ОБЛАСТЬ ТЕХНИКИ
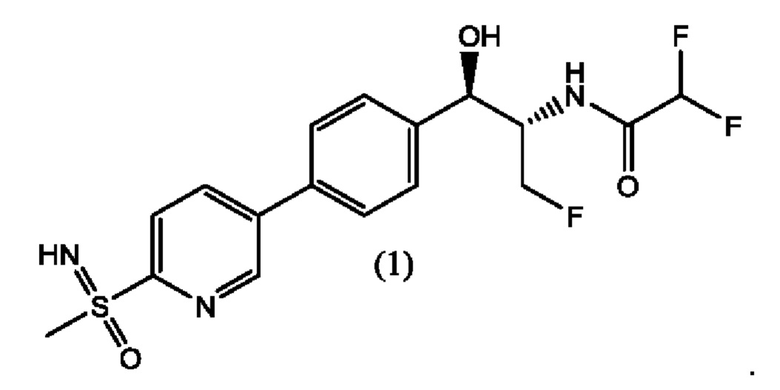
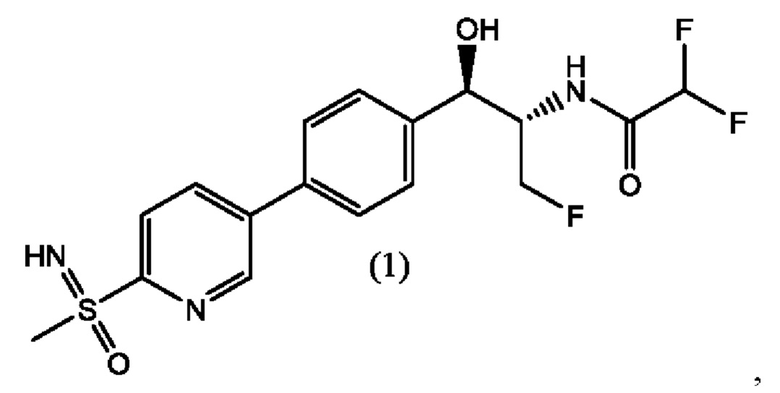
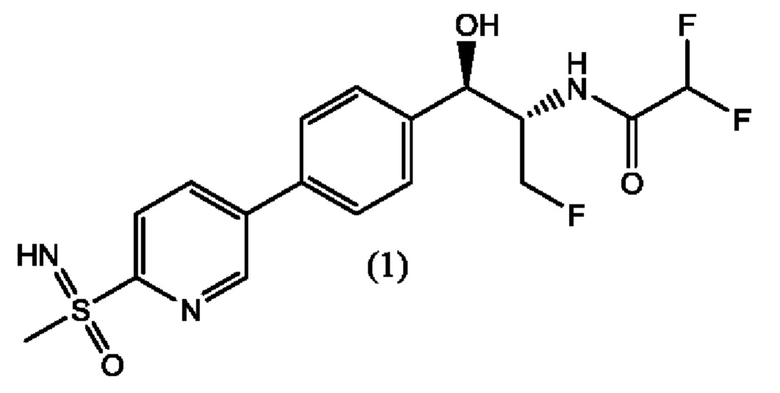
В настоящем изобретении предложен способ получения нового феникола формулы (1) и его диастереомеров
УРОВЕНЬ ТЕХНИКИ
Флорфеникол, также упоминаемый как 3-фтортиамфеникол, представляет собой ветеринарный хлорамфениколовый антибиотик широкого спектра действия, обладающий биологической активностью против различных грамположительных и грамотрицательных бактерий. По сравнению с тиамфениколом, флорфеникол имеет более высокую антибактериальную активность, более широкий антибактериальный спектр, улучшенное усвоение и меньше неблагоприятных реакций, причем, антибактериальная активность флорфеникола до 10 раз выше, чем у тиамфеникола. Флорфеникол можно применять для лечения крупного рогатого скота, свиней, птиц и других животных с бактериальными и грибковыми заболеваниями, а также для получения лекарственных средств для водных животных и рыб.
В последние годы многие роды и виды бактерий начали проявлять определенную резистентность к флорфениколу. Например, обнаружена резистентность Salmonella (Bolton, L.F. et al., Clin. Microbiol., 1999, 37, 1348); E. coli; (Keyes, K. et al., Antimicrob. Agents Chemother., 2000, 44, 421); Klebsiella pneumoniae (Cloeckaert, A. et al., Antimicrob. Agents Chemother., 2001, 45, 2381); и водно-патогенных организмов Photobacterium damselae subsp. piscicida (Kim, Е. et. al., Microbiol. Immunol., 1996, 40, 665) к флорфениколу. Появление резистентности к флорфениколу наряду с риском ее распространения обусловливают потребность в новых антибиотиках, которые могут сохранять или превосходить активность флорфеникола. В WO 2014172443 A1 описан новый фениколовый антибиотик формулы (1), имеющий структуру, представленную ниже:
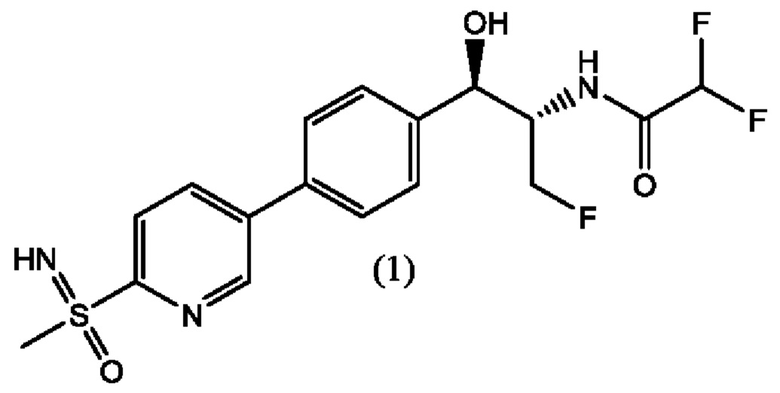
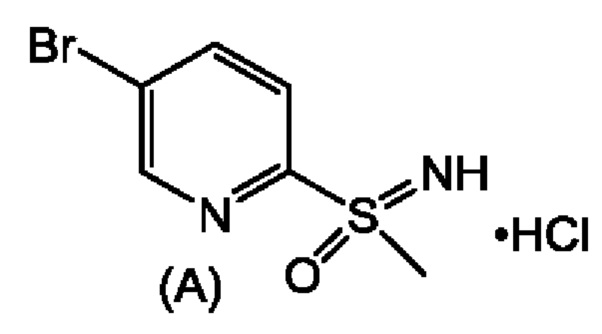
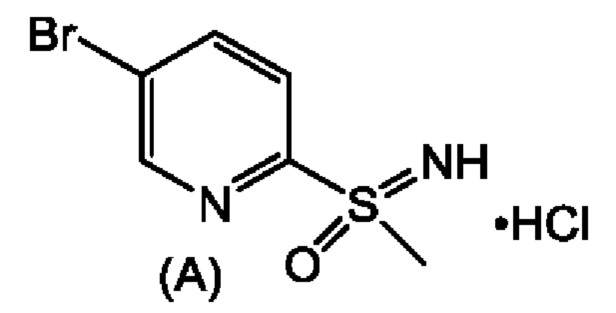
которая представляет собой 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(8-метилсульфонимидоил)пиридин-3-ил)фенил)пропан-2-ил)ацетамид. В настоящей заявке описан альтернативный способ получения соединения формулы (1), его диастереомеров и соединения формулы (А). Ключевым промежуточным соединением при получении феникола формулы (1) является соединение формулы (А), гидрохлорид (5-бромпиридин-2-ил)(имино)(метил)-λ6-сульфолана.
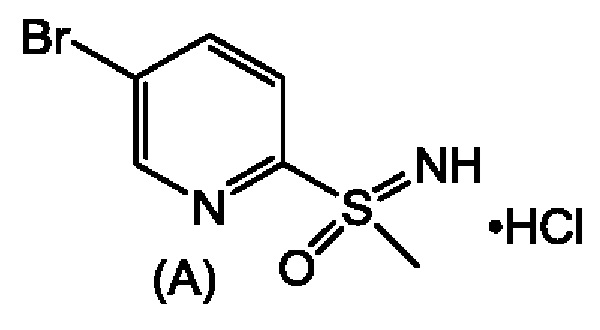
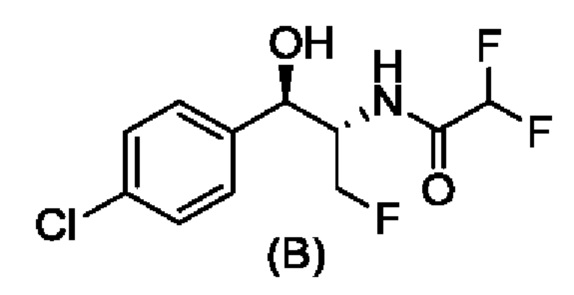
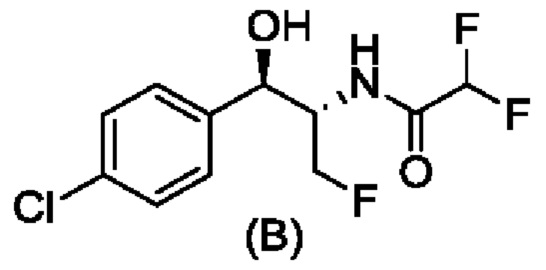
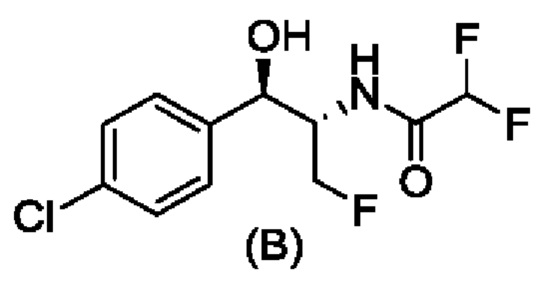
Свободное основание соединения формулы (А) описано ранее в WO 2014/172443. Вторым ключевым промежуточным соединением для получения соединения формулы (1) является соединение формулы (В), N-((1R,2S)-1-(4-хлорфенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамид. Получение хирального соединения формулы (В) описано в CN 106631872 А.
В настоящей заявке предложен новый способ химического синтеза для получения приблизительно 1:1 диастереомерной смеси формулы (1) посредством связывания ключевых промежуточных соединений (рацемической формулы (А) и энантиомерно чистой формулы (В)), который включает меньшее количество химических стадий и обеспечивает более высокий выход, чем описанные ранее способы. В настоящей заявке также предложен новый способ химического синтеза для получения формулы (А), который также включает меньшее количество химических стадий и обеспечивает более высокий выход.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Учитывая количество технологических стадий для получения соединения формулы (1) с низким выходом, необходим более надежный и эффективный способ, в частности, для получения приблизительно 1:1 диастереомерной смеси соединения формулы (1). Диастереомерная смесь 1:1 формулы (1) является предпочтительной комбинацией (формул 1a:1b) для получения, производства, нормативного регулирования и единообразия состава лекарственного средства для обеспечения антибактериальной клинической эффективности. Как описано в настоящем документе, соединение формулы (1) представляет собой диастереомерную смесь 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-((S)-S-метилсульфонимидоил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида (формулы (1а)) и 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-((R)-S-метилсульфонимидоил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида (формулы (1b)), изображенных ниже.
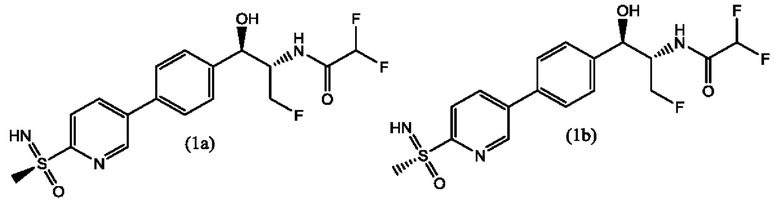
Предпочтительно, диастереомеры формулы (1а) и формулы (1b) получают в диастереомерной смеси в соотношении от приблизительно 48:52 (1a:1b) до 52:48 (1a:1b); и предпочтительно от приблизительно 49:51 (1a:1b) до 51:49 (1a:1b). Следующие технологические стадии представляют собой стадии синтеза для получения диастереомерной смеси формулы (1).
В одном аспекте настоящего изобретения предложен способ получения неочищенного соединения формулы (1) посредством связывания рацемической формулы (А) с энантиомерно чистой формулой (В). В другом аспекте настоящего изобретения предложен способ получения неочищенного соединения формулы (1) посредством связывания рацемической формулы (А) с энантиомерно чистой формулой (В); и с последующей очисткой неочищенного соединения формулы (1) с получением 1:1 диастереомерной смеси формулы (1). В другом аспекте настоящего изобретения предложен способ получения энантиомерно чистой формулы (В). В другом аспекте настоящего изобретения предложен способ получения рацемической формулы (А).
В одном аспекте настоящего изобретения предложен способ получения соединения формулы (1) (неочищенного)
включающий стадии:
а. предварительной активации соединения формулы (В) на палладиевом катализаторе в присутствии спирта, лиганда и буфера для борилирования;
б. борилирования предварительно активированного соединения формулы (В) со стадии (а) посредством добавления борилирующего агента в спирте к реагентам со стадии (а);
в. смешивания соединения формулы (А) с основанием в спирте, сорастворителе или с их смесями; и
г. смешивания реагентов со стадии (в) с реагентами со стадии (б) с получением соединения формулы (1) и его диастереомеров.
В другом аспекте настоящего изобретения предложен способ получения соединения формулы (1) (неочищенного), включающий стадии:
а. предварительной активации соединения формулы (В) на палладиевом катализаторе, выбранном из группы, состоящей из: Pd(OAc)2, PdCl2, Pd-G2-XPhos, Pd-XPhos-кротил-Cl, Pd(dppf)Cl2, Pd-G2-PCy3 и Pd2(dba)3, в присутствии спирта, лиганда, выбранного из группы, состоящей из: XPhos, SPhos, dppp, dppf, dba, PPh3 и РСу3, и буфера для борилирования, который содержит кислоту и основание, причем указанная кислота выбрана из группы, состоящей из: НОАс, лимонной кислоты, муравьиной кислоты, хлоруксусной кислоты и ацетата аммония;
б. борилирования предварительно активированного соединения формулы (В) со стадии (а) посредством добавления борилирующего агента в спирте, причем указанный борилирующий агент выбран из группы, состоящей из бисбороновой кислоты, бисбороновой кислоты и этиленгликоля, бисбороновой кислоты и пропиленгликоля, B2Pin2 и B2(NMe2)4;
в. смешивания соединения формулы (А) с основанием в спирте, сорастворителе или с их смесями; и
г. смешивания реагентов со стадии (в) с реагентами со стадии (б) с получением соединения формулы (1) и его диастереомеров.
В другом аспекте настоящего изобретения основание на стадии (а) и (в) выбрано из группы, состоящей из: KOAc, CsOAc, ТЭА, K2CO3, Na2CO3, Cs2CO3, DIPEA и K3PO4 и их смесей. В другом аспекте настоящего изобретения основание на стадии (а) и (в) выбрано из группы, состоящей из: KOAc, ТЭА, K2CO3, Na2CO3 и их смесей. В другом аспекте настоящего изобретения спирт на стадиях (а), (б) и (в) выбран из группы, состоящей из: метанола, этанола, 1-пропанола, 2-пропанола и 2-бутанола. В одном аспекте спирт на стадиях (а) и (б) является безводным, а спирт на стадии (в) является водным. В другом аспекте спирт на стадиях (а), (б) и (в) представляет собой этанол. В другом аспекте сорастворитель выбран из группы, состоящей из: iPrOAc, EtOAc, ДМФА, ДМЭ, ТГФ, Ме-ТГФ, ацетонитрила и их смесей. В одном аспекте растворы, добавляемые к реакционной смеси на стадиях (а-в), продувают азотом (N2) или аргоном (Ar), и реакции на стадиях (а-г) проводят в инертной атмосфере N2 или аргона (Ar).
В другом аспекте настоящего изобретения предложен способ получения соединения формулы (1) (неочищенного), включающий стадии:
а. предварительной активации соединения формулы (В) на палладиевом катализаторе Pd(OAc)2 в присутствии безводного этанола, лиганда, который представляет собой XPhos, и буфера для борилирования, содержащего кислоту, НОАс, и основание, KOAc;
б. борилирования предварительно активированного соединения формулы (В) со стадии (а) посредством добавления борилирующего агента бисбороновой кислоты и этиленгликоля в безводном этаноле;
в. смешивания соединения формулы (А) с K2CO3, Na2CO3 или ТЭА в водном этаноле; и
г. смешивания реагентов со стадии (в) с реагентами со стадии (б) с получением соединения формулы (1) и его диастереомеров.
