1. Область техники, к которой относится изобретение
Настоящее изобретение относится к области фармацевтической химии и, в частности, к противовоспалительному соединению, способу его получения и его применению для лечения воспалительных кожных заболеваний.
2. Описание предшествующего уровня техники
Аутоиммунное воспаление является основным фактором, вызывающим многие заболевания человека. У людей с дегенеративными заболеваниями обычно наблюдается повышенный уровень провоспалительных модуляторов в крови. Классом таких провоспалительных модуляторов являются цитокины. Цитокины включают провоспалительные цитокины (включая IL-1, IL-1β, IL-2, IL-3, IL-6, IL-7, IL-9, IL-12, IL-17, IL-18, IL-23, TNF-, LT, LIE, онкопротеины и IFN-); противовоспалительные цитокины (IL-4, IL-10, IL-11, IL-13 и TGF-β); и хемокины (IL-8, Gro-a, MIP-1, МСР-1, ENA-78 и RANTES).
Во многих ситуациях воспаления провоспалительные цитокины, особенно TNF-, IL-1β и IL-6, и противовоспалительный цитокин IL-10 играют важную роль в патогенезе различных заболеваний, связанных с воспалением, и поэтому могут использоваться в качестве потенциальных терапевтических агентов. Например, повышенные уровни провоспалительных цитокинов (TNF-, IFN, IL-1, IL-2, IL-6 и IL-12) и хемокинов (IL-8, МСР-1 и RANTES) наблюдались при заболеваниях, связанных с воспалением, таких как увеличением в них растворимых рецепторов TNF, антагонистов рецепторов IL-1 и противовоспалительного цитокина IL-10. Было подтверждено, что IL-10 ингибирует увеличение выработки провоспалительных цитокинов в культуре LPMC in vitro и у пациентов.
Изоферменты фосфодиэстеразы (PDE) участвуют в регуляции каскада трансдукции сигналов в клетках, регулируя уровень циклических нуклеотидов. К настоящему моменту идентифицировано семейство из 11 изоферментных генов PDE. Различие между этими изоферментами заключается в их клеточном распределении и биохимических функциях. Высокая активность PDE4 была обнаружена в лейкоцитах пациентов с атопическим дерматитом, особенно детей (Butle, JM, et al., J. Allergy Clin. Immunol. 1983, 71: 490-497). PDE4 является основным изоферментом в воспалительных клетках, таких как моноциты и макрофаги, происходящие из моноцитов (Gantner et al., Br. J. Pharmacol., 1997, 121: 221-2317), эозинофилы (Dent et al., J. Pharmacol. Exp. Ther., 1994, 271: 1167-1174) и В-лимфоциты (Cooper et al., J. Invest. Dermatol., 2985, 84: 477-482). Ингибиторы PDE4 проявляют сильное противовоспалительное действие за счет увеличения внутриклеточного уровня цАМФ. Подавляя расщепление цАМФ, ингибиторы PDE4 могут регулировать внутриклеточные функции (например, снижать выработку пероксида) и транскрипцию генов (например, ингибировать синтез и/или высвобождение воспалительных цитокинов). Так как PDE4 также экспрессируется в кератиноцитах, эти клетки можно использовать в качестве других возможных фармакологических мишеней для контроля воспалительных кожных заболеваний с использованием ингибиторов PDE4.
Ингибиторы PDE4 полезны при заболеваниях, связанных с активностью эозинофилов, при частности воспалительных заболеваниях трахеи, таких как бронхиальная астма, аллергический ринит, аллергический конъюнктивит, атопический дерматит, экзема, аллергический ангиит, воспалительных и пролиферативных кожных заболеваниях, таких как псориаз или кератоз. Эти соединения можно вводить, возможно, в форме пероральных, трансдермальных, местных, ингаляционных и интраназальных препаратов.
В последние годы в лечении воспалительных заболеваний произошли большие изменения. В связи с тем, что пациенты и врачи уделяют все больше внимания тяжести этих заболеваний и пониманию важной роли цитокинов в их иммуном патогенезе, многие лекарственные препараты, нацеленные на цитокины, доступны в клинике, а некоторые доступны на рынке для лечения этих заболеваний, связанных с аутоиммунной системой. Большинство усилий сосредоточено на нацеливании на TNF-α и IL-1. В настоящее время на рынке доступно множество лекарственных препаратов. Например, ингибиторы TNF-α, такие как этанерцепт (Энбрел), инфликсимаб (Ремикейд) и адалимумаб (Хумира), цертолизумаба пегол (Симзия), могут использоваться для лечения заболеваний, связанных с иммунной системой, таких как ревматический артрит, псориаз и раздражающий энтерит; и несколько препаратов, нацеленных на IL-1 (Анакинра (Кинерет)), IL-4 (Дупилумаб (Дупиксент)), IL-6 (Тоцилизумаб (Актемра) и Силтуксимаб (Силвант)) или IL-12/IL-23 (Устекинумаб (Стелара)) и Алефацепт (Амевив), ингибирующие Т-иммунные клетки, могут использоваться для лечения различных иммунных заболеваний.
Однако, когда эти продукты моноклональных антител вводятся системно, иммунитет пациентов подавляется, что приводит к инфекции или другим системным побочным эффектам. Следовательно, эти лекарственные препараты клинически одобрены для лечения пациентов с воспалительными заболеваниями средней и тяжелой степени. Большинство пациентов с псориазом и экземой относятся к легкой или средней степени тяжести, поэтому эти лекарственные препараты не могут им помочь.
Кризаборол, ингибитор PDE4, доказал свою эффективность и безопасность при лечении экземы легкой и средней степени тяжести у детей и взрослых в клинических испытаниях фазы III (исследовательские проекты №№: AD-301 и AD-302) экземы (также известной как аллергический дерматит, атопический дерматит или атопический дерматит) (Paller AS et al., J Am Acad Dermatol. 2016; 75(3): 494-503). PDE4 является ключевым регулятором производства воспалительных цитокинов при экземе, который в основном действует путем расщепления циклического аденозинмонофосфата. В циркулирующих воспалительных клетках у пациентов с экземой увеличивается активность PDE4, и тесты in vitro показывают, что при ингибировании PDE4 в моноцитах снижается высвобождение цитокинов, которые способствуют воспалительному процессу.
Доказано, что кризаборол обладает хорошей безопасностью, но ограниченным клиническим эффектом, о чем свидетельствует клинический эффект в отношении экземы, который только примерно на 10% выше, чем в случае пустой контрольной пробы. Кроме того, значительная часть (около 50%) пациентов не может получить пользу от кризаборола. Кризаборол имеет незначительный клинический эффект при лечении псориаза из-за низкой активности (где активность ингибирования ферментов составляет около 100 нМ, а активность клеток, ингибирующих высвобождение цитокинов, составляет около 500 нМ) (не одобрен для использования).
В связи с этим можно обнаружить, что существующие лекарственные препараты для лечения экземы и псориаза не могут удовлетворить потребности пациентов, особенно большинства пациентов с легкой и средней степенью тяжести, и все еще существует потребность в разработке более эффективных и безопасных местных лекарственных препаратов для лечения большинства воспалительных кожных заболеваний легкой и средней степени тяжести (например, псориаз, экзема и т.д.).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение предлагает новый противовоспалительный лекарственный препарат, который оказывает сильное ингибирующее действие на PDE4, важную мишень для аутоиммунной активации, легко проникает через кожу и легко разлагается.
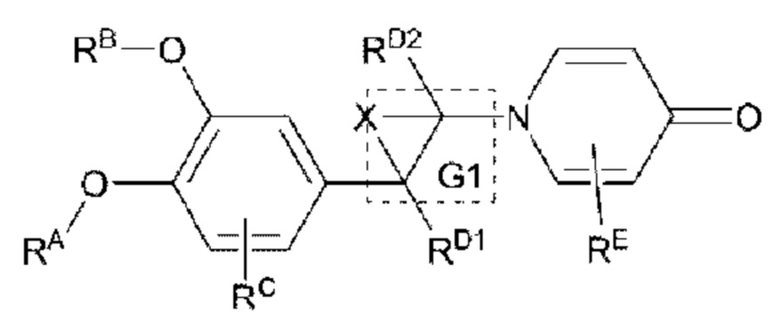
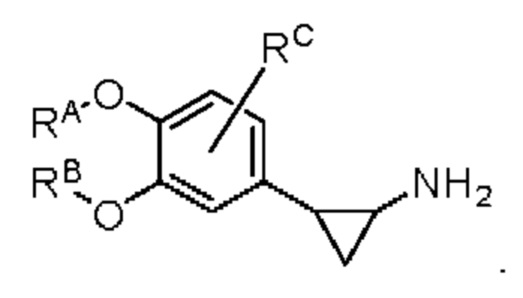
Настоящее изобретение предлагает противовоспалительное соединение, которое представляет собой соединение, имеющее структуру, показанную ниже:
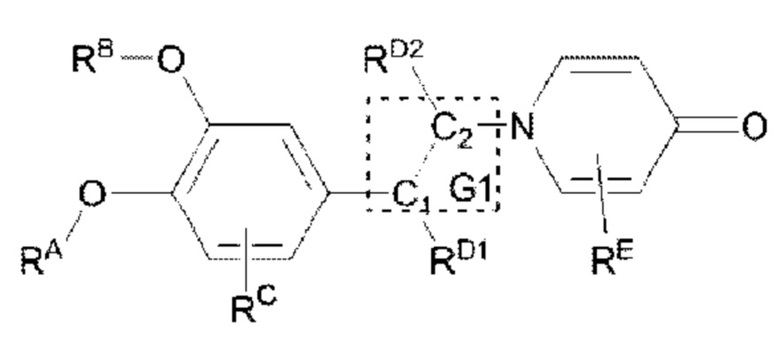
В структурной формуле RA представляет собой водород, алкил или арил, и в вышеуказанных группах один или несколько атомов водорода, присоединенных к углероду в этих группах, необязательно замещены алкилом, циклоалкилом, арилом или галогеном,
алкил представляет собой алкильную группу, содержащую от 1 до 15 атомов углерода, обычно группу, имеющую общую формулу CnH2n+1-, и предпочтительно линейную или разветвленную алкильную группу, в которой n не превышает 6;
арильная группа выбрана из любых ароматических групп, состоящих из чистого карбоциклического или гетероциклического кольца(колец), содержащего(их) от 5 до 24 атомов углерода, таких как циклопентадиенил, фенил, нафтил, хинолинил, пирролил, пиридинил, фурил и тому подобное; и
«один или несколько атомов водорода, присоединенных к углероду в группах, необязательно замещены алкилом (который обычно относится к алкильной группе с не более чем 10 атомами углерода), циклоалкилом (трех-, четырех-, пяти-, шести- или семичленный), гетероциклоалкилом (аза/тиа/окса- трех/четырех/пяти/шести/ семичленный), арилом (который обычно относится к арильной группе, имеющей от 5 до 20 атомов углерода) или галогеном (фторо, хлоро, бромо, йодо)» означает формы замещения водорода в алкильной группе, например, -CH2Cl, -CH2CH2Cl, -СН2(Br)СН3, -CH2CH2F, -CHF2, -CH(CH3)CHF2, -CH(Ph)CH3, 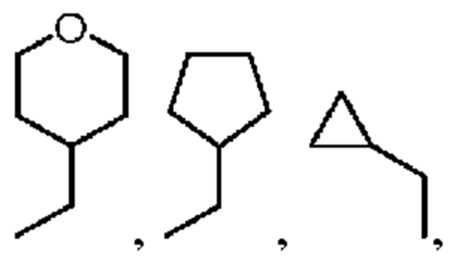 или -CH2Ph,
или -CH2Ph,
или формы замещения водорода в ароматической группе, например, 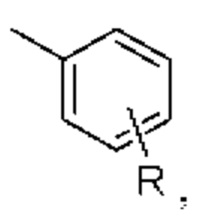
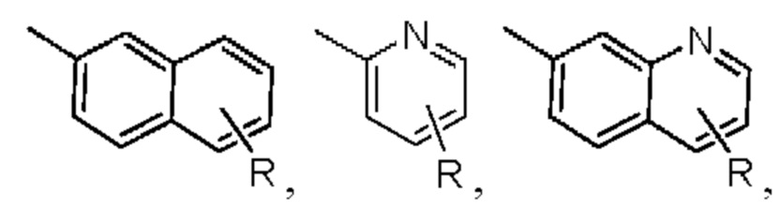 в которой R представляет собой одну или несколько алкильных групп (обычно содержащих не более 6 атомов углерода), циклоалкильных групп (обычно трех-, четырех-, пяти- или шестичленных, которые могут быть гетероциклоалкильными), арильных групп (которые обычно относятся к арильной группе, содержащей от 5 до 20 атомов углерода, и которая может быть гетероарильной), и галогенов, замещающих водород в ароматическом кольце.
в которой R представляет собой одну или несколько алкильных групп (обычно содержащих не более 6 атомов углерода), циклоалкильных групп (обычно трех-, четырех-, пяти- или шестичленных, которые могут быть гетероциклоалкильными), арильных групп (которые обычно относятся к арильной группе, содержащей от 5 до 20 атомов углерода, и которая может быть гетероарильной), и галогенов, замещающих водород в ароматическом кольце.
RB представляет собой водород, алкил, циклоалкил, гетероциклоалкил, арил, алкенил или алкинил, и в указанных выше группах один или несколько атомов водорода, присоединенных к углероду в этих группах, необязательно замещены алкилом, циклоалкилом, гетероциклоалкилом, арилом или галогеном, и один или несколько атомов углерода в этих группах необязательно замещены серой, сульфоксидом, сульфоном или сульфонилом; или RB представляет собой гидроксильную защитную группу,
в которой алкил такой, как определено в описании RA;
арил такой, как определено в описании RA;
циклоалкил представляет собой трехчленный, четырехчленный, пятичленный или шестичленный цикл;
гетероциклоалкил представляет собой окса/аза/тиа трехчленный, четырехчленный, пятичленный или шестичленный цикл;
алкенил может быть выбран из линейного или разветвленного алкенила, содержащего от 2 до 10 атомов углерода, такого как этенил (-=), 2-пропенил (-СН2-=), 2-бутенил (-СН2-=-СН3), 3-бутенил (-СН2-СН2-=), циклопентенил 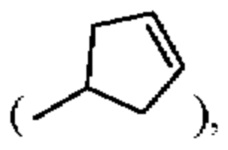 и тому подобное;
и тому подобное;
алкинил может быть выбран из линейного или разветвленного алкинила, содержащего от 2 до 10 атомов углерода, такого как этинил 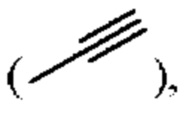 2-пропинил
2-пропинил 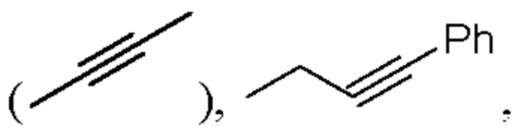 и тому подобное;
и тому подобное;
«один или несколько атомов водорода, присоединенных к углероду в этих группах, необязательно замещены алкилом, циклоалкилом, гетероциклоалкилом, арилом или галогеном» имеет то же значение, что и в описании RA;
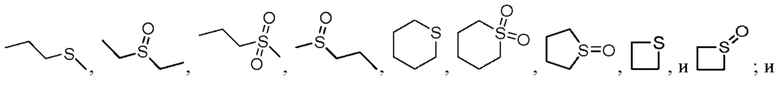
«один или несколько атомов углерода в этих группах необязательно замещены серой, сульфоксидом, сульфоном или сульфонилом» относится к ситуации, в которой один или несколько атомов углерода, таких как C1 и C2 в структуре 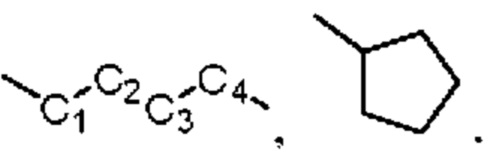 или
или  заменяются на S, (S=O) или (O=S=O), например,
заменяются на S, (S=O) или (O=S=O), например, 
гидроксильная защитная группа означает, что защитное вещество реагирует с гидроксильной группой, преобразуя гидроксильную группу в кремнийорганический эфир, бензиловый эфир, метиловый эфир, аллиловый эфир и другие формы так, что она не подвергается окислению, восстановлению или другим реакциям.
RC представляет собой водород, алкил, галоген, алкокси или циано,
где алкил выбран из линейной или разветвленной алкильной группы, содержащей от 1 до 20 атомоов углерода; и
алкокси относится к группе -O-R, в которой R представляет собой разветвленную или линейную алкильную группу, содержащую от 1 до 20 атомов углерода;
RD1 представляет собой водород, кислород, азот, гидрокси, циано, амино, амидо, алкил, арил, сложноэфирную группу, карбоксил, алкинил или алкенил, и в вышеуказанных группах один или несколько атомов водорода, присоединенных к углероду/кислороду (то есть водород в -ОН) /азоту (то есть водород в -NH2, -NH(R) или =NH) в группах, необязательно замещены алкилом, циклоалкилом, алкинилом, алкенилом, арилом, галогеном, сульфонилом, сульфоксидной группой или эфирной группой, и один или несколько атомов углерода в этих группах необязательно замещены серой, сульфоксидом, сульфоном или сульфонилом,
где алкил такой, как определено в описании RA;
амино относится к -NH2;
амидо относится к группе, обозначенной 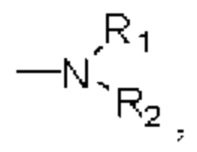 в которой R1 и R2 могут быть любым заместителем, таким как алкил, арил и тому подобное;
в которой R1 и R2 могут быть любым заместителем, таким как алкил, арил и тому подобное;
арил такой, как определено в описании RA;
алкенил такой, как определено в описании RB;
алкинил такой, как определено в описании RB;
сложноэфирная группа относится к группе, обозначенной 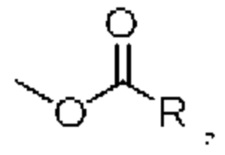 в которой R представляет собой алкил, циклоалкил, гетероциклоалкил, арил, гетероарил и тому подобное,
в которой R представляет собой алкил, циклоалкил, гетероциклоалкил, арил, гетероарил и тому подобное,
где алкил такой, как определено в описании RB, и
арил такой, как определено в описании RB;
в «один или несколько атомов водорода, присоединенных к углероду/кислороду/азоту в группах, необязательно замещены алкилом, циклоалкилом, алкинилом, алкенилом, арилом, галогеном, сульфонилом, сульфоксидной группой или эфирной группой», алкил, циклоалкил, арил и галоген в «замещенном эфире» такие, как определено в описании RA; алкинил обычно относится к алкинильному заместителю с не более чем 20 атомами углерода и может быть 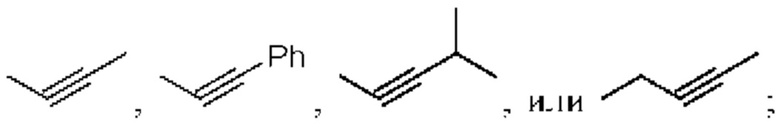 алкенил обычно относится к алкенильному заместителю с не более чем 20 атомами углерода и может быть
алкенил обычно относится к алкенильному заместителю с не более чем 20 атомами углерода и может быть 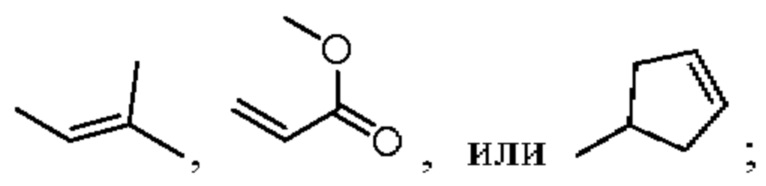 и эфирная группа обычно относится к эфирному заместителю с не более чем 20 атомами углерода и может быть (-O-R); и
и эфирная группа обычно относится к эфирному заместителю с не более чем 20 атомами углерода и может быть (-O-R); и
«один или несколько атомов углерода в группах необязательно замещены серой, сульфоксидом, сульфоном или сульфонилом» имеет то же значение, что и в описании RB.
Связь C1-RD1 представляет собой одинарную связь или двойную связь (например, С=O, C=N и тому подобное);
RD2 представляет собой водород, циано, алкил, циклоалкил, арил, сложноэфирную группу или карбоксил, и в вышеуказанных группах один или несколько атомов водорода, присоединенных к углероду/кислороду/азоту в группах, необязательно замещены алкилом, циклоалкилом, алкинилом, алкенилом, арилом, галогеном, сульфонилом, сульфоксидной группой или эфирной группой,
где амино, амидо, алкил, циклоалкил, арил, сложноэфирная группа, «один или несколько атомов водорода, присоединенных к углероду/кислороду/азоту в группах, необязательно замещены алкилом, циклоалкилом, алкинилом, алкенилом, арилом, галогеном, сульфонилом, сульфоксидной группой или эфирной группой» имеют то же значение, что и в описании RD1.
G1 представляет собой одинарную связь, двойную связь или циклическую структуру, содержащую C1 и С2,
где циклическая структура может быть продуктом раскрытия цикла двойных связей C1 и С2, например, циклопропаном и оксидом пропилена; или продуктом реакции циклизации соединения, имеющего двойную связь или тройную связь, такого как цис-бутадиен, циклопентен и т.п., с двойной связью C1=C2.
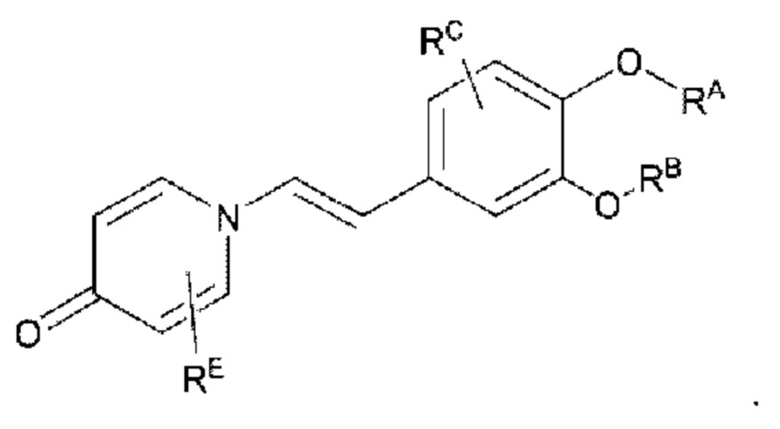
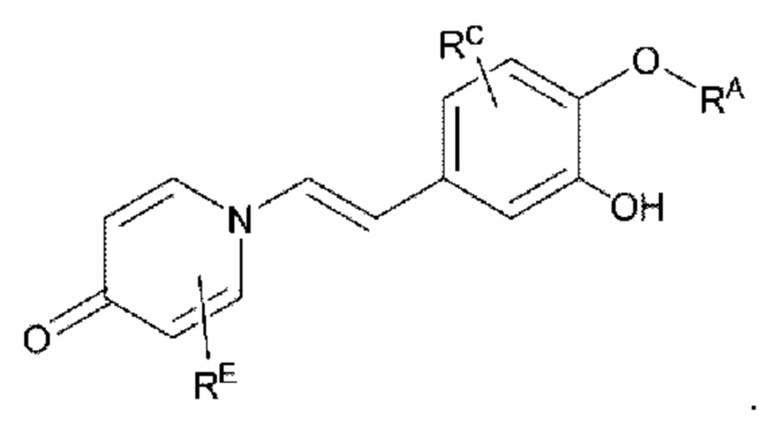
RE представляет собой группу, в которой один или несколько атомов водорода, присоединенных к углероду в пиридиноновом цикле G1, необязательно замещены алкилом, арилом, циано или галогеном,
где алкил, арил и галоген такие, как определено в описании RA.
Кроме того, противовоспалительное соединение, предлагаемое в настоящем изобретении, дополнительно характеризуется G1, который представляет собой трехчленный цикл и имеет специфическую структуру, показанную ниже:
Кроме того, настоящее изобретение также предлагает способ получения противовоспалительного соединения, который включает: применение производного 3-гидроксибензальдегида А в качестве исходного материала и замещение водорода в производном 3-гидроксибензальдегида А на RB для получения промежуточного продукта В;
взаимодействие промежуточного продукта B в присутствии триметилсилилцианида для получения промежуточного продукта С;
восстановление промежуточного продукта С для получения промежуточного продукта D, имеющего аминогруппу; и
взаимодействие аминогруппы промежуточного продукта D с шестичленным кислородсодержащим циклическим соединением с получением целевого продукта типа А,
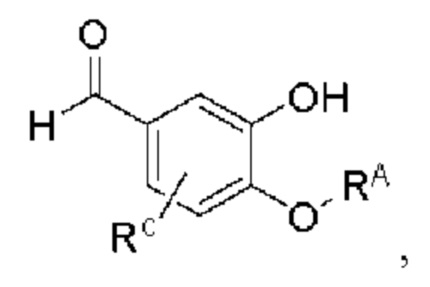
где производное 3-гидроксибензальдегида А представляет собой соединение, имеющее структуру, показанную ниже:
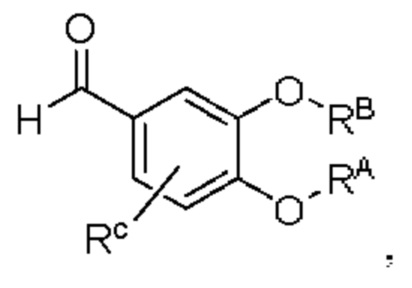
промежуточный продукт В представляет собой соединение, имеющее структуру, показанную ниже:
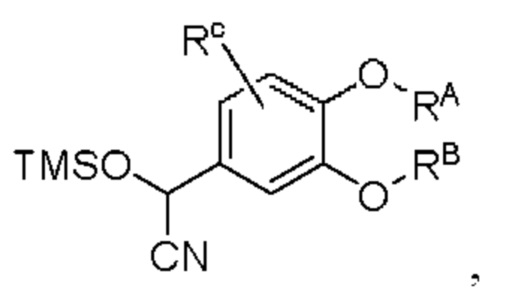
промежуточный продукт С представляет собой соединение, имеющее структуру, показанную ниже:
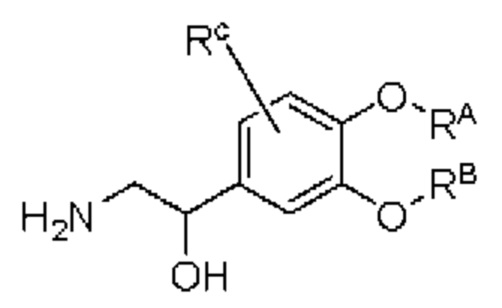
промежуточный продукт D представляет собой соединение, имеющее структуру, показанную ниже:
целевой продукт типа А представляет собой соединение, имеющее структуру, показанную ниже:
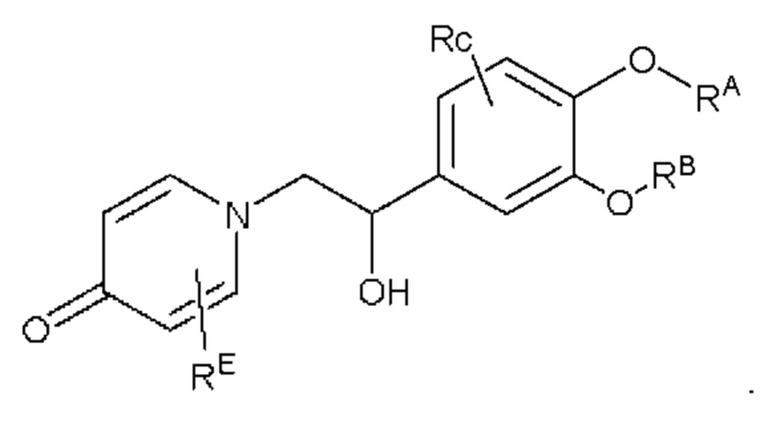
Кроме того, настоящее изобретение также предлагает способ получения противовоспалительного соединения, в котором гидроксильную группу на мостике в середине целевого продукта типа А подвергают реакции присоединения/замещения для получения целевого продукта типа А-1,
где целевой продукт типа А-1 представляет собой соединение, имеющее структуру, показанную ниже:
в которой Rd представляет собой алкильную, циклоалкильную или сложноэфирную группу, и в вышеуказанных группах один или несколько атомов водорода, присоединенных к углероду в группах, необязательно замещены алкилом, алкинилом, алкенилом, циклоалкилом, арилом, галогеном, гидроксилом, тио, циано или тиоалкилом.
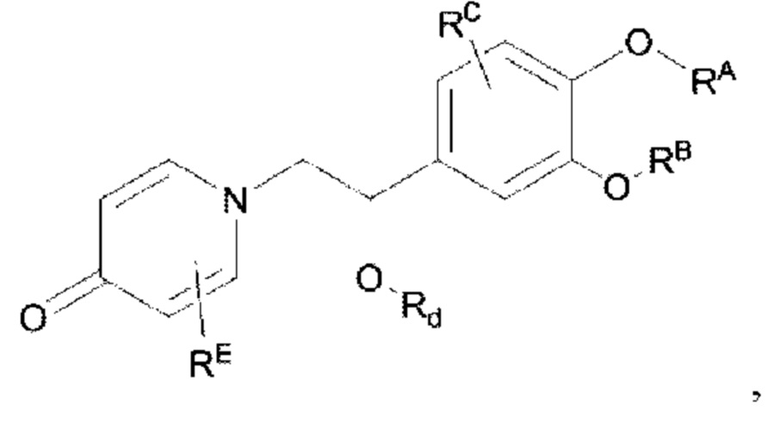
Кроме того, настоящее изобретение также предлагает способ получения противовоспалительного соединения, который включает окисление гидроксильной группы на соединительном мостике целевого продукта типа А для получения целевого продукта типа В,
где целевой продукт типа В представляет собой соединение, имеющее структуру, показанную ниже:
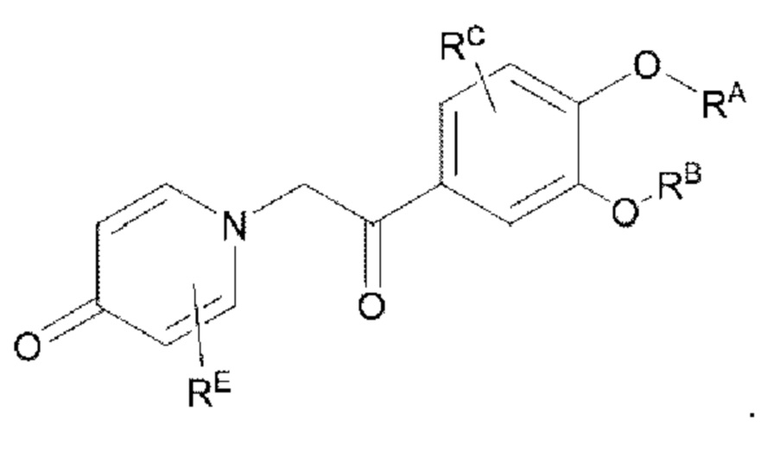
Кроме того, настоящее изобретение также предлагает способ получения противовоспалительного соединения, который включает снятие защиты группы RB, которая представляет собой гидроксильную защитную группу целевого продукта типа В, с получением целевого продукта типа В-1,
где целевой продукт типа В-1 представляет собой соединение, имеющее структуру, показанную ниже:
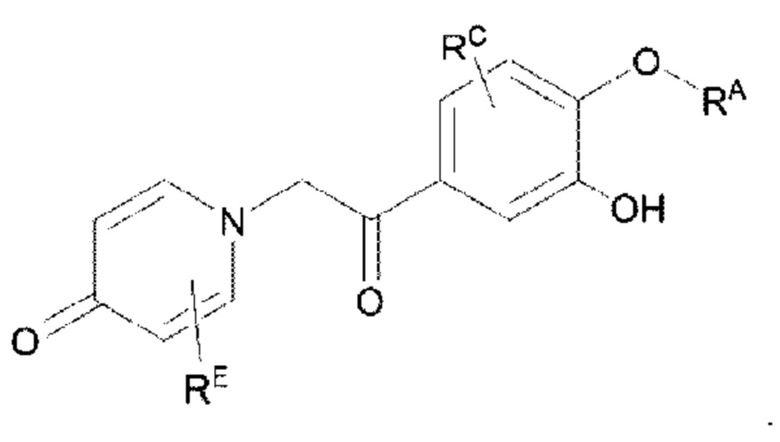
Кроме того, настоящее изобретение также предлагает способ получения противовоспалительного соединения, в котором гидроксильную группу фенильного кольца целевого продукта типа В-1 подвергают реакции присоединения/замещения для получения целевого продукта типа В-2,
где целевой продукт типа В-2 представляет собой соединение, имеющее структуру, показанную ниже:
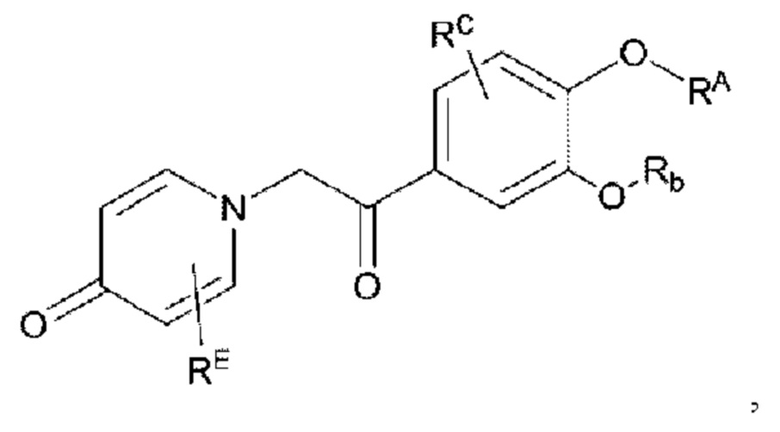
в которой Rb представляет собой алкил, циклоалкил или сложноэфирную группу, и один или несколько атомов водорода, присоединенных к углероду в вышеуказанных группах, необязательно замещены алкинилом, алкенилом, циклоалкилом, арилом, галогеном, гидроксилом, тио, циано или тиоалкилом.
Кроме того, настоящее изобретение также предлагает способ получения противовоспалительного соединения, который включает оксимирование карбонильной группы на мостике в середине целевого продукта типа В для получения целевого продукта типа В-3,
где целевой продукт типа В-3 представляет собой соединение, имеющее структуру, показанную ниже:
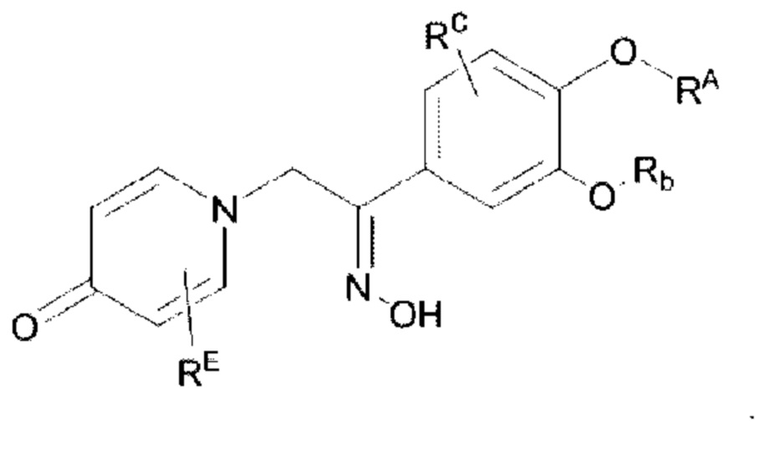
Кроме того, настоящее изобретение также предлагает способ получения противовоспалительного соединения, в котором гидроксильную группу оксима целевого продукта типа В-3 подвергают реакции присоединения/замещения для получения целевого продукта типа В-4,
где целевой продукт типа В-4 представляет собой соединение, имеющее структуру, показанную ниже:
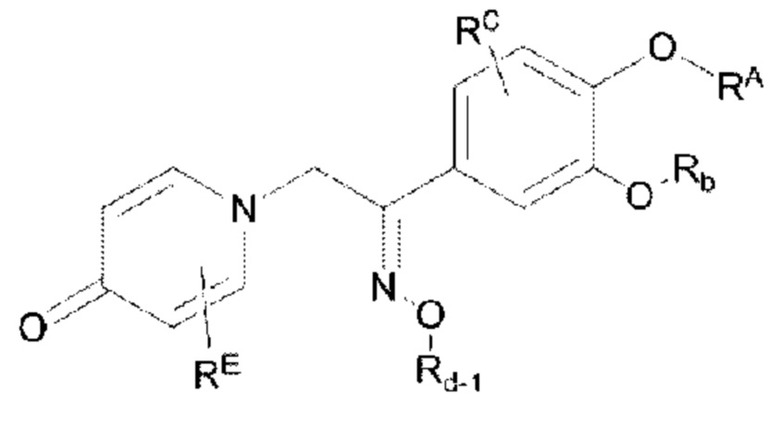
в которой Rd-1 представляет собой алкил, циклоалкил или сложноэфирную группу, и в вышеуказанных группах один или несколько атомов водорода, присоединенных к атому углерода в группе, необязательно замещены алкилом, алкинилом, алкенилом, циклоалкилом, гетероциклоалкилом, арилом, галогеном, гидрокси, тио, циано или тиоалкилом.
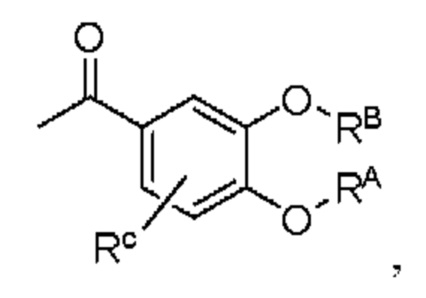
Кроме того, настоящее изобретение также предлагает способ получения противовоспалительного соединения, который включает: использование производного 3-гидроксиацетофенона I в качестве исходного материала и замещение водорода в производном 3-гидроксиацетофенона I на RB для получения промежуточного продукта II;
взаимодействие промежуточного продукта II в присутствии галогенирующего агента для получения промежуточного продукта III; и
взаимодействие галогена в промежуточном продукте III с шестичленным кислородсодержащим циклическим соединением для получения целевого продукта типа В,
где производное 3-гидроксиацетофенона I представляет собой соединение, имеющее структуру, показанную ниже:
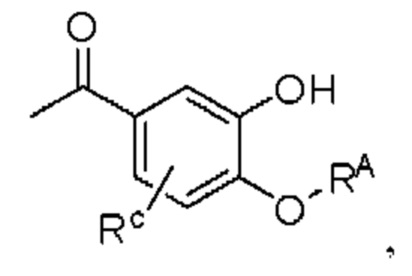
промежуточный продукт II представляет собой соединение, имеющее структуру, показанную ниже:
промежуточный продукт III представляет собой соединение, имеющее структуру, показанную ниже:
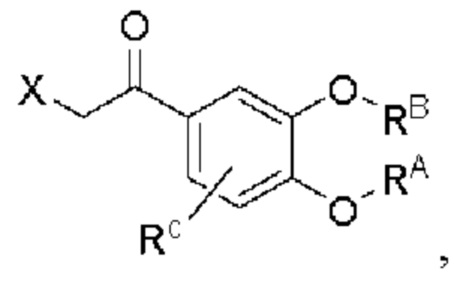 в которой X представляет собой галоген.
в которой X представляет собой галоген.
Кроме того, настоящее изобретение также предлагает способ получения противовоспалительного соединения, который включает удаление гидроксильной группы на соединительном мостике целевого продукта типа А для получения целевого продукта типа С,
или
восстановление, а затем удаление карбонильной группы на соединительном мостике целевого продукта типа В для получения целевого продукта типа С,
где целевой продукт типа С представляет собой соединение, имеющее структуру, показанную ниже:
Кроме того, настоящее изобретение также предлагает способ получения противовоспалительного соединения, который включает восстановление, а затем удаление карбонильной группы на соединительном мостике целевого продукта типа В-2 для получения целевого продукта типа С-1,
где целевой продукт типа С-1 представляет собой соединение, имеющее структуру, показанную ниже:
Кроме того, настоящее изобретение также предлагает способ получения противовоспалительного соединения, в котором гидроксильную группу фенильного кольца целевого продукта типа С-1 подвергают реакции присоединения/замещения для получения целевого продукта типа С-2,
где целевой продукт типа С-2 представляет собой соединение, имеющее структуру, показанную ниже:
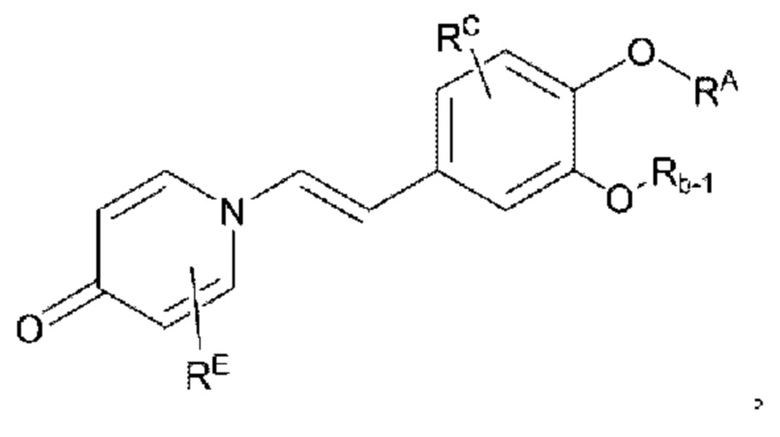
в которой Rb-1 представляет собой алкил, циклоалкил или сложноэфирную группу, и в вышеуказанных группах один или несколько атомов водорода, присоединенных к атому углерода в группах, необязательно замещены алкилом, алкинилом, алкенилом, циклоалкилом, гетероциклоалкилом, арилом, галогеном, гидрокси, тио, пиано или тиоалкилом.
Кроме того, настоящее изобретение также предлагает способ получения противовоспалительного соединения, который включает взаимодействие карбонильной группы на соединительном мостике целевого продукта типа В с галогенирующим реагентом для получения промежуточного продукта X1; и
замещение галогена в промежуточном продукте X1 для получения целевого продукта типа С-3,
где промежуточный продукт X1 представляет собой соединение, имеющее структуру, показанную ниже:
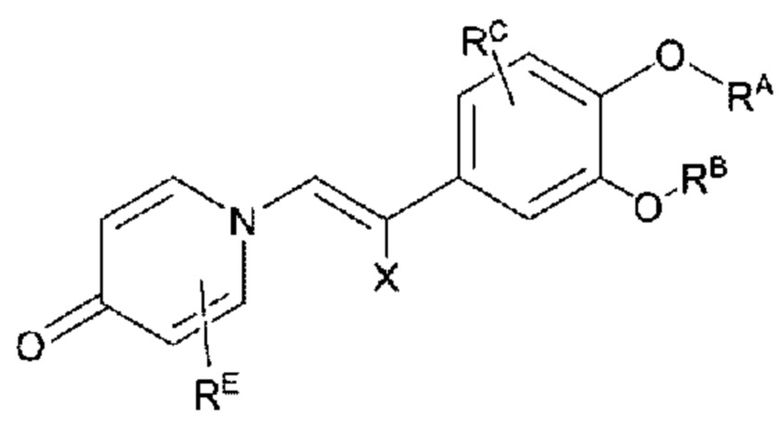 , в которой X представляет собой галоген; и
, в которой X представляет собой галоген; и
где целевой продукт типа С-3 представляет собой соединение, имеющее структуру, показанную ниже:
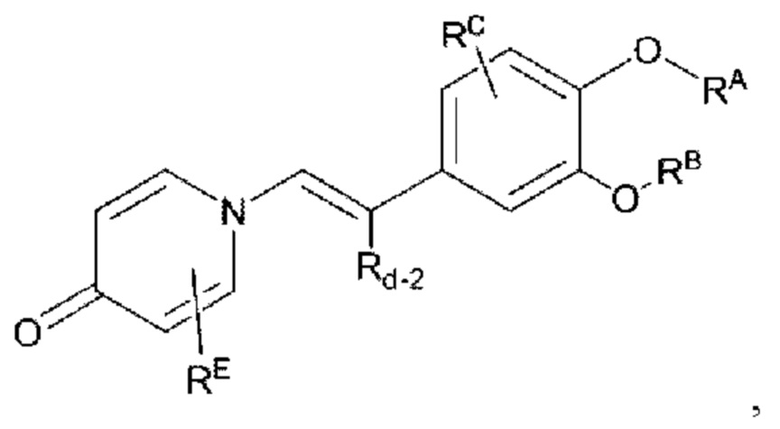
в которой Rd-2 представляет собой арил, алкил, циклоалкил, эфирную группу или сложноэфирную группу, где один или несколько атомов водорода, присоединенных к углероду в группах, необязательно замещены алкилом, алкинилом, алкенилом, циклоалкилом, арилом, галогеном, гидрокси, тио, циано или тиоалкилом.
Кроме того, настоящее изобретение также предлагает способ получения противовоспалительного соединения, который включает: взаимодействие производного ацетофенона 1 в качестве исходного материала с триметилсилилцианидом для получения промежуточного продукта 2;
восстановление промежуточного продукта 2 для получения промежуточного продукта 3; и
взаимодействие аминогруппы в промежуточном продукте 3 с шестичленным кислородсодержащим циклическим соединением для получения целевого продукта типа А',
где производное ацетофенона 1 представляет собой соединение, имеющее структуру, показанную ниже:
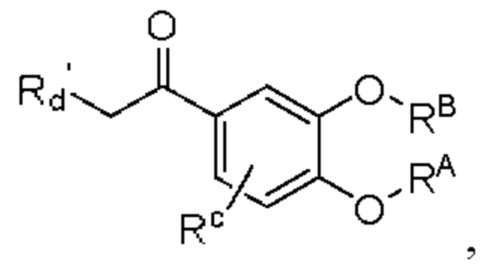
в которой Rd' представляет собой водород, алкил, арил, алкинил или алкенил, и в вышеуказанных группах один или несколько атомов водорода, присоединенных к атому углерода в группе, необязательно замещены алкилом, алкинилом, алкенилом, циклоалкилом, арилом, галогеном, гидрокси, тио, циано или тиоалкилом;
промежуточный продукт 2 представляет собой соединение, имеющее структуру, показанную ниже:
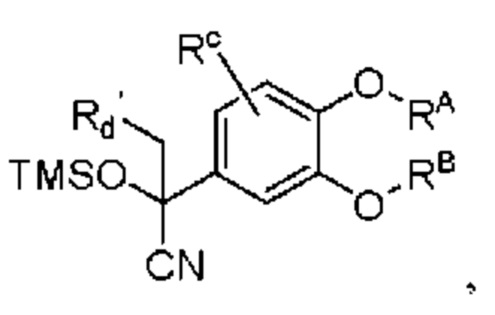
промежуточный продукт 3 представляет собой соединение, имеющее структуру, показанную ниже.
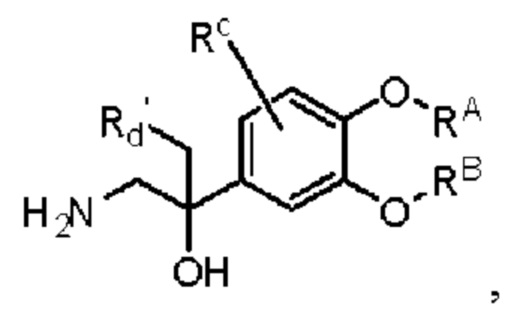
целевой продукт типа А' представляет собой соединение, имеющее структуру, показанную ниже:
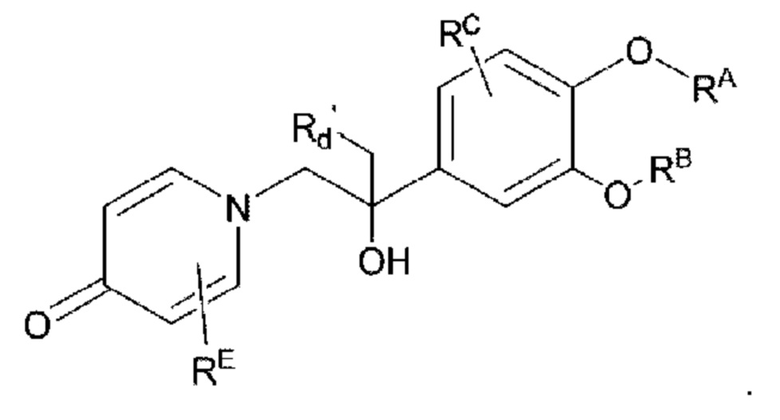
Кроме того, настоящее изобретение также предлагает способ получения противовоспалительного соединения, который включает удаление гидроксильной группы на соединительном мостике целевого продукта типа А' для получения целевого продукта типа С',
где целевой продукт типа С' представляет собой соединение, имеющее структуру, показанную ниже:
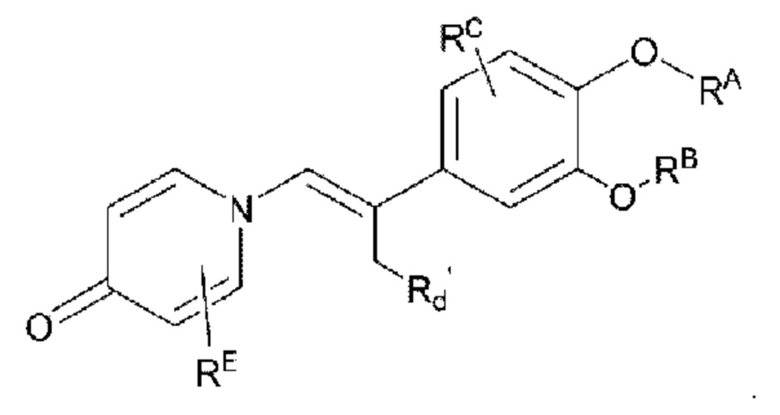
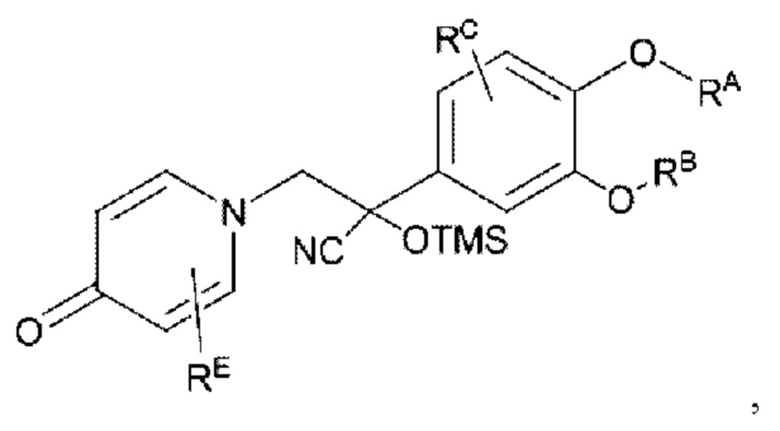
Кроме того, настоящее изобретение также предлагает способ получения противовоспалительного соединения, который включает взаимодействие карбонильной группы на соединительном мостике целевого продукта типа В с триметилсилилцианидом для получения промежуточного продукта Y1; и
подвергание промежуточного продукта Y1 восстановлению и отщеплению для получения целевого продукта типа С'',
где промежуточный продукт Y1 представляет собой соединение, имеющее структуру, показанную ниже:
целевой продукт типа С'' представляет собой соединение, имеющее структуру, показанную ниже:
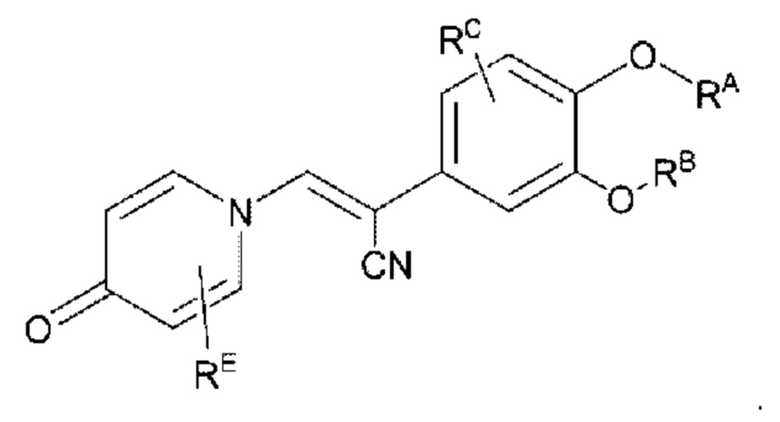
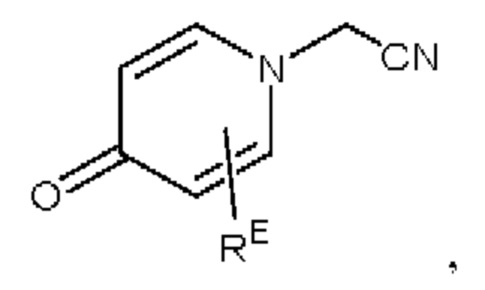
Кроме того, настоящее изобретение также предлагает способ получения противовоспалительного соединения, который включает взаимодействие шестичленного соединения N-ацетонитрила Z1 с производным бензальдегида Z2 для получения целевого продукта типа С,
где шестичленное соединение N-ацетонитрила Z1 представляет собой соединение, имеющее структуру, показанную ниже:
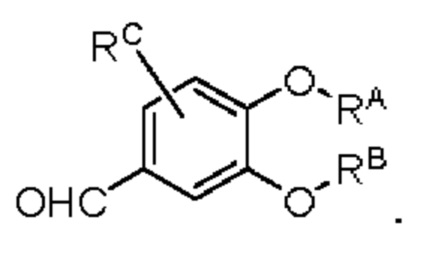
производное бензальдегида Z2 представляет собой соединение, имеющее структуру, показанную ниже:
Кроме того, настоящее изобретение также предлагает способ получения противовоспалительного соединения, который включает: этерификацию производного коричной кислоты 1 в качестве исходного материала для получения промежуточного продукта 2;
образование цикла из двойной связи в промежуточном продукте 2 для получения промежуточного продукта 3;
гидролиз концевой сложноэфирной группы промежуточного продукта 3 для получения промежуточного продукта 4, имеющего карбоксильную группу;
аминирование концевой карбоксильной группы промежуточного продукта 4 для получения промежуточного продукта 5; и
взаимодействие промежуточного продукта 5 с шестичленный кислородсодержащим циклическим соединением для получения целевого продукта типа D,
где производное коричной кислоты 1 представляет собой соединение, имеющее структуру, показанную ниже:
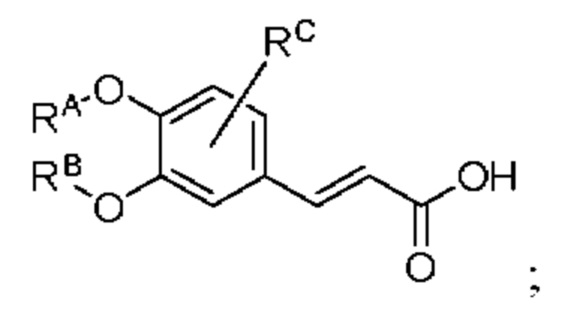
промежуточный продукт 2 представляет собой соединение, имеющее структуру, показанную ниже:
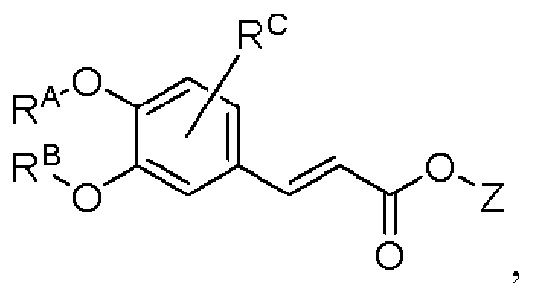 в которой Z представляет собой алкил;
в которой Z представляет собой алкил;
промежуточный продукт 3 представляет собой соединение, имеющее структуру, показанную ниже:
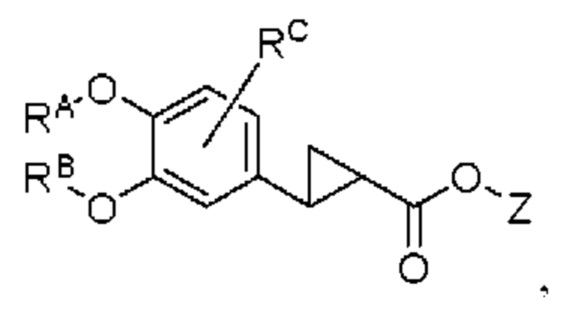
промежуточный продукт 4 представляет собой соединение, имеющее структуру, показанную ниже:
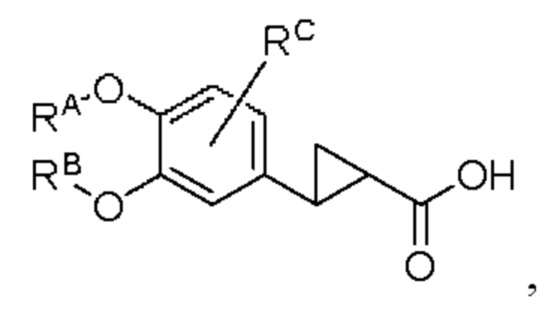
промежуточный продукт 5 представляет собой соединение, имеющее структуру, показанную ниже:
Кроме того, настоящее изобретение также предлагает применение противовоспалительного соединения в качестве ингибитора PDE4.
