ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к фармацевтической композиции для предупреждения или лечения волчанки, содержащей соединение, представленное формулой I, его оптический изомер или его фармацевтически приемлемую соль в качестве эффективного компонента, а также к способу лечения с использованием соединения, и к применению соединения дли изготовления лекарственного средства для лечения волчанки.
Уровень техники
Волчанка представляет собой аутоиммунное заболевание, обусловленное антителами, атакующими соединительные ткани, а также ассоциированное с продуцированием антинуклеарных антител, т.е. циркулирующего иммунного комплекса, а также с активацией системы комплемента. Это заболевание является системным, так что оно может возникнуть во всех системах органов и также может вызывать тяжелое повреждение тканей. У пациентов с волчанкой также могут продуцироваться аутоантитела, имеющие специфичность против ДНК, против Ro и против тромбоцитов, и они могут вызывать возникновение заболеваний, таких как гломерулонефрит, артрит, серозит, полная блокада сердца новорожденных или гематологическая аномалия.
В отсутствии лечения волчанка может стать смертельной, поскольку она начинает атаковать кожу и суставы и переходит в атаку даже внутренних органов, например легких, сердца и почек, из которых заболевание почек является наиболее опасным. Повреждение почек, которое определяют по величине протеинурии в моче, является одной из частей заболевания в виде острого повреждения, ассоциированных с патогенностью волчанки, и оно составляет 50% или более смертности и частоты случаев заболевания волчанкой.
В настоящее время отсутствует надежное лечение для пациентов с волчанкой. С практической точки зрения врачи обычно применяют ряд иммунодепрессантов, например, высокую дозу кортикостероида, преднизона, азатиоприна или циклофосфамида, где существует проблема, состоящая в том, что значительное количество таких лекарственных средств имеют потенциально вредоносные побочные эффекты для пациентов, проходящих лечение.
Ссылки уровня техники
Патентный документ
Публикация патентной заявки Кореи № 2014-0128886
Описание
Техническая проблема
Задачей настоящего изобретения является предоставление фармацевтической композиции для предупреждения или лечения волчанки, содержащей соединение, представленное следующей формулой I, его оптический изомер или его фармацевтически приемлемую соль в качестве эффективного компонента.
Другой задачей настоящего изобретения является предоставление способа лечения волчанки, где способ включает введение терапевтически эффективного количества указанного соединения.
Другой задачей настоящего изобретения является обеспечение применения соединения для изготовления лекарственного средства для лечения волчанки.
Техническое решение
Оно подробно описано ниже. Между тем, каждое описание и форма вариантов осуществления, описанных в настоящем описании, может быть использована для других описаний и форм вариантов осуществления, соответственно. Иными словами, все комбинации различных элементов, описанных в настоящем описании, входят в настоящего изобретения. Также не предусматривается, что объем настоящего изобретения ограничивается конкретным описанием, описанным ниже.
Настоящее изобретение относится к фармацевтической композиции для предупреждения или лечения волчанки, содержащей соединение, представленное следующей формулой I, его оптический изомер или его фармацевтически приемлемую соль в качестве эффективного компонента:
[Формула I]
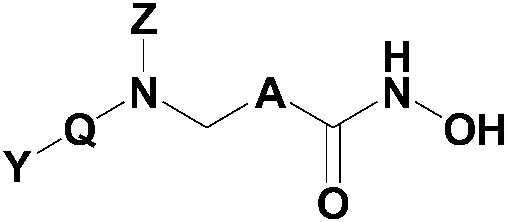
где
A представляет собой 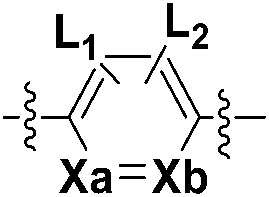
каждый из Xa и Xb независимо представляет собой CH или N,
каждый L1 и L2 независимо представляет собой водород, галоген, -CF3 или -C1-3 алкил с прямой или разветвленной цепью,
Q представляет собой C(=O), S(=O)2, S(=O) или C(=NH),
Y выбран из следующей группы:
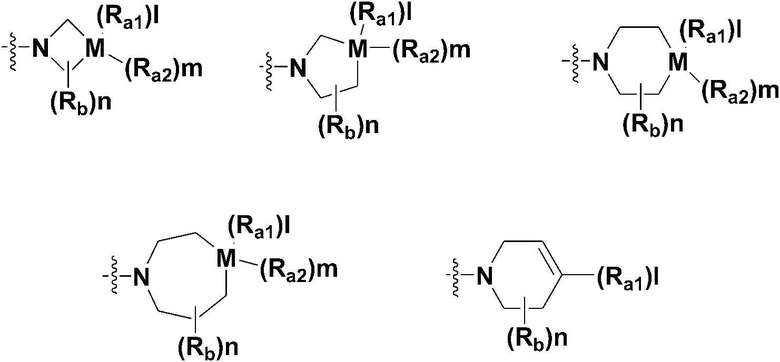
M представляет собой C, N, O, S или S(=O)2, где в этом случае, когда M представляет собой C, l и m равны 1; когда M представляет собой N, l равен 1 и m равен 0; и когда M представляет собой O, S или S(=O)2, l и m равны 0,
каждый из Ra1 и Ra2 независимо представляет собой водород; гидрокси; -C1-4 алкил с прямой или разветвленной цепью, который является незамещенным или замещен по меньшей мере одним галогеном; -C1-4 спирт с прямой или разветвленной цепью; бензгидрил; -C1-4 алкил с прямой или разветвленной цепью, который замещен насыщенным или ненасыщенным 5-7-членным гетероциклическим соединением, содержащим 1-3 гетероатома из N, O или S в качестве членов кольца, где в этом случае гетероциклическое соединение может быть незамещенным или по меньшей мере один атом водорода может быть необязательно замещен OH, OCH3, CH3, CH2CH3 или галогеном; насыщенное или ненасыщенное 5-7-членное гетероциклическое соединение, содержащее 1-3 гетероатома из N, O или S в качестве представителей кольца, где в этом случае гетероциклическое соединение может быть незамещенным или по меньшей мере один атом водорода может быть необязательно замещен OH, OCH3, CH3, CH2CH3 или галогеном; фенил, где он является незамещенным или по меньшей мере один атом водорода замещен галогеном, C1-4 алкокси, C1-2 алкилом или гидрокси; бензил, где он является незамещенным или по меньшей мере один атом водорода замещен галогеном, C1-4 алкокси, C1-2 алкилом или гидрокси; -S(=O)2CH3; галоген; -C1-6 алкокси с прямой или разветвленной цепью; -C2-6 алкоксиалкил; -C(=O)Rx, где Rx представляет собой C1-3 алкил с прямой или разветвленной цепью или C3-10 циклоалкил;  , где каждый из Rc и Rd независимо представляет собой водород, C1-3 алкил с прямой или разветвленной цепью; и
, где каждый из Rc и Rd независимо представляет собой водород, C1-3 алкил с прямой или разветвленной цепью; и  или
или 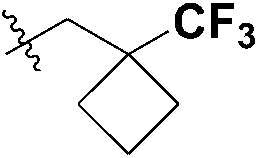
n представляет собой целое число, равное 0, 1 или 2,
Rb представляет собой водород; гидрокси; -C1-6 алкил с прямой или разветвленной цепью, где он является незамещенным или по меньшей мере один атом водорода замещен галогеном; -C(=O)CH3; -C1-4 гидроксиалкил с прямой или разветвленной цепью; -C1-6 алкокси с прямой или разветвленной цепью; -C2-6 алкоксиалкил с прямой или разветвленной цепью; -CF3; галоген или 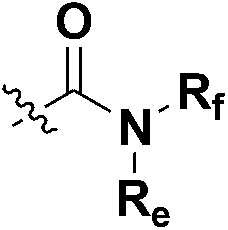 ,
,
каждый из Re и Rf независимо представляет собой водород или -C1-3 алкил с прямой или разветвленной цепью,
Z выбран из следующей группы:
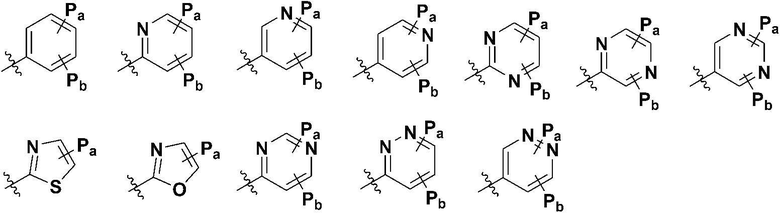
каждый из Pa и Pb независимо представляет собой  ; водород; гидрокси; -C1-4 алкил с прямой или разветвленной цепью, где он является незамещенным или по меньшей мере один атом водорода замещен галогеном; галоген; -CF3; -OCF3; -CN; -C1-6 алкокси с прямой или разветвленной цепью; -C2-6 алкилалкокси с прямой или разветвленной цепью; -CH2F или -C1-3 спирт,
; водород; гидрокси; -C1-4 алкил с прямой или разветвленной цепью, где он является незамещенным или по меньшей мере один атом водорода замещен галогеном; галоген; -CF3; -OCF3; -CN; -C1-6 алкокси с прямой или разветвленной цепью; -C2-6 алкилалкокси с прямой или разветвленной цепью; -CH2F или -C1-3 спирт,
где  представляет собой фенил, пиридин, пиримидин, тиазол, индол, индазол, пиперазин, хинолин, фуран, тетрагидропиридин, пиперидин или кольцо, выбранное из следующей группы:
представляет собой фенил, пиридин, пиримидин, тиазол, индол, индазол, пиперазин, хинолин, фуран, тетрагидропиридин, пиперидин или кольцо, выбранное из следующей группы:
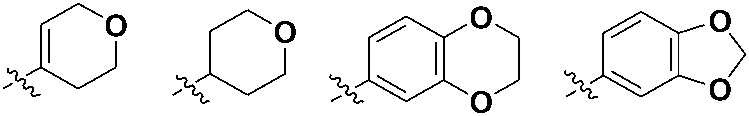
каждый из x, y и z независимо представляет собой целое число, равное 0 или 1,
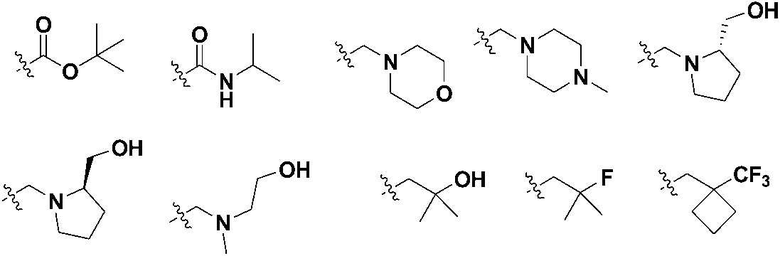
каждый из Rg1, Rg2 и Rg3 независимо представляет собой водород; гидрокси; -C1-3 алкил; -CF3; -C1-6 алкокси с прямой или разветвленной цепью; -C2-6 алкилалкокси с прямой или разветвленной цепью; -C(=O)CH3; -C1-4 гидроксиалкил с прямой или разветвленной цепью; -N(CH3)2; галоген; фенил; -S((=O)2)CH3; или выбран из следующей группы:
Соединение, представленное формулой I, в соответствии с настоящим изобретением может представлять собой соединение, представленное следующей формулой Ia:
[Формула Ia]
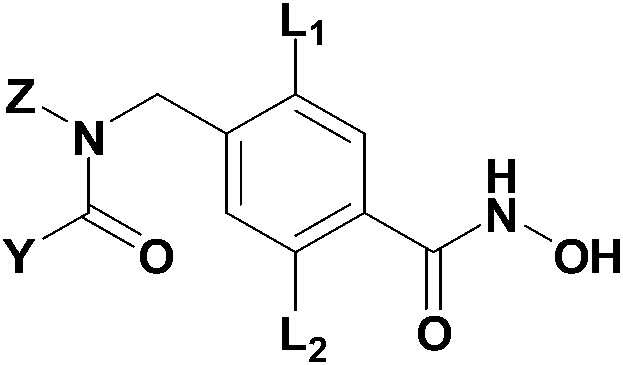
где
каждый из L1 и L2 независимо представляет собой водород или галоген,
Y представляет собой  ,
,  или
или  ,
,
Z представляет собой фенил или пиридинил, где по меньшей мере один атом водорода фенила или пиридинила может быть замещен галогеном, CF3 или CF2H.
В соответствии с конкретным вариантом осуществления настоящего изобретения соединение, представленное следующей формулой Ia, представляет собой соединение, описанное в таблице ниже:
Таблица 1
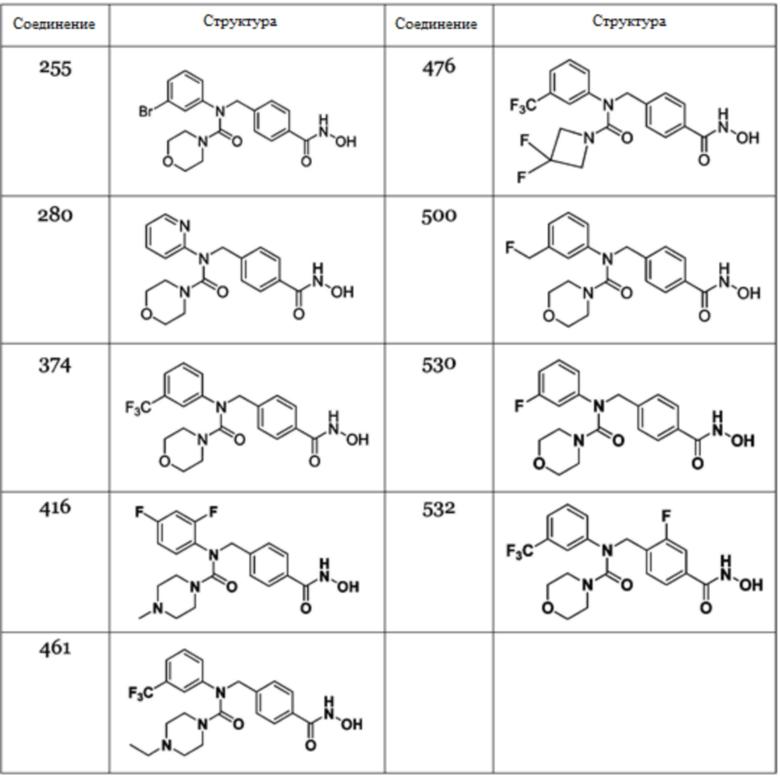
В рамках настоящего изобретения, соединение, представленное следующей формулой I, можно получать способом, описанным в публикации нерассмотренной патентной заявки Кореи № 2014-0128886, но он не ограничивается этим.
В рамках настоящего изобретения фармацевтически приемлемая соль означает соль, обычно используемую в медицинской промышленности, например, соль неорганического иона, полученную из кальция, калия, натрия, магния и т.п.; соль неорганической кислоты, полученную из хлористоводородной кислоты, азотной кислоты, фосфорной кислоты, бромной кислоты, йодной кислоты, перхлорной кислоты, серной кислоты и т.п.; соль органической кислоты, полученную из уксусной кислоты, трифторуксусной кислоты, лимонной кислоты, малеиновой кислоты, янтарной кислоты, щавелевой кислоты, бензойной кислоты, виннокаменной кислоты, фумаровой кислоты, миндальной кислоты, пропионовой кислоты, лимонной кислоты, молочной кислоты, гликолевой кислоты, глюконовой кислоты, галактуроновой кислоты, глутаминовой кислоты, глутаровой кислоты, глюкуроновой кислоты, аспарагиновой кислоты, аскорбиновой кислоты, угольной кислоты, ванилиновой кислоты, йодистоводородной кислоты и т.д.; соль сульфоновой кислоты, полученную из метансульфоновой кислоты, этансульфоновой кислоты, бензолсульфоновой кислоты, п-толуолсульфоновой кислоты, нафталинсульфоновой кислоты и т.п.; соль аминокислоты, полученную из глицина, аргинина, лизина и т.д.; соль амина, полученную из триметиламина, триэтиламина, аммиака, пиридина, пиколина и т.д., и т.п., однако типы солей в рамках настоящего изобретения не ограничиваются этими перечисленными солями.
Как используют в рамках изобретения, термин "волчанка" относится к аутоиммунному заболеванию, связанному с антителами, атакующими соединительные ткани, где оно включает хроническое воспалительное аутоиммунное заболевание, характеризующееся присутствием аутоантител, сыпью, язвами полости рта, серозитом, неврологическим нарушением, низким количество клеток крови, болью в суставах и опуханием. Если нет иных указаний, термин "волчанка" в рамках настоящего изобретения имеет общепринятое значение, используемое в области техники, к которой относится настоящее изобретение. В рамках настоящего изобретения волчанка включает системную красную волчанку (SLE), системную волчанку, дискоидную волчанку, индуцированную лекарственными средствами волчанку, волчанку новорожденных и т.п., однако, кроме того, она может включать различные дополнительные типы волчанки неограничивающим образом. Также волчанка может вызывать хронический нефрит, такой как волчаночный нефрит или гломерулонефрит.
В рамках настоящего изобретения термин "системная красная волчанка (SLE)" имеет общепринятое значение, используемое в области техники, к которой относится настоящее изобретение. SLE представляет собой полифилетическое аутоиммунное заболевание, при котором появляются антинуклеарные антитела, включающие антитела против дцДНК, и продуцируется комплекс антиген-антитело и оседает с мелких сосудах, таким образом, возможно вызывая воспаление и повреждение различных органов, включая базальную мембрану кожи или почек.
В одном варианте осуществления настоящего изобретения было идентифицировано, что соединения 255, 280, 374, 416, 461, 476, 500, 530 или 532, соответствующие формуле Ia, имели превосходный эффект подавления продуцирования in vitro молекул воспаления, таких как TNFα и т.д. (фиг.1), подавления пролиферации реактивных T-клеток (фиг.2) и улучшения функции регуляторных T-клеток (фиг.3).
Также было идентифицировано, что соединение 374 в соответствии с настоящим изобретением улучшало выживаемость мышей с заболеванием SLE (фиг.5), снижало встречаемость протеинурии (фиг.6), концентрацию антител против дцДНК (фиг.8) и уровень инфильтрации IgG и C3 в почках (фиг.12), снижало уровень сывороточных цитокинов IL-10, IL-12, IL-15, IL-17A, TNFα и IL-22 (фиг.13), снижало уровень CXCL10 и CCL2 (фиг.14), увеличивало уровень TGF-β (фиг.13) и снижало долю CD4-CD8- двойных негативных T-клеток, соотношение уровня CD4+CD8- клеток и уровня CD4-CD8+ клеток (фиг.15), соотношение CD4+T-bet+/CD4+GATA3+ (фиг.16), соотношение уровня CD4+CD25+ клеток и уровня CD4+ клеток (фиг.17) и уровня CD138+ клеток (фиг.19).
Фармацевтическая композиция в соответствии с настоящим изобретением, кроме того, может содержать по меньшей мере один тип фармацевтически приемлемого носителя, в дополнение к соединению, соответствующему формуле I выше, его оптическому изомеру или его фармацевтически приемлемой соли для цели введения. В качестве фармацевтически приемлемого носителя можно использовать физиологический раствор, стерилизованную воду, раствор Рингера, буферный солевой раствор, раствор декстрозы, раствор мальтодекстрина, глицерин, этанол и комбинацию по меньшей мере из одного их компонента, где также при необходимости могут быть добавлены другие общепринятые добавки, такие как антиоксидант, буферный раствор, бактериостатическое средство и т.д. Также фармацевтическая композиция в соответствии с настоящим изобретением может быть составлена в виде инъекционной дозированной формы, такой как водный раствор, суспензия, эмульсия и т.д., пилюля, капсула, гранула или таблетка, таким образом, чтобы к ним дополнительно добавлять разбавитель, диспергирующее средство, поверхностно-активное вещество, связующее вещество и смазывающее вещество. Таким образом, композиция в соответствии с настоящим изобретением может представлять собой пластырь, жидкое лекарственное средство, пилюлю, капсулу, гранулу, таблетку, суппозиторий и т.д. Эти препараты могут быть составлены посредством общепринятого способа, используемого для составления в области техники, к которой относится настоящее изобретение, в зависимости от каждого заболевания и/или компонента, или способа, описанного в Remington's Pharmaceutical Science (последняя версия), Mack Publishing Company, Easton PA.
