Согласно настоящей заявке испрашивается приоритет в соответствии с заявкой на выдачу патента Китая №201710395513.0, поданной 27 мая 2017 года в патентное ведомство Китая с названием изобретения «Лигандное соединение α7-никотинового ацетилхолинового рецептора и его применение», которая включена в настоящий документ посредством ссылки во всей своей полноте.
Область техники, к которой относится настоящее изобретение
Настоящее раскрытие относится к области медицинских технологий и в частности к лигандному соединению α7-никотинового ацетилхолинового рецептора и его применению.
Предшествующий уровень техники настоящего изобретения
Никотиновый ацетилхолиновый рецептор (nAChR) относится к классу нейромедиатор-зависимых ионных каналов, которые широко распространены в центральной нервной системе (ЦНС) и в периферической нервной системе (ПНС) и связаны с многообразием физиологических функций. Существуют различные подтипы nAChR, которые в общем состоят из α-субъединицы (например, α2-α10) и β-субъединицы (β2-β4). Среди них α7-никотиновый ацетилхолиновый рецептор (α7 nAChR) представляет собой гомопентамер, состоящий из 5 полностью идентичных α-субъединиц, и, главным образом, присутствует в важных областях, связанных с памятью, научением и т.д., таких как гиппокамп, таламус и кора головного мозга. В ходе недавних клинических исследований было обнаружено, что в головном мозге пациентов с нейродегенеративными заболеваниями, такими как болезнь Альцгеймера и болезнь Паркинсона, наблюдается сокращение плотности белка α7 nAChR, и исследование генного "нокаута", селективных в отношение подтипа лигандов и тому подобного показывает, что: нацеливание лиганда на рецептор α7 nAChR может оказывать положительное влияние на нарушение когнитивной способности и фильтрации сенсорной информации, например, высокоселективные агонисты α7 nAChR, такие как PNU-282987, РНА-543613 и А-582941, улучшают когнитивные функции на моделях, такие как нарушение фильтрации сенсорной информации, кратковременная оперативная память, консолидация памяти и тому подобное. Поэтому синтез агонистов и радиолигандов для α7-никотинового ацетилхолинового рецептора также привлекает все больше внимания.
Краткое раскрытие настоящего изобретения
Задача вариантов осуществления настоящего раскрытия состоит в обеспечении лигандного соединения α7-никотинового ацетилхолинового рецептора для того, чтобы получить радиоактивную или нерадиоактивную лигандную молекулу, имеющую высокое сродство к α7-никотиновому ацетилхолиновому рецептору. Специфическое техническое решение состояло в следующем:
Лигандное соединение α7-никотинового ацетилхолинового рецептора, представленное одной из следующих общих формул:
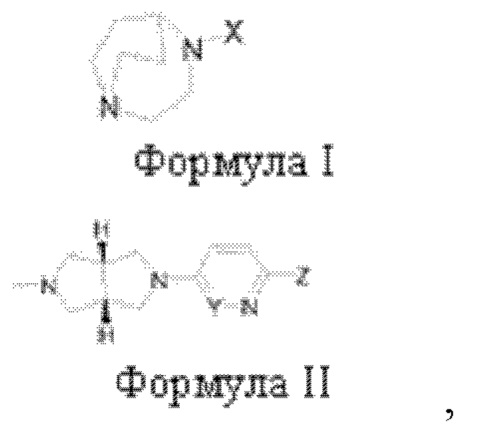
где
(1) X представляет собой 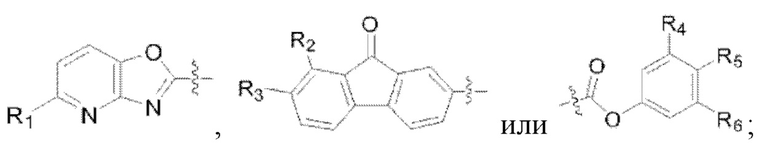
R1 представляет собой 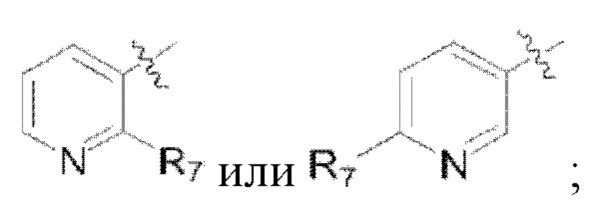 R7 представляет собой галоген;
R7 представляет собой галоген;
(2) R2 представляет собой водород, и R3 представляет собой галоген или аминогруппу; или R3 представляет собой водород, и R2 представляет собой галоген или аминогруппу;
(3) R6 представляет собой водород, и R4 и R5 объединены в  или R4 представляет собой водород, и R5 и R6 объединены в
или R4 представляет собой водород, и R5 и R6 объединены в 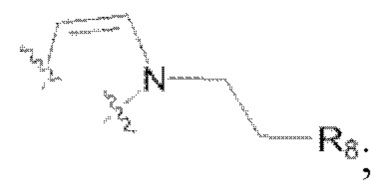 R8 представляет собой галоген;
R8 представляет собой галоген;
(4) Y представляет собой азот или углерод; Z представляет собой 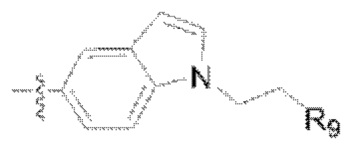 или
или 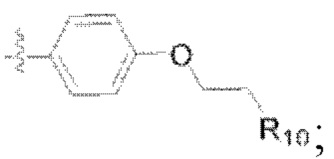 каждый R9, R10 представляет собой галоген, где
каждый R9, R10 представляет собой галоген, где  обозначает положение связывания группы с основной структурой.
обозначает положение связывания группы с основной структурой.
Термин «галоген» в контексте настоящего документа включает фтор, хлор, бром или иод.
В соответствии с некоторыми вариантами осуществления галоген является меченным радиоактивным изотопом или является немеченным радиоактивным изотопом.
В соответствии с некоторыми вариантами осуществления галоген представляет собой F или 18F.
В соответствии с некоторыми вариантами осуществления лигандное соединение α7-никотинового ацетилхолинового рецептора, представленное в настоящем раскрытии, может быть выбрано из следующих соединений:
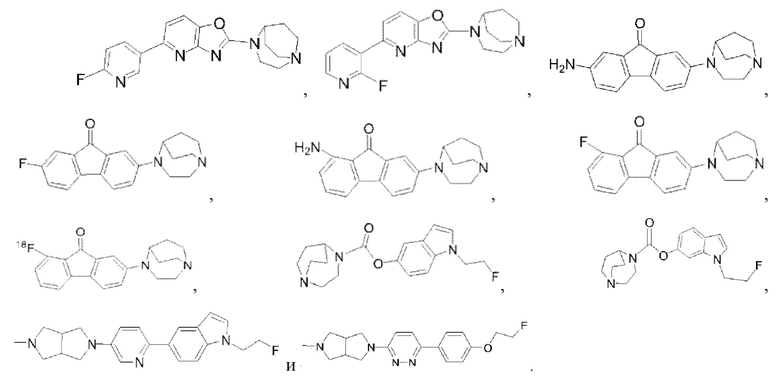
В соответствии с некоторыми вариантами осуществления, лигандное соединение α7-никотинового ацетилхолинового рецептора, представленное в настоящем раскрытии, может применяться в качестве агониста α7-никотинового ацетилхолинового рецептора.
В соответствии с некоторыми вариантами осуществления лигандное соединение α7-никотинового ацетилхолинового рецептора, представленное в настоящем раскрытии, может применяться в качестве частичного агониста α7-никотинового ацетилхолинового рецептора.
Термин "агонист" в контексте настоящего документа необходимо понимать в его самом широком значении, т.е. как любую молекулу, которая частично или полностью активирует по меньшей мере одну биологическую активность мишени (например, α7-никотинового ацетилхолинового рецептора). Например, лигандное соединение α7-никотинового ацетилхолинового рецептора, представленное в настоящем раскрытии, может специфически связываться с внеклеточным доменом α7-никотинового ацетилхолинового рецептора, так чтобы вызвать внутриклеточную сигнализацию, демонстрируя таким образом свою эффективность при профилактике или лечении когнитивных расстройств и неврологической реабилитации.
α7-никотиновый ацетилхолиновый рецептор, как известно, имеет огромное значение при улучшении когнитивных функций, таких как научение, память и внимание. Например, α7-никотиновый ацетилхолиновый рецептор вовлечен в следующие заболевания: легкие когнитивные расстройства, болезнь Альцгеймера, связанные с возрастом и другие когнитивные расстройства, шизофрения, синдром дефицита внимания, синдром дефицита внимания с гиперактивностью (ADHD), деменция, вызванная инъекцией или метаболическими нарушениями, деменция с тельцами Леви, судорожные припадки, такие как эпилептический припадок, множественные церебральные инфаркты, расстройства настроения, обсессивно-компульсивные и аддиктивные поведения, воспалительные заболевания и заболевания и нарушения, связанные с устранением боли, возникающей в результате этих нарушений. Активность α7-никотинового ацетилхолинового рецептора можно изменять или регулировать посредством введения лиганда α7- рецептора, и неограничивающие примеры лиганда α7-рецептора включают: антагонисты, агонисты, частичные агонисты и обратные агонисты. Лиганд α7-рецептора можно применять для лечения и профилактики этих и различных типов когнитивных расстройств и других состояний и заболеваний, и его агонисты и частичные агонисты, как известно, способны улучшать когнитивную функцию и внимание у грызунов, не являющихся человеком приматов и у людей.
На основе этого, согласно другому аспекту настоящее раскрытие обеспечивает применение лигандного соединения α7-никотинового ацетилхолинового рецептора при получении лекарственных препаратов для профилактики или лечения когнитивных расстройств.
Термин "когнитивное расстройство" в контексте настоящего документа относится к широкому диапазону нарушений когнитивных функций или когнитивных областей у животных, например, кратковременной памяти, внимания и активного внимания, вербального научения и памяти, визуального научения и памяти, мышления и решения проблем, и в частности, например, исполнительных функций, скорости обработки задач и/или социального познания. Когнитивное расстройство, как известно, проявляется в дефиците внимания, дезорганизованном мышлении, заторможенности мышления, трудности понимания, низком внимании, утрате способности решения проблем, неточной памяти, трудностях выражения мыслей и/или интеграции мыслей, восприятия и поведения или отказа от иррациональных мыслей.
Термин "лечение" можно рассматривать как включающий профилактику, подавление и облегчение (регрессию) заболевания, нарушения или состояния, связанных с когнитивным расстройством у животных, у которых никогда ранее не диагностировали такого расстройства, нарушения или состояния, обусловленных когнитивным расстройством, но которые являются подверженными такому заболеванию, нарушению или состоянию. Соответственно, термин "терапевтически эффективное количество" относится к клиническому обозначению эффективной дозы, необходимой для облегчения, уменьшения или профилактики симптомов подлежащего лечению заболевания, или эффективной дозе эффективного активного соединения, используемого для уменьшения или задержки начала появления таких симптомов, которую можно эмпирически определить в эксперименте на модели in vivo и/или in vitro подлежащего лечению заболевания.
Настоящее раскрытие, кроме того, обеспечивает лигандное соединение α7-никотинового ацетилхолинового рецептора, в котором галоген является меченным радиоактивным изотопом, для применения в качестве средства визуализации ПЭТ (позитронно-эмиссионной компьютерной томографии).
Настоящее раскрытие, кроме того, обеспечивает фармацевтическую композицию для профилактики или лечения когнитивных расстройств, где композиция содержит терапевтически эффективное количество описанного выше лигандного соединения α7-никотинового ацетилхолинового рецептора и фармацевтически приемлемый носитель.
В соответствии с некоторыми вариантами осуществления когнитивное расстройство выбрано из группы, состоящей из: пресенильной деменции, пресенильной болезни Альцгеймера, сенильной деменции, деменции альцгеймеровского типа, деменции с тельцами Леви, обусловленной микроинфарктами деменции, связанной со СПИДом деменции, связанной с ВИЧ деменции, связанной с тельцами Леви деменции, связанной с синдромом Дауна деменции, болезни Пика, умеренного когнитивного расстройства, связанного с возрастом нарушения памяти, нарушения краткосрочной памяти, связанного с возрастом когнитивного расстройства, связанного с лекарственными препаратами когнитивного расстройства, связанного с синдром иммунодефицита когнитивного расстройства, связанного с сосудистыми заболеваниями когнитивного расстройства, шизофрении, синдрома дефицита внимания, синдрома дефицита внимания с гиперактивностью (ADHD) и дефицита научения.
Различные лигандные соединения, представленные в настоящем раскрытие, обладают более высоким сродством к α7-никотиновому ацетилхолиновому рецептору, представляют собой отличные лигандные соединения α7-никотинового ацетилхолинового рецептора, и, кроме того, лигандное соединение α7-никотинового ацетилхолинового рецептора, представленное в настоящем раскрытие, после введения радиохимической метки, может быть использовано в качестве средства визуализации ПЭТ и отличается хорошим сродством, высокой специфичностью, высокой селективностью, умеренным накоплением в головном мозге и уровнем метаболизма, и обладает ценностью для клинического использования.
Краткое описание чертежей
Чтобы более ясно проиллюстрировать технические решения примеров настоящего раскрытия и предшествующего уровня техники, прилагаемые чертежи, которые должны быть использованы в примерах и уровне техники, кратко описаны ниже, и, очевидно, прилагаемые чертежи в последующем описании предназначены только для некоторых примеров настоящего раскрытия, но специалист в настоящей области техники может дополнительно получить другие сопроводительные чертежи согласно этим сопроводительным чертежам без творческих усилий.
На Фиг. 1 показана хроматограмма ВЭЖХ соединений [18F] II-15 и II-15.
На Фиг. 2 показана кривая специфического связывания для связывания между [125I] α-bgt и мембранным белком α7 nAChR.
На Фиг. 3 показан линейный график Хилла для связывания между [125I] α-bgt и мембранным рецепторным белком.
На Фиг. 4 показан линейный график Скэтчарда.
На Фиг. 5 показан график значений пробит Y и LogD(X).
На Фиг. 6 показана хроматограмма ВЭЖХ для анализа стабильности радиолиганда [18F] II-15 в фетальной бычьей сыворотке и физиологическом растворе.
На Фиг. 7 показаны результаты эксперимента по селективности соединения [18F] II-15 на головном мозге мышей.
На Фиг. 8 показаны виды изображений ПЭТ самок крыс CD-1.
Подробное описание вариантов осуществления
Настоящее раскрытие далее проиллюстрировано более подробно со ссылкой на прилагаемые чертежи и примеры, чтобы сделать задачи, технические решения и преимущества настоящего изобретения более ясными и понятными. Очевидно, что описанные примеры являются лишь некоторыми, но не всеми примерами настоящего раскрытия. Все другие примеры, полученные специалистом в настоящей области техники на основе примеров, приведенных в настоящем раскрытии, без применения творческих усилий, должны быть отнесены к объему защиты настоящего раскрытия.
Пример 1: Синтез 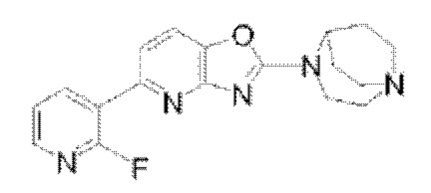 (обозначаемого как соединение I-9 для краткости)
(обозначаемого как соединение I-9 для краткости)
Путь синтеза соединения I-9 был следующим:
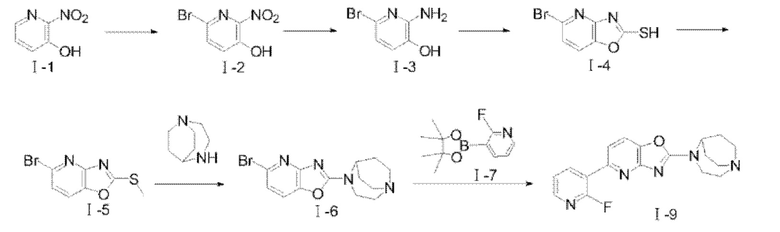
(1) Синтез соединения I-2
В чистую и сухую реакционную колбу добавляли соединение I-1 (5 г, 35,7 ммоля) и растворяли с помощью 50 мл метанола. После проведения трех замещений азотом метанольный раствор (15 мл) метоксида натрия (2,89 г, 53,5 ммоля) по каплям добавляли в реакционную систему с последующим перемешиванием при комнатной температуре в течение 30 мин; затем реакционную систему охлаждали до 0°С, по каплям добавляли бром (6,3 г, 39,4 ммоля), и температуру реакционной системы поддерживали при 0-5°С, и реакцию проводили в течение 4 часов; после того, как посредством ТСХ обнаружили, что реакция завершилась (CH2Cl2:СН3ОН=25:1), небольшое количество 10%-го водного раствора сульфита натрия использовали для прекращения реакции, и реакционную систему охлаждали до -15°С а затем перемешивали с выпадением в осадок твердого вещества желтого цвета. После фильтрования с отсасыванием фильтровальную лепешку промывали с применением холодного метанола (2×5 мл) и сушили с получением твердого вещества желтого цвета, а именно целевого продукта 1-2 (4,2 г, 53%). 1Н ЯМР (400 МГц, ДМСО) δ 7,85 (d, J=8,64 Гц, 1H), 7,66 (d, J=8,64 Гц, 1H); MS (М+Н+): m/z=216,97.