Кроме того, настоящее изобретение также предлагает применение противовоспалительного соединения для лечения воспалительных кожных заболеваний.
Кроме того, настоящее изобретение также предлагает лекарственное средство, содержащее от 0,01 до 10% противовоспалительного соединения, указанного выше; и
другие компоненты, выбранные из поверхностно-активного вещества, липидного соединения и вспомогательного средства;
где количество поверхностно-активного вещества составляет от 10 до 30% от общей массы лекарственного средства;
количество липидного соединения составляет от 50 до 85% от общей массы лекарственного средства; и
количество вспомогательного средства составляет от 10 до 30% от общей массы лекарственного средства.
Функция и эффекты:
Настоящее изобретение предлагает новое противовоспалительное соединение, которое оказывает сильное ингибирующее действие на PDE4, важную мишень для аутоиммунной активации, легко проникает через кожу и легко расщепляется. Оно является более эффективным, чем существующие лекарственные средства (например, Eucrisa), или имеет меньше побочных эффектов, чем существующие лекарственные средства (например, гормоны и такролимус), поэтому является местным лекарственным средством для лечения экземы с хорошими эффектами и без токсичных побочных эффектов.
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Пример 1
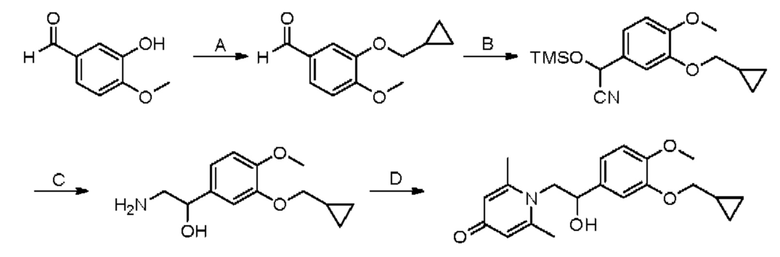
Конкретная схема реакции показана ниже:
Стадия А:
При комнатной температуре 3-гидрокси-4-метоксибензальдегид (3,04 г, 20 ммоль) растворяли в ацетонитриле (80 мл), и затем последовательно добавляли карбонат калия (5,52 г, 40 ммоль) и бромметилциклопропан (4,05 г, 30 ммоль), и нагревали при перемешивании до 80°С в течение 3 часов в атмосфере азота. После завершения реакции добавляли насыщенный водный раствор хлорида натрия (60 мл) и экстрагировали дихлорметаном (3×100 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, сушили на роторном испарителе и очищали колоночной хроматографией с получением 3-циклопропилметокси-4-метоксибензальдегида (3,50 г, 85%).
Стадия В:
3-циклопропилметокси-4-метоксибензальдегид (3,09 г, 15 ммоль) растворяли в дихлорметане (30 мл); добавляли триэтиламин (4,16 мл, 30 ммоль) и триметилсилилцианид (3,75 г, 30 ммоль) в атмосфере азота и перемешивали в течение 6 часов при комнатной температуре. Реакционную массу концентрировали и сушили на роторном испарителе с получением неочищенного продукта 2-(3-циклопропилметокси-4-метоксифенил)-2-триметилсилоксиацетонитрила, который сразу использовали в следующей стадии.
Стадия С:
2-(3-циклопропилметокси-4-метоксифенил)-2-триметилсилоксиацетонитрил (4,57 г, 15 ммоль), полученный на описанной выше стадии, растворяли в безводном тетрагидрофуране (50 мл), а затем порционно добавляли литийалюминийгидрид (1,14 г, 30 ммоль) на ледяной бане и перемешивали в течение ночи при комнатной температуре. После завершения реакции последовательно добавляли воду (1,2 мл), водный раствор гидроксида натрия (1,2 мл, 15%) и воду (3,6 мл), перемешивали в течение 30 минут, сушили над безводным сульфатом натрия, фильтровали и сушили на роторном испарителе с получением неочищенного продукта 2-амино-1-(3-циклопропилметокси-4-метоксифенил) этанола.
Стадия D:
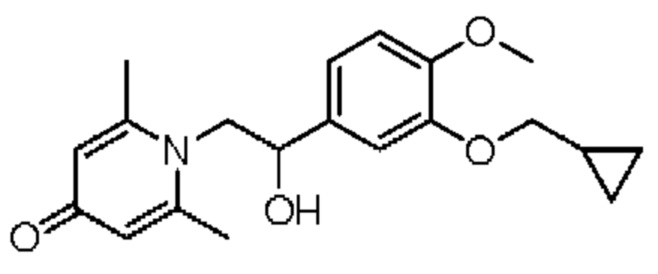
2-амино-1-(3-(циклопропилметокси)-4-метоксифенил)этанол, полученный в вышеупомянутой стадии, растворяли в этаноле (20 мл), затем добавляли 2,6-диметил-4H-пиран-4-он (1 г, 10,41 ммоль) и водный раствор гидроксида натрия (2 М, 20 мл), и перемешивали в течение ночи при 60°С. После завершения реакции реакционную массу сушили на роторном испарителе и очищали колоночной хроматографией с получением 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил)-2,6-диметилпиридин-4(1H)-она (2,01 г, выход 58%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,03 (д, J=1,6 Гц, 2Н), 6,90 (дд, J=8,0, 1,6 Гц, 2Н), 6,87 (д, J=8,0 Гц, 2Н), 6,05 (с, 2Н), 5,03 (дд, J=9,6, 3,2 Гц, 1Н), 4,03 (дд, J=15,2, 10,0 Гц, 1H), 3,93-3,84 (м, 6Н), 2,47 (с, 6Н), 1,39-1,26 (м, 1Н), 0,69-0,64 (м, 2Н), 0,40-0,36 (м, 2Н); LC-MS: m/z 344,2 [М+Н]+.
Пример 2
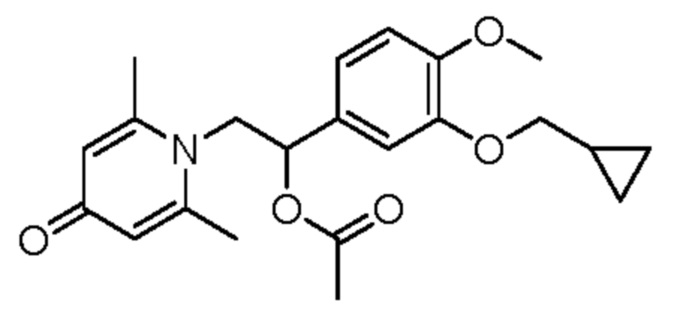
(1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4H)-ил)этил)ацетат
Конкретная схема реакции показана ниже:
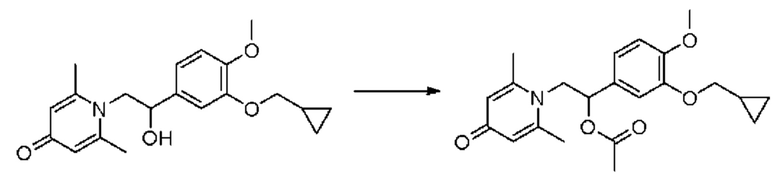
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил) пиридин-4(1H)-он (343 мг, 1,0 ммоль) растворяли в дихлорметане (8 мл), затем последовательно добавляли ацетилхлорид (118 мг, 1,5 ммоль) и триэтиламин (202 мг, 2,0 ммоль) и перемешивали в течение ночи при комнатной температуре. После завершения реакции реакционную массу сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4H)-ил) этил) ацетата (116 мг, выход 30%, бледно-желтое масло). 1H ЯМР (400 МГц, CD3OD) δ 7,05-6,96 (м, 5Н), 6,12-6,08 (м, 1Н), 4,82-4,75 (м, 1H), 4,62-4,56 (м, 1Н), 3,86-3,83 (м, 5Н), 2,75 (с, 6Н), 2,03 (с, 3Н) 1,29-1,21 (м, 1H), 0,64-0,60 (м, 2Н), 0,37-0,33 (м, 2Н); LC-MS: m/z 386,2 [М+Н]+.
Пример 3
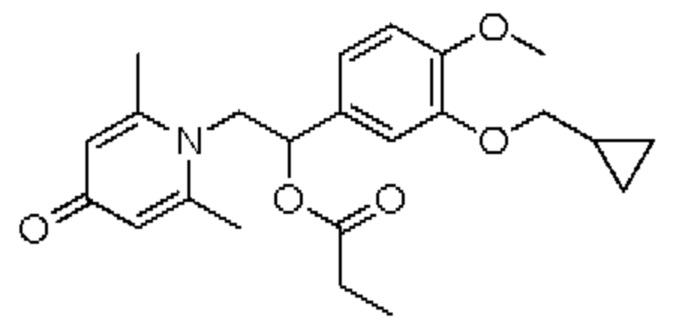
(1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)этил)пропионат
Конкретная схема реакции показана ниже:
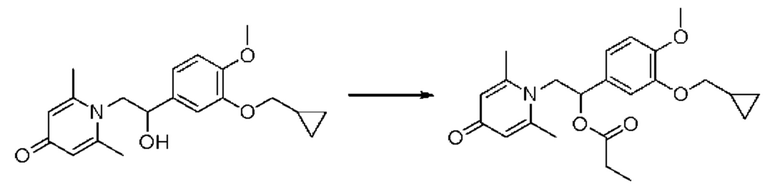
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил) пиридин-4(1H)-он (343 мг, 1,0 ммоль) растворяли в дихлорметане (8 мл), затем последовательно добавляли пропионилхлорид (139 мг, 1,5 ммоль) и триэтиламин (202 мг, 2,0 ммоль) и перемешивали в течение ночи при комнатной температуре. После завершения реакции реакционную массу сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4H)-ил) этил) пропионата (140 мг, выход 35%, бледно-желтое масло). 1H ЯМР (400 МГц, CD3OD) δ 7,05-6,97 (м, 5Н), 6,15-6,12 (м, 1H), 4,82-4,76 (м, 1H), 4,64-4,59 (м, 1H), 3,86-3,83 (м, 5Н), 2,77 (с, 6Н), 2,38-2,29 (м, 2Н), 1,29-1,24 (м, 1H), 1,05-1,01 (м, 3Н), 0,64-0,60 (м, 2Н), 0,36-0,33 (м, 2Н); LC-MS: m/z 399,9 [М+Н]+.
Пример 4
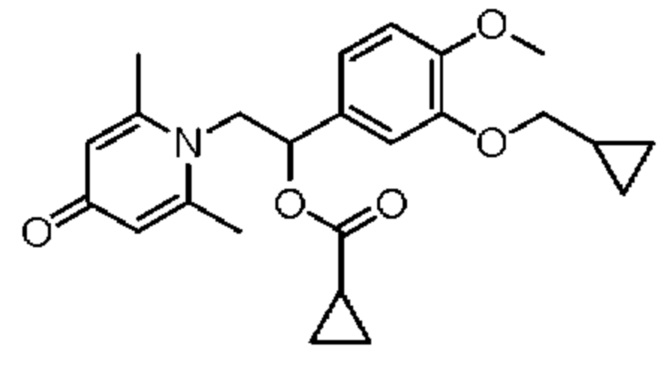
(1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1 (4H)-ил) этил) циклопропилкарбоксилат
Конкретная схема реакции показана ниже:
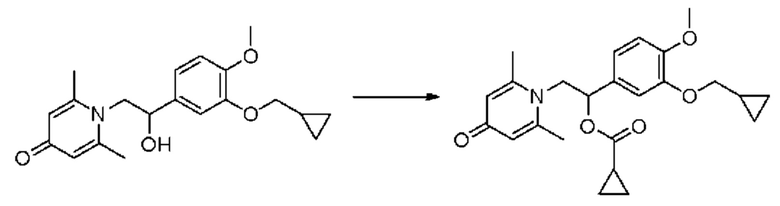
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил) пиридин-4(1H)-он (343 мг, 1,0 ммоль) растворяли в дихлорметане (8 мл), затем последовательно добавляли циклопропанкарбонилхлорид (157 мг, 1,5 ммоль) и триэтиламин (202 мг, 2,0 ммоль) и перемешивали в течение ночи при комнатной температуре. После завершения реакции реакционную массу сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4H)-ил) этил) циклопропилкарбоксилата (103 мг, выход 25%, бледно-желтое масло). 1Н ЯМР (400 МГц, CD3OD) δ 7,04-7,01 (м, 2Н), 6,95 (с, 1Н), 6,35 (с, 2Н), 6,11-6,06 (м, 1Н), 4,56-4,50 (м, 1Н), 4,37-4,32 (м, 1Н), 3,89-3,81 (м, 5Н), 2,53 (с, 6Н), 0,92-0,81 (м, 6Н), 0,66-0,62 (м, 2Н), 0,39-0,36 (м, 2Н); LC-MS: m/z 412,2 [М+Н]+.
Пример 5
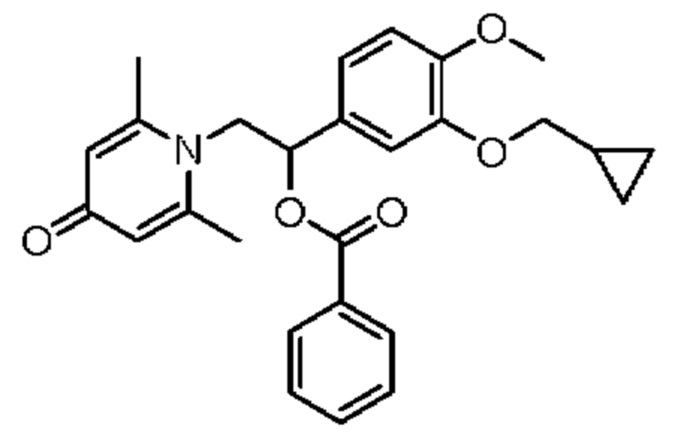
(1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)этил)бензоат
Конкретная схема реакции показана ниже:
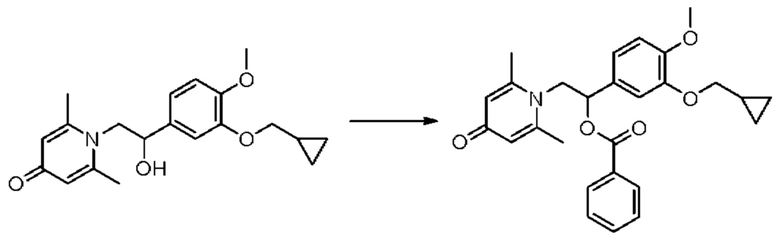
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил) пиридин-4(1H)-он (343 мг, 1,0 ммоль) растворяли в дихлорметане (8 мл), затем последовательно добавляли бензоилхлорид (211 мг, 1,5 ммоль) и триэтиламин (202 мг, 2,0 ммоль) и перемешивали в течение ночи при комнатной температуре. После завершения реакции реакционную массу сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4H)-ил)этил)бензоата (94 мг, выход 21%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 8,04-8,00 (м, 2Н), 7,70-7,65 (м, 1Н), 7,56-7,51 (м, 2Н), 7,22 (дд, J=8,4, 2,0 Гц, 1H), 7,12 (д, J=2,0 Гц, 1H), 7,08 (д, J=8,4 Гц, 1Н), 7,00 (с, 2Н), 6,48-6,44 (м, 1Н), 5,00-4,93 (м, 1Н), 4,81-4,75 (м, 1H), 3,91-3,87 (м, 5Н), 2,82 (с, 6Н), 2,03 (с, 3Н) 1,32-1,25 (м, 1H), 0,66-0,61 (м, 2Н), 0,39-0,35 (м, 2Н); LC-MS: m/z 448,2 [М+Н]+.
Пример 32
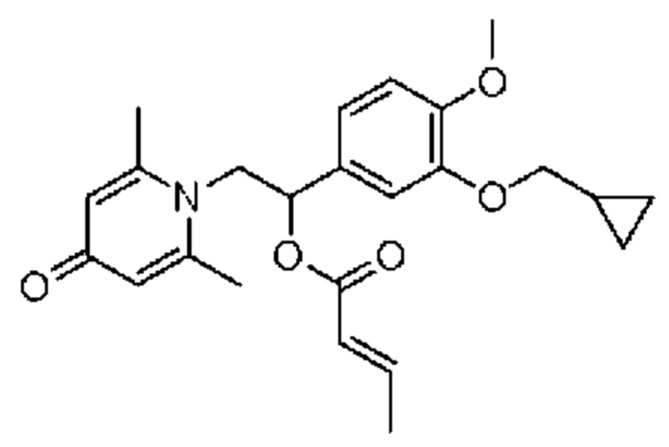
(1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)этил)кротонат
Конкретная схема реакции показана ниже:
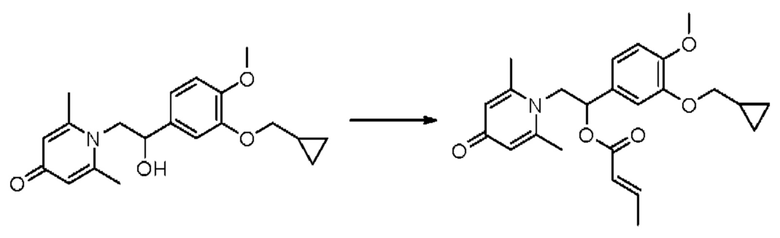
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил) пиридин-4(1H)-он (343 мг, 1,0 ммоль) растворяли в дихлорметане (8 мл), затем последовательно добавляли кротонилхлорид (157 мг, 1,5 ммоль) и триэтиламин (202 мг, 2,0 ммоль) и перемешивали в течение ночи при комнатной температуре. После завершения реакции реакционную массу сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)этил)кротоната (127 мг, выход 31%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 6,87 (д, J=8,4 Гц, 1Н), 6,82 (дд, J=8,4, 2,0 Гц, 1Н), 6,77 (д, J=2,0 Гц, 1Н), 6,33 (с, 2Н), 5,98-5,89 (м, 1H), 5,86-5,76 (м, 1H), 4,37-4,28 (м, 1H), 4,13-4,04 (м, 1Н), 3,88 (с, 3H), 3,84 (д, J=6,8 Гц, 2Н), 2,37 (с, 6Н), 1,33-1,28 (м, 1H), 0,69-0,64 (м, 2Н), 0,39-0,35 (м, 2Н); LC-MS: m/z 412,2 [М+Н]+.
Пример 33
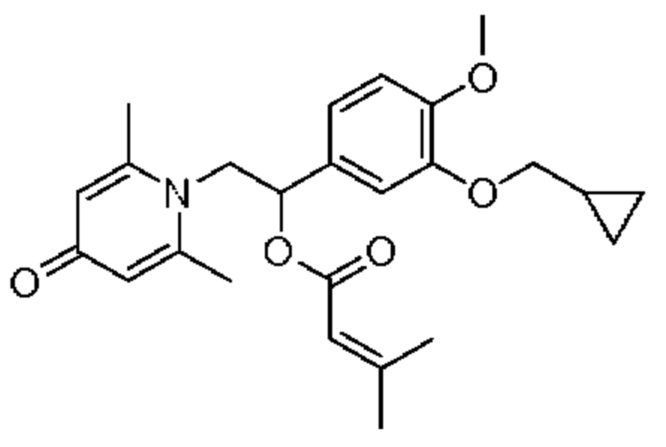
(1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)этил)3-метилкротонат
Конкретная схема реакции показана ниже:
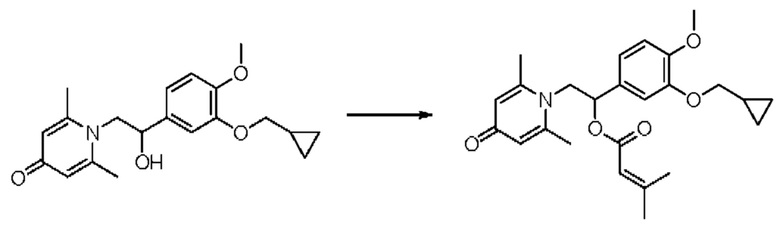
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил) пиридин-4(1H)-он (343 мг, 1,0 ммоль) растворяли в дихлорметане (8 мл), затем последовательно добавляли 3-метилкротонил хлорид (178 мг, 1,5 ммоль) и триэтиламин (202 мг, 2,0 ммоль) и перемешивали в течение ночи при комнатной температуре. После завершения реакции реакционную массу сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)этил)3-метилкротоната (137 мг, выход 32%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 6,87 (д, J=8,4 Гц, 1H), 6,83 (дд, J=8,4,2,0 Гц, 1H), 6,78 (д, J=2,0 Гц, 1H), 6,28 (с, 2Н), 5,90 (дд, J=9,2, 4,8 Гц, 1H), 4,91 (с, 1H), 4,30 (дд, J=15,2, 9,2 Гц, 1H), 4,05 (дд, J=15,2, 4,8 Гц, 1H), 3,88 (с, 3H), 3,83 (д, J=7,2 Гц, 2Н), 3,02 (с, 2Н), 2,37 (с, 6Н), 1,69 (с, 3H), 1,35-1,27 (м, 1H), 0,69-0,64 (м, 2Н), 0,39-0,35 (м, 2Н); LC-MS: m/z 426,2 [М+Н]+.
Пример 8
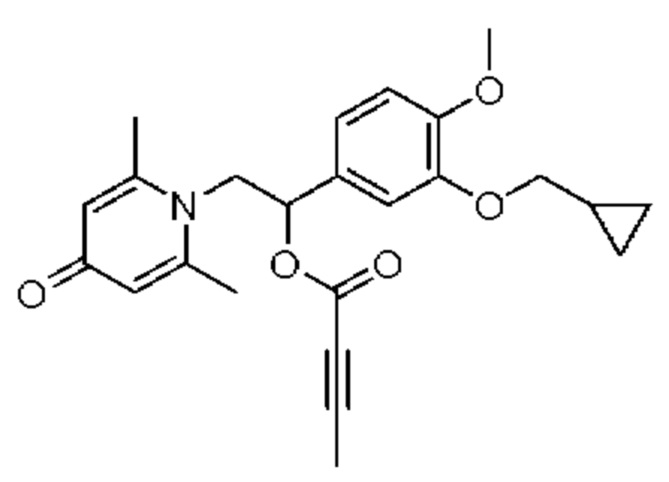
(1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)этил)бут-2-иноат
Конкретная схема реакции показана ниже:
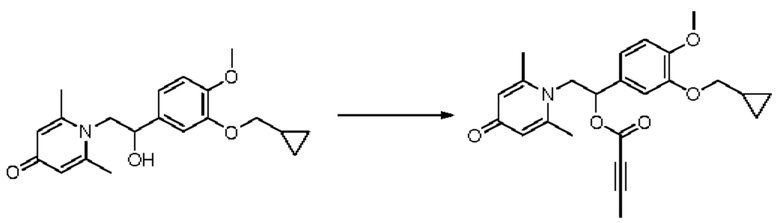
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил) пиридин-4(1H)-он (197 мг, 0,69 ммоль) и 2-бутиновую кислоту (72 мг, 0,86 ммоль) растворяли в дихлорметане (10 мл), а затем последовательно добавляли 4-диметиламинопиридин (105 мг, 0,86 ммоль) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (165 мг, 0,86 ммоль) и перемешивали в течение ночи при 30°С. После завершения реакции реакционную массу сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)этил)бут-2-иноата (37 мг, 9%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 6,88-6,82 (м, 2Н), 6,77 (д, J=1,6 Гц, 1Н), 6,22 (с, 2Н), 5,90 (дд, J=8,4, 5,2 Гц, 1Н), 4,31 (дд, J=15,2, 8,4 Гц, 1H), 4,04 (дд, J=15,2, 5,2 Гц, 1H), 3,88 (с, 3H), 3,84 (д, J=7,2 Гц, 2Н), 2,33 (с, 6Н), 2,01 (с, 3H), 1,32-1,28 (м, 1H), 0,69-0,64 (м, 2Н), 0,39-0,35 (м, 2Н); LC-MS: m/z 410,4 [М+Н]+.
Пример 9
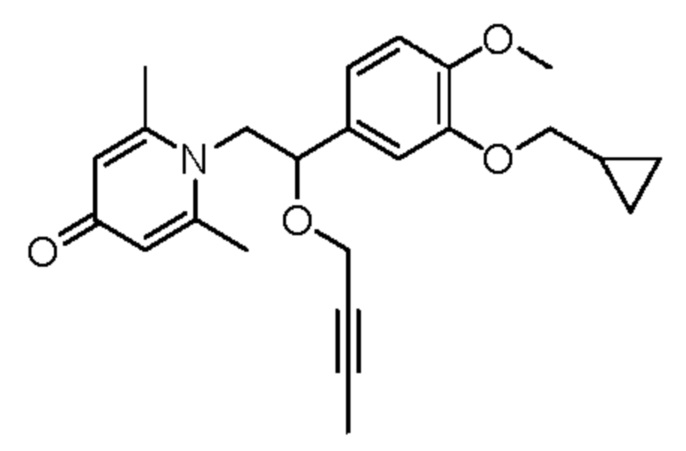
1-(2-(бут-2-ин-1-илокси)-2-(3-циклопропилметокси-4-метоксифенил)этил)-2,6-диметилпиридин-4-(1H)-он
Конкретная схема реакции показана ниже:
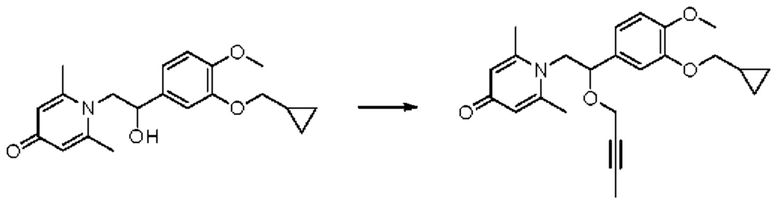
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил) пиридин-4(1H)-он (50 мг, 0,15 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли 1-бром-2-бутин (40 мг, 0,30 ммоль) и карбонат цезия (98 мг, 0,30 ммоль) и перемешивали в атмосфере азота в течение ночи при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(бут-2-ин-1-илокси)-2-(3-циклопропилметокси-4-метоксифенил)этил)-2,26-диметилпиридин-4-(1H)-она (19 мг, выход 32%, бледно-желтое масло). 1H ЯМР (400 МГц, CDCl3) δ 6,89 (d, J=7,6 Гц, 1H), 6,84-6,81 (м, 2H), 6,58 (с, 2Н), 4,70-4,66 (м, 1H), 4,30-4,24 (м, 1Н), 4,14-4,08 (м, 1H), 4,05-4,00 (м, 1H), 3,91-3,86 (м, 5Н), 3,82-3,77 (м, 1H), 2,45 (с, 6Н), 1,37-1,31 (м, 1H), 1,27 (с, 3H), 0,72-0,66 (м, 2Н), 0,42-0,38 (м, 2Н); LC-MS: m/z 396,1 [М+Н]+.
Пример 10
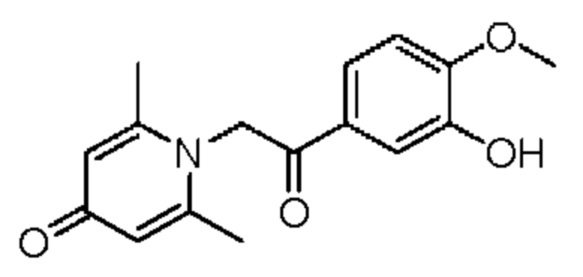
1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
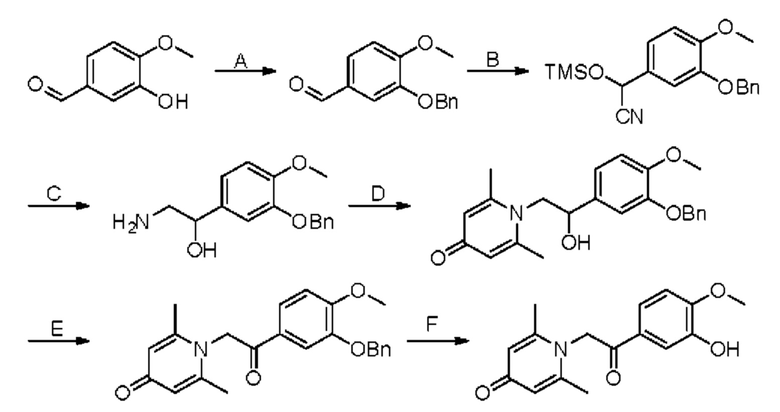
Стадия А:
3-Гидрокси-4-метоксибензальдегид (1,52 г, 10 ммоль) растворяли при комнатной температуре в ацетонитриле (40 мл), а затем последовательно добавляли карбонат калия (2,76 г, 20 ммоль) и бензилбромид (2,56 г, 15 ммоль) и нагревали при перемешивании в атмосфере азота до 80°С в течение 3 часов. После завершения реакции добавляли насыщенный водный раствор хлорида натрия (30 мл) и экстрагировали дихлорметаном (3×50 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, сушили на роторном испарителе и очищали колоночной хроматографией с получением 3-бензокси-4-метоксибензальдегида (2,30 г, 95%, белое твердое вещество).
Стадия В:
3-бензокси-4-метоксибензальдегид (2,30 г, 9,5 ммоль) растворяли в дихлорметане (30 мл), а затем последовательно добавляли триэтиламин (1,92 г, 19 ммоль) и триметилсилилцианид (2,82 г, 28,5 ммоль) в ледяной бане и перемешивали в атмосфере азота при комнатной температуре в течение 16 часов. После завершения реакции сушили на роторном испарителе с получением 2-(3-бензокси-4-метоксифенил) -2-триметилсилоксиацетонитрила, который использовали в следующей стадии без дополнительной очистки.
Стадия С:
2-(3-бензокси-4-метоксифенил)-2-триметилсилоксиацетонитрил, полученный на описанной выше стадии, растворяли в безводном тетрагидрофуране (40 мл), и затем порционно добавляли литийалюмогидрид (1,08 г, 28,5 ммоль) на ледяной бане и перемешивали при комнатной температуре в течение ночи. После завершения реакции последовательно добавляли воду (1,1 мл), водный раствор гидроксида натрия (1,1 мл, 15%) и воду (3,3 мл), перемешивали в течение 30 минут, сушили над безводным сульфатом натрия, фильтровали и сушили на роторном испарителе с получением неочищенного продукта 2-амино-1-(3-бензокси-4-метоксифенил)этанола, который сразу использовали в следующей стадии.
Стадия D:
2-Амино-1-(3-бензокси-4-метоксифенил)этанол, полученный на описанной выше стадии, растворяли в этаноле (60 мл), а затем последовательно добавляли 2,6-диметил-4H-пиран-4-он (1,24 г, 10 ммоль), гидроксид натрия (800 мг, 20 ммоль) и воду (10 мл), нагревали до 60°С и перемешивали в атмосфере азота в течение ночи. После завершения реакции реакционную массу сушили на роторном испарителе и очищали колоночной хроматографией с получением 1-(2-(3-бензокси-4-метоксифенил)-2-гидроксиэтил)-2,6-диметилпиридин-4(1H)-она (1,62 г, 45%, белое твердое вещество). LC-MS m/z 380,2 [М+Н]+.
Стадия Е:
1-(2-(3-бензокси-4-метоксифенил)-2-гидроксиэтил)-2,6-диметилпиридин-4(1H)-он (1,62 г, 4,26 ммоль) растворяли в дихлорметане (30 мл), а затем добавляли периодинан Десса-Мартина (2,16 г, 5,11 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Реакционную массу фильтровали и промывали насыщенным раствором соли. Органическую фазу сушили над безводным сульфатом натрия, фильтровали, сушили на роторном испарителе и очищали колоночной хроматографией с получением 1-(2-(3-бензокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-она (1,37 г, выход 85%, белое твердое вещество). LC-MS m/z 378,2 [М+Н]+.
Стадия F:
Соединение 1-(2-(3-бензокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он (1,37 г, 3,61 ммоль) растворяли в метаноле (50 мл), затем добавляли Pd/C (137 мг) и триэтиламин (1 мл) и вводили водород. Массу перемешивали в течение ночи при комнатной температуре. После завершения реакции реакционную массу фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он (660 мг, выход 64%, серое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7.73 (дд, J=8,4, 2,0 Гц, 1H), 7,53 (д, J=2,0 Гц, 1H), 7,12 (д, J=8,4 Гц, 1H), 7,04 (с, 2Н), 5,97 (с, 2Н), 3,98 (с, 3H), 2,51 (с, 6Н); LC-MS: m/z 288,2 [М+Н]+.
Пример 11
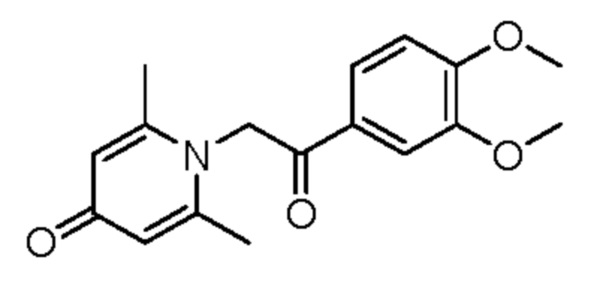
1-(2-(3,4-диметоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он (28 мг, 0,1 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли йодметан (19 мг, 0,13 ммоль) и карбонат калия (21 мг, 0,15 ммоль) и перемешивали при комнатной температуре в атмосфере азота в течение 1 часа. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3,4-диметоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-она (10 мг, выход 30%, бледно-желтое масло). 1H ЯМР (400 МГц, CD3OD) δ 7,85 (дд, J=8,4, 2,0 Гц, 1H), 7,60 (д, J=2,0 Гц, 1H), 7,15 (д, J=8,4 Гц, 1H), 7,05 (с, 2Н), 6,02 (с, 2Н), 4,07 (с, 3H), 3,96 (с, 3H), 2,52 (с, 6Н); LC-MS: m/z 302,1 [М+Н]+.
Пример 12
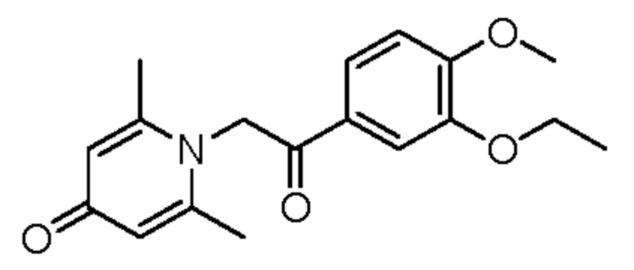
1-(2-(3-этокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
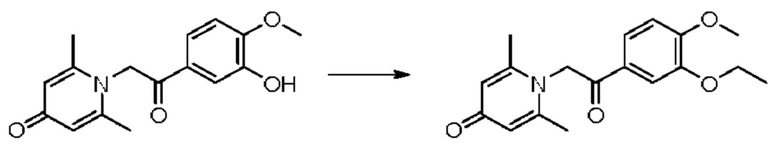
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он (28 мг, ОД ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли йодэтан (21 мг, 0,13 ммоль) и карбонат калия (21 мг, 0,15 ммоль) и перемешивали при комнатной температуре в атмосфере азота в течение 1 часа. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-этокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-она (15 мг, выход 48%, бледно-желтое масло). 1Н ЯМР (400 МГц, CDCl3) δ 8,04 (с, 1H), 7,65 (д, J=8,0 Гц, 1H), 7,02 (с, 2Н), 6,91 (д, J=8,0 Гц, 1H), 6,15 (с, 2Н), 4,32 (к, J=6,0 Гц, 2Н), 3,92 (с, 3H), 2,62 (с, 6Н), 1,51 (т, J=6,0 Гц, 1H); LC-MS m/z 316,1 [М+Н]+.
Пример 13
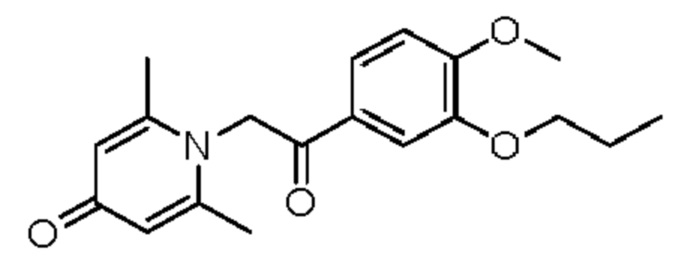
1-(2-(3-пропокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
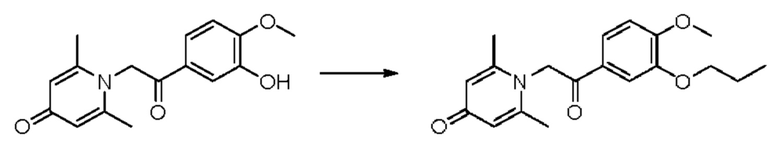
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он (20 мг, 0,07 ммоль) растворяли в N,N-диметилформамиде (5 мл), а затем добавляли бромпропан (10 мг, 0,08 ммоль) и карбонат калия (15 мг, 0,11 ммоль) и перемешивали при комнатной температуре в атмосфере азота в течение 2 часов. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-пропокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-она (8 мг, выход 34%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7.22 (д, J=2,0 Гц, 1Н), 7,14 (дд, J=8,4, 2,0 Гц, 1H), 7,01 (д, J=8,4 Гц, 1Н), 6,38 (с, 2Н), 6,02 (с, 2Н), 4,03 (т, J=6,4 Гц, 2Н), 3,89 (с, 3H), 2,37 (с, 6Н), 1,90-1,80 (м, 2Н), 1,08 (т, J=7,2 Гц, 3H); LC-MS: m/z 330,2 [М+Н]+.
Пример 14
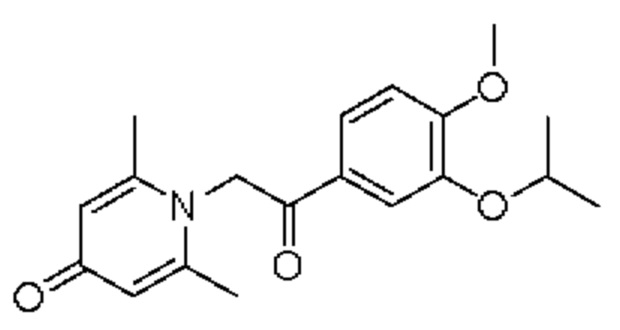
1-(2-(3-изопропокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
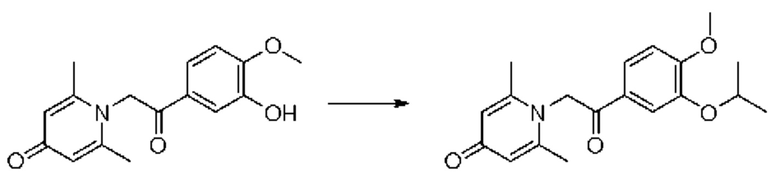
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он (35 мг, 0,12 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли бромизопропан (17 мг, 0,14 ммоль) и карбонат калия (25 мг, 0,18 ммоль) и перемешивали при комнатной температуре в атмосфере азота в течение 2 часов. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-изопропокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4 (1H)-она (10 мг, выход 63%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,02 (дд, J=8,4, 2,0 Гц, 1H), 6,98 (д, J=2,0 Гц, 1H), 6,91 (д, J=8,4 Гц, 1H), 6,76 (д, J=14,4 Гц, 1H), 6,60 (д, J=14,4 Гц, 1H), 6,30 (с, 2Н), 4,69-4,60 (м, 1Н), 3,90 (с, 3H), 2,57 (с, 6Н), 1,35 (д, J=6,0 Гц, 6Н); LC-MS: m/z 330,2 [М+Н]+.
Пример 15
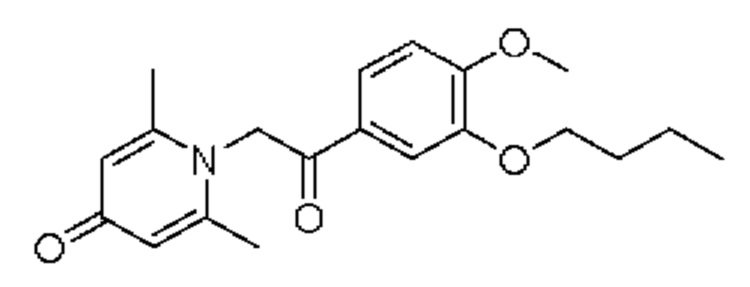
1-(2-(3-н-бутокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
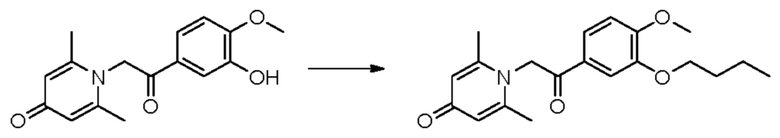
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он (50 мг, 0,17 ммоль) растворяли в N,N-диметилформамиде (5 мл), а затем добавляли бромизопропан (95 мг, 0,7 ммоль) и карбонат калия (37 мг, 0,7 ммоль) и перемешивали при комнатной температуре в атмосфере азота в течение 2 часов. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-н-бутокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он (26 мг, выход 42%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,85 (дд, J=8,4, 2,0 Гц, 1H), 7,60 (д, J=2,0 Гц, 1H), 7,15 (д, J=8,4 Гц, 1H), 7,05 (с, 2Н), 6,02 (с, 2Н), 4,07 (т, J=6,4 Гц, 2Н), 3,96 (с, 3H), 2,52 (с, 6Н), 1,84-1,77 (м, 2Н), 1,58-1,48 (м, 2Н), 0,95 (т, J=7,2 Гц, 3H); LC-MS: m/z 344,1 [М+Н]+.
Пример 16
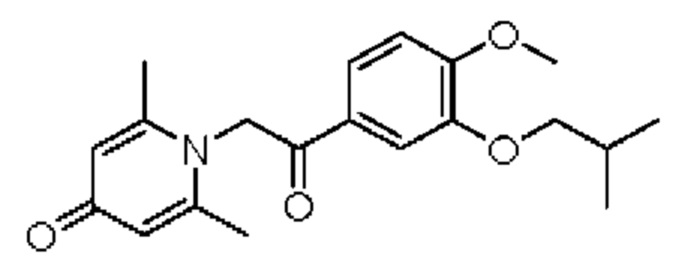
1-(2-(3-изобутокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
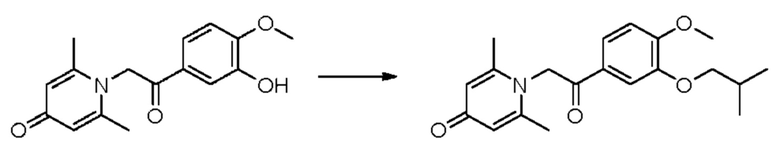
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он (50 мг, 0,17 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли бромизобутан (27 мг, 0,2 ммоль) и карбонат калия (37 мг, 0,27 ммоль) и перемешивали при комнатной температуре в атмосфере азота в течение 2 часов. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-изобутокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-она (25 мг, выход 40%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,02 (дд, J=8,4, 2,0 Гц, 1H), 6,98 (д, J=2,0 Гц, 1Н), 6,91 (д, J=8,4 Гц, 1H), 6,76 (д, J=14,4 Гц, 1Н), 6,60 (д, J=14,4 Гц, 1H), 6,30 (с, 2Н), 3,92 (с, 3H), 3,81 (д, J=6,8 Гц, 2Н), 2,27 (с, 6Н), 2,25-2,17 (м, 1H), 1,08 (д, J=6,8 Гц, 6Н); LC-MS: m/z 344,1 [М+Н]+.
Пример 17
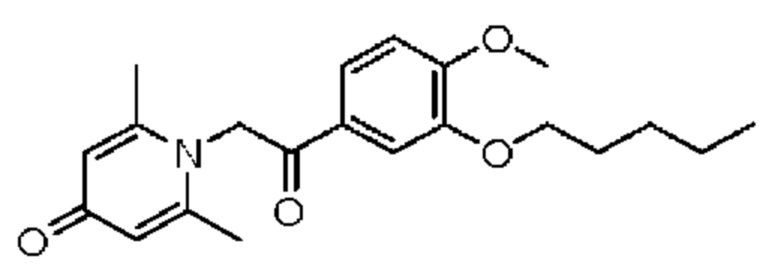
1-(2-(3-н-пентилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
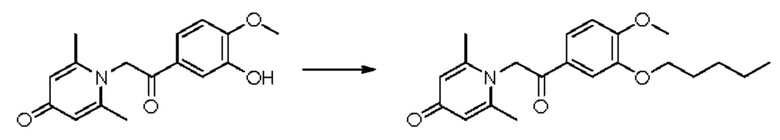
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он (50 мг, 0,17 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли 1-бромпентан (105 мг, 0,7 ммоль) и карбонат калия (97 мг, 0,7 ммоль) и перемешивали в атмосфере азота в течение 5 часов при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-н-пентилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-она (20 мг, выход 32%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,85 (дд, J=8,4, 2,0 Гц, 1Н), 7,60 (д, J=2,0 Гц, 1H), 7,15 (д, J=8,4 Гц, 1Н), 7,07 (с, 2Н), 6,03 (с, 2Н), 4,06 (т, J=6,4 Гц, 2Н), 3,96 (с, 3H), 2,53 (с, 6Н), 1,86-1,79 (м, 2Н), 1,52-1,36 (м, 4Н), 0,95 (т, J=7,2 Гц, 3H); LC-MS: m/z 358,1 [М+Н]+.
Пример 18
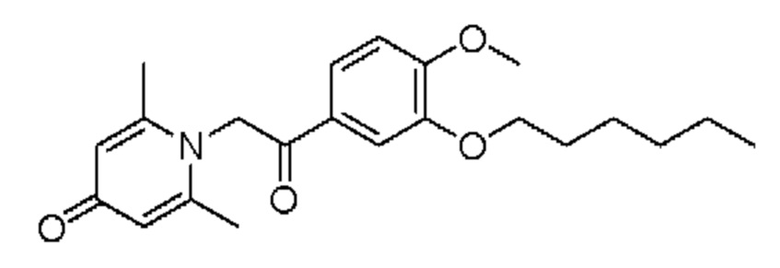
1-(2-(3-н-гексилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
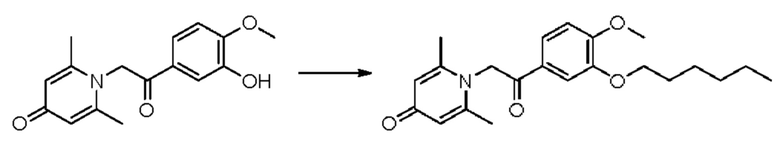
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он (50 мг, 0,17 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли 1-бромгексан (115 мг, 0,7 ммоль) и карбонат калия (97 мг, 0,7 ммоль) и перемешивали в атмосфере азота в течение 5 часов при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-н-гексилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-она (43 мг, выход 66%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,85 (дд, J=8,4, 2,0 Гц, 1H), 7,60 (д, J=2,0 Гц, 1H), 7,15 (д, J=8,4 Гц, 1H), 7,08 (с, 2Н), 6,04 (с, 2Н), 4,07 (т, J=6,4 Гц, 2Н), 3,96 (с, 3H), 2,53 (с, 6Н), 1,86-1,78 (м, 2Н), 1,54-1,46 (м, 2Н), 1,40-1,34 (м, 4Н), 0,93 (т, J=7,2 Гц, 3H); LC-MS: m/z 372,0 [М+Н]+.
Пример 19
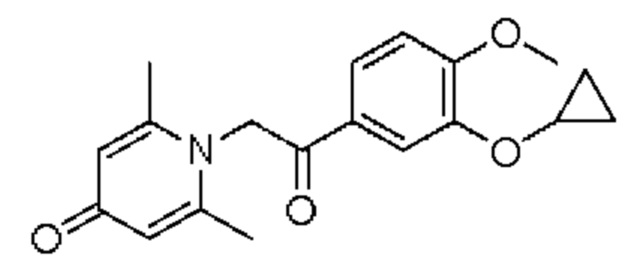
1-(2-(3-циклопропилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
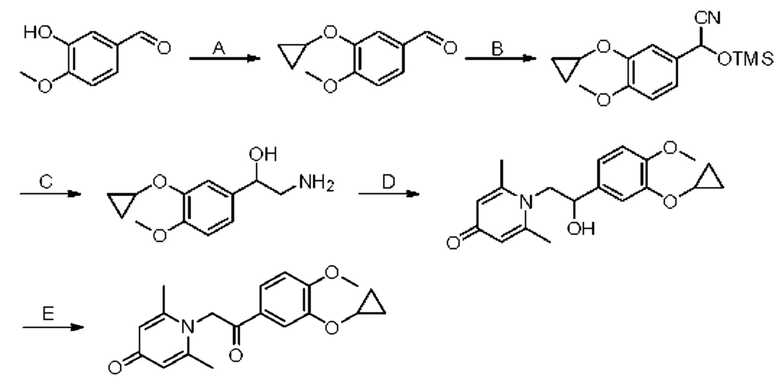
Стадия А:
Соединение 3-гидрокси-4-метоксибензальдегид (1 г, 6,6 ммоль), бромциклопропан (2,4 мг, 19,8 ммоль), карбонат цезия (6,5 г, 19,8 ммоль) и иодид калия (168 мг, 0,15 ммоль) растворяли в N,N-диметилформамиде (10 мл), перемешивали в течение 1 часа в запаянной трубке при 180°С в атмосфере азота, затем нагревали до 220°С и перемешивали еще 1 час. После завершения реакции добавляли насыщенный раствор соли (20 мл) и экстрагировали дихлорметаном (3×20 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали колоночной хроматографией с получением 3-циклопропилокси-4-метоксибензальдегида (170 мг, выход 13%, бледно-желтое масло). LC-MS: m/z 193,4 [М+Н]+.
Стадия В:
3-циклопропилокси-4-метоксибензальдегид (170 мг, 0,88 ммоль) растворяли в дихлорметане (4 мл); добавляли триэтиламин (356 мг, 3,52 ммоль) и триметилсилилцианид (349 мг, 3,52 ммоль) в атмосфере азота и перемешивали в течение 6 часов при комнатной температуре. После завершения реакции реакционную массу концентрировали и сушили на роторном испарителе с получением 2-(3-циклопропилокси-4-метоксифенил)-2-триметилсилоксиацетонитрила, который сразу использовали в следующей стадии.
Стадия С:
2-(3-циклопропилокси-4-метоксифенил)-2-триметилсилоксиацетонитрил, полученный на описанной выше стадии, растворяли в безводном тетрагидрофуране (20 мл), а затем порционно добавляли литийалюмогидрид (100 мг, 2,6 ммоль) на ледяной бане и перемешивали в течение ночи при комнатной температуре. После завершения реакции последовательно добавляли воду (0,1 мл), водный раствор гидроксида натрия (0,1 мл, 15%) и воду (0,3 мл), перемешивали в течение 30 минут, сушили над безводным сульфатом натрия и фильтровали. Фильтрат сушили на роторном испарителе с получением неочищенного продукта 2-амино-1-(3-циклопропилокси-4-метоксифенил)этанола (300 мг).
