Область техники, к которой относится изобретение
[0001] Изобретение относится к области доставки лекарственных средств и лекарственных форм с замедленным высвобождением. В частности, оно относится к композициям с замедленным высвобождением, которые доставляют стабилизированные формы агонистов ГПП-1 в течение одного месяца или больше.
Уровень техники
[0002] Эксенатид - пептид из 39 аминокислот, который является мощным агонистом рецептора ГПП-1, делая его средством, усиливающим секрецию инсулина и обладающим глюкорегуляторным действием. Его широко применяют при лечении диабета 2 типа в виде свободного пептида, выпускаемого под наименованием Баета® (AstraZeneca), при этом пептид вводят два раза в день из-за короткого периода полувыведения in vivo, 2,5 часа. Крайне желательно увеличить период полувыведения эксенатида и подобных пептидов агонистов ГПП-1, чтобы повысить их эффективность, уменьшить побочные эффекты и ослабить нагрузку при лечении у пациентов.
[0003] Период полувыведения пептидов, как правило, увеличивают с помощью одного или комбинации нескольких методов: (i) химической модификации пептида для замедления метаболизма; (ii) инкапсулирования с получением лекарственной депо-формы с замедленным высвобождением; и (iii) конъюгирования с макромолекулой для замедления выведения. См., например, Cai, et al., Drug Design, Development, and Therapy (2013) 7:963 970.
[0004] Химические модификации пептидов с целью увеличения периода полувыведения привели к созданию агонистов ГПП-1, которые можно вводить один раз в день, например ликсисенатида (Ликсумия®) и лираглутида (Виктоза®). Инкапсулирование пептида в микрочастицах ПЛГА (сополимера молочной и гликолевой кислот) использовали для получения лекарственной формы с замедленным высвобождением, выпускаемой под наименованием Bydureon® (AstraZeneca), который можно вводить путем подкожной инъекции раз в неделю. Попытки увеличить продолжительность действия Bydureon с возможностью введения раз в месяц при использовании триглицеридных лекарственных форм пока не оказались успешными. Конъюгирование агонистов ГПП-1 пептида с Fc-доменами антител или полипептидами со случайной последовательностью (XTEN) позволили увеличить период полувыведения лишь до 5-6 дней.
[0005] При исследовании требований к введению доз удобно определять период полувыведения из плазмы, время, необходимое для выведения половины лекарственного средства из системы. Частота введения определяется необходимостью поддерживать уровень лекарственного средства на определенном эффективном уровне или выше. Если введение дозы производят один раз через каждый период полувыведения, то доза должна быть такой, чтобы создавать начальный уровень лекарственного средства, который в 2 раза превышает эффективный уровень; аналогичным образом, если введение дозы производят один раз через каждые 2 периода полувыведения, то тогда доза должна быть такой, чтобы создавать начальный уровень лекарственного средства, который в 4 раза превышает эффективный уровень. Теоретически введение дозы можно производить так редко, как требуется, независимо от периода полувыведения, просто путем увеличения количества лекарственного средства, вводимого с каждой дозой. Многие лекарственные средства проявляют токсическое действие, которое связано с высокой начальной концентрацией, однако на практике они имеют ограничение частоты введения в зависимости от периода полувыведения. Как правило, интервалы между введением доз составляют приблизительно 1-2 периода полувыведения. Хотя было описано несколько препаратов пептидов-агонистов ГПП-1, имеющих подходящие эффективные периоды полувыведения для однократного ежемесячного введения, они имеют несколько недостатков. Инкапсулирующие микросферы и липидные композиции с фазовым переходом могут иметь недостаток, связанный с первоначальным импульсным высвобождением пептида, что подвергает пациента первоначальному нежелательно высокому уровню лекарственного средства. Имплантируемые насосы требуют хирургического вмешательства для имплантации и удаления.
[0006] Начальные импульсы при высвобождении лекарственного средства можно исключить путем применения ковалентно связанных конъюгатов. Тогда как растворимые, циркулирующие конъюгаты обычно не имеют достаточный период полувыведения, чтобы предоставлять возможность введения лекарственного средства один раз в месяц (максимальный период полувыведения полиэтиленгликоля у людей составляет около 1 недели), могут применяться нерастворимые имплантируемые конъюгаты, такие как гидрогели. Композиции, в которых лекарственные средства, такие как эксенатид, ковалентно связаны с различными матрицами через линкеры, с регулируемой скоростью высвобождения лекарственного средства, были ранее раскрыты, например, в патентах США 8,680,315; US 8,754,190; U.S. 8,703,907, включая нерастворимые матрицы, описанные в патенте США 8,946,405, и гидрогели, как описано в US 2014/0288190 ('190). В одном варианте осуществления в публикации '190 эксенатид связан с гидрогелевой матрицей, где после инъекции в подкожное пространство гидрогель создает депо, из которого эксенатид высвобождается при бета-элиминационном расщеплении линкера, обеспечивая длительно действующий источник лекарственного средства. Такие гидрогели по определению состоят в основном из воды, и поэтому эксенатид подвергается воздействию водной среды на протяжении всего действия депо. Хотя такая водная среда считается выгодной для поддержания сложной структуры белков, некоторые пептидные последовательности могут быть нестабильными при таких условиях длительного воздействия.
[0007] Было обнаружено, что скорость расщепления эксенатида в гидрогелях при физиологических условиях делает ежемесячное или даже еще менее частое введение таких лекарственных форм нецелесообразным. Эксенатид, как сообщали, демонстрировал химическую нестабильность в неблагоприятных условиях (pH 7,9, 40°C в течение 6 дней) в результате окисления в M14 и W25 и гидролиза амида боковой цепи в Q13 и N28 (заявка на патент США 2006/0194719; Zealand). Подробная кинетика нестабильности не была раскрыта. Хотя была описана подробная модель дезамидирования остатков Asn в пептидах (Geiger & Clarke, J. Biol. Chem. (1987) 262:785-794), скорость дезамидирования пептидов и белков при физиологических условиях, как известно, сильно различается (Robinson & Robinson, Proc Natl Acad Sci (2010) 98:12409-12413). Эксенатид сам по себе достаточно стабилен при введении два раза в день (Баета®) и при введении раз в неделю посредством имплантата ПЛГА (Bydureon®).
[0008] Были раскрыты стабилизированные формы эксенатида. Например, в заявке на патент WO2008/116294 (Matregen) раскрыты стабилизированные аналоги эксенатида, модифицированные по трем положениям: Q13, M14 и N28. Хотя в последовательности эксенатида допустимы множественные аминокислотные замены, было обнаружено, что замена N28 на более стабильный остаток достаточна для эффективной стабилизации пептида при pH 7,4, 37°C. Несколько продуктов дезамидирования эксенатида, например N28D и N28-изоD, были раскрыты в вышеуказанной заявке на патент США 2006/0194719 (Zealand) и, как обнаружили, сохраняли способность к активации рецептора ГПП-1. Впрочем, эти продукты формируют систему взаимного превращения и нестабильны к уравновешиванию в исходной смеси продуктов.
[0009] Подробное исследование поведения эксенатида в водных буферах при рН 7,4, 37°C (т.е. при физиологическом рН и температуре) показало, что он подвергается дезамидированию по Asn28 (N28) с периодом полувыведения от 8 до 14 дней, в зависимости от буфера. Таким образом, конъюгат гидрогеля, предназначенный для введения раз в месяц, высвобождает в основном продукты расщепления эксенатида через 8-14 дней. Таким образом, важно, чтобы стабилизированный агонист ГПП-1 обладал достаточной устойчивостью к химическому разложению, чтобы свести к минимуму количество расщепленных форм, высвобождаемых к концу периода введения. Это достигается, когда агонист ГПП-1 образует менее 10% продуктов разложения через один месяц при рН 7,4, 37°C, предпочтительно менее 5% продуктов разложения через один месяц при рН 7,4, 37°С.
Описание изобретения
[0010] Настоящее изобретение направлено на конъюгаты, которые обеспечивают замедленное высвобождение стабилизированных пептидов агонистов ГПП-1, которые предоставляют возможность введения один раз в месяц или еще менее частого введения таких пептидов и могут применяться при лечении нарушений обмена веществ и таких состояний, как метаболический синдром, диабет и ожирение. Конъюгаты объединяют в себе повышенную стабильность агониста ГПП-1 с контролируемым временем высвобождения, обеспечиваемым подходящим линкером с резервуарной матрицей, которая служит в качестве депо для высвобождения.
[0011] В одном аспекте настоящего изобретения предложены конъюгаты с замедленным высвобождением, включающие нерастворимую матрицу с множеством ковалентно присоединенных линкерных пептидов, где линкеры расщепляются при физиологических значениях pH и температуры, высвобождая свободный пептид, и где пептид является стабилизированным агонистом ГПП-1, который демонстрирует меньше 10% расщепления в течение одного месяца при pH 7,4, 37°C. Конъюгаты согласно изобретению могут быть схематично представлены формулой (1):
M-(L-E)x (1)
где M является нерастворимой матрицей, связанной с множеством (x) пептидов агонистов ГПП-1 E через расщепляемый линкер L. E является агонистом ГПП-1, стабилизированным от расщепления, которое происходит при физиологических значениях pH и температуры, демонстрируя меньше 10% расщепления в течение одного месяца. x является целым числом, которое обозначает количество групп L-E, которые создают подходящие концентрации в объеме матрицы. Подходящие концентрации составляют 1-1000 мг пептида на мл матрицы. Линкер L высвобождает свободный пептид с периодом полувыведения, подходящим для требуемого периода введения.
[0012] Во втором аспекте настоящего изобретения предложены линкер-пептиды L-E, имеющие формулу (4):

в которой по меньшей мере один или оба R1 и R2 независимо являются CN; NO2;
необязательно замещенным арилом;
необязательно замещенным гетероарилом;
необязательно замещенным алкенилом;
необязательно замещенным алкинилом;
COR3 или SOR3 или SO2R3, где
R3 является H или необязательно замещенным алкилом;
арилом или арилалкилом, каждый из которых необязательно замещен;
гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен; или R3
OR9 или NR92, где каждый R независимо является H или необязательно замещенным алкилом, или обе группы R9, взятые вместе с азотом, к которому они присоединены, образуют гетероциклическое кольцо;
SR4, где
R4 является необязательно замещенным алкилом;
арилом или арилалкилом, каждый из которых необязательно замещен; или
гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен;
где R1 и R2 могут быть соединены с образованием 3-8-членного кольца; и
где один и только один из R1 и R2 может быть H или алкилом, арилалкилом или гетероарилалкилом, каждый из которых необязательно замещен; и
где один из R5 является (CH2)yZ, (CH2CH2O)xCH2CH2Z или (CH2)yNH-CO-(CH2CH2O)xCH2CH2Z, где x является 1-100, y=1-6, и другой R5 является H, алкилом, алкенилалкилом, алкинилалкилом, арилом, арилалкилом, гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен;
Z является функциональной группой для опосредования связывания с нерастворимой матрицей, и NH является остатком аминогруппы агониста ГПП-1 E.
[0013] В одном варианте осуществления R1 является CN или R3SO2, где R3 является алкилом или R92N, где каждый R9 является H, алкилом или замещенным алкилом, один R5 является H, а другой R5 является (CH2)nZ, где n=1-6, и Z является функциональной группой, через которую линкер-пептид может быть присоединен к M.
[0014] В одном варианте осуществления E является [N28Q]эксенатидом (SEQ ID NO:2). Изобретение также включает этот пептид и его любые фармацевтически приемлемые соли и фармацевтические композиции, а также протокол для введения агониста ГПП-1, который включает введение субъекту, имеющему состояние, при котором эффективен агонист ГПП-1, композиция, в которой применяется этот пептид, или его соль.
[0015] В третьем аспекте изобретение направлено на протоколы введения конъюгата формулы (1). В одном варианте осуществления конъюгат получен в виде микросфер гидрогеля, подходящих для подкожной инъекции при использовании иглы малого диаметра. Ожидается, что конъюгаты изобретения можно будет применять для лечения нарушений обмена веществ и состояний у людей и у животных. Получены схемы с введением через 1-3 месяца.
Краткое описание чертежей
[0016] Фигуры 1-6 представляют собой схематические изображения различных вариантов осуществления конъюгатов с замедленным высвобождением согласно изобретению.
[0017] На Фигурах 1A и 1B показан общий вид конъюгата в форме схемы. На Фигуре 1A один из компонентов является 8-лучевым макромономером, а другой - 4-лучевым макромономером, и где линкер, присоединенный к агонисту ГПП-1, связан с лучами 8-лучевого макромономера. На Фигуре 1B структура предусматривает присоединение линкер-агониста к самим сшивающим агентам.
[0018] На Фигуре 2 более подробно показаны связи из Фигуры 1A.
[0019] Фигура 3 представляет собой схему варианта осуществления, где сшивающая группа, которая связывает различные макромономеры, имеет реакционноспособную группу для присоединения связанного с линкером агониста.
[0020] На Фигуре 4 показана матрица из Фигуры 3 с присоединенным линкер-пептидом.
[0021] На Фигурах 5 и 6 показаны определенные варианты осуществления, представленные в Примере 4 ниже.
[0022] На Фигурах 7A-7B показаны отношения между скоростью высвобождения лекарственного средства из депо конъюгата, частотой введения и дозой, требуемой для достижения установленной конечной концентрации (Cmin).
[0023] На Фигурах 8A-8C показана стабильность эксенатида и [N28Q]эксенатида в 200 мМ фосфатном буфере, pH 7,4, 37°C.
[0024] На Фигуре 9 показаны результаты анализа с пептид-изоаспартат-метилтрансферазов (PIMT) на содержание изоаспартата (изо-Asp) в изолированных пиках реакций разложения эксенатида в течение 56 дней, показанных на Фигурах 8А и 8B. Определение изо-Asp в реакционной смеси эксенатида проводили при t=0 и 56 дней, и выделенных компонентов смеси продуктов разложения - через 56 дней. Значения для содержащих L-Asp и D-изо-Asp пептидов корректировали с учетом небольших количеств L-изо-Asp пептида, обнаруженного с помощью ВЭЖХ в образцах. Остаточные PIMT-положительные пики при RV 9,9 и 10,4 связывали с примесями [L-изо-Asp28]эксенатида, присутствующими на низком уровне в выделенных фракциях ВЭЖХ. Планки погрешности соответствуют ±SD.
[0025] На Фигуре 10 показана фармакокинетика эксенатида у крысы после введения дозы гидрогель-связанного немодифицированного эксенатида (R1=CN; R2=H). Кривую ожидаемой концентрации в зависимости от времени строили (пунктирная линия) на основе результатов кинетики высвобождения in vitro (t1/2=1400 ч) и известных фармакокинетических свойств эксенатида. Экспериментальные данные (квадраты) лучше согласуются с моделью, учитывающей разложение эксенатида на гидрогеле (сплошная линия), где общее t1/2=190 ч.
[0026] На Фигуре 11 показана фармакокинетика [N28Q]эксенатида у крысы после п/к введения дозы гидрогель-связанного [N28Q]эксенатида. На панели A показан гидрогель, где пептид связан с использованием L с R1=MeSO2 и дает t1/2=350 ч. На панели B показан гидрогель, где пептид связан с L, в котором R1=CN, и дает t1/2=760 ч. Таким образом, уровни [N28Q]эксенатид в плазме могут сохраняться в течение по меньшей мере одного месяца после однократной дозы.
[0027] На Фигуре 12 показаны сравнительные результаты эксенатида и [N28Q]эксенатида в пероральном глюкозотолерантном тесте.
[0028] На Фигуре 13 показан анализ AUC для данных перорального глюкозотолерантного теста, показанного на Фигуре 7.
[0029] На Фигурах 14A-14E показаны результаты введения один раз в месяц препарата микросфер гидрогеля, включающего [N28Q]эксенатид ("PL-cmpd") у крыс ZDF с диабетом.
[0030] На Фигурах 15А-15B показана фармакокинетика [N28Q]эксенатида в сыворотке крыс после п/к введения препаратов микросфер гидрогеля, включающих [N28Q]эксенатид, как описано в Примере 8А.
[0031] На Фигурах 16А-16B показаны уровни [N28Q]эксенатида в сыворотке после п/к инъекции мышам микросфер конъюгатов, как описано в Примере 8B.
Варианты осуществления изобретения
[0032] Для того чтобы пептид можно было вводить один раз в месяц, пептид нужно доставлять в форме нерастворимой матрицы, которая не циркулирует в кровотоке, а действует в виде депо для высвобождения лекарственного средства. Циркулирующие конъюгаты лекарственных средств, содержащие макромолекулы, неудовлетворительны, поскольку сами конъюгаты по существу выводятся из кровотока, например плазмы. Поэтому пептид нужно доставлять в матрице, которая присутствует в макромасштабе, и где пептид (или другое лекарственное средство) присутствует в объеме матрицы в концентрации 1-1000 мг пептида/мл матрицы, предпочтительно 1-100 мг пептида/мл матрицы и более предпочтительно 1-50 мг пептида/мл матрицы. Таким образом, матрица имеет определенный объем и ее можно удобно и совокупно вводить в форме микросфер ("объем матрицы" является общим объемом дозы, вводимой при этом, включая дозу микросфер).