(2) Синтез соединения I-3
Соединение I-2 (1 г, 4,6 ммоля) добавляли в трехгорлую колбу и растворяли с помощью 20 мл этанола. После проведения трех замещений азотом порошок цинка (1,5 г, 22,9 ммоля) и хлорид аммония (2,46 г, 46 ммоля) добавляли в колбу, и снова проводили замещение азотом; реакцию проводили при 50°С в течение 16 ч. После того, как посредством ТСХ обнаружили, что реакция завершилась (CH2Cl2:СН3ОН=25:1), реакционный раствор фильтровали с проведением сбора фильтрата, и фильтрат концентрировали при пониженном давлении а затем очищали посредством колоночной хроматографии с применением простого петролейного эфира/этилацетата (5:1) с получением твердого вещества темно-серого цвета, а именно целевого продукта I-3 (524 мг, 60%). 1H ЯМР (400 МГц, ДМСО) δ 9,70 (s, 1H), 6,74 (d, J=7,8 Гц, 1H), 6,49 (d, J=7,8 Гц, 1H), 5,91 (s, 1H); MS (M+H+):m/z=l88,99.
(3) Синтез соединения I-5
В реакционную колбу добавляли соединение I-3 (0,3 г, 1,59 ммоля) и растворяли с помощью 10 мл этанола, а затем добавляли при перемешивании сероуглерод (2,42 г, 31,7 ммоля) и гидроксид калия (191 мг, 4,76 ммоля); реакцию проводили при 80-85°С в течение 16 часов. После того, как посредством ТСХ обнаружили, что реакция завершилась (CH2Cl2:СН3ОН=25:1), этанол и сероуглерод удаляли посредством концентрирования при пониженном давлении, добавляли воду для растворения, и значение рН системы доводили с помощью разбавленной соляной кислоты до приблизительно 3 с выпадением в осадок твердого вещества, которое фильтровали и сушили с получением 0,26 г неочищенного продукта соединения I-4, и непосредственно проводили последующую реакцию.
В трехгорлую реакционную колбу добавляли соединение I-4 (0,26 г, 1,13 ммоля) и карбонат калия (156 мг, 1,13 ммоля) и растворяли с помощью 10 мл DMF. После проведения трех замещений азотом реакционную систему охлаждали до 0°С; затем 176 мг метилиодида (1,24 ммоля) по каплям добавляли в реакционную колбу. После завершения добавления по каплям смесь перемешивали и подвергали реакции при 0°С в течение 10 мин а затем подвергали реакции при комнатной температуре в течение 3 ч; после того, как посредством ТСХ обнаружили, что реакция завершилась (CH2Cl2:СН3ОН=25:1), 60 мл воды и 20 мл этилацетата добавляли в реакционную систему, перемешивали в течение 5 мин а затем оставляли отстаиваться; органические фазы собирали, сушили с помощью безводного MgSO4, фильтровали и концентрировали при пониженном давлении с удалением растворителя и получением твердого вещества рыжевато-коричневого цвета I-5 (180 мг, 65%). 1Н ЯМР (400 МГц, ДМСО) δ 8,06 (d, J=8,4 Гц, 1H), 7,55 (d, J=8,4 Гц, 1H), 2,79 (s, 3Н); MS (M+H+):m/z=246,93.
(4) Синтез соединения I-6
В реакционную колбу 1,4-диазабицикло[3.2.2]нонан (200 мг, 1,58 ммоля), соединение I-5 (466 мг, 1,9 ммоля), триэтиламин (321 мг, 3,17 ммоля) и 10 мл изопропанола добавляли последовательно, систему нагревали до 100°С при перемешивании, и перемешивание продолжали в целях реакции всю ночь после того, как растворитель испарился; после того, как посредством ТСХ обнаружили, что реакция завершилась (этилацетат/метанол=10:1), этилацетат (содержащий 1% метанола и 1% триэтиламина) применяли в качестве элюента для очистки посредством колоночной хроматографии с получением твердого вещества светло-желтого цвета 1-6 (250 мг, 48,8%). 1Н ЯМР (400 МГц, ДМСО) δ 7,68 (d, J=8,12 Гц, 1H), 7,15 (d, J=8,12 Гц, 1H), 4,43-4,42 (m, 1H), 3,90 (t, J=5,56 Гц, 2Н), 3,09 (t, J=5,76 Гц, 2Н), 3,04-2,95 (m, 4Н), 2,10-2,07 (m, 2Н), 1,84-1,76 (m, 2Н); MS (M+H+):m/z=323,905.
(5) Синтез соединения I-9
В реакционную колбу добавляли соединение I-6 (100 мг, 0,31 ммоля), соединение I-7 (70 мг, 0,314 ммоля), карбонат цезия (303 мг, 0,93 ммоля) и Pd(dppf)Cl2 (45 мг, 0,062 ммоля). После того, как реакционную систему трижды подвергали замещению азотом, добавляли дважды перегнанный 1,4-диоксан (10 мл); реакцию проводили при 85-90°С, и после того, как реакция завершилась, растворитель удаляли посредством концентрирования при пониженном давлении, и этилацетат/метанол (10:1) применяли в качестве элюента для очистки посредством колоночной хроматографии с получением продукта в виде твердого вещества светло-желтого цвета, а именно соединения I-9 (89,4 мг, 85%). 1Н ЯМР (400 МГц, CDCl3) δ 8,72 (td, J=8,82 Гц, 2 Гц, 1H), 8,18 (dt, J=l,76 Гц, 4,32 Гц, 1H), 7,61 (dd, J=8,24 Гц, 1,32 Гц, 1H), 7,49 (d, J=8,2 Гц, 1H), 7,30 (td, J=5,94 Гц, 2,04 Гц, 1H), 4,61 (s, 1H), 3,99 (m, 2Н), 3,21-3,13 (m, 4Н), 3,06-2,99 (m, 2Н), 2,18-2,16 (m, 2Н), 1,88-1,80 (m, 2Н); 13С ЯМР (100 МГц, CDCl3) δ 163,54, 161,83, 159,45, 158,78, 146,72, 146,02, 141,72, 141,05, 122,63, 122,37, 122,01, 116,37, 114,92, 56,96, 50,73, 46,32, 44,33, 26,82; MS (M+H+):m/z=340,15.
Пример 2: Синтез 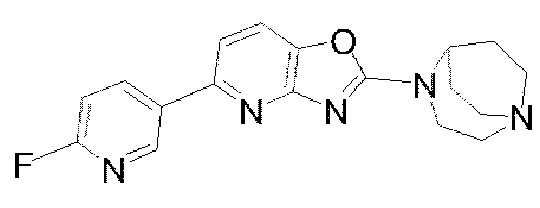 (обозначаемого как соединение I-10 для краткости)
(обозначаемого как соединение I-10 для краткости)
Путь синтеза соединения I-10 был следующим:
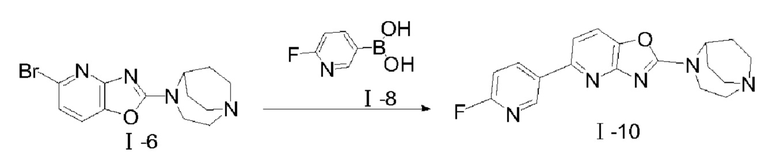
В реакционную колбу добавляли соединение I-6 (100 мг, 0,31 ммоля), соединение I-8 (44 мг, 0,313 ммоля), карбонат цезия (303 мг, 0,93 ммоля) и Pd(dppf)Cl2 (45 мг, 0,062 ммоля). После того, как реакционную систему трижды подвергали замещению азотом, добавляли дважды перегнанный 1,4-диоксан (10 мл); реакцию проводили при 85-90°С, после того, как реакция завершилась, растворитель удаляли посредством концентрирования при пониженном давлении, и этилацетат/метанол (10:1) применяли в качестве элюента для очистки посредством колоночной хроматографии с получением твердого продукта 1-10 желтого цвета (53 мг, 50%). 1Н ЯМР (400 МГц, CDCl3) δ 8,80 (d, J=2,32 Гц, 1H), 8,52 (td, J=7,8 Гц, 2,52 Гц, 1H), 7,50 (d, J=8,16 Гц, 1H), 7,35 (d, J=8,12 Гц, 1H), 6,99 (dd, J=8,52 Гц, 2,8 Гц, 1H), 4,65 (s, 1H), 4,03 (t, J=5,36 Гц, 2Н), 3,25-3,19 (m, 4Н), 3,10-3,04 (m, 2Н), 2,22 (m, 2Н), 1,93-1,86 (m, 2Н); 13С-ЯМР (100 МГц, CDCl3) δ 164,88, 163,48, 162,49, 158,88, 149,03, 145,90, 141,07, 139,96, 133,43, 115,37, 112,24, 109,51, 109,14, 55,83, 50,57, 46,30, 26,43, 8,70; MS (M+H+):m/z=340,15.
Пример 3: Синтез соединения 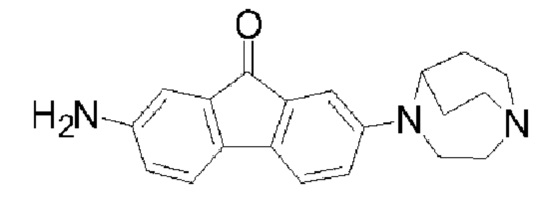 (обозначаемого как соединение II-5 для краткости)
(обозначаемого как соединение II-5 для краткости)
Путь синтеза соединения II-5 был следующим:
(1) Синтез соединения II-2
В реакционную колбу объемом 1000 мл добавляли 30,0 г 9-флуоренона (II-1) (0,17 моля) и 100 мл воды и нагревали до 80-85°С, затем бром (32,0 г, 0,2 моля) по каплям добавляли в реакционную колбу в течение 30 мин. После завершения добавления по каплям реакцию продолжали при 80-85°С в течение 4 ч; после того, как реакционную систему охлаждали до комнатной температуры, 300 мл воды добавляли в реакционную систему, чтобы погасить реакцию, затем 300 мл 10%-ого раствора NaHSO3 добавляли в реакционную систему, и после 30 мин перемешивания твердые вещества светло-желтого цвета получали посредством фильтрования с отсасыванием; твердые вещества светло-желтого цвета растворяли в 300 мл этанола, затем дефлегмировали и перемешивали при 80°С в течение около 16 ч, охлаждали до комнатной температуры и подвергали фильтрованию с отсасыванием с получением твердого вещества светло-желтого цвета, которое сушили в вакууме при 50°С с получением 2-бром-9-флуоренона (II-2) (39,1 г, 90,4%). 1H ЯМР (400 МГц, CDCl3) δ 7,76 (d, J=l,76 Гц, 1H), 7,66 (d, J=7,32 Гц, 1H), 7,61 (dd, J=7,88 Гц, 1,76 Гц, 1H), 7,51-7,50 (m, 2Н), 7,39 (d, J=7,88 Гц, 1H), 7,34-7,30 (m, 1H); MS (M+H+):m/z=260,98.
(2) Синтез соединения II-3
В реакционную колбу объемом 1000 мл добавляли 23,4 г 2-бром-9-флуоренона (II-2) (0,09 моля) и 180 мл воды и нагревали до 80-85°С, а затем перемешанный раствор 180 мл 65%-ого раствора азотной кислоты (2,6 моля) и 180 мл 98%-ого раствора серной кислоты (3,3 моля) по каплям добавляли в реакционную систему в течение приблизительно 30 мин; реакционную систему нагревали до 110-120°С, дефлегмировали в течение 4 ч, и после охлаждения до комнатной температуры добавляли к ней 300 мл воды, чтобы погасить реакцию, и посредством фильтрации получали твердое вещество желтого цвета; после того, как полученное твердое вещество промывали водой (3×100 мл), 200 мл метанола добавляли для взбивания, промывки и фильтрации со сбором твердого вещества, которое затем сушили в вакууме при 50°С с получением 2-бром-7-нитро-9-флуоренона (II-3) (21,0 г, 76,4%). 1Н ЯМР (400 МГц, CDCl3) δ 8,49 (s, 1H), 8,44 (d, J=8,16 Гц, 1H), 7,90 (s, 1H), 7,75 (d, J=7,92 Гц, 1H), 7,71 (d, J=8,16 Гц, 1H), 7,55 (d, J=7,92 Гц, 1H); MS (M+H+):m/z=306,02.
(3) Синтез соединения II-4
В реакционную колбу объемом 500 мл 2-бром-7-нитро-9-флуоренона (II-3) (9,0 г, 0,03 моля), Pd2(dba)3 (0,6 г, 0,655 ммоля), BINAP (1,3 г, 2,087 ммоля), трет-бутоксида натрия (3,3 г, 0,0343 моля), 1,4-диазабицикло[3.2.2]нонан (3,0 г, 0,024 моля) и 1,4-диоксан (150 мл) добавляли последовательно. После проведения трех замещений азотом реакционную систему нагревали до 80-85°С, и реакцию проводили в атмосфере азота в течение 16 ч; после того, как реакция завершилась, 150 мл этилацетата добавляли, а затем нерастворимые вещества удаляли посредством фильтрации через воронку, футерированную диатомитом; 10%-ый раствор K2CO3 добавляли к фильтрату для доведения значения рН до приблизительно 8,0, затем фильтрат экстрагировали с помощью CH2Cl2 до тех пор, пока водная фаза не стала бесцветной, и органические фазы объединяли, сушили с помощью безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении с получением продукта синевато-фиолетового цвета (II-4) (2,1 г, 20,4%). Этот продукт мог непосредственно применяться для следующей реакции. 1Н ЯМР (400 МГц, CDCl3) δ 7,56 (d, J=7,24 Гц, 1H), 7,38 (d, J=6,56 Гц, 1H), 7,34 (d, J=2,8 Гц, 1H), 7,14-7,09 (m, 2Н), 6,78 (d, J=8,16 Гц, 1H), 4,13-4,11 (m, 1H), 3,67-3,65 (m, 1H), 3,61-3,58 (m, 1H), 3,15-3,14 (m, 4Н), 3,04 (m, 2Н), 2,14 (m, 2Н), 1,78 (m, 2Н); MS (M+H+):m/z=350,16.
(4) Синтез соединения II-5
В реакционную колбу последовательно добавляли соединение II-4 (2,1 г, 6,02 ммоля), порошок железа (1,7 г, 0,03 моля), этанол (80 мл), воду (54 мл) и концентрированную соляную кислоту (1,8 мл) и нагревали до 80°С, и посредством ТСХ обнаруживали завершение реакции исходных веществ (приблизительно 5 ч); значение рН реакционной системы доводили до приблизительно 8,0 с помощью 10%-ого раствора K2CO3, 150 мл этилацетата добавляли, и смесь фильтровали с помощью воронки, футерированной диатомитом, фильтровальную лепешку промывали этилацетатом, органические фазы объединяли, растворитель удаляли посредством концентрирования при пониженном давлении, а затем твердое вещество растворяли в CH2Cl2 и промывали посредством добавления воды. Органические фазы получали посредством разделения, сушили с помощью безводного Na2SO4, фильтровали и концентрировали при пониженном давлении с получением конечного продукта II-5 (1,5 г, 80%). 1Н ЯМР (400 МГц, CDCl3) δ 7,12 (d, J=8,24 Гц, 1H), 7,08 (d, J=7,92 Гц, 1H), 7,01 (d, J=2,44 Гц, 1H), 6,88 (d, J=2,2 Гц, 1H), 6,70 (dd, J=8,24 Гц, 2,52 Гц, 1H), 6,64 (dd, J=7,88 Гц, 2,24 Гц, 1H), 4,01 (m, 2Н), 3,51-3,48 (m, 2Н), 3,13-3,06 (m, 4Н), 3,01-2,94 (m, 2Н), 2,12-2,05 (m, 2Н), 1,75-1,67 (m, 2Н); 13С ЯМР (101 МГц, CDCl3) δ 194,18, 148,14, 144,99, 135,02, 134,79, 134,56, 132,85, 119,00, 118,94, 117,11, 110,32, 108,94, 55,59, 50,91, 45,26, 43,25, 25,50; MS (M+H+):m/z=320,0.
Пример 4: Синтез соединения 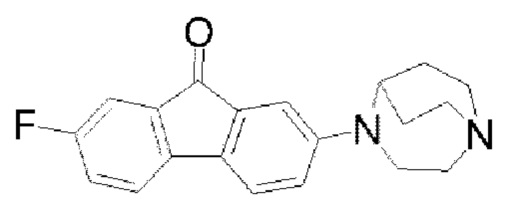 (обозначаемого как соединение II-6 для краткости)
(обозначаемого как соединение II-6 для краткости)
Путь синтеза соединения II-6 был следующим:
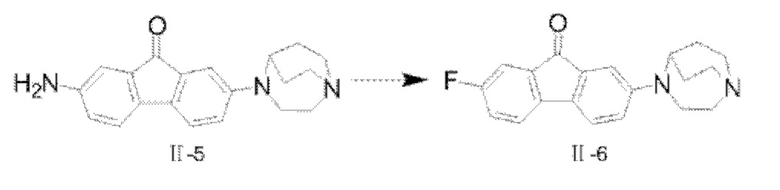
В реакционную колбу добавляли соединение II-5 (1,9 г, 6 ммоля) и тетрафтороборную кислоту (30 мл), и реакционную колбу помещали на ледяную баню с охлаждением реакционной системы до 0-5°С, а затем водный раствор нитрита натрия (0,5 г, 7,2 ммоля, 15 мл) по каплям добавляли в реакционную систему в течение 15 мин; после того как добавление по каплям было завершено, реакцию продолжали в течение 30 мин, затем перемешивание останавливали, и проводили фильтрование с отсасыванием. Фильтровальную лепешку промывали сначала с помощью 15 мл этанола, затем промывали с помощью 15 мл метил-трет-бутилового простого эфира, наконец взбивали и промывали с помощью метил-трет-бутилового простого эфира и фильтровали с получением твердого вещества; твердое вещество переносили в реакционную колбу и нагревали до 120°С. Проводили обнаружение посредством ТСХ, и после того, как исходные вещества были полностью превращены, реакционную систему охлаждали до комнатной температуры, 50 мл CH2Cl2. добавляли для растворения, смесь фильтровали с удалением нерастворимого твердого вещества, и фильтровальную лепешку взбивали и промывали с помощью 50 мл CH2Cl2. Органические фазы собирали, сушили с помощью безводного Na2SO4, а затем фильтровали и концентрировали при пониженном давлении с удалением растворителя; неочищенный продукт очищали с помощью колоночной хроматографии с применением дихлорметана и метанола для градиентного элюирования (CH2Cl2:СН3ОН=25:1) с получением твердого вещества фиолетового цвета, а именно конечного продукта II-6 (1,2 г, 67,3%). 1H ЯМР (400 МГц, CDCl3) δ 7,24-7,19 (m, 3Н), 7,05 (d, J=2,28 Гц, 1H), 7,02 (dd, J=8,4 Гц, 2,4 Гц, 1H), 6,74 (dd, J=8,32 Гц, 2,56 Гц, 1H), 4,0437-4,0391 (m, 1H), 3,55-3,52 (m, 2Н), 3,15-3,07 (m, 4Н), 3,01-2,94 (m, 2Н), 2,12-2,05 (m, 2Н), 1,77-1,68 (m, 2Н); 13СЯМР (101 МГц, CDCl3) δ 192,51 (d, J=2,2 Гц), 162,49 (s), 160,03 (s), 148,88 (s), 140,55 (d, J=2,9 Гц), 130,56 (s), 120,13 (s), 119,86 (s), 119,63 (s), 118,92 (s), 118,85 (s), 116,74 (s), 110,86 (s), 110,63 (s), 108,67 (s), 55,97 (s), 50,73 (s), 45,48 (s), 43,49 (s), 25,76 (s); 19FflMP (376 МГц, CDCl3) δ -115,21 (s); MS (M+H+):m/z=323,2.
