Настоящая заявка испрашивает приоритет на основании предыдущей заявки на патент №202110432773.7 под названием «PREPARATION METHOD OF L-NICOTINE», поданной в Национальное управление по интеллектуальной собственности Китая 21 апреля 2021 года, которая в полном объеме включена в настоящий документ посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к технической области химического синтеза и, в частности, к способу получения L-никотина.
УРОВЕНЬ ТЕХНИКИ
Никотин, имеющий химическое название 3-(1-метилпиррол-2-ил)пиридин, представляет собой природный жидкий алкалоид, обладающий сильной физиологической активностью. Никотин обычно содержится главным образом в натуральном табаке и имеет широкий спектр применений в области сельского хозяйства, фармацевтических промежуточных продуктов и электронных сигарет.
В настоящее время коммерческий никотин в основном извлекают и очищают из таких растений, как табак, при этом натуральный никотин находится преимущественно в левовращающей форме. Листья табака содержат различные алкалоиды, которые нелегко отделить друг от друга, поэтому левовращающий никотин (L-никотин), полученный экстракционным способом, имеет относительно низкую чистоту, в общем случае менее 95%, и содержит много других никотиновых примесей, вредных для систем человеческого организма, многие из которых, как показано, являются канцерогенными. Между тем, на извлечение и очистку никотина из растений, таких как табак, могут влиять различные факторы, например, исходные вещества, климат, земельные ресурсы и период. Таким образом, ориентированный синтез L-никотина химическим способом представляет собой актуальную область активных исследований, что может позволить избежать недостатков, связанных с низкой чистотой продукта и строгими ограничениями в отношении исходных веществ в традиционном процессе экстракции.
В литературе в Journal of Labelled Compounds and Radiopharmaceuticals, 1977, 9(4), 461-469 описан способ получения никотина путем реакции с триметилсилил-защищенным пирролидоном с применением пиридина в качестве исходного вещества:
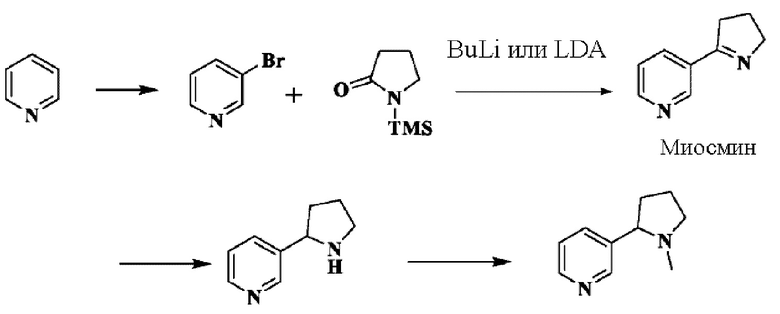
Указанный способ требует применения легковоспламеняющегося органического металлического лития и должен проводиться при температуре -78°С, при этом согласно такой схеме атом азота в пирролидоне защищен с помощью триметилсилана, поэтому стоимость материала является относительно высокой.
В литературе в Organic Syntheses, [J], 1998, 215-218 описан способ получения рацемического никотина посредством четырехстадийной реакции путем синтезирования с применением метилникотината в качестве исходного вещества:
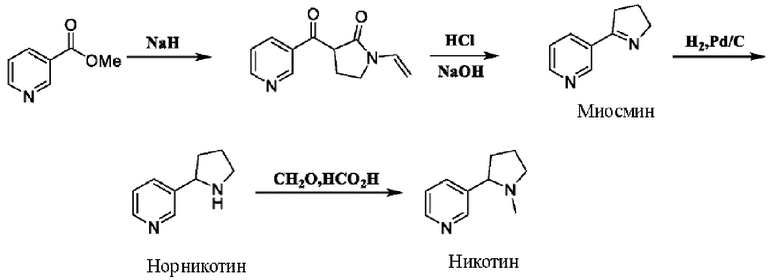
Первые две стадии запатентованной схемы синтеза миосмина приводят к выходу реакции примерно 40% и большому количеству смолы, что влияет на чистоту и внешний вид последующего готового продукта.
В патентах US 2013030188 А1 и CN 102633773 В описан способ получения рацемического никотина посредством четырехстадийной реакции путем синтезирования с применением метилникотината и N-бутенилпирролидона в качестве исходных веществ:
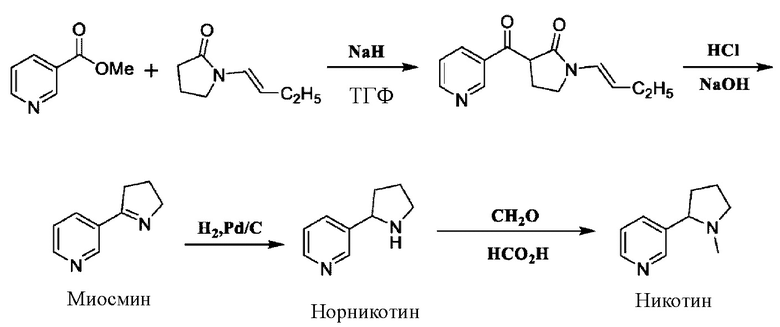
Согласно такой схеме N-бутенилпирролидон необходимо получать собственными силами, что приводит к относительно высокой стоимости материала. В качестве щелочи используют гидрид натрия с относительно высоким риском, так что в процессе реакции может образовываться большое количество водорода, и при увеличении масштабов производства возникает потенциальная угроза безопасности.
