Область техники, к которой относится изобретение
[1] Настоящее изобретение относится к новому кристаллическому твердому соединению, гидрохлориду 3-фенил-4-пропил-1-(пиридин-2-ил)-1Н-пиразол-5-ола, способу получения соединения и фармацевтической композиции, содержащей соединение в качестве активного ингредиента.
Уровень техники
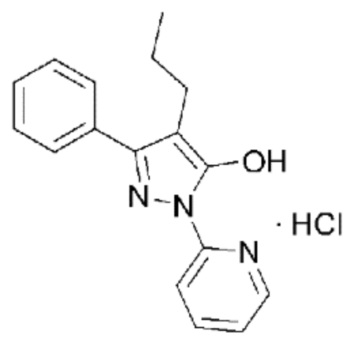
[2] Гидрохлорид 3-фенил-4-пропил-1-(пиридин-2-ил)-1Н-пиразол-5-ола, представленный следующей Формулой 1 (далее сокращенно называемый как «соединение гидрохлорида») или 3-фенил-4-пропил-1-(пиридин-2-ил)-1Н-пиразол-5-ол, представленный следующей Формулой 2 (далее сокращенно называемый как «соединение свободного основания»), впервые синтезировали и сообщили в корейском патенте №10-1280160 (Патентный документ 1).
[3] [Формула 1]
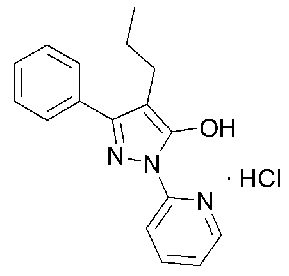
[4] [Формула 2]
[5] Кроме того, в корейском патенте №10-1280160 (Патентный документ 1), корейском патенте №10-1633957 (Патентный документ 2) и корейской патентной заявке №10-2017-0024083 (Патентный документ 3) установлено, что соединение, представленное Формулой 1 или 2, обладает превосходной ингибирующей активностью в отношении активных форм кислорода (АФК). Кроме того, в данных документах сообщалось, основываясь на фармакологическом механизме, что соединение, представленное Формулой 1 или 2, является эффективным в качестве активного ингредиента фармацевтических композиций для лечения остеопороза, заболеваний почек и глазных заболеваний.
[6] Кроме того, в корейском патенте №10-1633957 (Патентный документ 2) раскрыт способ получения соединения, представленного Формулой 1 или Формулой 2. В частности, в соответствии со способом получения, раскрытым в Патентном документе 2, этиловый эфир 2-пропил-3-оксо-3-фенилпропионовой кислоты и 2-гидразинопиридин нагревают и кипятят с обратным холодильником в этанольном растворителе, и полученное твердое вещество промывают гексаном и этилацетатом, и высушивают под вакуумом с получением соединения свободного основания, представленного Формулой 2. Кроме того, полученное соединение свободного основания растворяют в диэтиловом эфире, к нему по каплям добавляют раствор HCl/диэтиловый эфир при 0°С, и полученное твердое вещество промывают гексаном и этилацетатом, и высушивают под вакуумом с получением соединения гидрохлорида, представленного Формулой 1.
[7]
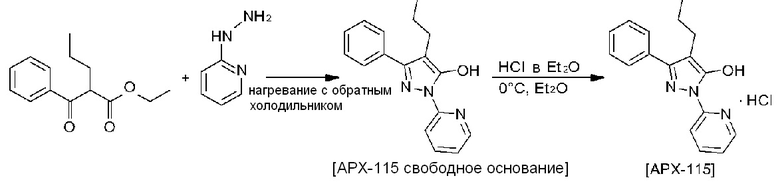
[8] В Патентных документах 1 и 2 соединение, представленное Формулой 1 или 2, получают в виде некристаллического твердого соединения, а не кристаллического соединения.
[9]
[10] В то же время аморфные или некристаллические соединения имеют большую площадь поверхности частиц, чем кристаллические соединения. Таким образом, аморфное или некристаллическое соединение обладает преимуществом превосходной кинетической растворимости в растворителе, но имеет недостаток низкой стабильности по сравнению с кристаллическими соединениями, поскольку оно не имеет энергии кристаллической решетки вследствие кристаллизации.
[11] Кроме того, кристаллическое соединение обладает определенной (уникальной) кристаллической структурой. Структура может включать единичную кристаллическую форму или полиморфную форму, включающую две или более из них. Кроме того, полиморфные соединения могут быть различными с точки зрения содержания воды (гигроскопичности) и подобного, также как физических свойств, таких как растворимость и температура плавления. Кроме того, когда фармацевтический ингредиент представляет собой полиморфное соединение, он может влиять на высвобождение и дезинтеграцию препарата (состава) вследствие изменения кристаллической формы, что также может влиять на абсолютную скорость пероральной абсорбции.
[12] Иными словами полиморфные соединения могут иметь различные кристаллические формы, даже если они имеют одинаковую химическую структуру и, таким образом, могут отличаться в отношении стабильности и физиологической активности соединений. В частности, полиморфные соединения, используемые для фармацевтических применений, могут оказывать большое влияние на удобство получения фармацевтических ингредиентов, растворимость, стабильность при хранении, удобство получения конечных лекарственных средств и фармакологическую активность in vivo в зависимости от кристаллической формы. Следовательно, очень важно выбрать кристаллическую форму, необходимую для фармацевтических ингредиентов, в соответствии с путем введения, дозировкой и подобным. Критерии выбора общих кристаллических лекарственных средств определяются в зависимости от физико-химических свойств кристаллической формы. Например, могут быть выбраны наиболее термодинамически стабильные кристаллические формы, могут быть выбраны кристаллические формы, оптимизированные для получения фармацевтических ингредиентов и лекарственных препаратов, или могут быть выбраны кристаллические формы, способные улучшить растворимость и скорость растворения лекарственных средств или изменить их фармакокинетические свойства.
[13] [Документ предшествующего уровня техники]
[14] [Патентный документ]
[15] (Патентный документ 1) Корейский патент №10-1280160
[16] (Патентный документ 2) Корейский патент №10-1633957
[17] (Патентный документ 3) Корейская заявка на патент №10-2017-0024083.
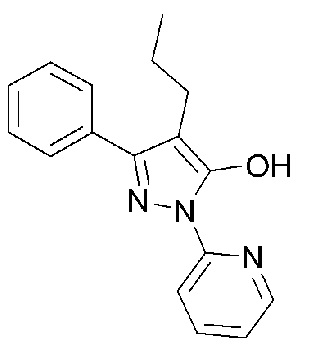
Раскрытие
Техническая задача
[18] Авторы настоящего изобретения завершили настоящее изобретение, получив новое кристаллическое соединение гидрохлорида, представленное Формулой 1, которое используется в качестве фармацевтического ингредиента, имеет превосходные физические свойства и стабильность и может быть термодинамически стабилизировано для предотвращения кристаллического (полиморфного) перехода вследствие изменений со временем при условиях хранения.
[19] Следовательно, настоящее изобретение было осуществлено с учетом вышеуказанных задач, и целью настоящего изобретения является создание нового кристаллического соединения гидрохлорида, представленного следующей Формулой 1:
[20] [Формула 1]
[21]
[22] Другой целью настоящего изобретения является создание способа получения кристаллического соединения гидрохлорида, представленного Формулой 1.
[23] Дополнительной целью настоящего изобретения является создание фармацевтической композиции, содержащей кристаллическое соединение гидрохлорида, представленное Формулой 1, в качестве активного ингредиента.
Техническое решение
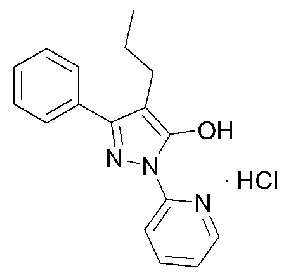
[24] В соответствии с аспектом настоящего изобретения вышеуказанные и другие цели могут быть достигнуты путем предоставления кристаллического соединения гидрохлорида, представленного следующей Формулой 1, имеющего максимальную эндотермическую температуру, измеренную с помощью дифференциального сканирующего калориметра (ДСК) 134,25±3°С.
[25] [Формула 1]
[26]
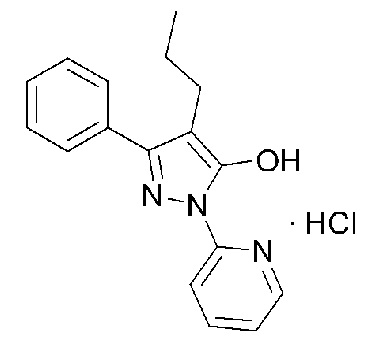
[27] В предпочтительном варианте осуществления настоящего изобретения кристаллическое соединение гидрохлорида, представленное Формулой 1, может иметь углы дифракции 2θ (2θ±0,2°), имеющие относительную интенсивность 15% или выше, полученные с помощью анализа порошковой рентгеновской дифракции, 7,15, 10,72, 13,36, 15,99, 16,39, 16,71, 17,14, 19,61, 21,50, 21,82, 23,46, 24,08, 25,91 и 27,36.