В другом аспекте настоящего изобретения предложен способ получения соединения формулы (1) (неочищенного), включающий стадии:
а. предварительной активации соединения формулы (В) на палладиевом катализаторе Pd(OAc)2 в присутствии безводного этанола, лиганда, который представляет собой XPhos, и буфера для борилирования, содержащего кислоту, НОАс, и основание, KOAc;
б. борилирования предварительно активированного соединения формулы (В) со стадии (а) посредством добавления борилирующего агента бисбороновой кислоты и этиленгликоля в безводном этаноле;
в. смешивания соединения формулы (А) с K2CO3, Na2CO3 или ТЭА в водном этаноле; и
г. смешивания реагентов со стадии (в) с реагентами со стадии (б) с получением соединения формулы (1) и его диастереомеров; и при этом растворы, добавляемые в реакционную смесь на стадиях (а-в), продувают N2 или Ar, и реакции на стадиях (а-г) проводят в инертной атмосфере N2 или Ar.
В другом аспекте настоящего изобретения предложен способ получения и очистки соединения формулы (1),
включающий стадии:
а) предварительной активации соединения формулы (В) на палладиевом катализаторе в присутствии спирта, лиганда и буфера для борилирования;
б) борилирования предварительно активированного соединения формулы (В) со стадии (а) посредством добавления борилирующего агента в спирте к реагентам со стадии (а);
в) смешивания соединения формулы (А) с основанием в спирте, сорастворителе или с их смесями;
г) добавления реагентов со стадии (в) к реагентам со стадии (б) с получением неочищенного соединения формулы (1) и его диастереомеров;
д) очистки неочищенного соединения формулы (1), полученного на стадии (г), посредством концентрирования продукта реакции со стадии (г) и экстракции соединения формулы (1) в экстрагирующий растворитель;
е) добавления промывочного водного раствора к экстрагирующему растворителю со стадии (д) при перемешивании; и отделения органического слоя;
ж) добавления поглотителя палладия в органический слой со стадии (е) или рециркуляции органического слоя со стадии (е) через картридж, содержащий поглотитель палладия, перемешивания и отфильтровывания твердых веществ, промывания твердых веществ экстрагирующим(и) растворителем(ями) и концентрирования фильтрата;
з) растворения полученного концентрата со стадии (ж) в органическом(их) растворителе(ях) при нагревании; охлаждения и внесения затравки соединения формулы (1) в полученную смесь;
и) охлаждения смеси со стадии (з) до приблизительно 5-25°С, необязательно добавления антирастворителя; сбора полученных твердых веществ фильтрованием, промывания твердых веществ антирастворителем и затем сушки твердых веществ с получением 1:1 диастереомерной смеси соединения формулы (1); растворы, добавляемые к реакционной смеси на стадиях (а-в), продувают азотом (N2) или аргоном (Ar) и реакции на стадиях (а-г) проводят в инертной атмосфере N2 или аргона (Ar).
В другом аспекте настоящего изобретения предложен способ получения соединения формулы (1), включающий стадии:
а) предварительной активации соединения формулы (В) на палладиевом катализаторе, выбранном из группы, состоящей из: Pd(OAc)2, PdCl2, Pd-G2-XPhos, Pd-XPhos-кротил-Cl, Pd(dppf)Cl2, Pd-G2-PCy3 и Pd2(dba)3, в присутствии спирта, лиганда, выбранного из группы, состоящей из: XPhos, SPhos, dppp, dppf, dba, PPh3 и РСу3, и буфера для борилирования, который содержит кислоту и основание, причем указанная кислота выбрана из группы, состоящей из: НОАс, лимонной кислоты, муравьиной кислоты, хлоруксусной кислоты и ацетата аммония;
б) борилирования предварительно активированного соединения формулы (В) со стадии (а) посредством добавления борилирующего агента в спирте, причем борилирующий агент выбран из группы, состоящей из бисбороновой кислоты; бисбороновой кислоты и этиленгликоля; бисбороновой кислоты и пропиленгликоля; B2Pin2; и B2(NMe2)4;
в) смешивания соединения формулы (А) с основанием в спирте, сорастворителе или с их смесями;
г) смешивания реагентов со стадии (в) с реагентами со стадии (б) с получением соединения формулы (1) и его диастереомеров;
д) очистки неочищенного соединения формулы (1) со стадии (г) посредством концентрирования продукта реакции со стадии (г) и экстракции соединения формулы (1) в экстрагирующий растворитель, выбранный из группы, состоящей из: ТГФ, EtOAc, МеОАс, метиленхлорида и Ме-ТГФ;
е) добавления промывочного водного раствора, выбранного из группы, состоящей из воды или солевого раствора, каждый из которых содержит соединение, образующее хелат с палладием, выбранное из группы, состоящей из: ЭДА, TMT-Na3, NH4OH, ТМТ, NaHSO3, тиомочевины, ДЭА, ЭДТК, Ac-L-цистеина, лимонной кислоты и их смесей, при перемешивании; и отделения органического слоя;
ж) добавления поглотителя палладия в органический слой со стадии (е), выбранного из группы, состоящей из: угля с ЭДА, силикагеля с ЭДА, Si-тиола, MP-TU, МР-ТМТ, Si-TMT, Si-DMT и Si-цистеина, и их смесей; или рециркуляции органического слоя со стадии (е) через картридж из угля с ЭДА, силикагеля с ЭДА, Si-тиола, MP-TU, МР-ТМТ, Si-TMT, Si-DMT и Si-цистеина при перемешивании, отфильтровывания твердых веществ, промывания твердых веществ экстрагирующим(и) растворителем(ями); и концентрирования фильтрата;
з) растворения при нагревании полученного концентрата со стадии (ж) в органическом(их) растворителе(ях), выбранном(ых) из группы, состоящей из: МЭК, iPrOAc, EtOAc, ацетона, 1-бутанола, 1-пропанола, 2-пропанола и их смесей, при нагревании выше 55°С; охлаждения до приблизительно 50-55°С и внесения затравки соединения формулы (1) в полученную смесь;
и) охлаждения смеси со стадии (з) до приблизительно 5-25°С, необязательно добавления антирастворителя, выбранного из группы, состоящей из: воды, МТБЭ, гексана, гептана и их смесей; сбора полученных твердых веществ фильтрованием, промывания твердых веществ антирастворителем и затем сушки твердых веществ с получением 1:1 диастереомерной смеси соединения формулы (1); причем растворы, добавляемые к реакционной смеси на стадиях (а-в), продувают азотом (N2) или аргоном (Ar) и реакции на стадиях (а-г) проводят в инертной атмосфере N2 или аргона (Ar).
Как описано выше, основание на стадиях (а) и (в) выбрано из группы, состоящей из: KOAc, CsOAc, ТЭА, K2CO3, Na2CO3, Cs2CO3, DIPEA и K3PO4 и их смесей. В другом аспекте настоящего изобретения основание на стадиях (а) и (в) выбрано из группы, состоящей из: KOAc, ТЭА, K2CO3, Na2CO3 и их смесей. В другом аспекте настоящего изобретения основание на стадиях (а) и (в) выбрано из группы, состоящей из: KOAc, ТЭА и K2CO3, и их смесей. В другом аспекте настоящего изобретения спирт на стадиях (а), (б) и (в) выбран из группы, состоящей из: метанола, этанола, 1-пропанола, 2-пропанола и 2-бутанола. В одном аспекте спирт на стадиях (а) и (б) является безводным, а спирт на стадии (в) является водным. В другом аспекте спирт на стадиях (а), (б) и (в) представляет собой этанол. В другом аспекте сорастворитель выбран из группы, состоящей из: iPrOAc, EtOAc, ДМФА, ДМЭ, ТГФ, Ме-ТГФ, ацетонитрила и их смесей. В другом аспекте сорастворитель выбран из группы, состоящей из: ТГФ, Ме-ТГФ, ацетонитрила и их смесей. В другом аспекте сорастворитель выбран из группы, состоящей из: ТГФ, Ме-ТГФ и их смесей. В другом аспекте сорастворитель представляет собой ТГФ. В одном аспекте растворы, добавляемые к реакционной смеси на стадиях (а-в), продувают азотом (N2) или аргоном (Ar), и реакции на стадиях (а-г) проводят в инертной атмосфере N2 или аргона (Ar). В другом аспекте промывочный водный раствор на стадии (е) представляет собой раствор, выбранный из группы, состоящей из воды или солевого раствора, каждый из которых содержит соединение, образующее хелат с палладием, выбранное из группы, состоящей из: ЭДА, TMT-Na3, NH4OH, ТМТ, NaHSO3, тиомочевины, ДЭА, ЭДТК, Ac-L-цистеина, лимонной кислоты и их смесей. В другом аспекте промывочный водный раствор на стадии (е) представляет собой солевой раствор, содержащий соединение, образующее хелат с палладием, выбранное из: ЭДА, NH4OH и их смесей. В другом аспекте органический слой на стадии (е) промывают 1х, 2х, 3х или 4х промывочным водным раствором. В другом аспекте органический слой на стадии (е) промывают 2х водным раствором. В другом аспекте органический слой на стадии (е) промывают 3х водным раствором. В другом аспекте органический слой на стадии (е) промывают 4х водным раствором.
В другом аспекте настоящего изобретения предложен способ получения соединения формулы (1), включающий стадии:
а) предварительной активации соединения формулы (В) на палладиевом катализаторе Pd(OAc)2 в присутствии безводного этанола, лиганда, который представляет собой XPhos, и буфера для борилирования, содержащего кислоту, которая представляет собой НОАс, и основание, которое представляет собой KOAc;
б) борилирования предварительно активированного соединения формулы (В) со стадии (а) посредством добавления борилирующего агента, который представляет собой бисбороновую кислоту и этиленгликоль в безводном этаноле;
в) смешивания соединения формулы (А) с K2CO3, Na2CO3, ТЭА или их смесями в водном этаноле или в сорастворителе, или в их смесях;
г) смешивания реагентов со стадии (в) с реагентами со стадии (б) с получением соединения формулы (1) и его диастереомеров;
д) очистки неочищенного соединения формулы (1) со стадии (г) посредством концентрирования продукта реакции со стадии (г) и экстракции соединения формулы (1) в экстрагирующий растворитель, ТГФ;
е) добавления промывочного водного раствора, состоящего из воды или солевого раствора, каждый из которых содержит соединение, образующее хелат с палладием, выбранное из группы, состоящей из: ЭДА, TMT-Na3, NH4OH, ТМТ, NaHSO2, тиомочевины, ДЭА, ЭДТК, Ac-L-цистеина, лимонной кислоты и их смесей; и отделения органического слоя;
ж) добавления поглотителя палладия, выбранного из группы, состоящей из: угля с ЭДА, силикагеля с ЭДА, Si-тиола, MP-TU, МР-ТМТ, Si-TMT, Si-DMT и Si-цистеина, и их смесей, к органическому слою со стадии (е) или рециркуляции органического слоя со стадии (е) через картридж из угля с ЭДА, силикагеля с ЭДА, Si-тиола, MP-TU, МР-ТМТ, Si-TMT, Si-DMT и Si-цистеина, перемешивания и отфильтровывания твердых веществ, промывания твердых веществ экстрагирующим(и) растворителем(ями), и концентрирования фильтрата;
з) растворения полученного концентрата со стадии (ж) в органическом(их) растворителе(ях), выбранном(ых) из группы, состоящей из: МЭК, iPrOAc, EtOAc, ацетона, 1-бутанола, 1-пропанола, 2-пропанола и их смесей, при нагреве выше 55°С, охлаждении до приблизительно 50-55°С и внесении затравки соединения формулы (1) в полученную смесь;
и) охлаждения смеси со стадии (з) до приблизительно 5-25°С, необязательно добавления антирастворителя, выбранного из группы, состоящей из: воды, МТБЭ, гексана, гептана и их смесей, сбора полученных твердых веществ фильтрованием, промывания твердых веществ антирастворителем и затем сушки твердых веществ с получением 1:1 диастереомерной смеси соединения формулы (1); причем растворы, добавляемые к реакционной смеси на стадиях (а-в), продувают азотом (N2) или аргоном (Ar) и реакции на стадиях (а-г) проводят в инертной атмосфере N2 или аргона (Ar).
В другом аспекте настоящего изобретения предложен способ получения соединения формулы (1), включающий стадии:
а) предварительной активации соединения формулы (В) на палладиевом катализаторе Pd(OAc)2 в присутствии безводного этанола, лиганда, который представляет собой XPhos, и буфера для борилирования, содержащего кислоту, которая представляет собой НОАс, и основание, которое представляет собой KOAc;
б) борилирования предварительно активированного соединения формулы (В) со стадии (а) посредством добавления борилирующего агента, который представляет собой бисбороновую кислоту и этиленгликоль в безводном этаноле;
в) смешивания соединения формулы (А) с K2CO3, Na2CO3, ТЭА или их смесями в водном этаноле или в сорастворителе, или в их смесях;
г) смешивания реагентов со стадии (в) с реагентами со стадии (б) с получением соединения формулы (1) и его диастереомеров; и при этом растворы на стадиях (а-в) продувают N2, и реакции на стадиях (а-г) проводят в инертной атмосфере N2 или Ar;
д) очистки неочищенного соединения формулы (1) со стадии (г) посредством концентрирования продукта реакции со стадии (г) и экстракции соединения формулы (1) в экстрагирующий растворитель, ТГФ;
е) добавления промывочного водного раствора, который представляет собой солевой раствор NaCl, содержащий соединение, образующее хелат с палладием, выбранное из: ЭДА, NH4OH и их смесей, к ТГФ со стадии (д) при перемешивании; и отделения органического слоя;
ж) добавления поглотителя палладия, угля с ЭДА, в органический слой со стадии (е) или рециркуляции органического слоя со стадии (е) через угольный картридж с ЭДА, перемешивания и отфильтровывания твердых веществ, промывания твердых веществ экстрагирующим растворителем, ТГФ, и концентрирования фильтрата;
з) растворения полученного концентрата со стадии (ж) в 1-пропаноле при нагреве выше 55°С, охлаждении до приблизительно 50-55°С и внесении затравки соединения формулы (1) в полученную смесь;
и) охлаждения смеси со стадии (з) до приблизительно 5-25°С, необязательно добавления гептана, сбора полученных твердых веществ фильтрованием, промывания твердых веществ гептаном, сушки твердых веществ с получением 1:1 диастереомерной смеси соединения формулы (1); причем растворы, добавляемые к реакционной смеси на стадиях (а-в), продувают азотом (N2) или аргоном (Ar) и реакции на стадиях (а-г) проводят в инертной атмосфере N2 или аргона (Аг).
В другом аспекте настоящего изобретения предложен способ получения соединения формулы (1), включающий стадии:
а) объединения Pd(OAc)2, XPhos, соединения формулы (В) и KOAc в атмосфере N2; получения раствора безводного EtOH и НОАс, продутого N2, и добавления его к палладиевой смеси; нагрева до приблизительно 72°С в течение приблизительно 30 минут и затем охлаждения до приблизительно 52°С;
б) борилирования соединения формулы (В) посредством добавления раствора бисбороновой кислоты и этиленгликоля в безводном EtOH, продутого N2, к смеси со стадии (а) на протяжении приблизительно 30 минут при приблизительно 52°С;
в) смешивания продутого N2 раствора соединения формулы (А) и K2CO3 в водном ТГФ при приблизительно 42°С;
г) смешивания реагентов со стадии (б) и стадии (в); затем нагрева до приблизительно 72°С при перемешивании до завершения связывания формулы (А) и (В), приблизительно 4 часа;
д) охлаждения смеси со стадии (г) до комнатной температуры, нейтрализации смеси до рН 7,0 концентрированной HCl; концентрирования продукта реакции, добавления воды и экстракции концентрата в экстрагирующий растворитель, ТГФ, и отделения экстракционного слоя;
е) промывания экстракционного слоя со стадии (д) промывочным водным раствором NaCl с соединением, образующим хелат с палладием, ЭДА, и отделения органического слоя;
ж) добавления поглотителя палладия, угля с ЭДА, в органический слой со стадии (е) или рециркуляции органического слоя со стадии (е) через картридж из угля с ЭДА при перемешивании, отфильтровывания твердых веществ, промывания твердых веществ экстрагирующим растворителем, ТГФ, и концентрирования фильтрата;
з) растворения полученного концентрата со стадии (ж) в 1-пропаноле при нагреве приблизительно выше 55°С, охлаждении до приблизительно 55°С и внесении затравки соединения формулы (1);
и) охлаждения смеси со стадии (з) до приблизительно 5-25°С, необязательно добавления антирастворителя гептана, сбора твердых веществ фильтрацией и промывания гептаном; высушивания твердых веществ в вакууме при приблизительно 57°C с получением 1:1 диастереомерной смеси соединения формулы (1).
