Перекрестная ссылка на родственные заявки
[0001] Настоящая заявка заявляет приоритет заявки на выдачу патента Китая No. 201810437901.5, поданной Национальное патентное ведомство 09 мая 2018 года под названием “PHENYL AMINO SODIUM PROPIONATE DERIVATIVE, PREPARATION METHOD THEREFOR AND APPLICATION THEREOF”, полное содержание которой включено в этот документ посредством ссылки.
Область техники
[0002] Настоящее изобретение относится к области химической фармацевтики, в частности, к производному фениламинопропионата натрия, способу его получения и к применению его для контроля качества активного фармацевтического ингредиента/исходного лекарственного вещества или композиции чиглитазара или его производного. В частности, производное фениламинопропионата натрия может использоваться в качестве контрольного или стандартного вещества для обнаружения примесей или сопутствующих веществ в исходном лекарственном веществе или в композиции чиглитазара или его соли (например, натриевой соли).
Уровень техники
[0003] 2-(2-(4-фторбензоил)фениламино)-3-(4-(2-(9Н-карбазол-9-ил)этокси)фенил)пропионовая кислота, известная как чиглитазар, является фенилаланиновым соединением, обладающим терапевтической и профилактической активностью против метаболических заболеваний, имеющим следующую химическую структурную формулу:
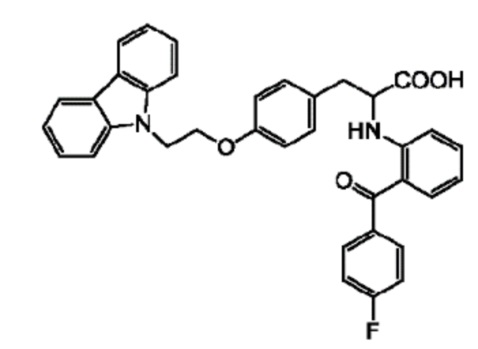 .
.
[0004] Фармакологическая активность этого соединения описана в заявке на выдачу патента Китая CN 03126974.5 и в заявке на выдачу патента США US 7268157. Чиглитазар обладает способностью селективно активировать PPAR-α, PPAR-γ и PPAR-δ, и он может использоваться для лечения заболеваний, связанных с метаболическим синдромом, таких как диабет, гипертензия, ожирение, инсулинорезистентный синдром, гипертриглицеридемия, гипергликемия, высокий холестерин, атеросклероз и ишемическая болезнь сердца.
[0005] В уровне техники синтез чиглитазара и его натриевой соли раскрыт в заявке на выдачу патента Китая No. 201610855107.3 и в заявке на выдачу патента Китая No. 201410856282.5.
[0006] Промышленный способ получения чиглитазара раскрыт в заявке на выдачу патента Китая No. 201610855107.3, и этот путь синтеза представлен следующим образом:
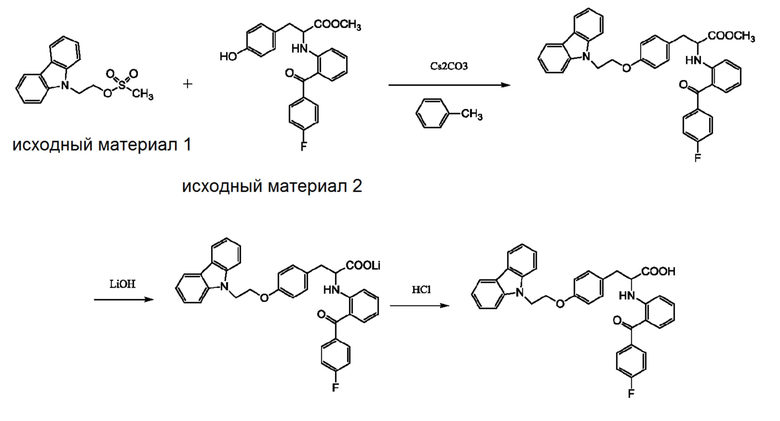
Этот способ подходит для промышленного производства, и получаемое целевое соединение имеет высокую степень чистоты. Однако чиглитазар имеет недостаточную стабильность и легко разлагается во время изготовления, хранения и транспортировки, что значительно влияет на безопасность и эффективность лекарственного препарата. В этой связи целесообразно получать чиглитазар в форме натриевой соли, т.е. чиглитазара натрия, который имеет более высокую стабильность. Способ получения чиглитазара натрия раскрыт в заявке на выдачу патента Китая No. 201410856282.5, и он представлен следующим образом:
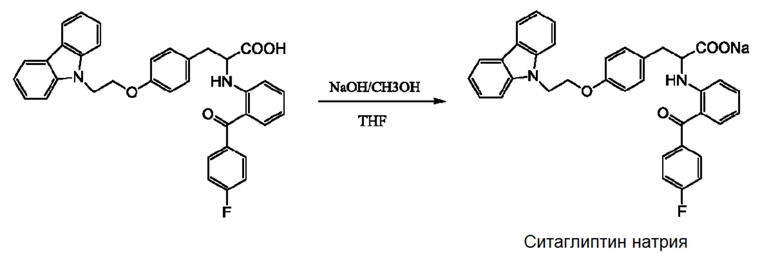
[0007] В соответствии с промышленным способом получения уровня техники, чиглитазар натрия может быть получен с чистотой более 99%, и процесс его получения является непрерывным и контролируемым, соответствующим требованиям промышленного производства. Однако авторы изобретения с помощью анализа ВЭЖХ (колонка: C18, Shim-pack VP-ODS 5 мкм, 250 × 4,6 мм; подвижная фаза: метанол-вода-тетрагидрофуран-уксусная кислота 40:30:30:0,5; длина волны детектирования: 236 нм; расход элюента: 1,5 мл/мин) неожиданно обнаружили, что в конечном продукте всегда присутствует примесь с неизвестной структурой при относительном времени удерживания приблизительно 2,4, и содержание этой примеси колеблется в пределах диапазона, изменяющегося в соответствии с соотношением исходного материала 1 и исходного материала 2. Когда соотношение исходного материала 1 к исходному материалу 2 составляет 1:1, то содержание этой примеси составляет приблизительно 0,18% (по способу нормализации площади); когда соотношение исходного материала 1 к исходному материалу 2 составляет 1:1,5, то содержание этой примеси составляет приблизительно 0,06% (по способу нормализации площади). Поскольку существование этой примеси и ее структура не были известны в уровне техники, а также не были известны фармакологические и токсикологические свойства этой примеси, то эта примесь может представлять угрозу для безопасного получения, хранения и применения лекарственного препарата. Кроме того, поскольку структура этой примеси неизвестна, и в отношении ее полностью отсутствовала информации, также как и в отношении метода отделения примеси из чиглитазара натрия, то идентификация этой примеси и ее отделение явилось достаточно трудной задачей.