[28]
[29] В соответствии с другим аспектом настоящего изобретения предоставлен способ получения кристаллического соединения гидрохлорида, представленного Формулой 1, включающий:
[30] а) взаимодействие этилового эфира 2-пропил-3-оксо-3-фенилпропионовой кислоты с 2-гидразинопиридином с получением неочищенного продукта;
[31] b) растворение неочищенного продукта в нормальном гексане и затем медленное охлаждение полученного раствора до от -20°С до -10°С с получением твердого вещества;
[32] с) фильтрование, промывание и сушка полученного твердого вещества с получением некристаллического соединения свободного основания;
[33] d) добавление некристаллического соединения свободного основания к смешанному растворителю, содержащему ацетонитрил и дистиллированную воду в одинаковом количестве, и энергичное перемешивание полученной смеси при от 20°С до 25°С с получением кристаллического вещества;
[34] е) фильтрование, промывание и сушка полученного кристаллического вещества с получением кристаллического соединения свободного основания, представленного Формулой 2;
[35] f) взаимодействие кристаллического соединения свободного основания с раствором хлористоводородная кислота-изопропиловый эфир с получением твердого вещества гидрохлорида;
[36] g) добавление твердого вещества гидрохлорида к смешанному растворителю, содержащему трет-бутиловый эфир и толуол в одинаковом количестве, и энергичное перемешивание полученной смеси при от 5 до 10°С с получением кристаллического вещества и
[37] h) фильтрование, промывание и сушка полученного кристаллического вещества с получением кристаллического соединения гидрохлорида, представленного следующей Формулой 1.
[38] [Формула 2]
[39]
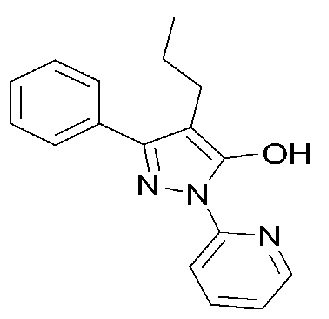
[40] [Формула 1]
[41]
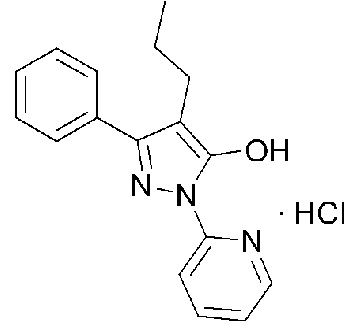
[42] В соответствии с другим аспектом настоящего изобретения предоставлена фармацевтическая композиция для профилактики или лечения заболевания, опосредованного активными формами кислорода (АФК), содержащая кристаллическое соединение гидрохлорида, представленное Формулой 1, в качестве активного ингредиента.
[43] В соответствии с другим аспектом настоящего изобретения предоставлен способ лечения, профилактики или облегчения заболевания, опосредованного активными формами кислорода (АФК), включающий введение эффективного количества кристаллического соединения гидрохлорида, представленного Формулой 1, объекту, нуждающемуся в таком лечении.
[44] В соответствии с другим аспектом настоящего изобретения предоставлено применение кристаллического соединения гидрохлорида, представленного Формулой 1, для получения лекарственного средства для лечения, профилактики или облегчения заболевания, опосредованного активными формами кислорода (АФК).