[0033] Чтобы представить природу структур, включенных в изобретение, на перспективу, настоящие заявители приводят Фигуры 1-6, на которых предоставлен обзор типичных вариантов осуществления, которые входят в объем изобретения.
[0034] Изображение типичной нерастворимой матрицы гидрогеля, включающей связанный пептид согласно изобретению, показано на Фигуре 1A. Это - упрощенная структура матрицы, сформированной при сшивании 8-лучевого макромономера P с 4-лучевым макромономером T, с такой стехиометрией, что половина лучей P сшита с лучами T. Остальные несшитые лучи P связаны с линкер-пептидом. Получение гидрогелей такого типа описано в публикации PCT WO2013/036847 и проиллюстрировано, как описано ниже в Получении E.
[0035] На Фигуре 1B показана альтернатива, в которой высвобождаемое линкер-лекарственное средство связано с разлагаемым сшивающим агентом. На Фигуре 1B показана упрощенная иллюстративная структура матрицы, сформированной при сшивании 4-лучевого макромономера A, где каждый луч A имеет концевую группу, включающую ортогональные первую и вторую функциональные группы, со вторым 4-лучевым макромономером B, где каждый луч B имеет концевую функциональную группу, которая способна реагировать только с одной из первой или второй функциональных групп макромономера A. Оставшаяся функциональная группа макромономера A доступна для реакции с линкер-пептидом, включающим функциональную группу, которая способна реагировать с оставшейся функциональной группой макромономера. Присоединение линкер-пептида может быть выполнено либо до формирования геля в реакции макромономера с линкер-пептидом, с последующим сшиванием с макромономером B, или после формирования геля при сшивании A и B с образованием гидрогеля, с последующей реакцией с линкер-пептидом. Как описано выше, реакция сшивания с образованием нерастворимой матрицы гидрогеля может быть проведена либо в виде объемного материала, либо в суспензии или эмульсии, с получением полимера в форме тонкодисперсных частиц, например микросфер, как описано в Примере 2 ниже.
[0036] На Фигуре 2 показана общая структура перекрестной связи между 8-лучевым макромономером P и 4-лучевым макромономером T в матрице, дополнительно включающей n линкер-пептидов на каждый P, как показано на Фигуре 1A. Альтернативный вариант, где линкер-агонист связан с группами сшивающего агента гидрогеля, представлен на Фигуре 1B и Примере 4 ниже.
[0037] Общее описание этой альтернативы представлено на Фигурах 3 и 4. На Фигуре 3 показан пример дериватизации нерастворимой матрицы, имеющей доступные аминогруппы, реагентом с введением циклооктиновых групп. В этом примере макромономер, используемый при получении матрицы, как показано в Получении D ниже, включает остаток лизина. При образовании матрицы путем сшивания с макромономером B, полученная матрица имеет доступные аминогруппы, подходящие для последующей функционализации, например, посредством реакции с реагентом, который вводит циклооктиновую группу.
[0038] На Фигуре 4 показана общая структура разлагаемого гидрогеля, включающего высвобождаемые линкер-пептиды. Два макромономера A и B (например, как показано в Получении D ниже) сшивают таким образом, что каждая перекрестная связь включает высвобождаемый линкер-пептид. На диаграмме каждый A и B соединен показанной перекрестной связью с образованием нерастворимой матрицы. Y и Z являются соединительными функциональными группами.
[0039] На Фигурах 5 и 6 показаны структуры связей в конъюгатах с замедленным высвобождением, полученных в Примере 4. На Фигуре 5 матрица гидрогеля включает перекрестные связи, расщепление которых регулируется модулятором бис(2-этокси)аминосульфонилом, тогда как высвобождение пептида SEQ ID NO: 2 из гидрогеля регулируется модулятором CN. Соединительные функциональные группы являются триазолами, образующимися в результате присоединения азидной группы к MFCO циклооктину.
[0040] На Фигуре 6 показана та же структура, как на Фигуре 5, за исключением того, что и расщепление перекрестных связей гидрогеля, и высвобождение пептида из гидрогеля регулируется модулятором CN, и соединительные функциональные группы являются триазолами, образующимися в результате реакции азидной группы с 5-гидроксициклооктином.
Матрица M:
[0041] Матрица M является нерастворимой подложкой, к которой присоединяют линкер-пептид L-E и которая служит резервуаром, из которого E высвобождается в течение курса лечения. M должен подходить для присоединения линкер-пептида L-E, или же включать функциональные группы, которые могут быть дериватизированы, чтобы обеспечить такое присоединение. M должен допускать свободную диффузию пептида E после его высвобождения при расщеплении линкера L. Кроме того, М должен быть биоразлагаемым до растворимых продуктов и достаточно медленно разлагаться, чтобы обеспечивать высвобождение E без образования чрезмерных количеств растворимых фрагментов M-L-E, но при этом достаточно быстро, чтобы минимизировать нагрузку от M-L, не содержащего лекарственных средств, оставшегося после высвобождения E в режиме введения многократных доз.
[0042] В одном варианте осуществления M является биоразлагаемым гидрогелем, полученным, как раскрыто в патентной публикации PCT WO2013/036847 и US2014/0288190, которые включены в настоящую заявку посредством отсылки в отношении описания в них таких гидрогелей. Эти гидрогели включают бета-элиминационные сшивающие агенты, которые обеспечивают контроль скорости расщепления. Таким образом, в некоторых вариантах осуществления, сшивающие агенты имеют следующую Формулу (1) или (2).
 (1)
(1)
где m является 0 или 1; и
где X и один из R1, R2 и R5 включают функциональную группу для связывания с полимером, и
при условии, что по меньшей мере один из R1 и R2 является CN; NO2;
необязательно замещенным арилом;
необязательно замещенным гетероарилом;
необязательно замещенным алкенилом;
необязательно замещенным алкинилом;
COR3 или SOR3 или SO2R3, где
R3 является H или необязательно замещенным алкилом;
арилом или арилалкилом, каждый из которых необязательно замещен;
гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен; или
OR9 или NR92, где каждый R9 независимо является H или необязательно замещенным алкилом, или обе группы R9, взятые вместе с азотом, к которому они присоединены, образуют гетероциклическое кольцо;
SR4, где
R4 является необязательно замещенным алкилом;
арилом или арилалкилом, каждый из которых необязательно замещен; или
гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен;
где R1 и R2 могут быть соединены с образованием 3-8-членного кольца; и
где любой оставшийся R1 и R2 является H или алкилом, арилалкилом или гетероарилалкилом, каждый из которых необязательно замещен; и
любой оставшийся R5 независимо является H или алкилом, алкенилалкилом, алкинилалкилом, (OCH2CH2)pO-алкилом, где p=1-1000, арилом, арилалкилом, гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен; или
указанный сшивающий агент имеет формулу (2)
 (2)
(2)
где два из R1, R2 и R5 включают функциональную группу для связывания с полимером;
m является 0-1;
n является 1-1000;
s является 0-2;
t является 2,4, 8, 16 или 32;
Q является основной группой, имеющей валентность t;
W является O(C=O)O, O(C=O)NH, O(C=O)S,  или
или 
где R6 является H, необязательно замещенным алкилом, необязательно замещенным арилом, необязательно замещенным гетероарилом, необязательно замещенным арилалкилом или необязательно замещенным гетероарилалкилом; и
при условии, что по меньшей мере один из R1 и R2 является CN; NO2;
необязательно замещенным арилом;
необязательно замещенным гетероарилом;
необязательно замещенным алкенилом;
необязательно замещенным алкинилом;
COR3 или SOR3 или SO2R3, где
R3 является H или необязательно замещенным алкилом;
арилом или арилалкилом, каждый из которых необязательно замещен;
гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен; или
OR9 или NR92, где каждый R9 независимо является H или необязательно замещенным алкилом, или обе группы R9, взятые вместе с азотом, к которому они присоединены, образуют гетероциклическое кольцо;
SR4, где
R4 является необязательно замещенным алкилом;
арилом или арилалкилом, каждый из которых необязательно замещен; или
гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен;
где R1 и R2 могут быть соединены с образованием 3-8-членного кольца; и
где любой оставшийся R1 и R2 является H или алкилом, арилалкилом или гетероарилалкилом, каждый из которых необязательно замещен; и
любой оставшийся R5 независимо является H или алкилом, алкенилалкилом, алкинилалкилом, (OCH2CH2)pO-алкил, где p=1-1000, арилом, арилалкилом, гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен.
Функциональные группы, используемые для соединения таких сшивающих агентов с матрицей, включают N3, NH2, NH-CO2tBu, SH, StBu, малеимид, CO2H, CO2tBu, 1,3-диен, циклопентадиен, фуран, алкин, циклооктин, акрилат, аминоокси, кето и акриламид. Две функциональных группы на Формулах (1) и (2) отличаются друг от друга, но не являются родственными. Например, если одна группа азидная, другая не является циклооктиновой или алкиновой.
[0043] При выборе бета-элиминационного сшивающего агента, который приводит к расщеплению M и солюбилизации со скоростью, в несколько раз более низкой, чем скорость высвобождения E при расщеплении L-E, образование растворимых фрагментов M-L-E сведено к минимуму, с одновременным обеспечением эффективной солюбилизации и выведения матрицы. В одном варианте осуществления изобретения такие гидрогели получают путем сшивания многолучевых полиэтиленгликолей. Кроме того, изобретение относится к другим полезным матрицам, включающим сшитые декстраны и гиалуроновые кислоты.
[0044] Такие матрицы предпочтительно могут быть получены в виде суспензии микросфер, которую можно вводить при использовании иглы молого диаметра. Такие суспензии могут быть получены при использовани известных методов, например, эмульгирования объемной фазы или более точно при микрофлюидном капельном эмульгировании преполимерных смесей. Распределение частиц по размеру при необходимости можно установить с помощью известных методов, например, при просеивании на ситах.
Пептид E:
[0045] Пептиды (E), которые доставляют согласно изобретению, являются агонистами ГПП-1, что подразумевает под собой пептид, способный связываться с и активировать рецептор ГПП-1. Примеры агонистов ГПП-1 включают природные эксендины, например, эксенатид (эксендин-4; SEQ ID NO:1), лираглутид (SEQ ID NO:13), ликсисенатид (SEQ ID NO:7), таспоглутид (SEQ ID NO:12) и их варианты с измененной последовательностью. Также рассматриваются синтетические последовательности, которые связывают и активируют рецептор ГПП-1, например, последовательности, полученные при скрининге и/или отборе in vitro (Zhang, et al., Nature Commun. (2015) 6:8918).
SEQ ID NO:1 HGEGTFTSDLSKQMEEEAVRLFIEWLKNGGPSSGAPPPS-NH2
[эксенатид]
SEQ ID NO:2 HGEGTFTSDL SKQMEEEAVR LFIEWLKQGG PSSGAPPPS-NH2
[N28Q]эксенатид
SEQ ID NO:3 HGEGTFTSDL SKQMEEEAVR LFIEWLKAGG PSSGAPPPS-NH2
[N28A]эксенатид
SEQ ID NO:4 HGEGTFTSDL SKQMEEEAVR LFIEWLKKGG PSSGAPPPS-NH2
[N28K]эксенатид
SEQ ID NO:5 HGEGTFTSDL SKQMEEEAVR LFIEWLKDGG PSSGAPPPS-NH2
[N28D]эксенатид
SEQ ID NO:7 HGEGTFTSDLSKQMEEEAVRLFIEWLKNGGPSSGAPPSKKKKKK-NH2
[ликсисенатид]
SEQ ID NO:8 HGEGTFTSDLSKQMEEEAVRLFIEWLKQGGPSSGAPPSKKKKKK-NH2
[N28Q]ликсисенатид
SEQ ID NO:9 HGEGTFTSDLSKQMEEEAVRLFIEWLKAGGPSSGAPPSKKKKKK-NH2
[N28A]ликсисенатид
SEQ ID NO:10 HGEGTFTSDLSKQMEEEAVRLFIEWLKKGGPSSGAPPSKKKKKK-NH2
[N28K]ликсисенатид
SEQ ID NO:11 ELVDNAVGGDL SKQMEEEAVR LFIEWLKQGG PSSGAPPPS-NH2
SEQ ID NO:12 HUEGTFTSDV SSYLEGQAAK EFIAWLVKUR-NH2
[таспоглутид] U=2-аминоизомасляная кислота
SEQ ID NO:13 HAEGTFTSDVSSYLEGQAAK*EFIAWLVRGRG-OH
K*=Lys(y-Glu-пальмитоил)
[0046] Важно, что пептид агонист E химически стабилен при физиологических условиях в течение периода введения. Хотя это может и не быть проблемой в случае прямого введения пептидов, учитывая их быстрый клиренс и частое введение, препараты с замедленным высвобождением предъявляют более жесткие требования к стабильности пептидов. Например, пептид, который разлагается при физиологических условиях с периодом полувыведения 14 дней, может идеально подходить для однократного введения, поскольку только 5% пептида будет разлагаться за 1 день. Тот же пептид в условиях замедленного высвобождения в течение 30 дней (то есть при введении один раз в месяц) разложится на 80% к концу периода введения и, следовательно, будет непригодным.
[0047] Механизм деградации пептидов, которые содержат последовательность Asn-Gly, например дипептид N28-G29 в эксенатиде, показан ниже. Как показано, сначала образуется L-сукцинимид, что приводит к превращению остатка аспарагина в аспарагиновую кислоту или изоаспарагиновую кислоту. Получены обе формы D и L таких модифицированных аминокислот.

[0048] Проверка вариантов последовательности самого эксенатида показала, что замена N28 другими аминокислотами может давать агонист с достаточной стабильностью, чтобы можно было производить введение раз в месяц с применением конъюгата с замедленным высвобождением при минимальном воздействии на способность пептида связывать и активировать рецептор ГПП-1. Таким образом, агонисты, имеющие SEQ ID NO:2-4, где N28 из SEQ ID NO:1 заменен остатками Q, A или K, соответственно, как обнаружили, связывали и активировали рецептор ГПП-1 с сопоставимой аффинностью и активностью, как и нативный эксенатид, демонтрируя <10% или <9% химического разложения в течение периода продолжительностью один месяц.
[0049] Настоящее изобретение также предусматривает применение надлежащим образом стабилизированных агонистов ГПП-1, отличающихся от эксенатида. Вышеописанная нестабильность, как предполагают, будет наблюдаться в случае с другими агонистами ГПП-1, содержащими последовательность дипептида Asn Gly, например, ликсисенатидом и другими синтетическими пептидными последовательностями. Такие пептиды предположительно будут подвергаться такой же последовательности реакций разложения, как показано выше. Надлежащим образом стабилизированные формы этих агонистов ГПП-1, применимых в настоящем изобретении, включают SEQ ID NOS:8-11. Таспоглутид (SEQ ID NO:13) и лираглутид (SEQ ID NO:14) не содержат нестабильный дипептид Asn-Gly и подходят для применения в изобретении. Другие причины нестабильности также можно устранить. В случае агониста согласно настоящему изобретению стабилизированная форма дает меньше 10% продуктов разложения через один месяц при pH 7,4, 37°C, предпочтительно меньше 9% продуктов разложения через один месяц при pH 7,4, 37°C.
Расщепляемый линкер L:
[0050] Расщепляемый линкер соединяет агонист ГПП-1 E с нерастворимой матрицей M и расщепляется при физиологических условиях с высвобождением свободного E. Скорость расщепления линкера определяет период полувыведения пептида и подбирается в зависимости от необходимой частоты введения. Также важно, чтобы скорость расщепления матрицы и скорость высвобождения пептида были скоординированы с требуемой частотой введения. Ранее не предпринимали никаких попыток уравновесить эти показатели, что необходимо для успешного применения композиций согласно изобретению при обеспечении возможности введения один раз в месяц или менее часто. Любой линкер, который обеспечивает достижение такого баланса, будет удовлетворительным.
Хотя не существует никаких строго определенных правил по поводу отношения между скоростью высвобождения лекарственного средства и скоростью разрушения геля матрицы, удобное практическое правило состоит в том, что скорость разрушения геля должна быть примерно в три раза выше, чем скорость высвобождения свободного пептида. Если пептид высвобождается слишком быстро, до того как происходит разрушения геля, у субъекта сохраняются остатки геля при введении последующей дозы. Если высвобождение происходит слишком медленно по сравнению со скоростью разрушения геля, пептид остается связанным с частями геля, которые высвобождаются в кровоток. Оба таких случая нежелательны. Это отношение описано в публикации Reid, R. et al., Macromolecules (2015) 48:7359-7369. Структурные свойства, которые определяют скорость разрушения геля различных матриц, зависят от сшивающих групп, при этом структурные корреляции могут использоваться для получения подходящей скорости разрушения геля конкретной матрицы.
[0051] Баланс между скоростью разложения и высвобождения можно представить следующим образом:
[0052] Высвобождение относительно быстровыводимого лекарственного средства из депо в результате расщепления ковалентного линкера сообщает период полувыведения, определяемый расщеплением линкера, периоду полувыведения лекарственного средства из плазмы. Поскольку частота введения зависит от периода полувыведения лекарственного средства из плазмы, также существует отношение между скоростью расщепления линкера и частотой введения. Как правило, желательно свести к минимуму различие между максимальной (Cmax) и минимальной (Cmin) концентрациями лекарственного средства в плазме, которым подвергается пациент, чтобы уменьшить вероятность токсического действия, которое является результатом чрезмерно высоких концентраций лекарственного средства, при одновременном поддержании количества, требуемого для обеспечения эффективности, между дозами.