Неограничивающим примером препарата для перорального введения с использованием фармацевтической композиции в соответствии с настоящим изобретением может быть таблетка, троше, пастилка, растворимая в воде суспензия, масляная суспензия, готовый порошок, гранула, эмульсия, твердая капсула, мягкая капсула, сироп, эликсир и т.п. Для составления фармацевтической композиции в соответствии с настоящим изобретением в препарат для перорального введения также можно использовать связующее вещество, такое как лактоза, сахароза, сорбит, маннит, крахмал, амилопектин, целлюлоза, желатин и т.п.; эксципиент, такой как дикальцийфосфат и т.д.; дезинтегрирующее средство, такое как кукурузный крахмал, сладкий картофельный крахмал и т.п.; смазывающее вещество, такое как стеарат магния, стеарат кальция, стеарилфумарат натрия, полиэтиленгликоль, воск и т.п., и т.д., и также можно использовать подсластитель, вкусовую добавку, сироп и т.д. Более того, в случае капсулы, кроме того, можно использовать жидкий носитель, такой как жирное масло и т.д., в дополнение к вышеупомянутым материалам.
Неограничивающим примером парентерального препарата с использованием фармацевтической композиции в соответствии с настоящим изобретением может быть инъекционный раствор, суппозиторий, порошок для ингаляции, препарат аэрозоля для распыления, мазь, порошок для нанесения, масло, крем и т.д. Для составления фармацевтической композиции в соответствии с настоящим изобретением в препарат для парентерального введения можно использовать стерилизованный водный раствор, неводный растворитель, суспензию, эмульсию, лиофилизированный препарат, наружный препарат и т.д. В качестве указанного неводного растворителя и суспензии можно использовать растительное масло, такое как пропиленгликоль, полиэтиленгликоль и оливковое масло; инъекционный сложный эфир, такой как этилолеат; и т.д.
В случае составления фармацевтической композиции в соответствии с настоящим изобретением в виде инъекционного раствора, фармацевтическую композицию в соответствии с настоящим изобретением можно получать в виде раствора или суспензии таким образом, чтобы смешивать композицию по изобретению со стабилизатором или буфером, а затем раствор или суспензию можно составлять в виде единичной дозированной формы в ампуле или флаконе.
В случае составления фармацевтической композиции в соответствии с настоящим изобретением в виде препарата аэрозоля, композицию по изобретению можно смешивать с добавками, включая пропеллент и т.д., так чтобы диспергированный в воде концентрат или влажный порошок мог диспергироваться.
В случае составления фармацевтической композиции в соответствии с настоящим изобретением в виде мази, крема, порошка для нанесения, масла, наружного препарата для кожи и т.д. композицию по изобретению можно составлять в виде препарата так, чтобы в качестве носителя использовался животный жир, растительное масло, воск, парафин, крахмал, трагакант, производное целлюлозы, полиэтиленгликоль, силикон, бентонит, диоксид кремния, тальк, оксид цинка, и т.д.
Суточная дозировка соединения, соответствующего формуле I, в соответствии с настоящим изобретением, его оптического изомера или его фармацевтически приемлемой соли может находиться в диапазоне, например, приблизительно от 0,1 до 10000 мг/кг, в диапазоне приблизительно от 1 до 8000 мг/кг, в диапазоне приблизительно от 5 до 6000 мг/кг, или в диапазоне приблизительно от 10 до 4000 мг/кг, предпочтительно в диапазоне приблизительно от 50 до 2000 мг/кг, но не ограничиваясь ими, где такую дозировку также можно вводить один раз в сутки или разделять для введения несколько раз в сутки.
Фармацевтически эффективное количество и эффективная дозировка фармацевтической композиции в соответствии с настоящим изобретением могут быть диверсифицированы посредством способа составления фармацевтической композиции в виде препарата, пути введения, времени введения и/или способа введения и т.д., и также они могут быть диверсифицированы в зависимости от различных факторов, включая тип и степень реакций, которые намереваются достигнуть посредством введения фармацевтической композиции, тип индивидуума, которому намереваются проводить введение, возраст, массу тела, общее состояние здоровья, симптом или тяжесть заболевания, пол, рацион, экскрецию компонент других лекарственных композиций, используемых вместе в то же время или в другое время для соответствующего индивидуума, и т.д., а также других известных факторов, хорошо известных в области медицины, где специалисты в данной области могут без труда определить и назначить дозировку, эффективную для данного лечения.
В случае введения фармацевтической композиции в соответствии с настоящим изобретением, ее можно вводить один раз в сутки или разделять для введения несколько раз в сутки. Фармацевтическую композицию в соответствии с настоящим изобретением можно вводить в качестве отдельного терапевтического средства или в комбинации с другими терапевтическими средствами, а также ее можно вводить последовательно или одновременно с общепринятым терапевтическим средством. Учитывая все факторы, описанные выше, фармацевтическую композицию в соответствии с настоящим изобретением можно вводить в количестве, которое может демонстрировать максимальный эффект при минимальном количестве без какого-либо побочного эффекта, где такое количество может быть без труда определено специалистами в области, к которой относится настоящее изобретение.
Фармацевтическая композиция в соответствии с настоящим изобретением может демонстрировать превосходный эффект, когда ее используют отдельно, однако, кроме того, ее можно использовать в комбинации с различными способами, такими как гормональная терапия, медикаментозное лечение и т.д. для повышения терапевтической эффективности.
Настоящее изобретение также относится к способу лечения волчанки, где способ включает введение терапевтически эффективного количества соединения, соответствующего формуле I выше, его изомера или его фармацевтически приемлемой соли индивидууму, нуждающемуся в этом.
Как используют в рамках изобретения, термин "терапевтически эффективное количество" относится к количеству соединения, соответствующего формуле I выше, которое является эффективным для лечения волчанки.
В способе лечения в соответствии с настоящим изобретением подходящую общую суточную дозировку соединения, соответствующего формуле I выше, его изомера или его фармацевтически приемлемой соли может определять лечащий врач в рамках корректного медицинского решения, и она может находиться в диапазоне, например, приблизительно от 0,1 до 10000 мг/кг, в диапазоне приблизительно от 1 до 8000 мг/кг, в диапазоне приблизительно от 5 до 6000 мг/кг, или в диапазоне приблизительно от 10 до 4000 мг/кг, и предпочтительно такую дозу в диапазоне приблизительно 50-2000 мг/кг можно вводить один раз в сутки или разделять для введения несколько раз в сутки. Однако для цели настоящего изобретения предпочтительно, чтобы определенное терапевтически эффективное количество для определенного пациента применялось по-разному в зависимости от различных факторов, включающих тип и степень реакций, которые намереваются достигнуть, конкретную композицию, включая в некоторых случаях наличие или отсутствие применения других препаратов, возраст пациента, массу тела, общее состояние здоровья, пол и рацион, время введения, путь введения и скорость секреции композиции, период лечения и лекарственное средство, используемое вместе или одновременно с конкретной композицией, а также другие сходные факторы, хорошо известные в области медицины.
Способ лечения волчанки в соответствии с настоящим изобретением включает не только борьбу с самим заболеванием перед проявлением его симптомов, но также ингибирование или предотвращение таких симптомов путем введения соединения, соответствующего формуле I выше. При управлении течением заболевания профилактическая или терапевтическая доза определенного активного компонента может варьироваться в зависимости от характеристик и тяжести заболевания или состояния, и пути, посредством которого активный компонент вводят. Дозы и их частота могут варьироваться в зависимости от возраста, массы тела и реакций конкретного пациента. Подходящая доза и способ применения могут быть без труда выбраны специалистами в данной области, естественно, с учетом таких факторов. Также способ лечения волчанки в соответствии с настоящим изобретением, кроме того, может включать введение терапевтически эффективной дозы дополнительного активного вещества, которое может быть полезным при лечении заболевания, вместе с соединением, соответствующим формуле I выше, где дополнительное активное вещество может демонстрировать синергический эффект или аддитивный эффект вместе с соединением формулы I выше.
Настоящее изобретение также относится к применению соединения формулы I выше, его изомера или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения волчанки.
Соединение, соответствующее формуле I, выше, для изготовления лекарственного средства можно комбинировать с фармацевтически приемлемым адъювантом, разбавителем, носителем и т.д., или можно получать в виде составного средства с другими активными веществами, таким образом, обеспечивая синергическое действие.
Положения, упомянутые в отношении фармацевтической композиции по изобретению, способа лечения и применения, применимы в равной степени, если они не противоречат друг другу.
Преимущественные эффекты
Фармацевтическая композиция, содержащая соединение, соответствующая формуле I в соответствии с настоящим изобретением, его оптический изомер или его фармацевтически приемлемую соль, может демонстрировать превосходный эффект лечения волчанки, так что фармацевтическую композицию можно широко использовать для предупреждения или лечения волчанки.
Описание чертежей
На фиг.1 представлены результаты определения эффекта фармацевтической композиции по изобретению на подавление секреции TNFα.
На фиг.2 представлены результаты определения эффекта фармацевтической композиции по изобретению на подавление пролиферации реактивных T-клеток.
На фиг.3 представлены результаты определения эффекта фармацевтической композиции по изобретению на изменение функции регуляторных T-клеток.
На фиг.4 представлен эффект фармацевтической композиции по изобретению на восстановление после снижения массы тела в модели заболевания на животных.
На фиг.5 представлен эффект фармацевтической композиции по изобретению на повышение выживаемости в модели заболевания на животных.
На фиг.6 представлен эффект фармацевтической композиции по изобретению на снижение встречаемости протеинурии в модели заболевания на животных.
На фиг.7 представлен эффект фармацевтической композиции по изобретению на снижение величины UP/C в модели заболевания на животных.
На фиг.8 представлен эффект фармацевтической композиции по изобретению на снижение сывороточной концентрации антител против дцДНК в модели заболевания на животных.
На фиг.9 представлен эффект фармацевтической композиции по изобретению на снижение сывороточной концентрации BUN и креатинина в модели заболевания на животных.
На фиг.10 представлено изображение окрашенных тканей почки в модели заболевания на животных.
На фиг.11 представлен эффект фармацевтической композиции по изобретению на гистологические изменения в почках в модели заболевания на животных.
На фиг.12 представлен эффект фармацевтической композиции по изобретению на снижение отложений IgG и C3 в почках в модели заболевания на животных.
На фиг.13 представлено изменение профилей цитокинов в сыворотке посредством фармацевтической композиции по изобретению в модели заболевания на животных.
На фиг.14 представлен эффект фармацевтической композиции по изобретению на снижение сывороточной концентрации CXCL10 и CCL2 в модели заболевания на животных.
На фиг.15 представлен эффект фармацевтической композиции по изобретению на соотношения CD4 и CD8 T-клеток в модели заболевания на животных.
На фиг.16 и 17 представлен эффект фармацевтической композиции по изобретению на CD4+ CD25+ T-клетки в модели заболевания на животных.
На фиг.18 представлен эффект фармацевтической композиции по изобретению на экспрессию основных регуляторов Treg, Th17, Th1, Th2 клеток в модели заболевания на животных.
На фиг.19 представлен эффект фармацевтической композиции по изобретению на CD138+ клетки в модели заболевания на животных.
На фиг.20 представлен эффект фармацевтической композиции по изобретению на пре-B и про-B клетки в костном мозге в модели заболевания на животных.
На фиг.21 представлен эффект фармацевтической композиции по изобретению на уровень изотипов иммуноглобулинов в модели заболевания на животных.
Способ осуществления изобретения
Далее настоящее изобретение будет описано подробнее в соответствии с примерами получения и вариантами осуществления. Однако эти примеры получения и варианты осуществления предоставлены только для иллюстрации настоящего изобретения и, таким образом, настоящее изобретение не ограничивается ими.
Соединения 255, 280, 374, 416, 461, 476, 500, 530 или 532 в соответствии с настоящим изобретением получали способом, описанным в публикации нерассмотренной патентной заявки Кореи № 2014-0128886, и конкретные примеры получения описаны ниже. Вновь названная формула в каждом примере получения упоминается в рамках только соответствующего примера получения, и формулы, упоминаемые по меньшей мере в двух примерах получения, используются независимо в каждом примере получения.
Пример получения 1. Синтез соединения 255 {N-(3-бромфенил)-N-(4-(гидроксикарбамоил)бензил)морфолин-4-карбоксамид}
[Стадия 1] Синтез метил 4-((N-(3-бромфенил)морфолин-4-карбоксамидо)метил)бензоата
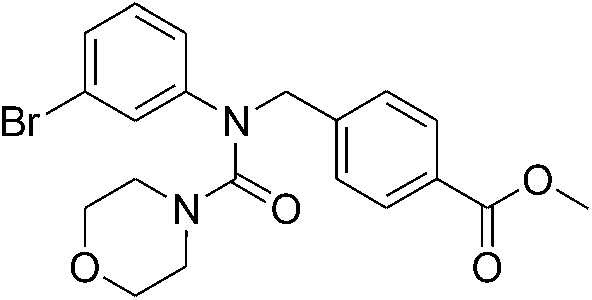
Метил 4-(((3-бромфенил)((4-нитрофенокси)карбонил)амино)метил)бензоат (1,5 г, 3,09 ммоль) растворяли в ацетонитриле (50 мл), а затем к нему медленно добавляли карбонат калия (1,28 г, 9,3 ммоль) и морфолин (0,40 мл, 4,64 ммоль). После этого температуру полученной смеси медленно повышали до 80°С, а затем полученную смесь перемешивали в течение трех часов при этой температуре. Температуру снижали до комнатной температуры, затем дополнительно добавляли диметилформамид (50 мл), затем температуру вновь повышали до 80°С, а затем полученную смесь перемешивали в течение пяти часов при этой температуре. После завершения реакции органический слой промывали насыщенным водным раствором хлорида аммония три раза, затем сушили с использованием сульфата натрия и фильтровали, а затем фильтрат концентрировали при пониженном давлении. Концентрат очищали посредством колоночной хроматографии (диоксид кремния; этилацетат/гексан=0-50%), с получением указанного в заголовке соединения (0,45 г, 33,6%) в виде прозрачного масла.
[Стадия 2] Синтез N-(3-бромфенил)-N-(4-(гидроксикарбамоил)бензил)морфолин-4-карбоксамида
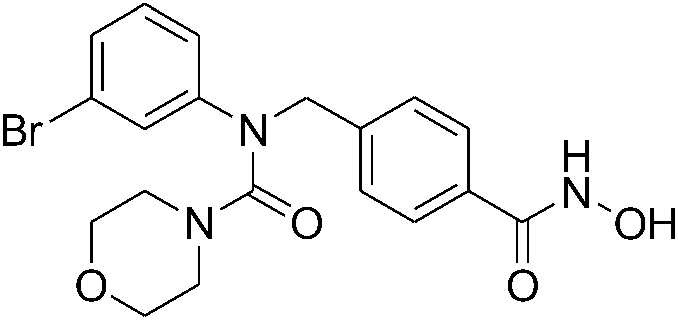
Метил 4-((N-(3-бромфенил)морфолин-4-карбоксамидо)метил)бензоат (0,05 г, 0,12 ммоль) растворяли в метаноле (2 мл), а затем медленно добавляли хлористоводородную соль гидроксиламина (0,040 г, 0,58 ммоль). После этого к полученной смеси добавляли гидроксид калия (0,065 г, 1,15 ммоль) и перемешивали при комнатной температуре в течение десяти минут, а затем добавляли гидроксиламин (50,0 масс.% водный раствор, 0,14 мл, 2,31 ммоль). Полученную смесь перемешивали при комнатной температуре в течение суток, затем органический растворитель концентрировали при пониженном давлении и нейтрализовывали добавлением 2 н хлористоводородной кислоты. Затем органический слой промывали насыщенным водным раствором хлорида натрия три раза, а затем сушили безводным сульфатом натрия и фильтровали. После этого фильтрат концентрировали при пониженном давлении, а затем концентрат очищали колоночной хроматографией (диоксид кремния; этилацетат/гексан=0-80%) с получением указанного в заголовке соединения (0,036 г, 72%) в белой твердой форме.
1H-ЯМР (400 МГц, CDCl3-d6) δ 7,63 (д, 2 H, J=7,8 Гц), 7,27-7,20 (м, 4 H), 7,13 (т, 1 H, J=7,8 Гц), 6,96 (д, 1 H, J=7,1 Гц), 4,83 (с, 2 H), 3,49 (уш. с, 4 H), 3,23 (уш. с, 4 H); MS (ESI) m/z 436 (M+ + H).
Пример получения 2. Синтез соединения 280 {N-(4-(гидроксикарбамоил)бензил)-N-(пиридин-2-ил)морфолин-4-карбоксамид}
[Стадия 1] Синтез метил 4-((пиридин-2-иламино)метил)бензоата
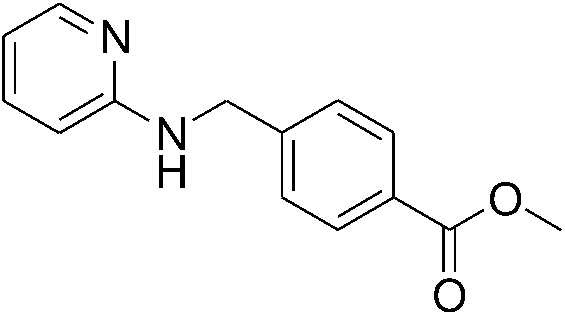
Пиридин-2-амин (0,2 г, 2,13 ммоль) растворяли в метаноле (10 мл), а затем к нему добавляли метил 4-формилбензоат (0,35 г, 2,13 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 20 минут, а затем к ней медленно добавляли цианоборгидрид натрия (0,13 г, 2,13 ммоль) и уксусную кислоту (0,12 мл. 2,13 ммоль), а затем перемешивали при комнатной температуре в течение пяти часов. Полученную смесь промывали насыщенным водным раствором хлорида натрия три раза, затем органический слой сушили сульфатом натрия и фильтровали, а затем фильтрат концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (диоксид кремния; этилацетат/гексан=0-30%) с получением указанного в заголовке соединения (0,10 г, 19%) в виде прозрачного масла.
1H-ЯМР (400 МГц, CDCl3) δ 8,17 (д, 1 H, J=5,8 Гц), 8,06 (д, 2 H, J=8,4 Гц), 7,66 (т, 1 H, J=7,8 Гц), 7,44 (д, 2 H, J=8,0 Гц), 6,76 (т, 1 H, J=6,7 Гц), 6,58 (д, 1 H, J=8,6 Гц), 4,67 (д, 2 H, J=6,0 Гц), 3,92 (с, 3 H).