В патентах ЕР 2484673, US 0197022, WO 121644 и CN 1124093293 описан способ получения L-никотина путем разделения с помощью дешевой хиральной кислоты с применением рацемического никотина в качестве исходного вещества.
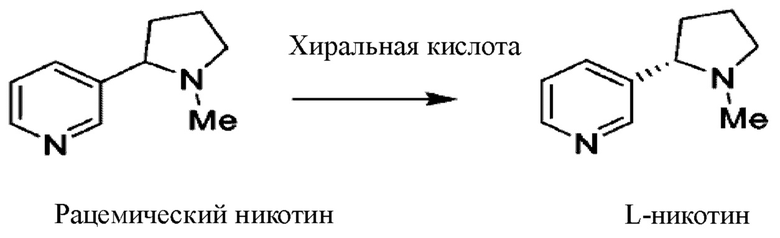
Согласно такой схеме рацемат разделяют с применением дешевой хиральной кислоты с получением L-никотина, при этом выход одностадийного разделения составляет примерно 40%. Такой процесс разделения имеет высокую стоимость и характеризуется потерей никотина в маточном разделяющем растворе, составляющей 60%; для разделения кислоту и основание необходимо заливать дважды, так что образуется более трех отходов.
В патенте CN 112409327 описан способ получения целевого L-никотина, в котором в качестве исходного вещества используют никотинат, под действием основания образуется амид Р-кетоновой кислоты, миосмин получают путем циклизации в кислых условиях, затем способом биологической ферментации получают L-норникотин и, наконец, целевой L-никотин получают путем добавления метила.
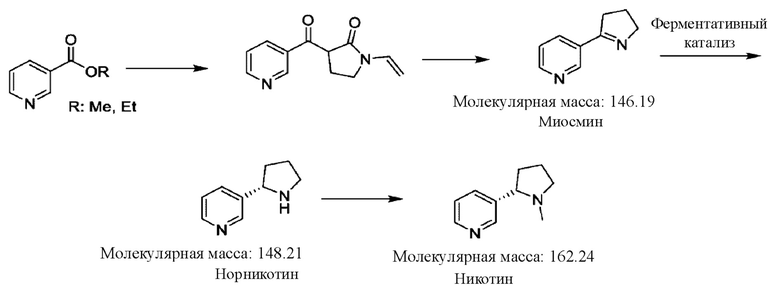
В таком способе L-никотин получают путем ферментативного катализа, стоимость которого на 50% ниже, чем стоимость разделения, но полученный L-никотин представляет собой биологический продукт, который не является полностью синтезированным продуктом вследствие введения следового количества белкового остатка в процессе ферментации.
Большая часть никотина, полученного в известном уровне техники, представляет собой рацемический никотин. Для получения гомохирального никотина с высокой оптической чистотой требуется способ химического разделения для выделения и очистки, который является слишком сложным; или для получения L-никотина используют биологический ферментативный катализ, который приводит к введению следового количества белкового остатка, при этом остаточные белки с трудом поддаются обнаружению и количественному определению.
Соответственно, для удовлетворения потребности современного рынка в отношении высокочистого L-никотина без загрязнения другими вредными соединениями необходимо разработать способ синтеза искусственного L-никотина, который характеризуется относительно высокой эффективностью разработки и относительно высокой чистотой продукта и подходит для крупномасштабного промышленного производства.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Для решения проблем, существующих в известном уровне техники, в настоящем документе предложен способ получения L-никотина согласно следующей схеме синтезирования:
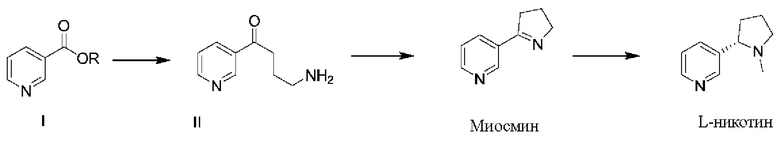
при этом указанный способ получения включает следующие стадии:
(1) проведение реакции замещения соединения формулы I с N-винилпирролидоном и проведение декарбоксилирования с получением соединения формулы II;
(2) проведение реакции циклизации соединения формулы II с получением миосмина; и
(3) проведение восстановления и метилирования миосмина с получением L-никотина;
при этом в соединении формулы I R выбран из C1-C8 алкила, фенила и бензила. Согласно одному из вариантов реализации настоящего изобретения C1-C8 алкил может быть выбран из метила, этила, н-пропила, изопропила, н-бутила, изобутила, трет-бутила, н-пентила, изопентила, неопентила, н-гексила и т.п.