[45] В соответствии с другим аспектом настоящего изобретения предоставлено кристаллическое соединение гидрохлорида, представленное Формулой 1, пригодное для лечения, профилактики или облегчения заболевания, опосредованного активными формами кислорода (АФК).
[46] В предпочтительном варианте осуществления настоящего изобретения заболевание, опосредованное активными формами кислорода (АФК), может представлять собой остеопороз.
[47] В предпочтительном варианте осуществления настоящего изобретения заболевание, опосредованное активными формами кислорода (АФК), может представлять собой, по меньшей мере, одно заболевание почек, выбранное из группы, состоящей из диабетической нефропатии, гипертензивной нефропатии, гломерулонефрита, пиелонефрита, интерстициального нефрита, волчаночного нефрита, поликистозной болезни почек и почечной недостаточности.
[48] В предпочтительном варианте осуществления настоящего изобретения заболевание, опосредованное активными формами кислорода (АФК), может представлять собой, по меньшей мере, одно глазное заболевание, выбранное из группы, состоящей из диабетической ретинопатии (ДР), диабетического макулярного отека, возрастной макулярной дегенерации, ретинопатии недоношенных (РН), полипоидной хориоидальной васкулопатии, ишемической пролиферативной ретинопатии, пигментного ретинита, колбочковой дистрофии, пролиферативной витреоретинопатии (ПВР), окклюзии артерии сетчатки, окклюзии вены сетчатки, птеригиума, ретинита, кератита, конъюнктивита, увеита, наследственной оптической нейропатии Лебера, отслойки сетчатки, отслойки пигментного эпителия сетчатки, неоваскулярной глаукомы, неоваскуляризации роговицы, неоваскуляризации сетчатки, хориоидальной неоваскуляризации (ХНВ) и вирусной инфекции.
[49] В предпочтительном варианте осуществления настоящего изобретения фармацевтическая композиция может быть получена в форме состава, выбранной из группы, состоящей из порошка, гранулы, таблетки, капсулы, суспензии, эмульсии, сиропа, аэрозоля, мази, крема, суппозитория, глазных капель и инъекции.
Положительные эффекты
[50] Кристаллическое соединение гидрохлорида, представленное Формулой 1, предоставляемое настоящим изобретением, является новым веществом, о котором не сообщалось в литературе, и обладает удивительно высокой стабильностью при воздействии тепла и влажности по сравнению с некристаллическими соединениями гидрохлорида.
[51] Кроме того, кристаллическое соединение гидрохлорида, предоставляемое настоящим изобретением, обладает превосходной стабильностью по сравнению с различными кристаллическими кислотно-аддитивными соединениями солей, полученными путем добавления кислоты, отличной от хлористоводородной кислоты.