[0053] Если частота введения будет установлена равной периоду полувыведения лекарственного средства из плазмы, то, например, между Cmax и Cmin будет 2-кратное различие, тогда как если лекарственное средство вводят один раз за 2 периода полувыведения из плазмы, то различие увеличивается до 4-кратного. Таким образом, обычно количество периодов полувыведения лекарственного средства между дозами сокращают до минимума, чтобы максимально снизить Cmax. В случае конъюгата с замедленным высвобождением это достигается при уменьшении скорости высвобождения.
[0054] Впрочем, в случае конъюгата с замедленным высвобождением в равновесном состоянии уровень лекарственного средства из депо обратно пропорционален скорости высвобождения. Хотя отношение Cmax/Cmin можно уменьшить до любого значения путем снижения скорости высвобождения лекарственного средства из конъюгата, потребность в поддержании определенного значения Cmin с одновременным применением приемлемой дозы накладывает ограничение на данный метод. В случае однократного введения доза может быть вычислена согласно следующему:
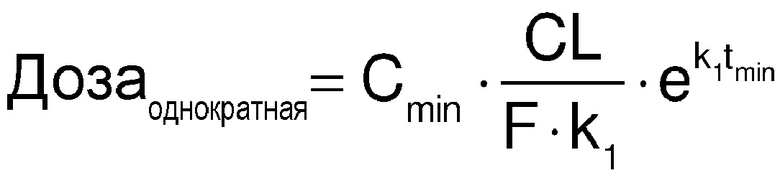
где CL=скорость выведения лекарственного средства, F=биодоступность, k1=скорость высвобождения лекарственного средства из депо конъюгата и tmin=время до достижения Cmin. Из этого уравнения можно показать, что наименьшая доза, необходимая для поддержания Cmin в течение данного tmin, достигается, когда k1=1/tmin. Иными словами, это когда период полувыведения высвобождения лекарственного средства (=ln(2)/k1)=ln(2)*tmin. Оптимальный период полувыведения высвобождения лекарственного средства для определенной частоты введения, таким образом, приводят как ln(2)*(интервал между введением доз). Когда период полувыведения высвобождения меньше оптимального, требуемая доза увеличивается из-за исчерпания депо до достижения tmin.
[0055] В общих словах, скорости высвобождения лекарственного средства, которые ниже оптимальных, являются более допустимыми, чем слишком высокие скорости. Таким образом, конъюгат со скоростью высвобождения лекарственного средства, которая предоставляет возможность введения один раз в месяц, также может применяться в схемах введения один раз в две недели или один раз в неделю. Кроме того, отношения между скоростью высвобождения лекарственного средства и дозой, требуемой для поддержания Cmin, таковы, что допускается некоторое отклонение от идеала. Это показано на Фигурах 7А и 7B в соответствии с параметрами, представленными выше. Как показано на Фигуре 7A, оптимальная доза (1) наблюдается, когда отношение периода полувыведения высвобождения к частоте введения составляет ln(2)=0,693. Это показано на Фигуре 7A, где указан диапазон скоростей высвобождения, которые можно допускать в зависимости от уровней дозы. Более высокий уровень дозы допускает как более высокую скорость высвобождения, поскольку концентрация выше минимально требуемой все еще сохраняется, так и более низкую скорость высвобождения, поскольку лекарственное средство предоставляется на более высоком уровне в течение такого более длительного периода. В частности, если дозу повышают до 10%, допускаются периоды полувыведения высвобождения в пределах 0,45×-1,1× частоты введения. Если дозу можно увеличить до 20%, допускаются периоды полувыведения высвобождения в пределах 0,4×-1,4× частоты введения. Если дозу можно увеличить до 50%, допускаются периоды полувыведения высвобождения в пределах 0,3×-2× частота введения.
[0056] Как подробно показано на Фигуре 7B: для однократной дозы, действующей в течение одного месяца (tmin=720 часов), оптимальная скорость высвобождения лекарственного средства составляет 500 часов, при этом также может использоваться любая скорость высвобождения от 320 до 800 часов при увеличении дозы на ~10%, от 280 до 1000 часов при увеличением дозы на ~20% или от 220 до 1440 часов при увеличении дозы на ~50%. Аналогичным образом, для однократной дозы, действующей в течение 3 месяцев (tmin=720 часов), оптимальная скорость высвобождения лекарственного средства составляет 1500 часов, при этом также может использоваться любая скорость высвобождения от 320 до 2400 часов при увеличении дозы на ~10%, от 840 до 3000 часов при увеличении дозы на ~20% или от 660 и 4350 часов при увеличении дозы на ~50%. Для однократной дозы, действующей 2 недели (tmin=336 часов), оптимальная скорость высвобождения лекарственного средства составляет 230 часов, при этом также может использоваться любая скорость высвобождения от 150 до 380 часов при увеличении дозы на ~10%, от 130 до 470 часов при увеличении дозы на ~20% или от 100 до 680 часов при увеличении дозы на ~50%.
[0057] Аналогичным образом, доза, требуемая для поддержания концентрации лекарственного средства выше Cmin в равновесном состоянии (ss) в режиме с повторным введением доз, приведена как:
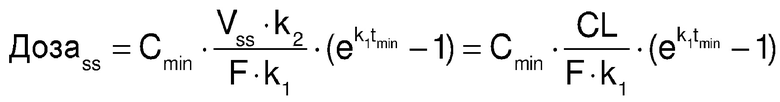
[0058] В режиме с повторным введением доз нет оптимальной дозы, как описано выше, а скорее необходимы снижения дозы при снижении скорости высвобождения, поскольку более высокий процент лекарственного средства остается от предыдущих доз, добавляясь к полному присутствующему депо лекарственного средства.
[0059] Существуют практические пределы для применения более низкой скорости высвобождения при уменьшении дозы, однако требуется свести к минимуму высвобождение несущих лекарственное средство фрагментов геля из биоразлагаемой матрицы, минимизировать общую нагрузку депо на пациента и увеличить время, требуемое для достижения уровней лекарственного средства в равновесном состоянии.
[0060] В одном варианте осуществления расщепляемый линкер L имеет формулу (3):
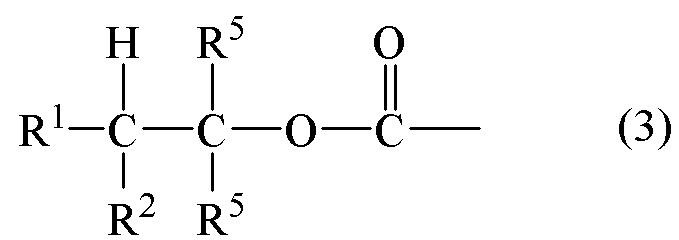
где по меньшей мере один или оба R1 и R2 независимо являются CN; NO2;
необязательно замещенным арилом;
необязательно замещенным гетероарилом;
необязательно замещенным алкенилом;
необязательно замещенным алкинилом;
COR3 или SOR3 или SO2R3, где
R3 является H или необязательно замещенным алкилом;
арилом или арилалкилом, каждый из которых необязательно замещен;
гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен; или R3
OR9 или NR92, где каждый R независимо является H или необязательно замещенным алкилом, или обе группы R9, взятые вместе с азотом, к которому они присоединены, образуют гетероциклическое кольцо;
SR4, где
R4 является необязательно замещенным алкилом;
арилом или арилалкилом, каждый из которых необязательно замещен; или
гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен;
где R1 и R2 могут быть соединены с образованием 3-8-членного кольца; и
где один и только один из R1 и R2 может быть H или алкилом, арилалкилом или гетероарилалкилом, каждый из которых необязательно замещен; и
где один из R5 является (CH2)yZ, (CH2CH2O)xCH2CH2Z или (CH2)yNH-CO-(CH2CH2O)xCH2CH2Z, где x является 1-100, y=1-6, и другой R5 является H, алкилом, алкенилалкилом, алкинилалкилом, арилом, арилалкилом, гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен; и
Z является функциональной группой для опосредования связывания с матрицей.
[0061] Такие линкеры расщепляются при бета-элиминировании. В предпочтительных вариантах осуществления изобретения R1 является CN или R3SO2, где R3 является замещенным или незамещенным алкилом или (R9)2N, где каждый R9 независимо является замещенным или незамещенным алкилом; R2 является H; один R5 является (CH2)yZ, и другой R5 является H. Z является N3, SH, NH-C(=O)CH2ONH2 или O-NH2. В наиболее предпочтительных вариантах осуществления R1 является CN или CH3SO2, и/или Z является N3.
[0062] Могут использоваться другие типы расщепляемых линкеров, например, линкеры, которые расщепляются путем ферментативного или неферментативного гидролиза, такие как линкеры в публикации PCT WO2006/136586, включенной в настоящую заявку посредством отсылки. Единственное требование состоит в том, чтобы скорость расщепления линкера соответствовала требуемому режиму введения, как описано выше.
[0063] В одном варианте осуществления настоящего изобретения линкер расщепляется и высвобождает E с периодом полувыведения при физиологических условиях, подходящим для введения один раз в месяц, т.е. линкер расщепляется и высвобождает E с периодом полувыведения от 220 до 1440 часов. В более предпочтительном варианте осуществления линкер расщепляется и высвобождает E с периодом полувыведения от 280 до 1000 часов, более предпочтительно от 320 до 800 часов. В других вариантах осуществления изобретения линкер расщепляется и высвобождает E с периодом полувыведения при физиологических условиях, подходящим для введения один раз в 3 месяца или раз в две недели.
[0064] В определенном варианте осуществления изобретения линкер L имеет формулу (5), где R1 является CN. Как продемонстрировано в Примере 5, этот линкер высвобождает пептид из гидрогеля у крысы с периодом полувыведения 760 ч.
[0065] В другом определенном варианте осуществления линкер L имеет формулу (5), где R1 является CH3SO2. Как продемонстрировано в Примере 5, этот линкер высвобождает пептид из гидрогеля у крысы с периодом полувыведения 350 ч.
[0066] Линкер L связан с пептидом E посредством образования карбаматной связи между группой C=O в L и аминогруппой в E. Аминогруппа может быть N-концевой альфа-аминогруппой или эпсилон-аминогруппой боковой цепи лизина. Способы их получения известны в уровне техники. В одном варианте осуществления L присоединяют к альфа-аминогруппе E в ходе твердофазного синтеза пептида.
[0067] Линкер L дополнительно включает группу Z, которая позволяет присоединять линкер-пептид L-E к матрице при использовании химии, которая является совместимой и селективной в присутствии функциональных групп на пептиде E. Z может быть азидом, в этом случае L-E связывают с матрицей при использовании реакции 1,3-диполярного циклоприсоединения с образованием 1,2,3-триазольной связи или фосфин-опосредованного лигирования по Штаудингеру с образованием амида; обе реакции хорошо известны в уровне техники. Реакция циклоприсоединения может быть либо катализируемым медью присоединением к алкин-дериватизированной матрице, либо вызванным напряжением присоединением к циклооктин-или бициклононин-дериватизированной матрице. Z также может быть аминоокси или аминоокси-ацетамидогруппой, в этом случае L-E связывают с кетодериватизированной матрицей при использовании реакции оксимирования. Или сам Z может быть кетогруппой, присоединяемой к аминооксигруппе на матрице. Z также может быть тиольной группой, в этом случае L-E связывают с галогенацетил-дериватизированной, малеимид-дериватизированной или эпокси-дериватизированной матрицей посредством образования тиоэфира.
[0068] Таким образом, функциональные группы, используемые для присоединения L к матрице, включают N3, NH2, NH-CO2tBu, SH, StBu, малеимид, CO2H, CO2tBu, 1,3-диен, циклопентадиен, фуран, алкин, циклооктин, акрилат, аминоокси, кето или акриламид.
Получение конъюгатов :
[0069] Конъюгаты получают при соединении пептида E, расщепляемого линкера L и матрицы M. В одном варианте осуществления связи образуются попарно, при этом порядок связи может изменяться. Таким образом, пептид E может быть сначала связан с линкером L, и полученный L-E связан с матрицей M. В альтернативе линкер L может быть связан с матрицей M и E, после чего связан с M-L. В случае, когда M является матрицей, полученной при полимеризации мономерных звеньев, M-L или M-L-E может быть результатом процесса полимеризации при использовании сшиваемого мономера-L или звена мономера-L-E в реакции.
[0070] Для биологического применения конъюгаты должны соответствовать строгим критериям стерильности и содержания эндотоксинов. Хотя в определенных случаях может применяться процесс конечной стерилизации, обычно конъюгаты согласно изобретению не подлежат такой обработке. Нерастворимые гидрогели, например, также не поддаются стерилизации фильтрованием. Таким образом, может быть предпочтительно, чтобы конъюгаты согласно изобретению были получены в асептических условиях. Конъюгаты могут быть получены либо в виде суспензий микросфер для инъекций, либо они могут получены in situ при совместной инъекции мономерных звеньев.
Лекарственные формы:
[0071] Конъюгаты могут быть включены в лекарственные формы с применением стандартных фармацевтически приемлемых буферов и вспомогательных веществ для улучшения стабильности при хранении и пригодности для инъекций. Типичные лекарственные формы включают буфер для поддержания pH в пределах 4-7, предпочтительно 5-6. Вспомогательные вещества могут включать стабилизирующие вещества для пептидного лекарственного средства, например, антибактериальные средства и/или антиоксиданты, такие как метакрезол, регуляторы тоничности, такие как полиол, такой как манит, и снижающие вязкость вещества, такие как таурин, теанин, саркозин, цитруллин и бетаин.
Способы применения:
[0072] Конъюгаты согласно изобретению могут применяться при лечении нарушений и заболеваний обмена веществ у людей и у животных, при которых введение агониста ГПП-1, как известно, является эффективным, включающих, без ограничения, диабет 2 типа, метаболический синдром и ожирение. Существенно увеличенный период полувыведения позволяет вводить дозы один раз в месяц, улучшая, таким образом, соблюдение пациентом режима (исключение пропущенных доз) и качество жизни пациента. Введение доз предпочтительно производят путем подкожной инъекции и могут выполнять при использовании автоинъектора.
[0073] Следующие примеры предложены для иллюстрации, но не ограничения, изобретения.
Получение A
Разложение эксенатида in vitro
[0074] Раствор 2,4 мМ эксенатида (1 мл), 0,1% NaN3 и 200 мкМ Lys(DNP)OH в качестве внутреннего стандарта в 200 мМ NaPi, pH 7,4, хранили при 37°C. С интервалами, аликвоты по 50 мкл отбирали и замораживали при 20°C до анализа. Различные образцы размораживали и исследовали: a) с помощью ВЭЖХ, b) на GLP1R агонистическую активность и c) на активность протеин-изоаспартат-метилтрансферазы. Образец инкубировали в течение 56 дней, подвергали ВЭЖХ и образцы при RV 9,9, 10,4 и 10,8 собирали и исследовали индивидуально на чистоту с помощью аналитической ОФ-ВЭЖХ, GLP1R агонистическую и PIMT активность.
[0075] (a) ВЭЖХ профили дезамидирования эксенатида в зависимости от времени показаны на Фигурах 8А-8C. Каждый пик из реакции через 56 дней очищали с помощью ОФ-ВЭЖХ. На панели A показаны кривые ВЭЖХ, полученные через 0 (сверху), 7, 28 и 56 (снизу) дней для нативного эксенатида. Начальный одиночный пик эксенатида постепенно заменялся пиками нескольких продуктов разложения. На панели B показана динамика разложения эксенатида, полученная на основе данных в панели A. Снижение эксенатида (квадраты) и увеличение основного продукта разложения показывает t1/2 приблизительно 10 дней. На панели C показаны кривые ВЭЖХ, полученные с [N28Q]эксенатидом, соответствующие кривым эксенатида на панели A. Аналитическая ВЭЖХ выделенных продуктов разложения показала, что пики L-Asp и D-изоAsp содержали ~7 и 12% контаминирующего изоAsp, соответственно; выделенный изоAsp показал одиночный пик.
[0076] (b) Точки времени исследовали на активацию рецептора ГПП-1 с помощью биоанализа GLP1R цАМФ HunterTM (DiscoverX). Значения EC50 приведены ниже в Таблице 1.
Таблица 1
Значения EC50 продуктов разложения эксенатида в анализе GLP1R цАМФ Hunter™.
Концентрации пептида вычисляли по A280 и ε80=5500 М-1см-1; значения L-Asp и D-изоAsp не корректировали с учетом небольших количеств примеси изоAsp.
[0077] Пептид Asp, выделенный из смеси дезамидирования, содержал небольшое количество потенциально мешающего пептида изоAsp, однако синтетический [Asp28]эксенатид показал агонистическую активность, сопоставимую с эксенатидом. Количество D-изоAsp, образовавшегося в реакции дезамидирования через 56 дней, настолько мало (~12%), что он не может оказывать значимое влияние на агонистическую активность смеси.
[0078] (c) Для анализа присутствия изоаспартата проводили анализы протеин-изоаспартат-метилтрансферазы (PIMT) при использовании набора для обнаружения изоаспартата ISOQUANT® согласно рекомендациям поставщика (Promega). Смеси образцов при t=0 и t=56 дней, а также отдельные образцы, очищенные из смеси t=56 дней, анализировали на пептиды изоAsp. На Фигуре 9 показана AdoHCys/пептидспецифическая активность, полученная в a) суммарной смеси и в b) выделенных пиках RV 9,9 и RV 10,4, с поправкой на присутствие небольших количеств контаминирующего изоAsp из пика с RV 10,8.