Согласно одному из вариантов реализации настоящего изобретения на стадии (1) соединение формулы I подвергают реакции замещения с N-винилпирролидоном под действием основания; основание выбрано из одного или более следующих оснований: гидроксида калия, гидроксида натрия, трет-бутоксида калия, этоксида натрия, карбоната калия, гидрида натрия, бутиллития и бромида метилмагния; в реакции замещения на стадии (1) в качестве реакционного растворителя используют один или более из следующих реагентов: толуол, ксилол, тетрагидрофуран, этанол, 2-метилтетрагидрофуран и н-гексан; реакцию замещения проводят в диапазоне температур от 30 до 150°С, предпочтительно от 60 до 120°С, например, при температуре, выбранной из 60°С, 70°С, 80°С, 90°С, 100°С и 110°С; в реакции замещения массовое соотношение при подаче реакционного растворителя и соединения формулы I составляет от 20:1 до 1:1, предпочтительно от 10:1 до 1:1, например, 8:1, 7:1, 6:1, 6:1, 4:1, 3:1 или 2:1; в реакции замещения массовое соотношение при подаче основания и соединения формулы I составляет от 2:1 до 1:5, например, 1:1, 1:1,5, 1:2 или 1:2,5; в реакции замещения массовое соотношение при подаче N-винилпирролидона и соединения формулы I составляет от 2:1 до 1:5, например, 1:1, 1:1,1, 1:1,2 или 1:1,3.
Согласно одному из вариантов реализации настоящего изобретения на стадии (1) для проведения реакции декарбоксилирования добавляют кислоту; кислота выбрана из одной или более следующих кислот: соляной кислоты, фосфорной кислоты, серной кислоты, муравьиной кислоты и уксусной кислоты; соляная кислота предпочтительно может быть выбрана из соляной кислоты с концентрацией 15%; реакцию декарбоксилирования проводят в диапазоне температур от 10 до 100°С, предпочтительно от 20 до 80°С, например, при температуре, выбранной из 30°С, 40°С, 50°С, 60°С и 70°С; в реакции декарбоксилирования в процессе добавления кислоты температуру предпочтительно регулируют в диапазоне от 40 до 60°С (время добавления можно регулировать в пределах от 2 до 4 часов), и после добавления кислоты полученную систему нагревают до температуры от 80 до 100°С для проведения реакции (время реакции можно регулировать в пределах от 1 до 3 часов).
Согласно одному из вариантов реализации настоящего изобретения стадия (1) дополнительно включает следующие стадии: после реакции декарбоксилирования доведение рН до 6-8 с помощью основного реагента и проведение экстракции с получением соединения формулы II; согласно одному из вариантов реализации настоящего изобретения основной реагент может быть выбран из раствора гидроксида натрия (например, 10% гидроксида натрия).
Согласно одному из вариантов реализации настоящего изобретения на стадии (1) в реакционный сосуд при температуре от 20 до 40°С добавляют ксилол, этилникотинат, трет-бутоксид калия и N-винилпирролидон; после нагревания указанной системы до температуры от 80 до 120°С для проведения реакции в течение от 3 до 5 часов добавляют по каплям 15% соляную кислоту в течение от 2 до 4 часов при температуре, регулируемой на уровне от 30 до 60°С; затем систему нагревают до температуры от 80 до 100°С для проведения реакции в течение от 1 до 3 часов и охлаждают до комнатной температуры; доводят указанную систему до рН 7 с помощью гидроксида натрия и подвергают последующей обработке с получением соединения формулы II.
Согласно одному из вариантов реализации настоящего изобретения на стадии (2) соединение формулы II подвергают реакции циклизации под действием основания; стадия (2) дополнительно включает следующие стадии: проведение экстракции с получением миосмина.
Согласно одному из вариантов реализации настоящего изобретения на стадии (2) в качестве реакционного растворителя используют один или более из следующих реагентов: воду, этилацетат, дихлорметан, N,N-диметилформамид, тетрагидрофуран, этанол, 2-метилтетрагидрофуран, н-гексан и метил-трет-бутиловый эфир.
Согласно одному из вариантов реализации настоящего изобретения на стадии (2) основание выбрано из гидроксида калия, гидроксида натрия, трет-бутоксида калия, этоксида натрия, карбоната калия, гидрида натрия, триэтиламина, бутиллития и бромида метилмагния.
Согласно одному из вариантов реализации настоящего изобретения на стадии (2) реакцию проводят в диапазоне температур от 30 до 100°С, например, при температуре, выбранной из 40°С, 50°С, 60°С, 70°С, 80°С и 90°С.
Согласно одному из вариантов реализации настоящего изобретения на стадии (2) массовое соотношение при подаче реакционного растворителя и соединения формулы II составляет от 20:1 до 2:1, предпочтительно от 10:1 до 3:1, например, 4:1; массовое соотношение при подаче основания и соединения формулы II составляет от 1:1 до 1:20, предпочтительно от 1:2 до 1:8, например, 1:3, 1:4 или 1:5.
Согласно одному из вариантов реализации настоящего изобретения на стадии (2) в реакционный сосуд при комнатной температуре добавляют реакционный растворитель и соединение формулы II, а затем добавляют гидроксид натрия при температуре системы, регулируемой на уровне от 30 до 50°С; после того, как система полностью прореагировала при температуре от 30 до 50°С, выполняют последующую обработку с получением миосмина.
Согласно одному из вариантов реализации настоящего изобретения на стадии (3) восстановление осуществляют в присутствии лиганда и металлического катализатора; в некоторых вариантах реализации из лиганда и металлического катализатора получают хиральный катализатор in situ.