[Стадия 2] Синтез метил 4-((((4-нитрофенокси)карбонил)(пиридин-2-ил)амино)метил)бензоата
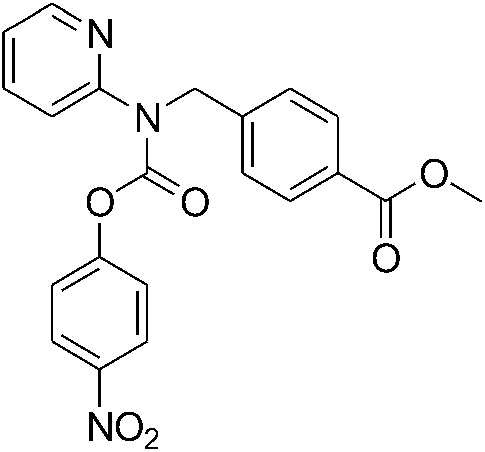
Метил 4-((пиридин-2-иламино)метил)бензоат (0,040 г, 0,16 ммоль) растворяли в диметилформамиде (3 мл), а затем медленно добавляли карбонат калия (0,046 г, 0,33 ммоль). После этого к полученной смеси добавляли 4-нитрофенилхлорформиат (0,037 г, 0,18 ммоль), а затем температуру полученной смеси медленно повышали до 50°C, а затем полученную смесь перемешивали в течение двух суток при этой температуре. После завершения реакции слой этилацетата промывали насыщенным водным раствором хлорида аммония три раза, а затем органический слой сушили сульфатом натрия и фильтровали, а затем фильтрат концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (диоксид кремния; этилацетат/гексан=0-50%) с получением указанного в заголовке соединения (0,048 г, 71%) в виде желтого масла.
1H-ЯМР (400 МГц, CDCl3) δ 8,49-8,48 (м, 1 H), 8,24 (дд, 2 H, J=7,0, 2,2 Гц), 8,17 (дд, 2 H, J=7,2, 2,0 Гц), 8,00 (д, 2 H, J=8,4 Гц), 7,78 (т, 1 H, J=3,8 Гц), 7,44 (д, 2 H, J=8,0 Гц), 6,91 (дд, 2 H, J=7,3, 2,1 Гц), 5,39 (уш. с, 2 H), 3,92 (с, 3 H); MS (ESI) m/z 408 (M+ + H).
[Стадия 3] Синтез метил 4-((N-(пиридин-2-ил)морфолин-4-карбоксамидо)метил)бензоата

Метил 4-((((4-нитрофенокси)карбонил)(пиридин-2-ил)амино)метил)бензоат (0,040 г, 0,098 ммоль) растворяли в диметилформамиде (5 мл), а затем медленно добавляли карбонат калия (0,040 г, 0,30 ммоль) и морфолин (0,013 мл, 0,15 ммоль). После этого температуру полученной смеси медленно повышали до 80°С, а затем полученную смесь перемешивали в течение трех часов при этой температуре. После завершения реакции полученную смесь промывали насыщенным водным раствором хлорида аммония три раза, затем органический слой сушили сульфатом натрия и фильтровали, а затем фильтрат концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (диоксид кремния; этилацетат/гексан=0-50%) с получением указанного в заголовке соединения (0,022 г, 63%) в форме светло-желтого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ 8,37-8,35 (м, 1 H), 7,95 (д, 2 H, J=8,4 Гц), 7,60-7,58 (м, 1 H), 7,47 (д, 2 H, J=8,4 Гц), 6,94-6,89 (м, 2 H), 5,13 (с, 2 H), 3,89 (с, 3 H), 3,53-3,51 (м, 4 H), 3,31-3,29 (м, 4 H).
[Стадия 4] Синтез N-(4-(гидроксикарбамоил)бензил)-N-(пиридин-2-ил)морфолин-4-карбоксамида
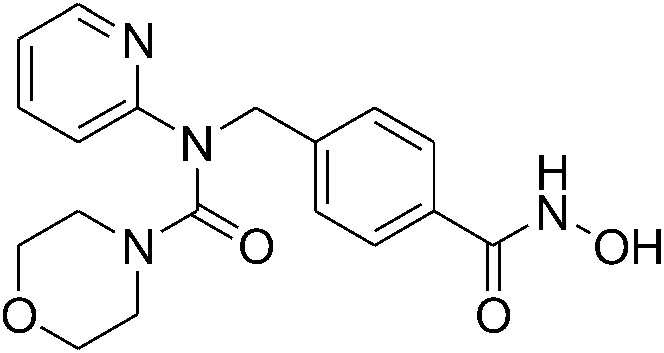
Метил 4-((N-(пиридин-2-ил)морфолин-4-карбоксамидо)метил)бензоат (0,022 г, 0,062 ммоль) растворяли в MeOH (2 мл), а затем медленно добавляли хлористоводородную соль гидроксиламина (0,022 г, 0,31 ммоль). После этого к полученной смеси добавляли гидроксид калия (0,035 г, 0,62 ммоль), затем перемешивали при комнатной температуре в течение десяти минут, а затем добавляли гидроксиламин (50,0 масс.% водный раствор, 0,082 мл, 1,24 ммоль). Полученную смесь перемешивали при комнатной температуре в течение суток, а затем органический растворитель концентрировали при пониженном давлении и нейтрализовывали добавлением 2 Н HCl, а затем промывали насыщенным водным раствором хлорида натрия три раза, а затем органический слой сушили безводным сульфатом натрия и фильтровали с получением указанного в заголовке соединения (0,007 г, 32%) в форме белого твердого вещества.
1H-ЯМР (400 МГц, MeOD-d3) δ 8,32 (д, 1 H, J=3,6 Гц), 7,72 (т, 1 H, J=6,6 Гц), 7,67 (д, 2 H, J=8,2 Гц), 7,48 (д, 2 H, J=8,2 Гц), 7,08-7,01 (м, 2 H), 5,08 (с, 2 H), 3,52 (т, 4 H, J=4,8 Гц), 3,29 (т, 4 H, J=4,8 Гц); MS (ESI) m/z 357 (M+ + H).
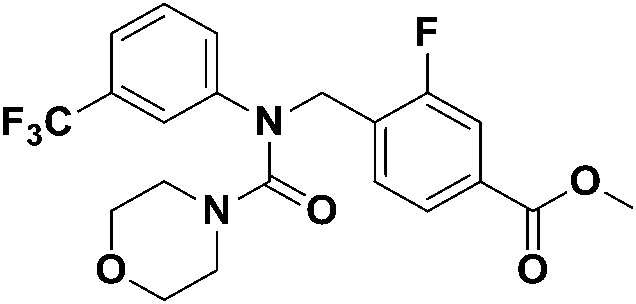
Пример получения 3. Синтез соединения 374 {N-(4-(гидроксикарбамоил)бензил)-N-(3-(трифторметил)фенил)морфолин-4-карбоксамид}
[Стадия 1] Синтез метил 4-((3-(трифторметил)фениламино)метил)бензоата
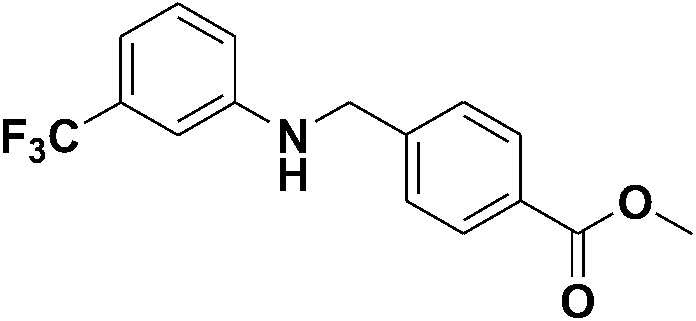
3-(трифторметил)бензоламин (0,30 г, 1,84 ммоль) и карбонат калия (0,76 г, 5,53 ммоль) растворяли в диметилформамиде (DMF) (5 мл), а затем добавляли метил 4-(бромметил)бензоат (0,42 г, 1,84 ммоль). Полученную смесь подвергали реакции при комнатной температуре в течение суток и разбавляли этилацетатом. Реагирующие вещества промывали водой и насыщенным водным раствором хлорида натрия, затем сушили безводным сульфатом магния и фильтровали, а затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (диоксид кремния; этилацетат/гексан=20%) с получением указанного в заголовке соединения (0,37 г, 65%).
1H-ЯМР (400 МГц, DMSO-d6) δ 7,93 (д, 2 H, J=8,3 Гц), 7,49 (д, 2 H, J=8,3 Гц), 7,24 (т, 1 H, J=7,9 Гц), 6,88-6,78 (м, 4 H), 4,42 (д, 2 H, J=6,1 Гц), 3,83 (с, 3H), MS (ESI) m/z 310 (M+ + H).
[Стадия 2] Синтез метил 4-((((4-нитрофенокси)карбонил)(3-(трифторметил)фенил)амино)метил)бензоата
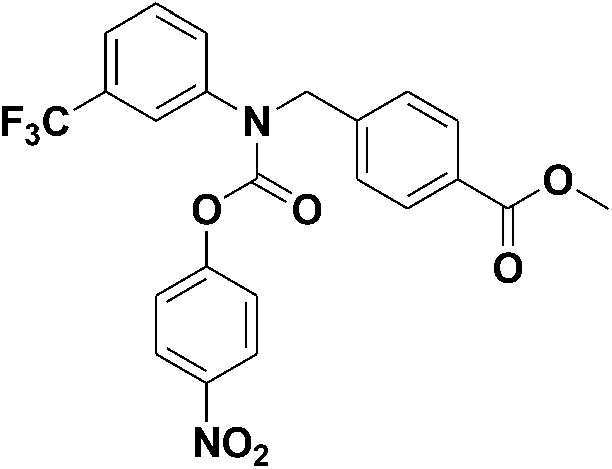
Метил 4-((3-(трифторметил)фениламино)метил)бензоат (0,26 г, 0,82 ммоль) и 4-нитрофенилкарбонохлоридат (0,33 г, 1,65 ммоль) растворяли в ацетонитриле (10 мл), а затем добавляли карбонат калия (0,34 г, 2,47 ммоль). Полученную смесь подвергали реакции при комнатной температуре в течение суток и разбавляли этилацетатом. Реагирующие вещества промывали насыщенным водным раствором хлорида натрия, затем сушили безводным сульфатом натрия и фильтровали, а затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (диоксид кремния; этилацетат/гексан=20%) с получением указанного в заголовке соединения (0,35 г, 89%) в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ 8,20 (д, 2 H, J=10,2 Гц), 8,01 (д, 2 H, J=7,8 Гц), 7,56-7,46 (м, 3H), 7,35 (д, 3 H, J=8,0 Гц), 7,26 (д, 2 H, J=8,1 Гц), 5,01 (уш. с, 2H), 3,90 (с, 3H).
[Стадия 3] Синтез метил 4-((N-(3-(трифторметил)фенил)морфолин-4-карбоксамидо)метил)бензоата
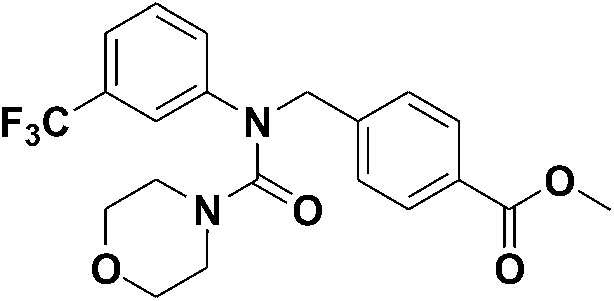
Метил 4-((((4-нитрофенокси)карбонил)(3-(трифторметил)фенил)амино)метил)бензоат (0,29 г, 0,60 ммоль) растворяли в диметилформамиде (10 мл), а затем добавляли карбонат калия (0,25 г, 1,81 ммоль) и морфолин (0,05 мл, 0,60 ммоль). Полученную смесь подвергали реакции при 60°C в течение двух суток, а затем разбавляли насыщенным раствором хлорида аммония. Проводили экстракцию этилацетатом, а затем экстракт сушили безводным сульфатом натрия и фильтровали, а затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (диоксид кремния; этилацетат/гексан=50%) с получением указанного в заголовке соединения (0,15 г, 60%).
1H-ЯМР (400 МГц, DMSO-d6) δ 7,97 (д, 2 H, J=8,2 Гц), 7,43-7,32 (м, 5H), 7,20 (д, 1 H, J=8,0 Гц), 4,94 (с, 2H), 3,90 (с, 3H), 3,50 (т, 4 H, J=4,8 Гц), 3,25 (т, 4 H, J=4,8 Гц); MS (ESI) m/z 423 (M+ + H).
[Стадия 4] Синтез N-(4-(гидроксикарбамоил)бензил)-N-(3-(трифторметил)фенил)морфолин-4-карбоксамида
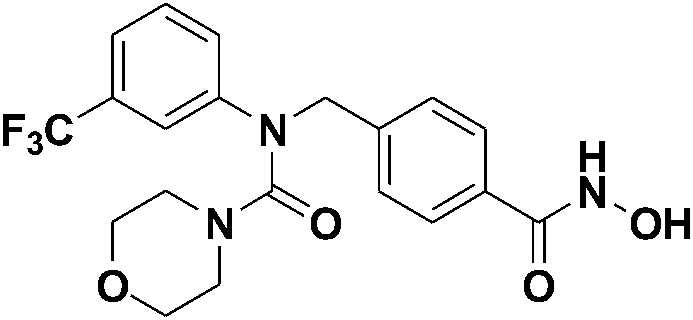
Метил 4-((N-(3-(трифторметил)фенил)морфолин-4-карбоксамидо)метил)бензоат (0,15 г, 0,36 ммоль) растворяли в метаноле (5 мл), затем добавляли водный раствор гидроксиламина (50 масс.%, 1 мл) и гидроксид калия (0,10 г, 1,81 ммоль), а затем перемешивали в течение ночи. После завершения реакции метанол отгоняли при пониженном давлении и удаляли, а затем проводили экстракцию этилацетатом и водой, а затем собирали. Полученный экстракт сушили безводным сульфатом натрия и фильтровали, а затем концентрировали при пониженном давлении. Остаток перемешивали в диэтиловом эфире, а затем получали твердый продукт, фильтровали и сушили с получением указанного в заголовке соединения (0,082 г, 54%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, MeOD-d3) δ 11,14 (уш. с, 1 H), 8,99 (уш. с, 1 H), 7,85 (д, 2 H, J=8,0 Гц), 7,66-7,27 (м, 6 H), 4,94 (с, 2 H), 3,41 (с, 2 H), 3,15 (с, 2 H). MS (ESI) m/z 424 (M+ + H).
Пример получения 4. Синтез соединения 416 {N-(2,4-дифторфенил)-N-(4-(гидроксикарбамоил)бензил)-4-метилпиперазин-1-карбоксамид}
[Стадия 1] Синтез метил 4-((N-(2,4-дифторфенил)-4-метилпиперазин-1-карбоксамидо)метил)бензоата
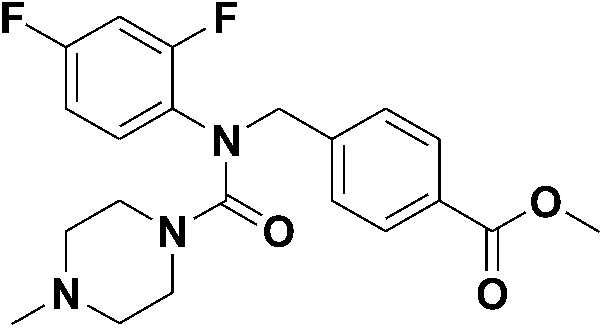
Метил 4-(((2,4-дифторфенил)((4-нитрофенокси)карбонил)амино)метил)бензоат (0,50 г, 1,13 ммоль) и 1-метилпиперазин (0,126 мл, 1,13 ммоль) растворяли в диметилформамиде (10 мл), а затем нагревали и перемешивали при 60°C в течение двух суток. Диметилформамид удаляли при пониженном давлении, затем в полученную реакционную смесь наливали воду, а затем проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем сушили посредством безводного сульфата магния, а затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (диоксид кремния; метанол/дихлорметан=5%) и концентрировали с получением указанного в заголовке соединения (0,46 г, 101%) в виде желтого масла.
[Стадия 2] Синтез N-(2,4-дифторфенил)-N-(4-(гидроксикарбамоил)бензил)-4-метилпиперазин-1-карбоксамида
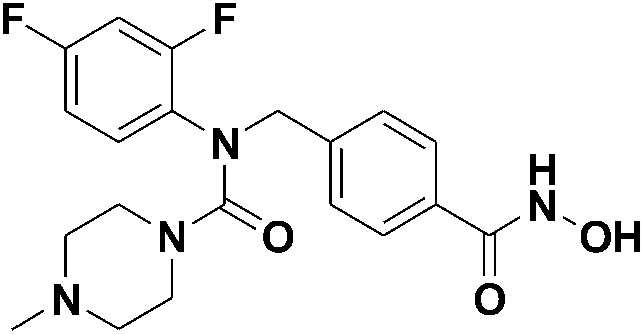
Метил 4-((N-(2,4-дифторфенил)-4-метилпиперазин-1-карбоксамидо)метил)бензоат (0,22 г, 0,545 ммоль) растворяли в метаноле (20 мл), затем добавляли хлористоводородную соль гидроксиламина (0,189 г, 2,73 ммоль) и гидроксид калия (0,306 г, 5,45 ммоль) и перемешивали, затем по каплям добавляли гидроксиламин (50 масс.% водный раствор; 0,701 мл, 10,9 ммоль), а затем перемешивали при комнатной температуре в течение трех часов. После завершения реакции метанол удаляли при пониженном давлении, затем в полученную смесь наливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем сушили безводным сульфатом магния, а затем концентрировали при пониженном давлении. После этого полученный концентрат растворяли в дихлорметане, затем добавляли гексан, затем твердое вещество выпадало в осадок, а затем его фильтровали и сушили с получение указанного в заголовке соединения (0,154 г, 70%) в форме желтого твердого вещества.
1H-ЯМР (400 МГц, MeOD-d3) δ 7,65 (д, 2 H, J=8,2 Гц), 7,40 (д, 2 H, J=8,2 Гц), 7,26-7,25 (м, 1 H), 7,04-6,96 (м, 2 H), 4,79 (с, 2 H), 3,25-3,23 (м, 4 H), 2,24-2,21 (м, 7 H); MS (ESI) m/z 405,1 (M+ + H).
Пример получения 5. Синтез соединения 461 {4-этил-N-(4-(гидроксикарбамоил)бензил)-N-(3-(трифторметил)фенил)пиперазин-1-карбоксамид}
[Стадия 1] Синтез метил 4-((4-этил-N-(3-(трифторметил)фенил)пиперазин-1-карбоксамидо)метил)бензоата
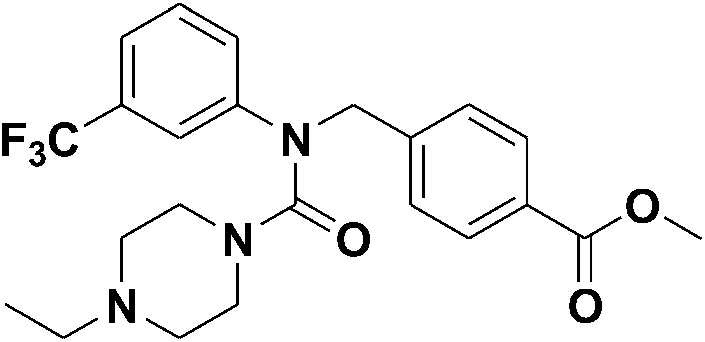
Метил 4-((((4-нитрофенокси)карбонил)(3-(трифторметил)фенил)амино)метил)бензоат (0,346 г, 0,73 ммоль) растворяли в диметилформамиде (10 мл), а затем добавляли карбонат калия (0,30 г, 2,19 ммоль) и 1-этилпиперазин (0,09 мл, 0,73 ммоль). Полученную смесь подвергали реакции при 60°С в течение суток, затем разбавляли этилацетатом, а затем промывали насыщенным раствором хлорида аммония. Полученную смесь сушили безводным сульфатом магния и фильтровали, а затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (диоксид кремния; этилацетат/гексан=50%) с получением указанного в заголовке соединения (0,15 г, 46%).