Пример 5: Синтез соединения 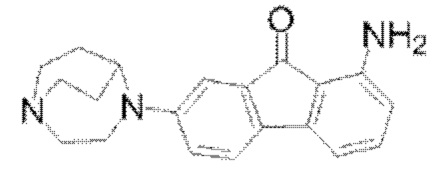 (обозначаемого как соединение II-14 для краткости)
(обозначаемого как соединение II-14 для краткости)
Путь синтеза соединения II-14 был следующим:
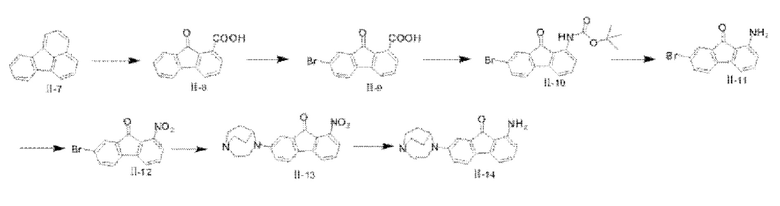
(1) Синтез соединения II-8
Триоксид хрома (138,0 г, 1,38 моля) растворяли в перемешанном растворе 120 мл воды и 80 мл уксусной кислоты и перемешивали до полного растворения для последующего использования; в реакционную колбу добавляли 40,0 г флуорантена (II-7) (0,2 моля) и 500 мл уксусной кислоты, и реакционную систему нагревали до 80-85°С, затем раствор триоксида хрома по каплям добавляли в реакционную систему, температуру системы поддерживали при 80-85°С. После того, как добавление по каплям было завершено, реакционную систему нагревали до 110-120°С, охлаждали до комнатной температуры после 2 часов реакции, и реакционный раствор переливали в 3 л воды, большое количество твердого вещества желтого цвета выпало в осадок. После фильтрования с отсасыванием твердое вещество растворяли в 600 мл 2 М раствора NaOH, подвергали фильтрованию с отсасыванием с удалением нерастворимых примесей, и фильтровальную лепешку промывали с помощью 500 мл воды; метил-трет-бутиловый простой эфир добавляли для промывания водной фазы, и водную фазу получали посредством разделения. Значение рН водной фазы доводили до приблизительно 1,0, применяя концентрированную соляную кислоту, твердое вещество желтого цвета снова выпадало в осадок, и его сушили в вакууме при 60°С после фильтрования с отсасыванием с получением целевого продукта II-8 (9-флуоренон-1-карбоновой кислоты) (29,5 г, 66,0%). 1Н ЯМР (400 МГц, CDCl3) δ 8,18 (d, J=7,52 Гц, 1H), 7,74-7,53 (m, 5Н), 7,36 (t, J=7,16 Гц, 1H); MS (M+H+):m/z=225,10.
(2) Синтез соединения II-9
В реакционную колбу 9-флуоренон-1-карбоновую кислоту (II-8) (15,0 г, 0,067 моля) и 300 мл воды добавляли. Реакционную систему нагревали до 80-85°С, затем 10 мл Br2 (0,23 моля) по каплям добавляли в реакционную систему. После того, как завершение по каплям было завершено, реакцию продолжали в течение 16 часов. После того, как посредством ТСХ обнаружили, что реакция завершилась (CH2Cl2:СН3ОН=10:1), 300 мл 10%-ого водного раствора бисульфита натрия добавляли в реакционную систему с последующим перемешиванием в течение 30 мин, с получением при фильтрации твердого вещества желтого цвета. Твердое вещество сушили в вакууме при 50°С с получением конечного продукта II-9 (19,5 г, 96,5%). 1H ЯМР (400 МГц, CDCl3) δ 8,22 (d, J=7,04 Гц, 1H), 7,85 (s, 1H), 7,73-7,66 (m, 3Н), 7,43 (d, J=7,84 Гц, 1H).
(3) Синтез соединения II-10
В условиях перемешивания в реакционную колбу последовательно добавляли соединение II-9 (6 г, 19,87 ммоля), толуол (60 мл), триэтиламин (4,1 мл, 29,8 ммоля), DPPA (6,4 мл, 29,8 ммоля) и трет-бутанол (10 мл), затем реакционную систему нагревали до 110°С и подвергали реакции дефлегмации. После того, как посредством ТСХ обнаружили, что реакция завершилась (исходные вещества, СН2С2:СН3ОН=5:1; промежуточное соединение, простой петролейный эфир : этилацетат = 5:1), растворитель удаляли с последующим очищением с помощью колонки с силикагелем (соотношение простой петролейный эфир: этилацетат составляло 30:1) с получением твердого вещества желтого цвета, а именно продукта II-10 (5,5 г, 74%). 1Н ЯМР (400 МГц, CDCl3) δ 8,15 (d, J=8,56 Гц, 1H), 7,71 (d, J=l,72 Гц, 1H), 7,59 (dd, J=7,88 Гц, 1,8 Гц, 1H), 7,42 (dd, J=7,44 Гц, 8,36 Гц, 1H), 7,37 (d, J=7,92 Гц, 1H), 7,09 (d, J=7,16 Гц, 1H), 1,55 (s, 9Н).
(4) Синтез соединения II-11
Соединение II-10 (808 мг, 2,16 ммоля) растворяли в 40 мл ацетонитрила, в раствор добавляли 22 мл 1 М соляной кислоты (21,6 ммоля) и нагревали для реакции при 82°С в течение 4 ч. После того, как посредством ТСХ обнаружили, что реакция завершилась, реакционную систему охлаждали до комнатной температуры, в реакционную систему добавляли количество 1 М раствора NaOH, необходимое для достижения сильнощелочного значения рН, с последующей экстракцией с помощью этилацетат со сбором органических фаз, которые сушили с помощью безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении с удалением растворителя и получением твердого вещества желтого цвета, а именно продукта II-11 (500 мг, 84,5%). 1Н ЯМР (400 МГц, CDCl3) δ 7,71 (d, J=1,4 Гц, 1H), 7,55 (dd, J=7,8 Гц, 1H), 7,35 (d, J=7,9 Гц, 1H), 7,22 (t, J=7,6 Гц, 1H), 6,81 (d, J=7,l Гц, 1H), 6,52 (d, J=8,4 Гц, 1H), 5,54 (s, 2Н); MS (M+H+):m/z=273,99.
(5) Синтез соединения II-12
В трехгорлую колбу объемом 25 мл добавляли 1,729 мл 30%-ого пероксида водорода (16,95 ммоля). После того, как трехгорлую колбу охлаждали до 0°С на ледяной бане, раствор дихлорметана (2 мл) и ангидрида трифторуксусной кислоты (2,736 мл, 19,68 ммоля) по каплям добавляли в колбу в атмосфере аргона. После того, как добавление по каплям было завершено, реакционную систему продолжали перемешивать при 0°С в течение 1,5 ч (реакционная смесь из бесцветной превратилась в смесь коричневого цвета); затем раствор дихлорметана (4 мл) и соединения II-11 (500 мг, 1,824 ммоля) по каплям добавляли в реакционную систему, и реакционную систему нагревали до комнатной температуры после завершения добавления по каплям, с последующим перемешиванием и проведением реакции в течение 1 ч. После того, как посредством ТСХ обнаружили, что реакция завершилась, необходимое количество воды добавляли, чтобы погасить реакцию, с последующей экстракцией с помощью дихлорметана со сбором органических фаз, которые сушили с помощью безводного сульфата натрия, фильтровали и подвергали ротационному выпариванию при пониженном давлении с удалением растворителя, с последующей очисткой с помощью колоночной хроматографии на силикагеле (простой петролейный эфир : этилацетат = 10:1) с получением твердого вещества желтого цвета II-12 (212 мг, 38,2%). 1H ЯМР (400 МГц, CDCl3) δ 8,32 (d, J=8,4 Гц, 1H), 7,75 (d, J=l,8 Гц, 1H), 7,64 (dd, J=7,9 Гц, 1H), 7,54 (t, J=7,6 Гц, 1H), 7,41 (d, J=7,9 Гц, 1H), 7,31 (d, J=7,4 Гц, 1H).
(6) Синтез соединения II-13
Соединение II-12 (100 мг, 0,329 ммоля), 1,4-диазабицикло[3.2.2]нонан (125 мг, 0,989 ммоля) и карбонат цезия (426 мг, 1,31 ммоля) растворяли в 2 мл дважды перегнанного безводного толуола для последующего применения; Pd2(dba)3 (30 мг, 0,033 ммоля) и (±)-BINAP (61 мг, 0,099 ммоля) растворяли в 2 мл дважды перегнанного безводного толуола в атмосфере аргона. После перемешивания при 90°С в течение 15 мин (реакционная смесь превратилась из мутной жидкости темно-фиолетового цвета в прозрачную жидкость оранжево-желтого цвета) смесь охлаждали до комнатной температуры; затем предшествующий раствор добавляли в реакционную систему после перемешивания при 60°С в течение 14 ч (реакционная смесь превратилась из прозрачной жидкости оранжево-желтого цвета в жидкость коричневато-красного цвета) в атмосфере аргона, реакционную систему охлаждали до комнатной температуры, реакцию гасили посредством добавления 10 мл воды, с последующей экстракцией с помощью дихлорметана со сбором органических фаз, которые сушили с помощью безводного сульфата натрия, фильтровали и подвергали ротационному выпариванию при пониженном давлении с удалением растворителя, с последующей очисткой с помощью колоночной хроматографии на силикагеле (CH2Cl2:СН3ОН=20:1) с получением твердого вещества фиолетово-черного цвета II-13 (48 мг, 41,8%). 1Н ЯМР (400 МГц, CDCl3) δ 7,49-7,52 (m, 2Н), 7,36 (d, J=7,9 Гц, 2Н), 7,10 (s, 1H), 6,82 (t, J=7,6 Гц, 1H), 4,13 (s, 1H), 3,62-3,63 (m, 2Н), 3,04-3,18 (m, 6Н), 2,31 (s, 2Н), 1,82-1,83 (m, 2Н); MS (M+H+):m/z=350,37.
(7) Синтез соединения II-14
В реакционную колбу соединение II-12 (2,1 г, 6,02 ммоля), порошок железа (1,7 г, 0,03 моля), этанол (80 мл), воду (54 мл) и концентрированную соляную кислоту (1,8 мл) добавляли последовательно и нагревали до 80°С для реакции в течение 5 ч; после того, как посредством ТСХ хроматографии обнаружили, что реакция исходных веществ завершилась, значение рН доводили до приблизительно 8,0 с применением 10%-ого раствора K2CO3, 150 мл этилацетата добавляли с последующей фильтрацией через воронку, футерированную диатомитом, объединением органических фаз и удалением растворителя посредством концентрирования при пониженном давлении, затем твердое вещество растворяли в CH2Cl2 и промывали посредством добавления воды с последующим разделением с получением органических фаз, которые сушили с помощью безводного Na2SO4, фильтровали и концентрировали при пониженном давлении с получением конечного продукта II-14 (1,5 г, 80%). 1Н ЯМР (400 МГц, CDCl3) δ 7,30 (d, J=8,28 Гц, 1H), 7,16-7,12 (m, 1H), 7,06 (d, J=2,48 Гц, 1H), 6,75 (dd, J=8,28 Гц, 2,52 Гц, 1H), 6,67 (d, J=7,04 Гц, 1H), 6,35 (d, J=8,28 Гц, 1H), 5,43 (s, 2Н), 4,10 (s, 1H), 3,58 (t, J=5,48 Гц, 2Н), 3,19-3,13 (m, 3Н), 3,07-3,00 (m, 2Н), 2,14-2,11 (m, 2Н), 1,80-1,74 (m, 3Н); 13С ЯМР (100 МГц, CDCl3) δ 195,66 (s), 150,20 (s), 147,18 (s), 137,02-136,73 (m), 136,37 (s), 131,22 (s), 121,42 (s), 116,55 (s), 115,39 (s), 108,58 (s), 108,48 (s), 57,06 (s), 51,87 (s), 46,56 (s), 44,64 (s), 26,91 (s); MS (M+H+):m/z=320,16.
Пример 6: Синтез соединения 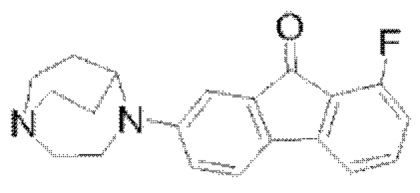 (обозначаемого как соединение II-15 для краткости)
(обозначаемого как соединение II-15 для краткости)
Путь синтеза соединения II-15 был следующим:
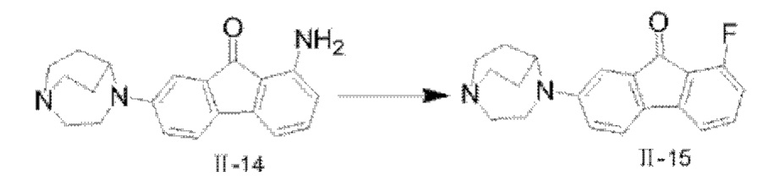
В реакционную колбу добавляли соединение II-14 (1,3 г, 4,07 ммоля) и тетрафтороборную кислоту (20 мл). Реакционную колбу помещали на ледяную баню, и после того, как температура понизилась до 0-5°С, раствор NaNO2 (0,4 г, 5,8 ммоля, 10 мл) по каплям добавляли в реакционную систему, и реакцию проводили в течение 30 мин после того, как добавление по каплям было завершено; перемешивание останавливали, проводили фильтрование с отсасыванием, фильтровальную лепешку промывали сначала с помощью 15 мл этанола, затем промывали с помощью 15 мл метил-трет-бутилового простого эфира, наконец взбивали и промывали с помощью метил-трет-бутилового простого эфира и фильтровали с получением твердого вещества; твердое вещество переносили в реакционную колбу, нагревали до 120°С, охлаждали до комнатной температуры после того, как посредством ТСХ обнаружили, что реакция завершилась. 50 мл дихлорметана добавляли для растворения, с последующим фильтрованием с удалением нерастворимых твердых веществ, фильтровальную лепешку взбивали и снова промывали с помощью 50 мл дихлорметана, с последующим объединением органических фаз, концентрированием при пониженном давлении с удалением растворителя и получением неочищенного продукта, который очищали посредством колоночной хроматографии с применением дихлорметана и метанола (25:1) с получением твердого вещества фиолетового цвета II-15 (446 мг, 34,0%). 1Н ЯМР (400 МГц, CDCl3) δ 7,39-7,34 (m, 1H), 7,32 (d, J=8,36 Гц, 1H), 7,11 (d, J=7,36 Гц, 1H), 7,09 (d, J=2,48 Гц, 1H), 6,80-6,74 (m, 2H), 4,10 (s, 1H), 3,60 (t, J=5,76 Гц, 2H), 3,19-3,12 (m, 4H), 3,05-2,99 (m, 2H), 2,15-2,10 (m, 2H), 1,82-1,73 (m, 2H); 13C ЯМР (101 МГц, CDCl3) δ 191,17 (s), 158,08 (s), 150,54 (s), 137,22 (s), 137,14 (s), 135,82 (s), 130,97 (s), 121,76 (s), 117,27 (s), 115,33 (s), 115,12 (s), 114,98 (s), 109,26 (s), 56,85 (s), 51,62 (s), 46,36 (s), 44,39 (s), 29,70 (s), 26,65 (s); 19F ЯМР (376 МГц, CDCl3) δ -114,11 (s); MS (M+H+):m/z=323,2.