В другом аспекте настоящего изобретения предложена ветеринарная композиция, содержащая соединение формулы (1), полученное способом, описанным в настоящем документе, или его приемлемую для ветеринарии соль. В другом аспекте настоящего изобретения ветеринарная композиция дополнительно содержит приемлемый для ветеринарии носитель.
В другом аспекте настоящего изобретения предложен способ сдерживания или лечения бактериальных инфекций у животного посредством введения нуждающемуся животному терапевтически эффективного количества соединения формулы (1), полученного способом, описанным в настоящем документе.
В другом аспекте настоящего изобретения предложен способ получения соединения формулы (А), включающий окисление сульфилиминного соединения, промежуточного соединения (с)
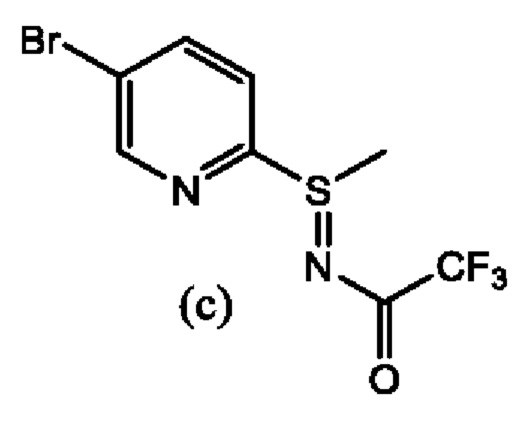
пероксидом водорода и карбонатом в растворе, содержащем ацетонитрил, спирт и необязательно воду. В другом аспекте спирт представляет собой метанол. Отношение ацетонитрила к метанолу составляет от приблизительно 75:25 до 50:50. В другом аспекте раствор содержит воду. В другом аспекте количество ацетонитрила, метанола и воды составляет приблизительно 126 мл, 74 мл и 4,3 мл, соответственно. В другом аспекте карбонат представляет собой карбонат калия.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
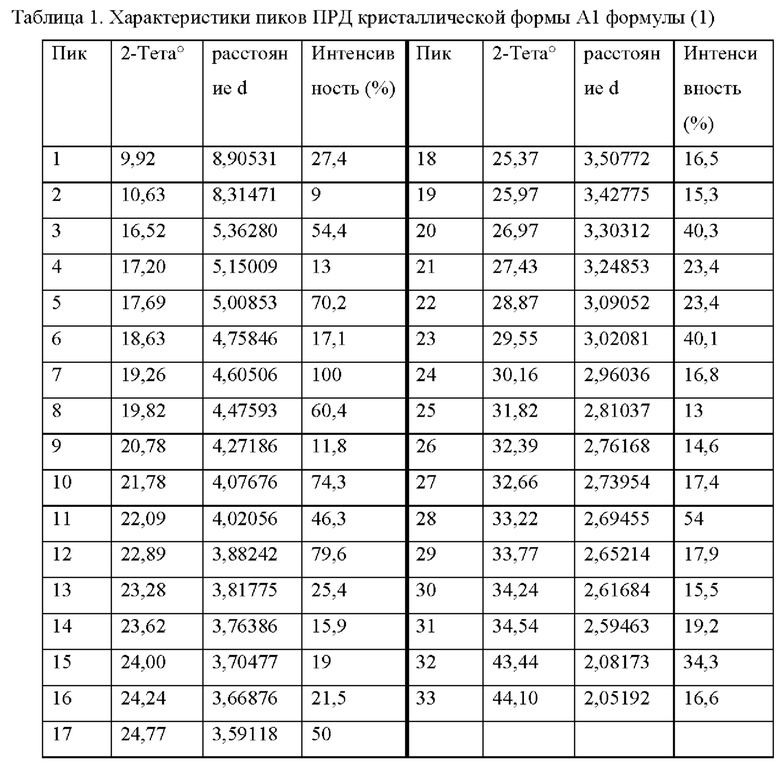
Фигура 1. Представлена иллюстративная диаграмма ПРД кристаллической формы А1; диастереомерной смеси формулы 1а и 1b [50,83:49,17; 1a/1b ~ 1,03].
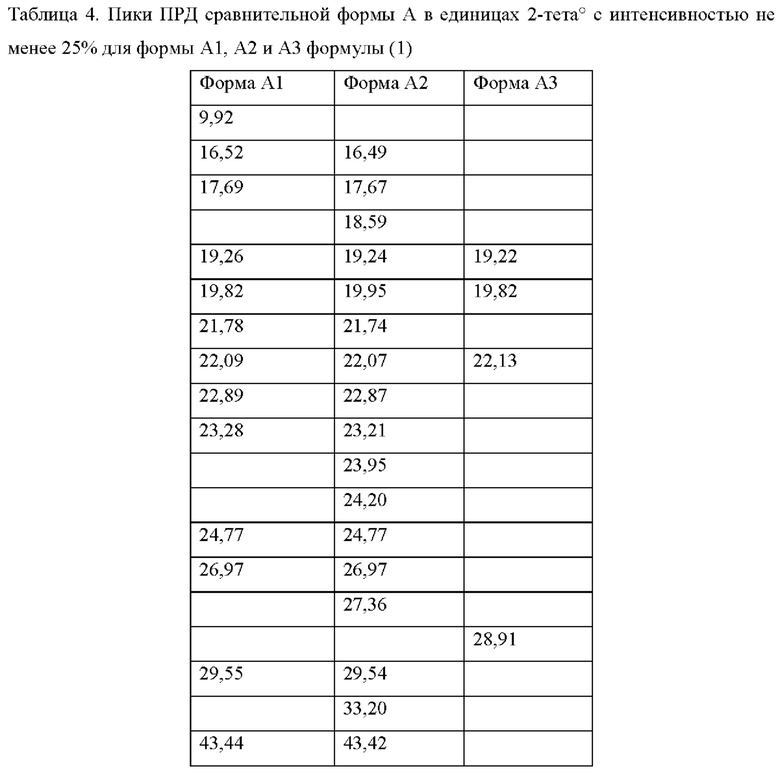
Фигура 2. Представлена иллюстративная диаграмма ПРД кристаллической формы А2; диастереомерной смеси формулы 1а и 1b [47,46:52,54; 1a/1b ~ 0,903].
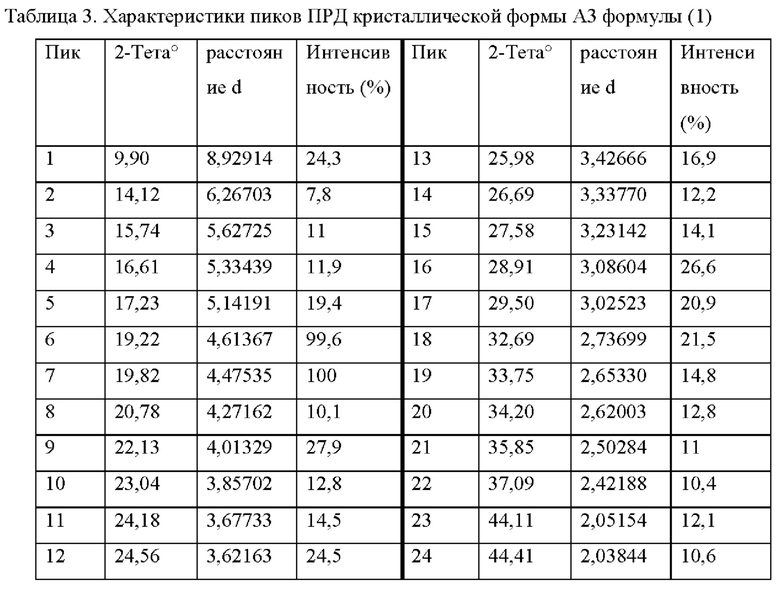
Фигура 3. Представлена иллюстративная диаграмма ПРД кристаллической формы A3; диастереомерной смеси формулы 1а и 1b [56,43:43,57; 1a/1b ~ 1,295].
Для целей настоящего изобретения, описанного и заявленного в данном документе, использованы следующие определения терминов и выражений:
«Приблизительно», при использовании в связи с измеримой числовой переменной, относится к указанному значению переменной и ко всем значениям указанной переменной, которые находятся в пределах экспериментальной погрешности указанного значения (например, в пределах 95% доверительного интервала для среднего значения) или в пределах ± 10 процентов от указанного значения, в зависимости от того, что больше.
«Животное» в данном контексте, если не указано иное, относится к отдельному животному, и указанное конкретное животное является млекопитающим. В частности, млекопитающее относится к позвоночному животному, которое является человеком или другим млекопитающим, и является членом таксономического класса Mammalia. Неограничивающие примеры млекопитающих, отличных от человека, включают домашних питомцев и сельскохозяйственных животных. Предпочтительными животными являются животные, не являющиеся людьми. Неограничивающие примеры домашних питомцев включают: собак, кошек и лошадей. Предпочтительные домашние животные представляют собой собак и лошадей. Более предпочтительными являются собаки. Неограничивающие примеры сельскохозяйственных животных включают: овец, коз, крупный рогатый скот и свиней. Предпочтительные сельскохозяйственные животные представляют собой крупный рогатый скот и свиней. Предпочтительные сельскохозяйственные животные представляют собой крупный рогатый скот.Предпочтительные сельскохозяйственные животные представляют собой свиней.
«Внесение затравки» или «введение затравки» в данном контексте, если не указано иное, относится к добавлению соединения формулы (1) в реакционную смесь для получения диастереоизомеров. Затравка формулы (1) может быть получена способами, описанными ранее в патенте США US 9422236; или может быть получена способами, описанными в настоящем документе, таким образом, что исходные «затравочные» диастереомеры получают по окончании реакции хроматографическим разделением и/или выделением с последующей кристаллизацией.
«Терапевтически эффективное количество» в данном контексте, если не указано иное, относится к такому количеству диастереомерного соединения формулы (1) согласно настоящему изобретению, которое (i) обеспечивает лечение или предупреждение конкретной бактериальной инфекции. Предусмотрено, что терапевтически эффективной дозой является доза в диапазоне от приблизительно 1 до 50 мг/кг. Предпочтительная доза составляет от приблизительно 10 до 40 мг/кг. Более предпочтительная доза составляет от приблизительно 15 до 35 мг/кг. Наиболее предпочтительная доза составляет приблизительно 20 мг/кг.
«Лечение», «лечить» и т.п. в данном контексте, если не указано иное, относится к реверсированию, облегчению или подавлению бактериальной инфекции. В данном контексте указанные термины также включают, в зависимости от состояния животного, предотвращение возникновения расстройства или патологического состояния, или симптомов, связанных с расстройством или патологическим состоянием, включая снижение тяжести расстройства или патологического состояния, или симптомов, связанных с ним, до заражения указанной инфекцией.
«Ветеринарно приемлемый» в данном контексте, если не указано иное, означает, что данное вещество или композиция должны быть совместимы химически и/или токсикологически с другими ингредиентами, входящими в состав композиции, и/или с организмом животного, подлежащего лечению. Ветеринарно приемлемый также охватывает фармацевтически приемлемый.
Помимо определений, описанных выше, установлены следующие термины:
«Промывочный водный раствор» используют для промывания соединения формулы (1) в экстрагирующем растворителе на стадии очистки предложенного способа получения 1:1 диастереомерной смеси соединения формулы (1). Иллюстративные промывочные водные растворы представляют собой воду или солевой раствор (водный раствор NaCl), каждый из которых содержит соединение(ия), образующее(ие) хелат с палладием. Иллюстративные соединения, образующие хелат с палладием, выбраны из группы, состоящей из: ЭДА, TMT-Na3, NH4OH, ТМТ, NaHSO3, тиомочевины, ДЭА, ЭДТК, Ac-L-цистеина, лимонной кислоты и их смесей. Солевой раствор может быть насыщенным.
«Органический растворитель» используют для растворения соединения формулы (1) на стадии очистки предложенного способа получения 1:1 диастереомерной смеси соединения формулы (1). Иллюстративные органические растворители включают: МЭК, iPrOAc, EtOAc, ацетон, 1-бутанол, 1-пропанол, 2-пропанол и их смеси.
«Экстрагирующий растворитель» используют для экстракции соединения формулы (1) на стадиях очистки предложенного способа получения 1:1 диастереомерной смеси соединения формулы (1). Иллюстративные экстрагирующие растворители включают: ТГФ, EtOAc, MeOAc, CH2Cl2 и Ме-ТГФ.
«Антирастворитель» используют для кристаллизации соединения формулы (1) на стадии очистки предложенного способа получения 1:1 диастереомерной смеси соединения формулы (1). Иллюстративные антирастворители включают воду, МТБЭ, гексан, гептан и их смеси.
Термин «отношение 1:1», используемый для описания соотношения двух диастереомеров формулы (1) (т.е. 1a:1b), относится к соотношению, которое находится в диапазоне от приблизительно 47:53 до приблизительно 53:47; предпочтительно от приблизительно 48:52 до приблизительно 52:48; и более предпочтительно от приблизительно 49:51 до приблизительно 51:49 формулы (1а) и формулы (1b), соответственно.
Соединение согласно настоящему изобретению содержит три хиральных центра. Таким образом, некоторые промежуточные соединения (f и g; схема 2) при получении формулы (В) состоят из рацемической смеси энантиомеров. Каждое из соответствующих энантиомерных промежуточных соединений имеет одинаковые химические и физические свойства, за исключением их способности вращать плоскополяризованный свет (+/-) на равные значения, но в противоположных направлениях. Энантиомеры также называют оптическими изомерами. Смесь равных частей оптически активного изомера и его энантиомера имеет суммарное нулевое вращение плоскополяризованного света, поскольку положительному вращению каждой (+) формы точно противодействует отрицательное вращение каждой (-) формы. Для получения энантиомерно чистого соединения формулы (В) приводят во взаимодействие промежуточное соединение (g) с оптически активной кислотой (например, (S)-миндальной кислотой) для образования кристаллов чистого (1R,2S) биологически активного (эутомерного) энантиомера (промежуточного соединения h). Биологически неактивный (дистомерный) энантиомер, который остается в растворе, отбрасывают. Полученное промежуточное соединение перерабатывают на следующих стадиях синтеза с получением энантиомерно чистой формулы (В). Разделение рацемической смеси промежуточного соединения (g) также может быть осуществлено стандартными хроматографическими способами на хиральных адсорбентах (например, на ацетилцеллюлозе). При связывании рацемической формулы (А) и энантиомерно чистой формулы (В) конечное соединение формулы (1) представляет собой смесь диастереомеров, формул (1а) и (1b). Смесь диастереомеров может быть очищена с получением 1:1 смеси диастереомеров.
Фармацевтические соли
Соединение формулы (1) может быть использовано в своей естественной форме (основания) или в форме соли. Соединение формулы (1) имеет основную функциональную группу и может образовывать соли присоединения с кислотами. Такие соли включены в объем настоящего изобретения в той степени, в которой они являются приемлемыми для ветеринарного применения. Подходящие соли присоединения кислот получают из кислот, которые образуют нетоксичные соли. Примеры включают ацетатные, адипатные, аспартатные, бензоатные, безилатные, бикарбонатные/карбонатные, бисульфатные/сульфатные, боратные, камзилатные, цитратные, цикламатные, эдизилатные, эзилатные, формиатные, фумаратные, глюцептатные, глюконатные, глюкуронатные, гексафторфосфатные, гибензатные, гидрохлоридные/хлоридные, гидробромидные/бромидные, гидройодидные/йодидные, изетионатные, лактатные, малатные, малеатные, малонатные, мезилатные, метилсульфатные, нафтилатные, 2-напзилатные, никотинатные, нитратные, оротатные, оксалатные, пальмитатные, памоатные, фосфатные/гидрофосфатные/дигидрофосфатные, пироглутаматные, сахаратные, стеаратные, сукцинатные, таннатные, тартратные, тозилатные, трифторацетатные и ксинофоатные соли.
