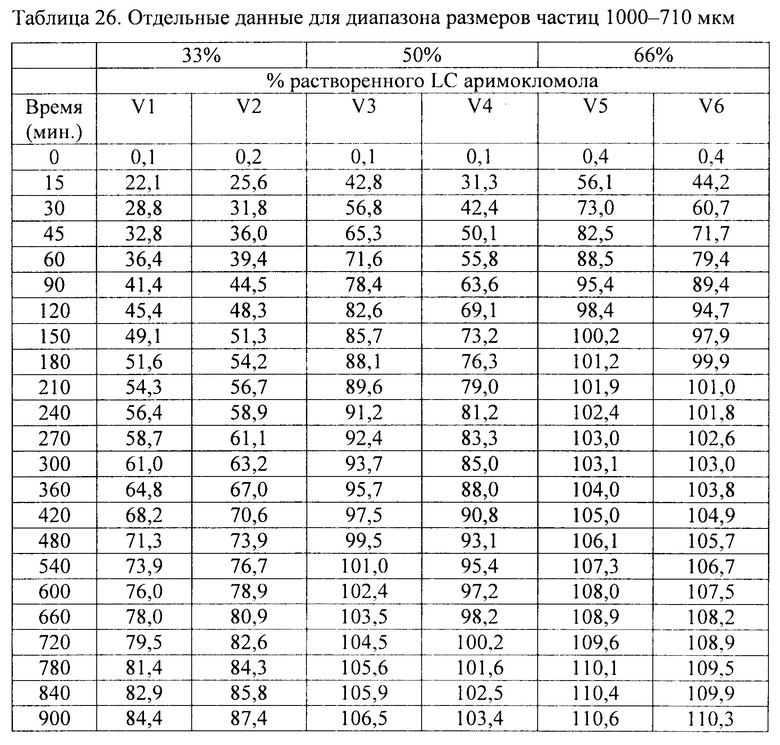
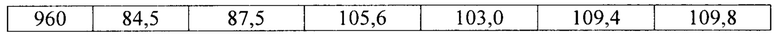
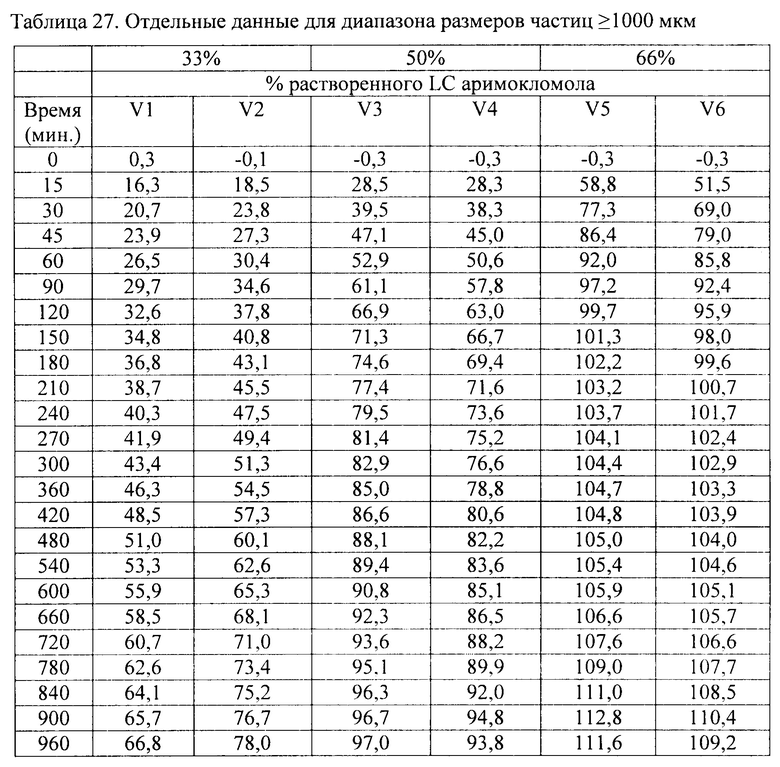
Пример 4. In vivo исследование состава с контролируемым высвобождением аримокломола
Состав с модифицированным высвобождением в соответствии с настоящим изобретением изучают на самцах карликовых свиней геттингенской породы в РО PK исследованиях (фармакокинетические исследования при пероральном приеме исследуемого лекарственного средства) с 1 исследуемым препаратом (аримокломол) в 5-факторном перекрестном исследовании: одна группа с заданным составом и 4 с модифицированными составами.
Исследования на живых организмах проводят в Charles River laboratories, Великобритания, UK, их осуществляли без соблюдения стандартов GLP. Уровни дозы нуждаются в подтверждении.
N=2 карликовых свиньи на условие (в общей сложности 10 карликовых свиней), самцы карликовых свиней геттингенской породы. Животных не кормили перед исследованием. Временные точки РО: 0 (перед введением дозы), 25 мин., 45 мин., 1, 2, 4, 6, 8, 12 и 24 часов (10 образцов крови).
Плазму крови получают посредством центрифугирования так скоро, насколько это практически возможно после сбора, и полученную в результате плазму крови хранят примерно при -80°С до отправки XenoGesis Ltd. на сухом льду для количественного биологического анализа в соответствии с отдельным протоколом.
Выполняют количественный биологический анализ с помощью LC-MS/MS всех образцов плазмы крови из исследования (100 исследуемых образцов, а также анализ дозы, если необходимо, и холостые пробы). Образцы плазмы крови получают посредством осаждения белка. Отдельные точные взвешивания (до 0,01 мг) каждого соединения для STD и QC.
Средние значения Cmax и AUC можно определить с использованием традиционных способов, см., например, Cudkowicz et al., Muscle & Nerve, July 2008, p. 837-844).