Стадия D:
2-Амино-1-(3-(циклопропилокси)-4-метоксифенил)этанол, полученный на описанной выше стадии, растворяли в этаноле (5 мл), затем добавляли 2,6-диметил-4H-пиран-4-он (124 мг, 1 ммоль) и водный раствор гидроксида натрия (2 М, 2 мл), и перемешивали в течение ночи при 60°С. Завершение реакции контролировали с помощью LCMS. Реакционную массу сушили на роторном испарителе и очищали колоночной хроматографией с получением 1-(2-(3-циклопропилокси-4-метоксифенил) -2-гидроксиэтил) -2,6-диметилпиридин-4 (1H) -она (100 мг, 34%, белое твердое вещество). LC-MS: m/z 330,1 [М+Н]+.
Стадия Е:
1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил)-2,6-диметилпиридин-4 (1Н)-он (30 мг, 0,1 ммоль) растворяли в дихлорметане (5 мл) и перемешивали в течение 2 часов при нормальной температуре. После завершения реакции реакционную массу фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-циклопропилокси-4-метоксифенил)-2-оксоэтил) -2,6-диметилпиридин- 4 (1Н) -она (15 мг, выход 45%, белое твердое вещество).
Пример 20
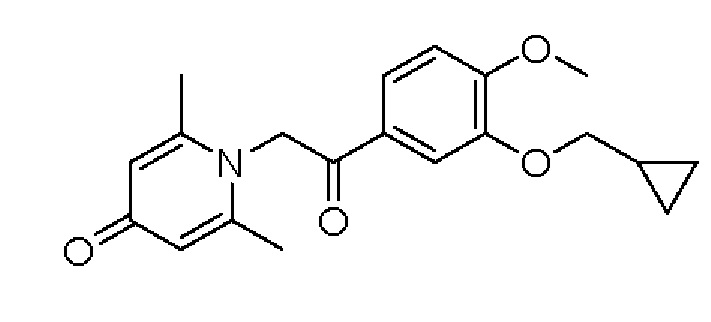
1-(2-(3-циклопропилметокси-4-метоксифенил)-2-оксо)этил-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
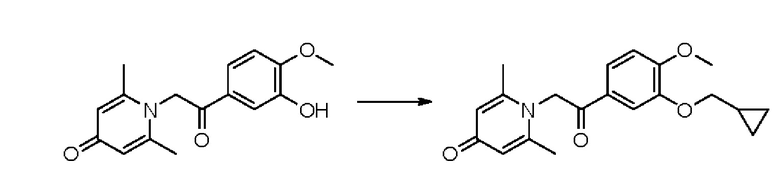
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил) -2,6-диметилпиридин -4(1Н)-он (50 мг, 0,17 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли бромметилциклопропан (27 мг, 0,2 ммоль) и карбонат калия (37 мг, 0,27 ммоль) и перемешивали в атмосфере азота в течение 2 часов при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-оксоэтил)-2,6-диметил пиридин -4 (1Н) -она (36 мг, выход 62%, белое твердое вещество). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7.79 (дд, J=8,4, 2,0 Гц, 1Н), 7,50 (д, J=2,0 Гц, 1Н), 7,16 (д, J=8,4 Гц, 1Н), 5,99 (с, 2Н), 5,62 (с, 2Н), 3,90 (с, 3Н), 3,53 (д, J=6,8 Гц, 2Н), 2,11 (с, 6Н), 1,26-1,21 (м, 1Н), 0,62-0,56 (м, 2Н), 0,36-0,31 (м, 2Н); LC-MS: m/z 342,1 [М+Н]+.
Пример 21
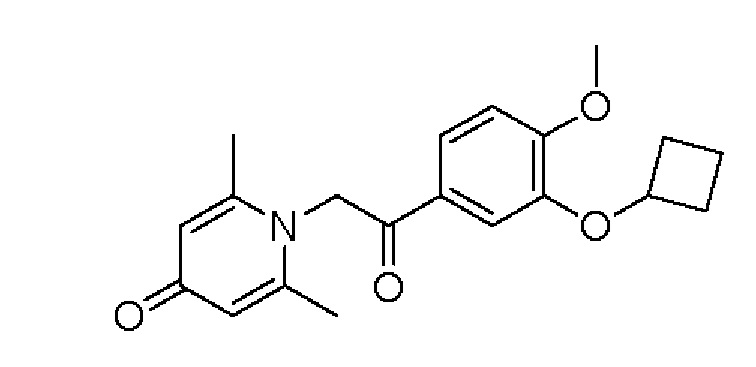
Конкретная схема реакции показана ниже:
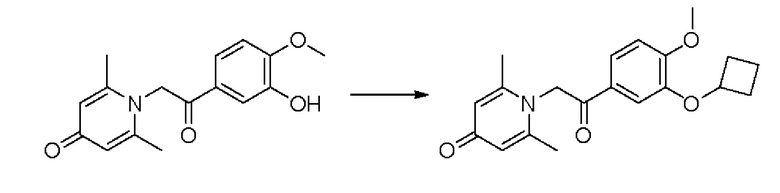
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил) -2,6-диметилпиридин -4(1Н)-он (50 мг, 0,17 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли циклобутилбромид (69 мг, 0,51 ммоль) и карбонат калия (71 мг, 0,51 ммоль) и перемешивали в атмосфере азота в течение 5 часов при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-циклобутилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметил пиридин-4(1H)-она (16 мг, выход 26%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,85 (д, J=8,4 Гц, 1Н), 7,45 (с, 1H), 7,14 (д, J=8,4 Гц, 1H), 7,04 (с, 2Н), 5,99 (с, 2Н), 4,79-4,73 (м, 1H), 3,96 (с, 3Н), 2,51 (с, 6Н), 2,51-2,45 (м, 1H), 2,23-2,13 (м, 1H), 1,91-1,83 (м, 1H), 1,79-1,70 (м, 1H); LC-MS: m/z 342,1 [М+Н]+.
Пример 22
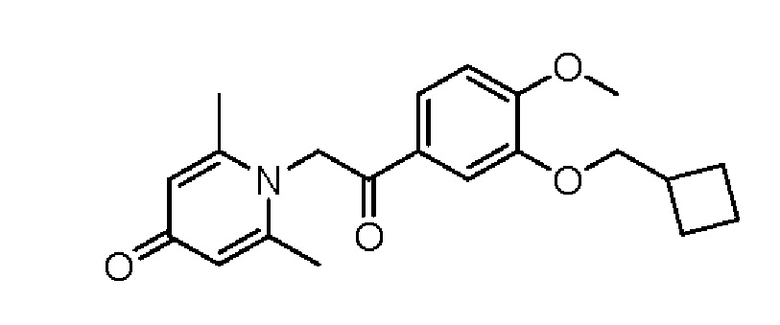
1-(2-(3-циклобутилметокси-4-метоксифенил)-2-оксо)этил-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
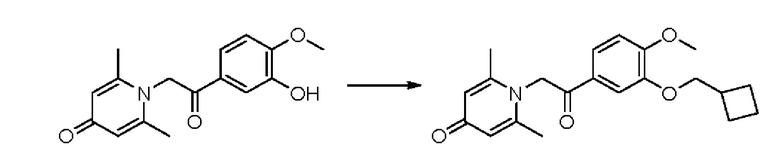
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил) -2,6-диметилпиридин -4(1H)-он (50 мг, 0,17 ммоль) растворяли в N, N-диметилформамиде (5 мл), затем добавляли бромметилциклобутан (76 мг, 0,51 ммоль) и карбонат калия (71 мг, 0,51 ммоль) и перемешивали в атмосфере азота в течение 4 часов при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-циклобутилметокси-4-метоксифенил)-2-оксоэтил)-2,6-диметил пиридин-4 (1Н) -она (16 мг, выход 24%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,84 (д, J=8,4 Гц, 1H), 7,61 (с, 1H), 7,14 (д, J=8,4 Гц, 1H), 6,91 (с, 2Н), 5,95 (с, 2Н), 4,04 (д, J=6,8 Гц, 2Н), 3,95 (с, 3Н), 2,84-2,78 (м, 1H), 2,47 (с, 6Н), 2,20-2,10 (м, 2Н), 2,02-1,86 (м, 4Н); LC-MS: m/z 356,2 [М+Н]+.
Пример 23
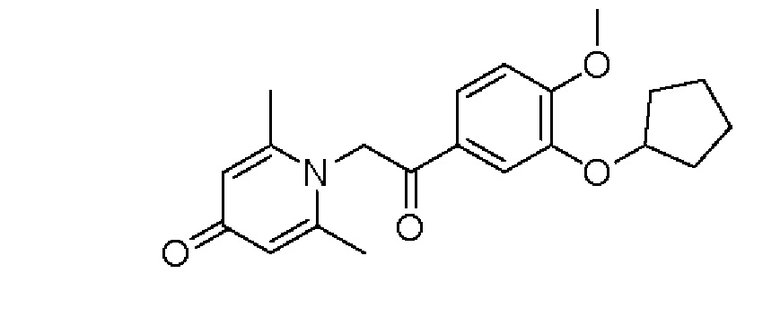
1-(2-(3-циклопентилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
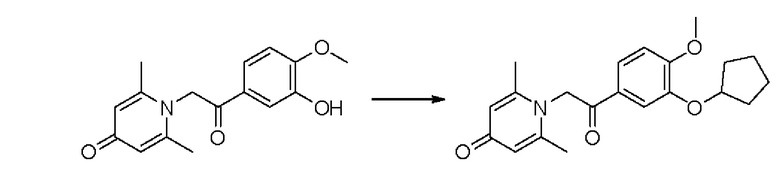
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин -4(1Н)-он (50 мг, 0,17 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли бромциклопентан (76 мг, 0,51 ммоль) и карбонат калия (71 мг, 0,51 ммоль) и перемешивали в атмосфере азота в течение 4 часов при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-циклопентил-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4 (1Н) -она (20 мг, выход 33%, белое твердое вещество). 1Н ЯМР (400 МГц, CD3OD) δ 7,84 (дд, J=8,4, 2,0 Гц, 1H), 7,60 (д, J=2,0 Гц, 1H), 7,14 (д, J=8,4 Гц, 1H), 6,39 (с, 2Н), 5,71 (с, 2Н), 4,95-4,91 (м, 1H), 3,96 (с, 3Н), 2,31 (с, 6Н), 2,01-1,80 (м, 6Н), 1,71-1,61 (м, 2Н); LC-MS m/z 356,2 [М+Н]+.
Пример 24
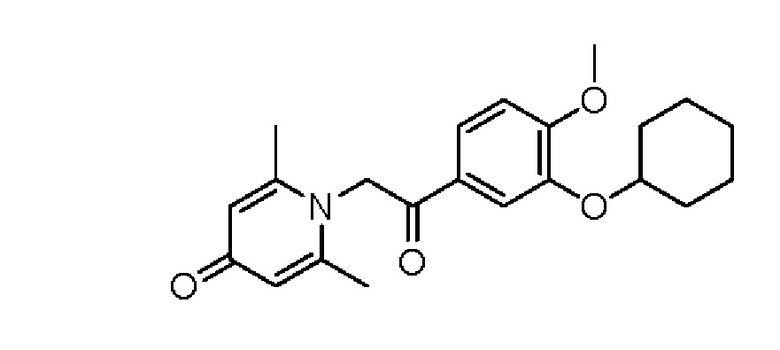
1-(2-(3-циклогексилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
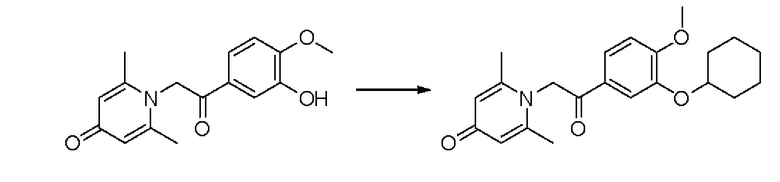
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил) -2,6-диметилпиридин -4(1Н)-он (50 мг, 0,17 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли бромциклогексан (83 мг, 0,51 ммоль) и карбонат калия (71 мг, 0,51 ммоль) и перемешивали в атмосфере азота в течение 4 часов при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-циклогексилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметил пиридин-4 (1H) -она (8 мг, выход 13%, белое твердое вещество). 1Н ЯМР (400 МГц, CD3OD) δ 7,86 (д, J=8,4 Гц, 1Н), 7,63 (с, 1H), 7,16 (д, J=8,4 Гц, 1H), 7,04 (с, 2Н), 6,00 (с, 2Н), 4,39-4,32 (м, 1H), 3,95 (с, 3Н), 2,52 (с, 6Н), 2,02-1,93 (м, 2Н), 1,86-1,77 (м, 2Н), 1,62-1,52 (м, 2Н), 1,45-1,28 (м, 4Н); LC-MS m/z 370,2 [М+Н]+.
Пример 25
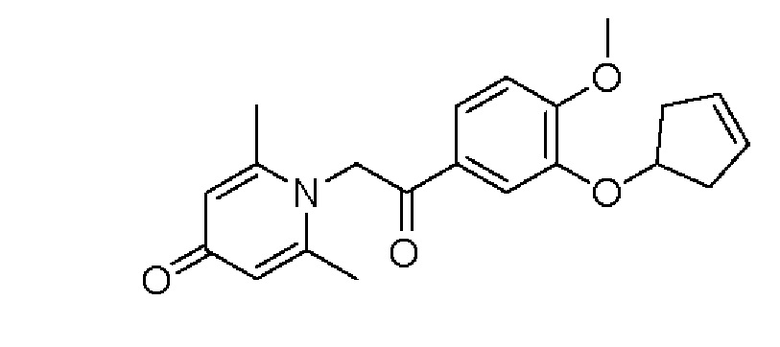
1-(2-(3-(циклопент-3-ен-1-илокси)-4-метоксифенил)-2-оксоэтил)-2,6-диметилпи-ридин-4(1H)-он
Конкретная схема реакции показана ниже:
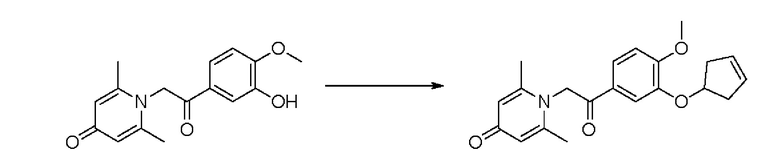
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил) -2,6-диметилпиридин -4(1H)-он (30 мг, 0,11 ммоль) растворяли в ацетонитриле (5 мл), затем добавляли циклопент-3-ен-1-илметансульфонат (68 мг, 0,42 ммоль) и карбонат калия (58 мг, 0,42 ммоль) и перемешивали в атмосфере азота в течение ночи при 80°С. После завершения реакции реакционную массу фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-(циклопент-3-ен-1-илокси) -4-метоксифенил)-2-оксоэтил) -2,6-диметилпиридин-4 (1H)-она (12 мг, выход 32%, белое твердое вещество). 1Н ЯМР (400 МГц, CDCl3) δ 7,85 (д, J=8,4 Гц, 1Н), 7,55 (с, 1Н), 7,18 (с, 2Н), 7,01 (д, J=8,4 Гц, 1Н), 6,04-5,91 (м, 2Н), 5,77 (с, 2Н), 5,17-5,11 (м, 1H), 3,96 (с, 3Н), 2,95-2,89 (м, 2Н), 2,63-2,60 (м, 2Н), 2,53 (с, 6Н); LC-MS m/z 354,2 [М+Н]+.
Пример 26
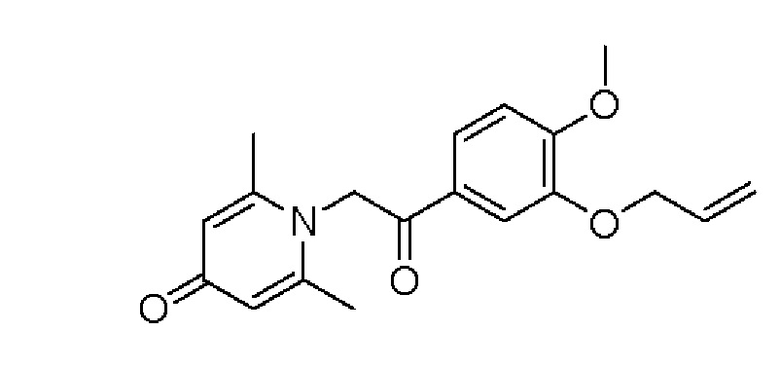
1-(2-(3-аллилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
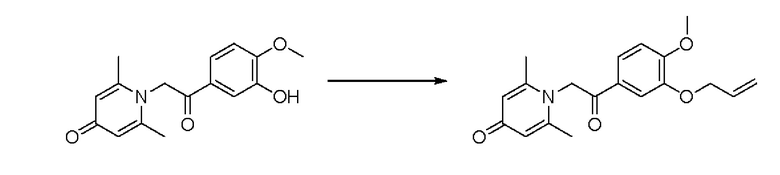
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил) -2,6-диметилпиридин -4(1Н)-он (50 мг, 0,18 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли 3-бромпропен (26 мг, 0,21 ммоль) и карбонат калия (37 мг, 0,27 ммоль) и перемешивали в атмосфере азота в течение ночи при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-аллилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н) -она (25 мг, выход 42%, белое твердое вещество). 1Н ЯМР (400 МГц, CD3OD) δ 7,83 (д, J=8,4 Гц, 1H), 7,61 (с, 1H), 7,13 (д, J=8,4 Гц, 1H), 6,34 (с, 2Н), 6,14-6,03 (м, 1H), 5,68 (с, 2Н), 5,43 (д, J=17,2 Гц, 1Н), 5,27 (д, J=10,4 Гц, 1Н), 4,64 (д, J=4,8 Гц, 2Н), 3,95 (с, 3Н), 2,26 (с, 6Н), LC-MS m/z 328,2 [М+Н]+.
Пример 27
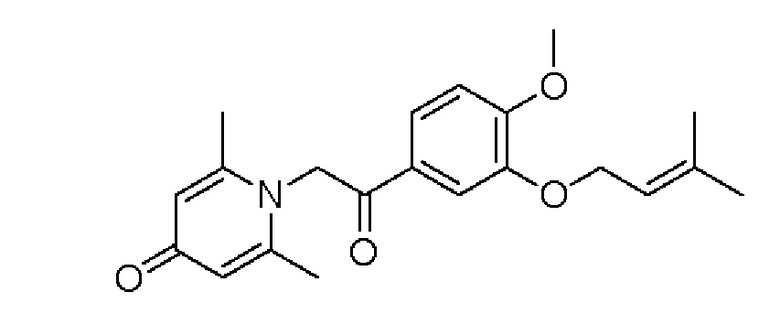
1-(2-(3-((3-метилбут-2-ен-1-ил)окси)-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
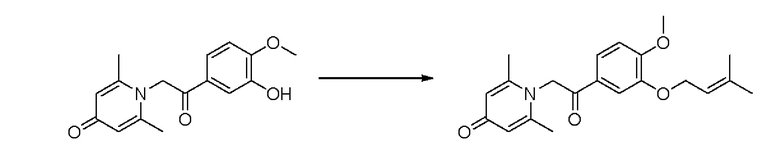
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил) -2,6-диметилпиридин -4(1Н)-он (50 мг, 0,18 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли 1-бром-3-метил-2-бутен (31 мг, 0,21 ммоль) и карбонат калия (37 мг, 0,27 ммоль) и перемешивали в атмосфере азота в течение 1 часа при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-((3-метилбут-2-ен-1-ил)окси)-4-метоксифенил)-2-оксоэтил)-2,6-ди-метилпиридин-4 (1H)-она (25 мг, выход 40%, белое твердое вещество). 1Н ЯМР (400 МГц, CDCl3) δ 7,63 (д, J=8,4 Гц, 1H), 7,55 (д, J=2,0 Гц, 1H), 6,96 (д, J=8,4 Гц, 1H), 6,32 (с, 2Н), 5,51 (д, J=6,8 Гц, 1Н), 5,32 (с, 2Н), 4,65 (д, J=6,8 Гц, 2Н), 3,98 (с, 3Н), 2,21 (с, 6Н), 1,79 (с, 3Н), 1,77 (с, 3Н); LC-MS m/z 356,2 [М+Н]+.
Пример 28
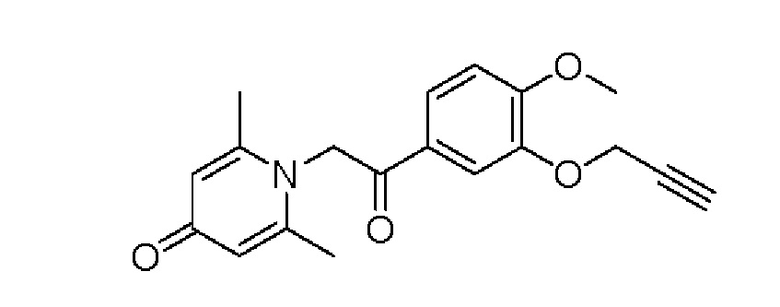
1-(2-(3-пропаргилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
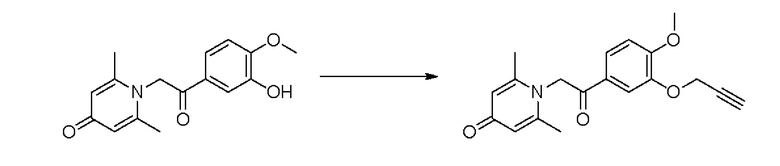
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил) -2,6-диметилпиридин -4(1Н)-он (50 мг, 0,18 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли пропаргилбромид (25 мг, 0,21 ммоль) и карбонат калия (37 мг, 0,27 ммоль) и перемешивали в атмосфере азота в течение 4 часов при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-пропаргилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4 (1Н) -она (21 мг, выход 35%, белое твердое вещество). 1Н ЯМР (400 МГц, CDCl3) δ 7,82 (д, J=8,0 Гц, 1H), 7,70 (с, 1H), 7,04 (д, J=8,0 Гц, 1H), 7,03 (с, 2Н), 5,78 (с, 2Н), 4,85 (д, J=2,4 Гц, 2Н), 4,00 (с, 3Н), 2,56 (т, J=2,4 Гц, 3Н), 2,46 (с, 6Н); LC-MS: m/z 326,3 [М+Н]+.
Пример 29
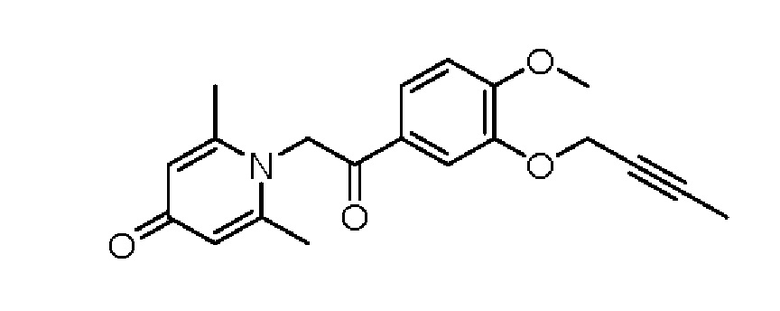
1-(2-(3-(бут-2-ин-1-илокси)-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
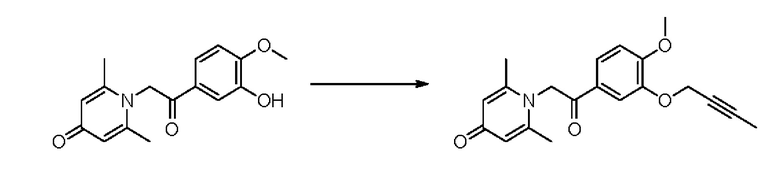
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил) -2,6-диметилпиридин -4(1Н)-он (50 мг, 0,18 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли 1-бром-2-бутин (83 мг, 0,70 ммоль) и карбонат калия (97 мг, 0,70 ммоль) и перемешивали в атмосфере азота в течение 2 часов при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-(бут-2-ин- 1-илокси)-4-метоксифенил)-2-оксоэтил)-2,6-диметил пиридин-4 (1Н) -она (14 мг, выход 23%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,90 (дд, J=8,4, 2,0 Гц, 1H), 7,72 (д, J=2,0 Гц, 1H), 7,18 (д, J=8,4 Гц, 1H), 7,09 (с, 2Н), 6,03 (с, 2Н), 4,78 (к, J=2,4 Гц, 2Н), 3,96 (с, 3Н), 2,54 (с, 6Н), 1,82 (т, J=2,4 Гц, 3Н); LC-MS: m/z 340,0 [М+Н]+.
Пример 30
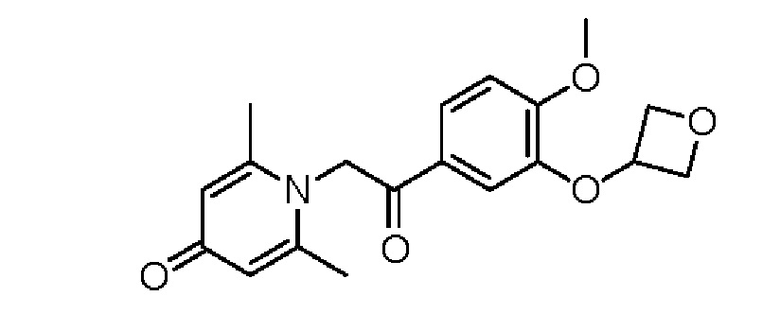
1-(2-(3-(оксициклобутан-3-ил-окси)-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он
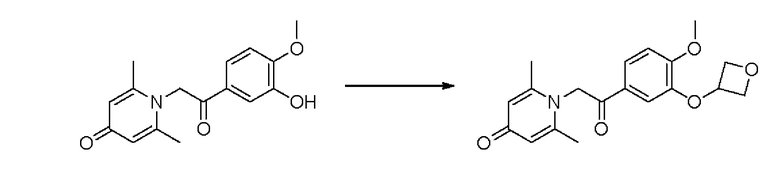
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил) -2,6-диметилпиридин -4(1H)-он (158 мг, 0,55 ммоль), оксициклобутан-3-ол (81,4 мг 1,1 ммоль) и трифенилфосфин (288 мг, 1,1 ммоль) растворяли в безводном тетрагидрофуране (10 мл), реакционную массу охлаждали на ледяной бане, затем добавляли по каплям диизопропилазодикарбоксилат (222 мг, 1,1 ммоль) в течение 5 мин в атмосфере азота, а затем проводили реакцию при комнатной температуре в течение 24 часов. После завершения реакции добавляли насыщенный раствор соли (20 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-(оксициклобутан-3-илокси) -4-метокси фенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он (45 мг, выход 24%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,84 (дд, J=8,4, 2,0 Гц, 1H), 7,31 (д, J=2,0 Гц, 1H), 7,23 (д, J=8,4 Гц, 1H), 7,13 (с, 2Н), 6,06 (с, 2Н), 5,38-5,32 (м, 1H), 5,06-5,02 (м, 2Н), 4,78-4,75 (м, 2Н), 4,00 (с, 3Н), 2,56 (с, 6Н); LC-MS: m/z 344,4 [М+Н]+.
Пример 31
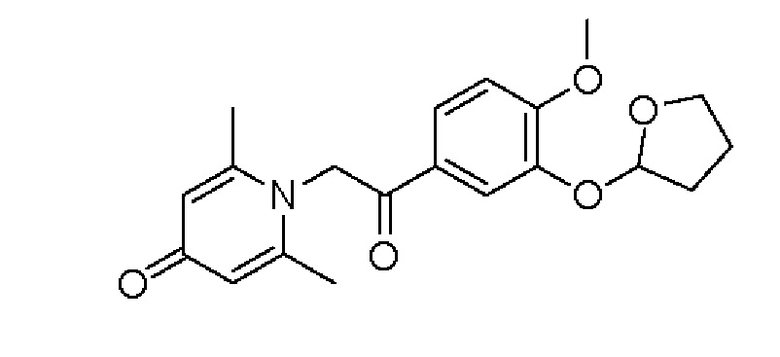
1-(2-(3-(тетрагидрофуран-2-ил)окси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
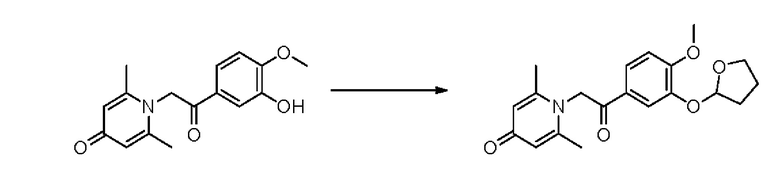
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил) -2,6-диметилпиридин -4(1Н)-он (50 мг, 0,17 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 2,3-дигидрофуран (68 мг, 0,42 ммоль) и толуол-4-сульфонат пиридиния (4,6 мг, 0,018 ммоль) и перемешивали в течение ночи при комнатной температуре. После завершения реакции реакционную массу фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-(тетрагидрофуран-2-ил) окси-4- метоксифенил)-2-оксоэтил) -2,6-диметилпиридин-4(1H)-он (28 мг, выход 44%, белое твердое вещество). 1Н ЯМР (400 МГц, CD3OD) δ 7,87 (дд, J=8,4, 2,0 Гц, 1H), 7,81 (д, J=2,0 Гц, 1Н), 7,15 (д, J=8,4 Гц, 1Н), 6,40 (с, 2Н), 5,87 (д, J=4,4 Гц, 1H), 5,70 (с, 2Н), 4,07-4,02 (м, 1Н), 3,97-3,92 (м, 1Н), 3,94 (с, 3Н), 2,31-2,11 (м, 4Н), 2,29 (с, 6Н); LC-MS m/z 358.1 [М+Н]+.
Пример 32
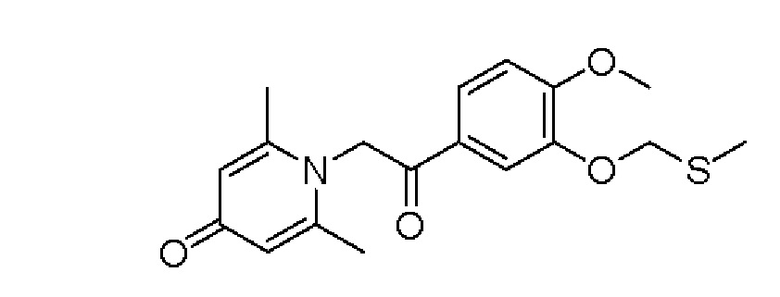
1-(2-(3-метилтиометокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
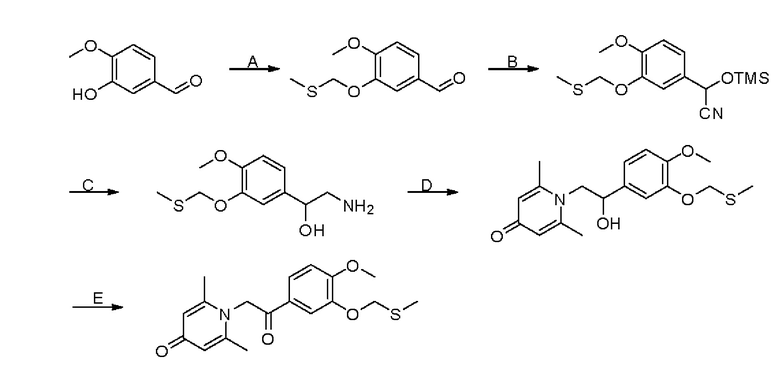
Стадия А:
3-Гидрокси-4-метоксибензальдегид (2 г, 13 ммоль) растворяли в N,N-диметилформамиде (20 мл) в атмосфере азота, затем добавляли хлорметил метил сульфид (1,5 г, 15,6 ммоль) и карбонат цезия (6 г, 19,5 ммоль) и перемешивали в атмосфере азота в течение ночи при комнатной температуре. После завершения реакции добавляли насыщенный раствор соли (40 мл) и экстрагировали дихлорметаном (3×30 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали колоночной хроматографией с получением 3-метилтиометокси-4-метоксибензальдегида (1,7 г, выход 61%, желтое масло). LC-MS: m/z 213,1 [М+Н]+.
Стадия В:
3-Метилтиометокси-4-метоксибензальдегид (1,7 г, 8 ммоль) растворяли в дихлорметане (20 мл); добавляли в атмосфере азота триэтиламин (1,6 г, 16 ммоль) и триметилсилилцианид (1,6 г, 16 ммоль) и перемешивали в течение 6 часов при комнатной температуре. Реакционную массу концентрировали и сушили на роторном испарителе с получением 2-(3-метилтиометокси-4-метоксифенил)-2-триметилсилоксиацетонитрила, который сразу использовали в следующей стадии.
Стадия С:
2-(3-Метилтиометокси- 4- метоксифенил) -2- триметилсилоксиацетонитрил, полученный на описанной выше стадии, растворяли в безводном тетрагидрофуране (50 мл), а затем порционно добавляли литийалюмогидрид (608 мг, 16 ммоль) на ледяной бане и перемешивали в течение ночи при комнатной температуре. После завершения реакции последовательно добавляли воду (0,6 мл), водный раствор гидроксида натрия (0,6 мл, 15%) и воду (1,8 мл), перемешивали в течение 30 минут, сушили над безводным сульфатом натрия и фильтровали. Фильтрат сушили на роторном испарителе с получением неочищенного продукта 2-амино-1- (3-метилтиометокси-4-метоксифенил) этанола.
Стадия D:
2-Амино-1-(3-(метилтиометокси)-4-метоксифенил) этанол, полученный на описанной выше стадии, растворяли в этаноле (10 мл), затем добавляли 2,6-диметил-4-пиранон (1,24 г, 10 ммоль) и водный раствор гидроксида натрия (2 М, 10 мл) и перемешивали в течение ночи при 60°С. Завершение реакции контролировали с помощью LCMS. Реакционную массу сушили на роторном испарителе и очищали колоночной хроматографией с получением 1-(2-(3-метилтиометокси-4-метоксифенил) -2-гидроксиэтил) -2,6-диметилпиридин-4 (1Н) -она (1 г, 35%). LC-MS: m/z 350,1 [М+Н]+.
Стадия Е:
1-(2-(3-Метилтиометокси-4-метоксифенил)-2-гидроксиэтил)-2,6-диметилпиридин-4(1Н)-он (50 мг, 0,14 ммоль) растворяли в диметилсульфоксиде (5 мл), затем медленно добавляли комплекс триоксида серы и пиридина (111 мг, 0,7 ммоль) в ДМСО (2,5 мл) и перемешивали в течение ночи при нормальной температуре. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-метилтиометокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин -4 (1Н) -она (25 мг, выход 51%, белое твердое вещество). 1Н ЯМР (400 МГц, CDCl3) δ 7,67 (дд, J=8,4, 2,0 Гц, 1H), 7,55 (д, J=2,0 Гц, 1H), 7,00 (д, J=8,4 Гц, 1H), 6,28 (с, 2Н), 5,31 (с, 2Н), 5,23 (с, 2Н), 3,93 (с, 3Н), 3,68 (с, 3Н), 2,12 (с, 6Н); LC-MS: m/z 348,2 [М+Н]+.
Пример 33
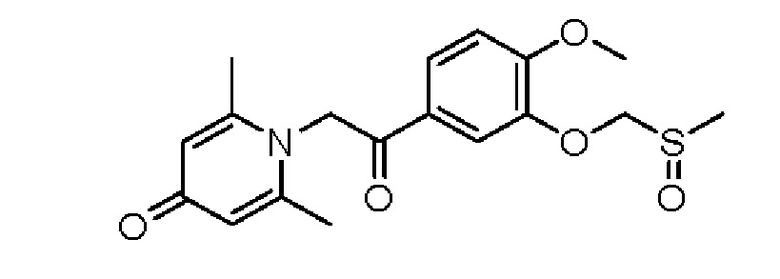
1-(2-(3-мети лсульфоксидметокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
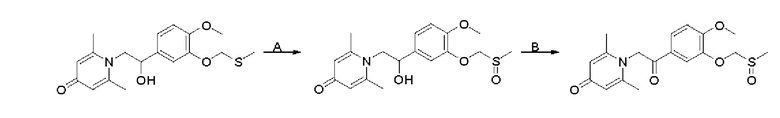
Стадия А:
1-(2-(3-Метилтиометокси-4-метоксифенил)-2-гидроксиэтил)-2,6-диметилпиридин-4(1Н)-он (100 мг, 0,29 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (56 мг, 0,28 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Затем добавляли насыщенный водный раствор сульфита натрия (5 мл) и перемешивали в течение 10 минут. Реакционную массу экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали колоночной хроматографией с получением 1-(2-(3-метилсульфоксидметокси-4-метоксифенил)-2-гидроксиэтил)-2,6-диметилпиридин-4(1Н)-она (38 мг, выход 36%, белое твердое вещество). LC-MS: m/z 366.2 [М+Н]+.
Стадия В:
1-(2-(3-Метилсульфоксидметокси-4-метоксифенил)-2-гидроксиэтил)-2,6-диметилпиридин-4(1Н)-он (38 мг, 0,1 ммоль) растворяли в дихлорметане (10 мл), затем добавляли периодинан Десса-Мартина (85 мг, 0,2 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Реакционную массу фильтровали и промывали насыщенным раствором соли. Органическую фазу сушили над безводным сульфатом натрия, фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-метилсульфоксидметокси-4-метоксифенил)-2-оксоэтил) -2,6-диметилпиридин-4(1Н)-она (10 мг, выход 37%, белое твердое вещество). 1Н ЯМР (400 МГц, CDCl3) δ 7,93 (д, J=2,0 Гц, 1H), 7,74 (дд, J=8,4, 2,0 Гц, 1H), 6,98 (д, J=8,4 Гц, 1H), 6,26 (с, 2Н), 5,25 (с, 2Н), 5,23 (с, 2Н), 3,93 (с, 3Н), 3,68 (с, 3Н), 2,12 (с, 6Н); LC-MS: m/z 364,2 [М+Н]+.
Пример 34
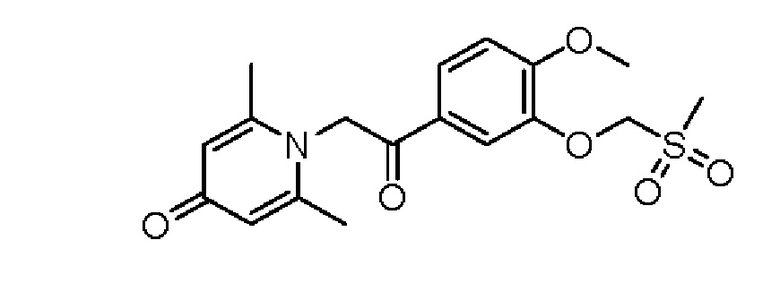
1-(2-(3-метилсульфонметокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
Стадия А:
1-(2-(3-Метилтиометокси-4-метоксифенил)-2-гидроксиэтил)-2,6-диметилпиридин-4 (1Н) -он (100 мг, 0,29 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (176 мг, 0,87 ммоль) и перемешивали в течение ночи при комнатной температуре. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл) и перемешивали в течение 10 минут. Реакционную массу экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали колоночной хроматографией с получением 1-(2-(3-метилсульфонметокси-4-метоксифенил) -2-гидроксиэтил)-2,6-диметилпиридин-4(1Н)-она (50 мг, выход 55%, белое твердое вещество). LC-MS: m/z 382,2 [М+Н]+.
Стадия В:
1-(2-(3-метилсульфонметокси-4-метоксифенил)-2-гидроксиэтил)-2,6-диметилпиридин-4 (1Н) -он (50 мг, 0,13 ммоль) растворяли в дихлорметане (10 мл), затем добавляли периодинан Десса-Мартина (110 мг, 0,26 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Реакционную массу фильтровали и промывали насыщенным раствором соли. Органическую фазу сушили над безводным сульфатом натрия, фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-метилсульфоксидметокси-4-метоксифенил)-2-оксоэтил) -2,6-диметилпиридин- 4(1H)-она (30 мг, выход 61%, белое твердое вещество). 1Н ЯМР (400 МГц, CDCl3) δ 7,84 (дд, J=8,4, 2,0 Гц, 1H), 7,70 (д, J=2,0 Гц, 1Н), 7,10 (д, J=8,4 Гц, 1H), 6,26 (с, 2Н), 5,35 (с, 2Н), 5,23 (с, 2Н), 3,98 (с, 3Н), 3,78 (с, 3Н), 2,25 (с, 6Н); LC-MS: m/z 380,1 [М+Н]+.
Пример 35
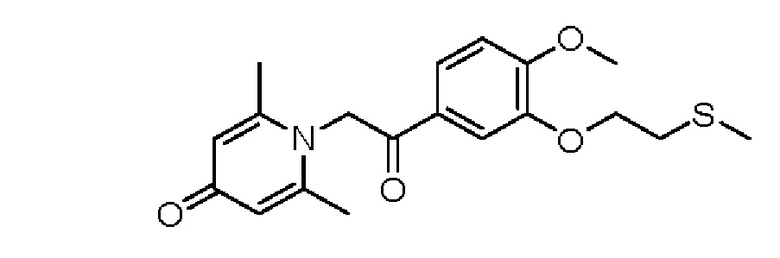
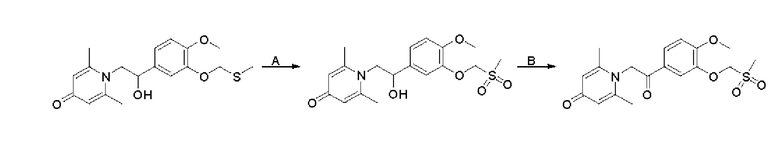
1-(2-(3-метилтиоэтокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
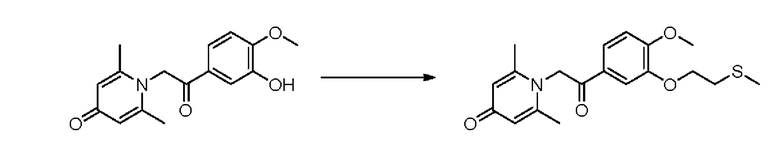
Соединение 1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин -4(1Н)-он (287 мг, 1 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли хлорэтил метил сульфид (166 мг, 1,5 ммоль) и карбонат калия (276 мг, 2 ммоль) и перемешивали при комнатной температуре в течение 2 часов. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-метилтиоэтокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4 (1Н)-она 200 мг, выход 55%, белое твердое вещество). 1Н ЯМР (400 МГц, CDCl3) δ 7,66 (дд, J=8,4, 2,0 Гц, 1H), 7,56 (д, J=2,0 Гц, 1H), 6,98 (д, J=8,4 Гц, 1H), 6,28 (с, 2Н), 5,31 (с, 2Н), 4,27 (т, J=6,8 Гц, 2Н), 3,98 (с, 3Н), 2,95 (т, J=6,8 Гц, 2Н), 2,24 (с, 3Н), 2,19 (с, 6Н); LC-MS: m/z 362,2 [М+Н]+.
Пример 36
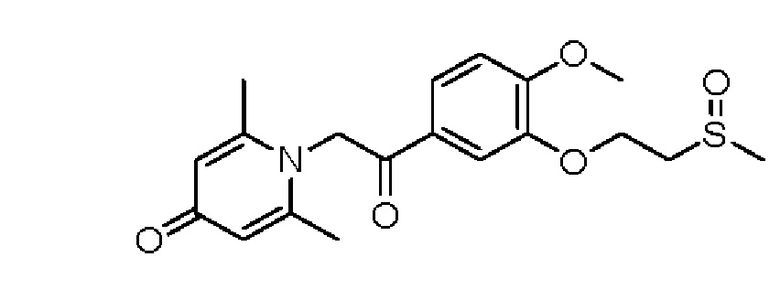
1-(2-(3-метилсульфоксидэтокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
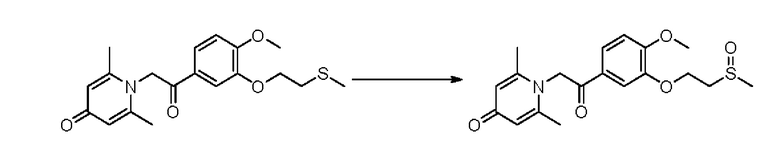
Соединение 1-(2-(3-метилтиоэтокси-4-метоксифенил)-2-оксоэтил)-2,6-диметил пиридин-4(1Н)-он (50 мг, 0,15 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (28 мг, 0,14 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (Е)-1-(3-метилсульфоксидэтокси -4-метоксистирил)-2,6-диметилпиридин-4(1Н)-она (20 мг, выход 40%, бесцветное масло). 1Н ЯМР (400 МГц, CDCl3) δ 7,71 (дд, J=8,4, 2,0 Гц, 1H), 7,66 (д, J=2,0 Гц, 1H), 6,99 (д, J=8,4 Гц, 1H), 6,26 (с, 2Н), 5,32 (с, 2Н), 4,57-4,54 (м, 2Н), 3,97 (с, 3Н), 3,35-3,31 (м, 1H), 3,15-3,09 (м, 1H), 2,75 (с, 3Н), 2,18 (с, 6Н); LC-MS: m/z 378,2 [М+Н]+.
Пример 37
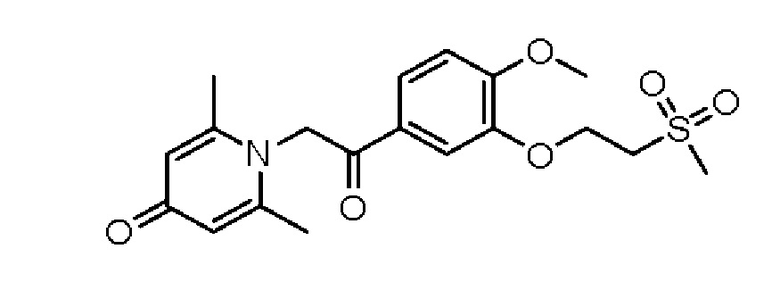
1-(2-(3-метилсульфонэтокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
Соединение 1-(2-(3-метилтиоэтокси-4-метоксифенил)-2-оксоэтил)-2,6-диметил пиридин-4(1Н)-он (50 мг, 0,15 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (71 мг, 0,35 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-метилсульфонэтокси-4-метоксистирил)-2,6-диметилпиридин-4 (1Н)-она (20 мг, выход 36%, белое твердое вещество). 1Н ЯМР (400 МГц, CDCl3) δ 7,74 (дд, J=8,4, 2,0 Гц, 1H), 7,60 (д, J=2,0 Гц, 1Н), 7,00 (д, J=8,4 Гц, 1H), 6,26 (с, 2Н), 5,30 (с, 2Н), 4,53 (т, J=4,2 Гц, 2Н), 3,95 (с, 3Н), 3,51 (т, J=4,2 Гц, 2Н), 3,18 (с, 3Н), 2,18 (с, 6Н); LC-MS: m/z 394,2 [М+Н]+.
Пример 38
(Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(гидроксиимидо)этил)-2,6-диметилпиридин-4(1Н)-он
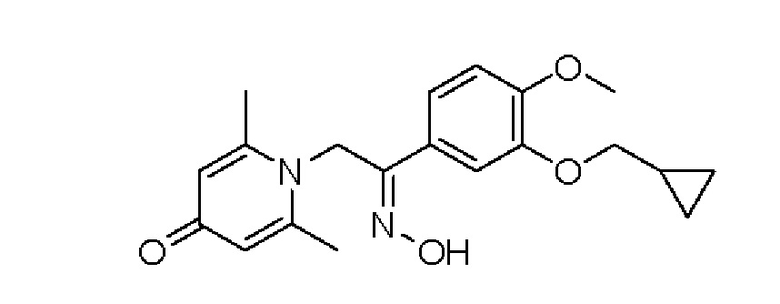
(E)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(гидроксиимидо)этил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
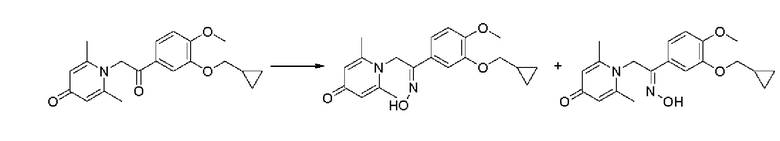
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он (341 мг, 1 ммоль) и гидрохлорид гидроксиламина (139 мг, 2 ммоль) растворяли в пиридине (10 мл) и перемешивали при кипении с обратным холодильником в атмосфере азота в течение 24 часов. После завершения реакции пиридин сушили на роторном испарителе и реакционную массу очищали обращенно-фазовой ВЭЖХ с получением (Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(гидроксиимидо)этил) -2,6-диметилпиридин-4(1Н)-она (96 мг, выход 27%, белое твердое вещество) и (Е)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(гидроксиимидо)этил)-2,6-диметилпиридин-4 (1H)-она (43 мг, выход 12%, белое твердое вещество).
(Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(гидроксиимидо)этил)-2,6-диметилпиридин-4(1Н)-он. 1Н ЯМР (400 МГц, CDCl3) δ 7,45 (д, J=2,0 Гц, 1Н), 7,30 (д, J=8,4 Гц, 1Н), 6,93 (дд, J=8,4, 2,0 Гц, 1H), 6,48 (с, 2Н), 5,00 (с, 2Н), 3,92 (с, 3Н), 3,89 (д, J=6,8 Гц, 2Н), 2,41 (с, 6Н), 1,42-1,32 (м, 1Н), 0,68-0,62 (м, 2Н), 0,40-0,35 (м, 2Н); LC-MS: m/z 357,4 [М+Н]+.
(Е)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(гидроксиимидо)этил)-2,6-диметилпиридин-4(1Н)-он. 1Н ЯМР (400 МГц, CDCl3) δ 7,50 (д, J=2,0 Гц, 1H), 7,13 (д, J=8,4 Гц, 1H), 6,93 (дд, J=8,4, 2,0 Гц, 1H), 6,49 (с, 2Н), 5,00 (с, 2Н), 3,92 (с, 3Н), 3,89 (д, J=6,8 Гц, 2Н), 2,41 (с, 6Н), 1,42-1,32 (м, 1H), 0,68-0,62 (м, 2Н), 0,40-0,35 (м, 2Н); LC-MS: m/z 357,4 [М+Н]+.
Пример 39
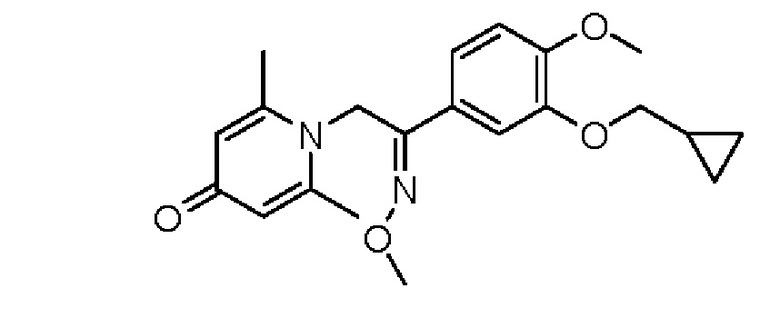
(Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(метоксиимидо)этил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
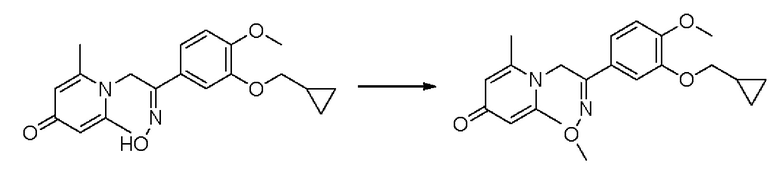
Соединение (Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(гидроксиимидо) этил)-2,6-диметилпиридин-4(1Н)-он (36 мг, 0,1 ммоль) растворяли в N,N-диметилформамиде (2 мл), затем добавляли 60% гидрид натрия (12 мг, 0,3 ммоль) при 0°С и перемешивали в течение 30 минут. Затем добавляли йодметан (28,4 мг, 0,2 ммоль) и перемешивали при комнатной температуре в течение 4 часов. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(метоксиимидо)этил)-2,6- диметилпиридин-4 (1Н)-она (20 мг, выход 55%, белое твердое вещество). 1Н ЯМР (400 МГц, CDCl3) δ 7,46 (д, J=2,0 Гц, 1H), 7,32 (д, J=8,4 Гц, 1H), 7,02 (дд, J=8,4, 2,0 Гц, 1Н), 6,52 (с, 2Н), 5,02 (с, 2Н), 3,93 (с, 3Н), 3,92 (с, 3Н), 3,89 (д, J=6,8 Гц, 2Н), 2,45 (с, 6Н), 1,32-1,23 (м, 1H), 0,70-0,65 (м, 2Н), 0,50-0,38 (м, 2Н); LC-MS: m/z 371,2 [М+Н]+.
Пример 40
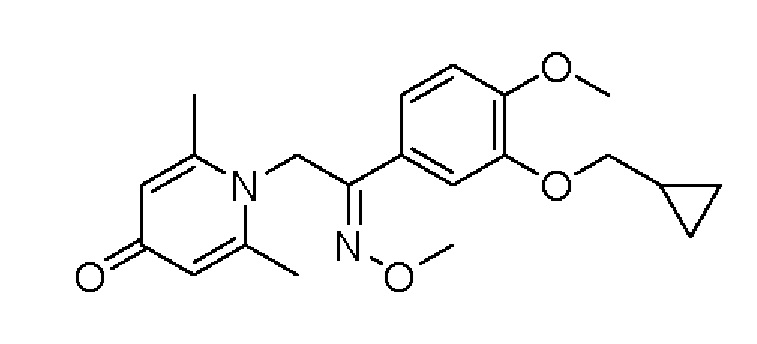
(Е)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(гидроксиметилимидо)этил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
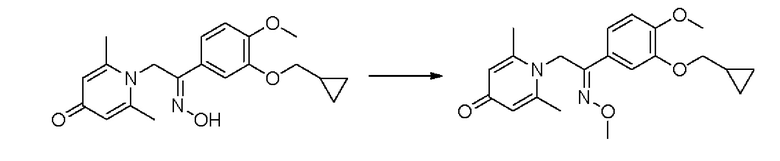
Соединение (Е)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(гидроксиимидо)этил)-2,6-диметилпиридин-4(1H)-он (36 мг, 0,1 ммоль) растворяли в N,N-диметилформамиде (2 мл), затем добавляли 60% гидрид натрия (12 мг, 0,3 ммоль) при 0°С и перемешивали в течение 30 минут. Затем добавляли йодметан (28,4 мг, 0,2 ммоль) и перемешивали при комнатной температуре в течение 4 часов. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(метоксиимидо)этил)-2,6-диметилпиридин-4(1Н)-она (18 мг, выход 49%, белое твердое вещество). 1Н ЯМР (400 МГц, CDCl3) δ 7,51 (д, J=2,0 Гц, 1H), 7,11 (д, J=8,4 Гц, 1H), 7,00 (дд, J=8,4, 2,0 Гц, 1H), 6,46 (с, 2Н), 5,00 (с, 2Н), 3,95 (с, 3Н), 3,93 (с, 3Н), 3,88 (д, J=6,8 Гц, 2Н), 2,49 (с, 6Н), 1,42-1,32 (м, 1H), 0,68-0,62 (м, 2Н), 0,40-0,35 (м, 2Н); LC-MS: m/z 371,2 [М+Н]+.
