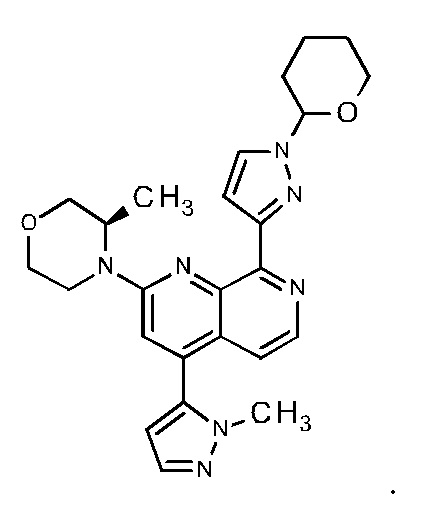
Настоящее изобретение охватывает способ получения 2-[(3R)-3-метилморфолин-4-ил]-4-(1-метил-1Н-пиразол-5-ил)-8-(1Н-пиразол-5-ил)-1,7-нафтиридина ("соединение формулы (I)" в дальнейшем), а также промежуточные соединения, пригодные для получения соединения формулы (I). Настоящее изобретение также охватывает полиморфную форму В соединения формулы (I) с очень высокой чистотой.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Пример 111 публикации WO 2016020320 A1 описывает способ синтеза 2-[(3R)-3-метилморфолин-4-ил]-4-(1-метил-1Н-пиразол-5-ил)-8-(1Н-пиразол-5-ил)-1,7-нафтиридина формулы (I) с использованием следующего пути синтеза:
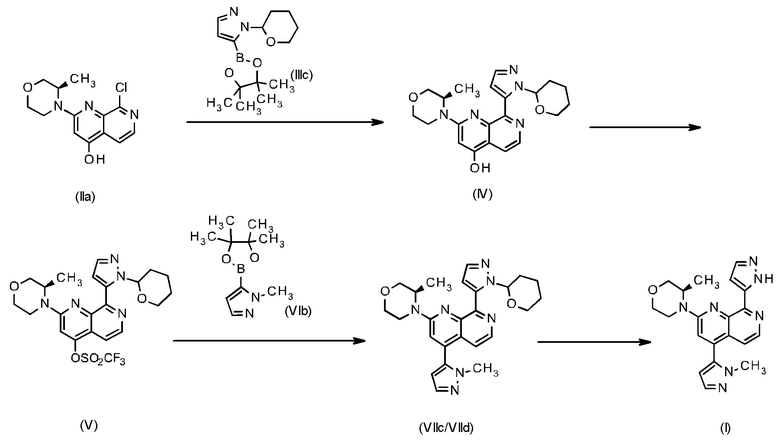
Синтез соединения (IIa) (=8-хлор-2-((R)-3-метилморфолин-4-ил)-[1,7]нафтиридин-4-ол) описан в публикации WO 2016020320 A1 в Примере "Промежуточное соединение-7", стадия с.
В соответствии с WO 2016020320 A1 "Промежуточное соединение-9", которое соответствует соединению (IV) (=2-[((R)-3-метилморфолин-4-ил)-8-[2-(тетрагидропиран-2-ил)-2Н-пиразол-3-ил]-[1,7]нафтиридин-4-ол), было получено по реакции сочетания Сузуки из соединения формулы (IIa) и тетрагидропиранил-защищенного сложного боронового эфира - соединения (IIIc) (=1-(тетрагидропиран-2-ил)-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-пиразол) в атмосфере аргона с использованием комплекса [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (1:1) в качестве катализатора и карбоната цезия в абсолютном 1,4-диоксане. Реакционную смесь перемешивали при 90°С в течение 16 ч. Коричневый реакционный раствор очищали с помощью колоночной хроматографии [силикагель 60; этилацетат)]. 506 мг (выход 72% от теоретического) 2-[(R)-3-метилморфолин-4-ил)-8-[2-(тетрагидропиран-2-ил)-2Н-пиразол-3-ил]-[1,7]нафтиридин-4-ола отделяли в виде желтого масла.
По следующим причинам, приводимым в качестве примера, способ получения соединения формулы (IV) в лабораторном масштабе, который описан в WO 2016020320 A1, непригоден для применения в качестве способа крупномасштабного производства:
• Применение абсолютного растворителя (например, 1,4-диоксана) является затруднительным с точки зрения обращения с ним в крупном масштабе.
• Карбонат цезия является относительно дорогим неорганическим основанием.
• Требуется большое время реакции (16 ч при 90°С).
• Соединение формулы (IV) представляет собой масло, поэтому оно не может быть очищено с помощью не требующей особых усилий стадии кристаллизации, которая является предпочтительным методом для производства в крупном масштабе.
• Очистка с помощью колоночной хроматографии требует много времени и больших затрат.
В соответствии с WO 2016020320 A1 "Промежуточное соединение 9", маслянистое соединение формулы (IV), затем превращают в "Промежуточное соединение 10", которое соответствует соединению формулы (V) (=2-[(3R)-3-метилморфолин-4-ил]-8-[1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-ил]-1,7-нафтиридин-4-ил трифторметансульфонат) путем обработки N-фенилбис-(трифторметансульфонимидом) и N,N-диизопропилэтиламином в атмосфере аргона в абсолютном дихлорметане. Время реакции при комнатной температуре составляло три дня. Растворитель отгоняли при пониженном давлении и остаток два раза хроматографировали [силикагель 60 (400 г); дихлорметан : метанол, 98:21 этилацетат]. После упаривания досуха соединение формулы (V) получали с выходом 2.6 г (42% от теоретического) в виде желтого твердого вещества.
Недостатками этой методики являются, например:
• Применение абсолютного растворитель (требует больших затрат).
• Очень большое время реакции (три дня при комнатной температуре), что требует больших затрат.
• Две стадии хроматографической очистки, которые требуют очень много времени и больших затрат.
• Очень низкий выход (42%) для этой стадии.
• Выделение путем упаривания хроматографических фракций. Такая операция не представляется возможной для увеличенного масштаба, поскольку она очень энергоемкая и дорогостоящая.
Соединение формулы (V) превращали в соединение формулы (VIIc/VIId) по реакции Сузуки, используя 1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (VIb), водн. раствор карбоната калия (1.4 мл, 2 М) и дихлорид бис(трифенилфосфин)палладия(II) (67 мг, 0.094 ммоль), которые растворяли в диметоксиэтане (60 мл). Реакционную смесь перемешивали в течение 20 минут при 130°С при микроволновом облучении. После охлаждения до комнатной температуры реакционную смесь фильтровали через силиконовый фильтр и концентрировали при пониженном давлении. Сырое вещество очищали с помощью колоночной флэш-хроматографии (используя смесь гексан/этилацетат/этанол). Целевые фракции концентрировали при пониженном давлении и растворяли в конц. серной кислоте (5 мл). Смесь перемешивали в течение 3 ч при комнатной температуре. Смесь затем выливали на лед и подщелачивали, используя твердый гидрокарбонат натрия. Суспензию фильтровали и твердое вещество перемешивали с этанолом при 40°С, фильтровали и сушили при пониженном давлении. Соединение формулы (I) получали с выходом 78% (0.28 г).
По меньшей мере следующие моменты являются критическими для увеличенного масштаба:
• Использование микроволнового реактора для увеличенного масштаба не представляется возможным. Проведение крупномасштабной реакции при 130°С всего за 20 минут является сложным и не может быть реализовано на мультикилограммовых масштабах.
• Выполнение хроматографии для выделения и очистки требует много времени и больших затрат в случае увеличенного масштаба.
• Упаривание содержащих соединение хроматографических фракций в крупном масштабе не представляется возможным, поскольку это очень энергоемкая и дорогостоящая операция.
• Чистота соединения формулы (I) не соответствует требованиям GMP (надлежащая практика производства лекарственных средств).
Подводя итог, следует отметить, что описанный способ получения целевого соединения (I) исходя из соединения формулы (IIa) (=8-хлор-2-((R)-3-метилморфолин-4-ил)-[1,7]нафтиридин-4-ол) является очень неэффективным, требует много времени и больших затрат, поскольку включает три хроматографических стадии и приводит к большому времени реакции и к очень низкому общему выходу (из соединения формулы (II) к соединению (I): теоретический выход 23.6%).
Таким образом, задача настоящего изобретения заключалась в обеспечении способа получения 2-[(3R)-3-метилморфолин-4-ил]-4-(1-метил-1Н-пиразол-5-ил)-8-(1Н-пиразол-5-ил)-1,7-нафтиридина формулы (I), который не обладает одним или несколькими из вышеупомянутых недостатков.
ОПРЕДЕЛЕНИЯ
Термин "замещенный" означает, что один или несколько атомов водорода на обозначенном(-й) атоме или группе замещен(-ы) заместителем(-ями), выбранным(-и) из указанной группы, при условии, что нормальная валентность обозначенного атома при существующих обстоятельствах не превышается. Допустимы комбинации заместителей и/или переменных.
Термин "необязательно замещенный" означает, что число заместителей может быть равно нулю или отличным от нуля. Если не указано иначе, необязательно замещенные группы могут быть замещены таким числом необязательных заместителей, какое можно разместить путем замены каждого атома водорода заместителем, не являющимся водородом, на любом доступном атоме углерода или азота. Обычно, число необязательных заместителей, если они присутствуют, может быть равно 1, 2, 3, 4 или 5, в частности, 1, 2 или 3.
В контексте настоящего документа, термин "один или несколько", например, в определении заместителей соединений общей формулы (I) настоящего изобретения, означает 1, 2, 3, 4 или 5, в частности, 1, 2, 3 или 4, более конкретно, 1, 2 или 3, еще более конкретно, 1 или 2.
Термин "включающий" при использовании в описании включает "состоящий из".
Если в настоящем тексте любой элемент упоминается в виде "как упомянуто в данной заявке", это означает, что он может быть упомянут в любом месте настоящего текста.
Термины, упомянутые в настоящем тексте, имеют следующие значений:
Термин "C1-С6-алкил" означает линейную или разветвленную, насыщенную, одновалентную углеводородную группу, содержащую 1, 2, 3, 4, 5 или 6 атомов углерода, например, метильную, этильную, пропильную, изопропильную, бутильную, втор-бутильную, изобутильную, трет-бутильную, пентильную, изопентильную, 2-метилбутильную, 1-метилбутильную, 1-этилпропильную, 1,2-диметилпропильную, нео-пентильную, 1,1-диметилпропильную, гексильную, 1-метилпентильную, 2-метилпентильную, 3-метилпентильную, 4-метилпентильную, 1-этилбутильную, 2-этилбутильную, 1,1-диметилбутильную, 2,2-диметилбутильную, 3,3-диметилбутильную, 2,3-диметилбутильную, 1,2-диметилбутильную или 1,3-диметилбутильную группу, или ее изомер. В частности, указанная группа содержит 1, 2, 3 или 4 атома углерода ("С1-С4-алкил"), например, представляет собой метильную, этильную, пропильную, изопропильную, бутильную, втор-бутильную, изобутильную или трет-бутильную группу, более конкретно, 1, 2 или 3 атома углерода ("С1-С3-алкил"), например, представляет собой метильную, этильную, н-пропильную или изопропильную группу.
Используемый в настоящем тексте термин "C1-C6", например, в контексте определения "C1-С6-алкила", означает алкильную группу, имеющую конечное число атомов углерода от 1 до 6, т.е. 1, 2, 3, 4, 5 или 6 атомов углерода.
В случаях, когда задан диапазон значений, указанный диапазон охватывает каждое значение и поддиапазон в пределах указанного диапазона.
Например:"С1-С6" охватывает C1, С2, С3, С4, С5, С6, C1-С6, С1-С5, С1-С4, C1-С3, С1-С2, С2-С6, С2-С5, С2-С4, С2-С3, С3-С6, С3-С5, С3-С4, С4-С6, С4-С5 и С5-С6.
Соединение формулы (I) может существовать в виде таутомера формулы (Ia)
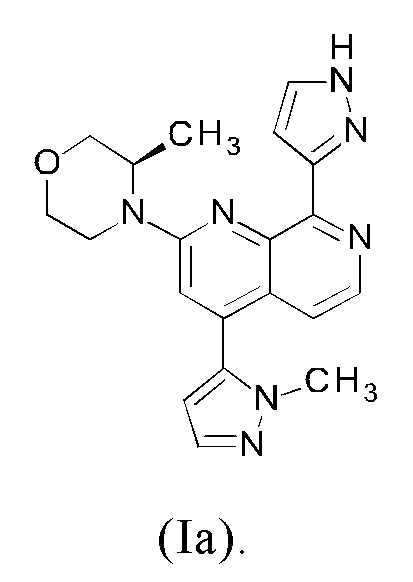
Специалисту в данной области также известно, что соединения формулы (I) и (Ia) могут существовать в виде смесей обоих таутомеров (I) и (Ia).
Соединения настоящего изобретения формулы (VIIIa) или (IXa) необязательно содержат более одного, в частности, два, асимметричных центра, в зависимости от расположения и природы различных требуемых заместителей. При этом, один или несколько асимметричных атомов углерода могут присутствовать в (R) или (S) конфигурации, что может привести к рацемическим смесям в случае одного асимметричного центра, и диастереомерным смесям в случае нескольких асимметричных центров.
Разделенные, чистые или частично очищенные изомеры и стереоизомеры или рацемические или диастереомерные смеси соединений настоящего изобретения также включены в объем настоящего изобретения. Очистку и разделение таких веществ можно выполнить с помощью стандартных методик, известных в данной области. Такие разделенные, чистые или частично очищенные изомеры или рацемические смеси соединений данного изобретения также включены в объем настоящего изобретения. Очистку и разделение таких веществ можно выполнить с помощью стандартных методик, известных в данной области.
Оптические изомеры можно получить путем разделения рацемических смесей в соответствии с обычными способами, например, путем образования диастереоизомерных солей с использованием оптически активной кислоты или основания, или образования ковалентных диастереомеров. Примерами подходящих кислот являются винная, диацетилвинная, дитолуоилвинная и камфорсульфоновая кислота. Смеси диастереоизомеров могут быть разделены на их отдельные диастереомеры на основе их физических и/или химических различий с помощью методов, известных в данной области, например, с помощью хроматографии или фракционной кристаллизации. Оптически активные основания или кислоты затем высвобождают из разделенных диастереоизомерных солей. Другой способ разделения оптических изомеров включает использование хиральной хроматографии (например, ВЭЖХ колонок с хиральной фазой), с обычной дериватизацией, оптимально выбранной для максимального разделения энантиомеров, или без нее. Подходящие ВЭЖХ колонки с хиральной фазой являются коммерчески доступными, в частности, такие колонки производит фирма Daicel, например, Chiracel OD и Chiracel OJ, например, среди многих других, обычно выбираемых. Также пригодны методы ферментативного разделения, с дериватизацией или без нее. Оптически активные соединения настоящего изобретения также можно получить с помощью хирального синтеза, используя оптически активные исходные вещества.
С целью разграничить друг от друга различные типы изомеров дается ссылка на правила IUPAC, раздел Е (Pure Appl Chem 45, 11-30, 1976).
Настоящее изобретение включает все возможные стереоизомеры соединений настоящего изобретения формулы (VIIIa) или (IXa) в виде отдельных стереоизомеров, или в виде любой смеси указанных стереоизомеров, например, (R)- или (S)- изомеров, в любом соотношении. Выделения отдельного стереоизомера, например, отдельного энантиомера или отдельного диастереомера, соединения настоящего изобретения достигают с помощью любого подходящего метода уровня техники, такого как, например, хроматография, в особенности хиральная хроматография.
Более того, соединения настоящего изобретения могут существовать в виде N-оксидов, которые определяются тем, что по меньшей мере один атом азота соединений настоящего изобретения окислен. Настоящее изобретение включает все такие возможные N-оксиды.
Настоящее изобретение также охватывает пригодные формы соединений настоящего изобретения формулы (VIIIa) или (IXa), такие как гидраты, сольваты, соли, в частности, фармацевтически приемлемые соли, и/или продукты совместного осаждения.
Более того, соединения настоящего изобретения формулы (VIIIa) или (IXa) могут существовать в свободной форме, например, в виде свободного основания, или в виде свободной кислоты, или в виде цвиттериона, или могут существовать в форме соли. Указанная соль может быть любой солью, либо органической, либо неорганической солью присоединения, в частности, любой фармацевтически приемлемой органической или неорганической солью присоединения, которую обычно используют в фармацевтике, или которую используют, например, для выделения или очистки соединений настоящего изобретения.
Термин "фармацевтически приемлемая соль" относится к соли присоединения неорганической или органической кислоты к соединению настоящего изобретения формулы (VIIIa) или (IXa). Например, см. S.М. Berge и др. "Pharmaceutical Salts," J. Pharm. Sci. 1977, 66, 1-19.
Подходящая фармацевтически приемлемая соль соединения настоящего изобретения формулы (VIIIa) или (IXa) может представлять собой, например, соль присоединения кислоты к соединению настоящего изобретения, несущему атом азота, в цепи или в кольце, например, который является достаточно основным, такую как соль присоединения неорганической кислоты, или "минеральной кислоты", такой как, например, хлористоводородная, бромистоводородная, йодистоводородная, серная, сульфаминовая, бисерная, фосфорная или азотная кислота, или органической кислоты, такой как, например, муравьиная, уксусная, ацетоуксусная, пировиноградная, трифторуксусная, пропионовая, масляная, гексановая, гептановая, ундекановая, лауриновая, бензойная, салициловая, 2-(4-гидроксибензоил)-бензойная, камфорная, коричная, циклопентанпропионовая, диглюконовая, 3-гидрокси-2-нафтойная, никотиновая, памоевая, пектиновая, 3-фенилпропионовая, пивалевая, 2-гидроксиэтансульфоновая, итаконовая, трифторметансульфоновая, додецилсерная, этансульфоновая, бензолсульфоновая, пара-толуолсульфоновая, метансульфоновая, 2-нафталинсульфоновая, нафталиндисульфоновая, камфорсульфоновая кислота, лимонная, винная, стеариновая, молочная, щавелевая, малоновая, янтарная, яблочная, адипиновая, альгиновая, малеиновая, фумаровая, D-глюконовая, миндальная, аскорбиновая, глюкогептановая, глицерофосфорная, аспарагиновая, сульфосалициловая или тиоциановая кислота.
Более того, другая подходящая фармацевтически приемлемая соль соединения настоящего изобретения формулы (VIIIa) или (IXa), которое является достаточно кислым, представляет собой соль щелочного металла, например, соль натрия или калия, соль щелочноземельного металла, например, соль кальция, магния или стронция, или соль алюминия или цинка, или соль аммония, полученную из аммиака или из органического первичного, вторичного или третичного амина, содержащего от 1 до 20 атомов углерода, такого как этиламин, диэтиламин, триэтиламин, этилдиизопропиламин, моноэтаноламин, диэтаноламин, триэтаноламин, дициклогексиламин, диметиламиноэтанол, диэтиламиноэтанол, трис(гидроксиметил)аминометан, прокаин, дибензиламин, N-метилморфолин, аргинин, лизин, 1,2-этилендиамин, N-метилпиперидин, N-метилглюкамин, N,N-диметилглюкамин, N-этилглюкамин, 1,6-гександиамин, глюкозамин, саркозин, серинол, 2-амино-1,3-пропандиол, 3-амино-1,2-пропандиол, 4-амино-1,2,3-бутантриол, или соль с четвертичным аммониевым ионом, содержащим от 1 до 20 атомов углерода, таким как тетраметиламмоний, тетраэтиламмоний, тетра(н-пропил)аммоний, тетра(н-бутил)аммоний, N-бензил-N,N,N-триметиламмоний, холин или бензалконий.
Специалистам в данной области также понятно, что соли присоединения кислоты к заявленным соединениям формулы (VIIIa) или (IXa) можно получить по реакции соединений с подходящей неорганической или органической кислотой с помощью любого из ряда известных методов. Альтернативно, соли щелочных и щелочноземельных металлов с кислыми соединениями настоящего изобретения формулы (VIIIa) или (IXa) получают по реакции соединений настоящего изобретения с подходящим основанием с помощью ряда известных методов.
Настоящее изобретение включает все возможные соли соединений настоящего изобретения формулы (VIIIa) или (IXa) в виде отдельных солей, или в виде любой смеси указанных солей, в любом соотношении.
Если не указано иное, дополнения к химическим названиям или структурным формулам, относящимся к солям, такие как "гидрохлорид", "трифторацетат", "натриевая соль" или "х HCl", "х CF3COOH", "х Na+", например, означают солевую форму, стехиометрия которой точно не определена.
Более того, настоящее изобретение включает все возможные кристаллические формы, или полиморфы, соединений настоящего изобретения формулы (VIIIa) или (IXa), либо в виде отдельных полиморфов, либо в виде смеси более чем одного полиморфа, в любом соотношении.
Соединения формулы (VIIIa) или (IXa) могут существовать в виде изотопных вариантов. Таким образом, изобретение включает один или несколько изотопных вариантов соединений формулы (VIIIa) или (IXa), в частности, содержащих дейтерий вариантов соединений формулы (VIIIa) или (IXa).
Термин "изотопный вариант" соединения или реагента определяют как соединение, демонстрирующее не природное относительное содержание одного или нескольких изотопов, которые составляют такое соединение.
Термин "изотопный вариант соединения формулы (VIIIa) или (IXa)" определяют как соединение формулы (VIIIa) или формулы (IXa), демонстрирующее не природное относительное содержание одного или нескольких изотопов, которые составляют такое соединение.
Выражение "не природное относительное содержание" означает относительное содержание такого изотопа, которое является более высоким, чем его распространенность в природе. Сведенья касательно распространенностей изотопов в природе, упоминаемых в данном контексте, описаны в документе "Isotopic Compositions of the Elements 1997", Pure Appl. Chem., 70(1), 217-235, 1998.
Примеры таких изотопов включают стабильные и радиоактивные изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, хлора, брома и йода, такие как 2Н (дейтерий), 3Н (тритий), 11C, 13С, 14С, 15N, 17О, 18O, 32Р, 33Р, 33S, 34S, 35S, 36S, 18F, 36Cl, 82Br, 123I, 124I, 125I, 129I и 131I, соответственно.
Что касается настоящего изобретения, изотопные варианты соединений общей формулы (VIIIa) или (IXa) предпочтительно содержат дейтерий ("содержащие дейтерий соединения формулы (VIIIa) или (IXa)").
Изотопные варианты соединений формулы (VIIIa) или (IXa) обычно можно получить методами, известными специалисту в данной области, такими как методы, описанные на схемах и/или примерах в данной заявке, путем замены реагента на изотопный вариант указанного реагента, предпочтительно на содержащий дейтерий реагент. В зависимости от желательных мест дейтерирования, в некоторых случаях дейтерий из D2O может быть введен либо непосредственно в соединения, либо в реагенты, которые являются пригодными для синтеза таких соединений. Газообразный дейтерий также является пригодным реагентом для введения дейтерия в молекулы. Быстрым путем для введения дейтерия является каталитическое дейтерирование олефиновых связей и ацетиленовых связей. Металлические катализаторы (т.е. Pd, Pt и Rh) в присутствии газообразного дейтерия можно использовать для прямого обмена водорода на дейтерий в функциональных группах, содержащих углеводородные группы. Множество дейтерированных реагентов и структурных элементов для синтеза являются коммерчески доступными от таких компаний, как, например, C/D/N Isotopes, Квебек, Канада; Cambridge Isotope Laboratories Inc., Андовер, Массачусетс, США; и CombiPhos Catalysts, Inc., Принстон, Нью-Джерси, США.
Термин "содержащее дейтерий соединение формулы (VIIIa) или (IXa)" определяют как соединение формулы (VIIIa) или формулы (IXa), в котором один или несколько атом(-ов) водорода заменен(-ы) на один или несколько атом(-ов) дейтерия и где относительное содержание дейтерия в каждом дейтерированном положении такого соединения формулы (VIIIa) или (IXa) выше, чем распространенность дейтерия в природе, которая составляет приблизительно 0.015%. В частности, в содержащем дейтерий соединении формулы (VIIIa) или (IXa) относительное содержание дейтерия в каждом дейтерированном положении соединения общей формулы (I) составляет выше 10%, 20%, 30%, 40%, 50%, 60%, 70% или 80%, предпочтительно выше 90%, 95%, 96% или 97%, еще более предпочтительно в указанном(-ых) положении(-ях) содержание дейтерия составляет выше 98% или 99%. Следует понимать, что относительное содержание дейтерия в каждом дейтерированном положении не зависит от относительного содержания дейтерия в другом(-их) дейтерированном(-ых) положении(-ях).
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Способ получения соединения формулы (I) в соответствии с изобретением характеризуется различными выгодными стадиями получения, а также промежуточными соединениями, что можно проиллюстрировать следующей схемой:
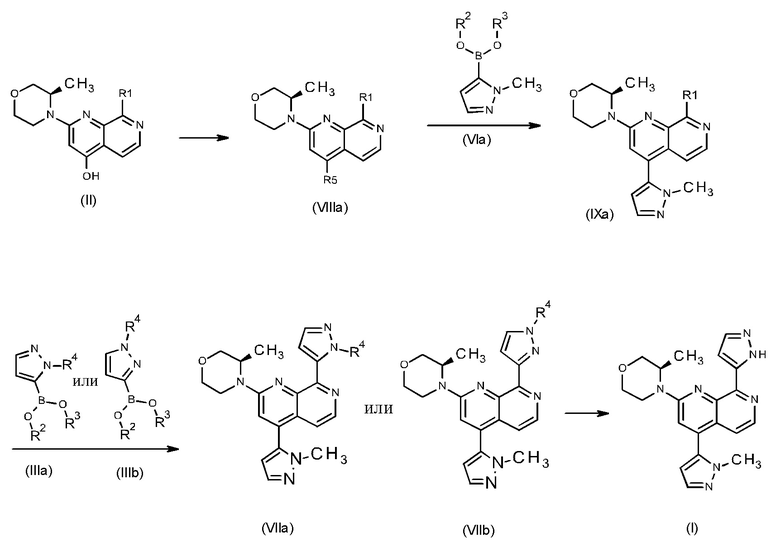
Новый путь синтеза из промежуточного соединения (II) к соединению формулы (I) имеет ряд преимуществ по сравнению с путем, описанным в WO 2016020320 A1. Новый путь обеспечивает по меньшей мере одно или несколько из следующих преимуществ по сравнению с ранее описанным способом:
• Нет необходимости в стадии хроматографической очистки, ни для промежуточных соединений формулы (VIIIa) или (IXa), ни для промежуточных соединений формулы (VIIa) или (VIIb).
• Микроволновый реактор не используют.
• Два новых промежуточных соединения (VIIIa)/(VIIIb) и (IXa)/(IXb) являются кристаллическими, могут быть легко выделены и очищены путем кристаллизации, в частности, путем использования безвредных для окружающей среды растворителей, таких как, например, изопропанол и н-бутанол.
• Более короткое время реакции.
• Общий выход значительно увеличен. Если принять во внимание наилучшие выходы на каждой стадии, общий выход увеличивается примерно в 2 раза, что приводит к приблизительно выходу 49.0% от теоретического для нового пути синтеза по сравнению с выходом 23.6% от теоретического для способа, описанного в WO 2016020320 A1.
• Чистота соединения формулы (I) значительно увеличена, например:
 Остаточное содержание палладия является очень низким (< 10 млн.ч.)
Остаточное содержание палладия является очень низким (< 10 млн.ч.)
Остаточное содержание бора является очень низким (< 10 млн.ч.)
• Остаточные растворители соответствуют нормативным требованиям.
• Новый способ позволяет крупномасштабное производство полиморфной формы В соединения формулы (I).
Способ в соответствии с изобретением получения соединения формулы (I)/(Ia) характеризуется по меньшей мере одной из следующих стадий:
1. Синтез соединения формулы (I) или (Ia) через промежуточное соединение формулы (VIIa) или (VIIb) исходя из промежуточного соединения формулы (IXa) или (IXb).
2. Синтез промежуточного соединения формулы (IXa) или (IXb) путем осуществления реакции промежуточного соединения формулы (VIIIa) или (VIIIb) с соединением формулы (VIa) или (VIb).
3. Синтез промежуточного соединения формулы (VIIIa) или (VIIIb), исходя из соединения формулы (II) или (IIa).
Способ в соответствии с изобретением получения соединения формулы (I) и соответствующие отдельные стадии способа дополнительно характеризуются новыми промежуточными соединениями (IXa)/(IXb) (см. раздел 4) и (VIIIa)/(VIIIb) (см. раздел 5), и обеспечивают соединение формулы (I) с высокой чистотой (см. раздел 6).
1. Синтез соединения формулы (I) или (Ia) через промежуточное соединение формулы (VIIa) или (VIIb) исходя из промежуточного соединения формулы (IXa) или (IXb)
В соответствии с одним аспектом, настоящее изобретение относится к способу получения соединения формулы (I)
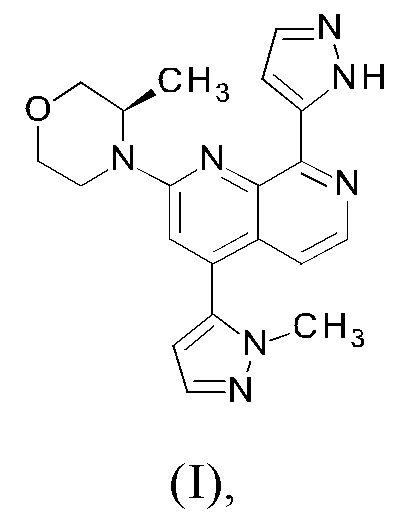
или его таутомера формулы (Ia)

или их смеси,
причем указанный способ включает следующие последовательные стадии: (а) осуществление реакции промежуточного соединения формулы (IXa)

