Область применения
Данное изобретение имеет отношение к липосомальным препаратам и содержащим наркотические вещества липосомальным фармацевтическим препаратам, в частности липосомальному фармацевтическому препарату митоксантрону. Более того, данное изобретение имеет отношение к способам изготовления липосомы, липосомального фармацевтического препарата и их использованию.
Основа изобретения.
Липосомы могут использоваться в качестве носителей многих лекарственных средств, в особенности противоопухолевых средств (в частности химиотерапевтических лекарственных средств). Липосомы могут уменьшить распределение лекарственного средства в нормальных тканях, однако увеличить накопление лекарственного средства в опухолевых тканях, повышая терапевтический индекс лекарственного средства таким образом. Причина того, почему липосома может пассивно нацеливаться на опухоль, имеет отношение к физиологическим свойствам опухолевой ткани. Размер пор кровеносных сосудов опухоли может достигать до 100-780 нм вследствие ее быстрого роста, тогда как стандартный размер нормальных эндотелиоцитов сосудистых стенок составляет около 2 нм. Вследствие этого, липосомы могут пассивно накапливаться в пораженной опухолью области, если они могут циркулировать в крови на протяжении относительно длительного периода времени и иметь размер менее чем 200 нм, потому что, после того, как липосомы маленького размера вводятся посредством внутривенной инъекции, они не могут попасть в нормальные ткани, но могут проникать в кровеносный сосуд пораженной опухолью области и достигать зоны лечения.
Однако нелегко добиться терапевтической пользы липосомы, должны быть удовлетворены следующие четыре требования: (1) лекарственное средство может быть инкапсулировано в липосому при высокой эффективности инкапсуляции и достаточной загрузке лекарственного средства; (2) лекарственное средство не должно выделяться из липосомы в течение периода хранения вне организма; (3) не должно быть значительной потери лекарственного средства во время циркуляции липосомального лекарственного средства в крови и (4) лекарственное средство может быть эффективно выведено и, таким образом, проявить свой терапевтический эффект, когда липосомы накапливаются в пораженной опухолью области. Что касается способов изготовления липосомных препаратов в настоящее время, три упомянутые выше задачи были успешно решены, в связи с этим, правильное выведение липосомального лекарственного средства в организме привлекает больше внимания. Одна особо важная техническая задача, которую необходимо решить для разработки некоторых липосомальных лекарственных средств, заключается в эффективном контролировании правильного выведения липосомальных лекарственных средств после нацеливания на пораженную опухолью область. Это особенно важно для некоторых лекарственных средств, таких как митоксантрон.
Группой по изучению липосом в Канаде было обнаружено, что липосомальная форма размером около 100 нм, которая была приготовлена с использованием гидрогенизированного соевого фосфатидилхолина (HSPC) и холестерина в качестве фосфолипидного бислоя и загрузки в лекарственное средство 300 ммоль градиента лимонной кислоты, уступала митоксантрону, не связанному с белками плазмы крови. С целью улучшить терапевтический эффект липосомы, исследовательская группа, в конце концов, изменила композицию фосфолипидного бислоя на димиристойл фосфатидилхолин (DMPC) и холестерин, и получила препарат с повышенными терапевтическими индексами. Однако утечка лекарственного средства может возрасти в течение периода хранения, так как температура фазового перехода DMPC - около 21°C, так что препарат может быть не стабильным (Дипосомалъные формы митоксантрона, US 5,858,397).
Американская корпорация Неофарм использовала другой способ разработки липосомальной формы митоксантрона, в котором кардиолипин, отрицательно заряженный, был прибавлен к фосфолипидному бислою. Благодаря интенсивному взаимодействию кардиолипина и митоксантрона, митоксантрон мог быть введен в фосфолипидный бислой в режиме пассивной загрузки. Данный способ пассивной загрузки отличается от способа активной загрузки. Посредством способа активной загрузки лекарственное средство откладывалось бы во внутрилипосомальной водной фазе в форме осадка. Фаза I клинических исследований продукта компании Неофарм показала, что липосомальные лекарственные средства могут увеличить вероятность случайного инфицирования по сравнению с лекарственным средством, не связанным с белками плазмы крови. Разработка данного продукта была прекращена ввиду безопасности (Липосомальный препарат митоксантрон, CN 01817424.8).
Тихоокеанский Институт Фармакологии (Чанчунь, Китай) также подал патентную заявку на липосомальный препарат митоксантрон (Дипосомалъное введение митоксантрона или гидрохлорида м итоксантрона и процесс его изготовления, CN 200410041612.1). В данной заявке был использован традиционный метод pH-градиента для загрузки лекарственных средств. Данная заявка собирается защитить форму особым коэффициентом, и не раскрывать влияние факторов, таких как композиция фосфолипидов, виды буферной соли в внутренней водной фазе, размер липосомы, соотношение лекарственного средства к липосоме, и т.п. на терапевтическую эффективность и токсичность липосомы.
Жирон Жан с соавторами из Фармацевтической школы Западного Китая, Сычуаньский Университет, также изучали липосомальный препарат митоксантрон. Они использовали соевый фосфатидилхолин с температурой фазового сдвига 0°C (которая продается под торговой маркой EPIKURON 200) для приготовления липосом размером около 60 нм. В данной статье, была изучена только фармакокинетика, не касаясь токсичности и терапевтической эффективности полученного липосомалыюго препарата. Информацию, имеющую отношение к данному вопросу, можно увидеть в "Изготовление липосом митоксантрона, циркулирующих в течение длительного времени, и их фармакокинетика", Жирон Жан, Ботао Ю и Исон Дуан, Акта Фармасьютика Синика (китайский журнал по фармацевтике), 2002, Том 37, Номер 6; Изучение приготовления липосом митоксантрона, циркулирующих в течение длительного времени, с трансмембранными градиентами сульфата аммония, Жирон Жан, Ботао Ю, Исон Дуан и Юан Хуан, Чайнис Фармасьютикал Джорнал (китайский журнал по фармацевтике), 2002 Том 37, Номер 12; и ”Изучение способов приготовления липосом митоксантрона”, Исон Дуан, Вест Чайна Джорнал оф Фармасьютикал Саенсес (Журнал по фармацевтике Западного Китая), 2001 Том 16, Номер 02.
В вышеупомянутых исследованиях размер липосом обычно регулируется на уровне 80~150 нм, так как существует всеобщее мнение в области исследований липосом, что липосома размером около 100 нм имеет наибольшую эффективность нацеливания (Фармакол. Рев. (фармакологическое обозрение) 1999 51: 691-744.). Однако как уже отмечалось, липосома должна иметь не только превосходную эффективность нацеливания, но и также достаточное выведение препарата из липосомы, чтобы проявить свой эффект.
Как отмечено выше, согласно предыдущему сегменту потеря лекарственного средства во время кровообращения должна, по сути, предотвращаться, чтобы лекарственное средство могло быть эффективно доставлено к опухолям, но это требование также приводит к затруднению выведения лекарственного средства из липосомы, когда оно нацелено на пораженную опухолью область. В обычных процессах изготовления липосом лекарственное средство обычно инкапсулируется способом активной загрузки, при котором лекарственное средство, инкапсулированное в липосому, представлено в форме коллоидного осадка, не обладающего биокативностью, так что только когда лекарственное средство эффективно выведено из липосомы, оно может превратиться в лекарственное средство с биоактивностью. Если скорость выведения лекарственного средства слишком медленная, данное лекарственное средство едва может проявлять свое терапевтическое действие, даже если оно было эффективно нацелено на пораженную опухолью область, и его терапевтический эффект может быть даже хуже чем у неинкапсулированного лекарственного средства.
Таким образом, существует насущная необходимость в отрасли в липосомальном препарате, способном доставить лекарственное средство с хорошей способностью нацеливания и эффективно вывести лекарственное средство в нацеленные ткани, и в соответствующем липосомальном фармацевтическом препарате.
Краткое описание изобретения
Авторы изобретения неожиданно случайно обнаружили, что некоторые лекарственные средства, имеющие множество диссоциирующихся групп и склонность к формированию плотных осадков с поливалентным противоионом, могут быть обработаны для создания маленького однослойного липосомального препарата с значительно повысившимся терапевтическим индексом, для того, чтобы вышеупомянутая техническая проблема могла быть решена.
Таким образом, с одной стороны, данное изобретение предоставляет липосомальный препарат размером около 30-80 нм, имеющий фосфолипид с температурой плавления выше, чем температура тела в фосфолипидном бислое, так что температура фазового сдвига липосомы выше температуры тела. Примеры вышеупомянутого фосфолипида включают, в том числе, фосфатидилхолин, гидрогенизированный соевый фосфатидилхолин (HSPC), гидрогенизированный яично-желточный фосфатидилхолин, дипальмитоил фосфатидилхолин (DPPC) или дистеароил фосфатидилхолин (DSPC), или их комбинации.
В одном из вариантов осуществления настоящего изобретения фосфолипид с температурой плавления выше, чем температура тела в фосфолипидном бислое, эквивалентен 50-100 моль/моль%, предпочтительно 55-95 моль/моль %, и еще более предпочтительнее 55-95 моль/моль % общего содержания фосфолипидов.
Дополнительно, фосфолипидный бислой липосомального препарата данного изобретения содержит, более того, дополнительные фосфолипиды, например, фосфолипид с температурой плавления не выше температуры тела, как например димиристойл фосфатидилхолин (DMPC) и тому подобные. Количество фосфолипида в липосомальных препаратах данного изобретения может быть условно определено теми, кто разбирается в данной области, при условии, что температура плавления липосомального препарата явно не понижается до значения ниже, чем температура тела.