Пример 7: Синтез соединения 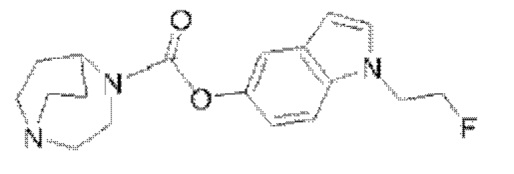 (обозначаемого как соединение III-5 для краткости)
(обозначаемого как соединение III-5 для краткости)
Путь синтеза соединения III-5 был следующим:
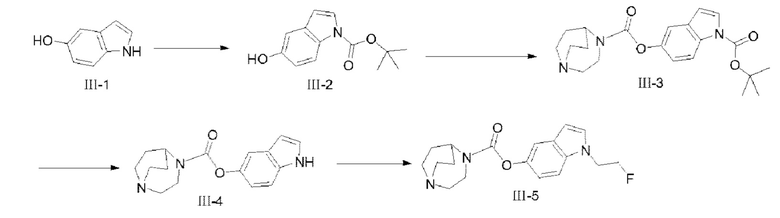
(1) Синтез соединения III-2
5-гидроксииндол (III-1) (10 г, 75,1 ммоля) растворяли в 100 мл ацетонитрила, и ди-трет-бутилдикарбонат (49,2 г, 225,3 ммоля) и DMAP (917 мг, 7,51 ммоля) добавляли в раствор с последующим перемешиванием при комнатной температуре в течение 15 ч; после того, как реакция завершилась, реакционный раствор концентрировали при пониженном давлении, 400 мл метанола добавляли для растворения, затем K2CO3 (51,9 г, 375,5 ммоля) добавляли, с последующим перемешиванием при комнатной температуре в течение 4 ч; после того, как реакция завершилась, значение рН доводили до нейтрального значения с применением уксусной кислоты, с последующим разбавлением посредством добавления Н2О, экстракцией органических фаз (этилацетат применяли в качестве экстрагирующего средства), удалением растворителя и разделением посредством колоночной хроматографии (простой петролейный эфир : этилацетат = 3:1) с получением продукта, т.е. соединения III-2 (12,5 г, 71%). 1Н ЯМР (400 МГц, CDCl3) δ 7,99 (d, J=6,96 Гц, 1H), 7,56 (d, J=2,84 Гц, 1H), 6,98 (d, J=2,44 Гц, 1H), 6,85-6,82 (m, 1H), 6,46 (d, J=3,64 Гц, 1H), 1,66 (s, 9Н); MS (M-H+):m/z=232,l 1.
(2) Синтез соединения III-3
Соединение III-2 (3,2 г, 13,7 ммоля) растворяли в 20 мл CH2Cl2, диизопропилэтиламин (1,77 г, 13,7 ммоля) растворяли в 120 мл тетрагидрофурана, оба раствора смешивали, затем смешанный раствор медленно добавляли к раствору CH2Cl2 (100 мл) трифосгена (1,3 г, 4,38 ммоля), и после перемешивания при комнатной температуре в течение 1 ч раствор CH2Cl2 и 1,4-диазабицикло[3.2.2]нонана (1,72 г, 13,7 ммоля) медленно добавляли в реакционную систему, и реакцию проводили при комнатной температуре в течение 4 ч; после того, как реакция завершилась, H2O добавляли для разбавления с последующей экстракцией с помощью CHCl3, сбором органических фаз, которые промывали насыщенным водным раствором NaCl, сушили с помощью безводного Na2SO4, с удалением растворителя и получением неочищенного продукта; разделение посредством колоночной хроматографии проводили с помощью CHCl3 и СН3ОН (90:10) в качестве элюентов с получением продукта III-3 (2,4 г, 45%). 1Н ЯМР (400 МГц, CDCl3) δ 8,11 (d, J=7,6 Гц, 1H), 7,60 (s, 1H), 7,30 (m, 1H), 7,05 (d, J=8,96 Гц, 1H), 6,52 (d, J=3,2 Гц, 1H), 4,52-4,41 (m, 1H), 3,89 (t, J=5,48 Гц, 1H), 3,78 (t, J=5,68 Гц, 1H), 3,20-3,06 (m, 6Н), 2,14-2,11 (m, 2Н), 1,82-1,72 (m, 2Н), 1,66 (s, 9Н); MS (M+H+):m/z=386,21.
(3) Синтез соединения III-4
Соединение III-3 (2,4 г, 6,23 ммоля) растворяли в 20 мл CH2Cl2, и после того, как температура реакционного раствора уменьшилась до 0°С, 10 мл трифторуксусной кислоты добавляли в реакционную систему, с последующим перемешиванием при 30°С в течение 2 ч; после того, как реакция завершилась, реакционный раствор концентрировали, разбавляли посредством добавления воды и экстрагировали с помощью CH2Cl2; значение рН водной фазы доводили до значения 8-9 с помощью насыщенного раствора NaHCO, затем экстрагировали с помощью CH2Cl2, сушили с помощью безводного сульфата натрия с удалением растворителя и получением продукта III-4 (360 мг, 20%). 1Н ЯМР (400 МГц, ДМСО) δ 11,16 (s, 1H), 7,38-7,26 (m, 3Н), 6,84 (d, J=8,52 Гц, 1H), 6,40 (s, 1H), 4,53-4,33 (m, 1H), 3,95 (m, 1H), 3,79 (m, 1H), 3,30 (m, 6Н), 2,20-2,10 (m, 2Н), 1,95-1,93 (m, 2Н); MS (M+H+):m/z=286,15.
(4) Синтез соединения III-5
При 0°С гидрид натрия (59 мг, 1,48 ммоля) добавляли в безводный раствор DMF (3 мл) и соединения III-4 (350 мг, 1,23 ммоля). Через 20 мин перемешивания безводный раствор DMF (3 мл) и 1-фтор-2-иодэтана (321 мг, 1,85 ммоля) медленно добавляли в реакционную систему, а затем реакцию проводили при комнатной температуре в течение 2 ч; после того, как реакция завершилась, раствор хлорида аммония добавляли в реакционную систему для разбавления с последующей экстракцией с помощью этилацетата, сбором органических фаз и очищением посредством колоночной хроматографии с применением CHCl3 и СН3ОН (95:5, с содержанием 20 капель водного раствора аммиака) с получением целевого продукта III-5 (50 мг, 12,3%). 1Н ЯМР (400 МГц, CDCl3) δ 7,36-7,34 (m, 1H), 7,26 (d, J=8,76 Гц, 1H), 7,14 (d, J=3,04 Гц, 1H), 6,98 (d, J=8,76 Гц, 1H), 6,48 (d, J=3,0 Гц, 1H), 4,72 (t, J=4,88 Гц, 1H), 4,61 (t, J=4,88 Гц, 1H), 4,51 (m, 1H), 4,39 (t, J=4,88 Гц, 1H), 4,32 (t, J=4,88 Гц, 1H), 3,87 (t, J=5,72 Гц, 1H), 3,76 (t, J=5,8 Гц, 1H), 3,17-3,01 (m, 6Н), 2,13-2,10 (m, 2Н), 1,80-1,69 (m, 2Н); MS (M+H+):m/z=332,19.
Пример 8: Синтез соединения 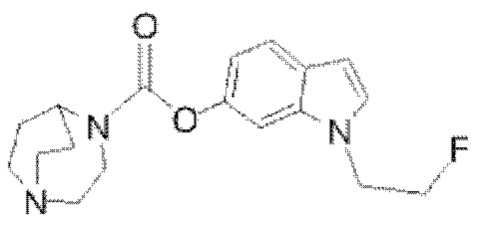 (обозначаемого как соединение III-12 для краткости)
(обозначаемого как соединение III-12 для краткости)
Путь синтеза соединения III-12 был следующим:
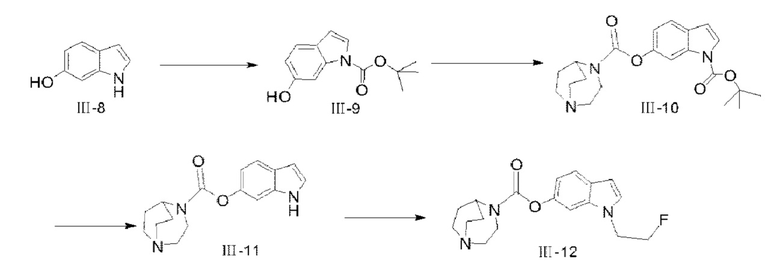
(1) Синтез соединения III-9
6-гидроксииндол (III-8) (10 г, 75,1 ммоля) растворяли в 100 мл ацетонитрила, и ди-трет-бутилдикарбонат (49,2 г, 225,3 ммоля) и DMAP (917 мг, 7,51 ммоля) добавляли в раствор с последующим перемешиванием при комнатной температуре в течение 15 ч; после того, как реакция завершилась, реакционный раствор концентрировали при пониженном давлении, 400 мл метанола добавляли для растворения, затем K2CO3 (51,9 г, 375,5 ммоля) добавляли с последующим перемешиванием при комнатной температуре в течение 4 ч; после того, как реакция завершилась, значение рН доводили с помощью СН3СООН до нейтрального значения, с последующим разбавлением посредством добавления Н2О и экстракцией со сбором органических фаз (этилацетат в качестве экстрагирующего средства); после удаления растворителя проводили разделение посредством колоночной хроматографии (простой петролейный эфир : этилацетат = 3:1) с получением продукта III-9 (12,5 г, 71%). 1Н ЯМР (400 МГц, CDCl3) δ 7,68 (s, 1H), 7,45 (d, J=3,16 Гц, 1H), 7,39 (d, J=8,4 Гц, 1H), 6,79 (dd, J=8,4 Гц, 2,2 Гц, 1H), 6,48 (d, J=3,68 Гц, 1H); MS (M-H+):m/z=232,11.
(2) Синтез соединения III-10
Соединение III-9 (3,2 г, 13,7 ммоля) растворяли в 20 мл CH2Cl2, диизопропилэтиламин (1,77 г, 13,7 ммоля) растворяли в 120 мл тетрагидрофурана, и оба раствора смешивали, затем смешанный раствор медленно добавляли к раствору CH2Cl2 (100 мл) трифосгена (1,3 г, 4,38 ммоля), и после перемешивания при комнатной температуре в течение 1 ч раствор CH2Cl2 и 1,4-диазабицикло[3.2.2]нонана (1,72 г, 13,7 ммоля) медленно добавляли в реакционную систему, и реакцию проводили при комнатной температуре в течение 4 ч; после того, как реакция завершилась, H2O добавляли для разбавления с последующей экстракцией с помощью CHCl3 со сбором органических фаз, которые промывали насыщенным водным раствором NaCl, сушили с помощью безводного NaSO4 с удалением растворителя и получением неочищенного продукта; разделение посредством колоночной хроматографии проводили с применением CHCl3 и СН3ОН (90:10) в качестве элюентов с получением продукта III-10 (2,4 г, 45%). 1Н ЯМР (400 МГц, CDCl3) δ 7,97 (s, 1H), 7,54 (d, J=3 Гц, 1H), 7,50 (d, J=8,44 Гц, 1H), 7,02 (d, J=8,44 Гц, 1H), 6,54 (d, J=3,64 Гц, 1H), 4,50-4,40 (m, 1H), 3,85 (t, J=5,64 Гц, 1H), 3,77 (t, J=5,8 Гц, 1H), 3,18-3,02 (m, 6Н), 2,13-2,10 (m, 2Н), 1,81-1,70 (m, 2Н), 1,65 (s, 9Н); MS (M+H+):m/z=386,21.
(3) Синтез соединения III-11
Соединение III-10 (2,4 г, 6,23 ммоля) растворяли в 20 мл CH2Cl2, после того, как реакцию охладили до 0°С, 10 мл трифторуксусной кислоты добавляли в реакционную систему с последующим перемешиванием при 30°С в течение 2 ч; после того, как реакция завершилась, реакционный раствор концентрировали, разбавляли посредством добавления воды и экстрагировали с помощью CH2Cl2; водную фазу доводили до значения рН 8-9 с помощью насыщенного раствора NaHCO3, затем экстрагировали с помощью CH2Cl2, сушили с помощью безводного NaSO4 с удалением растворителя и получением продукта III-11 (360 мг, 20%). 1Н ЯМР (400 МГц, CDCl3) δ 8,27 (s, 1H), 7,57 (d, J=8,52 Гц, 1H), 7,17 (d, J=2,36 Гц, 1H), 6,87 (d, J=8,48 Гц, 1H), 6,52 (m, 1H), 4,54-4,44 (m, 1H), 3,90 (t, J=5,72 Гц, 1H), 3,80 (t, J=5,72 Гц, 1H), 3,22-3,08 (m, 1H), 2,17-2,13 (m, 2Н), 1,84-1,74 (m, 2Н); MS (M+H+):m/z=286,16.
(4) Синтез соединения III-12
При 0°С гидрид натрия (59 мг, 1,48 ммоля) добавляли в безводный раствор DMF (3 мл) и соединения III-11 (350 мг, 1,23 ммоля), через 20 мин перемешивания безводный раствор DMF (3 мл) и 1-фтор-2-иодэтана (321 мг, 1,85 ммоля) медленно добавляли в реакционную систему, а затем реакцию проводили при комнатной температуре в течение 2 ч; после того, как реакция завершилась, раствор NH4Cl добавляли в реакционную систему для разбавления, с последующей экстракцией этилацетатом со сбором органических фаз и очищением посредством колоночной хроматографии с применением CHCl3 и СН3ОН (95:5, с содержанием 20 капель водного раствора аммиака) с получением целевого продукта III-12 (50 мг, 12,3%). 1H ЯМР (400 МГц, CDCl3) δ 7,57 (d, J=8,48 Гц, 1H), 7,13-7,11 (m, 2Н), 6,90-6,87 (m, 1H), 6,51 (d, J=3,04 Гц, 1H), 4,75 (t, J=4,84 Гц, 1H), 4,63 (t, J=4,92 Гц, 1H), 4,51 (m, 1H), 4,38 (t, J=4,92 Гц, 1H), 4,31 (t, J=4,88 Гц, 1H), 3,87 (t, J=5,64 Гц, 1H), 3,77 (t, J=5,68 Гц, 1H), 3,18-3,01 (m, 6Н), 2,13-2,10 (m, 2Н), 1,81-1,69 (m, 2Н); MS (M+H+):m/z=332,18.
Пример 9: Синтез соединения 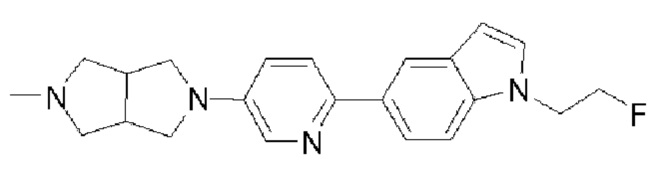 (обозначаемого как соединение III-19 для краткости)
(обозначаемого как соединение III-19 для краткости)
Путь синтеза соединения III-19 был следующим:
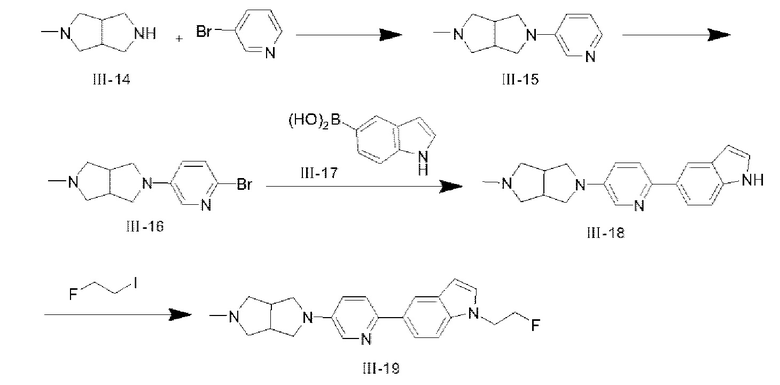
(1) Синтез соединения III-15
Соединение III-14 (2 г, 15,85 ммоля), 3-бромпиридин (3 г, 18,99 ммоля), Pd2(dba)3 (320 мг, 0,35 ммоля), rac-BINAP (660 мг, 1,06 ммоля) и трет-бутоксид натрия (1,83 г, 19,04 ммоля) растворяли в 20 мл толуола и нагревали для реакции при 85°С в течение 16 ч; после того, как реакция завершилась, реакционную систему охлаждали и концентрировали при пониженном давлении с удалением растворителя, и неочищенный продукт разделяли с помощью колоночной хроматографии с применением CH2Cl2:СН3ОН (0,1% NH3⋅H2O)=10:1 с получением твердого вещества коричневого цвета, а именно продукта III-15 (2,2 г, 68%). 1Н ЯМР (400 МГц, CDCl3) δ 8,05 (d, J=2,4 Гц, 1H), 7,98 (d, J=4,52 Гц, 1H), 7,12-7,09 (m, 1H), 6,90 (d, J=8,28 Гц, 1H), 3,43-3,39 (m, 2Н), 3,23-3,20 (m, 2Н), 3,0025-2,9960 (m, 2Н), 2,76-2,72 (m, 2Н), 2,50-2,47 (m, 2Н), 2,34 (s, 3Н); MS (M+H+):m/z=204,15.
(2) Синтез соединения III-16
При 0°С соединение III-15 (2,2 г, 10,82 ммоля) растворяли в 80 мл ацетонитрила, затем ацетонитрильный раствор (10 мл) N-бромсукцинимида (1,93 г, 10,82 ммоля) по каплям добавляли в реакционную систему в течение 30 мин с последующим перемешиванием в течение 30 мин, а затем реакционную систему нагревали до комнатной температуры; реакцию гасили с помощью 40 мл воды, СН2О2 (2×40 мл) добавляли для экстракции, органические фазы собирали, промывали насыщенным раствором NaCl, сушили с помощью безводного NaSO4, концентрировали при пониженном давлении с удалением растворителя, а затем неочищенный продукт очищали с помощью колоночной хроматографии с помощью CH2Cl2:СН3ОН (0,1% NH3⋅H2O)=15:1 с получением твердого вещества светло-коричневого цвета, а именно целевого продукта III-16 (1,4 г, 46%). 1Н ЯМР (400 МГц, CDCl3) δ 7,74 (s, 1H), 7,23 (d, J=8,68 Гц, 1H), 6,78 (d, J=8,68 Гц, 1H), 3,43-3,39 (m, 2Н), 3,18-3,16 (m, 2Н), 3,00 (m, 2Н), 2,69-2,66 (m, 2Н), 2,54-2,51 (m, 2Н), 2,34 (s, 3Н); MS (M+H+):m/z=282,06.