Пример 41
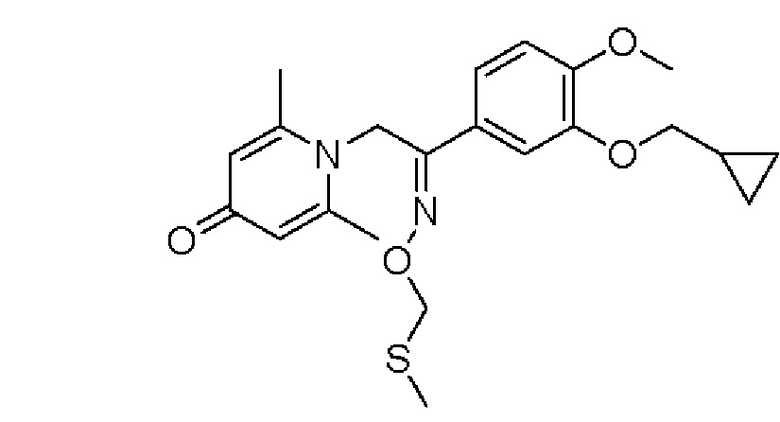
(Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(метилтиометоксиимидо)этил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
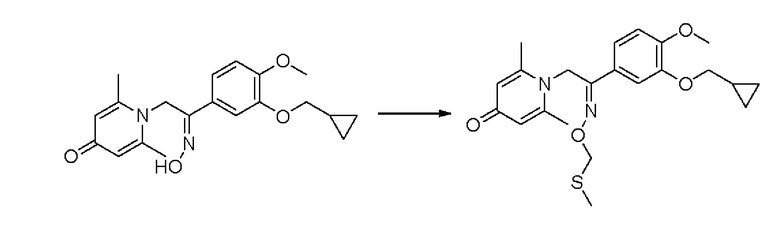
Соединение (Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(гидроксиимидо) этил)-2,6-диметилпиридин-4(1Н)-он (300 мг, 0,84 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли карбонат цезия (548 мг, 1,68 ммоль) и хлорметил метил сульфид (122 мг, 1,26 ммоль) и перемешивали при комнатной температуре в течение ночи. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (Z)-1-(2-(3-циклопропилметокси -4-метоксифенил)-2- (метилтиометоксиимидо) этил) -2,6-диметилпиридин-4 (1H)-она (200 мг, выход 57%, белое твердое вещество). 1Н ЯМР (400 МГц, CD3OD) δ 7,23 (д, J=8,4 Гц, 1Н), 7,18 (с, 1H), 7,08 (д, J=8,4 Гц, 1H), 6,96 (с, 2Н), 6,35 (с, 2Н), 5,18 (с, 2Н), 3,91 (с, 2Н), 3,85 (д, J=6,8 Гц, 2Н), 2,47 (с, 6Н), 2,40 (с, 3Н), 1,36-1,28 (м, 1Н), 0,69-0,64 (м, 2Н), 0,35-0,32 (м, 2Н); LC-MS: m/z 417,1 [М+Н]+.
Пример 42
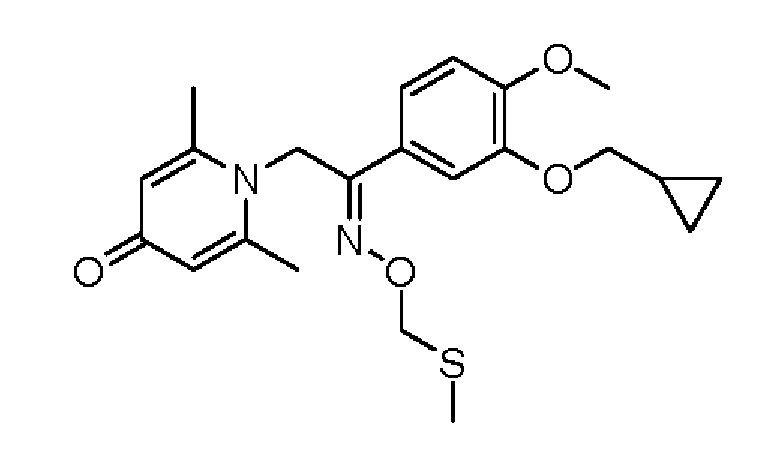
(Е)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(метилтиометоксиимидо)этил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
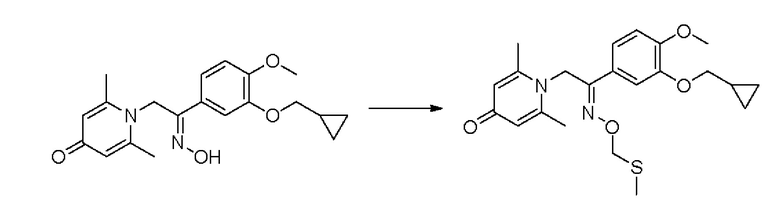
Соединение (Е)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(гидроксиимидо) этил)-2,6-диметилпиридин-4(1Н)-он (36 мг, 0,1 ммоль) растворяли в N,N-диметилформамиде (2 мл), затем добавляли карбонат цезия (65 мг, 0,2 ммоль) и хлорметил метил сульфид (15 мг, 0,15 ммоль) и перемешивали при комнатной температуре в течение ночи. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ дали (Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(метилтиометоксиимидо)этил)-2,6-диметилпиридин-4(1Н)-она (20 мг, выход 48%, белое твердое вещество). 1Н ЯМР (400 МГц, CD3OD) δ 7,36 (д, J=8,4 Гц, 1H), 7,25 (с, 1H), 6,88 (д, J=8,4 Гц, 1H), 6,86 (с, 2Н), 6,38 (с, 2Н), 5,10 (с, 2Н), 3,96 (с, 2Н), 3,90 (д, J=6,8 Гц, 2Н), 2,47 (с, 6Н), 2,40 (с, 3Н), 1,36-1,28 (м, 1H), 0,69-0,64 (м, 2Н), 0,35-0,32 (м, 2Н); LC-MS: m/z 417,1 [М+Н]+.
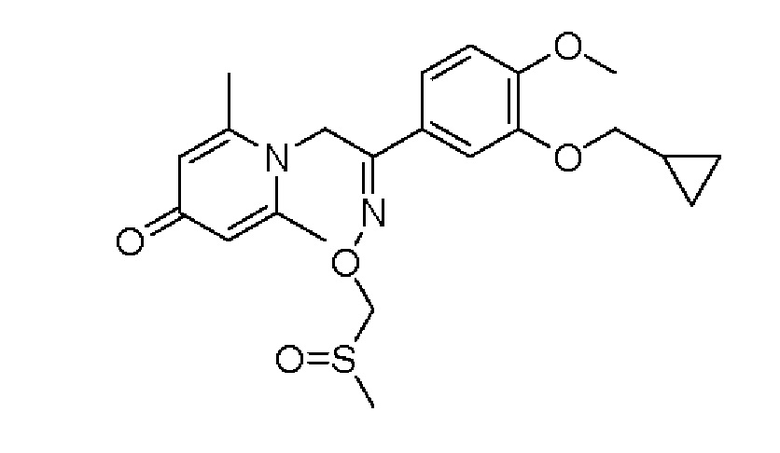
(Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(метилсульфоксидметоксиимидо)этил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
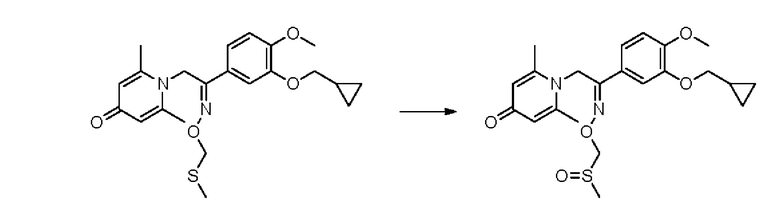
Соединение (Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(метилтиометоксиимидо)этил)-2,6-диметилпиридин-4(1H)-он (42 мг, 0,1 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (18 мг, 0,09 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (2)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(метилсульфоксидметоксиимидо) этил)-2,6-диметилпиридин-4(1H)-она (20 мг, выход 46%, белое твердое вещество). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,28 (д, J=8,4 Гц, 1H), 7,21 (с, 1Н), 6,96 (д, J=8,4 Гц, 1Н), 6,90 (с, 2Н), 6,36 (с, 2Н), 5,15 (с, 2Н), 3,94 (с, 2Н), 3,88 (д, J=6,8 Гц, 2Н), 2,46 (с, 6Н), 2,42 (с, 3Н), 1,38-1,30 (м, 1H), 0,69-0,65 (м, 2Н), 0,38-0,33 (м, 2Н); LC-MS: m/z 433,2 [М+Н]+.
Пример 44
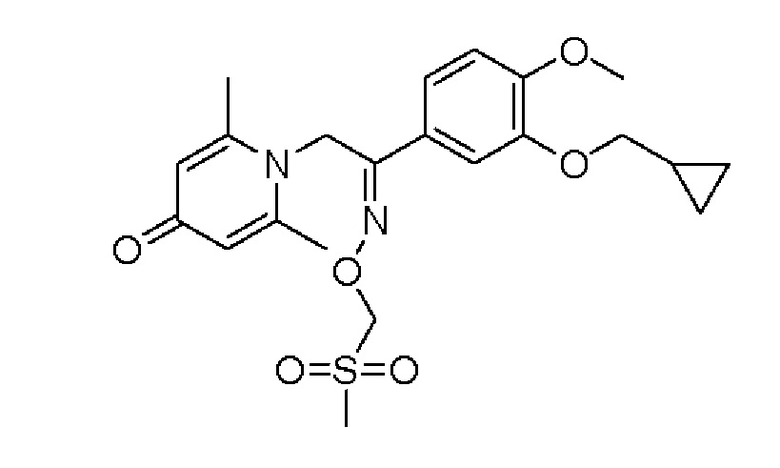
(Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(метилсульфонметоксиимидо)этил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
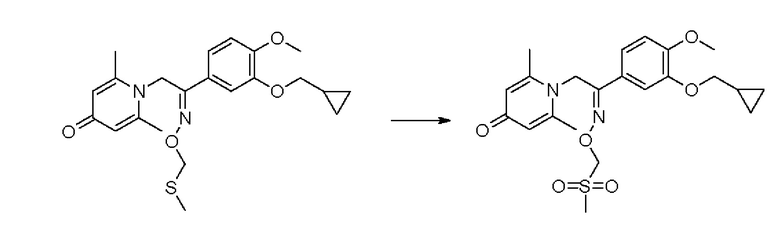
Соединение (Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(метилтиометоксиимидо)этил)-2,6-диметилпиридин-4(1H)-он (42 мг, 0,1 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (61 мг, 0,3 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(метилсульфонметоксиимидо)этил)-2,6-диметилпиридин -4 (1Н)-она (26 мг, выход 58%, белое твердое вещество). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,23 (д, J=8,4 Гц, 1H), 7,18 (с, 1H), 7,08 (д, J=8,4 Гц, 1H), 6,96 (с, 2Н), 6,35 (с, 2Н), 5,18 (с, 2Н), 3,91 (с, 2Н), 3,85 (д, J=6,8 Гц, 2Н), 2,47 (с, 6Н), 2,40 (с, 3Н), 1,36-1,28 (м, 1H), 0,69-0,64 (м, 2Н), 0,35-0,32 (м, 2Н); LC-MS: m/z 449,2 [М+Н]+.
Пример 45
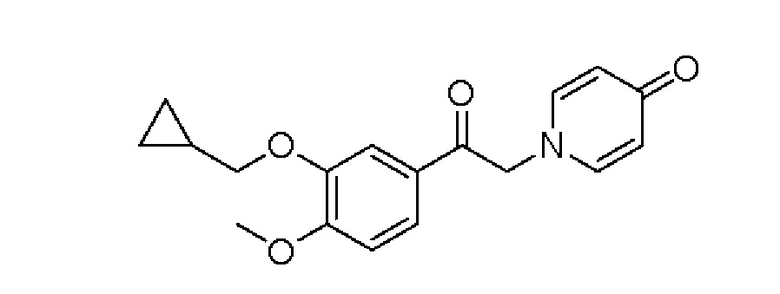
1-(2-(3-циклопропилметокси-4-метоксифенил)-2-оксоэтил)пиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
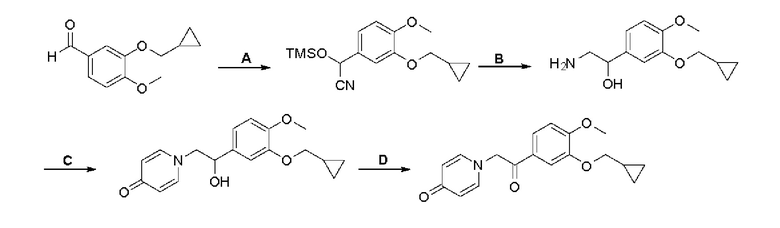
Стадия А:
3-Циклопропилметокси-4-метоксибензальдегид (3,09 г, 15 ммоль) растворяли в дихлорметане (30 мл); добавляли триэтиламин (4,16 мл, 30 ммоль) и триметилсилилцианид (3,75 г, 30 ммоль) в атмосфере азота и перемешивали при комнатной температуре в течение 6 часов. Реакционную массу концентрировали и сушили на роторном испарителе с получением 2-(3-циклопропилметокси-4-метоксифенил)-2-триметилсилоксиацетонитрила, который сразу использовали в следующей стадии.
Стадия В:
Соединение 2-(3-циклопропилметокси-4-метоксифенил)-2-триметилсилокси ацетонитрил (4,57 г, 15 ммоль) растворяли в безводном тетрагидрофуране (50 мл), затем порционно добавляли алюмогидрид лития (1,14 г, 30 ммоль) на ледяной бане и перемешивали при комнатной температуре в течение ночи. После завершения реакции последовательно добавляли воду (1,2 мл), водный раствор гидроксида натрия (1,2 мл, 15%) и воду (3,6 мл), перемешивали в течение получаса, сушили над безводным сульфатом натрия, фильтровали и сушили на роторном испарителе с получением неочищенного продукта 2-амино-1-(3-циклопропилметокси-4-метоксифенил) этанола.
Стадия С:
Соединение 2-амино-1-(3-(циклопропилметокси)-4-метоксифенил) этанол растворяли в этаноле (20 мл), затем добавляли пирон (1 г, 10,41 ммоль) и водный раствор гидроксида натрия (2М, 20 мл) и перемешивали в течение ночи при 60°С. Завершение реакции контролировали с помощью LCMS. Реакционную массу сушили на роторном испарителе и очищали колоночной хроматографией с получением 1-(2-(3-циклопропил метокси-4-метоксифенил)-2-гидроксиэтил) пиридин-4(1Н)-она (2 г, 63%).
Стадия D:
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил) пиридин-4(1Н)-он (280 мг, 0,88 ммоль) растворяли в дихлорметане (10 мл); добавляли периодинан Десса-Мартина (746 мг, 1,76 ммоль) в атмосфере азота и перемешивали при комнатной температуре в течение 3 часов. После завершения реакции реакционную массу концентрировали, сушили на роторном испарителе и очищали колоночной хроматографией с получением 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-оксоэтил) пиридин-4(1Н)-она (50 мг, выход 18%, белое твердое вещество). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,83 (д, J=7,6 Гц, 2Н), 7,68 (дд, J=8,4, 1,2 Гц, 1H), 7,45 (д, J=1,2 Гц, 1H), 7,16 (д, J=8,4 Гц, 1H), 6,47 (д, J=7,6 Гц, 2Н), 5,72 (с, 2Н), 3,89 (с, 3Н), 3,87 (д, J=6,8 Гц, 2Н), 1,26-1,21 (м, 1H), 0,62-0,56 (м, 2Н), 0,36-0,31 (м, 2Н); LC-MS: m/z 314,1 [М+Н]+.
Пример 46
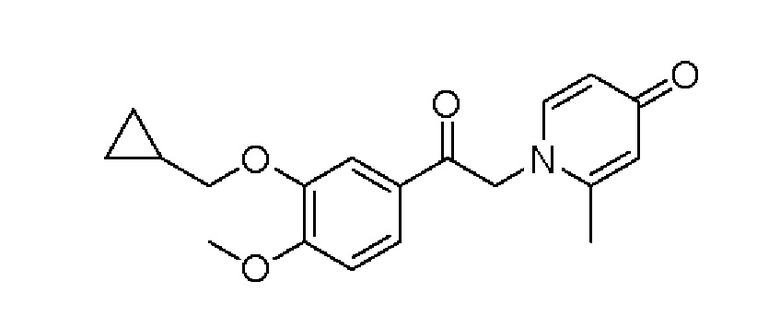
1-(2-(3-циклопропилметокси-4-метоксифенил)-2-оксоэтил)-2-метилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
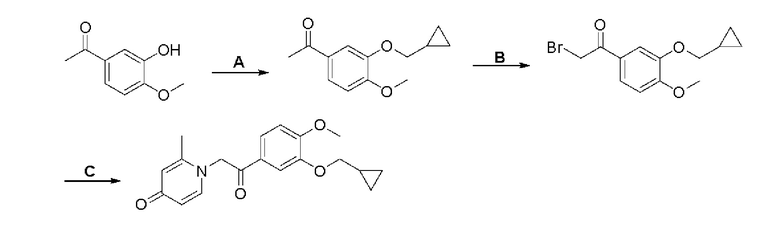
Стадия А:
3-Гидрокси-4-метоксиацетофенон (1,66 г, 10 ммоль) растворяли в ацетонитриле (30 мл); добавляли карбонат калия (2,76 г, 20 ммоль) и бромметилциклопропан (2,0 г, 15 ммоль) в атмосфере азота и перемешивали в течение 6 часов при 80°С. Реакционную массу фильтровали. Фильтрат концентрировали, сушили на роторном испарителе и очищали колоночной хроматографией с получением 1-(3-циклопропилметокси-4-метоксифенил) этанона (2,1 г, выход 95%, белое твердое вещество).
Стадия В:
Соединение 1-(3-циклопропилметокси-4-метоксифенил) этанон (2,2 г, 10 ммоль) растворяли в метаноле (30 мл), затем к раствору добавляли N-бромсукцинимид (2,14 г, 12 ммоль) и n-толуолсульфоновую кислоту (1,7 г, 10 ммоль) и проводили реакцию при 65°С в течение 12 часов. После завершения реакции реакционную массу концентрировали, сушили на роторном испарителе и очищали колоночной хроматографией с получением 2-бром-1-(3-циклопропилметокси-4-метоксифенил) этанона (800 мг, выход 27%, белое твердое вещество).
Стадия С:
Соединение 2-бром-1-(3-циклопропилметокси-4-метоксифенил)этанон (185 мг, 1,5 ммоль) и 4-метокси-2-метилпиридин (500 мг, 1,67 ммоль) растворяли в ацетонитриле (10 мл) и перемешивали при 80°С в течение 48 часов. После завершения реакции реакционную массу концентрировали, сушили на роторном испарителе и очищали колоночной хроматографией с получением 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-оксоэтил)-2-метилпиридин-4(1H)-она (300 мг, выход 61%, белое твердое вещество). 1H ЯМР (400 МГц, ДМСО-d6) δ 7,76 (дд, J=8,4, 2,0 Гц, 1H), 7,67 (д, J=8,4 Гц, 1H), 7,55 (д, J=1,2 Гц, 1H), 7,12 (д, J=8,8 Гц, 2Н), 6,42-6,38 (м, 2Н), 5,72 (с, 2Н), 3,95 (с, 3Н), 3,91 (д, J=6,8 Гц, 2Н), 2,24 (с, 3Н), 1,34-1,24 (м, 1H), 0,66-0,60 (м, 2Н), 0,39-0,34 (м, 2Н); LC-MS: m/z 328,1 [М+Н]+.
Пример 47
1-(2-(3-циклопропилметокси-4-метоксифенил)-2-оксоэтил)-2-хлорпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
2-бром-1-(3-циклопропилметокси-4-метоксифенил)этанон (594 мг, 2 ммоль) и 4-метокси-2-хлорпиридин (288 мг, 2 ммоль) растворяли в ацетонитриле (10 мл) и перемешивали при 80°С в течение 48 часов. После завершения реакции реакционную массу концентрировали, сушили на роторном испарителе и очищали колоночной хроматографией с получением 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-оксоэтил)-2-хлорпиридин-4 (1Н)-она (117 мг, выход 17%, белое твердое вещество). 1H ЯМР (400 МГц, ДМСО-d6) δ 7,73 (д, J=7,6 Гц, 2Н), 7,47 (д, J=2,0 Гц, 1H), 7,16 (д, J=8,4 Гц, 1H), 6,38 (д, J=2,4 Гц, 1H), 6,18 (дд, J=8,0, 2,4 Гц, 1H), 5,77 (с, 2Н), 3,89 (с, 3Н), 3,87 (д, J=6,8 Гц, 2Н), 1,27-1,22 (м, 1H), 0,62-0,56 (м, 2Н), 0,36-0,32 (м, 2Н); LC-MS: m/z 348.0 [М+Н]+.
Пример 48
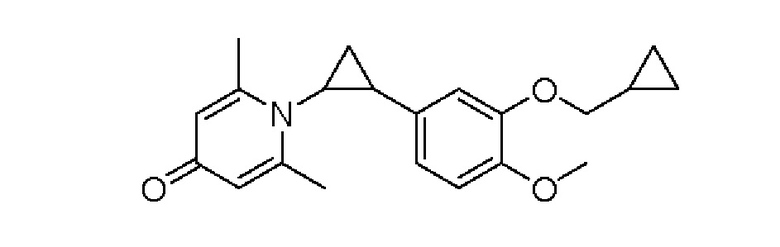
1-(2-(3-циклопропилметокси-4-метоксифенил)циклопропил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
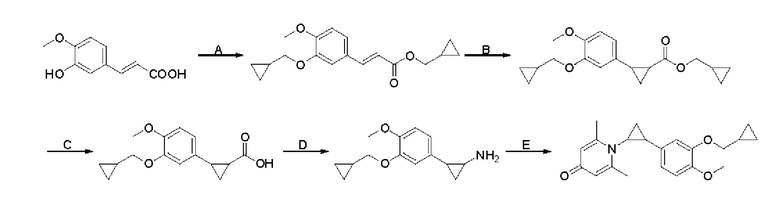
Стадия А:
3-Гидрокси-4-метоксикоричную кислоту (194 мг, 1 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли бромметилциклопропан (338 мг, 2,5 ммоль) и карбонат калия (414 мг, 3 ммоль). Реакционную массу перемешивали в атмосфере азота в течение ночи при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали этилацетатом (3×10 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, сушили на роторном испарителе и очищали колоночной хроматографией с получением циклопропилметил-3-циклопропилметокси-4-метоксициннамата (200 мг, выход 61%, желтая жидкость). LC-MS: m/z 303,3 [М+Н]+.
Стадия В:
Диметилсульфоксид (4 мл), 60% гидрид натрия (28,8 мг, 0,36 ммоль) и йодид триметилсульфоксония (160 мг, 0,36 ммоль) перемешивали при комнатной температуре в атмосфере азота в течение 20 минут. Затем медленно добавляли раствор (2 мл) циклопропилметил-3-циклопропилметил-4-метоксициннамата (200 мг, 0,66 ммоль) в тетрагидрофуране. После добавления по каплям реакционную массу перемешивали при комнатной температуре в течение 1 часа, затем нагревали до 50°С и непрерывно перемешивали в течение 1 часа. После завершения реакции реакционную массу выливали в ледяную воду и экстрагировали этилацетатом (3×10 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, сушили на роторном испарителе и очищали с помощью колоночной хроматографии с получением циклопропилметил 2-(3-циклопропилметокси-4-метоксифенил) циклопропилкарбоксилата (80 мг, выход 56%, белое твердое вещество). LC-MS: m/z 317,3 [М+Н]+.
Циклопропилметил 2-(3-циклопропилметокси-4-метоксифенил) циклопропил карбоксилат (80 мг, 0,25 ммоль) растворяли в метаноле (2 мл), затем добавляли раствор гидроксида натрия (2Н, 1 мл) и перемешивали при кипении с обратным холодильником в течение 2 часов. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали с получением 2-(3-циклопропилметокси-4-метоксифенил)циклопропил карбоновую кислоту (50 мг, выход 76%, белое твердое вещество).
Стадия D:
2-(3-Циклопропилметокси-4-метоксифенил)циклопропилкарбоновую кислоту (50 мг, 0,19 ммоль) растворяли в 1,4-диоксане (2 мл), затем добавляли триэтиламин (26 мг, 0,26 ммоль) и дифенилфосфорилазид (64 мг, 0,26 ммоль). Эту реакционную массу перемешивали в течение 16 часов при комнатной температуре, затем нагревали до 80°С и перемешивали в течение 1 часа. Затем добавляли смесь 10% соляной кислоты в 1,4-диоксане и непрерывно перемешивали при комнатной температуре в течение 18 часов. После завершения реакции реакционную массу гасили добавлением раствора гидроксида натрия (1 мл, 3Н) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали с получением 2-(3-циклопропилметокси-4-метоксифенил) циклопропиламина (12 мг, выход: 25%, желтое масло).
Стадия Е
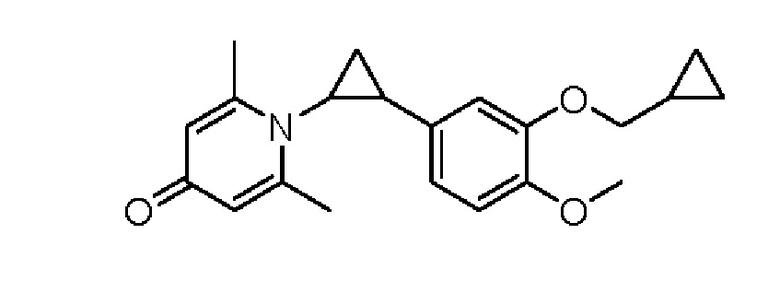
Соединение 2-(3-циклопропилметокси-4-метоксифенил) циклопропиламин (60 мг, 0,24 ммоль) растворяли в этаноле (3 мл), затем добавляли 2,6-диметил-4Н-пиран-4-он (45 мг, 0,36 ммоль) и гидроксид натрия (20 мг, 0,48 ммоль) и перемешивали в течение ночи при 60°С. Завершение реакции контролировали с помощью LCMS. Реакционную массу сушили на роторном испарителе и очищали с получением 1-(2-(3-циклопропил метокси-4-метоксифенил)циклопропил)-2,6-диметилпиридин-4(1Н)-она (10 мг, выход 11%, желтое масло). 1Н ЯМР (400 МГц, CD3OD) δ 6,83 (д, J=8,8 Гц, 1H), 6,75-6,58 (м, 2Н), 6,18 (с, 2Н), 3,76 (д, J=6,8 Гц, 2Н), 3,73 (с, 3Н), 2,40 (с, 6Н), 1,63-1,54 (м, 2Н), 1,29-1,12 (м, 2Н), 0,54-0,48 (м, 2Н), 0,26-0,22 (м, 2Н); LC-MS: m/z 340,0 [М+Н]+.
Пример 49
1-(2-(3-циклопропилметокси-4-дифторметоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
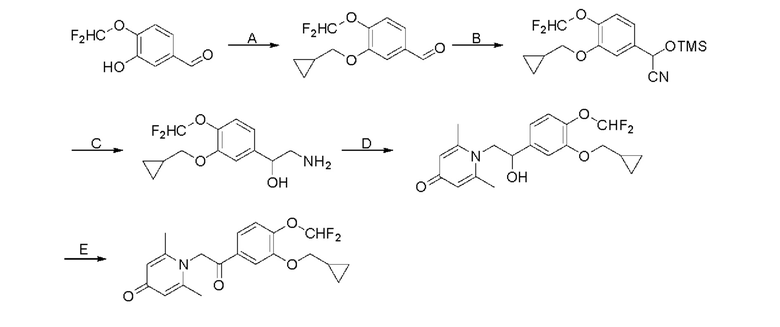
Стадия A:
3-Гидрокси-4-дифторметоксибензальдегид (1,88 г, 10,0 ммоль) растворяли при комнатной температуре в ацетонитриле (10 мл), затем последовательно добавляли карбонат калия (2,07 г, 15,0 ммоль) и бромметилциклопропан (1,76 г, 13,0 ммоль), и нагревали до 80°С при перемешивании в атмосфере азота в течение 3 часов. После завершения реакции добавляли насыщенный раствор соли (30 мл) и экстрагировали этилацетатом (3×60 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 3-циклопропилметокси -4-дифторметоксибензальдегида (2,20 г, 91%). LC-MS: m/z 243,2 [М+Н]+.
Стадия В:
Соединение 3-циклопропилметокси-4-дифторметоксибензальдегид (2,20 г, 9,1 ммоль) растворяли в дихлорметане (30 мл), затем последовательно добавляли триэтиламин (1,84 г, 18,2 ммоль) и триметилсилилцианид (2,7 г, 27,3 ммоль) на ледяной бане и перемешивали в атмосфере азота при комнатной температуре в течение 16 часов. После завершения реакции реакционную массу сразу сушили на роторном испарителе с получением продукта 2-(3-циклопропилметокси-4-дифторметоксифенил)-2-триметилсилоксиацетонитрила (3,1 г, 100%), который сразу использовали в следующей стадии.
Стадия С:
Соединение 2-(3-циклопропилметокси-4-дифторметоксифенил)-2-триметил силоксиацетонитрил (3,1 г, 9,1 ммоль) растворяли в безводном тетрагидрофуране (50 мл), затем порционно добавляли литийалюминийгидрид (1,04 г, 27,3 ммоль) на ледяной бане и перемешивали в течение ночи при комнатной температуре. После завершения реакции реакционную массу разбавляли безводным тетрагидрофураном (200 мл) и экстрагировали водой (1 мл). Затем добавляли водный раствор гидроксида натрия (3 мл, 1 М), а затем воду (3 мл), перемешивали в течение 30 минут, сушили над безводным сульфатом натрия, фильтровали и сушили на роторном испарителе с получением неочищенного продукта 2-амино-1-(3-циклопропилметокси-4-дифторметоксифенил) этанола (2,70 г, 100%). LC-MS: m/z 258,1 [М-18+1]+.
Стадия D:
Соединение 2-амино-1-(3-циклопропилметокси-4-дифторметоксифенил) этанол (2,70 г, 9,1 ммоль) растворяли в этаноле (30 мл), затем последовательно добавляли 2,6-диметил-4Н-пиран-4-он (1,86 г, 15,0 ммоль), гидроксид натрия (0,60 г, 15,0 ммоль) и воду (10 мл), нагревали до 60°С и перемешивали в атмосфере азота в течение 16 часов. После завершения реакции реакционную массу сушили на роторном испарителе и очищали с получением 1-(2-(3-циклопропилметокси-4-дифторметоксифенил)-2-гидрокси этил) -2,6-диметилпиридин-4 (1Н) -она (2,4 г, 69%). LC-MS: m/z 380,2 [М+Н]+.
Стадия Е
Соединение 1-(2-(3-циклопропилметокси-4-дифторметоксифенил)-2-гидрокси этил)-2,6-диметилпиридин-4(1H)-он (50 мг, 0,13 ммоль) растворяли в дихлорметане (10 мл), затем добавляли периодинан Десса-Мартина (110 мг, 0,26 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Реакционную массу фильтровали и промывали насыщенным раствором соли. Органическую фазу сушили над безводным сульфатом натрия, фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением 1-(2-(3-циклопропилметокси-4-дифторметокси фенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-она (35 мг, выход 72%, белое твердое вещество). 1Н ЯМР (400 МГц, CDCl3) δ 7,66 (с, 2Н), 7,64 (д, J=8,4 Гц, 1H), 7,33 (д, J=8,4 Гц, 1H), 6,79 (т, JH-F=74,4 Гц, 1H), 6,45 (с, 2Н), 5,50 (с, 2Н), 3,99 (д, J=7,2 Гц, 2Н), 2,26 (с, 6Н) 1,37-1,28 (м, 1H), 0,72-0,66 (м, 2Н), 0,43-0,37 (м, 2Н); LC-MS: m/z 378.3 [М+Н]+.
Пример 50
(E)-1-(3-циклопропилметокси-4-метоксистирил)пиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил) пиридин-4(1Н)-он (270 мг, 0,86 ммоль) и n-толуолсульфоновую кислоту (443 мг, 2,58 ммоль) растворяли в толуоле (5 мл) и перемешивали при кипении с обратным холодильником в течение ночи. После завершения реакции реакционную массу фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-циклопропилметокси-4-метоксистирил) пиридин-4(1Н)-она (50 мг, выход 20%, белое твердое вещество). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,02 (д, J=7,6 Гц, 2Н), 7,52 (д, J=14,4 Гц, 1H), 7,10 (с, 1H), 6,97-6,93 (м, 2Н), 6,85 (д, J=14,4 Гц, 1H), 6,21 (д, J=7,6 Гц, 2Н), 3,83 (д, J=7,2 Гц, 2Н), 3,77 (с, 3Н), 1,26-1,21 (м, 1H), 0,62-0,56 (м, 2Н), 0,36-0,31 (м, 2Н); LC-MS: m/z 298,1 [М+Н]+.
Пример 51
(Е)-1-(3-циклопропилметокси-4-метоксистирил)-2-метилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
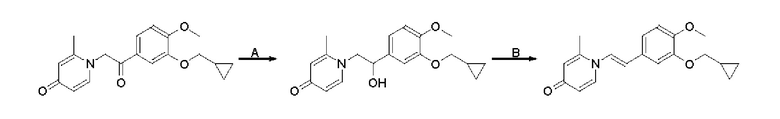
Стадия А:
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-оксоэтил)-2-метил пиридин-4(1Н)-он (654 мг, 2 ммоль) растворяли в метаноле (10 мл), затем порционно добавляли боргидрид натрия (114 мг, 3 ммоль) и перемешивали при комнатной температуре в течение 3 часов. После завершения реакции добавляли насыщенный раствор хлорида аммония (20 мл), и реакционную массу фильтровали, концентрировали и очищали колоночной хроматографией с получением 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил)-2-метилпиридин-4(1Н)-она (620 мг, выход 95%, белое твердое вещество). LC-MS: m/z 330,4 [М+Н]+
Стадия В:
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил) -2-метилпиридин-4(1Н)-он (620 мг, 1,88 ммоль) и n-толуолсульфоновую кислоту (970 мг, 5,64 ммоль) растворяли в толуоле (15 мл) и перемешивали при кипении с обратным холодильником в течение ночи. После завершения реакции реакционную массу фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-циклопропилметокси-4-метоксистирил)-2-метилпиридин-4(1H)-она (520 мг, выход 84%, белое твердое вещество). 1Н ЯМР (400 МГц, ДМСО-d6) 5 8,32 (д, J=7,2 Гц, 1H), 7,65 (д, J=14,0 Гц, 1H), 7,22 (д, J=1,6 Гц, 1H), 7,12 (дд, J=8,4, 1,6 Гц, 1H), 7,00 (д, J=8,4 Гц, 1H), 6,95 (д, J=14,0 Гц, 1H), 6,66-6,62 (м, 2Н), 3,86 (д, J=7,2 Гц, 2Н), 3,81 (с, 3Н), 2,51(с, 3Н), 1,28-1,22 (м, 1H), 0,62-0,57 (м, 2Н), 0,36-0,31 (м, 2Н); LC-MS: m/z 312,1 [М+Н]+.
Пример 52
(Е)-1-(3-гидрокси-4-метоксистирил)-2-метилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
Стадия А:
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-оксоэтил)-2-метил пиридин-4(1Н)-он (5,74 г, 20 ммоль) растворяли в метаноле (100 мл), а затем порционно добавляли боргидрид натрия (1,14 г, 30 ммоль) и перемешивали при комнатной температуре в течение 3 часов. После завершения реакции добавляли насыщенный раствор хлорида аммония (20 мл), и реакционную массу фильтровали, концентрировали и очищали колоночной хроматографией с получением 1-(2-(3-гидрокси-4-метоксифенил) -2 -гидроксиэтил) -2,6-диметилпиридин-4 (1H)-она (5,09 г, выход 88%, бесцветное масло). LC-MS: m/z 290,4 [М+Н]+.
Стадия В:
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил) -2-метилпиридин-4(1H)-он (5,09 г, 17,6 ммоль) и n-толуолсульфоновую кислоту (9,08 г, 52,8 ммоль) растворяли в толуоле (100 мл) и перемешивали при кипении с обратным холодильником в течение ночи. После завершения реакции реакционную массу фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-циклопропилметокси-4-метоксистирил)-2-метилпиридин-4(1Н)-она (3,05 г, выход 64%, серое твердое вещество). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,34 (с, 1H), 7,16 (с, 2Н), 7,05 (д, J=14,0 Гц, 1H), 6,91 (д, J=8,4 Гц, 1H), 6,84 (д, J=8,4 Гц, 1H), 6,62 (д, J=14,0 Гц, 1H), 3,92 (с, 2Н), 2,48(с, 6Н); LC-MS: m/z 272,1 [М+Н]+.
Пример 53
(Е)-1-(3-пропокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:

Соединение (E)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он (50 мг, 0,17 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли бромпропан (34,5 мг, 0,28 ммоль) и карбонат цезия (117 мг, 0,36 ммоль) и перемешивали в течение 2 часов при комнатной температуре. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (Е)-1-(3-пропокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-она (25 мг, выход 44%, белое твердое вещество). 1Н ЯМР (400 МГц, CD3OD) δ 7,22 (д, J=2,0 Гц, 1H), 7,14 (дд, J=8,4, 2,0 Гц, 1Н), 7,09 (д, J=14,2 Гц, 1Н), 7,01 (д, J=8,4 Гц, 1Н), 6,77 (д, J=14,2 Гц, 1H), 6,38 (с, 2Н), 4,03 (т, J=6,4 Гц, 2Н), 3,89 (с, 3Н), 2,37 (с, 6Н), 1,90-1,80 (м, 2Н), 1,08 (т, J=7,2 Гц, 3Н); LC-MS: m/z 314,2 [М+Н]+.
Пример 54
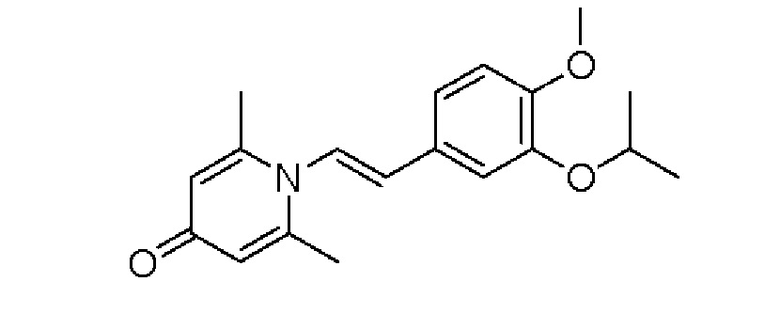
(Е)-1-(3-пропокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
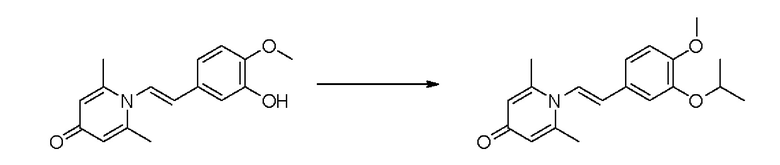
Соединение (E)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (50 мг, 0,17 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли бромизопропан (34,5 мг, 0,28 ммоль) и карбонат цезия (117 мг, 0,36 ммоль) и перемешивали в течение 2 часов при комнатной температуре. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-изопропокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-она (8 мг, выход 14%, бесцветное масло). 1Н ЯМР (400 МГц, CD3OD) δ 7,26 (д, J=2,0 Гц, 1H), 7,22 (дд, J=8,4, 2,0 Гц, 1H), 7,21 (д, J=14,4 Гц, 1H), 7,05 (д, J=8,4 Гц, 1H), 6,97 (с, 2Н), 6,89 (д, J=14,4 Гц, 1H), 4,69-4,60 (м, 1H), 3,90 (с, 3Н), 2,57 (с, 6Н), 1,35 (д, J=6,0 Гц, 6Н). LC-MS: m/z 314,2 [М+Н]+.
Привет 55
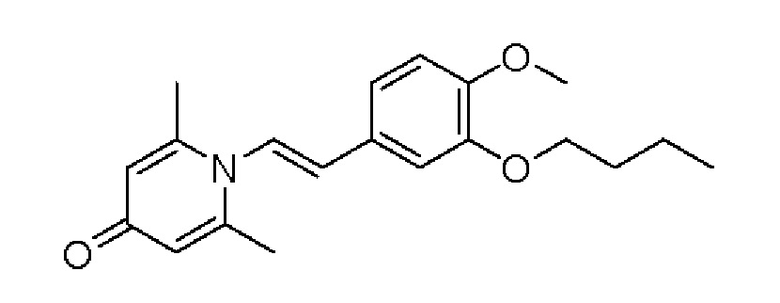
(Е)-1-((3-н-бутокси-4-метокси)-стирил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
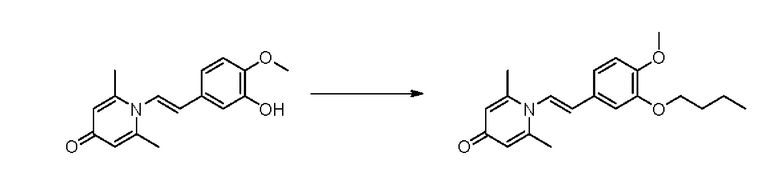
Соединение (E)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он (50 мг, 0,17 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли н-бутилбромид (38 мг, 0,28 ммоль) и карбонат цезия (117 мг, 0,36 ммоль) и перемешивали 6 течение 2 часов при комнатной температуре. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (Е)-1-(3-н-бутокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-она (20 мг, выход 34%, белое твердое вещество). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,57 (с, 2Н), 7,27 (д, J=14,4 Гц, 1H), 7,10 (д, J=1,6 Гц, 1H), 7,04-6,98 (м, 2Н), 6,95 (д, J=14,4 Гц, 1H), 4,34 (т, J=6,4 Гц, 2Н), 3,82 (с, 3Н), 2,60 (с, 6Н), 1,82-1,75 (м, 2Н), 1,50-1,40 (м, 2Н), 0,96 (т, J=7,2 Гц, 1H); LC-MS: m/z 328,2 [М+Н]+.
Пример 56
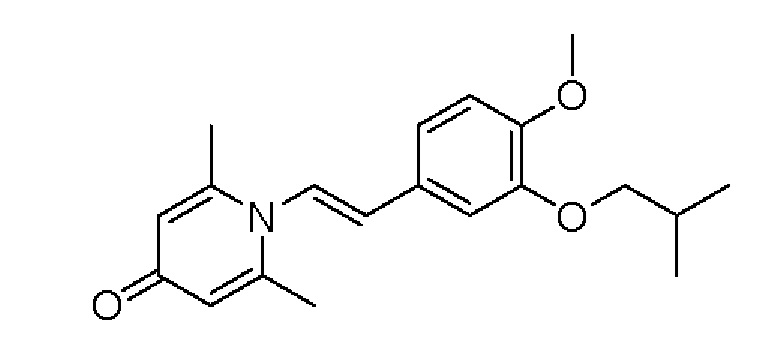
(Е)-1-(3-изо-бутокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
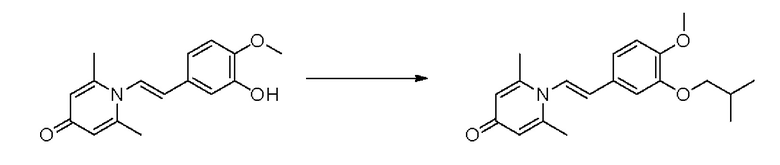
Соединение (E)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он (50 мг, 0,17 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли бромизобутан (38 мг, 0,28 ммоль) и карбонат цезия (117 мг, 0,36 ммоль) и перемешивали в течение 2 часов при комнатной температуре. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (Е)-1-(3-изобутокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-она (25 мг, выход 42%, белое твердое вещество). 1Н ЯМР (400 МГц, CDCl3) δ 7,02 (дд, J=8,4, 2,0 Гц, 1H), 6,98 (д, J=2,0 Гц, 1H), 6,91 (д, J=8,4 Гц, 1H), 6,76 (д, J=14,4 Гц, 1H), 6,60 (д, J=14,4 Гц, 1H), 6,30 (с, 2Н), 3,92 (с, 3Н), 3,81 (д, J=6,8 Гц, 2Н), 2,27 (с, 6Н), 2,24-2,17 (м, 1H), 1,08 (д, J=6,8 Гц, 6Н); LC-MS: m/z 328,2 [М+Н]+.
Пример 57
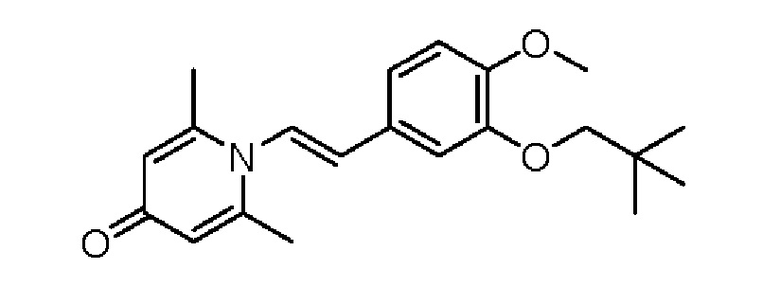
(Е)-1-(3-неопентилокси-4-метокси)-стирил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
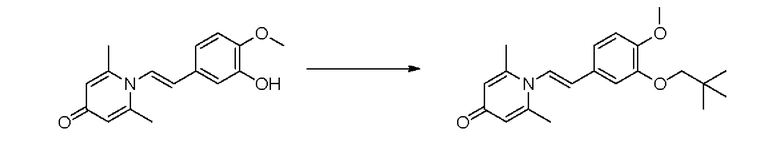
Соединение (E)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он (200 мг, 0,74 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли бромнеопентан (224 мг, 1,48 ммоль) и карбонат цезия (117 мг, 0,36 ммоль) и перемешивали в атмосфере азота в течение 24 часов при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (Е)-1-(3-неопентилокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-она (20 мг, выход 8%, белое твердое вещество). 1Н ЯМР (400 МГц, CDCl3) δ 7,25 (с, 2Н), 7,05-7,00 (м, 2Н), 6,97 (д, J=14,4 Гц, 1H), 6,90 (д, J=8,4 Гц, 1H), 6,70 (д, J=14,4 Гц, 1H), 3,91 (с, 3Н), 3,68 (с, 2Н), 2,54 (с, 6Н), 1,08 (с, 9Н); LC-MS m/z 342,3 [М+Н]+.
Пример 58
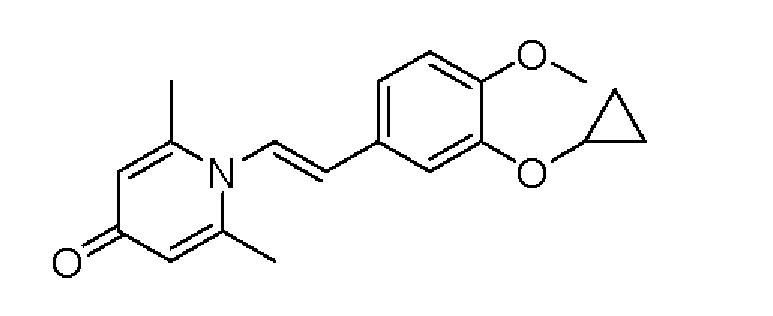
(Е)-1-(3-циклопропилокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он
Конкретная схема реакции показана ниже:
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил)-2,6-диметилпиридин-4(1H)-он (100 мг, 0,3 ммоль) и n-толуолсульфоновую кислоту (68,4 мг, 0,6 ммоль) растворяли в толуоле (5 мл), затем добавляли N,N-диметилформамид (1 мл) и перемешивали при кипении с обратным холодильником в течение ночи. После завершения реакции реакционную массу фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (Е)-1-(3-циклопропилокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-она (50 мг, выход 53%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,15 (с, 1H), 7,12 (д, J=8,4 Гц, 1H), 7,06 (д, J=14,4 Гц, 1H), 6,98 (д, J=8,4 Гц, 1H), 6,85 (д, J=14,4 Гц, 1H), 6,32 (с, 2Н), 4,02 (с, 3Н), 2,32 (с, 6Н), 2,23-2,19 (м, 1H), 0,94-0,90 (м, 4Н); LC-MS m/z 312,3 [М+Н]+.
Пример 59
(E)-1-(3-циклопропилметокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил) -2,6-диметилпиридин-4(1H)-он (186 мг, 0,54 ммоль) и n-толуолсульфоновую кислоту (140 мг, 0,81 ммоль) растворяли в толуоле (5 мл), затем добавляли N,N-диметилформамид (1 мл) и перемешивали при кипении с обратным холодильником в течение ночи. После завершения реакции реакционную массу фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (Е)-1-(3-циклопропил метокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-она (80 мг, выход 45%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,13 (дд, J=8,4, 2,0 Гц, 1H), 7,19 (д, J=2,0 Гц, 1Н), 7,07 (д, J=14,4 Гц, 1H), 7,11 (с, 2Н), 6,99 (д, J=8,4 Гц, 1H), 6,75 (д, J=14,4 Гц, 1H), 6,41 (с, 2Н), 3,89 (д, J=7,2 Гц, 2Н), 3,87 (с, 3Н), 2,36 (с, 6Н), 1,33-1,24 (м, 1H), 0,65-0,60 (м, 2Н), 0,38-0,34 (м, 2Н); LC-MS: m/z 326,1 [М+Н]+.
Пример 60
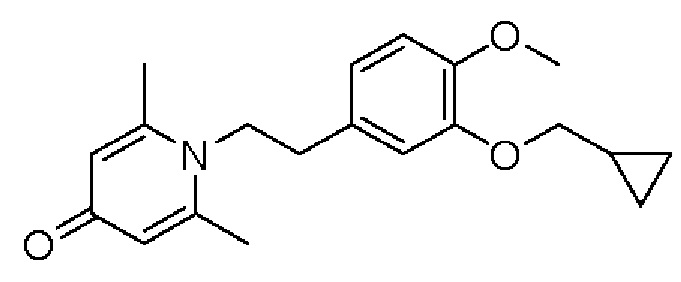
(E)-1-(3-циклопропилметокси-4-метоксифенэтил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
Соединение (E)-1-(3-циклопропилокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (10 мг, 0,03 ммоль) растворяли в этаноле (3 мл), затем добавляли Pd/C (50 мг) и перемешивали в течение 2 часов в атмосфере водорода при комнатной температуре. После завершения реакции реакционную массу фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-циклопропилметокси-4-метоксифенетил)-2,6-диметилпиридин-4(1H)-она (2,9 мг, выход 30%, бесцветное масло). 1H ЯМР (400 МГц, CD3OD) δ 7,13 (дд, J=8,4, 2,0 Гц, 1H), 7,19 (д, J=2,0 Гц, 1H), 7,11 (с, 2Н), 6,99 (д, J=8,4 Гц, 1H), 6,41 (с, 2Н), 3,89 (д, J=7,2 Гц, 2Н), 3,87 (с, 3Н), 2,89-2,83 (m, 2Н), 2,83-2,77 (m, 2Н), 2,36 (с, 6Н), 1,33-1,24 (m, 1Н), 0,65-0,60 (m, 2Н), 0,38-0,34 (m, 2Н); LC-MS: m/z 328,1 [М+Н]+.
Пример 61
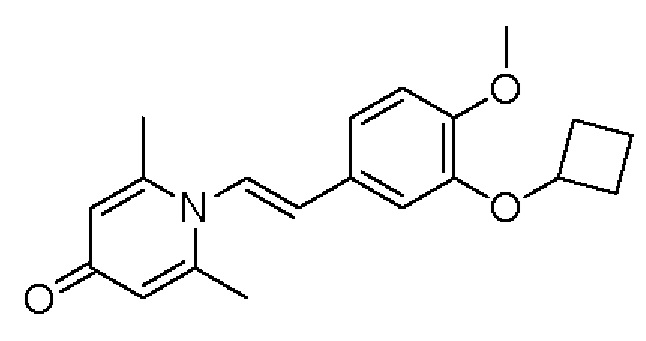
(E)-1-(3-циклобутилокси-4-метокси-стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
Соединение (Е)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (100 мг, 0,37 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли бромциклобутан (100 мг, 0,74 ммоль) и карбонат цезия (241 мг, 0,74 ммоль) и перемешивали в атмосфере азота в течение 4 часов при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-циклобутилокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-она (18 мг, выход 15%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,13 (дд, J=8,4, 2,0 Гц, 1Н), 7,05 (д, J=14,4 Гц, 1H), 7,03 (д, J=2,0 Гц, 1H), 6,99 (д, J=8,4 Гц, 1H), 6,75 (д, J=14,4 Гц, 1Н), 6,36 (с, 2Н), 4,78-4,71 (м, 1H), 3,86 (с, 3Н), 2,52-2,44 (м, 2Н), 2,34 (с, 6Н), 2,22-2,12 (м, 2Н), 1,89-1,81 (м, 1H), 1,77-1,65 (м, 1Н); LC-MS m/z 326,2 [М+Н]+.
