ПРИЛОЖЕНИЕ
[0001] Приложение включает библиографию потенциально релевантных источников, перечисленных в статье, написанной авторами настоящего изобретения. Объект этой статьи покрывают предварительные заявки США, на основании которых испрашивается приоритет по этой заявке. Доступ к этим заявкам можно получить по запросу у юрисконсульта или через систему Global Dossier. Эта статья приведена в списке источников первой.
ПРИОРИТЕТНЫЕ ЗАЯВКИ
[0002] Настоящая заявка испрашивает приоритет или преимущество предварительной заявки на патент США No. 62/573,144, озаглавленной “Training a Deep Pathogenicity Classifier Using Large-Scale Benign Training Data” (Обучение глубокого классификатора патогенности с применением большого объема доброкачественных обучающих данных”), авторы Hong Gao, Kai-How Farh, Laksshman Sundaram и Jeremy Francis McRae, поданной 16 октября 2017 г. (№ дела поверенного ILLM 1000-1/IP-1611-PRV), предварительной заявки на патент США № 62/573,149, озаглавленной “Pathogenicity Classifier Based On Deep Convolutional Neural Networks (CNNS)” (“Классификатор патогенности на основе глубоких сверточных нейронных сетей (CNNS)”), авторы Kai-How Farh, Laksshman Sundaram, Samskruthi Reddy Padigepati и Jeremy Francis McRae, поданной 16 октября 2017 г. (№ дела поверенного ILLM 1000-2/IP-1612-PRV), предварительной заяви на патент США № 62/573,153, озаглавленной “Deep Semi-Supervised Learning that Generates Large-Scale Pathogenic Training Data” (“Глубокое обучение с частичным привлечением учителя, которое генерирует большие объемы обучающих данных о патогенах”), авторы Hong Gao, Kai-How Farh, Laksshman Sundaram и Jeremy Francis McRae, поданной 16 октября 2017 г. (№ дела поверенного ILLM 1000-3 /IP-1613-PRV), и предварительной заявки на патент США № 62/582,898, озаглавленной ““Pathogenicity Classification of Genomic Data Using Deep Convolutional Neural Networks (CNNs)” (Классификация патогенности геномных данных с применением глубоких сверточных нейронных сетей ((CNN))), авторы Hong Gao, Kai-How Farh и Laksshman Sundaram, поданной 7 ноября 2г. 017 (№ дела поверенного ILLM 1000-4/IP-1618-PRV). Эти предварительные заявки включены в настоящий документ посредством ссылки для всех целей.
ВКЛЮЧЕНИЕ
[0003] Следующие документы полностью включены в настоящий текст посредством ссылки так, как если бы они были приведены здесь полностью:
[0004] Патентная заявка РСТ № PCT/US2018/_______, озаглавленная “DEEP CONVOLUTIONAL NEURAL NETWORKS FOR VARIANT CLASSIFICATION”(“ГЛУБОКИЕ СВЕРТОЧНЫЕ НЕЙРОННЫЕ СЕТИ ДЛЯ КЛАССИФИКАЦИИ ВАРИАНТОВ”) ("ГЛУБОКИЕ СВЕРТОЧНЫЕ НЕЙРОННЫЕ СЕТИ ДЛЯ КЛАССИФИКАЦИИ ВАРИАНТОВ"), авторы Laksshman Sundaram, Kai-How Farh, Hong Gao, Samskruthi Reddy Padigepati И Jeremy Francis McRae, поданная одновременно 15 октября 2018 г. (№ дела поверенного ILLM 1000-9/IP-1612-PCT), позже опубликованная как публикация PCT № WO ____________.
[0005] Патентная заявка РСТ № PCT/US2018/_______, озаглавленная “SEMI-SUPERVISED LEARNING FOR TRAINING AN ENSEMBLE OF DEEP CONVOLUTIONAL NEURAL NETWORKS” (“ОБУЧЕНИЕ С ЧАСТИЧНЫМ ПРИВЛЕЧЕНИЕМ УЧИТЕЛЯ ДЛЯ ТРЕНИРОВКИ АНСАМБЛЯ ГЛУБОКИХ СВЕРТОЧНЫХ НЕЙРОННЫХ СЕТЕЙ”), авторы Laksshman Sundaram, Kai-How Farh, Hong Gao и Jeremy Francis McRae, оданная одновременно 15 октября 2018 г.(№ дела поверенного ILLM 1000-10/IP-1613-PCT) , позже опубликованная как публикация PCT № WO ____________.
[0006] Непредварительная патентная заявка на патент США, озаглавленная “СПОСОБЫ ОБУЧЕНИЯ ГЛУБОКИХ СВЕРТОЧНЫХ НЕЙРОННЫХ СЕТЕЙ НА ОСНОВЕ ГЛУБОКОГО ОБУЧЕНИЯ”, авторы Hong Gao, Kai-How Farh, Laksshman Sundaram and Jeremy Francis McRae, (№ дела поверенного ILLM 1000-5/IP-1611-US), поданная одновременно.
[0007] Непредварительная патентная заявка на патент США, озаглавленная “DEEP CONVOLUTIONAL NEURAL NETWORKS FOR VARIANT CLASSIFICATION”(“ГЛУБОКИЕ СВЕРТОЧНЫЕ НЕЙРОННЫЕ СЕТИ ДЛЯ КЛАССИФИКАЦИИ ВАРИАНТОВ”), авторы Laksshman Sundaram, Kai-How Farh, Hong Gao и Jeremy Francis McRae, (№ дела поверенного ILLM 1000-6/IP-1612-US), поданная одновременно.
[0008] Непредварительная патентная заявка на патент США, озаглавленная “SEMI-SUPERVISED LEARNING FOR TRAINING AN ENSEMBLE OF DEEP CONVOLUTIONAL NEURAL NETWORKS” (“ОБУЧЕНИЕ С ЧАСТИЧНЫМ ПРИВЛЕЧЕНИЕМ УЧИТЕЛЯ ДЛЯ ТРЕНИРОВКИ АНСАМБЛЯ ГЛУБОКИХ СВЕРТОЧНЫХ НЕЙРОННЫХ СЕТЕЙ”), авторы Laksshman Sundaram, Kai-How Farh, Hong Gao и Jeremy Francis McRae, (№ дела поверенного ILLM 1000-7/IP-1613-US), поданная одновременно.
[0009] Документ 1 - A. V. D. Oord, S. Dieleman, H. Zen, K. Simonyan, O. Vinyals, A. Graves, N. Kalchbrenner, A. Senior, K. Kavukcuoglu, “WAVENET: A GENERATIVE MODEL FOR RAW AUDIO”, arXiv:1609.03499, 2016;
[0010] Документ 2 - S. Ö. Arik, M. Chrzanowski, A. Coates, G. Diamos, A. Gibiansky, Y. Kang, X. Li, J. Miller, A. Ng, J. Raiman, S. Sengupta, M. Shoeybi, “DEEP VOICE: REAL-TIME NEURAL TEXT-TO-SPEECH”, arXiv:1702.07825, 2017;
[0011] Документ 3 - F. Yu and V. Koltun, “MULTI-SCALE CONTEXT AGGREGATION BY DILATED CONVOLUTIONS”, arXiv:1511.07122, 2016;
[0012] Документ 4 - K. He, X. Zhang, S. Ren, J. Sun, “DEEP RESIDUAL LEARNING FOR IMAGE RECOGNITION”, arXiv:1512.03385, 2015;
[0013] Документ 5 - R.K. Srivastava, K. Greff, J. Schmidhuber, “HIGHWAY NETWORKS”, arXiv: 1505.00387, 2015;
[0014] Документ 6 - G. Huang, Z. Liu, L. van der Maaten, K. Q. Weinberger, “DENSELY CONNECTED CONVOLUTIONAL NETWORKS”, arXiv:1608.06993, 2017;
[0015] Документ 7 - C. Szegedy, W. Liu,Y. Jia, P. Sermanet, S. Reed, D. Anguelov, D. Erhan, V. Vanhoucke, A. Rabinovich, “GOING DEEPER WITH CONVOLUTIONS”, arXiv: 1409.4842, 2014;
[0016] Документ 8 - S. Ioffe, C. Szegedy, “BATCH NORMALIZATION: ACCELERATING DEEP NETWORK TRAINING BY REDUCING INTERNAL COVARIATE SHIFT”, arXiv: 1502.03167, 2015;
[0017] Документ 9 - J. M. Wolterink, T. Leiner, M. A. Viergever, I. Išgum, “DILATED CONVOLUTIONAL NEURAL NETWORKS FOR CARDIOVASCULAR MR SEGMENTATION IN CONGENITAL HEART DISEASE”, arXiv:1704.03669, 2017;
[0018] Документ 10 - L. C. Piqueras, “AUTOREGRESSIVE MODEL BASED ON A DEEP CONVOLUTIONAL NEURAL NETWORK FOR AUDIO GENERATION”, Tampere University of Technology (Технологиеский университет Тампере), 2016;
[0019] Документ 11 - J. Wu, “Introduction to Convolutional Neural Networks”, Nanjing University (Нанкинский университет), 2017;
[0020] Документ 12 - I. J. Goodfellow, D. Warde-Farley, M. Mirza, A. Courville, and Y. Bengio, “CONVOLUTIONAL NETWORKS”, Deep Learning, MIT Press, 2016; и
[0021] Документ 13 - J. Gu, Z. Wang, J. Kuen, L. Ma, A. Shahroudy, B. Shuai, T. Liu, X. Wang, G. Wang, “RECENT ADVANCES IN CONVOLUTIONAL NEURAL NETWORKS”, arXiv:1512.07108, 2017.
[0022] В документе 1 описаны архитектуры глубоких сверточных нейронных сетей, в которых используются группы остаточных блоков с фильтрами свертки, имеющими одинаковый размер окна свертки, слои пакетной нормализации, слои блоков линейной ректификации (сокращенно ReLU), меняющие размерность слои, слои разреженной (atrous) свертки с экспоненциально растущими показателями разрежения свертки, соединения с пропуском и слой классификации на основе функции softmax (логистической функции с многими переменными), которые принимают входные последовательности и выдают выходные последовательности, которые присваивают оценки записям во входной последовательности. В предложенном способе применяются компоненты нейронной сети и параметры, раскрытые в Документе 1. В одном варианте реализации раскрытая технология модифицирует параметры компонентов нейронной сети, описанных в Документе 1. Например, в отличие от Документа 1, показатель разряжения свертки в раскрытой технологии растет неэкспоненциально от нижней группы остаточных блоков к более высокой группе остаточных блоков. В другом примере в отличие от Документа 1, размер окна свертки в раскрытой технологии в разных группах остаточных блоков различаются.
[0023] Документ 2 описывает детали вариантов архитектуры глубоких сверточных нейронных сетей, описанных в Документе 1.
[0024] Документ 3 описывает разреженные свертки, используемые в раскрытой технологии. В настоящем документе свертки atrous (“дырчатые”) называются таже “разреженными свертками”. Atrous/разреженные свертки обеспечивают крупные рецептивные поля при меньшем количестве обучающихся параметров. Atrous/разреженная свертка представляет собой свертку, в которой ядро применяется на площади, большей, чем его длина, за счет того, что она пропускает входные значения с определенным шагом, называемым также показателем разрежения или фактором разрежения. Atrous/разреженные свертки увеличивают расстояние между элементами свертки фильтра свертки/ядра, в результате чего при осуществлении операции свертки используются соседние входные записи (например, нуклеотиды, аминокислоты) с большими интервалами. Это обеспечивает возможность введения во входные данные контекстуальных зависимостей дальнего действия. Дырчатые (atrous) свертки сохраняют расчет свертки для повторного использования при обработке соседних нуклеотидов.
[0025] Документ 4 описывает остаточные блоки и остаточные соединения (связи), применяемые в раскрытой технологии.
[0026] Документ 5 описывает соединения с пропуском, применяемые раскрытой технологией. В настоящем документе соединения с пропуском также называются “скоростными сетями”.
[0027] Документ 6 описана архитектура плотно соединенной (связанной) сверточной сети, применяемой в раскрытой технологии.
[0028] Документ 7 описаны меняющие размерность сверточные слои и модульные пайплайны (конвейеры) обработки, применяемые в раскрытой технологии. Одним из примеров свертки с изменением размерности является свертка 1х1.
[0029] Документ 8 описаны слои пакетной нормализации, применяемые в раскрытой технологии.
[0030] Документ 9 также описаны дырчатые (Atrous)/разреженные свертки, применяемые в раскрытой технологии.
[0031] Документ 10 описаны различные архитектуры глубоких нейронных сетей, которые могут применяться в раскрытой технологии, включая сверточные нейронные сети, глубокие сверточные нейронные сети с дырчатыми/разреженными свертками.
[0032] Документ 11 описывает детали сверточной нейронной сети, которая может применяться в раскрытой технологии, включая алгоритмы для обучения (тренировки) сверточной нейронной сети с субдисткретизирующими слоями (слоями подвыборки) (например, объединения) и полностью связанными слоями.
[0033] Документ 12 описывает детали различных операций свертки, которые могут применяться в раскрытой технологии.
[0034] Документ 13 описывает различные архитектуры сверточных нейронных сетей, которые могут применяться в раскрытой технологии.
ВКЛЮЧЕНИЕ ПУТЕМ ССЫЛКИ ТАБЛИЦ, ПОДАННЫХ В ЭЛЕКТРОННОМ ВИДЕ С НАСТОЯЩЕЙ ЗАЯВКОЙ
[0035] Указанные ниже файлы таблиц в формате ASCII поданы с настоящей заявкой и включены в нее посредством ссылки. Эти файлы имеют следующие имена, даты создания и размеры:
[0036] SupplementaryTable1.txt 2 октября 2018 г. 13 KB
[0037] SupplementaryTable2.txt 2 октября 2018 г. 13 KB
[0038] SupplementaryTable3.txt 2 октября 2018 г. 11 KB
[0039] SupplementaryTable4.txt 2 октября 2018 г. 13 KB
[0040] SupplementaryTable6.txt 2 октября 2018 г. 12 KB
[0041] SupplementaryTable7.txt 2 октября 2018 г. 44 KB
[0042] SupplementaryTable13.txt 2 октября 2018 г. 119 KB
[0043] SupplementaryTable18.txt 2 октября 2018 г. 35 KB
[0044] SupplementaryTable20.txt 2 октября 2018 г. 1027 KB
[0045] SupplementaryTable20Summary.txt 2 октября 2018 г. 9 KB
[0046] SupplementaryTable21.txt 2 октября 2018 г. 24 KB
[0047] SupplementaryTable21.txt 2 октября 2018 г. 24 KB
[0048] SupplementaryTable18.txt 4 октября 2018 г. 35 KB
[0049] DataFileS1.txt 4 октября 2018 г. 138 MB
[0050] DataFileS2.txt 4 октября 2018 г. 980 MB
[0051] DataFileS3.txt 4 октября 2018 г. 1.01 MB
[0052] DataFileS4.txt 4 октября 2018 г. 834 KB
[0053] Pathogenicity_prediction_model.txt 4 октября 2018 г. 8.24 KB
[0054] Дополнительная таблица 1: Детали вариантов каждого из видов, используемых для анализа. Эта таблица включает промежуточные результаты в пайплайне для каждого из этих источников данных. Обратите внимание, что эта таблица представлена в файле SupplementaryTable1.txt.
[0055] Дополнительная таблица 2: Истощение миссенс-вариантов, присутствующих в других вариантах при обычных частотах аллели у человека. Истощение рассчитывали на основании миссенс:синонимы в обычных вариантах (> 0.1%) в сравнении с редкими вариантами (< 0.1%), с использованием вариантов с идентичным состоянием у человека и других видов. Обратите внимание, что эта таблица представлена в файле SupplementaryTable2.txt.
[0056] Дополнительная таблица 3: Истощение миссенс-вариантов, присутствующих в других вариантах при обычных частотах аллели у человека, ограниченное только генами со средним показателем консервативных нуклеотидов между животным и другими млекопитающими > 50% . Истощение рассчитывали на основании миссенс:синонимы в обычных вариантах (> 0.1%) в сравнении с редкими вариантами (< 0.1%), с использованием вариантов с идентичным состоянием у человека и других видов. Обратите внимание, что эта таблица представлена в файле SupplementaryTable3.txt.
[0057] Дополнительная таблица 4: Истощение миссенс-вариантов, присутствующих в виде фиксированных замен с частотами обычных аллелей человека. Истощение рассчитывали на основании миссенс:синонимы в обычных вариантах (> 0.1%) в сравнении с редкими вариантами (< 0.1%), с использование вариантов, которые были идентичны по положениям в паре человека и родственного вида. Обратите внимание, что эта таблица представлена в файле SupplementaryTable4.txt.
[0058] Дополнительная таблица 6: Домен-специфичная аннотация гена SCN2A. Р-значения суммы рангов Вилкоксона указывают на расхождение оценок PrimateAI в конкретном домене по сравнению со всем белком. Домены, выделенные жирным шрифтом, покрывают примерно 7% белка, но содержат большинство патогенных аннотаций ClinVar. Это хорошо коррелирует со средними показателями PrimateAI для доменов и входит в топ-3 патогенных доменов согласно модели PrimateAI. Обратите внимание, что эта таблица представлена в файле SupplementaryTable6.txt.
[0059] Дополнительная таблица 7: Необработанные подсчеты, использованные при вычислении влияния частоты аллелей на ожидаемое отношение миссенс: синонимы. Ожидаемое количество синонимичных и миссенс-вариантов рассчитывали на основе вариантов в интронных областях с использованием тринуклеотидного контекста для контроля степени мутаций и конверсии генов. Обратите внимание, что эта таблица представлена в файле SupplementaryTables.xlsx.
[0060] Дополнительная таблица 13: Список названий белков из Protein DataBank (PDB), используемых для обучения моделей глубокого обучения для вторичной структуры с 3 состояниями и предсказания доступности растворителей с 3 состояниями. Столбец с меткой указывает, используются ли белки на этапах обучения / валидации / тестирования при обучении модели. Обратите внимание, что эта таблица представлена в файле SupplementaryTable13.txt.
[0061] Дополнительная таблица 18: Список из 605 генов, которые были номинально значимыми для ассоциации с заболеванием в исследовании DDD, рассчитанный только на основании варианта с укорочением белка (p <0,05). Обратите внимание, что эта таблица представлена в файле SupplementaryTable18.txt.
[0062] Дополнительная таблица 20: Результаты тестирования на обогащение мутациями de novo (DNM) на ген для всех генов с по меньшей мере одной наблюдаемой DNM. P-значения предоставляются при включении всех DNM и после удаления ошибочных DNM с оценками PrimateAI <0,803. Аналогичным образом представлены P-значения скорректированные FDR. Включены подсчеты наблюдаемого укорочения белка (PTV) и миссенс-DNM только из когорты DDD и из всей когорты метаанализа. Также включены аналогичные подсчеты наблюдаемых и ожидаемых миссенс-DNM, во-первых, при включении всех миссенс-DNM, а во-вторых, после удаления всех миссенс-DNM с показателем PrimateAI <0,803. Обратите внимание, что эта таблица представлена в файле SupplementaryTable20.txt и SupplementaryTable20Summary.txt.
[0063] Дополнительная таблица 21: Результаты тестирования обогащения мутациями de novo в генах с FDR <0,1. Включены подсчеты наблюдаемых мутаций укорочения белка (PTV) de novo и подсчеты других изменяющих белок мутаций de novo, однократно со всеми миссенс-мутациями de novo и однократно только с повреждающими миссенс-мутациями. Приведены P-значения при включении всех миссенс-сайтов в сравнении с P-значениями после исключения миссенс-сайтов с низким рейтингом. Обратите внимание, что эта таблица представлена в файле SupplementaryTable21.txt.
[0064] DataFileS1: Список всех вариантов, присутствующих у других видов. Столбец Значимость ClinVar содержит доступные неконфликтующие аннотации ClinVar. Обратите внимание, что эта таблица представлена в файле DataFileS1.txt.
[0065] DataFileS2: Список всех фиксированных замен из пар родственных видов. Обратите внимание, что эта таблица представлена в файле DataFileS2.txt.
[0066] DataFileS3: Список исключенных доброкачественных исследуемых (тестовых) вариантов IBS с приматами. Доброкачественные тестовые варианты - это нераспространенные (не обычные) человеческие варианты, которые являются IBS (идентичными по состоянию) с> = 1 видом приматов. Обратите внимание, что эта таблица представлена в файле DataFileS3.txt.
[0067] DataFileS4: Список немеченых вариантов IBS с приматами, сопоставленными с неустановленными доброкачественными тестовыми вариантами. Немеченые варианты сравниваются с доброкачественными тестовыми вариантами по степени мутаций, систематическим ошибкам охвата и сопоставимости с видами приматов. Обратите внимание, что эта таблица представлена в файле DataFileS4.txt.
[0068] Pathogenicity_prediction_model: Код на языке программирования Python, который позволяет использовать технологию, раскрытую согласно одной реализации. Обратите внимание, что этот файл кода представлен в Pathogenicity_prediction_model.txt.
ОБЛАСТЬ ТЕХНИКИ РАСКРЫТОЙ ТЕХНОЛОГИИ
[0069] Раскрытая технология относится к компьютерам и цифровым системам обработки данных, относящихся к типу искусственного интеллекта, и соответствующим способам обработки данных и продуктам для эмуляции интеллекта (т.е. системам, основанным на знаниях, системам построения рассуждений и системам приобретения знаний); включая системы для логических рассуждений в условиях неопределенности (например, системы нечеткой логики), адаптивным системам, системам машинного обучения и искусственным нейронным сетям. В частности, раскрытая технология относится к применению технологий глубокого обучения для обучения (тренировки) глубоких сверточных нейронных сетей.
Уровень техники
[0070] Не следует полагать, что аспекты, обсуждаемые в этом разделе, составляют часть уровня техники только потому, что они упоминаются в этом разделе. Аналогичным образом, не следует полагать, что задача, упоминающаяся в этом разделе или связанная с объектом, указанным в качестве предпосылки, является признанным уровнем техники. Предмет этого раздела лишь представляет различные подходы, которые сами по себе также могут соответствовать вариантам реализации заявленной технологии.
Машинное обучение
[0071] В машинном обучении входные переменные используются для предсказания выходных переменных. Входные переменные часто называют признаками и обозначают как X = (X1, X2, …, Xk), где each Xi, i 1, …, k представляет собой признак. Выходная переменная часто называется ответом или зависимой переменной и обозначается переменной Yi. Отношение между Y и соответствующей X можно зависать в общем виде:
Y = ƒ (X) + ∈
[0072] В приведенном выше уравнении f представляет собой функцию признаков (X1, X2, …, Xk) и представляет собой показатель случайной ошибки. Указанный показатель случайно ошибки не зависит от X и имеет среднее значение, равное нулю.
[0073] На практике признаки X доступны в отсутствие Y или без знания точного отношения X и Y. Поскольку среднее значение показателя ошибки равно нулю, задача заключается в том, чтобы оценить f.

[0074] В приведенном выше уравнении 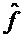
[0075] Функцию
Нейронные сети
[0076] ФИГ. 1A демонстрирует один из вариантов реализации полностью соединенной (связанной) нейронной сети с несколькими слоями. Нейронная сеть представляет собой систему взаимосвязанных искусственных нейронов (например, a1, a2, a3), которые обмениваются друг с другом сообщениями. Показанная нейронная сеть имеет три входа, два нейрона в скрытом слое и два нейрона в выходном слое. Скрытый слой имеет функцию активации, а выходной слой имеет функцию активации. Связи имеют численные веса (например, w11, w21, w12, w31, w22, w32, v11, v22), которые подстраиваются в процессе обучения (тренировки)таким образом, то обученная приемлемым образом сеть отвечает правильно при предъявлении образа для распознавания. Входной слой обрабатывает необработанные входные данные, скрытый слой обрабатывает данные, полученные на выходе входного слоя на основании весов связей между входным слоем и скрытым слоем. Выходной слой берет выход (выходные данные) скрытого слоя и обрабатывает на основании весов связей между скрытым слоем и выходным слоем. Сеть включает несколько слоев нейронов, детектирующих признаки. Каждый слой содержит много нейронов, которые отвечают на различные комбинации входов от предыдущих слоев. Слои сконструированы таким образом, что первый слой детектирует набор примитивных паттернов в данных входного образа, второй слой детектирует паттерны паттернов, а третий слой детектирует паттерны этих паттернов.
[0077] Обзор применения глубокого обучения в геномике можно найти в следующих публикациях:
• T. Ching et al., Opportunities And Obstacles For Deep Learning In Biology And Medicine, www.biorxiv.org:142760, 2017;
• [Angermueller C, Pärnamaa T, Parts L, Stegle O. Deep Learning For Computational Biology. Mol Syst Biol. 2016;12:878;
• Park Y, Kellis M. 2015 Deep Learning For Regulatory Genomics. Nat. Biotechnol. 33, 825-826. (doi:10.1038/nbt.3313);
• Min, S., Lee, B. & Yoon, S. Deep Learning In Bioinformatics. Brief. Bioinform. bbw068 (2016);
• Leung MK, Delong A, Alipanahi B et al. Machine Learning In Genomic Medicine: A Review of Computational Problems and Data Sets 2016; and
• Libbrecht MW, Noble WS. Machine Learning Applications In Genetics and Genomics. Nature Reviews Genetics 2015;16(6):321-32.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0078] На чертежах одинаковые ссылочные позиции обычно относятся к одинаковым деталям на разных видах. Кроме того, чертежи не обязательно выполнены в масштабе, вместо этого, как правило, делается акцент на иллюстрации принципов раскрытой технологии. В последующем описании различные реализации раскрытой технологии описаны со ссылкой на следующие чертежи, на которых:
[0079] ФИГ. 1A демонстрирует один из вариантов реализации многослойной нейронной сети прямого распространения.
[0080] ФИГ. 1B показывает один вариант реализации работы сверточной нейронной сети.
[0081] ФИГ. 1C показывает блок-схему тренировки сверточной нейронной сети в соответствии с одним из вариантов реализации раскрытой технологии.
[0082] ФИГ. 1D представляет собой один вариант реализации слоев субдискретизации (максимальный/средний пулинг) в соответствии с одним из вариантов реализации раскрытой технологии.
[0083] ФИГ. 1E показан один из вариантов реализации нелинейного слоя ReLU (с блоками линейной ректификации ) в соответствии с одним вариантом реализации раскрытой технологии.
[0084] ФИГ. 1F показывает один вариант осуществления двухслойной свертки сверточных слоев.
[0085] ФИГ. 1G показывает остаточную связь, которая снова вводит предварительную информацию ниже путем добавления карты признаков .
[0086] ФИГ. 1H показывает один вариант реализации остаточных блоков и связей с пропусками.
[0087] ФИГ. 1I демонстрирует прямой проход пакетной нормализации.
[0088] ФИГ. 1J иллюстрирует преобразование пакетной нормализации в момент теста.
[0089] ФИГ. 1K демонстрирует обратный проход пакетной нормализации.
[0090] ФИГ. 1L depicts use of a batch normalization layer after and before a convolutional or densely connected layer.
[0091] ФИГ. 1M демонстрирует один вариант реализации 1D-свертки.
[0092] ФИГ. 1N иллюстрирует работу глобального среднего объединения (GAP) .
[0093] ФИГ. 1O иллюстрирует разреженные свертки.
[0094] ФИГ. 1P демонстрирует один вариант реализации пакетных разреженных сверток.
[0095] ФИГ. 1Q демонстрирует пример вычислительной среды, в которой может работать раскрытая технология.
[0096] ФИГ. 2 демонстрирует пример архитектуры глубокой остаточной сети для прогнозирования патогенности, называемой здесь «PrimateAI».
[0097] На ФИГ. 3 схематично изображена PrimateAI, сетевая архитектура глубокого обучения для классификации патогенности.
[0098] ФИГ. 4A, 4B, и 4C представляют собой Дополнительную таблицу 16, в которой показаны детали архитектуры примера модели глубокого обучения модели PrimateAI для предсказания патогенности.
[0099] ФИГ. 5 и 6 иллюстрируют сетевую архитектуру глубокого обучения, используемую для прогнозирования вторичной структуры и доступности белков для растворителей.
[00100] ФИГ. 7A и 7B - это дополнительная таблица 11, в которой показан пример деталей архитектуры модели для модели глубокого обучения (DL) с предсказание вторичной структуры с 3 состояниями.
[001091] ФИГ. 8A и 8B - это дополнительная таблица 12, в которой показан пример деталей архитектуры модели для модели глубокого обучения предсказания доступности для растворителей с 3 состояниями.
[00102] ФИГ. 9 иллюстрирует одну реализацию создания референсных и альтернативных белковых последовательностей из доброкачественных и патогенных вариантов.
[00103] ФИГ. 10 демонстрирует один вариант реализации выравнивания референсных и альтернативных белковых последовательностей.
[00104] ФИГ. 11 представляет один из вариантов реализации генерации частотных матриц положения (сокращенно PFM), также называемых матрицами весовых коэффициентов положения (сокращенно PWM) или матрицей положение-специфичных весов (сокращенно PSSM).
[00105] ФИГ. 12, 13, 14 и 15 демонстрируют подсети обработки вторичной структуры и доступности для растворителей.
[00106] ФИГ. 16: работа классификации патогенности вариантов. В настоящем документе термин «вариант «также относится к однонуклеотидным полиморфизмам (сокращенно SNP) и, как правило, к однонуклеотидным вариантам (сокращенно SNV).
[00107] ФИГ. 17 иллюстрирует остаточный блок.
[00108] ФИГ. 18 изображает архитектуру нейронной сети вторичной структуры и подсетей доступности для растворителя.
[00109] ФИГ. 19 ФИГ. 19 иллюстрирует архитектуру нейросети классификатора патогенности вариантов.
[00110] ФИГ. 20 иллюстрирует предсказанную оценку патогенности в каждом положении аминокислоты в гене SCN2A с аннотациями для ключевых функциональных доменов.
[00111] ФИГ. 21D показано сравнение классификаторов при предсказании доброкачественных последствий для тестового набора из 10 000 распространенных вариантов приматов, которые не участвовали в обучении.
[00112] ФИГ. 21E иллюстрирует распределения бальных оценок предсказания PrimateAI для вариантов de novo миссенс-мутаций, встречающихся у пациентов с нарушениями развития (DDD), по сравнению с здоровыми братьями и сестрами, с соответствующим P-значением суммы рангов Вилкоксона.
[00113] На ФИГ. 21F показано сравнение классификаторов при выделении de novo миссенс-вариантов в случаях DDD по сравнению с контролями. Для каждого классификатора показаны P-значения критерия суммы рангов Вилкоксона.
[00114] ФИГ. 22А демонстрирует увеличение количества миссенс-мутаций de novo по сравнению с ожидаемым у пораженных индивидуумов из когорты DDD в пределах 605 связанных генов, которые были значимыми для вариантов с укорочением белка de novo (P <0,05).
[00115] ФИГ. 22B иллюстрирует распределение оценок прогноза PrimateAI для миссенс-вариантов de novo, встречающихся у пациентов с DDD, по сравнению со здоровыми братьями и сестрами в пределах 605 связанных генов с соответствующим P-значением суммы рангов по Вилкоксону.
[00116] ФИГ. 22C показано сравнение различных классификаторов при разделении миссенс-вариантов de в кейсах и в контроле в пределах 605 генов.
[00117] ФИГ. 22D изображает сравнение различных классификаторов, показанных на характеристической кривой оператора приемника, с площадью под кривой (AUC), указанной для каждого классификатора.
[00118] ФИГ. 22E иллюстрирует точность классификации и площадь под кривой (AUC) для каждого классификатора.
[00119] ФИГ. 23A, 23B, 23C и 23D демонстрируют влияние данных, используемых для обучения, на точность классификации.
[00120] ФИГ. 24 иллюстрирует поправку на влияние охвата секвенирования на определение распространенных вариантов приматов.
[00121] ФИГ. 25A, 25B, 25C и 26 показывают распознавание белковых мотивов описанными нейронными сетями. ФИГ. 26 включает линейный график, показывающий влияние нарушения каждого положения в варианте и вокруг него на предсказанную оценку глубокого обучения для этого варианта.
[00122] ФИГ. 27 иллюстрирует модели корреляции весов, имитирующих матрицы баллов BLOSUM62 и Grantham.
[00123] ФИГ. 28A, 28B и 28C демонстрируют оценку эффективности сети глубокого обучения PrimateAI и других классификаторов.
[00124] ФИГ. 29A и 29B иллюстрируют распределение оценок предсказания четырех классификаторов.
[00125] ФИГ. 30A, 30B и 30C сравнивают точность сети PrimateAI и других классификаторов при разделении патогенных и доброкачественных вариантов в 605 генах, связанных с заболеванием.
[00126] ФИГ. 31A и 31B иллюстрируют корреляцию между эффективностью классификатора на вариантах ClinVar, курируемых экспертами, и эффективностью на эмпирических наборах данных.
[00127] ФИГ. 32 - это дополнительная таблица 14, в которой показаны характеристики моделей вторичной структуры с 3 состояниями и моделей предсказания доступности для растворителей с 3 состояниями на аннотированных образцах из Protein DataBank.
[00128] ФИГ. 33 – это дополнительная таблица 15, в которой показано сравнение эффективности сети глубокого обучения с использованием аннотированных меток вторичной структуры человеческих белков из базы данных DSSP.
[00129] ФИГ. 34 - это дополнительная таблица 17, которая показывает значения точности для 10 000 удерживаемых вариантов приматов и p-значения для вариантов de novo в случаях DDD по сравнению с контролями для каждого из 20 оцениваемых нами классификаторов.
[00130] ФИГ. 35 - это дополнительная таблица 19, в которой показано сравнение эффективности различных классификаторов на вариантах de novo в случае DDD с контрольным набором данных, ограниченным 605 генами, связанными с заболеванием.
[00131] ФИГ. 36 демонстрирует вычислительную среду раскрытого полууправляемого ученика.
[00132] ФИГ. 37, 38, 39, 40 и 41 показывают различные циклы раскрытого полууправляемого обучения.
[00133] ФИГ. 42 представляет собой иллюстрацию итеративного процесса с сбалансированной выборкой.
[00134] ФИГ. 43 иллюстрирует один вариант реализации среды, используемый для создания доброкачественного набора данных.
[00135] ФИГ. 44 изображает один вариант реализации создания благоприятных человеческих миссенс- SNP.
[00136] ФИГ. 45 демонстрирует один вариант реализации человеческих ортологичных миссенс-SNP. Миссенс-SNP у не относящихся к человеку видов, которые имеют совпадающие контрольные и альтернативные кодоны с людьми.
[00137] ФИГ. 46 изображает один вариант реализации классификации как доброкачественных SNP видов приматов, не являющихся человеком (например, шимпанзе), с референсными кодонами, совпадающими с человеческими.
[00138] ФИГ. 47 ФИГ. 47 изображает один вариант реализации вычисления оценок обогащения и их сравнения.
[00139] ФИГ. 48 изображает один вариант реализации безопасного набора данных SNP.
[00140] ФИГ. 49A, 49B, 49C, 49D и 49E изображают отношения миссенс / синонимы в частотном спектре аллелей человека.
[00141] ФИГ. 50A, 50B, 50C и 50D демонстрируют очищающий отбор на миссенс-вариантах, идентичных по состоянию с другими видами.
[00142] ФИГ. 51 показывает ожидаемые отношения миссенс: синонимы по частотному спектру аллелей человека в отсутствие очищающего отбора.
[00143] ФИГ. 52A, 52B, 52C и 52D изображают отношения миссенс: синонимы для вариантов CpG и не-CpG.
[00144] ФИГ. 53, 54 и 55 иллюстрируют отношения миссенс: синонимы человеческих вариантов, идентичных по состоянию шести приматам.
[00145] ФИГ. 56 представляет моделирование, показывающее насыщение новыми распространенными миссенс-вариантами, обнаруженными при увеличении размера исследуемых когорт людей.
[00146] ФИГ. 57 показывает точность PrimateAI для различных профилей консервативности в геноме.
[00147] ФИГ. 58 - это Дополнительная таблица 5, которая показывает вклады в набор размеченных доброкачественных данных от распространенных человеческих вариантов и вариантов, присутствующих приматов, не являющихся человеком.
[00148] ФИГ. 59 - это дополнительная таблица 8, которая показывает влияние частоты аллелей на ожидаемое отношение миссенс: синонимы.
[00149] ФИГ. 60 - это Дополнительная таблица 9, в которой показан анализ ClinVar.
[00150] ФИГ. 61 - это дополнительная таблица 10, которая показывает количество миссенс-вариантов из других видов, обнаруженных в ClinVar, согласно одному варианту реализации.
[00159] ФИГ. 62 представляет собой Таблицу 1, которая демонстрирует один вариант реализации открытия 14 дополнительных генов-кандидатов при умственной отсталости.
[00160] ФИГ. 63 представляет собой Таблицу 2, демонстрирующую один вариант реализации средней разницы в баллах по Грэнтэму между патогенными и доброкачественными вариантами в ClinVar.
[00161] ФИГ. 64 демонстрирует один вариант реализации анализа обогащения по генам.
[00162] ФИГ. 65 демонстрирует один вариант реализации полногеномного анализа обогащения.
[00163] ФИГ. 66 представляет собой упрощенную блок-схему компьютерной системы, которую можно применять для реализации раскрытой технологии.
ПОДРОБНОЕ ОПИСАНИЕ
[00156] Приведенное ниже описание представлено для того, чтобы любой специалист в данной области техники мог осуществить и применить раскрытую технологию, и представлено в контексте конкретного случая применения и его требований. Различные модификации раскрытых вариантов осуществления будут очевидны для специалиста в данной области техники, а общие принципы, раскрытые в настоящем документе, могут быть применены к другим вариантам осуществления и областям применения без выхода за пределы идеи и объема раскрытой технологии. Таким образом, раскрытая технология не ограничена представленными вариантами осуществления, и она должна рассматриваться в соответствии с наиболее широким объемом, соответствующим принципам и признакам, раскрытым в настоящем документе.
Введение
Сверточные нейронные сети
[00157] Сверточная нейронная сеть представляет собой особый тип нейронной сети. Фундаментальная разница между плотно соединенным (связанным) слоем и сверточным слоем заключается в следующем: Соединенные слои изучают глобальные паттерны в своем пространстве входных признаков, в то время как сверточные слои изучают локальные паттерны: в случае образов паттерны находятся в малых двумерных окнах входных данных. Эта ключевая характеристика придает сверточным нейронным сетям две интересные особенности: (1) паттерны, которые они изучают, являются инвариантными относительно сдвига и (2) они могут изучать пространственные иерархии паттернов.
[00158] В отношении первого можно отметить, что после изучения конкретного паттерна в правом нижнем углу картинки сверточным слой может распознать его где угодно: например, в верхнем левом углу. Плотно соединенным нейронным сетям пришлось бы обучаться паттерну снова, если бы он появился в новом месте. Это делает сверточные нейронные сети эффективными в отношении данных, Поскольку им требуется меньшее количество тренировочных образцов для обучения способу задания функций в связи с тем, что они способны к обобщению.
[00159] В отношении второго можно отметить, что первый сверточный слой может изучать малые локальные паттерны, такие как края, второй сверточный слой будет изучать паттерны большего размера, выполненные из признаков первых слоев, и т.д. Это обеспечивает сверточным нейронным сетям возможность эффективного обучения существенно более сложным и абстрактным визуальным концептам.
[00160] Сверточная нейронная сеть обучена преобразованиям с высокой нелинейностью посредством взаимно соединенных слоев искусственных нейронов, расположенных во множестве различных слоев с функциями активации, которые делают слои зависимыми. Она содержит один или более сверточных слоев, перемежающихся с одним или более субдискретизирующих слоев, за которыми обычно следуют один или более плотно соединенные слоев. Каждый элемент сверточной нейронной сети принимает входные данные из совокупности признаков в предыдущем слое. Сверточная нейронная сеть обучена параллельно, поскольку нейроны в одной и той же карте признаков имеют идентичные весовые значения. Эти локальные общие весовые коэффициенты снижают сложность сети таким образом, что когда многомерные входные данные попадают в сеть, сверточная нейронная сеть избегает сложностей, связанных с реконструкцией данных при извлечении признаков и процессе регрессии или классификации.
[00161] Свертки осуществляют операции над трехмерными тензорами, называемыми картами признаков, с двумя пространственными осями (высота и ширина), а также с осью глубины (также называемой канальной осью). Для изображения RGB размер оси глубины составляет 3, поскольку изображение имеет три цветовых канала; красный, зеленый и синий. Для черно-белых картинок глубина составляет 1 (уровни серого). Операция свертки извлекает вставки из карты ее входных признаков и применяет то же преобразование ко всем вставкам с получением карты выходных признаков. Эта карта выходных признаков все еще является трехмерным тензором: она имеет ширину и высоту. Ее глубина может быть произвольной, Поскольку глубина выходных данных является параметром слоя, а различные каналы по указанной оси глубины больше не соответствуют конкретным цветам во входных данных RGB, наоборот, они соответствуют фильтрам. Фильтры кодируют определенные аспекты входных данных: например, на уровне высоты один фильтр может кодировать концепцию «наличие лица во входных данных».
[00162] Например, первый сверточный слой берет карту признаков размером (28, 28, 1) и выдает карту признаков размером (26, 26, 32): он вычисляет 32 фильтра по своим входным данным. Каждый из указанных 32 выходных каналов содержит сетку значений размером 26 х 26, которая представляет собой карту ответов фильтра на входные данные, указывающую ответ паттерна указанного фильтра в различных местах во входных данных. Иными словами, термин «карта признаков» обозначает следующее: каждая координата по оси глубины является признаков (или фильтром), а двумерный тензор выходных данных [:, :, n] представляет собой двумерную пространственную карту ответов указанного фильтра по входным данным.
[00163] Свертки заданы двумя ключевыми параметрами: (1) размер вставок, извлеченных из входных данных - они обычно составляют 1 х 1, 3 х 3 или 5 х 5, и (2) глубина карты выходных признаков - количество фильтров, вычисленных посредством свертки. Зачастую начинают с глубины 32, продолжают с глубиной 64 и заканчивают с глубиной 128 или 256.
[00164] Свертка работает посредством перемещения указанных окон размером 3 x 3 или 5 x 5 по трехмерной карте входных признаков с остановкой в каждом месте и извлечением трехмерной вставки окружающих признаков (shape (window_height, window_width, input_depth); форма (окно_высота, окно_ширина, входные данные_глубина)). Каждую такую трехмерную вставку затем преобразуют (посредством тензорного произведения с весовой матрицей, обученной таким же образом, называемой ядром свертки) в одномерный вектор формы (output_depth,; выходные данные_глубина,). Все из указанных векторов затем подвергают пространственной обратной сборке в трехмерную карту выходных данных формы (height, width, output_depth; высота, ширина, выходные данные_глубина). Каждое пространственное положение на карте выходных признаков соответствует тому же положению на карте входных признаков (например, нижний правый угол выходных данных содержит информацию о нижнем правом угле входных данных). Например, в случае окон 3 х 3, векторные выходные данные [i, j, :] происходят из входных данных [i-1: i+1, j-1:J+1, :] трехмерной вставки. Полностью процесс подробно показан на ФИГ. 1B.
[00165] Сверточная нейронная сеть содержит сверточные слои, которые выполняют операцию свертки между входными значениями и сверточными фильтрами (весовой матрицей), которые обучены на множестве итераций градиентного изменения во время обучения. Пусть (m, n) будет размером фильтра, а W - весовой матрицей, тогда сверточный слой выполняет свертку W с входными данными X посредством вычисления скалярного произведения W • x + b, где x представляет собой элемент из X, а b представляет собой смещение. Размер шага, на который сверточные фильтры перемещаются по входным данным, называют сдвигом, а область фильтрации (m × n) называют рецептивным полем. Один и тот же сверточный фильтр применяют к различным положениям входных данных, что позволяет снизить количество изученных весов. Это также обеспечивает возможность обучения с инвариантностью положений, т.е. если важный паттерн присутствует во входных данных, сверточные фильтры изучают его вне зависимости от его положения в последовательности
Обучение сверточной нейронной сети
[00166] На ФИГ. 1C показана блок-схема обучения сверточной нейронной сети в соответствии с одним из вариантов реализации раскрытой технологии. Сверточная нейронная сеть настроена или обучена таким образом, что входные данные ведут к особой выходной оценке. Сверточную нейронную сеть настраивают с использованием обратного распространения на основе сравнения выходной оценки и реальных данных до тех пор, пока выходная оценка прогрессивно совпадет или приблизится к реальным данным.
[00167] Сверточную нейронную сеть обучают посредством регулировки весов между нейронами на основании разницы между реальными данными и действительными выходными данными. Математически это можно описать следующим образом:
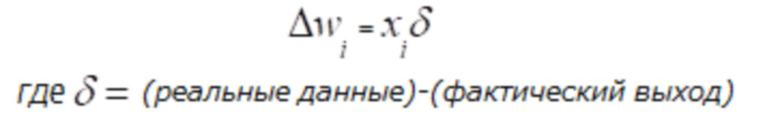
[00168] В одном варианте осуществления обучающее правило определено как:
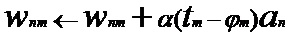
[00169] В представленном выше уравнении: стрелка указывает на изменение значения; 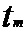


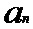

[00170] Промежуточный этап обучения включает выработку вектора признаков из входных данных с использованием сверточных слоев. Вычисляют градиент в отношении весов в каждом слое, начиная с выходных данных. Это называют обратным проходом или прохождением в обратном направлении. Веса в сети изменяют с использованием комбинации отрицательного градиента и предыдущих весов.
[00171] В одном варианте осуществления сверточная нейронная сеть использует алгоритм изменения со стохастическим градиентом (такой как ADAM), который выполняет обратное распространение ошибок посредством градиентного спуска. Один пример алгоритма обратного распространения на основе сигмоидной функции описан ниже:
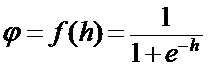
[00172] В приведенной выше сигмоидной функции, 
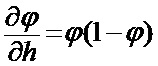
[00173] Алгоритм включает вычисление активации нейронов в сети, вырабатывая выходные данные для прямого прохода. Активация нейрона 
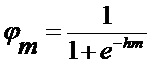
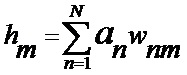
[00174] Это выполняется для всех скрытых слоев для получения активаций, описанных следующим образом:
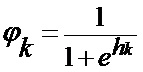

[00175] Затем для каждого слоя вычисляют ошибку и корректировочные веса. Ошибку в выходных данных вычисляют следующим образом:
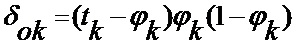
[00176] Ошибку в скрытых слоях вычисляют следующим образом:

[00177] Веса выходного слоя изменяют следующим образом:
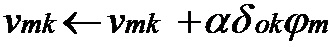
[00178] Веса скрытого слоя изменяют с использованием скорости обучения 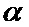
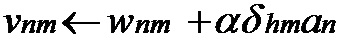
[00179] В одном варианте осуществления сверточная нейронная сеть использует оптимизацию с градиентным спуском для вычисления ошибки по всем слоям. При такой оптимизации для вектора x входных признаков и спрогнозированных выходных данных ŷ функция потерь определена как l в целях прогнозирования ŷ, когда целью является y, т.е. l (ŷ, y). Спрогнозированные выходные данные ŷ преобразуют из вектора x входных признаков с использованием функции f. Функция f параметризуется весами сверточной нейронной сети, т.е. ŷ = fw (x). Функция потерь описана как l (ŷ, y) = l (fw (x), y), или
Q (z, w) = l (fw (x), y) где z представляет собой пару (x, y) входных данных и выходных данных. Оптимизацию с градиентным спуском выполняют путем изменения весов в соответствии с:

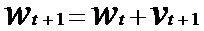
[00180] В приведенных выше уравнениях α представляет собой скорость обучения. Кроме того, потери вычисляют как среднее по совокупности n пар данных. Вычисление останавливают, когда скорость обучения α достаточно мала при линейном схождении. В других вариантах осуществления градиент вычисляют с использованием только выбранных пар данных, подаваемых в ускоренный градиент Нестерова и адаптивный градиент для обеспечения эффективности вычисления.
[00181] В одном варианте осуществления сверточная нейронная сеть использует стохастический градиентный спуск (SGD) для вычисления функции потерь (функции стоимости). SGD аппроксимирует градиент в отношении весов в функции потерь посредством его вычисления на основании только одной, выбранной в случайном порядке, пары данных, 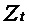
[00182] В приведенном выше уравнении: α представляет собой скорость обучения; μ представляет собой момент; а t представляет собой весовое значение перед изменением. Скорость схождения SGD составляет приблизительно 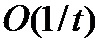
Сверточные слои
[00183] Сверточные слои сверточной нейронной сети служат в качестве экстракторов признаков. Сверточные слои функционируют как адаптивные экстракторы признаков, способные к обучению и декомпозиции входных данных на иерархические признаки. В одном варианте осуществления сверточные слои берут два изображения в качестве входных данных и выдают третье изображение в качестве выходных данных. В таком варианте осуществления свертка выполняется над двумя изображениями в двух измерениях (2D), причем одно изображение представляет собой входное изображение, а другое изображение, называемое «ядром» и применяемое в качестве фильтра к входному изображению, обеспечивает получение выходного изображения. Таким образом, для входного вектора f длиной n и ядра g длиной m, свертка f * g для f и g определяется как:
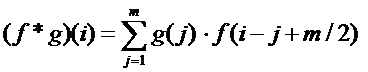
[00184] Операция свертки включает перемещение ядра по входному изображению. Для каждого положения ядра перекрывающиеся значения ядра и входного изображения умножаются и результаты складываются. Сумма произведений представляет собой значение выходного изображения в точке на входном изображении, в которой отцентровано ядро. Полученные в результате различные выходные данные от множества ядер называют картами признаков.
[00185] После того как сверточные слои обучены, их применяют для выполнения задач по распознаванию над новыми рассматриваемыми данными. Поскольку сверточные слои обучаются на тренировочных данных, они избегают извлечения признаков в явном виде и неявно обучаются на тренировочных данных. Сверточные слои используют сверточные веса ядра фильтрации, которые определяются и изменяются как часть процесса обучения. Сверточные слои извлекают различные признаки из входных данных, которые комбинируются на верхних слоях. Сверточная нейронная сеть использует различное количество сверточных слоев, каждый из которых имеет различные параметры свертки, такие как размер ядра, сдвиги, заполнение, количество карт признаков и веса.
Субдискретизирующие слои (слои подвыборок)
[00186] ФИГ. 1D показан один вариант осуществления субдискретизирующих слоев в соответствии с одним вариантом осуществления раскрытой технологии. Субдискретизирующие слои снижают разрешение признаков, извлеченных сверточными слоями, чтобы сделать извлеченные признаки или карты признаков устойчивыми к шуму и искажению. В одном варианте осуществления субдискретизирующие слои используют два типа объединяющих операций, среднее объединение и максимальное объединение. Объединяющие операции разделяют входные данные на неперекрывающиеся двумерные пространства. Для среднего объединения вычисляют среднее для четырех значений в области. Для максимального объединения выбирают максимальное значение из четырех значений.
[00187] субдискретизирующие слои включают объединяющие операции на совокупности нейронов в предыдущем слое посредством преобразования его выходных данных только до одних из входных данных при максимальном объединении и посредством преобразования его выходных данных до среднего из входных данных про среднем объединении. При максимальном объединении выходные данные объединяющего нейрона представляют собой максимальное значение, которое имеется во входных данных, что описано так:
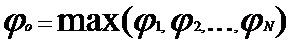
[00188] В приведенном выше уравнении N представляет собой общее количество элементов в совокупности нейронов.
00189] При среднем объединении выходные данные объединяющего нейрона представляют собой среднее значение входных данных, которое имеется во входной совокупности нейронов, что описано так:
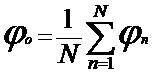
[00190] В приведенном выше уравнении представляет собой общее количество элементов во входной совокупности нейронов.
[00191] На ФИГ. 1D, входной размер составляет 4×4. Для субдискретизации 2×2 изображение 4×4 разделяют на четыре неперекрывающиеся матрицы размером 2×2. Для среднего объединения среднее для четырех значений является полностью целочисленными выходными данными. Для максимального объединения максимальное значение для четырех значений в матрице 2×2 является полностью целочисленными выходными данными.
Нелинейные слои
[00192] На фиг. 5 показан один вариант осуществления нелинейных слоев в соответствии с одним вариантом осуществления раскрытой технологии. Нелинейные слои используют различные пусковые функции для указания на явную идентификацию или наиболее вероятные признаки на каждом скрытом слое. Нелинейные слои используют множество особых функций для осуществления нелинейного запуска, включая блоки линейной ректификации (ReLU), гиперболический тангенс, абсолютную величину гиперболического тангенса, сигмоидную и непрерывную пусковые (нелинейные) функции. В одном варианте осуществления активация ReLU осуществляет функцию y = max(x, 0) и сохраняет размеры входных и выходных данных одинаковыми. Преимущество использования ReLU заключается в том, что сверточная нейронная сеть обучена во много раз быстрее. ReLU не является непрерывной, насыщающей функцией активации, которая является линейной относительно входных данных, если входные значения больше, чем ноль, и равна нулю в противном случае. С математической точки зрения функцию активации ReLU можно описать как:
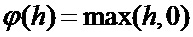
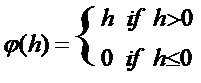
[00193] В других вариантах осуществления сверточная нейронная сеть использует функцию активации со степенным блоком, которая представляет собой непрерывную ненасыщающую функцию, описываемую как:
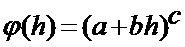
[00194] В приведенном выше уравнении a, b и c представляют собой параметры, управляющие смещением, масштабом и мощностью соответственно. Степенная функция активации может обеспечивать x и y - антисимметричную активацию, если c имеет нечетное значение, и y - симметричную активацию, если 
[00195] В других вариантах осуществления сверточная нейронная сеть использует функцию активации с сигмоидным блоком, которая представляет собой непрерывную ненасыщающую функцию, описываемую следующей логистической функцией:

[00196] В приведенном выше уравнении . Функция активации с сигмоидным блоком не обеспечивает отрицательную активацию и является только асимметричной по отношению к -оси.
Примеры свертки
[00197] На ФИГ. 1F показывает один вариант осуществления двухслойной свертки сверточных слоев. На ФИГ. 1F сворачивают входные данные размерностью 2048 измерений. При свертке 1 входные данные сворачивают посредством сверточного слоя, содержащего два канала с шестнадцатью ядрами размером 3×3. Полученные в результате шестнадцать карт признаков затем ректифицируют посредством функции активации ReLU при ReLU1, а затем объединяют в Pool 1 посредством среднего объединения с использованием объединяющего слоя с шестнадцатью каналами с ядрами размером 3×3. При свертке 2 входные данные из Pool 1 затем сворачивают посредством другого сверточного слоя, содержащего шестнадцать каналов с тридцатью ядрами размером 3×3. За этим следует другой ReLU2 и среднее объединение в Pool 2 с ядром размером 2×2. Сверточные слои используют переменное количество сдвигов и заполнений, например, ноль, два и три. Полученный в результате вектор признаков имеет пятьсот двенадцать (512) измерений в соответствии с одним вариантом осуществления.
[00198] В другом вариантах осуществления сверточная нейронная сеть использует различное количество сверточных слоев, субдискретизирующих слоев, нелинейных слоев и полностью соединенных слоев. В одном варианте осуществления сверточная нейронная сеть представляет собой неглубокую сеть с меньшим количеством слоев и большим количеством нейронов в каждом слое, например, с одним, двумя или тремя полностью соединенными слоями, содержащими сто (100) - двести (200) нейронов на слой. В другом варианте осуществления сверточная нейронная сеть представляет собой глубокую сеть с большим количеством слоев и меньшим количеством нейронов в каждом слое, например, с пятью (5), шестью (6) или восемью (8) полностью соединенными слоями, содержащими тридцать (30) - пятьдесят (50) нейронов на слой.
Прямой проход
[00199] Выходные данные нейрона в ряду x, столбце y в сверочном слое lth (l-uj)и карте kth (k-го) признаков для количества f ядер свертки на карте признаков определяют с помощью следующего уравнения:
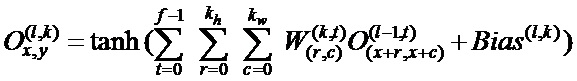
[00200] Выходные данные нейрона в ряду x, столбце y в субдискретизирующем слое lth и карте kth признаков определяют с помощью следующего уравнения:
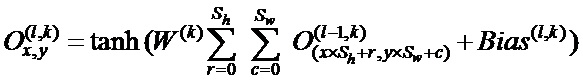
[00201] Выходные данные нейрона ith в выходном слое lth определяют с помощью следующего уравнения:

Обратное распространение
[00202] Выходное отклонение kth нейрона в выходном слое определяют с помощью следующего уравнения:
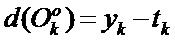
[00203] Входное отклонение kth нейрона в выходном слое определяют с помощью следующего уравнения:
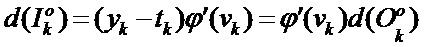
[00204] Вес и изменение смещения kth нейрона в выходном слое определяют с помощью следующего уравнения:
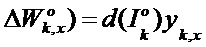
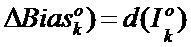
[00205] Выходное смещение kth нейрона в скрытом слое определяют с помощью следующего уравнения:
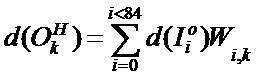
[00206] Входное смещение kth нейрона в скрытом слое определяют с помощью следующего уравнения:
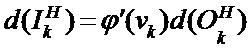
[00207] Вес и изменение смещения в ряду x, столбце y в mth карте признаков первичного слоя, принимающего входные данные от k нейронов в скрытом слое определяют с помощью следующего уравнения:

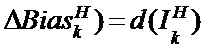
[00208] Выходное смещение в ряду x, столбце y в mth карте признаков субдискретизирующего слоя S определяют с помощью следующего уравнения:
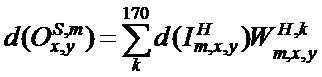
[00209] Входное смещение в ряду x, столбце y в mth карте признаков субдискретизирующего слоя S определяют с помощью следующего уравнения:
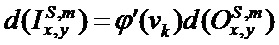
[00210] Вес и изменение смещения в ряду x, столбце y в mth карте признаков субдискретизирующего слоя S и сверточного слоя C определяют с помощью следующего уравнения:
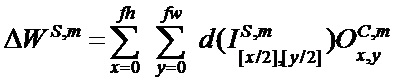

[00211] Выходное смещение в ряду x, столбце y в kth карте признаков сверточного слоя C определяют с помощью следующего уравнения:
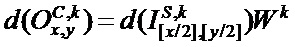
[00212] Входное смещение в ряду x, столбце y в kth карте признаков сверточного слоя C определяют с помощью следующего уравнения:
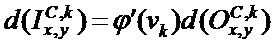
[00213] Вес и изменение смещения в ряду r, столбце c в mth ядре свертки kth карты признаков lth сверточного слоя C:
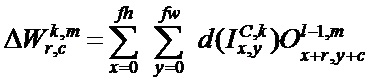
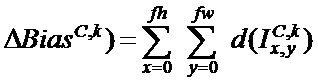
Остаточные соединения
[00214] На ФИГ. 1G показано остаточное соединение, которое повторно подает первичную информацию ниже по ходу потока посредством добавления карты признаков. Остаточное соединение включает повторную подачу предыдущих представлений в дальнейший поток данных посредством добавления тензора более ранних выходных данных к тензору более поздних выходных данных, что помогает предотвратить потери информации по ходу потока обработки данных. Остаточные соединения обладают двумя общими проблемами, которые наносят вред любой крупномасштабной модели глубокого обучения: исчезающие градиенты и узкие места, связанные со способом задания функций. В целом, добавление остаточных соединений в любую модель, имеющую более 10 слоев, наиболее вероятно обеспечит преимущество. Как описано выше, остаточное соединение включает обеспечение доступности выходных данных более раннего слоя в качестве входных данных более позднего слоя, что с фактически создает короткий путь в последовательной сети. Вместо того, чтобы быть конкатенированными к более поздним активациям, более ранние выходные данные суммируют с более поздними активациями, что предполагает, что обе активации имеют одинаковый размер. Если они имеют различные размеры. для изменения формы более ранней активации до целевой формы может быть использовано линейное преобразование.
Остаточное обучение и соединения с пропуском
[00215] ФИГ. 1H показан один вариант осуществления остаточных блоков и соединений с пропуском. Основная идея остаточного обучения заключается в том, что остаточное преобразование является более легким для обучения, чем первоначальное преобразование. Остаточная сеть собирает в стек некоторое количество остаточных блоков для того, чтобы уменьшить ухудшение точности обучения. Остаточные блоки используют особые дополнительные соединения с пропуском для борьбы с исчезающими градиентами в глубоких нейронных сетях. В начале остаточного блока поток данных разделяют на два потока: первый переносит неизменные входные данные блока, а второй применяет веса и нелинейности. В конце блока два потока сливаются с использованием поэлементного суммирования. Основным преимуществом таких структур является обеспечение более легкого протекания градиента через сеть. Дополнительную информацию об остаточных блоках и соединениях с пропуском можно найти в источнике A. V. D. Oord, S. Dieleman, H. Zen, K. Simonyan, O. Vinyals, A. Graves, N. Kalchbrenner, A. Senior и K. Kavukcuoglu, “WAVENET: A GENERATIVE MODEL FOR RAW AUDIO”, arXiv:1609.03499, 2016.
[00216] Пользуясь преимуществом остаточной сети глубокие сверточные нейронные сети (CNN) могут быть легко обучены и можно достичь повышенной точности для классификации изображений и обнаружения объектов. Сверточные сети с прямой связью соединяют выходные данные 


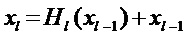
Разреженные свертки
[00217] ФИГ. 1O иллюстрирует разреженные свертки. Разреженные свертки иногда называются дырчатыми (atrous) свертками, что означает «с дырами» Французское название происходит от названия алгоритма «a trous», который вычисляет быстрое диадическое вейвлет-преобразование. В этом типе сверточных слоев входы, соответствующие принимающему полю фильтров, не являются соседними точками. Это проиллюстрировано в ФИГ. 1O. Расстояние между входами зависит от коэффициента разрежения.
WaveNet
[00218] WaveNet (WN) представляет собой глубокую нейронную сеть для выработки исходных звуковых сигналов. WaveNet отличается от других сверточных сетей, Поскольку она способна обрабатывать относительно сравнительно большие «визуальные области» малыми ресурсами. Более того, она способна приводить сигналы к требуемым условиям локально и глобально, что позволяет использовать WaveNet в качестве движка перевода текста в речь (TTS) со множеством голосов, при этом TTS дает локальное приведение к требуемым условиям, а конкретный голос - глобальное приведение к требуемым условиям.
[00219] Основные строительные блоки WaveNet представляют собой каузальные разреженные свертки. В качестве разрежения каузальных разреженных сверток WaveNet также позволяет собирать в стеки указанные сборки, как показано на фиг. 11. Для получения такой же рецептивной области с разреженными свертками на данной фигуре необходим другой расширяющий слой. Стеки представляют собой повторение разреженных сверток, что обеспечивает соединение выходных данных разреженного сверточного слоя в одни выходные данные. Это обеспечивает получение WaveNet большой «визуальной» области одного выходного узла с использованием сравнительно малых вычислительных ресурсов. Для сравнения, для получения визуальной области с 512 входными данными полностью сверточной сети (FCN) понадобится 511 слоев. В случае разреженной сверточной сети нам понадобится восемь слоев. Собранные в стек разреженные свертки потребуют только семь слоев с двумя стеками или шесть слоев с четырьмя стеками. Для получения представления о различиях в потребляемых вычислительных ресурсах, требуемых для покрытия одной и той же визуальной области, в приведенной ниже таблице показано количество весов, требуемое в сети с условием одного фильтра на слой и шириной фильтра равной двум. Кроме того, принято, что сеть использует двоичное восьмибитное кодирование.
[00220] WaveNet добавляет соединение с пропуском перед тем как выполнено остаточное соединение, что обеспечивает обход всех последующих остаточных блоков. Каждое из указанных соединений с пропусками суммируют перед их проходом через последовательности функций активации и сверток. Интуитивно это представляет собой сумму информации, извлеченной в каждом слое.
Пакетная нормализация (Batch normalization)
[00221] Пакетная нормализация представляет собой способ ускорения обучения глубоких сетей посредством того, что стандартизацию данных делают неотъемлемой частью архитектуры сети. Пакетная нормализация может адаптивным образом нормализовывать данные даже при изменении среднего и дисперсии со временем в процессе обучения. Это работает посредством внутреннего поддержания экспоненциально изменяющегося среднего значения среднего и дисперсии данных для каждого пакета данных, наблюдаемых во время обучения. Основной эффект нормализации пакетов данных заключается в том, что она помогает распространению градиента, наподобие остаточным соединениям, и тем самым обеспечивает получение глубоких сетей. Некоторые очень глубокие сети могут быть обучены, если они включают множество слоев с пакетной нормализаций.
[00222] Пакетная нормализация может выглядеть как еще один слой, который может быть вставлен в архитектуру модели, как плотно соединенный или сверточный слой. Слой с пакетной нормализацией (BatchNormalization layer) обычно используют после сверточного или плотно соединенного слоя. Также его могут использовать перед сверточным или Плотно соединенным слоем. Оба варианта осуществления могут быть использованы в раскрытой технологии и показаны на фиг. 15. Пакетная нормализация берет аргумент оси, который определяет ось признаков, которую необходимо нормализовать. Этот аргумент по умолчанию имеет значение -1, последняя ось во входном тензоре. Это корректное значение при использовании слоев Dense, слоев Conv1D, слоев RNN и слоев Conv2D с data_format (данные_формат), установленным на «channels_last» (каналы_последний). Однако при нишевом использовании слоев Conv2D с data_format установленном на “channels_first” (каналы_первый), признаки оси представляют собой axis 1 (ось 1); аргумент оси при BatchNormalization может быть установлен на 1.
[00223] Пакетная нормализация обеспечивает определение для прямой подачи входных данных и вычисления градиентов относительно параметров и их собственных входных данных посредством обратного прохода. На практике, слои с пакетной нормализацией вставлены после сверточного или плотно соединенного слоя, но перед подачей выходных данных в функцию активации. Для сверточных слоев различные элементы одной карты признаков, т.е. активации, в различных положениях нормализованы одинаковым образом для того, чтобы подчиняться свойству свертки. Таким образом, все активации в малом пакете данных (mini-batch) нормализованы по всем положениям, а не на каждую активацию.
[00224] Внутреннее ковариантное смещение представляет собой главную причину, почему глубокие архитектуры, как хорошо известно, нужно было долго обучать. Это вызвано тем фактом, что глубокие сети не только должны обучаться новому способу задания функций на каждом слое, но также должны учитывать изменение их распределения.
[00225] Ковариантное смещение в целом является известной проблемой в области глубокого обучения и часто встречается в проблемах в реальном мире. Известной проблемой ковариантного смещения является разница в распределении обучающего и тестового набора, что может привести к неоптимальной эффективности обобщения. Эту проблему обычно решают этапом стандартизации или предобработки выбеливанием. Однако особенно операция выбеливания является ресурсозатратной и, таким образом, непрактичной в онлайн системах, особенно, если ковариантное смещение происходит в различных слоях.
[00226] Внутреннее ковариантное смещение представляет собой феномен, при котором распределение активаций сети изменяется в слоях вследствие изменения параметров сети во время обучения. В идеале, каждый слой должен быть преобразован в пространство, в котором они имеют одинаковое распределение, но функциональное взаимодействие остается тем же. Для того, чтобы избежать ресурсозатратных вычислений ковариантных матриц для того, чтобы декоррелировать и выбелить данные на каждом слое и этапе, авторы изобретения нормализовали распределение каждого входного признака в каждом слое в каждом малом пакете данных для того, чтобы иметь его нулевое среднее и стандартное отклонение.
Прямой проход
[00227] Во время прямого прохода вычисляют среднее и дисперсию малого пакета данных. С такими статистическими показателями малого пакета данных данные нормализуют путем вычитания среднего и деления на стандартное отклонение. Наконец, данные масштабируют и смещают с изученными параметрами масштабирования и смещения. Прямой проход 
[00228] На фиг. 1I 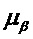

[00229] Поскольку нормализация является дифференцируемым преобразованием, ошибки распространяются в указанные изученные параметры и тем самым способны восстановить репрезентативную способность сути путем обучения тождественному преобразованию. В отличие от этого, путем изучения параметров масштабирования и смещения, которые идентичны соответствующим статистическим показателям пакета данных, преобразование с пакетной нормализацией не имело бы эффекта на сеть, если это было бы оптимальной операцией к выполнению. Во время тестирования среднее и дисперсия пакета данных заменены соответствующими статистическими показателями выборки, Поскольку входные данные не зависят от других образцов из малого пакета данных. Другой способ заключается в удержании скользящих средних значений статистических показателей пакета данных во время обучения и в использовании их для вычисления выходных данных сети во время тестирования. Во время тестирования преобразование с пакетной нормализацией может быть выражено как показано на фиг. 1J. На фиг. 1J μD и 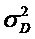
Обратный проход
[00230] Поскольку нормализация является дифференцируемой операцией, обратный проход может быть вычислен, как показано на фиг. 1K.
Одномерная (1D) свертка
[00231] Одномерные свертки извлекают локальные одномерные вставки или частичные последовательности из последовательностей, как показано на фиг. 16. Одномерная свертка получает каждый выходной шаг по времени из временной вставки во входной последовательности. Одномерные сверточные слои распознают локальные паттерны в последовательности. Поскольку та же самая входная информация выполняется над каждой вставкой, вставка, изученная в определенном положении во входной последовательности позже может быть распознана в другом положении, что делает одномерные сверточные слои инвариантными к перемещению для временных перемещений. Например, одномерный сверточный слой, обрабатывающий последовательности оснований с использованием окон свертки размером 5, должен быть способен изучать основания или последовательности оснований длиной 5 или менее, и он должен быть способен распознавать основные мотивы в любом контексте во входной последовательности. Одномерная свертка основного уровня таким образом способна учиться в отношении морфологии оснований.
Глобальное среднее объединение
[00232] На фиг. 1N показано, как работает глобальное среднее объединение (GAP). Глобальное среднее объединение может быть использовано для замены плотно соединенных (FC) слоев для классификации посредством взятия средних значений в последнем слое оценки. Это позволяет сократить обучающую нагрузку и обойти проблемы с переподгонкой. Глобальное среднее объединение применяет структурные априорные данные к модели и это эквивалентно линейному преобразованию с заданными весами. Глобальное среднее объединение уменьшает количество параметров и устраняет плотно соединенные слои. Плотно соединенные слои обычно являются наиболее загруженными с точки зрения параметров и соединений слоями, а глобальное среднее объединение обеспечивает значительно менее ресурсозатратный подход к достижению аналогичных результатов. Основная идея глобального среднего объединения заключается в создании среднего значения из каждой последней карты признаков слоя как коэффициента достоверности для оценки, подаваемого непосредственно в softmax слой.
[00233] Глобальное среднее объединение имеет три преимущества: (1) отсутствуют дополнительные параметры в слоях с глобальным средним объединением, тем самым позволяя избежать переподгонки в указанных слоях; (2) Поскольку выходные данные глобального среднего объединения является среднее всей карты признаков, то глобальное среднее объединение будет более устойчивым к пространственным перемещениям; и (3) вследствие огромного числа параметров в плотно соединенных слоях, которое обычно занимает более 50% всех параметров всей сети, их замена слоями с глобальным средним объединением может значительно уменьшить размер модели, и это делает глобальное среднее объединение очень полезным при сжатии моделей.
[00234] Глобальное среднее объединение является целесообразным, Поскольку ожидается, что более эффективные признаки в последнем слое будут иметь большее среднее значение. В некоторых вариантах осуществления глобальное среднее объединение может быть использовано в качестве посредника для оценки классификации. Карты признаков при глобальном среднем объединении могут быть интерпретированы как карты достоверности, и обеспечивают соответствие между картами признаков и категориями. Глобальное среднее объединение может быть особенно эффективным, если признаки последнего слоя достаточно абстрактны для прямой классификации, глобального среднего объединения в отдельности не достаточно, если многоуровневые признаки должны быть комбинированы в группы наподобие частичных моделей, что наилучшим образом выполняется путем добавления простого плотно соединенного слоя или другого классификатора после Глобального среднего объединения.
Глубокое Обучение в Геномике
[00235] Генетические вариации могут помочь объяснить многие заболевания. Каждое человеческое существо имеет уникальный генетический код, а в группе индивидуумов встречается множество генетических вариантов. Большинство вредоносных генетических вариантов были исключены из геномов в результате естественного отбора. Важно идентифицировать, генетические варианты, которые вероятно являются патогенными или вредоносными. Это поможет исследователям сосредоточиться на вероятно патогенных вариантах и ускорить диагностику и лечение многих заболеваний.
[00236] Моделирование свойств и функциональных эффектов (например, патогенности) вариантов - это важная, но сложная для ученых область геномики. Несмотря на быстрое развитие функциональных генных технологий секвенирования, интерпретация функциональных эффектов вариантов остается крайне непростой задачей из-за сложности специфичных для разных типов клеток систем регуляции транскрипции быстрое развитие функциональных генных технологий секвенирования.
[00237] Достижения в биохимических технологиях за последние десятилетия привели к появлению платформ секвенирования следующего поколения (NGS), которые быстро производят геномные данные с гораздо меньшими затратами, чем когда-либо прежде. Такие чрезвычайно большие объемы секвенированной ДНК по-прежнему трудно аннотировать. Алгоритмы контролируемого машинного обучения обычно хорошо работают, если доступны большие объемы размеченных данных. В биоинформатике и многих других дисциплинах, связанных с большим объемом данных, процесс маркировки экземпляров стоит дорого; тем не менее, неразмеченные экземпляры недороги и легко доступны. Для сценария, в котором количество размеченных данных относительно невелико, а количество неразмеченных данных значительно больше, полу-контролируемое обучение представляет собой экономичную альтернативу ручной маркировке.