Липосомальный препарат данного изобретения может также дополнительно содержать холестерин с целью регулирования текучести липосомальной мембраны.
Липосомальный препарат данного изобретения может также содержать дополнительные вспомогательные вещества, в частности вспомогательные вещества для дальнейшего изменения характеристик поверхности липосомы, чтобы наделить липосому большими функциональными возможностями в организме. Подобные вспомогательные вещества включают, например, липиды и тому подобные вещества, модифицированные гидрофильными полимерами.
С другой стороны, данное изобретение представляет Липосомальный фармацевтический препарат, который содержит изучаемое лекарственное средство, в частности лекарственное средство с поливалентными ионами, в липосомальном препарате данного изобретения. Таким образом, данное изобретение имеет отношение к липосомальному фармацевтическому препарату размером 30-80 нм, где: (1) Липосомальный фармацевтический препарат содержит лекарственное средство с поливалентными ионами в качестве активного ингредиента; (2) фосфолипидный бислой содержит фосфолипид с температурой плавления выше, чем температура тела, так что температура фазового сдвига липосомы выше, чем температура тела; и дополнительно (3) липосомальный фармацевтический препарат содержит дополнительные лекарственные средства и/или дополнительные вспомогательные вещества, допустимые в липосомальном фармацевтическом препарате. Предпочтительно, чтобы главные пики размера липосомального фармацевтического препарата были сосредоточены в 35-75 нм, в частности приблизительно 40-60 нм.
С другой стороны, данное изобретение представляет методику приготовления вышеупомянутого липосомального фармацевтического препарата, методику, включающую в себя следующие этапы: (1) приготовление липосомы с использованием фосфолипида с температурой плавления выше, чем температура тела, и, по выбору, дополнительных фосфолипидов и/или холестерина; и (2) инкапсулирование изучаемого лекарственного средства, в частности лекарственного средства с поливалентными ионами, в липосому.
Данное изобретение также представляет методику лечения заболевания, включающую в себя введение липосомального фармацевтического препарата данного изобретения пациенту, нуждающемуся в лечении. Желательно, чтобы таким пациентом выступало млекопитающее, в частности человек. Краткое описание чертежей.
На фиг.1 представлена фармакокинетика PLM60 в организме мыши из г.Кунымин (Китай) и ее сравнение с фармакокинетикой митоксантрона, не связанного с белками плазмы крови, в организме, где PLM означает пегилированный липосомальный митоксантрон, FM означает митоксантрон, не связанный с белками плазмы крови, на оси абсцисс показано время (часы) и на оси ординат показан уровень митоксантрона в плазме (мкг митоксантрона/мл плазмы).
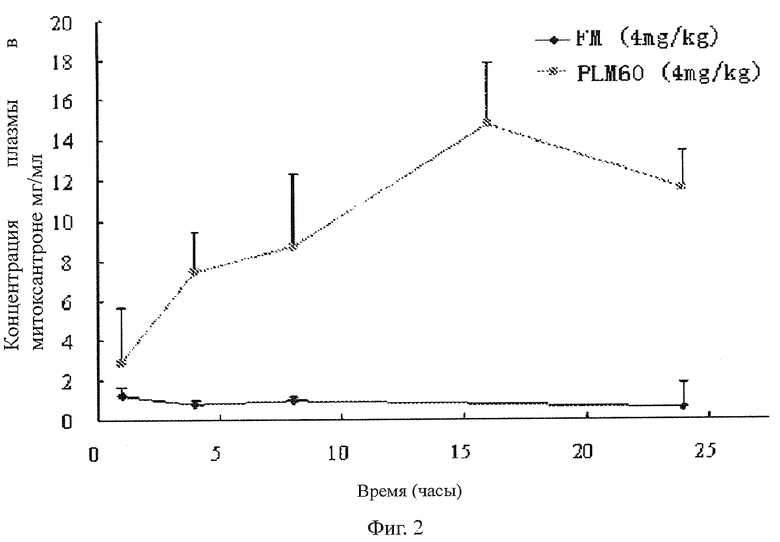
На фиг.2 представлен вид в разрезе PLM60 и FM в опухоли мыши, где PLM60 означает пегилированный липосомальный митоксантрон, FM означает митоксантрон, не связанный с белками плазмы крови, на оси абсцисс показано время (часы) и на оси ординат показана концентрация митоксантрона в опухолевых тканях (мкг митоксантрона/г опухолевой ткани).
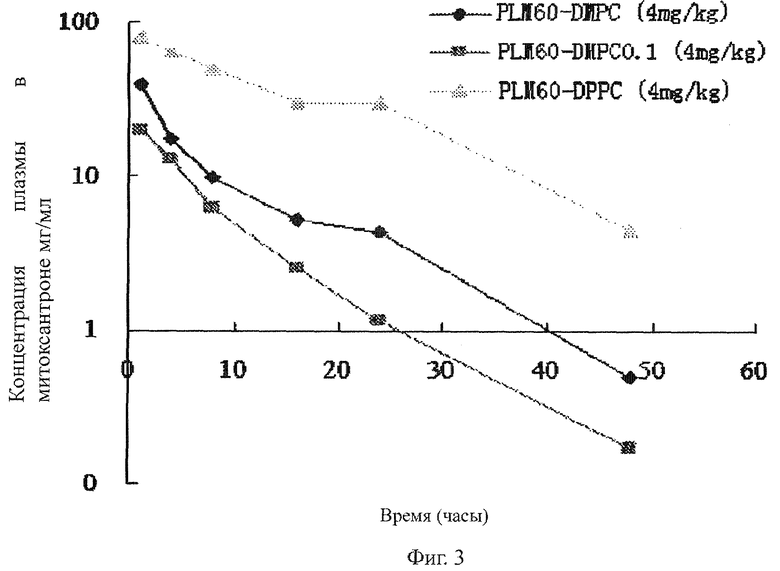
На фиг.3 представлено сравнение фармакокинетики различных форм в организме мыши, где на оси абсцисс показано время (часы) и на оси ординат показан уровень митоксантрона в плазме (мкг митоксантрона/мл плазмы), и дозировки различных форм составляют 4 мг/кг.
Описание предпочтительных вариантов изобретения
Обычно липосомы производятся из фосфолипидов и холестерина, являющихся мембранными материалами. Эти два ингредиента являются не только основными материалами создания липосомального бислоя, но также обладают очень важными физиологическими функциями.
Физические свойства липосомальной мембраны тесно связаны с температурой. Когда температура повышена, ацильные боковые цепи липидного бислоя меняют форму упорядоченной структуры на форму неупорядоченной структуры. Такой вид изменений может привести к множеству изменений физических свойств липидной мембраны. Например, «гелеобразное» состояние может превратиться в «жидкокристаллическое» состояние, поперечный срез мембраны может увеличиться, толщина бислоя может уменьшиться, текучесть мембраны может увеличиться. Температура, при которой происходят такие изменения, называется температурой фазового сдвига. Температура фазового сдвига липидной мембраны может быть определена Дифференциальной Сканирующей Калориметрией, Электронным Парамагнитным Резонансом (ЭПР) и тому подобным. Температура фазового сдвига липосомальной мембраны зависит от видов фосфолипидов. В общем, чем длиннее ацильная боковая цепь, чем выше температура фазового сдвига; и наоборот. Например, температура фазового сдвига димиристойл фосфатидилхолина составляет 24°C, в то время как дипальмитоил фосфатидилхолина и дистеароил фосфатидилхолина - 41°C и 58°C, соответственно. Текучесть мембраны является важным свойством липосомы. При температуре фазового сдвига текучесть мембраны увеличится, а лекарственное средство, инкапсулированное в липосому, будет иметь максимальную скорость выведения. Таким образом, текучесть мембраны оказывает непосредственное воздействие на стабильность липосомы.
В одном из вариантов осуществления настоящего изобретения данное изобретение представляет липосомальный препарат размером около 30-80 нм и фосфолипид с температурой плавления выше, чем температура тела в фосфолипидном бислое, так что температура фазового сдвига липосомы выше, чем температура тела.
Желательно, чтобы липосомальный фармацевтический препарат данного изобретения был изготовлен с использованием фосфолипидов с относительно высокой температурой фазового сдвига температуры плавления, как например фосфатидилхолина. Если температура плавления фосфатидилхолина выше, чем температура тела, желательно, чтобы длина его углеводородной цепи была не меньше 16 атомов углерода. Желательно, чтобы фосфолипиды данного изобретения содержали, но не ограничивались гидрогенизированным соевым фосфатидилхолином, гидрогенизированным яично-желточным фосфатидилхолином, дипальмитоил фосфатидилхолином (DPPC) или дистеароил фосфатидилхолином (DSPC), или их комбинацией.
В липосомальном препарате данного изобретения фосфолипиды с температурой плавления выше, чем температура тела в фосфолипидном бислое эквивалентны приблизительно 50-100 моль/моль%, предпочтительно около 55-95 моль/моль%, еще более предпочтительно около 60-90 моль/моль% относительно общего содержания фосфолипидов. Дополнительно, фосфолипидный бислой может содержать добавочные фосфолипиды, например, фосфолипиды с температурой плавления не выше, чем температура тела, как например димиристойл фосфатидилхолин (DMPC) и тому подобные. Такие фосфолипиды могут присутствовать в липосоме в любом количестве, отвечающем требованиям, при условии, что это не понижает температуру фазового сдвига липосомального препарата ниже температуры тела. Количество, отвечающее требованиям, может быть определено согласно обычным технологическим приемам тех, кто разбирается в данной области.