AUC, Tmax и Cmax можно определить, как описано, например, в патентном документе ЕР 2481400 В1.
| название | год | авторы | номер документа |
|---|---|---|---|
| АРИМОКЛОМОЛ ДЛЯ ЛЕЧЕНИЯ АССОЦИИРОВАННЫХ С ГЛЮКОЦЕРЕБРОЗИДАЗОЙ НАРУШЕНИЙ | 2017 |
|
RU2750154C2 |
| ПЕРОРАЛЬНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПОДХОДЯЩАЯ ДЛЯ ПОВЫШЕНИЯ ЭФФЕКТИВНОСТИ ЛЕЧЕНИЯ ДВИГАТЕЛЬНЫХ НАРУШЕНИЙ | 2013 |
|
RU2670272C2 |
| ЛЕКАРСТВЕННЫЕ ФОРМЫ ИНГИБИТОРА ГИСТОНДИАЦЕТИЛАЗЫ В КОМБИНАЦИИ С БЕНДАМУТИНОМ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2609833C2 |
| ЛЕКАРСТВЕННЫЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ГИПРОМЕЛЛОЗНЫЕ МАТРИЦЫ КОНТРОЛИРУЕМОГО ВЫСВОБОЖДЕНИЯ | 2006 |
|
RU2414241C2 |
| ТЕХНОЛОГИЯ ПРИГОТОВЛЕНИЯ ТАБЛЕТОК ДЛЯ CGRP-АКТИВНЫХ СОЕДИНЕНИЙ | 2015 |
|
RU2696578C1 |
| Твёрдая фармацевтическая композиция для изготовления перорального терапевтического средства для профилактики и/или лечения ВИЧ-инфекции | 2021 |
|
RU2759544C1 |
| ТВЕРДЫЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОР ИНТЕГРАЗЫ | 2010 |
|
RU2602865C2 |
| КОМПОЗИЦИИ С НЕМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ ЛЕКАРСТВЕННОГО СРЕДСТВА | 2010 |
|
RU2541807C2 |
| ПРОИЗВОДНЫЕ КАРБОКСАМИДИНА И ИХ ИСПОЛЬЗОВАНИЕ ПРИ ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ СОСУДОВ | 2003 |