Композиция/лекарственная форма
Фармацевтические композиции согласно настоящему изобретению могут быть получены способами, известными в данной области техники, например, традиционными способами смешивания, растворения, гранулирования, измельчения, эмульгирования, инкапсуляции, заливки, лиофилизации или распылительной сушки.
Фармацевтические композиции для применения в соответствии с настоящим изобретением могут быть составлены любым стандартным образом с использованием одного или более фармацевтически приемлемых носителей, включая эксципиенты и вспомогательные вещества, которые облегчают переработку активного соединения в препараты, которые могут быть использованы фармацевтически. Подходящая лекарственная форма зависит от выбранного способа введения. Фармацевтически приемлемые вспомогательные вещества и носители, в целом, известны специалистам в данной области техники и, следовательно, включены в настоящее изобретение. Такие вспомогательные вещества и носители описаны, например, в "Remington's Pharmaceutical Sciences", Mack Pub. Co., Нью-Джерси (1991). В одном аспекте композиция, содержащая соединение формулы (1), представляет собой композицию для инъекций. Инъекция может быть подкожной, внутримышечной, внутривенной. Предпочтительным способом инъекции является подкожный. Как описано в публикации Remington, композиция содержит вспомогательные вещества и носители, которые, как известно, обеспечивают растворимость соединения формулы (1) и которые приемлемы для фармацевтического применения в ветеринарии. Например, композиция для инъекций может содержать соединение формулы (1), ДМСО и ДМА. Другие обычно используемые вспомогательные вещества и/или носители могут включать глицерин, гликоли, диалкилгликолевые эфиры и т.п. Композиция может также содержать антиоксидант (например, ВНА, ВНТ, фенол и их смеси) и/или консерванты (например, бензиловый спирт, лимонную кислоту и т.п.). Композиция может содержать от приблизительно 100 мг/мл до приблизительно 600 мг/мл соединения формулы (1) на миллилитр носителя(ей). Предпочтительная композиция содержит от приблизительно 200 мг/мл до 500 мг/мл соединения формулы (1). Предпочтительная композиция содержит приблизительно 200 мг/мл соединения формулы (1). Предпочтительная композиция содержит приблизительно 300 мг/мл соединения формулы (1). Предпочтительная композиция содержит приблизительно 400 мг/мл соединения формулы (1). Предпочтительная композиция содержит приблизительно 500 мг/мл соединения формулы (1). Композиция также может содержать соединение формулы (1) в количестве приблизительно 250 мг/мл, 350 мг/мл и 450 мг/мл. В настоящем документе также предусмотрены композиции в другом количестве/объеме.
Композиции согласно настоящему изобретению могут быть разработаны как быстродействующие препараты, препараты с быстрым высвобождением, препараты длительного действия, препараты с продолжительным высвобождением или препараты с контролируемым высвобождением. В частности, лекарственная форма согласно настоящему изобретению может представлять собой форму с продолжительным высвобождением. Таким образом, фармацевтические лекарственные формы также могут быть составлены для контролируемого высвобождения или для медленного высвобождения. Предложенные фармацевтические лекарственные формы содержат соединение формулы (1) и также могут содержать фармацевтически приемлемую соль соединения формулы (1).
Доза
Фармацевтические композиции, подходящие для применения согласно настоящему изобретению, включают композиции, в которых активный ингредиент, т.е. соединение формулы (1), содержится в количестве, достаточном для достижения заданной цели, т.е. сдерживания или лечения инфекции. Более конкретно, терапевтически эффективное количество означает количество соединения, эффективное для предупреждения, облегчения или улучшения симптомов/признаков инфекции или продления продолжительности существования субъекта, проходящего лечение.
Количество активного компонента, который представляет собой соединение согласно настоящему изобретению, в фармацевтической композиции и ее единичную лекарственную форму можно варьировать или изменять в широком диапазоне в зависимости от способа введения, эффективности конкретного соединения и требуемой концентрации. Определение терапевтически эффективной дозы входит в объем навыков специалистов в данной области техники. В целом, количество активного компонента составляет от 0,01% до 99% по массе композиции.
В целом, терапевтически эффективное значение дозы соединения формулы (1) составляет от приблизительно 1 до 50 мг/кг массы тела/сутки; предпочтительно от приблизительно 10 до 40 мг/кг массы тела/сутки; и более предпочтительно от приблизительно 15 до 35 мг/кг массы тела/сутки; и наиболее предпочтительно приблизительно 20 мг/кг массы тела/сутки. Следует понимать, что дозы могут варьироваться в зависимости от требований каждого субъекта и тяжести бактериальной инфекции.
Требуемая доза может быть для удобства представлена в виде одной дозы или дробных доз, вводимых с соответствующими интервалами, например, в виде двух, трех, четырех или более субдоз в сутки. Кроме того, следует понимать, что первоначальная введенная доза может быть увеличена до значения выше верхнего предела с целью быстрого достижения требуемой концентрации в плазме. С другой стороны, первоначальная доза может быть меньше оптимальной, и суточную дозу можно постепенно увеличивать в течение курса лечения в зависимости от конкретной ситуации. При необходимости суточную дозу также можно разделять на несколько доз для введения, например, от двух до четырех раз в сутки.
Антибактериальные анализы
Соединения согласно настоящему изобретению тестировали против ряда грамотрицательных и грамположительных организмов, используя стандартные промышленные технологии, описанные в М31-А3. Performance Standards for Antimicrobial Disk and Dilution Susceptibility Tests for Bacteria Isolated from Animals; Институт клинических и лабораторных стандартов, утвержденный стандарт, третье издание. Соединения согласно настоящему изобретению демонстрируют высокую антибактериальную активность против патогенов, вызывающих BRD, например, M. haemolytica Р. multo., H. sommun и M. bovis.
Медицинское и ветеринарное применение
Соединение формулы (1) согласно настоящему изобретению представляет собой антибактериальный агент, используемый для лечения инфекций, вызывающих респираторные заболевания крупного рогатого скота, у крупного рогатого скота, вызванных грамотрицательными респираторными патогенами, такими как М. haemolytica, P. multocida H. somnus и M. bovis.
В одном исследовании здоровых молочных бычков (N=15/группа; 85-140 кг) транстрахеально инфицировали штаммом A3579 (OSU-012103-BH1) Mannheimia haemolytica на -2 - -1 день исследования. Провокационная доза составляла от приблизительно 2,9×109 КОЕ до 4,9×108 КОЕ. На 0 день животным с симптомами респираторного заболевания крупного рогатого скота (BRD) вводили однократную подкожную дозу солевого раствора (контроль) или экспериментального соединения (20 или 40 мг/кг). При вскрытии контрольные животные демонстрировали верхний предел повреждения легких приблизительно 45%. Животные, которым вводили дозу 20 и 40 мг/кг, демонстрировали существенно более низкий процент поражения легких со значением верхнего предела приблизительно 12% и 8%, соответственно. Таким образом, однократная доза соединения формулы (1), введенная подкожно, была эффективной для лечения BRD, вызванного М. Haemolytica, у молочных бычков.
Во втором исследовании бычков (смешанных мясных и/или молочных пород; 180-270 кг) лечили от возникшей естественным путем инфекции BRD. Животным (n=40/группа) вводили однократную подкожную дозу солевого раствора (контроль) или экспериментального соединения (15 мг/кг или 20 мг/кг). В целом, успех лечения на основании респираторных симптомов (степень, слизисто-гнойные выделения из носа или глаз, дыхание ртом) и ориентации в пространстве (тревожность, время реакции на раздражители, мышечная слабость, атаксия, покачивание) составлял приблизительно 37% и 47% для дозы 15 мг/кг и 20 мг/кг, соответственно. Напротив, успех у контрольных животных составлял приблизительно 20%. В целом, показано, что соединение формулы (1) обеспечивает существенный лечебный эффект у крупного рогатого скота с естественными инфекциями, вызывающими BRD.
В третьем исследовании гибридным коровам Holstein/Holstein возрастом приблизительно 6 месяцев и весом приблизительно 330 кг делали транстрахеальную провокацию, используя приблизительно 3×109 КОЕ/доза М. haemolytica (штамм 34195). Животным вводили дозу солевого раствора (Т01, отрицательный контроль), нуфлора (флорфеникол, Т02, положительный контроль, 40 мг/кг), байтрила (энрофлоксацин, Т03, положительный контроль), соединения формулы (1) (Т04, 20 мг/кг) и соединения формулы (1) (Т05, 40 мг/кг). Дозы вводили подкожно через 4-6 часов после провокации. По окончании исследования (6 дней) оценивали смертность и поражение легких. Смертность для Т01, Т02, Т03, Т04 и Т05 составила 53,3%, 46,7%, 0%, 0% и 0%, соответственно. Животные, которых лечили Т03, Т04 и Т05, демонстрировали существенное снижение смертности, связанной с BRD, по сравнению с Т01 и Т02. Процент повреждения легких, рассчитанный методом наименьших квадратов с обратным преобразованием(1), для Т01, Т02, Т03, Т04 и Т05 составил 35,1%, 32,1%, 9,1%, 13,3% и 6,8%, соответственно. Все животные, которым вводили Т03, Т04 и Т05, демонстрировали существенное снижение процента поражения легких по сравнению с животными, которым вводили Т01 и Т02. Т04 и Т05 не отличались от Т03.
Примеры
Способ получения соединения формулы (1) проиллюстрирован следующими схемами и технологическими стадиями. Исходные материалы и различные реагенты могут быть приобретены у коммерческих поставщиков или без труда получены из доступных в продаже органических соединений с применением способов, известных специалистам в данной области техники. Сокращения, использованные в настоящем документе, имеют следующие значения: тиометоксид натрия (NaSMe); этанол (EtOH); и 2-пропанол (изопропанол, IPA, 2-PrOH); 1-пропанол (н-пропанол, 1-PrOH), 2-бутанол (2-BuOH), изопропилацетат (iPrOAc), этилацетат (EtOAc), метилацетат (МеОАс), 2-метилтетрагидрофуран (Ме-ТГФ), N-метил-2-пирролидон (NMP); диметилформамид (ДМФА), гидрид натрия (NaH); метанол (МеОН); карбонат натрия (Na2CO3); сульфит натрия (Na2SO3); сульфат магния (MgSO4); тетрагидрофуран (ТГФ); солевой раствор (водный раствор NaCl), тионилхлорид (SOCl2); диметилсульфоксид (ДМСО); хлористоводородная кислота (HCl); бисбороновая кислота (ВВА); бис(пинаколато)дибор (B2Pin2), тетракис(диметиламино)дибор B2(NMe2)4, метил-трет-бутиловый эфир (МТБЭ); бензонитрил (PhCN); метиленхлорид (CH2Cl2); ацетонитрил (MeCN, CH3CN); диметоксиэтан (ДМЭ); диметилацетамид (DMAc/ДМА); триэтиламин (ТЭА); диизопропилэтиламин (DIPEA), пероксид водорода (Н2О2); метоксид натрия (NaOMe); метилэтилкетон (МЭК); карбонат калия (K2CO3); карбонат цезия (CS2CO3); ацетат цезия (CsOAc), боргидрид калия (KBH4); трехосновной фосфат калия (K3PO4), 1,3-дибром-5,5-диметилгидантоин (DBDMH); трет-бутоксид калия (KOtBu); уксусная кислота (НОАс); ацетат калия (KOAc); палладий (Pd); ацетат палладия (II) (Pd(OAc)2); хлорид палладия (II) (PdCl2), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (X-Phos); 1,3-бис(дифенилфосфино)пропан (dppp); предкатализатор хлорид аминобифенилпалладия (Pd-G2-XPhos), хлор(кротил)(2-дициклогексилфосфино-2,4,6-триизопропилбифенил)палладий (II) (Pd-XPhos-кротил-Cl), предкатализатор хлорид трициклогексилфосфинаминобифенилпалладия (Pd-G2-РСу3), бис(дифенилфосфино)ферроцендихлорпалладий (Pd(dppf)Cl2), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 2,2,2-трифторацетамид (CF3CONH2); этилдифторацетат (MeO2CCHF2); этиленгликоль (ЭГ), пропиленгликоль (ПГ); 2-дициклогексилфосфино-2,6-диметоксибифенил (SPhos), ферроцендиил-бис(дифенилфосфин (dppf); дибензилиденацетон (dba); трифенилфосфин (PPh3); трициклогексилфосфин (РСу3); этилендиамин (ЭДА), уголь (активированный уголь), силикагель (SiO2), Siliamets Thiol (Si-тиол), Quadrapure TU (MP-TU), тримеркаптотриазин, связанный с микропористым полистиролом (МР-ТМТ), связанный с диоксидом кремния тримеркаптотриазин (Si-TMT), Siliamets DMT (Si-DMT), Siliamets Cysteine (Si-цистеин), гидроксид аммония (NH4OH), тримеркаптотриазин (ТМТ), натриевая соль тримеркаптотриазина (TMT-Na3), бисульфит натрия (NaHSO3); тиомочевина (H2NCSNH2), диэтиламин (ДЭА), этилендиаминтетрауксусная кислота (ЭДТК), ацетил-L-цистеин (Ас-L-цистеин), водный (водн.); объем (V); эквивалент (экв.); и мегапаскаль (МПа).
Альтернативно, соединение формулы (1) может быть получено способами, впервые описанными в патенте США US 9422236. Способ, описанный в настоящем документе, имеет множество преимуществ по сравнению со способом, описанным в US 9422236. Например:
1) синтез формулы (А) имеет на 2 стадии меньше, исключает колоночную хроматографию и увеличивает выход на приблизительно 67%;
2) начинается с более дешевого хлорсодержащего ядра (формула В), а не с йодсодержащего ядра (стадия 6, пример 17);
3) основан на использовании незащищенного хлорсодержащего ядра, а не защищенного йодсодержащего ядра, что обеспечивает возможность проведения двух стадий (борилирования и Сузуки) в одном реакторе, а не проведения трех стадий (борилирования, Сузуки и снятия защиты) в трех реакторах;
4) борилирование обеспечивает более реакционноспособный и атомно-эффективный боронат этиленгликолевого ядра, по сравнению с боронатом пинаколового ядра (стадия 8), что приводит к образованию побочного этиленгликоля, легко поддающегося удалению, а не побочного пинакола в реакции Сузуки;
5) основан на использовании одного выделения и кристаллизации для более экономичного технологического процесса с получением выхода белого продукта 70-80% по сравнению с 3 стадиями выделения и 3 хроматографическими очистками с получением выхода коричневого твердого вещества 41% (стадия 10);
6) включает растворитель для борилирования 3 класса, этанол, а не диоксан 2 класса, и более дешевый катализатор Pd(OAc)2 по сравнению с Pd(PPh3)2Cl2;
7) основан на использовании а) более низких температур борилирования (ниже 60°С против 90°С) и более короткого времени (4 часа против 22 часов); и б) более низких температур при связывании Сузуки (ниже 70°С против 80°С) и более короткого времени (4 часа против 8 часов);
8) подразумевает использование экстрагирующего растворителя ТГФ, а не ДХМ, для улучшения производительности выделения и минимизации технологической обработки до одного реактора вместо двух реакторов;
9) основан на использовании стабильного на воздухе борилирующего реагента тетрагидроксидибора, а не гексаметилдиолова для получения реакционноспособного промежуточного бороната для реакции связывания Сузуки, по сравнению с промежуточным органостаннаном (стадия 2), который, вероятно, может подвергаться связыванию Стилле с промежуточным соединением реакции; и
10) основан на использовании ЭДА и угля для удаления остаточного палладия, который, вероятно, присутствует в исходном коричневом твердом веществе (по результатам внутренних исследований, хроматографическая очистка соединения формулы (1), полученного в соответствии с известным способом, не обеспечивает удаление палладия до уровня менее 100 м.д.).