Желательно, чтобы липосомальный препарат данного изобретения мог в дальнейшем содержать холестерин. Холестерин обладает функцией регулирования текучести мембраны. Когда липосомальная мембрана содержит 50% (моль/моль) холестерина, фазовый сдвиг липосомальной мембраны может исчезнуть. Папахадйопоулос с соавторами называют холестерин "буфером текучести", так как добавление холестерина к фосфолипидам ниже температуры фазового сдвига может уменьшить упорядоченность структуры мембраны и увеличить текучесть мембраны, тогда как добавление холестерина в фосфолипиды выше температуры фазового сдвига может увеличить упорядоченность структуры мембраны и уменьшить текучесть мембраны. В липосомальном препарате данного изобретения содержание холестерина может составлять 2-60 моль/моль %, 5-55 моль/моль % или 10-50 моль/моль % относительно общего количества ингредиентов липосомы. Точнее говоря, содержание холестерина может составлять 15-45 моль/моль %, например 20-40 моль/моль % относительно общего количества ингредиентов липосомы. Содержание холестерина в липосоме данного изобретения может быть легко определено согласно стандартным технологическим приемам тех, кто разбирается в данной области.
Следует учесть, что фосфолипидный бислой в липосоме данного изобретения может также содержать дополнительные вспомогательные вещества, в частности вспомогательные вещества для дальнейшего изменения характеристик поверхности липосомы, чтобы наделить липосому большими функциональными возможностями в организме. Такие вспомогательные вещества содержат, например, липидные вещества, модифицированные гидрофильными полимерами, и их примеры следующие: ПЭГ-модифицированный дистеароил фосфатидил этаноламин (DSPE-PEG), ПЭГ-модифицированный дистеароил фосфатидил глицерин (DSPG-PEG), ПЭГ-модифицированный холестерин (chol-PEG), поливидон модифицированный дистеароил фосфатидил этаноламин (DSPE-PVP), ПЭГ-модифицированный дистеароил фосфатидил глицерин (DSPG-PVP), или ПЭГ-модифицированный холестерин (chol-PVP). Вышеупомянутые вспомогательные вещества могут также быть мембранными материалами, модифицированными специфическим антителом или лигандом. Количество таких вспомогательных веществ в липосоме данного изобретения может быть определено согласно стандратным технологическим приемам тех, кто разбирается в данной области, например, может составлять 0.1-20 моль/моль %, предпочтительно 0.3-18 моль/моль %, еще более предпочтительно 0.5-15 моль/моль %, в частности 0.8-12 моль/моль %, например 1-10 моль/моль %, или 2-8 моль/моль %, 2.5-7 моль/моль %, 3-6 моль/моль %, и т.д. относительно числа молей фосфолипидов. В случаях использования ПЭГ-модифицированных липидов в качестве вспомогательных веществ, молекулярная масса доли ПЭГ может составлять, например, 400-20000 Дальтон, предпочтительно 600-15000 Дальтон, еще более предпочтительно 800-10000 Дальтон, в частности 1000-8000 Дальтон, например 1200-5000 Дальтон. Использование ПЭГ в данном изобретении может также быть легко определено на основании стандартных исследований тех, кто разбирается в данной области.
Липосомальный препарат данного изобретения является маленьким однослойным липосомальным препаратом, и должен иметь подходящий размер. Желательно, чтобы размер препарата был 30-80 нм, еще более предпочтительно 35-70 нм, в частности предпочтительно 40-60 нм. Размер липосомы может быть определен с помощью анализатора размеров частиц или электронного микроскопа или других средств. Следует понимать, что липосомные частицы в данном изобретении не могут иметь в точности одинаковый размер, они охватывают диапазон величин, благодаря природе самой липосомы и свойств способа изготовления. Таким образом, в липосомальном препарате данного изобретения, не исключается наличие липосомных частиц, выходящих за пределы установленного диапазона величин, при условии, что они не оказывают явного воздействия на свойства липосомального препарата или фармацевтического препарата данного изобретения.
Липосома в данном изобретении может быть приготовлена с помощью различных соответствующих методик, в том числе, например, методики пленочной дисперсии, методики введения, методики ультразвуковой дисперсии, методики замораживания-высушивания, методики замораживания-оттаивания и тому подобного. Согласно системам запуска, использованным для приготовления липосомы, данные методики могут быть поделены на: (1) методики, основанные на использовании высушенных липидных мембран, липидного экстракта; (2) методики, основанные на использовании эмульгирующих веществ; (3) методики приготовления липосомальных препаратов, основанные на использовании смешанных мицелл; и (4) методики приготовления липосомальных препаратов, основанные на использовании трехкомпонентной фазовой смеси этанола, фосфолипидов и воды. Инкапсуляция лекарственного средства может быть осуществлена как с помощью режима пассивной загрузки, так и режима активной загрузки. Данные методики могут быть найдены во многих обзорных статьях, посвященных липосомам.
Во время или после приготовления липосомального препарата многие соответствующие методики могут быть использованы для инкапсулирования лекарственного средства в липосому и создания липосомального фармацевтического препарата. Соответствующие методики включают в себя, например, методики активной загрузки и методики пассивной загрузки. Методика активной загрузки обычно приводится в исполнение с помощью градиентных методов, например метода градиента сульфата аммония, то есть, использования раствора сульфата аммония в качестве водяной фазы для того, чтобы, во-первых, приготовить липосому, содержащую сульфат аммония и в внутрилипосомной, и в внелипосомной фазе, затем создания градиента плотности сульфата аммония между внутрилипосомной и внелипосомной фазами, удаляя внелипосомный сульфат аммония. Внутрилипосомный NH4 + диссоциируется на NH3 и H+, что приводит к разности концентраций H+ (то есть градиента pH) между внутрилипосомной и внелипосомной фазами, так что после того, как внелипосомное лекарственное средство в молекулярном состоянии входит в внутрилипосомную водяную фазу, оно переходит в ионное состояние, таким образом, лекарственное средство не может вернуться в внелипосомную водяную фазу, и в липосоме происходит меньше потери лекарственного средства, и она оказывается более стабильной. Методика пассивной загрузки может быть приведена в исполнение с помощью методики введения органического растворителя, методики пленочной дисперсии, методики замораживания-оттаивания и тому подобного.
В данном изобретении, могут быть использованы любые подходящие ингредиенты лекарственного средства. Желательно, чтобы активным фармацевтическим ингредиентом в липосомальном фармацевтическом препарате данного изобретения было лекарственное средство с поливалентными ионами. Под термином "лекарственное средство с поливалентными ионами" подразумевается лекарственное средство, имеющее две и более диссоциирующихся группы с константой диссоциации КД 4.5~9.5, так что лекарственное средство имеет больше позитивных зарядов или больше негативных зарядов в диапазонах КД. Желательно, чтобы вышеупомянутая константа диссоциации была в диапазоне 5.0-9.5. Еще более предпочтительнее, чтобы вышеупомянутая константа диссоциации была в диапазоне 5.5-9.5. В частности желательно, чтобы, вышеупомянутая константа диссоциации была в диапазоне 6.0-9.0 м, в частности 6.5-9.0. Значение КД каждой диссоциирующейся группы лекарственного средства с ионами может быть легко определено согласно стандартным технологическим приемам тех, кто разбирается в данной области.
В данном изобретении, лекарственные средства с поливалентными ионами могут содержать, но не ограничиваются противоопухолевыми препаратами, например, препараты, применимые для предотвращения или лечения следующих видов рака: рак легкого (как например немелкоклеточный рак легкого), рак поджелудочной железы, рак груди, рак прямой кишки или множественная миелома, рак печени, рак шейки матки, рак желудка, карцинома предстательной железы, рак почки и/или карцинома мочевого пузыря. Таким образом, в одном из вариантов осуществления данного изобретения, лекарственным средством с поливалентными ионами является противоопухолевое лекарственное средство с поливалентными ионами. Желательно, чтобы в качестве лекарственного средства с поливалентными ионами выступал митоксантрон, винкристин, винорелбин или винбластин. Еще более предпочтительно, чтобы в качестве вышеупомянутого лекарственного средства с поливалентными ионами выступал митоксантрон и мог дополнительно комбинироваться, по крайней мере, с одним из вспомогательных лекарственных средств, которым может выступить, например, противоопухолевое лекарственное средство, такое как винкристин, винорелбин или винбластин, и тому подобное.
Необходимо особенно отметить, что в предотвращающем изобретении в качестве лекарственного средства с поливалентными ионами может также выступить комбинация любого одного или двух или более вышеупомянутых лекарственных средств, например, комбинация двух противоопухолевых лекарственных средств, комбинация одного или более противоопухолевых лекарственных средств с вспомогательными препаратами, такими как иммуностимулятор, и комбинация двух или более других видов лекарственных средств.
Также стоит отметить, что липосомальные лекарственные средства данного изобретения также могут дополнительно содержать одно или более вспомогательных лекарственных средств с неполивалентными ионами кроме вышеперечисленных лекарственных средств с поливалентными ионами, которые могут быть введены в сочетании с лекарственными средствами с поливалентными ионами, как указано выше. Комбинированное введение состоит из введения всех компонентов в одном препарате, также состоит из комбинированного введения в отдельной стандартной лекарственной форме.
