Область настоящего изобретения
Настоящее изобретение относится к сульфонилгидроксамовым кислотам в качестве селективных ингибиторов HDAC. В частности, настоящее изобретение относится к соединениям общей формулы I сульфонилгидроксамовой кислоты на основе индола
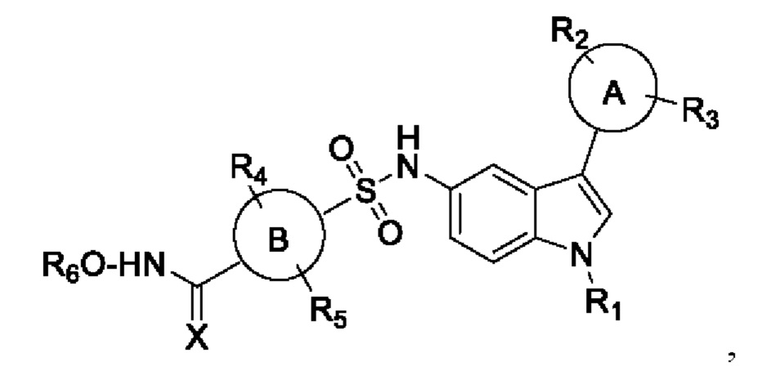
где
кольцо А и В представляет собой арил или гетероарил или циклоалкил или конденсированную арильную или конденсированную алкильную группу R1, R2, R3, R4, R5, R6 представляет собой водород, алкокси, арилокси, гидрокси, сложный эфир, амид, амино, алкил, арил, гетероарил, галоген, гидрокси, алкокси, арилокси, нитро, циано, сложный эфир, альдегид.
Предшествующий уровень техники настоящего изобретения
Сульфонилгидроксамовые кислоты представляют собой важные структурные мотивы среди ряда различных фармацевтически эффективных соединений. Было обнаружено, что эти соединения проявляют широкий спектр биологических свойств (EP0977745; JP2000500145; US3186992; US5804593; US5962481; US6437177; US6548524; US6583318; WO9816520; WO9831664; WO2009040517; CN1380288). Существует определенный класс производных сульфонилгидроксамовых кислот, которые, как сообщается, проявляют свойства ингибирования HDAC (US 7183298; US 2004092598; US 2004198830; US 2005085515; US 2005107445; US 2007004806; WO 0230879); в то же время, соединения, характеризующиеся селективным ингибированием HDAC, отмечаются очень редко. Таким образом, конструирование и разработка сульфонилгидроксамовых кислот, которые способны область свойствами селективного ингибирования HDAC, являются скрытой проблемой и наиболее востребованной задачей. Несмотря на то, что сообщалось о многих типах производных сульфонилгидроксамовых кислот с использованием различных стратегий конструирования архитектуры сульфонилгидроксамовых кислот, существуют определенные группы производных сульфонилгидроксамовых кислот, представляющие интерес, которые не были синтезированы и оценены в отношении биологических свойств. Индольные производные сульфонилгидроксамовых кислот по настоящему изобретению представляют собой примеры этого типа и характеризуются редкой встречаемостью. Таким образом, существует потребность в разработке способов синтеза и биологической оценки различным образом замещенных индольных соединений сульфонилгидроксамовых кислот. В этом направлении настоящее изобретение направлено на синтез и систематический скрининг структурно разнообразных сульфонилгидроксамовых кислот на основе индольного ядра.
В указанном контексте большое количество новых производных сульфонилгидроксамовых кислоь были синтезированы и оценены в отношении ингибирующей активности HDAC.
Цель настоящего изобретения
Основной целью настоящего изобретения является обеспечение новых производных сульфонилгидроксамовой кислоты в качестве применимых ингибиторов HDAC.
Основной целью настоящего изобретения является обеспечение новых производных сульфонилгидроксамовой кислоты в качестве применимых селективных ингибиторов HDAC.
Другой целью настоящего изобретения является обеспечение способа получения новых производных сульфонилгидроксамовой кислоты.
Краткое описание настоящего изобретения
Вышеуказанные и другие цели настоящего изобретения достигали обеспечением новых соединений сульфонилгидроксамовой кислоты, которые были синтезированы и испытаны на активность.
Соответственно, настоящее изобретение относится к новому классу производных сульфонилгидроксамовой кислоты общей формулы I.
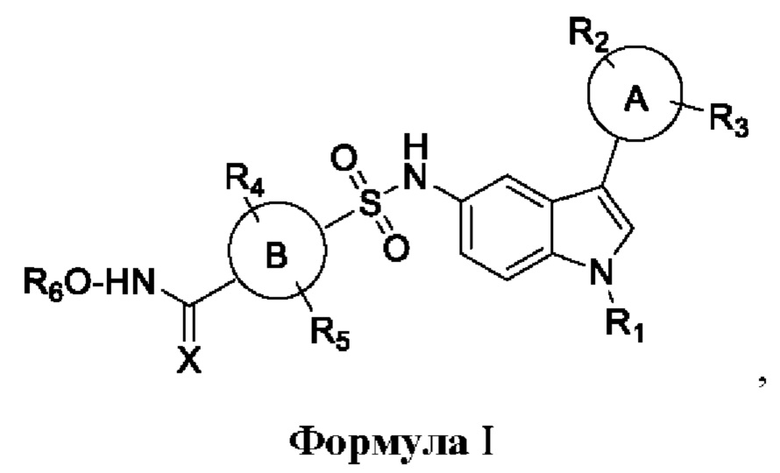
где
кольцо А и В представляет собой арил или гетероарил или циклоалкил или конденсированную арильную или конденсированную алкильную группу R1, R2, R3, R4, R5, R6 представляет собой водород, алкокси, арилокси, гидрокси, сложный эфир, амид, амино, алкил, арил, гетероарил, галоген, гидрокси, алкокси, арилокси, нитро, циано, сложный эфир, альдегид.
Структурные формулы характерных соединений следующие:
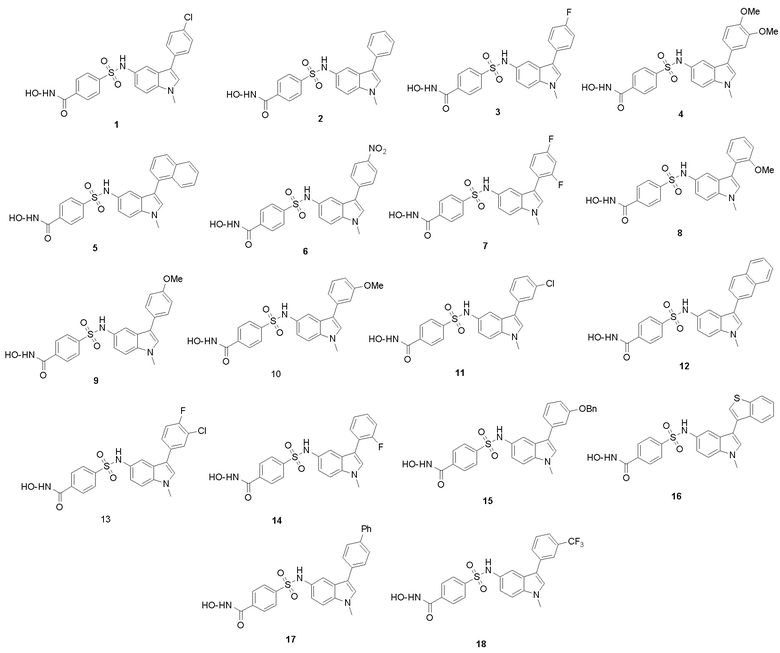
Согласно варианту осуществления настоящего изобретения описанные в настоящем изобретении новые производные сульфонилгидроксамовой кислоты представлены следующим:
N-гидрокси-4-(N-(1-метил-3-фенил-1Н-индол-5-ил)сульфамоил)бензамид (1)
4-(N-(3-(4-хлорфенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (2)
4-(N-(3-(4-фторфенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (3)
4-(N-(3-(3,4-диметоксифенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (4)
N-гидрокси-4-(N-(1-метил-3-(нафталин-1-ил)-1Н-индол-5-ил)сульфамоил)бензамид (5)
N-гидрокси-4-(N-(1-метил-3-(4-нитрофенил)-1Н-индол-5-ил)сульфамоил)бензамид (6)
4-(N-(3-(2,4-дифторфенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (7)
N-гидрокси-4-(N-(3-(2-метоксифенил)-1-метил-1Н-индол-5-ил)сульфамоил)бензамид (8)
N-гидрокси-4-(N-(3-(4-метоксифенил)-1-метил-1Н-индол-5-ил)сульфамоил)бензамид (9)
N-гидрокси-4-(N-(3-(3-метоксифенил)-1-метил-1Н-индол-5-ил)сульфамоил)бензамид (10)
4-(N-(3-(3-хлорфенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (11)
N-гидрокси-4-(N-(1-метил-3-(нафталин-2-ил)-1Н-индол-5-ил)сульфамоил)бензамид (12)
4-(N-(3-(3-хлор-4-фторфенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (13)
4-(N-(3-(2-фторфенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (14)
4-(N-(3-(3-(бензилокси)фенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (15)
4-(N-(3-(бензо[b]тиофен-3-ил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (16)
4-(N-(3-(бифенил-4-ил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (17)
N-гидрокси-4-(N-(1-метил-3-(3-(трифторметил)фенил)-1Н-индол-5-ил)сульфамоил)бензамид (18)
Настоящее изобретение также относится к способу получения производных сульфонилгидроксамовой кислоты, как описано в вышеуказанной общей формуле.