Схема 1. Получение соединения формулы (А)
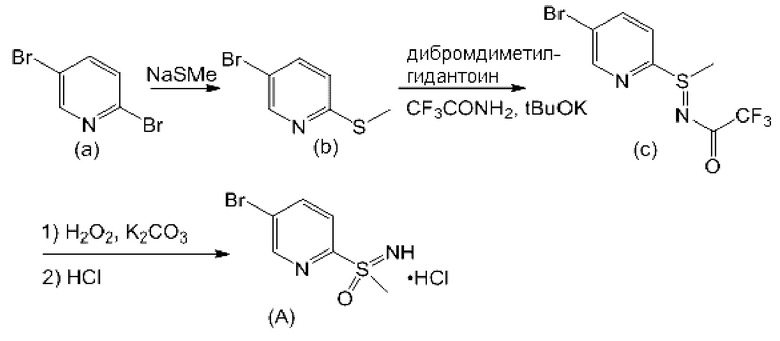
Формулу (А) получают в 3 стадии из 2,5-дибромпиридина. 2,5-Дибромпиридин (а) обрабатывают водным раствором тиометоксида натрия в полярном апротонном растворителе, таком как ДМФА, NMP, DMAc, ДМСО, предпочтительно ДМФА, с получением промежуточного соединения (b), которое осаждают посредством добавления воды. N-(трифторацетил)сульфилимин (с) синтезируют посредством обработки раствора сульфида (b) и трифторамида сильным основанием, таким как KOtBu, NaH, предпочтительно KOtBu, затем дибромдиметилгидантоином. Можно использовать различные растворители, включая МТБЭ, ацетонитрил, ТГФ, Ме-ПГФ, дихлорметан и 1,4-диоксан, с разным содержанием примеси сульфоксида, который также образуется. Сульфилимин (с) может быть выделен из систем на органической (IPA/гептан) или водной (спирт/вода, ТГФ/вода) основе. Окисление сульфилимина (с) до рацемического сульфоксимина формулы (А) осуществляют с использованием пероксида водорода, в присутствии карбонатного основания, предпочтительно порошкообразного карбоната калия, и в смеси ацетонитрила и короткоцепочечного спирта, предпочтительно метанола. Комбинация растворителей и соотношение ацетонитрила и спирта критичны для успешного окисления и регулирования образования примеси сульфона. Выделение HCl соли сульфоксиминного соединения формулы (А) приводит к удалению остаточного сульфона и других примесей.
Схема 2. Получение соединения формулы (В)
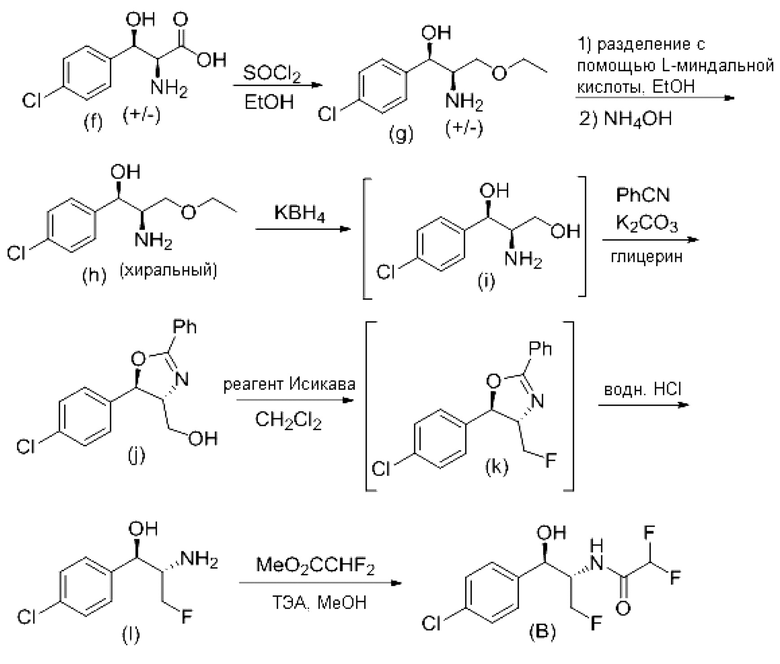
Получение энантиомерно чистой формулы (В) осуществляют в 8 стадий из 4-хлорбензальдегида и глицина. Взаимодействие глицина с 2 эквивалентами 4-хлорбензальдегида в основных условиях приводит к получению связанного кислотного рацемата промежуточного соединения (f) после нейтрализации. Этерификацию проводят с использованием тионилхлорида в этаноле с получением сложнее фирн ого рацемата (g). Классическое разделение (g) проводят с помощью L-миндальной кислоты. Сложноэфирную функциональную группу в промежуточном соединении (h) восстанавливают с помощью KBH4 и полученное промежуточное соединение (i) защищают в форме дигидрооксазола, без выделения, с использованием бензонитрила, глицерина и K2CO3 при высокой температуре с получением соединения (j). Промежуточное соединение (j) фторируют реагентом Исикава под давлением в дихлорметане при приблизительно 100°С. Затем полученное промежуточное соединение (k) гидролизуют водным раствором HCl. Наконец, соединение формулы (В) получают амидированием промежуточного соединения (l) метилтрифторацетатом и триэтиламином. Ph в (j) и (k) представляет собой фенил. Данная реакция описана также в CN 1Q6631S72 A.
Схема 3. Получение соединения формулы (1)
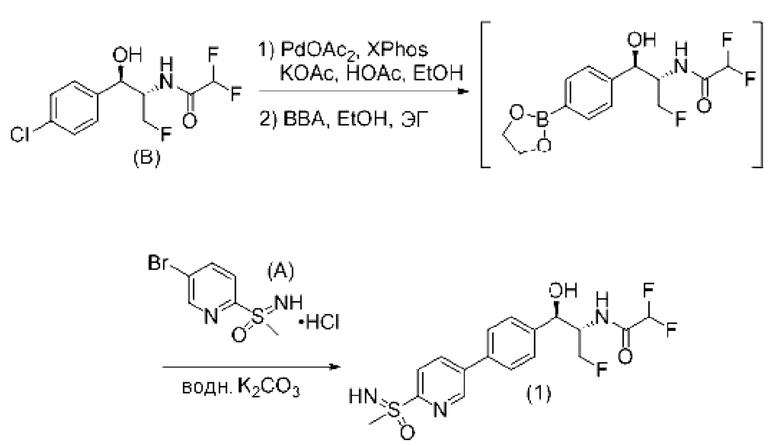
Соединение формулы (1) получали 2-стадийным сокращенным способом путем связывания промежуточных соединений (А) и (В). Промежуточное соединение борилировали посредством реакции на палладиевом катализаторе с использованием бисбороновой кислоты. Используемые соединения палладия, лиганд, протокол активации катализатора, система растворителей и буфер являются важными реагентами для данной реакции. Следует отдельно отметить, что добавление в реакционную смесь уксусной кислоты улучшает превращение в продукт по сравнению с отсутствием уксусной кислоты. После завершения борилирования добавляют водно-ТГФ раствор соединения формулы (А) в ТГФ и водном растворе K3CO3. Затем используют такой же палладиевый катализатор для связывания двух промежуточных соединений с получением соединения формулы (1).
Получение соединения формулы (А), (5-бромпиридин-2-ил)(имино)(метил)-λ6-сульфанона гидрохлорида.
Стадия 1А. Получение промежуточного соединения (b), 5-бром-2-метилсульфанилпиридина
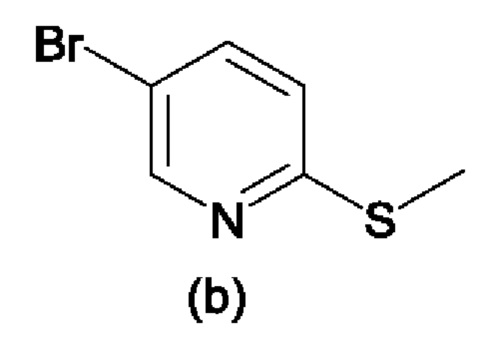
К перемешанному раствору 2,5-дибромпиридина (промежуточного соединения (а) (схема 1); 100 г, 0,49 моль) в ДМФА (800 мл) добавляли 20% раствор тиометоксида натрия в воде (41,2 г, 0,59 моль) при приблизительно 15-20°С. Температуру повышали и выдерживали при приблизительно 50-55°С в течение около 6 часов. После завершения реакции смесь охлаждали до приблизительно 15-20°С и добавляли воду (1,8 л). Смесь охлаждали до приблизительно 0-5°С и через приблизительно 1-2 часа собирали твердое вещество фильтрацией, промывали водой и сушили с получением указанного в заголовке соединения в виде бесцветного твердого вещества (75 г, выход 87%). 1H ЯМР (400 МГц, ДМСО) d: 2,49 (с, 3Н), 7,29 (д, 1Н, J=8,76 Гц), 7,85-7,88 (дд, 1Н, J1=2,44 Гц, J2=8,48 Гц), 8,55 (д, 1Н, J=2,4 Гц). ЖХ-МС (m/z): М+Н=206,1.
Альтернативно, указанное соединение может быть получено известными в данной области техники способами (WO 2014/172443 A1) или в небольшом количестве приобретено у поставщиков.
Стадия 2А. Получение промежуточного соединения (с), N-((5-бромпиридин-2-ил)(метил)-4-сульфанилиден)-2,2,2-трифторацетамида
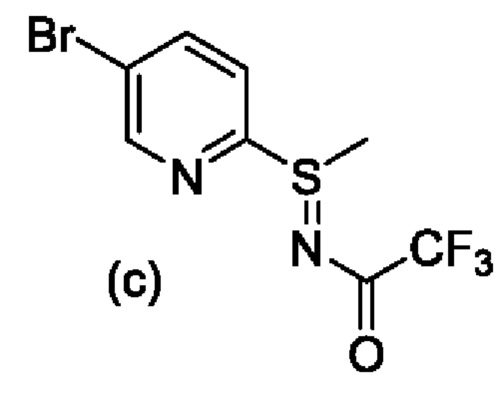
К раствору 5-бром-2-метилсульфанилпиридина (1,5 кг, 7,35 моль) и 2,2,2-трифторацетамида (961 г, 1,2 экв.) в МТБЭ (9 л) по частям добавляли трет-бутоксид калия (875 г, 1,10 экв.), поддерживая температуру смеси ниже 10°С. Отдельно растворяли 1,3-дибром-5,5-диметилгидантоин (DBDMH, 2230 г, 1,10 экв.) в ТГФ (7,2 л) и охлаждали раствор до приблизительно 0-5°С. Раствор DBDMH добавляли к полученной выше смеси, поддерживая температуру смеси от приблизительно минус 5 до 10°С. После завершения реакции добавляли раствор сульфита натрия в воде (1200 г Na2SO3/5,2 л воды), поддерживая температуру смеси при ниже 12°С. Добавляли воду (6 л) и нагревали смесь до приблизительно 20-25°С. Разделяли слои и промывали органический слой водой (4,5 л) и затем полунасыщенным солевым раствором (2×4,5 л). Органический слой концентрировали вакуумной перегонкой до густой суспензии (объем смеси приблизительно 1,5 л). В смесь добавляли изопропанол (1,5 л) и продолжали вакуумную перегонку до объема смеси приблизительно 1,5 л. К суспензии добавляли 1:1 раствор изопропанолтептан (1,5 л) и охлаждали смесь до приблизительно 0-5°С. Твердые вещества собирали и промывали осадок на фильтре 1:2 смесью изопропанолтептан (1,5-2,7 л). Продукт сушили в вакууме при приблизительно 50°C с получением указанного в заголовке соединения в виде бесцветного твердого вещества (2,09 кг, выход 90%). 1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 3,15 (с, 3Н), 7,90 (д, J=8,44 Гц, 1Н), 8,43 (шд, J=8,44 Гц, 1Н), 8,98 (с, 1Н). ЖХ-МС (m/z): М+Н = 315/317.
Альтернативно, к раствору трет-бутоксида калия (56,7 г, 1,02 экв.) в ТГФ (200 мл) добавляли раствор 5-бром-2-метилсульфанилпиридина (100 г, 0,49 моль) и 2,2,2-трифторацетамида (58,2 г, 1,05 экв.) в ТГФ (100 мл), поддерживая температуру смеси ниже 10°С. Отдельно растворяли 1,3-дибром-5,5-диметилгидантоин (DBDMH, 98,1 г, 0,70 экв.) в ТГФ (350 мл) и охлаждали раствор до 0-10°С. Раствор DBDMH добавляли к полученной выше смеси, поддерживая температуру смеси от приблизительно минус 5 до 5°С. После завершения добавления смесь нагревали до приблизительно 10°С. После завершения реакции добавляли раствор бисульфита натрия (13,7 г, 0,27 экв.), гидроксида натрия (13,7 г, 0,70 экв.) и хлорида натрия (50 г) в воде (200 мл) и нагревали смесь до 20-25°С. Добавляли воду (300 мл) и разделяли слои. Органический слой концентрировали вакуумной перегонкой до объема смеси приблизительно 200 мл. В смесь добавляли изопропанол (200 мл) и продолжали вакуумную перегонку до объема смеси приблизительно 300 мл. Смесь охлаждали до приблизительно 20°С и затем добавляли воду (400 мл) в течение приблизительно 30 минут. Твердые вещества собирали фильтрацией и промывали осадок на фильтре 1:4 смесью изопропанол : вода (200 мл). Продукт сушили в вакууме при приблизительно 50°C с получением указанного в заголовке соединения в виде бесцветного твердого вещества (134 г, выход 87%). 1Н ЯМР (600 МГц, ДМСО-d6) δ м.д. 3,15 (с, 3Н), 7,90 (д, J=8,44 Гц, 1Н), 8,43 (шд, J=8,44 Гц, 1Н), 8,98 (с, 1Н). ЖХ-МС (m/z): М+Н = 315/317.
Стадия 3А. Получение соединения формулы (А), (5-бромпиридин-2-ил)(имино)(метил)-λ6-сульфанона гидрохлорида
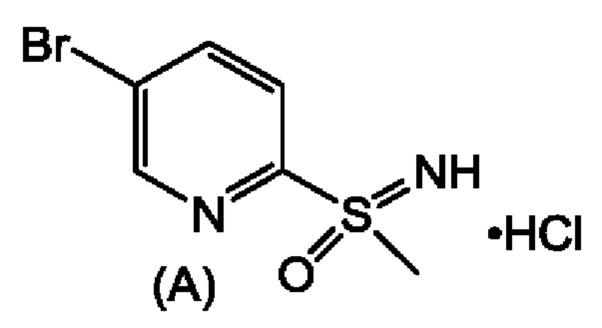
N-((5-бромпиридин-2-ил)(метил)-λ4-сульфанилиден)-2,2,2-трифторацетамид (50 г, 0,159 ммоль) и порошкообразный карбонат калия (26,3 г, 1,20 экв.) объединяли с ацетонитрилом (126 мл), метанолом (74 мл) и водой (4,3 мл). Медленно добавляли 30% пероксид водорода (19,4 мл, 1,20 экв.), поддерживая температуру смеси приблизительно 25-35°С. Смесь поддерживали при приблизительно 30°С до завершения реакции. К смеси добавляли раствор бисульфита натрия (3,3 г, 0,2 экв.) и воду (150 мл) и перемешивали до исчезновения пероксида по пероксидной индикаторной полоске. Смесь концентрировали вакуумной перегонкой для удаления органических растворителей. В смесь добавляли дихлорметан (150 мл) и разделяли слои. Водный слой экстрагировали дополнительным количеством дихлорметана (100 мл и затем 50 мл). Объединенный органический слой концентрировали атмосферной перегонкой до объема смеси приблизительно 250 мл. В смесь добавляли HCl в изопропаноле, поддерживая температуру смеси при ниже 35°С. Твердые вещества собирали при комнатной температуре и промывали изопропанолом (50 мл). Продукт сушили в вакууме при приблизительно 50°C с получением указанного в заголовке рацемического соединения в виде белого твердого вещества (40,4 г, выход 93%). 1H ЯМР (600 МГц, метанол-d4) δ м.д. 3,95 (с, 3Н), 8,30 (д, J=9,03 Гц, 1Н), 8,56 (дд, J=8,28, 2,26 Гц, 1Н), 9,08 (д, J=1,51 Гц, 1Н). ЖХ-МС (m/z): М+Н = 235/237.
Способы, описанные выше для стадий 2А и 3А, обеспечивают на 2 стадии меньше, исключают использование колоночной хроматографии и увеличивают выход на приблизительно 67% по сравнению с известным синтезом формулы (А), описанным в US 9422236.
Способ получения соединения формулы (В), N-((1R,2S)-1-(4-хлорфенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида.