Должен быть оценен по достоинству тот факт, что данное лекарственное средство в качестве активного ингредиента, как указано в настоящем документе, выполнено не только в своей первоначальной форме, но также в производных формах, например сольватов (таких как гидраты и продукты с добавлением спирта), пролекарств и других физиологически приемлемых производных форм, а также активных метаболитов, и тому подобного. Производные формы, пролекарства и другие физиологически приемлемые производные формы, а также активные метаболиты лекарственного средства хорошо известны тем, кто разбирается в данной области.
Липосомальный фармацевтический препарат данного изобретения может, более того, содержать два и более поливалентных противоиона с зарядами, противоположными тем, которые есть у активного ингредиента. Примеры поливалентных противоионов включают, но не ограничиваются анионами органических кислот, такими как анионы кислот следующих насыщенных или ненасыщенных органических кислот: лимонная кислота, винная кислота, фумаровая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, яблочная кислота и малеиновая кислота, и тому подобное;
анионы неорганических кислот, как например сульфатный анион, фосфат-анион и тому подобные. Среди них предпочтение оказывается аниону цитрата, аниону сульфата или фосфат-аниону. К тому же, в качестве вышеуказанных поливалентных противоионов могут также выступать аминокислоты, такие как цистин и тому подобные. Не будучи ограниченными рамками какой-либо определенной теории, существуют предположения, что поливалентный противоион способен создать нерастворимый осадок с изучаемым лекарственным средством (например, лекарственное средство с поливалентными ионами), инкапсулированным в липосому, таким образом обеспечивается наличие лекарственного средства с поливалентными ионами в липосоме.
Липосомальный фармацевтический препарат данного изобретения содержит, более того, дополнительные вспомогательные вещества и носители, общеизвестные в фармацевтической отрасли, такие как сахароза, гистидин, антиоксиданты, стабилизаторы, диспергирующие вещества, консерванты, разжижающие кровь вещества, растворители, соли для воздействия на осмотическое давление, и тому подобное.
В одном из вариантов осуществления настоящего изобретения, данное изобретение предусматривает методику приготовления липосомального фармацевтического препарата данного изобретения, включающую в себя: во-первых, приготовления липосомального препарата данного изобретения, как указано выше, а затем инкубирования изучаемого лекарственного средства с липосомальным препаратом при подходящих условиях. Точнее говоря, методика приготовления липосомального фармацевтического препарата данного изобретения включает в себя следующие этапы: (1) растворение липидных вспомогательных веществ, подходящих для приготовления липосомы в соответствующем органическом растворителе, как например трибутиловый спирт или циклогексан, затем лиофилизация для получения лиофилизированного порошка; (2) гидратирование лиофилизированного порошка с помощью раствора, содержащего противоион активного ингредиента изучаемого лекарственного средства для создания "пустой" липосомы; (3) извлечение внелипосомного противоиона с помощью соответствующих средств, таких как диализ или колоночная хроматография и тому подобных, с целью создания градиента противоиона между внутрилипосомной фазой и внелипосомной фазой; и (4) инкубирование лекарственного средства с липосомой для получения липосомального лекарственного средства. Характеристику фосфолипидов, холестерина, вспомогательных веществ и тому подобного см. выше для липосомального препарата.
Желательно, чтобы липидом был фосфолипид, в частности липидом с относительно высокой температурой фазового сдвига, например, фосфатидилхолин, гидрогенизированный соевый фосфатидилхолин, гидрогенизированный яично-желточный фосфатидилхолин, дипальмитоил фосфатидилхолин (DPPC) или дистеароил фосфатидилхолин (DSPC), или их комбинация. Дополнительно, вышеупомянутый липид может также содержать холестерин в количестве, например, 2-60 моль/моль%, 5-55 моль/моль % или 10-50 моль/моль %. Точнее говоря, количество холестерина может составлять 15-45 моль/моль %, например, 20-40 моль/моль % относительно общего количества молей всех ингредиентов в липосоме. Те, кто разбирается в данной области, могут определить количество холестерина в зависимости от особых требований по отношению к температуре фазового сдвига липосомы, которую предполагается получить, и заданных свойств.
Как только липосомальный фармацевтический препарат готов, эффективность инкапсулирования лекарственного средства в липосоме может быть определена с помощью обычных технологических приемов. Методики определения эффективности инкапсулирования липосомы включают в себя ультрафильтрацию, диализ, колоночную хроматографию, центрифугирование, и тому подобное. Ультрафильтрация не используется из-за высоких требований к экспериментальному устройству; колоночная хроматография не используется из-за того, что разбавление требует большого количества элюента, а содержание лекарственного средства очень низкое, так что сложно провести определение содержания, кроме того, разбавление большого количества элюента может также привести к потере лекарственного средства в липосоме, из данных исследования можно узнать, что эффективность инкапсулирования при диализе ниже (возможно из-за разрыва липосомы после разбавления), и времени для диализа требуется много, таким образом, методика не является подходящей. Определение эффективности инкапсуляции с помощью центрифугирования имеет следующие преимущества: небольшое количество затраченного времени, небольшая степень разбавления раствором липосомы, и отсутствие потребности в дорогостоящих инструментах.
Липосомальный фармацевтический препарат данного изобретения обеспечивает не только достаточную эффективность инкапсулирования и достаточную загрузку лекарственного средства, но также не допускает выделения лекарственного средства из липосомы во время хранения вне организма, значительной утечки лекарственного средства из липосомы во время кровообращения, чтобы увеличить токсичность. Важным полезным действием липосомального лекарственного средства данного изобретения является то, что уровень выведения лекарственного средства эффективно ускорен, терапевтический индекс липосомы повышен, период полураспада значительно пролонгирован, токсичность заметно снижена по сравнению с существующими продуктами в данной области, и, таким образом, достигнут значительный терапевтический эффект лекарственного средства. Например, для липосомального фармацевтического препарата, приготовленного с использованием гидрогенизированного соевого фосфатидилхолина (HSPC) и дипальмитоил фосфатидилхолина (DPPC), их токсичность явно снижена и их терапевтический индекс значительно повышен. Напротив, если фосфолипидный бислой состоит из димиристойл фосфатидилхолина (DMPC), выведение лекарственного средства будет слишком быстрым и вызовет значительную токсичность, даже безвредность для здоровья будет уступать лекарственному средству, не связанному с белками плазмы крови. Не будучи ограниченными рамками какой-либо определенной теории, существуют предположения, что небольшой однослойный липосомальный препарат данного изобретения может ускорить выведение лекарственного средства, так как небольшой однослойный липосомальный препарат может содержать больше липосомных частиц, в которых содержится осадок лекарственного средства с маленьким размером частиц, по сравнению с большим однослойным липосомальным препаратом, если соотношение лекарственное средство/липид неизменно. Осадок лекарственного средства с маленьким размером частиц имел бы относительно большую удельную площадь поверхности, и таким образом имел большую скорость растворения при таких же условиях.
Кроме того, липосомальный фармацевтический препарат данного изобретения следует готовить с использованием соответствующих фосфолипидов, чтобы достичь эффективного выведения лекарственного средства в целевые ткани, в частности в опухоли. Желательно, чтобы фосфолипидный бислой липосомального фармацевтического препарата данного изобретения состоял из фосфолипидов с относительно высокой температурой фазового сдвига. Во время экспериментов было выяснено, что токсичность препарата значительно уменьшилась бы, а терапевтический индекс значительно повысился бы, если бы гидрогенизированный соевый фосфатидилхолин (HSPC) и дипальмитоил фосфатидилхолин (DPPC) или тому подобные вещества были использованы в препарате. Если фосфолипидный бислой состоит из димиристойл фосфатидилхолина (DMPC), выведение лекарственного средства было бы слишком быстрым и вызвало бы значительную токсичность, даже безвредность для здоровья уступала бы неинкапсулированному лекарственному средству.
Липосомальный фармацевтический препарат данного изобретения может быть введен пациенту, нуждающемуся в нем, способом, широко используемым в данной отрасли. В одном из вариантов осуществления настоящего изобретения липосомальное лекарственное средство представляет собой препарат для парентерального введения. В одном из предпочтительных вариантов осуществления настоящего изобретения липосомальное лекарственное средство вводится посредством инъекции.
Данное изобретение также представляет методику лечения заболевания, в частности опухолей у пациента, методику, содержащую в себе введение липосомального фармацевтического препарата данного изобретения пациенту, нуждающемуся в лечении. Желательно, чтобы термотерапия (как например методика радиоактивной термотерапии) также могла быть применена в сочетании пациенту с опухолью с целью увеличения терапевтического эффекта липосомального фармацевтического препарата. В данном изобретении в качестве пациента может выступать млекопитающее, предпочтительнее человек.
Данное изобретение также имеет отношение к использованию липосомального препарата или липосомального фармацевтического препарата, как указанно выше, в производстве медикамента для лечения пациента с опухолью.
Данное изобретение далее иллюстрируется следующими примерами, которые являются лишь показательными, и их не следует толковать в качестве ограничения данного изобретения.