Согласно одному из вариантов реализации настоящего изобретения в реакции восстановления на стадии (3) массовое соотношение при подаче реакционного растворителя и миосмина составляет от 20:1 до 3:1, предпочтительно от 10:1 до 3:1, например, 9:1, 8:1, 7:1, 6:1 или 5:1; в реакции восстановления в качестве реакционного растворителя используют один или более из следующих реагентов: воду, 1,4-диоксан, тетрагидрофуран, метанол, диметиловый эфир гликоля, метил-трет-бутиловый эфир, диэтиловый эфир, хлороформ, дихлорметан и т.п.
Согласно одному из вариантов реализации настоящего изобретения на стадии (3) массовое соотношение при подаче лиганда и металлического катализатора составляет от 10:1 до 1:1, например, 10:1, 9:1, 8:1, 7:1,6:1, 5:1, 4:1,3:1, 2:1 или 1,5:1.
Согласно одному из вариантов реализации настоящего изобретения на стадии (3) реакция восстановления представляет собой восстановление путем гидрирования с введением в реакцию водорода, при этом указанную реакцию проводят при давлении от 0,5 до 2,0 МПа, например, 1,0 МПа, 1,1 МПа, 1,2 МПа, 1,3 МПа, 1,4 МПа или 1,5 МПа.
Согласно одному из вариантов реализации настоящего изобретения на стадии (3) реакцию восстановления проводят при температуре от 10 до 80°С, например, при температуре, выбранной из 20°С, 30°С, 40°С, 50°С, 60°С и 70°С.
Согласно одному из вариантов реализации настоящего изобретения на стадии (3) металлический катализатор выбран из Rh(COD)Cl2, Ir(COD)Cl2, Ru(COD)Cl2, PdCl(PPh3)3, PdCl2(PPh3)2, Ni(acac)2, NiCl2 и Ni(COD)2.
Согласно одному из вариантов реализации настоящего изобретения на стадии (3) лиганд выбран из следующих структур:
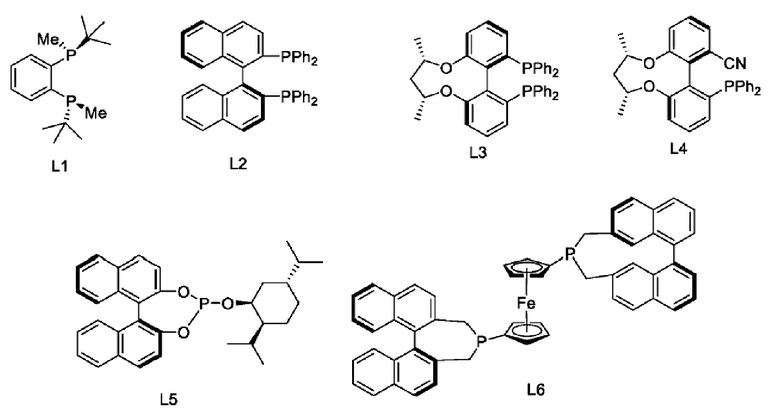
Согласно одному из вариантов реализации настоящего изобретения на стадии (3) реагент для метилирования выбран из одного или более следующих реагентов: формальдегида (например, водного раствора формальдегида), параформальдегида, йодметана и диметилсульфата.
Согласно одному из вариантов реализации настоящего изобретения, в реакции метилирования на стадии (3) указанную реакцию проводят в диапазоне температур от 40 до 120°С, предпочтительно от 50 до 100°С, например, при температуре, выбранной из 60°С, 70°С, 80°С, 90°С и 95°С.
Согласно одному из вариантов реализации настоящего изобретения на стадии (3) для метилирования используют систему реагентов, при этом указанная система реагентов для метилирования дополнительно содержит муравьиную кислоту в дополнение к одному или более реагентам, выбранным из формальдегида (например, водного раствора формальдегида), параформальдегида, йодметана и диметилсульфата; применяемая система реагентов для метилирования предпочтительно содержит параформальдегид и муравьиную кислоту; более предпочтительно, в системе реагентов для метилирования массовое соотношение при подаче параформальдегида и муравьиной кислоты составляет от 1:1 до 1:3, например, 1:2.
Согласно одному из вариантов реализации настоящего изобретения на стадии (3) массовое соотношение при подаче реагента для метилирования и миосмина составляет от 1:1 до 1:10, например, 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8 или 1:9.
Согласно одному из вариантов реализации настоящего изобретения на стадии (3) после реакции метилирования выполняют последующую обработку, которая может включать следующие стадии: регулирование значения рН с помощью основного реагента, осуществление экстракции и проведение дистилляции при пониженном давлении с получением чистого продукта; основной реагент выбран из водного раствора гидроксида натрия; значение рН составляет более или равно 8 и может быть выбрано из 9, 10, 11 и 12. Согласно одному из вариантов реализации настоящего изобретения на стадии (3) в автоклав при комнатной температуре добавляют реакционный растворитель, миосмин, лиганд и металлический катализатор; систему три раза продувают азотом, а затем загружают водород при давлении от 1 до 1,5 МПа; реакцию проводят при температуре от 20 до 40°С в течение от 3 до 5 часов; после опорожнения системы и продувки азотом от 1 до 3 раз добавляют параформальдегид и муравьиную кислоту для проведения реакции при кипячении с обратным холодильником в течение от 3 до 6 часов; после завершения реакции выполняют последующую обработку с получением чистого продукта.