Стадия 1В: Получение рацемического промежуточного соединения (f), (+/-)-(2S,3R)-2-амино-3-(4-хлорфенил)-3-гидроксипропановой кислоты
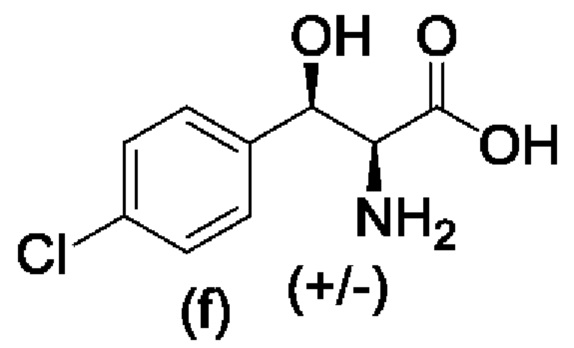
К раствору 4-хлорбензальдегида (1152 г, 2 экв.) в метаноле (8 л - 10 л) добавляли глицин (300 г), затем 30% NaOMe в метаноле (1439 г, 2 экв.) и перемешивали при комнатной температуре в течение ночи. Добавляли водный раствор HCl (810 г, 2 экв.) и перемешивали смесь в течение 1 часа. После охлаждения до приблизительно 5-10°С продукт собирали фильтрацией, промывали этанолом и сушили в вакууме при приблизительно 60°C с получением указанного в заголовке соединения (1,00 кг, количественный анализ 66%, выход 77%).
Стадия 2В. Получение рацемического промежуточного соединения (g), (+/-)-этил-(2S,3R)-2-амино-3-(4-хлорфенил)-3-гидроксипропаноата
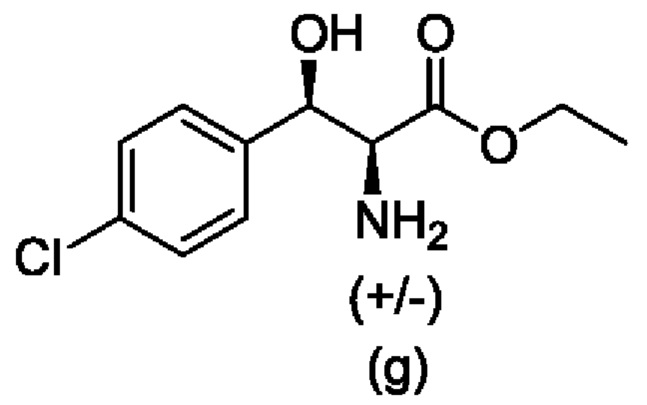
(+/-)-(2S,3R)-2-амино-3-(4-хлорфенил)-3-гидроксипропановую кислоту (1,00 кг) и этанол (4 л) объединяли и охлаждали до ниже 5°С. Добавляли тионилхлорид (740 г, 2 экв.) при приблизительно 0-5°С и затем нагревали смесь до приблизительно 50°С. После завершения реакции (10 часов) смесь концентрировали вакуумной перегонкой, разбавляли водой (1,5 л) и доводили до рН 8 гидроксидом аммония. Смесь охлаждали до приблизительно 5-10°С для осаждения продукта, который собирали фильтрацией, промывали водой и сушили с получением указанного в заголовке промежуточного соединения (725 г, выход 97%).
Стадия 3В. Хиральное разделение с получением промежуточного соединения (h), этил-(2S,3R)-2-амино-3-(4-хлорфенил)-3-гидроксипропаноата
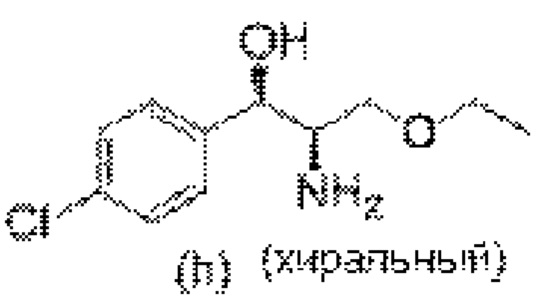
(+/-)-Этил-(2S,3R)-2-амино-3-(4-хлорфенил)-3-гидроксипропаноат (1100 г, 1 экв.) добавляли к раствору L-миндальной кислоты (1,05 экв.) в этаноле (7 л) и нагревали до приблизительно 35-40°С. Смесь охлаждали до приблизительно 20°С за 1 час и перемешивали в течение 1 часа, затем собирали твердые вещества фильтрацией. Осадок на фильтре растворяли в воде (7 л) и доводили до рН 8-9 гидроксидом аммония. Продукт выделяли фильтрованием, промывали водой и сушили в вакууме при приблизительно 45°C с получением соединения (h) (320 г, выход 29%). Вторую партию получали концентрированием маточного раствора для удаления 4 л этанола и затем охлаждали до приблизительно 20°С. Твердые вещества собирали фильтрованием и затем объединяли с L-миндальной кислотой (10 г) в 1200 мл этанола, и нагревали до приблизительно 40°С. После охлаждения до приблизительно 20°С за 1 час и выдерживания в течение 1 часа собирали твердые вещества фильтрованием и сушили в вакууме при приблизительно 45°С с получением промежуточного соединения (h) (40 г, выход 3,6%).
Стадия 4В. Получение промежуточного соединения (i), (1R,2R)-2-амино-1-(4-хлорфенил)пропан-1,3-диола
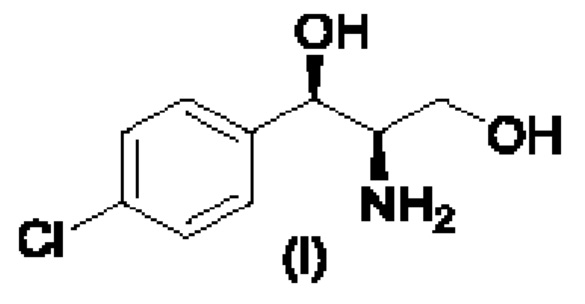
50 г этил-(2S,3R)-2-амино-3-(4-хлсрфенил)-3-гидроксипропаноата растворяли в 350 мл метанола и затем медленно добавляли 13,3 г боргидрида калия, и проводили реакцию при приблизительно 40°С в течение 6 часов. Добавляли подходящее количество разбавленной хлористоводородной кислоты и перемешивали в течение 30 минут. Растворитель выпаривали при пониженном давлении. Остаток растворяли в воде, доводили до рН более 10 с помощью 30% раствора гидроксида натрия и затем несколько раз экстрагировали дихлорметаном. Органические фазы объединяли, один раз промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением 39,7 г белого твердого вещества (выход 96%). Неочищенный продукт (i) напрямую использовали для получения соединения формулы (j) на следующей стадии, без очистки.
Стадия 5В. Получение промежуточного соединения (j), ((4R,5R)-5-(4-хлорфенил)-2-фенил-4,5-дигидрооксазол-4-ил)метанола
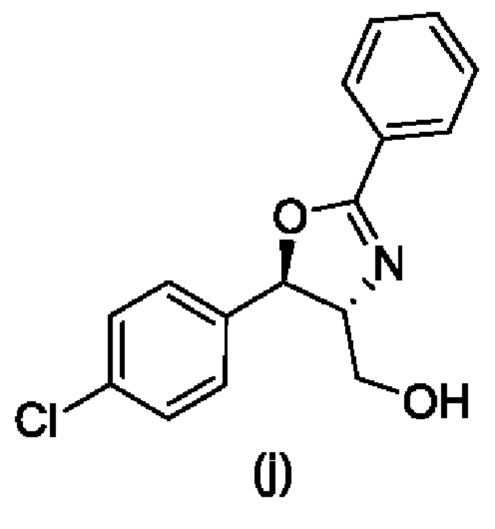
30 г (1R,2R)-2-амино-1-(4-хлорфенил)пропан-1,3-диола (i), 90 г глицерина и 6,8 г карбоната калия нагревали до приблизительно 105°С и затем по каплям добавляли 21,5 г бензонитрила в течение 20 минут, и проводили реакцию при приблизительно 105°С в течение 18 часов. После охлаждения до приблизительно 50°С добавляли 90 г воды, перемешивали при приблизительно 50°С в течение 30 минут и затем фильтровали в горячем состоянии. Осадок на фильтре один раз суспендировали в этаноле и затем фильтровали с получением 41,1 г белого твердого вещества (выход 96%), промежуточного соединения (j).
50 г промежуточного соединения (h) растворяли в 350 мл метанола и затем медленно добавляли 13,3 г боргидрида калия, и проводили реакцию при приблизительно 40°С в течение 6 часов. Добавляли 150 г глицерина, метанол выпаривали концентрированием при пониженном давлении и затем добавляли 11,3 г карбоната калия. После повышения температуры до приблизительно 105°С по каплям добавляли 33,8 г бензонитрила за 20 минут, и затем проводили реакцию при 105°С в течение 18 часов. После охлаждения до приблизительно 50°С добавляли 150 г воды, перемешивали при приблизительно 50°С в течение 30 минут и затем фильтровали в горячем состоянии. Осадок на фильтре один раз суспендировали в этаноле и затем фильтровали с получением 51,4 г промежуточного соединения (j) в виде белого твердого вещества (выход 87%).
Стадия 6В. Получение промежуточного соединения (k) (4S,5R)-5-(4-хлорфенил)-4-(фторметил)-2-фенил-4,5-дигидрооксазола

30 г ((4R,5R)-5-(4-хлорфенил)-2-фенил-4,5-дигидрооксазол-4-ил)метанола смешивали с 300 мл дихлорметана и перемешивали. По каплям добавляли 24,7 мл (0,136 моль) реагента Исикава при комнатной температуре в атмосфере азота, перемешивали до однородности и затем переносили в реактор для работы при высоком давлении, в котором давление реакции составляло 0,6 МПа. После проведения реакции при приблизительно 100°С в течение 2-3 часов реакционную смесь охлаждали до комнатной температуры и удаляли реакционную жидкость. Органическую фазу промывали водой, доводили до рН 6-8 с помощью 30% раствора гидроксида натрия, затем снова промывали водой, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и охлаждали с получением светло-желтого твердого вещества (k). Неочищенный продукт (k) можно напрямую использовать для следующей реакции без очистки с получением соединения формулы (l).
Стадия 7В. Получение промежуточного соединения (1), (1R,2S)-2-амино-1-(4-хлорфенил)-3-фторпропан-1-ола
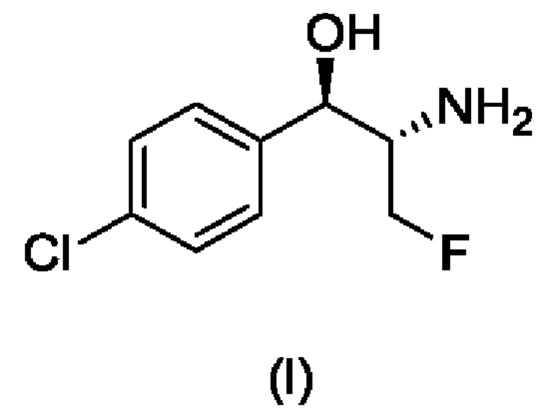
Неочищенный продукт (4S,5R)-5-(4-хлорфенил)-4-(фторметил)-2-фенил-4,5-дигидрооксазол добавляли к 300 мл 6 н. хлористоводородной кислоты, нагревали до приблизительно 100-105°С и проводили реакцию в течение 16 часов с обратным холодильником. После охлаждения до комнатной температуры отфильтровывали побочный продукт, бензойную кислоту. Фильтрат концентрировали при пониженном давлении с получением светло-желтого твердого вещества, которое растворяли в воде, доводили до рН более 12 с помощью 30% раствора гидроксида натрия, и затем дважды экстрагировали дихлорметаном. Органические фазы объединяли, один раз промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Растворитель в фильтрате выпаривали при пониженном давлении и перекристаллизовывали в изопропаноле и н-гексане с получением 17 г белого твердого вещества (выход 80%). Альтернативно, весь неочищенный продукт (k) добавляли к 300 мл 6 н. серной кислоты, нагревали до приблизительно 100-105°С и проводили реакцию в течение 16 часов с обратным холодильником. После охлаждения до комнатной температуры отфильтровывали побочный продукт, бензойную кислоту. Фильтрат концентрировали при пониженном давлении с получением светло-желтого твердого вещества, которое растворяли в воде, доводили до рН более 12 с помощью 30% раствора гидроксида натрия, и затем дважды экстрагировали дихлорметаном. Органические фазы объединяли, один раз промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Растворитель в фильтрате выпаривали при пониженном давлении, перекристаллизовывали в изопропаноле и н-гексане с получением 16,7 г белого твердого вещества (выход 78,5%).
Стадия 8В. Получение соединения формулы (В), N-((1R,2S)-1-(4-хлорфенил)-3-фтор-1-гидроксипропан-2-ил)-2,2-дифторацетамида
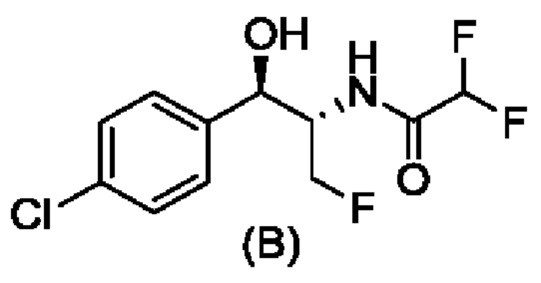
10 г (1R,2S)-2-амино-1-(4-хлорфенил)-3-фторпропан-1-ола растворяли в 350 мл метанола и затем добавляли 5 г триэтиламина и 30,5 г этилдифторацетата, и перемешивали в течение 12 часов при комнатной температуре. Растворитель выпаривали при пониженном давлении и перекристаллизовывали остаток в изопропаноле и воде с получением 12,7 г белого твердого вещества (выход 92%).
Способ получения соединения формулы (1): 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-(8-метилсульфонимидоил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида
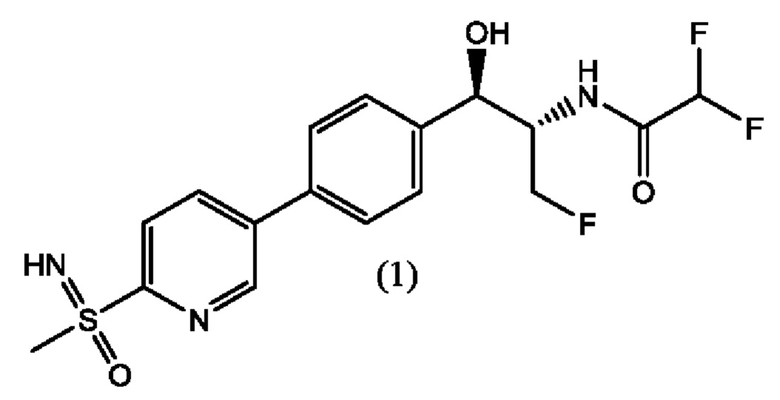
В атмосфере N2 объединяли Pd(OAc)2 (9 г, 1% мол.) XPhos (38,9 г, 2% мол.), соединение формулы (В) (1023 г, 1 экв.) и KOAc (891 г, 2,5 экв.). Пропускали N2, добавляли безводный EtOH 200 марки (5115 мл) и НОАс (109 г, 0,5 экв.) и нагревали смесь до приблизительно 72°С в течение приблизительно 30 минут, и затем охлаждали до приблизительно 52°С. Добавляли очищенный барботированием N2 раствор тетрагидроксидибора (бисбороновой кислоты, ВВА, 420 г, 1,25 экв.) и этиленгликоля (ЭГ, 564 г, 2,5 экв.) в безводном EtOH 200 марки (3070 мл) в течение приблизительно 30 минут при приблизительно 52°С и промывали EtOH (450 мл). После завершения борилирования, по данным ВЭЖХ (не более 1% формулы (В), 2 часа), к смеси с температурой приблизительно 52°С добавляли очищенный барботированием N2 раствор соединения формулы (А) (957 г, 0,97 экв.) в ТГФ (3069 мл) и K2CO3 (1258 г, 2,5 экв.) в воде (2046 мл) при приблизительно 42°С и промывали смесью ТГФ:вода (400 мл:50 мл). Смесь нагревали до приблизительно 72°С и перемешивали до завершения сочетания Сузуки (приблизительно 4 часа). После охлаждения до комнатной температуры смесь нейтрализовали до рН приблизительно 7,0 концентрированной HCl. Смесь концентрировали вакуумной перегонкой до приблизительно 7 объемов (температура смеси ниже 40°С, 60-70 мбар). Добавляли ТГФ (7,2 л) и повторно концентрировали смесь вакуумной перегонкой (температура смеси ниже 30°С, 60-70 мбар) до приблизительно 5 объемов. Добавляли ТГФ (5,2 л) и воду (3,1 л) и нагревали смесь при рН приблизительно 7 до приблизительно 60°С до получения двухфазного раствора, и затем охлаждали до 45-55°С. Нижний водный слой сливали, а органический слой перемешивали со смесью вода:насыщенный солевой раствор:ЭДА (1,3 л:1,3 л:0,6 экв.) при приблизительно 25°С в течение более 0,5 часа. Нижний водный слой сливали. Органический слой перемешивали с ЭДА (0,6 экв.) и углем (660 г, активированный уголь, тип С-941) в течение более 12 часов или рециркулировали с ЭДА (0,6 экв.) через картридж(и) из угля (тип С-941) в течение более 12 часов. Уголь отфильтровывали и промывали твердые вещества ТГФ (2,1). Фильтрат промывали насыщенным солевым раствором (1,3 л) с рН, доведенным до приблизительно 7 с помощью концентрированной HCl. Фильтрат промывали насыщенным солевым раствором (1,3 л) и концентрировали органический слой вакуумной перегонкой (температура смеси ниже 30°С, 60-70 мбар) до приблизительно 2 объемов. Добавляли 1-пропанол (1-PrOH, 2,1 л) и концентрировали смесь вакуумной перегонкой до приблизительно 2 объемов. Добавляли 1-пропанол (7,2 л) и нагревали смесь до приблизительно выше 55°C с получением раствора и затем охлаждали до 50-55°С и вносили затравку. Смесь охлаждали до 5-25°С и брали образец. Если хиральная ВЭЖХ твердого вещества показывала 49-51% диастереомерную смесь, то далее проводили фильтрование. Если хиральная ВЭЖХ твердого вещества показывала менее 49% - более 51% диастереомерную смесь, то необязательно добавляли гептан (1 л) и охлаждали смесь до приблизительно 0-10°С. Твердое вещество собирали фильтрацией и промывали гептаном (3 объема). Продукт сушили в вакууме при приблизительно 57°C с получением соединения (1) (выход 70-80%, площадь по ВЭЖХ более_97%, количественный анализ более_97% мас./мас., соотношение диастереомеров 49-51%). ЯМР (600 МГц, ДМСО) δ: 3,20 (с, 3Н), 4,40 (м, 2Н), 4,50 (шс, Ш), 4,60 (дк, 1H), 4,92 (шс, 1H), 5,99 (шс, 1H), 6,22 (т, 1Н), 7,52 (д, 2Н), 7,81 (д, 2Н), 8,13 (дд, 1Н), 8,39 (дд, 1Н), 8,90 (д, 1Н), 9,05 (д, 1Н). ЖХ-МС (m/z): М+Н = 402,1.