Было обнаружено, что большое число различных производных сульфонилгидроксамовой кислоты, которые обладают по-разному замещенной структурой, проявляют несколько биологических свойств. Их функциональные группы являются торчащими структурными мотивами новых лекарственных веществ от различных фармакологических групп. Развитие новых структурных скелетов строения сульфонилгидроксамовой кислоты является очень важным в процессе обнаружения лекарственного средства. В связи с этим большое количество производных сульфонилгидроксамовой кислоты обнаруживали как изображенные в вышеуказанной общей формуле I.
Способ получения производных сульфонилгидроксамовой кислоты, причем указанный способ включает в себя стадии:
a) бромирования нитроиндола с применением бромирующих реагентов в полярных не протонированных растворителях при от -5 до 5°С в течение 40-100 минут;
b) защиты индола NH с применением алкилгалогенидов и гидридного основания в полярном не протонированном растворителе при от -5 до 5°С в течение 40-100 минут;
c) восстановления нитрогруппы до амина с применением восстановителя металла в смеси полярного растворителя при от -5 до 5°С в течение 40-100 минут;
d) опосредованного основанием сочетания сульфонильных и аминовых функциональных групп в полярном растворителе при 25-40°С в течение 12-24 часов;
e) реакции Сузуки/сочетания с применением производного бороновой кислоты, палладиевого катализатора и фосфатной соли в полярном растворителе при 70-100°С в течение 5-10 часов;
f) введения гидроксамовой кислоты с применением гидроксиамина и основания в смеси полярных растворителей при 25-40°С в течение 12-24 часов.
Согласно еще одному варианту осуществления настоящего изобретения бромирующий реагент выбран из Br2 или NBS.
Согласно еще одному варианту осуществления настоящего изобретения полярный не протонированный растворитель выбран из DMF или DMSO.
Согласно еще одному варианту осуществления настоящего изобретения алкилгалогенид выбран из метилйодида, метилбромида, этилйодила или этилбромида.
Согласно еще одному варианту осуществления настоящего изобретения гидридное основание выбрано из NaH, KH или CsH; металл, используемый в качестве восстановителя, выбран из Zn или Fe.
Согласно еще одному варианту осуществления настоящего изобретения полярный растворитель/полярные растворители выбран/выбраны из THF, МеОН, EtOH, Н2О, CH3CN, 1,4-диоксана или Et2O.
Согласно еще одному варианту осуществления настоящего изобретения основание выбрано из NaOH, KOH, CsOH, NaHCO3 или KHCO3.
Согласно еще одному варианту осуществления настоящего изобретения производное бороновой кислоты выбрано из арила или гетероарила или циклоалкила или конденсированной арильной или конденсированной алкильной группы с замещениями R1 и/или R2, где R1 и/или R2 представляет собой водород, алкокси, арилокси, гидрокси, сложный эфир, амид, амино, алкил, арил, гетероарил, галоген, гидрокси, алкокси, арилокси, нитро, циано, сложный эфир или альдегид.
Согласно еще одному варианту осуществления настоящего изобретения палладиевый катализатор выбран из Pd(PPh3)2Cl2 или Pd(PPh3)4; фосфатная соль выбрана из K3PO4 или Na3PO4.
Согласно еще одному варианту осуществления настоящего изобретения полученные производные сульфонилгидроксамовой кислоты проверяли на их эффективность по отношению к ингибирующему свойству HDAC.
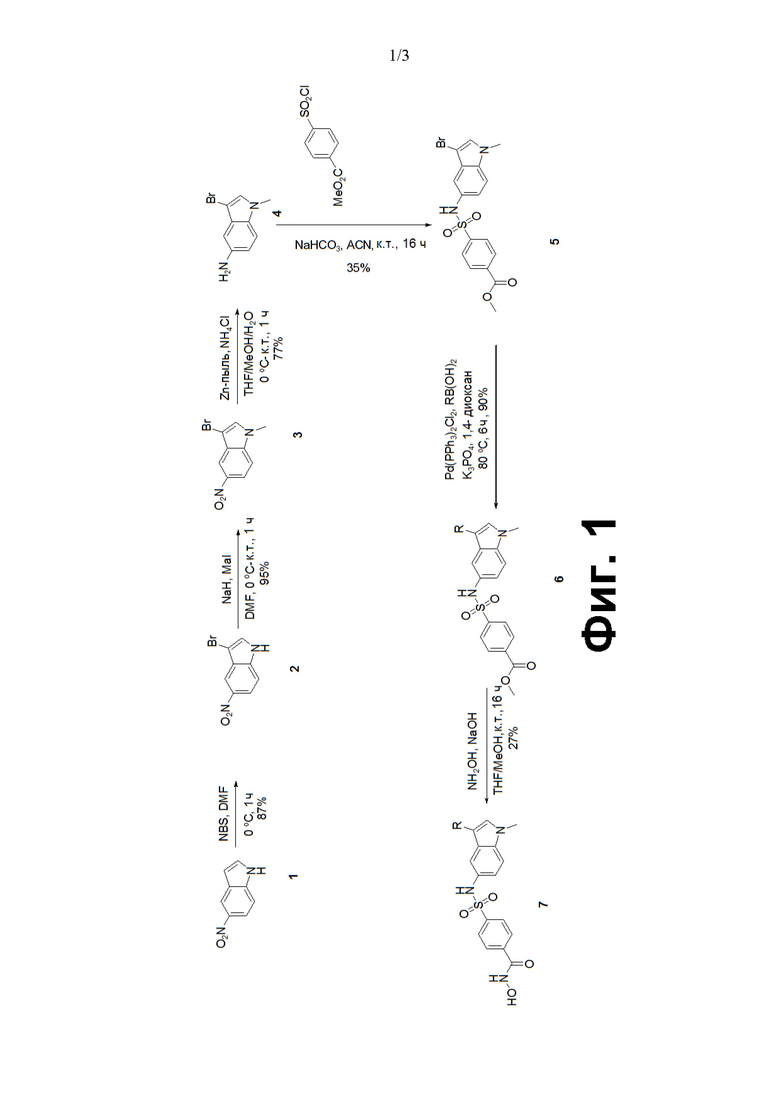
Таким образом, настоящее изобретение относится к новому классу производных сульфонилгидроксамовой кислоты, которые применимы в качестве селективных ингибиторов HDAC. В лаборатории была инициирована программа по проектированию и синтезу новых производных сульфонилгидроксамовой кислоты, которые могут служить в качестве новых химических структурных единиц для процесса обнаружения лекарственного препарата. В этих условиях были синтезированы новые производные сульфонилгидроксамовой кислоты и их оценивали на активность HDAC. Синтез таких соединений проводили как описано на следующих схемах с применением простых аналогов индола/оксиндола в качестве исходных субстратов.
Краткое описание чертежей
Фиг. 1. Синтез сульфонилгидроксамовой кислоты на основе индола.
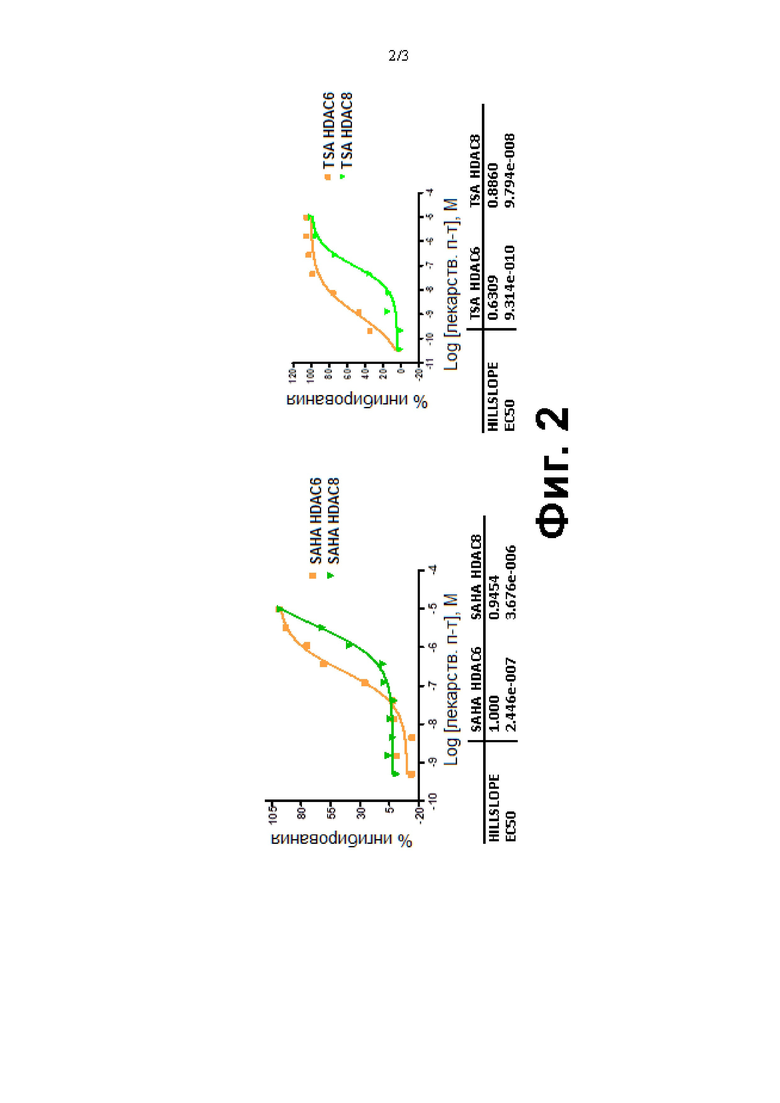
Фиг. 2. Сравнение SAHA [а] и TSA [b] в биохимическом анализе HDAC6 и HDAC8.
