ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка имеет приоритеты и преимущества Китайской заявки на патент на изобретение №201710667860.4, поданной в Ведомство интеллектуальной собственности Китая 7 августа 2017 года, и Китайской заявки на патент на изобретение №201810333652.5, поданной в Ведомство интеллектуальной собственности Китая 26 июня 2017 года, и Китайской заявки на патент на изобретение №201710867863.2, поданной в Ведомство интеллектуальной собственности Китая 13 апреля 2018 года. Полное содержание этих патентных заявок включено в данное описание посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Настоящая заявка относится к области медицины и конкретно относится к соединению формулы (I) или его фармацевтически приемлемой соли, к способу получения таковых, к фармацевтической композиции, содержащей соединение, и к применению его в приготовлении лекарственного средства для лечения опосредованного андрогеном заболевания.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Андрогеновый рецептор (AR) относится к стероидному рецептору надсемейства ядерных рецепторов. Связываясь с андрогеном (таким как тестостерон и дигидротестостерон), AR высвобождается из комплекса, образованного белками теплового шока, подвергается реакции фосфорилирования с образованием димера, который переносится в ядро и связывается с фрагментом ДНК, ассоциированным с ним, тем самым стимулируя транскрипцию его целевого гена. Транскрипционная активность андрогенового рецептора, активируемая связыванием с лигандом, достигается за счет координации белков ко-активаторов. Основная роль антагонистов AR заключается в том, чтобы непосредственно препятствовать связыванию тестостерона или дигидротестостерона с андрогеновым рецептором, блокировать воздействие андрогена на клетки, играть роль антиандрогена, ингибировать рост клеток и, в конечном счете, способствовать апоптозу и сыграть важную роль в лечении рака предстательной железы. В продаже имеется энзалутамид, антагонист андрогенового рецептора, разработанный Medivation & Astell.
Ввиду важной роли антагонистов андрогенового рецептора особенно важно разработать антагонисты андрогенового рецептора, подходящие в качестве терапевтических лекарственных средств. В общем, необходимо, чтобы соединение в качестве фармацевтического активного ингредиента, имело превосходные свойства в следующих аспектах: биологическая активность, безопасность, биодоступность, стабильность и т.п. Согласно настоящему изобретению предложено соединение диарилтиогидантоина, имеющее новую структуру, для применения в качестве антагониста андрогенового рецептора, и обнаружено, что соединение, имеющее такую структуру, демонстрирует превосходные противоопухолевые эффекты и обладает вышеупомянутыми превосходными свойствами.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В одном аспекте настоящая заявка относится к соединению формулы (I) или его фармацевтически приемлемой соли,

где
Т выбран из группы, состоящей из СН и N;
R1 выбран из группы, состоящей из водорода, галогена, С1-2алкила и замещенного галогеном С1-12алкила;
кольцо А выбрано из группы, состоящей из 
каждый из R2 и R3 независимо выбран из С1-12алкила, или вместе взятые R2 и R3 связаны друг с другом с образованием 3-6-членного циклоалкила;
каждый из X1, X2, X3 и X4 независимо выбран из группы, состоящей из СН и N, и по меньшей мере один из них представляет собой N;
n равно 0, 1, 2 или 3;
каждый R4 независимо выбран из С1-12алкила;
кольцо В представляет собой 

R5 выбран из группы, состоящей из водорода, С1-12алкила, С1-12алкокси и галогена;
R6 выбран из С1-12алкиламинокарбонила;
один из Х5, Х6 и Х7 представляет собой N(-Ra), а остальные представляют собой СН или N;
Ra выбран из 5-членного гетероциклоалкила, где гетероциклоалкил возможно замещен галогеном, С1-4алкилом, С2-4алкенилом, С2-4алкинилом, 3-6-членным циклоалкилом, 3-6-членным гетероциклоалкилом, С1-4алкокси, гидроксилом или амино;
каждый из X8, X9, X10 и X11 независимо выбран из группы, состоящей из СН, С(=O), N и NH, и три из X8, X9, X10 и X11 представляют собой С(=O), N и NH соответственно;
Rb выбран из С1-12алкила, где С1-12алкил возможно замещен галогеном;
каждый из Y8, Y9, Y10 и Y11 независимо выбран из группы, состоящей из СН и N, и по меньшей мере два из Y8, Y9, Y10 и Y11 представляют собой N;
m равно 0, 1 или 2;
каждый R7 независимо выбран из группы, состоящей из галогена, С1-12алкила, гидроксила, С1-12алкокси, амино, 3-10-членного циклоалкила, 3-10-членного гетероциклоалкила, 5-10-членного гетероарила и С1-12алкиламино, где С1-12алкил, 3-10-членный циклоалкил, 3-10-членный гетер оциклоалкил, 5-10-членный гетероарил или С1-12алкиламино возможно замещен галогеном, и где гидроксил замещен группой -С1-12алкил-ОН, -С1-12алкил-(3-10-членный гетероциклоалкил), -С1-12алкил-S(=O)2Rc, -C1-12алкил-NRdRe, -С1-12алкил- C(=O)NRfRg, -C1-12алкил-(3-10-членный циклоалкил), возможно замещенной галогеном или гидроксилом, или 3-10-членным гетероциклоалкилом, возможно замещенным галогеном или гидроксилом;
каждый из Z8, Z9, Z10 и Z11 независимо выбран из группы, состоящей из СН, С(=O) и N;
j равно 0, 1 или 2;
каждый R9 независимо выбран из группы, состоящей из галогена, С1-12алкила, С1-12алкокси и гидроксила, где С1-12алкил возможно замещен галогеном или С1-12алкокси, и где гидроксил возможно замещен группой -С1-12алкил-О-С1-12алкил, -C1-12алкил-ОН или -C1-12алкил-C(=O)NRfRg;
каждый из Rc, Rd, Re, Rf и Rg независимо выбран из группы, состоящей из водорода, С1-12алкила, 3-10-членного циклоалкила, 3-10-членного гетероциклоалкила, С1-12алкокси, гидроксила и амино;
два из X12, X13, X14, X15 и X16 представляют собой NH и С(=O) соответственно, а остальные представляют собой СН2, О или S;
q равно 0, 1, 2, 3 или 4; и
каждый R8 независимо выбран из группы, состоящей из галогена, С1-12алкила, гидроксила, амино, 3-10-членного циклоалкила, С1-12алкокси, 3-10-членного гетероциклоалкила и С1-12алкиламино;
при условии, что когда кольцо А выбрано из  тогда кольцо В не представляет собой
тогда кольцо В не представляет собой  и когда R7 выбран из С1-12алкокси, тогда R7 замещает водород на Y9, Y10 или Y11.
и когда R7 выбран из С1-12алкокси, тогда R7 замещает водород на Y9, Y10 или Y11.
В другом аспекте настоящая заявка относится к фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль согласно настоящей заявке. В некоторых воплощениях фармацевтическая композиция согласно настоящей заявке дополнительно содержит фармацевтически приемлемый эксципиент.
В еще одном другом аспекте настоящая заявка относится к способу лечения опосредованного андрогеном заболевания у млекопитающего, включающему введение млекопитающему, предпочтительно человеку, нуждающемуся в лечении, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли или фармацевтической композиции, и заболевание включает, без ограничения, клеточные пролиферагивные заболевания (например, рак).
В еще одном аспекте настоящая заявка относится к применению соединения формулы (I) или его фармацевтически приемлемой соли или фармацевтической композиции в приготовлении лекарственного средства для лечения опосредованного андрогеном заболевания, и заболевание включает, без ограничения, клеточные пролиферагивные заболевания (например, рак).
В еще одном аспекте настоящая заявка относится к применению соединения формулы (I) или его фармацевтически приемлемой соли или фармацевтической композиции в лечении опосредованного андрогеном заболевания, и заболевание включает, без ограничения, клеточные пролиферагивные заболевания (например, рак).
В другом аспекте настоящая заявка относится к соединению формулы (I) или его фармацевтически приемлемой соли или фармацевтической композиции для применения в предупреждении или лечении опосредованного андрогеном заболевания, и заболевание включает, без ограничения, клеточные пролиферагивные заболевания (например, рак).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном аспекте настоящая заявка относится к соединению формулы (I) или его фармацевтически приемлемой соли,

где
Т выбран из группы, состоящей из СН и N;
R1 выбран из группы, состоящей из водорода, галогена, С1-12алкила и замещенного галогеном С1-12алкила;
кольцо А выбрано из группы, состоящей из 
каждый из R2 и R3 независимо выбран из С1-12алкила, или вместе взятые R2 и R3 связаны друг с другом с образованием 3-6-членного циклоалкила;
каждый из X1, X2, X3 и X4 независимо выбран из группы, состоящей из СН и N, и по меньшей мере один из них представляет собой N;
n равно 0, 1, 2 или 3;
каждый R4 независимо выбран из С1-12алкила;
кольцо В представляет собой 

R5 выбран из группы, состоящей из водорода, С1-12алкила, С1-12алкокси и галогена;
R6 выбран из С1-12алкиламинокарбонила;
один из X5, X6 и X7 представляет собой N(-Ra), а остальные представляют собой СН или N;
Ra выбран из 5-членного гетероциклоалкила, где гетероциклоалкил возможно замещен галогеном, С1-4алкилом, С2-4алкенилом, С2-4алкинилом, 3-6-членным циклоалкилом, 3-6-членным гетероциклоалкилом, С1-4алкокси, гидроксилом или амино;
каждый из X8, X9, X10 и X11 независимо выбран из группы, состоящей из СН, С(=O), N и NH, и три из X8, X9, X10 и X11 представляют собой С(=O), N и NH соответственно;
Rb выбран из С1-12алкила, где С1-12алкил возможно замещен галогеном;
каждый из Y8, Y9, Y10 и Y11 независимо выбран из группы, состоящей из СН и N, и по меньшей мере два из Y8, Y9, Y10 и Y11 представляют собой N;
m равно 0, 1 или 2;
каждый R7 независимо выбран из группы, состоящей из галогена, С1-12алкила, гидроксила, С1-12алкокси, амино, 3-10-членного циклоалкила, 3-10-членного гетероциклоалкила, 5-10-членного гетероарила и С1-12алкиламино, где С1-12алкил, 3-10-членный циклоалкил, 3-10-членный гетер оциклоалкил, 5-10-членный гетероарил или С1-12алкиламино возможно замещен галогеном, и где гидроксил замещен группой -С1-12алкил-ОН, -С1-12алкил-(3-10-членный гетероциклоалкил), -С1-12алкил-S(=O)2Rc, -С1-12алкил-NRdRe, -C1-12алкил-C(=O)NRfRg, -С1-12алкил-(3-10-членный циклоалкил), возможно замещенной галогеном или гидроксилом, или 3-10-членным гетероциклоалкилом, возможно замещенным галогеном или гидроксилом;
каждый из Z8, Z9, Z10 и Z11 независимо выбран из группы, состоящей из СН, С(=O) и N;
j равно 0, 1 или 2;
каждый R9 независимо выбран из группы, состоящей из галогена, С1-12алкила, С1-12алкокси и гидроксила, где С1-12алкил возможно замещен галогеном или С1-12алкокси, и где гидроксил возможно замещен группой -С1-12алкил-О-С1-12алкил, -С1-12алкил-ОН или -С1-12алкил-C(=O)NRfRg;
каждый из Rc, Rd, Re, Rf и Rg независимо выбран из группы, состоящей из водорода, С1-12алкила, 3-10-членного циклоалкила, 3-10-членного гетероциклоалкила, С1-12алкокси, гидроксила и амино;
два из X12, X13, X14, X15 и X16 представляют собой NH и С(=O) соответственно, а остальные представляют собой СН2, О или S;
q равно 0, 1, 2, 3 или 4; и
каждый R8 независимо выбран из группы, состоящей из галогена, С1-12алкила, гидроксила, амино, 3-10-членного циклоалкила, С1-12алкокси, 3-10-членного гетероциклоалкила и С1-12алкиламино;
при условии, что когда кольцо А выбрано из  тогда кольцо В не представляет собой
тогда кольцо В не представляет собой 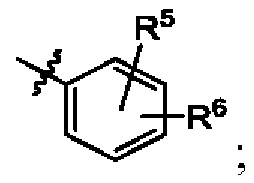 и когда R7 выбран из С1-12алкокси, тогда R7 замещает водород на Y9, Y10 или Y11.
и когда R7 выбран из С1-12алкокси, тогда R7 замещает водород на Y9, Y10 или Y11.
Гетероатомом(ами) в гетероциклоалкиле или гетероариле, описанных в данном документе, обычно являются 1, 2 или 3 гетероатома, независимо выбранных из группы, состоящей из серы, кислорода и/или азота; и в некоторых воплощениях гетероциклоалкил содержит 1 или 2 атома О, и гетероарил содержит 1 или 2 атома N.
В некоторых воплощениях R1 выбран из группы, состоящей из водорода, галогена, С1-6алкила и замещенного галогеном С1-6алкила; в некоторых воплощениях R1 выбран из группы, состоящей из галогена и замещенного галогеном С1-4алкила; в некоторых воплощениях R1 выбран из группы, состоящей из фтора, хлора, брома и замещенного фтором С1-4алкила; в некоторых воплощениях R1 выбран из группы, состоящей из фтора, хлора и замещенного фтором метила; и в некоторых воплощениях R1 выбран из группы, состоящей из фтора, хлора, дифторметила и трифторметила.
В некоторых воплощениях R1 выбран из замещенного галогеном С1-4алкила; в некоторых воплощениях R1 выбран из замещенного фтором С1-4алкила; в некоторых воплощениях R1 выбран из замещенного фтором метила; и в некоторых воплощениях R1 выбран из трифторметила.
В некоторых воплощениях каждый из X1, X2, X3 и X4 независимо выбран из группы, состоящей из СН и N, и один или два из X1, X2, X3 и X4 представляют собой N, а остальные представляют собой СН.
В некоторых воплощениях каждый из X1, X2, X3 и X4 независимо выбран из группы, состоящей из СН и N, и один из X1, X2, X3 и X4 представляет собой N, а остальные представляют собой СН.
В некоторых воплощениях кольцо А выбрано из группы, состоящей из 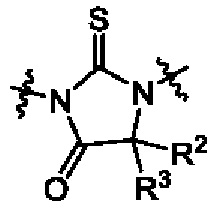 и
и  где каждый из X1 и X2 независимо выбран из группы, состоящей из СН и N, и по меньшей мере один из них представляет собой N, и n равно 0, 1, 2 или 3.
где каждый из X1 и X2 независимо выбран из группы, состоящей из СН и N, и по меньшей мере один из них представляет собой N, и n равно 0, 1, 2 или 3.
В некоторых воплощениях кольцо А выбрано из группы, состоящей из  и
и  и n равно 0 или 1.
и n равно 0 или 1.
В некоторых воплощениях кольцо А выбрано из группы, состоящей из  и
и  и n равно 0 или 1.
и n равно 0 или 1.
В некоторых воплощениях каждый из R2 и R3 независимо выбран из С1-6алкила, или вместе взятые R2 и R3 связаны друг с другом с образованием 3-6-членного циклоалкила; в некоторых воплощениях каждый из R2 и R3 независимо выбран из C1-4алкила, или вместе взятые R2 и R3 связаны друг с другом с образованием 3-4-членного циклоалкила; в некоторых воплощениях каждый из R2 и R3 независимо выбран из группы, состоящей из метила и этила, или вместе взятые R2 и R3 связаны друг с другом с образованием 3-4-членного циклоалкила; и в некоторых воплощениях вместе взятые R2 и R3 выбраны из метила, или R2 и R3 связаны друг с другом с образованием циклобутила.
В некоторых конкретных воплощениях кольцо А выбрано из группы, состоящей из 
В некоторых воплощениях каждый R4 независимо выбран из С1-6алкила; в некоторых воплощениях каждый R4 независимо выбран из С1-4алкила; и в некоторых воплощениях каждый R4 независимо выбран из метила.
В некоторых конкретных воплощениях кольцо А выбрано из группы, состоящей из 
В некоторых воплощениях кольцо В представляет собой 

В некоторых воплощениях R5 выбран из группы, состоящей из водорода, С1-6алкила, С1-6алкокси и галогена; в некоторых воплощениях R5 выбран из группы, состоящей из водорода, С1-4алкила, С1-4алкокси и галогена; в некоторых воплощениях R5 выбран из группы, состоящей из водорода, метила, метокси, фтора, хлора, брома и йода; и в некоторых воплощениях R5 выбран из группы, состоящей из водорода, метила, метокси, фтора и хлора.
В некоторых воплощениях R5 выбран из группы, состоящей из водорода и галогена.
В некоторых других воплощениях R5 выбран из группы, состоящей из водорода и фтора.
В некоторых воплощениях структурная единица  представляет собой
представляет собой  и в некоторых воплощениях структурная единица
и в некоторых воплощениях структурная единица  представляет собой
представляет собой 
В некоторых воплощениях R6 выбран из С1-6алкиламинокарбонила; в некоторых воплощениях R6 выбран из С1-4алкиламинокарбонила; и в некоторых воплощениях R6 выбран из метиламинокарбонила.
В некоторых конкретных воплощениях структурная единица  представляет собой
представляет собой 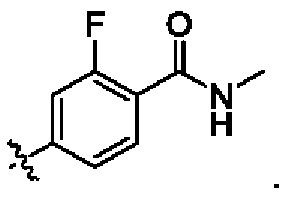
В некоторых воплощениях каждый из X5, X6 и X7 независимо выбран из группы, состоящей из СН, N и N(-Ra), и по меньшей мере два из X5, X6 и X7 представляют собой N и N(-Ra) соответственно, а остальные представляет собой СН или N.
В некоторых воплощениях каждый из X5, X6 и X7 независимо выбран из группы, состоящей из СН, N и N(-Ra), и два из X5, X6 и X7 представляют собой N и N(-Ra) соответственно, а остальной представляет собой СН или N.
В некоторых воплощениях X5, X6 и X7, каждый, независимо выбраны из группы, состоящей из СН, N и N(-Ra), и отличаются друг от друга.
В некоторых воплощениях структурная единица  выбрана из группы, состоящей из
выбрана из группы, состоящей из 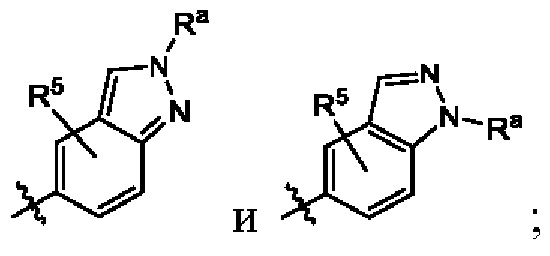 в некоторых воплощениях структурная единица
в некоторых воплощениях структурная единица  выбрана из группы, состоящей из
выбрана из группы, состоящей из  и в некоторых воплощениях структурная единица
и в некоторых воплощениях структурная единица  выбрана из группы, состоящей из
выбрана из группы, состоящей из 
В некоторых воплощениях Ra выбран из 3-7-членного гетероциклоалкила, где гетероциклоалкил возможно замещен галогеном, С1-6алкилом, С2-6алкенилом, С2-6алкинилом, 3-6-членным циклоалкилом, 3-6-членным гетероциклоалкилом, С1-6алкокси, гидроксилом или амино; в некоторых воплощениях Ra выбран из 5-членного гетероциклоалкила, где гетероциклоалкил возможно замещен галогеном, С1-4алкилом, С2-4алкенилом, С2-4алкинилом, 3-6-членным циклоалкилом, 3-6-членным гетероциклоалкилом, С1-4алкокси, гидроксилом или амино; в некоторых воплощениях Ra выбран из 5-членного оксациклоалкила, где оксациклоалкил замещен гидроксилом; и в некоторых воплощениях Ra выбран из 
В некоторых конкретных воплощениях структурная единица  представляет собой
представляет собой  и структурная единица
и структурная единица  представляет собой
представляет собой 
В некоторых конкретных воплощениях структурная единица  выбрана из группы, состоящей из
выбрана из группы, состоящей из 
В некоторых воплощениях X8, X9, X10 и X11, каждый, независимо выбраны из группы, состоящей из СН, С(=O), N и NH, и отличаются друг от друга.
В некоторых воплощениях Rb замещает водород на NH или СН.
В некоторых воплощениях Rb замещает водород на NH.
В некоторых воплощениях структурная единица  выбрана из группы, состоящей из
выбрана из группы, состоящей из  и в некоторых воплощениях структурная единица
и в некоторых воплощениях структурная единица  выбрана из группы, состоящей из
выбрана из группы, состоящей из 
В некоторых воплощениях структурная единица  выбрана из группы, состоящей из
выбрана из группы, состоящей из  и в некоторых воплощениях структурная единица
и в некоторых воплощениях структурная единица  выбрана из группы, состоящей из
выбрана из группы, состоящей из  и
и 
В некоторых воплощениях Rb выбран из C1-6алкила, где С1-6алкил возможно замещен фтором или хлором; в некоторых воплощениях Rb выбран из С1-4алкила, где С1-4алкил возможно замещен фтором; в некоторых воплощениях Rb выбран из этила, где этил возможно замещен фтором; и в некоторых воплощениях Rb выбран из группы, состоящей из -СН2СН3 и -CH2CF3.
В некоторых воплощениях Rb выбран из С1-6алкила, где С1-6алкил замещен фтором или хлором; в некоторых воплощениях Rb выбран из С1-4алкила, где С1-4алкил замещен фтором; в некоторых воплощениях Rb выбран из этила, где этил замещен фтором; и в некоторых воплощениях Rb выбран из -CH2CF3.
В некоторых конкретных воплощениях структурная единица 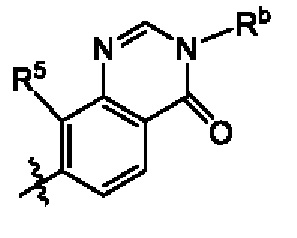 представляет собой
представляет собой  структурная единица
структурная единица 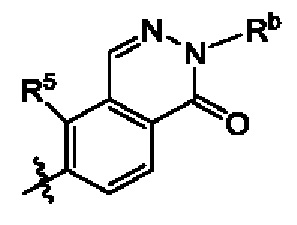 представляет собой
представляет собой  и структурная единица
и структурная единица  представляет собой
представляет собой 
В некоторых конкретных воплощениях структурная единица  выбрана из группы, состоящей из
выбрана из группы, состоящей из  и
и 
В некоторых воплощениях каждый из Y8, Y9, Y10 и Y11 независимо выбран из группы, состоящей из СН и N, и два из Y8, Y9, Y10 и Y11 представляют собой N, а остальные представляют собой СН.
В некоторых воплощениях структурная единица  представляет собой
представляет собой 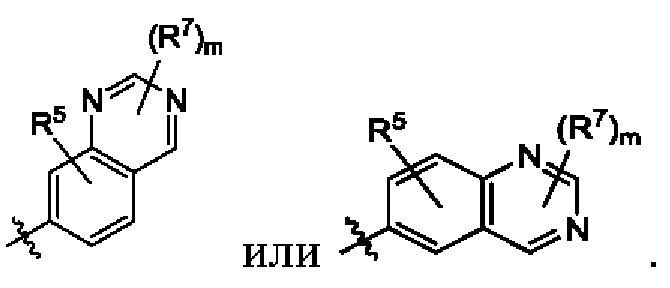
В некоторых воплощениях структурная единица  представляет собой
представляет собой  и в некоторых воплощениях структурная единица
и в некоторых воплощениях структурная единица  представляет собой
представляет собой 
В некоторых воплощениях структурная единица  представляет собой
представляет собой 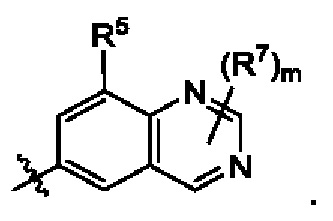
В некоторых воплощениях каждый из Rc, Rd и Re независимо выбран из группы, состоящей из водорода, С1-12алкила, 3-10-членного циклоалкила, 3-10-членного гетероциклоалкила, С1-12алкокси, гидроксила и амино. В некоторых воплощениях каждый из Rc, Rd и Re независимо выбран из группы, состоящей из водорода, С1-6алкила, 3-6-членного циклоалкила, 3-6-членного гетероциклоалкила, С1-6алкокси, гидроксила и амино; в некоторых воплощениях каждый из Rc, Rd и Re независимо выбран из С1-4алкила; и в некоторых воплощениях каждый из Rc, Rd и Re независимо выбран из метила.
В некоторых воплощениях каждый из Rc, Rd, Re, Rf и Rg независимо выбран из группы, состоящей из водорода, С1-6алкила, 3-6-членного циклоалкила, 3-6-членного гетероциклоалкила, С1-6алкокси, гидроксила и амино; в некоторых воплощениях каждый из Rc, Rd, Re, Rf и Rg независимо выбран из группы, состоящей из водорода и С1-4алкила; и в некоторых воплощениях каждый из Rc, Rd, Re, Rf и Rg независимо выбран из группы, состоящей из водорода и метила.
В некоторых воплощениях m равно 1 или 2.
В некоторых воплощениях R7 замещает водород на СН.
В некоторых воплощениях каждый R7 независимо выбран из группы, состоящей из галогена, С1-6алкила, гидроксила, С1-6алкокси, амино, 3-6-членного циклоалкила, 3-6-членного гетероциклоалкила, 5-6-членного гетероарила и С1-6алкиламино, где С1-6алкил, 3-6-членный циклоалкил, 3-6-членный гетероциклоалкил, 5-6-членный гетероарил или С1-6алкиламино возможно замещен галогеном, и где гидроксил возможно замещен группой -С1-6алкил-ОН, -С1-6алкил-(3-6-членный гетероциклоалкил), -С1-6алкил-S(=O)2Rc, -C1-6алкил-NRdRe, -C1-6алкил-C(=O)NRfRg, -С1-6алкил-(3-6-членный циклоалкил), возможно замещенной галогеном или гидроксилом, или 3-6-членным гетероциклоалкилом, возможно замещенным галогеном или гидроксилом, при условии, что когда R7 выбран из С1-6алкокси, тогда R7 замещает водород на Y9, Y10 или Y11. В некоторых воплощениях каждый R7 независимо выбран из группы, состоящей из галогена, С1-4алкила, гидроксила, С1-4алкокси, амино, 3-6-членного циклоалкила, 5-6-членного гетероарила и С1-4алкиламино, где С1-4алкил, 3-6-членный циклоалкил или С1-4алкиламино возможно замещен галогеном, и где гидроксил возможно замещен группой -С1-4алкил-ОН, -С1-4алкил-(3-6-членный гетероциклоалкил), -С1-4алкил-S(=O)2Rc, -C1-4алкил-NRdRe, -C1-4алкил-C(=O)NRfRg, -С1-4алкил-(3-6-членный циклоалкил), возможно замещенной галогеном или гидроксилом, или 5-6-членным гетероциклоалкилом, возможно замещенным галогеном или гидроксилом, при условии, что когда R7 выбран из С1-4алкокси, тогда R7 замещает водород на Y9, Y10 или Y11. В некоторых воплощениях каждый R7 независимо выбран из группы, состоящей из метила, этила, гидроксила, метокси, этокси, циклопропила, пиразолила, имидазолила и метиламино, где метил, этил или циклопропил возможно замещен фтором, и где гидроксил возможно замещен группой -этил-ОН, тетрагидропиранил, -метил-(оксетан), -пропил-S(=O)2Rc, -этил-NRdRe, -метил-C(=O)NRfRg, циклопропилметил-, возможно замещенной гидроксилом, или тетрагидрофуранилом, возможно замещенным гидроксилом, при условии, что когда R7 выбран из метокси или этокси, тогда R7 замещает водород на Y9, Y10 или Y11. В некоторых воплощениях каждый R7 независимо выбран из группы, состоящей из метила, этила, гидроксила, метокси, этокси, циклопропила, пиразолила, имидазолила и метиламино, где метил или этил возможно замещен фтором, и где гидроксил возможно замещен группой -этил-ОН,  -пропил-S(=O)2СН3, -CH2C(=O)NHCH3, -CH2C(=O)NH2, -этил-N(СН3)2,
-пропил-S(=O)2СН3, -CH2C(=O)NHCH3, -CH2C(=O)NH2, -этил-N(СН3)2,  возможно замещенной гидроксилом, или
возможно замещенной гидроксилом, или  возможно замещенным гидроксилом, при условии, что когда R7 выбран из группы, состоящей из метокси и этокси, тогда R7 замещает водород на Y9, Y10 или Y11. В некоторых воплощениях каждый R7 независимо выбран из группы, состоящей из метила, этила, циклопропила, гидроксила, метокси, этокси, пиразолила, имидазолила, дифторметила, дифторэтила и метиламино, где гидроксил возможно замещен группой -этил-ОН,
возможно замещенным гидроксилом, при условии, что когда R7 выбран из группы, состоящей из метокси и этокси, тогда R7 замещает водород на Y9, Y10 или Y11. В некоторых воплощениях каждый R7 независимо выбран из группы, состоящей из метила, этила, циклопропила, гидроксила, метокси, этокси, пиразолила, имидазолила, дифторметила, дифторэтила и метиламино, где гидроксил возможно замещен группой -этил-ОН,  -пропил-S(=O)2CH3, -CH2C(=O)NHCH3, -CH2C(=O)NH2 или -этил-N(СН3)2, при условии, что когда R7 выбран из группы, состоящей из метокси и этокси, тогда R7 замещает водород на Y9, Y10 или Y11.
-пропил-S(=O)2CH3, -CH2C(=O)NHCH3, -CH2C(=O)NH2 или -этил-N(СН3)2, при условии, что когда R7 выбран из группы, состоящей из метокси и этокси, тогда R7 замещает водород на Y9, Y10 или Y11.
В некоторых конкретных воплощениях каждый R7 независимо выбран из группы, состоящей из метила, этила, циклопропила, дифторметила,  метиламино,
метиламино,  метокси, этокси,
метокси, этокси, 
В некоторых более конкретных воплощениях каждый R7 независимо выбран из группы, состоящей из метила, этила, циклопропила, дифторметила,  метиламино,
метиламино, 
 метокси, этокси,
метокси, этокси, 
В некоторых воплощениях каждый R7 независимо выбран из группы, состоящей из галогена, С1-12алкила, гидроксила, амино, 3-10-членного циклоалкила, 3-10-членного гетероциклоалкила и С1-12алкиламино, где гидроксил замещен группой -С1-12алкил-ОН, 3-10-членный гетероциклоалкил, -C1-12алкил-S(=O)2Rc или -C1-12алкил-NRdRe, где С1-12алкил, 3-10-членный циклоалкил, 3-10-членный гетеро циклоалкил, или С1-12алкиламино возможно замещен галогеном.
В некоторых воплощениях каждый R7 независимо выбран из группы, состоящей из галогена, С1-6алкила, гидроксила, амино, 3-6-членного циклоалкила, 3-6-членного гетероциклоалкила и С1-6алкиламино, где гидроксил замещен группой -С1-6алкил-ОН, 3-6-членный гетероциклоалкил, -С1-6алкил-S(=O)2Rc или -C1-6алкил-NRdRe, где С1-6алкил, 3-6-членный циклоалкил, 3-6-членный гетероциклоалкил, или C1-6алкиламино возможно замещен галогеном. В некоторых воплощениях каждый R7 независимо выбран из группы, состоящей из галогена, С1-4алкила, гидроксила, амино, 3-6-членного циклоалкила и C1-4алкиламино, где гидроксил замещен группой -С1-4алкил-ОН, 5-6-членный гетероциклоалкил, -C1-4алкил-S(=O)2Rc или -C1-4алкил-NRdRc, где С1-4алкил, 3-6-членный циклоалкил или С1-4алкиламино возможно замещен галогеном. В некоторых воплощениях каждый R7 независимо выбран из группы, состоящей из метила, этила, гидроксила, циклопропила и метиламино, где гидроксил замещен группой -этил-ОН, тетрагидропиранил, -пропил-S(=O)2Rc или -этил-NRdRe, где метил, этил, циклопропил или метиламино возможно замещен фтором. В некоторых воплощениях каждый R7 независимо выбран из группы, состоящей из метила, этила, гидроксила, циклопропила и метиламино, где гидроксил замещен группой -этил-ОН,  -пропил-S(=O)2СН3 или -этил-N(СН3)2, где метил, этил, циклопропил или метиламино возможно замещен фтором. В некоторых воплощениях каждый R7 независимо выбран из группы, состоящей из метила, этила, циклопропила, гидроксила, дифторметила и метиламино, где гидроксил замещен группой -этил-ОН,
-пропил-S(=O)2СН3 или -этил-N(СН3)2, где метил, этил, циклопропил или метиламино возможно замещен фтором. В некоторых воплощениях каждый R7 независимо выбран из группы, состоящей из метила, этила, циклопропила, гидроксила, дифторметила и метиламино, где гидроксил замещен группой -этил-ОН,  -пропил-S(=O)2СН3 или -этил-N(CH3)2.
-пропил-S(=O)2СН3 или -этил-N(CH3)2.
В некоторых конкретных воплощениях каждый R7 независимо выбран из группы, состоящей из метила, этила, циклопропила, гидроксила, дифторметила, метиламино, 
В некоторых конкретных воплощениях структурная единица  представляет собой
представляет собой 
В некоторых более конкретных воплощениях структурная единица  выбрана из группы, состоящей из
выбрана из группы, состоящей из 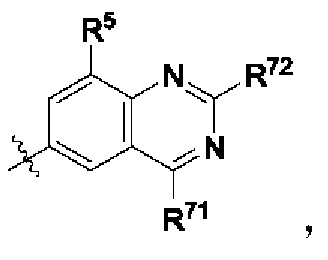
 где каждый R71 независимо выбран из группы, состоящей из гидроксила и 5-10-членного гетероарила, где гидроксил возможно замещен группой -С1-12алкил-ОН, -С1-12алкил-(3-10-членный гетероциклоалкил), -С1-12алкил-S(=O)2Rc, -C1-12алкил-NRdRe, -С1-12алкил- C(=O)NRfRg, -C1-12алкил-(3-10-членный циклоалкил), возможно замещенной галогеном или гидроксилом, или 3-10-членным гетероциклоалкилом, возможно замещенным галогеном или гидроксилом; и каждый R72 независимо выбран из группы, состоящей из С1-12алкила, гидроксила, С1-12алкокси, 3-10-членного циклоалкила и С1-12алкиламино, где С1-12алкил, 3-10-членный циклоалкил или С1-12алкиламино возможно замещен галогеном, и где гидроксил замещен группой -С1-12алкил-ОН, 3-10-членный гетероциклоалкил, -С1-12алкил-S(=O)2Rc или -С1-12алкил-NRdRe.
где каждый R71 независимо выбран из группы, состоящей из гидроксила и 5-10-членного гетероарила, где гидроксил возможно замещен группой -С1-12алкил-ОН, -С1-12алкил-(3-10-членный гетероциклоалкил), -С1-12алкил-S(=O)2Rc, -C1-12алкил-NRdRe, -С1-12алкил- C(=O)NRfRg, -C1-12алкил-(3-10-членный циклоалкил), возможно замещенной галогеном или гидроксилом, или 3-10-членным гетероциклоалкилом, возможно замещенным галогеном или гидроксилом; и каждый R72 независимо выбран из группы, состоящей из С1-12алкила, гидроксила, С1-12алкокси, 3-10-членного циклоалкила и С1-12алкиламино, где С1-12алкил, 3-10-членный циклоалкил или С1-12алкиламино возможно замещен галогеном, и где гидроксил замещен группой -С1-12алкил-ОН, 3-10-членный гетероциклоалкил, -С1-12алкил-S(=O)2Rc или -С1-12алкил-NRdRe.
В некоторых воплощениях каждый R71 независимо выбран из группы, состоящей из гидроксила и 5-6-членного гетероарила, где гидроксил возможно замещен группой -С1-6алкил-ОН, -С1-6алкил-(3-6-членный гетероциклоалкил), -С1-6алкил-S(=O)2Rc, -C1-6алкил-NRdRe, -C1-6алкил-C(=O)NRfRg, -С1-6алкил-(3-6-членный циклоалкил), возможно замещенной галогеном или гидроксилом, или 3-6-членным гетероциклоалкилом, возможно замещенным галогеном или гидроксилом. В некоторых воплощениях каждый R71 независимо выбран из группы, состоящей из гидроксила и 5-6-членного гетероарила, где гидроксил возможно замещен группой: -С1-4алкил-ОН, -С1-4алкил-(3-6-членный гетероциклоалкил), -С1-4алкил-S(=O)2Rc, -С1-4алкил-NRdRc, -C1-4алкил-С(=O)NRfRg, -С1-4алкил-(3-6-членный циклоалкил), возможно замещенной галогеном или гидроксилом, или 5-6-членным гетероциклоалкилом, возможно замещенным галогеном или гидроксилом. В некоторых воплощениях каждый R71 независимо выбран из группы, состоящей из гидроксила, пиразолила и имидазолила, где гидроксил возможно замещен группой -этил-ОН, тетрагидропиранил, -метил-(оксетан), -пропил-S(=O)2Rc, -этил-NRdRe, -метил-C(=O)NRfRg, циклопропилметил-, возможно замещенной гидроксилом, или тетрагидрофуранилом, возможно замещенным гидроксилом. В некоторых воплощениях каждый R71 независимо выбран из группы, состоящей из гидроксила, пиразолила и имидазолила, где гидроксил возможно замещен группой -этил-ОН,  -пропил-S(=O)2СН3, -CH2C(=O)NHCH3, -CH2C(=O)NH2, -этил-N(СН3)2,
-пропил-S(=O)2СН3, -CH2C(=O)NHCH3, -CH2C(=O)NH2, -этил-N(СН3)2,  возможно замещенной гидроксилом, или
возможно замещенной гидроксилом, или  возможно замещенным гидроксилом.
возможно замещенным гидроксилом.
В некоторых конкретных воплощениях каждый R71 независимо выбран из группы, состоящей из 

В некоторых более конкретных воплощениях каждый R71 независимо выбран из группы, состоящей из гидроксила, 

В некоторых воплощениях каждый R72 независимо выбран из группы, состоящей из С1-6алкила, гидроксила, С1-6алкокси, 3-6-членного циклоалкила и С1-6алкиламино, где C1-6алкил, 3-6-членный циклоалкил или С1-6алкиламино возможно замещен галогеном, и где гидроксил замещен группой -С1-6алкил-ОН, 5-6-членный гетероциклоалкил, -С1-6алкил-S(=O)2Rc или -С1-6алкил-NRdRe. В некоторых воплощениях каждый R72 независимо выбран из группы, состоящей из С1-4алкила, гидроксила, С1-4алкокси, 3-6-членного циклоалкила и С1-4алкиламино, где С1-4алкил, 3-6-членный циклоалкил или С1-4алкиламино возможно замещен галогеном, и где гидроксил замещен группой -С1-4алкил-ОН. В некоторых воплощениях каждый R72 независимо выбран из группы, состоящей из метила, этила, гидроксила, метокси, этокси, циклопропила и метиламино, где метил, этил или циклопропил возможно замещен фтором; где гидроксил замещен группой -этил-ОН. В некоторых воплощениях каждый R72 независимо выбран из группы, состоящей из метила, этила, циклопропила, гидроксила, метокси, этокси, дифторметила, дифторэтила и метиламино, где гидроксил замещен группой -этил-ОН.
В некоторых конкретных воплощениях каждый R72 независимо выбран из группы, состоящей из метила, этила, циклопропила, дифторметила,  метокси, этокси, метиламино и
метокси, этокси, метиламино и 
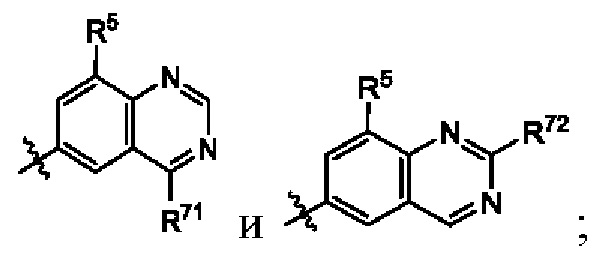
В некоторых воплощениях структурная единица  выбрана из группы, состоящей из
выбрана из группы, состоящей из  где каждый R71 независимо выбран из гидроксила, и гидроксил замещен группой -С1-12алкил-ОН, 3-10-членный гетероциклоалкил, -С1-12алкил-S(=O)2Rc или -C1-12алкил-NRdRc; и каждый R72 независимо выбран из группы, состоящей из С1-12алкила, гидроксила, 3-10-членного циклоалкила и С1-12алкиламино, где гидроксил замещен группой -С1-12алкил-ОН, 3-10-членный гетероциклоалкил, -С1-12алкил-S(=O)2Rc или -C1-12алкил-NRdRe, где С1-12алкил, 3-10-членный циклоалкил или С1-12алкиламино возможно замещен галогеном.
где каждый R71 независимо выбран из гидроксила, и гидроксил замещен группой -С1-12алкил-ОН, 3-10-членный гетероциклоалкил, -С1-12алкил-S(=O)2Rc или -C1-12алкил-NRdRc; и каждый R72 независимо выбран из группы, состоящей из С1-12алкила, гидроксила, 3-10-членного циклоалкила и С1-12алкиламино, где гидроксил замещен группой -С1-12алкил-ОН, 3-10-членный гетероциклоалкил, -С1-12алкил-S(=O)2Rc или -C1-12алкил-NRdRe, где С1-12алкил, 3-10-членный циклоалкил или С1-12алкиламино возможно замещен галогеном.
В некоторых воплощениях каждый R71 независимо выбран из гидроксила, и гидроксил замещен группой -С1-6алкил-ОН, 3-6-членный гетероциклоалкил, -С1-6алкил-S(=O)2Rc или -С1-6алкил-NRdRe. В некоторых воплощениях каждый R71 независимо выбран из гидроксила, где гидроксил замещен группой -С1-4алкил-ОН, 5-6-членный гетероциклоалкил, -С1-4алкил-S(=O)2Rc или -C1-4алкил-NRdRe. В некоторых воплощениях каждый R71 независимо выбран из гидроксила, где гидроксил замещен группой -этил-ОН, эпоксигексил, -пропил-S(=O)2Rc или -этил-NRdRe. В некоторых воплощениях каждый R71 независимо выбран из гидроксила, где гидроксил замещен группой -этил-ОН,  -пропил-S(=O)2СН3 или -этил-N(СН3)2.
-пропил-S(=O)2СН3 или -этил-N(СН3)2.
В некоторых конкретных воплощениях каждый R71 независимо выбран из группы, состоящей из 
В некоторых воплощениях каждый R72 независимо выбран из группы, состоящей из С1-6алкила, гидроксила, 3-6-членного циклоалкила и С1-6алкиламино, где С1-6алкил, 3-6-членный циклоалкил или С1-6алкиламино возможно замещен галогеном, и где гидроксил замещен группой -С1-6алкил-ОН, 5-6-членный гетероциклоалкил, -С1-6алкил-S(=O)2Rc или -С1-6алкил-NRdRe. В некоторых воплощениях каждый R72 независимо выбран из группы, состоящей из С1-4алкила, гидроксила, 3-6-членного циклоалкила и С1-4алкиламино, где С1-4алкил, 3-6-членный циклоалкил или С1-4алкиламино возможно замещен галогеном, где гидроксил замещен группой -С1-4алкил-ОН. В некоторых воплощениях каждый R72 независимо выбран из группы, состоящей из метила, этила, гидроксила, циклопропила и метиламино, где метил, этил или циклопропил возможно замещен фтором; и где гидроксил замещен группой -этил-ОН. В некоторых воплощениях каждый R72 независимо выбран из группы, состоящей из метила, этила, циклопропила, гидроксила, дифторметила и метиламино, где гидроксил замещен группой -этил-ОН.
В некоторых конкретных воплощениях каждый R72 независимо выбран из группы, состоящей из метила, этила, циклопропила, гидроксила, дифторметила, метиламино и 
В некоторых конкретных воплощениях структурная единица  или
или  выбрана из группы, состоящей из
выбрана из группы, состоящей из 

В некоторых конкретных воплощениях  выбран из группы, состоящей из
выбран из группы, состоящей из 
В некоторых конкретных воплощениях структурная единица  выбрана из группы, состоящей из
выбрана из группы, состоящей из 

В некоторых воплощениях каждый из Z8, Z9, Z10 и Z11 независимо выбран из группы, состоящей из СН, С(=O) и N, и по меньшей мере один из них выбран из N; в некоторых воплощениях по меньшей мере один из них выбран из С(=O), и по меньшей мере один из них выбран из N; и в некоторых воплощениях один из них выбран из С(=O), еще один из них выбран из N, а другие два, каждый, представляют собой СН.
В некоторых воплощениях структурная единица 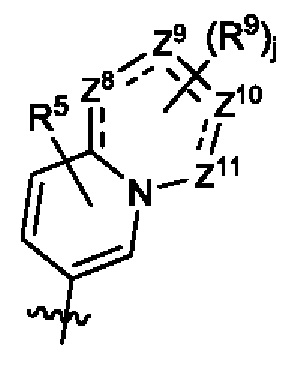 представляет собой
представляет собой  где каждый из Z9, Z10 и Z11 независимо выбран из группы, состоящей из СН, С(=O) и N; в некоторых воплощениях структурная единица
где каждый из Z9, Z10 и Z11 независимо выбран из группы, состоящей из СН, С(=O) и N; в некоторых воплощениях структурная единица  представляет собой
представляет собой  где каждый из Z9 и Z10 независимо выбран из группы, состоящей из СН и N; в некоторых воплощениях структурная единица
где каждый из Z9 и Z10 независимо выбран из группы, состоящей из СН и N; в некоторых воплощениях структурная единица  представляет собой
представляет собой  и в некоторых воплощениях структурная единица
и в некоторых воплощениях структурная единица  представляет собой
представляет собой 
В некоторых воплощениях j равно 1 или 2.
В некоторых воплощениях каждый R9 независимо выбран из группы, состоящей из галогена, С1-6алкила, С1-6алкокси и гидроксила, где С1-6алкил возможно замещен галогеном или С1-6алкокси, и где гидроксил возможно замещен группой -С1-6алкил-O-C1-6алкил, -C1-6алкил-ОН или -С1-6алкил-С(=O)NRfRg. В некоторых воплощениях каждый R9 независимо выбран из группы, состоящей из галогена, С1-4алкила, С1-4алкокси и гидроксила, где С1-4алкил возможно замещен галогеном или С1-4алкокси, и где гидроксил возможно замещен группой -С1-4алкил-О-С1-4алкил, -С1-4алкил-ОН, или -С1-4алкил-С(=O)NRfRg. В некоторых воплощениях каждый R9 независимо выбран из группы, состоящей из галогена, метила, этила, метокси, этокси и гидроксила, где метил или этил возможно замещен галогеном или метокси, и где гидроксил возможно замещен группой -этил-О-метил, -этил-ОН, или -метил-C(=O)NRfRg. В некоторых воплощениях каждый R9 независимо выбран из группы, состоящей из галогена, метила, этила, метокси, этокси и гидроксила, где метил или этил возможно замещен фтором или метокси, и где гидроксил возможно замещен группой -этил-О-метил, -этил-ОН, -CH2C(=O)NHCH3 или -CH2C(=O)NH2.
В некоторых конкретных воплощениях каждый R9 независимо выбран из группы, состоящей из этила, гидроксила, метокси,  фтор, этокси, дифторметил и
фтор, этокси, дифторметил и 
В некоторых воплощениях структурная единица  или
или  выбрана из группы, состоящей из
выбрана из группы, состоящей из  ,
,  - и
- и  , где R91 выбран из группы, состоящей из С1-12алкила, С1-12алкокси и гидроксила, где С1-12алкил возможно замещен С1-12алкокси или галогеном, и где гидроксил возможно замещен группой -С1-12алкил-ОН или -С1-12алкил-О-С1-12алкил, где R92 выбран из группы, состоящей из гидроксила, -С1-12галкокси и галогена, где гидроксил возможно замещен группой -С1-12алкил-ОН, -С1-12алкил-О-С1-12алкил или -C1-12алкил-C(=O)NRfRg.
, где R91 выбран из группы, состоящей из С1-12алкила, С1-12алкокси и гидроксила, где С1-12алкил возможно замещен С1-12алкокси или галогеном, и где гидроксил возможно замещен группой -С1-12алкил-ОН или -С1-12алкил-О-С1-12алкил, где R92 выбран из группы, состоящей из гидроксила, -С1-12галкокси и галогена, где гидроксил возможно замещен группой -С1-12алкил-ОН, -С1-12алкил-О-С1-12алкил или -C1-12алкил-C(=O)NRfRg.
В некоторых воплощениях R91 выбран из группы, состоящей из С1-6алкила, С1-6алкокси и гидроксила, где С1-6алкил возможно замещен С1-6алкокси или галогеном, и где гидроксил возможно замещен группой -С1-6алкил-ОН или -С1-6алкил-О-С1-6алкил. В некоторых воплощениях R91 выбран из группы, состоящей из С1-4алкила, С1-4алкокси и гидроксила, где С1-4алкил возможно замещен С1-4алкокси или галогеном, и где гидроксил возможно замещен группой -С1-4алкил-ОН или -С1-4алкил-О-С1-4алкил. В некоторых воплощениях R91 выбран из группы, состоящей из метила, этила, метокси, этокси и гидроксила, где метил или этил возможно замещен галогеном или метокси, и где гидроксил возможно замещен группой -этил-О-метил или -этил-ОН. В некоторых воплощениях R91 выбран из группы, состоящей из метила, этила, метокси, этокси и гидроксила, где метил или этил возможно замещен фтором или метокси, и где гидроксил возможно замещен группой -этил-О-метил или -этил-ОН.
В некоторых конкретных воплощениях R91 выбран из группы, состоящей из этила, метокси, этокси, дифторметила, 
В некоторых воплощениях R92 выбран из группы, состоящей из гидроксила, С1-6алкокси и галогена, где гидроксил возможно замещен группой -С1-6алкил-ОН, -С1-6алкил-О-С1-6алкил или -C1-6алкил-C(=O)NRfRg. В некоторых воплощениях R92 выбран из группы, состоящей из гидроксила, С1-4алкокси и галогена, где гидроксил возможно замещен группой -С1-4алкил-ОН, -С1-4алкил-О-С1-4алкил или -С1-4алкил-C(=O)NRfRg. В некоторых воплощениях R92 выбран из группы, состоящей из гидроксила, метокси и галогена, где гидроксил возможно замещен группой -этил-ОН, -этил-О-метил или -метил-C(=O)NRfRg. В некоторых воплощениях R92 выбран из группы, состоящей из гидроксила, метокси и галоген, где гидроксил возможно замещен группой -этил-ОН, -этил-О-метил, -CH2C(=O)NHCH3 или -CH2C(=O)NH2.
В некоторых конкретных воплощениях R92 выбран из группы, состоящей из гидроксила, метокси,  и фтора.
и фтора.
В некоторых конкретных воплощениях структурная единица  или
или  выбрана из группы, состоящей из
выбрана из группы, состоящей из 

В некоторых воплощениях два из X12, X13, X14, X15 и X16 представляют собой NH и С(=O) соответственно, а остальные представляют собой СН2 или О.
В некоторых воплощениях структурная единица  представляет собой
представляет собой  ; и в некоторых воплощениях структурная единица
; и в некоторых воплощениях структурная единица  представляет собой
представляет собой 
В некоторых воплощениях каждый R8 независимо выбран из группы, состоящей из водорода, С1-6алкила, гидроксила, амино, 3-6-членного циклоалкила, С1-6алкокси, 3-6-членного гетероциклоалкила и С1-6алкиламино; в некоторых воплощениях каждый R8 независимо выбран из С1-4алкила; и в некоторых воплощениях каждый R8 независимо выбран из этила.
В некоторых воплощениях q равно 0, 1 или 2; в некоторых воплощениях q равно 1 или 2; и в некоторых воплощениях q равно 1.
В некоторых воплощениях структурная единица 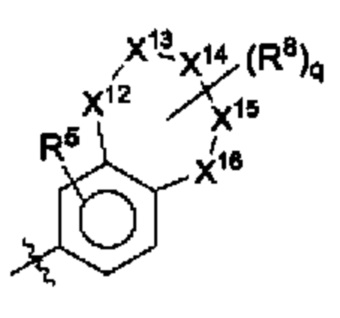 представляет собой
представляет собой 
В некоторых конкретных воплощениях структурная единица  представляет собой
представляет собой 
В еще одном аспекте настоящей заявки предложено соединение формулы (II) или его фармацевтически приемлемую соль:
 ,
,
где
R2 и R3 выбраны из метила, или вместе взятые R2 и R3 связаны друг с другом с образованием циклобутила; а
структурная единица 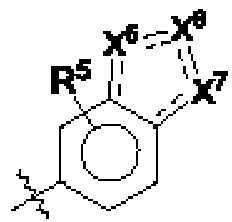 и X5, X6, X7, Т и R5 такие, как определено для соединения формулы (I).
и X5, X6, X7, Т и R5 такие, как определено для соединения формулы (I).
В некоторых воплощениях настоящего изобретения Т выбран из СН.
В еще одном другом аспекте настоящей заявки предложено соединение формулы (III-1) или соединение формулы (III-2) или его фармацевтически приемлемая соль:

где
R2 и R3 выбраны из метила, или вместе взятые R2 и R3 связаны друг с другом с образованием циклобутила; а
структурные единицы 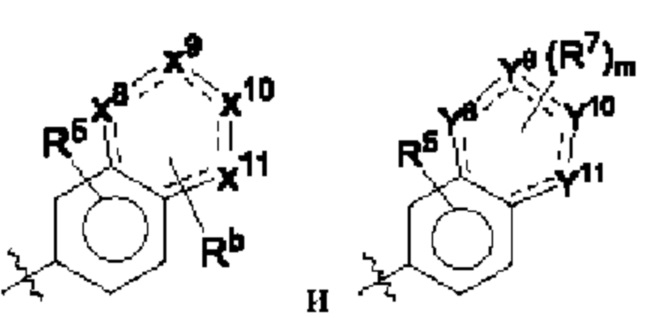 и X8, X9, X10, X11, Y8, Y9, Y10, Y11, Т, R5, Rb, R7 и m такие, как определено для соединения формулы (I).
и X8, X9, X10, X11, Y8, Y9, Y10, Y11, Т, R5, Rb, R7 и m такие, как определено для соединения формулы (I).
В еще одном другом аспекте настоящей заявки предложено соединение формулы (III-21) или соединение формулы (III-22) или его фармацевтически приемлемая соль:

где
m равно 1 или 2; R2 и R3 выбраны из метила, или вместе взятые R2 и R3 связаны друг с другом с образованием циклобутила; а
структурная единица  и Т, R5 и R7 такие, как определено для соединения формулы (I).
и Т, R5 и R7 такие, как определено для соединения формулы (I).
В некоторых воплощениях структурная единица  представляет собой
представляет собой  где определение структурной единицы
где определение структурной единицы  такое, как упомянуто выше.
такое, как упомянуто выше.
В еще одном другом аспекте настоящей заявки предложено соединение формулы (IV) или его фармацевтически приемлемая соль:
 ,
,
где
q равно 1 или 2; R2 и R3 выбраны из метила, или вместе взятые R2 и R3 связаны друг с другом с образованием циклобутила; а
структурная единица  и Т, X12, X13, X14, X15, X16, R5 и R8 такие, как определено для соединения формулы (I).
и Т, X12, X13, X14, X15, X16, R5 и R8 такие, как определено для соединения формулы (I).
В некоторых воплощениях настоящей заявки Т выбран из СН.
В другом аспекте настоящей заявки предложено соединение формулы (V) или его фармацевтически приемлемая соль:
 ,
,
где структурные единицы  и Т, R4, n, R5 и R6 такие, как определено для соединения формулы (I).
и Т, R4, n, R5 и R6 такие, как определено для соединения формулы (I).
В некоторых воплощениях настоящей заявки Т выбран из СН.
Предпочтительно, согласно настоящей заявке предложено соединение формулы (VI) или его фармацевтически приемлемая соль:
 ,
,
где
структурная единица  и T, R1, R5, R9, Z8, Z9, Z10, Z11 и j такие, как определено для соединения формулы (I); и
и T, R1, R5, R9, Z8, Z9, Z10, Z11 и j такие, как определено для соединения формулы (I); и
R2 и R3 выбраны из метила, или вместе взятые R2 и R3 связаны друг с другом с образованием циклобутила.
В некоторых конкретных воплощениях R1 выбран из группы, состоящей из фтора, хлора и трифторметила.
Предпочтительно, согласно настоящей заявке предложено соединение формулы (VI-1) или его фармацевтически приемлемая соль:
 ,
,
где
Т, R1, R5, R9 и j такие, как определено для соединения формулы (I); и R2 и R3 выбраны из метила, или вместе взятые R2 и R3 связаны друг с другом с образованием циклобутила.
В некоторых воплощениях структурная единица  представляет собой
представляет собой  где определение структурной единицы
где определение структурной единицы  такое, как упомянуто выше.
такое, как упомянуто выше.
В некоторых конкретных воплощениях R1 выбран из группы, состоящей из фтора, хлора и трифторметила.
В другом аспекте настоящей заявки предложено соединение формулы (VII) или его фармацевтически приемлемая соль:
 ,
,
где
T, R1, R4 и n такие, как определено для соединения формулы (I); и
кольцо В выбрано из группы, состоящей из 
 , где X8, X9, X10, X11, Y8, Y9, Y10, Y11, Z8, Z9, Z10, Z11, R5, R6, Rb, R7, R9, m, j и структурные единицы
, где X8, X9, X10, X11, Y8, Y9, Y10, Y11, Z8, Z9, Z10, Z11, R5, R6, Rb, R7, R9, m, j и структурные единицы  и
и  такие, как определено для соединения формулы (I).
такие, как определено для соединения формулы (I).
В еще одном аспекте настоящей заявки предложены следующие соединения или их фармацевтически приемлемые соли:




В еще одном другом аспекте настоящая заявка относится к фармацевтической композиции, содержащей соединение формулы (I), формулы (II), формулы (III-1), формулы (III-2), формулы (III-21), формулы (III-22), формулы (IV), формулы (V), формулы (VI), формулы (VI-1) или формулы (VII) или его фармацевтически приемлемую соль согласно настоящей заявке. В некоторых воплощениях фармацевтическая композиция по настоящей заявке дополнительно содержит фармацевтически приемлемый эксципиент.
В еще одном другом аспекте настоящая заявка относится к способу лечения опосредованного андрогеном заболевания у млекопитающего, включающему введение млекопитающему, предпочтительно человеку, нуждающемуся в лечении, терапевтически эффективного количества соединения формулы (I), формулы (II), формулы (III-1), формулы (III-2), формулы (III-21), формулы (III-22), формулы (IV), формулы (V), формулы (VI), формулы (VI-1) или формулы (VII) или его фармацевтически приемлемой соли или его фармацевтической композиции, и заболевание включает, без ограничения, клеточные пролиферативные заболевания (например, рак).
В другом аспекте настоящая заявка относится к применению соединения формулы (I), формулы (II), формулы (III-1), формулы (III-2), формулы (III-21), формулы (III-22), формулы (IV), формулы (V), формулы (VI), формулы (VI-1) или формулы (VII) или его фармацевтически приемлемой соли или его фармацевтической композиции в приготовлении лекарственного средства для лечения опосредованного андрогеном заболевания, и заболевание включает, без ограничения, клеточные пролиферативные заболевания (например, рак).
В еще одном другом аспекте настоящая заявка относится к соединению формулы (I), формулы (II), формулы (III-1), формулы (III-2), формулы (III-21), формулы (III-22), формулы (IV), формулы (V), формулы (VI), формулы (VI-1) или формулы (VII) или его фармацевтически приемлемой соли или его фармацевтической композиции для применения в лечении опосредованного андрогеном заболевания, и заболевание включает, без ограничения, клеточные пролиферативные заболевания (например, рак).
В еще одном другом аспекте настоящая заявка относится к соединению формулы (I), формулы (II), формулы (III-1), формулы (III-2), формулы (III-21), формулы (III-22), формулы (IV), формулы (V), формулы (VI), формулы (VI-1) или формулы (VII) или его фармацевтически приемлемой соли или его фармацевтической композиции для применения в предупреждении или лечении опосредованного андрогеном заболевания, и заболевание включает, без ограничения, клеточные пролиферативные заболевания (например, рак).
ОПРЕДЕЛЕНИЯ
Если не указано иное, нижеследующие термины, использованные в данном документе, имеют нижеследующие значения. Если конкретный термин специально не определен, он не может считаться неопределенным или неясным, и следует понимать, что он имеет значение, общепринятое в данной области. Если в данном документе приводится торговое наименование, то оно предназначено для обозначения соответствующего продукта или его активного ингредиента.
Термин "замещенный" означает, что один или более атомов водорода на конкретном атоме заменены заместителями при условии, что валентное состояние конкретного атома является нормальным, и замещенное соединение является стабильным. Когда заместитель представляет собой оксо или кето (то есть =O), тогда это означает, что замещены два атома водорода.
Термин "возможный" или "возможно" означает, что события или обстоятельства, описанные после него, могут иметь место или нет, и что данное описание включает в себя случаи, когда указанное событие или обстоятельство имеет место, и случаи, когда указанное событие или обстоятельство не имеет места. Например, когда этил "возможно" замещен фтором или хлором, тогда это означает, что этил может быть незамещенным (например, -СН2СН3), монозамещенным (например, -CH2CH2F, -CHFCH3), полизамещенным (например, -CHFCH2F, -CHClCH2F, -CH2CHCl2, -CH2CHF2 и т.п.) или полностью замещенным (-CFClCF3, -CF2CF3). Что касается любых групп, содержащих один или более заместителей, специалистам в данной области будет понятно, что такие группы не предназначены для введения каких-либо замещений или схем замещения, которые стерически не осуществимы на практике и/или синтетически не выполнимы. Если конкретно не указано иное, виды и количества заместителей могут быть произвольными, исходя из условия, что они химически доступны.
Когда заместитель может быть связан с более чем одним атомом на кольце, тогда такой заместитель может быть связан с любым атомом на кольце; например, структурная единица  включает
включает  , но не включает
, но не включает 
"Cm-n" в данном документе означает, что данная группировка имеет целое число атомов углерода в данном диапазоне. Например, "С1-6" означает, что группа может иметь 1 атом углерода, 2 атома углерода, 3 атома углерода, 4 атома углерода, 5 атомов углерода или 6 атомов углерода; и "С3-6" означает, что группа может иметь 3 атома углерода, 4 атома углерода, 5 атомов углерода или 6 атомов углерода.
Когда переменная (например, R7) встречается в составе или структуре соединения больше одного раза, тогда переменная в каждом случае определена независимо. Так, например, (R7)m представляет собой группу, замещенную m R7, и каждый R7 имеет независимые варианты; и конкретно, например, когда m=2, тогда одна группа замещена 2 R7, и каждый R7 имеет независимые варианты.
Термин "галоген" или "галогено" относится к фтору, хлору, брому и йоду.
Термин "гидроксил" относится к группе -ОН.
Термин "амино" относится к группе -NH2.
Термин "трифторметил" относится к группе -CF3.
Термин "алкил" относится к гидрокарбилу, имеющему общую формулу CnH2n+1, Алкил может быть прямым или разветвленным. Например, термин "С1-6алкил" относится к алкилу, содержащему от 1 до 6 атомов углерода (например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, неопентил, гексил, 2-метилпентил и т.п.). Аналогично, алкильная группировка (т.е. алкил) в алкокси имеет такое же определение, как указанное выше; и термин "С1-3алкил" относится к алкилу, содержащему от 1 до 3 атомов углерода (например, метил, этил, н-пропил или изопропил).
Термин "алкокси" относится к группе -О-алкил.
Термин "алкиламино" относится к группе -NH-алкил.
Термин "алкиламинокарбонил" относится к группе -С(=O)-NH-алкил.
Термин "алкенил" относится к прямому или разветвленному ненасыщенному алифатическому гидрокарбонилу, состоящему из атомов углерода и атомов водорода и имеющему по меньшей мере одну двойную связь. Не ограничивающие примеры алкенила включают, без ограничения, этенил, 1-пропенил, 2-пропенил, 1-бутенил, изобутенил, 1,3-бутадиенил и т.п. Например, термин "С2-6алкенил" относится к алкенилу содержащему от 2 до 6 атомов углерода и термин "С2-С3алкенил" относится к алкенилу содержащему от 2 до 3 атомов углерода (например, этенил, 1-пропенил или 2-пропенил).
Термин "алкинил" относится к прямому или разветвленному ненасыщенному алифатическому гидрокарбонилу, состоящему из атомов углерода и атомов водорода и имеющему по меньшей мере одну тройную связь. Не ограничивающие примеры алкинила включают, без ограничения, этинил (-С≡СН), 1-пропинил (-С≡С-СН3), 2-пропинил (-СН2-С≡СН), 1,3-бутадиенил (-C≡СС≡СН) и т.п.
Термин "циклоалкил" относится к полностью насыщенному углеродному кольцу, которое может существовать в виде моноциклического кольца, мостикового кольца или спиро кольца. Если конкретно не указано иное, углеродное кольцо обычно представляет собой 3-10-членное кольцо. Не ограничивающие примеры циклоалкила включают, без ограничения, циклопропил, циклобутил, циклопентил, циклогексил, норборнил (бицикло[2.2.1]гептил), бицикло[2.2.2]октил, адамантил и т.п. Циклоалкил предпочтительно представляет собой моноциклический циклоалкил, имеющий от 3 до 6 кольцевых атомов.
Термин "гетероциклоалкил" относится к полностью насыщенной циклической группе, которая может существовать в виде моноциклического кольца, мостикового кольца или спиро кольца. Если конкретно не указано иное, гетероциклическое кольцо обычно представляет собой 3-7-членное кольцо, содержащее от 1 до 3 гетероатомов (предпочтительно 1 или 2 гетероатома), независимо выбранных из группы, состоящей из атомов серы, кислорода и/или азота. Примеры 3-членного гетероциклоалкила включают, без ограничения, эпоксиэтил, циклотиоэтил и азиридинил; не ограничивающие примеры 4-членного гетероциклоалкила включают, без ограничения, азетидинил, оксетанил и тиетанил; примеры 5-членного гетероциклоалкила включают, без ограничения, тетрагидрофуранил, тетрагидротиенил, пирролидинил, изоксазолидинил, оксазолидинил, изотиазолидинил, тиазолидинил, имидазолидинил, тетрагидропиразолил и пирролинил; примеры 6-членного гетероциклоалкила включают, без ограничения, пиперидинил, тетрагидропиранил, тетрагидротиопиранил, морфолинил, пиперазинил, 1,4-тиоксанил, 1,4-диоксанил, тиоморфолинил, 1,2-дитиоалкил, 1,4-дитиоалкил и тетрагидропиранил; и примеры 7-членного гетероциклоалкила включают, без ограничения, азациклогептил, оксациклогептил и тиациклогептил. Гетероциклоалкил предпочтительно представляет собой моноциклический гетероциклоалкил, имеющий от 5 до 6 кольцевых атомов.
Термин "гетероарил" относится к моноциклической или конденсированной полициклической кольцевой системе, содержащей по меньшей мере один кольцевой атом, выбранный из группы, состоящей из N, О и S, и остальные кольцевые атомы, представляющие собой атомы С, и имеющей по меньшей мере одно ароматическое кольцо. Предпочтительный гетероарил имеет единственное 4-8-членное кольцо, и в частности, 5-8-членное кольцо, или множество конденсированных колец, содержащих от 6 до 14, и в частности, от 6 до 10 кольцевых атомов. Не ограничивающие примеры гетероарила включают, без ограничения, пирролил, фуранил, тиенил, имидазолил, оксазолил, пиразолил, пиридинил, пиримидинил, пиразинил, хинолил, изохинолил, тетразолил, триазолил, триазинил, бензофуранил, бензотиенил, индолил, изоиндолил и т.п.
"-С1-12алкил-(3-10-членный циклоалкил)" в данном документе представляет собой С1-12алкил, замещенный 3-10-членным циклоалкилом, и другие подобные выражения следует понимать аналогично.
В данном документе "-С1-12алкил-(3-10-членный циклоалкил), возможно замещенный галогеном или гидроксилом" означает, что любой атом водорода в группе -С1-12алкил-(3-10-членный циклоалкил) может быть замещен галогеном или гидроксилом, и другие подобные выражения следует понимать аналогично.
Структурная единица  представляет собой бензогетероциклическую кольцевую систему. Связь
представляет собой бензогетероциклическую кольцевую систему. Связь  соответственно представляет собой одинарную связь или двойную связь в соответствии с конкретным вариантом X5, X6, X7, X8, X9, X10, X11, Y8, Y9, Y10 или Y11 в настоящей заявке, и не должна нарушать теорию валентных связей. Например, когда X5 представляет собой СН, X6 представляет собой N(-Ra), и X7 представляет собой N, тогда структурная единица
соответственно представляет собой одинарную связь или двойную связь в соответствии с конкретным вариантом X5, X6, X7, X8, X9, X10, X11, Y8, Y9, Y10 или Y11 в настоящей заявке, и не должна нарушать теорию валентных связей. Например, когда X5 представляет собой СН, X6 представляет собой N(-Ra), и X7 представляет собой N, тогда структурная единица  представляет собой
представляет собой  Например, когда X5 представляет собой СН, X6 представляет собой N, и X7 представляет собой N(-Ra), тогда структурная единица
Например, когда X5 представляет собой СН, X6 представляет собой N, и X7 представляет собой N(-Ra), тогда структурная единица  представляет собой
представляет собой  . Например, когда X8 представляет собой N, X9 представляет собой СН, X10 представляет собой NH, и X11 представляет собой С(=O), тогда структурная единица
. Например, когда X8 представляет собой N, X9 представляет собой СН, X10 представляет собой NH, и X11 представляет собой С(=O), тогда структурная единица  представляет собой
представляет собой  Например, когда Y8 представляет собой N, Y9 представляет собой СН, Y10 представляет собой N, и Y11 представляет собой СН, тогда структурная единица
Например, когда Y8 представляет собой N, Y9 представляет собой СН, Y10 представляет собой N, и Y11 представляет собой СН, тогда структурная единица  представляет собой
представляет собой 
Структурная единица  представляет собой пиридиногетероциклическую кольцевую систему. Связь
представляет собой пиридиногетероциклическую кольцевую систему. Связь  соответственно представляет собой одинарную связь или двойную связь в соответствии с конкретным вариантом Z8, Z9, Z10 или Z11 в настоящей заявке, и не должна нарушать теорию валентных связей.
соответственно представляет собой одинарную связь или двойную связь в соответствии с конкретным вариантом Z8, Z9, Z10 или Z11 в настоящей заявке, и не должна нарушать теорию валентных связей.
Если не указано иное, клиновидная связь и пунктирная связь ( ) обозначают абсолютную конфигурацию стереоцентра, волнистая линия
) обозначают абсолютную конфигурацию стереоцентра, волнистая линия  обозначает одну из абсолютных конфигураций стереоцентра (например, одну из
обозначает одну из абсолютных конфигураций стереоцентра (например, одну из  ), и
), и  обозначают относительную конфигурацию стереоцентра. Когда соединения согласно настоящей заявке содержат олефиновые двойные связи или другие геометрически асимметрические центры, тогда они включают в себя Е и Z геометрические изомеры, если конкретно не указано иное. Аналогично, все таутомерные формы входят в объем настоящей заявки.
обозначают относительную конфигурацию стереоцентра. Когда соединения согласно настоящей заявке содержат олефиновые двойные связи или другие геометрически асимметрические центры, тогда они включают в себя Е и Z геометрические изомеры, если конкретно не указано иное. Аналогично, все таутомерные формы входят в объем настоящей заявки.
Соединения согласно настоящей заявке могут существовать в конкретных геометрических изомерных или стереоизомерных формах. Все такие соединения предусмотрены в настоящей заявке, в том числе таутомеры, цис- и транс-изомеры, (-)-и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереомеры, (D)-изомеры, (L)-изомеры и их рацемические смеси и другие смеси, такие как смеси, обогащенные энантиомером или диастереомером. Все такие смеси входят в объем настоящей заявки. Заместители, такие как алкильная группа, могут иметь дополнительные несимметрические атомы углерода. Все такие изомеры и их смеси входят в объем настоящей заявки.
Термин "проведение лечения" или "лечение" означает введение соединений или препаратов согласно настоящей заявке для предупреждения, ослабления или устранения заболевания или одного или более симптомов, ассоциированных с заболеванием, и охватывает:
(1) предупреждение возникновения заболевания или состояния у млекопитающего, особенно когда такое млекопитающее восприимчиво к этому состоянию, но это состояние не диагностировано;
(2) подавление заболевания или состояния, т.е. остановку его развития; и
(3) ослабление заболевания или состояния, т.е. вызывание регрессии заболевания или состояния.
Термин "терапевтически эффективное количество" относится к количеству соединения согласно настоящей заявке для (1) лечения или предупреждения конкретного заболевания, состояния или расстройства, (2) облегчения, ослабления или устранения одного или более симптомов конкретного заболевания, состояния или расстройства или (3) предупреждения или замедления возникновения одного или более симптомов конкретного заболевания, состояния или расстройства, описанных в данном документе. Количество соединения согласно настоящей заявке, составляющее "терапевтически эффективное количество", будет варьировать в зависимости от соединения, болезненного состояния и его тяжести, способа введения и возраста млекопитающего, подлежащего лечению, но может быть определено рутинно специалистами в данной области, исходя из их собственных знаний и настоящего описания изобретения.
Термин "фармацевтически приемлемый" относится к тем соединениям, веществам, композициям и/или лекарственным формам, которые являются подходящими для контактирования с тканями человека и животных в рамках объективного медицинского суждения, но не имеют излишней токсичности, стимуляции, аллергических реакций или не создают других проблем или осложнений, и соответствуют разумному соотношению польза/риск.
В качестве фармацевтически приемлемых солей могут быть упомянуты, например, соли металлов, аммониевые соли, соли органических оснований, соли неорганических кислот, соли органических кислот, соли щелочных или кислотных аминокислот.
Термин "фармацевтическая композиция" относится к смеси одного или более соединений или их солей согласно настоящей заявке и фармацевтически приемлемого эксципиента. Задача фармацевтической композиции заключается в облегчении введения соединения согласно настоящей заявке в организм.
Термин "фармацевтически приемлемый эксципиент" относится к эксципиентам, которые не вызывают очевидного раздражения и не оказывают неблагоприятного воздействия на биологическую активность и свойства активного соединения. Специалистам в данной области известны подходящие эксципиенты, такие как углеводы, воски, водорастворимые и/или набухающие в воде полимеры, гидрофильные или гидрофобные вещества, желатин, масла, растворители вода и т.п.
Слово "содержать" и его варианты (такие как "содержит" и "содержащий") следует понимать, как неограниченные и не эксклюзивные значения, т.е. "включают, без ограничения".
Промежуточные соединения и соединения согласно настоящей заявке могут также существовать в формах разных таутомеров, и все такие формы входят в объем настоящей заявки. Термин "таутомер" или "таутомерная форма" относится к структурным изомерам разной энергии, которые способны к взаимопревращению через низкий энергетический барьер. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимопревращения посредством миграции протона, такие как кето-енольная и имин-енаминнная изомеризации. Конкретным примером протонных таутомеров является имидазольная группировка, где протон может мигрировать между двумя атомами азота кольца. Валентные таутомеры включают взаимопревращения за счет перегруппировки некоторых связеобразующих электронов.
Настоящая заявка также охватывает меченные изотопами соединения согласно настоящей заявке, которые идентичны соединениям, описанным в данном документе, но у которых один или более атомов заменены атомами, имеющими атомную массу или массовое число, отличающиеся от атомной массы или массового числа, обычно существующих в природе. Примеры изотопов, которые могут быть введены в соединения согласно настоящей заявке, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, йода и хлора, например изотопы 2Н, 3Н, 11С, 13С, 14С, 13N 15N, 15O, 17O, 18O, 31P, 32P, 35S, 18F, 123I, 125I, 36Cl соответственно.
Некоторые меченные изотопом соединения согласно настоящей заявке (например, меченные 3Н и 14С) могут быть использованы в анализах распределения соединения и/или субстрата в тканях. Изотопы тритий (т.е. 3Н) и углерод-14 (т.е. 14С) являются особенно предпочтительным благодаря легкости их получения и обнаружения. Позитрониспускающие изотопы, такие как 15O, 13N, 11С и 18F могут быть использованы в исследованиях методом позитронно-эмиссионной томографии (PET) для определения занятости субстратом. Меченные изотопом соединения согласно настоящей заявке обычно могут быть получены по методикам, аналогичным методикам, описанным ниже на схемах и/или в примерах, путем замены немеченых реагентов реагентами, меченными изотопом.
Кроме того, замещение тяжелыми изотопами (такими как дейтерий, те 2Н) может давать некоторые терапевтические преимущества, благодаря более высокой метаболической стабильности, например увеличению периода полувыведения in vivo или снижению требований по дозировке, и поэтому в некоторых обстоятельствах может быть предпочтительным, причем замещение дейтерием может быть частичным или полным, и частичное замещение дейтерием означает, что по меньшей мере один атом водорода замещен по меньшей мере одним дейтерием.
Соединения согласно настоящей заявке могут быть асимметрическими, например имеющими один или более стереооизомеров. Если не утверждается иное, охвачены все стереоизомеры, такие как энантиомеры и диастереоизомеры. Соединения, содержащие асимметрические атомы углерода согласно настоящей заявке, могут быть выделены в оптически активных чистых формах или рацемических формах. Оптически активные чистые формы могут быть выделены из рацемических смесей или могут быть синтезированы с использованием хиральных исходных веществ или хиральных реагентов.
Фармацевтическая композиция согласно настоящей заявке может быть получена путем объединения соединения согласно настоящей заявке с подходящим фармацевтически приемлемым эксципиентом, и, например, она может быть приготовлена в виде твердых, полутвердых, жидких или газообразных препаратов, таких как таблетки, пилюли, капсулы, порошки, гранулы, мази, эмульсии, суспензии, суппозитории, инъекционные препараты, ингалируемые препараты, гели, микрочастицы, аэрозоли и т.п.
Типичные пути введения соединения или его фармацевтически приемлемой соли или фармацевтической композиции согласно настоящей заявке включают, без ограничения, пероральное, ректальное, местное, ингаляционное, парентеральное, сублингвальное, интравагинальное, интраназальное, внутриглазное, интраперитонеальное, внутримышечное, подкожное и внутривенное введение.
Фармацевтическая композиция согласно настоящей заявке может быть изготовлена с использованием способов, общеизвестных в данной области, таких как стандартный способ смешивания, способ растворения, способ гранулирования, способ для получения пилюль с сахарных покрытием, способом измельчения, способ эмульгирования, лиофилизация и т.п.
В некоторых воплощениях фармацевтическая композиция находится в пероральное форме. Для перорального введения фармацевтическая композиция может быть приготовлена путем смешивания активного соединения с фармацевтически приемлемым эксципиентом, известным в данной области. Эти эксципиенты делают возможным приготовление соединения согласно настоящей заявке в форме таблеток, пилюль, пастилок, драже, капсул, жидкостей, гелей, взвесей, суспензий и т.п. для перорального введения пациенту.
Твердая пероральная композиция может быть получена традиционными способами смешивания, заполнения или таблетирования. Например, она может быть получена следующим способом: смешивание активного соединения с твердым эксципиентом, возможно измельчение полученной смеси, добавление других подходящих эксципиентов, если необходимо, и затем переработка смеси в гранулы для получения ядра таблетки или драже. Подходящие эксципиенты включают, без ограничения, связующие, разбавители, разрыхлители, смазывающие вещества, глиданты, подсластители или корригенты.
Фармацевтическая композиция может быть также подходящей для парентерального введения, например в виде стерильного раствора, суспензии или лиофилизированного продукта в подходящих стандартных лекарственных формах.
При всех способах введения для соединений формулы (I), формулы (II), формулы (III-1), формулы (III-2), формулы (III-21), формулы (III-22), формулы (IV), формулы (V), формулы (VI), формулы (VI-1) или формулы (VII) согласно настоящей заявке суточная доза составляет от 0,01 до 200 мг/кг массы тела в форме отдельных или разделенных доз.
Соединения согласно настоящей заявке могут быть получены различными способами синтеза, известными специалистам в данной области, включая конкретные воплощения, перечисленные ниже, воплощения, достигаемые путем комбинирования конкретных воплощений, перечисленных ниже, с другими способами химического синтеза, и эквиваленты, известные специалистам в данной области. Предпочтительные воплощения включают, без ограничения, примеры, приведенные настоящей заявке.
Химические реакции в конкретных воплощениях согласно настоящей заявке проводят в подходящих растворителях, которые подходят для химических превращений согласно настоящей заявке и необходимых реагентов и веществ. Для получения соединений согласно настоящей заявке иногда необходимо модифицировать или выбирать стадии синтеза или реакционные схемы, исходя из существующих воплощений.
Одним из важных факторов для планирования схемы синтеза в данной области является выбор подходящих защитных групп для реакционноспособных функциональных групп (таких как гидроксильная группа согласно настоящей заявке). Например, можно сослаться на Greene's Protective Groups in Organic Synthesis (4th Ed). Hoboken, New Jersey: John Wiley & Sons, Inc. Все источники информации, процитированные в настоящей заявке, во всей их полноте включены в данный документ посредством ссылки.
В некоторых воплощениях соединения формулы (I) согласно настоящей заявке могут быть получены специалистами в области органического синтеза в результате осуществления следующих стадий и путей:
Синтез промежуточного соединения (I):
Стадия 1: 
Стадия 2:

Схема I Стадии 3:

Схема II Стадии 3:
Схема III Стадии 3:

Схема IV Стадии 3:

Синтез промежуточного соединения (II):
Стадия 1:

Схема I Стадии 2:

Схема II Стадии 2:

Получение целевого соединения (I)





Получение целевого соединения (II)


В вышеуказанных путях
каждый R71 независимо выбран из группы, состоящей из гидроксила и 5-10-членного гетероарила, и гидроксил возможно замещен группой -С1-12алкил-(3-10-членный гетероциклоалкил), -C1-12алкил-S(=O)2Rc, -C1-12алкил-NRdRe, -С1-12алкил-C(=O)NRfRg, -С1-12алкил-(3-10 -членный циклоалкил), возможно замещенной галогеном или гидроксилом, или 3-10-членный гетероциклоалкилом, возможно замещенным галогеном или гидроксилом; или каждый R71 независимо выбран из гидроксила, и гидроксил возможно замещен гетероциклоалкилом, группой -алкил-S(=O)2Rc или -алкил-NRdRe, где Rc, Rd и Re такие, как определено в настоящей заявке; и
R10 выбран из группы, состоящей из R72 и Н, и j, Т, R1, R5, R9 и R72 такие, как определено в настоящей заявке.
В настоящей заявке использованы следующие сокращения:
DMF означает М,М-диметилформамид, DMSO означает диметилсульфоксид, ЖХ/МС означает жидкостная хроматография/масс-спектрометрия, ТСХ означает тонкослойная хроматография, ВЭЖХ означает высокоэффективная жидкостная хроматография, Вое означает трет-бутоксикарбонил, TMSCHN2 означает триметилсилилдиазометан, TMSCN означает триметилсилилцианид, diBoc означает ди-трет-бутилдикарбонат, NBS означает N-бромсукцинимид, CDI означает 1,1'-карбонилдиимидазол, Boc-NH2 означает трет-бутилкарбамат, Вос2О означает ди-трет-бутилдикарбонат, EDTA-K2 означает дикалия этилендиаминтетраацетат, DAST означает диэтиламиносеры трифторид, NMP означает 1-метил-2-пирролидон, CMC означает карбоксиметил целлюлоза, HATU означает 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат; об./об. означает объемное соотношение; RLU означает относительная сила света; Solutol означает полиэтиленгликоль-15-гидроксистеарат; PEG400 означает полиэтиленгликоль 400; РО означает пероральное введение; QD означает частота введения; РМВ означает пара-метоксибензил; и DPPF означает 1,1'-бис(дифенилфосфино)ферроцен.
ПОДРОБНОЕ ОПИСАНИЕ ВОПЛОЩЕНИЙ
Настоящая заявка ниже описана подробно в сочетании с примерами, но они не предназначены для неприемлемого ограничения настоящего изобретения. Здесь настоящая заявка описана подробно, а также раскрыты конкретные воплощения. Для специалистов в данной области очевидно, что могут быть выполнены различные модификации и усовершенствования без отклонения от замысла и объема настоящей заявки. Все растворители, используемые в настоящей заявке, имеются в продаже и могут быть использованы без дополнительной очистки. Исходные вещества для синтеза соединений согласно настоящей заявке имеются в продаже или могут быть получены способами предшествующего уровня техники.
Ядерный магнитный резонанс (ЯМР) в настоящей заявке определен с использованием ядерного магнитного резонансного спектрометра BRUKER 400 с тетраметилсиланом (TMS = δ 0.00) в качестве внутреннего стандарта химического сдвига. Данные спектра протонного ядерного магнитного резонанса записаны в формате: паттерн пиков (s: синглет; d: дублет; t: триплет; q: квартет; m: мультиплет), константа взаимодействия (единица: герц (Гц)) и число протонов. В качестве прибора для масс-спектрометрии использован SHIMADUZU LCMS-2010.
Пример 1. Синтез Соединения 1

1) Синтез Соединения 1-2

0,5 мл дихлорсульфоксида добавляли по каплям в раствор Соединения 1-1 (300 мг, 1,33 ммоль) в 5 мл DMF. Реакционную смесь нагревали до 80°С и перемешивали при этой температуре в течение 6 ч. Реакционную смесь сушили в центробежной сушилке при пониженном давлении. Полученное твердое вещество растворяли в 30 мл этилацетата, промывали 20 мл воды и 20 мл насыщенного рассола в указанном порядке, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением Соединения 1-2. ЖХ/МС (ЭРИ (электрораспылительная ионизация)) m/z: 245 (М+3).
2) Синтез Соединения 1-3
В пробирке для микроволновой обработки Соединение 1-2 (150 мг, 616,04 мкмоль) и раствор метиламина в этаноле (750 мкл, чистота 20-30%) растворяли в трет-бутаноле (4 мл). Реакционную смесь выдерживали при 90°С для реакции под воздействием микроволнового излучения в течение 0,5 ч и затем концентрировали при пониженном давлении. Полученный остаток растворяли в 20 мл этилацетата, промывали 10 мл воды и 20 мл насыщенного рассола в указанном порядке, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением Соединения 1-3. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.45 (s, 1Н), 7.85(d, J=3.2 Гц, 2Н), 7.67 (s, 1Н), 3.07 (s, 3Н); ЖХ/МС (ЭРИ) m/z: 240 (М+3).
3) Синтез Соединения 1-5

В пробирке для микроволновой обработки Соединение 1-3 (250 мг, 961,86 мкмоль), Соединение 1-4 (149 мг, 1,44 ммоль), карбонат калия (332 мг, 2,40 ммоль), хлорид одновалентной меди (19 мг, 192,37 мкмоль) и 2-ацетилциклогексанон (27 мг, 192,37 мкмоль) растворяли в смешанном растворе DMF (5 мл) и воды (0,5 мл), и полученную смесь выдерживали при 130°С для реакции под воздействием микроволнового излучения в течение 1,5 ч. Реакционную смесь охлаждали и затем фильтровали. К фильтрату добавляли 12 мл воды, и полученную смесь затем экстрагировали этилацетатом (20 мл × 3), и водную фазу концентрировали при пониженном давлении с получением Соединения 1-5. ЖХ/МС (ЭРИ) m/z: 261 (М+1).
4) Синтез Соединения 53-6

Дихлорсульфоксид (500 мкл) добавляли по каплям в раствор Соединения 1-5 (300 мг, 1,15 ммоль) в метаноле (4 мл) в ледяной бане. Затем полученную смесь нагревали до 26°С и перемешивали при этой температуре в течение 6 ч. Реакционную смесь концентрировали при пониженном давлении, и к остатку добавляли 10 мл воды. Полученную смесь экстрагировали этилацетатом (15 мл × 3), и водную фазу доводили до рН=10 насыщенным раствором бикарбоната натрия. Выпавшее в осадок коричневое твердое вещество отфильтровывали. Осадок на фильтре собирали с получением Соединения 1-6. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.22 (s, 1Н), 7.80-7.78 (dd, J=4A, 8.8 Гц, 2Н), 6.78-6.76 (d, J=9.2 Гц, 1Н), 6.71(s, 1Н), 6.25 (s, 1Н), 3.59 (s, 3Н), 2.91 (s, 3Н), 1.49 (s, 6Н).
5) Синтез Соединения 1

Соединение 1-6 (150 мг, 546,81 мкмоль) и Соединение 1-7 (250 мг, 1,09 ммоль) растворяли в смешанном растворе DMF (500 мкл) и метилбензола (2 мл), и полученную смесь нагревали до 90°С и перемешивали при этой температуре в атмосфере азота в течение 48 ч. 3 мл метанола добавляли по каплям в реакционную смесь, и полученную смесь перемешивали при комнатной температуре в течение 0,5 ч. Затем реакционную смесь сушили в центробежной сушилке при пониженном давлении. Полученное твердое вещество растворяли в 15 мл этилацетата, затем промывали 15 мл воды и 30 мл насыщенного рассола в указанном порядке, сушили над безводным сульфатом натрия, фильтровали и сушили в центробежной сушилке. Неочищенный продукт очищали с использованием пластины для препаративной ТСХ и методом препаративной ВЭЖХ с получением Соединения 1. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.76 (s, 1Н), 8.02 (d, J=8 Гц, 2Н), 7.83-7.91 (m, 3Н), 7.44 (dd, J=2, 2.2 Гц, 1Н), 5.89 (d, J=4.4 Гц, 1Н), 3.27 (d, J=4.8 Гц, 3Н), 1.70 (s, 6Н); ЖХ/МС (ЭРИ) m/z: 471 (М+1).
Пример 54. Синтез Соединения 54

1) Синтез Соединения 2-2

Дихлорсульфоксид (1,96 г, 16,44 ммоль) добавляли по каплям в раствор Соединения 2-1 (850 мг, 5,48 ммоль) в метаноле (10 мл) при 0°С. После завершения капельного добавления полученную смесь перемешивали при 25°С в течение 12 ч. Реакционную смесь концентрировали, и полученный остаток подщелачивали насыщенным раствором бикарбоната натрия (30 мл) и экстрагировали дихлорметаном (20 мл × 2). Объединенную органическую фазу промывали водой (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением Соединения 2-2. ЖХ/МС (ЭРИ) m/z: 170 (М+1).
2) Синтез Соединения 2-4

Соединение 2-2 (300 мг, 1,77 ммоль), Соединение 2-3 (637 мг, 2,66 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (103 мг, 177,36 мкмоль), бис(дибензилиденацетон)палладий (102 мг, 177,36 мкмоль) и карбонат цезия (1,16 г, 3,55 ммоль) добавляли в пробирку для микроволновой обработки, заполненную метилбензолом (5 мл). Затем полученную смесь выдерживали при 120°С для реакции под воздействием микроволнового излучения в течение 2 ч. Реакционную смесь разбавляли дихлорметаном (5 мл) и фильтровали. Фильтрат концентрировали. Полученный неочищенный продукт очищали колоночной хроматографией с получением Соединения 2-4. ЖХ/МС (ЭРИ) m/z: 281 (М+1).
3) Синтез Соединения 2-6

Соединение 2-4 (200 мг, 712,56 мкмоль), Соединение 2-5 (159 мг, 855,07 мкмоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (41 мг, 71,26 мкмоль), карбонат цезия (464 мг, 1,43 ммоль) и бис(дибензилиденацетон)палладий (41 мг, 71,26 мкмоль) добавляли в пробирку для микроволновой обработки, заполненную метилбензолом (5 мл). Затем полученную смесь выдерживали при 120°С для реакции под воздействием микроволнового излучения в течение 2 ч. Реакционную смесь разбавляли дихлорметаном (20 мл) и фильтровали. Фильтрат концентрировали. Полученный неочищенный продукт очищали с использованием пластины для препаративной тонкослойной хроматографии с получением Соединения 2-6. ЖХ/МС (ЭРИ) m/z: 431 (М+1).
4) Синтез Соединения 2-7
Тиофосген (107 мг, 929,48 мкмоль) добавляли по каплям в раствор Соединения 2-6 (200 мг, 464,74 мкмоль) и трет-бутоксида натрия (223 мг, 2.32 ммоль) в тетрагидрофуране (2 мл) при 0°С. После завершения капельного добавления полученную смесь перемешивали при 60°С в течение 12 ч. Реакционную смесь охлаждали до 20°С и затем добавляли воду (10 мл), чтобы погасить реакцию. Полученную смесь разбавляли дихлорметаном (10 мл) и экстрагировали дихлорметаном (10 мл × 2). Объединенную органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный неочищенный продукт очищали с использованием пластины для препаративной тонкослойной хроматографии с получением Соединения 2-7. ЖХ/МС (ЭРИ) m/z: 473 (М+1).
5) Синтез Соединения 56-8

Водный раствор гидроксида лития (1М, 1 мл) добавляли по каплям в раствор Соединения 2-7 (40 мг, 84,67 мкмоль) в тетрагидрофуране (4 мл) при комнатной температуре (20°С). После завершения капельного добавления полученную смесь нагревали до 80°С и перемешивали в течение 2 ч. Реакционную смесь подкисляли до рН=5-6 разбавленной 1М соляной кислотой и экстрагировали дихлорметаном (10 мл × 3). Объединенную органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением Соединения 2-8. ЖХ/МС (ЭРИ) m/z: 459 (М+1).
6) Синтез Соединения 2

При комнатной температуре (20°С), метиламина гидрохлорид (5 мг, 78,54 мкмоль), триэтиламин (20 мг, 196,35 мкмоль) и O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (30 мг, 78,54 мкмоль) добавляли в раствор Соединения 2-8 (30 мг, 65,45 мкмоль) в дихлорметане (2 мл). Полученную смесь перемешивали при 20°С в течение 1 ч. В реакционную смесь добавляли воду (10 мл), и затем полученную смесь разбавляли дихлорметаном (10 мл) и экстрагировали дихлорметаном (10 мл × 2). Объединенную органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный неочищенный продукт очищали с использованием пластины для препаративной ТСХ и методом препаративной ВЭЖХ с получением Соединения 2. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.40-8.38 (m, 1Н), 8.27-8.24 (m, 2Н), 8.18-8.16 (m, 1Н), 8.10-8.09 (m, 1Н), 7.53-7.49 (m, 2Н), 7.38-7.36 (m, 1Н), 7.28-7.27 (m, 1Н), 3.11 (d, J=4.8 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 472 (М+1).
Пример 57. Синтез Соединения 57

1) Синтез Соединения 3-2

Порошок цинка (3,98 г, 60,84 ммоль) и хлорид аммония (3,25 г, 60,84 ммоль) добавляли в раствор Соединения 3-1 (2,10 г, 12,17 ммоль) в метаноле (20 мл) и дихлорметане (10 мл). Полученную реакционную смесь перемешивали при 15°С в течение 24 ч. Реакционную смесь фильтровали, и осадок на фильтре промывали дихлорметаном (40 мл). Полученный фильтрат концентрировали при пониженном давлении с получением Соединения 3-2. ЖХ/МС (ЭРИ) m/z: 143 (М+1).
2) Синтез Соединения 3-3

Дихлорсульфоксид (13,12 г, 110,31 ммоль) добавляли в раствор Соединения 3-9 (4,00 г, 18,26 ммоль) в безводном метаноле (40 мл) при 0°С. Полученную реакционную смесь перемешивали при 15°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении, и остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 3-3.
3) Синтез Соединения 3-4
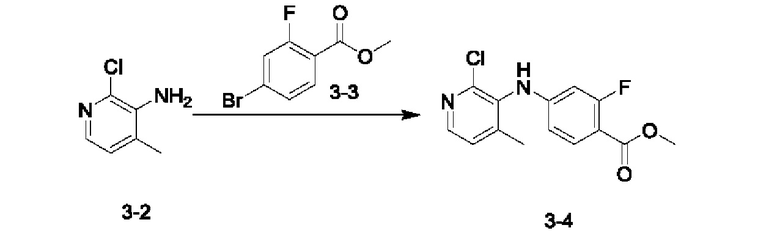
Смесь Соединения 3-3 (1,08 г, 4,63 ммоль), Соединения 3-2 (600 мг, 4,21 ммоль), бис(дибензилиденацетон)палладия (242 мг, 420,79 мкмоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантена (244 мг, 420,79 мкмоль), карбоната цезия (2,74 г, 8,42 ммоль) и метилбензола (15 мл) добавляли в пробирку для микроволновой обработки. Пробирку для микроволновой обработки герметично закрывали и затем нагревали до 130°С для реакции под воздействием микроволнового излучения в течение 2 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 3-4. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.23 (d, J=5.0 Гц, 1Н), 7.85 (t, J=8.4 Гц, 1Н), 7.22 (d, J=5.0 Гц, 1Н), 6.42 (dd, J=2.3, 8.5 Гц, 1Н), 6.21 (dd, J=2.3, 12.8 Гц, 1Н), 5.94 (s, 1Н), 3.90 (s, 3Н), 2.28 (s, 3Н); ЖХ/МС (ЭРИ) m/z: 295 (М+1).
4) Синтез Соединения 3-5

Соединение 3-4 (500 мг, 1,70 ммоль), Соединение 2-5 (348 мг, 1,87 ммоль), бис(дибензилиденацетон)палладий (98 мг, 170,00 мкмоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (98 мг, 170,00 мкмоль), карбонат цезия (1,11 г, 3,40 ммоль) и метилбензол (8 мл) добавляли в пробирку для микроволновой обработки. Пробирку для микроволновой обработки герметично закрывали и затем нагревали до 130°С для реакции под воздействием микроволнового излучения в течение 2 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 3-5. ЖХ/МС (ЭРИ) m/z: 445 (М+1).
5) Синтез Соединения 3-6

трет-Бутоксид калия (216 мг, 2,25 ммоль) и тиофосген (104 мг, 900,12 мкмоль) добавляли в раствор Соединения 3-5 (100 мг, 225,03 мкмоль) в тетрагидрофуране (2 мл) и метилбензоле (2 мл). Полученную реакционную смесь нагревали до 100°С и перемешивали в течение 16 ч. Этилацетат (20 мл) и воду (20 мл) добавляли в реакционную смесь для разделения жидких фаз. Органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ с получением Соединения 3-6. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.27 (d, J=1.5 Гц, 1Н), 8.21 (t, 3=7.9 Гц, 1Н), 8.17-8.10 (m, 2Н), 8.09-8.04 (m, 1Н), 7.43-7.32 (m, 2Н), 7.02 (d, J=5.0 Гц, 1Н), 4.01 (s, 3Н), 1.98 (s, 3Н); ЖХ/МС (ЭРИ) m/z: 487 (М+1)
6) Синтез Соединения 3-7

Гидроксид лития (9 мг, 205,58 мкмоль) добавляли в раствор Соединения 3-6 (50 мг, 102,79 мкмоль) в тетрагидрофуране (1,2 мл) и воде (0,3 мл). Полученную реакционную смесь перемешивали при 5°С в течение 16 ч. 1М раствор соляной кислоты (10 мл) и этилацетат (20 мл) добавляли в реакционную смесь для разделения жидких фаз. Органическую фазу промывали насыщенным рассолом (15 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением целевого Соединения 3-7. ЖХ/МС (ЭРИ) m/z: 473 (М+1).
7) Синтез Соединения 3

О-(7-Азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (39 мг, 101,60 мкмоль), триэтиламин (26 мг, 254,01 мкмоль) и метиламина гидрохлорид (9 мг, 127,01 мкмоль) добавляли в раствор Соединения 3-7 (40 мг, 84,67 мкмоль) в дихлорметане (1 мл). Полученную реакционную смесь перемешивали при 5°С в течение 16 ч. Дихлорметан (15 мл) и воду (10 мл) добавляли в реакционную смесь для разделения жидких фаз. Органическую фазу промывали насыщенным рассолом (10 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали методом препаративной ВЭЖХ с получением Соединения 3. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.37 (t, J=8.3 Гц, 1Н), 8.27 (s, 1Н), 8.18-8.10 (m, 2Н), 8.10-8.04 (m, 1Н), 7.45-7.33 (m, 2Н), 7.01 (d, J=5.0 Гц, 1Н), 6.76 (br s, 1Н), 3.10 (d, J=4.5 Гц, 3Н), 1.97 (s, 3Н); ЖХ/МС (3PH)m/z: 486 (М+1).
Пример 4. Синтез Соединения 4

1) Синтез Соединения 4-2

Триэтиламин (1,34 г, 13,22 ммоль) и гидроксиламина гидрохлорид (918 мг, 13,22 ммоль) добавляли в раствор Соединения 4-1 (2,00 г, 8,81 ммоль) в метаноле (30 мл). Полученную реакционную смесь перемешивали при 10°С в течение 16 ч, нагревали до 30°С и перемешивали в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении и затем добавляли этилацетат (50 мл) и воду (40 мл). Органическую фазу промывали насыщенным рассолом (40 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением Соединения 4-2. ЖХ/МС (ЭРИ) m/z: 242 (М+1).
2) Синтез Соединения 4-3

Смесь Соединения 4-2 (2,00 г, 8,26 ммоль) и полифосфорной кислоты (20 мл) нагревали до 95°С и перемешивали в течение 3 ч. 150 мл воды добавляли в реакционную смесь, и затем полученную смесь перемешивали в течение 30 мин и экстрагировали этилацетатом (100 мл × 3). Органическую фазу промывали насыщенным рассолом (150 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали препаративной ТСХ с получением Соединения 4-3. ЖХ/МС (ЭРИ) m/z: 242 (М+1).
3) Синтез Соединения 4-4

60% гидрид натрия (50 мг, 1,25 ммоль) добавляли в раствор Соединения 4-3 (200 мг, 826,21 мкмоль) в DMF (4 мл), и полученную смесь перемешивали при 10°С в течение 10 мин. Затем добавляли йодэтан (155 мг, 993,78 мкмоль). Полученную реакционную смесь перемешивали при 10°С в течение 30 мин. Реакционную смесь медленно вливали в воду (30 мл) и экстрагировали этилацетатом (20 мл × 3). Органическую фазу промывали насыщенным рассолом (40 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали методом препаративной ТСХ с получением Соединения 4-4. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.70 (d, J=8.3 Гц, 1Н), 7.26 (dd, J=1.8, 8.3 Гц, 1Н), 7.16 (d, J=1.8 Гц, 1Н), 4.38 (t, J=5.0 Гц, 2Н), 3.64 (q, J=7.3 Гц, 2Н), 3.50 (t, J=5.0 Гц, 2Н), 1.23 (t, J=7.2 Гц, 3Н).
4) Синтез Соединения 4-5

Мутную жидкость Соединения 4-4 (100 мг, 370,21 мкмоль), Соединения 1-4 (57 мг, 555,32 мкмоль), хлорида одновалентной меди (7 мг, 74,04 мкмоль), 2-ацетилциклогексанона (10 мг, 74,04 мкмоль) и карбоната калия (128 мг, 925,53 мкмоль) в DMF (1,5 мл) и воде (0,08 мл) добавляли в пробирку для микроволновой обработки и выдерживали при 130°С для реакции под воздействием микроволнового излучения в течение 1 ч. Реакционную смесь фильтровали через целит, промывали этилацетатом (5 мл) и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, растворяли в воде (10 мл) и экстрагировали этилацетатом (5 мл). Концентрированную соляную кислоту (0,5 мл) добавляли в водную фазу, и полученный мутный водный раствор концентрировали при пониженном давлении. Остаток, полученный после концентрирования, суспендировали в смеси дихлорметан/метанол (10/1, 20 мл) и фильтровали. Фильтрат концентрировали при пониженном давлении с получением Соединения 4-5. ЖХ/МС (ЭРИ) m/z: 293 (М+1).
5) Синтез Соединения 4-6

Дихлорсульфоксид (407 мг, 3,42 ммоль) осторожно добавляли по каплям (экзотермия) в мутную жидкость Соединения 4-5 (100 мг, 342,08 мкмоль) в метаноле (4 мл). Полученный желтый прозрачный раствор перемешивали при 40°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ с получением Соединения 4-6. ЖХ/МС (ЭРИ) m/z: 307 (М+1).
6) Синтез Соединения 4

Соединение 1-7 (186 мг, 816,05 мкмоль) добавляли в раствор Соединения 4-6 (50 мг, 163,21 мкмоль) в метилбензоле (1 мл) и DMF (0,02 мл). Полученную реакционную смесь нагревали до 100°С и перемешивали в течение 24 ч. Метанол (2 мл) добавляли в реакционную смесь, и полученную смесь перемешивали в течение 5 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ и затем очищали методом препаративной ВЭЖХ с получением Соединения 4. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.98 (d, J=8.3 Гц, 1Н), 7.95-7.84 (m, 2Н), 7.77 (dd, J=1.9, 8.2 Гц, 1Н), 7.01 (dd, J=2.0, 8.5 Гц, 1Н), 6.89 (d, J=2.0 Гц, 1Н), 4.40 (t, J=4.8 Гц, 2Н), 3.62 (q, J=7.2 Гц, 2Н), 3.56 (t, J=4.9 Гц, 2Н), 1.52 (s, 6Н), 1.20 (t, J=7.2 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 503 (М+1).
Пример 5. Синтез Соединения 5

64) Синтез Соединения 5-64

Карбонат цезия (869 мг, 2,66 ммоль) и 1,1,1-трифтор-2-йодэтан (933 мг, 4,44 ммоль) добавляли в раствор Соединения 1-1 (500 мг, 2,22 ммоль) в DMF (10 мл). Полученную реакционную смесь перемешивали при 100°С в течение 16 ч. Реакционную смесь охлаждали и затем фильтровали для удаления карбоната цезия. Фильтрат вливали в воду (40 мл) и экстрагировали этилацетатом (30 мл × 3). Органическую фазу промывали насыщенным рассолом (50 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, суспендировали в смеси петролейный эфир/этил ацетат (10 мл, 10/1) и фильтровали с получением Соединения 5-1. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.11 (d, J=8.5 Гц, 1Н), 7.96 (s, 1Н), 7.85 (d, J=1.8 Гц, 1Н), 7.60 (dd, J=1.8, 8.5 Гц, 1Н), 4.59 (q, J=8,4 Гц, 2Н).
2) Синтез Соединения 5-2

Мутную жидкость Соединения 5-1 (300 мг, 976,98 мкмоль), Соединения 1-4 (151 мг, 1,47 ммоль), хлорида одновалентной меди (19 мг, 195,40 мкмоль), 2-ацетилциклогексанона (27 мг, 195,40 мкмоль) и карбоната калия (338 мг, 2,44 ммоль) в DMF (3 мл) и воде (0,15 мл) добавляли в пробирку для микроволновой обработки и выдерживали при 130°С для реакции под воздействием микроволнового излучения в течение 1 ч. Реакционную смесь фильтровали через целит, промывали этилацетатом (10 мл) и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, растворяли в воде (30 мл) и экстрагировали этилацетатом (5 мл). Концентрированную соляную кислоту (1 мл) добавляли в водную фазу, и полученный мутный водный раствор концентрировали при пониженном давлении. Остаток, полученный после концентрирования, суспендировали в смеси дихлорметан/метанол (10/1, 20 мл) и фильтровали. Фильтрат концентрировали при пониженном давлении с получением Соединения 5-2. ЖХ/МС (ЭРИ) m/z: 330 (М+1).
3) Синтез Соединения 5-3

Дихлорсульфоксид (1,15 г, 9,65 ммоль, 0,7 мл) осторожно добавляли по каплям (экзотермия) в мутную жидкость Соединения 5-2 (500 мг, 945,42 мкмоль) в метаноле (5 мл). Полученный желтый прозрачный раствор перемешивали при 40°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ с получением Соединения 5-3. ЖХ/МС (ЭРИ) m/z: 344 (М+1).
4) Синтез Соединения 5

Соединение 1-7 (166 мг, 728,25 мкмоль) добавляли в раствор Соединения 5-3 (50 мг, 145,65 мкмоль) в метилбензоле (1 мл) и DMF (0,05 мл). Полученную реакционную смесь нагревали до 120°С и перемешивали в течение 48 ч. В реакционную смесь добавляли метанол (2 мл), и полученную смесь перемешивали в течение 5 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали методом препаративной ВЭЖХ с получением Соединения 5. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.42 (d, J=8.5 Гц, 1Н), 8.03 (s, 1Н), 7.95-7.89 (m, 2Н), 7.79 (dd, J=2.0, 8.3 Гц, 1Н), 7.65 (d, J=1.8 Гц, 1Н), 7.43 (dd, J=2.0, 8.5 Гц, 1Н), 4.64 (q, J=8.3 Гц, 2Н), 1.58 (s, 6Н); ЖХ/МС (ЭРИ) m/z: 540 (М+1).
Пример 6. Синтез Соединения 6

1) Синтез Соединения 6-2

1,1,1-Трифтор-2-йодэтан (933 мг, 4,44 ммоль) добавляли в раствор Соединения 6-1 (500 мг, 2,22 ммоль) и карбоната цезия (869 мг, 2,66 ммоль) в DMF (8 мл). Реакционную смесь перемешивали при 100°С в течение 16 ч. Реакционную смесь охлаждали и затем фильтровали для удаления карбоната цезия. Фильтрат вливали в воду (10 мл) и экстрагировали этилацетатом (10 мл × 3). Органическую фазу промывали насыщенным рассолом (15 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, суспендировали в смеси петролейный эфир/этилацетат (10 мл, 10/1) и фильтровали с получением Соединения 6-2. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.32 (d, J=8.3 Гц, 1Н), 8.14 (s, 1Н), 7.95-7.89 (m, 2Н), 4.87 (q, J=8.3 Гц, 2Н).
2) Синтез Соединения 6-3

Мутную жидкость Соединения 6-2 (300 мг, 976,98 мкмоль), Соединения 1-4 (151 мг, 1,47 ммоль), хлорида одновалентной меди (19 мг, 195,40 мкмоль), 2-ацетилциклогексанона (27 мг, 195,40 мкмоль) и карбоната калия (338 мг, 2,44 ммоль) в DMF (3 мл) и воде (0,15 мл) добавляли в пробирку для микроволновой обработки и выдерживали при 130°С для реакции под воздействием микроволнового излучения в течение 1 ч. Реакционную смесь фильтровали через целит, промывали этилацетатом (10 мл) и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, растворяли в воде (10 мл) и экстрагировали этилацетатом (5 мл). Концентрированную соляную кислоту (0,5 мл) добавляли в водную фазу, и полученный мутный водный раствор концентрировали при пониженном давлении. Остаток, полученный после концентрирования, суспендировали в смеси дихлорметан/метанол (10/1, 40 мл) и фильтровали. Фильтрат концентрировали при пониженном давлении с получением Соединения 6-3. ЖХ/МС (ЭРИ) m/z: 330 (М+1).
66) Синтез Соединения 6-4

Дихлорсульфоксид (1,31 г, 11,03 ммоль, 0,8 мл) осторожно добавляли по каплям (экзотермия) в мутную жидкость Соединения 6-3 (500 мг, 1,17 ммоль) в метаноле (5 мл). Полученный желтый прозрачный раствор перемешивали при 40°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ с получением Соединения 6-4. ЖХ/МС (ЭРИ) m/z: 344 (М+1).
4) Синтез Соединения 6

Соединение 1-7 (299 мг, 1,31 ммоль) добавляли в раствор Соединения 6-4 (90 мг, 262,16 мкмоль) в метилбензоле (2 мл) и DMF (0,1 мл). Полученную реакционную смесь нагревали до 120°С и перемешивали в течение 48 ч. В реакционную смесь добавляли метанол (2 мл), и полученную смесь перемешивали в течение 5 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ и затем очищали методом препаративной ВЭЖХ с получением Соединения 6. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.56 (d, J=8.3 Гц, 1Н), 8.17 (s, 1Н), 7.94 (d, J=8.3 Гц, 1Н), 7.90 (s, 1Н), 7.77 (dd, J=1.8, 8.3 Гц, 1Н), 7.69-7.62 (m, 2Н), 4.82 (q, J=8.5 Гц, 2Н), 1.59 (s, 6Н); ЖХ/МС (ЭРИ) m/z: 540 (М+1).
Пример 7. Синтез Соединения 7 и Соединения 8

1) Синтез Соединения 67-2

В пробирке для микроволновой обработки Соединение 7-1 (1,00 г, 5,08 ммоль), Соединение 1-4 (785 мг, 7,61 ммоль), 2-ацетилциклогексанон (142 мг, 1,02 ммоль), хлорид одновалентной меди (100 мг, 1,02 ммоль) и карбонат калия (1,75 г, 12,69 ммоль) растворяли в DMF (5 мл) и воде (90 мкл) при 15°С. Затем полученную смесь нагревали под воздействием микроволнового излучения до 130°С и перемешивали при этой температуре в течение 1,2 ч. Реакционную смесь напрямую фильтровали и сушили в центробежной сушилке. Добавляли 15 мл воды, и затем полученную смесь экстрагировали этилацетатом (15 мл × 2). Затем водную фазу концентрировали с получением Соединения 7-2. ЖХ/МС (ЭРИ) m/z: 220 (М+1).
2) Синтез Соединения 7-3

Дихлорсульфоксид (5,84 г, 49,08 ммоль) добавляли по каплям в раствор Соединения 7-2 (800 мг, 3,65 ммоль) в метаноле (10 мл) в бане лед-вода. После завершения капельного добавления полученную смесь перемешивали при 50°С в течение 12 ч. Реакционную смесь напрямую концентрировали с получением Соединения 7-3. ЖХ/МС (ЭРИ) m/z: 234 (М+1).
3) Синтез Соединения 7-4
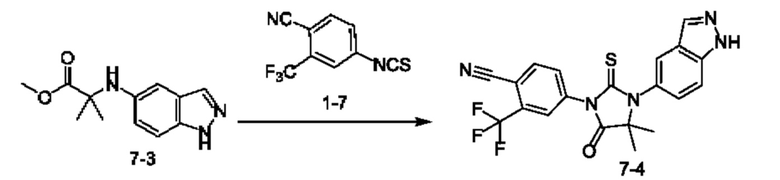
Соединение 7-3 (400 мг, 1,71 ммоль) и Соединение 1-7 (1,17 г, 5,14 ммоль) растворяли в DMF (1,5 мл) и метилбензоле (6 мл) при 15°С. После трехразового вытеснения воздуха азотом смесь нагревали до 100°С и перемешивали в течение 12 ч под защитой азота. В реакционную смесь добавляли 5 мл метанола, и затем полученную смесь перемешивали в течение 20 мин и концентрировали при пониженном давлении. Неочищенный продукт очищали методом колоночной флэш-хроматографии с получением Соединения 7-4. ЖХ/МС (ЭРИ) m/z: 430 (М+1).
69) Синтез Соединения 7 и Соединения 8

Соединение 7-4 (100 мг, 233 мкмоль), Соединение 7-5 (24 мг, 279,44 мкмоль) и карбонат цезия (114 мг, 349,30 мкмоль) вводили в N,N-диметилацетамид (5 мл) при 15°С, и полученную смесь нагревали до 120°С и перемешивали в течение 2,5 ч. Реакционную смесь фильтровали. В фильтрат добавляли 10 мл воды, и полученную смесь затем экстрагировали этилацетатом (10 мл × 2). Органическую фазу промывали 20 мл насыщенного рассола, сушили над безводным сульфатом натрия, фильтровали и сушили в центробежной сушилке. Неочищенный продукт очищали с использованием пластины для препаративной тонкослойной хроматографии и методом препаративной ВЭЖХ с получением Соединения 7 и Соединения 8.
1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.14 (s, 1Н), 8.04-7.99 (m, 2Н), 7.89 (br d, J=8.28 Гц, 1Н), 7.74-7.69 (m, 2Н), 7.34 (br d, J=8.78 Гц, 1Н), 5.14 (br s, 1Н), 4.81 (br s, 1 H), 4.55-4.49 (m, 1H), 4.40-4.29 (m, 2 H), 3.97 (br d, J=7.03 Гц, 1H), 2.15 (br d, J=5.27 Гц, 1H), 1.65 (s, 6 H); ЖХ/МС (ЭРИ) m/z: 516 (М+1) (Соединение 7).
1H ЯМР (400 МГц, CDCl3) δ м.д. 8.17 (s, 1H), 7.97-8.02 (m, 2H), 7.89-7.83 (m, 2H) 7.64 (s, 1 H), 7.16 (dd, J=9.03, 2.01 Гц, 1 H), 5.09 (br s, 1 H), 4.75 (br s, 1 H), 4.48 (dd, J=10.16, 6.15 Гц, 1 H), 4.39-4.31 (m, 2 H), 4.00-3.86 (m, 1 H), 2.51 (br s, 1 H), 1.63 (s, 6 H); ЖХ/МС (ЭРИ) m/z: 516 (М+1) (Соединение 8).
Пример 8. Синтез Соединения 9

1) Синтез Соединения 9-2

Смесь Соединения 9-1 (2,00 г, 9,26 ммоль), триэтилортоацетата (3,00 г, 25,00 ммоль) и аминометанола (20 мл) (15% масс.) добавляли в непроницаемый для воздуха сосуд и перемешивали при 110°С в течение 4 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, суспендировали в метаноле (30 мл) и фильтровали с получением белого твердого вещества. Фильтрат концентрировали при пониженном давлении, ресуспендировали в метаноле (15 мл) и фильтровали с получением белого твердого вещества. Две партии белого твердого вещества объединяли с получением Соединения 9-2. 1Н ЯМР (400 МГц, DMSO-d6) δ м.д. 12.38 (br s, 1Н), 8.14 (d, J=2.3 Гц, 1Н), 7.91 (dd, J=2.4, 8,7 Гц, 1Н), 7.53 (d, J=8.8 Гц, 1Н), 2.34 (s, 3Н).
2) Синтез Соединения 9-3

Соединение 9-2 (600 мг, 2,51 ммоль), Соединение 1-4 (388 мг, 3,76 ммоль), карбонат калия (867 мг, 6,28 ммоль), хлорид одновалентной меди (50 мг, 502,00 мкмоль), 2-ацетилциклогексанон (70 мг, 502,00 мкмоль), DMF (7 мл) и воду (350 мкл) добавляли в пробирку для микроволновой обработки на 20 мл. Полученную смесь выдерживали при 130°С для реакции под воздействием микроволнового излучения в течение 1 ч. Реакционную смесь фильтровали, и фильтрат концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, растворяли в воде (50 мл) и промывали дихлорметаном (30 мл × 3). Концентрированную соляную кислоту (1,5 мл) добавляли в водную фазу, так чтобы водная фаза стала кислотной (рН примерно 6), и затем водную фазу концентрировали при пониженном давлении. Остаток, полученный после концентрирования, суспендировали в смеси дихлорметан/метанол (30 мл/30 мл) при 29°С в течение 2 мин, фильтровали и концентрировали при пониженном давлении с получением Соединения 9-3. ЖХ/МС (ЭРИ) m/z: 262 (М+1).
3) Синтез Соединения 9-4

Соединение 9-3 (1,18 г, 2,58 ммоль) растворяли в безводном метаноле (20 мл) и по каплям добавляли дихлорсульфоксид (3,28 г, 27,58 ммоль, 2,00 мл) при 0°С.Полученную смесь нагревали до 50°С и перемешивали в течение 18 ч. Реакционную смесь концентрировали при пониженном давлении, добавляли насыщенный раствор бикарбоната натрия (50 мл), и полученную смесь экстрагировали дихлорметаном (30 мл × 3). Органические фазы объединяли, промывали насыщенным рассолом (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, суспендировали в смеси дихлорметан/этилацетат (10 мл/10 мл) при 29°С в течение 0,5 ч и фильтровали. Полученный осадок на фильтре представлял собой Соединение 9-4. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 10.94 (s, 1Н), 7.52-7.50 (d, J=9.2 Гц, 1Н), 7.30-7.29 (d, J=2.8 Гц, 1Н), 7.06-7.04(m, 1Н), 4.45 (s, 1Н), 3.76 (s, 3Н), 2.52 (s, 3Н), 1.65 (s, 6Н); ЖХ/МС (ЭРИ) m/z: 276 (М+1).
4) Синтез Соединения 9-5

Соединение 9-4 (300 мг, 933,89 мкмоль) и Соединение 1-7 (864 мг, 3,79 ммоль) растворяли в DMF (1,5 мл) и метилбензоле (6 мл). Полученную смесь нагревали до 120°С и перемешивали в течение 18 ч. Реакционную смесь концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной тонкослойной хроматографии с получением Соединения 9-5. ЖХ/МС (ЭРИ) m/z: 472 (М+1).
5) Синтез Соединения 9

Мутную жидкость Соединения 9-5 (130 мг, 275,74 мкмоль), 2-бромэтанола (83 мг, 661,78 мкмоль) и карбоната калия (100 мг, 722,44 мкмоль) в DMF (3 мл) перемешивали при 29°С в течение 67 ч. Реакционную смесь фильтровали напрямую. Фильтрат отделяли и очищали методом препаративной ВЭЖХ с получением Соединения 9. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.04 (d, J=2.4 Гц, 1Н), 7.96-7.92 (m, 3Н), 7.91-7.80 (dd, 3=45.6 Гц, 1Н), 7.65-7.64 (dd, J=2.8 Гц, 1Н), 4.721-4.71 (m, 2Н), 4.70 (s, 2Н), 2.69 (s, 3Н), 1.59 (s, 6Н); ЖХ/МС (ЭРИ) m/z: 516 (М+1).
Пример 9. Синтез Соединения 10

1) Синтез Соединения 10-2

Соединение 10-1 (20,00 г, 128,92 ммоль) растворяли в дихлорметане (200 мл) и добавляли NBS (22,95 г, 128,92 ммоль). Полученную смесь перемешивали при 20°С в течение 2 ч. Реакционную смесь фильтровали, и осадок на фильтре промывали дихлорметаном (75 мл × 3). Полученный осадок на фильтре сушили при пониженном давлении с получением Соединения 10-2. 1Н ЯМР (400 МГц, DMSO-d6) δ м.д. 7.66-7.61 (m, 1Н), 7.52 (dd, J=2.3, 10.8 Гц, 1Н); ЖХ/МС (ЭРИ) m/z: 234 (М+1).
2) Синтез Соединения 10-3

Аммиачную воду (51,52 г, 396,90 ммоль) (чистота: 27%) добавляли в раствор Соединения 10-2 (30,96 г, 132,30 ммоль), O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата (70,42 г, 185,22 ммоль) и триэтиламина (26,77 г, 264,60 ммоль) в DMF (500 мл). Полученную смесь перемешивали при 20°С в течение 4 ч. 2000 мл воды добавляли в реакционную смесь, и полученную смесь перемешивали в течение 1 ч и фильтровали. Осадок на фильтре промывали водой (50 мл × 3), и полученное белое твердое вещество сушили в инфракрасном шкафу. Фильтрат экстрагировали дихлорметаном (100 мл × 3). Полученную органическую фазу концентрировали при пониженном давлении. Остаток, полученный после концентрирования, суспендировали в воде (500 мл) при 20°С в течение 20 мин и фильтровали. Осадок на фильтре сушили в инфракрасном шкафу. Два высушенных белых твердых вещества объединяли с получением Соединения 10-3. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.35 (br d, J=7,0 Гц, 1H), 7.18-7.08 (m, 1H); ЖХ/МС (ЭРИ) m/z: 235 (М+3).
3) Синтез Соединения 10-4

Пропионилхлорид (28,39 г, 306,80 ммоль) добавляли в мутную жидкость Соединения 10-3 (14,30 г, 61,36 ммоль) в трихлорметане (200 мл). Полученную смесь перемешивали при 70°С в течение 16 ч. Реакционную смесь охлаждали до 15°С, добавляли метанол (5 мл), и затем полученную смесь концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, растворяли в насыщенном растворе бикарбоната натрия (100 мл) и экстрагировали смесью дихлорметан/метанол (10/1, 200 мл × 3). Органические фазы объединяли, промывали насыщенным рассолом (100 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, суспендировали в этилацетате (100 мл) при 15°С в течение 30 мин и фильтровали. Осадок на фильтре промывали этилацетатом (10 мл × 3) и затем сушили в инфракрасном шкафу с получением Соединения 10-4. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.00 (br d, J=1,3 Гц, 1H), 7.57-7.48 (m, 1H), 2.95 (s, 1H), 2.62 (q, J=7.5 Гц, 2H), 1.33-1.22 (m, 3H); ЖХ/МС (ЭРИ) m/z: 273 (М+3).
4) Синтез Соединения 10-5

Соединение 10-4 (1,50 г, 5,53 ммоль), Соединение 1-4 (855 мг, 8,30 ммоль), хлорид одновалентной меди (220 мг, 2,22 ммоль), 2-ацетилциклогексанон (310 мг, 2,22 ммоль), карбонат калия (1,91 г, 13,83 ммоль), DMF (10 мл) и воду (500 мкл) добавляли в пробирку для микроволновой обработки вместимостью 30 мл в указанном порядке. Полученную смесь выдерживали при 130°С для реакции под воздействием микроволнового излучения в течение 80 мин. Реакционную смесь фильтровали, и осадок на фильтре промывали DMF (10 мл × 3). Полученную водную фазу подкисляли до рН примерно 6 разбавленной соляной кислотой (2 моль/л). Водную фазу концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, суспендировали в смеси дихлорметан/метанол (10/1, 30 мл) при 15°С в течение 2 мин и фильтровали. Фильтрат концентрировали при пониженном давлении с получением Соединения 10-5. ЖХ/МС (ЭРИ) m/z: 294 (М+1).
5) Синтез Соединения 10-6

Дихлорсульфоксид (29,94 г, 251,64 ммоль) добавляли по каплям к раствору Соединения 10-5 (7,30 г, 24,89 ммоль) в метаноле (80 мл) при 0°С. После завершения капельного добавления полученную смесь нагревали до 50°С и перемешивали в течение 18 ч. Реакционную смесь охлаждали до 15°С и концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, растворяли в насыщенном растворе бикарбоната натрия (40 мл) и экстрагировали смесью дихлорметан/метанол (10/1, 60 мл × 4). Органические фазы объединяли, промывали насыщенным рассолом (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 10-6. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.01 (d, J=2.5 Гц, 1Н), 6.72 (dd, J=2.5, 12.3 Гц, 1H), 3.68 (s, 3H), 2.72 (q, J=7.6 Гц, 2Н), 1.56 (s, 6Н), 1.36-1.31 (m, 3H); ЖХ/МС (ЭРИ) m/z: 308 (М+1).
6) Синтез Соединения 10-7

Под защитой азота Соединение 10-6 (2,10 г, 6,83 ммоль) и Соединение 1-7 (6,23 г, 27,32 ммоль) растворяли в DMF (5 мл) и метилбензоле (50 мл), и полученную смесь нагревали до 120°С и перемешивали в течение 18 ч. Реакционную смесь концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 10-7. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.99-7.93 (m, 2Н), 7.78 (dd, J=1.6, 8.2 Гц, 1H), 7.58 (d, J=8.3 Гц, 1H), 7.40 (dd, J=2.1, 9.9 Гц, 1H), 2.86-2.77 (m, 3H), 1.59 (s, 6Н), 1.44-1.33 (m, 3H); ЖХ/МС (ЭРИ) m/z: 504 (М+1).
7) Синтез Соединения 10

Под защитой азота мутную жидкость Соединения 10-7 (1,32 г, 2,62 ммоль), 2-бромэтанола (4,10 г, 32,75 ммоль), карбоната калия (1,45 г, 10,48 ммоль) и DMF (50 мл) перемешивали при 40°С в течение 78 ч. Реакционную смесь фильтровали напрямую. Фильтрат отделяли и очищали методом препаративной ВЭЖХ и препаративной хроматографией с получением Соединения 10. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.94 (d, J=8.0 Гц, 1H), 7.90 (d, J=1.5 Гц, 1H), 7.85 (s, 1H), 7.78 (dd, J=1.8, 8.3 Гц, 1H), 7.39 (dd, 3=2.1, 9.9 Гц, 1H), 4.76-4.71 (m, 2Н), 4.03 (br d, J=3.8 Гц, 2Н), 2.98 (q, J=7.5 Гц, 2Н), 2.72 (br s, 1H), 1.60 (s, 6Н), 1.35 (t, J=7.7 Гц, 3H); ЖХ/МС (ЭРИ) m/z: 548 (М+1).
Пример 10. Синтез Соединения 11

1) Синтез Соединения 11-2

Циклопропилформилхлорид (8,55 г, 81,84 ммоль) добавляли по каплям в раствор Соединения 11-1 (4,40 г, 20,46 ммоль) в трихлорметане (100 мл) при 20°С. Реакционную смесь нагревали до 65°С и подвергали реакции в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали с получением Соединения 11-2. ЖХ/МС (ЭРИ) m/z: 285 (М+3).
2) Синтез Соединения 11-3

Метоксид натрия (4,12 г, 76,28 ммоль) добавляли в раствор Соединения 11-2 (5,40 г, 19,07 ммоль) в метаноле (100 мл) при 20°С. Реакционную смесь подвергали реакции при 20°С в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали с получением неочищенного продукта. Неочищенный продукт добавляли в воду (100 мл) и нейтрализовали 1М водным раствором соляной кислоты до рН=7. Большое количество серого твердого вещества выпало в осадок. После фильтрования осадок на фильтре собирали и сушили с получением Соединения 11-3. 1H ЯМР (400 МГц, DMSO-d6) δ м.д. 8.09 (d, J=2.26 Гц, 1Н), 7.68 (d, J=2.26 Гц, 1H), 7.41 (d, J=8.78 Гц, 1H), 7.24 (dd, J=8.78, 2.26 Гц, 1H), 2.00-1.86 (m, 1H), 1.13-0.97 (m, 4H); ЖХ/МС (ЭРИ) m/z: 267 (M+3).
3) Синтез Соединения 11-4

DMF (414 мг, 5,66 ммоль) добавляли по каплям к раствору Соединения 11-3 (1,50 г, 5,66 ммоль) и дихлорсульфоксида (20 мл), и полученную смесь перемешивали при 80°С в течение 3 ч. Реакционную смесь концентрировали досуха при пониженном давлении, и остаток растворяли в этандиоле (20 мл). Добавляли триэтиламин (344 мг, 3,40 ммоль), и полученную смесь дополнительно перемешивали при 80°С в течение 1 ч. Реакционную смесь охлаждали до 20°С. В полученную смесь добавляли дихлорметан (80 мл), и смесь промывали водой (50 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 11-4. ЖХ/МС (ЭРИ) m/z: 309 (М+1).
4) Синтез Соединения 11-76

Триэтиламин (275 мг, 2,72 ммоль), 4-диметиламинопиридин (22 мг, 181,14 мкмоль) и diBoc (495 мг, 2,72 ммоль) добавляли по каплям в раствор Соединения 11-4 (280 мг, 905,68 мкмоль) в дихлорметане (10 мл). Полученную смесь перемешивали при 15°С в течение 17 ч. Реакционную смесь разбавляли дихлорметаном (20 мл). В полученную смесь добавляли воду (20 мл), затем добавляли по каплям разбавленную 2 моль/л соляную кислоту (5 капель), и полученную смесь промывали три раза. Органическую фазу промывали насыщенным раствором карбоната натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением Соединения 11-5. ЖХ/МС (ЭРИ) m/z: 411 (М+3).
5) Синтез Соединения 11-6

Соединение 11-5 (250 мг, 610,84 мкмоль), Соединение 1-4 (95 мг, 916,27 мкмоль), карбонат калия (338 мг, 2,44 ммоль), хлорид одновалентной меди (12 мг, 122,17 мкмоль), 2-ацетилциклогексанон (17 мг, 122,17 мкмоль), DMF (5 мл) и воду (0,1 мл) добавляли в пробирку для микроволновой обработки вместимостью 10 мл. Полученную смесь выдерживали при 130°С для реакции под воздействием микроволнового излучения в течение 1 ч. Реакционную смесь фильтровали, и осадок на фильтре промывали DMF (5 мл × 2). Фильтрат концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, суспендировали в дихлорметане (20 мл) при 15°С в течение 2 мин и фильтровали. Фильтрат концентрировали при пониженном давлении с получением Соединения 11-6. ЖХ/МС (ЭРИ) m/z: 432 (М+1).
6) Синтез Соединения 11-77

Раствор TMSCHN2 в н-гексане (2М, 1,74 мл, 3,48 ммоль) добавляли по каплям в раствор Соединения 11-6 (500 мг, 1,16 ммоль) в дихлорметане (5 мл) и метаноле (500 мкл). Полученную смесь перемешивали при 20°С в течение 1,5 ч. Реакционную смесь концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной тонкослойной хроматографии с получением Соединения 11-7. ЖХ/МС (ЭРИ) m/z: 446 (М+1).
7) Синтез Соединения 11-8

Соединение 11-7 (85 мг, 190,79 мкмоль) и Соединение 1-7 (131 мг, 572,38 мкмоль) растворяли в DMF (500 мкл) и метилбензоле (3 мл). Полученную смесь перемешивали при 120°С в течение 16 ч под защитой азота. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной тонкослойной хроматографии с получением Соединения 11-8. ЖХ/МС (ЭРИ) m/z: 642 (М+1).
8) Синтез Соединения 11

Трифторуксусную кислоту (2 мл) добавляли в раствор Соединения 11-8 (50 мг, 77,92 мкмоль) в дихлорметане (6 мл), и затем полученную смесь перемешивали при 15°С в течение 3 ч. Реакционную смесь разбавляли дихлорметаном (10 мл) и промывали насыщенным водным раствором бикарбоната натрия (20 мл × 3) и водой (20 мл) в указанном порядке. Объединенную органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали методом препаративной ВЭЖХ с получением Соединения 11. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.02-7.97 (m, 1H), 7.95-7.88 (m, 3H), 7.79 (dd, J=8.28, 1.76 Гц, 1H), 7.59 (dd, J=8.91, 2.38 Гц, 1Н), 4.66-4.62 (m, 2 Н), 4.00 (br s, 2Н), 2.52 (br s, 1H), 2.26-2.17 (m, 1Н),1.58 (s, 6 Н), 1.19-1.12(m, 2Н), 1.09-1.01 (m, 2Н); ЖХ/МС (ЭРИ) m/z: 542 (М+1).
Пример 11. Синтез Соединения 12
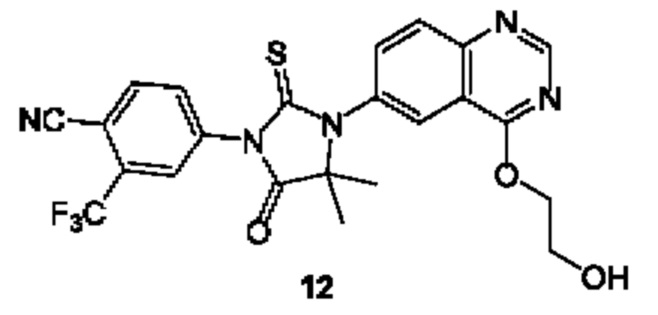
1) Синтез Соединения 12-1

пара-Метилбензолсульфоновую кислоту (218 мг, 1,40 ммоль) и Соединение 11-1 (3,00 г, 13,95 ммоль) добавляли к триметилортоформиату (29,10 г, 274,22 ммоль) при комнатной температуре (10°С). Реакционную смесь нагревали до 110°С и подвергали взаимодействию в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и напрямую концентрировали с получением Соединения 12-1. ЖХ/МС (ЭРИ) m/z: 225 (М+1).
2) Синтез Соединения 12-2

DMF (578 мг, 7,90 ммоль) добавляли в раствор Соединения 12-1 (2,00 г, 8,89 ммоль) в дихлорсульфоксиде (10 мл) при 20°С. Реакционную смесь нагревали до 80°С и подвергали реакции в течение 2 ч и затем концентрировали. Полученное желтое твердое вещество растворяли в этандиоле (10 мл), добавляли триэтиламин (4,00 г, 39,50 ммоль), и полученную смесь перемешивали при 80°С в течение 1 ч. Реакционную смесь разбавляли водой (50 мл) и экстрагировали дихлорметаном (50 мл × 3). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением Соединения 12-2. ЖХ/МС (ЭРИ) m/z: 269 (М+1).
3) Синтез Соединения 12-3

Триэтиламин (4,51 г, 44,58 ммоль) и diBoc (8,10 г, 37,15 ммоль) добавляли по каплям в раствор Соединения 12-2 (4,00 г, 14,86 ммоль) и 4-диметиламинопиридина (363 мг, 2,97 ммоль) в дихлорметане (30 мл). Полученную смесь перемешивали при 15°С в течение 3 ч. Реакционную смесь разбавляли дихлорметаном (20 мл). Полученную смесь промывали разбавленной соляной кислотой (30 мл × 4) (разбавленная соляная кислота была получена путем разбавления разбавленной 2 моль/л соляной кислоты (4 мл) водой (120 мл)). Органическую фазу промывали насыщенным раствором карбоната натрия (30 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 12-3. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.78 (s, 1H), 8.42-8.20 (m, 1Н), 7.98-7.75 (m, 2Н), 4.80 (br d, J=3.5 Гц, 2Н), 4.63-4.45 (m, 2Н), 1.50 (s, 9Н); ЖХ/МС (ЭРИ) m/z: 369 (М+1).
4) Синтез Соединения 12-4

Соединение 12-3 (1,00 г, 2,71 ммоль), Соединение 1-4 (419 мг, 4,07 ммоль), карбонат калия (562 мг, 4,07 ммоль), хлорид одновалентной меди (107 мг, 1,08 ммоль), 2-ацетилциклогексанон (152 мг, 1,08 ммоль), DMF (8 мл) и воду (800 мкл) добавляли в пробирку для микроволновой обработки на 30 мл. Полученную смесь выдерживали при 130°С для реакции под воздействием микроволнового излучения в течение 40 мин. Реакционную смесь фильтровали, и осадок на фильтре промывали DMF (3 мл × 3). Разбавленную соляную кислоту (2 моль/л) добавляли по каплям в фильтрат до доведения рН до примерно 7, и затем полученную смесь концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, суспендировали в смеси дихлорметан/метанол (10/1, 20 мл) при 15°С в течение 2 мин и фильтровали.
Фильтрат концентрировали при пониженном давлении с получением Соединения 12-4. ЖХ/МС (ЭРИ) m/z: 392 (М+1).
5) Синтез Соединения 12-5

Согласно способу синтеза соединения 11-7, Соединение 12-5 было получено с использованием Соединения 12-4 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 406 (М+1).
6) Синтез Соединения 12-6

Согласно Синтезу Соединения 11-8, Соединение 12-6 было получено с использованием Соединения 12-5 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 602 (М+1).
7) Синтез Соединения 12

Согласно Синтезу Соединения 11, Соединение 12 было получено с использованием Соединения 12-6 в качестве исходного вещества. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.90 (s, 1H), 8.19 (d, J=2.3 Гц, 1H), 8.15 (d, J=8.8 Гц, 1H), 8.05-7.99 (m, 2Н), 7.88 (dd, J=2.0, 8.3 Гц, 1H), 7.79 (dd, J=2.4, 8.9 Гц, 1H), 4.85-4.79 (m, 2Н), 4.15-4.08 (m, 2Н), 2.74 (br s, 1H), 1.69 (s, 6Н); ЖХ/МС (ЭРИ) m/z: 502 (М+1).
Пример 12. Синтез Соединения 13

1) Синтез Соединения 13-1
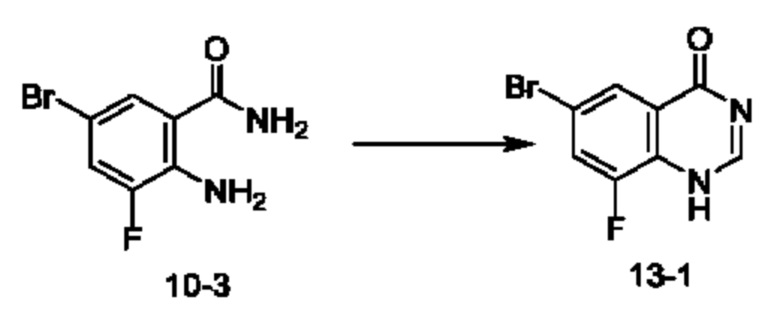
пара-Метилбензолсульфоновую кислоту (245 мг, 1,29 ммоль) добавляли в раствор Соединения 10-3 (3,00 г, 12,87 ммоль) и триметилортоформиата (30 мл). Полученную белую мутную жидкость нагревали до 110°С и перемешивали в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении с получением белого твердого вещества. Этилацетат (50 мл) добавляли к белому твердому веществу, и полученную смесь перемешивали в течение 30 мин и затем фильтровали. Полученный белый осадок на фильтре сушили при пониженном давлении с получением Соединения 13-1. 1Н ЯМР (400 МГц, DMSO-d6) δ м.д. 12.65 (brs, 1H), 8.19 (s, 1H), 8.07-7.98 (m, 2Н).
2) Синтез Соединения 13-2

DMF (15 мг, 206,0 мкмоль) добавляли в смешанный раствор Соединения 13-1 (500 мг, 2,06 ммоль) и дихлорсульфоксида (4,92 г, 41,36 ммоль, 3 мл). Полученную реакционную смесь нагревали до 80°С и перемешивали в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении. К остатку (светло-желтое твердое вещество), полученному после концентрирования, добавляли дихлорметан (3 мл), этандиол (1,11 г, 17,88 ммоль, 1 мл) и триэтиламин (657 мг, 6,49 ммоль, 0,9 мл). Полученную реакционную смесь нагревали до 80°С и перемешивали в течение 1 ч. Реакционную смесь фильтровали. Фильтрат вливали в воду (40 мл) и экстрагировали этилацетатом (30 мл). Органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением Соединения 13-2. ЖХ/МС (ЭРИ) m/z: 289 (М+3).
3) Синтез Соединения 13-3

diBoc (570 мг, 2,61 ммоль, 0,6 мл), триэтиламин (475 мг, 4,69 ммоль, 0,65 мл) и 4-диметиламинопиридин (27 мг, 221,00 мкмоль) добавляли в смешанный раствор Соединения 13-2 (630 мг, 2,19 ммоль) в дихлорметане (10 мл). Полученную реакционную смесь перемешивали при 10°С в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении, и остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 13-3. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.84 (s, 1H), 8.15 (t, J=1.5 Гц, 1H), 7.68 (dd, 3=2.0, 9.3 Гц, 1H), 4.86-4.78 (m, 2Н), 4.59-4.52 (m, 2Н), 1.51 (s, 9Н).
4) Синтез Соединения 13-4

Соединение 13-3 (300 мг, 774,79 мкмоль), Соединение 1-4 (120 мг, 1,16 ммоль), карбонат калия (268 мг, 1,94 ммоль), хлорид одновалентной меди (15 мг, 151,52 мкмоль), 2-ацетилциклогексанон (22 мг, 156,94 мкмоль), DMF (2 мл) и воду (0,1 мл) добавляли в пробирку для микроволновой обработки. Пробирку для микроволновой обработки герметично закрывали и выдерживали при 130°С для реакции под воздействием микроволнового излучения в течение 40 мин. Реакционную смесь фильтровали и промывали этилацетатом (20 мл). Фильтрат концентрировали при пониженном давлении. 1М соляную кислоту добавляли к остатку, полученному после концентрирования (рН=6-7), и полученную смесь экстрагировали этилацетатом (30 мл). Органическую фазу промывали насыщенным рассолом (30 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением Соединения 13-4. ЖХ/МС (ЭРИ) m/z: 410 (М+1).
5) Синтез Соединения 13-5

Раствор TMSCHN2 в н-гексане (2М, 1 мл) добавляли в раствор Соединения 13-4 (290 мг, 708,34 мкмоль) в дихлорметане (5 мл) и метаноле (0,5 мл). Полученную реакционную смесь перемешивали при 10°С в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ с получением Соединения 13-5. ЖХ/МС (ЭРИ) m/z: 424 (М+1).
6) Синтез Соединения 13-6
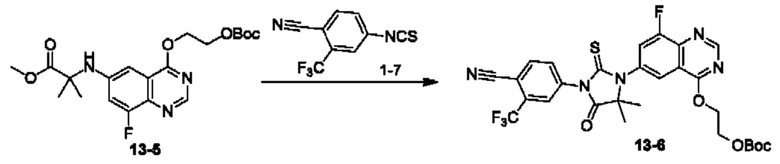
Смешанный раствор Соединения 13-5 (100 мг, 236,17 мкмоль), Соединения 1-7 (270 мг, 1,18 ммоль), метилбензола (2 мл) и DMF (0,5 мл) нагревали до 110°С и перемешивали в течение 16 ч. Соединение 1-7 (270 мг, 1,18 ммоль) добавляли в реакционную смесь. Реакционную смесь дополнительно перемешивали при 110°С в течение 16 ч. Метанол (2 мл) добавляли в реакционную смесь, и смесь перемешивали в течение 30 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ с получением Соединения 13-6. ЖХ/МС (ЭРИ) m/z: 620 (М+1).
7) Синтез Соединения 13

Трифторуксусную кислоту (0,4 мл) добавляли в раствор Соединения 13-6 (100 мг, 161,40 мкмоль) в дихлорметане (2 мл). Полученную реакционную смесь перемешивали при 10°С в течение 2 ч. Насыщенный водный раствор бикарбоната натрия добавляли в реакционную смесь (рН примерно 7), и смесь экстрагировали дихлорметаном (30 мл). Органическую фазу промывали насыщенным рассолом (30 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ и затем очищали методом препаративной ВЭЖХ с получением Соединения 13. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.94 (s, 1H), 8.06-7.94 (m, 3H), 7.86 (dd, J=1.9, 8.2 Гц, 1H), 7.53 (dd, J=2.1, 9.9 Гц, 1H), 4.86-4.76 (m, 2Н), 4.17-4.07 (m, 2Н), 2.47 (br t, J=5.5 Гц, 1H), 1.69 (s, 6H); ЖХ/МС (ЭРИ) m/z: 520 (М+1).
Пример 13. Синтез Соединения 84

1) Синтез Соединения 14-2

Оксихлорид фосфора (182 мг, 1,18 ммоль, 0,11 мл) и N,N-диизопропилэтиламин (29 мг, 224,40 мкмоль) добавляли в смешанный раствор Соединения 14-1 (50 мг, 222,18 мкмоль) и безводного метилбензола (1 мл). Полученную реакционную смесь перемешивали при 10°С в течение 0,5 ч, нагревали до 110°С и перемешивали в течение 5 ч. Реакционную смесь концентрировали при пониженном давлении. Этандиол (138 мг, 2,22 ммоль) и триэтиламин (73 мг, 722,08 мкмоль, 0,1 мл) добавляли к остатку, полученному после концентрирования. Полученный смешанный раствор перемешивали при 110°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, разбавляли дихлорметаном (20 мл) и промывали водой (10 мл) и насыщенным рассолом (15 мл). Органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением Соединения 14-2. ЖХ/МС (ЭРИ) m/z: 269 (М+1).
2) Синтез Соединения 14-3

diBoc (30 мг, 137,46 мкмоль), триэтиламин (23 мг, 227,30 мкмоль) и 4-диметиламинопиридин (2 мг, 16,37 мкмоль) добавляли в смешанный раствор Соединения 14-2 (30 мг, 111,49 мкмоль) в дихлорметане (1 мл). Полученную реакционную смесь перемешивали при 10°С в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ с получением Соединения 14-3. ЖХ/МС (ЭРИ) m/z: 391 (М+23).
3) Синтез Соединения 14-4

Соединение 14-3 (20 мг, 54,17 мкмоль), Соединение 1-4 (9 мг, 87,28 мкмоль), карбонат калия (20 мг, 144,63 мкмоль), хлорид одновалентной меди (2 мг, 20,20 мкмоль), 2-ацетилциклогексанон (2 мг, 14,27 мкмоль), DMF (1 мл) и воду (0,05 мл) добавляли в пробирку для микроволновой обработки. Пробирку для микроволновой обработки герметично закрывали и выдерживали при 130°С для реакции под воздействием микроволнового излучения в течение 30 мин. Реакционную смесь фильтровали и промывали этилацетатом (10 мл). Фильтрат концентрировали при пониженном давлении. 1 н. соляную кислоту добавляли к остатку, полученному после концентрирования (рН 6-7), и смесь экстрагировали этилацетатом (20 мл). Органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением Соединения 14-4. ЖХ/МС (ЭРИ) m/z: 392 (М+1).
4) Синтез Соединения 14-5

Раствор TMSCHN2 в н-гексане (2М, 0,1 мл) добавляли в раствор Соединения 14-4 (25 мг, 63,87 мкмоль) в дихлорметане (1 мл) и метаноле (0,1 мл). Полученную реакционную смесь перемешивали при 10°С в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ с получением Соединения 14-5. ЖХ/МС (ЭРИ) m/z: 428 (М+23).
5) Синтез Соединения 14-6

Смешанный раствор Соединения 14-5 (10 мг, 24,66 мкмоль), Соединения 1-7 (28 мг, 122,81 мкмоль), метилбензола (1 мл) и DMF (0,2 мл) нагревали до 110°С и перемешивали в течение 16 ч. Соединение 1-7 (28 мг, 122,81 мкмоль) добавляли в реакционную смесь, и полученную реакционную смесь дополнительно перемешивали при 110°С в течение 8 ч. Метанол (1 мл) добавляли в реакционную смесь, и смесь перемешивали в течение 30 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ с получением Соединения 14-6. ЖХ/МС (ЭРИ) m/z: 624 (М+23).
6) Синтез Соединения 14

Трифторуксусную кислоту (0,1 мл) добавляли в раствор Соединения 14-6 (10 мг, 16,62 мкмоль) в дихлорметане (1 мл). Полученную реакционную смесь перемешивали при 15°С в течение 1 ч. Насыщенный водный раствор бикарбоната натрия добавляли в реакционную смесь (рН примерно 8), и полученную смесь экстрагировали дихлорметаном (10 мл). Органическую фазу промывали насыщенным рассолом (10 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ с получением Соединения 14. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 9.33 (s, 1H), 8.04-7.96 (m, 3H), 7.90-7.83 (m, 2Н), 7.76 (dd, J=2.3, 8.8 Гц, 1Н), 4.76-4.65 (m, 2Н), 4.14-4.02 (m, 2Н), 2.67 (br s, 1Н), 1.67 (s, 6H); ЖХ/МС (ЭРИ) m/z: 502 (М+1).
Пример 14. Синтез Соединения 15

1) Синтез Соединения 15-1

При комнатной температуре (10°С) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (7,82 г, 20,55 ммоль) добавляли в раствор Соединения 11-1 (3,40 г, 15,81 ммоль), 2,2-дифторуксусной кислоты (3,04 г, 31,62 ммоль) и триэтиламина (4,80 г, 47,43 ммоль) в дихлорметане (50 мл). Реакционную смесь подвергали реакции при 10°С в течение 12 ч. Реакционную смесь напрямую концентрировали с получением неочищенного продукта. Неочищенный продукт очищали колоночной флэш-хроматографией с получением Соединения 15-1. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 12.72 (br s, 1 Н), 8.34 (br d, J=8.78 Гц, 1 H), 8.05 (br s, 1 H), 7.87 (br s, 1 H), 7.41 (br d, J=9.03 Гц, 1 H), 6.57 (br s, 1 H), 6.01-5.65 (m, 1 H).
2) Синтез Соединения 15-2

Метоксид натрия (1,77 г, 32,76 ммоль) добавляли в раствор Соединения 15-1 (3,20 г, 10,92 ммоль) в метаноле (10 мл) при комнатной температуре (10°С). Реакционную смесь подвергали реакции при 30°С в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали колоночной флэш-хроматографией с получением Соединения 15-2. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 12.69 (br s, 1 Н), 8.28 (s, 1 Н), 7.75 (br d, J=8.78 Гц, 1 H), 7.52 (d, J=8.53 Гц, 1 H), 6.51-6.18 (m, 1 Н); ЖХ/МС (ЭРИ) m/z: 277 (М+3).
3) Синтез Соединения 15-3

Согласно Синтезу Соединения 11-4, Соединение 15-3 было получено с использованием Соединения 15-2 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 321 (М+3).
4) Синтез Соединения 88-4

Согласно Синтезу Соединения 11-5, Соединение 15-4 было получено с использованием Соединения 15-3 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 419 (М+1).
5) Синтез Соединения 15-5

Согласно Синтезу Соединения 11-6, Соединение 15-5 было получено с использованием Соединения 15-4 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 442 (М+1).
6) Синтез Соединения 15-6

Согласно Синтезу Соединения 11-7, Соединение 15-6 было получено с использованием Соединения 15-5 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 456 (М+1).
7) Синтез Соединения 15-7

Согласно Синтезу Соединения 11-8, Соединение 15-7 было получено с использованием Соединения 15-6 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 652 (М+1).
8) Синтез Соединения 15

Согласно Синтезу Соединения 11, Соединение 15 было получено с использованием Соединения 15-7 в качестве исходного вещества. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.26-8.20 (m, 2Н), 8.06-8.00 (m, 2Н), 7.91-7.83 (m, 2Н), 6.85-6.52 (m, 1H), 4.92-4.87 (m, 2Н), 4.15 (br d, J=3.8 Гц, 2Н), 2.40 (br s, 1H), 1.70 (s, 6Н); ЖХ/МС (ЭРИ) m/z: 552 (М+1).
Пример 15. Синтез Соединения 16

1) Синтез Соединения 16-1

CDI (11,31 г, 69,75 ммоль) добавляли в мутную жидкость Соединения 11-1 (10 г, 46,50 ммоль) в тетрагидрофуране (100 мл). Полученную смесь перемешивали при 75°С в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры, и выпавшее в осадок твердое вещество отфильтровывали. Осадок на фильтре промывали тетрагидрофураном (10 мл × 3). Осадок на фильтре концентрировали досуха при пониженном давлении с получением Соединения 16-1. 1Н ЯМР (400 МГц, DMSO-d6) δ м.д. 11.47-11.16 (m, 2Н), 7.92 (d, J=2.3 Гц, 1H), 7.77 (dd, J=2.3, 8.8 Гц, 1H), 7.10 (d, J=8.5 Гц, 1Н).
2) Синтез Соединения 16-2

N,N-Диизопропилэтиламин (5,87 г, 45,43 ммоль) добавляли по каплям в раствор Соединения 16-1 (7,3 г, 30,29 ммоль) в оксихлориде фосфора (100 мл). Полученную смесь перемешивали при 110°С в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли дихлорметаном (400 мл), медленно добавляли в воду (500 мл) при перемешивании, экстрагировали дихлорметаном (50 мл × 3) и промывали насыщенным рассолом (50 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением Соединения 16-2. ЖХ/МС (ЭРИ) m/z: 279 (М+3).
3) Синтез Соединения 16-3

Гидрид натрия (173 мг, 4,32 ммоль, чистота 60%) добавляли в раствор Соединения 16-2 (1 г, 3,60 ммоль) и этандиола (268 мг, 4,32 ммоль) в тетрагидрофуране (50 мл). Смесь перемешивали при 10°С в течение 1 ч. Реакционную смесь гасили водой (20 мл), и полученную смесь экстрагировали дихлорметаном (20 мл × 3). Объединенную органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением Соединения 16-3. ЖХ/МС (ЭРИ) m/z: 305 (М+3).
4) Синтез Соединения 16-4

При 10°С триэтиламин (1,10 г, 10,87 ммоль) добавляли в смесь Соединения 16-3 (1,1 г, 3,62 ммоль), diBoc (1,19 г, 5,44 ммоль) и 4-диметиламинопиридина (88,55 мг, 724,78 мкмоль) в дихлорметане (20 мл). Реакционную смесь подвергали реакции при 10°С в течение 1 ч. Реакционную смесь промывали разбавленной 1М соляной кислотой (20 мл) и водой (20 мл × 2) в указанном порядке, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением Соединения 16-4. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.23 (d, J=2.26 Гц, 1H), 7.66 (d, J=9.03 Гц, 1H), 7.85 (dd, J=8.78, 2.26 Гц, 1H), 4.75 (dt, J=4.27, 2.38 Гц, 2Н), 4.48 (dt, J=4.20, 2.29 Гц, 2Н), 1.44 (s, 9Н).
5) Синтез Соединения 16-5

Раствор метиламина в тетрагидрофуране (2,0 М, 2,5 мл) добавляли в раствор Соединения 16-4 (1 г, 2,48 ммоль) в тетрагидрофуране (3 мл) при 10°С. Реакционную смесь выдерживали при 80°С для реакции под воздействием микроволнового излучения в течение 0,5 ч. Реакционную смесь напрямую концентрировали с получением неочищенного продукта. Неочищенный продукт очищали колоночной флэш-хроматографией с получением Соединения 16-5. ЖХ/МС (ЭРИ) m/z: 400 (М+3).
6) Синтез Соединения 16-6

Согласно Синтезу Соединения 11-6, Соединение 16-6 было получено с использованием Соединения 16-5 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 421 (М+1).
7) Синтез Соединения 16-7

Согласно Синтезу Соединения 11-7, Соединение 16-7 было получено с использованием Соединения 16-6 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 435 (М+1).
8) Синтез Соединения 16-8

Согласно Синтезу Соединения 11-8, Соединение 16-8 было получено с использованием Соединения 16-7 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 631 (М+1).
9) Синтез Соединения 16

Согласно Синтезу Соединения 11, Соединение 16 было получено с использованием Соединения 16-8 в качестве исходного вещества. 1H ЯМР (400 МГц, CDCl3) δ м.д. 7.99-7.87 (m, 2Н), 7.85-7.74 (m, 2Н), 7.58 (br d, J=8.03 Гц, 1Н), 7.42 (dd, J=8.78, 2.26 Гц, 1H), 4.59 (br s, 2Н), 4.09-3.85 (m, 2Н), 3.08-2.98 (m, 3H), 1.56 (s, 6Н); ЖХ/МС (ЭРИ) m/z: 531 (М+1).
Пример 16. Синтез Соединения 17

1) Синтез Соединения 17-1

Пропионилхлорид (12,91 г, 139,50 ммоль) добавляли в раствор Соединения 11-1 (10,00 г, 46,50 ммоль) в трихлорметане (200 мл) при 20°С. Реакционную смесь нагревали до 70°С и подвергали реакции в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали с получением неочищенного продукта. Этилацетат (100 мл) добавляли к неочищенному продукту, и полученную смесь перемешивали при 25°С в течение 0,5 ч и фильтровали. Собранный на фильтре осадок сушили сушильном шкафу с получением Соединения 17-1. ЖХ/МС (ЭРИ) m/z: 253 (М+1).
2) Синтез Соединения 17-2

2-Бромэтанол (1,24 г, 9,88 ммоль, 0,7 мл) добавляли в смешанный раствор Соединения 17-1 (1,00 г, 3,95 ммоль), карбоната калия (1,36 г, 9,88 ммоль), хлорида бензилтриэтиламмония (90 мг, 395,00 мкмоль) и диметоксиэтана (20 мл). Полученную реакционную смесь нагревали до 90°С и перемешивали в течение 16 ч. 2-Бромэтанол (1,24 г, 9,88 ммоль, 0,7 мл) и хлорид бензилтриэтиламмония (135 мг, 592,70 мкмоль) добавляли в реакционную смесь. Полученную реакционную смесь нагревали до 90°С и перемешивали в течение 16 ч. Реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 17-2. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.30 (d, J=2.0 Гц, 1H), 7.87 (dd, J=2.3, 8.8 Гц, 1H), 7.75 (d, J=8.8 Гц, 1H), 4.80-4.72 (m, 2Н), 4.08 (br d, J=3.5 Гц, 2Н), 3.31 (br s, 1H), 2.96 (q, J=7.5 Гц, 2H), 1.40 (t, J=7.7 Гц, 3H).
3) Синтез Соединения 17-3

diBoc (171 мг, 783,51 мкмоль), триэтиламин (139 мг, 1,37 ммоль, 0,19 мл) и 4-диметиламинопиридин (10 мг, 81,85 мкмоль) добавляли в смешанный раствор Соединения 17-2 (200 мг, 673,06 мкмоль) в дихлорметане (4 мл). Полученную реакционную смесь перемешивали при 10°С в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 17-3. ЖХ/МС (ЭРИ) m/z: 397 (М+1).
4) Синтез Соединения 17-4

Смесь Соединения 17-3 (130 мг, 327,24 мкмоль), Boc-NH2 (50 мг, 426,80 мкмоль), карбонат цезия (266 мг, 816,40 мкмоль), бис(дибензилиденацетон)палладия (20 мг, 34,78 мкмоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантена (20 мг, 34,56 мкмоль) и метилбензола (2 мл) добавляли в пробирку для микроволновой обработки и выдерживали при 120°С для реакции под воздействием микроволнового излучения в течение 0,5 ч. Реакционную смесь фильтровали через целит. Фильтрат разбавляли этилацетатом (30 мл), промывали водой (20 мл) и насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ с получением Соединения 17-4. ЖХ/МС (ЭРИ) m/z: 434 (М+1).
5) Синтез Соединения 17-5

Трифторуксусную кислоту (0,2 мл) добавляли в раствор Соединения 17-4 (85 мг, 196,08 мкмоль) в дихлорметане (2 мл). Полученную реакционную смесь перемешивали при 15°С в течение 12 ч. Насыщенный водный раствор бикарбоната натрия добавляли в реакционную смесь (рН примерно 8), и смесь экстрагировали дихлорметаном (20 мл). Органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Гидроксид лития (82 мг, 1,95 ммоль) добавляли в раствор полученного желтого масла (64 мг, 194,37 мкмоль) в тетрагидрофуране (2 мл) и воде (0,5 мл). Полученную реакционную смесь перемешивали при 15°С в течение 16 ч. Реакционную смесь сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением Соединения 17-5. ЖХ/МС (ЭРИ) m/z: 234 (М+1).
6) Синтез Соединения 17-6

Триметилсилилцианид (25 мг, 252,00 мкмоль) и хлорид цинка (4 мг, 29,32 мкмоль) добавляли в смешанный раствор Соединения 17-5 (20 мг, 85,74 мкмоль), циклобутанона (36 мг, 513,63 мкмоль), сульфата натрия (50 мг, 352,01 мкмоль) и тетрагидрофурана (2 мл). Полученную реакционную смесь перемешивали при 10°С в течение 19 ч. Водный раствор сульфита натрия (5 мл) добавляли в реакционную смесь, и смесь экстрагировали этилацетатом (5 мл × 3). Органическую фазу промывали насыщенным рассолом (10 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением Соединения 17-6. ЖХ/МС (ЭРИ) m/z: 313 (М+1).
7) Синтез Соединения 17-7

diBoc (23 мг, 105,39 мкмоль), триэтиламин (20 мг, 197,65 мкмоль) и 4-диметиламинопиридин (2 мг, 16,37 мкмоль) добавляли в смешанный раствор Соединения 17-6 (30 мг, 96,04 мкмоль) в дихлорметане (1 мл). Полученную реакционную смесь перемешивали при 10°С в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ с получением Соединения 17-7. ЖХ/МС (ЭРИ) m/z: 413 (М+1).
8) Синтез Соединения 17-8

Смешанный раствор Соединения 17-7 (20 мг, 48,49 мкмоль), Соединения 1-7 (28 мг, 122,70 мкмоль), метилбензола (1 мл) и DMF (0,2 мл) нагревали до 110°С и перемешивали в течение 16 ч. Соединение 1-7 (54 мг, 236,64 мкмоль) добавляли в реакционную смесь, и полученную смесь дополнительно перемешивали при 110°С в течение 4 ч. Метанол (1 мл) добавляли в реакционную смесь, и смесь перемешивали в течение 30 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ (петролейный эфир/этилацетат=1/1) с получением Соединения 17-8. ЖХ/МС (ЭРИ) m/z: 642 (М+1).
9) Синтез Соединения 17
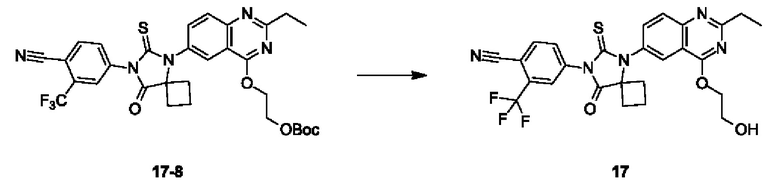
Трифторуксусную кислоту (0,2 мл) добавляли в раствор Соединения 17-8 (30 мг, 46,75 мкмоль) в дихлорметане (1 мл). Полученную реакционную смесь перемешивали при 10°С в течение 2 ч. Насыщенный водный раствор бикарбоната натрия добавляли в реакционную смесь (рН примерно 8), и смесь экстрагировали дихлорметаном (10 мл × 3). Органическую фазу промывали насыщенным рассолом (15 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ с получением Соединения 17. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.09 (d, J=2.3 Гц, 1Н), 8.01 (d, J=8.8 Гц, 1H), 7.92 (dd, J=3.1, 5.1 Гц, 2Н), 7.80 (dd, J=1.8, 8.3 Гц, 1Н), 7.64 (dd, J=2.5, 8.8 Гц, 1Н), 4.77-4.69 (m,2H), 4.07-3.98 (m, 2H), 3.12-2.81 (m, 3Н), 2.73-2.62 (m, 2H), 2.59-2.46 (m, 2H), 2.26-2.13 (m, 1H), 1.65-1.55 (m, 1H), 1.38-1.33 (m, 3Н); ЖХ/МС (ЭРИ) m/z: 542 (M+1).
Пример 17. Синтез Соединения 18

1) Синтез Соединения 18-1

Согласно Синтезу Соединения 11-2, Соединение 18-1 было получено с использованием Соединения 10-3 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 301 (М+1).
2) Синтез Соединения 18-2

Раствор трет-бутоксида калия в тетрагидрофуране (1М, 81 мл) добавляли в раствор Соединения 18-1 (8,1 г, 26,90 ммоль) в тетрагидрофуране (150 мл). Полученную смесь перемешивали при 30°С в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, растворяли в воде (40 мл) и доводили до рН примерно 7 разбавленной соляной кислотой (2 моль/л), и белое твердое вещество выпадало в осадок. Мутную жидкость фильтровали, и осадок на фильтре промывали водой (10 мл × 2). Полученный на фильтре осадок сушили в инфракрасном шкафу с получением Соединения 18-2. ЖХ/МС (ЭРИ) m/z: 285 (М+3).
3) Синтез Соединения 18-3

Согласно Синтезу Соединения 11-4, Соединение 18-3 было получено с использованием Соединения 18-2 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 329 (М+3).
4) Синтез Соединения 18-4

Согласно Синтезу Соединения 11-5, Соединение 18-4 было получено с использованием Соединения 18-3 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 429 (М+3).
5) Синтез Соединения 18-5
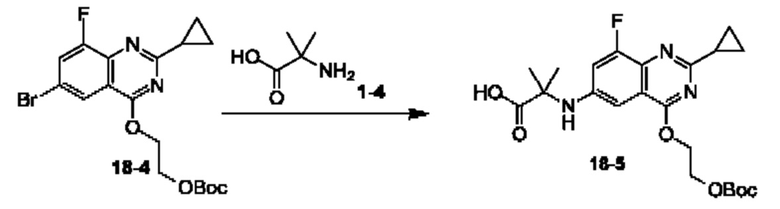
Согласно Синтезу Соединения 11-6, Соединение 18-5 было получено с использованием Соединения 18-4 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 450 (М+1).
6) Синтез Соединения 18-6

Согласно Синтезу Соединения 11-7, Соединение 18-6 было получено с использованием Соединения 18-5 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 464 (М+1).
7) Синтез Соединения 18-7

Согласно Синтезу Соединения 11-8, Соединение 18-7 было получено с использованием Соединения 18-6 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 660 (М+1).
8) Синтез Соединения 18

Согласно Синтезу Соединения 11, Соединение 18 было получено с использованием Соединения 18-7 в качестве исходного вещества. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.05-7.96 (m, 2Н), 7.90-7.84 (m, 2Н), 7.44 (dd, J=2.3, 10.0 Гц, 1H), 4.77-4.70 (m, 2Н), 4.10 (br s, 2Н), 2.44-2.36 (m, 1H), 2.32 (br s, 1H), 1.68 (s, 6Н), 1.31-1.24 (m, 2Н), 1.21-1.13 (m, 2Н); ЖХ/МС (ЭРИ) m/z: 560 (М+1).
Пример 18. Синтез Соединения 19

1) Синтез Соединения 19-1

пара-Метилбензолсульфоновую кислоту (1 г, 5,26 ммоль) добавляли в мутную жидкость Соединения 10-3 (10 г, 42,44 ммоль) и триметилортоформиата (60 мл). Полученную мутную жидкость нагревали до 120°С и перемешивали в течение 32 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 19-1. ЖХ/МС (ЭРИ) m/z: 271 (М+1).
2) Синтез Соединения 19-2

DMF (742 мг, 5,74 ммоль, 1 мл) добавляли в смешанный раствор Соединения 19-1 (1 г, 3,69 ммоль) и оксихлорида фосфора (19,3 г, 125,87 ммоль, 11,7 мл). Полученную реакционную смесь нагревали до 110°С и перемешивали в течение 4 ч. Реакционную смесь концентрировали при пониженном давлении, разбавляли дихлорметаном (100 мл) и затем медленно вливали в воду (80 мл). Полученную смесь экстрагировали дихлорметаном (50 мл × 2), и органическую фазу последовательно промывали насыщенным водным раствором бикарбоната натрия (рН примерно 7) и насыщенным рассолом (150 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением Соединения 19-2. ЖХ/МС (ЭРИ) m/z: 289 (М+1).
3) Синтез Соединения 19-3

Гидрид натрия (166 мг, 4,14 ммоль, чистота 60%) добавляли в раствор Соединения 19-2 (1 г, 3,45 ммоль) и тетрагидропиран-4-ола (423 мг, 4,14 ммоль) в тетрагидрофуране (30 мл). Смесь перемешивали при 13°С в течение 1 ч и дополнительно перемешивали при 10°С в течение 12 ч. Добавляли тетрагидропиран-4-ол (176 мг, 1,73 ммоль) и гидрид натрия (69 мг, 1,73 ммоль, чистота 60%), и полученную смесь дополнительно перемешивали при 14°С в течение 12 ч. Реакционную смесь гасили водой (20 мл) и экстрагировали дихлорметаном (20 мл × 3). Объединенную органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали колоночной флэш-хроматографией с получением Соединения 19-3. ЖХ/МС (ЭРИ) m/z: 355 (М+1).
4) Синтез Соединения 19-4

Согласно Синтезу Соединения 11-6, Соединение 19-4 было получено с использованием Соединения 19-3 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 378 (М+1).
5) Синтез Соединения 19-5

Согласно Синтезу Соединения 11-7, Соединение 19-5 было получено с использованием Соединения 19-4 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 392 (М+1).
6) Синтез Соединения 19

Согласно Синтезу Соединения 11-8, Соединение 19 было получено с использованием Соединения 19-5 в качестве исходного вещества. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.06-7.98 (m, 2Н), 7.92-7.83 (m, 2Н), 7.47 (dd, J=10.04, 2.26 Гц, 1H), 5.66 (tt, J=8.63, 4.30 Гц, 1H), 4.06 (dt, J=11.80, 4.52 Гц, 2Н), 3.70 (ddd, J=11.86, 9.10, 2.89 Гц, 2Н), 3.06 (q, J=7.70 Гц, 2Н), 2.26-2.16 (m, 2Н), 2.02-1.89 (m, 2Н), 1.69 (s, 6Н), 1.43 (t, J=7.53 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 588 (М+1).
Пример 19. Синтез Соединения 20
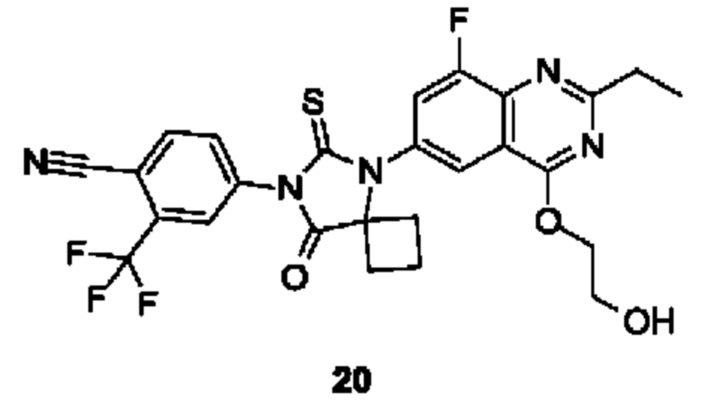
1) Синтез Соединения 20-1

Гидрид натрия (170 мг, 4,25 ммоль, 60%) добавляли в раствор Соединения 19-2 (1 г, 3,45 ммоль) и этандиола (266 мг, 4,29 ммоль) в тетрагидрофуране (10 мл). Полученную реакционную смесь перемешивали при 10°С в течение 6 ч. Реакционную смесь гасили насыщенным водным раствором хлорида аммония (100 мл) и затем экстрагировали дихлорметаном (100 мл × 2). Органическую фазу промывали насыщенным рассолом (100 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением Соединения 20-1. ЖХ/МС (ЭРИ) m/z: 315 (М+1).
2) Синтез Соединения 20-2

diBoc (835 мг, 3,83 ммоль), триэтиламин (654 мг, 6,47 ммоль, 0,9 мл) и 4-диметиламинопиридин (46 мг, 376,53 мкмоль) добавляли в смешанный раствор Соединения 20-1 (1 г, 3,17 ммоль) в дихлорметане (10 мл). Полученную реакционную смесь перемешивали при 10°С в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 20-2. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.12-8.06 (m, 1H), 7.62 (dd, J=2.0, 9.5 Гц, 1Н), 4.84-4.76 (m, 2Н), 4.58-4.51 (m, 2Н), 2.99 (q, J=7.5 Гц, 2Н), 1.51 (s, 9Н), 1.39 (t, J=7.7 Гц, 3Н).
3) Синтез Соединения 20-3

Смесь Соединения 20-2 (800 мг, 1,93 ммоль), Boc-NH2 (339 мг, 2,89 ммоль), карбоната цезия (1,57 г, 4,82 ммоль), бис(дибензилиденацетон)палладия (111 мг, 193,04 мкмоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантена (112 мг, 193,56 мкмоль) и метилбензола (10 мл) добавляли в пробирку для микроволновой обработки и выдерживали при 120°С для реакции под воздействием микроволнового излучения в течение 0,5 ч. Реакционную смесь фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 20-3. ЖХ/МС (ЭРИ) m/z: 452 (М+1).
4) Синтез Соединения 20-4

Трифторуксусную кислоту (2 мл) добавляли в раствор Соединения 20-3 (750 мг, 1,66 ммоль) в безводном дихлорметане (8 мл). Полученную реакционную смесь перемешивали при 10°С в течение 3 ч. Насыщенный водный раствор бикарбоната натрия добавляли в реакционную смесь (рН примерно 7), и смесь экстрагировали дихлорметаном (30 мл). Органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Гидроксид лития (520 мг, 12,39 ммоль) добавляли в раствор полученного желтого масла (430 мг, 1,24 ммоль) в тетрагидрофуране (6 мл) и воде (1,5 мл). Полученную реакционную смесь перемешивали при 10°С в течение 3 ч. Реакционную смесь сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением Соединения 20-4. ЖХ/МС (ЭРИ) m/z: 252 (М+1).
5) Синтез Соединения 20-5

Триметилсилилцианид (232 мг, 2,34 ммоль) и хлорид цинка (33 мг, 24,.98 мкмоль) добавляли в смешанный раствор Соединения 20-4 (200 мг, 796,00 мкмоль), циклобутанона (334 мг, 4,77 ммоль), сульфата натрия (453 мг, 3,19 ммоль) и тетрагидрофурана (5 мл). Полученную реакционную смесь перемешивали при 10°С в течение 16 ч. Водный раствор сульфита натрия (20 мл) добавляли в реакционную смесь, и полученную смесь экстрагировали этилацетатом (15 мл × 3). Органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением Соединения 20-5. ЖХ/МС (ЭРИ) m/z: 331 (М+1).
6) Синтез Соединения 20-6
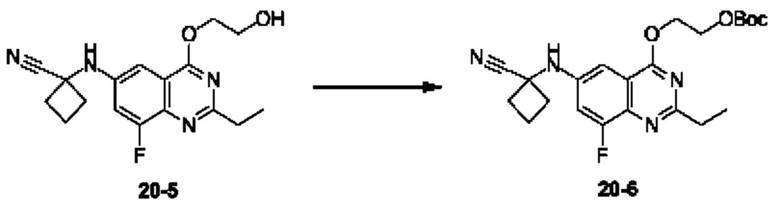
diBoc (237 мг, 1,09 ммоль), триэтиламин (189 мг, 1,87 ммоль) и 4-диметиламинопиридин (12 мг, 98,23 мкмоль) добавляли в смешанный раствор Соединения 20-5 (300 мг, 908,11 мкмоль) в дихлорметане (4 мл). Полученную реакционную смесь перемешивали при 10°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 20-6. ЖХ/МС (ЭРИ) m/z: 431 (М+1).
7) Синтез Соединения 20-7

Смешанный раствор Соединения 20-6 (80 мг, 185,84 мкмоль), Соединения 1-7 (170 мг, 744,98 мкмоль), метилбензола (2 мл) и DMF (0,5 мл) нагревали до 110°С и перемешивали в течение 16 ч. Метанол (1 мл) добавляли в реакционную смесь, и смесь перемешивали в течение 30 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ с получением Соединения 20-7. ЖХ/МС (ЭРИ) m/z: 660 (М+1).
8) Синтез Соединения 20

Трифторуксусную кислоту (0,4 мл) добавляли в раствор Соединения 20-7 (70 мг, 106,12 мкмоль) в безводном дихлорметане (2 мл). Полученную реакционную смесь перемешивали при 10°С в течение 1 ч. Насыщенный водный раствор бикарбоната натрия добавляли в реакционную смесь (рН примерно 8), и смесь экстрагировали дихлорметаном (10 мл × 3). Органическую фазу промывали насыщенным рассолом (15 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали методом препаративной ВЭЖХ с получением Соединения 20. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.98-7.86 (m, 3Н), 7.79 (br d, J=8.0 Гц, 1H), 7.40 (br d, J=9.8 Гц, 1H), 4.79-4.68 (m, 2H), 4.03 (br s, 2H), 2.99 (q, J=7.5 Гц, 2H), 2.76-2.60 (m, 3H), 2.59-2.46 (m, 2H), 2.30-2.14 (m, 1H), 1.52 (br s, 1H), 1.36 (t, J=7.5 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 560 (М+1).
Пример 20. Синтез Соединения 21

1) Синтез Соединения 21-1

При комнатной температуре (10°С) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (10.61 г, 27,89 ммоль) добавляли в раствор Соединения 10-3 (5 г, 21,46 ммоль), 2,2-дифторуксусной кислоты (6,18 г, 64,37 ммоль) и триэтиламина (8,68 г, 85,82 ммоль) в дихлорметане (10 мл). Реакционную смесь выдерживали при 10°С в течение 13 ч. Реакционную смесь концентрировали с получением неочищенного продукта. Неочищенный продукт очищали колоночной флэш-хроматографией с получением Соединения 21-1. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 12.90 (brs, 1Н),7.79 (s, 1Н), 7.38-7.29 (m, 1Н), 6.50-5.96 (m, 1Н).
2) Синтез Соединения 21-2

Согласно Синтезу Соединения 11-4, Соединение 21-2 было получено с использованием Соединения 21-1 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 337 (М+1).
3) Синтез Соединения 21-3

Согласно Синтезу Соединения 11-5, Соединение 21-3 было получено с использованием Соединения 21-2 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 439 (М+3).
4) Синтез Соединения 21-4
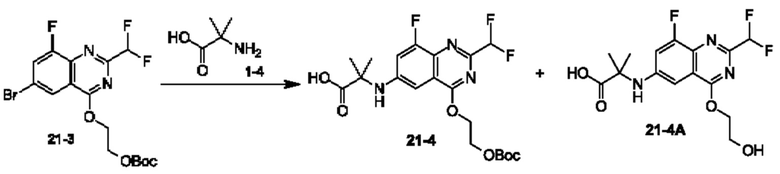
Соединение 21-3 (1,5 г, 3,43 ммоль), Соединение 1-4 (531 мг, 5,15 ммоль), хлорид одновалентной меди (34 мг, 343,09 мкмоль), 2-ацетилциклогексанон (48 мг, 343,09 мкмоль) и карбонат калия (948 мг, 6,86 ммоль) добавляли в пробирку для микроволновой обработки, заполненную DMF (15 мл) и водой (1,5 мл). После продувки азотом в течение 1 мин полученную смесь выдерживали при 130°С для реакции под воздействием микроволнового излучения в течение 20 мин. Реакционную смесь охлаждали до комнатной температуры и фильтровали. Осадок на фильтре промывали DMF (5 мл × 2). Фильтраты объединяли, подкисляли до рН=6-7 разбавленным водным раствором соляной кислоты (2М) и концентрировали. Полученное масло добавляли в смесь дихлорметан/метанол (30 мл/3 мл), перемешивали при комнатной температуре (20°С) в течение 10 мин и фильтровали для удаления нерастворимых веществ. Фильтрат концентрировали с получением смеси Соединения 21-4 и Соединения 21-4А. ЖХ/МС (ЭРИ) m/z: 360 (М+1); 460 (М+1).
5) Синтез Соединения 21-5

При 0°С раствор TMSCHN2 в н-гексане (2М, 5 мл) добавляли по каплям в раствор Соединения 21-4 (2,3 г, 5,01 ммоль) (смесь, содержащая Соединение 21-4А) в дихлорметане (20 мл) и метаноле (4 мл), и затем полученную смесь дополнительно перемешивали при 10°С в течение 2 ч. Реакционную смесь концентрировали с получением неочищенного продукта. Неочищенный продукт очищали колоночной флэш-хроматографией с получением Соединения 21-5. ЖХ/МС (ЭРИ) m/z: 374 (М+1).
6) Синтез Соединения 21-6

Согласно Синтезу Соединения 11-5, Соединение 21-6 было получено с использованием Соединения 21-5 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 474 (М+1).
7) Синтез Соединения 21-7

Согласно Синтезу Соединения 11-8, Соединение 21-7 было получено с использованием Соединения 21-6 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 670 (М+1).
8) Синтез Соединения 21

Согласно Синтезу Соединения 11, Соединение 21 было получено с использованием Соединения 21-7 в качестве исходного вещества. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.95 (s, 3Н), 7.78 (dd, J=8.28, 2.01 Гц, 1H), 7.52 (dd, J=9.79, 2.01 Гц, 1H), 6.84-6.36 (m, 1Н), 4.89-4.75 (m, 2Н), 4.17-4.00 (m, 2Н), 2.25-2.12 (m, 1H), 1.61 (s, 6Н); ЖХ/МС (ЭРИ) m/z: 570 (М+1).
Пример 21. Синтез Соединения 22

1) Синтез Соединения 22-1

Гидрид натрия (166 мг, 4,14 ммоль, чистота 60%) добавляли в раствор Соединения 19-2 (1 г, 3,45 ммоль) и 3-(метилсульфонил)-1-пропанола (573 мг, 4,14 ммоль) в тетрагидрофуране (20 мл). Смесь перемешивали при 16°С в течение 1 ч. В реакционную смесь добавляли воду (20 мл), и смесь экстрагировали дихлорметаном (20 мл × 2). Объединенную органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением Соединения 22-1. ЖХ/МС (ЭРИ) m/z: 393 (М+3).
2) Синтез Соединения 22-2

Согласно Синтезу Соединения 11-6, Соединение 22-2 было получено с использованием Соединения 22-1 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 414 (М+1).
3) Синтез Соединения 22-3

Согласно Синтезу Соединения 11-7, Соединение 22-3 было получено с использованием Соединения 22-2 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 428 (М+1).
4) Синтез Соединения 22

Согласно Синтезу Соединения 11-8, Соединение 22 было получено с использованием Соединения 22-3 в качестве исходного вещества. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.07-7.99 (m, 2Н), 7.94-7.83 (m, 2Н), 7.48 (dd, J=10.04, 2.26 Гц, 1Н), 4.81 (t, J=6.27 Гц, 2Н), 3.37-3.23 (m, 2Н), 3.07 (q, J=7.53 Гц, 2Н), 3.00 (s, 3Н), 2.57-2.47 (m, 2Н), 1.69 (s, 6Н), 1.44 (t, J=7.53 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 624 (М+1).
Пример 22. Синтез Соединения 23

1) Синтез Соединения 23-2

Соединение 23-1 (10 г, 61,31 ммоль) растворяли в смешанном растворе ацетонитрила (50 мл) и DMF (50 мл) и добавляли NBS (13,79 г, 61,31 ммоль). Полученную смесь нагревали до 80°С и перемешивали в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры. Раствор 1 моль/л бикарбоната натрия (62 мл) добавляли в реакционную смесь и перемешивали в течение 5 мин. Полученную смесь концентрировали досуха при пониженном давлении. В остаток, полученный после концентрирования, добавляли воду (80 мл), и полученную смесь экстрагировали дихлорметаном (100 мл × 4). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали досуха при пониженном давлении. При комнатной температуре дихлорметан (15 мл) добавляли к остатку, полученному после концентрирования. Полученную смесь перемешивали в течение 5 мин и фильтровали. Осадок на фильтре промывали дихлорметаном (5 мл), и осадок на фильтре сушили при пониженном давлении с получением Соединения 23-2. 1Н ЯМР (400 МГц, DMSO-d6) δ м.д. 12.57 (br s, 1H), 8.00 (s, 1H), 7.99-7.97 (m, 1H); ЖХ/МС (ЭРИ) m/z: 290 (М+1).
2) Синтез Соединения 23-3

DMF (8 мл) добавляли в раствор Соединения 23-2 (8,1 г, 28,03 ммоль) в оксихлориде фосфора (21,38 г, 139,58 ммоль). Полученную смесь перемешивали при 110°С в течение 80 мин. Реакционную смесь охлаждали до комнатной температуры и медленно добавляли по каплям в воду (150 мл), и смесь перемешивали при комнатной температуре. Полученную смесь доводили до рН примерно 8 насыщенным раствором бикарбоната натрия и экстрагировали дихлорметаном (40 мл × 4). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением Соединения 23-3. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.71 (d, J=1.8 Гц, 1H), 8.21 (d, J=2,0 Гц, 1Н).
3) Синтез Соединения 23-4

пара-Метоксибензиламин (3,25 г, 23,68 ммоль), бис(дибензилиденацетон)-палладий (2,72 г, 4,74 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (2,74 г, 4,74 ммоль) и трет-бутоксид натрия (3,41 г, 35,52 ммоль) добавляли в раствор Соединения 23-3 (7,28 г, 23,68 ммоль) в метилбензоле (100 мл). Полученную смесь продували азотом четыре раза, нагревали до 110°С и перемешивали в течение 1 ч под защитой азота. Реакционную смесь охлаждали до комнатной температуры, добавляли воду (100 мл) в реакционную смесь, и полученную смесь экстрагировали этилацетатом (80 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 23-4. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.84 (d, J=2.8 Гц, 1H), 7.18 (d, J=8.0 Гц, 2Н), 7.13-7.09 (m, 1H), 6.84-6.80 (m, 2Н), 4.20 (d, J=5.5 Гц, 2Н), 3.73 (s, 3Н); ЖХ/МС (ЭРИ) m/z: 317 (М+1).
4) Синтез Соединения 23-5

Цианид цинка (4,16 г, 35,42 ммоль) добавляли в раствор Соединения 23-4 (7,1 г, 22,42 ммоль) в DMF (100 мл). После трехразовой продувки азотом добавляли 1,1'-бис(дифенилфосфино)ферроцен (4,53 г, 4,48 ммоль) и затем добавляли бис(дибензилиденацетон)палладий (2,58 г, 4,48 ммоль) после трехразовой продувки азотом. После трехразовой продувки азотом полученную смесь нагревали до 150°С и перемешивали в течение 50 мин под защитой азота. Реакционную смесь охлаждали до комнатной температуры и концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 23-5. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.18 (d, J=2.5 Гц, 1H), 7.27 (d, J=8.5 Гц, 2Н), 7.09 (d, J=2.8 Гц, 1H), 6.93 (d, J=8.5 Гц, 2Н), 5.07 (br s, 1H), 4.38 (d, J=5.3 Гц, 2H), 3.83 (s, 3Н); ЖХ/МС (ЭРИ) m/z: 308 (М+1).
5) Синтез Соединения 23-6

Соединение 23-5 (3,4 г, 11,07 ммоль) растворяли в смешанном растворе дихлорметана (4 мл) и трифторуксусной кислоты (16 мл). Полученную смесь перемешивали при 10°С в течение 2 ч. Реакционную смесь концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, разбавляли этилацетатом (50 мл) и промывали насыщенным раствором бикарбоната натрия (50 мл × 3). Полученную органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, суспендировали в этилацетате (20 мл) при комнатной температуре в течение 20 мин и фильтровали. Осадок на фильтре концентрировали при пониженном давлении с получением Соединения 23-6. 1Н ЯМР (400 МГц, DMSO-d6) δ м.д. 8.19 (d, J=2.3 Гц, 1H), 7.29 (d, J=2.5 Гц, 1H), 7.00 (s, 2Н).
6) Синтез Соединения 23-7

Тиофосген (1,76 г, 15,28 ммоль) добавляли по каплям в воду (50 мл), и полученную смесь перемешивали при 10°С в течение 30 мин. Затем добавляли порциями Соединение 23-6 (1,43 г, 7,64 ммоль), и полученную смесь перемешивали при 10°С в течение 5 ч. Реакционную смесь экстрагировали дихлорметаном (40 мл × 2). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением Соединения 23-7.
7) Синтез Соединения 23-8

Согласно Синтезу Соединения 11-8, Соединение 23-8 было получено с использованием Соединения 18-6 и Соединение 23-7 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 661 (М+1).
8) Синтез Соединения 23

Согласно Синтезу Соединения 11, Соединение 23 было получено с использованием Соединения 23-8 в качестве исходного вещества. 1H ЯМР (400 МГц, CDCl3) δ м.д. 9.12 (d, J=2.01 Гц, 1H), 8.39 (d, J=2.26 Гц, 1H), 7.88 (s, 1H), 7.43 (dd, 3=9.91, 2.13 Гц, 1H), 4.79-4.68 (m, 2Н), 4.15-4.05 (m, 2Н), 2.45-2.35 (m, 1H), 2.30 (t, J=5.65 Гц, 1H), 1.75-1.73 (m, 1H), 1.70 (s, 5H), 1.31-1.25 (m, 2H), 1.20-1.15 (m, 2H); ЖХ/МС (ЭРИ) m/z: 561 (M+1).
Пример 23. Синтез Соединения 24

1) Синтез Соединения 24-1
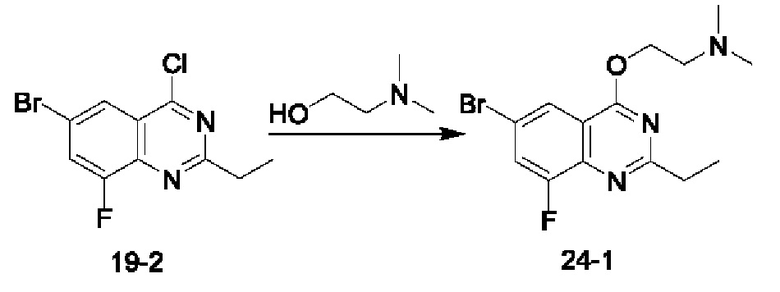
Гидрид натрия (166 мг, 4,15 ммоль, 60%) добавляли в раствор Соединения 19-2 (1 г, 3,45 ммоль) и N,N-диметилэтаноламина (370 мг, 4,15 ммоль) в тетрагидрофуране (20 мл). Полученную реакционную смесь перемешивали при 10°С в течение 1 ч. Реакционную смесь гасили насыщенным водным раствором хлорида аммония (50 мл) и затем экстрагировали дихлорметаном (40 мл × 2). Органическую фазу промывали насыщенным рассолом (50 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 24-1. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.10-8.04 (m, 1H), 7.60 (dd, J=2.1, 9.7 Гц, 1Н), 4.70 (t, J=5.8 Гц, 2Н), 2.99 (q, J=7.5 Гц, 2Н), 2.84 (t, J=5.8 Гц, 2Н), 2.38 (s, 6Н), 1.40 (t, J=7.7 Гц, 3Н).
2) Синтез Соединения 24-2

Соединение 24-1 (400 мг, 1,17 ммоль), Соединение 1-4 (180 мг, 1,75 ммоль), карбонат калия (404 мг, 2,92 ммоль), хлорид одновалентной меди (23 мг, 232,32 мкмоль), 2-ацетилциклогексанон (33 мг, 235,41 мкмоль), DMF (4 мл) и воду (0,2 мл) добавляли в пробирку для микроволновой обработки. Пробирку для микроволновой обработки герметично закрывали и выдерживали при 130°С для реакции под воздействием микроволнового излучения в течение 40 мин. Реакционную смесь фильтровали и промывали этилацетатом (20 мл). Фильтрат концентрировали при пониженном давлении. К остатку, полученному после концентрирования (рН 6-7), добавляли 1 н. соляную кислоту. Полученную смесь подвергали сублимационной сушке. Смесь дихлорметан/метанол (20 мл, 10/1) добавляли к полученному твердому веществу, и полученную смесь сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением Соединения 24-2. ЖХ/МС (ЭРИ) m/z: 365 (М+1).
3) Синтез Соединения 24-3

Раствор TMSCHN2 в н-гексане (2М, 0,4 мл) добавляли в раствор Соединения 24-2 (0,25 г, 686,03 мкмоль) в дихлорметане (2 мл) и метаноле (0,2 мл). Полученную реакционную смесь перемешивали при 10°С в течение 1 ч. В реакционную смесь вливали воду, и смесь экстрагировали дихлорметаном (20 мл). Органическую фазу промывали насыщенным рассолом (15 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ с получением Соединения 24-3. ЖХ/МС (ЭРИ) m/z: 379 (М+1).
4) Синтез Соединения 24

Смешанный раствор Соединения 24-3 (60 мг, 158,55 мкмоль), Соединения 1-7 (120 мг, 525,87 мкмоль), метилбензола (2 мл) и DMF (0,5 мл) нагревали до 110°С и перемешивали в течение 16 ч. Метанол (1 мл) добавляли в реакционную смесь, и полученную смесь перемешивали в течение 30 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ и затем очищали методом препаративной ВЭЖХ с получением Соединения 24. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.97-7.84 (m, 3Н), 7.79 (br d, J=10.0 Гц, 1H), 7.37 (dd, J=2.1, 10.2 Гц, 1H), 4.70 (t, J=5.9 Гц, 2Н), 2.98 (q, J=7.5 Гц, 2Н), 2.83 (br s, 2Н), 2.36 (br s, 6H), 1.59 (s, 6H), 1.35 (t, J=7.5 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 575,0 (М+1).
Пример 24. Синтез Соединения 25

1) Синтез Соединения 25-1

Смешанный раствор Соединения 20-6 (80 мг, 185,84 мкмоль), Соединения 23-7 (130 мг, 567,24 мкмоль), метилбензола (2 мл) и DMF (0,5 мл) нагревали до 110°С и перемешивали в течение 16 ч. Метанол (1 мл) добавляли в реакционную смесь, и полученную смесь перемешивали в течение 30 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали препаративной ТСХ с получением Соединения 25-1. ЖХ/МС (ЭРИ) m/z: 661 (М+1).
2) Синтез Соединения 25

Трифторуксусную кислоту (0,4 мл) добавляли в раствор Соединения 25-1 (90 мг, 136,23 мкмоль) в дихлорметане (2 мл). Полученную реакционную смесь перемешивали при 10°С в течение 1 ч. Насыщенный водный раствор бикарбоната натрия добавляли в реакционную смесь (рН примерно 8), и смесь экстрагировали дихлорметаном (10 мл × 3). Органическую фазу промывали насыщенным рассолом (15 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали методом препаративной ВЭЖХ с получением Соединения 25. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 9.03 (d, J=1.8 Гц, 1H), 8.30 (d, J=1.8 Гц, 1H), 7.91 (s, 1H), 7.39 (dd, J=2.0, 9.8 Гц, 1H), 4.78-4.69 (m, 2Н), 4.08-3.97 (m, 2Н), 2.99 (q, J=7.5 Гц, 2Н), 2.70 (br t, J=9.4 Гц, 2Н), 2.61-2.48 (m, 2Н), 2.30-2.13 (m, 1H), 1.73-1.55 (m, 2Н), 1.36 (t, J=7.7 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 561 (М+1). Пример 25. Синтез Соединения 26
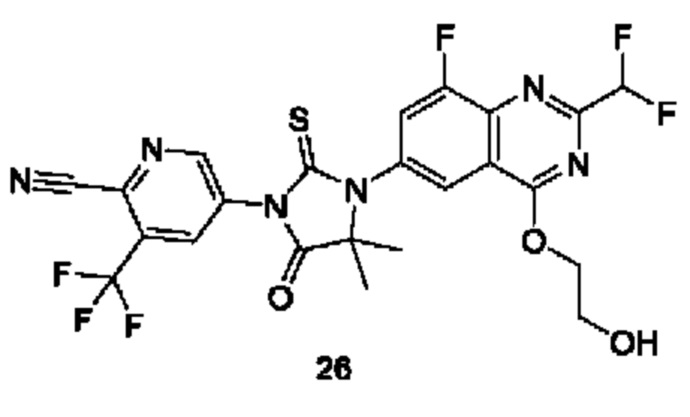
1) Синтез Соединения 26-1

Согласно Синтезу Соединения 11-8, Соединение 26-1 было получено с использованием Соединения 21-6 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 671 (М+1).
2) Синтез Соединения 26

Согласно Синтезу Соединения 11, Соединение 26 было получено с использованием Соединения 26-1 в качестве исходного вещества. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 9.03 (d, J=1.76 Гц, 1Н), 8.29 (d, J=2.01 Гц, 1H), 7.96 (s, 1H), 7.58-7.45 (m, 1Н), 6.85-6.44 (m, 1Н), 4.89-4.76 (m, 2Н), 4.07 (br d, J=3.76 Гц, 2Н), 2.16 (br s, 1Н), 1.64 (s, 6H); ЖХ/МС (ЭРИ) m/z: 571 (М+1).
Пример 26. Синтез Соединения 27

1) Синтез Соединения 27-1

Соединение 17-1 (1,50 г, 5,93 ммоль), Соединение 1-4 (917 мг, 8,89 ммоль), хлорид одновалентной меди (117 мг, 1,19 ммоль), 2-ацетилциклогексанон (166 мг, 1,19 ммоль), карбонат калия (2,05 г, 14,82 ммоль), N,N-диметилформамид (10 мл) и воду (2,5 мл) добавляли в пробирку для микроволновой обработки на 30 мл. Полученную смесь выдерживали при 130°С для реакции под воздействием микроволнового излучения в течение 1 ч. Реакционную смесь фильтровали, и осадок на фильтре промывали DMF (10 мл × 3). Разбавленную соляную кислоту (2 моль/л) добавляли по каплям в фильтрат, так чтобы фильтрат стал слабокислотным (рН примерно 6). Фильтрат концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, суспендировали в смеси дихлорметан/метанол (10/1, 30 мл) при 15°С в течение 2 мин и фильтровали. Фильтрат концентрировали досуха при пониженном давлении с получением Соединения 27-1. ЖХ/МС (ЭРИ) m/z: 276 (М+1).
2) Синтез Соединения 27-2

Соединение 27-1 (4,40 г, 15,98 ммоль) растворяли в метаноле (40 мл), и дихлорсульфоксид (19,01 г, 159,80 ммоль, 11,59 мл) добавляли по каплям при 0°С. Полученную смесь нагревали до 50°С и перемешивали в течение 18 ч. Реакционную смесь охлаждали до 15°С и концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, растворяли в насыщенном растворе бикарбоната натрия (50 мл) и экстрагировали смесью дихлорметаном/метанол (10:1, 80 мл × 4). Органические фазы объединяли, промывали насыщенным рассолом (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 27-2. ЖХ/МС (ЭРИ) m/z: 290 (М+1).
3) Синтез Соединения 27-3

Под защитой азота Соединение 27-2 (560 мг, 1,94 ммоль) и Соединение 1-7 (1,77 г, 7,74 ммоль) растворяли в N,N-диметилформамиде (2 мл) и метилбензоле (20 мл), и полученную смесь нагревали до 120°С и перемешивали в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 27-3. ЖХ/МС (ЭРИ) m/z: 486 (М+1).
4) Синтез Соединения 27
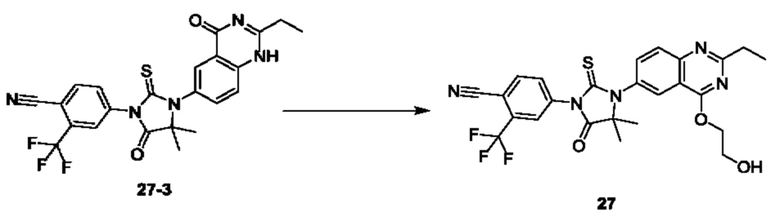
Под защитой азота мутную жидкость соединения 27-3 (789 мг, 1,63 ммоль), 2-бромэтанола (609 мг, 4,88 ммоль), карбоната калия (674 мг, 4,88 ммоль) и N,N-диметилформамида (10 мл) перемешивали при 30°С в течение 17 ч, добавляли 2-бромэтанол (609 мг, 4,88 ммоль) и дополнительно перемешивали при 30°С в течение 5 ч, и добавляли 2-бромэтанол (609 мг, 4,88 ммоль) и дополнительно перемешивали при 30°С в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры и затем напрямую фильтровали. Фильтрат отделяли и очищали методом препаративной ВЭЖХ с получением Соединения 27. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.13 (d, J=2.5 Гц, 1H), 8.08-7.99 (m, 3Н), 7.88 (dd, J=2.0, 8.3 Гц, 1H), 7.73 (dd, J=2.4, 8.9 Гц, 1Н), 4.85-4.78 (m, 2Н), 4.14-4.07 (m, 2Н), 3.16 (t, J=5.6 Гц, 1H), 3.03 (q, J=7.5 Гц, 2Н), 1.68 (s, 6Н), 1.44 (t, J=7.5 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 530 (М+1).
Пример 27. Синтез Соединения 28

1) Синтез Соединения 28-1

Гидрид натрия (166 мг, чистота 60%) добавляли в раствор Соединения 19-2 (1,00 г) и метил-2-гидроксиацетата (373 мг) в тетрагидрофуране (20 мл). Смесь перемешивали при 16°С в течение 1 ч. Реакционную смесь гасили водой (20 мл) и экстрагировали дихлорметаном (20 мл × 3). Объединенную органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением Соединения 28-1. ЖХ/МС (ЭРИ) m/z: 345 (М+3).
2) Синтез Соединения 28-2

Соединение 28-1 (600 мг), Соединение 1-4 (270 мг), хлорид одновалентной меди (17 мг), 2-ацетилциклогексанон (25 мг) и карбонат калия (604 мг) добавляли в пробирку для микроволновой обработки, заполненную DMF (10 мл) и водой (2 мл). После продувки азотом в течение 1 мин полученную смесь выдерживали при 130°С для реакции под воздействием микроволнового излучения в течение 1 ч и фильтровали, и осадок на фильтре промывали DMF (2 мл). Фильтрат доводили до рН=7 2М соляной кислотой и затем концентрировали. Смесь дихлорметан/метанол (10/1, 20 мл) добавляли в полученное масло для осаждения твердого вещества. После фильтрования полученный фильтрат концентрировали с получением Соединения 28-2. ЖХ/МС (ЭРИ) m/z: 352 (М+1).
3) Синтез Соединения 28-3

TMSCHN2 (2М, 4,70 мл) добавляли по каплям в раствор Соединения 28-2 (1,10 г) в дихлорметане (20 мл) и метаноле (2 мл). После завершения капельного добавления смесь подвергали реакции при 18°С в течение 2 ч. Реакционную смесь концентрировали, и концентрат очищали методом тонкослойной хроматографии с получением Соединения 28-3. ЖХ/МС (ЭРИ) m/z: 380 (М+1).
4) Синтез Соединения 28-4

Соединение 1-7 (230 мг) добавляли к Соединению 28-3 (130 мг) в смешанных растворителях метилбензоле (4 мл) и DMF (1 мл), и затем полученную смесь нагревали до 120°С и перемешивали в течение 28 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Концентрат очищали методом тонкослойной хроматографии с получением Соединения 28-4. ЖХ/МС (ЭРИ) m/z: 576 (М+1).
5) Синтез Соединения 28-5

Гидроксид лития (1М, 0,5 мл) добавляли в раствор Соединения 28-4 (90 мг) в тетрагидрофуране (3 мл), и полученную смесь перемешивали при 15°С в течение 1 ч. Затем реакционную смесь доводили до рН примерно 6 разбавленной 1М соляной кислотой и экстрагировали дихлорметаном (20 мл × 2). Объединенную органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением Соединения 28-5. ЖХ/МС (ЭРИ) m/z: 562 (М+1).
6) Синтез Соединения 28

HATU (75 мг) добавляли в раствор Соединения 28-5 (80 мг), метиламина гидрохлорида (16 мг) и триэтиламина (50 мг) в дихлорметане (5 мл). Затем полученную смесь перемешивали при 15°C в течение 1 ч. Реакционную смесь концентрировали, и концентрат очищали тонкослойной хроматографией. Полученный образец дополнительно очищали методом ВЭЖХ (щелочная) с получением Соединения 28. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.97-8.08 (m, 2Н), 7.94 (s, 1Н), 7.87 (dd, J=8.28, 1.76 Гц, 1Н), 7.52 (dd, J=10.04, 2.01 Гц, 1Н), 6.12 (br s, 1H), 5.17 (s, 2Н), 3.08 (q, J=7.53 Гц, 2Н), 2.94 (d, J=4.77 Гц, 3Н), 1.70 (s, 6Н), 1.42 (t, J=7.53 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 575 (М+1).
Пример 28. Синтез Соединения 29

1) Синтез Соединения 29-2

Согласно Синтезу Соединения 28-1, Соединение 29-2 было получено с использованием Соединения 19-2 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 343 (М+3).
2) Синтез Соединения 29-3

Согласно Синтезу Соединения 28-2, Соединение 29-3 было получено с использованием Соединения 29-2 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 364 (М+1).
3) Синтез Соединения 29-4

Согласно Синтезу Соединения 28-3, Соединение 29-4 было получено с использованием Соединения 29-3 в качестве исходного вещества. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 6.71 (dd, J=12.17, 2.64 Гц, 1H), 6.64 (d, J=2.01 Гц, 1H), 4.76 (dd, J=7.78, 6.27 Гц, 2Н), 4.62 (d, J=6.27 Гц, 2Н), 4.53 (t, J=6.15 Гц, 2Н), 4.29 (s, 1H), 3.58 (s, 3Н), 3.35-3.45 (m, 1H), 2.79 (q, J=7.53 Гц, 2Н), 1.47 (s, 6Н), 1.22 (t, J=7.65 Гц, 3 Н); ЖХ/МС (ЭРИ) m/z: 378 (М+1).
4) Синтез Соединения 29

Согласно Синтезу Соединения 28-4, Соединение 29 было получено с использованием Соединения 29-4 в качестве исходного вещества. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.88-7.98 (m, 2Н), 7.74-7.84 (m, 2Н), 7.39 (dd, J=10.04, 2.01 Гц, 1H), 4.85 (dd, J=7.78, 6.27 Гц, 2Н), 4.79 (d, J=6.27 Гц, 2Н), 4.59 (t, J=6.15 Гц, 2Н), 3.44-3.55 (m, 1Н), 2.98 (q, J=7.53 Гц, 2Н), 1.58 (s, 6Н), 1.32-1.40 (m, 3Н); ЖХ/МС (ЭРИ) m/z: 574 (М+1).
Пример 29. Синтез Соединения 30
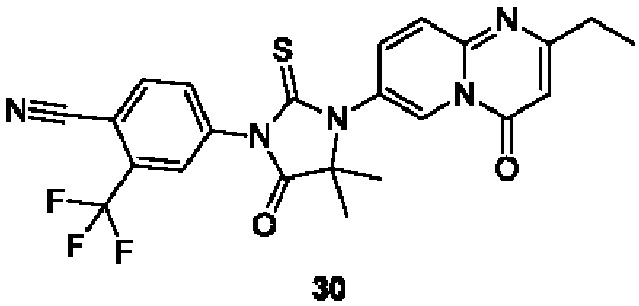
1) Синтез Соединения 30-3

Соединение 30-1 (20,00 г) и Соединение 30-2 (16,67 г) добавляли в уксусную кислоту (250 мл). Затем полученную смесь нагревали до 120°C и перемешивали при этой температуре в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении, разбавляли 200 мл воды и экстрагировали этилацетатом (200 мл × 3). После расслоения жидкости органические фазы собирали и объединяли. Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Концентрат суспендировали в петролейном эфире с получением Соединения 30-3. 1H ЯМР (400 МГц, CDCl3) δ м.д. 9.16 (d, J=1.98 Гц, 1H), 7.73 (dd, J=9.48, 2.21 Гц, 1H), 7.48 (d, J=9.48 Гц, 1Н), 6.39 (s, 1H), 2.72 (q, J=7.64 Гц, 2Н), 1.32 (t, Г=7.61 Гц, 3Н).
2) Синтез Соединения 30-4

Соединение 30-3 (1,50 г), Соединение 1-4 (916 мг), хлорид одновалентной меди (58 мг), 2-ацетилциклогексанон (83 мг) и карбонат калия (2,05 г) добавляли в пробирку для микроволновой обработки. Затем добавляли растворитель DMF (15 мл) и воду (3 мл), и полученную смесь выдерживали при 130°C для реакции под воздействием микроволнового излучения в течение 1 ч. Реакционную смесь фильтровали, и осадок на фильтре промывали DMF (10 мл × 3). Объединенный фильтрат концентрировали с получением Соединения 30-4. ЖХ/МС (ЭРИ) m/z: 276 (М+1).
3) Синтез Соединения 30-5
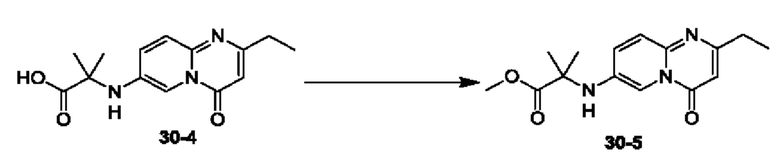
Раствор метанол/соляная кислота (100 мл) добавляли к Соединению 30-4 (3,76 г). Полученную смесь нагревали до 70°C и перемешивали при этой температуре в течение 16 ч. Реакционную смесь концентрировали, доводили до рН=7 насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом (25 мл × 3). Органические фазы объединяли и последовательно промывали насыщенным рассолом (25 мл × 3), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Концентрат очищали колоночной хроматографией с получением Соединения 30-5. ЖХ/МС (ЭРИ) m/z: 290 (М+1).
4) Синтез Соединения 30

Соединение 30-5 (200 мг) и Соединение 1-7 (315 мг) растворяли в смешанном растворе метилбензола (4 мл) и DMF (1 мл), и полученную смесь нагревали до 120°C и перемешивали при этой температуре в атмосфере азота в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении, растворяли в ацетонитриле и очищали препаративной ВЭЖХ с получением Соединения 30. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 9.02 (d, J=1.98 Гц, 1H), 8.02 (d, J=8.38 Гц, 1H), 7.96 (s, 1Н), 7.82-7.88 (m, 1H), 7.67-7.73 (m, 1H), 7.60-7.66 (m, 1H), 6.44 (s, 1H), 2.77 (q, J=7.57 Гц, 2Н), 1.35 (t, J=7.61 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 486 (М+1).
Пример 30. Синтез Соединения 31

1) Синтез Соединения 31-1

В сухую одногорлую колбу добавляли диацетат йодбензола (16,09 г) и метанол (250 мл), затем добавляли по каплям раствор диэтилэфират трифторида бора (7,09 г), и затем добавляли Соединение 30-2 (6,86 г). Полученную смесь перемешивали при 25°C в течение 28 ч. После завершения реакции реакционную смесь концентрировали. Добавляли 50 мл насыщенного водного раствора бикарбоната натрия, и полученную смесь экстрагировали этилацетатом (75 мл × 3). После разделения жидких фаз органическую фазу промывали 50 мл насыщенного рассола, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Концентрат очищали колоночной хроматографией с получением Соединения 31-1. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 4.27-4.18 (m, 3Н), 3.43 (s, 3Н), 2.67-2.51 (m, 2Н), 1.26 (t, J=7.2 Гц, 3Н), 1.02 (t, J=7.3 Гц, 3Н), ЖХ/МС (ЭРИ) m/z: 175 (М+1).
2) Синтез Соединения 31-4

В сухую реакционную колбу добавляли Соединение 31-1 (2,15 г) и Соединение 31-2 (2,14 г) и затем добавляли этанол (22 мл) и уксусную кислоту (2,2 мл). Полученную смесь нагревали до 90°C, перемешивали в течение 72 ч и концентрировали досуха для удаления растворителя. К остатку добавляли 50 мл воды, и полученную смесь основательно перемешивали и экстрагировали этилацетатом (30 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали, и концентрат очищали колоночной хроматографией с получением Соединения 31-4. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 9.08-9.07 (m, 1H), 7.59 (dd, J=2.2, 9.5 Гц, 1H), 7.44 (dd, J=0.7, 9.5 Гц, 1Н), 3.97 (s, 3Н), 2.81 (q, J=7.7 Гц, 2Н), 1.27 (t, J=7.6 Гц, 3Н).
3) Синтез Соединения 31-5

В пробирке для микроволновой обработки Соединение 31-4 (250 мг), Соединение 2-аминоизмасляную кислоту (149 мг), карбонат калия (332 мг), хлорид одновалентной меди (19 мг) и 2-ацетилциклогексанон (27 мг, 192 мкмоль) растворяли в смешанном растворителе DMF (5 мл) и воде (0,5 мл), и полученную смесь выдерживали при 130°C для реакции под воздействием микроволнового излучения в течение 1,5 ч. Реакционную смесь охлаждали и затем фильтровали. 12 мл воды добавляли в фильтрат, который затем экстрагировали этилацетатом (20 мл × 3), и водную фазу концентрировали при пониженном давлении с получением Соединения 31-5. ЖХ/МС (ЭРИ) m/z: 306 (М+1).
4) Синтез Соединения 31-6

В предварительно высушенную одногорлую колбу добавляли Соединение 31-5 (5,50 г) и раствор соляной кислоты в метаноле (4 н., 50 мл), и полученную смесь нагревали до 90°C и перемешивали в течение 12 ч под защитой азота. Концентрированием при пониженном давлении получали твердый остаток. Твердый остаток растворяли в 100 мл этилацетата и затем промывали насыщенным водным раствором бикарбоната натрия (50 мл × 1). После разделения жидких фаз органическую фазу собирали, промывали насыщенным рассолом (50 мл × 1), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали, и полученный концентрат очищали колоночной хроматографией с получением Соединения 31-6. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.15 (d, J=2.2 Гц, 1H), 7.44 (d, J=9.5 Гц, 1H), 7.15 (dd, J=2.8, 9.6 Гц, 1H), 4.28 (s, 1H), 3.95 (s, 3Н), 3.77 (s, 3Н), 2.78 (q, J=7.6 Гц, 2Н), 1.62 (s, 6Н), 1.26 (t, J=7.6 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 320 (М+1).
5) Синтез Соединения 31

В сухую одногорлую колбу добавляли Соединение 31-6 (200 мг), DMF (1,5 мл) и метилбензол (6 мл) и затем добавляли Соединение 1-7 (429 мг). Под защитой азота полученную смесь нагревали до 80°C и перемешивали при этой температуре в течение 3 ч. После концентрирования при пониженном давлении концентрат очищали методом препаративной ВЭЖХ с получением Соединения 31. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.97 (d, 3=2.2 Гц, 1H), 8.02 (d, J=8.2 Гц, 1H), 7.97 (s, 1H), 7.85 (br d, J=8.4 Гц, 1H), 7.68 (d, J=9.5 Гц, 1H), 7.51 (dd, J=2.2, 9.5 Гц, 1H), 4.02 (s, 3H), 2.88 (q, J=7.6 Гц, 2H), 1.69 (s, 6H), 1.32 (t, J=7.6 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 516 (M+1).
Пример 31. Синтез Соединения 32

1) Синтез Соединения 32-1

В сухую реакционную колбу добавляли Соединение 19-2 (1,00 г), метил 2-гидроксиацетат (466 мг) и тетрагидрофуран (10 мл) и затем порциями добавляли NaH (207 мг, чистота 60%). Реакционную смесь подвергали реакции при 20°C в течение 1 ч, затем разбавляли насыщенным водным раствором хлорида аммония (50 мл) и экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли и концентрировали. Полученный концентрат очищали колоночной хроматографией с получением Соединения 32-1. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.16-8.20 (m, 1H), 7.67 (dd, J=9.54, 2.13 Гц, 1H), 5.15 (s, 2Н), 3.82 (s, 3Н), 2.99 (q, J=7.57 Гц, 2Н), 1.37 (t, J=7.53 Гц, 3Н).
2) Синтез Соединения 32-2

В пробирку для микроволновой обработки добавляли Соединение 32-1 (500 мг), Соединение 1-4 (225 мг), хлорид одновалентной меди (14 мг), 2-ацетилциклогексанон (20 мг) и карбонат калия (402 мг) и затем добавляли DMF (4 мл) и воду (0,5 мл). После продувки азотом в течение 1 мин полученную смесь выдерживали при 130°C для реакции под воздействием микроволнового излучения в течение 1 ч. Реакционную смесь фильтровали в присутствии целита, и осадок на фильтре промывали DMF (5 мл × 2). Фильтрат собирали и концентрировали досуха при пониженном давлении, с получением Соединения 32-2. ЖХ/МС (ЭРИ) m/z: 352 (М+1).
3) Синтез Соединения 32-3
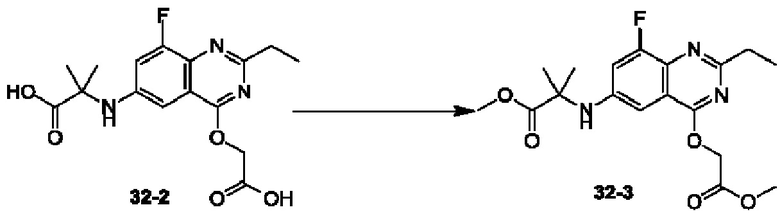
Раствор TMSCHN2 в н-гексане (2М, 4,70 мл) добавляли по каплям в раствор Соединения 32-2 (1,10 г) в дихлорметане (20 мл) и метаноле (2 мл). Полученную смесь подвергали реакции при 20°C в течение 2 ч. Добавляли TMSCHN2 (2М, 4,70 мл), и затем смесь перемешивали в течение 16 ч. Реакционную смесь концентрировали досуха. Остаток очищали колоночной хроматографией с получением Соединения 32-3. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 6.84-6.91 (m, 2Н) 5.04-5.12 (m, 2 Н) 4.48 (s, 1H) 3.79 (s, 3Н) 3.75 (s, 3Н) 2.91 (q, J=7.53 Гц, 2Н) 1.64 (s, 6Н) 1.32 (t, J=7.59 Гц, 3Н).
4) Синтез Соединения 32-4

В сухую одногорлую колбу добавляли Соединение 32-3 (630 мг) и Соединение 1-7 (378 мг) и затем добавляли метилбензол (4 мл) и DMF (1 мл). Полученную смесь подвергали реакции при 120°C в течение 24 ч. Реакционную смесь концентрировали при пониженном давлении. Полученный остаток очищали методом препаративной ВЭЖХ с получением Соединения 32-4. ЖХ/МС (ЭРИ) m/z: 576 (М+1).
5) Синтез Соединения 32-5

В предварительно высушенную одногорлую колбу добавляли Соединение 32-4 (55 мг) и тетрагидрофуран (4 мл) и затем добавляли раствор гидроксида лития моногидрата (6 мг) в воде (1 мл). Полученную смесь перемешивали при 25°C в течение 2 ч. Насыщенный раствор хлорида аммония (5 мл) и этилацетат (10 мл) добавляли в реакционную смесь для разделения жидких фаз. Водную фазу экстрагировали этилацетатом (5 мл × 3). Органические фазы объединяли, сушили над сульфатом натрия и концентрировали. Полученный остаток очищали методом препаративной ТСХ с получением Соединения 32-5. ЖХ/МС (ЭРИ) m/z: 562 (М+1).
6) Синтез Соединения 32

В сухую реакционную колбу вместимостью 10 мл добавляли Соединение 32-5 (37 мг) и дихлорметан (2 мл) и затем добавляли HATU (30 мг), хлорид аммония (5 мг), и триэтиламин (20 мг) при 0°C. Полученную смесь перемешивали при 25°C в течение 16 ч, затем разбавляли 4 мл воды, экстрагировали дихлорметаном (5 мл × 3), сушили и концентрировали. Полученный остаток отделяли методом препаративной ВЭЖХ с получением Соединения 32. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.99 (d, J=8.16 Гц, 1H), 7.95 (s, 1H), 7.90 (s, 1H), 7.83 (dd, J=8.27, 1.87 Гц, 1H), 7.48 (dd, J=9.81, 2.09 Гц, 1H), 6.03 (br s, 1Н), 5.59 (br s, 1H), 5.13 (s, 2Н), 3.05 (q, J=7.57 Гц, 2Н),1.66 (s, 6Н), 1.40 (t, J=7.61 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 561 (М+1).
Пример 32. Синтез Соединения 33

1) Синтез Соединения 33
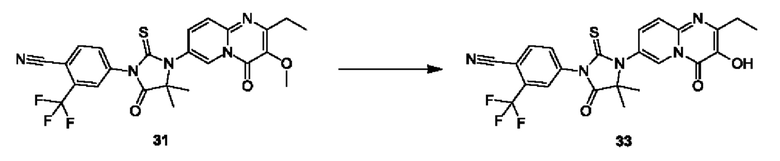
В сухую одногорлую колбу добавляли Соединение 31 (50 мг) и дихлорметан (2 мл) и затем добавляли трибромид бора (97 мг) в ледяной бане. Полученную смесь поддерживали при температуре 0°C и перемешивали в течение 2 ч. При 0°C 0,5 мл ледяной воды добавляли в реакционную смесь, чтобы погасить реакцию, и затем добавляли небольшое количество твердого бикарбоната натрия до доведения рН до 7-8. После удаления дихлорметана при пониженном давлении при 0°C 3 мл DMSO добавляли к остатку для растворения смеси. После фильтрования полученный раствор очищали методом препаративной ВЭЖХ с получением Соединения 33. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.83 (d, J=1.5 Гц, 1H), 8.02 (d, J=8.4 Гц, 1H), 7.97 (d, J=2.0 Гц, 1H), 7.85 (dd, J=2.0, 8.2 Гц, 1Н), 7.70-7.67 (m, 1H), 7.40 (dd, J=2.3, 9.6 Гц, 1H), 2.92 (q, J=7.6 Гц, 2Н), 1.69 (s, 6Н), 1.35 (t, J=7.6 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 502 (М+1).
Пример 33. Синтез Соединения 34

1) Синтез Соединения 34-2

Соединение 31-1 (5,03 г) добавляли в раствор Соединения 34-1 (2,00 г) в уксусной кислоте (20 мл), и полученную смесь нагревали до 110°C и перемешивали в течение 16 ч. Реакционную смесь концентрировали. Концентрат разбавляли дихлорметаном (30 мл), промывали насыщенным раствором бикарбоната натрия (30 мл × 2) и насыщенным рассолом (30 мл) в указанном порядке, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Концентрат очищали колоночной флэш-хроматографией (модель: ISCO-RF150) с получением Соединения 34-2. ЖХ/МС (ЭРИ) m/z: 301 (М+1).
2) Синтез Соединения 34-3

Согласно Синтезу Соединения 31-5, Соединение 34-3 было получено с использованием Соединения 34-2 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 324 (М+1).
3) Синтез Соединения 34-4

Согласно Синтезу Соединения 31-6, Соединение 34-4 было получено с использованием Соединения 34-3 в качестве исходного вещества. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.92 (dd, J=2.38, 0.88 Гц, 1 Н), 6.90 (dd, J=10.42, 2.38 Гц, 1 Н), 3.89-3.94 (m, 1 Н), 3.91 (s, 2 Н), 3.73 (s, 3 Н), 2.77 (q, J=7.70 Гц, 2 Н), 1.57 (s, 6 Н), 1.21 (t, J=7.53 Гц, 3 Н); ЖХ/МС (ЭРИ) m/z: 337,9 (М+1).
4) Синтез Соединения 34

Согласно Синтезу Соединения 31, Соединение 34 было получено с использованием Соединения 34-4 в качестве исходного вещества. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.74-8.85 (m, 1 Н), 8.02 (d, J=8.28 Гц, 1 Н), 7.95 (d, J=2.01 Гц, 1 H), 7.83 (dd, Г=8.28, 2.01 Гц, 1 Н), 7.27-7.31 (m, 1 Н), 4.03 (s, 3 Н), 2.91 (q, J=7.53 Гц, 2 Н), 1.69 (s, 6 Н), 1.33 (t, J=7.53 Гц, 3 Н); ЖХ/МС (ЭРИ) m/z: 534 (М+1).
Пример 34. Синтез Соединения 35

1) Синтез Соединения 35-1
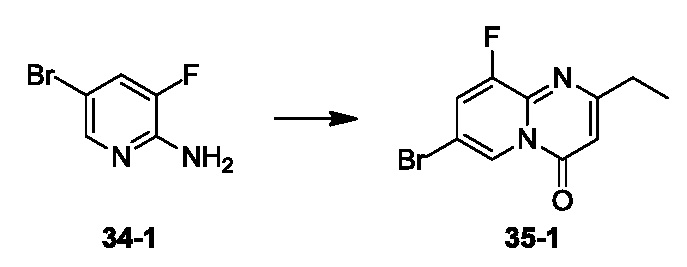
Метил пропионил ацетат (4,00 г) добавляли в раствор Соединения 34-1 (4,00 г) в уксусной кислоте (40 мл). Полученную смесь нагревали до 110°C и перемешивали в течение 94 ч. Метилпропионилацетат (8,26 г) добавляли в реакционную смесь, и реакционную смесь дополнительно перемешивали в течение 16 ч. Реакционную смесь концентрировали. Концентрат разбавляли этилацетатом (80 мл) и добавляли насыщенный водный раствор бикарбоната натрия (80 мл). После разделения жидких фаз органическую фазу промывали насыщенным рассолом (80 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 35-1. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.89 (s, 1H), 7.45 (dd, J=2.0, 8.0 Гц, 1Н), 6.36 (s, 1Н), 2.70 (q, J=7.5 Гц, 2Н), 1.25 (t, J=7.5 Гц, 3Н).
2) Синтез Соединения 35-2

Соединение 35-1 (400 мг), Соединение 1-4 (228 мг), карбонат калия (510 мг), хлорид одновалентной меди (30 мг), 2-ацетилциклогексанон (42 мг), N,N-диметилформамид (4 мл) и воду (0,2 мл) добавляли в пробирку для микроволновой обработки. Пробирку для микроволновой обработки герметично закрывали и выдерживали при 130°C для реакции под воздействием микроволнового излучения в течение 30 мин. Реакционную смесь фильтровали и промывали этилацетатом (20 мл). Фильтрат концентрировали. 1 н. соляную кислоту добавляли к остатку, полученному после концентрирования (для доведения рН до 6-7), и полученную смесь концентрировали. Смесь дихлорметан/метанол (40 мл, 10/1) добавляли к остатку, полученному после концентрирования, и полученную смесь сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением Соединения 35-2. ЖХ/МС (ЭРИ) m/z: 294 (М+1).
3) Синтез Соединения 35-3

Раствор триметилсилилдиазометана в н-гексане (2М, 1,2 мл) добавляли в раствор Соединения 35-2 (500 мг) в дихлорметане (10 мл) и метаноле (1 мл). Полученную смесь перемешивали при 15°C в течение 2 ч. Реакционную смесь концентрировали. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 35-3. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.01 (d, J=1.3 Гц, 1H), 7.04 (dd, J=2.4, 10.4 Гц, 1H), 6.26 (s, 1H), 4.43 (br s, 1H), 3.72 (s, 3Н), 2.66 (q, J=7.5 Гц, 2Н), 1.56 (s, 6Н), 1.23 (t, J=7.5 Гц, 3Н).
4) Синтез Соединения 35

Смешанный раствор Соединения 35-3 (100 мг) и Соединения 1-7 (297 мг) в N,N-диметилформамиде (0,5 мл) и метилбензоле (2 мл) нагревали до 120°C и перемешивали в течение 16 ч. Метанол (0,5 мл) добавляли в реакционную смесь, и смесь перемешивали в течение 30 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной тонкослойной хроматографии с получением Соединения 35. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.76 (d, J=1.8 Гц, 1H), 7.94 (d, J=8.3 Гц, 1H), 7.88 (d, J=2.0 Гц, 1H), 7.76 (dd, J=2.0, 8.3 Гц, 1H), 7.33 (dd, J=2.3, 8.8 Гц, 1H), 6.42 (s, 1H), 2.74 (q, J=7.7 Гц, 2Н), 1.62 (s, 6Н), 1.28 (t, J=7.5 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 504 (М+1).
Пример 35. Синтез Соединения 36

1) Синтез Соединения 36-1

В сухую одногорлую колбу вместимостью 50 мл добавляли Соединение 19-1 (1,00 г), Соединение 1-4 (571 мг), воду (2,5 мл) и DMF (10 мл), и затем добавляли хлорид одновалентной меди (36 мг), 2-ацетилциклогексанон (52 мг) и карбонат калия (1,02 г). Полученную смесь перемешивали при 130°C в течение 90 мин. Добавляли воду (10 мл), смесь доводили до рН=2-3 разбавленной 1М соляной кислотой и добавляли этилацетат (40 мл). Водную фазу экстрагировали этилацетатом (40 мл × 3). Органические фазы объединяли и концентрировали для удаления растворителя. В концентрат добавляли дихлорметан (5 мл) и перемешивали в течение 5 мин. Медленно по каплям добавляли петролейный эфир (10 мл), и полученную смесь перемешивали в течение 10 мин. Смесь фильтровали, и осадок на фильтре сушили с получением Соединения 36-1. ЖХ/МС (ЭРИ) m/z: 294 (М+1).
2) Синтез Соединения 36-2

В сухую одногорлую колбу вместимостью 50 мл добавляли Соединение 36-1 (3,00 г) и раствор соляная кислота/метанол (30 мл), и полученную смесь перемешивали при 60°C в течение 16 ч и затем концентрировали. К остатку, полученному после концентрирования, добавляли этилацетат (20 мл) и насыщенный раствор карбоната калия (20 мл). После разделения жидких фаз полученную органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением Соединения 36-2. ЖХ/МС (ЭРИ) m/z: 308 (М+1).
3) Синтез Соединения 36-3

В одногорлую колбу добавляли Соединение 36-2 (2,10 г), Соединение 1-7 (4,68 г), метилбензол (20 мл) и DMF (5 мл), и полученную смесь подвергали реакции при 120°C в течение 16 ч под защитой азота и затем концентрировали для удаления метилбензола и DMF. Остаток, полученный после концентрирования, очищали колоночной хроматографией с получением Соединения 36-3. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.97-8.06 (m, 4 Н), 7.86 (dd, J=8.22, 1.82 Гц, 1 Н), 7.48 (dd, J=9.79, 2.26 Гц, 1 Н), 2.85-2.91 (m, 2 Н), 1.67 (s, 6 Н), 1.47 (t, J=7.59 Гц, 3 Н).
4) Синтез Соединения 36-4

В одногорлую колбу добавляли Соединение 36-3 (200 мг) и оксихлорид фосфора (3,30 г). Под защитой азота добавляли N,N-диизопропилэтиламин (80 мг), и полученную смесь подвергали реакции при 110°C в течение 0,5 ч. Оксихлорид фосфора удаляли из реакционной смеси при пониженном давлении. Полученный остаток растворяли в 10 мл дихлорметана в бане лед-вода, и затем добавляли 30 мл насыщенного раствора бикарбоната натрия. После разделения жидких фаз водную фазу экстрагировали дихлорметаном (10 мл × 3). Органические фазы объединяли, промывали 10 мл насыщенного рассола, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении при 45°C с получением Соединения 36-4. ЖХ/МС (ЭРИ) m/z: 522 (М+1).
5) Синтез Соединения 36

В реакционную колбу добавляли Соединение 36-4 (100 мг), Соединение 36-5 (25 мг) и тетрагидрофуран (1 мл). Под защитой азота добавляли гидрид натрия (11 мг, чистота 60%) при 0°C, и полученную смесь подвергали реакции при 20°C в течение 0,5 ч. Реакционную смесь гасили насыщенным раствором хлорида аммония (3 мл). После экстрагирования этилацетатом (3 мл × 3) органическую фазу промывали насыщенным рассолом (1 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при 45°C. Остаток, полученный после концентрирования, очищали методом препаративной ВЭЖХ с получением Соединения 36. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.02 (d, J=8.16 Гц, 1H), 7.99 (s, 1H), 7.94 (s, 1H), 7.86 (br d, J=8.41 Гц, 1H), 7.48 (dd, J=10.04, 2.26 Гц, 1H), 4.76 (s, 2Н), 4.05 (s, 1H), 3.06 (q, J=7.49 Гц, 2Н), 1.68 (s, 6Н), 1.43 (t, J=7.59 Гц, 3Н), 0.98-1.02 (m, 2Н), 0.82-0.86 (m, 2Н). ЖХ/МС (ЭРИ) m/z: 574 (М+1).
Пример 36. Синтез Соединения 37

1) Синтез Соединения 37-1

Трибромид бора (2,40 г) добавляли по каплям в раствор Соединения 31-6 (300 мг) в дихлорметане (8 мл) при 0°C. Полученную смесь перемешивали при 0°C в течение 2 ч. Реакционную смесь гасили 3 г ледяной воды, и реакционную смесь концентрировали при пониженном давлении с получением Соединения 37-1. ЖХ/МС (ЭРИ) m/z: 292 (М+1).
2) Синтез Соединения 37-2

Смесь метанол/соляная кислота (4М, 100 мл) в качестве растворителя добавляли к Соединению 37-1 (2,02 г), и полученную смесь перемешивали при 70°C в течение 16 ч. Реакционную смесь концентрировали. Остаток, полученный после концентрирования, нейтрализовали до нейтрального рН насыщенным раствором бикарбоната натрия и фильтровали. Фильтрат подвергали сублимационной сушке с получением Соединения 37-2. ЖХ/МС (ЭРИ) m/z: 306 (М+1).
3) Синтез Соединения 37-3

Соединение 37-2 (200 мг) и Соединение 1-7 (448 мг) растворяли в смешанном растворе DMF (4 мл) и метилбензола (1 мл). Полученную смесь нагревали до 120°C и перемешивали в течение 16 ч. Реакционную смесь концентрировали, растворяли в ацетонитриле и очищали методом препаративной ВЭЖХ с получением Соединения 37-3. ЖХ/МС (ЭРИ) m/z: 502 (М+1).
4) Синтез Соединения 37

Соединение 37-3 (75 мг), Соединение 37-4 (25 мг) и карбонат калия (62 мг) растворяли в DMF (1 мл). Полученную смесь нагревали до 110°C и перемешивали при этой температуре в течение 3 ч. Соединение 37 было получено методом препаративной ВЭЖХ. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.95 (d, J=2.21 Гц, 1H), 8.02 (d, J=8.16 Гц, 1H), 7.97 (s, 1H), 7.82-7.88 (m, 1Н), 7.68 (d, J=9.26 Гц, 1H), 7.51 (dd, J=9.48, 2.43 Гц, 1H), 4.37-4.42 (m, 2Н), 3.72-3.77 (m, 2Н), 3.46 (s, 3Н), 2.93 (q, J=7.50 Гц, 2Н), 1.68 (s, 6Н), 1.33 (t, J=7.61 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 560 (М+1).
Пример 37. Синтез Соединения 38

1) Синтез Соединения 38

Соединение 37-3 (100 мг), Соединение 38-1 (41 мг) и карбонат калия (82 мг) добавляли в DMF (1 мл), и полученную смесь нагревали до 110°C и перемешивали при этой температуре в течение 3 ч. Соединение 38 было получено путем очистки методом препаративной ВЭЖХ. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.94 (d, J=1.98 Гц, 1H), 8.00 (d, J=8.38 Гц, 1H), 7.94 (s, 1H), 7.83 (br d, J=8.38 Гц, 1H), 7.69 (d, J=9.70 Гц, 1H), 7.54 (dd, J=9.59, 2.32 Гц, 1Н), 4.19-4.25 (m, 2Н), 4.05-4.12 (m, 1H), 3.87-3.94 (m, 2Н), 2.89 (q, J=7.50 Гц, 2Н), 1.67 (s, 6Н), 1.33 (t, J=7.61 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 546 (М+1).
Пример 38. Синтез Соединения 39

1) Синтез Соединения 39-1
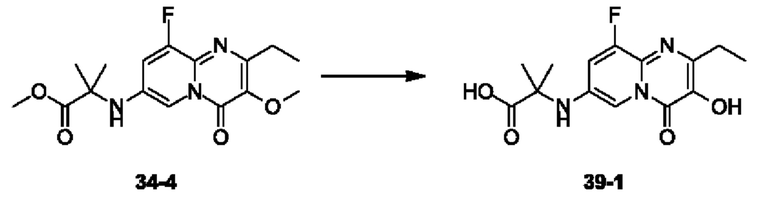
Трибромид бора (0,4 мл) добавляли в раствор Соединения 34-4 (320 мг) в безводном дихлорметане (8 мл) при 0°C. Полученную смесь перемешивали при 0°C в течение 2 ч, разбавляли дихлорметаном (40 мл), медленно вливали в воду (20 мл) и экстрагировали смесью дихлорметан/метанол (10/1, 40 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением Соединения 39-1. ЖХ/МС (ЭРИ) m/z: 310 (М+1).
2) Синтез Соединения 39-2

Раствор триметилсилилдиазометана в н-гексане (2М, 0,3 мл) добавляли в раствор Соединения 39-1 (120 мг) в дихлорметане (3 мл) и метаноле (0,3 мл). Полученную смесь перемешивали при 20°C в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной тонкослойной хроматографии с получением Соединения 39-2. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.73 (s, 1Н), 6.80 (br d, 3=9.5 Гц, 1H), 6.22 (br s, 1H), 4.21 (br s, 1H), 3.73 (s, 3H), 2.81 (q, J=7.5 Гц, 2H), 1.57 (s, 6H), 1.24 (t, J=7.7 Гц, 3Н).
3) Синтез Соединения 39

Смесь Соединения 39-2 (30 мг), Соединения 1-7 (100 мг), метилбензола (1 мл) и N,N-диметилформамида (0,2 мл) нагревали до 110°C и перемешивали в течение 16 ч. Метанол (1 мл) добавляли в реакционную смесь, и смесь перемешивали в течение 30 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, отделяли и очищали последовательно с использованием пластины для препаративной тонкослойной хроматографии и методом препаративной ВЭЖХ с получением Соединения 39. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.58 (s, 1H), 7.94 (d, J=8.3 Гц, 1Н), 7.88 (s, 1H), 7.76 (br d, J=8.0 Гц, 1H), 7.11 (dd, J=2.0, 9.0 Гц, 1Н), 2.88 (q, J=7.4 Гц, 2Н), 1.62 (s, 6Н), 1.28 (t, J=7.7 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 520 (М+1).
Пример 39. Синтез Соединения 40
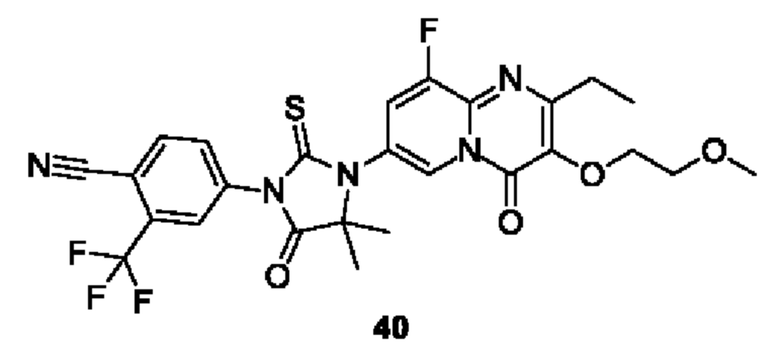
1) Синтез Соединения 40
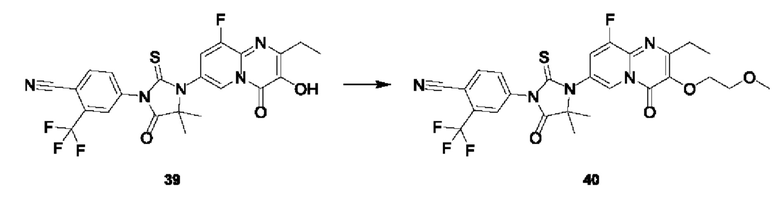
1-Бром-2-метокси-этан (41 мг) и карбонат калия (54 мг) добавляли в раствор Соединения 39 (100 мг) в N,N-диметилформамиде (2 мл). Полученную смесь перемешивали при 80°C в течение 1 ч и фильтровали. Фильтрат отделяли и очищали методом препаративной ВЭЖХ с получением Соединения 40. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.69 (br s, 1H), 8.03-7.69 (m, 3Н), 7.20 (br s, 1H), 4.33 (br s, 2Н), 3.66 (br s, 2H), 3.37 (br s, 3Н), 2.88 (br d, J=7.3 Гц, 2H), 1.61 (br s, 6H), 1.26 (br t, J=7.0 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 578 (М+1).
Пример 40. Синтез Соединения 41

1) Синтез Соединения 41

Карбонат калия (60 мг) добавляли в раствор Соединения 39 (100 мг) и 2-бромэтанола (40 мг) в DMF (2 мл), и полученную смесь нагревали до 100°C и перемешивали в течение 1 ч под защитой азота. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Концентрат очищали последовательно с использованием пластины для препаративной ТСХ и методом препаративной ВЭЖХ с получением Соединения 41. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.80 (d, J=1.5 Гц, 1Н), 8.04 (d, J=8.3 Гц, 1H), 7.97 (s, 1H), 7.85 (dd, J=8.3, 2.0 Гц, 1H), 7.35 (dd, J=8.8, 2.0 Гц, 1H), 4.23-4.35 (m, 2Н), 3.95 (br s, 2Н), 3.77 (br s, 1H), 2.97 (q, J=7.7 Гц, 2Н), 1.71 (s, 6Н), 1.38 м.д. (t, J=7.5 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 564 (М+1).
Пример 41. Синтез Соединения 42

1) Синтез Соединения 42-1

Карбонат калия (110 мг) добавляли в раствор Соединения 39 (200 мг) и этил-2-бромацетата (100 мг) в DMF (5 мл), и полученную смесь нагревали до 80°C и перемешивали в течение 1 ч под защитой азота. Реакционную смесь охлаждали до комнатной температуры и фильтровали. Осадок на фильтре промывали этилацетатом (2 мл). Фильтрат концентрировали с получением Соединения 42-1. ЖХ/МС (ЭРИ) m/z: 606 (М+1).
2) Синтез Соединения 42-2

Водный раствор гидроксида лития моногидрата (1М, 0,7 мл) добавляли в раствор Соединения 42-1 (200 мг) в тетрагидрофуране (5 мл), и полученную смесь перемешивали при 26°C в течение 1 ч под защитой азота. Реакционную смесь подкисляли до рН=5-6 разбавленным водным раствором соляной кислоты (1М) и экстрагировали этилацетатом (20 мл × 3). Объединенную органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением Соединения 42-2. ЖХ/МС (ЭРИ) m/z: 578 (М+1).
3) Синтез Соединения 42

Метиламина гидрохлорид (18 мг) добавляли в раствор Соединения 42-2 (100 мг), HATU (80 мг) и триэтиламина (50 мг, 494,12 мкмоль) в дихлорметане (5 мл), и полученную смесь перемешивали при 26°C в течение 1 ч. Реакционную смесь подкисляли до рН=5-6 разбавленным водным раствором соляной кислоты (1М) и экстрагировали этилацетатом (20 мл × 3). Объединенную органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток, полученный после концентрирования, очищали методом препаративной ТСХ и методом препаративной ВЭЖХ с получением Соединения 42. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.68 (s, 1Н), 7.95 (d, J=8.3 Гц, 1Н), 7.88 (d, J=1.5 Гц, 1H), 7.76 (dd, J=8.3, 1.8 Гц, 1H), 7.31-7.36 (m, 1H), 7.29 (dd, J=8.8, 2.0 Гц, 1H), 4.51 (s, 2Н), 2.90 (d, J=5.0 Гц, 3Н), 2.84 (q, J=7.7 Гц, 2Н), 1.62 (s, 6Н), 1.29 м.д. (t, J=7.5 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 591 (М+1).
Пример 42. Синтез Соединения 43

1) Синтез Соединения 43

Аммиачную воду (30 мг) добавляли в раствор Соединения 42-2 (100 мг), HATU (79 мг) и триэтиламина (53 мг) в DMF (5 мл), и полученную смесь перемешивали при 26°C в течение 1 ч. Реакционную смесь подкисляли до рН=5-6 разбавленным водным раствором соляной кислоты (1М) и экстрагировали этилацетатом (20 мл × 3). Объединенную органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Концентрат очищали методом препаративной ТСХ и методом препаративной ВЭЖХ с получением Соединения 43. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.69 (s, 1H), 7.91-7.99 (m, 1H), 7.88 (s, 1Н), 7.76 (dd, J=8.3, 1.8 Гц, 1Н), 7.29 (dd, J=8.8, 2.0 Гц, 2Н), 5.62 (br s, 1Н), 4.53 (s, 2H), 2.77-2.95 (m, 1H), 1.62 (s, 6H), 1.26-1.34 м.д. (m, 3Н); ЖХ/МС (ЭРИ) m/z: 577.0 (М+1).
Пример 43. Синтез Соединения 44

1) Синтез Соединения 44-2 и Соединение 44-3

В сухую одногорлую колбу добавляли Соединение 7-5 (5,00 г), бензойную кислоту (21,28 г) и метилбензол (5 мл). Под защитой азота добавляли дифенилфосфат (1,45 г), и полученную смесь подвергали реакции при 25°C в течение 16 ч. Реакционную смесь разделяли последовательно колоночной хроматографией и методом препаративной СФХ (сверхкритическая флюидная хроматография) (модель прибора: Thar SFC80 preparative SFC) с получением Соединения 44-2 и Соединения 44-3. ЖХ/МС (ЭРИ) m/z: 209(М+1).
2) Синтез Соединения 44-4

В сухую реакционную колбу добавляли Соединение 36-4 (200 мг), Соединение 44-2 (120 мг) и тетрагидрофуран (2 мл). Под защитой азота добавляли гидрид натрия (23 мг, чистота 60%), и полученную смесь подвергали реакции при 25°C в течение 0,5 ч. Реакционную смесь гасили насыщенным раствором хлорида аммония (2 мл). После экстрагирования дихлорметаном (3 мл × 3) органическую фазу промывали насыщенным рассолом (10 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении при 45°C. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной ТСХ с получением Соединения 44-4. ЖХ/МС (ЭРИ) m/z: 694 (М+1).
3) Синтез Соединения 44

В сухую реакционную колбу добавляли Соединение 44-4 (140 мг), LiOH.Н2О (13 мг), воду (0,8 мл) и тетрагидрофуран (1,5 мл), и полученную смесь подвергали реакции при 25°C в течение 3 ч под защитой азота. Реакционную смесь напрямую сушили в центробежной сушилке и очищали методом препаративной ВЭЖХ с получением Соединения 44. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.02 (d, J=8.38 Гц, 1Н), 7.98 (s, 1H), 7.83-7.88 (m, 2Н), 7.49 (dd, J=9.92, 2.21 Гц, 1H), 5.46-5.50 (m, 1H), 4.52 (br s, 1H), 4.36 (dd, J=11.03, 5.73 Гц, 1H), 4.23 (dd, J=10.03, 5.84 Гц, 1H), 4.16 (dd, J=10.58, 2.87 Гц, 1H), 3.82 (dd, J=9.81, 4.74 Гц, 1H), 3.40 (d, J=2.65 Гц, 1H), 3.09 (q, J=7.50 Гц, 2Н), 1.67 (s, 6Н), 1.45 (t, J=7.50 Гц, 3Н).
Пример 44. Синтез Соединения 45

1) Синтез Соединения 45-1

В сухую одногорлую колбу добавляли метанол (2 мл) и натрий (24 мг), и полученную смесь перемешивали при 25°C до исчезновения натрия. Затем свежеприготовленный раствор метоксида натрия в метаноле добавляли в сухую одногорлую колбу, заполненную Соединением 19-2 (300 мг), и полученную смесь перемешивали при 25°C в течение 2 ч. После фильтрования осадок на фильтре промывали этилацетатом (10 мл). Фильтрат собирали и концентрировали при пониженном давлении с получением Соединения 45-1. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.00 (dd, J=1.3, 2.0 Гц, 1H), 7.54 (dd, J=2.1, 9.6 Гц, 1H), 4.12-4.09 (m, 3Н), 2.93 (q, J=7.5 Гц, 2Н), 1.34 (t, J=7.6 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 285 (М+1).
2) Синтез Соединения 45-2

В сухую пробирку для микроволновой обработки добавляли Соединение 45-1 (888 мг), Соединение 3-2 (489 мг), карбонат цезия (2,54 г), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (180 мг), бис(дибензилиденацетон)палладий (179 мг) и метилбензол (18 мл). После продувки азотом в течение пяти минут полученную смесь подвергали воздействию микроволнового излучения и перемешивали при 130°C в течение 4 ч и затем концентрировали досуха при пониженном давлении для удаления растворителя. Остаток, полученный после концентрирования, очищали колоночной хроматографией с получением Соединения 45-2. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.19 (d, J=4.9 Гц, 1H), 7.20 (d, 3=4.9 Гц, 1H), 6.96 (dd, J=2.5, 11.6 Гц, 1H), 6.81-6.77 (m, 1H), 5.79 (s, 1H), 4.09 (s, 3H), 3.00-2.91 (m, 2H), 2.24 (s, 3H), 1.38 (t, J=7.5 Гц, 3Н). ЖХ/МС (ЭРИ)m/z: 347 (M+1).
3) Синтез Соединения 45-3

В сухую пробирку для микроволновой обработки добавляли Соединение 45-2 (190 мг), Соединение 2-5 (112 мг), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (31 мг), бис(дибензилиденацетон)палладий (31 мг) и карбонат цезия (357 мг) и затем добавляли метилбензол (5 мл). После продувки азотом в течение 5 минут пробирку для микроволновой обработки герметично закрывали. Полученную смесь подвергали воздействию микроволнового излучения и перемешивали при 130°С в течение 4 ч, и затем концентрировали досуха при пониженном давлении для удаления растворителя. Остаток, полученный после концентрирования, очищали колоночной хроматографией с получением неочищенного продукта соединения 45-3. Полученный неочищенный продукт суспендировали в 5 мл метил-трет-бутилового эфира с получением Соединения 45-3. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.27 (d, J=2.2 Гц, 1H), 8.15 (d, J=5.1 Гц, 1Н), 8.00 (dd, J=2.1, 8.7 Гц, 1H), 7.72 (d, J=8.8 Гц, 1H), 7.14 (dd, J=2.5, 12.5 Гц, 1H), 7.00 (d, J=5.7 Гц, 1H), 6.56-6.53 (m, 1Н), 4.02 (s, 3Н), 3.19 (s, 3Н), 2.85 (q, J=7.5 Гц, 2Н), 1.33 (t, J=7.6 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 497 (М+1).
4) Синтез Соединения 45-4

В сухую одногорлую колбу добавляли Соединение 45-3 (65 мг), тетрагидрофуран (0,5 мл) и метилбензол (0,5 мл) и затем добавляли трет-бутоксид натрия (50 мг). Полученную смесь перемешивали при 30°С в течение 1 ч, и затем добавляли тиофосген (45 мг) при 30°С. Полученную смесь перемешивали при 30°С в течение 1 ч под защитой азота и затем концентрировали досуха при пониженном давлении для удаления растворителя. Остаток, полученный после концентрирования, очищали колоночной хроматографией с получением Соединения 45-4. ЖХ/МС (ЭРИ) m/z: 539 (М+1).
5) Синтез Соединения 45

В сухую одногорлую колбу добавляли Соединение 45-4 (45 мг), соляную кислоту (2М, 2 мл) и тетрагидрофуран (2 мл). Полученную смесь перемешивали при 30°С в течение 1 ч под защитой азота и концентрировали досуха при пониженном давлении для удаления растворителя. Остаток, полученный после концентрирования, очищали методом препаративной ВЭЖХ с получением Соединения 45. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 10.55 (br s, 1H), 8.30 (br s, 1H), 8.17 (br d, J=19.6 Гц, 3Н), 8.09 (br s, 1H), 7.67 (br s, 1Н), 7.03 (br s, 1H), 2.87 (br s, 2Н), 1.99 (br s, 3Н), 1.47 (br s, 3Н). ЖХ/МС (ЭРИ) m/z: 525 (М+1).
Пример 45. Синтез Соединения 46

1) Синтез Соединения 46-1

В сухую реакционную колбу добавляли Соединение 36-4 (200 мг), Соединение 44-3 (120 мг) и тетрагидрофуран (2 мл). Под защитой азота добавляли гидрид натрия (23 мг, чистота 60%), и полученную смесь подвергали реакции при 25°С в течение 0,5 ч. Реакционную смесь гасили насыщенным раствором хлорида аммония (5 мл). После экстрагирования дихлорметаном (5 мл × 3) органическую фазу промывали насыщенным рассолом (10 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении при 45°С. Остаток очищали с использованием пластины для препаративной ТСХ с получением Соединения 46-1. ЖХ/МС (ЭРИ) m/z: 694 (М+1).
2) Синтез Соединения 46

В сухую реакционную колбу добавляли Соединение 46-1 (130 мг), гидроксида лития моногидрат (12 мг), воду (0,5 мл) и тетрагидрофуран (1 мл), и полученную смесь подвергали реакции при 25°С в течение 4 ч под защитой азота. Реакционную смесь напрямую концентрировали досуха, и остаток, полученный после концентрирования, очищали методом препаративной ВЭЖХ с получением Соединения 46. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.02 (d, J=8.38 Гц, 1H), 7.98 (s, 1H), 7.83-7.89 (m, 2Н), 7.49 (dd, J=9.81, 2.09 Гц, 1H), 5.45-5.51 (m, 1H), 4.52 (br s, 1Н), 4.36 (dd, J=10.80, 5.95 Гц, 1H), 4.23 (dd, J=9.92, 5.73 Гц, 1H), 4.16 (dd, J=10.80, 3.09 Гц, 1Н), 3.82 (dd, J=9.70, 4.85 Гц, 1H), 3.40 (d, J=2.65 Гц, 1H), 3.09 (q, J=7.57 Гц, 2Н), 1.67 (s, 6Н), 1.45 (t, J=7.61 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 590 (М+1).
Пример 46. Синтез Соединения 47

1) Синтез Соединения 47-2

Соединение 47-1 (23,00 г) и пропионилхлорид (50,14 г) последовательно добавляли в раствор этоксида натрия (22,13 г) и триэтиламина (1,10 г) в тетрагидрофуране (220 мл) при 5°С. После завершения добавления полученную смесь перемешивали при 25°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении и затем перегоняли при пониженном давлении с получением Соединения 47-2 путем сбора фракций при температурах 36°С, 40°С и 60°С в указанном порядке.
2) Синтез Соединения 47-3

Соединение 34-1 (5,00 г) и Соединение 47-2 (5,52 г) добавляли в полифосфорную кислоту (15,00 г), и полученную смесь нагревали до 110°С и перемешивали в течение 16 ч. Реакционную смесь разбавляли 50 мл воды, доводили до нейтрального рН насыщенным раствором гидроксида натрия и экстрагировали этилацетатом (30 мл × 3). Органическую фазу собирали, сушили, фильтровали и концентрировали. Остаток очищали колоночной хроматографией с получением Соединения 47-3. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.93 (t, J=1.65 Гц, 1Н), 7.49 (dd, J=8.16, 1.76 Гц, 1H), 2.90 (qd, J=7.61, 2.98 Гц, 2Н), 1.35 (t, J=7.61 Гц, 3Н).
3) Синтез Соединения 47-4

Соединение 47-3 (800 мг), трет-бутилкарбамат (972 мг), карбонат цезия (2,25 г), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (160,13 мг), и бис(дибензилиденацетон)-палладий (159 мг) добавляли в пробирку для микроволновой обработки, заполненную метилбензолом (8 мл). Полученную смесь подвергали реакции при 120°С для реакции под воздействием микроволнового излучения в течение 50 мин. Реакционную смесь концентрировали при пониженном давлении, и остаток, полученный после концентрирования, очищали колоночной хроматографией с получением Соединения 47-4. 1H ЯМР (400 МГц, CDCl3) δ м.д. 9.00 (s, 1H) 7.31-7.37 (m, 1H) 2.92 (qd, J=7.57, 2.87 Гц, 2 Н) 1.57 (s, 9Н) 1.36 (t, J=7.61 Гц, 3Н).
4) Синтез Соединения 148-5

Соединение 47-4 (400 мг) добавляли в раствор соляная кислота-метанол (4М, 20 мл), и полученную смесь перемешивали при 25°С в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении. К остатку, полученному после концентрирования, добавляли воду (25 мл), и полученную смесь экстрагировали этилацетатом (30 мл × 3). Органическую фазу собирали, сушили, фильтровали и концентрировали при пониженном давлении с получением Соединения 47-5. ЖХ/МС (ЭРИ) m/z: 226 (М+1).
5) Синтез Соединения 47-6

В реакционную колбу добавляли Соединение 47-5 (200 мг), хлорид цинка (36 мг), сульфат натрия (504 мг), ацетон (331 мг), TMSCN (264 мг) и тетрагидрофуран (2 мл), и полученную смесь подвергали реакции при 25°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении, и остаток, полученный после концентрирования, очищали с использованием пластины для препаративной тонкослойной хроматографии с получением Соединения 47-6. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.48 (s, 1H), 7.30 (dd, J=10.03, 2.32 Гц, 1H), 4.28 (s, 1H), 3.50 (s, 1H), 2.90 (qd, J=7.61, 2.98 Гц, 2Н), 1.80 (s, 5Н), 1.31-1.39 (m, 3Н).
6) Синтез Соединения 47-7

В реакционную колбу добавляли Соединение 47-6 (100 мг), Соединение 1-7 (312 мг), метилбензол (2 мл) и DMF (0,5 мл), и полученную смесь перемешивали при 25°С. Затем добавляли гидрид натрия (21 мг, чистота 60%), и полученную смесь подвергали реакции в течение 4 ч при перемешивании. Реакционную смесь концентрировали при пониженном давлении, и остаток, полученный после концентрирования, очищали методом препаративной ВЭЖХ с получением Соединения 47-7. ЖХ/МС (ЭРИ) m/z: 521 (М+1).
7) Синтез Соединения 47

В реакционную колбу добавляли Соединение 47-7 (20 мг), метилбензол (2 мл) и уксусную кислоту (0,5 мл), и полученную смесь кипятили с обратным холодильником при 120°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении, и остаток, полученный после концентрирования, очищали методом препаративной ВЭЖХ с получением Соединения 47. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.80 (s, 1Н), 8.02 (d, J=8.38 Гц, 1Н), 7.95 (s, 1Н), 7.83 (d, J=9.04 Гц, 1Н), 7.37 (d, J=7.28 Гц, 1Н), 2.95 (dd, J=7.72, 3.09 Гц, 2Н), 1.70 (s, 6Н), 1.38 (t, Г=7.61 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 522 (М+1).
Пример 47. Синтез Соединения 48

1) Синтез Соединения 48-2

Нитрат калия (4,10 г) добавляли в раствор Соединения 48-1 (5,00 г) в концентрированной серной кислоте (40 мл) при 0°С. Полученную смесь перемешивали при 0°С в течение 1 ч. Реакционную смесь медленно вливали в ледяную воду (150 мл) и перемешивали для разбавления, и твердое вещество выпадало в осадок. После фильтрования осадок на фильтре промывали водой (50 мл), растворяли в этилацетате (150 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением Соединения 48-2. 1H ЯМР (400 МГц, CDCl3) δ м.д. 10.27-10.35 (m, 1H), 8.65 (d, J=2.8 Гц, 1Н), 8.23 (dd, J=8.8, 2.8 Гц, 1H), 7.82 м.д. (d, J=8.5 Гц, 1Н).
2) Синтез Соединения 48-3
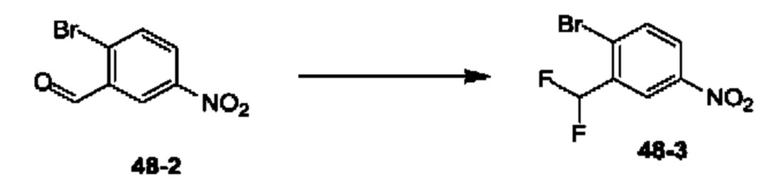
DAST (8,54 г) добавляли в раствор Соединения 48-2 (6 г) в дихлорметане (100 мл) при 0°С. Полученную смесь перемешивали при 0°С в течение 1 ч и затем охлаждали до 0°С. Добавляли насыщенный раствор бикарбоната натрия (50 мл), чтобы погасить реакцию. После экстрагирования дихлорметаном (50 мл) органическую фазу промывали насыщенным рассолом (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток, полученный после концентрирования, очищали колоночной флэш-хроматографией (модель: ISCO-RF150) с получением Соединения 48-3. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.45 (d, J=2.5 Гц, 1H), 8.14 (dd, J=8.8, 2.8 Гц, 1Н), 7.77 (d, J=8.8 Гц, 1H), 6.71-7.01 м.д. (m, 1H).
3) Синтез Соединения 48-4

Цианид цинка (4,00 г), порошок цинка (1,60 г, 24,47 ммоль), DPPF (1,76 г) и бис(дибензилиденацетон)палладий (1,83 г) последовательно добавляли в раствор Соединения 48-3 (4,00 г) в DMF (15 мл). После продувки азотом в течение 30 с полученную смесь нагревали до 130°С для реакции под воздействием микроволнового излучения в течение 1 ч, затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали, и остаток, полученный после концентрирования, очищали колоночной флэш-хроматографией (ISCO-RF150) с получением Соединения 48-4. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.41 (d, J=8.3 Гц, 1H), 6.58-6.90 (m, 3Н), 4.32 м.д. (br s, 2Н).
4) Синтез Соединения 48-5

При 29°С тиофосген (900 мг) добавляли в Н2О (10 мл) с образованием раствора, и полученную смесь перемешивали в течение получаса. Затем в вышеуказанную смесь добавляли Соединение 48-4 (500 мг), и смесь дополнительно перемешивали при 29°С в течение 2 ч. Реакционную смесь экстрагировали дихлорметаном (30 мл × 2). Объединенные органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением Соединения 48-5. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.69 (d, J=8.3 Гц, 1Н), 7.50 (s, 1H), 7.28-7.39 (m, 1H), 6.64-6.99 м.д. (m, 1H).
5) Синтез Соединения 48-6

Соединение 17-3 (200 мг), Соединение 1-4 (100 мг), хлорид одновалентной меди (10 мг), 2-ацетилциклогексанон (10 мг) и карбонат калия (180 мг) добавляли в пробирку для микроволновой обработки, заполненную DMF (5 мл) и водой (1 мл). После продувки азотом в течение 1 мин полученную смесь выдерживали при 130°С для реакции под воздействием микроволнового излучения в течение 1,5 ч. Реакционную смесь фильтровали, и осадок на фильтре промывали DMF (2 мл). Фильтрат нейтрализовали разбавленной 1М соляной кислотой до рН=7 и концентрировали. Остаток, полученный после концентрирования, добавляли в смесь DCM/MeOH (20 мл, об./об.=10/1), и выпавшее в осадок твердое вещество отфильтровывали. Фильтрат концентрировали с получением Соединения 48-6. ЖХ/МС (ЭРИ) m/z: 320 (М+1).
6) Синтез Соединения 48-7

Раствор TMSCHN2 в н-гексане (2М, 0,75 мл) добавляли по каплям в раствор Соединения 48-6 (320 мг) в дихлорметане (10 мл) и метаноле (1 мл) при 29°С. После завершения капельного добавления полученную смесь подвергали реакции при 29°С в течение 1 ч. Реакционную смесь концентрировали, и остаток, полученный после концентрирования, очищали методом препаративной ТСХ с получением Соединения 48-7. ЖХ/МС (ЭРИ) m/z: 334 (М+1).
7) Синтез Соединения 152-8

Вос2О (160 мг) добавляли в раствор Соединения 48-7 (220 мг), триэтиламина (170 мг) и DMAP (20 мг) в дихлорметане (10 мл) при 29°С. После завершения капельного добавления полученную смесь подвергали реакции при 29°С в течение 0,5 ч. Реакционную смесь концентрировали, и остаток, полученный после концентрирования, очищали методом препаративной ТСХ с получением Соединения 48-8. ЖХ/МС (ЭРИ) m/z: 434 (М+1).
8) Синтез Соединения 48-9

Соединение 48-5 (75 мг) добавляли к Соединению 48-8 (50 мг) в смешанном растворителе, состоящем из метилбензола (15 мл) и DMF (3 мл), и затем полученную смесь нагревали до 120°С и перемешивали в течение 12 ч под защитой азота. Реакционную смесь охлаждали до комнатной температуры и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали методом препаративной ТСХ с получением Соединения 48-9. ЖХ/МС (ЭРИ) m/z: 612 (М+1).
9) Синтез Соединения 48

Трифторуксусную кислоту (581 мг) добавляли по каплям в раствор Соединения 48-9 (35 мг) в дихлорметане (5 мл). Затем полученную смесь перемешивали при 28°С в течение 0,5 ч. Реакционную смесь нейтрализовали насыщенным раствором бикарбоната натрия до рН примерно 7 и экстрагировали дихлорметаном (20 мл × 2). Объединенную органическую фазу промывали насыщенным рассолом (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали методом препаративной ТСХ с получением Соединения 48. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.05 (d, J=2.0 Гц, 1H), 7.97 (d, J=9.0 Гц, 1Н), 7.86 (br d, J=4.5 Гц, 2H), 7.58-7.73 (m, 2H), 6.75-7.10 (m, 1H), 4.62-4.84 (m, 2H), 4.02 (br s, 2H), 3.10 (br s, 1H), 2.94 (q, J=7.7 Гц, 2H), 1.58 (s, 6H), 1.35 м.д. (t, J=7.7 Гц, 3H); ЖХ/МС (ЭРИ) m/z: 512 (М+1).
Пример 48. Синтез Соединения 49

1) Синтез Соединения 49-2

В сухую реакционную колбу добавляли Соединение 10-3 (10,00 г) и Соединение 49-1 (5,67 г) и затем добавляли триметилсилилполифосфат (42,91 ммоль). Полученную смесь нагревали до 130°С и перемешивали в течение 12 ч под защитой азота. 100 мл воды и 100 мл этилацетата добавляли в реакционную смесь. После разделения жидких фаз органическую фазу собирали, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток, полученный после концентрирования, очищали колоночной хроматографией с получением неочищенного продукта. Полученный неочищенный продукт суспендировали в смеси 20 мл этилацетата и 40 мл метил-трет-бутилового эфира с получением Соединения 49-2. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.09-8.00 (m, 2Н), 2.00 (t, J=19.3 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 307 (М+1).
2) Синтез Соединения 49-3

В сухую пробирку для микроволновой обработки добавляли Соединение 49-2 (1,00 г), Соединение 1-4 (1,01 г), DMF (20 мл), воду (4 мл), карбонат калия (2,25 г) и 2-ацетилциклогексанон (91 мг) и затем добавляли хлорид одновалентной меди (645 мг). После продувки азотом в течение пяти минут пробирку для микроволновой обработки герметично закрывали, и затем полученную смесь подвергали воздействию микроволнового излучения и перемешивали при 90°С в течение 1 ч. Смесь концентрировали при пониженном давлении с получением Соединения 49-3. ЖХ/МС (ЭРИ) m/z: 330 (М+1).
3) Синтез Соединения 49-4

В сухую одногорлую колбу добавляли Соединение 49-3 (8,58 г) и раствор соляная кислота-метанол (4М, 250 мл), и полученную смесь перемешивали при 80°С в течение 12 ч и затем концентрировали досуха при пониженном давлении для удаления растворителя и получения твердого вещества. Твердое вещество очищали колоночной хроматографией с получением Соединения 49-4. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 9.33 (br s, 1Н), 7.08 (d, J=1.8 Гц, 1Н), 6.77 (dd, J=2.8, 12.0 Гц, 1Н), 4.68 (s, 1H), 3.75 (s, 3Н), 2.09 (t, J=19.0 Гц, 3Н), 1.63 (s, 6Н). ЖХ/МС (ЭРИ) m/z: 344 (М+1).
4) Синтез Соединения 49-5
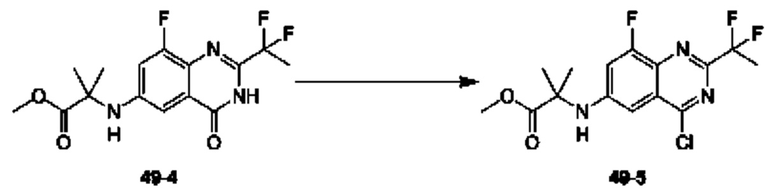
В сухую одногорлую колбу добавляли Соединение 49-4 (2,85 г, 8,30 ммоль) и оксихлорид фосфора (43,29 г) и затем добавляли диизопропилэтиламин (1,67 г). Полученную смесь перемешивали при 110°С в течение 4 ч и затем концентрировали при пониженном давлении для удаления растворителя. Твердое вещество, полученное после концентрирования, растворяли в 26 мл этилацетата, и полученный раствор медленно по каплям добавляли в смешанный раствор 31 мл метанола и 110 мл триэтиламина при контролируемой температуре 0-10°С. После завершения капельного добавления полученную смесь фильтровали. Фильтрат собирали и концентрировали досуха при пониженном давлении при 40°С для удаления растворителя. Остаток, полученный после концентрирования, очищали колоночной хроматографией с получением Соединения 49-5. ЖХ/МС (ЭРИ) m/z: 362 (М+1).
5) Синтез Соединения 155-6

В сухую одногорлую колбу добавляли Соединение 49-5 (4,00 г), затем добавляли метанол (100 мл) и затем добавляли метоксид натрия (2,99 г). Полученную смесь перемешивали при 30°С в течение 0,5 ч под защитой азота. 200 мл этилацетата добавляли в реакционную смесь, и полученную смесь фильтровали. Раствор соляная кислота-метанол (4М, 3 мл) добавляли в фильтрат. Полученную смесь концентрировали при пониженном давлении при 30°С до 100 мл и фильтровали. Фильтрат концентрировали досуха при пониженном давлении при 30°С, и остаток, полученный после концентрирования, очищали колоночной хроматографией с получением Соединения 49-6. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 6.89 (dd, J=2.5, 11.8 Гц, 1H), 6.78 (d, J=1.8 Гц, 1H), 4.60 (br s, 1H), 4.18 (s, 3Н), 3.73 (s, 3Н), 2.09 (t, J=18.5 Гц, 3Н), 1.67-1.63 (m, 6Н). ЖХ/МС (ЭРИ) m/z: 358 (М+1).
6) Синтез Соединения 49-7

В сухую одногорлую колбу добавляли Соединение 49-6 (2,66 г), Соединение 1-7 (3,40 г), DMF (3,6 мл) и метилбензол (18 мл). Под защитой азота полученную смесь перемешивали при 90°С в течение 48 ч и затем концентрировали при пониженном давлении для удаления растворителя. Остаток, полученный после концентрирования, очищали колоночной хроматографией с получением Соединения 49-7. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.00 (d, J=8.4 Гц, 1Н), 7.98-7.95 (m, 2Н), 7.84 (dd, J=1.9, 8.3 Гц, 1H), 7.54 (dd, J=2.2, 9.7 Гц, 1H), 4.28 (s, 3Н), 2.14 (t, J=18.5 Гц, 3Н), 1.67 (s, 6Н). ЖХ/МС (ЭРИ) m/z: 554 (М+1).
7) Синтез Соединения 49-8
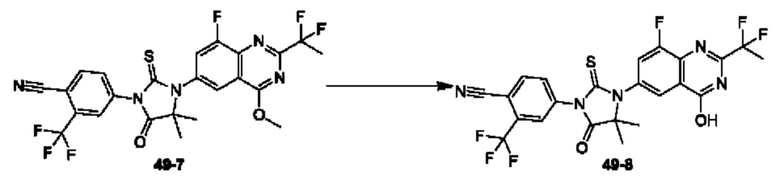
В сухую одногорлую колбу добавляли Соединение 49-7 (200 мг) и затем добавляли тетрагидрофуран (1 мл) и концентрированную соляную кислоту (12М, 1 мл), и полученную смесь перемешивали при 25°С в течение 5 мин. Дихлорметан (5 мл) добавляли в реакционную смесь. После разделения жидких фаз органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха при пониженном давлении для удаления растворителя с получением Соединения 49-8. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 9.73 (br s, 1H), 8.05 (s, 1H), 8.00 (d, J=8.4 Гц, 1H), 7.95 (s, 1Н), 7.83 (d, J=8.2 Гц, 1H), 7.53 (dd, J=2.2, 9.5 Гц, 1H), 2.15 (т, J=19.2 Гц, 3Н), 1.65 (s, 6Н). ЖХ/МС (ЭРИ) m/z: 540 (М+1).
8) Синтез Соединения 49-9

В сухую одногорлую колбу добавляли Соединение 49-8 (300 мг) и оксихлорид фосфора (2,90 г) и затем добавляли диизопропилэтиламин (112 мг). Полученную смесь нагревали до 110°С и перемешивали в течение 12 ч и затем концентрировали досуха при пониженном давлении для удаления растворителя. Дихлорметан (5 мл) добавляли к остатку, полученному после концентрирования, и полученную смесь промывали 10 мл ледяного насыщенного раствора бикарбоната натрия. После разделения жидких фаз органическую фазу промывали насыщенным рассолом, сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха при пониженном давлении для удаления растворителя, и остаток, полученный после концентрирования, очищали методом препаративной ТСХ с получением Соединения 49-9. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.11-8.08 (m, 1H), 8.02 (d, J=8.2 Гц, 1H), 7.97 (d, J=2.0 Гц, 1H), 7.84 (dd, J=2.1, 8.3 Гц, 1Н), 7.72 (dd, J=2.1, 9.2 Гц, 1H), 2.18 (t, J=18.5 Гц, 3Н), 1.70 (s, 6Н). ЖХ/МС (ЭРИ) m/z: 558 (М+1).
9) Синтез Соединения 49

В сухую одногорлую колбу добавляли Соединение 49-9 (44 мг), этандиол (45 мг) и тетрагидрофуран (0,5 мл) и затем добавляли гидрид натрия (9 мг, чистота 60%). Полученную смесь перемешивали при 25°С в течение 1 ч. Реакционную смесь очищали последовательно методом препаративной ТСХ и методом препаративной ВЭЖХ с получением Соединения 49. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.05-7.99 (m, 2Н), 7.98 (s, 1Н), 7.85 (br d, J=7.5 Гц, 1H), 7.58 (br d, J=9.5 Гц, 1H), 4.91-4.85 (m, 2Н), 4.13 (br d, J=3.5 Гц, 2Н), 2.47 (t, J=5.7 Гц, 1H), 2.15 (t, J=18.5 Гц, 3Н), 1.69 (s, 6H); ЖХ/МС (ЭРИ) m/z: 584.
Пример 49. Синтез Соединения 50
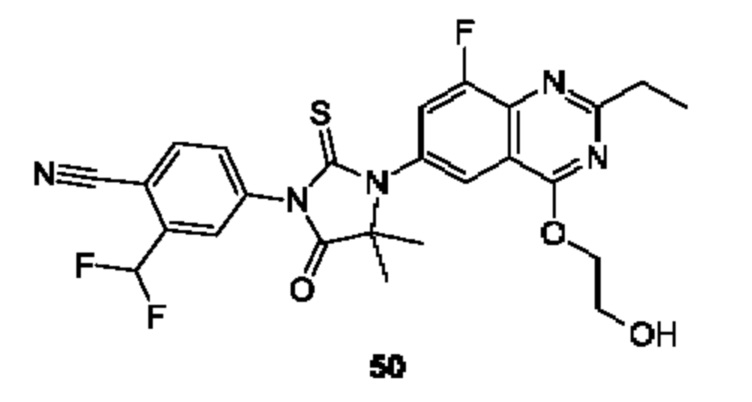
1) Синтез Соединения 50-1

Согласно Синтезу Соединения 48-6, Соединение 50-1 было получено с использованием Соединения 20-1 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 338 (М+1).
2) Синтез Соединения 50-2

Согласно Синтезу Соединения 48-7, Соединение 50-2 было получено с использованием Соединения 50-1 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 352 (М+1).
3) Синтез Соединения 158-3
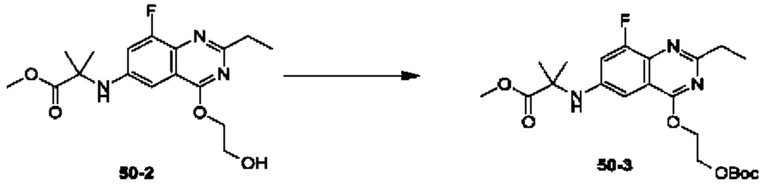
Согласно Синтезу Соединения 48-8, Соединение 50-3 было получено с использованием Соединения 50-2 в качестве исходного вещества. ЖХ/МС (ЭРИ) m/z: 452 (М+1).
4) Синтез Соединения 50-4
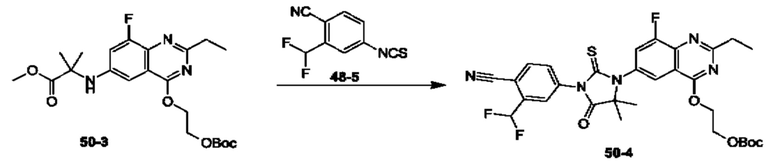
Согласно Синтезу Соединения 48-9, Соединение 50-4 было получено с использованием Соединения 50-3 и Соединение 48-5 в качестве исходных веществ. ЖХ/МС (ЭРИ) m/z: 630 (М+1).
5) Синтез Соединения 50

Согласно Синтезу Соединения 48, Соединение 50 было получено с использованием Соединения 50-4 в качестве исходного вещества. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.80-7.91 (m, 3Н), 7.69 (d, J=8.3 Гц, 1H), 7.40 (dd, J=10.0, 2.0 Гц, 1H), 6.75-7.08 (m, 1H), 4.68-4.78 (m, 2Н), 4.02 (br s, 2Н), 2.98 (q, J=7.6 Гц, 2Н), 2.74 (br s, 1H), 1.59 (s, 6Н), 1.35 м.д. (t, J=7.5 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 530 (М+1).
Пример 50. Синтез Соединения 51

1) Синтез Соединения 159
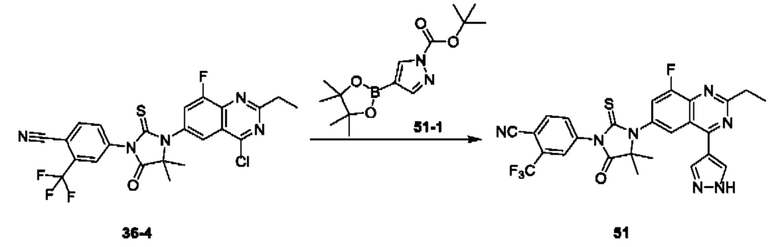
В сухую пробирку для микроволновой обработки добавляли Соединение 36-4 (500 мг), Соединение 55-1 (564 мг), карбонат натрия (2М, 800 мкл, водный раствор), 1,2-дихлорэтан (7 мл) и воду (3 мл). После продувки азотом добавляли дихлорбис(трифенилфосфин)палладий (67 мг), и полученную смесь подвергали реакции при 140°С в течение 10 мин. Реакционную смесь концентрировали, и остаток, полученный после концентрирования, очищали последовательно с использованием пластины для препаративной ТСХ и методом препаративной ВЭЖХ с получением Соединения 51. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.28 (s, 2Н), 8.08 (s, 1H), 8.03 (d, J=8.16 Гц, 1H), 7.99 (s, 1H), 7.86 (d, J=8.16 Гц, 1H), 7.51-7.55 (m, 1H), 3.23 (q, J=7.50 Гц, 2Н), 1.69 (s, 6Н), 1.51 (t, J=7.72 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 554 (М+1).
Пример 51. Синтез Соединения 52

1) Синтез Соединения 52

В сухую одногорлую колбу добавляли Соединение 36-4 (300 мг), Соединение 52-1 (129 мг), карбонат натрия (102 мг), 1,2-дихлорэтан (2,1 мл) и воду (0,9 мл). После продувки азотом добавляли дихлорбис(трифенилфосфин)палладий (40 мг), и полученную смесь кипятили с обратным холодильником при 100°С и подвергали реакции в течение 16 ч. Реакционную смесь концентрировали, и остаток, полученный после концентрирования, очищали последовательно с использованием пластины для препаративной ТСХ и методом препаративной ВЭЖХ с получением Соединения 52. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 9.52-9.55 (m, 1H), 8.93 (d, J=2.89 Гц, 1H), 7.98-8.04 (m, 2Н), 7.92 (d, J=0.88 Гц, 1Н), 7.86-7.91 (m, 1Н), 7.59 (dd, J=9.91, 2.13 Гц, 1Н), 6.60 (dd, J=2.89, 1.63 Гц, 1Н), 3.20 (q, J=7.53 Гц, 2Н), 1.73 (s, 5Н), 1.67-1.77 (m, 1Н), 1.50 (t, J=7.59 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 554 (М+1).
Пример 52. Синтез Соединения 53

1) Синтез Соединения 53

Смешанный раствор Соединения 35-4 (100 мг), Соединения 23-7 (299 мг), N,N-диметилформамида (0,5 мл) и метилбензола (2 мл) нагревали до 120°С и перемешивали в течение 16 ч. Метанол (5 мл) добавляли в реакционную смесь, и полученную смесь перемешивали в течение 30 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали последовательно с использованием силикагелевой колонки и методом препаративной ВЭЖХ с получением Соединения 53. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 9.01 (d, J=1.5 Гц, 1H), 8.76 (s, 1Н), 8.28 (d, J=1.8 Гц, 1H), 7.32 (dd, J=1.8, 8.8 Гц, 1H), 6.42 (s, 1H), 2.75 (q, J=7.5 Гц, 2Н), 1.64 (s, 6Н), 1.29 (t, J=7.5 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 505 (М+1).
Пример 53. Синтез Соединения 54

1) Синтез Соединения 54-1

В пробирку для микроволновой обработки добавляли Соединение 35-2 (500 мг), mpem-бутилкарбамат (324 мг), карбонат цезия (1,50 г), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (107 мг), бис(дибензилиденацетон)палладий (170 мг) и метилбензол (6 мл). Пробирку для микроволновой обработки герметично закрывали, и полученную смесь выдерживали при 120°С для реакции под воздействием микроволнового излучения в течение 30 мин. Реакционную смесь фильтровали, и промывали этилацетатом (20 мл). Фильтрат концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 54-1. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.90 (s, 1H), 8.15 (br s, 1H), 7.57 (br s, 1H), 6.32 (s, 1H), 2.71 (q, J=7.5 Гц, 2H), 1.49 (s, 9H), 1.26 (t, J=7.5 Гц, 3Н).
2) Синтез Соединения 54-2

Трифторуксусную кислоту (0,4 мл) добавляли в раствор Соединения 54-1 (200 мг) в дихлорметане (2 мл). Полученную реакционную смесь перемешивали при 26°С в течение 4 ч. Насыщенный водный раствор бикарбоната натрия (рН примерно 7) добавляли в реакционную смесь, и смесь экстрагировали дихлорметаном (20 мл). Органическую фазу промывали насыщенным рассолом (15 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением Соединения 54-2. ЖХ/МС (ЭРИ) m/z: 208 (М+1).
3) Синтез Соединения 54-3

Циклобутанон (115 мг) и хлорид цинка (12 мг) добавляли в смешанный раствор Соединения 54-2 (60 мг), триметилсилилцианида (81 мг), сульфата натрия (154 мг) и тетрагидрофурана (2 мл). Полученную реакционную смесь перемешивали при 25°С в течение 16 ч. Водный раствор сульфита натрия (10 мл) добавляли в реакционную смесь, и полученную смесь экстрагировали этилацетатом (10 мл × 3). Органическую фазу промывали насыщенным рассолом (10 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 54-3. ЖХ/МС (ЭРИ) m/z: 287 (М+1). 4) Синтез Соединения 54

Соединение 1-7 (224 мг) добавляли в раствор Соединения 54-3 (70 мг), N,N-диметилформамида (0,5 мл) и метилбензола (2 мл). Полученную смесь нагревали до 120°С и перемешивали в течение 16 ч. Соединение 1-7 (224 мг) добавляли в реакционную смесь, и смесь дополнительно перемешивали в течение 16 ч. Метанол (5 мл) добавляли в реакционную смесь, и смесь перемешивали в течение 30 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали последовательно с использованием силикагелевой колонки и методом препаративной ВЭЖХ с получением Соединения 54. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.82 (s, 1H), 7.94 (d, J=8.3 Гц, 1H), 7.89 (s, 1H), 7.76 (br d, J=8.3 Гц, 1H), 7.33 (dd, J=1.8, 8.8 Гц, 1H), 6.43 (s, 1H), 2.82-2.63 (m, 4H), 2.58-2.43 (m, 2H), 2.34-2.18 (m, 1H), 1.73 (q, J=10.5 Гц, 1H), 1.29 (t, J=7.5 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 516 (М+1).
Пример 54. Синтез Соединения 55

1) Синтез Соединения 163-1

В сухую одногорлую колбу добавляли Соединение 34-1 (15,00 г) и дихлорметан (150 мл) и охлаждали до -40°С. Затем добавляли малонилдихлорид (14,72 г). Полученную смесь медленно нагревали до 25°С и перемешивали в течение 24 ч. После фильтрования твердое вещество собирали. Полученное твердое вещество суспендировали в смешанном растворителе, состоящем из 100 мл метанола и 100 мл дихлорметана, и фильтровали с получением Соединения 55-1. 1Н ЯМР (400 МГц, DMSO-d6) δ м.д. 8.73 (t, J=1.7 Гц, 1H), 8.20 (dd, J=2.0, 9.3 Гц, 1Н), 5.54 (s, 1Н). ЖХ/МС (ЭРИ) m/z: 259 (М+1).
2) Синтез Соединения 55-2

В сухую одногорлую колбу добавляли Соединение 55-1 (1,00 г), карбонат калия (1,07 г), йодэтан (682 мг) и NMP (10 мл). Под защитой азота полученную смесь нагревали до 60°С и перемешивали в течение 48 ч. Добавляли йодэтан (682 мг), и полученную смесь нагревали до 60°С и кипятили с обратным холодильником в течение 12 ч. В реакционную смесь последовательно добавляли 100 мл насыщенного рассола и 100 мл этилацетата, и смесь фильтровали для удаления твердого вещества. После разделения жидких фаз фильтрата органическую фазу собирали, сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха при пониженном давлении. Остаток, полученный после концентрирования, последовательно очищали колоночной хроматографией и суспендировали в 20 мл метил-третбутилового эфира с получением Соединения 55-2. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.98 (t, J=1.7 Гц, 1H), 7.56 (dd, J=2.1, 7.8 Гц, 1H), 5.82 (s, 1H), 4.42 (q, J=7.1 Гц, 2Н), 1.42 (t, J=7.1 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 287 (М+1).
3) Синтез Соединения 164-3

В сухую пробирку для микроволновой обработки добавляли Соединение 55-2 (600 мг), 2-аминоизомасляную кислоту (646 мг), 2-ацетилциклогексанон (59 мг), карбонат калия (578 мг), DMF (12 мл) и воду (2,4 мл) и затем добавляли хлорид одновалентной меди (41 мг). После продувки азотом в течение 5 минут пробирку для микроволновой обработки герметично закрывали. Полученную смесь подвергали воздействию микроволнового излучения и перемешивали при 90°С в течение 2 ч. Добавляли 2-ацетилциклогексанон (59 мг) и хлорид одновалентной меди (41 мг), и полученную смесь подвергали воздействию микроволнового излучения и перемешивали в течение 1,5 ч и затем концентрировали при пониженном давлении с получением Соединения 55-3. ЖХ/МС (ЭРИ) m/z: 310 (М+1).
4) Синтез Соединения 55-4

В сухую одногорлую колбу добавляли Соединение 55-3 (1,20 г), раствор TMSCHN2 в н-гексане (2М, 3,88 мл), дихлорметан (20 мл) и метанол (3 мл). Под защитой азота полученную смесь перемешивали при 25°С в течение 12 ч. Добавляли раствор TMSCHN2 в н-гексане (2М, 3,88 мл), и полученную смесь перемешивали при 25°С в течение 6 ч; снова добавляли раствор TMSCHN2 в н-гексане (2М, 3,88 мл), и полученную смесь перемешивали при 25°С в течение 12 ч; снова добавляли раствор TMSCHN2 в н-гексане (2М, 3,88 мл), и полученную смесь перемешивали при 25°С в течение 6 ч; и снова добавляли раствор TMSCHN2 в н-гексане (2М, 7,76 мл), и полученную смесь перемешивали при 25°С в течение 12 ч. Полученную смесь концентрировали досуха при пониженном давлении для удаления растворителя. Полученный неочищенный продукт очищали колоночной хроматографией с получением Соединения 55-4. ЖХ/МС (ЭРИ) m/z: 324 (М+1).
5) Синтез Соединения 55

В сухую одногорлую колбу добавляли Соединение 55-4 (80 мг), DMF (0,4 мл) и метилбензол (2 мл) и добавляли затем Соединение 1-7 (624 мг). Под защитой азота полученную смесь нагревали до 90°С и перемешивали в течение 10 ч и затем концентрировали досуха при пониженном давлении для удаления растворителя. Неочищенный продукт очищали последовательно с использованием пластины для препаративной ТСХ и методом препаративной ВЭЖХ с получением Соединения 55. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.80 (s, 1H), 7.94 (d, J=8,4 Гц, 1H), 7.87 (s, 1Н), 7.75 (br d, J=7.7 Гц, 1H), 7.39 (br d, J=8.6 Гц, 1H), 5.82 (s, 1H), 4.41 (q, J=7.2 Гц, 2H), 1.61 (s, 6Н), 1.39 (t, J=7.1 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 520 (М+1).
Пример 55. Синтез Соединения 56

1) Синтез Соединения 56-1

В реакционную колбу медленно добавляли оксихлорид фосфора (50 мл), Соединение 17-1 (5,00 г) и затем добавляли М,К-диизопропилэтиламин (3,98 г). Полученную смесь подвергали реакции при 110°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт растворяли в 80 мл этилацетата, и 20 мл ледяной воды добавляли для разделения жидких фаз. 40 мл ледяного насыщенного раствора бикарбоната натрия добавляли в органическую фазу. После разделения жидких фаз органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением Соединения 56-1.
2) Синтез Соединения 56-2

В круглодонную колбу добавляли Соединение 56-1 (4,65 г), метоксид натрия (4,63 г) и метанол (47 мл) и проводили реакцию при 25°С в течение 60 мин. После завершения реакции полученную смесь концентрировали, и остаток, полученный после концентрирования, очищали колоночной флэш-хроматографией с получением Соединения 56-2. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.25 (d, J=2.21 Гц, 1H), 7.83 (dd, J=8.93, 2.32 Гц, 1H), 7.70-7.74 (m, 1H), 4.13-4.20 (m, 3Н), 2.96 (q, J=7.50 Гц, 2Н), 1.41 (t, J=7.50 Гц, 3Н).
3) Синтез Соединения 56-3

В реакционную колбу добавляли Соединение 56-2 (500 мг), Соединение 3-2 (801 мг), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (108 мг), карбонат цезия (1,52 г) и DMF (5 мл). После продувки азотом в течение 1 минуты добавляли бис(дибензилиденацетон)палладий (107 мг), и полученную смесь подвергали реакции при 80°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт отделяли и очищали колоночной флэш-хроматографией с получением Соединения 56-3. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.18 (d, J=4.85 Гц, 1H), 7.79 (d, Г=9.04 Гц, 1H), 7.29 (d, J=2.87 Гц, 1H), 7.20 (d, J=4.85 Гц, 1Н), 6.99 (d, J=2.65 Гц, 1H), 5.84 (s, 1Н), 4.10 (s, 3Н), 2.90-2.98 (m, 2Н), 2.23 (s, 3Н), 1.40 (t, J=7.61 Гц, 3Н).
4) Синтез Соединения 56-4

В пробирку для микроволновой обработки добавляли Соединение 56-3 (500 мг), Соединение 2-5 (311 мг), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (88 мг), карбонат цезия (991 мг) и метилбензол (5 мл). В условиях продувки азотом добавляли Pd(dba)2 (87 мг), и после продувки азотом в течение 1 мин полученную смесь выдерживали при 130°С для реакции под воздействием микроволнового излучения в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией с получением Соединения 56-4. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.23 (d, J=5.02 Гц, 1H), 8.16 (d, J=2.13 Гц, 1H), 7.98 (dd, J=8.66, 2.26 Гц, 1H), 7.79 (d, J=8.91 Гц, 1Н), 7.76 (s, 1H), 7.68 (d, J=8.66 Гц, 1H), 7.00 (d, J=2.64 Гц, 1Н), 6.91 (d, J=5.14 Гц, 1H), 5.31 (s, 1H), 5.27 (s, 1H), 4.07 (s, 3Н), 2.93 (q, J=7.57 Гц, 2Н), 2.21 (s, 3Н), 1.39 (t, J=7.59 Гц, 3Н).
5) Синтез Соединения 56-5

При 0°С в реакционную колбу добавляли Соединение 56-4 (79 мг) и тетрагидрофуран (0,8 мл) и основательно перемешивали, и затем добавляли гидрид натрия (22 мг, чистота 60%). После протекания реакции полученной смеси в течение 0,5 ч добавляли тиофосген (30 мг), и реакционную смесь перемешивали при 25°С в течение 15,5 ч. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали с использованием силикагелевой пластины для тонкослойной хроматографии с получением Соединения 56-5. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.29 (s, 1Н), 8.27 (d, J=2.43 Гц, 1H), 8.24-8.28 (m, 1H), 8.08-8.12 (d, J=5.07 Гц, 1H), 8.07 (s, 1H), 8.05 (s, 1H), 7.84 (dd, J=8.82, 2.43 Гц, 1H), 6.97-7.01 (m, 1H), 4.17 (s, 3Н), 2.98-3.07 (m, 2Н), 1.88 (s, 3Н), 1.44 (t, J=7.50 Гц, 3Н).
8) Синтез Соединения 56

В реакционную колбу добавляли Соединение 56-5 (70 мг), водный раствор соляной кислоты (2М, 1 мл) и тетрагидрофуран (1 мл) и перемешивали при 30°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали методом препаративной ВЭЖХ с получением Соединения 56. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 9.28 (brs, 1Н), 8.36 (d, m=1.98 Гц, 1Н), 8.31 (s, 1Н), 8.19 (d, J=8.16 Гц, 1Н), 8.13 (d, J=5.07 Гц, 1H), 8.08 (d, J=8.38 Гц, 1Н), 7.91-7.95 (m, 1H), 7.85-7.90 (m, 1H), 6.99-7.02 (m, 1H), 2.81 (q, J=7.64 Гц, 2Н), 1.93 (s, 3Н), 1.45 (t, J=7.61 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 507 (М+1).
Пример 56. Синтез Соединения 57

1) Синтез Соединения 57-2

Метилпропионилацетат (2,56 г) добавляли в раствор Соединения 57-1 (1,00 г) в уксусной кислоте (10 мл). Полученную смесь нагревали до 110°С и перемешивали в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, разбавляли этилацетатом (30 мл) и добавляли насыщенный водный раствор бикарбоната натрия (30 мл). После разделения жидких фаз органическую фазу промывали насыщенным рассолом (30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 57-2. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.83 (s, 1H), 7.05 (s, 1H), 6.44 (s, 1H), 4.08 (s, 3Н), 2.82 (q, J=7.5 Гц, 2Н), 1.34 (t, J=7.7 Гц, 3Н).
2) Синтез Соединения 57-3

Соединение 57-2 (300 мг), Соединение 1-4 (165 мг), карбонат калия (366 мг), 2-ацетилциклогексанон (30 мг), хлорид одновалентной меди (21 мг), N,N-диметилформамид (2 мл) и воду (0,1 мл) добавляли в пробирку для микроволновой обработки. Пробирку для микроволновой обработки герметично закрывали, и полученную смесь выдерживали при 130°С для реакции под воздействием микроволнового излучения в течение 30 мин. Реакционную смесь фильтровали, осадок на фильтре промывали этилацетатом (20 мл), и фильтрат концентрировали при пониженном давлении. 1 н. соляную кислоту добавляли к остатку, полученному после концентрирования (рН 6-7). Полученную смесь экстрагировали этилацетатом (20 мл ×3), и органическую фазу промывали насыщенным рассолом (30 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали. Смесь дихлорметан/метанол (10/1, 20 мл) добавляли к остатку, полученному после концентрирования. Полученную смесь фильтровали, и фильтрат концентрировали при пониженном давлении с получением Соединения 57-3. ЖХ/МС (ЭРИ) m/z: 306 (М+1).
3) Синтез Соединения 57-4

Раствор триметилсилилдиазометана в н-гексане (2М, 0,8 мл) добавляли в раствор Соединения 57-3 (300 мг) в дихлорметане (5 мл) и метаноле (0,5 мл). Полученную реакционную смесь перемешивали при 20°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 57-4. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.88 (d, J=2.3 Гц, 1H), 6.55 (d, J=1.8 Гц, 1H), 6.26 (s, 1H), 3.94 (s, 3H), 3.72 (s, 3H), 2.72 (q, J=7.4 Гц, 2H), 1.57 (s, 6H), 1.24 (t, J=7.5 Гц, 3H).
4) Синтез Соединения 57
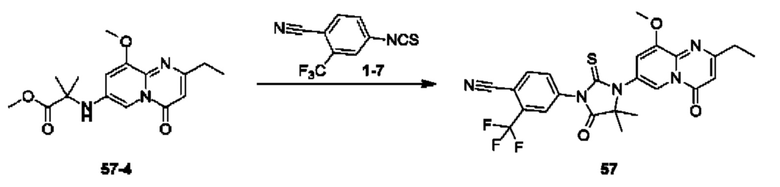
Смешанный раствор Соединения 57-4 (150 мг), Соединения 1-7 (430 мг), N,N-диметилформамида (0,5 мл) и метилбензола (2 мл) нагревали до 120°С и перемешивали в течение 16 ч. Метанол (5 мл) добавляли в реакционную смесь, и смесь перемешивали в течение 30 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали последовательно с использованием силикагелевой колонки и методом препаративной ВЭЖХ с получением Соединения 57. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.71 (d, J=2.0 Гц, 1H), 8.03 (d, J=8.3 Гц, 1H), 7.98 (d, J=1.5 Гц, 1H), 7.86 (dd, J=2.0, 8.3 Гц, 1H), 6.90 (d, J=2.0 Гц, 1H), 6.49 (s, 1Н), 4.09 (s, 3Н), 2.86 (q, J=7.5 Гц, 2Н), 1.72 (s, 6Н), 1.37 (t, J=7.5 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 516 (М+1).
Пример 57. Синтез Соединения 58

1) Синтез Соединения 58-1

В сухую реакционную колбу добавляли Соединение 54-2 (300 мг), хлорид цинка (59 мг), сульфат натрия (823 мг), ацетон (505 мг), триметилсилилцианид (431 мг) и тетрагидрофуран (3 мл) и проводили реакцию при 25°С в течение 4 ч под защитой азота. Реакционную смесь напрямую концентрировали, и остаток, полученный после концентрирования, очищали методом препаративной ТСХ с получением Соединения 58-1. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.52 (s, 1H), 7.33 (dd, J=9.98, 2.32 Гц, 1H), 6.46 (s, 1Н), 2.78 (q, J=7.65 Гц, 2H), 1.78 (s, 6H), 1.33 (t, J=7.59 Гц, 3H).
2) Синтез Соединения 58-3

Воду (10 мл) добавляли в одногорлую колбу и затем по каплям добавляли тиофосген (1,13 г). После перемешивания при 25°С в течение 0,5 ч под защитой азота порциями добавляли Соединение 58-2 (1,00 г), и полученную смесь дополнительно подвергали реакции при 25°С в течение 2 ч. Реакционную смесь экстрагировали дихлорметаном (10 мл ×3). Органическую фазу промывали насыщенным рассолом (15 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали. Остаток, полученный после концентрирования, очищали колоночной хроматографией с получением Соединения 58-3. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.67 (d, J=8.38 Гц, 1H), 7.37 (d, J=1.98 Гц, 1H) 7.21 (dd, J=8.38, 1.98 Гц, 1H).
3) Синтез Соединения 58-4

В сухую реакционную колбу добавляли Соединение 58-1 (200 мг), Соединение 58-3 (568 мг), метилбензол (2 мл) и DMF (0,5 мл). Под защитой азота добавляли гидрид натрия (44 мг, чистота 60%), и полученную смесь подвергали реакции при 25°С в течение 0,5 ч. Реакционную смесь концентрировали, и остаток, полученный после концентрирования, очищали колоночной хроматографией с получением Соединения 58-4. ЖХ/МС (ЭРИ) m/z: 469 (М+1).
4) Синтез Соединения 58

В сухую реакционную колбу добавляли Соединение 58-4 (110 мг), метилбензол (1,1 мл) и ледяную уксусную кислоту (1,1 мл) и выдерживали при 110°С в течение 16 ч под защитой азота. Реакционную смесь концентрировали, и остаток, полученный после концентрирования, очищали методом препаративной ВЭЖХ с получением Соединения 58. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.83 (s, 1H), 7.84 (d, J=8.16 Гц, 1H), 7.68 (d, J=1.98 Гц, 1H), 7.51 (dd, J=8.27, 2.09 Гц, 1H), 7.41 (dd, J=8.71, 2.09 Гц, 1H), 6.49 (s, 1H), 2.82 (q, J=7.57 Гц, 2Н),1.68 (s, 6Н), 1.36 (t, J=7.61 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 470 (М+1).
Пример 58. Синтез Соединения 59

1) Синтез Соединения 59-2
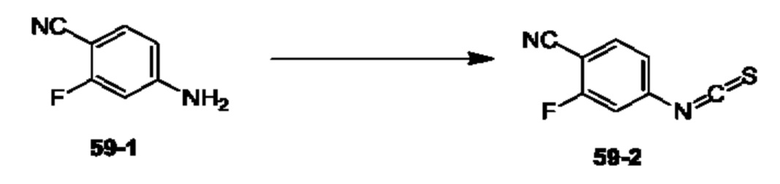
Воду (10 мл) добавляли в одногорлую колбу и затем по каплям добавляли тиофосген (1,27 г). После перемешивания при 25°С в течение 0,5 ч под защитой азота порциями добавляли Соединение 59-1 (1,00 г), и полученную смесь дополнительно подвергали реакции при 25°С в течение 2 ч. Реакционную смесь экстрагировали дихлорметаном (10 мл ×3). Органическую фазу промывали насыщенным рассолом (15 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали. Остаток, полученный после концентрирования, очищали колоночной хроматографией с получением Соединения 59-2. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.63 (dd, J=8.38, 7.06 Гц, 1H), 7.10-7.15 (m, 1H), 7.07 (dd, J=9.15, 1.87 Гц, 1H).
2) Синтез Соединения 59-3

В сухую реакционную колбу добавляли Соединение 58-1 (200 мг), Соединение 59-2 (520 мг), метилбензол (2 мл) и DMF (0,5 мл). Под защитой азота добавляли гидрид натрия (44 мг, чистота 60%), и полученную смесь подвергали реакции при 25°С в течение 0,5 ч. Реакционную смесь концентрировали, и остаток, полученный после концентрирования, очищали колоночной хроматографией с получением Соединения 59-3. ЖХ/МС (ЭРИ) m/z: 453 (M+1).
3) Синтез Соединения 59

В сухую реакционную колбу добавляли Соединение 59-3 (80 мг), метилбензол (0,8 мл) и ледяную уксусную кислоту (0,8 мл), и реакцию проводили при 110°С в течение 16 ч под защитой азота. Реакционную смесь концентрировали с получением неочищенного продукта, и неочищенный продукт очищали методом препаративной ВЭЖХ с получением Соединения 59. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.83 (s, 1H) 7.77-7.84 (m, 1H) 7.38-7.45 (m, 3Н) 6.49 (s, 1Н) 2.82 (q, J=7.35 Гц, 2Н) 1.68 (s, 6Н) 1.36 (t, J=7.61 Гц, 3Н).
Пример 59. Синтез Соединения 60
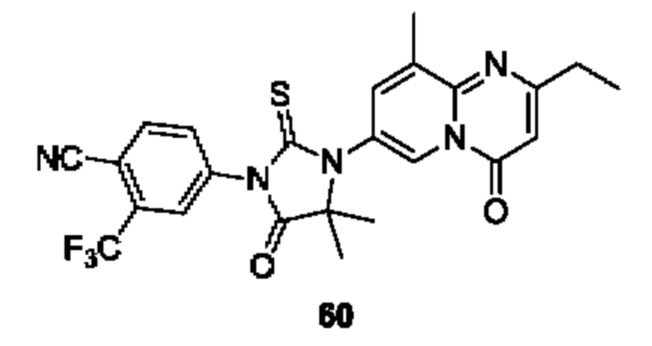
1) Синтез Соединения 60-2
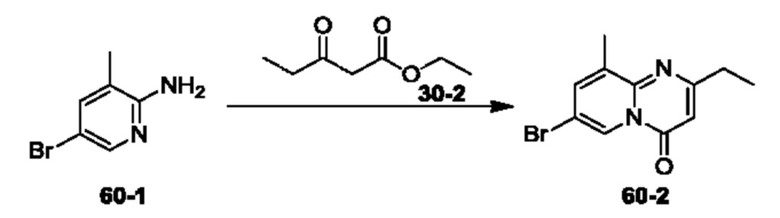
В сухую одногорлую колбу добавляли Соединение 60-1 (10,00 г), Соединение 30-2 (10,02 г) и полифосфорную кислоту (30 г), нагревали до 110°С и перемешивали в течение 16 ч под защитой азота. Реакционную смесь вливали в 200 мл ледяной воды и затем доводили до рН=7 твердым бикарбонатом натрия. Добавляли 200 мл этилацетата. После разделения жидких фаз органическую фазу собирали, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток, полученный после концентрирования, очищали колоночной хроматографией с получением Соединения 60-2. 1Н ЯМР (400 МГц, DMSO-d6) δ м.д. 8.81 (d, J=1.8 Гц, 1H), 7.94 (d, J=0,9 Гц, 1H), 6.31 (s, 1H), 2.62 (q, J=7.5 Гц, 2Н), 2.44 (s, 3Н), 1.21 (t, J=7.5 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 267 (М+1).
2) Синтез Соединения 60-3

В сухую пробирку для микроволновой обработки добавляли Соединение 60-2 (500 мг), 2-аминоизомасляную кислоту (579 мг), 2-ацетилциклогексанон (52 мг), карбонат калия (517 мг), DMF (4 мл) и воду (1 мл) и затем добавляли хлорид одновалентной меди (37 мг). После продувки азотом в течение 5 минут пробирку для микроволновой обработки герметично закрывали, и полученную смесь подвергали воздействию микроволнового излучения и перемешивали при 90°С в течение 4 ч. Реакционную смесь концентрировали при пониженном давлении с получением Соединения 60-3. ЖХ/МС (ЭРИ) m/z: 290 (М+1).
3) Синтез Соединения 60-4

В сухую одногорлую колбу добавляли Соединение 60-3 (800 мг) и раствор соляная кислота-метанол (4М, 25 мл), нагревали до 90°С и кипятили с обратным холодильником в течение 2 ч под защитой азота. Реакционную смесь концентрировали, и 20 мл насыщенного раствора бикарбоната натрия и 20 мл этилацетата добавляли к остатку, полученному после концентрирования. После разделения жидких фаз органическую фазу собирали, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией с получением Соединения 60-4. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.18 (d, J=2.6 Гц, 1H), 7.13 (d, J=1.5 Гц, 1H), 7.14-7.11 (m, 1H), 6.49-5.91 (m, 1H), 3.77 (s, 3Н), 2.68 (q, J=7.6 Гц, 2Н), 2.53 (s, 3Н), 1.61 (s, 6Н), 1.29 (t, J=7.5 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 304 (М+1).
4) Синтез Соединения 60

В сухую одногорлую колбу добавляли Соединение 60-4 (ПО мг), Соединение 1-7 (165 мг), DMF (0,2 мл) и метилбензол (1 мл), нагревали до 90°С и перемешивали в течение 48 ч под защитой азота. Реакционную смесь концентрировали досуха, и полученный неочищенный продукт очищали методом препаративной ВЭЖХ с получением Соединения 60. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.95 (d, J=2.0 Гц, 1Н), 8.03 (d, J=8.4 Гц, 1Н), 7.98 (s, 1H), 7.86 (br d, J=8.2 Гц, 1Н), 7.48 (s, 1Н), 6.44 (s, 1H), 2.79 (q, J=7.6 Гц, 2Н), 2.64 (s, 3Н), 1.69 (s, 6Н), 1.37 (t, J=7.5 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 500 (М+1).
Пример 60. Синтез Соединения 61

1) Синтез Соединения 61-2

В сухую одногорлую колбу добавляли Соединение 61-1 (5,00 г) и ацетонитрил (50 мл), поддерживая температуру 0°С, и в реакционную колбу медленно по каплям добавляли NBS (6,92 г), растворенный в ацетонитриле (50 мл), поддерживая температуру не более 10°С. После завершения капельного добавления полученную смесь перемешивали при 20°С в течение 20 ч и затем концентрировали досуха при пониженном давлении для удаления растворителя. К полученному твердому неочищенному продукту добавляли 200 мл насыщенного раствора бикарбоната натрия. После ультразвуковой обработки в течение 1 ч и последующего фильтрования осадок на фильтре промывали 100 мл воды с получением Соединения 61-2. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.06 (br s, 1H), 7.63 (d, J=2.0 Гц, 1H), 4.93 (br s, 2Н). ЖХ/МС (ЭРИ) m/z: 207 (М+1).
2) Синтез Соединения 61-3

В сухую одногорлую колбу добавляли Соединение 61-2 (5,00 г), Соединение 30-2 (4,52 г) и полифосфорную кислоту (15 г) и перемешивали при 110°С в течение 16 ч под защитой азота. 200 мл воды добавляли в реакционную смесь, и полученную смесь перемешивали до растворения. 200 мл этилацетата добавляли для экстракции. После разделения жидких фаз органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха при пониженном давлении для удаления растворителя. Полученный неочищенный продукт очищали колоночной хроматографией с получением Соединения 61-3. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 9.06 (d, J=2.0 Гц, 1Н), 7.89 (d, J=2.0 Гц, 1H), 6.40 (s, 1Н), 2.76 (q, J=7.6 Гц, 2Н), 1.32 (t, J=7.5 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 288 (М+1).
3) Синтез Соединения 61-4

В сухую пробирку для микроволновой обработки добавляли Соединение 61-3 (500 мг), 2-аминоизомасляную кислоту (538 мг), 2-ацетилциклогексанон (49 мг), карбонат калия (481 мг), DMF (4 мл) и воду (1 мл) и затем добавляли хлорид одновалентной меди (34 мг). После продувки азотом в течение 5 минут пробирку для микроволновой обработки герметично закрывали, и полученную смесь подвергали воздействию микроволнового излучения и перемешивали при 110°С в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении с получением Соединения 61-4. ЖХ/МС (ЭРИ) m/z: 310 (М+1).
4) Синтез Соединения 61-5

В сухую одногорлую колбу добавляли Соединение 61-4 (300 мг) и раствор соляная кислота-метанол (4М, 8,76 мл), нагревали до 90°С и кипятили с обратным холодильником в течение 2 ч под защитой азота. Реакционную смесь концентрировали досуха при пониженном давлении. 50 мл насыщенного раствора бикарбоната натрия и 50 мл этилацетата добавляли к остаточному твердому продукту. После разделения жидких фаз органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха для удаления растворителя. Полученный неочищенный продукт очищали колоночной хроматографией с получением Соединения 61-5. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.21 (d, J=2.6 Гц, 1H), 7.48 (d, J=2.6 Гц, 1H), 6.30 (s, 1H), 4.41 (s, 1Н), 3.77 (s, 3Н), 2.73 (q, J=7.6 Гц, 2Н), 1.61 (s, 6Н), 1.29 (t, J=7.6 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 323.77 (М+1).
5) Синтез Соединения 61

В сухую одногорлую колбу добавляли Соединение 61-5 (110 мг), Соединение 1-7 (155 мг), DMF (0,2 мл) и метилбензол (1 мл) и перемешивали при 90°С в течение 20 ч под защитой азота. Полученную смесь концентрировали досуха при пониженном давлении, и полученный неочищенный продукт очищали методом препаративной ВЭЖХ с получением Соединения 61. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.94 (d, J=2.4 Гц, 1H), 8.00 (d, J=8.4 Гц, 1H), 7.94 (s, 1H), 7.82 (d, J=8.2 Гц, 1H), 7.78 (d, J=2.2 Гц, 1H), 6.46 (s, 1H), 2.81 (q, J=7.4 Гц, 2Н), 1.68 (s, 6Н), 1.36 (t, J=7.5 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 520 (М+1).
Пример 61. Синтез Соединения 62

1) Синтез Соединения 62-2

В сухую одногорлую колбу добавляли Соединение 34-1 (5,00 г) и Соединение 62-1 (5,65 г) и затем добавляли полифосфорную кислоту (15,00 г). Полученную смесь нагревали до 110°С и перемешивали при этой температуре в течение 16 ч под защитой азота. Реакционную смесь медленно добавляли в ледяной водный раствор бикарбоната натрия (примерно 500 мл), доводили до рН=7 и экстрагировали этилацетатом (200 мл×4). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали колоночной хроматографией с получением Соединения 62-2. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 9.04 (t, J=1.57 Гц, 1H), 7.67 (dd, J=7.78, 2.01 Гц, 1H), 6.84 (s, 1Н), 6.37-6.70 (m, 1H).
2) Синтез Соединения 62-3

В сухую одногорлую колбу добавляли Соединение 62-2 (1,00 г), трет-бутилкарбамат (1,20 г), карбонат цезия (2,78 г), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (197 мг) и метилбензол (20 мл). Под защитой азота добавляли бис(дибензилиденацетон)палладий (196 мг), и полученную смесь кипятили с обратным холодильником при 120°С в течение 2 ч. Реакционную смесь разбавляли водой (30 мл) и экстрагировали этилацетатом (30 мл ×3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали колоночной хроматографией с получением Соединения 62-3. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 9.10 (s, 1H), 8.35 (br s, 1Н), 7.88 (br s, 1H), 6.77 (s, 1H), 6.40-6.70 (m, 1Н), 1.57 (s, 9Н).
3) Синтез Соединения 62-4

В сухую одногорлую колбу добавляли Соединение 62-3 (1,50 г) и раствор соляная кислота/метанол (4М, 576,92 мл) и перемешивали при 25°С в течение 16 ч. Реакционную смесь концентрировали досуха при пониженном давлении. Полученный остаток растворяли в 50 мл воды, доводили до рН=8-9 добавлением насыщенного раствора бикарбоната натрия, экстрагировали этилацетатом (50 мл ×3), промывали насыщенным рассолом (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Концентрат очищали колоночной хроматографией с получением Соединения 62-4. ЖХ/МС (ЭРИ) m/z: 230 (М+1).
4) Синтез Соединения 62-5

В сухую реакционную колбу добавляли Соединение 62-4 (450 мг), хлорид цинка (80 мг), сульфат натрия (1,12 г), ацетон (684 мг), триметилсилилцианид (584 мг) и тетрагидрофуран (4,5 мл) и подвергали реакции при 30°С в течение 4 ч под защитой азота. Реакционную смесь напрямую сушили в центробежной сушилке и очищали колоночной хроматографией с получением Соединения 62-5. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.55 (s, 1H), 7.38 (dd, J=9.79, 2.51 Гц, 1Н), 6.78 (s, 1H), 6.40-6.69 (m, 1Н), 2.02-2.21 (m, 1H), 1.82 (s, 6Н).
5) Синтез Соединения 62

В сухую реакционную колбу добавляли Соединение 62-5 (200 мг), Соединение 1-7 (616 мг), метилбензол (2 мл) и DMF (0,5 мл). Под защитой азота добавляли гидрид натрия (40 мг, чистота 60%), и полученную смесь подвергали реакции при 25°С в течение 0,5 ч. Реакционную смесь напрямую сушили в центробежной сушилке и очищали методом препаративной ВЭЖХ. После завершения разделения продукт оставляли стоять в системе разделения(вода (0,05% HCl)-ацетонитрил) еще в течение 16 ч, затем доводили до рН=8 насыщенным водным раствором бикарбоната натрия и экстрагировали дихлорметаном (10 мл ×3). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученное твердое вещество растворяли в воде (40 мл) и ацетонитриле (8 мл), и полученный раствор подвергали сублимационной сушке с получением Соединения 62. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.89 (s, 1H), 8.03 (d, J=8.38 Гц, 1Н), 7.95 (s, 1Н), 7.81-7.85 (m, 1H), 7.56 (dd, J=8.49, 2.09 Гц, 1H), 6.90 (s, 1H), 6.43-6.72 (m 1H), 1.71 (s, 6Н).
Пример 62. Синтез Соединения 63

1) Синтез Соединения 63-3
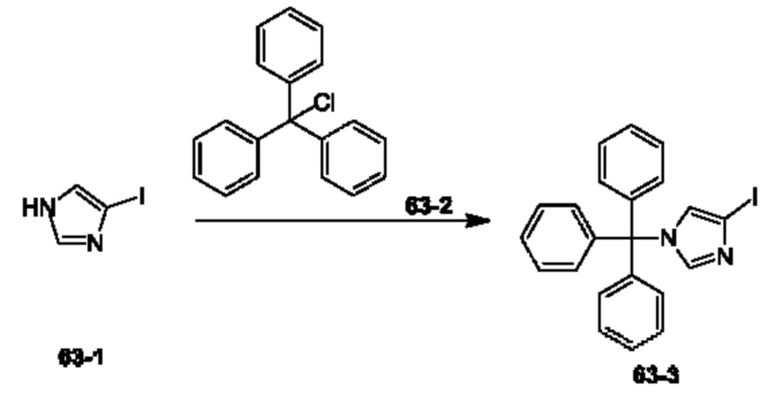
В сухую одногорлую колбу добавляли Соединение 63-1 (1,00 г), DMF (10 мл) и триэтиламин (620 мг). Под защитой азота добавляли Соединение 63-2 (1,44 г), и полученную смесь подвергали реакции при 20°С в течение 16 ч. Реакционную смесь разбавляли 10 мл ледяной воды и затем фильтровали. Осадок на фильтре собирали и суспендировали в 3 мл метил-трет-бутилового эфира для очистки. Твердое вещество собирали с получением Соединения 63-3. 1 ЯМР (400 МГц, CDCl3) δ м.д. 7.34-7.38 (m, 9 Н), 7.33 (d, J=1.38 Гц, 1 Н), 7.10-7.15 (m, 6 Н), 6.92 (d, J=1.38 Гц, 1 Н).
2) Синтез Соединения 63

В сухую реакционную колбу добавляли Соединение 63-3 (167 мг) и тетрагидрофуран (1,2 мл). Под защитой азота быстро добавляли раствор бромида этилмагния в тетрагидрофуране (3М, 147 мкл), и полученную смесь подвергали реакции при комнатной температуре (15°С) в течение 0,17 ч. Раствор хлорид цинка в диэтиловом эфире (1М, 766 мкл) быстро добавляли в реакционную систему, и полученную смесь дополнительно подвергали реакции при 15°С в течение 2 ч. Затем под защитой азота реакционную систему переносили в сухую реакционную колбу, заполненную Соединением 36-4 (0,10 г) и тетракис(трифенилфосфин)палладием (22 мг), нагревали до 95°С и кипятили с обратным холодильником в течение 12 ч. Реакционную смесь концентрировали, и полученный неочищенный продукт очищали последовательно с использованием пластины для препаративной ТСХ и методом препаративной ВЭЖХ с получением Соединения 63. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.29 (s, 1H), 8.02 (d, J=8.16 Гц, 1H), 7.97 (s, 1Н), 7.94 (d, J=1.54 Гц, 1Н), 7.85 (dd, J=8.27, 1.87 Гц, 1H), 7.69 (t, J=1.43 Гц, 1Н), 7.61 (dd, J=9.59, 2.09 Гц, 1H), 7.36 (s, 1H), 3.24 (q, J=7.57 Гц, 2Н), 1.68 (s, 6Н), 1.51 (t, J=7.61 Гц, 3Н).
Пример 63. Синтез Соединения 64

1) Синтез Соединения 64

В сухую одногорлую колбу добавляли Соединение 55-4 (5 мг), Соединение 23-7 (7 мг), DMF (0,1 мл) и метилбензол (0,5 мл), нагревали до 90°С и перемешивали в течение 48 ч под защитой азота. Реакционную смесь концентрировали досуха для удаления растворителя, и полученный неочищенный продукт очищали методом препаративной ВЭЖХ с получением Соединения 64. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 9.08 (s, 1H), 8.87 (s, 1H), 8.34 (s, 1H), 7.44 (br d, J=8.4 Гц, 1H), 5.89 (s, 1H), 4.48 (q, J=6.9 Гц, 2Н), 1.71 (s, 6Н), 1.46 (t, J=6.9 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 521 (М+1).
Пример 64. Синтез Соединения 65

1) Синтез Соединения 65-1

В сухую одногорлую колбу добавляли Соединение 55-1 (500 мг), карбонат калия (533 мг), 2-бромэтил-метиловый эфир (606 мг) и NMP (5 мл), нагревали до 70°С и перемешивали в течение 72 ч под защитой азота. 100 мл насыщенного рассола и 100 мл этилацетата последовательно добавляли в реакционную смесь. После разделения жидких фаз органическую фазу собирали, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией, суспендировали в 10 мл метил-трет-бутилового эфира и фильтровали с получением Соединения 65-1. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.99 (d, J=1.3 Гц, 1H), 7.57 (dd, J=1.8, 7.7 Гц, 1H), 5.90 (s, 1H), 4.56 (dd, J=4.0, 5.3 Гц, 2Н), 3.74 (dd, J=4.0, 5.3 Гц, 2Н), 3.43 (d, J=1.1 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 317 (М+1).
2) Синтез Соединения 65-2

В сухую пробирку для микроволновой обработки добавляли Соединение 65-1 (230 мг), 2-аминоизомасляную кислоту (224 мг), 2-ацетилциклогексанон (20 мг), карбонат калия (200 мг), DMF (2 мл) и воду (0,4 мл) и затем добавляли хлорид одновалентной меди (14 мг). После продувки азотом в течение 5 минут пробирку для микроволновой обработки герметично закрывали, и полученную смесь подвергали воздействию микроволнового излучения и перемешивали при 90°С в течение 4 ч. Реакционную смесь концентрировали, и полученный неочищенный продукт очищали колоночной хроматографией с получением Соединения 65-2. ЖХ/МС (ЭРИ) m/z: 340 (М+1).
3) Синтез Соединения 65-3

В сухую одногорлую колбу добавляли Соединение 65-2 (320 мг), дихлорметан (6 мл) и метанол (1 мл) и затем добавляли TMSCHN2 (2М, 1,89 мл). Под защитой азота полученную смесь перемешивали при 18°С в течение 1 ч. Добавляли TMSCHN2 (2М, 1,89 мл), и реакцию продолжали в течение еще 2 ч. Реакционную смесь концентрировали досуха при пониженном давлении для удаления растворителя, и полученный неочищенный продукт очищали методом препаративной ВЭЖХ с получением Соединения 65-3. ЖХ/МС (ЭРИ) m/z: 354 (М+1).
4) Синтез Соединения 65

В сухую одногорлую колбу добавляли Соединение 65-3 (30 мг), Соединение 1-7 (39 мг), DMF (0,1 мл) и метилбензол (0,5 мл), выдерживали при 90°С и перемешивали в течение 48 ч под защитой азота. Реакционную смесь концентрировали досуха при пониженном давлении. Полученный неочищенный продукт очищали методом препаративной ВЭЖХ с получением Соединения 65. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.88 (s, 1H), 8.01 (d, J=8.4 Гц, 1H), 7.94 (s, 1H), 7.82 (dd, J=2.0, 8.4 Гц, 1H), 7.47 (dd, J=2.1, 8.5 Гц, 1H), 5.96 (s, 1H), 4.64-4.60 (m, 2Н), 3.79-3.74 (m, 2Н), 3.45 (s, 3Н), 1.68 (s, 6Н). ЖХ/МС (ЭРИ) m/z: 550 (М+1).
Пример 65. Синтез Соединения 66

1) Синтез Соединения 66-1

В реакционную колбу добавляли Соединение 35-2 (500 мг), Соединение 3-2 (789 мг), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (107 мг), карбонат цезия (1,50 г) и метилбензол (5 мл). Под защитой азота добавляли бис(дибензилиденацетон)палладий (106 мг), и полученную смесь подвергали реакции при 90°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении, и полученный неочищенный продукт разделяли колоночной хроматографией с получением Соединения 66-1. ЖХ/МС (ЭРИ) m/z: 333 (М+1).
2) Синтез Соединения 66-2

В пробирку для микроволновой обработки добавляли Соединение 66-1 (200 мг), Соединение 2-5 (123 мг), карбонат цезия (392 мг), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (35 мг) и метилбензол (2 мл). Под защитой азота добавляли бис(дибензилиденацетон)палладий (34,56 мг), и полученную смесь выдерживали при 130°С для реакции под воздействием микроволнового излучения в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта, который очищали методом препаративной ВЭЖХ с получением Соединения 66-2. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.21-8.26 (m, 2Н), 8.07-8.12 (m, 2Н), 7.85 (s, 1Н), 7.70 (d, J=8.38 Гц, 1H), 7.08 (br d, J=7.50 Гц, 1Н), 6.90 (d, J=5.29 Гц, 1H), 6.33 (s, 1H), 5.63 (br s, 1H), 2.74 (q, J=7.64 Гц, 2H), 2.25 (s, 3H), 1.22-1.24 (t, J=7.61 Гц, 3Н).
3) Синтез Соединения 66

При 0°С в реакционную колбу добавляли Соединение 66-3 (40 мг) и тетрагидрофуран (0,4 мл) и основательно перемешивали, и затем добавляли NaH (11 мг, чистота 60%). После протекания реакции в течение 0,5 ч добавляли тиофосген (19 мг), и реакционную смесь перемешивали при 25°С в течение 15,5 ч. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного продукта, который очищали последовательно с использованием пластины для препаративной ТСХ и методом препаративной ВЭЖХ с получением Соединения 66. 1H ЯМР (400 МГц, CDCl3) δ м.д. 9.01 (s, 1H), 8.28 (s, 1H), 8.17 (d, J=5.07 Гц, 1H), 8.15 (d, J=1.98 Гц, 1H), 8.07-8.12 (m, 1H), 7.63 (dd, J=8.49, 2.32 Гц, 1H), 7.07 (d, J=5.29 Гц, 1Н), 6.52 (s, 1Н), 2.85 (q, J=7.50 Гц, 2Н), 2.15 (s, 3Н), 1.39 (t, J=7.50 Гц, 3Н). ЖХ/МС (ЭРИ) m/z: 525 (М+1).
Пример 66. Синтез Соединения 67

1) Синтез Соединения 67

В сухую реакционную колбу добавляли Соединение 65 (40 мг) и дихлорметан (0,5 мл). Под защитой азота добавляли трибромид бора (73 мг) при 0°С, и полученную смесь нагревали до 15°С и подвергали реакции в течение 2 ч. Реакцию гасили насыщенным раствором бикарбоната натрия (15 мл). Полученную смесь экстрагировали дихлорметаном (10 мл ×3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали. Остаток, полученный после концентрирования, очищали методом препаративной ВЭЖХ с получением Соединения 67. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.89 (s, 1H), 7.99-8.05 (m, 1H), 7.95 (br s, 1H), 7.83 (br d, J=8.16 Гц, 1H), 7.51 (br d, J=7.72 Гц, 1H), 5.96 (d, J=3.09 Гц, 1H), 4.59 (br d, J=4.41 Гц, 2H), 4.01 (br s, 2H), 2.53 (br s, 1H), 1.69 (d, J=2.87 Гц, 6H). ЖХ/МС (ЭРИ) m/z: 536 (М+1).
Пример 67. Синтез Соединения 68

1) Синтез Соединения 68-1
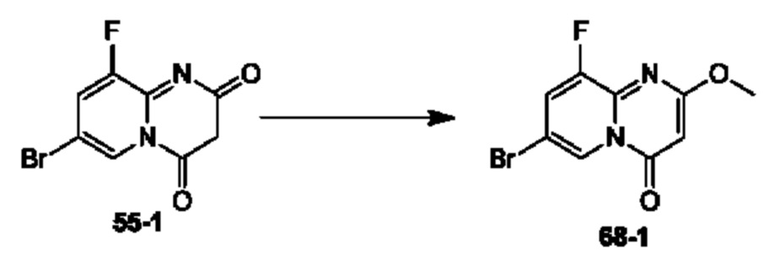
В сухую одногорлую колбу добавляли Соединение 55-1 (1,00 г), N-метилпирролидон (10 мл) и карбонат калия (1,07 г). Под защитой азота добавляли йодметан (1,24 г), и полученную смесь подвергали реакции при 40°С в течение 16 ч. Реакционную смесь разбавляли водой (200 мл) и экстрагировали этилацетатом (150 мл ×2). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали досуха с получением Соединения 68-1. 1H ЯМР (400 МГц, CDCl3) δ м.д. 9.01 (s, 1Н), 7.60 (dd, J=7.84, 2.07 Гц, 1H), 5.87 (s, 1H), 4.04 (s, 3Н).
2) Синтез Соединения 68-2

В пробирку для микроволновой обработки добавляли Соединение 68-1 (600 мг), Соединение 1-4 (340 мг), DMF (12 мл), воду (2,4 мл), карбонат калия (607 мг) и 2-ацетилциклогексанон (31 мг). В условиях продувки азотом добавляли хлорид одновалентной меди (22 мг), и полученную смесь выдерживали при 110°С для реакции под воздействием микроволнового излучения в течение 3 ч. Реакционную смесь напрямую концентрировали, и остаток, полученный после концентрирования, очищали колоночной хроматографией с получением Соединения 68-2. 1Н ЯМР (400 МГц, CD3OD) δ м.д. 7.98 (br s, 1H), 7.40-7.49 (m, 1H), 5.74 (s, 1H), 3.98 (s, 3Н), 1.60 (s, 6Н), 1.58-1.62 (m, 1H).
3) Синтез Соединения 68-3
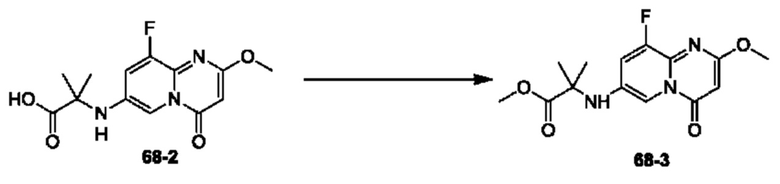
В сухую одногорлую колбу добавляли Соединение 68-2 (530 мг), дихлорметан (10 мл) и метанол (1,7 мл). Под защитой азота добавляли раствор триметилсилилдиазометана в н-гексане (2М, 3,59 мл), и полученную смесь подвергали реакции при 15°С в течение 16 ч. Реакционную смесь концентрировали, и остаток, полученный после концентрирования, очищали колоночной хроматографией с получением Соединения 68-3. 1H ЯМР (400 МГц, CDCl3) δ м.д. 8.16 (d, J=1.38 Гц, 1H), 7.14 (dd, J=10.16, 2.51 Гц, 1H), 5.80 (s, 1Н), 4.32 (br s, 1Н), 3.99 (s, 3Н), 3.80 (s, 3Н), 1.63 (s, 6Н).
4) Синтез Соединения 68

В сухую реакционную колбу добавляли Соединение 68-3 (120 мг), метилбензол (2,5 мл) и DMF (0,5 мл). Под защитой азота добавляли Соединение 1-7 (177 мг), и полученную смесь подвергали реакции при 90°С в течение 16 ч. Реакционную смесь очищали методом препаративной ВЭЖХ с получением Соединения 68. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.89 (s, 1H), 8.02 (d, J=8.38 Гц, 1H), 7.95 (s, 1H), 7.83 (d, J=7.72 Гц, 1H), 7.48 (br d, J=8.16 Гц, 1H), 5.92 (s, 1H), 4.08 (s, 3Н), 1.69 (s, 6H). ЖХ/МС (ЭРИ) m/z: 506 (М+1).
Пример 68. Синтез Соединения 69

1) Синтез Соединения 69-2
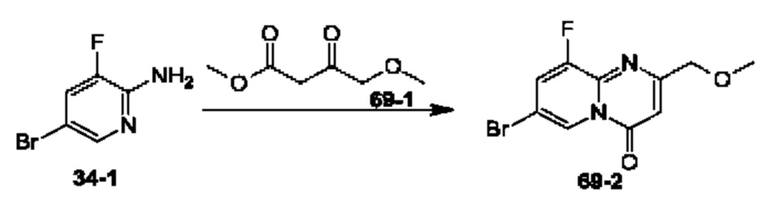
В сухую одногорлую колбу добавляли Соединение 34-1 (3,00 г) и полифосфорную кислоту (20 мл) и затем добавляли Соединение 69-1 (4,59 г, 31,41 ммоль). Под защитой азота полученную смесь нагревали до 110°С и перемешивали в течение 16 ч. В реакционную смесь добавляли 200 мл воды и затем добавляли 200 мл этилацетата. Полученную смесь фильтровали для удаления нерастворимых веществ. Фильтрат оставляли стоять для разделения фаз. Органическую фазу собирали, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали досуха при пониженном давлении для удаления растворителя. Полученный неочищенный продукт очищали колоночной хроматографией с получением Соединения 69-2. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 9.01 (t, 3=1.6 Гц, 1H), 7.57 (dd, J=2.0, 8.0 Гц, 1H), 6.74 (s, 1H), 4.53 (s, 2Н), 3.56-3.53 (m, 3Н). ЖХ/МС (ЭРИ) m/z: 287 (М+1).
2) Синтез Соединения 69-3
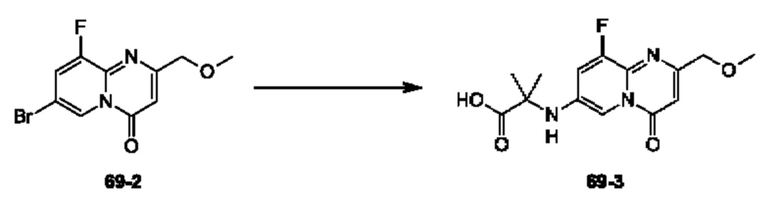
В сухую одногорлую колбу добавляли Соединение 69-2 (200 мг), 2-аминоизомасляную кислоту (216 мг), карбонат калия (193 мг), DMF (4 мл), воду (1 мл) и 2-ацетилциклогексанон (19 мг) и затем добавляли хлорид одновалентной меди (14 мг). После продувки азотом в течение 5 минут одногорлую колбу герметично закрывали, и затем полученную смесь подвергали воздействию микроволнового излучения и перемешивали при 90°С в течение 3 ч. Реакционную смесь концентрировали досуха при пониженном давлении для удаления растворителя, и полученный неочищенный продукт очищали колоночной хроматографией с получением Соединения 69-3. ЖХ/МС (ЭРИ) m/z: 310 (М+1).
3) Синтез Соединения 69-4

В сухую одногорлую колбу добавляли Соединение 69-3 (150 мг), дихлорметан (3 мл) и метанол (0,45 мл) и затем добавляли раствор TMSCHN2 в н-гексане (2М, 1,94 мл). Под защитой азота полученную смесь перемешивали при 18°С в течение 2 ч. Реакционную смесь концентрировали досуха при пониженном давлении для удаления растворителя, и полученный неочищенный продукт очищали колоночной хроматографией с получением Соединения 69-4. ЖХ/МС (ЭРИ) m/z: 324 (М+1).
4) Синтез Соединения 69

В сухую одногорлую колбу добавляли Соединение 69-4 (130 мг), DMF (0,2 мл) и метилбензол (1,5 мл) и затем добавляли Соединение 1-7 (276 мг). Под защитой азота полученную смесь нагревали до 90°С и перемешивали в течение 16 ч. Реакционную смесь концентрировали досуха при пониженном давлении. Полученный неочищенный продукт очищали методом препаративной ВЭЖХ с получением Соединения 69. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.87 (s, 1Н), 8.04 (d, J=8.3 Гц, 1Н), 7.97 (s, 1H), 7.85 (dd, J=1.9, 8.3 Гц, 1H), 7.45 (dd, J=2.2, 8.7 Гц, 1H), 6.81 (s, 1H), 4.58 (s, 2Н), 3.57 (s, 3Н), 1.72 (s, 6Н). ЖХ/МС (ЭРИ) m/z: 520 (М+1).
Пример 69. Синтез Соединения 70

1) Синтез Соединения 70-1

1,1'-Карбонилдиимидазол (1,04 г) добавляли в раствор Соединения 10-3 (1,00 г) в тетрагидрофуране (10 мл). Полученную смесь перемешивали при 70°С в течение 16 ч. Реакционную смесь фильтровали, и осадок на фильтре сушили при пониженном давлении с получением Соединения 70-1. 1H ЯМР (400 МГц, DMSO-d6) δ м.д. 11.59 (br s, 2Н), 7.91 (dd, J=2.0, 10.0 Гц, 1H), 7.80 (s, 1H).
2) Синтез Соединения 70-2
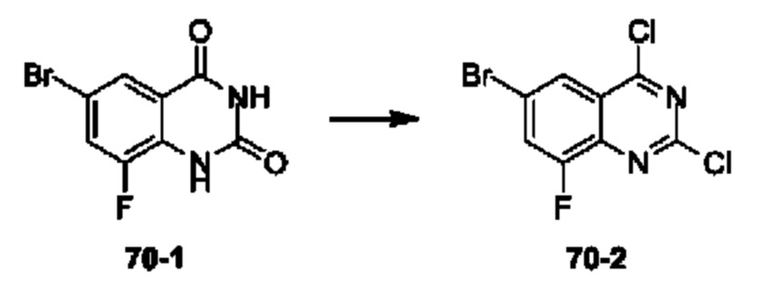
N,N-диизопропилэтиламин (7,18 г) добавляли по каплям в раствор Соединения 70-1 (9,60 г) в оксихлориде фосфора (50 мл). Полученную смесь перемешивали при 110°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении, разбавляли нагретым дихлорметаном (200 мл) и затем медленно вливали в ледяную воду (150 мл). После разделения жидких фаз водный слой экстрагировали дихлорметаном (100 мл ×2), и органические фазы объединяли, последовательно промывали насыщенным водным раствором бикарбоната натрия (200 мл) и насыщенным рассолом (200 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 70-2. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.17 (t, 3=1.6 Гц, 1H), 7.76 (dd, 3=1.9, 8.7 Гц, 1H); ЖХ/МС (ЭРИ) m/z: 297 (М+1).
3) Синтез Соединения 70-3