Пример 62
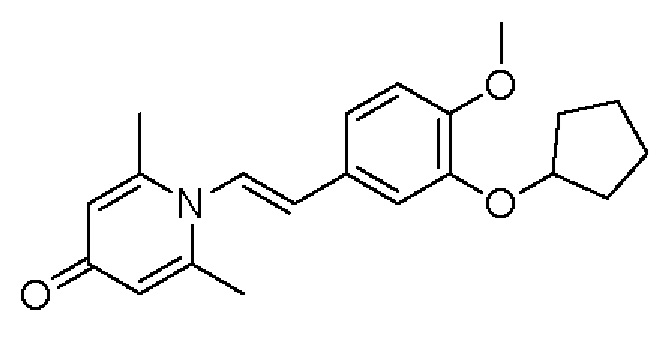
(E)-1-((3-циютопентилокси-4-метокси)-стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
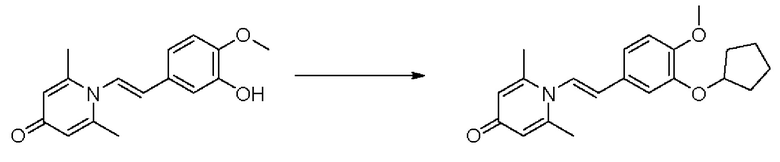
Соединение (Е)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (100 мг, 0,37 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли бромциклопентан (110 мг, 0,74 ммоль) и карбонат цезия (241 мг, 0,74 ммоль) и перемешивали в течение 4 часов при 80°С в атмосфере азота. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-циклопентилокси-4-метоксистирил)-2,6-диметилпиридин-4(1Л)-она (25 мг, выход 20%, бледно-желтое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,16 (с, 1H), 7,13 (д, J=8,4 Гц, 1H), 7,06 (д, J=14,4 Гц, 1H), 6,98 (д, J=8,4 Гц, 1H), 6,76 (д, J=14,4 Гц, 1H), 6,36 (с, 2Н), 3,35-3,83 (м, 1H), 3,86 (с, 3Н), 2,52-2,44 (м, 2Н), 2,35 (с, 6Н), 1,93-1,79 (м, 6Н), 1,69-1,58 (м, 2Н); LC-MS m/z 340,2 [М+Н]+.
Пример 63
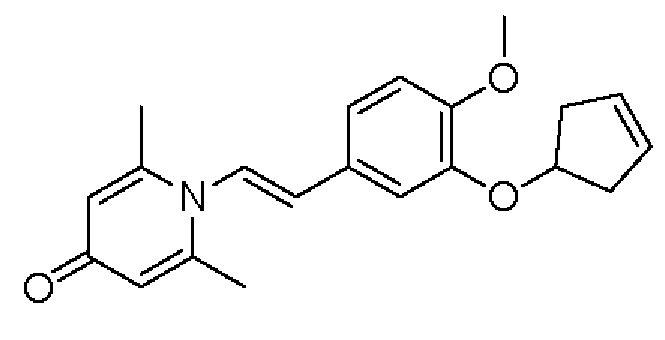
(E)-1-(3-(циклопент-3-ен-1-илокси)-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
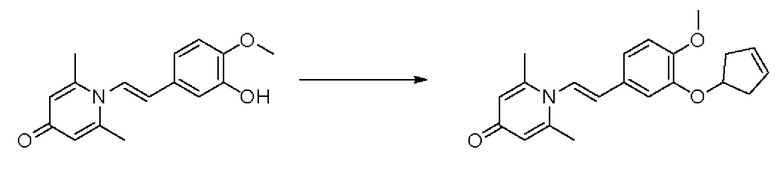
Соединение (E)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он (100 мг, 0,37 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли циклопент-3-ен-1-илметансульфонат (110 мг, 0,74 ммоль) и карбонат цезия (241 мг, 0,74 ммоль) и перемешивали в атмосфере азота в течение ночи при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-(циклопент-3-ен-1-илокси)-4-метоксистирил)-2,6-диметил пиридин-4(1H)-он (22 мг, выход 15%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,45 (с, 2Н), 7,08 (д, J=8,0 Гц, 1H), 7,02-6,99 (м, 2Н), 6,92 (д, J=8,0 Гц, 1H), 6,74 (д, J=14,4 Гц, 1H), 5,77 (с, 2Н), 5,12-5,05 (м, 1H), 3,90 (с, 3Н), 2,89-2,82 (м, 2Н), 2,69-2,63 (м, 2Н), 2,58 (с, 6Н); LC-MS m/z 338,1 [М+Н]+.
Пример 64
(E)-1-(3-аллилокси-4-метокси-стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
Соединение (Е)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (50 мг, 0,18 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли 3-бромпропен (44 мг, 0,36 ммоль) и карбонат цезия (117 мг, 0,36 ммоль) и перемешивали в атмосфере азота в течение 2 часов при 80°С.После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-аллилокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-она (12 мг, выход 20%, бледно-желтое масло). 1H ЯМР (400 МГц, CDCl3) δ 7,23 (с, 2Н), 7,09-6,92 (м, 4Н), 6,73-6,69 (м, 1H), 6,16-6,05 (м, 1H), 5,45 (д, J=17,2 Гц, 1H), 5,34 (д, J=10,4 Гц, 1H), 4,67 (д, J=4,0 Гц, 2Н), 3,94 (с, 3Н), 2,54 (с, 6Н); LC-MS m/z 312,2 [М+Н]+.
Пример 65
(E)-1-((3-(3-метилбут-2-ен-1-ил)окси-4-метокси)-стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
Соединение (E)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (50 мг, 0,18 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли 1-бром-3-метил-2-бутен (55 мг, 0,36 ммоль) и карбонат цезия (117 мг, 0,36 ммоль) и перемешивали в атмосфере азота в течение 1 часа при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-((3-(3-метилбут-2-ен-1-ил)окси-4-метокси)стирил)-2,6-диметил пиридин-4(1H)-она (15 мг, выход 18%, желтое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,08 (с, 2Н), 7,08-7,04 (м, 2Н), 6,92-6,90 (м, 1H), 6,91 (д, J=14,4 Гц, 1H), 6,69 (д, J=14,4 Гц, 1H), 5,54 (m, J=6,0 Гц, 1H), 4,62 (д, J=6,0 Гц, 2Н), 3,92 (с, 3Н), 2,51 (с, 6Н), 1,80 (с, 3Н), 1,72 (с, 3Н); LC-MS m/z 340,0 [М+Н]+.
Пример 66
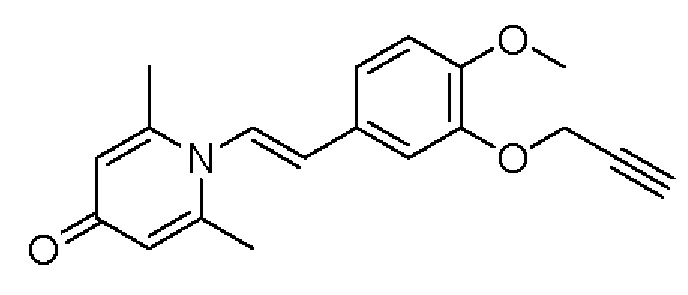
(E)-1-(3-(проп-2-ин-1-илокси)-4-метокси)стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
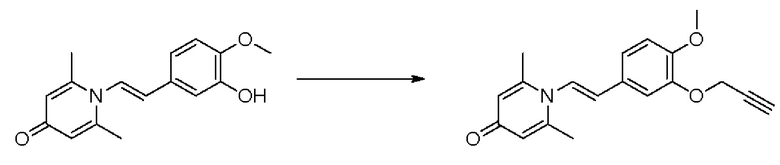
Соединение (Е)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (50 мг, 0,18 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли 3-бромпропин (44 мг, 0,36 ммоль) и карбонат цезия (117 мг, 0,36 ммоль) и перемешивали в атмосфере азота в течение 3 часов при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-(проп-2-ин-1-илокси)-4-метокси)стирил)-2,6-диметилпиридин-4(1H)-она (10 мг, выход 15%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,26-7,14 (м, 3Н), 7,13 (с, 2Н), 6,96 (д, J=8,4 Гц, 1H), 6,73 (д, J=14,4 Гц, 1H), 4,83 (с, 2Н), 3,94 (с, 3Н), 2,56 (с, 1H), 2,52 (с, 6Н); LC-MS m/z 310,1 [М+Н]+.
Пример 67
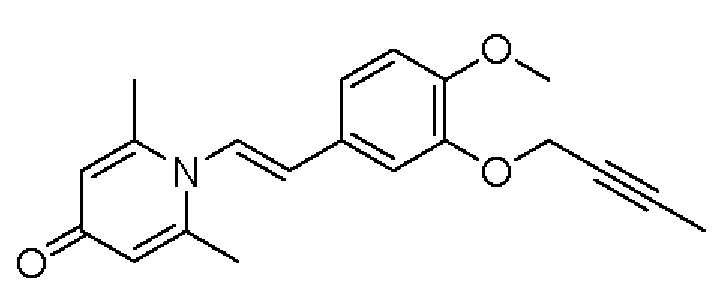
(E)-1-(3-(бут-2-ин-1-илокси)-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
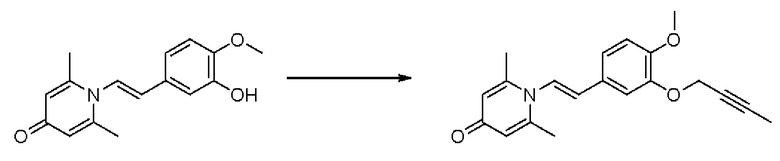
Соединение (E)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (50 мг, 0,18 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли 1-бром-2-бутин (49 мг, 0,36 ммоль) и карбонат цезия (117 мг, 0,36 ммоль) и перемешивали в атмосфере азота в течение 1 часа при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-(бут-2-ин-1-илокси)-4-метоксистирил)-2,6-диметилпиридин-4(1H)-она (15 мг, выход 25%, желтое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,17 (с, 1H), 7,13 (д, J=8,4 Гц, 1H), 6,09 (с, 2Н), 6,98 (д, J=14,4 Гц, 1H), 6,94 (д, J=8,4 Гц, 1H), 6,74 (д, J=14,4 Гц, 1H), 4,78 (с, 2Н), 3,93 (с, 3Н), 2,52 (с, 6Н), 1,86 (с, 3Н); LC-MS m/z 324,1 [М+Н]+.
Пример 68
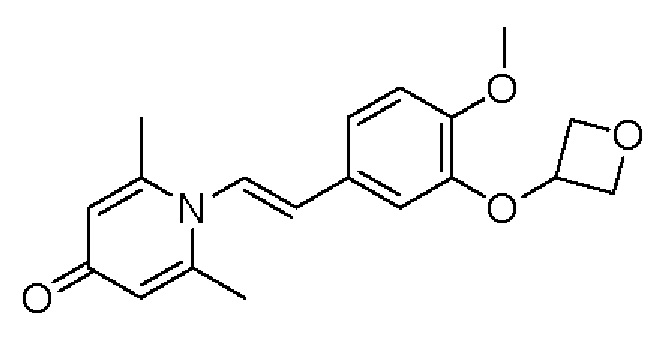
(E)-1-((3-(оксициклобутан-3-ил-окси)-4-метокси)-стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
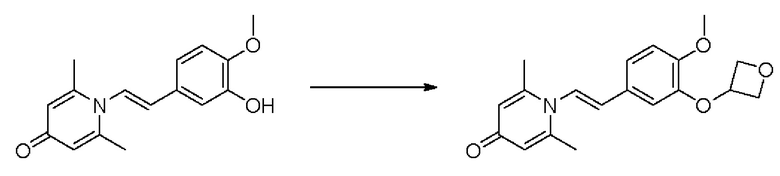
(E)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (150 мг, 0,55 ммоль), оксициклобутан-3-ол (81,4 мг, 1,1 ммоль) и трифенилфосфин (288 мг, 1,1 ммоль) растворяли в безводном тетрагидрофуране (10 мл), реакционную массу охлаждали на ледяной бане, затем добавляли по каплям в атмосфере азота диизопропилазодикарбоксилат (222 мг, 1,1 ммоль) в течение 5 минут, а затем проводили реакцию в течение 24 часов при комнатной температуре. После завершения реакции добавляли насыщенный раствор соли (20 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-((3-(оксициклобутан-3-илокси)-4-метокси)стирил)-2,6-диметил пиридин-4(1H)-она (40 мг, выход 22%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,15 (д, J=8,4 Гц, 1Н), 7,07 (с, 2Н), 6,96 (д, J=8,4 Гц, 1H), 6,94 (д, J=13,2 Гц, 1Н), 6,70 (с, 1Н), 6,69 (д, J=13,2 Гц, 1Н), 5,27 (м, 1H), 5,01 (м, 1H), 4,88 (м, 1H), 3,94 (с, 3Н), 2,53 (с, 6Н); LC-MS: m/z 328,2 [М+Н]+.
Пример 69
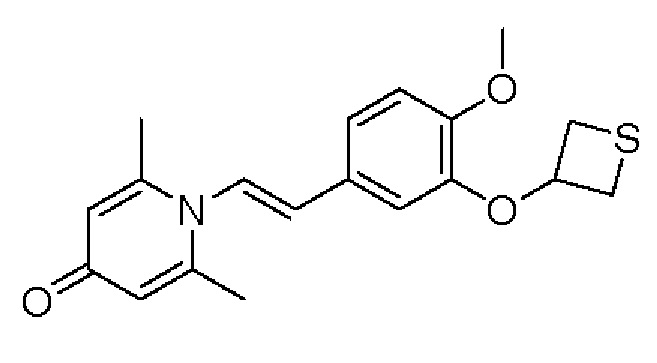
(E)-1-((3-(тиоциклобутан-3-ил-окси)-4-метокси)-стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
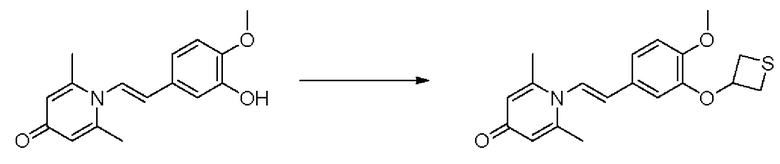
(E)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (150 мг, 0,55 ммоль), тиациклобутан-3-ол (99 мг, 1,1 ммоль) и трифенилфосфин (288 мг, 1,1 ммоль) растворяли в безводном тетрагидрофуране (10 мл), реакционную массу охлаждали на ледяной бане, добавляли по каплям в атмосфере азота диизопропилазодикарбоксилат (222 мг, 1,1 ммоль) в течение 5 минут, а затем проводили реакцию в течение 24 часов при комнатной температуре. После завершения реакции добавляли насыщенный раствор соли (20 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-((3-(тиациклобутан-3-илокси)-4-метокси)стирил)-2,6-диметилпиридин-4(1H)-она (30 мг, выход 16%, бледно-желтое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,37 (с, 2Н), 7,19 (д, J=14,4 Гц, 1H), 7,13 (д, J=2,0 Гц, 1H), 7,05 (дд, J=8,4, 2,0 Гц, 1H), 6,98 (д, J=8,4 Гц, 1H), 6,88 (д, J=14,4 Гц, 1H), 5,77-5,69 (м, 1H), 3,90 (с, 3Н), 3,65-3,61 (м, 2Н), 3,57-3,53 (м, 2Н), 2,66 (с, 6Н); LC-MS: m/z 344,2 [М+Н]+.
Пример 70
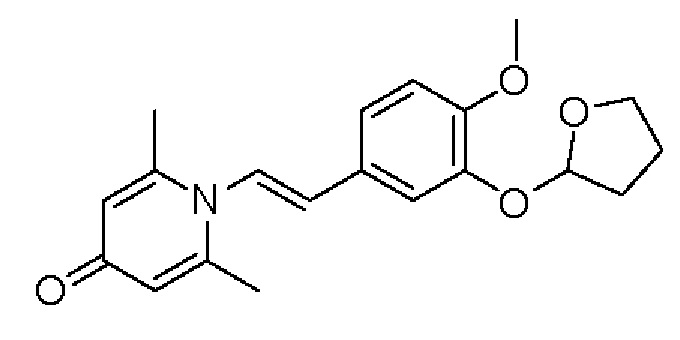
(E)-1-((3-(тетрагидрофуран-2-ил)окси-4-метокси)-стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
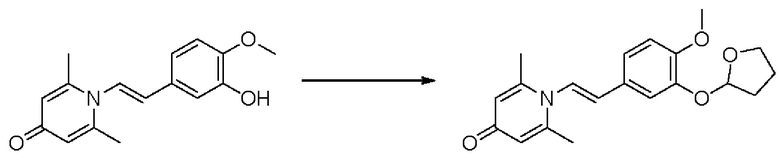
Соединение (Е)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (50 мг, 0,15 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 2,3-дигидрофуран (68 мг, 0,42 ммоль) и толуол-4-сульфонат пиридиния (4,6 мг, 0,018 ммоль) и перемешивали при комнатной температуре в течение 3 часов. После завершения реакции реакционную массу фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-((3-(тетрагидрофуран-2-ил)окси-4-метокси)стирил)-2,6-диметилпиридин-4 (1H)-она (16 мг, выход 24%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,31 (д, J=2,0 Гц, 1H), 7,26 (с, 2Н), 7,09 (дд, J=8,4, 2,0 Гц, 1H), 6,91 (д, J=8,4 Гц, 1H), 7,05 (д, J=14,4 Гц, 1H), 6,75 (д, J=14,4 Гц, 1H), 5,82 (д, J=4,4 Гц, 1H), 5,70 (с, 2Н), 4,13-4,08 (м, 1H), 4,01-3,95 (м, 1H), 3,90 (с, 3Н), 2,35 (с, 6Н), 2,24-2,14 (м, 2Н), 2,02-1,95 (м, 2Н); LC-MS m/z 342,3 [М+Н]+.
Пример 71
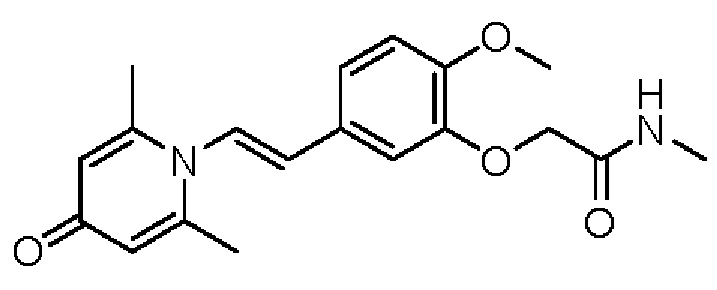
(Е)-2-(5-(2-(2,6-диметил-4-карбонилпиридин-1(4H)-ил)этенил)-2-метоксифенокси)-N-метилацетамид
Конкретная схема реакции показана ниже:
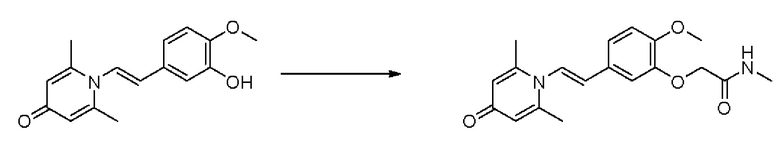
Соединение (E)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (200 мг, 0,74 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли 2-хлор-N-метилацетамид (119 мг, 1,11 ммоль) и карбонат калия (153 мг, 1,11 ммоль) и перемешивали в атмосфере азота в течение ночи при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-2-(5-(2-(2,6-диметил-4-карбонилпиридин-1(4H)-ил)этенил)-2-метоксифенокси)-N-метилацетамида (100 мг, выход 40%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,28 (д, J=2,0 Гц, 1H), 7,23 (дд, J=8,4, 2,0 Гц, 1H), 7,08 (д, J=14,2 Гц, 1H), 7,07 (д, J=8,4 Гц, 1H), 6,76 (д, J=14,2 Гц, 1H), 6,35 (с, 2Н), 4,55 (с, 2Н), 3,92 (с, 3Н), 2,84 (с, 3Н), 2,34 (с, 6Н); LC-MS m/z 343,1 [М+Н]+.
Пример 72
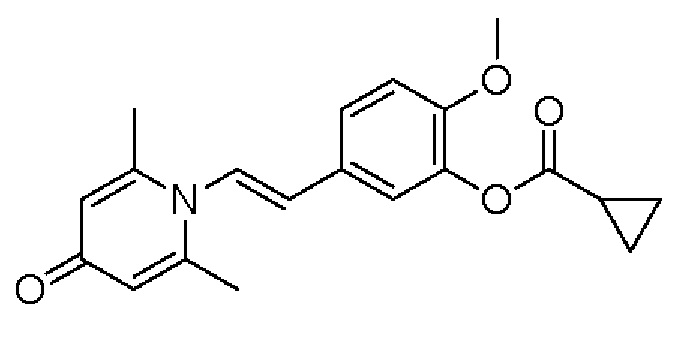
(E)-1-((3-циклопропилформилокси-4-метокси)-стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
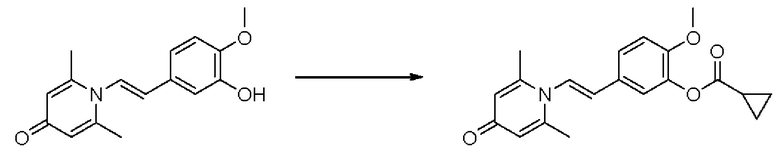
Соединение (E)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (50 мг, 0,17 ммоль) растворяли в дихлорметане (5 мл), затем добавляли циклопропанкарбонилхлорид (27 мг, 0,26 ммоль) и карбонат калия (36 мг, 0,26 ммоль) и перемешивали при комнатной температуре в течение 2 часов. После завершения реакции реакционную массу фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-((3-циклопропилформилокси-4-метокси)стирил)-2,6-диметилпиридин-4(1Н)-она (20 мг, выход 35%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,37 (дд, J=8,4, 2,0 Гц, 1H), 7,27 (д, J=2,0 Гц, 1H), 7,11 (с, 2Н), 7,04 (д, J=8,4 Гц, 1H), 6,96 (д, J=14,4 Гц, 1H), 6,72 (д, J=14,4 Гц, 1H), 3,92 (с, 3Н), 2,52 (с, 6Н), 1,96-1,89 (м, 1H), 1,25-1,21 (м, 2Н), 1,12-1,06 (м, 2Н); LC-MS: m/z 340,2 [М+Н]+.
Пример 73
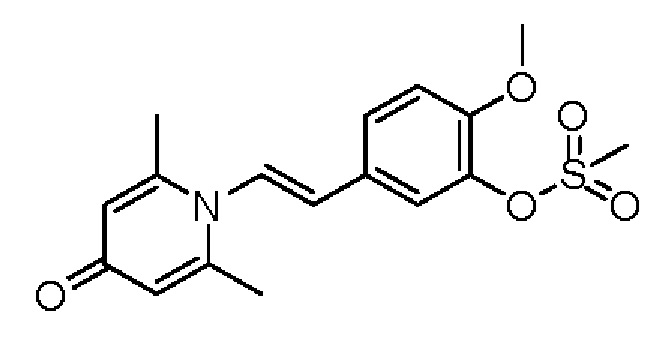
(E)-5-(2-(2,6-диметил-4-карбонилпиридин-1(4H)-ил)этенил)-2-метоксифенилметил сульфонат
Конкретная схема реакции показана ниже:
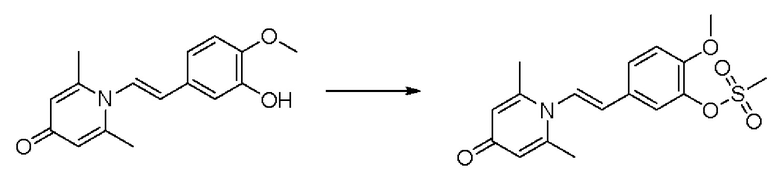
Соединение (Е)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (271 мг, 1,0 ммоль) растворяли в N,N-диметилформамиде (10 мл), затем добавляли триэтиламин (202 мг, 2,0 ммоль) и метилсульфонилхлорид (172 мг, 1,5 ммоль) и перемешивали при комнатной температуре в течение ночи. После завершения реакции добавляли насыщенный раствор соли (20 мл) и экстрагировали дихлорметаном (3×20 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-5-(2-(2,6-диметил-4-карбонилпиридин-1(4H)-ил)этенил)-2-метоксифенил метилсульфоната (130 мг., выход 40%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,61 (д, J=2,4 Гц, 1H), 7,58 (дд, J=8,8, 2,4 Гц, 1H), 7,28 (д, J=14,4 Гц, 1H), 7,25 (д, J=8,8 Гц, 1H), 7,05 (с, 2Н), 6,96 (д, J=14,4 Гц, 1H), 3,96 (с, 3Н), 2,58 (с, 6Н). LC-MS: m/z 350,1 [М+Н]+.
Пример 74
(E)-1-(3-метилтиометокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
Соединение (Е)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (70 мг, 0,26 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли хлорметил метил сульфид (50 мг, 0,52 ммоль) и карбонат цезия (170 мг, 0,52 ммоль) и перемешивали при комнатной температуре в течение ночи. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (Е)-1-(3-метилтиометокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-она (50 мг, выход 58%, белое твердое вещество). 1H ЯМР (400 МГц, CDC13) 5 7,31 (д, J=2,0 Гц, 1H), 7,25 (дд, J=8,4, 2,0 Гц, 1H), 7,08 (д, J=14,4 Гц, 1H), 7,05 (д, J=8,4 Гц, 1H), 6,78 (д, J=14,4 Гц, 1Н), 6,77 (с, 2Н), 5,03 (с, 2Н), 3,90 (с, 3Н), 2,36 (с, 6Н), 2,26 (с, 3Н); LC-MS: m/z 332,5 [М+Н]+.
Пример 75
(E)-1-(3-метилсульфоксидметокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
Соединение (E)-1-(3-метилтиометокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (50 мг, 0,15 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (28 мг, 0,14 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-метилсульфоксидметокси-4-метоксистирил)-2,6-диметилпиридин-4 (1H)-она (36 мг, выход 69%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,41 (д, J=2,0 Гц, 1H), 7,28 (дд, J=8,4, 2,0 Гц, 1Н), 7,09 (д, J=14,4 Гц, 1Н), 7,05 (д, J=8,4 Гц, 1Н), 7,03 (с, 2Н), 6,68 (д, J=14,4 Гц, 1Н), 5,26 (д, J=11,2 Гц, 1Н), 5,11 (д, J=11,2 Гц, 1H), 3,96 (с, 3Н), 2,36 (с, 6Н), 2,26 (с, 3Н); LC-MS: m/z 348,2 [М+Н]+.
Пример 76
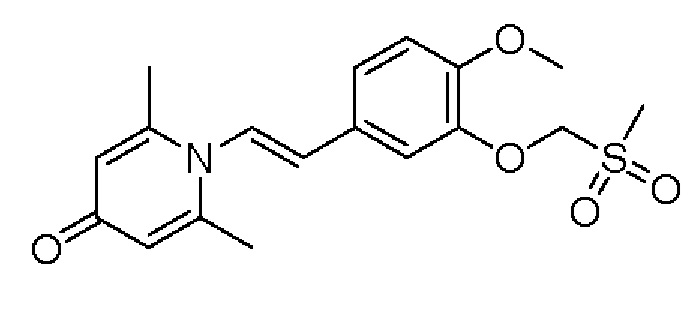
(E)-1-(3-метилсульфонметокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
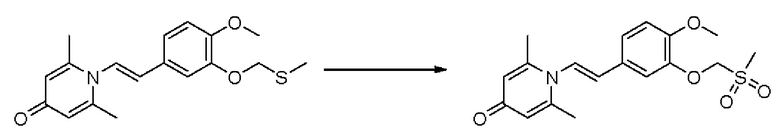
Соединение (Е)-1-(3-метилтиометокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (30 мг, 0,09 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (54 мг, 0,27 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-метилсульфонметокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-она (26 мг, выход 79%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,50 (д, J=2,0 Гц, 1H), 7,32 (дд, J=8,4, 2,0 Гц, 1Н), 7,11 (д, J=8,4, 1Н), 7,10 (д, J=14,0 Гц, 1Н), 6,79 (д, J=14,0 Гц, 1H), 6,37 (с, 2Н), 5,20 (с, 2Н), 3,93 (с, 3Н), 3,11 (с, 3Н), 2,35 (с, 6Н); LC-MS m/z 364,1 [М+Н]+.
Пример 77
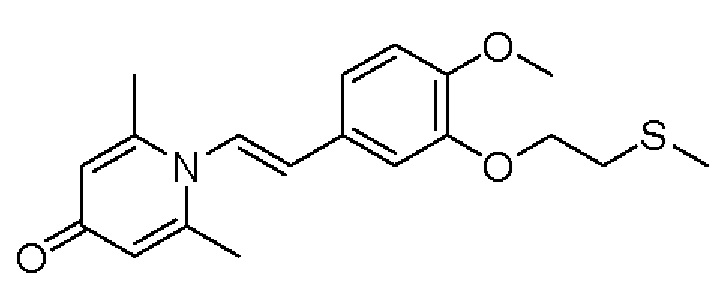
(E)-1-(3-метилтиоэтокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
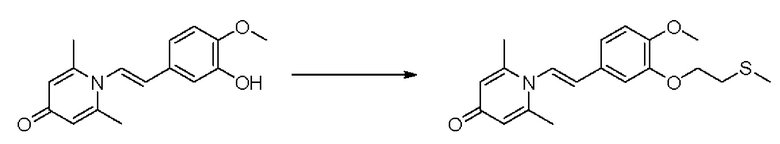
Соединение (E)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (200 мг, 0,73 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли хлорэтил метил сульфид (165 мг, 1,5 ммоль) и карбонат цезия (489 мг, 1,5 ммоль) и перемешивали при комнатной температуре в течение ночи. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-метилтиоэтокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-она (220 мг, выход 87%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,05 (дд, J=8,4, 2,0 Гц, 1H), 7,01 (д, J=2,0 Гц, 1H), 6,91 (д, J=8,4 Гц, 1H), 6,77 (д, J=14,4 Гц, 1H), 6,59 (д, J=14,4 Гц, 1H), 6,26 (с, 2Н), 4,24 (m, J=7,2 Гц, 2Н), 3,91 (с, 3Н), 2,96 (m, J=7,2 Гц, 2Н), 2,26 (с, 6Н), 2,24 (с, 3Н); LC-MS: m/z 346,5 [М+Н]+.
Пример 78
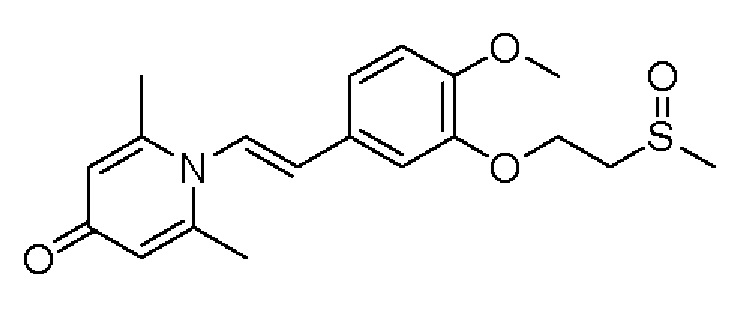
(E)-1-(3-метилсульфоксидэтокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
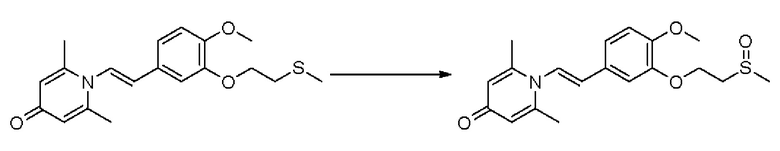
Соединение (E)-1-(3-метилтиоэтокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (50 мг, 0,15 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (28 мг, 0,14 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-метилсульфоксидэтокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-она (30 мг, выход 53%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,07 (д, J=2,0 Гц, 1H), 6,99 (дд, J=8,4, 2,0 Гц, 1H), 6,84 (д, J=8,4 Гц, 1H), 6,72 (д, J=14,4 Гц, 1H), 6,51 (д, J=14,4 Гц, 1H), 6,17 (с, 2Н), 4,50-4,41 (м, 2Н), 3,83 (с, 3Н), 3,27-3,20 (м, 1H), 3,07-3,01 (м, 1H), 2,68 (с, 3Н), 2,17 (с, 6Н); LC-MS: m/z 362,1 [М+Н]+.
Пример 79
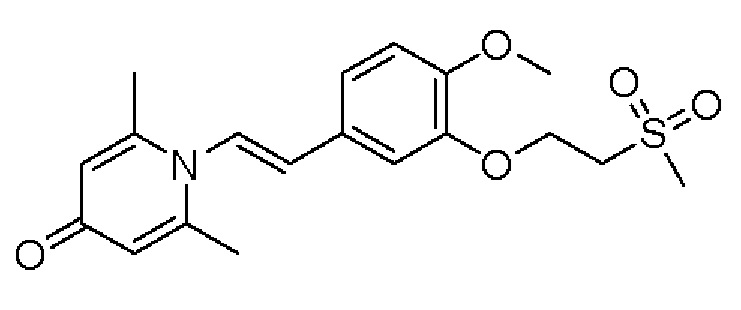
(E)-1-(3-метилсульфонэтокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
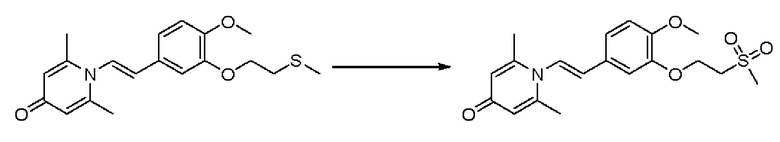
Соединение (E)-1-(3-метилтиоэтокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (30 мг, 0,09 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (54 мг, 0,54 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-метилсульфонэтокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-она (35 мг, выход 61%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,08 (дд, J=8,4, 2,0 Гц, 1H), 7,05 (д, J=2,0 Гц, 1H), 6,92 (д, J=8,4 Гц, 1H), 6,78 (д, J=14,4 Гц, 1H), 6,58 (д, J=14,4 Гц, 1H), 6,31 (с, 2Н), 4,50 (m, J=4,2 Гц, 2Н), 3,89 (с, 3Н), 3,50 (m, J=4,2 Гц, 2Н), 3,19 (с, 3Н), 2,26 (с, 6Н); LC-MS: m/z 378,1 [М+Н]+.
Пример 80
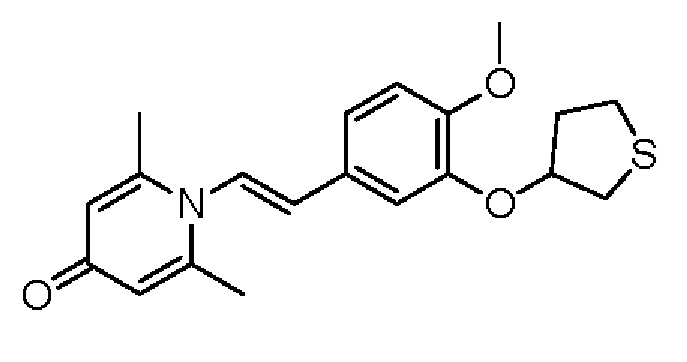
(E)-1-((3-(тетрагидротиофен-3-ил)окси-4-метокси)-стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
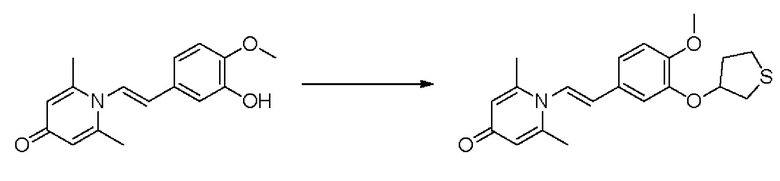
Соединение (Е)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (45 мг, 0,15 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли тетрагидротиофен-3-ил метансульфонат (53 мг, 0,29 ммоль) и карбонат цезия (95 мг, 0,29 ммоль) и перемешивали в атмосфере азота в течение ночи при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-(тетрагидротиофен-3-ил)окси-4-метоксистирил)-2,6-диметил пиридин-4(1H)-она (35 мг, выход 65%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,10 (дд, J=8,4, 2,0 Гц, 1H), 7,08 (д, J=2,0 Гц, 1H), 6,93 (д, J=8,4 Гц, 1H), 6,77 (д, J=14,4 Гц, 1H), 6,58 (д, J=14,4 Гц, 1H), 6,31 (с, 2Н), 5,16-5,13 (м, 1H), 3,90 (с, 3Н), 3,18-3,08 (м, 1H), 3,09 (д, J=3,6 Гц, 2Н), 2,98-2,93 (м, 1H), 2,49-2,42 (м, 1H), 2,27 (с, 6Н), 2,08-1,99 (м, 1H); LC-MS: m/z 358,2 [М+Н]+.
Пример 81
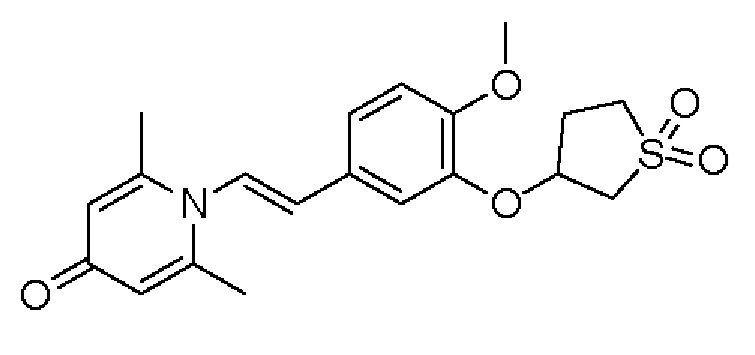
(E)-1-(3-((1,1-диоксотетрагидротиофен-3-ил)окси)-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
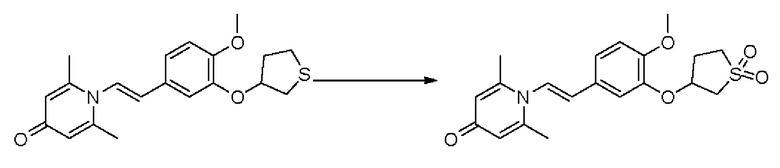
Соединение (E)-1-((3-(тетрагидротиофен-3-ил)окси-4-метокси)стирил)-2,6-диметилпиридин-4(1H)-он (68 мг, 0,19 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (116 мг, 0,57 ммоль) и перемешивали при комнатной температуре в течение 3 часов. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-((1,1-диоксотрагидротиофен-3-ил)окси)-4-метоксистирил)-2,6-диметилпиридин-4(1H)-она (62 мг, выход 86%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,28 (с, 1H), 7,19 (д, J=8,4 Гц, 1H), 7,13 (д, J=14,4 Гц, 1Н), 7,10 (с, 2Н), 6,97 (д,;=14,4 Гц, 1H), 5,34-5,27 (м, 1H), 3,91 (с, 3Н), 3,52-3,44 (м, 1H), 3,41-3,32 (м, 2Н), 3,23-3,17 (м, 1H), 2,70-2,61 (м, 1H), 2,58-2,47 (м, 1H), 2,54 (с, 6Н); LC-MS: m/z 390,1 [М+Н]+.
Пример 82
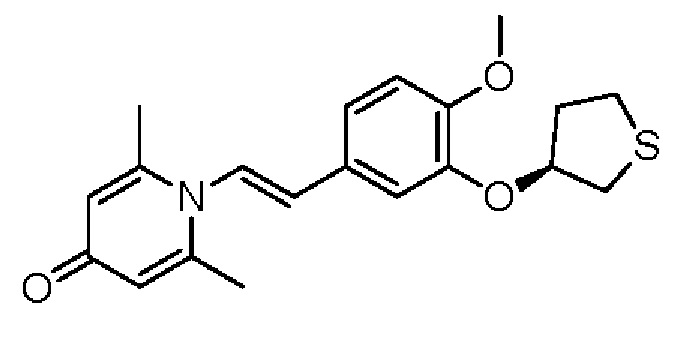
(S,E)-1-(3-(тетрагидротиофен-3-ил)окси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
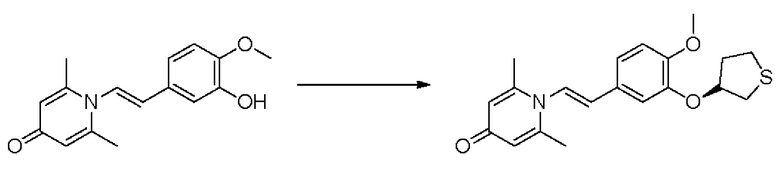
Соединение (Е)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (45 мг, 0,15 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли (R)-тетрагидротиофен-3-ил метансульфонат (53 мг, 0,29 ммоль) и карбонат цезия (95 мг, 0,29 ммоль) и перемешивали в атмосфере азота в течение ночи при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (S,E)-1-(3-(тетрагидротиофен-3-ил)окси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-она (30 мг, выход 56%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,10 (дд, J=8,4, 2,0 Гц, 1H), 7,08 (д, J=2,0 Гц, 1H), 6,93 (д, J=8,4 Гц, 1H), 6,77 (д, J=14,4 Гц, 1H), 6,58 (д, J=14,4 Гц, 1H), 6,31 (с, 2Н), 5,16-5,13 (м, 1H), 3,90 (с, 3Н), 3,18-3,08 (м, 1H), 3,09 (д, J=3,6 Гц, 2Н), 2,98-2,93 (м, 1H), 2,49-2,42 (м, 1H), 2,27 (с, 6Н), 2,08-1,99 (м, 1H); LC-MS: m/z 358,1 [М+Н]+.
Пример 83
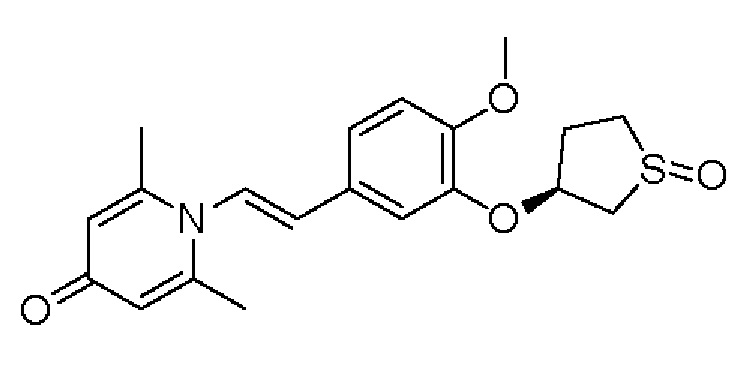
1-((E)-4-метокси-3-(((3S)-1-охотетрагидротиофен-3-ил)окси)стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
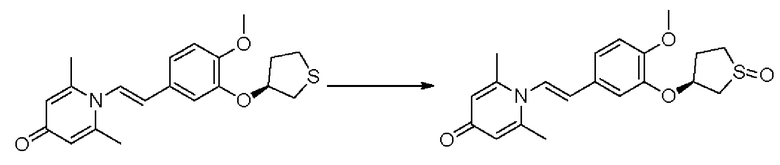
Соединение (S,E)-1-(3-(тетрагидротиофен-3-ил)окси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (81 мг, 0,23 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (41 мг, 0,20 ммоль) и перемешивали в течение 3 часов при 0°С. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением диастереоизомера 1-((E)-4-метокси-3-(((3S)-1-оксотрагидротиофен-3-ил)окси)стирил)-2,6-диметилпиридин-4(1H)-она, который показан ниже:
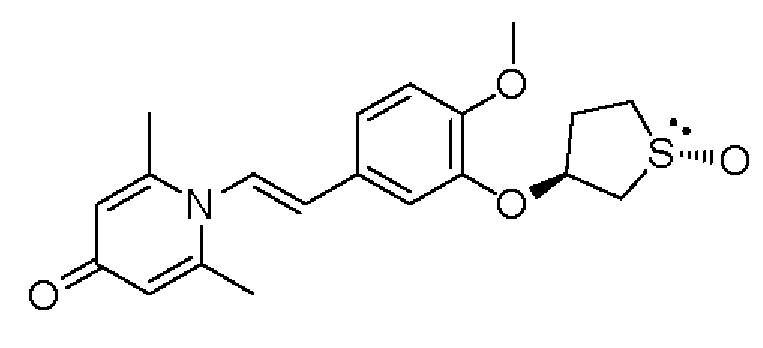
(25 мг, выход 29%, бледно-желтое масло). 1H ЯМР (400 МГц, CDCl3) δ 7,35 (д, J=1,6 Гц, 1H), 7,12 (дд, J=8,4, 1,6 Гц, 1H), 7,07 (д, J=14,4 Гц, 1H), 7,01 (с, 2Н), 6,94 (д, J=8,4 Гц, 1H), 6,68 (д, J=14,4 Гц, 1H), 5,28-5,24 (м, 1H), 3,91 (с, 3Н), 3,34-3,14 (м, 4Н), 2,93-2,85 (м, 1H), 2,51 (с, 6Н), 2,34-2,25 (м, 1Н); LC-MS: m/z 374,1 [М+Н]+.
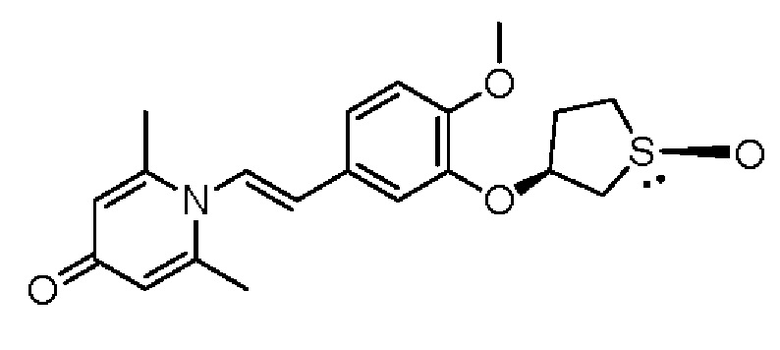
(20 мг, выход 23%, бледно-желтое масло). 1H ЯМР (400 МГц, CDCl3) δ 7,17 (д, J=8,4 Гц, 1Н), 7,15 (с, 1H), 7,07 (д, J=14,4 Гц, 1H), 7,03 (с, 2Н), 6,95 (д, J=8,4 Гц, 1H), 6,71 (д, J=14,4 Гц, 1H), 5,48-5,43 (м, 1H), 3,89 (с, 3Н), 3,64 (д, J=15,2 Гц, 1H), 3,21-3,07 (м, 3Н), 2,83-2,74 (м, 1H), 2,65-2,59 (м, 1H), 2,51 (с, 6Н); LC-MS: m/z 374,1 [М+Н]+.
Пример 84
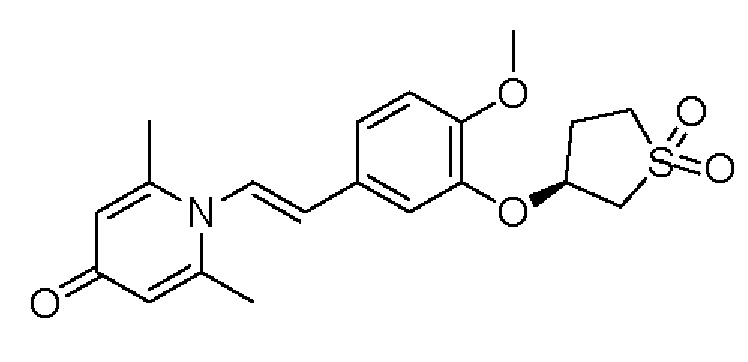
(S,E)-1-(3-((1,1-диоксотетрагидротиофен-3-ил)окси)-4-метокси-стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
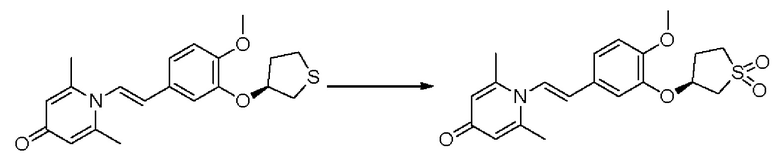
Соединение (E)-1-((3-(тетрагидротиофен-3-ил)окси-4-метокси)стирил)-2,6-диметилпиридин-4(1H)-он (81 мг, 0,23 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (140 мг, 0,69 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-((1,1-диоксотрагидротиофен-3-ил)окси)-4-метоксистирил)-2,6-диметилпиридин-4(1Л)-она (58 мг, выход 66%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,26 (с, 1H), 7,19 (д, J=8,4 Гц, 1H), 7,08 (д, J=14,4 Гц, 1H), 7,05 (с, 2Н), 6,97 (д, J=8,4 Гц, 1H), 6,71 (д, J=14,4 Гц, 1Н), 5,32-5,27 (м, 1H), 3,91 (с, 3Н), 3,52-3,43 (м, 1H), 3,41-3,31 (м, 2Н), 3,22-3,16 (м, 1H), 2,69-2,62 (м, 1H), 2,57-2,47 (м, 1H), 2,52 (с, 6Н); LC-MS m/z 390,2 [М+Н]+.
Пример 85
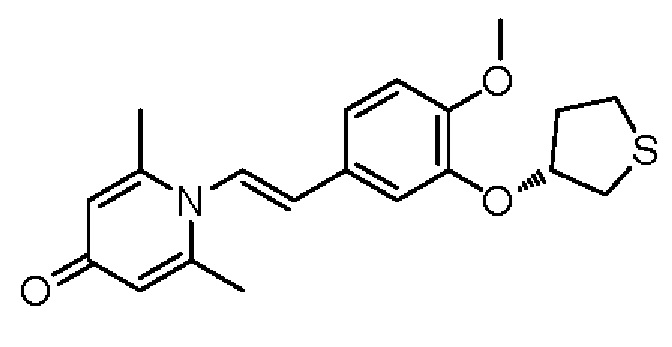
(R,E)-1-(3-(тетрагидротиофен-3-ил)окси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
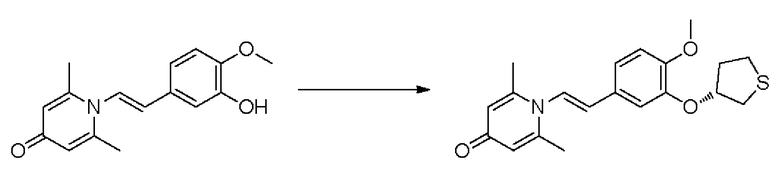
Соединение (Е)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (45 мг, 0,15 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли (5) -тетрагидротиофен-3-ил метансульфонат (53 мг, 0,29 ммоль) и карбонат цезия (95 мг, 0,29 ммоль) и перемешивали в атмосфере азота в течение ночи при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (S,E)-1-(3-(тетрагидротиофен-3-ил)окси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-она (30 мг, выход 56%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,10 (дд, J=8,4, 2,0 Гц, 1H), 7,08 (д, J=2,0 Гц, 1Н), 6,93 (д, J=8,4 Гц, 1H), 6,77 (д, J=14,4 Гц, 1Н), 6,58 (д, J=14,4 Гц, 1H), 6,31 (с, 2Н), 5,16-5,13 (м, 1Н), 3,90 (с, 3Н), 3,18-3,08 (м, 1H), 3,09 (д, J=3,6 Гц, 2Н), 2,98-2,93 (м, 1H), 2,49-2,42 (м, 1H), 2,27 (с, 6Н), 2,08-1,99 (м, 1H); LC-MS: m/z 358,1 [М+Н]+.
Пример 86
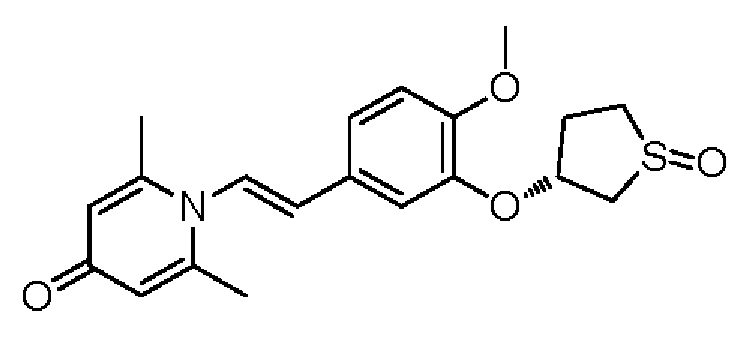
1-((E)-4-метокси-3-(((3R)-1-оксотетрагидротиофен-3-ил)окси)стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
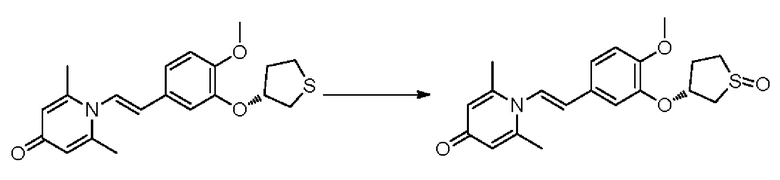
Соединение (R,E)-1-(3-(тетрагидротиофен-3-ил)окси-4-метоксистирил)-2,6-диметилпиридин-4(1H)-он (57 мг, 0,16 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (29 мг, 0,15 ммоль) и перемешивали в течение 3 часов при 0°С. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением диастереоизомера 1-((E)-4-метокси-3-(((3R)-1-оксотрагидротиофен-3-ил) окси)стирил)-2,6-диметилпиридин-4(1H)-она, который показан ниже:
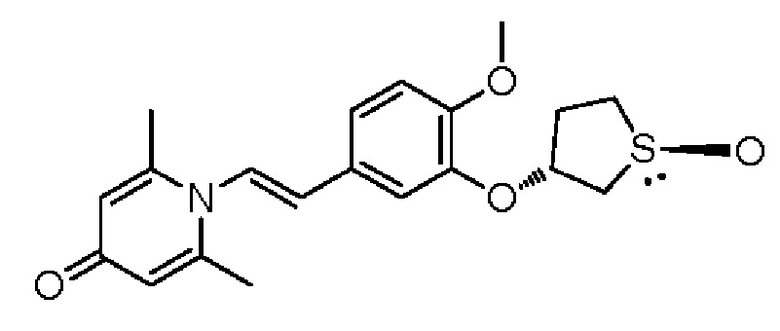
(15 мг, выход 25%, бледно-желтое масло). 1H ЯМР (400 МГц, CD3OD) δ 7,26 (д, J=2,0 Гц, 1H), 7,20 (дд, J=8,4, 2,0 Гц, 1H), 7,12 (д, J=14,4 Гц, 1H), 6,99 (д, J=7,6, 1H), 6,96 (с, 2Н), 6,80 (д, J=14,4, 1H), 5,18-5,13 (м, 1H), 3,80 (с, 3Н), 3,36-3,25 (м, 2Н), 3,09-3,00 (м, 2Н), 2,69-2,61 (м, 1H), 2,49 (с, 6Н), 2,27-2,19 (м, 1H); LC-MS: m/z 374,1 [М+Н]+.