|
RU2320330C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАПОЛНЕННОГО КОНТЕЙНЕРА ДЛЯ ИНГАЛЯТОРА | 2011 |
|
RU2562017C2 |
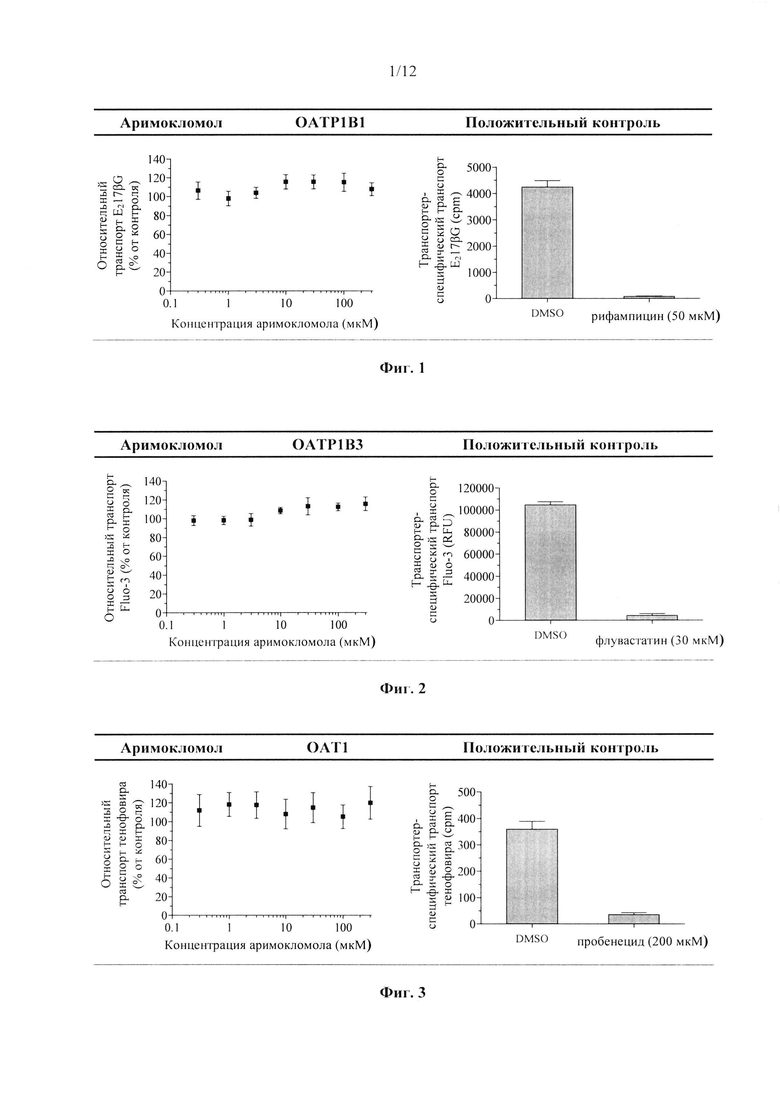
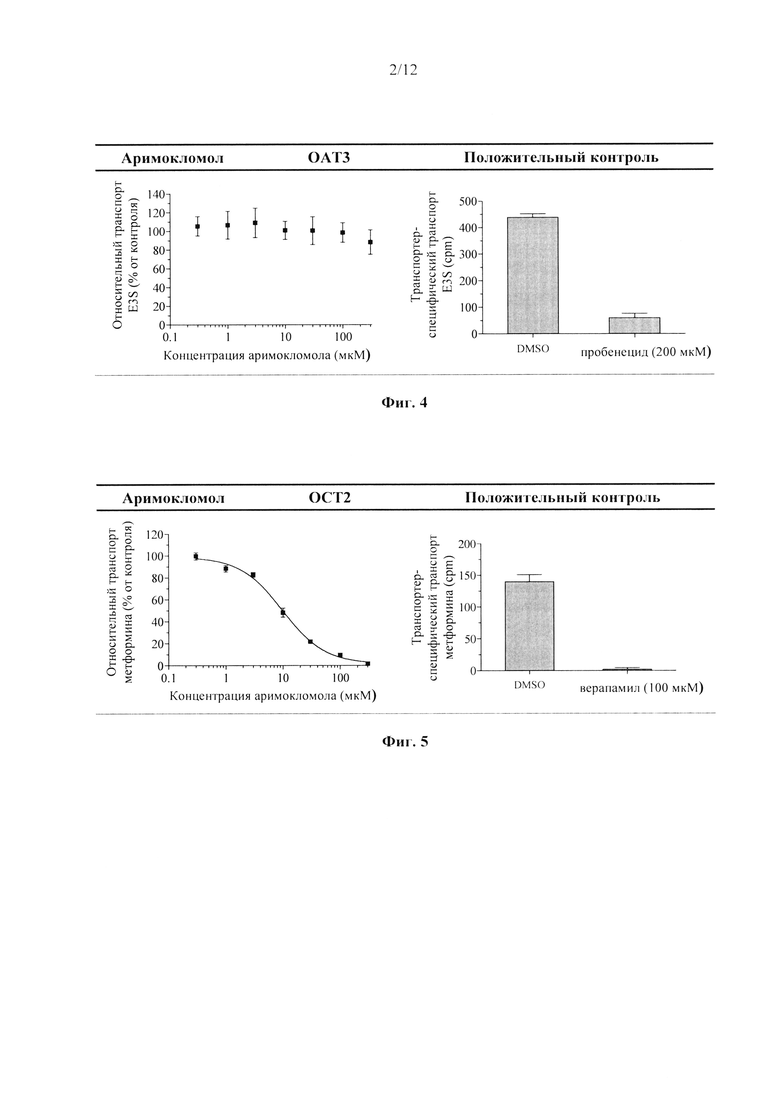

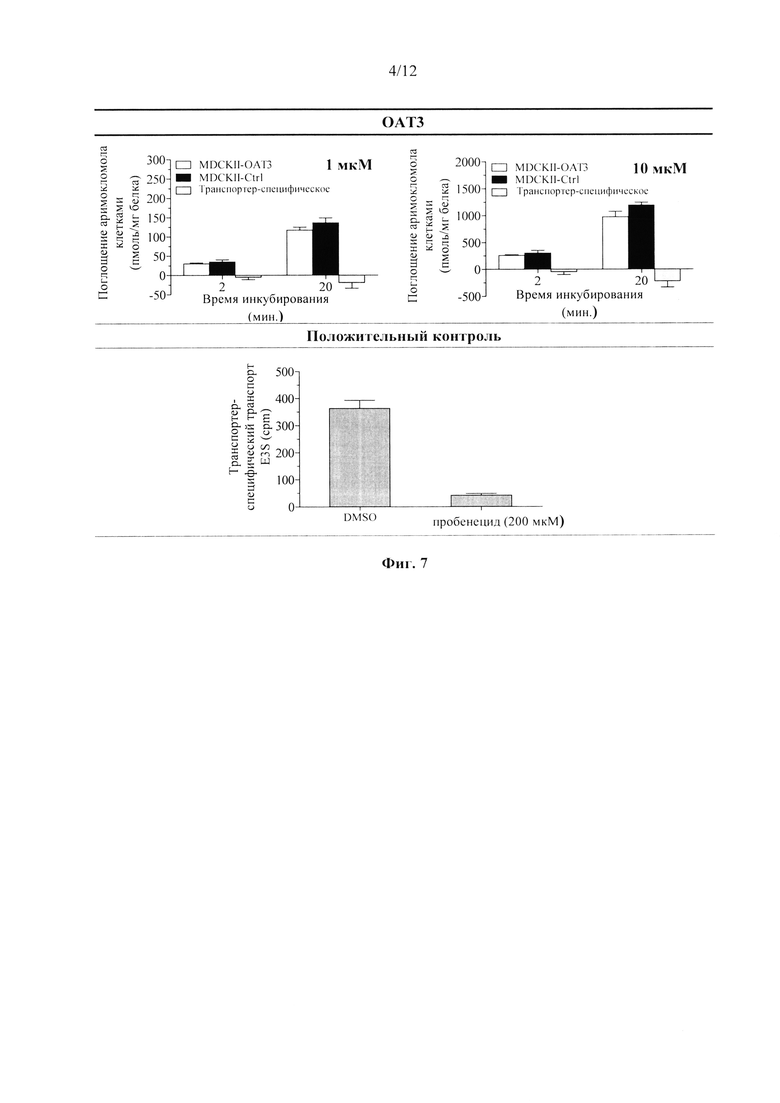

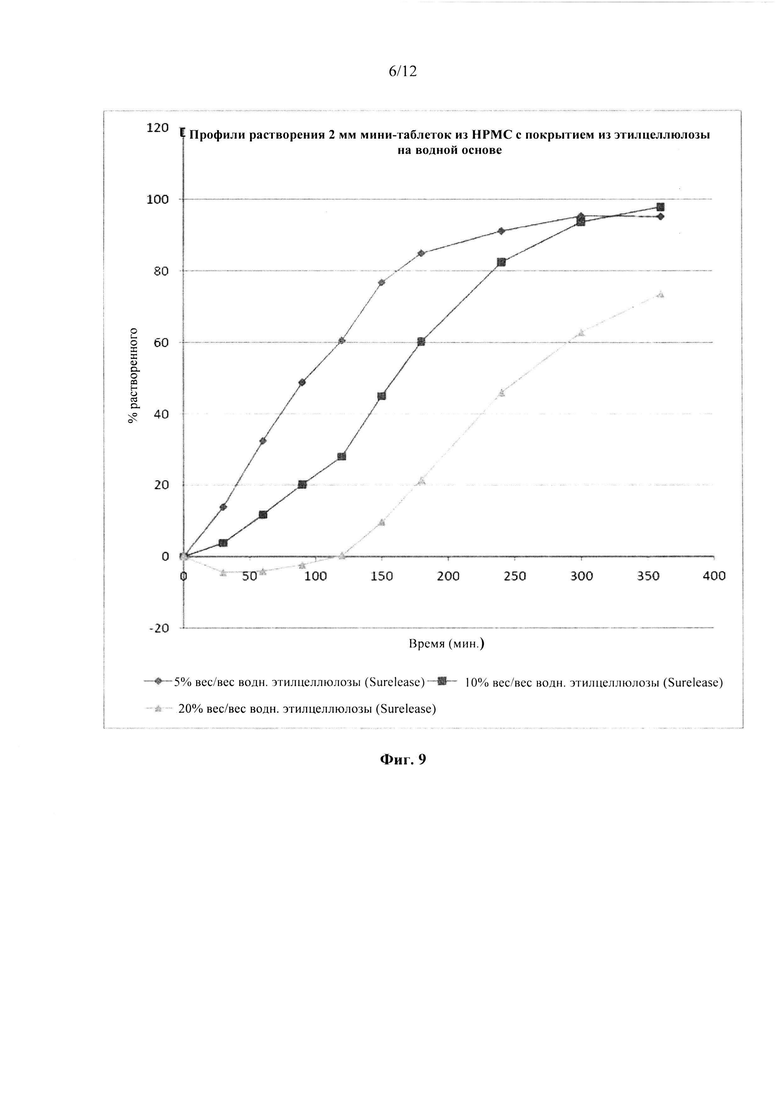

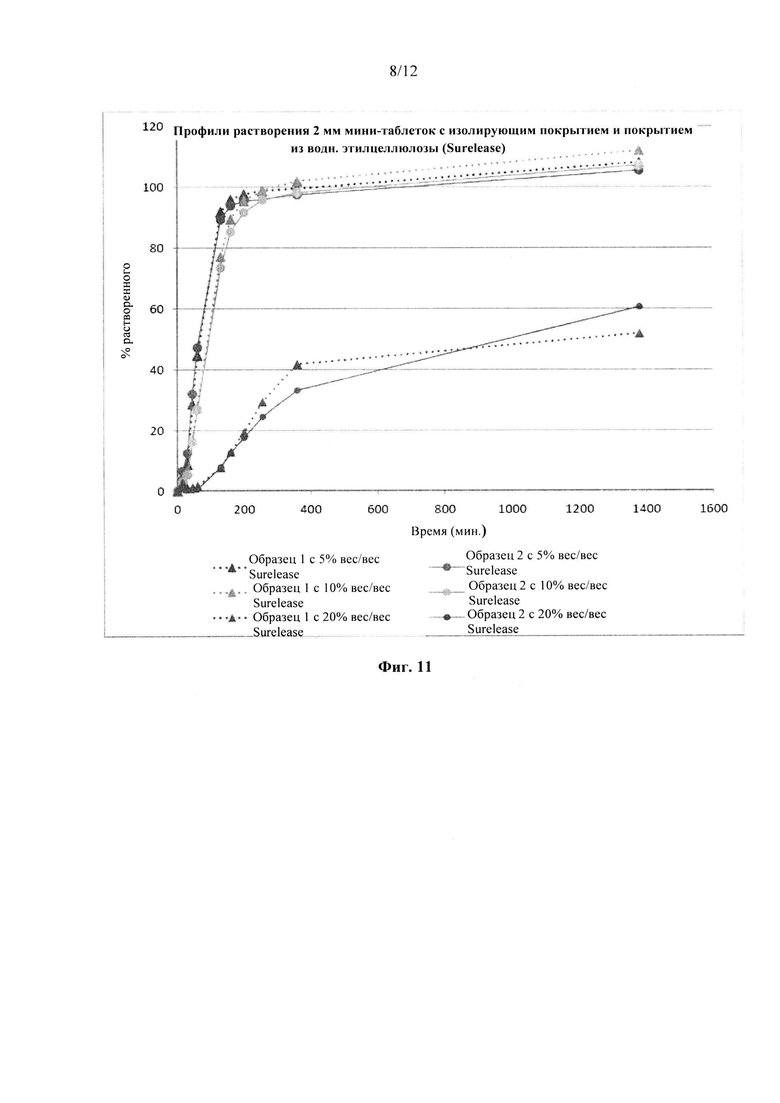
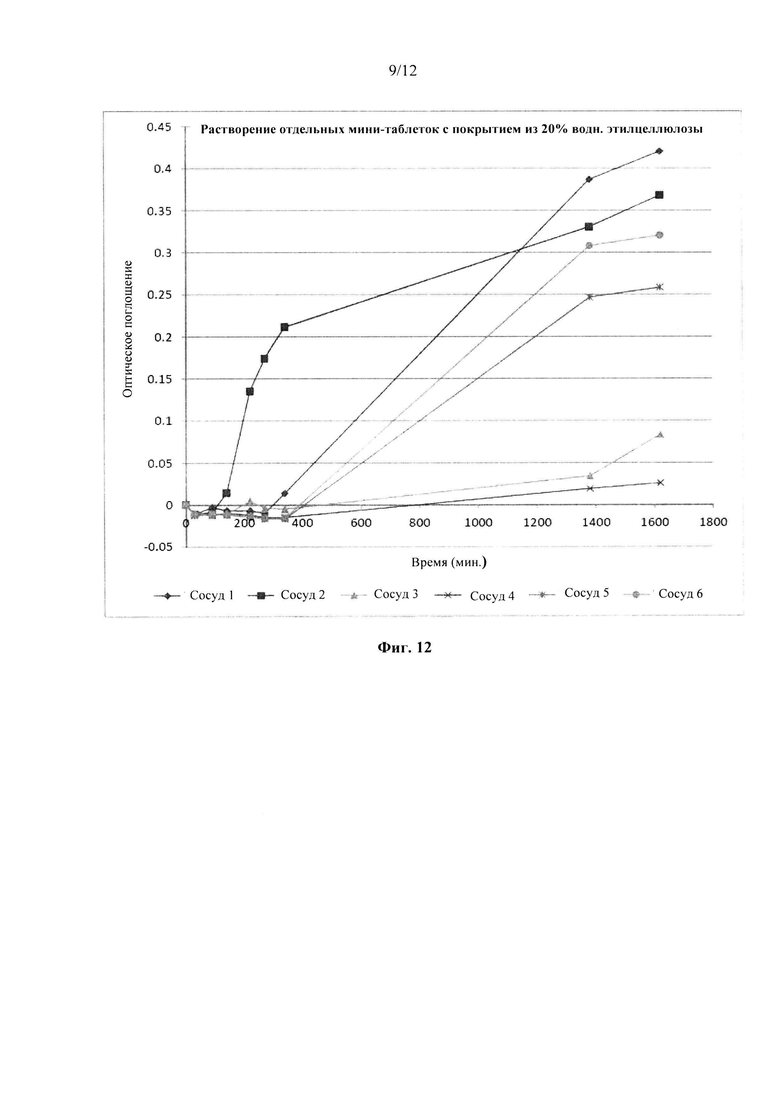
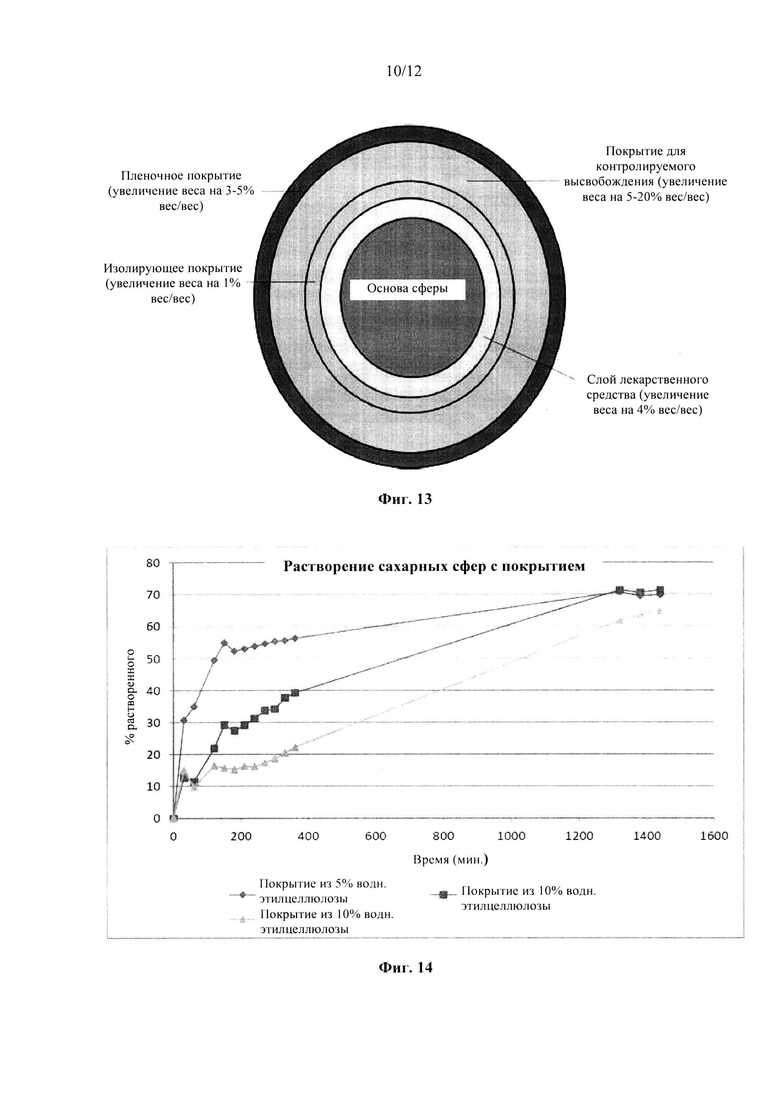
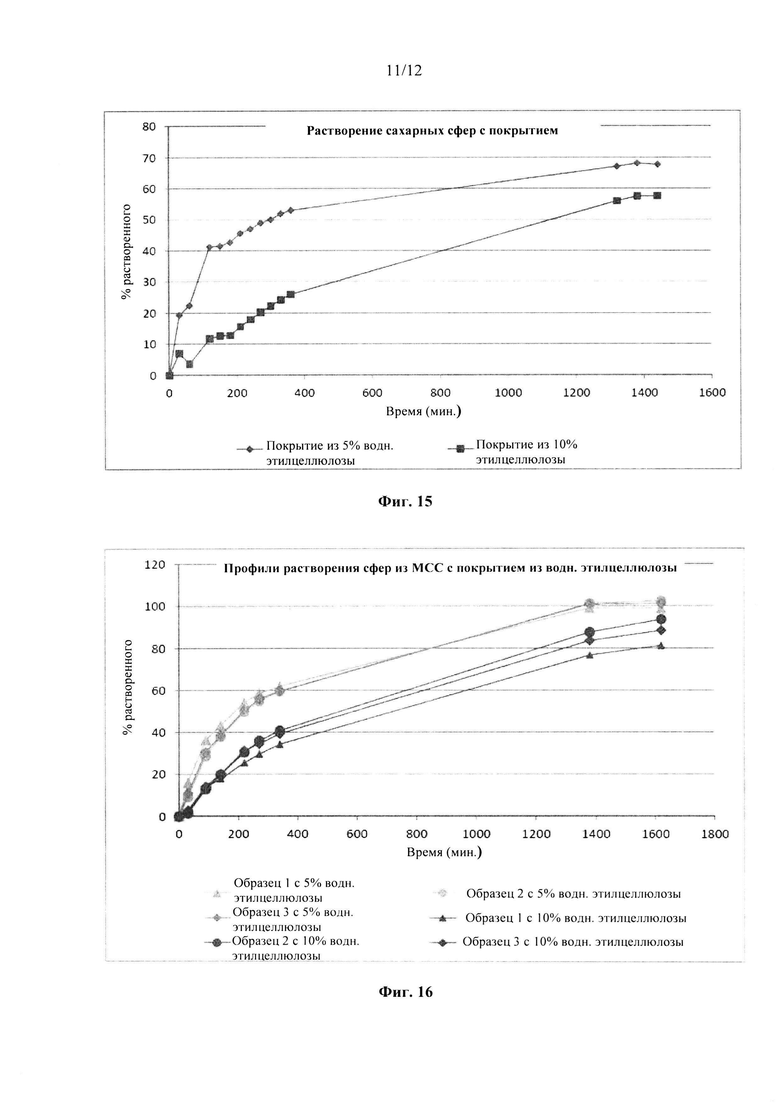
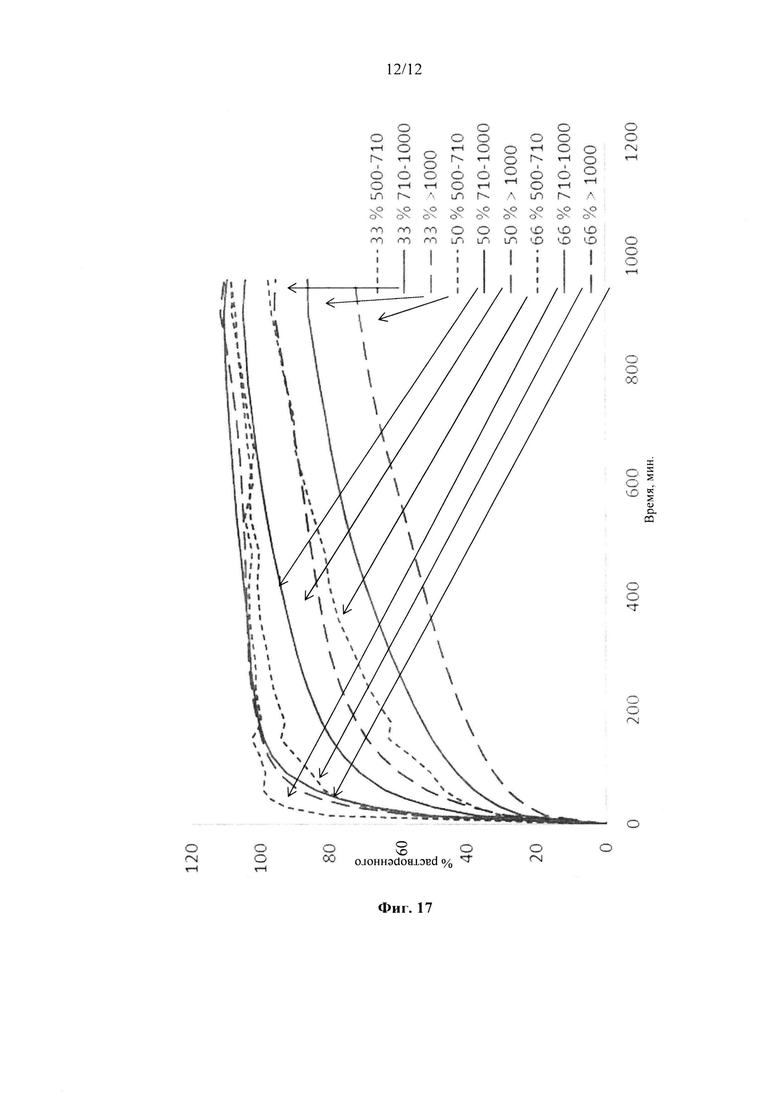
Изобретение относится к фармацевтической промышленности, а именно к составу на основе N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты. Фармацевтический состав с длительным высвобождением в пероральной лекарственной форме, содержащий: a. активный фармацевтический ингредиент, выбранный из N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, и b. контролирующее высвобождение вспомогательное вещество, причем указанное контролирующее высвобождение вспомогательное вещество обеспечивает длительное высвобождение указанного активного фармацевтического ингредиента, и указанный состав снижает ингибирование ОСТ2 по сравнению с эквивалентным количеством N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, вводимых с помощью пероральной лекарственной формы с немедленным высвобождением и/или с помощью болюсной IV инъекции, и где активный фармацевтический ингредиент присутствует в одной лекарственной форме или единице состава в общем количестве 5-1000 мг на дозу, причем указанный состав обеспечивает Сmax, меньшую или равную 10 мкМ. Вышеописанный состав эффективно снижает ингибирование ОСТ2 по сравнению с эквивалентным количеством N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, вводимых с помощью пероральной лекарственной формы с немедленным высвобождением и/или с помощью болюсной IV инъекции. 45 з.п. ф-лы, 27 табл., 4 пр., 17 ил.
1. Фармацевтический состав с длительным высвобождением в пероральной лекарственной форме, содержащий:
a. активный фармацевтический ингредиент, выбранный из N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, и
b. контролирующее высвобождение вспомогательное вещество,
причем указанное контролирующее высвобождение вспомогательное вещество обеспечивает длительное высвобождение указанного активного фармацевтического ингредиента, и
указанный состав снижает ингибирование ОСТ2 по сравнению с эквивалентным количеством N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, вводимых с помощью пероральной лекарственной формы с немедленным высвобождением и/или с помощью болюсной IV инъекции, и где активный фармацевтический ингредиент присутствует в одной лекарственной форме или единице состава в общем количестве 5-1000 мг на дозу,
причем указанный состав обеспечивает Cmax, меньшую или равную 10 мкМ.
2. Фармацевтический состав по п. 1, у которого Cmax понижена на величину по меньшей мере 10%, как например на величину по меньшей мере 20%, как например на величину по меньшей мере 30%, как например на величину по меньшей мере 40%, как например на величину по меньшей мере 50%, как например на величину по меньшей мере 60%, как например на величину по меньшей мере 70%, как например на величину по меньшей мере 80%, как например на величину по меньшей мере 90%, как например на величину по меньшей мере 100%, по сравнению с Cmax для их эквивалентного количества, вводимого с помощью пероральной лекарственной формы с немедленным высвобождением.
3. Фармацевтический состав по п. 1, у которого Cmax понижена при сохранении воздействия и/или AUC активного фармацевтического ингредиента.
4. Фармацевтический состав по п. 1, у которого Cmax понижена в сравнении с общим обеспечиваемым воздействием, что выражается площадью под кривой (AUC).
5. Фармацевтический состав по п. 1, у которого Cmax ниже, чем полумаксимальная ингибирующая концентрация (IC50) активного фармацевтического ингредиента для ОСТ2.
6. Фармацевтический состав по п. 1, у которого Tmax повышена на величину 10-20%, как например 20-30%, как например 30-40%, как например 40-50%, как например 50-60%, как например 60-70%, как например 70-80%, как например 80-90%, как например 90-100%, как например 100-125%, как например 125-150%, как например 150-200%, как например 200-250%, по сравнению с указанными величинами для их эквивалентного количества, вводимого с помощью пероральной лекарственной формы с немедленным высвобождением.
7. Фармацевтический состав по п. 1, у которого ингибирование ОСТ2 понижено на величину по меньшей мере 10%, как например на величину по меньшей мере 20%, как например на величину по меньшей мере 30%, как например на величину по меньшей мере 40%, как например на величину по меньшей мере 50%, как например на величину по меньшей мере 60%, как например на величину по меньшей мере 70%, как например на величину по меньшей мере 80%, как например на величину по меньшей мере 90%, как например на величину по меньшей мере 100%, по сравнению с указанными величинами для их эквивалентного количества, вводимого с помощью пероральной лекарственной формы с немедленным высвобождением.