Получение B
Получение аналогов эксенатида
[0079] Ala, Asp, Gln и Lys замены в Asn28 эксенатида вводили с помощью ТФСП. Все [Xaa28]эксенатиды имели значения EC50 (17-41 пМ), сопоставимые с эксенатидом (17 пМ), в анализе GLP-1RA. При долговременном инкубировании (~3 месяца) этих [Xaa28]эксенатидов в 200 мМ Pi, pH 7,4, 37°C, новых основных пиков не наблюдали, за исключением [Asp28]эксенатида, который медленно изомеризовался в [изоAsp28]эксенатид. При низких концентрациях пептида (~0,2 мМ) наблюдали небольшие снижения в A280, согласующиеся с неспецифической адсорбцией на поверхностях сосуда. При 2 мМ [Gln28]эксенатида, t1/2 для потери пептида оценивали на уровне >/= 30 недель. Следовательно, [Gln28]эксенатид обладает высокой стабильностью при физиологических условиях. t1/2 30 недель будет давать <9% потерю за один месяц.
Получение C
Связывание рецептора и активность аналогов эксендина in vitro
[0080] Способность различных аналогов эксенатида активировать рецептор ГПП-1 исследовали при использовании анализа цАМФ (биоанализ GLP1R цАМФ Hunter™ (DiscoverX)). Как обнаружили, аналоги N28D, N28A, N28K и N28Q обладали сопоставимой с эксенатидом активностью.
[0081] При исследовании смеси продуктов разложения (Фигура 8A, 56 дней) в анализе цАМФ, она имела EC50, эквивалентную исходным показателям эксенатида (Фигура 8A, 0 дней). Аналогичным образом, предполагаемые [ИзоAsp]28- и [Asp]28 эксенатиды, очищенные из смесей, а также синтетический [Asp]28 эксенатид показали значения EC50, подобные эксенатиду. Однако при исследовании в ИФА анализе (Peninsula Lab) выделенные продукты дезамидирования и синтетический [N28D]-эксенатид показали значительно сниженную аффинность (EC50 эксенатид, 0,2 нМ; N28A=6 нМ; N28Q=10 нМ; N28D >100 нМ; N28K >100 нМ). Следовательно, основные продукты разложения имеют эквивалентную агонистическую активность, как и эксенатид, но не были исследованы с помощью ЖХ-МС/МС или ИФА анализов, используемых для эксенатида в сыворотке.
Получение D
Получение разлагаемых микросфер
Подготовка макромономера A
[0082] (a) Раствор 1-(бис-(2-метоксиэтил)аминосульфонил)-7-азидо-2-гептил-сукцинимидилкарбоната (полученного с использованием способов, описанных в WO2013/036847; 1,12 ммоль) в 5 мл ацетонитрила добавляли к раствору H-Lys(Boc)-OH (300 мг, 1,22 ммоль) и NaHCO3 (420 мг, 5,0 ммоль) в 10 мл воды и 5 мл ацетонитрила. Через 0,5 ч раствор выпаривали в вакууме для удаления ацетонитрила, подкисляли 1 Н HCl и экстрагировали этилацетатом. Экстракт промывали водой и рассолом, затем сушили над MgSO4, фильтровали и выпаривали с получением неочищенного Na-[1-(бис-(2-метоксиэтил)аминосульфонил)-7-азидо-2-гептилокси)-карбонил]-Ne-(BOC)-лизина ("азидо-линкер[мод]-Lys(Boc)-OH").
[0083] Согласно этому способу получали азидо-линкер[мод]-Lys(Boc)-OH, где модулятором является бис(2-метоксиэтил)аминосульфонил, диметиламиносульфонил, циано, метилсульфонил, 4-метилпиперидинилсульфонил, морфолиносульфонил или фенилсульфонил.
[0084] (b) Указанный выше неочищенный азидо-линкер[мод]-Lys(Boc)-OH растворяли в 25 мл CH2Cl2 и обрабатывали N-гидроксисукцинимидом (138 мг, 1,2 ммоль) и дициклогексилкарбодиимидом (0,5 мл 60% по весу раствора в ксилолах) в течение 2 ч. Смесь фильтровали и подвергали хроматографии на SiO2 при использовании градиента ацетона в гексане с получением сукцинимидилового сложного эфира азидо-линкер[мод]-Lys(Boc)-OSu.
[0085] Согласно этому способу получали азидо-линкер[мод]-Lys(Boc)-OSu, где модулятором является бис(2-метоксиэтил)аминосульфонил, диметиламиносульфонил, циано, метилсульфонил, 4-метилпиперидинилсульфонил, морфолиносульфонил или фенилсульфонил.
[0086] (c) Раствор 20 кДа 4-лучевого ПЭГ-тетраамина в ацетонитриле (10 мл, 200 мг/мл, 40 мМ амина, 0,4 ммоль, 1 эквив.), содержащий N,N-диизопропилэтиламин (80 мМ, 0,8 ммоль, 2 эквив.), обрабатывали раствором азидо-линкер[мод]-Lys(Boc)-OSu в ацетонитриле (3,3 мл, 145,5 мМ, 0,48 ммоль, 1,2 эквив.). Полученную смесь выдерживали при комнатной температуре в течение 1 ч, после чего образец 0,010 мл оценивали на содержание амина с помощью анализа с TNBS при использовании ПЭГ-тетраамина в качестве стандарта; обычно остается <1% исходных аминов. Затем реакцию обрабатывали уксусным ангидридом (40,8 мг, 0,0378 мл, 0,4 ммоль, 1 эквив.) в течение 15 минут перед выпариванием в вакууме до вязкого сиропа (~4 мл), который медленно добавляли к МТБЭ (350 мл). Полученную суспензию перемешивали в течение 1 ч, затем осадок отделяли с помощью фильтрования, промывали МТБЭ (150 мл) и сушили в вакууме с получением макромономера в виде белого твердого вещества.
[0087] Согласно этому способу получали макромономер, где модулятором является бис(2-метоксиэтил)аминосульфонил, диметиламиносульфонил, циано, метилсульфонил, 4-метилпиперидинилсульфонил, морфолиносульфонил или фенилсульфонил.
Получение макромономера B
[0088] Флакон с завинчивающейся крышкой на 4 мл наполняли ПЭГ20кДа-[NH2]4 (SunBright PTE-200PA; 150 мг, 7,6 мкмоль ПЭГ, 30,2 мкмоль NH2, 1,0 эквив., конечная концентрация амина 20 мМ), MeCN (1,5 мл) и iPR2NEt (7 мкл, 40 мкмоль, 1,3 эквив., конечная концентрация 27 мМ). Добавляли раствор активированного сложного эфира циклооктина (39 мкмоль, 1,3 эквив., конечная концентрация 27 мМ) и перемешивали реакционную смесь при температуре окружающей среды. Ход реакций контролировали с помощью ВЭЖХ C18 (20-80% B за 11 минут) при использовании ИДС. После завершения в реакционную смесь добавляли Ac2O (3 мкл, 30 мкмоль, по 1 эквив. на исходный NH2) и перемешивали смесь в течение 30 минут. Затем реакционную смесь выпаривали до вязкого масла и суспендировали в МТБЭ (20 мл). Полученную суспензию энергично перемешивали в течение 10 мин. Полученное твердое вещество три раза растирали с МТБЭ (20 мл) при энергичном перемешивании, осаждали в центрифуге (2800 об/мин, 4°C, 10 мин) и удаляли супернатант пипеткой. Полученное твердое вещество сушили в вакууме при температуре окружающей среды в течение не больше чем 30 мин. Стоковые растворы приготавливали в 20 мМ NaOAc (pH 5) с концентрацией целевого амина 20 мМ. Затем концентрацию циклооктина подтверждали при обработке ПЭГ7-N3 (2 эквив.) и обратном титровании непрореагировавшего ПЭГ7-N3 с использованием DBCO-CO2H.
[0089] Макромономеры, полученные при использовании этой методики, включают такие, в которых циклооктиновой группой является MFCO, 5-гидроксициклооктин, 3-гидроксициклооктин, BCN, DIBO, 3-(карбоксиметокси)циклооктин и 3-(2-гидроксиэтокси)-циклооктин, полученные при использовании сложного пентафторфенилового эфира MFCO, 5-(4-нитрофенокси-карбонил)окси)циклооктин, 3-(4-нитрофеноксикарбонил)-оксициклооктин, BCN гидроксисукцинимидилкарбонат, DIBO 4-нитрофенилкарбонат, 3-(карбоксиметокси)циклооктин сукцинимидиловый сложный эфир или 3-(гидроксиэтокси)циклооктин 4-нитрофенилкарбонат.
Получение E
Получение иллюстративного гидрогеля из Фигуры 1A
[0090] Дериватизированный 8-лучевой макромономер P получали следующим образом: Макромономер P представляет собой 8-лучевой ПЭГ, каждый луч которого содержит концевой циклооктин. Раствор 200 мг 40 кДа 8-лучевой ПЭГ⋅амина HCl (JenKem Technologies; 40 мкмоль NH2), 20 мг BCN п-нитрофенилкарбоната (SynAffix; 63 мкмоль) и 20 мкл N,N-диизопропилэтиламина (115 мкмоль) в 2 мл ДМФА перемешивали 16 ч при температуре окружающей среды. После остановки реакции 0,5 мл 100 мМ таурина в 0,1 М KPi, pH 7,5, в течение 1 ч, смесь последовательно диализировали против воды, метанола/воды 1:1 и метанола при использовании 12 кДа мембраны. После выпаривания остаток растворяли в 2 мл ТГФ и осаждали 10 мл метилтретбутилового эфира. Продукт собирали и сушили (190 мг). Макромономеры P, включающие другие циклооктины, могут быть получены так же, при использовании подходящего активированного циклооктина.
[0091] Дериватизированный 4-лучевой макромономер T получали следующим образом: T включает 4-лучевой ПЭГ, каждый луч которого содержит на конце высвобождаемый линкер-азид. Раствор 25 мкмоль азидо-линкер-сукцинимидилкарбоната (подлученный при использовании способов, описанных в WO2013/036847) в 1 мл ACN, добавляли к смеси 5 мкмоль (100 мг) 20 кДа 4-лучевого ПЭГ-амина гидрохлорида (пентаэритритное ядро, JenKem Technologies) в 1 мл воды и 40 мкл 1,0 М NaHCO3 (40 мкмоль). Через 1 ч при температуре окружающей среды раствор диализировали (НОММ 12-14 кДа) против 1 л 50% метанола, затем 1 л метанола. После выпаривания остаток (109 мг) растворяли в 2,12 мл стерилизованного фильтрованием 10 мМ NaOAc, pH 5,0, и хранили в замороженном виде при -20°C. Концентрация азида, определенная в реакции с DBCO-кислотой, составила 9,5 мМ. Макромономеры T, включающие линкер-азиды, содержащие альтернативные модуляторы, могут быть получены аналогичным образом при помощи подходящих азид-линкер-сукцинимидилкарбонатов.
[0092] Высвобождающие пептид гидрогели могут быть получены из дериватизированных макромономеров по меньшей мере двумя разными способами.
[0093] (a) В одном варианте осуществления линкер-пептид присоединяют к макромономеру P до образования нерастворимой матрицы гидрогеля. Азидо-линкер-пептид формулы (4), такой как представленный в Примере 1 ниже, смешивают с макромономером P в такой стехиометрии, что линкер-пептид дериватизирует некоторую часть лучей P. Полученный материал затем сшивают при использовании достаточного количества макромономера T, чтобы лучи T прореагировали с оставшимися лучами P и, таким образом, образовали нерастворимую матрицу. Таким образом, n молей азидо-линкер-пептида формулы (4) смешивают с n/(8f) молей макромономера P, где f=требуемая фракционная нагрузка лучей линкер-пептидом (т.е. при 50% нагрузке лучей f=0,5), в подходящем растворителе, как правило, забуференной водной среде. После истечения времени, достаточного для реакции азидо-линкер-пептида, полученный раствор смешивают с n(1/f-1)/4 молей макромономера T, с образованием нерастворимой матрицы гидрогеля. Как правило, f подбирают таким образом, чтобы на каждый остаток P в матрице гидрогеля присутствовало >3 сшитых лучей (f<0,625). Реакцию сшивания с образованием нерастворимой матрицы гидрогеля могут проводить в виде объемного материала или в суспензии или эмульсии, с образованием полимера в форме тонкодисперсных частиц, например микросфер, как описано в Примере 2 ниже.
[0094] (b) В альтернативе нерастворимая матрица гидрогеля может быть получена с последующим присоединением линкер-пептида. Для конечного гидрогеля, включающего 8f эквивалентов линкер-пептида на каждый остаток макромономера P в матрице, нерастворимая матрица гидрогеля образуется в реакции n/(8f) молей макромономера P с n(1/f-1)/4 молей макромономера T. Реакцию сшивания с образованием нерастворимой матрицы гидрогеля могут проводить в виде объемного материала или в суспензии или эмульсии, с образованием полимера в форме тонкодисперсных частиц, например микросфер, как описано в Примере 2 ниже. Затем полимеризованной матрице позволяют реагировать с раствором по меньшей мере n молей азидо-линкер-пептида формулы (4), при этом линкер-пептид ковалентно присоединяется к матрице. Непрореагировавший азидо-линкер-пептид удаляют из матрицы при промывке с получением высвобождающего пептид гидрогеля.
Пример 1
Получение азидо-линкер-[N28Q]эксенатидов формулы (4)
где R1=CN или MeSO2; R2=H; один R5=H, и другой R5=(CH2)5N3; P=Nα-[N28Q]эксенатид
[0095] Пептиды синтезировали с помощью стандартной методики твердофазного синтеза при использовании амидной смолы Chemmatrix® Rink (0,5 мэкв/г) на синтезаторе пептидов Symphony®. Fmoc-аминокислоты (5 экв. на связывание) связывали в две стадии с N-концом пептидной цепи при использовании HCTU (4,9 экв. на связывание) и N,N-диизопропилэтиламина (10 экв. на связывание) в ДМФА при температуре окружающей среды. Fmoc-группы удаляли при использовании 20% 4-метилпиперидина в ДМФА. Удаление защитных групп и снятие [N28Q]эксенатида со смолы проводили при использовании смеси 95:2,5:2,5 трифторуксусной кислоты/триизопропилсилана/дитиотреитола.
[0096] Неочищенный [N28Q]эксенатид (22 мг) очищали в полупрепаративном масштабе при использовании системы Shimadzu™ LC-20AD, оборудованной 5 мкм колонкой C18 Peak Scientific HiQ® (50×20 мм в.д.), при элюировании в линейном градиенте 30-60% MeCN (0,1% ТФУ) в воде (0,1% ТФУ). Наиболее чистые фракции, согласно оценке с помощью аналитической ВЭЖХ C18, объединяли, выпаривали на ~40% для удаления MeCN и лиофилизировали с получением [N28Q]эксенатида (6,4 мг, 1,4 ммоль) в виде белого хлопьевидного вещества. Чистоту согласно ВЭЖХ C18 определяли при 280 нм: чистота 86% (RV=9,4 мин); [N28Q]эксенатид; m/z=4200.
[0097] N-концевое карбамоилирование пептидов на смоле было проведено при использовании модификации ранее описанного метода (Schneider, E. L. et al., Biocong. Chem (2016) 1 March, предварительная онлайн публикация) и представлено следующим примером.
[0098] Nα-(7-Азидо-1-циано-2-гептилоксикарбонил)-[Gln28]эксенатид. В закрытой колпачком с мембраной колбе Эрленмейера объемом 125 мл NH2[N28Q]эксенатид (свободный α-амин) на амидной смоле Chemmatrix® Rink (0,5 мэкв/г замещение, 0,48 ммоль пептида/г пептида-смола, 4,00 г пептида-смолы, 0,48 ммоль пептида) мягко перемешивали в 40 мл ДМФА в течение 30 минут при температуре окружающей среды под N2. Затем набухшую смолу обрабатывали 8 мл O-(7-азидо-1-циано-2-гептил)-O'-сукцинимидил-карбоната (0,18 М в ДМФА, 1,44 ммоль, конечная концентрация 30 мМ) и 4-метил-морфолина (158 мкл, 1,44 ммоль). Реакционную смесь мягко перемешивали под N2 в течение 2 ч, затем фильтровали на вакуумном фильтре. Смолу последовательно промывали ДМФА (3×30 мл) и CH2Cl2 (4×50 мл), затем сушили в глубоком вакууме. Тест Кайзера на свободные амины в промежуточной линкер-модифицированной смоле (3,84 г) был отрицательным. Затем смолу обрабатывали 40 мл 90:5:5 ТФУ:TIPS:H2O при перемешивании под N2. Через 2,5 ч смолу фильтровали на вакуумном фильтре и промывали ТФУ (2×10 мл). Фильтрат выпаривали до ~20 мл. Неочищенный линкер-пептид осаждали при добавлении по каплям концентрированного раствора в ТФУ к охлажденному во льду Et2O:гексану (2:1, 160 мл) в 4 взвешенных пробирках типа Falcon объемом 50 мл. После инкубирования на льду в течение 30 мин, неочищенный линкер-пептид осаждали с помощью центрифугирования (3 мин при 2000×g) и удаляли супернатант. Осадок растирали/встряхивали на вортексе с охлажденным во льду Et2O:гексаном (2:1, 160 мл), инкубировали на льду, центрифугировали и фильтровали, как указано выше. После сушки в глубоком вакууме неочищенный линкер-пептид выделяли в виде почти белого твердого вещества (1,76 г), которое затем растворяли в 5% AcOH. После нагревания при 40°C в течение 1 ч, неочищенный материал очищали с помощью препаративной ВЭЖХ. МС: m/z=4408.