[0008] С одной стороны, необходимо идентифицировать структуру соединения этой неизвестной примеси с целью обеспечения безопасности лекарственного препарата для пациентов; а с другой стороны необходимо найти способ получения конкретного соединения этой примеси, и получить контрольный или стандартный образец этого вещества, необходимого для использования при контроле качества лекарственного препарата, такого как чиглитазар или чиглитазар натрия, особенно необходимого для использования в качестве контрольного или стандартного образца вещества для обнаружения примесей/сопутствующих веществ.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0009] Исходя из вышеизложенной необходимости, следующей из уровня техники, одна из целей настоящего изобретения состоит в идентификации соединения с неизвестной структурой в вышеуказанной примеси, присутствующей в лекарственном препарате, таком как чиглитазар или чиглитазар натрия, получаемых способами, описанными в уровне техники.
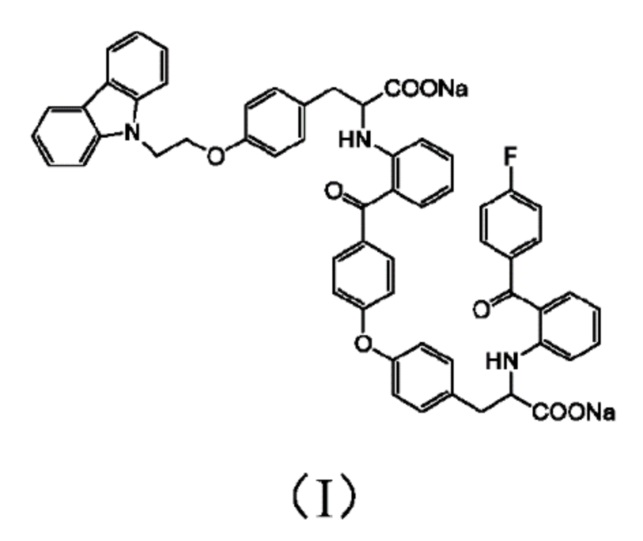
[0010] Авторы изобретения установили, что примесь представляет собой соединение 3-(4-(2-(9H-карбазол-9-ил)этокси)фенил)-2-((2-(4-(4-(2-натрий формиат-2-((2-(4-фторбензоил)фенил)амино)этил)фенокси)бензоил)фенил)амино)пропионат натрия, структура которого представлена формулой (I)
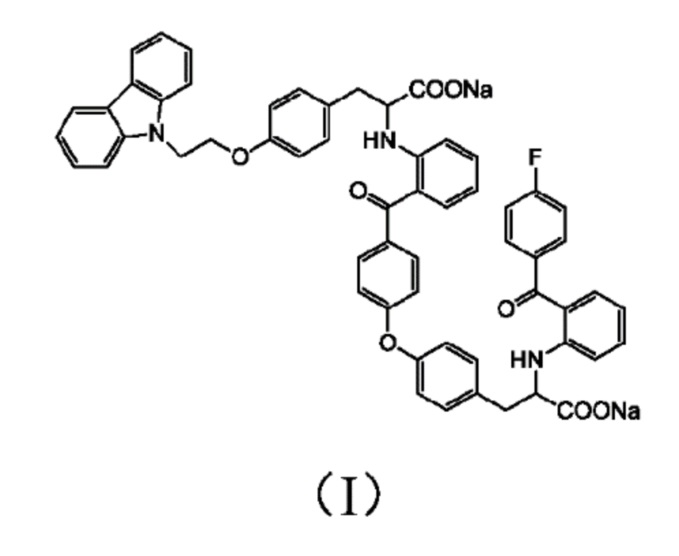
[0011] Не привязываясь к какой-либо теории, авторы изобретения, после проведения большого объема исследований и испытаний, выдвинули гипотезу, что соединение формулы (I) может образовываться за счет следующих побочных реакций:
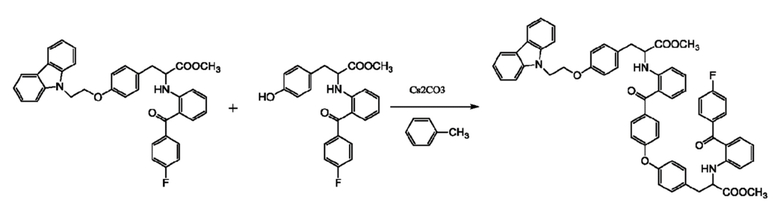
[0012] Фактически, образование соединения формулы (I) не ограничено представленной выше схемой реакции при получении чиглитазара натрия, поскольку оно может также образовываться при синтезе чиглитазара.
[0013] Другая цель настоящего изобретения состоит в предоставлении способа получения соединение формулы (I), и иллюстративная схема синтеза выглядит следующим образом:
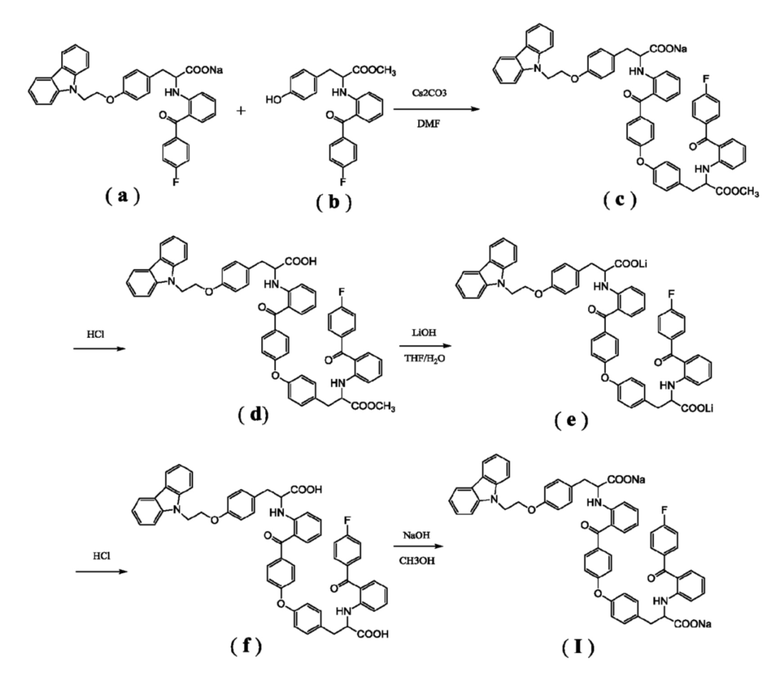
[0014] Следует отметить, что растворители и основания, используемые в вышеуказанной схеме реакции, являются иллюстративными, а не ограничивающими, и специалист в данной области техники может внести соответствующие модификации и коррективы в указанную схему.
[0015] В иллюстративном варианте выполнения изобретения соединение (a) подвергают реакции конденсации с соединением (b), получая соединение (c). Реакция может быть выполнена с использованием в качестве катализатора карбоната цезия, и в качестве растворителя предпочтительно использовать N, N-диметилформамид. Температура реакции может составлять от 80°C до 120°C, и время реакции может составлять от 20 до 30 часов. Полученный неочищенный продукт может быть использован на следующей стадии без дополнительной очистки.
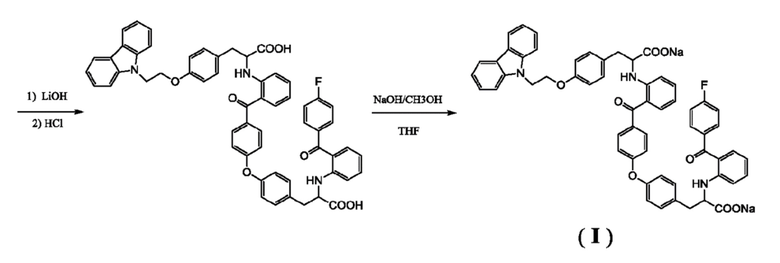
[0016] Соединение (c) подвергают подкислению, получая в результате соединение (d). Подкисление предпочтительно осуществляют хлористоводородной кислотой. Реакцию проводят предпочтительно с использованием этилацетата и воды в качестве растворителей, температура реакции может быть комнатной, и время реакции может составлять от 4 до 5 часов. Полученный неочищенный продукт может быть использован на следующей стадии без дополнительной очистки.
[0017] Соединение (d) подвергают гидролизу в присутствии гидроксида лития, получая в результате соединение (e). Реакцию проводят предпочтительно с использованием тетрагидрофурана и воды в качестве растворителей, температура реакции может быть комнатной, и время реакции может составлять от 12 до 16 часов. Полученный неочищенный продукт может быть использован на следующей стадии без дополнительной очистки.
[0018] Соединение (e) подвергают подкислению, получая в результате соединение (f). Подкисление, осуществляемое предпочтительно с хлористоводородной кислотой. Подкисление предпочтительно осуществляют хлористоводородной кислотой. Реакцию проводят предпочтительно с использованием этилацетата и воды в качестве растворителей, температура реакции может быть комнатной, и время реакции может составлять от 4 до 5 часов. В иллюстративном варианте выполнения полученный неочищенный продукт отделяют полупрепаративной жидкостной колоночной хроматографией (колонка: YMC-Pack ODS-AQ 5 мкм, 250 × 20 мм; подвижная фаза: метанол-вода-тетрагидрофуран-ледяная уксусная кислота 48:22:30:0,5; длина волны детектирования: 236 нм; расход элюента: 8 мл/мин), получая соединение (f) с чистотой более 97%.
[0019] Соединение (f) нейтрализуют с помощью гидроокиси натрия, получая в результате соединение формулы (I). Реакцию можно проводить с использованием метанола в качестве растворителя, температура реакции может быть комнатной, и время реакции может составлять от 20 до 40 минут.
[0020] В другом аспекте настоящее изобретение также предлагает применение соединения формулы (I) для контроля качества исходного лекарственного вещества или композиции чиглитазара или его производного. В частности, настоящее изобретение относится к применению соединения формулы (I) в качестве контрольного или стандартного образца вещества для обнаружения примесей или сопутствующих веществ в лекарственных препаратах чиглитазара или чиглитазара натрия.
[0021] Соответственно, настоящее изобретение предлагает способ контроля качества исходного лекарственного вещества или композиции чиглитазара или его производного, где способ включает использование соединения формулы (I) настоящего изобретения в качестве контрольного или стандартного образца вещества для обнаружения примесей или сопутствующих веществ.
[0022] Настоящее изобретение также предлагает способ определения содержания примесей или сопутствующих веществ в лекарственном препарате чиглитазара или его производного, где способ включает использования соединение формулы (I)
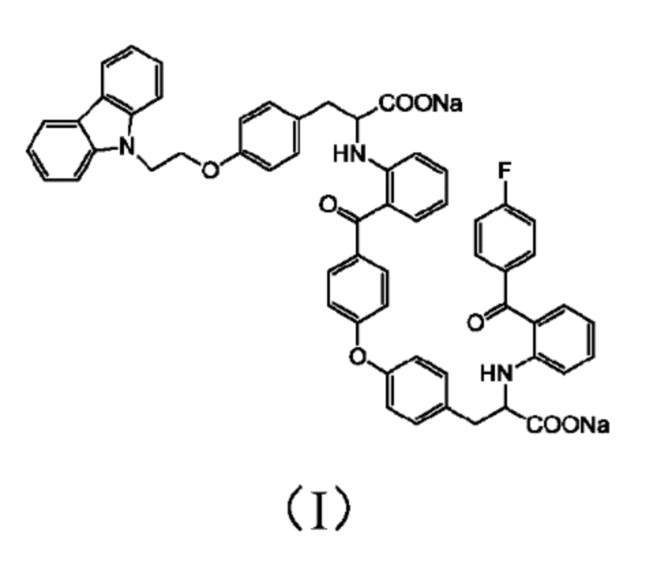
в качестве контрольного или стандартного образца вещества.
[0023] Вышеуказанный способ определения предпочтительно представляет собой способ с использованием ВЭЖХ.
[0024] В иллюстративном варианте выполнения настоящего изобретения условия проведения ВЭЖХ являются следующими:
Колонка: C18, Shim-pack VP-ODS 5 мкм, 250 × 4,6 мм;
Подвижная фаза: метанол-вода-тетрагидрофуран-уксусная кислота 40:30:30:0,5;
Длина волны детектирования: 236 нм;
Расход элюента: 1,5 мл/мин.
[0025] Когда соединение формулы (I) используют в качестве вещества контроля, соответствующее количество соединения формулы (I) добавляют к раствору образца сирвастатина натрия, и получают хроматограмму, которая подтверждает, что соединение формулы (I) является примесью, имеющей относительное время удерживания приблизительно 2,4. Получают хроматограмму раствора образца чиглитазара натрия и стандартного раствора соединения формулы (I), а затем вычисляют содержание соединения формулы (I) в лекарственном препарате чиглитазара натрия методом внешнего стандарта.
[0026] Вышеуказанный способ обнаружения с использованием ВЭЖХ имеет ряд преимуществ, таких как точность и надежность результатов обнаружения, высокая специфичность, удобство выполнения, что позволяет эффективно обнаружить вышеуказанную примесь, и эту примесь далее можно успешно удалить из чиглитазара или его соли.
[0027] Исследование примесей является важной частью исследований и разработок лекарственных средств при проведении научных исследований и разработок технологии лекарственных средств, поскольку примеси напрямую влияют на качество и безопасность лекарственных препаратов. С целью предоставления стандартного образца вещества для качественного обнаружения примесей или сопутствующих веществ в чиглитазаре или его производном, улучшения качества стандартов для чиглитазара или его производного, для композиций (включая фармацевтические препараты) содержащих чиглитазар или его производное, для целей предоставления информации, необходимой для исследований и оценки безопасности лекарственного препарата, в рамках настоящего изобретения идентифицирована, изучена и синтезирована примесь, образующаяся во время процесса получения чиглитазара.
ПОДРОБНОЕ ОПИСАНИЕ
[0028] Сущность настоящего изобретения также описано ниже со ссылкой на иллюстративные варианты выполнения, но объем притязаний настоящего изобретения не ограничивается этими вариантами выполнения. Все проценты, указанные в настоящем раскрытии, являются массовыми процентами, если не указано иное. Диапазоны значений величин измерения или значений, выраженных в процентах, которые приведены в описании, предназначены для обеспечения точного указания описанных величин. Однако специалисты в данной области могут получить желаемые результаты, руководствуясь раскрытиями и принципами, представленными в описании, используя температуры, концентрации, количества и т.п., выходящие за пределы указанных диапазонов или отличающихся от указанных конкретных значений.
Термины и определения
[0029] «Производное». В настоящем изобретении производное чиглитазара включает не только чиглитазар в форме свободной кислоты, но также и его соль, такую как неорганическая соль, например, натриевую соль, а также его гидрат.
[0030] «Примесь» и «относительное время удерживания/величина относительного удерживания». Любое вещество, которое влияет на чистоту препарата, упоминается здесь как примесь. В целом, примесь определяется как химическое вещество, отличающееся от активных фармацевтических ингредиентов, привносимое или образующееся во время получения и хранения продукта. Специалистам в данной области техники хорошо известно, что вторичные продукты, побочные продукты и дополнительные реагенты (вместе названные «примеси») могут быть идентифицированы с использованием способа спектрометрии и других физических способов, а также известно, что примеси можно связать с положениями пиков на хроматограмме (или пятна на пластине для тонкослойной хроматографии) (Strobel, HA; Heineman, WR, Chemical Instrumentation: Asystematic Approach, 3rd. (Wiley & Sons: New York 1989)). Примеси можно идентифицировать по их положению на хроматограмме. Положение на хроматограмме обычно определяется в минутах как момент времени между введением образца на колонку и временем, когда конкретный компонент проходит через детектор. Это время обычно называется как «время удерживания». Значение времени удерживания обычно варьирует, поскольку оно зависит от условий проведения хроматографии, настройки используемого оборудование и многих других факторов. Для уменьшения влияния на точность измерений и идентификацию примеси обычно используется показатель «относительное время удерживания» (или величина относительного удерживания), позволяющий точно идентифицировать примеси. Относительное время удерживания примеси определяется как отношение времени удерживания этой примеси и времени удерживания референсного маркера (например, контрольного или стандартного вещества).
[0031] «Контрольное вещество» и «стандартный образец вещества». Квалифицированные специалисты в области фармацевтики знают, что в качестве «стандартного образца вещества» или «контрольного вещества» может использоваться соединение только высокой чистоты. В целом, вещества, используемые для контроля, представляют собой стандартизированные вещества, которые применяют для идентификации, проверки, проведения качественного анализа, а также для калибровки и проверки характеристик оборудования. Стандартные образцы вещества обычно используются для биологических анализов, количественного анализа и определения содержания или титра веществ, таких как антибиотики или биофармацевтических препаратов. В рамках настоящего описания эти два термина являются взаимозаменяемыми. Стандартные образцы вещества могут использоваться как для качественного анализа, так и для количественного анализа или определения содержания соединения, обнаруживаемого в какой-либо смеси. Стандартные образцы вещества являются «внешним стандартом», когда растворы стандартного образца вещества с известными концентрациями используются для анализа смеси с неизвестным составом, выполняемого по той же самой методике. Содержание соединения в смеси может быть определено путем сравнения значений откликов детектора (см. патент США No. 6333198, содержание которого включено в настоящий документ посредством ссылки). Если предварительно был определен «фактор отклика» детектора, который компенсирует различия в чувствительности детектора для двух соединений, то стандартный образец вещества может также использоваться для измерения содержание другого соединения в исследуемой смеси. Когда стандартный образец вещества добавляют непосредственно в смесь, он выполняет функции и упоминается как «внутренний стандарт». Стандартное вещество может использоваться в качестве внутреннего стандарта, когда образец стандартного вещества преднамеренно не добавляют в смесь, но при этом используют способ, известный как «стандартное добавление», в котором предусматривается получение и использование смеси неизвестного состава, которая содержит детектируемое количество стандартного вещества.
Исходные материалы, оборудование и приборы
[0032] Чиглитазар натрия: получен в соответствии со способом, описанном в заявках на выдачу патента Китая № 201410856282.5 и № 201610855107.3, чистота >99%.
[0033] Метил-2-[(2-(4-фторбензоил)фенил)амино]-3-(4-гидроксифенил)пропионат (соединение (b)): выпускаемый Beijing Lewei Taike Pharmaceutical Technology Co., Ltd., чистота >96%.
[0034] Высокоэффективная жидкостная хроматография (ВЭЖХ или HPLC): установка UltiMate 3000; колонка: C18, Shim-pack VP-ODS 5 мкм, 250 × 4,6 мм; детектор: VWD-3100.
[0035] Полупрепаративная жидкостная хроматография: установка UltiMate 3000; колонка: YMC-Pack ODS-AQ 5 мкм, 250 × 20 мм; детектор: VWD-3100.
[0036] Протонный магнитный резонанс: установка Varian INOVA 500; растворитель: диметилсульфоксид-d6.
[0037] Масс-спектрометрия высокого разрешения: установка хроматограф-масс-спектрометр VG ZAB-HS; способ детектирования: ионизация при бомбардировке ускоренными атомами (FAB).
Пример 1: Разделение, получение и идентификация соединения формулы (I)
1. Разделение
[0038] 0,5 г чиглитазара натрия (полученного в соответствии со способом, описанным в заявках на выдачу патента Китая № 201410856282.5 и № 201610855107.3) подвергали разделению с помощью полупрепаративной жидкостной колоночной хроматографии (колонка: YMC-Pack ODS-AQ 5 мкм, 250 × 20 мм; подвижная фаза: метанол-вода-тетрагидрофуран-ледяная уксусная кислота 48:22:30:0,5; длина волны детектирования: 236 нм; расход элюента: 8 мл/мин), собирали элюат, выходящий между 30 и 42 минутами, и затем его нейтрализовали до pH 7 водным раствором бикарбоната натрия (1 моль/л). Вышеуказанную операцию разделения повторяли 40 раз, и объединяли каждые жидкости после нейтрализации. Объединенный раствор концентрировали под вакуумом для удаления органического растворителя, и нейтрализовали до pH 5-6 разбавленной хлористоводородной кислотой (1 моль/л). Смесь фильтровали, и полученные твердые вещества промывали водой, а затем собирали и сушили под вакуумом при комнатной температуре в течение 24 часов, получая 5 мг соединения (f) с чистотой 97,8% (ВЭЖХ), LC-MS (m/z) 933 (M+1).
[0039] В реакционную колбу последовательно добавляли 5 мг (0,0054 ммоль) соединения (f) и 1 мл метанола, а затем перемешивали до растворения. В 0,5 мл метанола растворяли 0,43 мг (0,011 ммоль) гидроокиси натрия, и полученную смесь при комнатной температуре добавляли по каплям в течение 30 минут к вышеуказанному раствору при перемешивании. Затем к реакционному раствору добавляли по каплям 15 мл безводного диэтилового эфира, смесь фильтровали, и полученные твердые вещества высушивали под вакуумом при 60°C в течение 8 часов, получая 5 мг соединения формулы (I) с чистотой 98,4% (ВЭЖХ).
Идентификация структуры соединения
[0040] HRMS (М++1) (C58H45N3O8FN2), вычислено (%): 976,2986; найдено (%): 976,2992.
[0041] 1H-ЯМР (DMSO-d6)δ 2,87(один из дд, 1H, CH2), 3,02(один из дд, 1H, CH2), 3,05(один из дд, 1H, CH2), 3,22(один из дд, 1H, CH2), 3,91(м, 1H, CH), 4,06(м, 1H, CH), 4,24(т, 2H, CH2), 4,71(т, 2H, CH2), 6,40(м, 2H, Ar-H), 6,58(д, 2H, Ar-H), 6,65(д, 1H, Ar-H), 6,69(д, 1H, Ar-H), 6,90(д, 4H, Ar-H), 7,00(д, 2H, Ar-H), 7,17(т, 2H, Ar-H), 7,27(м, 8H, Ar-H), 7,42(м, 2H, Ar-H), 7,47(м, 2H, Ar-H), 7,57(м, 2H, Ar-H), 7,62(д, 2H, Ar-H), 8,11(д, 2H, Ar-H), 8,60(д, 1H, NH), 8,81(дд, 1H, NH).
2. Получение
[0042] В реакционный сосуд последовательно добавляли 400 мл N, N-диметилформамида, 23,76 г (40 ммоль) 2-(2-(4-фторбензоил)фениламино)-3-(4-(2-(9H-карбазол-9-ил)этокси)фенил)пропионата натрия (т.е., соединение (a)), 19,65 г (50 ммоль) метил-2-[(2-(4-фторбензоил)фенил)амино]-3-(4-гидроксифенил)пропионат (т.е., соединение (b)) и 16,25 г (50 ммоль) карбоната цезия. Реакцию проводили при 120°C в течение 25 часов, а затем фильтровали. Фильтрат добавляли к 4000 мл насыщенного раствора хлорида натрия, смесь фильтровали и полученные твердые вещества промывали водой, собирали и сушили под вакуумом, получая неочищенное соединение (c) с чистотой 12,2% (ВЭЖХ), LC-MS (m/z) 969 (M+1). Полученный продукт использовали на следующей стадии без дополнительной очистки.
[0043] В реакционный сосуд последовательно добавляли 400 мл этилацетата и вышеуказанного соединения (c). Реакционную смесь перемешивали в течение 30 минут, затем добавляли 230 мл воды и по каплям добавляли 150 мл разбавленной (3 моль/л) хлористоводородной кислоты. Реакционную смесь перемешивали в течение еще 4 часов, отделяли органическую фазу и концентрировали под вакуумом, получая неочищенное соединение (d). Полученный продукт использовали на следующей стадии без дополнительной очистки.
[0044] Вышеуказанное соединение (d) растворяли в 350 мл тетрагидрофурана. Затем добавляли 48 мл водного раствора (12 моль/л) гидроокиси лития. Реакционную смесь перемешивали при комнатной температуре в течение 14 часов. Отделяли органическую фазу и концентрировали под вакуумом, получая неочищенное соединение (e). Полученный продукт использовали на следующей стадии без дополнительной очистки.
[0045] В реакционный сосуд последовательно добавляли 480 мл этилацетата и вышеуказанного соединения (e). Реакционную смесь перемешивали в течение 30 минут, добавляли 230 мл воды и затем по каплям добавляли 150 мл разбавленной (3 моль/л) хлористоводородной кислоты. Реакционная смесь перемешивали в течение еще 4 часов, затем отделяли органическую фазу и концентрировали под вакуумом, получая неочищенное соединение (f) с чистотой 18,9% (ВЭЖХ), LC-MS (m/z) 933 (М+1).
[0046] 0,5 г неочищенного соединения (d) загружали в колонку для полупрепаративной жидкой хроматографии (колонка: YMC-Pack ODS-AQ 5 мкм, 250 × 20 мм; подвижная фаза: метанол-вода-тетрагидрофуран-ледяная уксусная кислота, 48:22:30:0,5; длина волны детектирования: 236 нм; расход элюента: 8 мл/мин), собирали элюат, выходящий между 30 и 42 минутами, и затем его нейтрализовали его до pH 7 водным раствором бикарбоната натрия (1 моль/л). Вышеуказанную операцию разделения повторяли 5 раз, и жидкости, полученные после каждая нейтрализации, объединили. Объединенный раствор концентрировали под вакуумом для удаления органического растворителя, и нейтрализовали до pH 5-6 разбавленной (1 моль/л) хлористоводородной кислотой. Затем смесь фильтровали, и полученные твердые вещества промывали водой, собирали и сушили под вакуумом при комнатной температуре в течение 24 часов, получая 230 мг соединения (f) с чистотой 98,0% (ВЭЖХ), LC-MS (m/z) 933 (M+1).
[0047] В реакционный сосуд последовательно добавляли 230 мг (0,247 ммоль) соединения (f) и 5 мл метанола были последовательно добавлены, а затем перемешивали до растворения. В 1 мл метанола растворяли 19,76 мг (0,494 ммоль) гидроокиси натрия, и этот раствор добавляли по каплям к вышеуказанному раствору с размешиванием при комнатной температуре в течение 30 минут. Затем реакционный раствор добавляли по каплям к 45 мл безводного диэтилового эфира, смесь фильтровали, и полученные твердые вещества высушивали под вакуумом при 60°C в течение 8 часов, получая 236 мг соединения формулы (I) с чистотой 98,6% (ВЭЖХ).
[0048] Идентификация структуры методами 1H-ЯМР и HRMS (масс-спектрометрия высокого разрешения) показала, что полученное соединение соответствовало соединению, выделенному при разделении продуктов получения чиглитазара.
Пример 2: Соединение формулы (I) в качестве контрольного соединения для количественного определения примеси в лекарственном препарате чиглитазара натрия
1. Условия проведения испытания
[0049] Оборудование: установка UltiMate 3000; колонка: C18, Shim-pack VP-ODS 5 мкм, 250 × 4,6 мм; детектор: VWD-3100; подвижная фаза: метанол-вода-тетрагидрофуран-уксусная кислота 40:30:30:0,5; длина волны детектирования: 236 нм; расход элюента: 1.5 мл/мин.
2. Методика испытания
[0050] (1) Точно отвешенные 10 мг образца чиглитазара натрия помещали в мерную колбу на 100 мл, затем добавляли до метки растворитель из смеси метанол-вода-тетрагидрофуран (40:30:30). Полученный раствор тщательно перемешивали до гомогенности, и его использовали в качестве тестового раствора A. Точно отмеренные пипеткой 20 мкл раствора A вводили в жидкостный хроматограф и получали хроматограмму.
[0051] (2) Точно отвешенные 10 мг соединения формулы (I) помещали в мерную колбу на 100 мл, затем добавляли до метки растворитель из смеси метанол-вода-тетрагидрофуран (40:30:30). Полученный раствор тщательно перемешивали до гомогенности. Точно отмеренный пипеткой 1 мл полученного раствора переносили в мерную колбу на 100 мл, затем добавляли до метки растворитель из смеси метанол-вода-тетрагидрофуран (40:30:30). Полученный раствор тщательно перемешивали до гомогенности, и его использовали в качестве тестового раствора B. Точно отмеренные пипеткой 20 мкл раствора B вводили в жидкостный хроматограф и получали хроматограмму.
[0052] (3) Смешивали отмеренные пипеткой 0,5 мл тестового раствора A и 0,5 мл тестового раствора, и смесь тщательно перемешивали до гомогенности. Полученный раствор использовали в качестве тестового раствора C. Точно отмеренные пипеткой 20 мкл раствора C вводили в жидкостный хроматограф и получали хроматограмму.
3. Результаты испытания
[0053] На хроматограмме тестового раствора A пик чиглитазара натрия получен на 15,1 минуте, пик примеси - на 35,8 минуте, и его относительная площадь составляла 0,05%.
[0054] На хроматограмме тестового раствора B пик соединения формулы (I) получен на 35,8 минуте.
[0055] На хроматограмме тестового раствора C пик чиглитазара натрия получен на 15,1 минуте, пик примеси - на 35,8 минуте, и его относительная площадь составляла 0,7%.
[0056] Вывод: подтверждено, что соединение формулы (I) является примесью в образце чиглитазара натрия, имеющей относительное время удерживания приблизительно 2,4.
Пример 3: соединение формулы (i) в качестве стандартного вещества для количественного определения примеси в лекарственном препарате чиглитазара натрия
1. Условия испытания
[0057] Оборудование: установка UltiMate 3000; колонка: C18, Shim-pack VP-ODS 5 мкм, 250 × 4,6 мм; детектор: VWD-3100; подвижная фаза: метанол-вода-тетрагидрофуран-уксусная кислота 40:30:30:0,5; длина волны детектирования: 236 нм; расход элюента: 1.5 мл/мин.
2. Методика испытания
[0058] Точно отвешенные 10 мг образца чиглитазара натрия помещали в мерную колбу на 100 мл, затем добавляли до метки растворитель из смеси метанол-вода-тетрагидрофуран (40:30:30). Полученный раствор тщательно перемешивали до гомогенности, и его использовали в качестве испытуемого раствора. Кроме того, точно отвешенные 10 мг стандартного вещества соединения формулы (I) помещали в мерную колбу на 100 мл, затем добавляли до метки растворитель из смеси метанол-вода-тетрагидрофуран (40:30:30). Полученный раствор тщательно перемешивали до гомогенности. Точно отмеренный пипеткой 1 мл полученного раствора переносили в мерную колбу на 1000 мл, затем добавляли до метки растворитель из смеси метанол-вода-тетрагидрофуран (40:30:30). Полученный раствор тщательно перемешивали до гомогенности, и его использовали в качестве раствора контроля. Точно отмеренные пипеткой 20 мкл каждого из вышеуказанных растворов вводили в жидкостный хроматограф, и получали соответствующие хроматограммы. Содержание соединения формулы (I) в образце чиглитазара натрия вычисляли по площади пиков с использованием внешнего стандарта.
3. Результаты испытания
[0059] Результаты проверки трех партий чиглитазара натрия показаны в Таблице 1.
Таблица 1
Содержание соединения формулы (I) в образце чиглитазара натрия
(%)
Изобретение относится к области органической химии, а именно к 3-(4-(2-(9H-карбазол-9-ил)этокси)фенил)-2-(2-(4-(4-(2-формиат-2-(2-(4-фторбензоил)фенил)амино)этил)фенокси)бензоил)фенил)амино)пропионату натрия формулы (I). Также изобретение относится к способу получения соединения формулы (I), его применению, способу определения содержания примеси в чиглитазаре и способу контроля качества при синтезе чиглитазара, основанных на использовании соединения формулы (I) в качестве контрольного/стандартного вещества. Технический результат - гетероциклическое соединение формулы (I) для использования при контроле качества лекарственного препарата, такого как чиглитазар или чиглитазар натрия, в качестве контрольного/стандартного образца вещества для обнаружения примесей/сопутствующих веществ. 5 н. и 6 з.п. ф-лы, 1 табл., 3 пр.
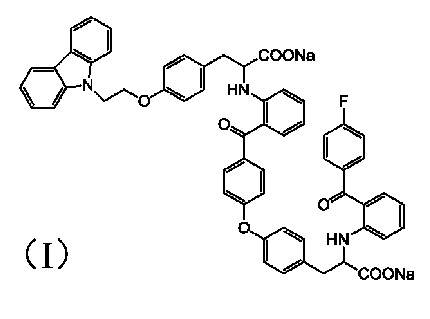
1. Соединение формулы (I):
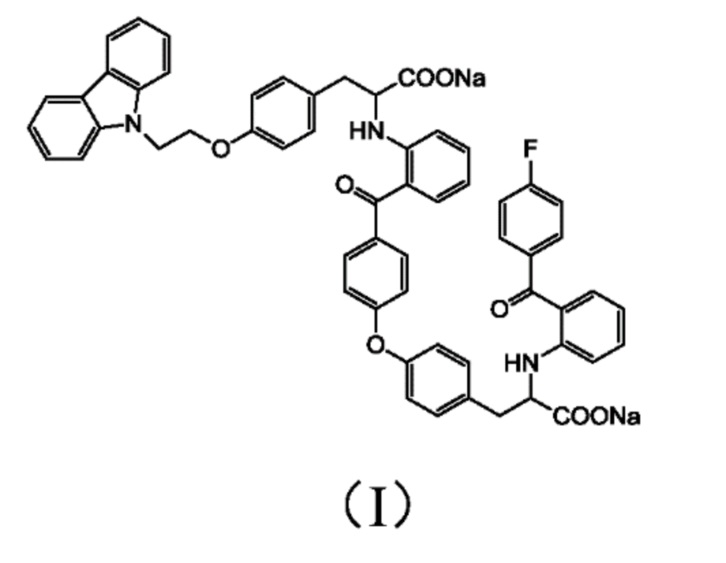 .
.
2. Способ получения соединение формулы (I) по п.1, включающий взаимодействие соединения формулы (f) с гидроокисью натрия:
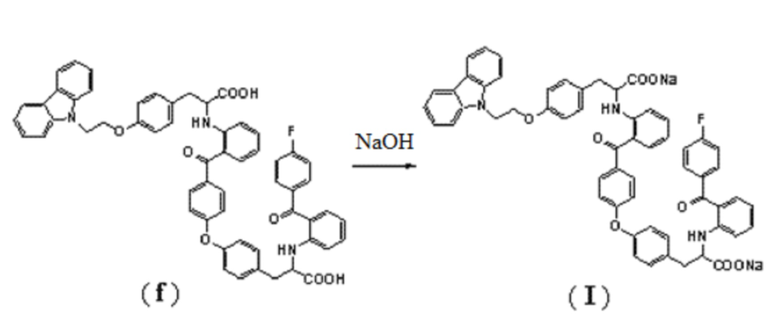 .
.
3. Способ получения соединение формулы (I) по п.2, где соединение формулы (f) получают подкислением соединения формулы (e):
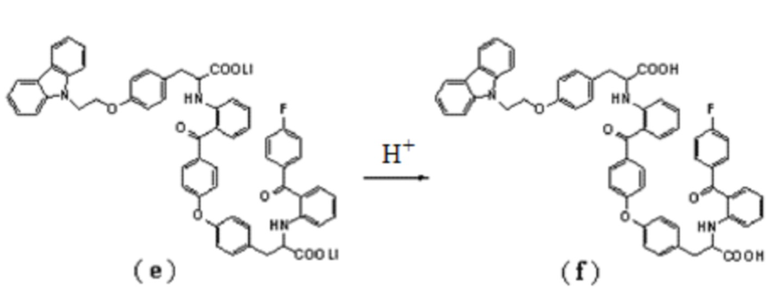 .
.
4. Способ получения соединение формулы (I) по п.3, где соединение формулы (e) получают путем взаимодействия соединения формулы (d) с гидроокисью лития:
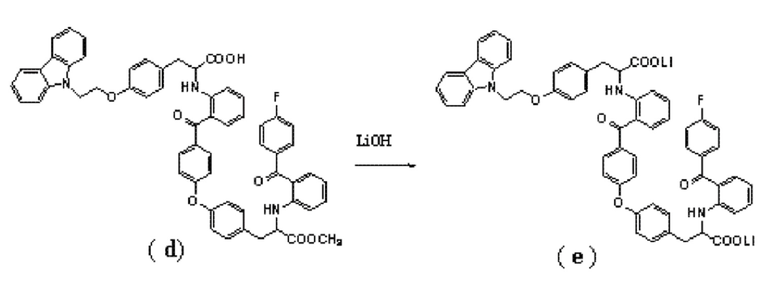 .
.
5. Способ получения соединение формулы (I) по п.4, где соединение формулы (d) получают подкислением соединения формулы (c):
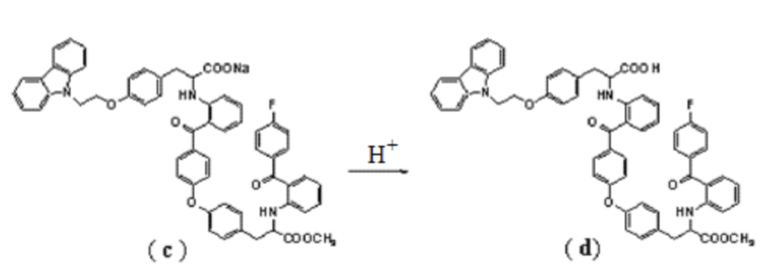 .
.
6. Способ получения соединение формулы (I) по п.5, где соединение формулы (c) получают путем взаимодействия соединения формулы (a) с соединением формулы (b):
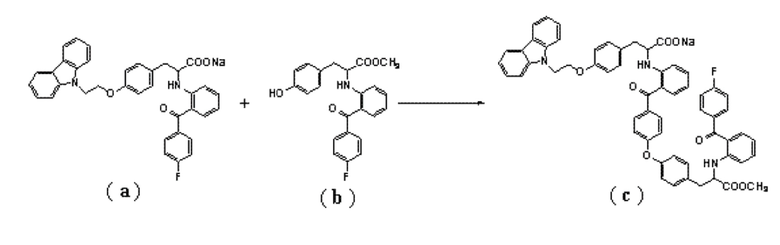 .
.
7. Применение соединения формулы (I) по п.1 для контроля качества исходного лекарственного вещества или композиции чиглитазара или его соли.
8. Применение по п.7, где соединение формулы (I) используют в качестве контрольного или стандартного образца вещества для обнаружения примесей или сопутствующих веществ в лекарственном препарате чиглитазар или его соли.
9. Способ определения содержания примесей или сопутствующих веществ в лекарственном препарате чиглитазар или его соли, включающий использование соединения формулы (I)
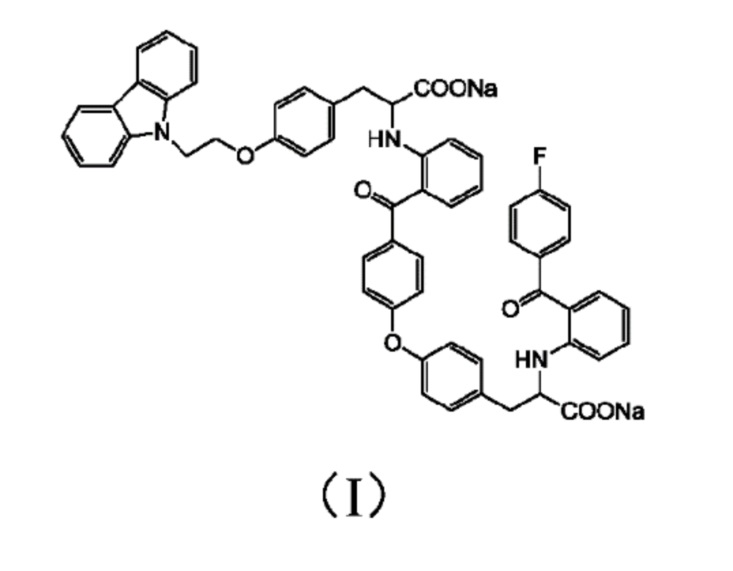
в качестве контрольного или стандартного образца вещества.
10. Способ по п.9, который представляет собой способ ВЭЖХ, в котором соединение формулы (I) используют в качестве внешнего стандарта и условия обнаружения являются следующими: колонка C18; подвижная фаза: метанол-вода-тетрагидрофуран-уксусная кислота (40:30:30:0,5); длина волны детектирования: 236 нм.
11. Способ контроля качества при синтезе чиглитазара или его соли, включающий использование соединения формулы (I)
для определения или контроля содержания примесей или сопутствующих веществ.
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |
| Способ преобразования угла поворота вала в код | 1988 |
|
SU1562970A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Токарный резец | 1924 |
|
SU2016A1 |
| ПРОИЗВОДНЫЕ ЦИКЛОАЛКАНО-ИНДОЛА И АЗАИНДОЛА, СМЕСЬ ИХ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫЕ ИЗОМЕРЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ПРОИЗВОДНЫЕ КАРБОНОВОЙ КИСЛОТЫ В КАЧЕСТВЕ ИСХОДНЫХ СОЕДИНЕНИЙ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИНГИБИРУЮЩАЯ ВЫСВОБОЖДЕНИЕ АССОЦИИРОВАННЫХ С АПОЛИПОПРОТЕИНОМ В-100 ЛИПОПРОТЕИНОВ | 1995 |
|
RU2157803C2 |