8. Фармацевтический состав по п. 1, причем указанный состав следует вводить один раз в сутки или два раза в сутки.
9. Фармацевтический состав по п. 1, причем указанный состав характеризуется скоростью растворения, при которой 85% активного фармацевтического ингредиента высвобождается в течение 3-5 часов.
10. Фармацевтический состав по п. 1, причем указанный состав характеризуется скоростью растворения, при которой 85% активного фармацевтического ингредиента высвобождается спустя по меньшей мере 6 часов.
11. Фармацевтический состав по п. 1, причем указанный состав представляет собой лекарственную форму, такую как перорально доступная лекарственная форма.
12. Фармацевтический состав по п. 1, причем указанное контролирующее высвобождение вспомогательное вещество контролирует скорость высвобождения указанного активного фармацевтического ингредиента из указанного состава.
13. Фармацевтический состав по п. 1, причем указанный состав содержит внутреннюю матрицу и по меньшей мере одно наружное покрытие, при этом указанная внутренняя матрица содержит активный фармацевтический ингредиент.
14. Фармацевтический состав по п. 13, причем указанный состав выбран из группы, состоящей из таблетки с покрытием, мини-таблетки с покрытием и микро-таблетки с покрытием.
15. Фармацевтический состав по п. 14, причем указанное контролирующее высвобождение вспомогательное вещество выбрано из группы, состоящей из гидроксипропилметилцеллюлозы (НРМС), этилцеллюлозы (ЕС), метилцеллюлозы, гидроксипропилцеллюлозы, гипромеллозы ацетата сукцината, фталата гипромеллозы, ацетата целлюлозы, глицеринмоностеарата, глицерилмоноолеата, глицерилпальмитата, глицерилбегената, гидрирогенизированного растительного масла, гуаровой камеди, поливинилового спирта, альгинатов, ксантановой камеди, карнаубского воска, желтого воска, белого воска, зеина, каррагенана, карбомеров и агара.
16. Фармацевтический состав по п. 13, причем указанная внутренняя матрица дополнительно содержит одно или более дополнительных вспомогательных веществ, выбранных из группы, состоящей из наполнителей, связующих, смазывающих средств, ионных полимеров, неионных полимеров и нерастворимых в воде полимеров.
17. Фармацевтический состав по п. 13, причем указанная внутренняя матрица дополнительно содержит микрокристаллическую целлюлозу (МСС), такую как Avicel РН 101 или Avicel РН 102.
18. Фармацевтический состав по п. 13, причем указанная внутренняя матрица содержит гидроксипропилметилцеллюлозу (НРМС), крахмал, этилцеллюлозу (ЕС), микрокристаллическую целлюлозу (МСС), диоксид кремния, стеарат магния и стеариновую кислоту.
19. Фармацевтический состав по п. 13, причем указанная внутренняя матрица подвергается прессованию с образованием таблетки, такой как таблетка с твердостью от 10 до 50 Кп (килопонд), как например от 10 до 15 Кп, например от 15 до 20 Кп, как например от 20 до 25 Кп, например от 25 до 30 Кп, как например от 30 до 35 Кп, например от 35 до 40 Кп, как например от 40 до 50 Кп.
20. Фармацевтический состав по п. 13, причем указанное наружное покрытие содержит одно или более вспомогательных веществ, таких как этилцеллюлоза (ЕС) на водной основе, ЕС на основе растворителя или полиметакрилат на водной основе.
21. Фармацевтический состав по п. 13, причем указанное наружное покрытие дополнительно содержит наружное изолирующее покрытие.
22. Фармацевтический состав по п. 1, причем указанный состав представляет собой сферу с покрытием, содержащую внутреннюю основу сферы и наружное покрытие, содержащее один или более отдельных слоев, при этом указанное наружное покрытие содержит активный фармацевтический ингредиент.
23. Фармацевтический состав по п. 22, причем указанное наружное покрытие содержит два или более отдельных слоев, как например три или более слоев, как например четыре или более слоев, как например пять или более отдельных слоев.
24. Фармацевтический состав по п. 23, причем первый или самый внутренний слой наружного покрытия содержит активный фармацевтический ингредиент.
25. Фармацевтический состав по любому из пп. 23, 24, содержащий: 1) основу сферы, 2) слой лекарственного средства, содержащий активный фармацевтический ингредиент, 3) необязательно изолирующее покрытие, 4) покрытие для контролируемого высвобождения и 5) необязательно пленочное покрытие.
26. Фармацевтический состав по п. 25, причем указанная основа сферы содержит или состоит из сахара, как например растворимая сахарная сфера.
27. Фармацевтический состав по п. 25, причем указанная основа сферы представляет собой нерастворимую сферу из микрокристаллической целлюлозы или растворимую сахарную сферу.
28. Фармацевтический состав по п. 25, причем указанный слой лекарственного средства содержит активный фармацевтический ингредиент и вспомогательное вещество, такое как НРМС.
29. Фармацевтический состав по п. 25, причем указанное покрытие для контролируемого высвобождения содержит или состоит из этилцеллюлозы (ЕС) на водной основе, этилцеллюлозы (ЕС) на неводной основе или водной дисперсии на основе полиакрилата.
30. Фармацевтический состав по п. 1, причем указанный состав находится в форме гранул с длительным высвобождением, содержащих N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорид, его стереоизомеры и их соли присоединения кислоты и контролирующее высвобождение вспомогательное вещество.
31. Фармацевтический состав по п. 30, причем указанное контролирующее высвобождение вспомогательное вещество выбрано из группы, состоящей из вспомогательного вещества для экструзии горячего расплава (НМЕ) (полимера для НМЕ), вспомогательного вещества для НМЕ с высокой способностью к солюбилизации, расплавленного липидного вспомогательного вещества, липидного вспомогательного вещества, липидной матрицы для длительного высвобождения, расплавленного покровного средства для составов с пролонгированным высвобождением лекарственного средства, глицеринбегената, глицериндибегената и смесей разных сложных эфиров бегеновой кислоты с глицерином.