[Стадия 2] Синтез 4-этил-N-(4-(гидроксикарбамоил)бензил)-N-(3-(трифторметил)фенил)пиперазин-1-карбоксамида
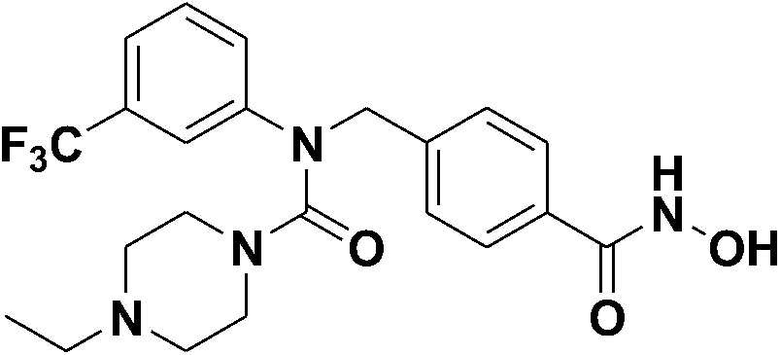
Метил 4-((4-этил-N-(3-(трифторметил)фенил)пиперазин-1-карбоксамидо)метил)бензоат (0,15 г, 0,33 ммоль) растворяли в метаноле (10 мл), затем добавляли гидроксиламин (50 масс.% водный раствор, 0,20 мл) и гидроксид калия (0,09 г, 1,67 ммоль), а затем перемешивали в течение ночи. После завершения реакции метанол отгоняли при пониженном давлении и удаляли, затем проводили экстракцию этилацетатом и водой, а затем собирали. Полученный экстракт сушили безводным сульфатом магния и фильтровали, а затем концентрировали при пониженном давлении. Остаток перемешивали в диэтиловом эфире, а затем образовывалось твердое вещество, и его фильтровали и сушили с получением указанного в заголовке соединения (0,09 г, 61%) в виде желтого твердого вещества.
1H-ЯМР (400 МГц, DMSO-d6) δ 11,1 (уш. с, 1 H), 7,65 (д, 2 H, J=8,2 Гц), 7,51 (т, 1 H, J=7,9 Гц), 7,41-7,36 (м, 5 H), 4,92 (с, 2 H), 3,17-3,14 (м, 4 H), 2,25, 2,22 (ABq, 2 H, J=12,4, 7,2 Гц), 2,18-2,15 (м, 4 H), 0,92 (т, 3 H, J=7,2 Гц); MS (ESI) m/z 451,1 (M+ + H).
Пример получения 6. Синтез соединения 476 {3,3-дифтор-N-(4-(гидроксикарбамоил)бензил)-N-(3-(трифторметил)фенил)азетидин-1-карбоксамид}
[Стадия 1] Синтез метил 4-((3,3-дифтор-N-(3-(трифторметил)фенил)азетидин-1-карбоксамидо)метил)бензоата

Метил 4-((((4-нитрофенокси)карбонил)(3-(трифторметил)фенил)амино)метил)бензоат (0,24 г, 0,51 ммоль) растворяли в диметилформамиде (5 мл), а затем добавляли карбонат калия (0,21 г, 1,52 ммоль) и гидрохлорид 3,3-дифторазетидина (0,13 г, 1,10 ммоль). Полученную смесь подвергали реакции при 60°С в течение двух суток, а затем разбавляли насыщенным раствором хлорида аммония. Проводили экстракцию этилацетатом, а затем полученный экстракт сушили безводным сульфатом натрия и фильтровали, а затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (диоксид кремния; этилацетат/гексан=30%) с получением указанного в заголовке соединения (0,14 г, 63%).
[Стадия 2] Синтез 3,3-дифтор-N-(4-(гидроксикарбамоил)бензил)-N-(3-(трифторметил)фенил)азетидин-1-карбоксамида
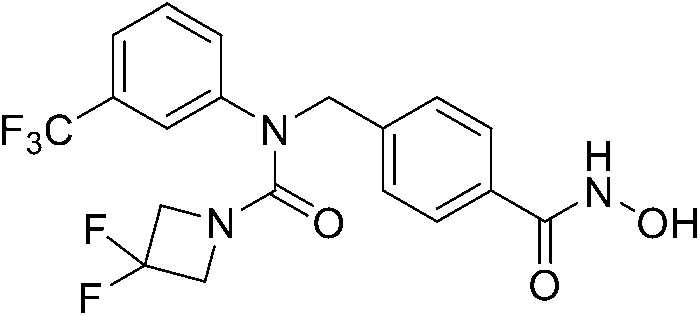
Метил 4-((3,3-дифтор-N-(3-(трифторметил)фенил)азетидин-1-карбоксамидо)метил)бензоат (0,14 г, 0,32 ммоль) растворяли в метаноле (10 мл), затем добавляли водный раствор гидроксиламина (50 масс.%, 0,2 мл) и гидроксид калия (0,09 г, 1,60 ммоль), а затем перемешивали в течение ночи. После завершения реакции метанол отгоняли при пониженном давлении и удаляли, а затем проводили экстракцию этилацетатом и водой, и собирали. Полученный экстракт сушили безводным сульфатом натрия и фильтровали, а затем концентрировали при пониженном давлении. Остаток перемешивали в диэтилового эфире, а затем образовывался твердый продукт, и его фильтровали и сушили с получением указанного в заголовке соединения (0,072 г, 52%) в форме белого твердого вещества.
Пример получения 7. Синтез соединения 500 {N-(3-(фторметил)фенил)-N-(4-(гидроксикарбамоил)бензил)морфолин-4-карбоксамид}
[Стадия 1] Синтез метил 4-((N-(3-(фторметил)фенил)морфолин-4-карбоксамидо)метил)бензоата
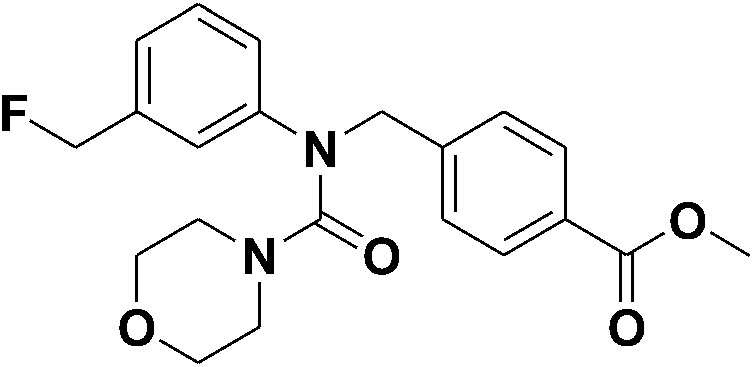
4-((N-(3-(гидроксиметил)фенил)морфолин-4-карбоксамидо)метил)бензойную кислоту (1,25 г, 3,25 ммоль) растворяли в дихлорметане (20 мл), затем добавляли трифторид диэтиламиносеры (DAST, 0,424 мл, 3,58 ммоль) при 0°C, затем перемешивали при той же температуре в течение одного часа, затем в реакционную смесь наливали водный раствор гидрокарбоната натрия, а затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем сушили безводным сульфатом магния, а затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (диоксид кремния; этилацетат/гексан=30-50%) и концентрировали с получением указанного в заголовке соединения (0,617 г, 49%) в виде бесцветной жидкости.
[Стадия 2] Синтез N-(3-(фторметил)фенил)-N-(4-(гидроксикарбамоил)бензил)морфолин-4-карбоксамида
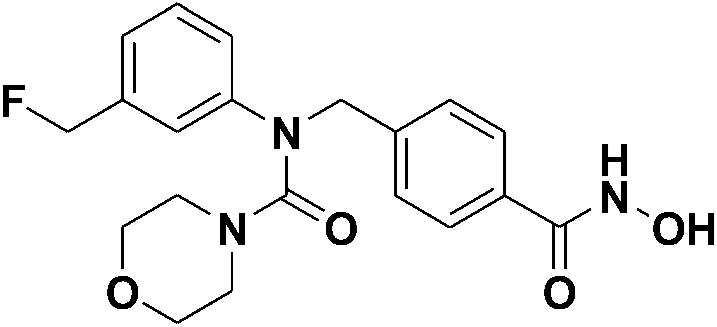
Метил 4-((N-(3-(фторметил)фенил)морфолин-4-карбоксамидо)метил)бензоат (0,100 г, 0,259 ммоль) растворяли в метаноле (10 мл), а затем добавляли гидроксиламин (50,0 масс.% водный раствор, 1,11 мл, 18,1 ммоль) при комнатной температуре. Затем к полученной смеси добавляли гидроксид калия (0,145 г, 2,59 ммоль) и перемешивали при этой же температуре в течение 30 минут. После этого из полученной реакционной смеси удаляли растворитель при пониженном давлении, затем к полученному концентрату добавляли насыщенный водный раствор гидрокарбоната натрия, а затем проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем сушили посредством безводного сульфата магния, а затем концентрировали при пониженном давлении. К полученному концентрату добавляли дихлорметан (5 мл) и гексан (30 мл) и перемешивали, а затем осажденное твердое вещество фильтровали и сушили с получением указанного в заголовке соединения (0,089 г, 89%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, DMSO-d6) δ 11,12 (уш. с, 1 H), 8,98 (уш. с, 1 H), 7,64 (д, 2 H, J=8,3 Гц), 7,36-7,32 (м, 3 H), 7,20 (с, 1 H), 7,15 (д, 1 H, J=7,5 Гц), 7,09 (д, 1 H, J=7,4 Гц), 5,36 (д, 2 H, J=47,5 Гц), 4,87 (с, 2 H), 3,39 (т, 4 H, J=4,6 Гц), 3,13 (т, 4 H, J=4,6 Гц). MS (ESI) m/z 388 (M+ + H).
Пример получения 8. Синтез соединения 530 {N-(3-фторфенил)-N-(4-(гидроксикарбамоил)бензил)морфолин-4-карбоксамид}
[Стадия 1] Синтез метил 4-((3-фторфениламино)метил)бензоата
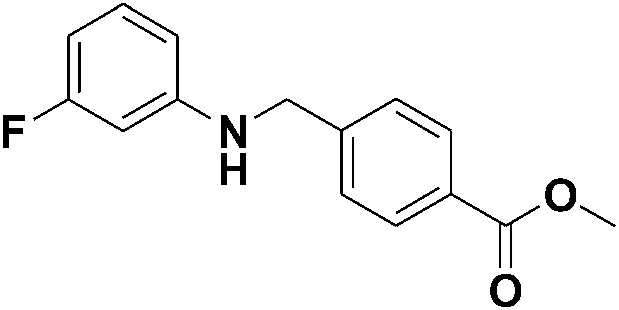
Метил 4-формилбензоат (1,47 г, 8,99 ммоль) растворяли в метаноле (50 мл), а затем добавляли 3-фторбензоламин (1,0 г, 8,99 ммоль). Полученную смесь подвергали реакции при комнатной температуре в течение трех часов, а затем добавляли цианоборгидрид натрия (NaCNBH3) (0,56 г, 8,99 ммоль) и уксусную кислоту (1,03 мл, 17,99 ммоль). После того, как компоненты реакции реагировали при комнатной температуре в течение суток растворитель с реагирующими компонентами помещали под пониженное давление и удаляли, затем в него наливали насыщенный водный раствор гидрокарбоната натрия, а затем проводили экстракцию этилацетатом. Органический слой сушили безводным сульфатом магния, а затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (диоксид кремния; этилацетат/гексан=20%) с получением указанного в заголовке соединения (1,84 г, 79%).
[Стадия 2] Синтез метил 4-(((3-фторфенил)((4-нитрофенокси)карбонил)амино)метил)бензоата
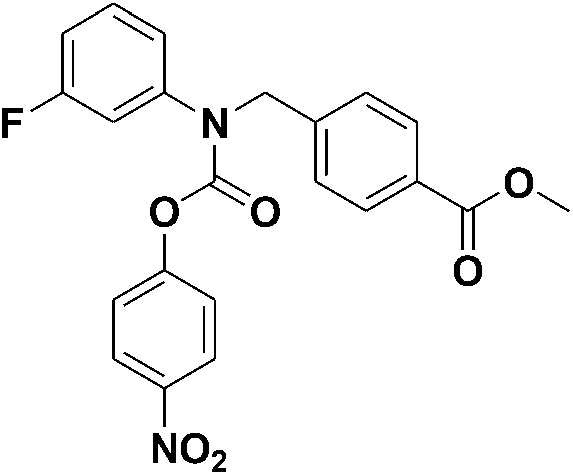
Метил 4-((3-фторфениламино)метил)бензоат (2,7 г, 10,4 ммоль) и 4-нитрофенилхлорформиат (4,20 г, 20,8 ммоль) растворяли в ацетонитриле (100 мл), а затем добавляли карбонат калия (4,32 г, 31,2 ммоль). Полученную смесь подвергали реакции при комнатной температуре в течение суток и разбавляли этилацетатом. Реакционную смесь промывали насыщенным водном раствором хлорида натрия, затем сушили безводным сульфатом натрия и фильтровали, а затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (диоксид кремния; этилацетат/гексан=20%) с получением указанного в заголовке соединения (2,65 г, 60%) в виде бесцветного масла.
[Стадия 3] Синтез метил 4-((N-(3-фторфенил)морфолин-4-карбоксамидо)метил)бензоата
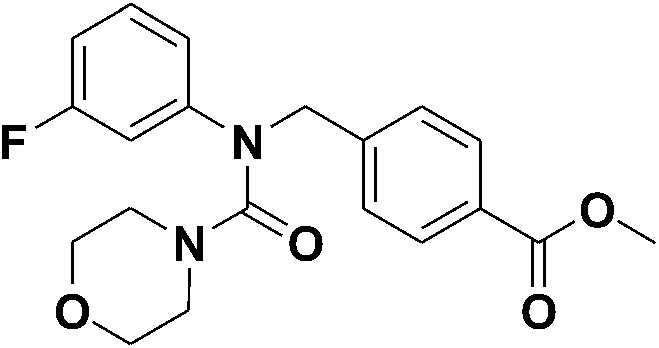
Метил 4-(((3-фторфенил)((4-нитрофенокси)карбонил)амино)метил)бензоат (0,32 г, 0,75 ммоль) растворяли в диметилформамиде (5 мл), а затем добавляли карбонат калия (0,31 г, 2,24 ммоль) и морфолин (0,13 мл, 1,49 ммоль). Полученную смесь подвергали реакции при 60°C в течение суток, а затем разбавляли насыщенным раствором хлорида аммония. Проводили экстракцию этилацетатом, а затем полученный экстракт сушили безводным сульфатом натрия и фильтровали, а затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (диоксид кремния; этилацетат/гексан=30%) с получением указанного в заголовке соединения (0,13 г, 45%).
[Стадия 4] Синтез N-(3-фторфенил)-N-(4-(гидроксикарбамоил)бензил)морфолин-4-карбоксамид

Метил 4-((N-(3-фторфенил)морфолин-4-карбоксамидо)метил)бензоат (0,108 г, 0,290 ммоль) растворяли в метаноле (10 мл), а затем добавляли гидроксиламин (50,0 масс.% водный раствор, 1,19 мл, 19,4 ммоль) при комнатной температуре. Затем к полученной смеси добавляли гидроксид калия (0,156 г, 2,78 ммоль) и перемешивали при той же температуре в течение 16 часов. После этого из полученной реакционной смеси удаляли растворитель при пониженном давлении, затем в полученный концентрат наливали насыщенный водный раствор гидрокарбоната натрия, а затем проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем сушили безводным сульфатом магния, а затем концентрировали при пониженном давлении. Осажденное твердое вещество фильтровали и сушили с получением указанного в заголовке соединения (0,062 г, 57%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, DMSO-d6) δ 11,14 (уш. с, 1 H), 8,99 (уш. с, 1 H), 7,65 (д, 2 H, J=7,0 Гц), 7,38-7,30 (м, 3H), 7,05-6,85 (м, 3H), 4,89 (с, 1H), 3,44-3,42 (м, 4H), 3,18-3,15 (м, 4H), 2,08 (с, 3H). MS (ESI) m/z 374 (M+ + H).
Пример получения 9. Синтез соединения 532 {N-(2-фтор-4-(гидроксикарбамоил)бензил)-N-(3-(трифторметил)фенил)морфолин-4-карбоксамид}
[Стадия 1] Синтез 3-фтор-4-(((3-(трифторметил)фенил)амино)метил)бензонитрила
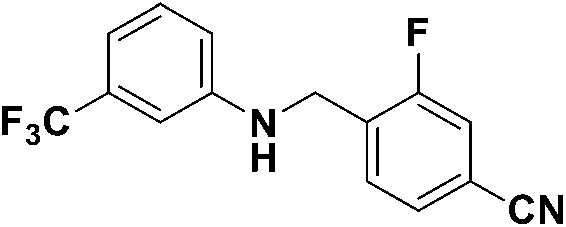
3-(трифторметил)анилин (0,998 мл, 8,068 ммоль) растворяли в ацетонитриле (60 мл), а затем добавляли 4-(бромметил)-3-фторбензонитрил (2,072 г, 9,682 ммоль) и DIPEA (2,143 мл, 12,102 ммоль) при комнатной температуре, а затем перемешивали при той же температуре в течение суток. После этого к полученной реакционной смеси добавляли насыщенный водный раствор гидрокарбоната натрия, а затем проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем сушили безводным сульфатом магния, а затем концентрировали при пониженном давлении. Полученный концентрат очищали колоночной хроматографией (диоксид кремния; этилацетат/гексан=5-20%) и концентрировали с получением указанного в заголовке соединения (2,380 г, 64,4%) в виде желтой жидкости.
[Стадия 2] Синтез 3-фтор-4-(((3-(трифторметил)фенил)амино)метил)бензойной кислоты
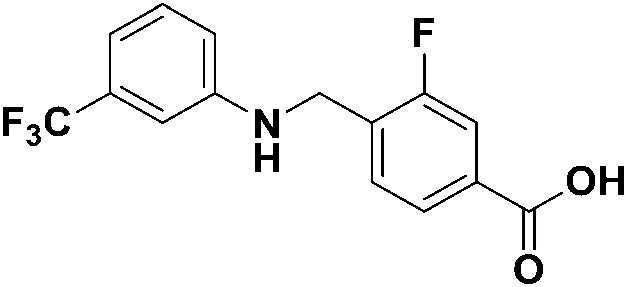
3-фтор-4-(((3-(трифторметил)фенил)амино)метил)бензонитрил (2,310 г, 7,850 ммоль) и гидроксид лития (3,294 г, 78,505 ммоль) перемешивали в метаноле (40 мл)/H2O (20 мл), затем полученную реакционную смесь нагревали при кипячении с обратным холодильником в течение 16 часов, затем охлаждали до комнатной температуры, а затем концентрировали при пониженном давлении. К полученной смеси добавляли 2 M водный раствор хлористоводородной кислоты для достижения pH=1, а затем осажденное твердое вещество отфильтровывали и сушили с получением указанного в заголовке соединения (1,700 г, 69,1%) в виде белого твердого вещества.
[Стадия 3] Синтез метил 3-фтор-4-(((3-(трифторметил)фенил)амино)метил)бензоата
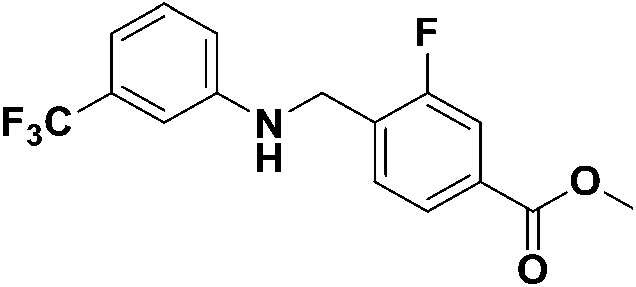
3-фтор-4-(((3-(трифторметил)фенил)амино)метил)бензойную кислоту (1,700 г, 5,427 ммоль), метанол (4,402 мл, 108,540 ммоль), EDC (2,081 г, 10,854 ммоль), HOBt (1,467 г, 10,854 ммоль) и DIPEA (2,883 мл, 16,281 ммоль) растворяли в тетрагидрофуране (50 мл) при комнатной температуре, затем полученный реакционный раствор перемешивали при той же температуре в течение 16 часов, затем в полученную реакционную смесь наливали насыщенный водный раствор гидрокарбоната натрия, а затем проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем сушили безводным сульфатом магния, а затем концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией (диоксид кремния; этилацетат/гексан=10-40%) и концентрировали с получением указанного в заголовке соединения (1,500 г, 84,5%) в виде бесцветной жидкости.