[52] Кроме того, кристаллическое соединение гидрохлорида, предоставляемое настоящим изобретением, не только обладает физическими свойствами, предпочтительными для получения лекарственного средства, но также обладает превосходной стабильностью при воздействии тепла и влажности, и полученное таким образом лекарственное средство стабильно сохраняется в течение периода времени, превышающего срок годности, не вызывая разложения активного ингредиента или полиморфного перехода.
[53] Следовательно, кристаллическое соединение гидрохлорида, предоставляемое настоящим изобретением, является пригодным в качестве активного ингредиента при получении лекарственных средств для профилактики или лечения остеопороза, заболеваний почек и глазных заболеваний. В частности, кристаллическое соединение гидрохлорида может быть легко использовано в качестве фармацевтического ингредиента лекарственного средства, входящего в состав препарата для перорального введения, препарата для инъекций или глазных капель.
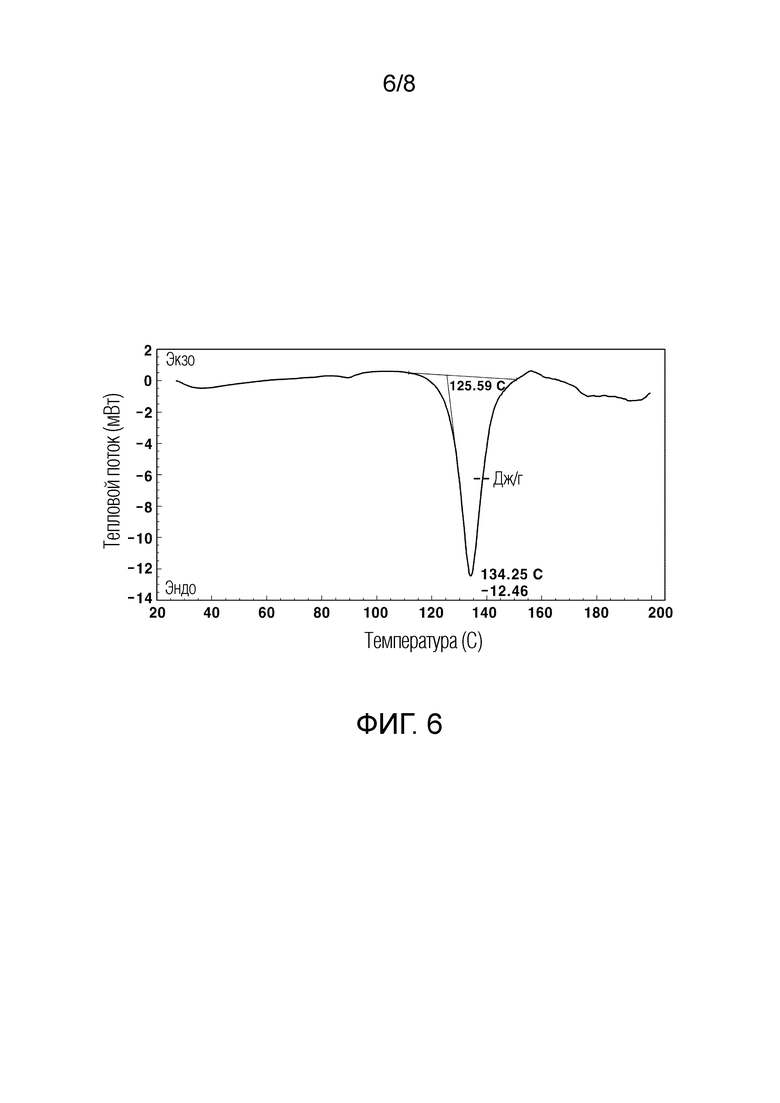
Описание чертежей
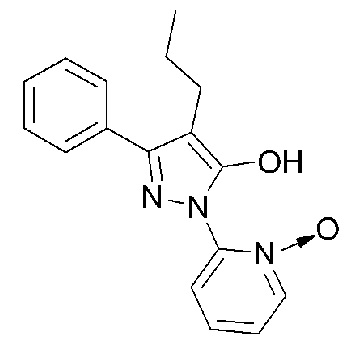
[54] ФИГ. 1 представляет собой изображение, показывающее молекулярную модель 3-фенил-4-пропил-1-(пиридин-2-ил)-1Н-пиразол-5-ола;
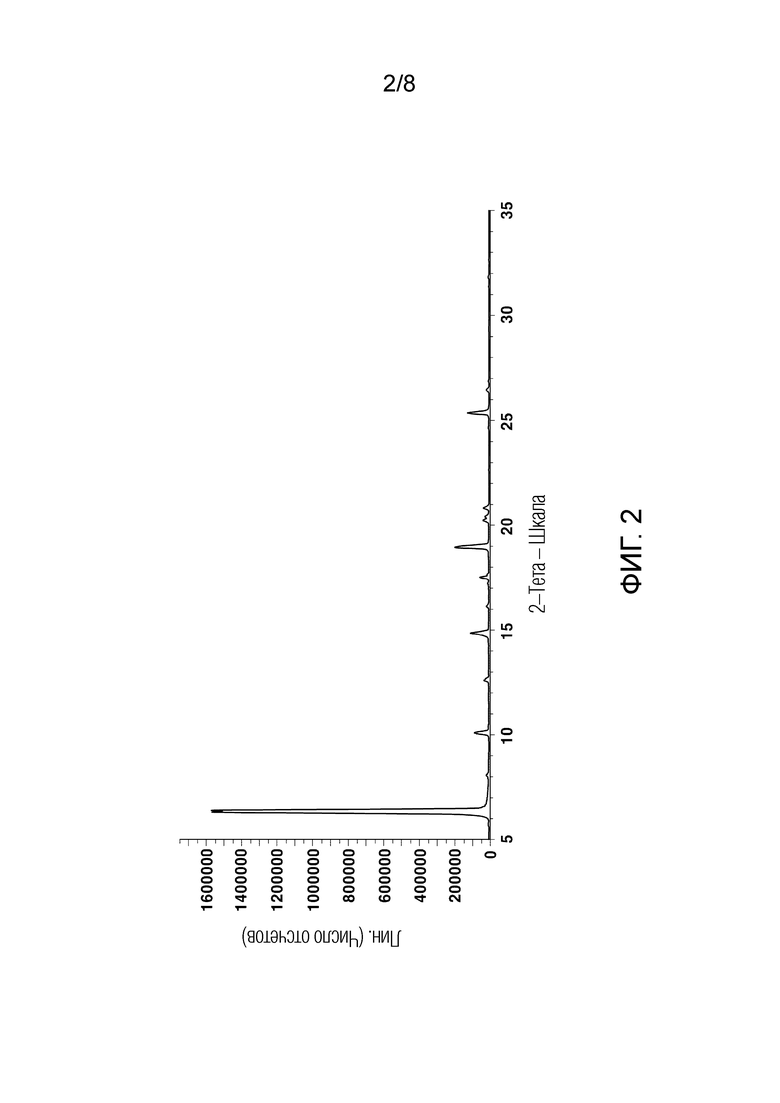
[55] На ФИГ. 2 показана порошковая рентгеновская дифрактограмма кристаллического 3-фенил-4-пропил-1-(пиридин-2-ил)-1Н-пиразол-5-ола (кристаллическое соединение свободного основания);
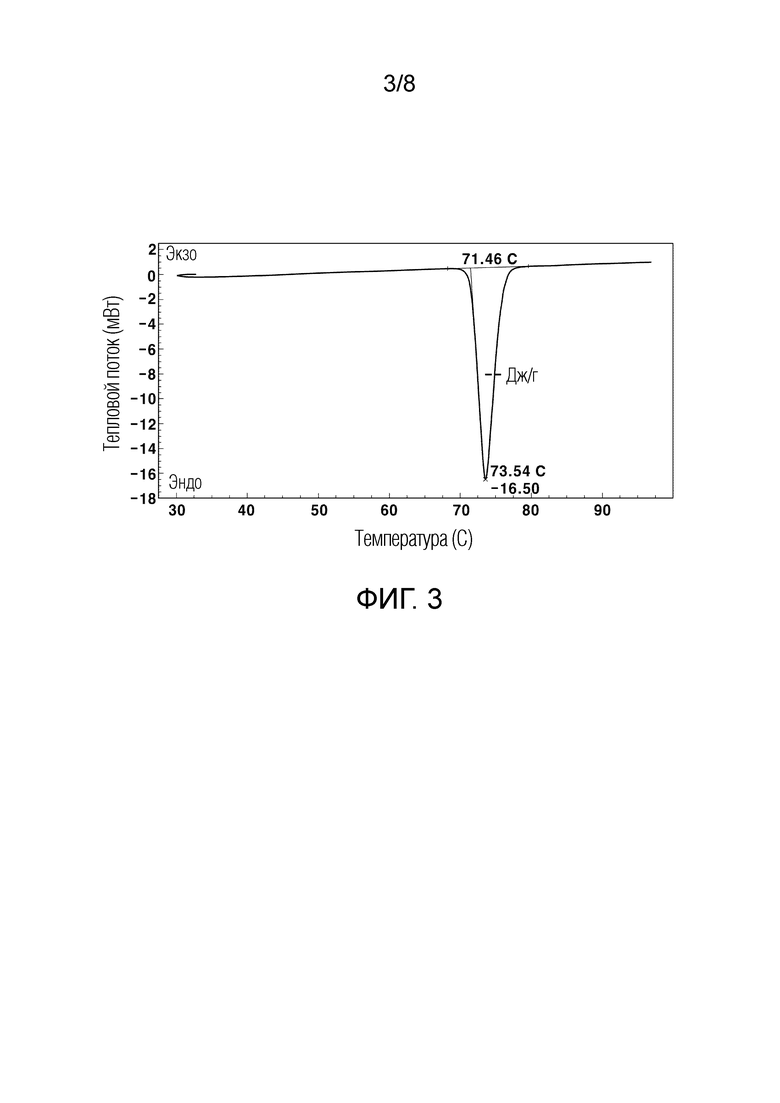
[56] На ФИГ. 3 показан график термического анализа ДСК кристаллического соединения свободного основания;
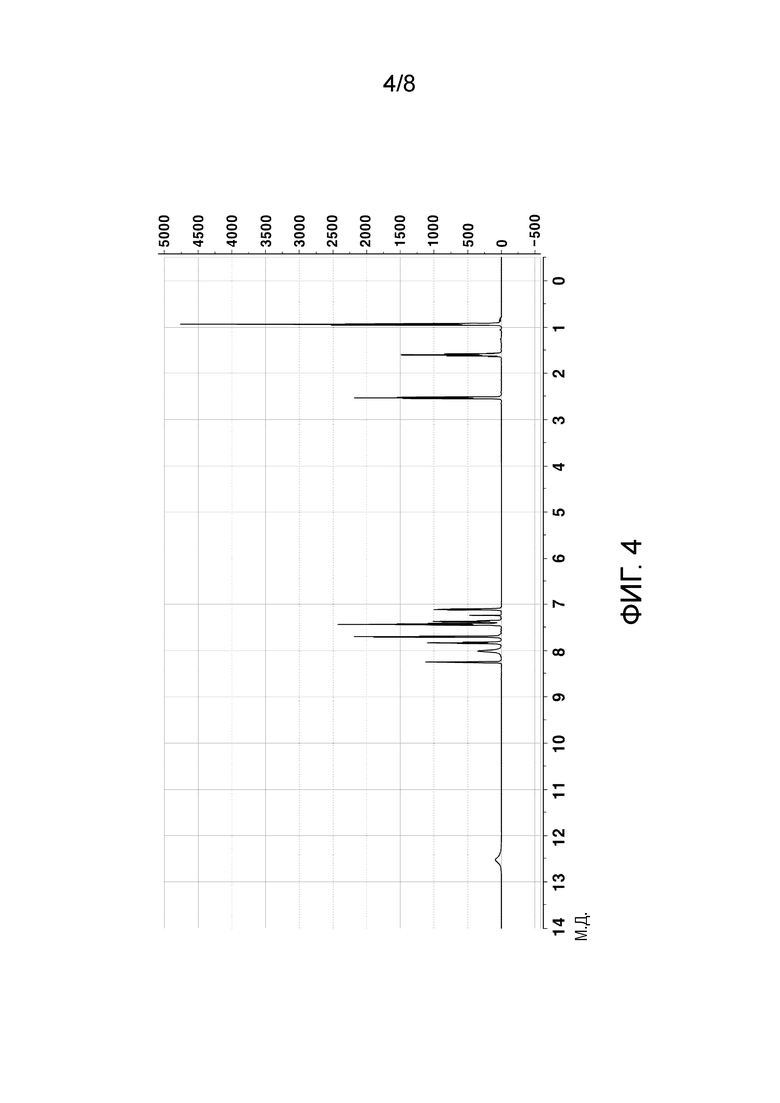
[57] На ФИГ. 4 показан 1H ЯМР-спектр кристаллического соединения свободного основания;
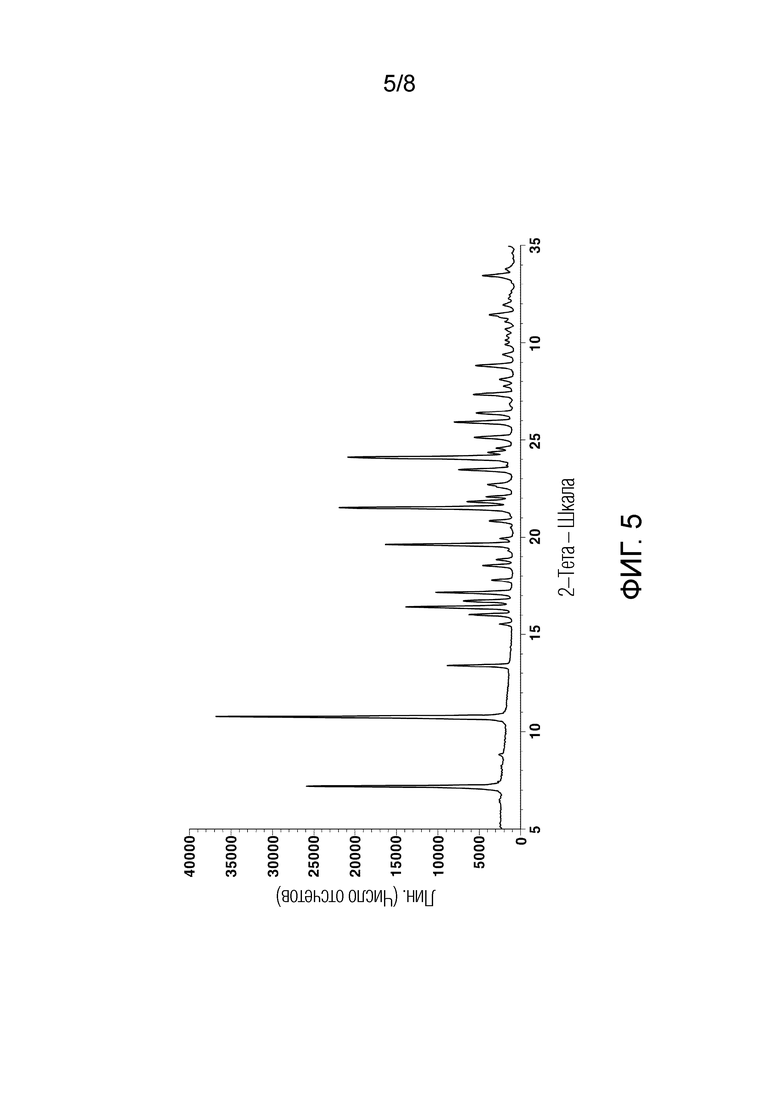
[58] На ФИГ. 5 показана порошковая рентгеновская дифрактограмма кристаллического 3–фенил–4–пропил–1–(пиридин–2–ил)–1Н–пиразол–5–ола гидрохлорида (кристаллическое соединение гидрохлорида);
[59] На ФИГ. 6 показан график термического анализа ДСК указанного кристаллического соединения гидрохлорида;
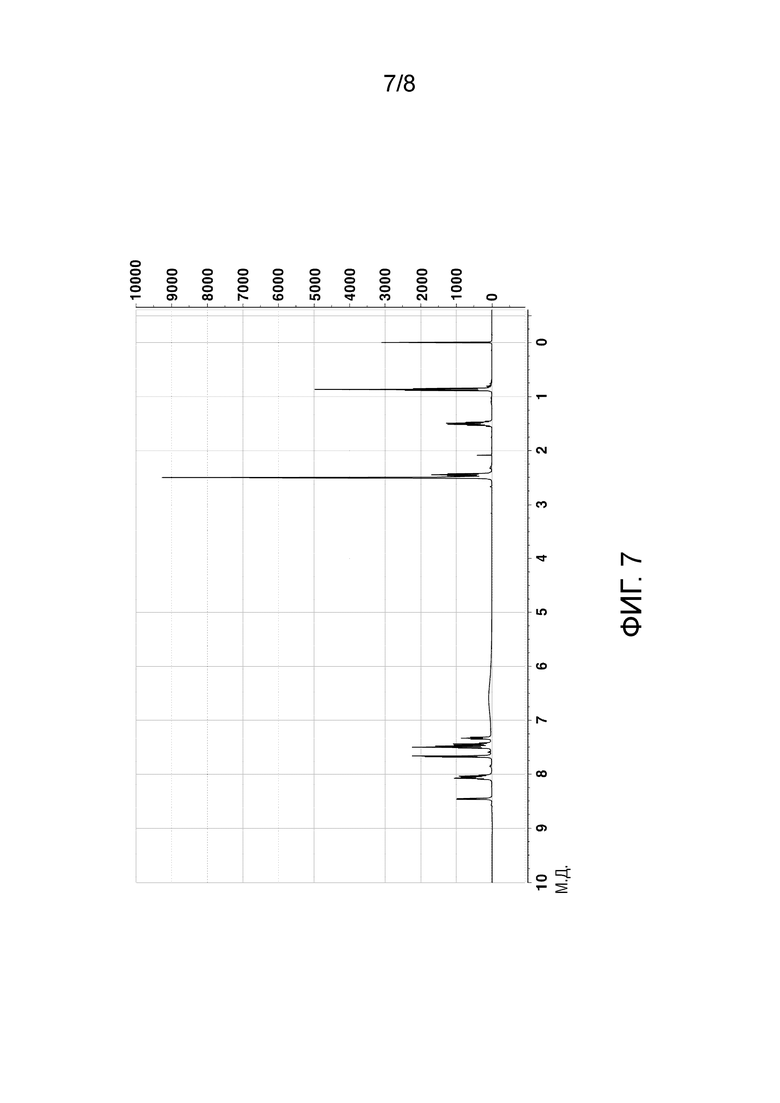
[60] На ФИГ. 7 показан 1H ЯМР–спектр кристаллического соединения гидрохлорида и
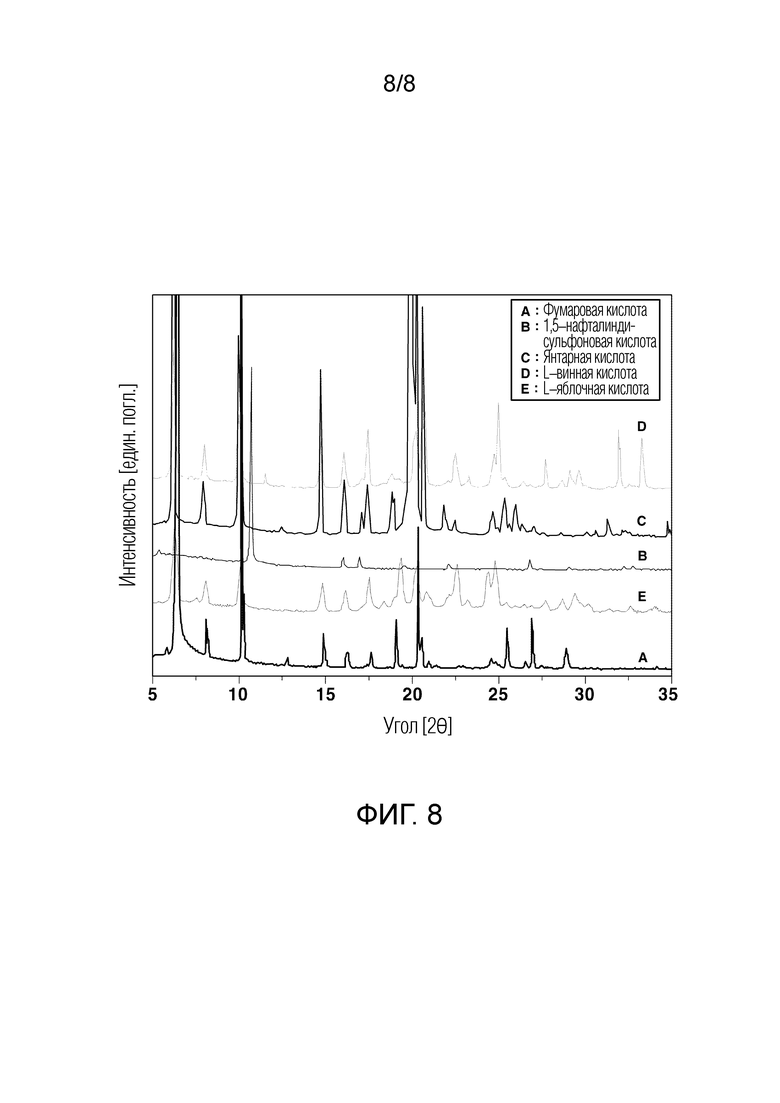
[61] На ФИГ. 8 показаны порошковые рентгеновские дифрактограммы различных кристаллических кислотно–аддитивных соединений солей.
Лучший вариант
[62] Настоящее изобретение относится к новому кристаллическому соединению гидрохлорида, способу его получения и фармацевтической композиции, содержащей соединение в качестве активного ингредиента.
[63] Кристаллическое соединение гидрохлорида, характеризуемое настоящим изобретением, является новым веществом, о котором не сообщалось в литературе, и обладает физическими свойствами, предпочтительными для получения лекарственного средства, является стабильным при воздействии тепла и влажности и обеспечивает достаточную стабильность для предотвращения разложения активного ингредиента или полиморфного перехода в течение хранения.
[64] Кристаллическое соединение гидрохлорида, предоставляемое настоящим изобретением, представляет собой кислотно-аддитивное соединение соли, в котором хлористоводородную кислоту добавляют к 3-фенил-4-пропил-1-(пиридин-2-ил)-1Н-пиразол-5-олу, исходной молекуле, как показано в следующей Формуле 1.
[65] [Формула 1]
[66]
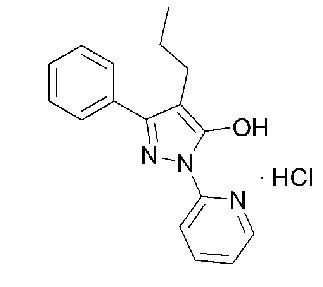
[67] Однако, пиридиновая группа исходной молекулы, которая образует кислотно-аддитивную соль, является относительно слабым основанием, и гидроксигруппа (-OH) в положении C5 пиразола может образовывать водородную связь с атомом азота (N) пиридина. По этой причине основность пиридина может быть слабее, чем в общем случае. Это может быть видно из молекулярной модели ФИГ. 1. ФИГ. 1 представляет собой изображение, показывающее молекулярную модель свободного основания 3-фенил-4-пропил-1-(пиридин-2-ил)-1Н-пиразол-5-ола, которое имеет наиболее стабильную форму, способную минимизировать молекулярную механическую энергию. Из изображения можно увидеть, что гидроксигруппа в положении C5 пиразола образует водородную связь с атомом азота пиридина.
[68] Результаты экспериментов, проведенных авторами настоящего изобретения, показали, что хлорид водорода (HCl) непрерывно отделяется от некристаллического соединения гидрохлорида, полученного стандартным способом получения. В результате соединение, представленное следующей Формулой 3, образуется в качестве продукта разложения.
[69] [Формула 3]
[70]
[71] Свободное основание 3-фенил-4-пропил-1-(пиридин-2-ил)-1Н-пиразол-5-ола имеет высокую вероятность создания продукта разложения, представленного Формулой 3, вследствие его более низкой температуры плавления по сравнению с фармацевтически приемлемыми соединениями соли. Кроме того, когда время воздействия влажности или тепла увеличивается, могут образовываться различные примеси, включая продукт разложения Формулы 3.
[72] Требования к кристаллическим соединениям гидрохлорида для применения в лекарственных средствах включают следующие. Во-первых, кристаллические соединения гидрохлорида должны быть физически стабильными для применения в процессе синтеза фармацевтически приемлемых солей или в процессе приготовления фармацевтических ингредиентов. Во-вторых, кристаллические соединения гидрохлорида не должны легко переходить в кристаллическую форму с течением времени во время хранения и распределения. В-третьих, кристаллические соединения гидрохлорида должны быть способны минимизировать образование примесей, включая N-O-соединение пиридина, представленное Формулой 3, во время хранения и распределения.
[73] Таким образом, авторы настоящего изобретения провели исследование, чтобы выбрать кислотно-аддитивное соединение соли, способное к дополнительной динамической стабилизации свободного основания 3-фенил-4-пропил-1-(пиридин-2-ил)-1Н-пиразол-5-ола. Иными словами, авторы настоящего изобретения приготовили различные кристаллические соединения гидрохлорида и различные кристаллические соединения гидрохлорида с добавлением фармацевтически приемлемых кислот, имеющих более высокую кислотность, чем хлористоводородная кислота (HCl) и не имеющих летучести, и провели эксперименты для сравнения стабильности между полученными кислотно-аддитивными соединениями солей. Можно предсказать, что по сравнению с кристаллическими соединениями гидрохлорида с добавлением с хлористоводородной кислоты кислотно-аддитивные соединения солей с добавлением кислоты, имеющей значение pKa, превышающее значения pKa пиридина и хлористоводородной кислоты, и будучи нелетучими, могут эффективно ингибировать высвобождение (десорбцию) кислоты из исходной молекулы. Однако сравнительные эксперименты авторов настоящего изобретения показали неожиданные результаты, заключающиеся в том, что среди различных кристаллических кислотно-аддитивных соединений солей на основе 3-фенил-4-пропил-1-(пиридин-2-ил)-1Н-пиразол-5-ола в качестве исходной молекулы кристаллическое соединение гидрохлорида является наиболее стабильным в условиях хранения лекарственного средства [См. следующий Экспериментальный пример 2].
[74] Следовательно, кристаллическое соединение гидрохлорида, предоставляемое настоящим изобретением, которое представляет собой соединение гидрохлорида с добавлением хлористоводородной кислотой, является наиболее стабильным среди различных кристаллических кислотно-аддитивных соединений солей. Таким образом, кристаллическое соединение гидрохлорида может быть использовано в качестве активного ингредиента лекарственного средства, поскольку активный ингредиент не разлагается, а стабильно сохраняется даже при хранении в течение длительного периода срока годности.
[75]
[76] Настоящее изобретение также предоставляет способ получения кристаллического соединения гидрохлорида. В частности, способ получения кристаллического соединения гидрохлорида в соответствии с настоящим изобретением включает:
[77] а) взаимодействие этилового эфира 2-пропил-3-оксо-3-фенилпропионовой кислоты с 2-гидразинопиридином с получением неочищенного продукта;
[78] b) растворение неочищенного продукта в нормальном гексане и затем медленное охлаждение полученного раствора до от -20 до -10°С с получением твердого вещества;
[79] c) фильтрование, промывание и сушка полученного твердого вещества с получением некристаллического соединения свободного основания;
[80] d) добавление некристаллического соединения свободного основания к смешанному растворителю, содержащему ацетонитрил и дистиллированную воду в одинаковом количестве, и энергичное перемешивание полученной смеси при от 20 до 25°С с получением кристаллического вещества;
[81] е) фильтрование, промывание и сушка полученного кристаллического вещества с получением кристаллического соединения свободного основания, представленного Формулой 2;
[82] f) взаимодействие кристаллического соединения свободного основания с раствором хлористоводородная кислота-изопропиловый эфир с получением твердого вещества гидрохлорида;
[83] g) добавление твердого вещества гидрохлорида к смешанному растворителю, содержащему трет-бутиловый эфир и толуол в одинаковом количестве, и энергичное перемешивание полученной смеси при от 5 до 10°С с получением кристаллического вещества и
[84] h) фильтрование, промывание и сушка полученного кристаллического вещества с получением кристаллического соединения гидрохлорида, представленного следующей Формулой 1.
[85] [Формула 2]
[86]
[87] [Формула 1]
[88]
[89] Способ получения кристаллического соединения гидрохлорида в соответствии с настоящим изобретением будет описан в каждой стадии более подробно.
[90] В стадии а) неочищенный продукт получают в соответствии со способом изготовления, раскрытым в корейском патенте №10-1633957 (Патентный документ 2).
[91] Стадия b) представляет собой процесс отверждения неочищенного продукта и включает растворение неочищенного продукта в нормальном гексане и затем охлаждение раствора для получения твердого вещества. В процессе растворения неочищенного продукта может потребоваться небольшое нагревание для полного растворения, и нагревание может быть соответствующим образом проведено в диапазоне температур от 50°С до 60°С. Охлаждение может быть соответствующим образом проведено в диапазоне температур от -20°С до -10°С. Когда температура охлаждения является чрезмерно высокой, скорость образования твердого вещества может быть низкой и выход может быть низким, а когда температура охлаждения является чрезвычайно низкой, кристаллическое вещество может не образовываться в последующем процессе кристаллизации.
[92] В стадии с) полученное твердое вещество отфильтровывают, промывают и высушивают с получением некристаллического соединения свободного основания. Промывание может проводиться с использованием в качестве растворителя нормального гексана, охлажденного до от 0°С до 10°С. Сушка может проводиться при комнатной температуре или может быть осуществлена с помощью вакуумной сушки при от 30°С до 40°С.
[93] Стадия d) представляет собой процесс кристаллизации некристаллического соединения свободного основания. В частности, некристаллическое соединение свободного основания добавляют к смешанному растворителю, содержащему ацетонитрил и дистиллированную воду в массовом соотношении 1:1, и энергично перемешивают при от 20°С до 25°С с получением кристаллического вещества. Массовое соотношение ацетонитрила и дистиллированной воды может составлять массовое соотношение от 1:0,5 до 1:2.
[94] Стадия е) представляет собой процесс фильтрования полученного кристаллического вещества, промывания кристаллического вещества растворителем и его сушки с получением кристаллического соединения свободного основания. Используемый в настоящем описании растворитель для промывания представляет собой смешанный растворитель, содержащий ацетонитрил и дистиллированную воду в массовом соотношении 1:1, и растворитель предпочтительно охлаждают до от 0 до 10°С. Сушка может быть осуществлена стандартным способом сушки, например, лиофилизацией, сушкой с роторным испарением, сушкой распылением, вакуумной сушкой или сушкой в псевдоожиженном слое и в частности, может быть осуществлена вакуумной сушкой. Предпочтительно, сушку можно проводить вакуумной сушкой при от 30°С до 40°С.
[95] Стадия f) представляет собой процесс взаимодействия кристаллического соединения свободного основания с хлористоводородной кислотой с получением твердого вещества аддитивной соли хлористоводородной кислоты. Реакцию получения гидрохлорида проводят в растворителе изопропиловом эфире. Хлористоводородная кислота может быть разбавлена изопропиловым эфиром для получения раствора, имеющего концентрацию от 0,5 до 2М. Температуру реакции предпочтительно поддерживают на уровне от -10°С до 10°С, более предпочтительно от 0°С до 5°С.
[96] Стадия g) представляет собой процесс кристаллизации твердого вещества гидрохлорида. В частности, кристаллическое соединение гидрохлорида добавляют к смешанному растворителю, содержащему трет-бутиловый эфир и толуол в массовом соотношении 1:1, и энергично перемешивают при от 5 до 10°С для получения кристаллического вещества.
[97] Стадия h) представляет собой процесс фильтрования, промывания и сушки полученного кристаллического вещества с получением кристаллического соединения гидрохлорида. Растворитель для промывания представляет собой смешанный растворитель, содержащий трет-бутиловый эфир и толуол в массовом соотношении 1:1, и растворитель предпочтительно охлаждают до от 0°С до 10°С. Сушка может быть осуществлена стандартным способом сушки, например, лиофилизацией, сушкой с роторным испарением, сушкой распылением, вакуумной сушкой или сушкой в псевдоожиженном слое и в частности, может быть осуществлена вакуумной сушкой. Предпочтительно, сушку можно проводить вакуумной сушкой при от 30°С до 40°С.
[98]
[99] Также настоящее изобретение предоставляет фармацевтическую композицию, содержащую кристаллическое соединение гидрохлорида в качестве активного ингредиента.
[100] В соответствии с Патентными документами 1-3 некристаллическое соединение гидрохлорида является эффективным в качестве активного ингредиента фармацевтической композиции для лечения заболевания, опосредованного активными формами кислорода (АФК), в частности, остеопороза, заболеваний почек и глазных заболеваний. Следовательно, кристаллическое соединение гидрохлорида, предоставляемое настоящим изобретением, также обладает активностью ингибирования образования активных форм кислорода (АФК) и, таким образом, на основе данного фармакологического механизма фармацевтическая композиция, содержащая кристаллическое соединение гидрохлорида в качестве активного ингредиента, может быть использована для лечения или профилактики остеопороза, заболевания почек и глазного заболевания.
[101] Заболевание почек может быть выбрано из группы, состоящей из диабетической нефропатии, гипертензивной нефропатии, гломерулонефрита, пиелонефрита, интерстициального нефрита, волчаночного нефрита, поликистозной болезни почек и почечной недостаточности.
[102] Глазное заболевание может быть выбрано из группы, состоящей из диабетической ретинопатии (ДР), диабетического макулярного отека, возрастной макулярной дегенерации, ретинопатии недоношенных (РН), полипоидной хориоидальной васкулопатии, ишемической пролиферативной ретинопатии, пигментного ретинита, колбочковой дистрофии, пролиферативной витреоретинопатии (ПВР), окклюзии артерии сетчатки, окклюзии вены сетчатки, птеригиума, ретинита, кератита, конъюнктивита, увеита, наследственной оптической нейропатии Лебера, отслойки сетчатки, отслойки пигментного эпителия сетчатки, неоваскулярной глаукомы, неоваскуляризации роговицы, неоваскуляризации сетчатки, хориоидальной неоваскуляризации (ХНВ) и вирусной инфекции.
[103] Фармацевтическая композиция настоящего изобретения содержит кристаллическое соединение гидрохлорида в качестве активного ингредиента. Содержание активного ингредиента может быть определено с учетом возраста, массы тела или подобного пациента и обычно может находиться в диапазоне от 0,01 до 10 масс. % исходя из общего количества фармацевтической композиции.
[104] Кроме того, фармацевтическая композиция настоящего изобретения может включать фармацевтически приемлемые вспомогательные вещества, такие как носители, разбавители, связующие вещества, дезинтегранты, скользящие вещества, регуляторы рН, антиоксиданты и вспомогательные вещества для растворения в пределах диапазона, который не ослабляет действие активного ингредиента. Примеры фармацевтически приемлемых вспомогательных веществ, которые можно использовать для приготовления фармацевтической композиции изобретения, включают микрокристаллическую целлюлозу, ксилит, эритрит, метилцеллюлозу, поливинилпирролидон, крахмал, камедь, альгинат, желатин, лактозу, декстрозу, сахарозу, пропилгидроксибензоат, целлюлозу, воду, метилгидроксибензоат, стеарат магния, тальк, сорбит, маннит, мальтит, фосфат кальция, силикат кальция, минеральное масло и подобные.
[105] Кроме того, фармацевтическая композиция настоящего изобретения может быть получена в форме состава, выбранной из группы, состоящей из порошка, гранулы, таблетки, капсулы, суспензии, эмульсии, сиропа, аэрозоля, мази, крема, суппозитория, глазных капель и инъекции в соответствии со стандартными способами получения. Нет конкретного ограничения в отношении формы состава в настоящем изобретении.
[106] Кроме того, вода для инъекций может быть использована для приготовления глазных капель или инъекционной композиции в соответствии с настоящим изобретением. Глазные капли или инъекции, содержащие фармацевтически приемлемые соли, могут необязательно содержать, но не ограничиваясь ими, изотонические агенты, буферные растворы, осмотические агенты и подобные, которые обычно используются в данной области техники.
Принцип изобретения
[107] В дальнейшем, конфигурации и эффекты настоящего изобретения будут описаны более подробно со ссылкой на примеры. Однако следующие примеры приведены только для иллюстрации и не должны рассматриваться как ограничивающие объем настоящего изобретения.
[108] [Пример]
[109] Пример 1: Получение кристаллического соединения свободного основания
[110] Этиловый эфир 2-пропил-3-оксо-3-фенилпропионовой кислоты (5,6 г, 51,4 ммоль) и 2-гидразинопиридин (11,5 г, 49 ммоль) вводили в круглую колбу с последующим перемешиванием в атмосфере азота при 150°С в течение 24 часов без реакционного растворителя. После охлаждения реакционного раствора до комнатной температуры остаток очищали с помощью колоночной хроматографии на силикагеле (30 г; н-гексан/EtOAc=5/1) и концентрировали при пониженном давлении. К полученному твердому соединению добавляли нормальный гексан (70 мл), и твердое соединение медленно растворяли при нагревании, и затем медленно охлаждали до -20°C в течение 1 часа. Полученное твердое вещество отфильтровывали при пониженном давлении и промывали нормальным гексаном, охлажденным до от 0 до 10°С. Промытое твердое вещество добавляли к смешанному растворителю (100 мл) ацетонитрила и дистиллированной воды (1:1) и затем энергично перемешивали при 25°C в течение 1 часа с образованием кристаллического вещества. Полученное кристаллическое вещество отфильтровывали, промывали смешанным растворителем из ацетонитрила и дистиллированной воды (1:1), который охлаждали до 10°С или ниже и высушивали под вакуумом при 40°С в течение 12 часов с получением кристаллического соединения свободного основания.
[111] График анализа порошковой рентгеновской дифракции (PXRD), график термического анализа ДСК и 1H ЯМР-спектры кристаллического соединения свободного основания, полученного в Примере 1, показаны на ФИГ. 2-4, соответственно.
[112] Пример 2. Получение кристаллического соединения гидрохлорида
[113] Кристаллическое соединение свободного основания (12,9 г, 46,2 ммоль) вводили в круглую колбу и растворяли в изопропиловом эфире (300 мл) в атмосфере азота и затем добавляли к нему 1 М раствор хлористоводородная кислота-изопропиловый эфир при от 0 до 5°С в течение 10 минут. Реакционный раствор перемешивали при от 0 до 5°С в течение 1 часа с получением твердого соединения. Полученное твердое соединение отфильтровывали при пониженном давлении в атмосфере азота и промывали изопропиловым эфиром (30 мл), который охлаждали до 10°С или ниже. Промытое твердое соединение добавляли к трет-бутиловому эфиру и толуолу (1:1, 50 мл) с последующим энергичным перемешиванием при от 5 до 10°С в атмосфере азота в течение 1 часа с образованием кристаллического вещества. Полученное кристаллическое вещество отфильтровывали, и промывали смешанным растворителем трет-бутилового эфира и толуола (1:1), который охлаждали до 10°С или ниже, и высушивали при 40°С в течение 12 часов с получением белого кристаллического соединения гидрохлорида.
[114] График анализа порошковой рентгеновской дифракции (PXRD), график термического анализа ДСК и 1H ЯМР-спектры кристаллического соединения свободного основания, полученного в Примере 2, показаны на ФИГ. 5-7, соответственно.
[115] Сравнительный пример 1: Получение некристаллического соединения свободного основания
[116] Этиловый эфир 2-пропил-3-оксо-3-фенилпропионовой кислоты (5,6 г, 51,4 ммоль) и 2-гидразинопиридин (11,5 г, 49 ммоль) вводили в круглую колбу с последующим перемешиванием в атмосфере азота при 150°С в течение 3 дней без реакционного растворителя. После охлаждения реакционного раствора до комнатной температуры остаток концентрировали при пониженном давлении, промывали гексаном и этилацетатом и высушивали под вакуумом с получением некристаллического соединения свободного основания.
[117] Сравнительный пример 2. Получение некристаллического соединения гидрохлорида
[118] Некристаллическое соединение свободного основания (12,9 г, 46,2 ммоль), полученное в Сравнительном примере 1, вводили в круглую колбу и растворяли в диэтиловом эфире (300 мл) и затем добавляли к нему 2М раствор хлористоводородная кислота-диэтиловый эфир при от 0 до 5°С в течение 10 минут. Полученное твердое соединение отфильтровывали при пониженном давлении, промывали гексаном и этилацетатом и высушивали под вакуумом с получением некристаллического соединения гидрохлорида.
[119] Сравнительный пример 3. Получение различных кристаллических кислотно-аддитивных соединений
[120] Кислотно-аддитивные соли получали путем добавления различных кислот к кристаллическому 3-фенил-4-пропил-1-(пиридин-2-ил)-1Н-пиразол-5-олу (кристаллическое свободное основание). Методы кристаллизации включают различные методы, известные в литературе, например, реакционную кристаллизацию, кристаллизацию с охлаждением, кристаллизацию вытяжкой и кристаллизацию с испарением. Кристаллические кислотно-аддитивные соединения солей получали с использованием приблизительно 20 типов кислот. Среди них кристаллические кислотно-аддитивные соединения солей, полученные кристаллизацией с испарением, показаны в следующей Таблице 1.
[121] В частности, кристаллическое соединение свободного основания (12,9 г, 46,2 ммоль) вводили в круглую колбу и добавляли в него кислоту, показанную в Таблице 1. 20 мл безводного метанола добавляли к каждому продукту реакции и продукт реакции полностью растворяли при нагревании до от приблизительно 50 до 60°С. Колбу открывали, ее воздухозаборник покрывали салфеткой Kimwipes™, чтобы обеспечить прохождение воздуха, и оборачивали резиновой лентой, и колбу хранили в вытяжном шкафу в течение 36 часов. В это время температуру внутри вытяжного шкафа поддерживали от 23 до 26°C. Полученное твердое вещество отфильтровывали, промывали нормальным гексаном (5 мл), который охлаждали до 10°C или ниже, и затем высушивали под вакуумом с получением кристаллической кислотно-аддитивной соли.
[122] [Таблица 1]
[123] Результаты анализа порошковой рентгеновской дифракции, проведенного с кристаллическим соединением гидрохлорида, полученным в Примере 2, и различными кристаллическими кислотно-аддитивными соединениями солей, полученными в Сравнительном Примере 3, показаны на ФИГ. 8.
[124] [Экспериментальный пример]
[125] Экспериментальный пример 1. Исследование гигроскопичности
[126] Эксперименты по сравнению гигроскопичности проводили для каждого из кристаллического соединения свободного основания (Пример 1), кристаллического соединения гидрохлорида (Пример 2), некристаллического соединения свободного основания (Сравнительный Пример 1), некристаллического соединения гидрохлорида (Сравнительный пример 2) и различных кристаллических кислотно-аддитивных соединений (Сравнительный пример 3).
[127] Исследуемые соединения подвергали воздействию в чашке Петри условий ускоренного хранения при температуре 40°С и относительной влажности 75% в течение 7 дней без герметизации. Содержание воды в каждом исследуемом соединении измеряли с использованием влагомера по методу Карла-Фишера. Таблица 2 суммирует содержание воды (%) каждого исследуемого соединения, измеренное в течение периода хранения.
[128] [Таблица 2]
[129] Результаты Таблицы 2 показывают, что в случае соединения свободного основания и соединения гидрохлорида кристаллическое соединение имеет более низкое исходное содержание воды и также имеет значительную разницу в содержании воды через 7 дней в условиях ускоренного хранения по сравнению с некристаллическим соединением. Следовательно, кристаллическое соединение свободного основания или кристаллическое соединение гидрохлорида значительно менее гигроскопично и становится насыщенным, а не с повышенным содержанием воды с течением времени, и, таким образом, является пригодным в качестве фармацевтического ингредиента для фармацевтического применения. Кроме того, можно видеть, что по сравнению с кристаллическими кислотно-аддитивными соединениями солей с добавлением с кислот, отличных от хлористоводородной кислоты (Сравнительный пример 3), кристаллические соединения гидрохлорида имеют превосходную гигроскопичность по сравнению с кристаллическими кислотно-аддитивными солями, за исключением кристаллического 1,5-нафталиндисульфоната и кристаллического малата.
[130] Экспериментальный пример 2. Сравнительное исследование стабильности
[131] Были проведены эксперименты по сравнению стабильности для каждого из кристаллического соединения свободного основания (Пример 1), кристаллического соединения гидрохлорида (Пример 2), некристаллического соединения свободного основания (Сравнительный Пример 1), некристаллического соединения гидрохлорида (Сравнительный пример 2) и различных кристаллических кислотно-аддитивных соединений (Сравнительный пример 3).
[132] Исследуемое соединение хранили в камере для измерения стабильности в условиях длительного хранения при температуре 25°С и относительной влажности 60% в течение 6 месяцев. Для хранения каждое исследуемое соединение помещали в двойной полиэтиленовый пакет, и полиэтиленовый пакет заполняли мешочком с силикагелем и затем помещали в небольшую бумажную коробку (фибровый барабан).
[133] Следующая Таблица 3 суммирует результаты измерения концентрации продукта разложения Формулы 3 и общей концентрации примесей с помощью анализа ВЭЖХ после хранения в условиях длительного хранения в течение 6 месяцев.
[134] [Аналитические условия жидкостной хроматографии (ВЭЖХ)]
[135] Колонка: 4,6 мм × 250 мм, 5 мкм, размер пор 100 Å
[136] Температура колонки: 35°С
[137] Детектор: УФ-детектор (246 нм)
[138] Скорость потока: 1,0 мл/мин
[139] Время: 55 минут
[140] Градиентное условие подвижной фазы:
[141]
Подвижная фаза B: Метанол
Подвижная фаза C: Ацетонитрил
[142] [Таблица 3]
(Пример 1)
(Пример 2)
(Сравнительный пример 3)
[143] Результаты Таблицы 3 показывают, что в случае соединения свободного основания по сравнению с кристаллическим соединением свободного основания (Пример 1) некристаллическое соединение свободного основания (Сравнительный пример 1) имеет значительно более высокую концентрацию продукта разложения, представленного Формулой 3. Можно видеть, что в случае общей концентрации примесей, измеренной через 6 месяцев, по сравнению с исходным состоянием, содержание некристаллического соединения свободного основания (Сравнительный пример 1) значительно увеличивается по сравнению с кристаллическим соединением свободного основания (Пример 1). Кроме того, в случае соединения гидрохлорида некристаллическое соединение гидрохлорида (Сравнительный пример 2) подвергается отделению хлористоводородной кислоты (HCl). В результате можно видеть, что некристаллическое соединение гидрохлорида (Сравнительный пример 2) имеет значительно более высокую концентрацию продукта разложения, представленного Формулой 3, по сравнению с кристаллическим соединением гидрохлорида (Пример 2). Кроме того, можно видеть, что некристаллическое соединение гидрохлорида (Сравнительный пример 2) имеет более высокую концентрацию примесей, чем концентрация кристаллического соединения гидрохлорида (Пример 2), поскольку некристаллическое соединение гидрохлорида (Сравнительный пример 2) имеет более низкую термодинамическую стабильность даже при сравнении концентрации примесей.
[144] Кроме того, по сравнению с кристаллическим кислотно-аддитивным соединением соли, к которому добавляют кислоту, отличную от хлористоводородной кислоты, кристаллическое соединение гидрохлорида (Пример 2) имеет значительно более низкую концентрацию продукта разложения, представленного Формулой 3, и значительно более низкую концентрацию примесей по сравнению с кристаллическим кислотно-аддитивным соединением соли (Сравнительный пример 3). Следовательно, кристаллическое соединение гидрохлорида является наиболее стабильным среди различных кристаллических кислотно-аддитивных соединений солей.
[145] Экспериментальный пример 3. Оценка полиморфного перехода
[146] Для определения происходит ли явление полиморфного перехода в условиях хранения в кристаллическом соединении свободного основания (Пример 1) и кристаллическом соединении гидрохлорида (Пример 2), исследовали результаты анализа PXRD в исходном состоянии и возможность полиморфного перехода во времени.
[147] В частности, исследуемое соединение хранили в полиэтиленовом пакете в камере для измерения стабильности при температуре 40°С и относительной влажности 75% в течение 4 недель. Также сравнивали результаты анализа PXRD и ДСК, измеренные в исходном состоянии и через 4 недели. Результаты сравнения приведены в Таблице 4 ниже.
[148] [Таблица 4]
[149] Кристаллическое соединение свободного основания (Пример 1) и кристаллическое соединение гидрохлорида (Пример 2) подвергали PXRD и ДСК через 4 недели после хранения. Результаты показали, что с течением времени полиморфный переход не происходил. Это указывает на то, что кристаллическое соединение свободного основания (Пример 1) и кристаллическое соединение гидрохлорида (Пример 2) являются стабильными соединениями.
[150] Экспериментальный пример 4. Анализ физических свойств
[151] (1) Анализ порошковой рентгеновской дифракции (PXRD)
[152] Анализ порошковой рентгеновской дифракции кристаллического соединения свободного основания и кристаллического соединения гидрохлорида, полученных в Примерах 1 и 2, проводили с использованием Cu-Kα-лучей на порошковом рентгеновском дифрактометре D8 Advance, выпускаемом Bruker Corporation. Дифрактометр был оборудован динамической оптимизацией потока (DBO), и величина тока была установлена на 45 кВ и 40 мА. Щели расхождения и рассеяния были установлены на 1°, и щель для приема света - на 0,2 мм. 2θ измеряли при 6°/мин от 5 до 35°. Результаты анализа PXRD показаны на ФИГ. 2 и 5.
[153] Кроме того, в следующей Таблице 5 показаны результаты анализа порошковой рентгеновской дифракции, в частности, угол дифракции 2θ (2θ±0,2°), имеющий относительную интенсивность 5% или более в случае кристаллического соединения свободного основания, и угол дифракции 2θ (2θ±0,2°), имеющий относительную интенсивность 15% или более в случае кристаллического соединения гидрохлорида.
[154] [Таблица 5]
[155] (2) Анализ температуры методом дифференциально-температурной сканирующей калориметрии (ДСК)
Температуры плавления кристаллического соединения свободного основания и кристаллического соединения гидрохлорида, полученных в Примерах 1 и 2, измеряли с помощью анализа температуры методом дифференциальной сканирующей калориметрии (ДСК).
[156] Измерения ДСК проводили с использованием DSC N-650, полученного от SCINCO в потоке азота в герметичной посуде со скоростью сканирования 10°C/мин от 20°C до 150°C. Результаты показаны на ФИГ. 3 и 6, соответственно.
[157] Как можно увидеть из ФИГ. 3 и 6, кристаллическое соединение свободного основания показало характерный эндотермический пик при 73,54±3°С, и кристаллическое соединение гидрохлорида показало характерный эндотермический пик при 134,25±3°С.
[158] Кроме того, можно увидеть, что некристаллическое соединение свободного основания, полученное в Сравнительном примере 1, имеет непостоянную температуру плавления и полностью плавится при 60°C или выше. Некристаллическое соединение гидрохлорида, полученное в Сравнительном примере 2, также имеет непостоянную температуру плавления, медленно плавится в широком диапазоне температур и полностью плавится при 160°C или выше. При рассмотрении данных аспектов можно увидеть, что кристаллическое соединение, предоставляемое настоящим изобретением, является выделенным соединением, имеющим совершенно отличные физико-химические свойства от свойств некристаллического соединения.
[159] Экспериментальные результаты, описанные выше, показывают, что кристаллическое соединение свободного основания, представленное Формулой 2, и кристаллическое соединение гидрохлорида, представленное Формулой 1, не обладают гигроскопичностью, обладают превосходной стабильностью и низкой вероятностью полиморфного перехода с течением времени и, таким образом, оптимизированы как фармацевтические ингредиенты по сравнению с другими кристаллическими кислотно-аддитивными солями и стандартными веществами.
[160] Определенные конфигурации настоящего изобретения были раскрыты, и специалисту в данной области техники должно быть понятно, что иллюстративное подробное описание предоставлено только для описания предпочтительных вариантов осуществления и не должно рассматриваться как ограничение объема настоящего изобретения.
[161] Следовательно, фактический объем настоящего изобретения определяется прилагаемой формулой изобретения и ее эквивалентами.
[162]
[163]
| название | год | авторы | номер документа |
|---|---|---|---|
| ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ПЕЧЕНИ | 2018 |
|
RU2723686C1 |
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ОСТРОГО ПОРАЖЕНИЯ ПОЧЕК, ВЫЗВАННОГО КОНТРАСТНЫМ ВЕЩЕСТВОМ | 2021 |
|
RU2817980C1 |
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ФИБРОЗА ЛЕГКИХ, СОДЕРЖАЩЕЕ ПРОИЗВОДНОЕ ПИРАЗОЛА | 2021 |
|
RU2817989C1 |
| НОВОЕ ПИРРОЛОПИРИДИНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2017 |
|
RU2733723C1 |
| СОЛИ ПРОИЗВОДНЫХ ПИРРОЛОПИРИМИДИНОНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2007 |
|
RU2448967C2 |
| НОВОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ ПИРИМИДИНА, ПРОЯВЛЯЮЩЕЕ ИНГИБИРУЮЩЕЕ ДЕЙСТВИЕ НА РОСТ РАКОВЫХ КЛЕТОК, И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕГО | 2020 |
|
RU2834201C1 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ МОДУЛЯЦИИ КИНАЗНОЙ АКТИВНОСТИ МУТАНТОВ EGFR | 2024 |
|
RU2838180C1 |
| СОЛИ ЭФИРОВ БЕНЗИМИДАЗОЛИЛПИРИДИЛА И ИХ СОДЕРЖАЩИЕ СОСТАВЫ | 2007 |
|
RU2457206C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИВАБРАДИНА ГИДРОХЛОРИДА И ЕГО ПОЛИМОРФНЫХ МОДИФИКАЦИЙ | 2008 |
|
RU2473544C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКООЧИЩЕННОГО ПРАСУГРЕЛЯ ИЛИ ЕГО КИСЛОТНО-АДДИТИВНОЙ СОЛИ | 2007 |
|
RU2424243C2 |
Изобретение относится к способу получения кристаллического соединения гидрохлорида 3-фенил-4-пропил-1-(пиридин-2-ил)-1Н-пиразола. Способ осуществляют путем а) взаимодействия этилового эфира 2-пропил-3-оксо-3-фенилпропионовой кислоты с 2-гидразинопиридином с получением неочищенного продукта; b) растворения неочищенного продукта в нормальном гексане и затем медленное охлаждение полученного раствора до от -20 до -10°С с получением твердого вещества; c) фильтрования, промывания и сушки полученного твердого вещества с получением некристаллического соединения свободного основания; d) добавления некристаллического соединения свободного основания к смешанному растворителю, содержащему ацетонитрил и дистиллированную воду в одинаковом количестве, и энергичного перемешивания полученной смеси при от 20 до 25°С с получением кристаллического вещества; е) фильтрования, промывания и сушки полученного кристаллического вещества с получением кристаллического соединения свободного основания, представленного Формулой 2; f) взаимодействия кристаллического соединения свободного основания с раствором хлористоводородная кислота – изопропиловый эфир с получением твердого вещества гидрохлорида; g) добавления твердого вещества гидрохлорида к смешанному растворителю, содержащему трет-бутиловый эфир и толуол в одинаковом количестве, и энергичного перемешивания полученной смеси при от 5 до 10°С с получением кристаллического вещества и h) фильтрования, промывания и сушки полученного кристаллического вещества с получением кристаллического соединения гидрохлорида, представленного следующей Формулой 1. Технический результат – получение гидрохлорида 3-фенил-4-пропил-1-(пиридин-2-ил)-1Н-пиразола в кристаллической форме, обладающей стабильностью, более низкой концентрацией примесей. 8 ил., 5 табл., 4 пр.
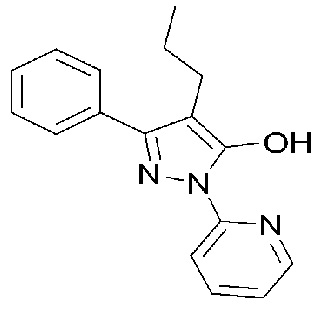
[Формула 2]
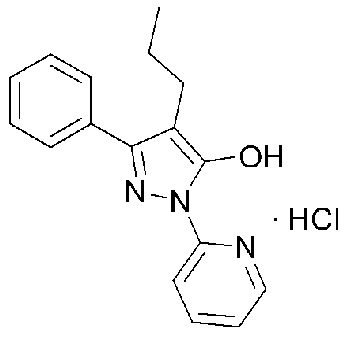
[Формула 1]
Способ получения кристаллического гидрохлорида 3-фенил-4-пропил-1-(пиридин-2-ил)-1Н-пиразол-5-ола, включающий:
а) взаимодействие этилового эфира 2-пропил-3-оксо-3-фенилпропионовой кислоты с 2-гидразинопиридином с получением неочищенного продукта;
b) растворение неочищенного продукта в нормальном гексане и затем медленное охлаждение полученного раствора до от -20 до -10°С с получением твердого вещества;
с) фильтрование, промывание и сушку полученного твердого вещества с получением некристаллического соединения свободного основания;
d) добавление некристаллического соединения свободного основания к смешанному растворителю, содержащему ацетонитрил и дистиллированную воду в одинаковом количестве, и энергичное перемешивание полученной смеси при от 20 до 25°С с получением кристаллического вещества;
е) фильтрование, промывание и сушку полученного кристаллического вещества с получением кристаллического соединения свободного основания, представленного Формулой 2;
f) взаимодействие кристаллического соединения свободного основания с раствором хлористоводородная кислота - изопропиловый эфир с получением твердого вещества гидрохлорида;
g) добавление твердого вещества гидрохлорида к смешанному растворителю, содержащему трет-бутиловый эфир и толуол в одинаковом количестве, и энергичное перемешивание полученной смеси при от 5 до 10°С с получением кристаллического вещества и
h) фильтрование, промывание и сушку полученного кристаллического вещества с получением кристаллического соединения гидрохлорида, представленного следующей Формулой 1
[Формула 2]
[Формула 1]
| WO 2014035070 A1, 06.03.2014 | |||
| US 20140088152 A1, 27.03.2014 | |||
| Joo, J | |||
| H., Huh, J.-E., Lee, J | |||
| H., Park, D | |||
| R., Lee, Y., Lee, S | |||
| G | |||
| Bae, Y | |||
| S | |||
| Токарный резец | 1924 |
|
SU2016A1 |
| A novel pyrazole derivative protects from ovariectomy-induced osteoporosis through the inhibition of NADPH oxidase | |||
| Scientific Reports, 6(1) | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| US 20140066404 A1, 06.03.2014 | |||
| WO | |||