(3) Синтез соединения III-18
Соединение III-16 (1,35 г, 4,78 ммоля), соединение III-17 (1,16 г, 7,21 ммоля) и Pd2(dba)3 (553 мг, 0,48 ммоля) растворяли в 40 мл 1,4-диоксана с последующим перемешиванием в течение 10 мин, затем 10 мл раствора карбоната натрия (1,93 г, 18,21 ммоля) добавляли в реакционную систему; реакционную систему нагревали до 115°С и дефлегмировали в течение 2 ч; после того, как реакция завершилась, реакционную систему охлаждали, и смешанный растворитель вода/этилацетат (50:50) добавляли в реакционную систему; твердое вещество собирали посредством фильтрации и промывали с помощью 30 мл этилацетата с получением твердого вещества светло-желтого цвета, а именно целевого продукта III-18 (1,0 г, 66%). 1H ЯМР (400 МГц, CDCl3) δ 8,23 (s, 1H), 8,16 (m, 2Н), 7,81 (d, J=8,48 Гц, 1H), 7,63 (d, J=8,68 Гц, 1H), 7,44 (d, J=8,52 Гц, 1H), 7,22 (s, 1H), 7,03-6,99 (m, 1H), 6,60 (s, 1H), 3,47-3,43 (m, 2Н), 3,29-3,27 (m, 2Н), 3,01 (m, 2Н), 2,79-2,75 (m, 2Н), 2,50-2,47 (m, 2Н), 2,35 (s, 3Н); MS (M+H+):m/z=319,19.
(4) Синтез соединения III-19
При 0°С 94 мг гидрида натрия (2,35 ммоля) добавляли к безводному раствору DMF (3 мл) и соединения III-18 (500 мг, 1,57 ммоля) с последующим перемешиванием в течение 30 мин, затем безводный раствор DMF (3 мл) и 1-фтор-2-иодэтана (546 мг, 3,14 ммоля) медленно добавляли в реакционную систему, ледяную баню удаляли, и реакционную систему нагревали до комнатной температуры, и реакцию проводили в течение 2 ч; после того, как реакция завершилась, реакционную систему разбавляли раствором NH4Cl, экстрагировали посредством добавления CH2Cl2 со сбором органических фаз и разделяли посредством колоночной хроматографии с применением CH2Cl2:СН3ОН (0,1%NH3⋅H2O)=15:1 с получением целевого продукта III-19 (180 мг, 31%). 1Н ЯМР (400 МГц, CDCl3) δ 8,16 (s, 2Н), 7,85 (d, J=8,6 Гц, 1H), 7,64 (d, J=8,64 Гц, 1H), 7,37 (d, J=8,64 Гц, 1H), 7,16 (d, J=2,8 Гц, 1H), 7,03-7,00 (m, 1H), 6,58 (d, J=2,88 Гц, 1H), 4,80 (t, J=4,84 Гц, 1H), 4,68 (t, J=4,84 Гц, 1H), 4,47 (t, J=4,92 Гц, 1H), 4,40 (t, J=4,8 Гц, 1H), 3,45-3,41 (m, 2Н), 3,31-3,29 (m, 2Н), 3,05 (m, 2Н), 2,88-2,87 (m, 2Н), 2,52-2,50 (m, 2Н), 2,40 (s, 3Н); 13С ЯМР (100 МГц, CDCl3) δ 147,67, 143,13, 135,99, 135,71, 131,92, 129,22, 128,68, 121,21, 120,68, 120,28, 118,62, 109,19, 102,66, 83,21, 81,53, 63,13, 54,49, 42,37, 41,80; MS (M+H+):m/z=365,21.
Пример 10: Синтез соединения 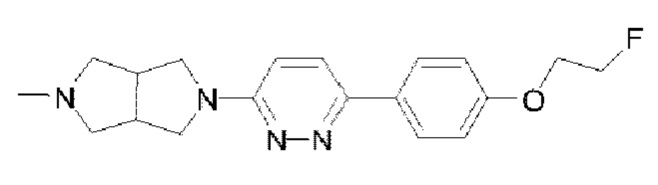 (обозначаемого как соединение III-24 для краткости)
(обозначаемого как соединение III-24 для краткости)
Путь синтеза соединения III-24 был следующим:
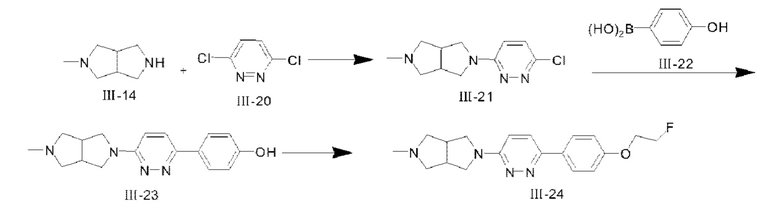
(1) Синтез соединения III-21
2,22 мл DIEPA (13,4 ммоля) и соединения III-14 (847 мг, 6,71 ммоля) добавляли к н-октанольному раствору (20 мл) соединения III-20 (1 г, 6,71 ммоля), и реакцию проводили при 120°С всю ночь; после того, как реакция завершилась, растворитель удаляли посредством концентрирования при пониженном давлении, и неочищенный продукт очищали посредством колоночной хроматографии с применением CH2Cl2:СН3ОН (2М NH3⋅Н2О)=4:1 в качестве элюента, с получением твердого вещества белого цвета, а именно целевого продукта III-21 (1,2 г, 75%). 1H ЯМР (400 МГц, CDCl3) δ 7,165 (d, J=9,36 Гц, 1H), 6,656 (d, J=9,4 Гц, 1H), 3,72-3,68 (m, 2Н), 3,47 (m, 2Н), 3,06 (m, 2Н), 2,76-2,72 (m, 2Н), 2,59-2,57 (m, 2Н), 2,366 (s, 3Н); MS (M+H+):m/z=239,11.
(2) Синтез соединения III-23
При комнатной температуре Pd2(dba)3 (421 мг, 0,46 ммоля) и 1,3-бис(2,6-диизопропилфенил)имидазолия хлорид (587 мг, 1,38 ммоля) добавляли к перемешанному 1,4-диоксановому раствору (80 мл) соединений III-21 (1,1 г, 4,61 ммоля) и 111-22 (763 мг, 5,53 ммоля); после проведения трех замещений азотом 20% Na2CO3 (10 мл, 18,4 ммоля) добавляли к раствору, и снова проводили замещение азотом (4 раза); реакцию проводили при 85°С в атмосфере азота в течение 19 ч, и после того, как реакция завершилась, реакционную систему охлаждали; 200 мл этилацетата добавляли в реакционную систему с последующей фильтрацией с применением диатомита; фильтрат собирали, и растворитель удаляли посредством концентрирования, и неочищенный продукт очищали посредством колоночной хроматографии с применением CH2Cl2:СН3ОН (2М NH3⋅H2O)=9:1 с получением твердого вещества рыжевато-коричневого цвета, а именно целевого продукта III-23 (850 мг, 62%). 1H ЯМР (400 МГц, CDCl3) δ 7,596 (d, J=8,2 Гц, 2Н), 7,231 (m, 1H), 6,722 (d, J=8,24 Гц, 2Н), 6,388 (d, J=9,44 Гц, 1H), 3,72-3,70 (m, 2Н), 3,68-3,59 (m, 2Н), 3,093 (m, 2Н), 2,88-2,86 (m, 2Н), 2,67-2,63 (m, 2Н), 2,407 (s, 3Н); MS (M+H+):m/z=297,16.
(3) Синтез соединения III-24
K2CO3 (652 мг, 4,72 ммоля) и соединение III-23 (400 мг, 1,35 ммоля) добавляли последовательно в N,N-диметилформамидный (10 мл) раствор 1-фтор-2-иодэтана (351 мг, 2,02 ммоля); смесь перемешивали при комнатной температуре всю ночь, и после того, как реакция завершилась, к смеси добавляли воду и этилацетат с разделением органических фаз и водной фазы. Водную фазу дважды экстрагировали этилацетатом, и органические фазы собирали, сушили с помощью безводного Na2SO4 и фильтровали с удалением растворителя; неочищенный продукт отделяли посредством колоночной хроматографии с применением CH2Cl2:СН3ОН (2М NH3⋅H2O)=9:1 с получением целевого продукта III-24 (160 мг, 34,6%). 1H ЯМР (400 МГц, CDCl3) δ 7,947 (d, J=8,68 Гц, 2Н), 7,587 (d, J=9,4 Гц, 1H), 7,014 (d, J=8,72 Гц, 2Н), 6,751 (d, J=9,4 Гц, 1H), 4,844 (t, J=3,92 Гц, 1H), 4,726 (t, J=4,08 Гц, 1H), 4,304 (t, J=3,96 Гц, 1H), 4,234 (t, J=4,16 Гц, 1H), 3,76-3,72 (m, 2Н), 3,60-3,57 (m, 2Н), 3,116 (m, 2Н), 2,96-2,90 (m, 2Н), 2,60-2,58 (m, 2Н), 2,431 (s, 3Н); 13С ЯМР (100 МГц, CDCl3) δ 158,21, 148,87, 139,36, 133,07, 121,35, 120,55, 120,09, 118,33, 110,13, 82,70, 81,01, 63,01, 54,79, 42,25, 41,70; MS (M+H+):m/z=343,19.
Пример 11: Синтез радиолиганда 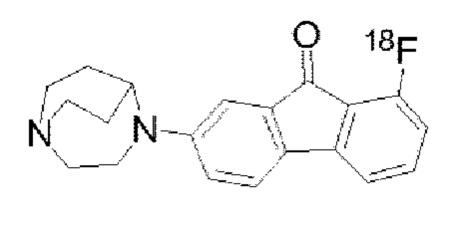 называемого [18F] II-15 для краткости)
называемого [18F] II-15 для краткости)
Путь синтеза [18F] II-15 был следующим:
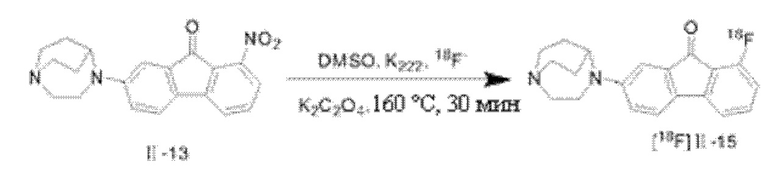
15 мг Kryptofix 222 растворяли в 0,7 мл безводного ацетонитрила, 2 мг K2C2O4 растворяли в 0,3 мл воды, а затем оба раствора однородно смешивали с получением 1,0 мл элюирующего раствора Kryptofix 222/K2C2O4; 18F-, зафиксированный на колонке QMA, элюировали в реакционную колбу, применяя элюирующий раствор, растворитель в реакционной колбе сушили продуванием потока N2 при 100°С, затем 0,5 мл безводного ацетонитрила добавляли в колбу, смесь сушили посредством повторного продувания, этот процесс трижды повторяли с обеспечением полного удаления влаги в реакционной колбе; безводный раствор ДМСО (0,3 мл) соединения II-13, подлежащего введению метки, (2 мг) быстро добавляли в вышеуказанную реакционную колбу, запаивали, и проводили реакцию при 160°С в течение 30 мин; после того, как реакция завершилась, дистиллированную воду (2×10 мл) добавляли, чтобы погасить реакцию, и реакционный раствор отсасывали шприцом, так чтобы пропустить через заранее активированную колонку для твердофазной экстракции Sep-Pak С18; затем продукт реакции элюировали с колонки С18 с помощью 2 мл ацетонитрила, элюат собирали и концентрировали при пониженном давлении с удалением растворителя, после того, как необходимое количество ацетонитрила добавляли для растворения, очистку посредством разделения с помощью радио-ВЭЖХ проводили с применением подвижной фазы ацетонитрил вода (с содержанием 0,2% ацетата аммония) = 28:72 при скорости потока 4 мл/мин и длине волны 280 нм. В качестве полупрепаративной колонки применяли полупрепаративную колонку Inertsil® с обращенной фазой ODS-3 типа С18 (GL Sciences, Inc. 5 мкм, 10 мм × 250 мм).
После очистки посредством разделения с помощью радио-ВЭЖХ, радиолиганд [18F] II-15 обладал радиохимической чистотой более 98% и степенью введения радиоактивной метки приблизительно 13,1% (не учитывая поправку на распад); очищенный радиолиганд [18F] II-15 и стабильное соединение без введенной метки II-15 совместно впрыскивали для ВЭЖХ анализа с подвижной фазой ацетонитрил : вода (с содержанием 0,2% ацетата аммония) = 30:70, скоростью потока 1 мл/мин, длиной волны 280 нм, аналитической колонкой Agela Technologies, Venusil ХВР C18 (L), 5 мкм, 150A, 4,6×250 мм. Результаты анализа показаны на Фиг. 1, где время удерживания [18F] II-15 и II-15 составляет 14,037 мин и 13,466 мин, соответственно, таким образом, время удерживания обоих соединений совпадает, подтверждая точность радиолиганда.
Пример 12: In vitro анализ конкурентного связывания лигандного соединения
(1) Эксперимент по насыщению связывания
а) Получение рецепторного белка и измерение его концентрации Все рецепторные белки, использованные в экспериментальной методике, были выделены и экстрагированы из головного мозга самок крыс линии Спрег-Доули (180-200 г). После того, как самок крыс линии Спрег-Доули (180-200 г) умерщвляли цервикальной дислокацией, их головной мозг быстро вынимали и помещали на ледяные блоки, а после того, как прожилки крови смывали ледяным физиологическим раствором, кору головного мозга вырезали (это область была обогащена рецепторными белками α7 nAChR) и помещали в ледяной 50 мМ буферный раствор трис-HCl 10-ти кратного объема (50 мМ Трис, 120 мМ NaCl, 5 мМ KCl, 2 мМ CaCl2, 1 мМ MgCl2, рН=7,4, 4°С), а затем лабораторный стакан помещали на ледяную баню, и смесь гомогенизировали с помощью ручного гомогенизатора тканей в течение 30 с (параметр №6). Гомогенизированный мембранный раствор разделяли на три равные части в центрифужные пробирки, объемом 50 мл, центрифугировали на низкотемпературной высокоскоростной центрифуге в течение 20 мин (4°С, 48000 g), супернатант отбрасывали после завершения центрифугирования, нижние осадки растворяли в ледяном 50 мМ буферном растворе трис-HCl 10-ти кратного объема, и смесь гомогенизировали, центрифугировали и промывали таким же образом. Нижние осадки, полученные после повторения стадий 3 раза, представляли собой мембранные рецепторные белки, которые растворяли в ледяном 50 мМ буферном растворе трис-HCl 10-ти кратного объема и гомогенизировали так, чтобы они были полностью и равномерно смешаны. Извлекали 10 мкл равномерно смешанного раствора мембранных рецепторных белков, измеряли концентрацию белков по методу Лоури, а оставшийся мембранный раствор снова упаковывали в центрифужную пробирку объемом 2 мл и хранили в холодильнике при -80°С для последующего использования.