[Стадия 4] Синтез метил 3-фтор-4-((((4-нитрофенокси)карбонил)(3-(трифторметил)фенил)амино)метил)бензоата
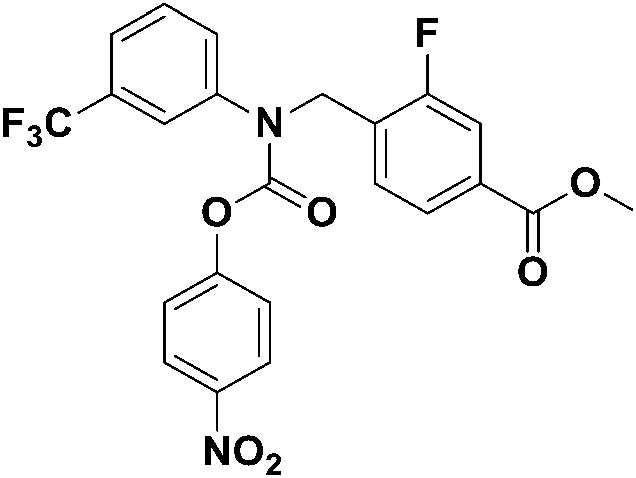
Метил 3-фтор-4-(((3-(трифторметил)фенил)амино)метил)бензоат (1,500 г, 4,583 ммоль), 4-нитрофенилкарбонохлоридат (1,848 г, 9,167 ммоль) и карбонат калия (1,900 г, 13,750 ммоль) растворяли в ацетонитриле (80 мл) при комнатной температуре, затем полученный реакционный раствор перемешивали при той же температуре в течение 16 часов, затем в полученную реакционную смесь наливали насыщенный водный раствор гидрокарбоната натрия, а затем проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем сушили безводным сульфатом магния, а затем концентрировали при пониженном давлении. Полученный концентрат очищали колоночной хроматографией (диоксид кремния; этилацетат/гексан=10-40%) и концентрировали с получением указанного в заголовке соединения (0,927 г, 41,1%) в виде бесцветной жидкости.
[Стадия 5] Синтез метил 3-фтор-4-((N-(3-(трифторметил)фенил)морфолин-4-карбоксамидо)метил)бензоата
Метил 3-фтор-4-((((4-нитрофенокси)карбонил)(3-(трифторметил)фенил)амино)метил)бензоат (0,129 г, 0,262 ммоль), морфолин (0,046 мл, 0,524 ммоль) и карбонат калия (0,109 г, 0,786 ммоль) растворяли в N, N-диметилформамиде (5 мл) при 60°С, затем полученный реакционный раствор перемешивали при той же температуре в течение двух суток, затем в полученную реакционную смесь наливали насыщенный водный раствор гидрокарбоната натрия, а затем проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем сушили безводным сульфатом магния, а затем концентрировали при пониженном давлении. Полученный концентрат очищали колоночной хроматографией (диоксид кремния; этилацетат/гексан=30-60%) и концентрировали с получением указанного в заголовке соединения (0,094 г, 81,5%) в виде бесцветной жидкости.
[Стадия 6] Синтез N-(2-фтор-4-(гидроксикарбамоил)бензил)-N-(3-(трифторметил)фенил)морфолин-4-карбоксамида
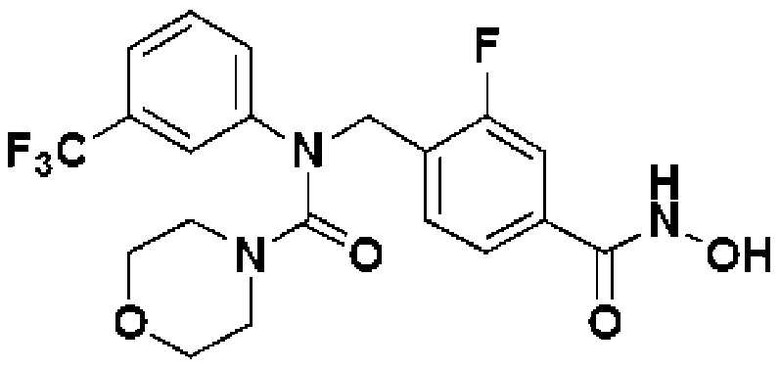
Метил 3-фтор-4-((N-(3-(трифторметил)фенил)морфолин-4-карбоксамидо)метил)бензоат (0,094 г, 0,213 ммоль) и гидроксиламин (50,0 масс.% водный раствор, 0,071 г, 2,134 ммоль) растворяли в метаноле (5 мл), затем добавляли гидроксид калия (0,060 г, 1,067 ммоль) при комнатной температуре, а затем перемешивали при той же температуре в течение двух часов, а затем полученную реакционную смесь концентрировали при пониженном давлении. В полученный концентрат добавляли диэтиловый эфир (10 мл) и перемешивали, а затем осажденное твердое вещество фильтровали и сушили с получением соединения 532 (0,068 г, 72,2%) в виде светло-желтого твердого вещества.
1H-ЯМР (400 МГц, DMSO-d6) δ 11,2 (уш. с, 1 H), 9,13 (уш. с, 1 H), 7,57-7,42 (м, 7 H), 4,94 (с, 2 H), 3,44-3,34 (м, 4 H), 3,18-3,12 (м, 4 H); MS (ESI) m/z 442,1 (M+ + H).
Пример получения 10. Получение модели SLE на мышах
Самок мышей 80 NZB/W F1 (в возрасте четырех недель) приобретали в Jackson Laboratory и содержали в помещении для разведения SPF количестве пяти мышей на клетку. Измерение их массы и уровня белка в моче проводили через 24 недели после рождения (непосредственно перед введением), а затем всех животных, за исключением животных, которые имели слишком много белка в моче, разделяли на группы, как показано в таблице 2 ниже, так чтобы уровень белка в моче и масса могли быть равномерно распределены между группами.
Таблица 2
Затем мышам проводили введение, как описано в таблице 2, один раз в сутки в течение приблизительно 19 недель с 24 недели после рождения по 42 неделю после рождения.
Свежую мочу мышей получали до и после введения каждые две недели, взятие лейкоцитов проводили до и после введения каждые четыре недели. Сыворотку получали центрифугированием взятой крови, а затем держали при -70°С.
Пример 1. Идентификация супрессивного эффекта на секрецию TNFα в иммунных клеточных линиях (in vitro)
Для идентификации эффективности соединения по изобретению в отношении подавления секреции TNFα при иммунном ответе, подавление продукции TNFα, которое достигали путем обработки соединениями 374, 461, 500, 530 и 532 в соответствии с настоящим изобретением стимулированных LPS клеточных линий моноцитов человека (THP-1), количественно определяли посредством иммуноферментного анализа (ELISA).
В частности, клеточные линии THP-1 (ATCC) культивировали в среде RPMI-1640, содержавшей 10% FBS. Клеточные линии распределяли в 24-луночный планшет в количестве 1×105 клеток на лунку, затем обрабатывали 100 нг/мл PMA (форбол 12-миристат 13-ацетат) в течение 24 часов, а затем подвергали дифференцировке в макрофаги. Затем культуральную среду заменяли новой средой, затем обрабатывали исследуемым лекарственным средством в течение 24 часов, а затем вновь обрабатывали 10 нг/мл LPS (E.Coli, O55:B5) в течение четырех часов для стимуляции. После этого супернатант отбирали и использовали для определения количества TNFα, секретированного из клеток, с использованием набора Human TNFα Instant ELISA kit (eBioscience, BMS223INST) в соответствии с протоколом, предоставленным изготовителем.
В результате, было показано, что уровень секреции TNFα снижался во всех экспериментальных группах по сравнению с контрольной группой, в которой воспалительный ответ индуцировали посредством LPS. На фиг.1 соединения, обозначаемые как SM374, SM461, SM500, SM530 и SM532, представляют собой соединения 374, 461, 500, 530 и 532, соответственно. В частности, в случае соединений 374, 461 и 500, уровень секреции TNFα был значительно снижен до уровня, при котором воспалительный ответ не индуцировался посредством LPS в концентрациях как 100 нМ, так и 300 нМ. Также в случае увеличения концентрации соединения при обработке от 100 нМ до 300 нМ для соединений 530 и 532, уровень секреции TNFα значительно снижался (фиг.1).
Результаты эксперимента показывают, что соединение в соответствии с настоящим изобретением очень эффективно подавляет секрецию TNFα, т.е. фактора воспалительного ответа, уровень которого репрезентативно возрастает при волчанке, таким образом, таким образом, эффективно подавляя воспалительные ответы, вызываемые волчанкой.
Пример 2. Идентификация супрессивного эффекта на пролиферацию реактивных T-клеток (in vitro)
Для идентификации эффективности соединений по изобретению в отношении подавления пролиферации реактивных T-клеток при иммунном ответе, соединения 255, 280, 374, 416 и 476 в соответствии с настоящим изобретением культивировали вместе с реактивными T-клетками и регуляторными T-клетками в стимулированных LPS клеточных линиях моноцитов человека (THP-1), а затем определяли эффективность супрессии регуляторных T-клеток.
В частности, самцов мышей C57BL6 в возрасте шести недель приобретали в Central Lab Animal Inc., затем позволяли им акклиматизироваться в течение одной недели, а затем использовали в эксперименте. Селезенку извлекали из мышей, а затем обрабатывали коллагеназой D (Roche, 11088866001), чтобы выделить из нее спленоциты. Treg (CD4+CD25-) и Teff (CD4+CD25+) выделяли с использованием набора для выделения CD4+CD25+ регуляторных T-клеток (Miltenyi Biotec, 130-091-041) в соответствии с протоколом, предоставляемым изготовителем. Клетки Teff культивировали при 37°С в течение десяти минут посредством eFluor®670 (Cell Proliferation Dye eFluor®670, eBioscience), чтобы окрасились клеточные мембраны. Teff и Treg распределяли в 96-луночный планшет в соотношении 2:1, а затем T-клетки активировали в течение трех суток посредством магнитных гранул с mAb против CD3ε и против CD28 (набор для активации/экспансии T-клеток, Miltenyi Biotec, 130-093627), и проводили анализ подавления Treg. Одновременно проводили обработку тестируемым лекарственным средством в течение трех суток, в ходе которых проводили анализ. Величину разведения eFluor®670, которым были мечены клеточные мембраны Teff, определяли, таким образом, оценивая степень пролиферации T-клеток. График разведения eFluor®670 строили с использованием проточного цитометра (FACS LSR Fortessa, BD bioscience). Способность к подавлению пролиферации T-клеток вычисляли с использованием следующего уравнения.
Относительное подавление = 
В результате было показано, что пролиферация реактивных T-клеток подавлялась во всех экспериментальных группах. На фиг.2 соединения, обозначаемые как SM255, SM280, SM374, SM416, SM476, представляют собой соединения 255, 280, 374, 416, 476, соответственно. Соединения в соответствии с настоящим изобретением, использованные в эксперименте, продемонстрировали уровень подавления пролиферации реактивных T-клеток, который был более высоким максимум в два раза, когда проводили обработку в концентрации 200 нМ, а также продемонстрировал выраженный эффект подавления пролиферации T-клеток, который максимально достигал четырех раз, при обработке в концентрации 500 нМ (фиг.2).
Результаты эксперимента, приведенные выше, демонстрируют, что соединения в соответствии с настоящим изобретением эффективно подавляют дифференцировку реактивных T-клеток, которые чрезмерно активировались при волчанке.
Пример 3. Идентификация регуляторного эффекта функции регуляторных T-клеток (in vitro)
Для идентификации того, регулируют ли соединения в соответствии с настоящим изобретением функцию регуляторных T-клеток при иммунном ответе, проводили обработку соединениями 255, 280, 374, 416 и 476, а затем определяли уровень экспрессии рецептора иммунной точки контроля CTLA4 (ассоциированный с цитотоксическими T-лимфоцитами белок 4) в регуляторных T-клетках посредством проточной цитометрии.
В частности, самцов мышей C57BL6 в возрасте шести недель получали в Central Lab Animal Inc., затем позволяли им акклиматизироваться в течение одной недели, а затем использовали в эксперименте. Селезенку извлекали из мыши, а затем обрабатывали коллагеназой D (Roche, 11088866001), выделяя из нее спленоциты. CD4+CD25- T-клетки выделяли с использованием набора для выделения CD4+CD25+ регуляторных T-клеток (Miltenyi Biotec, 130-091-041), а затем CD4+CD25- T-клетки (в количестве 5×105 клеток/лунка) обрабатывали гранулами с mAb против CD3ε/против CD28 (набор для активации/экспансии T-клеток, Miltenyi Biotec, 130-093627) и рекомбинантного TGF-β2 мыши в течение шести суток, так что они дифференцировались в iTreg. Одновременно проводили обработку тестируемым лекарственным средством в течение шести суток, в ходе которой клетки дифференцировались в iTreg. После этого клетки инкубировали с means mAb против CD4/против CD25 (eBioscience, 25-0042-82, 17-0251-82) при 4°С в течение 20 минут, а затем проводили мечение. Для внутрицитоплазматического окрашивания пермеабилизацию обеспечивали с использованием Fix/буфера для пермеабилизации (eBioscience, 00-5523-00), затем проводили мечение посредством антитела против FOXP3-Alexafluor488 (eBioscience, 53-5773-82) и антитела против CTLA4-PE (eBioscience, 12-1522-82), а затем проводили проточную цитометрию посредством FACS LSR Fortessa (BD bioscience).
В результате, было идентифицировано, что уровень экспрессии CTLA4 в T-клетках возрастал после обработки соединениями в соответствии с настоящим изобретением. На фиг.3., соединения, обозначаемые как SM255, SM280, SM374, SM416, SM476, представляют собой соединения 255, 280, 374, 416, 476, соответственно. В частности, в случае соединений 255, 374 и 476, было показано, что экспрессия CTLA4 в 40% или более T-клеток возрастала при концентрации 500 нМ или более. Соединение 255 продемонстрировало высокую цитотоксичность при обработке в концентрации 1000 нМ, так что анализ данных не проводили (фиг.3).
Результаты эксперимента, приведенные выше, демонстрируют, что соединения в соответствии с настоящим изобретением улучшают функцию регуляторных T-клеток, так что можно эффективно регулировать чрезмерную активность реактивных T-клеток при волчанке.
Пример 4. Эффект восстановления массы в модели на животных
В результате идентификации изменения массы, масса продемонстрировала общий паттерн снижения после введения в течение первых четырех недель (предположительно, что это действие было вызвано растворителем, состоящим из этанола+Kolliphor+солевой раствор), однако более выраженный эффект восстановления массы происходил в группе положительного контроля, чем в группе отрицательного контроля в возрасте после 36 недель. В случае экспериментальных групп, масса в группе, в которой дозировали 10 мг/кг, восстанавливалась до того же уровня, что и в группе положительного контроля в возрасте приблизительно 40 недель, в то время как еще лучший эффект восстановления массы был показан для групп, в которых проводили дозирование 30 и 50 мг/кг, чем в группе положительного контроля (фиг.4).
Пример 5. Эффект повышения выживаемости в модели на животных
В качестве модели заболевания SLE человека используют самок мышей NZB/W F1. Известно, что без лечения мыши в этой модели погибают в возрасте после 12 месяцев или около того (возраст приблизительно 52 недели) вследствие иммунокомплексного гломерулонефрита.
Для идентификации, того, демонстрирует ли фармацевтическая композиция в соответствии с настоящим изобретением эффект повышения выживаемости в модели SLE на животных, проводили определение выживаемости самок мышей NZB/W F1 каждые сутки в течение всего экспериментального периода от 24 недель после рождения до 42 недель после рождения.
В результате отсутствовали изменения выживаемости во всех группах до 32 недель или около того после рождения. В случае группы отрицательного контроля без какой-либо обработки лекарственным средством, выживаемость мышей значительно снижалась после 35 недель после рождения, так что 7 из 15 мышей погибли в возрасте 42 недель после рождения (выживаемость приблизительно 53,3%). Напротив, как группа положительного контроля, так и экспериментальная группа, продемонстрировала тенденцию к лучшему профилю выживаемости, чем группа отрицательного контроля. В частности, 4 из 15 мышей (выживаемость приблизительно 73,3%) погибли через 42 недели после рождения в группе дозирования 10 мг/кг, и неожиданно только одна мышь (выживаемость приблизительно 93,3%) погибла в группах дозирования 30 мг/кг и 50 мг/кг, таким образом, было идентифицировано, что фармацевтическая композиция в соответствии с настоящим изобретением продемонстрировала превосходный эффект повышения выживаемости в модели на животных с SLE (фиг.5).
Пример 6. Эффект снижения протеинурии в модели на животных
Для идентификации того, демонстрирует ли фармацевтическая композиция в соответствии с настоящим изобретением терапевтический эффект в отношении протеинурии, т.е. репрезентативного симптома заболевания SLE, проводили взятие мочи мышей каждые две недели, а затем определяли соотношение UP/C (соотношение белок в моче:креатин) способом окрашивания кумасси бриллиантовым синим (CBB). Концентрацию креатинина в моче получали путем разбавления мочи в 100 раз, затем определения его концентрации с использованием анализатора для биохимического анализа крови (колориметрический анализатор Dri-CHEM 3000, Fujifilm), а затем калибровки измеренной величины с использованием коэффициента разведения.
В результате, было идентифицировано, что встречаемость тяжелой протеинурии 300 мг/дл или более значительно возрастала при старении от 24 до 42 недель после рождения в группе отрицательного контроля, в то время как проблема с такой встречаемостью тяжелой протеинурии значительно улучшалось в экспериментальных группах. В частности, встречаемость тяжелой протеинурии 300 мг/дл или более в возрасте 42 недель составляла 80% в группе отрицательного контроля, в то время как такая встречаемость составляла 20% в группе положительного контроля. Кроме того, она также составляла 60%, 6,7% и 0% в группах дозирования 10, 30 и 50 мг/кг, соответственно, что говорило о превосходном эффекте снижения протеинурии (фиг.6).
Между тем, UP/C мышей в возрасте 36 недель значительно снижалось во всех экспериментальных группах. Даже в случае мышей в возрасте 42 недель, было выявлено, что величина UP/C была более сниженной во всех экспериментальных группах за исключением группы дозирования 10 мг/кг, чем в группе отрицательного контроля (фиг.7).
Пример 7. Эффект снижения сывороточной концентрации антител против дцДНК в модели на животных
Для идентификации того, может ли фармацевтическая композиция в соответствии с настоящим изобретением снижать увеличенную концентрацию антител против дцДНК, показанную в модели на животных с SLE, проводили измерение концентрации антител против дцДНК в сыворотке мышей посредством набора для ELISA (твердофазный иммуноферментный анализ) для антител против дцДНК (Shibayagi).
В результате, сывороточная концентрация антител против дцДНК после 28 недели после рождения имела тенденцию к большему снижению во всех экспериментальных группах, чем в группе отрицательного контроля. В частности, в случае дозирования 50 мг/кг, было выявлено, что сывороточная концентрация антител против дцДНК в большей степени снижалась максимум до двукратного уровня, чем в группе отрицательного контроля (фиг.8).
Пример 8. Эффект снижения BUN и концентраций сывороточного креатинина в модели на животных
Для оценки функции почек в модели SLE на мышах проводили определение BUN и концентраций креатинина в сыворотке мыши, которую собирали каждый месяц (в возрасте 24, 28, 32, 36 и 40 недель и в ходе аутопсии) посредством колориметрического анализатора Dri-CHEM 3000 (Fuji film).