Как описано в настоящем документе, соединение формулы 1 представляет собой диастереомерную смесь
2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-((S)-S-метилсульфонимидоил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида (1а) и 2,2-дифтор-N-((1R,2S)-3-фтор-1-гидрокси-1-(4-(6-((R)-S-метилсульфонимидоил)пиридин-3-ил)фенил)пропан-2-ил)ацетамида (1b)
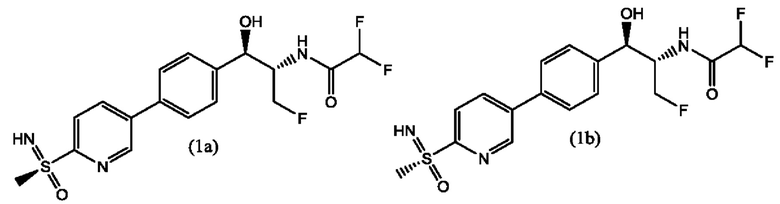
Предпочтительно, диастереомеры (формулы 1а и формулы 1b) получают в диастереомерной смеси в соотношении от приблизительно 48:52 (1a:1b) до 52:48 (1a:1b); и предпочтительно от приблизительно 49:51 (1a:1b) до 51:49 (1a:1b).
Различные твердые формы фармацевтического или ветеринарного соединения могут иметь существенно различные физические свойства. Такие различия физических свойств могут влиять, например, на способы получения фармацевтического или ветеринарного соединения, его переработки, составления лекарственных форм или введения. Например, кристаллические формы одного соединения могут иметь очень разные свойства: растворимость, скорость растворения, стабильность в суспензии, стабильность при измельчении, давление паров, оптические и механические свойства, гигроскопичность, размер кристаллов, свойства при фильтровании, высыхание, плотность, температура плавления, стойкость к разложению, стабильность против фазового превращения в другие кристаллические формы, цвет и даже химическая активность. В предпочтительном аспекте настоящего изобретения предложена определенная кристаллическая форма, предпочтительно форма А1 диастереомерной смеси соединения формулы (1).
Рентгеновский анализ кристаллов
Кристаллическую структуру, описанную в настоящем документе, анализировали методом порошковой рентгеновской дифракции (ПРД). Рентгеновские дифрактограммы получали на приборе Bruker D4 Endeavor, оснащенном детектором LynxEye, который эксплуатировали с щелью фиксированного размера и источником Cu, работающем при 40 кВ и 40 мА, длина волны K2a 1,5406 ангстрем. Дифрактограммы записывали в замкнутом режиме связанных колебаний, от 5 до 50 градусов два-тета. Размер шага составлял 0,020 градуса два-тета, а время записи одного шага составляло 0,5 секунды или 1 секунду. Щель расходимости устанавливали на 1,00 градус. Вычитали соответствующую холостую дифрактограмму. Во всех испытаниях использовали держатели с нулевым уровнем фона, образец распределяли по поверхности тонким плоским слоем. Все испытания проводили при контролируемых комнатных температуре и влажности в помещении (обычно 21-22°С, ОВ 25-50%). Во время записи держатель образца вращали при 20 об./мин. Данные анализировали в пакете программного обеспечения EVA компании Bruker.
Специалистам в области кристаллографии понятно, что относительные интенсивности различных пиков, указанные в таблицах 1-3 и на фигурах 1-3, соответственно, могут варьироваться вследствие многих факторов, таких как эффект ориентации кристаллов в рентгеновском луче или чистота анализируемого материала, или степень кристалличности образца. Положения пиков ПРД также могут смещаться в результате различной высоты образца, но положения пиков остаются по существу при указанных значениях. Специалистам в области кристаллографии также понятно, что измерения с использованием различных длин волн приводят к получению разных сдвигов в соответствии с уравнением Брэгга - nA, = 2ci sinΘ. Такие дополнительные диаграммы ПРД, полученные при использовании альтернативных длин волн, считаются альтернативными формами представления диаграмм ПРД кристаллических материалов согласно настоящему изобретению и, следовательно, входят в объем настоящего изобретения. Аналогично, изменение доли каждого диастереомера также влияет на интенсивность пика и потенциально на положение пика, если соотношение между двумя диастереомерами отклоняется от соотношения 1:1, как показано в таблице 4.
Диастереомерная смесь (формулы 1а и 1b) имеет уникальную трехмерную кристаллическую конфигурацию, которую можно охарактеризовать, среди прочего, дифракцией электромагнитного излучения (например, ПРД) данной кристаллической решеткой. Форма А1 (50,83 соединения 1а: 49,17 соединения 1b) представляет собой диастереомерную смесь соединений формулы (1а) и (1b) в соотношении 1:1; которая демонстрирует по существу такую диаграмму ПРД, как показано на фигуре 1. Характеристические пики формы 1А, выраженные в градусах 20 [2-тета°] (±0,2°), межплоскостные расстояния (расстояния d) и соответствующие интенсивности (%) представлены в таблице 1. Соответствующая диаграмма пиков ПРД и характеристики пиков формы А2 (47,46 соединения 1а: 52,54 соединения 1b) представлены на фигуре 2 и в таблице 2, соответственно. Соответствующая диаграмма пиков ПРД и характеристики пиков формы A3 (56,43 соединения 1а: 43,57 соединения 1b) представлены на фигуре 3 и в таблице 3, соответственно. Сравнительные пики ПРД в единицах 2-тета° с интенсивностью не менее 25% представлены в таблице 4. Как показано в таблицах и на фигурах, диаграмма пиков ПРД и интенсивность изменяются для формы А в зависимости от относительного количества соединений формулы 1а и 1b. Формы А1 и А2 имеют схожие характеристики ПРД, что объясняется относительной гомологией приблизительно 76%. В таблице 4 представлены аналогичные картины пиков с интенсивностью пиков не менее 25% для формы A1, А2 и A3 при приблизительно 19,24, 19,86 и 22,09 2-тета°. Как описано выше, соотношение диастереомеров в образце продукта обусловливает несколько различные диаграммы и интенсивности пиков ПРД, в соответствии с ожиданиями.

| название | год | авторы | номер документа |
|---|---|---|---|
| ТВЕРДЫЕ ФОРМЫ (R)-1-(2, 2-ДИФТОРБЕНЗО[d][1, 3]ДИОКСОЛ-5-ИЛ)-N-(1-(2, 3-ДИГИДРОКСИПРОПИЛ)-6-ФТОР-2-(1-ГИДРОКСИ-2-МЕТИЛПРОПАН-2-ИЛ)-1H-ИНДОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДА | 2011 |
|
RU2711481C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5-(1,2,4-ТРИАЗОЛ-1-ИЛ)ТРИПТАМИНОВЫХ СОЕДИНЕНИЙ И 2-[5-(1,2,4-ТРИАЗОЛ-1-ИЛ- МЕТИЛ)-1Н-ИНДОЛ-3-ИЛ/ЭТИЛОВЫЙ СПИРТ | 1995 |
|
RU2138496C1 |
| ПРОИЗВОДНЫЕ [4-(1-АМИНОЭТИЛ)ЦИКЛОГЕКСИЛ]МЕТИЛАМИНА И [6-(1-АМИНОЭТИЛ)ТЕТРАГИДРОПИРАН-3-ИЛ]МЕТИЛАМИНА | 2009 |
|
RU2515906C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-[1-ЭТИЛСУЛЬФОНИЛ-3-[4-(7H-ПИРРОЛО[2,3-D]ПИРИМИДИН-4-ИЛ)ПИРАЗОЛ-1-ИЛ]АЗЕТИДИН-3-ИЛ]АЦЕТОНИТРИЛА | 2024 |
|
RU2835443C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-[1-ЭТИЛСУЛЬФОНИЛ-3-[4-(7H-ПИРРОЛО[2,3-D]ПИРИМИДИН-4-ИЛ)ПИРАЗОЛ-1-ИЛ]АЗЕТИДИН-3-ИЛ]АЦЕТОНИТРИЛА | 2024 |
|
RU2835441C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-[1-ЭТИЛСУЛЬФОНИЛ-3-[4-(7H-ПИРРОЛО[2,3-D]ПИРИМИДИН-4-ИЛ)ПИРАЗОЛ-1-ИЛ]АЗЕТИДИН-3-ИЛ]АЦЕТОНИТРИЛА | 2024 |
|
RU2835445C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-[(3R)-3-МЕТИЛМОРФОЛИН-4-ИЛ]-4-(1-МЕТИЛ-1H-ПИРАЗОЛ-5-ИЛ)-8-(1H-ПИРАЗОЛ-5-ИЛ)-1,7-НАФТИРИДИНА | 2019 |
|
RU2802512C2 |
| СПОСОБ ПОЛУЧЕНИЯ (S)-N-(5-((R)-2-(2,5-ДИФТОРФЕНИЛ)ПИРРОЛИДИН-1-ИЛ)-ПИРАЗОЛО[1,5-A]ПИРИМИДИН-3-ИЛ)-3-ГИДРОКСИПИРРОЛИДИН-1-КАРБОКСАМИДА И ЕГО СОЛЕЙ | 2017 |
|
RU2745953C2 |
| ТВЕРДЫЕ ФОРМЫ (R)-1-(2,2-ДИФТОРБЕНЗО[d][1,3]ДИОКСОЛ-5-ИЛ)-N-(2,3-ДИГИДРОКСИПРОПИЛ)-6-ФТОР-2-(1-ГИДРОКСИ-2-МЕТИЛПРОПАН-2-ИЛ)-1H-ИНДОЛ-5-ИЛ)ЦИКЛОПРОПАНКАРБОКСАМИДА | 2011 |
|
RU2573830C2 |
| Способ получения соединений 7H-пирроло[2,3-d]пиримидина | 2017 |
|
RU2699034C1 |

Изобретение относится к способу получения соединения формулы (1) в виде смеси диастереомеров, которое может найти применение для лечения бактериальных инфекций у животных. Способ включает стадии а)-г). На стадии а) осуществляют предварительную активацию соединения формулы (В) на палладиевом катализаторе в присутствии спирта, лиганда, выбранного из группы, состоящей из 2-дициклогексилфосфино-2',4',6'-триизопропилбифенила, 2-дициклогексилфосфино-2,6-диметоксибифенила, 1,3-бис(дифенилфосфино)пропана, ферроцендиил-бис(дифенилфосфина), дибензилиденацетона, трифенилфосфина и трициклогексилфосфина, и буфера для борилирования, который содержит кислоту и основание, причем указанная кислота выбрана из группы, состоящей из уксусной кислоты, лимонной кислоты, муравьиной кислоты и хлоруксусной кислоты, а основание выбрано из группы, состоящей из ацетата калия, ацетата цезия, триэтиламина, карбоната калия, карбоната натрия, карбоната цезия, диизопропилэтиламина и трехосновного фосфата калия и их смесей. На стадии б) осуществляют борилирование предварительно активированного соединения формулы (В) со стадии а) посредством добавления борилирующего агента в спирте, причем борилирующий агент выбран из группы, состоящей из бисбороновой кислоты, бисбороновой кислоты и этиленгликоля, бисбороновой кислоты и пропиленгликоля, бис(пинаколато)дибора и тетракис(диметиламино)дибора. На стадии в) смешивают соединение формулы (А) с основанием буфера для борилирования в спирте, сорастворителе или с их смесями и на стадии г) добавляют реагенты со стадии в) к реагентам со стадии б) с получением соединения формулы (1). Предлагаемый способ включает меньшее количество химических стадий и обеспечивает более высокий выход. Изобретение относится также к варианту указанного способа. 2 н. и 10 з.п. ф-лы, 3 ил., 4 табл.