32. Фармацевтический состав по п. 31, причем указанное вспомогательное вещество для НМЕ характеризуется температурой плавления приблизительно 70°С, как например 50-55°С, как например 55-60°С, как например 60-65°С, как например 65-70°С, как например 70-75°С.
33. Фармацевтический состав по п. 30, причем указанные гранулы с длительным высвобождением получены или могут быть получены с помощью экструзии горячего расплава, предусматривающей стадии:
a. смешивания API, выбранного из N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида, его стереоизомеров и их солей присоединения кислоты, и вспомогательного вещества для НМЕ;
b. нагревания и экструзии указанных API и вспомогательного вещества для НМЕ с обеспечением экструдата, содержащего указанный API и вспомогательное вещество для НМЕ;
c. воздействия измельчением на указанный экструдат, как например с помощью размола, и необязательно фракционирования по размеру, как например с помощью просеивания.
34. Фармацевтический состав по п. 33, причем температура при экструзии составляет приблизительно 60-65°С, как например 65-70°С, как например 70-75°С, например 60-61°С, как например 61-62°С, как например 62-63°С, как например 63-64°С, как например 64-65°С, как например 65-66°С, как например 66-67°С, как например 67-68°С, как например 68-69°С, как например 69-70°С, как например 70-71°С.
35. Фармацевтический состав по любому из пп. 33, 34, причем давление экструзии составляет 0-8 бар, как например 0-1 бар, как например 1-2 бар, как например 2-3 бар, как например 3-4 бар, как например 4-5 бар, как например 5-6 бар, как например 6-7 бар, как например 7-8 бар.
36. Фармацевтический состав по п. 30, причем указанные гранулы с длительным высвобождением характеризуются размером частиц 500-710 мкм, 710-1000 мкм или более 1000 мкм.
37. Фармацевтический состав по п. 30, причем указанные гранулы с длительным высвобождением содержат 25-75 масс. % API, как например 30-65 масс. % API, как например 30-50 масс. % API.
38. Фармацевтический состав по п. 30, причем указанные гранулы с длительным высвобождением содержат 20-60 масс. % API, как например 25-50 масс. % API, и характеризуются размером частиц более 710 мкм, как например более 1000 мкм.
39. Фармацевтический состав по любому из пп. 1, 14-21, 23, 24, 26-29, 31-34, 36-38, причем указанный состав представляет собой пероральную лекарственную форму с множеством единиц.
40. Фармацевтический состав по любому из пп. 1, 14-21, 23, 24, 26-29, 31-34, 36-38, причем указанный состав содержится в капсуле, такой как капсула с твердой оболочкой, как например капсула, содержащая желатин.
41. Фармацевтический состав по любому из пп. 1, 14-21, 23, 24, 26-29, 31-34, 36-38, причем указанный состав содержится в пакетике или саше.
42. Фармацевтический состав по любому из пп. 1, 14-21, 23, 24, 26-29, 31-34, 36-38, причем указанный активный фармацевтический ингредиент выбран из группы, состоящей из:
a. рацемата N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида,
b. оптически активного стереоизомера N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида,
c. энантиомера N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида,
d. (+)-R-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида,
e. (-)-(S)-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида,
f. соли присоединения кислоты N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида,
g. N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида цитрата,
h. N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида малеата,
i. (+)-R-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида цитрата;
j. (-)-S-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида цитрата;
k. (+)-R-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида малеата и
l. (-)-S-N-[2-гидрокси-3-(1-пиперидинил)-пропокси]-пиридин-1-оксид-3-карбоксимидоил-хлорида малеата.
43. Фармацевтический состав по любому из пп. 1, 14-21, 23, 24, 26-29, 31-34, 36-38, причем указанный состав содержит указанный активный фармацевтический ингредиент в количестве от 0,1 мг до 100 мг, например от 0,1 до 0,5 мг, как например от 0,5 до 1 мг, например от 1 до 2 мг, как например от 2 до 3 мг, например от 3 до 4 мг, как например от 4 до 5 мг, например от 5 до 7,5 мг, как например от 7,5 до 10 мг, например от 10 до 15 мг, как например от 15 до 20 мг, например от 20 до 30 мг, как например от 30 до 40 мг, например от 40 до 50 мг, как например от 50 до 60 мг на дозу, например от 60 до 70 мг, как например от 70 до 80 мг, например от 80 до 90 мг, как например от 90 до 100 мг.
44. Фармацевтический состав по любому из пп. 1, 14-21, 23, 24, 26-29, 31-34, 36-38, причем указанный состав вводят в количестве от 75 до 1000 мг/сутки, как например 75-100, 100-150, 150-200, 200-300, 300-400, 400-500, 500-600, 600-700, 700-800, 800-900 или 900-1000 мг/сутки.
45. Фармацевтический состав по любому из пп. 1, 14-21, 23, 24, 26-29, 31-34, 36-38 для введения индивиду, выбранному из группы, состоящей из пациентов-детей; пациентов, у которых присутствует повышенный уровень креатинина в сыворотке крови; и пациента, имеющего заболевание, выбранное из заболевания почек (нефропатии), в том числе невоспалительной нефропатии (нефроза) и воспалительной нефропатии (нефрита); сахарного диабета типа I и сахарного диабета типа II и артериальной гипертензии.
46. Фармацевтический состав по любому из пп. 1, 14-21, 23, 24, 26-29, 31-34, 36-38 для применения в способе лечения лизосомной болезни накопления (LSD), лизосомной болезни накопления, выбранной из группы, состоящей из нарушений накопления липидов (или липидоза), в том числе сфинголипидозов, ганглиозидозов и лейкодистрофий; мукополисахаридозов, нарушений накопления гликопротеинов (или гликопротеиноза) и муколипидозов, или бокового амиотрофического склероза (ALS).
| Колосоуборка | 1923 |
|
SU2009A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| US 6649628 B1, 18.11.2003 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Mohamad A | |||
| Ibrahim et al., Employing Compritol in mixed matrix for sustaining chlorpheniramine maleat release: kinetic study., Digest Journal of Nanomaterials and Biostructures, Vol.8, No.2, 2013, pages 737-746 | |||
| US 5133974 A, | |||