В результате, было идентифицировано, что BUN и сывороточные концентрации креатинина были сниженными в экспериментальных группах по сравнению с контрольными группами, в частности, также было идентифицировано, что существовал превосходный эффект снижения BUN и концентраций креатинина в сыворотке в возрасте 40 недель (фиг.9).
Пример 9. Гистологическое изменение почек в модели на животных
Для идентификации эффекта фармацевтической композиции по изобретению на почки в модели заболевания SLE проводили гистопатологическую оценку.
Сначала половину левой почки мыши, извлеченную в ходе аутопсии, фиксировали 10% нейтральным забуференным формалином и приготавливали в виде парафинового блока. Затем проводили гистопатологическую оценку путем окрашивания гематоксилином и эозином (H&E, BBC Biochemicals, Mount Vernon, WA, США), окрашивание Шифф-йодной кислотой (PAS, BBC Biochemicals) и окрашиванием трихромом по Массону (BBC Biochemicals).
В результате, наблюдали данные, касающиеся инфильтрации ряда воспалительных клеток и пролиферации мезангия, в группе отрицательного контроля. В ходе окрашивания PAS наблюдали множество расширенных канальцев и цилиндров в канальцах. В ходе окрашивания трихромом по Массону паттерн фиброза был более выраженным (фиг.10).
Затем, после присвоения обозначений 0-4 (0: отсутствие инфильтрации, 4: тяжелая инфильтрация) в зависимости от степени инфильтрации воспалительных клеток, степень инфильтрации воспалительных клеток была снижена в почках мышей экспериментальной группы, в частности, было выявлено, что существовал превосходный эффект снижения инфильтрации воспалительных клеток в группах дозирования 30 и 50 мг/кг (фиг.11).
Пример 10. Идентификация эффекта снижения отложений IgG и C3 в почках в модели на животных
Для идентификации терапевтического эффекта фармацевтической композиции по изобретению на почки в модели заболевания SLE проводили определение степени отложений IgG и C3 в гломерулах.
Что касается другой половины левой почки, полученной в ходе аутопсии, проводимой согласно примеру 9, получали замороженный блок ткани для получения срезов замороженной ткани посредством реагента OCT, а затем проводили флуоресцентное иммунное окрашивание на IgG и C3 с использованием способа, известного из уровня техники, к которой относится настоящее изобретение.
В частности, получали срез замороженной ткани толщиной 4 мкм, затем ее фиксировали в холодном ацетоне в течение пяти минут, а затем промывали фосфатно-солевым буфером (PBS) два раза в течение пяти минут. Чтобы предотвратить неспецифическую реакцию, полученную смесь блокировали раствором PBS с добавлением 1% бычьего сывороточного альбумина и 0,05% tween-20 в течение 30 минут, а затем подвергали реакции при комнатной температуре в течение одного часа с использованием конъюгированных с FITC антител против IgG мыши (1:200, AP308F, Merck Millipore) или конъюгированных с FITC антител козы против C3 мыши (1:100, Cappel 55500, MP Bio). После этого полученную ткань промывали PBS в течение пяти минут три раза, затем накрывали покровным стеклом путем заливки средой, включавшей DAPI, а затем наблюдали посредством конфокальной лазерной сканирующей микроскопии (LSM 700, ZEISS).
В результате уровень отложений IgG и C3 был сниженным в экспериментальных группах, в частности, было выявлено, что уровень таких отложений был значительно снижен в группе, в которой дозировали 50 мг/кг (фиг.12).
Пример 11. Сывороточная концентрация цитокинов в модели на животных
Для анализа эффекта фармацевтической композиции по изобретению на изменение сывороточного уровня цитокинов у мышей с заболеванием SLE, определяли уровень GM-CSF, IFN-γ, IL-1α, IL-1β, IL-10, IL-12(p70), IL-15, IL-17a, IL-2, IL-4, IL-6, TNF-α, TGF-β, IL-22 и IL-23 в сыворотке с использованием набора Mouse Cytokine Milliplex MAP kit (Merck Millipore), и его результаты представлены в таблице 3.
Таблица 3
Данные, полученные для групп, сравнивали с использованием критерия Крускала-Уоллиса (†p < 0,05) *p < 0,05 против группы C (U-критерий Манна-Уитни).
Как представлено в таблице 3 выше, было выявлено, что IL-10, IL-12, IL-15, IL-17 и TNF-α имели тенденцию к снижению в группе положительного контроля по сравнению с группой отрицательного контроля, и сывороточная концентрация IL-10, IL-12, IL-15, IL-17, TNF-α и IL-22 была снижена у мышей в экспериментальных группах, в то время как уровень TGF-β возрастал. В частности, было выявлено, что уровни IL-10, IL-15, IL-17A, TNF-α и IL-22 были на значимом уровне более низкими и уровень TGF-β был на значимом уровне более высоким в экспериментальной группе, в которой вводили 50 мг/кг, по сравнению с контрольной группой (фиг.13).
В связи с этим, было описано, что сывороточная концентрация IL-6, IL-10, IL-17, IL-23 и IFN-γ была значительно более высокой, и сывороточная концентрация TGF-β была значительно более низкой у пациентов с SLE, чем у здоровых людей (Document [Zickert A et al., 2015]).
Между тем, когда измеряли уровень CXCL10 (IFN-индуцибельный белок 10, IP-10) и CCL2 (MCP-1) в сыворотке определяли в возрасте 36 недель, было идентифицировано, что уровень CXCL10 в сыворотке был снижен во всех экспериментальных группах, и уровень CCL2 в сыворотке был снижен в группе, в которой вводили 50 мг/кг (фиг.14).
Пример 12. Популяции T-клеток в селезенке в модели на животных
Для оценки паттерна популяций иммунных T-клеток в селезенке у мышей с заболеванием SLE, определяли соотношение экспрессии CD3, CD4, CD8a и CTLA4, а также соотношение Treg/Th1, Th2, Th17 и профили экспрессии основных регуляторов Treg (T-bet, GATA-3, ROR-γt, Foxp3).
Для этого сначала получали спленоциты из селезенки в ходе аутопсии мышей посредством клеточного сита с размером ячеек 70 мкм (BD), а затем эритроциты подвергали гемолизу посредством буфера EL и промывали буферным раствором для FACS (5% BSA/PBS). После этого проводили окрашивание в концентрации, рекомендованной изготовителем, посредством конъюгированного с PerCP-cy5.5 антитела против CD3 мыши (ebioscience, Sandiego, CA, США), конъюгированного с PE-Cyanine7 антитела против CD8a мыши (ebioscience), конъюгированного с FITC антитела против CD4 (BD), конъюгированного с APC антитела против CD25 мыши (BD), конъюгированного с PE антитела против FoxP3 мыши (BD), конъюгированного с PE антитела против RORγt мыши (ebioscience), конъюгированного с PE антитела против T-bet мыши (ebioscience), конъюгированного с PE антитела против GATA-3 мыши (ebioscience) и конъюгированного с PE антитела против CTLA4 мыши (ebioscience).
Спленоциты и маркерные антитела клеточной поверхности сразу подвергали реакции. Перед реакцией с антителами против FoxP3, RORγt, T-bet и GATA3, спленоциты фиксировали посредством набора буферов для окрашивания на FoxP3/факторы транскрипции, затем подвергали пермеабилизации, а затем подвергали реакции с антителами. Соответственно определяли соотношение CD4:CD8 в CD3-клетках, а также долю CD4+CD8-клеток, CD4-CD8+клеток, CD4+CD8+клеток и двойных негативных T-клеток и сравнивали между группами. Для определения доли FoxP3, ROR γt, T-bet и GATA3, которые были основными регуляторами Treg, Th17, Th1 и Th2, соответственно определяли долю CD4+CD25+FoxP3+ клеток, CD4+CD25+RORγt+ клеток, CD4+CD25+ T-bet+ клеток и CD4+CD25+ GATA3+ клеток.
Анализ профилей экспрессии CD4, CD8
Было описано, что доля двойных отрицательных CD4-CD8- T-клеток возрастала в моноцитах периферической крови пациентов с SLE, и что такие клетки продуцировали воспалительные цитокины IL-17 и IFN-γ, таким образом внося вклад в патогенез повреждений почек у пациентов с SLE (документ [Shivakumar S et al., 1989]; документ [Crispin JC et al., 2008]).
В результате гейтирования CD3+ клеток и определения экспрессии CD4 и CD8, доля CD4-CD8+ T-клеток возрастала в экспериментальных группах, однако доля CD4-CD8- T-клеток, напротив, снижалась. Было показано, что соотношение CD4+CD8- : CD4-CD8+ T-клеток имело тенденцию к снижению по мере возрастания дозы в экспериментальных группах (фиг.15).
Анализ паттернов экспрессии основных регуляторов Treg, Th17, Th1 и Th2
Когда долю CD4+CD25+Foxp3+, CD4+CD25+RORγt+, CD4+CD25+T-bet+ и CD4+CD25+GATA-3+ клеток в селезенке сравнивали и анализировали для определения степени экспрессии Foxp3, ROR-γt, T-bet и GATA-3, которые были основными регуляторами Treg, Th17, Th1 и Th2, еще не наблюдали значимых отличий между группами (фиг.16).
Между тем, доля CD25+ клеток была значимо более низкой и соотношение CD4+CD25+ клетки/CD4+ клетки также было значимо более низким в экспериментальной группе, в которой дозировали 50 мг/кг, по сравнению с контрольной группой (фиг.17).
В результате гейтирования в CD4+ клетках и исследования экспрессии T-bet, GATA-3, Foxp3 и RORγt, экспрессия T-bet была значимо более низкой в экспериментальной группе, в которой вводили 50 мг/кг, чем в контрольной группе. Более того, было выявлено, что соотношение CD4+T-bet+/CD4+GATA-3+ (соотношение экспрессии основных регуляторов Th1/Th2 в CD4-клетках) на более значимом уровне снижалось в экспериментальных группах, в которых вводили 30 мг/кг и 50 мг/кг, чем в контрольной группе (фиг.18).
Анализ паттерна экспрессии CD138
Было идентифицировано, что доля CD138+ клеток снижалась на 16,3% в группе положительного контроля по сравнению с группой отрицательного контроля, а также снижалась на 21,2% в группе, в которой вводили 10 мг/кг, на 25,2% в группе, в которой вводили 30 мг/кг, и на 31,6% в группе, в которой вводили 50 мг/кг, соответственно (фиг.19).
Пример 13. Доля пре-B-клеток и про-B-клеток в костном мозге в модели на животных
Долю про-B (B220+, CD43+) и пре-B (B220+, CD43-) в клетках костного мозга мыши определяли посредством FACS. Для этого клетки костного мозга получали путем промывания бедренной кости PBS, содержавшим 5% BSA. После этого проводили гемолиз эритроцитов посредством буфера EL, затем промывали буфером FACS (5% BSA/PBS), а затем окрашивали конъюгированными с FITC антителами против CD43 мыши (ebioscience) и конъюгированными с PE антителами против CD220 мыши (ebioscience), для определения доли пре-B (B220+CD43-) и про-B (B220+CD43+) клеток.
В результате, еще не наблюдали значимого изменения доли пре-B и про-B-клеток в экспериментальных группах по сравнению с контрольной группой (фиг.20).
Пример 14. Изменение изотипов иммуноглобулинов в сыворотке в модели на животных
Концентрацию изотипов иммуноглобулинов (IgG1, IgG2a, IgG2b, IgG3 и IgM) в сыворотке, полученной посредством аутопсии мышей, определяли с использованием набора для анализа Luminex изотипов антител мыши (R&D systems, Minneapolis, MN).
В результате, концентрация IgG1, IgG2a, IgG2b, IgG3 и IgM снижалась в группе, в которой проводили дозирование 50 мг/кг, по сравнению с группой отрицательного контроля, в частности, было выявлено, что концентрация IgM была значительно снижена (фиг.21).
Пример 15. Изменение биохимического анализа сыворотки в модели на животных
Для идентификации того, происходит ли какое-либо изменение биохимического анализа сыворотки при длительном введении лекарственного введения в модели SLE на животных, проводили определение концентрации ALT, AST, ALP, общего белка, альбумина, общего холестерина, TG, CPK, общего билирубина и Ca в сыворотке, полученной в ходе аутопсии, с использованием колориметрического анализатора Dri-CHEM 3000 (Fujifilm), где его результаты представлены в таблице 4.
Таблица 4
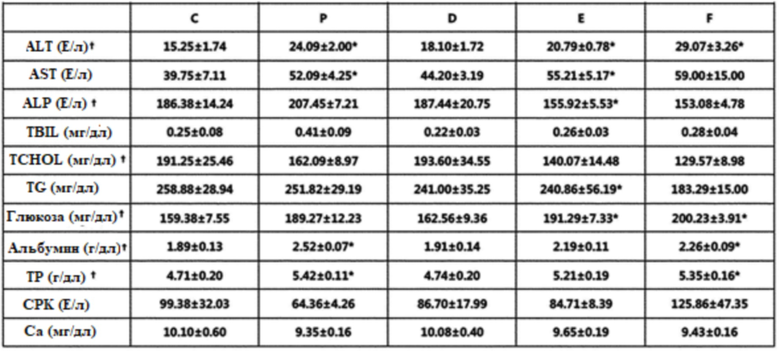
Данные, полученные для групп, сравнивали с использованием критерия Крускала-Уоллиса († p < 0,05), *p < 0,05 против группы C (U-критерий Манна-Уитни), ALT: аланинаминотрансфераза, AST: аспартатаминотрансфераза, ALP: щелочная фосфатаза, TBIL: общий билирубин, TCHOL: общий холестерин, TG: триглицерид, TP: общий белок, CPK: креатинфосфокиназа)
Как показано в описанных выше вариантах осуществления, было выявлено, что фармацевтическая композиция в соответствии с настоящим изобретением повышала выживаемость при заболевании SLE у мышей (фиг.5), снижала встречаемость протеинурии (фиг.6), концентрацию антител против дцДНК (фиг.8) и уровень инфильтрации IgG и C3 в почках (фиг.12), снижала уровень сывороточных цитокинов IL-10, IL-12, IL-15, IL-17a, TNFα и IL-22 (фиг.13) и уровень CXCL10 и CCL2 (фиг.14), увеличивала уровень TGF-β (фиг.13), и снижала долю CD4-CD8- двойных негативных T-клеток, соотношение уровня CD4+CD8- клеток и уровня CD4-CD8+ клеток (фиг.15), соотношение CD4+T-bet+/CD4+GATA3+ (фиг.16), соотношение уровня CD4+CD25+ клеток и уровня CD4+ клеток (фиг.17) и уровня CD138+ клеток (фиг.19). Исходя из приведенных выше результатов, можно видеть, что фармацевтическая композиция в соответствии с настоящим изобретением имеет превосходный эффект в отношении лечения волчанки, включая SLE.