[00238] Возникает возможность использовать полууправляемые (полуконтролируемые) алгоритмы для построения классификаторов патогенности на основе глубокого обучения, которые точно предсказывают патогенность вариантов. В результате можно получить базы данных патогенных вариантов, которые свободны от предвзятости, связанной с вмешательством человека.
[00239] Применительно к классификаторам патогенности глубокие нейросети - это тип искусственных нейронных сетей, которые используют множественные нелинейные и сложные преобразующие слои, чтобы последовательно моделировать высокоуровневые признаки. Глубокие нейросети обеспечивают обратную связь посредством алгоритма обратного распространения, который несет информацию о разнице между наблюдаемыми и ожидаемыми выходными данными, с целью коррекции параметров. Глубокие нейронные сети развивались по мере того, как становились доступны большие объемы данных для обучения, мощности параллельных и распределенных вычислений, и развитые алгоритмы обучения. глубокие нейросети способствовали существенному развитию в множестве областей, таких как компьютерное зрение, распознавание речи и обработка естественных языков.
[00240] Сверточные нейронные сети (CNN) и рекуррентные нейронные сети (RNN) являются компонентами нейронных сетей глубокого обучения (глубоких нейронных сетей). Сверточные нейронные сети особенно успешно выполняют задачи по распознаванию образов и имеют архитектуру, которая включает слои свертки, нелинейные слои, слои пулинга. Рекуррентные нейронные сети созданы для использования последовательных входных данных с циклическими связями между строительными блоками, перцептронами, единицами долгосрочной и краткосрочной памяти, и управляемые рекуррентные блоки. В дополнение было предложено много других новейших нейросетей глубокого обучения для ограниченных контекстов, например глубокие пространственно-временные нейронные сети, многомерные рекуррентные нейронные сети, и сверточные автоэнкодеры.
[00241] Цель обучения глубоких нейронных сетей заключается в оптимизации веса параметров в каждом слое, который постепенно комбинирует более простые признаки в сложные, что позволяет получить из данных наиболее подходящие иерархические представления. Отдельный цикл процесса оптимизации организован следующим образом. Сначала, на тренировочном (обучающем) наборе данных, прямой проход алгоритма последовательно вычисляет выходные данные в каждом слое, и распространяет сигналы функции вперед по сети. В конечном выходном слое (слое выходных данных), целевая функция потерь измеряет погрешность между выходными данными работы обученной нейронные сети и данными метками. Для минимизации ошибок обучения, при обратном проходе используется правило сложной производной (цепное правило) для обратного распространения сигналов ошибки и вычисления градиентов по всем весам по всей нейронные сети. В конце весовые параметры обновляются посредством алгоритмов оптимизации, основанных на стохастическом градиентном спуске. В то время как градиентный спуск осуществляет обновление параметров для каждого полного набора данных, стохастический градиентный спуск обеспечивает стохастическую аппроксимацию, проводя обновление для каждого небольшого набора семплированных данных (данных в выборке). На принципе стохастического градиентного спуска основаны несколько алгоритмов оптимизации. Например, обучающий алгоритм Адаграда и Адама проводит стохастический градиентный спуск с адаптивным изменением скорости обучения на основе частоты обновления моментов градиентов для каждого параметра, соответственно.
[00242] Другим базовым элементом обучения глубокой нейронной сети является регуляризация, понятие, относящееся к стратегиям, направленным на то, чтобы избежать переобучения нейронные сети, и таким образом добиться хорошей производительности генерализации. Например, сокращение весов добавляет штрафные слагаемые к целевой функции потерь, так что весовые параметры сходятся к меньшим абсолютным значениям. Метод исключения (dropout) случайным образом убирает скрытые узлы из нейронной сети во время обучения, и может рассматриваться как ансамбль возможных подсетей. Чтобы улучшить возможности метода исключения, была предложена новая функция активации, maxout, и определен вариант метода исключения для рекуррентных нейросетей - rnnDrop. Кроме того, пакетная нормализация обеспечивает новый метод регуляризации посредством нормализации скалярных признаков для каждой активации внутри мини-пакета с обучением каждого среднего и отклонения в качестве параметров.
[00243] Кроме того, пакетная нормализация обеспечивает новый метод регуляризации посредством нормализации скалярных признаков для каждой активации внутри мини-пакета с обучением каждого среднего и отклонения в качестве параметров. Сверточные нейронные сети адаптированы для решения задач геномики, основанных на последовательностях, таких как обнаружение мотива, идентификация патогенных вариантов и исследование экспрессии генов. Сверточные нейронные сети используют стратегию совместно используемых весов (weight-sharing), которая особенно полезна для изучения ДНК, потому что они могут регистрировать мотивы последовательностей, которые являются короткими, рекуррентными локальными паттернами в ДНК, имеющими, как полагают, важные биологические функции. Характерной чертой сверточных нейросетей является использование сверточных фильтров. В отличие от традиционных подходов к классификации, которые основаны на признаках, полученных в результате тщательной ручной работы, сверточные фильтры проводят адаптивное обучение признаков, аналогично процессу картирования необработанных входных данных на информативное представление знаний. В этом смысле, сверточные фильтры служат серией сканеров мотивов, поскольку набор таких фильтров способен опознать релевантные паттерны во входных данных, и адаптироваться в процессе обучения. Рекуррентные нейронные сети могут регистрировать дальномерные зависимости в последовательных данных различной длины, таких как белковые последовательности или ДНК.
[00244] Таким образом, мощная вычислительная модель, предсказывающая патогенность вариантов, может давать огромное преимущество как для фундаментальной, так и для прикладной науки.
[00245] Общие (обычные, распространенные) полиморфизмы представляют природные эксперименты, пригодность которых проверена поколениями естественного отбора. Сравнивая распределения частот аллелей для человеческих миссенс- и синонимичных замен, мы обнаруживаем, что присутствие миссенс-варианта с высокими частотами аллелей у приматов, отличных от человека, надежно предсказывает, что этот вариант также находится под нейтральным отбором в человеческой популяции. Напротив, распространенные варианты у более далеких видов подвергаются негативному отбору по мере увеличения эволюционной дистанции.
[00246] Мы используем распространённые варианты шести видов приматов, отличных от человека, для обучения полу-контролируемой сети глубокого обучения, которая точно классифицирует клинические миссенс-мутации de novo, используя только последовательность. Линия приматов, насчитывающая более 500 известных видов, содержит достаточно распространенных вариантов, чтобы систематически моделировать эффекты большинства человеческих вариантов неизвестного значения.
[00247] Контрольный геном человека содержит более 70 миллионов потенциальных изменяющих белок миссенс-замен, подавляющее большинство из которых являются редкими мутациями, влияние которых на здоровье человека не охарактеризовано. Эти варианты неизвестного значения представляют собой проблему для интерпретации генома в клинических применениях и препятствуют долгосрочному внедрению секвенирования для популяционного скрининга и индивидуализированной медицины.
[00248] Каталогизация распространенных вариантов среди различных человеческих популяций является эффективной стратегией для идентификации клинически доброкачественных вариантов, но распространенные вариации, доступные у современных людей, ограничены узкими местами далекого прошлого нашего вида. Люди и шимпанзе имеют 99% идентичности последовательностей, что позволяет предположить, что естественный отбор, действующий на варианты шимпанзе, имеет потенциал для моделирования эффектов вариантов у человека, которые идентичны им по состояниям. Среднее время слияния нейтральных полиморфизмов в человеческой популяции составляет часть времени расхождения вида, следовательно, естественная вариация шимпанзе в значительной степени исследует пространство мутаций, которое не пересекается с вариантами человека, за исключением редких случаев гаплотипов, поддерживаемых сбалансированным отбором.
[00249] То, что недавно стали доступны агрегированные данные экзома от 60 706 человек позволяет нам проверить эту гипотезу путем сравнения частотных спектров аллелей для миссенс и синонимичных мутаций. Одноэлементные варианты в ExAC близко соответствуют ожидаемому соотношению миссенс/синонимы 2,2: 1, предсказанному мутацией de novo после корректировки на степень мутаций с использованием тринуклеотидного контекста, но при более высоких частотах аллелей количество наблюдаемых миссенс-вариантов уменьшается из-за фильтрации вредных вариантов естественным отбором. Паттерн соотношений миссенс/синонимы в частотном спектре аллелей указывает на то, что большая часть миссенс-вариантов с популяционной частотой <0,1% являются умеренно вредными, то есть недостаточно патогенными, чтобы требовать немедленного удаления из популяции, и недостаточно нейтральными, чтобы им можно было позволить существовать с высокими частотами аллелей, что согласуется с предыдущими наблюдениями на более ограниченных данных о популяции. Эти результаты подтверждают широко распространенную эмпирическую практику диагностических лабораторий по фильтрации вариантов с частотой аллелей более 0,1% ~ 1% как вероятных доброкачественных для пенетрантного генетического заболевания, за исключением нескольких хорошо задокументированных исключений, вызванных балансирующим отбором и эффектами основателя.
[00250] Повторяя этот анализ с подмножеством вариантов человека, которые идентичны по состояниям с распространенными вариантами шимпанзе (наблюдаемыми более одного раза при секвенировании популяции шимпанзе), мы обнаруживаем, что соотношение миссенс: синонимы в значительной степени постоянно во всем частотном спектре аллелей. Высокая частота аллелей этих вариантов в популяции шимпанзе указывает на то, что они уже прошли через сито естественного отбора у шимпанзе, и их нейтральное влияние на приспособленность в человеческих популяциях является убедительным доказательством того, что селективное давление на миссенс-варианты очень согласовано в двух видах. Более низкое соотношение миссенс: синонимы, наблюдаемое у шимпанзе, согласуется с большим эффективным размером популяции в популяциях предков шимпанзе, что позволяет более эффективно фильтровать умеренно вредоносные варианты.
[00251] В отличие от этого, редкие варианты шимпанзе (наблюдавшиеся только один раз при секвенировании популяции шимпанзе) показывают умеренное снижение отношения миссенс: синонимы при более высоких частотах аллелей. Моделируя когорту одинакового размера на основе данных об изменениях у человека, мы оцениваем, что только 64% вариантов, наблюдаемых один раз в когорте такого размера, будут иметь частоту аллелей более 0,1% в общей популяции по сравнению с 99,8% для вариантов, которые наблюдались в когорте многократно, что указывает на то, что не все редкие варианты шимпанзе прошли через сито отбора. В целом, по нашим оценкам, 16% установленных миссенс-вариантов шимпанзе имеют частоту аллелей менее 0,1% в общей популяции и будут подвергаться отрицательному отбору при более высоких частотах аллелей.
[00252] Далее мы характеризуем варианты у человека, которые идентичны по состояниям с вариантами, наблюдаемыми у других видов приматов, отличных от человека (бонобо, гориллы, орангутанги, резусов и игрунок). Как и в случае шимпанзе, мы наблюдаем, что отношения миссенс: синонимы примерно эквивалентны по частотному спектру аллелей, за исключением небольшого истощения миссенс-вариантов при высоких частотах аллелей, чего можно было бы ожидать из-за включения небольшого числа редких вариантов ( ~ 5-15%). Эти результаты предполагают, что селективные силы отбора по миссенс-вариантам в значительной степени согласуются внутри линии приматов по меньшей мере до обезьян нового света, которые, по оценкам, отошли от линии происхождения человека примерно 35 миллионов лет назад.
[00253] Миссенс-варианты человека, которые идентичны по состояниям вариантам у других приматов, в ClinVar сильно обогащены доброкачественными последствиями. После исключения вариантов с неизвестными или противоречивыми аннотациями мы наблюдаем, что человеческие варианты с ортологами приматов с вероятностью примерно 95% будут аннотированы как доброкачественные или вероятно доброкачественные в ClinVar по сравнению с 45% для миссенс-вариации в целом. Небольшая доля вариантов ClinVar, которые классифицируются как патогенные у приматов, отличных от человека, сравнима с долей патогенных вариантов ClinVar, которые можно было бы наблюдать при выявлении редких вариантов из когорты здоровых людей аналогичного размера. Значительная часть этих вариантов, помеченных как патогенные или вероятно патогенные, указывает на то, что они получили свою классификацию до появления больших баз данных частот аллелей, и сегодня их можно определить иначе.
[00254] В области генетики человека долгое время полагались на модельные организмы чтобы делать выводы о клиническом влиянии мутаций человека, но большое эволюционное расстояние до большинства поддающихся генетическому изучению животных моделей вызывает опасения по поводу степени, в которой эти результаты могут быть обобщены на человека. Чтобы изучить соответствие естественного отбора на миссенс-варианты у человека и более отдаленных видов, мы расширяем наш анализ за пределы линии приматов, чтобы включить в основном распространенные вариации от четырех дополнительных видов млекопитающих (мыши, свиньи, козы, коровы) и двух видов более отдаленных позвоночных (курица, данио). В отличие от предыдущих анализов приматов, мы наблюдаем, что миссенс-варианты заметно снижается при частотах распространенных аллелей по сравнению с частотами редких аллелей, особенно на больших эволюционных дистанциях, что указывает на то, что значительная часть общих миссенс-вариантов у более отдаленных видов будет испытывать отрицательный отбор у в человеческих популяциях. Тем не менее, наблюдение миссенс-варианта у более отдаленных позвоночных по-прежнему увеличивает вероятность доброкачественных последствий, поскольку доля обычных миссенс-вариантов, истощенных естественным отбором, намного меньше, чем ~ 50% истощение миссенс-вариантов человека на исходном уровне. В соответствии с этими результатами, мы обнаружили, что миссенс-варианты человека, которые наблюдались у мышей, собак, свиней и коров, с вероятностью примерно 85% будут аннотироваться как доброкачественные или вероятные доброкачественные в ClinVar по сравнению с 95% для вариаций приматов и 45%. для базы данных ClinVar в целом.
[00255] Присутствие близкородственных пар видов на различных эволюционных дистанциях также дает возможность оценить функциональные последствия фиксированных миссенс-замен в человеческих популяциях. В пределах близкородственных пар видов (длина ветви <0,1) на генеалогическом дереве млекопитающих мы наблюдаем, что фиксированных миссенс-вариант истощается в частотах распространенных аллелей по сравнению с частотами редких аллелей, что указывает на то, что значительная часть фиксированных замен между видами не будет - нейтральной по отношению к человеку, даже по роду приматов. Сравнение величины истощения миссенс-варианта показывает, что фиксированные замены между видами значительно менее нейтральны, чем внутривидовые полиморфизмы. Интересно, что межвидовые варианты между близкородственными млекопитающими не являются существенно более патогенными в ClinVar (83% с вероятностью будут отмечены как доброкачественные или вероятно доброкачественные) по сравнению с общими внутривидовыми полиморфизмами, что позволяет предположить, что эти изменения не уничтожают функцию белка, а скорее отражают настройку функции белка, которая дает видоспецифичные адаптивные преимущества.
[00256] Большое количество возможных вариантов неизвестного значения и критическая важность точной классификации вариантов для клинических приложений обусловили множество попыток решить проблему с помощью машинного обучения, но эти усилия в значительной степени ограничивались недостаточным количеством распространенных человеческих вариантов. и сомнительным качеством аннотаций в тщательно подобранных базах данных. Варианты от шести приматов, отличных от человека, вносят вклад в более 300000 уникальных миссенс-вариантов, которые не пересекаются с распространенными человеческими вариантами и в значительной степени имеют благоприятные последствия, что значительно увеличивает размер обучающего набора данных, который может использоваться для подходов машинного обучения.
[00257] В отличие от более ранних моделей, которые используют большое количество созданных человеком функций и мета-классификаторов, мы применяем простую остаточную сеть глубокого обучения, которая принимает в качестве входных данных только аминокислотную последовательность, фланкирующую интересующий вариант, и выравнивания ортологичных последовательностей в других видах. Чтобы предоставить сети информацию о структуре белка, мы обучаем две отдельные сети для изучения вторичной структуры и доступности для растворителей только на основе последовательности и включаем их в качестве подсетей в более крупную сеть глубокого обучения для прогнозирования воздействия на структуру белка. Использование последовательности в качестве отправной точки позволяет избежать потенциальных смещений в структуре белка и аннотации функционального домена, которые могут быть не полностью установлены или применены непоследовательно.
[00258] Мы используем полууправляемое обучение чтобы преодолеть проблему обучающего набора, содержащего только варианты с доброкачественными метками, путем первоначального обучения ансамбля сетей для разделения вероятно доброкачественных вариантов приматов и случайных неизвестных вариантов, которые совпадают по степени мутаций и охвату секвенированием. Этот ансамбль сетей применяется для оценки полного набора неизвестных вариантов и влияет на выбор неизвестных вариантов для засева следующей итерации классификатора путем смещения в сторону неизвестных вариантов с более прогнозируемыми патогенными последствиями, с постепенными шагами на каждой итерации для предотвращения преждевременного схождения модели к неоптимальному результату.
[00259] Распространенные варианты приматов также обеспечивают чистый набор данных для валидации для оценки существующих методов, который полностью независим от ранее использованных обучающих данных, которые было трудно объективно оценить из-за распространения метаклассификаторов. Мы оценили производительность нашей модели вместе с четырьмя другими популярными алгоритмами классификации (Sift, Polyphen2, CADD, M-CAP), используя 10 000 распространенных вариантов приматов. Поскольку примерно 50% всех миссенс-вариантов человека будут удалены естественным отбором при распространенных частотах аллелей, мы вычислили 50-й процентиль для каждого классификатора на наборе случайно выбранных миссенс-вариантов, которые были сопоставлены с 10000 распространенными вариантами приматов по частоте мутаций и использовали этот порог для оценки распространенных вариантов приматов. Точность нашей модели глубокого обучения была значительно лучше, чем у других классификаторов на этом наборе данных независимой валидации, в которых использовались либо сети глубокого обучения, которые были обучены только на распространенных вариантах человека, либо с использованием как распространенных вариантов человека, так и вариантов приматов.
[00260] Недавние исследования по тройному секвенированию приведи к каталогизации тысячи мутаций de novo у пациентов с нарушениями развития нервной системы и их здоровых братьев и сестер, что позволило оценить эффективность различных алгоритмов классификации при разделении миссенс-мутаций de novo в случаях болезни (кейсах) по сравнению с контролем. Для каждого из четырех алгоритмов классификации мы оценивали каждый вариант de novo миссенс-мутации в случаях по сравнению с контролем и фиксировали p-значение критерия суммы рангов Вилкоксона для разницы между двумя распределениями, показывая, что метод глубокого обучения, обученный на вариантах приматов (p ~ 10-33), показал себя намного лучше, чем другие классификаторы (p ~ 10-13 - 10-19) в этом клиническом сценарии. Исходя из ~ 1,3-кратного увеличения количества миссенс-вариантов de novo по сравнению с ожиданиями, о которых ранее сообщалось для этой когорты, и предварительных оценок, согласно которым ~ 20% миссенс-вариантов вызывают эффекты потери функции, мы могли бы ожидать, что идеальный классификатор разделит два класса с p-значением p ~ 10-40, что указывает на то, что наш классификатор все еще нуждается в улучшении.
[00261] Точность классификатора глубокого обучения масштабируется в зависимости от размера обучающего набора данных, и данные об изменениях от каждого из шести видов приматов независимо вносят вклад в повышение точности классификатора. Большое количество и разнообразие существующих видов приматов, отличных от человека, наряду с доказательствами, показывающими, что селективное давление на варианты, изменяющие белок, в значительной степени согласованы в пределах линии приматов, предполагает систематическое секвенирование популяции приматов как эффективную стратегию для классификации миллионов вариантов человека. неизвестного значения, которые в настоящее время ограничивают клиническую интерпретацию генома. Из 504 известных видов приматов, отличных от человека, примерно 60% находятся под угрозой исчезновения из-за охоты и исчезновения среды обитания, что мотивирует безотлагательность всемирных усилий по сохранению, которые принесут пользу как этим уникальным и незаменимым видам, так и нашему собственному.
[00262] Хотя не так уж много агрегированных данных по полному геному доступно в форме экзомных данных, что ограничивает возможности обнаружения влияния естественного отбора в глубоких интронных областях, мы также смогли вычислить наблюдаемое и ожидаемое количество криптических сплайсинговых мутаций, далеких от экзонных областей. В целом, мы наблюдаем 60%-ное истощение криптических мутаций сплайсинга на расстоянии> 50нт от границы экзон-интрон. Ослабленный сигнал, вероятно, представляет собой комбинацию меньшего размера выборки с данными полного генома по сравнению с экзомом и большей трудностью прогнозирования влияния глубоких интронных вариантов.
Терминология
[00263] Все литературные источники и аналогичный материал, цитируемый в настоящей заявке, в том числе, но не ограничиваясь перечисленным, патенты, патентные заявки, статьи, книги, научные работы и веб-страницы, независимо от формата таких литературных источников и аналогичных материалов, явным образом и полностью включены в настоящий документ посредством ссылок. В тех случаях, когда один или более из включенных литературных источников и аналогичных материалов отличается от настоящей заявки или противоречит ей, в том числе, но не ограничиваясь перечисленным, определяемые термины, силу будет иметь настоящая заявка.
[00264] В настоящем документе следующие термины имеют указанные значения.
[00265] Основание относится к нуклеотидному основанию или нуклеотиду, A (аденину), C (цитозину), T (тимину) или G (гуанину).
[00266] В настоящей заявке взаимозаменяемо используются термины “белок” и “транслируемая последовательность”.
[00267] В настоящей заявке взаимозаменяемо используются термины “кодон” и “триплет оснований”.
[00268] В настоящей заявке взаимозаменяемо используются термины “аминокислота” и “транслируемая единица”.
[00269] В настоящей заявке взаимозаменяемо используются выражения “классификатор патогенности вариантов”, “классификатор на основе сверточной нейронной сети для классификации вариантов” и “классификатор на основе глубокой сверточной нейронной сети для классификации вариантов”.
[00270] Термин “хромосома” относится к носителю генов, передающих наследственные признаки, в живой клетке, происходящему из нитей хроматина, содержащих ДНК и белковые компоненты (в частности, гистоны). В настоящем документе используется стандартная международно признанная система нумерации индивидуальных хромосом генома человека.
[00271] Термин “сайт” относится к уникальному положению (например, идентификатору хромосомы, положению и ориентации хромосомы) на референсном геноме. В некоторых вариантах реализации сайт может представлять собой остаток, метку последовательности или положение сегмента на последовательности. Термин “локус” может применяться для обозначения специфической локализации последовательности нуклеиновой кислоты или полиморфизма на референсной хромосоме.
[00272] Термин “образец” в настоящем документе относится к образцу, как правило, происходящему из биологической жидкости, клетки, ткани, органа или организма, содержащего нуклеиновую кислоту или смесь нуклеиновых кислот, содержащую по меньшей мере одну последовательность нуклеиновой кислоты, подлежащую секвенированию и/или фазированию. Такие образцы включают, не ограничиваясь перечисленными, образцы мокроты/жидкости ротовой полости, амниотической жидкости, крови, фракции крови, тонкоигольной биопсии (например, хирургической биопсии, тонкоигольной биопсии и т.п.),мочи, жидкости брюшной полости, плевральной жидкости, эксплантата ткани, культуры органа и любого другого препарата ткани или клеток, или его фракции или производного, или выделенные из них образцы. Хотя образец часто получают от субъекта-человека (например, пациента), образцы могут быть взяты из любого организма, имеющего хромосомы, в том числе, но не ограничиваясь перечисленными, организма собак, кошек, лошадей, коз, овец, крупного рогатого скота, свиней и т.п. Образец может применяться непосредственно в полученном из биологического источника виде или после предварительной обработки для модификации характера образца. Например, такая предварительная обработка может включать получение плазмы из крови, разведение вязких текучих сред и т.д. Методы предварительной обработки могут также включать, не ограничиваясь перечисленными, фильтрацию, осаждение, разведение, дистилляцию, смешивание, центрифугирование, замораживание, лиофилизацию, концентрацию, амплификацию, фрагментацию нуклеиновых кислот, инактивацию мешающих компонентов, добавление реагентов, лизис и т.п.
[00273] Термин “последовательность” включает или обозначает цепь взаимно сопряженных нуклеотидов. Нуклеотиды могут быть основаны на ДНК или РНК. Следует понимать, что одна последовательность может включать несколько субпоследовательностей. Например, одна последовательность (например, ПЦР-ампликона) может содержать 350 нуклеотидов. Рид образца может включать несколько субпоследовательностей в пределах указанных 350 нуклеотидов. Например, рид образца может включать первую и вторую фланкирующие субпоследовательности, содержащие, например, 20-50 нуклеотидов. Указанные первая и вторая фланкирующие субпоследовательности могут быть локализованы на любой стороне повторяющегося сегмента, содержащего соответствующую субпоследовательность (например, 40-100 нуклеотидов). Каждая из фланкирующих субпоследовательностей может включать (или включать частично) субпоследовательность праймера (например, 10-30 нуклеотидов). Для простоты чтения вместо термина “субпоследовательность” используют “последовательность”, но следует понимать, что две последовательности не обязательно отделены одна от другой на общей цепи. Для различения различных последовательностей, описанных в настоящем документе, в указанные последовательности могут быть включены разные метки (например, целевая последовательность, праймерная последовательность, фланкирующая последовательность, референсная последовательность и т.п.). В другие объекты, такие как описываемые термином “аллель”, могут быть включены разные метки для дифференциации сходных объектов.
[00274] Термин “парно-концевое секвенирование” относится к способам секвенирования с секвенированием обоих концов целевого фрагмента. Парно-концевое секвенирование может облегчать детекцию геномных перестановок и повторяющихся сегментов, а также слитых генов и новых транскриптов. Методология парно-концевого секвенирования описана в PCT-публикации WO07010252, PCT-публикации сер. № PCTGB2007/003798 и опубликованной заявке на патент США US 2009/0088327, каждая из которых включена посредством ссылки в настоящий документ. Согласно одному примеру может быть выполнен следующий ряд операций; (a) генерация кластеров нуклеиновых кислот; (b) линеаризация указанных нуклеиновых кислот; (c) гибридизация первого праймера для секвенирования и проведение многократных циклов удлинения, сканирования и деблокирования согласно описанию выше; (d) “инверсия” целевых нуклеиновых кислот на поверхности проточной ячейки путем синтеза комплементарной копии; (e) линеаризация ресинтезированной цепи; и (f) гибридизация второго праймера для секвенирования и проведения многократных циклов удлинения, сканирования и деблокирования согласно описанию выше. Операция инверсии может быть проведена с доставкой реагентов согласно описанию выше для одного цикла мостиковой амплификации.
[00275] Термин “референсный геном” или “референсная последовательность” относится к любой конкретной известной последовательности генома, частичной или полной, любого организма, которая может быть использована в качестве референсной для идентифицированных последовательностей субъекта. Например, референсный геном, используемый для субъектов-людей, а также многих других организмов можно найти по ссылке ncbi.nlm.nih.gov от Национального центра биотехнологической информации. “Геном” относится к полной генетической информации организма или вируса, представленной в виде последовательностей нуклеиновых кислот. Геном включает как гены, так и некодирующие последовательности ДНК. Референсная последовательность может быть длиннее ридов, которые на нее выравнивают. Например, она может быть по меньшей мере приблизительно в 100 раз длиннее, или по меньшей мере приблизительно в 1000 раз длиннее, или по меньшей мере приблизительно в 10 000 раз длиннее, или по меньшей мере приблизительно в 105 раз длиннее, или по меньшей мере приблизительно 106 раз длиннее, или по меньшей мере приблизительно в 107 раз длиннее. В одном примере референсная последовательность генома представляет собой последовательность полноразмерного генома человека. В другом примере референсная последовательность генома ограничена специфической хромосомой человека, такой как хромосома 13. В некоторых вариантах реализации референсная хромосома представляет собой последовательность хромосомы из генома человек версии hg19. Такие последовательности могут называться референсными последовательностями хромосомы, хотя предполагается, что термин “референсный геном” охватывает такие последовательности. Другие примеры референсных последовательностей включают геномы других видов, а также хромосом, субхромосомных областей (например, цепей) и т.п., любых видов. В различных вариантах реализации референсный геном представляет собой консенсусную последовательность или другую комбинацию, полученную от нескольких индивидуумов. Однако в определенных вариантах применения референсная последовательность может быть получена от конкретного индивидуума.
[00276] Термин “рид” относится к совокупности данных о последовательности, описывающих фрагмент нуклеотидного образца или референсной последовательности. Термин “рид” может относиться к риду образца и/или референсному риду. Обычно, хотя не обязательно, рид представлен короткой последовательностью непрерывно расположенных пар оснований в образце или референсной последовательности. Рид может быть символически представлен последовательностью пар оснований (ATCG) образца или референсного фрагмента. Он может храниться в запоминающем устройстве и обрабатываться подходящим образом для определения того, совпадает ли рид с референсной последовательностью или отвечает ли другим критериям. Рид может быть получен непосредственно из аппарата для секвенирования или непрямо, из сохраненной информации о последовательности, касающейся указанного образца. В некоторых случаях рид представляет собой последовательность ДНК достаточной длины (например, по меньшей мере приблизительно 25 п.о.) которые могут применяться для идентификации последовательности или области большей длины, например, например, которая может быть выравнена и специфическим образом соотнесена с хромосомой, или геномной областью, или генов.
[00277] Методы секвенирования следующего поколения включают, например, технологию секвенирования путем синтеза (Illumina), пиросеквенирование (454), технологию ионного полупроводникового секвенирования (секвенирование Ion Torrent), одномолекулярное секвенирование в реальном времени (Pacific Biosciences) и секвенирование путем лигирования (секвенирование SOLiD). В зависимости от методов секвенирования длина каждого рида может варьировать от приблизительно 30 п.о. до более 10 000 п.о. Например, метод секвенирования Illumina с использованием секвенатора SOLiD генерирует риды нуклеиновых кислот длиной приблизительно 50 п.о. В другом примере секвенирование Ion Torrent генерирует риды нуклеиновых кислот длиной до 400 п.о., а пиросеквенирование 454 генерирует риды нуклеиновых кислот длиной приблизительно 700 п.о. В еще одном примере способы одномолекулярного секвенирования в реальном времени могут генерировать риды длиной от 10 000 п.о. до 15 000 п.о. Соответственно, в определенных вариантах реализации риды последовательностей нуклеиновых кислот имеют длину 30-100 п.о., 50-200 п.о. или 50-400 п.о.
[00278] Термины “рид образца”, “последовательность образца” или “фрагмент образца” относятся к данным представляющей интерес геномной последовательности из образца. Например, рид образца содержит данные о последовательности из ПЦР-ампликона, содержащего последовательности прямого и обратного праймера. Данные о последовательности могут быть получены с применением любого выбранного метода секвенирования. Рид образца может быть получен, например, в результате реакции секвенирования путем синтеза (SBS), реакции секвенирования путем лигирования или любого другого подходящего метода секвенирования, для которого требуется определение длины и/или идентичности повторяющегося элемента. Рид образца может представлять собой консенсусную (например, усредненную или взвешенную) последовательность, полученную из нескольких ридов образца. В некоторых вариантах реализации получение референсной последовательности включает идентификацию представляющего интерес локуса на основании последовательности праймера из ПЦР-ампликона.
[00279] Термин “необработанный фрагмент” относится к данным о последовательности части представляющей интерес геномной последовательности, которая по меньшей мере частично перекрывает заданное положение или представляющее интерес вторичное положение в риде образца или фрагменте образца. Неограничивающие примеры необработанных фрагментов включают дуплексный фрагмент со сшивкой, симплексный фрагмент со сшивкой, дуплексный фрагмент без сшивки и симплексный фрагмент без сшивки. Термин “необработанный” используют, чтобы показать, что необработанный фрагмент включает данные о последовательности, определенным образом связанные с данными о последовательности в риде образца, независимо от того, демонстрирует ли необработанный фрагмент подтверждающий вариант, который соответствует и удостоверяет или подтверждает потенциальный вариант в риде образца. Термин “необработанный фрагмент” не указывает на то, что указанный фрагмент обязательно включает подтверждающий вариант, валидирующий распознанный вариант в риде образца. Например, если приложением для распознавания вариантов определено, что рид образца демонстрирует первый вариант, указанное приложение для распознавания вариантов может определить, что в одном или более необработанных фрагментах отсутствует соответствующий тип “подтверждающего” варианта, наличие которого в ином случае можно ожидать на основании варианта в риде образца.
[00280] Термины “картирование”, “выравненный”, “выравнивание” относятся к процессу сравнения рида или метки с референсной последовательностью, с определением таким образом того, содержит ли указанная референсная последовательность содержит последовательность рида. Если референсная последовательность содержит рид, указанный рид может быть картирован на указанную референсную последовательность или, в определенных вариантах реализации, на конкретное место в референсной последовательности. В некоторых случаях выравнивание просто показывает, входит ли рид в состав конкретной референсной последовательности (т.е. присутствует или отсутствует указанный рид в референсной последовательности). Например, выравнивание рида на референсную последовательность хромосомы 13 человека показывает, присутствует ли указанный рид в указанной референсной последовательности хромосомы 13. Инструмент, который обеспечивает получение указанной информации, может называться тестировщиком принадлежности множеству. В некоторых случаях выравнивание, кроме того, указывает на место в референсной последовательности, куда картируется рид или метка. Например, если референсная последовательность представляет собой полную последовательность генома человека, выравнивание может показать, что рид присутствует на хромосоме 13, и может дополнительно показать, что рид располагается в конкретной цепи и/или сайте хромосомы 13.
[00281] Термин “индел” относится к инсерции и/или делеции оснований в ДНК организма. Микроиндел представляет собой индел, который приводит к чистому изменению 1-50 нуклеотидов. В кодирующих областях генома, за исключением случаев, когда длина индела кратна 3, он дает мутацию со сдвигом рамки. Инделы могут быть противопоставлены точечным мутациям. Индел инсертирует и делетирует нуклеотиды в последовательности, тогда как точечная мутация представляет собой форму замены, при которой один из нуклеотидов заменяют без изменения общего числа в ДНК. Инделы могут также быть противопоставлены тандемной мутации оснований (TBM), которая может быть определена как замена нуклеотидов в смежных положениях (“вариант” относится к последовательности нуклеиновой кислоты, отличающейся от референсной нуклеиновой кислоты).
[00282] Термин “variant” относится к последовательности нуклеиновой кислоты, которая отличается от референсной последовательности нуклеиновой кислоты. Типичный вариант последовательности нуклеиновой кислоты включает, без ограничения, однонуклеотидный полиморфизм (SNP), короткие делеционные и инсерционные полиморфизмы (индел), вариацию числа копий (CNV), микросателлитные маркеры или короткие тандемные повторы, и структурную вариацию. Распознавание соматических вариантов представляет собой попытку идентификации вариантов, присутствующих в образце ДНК с низкой частотой. Распознавание соматических вариантов представляет интерес в контексте лечения рака. Образец ДНК из опухоли обычно являются гетерогенным и включает некоторое число нормальных клеток, некоторое число клеток ранней стадии прогрессирования рака (с меньшим количеством мутаций) и некоторое число клеток поздней стадии (с большим количеством мутаций). Из-за указанной гетерогенности при секвенировании опухоли (например, из фиксированного формалином и залитого в парафин (FFPE) образца) соматические мутации часто появляется с низкой частотой. Например, однонуклеотидная вариация (SNV) наблюдается только в 10% ридов, захватывающих заданное основание. Вариант, который подлежит классификации как относящийся к соматической или зародышевой линии классификатором вариантов, также называется в настоящем документе “тестируемым вариантом”.
[00283] Термин “шум” относится к ошибочно распознанному варианту, полученному в результате одной или более ошибок в процессе секвенирования и/или в приложении для распознавания вариантов.
[00284] Термин “частота варианта” относится к относительной частоте аллеля (варианта гена) в конкретном локусе в популяции, выраженной в виде доли или процента. Например, указанные доля или процент могут быть представлены долей всех хромосом в популяции, несущих указанный аллель. Например, частота варианта в образце представляет собой относительную частоту аллеля/варианта в конкретном локусе/положении вдоль представляющей интерес геномной последовательности в “популяции”, соответствующей числу ридов и/или образцов, полученных для указанной представляющей интерес геномной последовательности от индивидуума. В другом примере исходная частота варианта представляет собой относительную частоту аллеля/варианта в конкретном локусе/положении вдоль одной или более исходных геномных последовательностей, где “популяция” соответствует числу ридов и/или образцов, полученных для одной или более исходных геномных последовательностей из популяции здоровых индивидуумов.
[00285] Термин “частота варианта аллеля (VAF)” относится к наблюдаемому проценту секвенированных ридов, совпадающих с указанным вариантом, разделенному на общее покрытие в целевом положении. VAF представляет собой показатель пропорции секвенированных ридов, несущих указанный вариант.
[00286] Термины положение”, “заданное положение” и “локус” относятся к месту или координатам одного или более нуклеотидов в составе последовательности нуклеотидов. Термины “положение”, “заданное положение” и “локус” также относятся к месту или координатам одной или более пар оснований в последовательности нуклеотидов.
[00287] Термин “гаплотип” относится к комбинации аллелей в смежных сайтах на хромосоме, наследуемых вместе. Гаплотип может быть представлен одним локусом, несколькими локусами или всей хромосомой в зависимости от числа событий рекомбинации, произошедших между локусами в определенном наборе локусов, если они вообще происходили.
[00288] Термин “порог” в настоящем документе относится к численному или не-численному значению, которое применяют в качестве значения отсечения для характеризации образца, нуклеиновой кислоты или их части (например, рида). Порог может варьировать на основании результатов эмпирического анализа. Порог можно сравнивать с измеренным или рассчитанным значением для определения того, должен ли источник таких предполагаемых значений быть классифицирован конкретным образом. Выбор порога зависит от уровня доверительности, с которым пользователь желает получить при осуществлении классификации. Порог может быть выбран с конкретной целью (например, для достижения баланса чувствительности и селективности). В настоящем документе порог” указывает на точку, в которой ход анализа может быть изменен, и/или точку, в которой может быть запущено действие. Порог не обязательно должен представлять собой заранее заданное число. Вместо этого порог может представлять собой, например, функцию, основанную на множестве факторов. Порог может быть адаптивно регулируемым с учетом обстоятельств. Кроме того, порог может задавать верхний предел, нижний предел или диапазон между пределами.
[00289] В некоторых вариантах реализации меру или оценку (балл, score), основанная(ый) на данных секвенирования, можно сравнивать с порогом. В настоящем документе термины “мера” или “оценка” могут включать значения или результаты, определенные исходя из данных секвенирования, или могут включать функции, основанные на значениях или результатах, определенных исходя из данных секвенирования. Как и порог, мера или оценка могут быть адаптивно регулироваться с учетом обстоятельств. Например, метрика или оценка может представлять собой нормированное значение. В качестве примера оценки или меры один или более вариантов реализации может задействовать показатели подсчитанных количеств при анализе данных. оценка подсчитанного количества может быть основан на числе ридов образца. Счетная оценка подсчитанного количества может быть основана на числе ридов образца. Риды образца могут быть подвергнуты одной или более стадий фильтрации, таким образом, чтобы они обладали по меньшей мере одной общей характеристикой или одним общим качеством. Например, каждый из ридов образца, который используют для определения оценки подсчитанного количества, может быть выравнен по референсной последовательности или может быть определен как потенциальный аллель. Может быть подсчитано число ридов образца, обладающих общей характеристикой, для определения подсчитанного количества ридов. Счетные оценки могут быть основаны на подсчитанном количестве ридов. В некоторых вариантах реализации счетная оценка может представлять собой значение, равное подсчитанному количеству ридов. Согласно другим вариантам реализации счетная оценка может быть основана на подсчитанном количестве ридов и другой информации. Например, счетная оценка может быть основана на подсчитанном количестве ридов для конкретного аллеля генетического локуса и общего числа ридов для генетического локуса. В некоторых вариантах реализации счетные оценки могут быть основаны на подсчитанном количестве ридов и ранее полученных данных для генетического локуса. В некоторых вариантах реализации счетные оценки могут представлять собой нормированные показатели между заранее заданными значениями. Счетная оценка может также представлять собой функцию от подсчитанных количеств ридов из других локусов образца или функцию от подсчитанных количеств ридов из других образцов, которые анализировали одновременно с представляющим интерес образцом. Например, счетная оценка может представлять собой функцию от подсчитанного количества ридов конкретного аллеля и подсчитанных количеств ридов других локусов в образце, и/или подсчитанных количества ридов из других образцов. В одном примере подсчитанные количества ридов из других локусов и/или подсчитанные количества ридов из других образцов могут быть использованы для нормирования оценки подсчитанного количества для конкретного аллеля.
[00290] Термины “покрытие” или “покрытие фрагмента” относятся к подсчитанному количеству или другой мере ряда ридов образца для одного и того же фрагмента последовательности. Подсчитанное количество ридов может представлять собой подсчитанное количество ридов, покрывающих соответствующий фрагмент. Как вариант, покрытие может быть определено путем умножения подсчитанного количества ридов на заданный коэффициент, основанный на ретроспективной информации, информации об образце, информации о локусе и т.п.
[00291] Термин “глубина считывания” (обычно в виде числа с последующим символом “×”) относится к числу секвенированных ридов, перекрывающихся при выравнивании в целевом положении. Его часто выражают через среднее значение или процент, превышающий значение отсечения на протяжении множества интервалов (таких как экзоны, гены или панели). Например, в клиническом заключении может быть сказано, что среднее покрытие панели составляет 1,105× при 98% покрытии целевых оснований >100×.
[00292] Термины “оценка качества распознавания оснований” или “оценка Q” относятся к вероятности по шкале PHRED в диапазоне от 0-20, обратно пропорциональной вероятности того, что отдельное секвенированное основание является корректным. Например, распознанное основание T с Q, равным 20, считают вероятно корректным с достоверностью, соответствующей P-значению 0,01. Любые распознанные основания с Q<20 должны считаться результатами низкого качества, и любой идентифицированный вариант с существенной пропорцией имеющих низкое качество секвенированных ридов, подтверждающих указанный вариант, должен считаться потенциально ложноположительным.
[00293] Термины “риды вариантов” или “число ридов вариантов” относятся к числу секвенированных ридов, свидетельствующих о присутствии указанного варианта.
Процесс секвенирования
[00294] Варианты реализации, представленные в данном документе, могут быть применимы к анализу последовательностей нуклеиновых кислот для идентификации вариаций последовательностей. Варианты реализации могут применяться для анализа потенциальных вариантов / аллелей генетического положения / локуса и определения генотипа генетического локуса или, другими словами, обеспечения распознавания генотипа для локуса. В качестве примера, последовательности нуклеиновой кислоты могут быть проанализированы в соответствии со способами и системами, описанными в публикации заявки на патент США № 2016/0085910 и публикации заявки на патент США № 2013/0296175, полное содержащие которых в явном виде включено в настоящий документ в полном объеме посредством ссылки.
[00295] В одном варианте реализации процесс секвенирования включает получение образца, который содержит или предположительно содержит нуклеиновые кислоты, такие как ДНК. Образец может быть из известного или неизвестного источника, такого как животное (например, человек), растение, бактерии или гриб. Образец может быть взят непосредственно из источника. Например, кровь или слюна могут быть взяты непосредственно от индивидуума. Как вариант, образец может не быть получен непосредственно из источника. Затем один или более процессоров дают системе команду на подготовку образца к секвенированию. Подготовка может включать удаление постороннего материала и / или выделение определенного материала (например, ДНК). Биологический образец может быть подготовлен для включения признаков для конкретного анализа. Например, биологический образец может быть подготовлен для секвенирования путем синтеза (SBS). В некоторых вариантах реализации подготовка может включать амплификацию определенных областей генома. Например, подготовка может включать амплификацию заранее определенных генетических локусов, которые, как известно, включают STR (короткие тандемные повторы)и / или SNP (однонуклеотидные полиморфизмы). Генетические локусы могут быть амплифицированы с использованием предварительно определенных последовательностей праймеров.
[00296] Затем, указанные один или более процессоров передают системе инструкцию секвенировать образец. Секвенирование может осуществляться в соответствии с различными известными протоколами секвенирования. В частных вариантах реализации секвенирование включает SBS. В SBS множество флуоресцентно меченых нуклеотидов используется для последовательности множества кластеров амплифицированной ДНК (возможно, миллионов кластеров), присутствующих на поверхности оптического субстрата (например, поверхности, которая по меньшей мере частично ограничивает канал в проточной ячейке). Проточные ячейки могут содержать образцы нуклеиновых кислот для секвенирования, причем проточные ячейки размещены в соответствующих держателях проточных ячеек.
[00297] Нуклеиновые кислоты могут быть подготовлены таким образом, чтобы они содержали известную последовательность праймера, которая соседствует с неизвестной целевой последовательностью. Чтобы инициировать первый цикл секвенирования SBS, один или несколько нуклеотидов, меченных различным образом, ДНК-полимеразу и т. Д., можно подать в проточную ячейку или через нее посредством подсистемы потока жидкости. Можно добавлять либо по одному типу нуклеотида, либо нуклеотиды, используемые в процедуре секвенирования, могут быть специально сконструированы так, чтобы обладать свойством обратимой терминации, что дает возможность одновременного проведения каждого цикла реакции секвенирования в присутствии нескольких типов меченых нуклеотидов (например, A, C, T, G). Нуклеотиды могут включать обнаруживаемые фрагменты-метки, такие как флуорофоры. Когда четыре нуклеотида смешаны вместе, полимераза может выбрать правильное основание для включения, и каждая последовательность удлиняется на одно основание. Невключенные нуклеотиды можно отмывать потоком промывочного раствора через проточную ячейку. Один или несколько лазеров могут возбуждать нуклеиновые кислоты и вызывать флуоресценцию. Флуоресценция, испускаемая нуклеиновыми кислотами, основана на флуорофорах включенного основания, и разные флуорофоры могут излучать света с разными длинами волн. Деблокирующий реагент может быть добавлен в проточную ячейку для удаления обратимых терминаторных групп из удлиненных и детектированных цепей ДНК. Деблокирующий реагент затем можно отмыть, пропуская промывочный раствор через проточную ячейку. После этого проточная ячейка готова к следующему циклу секвенирования, начиная с введения меченого нуклеотида, как описано выше. Операции с текучей средой и обнаружением могут повторяться несколько раз для завершения последовательности операций. Примеры способов секвенирования описаны, например, в Bentley et al., Nature 456: 53-59 (2008), международной публикации № WO 04/018497, патенте США № 7,057,026, международной публикации № WO 91/06678, международной публикации № WO 07/123744, патенте США № 7,329,492, патенте США № 7,211,414, патенте США № 7,315,019, патенте США № 7,405,281и публикации заявки на патент США № 2008/0108082, каждый (ая) из которых включен(а) в настоящий документ посредством ссылки.
[00298] В некоторых проявлениях реализации нуклеиновые кислоты могут быть присоединены к поверхности и амплифицированы до или во время секвенирования. Например, амплификация может быть проведена с использованием мостиковой амплификации с образованием кластеров нуклеиновых кислот на поверхности. Применимые методы амплификации описаны, например, в Патенте США № 5,641,658, патентной публикации США № 2002/0055100, патенте США № 7,115,400, патентной публикации США № 2004/0096853, патентной публикации США № 2004/0002090, патентной публикации США № 2007/0128624и публикации заявки на патент США № 2008/0009420, каждый из этих документов полностью включен в настоящую заявку посредством ссылки. Другим полезным способом амплификации нуклеиновых кислот на поверхности является амплификация по типу катящегося кольца (RCA), например, как описано в Lizardi et al., Nat. Genet. 19:225-232 (1998) и публикации заявки на патент США № 2007/0099208 A1, каждый (ая) из которых включен(а) в настоящий документ посредством ссылки.
[00299] Один пример протокола SBS использует модифицированные нуклеотиды, имеющие удаляемые 3’-блоки, например, как описано в международной публикации № WO 04/018497, публикации заявки на патент США № 2007/0166705A1 и патенте США № 7057026, каждый (ая) из которых включен(а) в настоящий документ посредством ссылки. Например, реагенты SBS могут доставляться повторяющимися циклами в проточную ячейку, к которой присоединены целевые нуклеиновые кислоты, например, по протоколу мостиковой амплификации. Кластеры нуклеиновых кислот могут быть преобразованы в одноцепочечную форму с использованием линеаризирующего раствора. Линеаризирующий раствор может содержать, например, эндонуклеазу рестрикции, способную расщеплять одну цепь каждого кластера. Линеаризирующий раствор может содержать, например, эндонуклеазу рестрикции, способную расщеплять одну цепь каждого кластера (например, расщепление диольной связи периодатом), расщепление сайтов без оснований путем расщепления эндонуклеазой (например, “USER”, которая поставляется компанией NEB, Ипсвич, штат Массачусетс, США, номер компонента (M5505S), (путем воздействия тепла или щелочи, расщепления рибонуклеотидов, включенных в продукты амплификации, в остальном состоящих из дезоксирибонуклеотидов, фотохимического расщепления или расщепления пептидного линкера. После операции линеаризации праймер для секвенирования может быть подан в проточную ячейку в условиях гибридизации праймера для секвенирования с целевыми нуклеиновыми кислотами, которые должны быть секвенированы.
[00300] Затем проточную клетку можно привести в контакт с реагентом-удлинителем SBS, имеющим модифицированные нуклеотиды с удаляемыми 3’-блоками и флуоресцентными метками в условиях, позволяющих удлинить праймер, гибридизованный с каждой целевой нуклеиновой кислотой путем добавления одного нуклеотида. К каждому праймеру добавляется только один нуклеотид, поскольку включение модицифированного нуклеотида в растущую полинуклеотидную , комплементарную секвенируемой области матрицы, обуславливает отсутствие свободной группы 3’-ОН, доступной для направления дальнейшего удлинения последовательности и, следовательно, полимераза. не может добавить дополнительные нуклеотиды. Удлиняющий реагент SBS можно удалить и заменить сканирующим реагентом, содержащим компоненты, которые защищают образец при возбуждении излучением. Примеры компонентов сканирующего реагента описаны в публикации заявки на патент США № 2008/0280773 А1 и заявке на патент США № 13/018,255, каждый (ая) из которых включен(а) в настоящий документ посредством ссылки. Затем удлиненные нуклеиновые кислоты могут быть детектированы флуоресцентно в присутствии сканирующего реагента. После детектирования флуоресценции 3’-блок может быть удален с использованием деблокирующего реагента, который соответствует используемой блокирующей группе. Примеры деблокирующих реагентов, которые можно применять для соответствующих блокирующих групп, описаны в WO 004018497, US 2007 / 0166705A1 и патенте США № 7057026, каждый (ая) из которых включен(а) в настоящий документ посредством ссылки. Деблокирующий реагент можно смыть, оставляя целевые нуклеиновые кислоты гибридизованными с удлиненными праймерами, имеющими 3’-ОН-группы, к которым теперь можно присоединять другие нуклеотиды. Соответственно, циклы добавления удлиняющего реагента, сканирующего реагента и деблокирующего реагента с необязательными промываниями между одной или несколькими операциями могут повторяться до тех пор, пока не будет получена необходимая последовательность. Вышеуказанные циклы могут быть выполнены с использованием одной операции доставки удлиняющего реагента на цикл, когда к каждому из модифицированных нуклеотидов прикреплена отличная от других метка, о которой известно, что она соответствует конкретному основанию. Различные метки облегчают различение нуклеотидов, добавляемых во время каждой операции включения. В качестве альтернативы, каждый цикл может включать в себя отдельные операции доставки удлиняющего реагента, за которыми следуют отдельные операции доставки и детектирования сканирующего реагента, и в этом случае два или более нуклеотида могут иметь одинаковую метку и могут различаться на основании известного порядка доставки.
[00301] Хотя операция секвенирования обсуждалась выше в отношении конкретного протокола SBS, следует понимать, что при желании могут выполнятся другие протоколы для секвенирования любого из множества других молекулярных анализов.
[00302] Затем указанные один или более процессоров системы получают данные секвенирования для последующего анализа. Данные секвенирования могут быть отформатированы различными способами, например, в файле .BAM. Данные секвенирования могут включать в себя, например, несколько ридов образцов. Данные секвенирования могут включать в себя множество ридов образцов, которые имеют соответствующие нуклеотидные последовательности образцов. Хотя обсуждается только один рид образца, следует понимать, что данные последовательности могут включать, например, сотни, тысячи, сотни тысяч или миллионы ридов образцов. Различные риды образцов могут содержать различное число нуклеотидов. Например, риды образцов может варьировать от 10 нуклеотидов до 500 нуклеотидов или более. Риды образцов могут охватывать весь геном источника (ов). В качестве одного примера, риды образцов направлены на заранее определенные генетические локусы, такие как генетические локусы, которые имеют подозрительные STR или предполагаемые SNP.
[00303] Каждый рид образца может включать последовательность нуклеотидов, которая может называться последовательностью образца, фрагментом образца или целевой последовательностью. Последовательность образца может включать, например, последовательности праймеров, фланкирующие последовательности и целевую последовательность. Количество нуклеотидов в последовательности образца может включать 30, 40, 50, 60, 70, 80, 90, 100 или более. В некоторых вариантах реализации один или более ридов образцов (или последовательностей образцов) включают по меньшей мере 150 нуклеотидов, 200 нуклеотидов, 300 нуклеотидов, 400 нуклеотидов, 500 нуклеотидов или более. В некоторых вариантах реализации риды образцов могут включать более 1000 нуклеотидов, 2000 нуклеотидов или более. Риды образцов (или последовательности образцов) могут включать последовательности праймеров на одном или обоих концах.
[00304] Затем, указанные один или более процессоров анализируют данные секвенирования, чтобы получить потенциальные распознавание (ия) варианта (ов) образца и частоту варианта образца для указанных распознавания (ий) варианта (ов) образца. Эта операция также может называться приложением распознавания вариантов или распознавателем (определителем) вариантом. Таким образом, распознаватель вариантов идентифицирует или обнаруживает варианты, а классификатор вариантов классифицирует обнаруженные варианты как соматические или зародышевые. Могут применяться альтернативные распознаватели вариантов в соответствии с приведенным в настоящем документе вариантами реализации, причем могут применяться различные распознаватели вариантов в зависимости от типа выполняемой операции упорядочения, на основе характеристик образца, которые представляют интерес, и т.п. Одним из неограничивающих вариантов такого приложения для распознавания вариантов является приложение Pisces™ от компании Illumina Inc., (San Diego, CA, США), размещенное по адресу и https://github.com/Illumina/Pisces и описанное в статье Dunn, Tamsen & Berry, Gwenn & Emig-Agius, Dorothea & Jiang, Yu & Iyer, Anita & Udar, Nitin & Strömberg, Michael. (2017). Pisces: An Accurate and Versatile Single Sample Somatic and Germline Variant Caller. 595-595. 10.1145/3107411.3108203, полное содержание которой в явном виде полностью включено в настоящий документ посредством ссылки.
[00305] Такое приложение для распознавание вариантов содержит четыре выполняемых последовательно модуля:
[00306] ((1) Pisces Read Stitcher (сшиватель ридов Pisces): снижает шум путем сшивания парных ридов в BAM (рида один и рида два одной молекулы) в консенсусные. На выходе сшитый BAM.
[00307] (2) Pisces Variant Caller (определитель вариантов Pisces): определяет небольшие SNV, вставки (инсерции) и делеции. Pisces включают в себя алгоритм свертки вариантов для объединения вариантов, разбитых по границам ридов, основные алгоритмы фильтрации и простой алгоритм оценки достоверности вариантов на основе пуассоновского процесса. На выходе - VCF.
[00308] (3) Pisces Variant Quality Recalibrator (Рекалибратор качества вариантов Pisces, VQR): В случае, если определения (вызовы) вариантов в подавляющем большинстве случаев следуют некоторому паттерну, связанному с термическим повреждением или дезаминированием FFPE, шаг VQR будет понижать оценку Q варианта для подозрительных определений (вызовов). На выходе- откорректированный VCF.
[00309] (4) Pisces Variant Phaser (Фазировщик фариантов Pisces -Scylla): использует жадный метод кластеризации на основе ридов для сборки небольших вариантов в сложные аллели из клональных субпопуляций. Это позволяет более точно определять функциональные последствия последующими инструментами. На выходе- откорректированный VCF.
[00310] В качестве дополнения или альтернативы для этой операции можно применять приложение для определения вариантов Strelka™ , от компании Illumina Inc., размещенное по адресу https://github.com/Illumina/strelka и описанное в статье T Saunders, Christopher & Wong, Wendy & Swamy, Sajani & Becq, Jennifer & J Murray, Lisa & Cheetham, Keira. (2012). Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics (Oxford, Англия). 28. 1811-7. 10.1093/bioinformatics/bts271, полное содержание которой в явном виде полностью включено в настоящий документ посредством ссылки. Далее, в качестве дополнения или альтернативы, для этой операции можно применять приложение Strelka2™ , от компании Illumina Inc., размещенное по адресу https://github.com/Illumina/strelka и описанное в статье Kim, S., Scheffler, K., Halpern, A.L., Bekritsky, M.A., Noh, E., Källberg, M., Chen, X., Beyter, D., Krusche, P., and Saunders, C.T. (2017). Strelka2: Fast and accurate variant calling for clinical sequencing applications, полное содержание которой в явном виде полностью включено в настоящий документ посредством ссылки. Более того, в качестве дополнения или альтернативы, для этой операции можно применять инструмент для аннотации/определения вариантов, такой как Nirvana™ , от компании Illumina Inc., размещенное по адресу https://github.com/Illumina/Nirvana/wiki и описанная в статье Stromberg, Michael & Roy, Rajat & Lajugie, Julien & Jiang, Yu & Li, Haochen & Margulies, Elliott. (2017). Nirvana: Clinical Grade Variant Annotator. 596-596. 10.1145/3107411.3108204, полное содержание которой в явном виде полностью включено в настоящий документ посредством ссылки.
[00311] Такой инструмент для аннотации/определения вариантов может применять различные алгоритмические методики, такие как описанные в Nirvana:
[00312] a. Идентификация всех перекрывающихся транскриптов с помощью массива интервалов: для функциональной аннотации мы можем идентифицировать все транскрипты, перекрывающие вариант, и можно применять дерево интервалов. Однако, поскольку набор (множество) интервалов может быть статическим, мы смогли дополнительно оптимизировать его в Массив Интервалов. Дерево интервалов возвращает все перекрывающиеся транскрипты за время O (min (n, k lg n)), где где n - количество интервалов в дереве, а k - количество перекрывающихся интервалов. На практике, поскольку k на самом деле мало по сравнению с n для большинства вариантов, эффективное время выполнения на дереве интервалов будет O (k lg n). Мы улучшили до O (lg n + k) за счет создания массива интервалов, в котором все интервалы хранятся в отсортированном массиве, так что нам нужно только найти первый перекрывающийся интервал, а затем пронумеровать оставшиеся (k-1).
[00313] b. CNV / SV (Yu): могут быть предоставлены аннотации для вариаций количества копий (CNV) и структурных вариантов (SV). Аналогично аннотациям небольших вариантов, транскрипты, перекрывающиеся с SV, а также ранее определенные структурные варианты могут быть аннотированы в онлайн-базах данных. В отличие от небольших вариантов, не обязательно все перекрывающиеся транскрипты аннотировать, Поскольку слишком много транскриптов будут перекрываться с большими SV. Вместо этого могут быть аннотированы все перекрывающиеся транскрипты, относящиеся к частичному перекрывающемуся гену. В частности, для этих транскриптов могут выявляться (включаться в отчет) затронутые интроны, экзоны и последствия, обусловленные структурными вариантами. Доступна опция, позволяющая выводить все перекрывающиеся транскрипты, но может быть представлена основная информация для этих транскриптов, такая как символ гена, отметка, является ли это каноническим перекрыванием или частичным перекрыванием с транскриптами. Для каждого SV / CNV также интересно знать, были ли изучены эти варианты и их частота в разных популяциях. Соответственно, мы регистрировали перекрывающиеся SV во внешних базах данных, таких как “1000 геномов”, DGV и ClinGen. Чтобы избежать применения произвольного отсечения для определения того, какой SV перекрывается, вместо этого можно применять все перекрывающиеся транскрипты и вычислять взаимное перекрывание, вместо этого можно применять все перекрывающиеся транскрипты и вычислять взаимное то есть длину перекрывания, деленную на минимум длины этих двух SV.
[00314] c. Регистрация дополнительных аннотаций: Дополнительные аннотации бывают двух типов: малые и структурные варианты (SV). SV можно моделировать как интервалы и использовать массив интервалов, описанный выше, для идентификации перекрывающихся SV. Небольшие варианты моделируются в виде точек и сопоставляются по положению и (необязательно) аллелю. Соответственно, их ищут с применением алгоритма, подобного бинарному поиску. Поскольку база данных дополнительных аннотаций может быть довольно большой, создают гораздо меньший индекс для картирования хромосомных положений на местоположения файлов, в которых находится дополнительная аннотация. Индекс - это отсортированный массив объектов (состоящих из хромосомного положения и расположения файла), по которым можно выполнять двоичный поиск с использованием положения. Чтобы размер индекса оставался небольшим, множество положений (до определенного максимального числа) сжимают в один объект, который хранит значения для первого положения и только дельты для последующих положений. Поскольку мы используем двоичный поиск, время выполнения - O (lg n), где n - количество элементов в базе данных.
[00315] d. Кэш-файлы VEP
[00316] e. База данных транскриптов: Файлы Transcript Cache (кэш транскриптов, кэш) и Supplementary database (дополнительная база данных, SAdb) представляют собой упорядоченное хранилище объектов данных, таких как транскрипты и дополнительные аннотации. Мы применяем кэш Ensembl VEP cache в качестве источника данных для кэша. Для создания кэша все транскрипты помещают в массив интервалов, а конечное состояние массива сохраняется в файлах кэша. Таким образом, в процессе аннотации нам нужно только загрузить предварительно вычисленный массив интервалов и выполнить поиск по нему. Поскольку кэш загружается в память, а поиск выполняется очень быстро (описано выше), поиск перекрывающихся транскриптов согласно Nirvana выполняется очень быстро (профилировано менее 1% от общего времени выполнения?).
[00317] f. Дополнительная база данных: источники данных для SAdb перечислены в дополнительных материалах. База данных SAdb для небольших вариантов создается путем k-направленного объединения всех источников данных, так что каждый объект в базе данных (идентифицируемый ссылочным именем и положением) содержит все соответствующие дополнительные аннотации. Проблемы, возникающие при парсировании файлов - источников данных, подробно описаны на домашней странице Nirvana. Чтобы ограничить использование памяти, в память загружается только индекс SA. Этот индекс позволяет осуществить быстрый поиск положения файла для дополнительной аннотации. Однако, поскольку данные должны быть извлечены с диска, добавление дополнительных аннотаций было определено как самое узкое место Nirvana (профилируется примерно как 30% от общего времени выполнения).
[00318] g. Последствия и онтология последовательности: Последствия и онтология последовательности. Иногда у нас была возможность выявить проблемы в текущей SO и сотрудничать с командой SO, чтобы улучшить состояние аннотации.
[00319] Такой инструмент вариантов аннотации может включать предварительную обработку. Например, Nirvana включала большое количество аннотаций из внешних источников данных, таких как ExAC, EVS, проект “1000 геномов”, dbSNP, ClinVar, Cosmic, DGV и ClinGen. Чтобы в полной мере использовать эти базы данных, мы должны очистить информацию из них. Мы реализовали разные стратегии для решения разных конфликтов, обусловленных разными источниками данных. Например, в случае нескольких записей dbSNP для одного и того же положения и другого аллеля, мы объединяем все идентификаторы в список идентификаторов, разделенных запятыми; если есть несколько записей с разными значениями CAF для одного и того же аллеля, мы используем первое значение CAF. Для конфликтующих записей ExAC и EVS мы учитываем количество образцов и используем запись с большим количеством образцов. В проекте “1000 геномов” мы удаляли частоту аллеля конфликтующего аллеля. Другая проблема - неточная информация. В основном мы брали информацию о частотах аллелей из проекта “1000 геномов”, однако мы заметили, что для GRCh38 частота аллелей, указанная в информационном поле, не исключала образцы с недоступным генотипом, что приводило к повышенным частотам для вариантов, которые доступны не для всех образцов. Чтобы гарантировать точность нашей аннотации, мы используем все генотипы индивидуального уровня для вычисления истинных частот аллелей. Как мы знаем, одни и те же варианты могут иметь разные представления на основе разных выравниваний. Чтобы быть уверенным, что мы можем точно получить (вывести) информацию об уже идентифицированных вариантах, мы должны предварительно обработать варианты из разных ресурсов, чтобы они имели единообразное представление. Для всех внешних источников данных мы удалили аллели, чтобы удалить дублированные нуклеотиды как в референсном аллеле, так и в альтернативном аллелях. Для ClinVar мы непосредственно парсировали xml-файл и выполнили пятизначное выравнивание для всех вариантов, которое часто используется в vcf-файле. Различные базы данных могут содержать одинаковый набор информации. Чтобы избежать ненужного дублирования, мы удалили часть повторяющейся информации. Чтобы избежать ненужного дублирования, мы удалили часть повторяющейся информации.
[00320] В соответствии с по меньшей мере некоторыми вариантами реализации, указанное приложение для определения вариантов выдает варианты с низкой частотой, определение зародышевой линии и т.п. В качестве неограничивающего примера, указанное приложение для определения вариантов может работать только с опухолевыми образцами и/или с парными образцами опухоль-норма. Приложение для определения вариантов может искать однонуклеотидные варианты(SNV), многонуклеотидные варианты (MNV), инделы и т.п.. Приложение определения вариантов идентифицирует варианты, одновременно фильтруя несоответствия из-за ошибок секвенирования или подготовки образца. Для каждого варианта определитель вариантов идентифицирует референсную последовательность, положение варианта и потенциальную последовательность (и) варианта (например, SNV от A до C или делеция из AG в A). Приложение определения вариантов идентифицирует последовательность образца (или фрагмент образца), референсную последовательность / фрагмент и определение варианта как показатель присутствия варианта. Приложение определения вариантов может идентифицировать необработанные фрагменты и выводить обозначение исходных фрагментов, подсчет числа необработанных фрагментов, которые верифицируют возможное определение варианта, положение в исходном фрагменте, в котором присутствует подтверждающий вариант, и другую важную информацию. Неограничивающие примеры необработанных фрагментов включают дуплексных сшитый фрагмент, симплекснный сшитый фрагмент, дуплексный несшитый фрагмент и симплексный несшитый фрагмент.
[00321] Приложение для определения вариантов может выводить определения (вызовы) в различных форматах, например, в файл .VCF или .GVCF. Только в качестве примера указанное приложение для определения вариантов может быть включено в пайплайн MiSeqReporter (например, когда оно реализовано в секвенаторе MiSeq®). При желании приложение может быть реализовано с различными рабочими процессами. Анализ может включать единый протокол или комбинацию протоколов, которые анализируют риды образца определенным образом для получения желаемой информации.
[00322] Затем указанные один или более процессоров осуществляют операцию валидации применительно к определению потенциальных вариантов. Операция валидации может быть основана на оценке качества и / или иерархии многоуровневых тестов, как объясняется ниже. Когда операция валидации (проверки) аутентифицирует или проверяет наличие потенциального определения варианта, операция проверки передает информацию об определенном варианте (из указанного приложения для определения вариантов) в генератор отчетов по образцам. В качестве альтернативы, когда операция проверки делает недействительным или дисквалифицирует потенциальное определение варианта, операция проверки передает соответствующий индикатор (например, отрицательный индикатор, индикатор отсутствия определения, индикатор недействительного определения) генератору отчетов по образцам. Операция проверки также может передавать оценку достоверности, связанную со степенью уверенности в том, что конкретное определение варианта правильно или определение варианта правильно обозначено как недействительное (невалидное).
[00323] Затем, указанные один или более процессоров генерируют и сохраняют отчет по образцу. Отчет по образцу может включать, например, информацию о множестве генетических локусов по отношению к образцу. Например, для каждого генетического локуса заранее определенного набора генетических локусов отчет по образцу может по меньшей мере одно из: определить генотип; указывать, что определение генотипа невозможно; предоставить оценку достоверности определения генотипа; или указать потенциальные проблемы с анализом в отношении одного или нескольких генетических локусов. В отчете по образцу также может быть указан пол человека, предоставившего образец, и / или указано, что образец включает несколько источников. В настоящем документе “отчет по образцу” (“отчет об образце”) может включать цифровые данные (например, файл данных) генетического локуса или заранее определенного набора генетических локусов и / или печатный отчет о генетическом локусе или наборе генетических локусов. Таким образом, создание или предоставление может включать в себя создание файла данных и / или печать отчета по образцу, или отображение отчета по образцу.
[00324] Отчет по образцу может указывать на то, что определение варианта было установлено, но не было подтверждено. Когда определение варианта определяется как недопустимое, отчет по образцу может указывать дополнительную информацию, касающуюся основания для решения не подтверждать определение варианта. Например, дополнительная информация в отчете может включать описание исходных фрагментов и степень (например, количество), в которой исходные фрагменты поддерживают определение варианта или противоречат ему. Дополнительно или в качестве альтернативы, дополнительная информация в отчете может включать оценку качества, полученную в соответствии с вариантами реализации, описанными в данном документе.
Применение определения вариантов
[00325] Варианты реализации, раскрытые в настоящем документе, включают анализ секвенированных данных для определения потенциальных вариаций. Распознавание вариантов может проводиться над сохраненными данными для выполненной ранее операции секвенирования. В качестве дополнения или альтернативы, его можно проводить в режиме реального времени одновременно с выполнением операции секвенирования. Каждый из ридов образцов ставится в соответствие соответствующим генетическим локусам. Риды образца могут быть поставлены в соответствие определенным генетическим локусам на основании последовательности нуклеотидов рида образца, или, другими словами, порядку нуклеотидов, входящих в рид (например, A, C, G, T). На основании этого анализа рид образца может быть охарактеризован как включающий возможную вариацию/аллель определенного генетического локуса. Рид образца можно собирать (или агрегировать или группировать) вместе с другими ридами образца, охарактеризованными как включающие возможную вариацию/аллель генетического локуса. Под операцией определения соответствия можно также понимать операцию распознавания, в которой рид образца определяется как как возможно ассоциированный с определенным генетическим положением/локусом. Риды образцов можно анализировать с целью локализовать идентифицирующие последовательности (например, последовательности праймеров) нуклеотидов, которые отличают данный рид образца от других ридов образца. Более конкретно, идентифицирующая последовательность(и) может идентифицировать рид образца среди других ридов образцов как ассоциированный с определенным генетическим локусом.
[00326] Операция определения соответствия (присваивания) может включать анализ серии n нуклеотидов идентифицирующей последовательности для определения, соответствует ли серия n нуклеотидов, идентифицирующих последовательности, одной или более выбранным последовательностям. В частных вариантах реализации, операция определения соответствия (присваивания) может включать анализ первых n нуклеотидов последовательности образца для определения, соответствуют ли первые n нуклеотидов последовательности образца одной или более выбранным последовательностям. Число n может принимать разнообразные значения, которые могут быть заложены в программу протокола или вводиться пользователем. Например, число n может быть определено как число нуклеотидов самой короткой выбранной последовательности в базе данных. Это заранее предопределенное число может составлять, например, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 или 30 нуклеотидов. Однако, в других вариантах реализации может применяться меньшее или большее число нуклеотидов. Число n может также быть выбрано человеком, например пользователем системы. Выбор числа n может быть основан на одном или более условиях. Например, число n может быть определено как число нуклеотидов самой короткой последовательности праймера в базе данных, или определенное число, смотря какое из них меньше. В некоторых вариантах реализации a для n может быть использовано минимальное значение, такое как 15, такое что любую последовательность праймера короче 15 нуклеотидов можно считать исключением.
[00327] В некоторых случаях, серия n нуклеотидов идентифицирующей последовательности может не соответствовать точно нуклеотидам последовательности выборки. Тем не менее, идентифицирующая последовательность может эффективно соответствовать последовательности выборки, если идентифицирующая последовательность почти идентична последовательности выборки. Например, рид образца может быть определен для генетического локуса, если серия n нуклеотидов (например, первые n нуклеотидов) идентифицирующей последовательности совпадают с последовательностью выборки с не более чем установленным числом несоответствий (например, 3) и/или установленным числом сдвигов (например, 2). Правила можно установить так, что каждое несоответствие или сдвиг может считаться как различие между ридом образца и последовательностью праймера. Если число различий меньше установленного значения, операция присваивания может быть применена к риду образца для соответствующего генетического локуса (то есть, рид присвоен соответствующему локусу). В некоторых вариантах реализации, вводится оценка совпадения, которая основана на количестве различий между идентифицирующей последовательностью рида образца и последовательностью выборки, ассоциированной с генетическим локусом. Если оценка совпадения превосходит установленный порог совпадения, генетический локус, соответствующий выбранной последовательности, можно считать потенциальным локусом рида образца. В некоторых вариантах реализации, может проводиться последующий анализ с целью определить, действительно ли рид образца соответствует генетическому локусу.
[00328] Если рид образца эффективно совпадает с одной из выбранных последовательностей в базе данных (т.е., в точности совпадает или совпадает в пределах критериев, описанных выше), то риду образца назначают или ставят в соответствие генетический локус, который коррелирует с выбранной последовательностью. Это можно назвать определением локуса или предварительным определением локуса, где рид образца определен для генетического локуса, который коррелирует с выбранной последовательностью. Однако, как описано выше, рид образца может быть определен для более одного генетического локуса В таких вариантах осуществления, может проводиться последующий анализ для определения или присваивания рида образца только одному из потенциальных генетических локусов. В некоторых вариантах реализации рид образца, который сравнивают с базой данных референсных последовательностей, представляет собой первый рид из секвенирования спаренных концов. При осуществлении секвенирования спаренных концов, получают второй рид (представляющий фрагмент необработанных данных) который коррелирует с ридом образца. После присваивания, последующий анализ, который проводится с присвоенными ридами, может быть основан на типе генетического локуса, который был определен для этого рида.
[00329] Затем, риды образца анализируют для идентификации потенциальных вариантов. Среди прочего, результаты этого анализа идентифицируют потенциальный вариант, частоту последовательности варианта, референсную последовательность и положение в исследуемой генетической последовательности, в которой встретился вариант. Например, если известно, что генетический локус включает однонуклеотидные полиморфизмы, то присвоенные риды, которые были определены для генетического локуса можно подвергать дополнительному анализу для идентификации однонуклеотидных полиморфизмов присвоенных ридов. Если известно, что генетический локус включает полиморфные повторяющиеся элементы ДНК, то присвоенные риды можно анализировать для того, чтобы идентифицировать или охарактеризовать полиморфные повторяющиеся элементы ДНК в составе ридов образцов. В некоторых вариантах реализации если присвоенный рид эффективно совпадает с STR-локусом и SNP-локусом, риду образца может быть присвоено предупреждение или флаг. Рид образца может быть определен и как STR-локус и как SNP-локус. Анализ может включать выравнивание присвоенных ридов в соответствии с алгоритмом выравнивания с целью определить последовательности и/или длины присвоенных ридов. Протокол выравнивания может включать метод, описанный в Международной Патентной Заявке № PCT/US2013/030867 (№ публикации WO 2014/142831), поданной 15 марта 2013, которая в полном объеме включена в данную заявку посредством ссылки.
[00330] Затем один или более процессов анализируют необработанный фрагмент с целью определить, существуют ли поддерживающие варианты в соответствующих положениях необработанных фрагментов. Можно идентифицировать различные типы необработанных фрагментов. Например, определитель вариантов может идентифицировать тип необработанного фрагмента, который имеет вариант, валидирующий (подтверждающий) исходно найденный вариант. Например, тип необработанного фрагмента может представлять двунитевый сшитый фрагмент, однонитевый сшитый фрагмент, двунитевый несшитый фрагмент или однонитевой несшитый фрагмент. Опционально можно идентифицировать другие необработанные фрагменты вместо или в дополнение к приведенным примерам. Вместе с идентификацией каждого типа необработанных фрагментов, пользователь также определяет положение в этом фрагменте, в котором встретился поддерживающий вариант, а также число необработанных фрагментов, в которых этот выявили поддерживающий вариант. Например, определитель вариантов может вывести индикацию того, что 10 ридов необработанных фрагментов идентифицированы как представляющие собой двунитевые сшитые фрагменты, содержащие поддерживающий вариант в определенном положении X. Определитель вариантов может также выводить индикацию того, что пять ридов необработанных фрагментов представляют собой однонитевые несшитые фрагменты, имеющие поддерживающий вариант в определенном положении Y. Определитель вариантов может также выводить число необработанных фрагментов, которые соответствуют референсной последовательности, и таким образом не включают поддерживающий вариант, который в ином случае был бы свидетельством поддерживающим определение потенциального варианта в исследуемой генной последовательности.
[00331] Далее, сохраняется число необработанных фрагментов, которые включают поддерживающие варианты, а также положения, в которых встретились поддерживающие варианты. В качестве дополнения или альтернативы, можно сохранять число необработанных фрагментов, которые не включают поддерживающие варианты в представляющем интерес положении (относительно положения определения потенциального вариантов рида образца или фрагмента образца). В качестве дополнения или альтернативы, может сохраняться число необработанных фрагментов, которые соответствуют референсной последовательности и не удостоверяют/подтверждают определение потенциального варианта. Полученная информация выводится в приложение валидации определения вариантов, включая количество и тип необработанных фрагментов, которые поддерживают определение потенциального варианта, положения поддерживающих вариантов в необработанных фрагментах, число необработанных фрагментов, которые не поддерживают потенциального определение варианта и т.п.
[00332] Когда потенциальный вариант идентифицирован, в выходных данных процесса появляется индикация определения потенциального варианта, последовательность варианта, положение варианта и референсная последовательность, ассоциированная с ним. Вариант обозначается как “потенциальный”, поскольку ошибки могут приводить к идентификации ложного варианта. В соответствии с приведенными здесь вариантами осуществления определение потенциального варианта анализируют, чтобы уменьшить или исключить ложные варианты и ложные совпадения. В качестве дополнения или альтернативы, процесс (способ) анализирует один или более необработанных фрагментов, ассоциированных с ридом образца ,и дополняет выходные данные соответствующим вариантом, ассоциированным с необработанными фрагментами.
Создание доброкачественного тренировочного набора
[00333] Секвенированы миллионы геномов и экзомов человека, но их клиническое применение остается ограниченным из-за сложности в различении вызывающих заболевания мутаций и доброкачественных генетических вариантов. Здесь мы демонстрируем, что распространенные миссенс-варианты у других видов приматов в значительной степени клинически доброкачественны у человека, что позволяет систематически идентифицировать патогенные мутации в процессе устранения. Используя сотни тысяч распространенных вариантов, полученных в результате популяционного секвенирования шести видов приматов, отличных от человека, мы обучаем глубокую нейронную сеть, которая идентифицирует патогенные мутации у пациентов с редкими заболеваниями с точностью 88% и позволяет обнаруживать 14 новых генов-кандидатов умственной отсталости со полногеномной значимостью. Каталогизация распространенных вариантов от дополнительных видов приматов улучшит интерпретацию миллионов вариантов неопределенной значимости, что еще больше повысит клиническую ценность секвенирования генома человека.
[00334] Клиническая эффективность диагностического секвенирования ограничена трудностью интерпретации редких генетических вариантов в человеческих популяциях и определения их влияния на риск заболевания. Из-за пагубного воздействия на приспособленность клинически значимые генетические варианты, как правило, чрезвычайно редки в популяции, и для подавляющего большинства их влияние на здоровье человека не определено. Большое количество и редкость этих вариантов с неопределенной клинической значимостью представляют собой серьезное препятствие на пути внедрения секвенирования для индивидуализированной медицины и скрининга здоровья населения в целом.
[00335] Большинство пенетрантных менделевских заболеваний имеют очень низкую распространенность в популяции, поэтому наблюдение варианта с высокой частотой в популяции является убедительным доказательством в пользу доброкачественности последствий. Анализ распространенных вариантов среди различных человеческих популяций является эффективной стратегией каталогизации доброкачественных вариантов, но общее количество распространенных вариантов у современных людей ограничено из-за узких мест в недавней истории нашего вида, во время которых большая часть наследственного разнообразия была потеряна. Популяционные исследования современных людей показывают заметную инфляцию при эффективном размере популяции (Ne) менее 10 000 особей в течение последних 15000-65000 лет, а небольшой пул распространенных полиморфизмов восходит к ограниченной способности к изменчивости популяции. такого размера. Из более чем 70 миллионов потенциальных изменяющих белок миссенс-замен в референсном геноме только примерно 1 из 1000 присутствует с частотой аллелей более 0,1% от общей популяции.
[00336] Вне современных человеческих популяций шимпанзе составляют следующий ближайший существующий вид и имеют 99,4% идентичности аминокислотных последовательностей с человеком. Почти полная идентичность кодирующих белок последовательностей у людей и шимпанзе указывает на то, что очищающий отбор, действующий на варианты, кодирующие белки у шимпанзе, может также моделировать последствия для приспособленности человеческих мутаций, которые идентичны по состоянию.
[00337] Поскольку среднее время, в течение которого нейтральные полиморфизмы сохраняются в линии происхождения человека (~ 4Ne поколений), составляет часть времени расхождения вида (~ 6 миллионов лет назад), естественный вариант шимпанзе исследует пространство мутаций, которое в значительной степени не перекрывается за исключением случайного перекрывания, за исключением редких случаев гаплотипов, поддерживаемых балансирующим отбором. Если полиморфизмы, которые идентичны по состоянию, одинаково влияют на приспособленность у двух видов, наличие варианта с высокой частотой аллелей в популяциях шимпанзе должно указывать на доброкачественные последствия для человека, расширяя каталог известных вариантов, доброкачественные последствия которых были зафиксированы очищающим отбором.
Результаты - Общие варианты у других приматов в значительной степени доброкачественны у человека
[00338] То, что недавно стали доступны агрегированные данные экзома, включающие 123136 человек, собранные в Консорциуме агрегации экзома (Exome Aggregation Consortium, ExAC) и базе Сборной базе данных геномов (Genome Aggregation Database, gnomAD), позволяет нам измерить влияние естественного отбора на миссенс- и синонимичные мутации по всему частотному спектру аллелей. Редкие синглетные варианты, которые наблюдаются в когорте только однократно, близко соответствуют ожидаемому соотношению миссенс / синонимы 2,2 / 1, предсказанному на основе мутаций de novo после поправки на влияние тринуклеотидного контекста на степень мутаций (ФИГ. 49A, ФИГ. 51 и ФИГ. 52A, 52B, 52C и 52D), но при более высоких частотах аллелей количество наблюдаемых миссенс-вариантов уменьшается из-за удаления вредных мутаций естественным отбором. Постепенное уменьшение соотношений миссенс/синонисы с увеличением частоты аллелей согласуется со значительной долей миссенс-вариантов с популяционной частотой <0,1%, имеющей умеренно пагубные последствия, несмотря на то, что они наблюдаются у здоровых людей. Эти результаты подтверждают широко распространенную эмпирическую практику диагностических лабораторий по отфильтровыванию вариантов с частотой аллелей от более 0,1% до ~ 1% как возможно доброкачественных для пенетрантного генетического заболевания, за исключением нескольких хорошо задокументированных исключений, связанных с уравновешиванием эффектов отбора и основателя.
[00339] Мы идентифицировали распространенные варианты шимпанзе, образцы которых отбирали два или более раз в когорте из 24 неродственных особей; по нашим оценкам, 99,8% этих вариантов распространены в общей популяции шимпанзе (частота аллелей (AF)> 0,1%), что указывает на то, что эти варианты уже прошли через сито очищающего отбора. Мы исследовали частотный спектр аллелей человека для соответствующих идентичных по состоянию человеческих вариантов (ФИГ. 49B), исключая расширенную область главного комплекса гистосовместимости как известную область балансирующего отбора, наряду с вариантами, не имеющими однозначного сопоставления в множественном выравнивании последовательностей. Для человеческих вариантов, которые идентичны по состояниям с распространенными вариантами шимпанзе, отношение миссенс / синонимы в основном постоянное в частотном спектре аллелей человека (P> 0,5 по критерию хи-квадрат (χ2)), что согласуется с отсутствием отрицательный отбор против распространенных вариантов шимпанзе в человеческой популяции и согласованные коэффициенты отбора по миссенс-вариантам у двух видов. Низкое соотношение миссенс / синонимы, наблюдаемое в человеческих вариантах, которые идентичны по состояниям с распространенными вариантами шимпанзе, согласуется с большим эффективным размером популяции у шимпанзе (Ne ~ 73000), что позволяет более эффективно отфильтровывать умеренно вредоносные варианты.
[00340] В отличие от этого, для единичных вариантов шимпанзе (определенных только один раз в когорте) мы наблюдаем значительное снижение отношения миссенс / синонимы при распростнаненных частотах аллелей (P <5,8 × 10-6; ФИГ. 49C), что указывает на то, что 24% единичных миссенс-вариантов шимпанзе будет отфильтровано очищающим отбором в человеческих популяциях с частотами аллелей более 0,1%. Это истощение указывает на то, что значительная часть единичных вариантов шимпанзе представляет собой редкие вредные мутации, повреждающее воздействие которых на приспособленность не позволяет им достичь распространенных частот аллелей у обоих видов. По нашим оценкам, в общей популяции шимпанзе распространены только 69% единичных вариантов (AF> 0,1%).
[00341] Затем мы идентифицировали человеческие варианты, которые идентичны по состоянию с вариантами, наблюдаемыми, по меньшей мере, у одного из шести видов приматов, отличных от человека. Варианты в каждом из шести видов были идентифицированы либо из проекта геномов высших обезьян (шимпанзе, бонобо, горилла и орангутан), либо были представлены в базе данных однонуклеотидных полиморфизмов (dbSNP) из проектов генома приматов (резус, мартышка) и в основном представляют собой распространенные варианты, основанные на ограниченном количестве секвенированных особей и низком соотношении миссенс: синонимы, наблюдаемом для каждого вида (Дополнительная таблица 1). Как и в случае с шимпанзе, мы обнаружили, что отношения миссенс / синонимы для вариантов из шести видов приматов, отличных от человека, примерно одинаковы по частотному спектру аллелей человека, за исключением умеренного истощения миссенс-вариантов на частотах распространенных аллелей (ФИГ. 49D, ФИГ 53, 54 и 55, файл дополнительных данных 1), что ожидалось из-за включения меньшинства редких вариантов (~ 16% с частотой аллелей менее 0,1% у шимпанзе, и меньше у других видов из-за меньшего количества секвенированных особей. ). Эти результаты позволяют предположить, что коэффициенты отбора для миссенс-вариантов, идентичных по состояниям, в пределах родословной приматов в меньшей степени совпадают с обезьянами Нового Света, которые, по оценкам, отошли от линии происхождения человека примерно 35 миллионов лет назад.
[00342] Мы обнаружили, что миссенс-варианты человека, которые идентичны по состоянию с наблюдаемыми вариантами приматов, сильно обогащены доброкачественными последствиями в базе данных ClinVar. После исключения вариантов с неопределенной значимостью и вариантов с противоречивыми аннотациями варианты ClinVar, которые присутствуют, по меньшей мере, у одного вида приматов, отличных от человека, аннотируются как доброкачественные или вероятно доброкачественные в среднем в 90% случаев по сравнению с 35% для миссенс-вариантов ClinVar. в целом (P <10-40; ФИГ. 49E). Патогенность аннотаций ClinVar для вариантов приматов немного выше, чем наблюдаемая при выборке из когорты здоровых людей близкого размера (~ 95% доброкачественные или вероятные доброкачественные последствия, P = 0,07), за исключением человеческих вариантов с частотой аллелей более 1% для снижения системного смещения, обусловленного курированием.
[00343] Область генетики человека долгое время опиралась на модельные организмы для выводов о клиническом влиянии мутаций человека, но большое эволюционное расстояние до большинства поддающихся генетическому изучению животных моделей вызывает опасения по поводу степени, в которой результаты, полученные на модельных организмах, могут быть обобщены на человека. . Мы расширили наш анализ за пределы родословной приматов, включив в него в основном распространенные варианты от четырех дополнительных видов млекопитающих (мыши, свиньи, козы и коровы) и двух видов более отдаленных позвоночных (куриц и рыбок данио). Мы отобрали виды с достаточным подтверждением вариабельности dbSNP по всему геному и подтвердили, что это в основном распространенные варианты, на основе отношений миссенс / синонимы, которые намного ниже, чем 2,2 / 1. В отличие от нашего анализа приматов, миссенс-мутации человека, которые идентичны по состояниям с вариантыми у более далеких видов, заметно истощаются на распространенных частотах аллелей (ФИГ. 50A), и величина этого истощения увеличивается при более длинных эволюционных расстояниях (ФИГ. 50B и дополнительные таблицы 2 и 3).
[00344] Миссенс-мутации, которые вредны для человека, но допускаются при высоких частотах аллелей у более отдаленных видов, указывают на то, что коэффициенты отбора миссенс-мутаций, идентичных по состоянию, существенно различаются между людьми и более отдаленными видами. Тем не менее, наличие миссенс-варианта у более далеких млекопитающих по-прежнему увеличивает вероятность доброкачественных последствий, поскольку доля миссенс-вариантов, истощенных естественным отбором при распространённых частотах аллелей меньше, чем ~ 50% -ное истощение, наблюдаемое для миссенс-вариантов человека в целом ( ФИГ.49А). В соответствии с этими результатами мы обнаружили, что миссенс-варианты ClinVar, которые наблюдались у мышей, свиней, коз и коров, с вероятностью 73% имеют доброкачественные или вероятно доброкачественные последствия, по сравнению с 90% для вариаций приматов (P <2 × 10-8; ФИГ. 50C) и 35% по базе ClinVar в целом.
[003945] Чтобы подтвердить, что эволюционное расстояние, а не артефакт одомашнивания, является основной движущей силой дивергенции коэффициентов отбора, мы повторили анализ, используя фиксированные замены между парами близкородственных видов вместо внутривидовых полиморфизмов в широком диапазоне эволюционных расстояний (ФИГ. 50D, Дополнительная таблица 4 и Supplementary Data File 2). Мы обнаружили, что истощение человеческих миссенс-вариантов, которые идентичны по состояниям с межвидовыми фиксированными заменами, увеличивается с увеличением длины эволюционной ветви без заметного отличия для диких видов по сравнению с теми, которые подверглись одомашниванию. Это согласуется с более ранними работами на мухах и дрожжах, в которых было выявлено, что количество фиксированных миссенс-замен идентичных состояний было ниже, чем ожидалось случайно в расходящихся линиях.
Сеть глубокого обучения для классификации патогенности вариантов
[00346] Раскрытая технология обеспечивает сеть глубокого обучения для классификации патогенности вариантов. Важность классификации вариантов для клинических приложений вдохновила многочисленные попытки использовать контролируемое машинное обучение для решения этой проблемы, но этим усилиям препятствовало отсутствие адекватного размера набора достоверных данных, содержащего надежно помеченные доброкачественные и патогенные варианты для обучения.
[00347] Существующие базы данных вариантов, отобранных экспертами-людьми, не представляют весь геном, при этом ~ 50% вариантов в базе данных ClinVar получены из всего только 200 генов (~ 1% генов, кодирующих белки человека). Более того, систематические исследования показывают, что многие комментарии экспертов-людей имеют сомнительные подтверждающие доказательства, что подчеркивает сложность интерпретации редких вариантов, которые могут наблюдаться только у одного пациента. Хотя интерпретация экспертов - людей становится все более строгой, рекомендации по классификации в основном сформулированы на основе консенсусных практик и рискуют усилить существующие тенденции. Чтобы уменьшить обусловленные человеческим фактором ошибки интерпретации, более новые классификаторы были обучены на распространенных человеческих полиморфизмах или фиксированных заменах человек-шимпанзе, но эти классификаторы также используют в качестве входных данных оценки предсказаний более ранних классификаторов, которые были обучены на основе данных, подготовленных (курируемых) людьми. Объективный сравнительный анализ эффективности этих различных методов был труднодостижим из-за отсутствия независимого, беспристрастного набора истинных данных.
[00348] Варианты от шести нечеловеческих приматов (шимпанзе, бонобо, горилла, орангутанг, резусов и игрунок) вносит вклад в более 300000 уникальных миссенс-вариантов, которые не перекрываются с общими человеческими вариантами и в значительной степени представляют собой распространенные варианты с доброкачественными последствиями, которые прошли через сито очищающего отбора, значительно увеличив набор обучающих данных, доступных для подходов машинного обучения. В среднем каждый вид приматов вносит больше вариантов, чем вся база данных ClinVar (~ 42 000 миссенс-вариантов по состоянию на ноябрь 2017 г., после исключения вариантов с неопределенной значимостью и вариантов с противоречивыми аннотациями). Кроме того, этот контент свободен от предвзятой интерпретации человеком.
[00349] Используя набор данных, включающий распространенные варианты человека (AF> 0,1%) и вариации приматов (Дополнительная таблица 5 (ФИГ. 58)), мы обучили новую глубокую остаточную сеть PrimateAI, которая принимает в качестве входных данных аминокислотную последовательность, фланкирующую вариант, представляющий интерес, и выравнивания ортологичных последовательностей других видов (ФИГ. 2 и ФИГ. 3). В отличие от существующих классификаторов, которые используют функции, созданные человеком, наша сеть глубокого обучения учится извлекать признаки непосредственно из первичной последовательности. Чтобы включить информацию о структуре белка мы обучили отдельные сети предсказывать вторичную структуру и доступность для растворителя только по последовательности, а затем включили их в качестве подсетей в полную модель (ФИГ. 5 и ФИГ. 6). Учитывая небольшое количество белков человека, которые были успешно кристаллизованы, определение структуры из первичной последовательности имеет то преимущество, что позволяет избежать смещений из-за неполной структуры белка и аннотации функционального домена. Общая глубина сети ork, с включенной структурой белка, составляла 36 слоев сверток, содержащих примерно 400 000 обучаемых параметров.
[00350] Чтобы обучить классификатор, используя только варианты с доброкачественными метками, мы сформулировали проблему прогнозирования как вероятность того, что данная мутация будет наблюдаться как распространенный вариант в популяции. Несколько факторов влияют на вероятность наблюдения варианта при высоких частотах аллелей, из которых нас интересует только вредоносность; другие факторы включают частоту мутаций, технические артефакты, такие как охват секвенированием, и факторы, влияющие на нейтральный генетический дрейф, такие как конверсия генов.
[00352] Мы сопоставили каждый вариант в доброкачественной обучающей выборке с миссенс-мутацией, которая отсутствовала в 123136 экзомах из базы данных ExAC, контролируя каждый из этих факторов смещения, и обучили сеть глубокого обучения различать доброкачественные варианты и соответствующие контроли ( ФИГ.24). Поскольку количество неразмеченных вариантов значительно превышает размер помеченного доброкачественного набора обучающих данных, мы обучили восемь сетей параллельно, каждая из которых использовала свой набор неразмеченных вариантов, сопоставленных с доброкачественным обучающим набором данных, чтобы получить согласованное предсказание.
[00353] Используя только первичную аминокислотную последовательность в качестве входных данных, сеть глубокого обучения точно присваивает высокие баллы патогенности остаткам в функциональных доменах полезных белков, как показано для потенциалзависимого натриевого канала SCN2A (ФИГ.20), основного гена заболевания для эпилепсии, аутизма и умственной отсталости. Структура SCN2A включает четыре гомологичных повтора, каждый из которых содержит шесть трансмембранных спиралей (S1 - S6). При деполяризации мембраны положительно заряженная трансмембранная спираль S4 перемещается к внеклеточной стороне мембраны, заставляя порообразующие домены S5 / S6 открываться через линкер S4-S5. Мутации в доменах S4, S4 - S5 линкера и S5, которые клинически связаны с ранним началом эпилептической энцефалопатии, по прогнозам сети, имеют наивысшие оценки патогенности в гене, и их количество в здоровой популяции истощается ( таблица 6). Мы также обнаружили, что сеть распознает важные аминокислотные положения внутри доменов и присваивает наивысшие оценки патогенности мутациям в этих положениях, таких как ДНК-контактирующие остатки факторов транскрипции и каталитические остатки ферментов (ФИГ. 25A, 25B, 25C и 26).
[00354] Чтобы лучше понять, как сеть глубокого обучения извлекает сведения о структуре и функции белка из первичной последовательности, мы визуализировали обучаемые параметры из первых трех уровней сети. Внутри этих слоев мы наблюдали, что сеть изучает корреляции между весами различных аминокислот, которые приблизительно соответствуют существующим измерениям аминокислотного расстояния, таким как оценка Грэнтэма (ФИГ. 27). Выходные данные этих начальных уровней становятся входами для последующих уровней, позволяя сети глубокого обучения создавать представления данных более высокого порядка.
[00355] Мы сравнили эффективность нашей сети с существующими алгоритмами классификации, используя 10 000 распространенных вариантов приматов, которые не участвовали в обучении. Поскольку ~ 50% всех вновь возникающих миссенс-вариантов человека фильтруются очищающим отбором при распространенных частотах аллелей (ФИГ. 49A), мы определили оценку 50-го процентиля для каждого классификатора по набору из 10000 случайно выбранных вариантов, которые были сопоставлены с 10000 распространенных вариантов приматов по степени мутаций и охвату секвенированием, и оценивали точность каждого классификатора при этом пороге (ФИГ. 21D, ФИГ. 28A и файл дополнительных данных 4). Наша сеть глубокого обучения (точность 91%) превзошла производительность других классификаторов (точность 80% для следующей лучшей модели) при назначении благоприятных последствий для 10 000 отложенных распространенных вариантов приматов.
[00356] Примерно половина улучшений по сравнению с существующими методами достигается за счет применения сети глубокого обучения, а половина - за счет дополнения набора обучающих данных вариантами приматов по сравнению с точностью сети, обученной только на данных по вариантам человека (ФИГ. 21D). Чтобы проверить классификацию вариантов с неопределенной значимостью в клиническом сценарии, мы оценили способность сети глубокого обучения различать мутации de novo, возникающие у пациентов с нарушениями развития нервной системы, и здоровых люди из контрольной группы. По распространенности нарушения развития нервной системы составляют одну из крупнейших категорий редких генетических заболеваний, и недавние исследования тройного секвенирования выявили центральную роль de novo миссенс-мутаций и мутаций, приводящих к укорочению белка.
[00357] Мы классифицировали каждый надежно определенных de novo миссенс-вариант у 429 больных лиц из когорты «Расшифровка нарушений развития» (DDD) по сравнению с de novo миссенс-вариантами от 2517 здоровых братьев и сестер в когорте Simon's Simplex Collection (Коллекция симплексов Саймона, SSC), и оценили разницу в предсказаниях оценки между двумя распределениями с помощью теста суммы рангов Вилкоксона (ФИГ. 21E и ФИГ. 29A и 29B). Сеть глубокого обучения явно превосходит другие классификаторы в этой задаче (P <10-28; ФИГ. 21F и ФИГ. 28B). Более того, эффективность различных классификаторов на наборе данных удержанных вариантов приматов и коррелировала со случаями DDD по сравнению с контрольным набором данных (Spearman ρ = 0,57, P <0,01), что указывает на хорошее согласие между двумя наборами данных для оценки патогенности, несмотря на использование совершенно разных источников и методики (ФИГ. 30А).
[00358] Затем мы попытались оценить точность сети глубокого обучения при классификации доброкачественных и патогенных мутаций в одном и том же гене. Учитывая, что популяция DDD в основном включает индексные случаи больных детей без больных родственников первой степени родства, важно показать, что классификатор не преувеличил свою точность, отдав предпочтение патогенности генов с de novo доминантными путями наследования. Мы ограничили анализ 605 генами, которые были имели номинальную значимость для связи с заболеванием в исследовании DDD, рассчитанном только по вариантам, укорачивающим белок (P <0,05). В пределах этих генов количество миссенс-мутаций de novo на 3/1 больше, чем ожидалось (ФИГ. 22A), что указывает на то, что ~ 67% являются патогенными.
[00359] Сеть глубокого обучения смогла различать патогенные и доброкачественные варианты de novo в одном и том же наборе генов (P <10-15; ФИГ. 22B), значительно превосходя другие методы (ФИГ. 22C и 28C). При бинарном пороге ≥ 0.803 (ФИГ. 22D и 30B) 65% миссенс-мутаций de novo в случаях классифицируются сетью глубокого обучения как патогенные, по сравнению с 14% миссенс-мутаций de novo в контроле, что соответствует точности классификации 88% (ФИГ. 22E и 30C). Учитывая частую неполную пенетрантность и переменную выраженность при нарушениях нервного развития, эта цифра, вероятно, недооценивает точность нашего классификатора из-за включения частично пенетрантных патогенных вариантов в контроль.
Новый способ обнаружения кандидатных генов
[00359] Применение порогового значения ≥ 0.803 чтобы удовлетворить условию миссенс-мутации увеличивает обогащение de novo миссенс-мутаций у пациентов с DDD с кратности 1,5-до 2,2, близкой к мутациям, приводящим к укорочению белка (кратность 2.5), в то же время оставляя менее одной трети от всего числа вариантов, обогащенных сверх ожиданий. Это существенно улучшает статистическую мощность способа, позволяя обнаруживать 14 дополнительных кандидатных генов умственной отсталости, которые до этого не достигали порога значимости для всего генома в исходном исследовании DDD (Таблица 1).
Сравнение с курированием экспертом-человеком
[00360] Мы испытали эффективность различных новых классификаторов на базе данных вариантов ClinVar, курируемых человеческой экспертизой, но обнаружили, что эффективность классификаторов на наборе данных ClinVar не имеет существенных корреляций как c удержанным набором вариантов приматов, так и наборами данных случаев DDD в сравнении с контрольным набором данных (P = 0.12 и P = 0.34, соответственно) (ФИГ.. 31A и 31B). Мы предполагаем, что существующие классификаторы имеют отклонения от курируемых человеческой экспертизой, и, хотя человеческие эвристики склонны иметь правильное направление, они могут быть неоптимальны. Один из примеров - это среднее отклонение расстояния Грантама между патогенными и доброкачественными вариантами в ClinVar, которое вдвое больше, чем разница между de novo вариантами в случаях DDD и у контрольного набора, в рамках 605 генов, ассоциированных с заболеванием (Таблица 2). Для сравнения, человеческая экспертиза как оказалось, недостаточно использует структуру белков, особенно важность остатка, который обращен на поверхность и может взаимодействовать с другими молекулами. Мы наблюдаем, что и патогенные мутации из набора ClinVar, и de novo мутации DDD ассоциированы с заранее предсказанными контактирующими с растворителем остатками, однако имеется разница в доступности растворителя: для доброкачественных и патогенных вариантов ClinVar - только половина наблюдаемых в случаях DDD по сравнению с контрольной группой. Эти открытия указывают на искажение при принятии решений, в пользу более прямолинейных факторов, которые человеку легче интерпретировать, таких как расстояние Грантама и консервация. Классификаторы машинного обучения, натренированные на базах данных, курируемых людьми, вероятно, будут склонны усиливать эти тенденции.
[00361] Наши результаты указывают на то, что семантическое секвенирование популяции приматов является эффективной стратегией классификации миллионов вариантов человеческого генома неясной значимости, которые сейчас ограничивают клиническую интерпретацию генома. Точность нейросети глубокого обучения, как на удержанных общих вариантах приматов, так и на клинических вариантах увеличивается с количеством доброкачественных вариантов, использованных для обучения нейросети (ФИГ. 23A). Более того, обучение на вариантах каждого из шести видов приматов, не являющихся людьми, независимо вносит вклад в эффективность нейросети, в то время как обучение на вариантах более отдаленных млекопитающих отрицательно сказывается на эффективности нейросети (ФИГ. 23B и 23C). Эти результаты говорят в пользу предположения, что распространенные варианты приматов являются по большей части доброкачественными у людей по отношению к пенетрантным менделевским заболеваниям, но того же нельзя сказать о вариантах более отдаленных видов.
[00362] Хотя число геномов приматов, не являющихся людьми, изученных в этом исследовании, мало в сравнении с числом человеческих геномов и экзомов, которые были секвенированы, важно обратить внимание на то, что дополнительный вклад генома приматов делает непропорциональным количество информации о распространенных доброкачественных вариантах. Компьютерное моделирование на ExAC показывает, что обнаружение распространенных человеческих вариантов (частота аллеля > 0.1%) быстро выходит на плато после проверки всего нескольких сотен индивидуумов (ФИГ. 56), и дальнейшее секвенирование миллионов образцов здоровой популяции в основном вносит дополнительные редкие варианты. В отличие от распространенных вариантов, клиническая доброкачественность которых хорошо известна на основании частоты аллеля, редкие варианты в здоровых популяциях могут вызывать рецессивные генетические заболевания или доминантные генетические заболевания с неполной пенетрацией. Из-за того, что каждый вид приматов несет свой пул распространенных вариантов, секвенирование генома нескольких дюжин представителей каждого вида эффективно для семантической каталогизации доброкачественных миссенс-вариантов в линии приматов. Действительно, 134 индивидуума из 6 видов приматов, не являющихся людьми, изученные в этом исследовании, вносят почти в 4 раза больше общих миссенс-вариантов, чем 123136 людей из исследования ExAC (Дополнительная таблица 5 (ФИГ. 58)). Исследования секвенирования популяции приматов, включающие сотни индивидуумов могут быть практически полезны даже с относительно небольшой выборкой несвязанных друг с другом индивидуумов, обитающих в заповедниках дикой природы и зоопарках, таким образом минимизируя вмешательство в дикие популяции, что важно с точки зрения консервации и этичного обращения с приматами, не являющимися людьми.
[00363] Современная человеческая популяция несет намного меньшее генетическое разнообразие, чем большинство видов приматов, не являющихся людьми, по грубой оценке, в два раза меньше однонуклеотидных вариантов на индивидуума, по сравнению с шимпанзе, гориллами и гиббонами, и в три раза меньше вариантов на индивидуума по сравнению с орангутанами. Хотя уровень генетического разнообразия для большинства видов приматов, не являющихся людьми неизвестен, большое число дошедших до наших дней видов приматов, не являющихся людьми, дают возможность обобщить, что большинство возможных доброкачественных человеческих миссенс-положений, вероятно, покрываются распространенными вариантами у по меньшей мере одного вида приматов, что позволяет систематически выявлять патогенные варианты процессом удаления (ФИГ. 23D). Даже при доступности лишь подмножества секвенированных геномов этих видов, увеличение объема данных для обучения позволит более точно предсказывать миссенс-последовательности с помощью машинного обучения. Наконец, в то время как наши открытия сфокусированы на миссенс-вариантах, эта стратегия может также быть применима для предсказания последствий некодирующих вариантов, в особенности в консервативных регуляторных областях, где имеется достаточное выравнивание между геномами человека и примата, для того, чтобы однозначно определить, является ли вариант идентичным по состоянию.
[00364] Из 504 известных видов приматов, не являющихся людьми, по грубой оценке, 60% вымирают из-за браконьерства и потери территорий среды обитания. Уменьшение популяции и потенциальное вымирание этих видов представляет невосполнимую потерю разнообразия генов, подталкивая к тому, чтобы как можно быстрее предпринять шаги, направленные на сохранение этих видов, для их и нашей собственной пользы.
Генерация данных и Выравнивание
[00365] Координаты в приложении представляют сборку человеческого генома в системе UCSC hg19/GRCh37, включая координаты для вариантов других видов, картированные на hg19 с помощью различного выравнивания последовательностей. Каноническое транскрипты для белок-кодирующих последовательностей ДНК и множества выравниваний последовательностей геномов 99 видов позвоночных и длины ветвей скачивали из геномного браузера UCSC.
[00366] Мы получили данные полиморфизмом человеческих экзомов из Консорциума Агрегации Экзомов (Exome Aggregation Consortium ExAC)/Базы данных агрегации геномов (gnomAD exomes) версии 2.0. Мы получили данные вариантов приматов из проекта по секвенированию генома высших приматов, который включает полные данные секвенирования генома и генотипов 24 шимпанзе, 13 бонобо, 27 горилл, и 10 орангутанов. Мы также включили варианты 35 шимпанзе из другого исследования шимпанзе и бонобо, но из-за различия в методологии поиска вариантов, мы исключили их из анализа популяции, и использовали их только для обучения модели нейросети. Дополнительно, мы использовали данные 16 представителей макак резус и 9 особей мартышек для анализа вариантов в исходном геноме этих видов, однако у нас не было доступа к информации на уровне отдельных особей. Мы получили данные вариантов для резусов, мартышек, свиней, коров, козлов, мышей, куриц и данио-рерио из dbSNP. База dbSNP также включали дополнительные варианты орангутанов, которые мы использовали только для тренировки модели глубокого обучения, поскольку информация о генотипе индивидуума не была доступна при анализе популяций. Чтобы избежать эффектов, связанных с балансирующим отбором, мы также исключили варианты, находящиеся в пределах расширенной области главного комплекса гистосовместимости (chr6: 28,477,797-33,448,354) при анализе популяции.
[00367] Мы использовали многовидовое выравнивание для 99 видов позвоночных , чтобы убедиться в правильности картирования один к одному ортологических генов на белок-кодирующе участки, и избежать картирования на псевдогены. Мы считали варианты идентичными по состоянию, если они встречаются в референсной/альтернативной ориентации. Чтобы убедиться, что у варианта одна и та же предсказанная белок-кодирующая последовательность как у человека, так и у другого вида, мы добивались, чтобы другие два нуклеотида в кодоне были идентичными для разных видов, и в миссенс-вариантах и в синонимических вариантах. Полиморфизмы каждого вида, включенного в анализ, перечислены в Файле Дополнительных Данных 1, а детальные метрики приведены в Дополнительной Таблице 1.
[00368] Для каждой из четырех категорий частоты аллеля (ФИГ. 49A), мы использовали варианты интронных участков чтобы оценить ожидаемое число синонимических и миссенс-вариантов в каждом из 96 возможных тринуклеотидных контекстов, и чтобы скорректировать по частоте мутаций (ФИГ. 51 и Дополнительные таблицы 7, 8 (ФИГ. 59). Мы также отдельно анализировали идентичные по состоянию CpG нуклеотидные и не-CpG нуклеотидные варианты, и подтвердили, что соотношение миссенс/синонимичных вариантов остается одинаковым для всего спектра частот аллелей для обоих классов, что указывает на то, что наш анализ работает и для CpG, и для не-CpG вариантов, несмотря на большое различие в частоте мутаций (ФИГ. 52A, 52B, 52C, и 52D).
Истощение человеческих миссенс-вариантов, идентичных по состоянию с полиморфизмами других видов
[00369] Чтобы оценить, насколько хорошо люди будут переносить варианты, представленные у других видов, при высокой частоте аллеля (> 0.1%), мы идентифицировали человеческие варианты, которые были идентичным по состоянию вариантам у других видов. Для каждого из вариантов, мы ставили ему в соответствие одну из четырех категорий на основании частоты аллеля в человеческой популяции (единичный, более одного ~0.01%, от 0.01% до ~0.1%, > 0.1%), и оценивали снижение соотношения миссенс/синонимы (MSR) между редкими (< 0.1%) и распространенными (> 0.1%) вариантами. Истощение идентичных по состоянию миссенс-вариантов при распространенных частотах аллеля для человека (> 0.1%) показывает ту часть вариантов из других видов, которые весьма вредоносны, и в человеческой популяции исчезли бы из-за естественного отбора при распространенных частотах аллеля:
% истощения =(MSредких -MSRраспространенных)/(MSRредких)
[00370] Соотношение миссенс/синонимических вариантов и процент истощения на вид показаны на ФИГ. 50B и в Дополнительной Таблице 2. Дополнительно, для распространенных вариантов шимпанзе (ФИГ. 49B), единичных вариантов шимпанзе (ФИГ. 49C), вариантов у млекопитающих (ФИГ. 50A), мы применили критерий хи-квадрат (χ 2) гомогенности к 2×2 таблице сопряженности, чтобы проверить, что разница в соотношении миссенс/синонимичных вариантов между распространенными и частыми вариантами была статистически значима.
[00371] Поскольку секвенирование проводили на ограниченном количестве индивидуумов из проекта генома высших приматов, мы использовали человеческий спектр частот аллеля из ExAC. чтобы оценить часть семплированных вариантов, которые были редкими (< 0.1%) или распространенными (> 0.1%) в общей популяции шимпанзе. Мы семплировали когорту из 24 человек на основании данных частоты аллеля ExAC и выявляли миссенс варианты, которые наблюдались один раз или более одного раза в этой когорте. Варианты, которые наблюдали более одного раза, с вероятностью 99.8% были распространенными (> 0.1%) в общей популяции, в то время, как варианты, наблюдаемые только один раз в когорте, с вероятностью 69% были распространенными в общей популяции. Чтобы убедиться, что наблюдаемое истощение миссенс-вариантов у более отдаленных млекопитающих не было вызвано затрудняющим интерпретацию эффектом от генов, которые более консервативны, и соответственно, для которых доступно более точное выравнивание, мы повторили описанный анализ, сузив рассмотрение до генов с >50% средней нуклеотидной идентичностью в выравнивании множества последовательностей 11 приматов и 50 млекопитающих в сравнении с людьми (см. Дополнительную Таблицу 3).
[00372] Это сужение удалило из рассмотрения ~7% человеческих белок-кодирующих генов, без существенного влияния на результат. Дополнительно, чтобы удостовериться, что наши результаты не были затронуты ошибками поиска вариантов, или артефактами одомашнивания (поскольку большинство видов из базы данных dbSNP - одомашнены), мы повторили анализ для фиксированного числа замен из пар близких видов in lieu межвидовых полиморфизмов (ФИГ. 50D, Дополнительная Таблица 4, и Файл Дополнительных Данных 2).
Анализ полиморфизма данных ClinVar для людей, приматов, млекопитающих и других позвоночных
[00373] Чтобы проверить клинический эффект вариантов, идентичных по состоянию с другими видами, мы скачали базу данных ClinVar, исключая те варианты, которые имели конфликтующие аннотации патогенности, или были только помечены как варианты неустановленной значимости. Согласно пошаговой фильтрации, как показано в Дополнительной Таблице 9, всего имеется 2,853 миссенс вариантов в патогенной категории и 17,775 миссенс вариантов в доброкачественной категории.
[00374] Мы подсчитали патогенные и доброкачественные варианты ClinVar, которые были идентичны по состоянию вариантам человека, приматов, не являющихся людьми, млекопитающих, и других позвоночных. В случае людей, мы проводили компьютерное моделирование когорты из 30 индивидуумов, семплированных из базы ExAC частот аллелей. Количество доброкачественными и патогенных вариантов для каждого вида приведены в Дополнительной Таблице 10.
Генерация наборов доброкачественных и непомеченных вариантов для обучения модели
[00375] Мы построили набор для тренировки нейросети из доброкачественных распространенных вариантов для обучения модели нейросети по данным людей и приматов, не являющихся людьми. Набор данных включает частые человеческие варианты (> 0.1% частоты аллеля; 83546 вариантов), и варианты шимпанзе, бонобо, гориллы и орангутана, резусов и мартышек (301690 уникальных вариантов приматов). Количество доброкачественных вариантов для обучения от каждого источника данных приведено в Дополнительной Таблице 5.
[00376] Мы обучили нейросеть глубокого обучения отличать набор вариантов, обозначенных как доброкачественные, от н помеченных вариантов, которым было найдено соответствие в контролях в тринуклеотидном контексте, с покрытием секвенирования, и возможностью выравнивания геномов других видов и человека. Чтобы получить набор непомеченных данных для обучения, мы начинали со всех возможных миссенс-вариантов в канонических кодирующих участках. Мы исключили варианты, которые наблюдались в 123136 экзомах в данных базы ExAC, и варианты в инициирующем и стоп-кодонах. Всего было сгенерировано 68258623 непомеченных миссенс вариантов. В этом наборе проводили фильтрацию, чтобы сделать поправку на участки низкого покрытия секвенированием, и участки, где не было соответствия один-к-одному между человеческим геномом и геномом приматов, при выборе имеющих соответствие непомеченных вариантов из набора данных приматов.
[00377] Мы получили согласованное предсказание, обучив восемь моделей, которые используют один тот же набор помеченных как доброкачественные вариантов и восемь случайным образом семплированных наборов из непомеченных вариантов, и беря среднее от их предсказаний. Мы также оставили в стороне два случайным образом семплированных набора из 10,000 вариантов приматов для валидации и тестирования, которые не принимали участие в обучении (Файл Дополнительных Данных 3). Для каждого из этих наборов, мы семплировали 10,000 непомеченных вариантов, которым нашлось соответствие по тринуклеотидному контексту, которые мы использовали для нормализации порогового значения каждого классификатора при сравнении различных алгоритмов классификации (Дополнительный Файл Данных 4). В других вариантах реализации, может использоваться ансамбль из меньшего или большего числа моделей, от 2 до 500.
[00378] Мы оценили точность классификации двух вариантов нейросети глубокого обучения: одной, обученной на только человеческих вариантах, и одной, обученной на помеченном полностью доброкачественным наборе, включающем как данные вариантов людей, так данные вариантов приматов.
Архитектура сети глубокого обучения
[00379] Для каждого варианта сеть предсказания патогенности принимает в качестве входных данных аминокислотную последовательность длиной 51 с центром в целевом варианте, а также выходные данные сетей вторичной структуры и доступности для растворителя (ФИГ. 2 и ФИГ. 3) с заменой миссенс-варианта заменен в центральном положении. Три частотные матрицы положений для длины 51 генерируются из множественных выравниваний последовательностей 99 позвоночных, в том числе одна для 11 приматов, одна для 50 млекопитающих, исключая приматов, и одна для 38 позвоночных, исключая приматов и млекопитающих.
[00380] Сеть глубокого обучения вторичной структуры предсказывает вторичную структуру с тремя состояниями в каждом аминокислотном положении: альфа-спираль (H), бета-складчатость (B) и спирали (C) (Дополнительная таблица 11). Сеть доступности для растворителя предсказывает доступность для растворителя в тремя состояниями для каждого положения аминокислоты: скрытая (заглубленная) (B), промежуточная (I) и открытая (экспонированная) (E) (Дополнительная таблица 12). Обе сети принимают только фланкирующую аминокислотную последовательность в качестве входных данных и обучены с использованием меток по известным неизбыточным кристаллическим структурам в Protein DataBank (Дополнительная таблица 13). В качестве входа для предварительно обученной сети вторичной структуры с тремя состояниями и сети доступности для растворителя с тремя состояниями мы использовали частотную матрицу единичной длины, сгенерированную по нескольким выравниваниям последовательностей для всех 99 позвоночных, также с длиной 51 и глубиной 20. После предварительного обучения сети на известных кристаллических структурах из Protein DataBank, последние два слоя для моделей вторичной структуры и растворителя были удалены, а выход сети был напрямую связан с входом модели патогенности. Наилучшая точность тестирования для модели предсказания вторичной структуры с тремя состояниями составила 79,86% (Дополнительная таблица 14). Не было существенной разницы при сравнении предсказаний нейронной сети при использовании DSSP-аннотированных (Define Secondary Structure of Proteins) структурных меток для примерно ~ 4000 белков человека, которые имели кристаллические структуры, по сравнению с использованием только предсказанных структурных меток (Дополнительная таблица 15).
[00381] И наша сеть глубокого обучения для предсказания патогенности (PrimateAI), и сети глубокого обучения для предсказания вторичной структуры и доступности для растворителя имели архитектуру остаточных блоков. Подробная архитектура PrimateAI описана на (ФИГ. 3) и в Дополнительной таблице 16 (ФИГ. 4A, 4B и 4C). Подробная архитектура сетей для предсказания вторичной структуры и доступности для растворителя описана на ФИГ. 6 и в дополнительных таблицах 11 (ФИГ. 7A и 7B) и 12 (ФИГ. 8A и 8B).
Сравнительный анализ производительности классификатора на отложенном тестовом наборе из 10 000 вариантов приматов
[00382] Мы использовали 10000 отложенных вариантов приматов в наборе тестовых данных для тестирования сети глубокого обучения, а также других 20 ранее опубликованных классификаторов, для которых мы получили оценки предсказаний из базы данных dbNSFP. Показатели эффективности для каждого из классификаторов на 10 000 отложенных тестовых вариантах приматов также представлены в ФИГ. 28А. Поскольку разные классификаторы имели сильно различающиеся распределения оценок, мы использовали 10000 случайно выбранных неразмеченных вариантов, которые сопоставлялись с тестовым набором по тринуклеотидному контексту, чтобы определить порог 50-го процентиля для каждого классификатора. Мы сравнили каждый классификатор с долей вариантов в наборе из 10 000 отложенных вариантов приматов, которые были классифицированы как доброкачественные при пороге 50-го процентиля для этого классификатора, чтобы обеспечить справедливое сравнение методов.
[00383] Для каждого из классификаторов доля отложенных тестовых вариантов приматов, предсказанных как доброкачественные с использованием порога, соответствующего 50-му процентилю, также показана на ФИГ. 28A и в Дополнительной таблице 17 (ФИГ. 34). Мы также показываем, что производительность PrimateAI устойчива по отношению к количеству выровненных видов в вариантном положении и, как правило, работает хорошо, при условии, что имеется достаточная информация о консервативности (сохранении) у млекопитающих, которое выполняется для большинства кодирующих белок последовательностей (ФИГ. 57).
Анализ de novo вариантов из исследования нарушений развития (DDD)
[00384] Мы получили опубликованные варианты de novo из исследования нарушений развития (DDD) и de novo варианты от здоровых братьев и сестер в контрольной группе в исследовании SSC (Simons Simplex Collection) для аутизма. Исследование DDD обеспечивает уровень достоверности для вариантов de novo, и мы исключили варианты из набора данных DDD с порогом <0,1 как потенциально ложные срабатывания из-за ошибок определения вариантов. В одном варианте реализации всего у нас было 3512 миссенс-вариантов de novo от индивидуумов, пораженных DDD, и 1208 миссенс-вариантов de novo от здоровых людей. Аннотации канонических транскриптов, используемые в UCSC для выравнивания множественных последовательностей 99 позвоночных, немного отличались от аннотаций транскриптов, используемых в исследовании DDD, что приводило к небольшой разнице в общем количестве миссенс-вариантов. Мы оценили методы классификации по их способности различать de novo миссенс-варианты у лиц, пораженных DDD, по сравнению с de novo миссенс-вариантами у здоровых братьев и сестер из контрольной группы исследований аутизма. Для каждого классификатора мы сообщали P-значение из теста суммы рангов Уилкоксона для разницы между оценками предсказаний для двух распределений (Дополнительная таблица 17 (ФИГ. 34)).
[00385] Чтобы измерить точность различных классификаторов в различении доброкачественных и патогенных вариантов в пределах одного и того же гена заболевания, мы повторили анализ на подмножестве из 605 генов, которые были обогащены de novo вариантами, укорачивающими белок, в когорте DDD (P <0,05 , точный критерий Пуассона) (Дополнительная таблица 18). В пределах этих 605 генов, по нашим оценкам, две трети вариантов de novo в наборе данных DDD были патогенными, а одна треть - доброкачественными, исходя из обогащения 3/1 миссенс-мутаций de novo над ожиданием. Исходили из предположения минимальной неполной пенетрантности и того, что миссенс-мутации de novo у здоровых контролей были доброкачественными. Для каждого классификатора мы определили порог, который дал такое же количество доброкачественных или патогенных предсказаний, что и эмпирические пропорции, наблюдаемые в этих наборах данных, и использовали этот порог в качестве двоичного порога для оценки точности каждого классификатора в различении мутаций de novo в случаях заболевания по сравнению с контролями. Чтобы построить кривую характеристик оператора-приемника, мы рассматривали патогенную классификацию de novo вариантов нарушений развития (DDD) как истинно положительные определения, а классификацию вариантов de novo в здоровых контрольных группах в качестве патогенных - как ложноположительные определения. Поскольку набор данных DDD содержит одну треть доброкачественных de novo вариантов, площадь под кривой (AUC) для теоретически совершенного классификатора меньше единицы. Следовательно, классификатор с идеальным разделением доброкачественных и патогенных вариантов классифицировал бы 67% вариантов de novo у пациентов с DDD как истинно положительные, 33% вариантов de novo у пациентов с DDD как ложноотрицательные и 100% вариантов de novo в контроле как истинно отрицательные, что дает максимально возможную AUC 0,837 (ФИГ. 29A и 29B и Дополнительная таблица 19 (ФИГ. 35)).
Обнаружение новых кандидатных генов
[00386] Мы протестировали обогащение количества мутаций de novo в генах, сравнивая наблюдаемое количество мутаций de novo с количеством, ожидаемым в рамках модели нулевой мутации. Мы повторили анализ обогащения, проведенный в исследовании DDD, и определили гены, которые являются новыми значимыми для всего генома, при подсчете только миссенс-мутаций de novo с показателем PrimateAI> 0,803. Мы скорректировали ожидание по всему геному для de novo повреждающей миссенс-вариации на долю миссенс-вариантов, которые соответствуют порогу PrimateAI> 0,803 (примерно пятая часть всех возможных миссенс-мутаций в масштабах всего генома). Согласно исследованию DDD, для каждого гена требовалось четыре теста, один тест на обогащение вариантов, приводящих у укорочению белка, и один тест на обогащение изменяющими белок мутациями de novo, причем оба теста проводились только для когорты DDD и для более крупного метаанализа когорт секвенирования на развитие нервной системы. Обогащение изменяющих белок мутаций de novo было объединено по методу Фишера с тестом кластеризации de novo миссенс-мутаций в кодирующей последовательности (дополнительные таблицы 20, 21). P-значение для каждого гена было взято из минимума по четырем тестам, и значимость в масштабах всего генома определялась как P <6,757 × 10-7 (α = 0,05, 18 500 генов с четырьмя тестами).
Точность классификации ClinVar
[00387] Поскольку большинство существующих классификаторов обучаются прямо или косвенно на содержимом ClinVar, например, с использованием оценок предсказаний от классификаторов, обученных на ClinVar, мы ограничили анализ набора данных ClinVar: мы использовали только варианты ClinVar, которые были добавленые с 2017 года. Между недавними вариантами ClinVar и другими базами данных наблюдалось существенное совпадение, поэтому мы дополнительно отфильтровали варианты, встречающиеся с распространенными частотами аллелей (> 0,1%) в ExAC или присутствующие в HGMD (База данных мутаций генов человека), LOVD ( Leiden Open Variation Database) или Uniprot (Universal Protein Resource). После исключения вариантов, имеющих только неопределенное значение, и вариантов с противоречивыми аннотациями, у нас осталось 177 миссенс-вариантов с доброкачественной аннотацией и 969 миссенс-вариантов с патогенной аннотацией. Мы оценили эти варианты ClinVar, используя как сеть глубокого обучения, так и другие методы классификации. Для каждого классификатора мы определили порог, который дает такое же количество доброкачественных или патогенных прогнозов, что и эмпирические пропорции, наблюдаемые в этих наборах данных, и использовали этот порог в качестве двоичного порога для оценки точности каждого классификатора (ФИГ. 31A и 31B).
Влияние увеличения размера обучающих данных и использования различных источников обучающих данных
[00388] Чтобы оценить влияние размера обучающих данных на показатели работы сети глубокого обучения, мы случайным образом отобрали подмножество вариантов из размеченного доброкачественного обучающего набора от 385 236 приматов и распространенных человеческих вариантов и сохранили базовую архитектуру сети глубокого обучения такой же. Чтобы показать, что варианты от каждого отдельного вида приматов вносят свой вклад в точность классификации, тогда как варианты от каждого отдельного вида млекопитающих снижают точность классификации, мы обучили сети глубокого обучения, используя обучающий набор данных, содержащий 83546 человеческих вариантов плюс постоянное количество случайно выбранных вариантов для каждого вида, снова сохранив базовой сетевой архитектуры неизменной в соответствии с одним вариантом реализации. Постоянное количество вариантов, которые мы добавили в обучающий набор (23380), было общим количеством вариантов, доступных для видов с наименьшим количеством миссенс-вариантов, то есть бонобо. Мы повторили процедуры обучения пять раз, чтобы получить средние показатели работы каждого классификатора.
Предельное значение (насыщение) всех возможных миссенс-мутаций человека при увеличении числа секвенированных популяций приматов
[00389] Мы исследовали ожидаемое насыщение всех ~ 70 миллионов возможных миссенс-мутаций человека распространенными вариантами, присутствующими у 504 существующих видов приматов, путем моделирования вариантов на основе тринуклеотидного контекста распространенных миссенс-вариантов человека (частота аллелей> 0,1%), присутствующих в ExAC. Для каждого вида приматов мы смоделировали в четыре раза большее количество распространенных миссенс-вариантов, наблюдаемых у человека (~ 83500 миссенс-вариантов с частотой аллелей> 0,1%), потому что у людей примерно вдвое меньше вариантов на человека, чем у других видов приматов, и примерно на ~ 50% миссенс-вариантов человека были отфильтрованы очищающим отбором при частоте аллелей> 0,1% (ФИГ. 49A).
[00390] Чтобы смоделировать долю распространенных миссенс-вариантов человека (частота аллелей> 0,1%), обнаруженных при увеличении размера исследованных когорт людей (ФИГ. 56), мы взяли выборку генотипов в соответствии с частотами аллелей ExAC и определили долю распространенных вариантов, которые были наблюдалось по меньшей мере один раз в этих модельных когортах.
[00391] В одном варианте реализации для практического применения оценок PrimateAI порог> 0,8 является предпочтительным для вероятно патогенной классификации, <0,6 для вероятно доброкачественной и 0,6-0,8 - как промежуточный для генов с доминантными типами наследования на основе обогащения вариантами de novo в случаях заболевания по сравнению с контролем (ФИГ. 21D) и порог> 0,7 для вероятно патогенных и <0,5 для вероятно доброкачественных генов в генах с рецессивными способами наследования.
[00392] На ФИГ. 2 показан пример архитектуры глубокой остаточной сети для предсказания патогенности, называемой здесь «PrimateAI». На ФИГ. 2, 1D относится к одномерному сверточному слою. Предсказанная патогенность оценивается по шкале от 0 (доброкачественная) до 1 (патогенная). Сеть принимает в качестве входных данных референсную человеческую аминокислоту (AA) и альтернативную последовательность (51 AA) с центром в варианте, матрицы весовых коэффициентов профилей консервативности (PWM), рассчитанные по 99 видам позвоночных, а также результаты предсказания вторичной структуры и доступности для растворителя. Сети глубокого обучения, которые предсказывают вторичную структуру белка с тремя состояниями (спираль - H, бета-лист - B и скрученная спираль (coil) - C) и доступность для растворителя с тремя состояниями (скрытый - B, промежуточный - I и открытый - E).
[00393] На ФИГ. 3 схематично изображена PrimateAI, сетевая архитектура глубокого обучения для классификации патогенности. Входные данные для модели включают 51 аминокислоту (а.к.) фланкирующей последовательности как для референсной последовательности, так и для последовательности с вариантом-заменой, консервация представлена тремя взвешенными по положению матрицами длиной 51-а.к. из выравниваний приматов, млекопитающих и позвоночных, а также выходы предварительно обученной сети вторичной структуры и сети доступности для растворителя (также длиной 51 а.к).
[00394] ФИГ. 4A, 4B и 4C - Дополнительная таблица 16, в которой показаны детали архитектуры примера модели глубокого обучения модели PrimateAI для предсказания патогенности. Форма определяет форму выходного тензора на каждом слое модели, а активация - это активация, переданная нейронам этого слоя. Входными данными в модели являются частотные матрицы положений (длина 51 а.к., глубина 20) для фланкирующей аминокислотной последовательности вокруг варианта, закодированные кодированием с одним активным состоянием референсные и альтернативные последовательности человека (длина 51 а.к., глубина 20) и выход из вторичной моделей структуры и доступности для растворителя (длина 51 а.к., глубина 40).
[00395] В показанном примере используются одномерные свертки. В других вариантах реализации модель может использовать различные типы сверток, такие как двумерные свертки, трехмерные свертки, разреженные или дырчатые свертки, транспонированные свертки, разделяемые свертки и свертки по глубине. Некоторые слои также используют функцию активации ReLU, которая значительно ускоряет сходимость стохастического градиентного спуска по сравнению с насыщающими нелинейностями, такими как сигмоидальный или гиперболический тангенс. Другие примеры функций активации, которые могут использоваться раскрытой технологией, включают параметрическое ReLU, утечку ReLU и экспоненциальный линейный блок (ELU).
[00396] Некоторые слои также используют пакетную нормализацию (Ioffe, Szegedy, 2015). Что касается пакетной нормализации, распределение каждого слоя в сверточной нейронной сети (CNN) изменяется во время обучения, и оно меняется от одного уровня к другому. Это снижает скорость сходимости алгоритма оптимизации. Пакетная нормализация - это метод решения этой проблемы. Если обозначить вход слоя пакетной нормализации через x, а его выход через z, пакетная нормализация применяет следующее преобразование к x:
 [
[
[00397] Пакетная нормализация применяет нормализацию среднего отклонения на входе x с использованием μ и σ и линейно масштабирует и сдвигает его, используя γ и β. Параметры нормализации μ и σ вычисляются для текущего слоя по обучающему набору с использованием метода, называемого экспоненциальным скользящим средним. Другими словами, это не обучаемые параметры. Напротив, γ и β являются обучаемыми параметрами. Значения μ и σ, вычисленные во время обучения, используются при прямом проходе во время логического вывода.
[00398] На ФИГ. 5 и 6 показана архитектура сети глубокого обучения, используемая для предсказания вторичной структуры и доступности белков для растворителей. Входными данными для модели является взвешенная по положению матрица с использованием консервации, созданная с помощью программного обеспечения RaptorX (для обучения последовательностям банка данных белков) или выравниваний 99 позвоночных (для обучения и вывода последовательностей белков человека). Выходные данные предпоследнего слоя, длина которого составляет 51 а.к., становятся входными данными для сети глубокого обучения для классификации патогенности.
[00399] ФИГ. 7A и 7B - это дополнительная таблица 11, в которой показан пример подробностей архитектуры модели для модели глубокого обучения (DL) с предсказанием вторичной структуры с 3 состояниями. Форма определяет форму выходного тензора на каждом слое модели, а активация - это активация, переданная нейронам этого слоя. Входами в модель служили частотные матрицы, зависящие от положения (длина 51 а.к., глубина 20) для фланкирующей аминокислотной последовательности вокруг варианта.
[00400] ФИГ. 8A и 8B - это дополнительная таблица 12, в которой показан пример подробностей архитектуры модели для модели глубокого обучения для предсказанием доступности для растворителя с 3 состояниями. Форма определяет форму выходного тензора на каждом слое модели, а активация - это активация, переданная нейронам этого слоя. Входами в модель служили частотные матрицы, зависящие от положения (длина 51 а.к., глубина 20) для фланкирующей аминокислотной последовательности вокруг варианта.
[00401] На ФИГ. 20 показана предсказанная оценку патогенности для каждого аминокислотного положения в гене SCN2A с аннотациями для ключевых функциональных доменов. Вдоль гена нанесена средняя оценка PrimateAI для миссенс-замен в каждом аминокислотном положении.
[00402] На ФИГ. 21D показано сравнение классификаторов при предсказании доброкачественных последствий для тестового набора из 10 000 распространенных вариантов приматов, которые не участвовали в обучении. Ось y представляет процент вариантов приматов, правильно классифицированных как доброкачественные, после нормировки порога каждого классификатора до его 50-го процентиля на наборе из 10000 случайных вариантов, которые были сопоставлены по степени мутаций.
[00403] На ФИГ. 21E показаны распределения оценок предсказания PrimateAI для миссенс-вариантов de novo, встречающихся у пациентов с нарушениями развития (DDD), по сравнению ос здоровыми братьями и сестрами, с соответствующим P-значением суммы рангов Вилкоксона.
[00404] На ФИГ. 21F показано сравнение классификаторов в разделении de novo миссенс-вариантов в случаях DDD по сравнению с контролями. Для каждого классификатора показаны Р-значения критерия суммы рангов Вилкоксона.
[00405] ФИГ. 22A, 22B, 22C, 22D и 22E иллюстрируют точность классификации в пределах 605 генов DDD с P <0,05. ФИГ. 22А демонстрирует увеличение количества миссенс-мутаций de novo по сравнению с ожидаемым у пораженных индивидуумов из когорты DDD в пределах 605 связанных генов, которые были значимыми для de novo вариантов, приводящих к усечению белка (P <0,05). На ФИГ. 22B показано распределение оценок прогноза PrimateAI для de novo миссенс-вариантов, встречающихся у пациентов с DDD, по сравнению с незатронутыми братьями и сестрами в пределах 605 связанных генов с соответствующим Р-значением суммы рангов по Вилкоксону.
[00406] На ФИГ. 22C показано сравнение различных классификаторов в разделении миссенс-вариантов de novo в случаях заболевания и в контроле в пределах 605 генов. Ось y показывает P-значения критерия суммы рангов Вилкоксона для каждого классификатора.
[00407] На ФИГ. 22D показано сравнение различных классификаторов, показанных на характеристической кривой оператор-приемник с указанной для каждого классификатора AUC.
[00408] ФИГ. 22E иллюстрирует точность классификации и AUC для каждого классификатора. Показанная точность классификации представляет собой среднее значение истинно положительных и истинно отрицательных показателей ошибок с использованием порога, при котором классификатор предсказывает такое же количество патогенных и доброкачественных вариантов, как и ожидание на основе обогащения, показанное на ФИГ. 22А. Чтобы учесть тот факт, что 33% миссенс-вариантов de novo, связанных с DDD, представляют собой фон, максимально достижимая AUC для идеального классификатора обозначена пунктирной линией.
[00409] На ФИГ. 23A, 23B, 23C и 23D показано влияние данных, используемых для обучения, на точность классификации. Сети глубокого обучения обучаются на увеличивающемся количестве распространенных вариантов приматов и людей, вплоть до полного набора данных (385 236 вариантов). На ФИГ. 23А, эффективность классификации для каждой из сетей оценивается по точности для 10 000 отложенных вариантов приматов и вариантов de novo в случаях DDD по сравнению с контрольными.
[00410] На ФИГ. 23B и 23C показаны характеристики сетей, обученных с использованием наборов данных, содержащих 83546 распространенных вариантов для человека плюс 23380 вариантов от одного вида приматов или млекопитающих, согласно одному варианту реализации. Результаты показаны для каждой сети, обученной с разными источниками распространенных, со сравнительном анализе на 10 000 отложенных вариантов приматов (ФИГ. 23B) и на de novo миссенс-вариантах (ФИГ. 23C) в случаях DDD по сравнению с контролем.
[00411] ФИГ. 23D показывает ожидаемое насыщение всех возможных доброкачественных миссенс-положений человека распространенными вариантами, идентичными по состоянию (> 0,1%) у 504 существующих видов приматов. Ось y показывает долю миссенс-вариантов человека, наблюдаемую, по меньшей мере, у одного вида приматов, причем миссенс-варианты CpG показаны зеленым, а все миссенс-варианты показаны синим. Чтобы смоделировать распространенные варианты у каждого вида приматов, мы взяли образцы из набора всех возможных однонуклеотидных замен с заменой, с соответствующим распределением тринуклеотидного контекста, наблюдаемого для респространенных вариантов человека (частота аллелей> 0,1%) в ExAC.
[00412] ФИГ. 24 иллюстрирует поправку на влияние покрытия секвенированием на установление распространенных вариантов приматов. Вероятность наблюдения данного варианта у приматов, за исключением человека, обратно коррелирует с глубиной секвенирования в этом положении в наборе данных экзома ExAC / gnomAD. Напротив, более низкая глубина считывания gnomAD не повлияла на вероятность наблюдения общего человеческого варианта в этом положении (частота аллелей> 0,1%), потому что большое количество секвенированных экзомов человека делает установление распространенного варианта почти гарантированным. При выборе подходящих вариантов для каждого из вариантов приматов для обучения сети вероятность выбора варианта была скорректирована с учетом эффектов глубины секвенирования, в дополнение к сопоставлению по тринуклеотидному контексту для контроля степени мутаций и конверсии генов.
[00413] На ФИГ. 25A, 25B, 25C и 26 показано распознавание белковых мотивов описанными нейронными сетями. В отношении ФИГ. 25A, 25B и 25C, чтобы проиллюстрировать распознавание нейронными сетями белковых доменов, мы показываем средние оценки PrimateAI для вариантов в каждом аминокислотном положении в трех разных белковых доменах. В ФИГ. 25А выделена коллагеновая цепь COL1A2 с глицином в повторяющемся мотиве GXX. Клинически идентифицированные мутации в генах коллагена в значительной степени связаны с миссенс-мутациями глицина в повторах GXX, поскольку они мешают нормальной сборке коллагена и оказывают сильные доминантно-отрицательные эффекты. На ФИГ. 25В выделен активный сайт фермента IDS-сульфатазы, который содержит цистеин в активном центре, который посттрансляционно модифицирован в формилглицин. На ФИГ. 25C показан домен bHLHzip фактора транскрипции MYC. Основной домен связывается с ДНК через положительно заряженные остатки аргинина и лизина (выделены), которые взаимодействуют с отрицательно заряженным сахарно-фосфатным остовом. Домен лейциновой молнии содержит остатки лейцина, разделенные на семью аминокислотами (выделены), которые имеют решающее значение для димеризации.
[00414] ФИГ. 26 включает линейный график, показывающий влияние изменения каждого положения в варианте и вокруг него на предсказываемую оценку глубокого обучения для этого варианта. Мы систематически обнуляли входные данные для близлежащих аминокислот (положения от -25 до +25) вокруг варианта и измеряли изменение в предсказываемой нейронной сетью патогенности варианта. На графике показано среднее изменение предсказываемой оценки патогенности изменений в каждой соседней аминокислотной позиции для 5000 случайно выбранных вариантов.
[00415] ФИГ. 27 иллюстрирует модели корреляции весов, имитирующие матрицы оценок BLOSUM62 и Grantham. Паттерны корреляции весов из первых трех уровней сети глубокого обучения для вторичной структуры показывают корреляции между аминокислотами, которые аналогичны матрицам BLOSUM62 и Grantham Score. Левая тепловая карта показывает корреляцию весов параметров из первого сверточного слоя после двух начальных уровней повышающей дискретизации сети глубокого обучения для вторичной структуры между аминокислотами, закодированными с использованием кодирования с одним горячим состояние. Средняя тепловая карта показывает оценки BLOSUM62 между парами аминокислот. На правой тепловой карте показано расстояние Грэнтэма между аминокислотами. Корреляция Пирсона между весами глубокого обучения и оценками BLOSUM62 составляет 0,63 (P = 3,55 × 10-9). Корреляция между весами глубокого обучения и оценками по Грэнтэму составляет -0,59 (P = 4,36 × 10-8). Корреляция между оценками BLOSUM62 и Грэнтэма составляет -0,72 (P = 8,09 × 10-13).
[00416] ФИГ. 28A, 28B и 28C демонстрируют оценку показателей работы сети глубокого обучения PrimateAI и других классификаторов. На ФИГ. 28A показана точность сети глубокого обучения PrimateAI при предсказывании доброкачественных последствий для тестового набора из 10000 вариантов приматов, которые были исключены из обучения, и сравнения с другими классификаторами, включая SIFT, PolyPhen-2, CADD, REVEL, M-CAP, LRT, MutationTaster, MutationAssessor, FATHMM, PROVEAN, VEST3, MetaSVM, MetaLR, MutPred, DANN, FATHMM-MKL_coding, Eigen, GenoCanyon, Integrated_fitCons и GERP. Ось y представляет процент вариантов у приматов, классифицированных как доброкачественные, на основе нормализации порога для каждого классификатора до его 50-го процентиля с использованием набора из 10000 случайно выбранных вариантов, которые сопоставлялись с вариантами приматов для тринуклеотидного контекста, чтобы контролировать степень мутаций и конверсию генов.
[00417] На ФИГ. 28B показано сравнение показателей работы сети PrimateAI при разделении de novo миссенс-вариантов в случаях DDD и в контроле, а также 20 существующих методов, перечисленных выше. Ось y показывает P-значения критерия суммы рангов Вилкоксона для каждого классификатора.
[00418] На фиг.28C показано сравнение показателей работы сети PrimateAI при разделении de novo миссенс-вариантов в случаях DDD и здоровых контролей в пределах 605 генов, связанных с заболеванием, с помощью 20 методов, перечисленных выше. Ось y показывает P-значения критерия суммы рангов Вилкоксона для каждого классификатора.
[00419] ФИГ. 29A и 29B иллюстрируют распределение оценок предсказания четырех классификаторов. Гистограммы оценок предсказаний четырех классификаторов, включая SIFT, PolyPhen-2, CADD и REVEL, для de novo миссенс-вариантов, встречающихся в случаях DDD, по сравнению с незатронутыми контролями, с соответствующими P-значениями суммы рангов Вилкоксона.
[00420] На ФИГ. 30A, 30B и 30C сравнивается точность сети PrimateAI и других классификаторов при разделении патогенных и доброкачественных вариантов в 605 генах, связанных с заболеванием. Диаграмма рассеяния в ФИГ. 30A показывает эффективность каждого из классификаторов в случаях DDD по сравнению с контролями (ось y) и точность предсказания доброкачественности для набора отложенных данных приматов (ось x). На ФИГ. 30B сравниваются различные классификаторы в разделении de novo миссенс-вариантов в случаях заболевания по сравнению с контролем в пределах 605 генов, показанных на кривой рабочей характеристики приемника (ROC), с площадью под кривой (AUC), указанной для каждого классификатора. ФИГ. 30C показывает точность классификации и AUC для сети PrimateAI и 20 классификаторов, перечисленных в ФИГ. 28A, 28B и 28C. Показанная точность классификации представляет собой среднее значение истинно положительных и истинно отрицательных показателей с использованием порога, при котором классификатор может предсказать такое же количество патогенных и доброкачественных вариантов, как ожидалось на основе обогащения ФИГ. 22А. Максимально достижимая AUC для идеального классификатора обозначена пунктирной линией, исходя из предположения, что de novo миссенс-варианты в случаях DDD являются в 67% патогенными вариантами, и в 33%доброкачественными, а de novo миссенс-варианты в контроле являются на 100% доброкачественными.
[00421] ФИГ. 31A и 31B иллюстрируют корреляцию между показателями работы классификатора на вариантах ClinVar, курируемых людьми, и показателями работы на эмпирических наборах данных. Диаграмма рассеяния в ФИГ. 31A показывает точность классификации (ось y) вариантов ClinVar для 10000 отложенных вариантов приматов (ось x) для каждого из 20 других классификаторов и сети PrimateAI, обученной с использованием данных только для человека или данных человека + приматы. Показаны коэффициент корреляции Спирмена rho и соответствующее значение P. Чтобы ограничить оценку данными, которые не использовались для обучения классификаторов, мы использовали только варианты ClinVar, которые были добавлены в период с января 2017 года по ноябрь 2017 года, и исключили распространенные человеческие варианты из ExAC / gnomAD (частота аллелей> 0,1%). Показанная точность классификации ClinVar представляет собой среднее значение истинно положительных и истинно отрицательных показателей с использованием порогового значения, при котором классификатор предсказывает такое же количество патогенных и доброкачественных вариантов, как и наблюдаемое в наборе данных ClinVar.
[00422] Диаграмма рассеяния в ФИГ. 31B показывает точность классификации (ось y) вариантов ClinVar, случаи DDD по сравнению с контрольным набором данных (ось x) для каждого из 20 других классификаторов и сети PrimateAI, обученной с использованием данных только для человека или человека + приматы.
[00423] ФИГ. 32 - это Дополнительная таблица 14, которая демонстрирует показатели работы моделей прогнозирования доступности для растворителей и вторичной структуры с 3 состояниями на аннотированных образцах из Protein DataBank с использованием 6367 несвязанных последовательностей белков для обучения, 400 для валидации и 500 для тестирования. Из Protein DataBank были выбраны только белки с <25% сходством последовательностей. Мы приводим значения точности сетей глубокого обучения как метрики качества работы, поскольку эти три класса не сильно разбалансированы по вторичной структуре или по доступности для растворителей.
[00424] ФИГ. 33 - это Дополнительная таблица 15, в которой показано сравнение показателей работы сети глубокого обучения с использованием аннотированных меток вторичной структуры человеческих белков из базы данных DSSP, если они доступны в сети глубокого обучения с использованием предсказанных меток вторичной структуры.
[00425] ФИГ. 34 - это Дополнительная таблица 17, которая показывает значения точности для 10 000 отложенных вариантов приматов и p-значения для вариантов de novo в случаях DDD по сравнению с контролями для каждого из 20 оцениваемых нами классификаторов. Модель PrimateAI с данными только о человеке - это наша сеть глубокого обучения, которая была обучена с использованием помеченного доброкачественного набора данных для обучения, включающего только распространенные человеческие варианты (83.5 тыс. Вариантов с> 0.1% в популяции), в то время как модель PrimateAI с данными о человеке + приматах - это наша сеть глубокого обучения, обученная на полном наборе из 385 тыс. размеченных доброкачественных вариантов, включающих как распространенные варианты человека, так и варианты приматов.
[00426] ФИГ. 35 - это Дополнительная таблица 19, в которой показано сравнение показателей работы различных классификаторов для вариантов de novo для случаев DDD в сравнении с контрольным набором данных, ограниченным 605 генами, связанными с заболеванием. Для нормализации между различными методами для каждого классификатора мы определили порог, при котором классификатор будет предсказывать такое же количество патогенных и доброкачественных вариантов, как и ожидаемое на основе обогащения DDD и контрольного набора. Показанная точность классификации представляет собой среднее значение показателей ошибок истинно положительных и истинно отрицательных определений при этом пороге.
[00427] На ФИГ. 49A, 49B, 49C, 49D и 49E показаны отношения миссенс / синонимы в частотном спектре аллелей человека. ФИГ. 49A показывает миссенс и синонимичные варианты, наблюдаемые у 123 136 человек из базы данных ExAC / gnomAD, которые были разделены на четыре категории по частоте аллелей. Заштрихованные серые столбики представляют собой количество синонимичных вариантов в каждой категории; темно-зеленые столбики представляют собой миссенс-варианты. Высота каждого столбика масштабируется по количеству синонимичных вариантов в каждой категории частоты аллелей, и после корректировки на частоту мутаций отображаются числа и отношения миссенс / синонимы. ФИГ. 49B и 49C иллюстрируют частотный спектр аллелей для человеческих миссенс- и синонимичных вариантов, которые идентичны по состоянию (IBS) с распространенным вариантами шимпанзе (ФИГ. 49B) и одиночными вариантами (синглтонами) шимпанзе (ФИГ. 49C). Истощение миссенс-вариантов шимпанзе при распространенных частотах аллелей человека (> 0,1%) по сравнению с частотами редких аллелей человека (<0,1%) показано красным прямоугольником вместе с соответствующими P-значениями теста хи-квадрат (χ2).
[00428] На ФИГ. 49D показаны человеческие варианты, которые наблюдаются, по меньшей мере, у одного из видов приматов, кроме человека. ФИГ. 49E иллюстрирует количество доброкачественных и патогенных миссенс-вариантов в общей базе данных ClinVar (верхний ряд) по сравнению с вариантами ClinVar в когорте из 30 человек, отобранных по частотам аллелей ExAC / gnomAD (средний ряд), по сравнению с вариантами, наблюдаемыми у приматов (нижний ряд ). Были исключены вариантами, противоречиво определенные как доброкачественные и патогенные, аннотированные только неопределенным значением.
[00429] На ФИГ. 50A, 50B, 50C и 50D показан очищающий отбор на миссенс-вариантах, идентичных по состоянию другим видам. ФИГ. 50А изображает частотный спектр аллелей для миссенс-вариантов человека и синонимичных вариантов, которые идентичны по состоянию, на варианты, присутствующие у четырех видов млекопитающих, не относящихся к приматам (мыши, свиньи, козы и коровы). Истощение миссенс-вариантов при распространенных частотах аллелей человека (> 0,1%) показано красным прямоугольником вместе с соответствующим Р-значением критерия хи-квадрат (χ2).
[00430] ФИГ. 50B представляет собой диаграмму рассеяния, показывающую истощение миссенс-вариантов, наблюдаемое у других видов при распространенных частотах аллелей человека (> 0,1%), в зависимости от эволюционного расстояния вида от человека, выраженного в единицах длины ветви (среднее количество замен на положение нуклеотида). Общая длина ветви между каждым видом и человеком указана рядом с названием вида. Значения истощения для одноэлементных и обычных вариантов показаны для видов, для которых были доступны частоты вариантов, за исключением горилл, среди которых были родственные особи.
[00431] ФИГ. 50C иллюстрирует количество доброкачественных и патогенных миссенс-вариантов в когорте из 30 человек, отобранных по частотам аллелей ExAC / gnomAD (верхний ряд), по сравнению с вариантами, наблюдаемыми у приматов (средний ряд), и с вариантами, наблюдаемыми у мышей, свиней, коз, и корова (нижний ряд). Были исключены варианты с противоречащими доброкачественными и патогенными метками утверждения и варианты, аннотированные только с неопределенной значимостью.
[00432] ФИГ. 50D представляет собой диаграмму рассеяния, показывающую истощение фиксированных миссенс-замен, наблюдаемое в парах близкородственных видов при распространенных частотах аллелей человека (> 0,1%), в зависимости от эволюционного расстояния вида от человека (выраженного в единицах средней длины ветви).
[00433] На ФИГ. 51 показаны ожидаемые отношения миссенс / синонимы по частотному спектру аллелей человека в отсутствие очищающего отбора. Заштрихованные серые столбики представляют количество синонимичных вариантов, а темно-зеленые столбики представляют количество пропущенных вариантов. Пунктирная линия показывает базовую линию, образованную синонимичными вариантами. Отношения миссенс: синонимы указаны для каждой категории частоты аллелей. В соответствии с одной реализацией ожидаемое количество миссенс-вариантов и синонимов в каждой категории частот аллелей было рассчитано путем взятия интронных вариантов из набора данных ExAC / gnomAD, содержащего 123 136 экзомов, и их использования для оценки доли вариантов, которые, как ожидается, попадут в каждую из четырех частот аллелей. категории, на основе тринуклеотидного контекста варианта, который определяет скорость мутаций и смещение GC при конверсии гена.
[00434] На ФИГ. 52A, 52B, 52C и 52D изображают отношения миссенс / синонимы для вариантов CpG и не-CpG. ФИГ. 52A и 52B показывают отношения миссенс / синонимы для вариантов CpG (ФИГ. 52A) и вариантов, не являющихся CpG (ФИГ. 52A), по частотному спектру аллелей человека, с использованием всех вариантов из экзомов ExAC / gnomAD. ФИГ. 52C и 52D показывают отношения миссенс / синонимы для вариантов CpG (ФИГ. 52C) и вариантов не-CpG (ФИГ. 52D) в частотном спектре аллелей человека, ограниченные только вариантами человека, которые идентичны по состоянию с распространенными полиморфизмами шимпанзе.
[00435] ФИГ. 53, 54 и 55 иллюстрируют отношения миссенс: синонимы человеческих вариантов, идентичных по состоянию шести приматам. Паттерны отношений миссенс / синонимы в частотном спектре аллелей человека для вариантов ExAC / gnomAD, которые идентичны по состоянию с вариантами, присутствующими у шимпанзе, бонобо, гориллы, орангутанга, резусов и игрунок.
[00436] ФИГ. 56 - моделирование, показывающее насыщение новыми распространенными миссенс-вариантами, обнаруженными в результате увеличения размера исследуемых когорт людей. При моделировании генотипы каждого образца отбирались в соответствии с частотами аллелей gnomAD. Доля обнаруженных общих вариантов gnomAD усредняется по 100 моделированиям для каждого размера выборки от 10 до 100 000.
[00437] ФИГ. 57 показывает точность PrimateAI для различных профилей сохранения в геноме. Ось x представляет процентную вероятность выравнивания 51 А.к. вокруг последовательности с выравниванием 99 позвоночных. Ось y представляет собой классификацию точности PrimateAI для вариантов в каждом из сегментов консервации, измеренную на тестовом наборе данных из 10 000 отложенных вариантов приматов.
[00438] ФИГ. 58 - это Дополнительная таблица 5, которая показывает вклады в размеченный набор данных распространенных вариантов человека и вариантов, присутствующих у приматов, отличных от человека.
[00439] ФИГ. 59 - это Дополнительная таблица 8, которая показывает влияние частоты аллелей на ожидаемое отношение миссенс: синонимы. Ожидаемое количество синонимичных и миссенс-вариантов было рассчитано на основе частотного спектра аллелей вариантов в интронных областях, в меньшей степени на 20-30 нуклеотидов от границ экзонов, с использованием тринуклеотидного контекста для контроля системных ошибок степени мутаций и ошибок генов.
[00440] ФИГ. 60 - это Дополнительная таблица 9, в которой показан анализ ClinVar. Согласно одной реализации варианты, загруженные из базы данных сборок ClinVar за ноябрь 2017 г., были отфильтрованы и удалены варианты с конфликтующими аннотациями и исключены варианты с неопределенным значением, в результате чего осталось 17 775 доброкачественных вариантов и 24 853 патогенных варианта.
[00441] ФИГ. 61 - это Дополнительная таблица 10, которая показывает количество миссенс-вариантов из других видов, обнаруженных в ClinVar, согласно одному варианту реализации. Варианты должны были быть идентичными по состоянию с соответствующим человеческим вариантом и иметь идентичные нуклеотиды в двух других положениях в рамке считывания, чтобы гарантировать те же последствия кодирования.
[00442] ФИГ. 62 представляет собой Таблицу 1, который демонстрирует один вариант реализации обнаружения 14 дополнительных кандидатных генов умственной отсталости, которые ранее не достигли порога общегеномной значимости в первоначальном исследовании DDD.
[00443] ФИГ. 63 представляет собой Таблицу 2, демонстрирующую один вариант реализации средней разницы в баллах по Грэнтэму между патогенными и доброкачественными вариантами в ClinVar, которая в два раза больше, чем разница между de novo вариантами в случаях DDD по сравнению с контролем в пределах 605 генов, связанных с заболеванием.
Генерация данных
[00444] Все координаты, используемые в статье, относятся к построению человеческого генома UCSC hg19 / GRCh37, включая координаты для вариантов у других видов, которые были картированы на hg19 с использованием множественного выравнивания последовательностей с использованием процедуры, описанной в этом разделе. Кодирующая белок последовательность ДНК и множественные выравнивания последовательностей 99 геномов позвоночных с человеческими были загружены из браузера генома UCSC для сборки hg19. (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/multiz100way/alignments/knownCanonical.exonNuc.fa.gz). Для генов с множественными каноническими аннотациями генов был выбран самый длинный кодирующий транскрипт.
[00445] Мы загрузили данные полиморфизмов экзома человека из Консорциума агрегации экзома (ExAC) / базы данных агрегирования генома (gnomAD) v2.0, в котором собраны данные полного секвенирования экзома (WES) 123 136 человек из восьми субпопуляций по всему миру (http : //gnomad.broadinstitute.org/). Мы исключили варианты, которые не соответствовали фильтрам контроля качества по умолчанию, как аннотировано в файле VCF базы ExAC, или выпадали за пределы областей канонического кодирования. Чтобы избежать эффектов сбалансированного отбора, мы также исключили варианты из расширенной области MHC (chr6: 28 477 797-33 448 354) для анализа приматов. Проект секвенирования генома большой обезьяны предоставляет данные и генотипы всего генома для 24 шимпанзе, 13 бонобо, 27 горилл и 10 орангутанов (в том числе 5 из суматранского подвида и 5 из калимантанского подвида, которые мы свернули для последующего анализа). Исследование шимпанзе и бонобо предоставляет последовательности генома еще 35 шимпанзе. Однако, поскольку варианты этих дополнительных шимпанзе не определялись с использованием тех же методов, что и проект секвенирования генома человекообразных обезьян, мы исключили их из анализа частотного спектра аллелей и использовали их только для обучения модели глубокого обучения. Вариации этих исследований разнообразия приматов уже были сопоставлены с эталоном человека (hg19). Кроме того, для мартышек и резусов 16 особей-резусов и 9 особей мартышек были использованы для анализа вариабельности исходного секвенирования геномов этих видов, но информация на индивидуальном уровне недоступна.
[00446] Проект секвенирования генома человекообразных обезьян 4 предоставляет данные секвенирования полного генома и генотипы для 24 шимпанзе, 13 бонобо, 27 горилл и 10 орангутанов (включая 5 из суматранского подвида и 5 из калимантанского подвида, которые мы свернули для последующего анализа). Исследование шимпанзе и бонобо предоставляет последовательности генома еще 35 шимпанзе. Однако, поскольку варианты этих дополнительных шимпанзе не вызывались с использованием тех же методов, что и проект секвенирования генома человекообразных обезьян, мы исключили их из анализа частотного спектра аллелей и использовали их только для обучения модели глубокого обучения. Варианты из этих исследований разнообразия приматов уже были выровнены с референсом человека (hg19). Кроме того, для мартышек и резусов 16 особей-резусов и 9 особей мартышек были использованы для анализа вариабельности исходного секвенирования геномов этих видов, но информация на индивидуальном уровне была недоступна.
[00447] Для сравнения с другими приматами и млекопитающими мы также загрузили SNP других видов из dbSNP, включая резусов, мартышку, свинью, корову, козу, мышь, курицу и рыбок данио. dbSNP также включала дополнительные варианты орангутана, которые мы использовали только для обучения модели глубокого обучения, поскольку информация об индивидуальном генотипе не была доступна для анализа частотного спектра аллелей. Мы отказались от других видов, таких как собаки, кошки или овцы, поскольку dbSNP предоставляет ограниченное количество вариантов для этих видов.
[00448] Чтобы картировать варианты с человеческими, мы использовали выравнивание 99 видов позвоночных, чтобы обеспечить ортологичное картирование 1:1 на участки, кодирующие белки человека. Картирование вариантов с использованием ортологичных выравниваний нескольких видов было важно для удаления артефактов, вызванных псевдогеном или ретротранспонированными последовательностями, которые возникают при прямом картировании SNP между видами с использованием таких инструментов, как liftOver, которые позволяют картировать многих к 1. В случаях, когда сборка генома вида в dbSNP не соответствовала сборке генома вида при выравнивании множественных последовательностей 99 позвоночных, мы использовали liftOver для обновления вариантов сборки генома, используемого при выравнивании множественных последовательностей. Мы принимали варианты как идентичные по состоянию, если они встречались в любой ориентации референсной / альтернативной, например, если человеческий референс был G, а альтернативный аллель был A, это считалось идентичным по состоянию с вариантом в других видах, где референс представлял собой A, а альтернативный аллель - G. Чтобы гарантировать, что вариант имел такие же предсказанные последствия для кодирования белка, как у человека, так и у других видов, мы требовали, чтобы два других нуклеотида в кодоне были идентичны для разных видов, как для миссенс-варианта, так и для синонимичного варианта. Полиморфизмы каждого вида, включенного в анализ, перечислены в файле дополнительных данных 1, а подробные показатели показаны в дополнительной таблице 1.
[00449] Чтобы гарантировать, что варианты из каждой отправляемой партии dbSNP были высокого качества и правильно выровнены с человеческими, мы вычисляли отношение миссенс: синонимы для каждой партии, подтверждая, что оно меньше ожидаемого отношения 2.2: 1; у большинства видов соотношение было ниже 1: 1, особенно у рыбок данио и мышей, которые, как ожидается, будут иметь очень большие эффективные размеры популяции. Мы исключили две партии SNP коров с необычно высокими отношениями миссенс: синонимы из дальнейшего анализа (snpBatch_1000_BULL_GENOMES_1059190.gz с соотношением 1,391 и snpBatch_COFACTOR_GENOMICS_1059634.gz с соотношением 2,568). Среднее соотношение миссенс: синонимы для остальных партий коров составляло 0,8: 1.
Коррекция влияния частоты аллелей на миссенс: соотношение синонимов, скорость мутаций, генетический дрейф и конверсия генов, обусловленная GC
[00450] В дополнение к действию очищающего отбора, наблюдаемое истощение человеческих миссенс-вариантов с высокими частотами аллелей также может зависеть от факторов, не связанных с естественным отбором. Вероятность появления нейтральной мутации с определенной частотой аллеля в популяции является функцией степени мутаций, конверсии генов и генетического дрейфа, и эти факторы потенциально могут вносить систематическую ошибку в отношение миссенс / синонимы по спектру частот аллелей даже в отсутствие сил отбора.
[00451] Чтобы вычислить ожидаемые отношения миссенс:синонимы в каждой категории частоты аллелей в отсутствие отбора, влияющего на кодирование белка, мы выбрали варианты в интронных областях на 31-50 п.о. выше и 21-50 п.о. ниже каждого экзона. Эти области были выбраны достаточно удаленными, чтобы избежать влияния протяженных мотивов сплайсинга. Поскольку эти области находятся рядом с краями последовательности захвата экзома для экзомов ExAC / gnomAD, для обеспечения точного определения вариантов мы удалили все области chrX и исключили области со средней глубиной прочитывания <30. Каждый вариант и непосредственно следующие за ним в направлении 5 'и в направлении 3 'нуклеотиды попадают в один из 64 тринуклеотидных контекстов. Если мы мутируем средний нуклеотид в три других основания, всего возможно 64 × 3 = 192 тринуклеотидных конфигурации. Поскольку тринуклеотидные конфигурации и их обратные комплементы эквивалентны, фактически существует 96 тринуклеотидных контекстов. Мы заметили, что тринуклеотидный контекст имеет очень сильное влияние на скорость мутаций и меньшее влияние на конверсию генов, обусловленную GC, что делает тринуклеотидный контекст эффективным для моделирования этих переменных.
[00452] Внутри этих интронных областей мы взяли каждый вариант из 126 136 экзомов ExAC / gnomAD и разделили их на 4 × 192 категории на основе четырех категорий частоты аллелей (одиночный, более чем одиночный ~ 0,01%, 0,01% ~ 0,1% ,> 0,1%) и 192 тринуклеотидных контекста. Мы нормализовали количество вариантов, наблюдаемых в каждой из 4 × 192 категорий (частота аллелей × тринуклеотидный контекст), разделив на общее количество возможных вариантов с этим тринуклеотидным контекстом (полученным путем замены каждого нуклеотида в интронной последовательности на три различные пути). Таким образом, для каждого из 192 тринуклеотидных контекстов мы получили ожидаемую долю вариантов, которые попадут в каждую из 4 категорий частот аллелей в отсутствие отбора, кодирующего белок. Это косвенно моделирует эффекты степени мутаций, конверсии генов, обусловленной GC, и генетического дрейфа, которые возникают из-за различий в тринуклеотидном контексте (Дополнительная таблица 7)..
[00453] Чтобы получить ожидаемое соотношение миссенс: синонимы в каждой частотной категории аллелей, мы подсчитали общее количество возможных синонимичных и миссенс-мутаций в геноме человека, доступных для однонуклеотидных замен, и присвоили каждую из них одному из 192 тринуклеотидных контекстов. Для каждого контекста мы использовали таблицу 4 × 192, чтобы вычислить количество вариантов, которые, как ожидается, попадут в каждую из 4 категорий частот аллелей. Наконец, мы суммировали количество синонимичных и миссенс-вариантов в 192 тринуклеотидных контекстах, чтобы получить общее ожидаемое количество синонимических и миссенс-вариантов в каждой из четырех категорий частот аллелей (ФИГ.51 и Дополнительная таблица 8 (ФИГ. 59)).
[00454] Ожидаемые отношения миссенс: синонимы были почти постоянными по частотному спектру аллелей и близкими к соотношению 2,23: 1, которое можно было бы ожидать для вариантов de novo в отсутствие естественного отбора, за исключением одиночных вариантов, ожидаемое соотношение миссенс: синонимы для которых было 2,46: 1. Это указывает на то, что из-за действия факторов, не зависящих от давления отбора на кодирование белков (степень мутаций, конверсия генов, генетический дрейф), варианты с категорией частоты одиночного аллелей в ExAC / gnomAD, как ожидается, будут иметь отношение миссенс: синонимы около 10%, по умолчанию выше, чем у мутаций de novo. Чтобы исправить это, мы скорректировали соотношение миссенс: синонимы для синглтонов на 10% в частотном анализе аллелей (ФИГ. 49A, 49B, 49C, 49D и 49E и ФИГ. 50A, 50B, 50C и 50D). Эта небольшая корректировка снизила расчетное истощение миссенс-вариантов по распространенным человеческим вариантам, присутствующим у приматов и других млекопитающих (показано на ФИГ.49A, 49B, 49C, 49D и 49E и ФИГ.50A, 50B, 50C и 50D) примерно на ~ 3,8%. . Более высокое соотношение миссенс: синонимы для одиночных вариантов обусловлено мутациями-транзициями (которые с большей вероятностью создают синонимичные изменения), имеющими более высокие частоты аллелей из-за более высокой скорости мутаций, чем мутации-трансверсии (которые с большей вероятностью создают миссенс-изменения).
[00455] Кроме того, это объясняет наблюдаемое соотношение миссенс: синонимы 2,33: 1 для одиночных вариантов в ExAC / gnomAD, которое превышает ожидаемое соотношение для мутаций de novo 2,23: 1. После учета влияния частотного спектра аллелей на соотношение миссенс: синонимы это фактически отражает истощение одиночных вариантов на 5,3% по сравнению с ожидаемым, что, предположительно, было бы связано с отбором против патогенных миссенс-мутаций с доминантными формами наследования de novo. Действительно, когда мы рассматриваем только гаплонедостаточные гены с высокой вероятностью потери функции (pLI> 0,9), соотношение миссенс: синонимы для одиночных вариантов ExAC / gnomAD составляет 2,04: 1, что указывает на истощение примерно на 17% внутри гаплонедостаточных генов. Этот результат согласуется с предыдущими оценками, согласно которым 20% миссенс-мутаций эквивалентны мутациям с потерей функции, если предположить некоторую степень неполной пенетрантности.
[00456] Мы также специально исследовали отношения миссенс:синонимы для вариантов CpG и не-CpG по частотному спектру аллелей человека из-за больших различий в частотах их мутаций (ФИГ. 52A, 52B, 52C и 52D). Мы подтвердили, что как для мутаций CpG, так и для не-CpG, человеческие варианты, которые идентичны по состоянию с распространенными полиморфизмами шимпанзе, имеют почти постоянные отношения миссенс:синонимы по частотному спектру аллелей.
Истощение человеческих миссенс-вариантов, которые идентичны по состояниям с полиморфизмами у других видов
[00457] Чтобы оценить, будут ли варианты других видов переноситься при распространенных частотах аллелей (> 0,1%) у человека, мы идентифицировали варианты человека, которые были идентичны по состоянию с вариантами у других видов. Для каждого из вариантов мы отнесли его к одной из четырех категорий на основании частот его аллелей в человеческих популяциях (синглтон (одиночный вариант), более, чем одиночный ~ 0,01%, 0,01% ~ 0,1%,> 0,1%) и оценили снижение отношения миссенс : синонимы (MSR) между редким (<0,1%) и распространенными (> 0,1%) вариантами. Истощение количества миссенс-вариантов, идентичных по состояниям, при распространенных частотах аллелей человека (> 0,1%) указывает на долю вариантов из других видов, которые являются достаточно вредными, чтобы они могли быть отфильтрованы естественным отбором при распространенных частотах аллелей у человека.
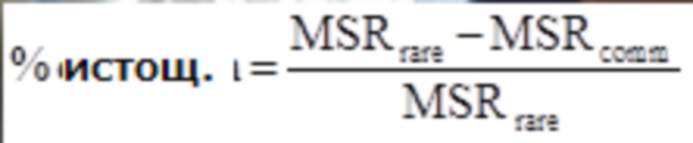
rare = редкие; comm =распространенные
[00458] Отношения миссенс: синонимы и проценты истощения были рассчитаны для каждого вида и показаны на ФИГ. 50B и в Дополнительной таблице 2. Кроме того, для распространенных вариантов шимпанзе (ФИГ. 49A), одиночных вариантов шимпанзе (ФИГ. 49C) и вариантов млекопитающих (ФИГ. 50A) мы применили критерий однородности хи-квадрат (χ2) на Таблице сопряженности 2×2, чтобы проверить, были ли различия в соотношениях миссенс: синонимы между редкими и распространенными вариантами значимыми.
[00459] Поскольку секвенирование было выполнено только на ограниченном количестве особей из проекта по разнообразию человекообразных обезьян, мы использовали частотный спектр аллелей человека из ExAC / gnomAD для оценки доли отобранных вариантов, которые были редкими (<0,1%) или распространенными (> 0,1%) в общей популяции шимпанзе. Мы отобрали когорту из 24 человек на основе частот аллелей ExAC / gnomAD и определили миссенс-варианты, которые наблюдались один или более раз в этой когорте. Варианты, которые наблюдались более одного раза, имели шанс 99,8% быть распространенными (> 0,1%) в общей популяции, тогда как варианты, которые наблюдались только один раз в когорте, имели шанс 69% быть распространенными в общей популяции. В ФИГ. 49B и 49C, мы показываем, что вследствие того, что некоторые из однотипных вариантов шимпанзе являются редкими вредными мутациями, мы наблюдаем истощение одиночных вариантов шимпанзе с высокими частотами аллелей у человека, но не для распространенных вариантов шимпанзе. Примерно половина вариантов шимпанзе, наблюдавшихся в когорте из 24 особей, наблюдалась только один раз, а примерно половина - более одного раза.
[00460] Чтобы подтвердить, что наблюдаемое истощение миссенс-вариантов у более удаленных млекопитающих не было связано с мешающим эффектом генов, которые более консервативны, и, соответственно, более точно выравниваются, мы повторили вышеупомянутый анализ, ограничиваясь только генами с > 50% средней идентичностью нуклеотидов при множественном выравнивании последовательностей 11 приматов и 50 млекопитающих по сравнению с человеком (см. дополнительную таблицу 3). Это удалило из анализа ~ 7% генов, кодирующих человеческий белок, без существенного влияния на результаты.
Фиксированные замены среди приматов, млекопитающих и далеких позвоночных
[00461] Чтобы гарантировать, что на наши результаты с использованием вариантов dbSNP не повлияли проблемы с данными вариантов или артефакты одомашнивания (поскольку большинство видов, выбранных из dbSNP, были одомашненными), мы также повторили анализ с использованием фиксированных замен из пар близко родственных виды вместо внутривидовых полиморфизмов. Мы загрузили филогенетическое дерево 100 видов позвоночных (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/multiz100way/hg19.100way.commonNames.nh) из геномного браузера UCSC с филогенетическим расстоянием, измеренным в длине ветвей (среднее количество нуклеотидных замен на положение). Для дальнейшего анализа мы выбрали пары близкородственных видов (длина ветви <0,25). Чтобы идентифицировать фиксированные замены между близкородственными парами видов, мы загрузили кодирующие области для множественных выравниваний последовательностей 99 геномов позвоночных с человеческими, а также выравнивания геномов 19 млекопитающих (16 приматов) с человеческими из геномного браузера UCSC. Дополнительное сопоставление 19 млекопитающих с несколькими видами было необходимо, потому что некоторые виды приматов, такие как бонобо, отсутствовали в сопоставлении 99 позвоночных (http://hgdownload.soe.ucsc.edu/goldenPath/hg38/multiz20way/alignments/knownCanonical. exo nNuc.fa.gz). Всего было получено 15 пар близкородственных видов, в том числе 5 пар приматов, перечисленных на ФИГ. 50D и в Дополнительной таблице 4.
[00462] Мы провели множественные выравнивания последовательностей 19 млекопитающих или геномов 99 позвоночных с человеческими в пределах канонических кодирующих областей и получили замены нуклеотидов между каждой выбранной парой позвоночных, перечисленных в файле дополнительных данных 2. Эти замены были выровнены с геномом человека при требовании, чтобы два других нуклеотида в кодоне оставались неизменными между человеком и другими видами, и с приемом варианта либо в референсной, либо в альтернативной ориентации. Используя человеческие варианты, которые были идентичны по состояниям с фиксированными заменами из пар родственных видов, мы рассчитали отношения миссенс:синонимы для вариантов в категориях частот редких (<0,1%) и общих (> 0,1%) аллелей, чтобы получить долю фиксированных замен при отрицательном отборе, как показано в Дополнительной таблице 4.
Анализ данных полиморфизма для человека, приматов, млекопитающих и других позвоночных по ClinVar
[00463] Чтобы изучить клиническое влияние вариантов, которые идентичны по состоянию с другими видами, мы загрузили релиз сводки вариантов для базы данных ClinVar (ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/ Clinvar_20171029.vcf.gz от 2 ноября 2017 г.) 12. База данных содержала 324 698 вариантов построения генома hg19, из которых 122 884 были несенсорными однонуклеотидными вариантами, отображаемыми в нашем списке генов, кодирующих белок (Дополнительная таблица 9). Большинство вариантов в базе данных ClinVar не имели миссенс-последствий и были исключены. Затем мы отфильтровали варианты с противоречивыми интерпретациями патогенности и оставили только те, которые имели доброкачественные, вероятно доброкачественные, патогенные и вероятно патогенные аннотации. Мы объединили варианты с аннотациями «Доброкачественные» или «Вероятно доброкачественные» в одну категорию, а также объединили варианты с аннотациями «патогенные» или «вероятные патогенные». Следуя шагам фильтрации, приведенным в дополнительной таблице 9, всего 24 853 варианта отнесены к категории патогенных и 17 775 вариантов - к доброкачественным; остальные были исключены, поскольку они представляют собой варианты неизвестного значения или противоречивые аннотации.
[00464] Чтобы получить исходный (фоновый) уровень для миссенс-вариантов ClinVar в человеческой популяции, мы исследовали миссенс-варианты ClinVar в когорте из 30 человек, отобранных по частотам аллелей ExAC / gnomAD. Этот размер когорты был выбран, чтобы примерно отразить количество особей, секвенированных в проекте исследования разнообразия приматов. Мы определили среднее количество патогенных и доброкачественных вариантов в когорте из 30 человек (ФИГ. 49E) из 100 таких симуляций. Поскольку кураторы систематически аннотировали общие человеческие варианты с доброкачественными последствиями в ClinVar, мы исключили варианты с частотой аллелей более 1%, чтобы избежать этой систематической ошибки курирования.
[00465] Мы проанализировали варианты ClinVar, которые были идентичны по состоянию с вариантам у приматов, млекопитающих и других позвоночных. Количество доброкачественных и патогенных вариантов для каждого вида показано в дополнительной таблице 10. Сводная информация о количестве вариантов ClinVar, которые присутствовали у людей, приматов и более отдаленных млекопитающих, показаны в ФИГ. 49E и 50B, а также результаты теста однородности по критерию хи-квадрат (χ2) для различий в соотношении доброкачественных и патогенных вариантов.
Генерация доброкачественных вариантов для обучения моделей
[00466] Варианты, которые распространены в человеческой популяции, в значительной степени нейтральны, за исключением редких случаев эффектов основателя или балансирующего выбора, что делает их подходящими в качестве удобного обучающего набора данных для машинного обучения, на который не влияют ошибки интерпретации человеком. Мы использовали данные частоты аллелей из 123136 экзомов из базы данных ExAC / gnomAD (выпуск v2.0), исключая варианты, которые не прошли фильтры, в результате чего у нас осталось 83546 миссенс-вариантов с общей популяционной аллельной частотой> = 0,1% в рамках канонического белок-кодирующих транскриптов.
[00467] На основании наших более ранних результатов, показывающих, что варианты, присутствующие у приматов, в значительной степени являются доброкачественными для человека, мы создали обучающий набор доброкачественных данных для машинного обучения, включающий распространенные человеческие варианты (частота аллелей> 0,1%), варианты шимпанзе, бонобо, гориллы и орангутана из проекта по разнообразию обезьян, и дополнительного секвенирования приматов, а также варианты резусов, орангутанов и мартышек из dbSNP. В общей сложности 301 690 уникальных вариантов приматов были добавлены к доброкачественному обучающему набору согласно одному варианту реализации. Количество доброкачественных обучающих вариантов, предоставленных каждым источником, показано в Дополнительной таблице 5.
[00468] Необходимо оговориться, что, хотя большинство вариантов приматов распространены в соответствующих популяциях, лишь меньшая их часть является редкими вариантами. Поскольку у видов приматов, отличных от человека, было секвенировано ограниченное количество особей, мы ожидаем, что набор установленных вариантов обычно представляет собой распространенные варианты. Действительно, мы обнаружили, что отношение миссенс: синонимы для вариантов от каждого вида приматов составляет менее половины ожидаемого соотношения 2,23: 1 для мутации de novo, что указывает на то, что это в основном распространенные варианты, которые уже прошли через сито отбора. Более того, для когорты шимпанзе мы подсчитали, что ~ 84% установленных вариантов присутствуют с общими частотами аллелей (> 0,1%) в соответствующих популяциях. Поскольку ~ 50% вновь возникающих миссенс-мутаций отфильтровываются очищающим отбором при распространенных частотах аллелей человека (> 0,1%) (ФИГ. 49A), эта цифра согласуется с ~ 16% редких вариантов, составляющих наблюдаемое истощение 8,8% человеческих аллелей миссенс-вариантов, которые идентичны по состояниям наблюдаемым вариантам изменчивости (ФИГ. 49D).
[00469] Применяя оценку, согласно которой ~ 20% миссенс-мутаций человека соответствуют потере функции, можно ожидать, что варианты приматов будут включать 3,2% полностью патогенных мутаций, 91,2% доброкачественных мутаций (допускаются при частоте аллелей> 0,1%) и 5,6% промежуточные мутации, которые не полностью уничтожают функцию гена, но достаточно опасны, чтобы отфильтровать их при распространенных частотах аллелей (> 0,1%). Несмотря на известные недостатки в этом наборе обучающих данных, точность классификации сети глубокого обучения была намного лучше при обучении на доброкачественном наборе обучающих данных, включающем как общие человеческие варианты, так и варианты приматов, по сравнению с обычными человеческими вариантами. Следовательно, видимо, при текущей точности классификации количество доступных обучающих данных является более сильным ограничением. По мере того как большее количество особей будет секвенировано в каждом виде приматов, появится возможность подготовить наборы обучающих данных, которые будут содержать более высокую долю распространенных вариантов приматов, при снижении загрязнения патогенными вариантами в наборе обучающих данных и дополнительном улучшении эффективности классификации.
Создание неразмеченных вариантов для дополнения доброкачественного набора обучающих данных
[00470] Все возможные миссенс-варианты были получены из каждого положения основания канонических кодирующих областей путем замены нуклеотида в этом положении тремя другими нуклеотидами. Мы исключили варианты, которые наблюдались в 123 136 экзомах из ExAC / gnomAD, и варианты в старт- или стоп-кодонах. Всего было создано 68 258 623 неразмеченных варианта. Мы отнесли каждый из неразмеченных вариантов к одной из 96 различных категорий тринуклеотидного контекста. Мы обучили сеть глубокого обучения, используя полуконтролируемый подход, путем выборки вариантов из этого неразмеченного набора данных, которые соответствуют вариантам в доброкачественном наборе данных по тринуклеотидному контексту, и обучения классификатора различать доброкачественные и неразмеченные обучающие примеры.
Дополнительная фильтрация неразмеченных вариантов
[00471] Представляя примеры доброкачественных и неразмеченных вариантов вместе с фланкирующей аминокислотной последовательностью, сеть глубокого обучения изучает области белков, которые крайне нетерпимы к мутациям. Однако отсутствие общих вариантов в области белковой последовательности может быть связано с сильным очищающим отбором или может быть связано с техническими артефактами, которые препятствуют определению вариантов в этой области. Чтобы исправить последнее, мы удалили варианты как из доброкачественных, так и из неразмеченных наборов данных из областей, где набор данных ExAC / gnomAD имел среднее покрытие <1. Аналогичным образом, при сопоставлении неразмеченных вариантов с вариантами приматов в доброкачественном наборе данных во время обучения мы исключили неразмеченные варианты из областей, в которых этот примат не имел ортологичной выровненной последовательности с человеческой при множественном выравнивании последовательностей.
Варианты приматов, отложенные для валидации и тестирования, а также варианты de novo от больных и здоровых лиц
[00472] Для валидации и тестирования сети глубокого обучения мы случайным образом отобрали два набора из 10 000 вариантов приматов для валидации и тестирования, которые мы не использовали обучения. Остальные варианты приматов, наряду с распространенным человеческими вариантами (частота аллелей> 0,1%), использовались в качестве доброкачественного набора данных для обучения сети глубокого обучения. Кроме того, мы также отобрали два набора из 10000 неразмеченных вариантов, которые были выровнены с отложенными вариантами приматов для набора для валидации и набора для тестирования.