[0099] Nα-[1-(метилсульфонил)-7-азидо-2-гептилоксикарбонил]-эксенатид получали так же, при использовании O-(7-азидо-1-(метилсульфонил)-2-гептил)-O'-сукцинимидилкарбоната. МС: m/z=4461.
Пример 2
Получение микросфер ПЭГ гидрогеля
[00100] Использовали микрофлюидный 2-реагентный гидрофобный смесительный чип Telos® (Dolomite) с семью параллельными 50 мкм каплеформирующими каналами. Поток жидкости регулировали пневматическим нагнетательным насосом, подобным по функции нагнетательным насосам Mitos производства Dolomite Microfluidics. В этих насосах используется сжатый газ для направления потока жидкости через микрофлюидный чип. Рабочее давление регулируется компьютером при помощи пропорциональных регуляторов давления (Proportion Air, серия MPV) для поддержания стабильной скорости потока при помощи контура обратной связи от датчика потока жидкости (Sensirion, SLI-0430). Этот тип регулирования потока можно масштабировать для подачи жидкости из многолитровых резервуаров и получать скорости потока со стандартным отклонением ~1%, что превосходит шприцевые насосы, которые часто дают до 20% колебания скорости потока. Такую систему использовали для подачи двух растворов преполимера гидрогеля и непрерывной фазы. Типичные скорости потока составляли 2,1 мл/ч для каждого раствора преполимера и 14 мл/ч для непрерывной фазы. Непрерывная фаза состояла из декана, содержащего 1% в./об. Abil® EM90 (Evonik) и 1% в./об. PGPR (Danisco). Выпускная трубка устройства была соединена с коллектором фракций (Gilson FC203B), и фракции отбирали с 10-минутными интервалами. Контроль качества осуществляли при фотографировании чипа при 5× увеличении с помощью высокоскоростной камеры (UniBrain®, Fire I 580b), установленной на микроскоп (Nikon™, EQ 51436), оборудованный автоматическим столиком, для визуализации семи каналов чипа. Изображения каждого канала регистрировали каждые 5 минут. Фракции, содержащие крупные частицы, образовавшиеся в результате отказа устройства, можно было устранить из партии.
[0100] Промывку микросфер проводили в центрифужных пробирках объемом 50 мл из Teflon® (ФЭП) (Oak Ridge, 3114 0050). После промывки, микросферы собирали с помощью центрифугирования. Центрифугирование проводили в течение 5 мин при 3000g для разделения органических фаз, и 20 мин для разделения водных фаз. Все растворы и растворители для промывки фильтровали через 0,2 мкм фильтры из Nylon 66 (Tisch, SPEC17984).
[0101] Суспензию микросфер из микрофлюидной жидкости (30 мл) в декансодержащем поверхностно-активном веществе оставляли для отверждения при комнатной температуре в течение 24 ч. Слой декана удаляли и микросферы делили между 0,1% (в./об.) водным NaN3 (15 мл) и пентаном. Смесь перемешивали в течение 30 мин, затем пентановую фазу отделяли центрифугированием. Затем суспензию микросфер обрабатывали водой (30 мл) и промывали пятью последовательными порциями (39 мл) пентана. После центрифугирования удаляли избыток водной фазы и суспензию микросфер обрабатывали равным объемом 50% в/об ТФУ в течение 30 мин для стерилизации. Микросферы собирали центрифугированием при 1000 g (примечание: сферы сжимаются в ТФУ и образуют компактный осадок, поэтому следует избегать чрезмерной силы). Осадок обрабатывали 0,125 М Na2HPO4 (150 мл) с получением суспензии с pH ~6,5. После набухания в течение 18 ч сферы собирали центрифугированием, затем промывали пятью порциями по 100 мл воды и, наконец, пятью порциями по 100 мл 70% этанола. Суспензию осаждали до конечной концентрации при 3000g в течение 30 мин. После аспирации супернатанта суспензию микросфер переносили в 60 мл шприц (BD 309653), который был соединен через коннектор Луэр с другим 60 мл шприцем и гомогенизировали с помощью нескольких перекачиваний вперед и назад для диспергирования небольших комков. Шприц, содержащий суспензию, использовали для наполнения отдельных шприцев на 10 мл через соединение Луэр, которые хранили при 4°C до применения.
[0102] Три порции по 0,100 мл суспензии амино-микросфер в ацетонитриле взвешивали для определения их плотности (0,79±0,2 г/мл), затем каждую порцию обрабатывали 0,900 мл 50 мМ NaOH в течение 18 ч при комнатной температуре для расщепления сшивок и образования [H2N-Lys(NH2)-NH]4-ПЭГ20кДа мономеров. Каждый образец исследовали на общую концентрацию амина с помощью анализа с TNBS при разбавлении 0,030 мл до 0,120 мл боратным буфером (100 мМ, рН 9,3), затем обрабатывали 0,150 мл боратного буфера, содержащего 0,04% в/об 2,4,6-тринитробензолсульфоната в микротитровальном планшете. Изменение оптической плотности реакций с TNBS при 420 нм контролировали в течение 3 ч в спектрофотометре для планшетов при 25°C, после чего регистрировали конечную оптическую плотность при 420 нм. Эквивалентные реакции, содержащие только TNBS, использовали для вычитания фона, и реакции, содержащие 40, 20 или 10 мкМ лизина, использовали в качестве стандартных концентраций амина. Общая концентрация амина/2 гидролизатов микросфер обеспечивает свободное содержание e-амина в геле.
Пример 3
Получение циклооктин-микросфер
[0103] Реакцию проводили в реакционном сосуде-шприце следующим образом. На каждые 4 мл уплотненной суспензии амино-микросфер в MeCN, содержащей 2 мкмоль амина/мл суспензии геля, добавляли 32 мкмоль DIPEA (4 эквивалента) в 1 мл MeCN и 9,6 мкмоль (1,2 эквивалента) 1-фтор-2-циклооктин-1-карбоксилата пентафторфенилового эфира (MFCO-PFP) в 1 мл MeCN. Через 1 ч качания при температуре окружающей среды небольшое количество (~50 мкл) микросфер вытесняли из выходного отверстия шприца и обрабатывали 0,5 мл 0,04% в/об TNBS в 0,1 М борате натрия, рН 9,3 (1), в течение 30 мин; на завершение реакции указывает цвет микросфер, совпадающий с раствором TNBS, по сравнению с исходными амино-микросферами, которые имеют интенсивный оранжевый цвет. После реакции микросферы блокировали добавлением 8 мкмоль (1 эквивалента) Ac2O в 1 мл MeCN в течение 10 мин. После удаления супернатанта ~2 мл микросфер переносили во второй 10 мл шприц, каждую суспензию промывали 4×3 объемами MeCN на объем уплотненной суспензии и объединяли суспензии.
Пример 4
Получение пептид-высвобождающих микросфер ПЭГ гидрогеля
[0104] Связывание азидо-линкер-[N28Q]эксенатидов, полученных в Примере 1, проводили в реакционном сосуде-шприце, описанном в публикации Schneider, et al., выше. К суспензии 2,4 г суспензии MFCO-дериватизированной микросферы Примера 3 (11,2 мкмоль MFCO) в 30% MeCN в 10 мл шприце добавляли раствор 46 мг (10,4 мкмоль) Nα-[1-(метилсульфонил)-7-азидо-2-гептилоксикарбонил]-[N28Q]эксенатида в 2 мл 30% MeCN. Смесь медленно вращали до тех пор, пока OD280 аликвоты не станет постоянной, в течение ~24 ч. Около 50% суспензии переносили во второй шприц и оба образца промывали 4×2 мл 30% MeCN, а затем 5×5 мл 10 мМ NaPi, 0,04% Tween® 20, pH 6,2. Затем микросферы, нагруженные [N28Q]эксенатидом, переносили из шприца в шприц, в несколько 1,0 мл дозирующих шприцев. Общая нагрузка микросферы составляла 1,9 мкмоль пептида г-1 суспензии, как определяли по общему количеству пептида, высвобождаемого при рН 8,4.
[0105] Высвобождение свободного [N28Q]эксенатида из микросфер измеряли in vitro при суспендировании образца конъюгата в 0,1 М борате, рН 9,4, и последующей солюбилизации по увеличению OD280 при 37°C (Фигура 5). Наблюдали высвобождение первого порядка с периодом полувыведения =6,7 ч при рН 9,4. Это экстраполировали в 670 ч при рН 7,4. Аналогичным образом получали микросферы, нагруженные Nα-[1-циано-7-азидо-2-гептилоксикарбонил]-[N28Q]эксенатидом. Высвобождение свободного [N28Q]эксенатида из этих микросфер измеряли in vitro при суспендировании образца конъюгата в 0,1 М борате, рН 9,4, и последующей солюбилизации по увеличению OD280 при 37°С. Наблюдали высвобождение первого порядка с периодом полувыведения =16,5 ч при рН 9,4. Это экстраполировали в 1650 ч при рН 7,4.
[0106] Микросферы, включающие [N28Q]эксенатид, присоединенный через линкер, где R1=CN и R2=H, получали аналогичным образом, при использовании Nα-[1-циано-7-азидо-2-гептилоксикарбонил]-[N28Q]эксенатида (Пример 1). Конечный препарат включал 2,2 мкмоль пептида г-1 в изотоническом ацетатном буфере (10 мМ ацетата, 120 мМ NaCl, pH 5,0) с 0,05% Tween® 20.
Пример 5
Фармакокинетика у крыс
[0107] Содержимое взвешенных 1 мл дозирующих шприцев, содержащих суспензии микросфер, полученные в Примере 4, вводили через иглу 27G п/к в бок канюлированным самцам крыс Спрег-Доули, ~350 г. Каждый шприц содержал 0,45 или 0,98 мкмоль пептида при концентрации пептида 1,9 мкмоль г-1 суспензии. Шприцы взвешивали до и после введения для проверки массы, доставленной каждой крысе. Кровь (300 мкл) забирали и сыворотку замораживали при -80°C до анализа.
[0108] Концентрации эксенатида измеряли с помощью ИФА согласно протоколу производителя (Peninsula Laboratories Inc., #S-1311). Замороженные образцы сыворотки размораживали во льду и разводили в 5-20 раз в полученной крысиной сыворотке. Стандартный эксенатид показал EC50=0,22 нМ (по сообщениям 0,19 нМ). Повторности данных усредняли и аппроксимировали в соответствии с подходящими фармакокинетическими моделями. Концентрации [N28Q]эксенатида в сыворотке измеряли с помощью ЖХ-МС/МС.
[0109] Результаты показаны на Фигурах 10 и 11.
[0110] На Фигуре 10 показана фармакокинетика эксенатида у крысы после введения дозы гидрогель-линкера (такого же, как в Примере 4, за исключением использования эксенатида, который находился в нативной форме, и где R1=MeSO2). Кривую зависимости ожидаемой концентрации от времени строили (пунктирная линия) на основе результатов кинетики высвобождения in vitro (t1/2=1400 ч) и известных фармакокинетических параметров эксенатида. Экспериментальные данные (квадраты) показали наилучшее соответствие модели, учитывающей разложение эксенатида на гидрогеле (сплошная линия), где общее t1/2=190 ч.
[0111] На Фигуре 11 показана фармакокинетика [N28Q]эксенатидов Примера 4 у крысы после п/к введения. На панели A показан гидрогель, в котором пептид связан при использовании L, где R1=MeSO2, и дает t1/2=350 ч. На панели B показан гидрогель, в котором пептид связан при использовании L, где R1=CN, и дает t1/2=760 ч. Таким образом, уровни [N28Q]эксенатида в плазме могут сохраняться в течение по меньшей мере одного месяца после введения однократной дозы, тогда как данные на Фигуре 11 показывают намного более короткий t1/2.
Пример 6
Пероральный глюкозотолерантный тест
[0112] Способность [N28Q]эксенатида обеспечивать толерантность при однократном пероральном введении глюкозы по отношению к эксенатиду определяли у мышей. В общей сложности 54 самца мышей C57BL/6J возрастом 8 недель (JanVier France) выдерживали для акклиматизации в течение 2 недель, затем распределяли в 9 групп (n=6). В день 0 мышам не давали корм в течение 4 ч, затем вводили исследуемый продукт путем подкожной инъекции в t=15 минут, затем глюкозу в 0 минут. Глюкозу в крови измеряли через 60, 15, 0, 15, 30, 60 и 120 минут. Образцы крови для измерения инсулина забирали в 0 и 15 минут. Результаты показаны на Фигурах 12 и 13. Эксенатид и [N28Q]эксенатид показали сопоставимую активность в пероральном глюкозотолерантном тесте.
[0113] На Фигуре 12 показаны сравнительные результаты эксенатида и [N28Q]эксенатида в пероральном глюкозотолерантном тесте. Очевидный и подобный дозозависимый эффект наблюдали для эксенатида и [N28Q]эксенатида в пероральном глюкозотолерантном тесте у мышей C57BL/J, получавших в 30 мин эксендин-4 и [N28Q]эксенатид в 5 различных концентрациях. Каждая линия представляет собой значимое отличие от растворителя. Двухфакторный дисперсионный анализ с повторными измерениями в сравнении с растворителем. P<0,05 апостериорный критерий Бонферрони. Доза глюкозы составила 2 г/кг в 10 мл перорально.
[0114] На Фигуре 13 показан анализ AUC для данных перорального глюкозотолерантного теста, показанного на Фигуре 7. Очевидный и подобный дозозависимый эффект наблюдали для эксенатида и [N28Q]эксенатида по данным площади под кривой после перорального глюкозотолерантного теста (ПГТТ). Однофакторный дисперсионный анализ, апостериорный критерий Бонферрони в сравнении с растворителем. ***p<0,001.
Пример 7
Длительное глюкорегуляторное действие конъюгированного с микросферами гидрогеля [N28Q]эксенатида, вводимого раз в 4 недели, в сравнении с постоянной инфузией эксенатида
[0115] В общей сложности 55 самцов крыс линии Цукер с диабетом и ожирением (ZDF-Leprfa/Crl), возрастом 6 недель и весом 180-200 граммов (Charles River, USA) содержали в отдельных боксах и глюкозу в крови и массу тела контролировали раз в две недели в течение 2-4 недель. В зависимости от глюкозы в крови утром после еды, резко отличавшихся животных исключали и 40 крыс с диабетом (средний вес 340 г, глюкоза в крови 9,7-22,5 мМ, среднее значение 16,2 мМ) распределяли в 4 группы n=10.
При t=0:
Группа I получила растворитель в насосе Alzet® (2ML4);
Группа II получила эксенатид, насос Alzet® (30 мкг/кг/день, в AcOH, pH 4,5);
Группа III получала п/к микросферы гидрогеля-[N28Q]эксенатида, имеющие модулятор высвобождения лекарственного средства R1=CN, описанный в Примере 4, 0,37 мг пептида) плюс растворитель при использовании насоса;
Группа IV получала п/к микросферы гидрогеля-[N28Q]эксенатида (3,7 мг пептида) плюс растворитель при использовании насоса.
[0116] В день 29, через два дня после 4 недель ПГТТ, насосы удаляли и повторно п/к вводили микросферы гидрогеля-[N28Q]эксенатида в тех же уровнях. В день 56 насосы удаляли и оставляли животных для восстановления в течение 4 недель. Шесть крыс, у которых впоследствии развился диабет, получали [N28Q]эксенатид в осмотических насосах (30 мкг/кг/день, в AcOH, pH 4,5) в день 28, введение которого прекращали в день 56, после чего животных оставляли для восстановления в течение 4 недель.
[0117] Массу тела ежедневно контролировали со дня 3 в ходе исследования. Потребление пищи и воды проверяли в день 3 и ежедневно, в течение первых 11 дней после введения первой дозы, затем каждые две недели в течение остальной части периода исследования. Забор крови производили для фармакокинетических исследований через 2 дня после введения первой дозы, а затем один раз в неделю. HbA1c измеряли в день -3 и дни 26 и 55 до ПГТТ, и тест опорожнения желудка выполняли в день 26. Для ПГТТ крысам оставляли без корма на половину ночи (60%), после чего глюкозу вводили внутрь (2 г/кг в 10 мл) в t=0. Глюкозу в крови и инсулин измеряли в t=-60, -15, 0, 15, 30, 60 и 120 минут после глюкозной нагрузки. Опорожнение желудка измеряли путем введения ацетаминофена (100 мг/кг) с ПГТТ в день 26, и уровни в крови измеряли в 15, 30, 60 и 120 минут.