В то время как конкретные части настоящего изобретения подробно описаны выше, специалистам в данной области понятно, что такое подробное описание приведено только для иллюстрации предпочтительных иллюстративных вариантов осуществления, и его не следует истолковывать как ограничивающее объем настоящего изобретения. Таким образом, следует понимать, что основной объем настоящего изобретения определяется прилагаемой формулой изобретения и ее эквивалентами.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИИ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ УВЕИТА | 2019 |
|
RU2757273C1 |
| КОМПОЗИЦИИ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ ХРОНИЧЕСКИХ ОБСТРУКТИВНЫХ ЗАБОЛЕВАНИЙ ЛЕГКИХ (COPD) | 2020 |
|
RU2798874C1 |
| НОВЫЕ СОЕДИНЕНИЯ ДЛЯ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ГИСТОНДЕАЦЕТИЛАЗЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ ТАКИЕ СОЕДИНЕНИЯ | 2014 |
|
RU2634694C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ ИНГИБИТОР ГИСТОНДЕАЦЕТИЛАЗЫ И МЕТОТРЕКСАТ | 2019 |
|
RU2772018C1 |
| КОМПОЗИЦИИ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ СУХОСТИ ГЛАЗ | 2019 |
|
RU2763423C1 |
| СОЕДИНЕНИЯ ЗАМЕЩЕННОЙ ТРИАЗОЛБОРОНОВОЙ КИСЛОТЫ | 2013 |
|
RU2625801C2 |
| НОВЫЕ ИНГИБИТОРЫ S-НИТРОЗОГЛУТАТИОНРЕДУКТАЗЫ | 2011 |
|
RU2585763C2 |
| РАСТВОРИМЫЕ С5аR АНТАГОНИСТЫ | 2017 |
|
RU2748260C2 |
| ИНГИБИТОРЫ БРОМОДОМЕНА | 2017 |
|
RU2741808C2 |
| АНТАГОНИСТЫ ЕР4 | 2016 |
|
RU2761341C2 |
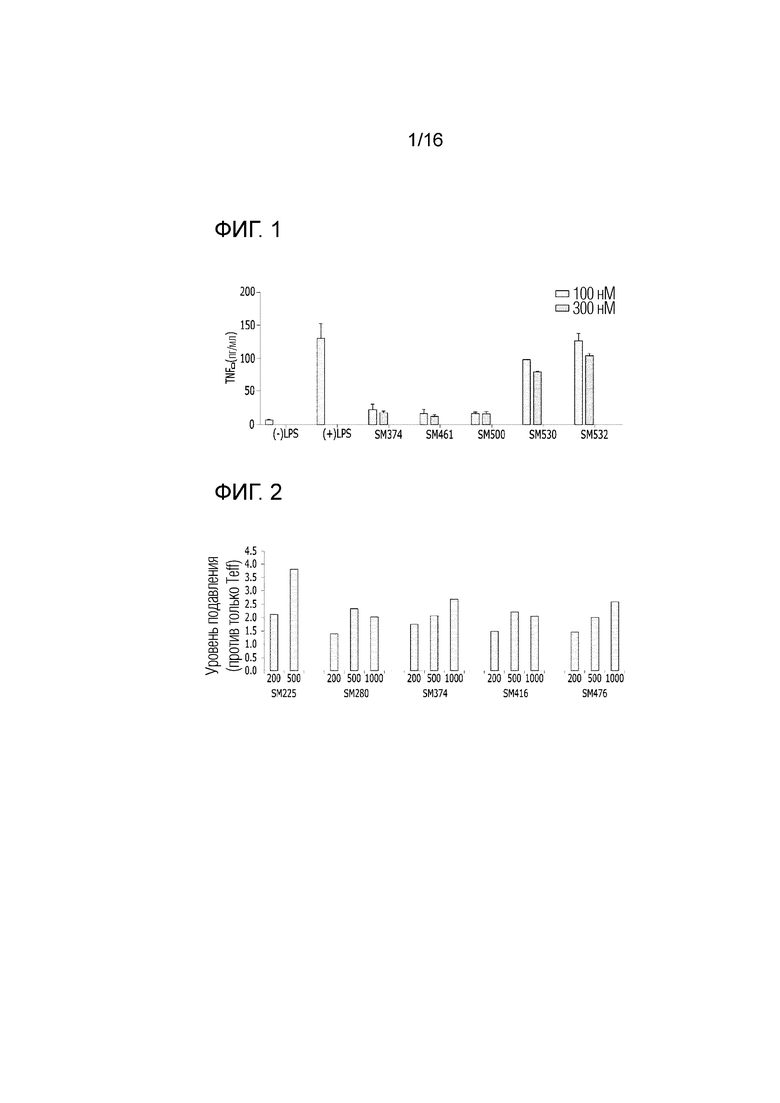


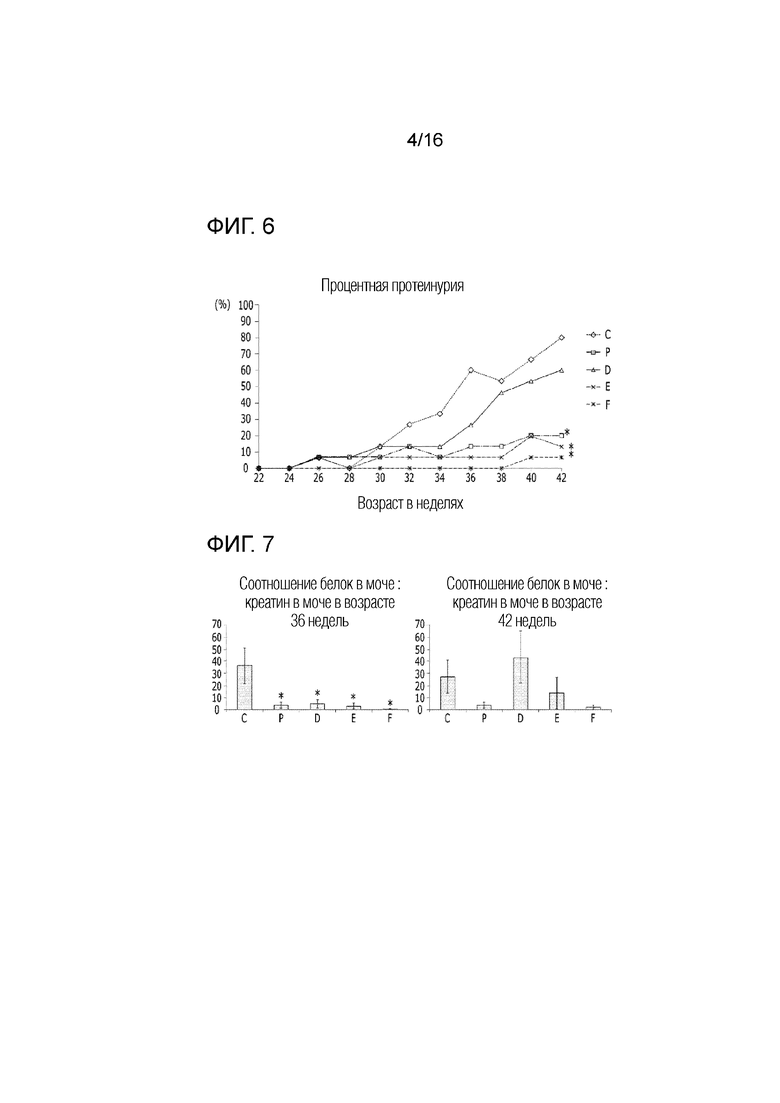
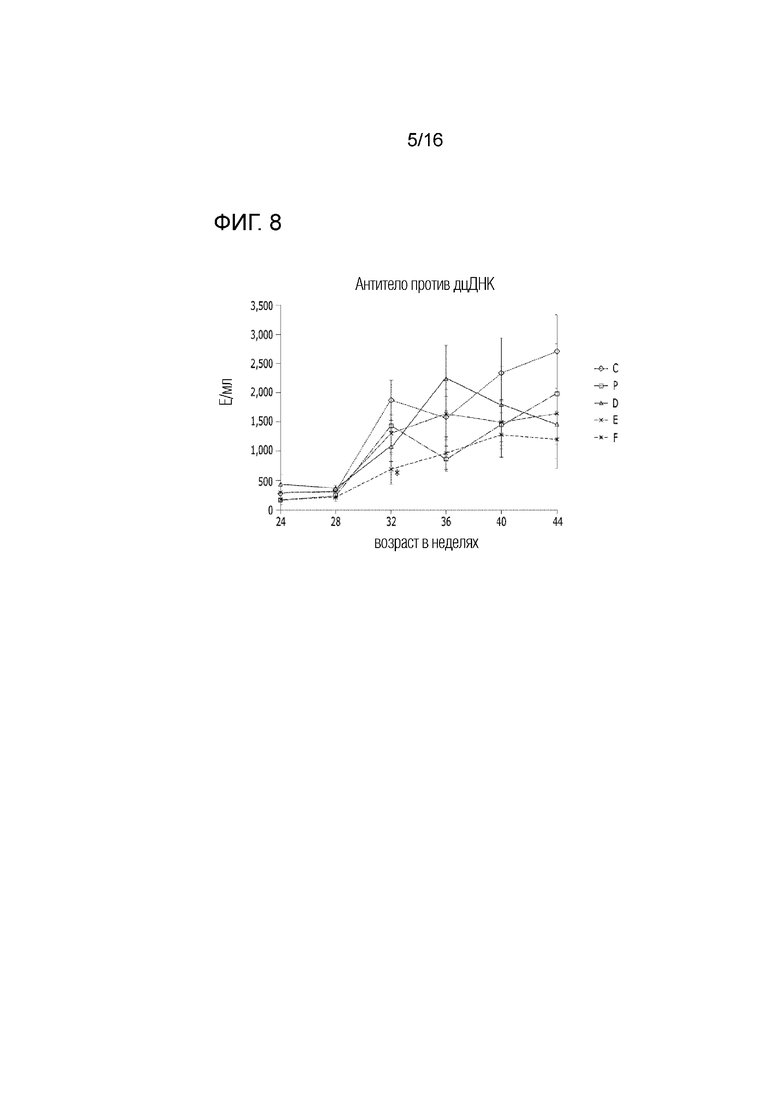
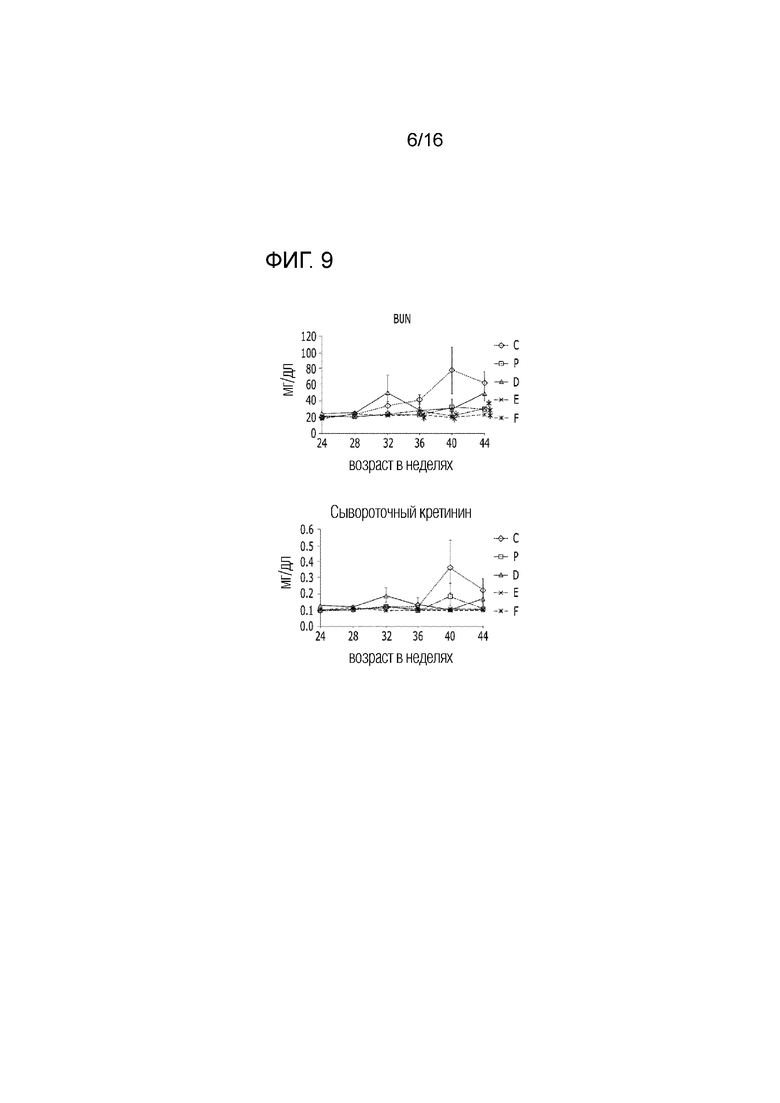

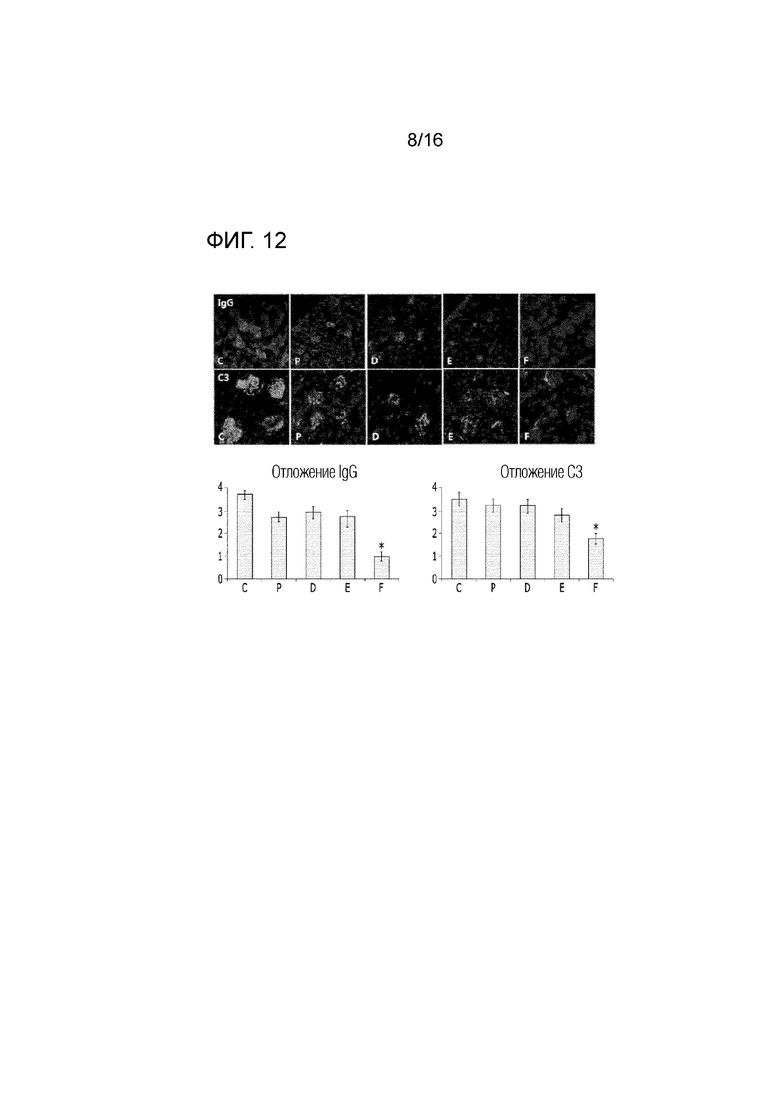

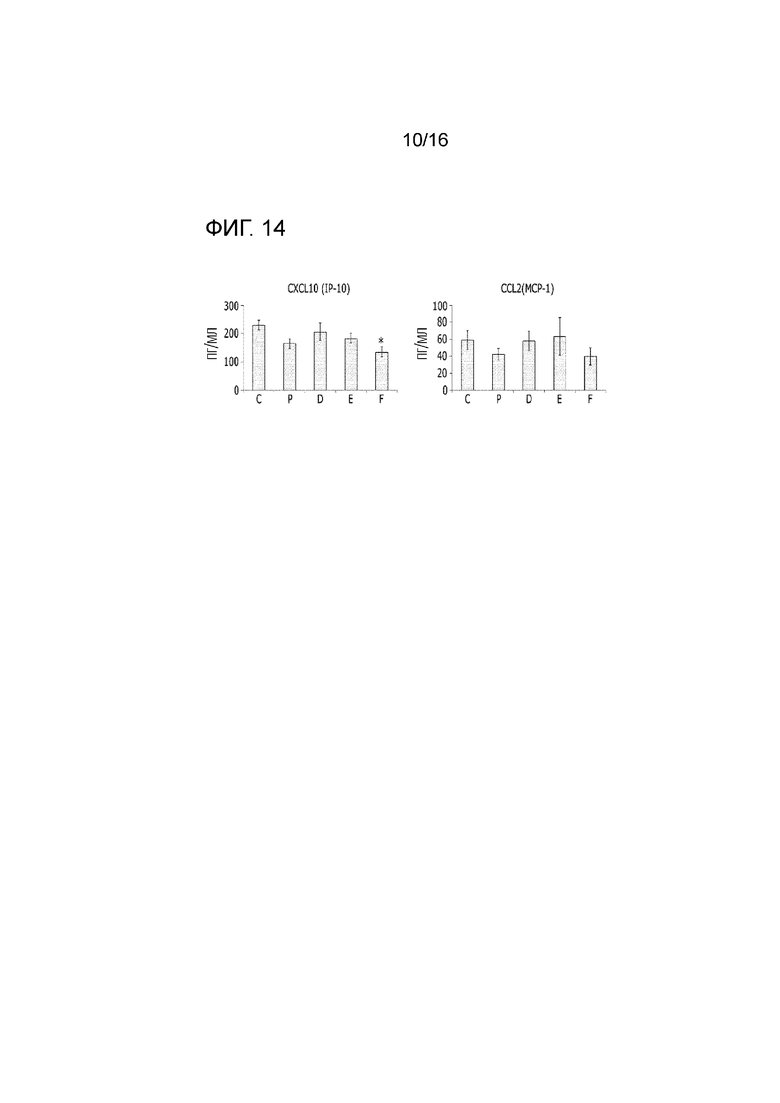
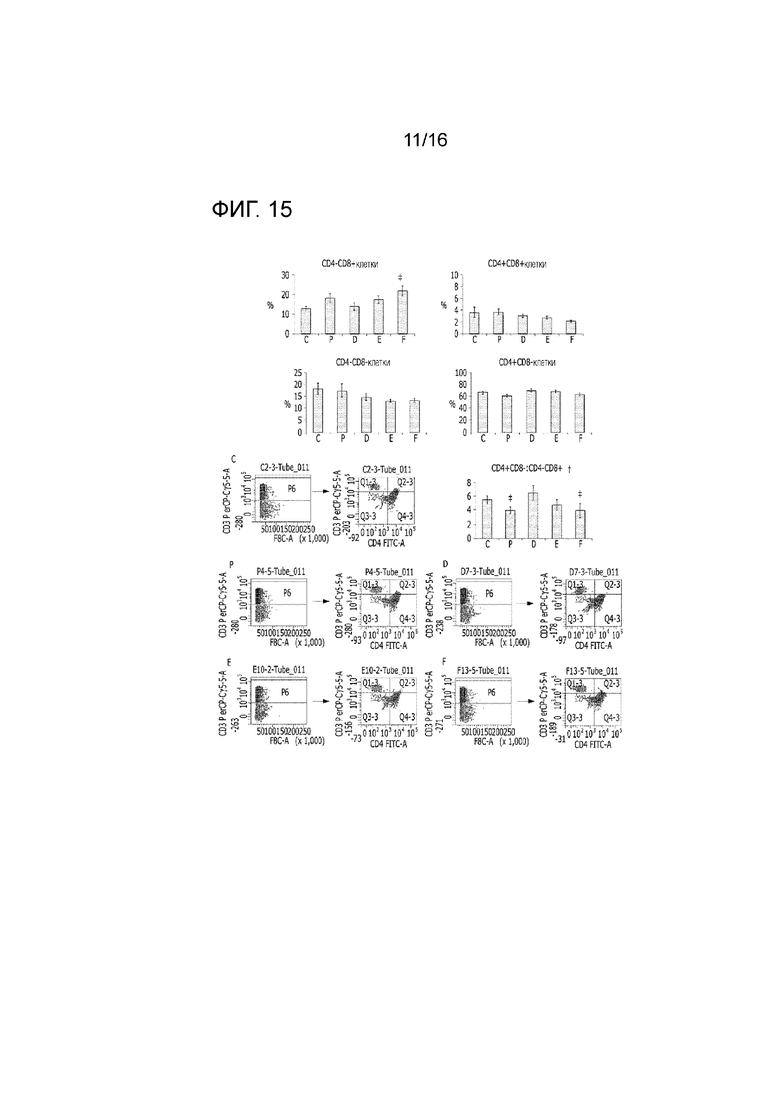
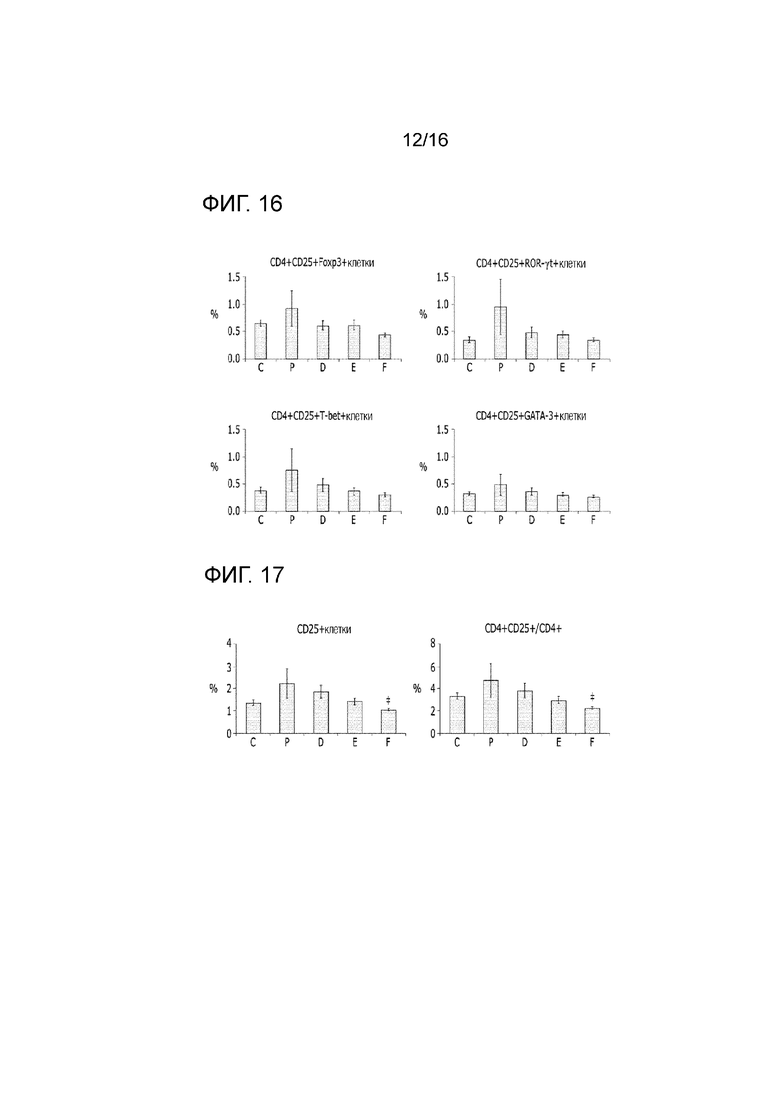
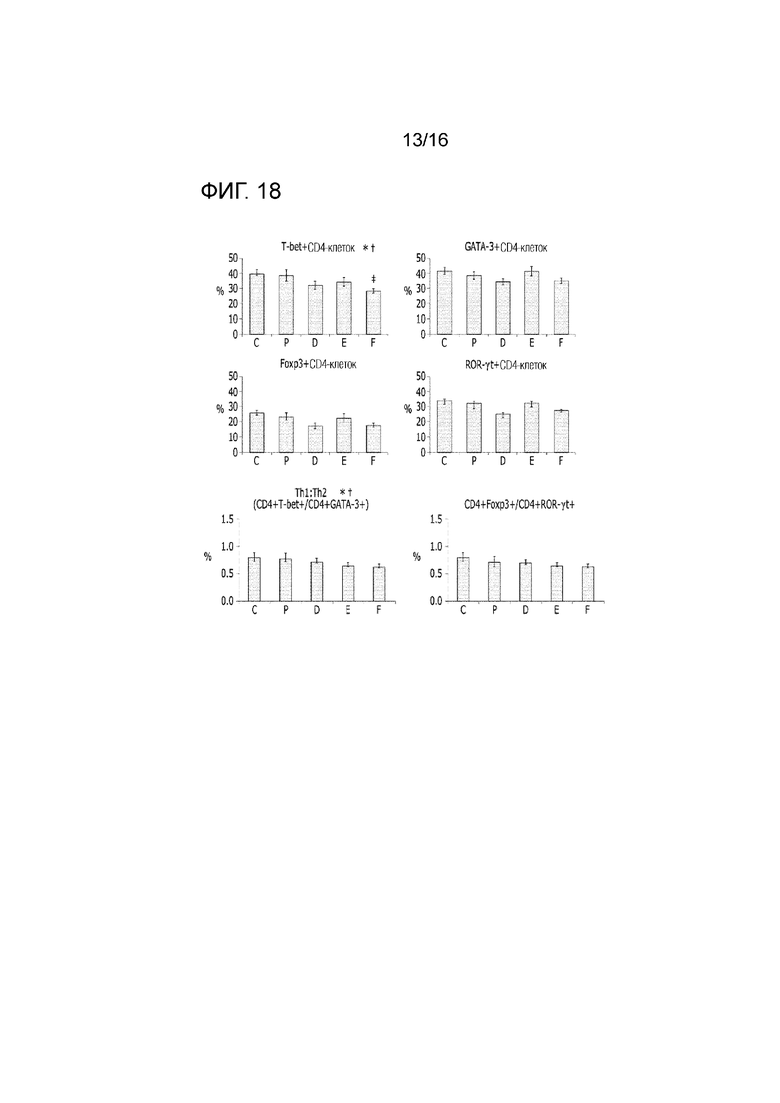
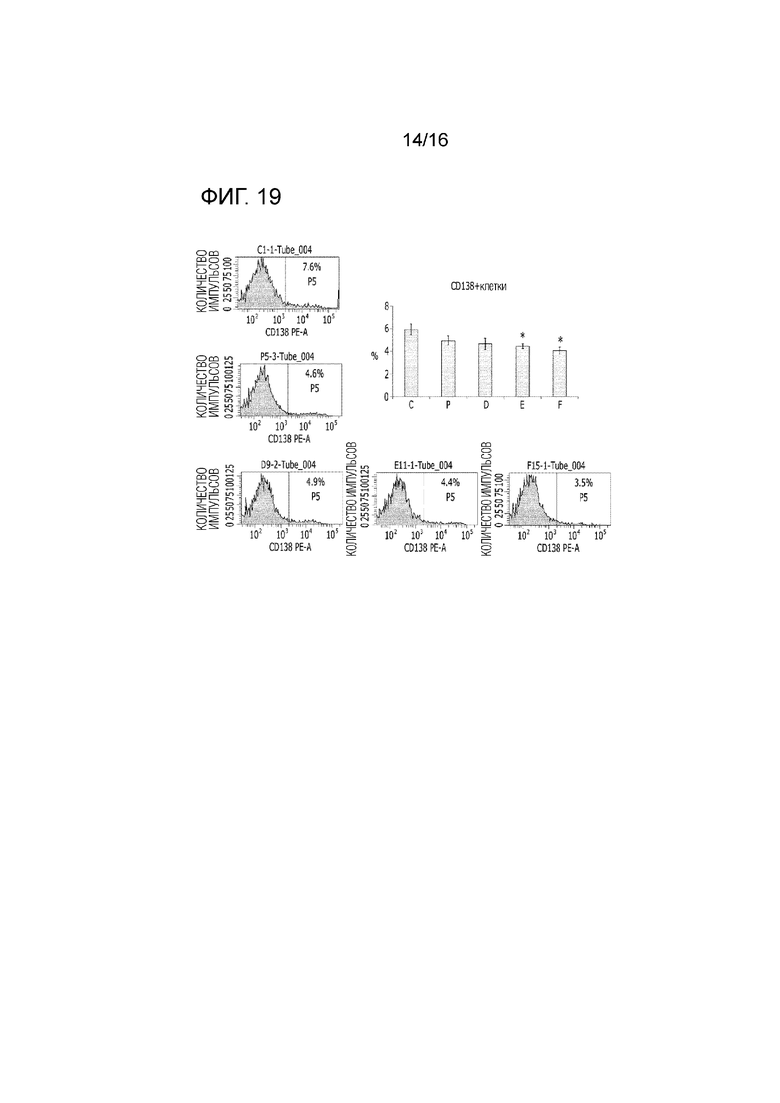

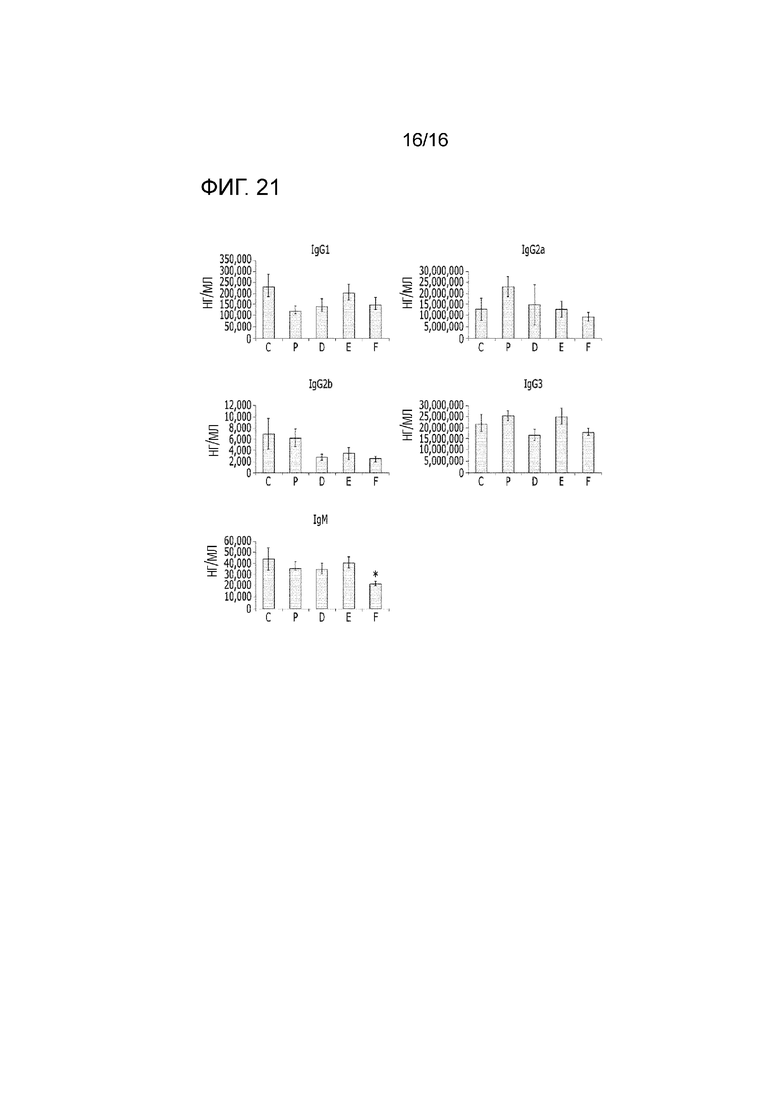
Представленная группа изобретений относится к области медицины и фармацевтики, а именно, к применению соединения, представленного формулой I или его фармацевтически приемлемой соли в эффективном количестве для предупреждения или лечения волчанки: где A представляет собой 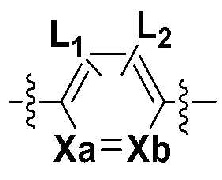 , каждый из Xa и Xb независимо представляет собой CH, каждый из L1 и L2 независимо представляет собой водород или галоген, Q представляет собой C(=O), Y выбран из следующей группы:
, каждый из Xa и Xb независимо представляет собой CH, каждый из L1 и L2 независимо представляет собой водород или галоген, Q представляет собой C(=O), Y выбран из следующей группы:  , M представляет собой C, N или O, где в этом случае, когда M представляет собой C, l и m равны 1; когда M представляет собой N, l равен 1 и m равен 0; и когда M представляет собой O, l и m равны 0, каждый из Ra1 и Ra2 независимо представляет собой водород; -C1-4 алкил с прямой или разветвленной цепью; или галоген; n представляет собой целое число, равное 0, 1 или 2, Rb представляет собой водород; Z выбран из следующей группы:
, M представляет собой C, N или O, где в этом случае, когда M представляет собой C, l и m равны 1; когда M представляет собой N, l равен 1 и m равен 0; и когда M представляет собой O, l и m равны 0, каждый из Ra1 и Ra2 независимо представляет собой водород; -C1-4 алкил с прямой или разветвленной цепью; или галоген; n представляет собой целое число, равное 0, 1 или 2, Rb представляет собой водород; Z выбран из следующей группы: 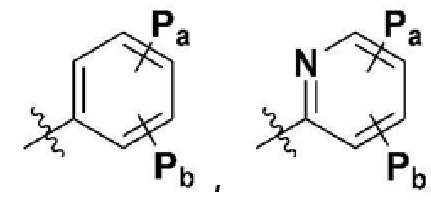 , каждый из Pa и Pb независимо представляет собой водород; -C1-4 алкил с прямой или разветвленной цепью, где он является незамещенным или по меньшей мере один атом водорода замещен галогеном; галоген; или -CF3. Кроме того, описан способ лечения волчанки, включающий введение терапевтически эффективного количества соединения, представленного следующей формулой I, или его фармацевтически приемлемой соли, а также применение соединения, представленного следующей формулой I, или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения волчанки. Технический результат: обеспечено применение соединений, демонстрирующих превосходный эффект в предупреждении или лечении волчанки. 3 н. и 7 з.п. ф-лы, 21 ил., 4 табл., 15 пр.
, каждый из Pa и Pb независимо представляет собой водород; -C1-4 алкил с прямой или разветвленной цепью, где он является незамещенным или по меньшей мере один атом водорода замещен галогеном; галоген; или -CF3. Кроме того, описан способ лечения волчанки, включающий введение терапевтически эффективного количества соединения, представленного следующей формулой I, или его фармацевтически приемлемой соли, а также применение соединения, представленного следующей формулой I, или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения волчанки. Технический результат: обеспечено применение соединений, демонстрирующих превосходный эффект в предупреждении или лечении волчанки. 3 н. и 7 з.п. ф-лы, 21 ил., 4 табл., 15 пр.
 [Формула I]
[Формула I]