где
R1 представляет собой атом хлора, брома или йода или представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси;
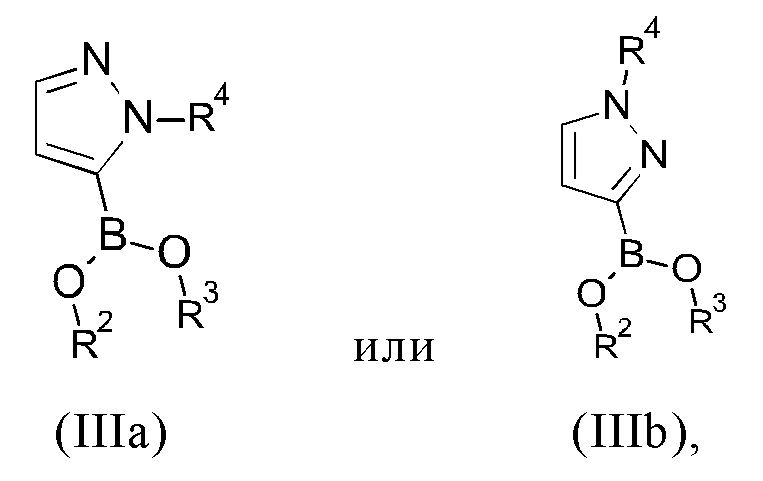
с соединением формулы (IIIa) или (IIIb)
или их смесью,
где
R2 и R3 представляют собой, независимо друг от друга, атом водорода или C1-С6-алкильную группу;
или
R2 и R3 вместе представляют собой -СН2-СН2- группу или -СН2-СН2-СН2- группу, где указанная -СН2-СН2- группа или -СН2-СН2-СН2- группа необязательно замещена один, два, три или четыре раза группой, выбранной из метила и этила;
или
R2 и R3 вместе представляют собой группу

где "*" представляет собой место присоединения к остальной части молекулы; и
R4 представляет собой группу, выбранную из тетрагидро-2Н-пиран-2-ила, 1-метил-1-метоксиэтила, 1-метил-1-феноксиэтила, 1-метил-1-бензилоксиэтила;
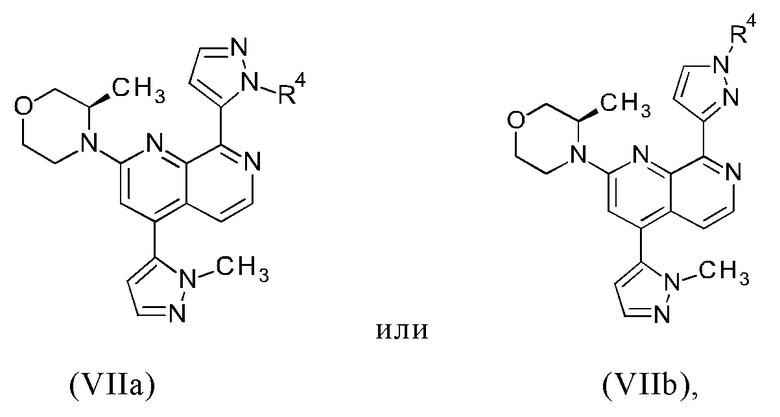
с получением промежуточного соединения формулы (VIIa) или (VIIb)
или их смеси,
где
R4 представляет собой группу, выбранную из тетрагидро-2Н-пиран-2-ила, 1-метил-1-метоксиэтила, 1-метил-1-феноксиэтила, 1-метил-1-бензилоксиэтила; и
(b) удаление группы R4 с промежуточного соединения формулы (VIIa) или (VIIb), таким образом обеспечивая соединение формулы (I), или его таутомер формулы (Ia), или их смесь.
В другом варианте осуществления способа в соответствии с настоящим изобретением R1 соединения формулы (IXa) представляет собой атом хлора или брома, предпочтительно атом хлора.
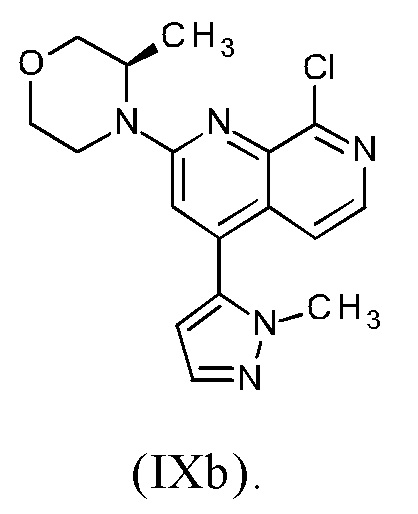
Соединение формулы (IXa), где R1 представляет собой атом хлора, является предпочтительным соединением формулы (IXb):
В другом варианте осуществления способа в соответствии с настоящим изобретением R1 соединения формулы (IXa) представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси.
В другом варианте осуществления способа в соответствии с настоящим изобретением R1 соединения формулы (IXa) представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси и (п-толуолсульфонил)окси.
В другом варианте осуществления настоящего изобретения R2 и R3 соединения формулы (IIIa) или (IIIb) представляют собой, независимо друг от друга, атом водорода или С1-С3 алкильную группу, в частности, метильную или этильную группу.
В другом варианте осуществления настоящего изобретения R2 и R3 соединения формулы (IIIa) или (IIIb) вместе представляют собой -СН2-СН2- группу или -СН2-СН2-СН2- группу, где указанная -СН2-СН2- группа или -СН2-СН2-СН2- группа необязательно замещена один, два, три или четыре раза группой, выбранной из метила и этила.
Если R2 и R3 соединения формулы (IIIa) или (IIIb) вместе представляют собой -СН2-СН2- группу или -СН2-СН2-СН2- группу, указанная -СН2-СН2- группа или указанная -СН2-СН2-СН2- группа вместе с атомом бора и атомами кислорода, к которым указанная группа присоединена, образует 5- или 6-членное кольцо.
В другом варианте осуществления настоящего изобретения R2 и R3 соединения формулы (IIIa) или (IIIb) вместе представляют собой группу
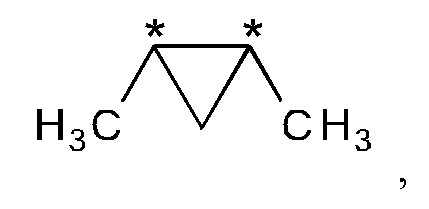
где "*" представляет собой место присоединения к остальной части молекулы.
В другом варианте осуществления настоящего изобретения R2 и R3 соединения формулы (IIIa) или (IIIb) вместе представляют собой -С(СН3)2-С(СН3)2- или -СН2-С(СН3)2-СН2- группу.
В предпочтительном варианте осуществления настоящего изобретения R2 и R3 соединения формулы (IIIa) или (IIIb) вместе представляют собой -С(СН3)2-С(СН3)2- группу.
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (IIIa) представляет собой сложный пинаколовый эфир 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (формулы IIIc):

Соединения формулы (IIIa), (IIIb) или (IIIc) являются коммерчески доступными или могут быть синтезированы способами, известными специалисту в данной области техники.
В другом варианте осуществления настоящего изобретения R4 соединения формулы (IIIa), (IIIb), (VIIa) или (VIIb) представляет собой группу, выбранную из тетрагидро-2Н-пиран-2-ила, 1-метил-1-метоксиэтила, 1-метил-1-феноксиэтила, 1-метил-1-бензилоксиэтила.
В предпочтительном варианте осуществления настоящего изобретения R4 соединения формулы (IIIa), (IIIb), (VIIa) или (VIIb) представляет собой тетрагидро-2Н-пиран-2-ильную группу.
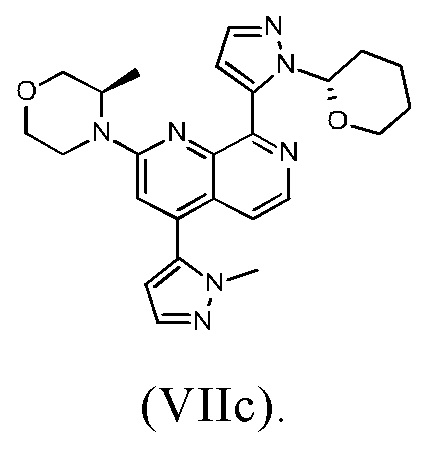
В другом предпочтительном варианте осуществления настоящего изобретения промежуточное соединение формулы (VIIa) представляет собой соединение формулы (VIIc)
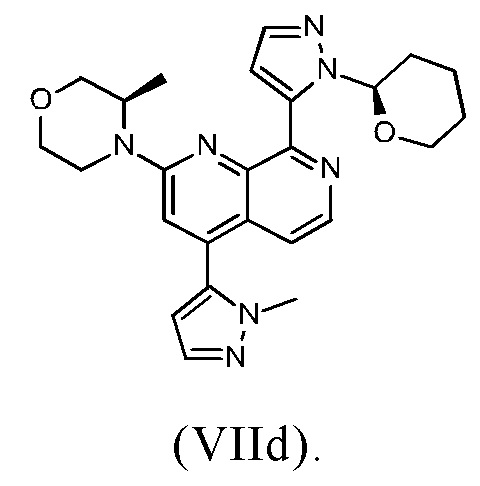
В другом предпочтительном варианте осуществления настоящего изобретения промежуточное соединение формулы (VIIa) представляет собой соединение формулы (VIId)
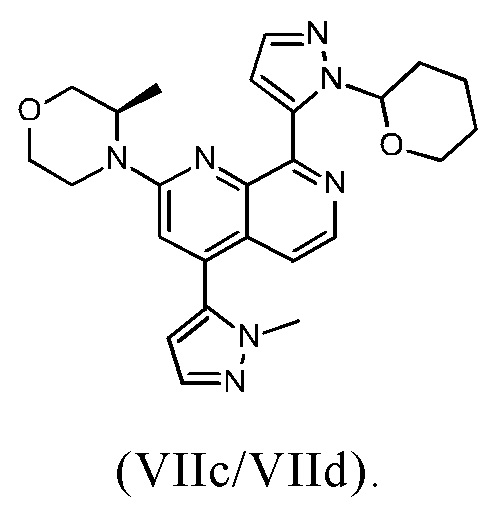
В другом предпочтительном варианте осуществления настоящего изобретения промежуточное соединение формулы (VIIa) представляет собой смесь, в частности, 1:1 смесь, соединений формулы (VIIc) и (VIId). Указанная 1: 1 смесь представляет собой (3R)-3-метил-4-(4-(1-метил-1Н-пиразол-5-ил)-8-(1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)морфолин, который также называют соединением формулы (VIIc/VIId) в дальнейшем:
В другом варианте осуществления способа в соответствии с настоящим изобретением промежуточное соединение формулы (IXa) подвергают реакции с соединением формулы (IIIa).
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолин (IXb) подвергают реакции со сложным пинаколовым эфиром 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIc).
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением соединение формулы (IXa) представляет собой (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолин; и/или соединение формулы (IIIa) представляет собой сложный пинаколовый эфир 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты; и/или соединение формулы (VIIa) представляет собой (3R)-3-метил-4-(4-(1-метил-1Н-пиразол-5-ил)-8-(1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)морфолин.
В другом варианте осуществления способа в соответствии с настоящим изобретением промежуточное соединение формулы (IXa) подвергают реакции с 0.95-2.0 молярными эквивалентами соединения формулы (IIIa) или (IIIb), предпочтительно с 1.0-1.7 молярными эквивалентами соединения формулы (IIIa) или (IIIb), наиболее предпочтительно с 1.2-1.5 молярными эквивалентами соединения формулы (IIIa) или (IIIb).
В другом варианте осуществления способа в соответствии с настоящим изобретением промежуточное соединение формулы (IXa) подвергают реакции с 0.95-2.0 молярными эквивалентами сложного пинаколового эфира 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIc), предпочтительно с 1.0-1.7 молярными эквивалентами сложного пинаколового эфира 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIc), наиболее предпочтительно с 1.2-1.5 молярными эквивалентами сложного пинаколового эфира 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIc).
В другом варианте осуществления способа в соответствии с настоящим изобретением (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолин (IXb) подвергают реакции с 0.95-2.0 молярными эквивалентами сложного пинаколового эфира 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIc), предпочтительно с 1.0-1.7 молярными эквивалентами сложного пинаколового эфира 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIc), наиболее предпочтительно с 1.2-1.5 молярными эквивалентами сложного пинаколового эфира 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIc).
В другом варианте осуществления способа в соответствии с настоящим изобретением соединение формулы (IIIa) или (IIIb), в частности, сложный пинаколовый эфир 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIc), растворяют в растворителе, например, в изопропилацетате, этилацетате, 1,2-диметоксиэтане, диоксане, N,N-диметилформамиде (=ДМФА), 1,2-диметоксиэтане (=DME), тетрагидрофуране (=ТГФ), 2-метилтетрагидрофуране (=2-Ме-ТГФ) или изопропаноле.
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением соединение формулы (IIIa) или (IIIb), в частности, сложный пинаколовый эфир 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIc), растворяют в изопропилацетате или этилацетате, наиболее предпочтительно в этилацетате.
В другом варианте осуществления способа в соответствии с настоящим изобретением соединение формулы (IXa) или (IXb) растворяют в растворителе, например, в изопропилацетате, этилацетате, 1,2-диметоксиэтане, диоксане, N,N-диметилформамиде (=ДМФА), 1,2-диметоксиэтане (=DME), тетрагидрофуране (=ТГФ), 2-метилтетрагидрофуране (=2-Ме-ТГФ) или изопропаноле.
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением соединение формулы (IXa) или (IXb), в частности, (IXb), растворяют в изопропилацетате или этилацетате, наиболее предпочтительно в этилацетате.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (IXa), в частности, (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина, с соединением формулы (IIIa) или (IIIb), в частности, со сложным пинаколовым эфиром 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты, осуществляют в течение 1-36 часов, в частности, в течение 1.5-5 часов, предпочтительно в течение 1.5-3 часов, наиболее предпочтительно в течение 100-140 минут.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (IXa), в частности, (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина, с соединением формулы (IIIa) или (IIIb), в частности, со сложным пинаколовым эфиром 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты, осуществляют в присутствии подходящей каталитической системы, такой как, например, палладиевый катализатор.
В другом варианте осуществления способа в соответствии с настоящим изобретением палладиевый катализатор выбирают из группы, состоящей из [1,1'-бис(дифенилфосфино)ферроцен]дихлор палладия(II), ацетата палладия(II), дихлорида бис(трифенилфосфин)палладия(II), дихлорбис(трициклогексилфосфин)палладия(II), хлорида (2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2-аминоэтил)фенил)]палладия(II), хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладия(II), тетракис(трифенилфосфин)палладия(0), бис(три-трет-бутилфосфин)палладия(0), бис[трис(2-метилфенил)фосфин]палладия(0), трис(дибензилиденацетон)дипалладия(0).
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением палладиевый катализатор представляет собой [1,1'-бис(дифенилфосфино)ферроцен]дихлор палладий(II).
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (IXa), в частности, (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина, с соединением формулы (IIIa) или (IIIb), в частности, со сложным пинаколовым эфиром 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты, осуществляют в присутствии 0.001-0.1 молярных эквивалентов, предпочтительно 0.005-0.05 молярных эквивалентов, наиболее предпочтительно 0.01-0.03 молярных эквивалентов палладиевого катализатора, предпочтительно [1,1'-бис(дифенилфосфино)ферроцен]дихлор палладия(II).
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (IXa), в частности, (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина (IXb), с соединением формулы (IIIa) или (IIIb), в частности, со сложным пинаколовым эфиром 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIc), осуществляют в органическом растворителе, где растворитель включает изопропилацетат, этилацетат, 1,2-диметоксиэтан, 1,4-диоксан, диметилформамид, тетрагидрофуран, 2-метилтетрагидрофуран, метанол, этанол, 1-пропанол, изопропанол, 1-бутанол или 2-бутанол; или где указанную реакцию осуществляют в смеси растворителей, включающей один или несколько из указанных растворителей и воду.
В другом варианте способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (IXa), в частности, (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина, с соединением формулы (IIIa) или (IIIb), в частности, со сложным пинаколовым эфиром 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты, осуществляют в органическом растворителе, где растворитель включает изопропилацетат и этилацетат; или где указанную реакцию осуществляют в смеси растворителей, включающей изопропилацетат и воду, или включающей этилацетат и воду. Предпочтительно смесь растворителей включает этилацетат и воду.
В другом варианте осуществления способа в соответствии с настоящим изобретением в случае реакции промежуточного соединения формулы (IXa), в частности, (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина, с соединением формулы (IIIa) или (IIIb), в частности, со сложным пинаколовым эфиром 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты, на кг соединения формулы (IXa) используют 5-20 кг органического растворителя, предпочтительно 6-15 кг органического растворителя, наиболее предпочтительно 7-11 кг органического растворителя. Предпочтительный органический растворитель включает изопропилацетат или этилацетат, предпочтительная смесь растворителей включает изопропилацетат и воду, или она включает этилацетат и воду.
В другом варианте осуществления способа в соответствии с настоящим изобретением в случае реакции промежуточного соединения формулы (IXa), в частности, (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина (IXb), с соединением формулы (IIIa) или (IIIb), в частности, со сложным пинаколовым эфиром 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIc), на кг соединения формулы (IXa) или (IXb) используют 5-20 кг органического растворителя и 1-5 кг воды, предпочтительно 6-15 кг органического растворителя и 1-4 кг воды, наиболее предпочтительно 7-11 кг органического растворителя и 1.5-2.5 кг воды.
В другом варианте способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (IXa), в частности, (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина (IXb), с соединением формулы (IIIa) или (IIIb), в частности, со сложным пинаколовым эфиром 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIc), осуществляют в присутствии основания. Можно использовать такие основания, как фосфат калия, карбонат калия, гидрокарбонат калия, фосфат натрия, карбонат натрия, гидрокарбонат натрия, гидроксид бария, карбонат бария, карбонат цезия или карбонат лития. Фосфат калия или фосфат натрия являются предпочтительными, наиболее предпочтительным является фосфат калия.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (IXa), в частности, (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина (IXb), с соединением формулы (IIIa) или (IIIb), в частности, со сложным пинаколовым эфиром 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIc), осуществляют в присутствии 1-15 молярных эквивалентов, предпочтительно 2-11 молярных эквивалентов, наиболее предпочтительно 3-10 молярных эквивалентов основания.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (IXa), в частности, (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина (IXb), с соединением формулы (IIIa) или (IIIb) осуществляют в присутствии палладиевого катализатора и/или основания.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (IXa), в частности, (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина (IXb), с соединением формулы (IIIa) или (IIIb), в частности, со сложным пинаколовым эфиром 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIc), осуществляют при температурах в диапазоне от комнатной температуры до температуры кипения растворителя.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (IXa), в частности, (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина (IXb), с соединением формулы (IIIa) или (IIIb), в частности, со сложным пинаколовым эфиром 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIc), осуществляют под давлением при температурах выше температуры кипения.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (IXa), в частности, (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина, с соединением формулы (IIIa) или (IIIb), в частности, со сложным пинаколовым эфиром 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты, осуществляют в изопропилацетате или в изопропилацетате и воде при температуре 55-75°С, предпочтительно при 60-70°С, наиболее предпочтительно при 65°С.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (IXa), в частности, (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина, с соединением формулы (IIIa) или (IIIb), в частности, со сложным пинаколовым эфиром 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты, осуществляют в этилацетате или в этилацетате и воде при температуре 45-65°С, предпочтительно при 50-60°С, наиболее предпочтительно при 55°С.
В частности, если реакцию промежуточного соединения формулы (IXa), предпочтительно (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина, с соединением формулы (IIIa) или (IIIb), предпочтительно со сложным пинаколовым эфиром 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты, осуществляют в смеси этилацетата и воды, указанный этилацетат и указанную воду перемешивают, в частности, энергично перемешивают. Предпочтительно этилацетат и воду перемешивают в условиях, которые гарантируют достаточное смешивание этилацетатной и водной фаз.
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (IXa), в частности, (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина (IXb), с соединением формулы (IIIa) или (IIIb), в частности, со сложным пинаколовым эфиром 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIc), осуществляют в атмосфере инертного газа, где инертный газ представляет собой азот или аргон, предпочтительно азот.
1.1 Дальнейшая обработка сырого промежуточного соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId)
В другом варианте осуществления способа в соответствии с настоящим изобретением промежуточное соединение формулы (VIIa) или (VIIb), которое получают по реакции промежуточного соединения формулы (IXa) с промежуточным соединением формулы (IIIa) или (IIIb), не выделяют после указанной реакции ("сырое промежуточное соединение формулы (VIIa) или (VIIb)" в дальнейшем) и/или промежуточное соединение формулы (VIIa) или (VIIb) не очищают.
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением промежуточное соединение формулы (VIIc/VIId), которое получают по реакции промежуточного соединения формулы (IXb) с промежуточным соединением формулы (IIIc), не выделяют после указанной реакции ("сырое промежуточное соединение формулы (VIIc/VIId)" в дальнейшем) и/или промежуточное соединение формулы (VIIc/VIId) не очищают.
В другом варианте осуществления способа в соответствии с настоящим изобретением промежуточное соединение формулы (VIIa) или (VIIb) не выделяют и/или не очищают перед удалением группы R4 с промежуточного соединения формулы (VIIa) или формулы (VIIb). Сырое промежуточное соединение формулы (VIIa) или (VIIb) сразу превращают в соединение формулы (I) или в его таутомер формулы (Ia), путем удаления группы R4 с соединения формулы (VIIa) или (VIIb).
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением промежуточное соединение формулы (VIIc), (VIId) или (VIIc/VIId) не выделяют и/или не очищают перед удалением группы R4 с промежуточного соединения формулы (VIIc), (VIId) или (VIIc/VIId). Сырое промежуточное соединение формулы (VIIc), (VIId) или (VIIc/VIId) сразу превращают в соединение формулы (I) или в его таутомер формулы (Ia), путем удаления группы R4 с соединения формулы (VIIc), (VIId) или (VIIc/VIId).
В другом варианте осуществления способа в соответствии с настоящим изобретением промежуточное соединение формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId) не выделяют и/или не очищают перед удалением группы R4 с промежуточного соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId), и после реакции промежуточного соединения формулы (IXa) или (IXb) с соединением формулы (IIIa), (IIIb) или (IIIc) и перед удалением группы R4 с промежуточного соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId), растворитель или смесь растворителей указанной реакции заменяют на другой растворитель ("растворитель X" в дальнейшем), где растворитель X включает растворитель, выбранный из дихлорметана, этилацетата, изопропилацетата, тетрагидрофурана, 2-метилтетрагидрофурана, толуола, хлороформа или их смесей; и, необязательно,
a) растворитель X промывают водным раствором основания, выбранного из гидроксида калия, карбоната калия, гидрокарбоната калия, гидроксида калия, трет-бутоксида калия, гидроксида натрия, фосфата натрия, карбоната натрия, гидроксида натрия, трет-бутоксида натрия, гидроксида бария, карбоната цезия, триэтиламина; предпочтительно растворитель X промывают гидроксидом калия; и, необязательно,
b) растворитель X обрабатывают адсорбентом; предпочтительно адсорбент представляет собой активированный уголь; и необязательно
c) адсорбент, в частности, активированный уголь, отфильтровывают;
с получением раствора соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId) в растворителе X.
Подходящие адсорбенты, такие как, например, активированный уголь (= активированный углерод), оксиды, такие как оксиды Al, Mg, Th, Ti, Zr и В, и их смеси, кремниевая кислота, борная кислота, силикаты, такие как диатомовая земля, кизельгур и силикагель; фуллерова земля, флоридин, и глины, такие как бентониты, монтмориллониты и обработанные кислотой глины, смолы, активированный глинозем или цеолиты, известны специалисту в данной области техники.
В другом варианте осуществления способа в соответствии с настоящим изобретением промежуточное соединение формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId) не выделяют и/или не очищают перед удалением группы R4 с промежуточного соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId), и после реакции промежуточного соединения формулы (IXa) или (IXb) с соединением формулы (IIIa), (IIIb) или (IIIc) и перед удалением группы R4 с промежуточного соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId), растворитель или смесь растворителей указанной реакции, в частности, этилацетат с водой/без воды, или изопропилацетат с водой/без воды, заменяют на другой растворитель, включающий дихлорметан; и, необязательно,
a) полученный в результате дихлорметановый раствор промывают водным раствором основания, выбранного из гидроксида калия, карбоната калия, гидрокарбоната калия, гидроксида калия, трет-бутоксида калия, гидроксида натрия, фосфата натрия, карбоната натрия, гидроксида натрия, трет-бутоксида натрия, гидроксида бария, карбоната цезия, триэтиламина; предпочтительно дихлорметан промывают гидроксидом калия; и, необязательно,
b) полученный в результате дихлорметановый раствор обрабатывают адсорбентом; предпочтительно адсорбент представляет собой активированный уголь; и необязательно
c) адсорбент, в частности, активированный уголь, отфильтровывают;
с получением раствора соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId) в дихлорметане.
В другом варианте осуществления способа в соответствии с настоящим изобретением промежуточное соединение формулы (VIIc/VIId) не выделяют и/или не очищают перед удалением группы R4 с промежуточного соединения формулы (VIIc/VIId), и после реакции (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина (IXb) со сложным пинаколовым эфиром 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIc) и перед удалением группы R4 с промежуточного соединения формулы (VIIc/VIId), растворитель или смесь растворителей указанной реакции, в частности, изопропилацетат с водой/без воды или этилацетат с водой/без воды, заменяют на другой растворитель, включающий дихлорметан; и,
а) полученный в результате дихлорметановый раствор промывают водным раствором основания, выбранного из гидроксида калия, карбоната калия, гидрокарбоната калия, гидроксида калия, трет-бутоксида калия, гидроксида натрия, фосфата натрия, карбоната натрия, гидроксида натрия, трет-бутоксида натрия, гидроксида бария, карбоната цезия, триметиламина, предпочтительно дихлорметан промывают гидроксидом калия; и/или
b) полученный в результате дихлорметановый раствор обрабатывают адсорбентом; предпочтительно адсорбент представляет собой активированный уголь; и адсорбент отфильтровывают;
с получением очищенного раствора промежуточного соединения формулы (VIIc/VIId) в дихлорметане ("очищенный раствор промежуточного соединения формулы (VIIc/VIId) в дихлорметане" в дальнейшем).
В другом варианте осуществления способа в соответствии с настоящим изобретением промежуточное соединение формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId) не выделяют и/или не очищают перед удалением группы R4 с промежуточного соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId), и после реакции промежуточного соединения формулы (IXa) или (IXb) с соединением формулы (IIIa), (IIIb) или (IIIc) и перед удалением группы R4 с промежуточного соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId), растворитель указанной реакции, в частности, этилацетат с водой/без воды или изопропилацетат с водой/без воды, предпочтительно этилацетат и воду, промывают водой и/или обрабатывают адсорбентом, определенным выше, в частности, активированным углем, с получением очищенного раствора промежуточного соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId), в частности, очищенного раствора в этилацетате или изопропилацетате ("очищенный раствор промежуточного соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId) в этилацетате или изопропилацетате" в дальнейшем).
В другом варианте осуществления способа в соответствии с настоящим изобретением промежуточное соединение формулы (VIIc/VIId) не выделяют и/или не очищают перед удалением группы R4 с промежуточного соединения формулы (VIIc/VIId), и после реакции промежуточного соединения формулы (IXb) с соединением формулы (IIIc) и перед удалением группы R4 с промежуточного соединения формулы (VIIc/VIId), растворитель указанной реакции, в частности, этилацетат с водой/без воды или изопропилацетат с водой/без воды, предпочтительно этилацетат и воду, промывают водой и/или обрабатывают адсорбентом, определенным выше, в частности, активированным углем, с получением очищенного раствора промежуточного соединения формулы (VIIc/VIId) ("очищенный раствор промежуточного соединения формулы (VIIc/VIId)"), в частности, раствора (VIIc/VIId) в этилацетате или в изопропилацетате ("очищенный раствор промежуточного соединения формулы (VIIc/VIId) в этилацетате или изопропилацетате" в дальнейшем), предпочтительно раствора промежуточного соединения формулы (VIIc/VIId) в этилацетате ("очищенный раствор промежуточного соединения формулы (VIIc/VIId) в этилацетате" в дальнейшем).
1.2 Удаление группы R4 с промежуточного соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId) с получением сырого соединения формулы (I)
В другом варианте осуществления способа в соответствии с настоящим изобретением группу R4 удаляют с промежуточного соединения формулы (VIIa) или формулы (VIIb) путем осуществления реакции промежуточного соединения формулы (VIIa) или формулы (VIIb) с кислотой, такой как, например, водный раствор хлористоводородной кислоты, водный раствор хлористоводородной кислоты с метанолом, хлористоводородная кислота с метанолом, водный раствор хлористоводородной кислоты с этанолом, хлористоводородная кислота с этанолом, водный раствор хлористоводородной кислоты с 1-пропанолом, хлористоводородная кислота с 1-пропанолом, водный раствор хлористоводородной кислоты с изопропанолом, хлористоводородная кислота с изопропанолом, водный раствор хлористоводородной кислоты с 1-бутанолом, хлористоводородная кислота с 1-бутанолом, водный раствор хлористоводородной кислоты с 2-бутанолом, хлористоводородная кислота с 2-бутанолом, водный раствор серной кислоты, метансульфоновая кислота, п-толуолсульфоновая кислота, трифторуксусная кислота или фосфорная кислота, или смеси одной или нескольких указанных кислот (хлористоводородной кислоты, водного раствора серной кислоты, метансульфоновой кислоты, п-толуолсульфоновой кислоты, трифторуксусной кислоты и/или фосфорной кислоты) с одним или несколькими спиртами, такими как метанол, этанол, 1-пропанол, изопропанол, 1-бутанол, 2-бутанол. Предпочтительно кислота включает водный раствор хлористоводородной кислоты с метанолом.
В другом варианте осуществления способа в соответствии с настоящим изобретением группу R4 удаляют с промежуточного соединения формулы (VIIc), (VIId) или (VIIc/VIId), путем осуществления реакции промежуточного соединения формулы (VIIc), (VIId) или (VIIc/VIId), с кислотой, как определено выше.
Можно использовать 0.7-10 молярных эквивалентов кислоты, предпочтительно 1-7.5 молярных эквивалентов, наиболее предпочтительно 1.5-5 молярных эквивалентов. Водный раствор хлористоводородной кислоты является предпочтительным, в частности, водный раствор хлористоводородной кислоты с метанолом или водный раствор хлористоводородной кислоты с метанолом с этилацетатом.
В частности, при удалении группы R4 в соответствии со способом настоящего изобретения, рН составляет менее 3 (рН<3), предпочтительно менее 2 (рН<2), наиболее предпочтительно менее 1.5 (рН<1.5).
В другом варианте осуществления способа в соответствии с настоящим изобретением при удалении группы R4 с промежуточного соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId) с помощью кислоты в растворителе, растворитель является протонным или апротонным растворителем, таким как, например, метанол, этанол, пропанол, бутанол, дихлорметан, тетрагидрофуран (=ТГФ), 2-метилтетрагидрофуран (=2-Ме-ТГФ), 1,4-диоксан, 1,2-диметоксиэтан, этилацетат, изопропилацетат; или растворитель представляет собой смесь растворителей, включающую указанный(-е) растворитель(-и) и необязательно дополнительно включающую воду. Предпочтительный растворитель включает дихлорметан, предпочтительная смесь растворителей включает дихлорметан с метанолом, или метанол с этилацетатом, или метанол с этилацетатом и водой, или метанол с изопропилацетатом, или метанол с изопропилацетатом и водой.
В другом варианте осуществления способа в соответствии с настоящим изобретением при удалении группы R4 с промежуточного соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId) кислоту выбирают из группы, состоящей из хлористоводородной кислоты, водного раствора хлористоводородной кислоты, водного раствора хлористоводородной кислоты с метанолом, водного раствора хлористоводородной кислоты с изопропанолом, водного раствора хлористоводородной кислоты с метанолом и дихлорметаном, и водного раствора хлористоводородной кислоты с метанолом и этилацетатом.
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением при удалении группы R4 с промежуточного соединения формулы (VIIc/VIId) кислота представляет собой водный раствор хлористоводородной кислоты и смесь растворителей включает дихлорметан и метанол, в частности, дихлорметан, метанол и воду.
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением при удалении группы R4 с промежуточного соединения формулы (VIIc/VIId) в очищенном растворе в дихлорметане, как определено выше, кислота представляет собой водный раствор хлористоводородной кислоты и смесь растворителей включает дихлорметан, метанол и воду.
При использовании водного раствора хлористоводородной кислоты с метанолом и дихлорметаном, на кг соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId) используют 1-20 кг 1 н. водного раствора хлористоводородной кислоты в 1-20 кг метанола и 1-20 кг дихлорметана. Предпочтительно используют 5-15 кг 1 н. водного раствора хлористоводородной кислоты в 2-15 кг метанола и 2-15 кг дихлорметана.
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением при удалении группы R4 с промежуточного соединения формулы (VIIc/VIId) кислота представляет собой водный раствор хлористоводородной кислоты и смесь растворителей включает метанол с этилацетатом, или метанол с изопропилацетатом, предпочтительно смесь растворителей включает метанол с этилацетатом и воду.
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением при удалении группы R4 с промежуточного соединения формулы (VIIc/VIId) в очищенном растворе в этилацетате или изопропилацетате, как определено выше, кислота представляет собой водный раствор хлористоводородной кислоты, в частности, 1 н. водный раствор хлористоводородной кислоты, и смесь растворителей включает метанол с этилацетатом с водой/без воды, или метанол с изопропилацетатом с водой/без воды, предпочтительно смесь растворителей включает метанол с этилацетатом и воду.
Применение смесей растворителей, включающих метанол, является особенно предпочтительным для того, чтобы предотвратить образование побочных продуктов, таких как, например,
При использовании водного раствора хлористоводородной кислоты с метанолом и этилацетатом, на кг соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId) используют 1-20 кг 1 н. водного раствора хлористоводородной кислоты с 1-20 кг метанола и 1-20 кг этилацетата. Предпочтительно используют 5-15 кг 1 н. водного раствора хлористоводородной кислоты с 2-15 кг метанола и 2-15 кг этилацетата.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (VIIa) или формулы (VIIb), предпочтительно соединения формулы (VIIc) или (VIId) или смеси соединений формулы (VIIc) и (VIId), с кислотой осуществляют при температуре -10-40°С, предпочтительно при 0-30°С, наиболее предпочтительно при 10-25°С. Время реакции составляет 2-60 мин, предпочтительно 2-30 мин, наиболее предпочтительно 5-20 мин.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию удаления группы R4 с промежуточного соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId) с помощью кислоты осуществляют в атмосфере инертного газа, где инертный газ представляет собой азот или аргон, предпочтительно азот.
Сразу после завершения реакции промежуточного соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId) с кислотой получают сырое соединение формулы (I) ("сырое соединение формулы (I)" в дальнейшем).
После удаления группы R4 с промежуточного соединения формулы (VIIc/VIId) с помощью водного раствора хлористоводородной кислоты в смеси растворителей, включающей дихлорметан, метанол и воду, сырое соединение формулы (I) растворяют в подкисленной водной фазе (в дальнейшем "подкисленный водный раствор сырого соединения формулы (I)"), где рН полученного в результате подкисленного водного раствора составляет менее 3 (рН<3), предпочтительно менее 2 (рН<2), наиболее предпочтительно менее 1.5 (рН<1.5).
Сырое соединение формулы (I) может быть дополнительно обработано (раздел 1.3) и/или кристаллизовано (раздел 1.4).
1.3 Дальнейшая обработка сырого соединения формулы (I)
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I),
a) подкисленный водный раствор сырого соединения формулы (I), описанного выше, экстрагируют один или несколько раз растворителем А, определенным ниже, и/или обрабатывают один или несколько раз поглотителем Pd, определенным ниже;
b) подкисленный водный раствор сырого соединения формулы (I), полученный на предыдущей стадии а), обрабатывают растворителем А и водным раствором основания, определенным ниже, с получением двухфазной системы, где водная фаза указанной двухфазной системы имеет рН>12;
c) водную фазу отделяют от указанной двухфазной системы с получением раствора сырого соединения формулы (I) в растворителе А; и, необязательно,
d) производят замену растворителя А раствора сырого соединения формулы (I) в растворителе А на растворитель В, определенный ниже, с получением раствора сырого соединения формулы (I) в растворителе В.
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I),
a) подкисленный водный раствор сырого соединения формулы (I), описанного выше, экстрагируют один или несколько раз дихлорметаном и/или обрабатывают один или несколько раз поглотителем Pd, определенным ниже;
b) подкисленный водный раствор сырого соединения формулы (I), полученный на предыдущей стадии а), обрабатывают дихлорметаном и водным раствором гидроксида калия, в частности, 5 н. водным раствором гидроксида калия, с получением двухфазной системы, где водная фаза указанной двухфазной системы имеет рН>12;
c) водную фазу отделяют от указанной двухфазной системы с получением раствора сырого соединения формулы (I) в дихлорметане; и, необязательно,
d) осуществляют замену дихлорметана раствора сырого соединения формулы (I) в дихлорметане на н-бутанол с получением раствора сырого соединения формулы (I) в н-бутаноле.
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) сырое соединение формулы (I) растворяют в растворителе ("растворитель А" в дальнейшем), где растворитель А включает растворитель, выбранный из дихлорметана, этилацетата, изопропилацетата, тетрагидрофурана, 2-метилтетрагидрофурана, толуола, хлороформа; или в смеси растворителей, включающей один или несколько из указанных растворителей А ("смесь растворителей А"); предпочтительный растворитель А представляет собой дихлорметан.
Предпочтительно растворитель А включает тот же самый растворитель или ту же самую смесь растворителей, что и растворитель/смесь растворителей, который(-ую) использовали для реакции удаления группы R4 с промежуточного соединения формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId).
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) сырое соединение формулы (I) растворяют в дихлорметане.
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) сырое соединение формулы (I), растворенное в растворителе А, обрабатывают водным раствором основания, выбранного из гидроксида калия, карбоната калия, гидрокарбоната калия, гидроксида калия, трет-бутоксида калия, гидроксида натрия, фосфата натрия, карбоната натрия, гидроксида натрия, трет-бутоксида натрия, гидроксида бария, карбоната цезия, триэтиламина; предпочтительно дихлорметан промывают гидроксидом калия. Предпочтительно сырое соединение формулы (I), растворенное в растворителе А, в частности, дихлорметане, обрабатывают гидроксидом калия.
В частности, при обработке сырого соединения формулы (I), растворенного в растворителе А, в частности, в дихлорметане, водным раствором основания, определенным выше, рН является большим чем 11 (рН>11), предпочтительно большим чем 12 (рН>12), наиболее предпочтительно рН=12-14, в частности, рН=12,5-13,5.
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) сырое соединение формулы (I), растворенное в растворителе А, предпочтительно растворенное в дихлорметане, обрабатывают поглотителем палладия, определенным ниже.
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) сырое соединение формулы (I), растворенное в растворителе А, предпочтительно растворенное в дихлорметане, обрабатывают водным раствором основания, определенным выше, и затем обрабатывают поглотителем палладия, определенным ниже.
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) сырое соединение формулы (I), растворенное в растворителе А, предпочтительно растворенное в дихлорметане, обрабатывают поглотителем палладия, определенным ниже, и затем обрабатывают водным раствором основания, определенным выше.
Поглотитель палладия представляет собой реагент, который можно использовать для отделения палладия из палладиевого катализатора от сырого соединения формулы (I). Различные поглотители палладия, например, дополнительно описаны Garret и Prasad (Advanced Synthesis & Catalysis (2004), 346 (8), 889-900CODEN: ASCAF7; ISSN: 1615-4150, Wiley-VCH Verlag GmbH & Co. KGaA). Они включают, например, тримеркаптотриазин (ТМТ), полистирол-связанный ТМТ, МР-ТМТ (макропористая полистирол-связанная тримеркаптотриазиновая смола с высокой степенью сшивки), полистирол-связанный этилендиамин, активированный углерод, губки из стеклянных бусин, смопекс (= волокна на основе полиэтилена или целлюлозы, содержащие привитые боковые цепи с соответствующими функциональными группами для образования комплекса с металлами), полимер-связанные лиганды и силикагель-связанные лиганды.
В одном варианте осуществления настоящего изобретения поглотитель палладия выбирают из группы, состоящей из N-ацетилцистеина, квадрасил-меркаптопропила (CAS-номер 1225327-73-0) и изолюта Si-TMT (поглотитель Pd от Biotage АВ, Швеция, № по каталогу 9538-1000) - силикагель-связанного эквивалента 2,4,6-тримеркаптотриазина, или представляет собой их смесь.
В другом варианте осуществления настоящего изобретения поглотитель палладия включает смесь N-ацетилцистеина с квадрасил-меркаптопропилом, N-ацетилцистеина с изолютом Si-TMT, или изолюта Si-TMT с квадрасил-меркаптопропилом.
Наиболее предпочтительный поглотитель палладия включает смесь N-ацетилцистеина, квадрасил-меркаптопропила и изолюта Si-TMT.
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) растворитель А, определенный выше, заменяют на растворитель ("растворитель В" в дальнейшем), выбранный из этанола, н-пропанола, н-бутанола, 2-бутанола, изопропанола, предпочтительно растворитель А заменяют на н-бутанол.
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) дихлорметан (растворитель А) заменяют на н-бутанол (растворитель В).
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) растворитель А заменяют на растворитель В после обработки растворителя А поглотителем Pd, и/или после обработки растворителя А водным раствором основания.
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) дихлорметан заменяют на н-бутанол после обработки дихлорметана поглотителем Pd и/или после обработки дихлорметана водным раствором основания.
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) растворитель А заменяют на растворитель В после обработки растворителя А поглотителем Pd и/или после обработки растворителя А водным раствором основания; и растворитель В затем обрабатывают поглотителем Pd.
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) дихлорметан заменяют на н-бутанол после обработки дихлорметана поглотителем Pd и/или после обработки дихлорметана водным раствором основания; и н-бутанол затем обрабатывают поглотителем Pd.
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) растворитель А заменяют на растворитель В перед обработкой растворителя А поглотителем Pd, и растворитель В затем обрабатывают поглотителем Pd.
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) дихлорметан заменяют на н-бутанол перед обработкой дихлорметана поглотителем Pd, и н-бутанол затем обрабатывают поглотителем Pd.
1.4 Кристаллизация сырого соединения формулы (I) с получением его полиморфной формы В
Стадия кристаллизации обеспечивает полиморфную форму В соединения формулы (I) (= Мод В) посредством воспроизводимого и надежного способа, который можно охарактеризовать, во-первых, заменой растворителя, а именно растворителя А на растворитель В, и, во-вторых, последующей стадией кристаллизации.
1.4.1 Замена растворителя
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) сырое соединение формулы (I), растворенное в растворителе А, предпочтительно в дихлорметане, кристаллизуют в растворителе В с получением полиморфной формы В соединения формулы (I).
Для того, чтобы кристаллизовать соединение формулы (I) в его полиморфную форму В растворитель А сначала необходимо заменить на растворитель В ("замена растворителя"). Следовательно, в другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) растворитель А, предпочтительно дихлорметан, заменяют на растворитель В, предпочтительно на н-бутанол.
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) растворитель А, предпочтительно дихлорметан, заменяют на растворитель В, предпочтительно на н-бутанол, с помощью, во-первых, смешивания растворителя А и растворителя В; и, во-вторых, отгонки растворителя А при нормальном давлении или при пониженном давлении.
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) растворитель А заменяют на растворитель В с помощью, во-первых, смешивания растворителя А и растворителя В с образованием азеотропной смеси; и, во-вторых, отделения растворителя А от растворителя В обычными методами, которые известны специалисту в данной области техники, с получением "соединения формулы (I), растворенного в растворителе В".
В другом варианте осуществления "соединение формулы (I), растворенное в растворителе В", нагревают с получением "соединения формулы (I), растворенного в нагретом растворителе В". В частности, растворитель В нагревают до температуры по меньшей мере 40°С, предпочтительно до температуры 60-120°С, предпочтительно до температуры 90-110°C с получением "соединения формулы (I), растворенного в нагретом растворителе В".
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) дихлорметан (растворитель А) заменяют на н-бутанол (растворитель В) с помощью, во-первых, смешивания дихлорметана и н-бутанола; и, во-вторых, отгонки растворителя А.
Для того, чтобы отогнать дихлорметан, смесь дихлорметана и н-бутанола нагревают до температуры по меньшей мере 60°С, в частности, до температуры 60-120°С, предпочтительно до температуры 90-110°C с получением "соединения формулы (I), растворенного в нагретом н-бутаноле".
1.4.2 Кристаллизация полиморфной формы В соединения формулы (I)
Для получения полиморфной формы В соединения формулы (I), соединение формулы (I), растворенное в растворителе В, в частности, в нагретом растворителе В, предпочтительно в нагретом н-бутаноле, охлаждают.
Рентгеновская порошковая дифрактограмма полиморфной формы В соединения (I) показана на Фигуре 1.
В другом варианте осуществления настоящего изобретения после нагревания смеси растворителя А и растворителя В и после замены растворителя A, предпочтительно дихлорметана, на нагретый растворитель В, предпочтительно на нагретый н-бутанол, нагретый растворитель В, предпочтительно нагретый н-бутанол, охлаждают до 0-30°С.
В частности, выражение "после замены растворителя А, предпочтительно дихлорметана, на растворитель В, предпочтительно на н-бутанол" означает, что растворителя А, предпочтительно дихлорметана, не остается, когда растворитель B, предпочтительно н-бутанол, охлаждают.
В другом варианте осуществления настоящего изобретения после замены растворителя А, предпочтительно дихлорметана, на растворитель В, предпочтительно на н-бутанол, растворитель В нагревают до температуры по меньшей мере 60°С, в частности, до температуры 60-120°С, предпочтительно до температуры 90-110°С, и затем охлаждают до 0-30°С.
Предпочтительно растворитель В на первой стадии охлаждают до 15-30°С, предпочтительно до 20-30°С, и затем на второй стадии далее охлаждают до 0-5°С, предпочтительно до 2-4°С.
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) растворитель В охлаждают до 0-30°С, предпочтительно до 20-30°С, в течение 2-36 часов, в частности, в течение 3-24 часов, предпочтительно в течение 4-12 часов, наиболее предпочтительно в течение 6-10 часов.
Предпочтительно растворитель В, во-первых, в течение 2-36 часов, в частности, в течение 3-24 часов, предпочтительно в течение 4-12 часов, наиболее предпочтительно в течение 6-10 часов, охлаждают от температуры 60-120°С, предпочтительно от температуры 90-110°С, до температуры 15-30°С, предпочтительно до температуры 20-30°С, и, во-вторых, растворитель В затем далее охлаждают до температуры 0-5°С, предпочтительно до температуры 2-4°С, и температуру поддерживают постоянной в течение по меньшей мере 0.5 часа, в частности, в течение 0.5-12 часов, предпочтительно в течение 1-8 часов.
Предпочтительно растворитель В,
a) во-первых, в течение 2-36 часов, в частности, в течение 3-24 часов, предпочтительно в течение 4-12 часов, наиболее предпочтительно в течение 6-10 часов, охлаждают до температуры 15-30°С, предпочтительно до температуры 20-30°С; и
b) во-вторых, температуру растворителя В поддерживают постоянной в течение по меньшей мере 0.5 часа, в частности, в течение 1-12 часов, предпочтительно в течение 1-8 часов, наиболее предпочтительно в течение 1-2 часов; и
c) в-третьих, растворитель B в течение 2-36 часов, в частности, в течение 3-24 часов, предпочтительно в течение 4-12 часов, наиболее предпочтительно в течение 6-10 часов, далее охлаждают до температуры 0-5°С, предпочтительно до температуры 2-4°С; и
d) в-четвертых, температуру растворителя В поддерживают постоянной в течение по меньшей мере 0.5 часа, в частности, в течение 0.5-12 часов, предпочтительно в течение 0.5-8 часов, наиболее предпочтительно в течение 0.5-2 часов.
В другом варианте осуществления, например, для улучшения фильтрационных свойств суспензии, после охлаждения растворителя В, в частности, охлаждения до определенных выше температур, растворитель В снова нагревают до температуры по меньшей мере 40°С, предпочтительно до температуры 60-120°С, предпочтительно до температуры 90-110°С, например, до температуры 60-100°С, в частности, в течение периода времени 0.5-10 часов, предпочтительно в течение 1-4 часов, и затем снова охлаждают до температуры 0-30°С, предпочтительно до температуры 20-30°С, в течение 2 -36 часов, в частности, в течение 3-24 часов, предпочтительно в течение 4-12 часов, наиболее предпочтительно в течение 6-10 часов.
Кристаллизацию соединения формулы (I) в растворителе В, предпочтительно в н-бутаноле, осуществляют с использованием на кг соединения формулы (I) 2-10 кг, предпочтительно 3-6 кг, наиболее предпочтительно 3.5-5 кг растворителя В, предпочтительно н-бутанола.
После охлаждения растворителя В, в частности, в соответствии с описанной выше методикой охлаждения, полиморфную форму В соединения формулы (I) получают в кристаллической форме.
В другом варианте осуществления способа в соответствии с настоящим изобретением кристаллы полиморфной формы В соединения формулы (I) выделяют, в частности, их выделяют с помощью фильтрования.
В другом варианте осуществления способа в соответствии с настоящим изобретением выделенные кристаллы полиморфной формы В соединения формулы (I) можно дополнительно очистить путем растворения выделенных кристаллов полиморфной формы В соединения формулы (I) в растворителе А, предпочтительно в дихлорметане, и путем повторения один или несколько раз, в частности, один раз, замены растворителя, описанной в разделе 1.4.1, и/или путем повторения один или несколько раз, в частности, один раз, кристаллизации полиморфной формы В соединения формулы (I), описанной в раздел 1.4.2.
Выделенную полиморфную форму В соединения формулы (I) можно дополнительно очистить, в частности, ее можно промыть с помощью 1-10 кг, в частности, 1-7 кг, предпочтительно 1-3 кг, н-бутанола, в перерасчете на кг полиморфной формы В соединения формулы (I), предпочтительно с помощью охлажденного н-бутанола, имеющего температуру -5-10°С, предпочтительно -3°-5°С.
Полиморфную форму В соединения формулы (I) можно сушить при температуре 35-75°С, предпочтительно при температуре 50-75°С, наиболее предпочтительно при температуре 40-60°С.
Полиморфную форму В соединения формулы (I) можно сушить в вакууме при 20-100 мбар, предпочтительно при 30-50 мбар, и в вакууме при 20-100 мбар, предпочтительно при 30-50 мбар, и при температурах 35-75°С, предпочтительно в вакууме при 20-100 мбар, предпочтительно при 30-50 мбар, и при температурах 50-75°С, наиболее предпочтительно в вакууме при 20-100 мбар, предпочтительно при 30-50 мбар, и при температурах 40-60°С.
Способ кристаллизации для получения полиморфной формы В соединения формулы (I) очень эффективен в отношении обеспечения чистоты соединения формулы (I).
2. Синтез промежуточного соединения формулы (IXa) или (IXb) путем осуществления реакции промежуточного соединения формулы (VIIIa) или (VIIIb) с соединением формулы (VIa) или (VIb)
В соответствии с другим аспектом настоящее изобретение охватывает способ получения соединения формулы (IXa) или (IXb), включающий стадию осуществления реакции промежуточного соединения формулы (VIIIa)
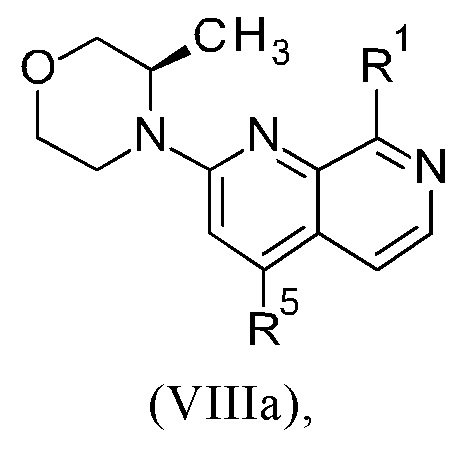
где
R1 представляет собой атом хлора, брома или йода или представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси; и
R5 представляет собой атом хлора, брома или йода или представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси;
с соединением формулы (VIa)