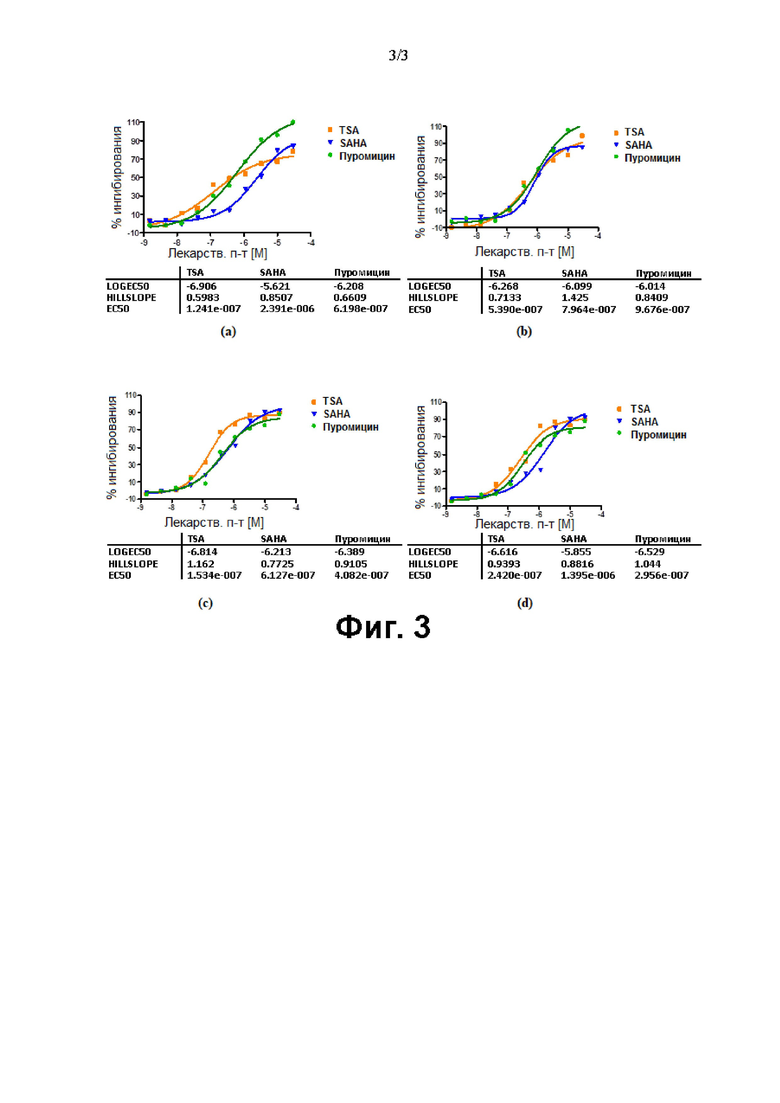
Фиг. 3. Сравнение SAHA, TSA и пуромицина в клеточных линиях HeLa [a], DU-145 [b], MCF-7 [с] и SKOV3 [d].
Подробное раскрытие настоящего изобретения
Производные сульфонилгидроксамовых кислот представляют собой эффективные структурные мотивы, способные проявлять различные виды биологической активности. Это привело к конструированию и синтезу большого количества производных сульфонилгидроксамовых кислот, как проиллюстрировано на Фиг. 1. Эти производные нового класса сульфонилгидроксамовых кислот являются пригодными в качестве селективных ингибиторов HDAC.
ПРИМЕРЫ
Настоящее изобретение будет более конкретно объяснено следующими примерами. Тем не менее, объем настоящего изобретения не ограничен объемом представленных ниже примеров.
Стадия 1: Синтез 3-бром-5-нитро-1Н-индола:
К перемешиваемому раствору 5-нитро-1H-индола (1) (100 г, 0,61 моль) в DMF (1 л) добавляли NBS (131,1 г, 0,74 моль) при 0°С и раствор перемешивали в течение 1 ч. После завершения реакции смесь разбавляли холодной водой, фильтровали. Твердое вещество промывали гексаном и продукт (130,0 г, 87%) использовали сразу на следующей стадии без дополнительной очистки.
Стадия 2: Синтез 3-бром-1-метил-5-нитро-1Н-индола:
К перемешиваемому раствору 3-бром-5-нитро-1H-индола (2) (100,0 г, 0,414 моль) в DMF (1 л) добавляли NaH (19,9 г, 0,829 моль) при 0°С и раствор перемешивали в течение 0,5 ч. Затем к реакционной смеси добавляли метилйодид (87,7 г, 0,622 моль) и раствор перемешивали при к. т.еще 2 ч. После завершения реакции смесь гасили водой и экстрагировали этилацетатом. Органический экстракт сушили над безводным сульфатом натрия, фильтровали и растворители выпаривали из фильтрата при пониженном давлении с получением неочищенного вещества, которое очищали методом колоночной хроматографии на силикагеле (100-200 меш), элюировали 10-15% градиентом EtOAc в петролейном эфире с получением 3-бром-1-метил-5-нитро-1Н-индола (101,0 г, 95%).
Стадия 3: Синтез 3-бром-1-метил-1Н-индол-5-амина:
К перемешиваемому раствору 3-бром-1-метил-5-нитро-1Н-индола (3) (7,0 г, 27,45 ммоль) в THF : MeOH : H2O (1:1:1, 80 мл) добавляли Zn пыль (17,9 г, 274,5 ммоль) и NH4Cl (14,8 г, 274,5 ммоль) при 0°С. Реакционную смесь оставляли нагреваться до к.т. в течение 1 ч. После завершения реакции смесь фильтровали через целитный слой и экстрагировали этилацетатом. Органический экстракт сушили над безводным сульфатом натрия, фильтровали и растворители выпаривали из фильтрата при пониженном давлении с получением неочищенного соединения (4,8 г, 77%), которое использовали сразу на следующей стадии без дополнительной очистки.
Стадия 4: Синтез метал-4-(N-(3-бром-1-метил-1Н-индол-5-ил)сульфамоил)бензоата:
К перемешиваемому раствору 3-бром-1-метил-1Н-индол-5-амина (4) (4,8 г, 21,3 ммоль) в ACN (30 мл) добавляли NaHCO3 (1,79 г, 21,3 ммоль) и метил-4-(хлорсульфонил)бензоат (6,0 г, 25,5 ммоль) при 0°С и раствор перемешивали при к.т. в течение 16 ч. После завершения реакции смесь гасили водой и экстрагировали этилацетатом. Органический экстракт сушили над безводным сульфатом натрия, фильтровали и растворители выпаривали из фильтрата при пониженном давлении с получением неочищенного соединения, которое очищали методом колоночной хроматографии на силикагеле (100-200 меш), элюировали 0-40% градиентом EtOAc в петролейном эфире с получением метил-4-(N-(3-бром-1-метил-1Н-индол-5-ил)сульфамоил)бензоата (5) (3,2 г, 35%).
Стадия 5: Синтез метил-4-(N-(1-метил-3-арил-1Н-индол-5-ил)сульфамоил)бензоата:
Фенилбороновую кислоту (0,90 ммоль) добавляли к раствору метил-4-(N-(3-бром-1-метил-1Н-индол-5-ил)сульфамоил)бензоата (5) (0,4 ммоль) в 1,4-диоксане (5 мл), а затем добавляли K3PO4 (1,4 ммоль) и смесь продували аргоном в течение 20 мин. К смеси добавляли Pd(PPh3)2Cl2 (0,06 ммоль), продували еще 5 мин аргоном и нагревали при 80°С в течение 6 ч. После завершения реакции смесь охлаждали до температуры окружающей среды и фильтровали через слой целита. Растворители выпаривали из фильтрата при пониженном давлении и полученное неочищенное вещество очищали методом колоночной хроматографии на силикагеле (100-200 меш) с получением метил-4-(N-(1-метил-3-фенил-1Н-индол-5-ил)сульфамоил)бензоата (6) (65-90%).