Согласно одному из вариантов реализации настоящего изобретения применяемый в реакции экстракционный растворитель (например, применяемый при последующей обработке на стадии (1), стадии (2) и стадии (3)) может быть выбран из одного или более из следующих реагентов: этилацетата, метил-трет-бутилового эфира, дихлорметана и т.п.
В настоящем изобретении также предложен катализатор, полученный in situ из лиганда и металлического катализатора, при этом указанный металлический катализатор выбран из Rh(COD)Cl2, Ir(COD)Cl2, Ru(COD)Cl2, PdCl(PPh3)3, PdCl2(PPh3)2, Ni(acac)2, NiCl2 и Ni(COD)2; указанный лиганд выбран из следующих структур:
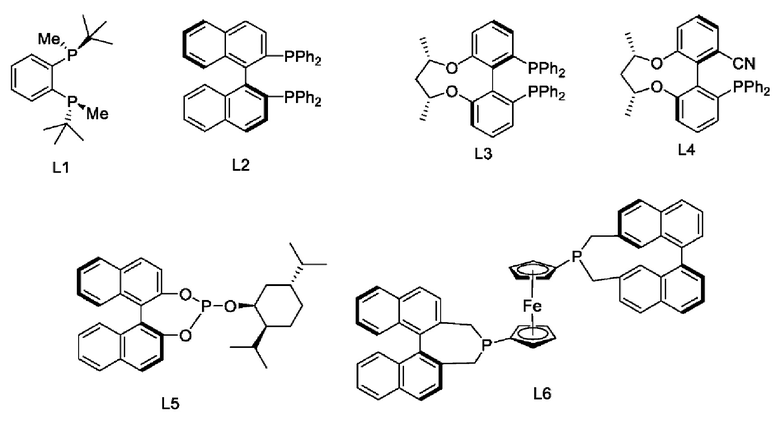
В настоящем изобретении также предложено применение катализатора в реакции восстановления, при этом указанный катализатор предпочтительно используют в реакции восстановления карбонильной группы и, более предпочтительно, в реакции асимметрического восстановления карбонильной группы, например, в реакции на стадии (3), описанной выше.
Преимущества
1) При применении способа синтеза согласно настоящему изобретению можно получать L-никотин с оптической чистотой более 99,9%, что намного выше чистоты аналогичных продуктов на современном рынке, при этом общий выход синтеза достигает от 50% до 60%; реакционные материалы являются дешевыми, и их просто получить; предложенный способ является простым при осуществлении, экологически безопасным и подходит для крупномасштабного промышленного производства.
2) В настоящем изобретении для проведения реакции восстановления используют конкретный лиганд и металлический катализатор, что может значительно улучшить качество реакции.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
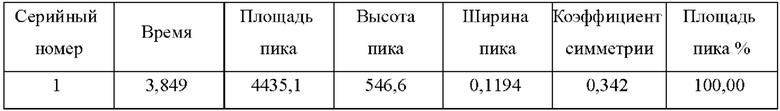
На фиг. 1 показан масс-спектр L-никотина согласно настоящему изобретению;
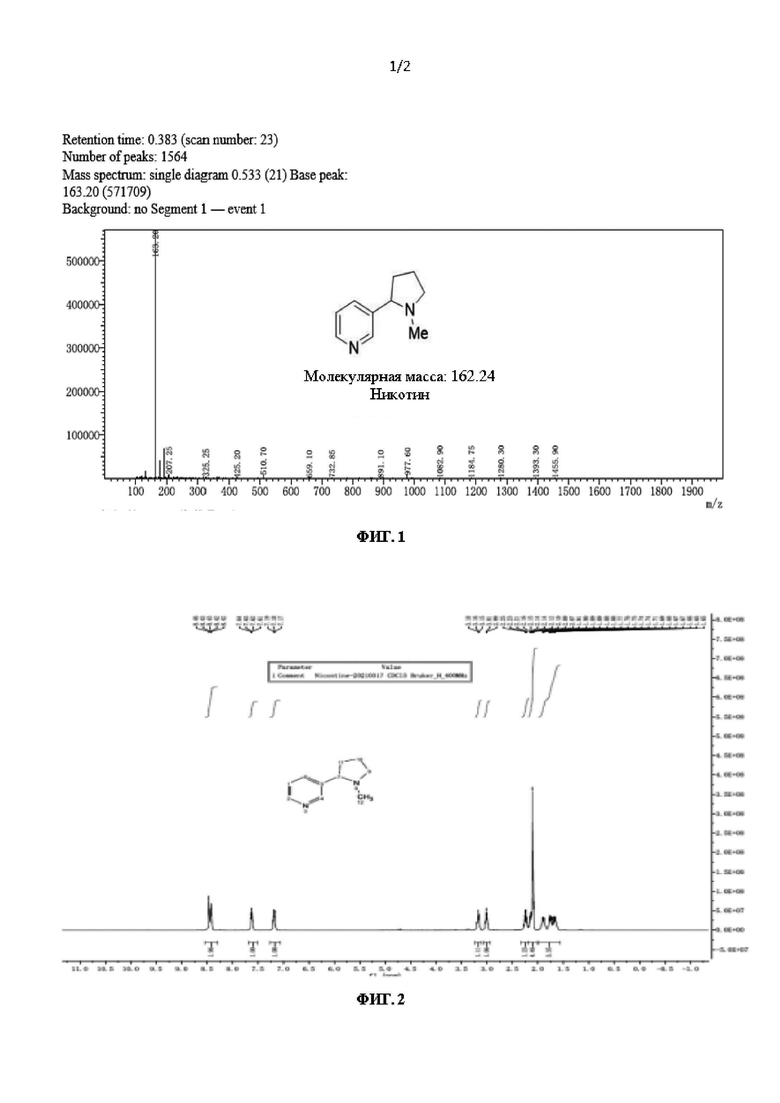
На фиг. 2 показан спектр ядерного магнитного резонанса L-никотина согласно настоящему изобретению;
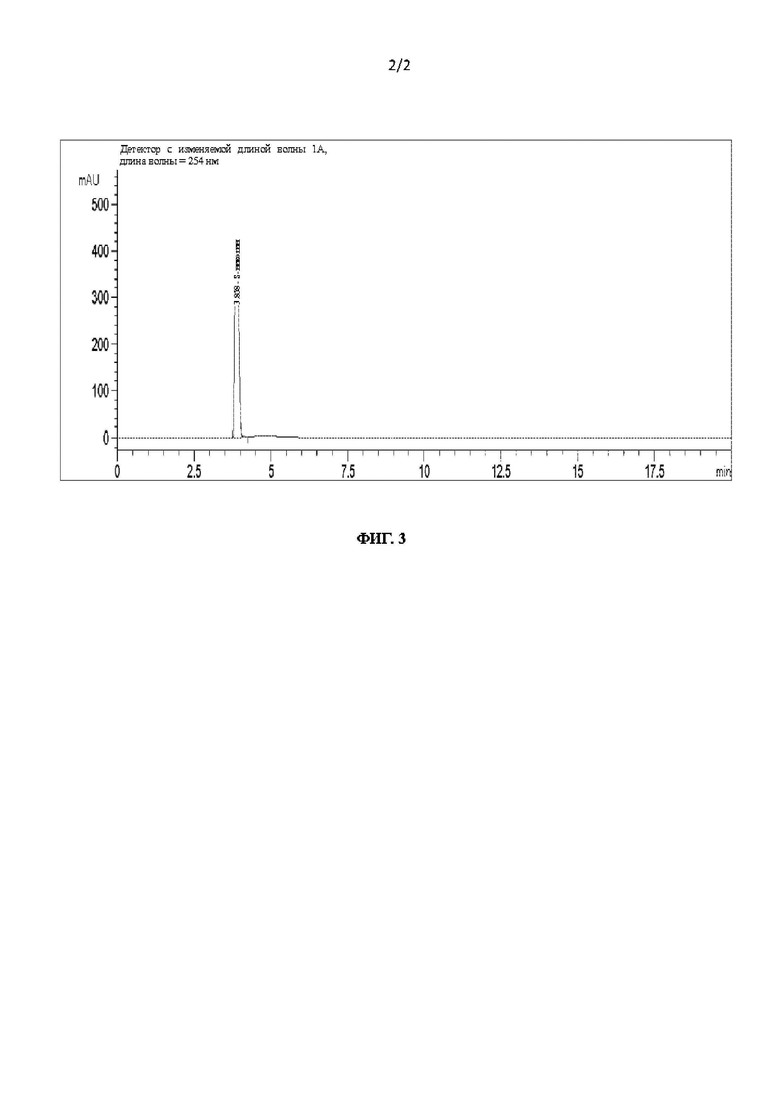
На фиг. 3 показана диаграмма оптической чистоты L-никотина согласно настоящему изобретению.
ПОДРОБНОЕ ОПИСАНИЕ