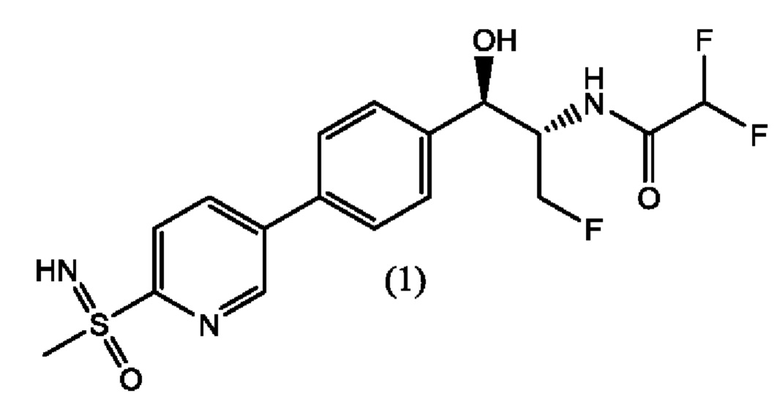
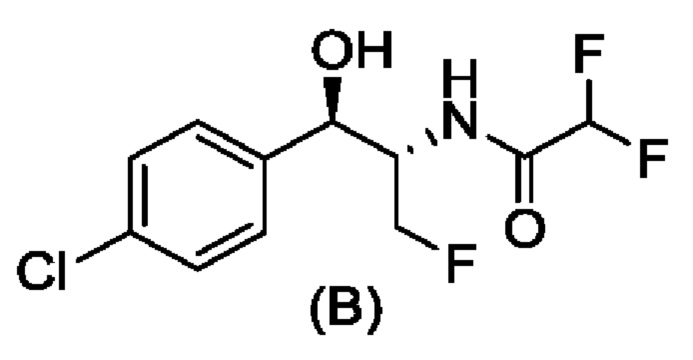
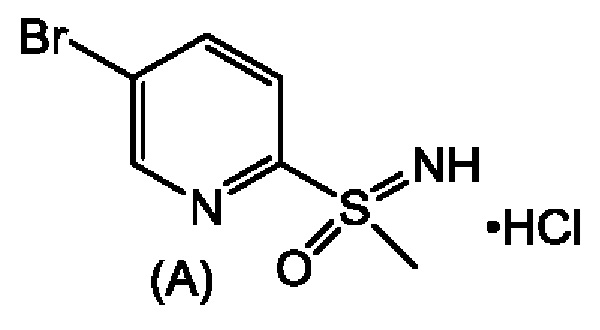
1. Способ получения соединения формулы (1) в виде смеси диастереомеров
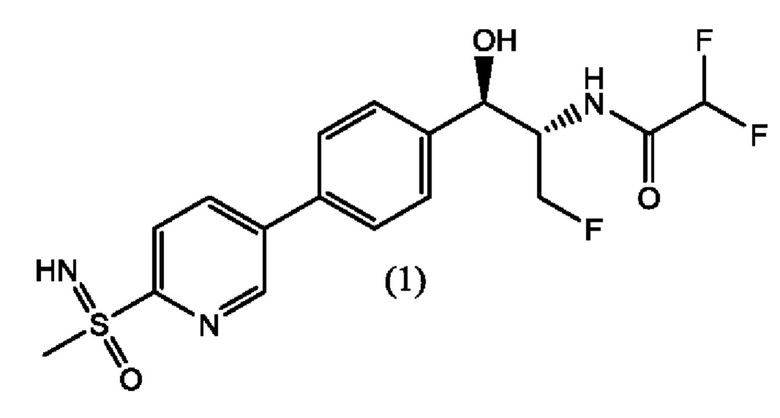 ,
,
включающий стадии:
а) предварительной активации соединения формулы (В) на палладиевом катализаторе в присутствии спирта, лиганда, выбранного из группы, состоящей из 2-дициклогексилфосфино-2',4',6'-триизопропилбифенила, 2-дициклогексилфосфино-2,6-диметоксибифенила, 1,3-бис(дифенилфосфино)пропана, ферроцендиил-бис(дифенилфосфина), дибензилиденацетона, трифенилфосфина и трициклогексилфосфина, и буфера для борилирования, который содержит кислоту и основание, причем указанная кислота выбрана из группы, состоящей из уксусной кислоты, лимонной кислоты, муравьиной кислоты и хлоруксусной кислоты, а основание выбрано из группы, состоящей из ацетата калия, ацетата цезия, триэтиламина, карбоната калия, карбоната натрия, карбоната цезия, диизопропилэтиламина и трехосновного фосфата калия, и их смесей;
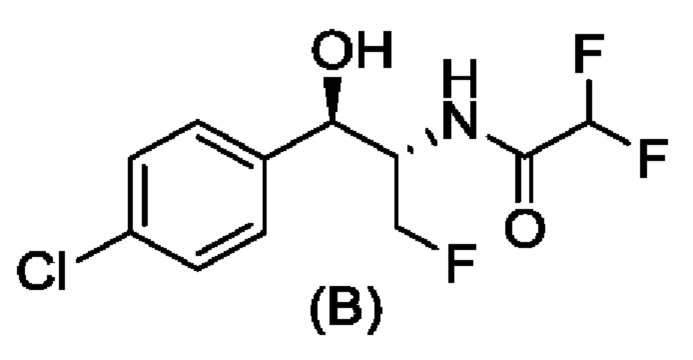
б) борилирования предварительно активированного соединения формулы (В) со стадии а) посредством добавления борилирующего агента в спирте, причем борилирующий агент выбран из группы, состоящей из бисбороновой кислоты, бисбороновой кислоты и этиленгликоля, бисбороновой кислоты и пропиленгликоля, бис(пинаколато)дибора и тетракис(диметиламино)дибора;
в) смешивания соединения формулы (А) с основанием буфера для борилирования в спирте, сорастворителе или с их смесями и
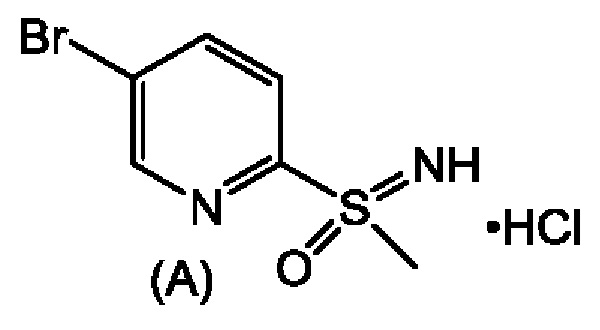
г) добавления реагентов со стадии в) к реагентам со стадии б) с получением соединения формулы (1).
2. Способ по п. 1, включающий стадии:
а) предварительной активации соединения формулы (В) на палладиевом катализаторе, выбранном из группы, состоящей из ацетата палладия (II), хлорида палладия (II), предкатализатора хлорид аминобифенилпалладия, хлор(кротил)(2-дициклогексилфосфино-2,4,6-триизопропилбифенил)палладия (II), бис(дифенилфосфино)ферроцендихлорпалладия, предкатализатора хлорид трициклогексилфосфинаминобифенилпалладия и трис(дибензилиденацетон)дипалладия, в присутствии спирта, лиганда и буфера для борилирования;
б) борилирования предварительно активированного соединения формулы (В) со стадии а) посредством добавления борилирующего агента в спирте;
в) смешивания соединения формулы (А) с основанием буфера для борилирования в спирте, сорастворителе или с их смесями и
г) смешивания реагентов со стадии в) с реагентами со стадии б) с получением соединения формулы (1).
3. Способ по п. 1, отличающийся тем, что спирт на стадиях а), б) и в) выбран из группы, состоящей из метанола, этанола, 1-пропанола, 2-пропанола и 2-бутанола.
4. Способ по п. 3, отличающийся тем, что спирт на стадиях а) и б) представляет собой безводный этанол и спирт на стадии в) представляет собой водный этанол.
5. Способ по п. 1, отличающийся тем, что сорастворитель на стадии в) выбран из группы, состоящей из изопропилацетата, этилацетата, диметилформамида, диметоксиэтана, тетрагидрофурана, 2-метилтетрагидрофурана, ацетонитрила и их смесей.
6. Способ по п. 1, включающий стадии:
а) предварительной активации соединения формулы (В) на палладиевом катализаторе, который представляет собой ацетат палладия (II), в присутствии безводного этанола, лиганда, который представляет собой 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил, и буфера для борилирования, содержащего кислоту, уксусную кислоту, и основание, ацетат калия;
б) борилирования предварительно активированного соединения формулы (В) со стадии а) посредством добавления борилирующего агента бисбороновой кислоты и этиленгликоля в безводном этаноле;
в) смешивания соединения формулы (А) с карбонатом калия или карбонатом натрия в водном тетрагидрофуране и
г) смешивания реагентов со стадии в) с реагентами со стадии б) с получением соединения формулы (1) и его диастереомеров; и при этом растворы, добавляемые в реакционную смесь на стадиях а)-в), продувают N2 или Ar и реакции на стадиях а)-г) проводят в инертной атмосфере N2 или Ar.
7. Способ получения соединения формулы (1) в виде смеси диастереомеров
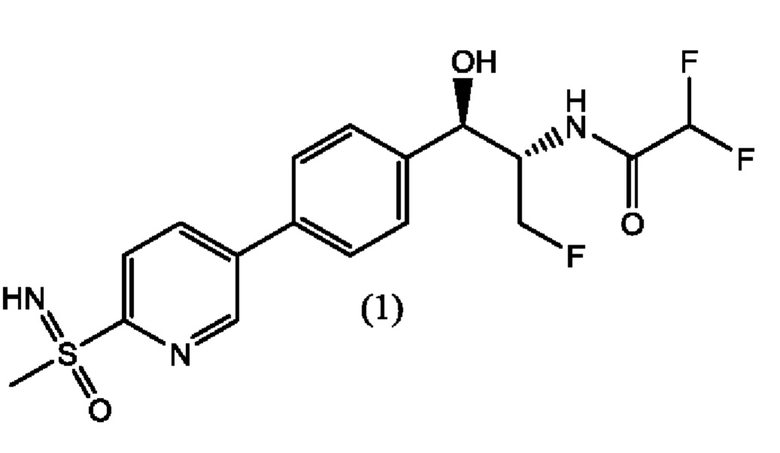 ,
,
включающий стадии:
а) предварительной активации соединения формулы (В) на палладиевом катализаторе в присутствии спирта, лиганда, выбранного из группы, состоящей из 2-дициклогексилфосфино-2',4',6'-триизопропилбифенила, 2-дициклогексилфосфино-2,6-диметоксибифенила, 1,3-бис(дифенилфосфино)пропана, ферроцендиил-бис(дифенилфосфина), дибензилиденацетона, трифенилфосфина и трициклогексилфосфина, и буфера для борилирования, который содержит кислоту и основание, причем указанная кислота выбрана из группы, состоящей из уксусной кислоты, лимонной кислоты, муравьиной кислоты и хлоруксусной кислоты, а основание выбрано из группы, состоящей из ацетата калия, ацетата цезия, триэтиламина, карбоната калия, карбоната натрия, карбоната цезия, диизопропилэтиламина и трехосновного фосфата калия, и их смесей;
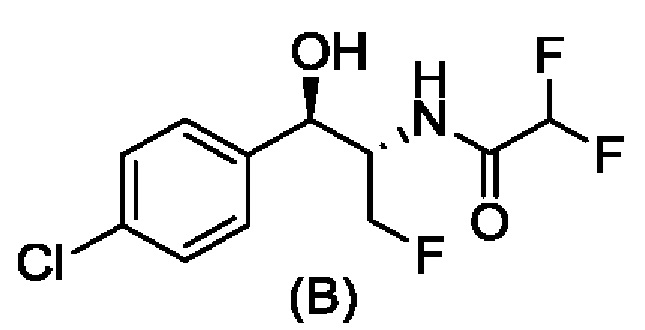
б) борилирования предварительно активированного соединения формулы (В) со стадии а) посредством добавления борилирующего агента, выбранного из группы, состоящей из бисбороновой кислоты, бисбороновой кислоты и этиленгликоля, бисбороновой кислоты и пропиленгликоля, бис(пинаколато)дибора и тетракис(диметиламино)дибора, в спирте к реагентам со стадии а);
в) смешивания соединения формулы (А) с основанием буфера для борилирования в спирте, сорастворителе или с их смесями;
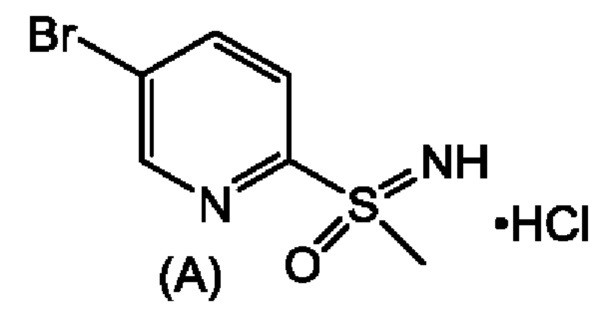
г) добавления реагентов со стадии в) к реагентам со стадии б) с получением соединения формулы (1);
д) очистки соединения формулы (1) со стадии г) посредством концентрирования продукта реакции со стадии г) и экстракции соединения формулы (1) в экстрагирующий растворитель;
е) добавления промывочного водного раствора к экстрагирующему растворителю со стадии д), перемешивания экстракционной смеси, промывания и отделения органического слоя;
ж) добавления поглотителя палладия в органический слой со стадии е) или рециркуляции органического слоя со стадии е) через картридж из поглотителя металла, перемешивания и отфильтровывания твердых веществ, промывания твердых веществ экстрагирующим(и) растворителем(ями) и концентрирования фильтрата;
з) растворения полученного концентрата со стадии ж) в органическом(их) растворителе(ях) при нагревании, охлаждения и внесения затравки соединения формулы (1) в полученную смесь;
и) охлаждения смеси со стадии з), добавления необязательного антирастворителя, сбора полученных твердых веществ фильтрованием, промывания твердых веществ антирастворителем и затем высушивания твердых веществ.
8. Способ по п. 7, включающий стадии:
а) предварительной активации соединения формулы (В) на палладиевом катализаторе, выбранном из группы, состоящей из ацетата палладия (II), хлорида палладия (II), предкатализатора хлорид аминобифенилпалладия, хлор(кротил)(2-дициклогексилфосфино-2,4,6-триизопропилбифенил)палладия (II), бис(дифенилфосфино)ферроцендихлорпалладия, предкатализатора хлорид трициклогексилфосфинаминобифенилпалладия и трис(дибензилиденацетон)дипалладия, в присутствии спирта, лиганда и буфера для борилирования, который содержит кислоту и основание;
б) борилирования предварительно активированного соединения формулы (В) со стадии а) посредством добавления борилирующего агента в спирте;
в) смешивания соединения формулы (А) с основанием буфера для борилирования в спирте, сорастворителе или с их смесями; и при этом спирт на стадиях а) и б) представляет собой безводный этанол;
г) смешивания реагентов со стадии в) с реагентами со стадии б) с получением соединения формулы (1);
д) очистки соединения формулы (1) со стадии г) посредством концентрирования продукта реакции со стадии г) и экстракции соединения формулы (1) в экстрагирующий растворитель, выбранный из группы, состоящей из тетрагидрофурана, этилацетата, метилацетата, метиленхлорида и 2-метилтетрагидрофурана;
е) добавления к экстракционной смеси со стадии д) промывочного водного раствора, выбранного из воды или солевого раствора, каждый из которых содержит соединение, образующее хелат с палладием, выбранное из группы, состоящей из этилендиамина, натриевой соли тримеркаптотриазина, гидроксида аммония, тримеркаптотриазина, бисульфита натрия, тиомочевины, диэтиламина, этилендиаминтетрауксусной кислоты, ацетил-L-цистеина, лимонной кислоты и их смесей, и отделения органического слоя;
ж) добавления поглотителя палладия в органический слой со стадии е) или рециркуляции органического слоя со стадии е) через картридж из поглотителя металла, выбранный из группы, состоящей из угля с этилендиамином, силикагеля с этилендиамином, Siliamets тиола, Quadrapure TU, тримеркаптотриазина, связанного с микропористым полистиролом, тримеркаптотриазина, связанного с диоксидом кремния, Siliamets димеркаптотриазина и Siliamets цистеина, отфильтровывания твердых веществ, промывания твердых веществ экстракционным(и) растворителем(ями) и концентрирования фильтрата;
з) растворения полученного концентрата со стадии ж) в органическом растворителе, выбранном из группы, состоящей из метилэтилкетона, изопропилацетата, этилацетата, ацетона, 1-бутанола, 1-пропанола, 2-пропанола и их смесей, при нагревании; охлаждения и внесения затравки соединения формулы (1) в полученный раствор и
и) охлаждения смеси со стадии з), необязательно добавления антирастворителя, выбранного из группы, состоящей из воды, метил-трет-бутилового эфира, гексана, гептана и их смесей; сбора полученных твердых веществ фильтрованием, промывания твердых веществ антирастворителем и затем высушивания твердых веществ с получением диастереомерной смеси 1:1 соединения формулы (1).
9. Способ по п. 8, отличающийся тем, что сорастворитель выбран из группы, состоящей из изопропилацетата, этилацетата, диметилформамида, диметоксиэтана, тетрагидрофурана, 2-метилтетрагидрофурана, ацетонитрила и их смесей; и при этом палладиевый катализатор на стадии а) представляет собой ацетат палладия (II); и при этом лиганд на стадии а) представляет собой 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил, и буфер для борилирования на стадии а) представляет собой спиртовой раствор, содержащий уксусную кислоту и ацетат калия; и при этом борилирующий агент на стадии б) представляет собой бисбороновую кислоту и этиленгликоль в безводном этаноле.
10. Способ по п. 9, отличающийся тем, что основание на стадии в) выбрано из группы, состоящей из карбоната калия, карбоната натрия, триэтиламина и их смесей.
11. Способ по п. 10, отличающийся тем, что экстрагирующий растворитель на стадии д) представляет собой тетрагидрофуран; и при этом органические слои со стадии д) промывают 1, 2, 3 или 4 раза промывочным водным раствором; и при этом поглотитель палладия на стадии ж) представляет собой уголь с этилендиамином, а органический растворитель выбран из группы, состоящей из метилэтилкетона, изопропанола, этилацетата, ацетона, 1-бутанола, 1-пропанола, 2-пропанола и их смесей; и при этом антирастворитель на стадии з) выбран из группы, состоящей из воды, метил-трет-бутилового эфира, гексана, гептана и их смесей; и растворы, добавляемые в реакционную смесь на стадиях а)-в), продувают N2 или Ar, и реакции на стадиях а)-г) проводят в атмосфере N2 или Ar.
12. Способ по п. 11, отличающийся тем, что органический растворитель представляет собой метилэтилкетон, 1-пропанол или их смесь и антирастворитель представляет собой гептан.
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| ФЕНИКОЛОВЫЕ ПРОТИВОБАКТЕРИАЛЬНЫЕ СРЕДСТВА | 2013 |
|
RU2593204C2 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |