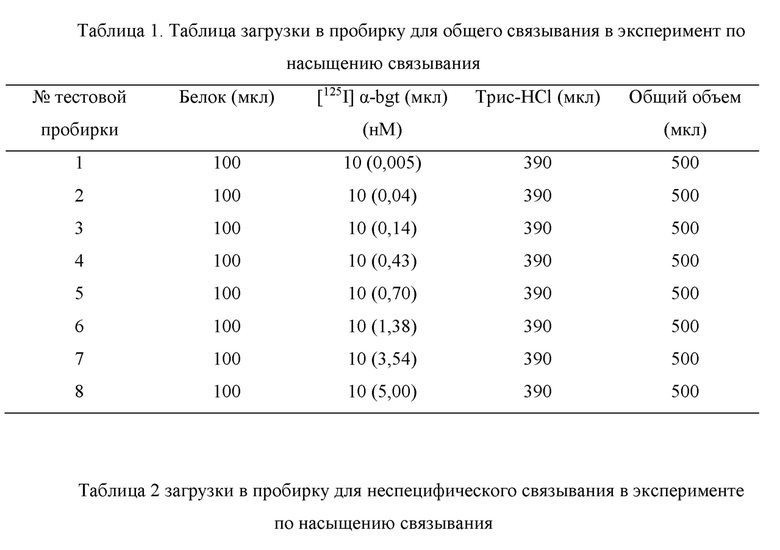
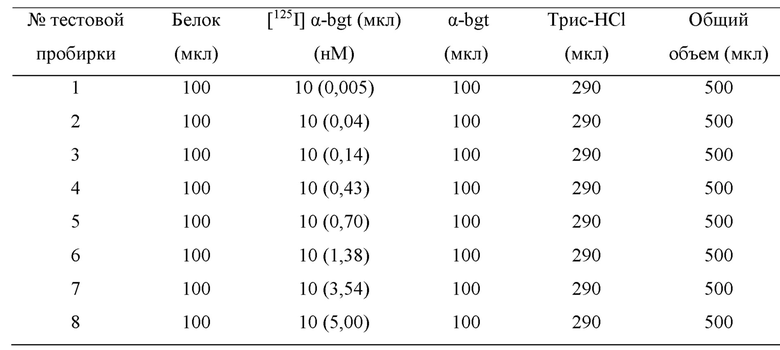
b) Эксперимент по насыщению связывания мембранных рецепторных белков Эксперимент по насыщению связывания проводили посредством измерения связывания радиолиганда [1251] α-бунгаротоксин (обозначаемого [125I] α-bgt для краткости) с мембранными белками головного мозга мышей. В этом эксперименте радиолиганд [1251] α-bgt применяли при 8 различных точках концентрации (0,005-5 нМ), причем каждая точка концентрации имела 3 параллельные группы. Акцепторные мембранные белки, которые хранились при -80°С в холодильнике, вынимали, подвергали замораживанию-оттаиванию при 4°С, и после замораживания-оттаивания подходящий объем ледяного 50 мМ буферного раствора Трис-HCl (50 мМ Трис, 120 мМ NaCl, 5 мМ KCl, 2 мМ CaCl2, 1 мМ MgCl2, рН 7,4, 4°С) добавляли для разбавления в зависимости от измеряемой концентрации белка. Общий объем реакционной смеси в пробирке для общего связывания составлял 500 мкл, включая 100 мкл раствора мембранного белка (конечное количество белков в каждой пробирке составляло 1,5 мг), 10 мкл радиолиганда [125I] α-bgt при различных концентрациях и 390 мкл ледяного 50 мМ буферного раствора Трис-HCl, загруженных в следующем порядке: мембранные белки, буферный раствор Трис-HCl и [125I] α-bgt (Таблица 1). Неспецифическое связывание определяли посредством 2 мкМ немеченого α-bgt, и реакционная смесь в тестовой пробирке включала 100 мкл раствора мембранного белка (конечное количество белка в каждой тестовой пробирке составляло 1,5 мг), 10 мкл радиолиганда [125I] α-bgt при различных концентрациях, 100 мкл 2 мкМ α-bgt и 290 мкл ледяного 50 мМ буферного раствора Трис-HCl, в общем объеме 500 мкл, загруженных в следующем порядке: мембранные белки, буферный раствор Трис-HCl, α-bgt и [125I] α-bgt (Таблица 2). После завершения загрузки тестовую пробирку закрывали герметизирующей пленкой, встряхивали в течение нескольких секунд, чтобы хорошо перемешать содержимое, а затем помещали в инкубатор с постоянной температурой 37°С для инкубации в течение 2,5 часов. После завершения инкубации тестовую пробирку вынимали и помещали на ледяную баню, чтобы прекратить связывание между рецепторным белком и лигандом, затем смешанный раствор фильтровали с применением 48-луночного коллектора клеток на фильтровальную бумагу Whatman GF/B (предварительно пропитанную 0,5%-ным раствором полиэтиленимина в течение 2,5 ч), фильтровальную бумагу промывали 5 мл ледяного 50 мМ буферного раствора Трис-HCl 3 раза, фильтровальную бумагу удаляли, и лист фильтровальной бумаги отрезали и помещали в РЕ пробирку для измерения, чтобы измерить отчеты с помощью γ-счетчика. Разницей между общим связыванием (ТВ) и неспецифическим связыванием (NSB) было специфическое связывание (SB), т.е. SB (cpm) = ТВ (cpm) -NSB (cpm).
с) Экспериментальные результаты
В эксперименте по насыщению связывания 8 различных концентраций (0,005-5 нМ) были установлены для радиолиганда [125I] α-bgt, и 3 группы измеряли параллельно для каждой концентрации. Экспериментальные результаты показали, что в измеренном диапазоне концентраций специфическое связывание радиолиганда [125I] α-bgt с мембранными рецепторными белками быстро достигало насыщения, и кривая специфического связывания является такой, как показано на Фиг. 2; линейный график Хилла получали путем нанесения log[B/(Bmax-B)] на график относительно log[L] согласно кривой специфического связывания и уравнению log[B/(Bmax-B)]=nHlog[L]-logKd (как показано на Фиг. 3): у=1,05828х+0,08731 (R=0,99031), ее наклон представлял собой коэффициент Хилла nH=1,058, указывающий на то, что связыванием между [125I] α-bgt и акцепторными мембранными белками представляло собой простую систему моносайтового действия.
Линейное уравнение регрессии у=46,42857-1,2987х (R=1) получали посредством нанесения на график B/F относительно количества специфического связывания В согласно уравнению Скэтчарда системы моносайтового действия: B/F=-B/Kd+Bmax/Kd (как показано на Фиг. 4). Согласно этому уравнению было получено, что при данных экспериментальных условиях равновесная константа диссоциации радиолиганда [1251] α-bgt составляла Kd=0,77±0,088 нМ (95%-ый доверительный интервал составлял 0,36-1,172 нМ), максимальное количество связывания составляло Bmax=35,75±4,64 фмоль/мг белка (95%-ый доверительный интервал 29,88-41,62 фмоль/мг белка). Экспериментальный результат соответствовал данным, представленным в литературе (Kd=1,5±0,7 нМ, Bmax=63±17 пмоль/мг белка), указывая на то, что используемый здесь метод измерения заслуживает доверия и может использоваться для измерения биологической активности соединения, подлежащего измерению.
(2) Эксперимент по конкурентному связыванию
а) Экспериментальный метод
Чтобы количественно оценить сродство лигандного соединения к рецепторам α7 nAChR, провели in vitro эксперимент по конкурентному связыванию с [125I] α-bgt в качестве радиоактивного стандарта. В эксперименте раствор мембранного белка (количество белков в каждой реакционной пробирке составляло 1,5 мг) инкубировали вместе с 0,4 нМ раствором радиолиганда [125I] α-bgt и серией растворов лиганда без введенной метки при 8 различных концентрациях (для каждой концентрации были измерены 3 группы параллельно) в инкубаторе с постоянной температурой 37°С в течение 2,5 ч, и после того, как инкубация была завершена, выполнялись та же процедура, которая описана в предыдущем эксперименте по насыщению связывания, и подсчет с помощью γ-счетчика. В то же время, чтобы обеспечить точность и надежность этой экспериментальной системы, было измерено сродство MLA (известный высокоселективный, высокоаффинный лиганд для рецепторов α7 nAChR) в качестве эталонного лиганда с применением рецепторов α7 nAChR головного мозга мышей. Способ приготовления и способ загрузки лигандного соединения показаны в следующей Таблице 3 и Таблице 4.


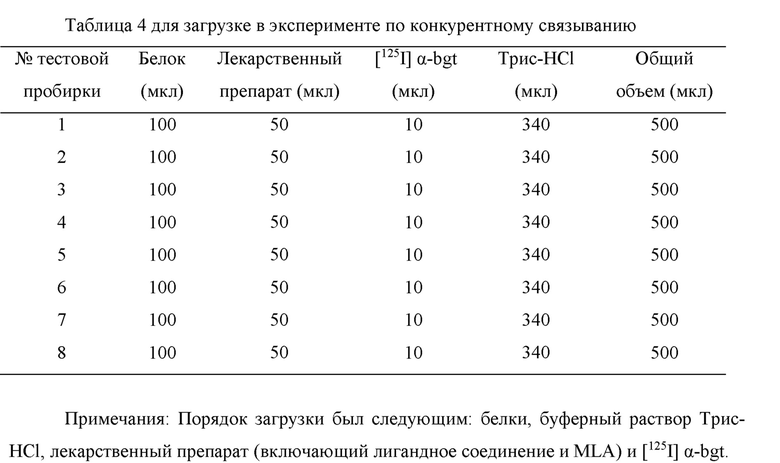
b) Экспериментальные результаты
Чтобы провести сравнение, MLA был выбран в качестве эталонного лиганда, и сродство MLA и разработанного соединения, подлежащего тестированию, к рецепторам α7 nAChR было измерено одновременно при одних и тех же экспериментальных условиях.
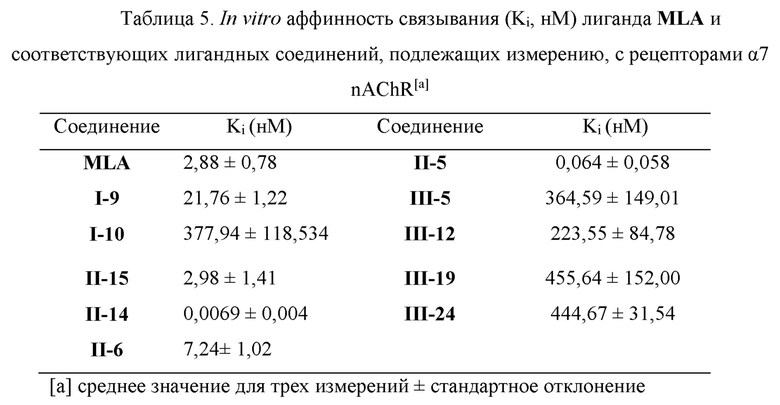
В эксперименте по конкурентному связыванию эталонный лиганд MLA и ряд соединений, подлежащих тестированию, получали при 8 различных концентрациях (10-6-10-14 моль/л для соединения II-14 и 10-5-10-13 моль/л для соединения П-5, и 10-3-10-10 моль/л для оставшихся соединений), их значение IC50 измеряли посредством ингибирования связывания 0,4 нМ [125I] α-bgt (Kd=0,77±0,088 нМ) с рецепторами α7 nAChR, и соответствующие значения Ki получали посредством вычисления по формуле Ченга-Прусоффа (Ki=IC50/(1+[L]/Kd)). Результаты измерения показаны в Таблице 5. При этих экспериментальных условиях константа ингибирования Ki лиганда MLA составляла Ki=2,88±0,78 нМ, что по существо близко к значению Ki (1,09±0,09 нМ), представленному в литературе, указывая на осуществимость экспериментального способа, принятого в настоящем документе.
На основании Таблицы 5 видно, что каждое лигандное соединение показало сродство к мембранным белкам α7 nAChR с константой ингибирования (Ki), распределенной в диапазоне 0,005-450 нМ, где соединения II-14, II-5, II-15, II-6 и I-9 показали довольно сильный ингибирующий эффект на [125I] α-бунгаротоксин, значения Ki которых составляли 0,0069±0,004 нМ, 0,064±0,058 нМ, 2,98±1,41 нМ, 7,24±1,02 нМ и 21,76±1,22 нМ соответственно, что указывает на что они имеют довольно высокое сродство к α7 nAChR, особенно сродство соединения II-14 (Ki=0,0069±0,004 нМ) превысило самое высокое значение (Ki=0,023 нМ) лигандных молекул того же типа, которые существуют в настоящее время.
Пример 13. In vitro эксперимент по ингибированию калиевого канала hERG
а) Экспериментальный метод
В настоящее время в основном существует три метода измерения ингибирующего действия лекарственного препарата на калиевый канал hERG: полностью автоматический метод локальной фиксации потенциала, традиционный метод локальной фиксации потенциала и анализ Thallium FluxORTM. В настоящем документе используется традиционный метод локальной фиксации потенциала для измерения ингибирующего действия соединений на калиевый канал hERG, который является общепринятым стандартным методом для исследований кардиотоксичности и наиболее точным методом измерения. В этом эксперименте цизаприд (соединение, которое, как известно, обладает сильным ингибирующим действием на калиевый канал hERG) используется в качестве стандартного соединения для оценки ингибирующего действия лигандного соединения, подлежащего измерению, на калиевый канал hERG.
При 37°С клеточную линию НЕК293 (Creacell), стабильно экспрессирующую калиевые каналы hERG, культивировали (5% СО2) с применением среды DMEM (содержащей 10% фетальной телячьей сыворотки и 0,8 мг/М1 G418); клетки отделяли посредством раствора TrypLE™ Express после субкультивирования, затем 3×103 клеток наносили на покровное стекло и культивировали в 24-луночном планшете в течение 18 часов, а затем обнаруживали по электрофизиологической активности, и обнаружение лекарственного препарата начинали после того, как ток калиевого канала hERG, зарегистрированный для целых клеток, был стабильным; в эксперименте для каждого измеряемого лигандного соединения были установлены 4 градиента концентрации (0,4 мкМ - 50 мкМ), соотношение между соседними концентрациями составляло 5, концентрация стандартного соединения цизаприда составляла 1 нМ - 1 мкМ, соотношение между соседними концентрациями составляло 10; при обнаружении концентрацию каждого лекарственного препарата (включая лигандное соединение и цизаприд) поддерживали в течение 5 мин (или продолжали действовать до тех пор, пока ток был стабильным), а затем обнаруживали следующую концентрацию, раствор лекарственного препарата последовательно пропускали через регистрирующую камеру от низкой концентрации до высокой концентрации методом самотечной перфузии для воздействия на клетки с осуществлением жидкостного обмена в регистрирующей камере; перед обнаружением лекарственного препарата была установлена холостая контрольная группа, и все экспериментальные группы и контрольная группа были независимо и повторно подвергнуты обнаружению 3 раза.
b) Экспериментальные результаты
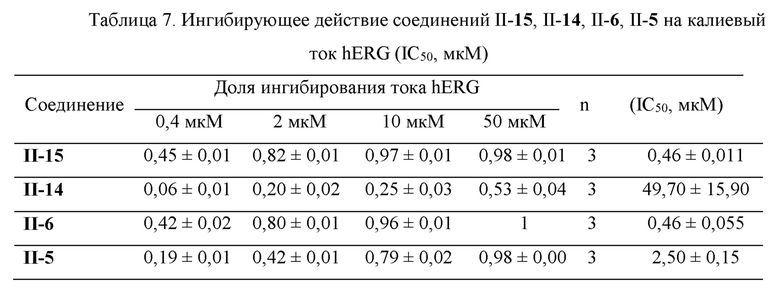
В ходе обработки данных ток действия каждой концентрации подлежащего измерению лекарственного препарата сначала калибровали с использованием записанного тока холостой контрольной группы (пиковое значение следового тока измеряемого лекарственного препарата/пиковое значение следового тока холостого контроля), затем вычисляли степень ингибирования, соответствующую каждой концентрации измеряемого лекарственного препарата (1-пиковое значение следового тока измеряемого лекарственного препарата/пиковое значение следового тока холостого контроля), и после того, как среднее значение и относительное стандартное отклонение для трижды повторенных экспериментов были получены, значение концентрации полумаксимального ингибирования IC50 для каждого соединения, подлежащего измерению, вычисляли с использованием следующего уравнения:
степень ингибирования = 1/[1+(IC50/c)h]
В приведенном выше уравнении с представляет собой концентрацию лекарственного препарата, подлежащего измерению, h представляет собой коэффициент Хилла; подбор кривой по точкам и вычисление значения IC50 были выполнены с помощью программного обеспечения IGOR.
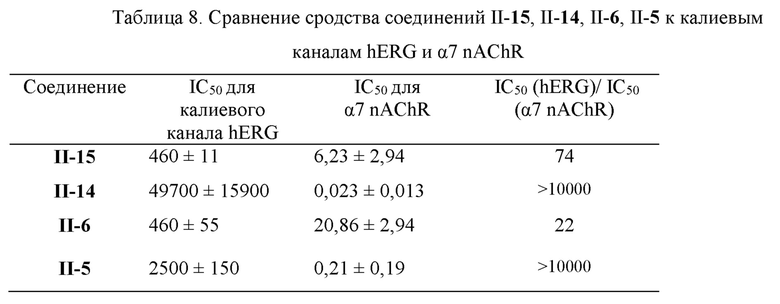
Результаты ингибирующего действия стандартного соединения цизаприда и соединений II-15, II-14, II-6 и II-5, подлежащих измерению, на калиевый канал hERG, представлены в Таблицах 6 и 7. Экспериментальные результаты показывают, что соединение II-14 практически не оказывает ингибирующего действия (IC50>10 мкМ) на калиевый канал hERG, соединение П-5 оказывает умеренное ингибирующее действие (1 мкМ≤IC50≤10 мкМ) на калиевый канал hERG, соединения II-15 и II-6 обладают более сильным ингибирующим действием (0,1 мкМ≤IC50≤1 мкМ) на калиевой канал hERG, но их сродство (IC50 находилось в диапазоне 460 нМ-2500 нМ) к белкам ионных каналов намного меньше, чем их сродство к мембранному белку α7 nAChR (значение IC50 находилось в диапазоне 0,21 нМ-21 нМ) (см. Таблицу 8). Кроме того, хотя этот метод измерения является наиболее чувствительным методом для исследования токсичности hERG, in vivo ингибирующее действие лекарственного препарата на калиевый ток hERG также связано с физиологическими факторами, такими как концентрация в крови, поэтому эти лигандные молекулы могут быть дополнительно изучены в качестве потенциальных агонистов α7 nAChR.