где
R2 и R3 представляют собой, независимо друг от друга, атом водорода или C1-С6-алкильную группу;
или
R2 и R3 вместе представляют собой -СН2-СН2- группу или -СН2-СН2-СН2-группу, где указанная -СН2-СН2- группа или -СН2-СН2-СН2- группа необязательно замещена один, два, три или четыре раза группой, выбранной из метила и этила;
или
R2 и R3 вместе представляют собой группу
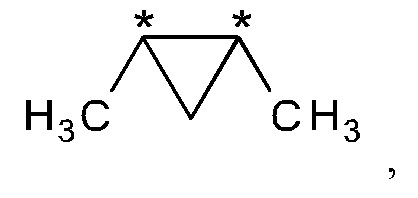
где "*" представляет собой место присоединения к остальной части молекулы;
с получением промежуточного соединения формулы (IXa)

где R1 представляет собой атом хлора, брома или йода или представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси.
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I) способ дополнительно включает (перед реакцией промежуточного соединения формулы (IXa) или (IXb) с соединением формулы (IIIa), (IIIb) или (IIIc)) следующую стадию:
(а) осуществление реакции промежуточного соединения формулы (VIIIa)
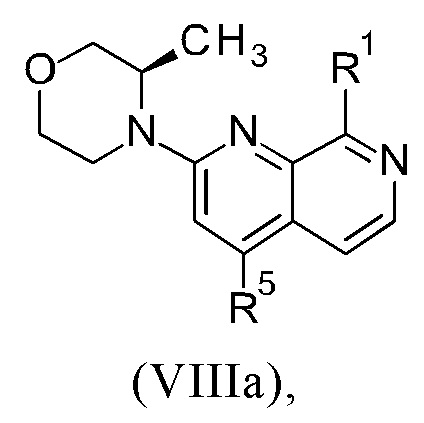
где
R1 представляет собой атом хлора, брома или йода или представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси; и
R5 представляет собой атом хлора, брома или йода или представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси;
с соединением формулы (VIa)
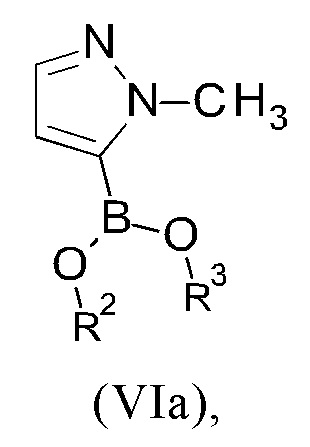
где
R2 и R3 представляют собой, независимо друг от друга, атом водорода или C1-С6-алкильную группу;
или
R2 и R3 вместе представляют собой -СН2-СН2- группу или -СН2-СН2-СН2-группу, где указанная -СН2-СН2- группа или -СН2-СН2-СН2- группа необязательно замещена один, два, три или четыре раза группой, выбранной из метила и этила;
или
R2 и R3 вместе представляют собой группу
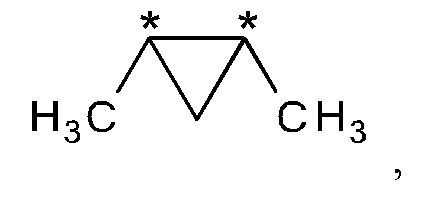
где "*" представляет собой место присоединения к остальной части молекулы;
с получением промежуточного соединения формулы (IXa)

где R1 представляет собой атом хлора, брома или йода или представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси.
В другом варианте осуществления способа в соответствии с настоящим изобретением R1 соединения формулы (VIIIa) представляет собой атом хлора или брома, предпочтительно атом хлора.
В другом варианте осуществления способа в соответствии с настоящим изобретением R1 соединения формулы (IXa) представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси.
В другом варианте осуществления способа в соответствии с настоящим изобретением R1 соединения формулы (VIIIa) представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси и (п-толуолсульфонил)окси.
В другом варианте осуществления способа в соответствии с настоящим изобретением R5 соединения формулы (VIIIa) представляет собой атом хлора, брома или йода.
В другом варианте осуществления способа в соответствии с настоящим изобретением R5 соединения формулы (IXa) представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси.
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением R5 соединения формулы (VIIIa) представляет собой [(трифторметил)сульфонил]окси группу.
В другом варианте осуществления способа в соответствии с настоящим изобретением R1 соединения формулы (VIIIa) представляет собой атом хлора или брома, предпочтительно атом хлора, и R5 представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси, предпочтительно [(трифторметил)сульфонил]окси группы.
Соединение формулы (VIIIa), где R1 представляет собой атом хлора и где R5 представляет собой [(трифторметил)сульфонил]окси группу, представляет собой (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфонат, который является предпочтительным соединением формулы (VIIIb):
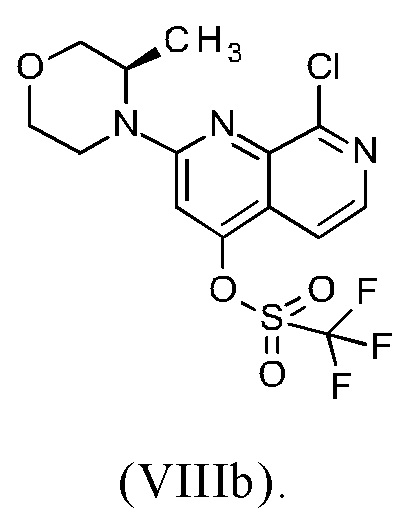
В другом варианте осуществления настоящего изобретения R2 и R3 соединения формулы (VIa) представляют собой, независимо друг от друга, атом водорода или С1-С3 алкильную группу, в частности, метильную или этильную группу.
В другом варианте осуществления настоящего изобретения R2 и R3 соединения формулы (VIa) вместе представляют собой -СН2-СН2- группу или -СН2-СН2-СН2- группу, где указанная -СН2-СН2- группа или -СН2-СН2-СН2- группа необязательно замещена один, два, три или четыре раза группой, выбранной из метила и этила.
Если R2 и R3 соединения формулы (VIa) вместе представляют собой -СН2-СН2- группу или -СН2-СН2-СН2- группу, указанная -СН2-СН2- группа или указанная -СН2-СН2-СН2- группа вместе с атомом бора и атомами кислорода, к которым указанная группа присоединена, образует 5- или 6-членное кольцо.
В другом варианте осуществления настоящего изобретения R2 и R3 соединения формулы (VIa) вместе представляют собой группу
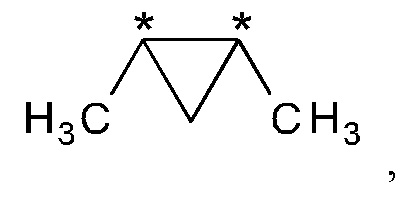
где "*" представляет собой место присоединения к остальной части молекулы.
В другом варианте осуществления настоящего изобретения R2 и R3 соединения формулы (VIa) вместе представляют собой -С(СН3)2-С(СН3)2- или -СН2-С(СН3)2-СН2- группу.
В предпочтительном варианте осуществления настоящего изобретения R2 и R3 соединения формулы (VIa) вместе представляют собой -С(СН3)2-С(СН3)2-группу.
В другом варианте осуществления настоящего изобретения соединение формулы (VIa) представляет собой 1-метил-1Н-пиразол-5-бороновую кислоту.
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (VIa) представляет собой сложный пинаколовый эфир 1-метил-1Н-пиразол-5-бороновой кислоты (формулы VIb):
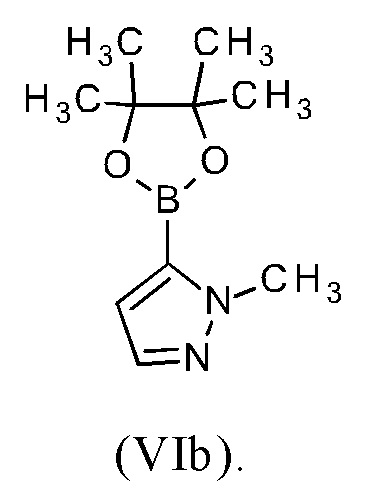
Соединения формулы (VIa) или (VIb) являются коммерчески доступными или могут быть синтезированы способами, известными специалисту в данной области техники.
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (VIIIa) представляет собой (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфонат (VIIIb) и соединение формулы (VIa) представляет собой сложный пинаколовый эфир 1-метил-1Н-пиразол-5-бороновой кислоты (VIb).
В другом варианте осуществления способа в соответствии с настоящим изобретением промежуточное соединение формулы (VIIIa) подвергают реакции с 0.7-2.0 молярными эквивалентами соединения формулы (VIa), предпочтительно с 0.8-1.5 молярными эквивалентами соединения формулы (VIa), наиболее предпочтительно с 0.9-1.2 молярными эквивалентами соединения формулы (VIa).
В другом варианте осуществления способа в соответствии с настоящим изобретением промежуточное соединение формулы (VIIIb) подвергают реакции с 0.7-2.0 молярными эквивалентами соединения формулы (VIb), предпочтительно с 0.8-1.5 молярными эквивалентами соединения формулы (VIb), наиболее предпочтительно с 0.9-1.2 молярными эквивалентами соединения формулы (VIb).
В другом варианте осуществления способа в соответствии с настоящим изобретением соединение формулы (VIa) или (VIb), в частности, сложный пинаколовый эфир 1-метил-1Н-пиразол-5-бороновой кислоты (VIb), растворяют в растворителе, например, в изопропилацетате, этилацетате, 1,2-диметоксиэтане, диоксане, N,N-диметилформамиде (=ДМФА), 1,2-диметоксиэтане (=DME), тетрагидрофуране (=ТГФ), 2-метилтетрагидрофуране (=2-Ме-ТГФ) или изопропаноле.
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением соединение формулы (VIa) или (VIb), в частности, сложный пинаколовый эфир 1-метил-1Н-пиразол-5-бороновой кислоты (VIb), растворяют в изопропилацетате или этилацетате, наиболее предпочтительно в этилацетате.
В другом варианте осуществления способа в соответствии с настоящим изобретением соединение формулы (VIIIa) или (VIIIb) растворяют в растворителе, например, в изопропилацетате, этилацетате, 1,2-диметоксиэтане, диоксане, N,N-диметилформамиде (=ДМФА), 1,2-диметоксиэтане (=DME), тетрагидрофуране (=ТГФ), 2-метилтетрагидрофуране (=2-Ме-ТГФ) или изопропаноле.
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением соединение формулы (VIIIa) или (VIIIb), в частности, (VIIIa), растворяют в изопропилацетате или этилацетате, наиболее предпочтительно в этилацетате.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (VIIIa) или (VIIIb), в частности, (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфоната (VIIIb), с соединением формулы (VIa) или (VIb), в частности, сложным пинаколовым эфиром 1-метил-1Н-пиразол-5-бороновой кислоты (VIb), осуществляют в органическом растворителе, включающем растворитель, который выбирают из группы, состоящей из изопропилацетата, этилацетата, 1,2-диметоксиэтана, 1,4-диоксана, диметилформамида, тетрагидрофурана, 2-метилтетрагидрофурана и изопропанола; или в смеси растворителей, включающей один или несколько из указанных органических растворителей и воду.
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (VIIIa) или (VIIIb), в частности, (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфоната (VIIIb), с соединением формулы (VIa) или (VIb), в частности, со сложным пинаколовым эфиром 1-метил-1Н-пиразол-5-бороновой кислоты (VIb), осуществляют в растворителе, включающем этилацетат или изопропилацетат, или в смеси растворителей, включающей этилацетат и воду или изопропилацетат и воду, наиболее предпочтительно в смеси растворителей, включающей этилацетат и воду.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (VIIIa) или (VIIIb), в частности, (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфоната (VIIIb), с соединением формулы (VIa) или (VIb), в частности, сложным пинаколовым эфиром 1-метил-1Н-пиразол-5-бороновой кислоты (VIb), осуществляют в течение 1-12 часов, в частности, в течение 1-5 часов, предпочтительно в течение 1-3 часов, наиболее предпочтительно в течение 60-90 минут.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (VIIIa) или (VIIIb), в частности, (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфоната (VIIIb), с соединением формулы (VIa) или (VIb), в частности, со сложным пинаколовым эфиром 1-метил-1Н-пиразол-5-бороновой кислоты (VIb), осуществляют в присутствии подходящей каталитической системы, такой как, например, палладиевый катализатор.
В другом варианте осуществления способа в соответствии с настоящим изобретением палладиевый катализатор выбирают из группы, состоящей из [1,1'-бис(дифенилфосфино)ферроцен]дихлор палладия(II), ацетата палладия(II), дихлорида бис(трифенилфосфин)палладия(II), дихлорбис(трициклогексилфосфин)палладия(II), хлорида (2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2-аминоэтил)фенил)]палладия(II), хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладия(II), тетракис(трифенилфосфин)палладия(0), бис(три-трет-бутилфосфин)палладия(0), бис[трис(2-метилфенил)фосфин]палладия(0), трис(дибензилиденацетон)дипалладия(0).
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением палладиевый катализатор представляет собой [1,1'-бис(дифенилфосфино)ферроцен]дихлор палладий(II).
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (VIIIa) или (VIIIb), в частности, (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфоната (VIIIb), с соединением формулы (VIa) или (VIb), в частности, со сложным пинаколовым эфиром 1-метил-1Н-пиразол-5-бороновой кислоты (VIb), осуществляют в присутствии 0.001-0.1 молярных эквивалентов, предпочтительно 0.005-0.05 молярных эквивалентов, наиболее предпочтительно 0.0-0.03 молярных эквивалентов, палладиевого катализатора, предпочтительно [1,1'-бис(дифенилфосфино)ферроцен]дихлор палладия(II).
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (VIIIa) или (VIIIb), в частности, (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфоната (VIIIb), с соединением формулы (VIa) или (VIb), в частности, со сложным пинаколовым эфиром 1-метил-1Н-пиразол-5-бороновой кислоты (VIb), осуществляют в присутствии основания. Можно использовать такие основания, как фосфат калия, карбонат калия, гидрокарбонат калия, фосфат натрия, карбонат натрия, гидрокарбонат натрия, гидроксид бария, карбонат бария, карбонат цезия или карбонат лития. Карбонат калия или гидрокарбонат калия являются предпочтительными, наиболее предпочтительным является гидрокарбонат калия.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (VIIIa) или (VIIIb), в частности, (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфоната (VIIIb), с соединением формулы (VIa) или (VIb), в частности, со сложным пинаколовым эфиром 1-метил-1Н-пиразол-5-бороновой кислоты (VIb), осуществляют в присутствии 1-15 молярных эквивалентов, предпочтительно 2-8 молярных эквивалентов, наиболее предпочтительно 3-5 или 3.5-4.5 молярных эквивалентов основания.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (VIIIa) или (VIIIb), в частности, (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфоната (VIIIb), с соединением формулы (VIa) или (VIb), в частности, со сложным пинаколовым эфиром 1-метил-1Н-пиразол-5-бороновой кислоты (VIb), осуществляют при температурах в диапазоне от комнатной температуры до температуры кипения растворителя.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (VIIIa) или (VIIIb), в частности, (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфоната (VIIIb), с соединением формулы (VIa) или (VIb), в частности, со сложным пинаколовым эфиром 1-метил-1Н-пиразол-5-бороновой кислоты (VIb), осуществляют под давлением при температурах выше температуры кипения.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (VIIIa) или (VIIIb), в частности, (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфоната (VIIIb), с соединением формулы (VIa) или (VIb), в частности, со сложным пинаколовым эфиром 1-метил-1Н-пиразол-5-бороновой кислоты (VIb), осуществляют в изопропилацетате или в изопропилацетате и воде при температуре 35-75°С, предпочтительно при 40-60°С, наиболее предпочтительно при 45-55°С.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (VIIIa) или (VIIIb), в частности, (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфоната (VIIIb), с соединением формулы (VIa) или (VIb), в частности, со сложным пинаколовым эфиром 1-метил-1Н-пиразол-5-бороновой кислоты (VIb), осуществляют в этилацетате или в этилацетате и воде, предпочтительно в этилацетате и воде, при температуре 30-60°С, предпочтительно при 35-50°С, наиболее предпочтительно при 38-45°С.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (VIIIa) или (VIIIb), в частности, (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфоната (VIIIb), с соединением формулы (VIa) или (VIb), в частности, со сложным пинаколовым эфиром 1-метил-1Н-пиразол-5-бороновой кислоты (VIb), осуществляют в атмосфере инертного газа, где инертный газ представляет собой азот или аргон, предпочтительно азот.
2.1 Дальнейшая обработка сырого соединения формулы (IXa) или (IXb) и кристаллизация соединения формулы (IXa) или (IXb)
В другом варианте осуществления способа в соответствии с настоящим изобретением в случае реакции промежуточного соединения формулы (VIIIa) или (VIIIb), в частности, (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфоната (VIIIb), с соединением формулы (VIa) или (VIb), в частности, со сложным пинаколовым эфиром 1-метил-1Н-пиразол-5-бороновой кислоты (VIb), органический растворитель или смесь растворителей, в частности, этилацетат с водой/без воды, или изопропилацетат с водой/без воды, необязательно
a) промывают водным раствором основания, выбранного из гидроксида калия, карбоната калия, гидрокарбоната калия, гидроксида калия, трет-бутоксида калия, гидроксида натрия, фосфата натрия, карбоната натрия, гидроксида натрия, трет-бутоксида натрия, гидроксида бария, карбоната цезия, триэтиламина; предпочтительно гидроксидом калия; и/или, необязательно,
b) промывают водой; и/или, необязательно
c) обрабатывают адсорбентом; предпочтительно адсорбент представляет собой активированный уголь; и/или необязательно
d) адсорбент, в частности, активированный уголь, отфильтровывают;
с получением раствора соединения формулы (IXa) или (IXb) в указанном органическом растворителе или смеси растворителей, предпочтительно раствора соединения формулы (IXa) или (IXb) в этилацетате или в изопропилацетате, наиболее предпочтительно в этилацетате.
В другом варианте осуществления способа в соответствии с настоящим изобретением растворитель или смесь растворителей указанного раствора соединения формулы (IXa) или (IXb), предпочтительно этилацетат или изопропилацетат, наиболее предпочтительно этилацетат, заменяют на другой растворитель (растворитель С в дальнейшем). Растворитель С включает растворитель, выбранный из метанола, этанола, н-пропанола, н-бутанола, 2-бутанола, изопропанола, предпочтительно растворитель заменяют на изопропанол.
Кристаллизацию соединения формулы (IXa) или (IXb) предпочтительно осуществляют в растворителе С, предпочтительно в изопропаноле.
Кристаллизацию соединения формулы (IXa) или (IXb) в растворителе С, предпочтительно в изопропаноле, выполняют, используя на кг соединения формулы (IXa) или (IXb) 2-20 кг, предпочтительно 2.5-10 кг, наиболее предпочтительно 2.5-6 кг, растворителя С, предпочтительно изопропанола, и продукт затем можно выделить.
Выделенное соединение формулы (IXa) или (IXb) можно сушить при температуре 35-75°С, предпочтительно при 40-65°С, наиболее предпочтительно при 45-55°С.
Выделенное соединение формулы (IXa) или (IXb) можно сушить в вакууме при 20-100 мбар, предпочтительно при 30-50 мбар, и в вакууме при 20-100 мбар, предпочтительно при 30-50 мбар, и при температурах 35-75°С, предпочтительно в вакууме при 20-100 мбар, предпочтительно при 30-50 мбар, и при температурах 50-75°С, наиболее предпочтительно в вакууме при 20-100 мбар, предпочтительно при 30-50 мбар, и при температурах 40-60°С.
3. Синтез промежуточного соединения формулы (VIIIa) или (VIIIb), исходя из соединения формулы (II) или (IIa)
В соответствии с другим аспектом настоящее изобретение охватывает способ получения соединения формулы (VIIIa), включающий стадию осуществления реакции промежуточного соединения формулы (II)
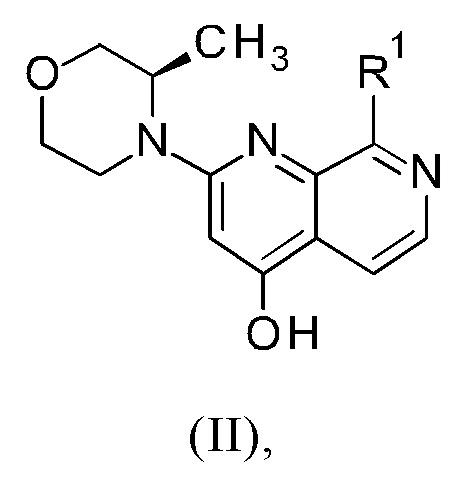
где
R1 представляет собой атом хлора, брома или йода или представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси;
с соединением, выбранным из N-фенил-бис(трифторметансульфонимида), трифторметансульфонового ангидрида, хлорангидрида метансульфоновой кислоты, п-толуолсульфонилхлорида, нонафторбутансульфонилхлорида, нонафторбутансульфонилфторида, бензолсульфонилхлорида, 4-бромбензолсульфонилхлорида, 4-нитробензолсульфонилхлорида, 2-нитробензолсульфонилхлорида, 4-изопропилбензолсульфонилхлорида, 2,4,6-триизопропилбензолсульфонилхлорида, 2-мезитиленсульфонилхлорида (=2,4,6-триметилбензолсульфонилхлорид), 4-трет-бутилбензолсульфонилхлорида и 4-метоксибензолсульфонилхлорида;
с получением промежуточного соединения формулы (VIIIa)

где
R1 представляет собой атом хлора, брома или йода или представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси;
R5 представляет собой атом хлора, брома или йода или представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси.
В другом варианте осуществления способа в соответствии с настоящим изобретением получения соединения формулы (I), способ дополнительно включает (перед реакцией промежуточного соединения формулы (VIIIa) или (VIIIb) с соединением формулы (VIa) или (VIb)) стадию осуществления реакции промежуточного соединения формулы (II)

где
R1 представляет собой атом хлора, брома или йода или представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси;
с соединением, выбранным из N-фенил-бис(трифторметансульфонимида), трифторметансульфонового ангидрида, хлорангидрида метансульфоновой кислоты, п-толуолсульфонилхлорида, нонафторбутансульфонилхлорида, нонафторбутансульфонилфторида, бензолсульфонилхлорида, 4-бромбензолсульфонилхлорида, 4-нитробензолсульфонилхлорида, 2-нитробензолсульфонилхлорида, 4-изопропилбензолсульфонилхлорида, 2,4,6-триизопропилбензолсульфонилхлорида, 2-мезитиленсульфонилхлорида (=2,4,6-триметилбензолсульфонилхлорид), 4-трет-бутилбензолсульфонилхлорида и 4-метоксибензолсульфонилхлорида;
с получением промежуточного соединения формулы (VIIIa)
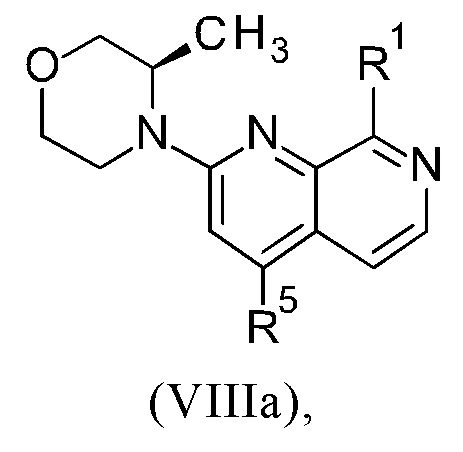
где
R1 представляет собой атом хлора, брома или йода или представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси;
R5 представляет собой атом хлора, брома или йода или представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси.
В другом варианте осуществления способа в соответствии с настоящим изобретением R1 соединения формулы (II) представляет собой атом хлора или брома, предпочтительно атом хлора. Соединение формулы (II), где R1 представляет собой атом хлора, является предпочтительным соединением формулы (IIa) (=(R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ол):
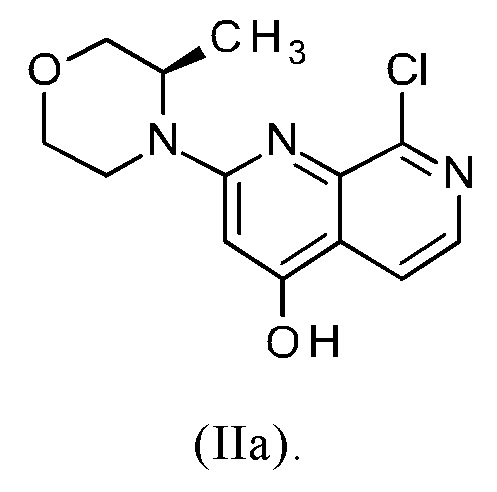
В другом варианте осуществления способа в соответствии с настоящим изобретением R1 соединения формулы (II) представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси.
В другом варианте осуществления способа в соответствии с настоящим изобретением R1 соединения формулы (II) представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси и (п-толуолсульфонил)окси.
В другом варианте осуществления способа в соответствии с настоящим изобретением R1 соединения формулы (VIIIa) представляет собой атом хлора или брома, предпочтительно атом хлора.
В другом варианте осуществления способа в соответствии с настоящим изобретением R1 соединения формулы (VIIIa) представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси.
В другом варианте осуществления способа в соответствии с настоящим изобретением R1 соединения формулы (VIIIa) представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси и (п-толуолсульфонил)окси.
В другом варианте осуществления способа в соответствии с настоящим изобретением R5 соединения формулы (VIIIa) представляет собой атом хлора, брома или йода.
В другом варианте осуществления способа в соответствии с настоящим изобретением R5 соединения формулы (IXa) представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси.
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением R5 соединения формулы (VIIIa) представляет собой [(трифторметил)сульфонил]окси группу.
В другом варианте осуществления способа в соответствии с настоящим изобретением R1 соединения формулы (VIIIa) представляет собой атом хлора или брома, предпочтительно атом хлора, и R5 представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (п-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси, предпочтительно [(трифторметил)сульфонил]окси группу.
Соединение формулы (VIIIa), где R1 представляет собой атом хлора и где R5 представляет собой [(трифторметил)сульфонил]окси группу, представляет собой (R)-8-хлор-2-(3-метил морфолино)-1,7-нафтиридин-4-ил трифторметансульфонат, который является предпочтительным соединением формулы (VIIIb):
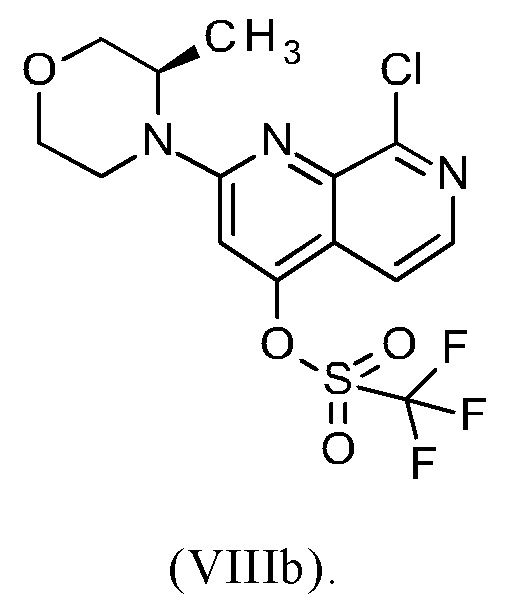
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (II) представляет собой ^)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ол (IIa) и соединение формулы (VIIIa) представляет собой (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфонат (VIIIb).
В другом варианте осуществления способа в соответствии с настоящим изобретением промежуточное соединение формулы (II), предпочтительно соединение формулы (IIa), подвергают реакции с 0.8-2.0, предпочтительно с 0.9-1.7, наиболее предпочтительно с 1.0-1.5, молярными эквивалентами соединения, выбранного из N-фенил-бис(трифторметансульфонимида), трифторметансульфонового ангидрида, хлорангидрида метансульфоновой кислоты, п-толуолсульфонилхлорида, нонафторбутансульфонилхлорида, нонафторбутансульфонилфторида, бензолсульфонилхлорида, 4-бромбензолсульфонилхлорида, 4-нитробензолсульфонилхлорида, 2-нитробензолсульфонилхлорида, 4-изопропилбензолсульфонилхлорида, 2,4,6-триизопропилбензолсульфонилхлорида, 2-мезитиленсульфонилхлорида (=2,4,6-триметилбензолсульфонилхлорид), 4-трет-бутилбензолсульфонилхлорида и 4-метоксибензолсульфонилхлорида.
Предпочтительно соединение формулы (IIa) подвергают реакции с 0.8-2.0, предпочтительно с 0.9-1.7, наиболее предпочтительно с 1.0-1.5, молярными эквивалентами трифторметансульфонового ангидрида или N-фенил-бис(трифторметансульфонимида), наиболее предпочтительно с 0.8-2.0, предпочтительно с 0.9-1.7, наиболее предпочтительно с 1.0-1.5, молярными эквивалентами трифторметансульфонового ангидрида.
В другом варианте осуществления способа, вышеупомянутую реакцию осуществляют в апротонном растворителе, таком как дихлорметан, тетрагидрофуран, пиридин, этилацетат, изопропилацетат, ацетонитрил, 2-метилтетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, ацетон, 2-бутанон, бутилацетат, циклопентилметиловый эфир, метил-трет-бутиловый эфир, толуол, пропионитрил, хлорбензол, анизол, хлороформ, предпочтительный растворитель представляет собой дихлорметан.
На кг соединения формулы (II) или (IIa) используют 4-20 кг растворителя, предпочтительно 6-15 кг растворителя, наиболее предпочтительно 7-9 кг растворителя, предпочтительно дихлорметана.
В другом варианте осуществления реакцию промежуточного соединения формулы (II) с соединением, выбранным из N-фенил-бис(трифторметансульфонимида), трифторметансульфонового ангидрида, хлорангидрида метансульфоновой кислоты, п-толуолсульфонилхлорида, нонафторбутансульфонилхлорида, нонафторбутансульфонилфторида, бензолсульфонилхлорида, 4-бромбензолсульфонилхлорида, 4-нитробензолсульфонилхлорида, 2-нитробензолсульфонилхлорида, 4-изопропилбензолсульфонилхлорида, 2,4,6-триизопропилбензолсульфонилхлорида, 2-мезитиленсульфонилхлорида (=2,4,6-триметилбензолсульфонилхлорид), 4-трет-бутилбензолсульфонилхлорида и 4-метоксибензолсульфонилхлорида, предпочтительно с трифторметансульфоновым ангидридом или N-фенил-бис(трифторметансульфонимидом), осуществляют в присутствии основания, в частности, органического основания или неорганического основания или смесей одного или нескольких органических оснований с одним или несколькими неорганическими основаниями.
Для вышеупомянутой реакции можно использовать органические основания, такие как пиридин, N,N-диэтилэтанамин (=триэтиламин), N,N-ди(пропан-2-ил)пропан-2-амин (=триизопропиламин), N,N-дибутилбутан-1-амин (=трибутиламин), 2,6-диметилпиридин (=2,6-лутидин), N-этил-N-(пропан-2-ил)пропан-2-амин (=N,N-диизопропилэтиламин или основания Хюнига), N-метилморфолин предпочтительными основаниями являются N,N-диэтилэтанамин (=триэтиламин) и пиридин, наиболее предпочтительным является пиридин.
Применение пиридина, в частности, является предпочтительным, чтобы избежать нежелательного изменения окраски соединения формулы (VIIIa) или (VIIIb).
Неорганическими основаниями, которые можно использовать для вышеупомянутой реакции, являются, например, карбонат калия, гидрокарбонат калия, фосфат натрия, карбонат натрия, гидрокарбонат натрия, карбонат кальция, гидрокарбонат кальция или карбонат цезия.
В другом варианте осуществления способа в соответствии с настоящим изобретением промежуточное соединение формулы (II), предпочтительно соединение формулы (IIa), подвергают реакции с соединением, выбранным из N-фенил-бис(трифторметансульфонимида), трифторметансульфонового ангидрида, хлорангидрида метансульфоновой кислоты, п-толуолсульфонилхлорида, нонафторбутансульфонилхлорида, нонафторбутансульфонилфторида, бензолсульфонилхлорида, 4-бромбензолсульфонилхлорида, 4-нитробензолсульфонилхлорида, 2-нитробензолсульфонилхлорида, 4-изопропилбензолсульфонилхлорида, 2,4,6-триизопропилбензолсульфонилхлорида, 2-мезитиленсульфонилхлорида (=2,4,6-триметилбензолсульфонилхлорид), 4-трет-бутилбензолсульфонилхлорида и 4-метоксибензолсульфонилхлорида, предпочтительно с трифторметансульфоновым ангидридом или N-фенил-бис(трифторметансульфонимидом), в присутствии 0.7-4.0, предпочтительно 0.9-3.0, наиболее предпочтительно 1.0-2.0, молярных эквивалентов основания, предпочтительно N,N-диэтилэтанамина или пиридина, наиболее предпочтительно пиридина.
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением промежуточное соединение формулы (II), предпочтительно соединение формулы (IIa), подвергают реакции с трифторметансульфоновым ангидридом в присутствии 0.7-4.0, предпочтительно 0.9-3.0, наиболее предпочтительно 1.0-2.0, молярных эквивалентов пиридина.
В другом варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (II) с соединением, выбранным из]^-фенил-бис(трифторметансульфонимида), трифторметансульфонового ангидрида, хлорангидрида метансульфоновой кислоты, п-толуолсульфонилхлорида, нонафторбутансульфонилхлорида, нонафторбутансульфонилфторида, бензолсульфонилхлорида, 4-бромбензолсульфонилхлорида, 4-нитробензолсульфонилхлорида, 2-нитробензолсульфонилхлорида, 4-изопропилбензолсульфонилхлорида, 2,4,6-триизопропилбензолсульфонилхлорида, 2-мезитиленсульфонилхлорида (=2,4,6-триметилбензолсульфонилхлорид), 4-трет-бутилбензолсульфонилхлорида и 4-метоксибензолсульфонилхлорида, предпочтительно с трифторметансульфоновым ангидридом или N-фенил-бис(трифторметансульфонимидом), осуществляют при температурах -30-30°С, предпочтительно -20-20°С, наиболее предпочтительно при -15-0°С. Время реакции может составлять от 1 до 24 часов, предпочтительное время составляет 1-4 часа.
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением промежуточное соединение формулы (II), предпочтительно соединение формулы (IIa), подвергают реакции с трифторметансульфоновым ангидридом в присутствии 0.7-4.0, предпочтительно 0.9-3.0, наиболее предпочтительно 1.0-2.0, молярных эквивалентов пиридина при температурах -30-30°С, предпочтительно -20-5°С, наиболее предпочтительно от -15 до -5°С. Время реакции может составлять от 1 до 24 часов, предпочтительное время составляет 1-4 часа.
3.1 Дальнейшая обработка сырого соединения формулы (VIIIa) или (VIIIb) и кристаллизация соединения формулы (VIIIa) или (VIIIb)
В другом варианте осуществления способа в соответствии с настоящим изобретением в случае реакции промежуточного соединения формулы (II) с соединением, выбранным из N-фенил-бис(трифторметансульфонимида), трифторметансульфонового ангидрида, хлорангидрида метансульфоновой кислоты, п-толуолсульфонилхлорида, нонафторбутансульфонилхлорида, нонафторбутансульфонилфторида, бензолсульфонилхлорида, 4-бромбензолсульфонилхлорида, 4-нитробензолсульфонилхлорида, 2-нитробензолсульфонилхлорида, 4-изопропилбензолсульфонилхлорида, 2,4,6-триизопропилбензолсульфонилхлорида, 2-мезитиленсульфонилхлорида (=2,4,6-триметилбензолсульфонилхлорид), 4-трет-бутилбензолсульфонилхлорида и 4-метоксибензолсульфонилхлорида, предпочтительно с трифторметансульфоновым ангидридом или N-фенил-бис(трифторметансульфонимидом), органический растворитель или смесь растворителей, в частности, дихлорметан, необязательно
a) промывают водой; и/или, необязательно
b) промывают водным раствором основания, выбранного из гидроксида калия, карбоната калия, гидрокарбоната калия, гидроксида калия, трет-бутоксида калия, гидроксида натрия, фосфата натрия, карбоната натрия, гидроксида натрия, трет-бутоксида натрия, гидроксида бария, карбоната цезия, триэтиламина; предпочтительно водным раствором карбоната калия; и/или, необязательно,
c) после промывания водным раствором основания снова промывают водой;
d) обрабатывают адсорбентом; предпочтительно адсорбент представляет собой активированный уголь; и/или необязательно
e) адсорбент, в частности, активированный уголь, отфильтровывают;
с получением раствора соединения формулы (VIIIa) или (VIIIb) в указанном органическом растворителе или смеси растворителей, предпочтительно раствора соединения формулы (VIIIa) или (VIIIb) в дихлорметане.
Последовательность выполнения операций дальнейшей обработки сырого соединения формулы (VIIIa) или (VIIIb) в соответствии со стадиями а) (промывание водой), b) (промывание водным раствором основания), с) (промывание водой после промывания водным раствором основания), d) (обработка адсорбентом) и/или е) (отфильтровывание адсорбента), как описано выше, может быть изменена. Например, стадии d) и е) можно выполнить до стадий а), b) и с).
В другом варианте осуществления способа в соответствии с настоящим изобретением растворитель или смесь растворителей указанного раствора соединения формулы (VIIIa) или (VIIIb), предпочтительно дихлорметан, заменяют на другой растворитель (растворитель D в дальнейшем). Растворитель D включает растворитель, выбранный из метанола, этанола, н-пропанола, н-бутанола, 2-бутанола, изопропанола, предпочтительно растворитель заменяют на изопропанол.
В предпочтительном варианте осуществления способа в соответствии с настоящим изобретением реакцию промежуточного соединения формулы (II) с соединением, выбранным из N-фенил-бис(трифторметансульфонимида), трифторметансульфонового ангидрида, хлорангидрида метансульфоновой кислоты, n-толуолсульфонилхлорида, нонафторбутансульфонилхлорида, нонафторбутансульфонилфторида, бензолсульфонилхлорида, 4-бромбензолсульфонилхлорида, 4-нитробензолсульфонилхлорида, 2-нитробензолсульфонилхлорида, 4-изопропилбензолсульфонилхлорида, 2,4,6-триизопропилбензолсульфонилхлорида, 2-мезитиленсульфонилхлорида (=2,4,6-триметилбензолсульфонилхлорид), 4-трет-бутилбензолсульфонилхлорида и 4-метоксибензолсульфонилхлорида осуществляют в атмосфере инертного газа, где инертный газ представляет собой азот или аргон, предпочтительно азот.
Кристаллизацию соединения формулы (VIIIa) или (VIIIb) предпочтительно осуществляют в растворителе D, предпочтительно в изопропаноле.
На кг соединения формулы (VIIIa) или (VIIIb) используют 2 - 6 кг органического растворителя, предпочтительно 3 - 5 кг органического растворителя, наиболее предпочтительно 3 - 4 кг органического растворителя, предпочтительно изопропанола.
Например, продукт можно выделить с помощью фильтрования или путем центрифугирования. Его можно сушить при температуре 25 - 60°С, предпочтительно при 40 - 50°С. Его можно сушить в течение 1-24 часов, предпочтительно 8-15 часов, наиболее предпочтительно 10 - 14 часов.
Обработка сырого соединения формулы (VIIIa) или (VIIIb) и его последующая кристаллизация из изопропанола дает соединение формулы (VIIIa) или (VIIIb) с высокой чистотой и выходом 64 - 87.7% от теоретического без использования стадии какой-либо хроматографической очистки.
Реакцию с получением соединения формулы (VIIIa) или (VIIIb) можно выполнить в больших масштабах (например, мультикилограммовых масштабах). Чистота полученного продукта была очень высокой (например, >98% (СВЭЖХ, % площади)).
4. Промежуточные соединения формулы (IХа) или (IXb)
В соответствии с другим аспектом настоящее изобретение охватывает промежуточное соединение формулы (IХа)

где
R1 представляет собой атом хлора, брома или йода или представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (n-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси.
В другом варианте осуществления настоящее изобретение относится к промежуточному соединению формулы (IХа), где R1 представляет собой атом хлора или брома, предпочтительно атом хлора.
В другом варианте осуществления настоящее изобретение относится к промежуточному соединению формулы (IХа), где R1 представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (n-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси.
В другом варианте осуществления настоящее изобретение относится к промежуточному соединению формулы (IХа), где R1 представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси и (n-толуолсульфонил)окси.
В предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы (IXb):
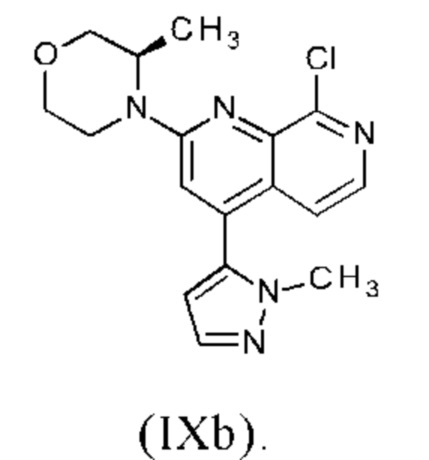
5. Промежуточные соединения формулы (VIIIа) или (VIIIb) В соответствии с другим аспектом настоящее изобретение охватывает промежуточное соединение формулы (VIIIa)
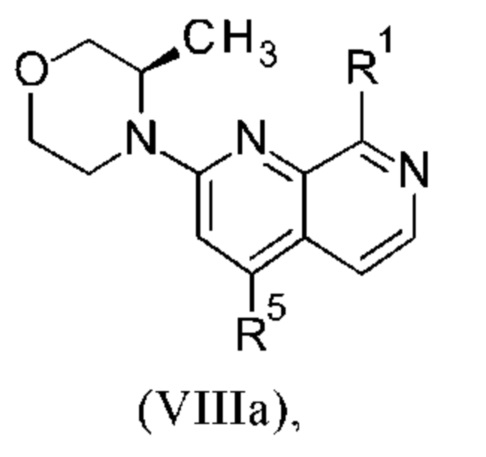
где
R1 представляет собой атом хлора, брома или йода или представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (n-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси; и
R5 представляет собой атом хлора, брома или йода или представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (n-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси.
В другом варианте осуществления настоящее изобретение относится к промежуточному соединению формулы (VIIIa), где R1 представляет собой атом хлора или брома, предпочтительно атом хлора.
В другом варианте осуществления настоящее изобретение относится к промежуточному соединению формулы (VIIIa), где R1 представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (n-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси.
В другом варианте осуществления настоящее изобретение относится к промежуточному соединению формулы (VIIIa), где R1 представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси и (n-толуолсульфонил)окси.
В другом варианте осуществления настоящее изобретение относится к промежуточному соединению формулы (VIIIa), где R5 представляет собой атом хлора, брома или йода.
В другом варианте осуществления настоящее изобретение относится к промежуточному соединению формулы (VIIIa), где R5 представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (n-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси.
В предпочтительном варианте осуществления настоящее изобретение относится к промежуточному соединению формулы (VIIIa), где R5 представляет собой [(трифторметил)сульфонил]окси группу.
В другом варианте осуществления настоящее изобретение относится к промежуточному соединению формулы (VIIIa), где R1 представляет собой атом хлора или брома, предпочтительно атом хлора, и где R5 представляет собой группу, выбранную из [(трифторметил)сульфонил]окси, [(нонафторбутил)сульфонил]окси, (метилсульфонил)окси, (n-толуолсульфонил)окси, (фенилсульфонил)окси, [(4-бромфенил)сульфонил]окси, [(4-нитрофенил)сульфонил]окси, [(2-нитрофенил)сульфонил]окси, [(4-изопропилфенил)сульфонил]окси, [(2,4,6-триизопропилфенил)сульфонил]окси, [(2,4,6-триметилфенил)сульфонил]окси, [(4-трет-бутилфенил)сульфонил]окси и [(4-метоксифенил)сульфонил]окси, предпочтительно [(трифторметил)сульфонил]окси группы.
В наиболее предпочтительном варианте осуществления настоящее изобретение относится к промежуточному соединению формулы (VIIIa), где R1 представляет собой атом хлора и где R5 представляет собой [(трифторметил)сульфонил]окси группу, с получением соединения формулы (VIIIb) (=(R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфонат):
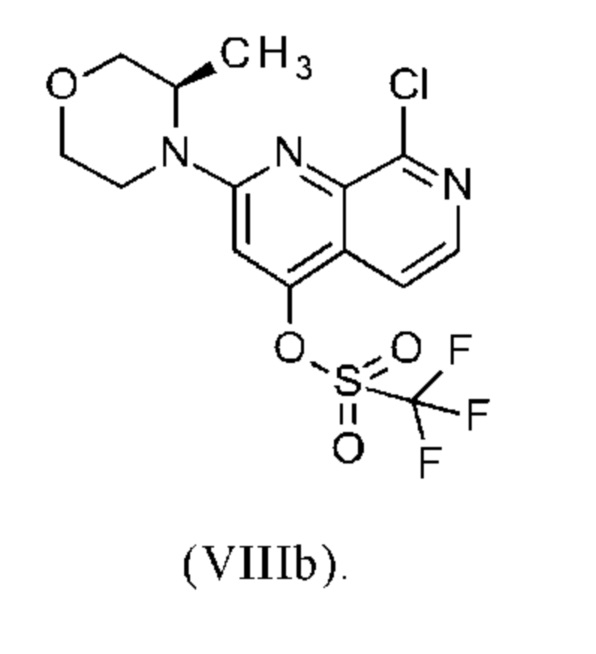
6. Полиморфная форма В соединения формулы (I) с высокой чистотой
Способ получения соединения формулы (I), описанного в WO2016020320 A1, приводит к полиморфной форме В соединения формулы (I). Однако несколько попыток масштабирования этого способа для получения полиморфной формы В соединения формулы (I) со степенью чистоты, которая приемлема для фармацевтического применения, не увенчались успехом. Кроме того, при попытке масштабирования способа, описанного в WO2016020320 A1, в результате получали продукт соединения формулы (I), который содержал более 0.15% одного или нескольких побочных продуктов, например, он содержал более 0.15% следующего соединения:
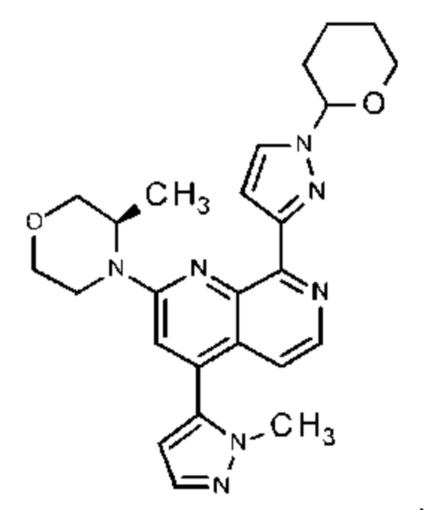
В противоположность этому, способ получения соединения формулы (I) в соответствии с настоящим изобретением теперь обеспечивает полиморфную форму В соединения формулы (I) с достаточно высокой степенью чистоты. Чистота полученных партий (в лабораторном масштабе, в килограммовом лабораторном масштабе и на опытно-промышленной установке) очень высока. Все отдельные побочные продукты известны, точно определены и токсикологически охарактеризованы. Было обнаружено, что содержание палладия (Pd) в готовом лекарственном веществе всегда составляло <10 млн.ч. Содержание бора (В) также было менее 10 млн.ч.
Таким образом, в соответствии с дополнительным аспектом, настоящее изобретение охватывает полиморфную форму В соединения формулы (I), которую можно получить способом получения соединения формулы (I) в соответствии с изобретением, в частности, с помощью способа обработки, описанного в разделе 1.3. ("Дальнейшая обработка сырого соединения формулы (I)") в комбинации со способом кристаллизации, описанным в разделе 1.4. ("Кристаллизация сырого соединения формулы (I) с получением его полиморфной формы В").
В другом варианте осуществления полиморфная форма В соединения формулы (I) в соответствии с настоящим изобретением характеризуется чистотой по меньшей мере 99,6% (=% площади), в частности, по меньшей мере 99.7% (=% площади), предпочтительно по меньшей мере 99.8% (=% площади), наиболее предпочтительно по меньшей мере 99.9% (=% площади), определенной с помощью СВЭЖХ.
В другом варианте осуществления настоящее изобретение охватывает полиморфную форму В соединения формулы (I) (=2-[(3R)-3-метилморфолин-4-ил]-4-(1-метил-1Н-пиразол-5-ил)-8-(1Н-пиразол-5-ил)-1,7-нафтиридин), которая характеризуется чистотой по меньшей мере 99,6% (=% площади), в частности, по меньшей мере 99.7% (=% площади), предпочтительно по меньшей мере 99.8% (=% площади), наиболее предпочтительно по меньшей мере 99.9% (=% площади), определенной с помощью СВЭЖХ.
Чистоту полиморфной формы В соединения формулы (I) определяют с помощью СВЭЖХ, предпочтительно с помощью метода, описанного в Экспериментальном разделе - Общей части ("СВЭЖХ - метод определения химической чистоты и анализа соединения формулы (I)").
Чистоту в "% площади" рассчитывают в процентном выражении площади пиков СВЭЖХ под пиком СВЭЖХ полиморфной формы В соединения формулы (I) относительно общей площади пиков всех СВЭЖХ пиков.
Полиморфная форма В соединения формулы (I) дополнительно характеризуется рентгеновской порошковой дифрактограммой (РПД), которая отображает по меньшей мере 3, в частности, по меньшей мере 5, предпочтительно по меньшей мере 7, более предпочтительно по меньшей мере 10, наиболее предпочтительно по меньшей мере 12 из следующих отражений, приведенных в виде 2θ значений: 8.3, 9.3, 13.8, 14.0, 18.0, 18.7, 19.6, 19.9, 20.1, 22.1, 23.9, 27.4.
В другом варианте осуществления полиморфная форма В соединения формулы (I) дополнительно характеризуется рентгеновской порошковой дифрактограммой (РПД), включающей следующие отражения, приведенные в виде 2θ значений: 8.3, 18.0, 19.9.
В другом варианте осуществления полиморфная форма В соединения формулы (I) дополнительно характеризуется рентгеновской порошковой дифрактограммой (РПД), включающей следующие отражения, приведенные в виде 2θ значений: 8.3, 9.3, 18.0, 19.9, 20.1.
В другом варианте осуществления полиморфная форма В соединения формулы (I) дополнительно характеризуется рентгеновской порошковой дифрактограммой (РПД), включающей следующие отражения, приведенные в виде 2θ значений: 8.3, 9.3, 13.8, 14.0, 18.0, 19.9, 20.1.
В другом варианте осуществления полиморфная форма В соединения формулы (I) дополнительно характеризуется рентгеновской порошковой дифрактограммой (РПД), по существу такой, как показано на Фигуре 1.
В другом варианте осуществления полиморфная форма В соединения формулы (I) характеризуется рентгеновской порошковой дифрактограммой (РПД), описанной в Таблице А1 (см. Экспериментальный раздел - Oбщую часть).
В другом варианте осуществления настоящее изобретение охватывает полиморфную форму В соединения формулы (I), которая характеризуется рентгеновской порошковой дифрактограммой (РПД), включающей определенные выше отражения, приведенные в виде 2θ значений, и которая дополнительно характеризуется чистотой по меньшей мере 99,6% (=% площади), в частности, по меньшей мере 99.7% (=% площади), предпочтительно по меньшей мере 99.8% (=% площади), наиболее предпочтительно по меньшей мере 99.9% (=% площади), определенной с помощью СВЭЖХ.
В другом варианте осуществления настоящее изобретение охватывает полиморфную форму В соединения формулы (I), которая характеризуется чистотой по меньшей мере 99,6% (=% площади), в частности, по меньшей мере 99.7% (=% площади), предпочтительно по меньшей мере 99.8% (=% площади), наиболее предпочтительно по меньшей мере 99.9% (=% площади), определенной с помощью СВЭЖХ, где полиморфная форма В соединения формулы (I) содержит
a) менее 15 мг бора, определенного с помощью ИСП-МС, предпочтительно менее 10 мг бора, на кг полиморфной формы В соединения формулы (I); и/или
b) менее 0.4 мг палладия, определенного с помощью ИСП-МС, предпочтительно 0.3 мг палладия или менее 0.3 мг палладия; и/или
c) менее 0.05% (=% площади) соединения формулы (IXb), определенного с помощью СВЭЖХ; и/или
d) менее 0.05% (=% площади) соединения формулы (VIb), определенного с помощью СВЭЖХ; и/или
e) менее 0.05% (=% площади) дигидропирана, определенного с помощью СВЭЖХ; и/или
f) менее 0.05% (=% площади) соединения формулы (IIIс), определенного с помощью СВЭЖХ; и/или
g) менее 0.05% (=% площади) пинакола, определенного с помощью IХ; и/или
h) менее 0.05% (=% площади) соединения формулы (VIIc/VIId), определенного с помощью СВЭЖХ; и/или
i) менее 0.05% (=% площади) соединения формулы

определенного с помощью СВЭЖХ; и/или
j) менее 0.05% (=% площади) соединения формулы

определенного с помощью СВЭЖХ.
Содержание бора и/или палладия в соединении формулы (I) определяют с помощью ИСП-МС, предпочтительно с помощью метода, описанного в Экспериментальном разделе - Общей части ("ИСП-МС - метод определения суммы элементов, бора, палладия, железа, калия, натрия").
Содержание соединений формул (IXb), (VIb), (IIIс), (VIIc/VIId), дигидропирана,
определяют с помощью СВЭЖХ, предпочтительно с помощью метода, описанного в Экспериментальном разделе - Общей части ("СВЭЖХ - метод определения химической чистоты и анализа соединения формулы (I)").
Содержание пинакола определяют с помощью ГХ, предпочтительно с помощью метода, описанного в Экспериментальном разделе - Общей части ("ГХ - метод определения пинакола").
В соответствии с дополнительным аспектом, настоящее изобретение охватывает фармацевтическую композицию, в частности, лекарственное средство, содержащую(-ее) полиморфную форму В соединения формулы (I) в соответствии с настоящим изобретением, которая характеризуется чистотой по меньшей мере 99,6% (=% площади), в частности, по меньшей мере 99.7% (=% площади), предпочтительно по меньшей мере 99.8% (=% площади), наиболее предпочтительно по меньшей мере 99.9% (=% площади), определенной с помощью СВЭЖХ, и один или несколько наполнителей, в частности, один или несколько фармацевтически подходящих наполнителей.
Можно использовать общепринятые методики приготовления таких фармацевтических композиций в подходящих дозированных формах.
Соединение формулы (I) в соответствии с изобретением может проявлять системную и/или местную активность. Для этой цели, оно может быть введено подходящим образом, как, например, пероральным, парентеральным, пульмональным, назальным, сублингвальным, лингвальным, буккальным, ректальным, вагинальным, дермальным, трансдермальным, конъюнктивальным, ушным путем или в виде имплантата или стента.
Для этих путей введения, соединение формулы (I) в соответствии с изобретением можно вводить в подходящих лекарственных формах.
Для перорального введения соединение формулы (I) в соответствии с изобретением можно ввести в дозированные формы, известные в уровне техники, которые доставляют соединение изобретения быстро и/или модифицированным образом, такие как, например, таблетки (непокрытые или покрытые оболочкой таблетки, например, с покрытиями, устойчивыми к желудочному соку или обеспечивающими контролируемое высвобождение, которые растворяются с задержкой или нерастворимы), таблетки, растворяющиеся во рту, пленки/облатки, пленки/лиофилизаты, капсулы (например, твердые или мягкие желатиновые капсулы), таблетки с сахарным покрытием, гранулы, пеллеты, порошки, эмульсии, суспензии, аэрозоли или растворы. В указанные дозированные формы соединение формулы (I) в соответствии с изобретением можно включать в кристаллической и/или аморфизированной и/или растворенной форме.
Парентеральное введение можно выполнить, избегая стадии абсорбции (например, внутривенно, внутриартериально, внутрисердечно, интраспинально или интралюмбально) или с включением абсорбции (например, внутримышечно, подкожно, внутрикожно, чрескожно или внутрибрюшинно). Лекарственными формами, которые являются подходящими для парентерального введения, являются, среди прочего, препараты для инъекции и инфузии в форме растворов, суспензий, эмульсий, лиофилизатов или стерильных порошков.
Примерами, которые являются подходящими для других путей введения, являются фармацевтические формы для ингаляции [среди прочего, включающие применение порошковых ингаляторов, небулайзеров], капли в нос, растворы для носа, спреи для носа, таблетки/пленки/облатки/капсулы для лингвального, сублингвального или буккального введения, суппозитории, глазные капли, глазные мази, глазные ванночки, глазные вставки, ушные капли, ушные спреи, ушные порошки, препараты для промывания ушей, ушные тампоны; вагинальные капсулы, водные суспензии (лосьоны, взбалтываемые смеси), липофильные суспензии, эмульсии, мази, кремы, трансдермальные терапевтические системы (такие как, например, пластыри), молочко, пасты, пены, присыпки, имплантаты или стенты.
Соединение формулы (I) в соответствии с изобретением можно включать в указанные лекарственные формы. Это можно осуществить способом, известным как таковым, путем смешивания с фармацевтически подходящими наполнителями. Фармацевтически подходящие наполнители включают, среди прочего,
 филлеры и носители (такие как, например, целлюлоза, микрокристаллическая целлюлоза (такая как, например, Avicel®), лактоза, маннит, крахмал, фосфат кальция (такой как, например, Di-Cafos®)),
филлеры и носители (такие как, например, целлюлоза, микрокристаллическая целлюлоза (такая как, например, Avicel®), лактоза, маннит, крахмал, фосфат кальция (такой как, например, Di-Cafos®)),
 мазевые основы (такие как, например, вазелиновое масло, парафины, триглицериды, воски, воск шерсти, спирты воска шерсти, ланолин, гидрофильная мазь, полиэтиленгликоли),
мазевые основы (такие как, например, вазелиновое масло, парафины, триглицериды, воски, воск шерсти, спирты воска шерсти, ланолин, гидрофильная мазь, полиэтиленгликоли),
 основы для суппозиториев (такие как, например, полиэтиленгликоли, какао-масло, твердый жир),
основы для суппозиториев (такие как, например, полиэтиленгликоли, какао-масло, твердый жир),
 растворители (такие как, например, вода, этанол, изопропанол, глицерин, пропиленгликоль, триглицериды со средней длиной цепи, жирные масла, жидкие полиэтиленгликоли, парафины),
растворители (такие как, например, вода, этанол, изопропанол, глицерин, пропиленгликоль, триглицериды со средней длиной цепи, жирные масла, жидкие полиэтиленгликоли, парафины),
 поверхностно-активные вещества, эмульгаторы, диспергаторы или смачиватели (такие как, например, додецилсульфат натрия, лецитин, фосфолипиды, жирные спирты (такие как, например, Lanette®), сложные эфиры сорбита и жирных кислот (такие как, например, Span®), полиоксиэтиленовые сложные эфиры сорбита и жирных кислот (такие как, например, Tween®), полиоксиэтиленовые глицериды жирных кислот (такие как, например, Cremophor®), полиоксиэтиленовые сложные эфиры жирных кислот, полиоксиэтиленовые простые эфиры жирных спиртов, сложные эфиры глицерина и жирных кислот, полоксамеры (такие как, например, Pluronic®),
поверхностно-активные вещества, эмульгаторы, диспергаторы или смачиватели (такие как, например, додецилсульфат натрия, лецитин, фосфолипиды, жирные спирты (такие как, например, Lanette®), сложные эфиры сорбита и жирных кислот (такие как, например, Span®), полиоксиэтиленовые сложные эфиры сорбита и жирных кислот (такие как, например, Tween®), полиоксиэтиленовые глицериды жирных кислот (такие как, например, Cremophor®), полиоксиэтиленовые сложные эфиры жирных кислот, полиоксиэтиленовые простые эфиры жирных спиртов, сложные эфиры глицерина и жирных кислот, полоксамеры (такие как, например, Pluronic®),
 буферы, кислоты и основания (такие как, например, фосфаты, карбонаты, лимонная кислота, уксусная кислота, хлористоводородная кислота, раствор гидроксида натрия, карбонат аммония, трометамол, триэтаноламин),
буферы, кислоты и основания (такие как, например, фосфаты, карбонаты, лимонная кислота, уксусная кислота, хлористоводородная кислота, раствор гидроксида натрия, карбонат аммония, трометамол, триэтаноламин),
 изотонические средства (такие как, например, глюкоза, хлорид натрия),
изотонические средства (такие как, например, глюкоза, хлорид натрия),
 адсорбенты (такие как, например, высокодисперсный силикагель),
адсорбенты (такие как, например, высокодисперсный силикагель),
 повышающие вязкость средства, гелеобразователи, загустители и/или связующие (такие как, например, поливинилпирролидон, метилцеллюлоза, гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза, натрий-карбоксиметилцеллюлоза, крахмал, карбомеры, полиакриловые кислоты (такие как, например, Carbopol®), альгинаты, желатин),
повышающие вязкость средства, гелеобразователи, загустители и/или связующие (такие как, например, поливинилпирролидон, метилцеллюлоза, гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза, натрий-карбоксиметилцеллюлоза, крахмал, карбомеры, полиакриловые кислоты (такие как, например, Carbopol®), альгинаты, желатин),
разрыхлители (такие как, например, модифицированный крахмал, натрий-карбоксиметилцеллюлоза, натрия крахмалгликолят (такой как, например, Explotab®), поперечно сшитый поливинилпирролидон, натрий кроскармеллоза (такая как, например, AcDiSol®)),
 регуляторы потока, смазывающие вещества, скользящие вещества и разделительные смазки для пресс-форм (такие как, например, стеарат магния, стеариновая кислота, тальк, высокодисперсный силикагель (такой как, например, Aerosil®)),
регуляторы потока, смазывающие вещества, скользящие вещества и разделительные смазки для пресс-форм (такие как, например, стеарат магния, стеариновая кислота, тальк, высокодисперсный силикагель (такой как, например, Aerosil®)),
 покрывающие вещества (такие как, например, сахар, шеллак) и пленкообразователи для пленок или диффузионных мембран, которые растворяются быстро или модифицированным образом (например, поливинилпирролидоны (такие как, например, Kollidon®), поливиниловый спирт, гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза, этилцеллюлоза, фталат гидроксипропилметилцеллюлозы, ацетат целлюлозы, ацетатфталат целлюлозы, полиакрилаты, полиметакрилаты, такие как, например, Eudragit®)),
покрывающие вещества (такие как, например, сахар, шеллак) и пленкообразователи для пленок или диффузионных мембран, которые растворяются быстро или модифицированным образом (например, поливинилпирролидоны (такие как, например, Kollidon®), поливиниловый спирт, гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза, этилцеллюлоза, фталат гидроксипропилметилцеллюлозы, ацетат целлюлозы, ацетатфталат целлюлозы, полиакрилаты, полиметакрилаты, такие как, например, Eudragit®)),
 вещества для капсул (такие как, например, желатин, гидроксипропилметилцеллюлоза),
вещества для капсул (такие как, например, желатин, гидроксипропилметилцеллюлоза),
 синтетические полимеры (такие как, например, полилактиды, полигликолиды, полиакрилаты, полиметакрилаты (такие как, например, Eudragit®), поливинилпирролидоны (такие как, например, Kollidon®), поливиниловые спирты, поливинилацетаты, полиэтиленоксиды, полиэтиленгликоли и их сополимеры и блок-сополимеры),
синтетические полимеры (такие как, например, полилактиды, полигликолиды, полиакрилаты, полиметакрилаты (такие как, например, Eudragit®), поливинилпирролидоны (такие как, например, Kollidon®), поливиниловые спирты, поливинилацетаты, полиэтиленоксиды, полиэтиленгликоли и их сополимеры и блок-сополимеры),
 пластификаторы (такие как, например, полиэтиленгликоли, пропиленгликоль, глицерин, триацетин, триацетилцитрат, дибутилфталат),
пластификаторы (такие как, например, полиэтиленгликоли, пропиленгликоль, глицерин, триацетин, триацетилцитрат, дибутилфталат),
вещества, способствующие проникновению,
 стабилизаторы (такие как, например, антиоксиданты, такие как аскорбиновая кислота, аскорбилпальмитат, аскорбат натрия, бутилгидроксианизол, бутилгидрокситолуол, пропилгаллат),
стабилизаторы (такие как, например, антиоксиданты, такие как аскорбиновая кислота, аскорбилпальмитат, аскорбат натрия, бутилгидроксианизол, бутилгидрокситолуол, пропилгаллат),
 консерванты (такие как, например, парабены, сорбиновая кислота, тиомерсал, хлорид бензалкония, ацетат хлоргексидина, бензоат натрия),
консерванты (такие как, например, парабены, сорбиновая кислота, тиомерсал, хлорид бензалкония, ацетат хлоргексидина, бензоат натрия),
 красители (такие как, например, неорганические пигменты, такие как оксиды железа, диоксид титана),
красители (такие как, например, неорганические пигменты, такие как оксиды железа, диоксид титана),
 ароматизирующие вещества, подсластители, вещества, корригирующие вкус и/или запах.
ароматизирующие вещества, подсластители, вещества, корригирующие вкус и/или запах.
ЭКСПЕРИМЕНТАЛЬНЫЙ РАЗДЕЛ
Формы ЯМР пиков указаны в том виде, в каком они проявляются в спектрах, возможные эффекты более высокого порядка не рассматривались.
Химические названия генерировали с использованием программного обеспечения ACD/Name от ACD/Labs. В некоторых случаях вместо названий, сгенерированных ACD/Name, использовали общепринятые названия коммерчески доступных реагентов.
В следующей таблице 1 перечислены сокращения, используемые в этом параграфе и в разделе "Примеры", поскольку они не поясняются в основном тексте. Другие сокращения имеют значения, по существу обычные для специалиста в данной области.


Другие сокращения имеют значения, по существу обычные для специалиста в данной области.
Различные аспекты изобретения, описанного в данной заявке, иллюстрируются следующими примерами, которые не предназначены для ограничения изобретения каким-либо образом.
Иллюстративные тестовые эксперименты, описанные в данной заявке, служат для иллюстрации настоящего изобретения и изобретение не ограничено приведенными примерами.
ЭКСПЕРИМЕНТАЛЬНЫЙ РАЗДЕЛ - ОБЩАЯ ЧАСТЬ
Все реагенты, для которых синтез не описан в экспериментальной части, являются либо коммерчески доступными, либо являются известными соединениями, либо могут быть образованы известными методами из известных соединений специалистом в данной области.
Аналитические методы и оборудование
1Н-ЯМР спектры регистрировали в CDCl3, ДМСО-d6 или D2O, используя ЯМР прибор Bruker Biospin с 400 МГц магнитом, химический сдвиг (8) представлен в сравнении с TMS в качестве внутреннего стандарта.
ВЭЖХ хроматограммы
ВЭЖХ Метод - Скан. основание: Система Agilent (бинарный насос 1260, G1312B, дегазатор; автодозатор, ColCom, ДМД детектор: Agilent G1315C, 220-320 нм); Колонка: Waters XSelect™ CSH (50×2.1 мм, 3.5 мкм); Темп. колонки: 3 5°С; Поток: 0.8 мл/мин; Вводимый объем: 1 мкл; Градиент: t-0мин=2% В, t3,5мин =98% В, t6мин=98% В, Время перерыва: 3 мин, 2% В. Элюент А: 10 мМ бикарбонат аммония в воде, рН 9.5; Элюент В: 95% ацетонитрила +5% 10 мМ бикарбоната аммония в воде (рН 9.5);
ВЭЖХ Метод - Скан. кислота: Система Agilent (1100, бинарный насос, G1312A, дегазатор; автодозатор, ColCom, ДМД детектор: Agilent G1315B, 220-320 нм); Колонка: Waters XSelect™ CSH (50×2.1 мм, 3.5 мкм); Темп. колонки: 35°С; Поток: 0.8 мл/мин; Вводимый объем: 1 мкл; Градиент: t0=2% В, t3.5мин=98% В, t6мин=98% В, Время перерыва: 3 мин; Элюент А: 0.1% муравьиная кислота в воде. Элюент В: 0.1% муравьиная кислота в ацетонитриле.
ВЭЖХ Метод - Скан. кислота: Система Agilent (1100, бинарный насос, G1312A, дегазатор; автодозатор, ColCom, ДМД детектор: Agilent G1315B, 220-320 нм); Колонка: Phenomex Kinetex С18 100А (100*4.6 мм 2.6 мкм); Темп, колонки: 35°С; Поток: 1.5 мл/мин; Вводимый объем: 1 мкл; Градиент: t0=2% D, t8мин=98%) D, t11мин=98%) D, Время перерыва: 3 мин.; Элюент С: 0.1% муравьиная кислота в воде. Элюент D: 0.1% муравьиная кислота в ацетонитриле.
ЖХ-МС хроматограммы
ЖХ-МС Метод - Скан. основание
Agilent 1100, бин. насос: G1312A, дегазатор; автодозатор, ColCom, ДМД: Agilent G1315B, 220-320 нм, МСД: Agilent LC/MSD G6130B, ЭРИ, положит, /отрицат.100-800; Колонка: Waters XSelect™ С18, 30×2.1 мм, 3.5 мк, Темп: 25°С, Поток: 1 мл/мин, Градиент: t0=2% A, t1.6мин=98% А, t3мин=98% А, Время перерыва: 1.3 мин, Элюент А: 95% ацетонитрила +5% 10 мМ бикарбоната аммония в воде в ацетонитриле, Элюент В: 10 мМ бикарбонат аммония в воде (рН=9.5).
ЖХ-МС Метод - Скан. кислота
Прибор: Agilent 1260, бин. насос: G1312B, дегазатор; автодозатор, ColCom, ДМД: Agilent G1315C, 220-320 нм, МСД: Agilent LC/MSD G6130B, ЭРИ, положит./отрицат.100-800, Колонка: Waters XSelect™ CSH С18, 30×2.1 мм, 3.5 мк, Темп: 35°С, Поток: 1 мл/мин, Градиент: t0=5% A, t1.6мин=98% A, t3мин =98% А, Время перерыва: 1.3 мин, Элюент А: 0.1% муравьиная кислота в ацетонитриле, Элюент В: 0.1% муравьиная кислота в воде.





 ГХ - метод определения 1-бутанола, дихлорметана, этилацетата, изопропанола, метанола: Парофазная ГХ - USP, версия 41, глава <467> "остаточные растворители".
ГХ - метод определения 1-бутанола, дихлорметана, этилацетата, изопропанола, метанола: Парофазная ГХ - USP, версия 41, глава <467> "остаточные растворители".
Ионная хроматография (ИХ) - метод определения хлорида, фосфата, сульфата: USP, версия 41, глава <1065> "ионная хроматография".
ИСП-МС - метод определения суммы элементов, бора, палладия, железа, калия, натрия: USP, версия 41, глава <233> "примеси элементов", или Ph. Eur. (9-с издание с дополнениями 9.1 - 9.5), глава 2.2.58.
Кулонометрическое титрование для определения воды: Карл Фишер, кулонометрическое титрование, Ph. Eur. (9-с издание с дополнениями 9.1 - 9.5), глава 2.5.32.
Рентгеновская порошковая дифрактометрия (РПД):
РПД анализы выполняли с использованием дифрактометра "X'Pert Pro" от PANalytical B.V., Нидерланды, оснащенного испускающей Сu-рентгеновской трубкой (излучение Сu K альфа 1, длина волны 1.5406  ) и детекторной системой Pixcel. Образцы анализировали при 25°С в трансмиссионном режиме и помещали между пленками полиэтилена низкой плотности. Использовали программное обеспечение HighScore Plus, версия 2.2 с, от PANalytical B.V., применяя следующие параметры: диапазон 3 - 40° 2θ, размер шага 0.013°, время счета 99 с, время анализа ~ 22 мин. Все отражения рентгеновских лучей приведены в виде значений ° 2θ (тета) с разрешением ±0.1°.
) и детекторной системой Pixcel. Образцы анализировали при 25°С в трансмиссионном режиме и помещали между пленками полиэтилена низкой плотности. Использовали программное обеспечение HighScore Plus, версия 2.2 с, от PANalytical B.V., применяя следующие параметры: диапазон 3 - 40° 2θ, размер шага 0.013°, время счета 99 с, время анализа ~ 22 мин. Все отражения рентгеновских лучей приведены в виде значений ° 2θ (тета) с разрешением ±0.1°.
Перечень отражений РПД (значения 2θ) Полиморфной формы В (=Мод В) соединения формулы (I)


ЭКСПЕРИМЕНТАЛЬНЫЙ РАЗДЕЛ - ПРИМЕРЫ
Если не указано иное, реакции проводили в атмосфере азота.
Пример 1а
(R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфонат (VIIIb)
К смеси (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ола (IIа) (20 г, 71.5 ммоль) и триэтиламина (19.88 мл, 143 ммоль, 2 экв.) в дихлорметане (100 мл) по каплям добавляли раствор] N-фенил-бис(трифторметансульфонимида) (25.5 г, 71.5 ммоль, 1 экв.) в дихлорметане (160 мл). Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение ночи, после чего ВЭЖХ анализ образца указывал на то, что все исходное вещество израсходовалось. Реакционную смесь выливали в воду (200 мл) и полученную в результате смесь подкисляли уксусной кислотой (110 мл) до рН~3.5. После разделения органический слой перемешивали с водным раствором уксусной кислоты (100 мл, рН~3) в течение 5 мин. Органический слой отделяли, промывали водой (200 мл) и затем 2М раствором карбоната калия (5 × 200 мл) до тех пор, пока ЖХМС анализ органического слоя не указывал на то, что весь сульфонамид не был удален. Затем органический слой последовательно промывали водой (200 мл) и соляным раствором (200 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток (23 г) перекристаллизовывали из горячего 2-пропанола (2 объема, 46 мл). На следующее утро твердые вещества отфильтровывали и промывали 2-пропанолом (5 мл) с получением (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-илтрифторметансульфоната (VIIIb). 18.91 г (выход от теор. 64%) в виде желтого твердого вещества.
1Н ЯМР (Хлороформ-d) δ: 8.17 (d, J=5.5 Гц, 1H), 7.53 (d, J=5.5 Гц, 1H), 7.08 (s, 1H), 4.54 - 4.36 (m, 1H), 4.27 (d, J=12.0 Гц, 1H), 4.11 (dd, J=11.5, 3.8 Гц, 1H), 3.89 (d, J=11.5 Гц, 1H), 3.80 (dd, J=11.6, 3.1 Гц, 1H), 3.66 (td, J=12.0, 3.0 Гц, 1H), 3.42 (td, J=12.9, 3.9 Гц, 1H), 1.39 (d, J=6.8 Гц, 3Н).
ЖХ-МС (способ: Скан. основание): Rt 2.33 мин;
МС (ЭРИ положит.) m/z=412.1 [М+Н]+.
ВЭЖХ (способ: Скан. основание): Rt 3.88 мин.
Пример 1b
(R)-4-(8-хлор-4-(1-метил-1H-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолин (IХb)
К раствору (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфоната (VIIIb) (125.0 г, 304 ммоль) в 300 мл дегазированного изопропилацетата добавляли PdCl2(dppf) (6.66 г, 9.1 ммоль, 3 мол.%). Смесь нагревали до 50°С, после чего к реакционной смеси добавляли дегазированный раствор гидрокарбоната калия (122 г, 4 экв.) в воде (750 мл) при 50°С. Сразу же после этого добавляли раствор сложного пинаколового эфира 1-метил-1Н-пиразол-5-бороновой кислоты (VIb) (63.2 г, 304 ммоль, 1.0 экв.) в 750 мл дегазированного изопропилацетата на протяжении 1.5 ч. После перемешивания в течение дополнительного часа при 50°С, ВЭЖХ анализ указывал на полное превращение.
Реакционную смесь фильтровали через диатомовую землю и осадок на фильтре промывали изопропилацетатом (300 мл). После выполнения этого фильтрования два слоя разделяли. Водную фазу экстрагировали изопропилацетатом (1×600 мл). Объединенные органические фазы промывали водным 1 н. раствором гидроксида натрия (4×600 мл) для удаления следов гидролизованного трифлата (в соответствии с ВЭЖХ было образовано только ~1% площади). Органическую фазу последовательно промывали водой (600 мл) и соляным раствором (600 мл), и сушили над сульфатом натрия. Через сорок минут добавляли активированный уголь (~10 г на 100 г исходного вещества) и полученную в результате суспензию перемешивали в течение ночи. На следующее утро твердые вещества отфильтровывали на диатомовой земле, осадок на фильтре промывали изопропилацетатом (200 мл) и фильтрат концентрировали с получением сырого (R)-4-(8-хлор-4-(1-метил-1H-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина (промежуточное соединение (IXb)) в виде коричневой пены (100.5 г, 96%). Сырой продукт очищали с помощью кристаллизации:
В 2 л круглодонную колбу загружали сырой (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолин (IXb) (98 г, 268 ммоль) и изопропанол (500 мл). При нагревании до 82°С образовывался прозрачный коричневый раствор. Добавляли активированный уголь (10 г, 833 ммоль) и черную суспензию перемешивали при 82°С в течение 3 часов. Уголь удаляли с помощью фильтрования через диатомовую землю с использованием предварительно нагретой фильтрационной установки. Фильтр промывали 50 мл изопропанола и фильтрат концентрировали при пониженном давлении с получением 92.87 г светло-коричневого твердого вещества. Фильтр, использованный для удаления активированного угля, промывали ДХМ и желтый фильтрат концентрировали в вакууме. Вещество повторно растворяли в 400 мл изопропанола при 83°С, что приводило к прозрачному темно-коричневому раствору. При охлаждении осаждалось светло-коричневое твердое вещество. Суспензию перемешивали в течение 3 часов при комнатной температуре. Твердое вещество собирали с помощью фильтрования и промывали на фильтре изопропанолом (2 × 30 мл). После сушки собирали 72.5 г очищенного (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина (промежуточное соединение (IXb)) (72.5 г, выход от теор. 79%).
Маточную жидкость, которая все еще содержала большие количества целевого продукта, концентрировали в вакууме с получением 20.8 г твердого коричневого вещества.
1H ЯМР (Хлороформ-d) δ: 8.06 (d, J=5.5 Гц, 1H), 7.65 (d, J=1.9 Гц, 1H), 7.15 (d, J=5.4 Гц, 1H), 7.07 (s, 1H), 6.43 (d, J=1.9 Гц, 1H), 4.51 (dt, J=9.3, 4.5 Гц, 1H), 4.34 (dd, J=13.4, 2.8 Гц, 1H), 4.11 (dd, J=11.5, 3.8 Гц, 1H), 3.88 (d, J=11.4 Гц, 1H), 3.81 (dd, J=11.5, 3.2 Гц, 1H), 3.71 (s, 3Н), 3.67 (td, J=11.8, 3.0 Гц, 1H), 3.42 (ddd, J=13.6, 12.4, 4.0 Гц, 1H), 1.40 (d, J=6.8 Гц, 3Н).
ЖХ-МС (способ: Скан. основание): Rt 2.02 мин; МС (ЭРИ положит.) m/z=344.2 [М+Н]+.
ВЭЖХ (способ: Скан. основание): Rt 3.24 мин.
Пример 1с
(3R)-3-метил-4-(4-(1-метил-1H-пиразол-5-ил)-8-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)-1,7-нафтиридин-2-ил)морфолин (VIIc/VIId)
В 100 мл трехгорлую круглодонную колбу загружали промежуточное соединение (IXb) (3.0 г, 8.73 ммоль) и PdCl2(dppf) (0.192 г, 0.262 ммоль). Добавляли дегазированный этилацетат (12 мл) с последующим добавлением дегазированной воды (6.0 мл) и безводного фосфата калия (5.56 г, 26.2 ммоль). Полученную в результате двухфазную реакционную смесь нагревали до 65°С, после чего к реакционной смеси по каплям добавляли раствор сложного пинаколового эфира 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIс) (2.91 г, 10.5 ммоль) в дегазированном этилацетате (15 мл) на протяжении 1 ч (для добавления использовали шприцевой насос). Почти полное превращение было достигнуто непосредственно после добавления всего количества сложного эфира бороновой кислоты. Реакционную смесь охлаждали и концентрировали для удаления этилацетата. Добавляли дихлорметан (30 мл) с последующим добавлением воды (30 мл, 10.0 объемов). Слои разделяли и органическую фазу промывали 2 М раствором карбоната калия (30 мл). Затем органическую фазу промывали водой (4 × 30 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Это приводило к 4.16 г сырого (3R)-3-метил-4-(4-(1-метил-1H-пиразол-5-ил)-8-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)-1,7-нафтиридин-2-ил)морфолина (промежуточное соединение VIIc/VIId) в виде коричневого твердого вещества.
В соответствии с 1H-ЯМР сырой продукт состоял из 1:1 смеси диастереоизомеров VIIc и VIId.
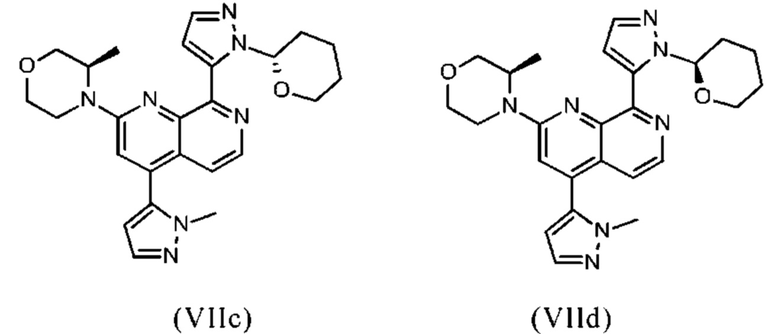
1Н ЯМР (Хлороформ-d) δ: 8.41 (dd, J=5.5, 2.2 Гц, 1H), 7.71 (d, J=1.8 Гц, 1H), 7.67 (d, J=1.9 Гц, 1H), 7.22 (dd, J=5.4, 1.4 Гц, 1H), 7.06 (d, J=3.8 Гц, 1H), 7.00 (dd, J=13.3, 1.8 Гц, 1H), 6.45 (t, J=1.7 Гц, 1H), 6.11 (dt, J=9.8, 2.7 Гц, 1H), 4.55 - 4.42 (m, 0.5Н), 4.34 (dd, J=7.4, 2.7 Гц, 0.5Н), 4.23 (dd, J=13.4, 2.8 Гц, 0.5Н), 4.09 - 4.01 (m, 1.5Н), 4.01 - 3.91 (m, 1H), 3.83 (dd, J=11.5, 5.7 Гц, 1H), 3.76 (s, 4H), 3.59 (tdd, J=11.5, 7.9, 2.9 Гц, 1H), 3.47 (tdd, J=11.1, 7.7, 2.5 Гц, 1H), 3.34 (dtd, J=21.7, 12.9, 3.9 Гц, 1H), 2.63 - 2.48 (m, 1H), 2.11 (td, J=12.9, 3.3 Гц, 2H), 1.81 - 1.58 (m, 3H), 1.35 (dd, J=6.8, 3.3 Гц, 3H).
ЖХ-МС (способ: Скан. основание): Rt 1.98 мин;
МС (ЭРИ положит.) m/z=460.3 [М+Н]+.
ВЭЖХ (способ: Скан. основание): Rt 3.16 мин.
Сырую смесь диастереоизомеров VIIc и VIId непосредственно превращали в целевое соединение (I) (см. Пример Id). Пример 1d
2-[(3R)-3-мeтилмopфoлин-4-ил]-4-(1-метил-1Н-пиразол-5-ил)-8-(1Н-пиразол-5-ил)-1,7-нафтиридин (I)
К раствору сырого промежуточного соединения (VIIc/VIId) (503 мг, 1.095 ммоль) в смеси дихлорметана (1.50 мл) и метанола (0.15 мл) добавляли водный раствор хлористоводородной кислоты (1 н., 1.50 мл, 1.5 ммоль). Полученную в результате темно-коричневую двухфазную смесь перемешивали при комнатной температуре в течение 30 минут, после чего ВЭЖХ анализ органической и водной фаз указывал на то, что было достигнуто полное превращение. Двухфазную смесь переносили в делительную воронку, используя водный раствор хлористоводородной кислоты (1 н., 10 мл) и дихлорметан (10 мл). После энергичного встряхивания водный слой отделяли. Органическую фазу дополнительно экстрагировали водным раствором хлористоводородной кислоты (1 н., 10 мл). Объединенные водные фазы промывали дихлорметаном (3×10 мл) и затем выливали в 25 мл 10% раствора карбоната калия, что приводило к образованию сине-зеленой суспензии. Суспензию экстрагировали дихлорметаном (4×10 мл). Объединенные органические слои промывали соляным раствором, сушили над сульфатом натрия и концентрировали при пониженном давлении с получением 337 мг ярко-желтой хрупкой пены. Это вещество переносили в дихлорметан (~8 мл), добавляли н-бутанол (10 мл) и полученный в результате раствор концентрировали при пониженном давлении при 40°С для удаления низкокипящего дихлорметана, оставляя в колбе приблизительно 6 мл зеленовато-желтого раствора. Оставшийся раствор перемешивали при комнатной температуре в течение ночи. На следующее утро кристаллизовавшийся продукт отфильтровывали, используя стеклянный фильтр, ярко-желтое твердое вещество промывали 1-бутанолом (3×2 мл) и сушили в потоке воздуха с получением 203 мг 2-[(3R)-3-метилморфолин-4-ил]-4-(1-метил-1Н-пиразол-5-ил)-8-(1Н-пиразол-5-ил)-1,7-нафтиридина (соединение (I)) в виде ярко-желтого (микро)-кристаллического порошка. Концентрирование фильтрата давало еще 67 мг соединения (I) в виде желто-коричневого порошка.
Выход: 203 мг + 67 мг=270 мг (суммарно 65.70% от теор.).
1Н ЯМР (Хлороформ-d) δ: 12.81 (s, 1Н, широкий сигнал), 8.41 (d, J=5.5 Гц, 1H), 7.73 (d, J=1.9 Гц, 1H), 7.67 (d, J=1.9 Гц, 1H), 7.34 (d, J=1.8 Гц, 1H), 7.17 (t, J=2.7 Гц, 2Н), 6.46 (d, J=1.9 Гц, 1H), 4.43 (tt, J=9.2, 4.4 Гц, 1H), 4.19 (dd, J=11.4, 3.9 Гц, 1H), 4.04 (dd, J=12.8, 2.8 Гц, 1H), 3.93 (d, J=11.6 Гц, 1H), 3.86 (dd, J=11.5, 3.1 Гц, 1H), 3.74 (s, 4Н), 3.57 (td, J=12.4, 3.9 Гц, 1H), 1.47 (d, J=6.8 Гц, 3Н).
ЖХ-МС (способ: Скан. основание): Rt 1.88 мин;
МС (ЭРИ положит.) m/z=376.2 [М+Н]+.
ЖХ-МС (способ: Скан. кислота): Rt 1.65 мин;
МС (ЭРИ положит.) m/z=376.2 [М+Н]+.
ВЭЖХ (способ: Скан. кислота): Rt 2.46 мин.
ВЭЖХ (способ: Скан. кислота): Rt 3.41 мин.
Пример 1е
(3R)-3-метил-4-(4-0-метил-1H-пиразол-5-ил)-8-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)-1,7-нафтиридин-2-ил)морфолин (VIIc/VIId)
В 100 мл трехгорлую круглодонную колбу загружали промежуточное соединение (IXb) (3.0 г, 8.73 ммоль) и PdCl2(dppf) (0.192 г, 0.262 ммоль). Добавляли дегазированный изопропилацетат (12 мл) с последующим добавлением дегазированной воды (6.0 мл) и безводного фосфата калия (2.86 г, 17.5 ммоль). Полученную в результате двухфазную реакционную смесь нагревали до 55°С, после чего к реакционной смеси по каплям добавляли раствор сложного пинаколового эфира 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIс) (2.91 г, 10.5 ммоль) в дегазированном изопропилацетате (15 мл) на протяжении 1 ч (для добавления использовали шприцевой насос). После завершения добавления реакционную смесь продолжали перемешивать при 55°С в течение ночи (16 часов). Реакционную смесь последовательно охлаждали до комнатной температуры и фильтровали через набивку целита для удаления вещества на границе раздела фаз. Слои разделяли и органическую фазу промывали 2 М раствором карбоната калия (30 мл). К органической фазе добавляли воду (30 мл) и слоям давали разделиться. Перед полным разделением двух фаз, смесь фильтровали через набивку целита для удаления вещества на границе раздела фаз. Органическую фазу еще 3 раза промывали водой (3 × 30 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Это приводило к 2.88 г сырого (3R)-3-метил-4-(4-(1-метил-1H-пиразол-5-ил)-8-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)-1,7-нафтиридин-2-ил)морфолина (промежуточное соединение VIIc/VIId) в виде коричневого твердого вещества. В соответствии с 1H-ЯМР сырой продукт состоял из 1: 1 смеси диастереоизомеров.
Пример 2а
(R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфонат (VIIIb)
100 г (357.49 ммоль) (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ола (IIа) суспендировали в 800 мл дихлорметана при комнатной температуре (~22°С). Затем добавляли 40.48 мл (500.485 ммоль) пиридина и смесь охлаждали до -10°С. При -10°С к смеси добавляли 84.2 мл (500.485 ммоль) трифторметансульфонового ангидрида, растворенного в 250 мл дихлорметана (30 мин, температура повышалась до -6°С). После завершения добавления смесь перемешивали в течение 1 ч при -10°С. Медленно добавляли 400 мл воды, поддерживая температуру между 0 и 4°С. Фазы разделяли. Органическую фазу промывали 400 мл воды. Органическую фазу два раза промывали 0.5 М водным раствором карбоната калия, каждый раз порцией 200 мл, и один раз 150 мл воды. Органическую фазу фильтровали через фильтр с активированным углем и объем фильтрата уменьшали до -100 мл путем отгонки дихлорметана при пониженном давлении. Добавляли 400 мл изопропанола и снова выполняли отгонку при пониженном давлении до объема ~ 100 мл (при 50°С). Эту операцию выполняли еще раз. В заключение добавляли 240 мл изопропанола и нагревали до 50°С. Смесь перемешивали в течение выходных и в заключение охлаждали до 0-3°С. Кристаллы собирали с помощью фильтрования и промывали 100 мл изопропанола. Продукт сушили в вакууме (20 мбар) при 45°С в течение ночи. Выход: 113.87 г (77.35% от теор.) желтых кристаллов.
ВЭЖХ: 100% (% площади) на 7.4 мин.
Пример 2b
(R)-4-(8-хлор-4-(1-метил-1H-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолин (IХb)
100 г (242.842 ммоль) (Л)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфоната (VHIb) и 5.3 г (7.285 ммоль) PdCl2(dppf) растворяли в 400 мл этилацетата и нагревали до 50°С. К этой смеси добавляли раствор 97.2 г (971.370 ммоль) гидрокарбоната калия в 600 мл воды, и температуру поддерживали на уровне 40°С. К этой смеси добавляли раствор 48.00 г (230.70 ммоль) сложного пинаколового эфира 1-метил-1Н-пиразол-5-бороновой кислоты (VIb) в 300 мл этилацетата в течение 3 ч (при внутренней температуре 40°С). Реакционную смесь охлаждали до комнатной температуры (+22°С) и фазы разделяли. Водную фазу экстрагировали 300 мл этилацетата и объединенные органические фазы два раза промывали 1 н. водным раствором гидроксида калия, каждый раз порцией 750 мл, и два раза водой, каждый раз порцией 500 мл. При выполнении последней промывки добавляли 10 мл соляного раствора для лучшего разделения фаз. Органическую фазу сушили над 100 г сульфата натрия, фильтровали и осадок на фильтре промывали 200 мл этилацетата. Затем к фильтрату добавляли 70 г активированного угля и суспензию перемешивали в течение 2 часов при комнатной температуре. Уголь удаляли с помощью фильтрования через диатомовую землю (50 г) и фильтр промывали 100 мл этилацетата. Замену растворителя выполняли путем добавления изопропанола и отгонки этилацетата при 85°С. Когда объем изопропилацетата достигал ~ 200 мл (внутренняя температура 85°С), смесь охлаждали до комнатной температуры (путем перемешивания в течение ночи). Кристаллы отделяли с помощью фильтрования и промывали 20 мл изопропанола. Продукт сушили в вакууме (20 мбар) при 45°С в течение ночи. Выход: 63.2 г (75.7% от теор.) желтых кристаллов.
ВЭЖХ: 99.7% (% площади) на 11.9 мин
Пример 2 с
2-[(3R-3-метилморфолин-4-ил]-4-(1-метил-1Н-пиразол-5-ил)-8-(1Н-пиразол-5-ил)-1,7-нафтиридин (I)
120 г сырого (3R)-3-метил-4-(4-(1-метил-1H-пиразол-5-ил)-8-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)-1,7-нафтиридин-2-ил)морфолина (VIIc/VIId) растворяли в 480 мл дихлорметана и 480 мл метанола и охлаждали до 0-5°С. Затем добавляли 1200 мл 1 н. водного раствора хлористоводородной кислоты, во время чего температура повышалась до 20°С. Смесь перемешивали в течение 5 мин при 20°С и затем фазы разделяли, и водную фазу экстрагировали два раза дихлорметаном, каждый раз порцией 480 мл. К водной фазе добавляли 46.6 г N-ацетилцистеина и раствор перемешивали при комнатной температуре в течение ночи. Затем добавляли 960 мл дихлорметана и смесь охлаждали до 10°С. Значение рН доводили до рН=13 путем добавления 480 мл 5 н. водного раствора гидроксида калия. Раствор перемешивали в течение 30 мин. Фазы разделяли и водную фазу экстрагировали 252 мл дихлорметана. Органические фазы объединяли и выполняли замену растворителя - дихлорметана на н-бутанол: к 631.5 мл н-бутанола при 110°С медленно добавляли фильтрат, и дихлорметан отгоняли. Смесь охлаждали до 22°С и перемешивали в течение ночи, затем охлаждали до 0-3°С и перемешивали в течение 1 ч при этой температуре. Продукт выделяли с помощью фильтрования и кристаллы два раза промывали холодным н-бутанолом, каждый раз порцией 160 мл. Продукт сушили в вакууме (20 мбар) при 45°С в течение ночи. Выход: 62.87 г (64.13% от теор.) желтых кристаллов.
ВЭЖХ: 99.48% (% площади) на 4.3 мин.
Пример 2d
2-[(3R)-3-метилморфолин-4-ил]-4-(1-метил-1Н-пиразол-5-ил)-8-(1Н-пиразол-5-ил)-1,7-нафтиридин (I)
В атмосфере азота 30 г (87.255 ммоль) (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина (IХb) растворяли в 105.8 мл этилацетата и добавляли 1.91 г (2.618 ммоль) PdCl2(dppf). Затем добавляли 53.1 мл воды и 55.56 г фосфата калия. Эту смесь перемешивали в течение 30 мин при 22°С. Внутреннюю температуру повышали до 55°С. При 5°С добавляли раствор 31.55 г (113.43 ммоль) сложного пинаколового эфира 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIс) в 162.2 мл этилацетата в течение 120 мин, поддерживая постоянную температуру на уровне 55°С. Смесь охлаждали до 35°С и этилацетат отгоняли в вакууме (при 110 мбар, ~100 мл этилацетата). Затем добавляли 164.4 мл дихлорметана и 255.6 мл воды. Фазы разделяли и водную фазу экстрагировали 84.5 мл дихлорметана. Объединенные органические фазы два раза экстрагировали 1 н. водным раствором гидроксида калия, каждый раз порцией 147 мл. Фазы разделяли и органическую фазу обрабатывали активированным углем (добавляли 21 г активированного угля и суспензию перемешивали в течение ночи при 22°С). Уголь удаляли с помощью фильтрования через диатомовую землю и осадок на фильтре промывали 40 мл дихлорметана. К фильтрату добавляли смесь 14.23 г N-ацетилцистеина и 7.8 г гидроксида калия, растворенных в 217.1 мл воды (рН=9). Двухфазную смесь перемешивали в течение 8 ч при 20°С. Фазы разделяли и органическую фазу сушили над 35.3 г сульфата натрия, который затем отфильтровывали и два раза промывали дихлорметаном, каждый раз порцией 35 мл. Раствор хранили в холодильнике и использовали затем на следующей стадии способа. К этому раствору (~ 350 мл) добавляли 160 мл метанола, и раствор охлаждали до 0-5°С. Затем добавляли 400 мл 1 н. водного раствора хлористоводородной кислоты и перемешивали в течение 30 мин при комнатной температуре. Фазы разделяли и водную фазу два раза экстрагировали дихлорметаном, каждый раз порцией 160 мл. К водной фазе (рН=1) добавляли 14.2 г N-ацетилцистеина и раствор перемешивали в течение ночи при 20°С. Затем добавляли 320 мл дихлорметана и рН доводили до рН=13 путем добавления 160 мл 5 н. водного раствора гидроксида калия. Раствор перемешивали в течение 30 мин. Фазы разделяли и водную фазу промывали 84 мл дихлорметана. Выполняли замену растворителя дихлорметана на н-бутанол: к 212 мл н-бутанола при 110°С медленно добавляли фильтрат и дихлорметан отгоняли. Смесь охлаждали в течение ночи до 22°С, затем охлаждали до 0-3°С и перемешивали в течение 1 ч при этой температуре. Продукт выделяли с помощью фильтрования и кристаллы промывали 20 мл холодного н-бутанола. Продукт сушили в вакууме (20 мбар) при 45°С в течение ночи. Выход: 25.60 г (78.34% от теор.) желтых кристаллов.
ВЭЖХ: 99.48% (% площади) на 4.3 мин.
Пример 2 е
2-[(3R)-3-метилморфолин-4-ил]-4-(1-метил-1Н-пиразол-5-ил)-8-(1Н-пиразол-5-ил)-1,7-нафтиридин (I)
22.5 г (59.93 ммоль) 2-[(3R)-3-метилморфолин-4-ил]-4-(1-метил-1Н-пиразол-5-ил)-8-(1Н-пиразол-5-ил)-1,7-нафтиридина (I) из примера 2d растворяли в 156.4 мл дихлорметана, добавляли 6.75 г квадрасил-меркаптопропила (поглотитель Pd от Johnson Matthey, CAS-номер 1225327-73-0) и 6.75 г изолюта Si-TMT (силикагель-связанный эквивалент 2,4,6-тримеркаптотриазина (ТМТ), поглотитель Pd от Biotage АВ, Швеция, №по каталогу 9538-1000) и суспензию перемешивали в течение 22 ч при 20°С. Суспензию фильтровали и осадок на фильтре два раза промывали дихлорметаном, каждый раз порцией 46.8 мл. Выполняли замену растворителя -дихлорметана на н-бутанол: к 85 мл н-бутанола при 105°С медленно добавляли фильтрат, и дихлорметан отгоняли. В заключение температуру повышали до 105°С (внутренняя температура) и затем весь дихлорметан удаляли. Смесь охлаждали в течение ночи до 22°С, затем охлаждали до 0-3°С и перемешивали в течение 1 часа при этой температуре. Продукт выделяли с помощью фильтрования и кристаллы промывали 24 мл холодного н-бутанола. Продукт сушили в вакууме (20 мбар) при 45°С в течение ночи. Выход: 19.50 г (86.67% от теор.) желтых кристаллов.
ВЭЖХ: 99.97% (% площади).
Содержание бора: <1 млн.ч.
Содержание палладия: <1 млн.ч.
Полиморфная форма соединения (I): В.
Пример 3а
(R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфонат (VIIIb)
К суспензии (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ола (IIа) (100 г, в перерасчете на кЯМР-чистоту 90 мас. %) в дихлорметане (1.00 л) при комнатной температуре добавляли триэтиламин (63 мл, 450 ммоль), что приводило к образованию прозрачного коричневого раствора. Реакционную смесь охлаждали до 0°С (внутренняя температура), используя баню со льдом и солью. При энергичном перемешивании по каплям добавляли трифторметансульфоновый ангидрид (0.076 л, 450 ммоль) на протяжении 10 мин, что приводило к повышению температуры на 10°С. К тому времени, когда было добавлено последнее количество трифторметансульфонового ангидрида, внутренняя температура уже снизилась. Образец (s1, взятый через 5 минут после завершения добавления трифторметансульфонового ангидрида) анализировали с помощью ВЭЖХ, которая указывала, что произошло почти полное превращение исходного вещества. Баню со льдом и солью удаляли и добавляли воду (500 мл). Полученную в результате двухфазную смесь энергично перемешивали в течение 1 минуты. Водную фазу отделяли и органическую фазу дополнительно промывали водой (2×500 мл) для удаления всех остатков триэтиламина и трифторметансульфонового ангидрида. Затем органический слой промывали насыщенным водн. раствором карбоната калия (500 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении до объема 250 мл. Добавляли изопропанол (300 мл), и смесь дополнительно концентрировали до объема -300 мл (отвешивали 290 г раствора продукта). Добавляли дополнительное количество изопропанола (100 мл, до запланированных 2 объемов) и полученную в результате смесь оставляли стоять в течение ночи для того, чтобы кристаллизовался продукт.
На следующее утро образовался твердый осадок кристаллического вещества. Твердое вещество разрыхляли, расцарапывая его шпателем, после чего смесь перемешивали магнитной мешалкой, чтобы разбить крупные комки на более мелкие частицы (2 часа). Твердые вещества отфильтровывали и сушили в потоке воздуха с получением 110.3 г (83%) (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфоната (промежуточное соединение (VIIIb)) в виде темно-желтого кристаллического порошка, который все еще содержал несколько больших комков. ВЭЖХ чистота 98% площади. Маточную жидкость концентрировали до тех пор, пока в колбе не оставалось 80 г раствора, что позволяло выделить вторую порцию продукта массой 6.26 г (4.7%).
Пример 3b
(R)-4-(8-хлор-4-(1-метил-1H-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолин (IXb)
К раствору (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфоната (VIIIb) (125.0 г, 304 ммоль) в 300 мл дегазированного изопропилацетата добавляли PdCl2(dppf) (6.66 г, 9.1 ммоль, 3 мол.%). Смесь нагревали до 50°С, после чего к реакционной смеси добавляли дегазированный раствор гидрокарбоната калия (122 г, 4 экв.) в воде (750 мл) при 50°С. Сразу же после этого добавляли раствор сложного пинаколового эфира 1-метил-1Н-пиразол-5-бороновой кислоты (VIb) (63.2 г, 304 ммоль, 1.0 экв.) в 750 мл дегазированного изопропилацетата на протяжении 1.5 часов. После перемешивания в течение дополнительного часа при 50°С ВЭЖХ анализ указывал на полное превращение.
Реакционную смесь фильтровали через диатомовую землю и осадок на фильтре промывали изопропилацетатом (300 мл). После выполнения этого фильтрования два слоя разделяли. Водную фазу экстрагировали изопропилацетатом (1*600 мл). Объединенные органические фазы промывали 1 н. водным раствором гидроксида натрия (4 × 600 мл) для удаления следов гидролизованного трифлата (в соответствии с ВЭЖХ только ~1% площади было образовано). Органическую фазу последовательно промывали водой (600 мл) и соляным раствором (600 мл) и сушили над сульфатом натрия. Через сорок минут добавляли активированный уголь (~10 г на 100 г исходного вещества) и полученную в результате суспензию перемешивали в течение ночи. На следующее утро твердые вещества отфильтровывали на диатомовой земле, осадок на фильтре промывали изопропилацетатом (200 мл) и фильтрат концентрировали с получением сырого (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина (IХb) в виде коричневой пены (100.5 г, 96%).
Очистка сырого промежуточного соединения (IХb)
В 2 л круглодонную колбу загружали сырой (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолин (IХb) (98 г, 268 ммоль) и изопропанол (500 мл). При нагревании до 82°С образовывался прозрачный коричневый раствор. Добавляли активированный уголь (10 г, 833 ммоль) и черную суспензию перемешивали при 82°С в течение 3 часов. Уголь удаляли с помощью фильтрования через диатомовую землю с использованием предварительно нагретой фильтрационной установки. Фильтр промывали 50 мл изопропанола и фильтрат концентрировали при пониженном давлении с получением 92.87 г светло-коричневого твердого вещества. Фильтр, использованный для удаления активированного угля, промывали дихлорметаном и желтый фильтрат концентрировали в вакууме. Вещество повторно растворяли в 400 мл изопропанола при температуре 83°С, что приводило к прозрачному темно-коричневому раствору. При охлаждении осаждалось светло-коричневое твердое вещество. Суспензию перемешивали в течение 3 часов при комнатной температуре. Твердое вещество собирали с помощью фильтрования и промывали на фильтре изопропанолом (2 × 30 мл). После сушки собирали 72.5 г очищенного (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина (IXb) (72.5 г, выход 79%). Маточную жидкость, которая все еще содержала большие количества целевого продукта, концентрировали в вакууме с получением 20.8 г твердого коричневого вещества.
Пример 4а
(R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил-трифторметансульфонат (VIIIb)
3.00 кг (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ола (IIа) растворяли в 31.9 кг дихлорметана при 20°С. Затем добавляли 1190 г пиридина. Раствор охлаждали до -10°С (внутренняя температура) и добавляли раствор 4.236 кг бис(трифторметансульфонового ангидрида) в 10 кг дихлорметана, поддерживая внутреннюю температуру при -10°С. Время добавления приблизительно составляло 1 час. После завершения реакции добавляли 12.0 кг воды, поддерживая внутреннюю температуру в диапазоне 0°С - 15°С.После добавления смесь перемешивали в течение 5 мин. Органическую фазу отделяли и промывали 12.0 кг воды. Органическую фазу промывали 2 раза 0.5 н. водным раствором карбоната калия, каждый раз порцией 6 кг, и в заключение 4.5 кг воды. Органическую фазу фильтровали через активированный уголь (пластины фильтра из активированного угля Seitz) и фильтрат отгоняли до изопропанола (замена растворителя). Сначала дихлорметан отгоняли в вакууме (100 мбар) до получения концентрированного раствора (до тех пор, пока он поддавался перемешиванию), затем добавляли 9.5 кг изопропанола и снова выполняли отгонку (100 мбар) (до тех пор, пока раствор поддавался перемешиванию). Добавляли еще 9.5 кг изопропанола и снова выполняли отгонку (до тех пор, пока раствор поддавался перемешиванию), и в заключение добавляли 1 кг изопропанола (всего ~ 8- 9 кг изопропанола).
Для кристаллизации температуру снижали от 50°С до 18°С в течение 90 мин (линейно). Смесь перемешивали в течение 12 мин при 18°С и затем охлаждали до 0°С (линейно, в течение 180 мин). Затем смесь перемешивали в течение 60 мин при 0°С. Кристаллы отделяли с помощью фильтрования и промывали 2.4 кг холодного изопропанола. Продукт сушили в вакууме при 45°С в течение по меньшей мере 12 ч (до постоянной массы).
В соответствии с этим протоколом получали четыре партии:
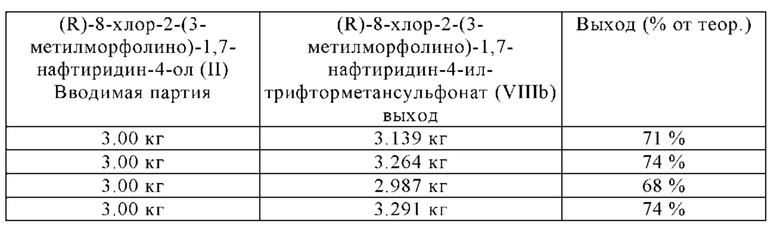
Пример 4b
(R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолин (IХb)
В атмосфере азота 2.1 кг (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-илтрифторметансульфоната (VIIIb) растворяли в 8.4 л этилацетата и добавляли 112 г PdCl2(dppf). Раствор нагревали до 50°С (внутренняя температура) и затем добавляли 2.042 кг гидрокарбоната калия (KНСО3), растворенного в 11.0 л воды, с последующим добавлением 1.6 л воды (для очистки трубок). Внутреннюю температуру устанавливали на 43°С и затем добавляли 1.061 кг сложного пинаколового эфира 1-метил-1Н-пиразол-5-бороновой кислоты, растворенного в 6.0 л этилацетата, в течение 3 ч, поддерживая внутреннюю температуру равной 43°С. После завершения добавления смесь перемешивали в течение 75 мин при 43°С.
Выделение продукта реакции: смесь охлаждали до 22°С и органическую фазу отделяли (~15.4 л). Водную фазу экстрагировали 6.3 л этилацетата. Органические фазы объединяли и охлаждали до 10°С и затем промывали 15.75 л 5.3% водного раствора гидроксида калия (выполняли перемешивание в течение 15 мин при 10°С). Фазы разделяли и органическую фазу снова промывали при 10°С с помощью 15.75 л 5.3% водного раствора гидроксида калия. После этого органическую фазу отделяли и промывали два раза водой, каждый раз порцией 10.5 л. К органической фазе добавляли 2.10 кг сульфата магния и перемешивали в течение 70 мин при 22°С. Затем сульфат магния отфильтровывали, два раза промывали этилацетатом, каждый раз порцией 4.2 л. К фильтрату добавляли 1.47 кг активированного угля и суспензию перемешивали в течение 2.5 ч при 22°С. Активированный уголь удаляли с помощью фильтрования через диатомовую землю и промывали этилацетатом (два раза, каждый раз порцией 4.2 л). Получали 27.0 кг фильтрата (продукт в этилацетате).
В соответствии с этим протоколом получали пять партий:
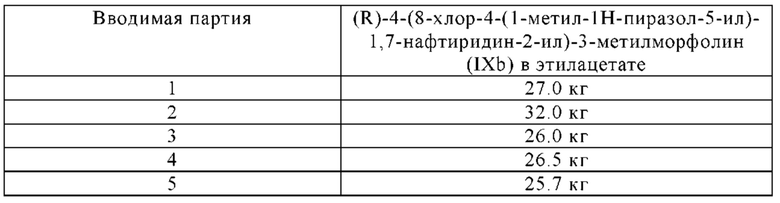
Кристаллизация из изопропанола:
27.0 кг (введение 1) и 32.0 кг (введение 2) (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина (IXb) в этилацетате (всего: 59.0 кг) объединяли и выполняли замену растворителя на изопропанол. Этилацетат отгоняли при нормальном давлении (внутренняя температура: 73°С в начале, 78°С в конце). Когда было отогнано 57.5 л, добавляли 10.5 кг изопропанола и 13.5 л жидкости отгоняли (при нормальном давлении, внутренняя температура: 81°С в начале, 83°С в конце). Добавляли еще 10.5 кг изопропанола и 13.5 л жидкости отгоняли (при нормальном давлении, внутренняя температура: 80°С в начале 83°С в конце). Затем добавляли еще 10.5 кг изопропанола и 14.0 л жидкости отгоняли (при нормальном давлении, внутренняя температура: 82°С в начале 84-85°С в конце). Раствор охлаждали до 18°С (линейно в течение 420 мин). Суспензию перемешивали в течение 1 ч при 20°С. Продукт выделяли с помощью фильтрования, промывали изопропанолом (всего 2.4 кг). Продукт сушили в вакууме при 50°С в течение по меньшей мере 12 ч (до постоянной массы). Выход: 2.69 кг (77% от теор.)
Подобным образом вводимые партии 3-5 объединяли (всего 78.2 кг) и выполняли такую же самую замену растворителя с получением 4.042 кг (77% от теор.) продукта.
В следующей таблице обобщены результаты:

В следующей таблице обобщены аналитические результаты двух партий (2.69 кг партия IXb и 4.042 кг партия IXb):
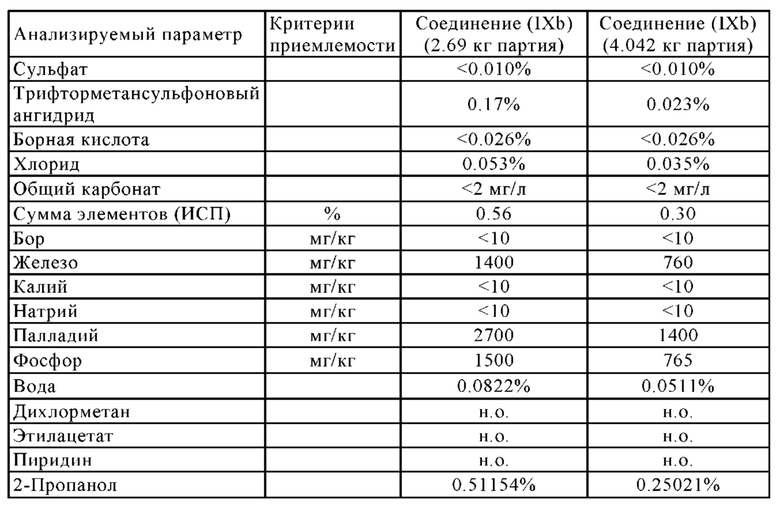
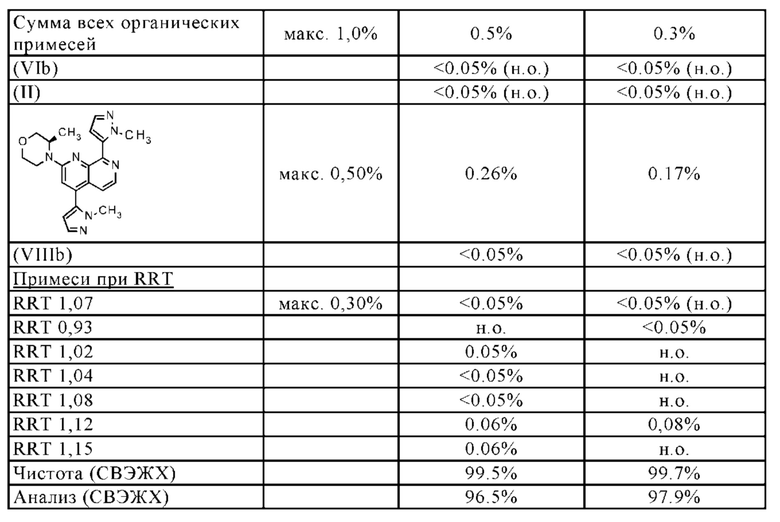
Пример 4с
2-[(3R)-3-метилморфолин-4-ил]-4-(1-метил-1Н-пиразол-5-ил)-8-(1Н-пиразол-5-ил)-1,7-нафтиридин (I)
В атмосфере азота 3.40 кг (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина (IXb) растворяли в 10.8 кг этилацетата и добавляли 221 г PdCl2(dppf). Затем добавляли 6.0 кг воды и 6.2 кг фосфата калия. Эту смесь перемешивали в течение 30 мин при 22°С. Внутреннюю температуру повышали до 55°С. При 55°С добавляли раствор 3.58 кг сложного пинаколового эфира 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIс) в 16.4 кг этилацетата в течение 120 мин, поддерживая постоянную температуру на уровне 55°С, и полученную в результате смесь перемешивали при 200 об/мин. Добавляли 2 кг этилацетата (использовали для промывки трубок). После добавления смесь перемешивали в течение 10 мин при 55°С.
Для выделения продукта реакции температуру снижали до 35°С и в вакууме отгоняли 26.6 кг этилацетата. Смесь охлаждали до 20°С и добавляли 24.7 кг дихлорметана и 29.0 кг воды. Смесь перемешивали в течение 10 мин. Органическую фазу отделяли и водную фазу экстрагировали 12.8 кг дихлорметана. Объединенные органические фазы промывали 17.7 кг 1 н. водного раствора гидроксида калия (выполняли перемешивание в течение 10 мин). Фазы разделяли и органическую фазу снова промывали 17.7 кг 1 н. водного раствора гидроксида калия (перемешивая в течение 10 мин). Органическую фазу отделяли и фильтровали через активированный уголь (пластины фильтра из активированного угля Seitz) в течение 3 ч (прокачивая фильтрат по кругу). Активированный уголь два раза промывали дихлорметаном, каждый раз порцией 13.3 кг, и один раз 6.6 кг дихлорметана. Объединенные фильтраты добавляли к раствору 1.61 N-ацетилцистеина в 9.0 кг водного раствора гидроксида калия (1099 г гидроксида калия, растворенного в 8.0 кг воды). Смесь перемешивали в течение 18 ч при 20°С. Фазы разделяли. Получали 35.72 кг органической фазы.
Подобным образом суммарно 3 партии, исходя из 3.40 кг (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина (IXb), превращали в (3R)-3-метил-4-(4-(1-метил-1Н-пиразол-5-ил)-8-(1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)морфолин (VIIc/VIId) в дихлорметане:

Три партии разделяли на 6 порций (по половине каждой партии) и каждую партию обрабатывали на следующей стадии реакции.
Подход описан для разделенной партии массой 17.8 кг (VIIc/VIId) в дихлорметане.
17.8 кг (VIIc/VIId) в дихлорметане брали из бочки и бочку промывали 800 г дихлорметана. Затем 9 л дихлорметана отгоняли при нормальном давлении (60°С). Раствор охлаждали до 22°С и добавляли 3.3 кг дихлорметана и 7.2 кг метанола. Смесь охлаждали до 0-5°С и добавляли 16.8 кг водного раствора хлористоводородной кислоты, поддерживая температуру между 0 и 20°С. Смесь перемешивали в течение 10 мин при 20-22°С. Фазы разделяли и водную фазу два раза экстрагировали дихлорметаном, каждый раз порцией 8.8 кг. Водную фазу отделяли и добавляли 800 г N-ацетилцистеина, и раствор перемешивали в течение 12 ч при 20°С. Затем добавляли 17.6 кг дихлорметана и добавляли 8246 г 5 н. водного раствора гидроксида калия, и смесь перемешивали в течение 30 мин. Конечное значение рН водной фазы составляло рН=13.6 (в этот момент рН должно составлять 12-14). Фазы разделяли, водную фазу экстрагировали 5.2 кг дихлорметана и органические фазы объединяли. Получали 22.7 кг соединения (I) в дихлорметане.
В следующей таблице обобщены результаты 6 партий, полученных описанным способом:
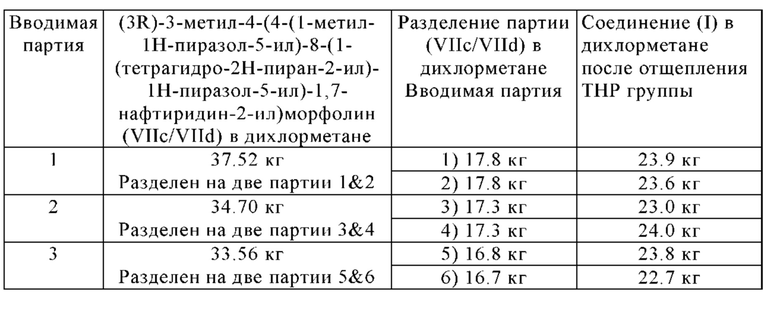
Для следующей стадии способа объединяли дважды по 3 партии из предыдущих полученных партий и выполняли превращение в конечный продукт:
Замена растворителя - дихлорметана на н-бутанол: объединяли 23.9 кг, 23.6 кг и 23.0 кг соединения (I) (всего 70.5 кг+5.0 кг промывки) и медленно добавляли к 16.6 кг н-бутанола, который предварительно нагревали до 98°С. При нормальном давлении отгоняли 59 л дихлорметана. Внутренняя температура во время отгонки составляла 81 - 90°С. Затем 5.0 кг дихлорметана, который использовали для очистки трубок, также отгоняли (5 л). Добавляли 15.5 кг н-бутанола и отгоняли 10 л жидкости при нормальном давлении (88 - 93°С). Температуру повышали до 108°С (внутренняя температура, отгоняли 100 мл н-бутанола). В этот момент дихлорметана уже не оставалось. Раствор охлаждали до 20-22°С (в течение 7 ч (линейно)). Суспензию перемешивали в течение 1 ч при 20°С, затем охлаждали до 2-3°С, и перемешивали в течение 1 ч при этой температуре. Кристаллы отделяли с помощью фильтрования и промывали 8.0 кг холодного н-бутанола. Сырой осадок на фильтре, содержащий продукт (I), растворяли непосредственно на фильтре (без выделения), используя 47.5 кг теплого 30°С дихлорметана (в случае, если этого количества недостаточно, можно использовать больше дихлорметана, а затем отогнать его). Раствор охлаждали до 20°С и добавляли 1020 г квадрасил-меркаптопропила (поглотитель Pd от Johnson Matthey, CAS-номер 1225327-73-0) и 1020 г изолюта Si-TMT (силикагель-связанный эквивалент 2,4,6-тримеркаптотриазина (ТМТ), поглотитель Pd от Biotage АВ, Швеция, №по каталогу 9538-1000). Суспензию перемешивали в течение 12 ч при 20-21°С.Суспензию фильтровали и осадок на фильтре два раза промывали дихлорметаном, каждый раз порцией 13.3 кг. Фильтрат фильтровали снова (фильтрация частиц согласно GMP).
Конечный способ кристаллизации
Замена растворителя - дихлорметана на н-бутанол: фильтрат соединения (I) медленно добавляли к 16.5 кг н-бутанола, который предварительно нагревали до 98°С. При нормальном давлении отгоняли 53 л дихлорметана. Внутренняя температура во время отгонки составляла 93 - 98°С. Затем 5.0 кг дихлорметана, который использовали для очистки трубок, также отгоняли (4 л). Добавляли 9.0 кг н-бутанола и отгоняли 10,5 л при нормальном давлении (90 - 91°С). Температуру повышали до 109°С (внутренняя температура, отгоняли 100 мл н-бутанола). В этот момент дихлорметана уже не оставалось. Раствор охлаждали до 20-22°С (в течение 7 ч (линейно)). Суспензию перемешивали в течение 1 ч при 20°С, затем охлаждали до 2-3°С, и перемешивали в течение 1 ч при этой температуре. Кристаллы отделяли с помощью фильтрования и промывали 8.0 кг холодного н-бутанола. Продукт сушили в вакууме (30 мбар) при 50°С в течение по меньшей мере 12 ч (до постоянной массы).
Получали 3.753 кг (67% от теор.) желтых кристаллов.

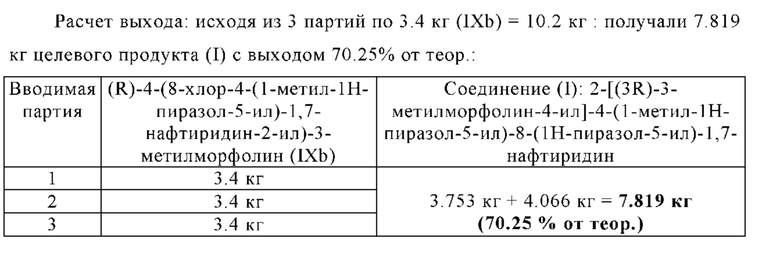
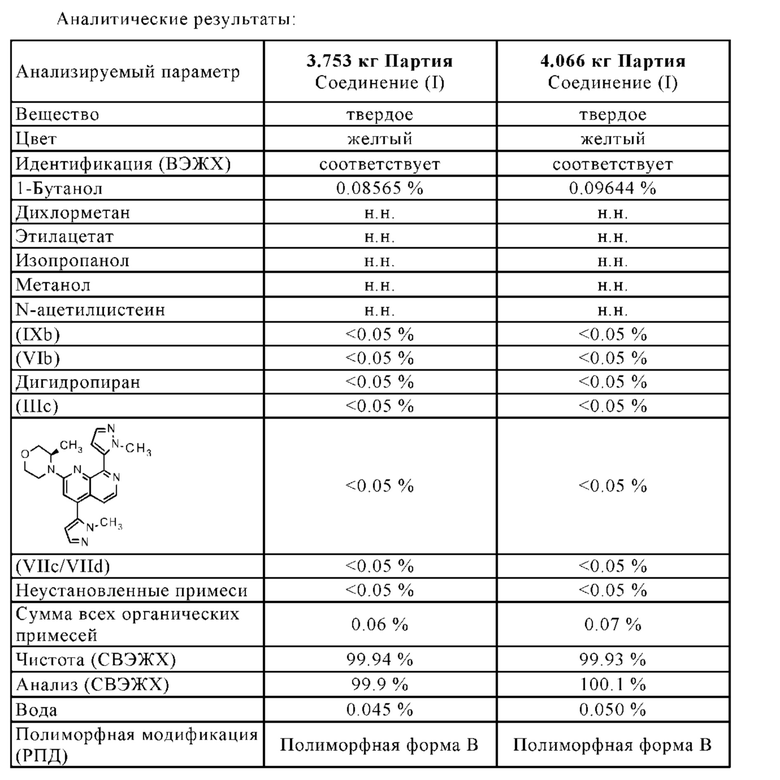
Пример 5
Производственный цикл опытно-промышленной установки
Осуществляли производственный цикл опытно-промышленной установки. Шесть партий (каждая по 16.3 кг) превращали таким же образом, как описано в примере 4с (без очистки реакторов и фильтровальных установок во время производственного цикла). Суммарно 97.8 кг (ГХЬ) давали 75.4 кг целевого соединения (I)
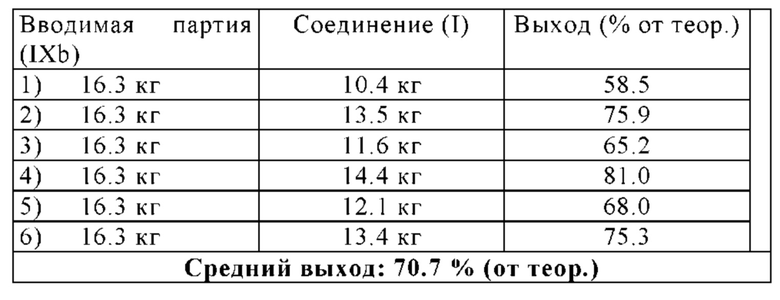
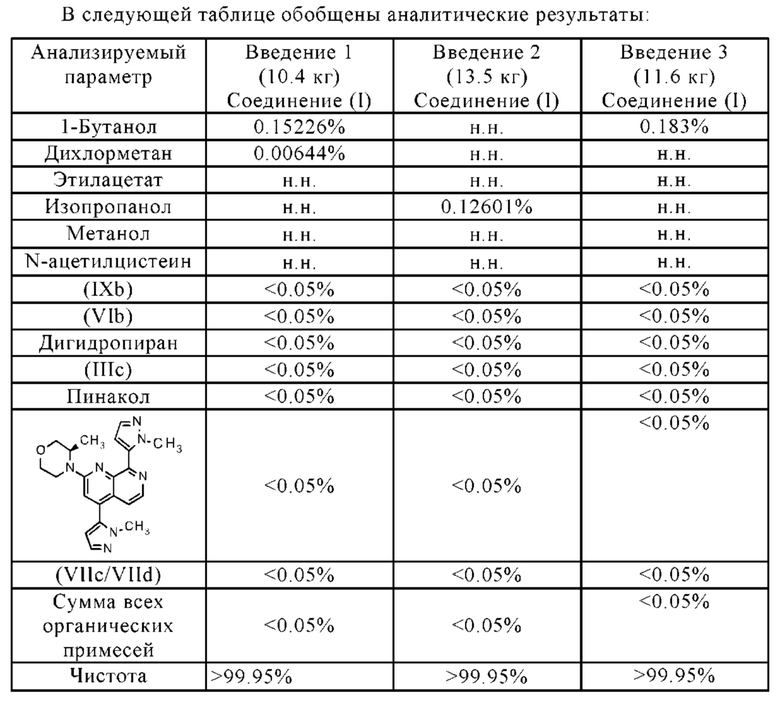