Стадия 6: Синтез N-гидрокси-4-(N-(1-метал-3-арил-1Н-индол-5-ил)сульфамоил)бензамида:
К перемешиваемому раствору 50% водн. гидроксиламина (3 мл) и метил-4-(N-(1-метил-3-фенил-1Н-индол-5-ил)сульфамоил)бензоата (6) (0,7 ммоль) в THF : MeOH (1:1, 5 мл) добавляли 50% водн. KOH (0,5 мл) и смесь перемешивали при к.т. в течение 5 ч. После завершения реакции раствор сушили в вакууме. Полученные полутвердые соединения промывали этилацетатом и диэтиловым эфиром с удалением всех неполярных отходов и остаток подкисляли 0,1 н HCl. Полученное твердое вещество собирали фильтрацией и промывали водой с получением неочищенного вещества, которое очищали методом колоночной хроматографии на силикагеле (230-400 меш) с получением N-гидрокси-4-(N-(1-метил-3-арил-1Н-индол-5-ил)сульфамоил)бензамида (15-45%).
Пример 1: N-гидрокси-4-(N-(1-метил-3-фенил-1Н-индол-5-ил)сульфамоил)бензамид (1): 1Н ЯМР (400 МГц, dmso) δ 11,35 (br s, 1H), 10,00 (br s, 1H), 9,19 (br s, 1H), 7,83-7,88 (m, 2H), 7,75 (d, J=8,31 Гц, 2H), 7,62-7,65 (m, 1H), 7,35-7,46 (m, 6H), 7,23 (td, J=4,40, 8,80 Гц, 1H), 6,94 (dd, J=1,96, 8,80 Гц, 1H), 3,77 (s, 3H). LC-MS чистота: 97,29%; (ES+): m/z 422,42 (M+H+); tr = 1,91 мин.
Пример 2: 4-(N-(3-(4-хлорфенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (2) 1Н ЯМР (400 МГц, dmso) δ 11,35 (br s, 1H), 10,02 (br s, 1H, 9,20 (br s, 1H), 7,80-7,87 (m, 2H), 7,72-7,80 (m, 2H), 7,69 (s, 1H), 7,36-7,50 (m, 6H), 6,94 (dd, J=1,71, 8,56 Гц, 1H), 3,77 (s, 3H). LC-MS чистота: 98,81%; (ES+): m/z 456,35 (M+H+); tr = 2,06 мин.
Пример 3: 4-(N-(3-(4-фторфенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (3): 1H ЯМР (400 МГц, dmso) δ 9,05-9,20 (br s, 3Н), 7,85 (d, J=8,31 Гц, 2H), 7,75 (d, J=8,31 Гц, 2Н), 7,62 (s, 1Н), 7,35-7,44 (m, 4H), 7,26 (br t, J=8,80 Гц, 2H), 6,95 (s, 1H), 3,76 (s, 3H). LC-MS чистота: 98,20%; (ES+): m/z 440,38 (M+H+); tr = 1,95 мин.
Пример 4: 4-(N-(3-(3,4-диметоксифенил)-1-метил-1H-индол-5-ил)сульфамоил)-N-гидроксибензамид (4): 1H ЯМР (400 МГц, dmso) δ 9,10-10,95 (m, 3Н), 7,81-7,86 (m, 2Н), 7,74 (d, J=8,31 Гц, 2Н), 7,69 (br d, J=8,80 Гц, 1H), 7,57 (s, 1H), 7,45-7,48 (m, 1H), 7,35 (d, J=8,80 Гц, 1H), 6,93-7,06 (m, 3Н), 6,89 (dd, J=1,71, 8,07 Гц, 1H), 3,79 (d, J=3,42 Гц, 6H), 3,75 (s, 3H). LC-MS чистота: 95,57%; (ES+): m/z 482,26 (M+H+); tr = 1,69 мин.
Пример 5: N-гидрокси-4-(N-(1-метил-3-(нафталин-1-ил)-1Н-индол-5-ил)сульфамоил)бензамид (5): 1H ЯМР (400 МГц, dmso) δ 11,34 (br s, 1H), 9,89 (br s, 1H), 9,23 (br s, 1H), 7,98 (d, J=7,82 Гц, 1H), 7,81-7,92 (m, 4H), 7,65-7,70 (m, 2H), 7,50-7,59 (m, 3H), 7,35-7,46 (m, 2H), 7,33 (d, J=6,36 Гц, 1H), 6,97-7,06 (m, 2H), 3,85 (s, 3H) LC-MS чистота: 98,55%; (ES+): m/z 472,42 (M+H+); tr = 2,08 мин.
Пример 6: N-гидрокси-4-(N-(1-метил-3-(4-нитрофенил)-1Н-индол-5-ил)сульфамоил)бензамид (6): 1Н ЯМР (400 МГц, dmso) δ 11,35 (br s, 1Н), 10,14 (br s, 1H), 9,19 (s, 1H), 8,30 (br d, J=8,80 Гц, 2H), 7,98-8,04 (m, 1H), 7,81-7,88 (m, 2H), 7,69-7,81 (m, 4H), 7,60 (s, 1H), 7,44 (br d, J=8,80 Гц, 1H), 6,97 (br d, J=8,31 Гц, 1H), 3,81 (s, 3H). LC-MS чистота: 97,55%; (ES+): m/z 467,19 (M+H+); tr = 1,84 мин.
Пример 7: 4-(N-(3-(2,4-дифторфенил)-1-метил-1H-индол-5-ил)сульфамоил)-N-гидроксибензамид (7): 1H ЯМР (400 МГц, dmso) δ 11,36 (br s, 1H), 9,99-10,05 (m, 1Н), 9,86 (s, 1H), 7,71-7,87 (m, 2H), 7,55-7,68 (m, 2H), 7,28-7,46 (m, 3H), 7,13-7,28 (m, 2H), 6,85-6,98 (m, 1H), 3,79 (s, 3H). LC-MS чистота: 96,17%; (ES+): m/z 458,16 (M+H+); tr = 3,10 мин.
Пример 8: N-гидрокси-4-(N-(3-(2-метоксифенил)-1-метил-1H-индол-5-ил)сульфамоил)бензамид (8): 1H ЯМР (400 МГц, dmso) δ 11,34 (br s, 1H), 9,95 (br s, 1H), 9,19 (br s, 1H), 7,81-7,87 (m, 2H), 7,73 (d, J=8,31 Гц, 2H), 7,50 (s, 1H), 7,31-7,35 (m, 1H), 7,18-7,28 (m, 3H), 7,06-7,11 (m, 1H), 6,98 (brt, J=7,34 Гц, 2H), 6,91 (dd, J=1,71, 8,56 Гц, 1H), 3,76 (s, 3H), 3,72 (s, 3H). LC-MS чистота: 95,83%; (ES+): m/z 450,46 (M-H+); tr = 4,05 мин.
Пример 9: N-гидрокси-4-(N-(3-(4-метоксифенил)-1-метил-1Н-индол-5-ил)сульфамоил)бензамид (9): 1H ЯМР (400 МГц, dmso) δ 11,39 (br s, 1H), 9,97 (br s, 1H), 9,21 (br s, 1H), 7,83-7,89 (m, 2H), 7,75 (d, J=8,31 Гц, 2H), 7,50-7,54 (m, 1H), 7,27-7,37 (m, 4H), 6,99 (d, J=8,80 Гц, 2H), 6,92 (dd, J=1,96, 8,80 Гц, 1H), 3,79 (s, 3H), 3,75 (s, 3H). LC-MS чистота: 95,78%; (ES+): m/z 452,22 (M+H+); tr = 1,83 мин.
Пример 10: N-гидрокси-4-(N-(3-(3-метоксифенил)-1-метил-1H-индол-5-ил)сульфамоил)бензамид (10): 1H ЯМР (400 МГц, dmso) δ 11,33 (s, 1H), 10,02 (s, 1H), 9,16 (br s, 1H), 7,81-7,85 (m, 2Н), 7,75 (d, J=8,80 Гц, 2Н), 7,64-7,68 (m, 1H), 7,51 (d, J=1,96 Гц, 1H), 7,29-7,39 (m, 2Н), 6,93-7,04 (m, 3Н), 6,80 (dd, J=1,96, 8,31 Гц, 1H), 3,80 (s, 3Н), 3,76 (s, 3Н). LC-MS чистота: 99,83%; (ES+): m/z 452,16 (М+Н+); tr = 1,84 мин.
Пример 11: 4-(N-(3-(3-хлорфенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (11): 1Н ЯМР (400 МГц, dmso) δ 11,32 (br s, 1H), 10,05 (br s, 1H), 9,16 (br s, 1H), 7,82-7,87 (m, 2H), 7,73-7,78 (m, 3H), 7,36-7,51 (m, 5H), 7,25-7,30 (m, 1H), 6,97 (dd, J=1,96, 8,80 Гц, 1H), 3,77 (s, 3H). LC-MS чистота: 96,56%; (ES+): m/z 454,16 (M+H+); tr = 2,0 мин.
Пример 12: N-гидрокси-4-(N-(1-метил-3-(нафталин-2-ил)-1Н-индол-5-ил)сульфамоил)бензамид (12): 1H ЯМР (400 МГц, dmso) δ 11,35-11,94 (m, 2Н), 9,19 (br s, 1H), 7,83-7,97 (m, 6H), 7,74-7,82 (m, 3H), 7,61-7,66 (m, 2H), 7,38-7,59 (m, 3H), 6,98 (dd, J=1,47, 8,80 Гц, 1H), 3,81 (s, 3H). LC-MS чистота: 96,03%; (ES+): m/z 472,22 (M+H+); tr = 2,06 мин.