[0118] Результаты показаны на Фигурах 14А-14E. На этих фигурах для контрольного растворителя (чернй), эксендин-4 вводили путем постоянной инфузии при использовании подкожного насоса в дозе 30 мкг/кг/день (розовый), PL-cmpd вводили в дозе 220 нмоль пептида/кг (серый), PL-cmpd вводили в дозе 2200 нмоль пептида/кг (синий) и [N28Q]эксенатид в форме свободного пептида вводили путем постоянной инфузии при использовании подкожного насоса в дозе 30 мкг/кг/день (зеленый).
[0119] На Фигуре 14A показана масса тела.
[0120] На Фигуре 14B показана глюкоза в крови в ммоль/л.
[0121] На Фигуре 14C показаны результаты пероральных глюкозотолерантных тестов, проводимых в 4 и 8 недель.
[0122] На Фигуре 14D показаны уровни гликилированного гемоглобина HbA1c в 4 и 8 недель.
[0123] На Фигуре 14E показаны уровни пептидов в плазме в зависимости от времени.
[0124] Однократная доза препарата микросфер гидрогеля, включающего [N28Q]эксенатид, была эффективной при контроле глюкозы в крови в течение по меньшей мере одного месяца.
Пример 8
Фармакокинетика конъюгатов микросфер гидрогеля у крысы и мыши
[0125] A. Шприцы (шприц для инсулина U-100 на 0,5 мл с фиксированной иглой 29G×12,7 мм, BD) наполняли в стерильных условиях суспензией [N28Q]эксенатид-микросфер, полученной в Примере 4, в изотоническом ацетате (10 мМ ацетата Na, 143 мМ NaCl), pH 5,0, 0,05% Tween® 20. Микросферы с использованием модулятора высвобождения лекарственного средства R1=MeSO2 содержали 1,3 мкмоль пептида/г суспензии, и микросферы с использованием модулятора высвобождения лекарственного средства R1=CN содержали 1,4 мкмоль пептида/г суспензии. Содержимое каждого шприца вводили п/к в бок шести канюлированным самцам крыс Спрег-Доули (средний вес 270 г).
[0126] Иглу в сборе со шприцем освобождали от воздуха и взвешивали до и после введения для определения массы суспензии, введенной каждой крысе; с модулятором MeSO2 130 мг суспензии, содержащей 0,7 мг пептида (170 нмоль), вводили каждой крысе, и с модулятором CN вводили 400 мг суспензии, содержащей 2,5 мг пептида (580 нмоль).
[0127] Образцы крови (300 мкл) забирали в 0, 1, 2, 4, 8, 24, 48, 72, 120, 168, 240, 336, 432, 504, 600, 672 часа для обоих линкеров, и дополнительные образцы получали в 840, 1008, 1176, 1344, 1512, 1680, 1848 и 2016 часов для линкера с модулятором CN. Сыворотку получали и замораживали при -80°C до анализа. [N28Q]эксенатид в сыворотке определяли с помощью ЖХ/МС/МС.
[0128] Результаты показаны на Фигурах 15А и 15B. На Фигуре 15A показаны результаты после инъекции микросфер гидрогеля-[N28Q]эксенатида с модулятором высвобождения лекарственного средства R1=MeSO2 (170 нмоль [N28Q]эксенатида/крыса или 2,6 мг/кг); t1/2,β составил 310 часов. На Фигуре 15B показаны результаты после инъекции микросфер гидрогеля-[N28Q]эксенатида с модулятором высвобождения лекарственного средства R1=CN (600 нмоль [N28Q]эксенатида/крыса или 8,9 мг/кг); t1/2,β составил 880 часов. Планки погрешности соответствуют ±SEM.
[0129] B. Небольшие количества микросфер, необходимые для введения мыши, требовали разбавителя для точного дозирования. При использовании двойного реакционного сосуда на основе шприца {Schneider, 2016 # 24} буфер суспензии микросфер-[N28Q]эксенатида (~1 мл для модулятора высвобождения лекарственного средства R1=MeSO2, 4 мл для R1=CN) асептически заменяли изотоническим раствором ацетата, pH 5,0 (10 мМ NaOAc, 143 мМ NaCl), 25% глицерина и 0,05% Tween® 20. Этот разбавитель служил для хранения микросфер в гомогенной суспензии до и во время введения и позволял дозировать удобные объемы. Затем суспензию разбавляли той же смесью, получив 264 нмоль пептида/мг суспензии (R1=MeSO2) или 720 нмоль пептида/мг суспензии (R1=CN). Шприцы (шприц для инсулина U-100 на 0,5 мл с фиксированной иглой 29G×12,7 мм, BD) наполняли суспендированными микросферами в асептических условиях. Иглу в сборе на каждом шприце освобождали от воздуха и взвешивали до и после введения дозы для определения средней массы суспензии, доставленной каждой мыши.
[0130] В случае R1=MeSO2, 120 мг суспензии, содержащей 130 мкг пептида (30 нмоль), вводили п/к в бок каждой из 18 мышей CD-1 (средний вес 30 г). Кровь (100 мкл) забирали из орбитального синуса в 8, 24, 48, 72, 96, 120, 168, 240, 336, 408, 504, 576 и 672 часа согласно ступенчатой схеме, получив 6 повторностей на каждую точку времени, при этом получали сыворотку для каждой точки. В случае R1=CN, 200 мг суспензии, содержащей 605 мкг пептида (144 нмоль), аналогично вводили п/к 24 мышам CD-1. Кровь (100 мкл) забирали из орбитального синуса в те же точки времени, как указано выше, а также в 840, 1008, 1176, 1344, 1512, 1680, 1848 и 2016 часов, согласно ступенчатой схеме, получив 6 повторностей на каждую точку времени. Сыворотку получали и замораживали при -80°C до анализа. [N28Q]эксенатид в сыворотке определяли с помощью ЖХ/МС/МС.
[0131] Образцы сыворотки обрабатывали 3 объемами ACN и центрифугировали. Супернатант сушили, восстанавливали и вводили в систему ВЭЖХ МС/МС. Образец элюировали в градиенте воды/ACN, содержащей 0,1% муравьиной кислоты. Калибровочная кривая для [N28Q]эксенатида была линейной в диапазоне 0,25-100 нг/мл. Анализы ВЭЖХ-МС/МС выполняли на масс-спектрометре Sciex 5500 QTrap®, соединенном с системой ВЭЖХ Shimadzu. Система ВЭЖХ Shimadzu состояла из двух ВЭЖХ насосов LC-30AD и автодозатора SIL-30AC с установленной петлей на 100 мкл. Хроматографические разделения выполняли на ВЭЖХ колонке C18 3 мкм, 2,1×50 мм, в градиентах подвижной фазы. Масс-спектрометр работал в режиме электрораспыления с положительной ионизацией, и используемые настройки разрешения устанавливали на единицу измерения для Q1 и для Q3. Переход мониторинга множественных реакций (ММР) составлял m/z=841,1→396,3 для [N28Q]эксенатида. Интегрирования площади пиков выполняли при использовании программы Analyst (версии 1.5.2) от Sciex.
[0132] Результаты показаны на Фигурах 16А и 16B. На Фигуре 16A показаны результаты после инъекции микросфер гидрогеля-[N28Q]эксенатида с модулятором высвобождения лекарственного средства R1=MeSO2 (30 нмоль [N28Q]эксенатид/мышь или 4,2 мг/кг). На Фигуре 16B показаны результаты после инъекции микросфер гидрогеля-[N28Q]эксенатида с модулятором высвобождения лекарственного средства R1=CN (144 нмоль [N28Q]эксенатид/мышь или 20,2 мг/кг). Ранние точки были недостаточными для вычисления кинетики в фазе абсорбции и не использовались в показанном выравнивании β-фазы. Планки погрешности соответствуют ±SEM.
[0133] Кривые зависимости C от t (текст) анализировали с помощью GraphPad Prism при использовании нелинейной регрессией eq X (текст) с взвешиванием по 1/SD2; поскольку ka>>k1, моделировали одну экспоненциальную фазу периода полувыведения в конечной фазе при использовании точек данных, полученных после первых нескольких дней.
Таблица 2
Фармакокинетические свойства микросфер гидрогеля-[N28Q]эксенатида у мыши и крысы
--->
СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> PROLYNX LLC
SCHNEIDER, Eric L.
HEARN, Brian
HENISE, Jeffrey C.
ASHLEY, Gary W.
SANTI, Daniel V.
<120> КОНЪЮГАТЫ АНАЛОГОВ ЭКСЕНАТИДА С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ
<130> 670572001640
<140> PCT/US2017/022791
<141> 2017-03-16
<150> US 62/416,058
<151> 2016-11-01
<150> US 62/309,330
<151> 2016-03-16
<160> 12
<170> FastSEQ для Windows версия 4.0
<210> 1
<211> 39
<212> Белок
<213> Искусственная последовательность
<220>
<223> Синтетический
<220>
<221> АМИДИРОВАНИЕ
<222> 39
<223> модифицирован введением NH2
<400> 1
His Gly Glu Gly Thr Phe Thr Ser Asp Leu Ser Lys Gln Met Glu Glu
1 5 10 15
Glu Ala Val Arg Leu Phe Ile Glu Trp Leu Lys Asn Gly Gly Pro Ser
20 25 30
Ser Gly Ala Pro Pro Pro Ser
35
<210> 2
<211> 39
<212> Белок
<213> Искусственная последовательность
<220>
<223> Синтетический
<220>
<221> АМИДИРОВАНИЕ
<222> 39
<223> модифицирован введением NH2
<400> 2
His Gly Glu Gly Thr Phe Thr Ser Asp Leu Ser Lys Gln Met Glu Glu
1 5 10 15
Glu Ala Val Arg Leu Phe Ile Glu Trp Leu Lys Gln Gly Gly Pro Ser
20 25 30
Ser Gly Ala Pro Pro Pro Ser
35
<210> 3
<211> 39
<212> Белок
<213> Искусственная последовательность
<220>
<223> Синтетический
<220>
<221> АМИДИРОВАНИЕ
<222> 39
<223> модифицирован введением NH2
<400> 3
His Gly Glu Gly Thr Phe Thr Ser Asp Leu Ser Lys Gln Met Glu Glu
1 5 10 15
Glu Ala Val Arg Leu Phe Ile Glu Trp Leu Lys Ala Gly Gly Pro Ser
20 25 30
Ser Gly Ala Pro Pro Pro Ser
35
<210> 4
<211> 39
<212> Белок
<213> Искусственная последовательность
<220>
<223> Синтетический
<220>
<221> АМИДИРОВАНИЕ
<222> 39
<223> модифицирован введением NH2
<400> 4
His Gly Glu Gly Thr Phe Thr Ser Asp Leu Ser Lys Gln Met Glu Glu
1 5 10 15
Glu Ala Val Arg Leu Phe Ile Glu Trp Leu Lys Lys Gly Gly Pro Ser
20 25 30
Ser Gly Ala Pro Pro Pro Ser
35
<210> 5
<211> 39
<212> Белок
<213> Искусственная последовательность
<220>
<223> Синтетический
<220>
<221> АМИДИРОВАНИЕ
<222> 39
<223> модифицирован введением NH2
<400> 5
His Gly Glu Gly Thr Phe Thr Ser Asp Leu Ser Lys Gln Met Glu Glu
1 5 10 15
Glu Ala Val Arg Leu Phe Ile Glu Trp Leu Lys Asp Gly Gly Pro Ser
20 25 30
Ser Gly Ala Pro Pro Pro Ser
35
<210> 6
<211> 43
<212> Белок
<213> Искусственная последовательность
<220>
<223> Синтетический
<220>
<221> АМИДИРОВАНИЕ
<222> 43
<223> модифицирован введением NH2
<400> 6
His Gly Glu Gly Thr Phe Thr Ser Asp Leu Ser Lys Gln Met Glu Glu
1 5 10 15
Glu Ala Val Arg Leu Phe Ile Glu Trp Leu Lys Gly Gly Pro Ser Ser
20 25 30
Gly Ala Pro Pro Ser Lys Lys Lys Lys Lys Lys
35 40
<210> 7
<211> 44
<212> Белок
<213> Искусственная последовательность
<220>
<223> Синтетический
<220>
<221> АМИДИРОВАНИЕ
<222> 44
<223> модифицирован введением NH2
<400> 7
His Gly Glu Gly Thr Phe Thr Ser Asp Leu Ser Lys Gln Met Glu Glu
1 5 10 15
Glu Ala Val Arg Leu Phe Ile Glu Trp Leu Lys Gln Gly Gly Pro Ser
20 25 30
Ser Gly Ala Pro Pro Ser Lys Lys Lys Lys Lys Lys
35 40
<210> 8
<211> 44
<212> Белок
<213> Искусственная последовательность
<220>
<223> Синтетический
<220>
<221> АМИДИРОВАНИЕ
<222> 44
<223> модифицирован введением NH2
<400> 8
His Gly Glu Gly Thr Phe Thr Ser Asp Leu Ser Lys Gln Met Glu Glu
1 5 10 15
Glu Ala Val Arg Leu Phe Ile Glu Trp Leu Lys Ala Gly Gly Pro Ser
20 25 30
Ser Gly Ala Pro Pro Ser Lys Lys Lys Lys Lys Lys
35 40
<210> 9
<211> 44
<212> Белок
<213> Искусственная последовательность
<220>
<223> Синтетический
<220>
<221> АМИДИРОВАНИЕ
<222> 44
<223> модифицирован введением NH2
<400> 9
His Gly Glu Gly Thr Phe Thr Ser Asp Leu Ser Lys Gln Met Glu Glu
1 5 10 15
Glu Ala Val Arg Leu Phe Ile Glu Trp Leu Lys Lys Gly Gly Pro Ser
20 25 30
Ser Gly Ala Pro Pro Ser Lys Lys Lys Lys Lys Lys
35 40
<210> 10
<211> 40
<212> Белок
<213> Искусственная последовательность
<220>
<223> Синтетический
<220>
<221> АМИДИРОВАНИЕ
<222> 40
<223> модифицирован введением NH2
<400> 10
Glu Leu Val Asp Asn Ala Val Gly Gly Asp Leu Ser Lys Gln Met Glu
1 5 10 15
Glu Glu Ala Val Arg Leu Phe Ile Glu Trp Leu Lys Gln Gly Gly Pro
20 25 30
Ser Ser Gly Ala Pro Pro Pro Ser
35 40
<210> 11
<211> 30
<212> Белок
<213> Искусственная последовательность
<220>
<223> Синтетический
<220>
<221> АМИДИРОВАНИЕ
<222> 30
<223> модифицирован введением NH2
<220>
<221> ВАРИАНТ
<222> 2, 29
<223> Xaa = Aib
<400> 11
His Xaa Glu Gly Thr Phe Thr Ser Asp Val Ser Ser Tyr Leu Glu Gly
1 5 10 15
Gln Ala Ala Lys Glu Phe Ile Ala Trp Leu Val Lys Xaa Arg
20 25 30
<210> 12
<211> 31
<212> Белок
<213> Искусственная последовательность
<220>
<223> Синтетический
<400> 12
His Ala Glu Gly Thr Phe Thr Ser Asp Val Ser Ser Tyr Leu Glu Gly
1 5 10 15
Gln Ala Ala Lys Glu Phe Ile Ala Trp Leu Val Arg Gly Arg Gly
20 25 30
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОЛЕКАРСТВА, СОДЕРЖАЩИЕ КОНЬЮГАТ ГИАЛУРОНОВОЙ КИСЛОТЫ, ЛИНКЕРА И ДВОЙНОГО АГОНИСТА GLP-1/ГЛЮКАГОНА | 2016 |
|
RU2719482C2 |
| ПРИМЕНЕНИЕ ДОЛГОДЕЙСТВУЮЩИХ ПЕПТИДОВ GLP-1 | 2013 |
|
RU2657573C2 |
| ПРОЛЕКАРСТВЕННЫЕ КОМПОЗИЦИИ НА ОСНОВЕ ГИДРОГЕЛЯ ПОПЕРЕЧНО-СШИТОЙ ГИАЛУРОНОВОЙ КИСЛОТЫ И СПОСОБЫ | 2018 |
|
RU2812787C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ДИАБЕТА И СНИЖЕНИЯ МАССЫ ТЕЛА | 2006 |
|
RU2421237C2 |
| КОМБИНАТОРНАЯ ТЕРАПИЯ С АГОНИСТАМИ CNP КОНТРОЛИРУЕМОГО ВЫСВОБОЖДЕНИЯ | 2017 |
|
RU2768747C2 |
| АНАЛОГ ЭКСЕНАТИДА | 2020 |
|
RU2823623C1 |
| ДВОЙНОЙ АГОНИСТ РЕЦЕПТОРОВ ГЛЮКАГОНОПОДОБНОГО ПЕПТИДА-1 И ГЛЮКАГОНА ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2021 |
|
RU2799327C1 |
| ПТГ-СОЕДИНЕНИЯ С НИЗКИМИ СООТНОШЕНИЯМИ ПИКА И МИНИМУМА | 2017 |
|
RU2766959C2 |
| СЛИТЫЕ КОНСТРУКЦИИ ЛЕКАРСТВЕННОГО СРЕДСТВА И КОНЪЮГАТЫ | 2005 |
|
RU2428431C2 |
| КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ДИАБЕТА, СОДЕРЖАЩАЯ КОНЪЮГАТ ИНСУЛИНА ДЛИТЕЛЬНОГО ДЕЙСТВИЯ И КОНЪЮГАТ ИНСУЛИНОТРОПНОГО ПЕПТИДА ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2012 |
|
RU2606840C2 |
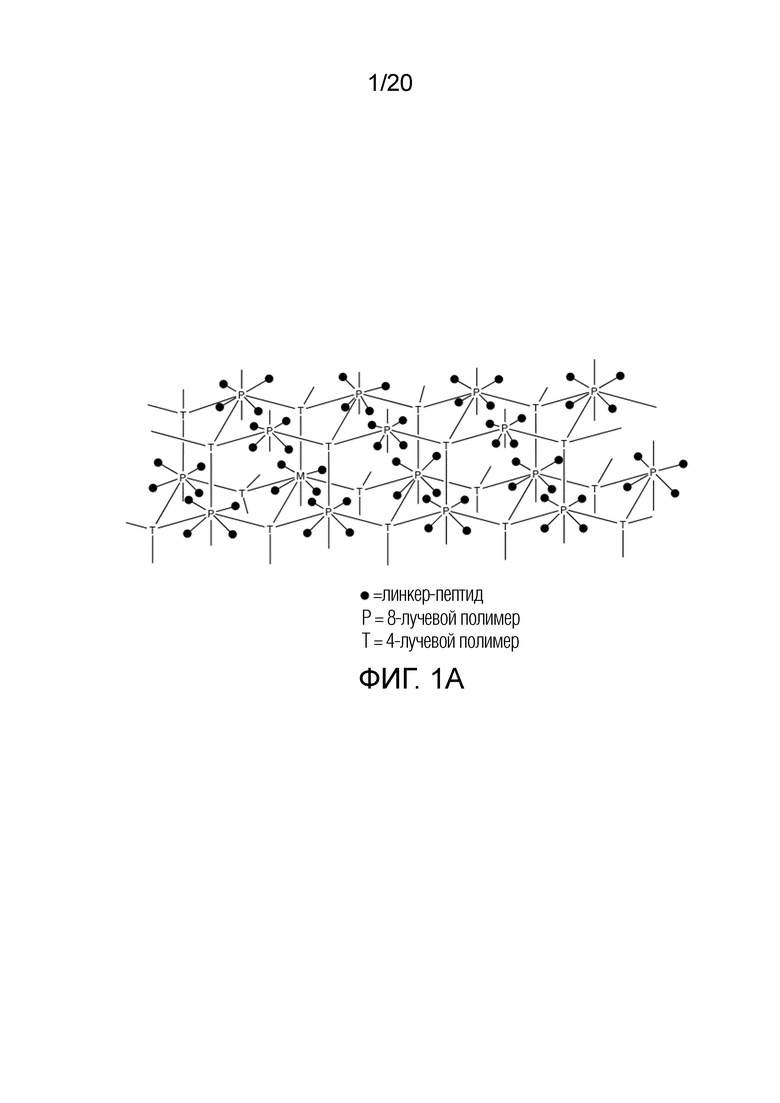
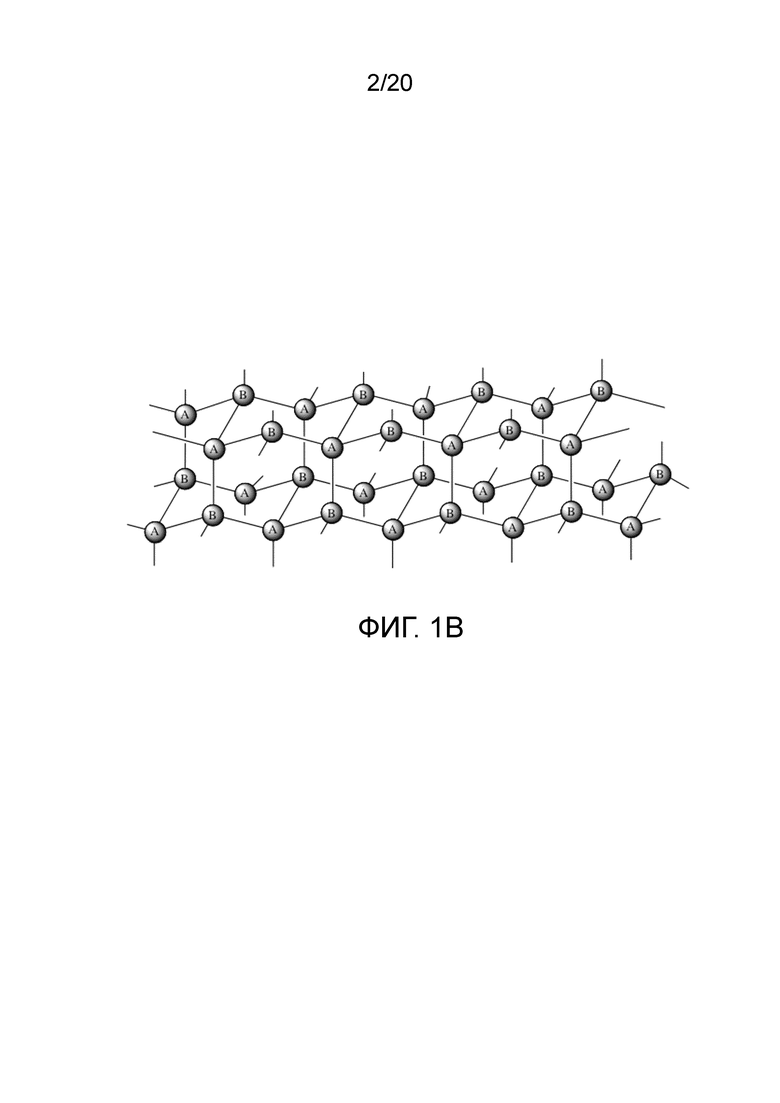

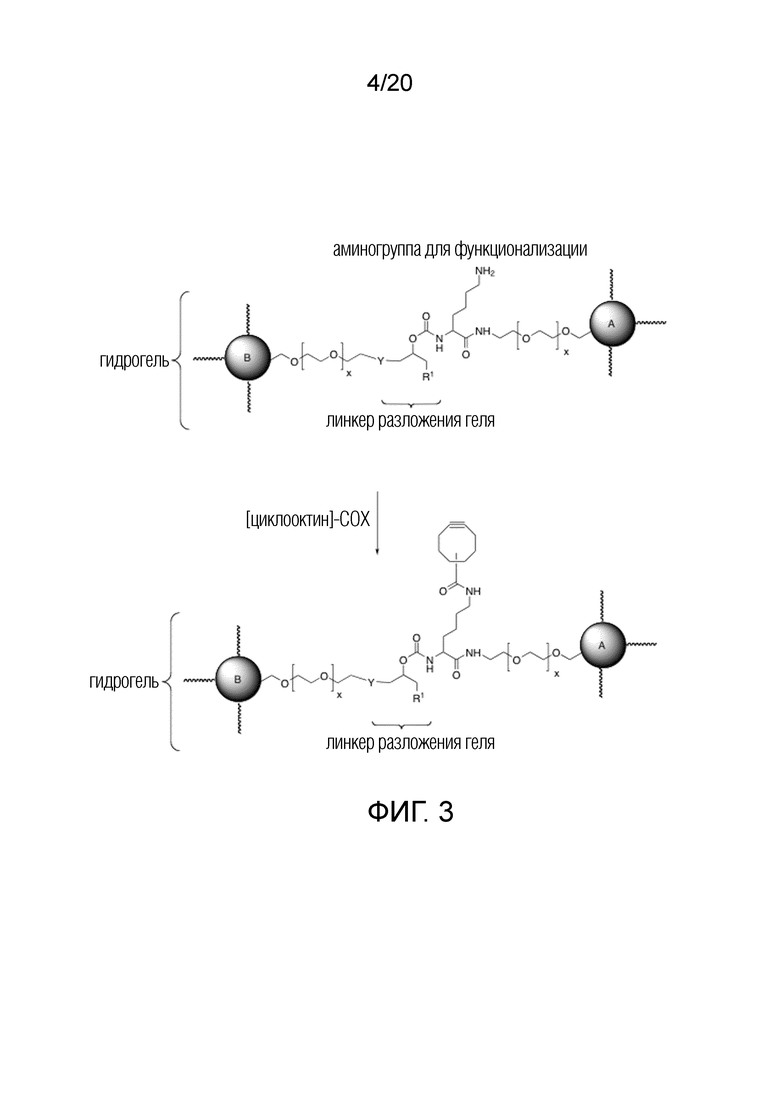



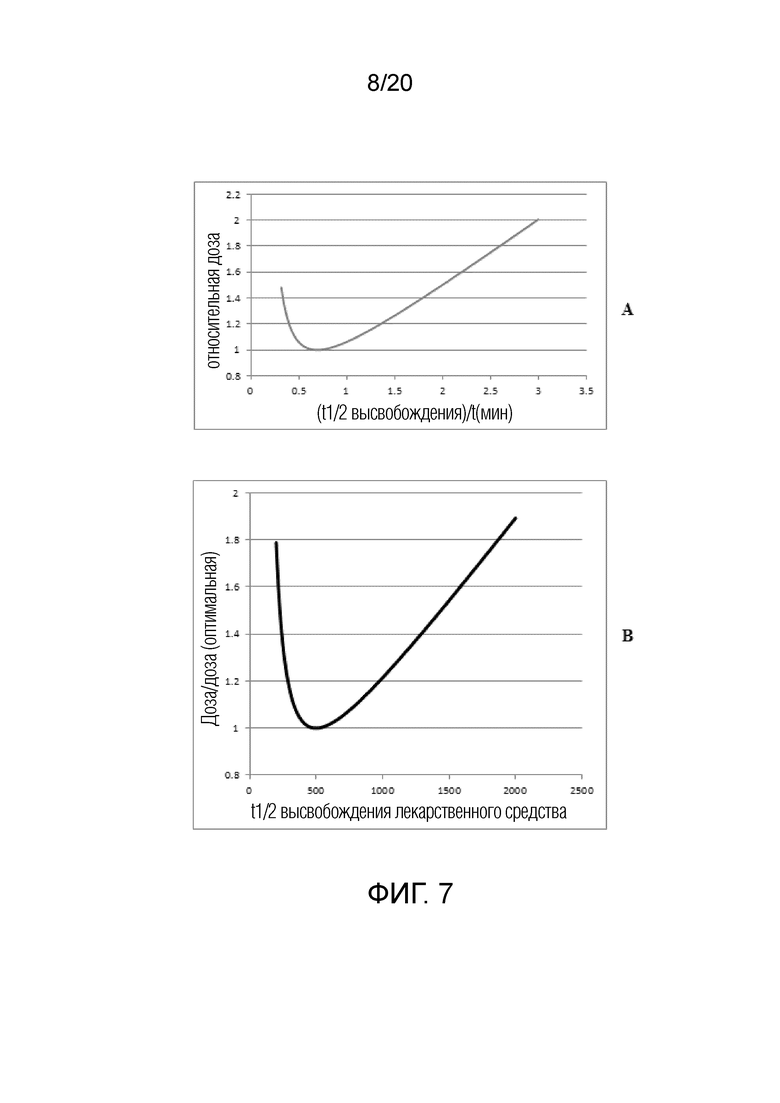
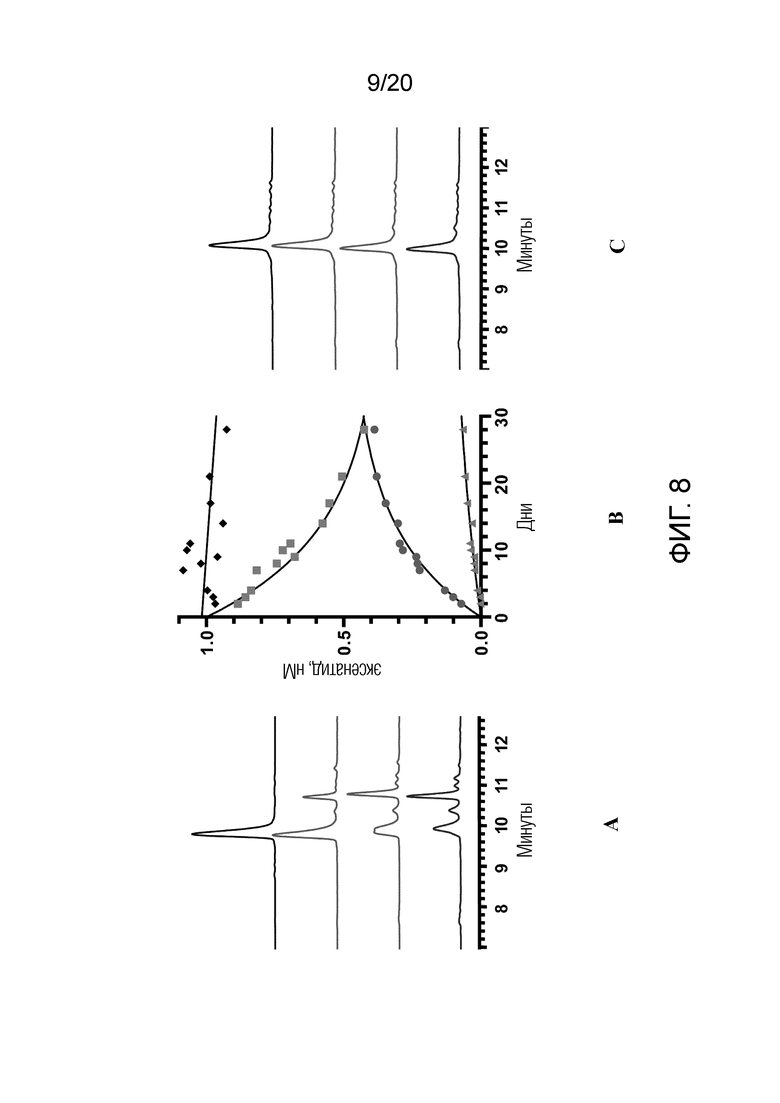

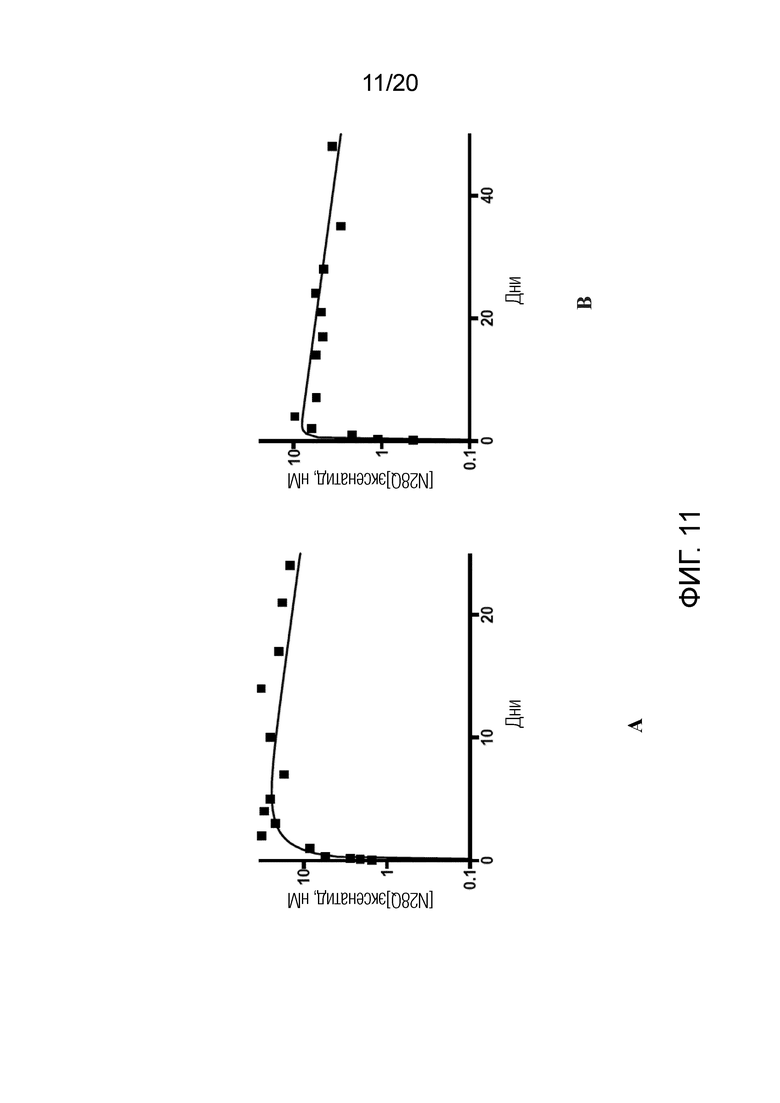


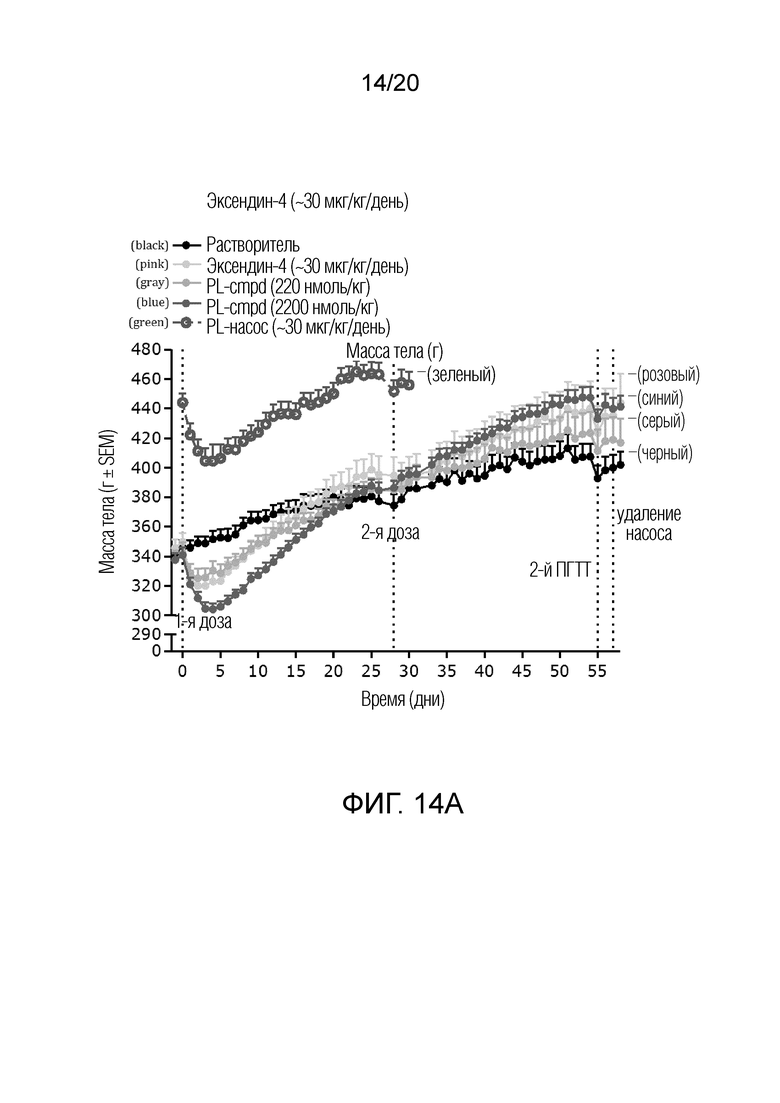



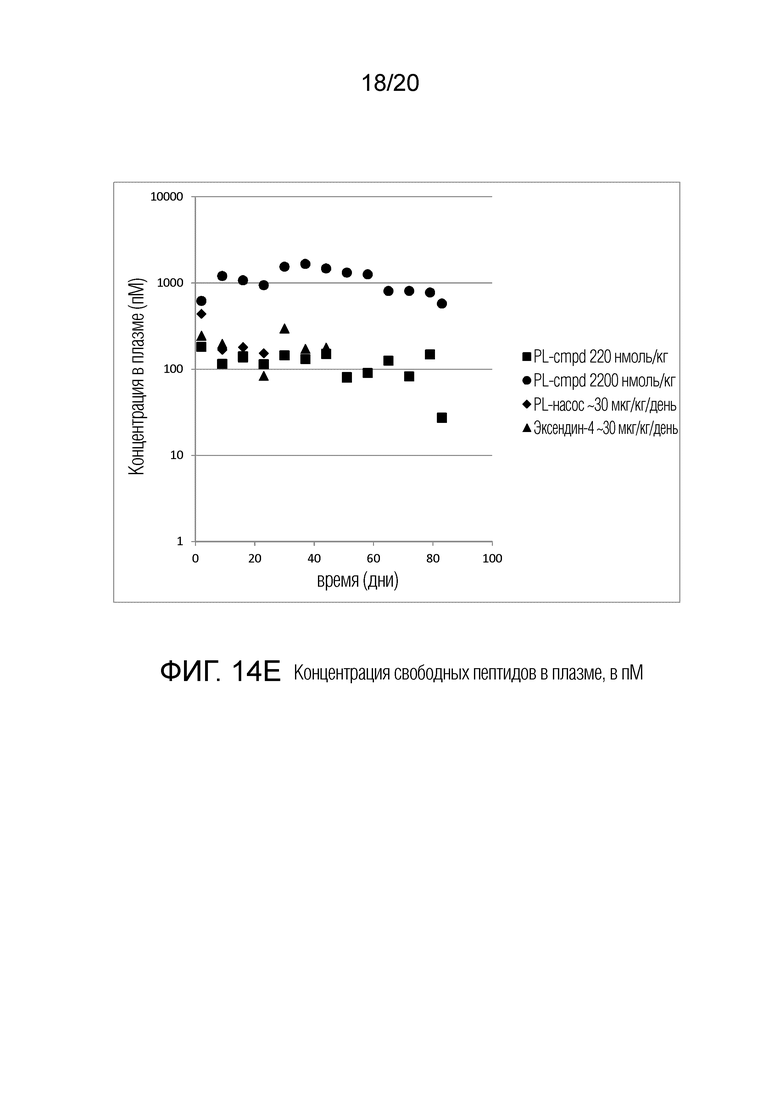

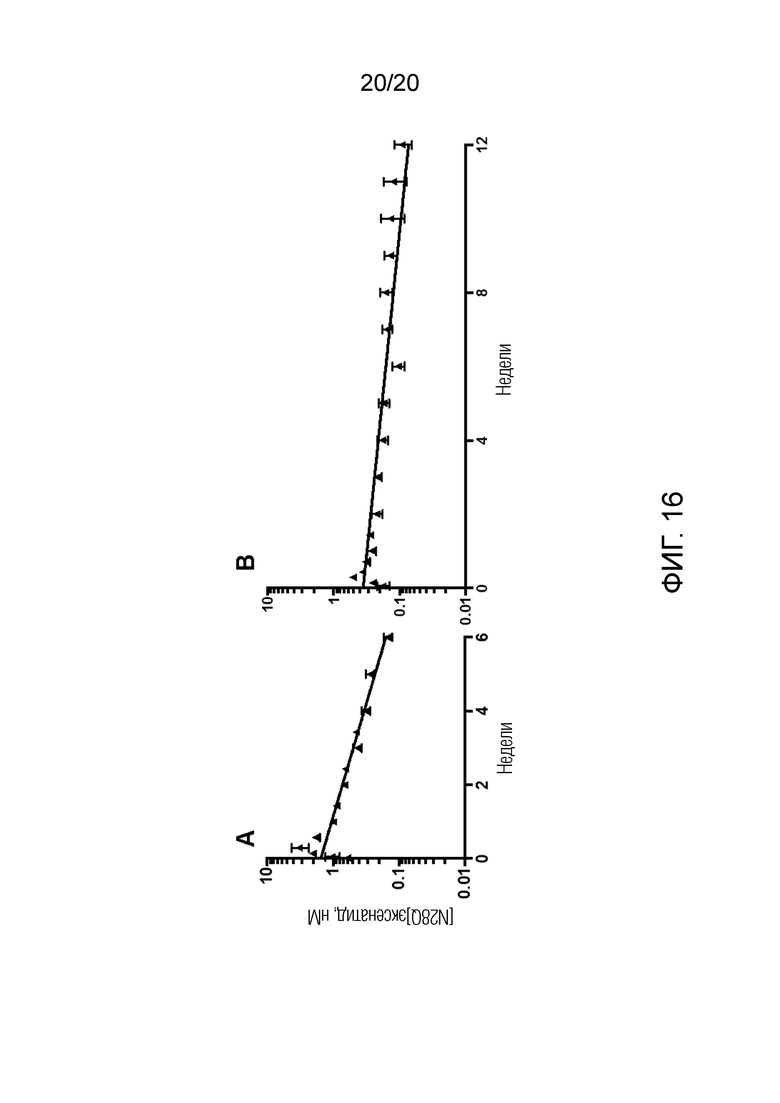
Настоящее изобретение относится к области биотехнологии, конкретно к новому конъюгату стабилизированного агониста рецептора глюкагоноподобного пептида-1 (ГПП-1), и может быть использовано в медицине. Изобретение позволяет получить конъюгат с улучшенными фармакологическими свойствами: замедленное высвобождение уравновешивает стабильность агониста со скоростью высвобождения в среде организма, обеспечивая его более длительное действие в сравнении с известными аналогами. Изобретение может быть использовано в медицинской практике при лечении пациентов, страдающих от диабета или иных подобных метаболических нарушений с применением схем, включающих введение дозы один раз в месяц или реже. 4 н. и 12 з.п. ф-лы, 16 ил., 2 табл., 8 пр.
1. Конъюгат агониста глюкагоноподобного пептида 1 (ГПП-1) с замедленным высвобождением, имеющий формулу:
M-(L-E)x,
где M является биоразлагаемым сшитым гидрогелем, включающим полимеры, соединенные сшивающими агентами;
x является целым числом, обозначающим количество единиц L-E, которые обеспечивают 1-1000 мг E в 1 мл гидрогеля;
L является расщепляемым линкером, имеющим Формулу (3)

где R1 представляет собой CN или R3SO2, где R3 представляет собой алкил, арил, гетероарил или (R9)2N, где каждый R9 независимо представляет собой H или алкил;
R2 представляет собой H, алкил, арилалкил или гетероарилалкил;
где один из R5 представляет собой (CH2)yZ, (CH2CH2O)xCH2CH2Z или (CH2)yNH-CO-(CH2CH2O)x CH2CH2Z, где x представляет собой 1-100, y=1-6, и другой R5 представляет собой H, алкил, алкенилалкил, алкинилалкил, арил, арилалкил, гетероарил или гетероарилалкил, причем каждый необязательно замещен, и Z содержит остаток функциональной группы, связанной с сшитым гидрогелем; и
где Е представляет собой агонист ГПП-1, и
агонист ГПП-1 является стабилизированным эксендином, включающим аминокислотную замену в положении, соответствующем N28, в последовательности нативного эксендина,
где нативный эксендин состоит из SEQ ID NO:1 или SEQ ID NO:7.
2. Конъюгат с замедленным высвобождением по п.1, где агонистом ГПП-1 является N29D, N28A, N28K или N28Q замещенный SEQ ID NO:1 или SEQ ID NO:7.
3. Конъюгат с замедленным высвобождением по п.2, где агонистом ГПП-1 является SEQ ID NO:2 или SEQ ID NO:8.
4. Конъюгат с замедленным высвобождением по п.1, где гидрогель является биоразлагаемым сшитым полиэтиленгликолем.
5. Конъюгат с замедленным высвобождением по п.1, где гидрогель находится в форме микросфер.
6. Конъюгат с замедленным высвобождением по п.1, где полимеры, включающие гидрогель, сшиты сшивающими агентами, которые расщепляются при бета-элиминировании.
7. Конъюгат с замедленным высвобождением по п.6, где указанные сшивающие агенты, содержащиеся в гидрогеле, имеют формулу (1):
 (1)
(1)
в которой m является 0 или 1; и
где X и один из R1, R2 и R5 соединен с полимером, содержащимся в гидрогеле,
при условии, что по меньшей мере один из R1 и R2 является CN; NO2;
необязательно замещенным арилом;
необязательно замещенным гетероарилом;
необязательно замещенным алкенилом;
необязательно замещенным алкинилом;
COR3 или SOR3 или SO2R3, где
R3 является H или необязательно замещенным алкилом;
арилом или арилалкилом, каждый из которых необязательно замещен;
гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен; или
OR9 или NR92, где каждый R9 независимо является H или необязательно замещенным алкилом, или обе группы R9, взятые вместе с азотом, к которому они присоединены, образуют гетероциклическое кольцо;
SR4, где
R4 является необязательно замещенным алкилом;
арилом или арилалкилом, каждый из которых необязательно замещен; или
гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен;
где R1 и R2 могут быть соединены с образованием 3-8-членного кольца; и
где любой оставшийся R1 и R2 является H или алкилом, арилалкилом или гетероарилалкилом, каждый из которых необязательно замещен; и
любой оставшийся R5 независимо является H или алкилом, алкенилалкилом, алкинилалкилом, (OCH2CH2)pO-алкилом, где p=1-1000, арилом, арилалкилом, гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен; или
указанные сшивающие агенты, когда они содержатся в гидрогеле, имеют формулу (2):
 (2)
(2)
где два из R1, R2 и R5 соединены с полимером, содержащимся в гидрогеле;
m является 0-1;
n является 1-1000;
s является 0-2;
t является 2, 4, 8, 16 или 32;
Q является кор-группой, имеющей валентность t;
W является O(C=O)O, O(C=O)NH, O(C=O)S,  или
или 
где R6 является H, необязательно замещенным алкилом, необязательно замещенным арилом, необязательно замещенным гетероарилом, необязательно замещенным арилалкилом или необязательно замещенным гетероарилалкилом; и
при условии, что по меньшей мере один из R1 и R2 является CN; NO2;
необязательно замещенным арилом;
необязательно замещенным гетероарилом;
необязательно замещенным алкенилом;
необязательно замещенным алкинилом;
COR3 или SOR3 или SO2R3, где
R3 является H или необязательно замещенным алкилом;
арилом или арилалкилом, каждый из которых необязательно замещен;
гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен; или
OR9 или NR92, где каждый R9 независимо является H или необязательно замещенным алкилом, или обе группы R9, взятые вместе с азотом, к которому они присоединены, образуют гетероциклическое кольцо;
SR4, где
R4 является необязательно замещенным алкилом;
арилом или арилалкилом, каждый из которых необязательно замещен; или
гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен;
где R1 и R2 могут быть соединены с образованием 3-8-членного кольца; и
где любой оставшийся R1 и R2 является H или алкилом, арилалкилом или гетероарилалкилом, каждый из которых необязательно замещен; и
любой оставшийся R5 независимо является H или алкилом, алкенилалкилом, алкинилалкилом, (OCH2CH2)pO-алкилом, где p=1-1000, арилом, арилалкилом, гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен.
8. Конъюгат с замедленным высвобождением по п.1, где R1 является CN или CH3SO2; R2 является H; один R5 является (CH2)nZ*, где y является 5; и другой R5 является H.
9. Конъюгат с замедленным высвобождением по п.1, где линкер-агонистический пептид L-E ковалентно связан с сшивающими агентами.
10. Соединение формулы (4) для применения при получении конъюгата по пп.1-9:
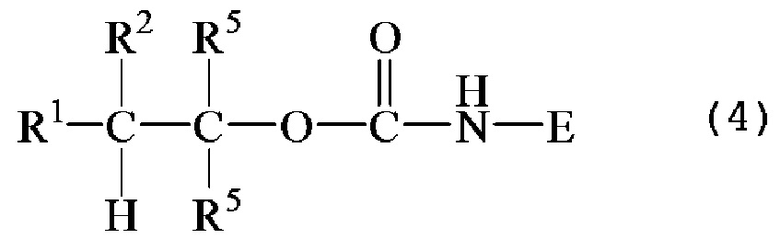
для получения конъюгата по п.1,
где E является агонистом глюкагоноподобного пептида 1 (ГПП-1), где агонист ГПП-1 является стабилизированным эксендином, включающим аминокислотную замену в положении, соответствующем N28, в последовательности нативного эксендина, где нативный эксендин состоит из SEQ ID NO:1 или SEQ ID NO:7,
где R1 представляет собой CN или R3SO2, где R3 представляет собой алкил, арил, гетероарил или (R9)2N, где каждый R9 независимо представляет собой H или алкил;
R2 представляет собой H, алкил, арилалкил или гетероарилалкил;
где один из R5 является (CH2)yZ, (CH2CH2O)xCH2CH2Z или (CH2)yNH-CO-(CH2CH2O)xCH2CH2Z, где x является 1-100, y=1-6, и другой R5 является H, алкилом, алкенилалкилом, алкинилалкилом, арилом, арилалкилом, гетероарилом или гетероарилалкилом, каждый из которых необязательно замещен; и
Z является функциональной группой для связывания с указанным биоразлагаемым сшитым гидрогелем.
11. Соединение по п.10, где указанная функциональная группа включает N3, NH2, NH-CO2tBu, SH, StBu, малеимид, CO2H, CO2tBu, 1,3-диен, циклопентадиен, фуран, алкин, циклооктин, акрилат, аминоокси, кето или акриламид.
12. Соединение по п.10, где R1 является CN или CH3SO2; R2 является H; один R5 является (CH2)yZ, где y является 5; и другой R5 является H.
13. Соединение по п.10, где E является N29D, N28A, N28K или N28Q замещенным SEQ ID NO:1 или SEQ ID NO:7.
14. Соединение по п.13, где E является SEQ ID NO:2.
15. Способ введения агониста глюкагоноподобного пептида 1 (ГПП-1), который включает введение субъекту, имеющему состояние, при котором эффективен агонист ГПП-1, конъюгата по любому из пп.1-9 в количестве одной дозы в один-три месяца.
16. Применение пептида формулы:
HGEGTFTSDL SKQMEEEAVR LFIEWLKQGG PSSGAPPPS-NH2 (SEQ ID NO:2) или его фармацевтически приемлемой соли для получения конъюгата по любому из пп.1-9, где пептид представляет собой агонист глюкагоноподобного пептида 1 (ГПП-1).
| US 20130123487 A1, 16.05.2013 | |||
| WO 2004022004 A2, 18.03.2004 | |||
| US 20110268807 A1, 03.11.2011 | |||
| US 20130116407 A1, 09.05.2013 | |||
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ АГОНИСТ ГПП-1 И МЕТИОНИН | 2010 |
|
RU2573995C2 |
| US 8946405 B2, 03.02.2015 | |||
| WO 2008116294 A1, 02.10.2008. | |||