1. Применение соединения, представленного следующей формулой I, или его фармацевтически приемлемой соли в эффективном количестве для предупреждения или лечения волчанки:
[Формула I]
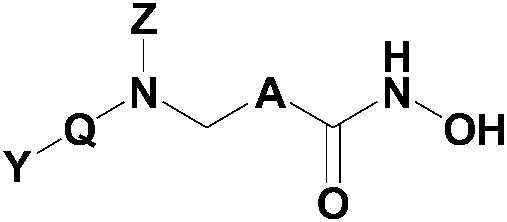
где
A представляет собой 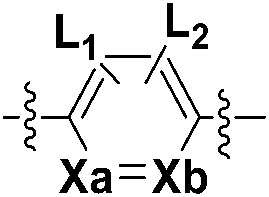 ,
,
каждый из Xa и Xb независимо представляет собой CH,
каждый из L1 и L2 независимо представляет собой водород или галоген,
Q представляет собой C(=O),
Y выбран из следующей группы:
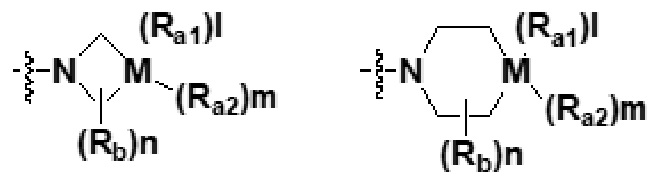
M представляет собой C, N или O, где в этом случае, когда M представляет собой C, l и m равны 1; когда M представляет собой N, l равен 1 и m равен 0; и когда M представляет собой O, l и m равны 0,
каждый из Ra1 и Ra2 независимо представляет собой водород; -C1-4 алкил с прямой или разветвленной цепью; или галоген;
n представляет собой целое число, равное 0, 1 или 2,
Rb представляет собой водород;
Z выбран из следующей группы:

каждый из Pa и Pb независимо представляет собой водород; -C1-4 алкил с прямой или разветвленной цепью, где он является незамещенным или по меньшей мере один атом водорода замещен галогеном; галоген; или -CF3.
2. Применение по п.1, где соединение, представленное следующей формулой I, представляет собой соединение, представленное следующей формулой Ia:
[Формула Ia]

где
каждый L1 и L2 независимо представляет собой водород или галоген,
Y представляет собой 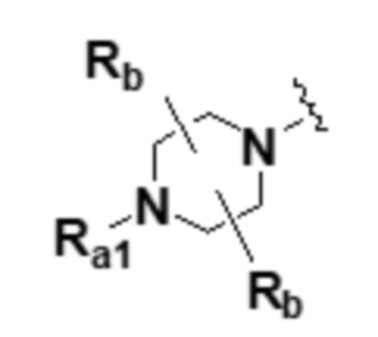 ,
, 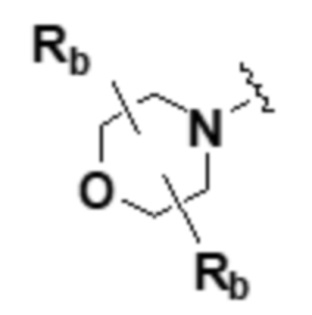 или
или 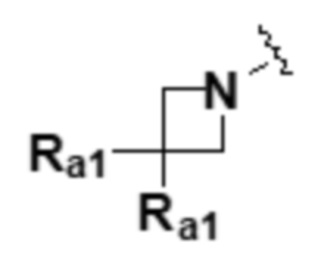 ,
,
где Ra1 и Rb такие как определено в п.1,
Z представляет собой фенил или пиридинил,
где по меньшей мере один водород фенила или пиридинила может быть замещен галогеном, CF3 или CH2F.
3. Применение по п.2, где соединение, представленное следующей формулой Ia, представляет собой соединение, приведенное в следующей таблице:
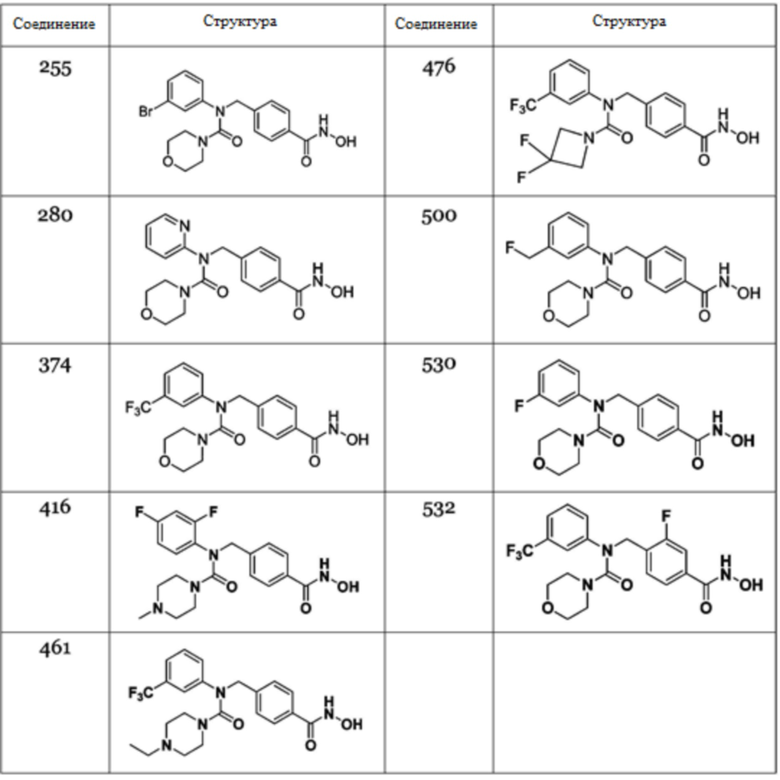
4. Применение по п.1, где указанная волчанка выбрана из группы, состоящей из системной красной волчанки (SLE), системной волчанки, дискоидной волчанки, индуцированной лекарственным средством волчанки, волчанки новорожденных и хронического нефрита.
5. Применение по п.4, где указанный хронический нефрит представляет собой волчаночный нефрит или гломерулонефрит.
6. Применение по п.1, где соединение или его фармацевтически приемлемая соль снижает сывороточную концентрацию антител против дцДНК.
7. Применение по п.1, где соединение или его фармацевтически приемлемая соль подавляет протеинурию.
8. Применение по п.1, где соединение или его фармацевтически приемлемая соль подавляет продуцирование TNFα в воспалительной реакции.
9. Способ лечения волчанки, включающий введение терапевтически эффективного количества соединения, представленного следующей формулой I, или его фармацевтически приемлемой соли:
[Формула I]
где
A представляет собой ,
каждый из Xa и Xb независимо представляет собой CH,
каждый из L1 и L2 независимо представляет собой водород или галоген,
Q представляет собой C(=O),
Y выбран из следующей группы:
M представляет собой C, N или O, где в этом случае, когда M представляет собой C, l и m равны 1; когда M представляет собой N, l равен 1 и m равен 0; и когда M представляет собой O, l и m равны 0,
каждый из Ra1 и Ra2 независимо представляет собой водород; -C1-4 алкил с прямой или разветвленной цепью; или галоген;
n представляет собой целое число, равное 0, 1 или 2,
Rb представляет собой водород;
Z выбран из следующей группы:
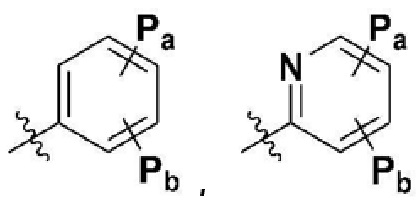 ,
,
каждый из Pa и Pb независимо представляет собой водород; -C1-4 алкил с прямой или разветвленной цепью, где он является незамещенным или по меньшей мере один атом водорода замещен галогеном; галоген; или -CF3.
10. Применение соединения, представленного следующей формулой I, или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения волчанки:
[Формула I]
где
A представляет собой ,
каждый из Xa и Xb независимо представляет собой CH,
каждый из L1 и L2 независимо представляет собой водород или галоген,
Q представляет собой C(=O),
Y выбран из следующей группы:
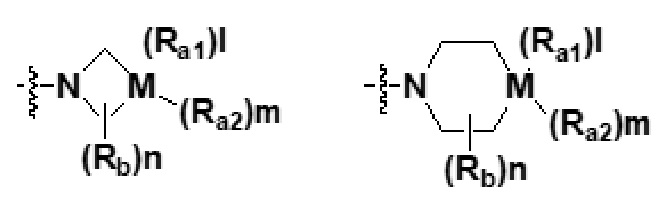
M представляет собой C, N или O, где в этом случае, когда M представляет собой C, l и m равны 1; когда M представляет собой N, l равен 1 и m равен 0; и когда M представляет собой O, l и m равны 0,
каждый из Ra1 и Ra2 независимо представляет собой водород; -C1-4 алкил с прямой или разветвленной цепью; или галоген;
n представляет собой целое число, равное 0, 1 или 2,
Rb представляет собой водород;
Z выбран из следующей группы:
каждый из Pa и Pb независимо представляет собой водород; -C1-4 алкил с прямой или разветвленной цепью, где он является незамещенным или по меньшей мере один атом водорода замещен галогеном; галоген; или -CF3.
| Токарный резец | 1924 |
|
SU2016A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| БЕНЗАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ДЕАЦЕТИЛАЗЫ ГИСТОНОВ | 2006 |
|
RU2448965C2 |