Пример 13: 4-(N-(3-(3-хлор-4-фторфенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (13): 1H ЯМР (400 МГц, dmso) δ 11,17-11,70 (m, 2Н), 9,18 (br s, 1H), 7,81-7,88 (m, 2Н), 7,72-7,79 (m, 3Н), 7,57-7,61 (m, 1Н), 7,36-7,51 (m, 4H), 6,93-7,00 (m, 1H), 3,76 (s, 3H). LC-MS чистота: 98,34%; (ES+): m/z 474,13 (M+H+); tr = 2,02 мин.
Пример 14: 4-(N-(3-(2-фторфенил)-1-метил-1H-индол-5-ил)сульфамоил)-N-гидроксибензамид (14): 1H ЯМР (400 МГц, dmso) δ 11,31 (br s, 1Н), 9,99 (br s, 1H), 9,17 (br s, 1H), 7,84 (br d, J=8,31 Гц, 2H), 7,74 (br d, J=8,31 Гц, 2H), 7,61 (s, 1H), 7,36-7,42 (m, 2H), 7,24-7,33 (m, 4H), 6,95 (br d, J=8,31 Гц, 1H), 3,79 (s, 3H). LC-MS чистота: 95,81%; (ES+): m/z 440,17 (M+H+); tr = 1,86 мин.
Пример 15: 4-(N-(3-(3-(бензилокси)фенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (15): 1Н ЯМР (400 МГц, dmso) δ 11,32 (s, 1H), 10,03 (s, 1H), 9,16 (br s, 1H), 7,79-7,85 (m, 1Н), 7,72-7,79 (m, 2H), 7,64-7,70 (m, 2H), 7,47-7,56 (m, 3H), 7,28-7,44 (m, 5H), 7,13-7,21 (m, 1H), 6,92-7,08 (m, 2H), 6,87 (br d, J=6,85 Гц, 2H), 5,14-5,20 (m, 2H), 3,72-3,79 (m, 3H). LC-MS чистота: 95,36%; (ES+): m/z 426,27 (M-H+); tr = 2,15 мин.
Пример 16: 4-(N-(3-(бензо[b]тиофен-3-ил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (16): 1Н ЯМР (400 МГц, dmso) δ 9,90 (br s, 1H), 8,01-8,08 (m, 1H), 7,80 (br d, J=7,82 Гц, 3Н), 7,54-7,64 (m, 3Н), 7,35-7,50 (m, 4H), 7,26-7,33 (m, 2H), 6,91 (br d, J=8,80 Гц, 1H), 3,77-3,84 (m, 3H). LC-MS чистота: 99,75%; (ES+): m/z 460,21 (M+H+); tr = 1,85 мин.
Пример 17: 4-(N-(3-(бифенил-4-ил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (17): 1H ЯМР (400 МГц, dmso) δ 11,37 (br s, 1H), 10,01 (br s, 1H), 9,18 (s, 1H), 7,85-7,90 (m, 2H), 7,69-7,80 (m, 7H), 7,46-7,54 (m, 5H), 7,34-7,41 (m, 2H), 6,96 (br d, J=8,80 Гц, 1H), 3,79 (s, 3H). LC-MS чистота: 99,81%; (ES+): m/z 478,08 (M+H+); tr = 2,02 мин.
Пример 18: N-гидрокси-4-(N-(1-метил-3-(3-(трифторметил)фенил)-1Н-индол-5-ил)сульфамоил)бензамид (18): 1H ЯМР (400 МГц, dmso) δ 10,46 (br s, 2Н), 9,14 (br s, 1H), 7,71-7,86 (m, 7H), 7,66 (br t, J=7,82 Гц, 1H), 7,52-7,59 (m, 2H), 7,40 (d, J=8,80 Гц, 1H), 6,97 (dd, J=1,47, 8,80 Гц, 1H), 3,78 (s, 3H). LC-MS чистота: 95,02%; (ES+): m/z 490,14 (M+H+); tr = 2,05 мин.
Биологическая активность
Химические соединения и реагенты: биохимический анализ был разработан с использованием люминесцентной платформы в формате 384-луночного планшета. Все рекомбинантные ферменты HDAC6 (№ по кат. BML-SE508-0050), HDAC8 (№ по кат. BML-SE145-0100) и референтное соединение трихостатин A (TSA, № по кат. BML-GR309-0005) приобретали в Enzo Life Sciences. Аналитический набор HDAC-Glo™ I/II (№ по кат. G6421) приобретали в Promega для измерения активности ингибитора HDAC I и II класса. В данном наборе ацетилированный пептид предлагали в качестве субстрата HDAC. Для осуществления эксперимента использовали луночные планшеты Optiplate-384 (№ по кат. 6007299, от Perkin Elmer).
Биохимичский анализ
С целью скрининга ингибиторов HDAC разведения неизвестных соединений и известного ингибитора HDAC трихостатина А готовили согласно требуемым концентрациям в буфере HDAC-Glo™ I/II. Конечный объем разведенных соединений поддерживали на уровне 10 мкл. Ферменты HDAC разбавляли с помощью буфера HDAC-Glo™ I/II до необходимых концентраций (25 нг HDAC6 на лунку и 0,125 ЕД HDAC8 на лунку) и 10 мкл каждого фермента HDAC распределяли в каждую лунку с разведениями ингибиторов. Фермент и соединение инкубировали в течение 15 минут при комнатной температуре. Ампулу с субстратом HDAC Glo разводили 10 мл буфера HDAC и смешивали с 10 мл реагента для проявления. 20 мкл приготовленного реагена HDAC Glo добавляли к каждой лунке и центрифугировали планшет при комнатной температуре в течение 30-60 секунд для обеспечения гомогенности. Планшет считывали на люминесцентном планшет-ридере (многоканальный планшет-ридер EnVision® от Perkin Elmer) через 15 минут после инкубирования при комнатной температуре. Полученные данные экспортировали в файл Excel и данные анализировали с помощью Graph Pad Prism для расчета значения IC50 ингибитор.
Клеточный скрининг
Культура клеток и реагенты: все клеточные линии, используемые для антипролиферативного скрининга, получали из Американской коллекции типовых культур (АТСС). HeLa (линия клеток рака шейки матки), MCF-7 (линия клеток рака молочной железы) и DU-145 (линия клеток рака предстательной железы) культивировали в среде Игла в модификации Дульбекко (DMEM, GIBCO), содержащей 10% фетальную бычью сыворотку (FBS; Life Technologies), пенициллин и стрептомицин (10000 ЕД/мл), при 37°С и в 5% CO2. Набор для анализа жизнеспособности клеток Cell Titer-Glo® Luminescent (№ по кат. G7573) приобретали в Promega для определения количества жизнеспособных клеток в культуре на основе количественной оценки присутствующей АТФ, которая свидетельствует о наличии метаболически активных клеток.
Антипролиферативный анализ: для антипролиферативного скрининга 2500 клеток на лунку (в случае клеточных линий HeLa и DU-145) и 5000 клеток на лунку (в случае клеточной линии MCF-7) высевали в белый непрозрачный планшет и инкубировали в течение 24 часов в 5% СО2 инкубаторе при 37°С. Через 24 часа готовили разведения соединений, согласно требуемым концентрациям, в среде для культивирования клеток (DMEM) и разведенные соединения добавляли и инкубировали с клетками в течение 72 часов при 37°С в 5% CO2 инкубаторе в том же самом 96-луночном планшете. Через 72 часа ампулу с реагентом Cell Titer-Glo® разводили 100 мл буфера Cell Titer Glo и 100 мкл полученного реагента добавляли в каждую лунку. После инкубирования при комнатной температуре в течение 30 минут люминесценцию регистрировали с помощью люминесцентного планшет-ридера (многоканальный планшет-ридер EnVision® от Perkin Elmer). Полученные данные экспортировали в файл Excel и данные анализировали с помощью Graph Pad Prism для расчета значения IC50 неизвестного ингибитора.
Скрининг ADME in vitro
Стабильность гепатоцитов: Стабильность гепатоцитов исследуемых соединений определяли в криоконсервированных гепатоцитах человека и мыши. 10 мМ исходного раствора исследуемого соединения готовили в DMSO. 1 мМ рабочего раствора исследуемого соединения готовили путем разведения 20 мкл 10 мМ исходного раствора в 180 мкл смеси ацетонитрил : вода (50:50). 2 мкМ конечного рабочего раствора готовили путем разведения 4 мкл 1 мМ рабочего раствора в 1996 мкл среды для инкубирования. 200 мкл суспензии гепатоцитов (2×106 клеток/мл) добавляли в 48-луночный планшет и преинкубировали в течение 30 минут при 37°С в инкубаторе. 200 мкл исследуемого соединения в рабочей концентрации 2 мкМ добавляли к клеточной суспензии и инкубировали при 37°С в инкубаторе. Реакцию останавливали через 0, 15, 30, 60, 90 и 120 минут с помощью осаждения 50 мкл инкубационной смеси 200 мкл ацетонитрила, содержащего внутренний стандарт. После осаждения образцы перемешивали на вортексе в течение 5 минут при 1200 об./мин. и центрифугировали при 4000 об./мин в течение 10 минут. Супернатант переносили в планшет для анализа, разбавляли в 2 раза водой и анализировали на LC-MS/MS.
Проницаемость: проницаемость исследуемых соединений определяли в монослое клеток Сасо-2 (культивированных в течение 21 дня). 5 мл 100 мМ пирувата натрия, 5 мл 100× заменимых аминокислот, 5 мл Pen-strep добавляли к 100 мл термоинактивированной фетальной бычьей сыворотки до 385 мл DMEM асептически и тщательно смешивали. Одну ампулу сбалансированного солевого раствора Хэнкса (Sigma-H1387) растворяли в 900 мл воды Milli Q; доводили до рН 7,4 и доводили объем до 1000 мл с помощью того же самого. Раствор стерилизовали посредством фильтрации и хранили при 4°С. 0,42 г гидроксида натрия (гранулы), 3,95 г дигидрофосфата калия и 6,18 г хлорида натрия растворяли в 500 мл очищенной воды в контейнере объемом 1 л и рН доводили в точности до 7,4 с помощью либо 1 н. гидроксида натрия, либо 1 н. гидрохлорида и доводили до указанного объема водой. В колбе объемом 1 л 2,24 г порошка SIF Phares растворяли в 500 мл фосфатного буфере FaSSIF при комнатной температуре. Перемешивали при комнатной температуре до тех пор, пока порошок SIF Phares не диспергировал, и после получения раствора доводили до объема (1 л) с помощью фосфатного буфера FaSSIF. Среду FaSSIF уравновешивали в течение 2 часов при комнатной температуре до появления опалесцентности. 10 мМ исходного раствора исследуемого соединения готовили в DMSO. 10 мМ исходного раствора разбавляли буфером FaSSIF до конечной концентрации 10 мкМ. 250 мкл DMEM добавляли к исходному компартменту 96-луночного многоэкранного планшета с Сасо-2 и высевали 12000 клеток/лунка (0,16×106 клеток/мл) во все необходимые апикальные лунки и одну лунку только со средой в качестве холостого раствора без клеток. Планшет с Сасо-2 помещали в СО2 инкубатор при 37°С для пролиферации клеток.
В день анализа среду удаляли, дважды промывали буфером HBSS, инкубировали с буфером HBSS в течение 30 минут в инкубаторе и лунки со значениями TEER выше 230 Ом.см2 отбирали для инкубации. 75 мкл исследуемого соединения добавляли в апикальные лунки и 250 мкл буфера HBSS с 2% BSA добавляли в базальные лунки. 25 мкл базальных образцов собирали в определенные временные точки (Т=120 мин.) и разбавляли 25 мкл буфера FaSSIF. 250 мкл исследуемого соединения добавляли в базальные лунки и 75 мкл буфера HBSS с 2% BSA добавляли в апикальные лунки. 25 мкл апикальных образцов собирали через 120 минут и разбавляли 25 мкл буфера FaSSIF. Использовали кривую с калибровкой по одной точке в буфере HBSS с 2% BSA. Образцы-доноры разбавляли 1:1 HBSS, содержащим 2% BSA, а образцы-приемники разбавляли 1:1 буфером FaSSIF, осаждали 200 мкл ацетонитрила, содержащего внутренний стандарт, перемешивали на вортексе в течение 5 минут при 1000 об./мин., центрифугировали при 4000 об./мин. в течение 10 минут. 100 мкл супернатанта разбавляли 200 мкл воды и передавали на анализ LC-MS/MS.
Стабильность в плазме крови
Стабильность исследуемых соединений в плазме крови определяли в плазме крови мыши, человека и крысы. 1 мМ исходного раствора исследуемого соединения готовили в смеси ацетонитрил : вода в результате разведения от 10 мМ исходного раствора (т.е., 10 мкл 10 мМ исходного раствора добавляли к 90 мкл смеси ацетонитрил : вода (50:50)). 25 мкМ исходного раствора исследуемого соединения готовили в смеси ацетонитрил : вода в результате разведения от 1 мМ исходного раствора (т.е., 2,5 мкл 1 мМ исходного раствора добавляли к 97,5 мкл смеси ацетонитрил : вода (50:50)). Замороженную плазму крови мыши, крысы и человека размораживали при комнатной температуре и центрифугировали при 1400× RCF 4°С в течение 15 минут. Примерно 90% прозрачной фракции супернатанта переносили в отдельную пробирку и использовали для анализа. В случае образцов, полученных через 0 минут, плазму крови термоинактивировали при 56°С в течение 5 минут. К 72 мкл термоинактивированной плазмы крови добавляли 3 мкл 25 мкМ исследуемого соединения. 25 мкл аликвоты смеси отбирали и измельчали с использованием 200 мкл ацетонитрила, содержащего внутренний стандарт, и дополнительно обрабатывали в сочетании с другими временными точками. В случае образцов в других временных точках конечный рабочий раствор в количестве 1 мкМ готовили путем разведения в плазме крови (т.е., 8 мкл 25 мкМ рабочего раствора ацетонитрил : вода добавляли к 192 мкл плазмы крови). 200 мкл плазмы крови, содержащей исследуемое соединение, инкубировали в течение 120 мин при 37°С в водяной бане с шейкером при осторожном встряхивании. Аликвоту 25 мкл образца через 60 и 120 минут немедленно осаждали 200 мкл ацетонитрила, содержащего внутренний стандарт, и центрифугировали при 4000× RCF, 4°С, в течение 20 минут. 150 мкл супернатанта разбавляли 150 мкл воды и анализировали на LC-MS/MS.
Метаболическая стабильность: метаболическую стабильность исследуемых соединений определяли в микросомах печени человека, крысы и мыши. 10 мМ исходного раствора исследуемого соединения готовили в DMSO и разбавляли смесью вода : ацетонитрил (1:1) до концентрации 1 мМ. Рабочую концентрацию 100 мкМ готовили путем дополнительного разбавления смесью вода : ацетонитрил (1:1). 100 мл воды Milli Q добавляли к K2HPO4 (1,398 г) и KH2PO4 (0,27 г) с получением конечного рН 7,4. 2,5 мкл исследуемого соединения преинкубировали в течение 10 минут при 37°C с 75 мкл HLM или RLM или MLM в концентрации 3,33 мг на мл и 85 мкл 100 мМ калий-фосфатного буфера. 32,5 мкл преинкубированной смеси инкубировали в течение 60 минут без кофактора (NADPH) при 37°C с 17,5 мкл 100 мМ калий-фосфатного буфера. В случае образца, полученного через 0 минут, 16,25 мкл пре инкубированной смеси добавляли к 200 мкл ацетонитрила, содержащего внутренний стандарт и 8,75 мкл кофактора (NADPH). 62 мкл кофактора (NADPH, 2,5 мМ) добавляли к оставшейся смеси для инкубации и инкубировали в течение 60 минут при 37°С. 25 мкл смеси для инкубации добавляли к 200 мкл ацетонитрила, содержащего внутренний стандарт, перемешивали на вортексе в течение 5 минут при 1200 об./мин. и центрифугировали в течение 10 минут при 4000 об./мин. Супернатант разбавляли в 2 раза водой, вводили и анализировали на LC-MS/MS.
Фармакокинетическое исследование: самцов крыс Sprague-Dawley (SD) использовали для фармакокинетического исследования исследуемого соединения. Однократную дозу исследуемого соединения 1 мг/кг использовали с объемом дозы 5 мл/кг и концентрацией дозы 0,2 мг/мл для внутривенного способа введения. Аналогично в случае перорального и подкожного способа введения использовали дозу 10 мг/кг с объемом дозы 5 мл/кг и концентрацией дозы 2 мг/мл. Исследуемое соединение готовили в различных основах для различных способов введения. В случае внутривенного и подкожного способа введения использовали DMSO (5%), 15% PEG-400 в воде (95%) в качестве контроля-основы. 0,2% Tween 80, 0,4% НРМС в воде использовали в качестве контроля-основы для приготовления исследуемого соединения для введения подкожно. Образцы собирали в различные временные точки и анализировали с помощью биоаналитического способа.
Результаты и обсуждение
• Анализы HDAC6 и HDAC8 успешно выполняли как в формате анализа флуоресценции, так и в формате анализа люминесценции.
• Исследование соединений выполняли в анализе люминесценции с целью поддержания SAR.
• Оптимизацию клеточного анализа выполняли на линиях раковых клеток, например, HeLa, DU145, MCF-7 и SKOV3, с целью исследования антипролиферативной активности соединений.
• Выполняли анализ Pan-HDAC (печени крысы) для исследования соединений в отношении их HDAC.
• Анализ ядерного экстракта клеток HeLa выполняли для проверки активности тех соединений, которые не являются активными в клеточных анализах.
• Основные соединения подвергали скринингу в отношении ADME и PK
Сравнение SAHA и TSA в биохимическом анализе HDAC6 и HDAC8
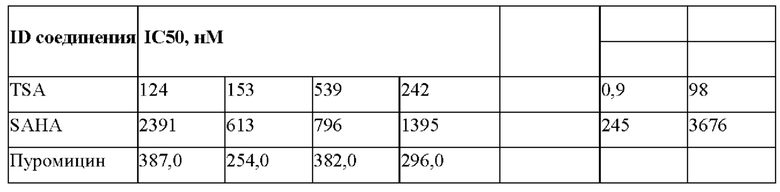
Исследуемые соединения подвергали скринингу в отношении HDAC6 и HDAC8 с полной DRC с получением точных значений IC50. Результаты скрининга для всех соединений, исследуемых в отношении HDAC6 и HDAC8, приведены ниже. Известные лекарственные препараты TSA и SAHA, используемые для скрининга в отношении HDAC6 и HDAC8, показаны на Фиг. 2.
Соединения SAHA и TSA (трихостатин А) использовали в качестве известных референтных соединений в биохимическом анализе. Поскольку TSA был более эффективным, чем SAHA, его использовали в качестве контроля анализа для всех биохимических анализов. Кроме того, по сравнению с SAHA, TSA был в 270 раз и 38 раз более селективным по отношению к HDAC6 и HDAC8 соответственно.
Сравнение SAHA,TSA и пуромицина в различных линиях раковых клеток (HeLa, DU-145, MCF-7 и SKOV3)
Соединения, подвергающиеся скринингу против различных линий раковых клеток [HeLa, DU-145, MCF-7 и SKOV3] в отношении антипролиферативной активности с помощью анализа пролиферации клеток на основе люминесценции с использованием набора Promega Glo, показаны на Фиг. 3.
Значения IC50 (нМ) для TSA, SAHA и пуромицина в отношении линий раковых клеток HeLa (клетки рака шейки матки), MCF-7 (клетки рака молочной железы), DU-145 (клетки рака предстательной железы) и SKOV3 (клетки рака яичника) в клеточных анализах представлены в табличной форме в Табл. 1.
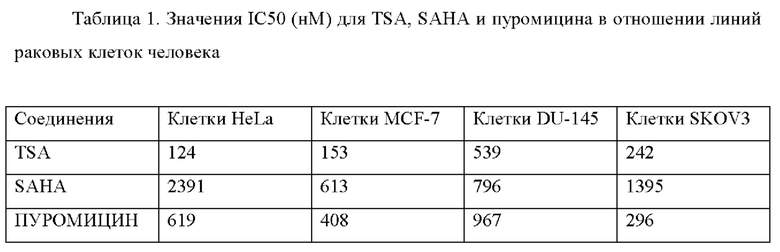
На основании этих данных было сделано заключение о применении либо пуромицина либо TSA в качестве референтной молекулы для анализа пролиферации. Молекула SAHA представляла собой не очень эффективное соединение, а значение IC50 наблюдали в микромолярном диапазоне.
Значения IC50 (нМ) при биохимическом анализе и IC50 (мкМ) при клеточном анализе некоторых основных исследуемых соединений с использованием 4 линий раковых клеток HeLa (клетки рака шейки матки), MCF-7 (клетки рака молочной железы), DU-145 (клетки рака предстательной железы), SKOV3 (клетки рака яичника) и не относящейся к раковым линии клеток AD293, а также данные представлены в табличном виде в Табл. 2.
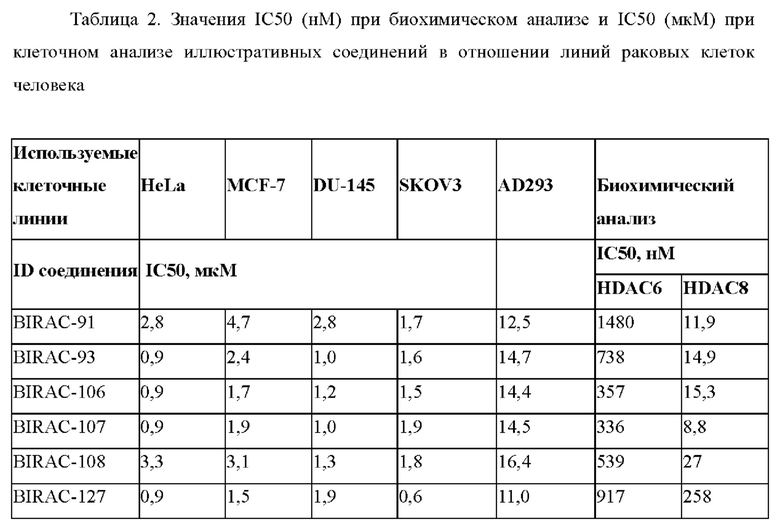
Основные соединения исследовали в отношении линий раковых клеток в сочетании с клетками AD293 (не относящимися к раковым) и они характеризовались антипролиферативной активностью в отношении раковых клеток, однако не характеризовались антипролиферативной активностью в отношении клеток AD293.
В случае клеточных анализов 30 мкМ использовали в качестве максимальной концентрации для скрининга соединений. Если соединения не характеризовались ингибированием при 30 мкМ, ее рассматривали в качестве нетоксичной концентрации, а их характеризовали как не оказывающие влияния на пролиферацию клеток.
Имеются данные о селективности HDAC в биохимическом анализе для некоторых исследуемых соединений в отношении HDAC1, HDAC2, HDAC3, HADC10 и HDAC11.
Некоторые соединения также подвергали скринингу в отношении панели HDAC (в том числе HDAC1, HDAC2, HADC3, HDAC10 и HDAC11) для проверки селективности соединения, а данные представлены в табличном виде в Табл. 3.
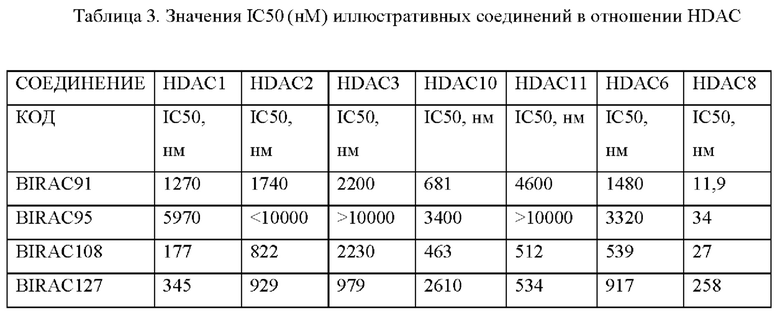
BIRAC-108 был более селективным в отношении HDAC-8 и почти в 80 раз менее селективным в отношении HDAC-3; референтное соединение TSA (трихостатин А) не было селективным в отношении HDAC и характеризовалось очень высокой эффективностью в отношении всех HDAC.
Преимущества настоящего изобретения
Преимущества способа представлены ниже.
1. Основное преимущество настоящего изобретения заключается в том, что в нем предусмотрены новые производные сульфонилгидроксамовых кислот.
2. Преимущество настоящего изобретения заключается в том, что в нем предусмотрен эффективный способ получения различным образом замещенных новых производных сульфонилгидроксамовых кислот.
3. Способ синтеза указанных новых производных сульфонилгидроксамовых кислот включает эксплуатационно простой и высокоэффективный протокол синтеза, приводящий к получению необходимых продуктов с высоким выходом.
4. Другое преимущество настоящего изобретения заключается в применении указанных сульфонилгидроксамовых кислот в качестве ингибиторов HDAC.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ЛЕЧЕНИЯ | 2012 |
|
RU2621148C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PRMT5 | 2018 |
|
RU2797822C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| Способ получения гидроксамовых кислот, производных 2-арил-2,3-дигидрохиназолин-4(1Н)-онов | 2020 |
|
RU2744750C1 |
| ПРОИЗВОДНЫЕ 2,3-ДИГИДРО-1Н-ИНДЕН-1-ИЛ-2,7-ДИАЗАСПИРО[3.5]НОНАНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ ИЛИ ОБРАТНЫХ АГОНИСТОВ ГРЕЛИНОВОГО РЕЦЕПТОРА | 2011 |
|
RU2524341C2 |
| ЗАМЕЩЕННЫЕ ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2827641C1 |
| ТИАЗОЛОПИРИМИДИНЫ | 2012 |
|
RU2610840C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| Соединения, активные по отношению к бромодоменам | 2015 |
|
RU2743074C2 |
| НОВЫЕ 4-АМИНО-N-ГИДРОКСИБЕНЗАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ HDAC ДЛЯ ЛЕЧЕНИЯ РАКА | 2012 |
|
RU2591190C2 |
Изобретение относится к соединениям сульфонилгидроксамовой кислоты общей формулы I, которые могут найти применение в качестве селективных ингибиторов деацетилаз гистонов (HDAC). В общей формуле I кольцо А представляет собой фенил, нафталинил и бензотиофенил; кольцо В представляет собой фенил; R1 представляет собой алкил; R2, R3 представляют собой водород, алкокси, галоген, нитро, бензилокси, фенил, CF3; R4, R5, R6 представляют собой водород; Х=O. Изобретение относится также к способу получения указанных соединений и их применению в качестве селективных ингибиторов HDAC. 3 н. и 12 з.п. ф-лы, 3 табл., 18 пр., 3 ил.