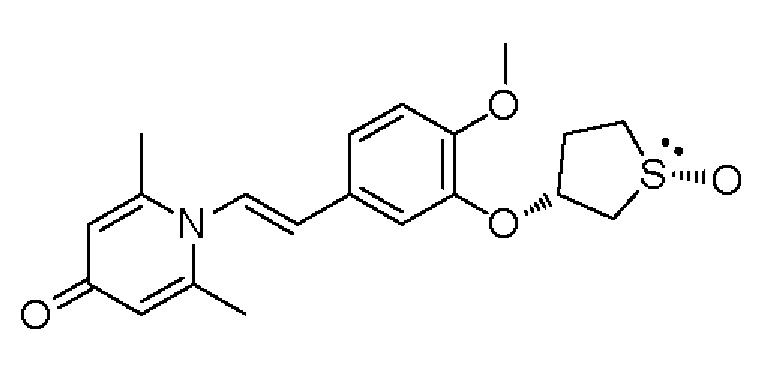
(10 мг, выход 17%, бледно-желтое масло). 1H ЯМР (400 МГц, CD3OD) δ 7,33 (д, J=2,0 Гц, 1Н), 7,30 (дд, J=8,4, 2,0 Гц, 1Н), 7,26 (д, J=14,4 Гц, 1H), 7,10 (д, J=7,6, 1H), 7,09 (с, 2Н), 6,93 (д, J=14,4, 1H), 5,52-5,49 (м, 1Н), 3,89 (с, 3Н), 3,76-3,71 (м, 1H), 3,30-3,22 (м, 2Н), 3,76-3,71 (м, 1H), 2,77-2,67 (м, 1H), 2,65-2,63 (м, 1H), 2,61 (с, 6Н); LC-MS: m/z 374,1 [М+Н]+.
Пример 87
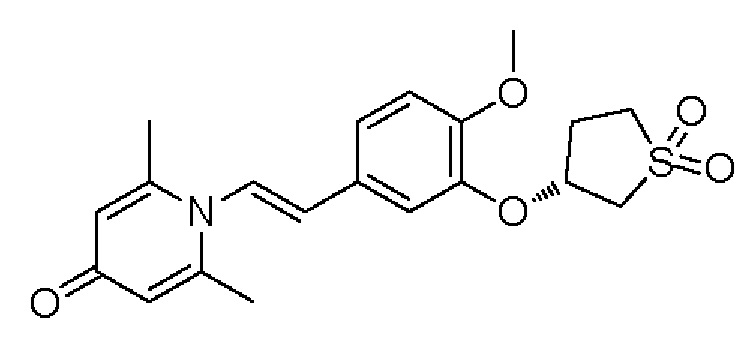
(R,E)-1-(3-((1,1-диоксотетрагидротиофен-3-ил)окси)-4-метокси-стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
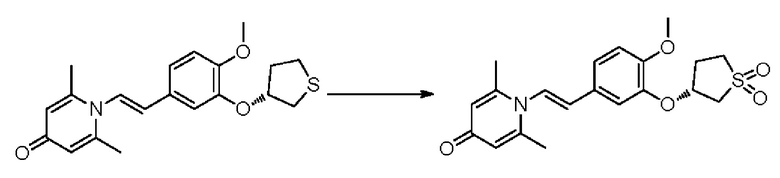
Соединение (Е)-1-((3-(тетрагидротиофен-3-ил)окси-4-метокси)стирил)-2,6-диметилпиридин-4(1H)-он (30 мг, 0,08 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (49 мг, 0,24 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-((1,1-диоксотрагидротиофен-3-ил)окси)-4-метоксистирил)-2,6-диметилпиридин-4(1H)-она (12 мг, выход 40%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,23 (с, 1H), 7,22 (дд, J=8,4, 2,0 Гц, 1H), 7,12 (д, J=14,4 Гц, 1H), 7,10 (дд, J=8,4, 2,0 Гц, 1H), 6,96 (с, 2Н), 6,81 (д, J=14,4, 1H), 5,20-5,16 (м, 1H), 3,80 (с, 3Н), 3,38-3,28 (м, 2Н), 3,25-3,24 (м, 1H), 3,13-3,07 (м, 1H), 2,49 (с, 6Н), 2,47-2,34 (м, 2Н); LC-MS: m/z 390,0 [М+Н]+.
Пример 88
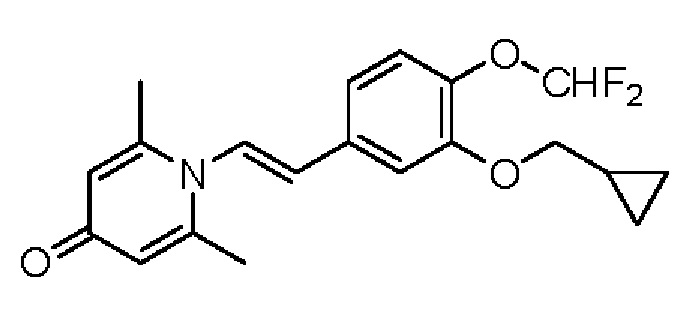
(E)-1-(3-(циклопропилметокси)-4-(дифторметокси)стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
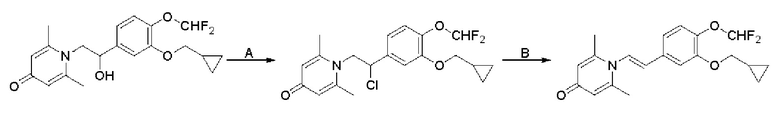
Стадия А:
Соединение 1-(2-(3-циклопропилметокси-4-дифторметоксифенил)-2-гидрокси этил)-2,6-диметилпиридин-4(1H)-он (80 мг, 0,21 ммоль) растворяли в трихлорметане (10 мл), и нагревают до 80°С. Затем добавляли тионилхлорид (0,5 мл) и перемешивали при 80°С в течение 15 минут. После завершения реакции реакционную массу сразу сушили на роторном испарителе с получением неочищенного продукта 1-(2-хлор-2-(3-циклопропил метокси-4-дифторметоксифенил)этил)-2,6-диметилпиридин-4(1H)-она (85 мг, 100%). LC-MS: m/z 398,2 [М+Н]+.
Стадия В:
Соединение 1-(2-хлор-2-(3-циклопропилметокси-4-дифторметоксифенил)этил)-2,6-диметилпиридин-4(1H)-он (85 мг, 0,21 ммоль) растворяли в этаноле (10 мл), затем последовательно добавляли гидроксид натрия (64 мг, 1,60 ммоль) и воду (2 мл), нагревали до 100°С и перемешивали в атмосфере азота в течение 16 часов. После завершения реакции реакционную массу сушили на роторном испарителе и очищали с получением (E)-1-(3-(циклопропилметокси)-4-(дифторметокси)стирил)-2,6-диметилпиридин-4(1Н)-она (20 мг, 26%, белое твердое вещество). 1H ЯМР (400 МГц, ДМСО-d6) δ 7,41 (д, J=2,0 Гц, 1Н), 7,34 (д, J=14,4 Гц, 1H), 7,23 (д, J=8,4 Гц, 1H), 7,17 (дд, J=8,4, 2,0 Гц, 1Н), 7,12 (m, Jh-f=74,4 Гц, 1Н), 6,91 (д, J=14,4 Гц, 1H), 6,49 (с, 2Н), 3,95 (д, J=7,2 Гц, 2Н), 2,34 (с, 6Н), 1,31-1,21 (м, 1H), 0,62-0,57 (м, 2Н), 0,37-0,33 (м, 2Н); LC-MS: m/z 362,2 [М+Н]+.
Пример 89
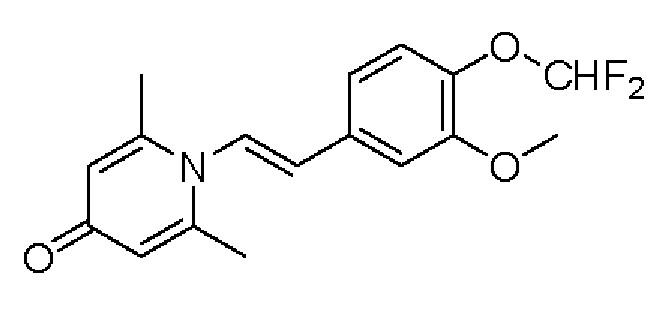
(E)-1-(3-метокси-4-дифторметокси-стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
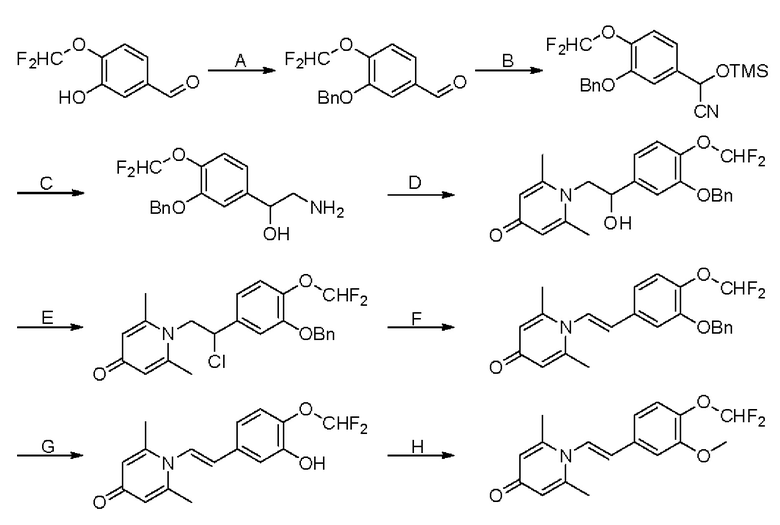
Стадия А:
3-Гидрокси-4-дифторметоксибензальдегид (2,36 г, 12,5 ммоль) растворяли в ацетонитриле (40 мл) при комнатной температуре, затем последовательно добавляли карбонат калия (3,46 г, 25,1 ммоль) и бензилбромид (2,79 г, 16,3 ммоль) и нагревали при перемешивании в атмосфере азота до 80°С в течение 3 часов. После завершения реакции добавляли насыщенный водный раствор хлорида натрия (30 мл) и экстрагировали дихлорметаном (3×120 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, сушили на роторном испарителе и очищали колоночной хроматографией с получением 3-бензокси-4-дифторметоксибензальдегида (3,30 г, 95%).
Стадия В:
3-Бензокси-4-дифторметоксибензальдегид (3,30 г, 11,9 ммоль) растворяли в дихлорметане (30 мл), затем последовательно добавляли триэтиламин (2,40 г, 23,7 ммоль) и триметилсилилцианид (3,53 г, 35,6 ммоль) в ледяной бане и перемешивали в атмосфере азота при комнатной температуре в течение 16 часов. После завершения реакции реакционную массу сразу сушили на роторном испарителе с получением 2-(3-бензокси-4-дифторметоксифенил)-2-триметилсилоксиацетонитрила, который сразу использовали в следующей стадии.
Стадия С:
Соединение 2-(3-бензокси-4-дифторметоксифенил)-2-триметилсилокси ацетонитрил (4,47 г, 11,9 ммоль) растворяли в безводном тетрагидрофуране (40 мл), затем порционно добавляли алюмогидрид лития (1,35 г, 35,6 ммоль) на ледяной бане и перемешивали при комнатной температуре в течение ночи. После завершения реакции реакционную массу разбавляли безводным тетрагидрофураном (200 мл), затем последовательно добавляли воду (1,35 мл), водный раствор гидроксида натрия (1,35 мл, 15%) и воду (4,05 мл), перемешивали в течение 30 минут, сушили над безводным сульфатом натрия, фильтровали и сушили на роторном испарителе с получением неочищенного продукта 2-амино-1-(3-бензокси-4-дифторметоксифенил)этанола, который сразу использовали в следующей стадии.
Стадия D:
Соединение 2-амино-1-(3-бензокси-4-дифторметоксифенил)этанол (3,67 г, 11,9 ммоль) растворяли в этаноле (60 мл), затем последовательно добавляли 2,6-диметил-4H-пиран-4-он (2,19 г, 17,7 ммоль), гидроксид натрия (708 мг, 17,7 ммоль) и воду (10 мл), нагревали до 60°С и перемешивали в атмосфере азота в течение 16 часов. После завершения реакции реакционную массу сушили на роторном испарителе и очищали колоночной хроматографией с получением 1-(2-(3-бензокси-4-дифтор метоксифенил)-2-гидроксиэтил)-2,6-диметилпиридин-4(1H)-она(2,3 г, 47%). LC-MS m/z 416,2 [М+Н]+.
Стадия Е:
Соединение 1-(2-(3-бензокси-4-дифторметоксифенил)-2-гидроксиэтил)-2,6-диметилпиридин-4(1H)-он (2,3 г, 5,3 ммоль) растворяли в трихлорметане (80 мл) и нагревали до 90°С.Затем добавляли тионилхлорид (3 мл) и перемешивали при 90°С в течение 15 минут.После завершения реакции реакционную массу сразу сушили на роторном испарителе с получением неочищенного продукта 1-(2-(3-бензокси-4-дифтор метоксифенил)-2-хлорэтил)-2,6-диметилпиридин-4(1H)-она (2,15 г, 93%). LC-MS m/z 434,2 [М+Н]+.
Стадия F:
Соединение 1-(2-(3-бензокси-4-дифторметоксифенил)-2-хлорэтил)-2,6-диметил пиридин-4(1H)-он (2,15 г, 5,0 ммоль) растворяли в этаноле (40 мл), затем последовательно добавляли гидроксид натрия (1,2 г, 30 ммоль) и воду (15 мл), нагревали до 90°С и перемешивали в атмосфере азота в течение 16 часов. После завершения реакции реакционную массу сушили на роторном испарителе и очищали с получением (Е)-1-(3-бензокси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-она (1,5 г, 75%). LC-MS: m/z 398,2 [M+H]+.
Стадия G:
Соединение (E)-1-(3-бензокси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-он (400 мг, 1,0 ммоль) растворяли в безводном дихлорметане (100 мл), затем медленно по каплям добавляли тетрахлорид титана (1 М, 2 мл, 2,0 ммоль) на ледяной бане и перемешивали в течение ночи при комнатной температуре. После завершения реакции реакционную массу сушили на роторном испарителе и очищали с получением (Е)-1-(3-гидрокси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-она (50 мг, 16%). LC-MS: m/z 308,2 [М+Н]+.
Стадия Н:
Соединение (E)-1-(3-гидрокси-4-дифторметокси-стирил)-2,6-диметилпиридин-4(1H)-он (50 мг, 0,16 ммоль) растворяли в ацетонитриле (5 мл), затем добавляли йодметан (30 мг, 0,21 ммоль) и карбонат калия (90 мг, 0,65 ммоль) и перемешивали в течение 2 часов при 80°С в атмосфере азота. После завершения реакции реакционную массу фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-метокси-4-дифторметокси-стирил)-2,6-диметилпиридин-4(1H)-она (38 мг, выход 71%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,21 (д, J=8,0 Гц, 1H), 7,08-7,05 (м, 2Н), 6,96 (д, J=14,0 Гц, 1Н), 6,69 (д, J=14,0 Гц, 1H), 6,59 (m, JH-f=74,8 Гц, 1Н), 6,41 (с, 2Н), 3,95 (с, 3Н), 2,31 (с, 6Н); LC-MS: m/z 322,2 [М+Н]+.
Пример 90
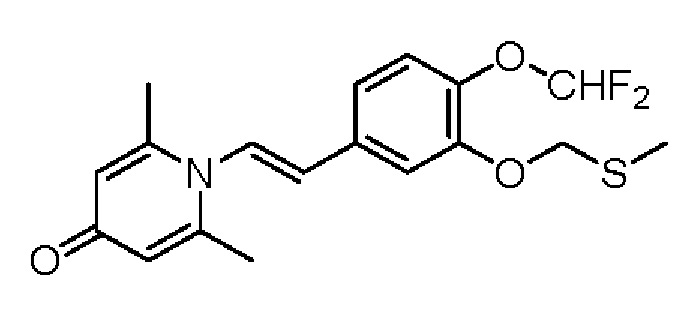
(E)-1-(3-метилтиометокси-4-дифторметокси-стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
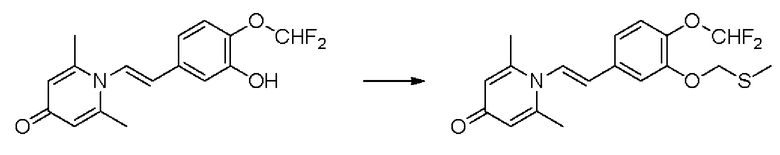
Соединение (E)-1-(3-гидрокси-4-дифторметокси-стирил)-2,6-диметилпиридин-4(1H)-он (35 мг, 0,11 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли хлорметил метил сульфид (22 мг, 0,23 ммоль) и карбонат цезия (150 мг, 0,46 ммоль) и перемешивали в атмосфере азота в течение 1 часа при 80°С. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-метилтиометокси-4-дифторметокси-стирил)-2,6-диметилпиридин-4 (1H)-она (30 мг, выход 71%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,24-7,22 (м, 2Н), 7,16 (дд, J=8,4, 2,0 Гц, 1H), 7,08 (д, J=14,4 Гц, 1H), 6,77 (с, 2Н), 6,76 (д, J=14,4 Гц, 1H), 6,59 (т, JH-F=74,0 Гц, 1H), 5,29 (с, 2Н), 2,41 (с, 6Н), 2,29 (с, 3Н); LC-MS: m/z 368,4 [М+Н]+.
Пример 91
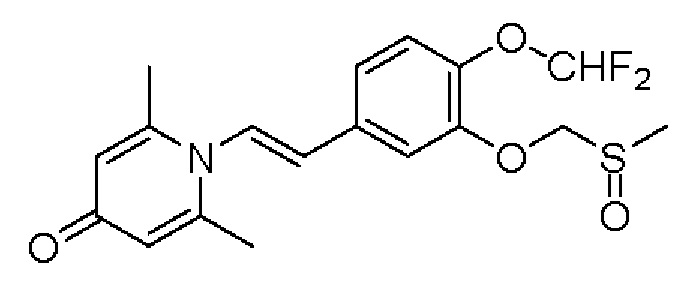
(Е)-1-(3-метилсульфоксидметокси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:

Соединение (E)-1-(3-метилтиометокси-4-дифторметоксистирил)-2,6-диметил пиридин-4(1H)-он (78 мг, 0,21 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (39 мг, 0,19 ммоль) и перемешивали в течение 2 часов при комнатной температуре. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (Е)-1-(3-метилсульфоксидметокси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-она (64 мг, выход 78%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,72 (с, 1H), 7,27 (д, J=14,8 Гц, 1H), 7,26 (д, J=8,4 Гц, 1H), 7,18 (д, J=8,4 Гц, 1H), 7,03 (с, 2Н), 6,80 (д, J=14,8 Гц, 1H), 6,64 (т, JH-F=73,6 Гц, 1H), 5,26 (д, J=11,2 Гц, 1H), 5,11 (д, J=11,2 Гц, 1H), 2,77 (с, 3Н), 2,52 (с, 6Н); LC-MS: m/z 384,0 [М+Н]+.
Пример 92
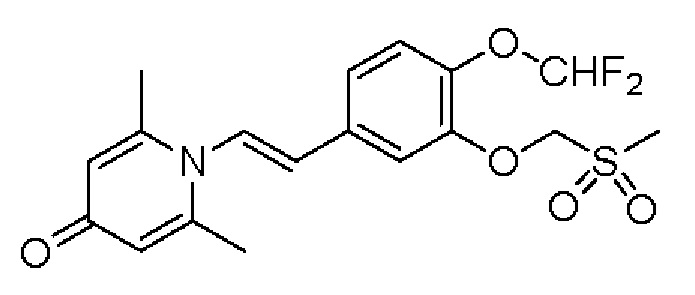
(E)-1-(3-метилсульфонметокси-4-дифторметокси-стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
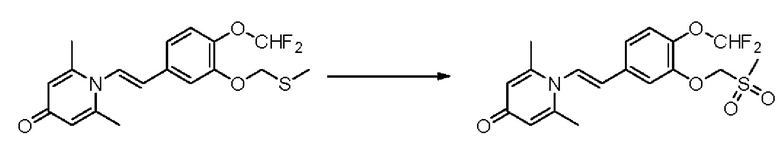
Соединение (E)-1-(3-метилтиометокси-4-дифторметоксистирил)-2,6-диметил пиридин-4(1H)-он (78 мг, 0,21 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (128 мг, 0,63 ммоль) и перемешивали в течение 2 часов при комнатной температуре. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (Е)-1-(3-метилсульфонметокси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-она (35 мг, выход 41%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,56 (д, J=2,0 Гц, 1Н), 7,27 (д, J=8,4, 2,0 Гц, 1Н), 7,26 (д, J=14,4 Гц, 1H), 7,21 (д, J=8,4 Гц, 1H), 6,97 (с, 2Н), 6,91 (д, J=14,4 Гц, 1H), 6,79 (т, JH-F=73,6 Гц, 1Н), 5,23 (с, 2Н), 3,01 (с, 3Н), 2,49 (с, 6Н); LC-MS: m/z 399,9 [М+Н]+.
Пример 93
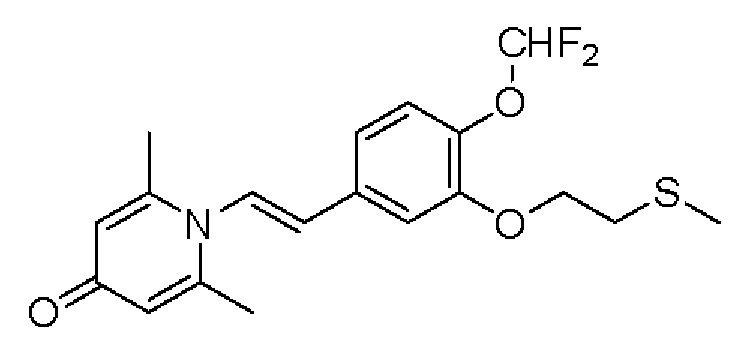
(E)-1-(3-метилтиоэтокси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
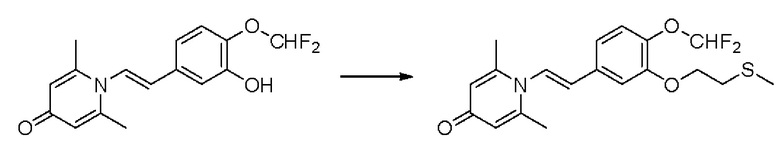
(E)-1-(4-(дифторметокси)-3-гидроксистирил)-2,6-диметилпиридин-4(1H)-он (50 мг, ОД 6 ммоль) растворяли в N,N-диметилформамиде (2 мл) при комнатной температуре, затем последовательно добавляли карбонат калия (34 мг, 0,24 ммоль) и хлорэтил метил сульфид (36 мг, 0,33 ммоль) и нагревали при перемешивании в атмосфере азота до 80°С в течение 16 часов. После завершения реакции реакционную массу фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-метилтиоэтокси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1Н)-она (8 мг, 14%). 1H ЯМР (400 МГц, CDCl3) δ 7,18 (д, J=8,4 Гц, 1H), 7,06-6,99 (м, 2Н), 6,84 (д, J=14,0 Гц, 1H), 6,59 (т, J=14,0 Гц, 1H), 6,58 (д, JH-F=75,2 Гц, 1Н), 6,19 (с, 2Н), 4,20 (т, J=6,4 Гц, 2Н), 2,87 (m, J=6,4 Гц, 2Н), 2,19 (с, 6Н), 2,16 (с, 3Н); LC-MS: m/z 382,2 [М+Н]+.
Пример 94
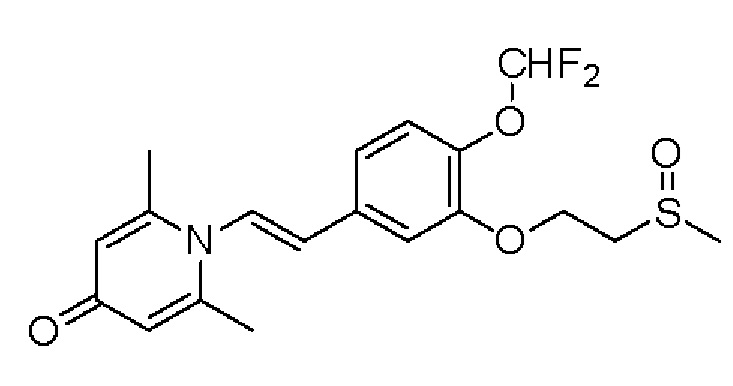
(Е)-1-(3-метилсульфоксидэтокси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
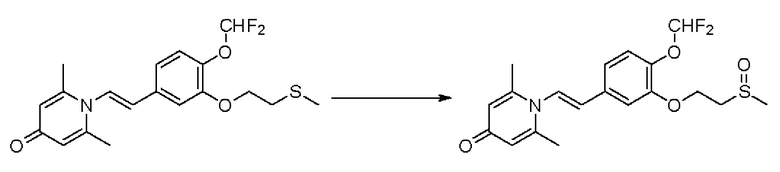
Соединение (E)-1-(3-метилтиоэтокси-4-дифторметоксистирил)-2,6-диметил пиридин-4(1Н)-он (55 мг, 0,14 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (27 мг, 0,13 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (Е)-1-(3-метилсульфоксидэтокси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1Н)-она (16 мг, выход 28%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,38 (с, 1Н), 7,26 (д, J=14,4 Гц, 1H), 7,22 (д, J=8,4 Гц, 1H), 7,10 (д, J=8,4 Гц, 1H), 6,97 (с, 2Н), 6,79 (д, J=14,4 Гц, 1H), 6,58 (т, JH-F=74,0 Гц, 1H), 4,60 (т, J=8,8 Гц, 1H), 3,33-3,26 (м, 1Н), 3,16-3,10 (м, 1H), 2,75 (с, 3Н), 2,51 (с, 6Н); LC-MS: m/z 398,0 [М+Н]+.
Пример 95
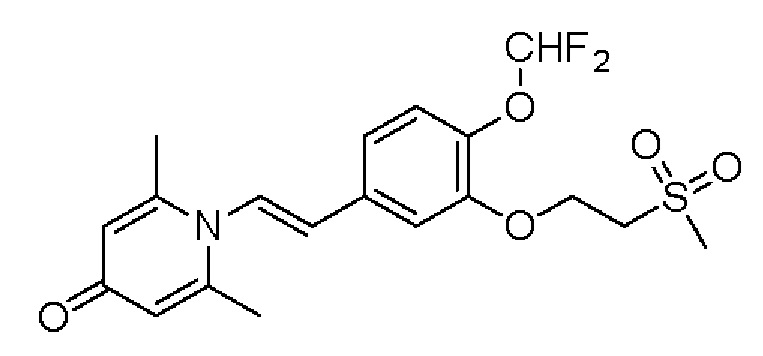
(E)-1-(3-метилсульфонэтокси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
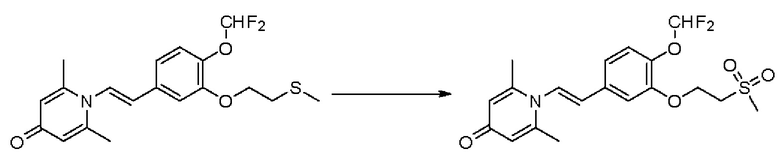
Соединение (E)-1-(3-метилтиоэтокси-4-дифторметоксистирил)-2,6-диметил пиридин-4(1H)-он (55 мг, 0,14 ммоль) растворяли в дихлорметане (5 мл), затем добавляли м-хлорпероксибензойную кислоту (85 мг, 0,42 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-метилсульфонэтокси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1Н)-она (33 мг, выход 75%, бело твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,45 (с, 1H), 7,36 (д, J=14,4 Гц, 1H), 7,30-7,25 (м, 2Н), 7,04 (с, 2Н), 7,00 (д, J=14,4 Гц, 1H), 6,58 (т, JH-F=74,4 Гц, 1H), 4,55 (т, J=5,2 Гц, 1H), 3,66 (т, J=5,2 Гц, 1H), 3,15 (с, 3Н), 2,59 (с, 6Н); LC-MS: m/z 414,0 [М+Н]+.
Пример 96
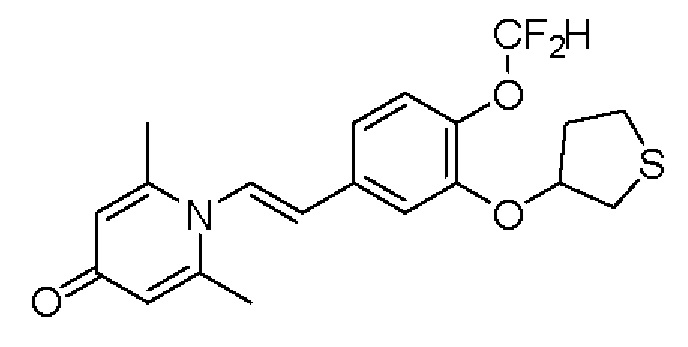
(Е)-1-((3-(тетрагидротиофен-3-ил)окси-4-дифторметокси)-стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
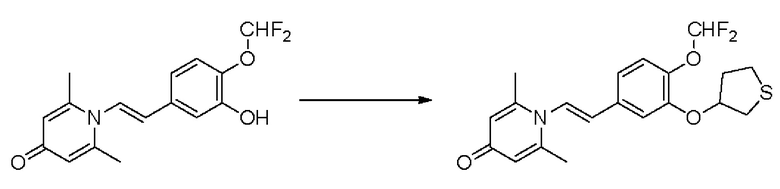
Соединение (E)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он (42 мг, 0,14 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли тетрагидротиофен-3-илметансульфонат (53 мг, 0,29 ммоль) и карбонат цезия (95 мг, 0,29 ммоль) и перемешивали в течение 1 часа при 80°С в атмосфере азота. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(3-(тетрагидротиофен-3-ил)окси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-она (30 мг, выход 54%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,41 (Д, J=2,0 Гц, 1H), 7,27 (д, J=14,4 Гц, 1H), 7,26 (дд, J=8,4, 2,0 Гц, 1Н), 7,21 (д, J=8,4 Гц, 1Н), 6,91 (д, J=14,4 Гц, 1Н), 6,79 (т, JH-F=75,0 Гц, 1H), 6,65 (с, 2Н), 5,36-5,32 (м, 1Н), 3,16 (дд, J=12,0, 4,4 Гц, 1Н), 3,11-3,02 (м, 2Н), 2,99-2,94 (м, 1H), 2,49-2,41 (м, 1H), 2,45 (с, 6Н), 2,10-2,01 (м, 1Н); LC-MS: m/z 394,2 [М+Н]+.
Пример 97
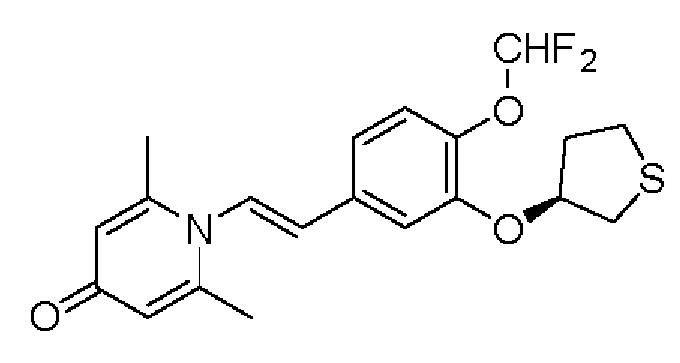
(S,E)-1-(3-(тетрагидротиофен-3-ил)окси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
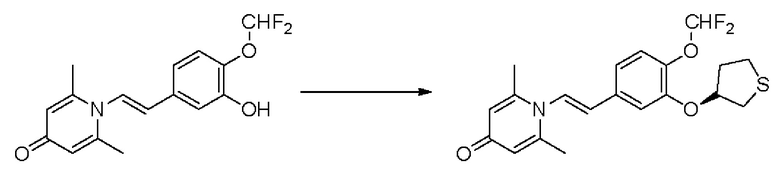
Соединение (E)-1-(3-гидрокси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1Н)-он (50 мг, 0,15 ммоль) растворяли в N,N-диметилформамиде (5 мл), затем добавляли (R)-тетрагидротиофен-3-илметансульфонат (53 мг, 0,29 ммоль) и карбонат цезия (95 мг, 0,29 ммоль) и перемешивали в течение ночи при 80°С в атмосфере азота. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (S,E)-1-(3-(тетрагидротиофен-3-ил)окси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-она (10 мг, выход 17%, бледно-желтое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,23 (д, J=8,4 Гц, 1H), 7,11 (дд, J=8,4, 2,0 Гц, 1Н), 7,08 (д, J=2,0 Гц, 1H), 6,87 (д, J=14,4 Гц, 1Н), 6,77 (д, J=14,4 Гц, 1H), 6,64 (д, J=14,4 Гц, 1H), 6,61 (т, JH-F=74,8 Гц, 1H), 6,29 (с, 2Н), 5,21-5,16 (м, 1Н), 3,16-3,09 (м, 3Н), 3,03-2,97 (м, 1H), 2,52-2,47 (м, 1H), 2,26 (с, 6Н), 2,11-2,02 (м, 1Н); LC-MS: m/z 394,2 [М+Н]+.
Пример 98
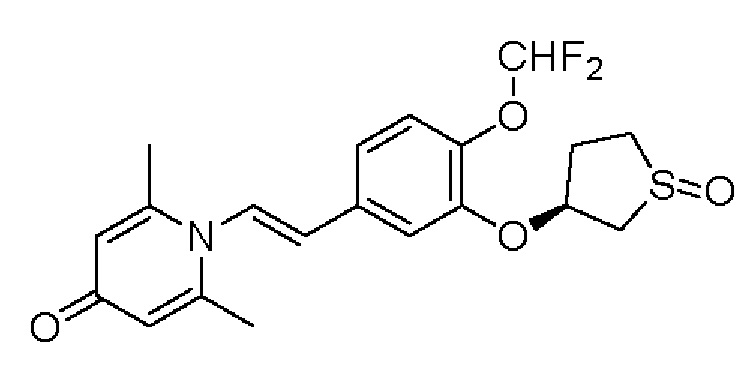
Конкретная схема реакции показана ниже:
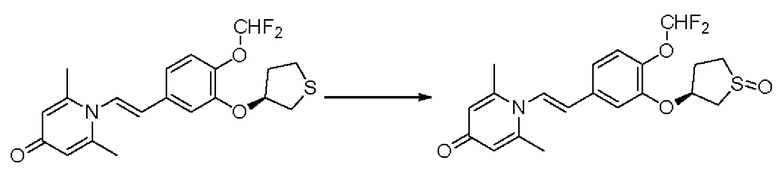
Соединение (E)-1-((3-(тетрагидротиофен-3-ил)окси-4-дифторметокси)стирил)-2,6-диметилпиридин-4(1Н)-он (103 мг, 0,26 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (48 мг, 0,24 ммоль) и перемешивали в течение 2 часов при 0°С. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением диастереоизомера 1-((E)-4-дифторметокси-3-(((3S)-1-оксотетрагидротиофен-3-ил)окси)стирил)-2,6-диметилпиридин-4(1H)-она, который показан ниже:
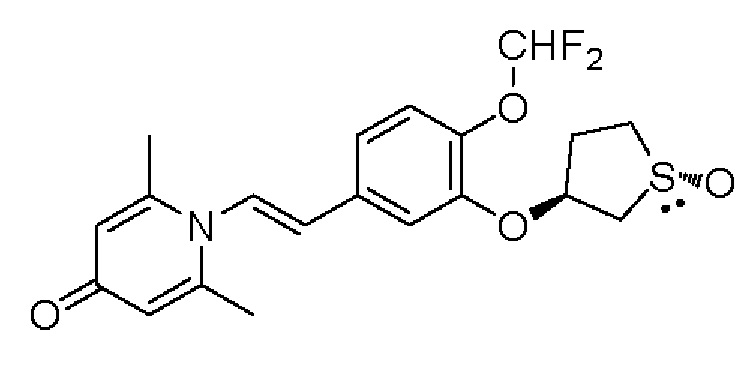
(42 мг, выход 39%, бледно-желтое масло). 1H ЯМР (400 МГц, CDCl3) δ 7,48 (с, 1Н), 7,37 (д, J=14,4 Гц, 1Н), 7,24 (д, J=8,4 Гц, 1H), 7,08 (д, J=8,4 Гц, 1H), 6,94 (с, 2Н), 6,81 (д, J=14,4 Гц, 1Н), 6,80 (т, JH-F=72,4 Гц, 1Н), 5,46-5,38 (м, 1H), 3,46-3,38 (м, 1H), 3,33-3,17 (м, 3Н), 2,94-2,85 (м, 1H), 2,54 (с, 6Н), 2,41-2,32 (м, 1H); LC-MS: m/z 410,1 [М+Н]+.
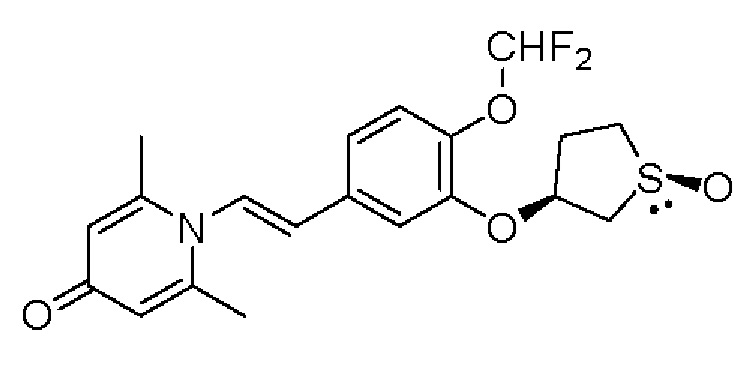
(30 мг, выход 28%, бледно-желтое масло). 1H ЯМР (400 МГц, CDCl3) δ 7,35 (с, 1Н), 7,30 (д, J=14,4 Гц, 1Н), 7,23 (д, J=8,4 Гц, 1H), 7,16 (дд, J=8,4, 1,2 Гц, 1H), 6,95 (с, 2Н), 6,82 (д, J=14,4 Гц, 1H), 6,48 (т, JH-F=73,6 Гц, 1H), 5,58-5,53 (м, 1H), 3,60 (д, J=15,2 Гц, 1H), 3,17 (дд, J=15,2, 4,8 Гц, 1H), 3,11-3,07 (м, 2Н), 2,90-2,80 (м, 1H), 2,65-2,61 (м, 1Н), 2,51 (с, 6Н); LC-MS: m/z 410,1 [М+Н]+.
Пример 99
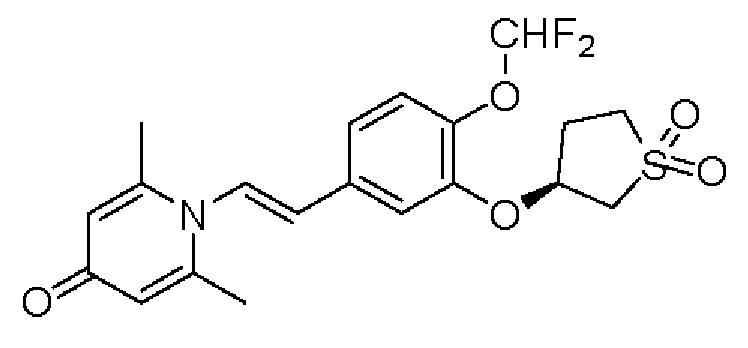
(S,E)-1-(3-(1,1-диоксотетрагидротиофен-3-ил)окси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
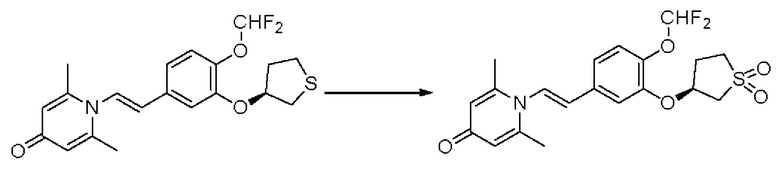
Соединение (E)-1-((3-(тетрагидротиофен-3-ил)окси-4-метокси)стирил)-2,6-диметилпиридин-4(1H)-он (55 мг, 0,14 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (85 мг, 0,42 ммоль) и перемешивали при комнатной температуре в течение 3 часов. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (S,Е)-1-(3-(1,1-диоксотрагидротиофен-3-ил)окси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-она (52 мг, выход 86%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,44 (с, 1H), 7,36 (д, J=14,0, Гц, 1Н), 7,27 (д, J=8,4 Гц, 1Н), 7,14 (д, J=8,4 Гц, 1Н), 6,96 (с, 2Н), 6,81 (д, J=14,0 Гц, 1H), 6,60 (т, JH-F=73,6 Гц, 1H), 5,45-5,40 (м, 1H), 3,50-3,35 (м, 3Н), 3,27-2,21 (м, 1H), 2,70-2,58 (м, 2Н), 2,53 (с, 6Н); LC-MS m/z 426,0 [М+Н]+.
Пример 100
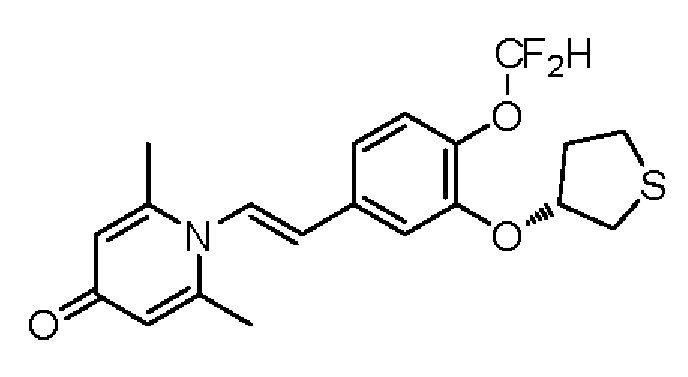
(R,E)-1-(3-(тетрагидротиофен-3-ил)окси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
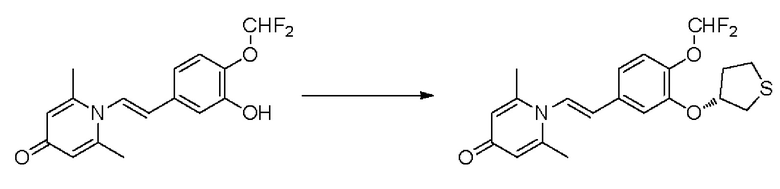
Соединение (E)-1-(3-гидрокси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-он (200 мг, 0,58 ммоль) растворяли в N,N-диметилформамиде (10 мл), затем добавляли (S)-тетрагидротиофен-3-ил метансульфонат (212 мг, 1,16 ммоль) и карбонат цезия (758 мг, 2,3 ммоль) и перемешивали в течение ночи при 80°С в атмосфере азота. После завершения реакции добавляли насыщенный раствор соли (20 мл) и экстрагировали дихлорметаном (3×20 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (R,E)-1-(3-(тетрагидротиофен-3-ил)окси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-она (30 мг, выход 13%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,23 (д, J=8,4 Гц, 1H), 7,11 (дд, J=8,4 2,0 Гц, 1H), 7,08 (д, J=2,0 Гц, 1H), 6,89 (д, J=14,4 Гц, 1H), 6,65 (д, J=14 Гц, 1H), 6,60 (м, J=14,4 Гц, 1Н), 6,63 (m, J=74,8 Гц, 1H), 6,31 (с, 2Н), 5,21-5,16 (м, 1H), 3,16-3,09 (м, 3Н), 3,03-2,97 (м, 1H), 2,52-2,47 (м, 1H), 2,26 (с, 6Н), 2,11-2,02 (м, 1H); LC-MS m/z 394,0 [М+Н]+.
Пример 101
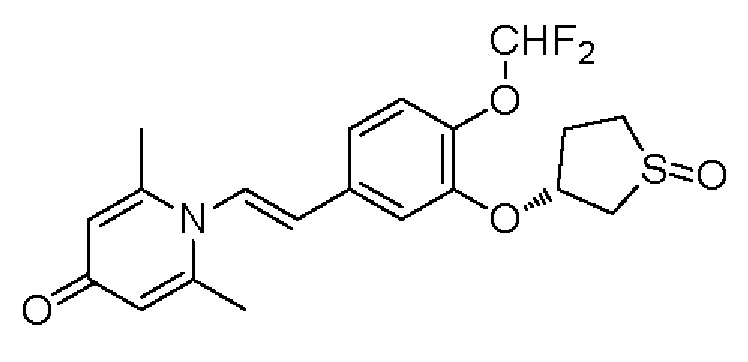
1-((E)-4-дифторметокси-3-(((3R)-1-оксотетрагидротиофен-3-ил)окси)стирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
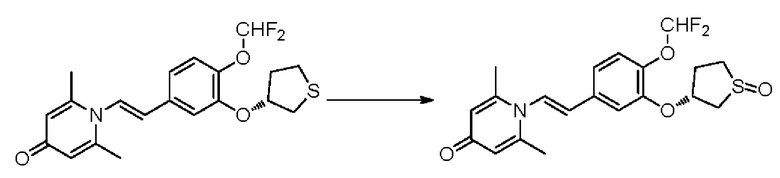
Соединение (R,E)-1-(3-(тетрагидротиофен-3-ил)окси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-он (55 мг, 0,14 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (26 мг, 0,13 ммоль) и перемешивали в течение 3 часов при 0°С. Затем добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением диастереоизомера 1-((Е)-дифторметокси-3-(((3R)-1-оксотрагидротиофен-3-ил)окси)стирил)-2,6-диметилпиридин-4(1H)-она, который показан ниже:
(20 мг, выход 35%, бледно-желтое масло). 1H ЯМР (400 МГц, CD3Cl) δ 7,50 (с, 1H), 7,20 (д, J=12,8 Гц, 1H), 7,24 (д, J=8,4 Гц, 1H), 7,07 (д, J=8,4 Гц, 1H), 6,95 (с, 2Н), 6,82 (с, 1H), 6,80 (м, J=76,4 Гц, 1H), 5,43 (м, 1H), 3,44-3,38 (м, 2Н), 3,29-3,16 (м, 2Н), 2,92-2,89 (м, 1H), 2,54 (с, 6Н), 2,41-2,33 (м, 1H); LC-MS: m/z 410,0 [М+Н]+.
(10 мг, выход 17%, бледно-желтое масло). 1H ЯМР (400 МГц, CD3Cl) δ 7,34 (с, 1H), 7,30 (д, J=14,4 Гц, 1H), 7,23 (д, J=8,4 Гц, 1H), 7,15 (д, J=8,4 Гц, 1H), 6,94 (с, 2Н), 6,82 (д, J=14,4 Гц, 1H), 6,48 (m, J=73,6 Гц, 1H), 5,55 (м, 1H), 3,59 (д, J=14,8 Гц, 1H), 3,19-3,14 (м, 1H), 3,11-3,08 (м, 2Н), 2,90-2,81 (м, 1H), 2,64-2,61 (м, 1H), 2,54 (с, 6Н); LC-MS: m/z 410,0 [М+Н]+.
Пример 102
(R,E)-1-(3-(1,1-диоксотетрагидротиофен-3-ил)окси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
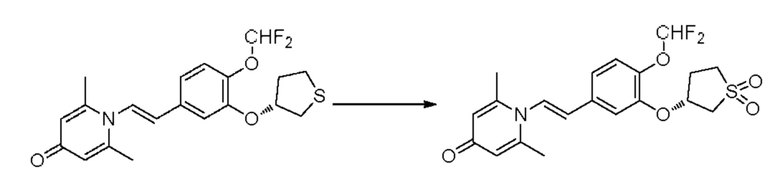
Соединение (R,E)-1-(3-(тетрагидротиофен-3-ил)окси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-он (40 мг, 0,1 ммоль) растворяли в дихлорметане (5 мл), затем добавляли 85% м-хлорпероксибензойную кислоту (61 мг, 0,3 ммоль) и перемешивали при комнатной температуре в течение 2 часов. После завершения реакции добавляли насыщенный водный раствор сульфита натрия (10 мл), перемешивали в течение 10 минут и экстрагировали дихлорметаном (3 × 10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (R,Е)-1-(3-(1,1-диоксотетрагидро тиофен-3-ил)окси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1H)-она (33 мг, выход 75%, белое твердое вещество). 1H ЯМР (400 МГц, CD3OD) δ 7,43 (д, J=1,6 Гц, 1H), 7,38 (дд, J=8,4, 2,0 Гц, 1H), 7,36 (д, J=2,0 Гц, 1H), 7,31 (д, J=8,4 Гц, 1H), 7,06 (с, 2Н), 7,01 (д, J=14,4 Гц, 1H), 6,87 (т, J=75,2 Гц, 1H), 5,43-5,39 (м, 1H), 3,56-8,51 (м, 1H), 3,44-3,36 (м, 2Н), 3,31-3,25 (м, 1H), 2,66-2,62 (м, 2Н), 2,61 (с, 6Н); LC-MS: m/z 426,0 [М+Н]+.
Пример 103
(E)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-метилэтенил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
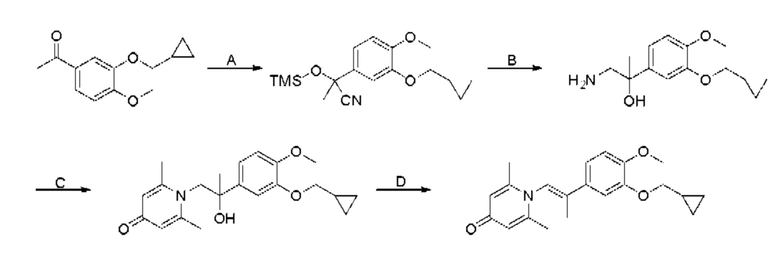
Стадия А:
3-Циклопропилметокси-4-метоксифенилацетофенон (220 мг, 1,0 ммоль) растворяли в ацетонитриле (2 мл); добавляли фторид цезия (76 мг, 0,50 ммоль) в атмосфере азота, затем медленно по каплям добавляли триметилсилилцианид (149 мг, 1,50 ммоль) при 0°С и перемешивали в течение 5 часов. Реакционную массу фильтровали и сушили на роторном испарителе с получением неочищенного продукта 2-(3-циклопропилметокси-4-метоксифенил)-2-триметилсилоксипропионитрила. LC-MS: m/z 320,2 [М+Н]+.
Стадия В:
Соединение 2-(3-циклопропилметокси-4-метоксифенил)-2-триметилсилокси пропионитрил (319 мг, 1,0 ммоль) растворяли в безводном тетрагидрофуране (3 мл); в атмосфере азота медленно по каплям добавляли алюмогидрид лития (96 мг, 2,0 ммоль) при 0°С, и раствор перемешивали при комнатной температуре в течение 1 часа. После завершения реакции добавляли декагидрат сульфата натрия для того, чтобы погасить реакцию. Затем реакционную массу фильтровали и фильтрат сушили на роторном барабане с получением неочищенного продукта 1-амино-2-(3-(циклопропилметокси)-4-метоксифенил)-2-пропанола. LC-MS: m/z 252,2 [М+Н]+.
Стадия С:
Соединение 1-амино-2-(3-(циклопропилметокси)-4-метоксифенил)-2-пропанол (251 мг, 1,0 ммоль) растворяли в этаноле (3 мл), затем добавляли 2,6-диметил-4Н-пиран-4-он (186 мг, 1,5 ммоль) и гидроксид натрия (80 мг, 2,0 ммоль) и перемешивали в течение ночи при 60°С. Завершение реакции контролировали с помощью LCMS. Реакционную массу сушили на роторном испарителе и очищали с получением 1-(2-(3-(циклопропилметокси)-4-метоксифенил)-2-гидроксипропил)-2,6-диметилпиридин-4(1Н)-она (40 мг, выход 11%). LC-MS: m/z 358,0 [М+Н]+.
Стадия D:
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксипропил)-2,6-диметилпиридин-4(1H)-он (36 мг, 0,1 ммоль) растворяли в толуоле (3 мл), затем добавляли n-толуолсульфоновую кислоту (29 мг, 0,15 ммоль) и перемешивали в течение ночи при 110°С. Завершение реакции контролировали с помощью ТСХ. Реакционную массу сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-метилэтенил)-2,6-диметилпиридин-4(1H)-она (27 мг, выход 80%, белое твердое вещество). 1H ЯМР (400МГц, CDCl3) δ 7,05-7,03 (м, 1H), 6,92-6,90 (м, 2Н), 6,68 (с, 2Н), 6,10 (с, 1H), 3,92 (с, 3Н), 3,88 (д, J=6,8 Гц, 2Н), 2,42 (с, 3Н), 2,52 (с, 6Н), 1,12-1,06 (м, 1H), 0,72-0,68 (м, 2Н), 0,40-0,36 (м, 2Н); LC-MS: m/z 340,2 [М+Н]+.