Часть 1: Получение липосом
Пример 1
Общие методы получения липосом
1. Общий метод 1
Фосфолипид (например, гидрогенизованный соевый фосфатидилхолин (HSPC), дипальмитойл фосфатидилхолин (DPPC) или димиристойл фосфатидилхолин (DMPC)) и холестерин (молярное соотношение от 1:1 до 10:1) растворяются в органическом растворителе таком, как трет-бутиловый спирт или циклогексан, для образования прозрачного раствора. Раствор обрабатывается методом обычной лиофилизации для получения лиофилизированного порошка. Лиофилизированный порошок гидратируется при 60-65°C с (50-1000 мм) раствором аммония сульфата, раствором лимонной кислоты или раствором сульфата переходного металла (например, сульфат никеля), и взбалтывается в течение около часа для получения неоднородных многоламинарных пузырьков. Размер полученных пузырьков уменьшается микрофлюидизатором или экструзийным аппаратом высокого давления для получения липосом. Образец полученных липосом разбавляется в 200 раз 0,9% раствора NaCl и выявляется НаноZS. Экстралипосомальный буферный раствор удаляется посредством ультрафильтрационного аппарата для образования динамического трансмембранного градиента. Раствор гидрохлорида митоксантрона (10 мг/мл) добавляется в пустые липосомы при соответствующем соотношении липосомы/лекарство, и загрузка лекарственным препаратом проводится при 60-65°C. После инкубации в течение около часа, применяется желевая эксклюзионная хроматография для определения эффективности включения вещества в желатиновую капсулу (EE).
2. Общий метод 2
Фосфолипид (например, гидрогенизированный соевый фосфатидилхолин (HSPC), дипальмитойл фосфатидилхолин (DPPC) или димиристойл фосфатидилхолин (DMPC)) и холестерин (молярное соотношение от 1:1 до 10:1) смешиваются, и в то же время добавляется дистеаройл фосфатидодутаноламин модифицированного полиэтиленгликоля (DSPE-PEG) в 0,1-20% посредством заноса фофолипида. Полученная смесь растворяется в органическом растворителе таком, как трет-бутиловый спирт или циклогексан, для образования прозрачного раствора. Раствор обрабатывается методом обычной лиофилизации для получения лиофилизированного порошка. Лиофилизированный порошок гидратируется при 60-65°C с (50-1000 мм) раствором аммония сульфата, раствором лимонной кислоты или раствором сульфата переходного металла (например, сульфат никеля), и взбалтывается в течение около часа для получения неоднородных многоламинарных пузырьков. Размер полученных пузырьков уменьшается микрофлюидизатором или экструзийным аппаратом высокого давления для получения липосом. Образец полученных липосом разбавляется в 200 раз 0,9% раствора Nad и выявляется HanoZS. Экстралипосомальный буферный раствор удаляется посредством ультрафильтрационного аппарата для образования динамического трансмембранного градиента. Раствор гидрохлорида митоксантрона (10 мг/мл) добавляется в пустые липосомы при соответствующем соотношении липосомы/лекарство, и загрузка лекарственным препаратом проводится при 60-65°C. После инкубации в течение около часа, применяется желевая эксклюзионная хроматография для определения эффективности включения вещества в желатиновую капсулу (EE).
Пример 2
Получение липосомы митоксантрона PLM60
HSPC, холестерин и DSPE-PEG2000 в весовом соотношении 3:1:1 были растворены в 95% трет-бутилового спирта для образования прозрачного раствора. Раствор обработан методом лиофилизации для получения лиофилизированного порошка. Лиофилизированный порошок гидратируется при 60-65°C с (50-1000 мм) раствором аммония сульфата (300 mM) и взбалтывается в течение около часа для получения неоднородных многоламинарных пузырьков, имеющих конечную концентрацию фосфолипида в количестве 96 мг/мл. Размер полученных пузырьков был уменьшен микрофлюидизатором для получения липосом. Образец полученных липосом разбавляется в 200 раз 0,9% раствором NaCl и выявляется HanoZS, имеющим средний размер около 60 нм и основной пик между 40 нм и 60 нм. Экстралипосомальный раствор аммония сульфата был удален посредством ультрафильтрационного аппарата и замещен раствором с 250 мм сахарозы и 50 мм глицина для образования динамического трансмембранного градиента. В пустые липосомы добавлен раствор гидрохлорида митоксантрона (10 мг/мл) при соотношении 16:1 липосомы/лекарство, и проведена загрузка лекарственным препаратом при 60-65°C. После инкубации в течение около часа, эффективность включения вещества в желатиновую капсулу (ЕЕ) посредством желевой эксклюзионной хроматографии была определена как 100%. Полученные липосомы названы PLM60.
Пример 3
Получение липосомы митоксантрона PLM85
HSPC, холестерин и DSPE-PEG2000 в весовом соотношении 3:1:1 были растворены в 95% трет-бутилового спирта для образования прозрачного раствора. Раствор был обработан методом лиофилизации для получения лиофилизированного порошка. Лиофилизированный порошок гидратируется при 60-65°C с (300 мм) раствором аммония сульфата (300 мм) и взбалтывается в течение около часа для получения неоднородных многоламинарных пузырьков, имеющих конечную концентрацию фосфолипида в количестве 96 мг/мл. Размер полученных пузырьков уменьшен экструзийным аппаратом высокого давления для получения липосом. Образец полученных липосом разбавляется в 200 раз раствором NaCl и выявляется HaHoZS, имеющим средний размер около 85 нм. Экстралипосомальный раствор аммония сульфата удален посредством ультрафильтрационного аппарата и замещен раствором с 250 мм сахарозы и 50 мм глицина для образования динамического трансмембранного градиента. В пустые липосомы был добавлен раствор гидрохлорида митоксантрона (10 мг/мл) при соотношении 16:1 липосомы/лекарство, и была проведена загрузка лекарственным препаратом при 60-65°C. После инкубации в течение около часа, эффективность включения вещества в желатиновую капсулу (ЕЕ) посредством желевой эксклюзионной хроматографии была определена как 100%. Полученные липосомы названы PLM85.
Пример 4
Получение липосомы митоксантрона PLM100
Для получения липосомы гидрохлорид митоксантрона PLM100, имеющего идентичный состав, как и PLM60 и PLM85, но размер липосом 100 нм, использовался такой же метод, который описан в Примере 3.
Пример 5
Получение липосомы митоксантрона PLM60-dppc
DPPC, холестерин и DSPE-PEG2000 в весовом соотношении 3:1:1 были смешаны, остальные шаги такие же, как в Примере 2. Полученные липосомы названы PLM60-dppc.
Пример 6
Получение липосомы митоксантрона PLM60-dmpc
DMPC, холестерин и DSPE-PEG2000 в весовом соотношении 3:1:1 были смешаны, остальные шаги такие же, как в Примере 2. Полученные липосомы названы PLM60-dmpc.
Пример 7
Получение липосомы митоксантрона PLM60-dmpc-0.1
DMPC, холестерин и DSPE-PEG2000 в весовом соотношении 3:1:0.1 были смешаны, остальные шаги такие же, как в Примере 2. Полученные липосомы названы PLM60-dmpc-0.1.
Пример 8
Получение липосом адриамицина PLD60
Адриамицин был замещен на митоксантрон во время нагрузки лекарства, остальные шаги такие же, как в Примере 2. Полученные липосомы названы PLD60.
Часть 2: Высвобождение лекарственного средства различных составов липосом
Пример 9
Различия высвобождения лекарственного препарата между липосомой адриамицина PLD60 и липосомой митоксантрона PLM60
В данном примере митоксантрон и адриамицин были загружены под градиентом pH. Если в высвобожденное средство добавляется определенная концентрация хлорид аммония, свободные молекулы аммония диффундируют во внутреннюю фазу липосомы при помощи градиента, так что pH внутренней фазы будет повышен и протонированный лекарственный препарат во внутренней фазе будет превращен в нейтральную форму, которая может диффундировать через мембрану. Данный процесс сможет ускорить в то же время растворение осадка во внутренней фазе липосомы. Скорость высвобождения лекарственного препарата регулировалась как растворением осадка, так и проницаемостью мембраны липосомы. Условия высвобождения лекарственного препарата были следующими. Липосомы были разбавлены в 25 раз высвобожденным средством. Высвобожденное средство было изотоническим, с 7,4 pH, и концентрацией 2, 10 и 40 мм хлорид аммония соответственно. Разбавленные липосомы были помещены в диализные трубки, и на 2 мл разбавленной липосомы выполнен диализ посредством 400 мл высвобожденного средства при 37°C. Спустя 96 часов для анализа в разные моменты времени взяты образцы.
Полученные данные были подвергнуты регрессионному анализу. В высвобожденном средстве с 2, 10 и 40 мм хлорид аммония периоды полураспада высвобождения лекарственного препарата PLD60 составили 94.3, 31.9 и 11.2 часов соответственно. В отношении PLM60 явного высвобождения в трех высвобожденных средствах не наблюдалось. Так как PLD60 и PLM60 не имеют различий по составу и размеру, различие динамической характеристики высвобождения лекарственного препарата может объясняться их различными фармацевтическими свойствами. Адриамицин, как и митоксантрон, является антрациклинным антибиотиком, и их различия заключаются в том, что адриамицин содержит одну разделимую группу при нормальном pH, в то время как митоксантрон содержит две разделимые группы (pKa=8.15) при нормальном pH. Пример иллюстрирует, что лекарственный препарат с мультиразделимыми группами такой, как митоксантрон, во время применения метода загрузки может образовывать смешанный осадок с противоионами, так что высвобождение лекарственного препарата в лабораторных условиях значительно замедляется. С другой стороны, лекарственный препарат с одноразделимой группой такой, как адриамицин, может быть высвобожден слишком быстро даже в высвобожденном средстве без плазмы во время применения липосомы маленького размера.
Пример 10
Характер высвобождения липосом митоксантрона различных размеров
Для сравнения характера высвобождения липосом митоксантрона различных размеров были взяты два условия высвобождения.
Условие высвобождения 1: липосома была разбавлена в 25 раз высвобожденным средством. Высвобожденное средство содержало в себе 50% человеческой плазмы, с помощью глюкозы стало изотоническим и имело 7,4 pH. Другие состояния идентичны описанным в Примере 9. Полученные данные подвергнуты регрессионному анализу. Результат показал, что период полураспада высвобождения PLM60 составил 56.4 часа, в то время как PLM85 не был значительно высвобожден при тех же условиях.
Условие высвобождения 2: использовалось высвобожденное средство, содержащее 50% человеческой плазмы и 20 мм хлорид аммония, другие условия идентичны описанным в Примере 9. Полученные данные подвергнуты регрессионному анализу. Результат показал, что период полураспада высвобождения PLM60 составил 26.2 часа, в то время как период полураспада высвобождения PLM85 составил 36.7 часов.
Данный пример в достаточной мере показал, что высвобождение лекарственного препарата может значительно увеличиться путем уменьшения размера липосомы.
Пример 11
Характер высвобождения липосом митоксантрона с различными составами мембраны
Были использованы такие же условия освобождения, как описано в Примере 9. Результат показал, что период полураспада высвобождения PLM60-DPPC составил 116 часов, период полураспада высвобождения PLM60-DMPC составил 26 часов, и период полураспада высвобождения PLM60-DMPC-0.1 составил 15 часов. Данный пример показал, что использование фосфолипида при более низкой температуре фазового сдвига Tm может ускорить высвобождение лекарственного препарата. Однако если высвобождение лекарственного препарата было ускорено чрезмерно, так же может увеличиться токсичность, и это далее подтверждено следующими примерами.
Часть 3: Фармакокинетика in vivo
Пример 12
Фармакокинетический характер PLM60 в мышах Кунымина и сравнение PLM60 и свободного митоксантрона
Данный пример проводился на мышах самцах Кунымина с весом тела около 20 г.В вену хвостов мышей были введены различные уровни доз митоксантрона. Дозирование PLM60 составило 1, 2 и 4 мг/кг, и дозирование свободного митоксантрона (FM) составило 2 мг/кг. В различные моменты времени были взяты образцы плазмы. Методы обработки и диагностирования образцов плазмы описаны в документе: Методы в энзимологии, Том: 391, стр.176-185. Результаты отображены в Таблице 1 и на Рис.1, где четко видно, что период полураспада митоксантрона значительно увеличился посредством инкапсуляции липосом. При такой же дозировке время задержки PLM60 в кровообращении составило 32 раза от времени задержки FM, и AUC 3700 раз от времени задержки FM. Зависимость AUC от дозы показала, что PLM60 имеет линейную фармакокинетику in vivo.
Пример 13
Распределение PLM60 и FM в тканях мышей-опухоленосителей
Наблюдались очевидные различия между распределением в тканях PLM60 и FM в мышах-опухоленосителях. В данном примере использовались мыши самцы Кунымина с весом тела около 20 г. Мыши были привиты в правую подмышку клетками саркомы S-180 в дозе 5×105. Лекарственные препараты были введены в вену мышей, когда опухоль увеличилась до 0,4-0,7 г.После введения лекарственного препарата, мыши были умерщвлены в разные моменты времени и их ткани были вынуты для определения концентрации митоксантрона. Ткани включали в себя сердце, печень, селезенку, легкие, почки, кишки, костный мозг и опухоли. Результаты показали, что PLM60 четко нацеливался на опухолевые ткани. Подробные данные отображены в Таблице 2 и на фиг.2.
Пример 14
Фармакокинетическое сравнение различных составов липосом
Использованные в данном примере животные сходны с теми, которые использовались в Примере 12. PLM60-DPPC, PLM60-DMPC-0.1 и PLM60-DMPC в дозе 4 мг/кг были введены в вену в хвосте мышей. Данные отображены в Таблице 3 и на Рис.3. Показано, что фармакокинетика липосомальных лекарственных препаратов значительно изменилась с изменением состава мембраны липосомы. MRT значения по PLM60-DPPC, PLM60-DMPC-0.1 и PLM60-DMPC in vivo составили 14.22, 7.914 и 10.123 часов, соответственно. Различие между PLM60-DPPC и PLM60-DMPC заключалось в длине углеводородных цепочек фосфолипидов, которые составляли 16 и 14 углеродов соответственно. Длина ациловой цепочки могла существенно повлиять на проницаемость мембраны фосфолипидного бислоя. Температура фазового сдвига DPPC составила 41°C и температуре фазового сдвига DMPC - 23°С. Различие между PLM60-DMPC-0.1 и PLM60-DMPC заключалось в уровне PEG. Высвобождение липосомального лекарственного препарата в плазме зависит от двух факторов: первый это высвобождение липосомального лекарственного препарата через фосфолипидный бислой, и другой это клиренс посредством липопротеина и ретикулоэндотелиальной системы (RES). Так как PEG препарата PLM60-DMPC-0.1 не был полным, высвобождение, вызванное компонентами плазмы, имело на него большее влияние.
Часть 4: Сравнение токсичности различных составов
Пример 15 Сравнение кратковременного токсичного эффекта между PLM60 и FM
100 мышей Кунымина (половина самцов и половина самок) с весом тела 18-22 г были поделены на две группы, в каждой группе по 10 мышей, половина самцов и половина самок. К мышам 1-5 групп были применены различные уровни дозировки FM, в то время как к мышам 6-10 групп был применен равнозначный уровень дозировки липосомального лекарственного препарата. Наблюдались изменения веса тела, и записывалось время смерти каждого животного. Умершие животные были вскрыты. Результаты всех групп отображены в Таблице 4, которые показали, что кратковременный токсичный эффект PLM60 была намного меньше, чем токсичность FM.
Таблица 4: Сравнение кратковременного токсичного эффекта PLM60 FM на мышах Кунымена
Пример 16
Сравнение кратковременного токсичного эффекта различных составов липосом
90 мышей самцов Balb/c с весом тела 18-22 г были поделены на 9 групп, в каждой по 10 мышей. Мышам 1 группы было назначено 6 мг/кг FM, в то время как мышам остальных 8 групп было назначено 6 и 12 мг/кг PLM60, PLM60-DPPC и PLM60-DMPC-0.1 и PLM60-DMPC соответственно. Наблюдались изменения веса тела, и записывалось время смерти каждого животного. Умершие животные были вскрыты. Результаты смерти мышей группы, принимавших FM, и групп, принимавших липосомальный лекарственный препарат, отображены в Таблице 5. Данный эксперимент показал следующий порядок кратковременного токсичного эффекта: PLM60<PLM60-DPPC<PLM60-DMPC-0.1 FM<PLM60-DMPC. Данный эксперимент также подтвердил, что высвобождение лекарственного препарата может быть далее ускорено использованием небольших униламинарных пузырьков и фосфолипида с меньшим Tm как композиция бислоя, такого как PLM60-DMPC, таким образом, вызывая большую токсичность в организме. Следует отметить, что токсичность липосом с неполным PEG была ниже, чем токсичность липосом с более полным PEG. Это может быть отнесено к тому, что под воздействием липопротеина и разрушающего действия иммунной системы во время кровообращения, PLM60-DMPC-0.1 с неполным PEG будет высвобождать лекарственный препарат раньше по сравнению с PLM60-DMPC и не будет высвобождать внезапно в важных тканях, таким образом, проявляя меньшую токсичность, но токсичность PLM60-DMPC-0.1 являлась все еще приблизительно равной токсичности свободного митоксантрона.
Пример 17
Сравнение токсичности составов липосом различных размеров
В сравнении токсичности PLM60, PLM85 и PLM100 использовались мыши самцы С57 с весом тела 18-22 г. Доза составляла 9 мг/кг. Результаты показали, что разности веса тела, вызванные составами трех липосом, были равнозначны, что подтверждает отсутствие значительного различия токсичности составов трех липосом при экспериментальных условиях. У мышей группы, получавшей FM, вес тела уменьшился на 30% и около 20% мышей умерли.
Часть 5: Антиопухолевые действия в организме
Пример 18
Сравнение лечебных действий PLM60 и FM на саркому S-180
Мыши носители асцитной опухоли, которые 7 дней назад были привиты опухолевыми клетками S180, были умерщвлены обезглавливанием, и была извлечена и разбавлена RPMI 1640 средством молочная вязкая асцитическая жидкость. После разбавления число опухолевых клеток было установлено до уровня 2,5×106 клетки/мл. 0,2 мл суспензии опухолевых клеток, содержащих около 5×105 опухолевых клеток, было привито в подмышку передней конечности мышей самцов КМ с весом тела 18-22 г. После инокуляции клетки в суспензии клеток резидуальной опухоли были подсчитаны под оптическим микроскопом; живые опухолевые клетки были больше, чем 95%. Число привитых мышей составило 80.
Через семь дней после инокуляции было выбрано и поделено на 5 групп по размеру опухоли и весу тела 39 мышей с четко выраженными опухолями, имеющими диаметр около 5 мм; то есть 7 мышей в полной контрольной группе, по 8 мышей в каждой из групп, получавших 4 мг/кг PLM60, или 6 мг/кг PLM60, или 4 мг/кг FM, или 6 мг/кг FM. Мышам делали внутривенное введение.
