ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к фармацевтически приемлемым солям конъюгатов, содержащих химиотерапевтическое лекарственное средство и аминокислоту или ее производное, которые легко усваиваются раковой клеткой. В частности, настоящее изобретение относится к фармацевтически приемлемым солям конъюгатов лекарственных средств, аналогов цитидина, и аспарагиновой или глутаминовой кислоты и их аналогов, фармацевтическим композициям, содержащим указанные конъюгаты, а также к их применению для лечения рака или предракового состояния или расстройства.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Антипролиферативные лекарственные средства
Антипролиферативные лекарственные средства, также известные как антиметаболиты, противоопухолевые агенты и лекарственные средства, ковалентно связывающие ДНК, действуют путем ингибирования основных метаболических путей и обычно применяются для лечения злокачественных заболеваний. Однако их применение в качестве терапевтических агентов ограничено из-за их высокой токсичности для нормальных клеток и серьезных нежелательных явлений. Нежелательные явления включают анемию, рвоту и облысение из-за цитотоксического воздействия на быстро делящиеся нормальные клетки, такие как стволовые клетки в костном мозге, эпителиальные клетки кишечного тракта, клетки волосяных фолликулов и т.д.
Другой серьезной проблемой, связанной с антипролиферативными лекарственными средствами, является устойчивость опухолей к лекарственным средствам, как первичная, так и приобретенная. Например, хотя начальная доля пациентов, выходящих после лечения L-аспарагиназой в полную клиническую ремиссию, довольно высока у пациентов с острым лимфобластным лейкозом (ОЛЛ), рецидив заболевания и связанная с этим резистентность к лекарственным средствам представляют собой значительную клиническую проблему. Исследования продемонстрировали увеличение экспрессии аспарагинсинтетазы (AS) в резистентных к аспарагиназе клетках, что привело к возникновению гипотезы, что повышенная активность AS обеспечивает выживаемость злокачественных клеток за счет резистентности к лекарственным средствам.
Нуклеотидные/нуклеозидные аналоги
Нуклеозидные аналоги конкурируют со своими физиологическими «эквивалентами» за включение в нуклеиновые кислоты и играют важную роль в лечении острого лейкоза. Наиболее важными из них являются арабинозные нуклеозиды, это уникальный класс антиметаболитов, первоначально выделенных из губки Cryptothethya crypta, которые теперь получают синтетическим путем. Они отличаются от физиологических дезоксирибонуклеозидов присутствием 2'-ОН-группы в цис-конфигурации относительно N-гликозильной связи между цитозином и сахаром арабинозидом. Несколько арабинозных нуклеозидов оказывают полезное противоопухолевое и противовирусное действие. Наиболее активным цитотоксическим агентом этого класса является цитозин-арабинозид (цитарабин). Родственный нуклеозид, аденин-арабинозид, также оказывает противоопухолевую активность, а его аналог, флударабинфосфат (2-фтор-ара-аденозинмонофосфат) обладает сильной противоопухолевой активностью в отношении лимфом и хронического лимфоцитарного лейкоза (Warrell & Berman, 1986). Другим членом группы является арабинозил-5-азацитидин, синтетический аналог, который показал неудовлетворительные результаты в клиническом исследовании (Dalai et al, 1986).
Одной из задач разработки аналогов в области антиметаболитов цитидина был поиск соединений, которые сохраняют ингибирующую активность ара-С, но устойчивы к дезаминированию. Был разработан ряд деаминаз-устойчивых аналогов, в том числе цикло-цитидин (Но DHW, 1974) и N4-бегеноил ара-С (Kodama et al., 1989), которые показали противолейкемическую активность в некоторых клинических испытаниях, но при этом имели нежелательные побочные эффекты (Woodcock et al., 1980). Другими репрезентативными соединениями являются N4-пальмитоил-ара, 2'-азидо-2'-дезокси-ара-С, 5'-(кортизон 21-фосфориловый) сложный эфир ара-С, 5'-ациловые эфиры ара-С (например, сложный эфир 5'-пальмитата), N4-бегеноил-ара-С, конъюгат ара-С с поли-Н5(2-гидроксиэтил)-L-глутамином, дигидро-5-азацитидином, 5-аза-арабинозилцитозином, 5-аза-2'-дезоксицитидином и 2'-2'-дифторооксицитидином (Hartel et al., 1990; Heineman et al., 1988).
Гемцитабин (2,2-дифтородеоксицитидин, dFdC) является наиболее важным аналогом цитидина, который вводили в клинические испытания после ара-С. Он включен в стандартную терапию первой линии для пациентов с раком поджелудочной железы, раком легкого и переходно-клеточным раком мочевого пузыря.
Нуклеотидные аналоги также имеют и другие применения, не относящиеся к раку. Например, флуцитозин (Flucytosine), аналог фторированного цитозина, применяется в качестве противогрибкового агента.
Аминокислоты и пролиферативное заболевание
Аспарагин - это заменимая аминокислота, в которой нуждаются быстро пролиферирующие клетки. Клетки млекопитающих могут синтезировать аспарагин из аспартата, используя АТФ-зависимый фермент аспарагинсинтетазу (СЕ 6.3.5.4), которая переносит аминогруппу из амида глутамина в β-карбоксил аспартата в ходе взаимодействия, которое может быть представлено следующим образом: Глутамин + аспартат + АТФ + H2O = глутамат + аспарагин + AMP + PPi.
Дефицит аспарагинасинтетазы встречается в некоторых опухолях, что заставляет их полагаться на поступление аспарагина из других источников, таких как сыворотка. Это наблюдение привело к применению фермента L-аспарагиназы (тип СЕ -2, СЕ 3.5.1.1) в качестве химиотерапевтического агента. L-аспарагиназа гидролизует L-аспарагин до аспартата и аммиака, что приводит к обеднению сыворотки в отношении L-аспарагина и ингибированию роста опухоли. L-аспарагиназу применяют главным образом для лечения острого лимфобластного лейкоза (ОЛЛ), также она проявляет некоторую активность против других опухолей системы крови, включая острый нелимфоцитарный лейкоз.
L-аспарагиназа для применения в клинических условиях доступна в двух немодифицированных (нативных) формах, полученных очисткой из бактериальных источников, и в качестве ПЭГилированного соединения. В патенте США №4179377 описана ПЭГилированная L-аспарагиназа, где фермент связан с ПЭГ, имеющим молекулярную массу от 500 до 20000 дальтон.
Пониженная регуляция аспарагиновой синтетазы in vivo может обеспечить эффективный механизм ингибирования роста опухоли. Однако клетки реагируют на нехватку аминокислот путем согласованного увеличения мРНК аспарагинасинтетазы, белковой и ферментативной активности, которая включает в себя транскрипционный контроль гена аспарагинсинтетазы.
Первоначально метаболический подход использовали для ингибирования активности аспарагинсинтетазы путем образования аналогов L-аспарагина и L-аспарагиновой кислоты. Аналоги, включая 5-карбоксамидо-4-амино-3-изоксазолидон (Stammer et al., 1978), N-замещенные сульфонамиды и N'-замещенные сульфонилгидразиды, были получены в виде серных аналогов L-аспарагина (Brynes S et al., 1978a, Brynes S et al., 1978b). В патенте США №4348522 описана соль PALA, N-фосфонацетил-L-аспарагиновой кислоты, которая, как было показано, проявляет противоопухолевую активность и в настоящее время находится в клинических испытаниях в качестве комбинированной химиотерапии колоректального рака и рака поджелудочной железы.
Аспарагиновую кислоту, аналог ара-С, применяли в качестве исходного материала для синтеза конъюгатов пептида Т Ара-С для направленного взаимодействия на CD4-позитивные клетки (Manfredini et al., 2000).
Применение пролекарств для получения желаемых характеристик, таких как повышенная биодоступность или повышенная сайт-специфичность, является признанной концепцией в области фармацевтического развития. Например, прямое или косвенное конъюгирование лекарственного средства с антителом создает стабильный конъюгат, который можно доставить в определенное место с минимальной диссоциацией лекарственного средства. Направленное взаимодействие лекарственных средств для максимальной эффективности может сочетаться с механизмом селективного высвобождения указанного лекарственного средства.
В патенте США №4296105 описаны производные доксорубицина, связанные с необязательно замещенной аминокислотой в гидроксигруппе аминокислотного остатка, которые in vitro обладают более высокой противоопухолевой активностью и меньшей токсичностью, чем доксорубицин.
В патенте США №5962226 описаны активируемые опухолями пролекарства, которые не способны проникнуть в клетку до тех пор, пока они не расщепляются фактором или факторами, выделяемыми клеткой-мишенью.
В патенте США №5650386 представлены композиции, содержащие по меньшей мере один активный агент и по меньшей мере одну модифицированную не-альфа аминокислоту или полиаминокислоту, которая действует как носитель активного агента. Модификация аминокислот включает ацилирование или сульфирование по меньшей мере одной свободной аминогруппы.
В патентах США №№6623731, 6428780 и 6344213 описаны нековалентные смеси, содержащие модифицированные аминокислоты в качестве носителей для биологически активных агентов.
В патенте США №5106951 описан конъюгат, содержащий ароматическое лекарственное средство, нековалентно интеркалированное между двумя ароматическими боковыми цепями на олигопептиде, и антитело или фрагмент антитела, ковалентно присоединенный к олигопептиду, для направленного взаимодействия с раковыми клетками.
В патенте США №6617306 описан носитель для доставки in vivo терапевтического агента, при этом указанные носитель и терапевтический агент связаны дисульфидной связью. В этом патенте описано, что указанный носитель содержит полимер и по меньшей мере одно тиольное соединение, конъюгированное с полимером, так что тиольная группа тиольного соединения и тиольная группа терапевтического агента образуют дисульфидную связь.
В публикации международной патентной заявки № WO 00/33888 описаны расщепляемые противоопухолевые и противовоспалительные соединения, содержащие терапевтический агент, способный проникать в клетку-мишень, олигопептид, стабилизирующую группу и необязательный линкер.
В публикации международной патентной заявки № WO 2005/072061 и в патенте США №8993278 некоторые из изобретателей настоящего изобретения описывают соединения, содержащие лекарственное средство, ковалентно связанное с аминокислотой через боковую цепь с функциональной группой, которые применяют для направленного взаимодействия лекарственных средств с неопластическими клетками.
Фармацевтические соли
Выбор оптимальных фармацевтических солей лекарственного средства может повысить эффективность и улучшить доставку лекарственного средства. Создание таких активных солей лекарственных средств не является тривиальной задачей, а разработка и исследование таких солей требует применения уникальных методик.
Существует неудовлетворенная медицинская потребность в соединениях и композициях, способных направленно взаимодействовать с опухолями, которые в то же время не вызывают цитотоксического повреждения нормальных тканей.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Задачей настоящего изобретения является направленное взаимодействие лекарственных средств на злокачественные и пред-злокачественные клетки при одновременном снижении побочных эффектов, связанных с лекарственной терапией.
Настоящее изобретение относится к фармацевтически приемлемым солям конъюгатов, содержащих первый химический фрагмент, ковалентно связанный со вторым химическим фрагментом, где указанный первый химический фрагмент представляет собой аминокислоту, содержащую на своей боковой цепи по меньшей мере одну функциональную группу, выбранную из группы, состоящей из аминогруппы, карбоксильной группы, сульфгидрильной группы и гидроксильной группы, а указанный второй химический фрагмент представляет собой противораковое лекарственное средство, выбранное из группы, состоящей из цитотоксического агента, цитостатического агента и химиотерапевтического агента. Указанное противораковое лекарственное средство присоединено к аминокислоте через функциональную группу боковой цепи аминокислоты. Указанная аминокислота предпочтительно представляет собой аспарагиновую кислоту или глутаминовую кислоту, или их производные или аналоги, ковалентно связанные с указанным противоопухолевым лекарственным средством, предпочтительно нуклеотидным/нуклеозидным аналогом, таким как цитарабин или гемцитабин. Указанная фармацевтически приемлемая соль предпочтительно выбрана из органической или неорганической кислоты или кислотного остатка (то есть конъюгатного основания). Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим соли конъюгатов, и их применению для лечения рака и предраковых состояний или расстройств. Кроме того, настоящее изобретение относится к способам получения солевых форм конъюгатов согласно настоящему изобретению, в частности, путем взаимодействия защищенного соединения-предшественника с кислотой с получением соответствующей соли.
Солевые формы согласно настоящему изобретению могут служить в качестве носителя для доставки лекарственного средства или пролекарства, в котором лекарственное средство быстро поглощается раковыми клетками. Солевые формы также могут служить для повышения стабильности и растворимости конъюгата.
Конъюгаты согласно настоящему изобретению обычно получают из соединений-предшественников, содержащих одну или более защитных групп на восприимчивых фрагментах, таких как амины или карбоновые кислоты. Например, аминогруппа аминокислоты обычно защищена трет-бутоксикарбонильной группой (ВОС). Хорошо известно, что ВОС-группы обычно удаляются с аминогрупп в кислотных средах, причем наиболее предпочтительным средством для удаления защитных групп является трифторуксусная кислота (ТФУ). Кроме того, известно, что ТФУ трудно удалить, и поэтому амины со снятыми защитными группами обычно содержат остаточные количества ТФУ. Необходимо, чтобы соединения для фармацевтического применения не содержали ТФУ, поскольку ТФУ оказывает токсическое воздействие. В некоторых вариантах реализации настоящее изобретение позволяет избежать необходимости использования ТФУ в качестве агента для удаления защитных групп. Неожиданно было обнаружено, что соли конъюгатов согласно настоящему изобретению можно получать путем удаления защитных групп ВОС с применением того же реагента, который образует указанную соль. В настоящем документе описано, что некоторые фармацевтически приемлемые кислоты могут одновременно удалять защитную группу (защитные группы) и образовывать соль конъюгата согласно настоящему изобретению на одной стадии, тем самым промежуточная стадия снятия защиты с помощью ТФУ до образования соли оказывается устранена. Таким образом, в некоторых вариантах реализации конъюгаты согласно настоящему изобретению не подвергаются воздействию ТФУ для удаления защитных групп, и поэтому соли конъюгатов согласно настоящему изобретению не содержат остаточной ТФУ. Таким образом, полученные соли обладают большим преимуществом из-за того, что они не содержат токсичной ТФУ и, следовательно, пригодны для фармацевтического применения.
Согласно первому аспекту настоящее изобретение относится к фармацевтически приемлемой соли соединения, указанная соль представлена структурой формулы (I)

где
А обозначает аминокислоту, содержащую на своей боковой цепи по меньшей мере одну функциональную группу, выбранную из группы, состоящей из карбоксильной группы, аминогруппы, сульфгидрильной группы и гидроксильной группы;
D обозначает остаток лекарственного средства, выбранного из группы, состоящей из цитотоксического агента, цитостатического агента и химиотерапевтического агента, где D присоединен к А через функциональную группу боковой цепи А; и
Y представляет собой фармацевтически приемлемую органическую или неорганическую кислоту или кислотный остаток (т.е. конъюгатное основание указанной кислоты), при этом указанная кислота выбрана из группы, состоящей из соляной кислоты, уксусной кислоты, метансульфоновой кислоты, фосфорной кислоты, лимонной кислоты, молочной кислоты, янтарной кислоты, винной кислоты, борной кислоты, бензойной кислоты, толуолсульфоновой кислоты, бензолсульфоновой кислоты, аскорбиновой кислоты, серной кислоты, малеиновой кислоты, муравьиной кислоты, малоновой кислоты, никотиновой кислоты и щавелевой кислоты, при этом указанная фармацевтически приемлемая соль содержит 0,1% или менее трифторуксусной кислоты (ТФУ) в качестве примеси. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Указанная аминокислота (А) может представлять собой α-аминокислоту (D или L), β-аминокислоту, γ-аминокислоту, δ-аминокислоту или ε-аминокислоту. В одном предпочтительном в настоящее время варианте реализации указанная аминокислота (А) представляет собой α-аминокислоту (D или L). В некоторых вариантах реализации указанная аминокислота (А) выбрана из группы, состоящей из аспарагиновой кислоты (Asp), глутаминовой кислоты (Glu), аспарагина (Asn), глутамина (Gln), α-аминоадипиновой кислоты (Aad), α-аминопимелиновой, γ-карбокси-глутаминовой кислоты, γ-гидрокси-глутаминовой кислоты, аминоглицина, аминоизомасляной кислоты (Aib), аргинина (Arg), цитруллина (Cit), цистеина (Cys), цистина, диаминобутановой кислоты, диаминомасляной кислоты (Dab), диаминопропионовой кислоты (Dpr), дигидроксифенилаланина, диметиларгинина, пироглутаминовой кислоты (p-Glu), гистидина (His), 1-метилгистидина, 3-метилгистидина, гомосерина (Hse), гомоцитруллина, гидроксипролина (Hyp), лизина (Lys), метиллизина, диметиллизина, триметилизина, азидолизина, метионина (Met), метионинсульфоксида, метионинсульфона, орнитина (Orn), саркозина (Sar), селеноцистеина (Sec), серина (Ser), фосфорсерина, метилсерина, аминосерина (Ams), тиенилаланина (Thi), треонина (Thr), фосфотреонина, триптофана (Trp), тирозина (Tyr), метилтирозина, фосфортирозина, сульфотирозина, α-аминосубериновой кислоты, 3,5-дийодтирозина, пеницилламина (Pen), 4-этиламинфенилглицина, 4-аминофенилглицина, 4-сульфофенилаланина, 4-аминофенилаланина и 2-амино-4[4-(2-амино)-пиримидинил]бутановой кислоты, 3-аминопропионовой кислоты, 6-аминокапроновой кислоты (ε-Ahx), п-аминобензойной кислоты, изонипекотиновой кислоты, статина (STA), 2-аминомасляной (Abu) и 4-аминомасляной кислоты, а также их производных и аналогов. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
В некоторых вариантах реализации указанная аминокислота (А) выбрана из группы, состоящей из аспарагиновой кислоты, глутаминовой кислоты, аспарагина, глутамина и их производных и аналогов. В одном предпочтительном в настоящее время варианте реализации указанная аминокислота (А) представляет собой аспарагиновую кислоту. В другом предпочтительном в настоящее время варианте реализации указанная аминокислота (А) представляет собой глутаминовую кислоту.
Группа (D) может быть получена из любого лекарственного средства, выбранного из группы, состоящей из цитотоксического агента, цитостатического агента и химиотерапевтического агента. В некоторых вариантах реализации указанный цитотоксический, цитостатический или химиотерапевтический агент D представляет собой остаток лекарственного средства, выбранного из группы, состоящей из аналога пиримидина, аналога пурина, антифолата, гидроксимочевины, антимикротубулинового агента, алкилирующего агента, противоопухолевого антибиотика, агента, направленно взаимодействующего с топоизомеразой, антиметаболита, связывающего ДНК агента и антагониста ДНК.
В некоторых вариантах реализации указанный цитотоксический, цитостатический или химиотерапевтический агент D представляет собой остаток аналога пиримидина, выбранного из цитозин-арабинозида (ара-С, цитарабина), гемцитабина, фторурацила, 5-фтордезоксиридина (5-FUDR), фторафура, капецитабина, кармофура, BOF-A2, 5-хлор-2,4-дигидроксипиридина, децитабина, лефлуномида, трифлуридина, идоксуридина, зидовудина, телбивудина, триметоприма, фторцитозина, дезокситимидина и 5-амино-6-нитроурацила. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения. В одном из предпочтительных в настоящее время вариантов реализации настоящего изобретения указанный цитотоксический, цитостатический или химиотерапевтический агент D представляет собой остаток цитарабина. В другом из предпочтительных в настоящее время вариантов реализации настоящего изобретения указанный цитотоксический, цитостатический или химиотерапевтический агент D представляет собой остаток гемцитабина.
В других вариантах реализации указанный цитотоксический, цитостатический или химиотерапевтический агент D представляет собой остаток аналога пурина, выбранного из клофарабина, декарбазина, кладрибина, меркаптопурина, неларабина, пентостатина, тиогуанина, гефитиниба, азатиопурина, 8-азагуанина, иммуциллина-G, 2-фтор-2'-дезоксиаденозина, 9-бета-D-ксилофуранозил-аденина, 3-дезоксигуанина, 6-метил-формицина А, 2-фтораденозина, 1-деаза-аденозина, N-этил-5-карбоксамидоаденозина, 3'-оксоаденозина, 2-аминоаденозина, 6-О-циклометилгуанина, флударабина и 8-йод-гуанина. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
В другом варианте реализации указанный цитотоксический, цитостатический или химиотерапевтический агент D представляет собой остаток лекарственного средства, выбранного из группы, состоящей из 2,4-диоксо-5-фторпиримидина (5-FU), азацитидина (5-AZC), 6-меркаптопурина (6-MP), 6-тиогуанина (6-TG), хлородеоксиаденозина (2-CDA) и пентастатина (dCF). Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
В некоторых вариантах реализации соли формулы (I) получают в по существу чистой форме. Термин «по существу чистый» относится к чистоте по меньшей мере 95%, определенной по ВЭЖХ, предпочтительно к чистоте по меньшей мере примерно 97%, более предпочтительно к чистоте по меньшей мере 98% и наиболее предпочтительно к чистоте по меньшей мере 99%.
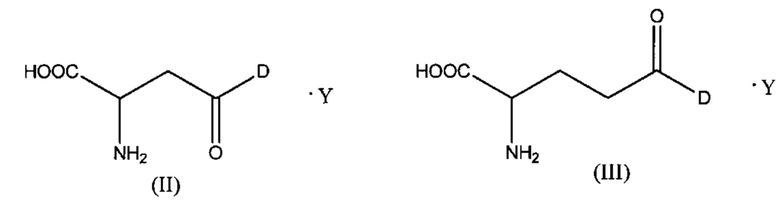
Как отмечено выше, в конъюгатах согласно настоящему изобретению указанный цитотоксический, цитостатический или химиотерапевтический агент D присоединен к аминокислоте (А) через функциональную группу боковой цепи (А). Некоторые неограничивающие варианты реализации таких конъюгатов описаны ниже. Согласно некоторым вариантам реализации указанная аминокислота (А) выбрана из группы, состоящей из аспарагиновой кислоты и глутаминовой кислоты, и указанное соединение представлено структурой формулы (II) или (III):
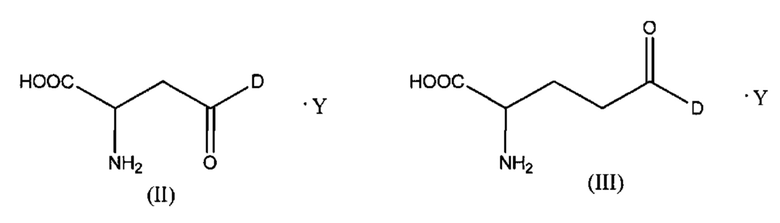
где D и Y такие, как определено выше.