Пример 14: Измерение полулетальной дозы LD50 лигандного соединения II-15
а) Экспериментальный метод
После того, как 60 мышей Кунымин (18-20 г) в общем, половина самки и половина самцы, кормили при экспериментальных условиях в течение 3 дней, они голодали, но не были лишены воды в течение 12 ч, затем мышей взвешивали и регистрировали одну за другой и случайным образом разделяли на 6 групп по весу, в общей сложности 10 мышей (половина самки и половина самцы) в каждой группе; 5 растворов с различными концентрациями и холостой контрольный раствор были приготовлены в соответствии с диапазоном концентраций, исследованным в предварительном эксперименте, где соотношение соседних концентраций экспериментальных групп составляло 0,90-0,95, и 0,1 мл раствора лекарственного препарата или холостого контрольного раствора инъецировали каждой группе мышей в режиме введения путем инъекции в хвостовую вену; после того, как инъекция была завершена, самок и самцов мышей раздельно помещали и кормили в соответствии с концентрациями, и ответы мышей тщательно наблюдали и регистрировали через 0,5 ч, 1 ч, 3 ч, 6 ч, 12 ч и 24 ч после введения (в том числе, было ли поведение активным, реакции на стимуляцию, условия питания, появились ли такие симптомы, как судороги, маниакальный синдром, гематемезис и покачивание), и мертвых мышей немедленно рассекали для наблюдения аномальных состояний каждого органа; регулярное наблюдение проводили каждый последующий день, непрерывно в течение 14 дней, и веса каждой группы мышей регистрировали через 2, 4, 6, 8, 10, 12 и 14 дней после введения; смертность каждой группы мышей рассчитывали в течение периода наблюдения, а полулетальную дозу LD50 соединения для мышей рассчитывали на основании экспериментальной концентрации и смертности в каждой группе.
b) Экспериментальные результаты
1. Ответы мышей в период наблюдений
В эксперименте все мыши, которые умерли после введения, проявляли конвульсии на стадии наблюдения, а часть мышей проявляла дрожание, гематемезис, покачивание и маниакальный синдром; мертвых мышей рассекали для наблюдения каждой ткани и органа мышей, и никаких отклонений от нормы не было обнаружено; все мыши, подвергнутые инъекции, показали отсутствие аппетита в течение 1 часа после введения, а затем возвращались к нормальному состоянию; вес мышей, выживших в экспериментальной группе, и мышей холостой контрольной группы увеличивался нормальным образом в период наблюдений, а изменение веса мышей каждой группы не имело очевидной разницы между экспериментами с различными концентрациями или разницы между самками и самцами. Ответы мышей в течение периода наблюдения приведены в Таблице 9:

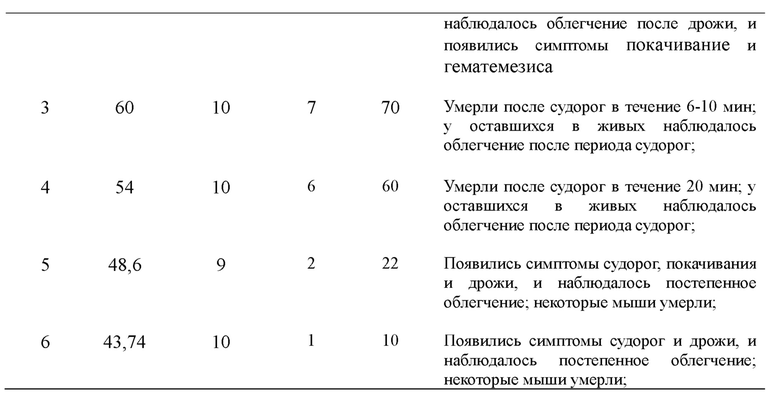
2. Вычисление полулетальной дозы LD50
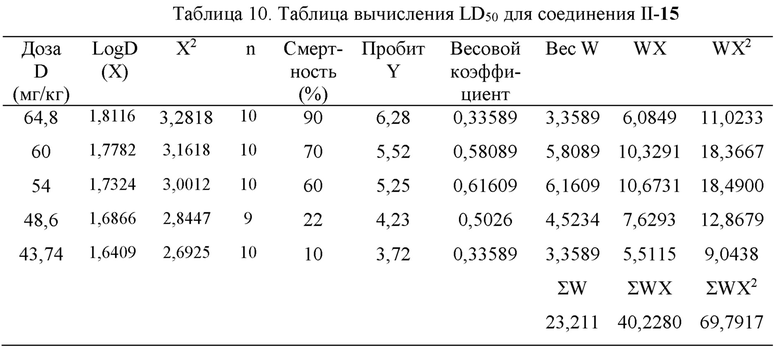
Существует много методов вычисления LD50, среди которых метод взвешенного пробит-анализа, созданный Блиссом, и затем разработанный последующими исследователями, был наиболее точным и строгим, и был стандартным методом вычисления LD50, признанным исследователями. Этот метод вычисления будет использован в настоящем документе для определения значения LD50 соединения, подлежащего измерению, для мышей.
Примечания: вес = весовой коэффициент * число животных в каждой группе (n);
Пробит Y наносили на график относительно LogD (X) с получением уравнения у=14,76х-20,53 (как показано на Фиг. 5). Когда уровень смертности составлял 50%, пробит Y составлял 5,00 в соответствии с поиском по таблице, подставляя в уравнение получали х=m=LogD=1,73, и D=53,70 мг/кг.
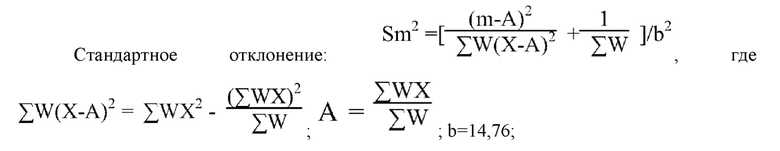
Согласно вышеприведенному выражению вычисленное стандартное отклонение составляло Sm2=0,0001977577, Sm=0,014;
m±1,96 Sm=1,73±0,02744, т.е. 1,70256-1,75744; обращенный логарифм вычисляли соответствующим образом, и принимали 95%-ый доверительный интервал LD50: 50,4150-57,2058 мг/кг; таким образом, значение LD50 соединения II-15 составляло 53,70 мг/кг.
Как видно из вышеописанного способа, значение полулетальной дозы LD50 соединения II-15 было порядка мг/кг, что значительно далеко от дозы, клинически вводимой для одной ПЭТ визуализации, и, следовательно, in vivo применение радиолиганда [18F] II-15, полученного путем введения метки 18F в соединение II-15, было безопасным.
Пример 15: Измерение коэффициента распределения [18F] II-15 в системе липиды/вода
Исследования и разработка большинства неврологических лекарственных препаратов должны учитывать их способность преодолевать гематоэнцефалический барьер (ВВВ), и то, могут ли молекулы лекарственного препарата преодолевать гематоэнцефалический барьер, тесно связано с их растворимостью в липидах. В общем считается, что лигандная молекула способна преодолевать гематоэнцефалический барьер, когда ее значение растворимости в липидах (log Р) составляет от 1,0 до 3,0.
Коэффициент распределения в системе липиды/вода (log Р) для [18F] II-15 измеряли в смешанной системе н-октанола и PBS с рН 7,4, и специальный экспериментальный метод состоял в следующем: добавление в центрифужную пробирку объемом 10 мл, содержащую 900 мкл PBS (предварительно насыщенного н-октанолом) и 1000 мкл н-октанола (предварительно насыщенного PBS), 100 мкл 10 мкКи радиолиганда (физиологический раствор), после встряхивания смешанного раствора в течение 5 минут центрифугирование раствора на центрифуге в течение 5 мин (7000 оборотов в минуту), после центрифугирования помещение 100 мкл органической фазы и водной фазы в РЕ тестовую пробирку, соответственно, для измерения количества радиоактивности, и отбор 100 мкл органической фазы из каждой группы для помещения в центрифужную пробирку объемом 10 мл, добавление 900 мкл н-октанола и 1000 мкл PBS, проведение встряхивания и центрифугирования, как указано выше, с измерением количества радиоактивности, и повторение операций 3 раза таким образом с вычислением среднего значения, log Р=log (количество органической фазы/количество водной фазы)
Посредством измерения было получено, что значение logP коэффициента распределения в системе липиды/вода радиолиганда [18F] II-15 составляло 1,64±0,12, что соответствует диапазону растворимости в липидах для лекарственного препарата для преодоления гематоэнцефалического барьера (ВВВ) с проникновением в головной мозг.
Пример 16: In vitro эксперимент по стабильности радиолиганда [18F] II-15
In vitro стабильность радиолиганда имеет большое значение для его дальнейших исследований in vivo. Как правило, исследование стабильности in vitro проводят в физиологическом растворе и сыворотке животных. Конкретный метод состоял в следующем: соответственно инкубирование 10 мкКи очищенного посредством ВЭЖХ радиолиганда [18F] II-15 и 100 мкл фетальной бычьей сыворотки при 37°С в течение 1 ч и 2 ч, добавление 200 мкл ацетонитрила после окончания инкубации с полным осаждением белков, затем центрифугирование при 40°С в течение 5 мин (7000 оборотов в минуту), сбор супернатант, фильтрация супернатанта с применением фильтрующей мембраной, а затем осуществление анализа ВЭЖХ на 100 мкл отфильтрованного супернатанта; инкубирование 10 мкКи очищенного посредством ВЭЖХ радиолиганда [18F] II-15 со 100 мкл физиологического раствора при комнатной температуре в течение 1 ч и 2 ч, соответственно, а затем непосредственно анализ с помощью ВЭЖХ.
Результаты in vitro эксперимента по стабильности радиолиганда [18F] II-15 показаны на Фиг. 6. На Фиг. 6 можно увидеть, что [18F] II-15 демонстрирует очень хорошую стабильность как в физиологическом растворе, так и в фетальной телячьей сыворотке. Через 1 ч (А на Фиг. 6) и 2 ч (В на Фиг. 6) инкубации в фетальной телячьей сыворотке при 37°С его радиохимическая чистота составляла в обоих случаях более 98%; после 1 ч (С на Фиг. 6) и 2 ч (D на Фиг. 6) инкубации в физиологическом растворе при комнатной температуре его радиохимическая чистота оставалась выше 98%.
Пример 17: In vivo эксперимент по распределению [18F] II-15 в тканях животных Очищенный посредством ВЭЖХ радиолиганд [18F] II-15 (10 мкКи, растворенный в 0,1 мл физиологического раствора, содержащего 5% ДМСО) вводили посредством инъекции нормальным мышам Кунымин (18-22 г, самки, n=5) путем инъекции в хвостовую вену, мышей умертвляли отсечением головы по истечении 5 мин, 15 мин, 30 мин, 60 мин и 90 мин, соответственно, и иссекали для получения крови, головного мозга, сердца, легкого, печени, селезенки, почки, мышцы, кости и хвоста, и вес каждого органа во влажном состоянии взвешивали и измеряли с использованием γ-счетчика. Накопление в каждой ткани было, наконец, выражено как % ID/г, % ID/г=ID/г÷1%, где ID/г = количество радиоактивности ткани (отсчеты)÷масса ткани (мг), 1% = среднее значение 1% Ш за временную фазу- количество радиоактивности хвоста/100, и результаты распределения in vivo этого радиолиганда являются такими, как показано в Таблице 11.
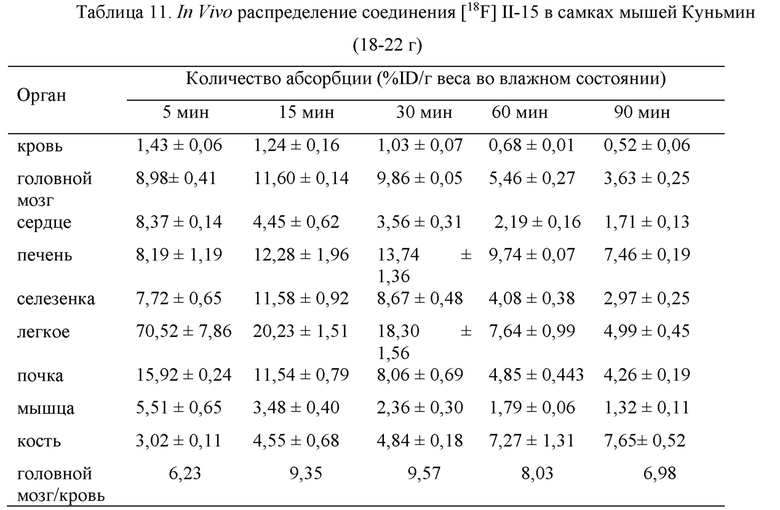
Данные в таблице представляют собой среднее значение для пяти измерений ± стандартное отклонение;
Из Таблицы 11 видно, что 18F-меченный радиолиганд [18F] II-15 имел очень высокое начальное накопление в головном мозге мышей, достигающее значения накопления 8,98±0,41% ID/г через 5 минут после инъекции, показывая самое высокое значение накопления в головном мозге 11,60±0,14% ID/г через 15 мин; между тем, радиолиганд показал надлежащую скорость выведения из головного мозга, и через 60 и 90 минут после введения значения накопления в головном мозге были соответственно снижены до 5,46±0,27% ID/г и 3,63±0,25% ID/г, что указывает на то, что соединение обладает надлежащими внутрицеребральными кинетическими свойствами; этот результат показал значительное преимущество по сравнению с [18F] ASEM (самое высокое значение накопления в головном мозге, составляющее 7,5% ID/г, появляется через 5 минут после введения), которое, как сообщалось, вступило в клинические испытания. Кроме того, [18F] II-15 имел очень низкое значение накопления в крови, демонстрируя очень высокое соотношение головной мозг/кровь, составляющее 9,35 и 9,57 через 5 минут и 15 минут, соответственно.
Явление дефторирования in vivo радиолиганда представляет собой проблему, которую необходимо учитывать в процессе исследования средства визуализации, меченного F-18. Из экспериментальных результатов в приведенной выше таблице видно, что значение накопления [18F] II-15 в кости увеличилось во временном интервале исследования, но скорость увеличения и амплитуда увеличения были не очень большими, что указывает на то, что явление дефторирования in vivo этого соединения было относительно слабым, и не ожидают, что это явление будет слишком сильно мешать исследованиям радиолигандов в отношении in vivo визуализации. Кроме того, существует также высокое накопление радиоактивности в других тканях и органах, например, в почке и легком, первоначальное значение накопления радиолигандов довольно высокое, но оно постепенно снижается с течением времени, и скорость выведения является очень высокой. Этот радиолиганд обладает лучшими свойствами накопления в головном мозге, чем [18F] ASEM.
Пример 18: Эксперимент по распределению [18F] II-15 в области головного мозга Очищенный посредством ВЭЖХ [18F] II-15 (10 мкКи, растворенный в 0,1 физиологического раствора, содержащего 5% ДМСО) вводили посредством инъекции нормальным мышам Кунымин (28-32 г, самки, n=5) путем инъекции в хвостовую вену, мышей умертвляли посредством смещения шейных позвонков через 5 мин, 15 мин, 30 мин, 60 мин и 90 мин после введения, соответственно, и головной мозг быстро иссекали и помещали на лед с удалением прожилок крови ледяным физиологическим раствором, затем кору головного мозга, полосатое тело, гиппокамп, верхнее двухолмие и нижнее двухолмие, таламус, мозжечок и остаток головного мозга иссекали по областям, взвешивали вес каждой области мозга во влажном состоянии и измеряли ее радиоактивность с помощью γ-счетчика. Накопление радиолиганда в каждой области в конечном итоге выражалось в % ID/г, % ID/г=ID ÷ 1%, где ID/г = количество радиоактивности ткани (отсчеты) + масса ткани (мг), 1% = среднее значение 1% ID за временную фазу. Распределение этого радиолиганда в области головного мозга показано в Таблице 12.
Из таблицы 12 видно, что после инъекции 10 мкКи радиолиганда [18F] II-15 в тела мышей радиолиганд показал более высокое накопление в коре головного мозга, полосатом теле и гиппокампе, где рецепторы α7 nAChR были наиболее многочисленным, и достигал пика при 9,39.±0,24% ID/г, 8,37±0,27% ID/г и 6,31±0,82% ID/г, соответственно, через 30 мин после введения, и значения накопления в этих областях постепенно снижались в течение следующего периода наблюдения; областями с умеренным накоплением были верхнее двухолмие и нижнее двухолмие и таламус, а областью с наименьшим накоплением был мозжечок (область, где распределение α7 nAChR в головном мозге мышей было наименьшим). Характеристики распределения в области головного мозга были сходны с [18F] ASEM и соответствовали in vitro и in vivo распределению α7 nAChR, приведенному в литературе, а значение накопления в области, где наблюдается плотность α7 nAChR, имела определенные преимущества по сравнению с [18F] ASEM: [18F] ASEM достигал наивысшего значения накопления через 5 минут после введения, и значения накопления в коре головного мозга, полосатом теле и гиппокампе составляли 7,2% ID/г, 6,0% ID/г и 5,0% ID/г соответственно. Соотношения ткань/мозжечок постепенно увеличивалось на протяжении всего эксперимента и достигали значения 2,5 (кора головного мозга), 2,9 (полосатое тело) и 3,6 (гиппокамп) через 90 минут после введения (Таблица 13), что указывает на то, что радиолиганд не только имеет относительно более высокое накопление в головном мозге, но также имеет хорошую региоселективность в головном мозге.