Технические решения настоящего изобретения будут дополнительно подробно проиллюстрированы со ссылкой на следующие конкретные примеры. Следует понимать, что приведенные ниже примеры представляют собой лишь типичные иллюстрации и объяснения настоящего изобретения и не должны рассматриваться как ограничивающие объем охраны настоящего изобретения. Все способы, осуществленные на основе содержания описанного выше настоящего изобретения, включены в объем защиты настоящего изобретения.
Если не указано иное, исходные вещества и реагенты, применяемые в следующих примерах, являются коммерчески доступными продуктами или могут быть получены с применением известных способов.
Прибор и способ оптического детектирования: прибор А для детектирования (ультрафиолетовый детектор); используют колонку для хиральной хроматографии Daicel OD-H, подвижная фаза представляет собой изопропанол/н-гептан=5:95, скорость потока составляет 0,9 мл/мин, объем вводимой пробы составляет 10 мкл, длина волны детектирования составляет 254 нм, и температура колонки составляет 25°С.
Общая схема реакции:
Пример 1: Получение промежуточного соединения формулы II В трехгорлую колбу объемом 50 л при температуре 30°С добавляли 20 кг ксилола, 5 кг этилникотината (т.е. R=этил), 2,5 кг трет-бутоксида калия и 4 кг N-винилпирролидона. Систему нагревали до 100°С для проведения реакции в течение 4 часов, а затем добавляли по каплям 3 кг 15% соляной кислоты в течение 3 часов при температуре, регулируемой на уровне 50°С. После добавления по каплям полученную систему нагревали до 90°С для проведения реакции в течение 2 часов, а затем охлаждали до 30°С. рН системы доводили до 7 с помощью 10% гидроксида натрия, а затем экстрагировали этилацетатом (20 кг ×2). Органические фазы объединяли, высушивали и концентрировали при пониженном давлении при 40°С с получением 4,11 кг соединения формулы II с чистотой жидкой фазы
97,3% и выходом 76,7%, которое непосредственно использовали на следующей стадии. 1Н ЯМР (CDCl3, 400 МГц) δ: 8,91 (d, J=7,8 Гц, 1H), 8,51 (d, J=8,0 Гц, 1H), 8,35 (d, J=7,8 Гц, 1H), 7,68 (d, J=8,0 Гц, 1H), 3,03 (t, J=8,0 Гц, 2Н), 2,81 (t, J=8,0 Гц, 2Н), 2,22-2,20 (m, 2Н), 1,90-1,88 (br, 2Н), Расчет ЖХ-МС: 164,21, детектирование М+1: 167,2.