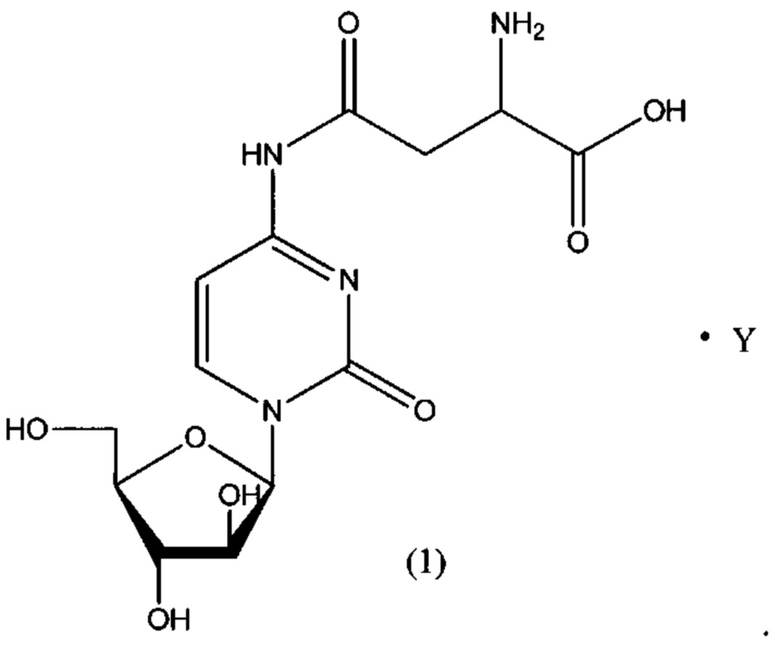
В определенном варианте реализации формулы (II) А представляет собой аспарагиновую кислоту, a D представляет собой остаток цитарабина. В этом варианте реализации указанное соединение представляет собой соль, представленную структурой формулы (1):
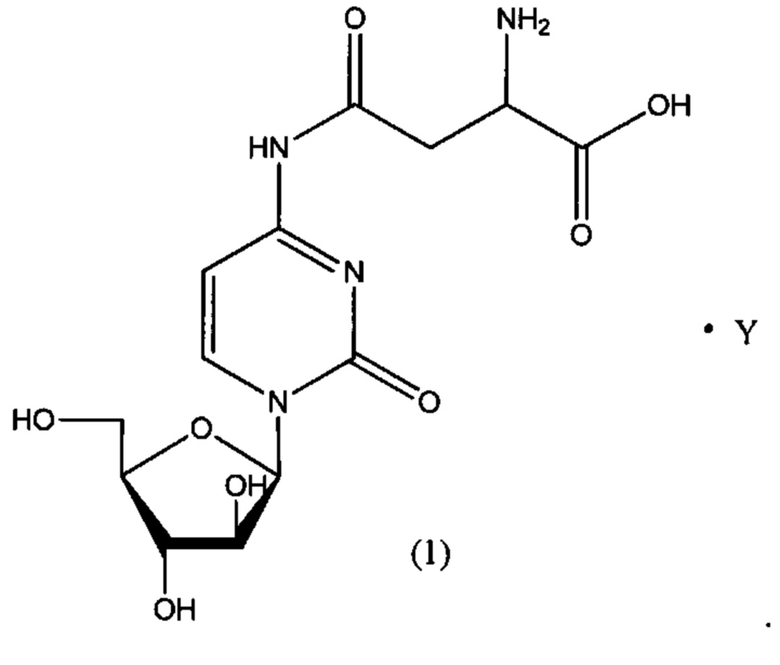
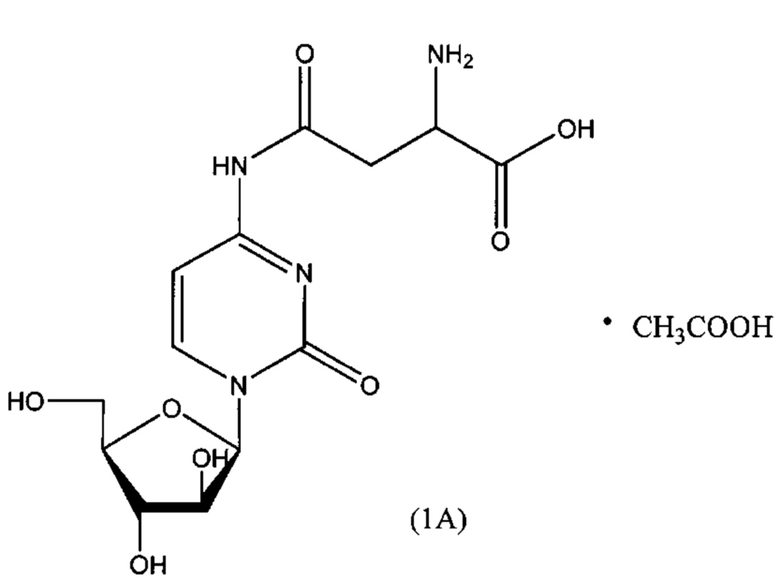
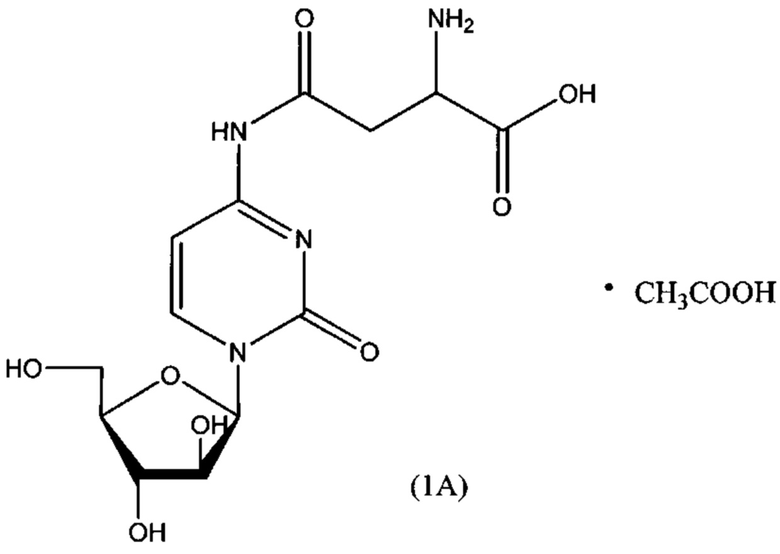
В одном варианте реализации формулы (1) указанное соединение представляет собой соль уксусной кислоты (то есть ацетатную соль), которая представлена структурой формулы (1А):
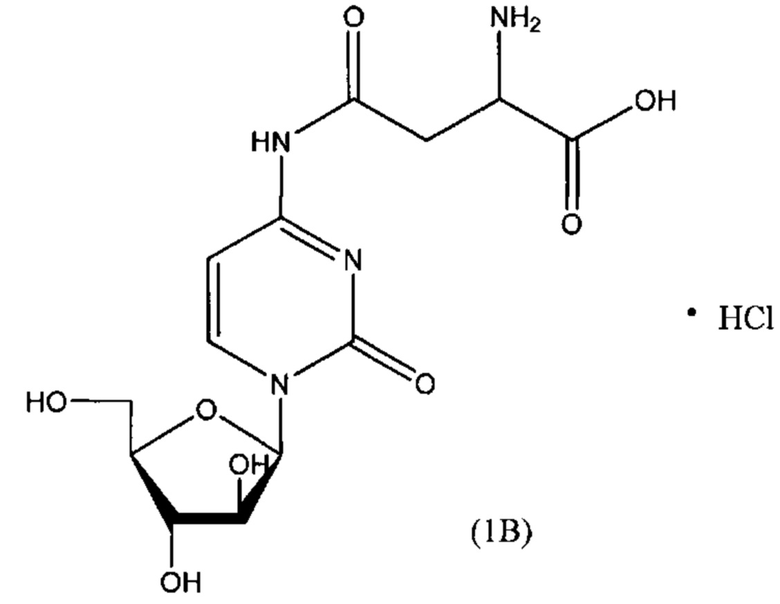
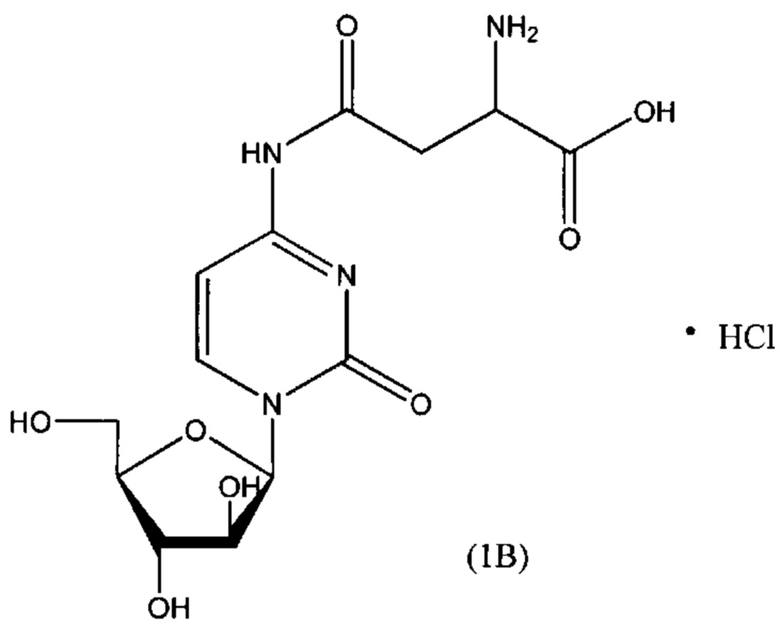
В другом варианте реализации формулы (1) указанное соединение представляет собой соль соляной кислоты (то есть гидрохлоридную соль), которая представлена структурой формулы (1В):
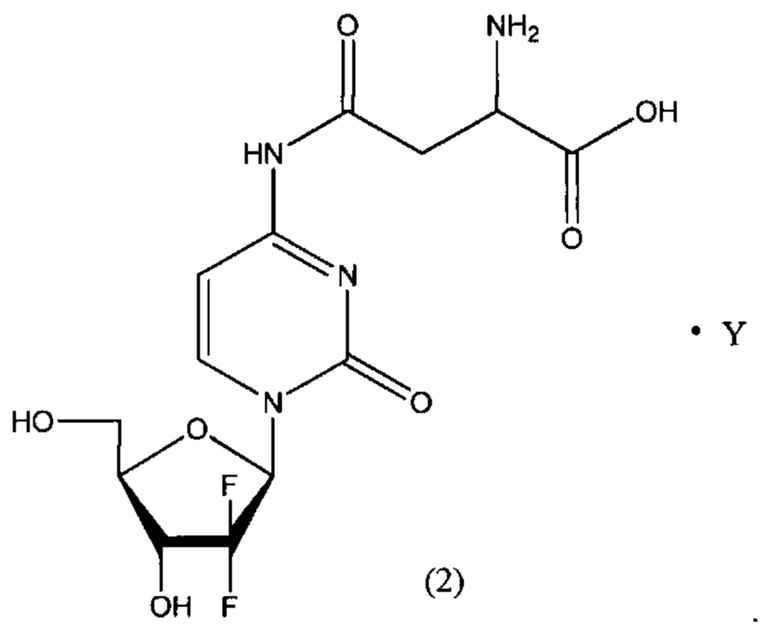
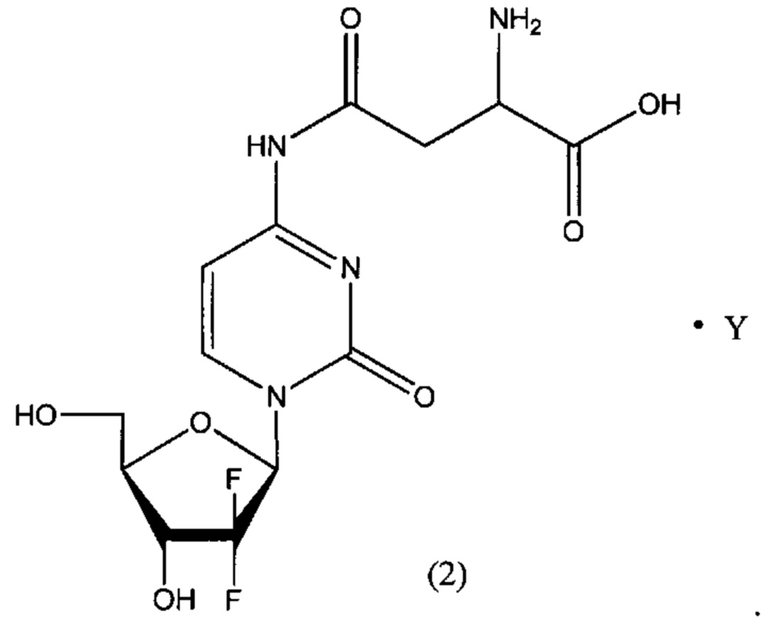
В другом определенном варианте реализации формулы (II) А представляет собой аспарагиновую кислоту, a D представляет собой остаток гемцитабина, и указанное соединение представлено структурой формулы (2):
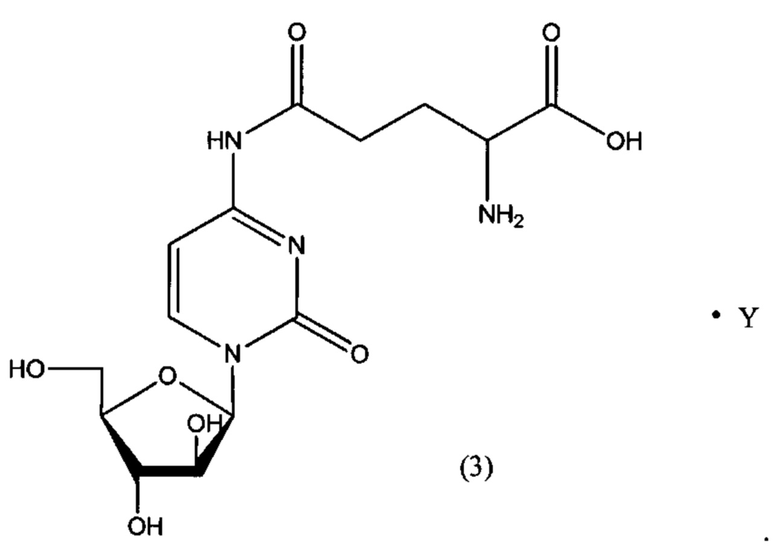
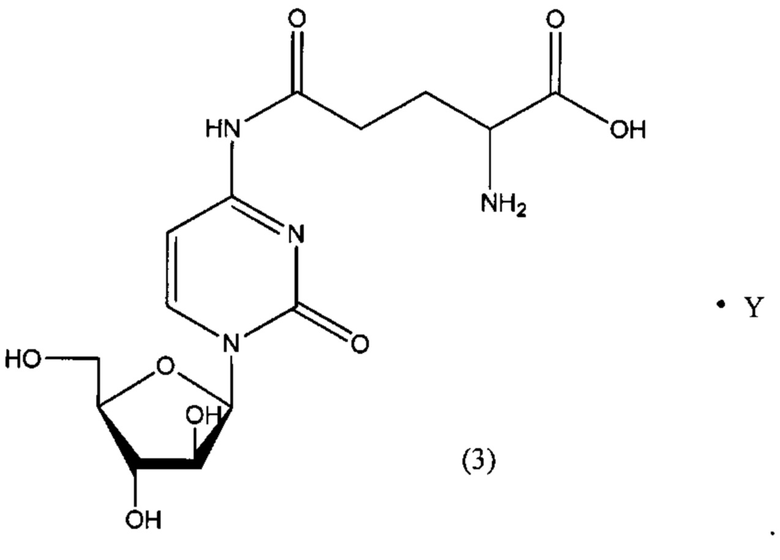
В определенном варианте реализации формулы (III) А представляет собой глутаминовую кислоту, a D представляет собой остаток цитарабина, и указанное соединение представлено структурой формулы (3):
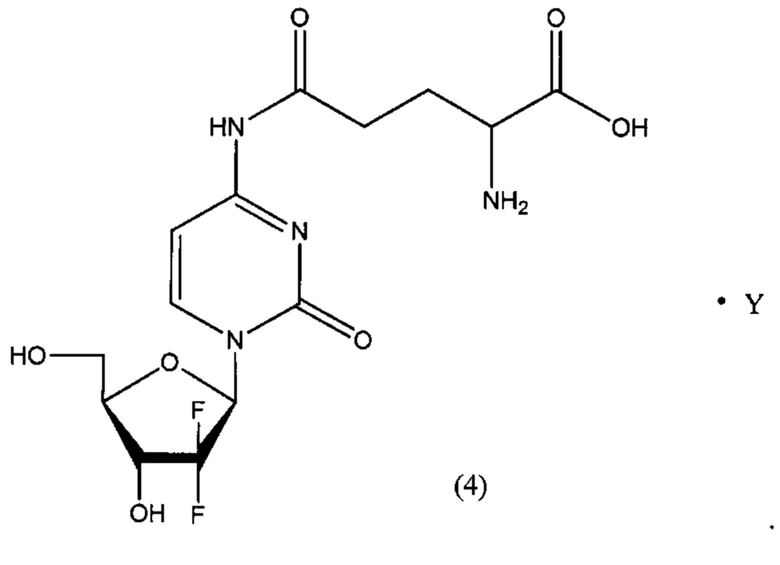
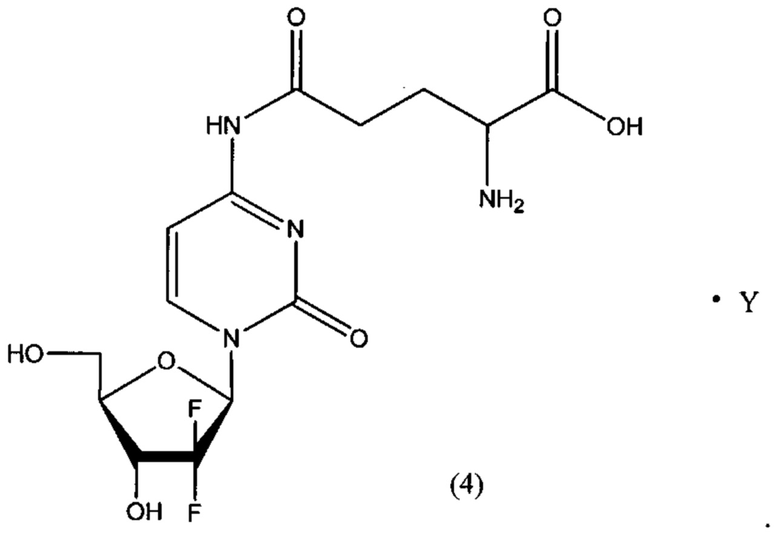
В другом определенном варианте реализации формулы (III) А представляет собой глутаминовую кислоту, a D представляет собой остаток гемцитабина, и указанное соединение представлено структурой формулы (4):
В любом из упомянутых выше соединений формул (I), (II), (III), (1), (2), (3) и (4) Y представляет собой фармацевтически приемлемую кислоту, выбранную из группы, состоящей из уксусной кислоты, соляной кислоты, метансульфоновой кислоты, фосфорной кислоты, лимонной кислоты, молочной кислоты, янтарной кислоты, винной кислоты, борной кислоты, бензойной кислоты, толуолсульфоновой кислоты, бензолсульфоновой кислоты, аскорбиновой кислоты, серной кислоты, малеиновой кислоты, муравьиной кислоты, малоновой кислоты, никотиновой кислоты и щавелевой кислоты. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
В одном предпочтительном в настоящее время варианте реализации указанная фармацевтически приемлемая кислота Y представляет собой уксусную кислоту, то есть органическую кислоту. В одном предпочтительном в настоящее время варианте реализации указанная фармацевтически приемлемая кислота Y представляет собой соляную кислоту (HCl), то есть неорганическую кислоту.
Специалисту в данной области понятно, что любая из вышеуказанных фармацевтически приемлемых кислот может образовывать фармацевтически приемлемую соль с соединениями согласно настоящему изобретению. В некоторых вариантах реализации указанная фармацевтически приемлемая соль выбрана из группы, состоящей из: ацетата, гидрохлорида, метансульфоната, фосфата, цитрата, лактата, сукцината, тартрата, бората, бензоата, толуолсульфоната, бензолсульфоната, аскорбата, сульфата, малеата, формиата, малоната, никотината и оксалата. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
В одном предпочтительном в настоящее время варианте реализации указанная фармацевтически приемлемая кислота представляет собой уксусную кислоту (СН3СООН). В этом варианте реализации указанная соль представляет собой ацетатную соль. В другом варианте реализации указанная фармацевтически приемлемая соль представляет собой гидрохлоридную соль (HCl). В этом варианте реализации указанная соль представляет собой гидрохлоридную соль.
В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей в качестве активного ингредиента соль формулы (I) или любое соединение, охватываемое такой формулой, например, соединения (II), (III), (1), (1А), (1В), (2), (3) или (4), описанные в настоящем документе, и фармацевтически приемлемый носитель или разбавитель.
В другом аспекте настоящее изобретение относится к способу лечения рака, включая его метастазы, включающему стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соли формулы (I) или любого соединения, охватываемого такой формулой, например, соединения (II), (III), (1), (1А), (1В), (2), (3) или (4), описанных в настоящем документе. В другом аспекте настоящее изобретение относится к соли формулы (I) или любому соединению, охватываемому такой формулой, например, соединениям (II), (III), (1), (1А), (1В), (2), (3) или (4), описанным в настоящем документе, для применения для лечения рака, включая его метастазы. В некоторых вариантах реализации указанный рак характеризуется несолидной опухолью или солидной опухолью или их комбинацией.
В других вариантах реализации указанный рак представляет собой опухоль системы крови. В некоторых других вариантах реализации настоящего изобретения указанная опухоль системы крови выбрана из группы, состоящей из лейкозов, лимфом и множественных миелом. В некоторых вариантах реализации указанный лейкоз выбран из группы, состоящая из острого миелоидного лейкоза (ОМЛ) и острого лимфобластного лейкоза (ОЛЛ). В некоторых вариантах реализации указанный лейкоз представляет собой рецидивный/рефрактерный лейкоз. В других вариантах реализации указанный лейкоз представляет собой впервые диагностированный лейкоз.
В некоторых определенных вариантах реализации указанный рак характеризуется солидной опухолью, выбранной из группы, состоящей из опухолей центральной нервной системы (ЦНС), рака печени, колоректальной карциномы, рака молочной железы, рака желудка, рака поджелудочной железы, карциномы мочевого пузыря, карциномы шейки матки, опухоли головы и шеи, рака вульвы и дерматологических новообразований, включая меланому, плоскоклеточную карциному и базальноклеточные карциномы.
В другом аспекте настоящее изобретение относится к способу лечения предракового состояния или расстройства, включающему стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соли формулы (I) или любого соединения, охватываемого такой формулой, например, соединения (II), (III), (1), (1А), (1В), (2), (3) или (4), описанных в настоящем документе.
В другом аспекте настоящее изобретение относится к соли формулы (I) или любому соединению, охватываемому такой формулой, например, соединениям (II), (III), (1), (1А), (1В), (2), (3) или (4), описанным в настоящем документе, для применения для лечения предракового состояния или заболевания.
В некоторых вариантах реализации указанное предраковое состояние или расстройство представляет собой миелодиспластические синдромы (МДС).
В других вариантах реализации настоящее изобретение относится к способу лечения индивидуума, который является кандидатом на трансплантацию костного мозга, включающему стадию предварительного лечения указанного индивидуума до трансплантации при помощи соли формулы (I), или любого соединения, охватываемого такой формулой, например, соединениями (II), (III), (1), (1А), (1В), (2), (3) или (4), описанными в настоящем документе.
В других вариантах реализации настоящее изобретение относится к соли формулы (I) или любому соединению, охватываемому такой формулой, например, соединениям (II), (III), (1), (1А), (1В), (2), (3) или (4), описанным в настоящем документе, для применения для предварительного лечения перед трансплантацией костного мозга.
Солевые формы согласно настоящему изобретению можно применять для лечения любого млекопитающего, предпочтительно человека.
Дополнительно описан способ получения фармацевтически приемлемой соли соединения, представленной формулой (I) согласно настоящему изобретению, включающий стадию
(а) взаимодействие соединения формулы (IV):
где
А и D такие, как определено выше;
n представляет собой целое число 1-6; и
каждый Р независимо в каждом случае представляет собой защитную группу; где каждый Р присоединен к любой функциональной группе, доступной для защиты, на аминокислоте (А), лекарственном остатке (D) или к аминокислоте (А) и к лекарственному остатку (D);
с реагентом, способным удалять защитную группу (защитные группы) Р; и (b) добавление реагента, способного вводить группу Y, с получением соли, представленной формулой:

где Y такой, как описано выше.
В одном варианте реализации неожиданно было обнаружено, что соли согласно настоящему изобретению можно получать путем удаления защитных групп с соединения-предшественника с применением той же фармацевтически приемлемой кислоты, которая образует указанную соль. Таким образом, в соответствии с принципами настоящего изобретения фармацевтически приемлемая кислота Y, способная вводить группу Y, может одновременно удалять защитную группу (защитные группы) Р и образовывать соль формулы (I) за одну стадию, тем самым устраняя промежуточную стадию снятия защиты перед образованием соли. Таким образом, чистота указанного солевого продукта улучшается, поскольку можно удается избежать появления остаточных количеств агента для удаления защитных групп (то есть примесей). Например, защитные группы, такие как трет-бутоксикарбонил (ВОС), обычно удаляют в кислой среде, например, при помощи трифторуксусной кислоты (ТФУ). Однако ТФУ трудно удалить, и поэтому продукт со снятыми защитными группами содержат остаточные количества этой кислоты. Это неизбежно приводит к увеличению уровня примеси ТФУ в конечных солевых формах, что неприемлемо с точки зрения разработки фармацевтического продукта. Настоящее изобретение обеспечивает решение этой проблемы за счет проведения заключительной стадии синтеза с применением того же реагента (например, кислоты), который в конечном итоге образует соль.
Таким образом, в одном варианте реализации способ согласно настоящему изобретению включает применение реагента, способного удалять защитную группу (защитные группы) Р и одновременно вводить группу Y, например, кислоту. В соответствии с этим вариантом реализации защитную группу (защитные группы) Р удаляют и вводят группу Y за одну стадию.
В соответствии с некоторыми вариантами реализации указанная фармацевтически приемлемая соль конъюгата согласно настоящему изобретению содержит примерно 5% или менее остаточной ТФУ. В соответствии с дополнительными вариантами реализации указанная фармацевтически приемлемая соль конъюгата согласно настоящему изобретению содержит примерно 1% или менее остаточной ТФУ. В соответствии с дополнительными вариантами реализации указанная фармацевтически приемлемая соль конъюгата согласно настоящему изобретению содержит примерно 0,1% или менее остаточной ТФУ. В соответствии с определенными вариантами реализации указанная фармацевтически приемлемая соль конъюгата согласно настоящему изобретению по существу свободна от примесной ТФУ. В соответствии с определенными вариантами реализации указанная фармацевтически приемлемая соль конъюгата согласно настоящему изобретению полностью не содержит примесной ТФУ.
Соединения формулы (I) обычно можно получить при сочетании фрагмента лекарственного средства с аминокислотой, предпочтительно в присутствии агента для реакции сочетания, и превращении в солевую форму в соответствии со способом, описанным выше. Этот способ проиллюстрирован в настоящем документе для соединений формулы (II) и (III).
В некоторых вариантах реализации настоящее изобретение относится к способу получения соединения формулы (II), включающему стадию:
(a) сочетание соединения формулы (i) с лекарственным средством формулы D или его защищенным производным формулы D-(P3)n в присутствии реагента для реакции сочетания с получением промежуточного соединения формулы (ii); и
(b) удаление защитных групп Р1, Р2 и Р3 (если присутствуют) при помощи кислоты (Y) или агента для снятия защитных групп и затем кислоты (Y), с получением соли, представленной формулой (II):
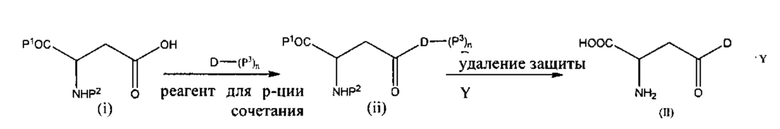
где Р1 представляет собой карбоксизащитную группу, Р2 представляет собой аминозащитную группу, Р3 представляет собой защитную группу, расположенную на одной или более функциональных группах на лекарственном средстве D, и n равно 0, 1, 2 или 3.
В другом варианте реализации настоящее изобретение относится к способу получения соединения формулы (III), включающему стадию:
(a) сочетание соединения формулы (iii) с лекарственным средством формулы D или его защищенным производным формулы D-(P3)n в присутствии реагента для реакции сочетания с получением промежуточного соединения формулы (iv); и
(b) удаление защитных групп Р1, Р2 и Р3 (если присутствуют) при помощи кислоты (Y) или агента для снятия защитных групп и затем кислоты (Y), с получением соли, представленной формулой (II)
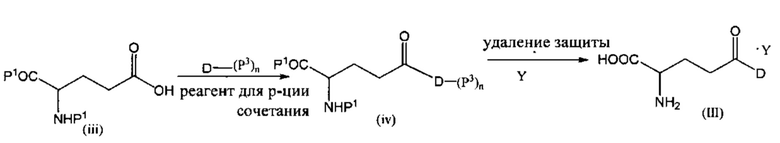
где Р1 представляет собой карбоксизащитную группу, Р2 представляет собой аминозащитную группу, Р3 представляет собой защитную группу, расположенную на одной или более функциональных группах на лекарственном средстве D, и n равно 0, 1, 2 или 3.
Согласно одному варианту реализации настоящего изобретения указанное лекарственное средство представляет собой цитарабин. В других вариантах реализации указанное лекарственное средство представляет собой защищенный цитарабин, содержащий защитные группы на 2', 3' и/или 5' гидроксильных фрагментах, такой как трифенилметил-цитарабин (CAS 7075-13-0), бензоил-цитарабин (CAS 34270-10-5), адмантоил-цитарабин (CAS 23113-01-1) и триметилсилил-цитарабин. Согласно другому варианту реализации настоящего изобретения указанное лекарственное средство представляет собой гемцитабин. В других вариантах реализации указанное лекарственное средство представляет собой защищенный гемцитабин, содержащий защитные группы на 3' и/или 5' гидроксильных фрагментах, такой как трифенилметил-гемцитабин (CAS 1642862-24-5).
В некоторых вариантах реализации защитные группы удаляют одновременно с введением группы Y, применяя кислоту, которая способна удалять защитные группы и вводить группу Y.
Эти и другие варианты реализации настоящего изобретения станут понятны в сочетании с приведенными ниже фигурами, описанием и формулой изобретения.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фиг. 1: Масс-спектр Asp(Цитарабин)⋅ацетатной соли (Y=СН3СООН).
Фиг. 2: Масс-спектр Asp(Цитарабин)⋅метансульфонатной соли (Y=CH3SO3H).
Фиг. 3: Масс-спектр Asp(Цитарабин)⋅бензолсульфонатной соли (Y=C6H5SO3H).
Фиг. 4: 1Н-ЯМР очищенного BOC-Asp-OtBu-(Цитарабин).
Фиг. 5: 1Н-ЯМР Asp(Цитарабин)⋅HCl.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к фармацевтически приемлемым солевым формам конъюгатов аминокислот, ковалентно связанных с терапевтическими агентами. В частности, настоящее изобретение относится к фармацевтически приемлемым солям конъюгатов, содержащих лекарственные средства, аналоги цитидина, и аспарагиновую или глутаминовую кислоту и их аналоги, фармацевтическим композициям, содержащим указанные конъюгаты, а также к их применению для лечения рака или предракового состояния или расстройства.
В уровне техники не описаны способы создания кислотно-аддитивных солевых форм лекарственного средства, ковалентно связанного с аминокислотой через функциональную группу боковой цепи с использованием методов непосредственного преобразования. В настоящем изобретении описано, что фармацевтические соли можно получать из предшественника лекарственного конъюгата, содержащего защитные группы, удаляемые тем же самым соединением (например, кислотой), которое применяют для получения фармацевтической солевой формы.
Конъюгаты
Конъюгаты согласно настоящему изобретению представлены в виде фармацевтически приемлемых солевых форм конъюгатов, которые биологически активны в качестве ингибиторов пролиферации раковых клеток. Солевые формы представлены структурой формулы (I):

где
А обозначает аминокислоту, содержащую на своей боковой цепи по меньшей мере одну функциональную группу, выбранную из группы, состоящей из карбоксильной группы, аминогруппы, сульфгидрильной группы и гидроксильной группы;
D обозначает остаток лекарственного средства, выбранного из группы, состоящей из цитотоксического агента, цитостатического агента и химиотерапевтического агента, где D присоединен к А через функциональную группу боковой цепи А; и
Y выбран из группы, состоящей из: фармацевтически приемлемой органической или неорганической кислоты или кислотного остатка (т.е. конъюгатного основания указанной кислоты), фармацевтически приемлемого неорганического катиона, фармацевтически приемлемого органического амина; и аминокислоты.
Фармацевтические соли
Используемый в настоящем документе термин «фармацевтическая соль» относится к «фармацевтически приемлемым солям» лекарственных веществ в соответствии с принципами ИЮПАК. Фармацевтически приемлемая соль представляет собой неактивный ингредиент в форме соли в сочетании с лекарственным средством. Используемый в настоящем документе термин «фармацевтически приемлемая соль» относится к солям соединений общей формулы (I), формулы (II) и формулы (III), например, к соединениям (1), (1А), (1В), (2), (3) или (4), или к любой другой соли, охватываемой общий формулой, которая по существу нетоксична для живых организмов. Типичные фармацевтически приемлемые соли включают соли, полученные при взаимодействии соединений согласно настоящему изобретению с фармацевтически приемлемым минералом, основанием, кислотой или солью, как описано в настоящем документе. Соли кислот также известны как соли присоединения кислоты.
Фармацевтические соли, такие как известные в данной области (Stahl and Wermuth, 2011, Handbook of pharmaceutical salts, Second edition, содержание включено в настоящее описание посредством ссылки во всей полноте), приведены в настоящем документе ниже в некоторых неограничивающих вариантах осуществления.
В соединениях согласно настоящему изобретению группа Y представляет собой солевую форму. Y может представлять собой любую фармацевтически приемлемую органическую или неорганическую кислоту или основание (или Y может быть получен из них), неограничивающие примеры которых приведены ниже:
(i) Соли присоединения кислот:
В одном варианте реализации Y представляет собой фармацевтически приемлемую органическую или неорганическую кислоту или кислотный остаток, при этом указанная кислота выбрана из группы, состоящей из уксусной кислоты, соляной кислоты, метансульфоновой кислоты, фосфорной кислоты, лимонной кислоты, молочной кислоты, янтарной кислоты, винной кислоты, борной кислоты, бензойной кислоты, 2-(4-гидроксибензоил)-бензойной кислоты, толуолсульфоновой кислоты, бензолсульфоновой кислоты, аскорбиновой кислоты, серной кислоты, малеиновой кислоты, муравьиной кислоты, малоновой кислоты, никотиновой кислоты, щавелевой кислоты, камфорсульфоновой кислоты, цикламовой кислоты, 2,2-дихлоруксусной кислоты, ди(трет-бутил)нафталинсульфоновой кислоты, ди(трет-бутил)нафталиндисульфоновой кислоты, додецилсерной кислоты, этан-1,2-дисульфоновой кислоты, этансульфоновой кислоты, фумаровой кислоты, галактаровой (муциновой) кислоты, гентизиновой кислоты, глюкаровой кислоты, глюконовой кислоты, глицерофосфорной кислоты, бромистоводородной кислоты, йодистоводородной кислоты, 2-гидроксиэтансульфоновой (изэтионовой) кислоты, 1-гидрокси-2-нафтойной кислоты, медроновой (бисфосфоновой) кислоты, метафосфорной кислоты, метилбороновой кислоты, нафталин-1,5-дисульфоновой кислоты, нафталин-2-сульфоновой кислоты, азотной кислоты, оротовой кислоты, 2-оксоглутаровой (кетоглутаровой) кислоты, памовой (эмбоновой) кислоты, пировиноградной кислоты, сахариновой кислоты, салициловой кислоты, 4-аминосалициловой кислоты и тиоциановой кислоты. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно некоторым предпочтительным в настоящее время вариантам реализации настоящего изобретения Y представляет собой фармацевтически приемлемую кислоту, выбранную из группы, состоящей из уксусной кислоты, соляной кислоты, метансульфоновой кислоты, фосфорной кислоты, лимонной кислоты, молочной кислоты, янтарной кислоты, винной кислоты, борной кислоты, бензойной кислоты, толуолсульфоновой кислоты, бензолсульфоновой кислоты, аскорбиновой кислоты, серной кислоты, малеиновой кислоты, муравьиной кислоты, малоновой кислоты, никотиновой кислоты и щавелевой кислоты. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Другие варианты реализации форм фармацевтической кислотной соли могут быть получены из кислот, включая ацетуровую, 4-ацетамидобензойную, адипиновую, аминогиппуровую, 4-аминосалициловую, альгиновую, аспарагиновую, борную, масляную, каприновую (декановую), капроновую (гексановую), угольную, камфорную, камфорсульфоновую, каприловую (октановую), цикламовую, коричную, 2,2-дихлоруксусную, ди(трет-бутил)нафталинсульфоновую, ди(трет-бутил)нафталиндисульфоновую, дегидроуксусную, диатризоевую, додецилсерную, этан-1,2-дисульфоновую, эдетовую, этансульфоновую, 2-этилгексановую, эриторбиновую, муравьиную, фумаровую, галактаровую (муциновую), гентизиновую, глюкогептановую, глюконовую, глюкуроновую, глутаминовую, глутаровую, глицерофосфорную, гликолевую, гиппуриновую, соляную, бромистоводородную, йодисто водородную, 2-(4-гидроксибензоил)-бензойную, 2-гидроксиэтансульфоновую (изетионовую), 1-гидрокси-2-нафтоновую, изомасляную, молочную, лактобионовую, лауриновую, йодоксамовую, изостеариновую, малеиновую, яблочную, малоновую, миндальную, медроновую, метансульфоновую, метафосфорную, метилбороновую, миристиновую, нафталин-1,5-дисульфоновую, нафталин-2-сульфоновую, никотиновую, олеиновую, щавелевую, пальмитиновую, диэтилентриамин-пентауксусную, пропионовую, пропановую, пироглутаминовую, пировиноградную, фосфорную, себациновую, сорбиновую, стеариновую (октадекановую), субериновую, янтарную, серную, винную, тиазолуксусную, тиоциановую, толуолсульфоновую, трифторуксусную и ундециленовую (ундек-10-еноевую) кислоты. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
(ii) Соли присоединения оснований:
В одном варианте реализации Y представляет собой фармацевтически приемлемое органическое или неорганическое основание или остаток основания, выбранного из группы, состоящей из щелочных металлов, щелочноземельных металлов, алюминия, цинка и аммония.
В некоторых предпочтительных в настоящее время вариантах реализации Y представляет собой фармацевтически приемлемый неорганический катион, выбранный из группы, состоящей из лития, натрия, калия, кальция, магния, алюминия, цинка и аммония. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
В другом предпочтительном в настоящее время варианте реализации Y представляет собой фармацевтически приемлемый органический амин, выбранный из группы, состоящей из аммония, первичного амина, вторичного амина, третичного амина, четвертичного аммониевого соединения, аминоспирта и аминосахара. В некоторых предпочтительных в настоящее время вариантах реализации Y представляет собой органическое аминное основание, выбранное из группы, состоящей из бенетамина, бензатина, бетаина, трет-бутиламина (эрбумина), деанола, дициклогексиламина, диэтиламина, 2-диэтиламиноэтанола, диэтаноламина, этаноламина, этилендиамина, гидрабамина, морфолина, 4-(2-гидроксиэтил)морфолина, 1-(2-гидроксиэтил)-пирролидина (эполамина), имидазола, N-метилглюкамина (меглумина), 4-фенилциклогексиламина, пиперазина и трометамина. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Другие основные фармацевтические солевые формы можно получать из оснований, включая гидроксид алюминия, аммиак, аргинин, бенетамин, бензатин, бетаин, трет-бутиламин (эрбумин), гидроксид кальция, холингидроксид, деанол, диэтиламин, 2-диэтиламиноэтанол, диэтаноламин, этаноламин, этилендиамин, гидрабамин, 4-(2-гидроксиэтил)морфолин, 1-(2-гидроксиэтил)пирролидин (эполамин), имидазол, гидроксид лития, лизин, N-метилглюкамин (меглумин), гидроксид магния, 4-фенилциклогексиламин, пиперазин, гидроксид калия, гидроксид натрия, трометамин и гидроксид цинка. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Как понятно специалисту в данной области техники, упомянутые выше кислоты могут реагировать с основным фрагментом (например, аминогруппой) в конъюгате аминокислоты и лекарственного средства с образованием соли. Например, когда кислота представляет собой карбоновую кислоту, и конъюгат содержит основной аминный фрагмент, соль может быть представлена структурой R-COO- R'-NH3+. Подобные продукты реакции кислоты с основанием могут образовываться с любой из других кислот, описанных в настоящем документе.
(iii) Соли с аминокислотами
В другом предпочтительном в настоящее время варианте реализации Y представляет собой аминокислоту, способную образовывать солевые формы, такую как аргинин или лизин. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
(iv) Другие соли
Другие формы фармацевтических солей могут быть получены из солей, включая ацетат аммония, сульфат аммония, ацетат кальция, карбонат кальция, хлорид кальция, глюцептат кальция, лактат кальция, фосфат кальция, сульфат кальция, натрия кальция эдетат, динатрия эдетат, эдетат натрия, йод, карбонат магния, хлорид магния, нитрат магния, сульфат магния, ацетат калия, карбонат калия, цитрат калия, метабисульфит калия, фосфат калия, сорбат калия, ацетат натрия, кислый пирофосфат натрия, альгинат натрия, алкилсульфат натрия, аминобензоат натрия, аскорбат натрия, бензоат натрия, бикарбонат натрия, бисульфат натрия, бисульфат натрия, борат натрия, карбонат натрия, каррагенат натрия, хлорат натрия, хлорид натрия, цитрат натрия, дитионат натрия, гипохлорид натрия, йодид натрия, лактат натрия, метабисульфит натрия, фосфат натрия, пирофосфат натрия, сукцинат натрия, сульфат натрия, сульфит натрия, тартрат натрия, тиогликолят натрия, тиосульфат натрия, хлорид олова, ацетат цинка, хлорид цинка и сульфат цинка. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Аминокислоты (А)
Согласно одному варианту реализации аминокислота (А) находится в L-конфигурации, D-конфигурации или в виде их смеси. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Указанная аминокислота А может представлять собой α-аминокислоту, β-аминокислоту, γ-аминокислоту, δ-аминокислоту или ε-аминокислоту, при этом каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения. В настоящее время предпочтительными аминокислотами являются α-аминокислоты.
В некоторых вариантах реализации указанная аминокислота (А) выбрана из группы, состоящей из аминоизомасляной кислоты (Aib), аргинина (Arg), аспарагина (Asn), аспарагиновой кислоты (Asp), цитруллина (Cit), цистеина (Cys), цистина, диаминобутановой кислоты, диаминомасляной кислоты (Dab), диаминопропионовой кислоты (Dpr), дигидроксифенилаланина, диметиларгинина, глутаминовой кислоты (Glu), пироглутаминовой кислоты (p-Glu), глутамина (Gln), гистидина (His), 1-метилгистидина, 3-метилгистидина, гомосерина (Hse), гомоцитруллина, гидроксипролина (Hyp), лизина (Lys), метиллизина, диметиллизина, триметилизина, азидолизина, метионина (Met), метионинсульфоксида, метионинсульфона, орнитина (Orn), саркозина (Sar), селеноцистеина (Sec), серина (Ser), фосфорсерина, метилсерина, аминосерина (Ams), тиенилаланина (Thi), треонина (Thr), фосфотреонина, триптофана (Trp), тирозина (Tyr), метилтирозина, фосфортирозина, сульфотирозина, α-аминосубериновой кислоты, 3,5-дийодтирозина, пеницилламина (Pen), 4-этиламинфенилглицина, 4-аминофенилглицина, 4-сульфофенилаланина, 4-аминофенилаланина и 2-амино-4[4-(2-амино)-пиримидинил]бутановой кислоты, 3-аминопропионовой кислоты, 6-аминокапроновой кислоты (ε-Ahx), п-аминобензойной кислоты, изонипекотиновой кислоты, статина (STA), 2-аминомасляной (Abu) и 4-аминомасляной кислоты, α-аминоадипиновой кислоты (Aad), α-аминопимелиновой, γ-карбокси-глутаминовой кислоты, γ-гидрокси-глутаминовой кислоты, аминоглицина, а также их производных и аналогов. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
В некоторых вариантах реализации указанная аминокислота (А) выбрана из группы, состоящей из аспарагина, аспарагиновой кислоты, глутамина, глутаминовой кислоты и их производных и аналогов. В одном предпочтительном в настоящее время варианте реализации указанная аминокислота (А) представляет собой глутаминовую кислоту. В другом предпочтительном в настоящее время варианте реализации указанная аминокислота (А) представляет собой аспарагиновую кислоту.
Лекарственное средство (D)
Группа D может быть получена из любого лекарственного средства, выбранного из группы, состоящей из цитотоксического агента, цитостатического агента и химиотерапевтического агента. В некоторых вариантах реализации указанный цитотоксический, цитостатический или химиотерапевтический агент D представляет собой остаток лекарственного средства, выбранного из группы, состоящей из аналога пиримидина, аналога пурина, антифолата, гидроксимочевины, антимикротубулинового агента, алкилирующего агента, противоопухолевого антибиотика, агента, направленно взаимодействующего с топоизомеразой, антиметаболита, связывающего ДНК агента и антагониста ДНК.
В некоторых вариантах реализации указанный цитотоксический, цитостатический или химиотерапевтический агент D представляет собой остаток аналога пиримидина, выбранного из группы, состоящей из цитозин-арабинозида (ара-С, цитарабина), гемцитабина, фторурацила, 5-фтордезоксиридина, фторафура, капецитабина, кармофура, BOF-A2, 5-хлор-2,4-дигидроксипиридина, децитабина, лефлуномида, трифлуридина, идоксуридина, зидовудина, телбивудина, триметоприма, фторцитозина, дезокситимидина и 5-амино-6-нитроурацила. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
В других вариантах реализации указанный цитотоксический, цитостатический или химиотерапевтический агент D представляет собой остаток аналога пурина, выбранного из клофарабина, декарбазина, кладрибина, меркаптопурина, неларабина, пентостатина, тиогуанина, гефитиниба, азатиопурина, 8-азагуанина, иммуциллина-G, 2-фтор-2'-дезоксиаденозина, 9-бета-D-ксилофуранозил-аденина, 3-дезоксигуанина, 6-метил-формицина А, 2-фтораденозина, 1-деаза-аденозина, N-этил-5-карбоксамидоаденозина, 3'-оксоаденозина, 2-аминоаденозина, 6-О-циклометилгуанина, флударабина и 8-йод-гуанина. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
В другом варианте реализации указанный цитотоксический, цитостатический или химиотерапевтический агент D представляет собой остаток противоопухолевого лекарственного средства, выбранного из группы, состоящей из 2,4-диоксо-5-фторпиримидина (5-FU), 5-фтордезоксиридина (5-FUDR), азацитидина (5-AZC), 6-меркаптопурина (6-МР), 6-тиогуанина (6-TG), хлородеоксиаденозина (2-CDA) и пентастатина (dCF). Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
В одном из предпочтительных в настоящее время вариантов реализации настоящего изобретения указанный цитотоксический, цитостатический или химиотерапевтический агент D представляет собой остаток цитарабина. В другом из предпочтительных в настоящее время вариантов реализации настоящего изобретения указанный цитотоксический, цитостатический или химиотерапевтический агент D представляет собой остаток гемцитабина.
Настоящее изобретение также относится к конъюгату аспарагин-лекарственное средство, в котором указанный лекарственный цитотоксический, цитостатический или химиотерапевтический агент D представляет собой антиметаболит, который может ингибировать ферменты, участвующие в метаболизме аспарагина. Одним из важных ферментов, которые могут быть ингибированы конъюгатом аспарагин-лекарственное средство, является аспарагинсинтетаза, которая необходима для синтеза аспарагина в клетках млекопитающих. Другие ферменты, участвующие в метаболизме гликопротеинов, особенно те, которые содержат N-связанные сахара, связанные с аспарагиновой аминокислотой в белке, включая, но не ограничиваясь ими, глюкозидазу I, глюкозидазу II, кальнексин и альфа-глюкозилтрансферазу, потенциально могут быть ингибированы конъюгатом аспарагин-лекарственное средство. N-гликозилирование в N-гликановых белках происходит на аспарагине на консенсусной последовательности Asn-X-Ser/Thr, а вмешательство в метаболизм гликозилирования нарушает фолдинг и секрецию гликопротеинов. Ингибирование гликозилирования основных гликопротеинов вызовет остановку роста клеток и гибель клеток.
Конъюгат аспарагин-лекарственное средство также может влиять на метаболизм других аминокислот, таких как орнитин, поскольку, как было показано, аспарагин принимает участие в мембранном антипорте Na+/H+ в индукции орнитиндекарбоксилазы (Fong and Law, 1988).
Конъюгат аспарагин-лекарственное средство может быстро поглощаться транспортерами аспартата и глутамата, которые опосредуют доставку глутамина/глутаминовой аминокислоты и аспарагина/аспарагиновой аминокислоты через гематоэнцефалический барьер (ГЭБ) и его можно применять в качестве эффективной системы доставки лекарственных средств для транспортировки химиотерапевтических препаратов для лечения опухолей ЦНС.Было показано, что уровень цереброспинальной жидкости (ЦСЖ) и уровень аспарагина в плазме значительно ниже у пациентов с первичной и вторичной опухолями центральной нервной системы (ЦНС) (Piek et al., 1987).
Было показано, что раковые клетки гепатомы экспрессируют транспортер глутамина, который обладает гораздо более высокой скоростью поглощения глутамина в клетках гепатомы человека, а не в нормальных гепатоцитах, что указывает на то, что конъюгат глютамин-лекарственное средство можно применять для лечения рака печени.
Как отмечено выше, в конъюгатах согласно настоящему изобретению указанный остаток лекарственного средства (D) присоединен к аминокислоте (А) через функциональную группу боковой цепи (А). Некоторые неограничивающие варианты реализации таких конъюгатов описаны ниже.
В определенных вариантах реализации указанное соединение содержит аминокислотный остаток аспарагиновой кислоты или глутаминовой кислоты, конъюгированный с лекарственным средством, выбранным из группы, состоящей из аналогов пурина и аналогов пиримидина. В определенных вариантах реализации указанное лекарственное средство выбрано из цитарабина и гемцитабина. В определенных вариантах реализации указанная солевая форма конъюгата аминокислотота-лекарственное средство представляет собой кислотную солевую форму, выбранную из уксусной кислоты и соляной кислоты.
Без связи с какой-либо теорией, соли аминокислотных конъюгатов согласно настоящему изобретению переносятся в клетку транспортерами аминокислот и тем самым обходят механизмы множественной лекарственной резистентности (МЛР) и останавливают рост клеток или убивают клетку изнутри.
Согласно некоторым вариантам реализации указанная аминокислота (А) выбрана из группы, состоящей из аспарагиновой кислоты и глутаминовой кислоты, и указанное соединение представлено структурой формулы (II) или (III)
где D и Y такие, как определено выше.
В определенном варианте реализации формулы (II) А представляет собой аспарагиновую кислоту, a D представляет собой остаток цитарабина, и указанное соединение представлено структурой формулы (1):
В одном варианте реализации формулы (1) указанное соединение представляет собой соль уксусной кислоты (то есть ацетатную соль), которая представлена структурой формулы (1А):
В другом варианте реализации формулы (1) указанное соединение представляет собой соль соляной кислоты (то есть гидрохлоридную соль), которая представлена структурой формулы (1В):
В другом определенном варианте реализации формулы (II) А представляет собой аспарагиновую кислоту, a D представляет собой остаток гемцитабина, и указанное соединение представлено структурой формулы (2):
В определенном варианте реализации формулы (III) А представляет собой глутаминовую кислоту, a D представляет собой остаток цитарабина, и указанное соединение представлено структурой формулы (3):
В другом определенном варианте реализации формулы (III) А представляет собой глутаминовую кислоту, a D представляет собой остаток гемцитабина, и указанное соединение представлено структурой формулы (4):
В любом из упомянутых выше соединений формул (1), (2), (3) и (4) Y может представлять собой любой из описанных выше вариантов, может предпочтительно представлять собой фармацевтически приемлемую кислоту, выбранную из группы, состоящей из уксусной кислоты, соляной кислоты, метансульфоновой кислоты, фосфорной кислоты, лимонной кислоты, молочной кислоты, янтарной кислоты, винной кислоты, борной кислоты, бензойной кислоты, толуолсульфоновой кислоты, бензолсульфоновой кислоты, аскорбиновой кислоты, серной кислоты, малеиновой кислоты, муравьиной кислоты, малоновой кислоты, никотиновой кислоты и щавелевой кислоты.
Определения
Для удобства и ясности здесь приведены некоторые термины, используемые в описании, примерах и формуле изобретения.
Термин «остаток лекарственного средства» относится к лекарственному средству, исключая функциональную группу, через которую присоединена указанная аминокислота при образовании конъюгата аминокислота-лекарственное средство A-D.
Термин «кислотный остаток» относится к конъюгатному основанию этой кислоты. Например, конъюгатное основание соляной кислоты (HCl) представляет собой хлорид (Cl-). Конъюгатное основание уксусной кислоты (СН3СООН) представляет собой ацетат (СН3СОО-).
Термин «лекарственное средство» обозначает любой фармакологически активный агент, способный останавливать рост клеток или индуцировать гибель гиперпролиферативной клетки, в которой он находится, и включает в себя известные цитотоксические, цитостатические, антипролиферативные лекарственные средства, такие как известные в данной области, как проиллюстрировано выше. Классификация лекарственных средств приводится в настоящем документе только ради удобства и не предназначена для ограничения какого-либо компонента конкретным перечисленным применением или применениями.
Используемый в настоящем документе термин «цитотоксический агент» относится к агенту, который токсичен для клеток, например, для раковых клеток, повреждает и/или разрушает их. Химиотерапия, проводимая при помощи химиотерапевтических препаратов, является формой цитотоксической терапии.
Используемый в настоящем документе термин «цитостатическое лекарственное средство» относится к любому веществу, которое ингибирует рост и деление клеток.
Используемый в настоящем документе термин «химиотерапевтический агент» относится к лекарственному средству, применяемому при химиотерапии.
Используемый в настоящем документе термин «остановка роста клеток» и «индуцирование смерти» гиперпролиферативной клетки, например, неопластической клетки, относится к замедлению, прерыванию или остановке роста и метастазирования и не обязательно указывает на полное устранение неопластического роста.
В настоящем изобретении применяют аминокислоты, которые доступны коммерчески или могут быть получены обычными методами синтеза. Природные аминокислоты и их производные обозначены трехбуквенными кодами согласно ИЮПАК. В тех случаях, когда нет прямого указания, можно применять L- или D-изомер. D-изомеры обозначены буквой «D» перед аббревиатурой остатка.
Используемый в настоящем документе термин «стереоизомер» относится к соединению, состоящему из тех же атомов, связанных одними и теми же связями, но имеющих разные трехмерные структуры, которые не являются взаимозаменяемыми. Трехмерные структуры называются конфигурациями. Используемый в настоящем документе термин «энантиомер» относится к двум стереоизомерам, молекулы которых являются не совпадающими при наложении зеркальными изображениями друг друга. Термин «хиральный центр» относится к атому углерода, к которому присоединены четыре разные группы. Используемый в настоящем документе термин «диастереомеры» относится к стереоизомерам, которые не являются энантиомерами. Кроме того, два диастереомера, которые имеют различную конфигурацию только в одном хиральном центре, в настоящем документе называются «эпимерами». Термины «рацемат», «рацемическая смесь» или «рацемическая модификация» относятся к смеси равных частей энантиомеров. Настоящее изобретение включает энантиомеры соединений, имеющих общую формулу (I), (II) и (III), и конкретные описанные в настоящем документе соединения.
Способы синтеза
В конъюгате согласно настоящему изобретению лекарственное средство ковалентно присоединено к аминокислоте или аналогу аминокислоты. Специалист в данной области сможет оптимизировать соответствующую связь и положение лекарственного фрагмента внутри соединения. При принятии решения специалист в данной области техники также учитывает различные задачи, такие как выбор конкретного лекарственного средства, выбор производных, выбор положения присоединения к лекарственному средству и требования, касающиеся внутриклеточных ферментов хозяина для активации лекарственного средства.