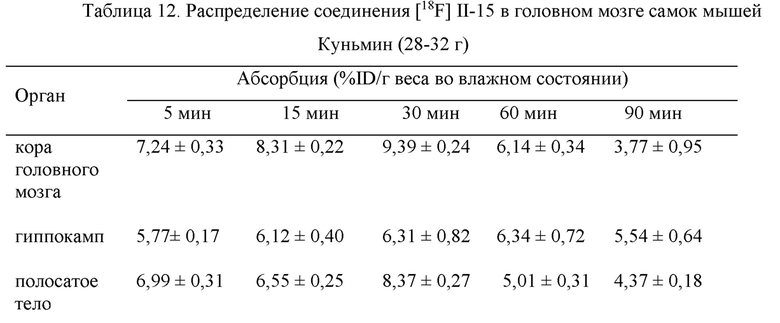
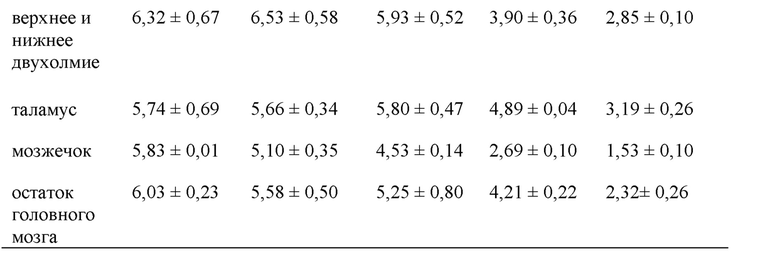
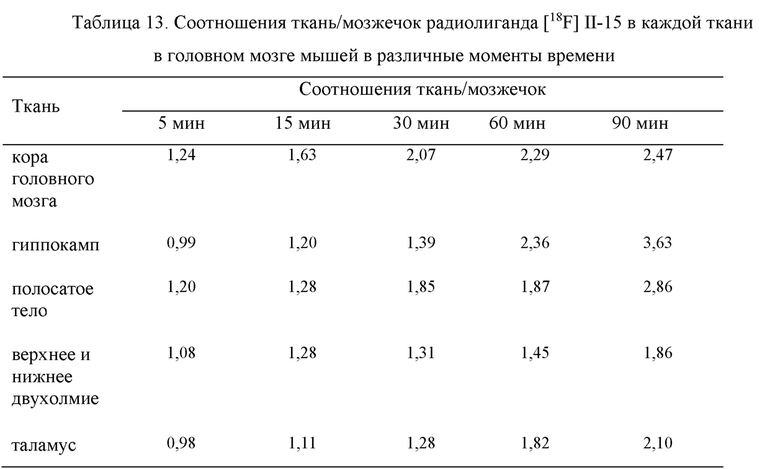
Пример 19: Эксперимент по селективности радиолиганда [18F] II-15 в головном мозге мышей
Селективность радиолиганда в отношении α4β2 nAChR исследовали путем подкожной инъекции 1 мг/кг цитизина (0,1 мл с равным объемом физиологического раствора и 1,2-пропандиола в качестве растворителей) заранее за 5 минут, тогда как селективность в отношении рецептора 5-гидрокситриптамина исследовали путем подкожной инъекции 2 мг/кг ондансетрона (0,1 мл с применением солевого раствора : ДМСО=5:1 (об./об.) в качестве растворителей) заранее за 10 минут. В эксперименте двум группам мышей (п=5, самки мышей Кунымин, 28-30 г) инъецировали 0,1 мл (60 мкКи) радиолиганда [18F] II-15 путем инъекции в хвостовую вену, мышам в холостой контрольной группе таким же образом инъецировали 0,1 мл соответствующего растворителя. Через 60 мин после введения мышей умерщвляли посредством цервикальной дислокации, головной мозг быстро иссекали и помещали на лед, после того, как прожилки крови вымывали ледяным физиологическим раствором, кору головного мозга, полосатое тело, гиппокамп, верхнее двухолмие и нижнее двухолмие, таламус, мозжечок и остаток головного мозга иссекали по областям для измерения веса и радиоактивности каждой области, и конечное накопление было выражено как % ID/г.
Цитизин представлял собой селективный агонист α4β2 nAChR, в то время как ондансетрон представлял собой селективный антагонист рецептора 5-гидрокситриптамина. Как видно на Фиг. 7, не наблюдалось значительного различия в накоплении радиолиганда [18F] II-15 между экспериментальными группами и контрольной группой, что указывает на то, что [18F] II-15 имел слабое связывание с рецептором α4β2 nAChR или рецептором 5-гидрокситриптамина, и радиолиганд имел хорошую селективность в отношении α7 nAChR
Пример 20: ПЭТ визуализация крыс посредством [18F] II-15
Радиолиганд [18F] II-15 (0,3 мл, 200 мкКи) вводили посредством инъекции в тела самок крыс CD-I (180-200 г) путем инъекции в хвостовую вену, затем крыс анестезировали с помощью 3% изофлурана до состояния комы, и крыс фиксировали в положение тела вытянувшись, лицом вниз на устройстве для микро-ПЭТ/КТ-визуализации (Super Argus PET 4R L/CT 180) для небольших животных, поддерживая крыс в состояние анестезии путем использования 1% изофлурана в процессе сканирования изображения. Получение изображения проводили соответственно через 15 мин, 30 мин и 60 мин после введения, чтобы наблюдать состояние распределения радиолиганда [18F] II-15 в головном мозге крыс.
На фиг. 8 показаны фронтальные, сагиттальные и аксиальные микро-ПЭТ-изображения головного мозга самок крыс CD-I через 15 мин, 30 мин и 60 мин после введения [18F] II-15, соответственно. Как видно на Фиг. 8 радиолиганд [18F] II-15 имеет более высокое накопление в головном мозге крыс, его распределение в головном мозге по существу согласуется с результатом эксперимента по распределению in vivo на животных. Наибольшее накопление достигается через 15 минут, а концентрация радиолиганда постепенно снижается с течением времени, при этом удерживание в головном мозге является более правильным, и обогащение определенной концентрации все еще можно наблюдать через 60 минут после введения. Согласно приведенному выше хорошему результату визуализации [18F] II-15 подходит для использования в качестве средства визуализации ПЭТ для α7 nAChR.
Выше приводится описание только предпочтительных примеров настоящего раскрытия, что не предназначается для ограничения настоящего раскрытия. Любая модификация, эквивалентная замена, усовершенствование и т.д., выполненные в рамках сущности и принципов настоящего раскрытия, должны охватываться объемом защиты настоящего раскрытия.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛИГАНДЫ НИКОТИНОВЫХ АЦЕТИЛХОЛИНОВЫХ РЕЦЕПТОРОВ | 2007 |
|
RU2441007C2 |
| НОВЫЕ ПРЕДШЕСТВЕННИКИ ПРОИЗВОДНЫХ ГЛУТАМАТА | 2012 |
|
RU2600981C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ПРОИЗВОДНЫЕ ПИРИДОНА | 2012 |
|
RU2585287C2 |
| АЗАБИЦИКЛИЧЕСКИЕ АЛКАНОВЫЕ ПРОИЗВОДНЫЕ, ЗАМЕЩЕННЫЕ КОНДЕНСИРОВАННЫМ БИЦИКЛОГЕТЕРОЦИКЛОМ | 2007 |
|
RU2437884C2 |
| ПРОИЗВОДНЫЕ БЕНЗОТИАЗОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ СВОЙСТВОМ СВЯЗЫВАТЬ АМИЛОИД, И СПОСОБ ДЕТЕКЦИИ ОТЛОЖЕНИЙ АМИЛОИДА У МЛЕКОПИТАЮЩЕГО | 2004 |
|
RU2440995C2 |
| АЗААДАМАНТАНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБЫ ПРИМЕНЕНИЯ | 2007 |
|
RU2450002C2 |
| ПРОИЗВОДНЫЕ ГЕТЕРОАРЕНКАРБОКСАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРИМЕНЕНИЕ | 2003 |
|
RU2320656C2 |
| ПОЗИТИВНЫЕ АЛЛОСТЕРИЧЕСКИЕ МОДУЛЯТОРЫ НИКОТИНОВОГО РЕЦЕПТОРА АЦЕТИЛХОЛИНА | 2012 |
|
RU2604737C2 |
| ПРОТИВОВОСПАЛИТЕЛЬНОЕ СОЕДИНЕНИЕ, ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2019 |
|
RU2759626C1 |
| СЕЛЕКТИВНЫЕ К ПОДТИПУ РЕЦЕПТОРА АЗАБИЦИКЛОАЛКАНОВЫЕ ПРОИЗВОДНЫЕ | 2008 |
|
RU2417984C1 |
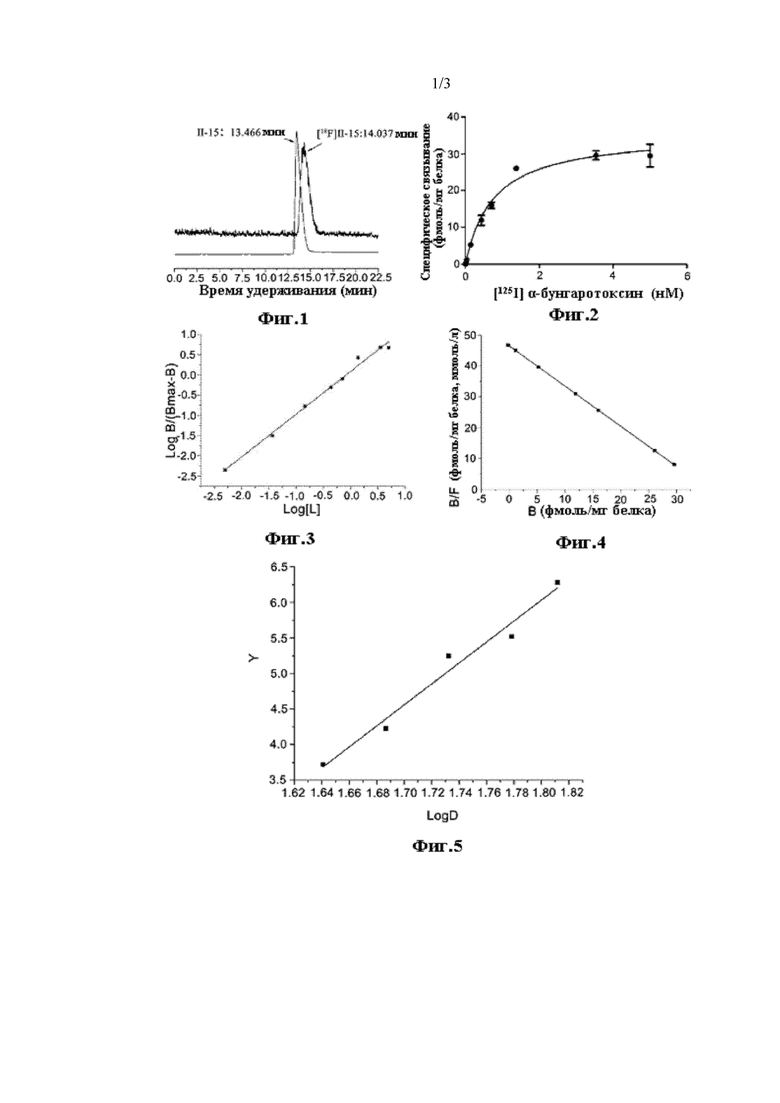
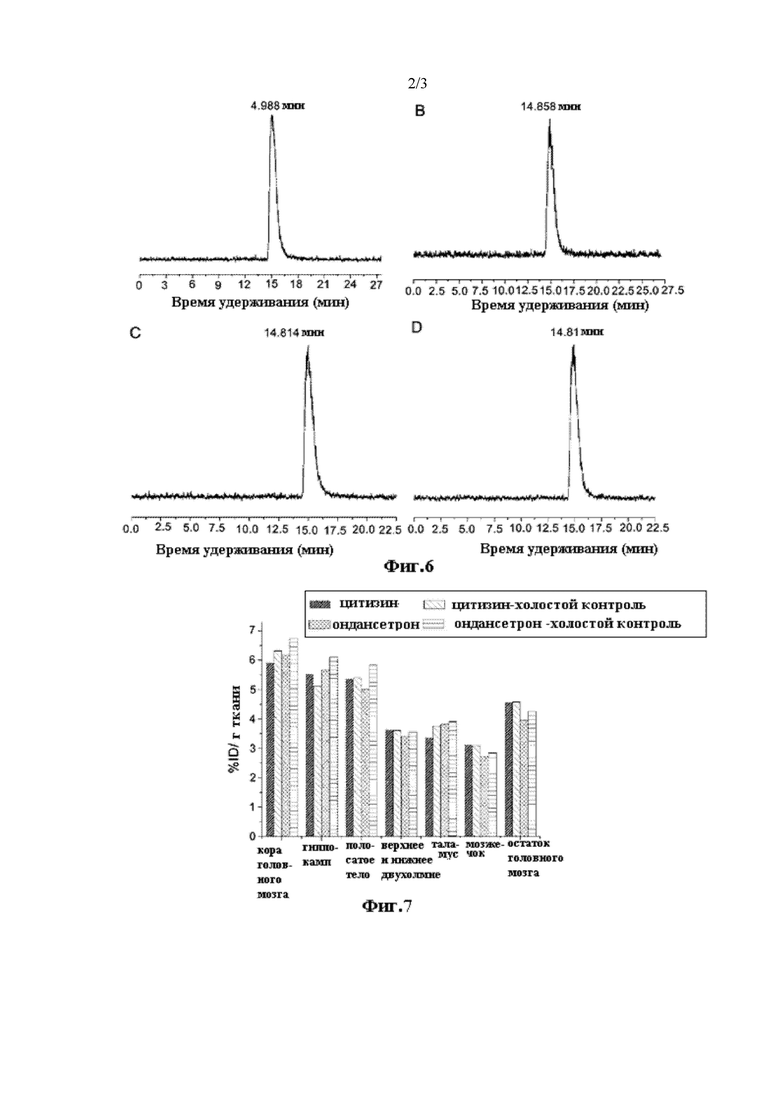
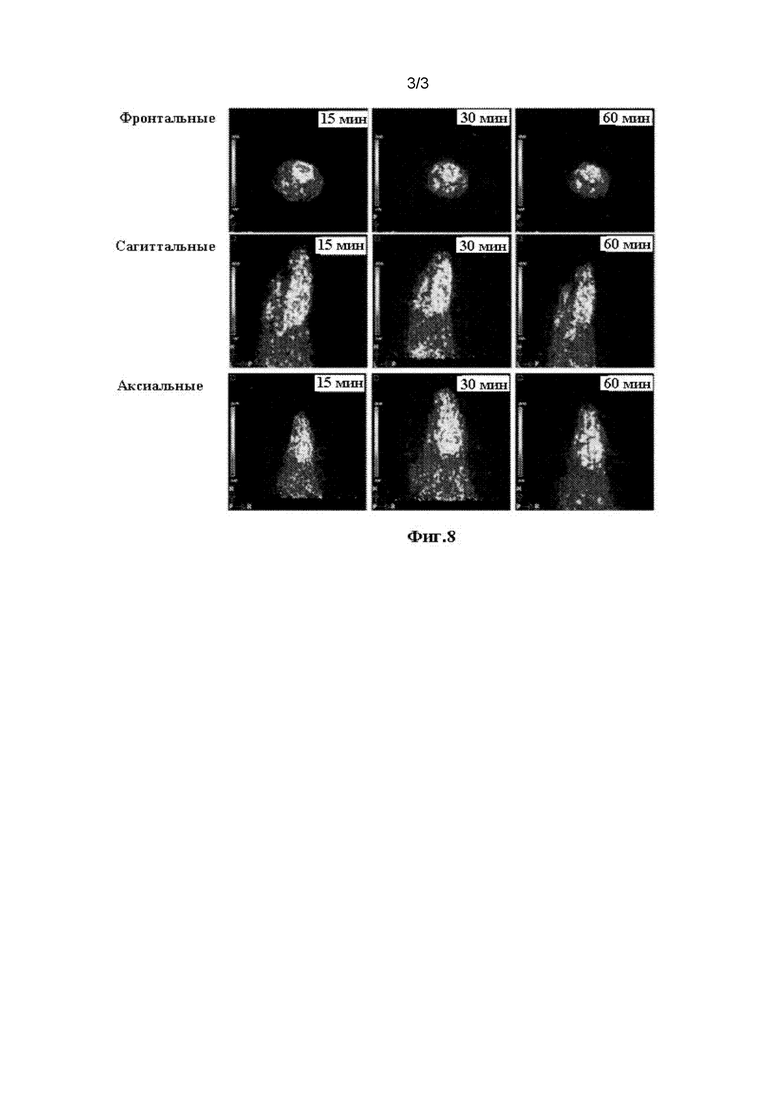
Изобретение относится к применению лигандного соединения α7-никотинового ацетилхолинового рецептора в качестве средства визуализации ПЭТ, где структурная формула указанного лигандного соединения представлена формулой I. 8 ил., 13 табл., 19 пр.
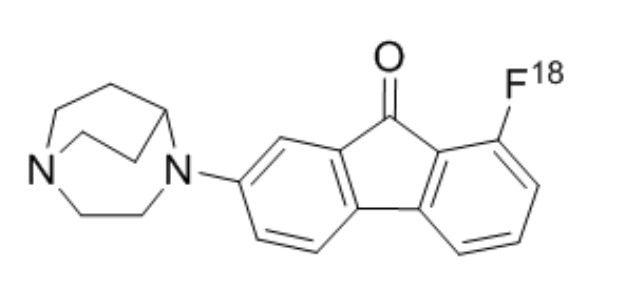 I
I
Применение лигандного соединения α7-никотинового ацетилхолинового рецептора в качестве средства визуализации ПЭТ, где структурная формула указанного лигандного соединения является следующей:
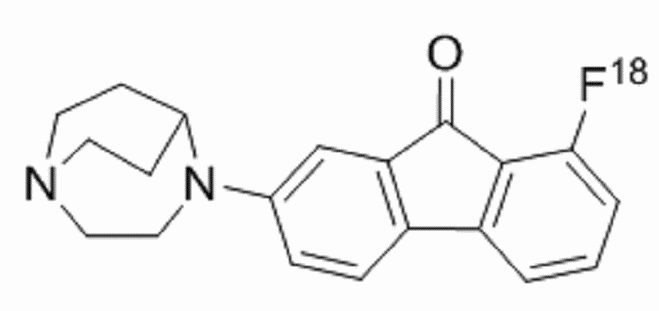 .
.
| WO 2006051394 A1, 18.05.2006 | |||
| US 20050171079 A1, 04.08.2005 | |||
| WO 2006051407 A1, 18.05.2006 | |||
| US 20050065178 A1, 24.03.2005 | |||
| US 20050234031 A1, 20.10.2005 | |||
| US 20050101602 A1, 12.05.2005 | |||
| Состав для гидрофобизации древесно-стружечных плит | 1983 |
|
SU1219622A1 |
| Michael R | |||
| Schrimpf et al | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