Пример 2: Получение миосмина
В трехгорлую колбу объемом 50 л при температуре 20°С добавляли 15 кг тетрагидрофурана, 5 кг воды и 5 кг соединения формулы II. При температуре указанной системы, регулируемой на уровне 40°С, добавляли 1,5 кг гидроксида натрия. Реакцию проводили при 40°С до полного расходования соединения формулы II. Систему экстрагировали дихлорметаном (10 кг ×2). Органические фазы объединяли и концентрировали при пониженном давлении при температуре 30°С с получением 4,35 кг миосмина с чистотой жидкой фазы 97,1% и выходом 97,8%, который непосредственно использовали на следующей стадии. 1Н ЯМР (CDCl3, 400 МГц) δ: 8,58(d, J=8,0 Гц, 1Н), 8,45 (d, J=7,8 Гц, 1H), 7,68 (d, J=8,0 Гц, 1H), 7,24-7,20 (m, 1Н), 4,13 (t, J=4,0 Гц, 1H), 3,20-3,17 (m, 1Н), 3,06-3,01 (m, 1Н), 2,22-2,20 (m, 1H), 1,90-1,88 (m, 1H), 1,80-1,60 (m, 1Н); Расчет ЖХ-МС: 146,19, детектирование М+1: 147,2.
Пример 3: Получение L-никотина
В автоклав объемом 50 л при температуре 25°С добавляли 20 кг тетрагидрофурана, 3 кг миосмина, 1,5 г лиганда L2 и 1 г Ir(COD)Cl2. Систему три раза продували азотом при давлении 0,3 МПа, а затем загружали водород при давлении 1,2 МПа. Полученную систему подвергали реакции при 30°С в течение 4 часов. После опорожнения системы и продувки азотом 2 раза при давлении 0,2 МПа добавляли 500 г параформальдегида и 1 кг муравьиной кислоты для проведения реакции при кипячении с обратным холодильником при 95°С в течение 5 часов. Систему концентрировали для удаления органического растворителя, доводили рН до 11 с помощью водного раствора гидроксида натрия и экстрагировали этилацетатом (9 кг ×3). Органические фазы объединяли и концентрировали с получением неочищенного продукта в виде никотина, который затем перегоняли при пониженном давлении с получением продукта, представляющего собой чистый L-никотин, в виде бесцветной прозрачной жидкости с оптической чистотой, составляющей более 99,9% ее (энантиомерный избыток). Фракционная масса чистого продукта составляла 2,46, при этом выход чистого продукта составлял 73,9%. 1Н ЯМР (400 МГц, CDCl3) δ: 8,47-8,44 (br, 2Н), 7,61 (d, J=8,0 Гц, 1H), 7,20-7,16 (m, 1H), 3,18 (t, J=8,0 Гц, 1Н), 3,11 (t, J=8,0 Гц, 1H), 2,25-2,19 (m, 1H), 2,13-2,09 (m, 1H), 2,08 (s, 3H), 1,89-1,87 (m, 1H), 1,76-1,66 (m, 2H); Расчет ЖХ-МС: 162,24, детектирование М+1: 163,20.
Полученный чистый L-никотин подвергали оптическому детектированию. Результаты показаны на фиг. 3 и ниже в таблице:
Выше были описаны варианты реализации настоящего изобретения. Однако настоящее изобретение не ограничено описанными выше вариантами реализации. Любая модификация, эквивалент, усовершенствование и т.п., выполненные без отступления от сущности и принципов настоящего изобретения, подпадают под объем защиты настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КУМАРИНОПОДОБНОЕ ЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА МЕК И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2742234C1 |
| СПОСОБ СИНТЕЗА (S)-НИКОТИНА | 2021 |
|
RU2803744C1 |
| СПОСОБ ПРОИЗВОДСТВА НИКОТИНА | 2018 |
|
RU2753548C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФЕНИЛИЗОКСАЗОЛИНОВОГО СОЕДИНЕНИЯ | 2021 |
|
RU2818767C1 |
| ПОЛИДЕНТАТНЫЙ ФУНКЦИОНАЛЬНЫЙ МОНОМЕР N,O-ТИПА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ В МАТЕРИАЛЕ С ИОННЫМ ОТПЕЧАТКОМ | 2020 |
|
RU2839651C1 |
| СПОСОБ СИНТЕЗА ПРОИЗВОДНЫХ 9-АЛЛИЛКАМПТОТЕЦИНА | 2014 |
|
RU2658017C2 |
| ПРОИЗВОДНОЕ СУЛЬФОНИЛБЕНЗАМИДА И ЕГО КОНЬЮГАТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2021 |
|
RU2839468C1 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2800153C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХИРАЛЬНОГО СИНТЕТИЧЕСКОГО НИКОТИНА | 2021 |
|
RU2780283C1 |
| СПОСОБ ПОЛУЧЕНИЯ S-НИКОТИНА | 2021 |
|
RU2778789C1 |
Предложен способ получения L-никотина, включающий реакцию замещения соединения формулы I с N-винилпирролидоном в присутствии основания и реакционного растворителя и проведение декарбоксилирования в присутствии кислоты с получением соединения формулы II, циклизацию соединения формулы II в растворителе под действием основания с получением миосмина и восстановление миосмина с получением L-норникотина с последующим метилированием полученного L-норникотина до L-никотина. При этом в соединении формулы I R представляет собой C1-C8 алкил, восстановление миосмина осуществляют в присутствии лиганда L2 и металлического катализатора Ir(COD)Cl2, а реагент для метилирования выбран из формальдегида, параформальдегида, йодметана и диметилсульфата. Технический результат изобретения заключается в предоставлении эффективного для крупномасштабного промышленного производства способа получения целевого продукта с высокой чистотой. 1 з.п. ф-лы, 3 ил., 1 табл., 3 пр.