Таким образом, в соответствии с другим аспектом, настоящее изобретение относится к способу получения фармацевтически приемлемой соли, представленной структурой формулы (I), включающий стадию
(а) взаимодействие соединения формулы (IV):

где А и D такие, как определено выше, n представляет собой целое число 1-6; и
каждый Р независимо в каждом случае представляет собой защитную группу; где каждый Р присоединен к любой функциональной группе, доступной для защиты, на аминокислоте (А), лекарственном остатке (D) или к аминокислоте (А) и к лекарственному остатку (D);
с реагентом, способным удалять защитную группу (защитные группы) Р; и
(b) добавление реагента, способного вводить группу Y, с получением соли, представленной формулой:

где Y такой, как описано выше.
Как упомянуто выше, неожиданно было обнаружено, что соли согласно настоящему изобретению можно получать путем удаления защитных групп с соединения-предшественника, содержащего одну или более защитных групп (Р) с применением того же реагента, который образует указанную соль. Таким образом, в соответствии с принципами настоящего изобретения реагент, способный вводить группу Y, может одновременно удалять защитную группу (защитные группы) Р и образовывать соль формулы (I) за одну стадию, тем самым устраняя промежуточную стадию снятия защиты перед образованием соли, что в конечном итоге приводит к повышению чистоты фармацевтического продукта. В частности, избегают применения ТФУ для снятия защитных групп ВОС, что приводит к образованию соли конъюгата согласно настоящему изобретению, представленной формулой I, при этом указанная соль свободна от токсичной ТФУ. Определенные варианты реализации указанного способа описаны ниже. Соединения, описанные в настоящем документе, включают фармацевтическую солевую форму противоопухолевого лекарственного средства, конъюгированного с аминокислотой, производными аминокислот или аналогами аминокислот. Соединения согласно настоящему изобретению можно легко получить из аминокислот способами, известными специалисту в данной области. Например, конъюгаты или их предшественники можно получить в соответствии со способами, описанными в публикации международной патентной заявки № WO 2005/072061 и в патенте США №7989188, содержание которых включено в настоящее описание посредством ссылки. Конъюгаты (или их предшественники с защитными группами) могут затем взаимодействовать с реагентом, способным вводить группу Y (например, указанный реагент представляет собой кислоту или основание), с получением соединения формулы (I). Дополнительные способы, пригодные для получения соединений согласно настоящему изобретению, представляют собой, например, способы, описанные в публикациях международных патентных заявок № WO 96/30036 и WO 97/36480 и в патентах США №№5643957 и 5650386, среди прочих. Например, указанные соединения можно получать путем взаимодействия единственной аминокислоты с соответствующим катализирующим агентом, который вступает во взаимодействие со свободным фрагментом, присутствующим в аминокислоте, и конъюгируя его со свободным фрагментом на лекарственном средстве с образованием сложного эфира или амида. Защитные группы можно использовать для предотвращения нежелательных побочных реакций, как известно специалистам в данной области техники. Содержание каждой из приведенных выше ссылок включено в настоящий документ посредством ссылки.
Например, в качестве иллюстрации, а не для ограничения, конъюгаты формулы (II) (конъюгаты аспарагиновая кислота-лекарственное средство) или формулы (III) (конъюгаты глутаминовая кислота-лекарственное средство) можно получать путем взаимодействия аспарагиновой кислоты или глутаминовой кислоты, защищенных на амине и альфа-карбоксиле (соединение (i) и (iii) на схемах 1А и 1В), с лекарственным средством или его активированным и/или защищенным производным в присутствии реагента для реакции сочетания с последующим снятием защиты. Лекарственное средство (D) может быть незащищенным или защищенным на любой доступной функциональной группе (например, ОН, NH2, SH, карбоксил и т.д.) любой из защитных групп, описанных в настоящем документе. Примерные процессы представлены на схемах 1А и 1В:
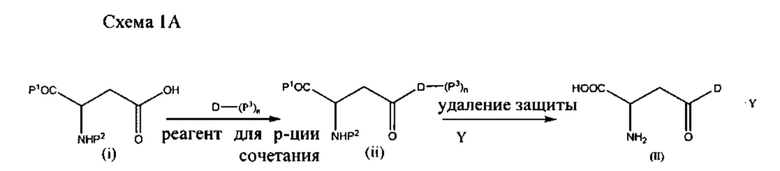
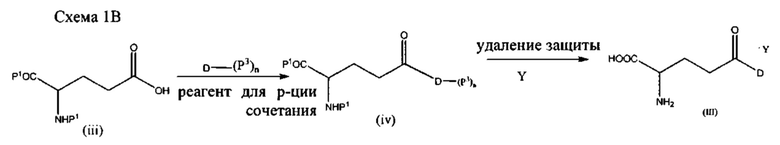
где Р1 представляет собой карбоксизащитную группу, Р2 представляет собой аминозащитную группу, Р3 представляет собой защитную группу, расположенную на одной или более функциональных группах на лекарственном средстве D, и n равно 0, 1, 2 или 3. В зависимости от количества функциональных групп на лекарственном средстве n также может составлять 4, 5, 6 и т.д.
Согласно одному варианту реализации настоящего изобретения указанное лекарственное средство представляет собой цитарабин. В других вариантах реализации указанное лекарственное средство представляет собой защищенный цитарабин, содержащий защитные группы на 2', 3' и/или 5' гидроксильных фрагментах, такой как трифенилметил-цитарабин (CAS 7075-13-0), бензоил-цитарабин (CAS 34270-10-5), адмантоил-цитарабин (CAS 23113-01-1) и триметилсилил-цитарабин. Согласно другому варианту реализации настоящего изобретения указанное лекарственное средство представляет собой гемцитабин. В других вариантах реализации указанное лекарственное средство представляет собой защищенный гемцитабин, содержащий защитные группы на 3' и/или 5' гидроксильных фрагментах, такой как трифенилметил-гемцитабин (CAS 1642862-24-5).
Неограничивающим примером защищенного лекарственного средства является защищенный цитарабин, где по меньшей мере один из 2', 3' или 5' гидроксилов защищен группой Р3, как описано выше. В одном неограниченном варианте реализации, описанном ниже для целей иллюстрации, указанное соединение представлено формулой Е, где каждый из 2', 3' или 5' гидроксилов защищен группой Р3.
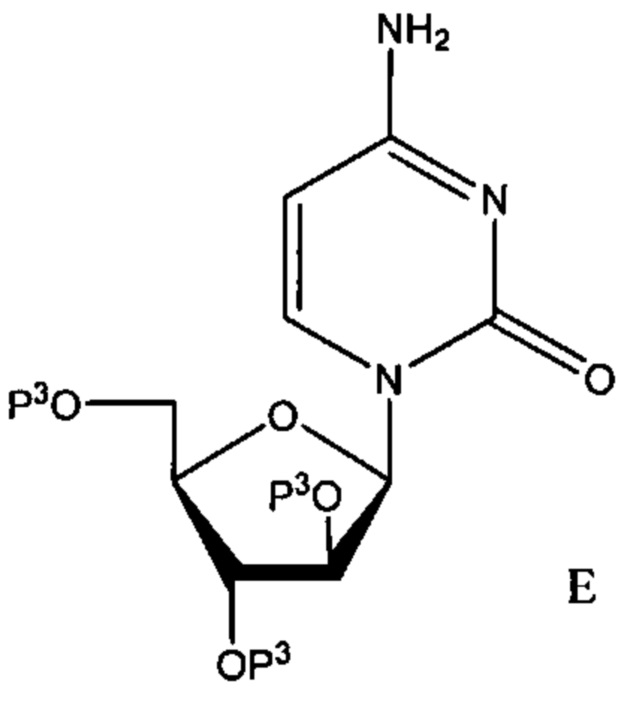
В других вариантах реализации защищена только группа 5'-ОН, тогда как 2' и 3' гидроксилы не защищены.
Другим неограничивающим примером защищенного лекарственного средства является защищенный гемцитабин, где по меньшей мере один из 3' или 5' гидроксилов защищен группой Р3, как определено выше. В одном неограничивающем варианте реализации, описанном ниже для целей иллюстрации, указанное соединение представлено формулой F, где каждый из 3' или 5' гидроксилов защищен группой Р3.
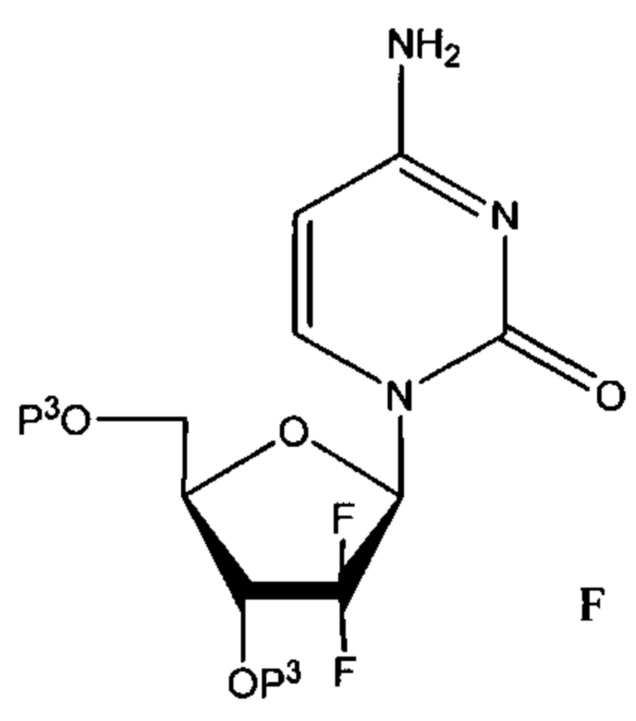
В других вариантах реализации защищена только группа 5'-ОН, тогда как 3' гидроксил не защищен.
В одном варианте реализации, обозначенном в настоящем документе как «Путь А», стадию снятия защитных групп проводят в присутствии трифторуксусной кислоты (ТФУ) с последующим взаимодействием с кислотой с введением группы Y. Согласно этому варианту реализации настоящее изобретение относится к фармацевтически приемлемой соли, предпочтительно содержащей менее чем примерно 5% ТФУ в качестве остаточной примеси, предпочтительно менее чем примерно 1% ТФУ, более предпочтительно менее чем примерно 0,1% ТФУ. Наиболее предпочтительно соль согласно настоящему изобретению по существу свободна от примесной ТФУ.
В другом варианте реализации, обозначенном в настоящем документе как «Путь В», стадию снятия защитных групп проводят непосредственно с реагентом, способным вводить группу Y (т.е., с кислотой), чтобы непосредственно получить соль согласно настоящему изобретению. В этом варианте реализации защитные группы удаляют одновременно с введением группы Y, и полученная в результате фармацевтически приемлемая соль полностью свободна от примесной ТФУ.
Защитные группы (Р)
Предшественники соединения (I) могут содержать одну или более защитных групп (Р), приведенных в качестве примера выше как Р1, Р2 и Р3, которые могут представлять собой любую защитную группу, известную специалисту в данной области. Термин «защитная группа» относится к химическим остаткам, используемым для блокирования реакционноспособных участков во время химического синтеза, что позволяет проводить химическую реакцию селективно на одном реакционноспособном участке в соединении, содержащем несколько функциональных групп, при этом другие реактивные сайты должны быть временно заблокированы. Остатки, используемые для блокирования этих реакционноспособных участков, называются защитными группами.
Защитная группа может представлять собой гидроксильную защитную группу, аминозащитную группу, карбоксизащитную группу и т.д. Используемый в настоящем документе термин «защитная группа ОН» или «гидроксизащитная группа» относится к легко расщепляемой группе, связанной с гидроксильными группами. Используемый в настоящем документе термин «защитная группа NH» или «аминозащитная группа» относится к легко расщепляемой группе, связанной с аминогруппами. Используемый в настоящем документе термин «карбоксизащитная группа» относится к легко расщепляемой группе, связанной с карбоксильными группами.
Согласно одному варианту реализации защитная группа Р выбрана из группы ацетамидометила (Acm), ацетила (Ас), ацетонида, адамантилокси (AdaO), альфа-аллила (OAll), Alloc, бензоила (Bz), бензила (Bzl), бензилокси (BzlO), бензилоксикарбонила (Z), бензилоксиметила (Bom), бис-диметиламино (NMe2), 2-бромбензилоксикарбонила (2-Br-Z), трет-бутокси (tBuO), трет-бутоксикарбонила (Boc), трет-бутоксиметила (Bum), трет-бутила (tBu), трет-бутилтио (tButhio), 2-хлорбензилоксикарбонила (2-Cl-Z), 2-хлортритила (2-Cl-Trt), циклогексилокси (сНхО), 1-циклопропил-1-метилэтила (Dmcp), 2,6-дихлорбензила, 4,4'-диметоксибензгидрила (Mbh), 1-(4,4-диметил-2,6-диоксоциклогексилиден)-3-метилбутила (ivDde), 4 {N-[1-(4,4-диметил-2,6-диоксоциклогексилиден)-3-метилбутил]амино}бензилокси (ODmab), 2,4-динитрофенила (Dnp), флуоренилметоксикарбонила (Fmoc), формила (For), мезитилен-2-сульфонила (Mts), 4-метоксибензила, 4-метокси-2,3,6-триметилбензолсульфонила (Mtr), 4-метокситритила (Mmt), 4-метилбензила (MeBzl), 3-метилпент-3-ила (Mpe), 1-метил-1-фенилэтила (PhiPr), метила, 4-метилтритила (Mtt), 3-нитро-2-пиридинсульфенила (Npys), 2,2,4,6,7-пентаметилдигидробензофуран-5-сульфонила (Pbf), 2,2,5,6,8-пентаметилхроман-6-сульфонила (Pmc), тозила (Tos), трифторацетила (Tfa), триметилацетамидометила (Tacm), трифенилметила (тритил, Trt) и ксантила (Xan). Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Неограничивающим примером гидроксизащитной группы является ацильная группа (COR, где R = алкил, ар ил и т.д.). Предпочтительная в настоящее время ацильная группа представляет собой ацетильную группу (т.е., OR'=ацетат, ОАс). Другим примером гидроксизащитной группы является силильная группа, которая может быть замещена алкилом (триалкилсилил), арилом (триарилсилил) или их комбинацией (например, диалкилфенилсилил). Предпочтительным примером силильной защитной группы является триметилсилил (TMS) или ди-трет-бутилдиметилсилил (TBDMS), триизопропилсилил (TIPS), триэтилсилил (TES). Другие примеры гидроксизащитных групп включают, например, С1-С4-алкил, -CO-(C1-C6-алкил), -SO2-(C1-С6-алкил), -SO2-арил, -СО-Ar, где Ar представляет собой арильную группу, определенную выше, и -СО- (C1-C6-алкил)Ar (например, карбоксибензил (Bz)).
Примеры аминозащитных групп включают трет-бутоксикарбонил (ВОС), бензилоксикарбонил, ацетил, фенилкарбонил или силильную группу, которая может быть замещена алкилом (триалкилсилил), арилом (триарилсилил) или их комбинацией (например, диалкилфенилсилил), например, триметилсилил (TMS) или трет-бутилдиметилсилил (TBDMS). Другие примеры гидроксизащитных групп включают, например, С1-С4-алкил (например, метил, этил, пропил, бутил и т.п.), -CH2Ph (бензил или bzl), аллил (All), (аллил)-СО-(C1-С6-алкил), -SO2-(С1-С6-алкил), -SO2-арил, -СО-Ar, где Ar представляет собой арильную группу, определенную выше, и -CO-(C1-С6-алкил)Ar (например, карбоксибензильную группу (Bz)). Другие примеры гидроксизащитных групп включают чувствительные к воздействию кислоты защитные группы, такие как тетрагидропиранил (ТНР), метоксиметил (MOM), трифенилметил (тритил) и диметокситритил (DMT). Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Типичные карбоксизащитные группы включают, но не ограничиваются ими, метил, этил, трет-бутил, бензил (Bn), п-метоксибензил (РМВ), 9-флуоренилметил (Fm), триметилсилил (ТМС), трет-бутилдиметилсилил (TBDMS), дифенилметил (бензгидрил, DPM) и т.п. Защитные группы можно удалять при помощи агентов для удаления защитных групп, которые в соответствии с принципами настоящего изобретения предпочтительно являются такими же, как фрагменты фармацевтической соли. В некоторых вариантах реализации фармацевтические кислоты, подходящие для снятия защитных групп, восприимчивым к кислотам, представляют собой уксусную кислоту, ацетуровую, 4-ацетамидобензойную, адипиновую, аминогиппуровую, 4-аминосалициловую, аскорбиновую, альгиновую, аспарагиновую, бензолсульфоновую, бензойную, борную, масляную, каприновую (декановую), капроновую (гексановую), угольную, лимонную, камфорную, камфорсульфоновую, каприловую (октановую), цикламовую, коричную, 2,2-дихлоруксусную, ди(трет-бутил)нафталинсульфоновую, ди(трет-бутил)нафталиндисульфоновую, дегидроуксусную, диатризоевую, додецилсерную, этан-1,2-дисульфоновую, эдетовую, этансульфоновую, 2-этилгексановую, эриторбиновую, муравьиную, фумаровую, галактаровую (муциновую), гентизиновую, глюкаровую, глюкогептановую, глюконовую, глюкуроновую, глутамовую, глутаровую, глицерофосфорную, гликолевую, гиппуровую, соляную, бромистоводородную, йодистоводородную, 2-(4-гидроксибензоил)-бензойную, 2-гидроксиэтансульфоновую (изетионовую), 1-гидрокси-2-нафтоновую, изомасляную, молочную, лактобионовую, лауриновую, йодоксамовую, изостеариновую, малеиновую, яблочную, малоновую, миндальную, медроновую, метансульфоновую, метафосфорную, метилбороновую, миристиновую, нафталин-1,5-дисульфоновую, нафталин-2-сульфоновую, никотиновую, азотную, олеиновую, оротовую, щавелевую, 2-оксоглутаровую (кетоглутаровую), пальмитиновую, памоевую (эмбоновую), диэтилентриамин-пентауксусную, пропионовую, пропановую, пироглутаминовую, пировиноградную, фосфорную, сахариновую, салициловую, себациновую, сорбиновую, стеариновую (октадекановую), субериновую, янтарную, серную, винную, тиазолуксусную, тиоциановую, толуолсульфоновую, трифторуксусную и ундециленовую (ундек-10-еноевую) кислоты. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
В определенных вариантах реализации фармацевтически приемлемая кислота, которая вступает во взаимодействие с соединением-предшественником формулы (IV) и снимает защитную группу, выбрана из группы, состоящей из уксусной кислоты, соляной кислоты, метансульфоновой кислоты, фосфорной кислоты, лимонной кислоты, молочной кислоты, янтарной кислоты, винной кислоты, борной кислоты, бензойной кислоты, толуолсульфоновой кислоты, бензолсульфоновой кислоты, аскорбиновой кислоты, серной кислоты, малеиновой кислоты, муравьиной кислоты, малоновой кислоты, никотиновой кислоты и щавелевой кислоты. Согласно этим вариантам реализации полученная соль выбрана из группы, состоящей из ацетата, гидрохлорида, метансульфоната, фосфата, цитрата, лактата, сукцината, тартрата, бората, бензоата, толуолсульфоната, бензолсульфоната, аскорбата, сульфата, малеата, формиата, малоната, никотината и оксалата. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
В определенных вариантах реализации фармацевтические основания, пригодные для снятия защитных групп, восприимчивых к основаниям, включают: гидроксид алюминия, аммиак, аргинин, бенетамин, бензатин, бетаин, трет-бутиламин (эрбумин), гидроксид кальция, холингидроксид, деанол, диэтиламин, 2-диэтиламиноэтанол, диэтаноламин, этаноламин, этилендиамин, гидрабамин, 4-(2-гидроксиэтил)морфолин, 1-(2-гидроксиэтил)пирролидин (эполамин), имидазол, гидроксид лития, лизин, N-метилглюкамин (меглумин), гидроксид магния, 4-фенилциклогексиламин, пиперазин, гидроксид калия, гидроксид натрия, трометамин и гидроксид цинка. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
В других вариантах реализации H2 Pd/C можно применять в качестве агента, снимающего защитные группы, когда защитной группой является, например, бензил.
Соединения-предшественники, содержащие защитные группы, в целом описаны на схемах 1А и 1В выше.
В определенных вариантах реализации предшественником является Вос-Asp(Цитарабин)-OtBu (формула А), тогда защитные группы удаляют кислотой с получением Asp(Цитарабин)⋅Y (формула 1), и Y представляет собой кислотную солевую форму фармацевтической кислоты.
В определенных вариантах реализации предшественником является Вос-Asp(Гемцитабин)-OtBu (формула В), тогда защитные группы удаляют кислотой с получением Asp(Гемцитабин)⋅Y (формула 2), и Y представляет собой кислотную солевую форму фармацевтической кислоты.
В других определенных вариантах реализации предшественником является Вос-Glu(Цитарабин)-OtBu (формула С), тогда защитные группы удаляют кислотой с получением Clu(Цитарабин)⋅Y (формула 3), и Y представляет собой кислотную солевую форму фармацевтической кислоты.
В других определенных вариантах реализации предшественником является Вос-Clu(Гемцитабин)-OtBu (формула D), тогда защитные группы удаляют кислотой с получением Clu(Гемцитабин)⋅Y (формула 4), и Y представляет собой кислотную солевую форму фармацевтической кислоты. Другие подходящие защитные группы описаны, например, в Wuts and Greene, 2007, "Greene's protective groups in organic synthesis", Fourth edition, содержание которого включено в настоящий документ посредством ссылки.
Хотя предпочтительно, чтобы защитную группу удаляли с помощью соединения, образующего фармацевтическую соль, настоящее изобретение не ограничивается такими вариантами реализации. Специалисту в данной области очевидно, что защитную группу можно удалить при помощи других агентов, удаляющих защитную группу, и солевую форму лекарственного средства можно получать на отдельной последующей стадии.
Лекарственное средство можно применять как есть или в качестве его активированного или защищенного производного. Например, один вариант реализации настоящего изобретения включает применение цитарабина или гемцитабина, у которых защищены гидроксильные группы сахарных фрагментов (С2, С3 и/или С5), для того, чтобы предотвратить или свести к минимуму образование побочных продуктов диаминокислота-цитарабин или диаминокислота-гемцитабин (т.е. вторая аминокислота, присоединенная к 5'-ОН арабинозной части). Защитной группой на сахарном фрагменте может быть любая из защитных групп, описанных выше, и их можно удалить с помощью соответствующего агента для удаления защитных групп, как описано в настоящем документе.
Принципы, относящиеся к выбору места присоединения органических кислот, аминокислот, защитных групп и формы фармацевтической соли и т.д., будут подробно описаны в настоящем документе для примерных соединений. Принципы можно обобщить следующим образом:
a) Выбор органической кислоты/аминокислоты или производного аминокислоты или аналога аминокислоты: подходящими являются аспартат/аспарагин и глутамат/глутамин и любые их производные или аналоги, которые могут быстро поглощаться злокачественными или раковыми клетками, а также другие органические кислоты, которые улучшают растворимость лекарственного средства, стабильность, биодоступность и приверженности пациента терапии;
b) Выбор защитных групп органической кислоты/аминокислоты: группы могут быть выбраны из защитных групп, подходящих для химически функциональных групп на органической кислоте/аминокислоте, которые можно удалить при помощи соединения, которое образует фармацевтическую соль, как и другие удаляемые защитные группы;
c) Выбор лекарственного средства: лекарственное средство может быть выбрано из антипролиферативных агентов, цитотоксических агентов и цитостатических агентов;
d) Выбор защитных групп лекарственного средства: группы могут быть выбраны из защитных групп, подходящих для химически функциональных групп на лекарственном средстве, которые можно удалить при помощи соединения, которое образует фармацевтическую соль, а также другие удаляемые защитные группы;
e) Выбор условий реакции для конъюгирования лекарственного средства с органической кислотой: Оптимальные растворители, катализаторы, добавки, время и температура различных стадий реакции.
f) Выбор условий для удаления защитных групп: при помощи фармацевтической соли или другого соединения, подходящего для удаления защитных групп и акцепторов, как известно специалистам в данной области техники (Wuts and Greene, 2007).
g) Выбор фармацевтической соли: соль может быть выбрана из минерального вещества, соли присоединения кислоты, соли присоединения основания и их комбинации.
h) Выбор условий для получения фармацевтической соли: Оптимальные растворители/антирастворители, концентрации, время и температура реакции.
В качестве примеров соединений и условий для реакции конъюгирования приведены следующие компоненты, без ограничения:
Растворители: диметилформамид (ДМФА), диметилацетамид (ДМА), этанол, диэтиловый эфир, этилацетатдиметилсульфоксид (ДМСО), дихлорметан (ДХМ), дихлорэтан (DCE), гептан, гексан, пиридин, 2-пропанол, метанол, трет-бутилметиловый эфир, N-метилпирролидон (NMP), тетрагидрофуран (ТГФ), трифторэтанол (ТФЭ), вода и их комбинация;
Катализаторы/реагенты для реакции сочетания: ВОР (бензотриазол-1-ил-окси-трис-(диметиламино)фосфоний гексафторфосфат), CDI (N,N'-карбонилдиимидазол), COMU (1-[(1-(циано-2-этокси-2-оксоэтилиденаминоокси)диметиламиноморфолино)]урония гексафторфосфат), DCC (N,N'-дициклогексилкарбодиимид), DIC (N,N'-диизопропилкарбодиимид), EDC (1-этил-3-(3-диметиламинопропил)карбодиимид), DEPBT (3-(диэтоксифосфорилокси)-1,2,3-бензо[(1]триазин-4(3Н)-он), HATU, HBTU (2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат), HCTU (O-(1Н-6-хлоробензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат), MSNT (1-(мезитилен-2-сульфонил)-3-нитро-1Н-1,2, 4-триазол), РуВОР (бензотриазол-1-ил-окси-триспирролидинофосфония гексафторфосфат), PyBroP (бромо-трис-пирролидинофосфония гексафторфосфат), TBTU (2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметиламиния тетрафторборат), TFFH (тетраметилфторформамидиния гексафторфосфат), TOTU (O-[(этоксикарбонил)цианометиленамино]-N,N,N',N',-тетраметилуроний);
Добавки: DIPEA (диизопропилэтиламин), DMAP (диметиламинопиридин), пиридин, триэтиламин, HOAt (N-гидрокси-9-азабензотриазол), HOBt (N-гидроксибензотриазол), Oxyma pure, NaclO4, LiCl, KSCN и Triton.
Температуры реакции составляют от примерно 0°C до примерно 80°C, предпочтительно от примерно 4°C до примерно 25°C;
Время реакции составляет от примерно 3 минут до примерно 72 часов, предпочтительно от примерно 3 до примерно 24 часов;
Соединения и условия для удаления защитных групп проиллюстрированы, но не ограничиваются следующими компонентами:
Фармацевтические соли: включая фармацевтически приемлемые минеральные вещества, основания, кислоты или соли; предпочтительная кислота и соль для удаления защитных групп имеют pKa≤3, а предпочтительное основание и соль имеют pKa>9.
Предпочтительная кислота для удаления защитных групп, имеющая pKa≤3, выбрана из группы, состоящей из: бензолсульфоновой, камфорсульфоновой, цикламовой, 2,2-дихлоруксусной, ди(трет-бутил)нафталинсульфоновой, ди(трет-бутил)нафталиндисульфоновой, додецилсерной, этан-1,2-дисульфоновой, этансульфоновой, фумаровой, галактаровой (муциновой), гентизиновой, глюкаровой, глюконовой, глицерофосфорной, соляной, бромистоводородной, йодистоводородной, 2-гидроксиэтансульфоновой (изетионовой), 1-гидрокси-2-нафтоновой, малеиновой, малоновой, медроновой (бисфосфоновой), метансульфоновой, метафосфорной, метилбороновой, нафталин-1,5-дисульфоновой, нафталин-2-сульфоновой, азотной, оротовой, щавелевой, 2-оксоглутаровой (кетоглутаровой), памоевой (эмбоновой), пировиноградной, фосфорной, сахариновой, салициловой, серной, винной, тиоциановой, толуолсульфоновой и трифторуксусной кислот.
Другие соединения, удаляющие защитную группу, включают, но не ограничиваются ими: TMSBr, HBF4, HF, пиперидин, Pd(Ph3P)4, NH2NH2, I2 и соединения Tl, Hg, Ag и Pd;
Акцепторы: Н2О, TIS, TES, EDT, м-крезол, тиоанизол, анизол, фенол;
Температуры составляют от -20°C до 80°C, предпочтительно от 0°C до 25°C;
Время составляет от 1 минуты до 24 часов, предпочтительно 1-3 часа;
Соединения и условия для образования фармацевтической соли выбирают из:
Растворители для растворения соединения с фармацевтической кислотой: Н2О, диоксан, этилацетат, метанол, этанол, изопропанол, ДМСО, тетрагидрофуран (ТГФ) и трифторэтанол (TFE) или любую их комбинацию. В одном из предпочтительных в настоящее время вариантов реализации настоящего изобретения растворитель представляет собой H2O. В другом предпочтительном в настоящее время варианте реализации настоящего изобретения растворитель представляет собой диоксан;
Антирастворитель для осаждения продукта, обозначенного формулой (I): диэтиловый эфир, трет-бутилметиловый эфир (tBME), гексан, гептан, ДХМ.
Молярное отношение от 1:1 до 1:30 соединения к фармацевтической соли, предпочтительно от 1:1,1 до 1:3 при комнатной температуре (25°C) инкубируют в течение 1 часа.
Данное изобретение дополнительно относится к гидратам фармацевтически приемлемых солей общей формулы (I).
В одном определенном варианте реализации фармацевтически приемлемая кислота представляет собой HCl и в качестве растворителя применяют Н2О или диоксан. Неожиданно было обнаружено, что при применении диоксана/HCl продукт выпадает в осадок во время реакции удаления защитных групп, и нет необходимости в проведении отдельной стадии осаждения.
Фармацевтические композиции
Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим по меньшей мере одно из соединений по настоящему изобретению и одно или более фармацевтически приемлемых вспомогательных веществ или разбавителей.
Композиции, содержащие соединения согласно настоящему изобретению, можно применять для доставки активных агентов в отдельные биологические системы и для обеспечения повышенной или улучшенной биодоступности активного агента по сравнению с введением активного агента без агента для дост осадок во время реакции удаления защитных групп, и нет необходимости в проведении отдельной стадии осаждения.
Фармацевтические композиции
Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим по меньшей мере одно из соединений по настоящему изобретению и одно или более фармацевтически приемлемых вспомогательных веществ или разбавителей.
Композиции, содержащие соединения согласно настоящему изобретению, можно применять для доставки активных агентов в отдельные биологические системы и для обеспечения повышенной или улучшенной биодоступности активного агента по сравнению с введением активного агента без агента для доставки. Доставку можно улучшить за счет доставки большего количества активного агента в течение определенного периода времени или при доставке активного агента за определенный период времени (например, для обеспечения более быстрой или отсроченной доставки) или в течение определенного периода времени (например, пролонгированная доставка).
Фармацевтические композиции согласно настоящему изобретению можно получать способами, хорошо известными в данной области, например, при помощи обычных процессов смешивания, растворения, гранулирования, измельчения, пульверизации, дражирования, левигации, эмульгирования, инкапсуляции, захвата или лиофилизации.
Используемый в настоящем документе термин «фармацевтическая композиция» относится к составу одного или более описанных в настоящем документе соединений или их физиологически приемлемых солей или растворителей с другими химическими компонентами, такими как физиологически приемлемые носители и вспомогательные вещества. Задача фармацевтической композиции состоит в том, чтобы облегчить введение соединения субъекту.
Фармацевтические композиции согласно настоящему изобретению можно вводить любым подходящим способом введения, выбранным из группы, состоящей из парентерального и перорального введения. Согласно некоторым вариантам реализации введение осуществляется посредством парентерального введения. В различных вариантах реализации способ введения представляет собой внутривенный, внутриартериальный, внутримышечный, подкожный, внутрибрюшинный, интрацеребральный, интрацеребровентрикулярный, интратекальный или внутрикожный путь введения. Например, фармацевтические композиции можно вводить системно, например, путем внутривенной (в/в) или внутрибрюшинной (в/б) инъекции или инфузии. Согласно определенному варианту реализации настоящего изобретения указанную фармацевтическую композицию вводят при помощи внутривенной инфузии в течение периода времени от 30 минут до 2 часов, например, в течение 1 часа. Композиции согласно настоящему изобретению можно вводить локально и они могут дополнительно содержать дополнительный активный агент и/или вспомогательное вещество.
Таким образом, фармацевтические композиции для применения в соответствии с настоящим изобретением могут быть составлены обычным способом с применением одного или более физиологически приемлемых носителей, содержащих эксципиенты и вспомогательные вещества, которые облегчают переработку активных соединений для получения составов, которые могут быть использованы фармацевтически. Подходящий состав зависит от выбранного способа введения.
Для инъекций соединения согласно настоящему изобретению можно приготовить в водных растворах, предпочтительно в физиологически совместимых буферах, таких как раствор Хэнкса или Рингера, или в физиологическом солевом буфере. Для трансмукозального и трансдермального введения в состав можно включать вещества, обеспечивающие проникновение (пенетранты), соответствующие барьеру, через который необходимо проникнуть. В данной области известны такие обеспечивающие проникновение вещества, включая, например, ДМСО или полиэтиленгликоль.
Для перорального введения соединения можно легко приготовить путем объединения активных соединений с фармацевтически приемлемыми носителями, хорошо известными в данной области. Такие носители позволяют приготовить соединения согласно настоящему изобретению в виде таблеток, пилюль, драже, капсул, жидкостей, гелей, сиропов, взвесей, суспензий и т.п. для перорального приема пациентом. Фармакологические составы для перорального применения можно получать с применением твердого вспомогательного вещества, необязательно измельчения полученной смеси и обработки смеси гранул после добавления подходящих вспомогательных веществ, если оно требуется, с получением таблеток или драже. Подходящими вспомогательными веществами являются, в частности, наполнители, такие как сахара, включая лактозу, сахарозу, маннит или сорбит; целлюлозные составы, такие как, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакантовая камедь, метилцеллюлоза, гидроксипропилметилцеллюлоза, карбометилцеллюлоза натрия; и/или физиологически приемлемые полимеры, такие как поливинилпирролидон (ПВП). При желании можно добавлять дезинтегрирующие агенты, такие как поперечно-сшитый поливинилпирролидон, агар или альгиновую кислоту или ее соль, такую как альгинат натрия.
Кроме того, поскольку желательно предотвратить воздействие соединений согласно настоящему изобретению на желудочную среду, энтеросолюбильное покрытие может быть полезным.
Фармацевтические композиции для перорального применения, включают твердые капсулы, изготовленные из желатина, а также мягкие герметичные капсулы, изготовленные из желатина и пластификатора, такого как глицерин или сорбит. Твердые капсулы могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связующими, такими как крахмалы, смазывающими веществами, такими как тальк или стеарат магния, и, необязательно, стабилизаторами.
В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как жирные масла, жидкий парафин или жидкие полиэтиленгликоли. Кроме того, можно добавлять стабилизаторы. Все составы для перорального введения должны быть в дозах, подходящих для выбранного способа введения.
Для трансбуккального введения композиции могут принимать форму таблеток или пастилок для рассасывания, составленных обычным способом.
Для введения путем ингаляции активное соединение для применения в соответствии с настоящим изобретением доставляют в виде распыляемого аэрозоля из находящейся под давлением упаковки или из небулайзера с использованием подходящего пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана или диоксида углерода. В случае аэрозоля под давлением лекарственную дозу можно отмерить с помощью клапана для доставки отмеренного количества. Капсулы и картриджи из, например, желатина, для применения в ингаляторе или инсуффляторе могут содержать порошковую смесь пептида и подходящую порошковую основу, такую как лактоза или крахмал.
Фармацевтические композиции для парентерального введения включают водные и растворы активных ингредиентов в водорастворимой форме. Кроме того, суспензии активных соединений можно получать в виде подходящих масляных суспензий для инъекций. Подходящие липофильные растворители или носители включают жирные масла, такие как кунжутное масло, или эфиры синтетических жирных кислот, такие как этилолеат, триглицериды или липосомы. Водные суспензии для инъекций могут содержать вещества, которые увеличивают вязкость суспензии, такие как натрийкарбоксиметилцеллюлоза, сорбит или декстран. Указанная суспензия необязательно может также содержать подходящие стабилизаторы или агенты, которые увеличивают растворимость соединений, чтобы обеспечить получение высококонцентрированных растворов.
Активные соединения согласно настоящему изобретению также можно приготовить в виде ректальных композиций, таких как суппозитории или микроклизмы с удержанием, с применением, например, обычных основ для суппозиториев, таких как масло какао или другие глицериды.
В зависимости от тяжести и восприимчивости состояния, подлежащего лечению, дозирование может также представлять собой однократное введение композиции с замедленным высвобождением, причем курс лечения длится от нескольких дней до нескольких недель или до тех пор, пока не будет осуществлено лечение или пока заболевание не перейдет в более легкую форму. Количество вводимой композиции будет, конечно, зависеть от многих факторов, включая субъекта, подлежащего лечению, тяжести поражения, пути введения и оценки врача, назначающего лечение.
Согласно определенному варианту реализации составы фармацевтически приемлемых солей согласно настоящему изобретению представляют собой водный изотонический раствор с осмолярностью примерно 300 мОсм и рН 4-8. Таким образом, фармацевтически приемлемый носитель для соединения согласно настоящему изобретению может представлять собой буферизированный физиологический раствор, буферизированный раствор декстрозы или буферизированный раствор глицерина с осмолярностью примерно 300 мОсм и рН 4-8.
В другом варианте реализации буферный раствор может представлять собой фармацевтически приемлемую моноионную буферную систему или полиионную буферную систему, имеющую pK ионизации в диапазоне 4-8.
Для регулирования рН раствора, содержащего фармацевтически приемлемую соль согласно настоящему изобретению, можно применять различные буферы с pK 4-8, такие как, например, ACES (N-(ацетамидо)-2-аминоэтансульфоновая кислота); ацетат; N-(2-ацетамидо)-2-иминодиуксусная кислота; BES (N,N-бис[2-гидроксиэтил]-2-аминоэтансульфоновая кислота); бицин (2-(бис(2-гидроксиэтил)амино)уксусная кислота); бис-трисметан (2-[бис(2-гидроксиэтил)амино]-2-(гидроксиметил)пропан-1,3-диол); бис-триспропан (1,3-бис(трис(гидроксиметил)метиламино)пропан); карбонат; цитрат; 3,3-диметилглутарат; DIPSO (3-[N,N-бис(2-гидроксиэтил)амино]-2-гидроксипропансульфоновая кислота); N-этилморфолин; глицерин-2-фосфат; глицин; глицин-амид; HEPBS (N-(2-гидроксиэтил)пиперазин-N'-4-бутансульфоновая кислота); HEPES (N-(2-гидроксиэтил)пиперазин-N'-2-этансульфоновая кислота); HEPPS (N-(2-гидроксиэтил)пиперазин-N'-(3-пропансульфоновая кислота)); HEPPSO (N-(2-гидроксиэтил)пиперазин-N'-(2-гидроксипропансульфоновая кислота); гистидин; гидразин; имидазол; малеат; 2-метилимидазол; MES (2-(N-морфолино)этансульфоновая кислота); MOBS (4-(N-морфолино)-бутансульфоновая кислота); MOPS (3-(N-морфолино)-пропансульфоновая кислота, MOPSO (3-(N-морфолино)-2-гидропропансульфоновая кислота), оксалат, фосфат; пиперазин; PIPES (1,4-пиперазин-диэтансульфоновая кислота); POPSO (пиперазин-N,N'-бис(2-гидроксипропансульфоновая кислота)); сукцинат; сульфит; TAPS (3-[[1,3-дигидрокси-2-(гидроксиметил)пропан-2-ил]амино]пропан-1-сульфоновая кислота); TAPSO (3-[[1,3-дигидрокси-2-(гидроксиметил)пропан-2-ил]амино]-2-гидроксипропан-1-сульфоновая кислота); винная кислота; TES (2-[[1,3-дигидрокси-2-(гидроксиметил)пропан-2-ил]амино]этансульфоновая кислота); THAM (Tris) (2-амино-2-гидроксиметилпропан-1,3-диол); и трицин (N-(2-гидрокси-1,1-бис(гидроксиметил)этил)глицин).
Согласно некоторым вариантам реализации указанный буфер представляет собой буфер производного сульфоновой кислоты, включая, но не ограничиваясь ими, ACES, BES, DIPSO, HEPBS, HEPES, HEPPS, HEPPSO, MES, MOBS, MOPS, MOPSO, PIPES, POPSO, сульфит, TAPS, TAPSO и TES.
Согласно дополнительным вариантам реализации указанный буфер представляет собой буфер производного карбоновой кислоты, включая, но не ограничиваясь ими, ацетат, N-(2-ацетамидо)-2-иминодиуксусную кислоту, 2-(бис(2-гидроксиэтил)амино)уксусную кислоту, карбонат, цитрат, 3,3-диметилглутарат, лактат, малеат, оксалат, сукцинат и винную кислоту.
Согласно дополнительным вариантам реализации указанный буфер представляет собой буфер производного аминокислоты, включая, но не ограничиваясь ими, бициновый, глициновый, глицин-амидный, гистидиновый и трициновый буфер.
Согласно другим вариантам реализации указанный буфер представляет собой буфер производного фосфорной кислоты, включая, но не ограничиваясь ими, глицерол-2-фосфатный и фосфатный буфер.
Альтернативно, буферизированный физиологический раствор для состава может представлять собой, например, сбалансированный солевой раствор Хэнкса, сбалансированный солевой раствор Эрла, сбалансированный солевой раствор Гейза, буферизированный HEPES физиологический раствор, буферизированный фосфатом физиологический раствор, Plasma-lyte, раствор Рингера, Рингера ацетат, Рингера лактат, цитратно-солевой буфер или трис-буферизированный физиологический раствор.
Буферизованный раствор декстрозы для состава может представлять собой, например, кислый цитратный раствор на основе декстрозы или раствор Эллиотта В.
Согласно иллюстративным вариантам реализации растворы для инъекции представляют собой Plasma-Lyte А или соединение лактат натрия, приобретенное у Baxter.
Терапевтическое применение
Соединения согласно настоящему изобретению способны останавливать рост клеток или индуцировать гибель гиперпролиферативной клетки, в которой они находятся, и включают известные цитотоксические, цитостатические, антипролиферативные лекарственные средства, такие как известные в данной области. Таким образом, соединения согласно настоящему изобретению пригодны для лечения и предотвращения различных видов рака, а также для предварительного лечения перед трансплантацией костного мозга.
Таким образом, в некоторых вариантах реализации настоящее изобретение относится к способу лечения рака, включая его метастазы, или предракового состояния или расстройства, включающему стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соли формулы (I) или любого соединения, охватываемого такой формулой, например, соединения (II), (III), (1), (1А), (1В), (2), (3) или (4), описанных в настоящем документе.
В других вариантах реализации настоящее изобретение относится к соли формулы (I) или любому соединению, охватываемому такой формулой, например, соединениям (II), (III), (1), (1А), (1В), (2), (3) или (4), описанным в настоящем документе, для применения для лечения рака, включая его метастазы, или предракового состояния или заболевания.
В некоторых вариантах реализации указанный рак характеризуется не солидной опухолью или солидной опухолью или их комбинацией. В других вариантах реализации указанный рак представляет собой опухоль системы крови.
В некоторых определенных вариантах реализации указанный рак характеризуется не солидной опухолью, выбранной из лимфопролиферативных расстройств, включая лейкозы и лимфомы.
В других определенных вариантах реализации указанный рак характеризуется солидной опухолью, выбранной из группы, состоящей из опухолей центральной нервной системы (ЦНС), рака печени, колоректальной карциномы, рака молочной железы, рака желудка, рака поджелудочной железы, карциномы мочевого пузыря, карциномы шейки матки, опухоли головы и шеи, рака вульвы и дерматологических новообразований, включая меланому, плоскоклеточную карциному и базальноклеточные карциномы.
Солевые формы согласно настоящему изобретению можно применять для лечения любого млекопитающего, предпочтительно человека. В других вариантах реализации настоящее изобретение относится к способу лечения индивидуума, который является кандидатом на трансплантацию костного мозга, включающему стадию предварительного лечения указанного индивидуума до трансплантации при помощи соли формулы (I), или любого соединения, охватываемого такой формулой, например, соединениями (II), (III), (1), (1А), (1В), (2), (3) или (4), описанными в настоящем документе. В других вариантах реализации настоящее изобретение относится к соли формулы (I) или любому соединению, охватываемому такой формулой, например, соединениям (II), (III), (1), (1А), (1В), (2), (3) или (4), описанным в настоящем документе, для применения для предварительного лечения перед трансплантацией костного мозга.
В некоторых вариантах реализации настоящего изобретения рак может быть выбран из опухолей системы крови или опухолей, не относящихся к системе крови.
Опухоли системы крови включают лейкозы, лимфомы и миеломы, включая, но не ограничиваясь ими, миелоидный лейкоз, лимфоцитарный лейкоз, например, острый миелоидный лейкоз (ОМЛ), хронический миелоидный лейкоз (ХМЛ), острый лимфоцитарный лейкоз (ОЛЛ), хронический лимфоцитарный лейкоз (ХЛЛ), лимфому Ходжкина, неходжкинскую лимфому, множественную миелому и макроглобулинемию Вальденстрема. Указанная опухоль системы крови может представлять собой рефрактерный/рецидивирующий рак или впервые диагностированный рак.
Опухоли, не относящиеся к системе крови, также известные как солидные опухоли, включают, но не ограничиваются ими, следующие: саркома, карцинома, фибросаркома, миксосаркома, липосаркома, хондросаркома, остеогенная саркома, хордома, ангиосаркома, эндотелиосаркома, мезотелиома, опухоль Юинга лейомиосаркома, рабдомиосаркома, плоскоклеточная карцинома, базальноклеточная карцинома, аденокарцинома, карцинома потовых желез, карцинома сальных желез, папиллярная карцинома, папиллярная аденокарцинома, цистаденокарцинома, медуллярная карцинома, бронхогенная карцинома, почечно-клеточная карцинома, гепатома, карцинома желчных протоков, хориокарцинома, семинома, эмбриональная карцинома, опухоль Вильмса, рак шейки матки, опухоль яичка, карцинома легкого, мелкоклеточная карцинома легкого, карцинома мочевого пузыря, эпителиальная карцинома, астроцитома, саркома Капоши и меланома. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Опухоли, не относящиеся к системе крови включают рак органов, где рак органа включает, но не ограничивается ими, рак молочной железы, рак мочевого пузыря, рак толстой кишки, рак прямой кишки, рак эндометрия, рак почки, рак легких, рак шейки матки, рак поджелудочной железы, рак предстательной железы, рак яичка, рак щитовидной железы, рак яичников, рак головного мозга, включая эпендимому, глиому, глиобластому, медуллобластому, краниофарингиому, пинеалому, неврому слухового нерва, гемангиобластому, олигодендроглиому, менангиому, нейробластому, ретинобластому и их метастазы. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
В другом аспекте настоящее изобретение относится к способу лечения предракового состояния или расстройства, включающему стадию введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соли формулы (I) или любого соединения, охватываемого такой формулой, например, соединения (II), (III), (1), (1А), (1В), (2), (3) или (4), описанных в настоящем документе.
В другом аспекте настоящее изобретение относится к соли формулы (I) или любому соединению, охватываемому такой формулой, например, соединениям (II), (III), (1), (1А), (1В), (2), (3) или (4), описанным в настоящем документе, для применения для лечения предракового состояния или заболевания.
Термин «предраковое расстройство или состояние» включает миелодиспластические синдромы (МДС). Термин «миелодиспластические синдромы» (МДС) относится к гетерогенной группе гемопоэтических пред-злокачественных новообразований, характеризующихся цитопениями крови, неэффективным гематопоэзом и гиперпластическим костным мозгом. МДС - это прелейкемические состояния, которые примерно в 30-40% случаев трансформируются в острый миелоидный лейкоз (ОМЛ). Если не предлагается аллогенная трансплантация стволовых клеток, ДМС обычно считается неизлечимым состоянием.
Кроме того, настоящее изобретение относится к способу лечения инфекции, вызванной вирусным агентом, представляющим собой вирус, вызывающий рак, включающему стадию введения соли формулы (I) или любого соединения, охватываемого такой формулой, например, соединения (II), (III), (1), (1А), (1В), (2), (3) или (4), описанных в настоящем документе. Таким образом, настоящее изобретение относится к способу лечения вирусной инфекции, вызванной вирусным онкогеном. Неограничивающие примеры таких вирусов включают вирус папилломы человека, вирус гепатита В, вирус гепатита С, вирус Эпштейна-Барр, Т-лимфотропный вирус человека, герпесвирус, связанный с саркомой Капоши и полиомавирус клеток Меркель. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Вирусное заболевание может быть вызвано другими вирусами, включая, но не ограничиваясь ими, вирусом иммунодефицита человека (ВИЧ), вирусом простого герпеса (ВПГ), цитомегаловирусом (ЦМВ) и вирусом ветряной оспы (VZV). Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения. Согласно одному варианту реализации указанное вирусное заболевание представляет собой вирусный герпетический энцефалит.
Способ согласно настоящему изобретению может быть полезен для лечения неопластического заболевания у субъекта, имеющего иммунологическое заболевание или расстройство. Иммунологические заболевания или расстройства включают, но не ограничиваются ими, ревматоидный артрит (РА), псориатический артрит, системную красную волчанку (СКВ), волчаночный нефрит, воспалительное заболевание кишечника (ВЗК), синдром раздраженной толстой кишки, диабет I типа, иммунную тромбоцитопеническую пурпуру, рассеянный склероз, синдром Шегрена, тиреоидит Хашимото, болезнь Грейвса, первичный билиарный цирроз, гранулематоз Вегенера, туберкулез и макроглобулемию Вальденстрема.
Способ согласно настоящему изобретению может быть полезен для лечения неопластического заболевания у субъектов с дисфункцией органов, таких как дисфункция печени, дисфункция почек, дисфункция поджелудочной железы, дисфункция костного мозга и дисфункция мозжечка.
Термин «дисфункция печени» относится к состоянию, в котором функция печени снижается по сравнению с нормальным состоянием. В целом, дисфункция печени представляет собой состояние, характеризующееся тем, что любое одно или более значений контрольных параметров для оценки функции печени (например, уровни в крови AST, ALT, ALP, ТТТ, ZTT, общий билирубин, общий белок, альбумин, лактатдегидрогеназа, холин эстераза и тому подобное) находятся вне диапазона нормальных значений (контрольных значений). Дисфункция печени характерна для таких заболеваний, как, например, молниеносный гепатит, хронический гепатит, вирусный гепатит, алкогольный гепатит, фиброз печени, цирроз печени, рак печени, аутоиммунный гепатит, аллергическая гепатопатия и первичный билиарный цирроз.
Дисфункция почек характерна для таких заболеваний, как, например, острая почечная недостаточность, гломерулонефрит, хроническая почечная недостаточность, азотемия, уремия, иммунное почечное заболевание, острый нефритический синдром, быстро прогрессирующий нефритический синдром, нефротический синдром, болезнь Бергера, хронический нефритический/протеинурический синдром, тубулоинтерстициальное заболевание, нефротоксические расстройства, некроз почек, атероэмболическая болезнь почек, кортикальный некроз почек, злокачественный нефроангиосклероз, тромбоз почечной вены, почечный тубулярный ацидоз, почечная глюкозурия, нефрогенный несахарный диабет, синдром Барттера, синдром Лиддла, поликистозная болезнь почек, интерстициальный нефрит, острый гемолитический уремический синдром, ювенильный нефронофтиз, спонгиозная почка, наследственный нефрит и наследственная онихоартроостеодисплазия.
Дисфункция поджелудочной железы характерна для таких заболеваний, как, например, диабет, гипергликемия, нарушение толерантности к глюкозе и резистентность к инсулину.
Дисфункция костного мозга характерна для таких заболеваний, как, например, остеомиелит, нарушение кроветворения, ион-дефицитная анемия, пернициозная анемия, мегалобластоз, гемолитическая анемия и апластическая анемия.
Дисфункция мозжечка характерна для моторных и нейро-поведенческих расстройств, таких как, например, гипотония, дизартрия, дизметрия, дисдиадохокинезия, нарушение рефлексов и дрожание при произвольных движениях.
В одном варианте реализации настоящего изобретения указанный субъект представляет собой субъекта с ослабленным здоровьем, который не подлежит лечению противоопухолевым агентом, например, цитарабином. Указанный субъект с ослабленным здоровьем может быть выбран из группы, состоящей из пожилых пациентов, субъектов с дисфункцией печени, субъектов с дисфункцией почек, субъектов с дисфункцией поджелудочной железы, субъектов с дисфункцией костного мозга, субъектов с дисфункцией мозжечка, субъектов с иммунологическим расстройством, субъектов с рецидивной или рефрактерной опухолью системы крови, и любой их комбинации.
Токсичность и терапевтическая эффективность соединений, описанных в настоящем документе, можно определять при помощи стандартных фармацевтических процедур в культурах клеток или у подопытных животных, например, путем определения IC50 (концентрация, обеспечивающая 50% ингибирование роста клеток) и МПД (максимальная переносимая доза у тестируемых животных) для требуемого соединения. Данные, полученные с помощью анализов клеточной культуры и исследований на животных, можно использовать при разработке диапазона доз для применения у людей. Доза может варьироваться в зависимости от используемой лекарственной формы и используемого способа введения. Точный состав, способ введения и дозировка могут быть выбраны лечащим врачом с учетом состояния пациента. (См., например, Fingl, et al., 1975, in "The Pharmacological Basis of Therapeutics", Ch. 1 pp. 1).
Количество активного агента, применяемого в композиции согласно настоящему изобретению, представляет собой такое количество, которое способно обеспечить достижение назначения конкретного активного агента. Количество активного агента в композициях обычно является терапевтически эффективным количеством, фармакологически эффективным количеством, биологически эффективным количеством или химически эффективным количеством. Однако количество может быть меньше этого количества в случае, когда композицию применяют в единичной дозированной форме, поскольку единичная дозированная форма может содержать множество соединений или активных агентов в одной композиции или может содержать разделенное фармакологически эффективное количество, биологически эффективное количество, терапевтически эффективное количество или химически эффективное количество. Общее эффективное количество можно вводить в кумулятивных единицах, содержащих в целом эффективное количество активного агента.
Соль соединения согласно настоящему изобретению можно вводить в суточной дозе в пределах от примерно 0,3 г/м2 до примерно 10 г/м2 площади поверхности тела субъекта. Согласно некоторым вариантам реализации указанную соль можно вводить в суточной дозе в пределах от примерно 0,5 г/м2 до примерно 5 г/м2 площади поверхности тела субъекта. Согласно некоторым вариантам реализации указанную соль можно вводить в суточной дозе в пределах от примерно 0,5 г/м2 до примерно 4,5 г/м2 площади поверхности тела субъекта. Согласно некоторым вариантам реализации указанную соль вводят в суточной дозе, составляющей примерно 0,3, 0,5, 0,8, 1, 1,5, 2, 2,3, 2,5, 3, 3,5, 4, 4,5, 5, 6, 7, 8, 9, 10 г/м2 площади поверхности тела субъекта. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно некоторым вариантам реализации указанную соль согласно настоящему изобретению вводят при помощи внутривенной инфузии в суточной дозе в пределах от 0,3 г/м2 до 4.5 г/м2 площади поверхности тела субъекта.
Согласно некоторым вариантам реализации указанную фармацевтическую композицию вводят по меньшей мере один раз в месяц. Согласно дополнительным вариантам реализации указанную фармацевтическую композицию вводят по меньшей мере два раза в месяц. Согласно дополнительным вариантам реализации указанную фармацевтическую композицию вводят по меньшей мере один раз в неделю. Согласно дополнительным вариантам реализации указанную фармацевтическую композицию вводят по меньшей мере два раза в неделю. Согласно дополнительным вариантам реализации настоящего изобретения указанную фармацевтическую композицию вводят один раз в сутки в течение по меньшей мере одной недели. Согласно дополнительным вариантам реализации указанную фармацевтическую композицию вводят по меньшей мере один раз в сутки в течение по меньшей мере одной недели или до тех пор, пока субъект не будет излечен.
Согласно некоторым вариантам реализации указанную фармацевтическую композицию вводят один раз в сутки в течение по меньшей мере 2, 3, 4, 5, 6, 8, 10, 12 или по меньшей мере 14 последовательных дней один раз в месяц. Альтернативно указанную фармацевтическую композицию вводят один раз в сутки в течение по меньшей мере 2, 3, 4, 5, 6 или 12 дней два раза в месяц или, альтернативно, указанную фармацевтическую композицию вводят каждый день или два раза в неделю, пока пациент не будет излечен.
В некоторых вариантах реализации, в которых фармацевтическую композицию применяют для предотвращения рецидива рака, указанную фармацевтическую композицию можно регулярно вводить в течение продолжительных периодов времени в соответствии с инструкциями врача.
В некоторых случаях может быть целесообразным вводить большую ударную дозу с последующими периодическими (например, вводимыми каждую неделю) поддерживающими дозами в течение периода лечения. Соединения можно также доставлять при помощи систем доставки с медленным высвобождением, помпами и другими известными системами доставки для непрерывной инфузии. Режимы дозирования могут варьироваться для обеспечения желаемых уровней циркуляции конкретного соединения на основе его фармакокинетики. Таким образом, дозы рассчитывают так, чтобы поддерживать желаемый уровень циркуляции терапевтического агента.
Обычно эффективная доза определяется активностью и эффективностью соединения и состоянием субъекта, а также массой тела или площадью поверхности тела подлежащего лечению субъекта. Доза и режим дозирования также определяются наличием, характером и степенью любых неблагоприятных побочных эффектов, которые сопровождают введение соединений у конкретного субъекта.
Согласно дополнительным вариантам реализации настоящего изобретения субъект, получающий лечение, представляет собой субъекта в возрасте 50 или более лет, например 60, 70, 75 или более лет. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения.
Согласно дополнительным вариантам реализации соединение согласно настоящему изобретению вводят в суточной дозе по меньшей мере в 2, 3, 5, 10, 15, 20 или по меньшей мере в 30 раз больше, чем максимальный стандартный уровень дозы цитарабина. Каждый из этих вариантов относится к отдельному варианту реализации настоящего изобретения. Следующие примеры следует рассматривать просто как иллюстративные и не ограничивающие настоящее изобретение. Специалисту в данной области техники будет очевидно, что можно произвести множество модификаций, перестановок и вариантов, не выходящих за рамки объема настоящего изобретения.
ПРИМЕРЫ
В примерах используются следующие сокращения:
ВОС: трет-бутилоксикарбонил
DCC: N,N'-дициклогексилкарбодиимид
ДМФА: диметилформамид
DCM: дихлорметан
DIC: диизопропил карбодиимид
DIEA: диизопропилэтиламин
DMAP: диметиламинопиридин
Fmoc: 9-флуоренилметилоксикарбонил
HBTU: 2-(1Н-бензотиазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат
HOBt: N-гидроксибензотриазол
ВЭЖХ: Высокоэффективная жидкостная хроматография
MS: Масс-спектрометрия
NMP: N-метил-2-пирролидон
OtBu: трет-бутиловый эфир
ТФУ: трифторуксусная кислота
Пример 1: Синтез ВОС-Asp(Цитарабин)-OtBu(А) - защищенного предшественника солевой формы Asp(Цитарабин)⋅Y(1)
BOC-Asp-OtBu (23,7 г), EDC (15,7 г), HOBT (11,1 г) и Ара-С (20 г) растворяли в 400 мл ДМФА и перемешивали в течение 24 часов при комнатной температуре. Смесь выпаривали при температуре 80°С с получением маслянистого остатка. Остаток растворяли в 300 мл этилацетата и последовательно экстрагировали 5% NaHCO3, 0,1 М HCl, 5 М NaCl. Указанную смесь полностью выпаривали с получением твердого продукта, представлявшего собой защищенный предшественник BOC-Asp(Цитарабин)-OtBu. Предшественник анализировали с помощью ВЭЖХ хроматографии со смешанным режимом на колонке Primesep 100 с градиентом ацетинонитрил/H2O 0,1% ТФУ, с получением пика при времени удерживания 29,6 мин. Выход продукта предшественника составлял 80-90% в виде беловатых кристаллов с чистотой по ВЭЖХ≥95%.
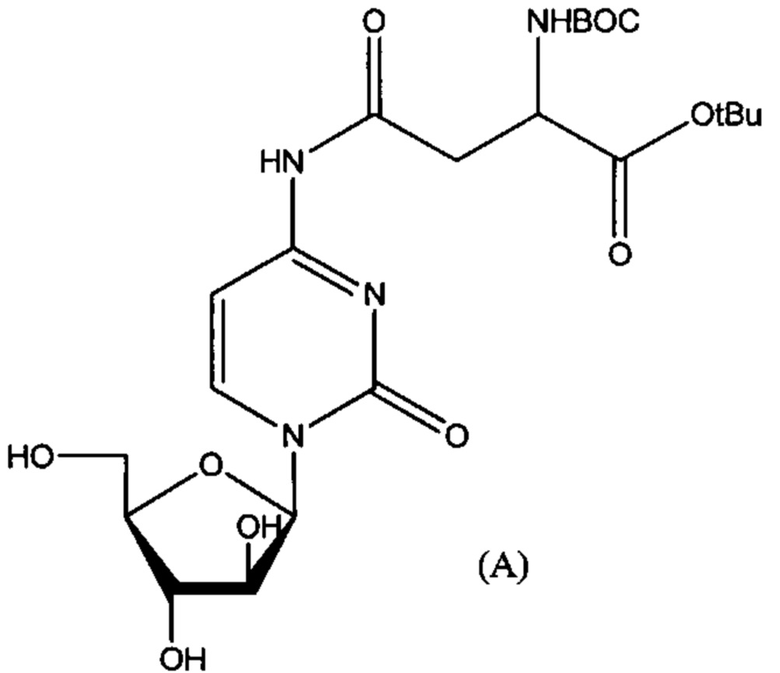
Защищенный предшественник ВОС-Asp(Цитарабин)-OtBu (А) далее используют для получения нескольких солевых форм конечного продукта Asp(Цитарабин)⋅Y (Y = соль присоединения кислоты):
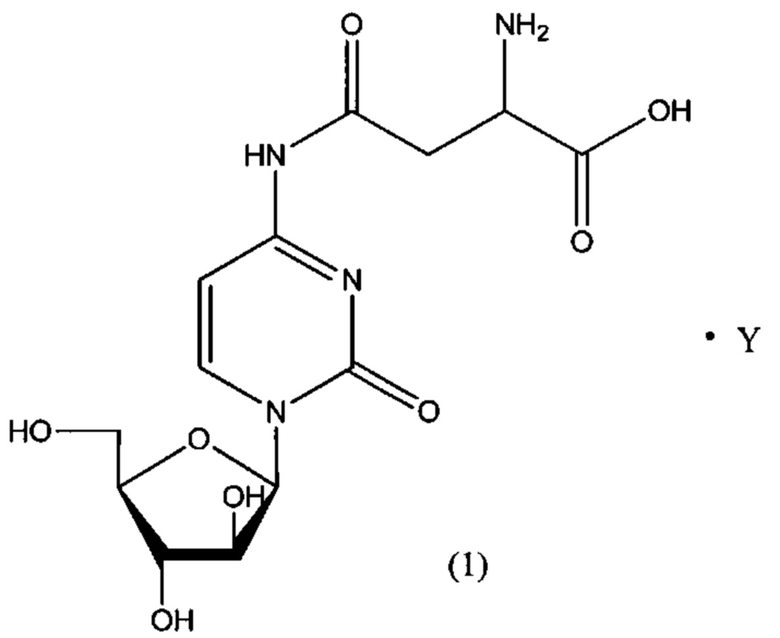
Пример 2: Синтез Asp(Цитарабин)⋅ацетата (Y=CH3COOH). (формула IV) - Путь А с применением ТФУ для удаления защитных групп
Защищенный предшественник ВОС-Asp(Цитарабин)-OtBu (из примера 1) подвергали расщеплению при помощи ТФУ. Расщепление защитных групп (ВОС, OtBu) проводили в 400 мл ТФУ + 2,5% H2O в течение 2,5 часов. Раствор переносили в 1,6 л tBME для осаждения продукта. Осадок сушили с получением беловатых кристаллов Asp(Цитарабин)⋅ТФУ, соли ТФУ (молекулярная масса = 472 г/моль). Asp(Цитарабин)⋅ТФУ дополнительно растворяли в концентрированной уксусной кислоте до полного растворения. Продукт осаждали при помощи tBME и сушили с получением Asp(Цитарабин)⋅Ацетата. Выход продукта составлял 75-85% (белые кристаллы). Неочищенный продукт анализировали с помощью ВЭЖХ хроматографии со смешанным режимом на колонке Primesep 100 с градиентом ацетинонитрил/H2O 0,1% ТФУ, при 240 нм, с получением пика при времени удерживания 9,2 мин, чистота 85,3%. По данным МС молекулярная масса M.W. = 359 г/моль (МС + 1H). Данные МС приведены на фиг. 1. Оптическая плотность составляет 213, 245 и 298 нм. Данные ЯМР: 1Н ЯМР (600 МГц, ДМСО-6d) δ ppm 2,98 (dd, 1Н), 3,08 (dd, 1Н), 3,61 (d, 1Н), 3,84 (ddd, 1Н), 3,93 (td, 1Н), 4,06 (td, 1Н), 4,23 (dtd, 1Н), 5,50 (s, 1H), 6,05 (d, 1H), 7,12 (d, 1H), 8,09 (d, 1H), 11,10 (s, 1H). 13C ЯМР (151 МГц, ДМСО-6d) δ ppm 36,61 (s, 1С), 48,23 (s, 1С), 61,02 (s, 1С), 74,57 (s, 1С), 76,15 (s, 1С), 85,82 (s, 1С), 87,06 (s, 1С), 94,30 (s, 1С), 147,01 (s, 1С), 154,33 (s, 1С), 161,84 (s, 1С), 170,00 (s, 1С), 170,04 (s, 1С). Данные ЯМР соответствуют ожидаемой структуре.
Очистку Asp(Цитарабин)⋅Ацетата до 95-100% проводили следующим образом: 7 г неочищенного продукта (85,3%) растворяли в 15 мл Н2О и разделяли при помощи препаративной ВЭЖХ так, как описано в таблице 1. Раствор А - 0,1% ТФУ или 1% уксусной кислоты растворяли в воде. Раствор В - 0,1% ТФУ или 1% уксусной кислоты растворяли в ацетонитриле/воде = 9/1. Объем впрыска: 15 мл, Длина волны: 220 нм, скорость потока: 40 мл/мин, Колонка: primesep100, размер: 60×300 мм, размер частиц: 10 мкм 100 А. Объем сбора фракций: 30 мл. Фракции, собранные с 18 до 36 минуту, показывали чистоту Asp(Цитарабин)⋅Ацетата≥95%. Фракции сушили вымораживанием с получением порошка очищенного Asp(Цитарабин)⋅Ацетата с выходом 3,18 г.
Пример 3: Синтез Asp(Цитарабин)⋅метансульфоната путем кислотной замены - Путь А с применением ТФУ для удаления защитных групп
Два грамма защищенного предшественника ВОС-Asp(Цитарабин)⋅OtBu подвергали расщеплению с помощью ТФУ, как описано в примере 1. 1 г Asp(Цитарабин)⋅ТФУ растворяли в 70% метансульфоновой кислоте в Н2О до полного растворения. Продукт Asp(Цитарабин)⋅Метансульфонат осаждали изопропанолом/tBME и осадок сушили. Выход продукта составлял 51% (белые кристаллы). Продукт анализировали с помощью ВЭЖХ и МС, как описано в примере 2. Продукт дает пик при времени удерживания 10,6 мин, чистота - 80,6%.
Очистку Asp(Цитарабин)⋅Метансульфоната до 95-100% проводили следующим образом: 20 г смолы G-10 оставляли для набухания в 100 мл Н2О в течение 1 часа. После набухания смолу G-10 переносили на спеченное стекло и промывали 20% этанолом в Н2О (объем колонки ×3), Н2О и 0,1% метансульфоновой кислоты в Н2О. 1 г неочищенного порошка Asp(Цитарабин)⋅Метансульфоната (80,6%) растворяли в 5 мл Н2О. Указанный раствор загружали в смолу G-10 и экстрагировали 0,1% метансульфоновой кислотой в Н2О. Собирали фракции по 5 мл. Очищенные фракции Asp(Цитарабин)⋅Метансульфоната показывали чистоту выше 95%.
Пример 4: Синтез нескольких солевых форм Asp(Цитарабин)⋅Y путем кислотной замены
Путь А можно применять для получения нескольких солевых форм путем замены ТФУ другими кислотами так же, как описано в примерах 2 и 3. Можно применять такие кислоты как: бензолсульфоновая, бензойная, борная, лимонная, этансульфоновая, муравьиная, соляная, молочная, малеиновая, малоновая, метансульфоновая, метафосфорная, никотиновая, щавелевая, фосфорная, салициловая, янтарная, серная, винная и толуолсульфоновая. Для реакций замены анализировали различные кислоты с выбранными концентрациями и соотношением от 1:1 до 1:20. После осаждения продукты анализировали с помощью ВЭЖХ и МС. Условия и результаты приведены в таблице 2. Продукты Asp(Цитарабин)⋅Y дополнительно очищали с использованием способов очистки, описанных в примерах 2 и 3.
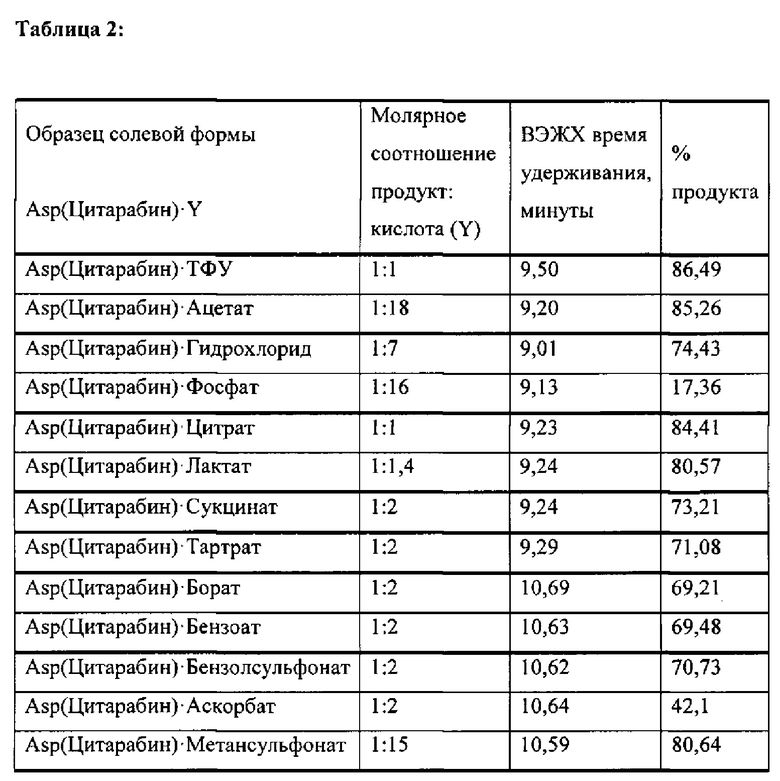
Пример 5: Синтез Asp(Цитарабин)⋅метансульфонатной солевой формы путем расщепления метансульфоновой кислотой - путь В
0,25 г предшественника ВОС-Asp(Цитарабин)-OtBu (из примера 1) подвергали расщеплению при помощи метансульфоновой кислоты. 0,25 г предшественника добавляли к 1 мл 70% MSA в Н2Ою После расщепления в течение 2 ч указанный раствор переносили в 10 мл tBME, осаждали продукт Asp(Цитарабин)⋅метансульфонат. Продукт промывали tBME и сушили. Выход продукта составлял 65% (белые кристаллы). Продукт анализировали с помощью ВЭЖХ хроматографии со смешанным режимом на колонке Primesep 100 с градиентом ацетинонитрил/Н2О 0,1% ТФУ, при 220 нм, с получением пика при времени удерживания 9,6 мин, чистота 56%. По данным МС молекулярная масса Asp(Цитарабин)⋅метансульфоната M.W. = 453 и 454 г/моль (MS±1H). Данные МС приведены на фиг. 2. Asp(Цитарабин)⋅Метансульфонат очищали так, как описано в примерах 2 и 3. Если используется способ очистки из примера 2, кислота, используемая в растворах А и В, заменяется на соль присоединения кислоты. Раствор А - 0,1% метансульфоновой кислоты растворяли в воде. Раствор В - 0,1% метансульфоновой кислоты растворяли в ацетонитриле/воде = 9/1.
Пример 6: Синтез Asp(Цитарабин)⋅бензолсульфонатной солевой формы путем расщепления бензолсульфоновой кислотой - путь В
Asp(Цитарабин)⋅бензолсульфонатная солевая форма была синтезирована из предшественника BOC-Asp(Цитарабин)-OtBu путем расщепления бензолсульфоновой кислотой, как описано в примере 5. Выход продукта составлял 31% (белые кристаллы). Продукт анализировали, как описано в примере 5. Данные ВЭЖХ показывают два пика при времени удерживания 9,3 и 14,7 мин, чистота 20 и 44%, соответственно. По данным МС молекулярная масса Asp(Цитарабин)⋅бензолсульфоната M.W. = 515 и 516 г/моль (MS±1H). Данные МС приведены на фиг. 3. Asp(Цитарабин)⋅Бензолсульфонат очищали так, как описано в примерах 2, 3 и 5.
Пример 7: Синтез Asp(Цитарабин)⋅фосфатной солевой формы путем расщепления фосфорной кислотой - путь В
Asp(Цитарабин)⋅фосфатная солевая форма была синтезирована из предшественника ВОС-Asp(Цитарабин)-OtBu путем расщепления концентрированной фосфорной кислотой, как описано в примере 5. Выход продукта составлял 34% (белые кристаллы). Продукт анализировали, как описано в примере 5. Данные ВЭЖХ показывают пик при времени удерживания 9,0 мин, чистота - 84,3%. По данным МС молекулярная масса Asp(Цитарабин)⋅фосфата M.W. = 455 и 456 г/моль (MS±1H). Asp(Цитарабин)⋅фосфат очищали так, как описано в примерах 2, 3 и 5.
Пример 8: Синтез Asp(Цитарабин)⋅сульфатной солевой формы путем расщепления серной кислотой - путь В
Asp(Цитарабин)⋅сульфатная солевая форма была синтезирована из предшественника BOC-Asp(Цитарабин)-OtBu путем расщепления концентрированной серной кислотой, как описано в примере 5. Выход продукта составлял 77%, белые кристаллы, молекулярная масса M.W. = 456 г/моль. Продукт анализировали, как описано в примере 5. Данные ВЭЖХ показывают пик при времени удерживания 9,3 мин, чистота - 53%. По данным МС молекулярная масса Asp(Цитарабин)⋅сульфата M.W. = 455 и 456 г/моль (MS±1H). Asp(Цитарабин)⋅сульфат очищали так, как описано в примерах 2, 3 и 5.
Пример 9: Синтез Asp(Цитарабин)⋅гидрохлоридной солевой формы путем расщепления соляной кислотой - путь С
1 г предшественника BOC-Asp(Цитарабин)-OtBu (из примера 1) подвергали расщеплению при помощи HCl. К предшественнику добавляли 3 мл 37% HCl/Н2О и инкубировали в течение 1 часа с удалением защитных групп ВОС и OtBu и образованием солевой формы. Раствор титровали 1N NaOH до рН 5-6. Во время титрования образовывался осадок. Полученный осадок сушили, растворяли в 2 мл Н2О и лиофилизировали. Продукт анализировали, как описано в примере 5. Данные ВЭЖХ показывают пик при времени удерживания 9,46 мин, чистота - 75,9%. Asp(Цитарабин)⋅гидрохлорид очищали так, как описано в примерах 2, 3 и 5.
Пример 10: Синтез нескольких солевых форм Asp(Цитарабин)⋅Y путем кислотного расщепления
Пути В и С можно применять для получения различных солевых форм путем расщепления кислотой, которая образует солевую форму, теми же способами, что описаны в примерах 5 и 9. Предпочтительные кислоты как для удаления защитных групп, так и для образования солевых форм, имеют pKa≤3 и отношение кислота:предшественник от 1:1 до 30:1. Предпочтительная кислота выбрана из группы, состоящей из: бензолсульфоновой, камфорсульфоновой, цикламовой, 2,2-дихлоруксусной, ди(трет-бутил)нафталинсульфоновой, ди(трет-бутил)нафталиндисульфоновой, додецилсерной, этан-1,2-дисульфоновой, этансульфоновой, фумаровой, галактаровой, гентизиновой, глюкаровой, глюконовой, глицерофосфорной, соляной, бромистоводородной, йодистоводородной, 2-гидроксиэтансульфоновой (изетионовой), 1-гидрокси-2-нафтоновой, малеиновой, малоновой, медроновой (бисфосфоновой), метансульфоновой, метафосфорной, метилбороновой, нафталин-1,5-дисульфоновой, нафталин-2-сульфоновой, азотной, оротовой, щавелевой, 2-оксоглутаровой (кетоглутаровой), памоевой (эмбоновой), пировиноградной, фосфорной, сахариновой, салициловой, серной, винной, тиоциановой, толуолсульфоновой и трифторуксусной кислот и т.п. Различные кислоты с выбранными концентрациями от 10 до 100% (концентрированная кислота) применяли для расщепления предшественника ВОС-Asp(Цитарабин)-OtBu для удаления защитной группы и образования солевых форм. Далее продукты анализировали при помощи ВЭЖХ и МС. Условия и результаты приведены в таблице 3. Продукты Asp(Цитарабин)⋅Y очищали так, как описано в примерах 2, 3 и 5.
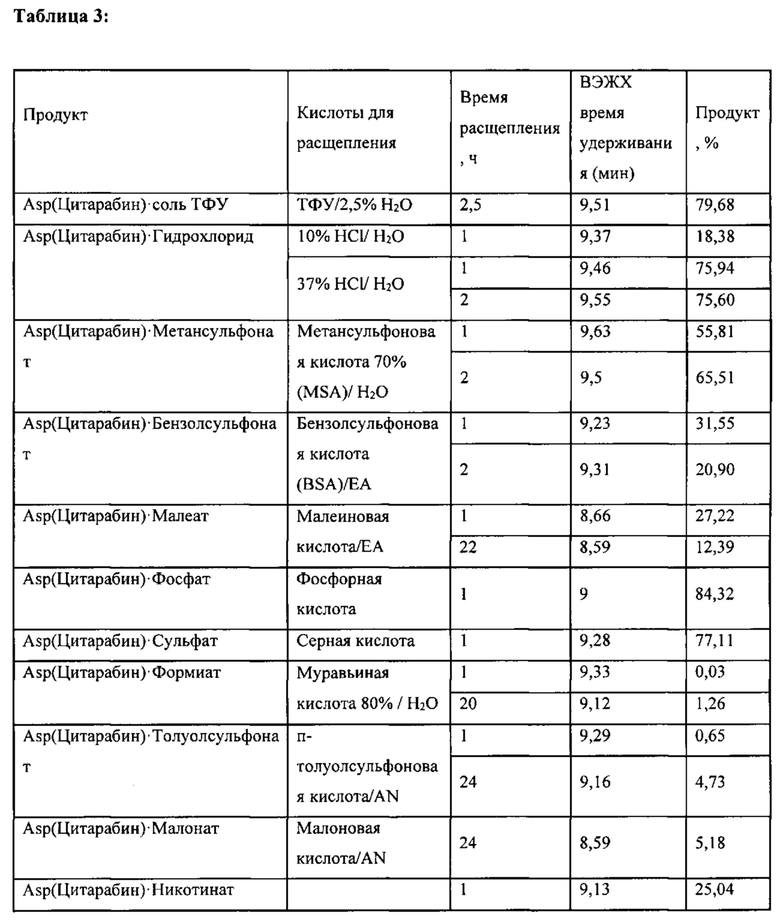

Пример 11: Получение соли присоединения Asp(Цитарабин)⋅Y с солями (такими как NaH2PO4)
Защищенный предшественник ВОС-Asp(Цитарабин)-OtBu синтезировали так, как описано в примере 1. Asp(Цитарабин)-ТФУ синтезировали так, как описано в примере 2. Затем 0,25 г Asp(Цитарабин)⋅ТФУ растворяли в 1 мл 1 NaH2PO4 до полного растворения. Продукт осаждали при помощи tBME и сушили с получением продукта Asp(Цитарабин)⋅фосфат.
Эту реакцию можно также применять с другими солями, такими как KH2PO4 и подобные.
Пример 12: Синтез ВОС-Asp(Гемцитабин)-OtBu (В) - защищенного предшественника солевой формы Asp(Гемцитабин)⋅Y (2)
Защищенный предшественник ВОС-Asp(Гемцитабин)-OtBu синтезировали так, как описано в примере 1.
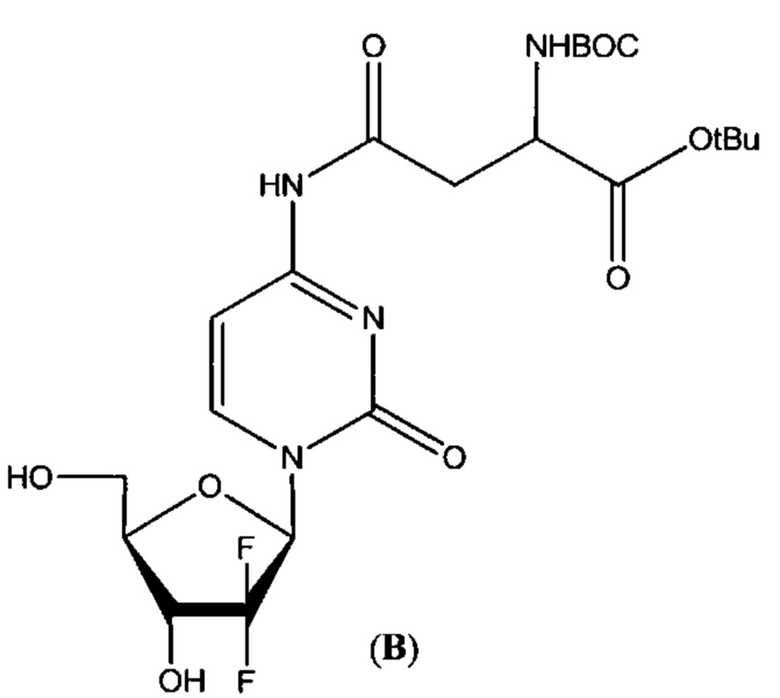
Защищенный предшественник далее использовали для получения нескольких солевых форм конечного продукта Asp(гемцитабин)⋅Y (Y = соль присоединения кислоты) (2).
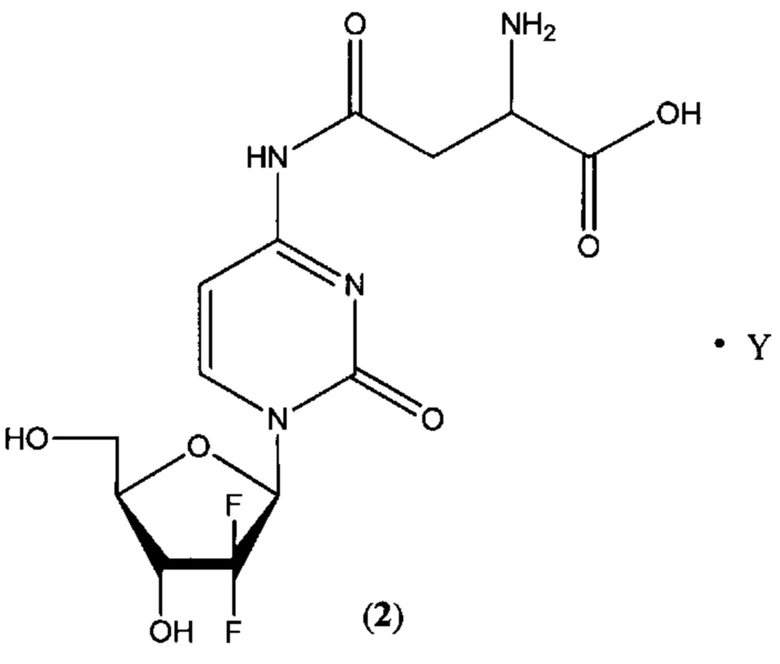
Пример 13: Синтез Asp(гемцитабин)⋅ацетатной солевой формы
Asp(гемцитабин)⋅ацетатную солевую форму (Y=СН3СООН) синтезировали из предшественника В кислотной заменой, как описано в примере 2.
Пример 14: Синтез Asp(гемцитабин)⋅метансульфонатной солевой формы
Asp(гемцитабин)⋅метансульфонатную солевую форму (Y=CH3SO3H) синтезировали из предшественника В расщеплением метансульфоновой кислотой, как описано в примере 5.
Пример 15: Синтез ВОС-Glu(Цитарабин)-OtBu (С) - защищенного предшественника солевой формы Glu(Цитарабин)⋅Y (3)
Защищенный предшественник ВОС-Glu(Цитарабин)-OtBu синтезировали так, как описано в примере 1.
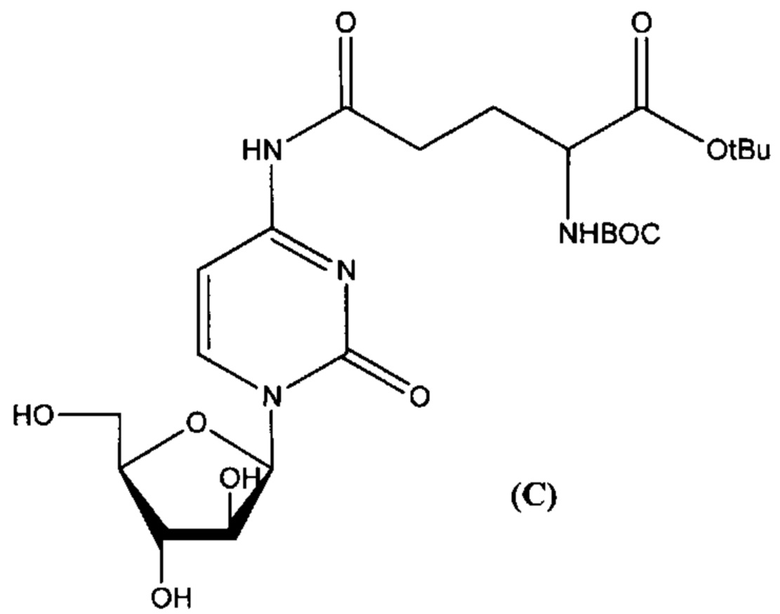
Защищенный предшественник далее использовали для получения нескольких солевых форм конечного продукта Glu(цитарабин)⋅Y (Y = соль присоединения кислоты) (3).
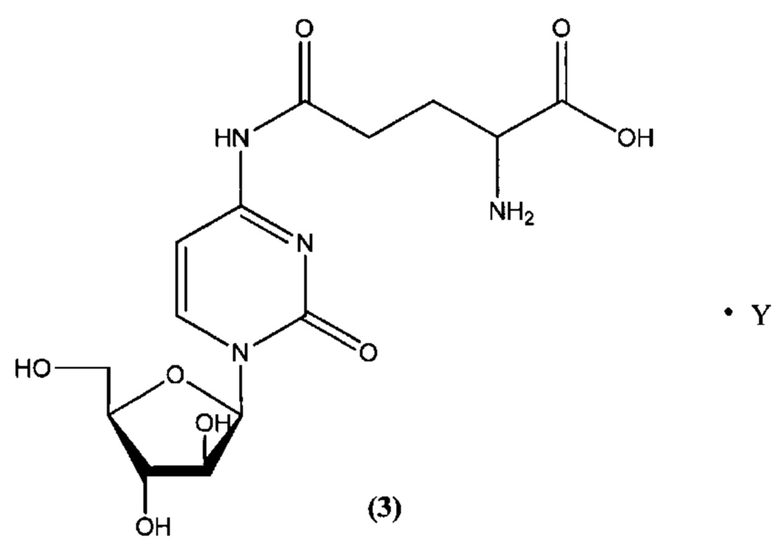
Пример 16: Синтез Glu(цитарабин)⋅ацетатной солевой формы
Glu(цитарабин)⋅ацетатную солевую форму (Y=СН3СООН) синтезировали из предшественника С кислотной заменой, как описано в примере 2.
Пример 17: Синтез Glu(цитарабин)⋅метансульфонатной солевой формы
Glu(цитарабин)⋅метансульфонатную солевую форму (Y=CH3SO3H) синтезировали из предшественника С расщеплением метансульфоновой кислотой, как описано в примере 5.
Пример 18: Синтез ВОС-Glu(Гемцитабин)-OtBu (D) - защищенного предшественника солевой формы Glu(Гемцитабин)⋅Y (4)
Защищенный предшественник ВОС-Glu(Гемцитабин)-OtBu синтезировали так, как описано в примере 1.
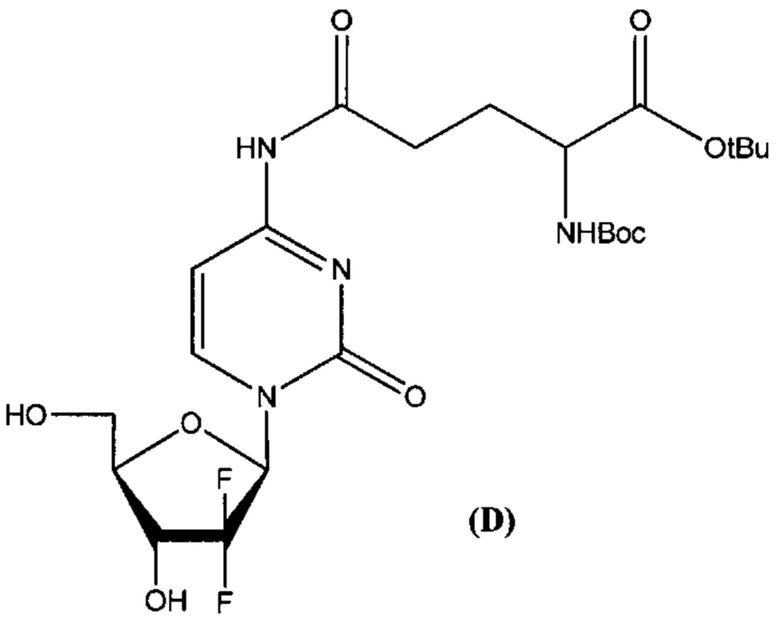
Защищенный предшественник BOC-Glu(Гемцитабин)-OtBu (D) далее применяют для получения нескольких солевых форм конечного продукта Glu(Гемцитабин)⋅Y (4) (Y = соль присоединения кислоты).
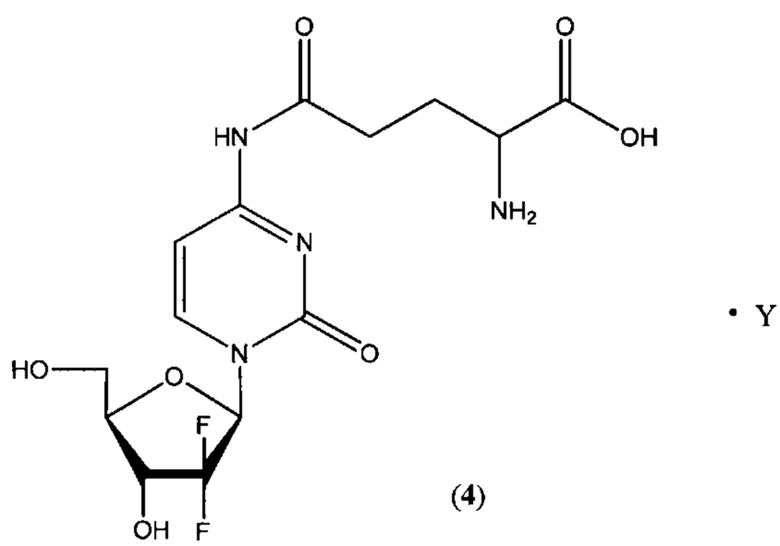
Пример 19: Синтез Glu(гемцитабин)⋅ацетатной солевой формы
Glu(Гемцитабин)⋅ацетатную солевую форму (Y=СН3СООН) синтезировали из предшественника D кислотной заменой, как описано в примере 2.
Пример 20: Синтез Glu(гемцитабин)⋅метансульфонатной солевой формы
Glu(гемцитабин)⋅метансульфонатную солевую форму (Y=CH3SO3H) синтезировали из предшественника D расщеплением метансульфоновой кислотой, как описано в примере 5.
Пример 21: Синтез Asp(Цитарабин)⋅HCl солевой формы (Y = HCl)
Стадия 1: Синтез Вос-Asp(Цитарабин)-OtBu
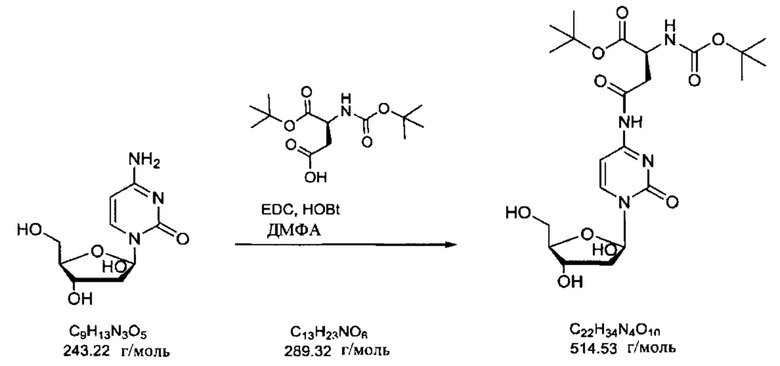
Цитарабин (0,50 г, 2,1 ммоль) суспендировали в 5 мл сухого ДМФА. Указанную смесь нагревали до температуры 70°С до получения прозрачного раствора. Boc-Asp-OtBu (0,40 г, 1,4 ммоль) растворяли при комнатной температуре в 5 мл сухого ДМФА. Добавляли и растворяли HOBt (0,22 г, 1,5 ммоль), затем EDC (0,28 г, 1,5 ммоль). Раствор перемешивали при комнатной температуре в течение 30 мин.
К раствору цитарабина по каплям добавляли активированную аминокислоту при температуре 45°С в течение 2 часов и затем перемешивали при температуре 45°С в течение ночи.
Растворитель выпаривали при пониженном давлении при температуре 60°С. Маслянистый остаток растворяли в этилацетате (55 мл) и промывали 3×5% NaHCO3, 3×0,1 М HCl и 3 × рассолом (каждая промывка 15 мл). После сушки над сульфатом натрия и фильтрации растворитель выпаривали и получали неочищенный продукт в виде пенящегося твердого вещества. Неочищенный продукт содержал ~5-15% примеси(Вос-Asp-OtBu)2-(цитарабина).
Стадия 2: Очистка Boc-Asp(Цитарабин)-OtBu
Неочищенный продукт очищали с использованием системы очистки Biotage Isolera®. 3,97 г неочищенного продукта (содержит менее 1% примеси (Boc-Asp-OtBu)2-цитарабина) очищали при помощи картриджа SNAP, KP-SIL 100 г колонки на силикагеле со следующим градиентом при скорости потока 40 мл/мин (мониторинг при 254 нм).

Фракции продукта объединяли и выпаривали досуха с получением 3,78 г очищенного продукта. Спектр 1Н-ЯМР (MeOD) очищенного Вос-Asp-OtBu-(Цитарабина) приведен на фиг. 4.
Стадия 3: Синтез Asp(Цитарабин)⋅HCl
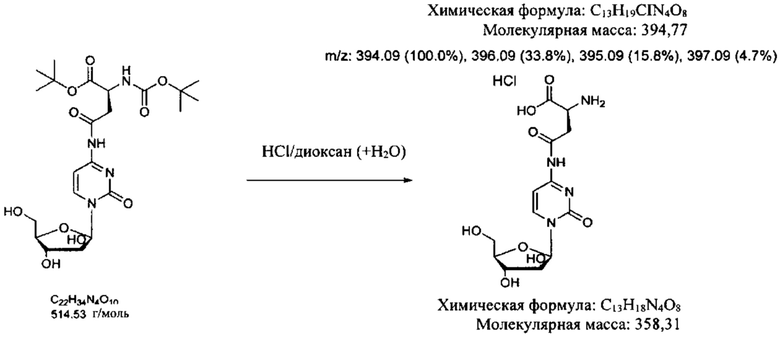
Boc-Asp-OtBu-(Цитарабин)⋅(3,6 г, 7,0 ммоль) растворяли в 8,8 мл сухого диоксана. После получения прозрачного раствора добавляли HCl в диоксане (4 М, 17,5 мл, 70 ммоль), смесь перемешивании в течение ночи (через 1 мин образовалось белое твердое вещество) при комнатной температуре. По каплям добавляли H2O (0,26 мл, 7,0 ммоль) и перемешивание продолжали в течение 3 часов. Осадок фильтровали (фильтр из спеченного стекла 4) и промывали сухим диоксаном (2×25 мл). Полученное белое твердое вещество сушили в вакууме при комнатной температуре с получением белого порошка (3,34 г). Спектр 1Н-ЯМР (MeOD) Asp(Цитарабин)⋅HCl приведен на фиг. 5.
Пример 22: Синтез Asp(Цитарабин)⋅HCl солевой формы (Y=HCl) из защищенного цитарабина.
Стадия 1: Защита цитарабина
2,44 г цитарабина и 1,5 г трет-бутилдиметилсилилхлорида (TBDMS-Cl) растворяли в 20 мл пиридина, указанную смесь перемешивали в течение 10 часов. Раствор выпаривали, осадок промывали водой и этилацетатом с получением белого твердого вещества, TBDMS-цитарабина (защищенный по 5'-ОН).
Стадия 2: Синтез Boc-Asp(TBDMS-Цитарабин)-OtBu
1 г TBDMS-цитарабина растворяли в 10 мл ДМФА. 0,8 г Boc-Asp-OtBu растворяли в 5 мл ДМФА. Добавляли и растворяли 0,44 г HOBt, а затем 0,56 г EDC. Раствор перемешивали при комнатной температуре в течение 30 мин. К раствору TBDMS-цитарабина по каплям добавляли активированную аминокислоту, затем перемешивали в течение ночи. Растворитель выпаривали при пониженном давлении при температуре 60°С. Маслянистый остаток растворяли в этилацетате и промывали 5% NaHCO3, 0,1 М HCl и рассолом. 2/3 растворителя выпаривали и неочищенный продукт осаждали гексаном с получением белого твердого вещества. Неочищенный продукт не содержал примесного (Boc-Asp-OtBu)2-(TBDMS-цитарабин), очистка на колонке не требовалась.
Стадия 3: Синтез Asp(Цитарабин)⋅HCl
Для синтеза Asp(Цитарабин)⋅HCl применяли Boc-Asp(TBDMS-Цитарабин)-OtBu, применяя тот же способ, что и на стадии 3 примера 22.
Пример 23: Биологическая активность солевых форм Asp(Цитарабин)⋅Y, влияние на пролиферацию лейкозных раковых клеток
Раковые клетки лейкоза L1210 выращивали в среде DMEM с 10% донорской лошадиной сывороткой, 1% глутамина, 1% незаменимых аминокислот, 0,1% амфотерицина и 0,1% гентамицина. Клетка разводили и высевали в 96-луночные планшеты 2000 клеток/мл, 400 клеток/лунку, в объеме 0,2 мл на лунку. Планшеты инкубировали при температуре 37°С в течение 180 мин. После инкубации добавляли исследуемые вещества в возрастающих концентрациях от 1 мкМ до 100 нМ, приготовленных в физиологическом растворе, по 6 повторов каждый. Исследуемыми веществами были следующие: Asp(цитарабин)⋅ацетатная солевая форма, Asp(цитарабин)⋅гидрохлоридная солевая форма, Asp(цитарабин)⋅метансульфонатная солевая форма, Asp(цитарабин)⋅бензолсульфонатная солевая форма и Asp(цитарабин)⋅фосфатная солевая форма. Планшеты также содержали 6 лунок с солевым раствором в качестве контроля. До проведения анализа планшеты инкубировали при температуре 37°С в течение 72 ч. В конце периода воздействия проводили анализ МТТ [3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолийбромид]. В каждую лунку добавляли МТТ в концентрации 5 мг/мл объемом 0,02 мл. Планшеты инкубировали при температуре 37°С в течение 3 часов, затем центрифугировали при 3500 об/мин в течение 6 минут и супернатанты отсасывали. Каждую из пеллет, содержащих кристаллы МТТ, растворяли в 0,2 мл ДМСО. Абсорбцию определяли с использованием считывателя ELISA при длине волны 570 нм. Полученные значения IC50 обобщены в таблице 4.
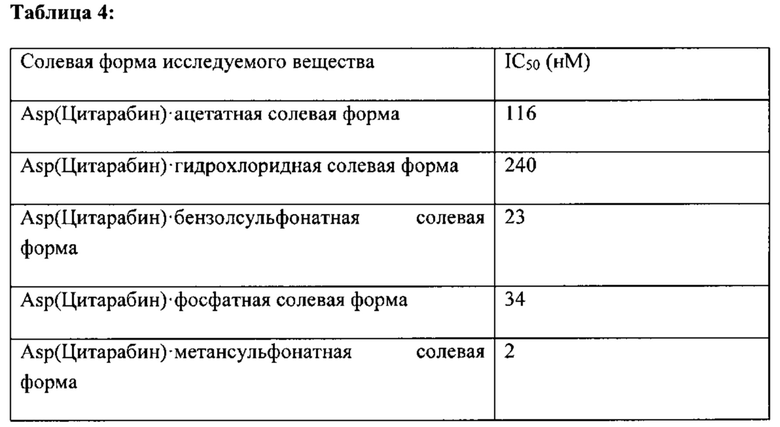
Пример 24: Биологическая активность Asp(Цитарабин)⋅ацетатной солевой формы, влияние на пролиферацию различных клеточных линий
Разнообразные раковые клетки лейкоза человека выращивали в среде RPMI с 10% фетальной бычьей сывороткой, 1% глутамина, 0,1% амфотерицина и 0,1% гентамицина. Клетки разбавляли и высевали, как описано в примере 23. Добавляли Asp(Цитарабин)⋅ацетатную солевую форму в увеличивающихся концентрациях от 1 мкМ до 100 нМ в физиологическом растворе, каждый в трех повторах. Проводили анализ МТТ так, как описано в примере 23. Полученные значения IC50 обобщены в таблице 5.


Специалистам в данной области техники понятно, что настоящее изобретение не ограничено настоящим описанием. Скорее, объем настоящего изобретения включает в себя как комбинации, так и подкомбинации различных признаков, описанных выше, а также изменения и модификации. Следовательно, настоящее изобретение не ограничено описанными вариантами реализации, и объем и суть изобретения будут более понятны из прилагаемой формулы изобретения.
Список литературы:
Brynes, S, Burckart, GJ and Mokotoff, M, 1978a. Potential inhibitors of L-asparagine biosynthesis. 4. Substituted sulfonamide and sulfonylhydrazide analogs of L-asparagine. J Med Chem. 21(1): 45-9.
Brynes S, Fiorina VJ, Cooney DA, Milman HA. 1978b. Potential antitumor agents via inhibitors of L-asparagine synthetase: substituted sulfonamides and sulfonyl hydrazides related to glutamine. J Pharm Sci. 67(11): 1550-3.
Dalal M, Plowman J, Breitman TR, et al. 1986. Arabinofuranosyl-5-azacytosine: antitumor and cytotoxic properties. Cancer Res.; 46:831.
Hartel LW, Boder GB, Kroin JS, et al. 1990. Evaluation of the antitumor activity of gemcitabine (2',2'-difluoro-2'deoxycytidine). Cancer Res.; 50:4417.
Heineman V, Hartel LW, Grindey GB, et al. 1988. Comparison of the cellular pharmacokinetics and toxicology of 2',2'-difluoro-2'deoxycytidine and 1-β-D-arabinofuranosylcytocsine. Cancer Res 1988; 48:4024.
Ho DHW, 1974. Biochemical studies on new antitumor agent, O2-2'-cyclocyttidine. Biochem Pharmacol.; 23:1235.
Kodama K, Morozumi M, Saitoh K, et al. 1989. Antitumor activity and pharmacology of 1-β-D-arabinofuranosylcytocsine. Jpn J Cancer Res.; 80:679.
Manfredini et al., 2000, Peptide T-araC conjugates: solid phase synthesis and biological activity of N4-(Acylpeptidyl)araC, Bioorg. Med. Chem. 8: 539-547.
Piek J, Adelt T, Huse K, Bock WJ. [Cerebrospinal fluid and plasma aminogram in patients with primary and secondary tumors of the CNS], [article in German], Infusionsther Klin Ernahr, 1987 Apr; 14(2): 73-77.
Stammer, CH and Sato, M, 1978. 5-Carboxamido-4-amino-3-isoxazolidone, an asparagine analog, J Med Chem. 21(7):709-712.
Warrell RP Jr, and Berman E, Phase I and II study of fludarabine phosphate in leukemia: efficacy with delayed central nervous system toxicity. J Clin Oncol, 1986 Jan; 4(1): 74-79.
Woodcock TM, Chou ТС, Tan CTC, et al. 1980. Biochemical, pharmacological, and phase I clinical evaluation of pseudoisocytidine. Cancer Res.; 40: 4242.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТЫ ЦИТАРАБИНА ДЛЯ ЛЕЧЕНИЯ РАКА | 2016 |
|
RU2708672C1 |
| СИСТЕМЫ ДОСТАВКИ ДЛЯ КОНТРОЛИРУЕМОГО ВЫСВОБОЖДЕНИЯ ЛЕКАРСТВЕННОГО СРЕДСТВА | 2016 |
|
RU2748992C2 |
| КОНЪЮГАТЫ ЦИТОТОКСИЧЕСКИХ СРЕДСТВ С ПЕПТИДАМИ | 2007 |
|
RU2457218C2 |
| ПРОИЗВОДНЫЕ ВИТАМИНА В6 НУКЛЕОТИДОВ, АЦИКЛИЧЕСКИХ НУКЛЕОТИДОВ И АЦИКЛИЧЕСКИХ НУКЛЕОЗИДНЫХ ФОСФОНАТОВ | 2012 |
|
RU2640582C2 |
| НОВЫЕ ПСА-СВЯЗЫВАЮЩИЕ АГЕНТЫ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2787105C2 |
| ГЛИКОЗИЛИРОВАННЫЙ ПЕПТИД GLP-1 | 2009 |
|
RU2543157C2 |
| КОНЪЮГАТЫ СКОНСТРУИРОВАННЫХ АНТИТЕЛ С ЦИСТЕИНОВЫМИ ЗАМЕНАМИ | 2016 |
|
RU2834224C1 |
| КОНЪЮГАТЫ ЛИГАНДА С НЕСКОЛЬКИМИ ЛЕКАРСТВЕННЫМИ СРЕДСТВАМИ | 2006 |
|
RU2470668C2 |
| НАЦЕЛИВАЮЩИЕ АМИНОКИСЛОТНЫЕ ЛИПИДЫ | 2013 |
|
RU2654210C2 |
| КОМПОЗИЦИИ И СПОСОБЫ, ОТНОСЯЩИЕСЯ К КОНЪЮГАТАМ ЛЕКАРСТВЕННЫХ СРЕДСТВ С АНТИТЕЛАМИ ПРОТИВ CD19 | 2019 |
|
RU2806333C2 |
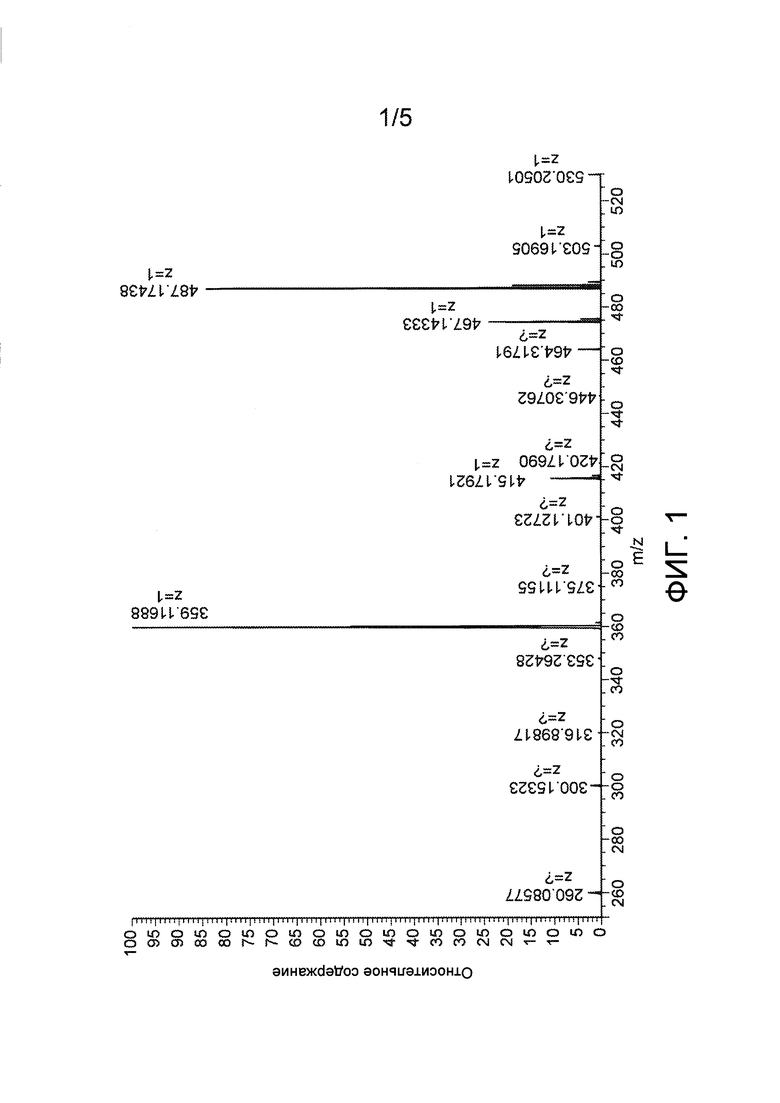
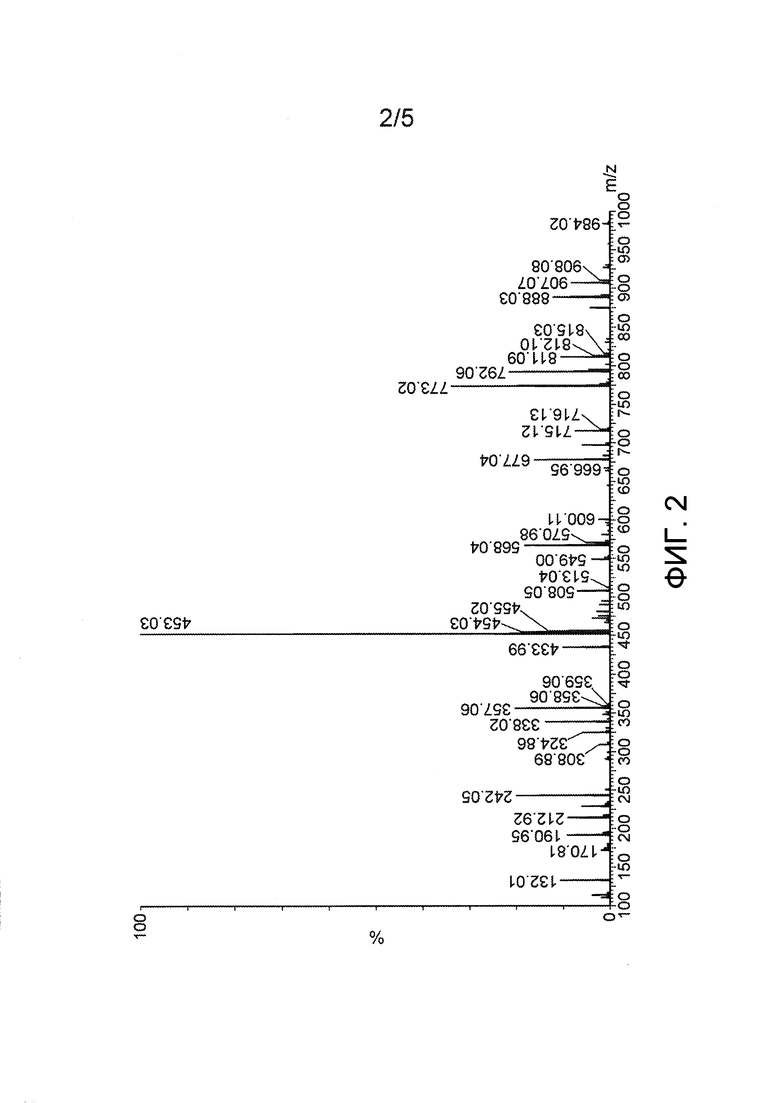
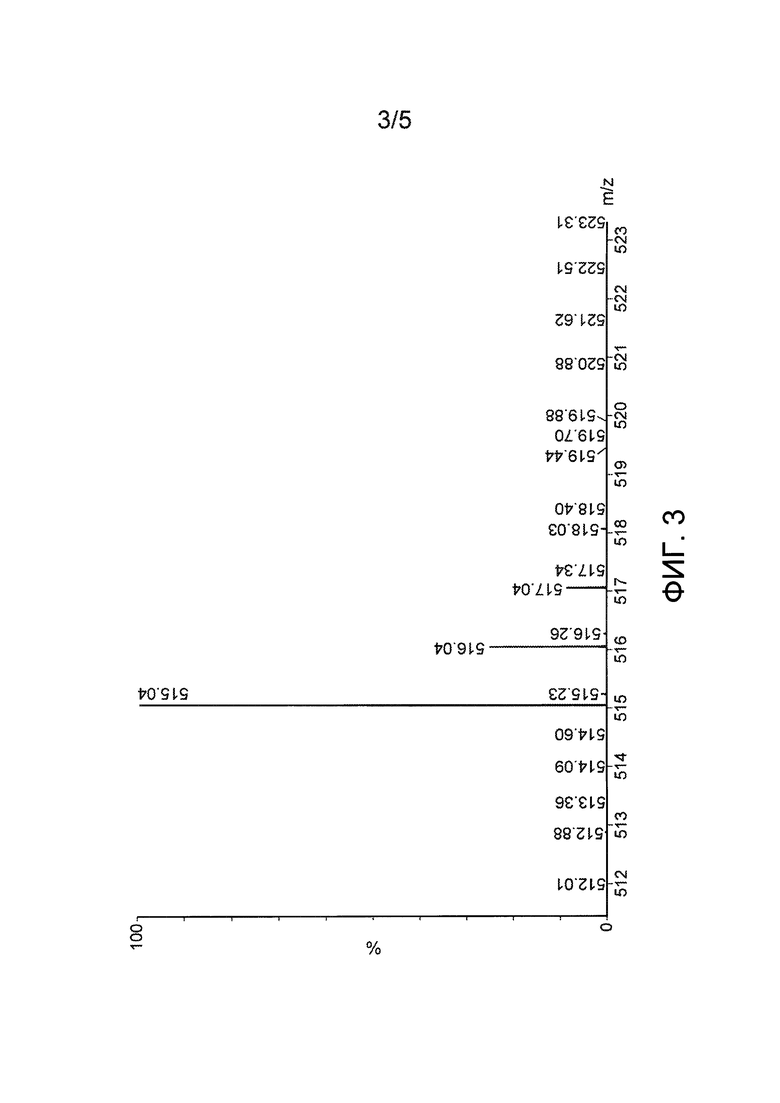
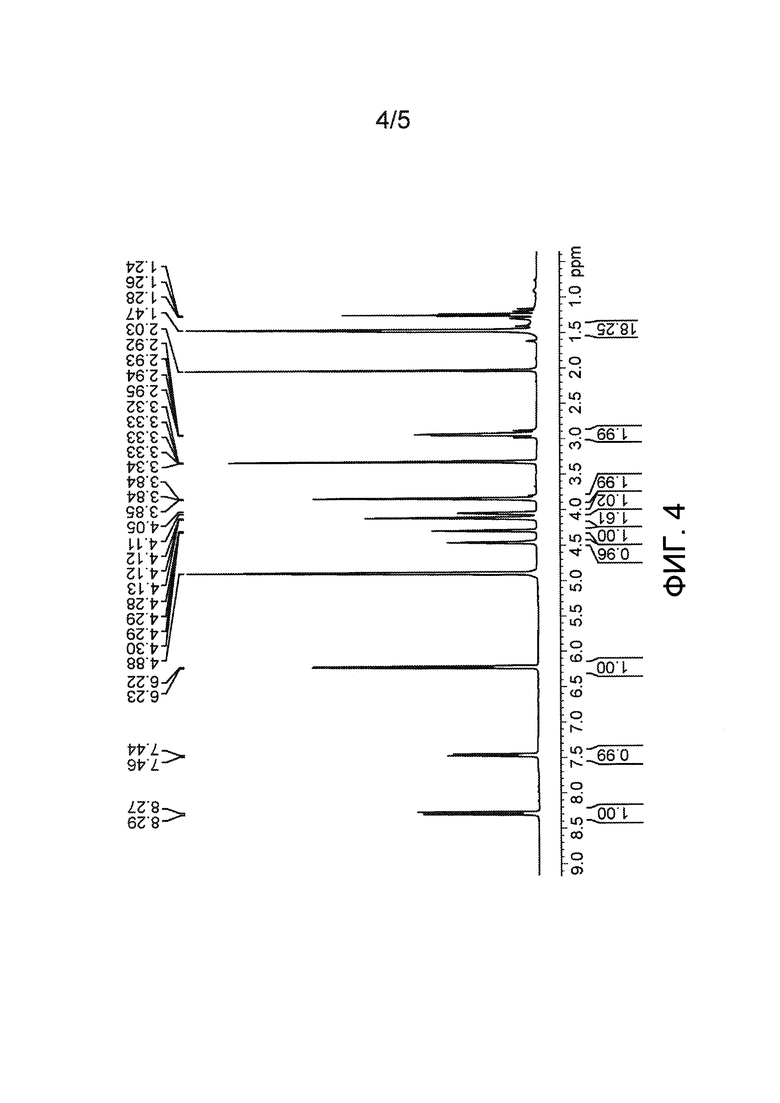
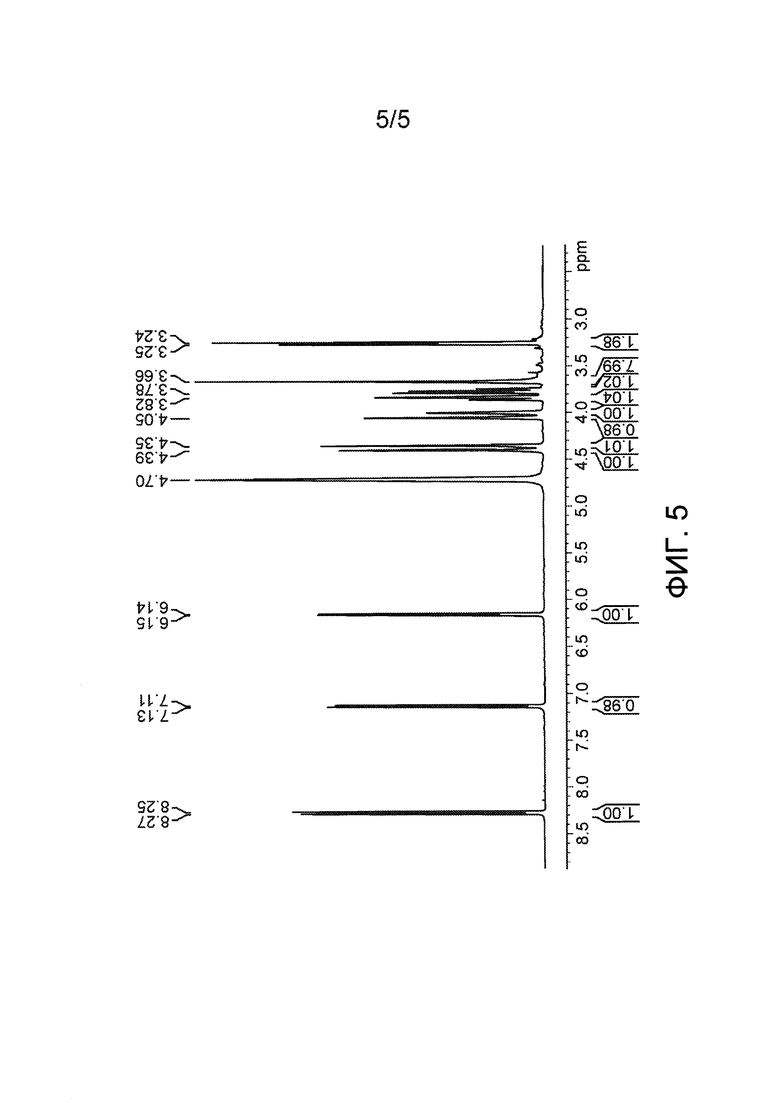
Изобретение относится к фармацевтически приемлемым солям, представленным структурой формулы (1), которые могут найти применение для лечения рака или предракового состояния или расстройства. В формуле (1) Y представляет собой фармацевтически приемлемую органическую или неорганическую кислоту или кислотный остаток, при этом указанная кислота выбрана из HCl, метансульфоновой кислоты, трифторуксусной кислоты (ТФУ), H2SO4 и H3PO4. Изобретение относится также к фармацевтической композиции, содержащей указанные соли, и способам лечения рака или предракового состояния, в том числе у субъекта, имеющего иммунологическое заболевание или расстройство или дисфункцию органов. 6 н. и 16 з.п. ф-лы, 5 ил., 5 табл., 24 пр.
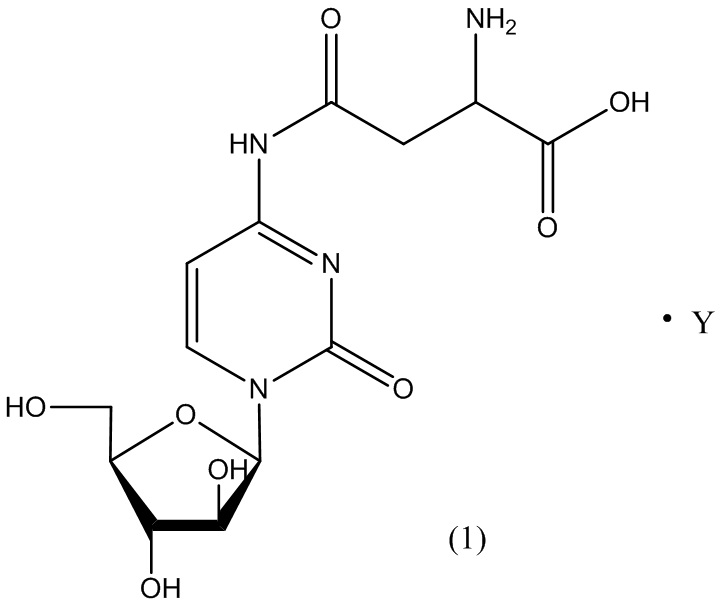
1. Фармацевтически приемлемая соль, представленная структурой формулы (1)
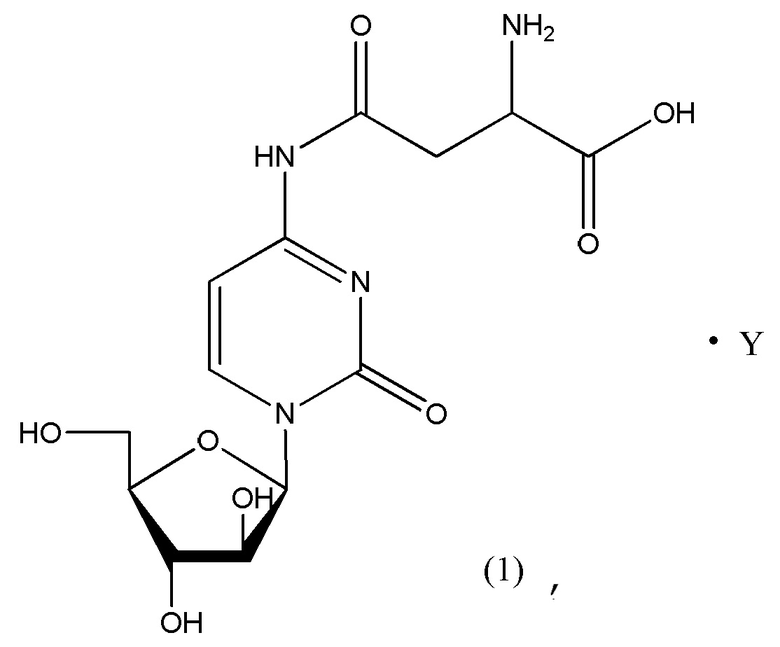
где Y представляет собой фармацевтически приемлемую органическую или неорганическую кислоту или кислотный остаток, при этом указанная кислота выбрана из HCl, метансульфоновой кислоты, трифторуксусной кислоты (ТФУ), H2SO4 и H3PO4.
2. Фармацевтически приемлемая соль по п. 1, отличающаяся тем, что Y представляет собой HCl, представленная структурой
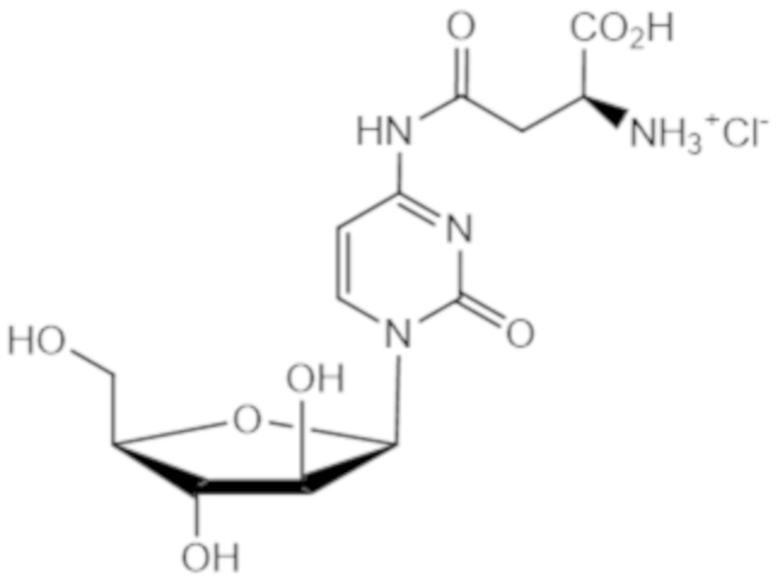 .
.
3. Фармацевтически приемлемая соль по п. 1, отличающаяся тем, что Y представляет собой метансульфоновую кислоту, представленная структурой
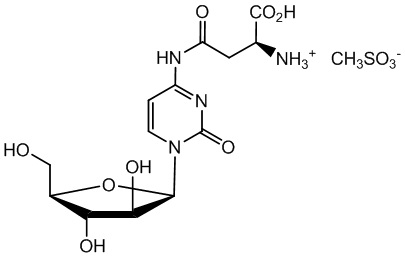 .
.
4. Фармацевтически приемлемая соль по п. 1, отличающаяся тем, что Y представляет собой ТФУ, представленная структурой
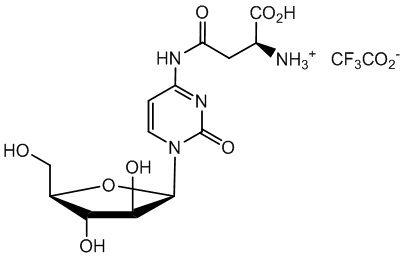 .
.
5. Фармацевтически приемлемая соль по п. 1, отличающаяся тем, что Y представляет собой H2SO4, представленная структурой
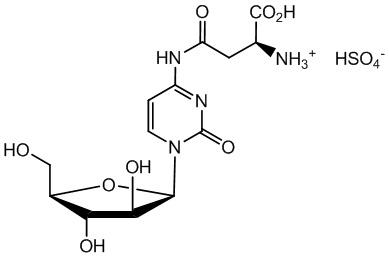 .
.
6. Фармацевтически приемлемая соль по п. 1, отличающаяся тем, что Y представляет собой H3PO4, представленная структурой
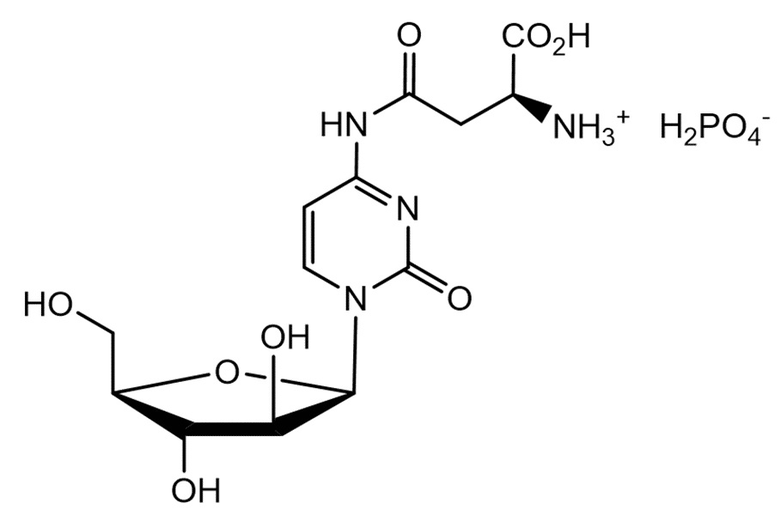 .
.
7. Фармацевтическая композиция для лечения рака или предракового состояния или расстройства, содержащая терапевтически эффективное количество фармацевтически приемлемой соли по п. 1 и фармацевтически приемлемое вспомогательное вещество.
8. Способ лечения рака или предракового состояния или расстройства, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества фармацевтически приемлемой соли по п. 1.
9. Способ по п. 8, отличающийся тем, что рак представляет собой метастазы рака.
10. Способ по п. 8, отличающийся тем, что рак представляет собой несолидную опухоль или солидную опухоль или их комбинацию.
11. Способ по п. 10, отличающийся тем, что несолидная опухоль включает лейкозы, лимфомы и множественную миелому.
12. Способ по п. 11, отличающийся тем, что лейкоз включает острый миелоидный лейкоз (ОМЛ) и острый лимфобластный лейкоз (ОЛЛ).
13. Способ по п. 10, отличающийся тем, что солидная опухоль включает опухоли центральной нервной системы (ЦНС), рак печени, колоректальную карциному, рак молочной железы, рак желудка, рак поджелудочной железы, карциному мочевого пузыря, карциному шейки матки, опухоли головы и шеи, рак вульвы и дерматологические новообразования.
14. Способ по п. 13, отличающийся тем, что дерматологические новообразования включают меланому, плоскоклеточную карциному и базальноклеточные карциномы.
15. Способ по п. 8, отличающийся тем, что рак представляет собой опухоль системы крови.
16. Способ по п. 15, отличающийся тем, что опухоль системы крови представляет собой лейкоз, лимфому и миелому.
17. Способ по п. 16, отличающийся тем, что опухоль системы крови представляет собой миелоидный лейкоз, лимфоцитарный лейкоз, острый миелоидный лейкоз (ОМЛ), хронический миелоидный лейкоз (ХМЛ), острый лимфоцитарный лейкоз (ОЛЛ), хронический лимфоцитарный лейкоз (ХЛЛ), лимфому Ходжкина, неходжкинскую лимфому, множественную миелому и макроглобулинемию Вальденстрема.
18. Способ по п. 8, отличающийся тем, что указанное предраковое состояние или расстройство представляет собой миелодиспластические синдромы (МДС).
19. Способ по п. 8, отличающийся тем, что указанный субъект представляет собой человека.
20. Способ лечения индивидуума, который является кандидатом на трансплантацию костного мозга, включающий введение указанному индивидууму до трансплантации костного мозга фармацевтически приемлемой соли по п. 1.
21. Способ лечения рака у субъекта, имеющего иммунологическое заболевание или расстройство, включающий введение фармацевтически приемлемой соли по п. 1.
22. Способ лечения рака у субъекта с дисфункцией органов, включающий введение указанному субъекту фармацевтически приемлемой соли по п. 1.
| S | |||
| HASABELNABY et al | |||
| Synthesis, chemical and enzymatic hydrolysis, and aqueous solubility of amino acid ester prodrugs of 3-carboranyl thymidine analogs for boron neutron capture therapy of brain tumors, EUR | |||
| J | |||
| MED | |||
| CHEM., 2012, vol | |||
| Устройство двукратного усилителя с катодными лампами | 1920 |
|
SU55A1 |
| Водяной двигатель | 1921 |
|
SU325A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| CN 101812105 А, 25.08.2010 | |||
| RU | |||