После введения мыши размножались нормально. Диаметр опухолей измерялся 3 раза в неделю при помощи штангенциркуля, в то же время измерялся вес тела. Размер опухоли (TV) был рассчитан по следующей формуле: V=1/2×а×b2, где а и b означают длину и ширину соответственно. Размеры опухолей были рассчитаны с использованием результатов измерения. Мышей умертвили обезглавливанием на 21-ый день после инокуляции, опухоли вынули и взвесили. Уровень подавления опухоли (%) рассчитан по следующей формуле: уровень подавления опухоли = (1 - средний вес опухоли в группе, принимавшей лекарственный препарат / средний вес опухоли в контрольной группе)×100%. Результаты эксперимента были протестированы по t-критерию.
Результаты, представленные в таблице 6 показали, что рост солидной опухоли S180 был значительно подавлен в группе, принимавшей 4 мг/кг LM60, и группе, принимавшей 6 мг/кг PLM60.
Пример 19
Лечебное действие PLM60 и FM на L1210 образец асцита
Мыши носители асцитной опухоли BDF1, привитые клетками асцитной опухоли L1210 семь дней назад, были умерщвлены обезглавливанием, и была извлечена при асептическом условии и разбавлена RPMI 1640 средством молочная вязкая асцитическая жидкость. После разбавления число опухолевых клеток было установлено до уровня 2,5×106 клетки/мл. 0.2 мл суспензии опухолевых клеток, содержащих около 5×105 опухолевых клеток, было привито в брюшную полость 7-8 недельных мышей самок BDF1. После инокуляции клетки в суспензии клеток резидуальной опухоли были подсчитаны под оптическим микроскопом; живые опухолевые клетки были больше, чем 95%.
Спустя 24 часа мышей поделили на 8 групп по весу тела и назначили FM в дозах 2, 4 и 8 мг/кг и PLM60 в дозах 2, 4, 6 и 8 мг/кг путем введения в объеме 20 мл/кг в вену в хвосте мышей соответственно. После введения мыши размножались нормально. Вес тела измерялся 3 раза в неделю, наблюдалось и записывалось время смерти каждого животного, и было рассчитано время выживания. Для оценки времени выживания каждой из групп были использованы среднее время выживания (MST) и срединное время выживания. Экспериментальное исследование проводилось в течение 60 дней после инокуляции.
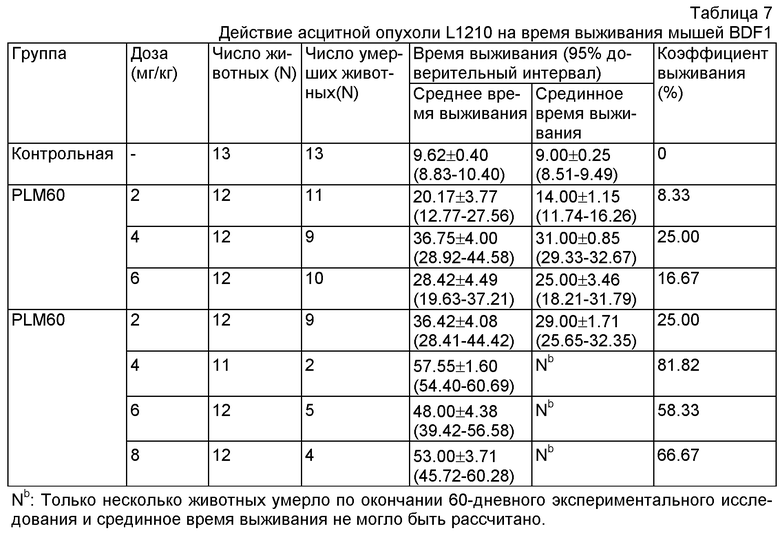
Данные были проанализированы с помощью статистической программы SPSS 11.5. Результаты показали, что все у всех административных групп проявилось значительное увеличение времени выживания в сравнении с контрольной группой, и у группы, принимавшей PLM60 (8 мг/кг), проявилось значительное улучшение лечения в сравнении с группой, принимавшей FM в той же дозе (P<0.05). Результаты отображены в Таблице 7.
Пример 20
Лечебные действия PLM60 и FM на образец метастаза печени L1210
Мыши носители асцитной опухоли BDF1, привитые клетками асцитной опухоли L1210 семь дней назад, были умерщвлены обезглавливанием, и была извлечена при асептическом условии и разбавлена RPMI 1640 средством молочная вязкая асцитическая жидкость. После разбавления число опухолевых клеток было установлено до уровня 2,5×105 клетки/мл. 0,2 мл суспензии опухолевых клеток, содержащих около 5×104 опухолевых клеток, было привито внутривенно 7-8 недельным мышам самцам BDF1. После инокуляции клетки в суспензии клеток резидуальной опухоли были подсчитаны под оптическим микроскопом; живые опухолевые клетки были больше, чем 95%. Всего было привито 62 мыши.
Спустя 24 часа, мышей поделили на группы и назначили введение лекарства. После введения мыши размножались нормально. Вес тела мышей измерялся три раза в неделю, и наблюдалось и записывалось время смерти каждой мыши, также было рассчитано время выживания. Экспериментальное исследование выполнялось в течение 60 дней после инокуляции.
Результат показал, что все мыши в контрольной группе умерли между 11-ым и 14-ым днем после инокуляции, все мыши в группах, принимавших три степени дозировки FM, умерли между 11-ым и 17-ым днем после инокуляции, все мыши в группе, принимавшей 2 мг/кг PLM60, умерли на 39-ый день после инокуляции, и ни одна мышь в группе, принимавшей 8 мг/кг PLM60, не умерла во время исследования.
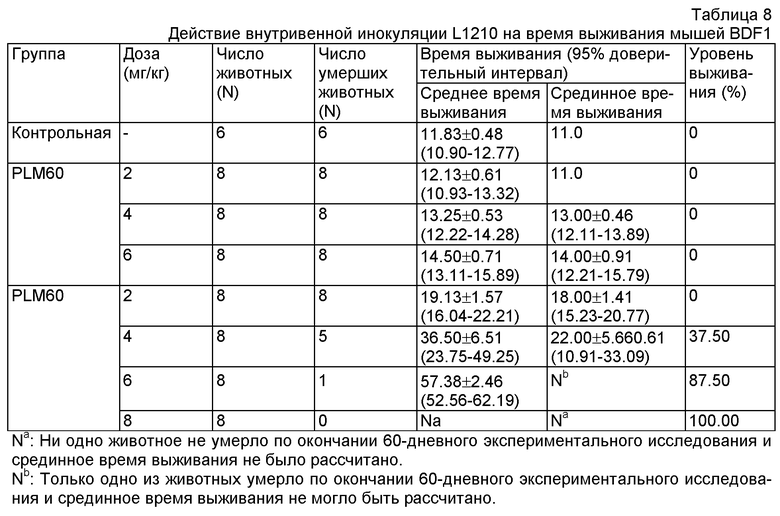
Данные были проанализированы с помощью статистической программы SPSS 11.5. Результаты показали, что у группы, принимавших 6 мг/кг FM, и всех групп, принимавших липосомный лекарственный препарат, проявилось значительное увеличение времени выживания мышей в сравнении со свободным митоксантроном. Результаты показаны в Таблице 8.
Пример 21
Лечебное действие липосомального митоксантрона различного размера на асцитную опухоль L1210
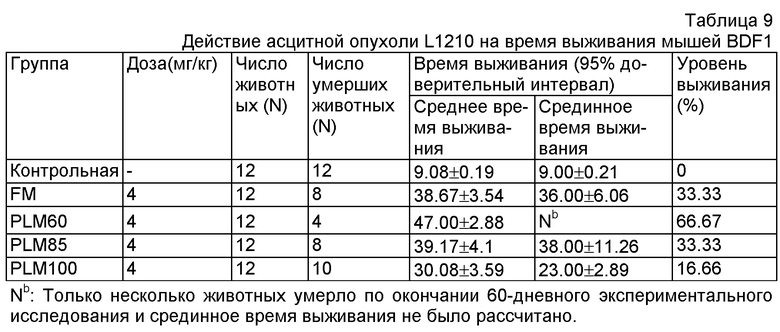
Схема эксперимента и метод обработки данных такие же, как в Примере 19. Было установлено 5 групп, включая контрольную группу, группу, принимавшую FM, группу, принимавшую PLM60, группу, принимавшую PLM85, и группу, принимавшую PLM100. Дозировка введения для мышей каждой группы составила 4 мг/кг. Результаты отображены в Таблице 9. Результаты показали, что липосома меньшего размера оказала лучшее лечебное действие.
Некоторые предпочтительные примеры данного изобретения описаны выше, но данные примеры никоим образом не направлены на ограничение объема изобретения. Кроме того то, что было описано и проиллюстрировано в данном тексте, обычный квалифицированный специалист в данной области ясно поймет другие модификации, вариации и изменения данного изобретения после прочтения раскрытия данного изобретения, и все из них должны подпадать под рамки защиты данного изобретения. Все патенты, опубликованные патентные заявки и публикации, цитированные в данном документе, включены посредством ссылки, как и их полные тексты включены в документ.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРИМЕНЕНИЕ ЛИПОСОМЫ МИТОКСАНТРОНА ДЛЯ ЛЕЧЕНИЯ НЕХОДЖКИНСКОЙ ЛИМФОМЫ | 2019 |
|
RU2804477C2 |
| Применение липосомы митоксантрона гидрохлорида | 2021 |
|
RU2821030C1 |
| ПРИМЕНЕНИЕ ЛИПОСОМЫ МИТОКСАНТРОНА ГИДРОХЛОРИДА ДЛЯ ЛЕЧЕНИЯ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ | 2021 |
|
RU2806277C1 |
| ЛИПОСОМА, ИМЕЮЩАЯ ВНУТРЕННЮЮ ВОДНУЮ ФАЗУ, СОДЕРЖАЩУЮ СОЛЬ СУЛЬФОБУТИЛОВОГО ЭФИРА ЦИКЛОДЕКСТРИНА | 2010 |
|
RU2575793C2 |
| ЗАМЕДЛЕННОЕ ВЫСВОБОЖДЕНИЕ ПРОТИВОИНФЕКЦИОННЫХ АГЕНТОВ | 2006 |
|
RU2438655C2 |
| НОВЫЕ КОМПОЗИЦИИ ЛИПОСОМ | 2006 |
|
RU2454229C2 |
| СТАБИЛИЗИРУЮЩИЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ КАМПТОТЕЦИНА | 2016 |
|
RU2833053C2 |
| СТАБИЛИЗИРОВАННЫЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ КАМПТОТЕЦИНА | 2016 |
|
RU2732567C2 |
| СРЕДСТВО, УЛУЧШАЮЩЕЕ ПРОТИВООПУХОЛЕВЫЙ ЭФФЕКТ, СОДЕРЖАЩЕЕ ЛИПОСОМАЛЬНОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ ОКСАЛИПЛАТИН, И ПРОТИВООПУХОЛЕВОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ ЛИПОСОМАЛЬНОЕ СРЕДСТВО | 2009 |
|
RU2492863C2 |
| КОМБИНАЦИОННЫЕ ЛИПОСОМАЛЬНЫЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ РАКА | 2012 |
|
RU2640934C2 |
Группа изобретений относится к медицине и касается липосомного лекарственного препарата, содержащего мультивалентный ионный лекарственный препарат в качестве активного вещества с двумя или несколькими диссоциирующимися группами с константой диссоциации 4,5-9,5, где липосома имеет размер около 30-80 мм и фосфолипидный бислой имеет фосфолипид с температурой фазового перехода выше температуры тела, так что температура фазового перехода липосомы выше температуры тела; способа лечения опухоли у пациента, включающего введение указанного липосомного лекарственного препарата. Группа изобретений обеспечивает повышение эффективности терапевтического индекса липосомного препарата, особенно повышение эффективности контролирования выведения липосомального лекарственного средства после нацеливания на пораженную опухолью область, снижение потери лекарственного средства во время циркуляции липосомального лекарственного средства в крови и повышение эффективности высвобождения лекарства в целевые ткани. 5 н. и 12 з.п. ф-лы, 21 пр., 3 ил., 9 табл.
1. Липосомный лекарственный препарат, в котором (1) липосомный лекарственный препарат содержит мультивалентный ионный лекарственный препарат в качестве активного вещества с двумя или несколькими диссоциирующимися группами с константой диссоциации 4,5-9,5; (2) липосома липосомного лекарственного препарата имеет размер 30-80 нм; (3) двухслойная структура липосомы содержит фосфолипид с температурой фазового перехода (Tm) выше, чем температура тела, холестерин и гидрофильный модифицированный полимером липид; (4) интралипосомная фаза липосомы содержит мультивалентный противоион; и (5) температура фазового перехода (Tm) липосомы выше температуры тела, где содержание фосфолипида с температурой фазового перехода (Tm) выше, чем температура тела, составляет около 50-100 мол./мол.%, предпочтительно 55-95 мол./мол.%, более предпочтительно 60-90 мол./мол.% относительно общего содержания фосфолипидов в фосфолипидной двухслойной структуре.
2. Липосомный препарат по п.1, отличающийся тем, что размер липосомы составляет 35-75 нм, предпочтительно 40-70 нм, особенно 40-60 нм.
3. Липосомный препарат по любому из п.1 или 2, отличающийся тем, что константа диссоциации pKa действующего вещества составляет 5,0-9,5, предпочтительно 5,5-9,5, более предпочтительно 6,0-9,0, особенно 6,5-9,0.
4. Липосомный препарат по любому из пп.1 или 2, отличающийся тем, что мультивалентный ионный лекарственный препарат является противораковым препаратом, выбранным из митоксантрона, винкристина, винорелбина, винбластина или любой их комбинации, предпочтительно митоксантрона.
5. Липосомный препарат по любому из пп.1 или 2, отличающийся тем, что мультивалентный противоион несет два или более зарядов, противоположных действующему веществу лекарственного препарата, где мультивалентный противоион выбирают из аниона насыщенной или ненасыщенной органической кислоты, аниона неорганической кислоты или ионной формы аминокислоты и где органическую кислоту выбирают из лимонной кислоты, винной кислоты, фумаровой кислоты, щавелевой кислоты, малоновой кислоты, янтарной кислоты, яблочной кислоты и малеиновой кислоты; неорганическую кислоту выбирают из сульфат- и фосфат-анионов; аминокислоту выбирают из цистина; предпочтительным мультивалентным противоионом является цитрат-анион, сульфат- или фосфат-анион.
6. Липосомный препарат по п.5, отличающийся тем, что мультивалентым противоионом является сульфат-анион.
7. Липосомный препарат по любому из пп.1 или 2, отличающийся тем, что фосфолипид с температурой фазового перехода (Tm) выше, чем температура тела в фосфолипидной двухслойной структуре, выбирают из фосфатидилхолина, гидрогенизированного фосфатидилхолина сои, гидрогенизированного фосфатидилхолина яичного желтка, дипалмитоилфосфатидилхолина, дистеароилфосфатидилхолина или любой их комбинации.
8. Липосомный препарат по п.7, отличающийся тем, что двухслойную структуру фосфолипида, кроме того, содержит другой фосфолипид с температурой фазового перехода (Tm) выше, чем температура тела.
9. Липосомный препарат по любому из пп.1 или 2, отличающийся тем, что содержание холестерина составляет около 2-60 мол./мол.%, предпочтительно 5-55 мол./мол.%, особенно 10-50 мол./мол.%, в частности 15-45 мол./мол.%, более конкретно 20-40 мол./мол.% относительно общего содержания ингредиентов в фосфолипидной двухслойной структуре.
10. Липосомный препарат по любому из пп.1 или 2, отличающийся тем, что липиды, модифицированные с гидрофильными полимерами, выбирают из дистеароилфосфатидил этаноламина, модифицированного ПЭГ (DSPE-PEG), дистеароилфосфатидилглицерола, модифицированного ПЭГ (DSPG-PEG), холестерин, модифицированный ПЭГ (cholo-PEG), дистеароилфосфатидилэтаноламина, модифицированного поливидоном (DSPE-PVP), дистеароилфосфатидилглицерола, модифицированного поливидоном (DSPG-PVP), или холестерин, модифицированный поливидоном (chol-PVP) или любой их комбинации, в количестве 0,1-20 мол./мол.%, предпочтительно 0,3-18 мол./мол.%, более предпочтительно 0,5-15 мол./мол.%, еще более предпочтительно 0,8-12 мол./мол.%, наиболее предпочтительно 3-6 мол./мол.% относительно общего содержания фосфолипидов в фосфолипидной двухслойной структуре.
11. Липосомный препарат по п.10, отличающийся тем, что содержит гидрогенизированный фосфатидилхолин сои, холестерин и дистеароил фосфатидилэтаноламин, модифицированный ПЭГ в весовом соотношении 3:1:1, в котором дистеароилфосфатидилэтаноламин, модифицированный ПЭГ, является предпочтительно дистеароилфосфатидилэтаноламином модифицированным ПЭГ2000.
12. Способ получения липосомного лекарственного препарата по любому из пп.1-11, отличающийся тем, что включает следующие стадии: (1) приготовление липосомы размером 30-80 нм с использованием фосфолипида с температурой фазового перехода (Tm) выше, чем температура тела, холестерина и гидрофильного модифицированного полимером липида; и (2) инкапсулирование мультивалентного ионного лекарственного препарата в липосоме, где мультивалентный ионный препарат имеет две или несколько диссоциируемых групп с константой диссоциации pKa 4,5-9,5, и интралипосомная фаза липосомы содержит мультивалентный противоион.
13. Липосомный фармацевтический препарат, содержащий липосомный лекарственный препарат по любому из пп.1-11, и необязательно фармацевтически приемлемый носитель и/или вспомогательное вещество.
14. Липосомный фармацевтический препарат по п.13, отличающийся тем, что содержит соль для изменяющегося осмотического давления, буферный агент и/или антиоксидант.
15. Способ лечения опухоли у пациента, включающий введение липосомного лекарственного препарата по любому из пп.1-11, или липосомного фармацевтического препарата по любому одному из пп.13-14 пациенту, нуждающемуся в таком лечении.
16. Способ по п.15, отличающийся тем, что дополнительно лечение включает применение к пациенту термотерапии, предпочтительно радиоактивной термотерапии.
17. Применение липосомного лекарственного препарата по любому из пп.1-11 или липосомного фармацевтического препарата по любому одному из пп.13 и 14 для изготовления лекарственного средства для лечения опухоли у пациента.
| Adams DJ., "The impact of tumor physiology on camptothecin-based drug development." Curr Med Chem Anticancer Agents | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Роторно-дисковый экстрактор | 1980 |
|
SU912198A1 |
| СПОСОБ ПРОФИЛАКТИКИ И КОРРЕКЦИИ ИЗМЕНЕНИЙ КОЖИ | 2004 |
|
RU2258530C1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |