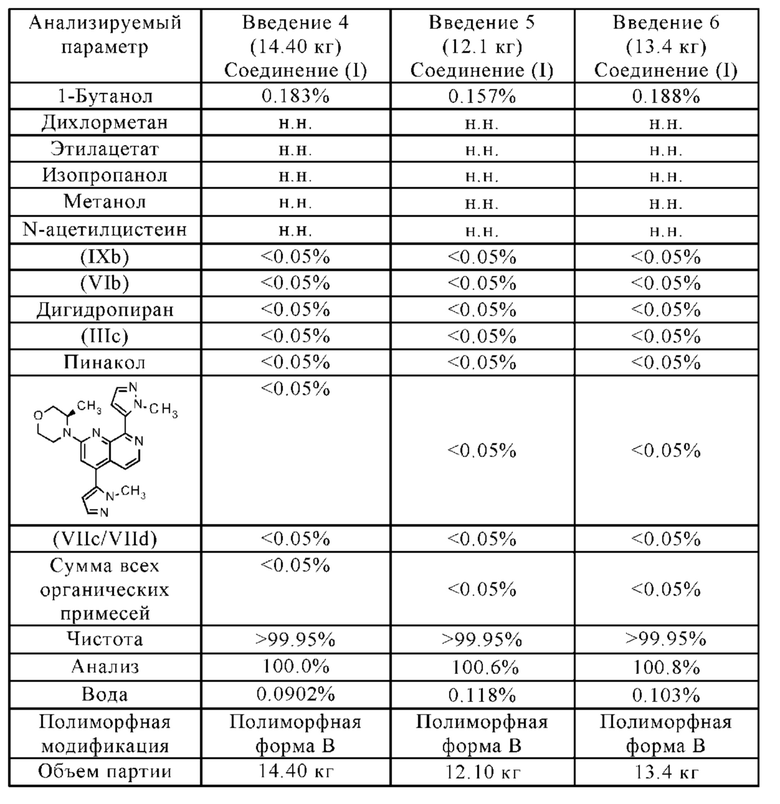
Две из этих партий (введение 1 - 10.40 кг I и введение 2 - 13.50 кг I) объединяли и анализировали.
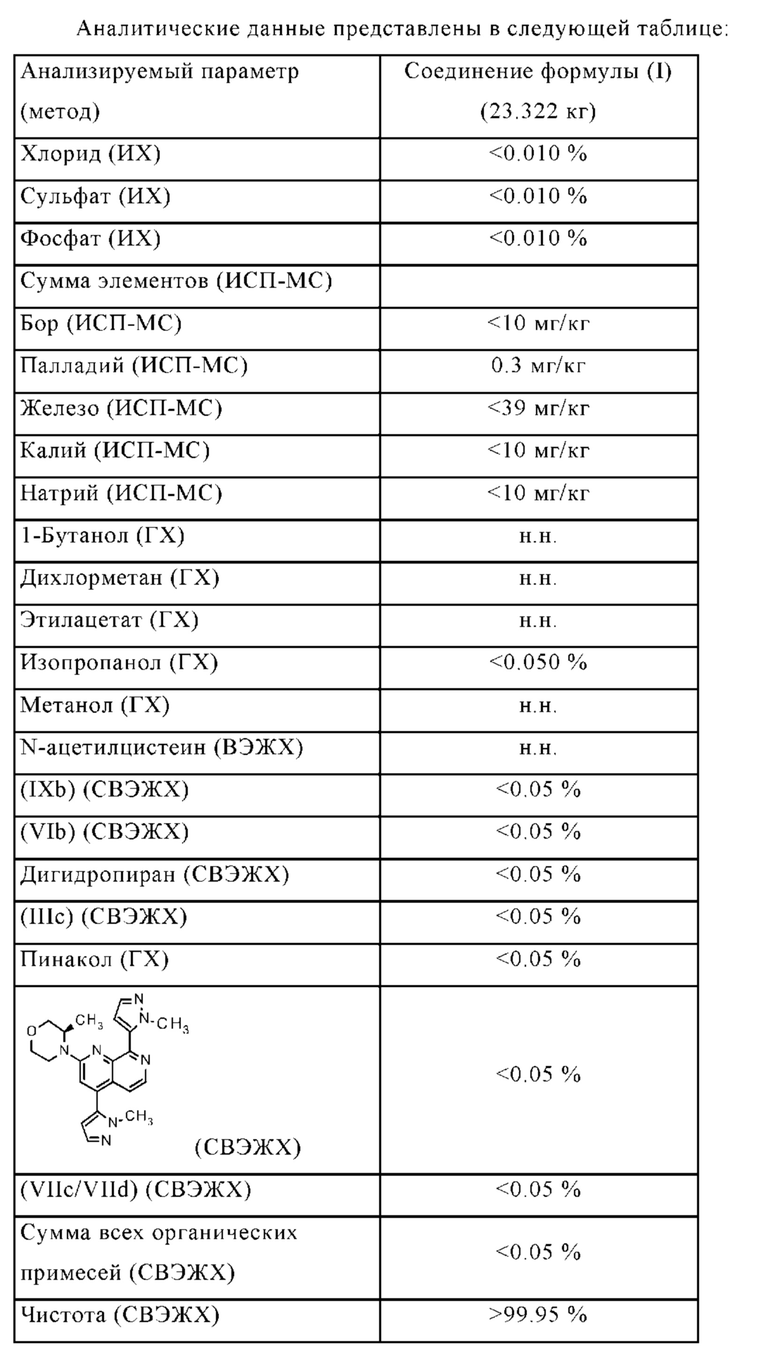
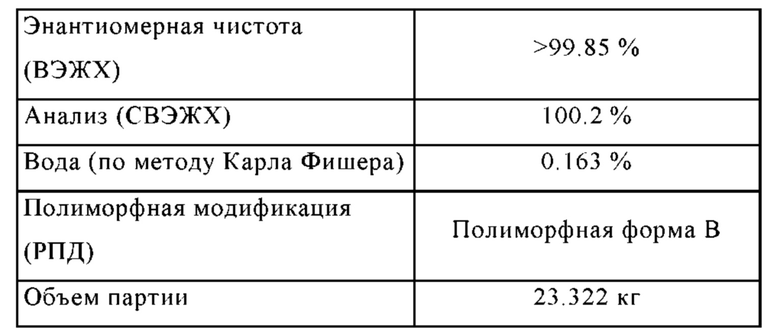
Пример 6
2-[(3R-3-метилморфолин-4-ил1-4-(1-метил-1Н-пиразол-5-ил)-8-(1Н-пиразол-5-ил)-1,7-нафтиридин (I)
В атмосфере азота 30 г (87.255 ммоль) (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолина (IХb) растворяли в 105.8 мл этилацетата и добавляли 1.91 г (2.618 ммоль) PdCl2(dppf). Затем добавляли 53.1 мл воды и 55.56 г фосфата калия. Эту смесь перемешивали в течение 30 мин при 22°С. Внутреннюю температуру повышали до 55°С. При 55°С добавляли раствор 31.55 г (113.43 ммоль) сложного пинаколового эфира 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты (IIIс) в 162.2 мл этилацетата в течение 120 мин, поддерживая постоянную температуру на уровне 55°С. Смесь охлаждали до комнатной температуры (~ 22°С) и добавляли 150 мл воды. Фазы разделяли и водную фазу экстрагировали 100 мл этилацетата. Объединенные органические фазы обрабатывали активированным углем (добавляли 21 г активированного угля и суспензию перемешивали в течение ночи при 22°С). Уголь удаляли с помощью фильтрования через диатомовую землю и осадок на фильтре промывали 30 мл этилацетата. К фильтрату добавляли 100 мл метанола и 300 мл 1 н. водного раствора хлористоводородной кислоты и перемешивали в течение 30 мин при комнатной температуре. Фазы разделяли и водную фазу экстрагировали 100 мл этилацетата. К водной фазе (рН=1) добавляли 14.82 г N-ацетилцистеина и раствор перемешивали в течение ночи при 20°С.Добавляли 300 мл дихлорметана и рН доводили до рН=13 путем добавления 190 мл 5 н. водного раствора гидроксида калия. Раствор перемешивали в течение 30 мин.
Фазы разделяли и органическую фазу промывали 150 мл воды. К органической фазе добавляли 6 г квадрасил-меркаптопропила (поглотитель Pd от Johnson Matthey, CAS-номер 1225327-73-0) и 6 г изолюта Si-TMT (силикагель-связанный эквивалент 2,4,6-тримеркаптотриазина (ТМТ), поглотитель Pd от Biotage АВ, Швеция, № по каталогу 9538-1000), и суспензию перемешивали в течение ночи при 20°С. Суспензию фильтровали и осадок на фильтре два раза промывали дихлорметаном, каждый раз порцией 25 мл. Выполняли замену растворителя -дихлорметана на н-бутанол: к 60 мл н-бутанола при 85°С медленно добавляли фильтрат и дихлорметан отгоняли. В заключение температуру повышали до 105°С (внутренняя температура) и затем весь дихлорметан удаляли. Смесь охлаждали в течение ночи до 22°С, затем охлаждали до 0-3°С, и перемешивали в течение 1 ч при этой температуре. Продукт выделяли с помощью фильтрования и кристаллы промывали 20 мл холодного н-бутанола. Продукт сушили в вакууме (20 мбар) при 45°С в течение ночи. Выход: 21.27 г (64.93% от теор.) желтых кристаллов.
ВЭЖХ: 99.19% (% площади).
Содержание бора: <1 млн.ч.
Содержание палладия: <1 млн.ч.
Модификация (Мод.): В.
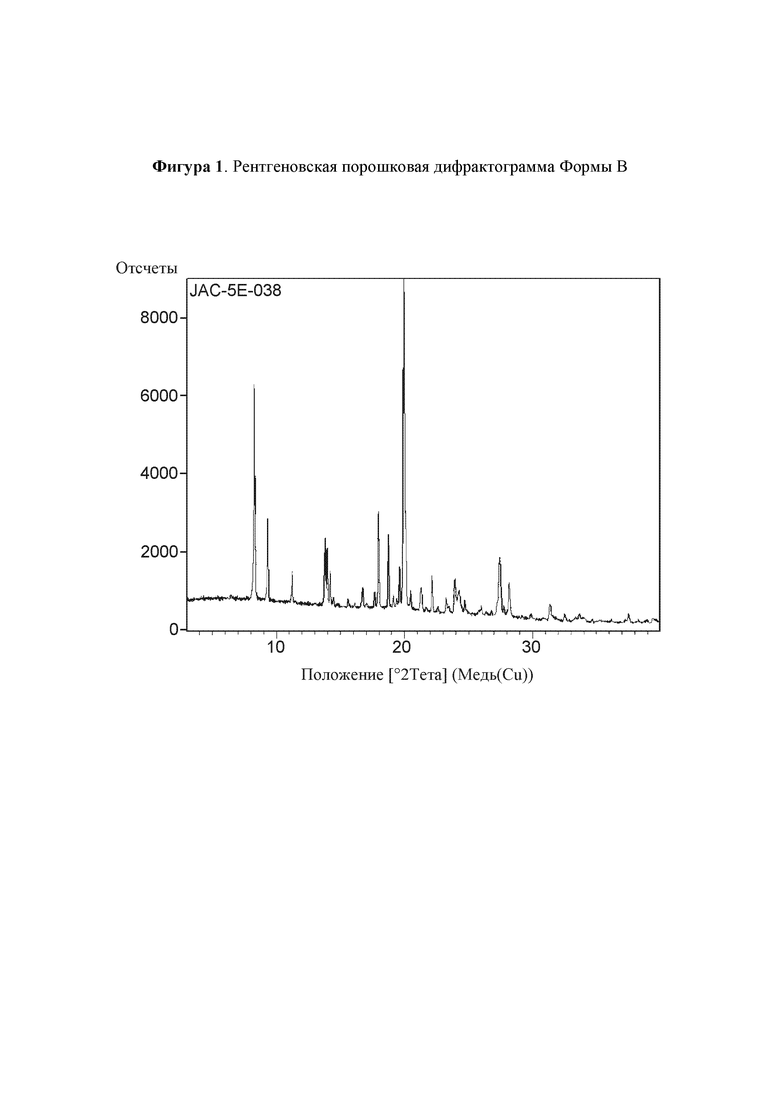
Изобретение относится к способу получения 2-[(3R)-3-метилморфолин-4-ил]-4-(1-метил-1Н-пиразол-5-ил)-8-(1Н-пиразол-5-ил)-1,7-нафтиридина ("соединение формулы (I)", а также промежуточным соединениям, пригодным для получения соединения формулы (I). Технический результат: разработан новый эффективный способ получения соединения формулы (I), позволяющий получить продукт с высоким выходом и чистотой. 3 н. и 17 з.п. ф-лы, 6 пр., 1 ил., 7 табл.
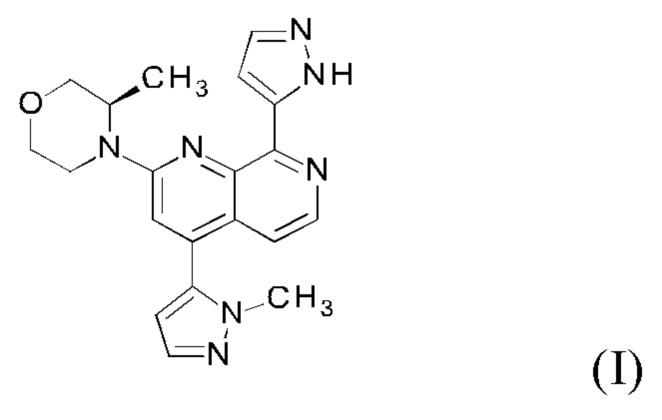
1. Способ получения соединения формулы (I)
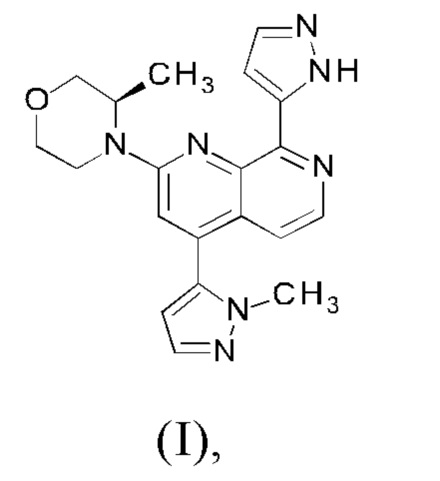
или его таутомера формулы (Ia)

или их смеси,
причем указанный способ включает следующие последовательные стадии:
(а) осуществление реакции промежуточного соединения формулы (IXa)
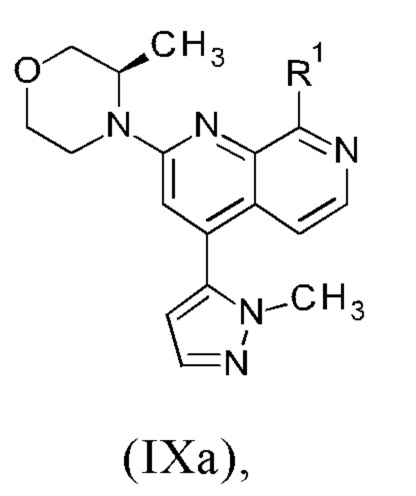
где
R1 представляет собой атом хлора, брома или йода;
с соединением формулы (IIIa) или (IIIb)
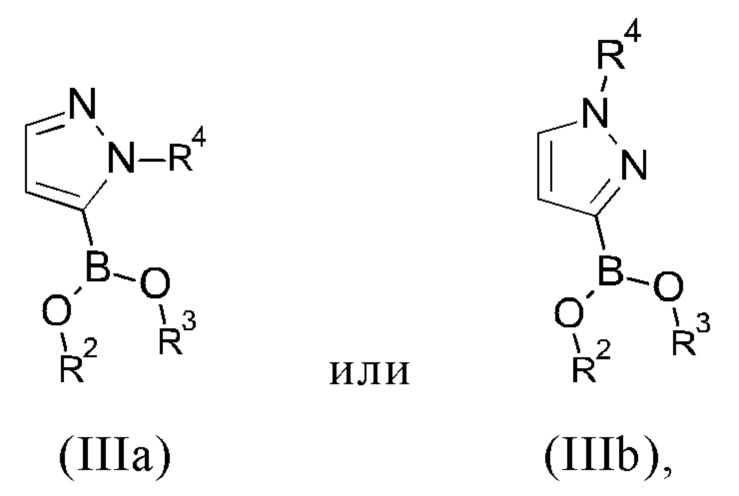
или их смесью, где
R2 и R3 представляют собой, независимо друг от друга, атом водорода или C1-С6-алкильную группу; или
R2 и R3 вместе представляют собой -СН2-СН2- группу или -СН2-СН2-СН2-группу, где указанная -СН2-СН2- группа или -СН2-СН2-СН2- группа необязательно замещена один, два, три или четыре раза группой, выбранной из метила и этила;
или
R2 и R3 вместе представляют собой группу
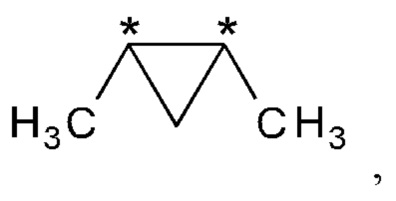
где "*" представляет собой место присоединения к остальной части молекулы;
и
R4 представляет собой группу тетрагидро-2Н-пиран-2-ила;
с получением промежуточного соединения формулы (VIIa) или (VIIb)
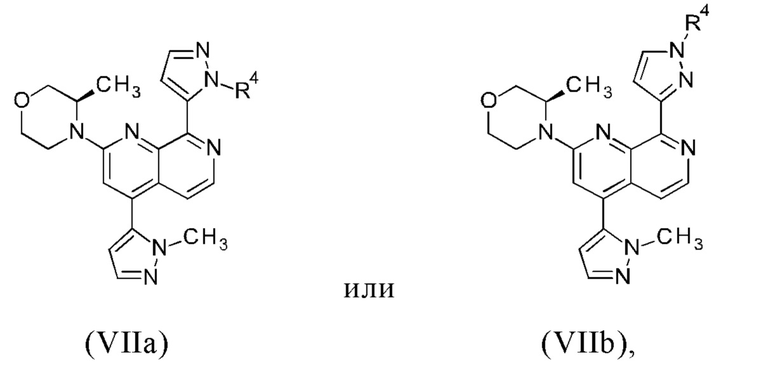
или их смеси, где
R4 представляет собой группу тетрагидро-2Н-пиран-2-ила; и
(b) удаление группы R4 с промежуточного соединения формулы (VIIa) или (VIIb), таким образом обеспечивая соединение формулы (I), или его таутомер формулы (Ia), или их смесь.
2. Способ по п. 1,
где соединение формулы (IXa) представляет собой (R)-4-(8-хлор-4-(1-метил-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)-3-метилморфолин; и/или
где соединение формулы (IIIa) представляет собой сложный пинаколовый эфир 1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-бороновой кислоты; и/или
где соединение формулы (VIIa) представляет собой (3R)-3-метил-4-(4-(1-метил-1Н-пиразол-5-ил)-8-(1-(тетрагидро-2Н-пиран-2-ил)-1Н-пиразол-5-ил)-1,7-нафтиридин-2-ил)морфолин.
3. Способ по п. 1 или 2,
где реакцию промежуточного соединения формулы (IXa) с соединением формулы (IIIa) или формулы (IIIb) осуществляют в присутствии палладиевого катализатора и/или основания.
4. Способ по любому из пп. 1-3, где реакцию промежуточного соединения формулы (IXa) с соединением формулы (IIIa) или (IIIb) осуществляют в органическом растворителе, где растворитель включает изопропилацетат, этилацетат, 1,2-диметоксиэтан, 1,4-диоксан, диметилформамид, тетрагидрофуран, 2-метилтетрагидрофуран, метанол, этанол, 1-пропанол, изопропанол, 1-бутанол или 2-бутанол; или где указанную реакцию осуществляют в смеси растворителей, включающей один или несколько из указанных растворителей и воду.
5. Способ по любому из пп. 1-4, где группу R4 удаляют с промежуточного соединения формулы (VIIa) или (VIIb) путем осуществления реакции промежуточного соединения формулы (VIIa) или (VIIb) с кислотой в растворителе или в смеси растворителей.
6. Способ по любому из пп. 1-5, где промежуточное соединение формулы (VIIa) или (VIIb) представляет собой соединение формулы
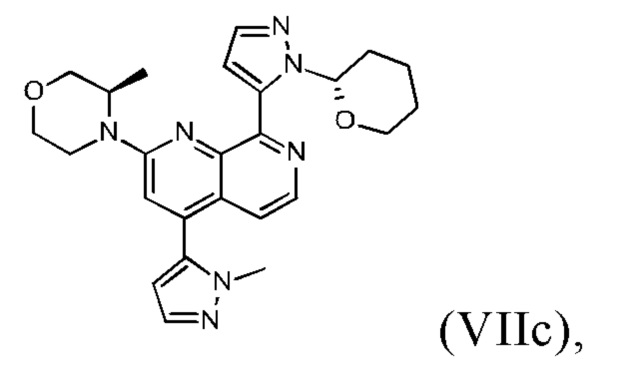
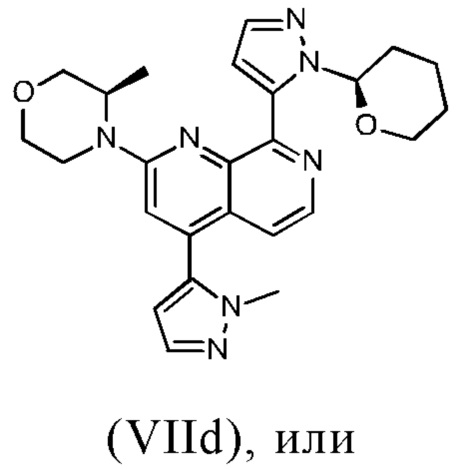
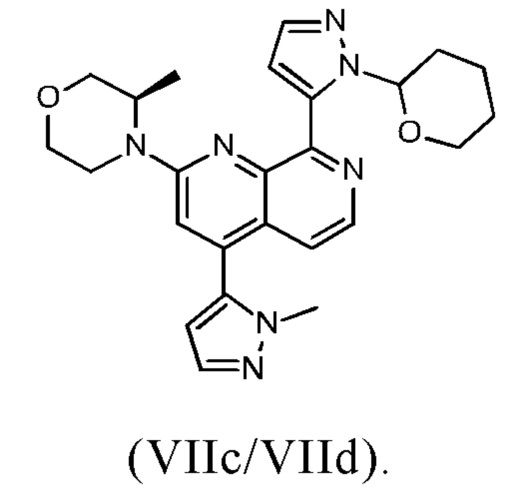
7. Способ по любому из пп. 1-6, где промежуточное соединение формулы (VIIa), (VIIb), (VIIc), (VIId) или (VIIc/VIId) не выделяют и/или не очищают перед стадией удаления группы R4 с промежуточного соединения формулы (VIIa), VII(b), (VIIc), (VIId) или (VIIc/VIId).
8. Способ по любому из пп. 1-7, где группу R4 удаляют с промежуточного соединения формулы (VIIa) или (VIIb) путем осуществления реакции промежуточного соединения формулы (VIIa) или (VIIb) с водным раствором хлористоводородной кислоты в смеси растворителей, включающей дихлорметан, метанол и воду, с получением сырого соединения формулы (I), растворенного в подкисленной водной фазе, где рН полученного в результате подкисленного водного раствора составляет менее 3 (рН<3).
9. Способ по п. 8, где
(а) подкисленный водный раствор сырого соединения формулы (I) экстрагируют один или несколько раз растворителем А, где растворитель А включает дихлорметан, этилацетат, изопропилацетат, тетрагидрофуран, 2-метилтетрагидрофуран, толуол или хлороформ;
и/или подкисленный водный раствор сырого соединения формулы (I) обрабатывают один или несколько раз поглотителем Pd;
(b) подкисленный водный раствор сырого соединения формулы (I), полученный на предыдущей стадии (а), обрабатывают растворителем А и водным раствором основания с получением двухфазной системы, где водная фаза указанной двухфазной системы имеет рН>12;
(c) водную фазу с рН>12, полученную на стадии (b), отделяют от двухфазной системы с получением раствора сырого соединения формулы (I) в растворителе А; и, необязательно,
(d) производят замену растворителя А раствора сырого соединения формулы (I) в растворителе А на растворитель В, где растворитель В включает этанол, н-пропанол, н-бутанол, 2-бутанол или изопропанол; с получением раствора сырого соединения формулы (I) в растворителе В.
10. Способ по п. 9, где растворитель А включает дихлорметан, и где водный раствор основания включает водный раствор гидроксида калия, и где растворитель В включает н-бутанол.
11. Способ по любому из пп. 8-10, где сырое соединение формулы (I) кристаллизуют в растворителе В, предпочтительно в н-бутаноле, с получением полиморфной формы В соединения формулы (I).
12. Способ по любому из пп. 1-11, который перед стадией (а) в соответствии с п. 1 дополнительно включает следующую стадию:
(а) осуществление реакции промежуточного соединения формулы (VIIIa)
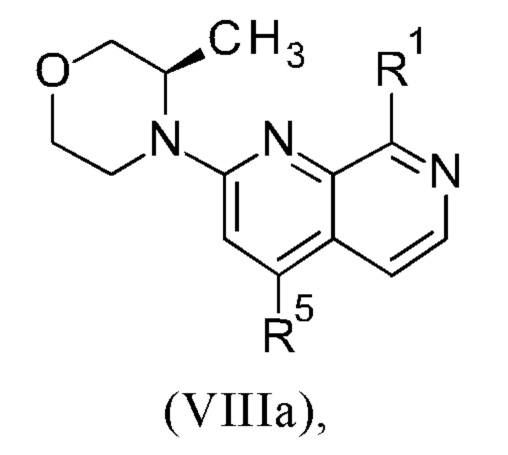
где
R1 представляет собой атом хлора, брома или йода; и
R5 представляет собой группу [(трифторметил)сульфонил]окси;
с соединением формулы (VIa)

где
R2 и R3 представляют собой, независимо друг от друга, атом водорода или C1-С6-алкильную группу; или
R2 и R3 вместе представляют собой -СН2-СН2- группу или -СН2-СН2-СН2-группу, где указанная -СН2-СН2- группа или -СН2-СН2-СН2- группа необязательно замещена один, два, три или четыре раза группой, выбранной из метила и этила;
или
R2 и R3 вместе представляют собой группу
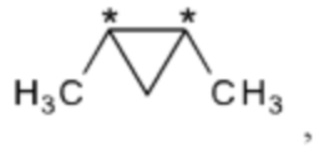
где "*" представляет собой место присоединения к остальной части молекулы;
с получением промежуточного соединения формулы (IXa)
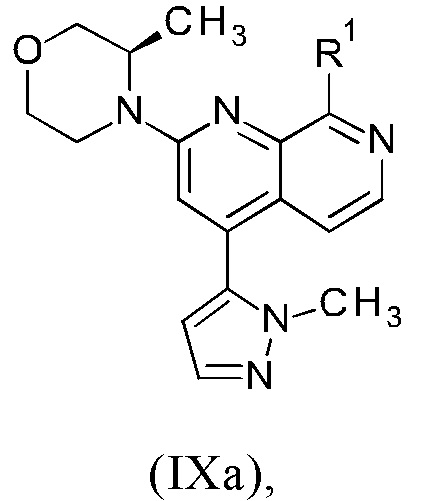
где
R1 представляет собой атом хлора, брома или йода.
13. Способ по п. 12, где соединение формулы (VIIIa) представляет собой (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ил трифторметансульфонат; и/или где соединение формулы (VIa) представляет собой сложный пинаколовый эфир 1-метил-1Н-пиразол-5-бороновой кислоты.
14. Способ по любому из пп. 1-13, где реакцию промежуточного соединения формулы (VIIIa) с соединением формулы (VIa) осуществляют в присутствии палладиевого катализатора и/или основания.
15. Промежуточное соединение формулы (IXa)
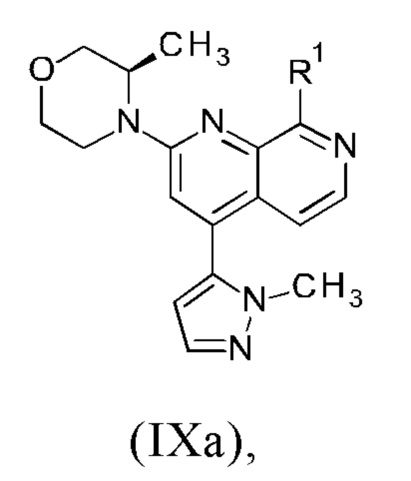
где
R1 представляет собой атом хлора, брома или йода.
16. Промежуточное соединение по п. 15, которое представляет собой соединение формулы (IXb)
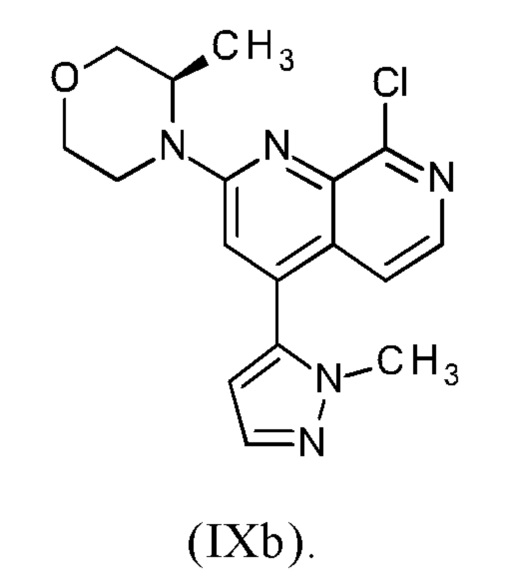
17. Промежуточное соединение формулы (VIIIa)
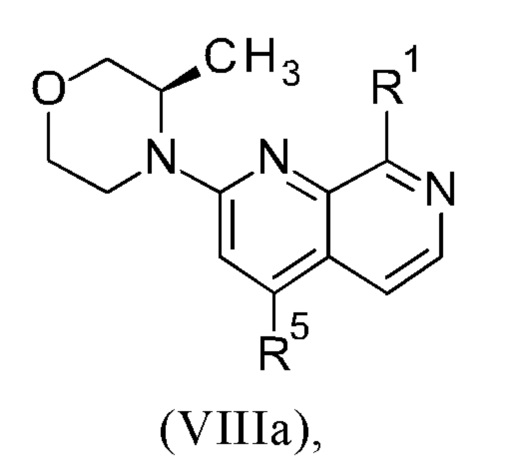
где
R1 представляет собой атом хлора или брома, предпочтительно атом хлора,
и
R5 представляет собой группу [(трифторметил)сульфонил]окси.
18. Промежуточное соединение по п. 17, которое представляет собой соединение формулы (VIIIb)
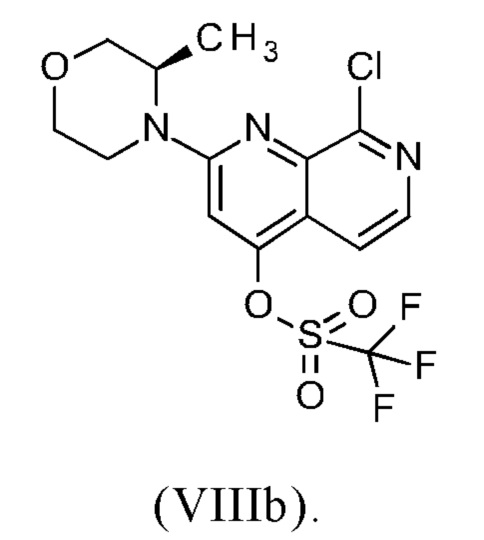
19. Способ по любому из пп. 1-14, который перед стадией (а) в соответствии с пунктом 12 дополнительно включает стадию осуществления реакции промежуточного соединения формулы (II)
где
R1 представляет собой атом хлора, брома или йода;
с соединением трифторметансульфонового ангидрида;
с получением промежуточного соединения формулы (VIIIa)
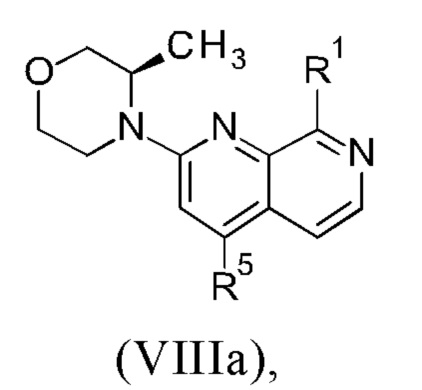
где
R1 представляет собой атом хлора, брома или йода;
R5 представляет собой группу [(трифторметил)сульфонил]окси.
20. Способ по п. 19, где соединение формулы (II) представляет собой (R)-8-хлор-2-(3-метилморфолино)-1,7-нафтиридин-4-ол, который подвергают реакции с трифторметансульфоновым ангидридом с получением промежуточного соединения формулы (VIIIb)

| WO 2016020320 A1, 11.02.2016 | |||
| WO 2011163527 A1, 29.12.2011. |