Водный раствор гидроксида натрия (1М, 40 мл) добавляли в раствор Соединения 70-2 (4,00 г) в тетрагидрофуране (50 мл), и полученную смесь дополнительно перемешивали при 10°С в течение 3 ч. Реакционную смесь вливали в водный раствор соляной кислоты (1 н.) (рН примерно 7) и экстрагировали этилацетатом (100 мл ×3). Органическую фазу промывали насыщенным рассолом (150 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением Соединения 70-3. ЖХ/МС (ЭРИ) m/z: 279 (М+1).
4) Синтез Соединения 70-4
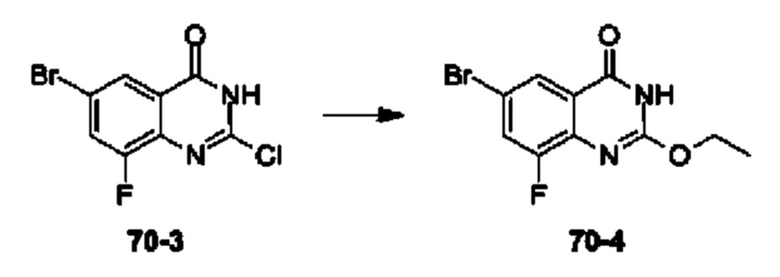
В пробирке для микроволновой обработки этоксид натрия (660 мг) добавляли в смешанный раствор Соединения 70-3 (900 мг) и этанола (12 мл). Полученную смесь выдерживали при 110°С для реакции под воздействием микроволнового излучения в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, растворяли в воде (30 мл) и экстрагировали этилацетатом (30 мл ×3). Затем водную фазу экстрагировали смесью дихлорметан/метанол (об./об. = 10/1, 30 мл ×3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением Соединения 70-4. 1H ЯМР (400 МГц, DMSO-d6) δ м.д. 7.84 (s, 1H), 7.69 (dd, J=2.0, 10.3 Гц, 1Н), 4.35 (q, J=7.0 Гц, 2Н), 1.30 (t, J=7.0 Гц, 3Н).
5) Синтез Соединения 70-5

N,N-Диизопропилэтиламин (742 мг) добавляли по каплям в раствор Соединения 70-4 (1,10 г) в оксихлориде фосфора (8 мл), и полученную смесь перемешивали при 110°С в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, разбавляли дихлорметаном (150 мл) и вливали в ледяную воду. После разделения жидких фаз органическую фазу последовательно промывали насыщенным водным раствором бикарбоната натрия (150 мл) и насыщенным рассолом (150 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением Соединения 70-5. ЖХ/МС (ЭРИ) m/z: 307 (М+3).
6) Синтез Соединения 70-6

Гидрид натрия (190 мг, чистота 60%) добавляли в раствор Соединения 70-5 (1,20 г) и этандиола (360 мг) в тетрагидрофуране (20 мл), и полученную смесь перемешивали при 10°С в течение 24 ч. Реакционную смесь вливали в насыщенный водный раствор хлорида аммония (50 мл) и экстрагировали этилацетатом (50 мл ×3). Органическую фазу промывали насыщенным рассолом (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением Соединения 70-6. ЖХ/МС (ЭРИ) m/z: 333 (М+3).
7) Синтез Соединения 70-7

Триэтиламин (1,19 г) и 4-диметиламинопиридин (48 мг) добавляли в смешанный раствор Соединения 70-6 (1,30 г), ди-трет-бутилдикарбоната (1,03 г) и дихлорметана (20 мл), и полученную смесь перемешивали при 15°С в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 70-7. 1H ЯМР (400 МГц, CDCl3) δ м.д. 7.96-7.92 (m, 1H), 7.49 (dd, J=2.1, 9.7 Гц, 1H), 4.73-4.69 (m, 2Н), 4.52-4.44 (m, 4Н), 1.44 (s, 9Н), 1.40 (t, J=7.0 Гц, 3Н).
8) Синтез Соединения 70-8

Соединение 70-7 (600 мг), Соединение 1-4 (215 мг), карбонат калия (480 мг), хлорид одновалентной меди (28 мг), 2-ацетилциклогексанон (39 мг), N,N-диметилформамид (6 мл) и воду (0,3 мл) добавляли в пробирку для микроволновой обработки. Пробирку для микроволновой обработки герметично закрывали, и полученную смесь выдерживали при 120°С для реакции под воздействием микроволнового излучения в течение 1 ч. Реакционную смесь фильтровали и промывали этилацетатом (10 мл). Фильтрат концентрировали при пониженном давлении. 1 н. соляную кислоту добавляли к остатку, полученному после концентрирования (рН=6-7), и полученную смесь экстрагировали смесью тетрагидрофуран/этилацетат (1/3, 20 мл × 3) для разделения жидких фаз. Органическую фазу промывали насыщенным рассолом (30 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением Соединения 70-8. ЖХ/МС (ЭРИ) m/z: 454 (М+1).
9) Синтез Соединения 70-9

Раствор триметилсилилдиазометана в н-гексане (2М, 1,1 мл) добавляли в раствор Соединения 70-8 (650 мг), дихлорметана (10 мл) и метанола (1 мл), и полученную смесь перемешивали при 10°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 70-9. ЖХ/МС (ЭРИ) m/z: 468 (М+3).
10) Синтез Соединения 70-10

Смешанный раствор Соединения 70-9 (100 мг), Соединения 1-7 (196 мг), N,N-диметилформамида (0,5 мл) и метилбензола (2 мл) нагревали до 120°С и перемешивали в течение 16 ч. В реакционную смесь добавляли метанол (2 мл), и смесь перемешивали в течение 30 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной тонкослойной хроматографии с получением Соединения 70-10. ЖХ/МС (ЭРИ) m/z: 664 (М+1).
11) Синтез Соединения 70

Трифторуксусную кислоту (1 мл) добавляли в раствор Соединения 70-10 (180 мг) в дихлорметане (4 мл), и полученную смесь перемешивали при 10°С в течение 2 ч. Насыщенный водный раствор бикарбоната натрия добавляли в реакционную смесь (рН примерно 8), и смесь экстрагировали дихлорметаном (10 мл × 3). Органическую фазу промывали насыщенным рассолом (15 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, отделяли и очищали последовательно с использованием пластины для препаративной тонкослойной хроматографией и методом препаративной ВЭЖХ с получением Соединения 70. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.93 (d, J=8.3 Гц, 1Н), 7.90 (d, J=1.5 Гц, 1Н), 7.81-7.74 (m, 2Н), 7.33 (dd, J=2.0, 10.0 Гц, 1H), 4.73-4.65 (m, 2Н), 4.54 (q, J=7.0 Гц, 2Н), 4.01 (br d, J=3.3 Гц, 2Н), 2.17 (br s, 1H), 1.58 (s, 6Н), 1.43 (t, J=7.2 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 564 (М+1).
Пример 70. Синтез Соединения 71
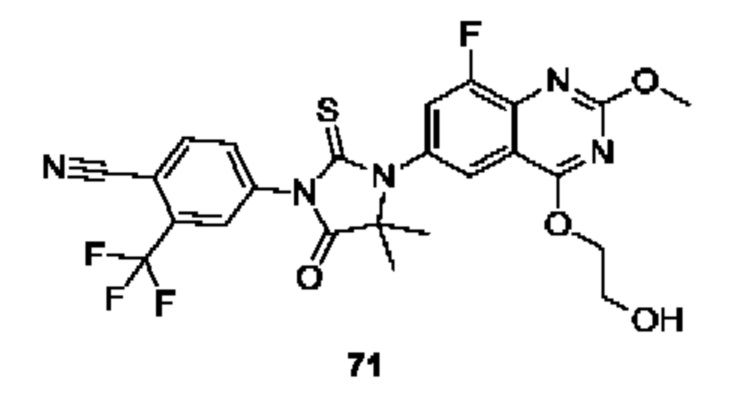
1) Синтез Соединения 71-1

В пробирке для микроволновой обработки метоксид натрия (520 мг) добавляли в раствор Соединения 70-3 (900 мг) в метаноле (10 мл). Полученную смесь выдерживали при 100°С для реакции под воздействием микроволнового излучения в течение 1,5 ч. Реакционную смесь концентрировали при пониженном давлении. Тетрагидрофуран (100 мл) добавляли к остатку, полученному после концентрирования. Полученную смесь перемешивали в течение 20 минут и затем фильтровали. Фильтрат концентрировали при пониженном давлении с получением Соединения 71-1. 1Н ЯМР (400 МГц, DMSO-d6) δ м.д. 7.78-7.73 (m, 1H), 7.42 (dd, J=2.3, 10.3 Гц, 1H), 3.72 (s, 3Н).
2) Синтез Соединения 71-2

N,N-Диизопропилэтиламин (618 мг) добавляли по каплям в раствор Соединения 71-1 (870 мг) в оксихлориде фосфора (6 мл), и полученную смесь перемешивали при 110°С в течение 4 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, разбавляли дихлорметаном (20 мл) и вливали в ледяную воду. После разделения жидких фаз органическую фазу последовательно промывали насыщенным водным раствором бикарбоната натрия (15 мл) и насыщенным рассолом (15 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением Соединения 71-2. ЖХ/МС (ЭРИ) m/z: 293 (М+3).
3) Синтез Соединения 71-3

Гидрид натрия (165 мг, чистота 60%) добавляли в раствор Соединения 71-2 (1,00 г) и этандиола (320 мг) в тетрагидрофуране (20 мл), и полученную смесь перемешивали при 10°С в течение 4 ч. Реакционную смесь вливали в насыщенный водный раствор хлорида аммония (40 мл) и экстрагировали этилацетатом (50 мл × 3). Органическую фазу промывали насыщенным рассолом (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением Соединения 71-3. ЖХ/МС (ЭРИ) m/z: 319 (М+3).
4) Синтез Соединения 71-4

Триэтиламин (1,05 г) и 4-диметиламинопиридин (43 мг) добавляли в смешанный раствор Соединения 71-3 (1,10 г), ди-трет-бутилдикарбоната (908 мг) и дихлорметана (20 мл), и полученную смесь перемешивали при 15°С в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 71-4. 1H ЯМР (400 МГц, CDCl3) δ м.д. 7.98-7.93 (m, 1H), 7.51 (dd, J=2.0, 9.5 Гц, 1Н), 4.75-4.67 (m, 2Н), 4.49-4.43 (m, 2Н), 4.05 (s, 3Н), 1.44 (s, 9Н).
5) Синтез Соединения 71-5

Соединение 71-4 (500 мг), Соединение 1-4 (185 мг), карбонат калия (414 мг), хлорид одновалентной меди (24 мг), 2-ацетилциклогексанон (34 мг), N,N-диметилформамид (4 мл) и воду (0,2 мл) добавляли в пробирку для микроволновой обработки. Пробирку для микроволновой обработки герметично закрывали, и полученную смесь выдерживали при 120°С для реакции под воздействием микроволнового излучения в течение 1 ч. Реакционную смесь фильтровали и промывали этилацетатом (10 мл). Фильтрат концентрировали при пониженном давлении. 1 н. соляную кислоту добавляли к остатку, полученному после концентрирования (рН 6-7), и полученную смесь экстрагировали смесью тетрагидрофуран/этилацетат (1/3, 20 мл × 3) для разделения жидких фаз. Органическую фазу промывали насыщенным рассолом (30 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением Соединения 71-5. ЖХ/МС (ЭРИ) m/z: 440 (М+1).
6) Синтез Соединения 71-6

Раствор триметилсилилдиазометана в н-гексане (2М, 1,1 мл) добавляли в раствор Соединения 71-5 (470 мг) в дихлорметане (5 мл) и метаноле (1 мл), и полученную смесь перемешивали при 10°С в течение 4 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 71-6. ЖХ/МС (ЭРИ) m/z: 454 (М+1).
7) Синтез Соединения 71-7

Смешанный раствор Соединения 71-6 (100 мг), Соединения 1-7 (202 мг), N,N-диметилформамида (0,5 мл) и метилбензола (2 мл) нагревали до 120°С и перемешивали в течение 16 ч. Метанол (2 мл) добавляли в реакционную смесь, и смесь перемешивали в течение 30 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной тонкослойной хроматографии с получением Соединения 71-7. ЖХ/МС (ЭРИ)m/z: 650 (М+1).
8) Синтез Соединения 71

Трифторуксусную кислоту (0,5 мл) добавляли в раствор Соединения 71-7 (100 мг) в дихлорметане (2 мл), и полученную смесь перемешивали при 10°С в течение 2 ч. Насыщенный водный раствор бикарбоната натрия добавляли в реакционную смесь (рН примерно 8), и полученную смесь экстрагировали дихлорметаном (10 мл × 3). Органическую фазу промывали насыщенным рассолом (15 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали последовательно с использованием пластины для препаративной тонкослойной хроматографией и методом препаративной ВЭЖХ с получением Соединения 71. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 7.97-7.87 (m, 2Н), 7.83-7.73 (m, 2Н), 7.35 (dd, 3=23, 10.0 Гц, 1Н), 4.73-4.65 (m, 2Н), 4.09 (s, 3Н), 4.02 (br d, J=3.5 Гц, 2Н), 2.21 (brt, J=5.4 Гц, 1H), 1.59 (s, 6Н); ЖХ/МС (ЭРИ)m/z: 550 (М+1).
Пример 71. Синтез Соединения 72

1) Синтез Соединения 72-1

Смешанный раствор Соединения 70-9 (60 мг), Соединения 23-7 (118 мг), N,N-диметилформамида (0,5 мл) и метилбензола (2 мл) нагревали до 120°С и перемешивали в течение 16 ч. Метанол (2 мл) добавляли в реакционную смесь, и смесь перемешивали в течение 30 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной тонкослойной хроматографии с получением Соединения 72-1. ЖХ/МС (ЭРИ) m/z: 665 (М+1).
2) Синтез Соединения 72

Трифторуксусную кислоту (0,2 мл) добавляли в раствор Соединения 72-1 (45 мг) в дихлорметане (1 мл). Полученную реакционную смесь перемешивали при 10°С в течение 2 ч. Насыщенный водный раствор бикарбоната натрия добавляли в реакционную смесь (рН примерно 8), и полученную смесь экстрагировали дихлорметаном (10 мл × 3). Органическую фазу промывали насыщенным рассолом (15 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, отделяли и очищали методом препаративной ВЭЖХ с получением Соединения 72. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 9.03 (d, J=2.3 Гц, 1H), 8.29 (d, J=2.3 Гц, 1H), 7.77 (d, J=1.5 Гц, 1H), 7.32 (dd, J=2.3, 10.0 Гц, 1H), 4.70 (dd, J=3.8, 5.3 Гц, 2Н), 4.54 (q, J=7.2 Гц, 2Н), 4.01 (br s, 2Н), 2.15 (br s, 1H), 1.61 (s, 6H), 1.43 (t, J=7.2 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 565 (М+1).
Пример 72. Синтез Соединения 73

1) Синтез Соединения 73-1

Смешанный раствор Соединения 71-6 (55 мг), Соединения 23-7 (112 мг), N,N-диметилформамида (0,5 мл) и метилбензола (2 мл) нагревали до 120°С и перемешивали в течение 16 ч. Метанол (2 мл) добавляли в реакционную смесь, и смесь перемешивали в течение 30 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием пластины для препаративной тонкослойной хроматографии с получением Соединения 73-1. ЖХ/МС (ЭРИ) m/z: 651 (М+1).
2) Синтез Соединения 73

Трифторуксусную кислоту (0,2 мл) добавляли в раствор Соединения 73-1 (50 мг) в дихлорметане (1 мл). Полученную реакционную смесь перемешивали при 10°С в течение 2 ч. Насыщенный водный раствор бикарбоната натрия добавляли в реакционную смесь (рН примерно 8), и полученную смесь экстрагировали дихлорметаном (10 мл × 3). Органическую фазу промывали насыщенным рассолом (15 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток, полученный после концентрирования, отделяли и очищали методом препаративной ВЭЖХ с получением Соединения 73. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 9.02 (d, J=2.0 Гц, 1H), 8.29 (d, J=2.0 Гц, 1H), 7.78 (s, 1H), 7.33 (dd, J=2.3, 10.0 Гц, 1H), 4.73-4.65 (m, 2H), 4.10 (s, 3Н), 4.02 (br s, 2H), 2.21 (br s, 1H), 1.61 (s, 6H); ЖХ/МС (ЭРИ) m/z: 551 (M+l).
Пример 73. Синтез Соединения 74

1) Синтез Соединения 74

В сухую реакционную колбу добавляли Соединение 62-5 (100 мг), Соединение 23-7 (309 мг), DMF (0.25 мл) и метилбензол (1 мл). Под защитой азота добавляли гидрид натрия (20 мг, чистота 60%), и полученную смесь подвергали реакции при 25°С в течение 0,5 ч. Реакционную смесь концентрировали. Остаток, полученный после концентрирования, очищали методом кислотной препаративной ВЭЖХ. Затем продукт оставляли стоять в элюенте для разделения (вода (0,05% HCl)-ацетонитрил), затем доводили до рН=8 насыщенным водным раствором бикарбоната натрия и экстрагировали дихлорметаном (10 мл × 3). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный концентрат растворяли в воде (20 мл) и ацетонитриле (8 мл) и затем подвергали сублимационной сушке с получением Соединения 74. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 9.09 (d, J=2.13 Гц, 1H), 8.89 (s, 1H), 8.35 (d, J=2.01 Гц, 1Н), 7.54 (dd, J=8.34, 2.07 Гц, 1Н), 6.91 (s, 1H), 6.42-6.75 (m, 1Н), 1.74 (s, 6Н). ЖХ/МС (ЭРИ) m/z: 527 (М+1).
Пример 74. Синтез Соединения 75

1) Синтез Соединения 75-2

Трис(дибензилиденацетон)дипалладий (421 мг) и 4,5-бис(дифенилфосфино)-9,9-диметилксантен (532 мг) добавляли в смешанный раствор Соединения 75-1 (2,00 г), трет-бутилкарбамата (1,08 г), трет-бутоксида натрия (2,21 г) и метилбензола (40 мл). Под защитой азота полученную смесь перемешивали при 100°С в течение 3 ч. Реакционную смесь фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 75-2. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.40 (s, 1H), 8.33 (d, J=2.3 Гц, 1H), 6.98 (br s, 1H), 1.55 (s, 9Н).
2) Синтез Соединения 75-3

Трифторуксусную кислоту (8 мл) добавляли в смешанный раствор Соединения 75-2 (1,70 г) и дихлорметана (20 мл). Полученную реакционную смесь перемешивали при 10°С в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток, полученный после концентрирования, разбавляли этилацетатом (60 мл) и промывали насыщенным раствором бикарбоната натрия (50 мл × 3). Полученную органическую фазу промывали насыщенным рассолом (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали с использованием силикагелевой колонки с получением Соединения 75-3. ЖХ/МС (ЭРИ) m/z: 154 (М+1).
3) Синтез Соединения 75-4

При 10°С тиофосген (900 мг) добавляли по каплям в воду (20 мл) и затем порциями добавляли Соединение 75-3 (920 мг). Полученную смесь перемешивали при 10°С в течение 1 ч. Реакционную смесь экстрагировали дихлорметаном (30 мл × 2). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали досуха при пониженном давлении с получением Соединения 75-4.
4) Синтез Соединения 75

Смешанный раствор Соединения 35-5 (300 мг) и Соединения 75-4 (764 мг) в N,N-диметилформамиде (1,5 мл) и метилбензоле (6 мл) нагревали до 120°С и перемешивали в течение 16 ч. Метанол (5 мл) добавляли в реакционную смесь, и смесь перемешивали в течение 30 мин и затем концентрировали при пониженном давлении. Остаток, полученный после концентрирования, очищали последовательно с использованием силикагелевой колонки и препаративной ВЭЖХ с получением Соединения 75. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8.82 (s, 1H), 8.78 (d, J=2.0 Гц, 1H), 8.10 (d, J=2.0 Гц, 1H), 7.39 (dd, J=2.1, 8.7 Гц, 1H), 6.49 (s, 1H), 2.82 (q, J=7.5 Гц, 2Н), 1.69 (s, 6Н), 1.36 (t, J=7.5 Гц, 3Н); ЖХ/МС (ЭРИ) m/z: 471 (М+1).
Пример 75. Тестирование антагонизма соединений по отношению к ядерной транслокации андрогенового рецептора (AR)
1. PathHunter NHR клеточные линии выделяли и культивировали для амплификации.
2. Клетки инокулировали на 384-луночный планшет для тестирования и инкубировали при 37°С. Сыворотку для культуры фильтровали со смесью древесный уголь-декстран для снижения в ней уровня гормона.
3. Для обнаружения антагонистической функции соединение добавляли к клеткам и инкубировали в течение 60 мин. Рабочую концентрацию тестируемого соединения разводили, начиная с 10 мкМ, с 3-кратным градиентом концентраций, соответственно включающим: 10000; 3333,3; 1111,1; 370,4; 123,5; 41,2; 13,7; и 4,67 нМ. Затем добавляли агонист 6а-фтортестостерон 0,06 мкМ (концентрация: ЕС80, т.е. концентрация соединения, вызывающая агонистический эффект, равный 80% от максимально возможного), и смесь инкубировали при 37°С или при комнатной температуре в течение 3-16 ч.
4. Детектирование сигнала: 12,5 мкл или 15 мкл (50%, об./об.) смешанный раствор для детектирования PathHunter (набор: DiscoverX Product, номер по каталогу: серия 93-0001) добавляли и инкубировали при комнатной температуре в течение 1 ч. Хемилюминисцентный сигнал регистрировали с использованием прибора PerkinElmerEnvision™.
5. Анализ данных: Активность соединения анализировали с использованием программного обеспечения для анализа данных SBIS (Chemlnnovation, СА). Формула для вычисления ингибирующего процента антагониста следующая: показатель ингибирования IC50 (%)=100%×(1-(среднее значение RLU тестируемого соединения-среднее значение RLU пустой контрольной группы)/(среднее значение RLU контрольной группы ЕС80-среднее значение RLU пустой контрольной группы)).
Результаты тестирования антагонизма соединений по отношению к ядерной транслокации андрогенового рецептора (AR) показаны в Таблице 1 ниже.


Пример 76. Фармакокинетическое тестирование Соединения 10
1. Краткое изложение
Используя самцов мышей CD-1 в качестве животных для тестирования, концентрации лекарственного средства в плазме крови в разные моменты времени после внутривенного и внутрижелудочного введения мышам соединения 10 определяли методом ЖХ/МС/МС. Фармакокинетическое поведение соединения 10 у мышей исследовали, и его фармакокинетический профиль оценивали.
2. Протокол эксперимента
2.1. Тестируемое лекарственное средство: Соединение 10
2.2. Животные для тестирования: Четырех здоровых самцов мышей CD-1 разделяли на 2 группы по 2 мыши в каждой группе в соответствии с принципом сходной массы тела. Животные были приобретены у Shanghai Super-BK Laboratory Animal Co., Ltd., имеющей лицензию на производство животных Animal Production License No.: SCXK (Shanghai) 2013-0016.
2.3. Приготовление лекарственного средства
Отвешивали соответствующее количество образца и последовательно добавляли соответствующее количество DMSO, PEG400 и воды в объемном соотношении 10:40:50. После перемешивания и ультразвуковой обработки полученная смесь достигала состояния прозрачного раствора 0,4 мг/мл для внутривенного введения.
Отвешивали соответствующе количество образца и растворяли в растворе 0,5% CMC+0,2% Tween 80. После перемешивания и ультразвуковой обработки полученная смесь достигала состояния однородной суспензии 0,4 мг/мл для внутрижелудочного введения.
2.4. Введение
Четырех самцов мышей CD-1 разделяли на 2 группы и не кормили в течение ночи. Первой группе внутривенно вводили в объеме для введения 2,5 мл/кг в дозе 1 мг/кг; а второй группе внутрижелудочно вводили в объеме для введения 5 мл/кг в дозе 2 мг/кг.
3. Операции
После внутривенного введения Соединения 10 самцам мышей CD-1 по 30 мкл крови собирали через 0,0833, 0,25, 0,5, 1, 2, 4, 8 и 24 ч соответственно и помещали в пробирки, содержащие 2 мкл of EDTA-K2. После введения Соединения 10 мышам группы внутрижелудочного введения по 30 мкл крови собирали через 0,25, 0,5, 1, 2, 4, 8 и 24 ч соответственно и помещали в пробирки, содержащие 2 мкл of EDTA-K2. Пробирки центрифугировали при 3000 g в течение 15 мин для отделения плазмы крови, и эту плазму хранили при -60°С. Животным давали корм через 2 часа после введения.
После внутривенного и внутрижелудочного введения мышам содержание тестируемого соединения в плазме крови определяли методом ЖХ/МС/МС. Линейный диапазон метода составлял 2,00-6000 нмоль/л; и образцы плазмы крови анализировали после обработки белком, осажденным ацетонитрилом. Результаты фармакокинетического тестирования соединения 10 показаны в Таблице 2 ниже.

Пример 77. Фармакокинетическое тестирование Соединения 35 и Соединения 58
1. Краткое изложение
Согласно Примеру 76 фармакокинетическое поведение Соединения 35 и Соединения 58 у мышей исследовали, и их фармаконетические профили оценивали.
2. Протокол эксперимента смотри в Примере 76.
3. Операции
После внутривенного введения Соединения 35 и Соединения 58 самцам мышей CD-I по 30 мкл крови перекрестно собирали через 0,0833, 0,25, 0,5, 1, 2, 4, 8, 24 и 48 ч соответственно и помещали в пробирки, содержащие 2 мкл EDTA-K2. После внутрижелудочного введения Соединения 35 и Соединения 58 мышам группы внутрижелудочного введения по 30 мкл крови перекрестно собирали через 0,25, 0,5, 1, 2, 4, 8, 24 и 48 ч соответственно и помещали в пробирки, содержащие 2 мкл EDTA-K2. Пробирки центрифугировали при 3000 g в течение 15 мин для отделения плазмы крови, и эту плазму хранили при -60°С. Животным давали корм через 4 часа после введения.
После внутривенного и внутрижелудочного введения мышам содержание тестируемого соединения в плазме крови определяли методом ЖХ/МС/МС. Линейный диапазон метода составлял 2,00-6000 нмоль/л; и образцы плазмы крови анализировали после обработки белком, осажденным ацетонитрилом.
Результаты фармакокинетического тестирования Соединения 35 и Соединения 58 показаны в Таблице 3 ниже.

Пример 78. Тестирование распределения в тканях Соединения 27 и Соединения 10
1. Краткое изложение
Используя самцов мышей CD-1 в качестве животных для тестирования, концентрации лекарственного средства в плазме крови и в головном мозге после внутрижелудочного введения Соединения 27 и Соединения 10 мышам определяли методом ЖХ/МС/МС.
2. Протокол эксперимента
2.1. Тестируемое лекарственное средство: Соединение 27 и Соединение 10
2.2. Животные для тестирования: Шесть здоровых взрослых самцов мышей CD-1 разделяли на 2 группы по 3 мыши в каждой группе в соответствии с принципом сходной массы тела. Животные были приобретены у Shanghai Super-BK Laboratory Animal Co., Ltd., имеющей лицензию на производство животных Animal Production License No.: SCXK (Shanghai) 2013-0016.
2.3. Приготовление лекарственного средства
Отвешивали соответствующее количество образца и добавляли соответствующее количество DMSO, PEG400 и воды последовательно в объемном соотношении 10:40:50. После перемешивания и ультразвуковой обработки полученная смесь достигала состояния прозрачного раствора 0,4 мг/мл.
2.4. Введение
Шесть самцов мышей CD-1 разделяли на 2 группы, не давали им корм в течение ночи и внутрижелудочно вводили им в объеме для введения 5 мл/кг в дозе 2 мг/кг.
3. Операции
После внутрижелудочного введения Соединения 27 и Соединения 10 самцам мышей CD-1 100 мкл крови собирали посредством пункции сердца через 2 ч, помещали в пробирку, содержащую 2 мкл EDTA-K2, и центрифугировали при 3000 g в течение 15 мин для отделения 50 мкл плазмы крови, которую хранили при -60°С. Вместе с тем, ткани головного мозга собирали, промывали, затем гомогенизировали 5-кратным 15 мМ PBS/MeOH (об.:об., 2:1) и хранили при -60°С. Животным давали корм через 2 часа после введения.
После внутрижелудочного введения мышам содержание тестируемого соединения в плазме крови определяли методом ЖХ/МС/МС. Линейный диапазон метода составлял 2,00-6000 нмоль/л; и образцы плазмы крови анализировали после обработки белком, осажденным ацетонитрилом.
Результаты по параметрам распределения в тканях показаны в Таблице 4 ниже.

Пример 79. Тестирование распределения в тканях Соединения 35 и Соединения 58
1. Краткое изложение
Используя самцов мышей CD-1 в качестве животных для тестирования, концентрации лекарственного средства в плазме крови и в головном мозге после внутрижелудочного введения Соединения 35 и Соединения 58 мышам определяли методом ЖХ/МС/МС.
2. Протокол эксперимента
2.1. Тестируемое лекарственное средство: Соединение 35 и Соединение 58
2.2. Животные для тестирования: Два здоровых взрослых самца мышей CD-1 Животные были приобретены у Shanghai Super-BK Laboratory Animal Co., Ltd.
2.3 Приготовление лекарственного средства
Отвешивали соответствующее количество образца и добавляли в раствор 0,5% СМС+0,2% Tween в воде. После перемешивания и ультразвуковой обработки полученная смесь достигала состояния суспензии 0,4 мг/мл.
2.4. Введение лекарственного средства
Двух самцов мышей CD-1 не кормили в течение ночи, и им внутрижелудочно вводили в объеме для введения 5 мл/кг в дозе 2 мг/кг.
3. Операции
После внутрижелудочного введения Соединения 35 и Соединения 58 самцам мышей CD-1 100 мкл крови собирали посредством пункции сердца через 4 ч, помещали в пробирку, содержащую 2 мкл EDTA-K2 и центрифугировали при 3000 g в течение 15 мин для отделения 30 мкл плазмы крови, которую хранили при -60°С. Вместе с тем, ткани головного мозга собирали, промывали, затем гомогенизировали 9-кратным 15 мМ PBS/MeOH (об.:об., 2:1) и хранили при -60°С. Животным давали корм через 4 часа после введения.
После внутрижелудочного введения мышам содержание тестируемого соединения в плазме крови и головном мозге определяли методом ЖХ/МС/МС. Линейный диапазон метода составлял 2,00-6000 нмоль/л; и образцы плазмы крови анализировали после обработки белком, осажденным ацетонитрилом.
Результаты тестирования распределения в тканях показаны в Таблице 5.

Пример 80. Фармакодинамическое исследование in vivo Соединения 27 и Соединения 10 на модели подкожного ксенотрасплантата опухоли из клеток рака предстательной железы человека LNCaP-FGC
1. План эксперимента



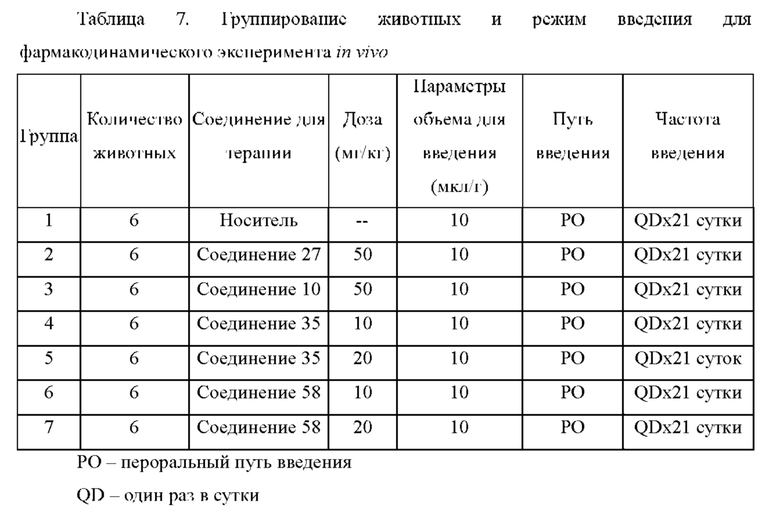
2. Экспериментальные материалы
2.1. Экспериментальные животные
Вид: Мыши
Штамм: СВ-17 SCID мышиный
Возраст в неделях и масса тела: 6-8 недель, масса тела 18-22 г
Пол: самцы
Поставщик: Beijing Vital River Laboratory Animal Technology Co., Ltd Сертификат на животных №11400700184227
3. Метод эксперимента и стадии
3.1. Клеточная культура
Клетки рака предстательной железы человека LNCaP-FGC (АТСС, Manassas, Virginia) культивировали in vitro в монослое в следующих условиях культивирования: среда RPMI1640, дополненная 10% фетальной бычьей сыворотки, при 37°С с 5% СО2. Рутинное расщепление путем обработки трипсином-EDTA осуществляли дважды в неделю для пассажа. Когда клеточное насыщение составляло 80%-90%, клетки собирали, подсчитывали и инокулировали.
3.2. Инокуляция опухолевых клеток
0,2 мл (10×106) клеток LNCaP-FGC (10×106+Matrigel, 1:1) инокулировали подкожно в правую заднюю лапу каждой мыши СВ-17 SCID. Когда средний объем опухоли достигал 100-150 мм, тогда мышам в группах начинали введение.
3.3. Измерение опухолей
Диаметры опухолей измеряли кронциркулем дважды в неделю. Формула для вычисления объема опухоли следующая: V=0,5а×b2, где а и b означают длинный диаметр и короткий диаметр опухоли соответственно. Противоопухолевую активность соединений оценивали по TGI (ингибирование роста опухоли) (%) или относительной скорости пролиферации опухоли Т/С (%). TGI (%)=[(1-(средний объем опухоли в группе лечения в конце введения лекарственного средства-средний объем опухоли в группе лечения в начале введения лекарственного средства))/(средний объем опухоли в контрольной группе, получавшей носитель, в конце лечения-средний объем опухоли в контрольной группе, получавшей носитель, в начале лечения)]×100%. Формула для вычисления относительной скорости пролиферации опухоли Т/С (%) следующая: Т/С%=Trtv/CRTV × 100% (TRTV: RTV в группе лечения; CRTV: RTV в отрицательной контрольной группе). Относительный объем опухоли (RTV) вычисляют, исходя из результатов измерений опухоли, и формула для вычисления следующая: RTV=Vt/V0, где V0 означает средний объем опухоли, измеренный в момент введения в группах (т.е. d0), Vt означает средний объем опухоли при одном измерении, и TRTV и CRTV представляют собой данные, полученные в один и тот же день.
3.4. Статистический анализ
Статистический анализ включает в себя среднее значение и стандартную ошибку среднего (SEM) объема опухоли в каждую точку времени для каждой группы. Группа лечения показала наилучший терапевтический эффект на 21-й день после введения в конце тестирования, и, следовательно, статистический анализ осуществляли с целью оценки различий между группами на основе данных. Сравнение между двумя группами анализировали с использованием критерия Стьюдента, и сравнение межу тремя или более группами анализировали методом однофакторного дисперсионного анализа ANOVA. Если имело место значительное различие в значении F, то использовали метод Games-Howell для тестирования. Если значительное различие отсутствовало в значении F, то для анализа использовали метод Dunnet (2-сторонний). Анализ всех данных осуществляли, используя SPSS 17.0. "р<0,05" считали значимым различием.
4. Результаты эксперимента
Через 21 сутки после введения тестируемое Соединение 10 имело значительный противоопухолевый эффект в группе 50 мг/кг по сравнению с контрольной группой, получавшей растворитель (Т/С=23,8%, TGI=83,0%, p≤0,001); и тестируемое Соединение 27 имело значительный противоопухолевый эффект в группе 50 мг/кг по сравнению с контрольной группой, получавшей растворитель (Т/С=53,1%, TGI=51,0%, р=0,002). В то же время животные имели хорошую переносимость вышеуказанных протестированных соединений.
Через 21 сутки после введения тестируемое Соединение 35 имело значительный противоопухолевый эффект в группе 10 мг/кг и в группе 20 мг/кг по сравнению с контрольной группой, получавшей растворитель (Т/С=47,39% и 32,47% соответственно; TGI=59,36% и 76,00% соответственно; р=0,006 и р<0,001 соответственно); и Соединение 58 имело значительный противоопухолевый эффект в группе 10 мг/кг и в группе 20 мг/кг по сравнению с контрольной группой, получавшей растворитель (Т/С=43,93% и 32,37% соответственно; TGI=62,75% и 76,16% соответственно; р=0,003 и р<0,001 соответственно). В то же время животные имели хорошую переносимость вышеуказанных протестированных соединений.
| название | год | авторы | номер документа |
|---|---|---|---|
| 3,6-ДИАМИНОПИРИДАЗИН-3-ИЛЬНЫЕ ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОАПОПТОТИЧЕСКИХ СРЕДСТВ | 2020 |
|
RU2830186C2 |
| Ингибиторы ErbB/BTK | 2019 |
|
RU2764069C1 |
| 6,7-ДИГИДРО-5H-ПИРИДО[2,3-C]ПИРИДАЗИНОВЫЕ ПРОИЗВОДНЫЕ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛКОВ BCLXL И ПРОАПОПТОТИЧЕСКИХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ | 2020 |
|
RU2832191C2 |
| ИНГИБИТОРЫ РЕЦЕПТОРОВ ERBB | 2019 |
|
RU2810215C2 |
| Производное индола, используемое в качестве ингибитора CRTH2 | 2017 |
|
RU2756270C2 |
| АНТАГОНИСТЫ TLR7/8 И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2758686C2 |
| КЛАСС БИФУНКЦИОНАЛЬНЫХ ХИМЕРНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ДЛЯ НАПРАВЛЕННОГО РАЗРУШЕНИЯ АНДРОГЕННЫХ РЕЦЕПТОРОВ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2825000C2 |
| C-3 и C-17 модифицированные тритерпеноиды в качестве ингибиторов ВИЧ-1 | 2017 |
|
RU2716502C2 |
| ПРОИЗВОДНЫЕ 7-ОКСО-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТ-3-ЕНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2013 |
|
RU2645678C2 |
| Новые 3-индол замещенные производные, фармацевтические композиции и способы применения | 2016 |
|
RU2672252C1 |
Изобретение относится к соединению диарилтиогидантоина формулы (I), где R1, T, кольца A и B определены в формуле изобретения, в качестве антагониста андрогенового рецептора или его фармацевтически приемлемой соли, к фармацевтической композиции, содержащей соединение, и к применению его в лечении клеточного пролиферативного заболевания, опосредованного андрогенами. Соединение настоящей заявки оказывает хорошее антагонистическое воздействие на андрогеновый рецептор и проявляет превосходный противоопухолевый эффект. 3 н. и 17 з.п. ф-лы, 7 табл., 80 пр.

1. Соединение формулы (I) или его фармацевтически приемлемая соль,
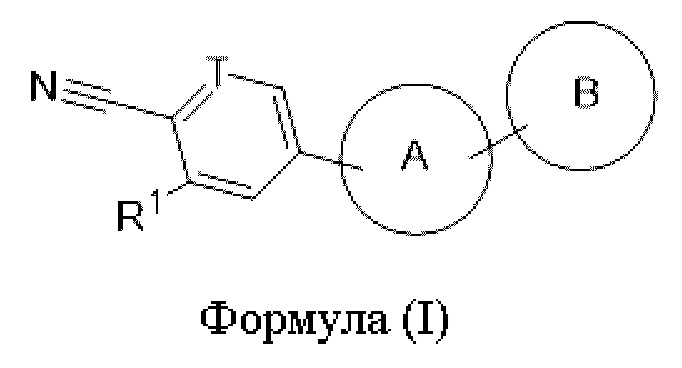
где
Т выбран из группы, состоящей из СН и N;
R1 выбран из группы, состоящей из водорода, галогена, С1-6алкила и замещенного галогеном С1-6алкила;
кольцо А выбрано из группы, состоящей из 
каждый из R2 и R3 независимо выбран из С1-6алкила, или вместе взятые R2 и R3 связаны друг с другом с образованием 3-4-членного циклоалкила;
n равно 0, 1 или 2;
каждый R4 независимо выбран из C1-6алкила;
кольцо В представляет собой 
 или
или  ;
;
R5 выбран из группы, состоящей из водорода, C1-6алкила, С1-6алкокси и галогена;
R6 выбран из С1-6алкиламинокарбонила;
один из X5, X6 и X7 представляет собой N(-Ra), а остальные представляют собой СН или N и отличаются друг от друга;
Ra выбран из 5-членного оксациклоалкила, где оксациклоалкил замещен гидроксилом;
где каждый R71 независимо выбран из группы, состоящей из гидроксила и 5-10-членного гетероарила, и гидроксил замещен группой -С1-6алкил-ОН, -С1-6алкил-(3-10-членный гетероциклоалкил), -С1-6алкил-S(=O)2Rc, -С1-6алкил-NRdRe, -С1-6алкил-C(=O)NRfRg, -С1-6 алкил-(3-10-членный циклоалкил), возможно замещенной галогеном или гидроксилом, или 3-10-членным гетероциклоалкилом, возможно замещенным галогеном или гидроксилом;
каждый R72 независимо выбран из группы, состоящей из С1-6алкила, гидроксила, С1-6алкокси, 3-6-членного циклоалкила, и С1-6алкиламино, где С1-6алкил возможно замещен галогеном, и где гидроксил замещен группой -С1-6 алкил-ОН;
каждый R91 независимо выбран из группы, состоящей из С1-6алкила, С1-6алкокси и гидроксила, где С1-6алкил возможно замещен С1-6алкокси или галогеном, и где гидроксил возможно замещен группой -С1-6алкил-ОН или -С1-6алкил-О-С1-6алкил; где каждый R92 независимо выбран из группы, состоящей из гидроксила, -С1-6алкокси и галогена, где гидроксил возможно замещен группой -С1-6алкил-ОН, -С1-6алкил-О-С1-6алкил или -С1-6алкил-С(=O)NRfRg;
каждый из Rc, Rd, Re, Rf и Rg независимо выбран из группы, состоящей из водорода и С1-6алкила; и
каждый R8 независимо выбран из С1-6алкила;
при условии, что когда кольцо А выбрано из  , тогда кольцо В не представляет собой
, тогда кольцо В не представляет собой  ;
;
где гетероциклоалкил или гетероарил содержит 1, 2 или 3 гетероатома, независимо выбранных из группы, состоящей из серы, кислорода и азота.
2. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где кольцо А выбрано из группы, состоящей из  , и n равно 0 или 1.
, и n равно 0 или 1.
3. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где каждый из R2 и R3 независимо выбран из группы, состоящей из метила и этила, или вместе взятые R2 и R3 связаны друг с другом с образованием 3-4-членного циклоалкила; и/или
каждый из R2 и R3 независимо выбран из метила, или вместе взятые R2 и R3 связаны друг с другом с образованием циклобутила.
4. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где каждый R4 независимо выбран из метила.
5. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где
R1 выбран из группы, состоящей из фтора, хлора, брома и замещенного фтором С1-4алкила; и/или
R1 выбран из группы, состоящей из фтора, хлора и замещенного фтором метила; и/или
R1 выбран из группы, состоящей из фтора, хлора, дифторметила и трифторметила.
6. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где R5 выбран из группы, состоящей из водорода, метила, метокси, фтора, хлора, брома и йода.
7. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где R6 выбран из метиламинокарбонила.
8. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где структурная единица  выбрана из группы, состоящей из
выбрана из группы, состоящей из  и
и  и/или
и/или
Ra выбран из  .
.
9. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где каждый R71 независимо выбран из группы, состоящей из гидроксила, пиразолила и имидазолила, где гидроксил замещен группой -этил-ОН,  , -пропил-S(=O)2СН3, -CH2C(=O)NHCH3, -CH2C(=O)NH2, -этил-N(СН3)2,
, -пропил-S(=O)2СН3, -CH2C(=O)NHCH3, -CH2C(=O)NH2, -этил-N(СН3)2,  , замещенной гидроксилом, или
, замещенной гидроксилом, или  , замещенной гидроксилом.
, замещенной гидроксилом.
10. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где каждый R72 независимо выбран из группы, состоящей из метила, этила, циклопропила, гидроксила, метокси, этокси, дифторметила, дифторэтила и метиламино, и где гидроксил замещен группой -этил-ОН.
11. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где каждый из Rc, Rd, Re, Rf и Rg независимо выбран из группы, состоящей из водорода и C1-4алкила; и/или
каждый из Rc, Rd, Re, Rf и Rg независимо выбран из группы, состоящей из водорода и метила.
12. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где
каждый R71 независимо выбран из группы, состоящей из гидроксила и 5-6-членного гетероарила, и гидроксил замещен группой -С1-6алкил-ОН, -С1-6алкил-(3-6-членный гетероциклоалкил), -C1-6алкил-S(=O)2Rc, -C1-6алкил-NRdRe, -C1-6алкил-C(=O)NRfRg, -С1-6алкил-(3-6-членный циклоалкил), возможно замещенной галогеном или гидроксилом, или 3-6-членным гетероциклоалкилом, возможно замещенным галогеном или гидроксилом; и/или
каждый R71 независимо выбран из группы, состоящей из гидроксила и 5-6-членного гетероарила, где гидроксил замещен группой -С1-4алкил-ОН, -С1-4алкил-(3-6-членный гетероциклоалкил), -C1-4алкил-S(=O)2Rc, -C1-4алкил-NRdRe, -C1-4алкил-C(=O)NRfRg, -С1-4алкил-(3-6-членный циклоалкил), возможно замещенной галогеном или гидроксилом, или 5-6-членным гетероциклоалкилом, возможно замещенным галогеном или гидроксилом; и/или
каждый R71 независимо выбран из группы, состоящей из гидроксила, пиразолила и имидазолила, где гидроксил замещен группой -этил-ОН, тетрагидропиранил, -метил-(оксетан), -пропил-S(=O)2Rc, -этил-NRdRe, -метил-C(=O)NRfRg, циклопропилметил-, возможно замещенной гидроксилом, или тетрагидрофуранилом, возможно замещенным гидроксилом; и/или
каждый R72 независимо выбран из группы, состоящей из С1-6алкила, гидроксила, С1-6алкокси, 3-6-членного циклоалкила и С1-6алкиламино, где С1-6алкил возможно замещен галогеном, и где гидроксил замещен группой -С1-6алкил-ОН; и/или
каждый R72 независимо выбран из группы, состоящей из С1-4алкила, гидроксила, С1-4алкокси, 3-6-членного циклоалкила и С1-4алкиламино, где С1-4алкил возможно замещен галогеном, и где гидроксил замещен группой -С1-4алкил-ОН; и/или
каждый R72 независимо выбран из группы, состоящей из метила, этила, гидроксила, метокси, этокси, циклопропила и метиламино, где метил, этил возможно замещен фтором; и где гидроксил замещен группой -этил-ОН.
13. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, каждый R91 независимо выбран из группы, состоящей из С1-4алкила, С1-4алкокси и гидроксила, где С1-4алкил возможно замещен С1-4алкокси или галогеном, и где гидроксил возможно замещен группой -С1-4алкил-ОН или -С1-4алкил-О-С1-4алкил; и/или
каждый R91 независимо выбран из группы, состоящей из метила, этила, метокси, этокси и гидроксила, где метил или этил возможно замещен фтором или метокси, и где гидроксил возможно замещен группой -этил-О-метил или -этил-ОН; и/или
каждый R92 независимо выбран из группы, состоящей из гидроксила, С1-4алкокси и галогена, и где гидроксил возможно замещен группой -С1-4алкил-ОН, -С1-4алкил-О-С1-4алкил или -C1-4алкил-C(=O)NRfRg; и/или
каждый R92 независимо выбран из группы, состоящей из гидроксила, метокси и галогена, где гидроксил возможно замещен группой -этил-ОН, -этил-О-метил или -метил-C(=O)NRfRg.
14. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где структурная единица  выбрана из группы, состоящей из
выбрана из группы, состоящей из
 ,
, 
15. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где каждый R8 независимо выбран из С1-4алкила; и/или
каждый R8 независимо выбран из этила.
16. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где каждый R91 независимо выбран из группы, состоящей из метила, этила, метокси, этокси и гидроксила, где метил или этил возможно замещен фтором или метокси, и где гидроксил возможно замещен группой -этил-О-метил или -этил-ОН; и/или
каждый R92 независимо выбран из группы, состоящей из гидроксила, метокси и галогена, где гидроксил возможно замещен группой -этил-ОН, -этил-О-метил, -CH2C(=O)NHCH3 или -СН2С(=O)NH2.
17. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где соединение формулы (I) представляет собой соединение формулы (VII)

где кольцо В выбрано из группы, состоящей из  ,
,
18. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, выбранное из группы, состоящей из следующих соединений:



или их фармацевтически приемлемых солей.
19. Фармацевтическая композиция для лечения опосредованного андрогеном заболевания, содержащая эффективное количество соединения формулы (I) или его фармацевтически приемлемую соль по любому из пп. 1-18.
20. Способ лечения опосредованного андрогеном заболевания, возможно рака предстательной железы, у млекопитающего, включающий введение млекопитающему, нуждающемуся в лечении, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1-18.
| CN 104341396 A, 11.02.2015 | |||
| CN 103804358 B, 29.06.2016 | |||
| CN 106146474 A, 23.11.2016 | |||
| Ivachtchenko, Alexandre V | |||
| et al | |||
| "Design, synthesis and biological evaluation of novel 5-oxo-2-thioxoimidazolidine derivatives as potent androgen receptor antagonists", European Journal of Medicinal Chemistry, 2015, vol | |||
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Способ запрессовки не выдержавших гидравлической пробы отливок | 1923 |
|
SU51A1 |
| EA 201270720 A1, 29.03.2013. | |||