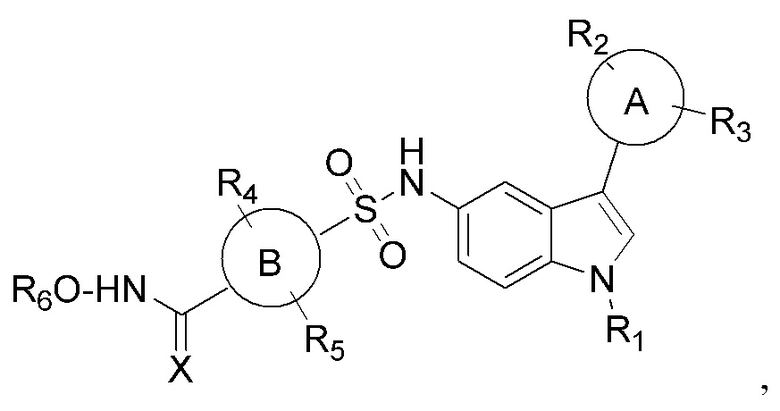
1. Соединения сульфонилгидроксамовой кислоты общей формулы I
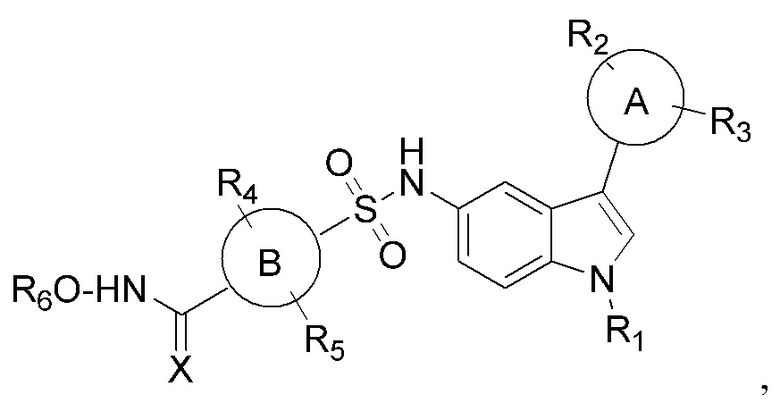
где кольцо А представляет собой фенил, нафталинил и бензотиофенил;
кольцо В представляет собой фенил;
R1 представляет собой алкил;
R2, R3 представляют собой водород, алкокси, галоген, нитро, бензилокси, фенил, CF3;
R4, R5, R6 представляют собой водород;
Х=O.
2. Соединения сульфонилгидроксамовой кислоты по п. 1, где структурные формулы характерных соединений включают в себя
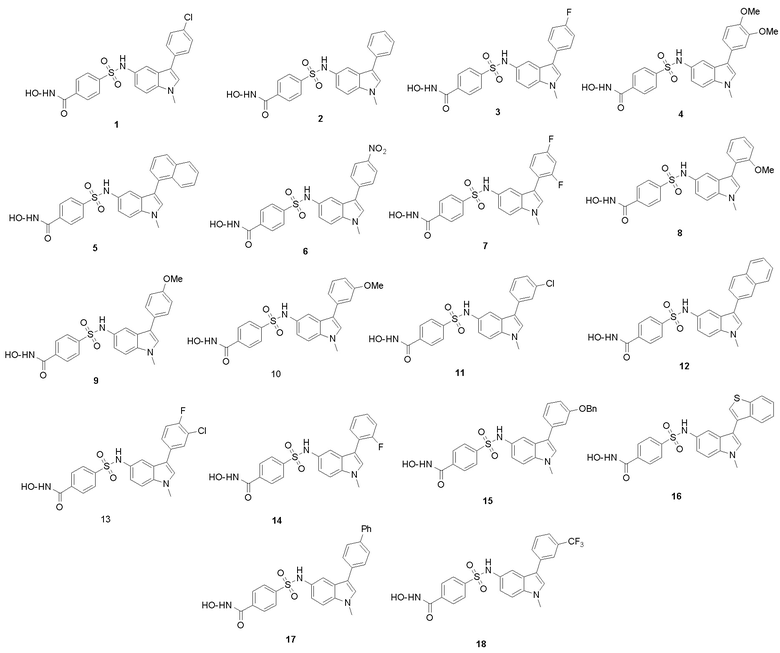
3. Соединения сульфонилгидроксамовой кислоты по п. 1, где соединения сульфонилгидроксамовой кислоты представлены следующими:
N-гидрокси-4-(N-(1-метил-3-фенил-1Н-индол-5-ил)сульфамоил)бензамид (1),
4-(N-(3-(4-хлорфенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (2),
4-(N-(3-(4-фторфенил)-1-метил-1H-индол-5-ил)сульфамоил)-N-гидроксибензамид (3),
4-(N-(3-(3,4-диметоксифенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (4),
N-гидрокси-4-(N-(1-метил-3-(нафталин-1-ил)-1Н-индол-5-ил)сульфамоил)бензамид (5),
N-гидрокси-4-(N-(1-метил-3-(4-нитрофенил)-1Н-индол-5-ил)сульфамоил)бензамид (6),
4-(Н-(3-(2,4-дифторфенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (7),
N-гидрокси-4-(N-(3-(2-метоксифенил)-1-метил-1Н-индол-5-ил)сульфамоил)бензамид (8),
N-гидрокси-4-(N-(3-(4-метоксифенил)-1-метил-1Н-индол-5-ил)сульфамоил)бензамид (9),
N-гидрокси-4-(N-(3-(3-метоксифенил)-1-метил-1Н-индол-5-ил)сульфамоил)бензамид (10),
4-(N-(3-(3-хлорфенил)-1-метил-1H-индол-5-ил)сульфамоил)-N-гидроксибензамид (11),
N-гидрокси-4-(N-(1-метил-3-(нафталин-2-ил)-1Н-индол-5-ил)сульфамоил)бензамид (12),
4-(N-(3-(3-хлор-4-фторфенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (13),
4-(N-(3-(2-фторфенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (14),
4-(N-(3-(3-(бензилокси)фенил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (15),
4-(N-(3-(бензо[b]тиофен-3-ил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (16),
4-(N-(3-(бифенил-4-ил)-1-метил-1Н-индол-5-ил)сульфамоил)-N-гидроксибензамид (17),
N-гидрокси-4-(N-(1-метил-3-(3-(трифторметил)фенил)-1Н-индол-5-ил)сульфамоил)бензамид (18).
4. Способ получения соединений сульфонилгидроксамовой кислоты формулы I по п. 1, который предусматривает стадии:
a) бромирования нитроиндола с применением NBS в полярных непротонированных растворителях при температуре от -5 до 5°С в течение 40-100 мин;
b) защиты NH-группы индола с применением алкилгалогенидов и гидридного основания в полярном непротонированном растворителе при температуре от -5 до 5°С в течение 40-100 мин;
c) восстановления нитрогруппы до амина с применением Zn пыли в смеси полярного растворителя при температуре от -5 до 5°С в течение 40-100 мин;
d) опосредованного основанием сочетания сульфонильных и аминовых функциональных групп в полярном растворителе при 25-40°С в течение 12-24 ч;
e) реакции Сузуки/сочетания с применением производного бороновой кислоты, палладиевого катализатора и фосфатной соли в полярном растворителе при 70-100°С в течение 5-10 ч;
f) введения гидроксамовой кислоты с применением гидроксиамина и основания в смеси полярных растворителей при 25-40°С в течение 12-24 ч.
5. Способ по п. 4, при котором бромирующий реагент представляет собой N-бромсукцинимид (NBS).
6. Способ по п. 4, при котором полярный непротонированный растворитель выбран из диметилформамида (DMF) или диметилсульфоксида (DMSO).
7. Способ по п. 4, при котором алкилгалогенид выбран из метилйодида, метилбромида, этилйодида или этилбромида.
8. Способ по п. 4, при котором гидридное основание выбрано из гидрида натрия (NaH), гидрида калия (КН) или гидрида цезия (CsH).
9. Способ по п. 4, при котором металл, используемый в качестве восстановителя, представляет собой цинк (Zn).
10. Способ по п. 4, при котором полярные растворители выбраны из тетрагидрофурана (THF), метанола (МеОН), этанола (EtOH), воды (H2O), ацетонитрила (CH3CN), 1,4-диоксана или диэтилового эфира (Et2O).
11. Способ по п. 4, при котором основание выбрано из гидроксида натрия (NaOH), гидроксида калия (КОН), гидроксида цезия (CsOH), бикарбоната натрия (NaHCO3) или бикарбоната калия (КНСО3).
12. Способ по п. 4, при котором производное бороновой кислоты выбрано из соединений со следующими заместителями:
кольцо А представляет собой фенил, нафталинил и бензотиофенил;
кольцо В представляет собой фенил;
R1 представляет собой алкил;
R2 представляет собой водород, алкокси, галоген, нитро, бензилокси, фенил, CF3.
13. Способ по п. 4, при котором палладиевый катализатор выбран из бис(трифенилфосфин)палладия (II) дихлорида (Pd(PPh3)2Cl2) или тетракис(трифенилфосфин)палладия (Pd(PPh3)4).
14. Способ по п. 4, при котором фосфатная соль выбрана из фосфата калия (К3РО4) или фосфата натрия (Na3PO4).
15. Применение соединений сульфонилгидроксамовой кислоты по п. 1 в качестве селективных ингибиторов деацетилаз гистонов (HDAC).
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| СУЛЬФОНАМИДСОДЕРЖАЩИЕ СОЕДИНЕНИЯ ИНДОЛА | 2000 |
|
RU2208607C2 |