Пример 104
(Е)- 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-фенилэтенил)-2,6-диметилпиридин-4(1H)-он
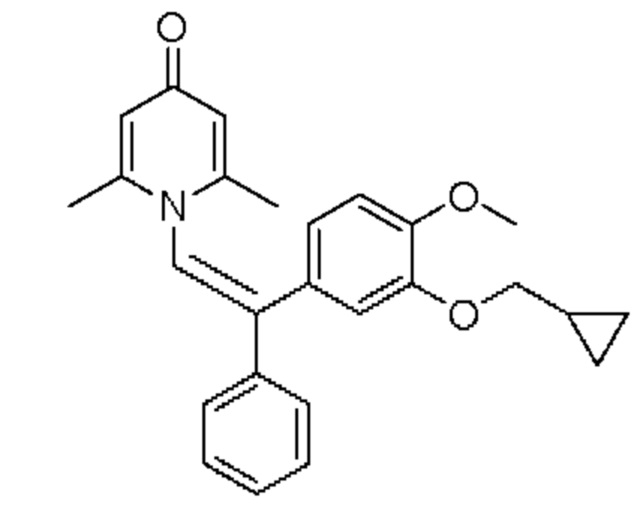
(Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-фенилэтенил)-2,6-диметилпиридин-4(1H)-он
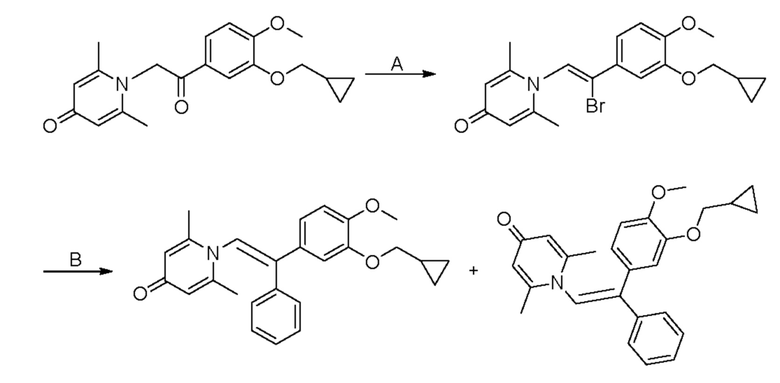
Стадия А:
Трисфенилфосфит (1,49 г, 4,80 ммоль) растворяли в безводном тетрагидрофуране (15 мл), затем триэтиламин (0,53 мг, 5,20 ммоль) и соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-оксоэтил)пиридин-2,6-диметил-4(1H)-он (1,36 мг, 4,00 ммоль) медленно добавляли к реакционной массе при -60°С и перемешивали в течение 15 минут. Затем к реакционной массе медленно добавляли бром (0,77 мг, 4,80 ммоль). После завершения реакции реакцию гасили насыщенным водным раствором хлорида аммония, затем реакционную массу сразу сушили на роторном испарителе и очищали колоночной хроматографией с получением 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-бромэтенил)-2,6-диметилпиридин-4(1H)-она (708 мг, выход 44%). LC-MS: m/z 403,9 [М+Н]+.
Стадия В:
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-бромэтенил)-2,6-диметилпиридин-4(1H)-он (200 мг, 0,50 ммоль) растворяли в тетрагидрофуране (2 мл) и воде (1 мл). В атмосфере азота к реакционной массе добавляли ацетат палладия (5,5 мг, 5 мол. %), карбонат натрия (5 мг, 0,50 ммоль) и фенилборную кислоту (73,0 мг, 0,60 ммоль) и перемешивали при 80°С в течение 1 часа. После завершения реакции добавляли насыщенный раствор хлорида аммония (10 мл) и экстрагировали дихлорметаном (3 × 10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Реакционную массу фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-1-(2-(3-циклопропилметокси-4-метокси фенил)-2-фенилэтенил)-2,6-диметилпиридин-4(1H)-она (69 мг, выход 34%, белое твердое вещество) и (Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-фенилэтенил)-2,6-диметилпиридин-4(1H)-она (30,4 мг, выход 15%, белое твердое вещество).
(Е)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-фенилэтенил)-2,6-диметилпиридин-4(1H)-он: 1H ЯМР (400 МГц, CDCl3) δ 8,40 (с, 1H), 8,35 (с, 1H), 8,06-8,04 (м, 2Н), 7,68-7,66 (м, 3Н), 7,26-7,20 (м, 2Н), 7,04 (д, J=8,4 Гц, 1H), 6,33 (м, 1H), 3,85 (д, J =6,8 Гц, 2Н), 3,81 (с, 3Н), 2,83 (с, 6Н), 0,89-0,82 (м, 1H), 0,62-0,58 (м, 2Н), 0,34-0,30 (м, 2Н); LC-MS: m/z 401,9 [М+Н]+.
(Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-фенилэтенил)-2,6-диметилпиридин-4(1H)-он: 1H ЯМР (400 МГц, CDCl3) δ 8,50 (с, 1H), 8,39 (с, 1H), 8,12-8,08 (м, 2Н), 7,65-7,62 (м, 3Н), 7,32 (д, J=8,4 Гц, 1H), 7,10-7,00 (м, 2Н), 6,13 (м, 1H), 3,86 (д, J=6,8 Гц, 2Н), 3,81 (с, 3Н), 2,83 (с, 6Н), 0,89-0,82 (м, 1Н), 0,62-0,58 (м, 2Н), 0,34-0,30 (м, 2Н); LC-MS: m/z 401,9 [М+Н]+.
Пример 105
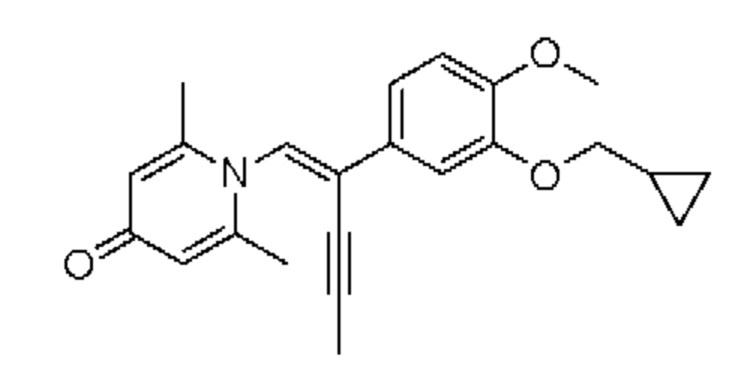
(Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)пент-1-ен-3-ин-1-ил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
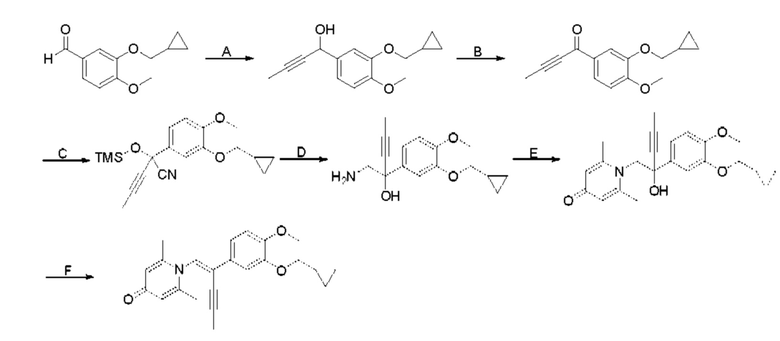
Стадия А:
3-(Циклопропилметокси)-4-метоксибензальдегид (1,03 г, 5 ммоль) растворяли в безводном тетрагидрофуране (5 мл); в атмосфере азота по каплям добавляли 1-этинилмагнийбромид (0,5 М, 12 мл, 6 ммоль) при 0°С, нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 3 часов. Реакцию гасили насыщенным водным раствором хлорида аммония и экстрагировали дихлорметаном (3 × 30 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и сушили на роторном испарителе с получением неочищенного продукта 1-(3-циклопропилметокси-4-метоксифенил)-2-алкинил-1-бутанола (1,50 г). LC-MS: m/z 228,9 [М - 17]+.
Стадия В:
Соединение 1-(3-циклопропилметокси-4-метоксифенил)-2-алкинил-1-бутанол (1,50 г, 5 ммоль) растворяли в дихлорметане (20 мл); затем добавляли периодинан Десса-Мартина (3,18 г, 7,5 ммоль) и твердый бикарбонат натрия (6,30 г, 75 ммоль) при 35°С и перемешивали в течение 1 часа. После завершения реакции добавляли насыщенный раствор бикарбоната натрия и экстрагировали дихлорметаном (3 × 30 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали, сушили на роторном испарителе и очищали с получением неочищенного продукта 1-(3-циклопропилметокси-4-метоксифенил)-2-алкинил-1-бутанола, который сразу использовали в следующей стадии. LC-MS: m/z 249,9 [М+Н]+.
Стадия С:
Соединение 1-(3-циклопропилметокси-4-метоксифенил)-2-алкинил-1-бутанон (336 мг, 1,5 ммоль) растворяли в ацетонитриле (5 мл); в атмосфере азота добавляли фторид цезия (114 мг, 0,75 ммоль), медленно по каплям добавляли триметилсилилцианид (225 мг, 2,25 ммоль) при 0°С и перемешивали в течение 1,5 часов. После завершения реакции реакционную массу фильтровали и сушили на роторном испарителе с получением неочищенного продукта 2-(3-циклопропилметокси-4-метоксифенил)-2-(триметилсилокси)пент-3-инил нитрила. LC-MS: m/z 344,2 [М+Н]+.
Стадия D:
Соединение 2-(3-циклопропилметокси-4-метоксифенил)-2-(триметилсилокси)-3-алкинилпентил нитрил (413 мг, 1,5 ммоль) растворяли в безводном тетрагидрофуране (4 мл); в атмосфере азота медленно по каплям добавляли алюмогидрид лития (114 мг, 3,0 ммоль) при 0°С, и раствор перемешивали при комнатной температуре в течение 1 часа. После завершения реакции добавляли декагидрат сульфата натрия для того, чтобы погасить реакцию. Реакционную массу фильтровали, фильтрат сушили на роторном испарителе с получением неочищенного продукта 1-амино-2-(3-циклопропилметокси) 4-метоксифенил)-3-алкинил-2-пентанола. LC-MS: m/z 276,2 [М+Н]+.
Стадия Е:
Соединение 1-амино-2-(3-(циклопропилметокси)-4-метоксифенил)-3-алкинил -2-пентанол (413 мг, 1,5 ммоль) растворяли в этаноле (5 мл), затем добавляли 2,6-диметилпиран-4-он (279 мг, 2,25 ммоль) и гидроксид натрия (120 мг, 3,0 ммоль) и перемешивали в течение ночи при 60°С. Завершение реакции контролировали с помощью LCMS. Реакционную массу сушили на роторном испарителе и очищали с получением 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксипент-3-ин-1-ил)-2,6-диметилпиридин-4(1H)-она (20 мг, выход 3%). 1H ЯМР(400 МГц, CDCl3) δ 7,12-7,07 (м, 2Н), 6,86 (д, J=8,4 Гц, 1H), 6,20 (с, 1H), 6,10 (с, 1H), 4,22-4,12 (м, 2Н), 3,88 (с, 3H), 3,83 (д, J=7,2 Гц, 2Н), 2,42 (с, 3H), 2,20 (с, 3H), 1,88 (с, 3H), 1,34-1,28 (м, 1H), 0,67-0,62 (м, 2Н), 0,39-0,35 (м, 2Н); LC-MS: m/z 381,9 [М+Н]+.
Стадия F:
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксипент-3-ин-1-ил)-2,6-диметилпиридин-4(1H)-он (20 мг, 0,05 ммоль) растворяли в толуоле (3 мл), затем добавляли n-толуолсульфоновую кислоту (15 мг, 0,08 ммоль) и перемешивали в течение ночи при 60°С. Завершение реакции контролировали с помощью ТСХ. Реакционную массу сушили на роторном испарителе и очищали с получением (Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)пент-1-ен-3-ин-1-ил)-2,6-диметилпиридин-4(1Н)-она (18 мг, выход 90%, белое твердое вещество). 1H ЯМР(400 МГц, CDCl3) δ 7,27 (дд, J=8,4, 2,0 Гц, 1H), 7,16 (J=8,4 Гц, 1H), 6,92-6,90 (м, 2Н), 6,31 (с, 2Н), 3,92-3,90 (м, 5Н), 2,25 (с, 6Н), 1,98 (с, 3H), 1,40-1,31 (м, 1H), 0,70-0,65 (м, 2Н), 0,41-0,37 (м, 2Н); LC-MS: m/z 363,9 [М+Н]+.
Пример 106
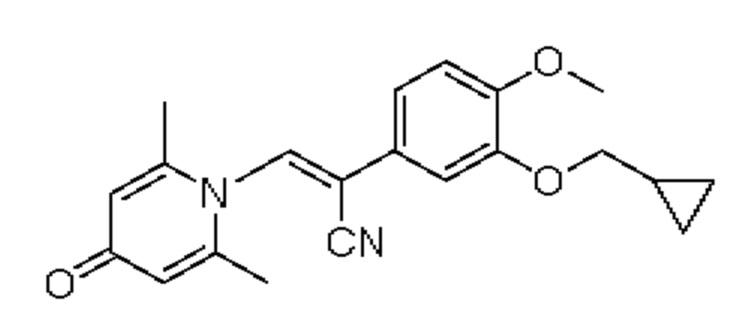
(Е)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-цианоэтенил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
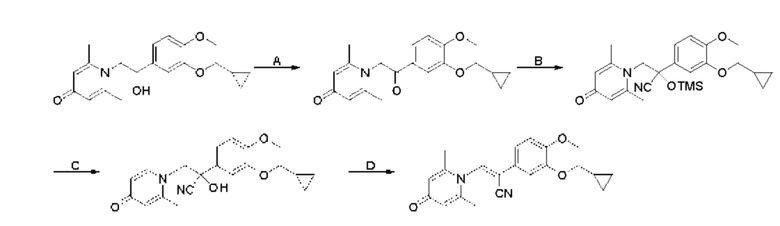
Стадия А:
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил)-2,6-диметилпиридин-4(1H)-он (2,50 г, 7,3 ммоль) растворяли в дихлорметане (50 мл); медленно добавляли периодинан Десса-Мартина (4,80 г, 11,0 ммоль) и бикарбонат натрия (9,00 г, ПО ммоль) при 35°С и перемешивали в течение 1 часа. После завершения реакции добавляли насыщенный раствор бикарбоната натрия (50 мл) и экстрагировали дихлорэтаном (3 × 30 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали с получением 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-она (1,98 г, выход 80%, белое твердое вещество). LC-MS: m/z 342,4 [М+Н]+.
Стадия В:
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1H)-он (500 мг, 1,5 ммоль) растворяли в ацетонитриле (25 мл); в атмосфере азота добавляли триэтиламин (456 мг, 4,5 ммоль), медленно по каплям добавляли триметилсилилцианид (600 мг, 6,0 ммоль) при 0°С и перемешивали в течение 4 часов. Реакционную массу сушили на роторном испарителе с получением неочищенного продукта 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-циано-2-(триметилсилилокси)этил)-2,6-диметилпиридин-4(1H)-она. LC-MS: m/z 441,4 [М+Н]+.
Стадия С:
Соединение 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-циано-2-(триметилсилилокси)этил)-2,6-диметилпиридин-4(1H)-он (660 мг, 1,5 ммоль) растворяли в ацетонитриле (25 мл), добавляли водную соляную кислоту (3 М, 1,5 мл) и перемешивали при 80°С в течение 30 минут. Завершение реакции контролировали с помощью LCMS.
Реакционную массу сушили на роторном испарителе с получением неочищенного продукта 1-(2-(3-циклопропилметокси-4-метоксифенил)-2-циано-2-гидроксиэтил)-2,6-диметилпиридин-4(1H)-она. LC-MS: m/z 369,4 [M+H]+.
Стадия D:
Соединение 1 -(2-(3-циклопропилметокси-4-метоксифенил)-2-циано-2-гидроксиэтил)-2,6-диметилпиридин-4(1H)-он (552 мг, 1,5 ммоль) растворяли в хлороформе (25 мл), затем добавляли тионилхлорид (1,0 мл) и перемешивали в течение 30 минут при 80°С. Завершение реакции контролировали с помощью ТСХ. Реакционную массу сушили на роторном испарителе и очищали с получением (Е)-1-(2-(3-циклопропил метокси-4-метоксифенил)-2-цианоэтенил)-2,6-диметилпиридин-4(1H)-она (283 мг, 54%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,44 (с, 1H), 7,29 (д, J=2,0 Гц, 1H), 7,14 (д, J=1,2 Гц, 1H), 6,97 (д, J=8,4 Гц, 1H), 6,36 (с, 2Н), 3,95-3,92 (м, 5Н), 2,30(с, 6Н), 1,38-1,30 (м, 1H), 0,71-0,67 (м, 2Н), 0,42-0,38 (м, 2Н). LC-MS: m/z 351,4 [М+Н]+.
Пример 107
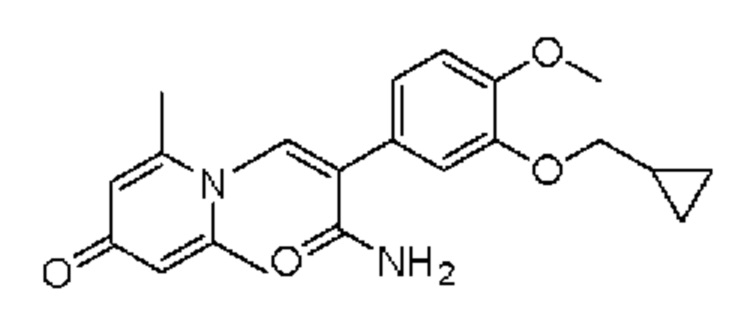
(Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4H)-ил)-акриламид
Конкретная схема реакции показана ниже:

(E)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-цианоэтенил)-2,6-диметилпиридин-4(1Н)-он (500 мг, 1,4 ммоль) растворяли в ацетонитриле (10 мл), затем добавляли гидроксид калия (10 мас. %, 10 мл) при комнатной температуре и перемешивали при 80°С в течение 2 часов. Завершение реакции контролировали с помощью ТСХ. Реакционную массу сразу сушили на роторном испарителе и очищали колоночной хроматографией с получением (Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонил пиридин-1(4H)-ил)-акриламида (140 мг, 27%, белое твердое вещество). 1H ЯМР (400 МГц, ДМСО-d6) δ 7,73 (с, 1Н), 7,56 (с, 1H), 7,13 (с, 1Н), 7,04 (с, 2Н), 6,99 (с, 1H), 5,96 (с, 2Н), 3,85 (д, J=7,2 Гц, 2Н), 3,80 (с, 3H), 2,27 (м, 6Н), 0,87-0,84 (м, 1H), 0,61-0,56 (м, 2Н), 0,35-0,31 (м, 2Н); LC-MS: m/z 369,1 [М+Н]+.
Пример 108
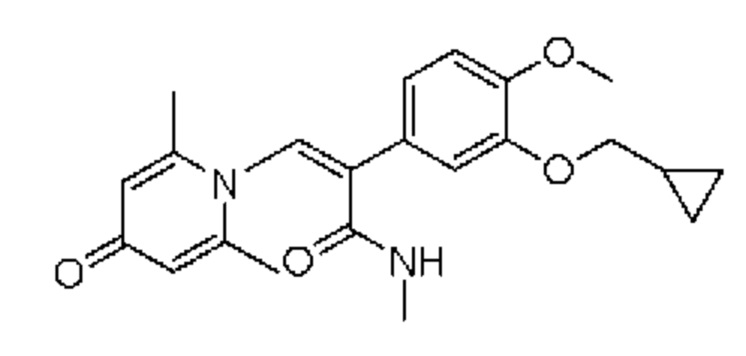
(Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1 (4H)-ил)-N-метилакриламид
Конкретная схема реакции показана ниже:
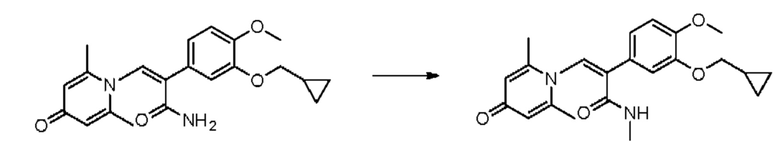
Соединение (Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4H)-ил)-акриламид (30 мг, 0,08 ммоль) растворяли в N,N-диметилформамиде (0,5 мл), затем добавляли 60% гидрид натрия (16 мг, 0,4 ммоль) и перемешивали при 70°С в течение 30 минут. К реакционной массе добавляли йодметан (12 мг, 0,08 ммоль) и перемешивали при 70°С в течение еще 1 часа. Реакционную массу гасили насыщенным водным раствором хлорида аммония, и затем сразу сушили на роторном испарителе и очищали колоночной хроматографией с получением (Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4H)-ил)-N-метилакриламида (5 мг, 17%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,04-7,02 (м, 1Н), 6,93-6,91 (м, 2Н), 6,63 (с, 1Н), 6,28 (с, 2Н), 6,02 (с, 1Н), 3,92 (с, 3H), 3,88 (д, J=6,8 Гц, 2Н), 2,81 (т, J=4,8 Гц, 3H), 2,34 (с, 6Н), 0,97-0,83 (м, 1Н), 0,76-0,62 (м, 2Н), 0,40-0,36 (м, 2Н); LC-MS: m/z 383,1 [М+Н]+.
Пример 109
(Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4H)-ил)-N,N-диметилакриламид
Конкретная схема реакции показана ниже:
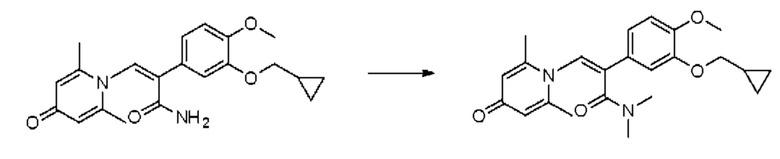
Соединение (Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4H)-ил)акриламид (30 мг, 0,08 ммоль) растворяли в N,N-диметилформамиде (0,5 мл), затем добавляли 60% гидрид натрия (16 мг, 0,4 ммоль) и перемешивали при 70°С в течение 30 минут. К реакционной массе добавляли йодметан (24 мг, 0,16 ммоль) и перемешивали при 70°С в течение еще 1 часа. Реакцию гасили насыщенным водным раствором хлорида аммония, и реакционную массу сразу сушили на роторном испарителе и очищали колоночной хроматографией с получением (Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4H)-ил)-N,N-диметилакриламида (10 мг, 50%, белое твердое вещество). 1H ЯМР (400МГц,CDCl3) δ 6,70-6,97 (м, 1Н), 6,93-6,91 (м, 2Н), 6,70 (с, 1Н), 6,27 (с, 2Н), 3,92 (с, 3H), 3,88 (д, J=6,8 Гц, 2Н), 2,92 (с, 3H), 2,78 (с, 3H), 2,37 (с, 6Н), 1,37-1,29 (м, 1Н), 0,69-0,65 (м, 2Н), 0,40-0,36 (м, 2Н). LC-MS: m/z 397,1 [М+Н]+.
Пример 110
(Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1 (4H)-ил)-N,N-диэтилакриламид
Конкретная схема реакции показана ниже:
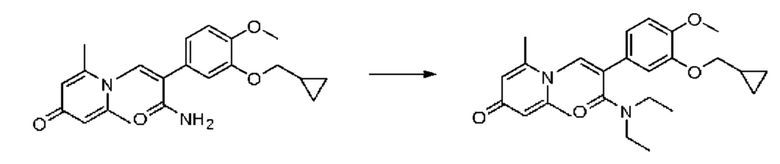
Соединение (Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4H)-ил) акриламид (20 мг, 0,06 ммоль) растворяли в N,N-диметилформамиде (0,5 мл), затем добавляли 60% гидрид натрия (12 мг, 0,3 ммоль) и перемешивали при 70°С в течение 30 минут. К реакционной массе добавляли йодэтан (19 мг, 0,12 ммоль) и перемешивали при 70°С в течение еще 1 часа. Реакцию гасили насыщенным водным раствором хлорида аммония, и реакционную массу сразу сушили на роторном испарителе и очищали колоночной хроматографией с получением (Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин)-1(4H-ил)-N,N-диэтилакриламида (6 мг, 30%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,04-7,01 (м, 1Н), 6,95-6,91 (м, 2Н), 6,72 (с, 1Н), 6,41 (с, 2Н), 3,92 (с, 3H), 3,88 (д, J=7,2 Гц, 2Н), 3,34-3,31 (м, 2Н), 3,18 (к, J=6,8 Гц, 2Н), 1,01 (т, J=7,2 Гц, 3H), 0,90-0,82 (м, 1Н), 0,81 (т, J=7,2 Гц, 3H), 0,69-0,65 (м, 2Н), 0,40-0,36 (м, 2Н); LC-MS: m/z 425,1 [М+Н]+.
Пример 111
(Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4H)-ио)-акриловая кислота
Конкретная схема реакции показана ниже:
Стадия А:
Соединение (Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4H)-ил)-N,N-диэтилакриламид (60,0 мг, 0,16 ммоль) растворяли в N,N-диметилформамиде (5 мл). 4-Диметиламинопиридин (4,0 мг, 0,03 ммоль) и триэтиламин (36 мг, 0,36 ммоль) добавляли при 35°С, затем к реакционной массе медленно добавляли ди-трет-бутилдикарбонат (71 мг, 0,33 ммоль) и перемешивали в течение 2 часов. После завершения реакции реакционную массу сушили на роторном испарителе с получением неочищенного продукта (2)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1 (4H)-ил)-N,N-ди-трет-бутилоксикарбонилакриламида. LC-MS: m/z 569,2 [M+H]+.
Стадия В:
Соединение (Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4H)-ил)-N,N-ди-трет-бутилоксикарбонилакриламид (90 мг 0,16 ммоль) растворяли в метаноле (2 мл), и к реакционной массе добавляли раствор гидроксида натрия (2 Н, 1 мл) при 35°С и перемешивали в течение 2 часов. После завершения реакции добавляли воду (10 мл) и экстрагировали дихлорметаном (3 × 10 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, сушили на роторном испарителе и очищали с получением (Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4H)-ил) акриловой кислоты (23,1 мг, выход 39%, белое твердое вещество). 1H ЯМР (400МГц, CDCl3) δ 7,18-7,16 (д, J=8,0 Гц, 1H), 7,11 (с, 2Н), 6,94 (д, J=8,0 Гц, 1H), 6,82 (с, 1H), 3,92 (с, 3H), 3,90(д, J=6,4 Гц, 2Н), 2,6 (с, 6Н), 1,33-1,27 (м, 1H), 0,68-0,63 (м, 2Н), 0,41-0,35 (м, 2Н), LC-MS: m/z 370,1 [М+Н]+.
Пример 112
(Z)-метил-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1 (4H)-ил)-акрилат
Конкретная схема реакции показана ниже:
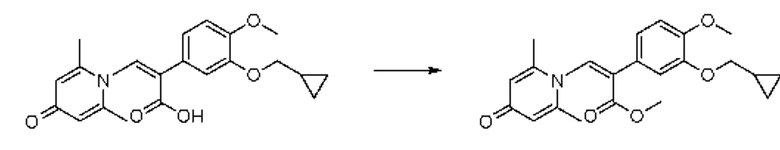
Соединение (Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4H)-ил) акриловую кислоту (10 мг, 0,03 ммоль) растворяли в дихлорметане (0,5 мл), и к реакционной массе добавляли одну каплю N,N-диметилформамида. Затем к реакционной массе медленно добавляли оксалилхлорид (8 мг, 0,03 ммоль) и перемешивали в течение 1 часа. Затем к реакционной массе добавляли метанол (0,5 мл). После завершения реакции реакцию гасили насыщенным водным раствором хлорида аммония, и реакционную массу сразу сушили на роторном испарителе и очищали с помощью колоночной хроматографии с получением (Z)-метил-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4H)-ил)-акрилата (3 мг, выход 26%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 8,02 (с, 1Н),7,95 (с, 2Н), 7,15 (с, 1Н), 7,09(д, J=8,4 Гц, 1Н), 6,94 (д, J=8,4 Гц, 1H), 3,94-3,93 (м, 5Н), 3,70 (с, 3H), 2,83 (с, 6Н), 1,70-1,59 (м, 1H), 0,68-0,63 (м, 2Н), 0,40-0,39 (м, 2Н); LC-MS: m/z 384,3 [М+Н]+.
Пример 113
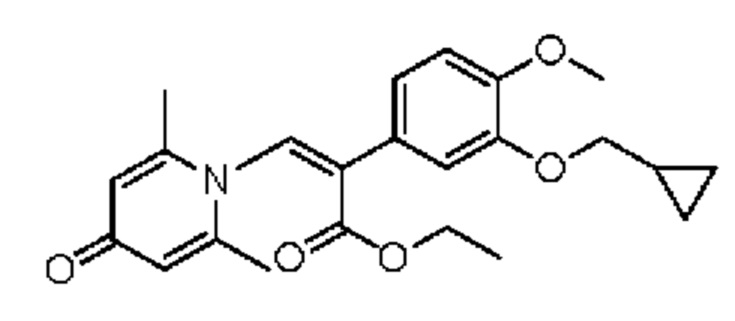
(Z)-этил-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонил пиридин-1 (4H)-ил)-акрилат
Конкретная схема реакции показана ниже:
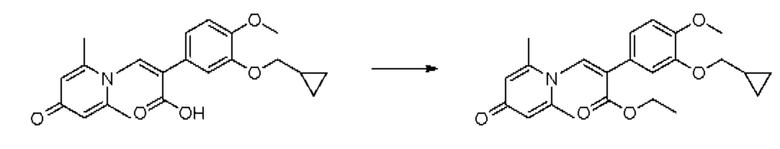
(Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4H)-ил) акриловую кислоту (10 мг, 0,03 ммоль) растворяли в дихлорметане (0,5 мл), и к реакционной массе добавляли одну каплю N,N-диметилформамида. К реакционной массе медленно добавляли оксалилхлорид (8 мг, 0,03 ммоль) и перемешивали в течение 1 часа. Затем к реакционной массе добавляли этанол (0,5 мл). После завершения реакции реакцию гасили насыщенным водным раствором хлорида аммония, и реакционный раствор сразу сушили на роторном испарителе и очищали колоночной хроматографией с получением (Z)-этил-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4H)-ил) акрилата (4 мг, выход 34%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 8,64 (с, 1Н), 7,65 (с, 2Н), 7,28 (с, 1Н), 7,16 (д, J=8,0 Гц, 1Н), 6,92 (д, J=8,4 Гц, 1Н), 4,18 (к, J=7,2 Гц, 2Н), 4,00 (д, J=6,8 Гц, 2Н), 3,92 (м, 3H), 2,92 (с, 6Н), 1,45-1,32 (м, 1H), 1,15 (т, J=14,4 Гц, 3H), 0,67-0,63 (м, 2Н), 0,44-0,40 (м, 2Н); LC-MS: m/z 398,3 [М+Н]+.
Пример 114
(Z)-1-(2-(3-циклопропилметокси-4-дифторметоксифенил)пент-1-ен-3-ин-1-ил)-2,6-диметилпиридин-4(1H)-он
Конкретная схема реакции показана ниже:
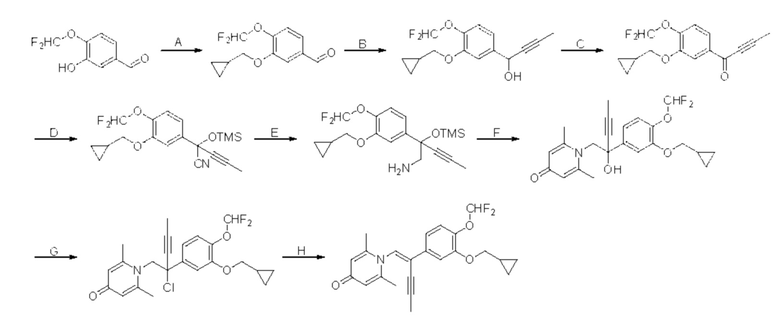
Стадия А:
4-(Дифторметокси)-3-гидроксибензальдегид (0,964 г, 5,13 ммоль) растворяли в ацетонитриле (10 мл) при комнатной температуре, затем последовательно добавляли карбонат калия (1,06 г, 7,69 ммоль) и бромметилциклопропан (0,9 г, 6,67 ммоль) и нагревали при перемешивании в атмосфере азота до 80°С в течение 3 часов. После завершения реакции добавляли воду (30 мл) и экстрагировали дихлорметаном (3 × 30 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, сушили на роторном испарителе и очищали с получением 3-циклопропилметокси-4-дифторметоксибензальдегида (1,16 г, выход 93%). LC-MS: m/z 343,2 [М+Н]+.
Стадия В:
Соединение 3-циклопропилметокси-4-дифторметоксибензальдегид (1,16 г, 4,79 ммоль) растворяли в безводном тетрагидрофуране (30 мл), затем медленно добавляли проп-1-ин-1-илмагнийбромид (0,5 Н, 19,2 мл, 9,6 ммоль) на ледяной бане и перемешивали при комнатной температуре в атмосфере азота в течение 16 часов. После завершения реакции реакционную массу промывали водой (30 мл) и экстрагировали этилацетатом (3 × 60 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали, сушили на роторном испарителе и очищали с получением 1-(3-циклопропилметокси-4-дифторметоксифенил) бут-2-ин-1-ола (1,01 г, 74%).
Стадия С:
Соединение 1-(3-циклопропилметокси-4-дифторметоксифенил)бут-2-ин-1-ол (1,01 г, 3,58 ммоль) растворяли в дихлорметане (50 мл), затем добавляли периодинан Десса-Мартина (3,04 г, 7,16 ммоль) и перемешивали в течение ночи при комнатной температуре. После завершения реакции реакционную массу фильтровали, сушили на роторном испарителе и очищали с получением 1-(3-циклопропилметокси-4-дифторметоксифенил) бут-2-ин-1-она (0,96 г, выход 96%).
Стадия D:
Соединение 1-(3-циклопропилметокси-4-дифторметоксифенил)бут-2-ин-1-он (0,96 г, 3,43 ммоль) растворяли в дихлорметане (10 мл), затем последовательно добавляли триэтиламин (0,693 г, 6,86 ммоль) и триметилсилилцианид (1,02 г, 10,29 ммоль) на ледяной бане и перемешивали в атмосфере азота при комнатной температуре в течение 16 часов. После завершения реакции реакционную массу сразу сушили на роторном испарителе с получением неочищенного продукта 2-(3-циклопропилметокси-4-дифторметоксифенил)-2-(триметилсилокси)пент-3-инил нитрила, который сразу использовали в следующей стадии.
Стадия Е:
Соединение 2-(3-циклопропилметокси-4-дифторметоксифенил)-2-(триметилсилокси)пент-3-инилнитрил (1,30 г, 3,43 ммоль) растворяли в безводном тетрагидрофуране (40 мл); затем порционно добавляли алюмогидрид лития (0,391 г, 10,29 ммоль) на ледяной бане и перемешивали в течение ночи при комнатной температуре. После завершения реакции последовательно добавляли воду (0,4 мл), 15% водный раствор гидроксида натрия (0,4 мл) и воду (1,2 мл), перемешивали в течение 30 минут, сушили над безводным сульфатом натрия, фильтровали и сушили на роторном испарителе с получением неочищенного продукта 1-амино-2-(3-циклопропилметокси-4-дифторметоксифенил)пент-3-ин-2-ола, который сразу использовали в следующей стадии.
Стадия F:
Соединение 1-амино-2-(3-циклопропилметокси-4-дифторметоксифенил) пент-3-ин-2-ол (1,07 г, 3,43 ммоль) растворяли в этаноле (40 мл), затем последовательно добавляли 2,6-диметил-4H-пиран-4-он (0,553 г, 4,46 ммоль), гидроксид натрия (206 мг, 5,15 ммоль) и воду (6 мл), нагревали до 60°С и перемешивали в течение 72 часов в атмосфере азота. После завершения реакции реакционную массу сушили на роторном испарителе и очищали колоночной хроматографией с получением 1-(2-(3-циклопропилметокси-4-дифторметоксифенил)-2-гидроксипент-3-ин-1-ил)-2,6-диметилпиридин-4(1H)-она (120 мг, выход 8%). LC-MS: m/z 418,2 [М+Н]+.
Стадия G:
Соединение 1-(2-(3-циклопропилметокси-4-дифторметоксифенил)-2-гидрокси пент-3-ин-1-ил)-2,6-диметилпиридин-4(1H)-он (120 мг, 0,29 ммоль) растворяли в трихлорметане (10 мл) и нагревали до 90°С. Затем добавляли тионилхлорид (0,5 мл) и перемешивали при 90°С в течение 15 минут. После завершения реакции реакционную массу сразу сушили на роторном испарителе с получением неочищенного продукта 1-(2-хлор-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)пент-3-ин-1-ил)-2,6-диметилпиридин-4(1H)-она. LC-MS: m/z 436,2 [М+Н]+.
Стадия Н:
Соединение 1-(2-хлор-2-(3-(циклопропилметокси)-4-(дифторметокси)фенил)пент-3-ин-1-ил)-2,6-диметилпиридин-4(1H)-он (125 мг, 0,29 ммоль) растворяли в этаноле (10 мл), затем последовательно добавляли гидроксид натрия (72 мг, 1,8 ммоль) и воду (2 мл), нагревали до 90°С и перемешивали в течение 16 часов в атмосфере азота. После завершения реакции реакционную массу сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением продукта (Z)-1-(2-(3-циклопропилметокси-4-дифторметоксифенил)пент-1-ен-3-ин-1-ил)-2,6-диметилпиридин-4(1H)-она (2,3 мг, выход 2%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,38-7,28 (м, 3H), 7,21 (д, J=8,0 Гц, 1Н), 7,01 (с, 2Н), 6,69 (т, J=75,2 Гц, 1H), 3,97 (д, J=6,4 Гц, 2Н), 2,48 (с, 6Н), 1,95 (с, 3H), 1,35-1,25 (м, 1Н), 0,70-0,65 (м, 2Н), 0,42-0,36 (м, 2Н); LC-MS: m/z 400,1 [М+Н]+.
Пример 115
(Z)-2-(3,4-диметоксифенил)-3-(2,6-диметил-4-карбонилпиридин)-1(4H)-ил)акрилонитрил
Конкретная схема реакции показана ниже:
Соединение (Z)-3-(2,6-диметил-4-карбонилпиридин)-1(4H)-ил)-2-(3-гидрокси-4-метоксифенил)акрилонитрил (100 мг, 0,34 ммоль) растворяли в ацетонитриле (10 мл), затем добавляли йодметан (96 мг, 0,68 ммоль) и карбонат калия (187 мг, 1,35 ммоль) и перемешивали в течение 5 часов при 85°С в атмосфере азота. После завершения реакции реакционную массу фильтровали, фильтрат сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (Z)-2-(3,4-диметоксифенил)-3-(2,6-диметил-4-карбонилпиридин)-1(4H)-ил) акрилонитрила (21 мг, выход 33%, белое твердое вещество). 1H ЯМР (400 МГц, CDCl3) δ 7,65 (с, 1H), 7,29 (дд, J=8,4, 2,0 Гц, 1Н), 7,18 (д, J=2,0 Гц, 1Н), 6,97 (д, J=8,4 Гц, 1H), 6,51 (с, 2Н), 3,98 (с, 3H), 3,96 (с, 3H), 2,37 (с, 6Н); LC-MS m/z 311,2 [М+Н]+.
Пример 116
(E)-3-(3-циклопропилметокси-4-метоксифенил)-2(2,6-диметил-4-карбонилпиридин-1 (4H)-ил)акрилонитрил
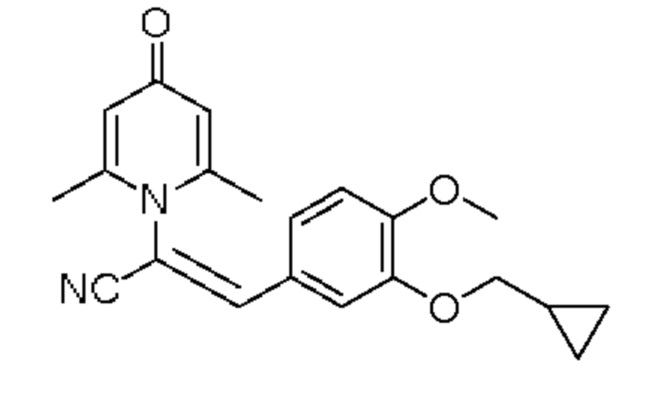
(Z)-3-(3-циклопропилметокси-4-метоксифенил)-2(2,6-диметил-4-карбонилпиридин-1(4H)-ил)акрилонитрил
Конкретная схема реакции показана ниже:
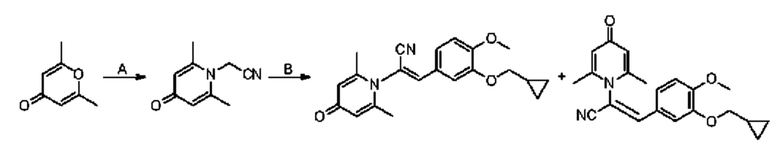
Стадия А
2,6-диметил-4H-пиран-4-он (620 мг, 5 ммоль) и гидрохлорид аминоацетонитрила (463 мг, 6 ммоль) растворяли в пиридине (10 мл) и перемешивали в течение 46 часов при 80°С в атмосфере азота. После завершения реакции реакционную массу сушили на роторном испарителе и очищали колоночной хроматографией с получением 2-(2,6-диметил-4-карбонилпиридин-1(4H)-ил) ацетонитрила (583 мг, выход 72%, белое твердое вещество). LC-MS: m/z 163,1 [М+Н]+.
Стадия В
2-(2,6-диметил-4-карбонилпиридин-1(4H)-ил) ацетонитрил (162 мг, 1 ммоль) и 3-циклопропилметокси-4-метоксибензальдегид (206 мг, 1 ммоль) растворяли в пиридине (10 мл) и перемешивали в течение ночи при 100°С в атмосфере азота. После завершения реакции реакционную массу сушили на роторном испарителе для удаления пиридина и очищали обращенно-фазовой ВЭЖХ с получением (Е)-3-(3-циклопропилметокси-4-метоксифенил)-2(2,6-диметил-4-карбонилпиридин)-1(4H)-ил) акрилонитрила (126 мг, выход 36%, белое твердое вещество) и (Z)-3-(3-циклопропилметокси-4-метоксифенил)-2 (2,6-диметил-4-карбонилпиридин-1(4Н)-ил) акрилонитрила (20 мг, выход 6%, белое твердое вещество).
(E)-3-(3-циклопропилметокси-4-метоксифенил)-2(2,6-диметил-4-карбонилпиридин-1(4H)-ил) акрилонитрил. 1H ЯМР (400 МГц, ДМСО-d6) δ 8,85 (с, 1H), 7,24 (д, J=8,4 Гц, 1H), 7,07 (дд, J=8,4, 2,0 Гц, 1H), 6,65 (д, J=2,0 Гц, 1H), 6,38 (с, 2Н), 3,92 (с, 3H), 3,88 (д, J=6,8 Гц, 2Н), 2,22 (с, 6Н), 1,26-1,17 (м, 1Н), 0,65-0,58 (м, 2Н), 0,32-0,26 (м, 2Н); LC-MS: m/z 351,2 [М+Н]+.
(Z)-3-(3-циклопропилметокси-4метоксифенил)-2(2,6-диметил-4-карбонилпиридин-1(4H)-ил) акрилонитрил. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,58 (с, 1H), 7,23 (д, J=8,4 Гц, 1H), 6,97 (дд, J=8,4, 1,6 Гц, 1H), 6,69 (д, J=1,6 Гц, 1H), 6,09 (с, 2Н), 3,80 (с, 3H), 3,53 (д, J=6,8 Гц, 2Н), 3,34 (с, 6Н), 2,02 (с, 6Н), 1,16-1,11 (м, 1H), 0,56-0,52 (м, 2Н), 0,27-0,23 (м, 2Н); LC-MS: m/z 351,2 [М+Н]+.
Пример 117
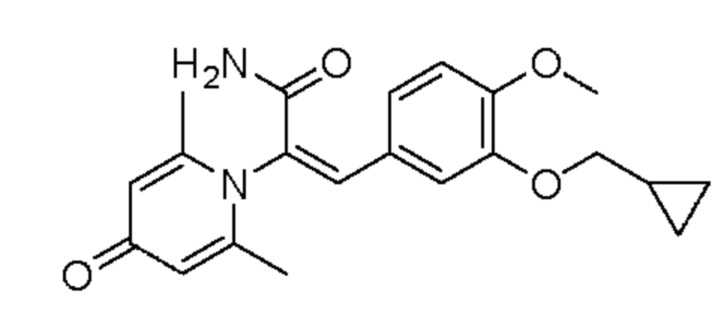
(E)-3-(3-пропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4H)-ил) акриламид
Конкретная схема реакции показана ниже:
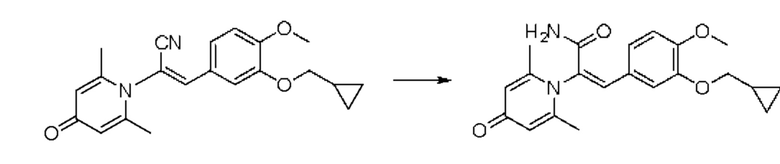
(E)-3-(3-циклопропилметокси-4-метоксифенил)-2(2,6-диметил-4-карбонилпиридин-1(4H)-ил) акрилонитрил (350 мг, 1 ммоль) растворяли в ацетонитриле (10 мл), и затем добавляли гидроксид калия (10 мас. %, 10 мл) при комнатной температуре и перемешивали при 80°С в течение 2 часов. После завершения реакции реакционную массу сразу сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4H)-ил) акриламида (247 мг, 67%, белое твердое вещество). 1H ЯМР (400 МГц, ДМСО-d6) δ 7,82 (с, 1H), 7,75 (с, 1H), 7,54 (с, 1H), 7,04 (д, J=8,4 Гц, 1H), 6,87 (дд, J=8,4, 2,0 Гц, 1H), 6,38 (д, J=2,0 Гц, 1H), 6,14 (с, 2Н), 3,80 (с, 3H), 3,51 (д, J=6,8 Гц, 2Н), 1,94 (с, 6Н), 1,16-1,07 (м, 1H), 0,56-0,51 (м, 2Н), 0,27-0,24 (м, 2Н); LC-MS: m/z 369,4 [М+Н]+.
Пример 118
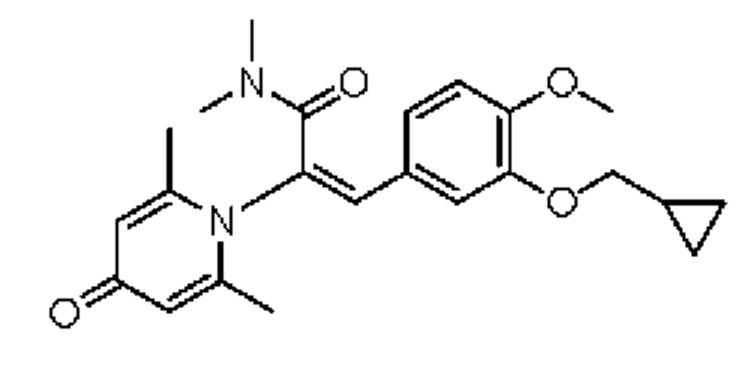
(E)-3-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин)-1 (4H)-ил)-N,N-диметилакриламид
Конкретная схема реакции показана ниже:
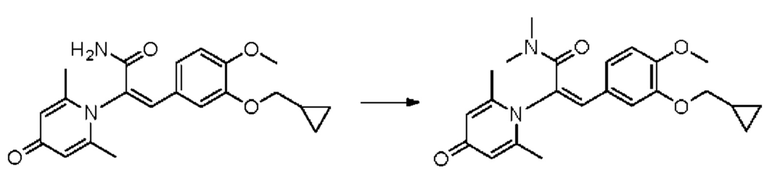
Соединение (E)-3-(3-(циклопропилметокси)-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин)-1(4H) акриламид (37 мг, 0,1 ммоль) растворяли в N,N-диметилформамиде (2 мл), затем добавляли 60% гидрид натрия (24 мг, 0,6 ммоль) при 0°С и перемешивали в течение 30 минут. Затем добавляли йодметан (43 мг, 0,3 ммоль) и перемешивали при комнатной температуре в течение 4 часов. После завершения реакции добавляли насыщенный раствор соли (10 мл) и экстрагировали дихлорметаном (3 × 10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия. Затем фильтровали, сушили на роторном испарителе и очищали обращенно-фазовой ВЭЖХ с получением (E)-3-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин)-1(4H)-ил)-N,N-диметилакриламида (22 мг, выход 55%, белое твердое вещество). 1H ЯМР (400 МГц, ДМСО-d6) δ 7,28 (с, 1H), 7,03 (д, J=8,4 Гц, 1H), 6,87 (дд, J=8,4, 1,6 Гц, 1H), 6,49 (д, J=1,6 Гц, 1H), 6,09 (с, 2Н), 3,80 (с, 3H), 3,53 (д, J=6,8 Гц, 2Н), 3,34 (с, 6Н), 2,02 (с, 6Н), 1,16-1,11 (м, 1H), 0,56-0,52 (м, 2Н), 0,27-0,23 (м, 2Н); LC-MS: m/z 397,2 [М+Н]+.
Пример 119
Определение ингибирования соединениями активности PDE4B1 с использованием анализа цАМФ HTRF®
Ингибирующее действие соединений в отношении активности фермента человека PDE4B1 определяли путем количественного определения 5'-аденозинмонофосфата (5'-АМФ), образованного из 3', 5'-циклического аденозинмонофосфата (цАМФ).
Тестируемое соединение или воду (контрольный образец) и рекомбинантный фермент человека PDE4B1 (4,8 Ед.) смешивали в буферном растворе (рН 7,4), состоящем из 1х сбалансированного солевого раствора Хэнка (HBSS), 5 мМ HEPES, 3 мМ MgCl2 и 0,1% BSA и инкубировали 10 мин. Добавляли субстрат фермента цАМФ (конечная концентрация 40 нМ), смесь инкубировали при комнатной температуре в течение 60 мин. Затем добавляли флуоресцентный акцептор (Dye2, меченный цАМФ), флуоресцентный донор (антитело против цАМФ, меченное криптатом европия) и неспецифический ингибитор фосфодиэстеразы IBMX (3-изобутил-1-метилксантин; конечная концентрация 1 мМ). Через 60 мин перенос флуоресценции, связанный с количеством оставшегося цАМФ, измеряли на микропланшетном анализаторе (Rubystar, BMG) при λех=337 нм, λem=620 нм и λem=665 нм. Активность фермента рассчитывают на основании отношения сигналов, измеренных при 665 нм и 620 нм. Результаты выражены в процентах ингибирования ферментативной активности контрольного образца (без ингибитора PDE4). Фермент не использовали для измерения основного контрольного образца. Значение IC50 (IC50 = концентрация, которая вызвала половину максимального ингибирования специфической активности контрольного образца) получено на основании измерений зависимости отклика от дозы с восемью различными концентрациями (n=2; повторяется 2 раза).
Полученные экспериментальные результаты приведены в таблице 1.
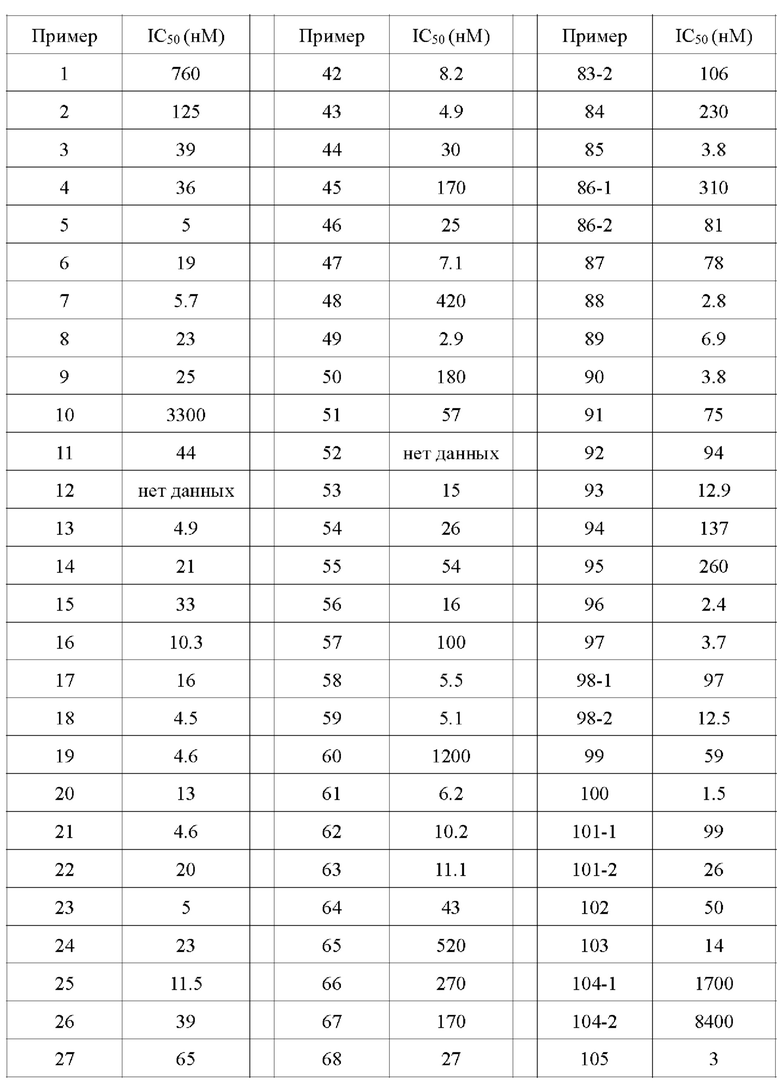
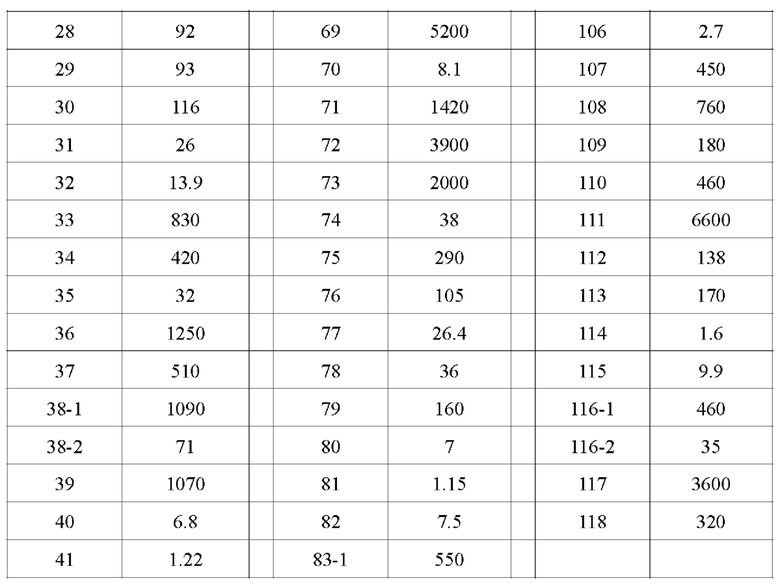
Пример 120
Ингибирование соединений высвобождения провоспалительных цитокинов мононуклеарными клетками периферической крови (РВМС)
Определяли влияние соединений на характеристики высвобождения цитокинов TNFα в замороженных мононуклеарных клетках периферической крови (РВМС) человека.
Выделяли свежие человеческие РВМС и суспендировали в свежей среде. Среда РВМС (СМ) представляет собой RPMI1640, содержащую 10% FBS, 1% PBS и 2 мМ L-глутамина. Клетки высевали при плотности 2*10^5 клеток/лунку (100 мкл). Тестируемое соединение растворяли в ДМСО с образованием 10 мМ образца (ДМСО, 100%). Соединение разбавляли средой РВМС до необходимых концентраций. Образец (50 мкл) инкубировали в течение 1 часа с клетками при 37°С, 5% СО2, после чего добавляли стимулятор (LPS, 1 мкг/мл). Стимулятор + наполнитель (LPS+0,05% ДМСО) использовали в качестве контроля в этом эксперименте. Носитель без стимулятора использовали в качестве негативного контроля. Кризаборол использовали в качестве положительного контроля. После 24 часов инкубации надосадочную жидкость экстрагировали и хранили при -80°С. Надосадочную жидкость растапливали, и уровень TNFα в надосадочной жидкости измеряли с помощью анализа Luminex4-plex.
Результат:
Используя этот экспериментальный метод, определено, что ЕС50 для Примера 77 составляет = 11 нМ, ЕС50 для Примера 80 составляет <5 нМ; и ЕС50 для Примера 88 составляет <5 нМ.
Пример 121
Тест ингибирования аллергического дерматита на ухе мыши, вызванного местным введением форболового эфира
Тестируемые соединения используются для лечения различных кожных заболеваний, таких как зуд, покраснение, сухость, образование корост, шелушение, воспаление и дискомфорт.
Тестируемое соединение наносили местно на правое ухо тестируемого животного за 30 минут до и через 15 минут после введения (12-) форболмирист (-13-) ацетата (РМА). Объем дозы составлял 20 мкл/ухо для растворителя-наполнителя или 20 мг/ухо для крема.
Использовали мышей-самцов CD-I массой 24±2 г. Всех животных содержали в среде с контролируемой температурой (22°С-23°С) и влажностью (70-80%) с 12-часовым световым и 12-часовым темным периодом. Животных разводили в помещении для животных в течение не менее 1 недели перед использованием в экспериментальных испытаниях. Мышам был предоставлен свободный доступ к пище и питьевой воде. В целом, все аспекты этой работы выполнялись в соответствии с руководящими принципами по уходу и использованию лабораторных животных (National Academy Press, Washington, D.C., 1996), включая разведение животных, эксперименты и обращение с ними.
В каждой группе было по 5 мышей. РМА (4 мкг в 20 мкл ацетона) наносили локально на переднюю и заднюю поверхности правого уха каждого животного. Наполнитель (этанол:ацетон / 1:1, 20 мкл/ухо) или тестируемое соединение (указанная доза в 20 мкл смеси 1:1 ацетон:этанол для каждого уха) наносили за 30 минут до и через 15 минут после введения РМА, и кризаборол вводили в качестве положительного контроля. Затем, через 6 часов после введения РМА, отек уха измеряли в качестве индикатора воспаления с помощью окрашивающего микрометра. Процент ингибирования рассчитывается по следующей формуле: ([IC-IT]/IC)×100%, где IC и IT означают увеличение толщины уха (мм) у мышей в контрольной и экспериментальной группах соответственно. Считается, что 30% или более ингибирования обладают значительной противовоспалительной активностью.
Пример 123
Технология изготовления местных препаратов.
Соединения по настоящему изобретению, такие как соединение 88, можно вводить в виде геля, лосьона, мази и раствора, путь введения включает, но не ограничивается ими, местное введение, инсталляцию, аэрозоль, трансдермальный пластырь, инсерцию, или пероральное введение.
Далее приведен способ приготовления 1% мази (весовой процент):
1% соединения, 15% PEG400, 0,02% бутилированного гидрокситолуола, 2% Span 80, 10% белого воска и 71,98% белого петролатума механически перемешивали до образования мази.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТЕТРАГИДРОИМИДАЗО[1,5-d][1,4]ОКСАЗЕПИНОВОЕ ПРОИЗВОДНОЕ | 2014 |
|
RU2659219C2 |
| ХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ SMO | 2015 |
|
RU2695815C2 |
| Ингибиторы ErbB/BTK | 2019 |
|
RU2764069C1 |
| ИНГИБИТОРЫ ФАКТОРА XIa | 2016 |
|
RU2728783C2 |
| ИМИДАЗОПИРАЗИНОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE1 | 2016 |
|
RU2712219C2 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2685417C1 |
| МОДУЛЯТОРЫ АКТИВНОСТИ НЕС1 И СПОСОБЫ ДЛЯ НИХ | 2011 |
|
RU2576036C2 |
| АМИДНОЕ ПРОИЗВОДНОЕ ПИРАЗОЛА | 2015 |
|
RU2658827C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРА МИНЕРАЛОКОРТИКОИДОВ | 2012 |
|
RU2598842C2 |
| ПРОИЗВОДНОЕ ЦИКЛОГЕКСАНА И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2009 |
|
RU2478621C2 |
Группа изобретений относится к области фармацевтической химии, в частности к противовоспалительному соединению, имеющему структуру (I), в которой RA представляет собой водород, алкил, содержащий от 1 до 6 атомов углерода, и в вышеуказанных группах один или несколько атомов водорода, присоединенных к углероду в этих группах, необязательно замещены алкилом содержащим от 1 до 6 атомов углерода, циклоалкилом, представляющим собой трехчленный, четырехчленный, пятичленный или шестичленный цикл, или галогеном; RB представляет собой водород, алкил, содержащий от 1 до 6 атомов углерода, циклоалкил, представляющий собой трехчленный, четырехчленный, пятичленный или шестичленный цикл, гетероциклоалкил, представляющий собой оксо/тио трехчленный, четырехчленный, пятичленный или шестичленный цикл, алкенил, содержащий от 2 до 4 атомов углерода, или алкинил, содержащий от 2 до 4 атомов углерода, и в вышеуказанных группах один или несколько атомов водорода, присоединенных к углероду в этих группах, необязательно замещены алкилом, содержащим от 1 до 6 атомов углерода, циклоалкилом, представляющим собой трехчленный, четырехчленный, пятичленный или шестичленный цикл, или галогеном, и один или несколько атомов углерода в этих группах необязательно замещены атомом серы, сульфоксидом, сульфоном или сульфонилом; RC представляет собой водород; RD1 представляет собой водород, кислород, гидрокси, циано, алкил, содержащий от 1 до 6 атомов углерода, фенил, сложноэфирную группу, карбоксил, или алкинил, содержащий от 2 до 4 атомов углерода, и в вышеуказанных группах один или несколько атомов водорода, присоединенных к углероду/кислороду в этих группах, необязательно замещены алкилом, содержащим от 1 до 6 атомов углерода, циклоалкилом, представляющим собой трехчленный, четырехчленный, пятичленный или шестичленный цикл, алкинилом, содержащим от 2 до 4 атомов углерода, алкенилом, содержащим от 2 до 4 атомов углерода, или фенилом, и один или несколько атомов углерода в этих группах необязательно замещены атомом серы; связь C1-RD1 представляет собой одинарную или двойную связь; RD2 представляет собой водород или циано; G1 представляет собой одинарную связь, двойную связь или циклопропан, содержащий C1 и C2; и RE представляет собой алкил, содержащий от 1 до 6 атомов углерода, или галоген. Кроме того, описаны способы получения указанных выше соединений, а также их применение для лечения кожных заболеваний, связанных с PDE4, и лекарственное средство для лечения воспалительных кожных заболеваний на основе соединений, имеющих формулу (I). Технический результат: получены и описаны соединения, которые оказывают сильное ингибирующее действие на PDE4, легко проникающие через кожу и которые могут быть применимы в лечении воспалительных кожных заболеваний. 18 н. и 2 з.п. ф-лы, 1 табл., 123 пр.
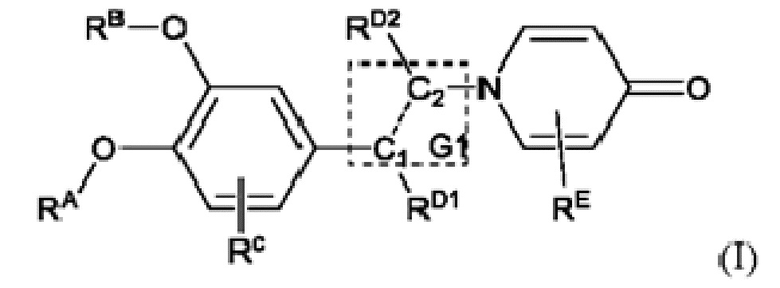
1. Противовоспалительное соединение, которое представляет собой соединение, имеющее структуру, показанную ниже:
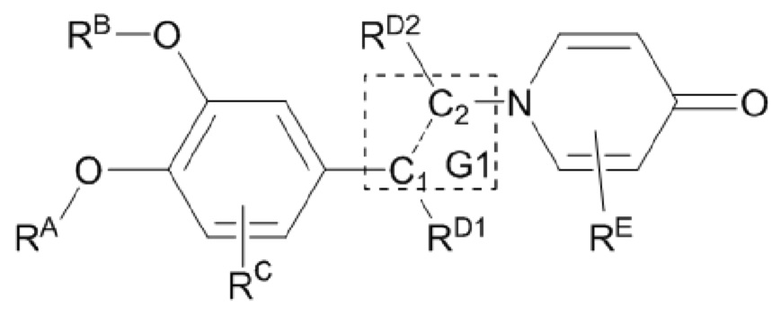
в которой RA представляет собой водород, алкил, содержащий от 1 до 6 атомов углерода, и в вышеуказанных группах один или несколько атомов водорода, присоединенных к углероду в этих группах, необязательно замещены алкилом, содержащим от 1 до 6 атомов углерода, циклоалкилом, представляющим собой трехчленный, четырехчленный, пятичленный или шестичленный цикл, или галогеном;
RB представляет собой водород, алкил, содержащий от 1 до 6 атомов углерода, циклоалкил, представляющий собой трехчленный, четырехчленный, пятичленный или шестичленный цикл, гетероциклоалкил, представляющий собой оксо/тио трехчленный, четырехчленный, пятичленный или шестичленный цикл, алкенил, содержащий от 2 до 4 атомов углерода, или алкинил, содержащий от 2 до 4 атомов углерода, и в вышеуказанных группах один или несколько атомов водорода, присоединенных к углероду в этих группах, необязательно замещены алкилом, содержащим от 1 до 6 атомов углерода, циклоалкилом, представляющим собой трехчленный, четырехчленный, пятичленный или шестичленный цикл, или галогеном, и один или несколько атомов углерода в этих группах необязательно замещены атомом серы, сульфоксидом, сульфоном или сульфонилом;
RC представляет собой водород;
RD1 представляет собой водород, кислород, гидрокси, циано, алкил, содержащий от 1 до 6 атомов углерода, фенил, сложноэфирную группу, карбоксил или алкинил, содержащий от 2 до 4 атомов углерода, и в вышеуказанных группах один или несколько атомов водорода, присоединенных к углероду/кислороду в этих группах, необязательно замещены алкилом, содержащим от 1 до 6 атомов углерода, циклоалкилом, представляющим собой трехчленный, четырехчленный, пятичленный или шестичленный цикл, алкинилом, содержащим от 2 до 4 атомов углерода, алкенилом, содержащим от 2 до 4 атомов углерода, или фенилом, и один или несколько атомов углерода в этих группах необязательно замещены атомом серы;
связь C1-RD1 представляет собой одинарную или двойную связь;
RD2 представляет собой водород или циано;
G1 представляет собой одинарную связь, двойную связь или циклопропан, содержащий C1 и С2; и
RE представляет собой алкил, содержащий от 1 до 6 атомов углерода, или галоген.
2. Противовоспалительное соединение по п. 1, в котором
G1 представляет собой трехчленный цикл, имеющий специфическую структуру, показанную ниже:
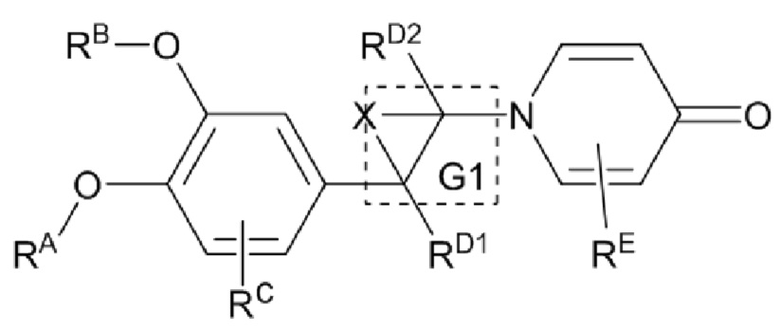
в которой X представляет собой углерод.
3. Способ получения противовоспалительного соединения по п. 1, который включает:
использование производного 3-гидроксибензальдегида А в качестве исходного материала и замещение водорода в производном 3-гидроксибензальдегида А на RB для получения промежуточного продукта В;
взаимодействие промежуточного продукта В в присутствии триметилсилилцианида для получения промежуточного продукта С;
восстановление промежуточного продукта С для получения промежуточного продукта D, имеющего аминогруппу; и
взаимодействие аминогруппы в промежуточном продукте D с шестичленным кислородсодержащим циклическим соединением с получением целевого продукта типа А,
где производное 3-гидроксибензальдегида А представляет собой соединение, имеющее структуру, показанную ниже:
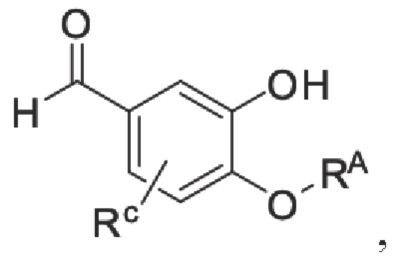
промежуточный продукт В представляет собой соединение, имеющее структуру, показанную ниже:
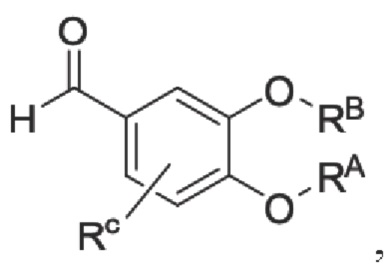
промежуточный продукт С представляет собой соединение, имеющее структуру, показанную ниже:
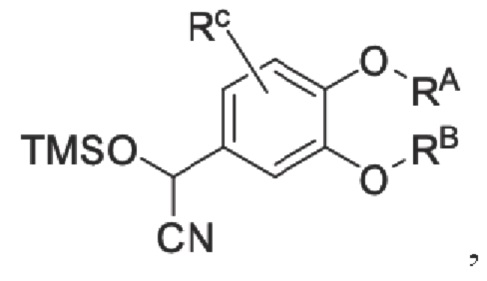
промежуточный продукт D представляет собой соединение, имеющее структуру, показанную ниже:
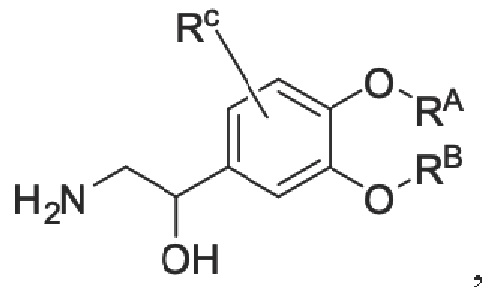 и
и
целевой продукт типа А представляет собой соединение, имеющее структуру, показанную ниже:
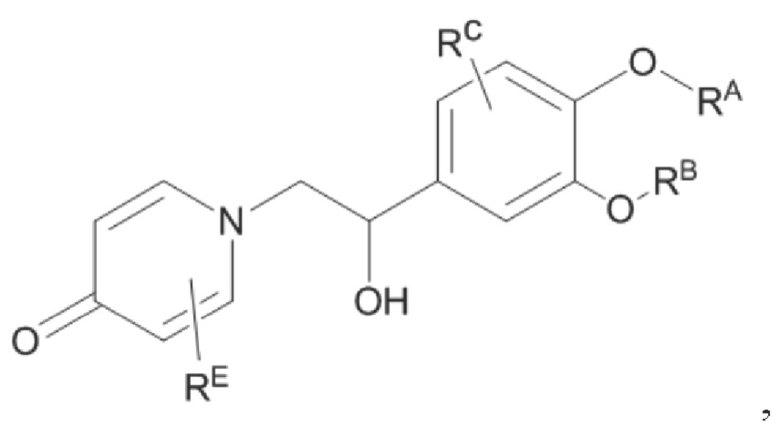
где значения RA, RB, Rc и RE соответствуют значениям, указанным в п. 1.
4. Способ получения противовоспалительного соединения по п. 1, в котором:
гидроксильную группу на соединительном мостике целевого продукта типа А по п. 3 подвергают реакции присоединения/замещения для получения целевого продукта типа А-1,
где целевой продукт типа А-1 представляет собой соединение, имеющее структуру, показанную ниже:
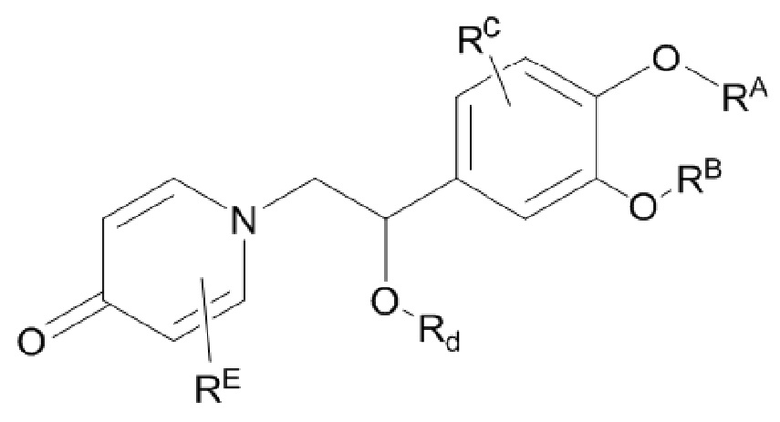
в которой Rd представляет собой алкил, содержащий от 1 до 6 атомов углерода, и в вышеуказанной группе один или несколько атомов водорода, в присоединенных к углероду в группе, необязательно замещены алкинилом, содержащим от 2 до 4 атомов углерода,
где значения RA, RB, Rc и RE соответствуют значениям, указанным в п. 1.
5. Способ получения противовоспалительного соединения по п. 1, включающий:
окисление гидроксильной группы на соединительном мостике целевого продукта типа А по п. 3 для получения целевого продукта типа В,
где целевой продукт типа В представляет собой соединение, имеющее структуру, показанную ниже:
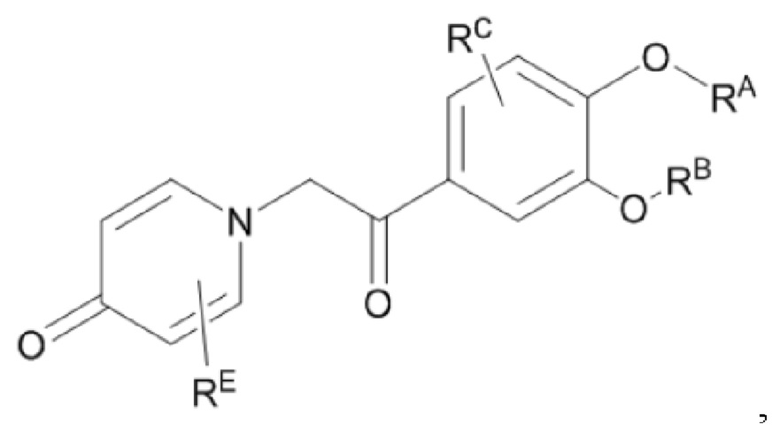
где значения RA, RB, Rc и RE соответствуют значениям, указанным в п. 1.
6. Способ получения противовоспалительного соединения по п. 1, который включает:
снятие защиты группы RB, которая представляет собой гидроксильную защитную группу целевого продукта типа В по п. 5, с получением целевого продукта типа В-1,
где целевой продукт типа В-1 представляет собой соединение, имеющее структуру, показанную ниже:
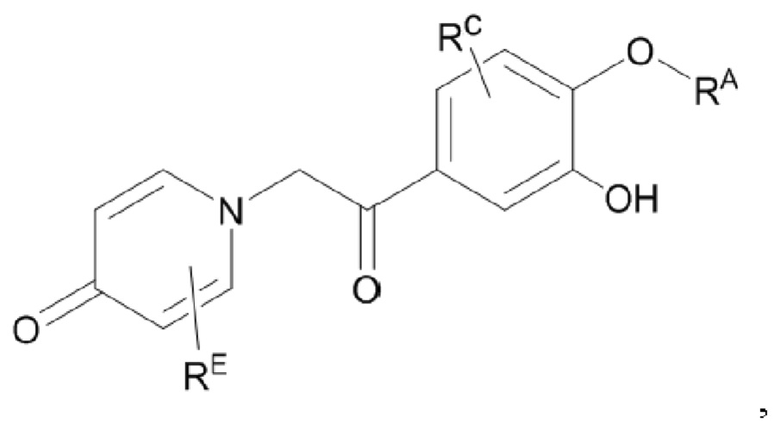
где значения RA, Rc и RE соответствуют значениям, указанным в п. 1.
7. Способ получения противовоспалительного соединения по п. 1, в котором:
гидроксильную группу бензольного кольца целевого продукта типа В-1 по п. 6 подвергают реакции присоединения/замещения для получения целевого продукта типа В-2,
где целевой продукт типа В-2 представляет собой соединение, имеющее структуру показанную ниже:
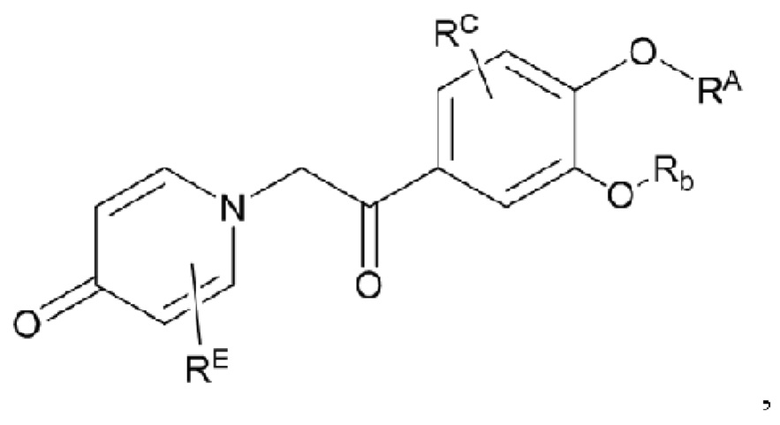
в которой Rb представляет собой алкил, содержащий от 1 до 6 атомов углерода, или циклоалкил, представляющий собой трехчленный, четырехчленный, пятичленный или шестичленный цикл, и в вышеуказанных группах один или несколько атомов водорода, присоединенных к углероду в группах, необязательно замещены алкилом, содержащим от 1 до 6 атомов углерода, циклоалкилом, представляющим собой трехчленный, четырехчленный, пятичленный или шестичленный цикл, или галогеном,
где значения RA, Rc и RE соответствуют значениям, указанным в п. 1.
8. Способ получения противовоспалительного соединения по п. 1, включающий:
использование производного 3-гидроксиацетофенона I в качестве исходного материала и замещение водорода в производном 3-гидроксиацетофенона I на RB для получения промежуточного продукта II;
взаимодействие промежуточного продукта II в присутствии галогенирующего агента для получения промежуточного продукта III; и
взаимодействие галогена в промежуточном продукте III с шестичленным кислородсодержащим циклическим соединением для получения целевого продукта типа В,
где производное 3-гидроксиацетофенона I представляет собой соединение, имеющее структуру, показанную ниже:
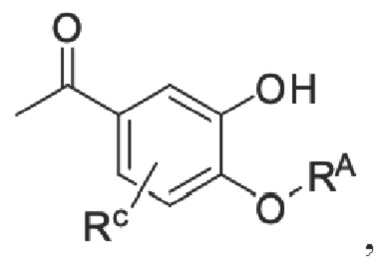
промежуточный продукт II представляет собой соединение, имеющее структуру, показанную ниже:
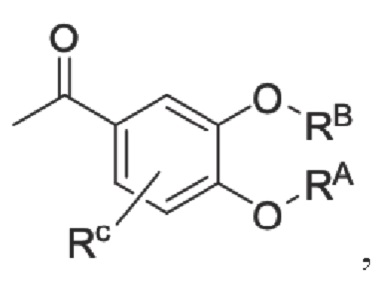 и
и
промежуточный продукт III представляет собой соединение, имеющее структуру, показанную ниже:
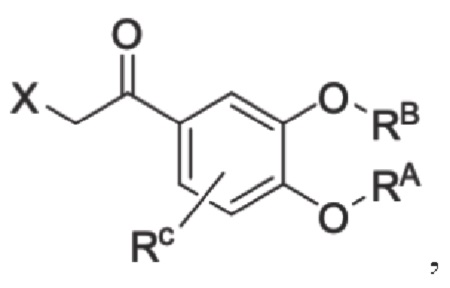 в которой X представляет собой галоген,
в которой X представляет собой галоген,
где значения RA, RB и Rc соответствуют значениям, указанным в п. 1.
9. Способ получения противовоспалительного соединения по п. 1, который включает:
удаление гидроксильной группы на соединительном мостике целевого продукта типа А по п. 3 для получения целевого продукта типа С,
или
восстановление, а затем удаление карбонильной группы на соединительном мостике целевого продукта типа В по п. 7 для получения целевого продукта типа С,
где целевой продукт типа С представляет собой соединение, имеющее структуру, показанную ниже:
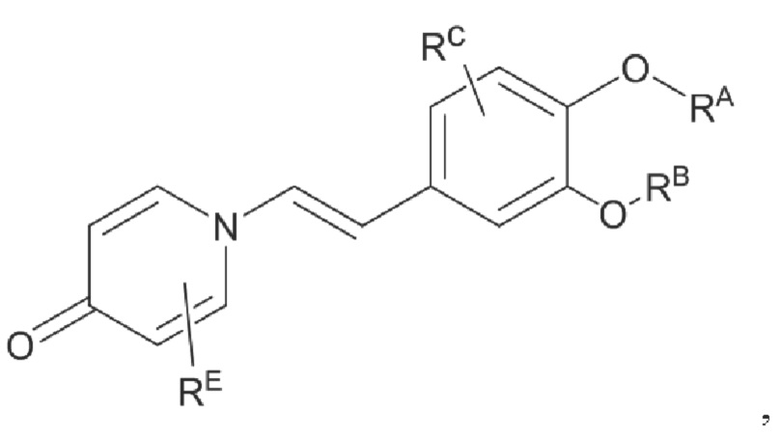
где значения RA, RB, Rc и RE соответствуют значениям, указанным в п. 1.
10. Способ получения противовоспалительного соединения по п. 1, включающий:
восстановление, а затем удаление карбонильной группы на соединительном мостике целевого продукта типа В-2 по п. 7 для получения целевого продукта типа С-1,
где целевой продукт типа С-1 представляет собой соединение, имеющее структуру, показанную ниже:
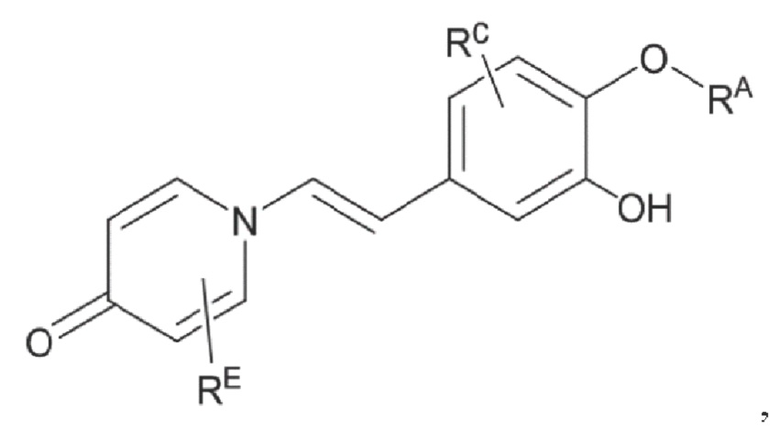
где значения RA, Rc и RE соответствуют значениям, указанным в п. 1.
11. Способ получения противовоспалительного соединения по п. 1, в котором:
гидроксильную группу фенильного кольца целевого продукта типа С-1 по п. 10 подвергают реакции присоединения/замещения для получения целевого продукта типа С-2,
где целевой продукт типа С-2 представляет собой соединение, имеющее структуру, показанную ниже:
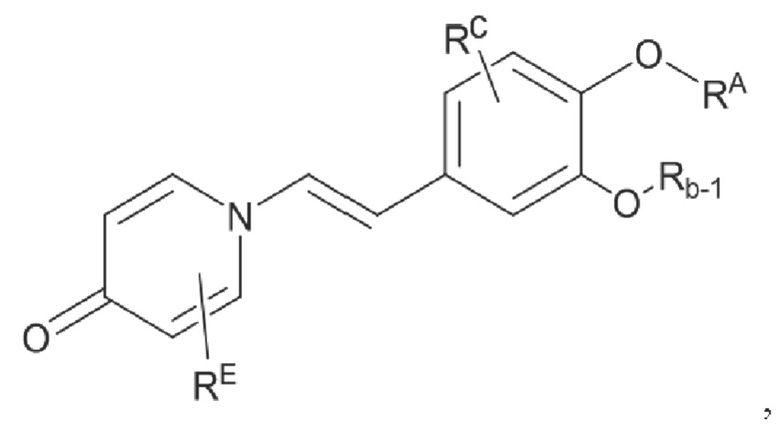
в которой Rb-1 представляет собой алкил, содержащий от 1 до 6 атомов углерода, или циклоалкил, представляющий собой трехчленный, четырехчленный, пятичленный или шестичленный цикл, и в вышеуказанных группах один или несколько атомов водорода, присоединенных к атому углерода в группах, необязательно замещены алкилом, содержащим от 1 до 6 атомов углерода, алкинилом, содержащим от 2 до 4 атомов углерода, алкенилом, содержащим от 2 до 4 атомов углерода, циклоалкилом, представляющим собой трехчленный, четырехчленный, пятичленный или шестичленный цикл, или галогеном,
где значения RA, Rc и RE соответствуют значениям, указанным в п. 1.
12. Способ получения противовоспалительного соединения по п. 1, включающий:
взаимодействие производного ацетофенона 1 в качестве исходного материала с триметилсилилцианидом для получения промежуточного продукта 2;
восстановление промежуточного продукта 2 для получения промежуточного продукта 3; и
взаимодействие аминогруппы в промежуточном продукте 3 с шестичленным кислородсодержащим циклическим соединением для получения целевого продукта типа А',
где производное ацетофенона 1 представляет собой соединение, имеющее структуру, показанную ниже:
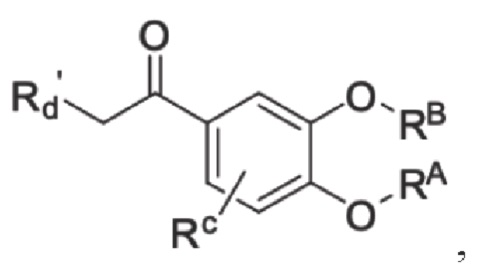
в которой Rd' представляет собой водород, алкил, содержащий от 1 до 6 атомов углерода, или алкинил, содержащий от 2 до 4 атомов углерода;
промежуточный продукт 2 представляет собой соединение, имеющее структуру, показанную ниже:
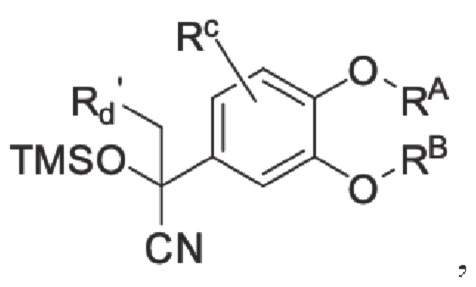
промежуточный продукт 3 представляет собой соединение, имеющее структуру, показанную ниже:
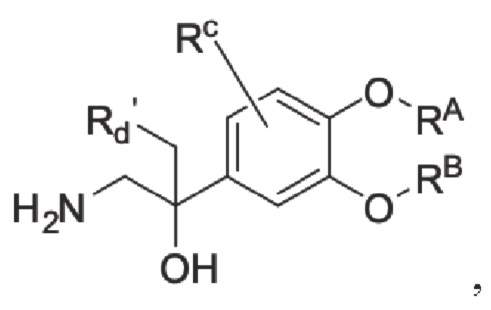 и
и
целевой продукт типа А' представляет собой соединение, имеющее структуру, показанную ниже:
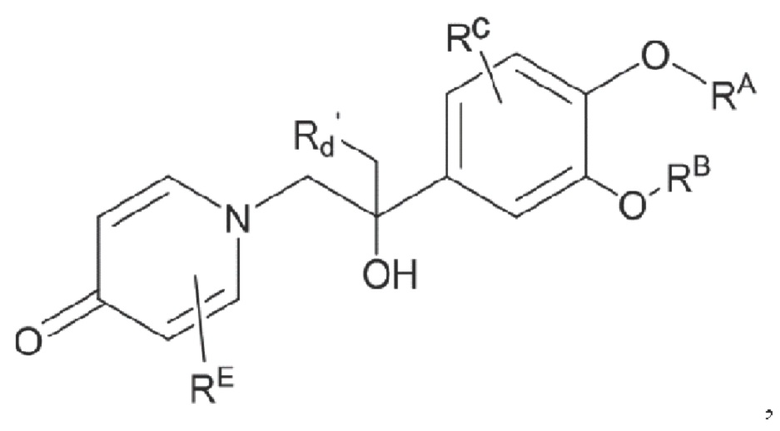
где значения RA, RB, Rc и RE соответствуют значениям, указанным в п. 1.
13. Способ получения противовоспалительного соединения по п. 1, который включает:
удаление гидроксильной группы на соединительном мостике целевого продукта типа А' по п. 12 для получения целевого продукта типа С’,
где целевой продукт типа С’ представляет собой соединение, имеющее структуру, показанную ниже:
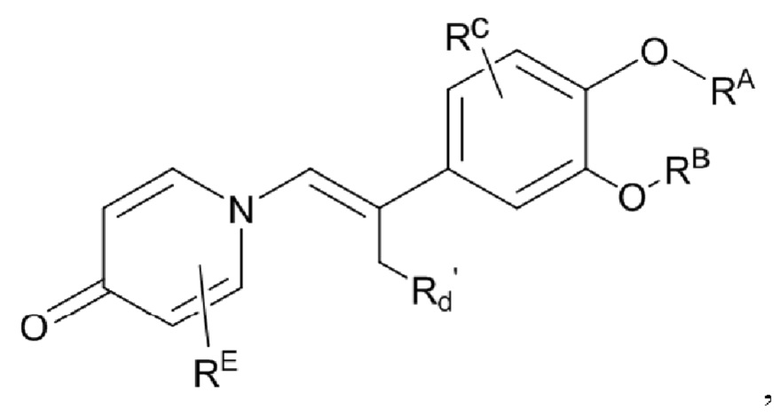
где значения RA, RB, Rc и RE соответствуют значениям, указанным в п. 1.
14. Способ получения противовоспалительного соединения по п. 1, который включает:
взаимодействие карбонильной группы на соединительном мостике целевого продукта типа В по п. 5 с триметилсилилцианидом для получения промежуточного продукта Y1; и
подвергание промежуточного продукта Y1 восстановлению и отщеплению для получения целевого продукта типа С'',
где промежуточный продукт Y1 представляет собой соединение, имеющее структуру, показанную ниже:
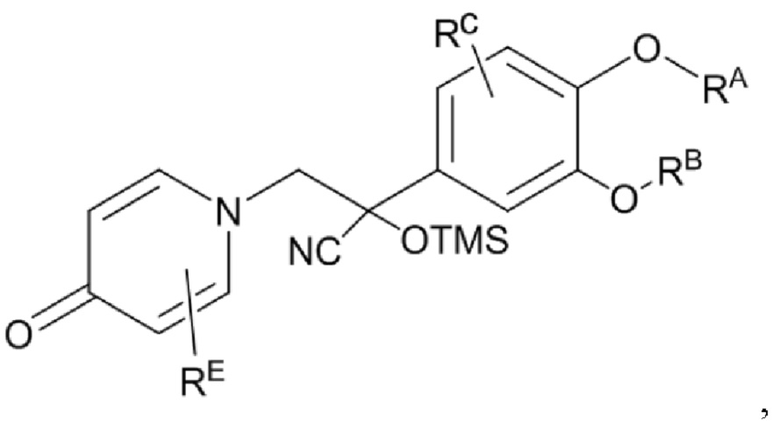
целевой продукт типа С'' представляет собой соединение, имеющее структуру, показанную ниже:
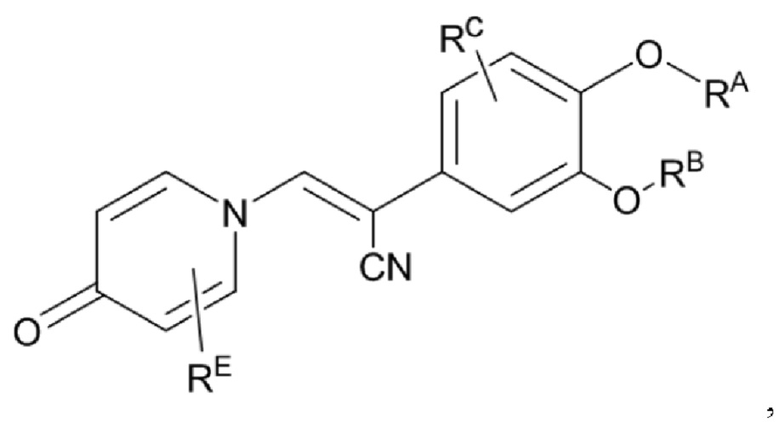
где значения RA, RB, Rc и RE соответствуют значениям, указанным в п. 1.
15. Способ получения противовоспалительного соединения по п. 1, который включает:
взаимодействие шестичленного соединения N-ацетонитрила Z1 с производным бензальдегида Z2 для получения целевого продукта типа С''.
где шестичленное соединение N-ацетонитрила Z1 представляет собой соединение, имеющее структуру, показанную ниже:
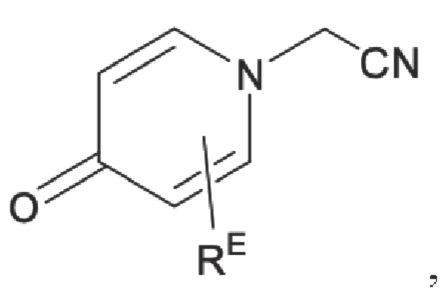 и
и
производное бензальдегида Z2 представляет собой соединение, имеющее структуру, показанную ниже:
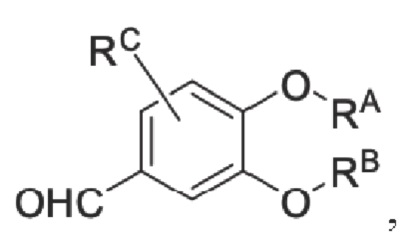
где значения RA, RB, Rc и RE соответствуют значениям, указанным в п. 1.
16. Способ получения противовоспалительного соединения по п. 1, который включает:
использование производного коричной кислоты 1 в качестве исходного материала и этерификацию производного коричной кислоты 1 для получения промежуточного продукта 2;
образование цикла из двойной связи в промежуточном продукте 2 для получения промежуточного продукта 3;
гидролиз концевой сложноэфирной группы промежуточного продукта 3 для получения промежуточного продукта 4, имеющего карбоксильную группу;
аминирование концевой карбоксильной группы промежуточного продукта 4 для получения промежуточного продукта 5; и
взаимодействие промежуточного продукта 5 с шестичленным кислородсодержащим циклическим соединением для получения целевого продукта типа D,
где производное коричной кислоты 1 представляет собой соединение, имеющее структуру, показанную ниже:
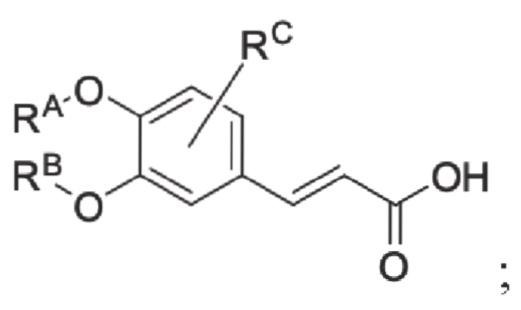
промежуточный продукт 2 представляет собой соединение, имеющее структуру, показанную ниже:
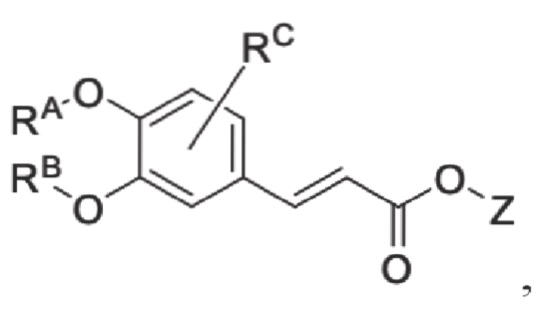 в которой Ζ представляет собой алкил;
в которой Ζ представляет собой алкил;
промежуточный продукт 3 представляет собой соединение, имеющее структуру, показанную ниже:
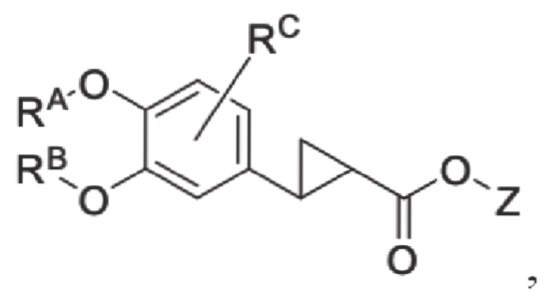
промежуточный продукт 4 представляет собой соединение, имеющее структуру, показанную ниже:
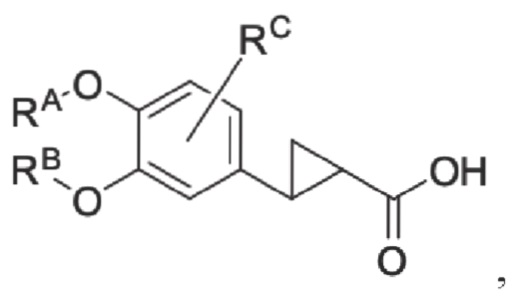 и
и
промежуточный продукт 5 представляет собой соединение, имеющее структуру, показанную ниже:
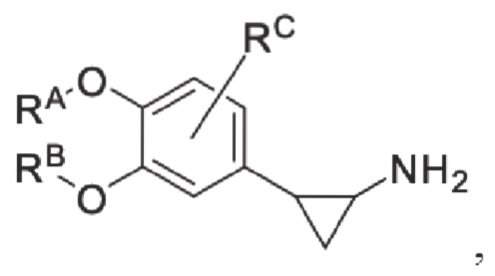
где значения RA, RB и Rc соответствуют значениям, указанным в п. 1.
17. Противовоспалительное соединение по п. 1, в котором соединение выбрано из группы, состоящей из:
1-(2-(3-циклопропилметокси-4-метоксифенил)-2-гидроксиэтил)-2,6-диметилпиридин-4(1Н)-он;
(1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)этил)ацетат;
(1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)этил)пропионат;
(1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)этил)циклопропилкарбоксилат;
(1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)этил)бензоат;
(1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)этил)кротонат;
(1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)этил)3-метилкротонат;
(1-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)этил)бут-2-инноат;
1-(2-(бут-2-ин-1-илокси)-2-(3-циклопропилметокси-4-метоксифенил)этил)-2,6-диметилпиридин-4-(1Н)-он;
1-(2-(3-гидрокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3,4-диметоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-этокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-пропокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-изопропокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-н-бутокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-изобутокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-н-пентилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-н-гексилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-циклопропилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-циклопропилметокси-4-метоксифенил)-2-оксо)этил-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-циклобутилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-циклобутилметокси-4-метоксифенил)-2-оксо)этил-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-циклопентилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-циклогексилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-(циклопент-3-ен-1-илокси)-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-аллилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-((3-метилбут-2-ен-1-ил)окси)-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-пропаргилокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-(бут-2-ин-1-илокси)-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-(оксациклобутан-3-илокси)-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-(тетрагидрофуран-2-ил)окси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-метилтиометокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-метилсульфинилметокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-метилсульфонилметокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-метилтиоэтокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-метилсульфинилэтокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-метилсульфонилэтокси-4-метоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
(Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(гидроксиимидо)этил)-2,6-диметилпиридин-4(1Н)-он;
(Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(метоксиимидо)этил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(метоксиимидо)этил)-2,6-диметилпиридин-4(1Н)-он;
(Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(метилтиометоксиимидо)этил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(метилтиометоксиимидо)этил)-2,6-диметилпиридин-4(1Н)-он;
(Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(метилсульфинилметоксиимидо)этил)-2,6-диметилпиридин-4(1Н)-он;
(Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-(метилсульфонилметоксиимидо)этил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-циклопропилметокси-4-метоксифенил)-2-оксоэтил)пиридин-4(1Н)-он;
1-(2-(3-циклопропилметокси-4-метоксифенил)-2-оксоэтил)-2-метилпиридин-4(1Н)-он;
1-(2-(3-циклопропилметокси-4-метоксифенил)-2-оксоэтил)-2-хлорпиридин-4(1H)-он;
1-(2-(3-циклопропилметокси-4-метоксифенил)циклопропил)-2,6-диметилпиридин-4(1Н)-он;
1-(2-(3-циклопропилметокси-4-дифторметоксифенил)-2-оксоэтил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-циклопропилметокси-4-метоксистирил)пиридин-4(1Н)-он;
(Е)-1-(3-циклопропилметокси-4-метоксистирил)-2-метилпиридин-4(1Н)-он;
(Е)-1-(3-гидрокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-пропокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-изопропокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-((3-н-бутокси-4-метокси)-стирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-изобутокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-((3-неопентилокси-4-метокси)-стирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-циклопропилокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-циклопропилметокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-циклопропилметокси-4-метоксифенетил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-циклобутилокси-4-метокси-стирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-((3-циклопентилокси-4-метокси)-стирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-(циклопент-3-ен-1-илокси)-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-аллилокси-4-метокси-стирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-((3-(3-метилбут-2-ен-1-ил)окси-4-метокси)-стирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-(проп-2-ин-1-илокси)-4-метокси)стирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-(бут-2-ин-1-илокси)-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-((3-(оксациклобутан-3-илокси)-4-метокси)-стирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-((3-(тиоциклобутан-3-илокси)-4-метокси)-стирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-((3-(тетрагидрофуран-2-ил)окси-4-метокси)-стирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-2-(5-(2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)этенил)-2-метоксифенокси)-N-метилацетамид;
(Е)-1-((3-циклопропилформилокси-4-метокси)-стирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-5-(2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)этенил)-2-метоксифенилметилсульфонат;
(Е)-1-(3-метилтиометокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-метилсульфинилметокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-метилсульфонилметокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-метилтиоэтокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-метилсульфинилэтокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-метилсульфонилэтокси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-((3-(тетрагидротиофен-3-ил)окси-4-метокси)-стирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-((1,1-диоксотетрагидротиофен-3-ил)окси)-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(S,Е)-1-(3-(тетрагидротиофен-3-ил)окси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он;
1-((Е)-4-метокси-3-(((3S)-1-оксотетрагидротиофен-3-ил)окси)стирил)-2,6-диметилпиридин-4(1Н)-он;
(S,E)-1-(3-((1,1-диоксотетрагидротиофен-3-ил)окси)-4-метокси-стирил)-2,6-диметилпиридин-4(1Н)-он;
(R,Е)-1-(3-(тетрагидротиофен-3-ил)окси-4-метоксистирил)-2,6-диметилпиридин-4(1Н)-он;
1-((Е)-4-метокси-3-(((3R)-1-оксотетрагидротиофен-3-ил)окси)стирил)-2,6-диметилпиридин-4(1Н)-он;
(R,Е)-1-(3-((1,1-диоксотетрагидротиофен-3-ил)окси)-4-метокси-стирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-(циклопропилметокси)-4-(дифторметокси)стирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-метокси-4-дифторметокси-стирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-метилтиометокси-4-дифторметокси-стирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-метилсульфинилметокси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-метилсульфонилметокси-4-дифторметокси-стирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-метилтиоэтокси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-метилсульфинилэтокси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(3-метилсульфонилэтокси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-((3-(тетрагидротиофен-3-ил)окси-4-дифторметокси)-стирил)-2,6-диметилпиридин-4(1Н)-он;
(S,E)-1-(3-(тетрагидротиофен-3-ил)окси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1Н)-он;
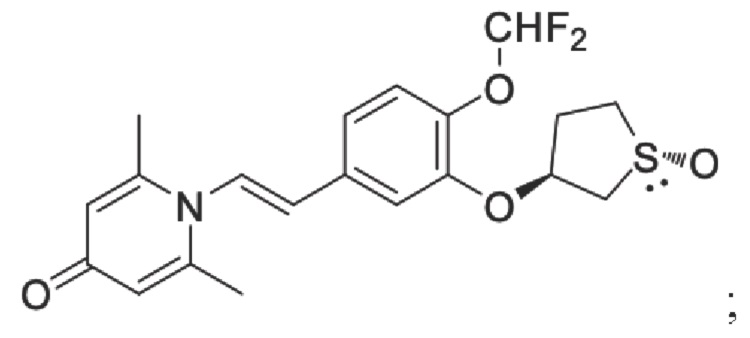
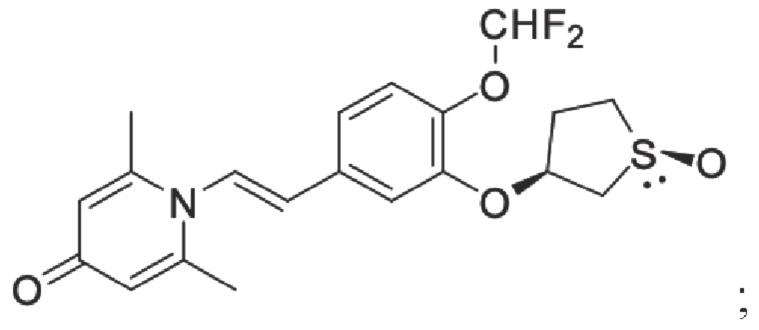
(S,Е)-1-(3-(1,1-диоксотетрагидротиофен-3-ил)окси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(R,E)-1-(3-(тетрагидротиофен-3-ил)окси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1Н)-он;
1-((Е)-4-дифторметокси-3-(((3R)-1-оксотетрагидротиофен-3-ил)окси)стирил)-2,6-диметилпиридин-4(1Н)-он;
(R,Е)-1-(3-(1,1-диоксотетрагидротиофен-3-ил)окси-4-дифторметоксистирил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-метилэтенил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-фенилэтенил)-2,6-диметилпиридин-4(1Н)-он;
(Z)-1-(2-(3-циклопропилметокси-4-метоксифенил)пент-1-ен-3-ин-1-ил)-2,6-диметилпиридин-4(1Н)-он;
(Е)-1-(2-(3-циклопропилметокси-4-метоксифенил)-2-цианоэтенил)-2,6-диметилпиридин-4(1Н)-он;
(Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)-акриламид;
(Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)-N-метилакриламид;
(Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)-N, N-диметилакриламид;
(Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4Η)-ил)-Ν, N-диэтилакриламид;
(Z)-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)-акриловая кислота;
(Z)-метил-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)-акрилат;
(Z)-этил-2-(3-циклопропилметокси-4-метоксифенил)-3-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)-акрилат;
(Z)-1-(2-(3-циклопропилметокси-4-дифторметоксифенил)пент-1-ен-3-ин-1-ил)-2,6-диметилпиридин-4(1Н)-он;
(Z)-2-(3,4-диметоксифенил)-3-(2,6-диметил-4-карбонилпиридин)-1(4Н)-ил)акрилонитрил;
(Е)-3-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)акрилонитрил;
(Е)-3-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин-1(4Н)-ил)акриламид; и
(Е)-3-(3-циклопропилметокси-4-метоксифенил)-2-(2,6-диметил-4-карбонилпиридин)-1(4H)-ил)-N, N-диметилакриламид.
18. Применение противовоспалительного соединения по п. 1 в качестве ингибитора PDE4.
19. Применение противовоспалительного соединения по п. 1 для лечения воспалительных кожных заболеваний, связанных с PDE4.
20. Лекарственное средство для лечения воспалительных кожных заболеваний, содержащее:
от 0,01 до 10% противовоспалительного соединения по п. 1 от общей массы лекарственного средства; и
другие компоненты, выбранные из поверхностно-активного вещества, липидного соединения и вспомогательного средства;
где количество поверхностно-активного вещества составляет от 10 до 30% от общей массы лекарственного средства;
количество липидного соединения составляет от 50 до 85% от общей массы лекарственного средства; и
количество вспомогательного средства составляет от 10 до 30% от общей массы лекарственного средства.
| Buzhe Xu et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Устройство для устранения мешающего действия зажигательной электрической системы двигателей внутреннего сгорания на радиоприем | 1922 |
|
SU52A1 |
| CAS 1408687-07-9 REGITRY, CA REGISTRY, published on 02.12.2012 | |||
| ДАТЧИК УГЛОВОЙ СКОРОСТИ | 0 |
|
SU376724A1 |
| CN 10374873 A, 09.08.2011 | |||
| WO 9923075 A1, 31.10.1997 | |||
| RU 2009113585 A, 20.10.2010. | |||