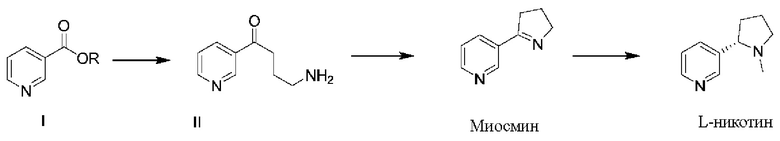
1. Способ получения L-никотина согласно следующей схеме синтеза:
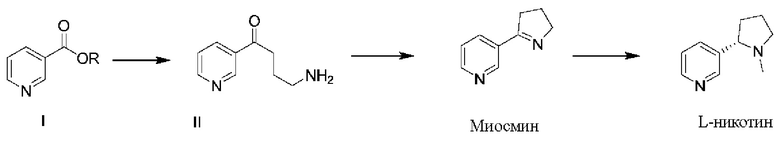
включающий следующие стадии:
(1) проведение реакции замещения соединения формулы I с N-винилпирролидоном и проведение декарбоксилирования с получением соединения формулы II;
(2) проведение реакции циклизации соединения формулы II с получением миосмина; и
(3) проведение восстановления миосмина с получением L-норникотина и затем проведение реакции метилирования полученного L-норникотина с получением L-никотина;
при этом в соединении формулы I R представляет собой C1-C8 алкил,
при этом на стадии (1) соединение формулы I подвергают реакции замещения с N-винилпирролидоном под действием основания, выбранного из одного или более следующих оснований: гидроксида калия, гидроксида натрия, трет-бутоксида калия, этоксида натрия, карбоната калия, гидрида натрия, бутиллития и бромида метилмагния; и в реакции замещения на стадии (1) в качестве реакционного растворителя используют один или более из следующих реагентов: толуол, ксилол, тетрагидрофуран, этанол, 2-метилтетрагидрофуран и н-гексан; и на стадии (1) для проведения реакции декарбоксилирования добавляют кислоту, выбранную из одной или более следующих кислот: соляной кислоты, фосфорной кислоты, серной кислоты, муравьиной кислоты и уксусной кислоты;
при этом на стадии (2) соединение формулы II подвергают реакции циклизации под действием основания; при этом основание выбрано из одного или более следующих оснований: гидроксида калия, гидроксида натрия, трет-бутоксида калия, этоксида натрия, карбоната калия, гидрида натрия, триэтиламина, бутиллития и бромида метилмагния; при этом на стадии (2) в качестве реакционного растворителя используют один или более из следующих реагентов: воду, этилацетат, дихлорметан, N,N-диметилформамид, тетрагидрофуран, этанол, 2-метилтетрагидрофуран, н-гексан и метил-трет-бутиловый эфир; и на стадии (2) массовое соотношение при подаче реакционного растворителя и соединения формулы II составляет от 20:1 до 2:1; массовое соотношение при подаче основания и соединения формулы II составляет от 1:1 до 1:20;
при этом на стадии (3) восстановление осуществляют в присутствии лиганда и металлического катализатора; при этом металлический катализатор представляет собой Ir(COD)Cl2, при этом лиганд выбран из следующей структуры:
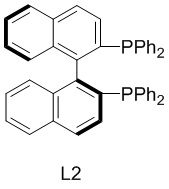 ;
;
на стадии (3) реагент для метилирования выбран из одного или более следующих реагентов: формальдегида, параформальдегида, йодметана и диметилсульфата.
2. Способ получения L-никотина по п. 1, в котором на стадии (3) используют систему реагентов для метилирования, при этом указанная система реагентов для метилирования дополнительно содержит муравьиную кислоту в дополнение к одному или более реагентов, выбранных из формальдегида, параформальдегида, йодметана и диметилсульфата.
| US 2017112182 A1, 27.04.2017 | |||
| US 20170189388 B2, 06.07.2017 | |||
| EP 3209653 B1, 30.08.2017 | |||
| WO 2019121649 A1, 27.06.2019 | |||
| US 20030163003 A1, 28.08.2003. |