[00473] Мы использовали 10000 отложенных вариантов приматов в валидационном наборе и выровненные 10000 неразмеченных вариантов для мониторинга эффективности сети глубокого обучения в ходе обучения путем измерения способности сети различать варианты в двух наборах. Это позволило нам определить точку остановки для обучения и избежать переобучения после насыщения сети.
[00474] Мы использовали 10000 отложенных вариантов приматов в наборе тестовых данных для тестирования сети глубокого обучения, а также других 20 классификаторов. Поскольку разные классификаторы имели сильно различающиеся распределения оценок, мы использовали эти неразмеченные варианты, чтобы определить порог 50-го процентиля для каждого классификатора. Мы сравнили каждый классификатор с долей вариантов в наборе из 10 000 отложенных вариантов приматов, которые были классифицированы как доброкачественные при пороге 50-го процентиля для этого классификатора, чтобы обеспечить справедливое сравнение методов.
[00475] Чтобы оценить эффективность сети глубокого обучения в клинических условиях с использованием вариантов de novo у больных лиц с нарушениями развития нервной системы и вариантов de novo у здоровых людей, мы загрузили варианты de novo из исследования Deciphering Developmental Disorders (DDD), и de novo варианты из контрольной группы здоровых братьев и сестер в исследовании аутизма Simons Simplex Collection (SSC). Исследование DDD обеспечивает уровень достоверности для вариантов de novo, и мы исключили варианты из набора данных DDD с порогом <0,1 как потенциально ложные определения, обусловленные ошибками определения вариантов. Всего у нас было 3512 миссенс-вариантов de novo от людей, пораженных DDD, и 1208 миссенс-вариантов de novo от здоровых людей.
[00476] Чтобы лучше смоделировать реальный клинический сценарий различения доброкачественных и патогенных вариантов неопределенной значимости в группе кандидатных заболевания, мы ограничили анализ только вариантами de novo в пределах 605 генов, которые были связаны с заболеванием в исследовании DDD (p <0,05) на основании расчета только для вариантов, приводящих к укорочению белка (Дополнительная таблица 18). Мы оценили ген-специфическое обогащение мутаций de novo, приводящих к укорочению белка, путем вычисления статистической значимости при нулевой гипотезе ожидаемого количества мутаций de novo с учетом степени ген-специфических мутаций и количества рассматриваемых хромосом. Мы выбрали 605 генов с номинальным значением P <0,05. Мы рассчитали избыток синонимичных и миссенс-мутаций de novo в 605 генах (ФИГ. 22A) как отношение количества наблюдаемых мутаций de novo к ожидаемым мутациям de novo, а также разницу наблюдаемых мутаций de novo минус ожидаемые de novo мутации. В пределах этих 605 генов мы наблюдали 380 de novo миссенс-мутаций у индивидуумов, пораженных DDD (ФИГ. 22A). Для каждого из классификаторов, включая наш собственный, небольшая часть вариантов не имела прогнозов, как правило, потому, что они не соответствовали тем же моделям транскрипции, которые использовались классификатором. Соответственно, для нашей сети глубокого обучения мы выполнили последующий анализ в ФИГ. 22A, 22B, 22C, 22D и 22E с использованием 362 миссенс-мутаций de novo от людей, пораженных DDD, и 65 миссенс-мутаций de novo от здоровых контролей.
Предельное значение (насыщение) всех возможных человеческих миссенс - мутаций при увеличении числа секвенированных приматов
[00477] Мы исследовали ожидаемое предельное значение всех 70 миллионов возможных миссенс-мутаций по распространенным вариантам, присутствующим в 49А) ныне существующих видов приматов. Для каждого вида приматов мы моделировали четырехкратное количество распространенных миссенс-вариантов, встречающихся у человека (83500 миссенс-вариантов с частотой встречаемости аллеля > 0,1%), Поскольку относительное количество вариантов на одну особь у человека примерно в два раза меньше, чем у других видов приматов, и примерно 50% миссенс-вариантов человека были отфильтрованы отбором по порогу частоты встречаемости аллеля > 0,1% (Фиг. 49А). Мы определяли моделируемые варианты на основании существующего распределения известных распространенных миссенс-вариантов человека в контексте последовательности 96 тринуклеотидов. Например, если 2% распространенных миссенс-вариантов человека находились в области CCG>CTG распределения по тринуклеотидам, то нам требовалось, чтобы 2% имитированных вариантов являлись случайным образом выбранными мутациями из области CCG>CTG. Это позволяло управлять эффектами частоты мутаций, генетического дрейфа и отклонений, связанных с конверсией генов, с использованием тринуклеотидного контекста.
[00478] Кривые, представленные на Фиг. 23D, демонстрируют достижение совокупного предельного значения 70 миллионов возможных миссенс-мутаций по распространенным вариантам, присутствующим в любом из 504 видов приматов, при предположении, что мы выявили все распространенные варианты для каждого вида примата (частота встречаемости аллеля > 0,1%). На Фиг. 49А показано, что примерно 50% миссенс-мутаций человека являются достаточно опасными как для человека, так и для других приматов, и их появление в числе распространенных частот встречаемости аллеля (> 0,1%) предотвращается, и, следовательно, кривые, представленные на Фиг. 23D, отображают относительное количество неопасных миссенс-мутаций человека, достигающее предельного значения в условиях распространенной изменчивости приматов при увеличении числа видов приматов. Мы показали, что для 504 видов приматов большинство неопасных миссенс-мутаций человека будут достигать предельного значения, при этом неопасные мутации CpG-богатых участков будут достигать предельного значения при гораздо меньшем числе видов из-за более высокой частоты мутаций.
[00479] При моделировании относительного количества миссенс-вариантов человека (с частотой встречаемости аллеля > 0,1%), исследуемого при увеличивающемся размере изученной группы людей (Фиг. 36), мы производили выборку генотипов в соответствии с частотами встречаемости аллелей из gnomAD. Относительное количество распространенных миссенс-вариантов, выбранных из gnomAD, усреднялось по 100 модельным тестам для каждого размера выборки от 10 до 100 тысяч.
Предсказание вторичной структуры и доступности для растворителя
[00480] Нейронная сеть глубокого обучения для предсказания патогенности содержит всего 36 сверточных слоев, включая 19 сверточных слоев для сетей предсказания вторичной структуры и доступности для растворителя и 17 слоев для основной сети предсказания патогенности, входными данными которой являются результаты работы сетей предсказания вторичной структуры и доступности для растворителя. Поскольку кристаллическая структура большинства белков человека неизвестна, мы проводили тренировку 2 моделей для того, чтобы сделать возможным обучение нейронной сети построению белковой структуры из первичной последовательности. Обе модели использовали одни и те же архитектуру сети и входные данные, показанные на Фиг. 6. Входные данные для сетей предсказания вторичной структуры и доступности для растворителя представляют собой позиционную матрицу частот аминокислот размером 51 x 20, в которой содержится информация, полученная в результате множественного выравнивания последовательности человека и 99 других позвоночных.
[00481] Сеть вторичной структуры тренируется (обучается) для предсказания трех состояний вторичной структуры: альфа-спирали (H), бета-складчатой структуры (B) и петлей (C). Сеть доступности для растворителя тренируется для предсказания трех состояний доступности для растворителя: скрытой (B), промежуточной (I) и открытой (E). Обе сети используют только первичную последовательность в качестве входных данных, и тренируются с использованием меток известных кристаллических структур из Protein Databank (Банка Данных Белков). Модели предсказывают одно состояние для каждого остатка аминокислоты.
Подготовка данных для предсказания вторичной структуры и доступности для растворителя
[00482] Мы использовали не связанные друг с другом кристаллические структуры из Protein Databank (Банка Данных Белков) для тренировки моделей. Аминокислотные последовательности с более чем 25% подобием удалялись. В итоге, 6367 белковых последовательностей были использованы для тренировки, 400 для валидации и 500 для тестирования (дополнительная Таблица 13). Данные, использовавшиеся при тренировке, включая последовательность аминокислот и меток вторичной структуры и доступности для растворителя, находятся в доступе на сайте http://raptorx.uchicago.edu/download/.
[00483] Поскольку большинство рассчитанных кристаллических структур являются структурами белков организмов, не являющихся людьми, для предварительной тренировки моделей вторичной структуры и доступности для растворителя мы использовали пакет Raptor-X (основанный на PSI-BLAST) с целью получения соответствующих последовательностей, поскольку многократные выравнивания последовательностей генома человека обычно не были доступны. Мы генерировали многократные выравнивания последовательностей белков, используя инструментальное средство CNFsearch1.66_release из пакета RaptorX, и вели подсчет аминокислот в каждом положении из 99 ближайших выравниваний с целью формирования позиционной матрицы частот. Например, для получения многократного выравнивания последовательности 1u7lA.fasta применялись следующие специальные команды с использованием RaptorX:
% ./buildFeature -i 1u7lA.fasta -c 10 -o ./TGT/1u7lA.tgt
% ./CNFsearch -a 30 -q 1u7lA
[00484] Для каждого положения аминокислоты в наборе данных мы выбирали окно в частотной матрице положения, соответствующее 51 фланкирующей аминокислоте, и использовали его для предсказания маркера вторичной структуры или доступности для растворителя для аминокислоты, находящейся в центре последовательности, состоящей из 51 аминокислоты. При этом маркеры вторичной структуры и доступности для растворителя брались непосредственно из известной трехмерной кристаллической структуры белка с помощью программы DSSP и не требовали предсказания по первичной последовательности. Для включения нейронных сетей вторичной структуры и доступности для растворителя в качестве составных частей нейронной сети предсказания патогенности мы вычислили позиционные матрицы частот по данным многократных выравниваний последовательностей человека и 99 позвоночных. Несмотря на то, что матрицы сохранения, полученные этими двумя способами, в общем, похожи, мы применяли метод обратного распространения ошибки обучения к моделям вторичной структуры и доступности для растворителя в процессе тренировки предсказания патогенности, чтобы обеспечить тонкую настройку весовых коэффициентов.
Архитектура и тренировка модели
[00485] Мы проводили тренировку двух отдельных моделей вторичной структуры и доступности для растворителя белков на основе сверточных нейронных сетей глубокого обучения. Эти две модели имеют идентичную архитектуру и входные данные, но различаются тем, какие состояния предсказывают. Мы провели подробный гиперпараметрический поиск с целью оптимизации моделей для их наилучшего функционирования. При построении обеих сетей глубокого обучения для предсказания патогенности и предсказания вторичной структуры и доступности для растворителя была заимствована архитектура остаточных блоков, которая широко используется благодаря ее успешному применению в классификации изображений.
Остаточные блоки включают в себя повторяющиеся модули свертки, перемежающиеся с обходными соединениями, что позволяет информации с более ранних слоев обходить остаточные блоки. В каждом остаточном блоке входной слой нормализуется по первому пакету данных, за которым следует слой активации, использующий выпрямленные линейные единицы (ReLU). Затем активация проходит через слой одномерной свертки. Промежуточные данные из слоя одномерной свертки снова нормализуются по пакету данных и активируются с использованием ReLU, после чего следует другой слой одномерной свертки. После второй одномерной свертки мы складываем ее выходные данные с начальными входными данными в остаточный блок, который функционирует в качестве обходного соединения, позволяя начальной входной информации обойти остаточный блок. В такой архитектуре, названной ее авторами «сетью глубокого обучения с остатками», входные данные сохраняются в их начальном состоянии, а остаточные соединения не подвержены нелинейным активациям, что обеспечивает эффективное обучение более глубоких сетей. Более подробно архитектура представлена на Фиг. 6 и в дополнительных таблицах 11 (Фиг. 7А и 7Б) и 12 (Фиг. 8А и 8Б).
[00486] Идущий за остаточными блоками слой многопеременных логистических функций (softmax) для каждой аминокислоты рассчитывает вероятность нахождения в каждом из трех состояний, при этом наибольшее из значений вероятностей определяет состояние аминокислоты. Модель тренируется с помощью суммарной качественной функции уменьшения перекрестной энтропии для полной последовательности белка с использованием оптимизатора ADAM. Сразу после того, как проведена предварительная тренировка сетей вторичной структуры и доступности для растворителя, вместо того, чтобы непосредственно использовать их выходные данные в качестве входных данных в сети предсказания патогенности, мы использовали слой, находящийся перед softmax-слоем, чтобы больше информации проходило через сеть предсказания патогенности.
[00487] При тестировании модели предсказания 3 состояний вторичной структуры, наибольшее полученное значение точности было равным 79.86 % (дополнительная Таблица 14), что близко к точности предсказания, получаемого с использованием известной из уровня техники модели DeepCNF model30. При тестировании модели предсказания 3 состояний доступности для растворителя, наибольшее полученное значение точности было равным 60.31% (дополнительная Таблица 14), что близко к наиболее точному предсказанию, получаемому с использованием RaptorX для похожего набора данных. Мы также провели сравнение предсказаний нейронной сети при использовании структурных меток с аннотациями по алгоритму оценивания водородных связей (DSSP) для приблизительно 4000 имеющих кристаллическую структуру белков человека, со стандартной PrimateAI моделью, работающей только по предсказанным структурным меткам. Использование меток с аннотациями по алгоритму оценивания водородных связей (DSSP) не привело к увеличению точности предсказания патогенности (дополнительная Таблица 15).
Особенности входных данных в моделях глубокого обучения для предсказания патогенности
[00488] Тренировочный набор данных для сети предсказания патогенности содержит 385236 размеченных доброкачественных вариантов и 68258623 вариантов после фильтрации. Для каждого из вариантов мы внесли во входные данные следующие особые элементы. Первым особым элементом входных данных была последовательность, состоявшая из набора 51 фланкирующей к варианту аминокислоты, т.е. по 25 аминокислот с каждой из сторон варианта, полученная из референсной последовательности hg19, для предоставления моделям глубокого обучения контекста варианта. Такая примыкающая референсная последовательность была длиной всего в 51 аминокислоту. Исходя из эмпирического опыта, мы решили, что представление белка в виде последовательности аминокислот является более эффективным, чем представление в виде кодирующей последовательности с использованием нуклеотидов.
[00489] Вторым особым элементом была последовательность, состоявшая из 51 фланкирующей аминокислоты, с альтернативной аминокислотой, которая была заменена вариантом в центральном положении. Такая альтернативная примыкающая последовательность ничем не отличалась от фланкирующей референсной последовательности первого особого элемента входных данных, кроме того, что в центральном положении находилась альтернативная аминокислота вместо референсной аминокислоты. И референсная и альтернативная последовательности аминокислот преобразовывались в вектора в унитарном коде размером 51x20, где каждая аминокислота представлена в виде вектора, в котором 19 аминокислотам приписано значение 0, а единственной аминокислоте приписано значение 1.
[00490] Три частотные матрицы положений (PFM) создаются по множественным выравниваниям последовательностей 99 позвоночных для варианта: одна по 11 приматам, одна по 50 млекопитающим, не являющихся приматами, и одна по 38 позвоночным, не являющихся приматами и млекопитающими. Каждая PFM имеет размер Lx20, где L - длина фланкирующих последовательностей вокруг варианта (в нашем случае L относится к 51 аминокислоте).
[00491] В качестве входных данных в прошедших предварительную тренировку сетях предсказания 3 состояний вторичной структуры и 3 состояний доступности для растворителя мы использовали одну матрицу PFM, созданную по множественным выравниваниям последовательностей всех 99 позвоночных, имевшую длину 51 и ширину 20. После предварительной тренировки сетей на известных кристаллических структурах, взятых из Protein Databank (Банка Данных Белков), два последних слоя моделей вторичной структуры и доступности для растворителя удалялись (итоговый слой объединения с определением максимального значения (maxpool) и выходной слой), а выходные данные с предыдущего слоя, размером 51 х 40, использовались в качестве входных данных в сети предсказания патогенности.
Полуконтролируемое обучение
[00492] Поскольку в алгоритмах полуконтролируемого обучения используются и размеченные и неразмеченные экземпляры для тренировки, получаемые классификаторы могут обладать лучшими функциональными характеристиками по сравнению с алгоритмами полностью контролируемого обучения, в которых используется только ограниченное число доступных для тренировки размеченных данных. Основная идея полуконтролируемого обучения состоит в том, что предсказательная способность контролируемой модели, использующей только размеченные экземпляры, усиливается за счет использования объективных знаний, содержащихся в неразмеченных данных, обеспечивая, таким образом, потенциальное преимущество полуконтролируемого обучения. Параметры модели, обученной контролируемыми классификаторами на основе небольшого количества размеченных данных, могут быть сдвинуты в направлении более реалистичного распределения (больше напоминающего распределение реальных данных) с помощью неразмеченных данных.
[00493] Другой распространенной проблемой биоинформатики является разбалансировка данных. Проблема разбалансировки данных возникает, когда данных, представляющих один из предсказываемых классов, недостаточно, Поскольку экземпляры, принадлежащие такому классу, встречаются редко (из ряда вон выходящие случаи), или существуют сложности с их получением.
[00494] Алгоритмический подход для работы с разбалансированными данными основывается на ансамблях классификаторов. Ограниченное количество размеченных данных естественным образом приводит к ослаблению классификаторов, однако, ансамбль слабых классификаторов может по своим функциональным характеристикам превосходить любой отдельный входящий в него классификатор. Более того, ансамбли обычно увеличивают точность предсказания, достигаемую с помощью одиночного классификатора, что оправдывает усилия и стоимость ресурсов, связанных с обучением нескольких моделей. Интуитивно понятно, что объединение нескольких классификаторов позволяет лучше контролировать возможное переобучение, поскольку усреднение сильно варьирующихся параметров отдельных классификаторов также приводит к усреднению переобучения классификаторов.
[00495] Мы придерживались стратегии полуконтролируемого обучения из-за недостаточного количества имеющихся наборов данных необходимого размера, содержавших достоверно размеченные патогенные варианты. Не смотря на то, что база данных ClinVar содержит более 300000 образцов, после удаления из нее неоднозначных вариантов, в ней содержится всего 42000 миссенс-вариантов с непротиворечиво интерпретируемой патогенностью.
[00496] Систематические обзоры имеющейся информации позволили установить, что приписываемая указанным экземплярам патогенность зачастую не имеет достаточного клинического подтверждения. Более того, большая часть вариантов в составляемых человеком наборах данных зачастую оказывается из очень небольшого набора генов, что приводит к их несовпадению с вариантами из доброкачественных тренировочных наборов данных, которые определяются по всему геному с использованием распространенных вариантов человека или закрепленных замещений геномом шимпанзе. Принимая во внимание то, как по-разному определяются наборы данных, тренировка модели контролируемого обучения с использованием составляемых человеком наборов данных в качестве патогенного набора и распространенных вариантов, определяемых по всему геному, в качестве доброкачественного набора, вероятно, будет вносить систематическую ошибку.
[00497] Мы провели тренировку сети глубокого обучения для различения между набором размеченных доброкачественных вариантов и неразмеченным набором вариантов, которые были аккуратно подогнаны для удаления систематической ошибки. В одном из вариантов реализации набор из 385236 размеченных доброкачественных вариантов содержал распространенные варианты человека (частота встречаемости аллеля > 0,1%) из базы данных ExAC/gnomAD и варианты из шести видов приматов, среди которых не было человека.
[00498] Мы производили выборку набора неразмеченных вариантов, так чтобы обеспечивалось совпадение с доброкачественными вариантами в тринуклеотидном контексте (для отслеживания частоты мутаций, генетического дрейфа и конверсии генов), с учетом влияния выравниваемости и глубины покрытия на определение варианта. Поскольку число неразмеченных вариантов существенно превосходило число размеченных вариантов, мы получали оптимальное предсказание, проводя тренировку восьми моделей, использующих один и тот же набор размеченных доброкачественных вариантов и восемь выбранных случайным образом наборов неразмеченных вариантов, и усредняя их предсказания.
[00499] Выбор полуконтролируемого обучения обоснован тем, что составляемые человеком наборы данных вариантов являются недостоверными и зашумленными, в частности, содержат недостаточное количество патогенных вариантов. Мы получали набор надежных доброкачественных вариантов из распространенных вариантов человека из gnomAD и вариантов приматов. Что касается патогенных вариантов, мы использовали принцип сбалансированной итерационной выборки при начальной выборке патогенных вариантов из набора неизвестных вариантов (вариантов VUS без приписываемой им клинической значимости).
[00500] С целью уменьшения систематической ошибки выборки мы проводили тренировку ансамбля из восьми моделей с использованием одного и того же набора доброкачественных вариантов и восьми различных наборов патогенных вариантов. В начале, мы проводили случайную выборку неизвестных вариантов, играющих роль патогенных вариантов. Затем, несколько раз использовался ансамбль моделей для выставления балльной оценки набору неизвестных вариантов, не использовавшихся в цикле предварительной тренировки. Получившие наибольшую оценку патогенные варианты затем отбирались с целью замещения 5% случайно отобранных неизвестных вариантов в предыдущем цикле. Отметим, что мы оставляли на 25% больше получивших наибольшую оценку патогенных вариантов, чем требовалось, так что мы могли производить выборку восьми различных наборов получивших оценку патогенных вариантов для замены неизвестных вариантов, что увеличивало степень случайности выборки при работе с восьмью моделями. После этого, формировался новый тренировочный патогенный набор, и запускался новый цикл тренировки. Этот процесс продолжался до тех пор, пока первоначальные выбранные случайным образом неизвестные варианты не были полностью замещены патогенными вариантами, имевшими высокую степень достоверности, предсказанными с помощью моделей ансамблей. Процесс сбалансированной итерационной выборки показан на Фиг. 42.
Балансировка доброкачественных и неизвестных тренировочных наборов
[00501] Схема, по которой производилась выборка неизвестных вариантов, совпадавших с доброкачественными вариантами, эффективна для уменьшения систематической ошибки при тренировке нашей модели. При случайной выборке неизвестных вариантов, модели глубокого обучения часто выдают информацию, которая содержит систематическую ошибку, и тривиальные решения. Например, если замена аминокислот K->M встречается чаще в неизвестных, чем в доброкачественных вариантах, то модели глубокого обучения всегда имеют тенденцию относить замены K->M к классу патогенных. Таким образом, важно производить балансировку распределения замещенных аминокислот между двумя тренировочными наборами.
[00502] Классы, склонные к более высокой мутагенности, такие как CpG-транзиции, имеют огромную систематическую ошибку при представлении данных распространенных доброкачественных вариантов. Ортологические варианты других приматов имеют такую же частоту мутаций, как у человека, связанную с наличием большого числа классов, склонных к более высокой мутагенности, во всем доброкачественном тренировочном наборе. Если процедура выборки неизвестных вариантов не достаточно хорошо контролируется и балансировка не проводится , модели глубокого обучения будут относить CpG-транзиции скорее к классу доброкачественных, чем к менее выраженным классам, таким как класс трансверсии или класс не-CpG-транзиций.
[00503] Для предотвращения сходимости моделей глубокого обучения к тривиальному небиологическому решению мы предлагаем производить балансировку доброкачественных и неизвестных вариантов в контексте тринуклеотидной последовательности. тринуклеотид состоит из основания, находящегося перед вариантом, референсного основания варианта и основания, находящегося за вариантом. При этом референсное основание может быть заменено на другие три нуклеотида. Всего, получается 64х3 элемента в тринуклеотидном контексте.
Итерирующая сбалансированная выборка
Цикл 1
[00504] Мы производили выборку неизвестных вариантов, так чтобы их число точно совпадало с числом доброкачественных вариантов для каждого тринуклеотидного контекста. Другими словами, в течение первого цикла мы зеркально отображали доброкачественные и патогенные тренировочные наборы вариантов в тринуклеотидном контексте. Такая методология проведения выборки основана на интуитивном понимании того, что варианты с одинаковой частотой мутаций представлены одинаково в неизвестных и доброкачественных вариантах. Это позволяет предотвратить сходимость модели к тривиальному решению, основанному на частоте мутаций.
Циклы с 2 по 20
[00505] Во втором цикле мы применяли тренированную модель из цикла 1 для выставления балльной оценки набору неизвестных вариантов, которые не участвовали в цикле 1, и заменяли 5% неизвестных вариантов наиболее хорошо предсказанными патогенными вариантами. Такой набор был полностью сформирован моделью, и к этому набору не применялась балансировка в тринуклеотидном контексте. Выборка оставшихся 95% неизвестных вариантов, необходимых для тренировки, производилась так, чтобы использовать 95% из числа каждого тринуклеотидного контекста в доброкачественных вариантах.
[00506] Поскольку в цикле 1 использовались полностью совпадавшие тренировочные наборы, интуитивно понятно, что наиболее хорошо предсказанные патогенные варианты были определены без какой-либо систематической ошибки, связанной с частотой мутаций. Таким образом, для этого набора отсутствовала необходимость учета какой-либо систематической ошибки. Частота мутаций в тринуклеотидном контексте для оставшихся 95% данных по-прежнему контролировалась для предотвращения сходимости модели к тривиальному решению.
[00507] В каждом следующем цикле процент замещенных неизвестных вариантов увеличивался на 5%. В цикле 3 мы заменили 5% неизвестных вариантов наиболее хорошо предсказанными патогенными вариантами, определенными моделью в цикле 3. В итоге, доля патогенных вариантов возросла на 10%, а доля зеркально отраженных неизвестных вариантов в тринуклеотидном контексте уменьшилась до 90%. Процесс выборки был схожим с другими циклами.
Цикл 21
[00508] В цикле 21, последнем цикле, патогенный тренировочный набор полностью состоял из наиболее хорошо предсказанных моделями глубокого обучения патогенных вариантов. Поскольку мы непосредственно контролировали в каждом цикле систематическую ошибку, связанную с частотой мутаций, патогенные варианты были достаточно достоверными, чтобы использовать их в качестве тренировочных данных, и не содержали систематической ошибки, связанной с частотой мутаций. Таким образом, в последнем цикле тренировки была создана окончательная модель глубокого обучения для предсказания патогенности.
Согласование размеченных доброкачественных и неразмеченных тренировочных наборов
[00509] Проведение сбалансированной выборки неразмеченных вариантов очень важно для удаления систематических ошибок, не связанных с вредоносностью варианта. В отсутствие надлежащего контроля над эффектами, затрудняющими интерпретацию, глубокое обучение может использовать случайно возникшие ошибки для определения границ между классами. Распространенные варианты человека могут содержать большое количество вариантов из классов с высокой мутагенностью, таких как, например, CpG-богатые последовательности. Кроме того, полиморфизмы приматов имеют такую же частоту мутаций, как у человека, связанную с наличием большого числа классов, склонных к более высокой мутагенности, во всем доброкачественном тренировочном наборе. Если процедура выборки неизвестных вариантов не достаточно хорошо контролируется и балансировка не производится, сети глубокого обучения будут подвержены влиянию частоты мутаций при классификации вариантов, и, таким образом, будут стремиться относить CpG-транзиции скорее к классу доброкачественных, чем к менее выраженным классам, таким как класс трансверсии или класс не-CpG-транзиций. Мы производили выборку, содержащую в точности одинаковое число неразмеченных вариантов и размеченных доброкачественных вариантов для каждоой из 96 тринуклеотидных контекстных последовательностей (см. выше).
[00510] При согласовании неразмеченных вариантов с вариантами приматов в размеченном доброкачественном наборе мы ввели запрет на отбор неразмеченных вариантов из тех участков генома человека, где такой вид примата не выравнивался путем многократного выравнивания последовательностей, Поскольку определение варианта для такого вида примата в таком положении не представлялось возможным.
[00511] Для каждого из 96 контекстов последовательности тринуклеотидов мы проводили корректировку с учетом глубины секвенирования для вариантов приматов. Поскольку секвенированиие проведено для большого числа людей, распространенные варианты в популяции человека встречаются достаточно часто и хорошо определены даже в участках с небольшой глубиной секвенирования. Это неверно для вариантов приматов, Поскольку секвенирование проведено только для небольшого числа особей. Мы разбили геном на 10 интервалов по глубине секвенирования экзомов из ExAC/gnomAD. Мы измеряли долю вариантов приматов в размеченном доброкачественном наборе данных относительно неразмеченного набора данных для каждого из интервалов. Мы рассчитали вероятность того, что вариант примата принадлежит размеченному доброкачественному набору данных, основываясь только на глубине секвенирования, с помощью линейной регрессии (Фиг. 24). При отборе неразмеченных вариантов для согласования вариантов приматов с размеченным доброкачественным набором данных мы использовали весовые коэффициенты, связанные с вероятностью того, что вариант попадет в выборку, на основании глубины покрытия в заданном положении, используя коэффициенты регрессии.
Генерация доброкачественных и неизвестных вариантов
Генерация доброкачественных и неизвестных вариантов
[00512] Недавние исследования показали, что распространенные варианты в человеческих популяциях, как правило, являются доброкачественными. gnomAD предоставляет 90 958 несинонимичных SNP с частотой минорных аллелей (MAF)> = 0,1% в пределах канонических кодирующих областей, согласно одному варианту реализации. Варианты, прошедшие фильтры, сохраняются. Инделы исключаются. Удаляются варианты, которые встречаются в стартовых или стоп-кодонах, а также варианты, приводящие к укорочению белка. При тщательном изучении субпопуляций общее количество миссенс-вариантов с MAF> = 0,1% в каждой субпопуляции увеличивается до 245360 согласно одному варианту реализации. Эти варианты составляют часть обучающей выборки доброкачественных вариантов.
Распространенные полиморфизмы у человекообразных обезьян
[00513] Поскольку известно, что кодирующие области являются высококонсервативными, легко предположить, что если полиморфизм сегрегируется в популяции человекообразных обезьян с высокой частотой, он также может иметь умеренное влияние на приспособленность человека. Данные полиморфизмов бонобо, шимпанзе, гориллы и орангутана из проектов генома человекообразных обезьян и других исследований были объединены с SNP резуса и мартышки из dbSNP.
Генерация неизвестных вариантов
[00514] Все возможные варианты генерируются из каждого положения основания канонических кодирующих областей путем замены нуклеотида в этом положении на три других нуклеотида. Формируются новые кодоны, что приводит к потенциальным изменениям аминокислот в положениях. Синонимичные изменения фильтруются.
[00515] Варианты, наблюдаемые в наборе данных gnomAD, удалены. Удаляются варианты, которые встречаются в старт-кодонах или стоп-кодонах, а также варианты, которые образуют стоп-кодоны. Для SNP с несколькими аннотациями генов выбирается каноническая аннотация гена, представляющая аннотацию SNP. Всего генерируется 68 258 623 неизвестных варианта согласно одному варианту реализации.
Дополнительная фильтрация вариантов
[00516] В некоторых областях генома человека, как известно, трудно выровнять риды. Включение этих областей оказывает смешанные эффекты в отношении наборов данных для обучения и тестирования. Например, области под высоким давлением отбора, как правило, имеют ограниченное количество полиморфизмов. Принимая во внимание, что области, которые трудно секвенировать, также имеют меньше полиморфизмов. Чтобы избежать такой вводящей в заблуждение информации для наших моделей, мы удалили варианты из генов, которые не были секвенированы в исследовании gnomAD.
[00517] Обычно доброкачественные варианты обнаруживаются в хорошо секвенированных областях, которые имеют тенденцию быть консервативными для многих видов. В то время как неизвестные варианты случайным образом отбираются по геномам, которые включают некоторые плохо покрытые области. Это вызывает расхождение в определении между доброкачественными и неизвестными наборами. Чтобы уменьшить систематическую ошибку, мы отфильтровали варианты с глубиной чтения <10 в gnomAD. Мы также отфильтровали все варианты, у которых отсутствует более 10% данных при выравнивании фланкирующих последовательностей для всех видов млекопитающих.
Данные для валидации и тестирования
[00518] Для валидации и тестирования моделей патогенности мы случайным образом отобрали из большого пула доброкачественных вариантов два набора из 10 000 доброкачественных вариантов для валидации и тестирования, соответственно, согласно одному варианту реализации. Остальные доброкачественные варианты используются для обучения моделей глубокого обучения. Эти варианты специально отбираются из ортологичных вариантов приматов, чтобы обеспечить справедливое сравнение между методами, поскольку некоторые методы обучаются на общих вариантах человека. Мы также случайным образом отобрали два набора из 10 000 неизвестных вариантов для проверки и тестирования отдельно в соответствии с одним вариантом реализации. Мы гарантируем, что количество неизвестных вариантов в каждом из 192 тринуклеотидных контекстов соответствует количеству доброкачественных вариантов для наборов для валидации и тестирования, соответственно.
[00519] Мы оценили эффективность нескольких методов в клинических условиях, используя de novo варианты детей с аутизмом или нарушениями развития (DDD) и их здоровых братьев и сестер. Всего, согласно одному варианту реализации, имеется 3821 миссенс-варианты de novo из случаев с DDD и 2736 миссенс-вариантов de novo из случаев с аутизмом. Согласно одной реализации, существует 1231 миссенс-вариант de novo для здоровых братьев и сестер.
Сетевая архитектура глубокого обучения
[00520] Сеть предсказания патогенности получает пять прямых входов и два косвенных входа через сети вторичной структуры и доступности для растворителей. Пять прямых входов представляют собой аминокислотные последовательности длиной 51 × глубину 20 (кодирующие 20 различных аминокислот) и включают референсную аминокислотную последовательность человека без варианта (1a), альтернативную аминокислотную последовательность человека с замененным вариантом в (1b) PFM из множественного выравнивания последовательностей видов приматов (1c), PFM из множественного выравнивания последовательностей видов млекопитающих (1d) и PFM из множественного выравнивания последовательностей более отдаленных видов позвоночных (1e). Каждая сеть вторичной структуры и доступности для растворителя получает в качестве входов PFM от множественного выравнивания последовательностей (1f) и (1g) и предоставляет свои выходы в качестве входов в основную сеть предсказания патогенности. Сети вторичной структуры и доступности для растворителей были предварительно обучены на известных кристаллических структурах белков для Protein DataBank и допускают обратное распространение во время обучения модели патогенности.
[00521] Пять каналов прямого ввода проходят через сверточный слой с повышающей дискретизацией из 40 ядер с линейными активациями. Референсная аминокислотная последовательность человека (1a) объединяется с PFM из множественного выравнивания последовательностей приматов, млекопитающих и позвоночных (слияние 1a). Точно так же альтернативная аминокислотная последовательность человека (1b) объединяется с PFM из множественных выравниваний последовательностей приматов, млекопитающих и позвоночных (слияние 1b). Это создает две параллельные дорожки, одну для референсной последовательности, а другую с альтернативной последовательностью с замененным вариантом.
[00522] Объединенную карту признаков референсного канала и альтернативного канала (объединение 1a и 1b) пропускают через серию из шести остаточных блоков альтернативного канала Объединение (Слои 2а-7а, Объединение 2a и 2b, слои 7b, объединение 2b). Выход остаточных блоков (Объединение 2a и Объединение 2b) объединяются вместе, чтобы сформировать карту признаков размера (51,80) (Объединение 3a, 3b), которая полностью перемешивает данные из референсных и альтернативных каналов. Затем, данные имеют два пути для параллельного прохождения через сеть, либо через серию из шести остаточных блоков, содержащих по два сверточных слоя каждый, как определено в разделе 2.1 (Объединение 3-9, уровни 9-46, исключая слой 21,34) или через соединение с пропуском, которые объединяют выходя двух остаточных блоков после прохождения одномерной свертки (уровень 21, уровень 37, уровень 47). Наконец, объединенные активации (Объединение 10) подаются в другой остаточный блок (слои с 48 по 53, Объединение 11). Активации из Объединения 11 передаются одномерной свертке с размером фильтра 1 и сигмовидной активацией (уровень 54), а затем проходят через глобальный максимальный уровень объединения, который выбирает одно значение, представляющее предсказания сети для патогенности варианта. Схематическое изображение модели приведено на ФИГ. 3 и в Дополнительной таблице 16 (ФИГ. 4A, 4B, 4C).
Обзор модели
[00523] Мы разработали полуконтролируемые (полууправляемые) модели глубокой сверточной нейронной сети (CNN) для предсказания патогенности вариантов. Входные признаки моделей включают последовательности белков и профили консервации, фланкирующие варианты, и истощение миссенс-вариантов в определенных областях гена. Мы также предсказали изменения, вызванные вариантами в вторичной структуре и доступности для растворителей, с помощью моделей глубокого обучения и интегрировали их в нашу модель предсказания патогенности. Для обучения модели мы сгенерировали доброкачественные варианты из распространенных вариантов субпопуляций человека и ортологические варианты приматов. Однако у нас по-прежнему отсутствуют надежные источники патогенных вариантов. Первоначально мы обучили модель на доброкачественных и неизвестных вариантах, а затем использовали алгоритм итеративной сбалансированной выборки (IBS) с полууправляемым контролем, чтобы постепенно заменять неизвестные варианты набором патогенных вариантов, предсказанных с высокой степенью уверенности. Наконец, мы продемонстрировали, что наша модель превосходит существующие методы в различении вариантов de novo, вызывающих нарушение развития у людей, от доброкачественных.
Принятие остаточного блока
[00524] На ФИГ. 17 показан остаточный блок. И наша модель глубокого обучения для предсказания патогенности, и модели глубокого обучения для предсказания вторичной структуры и доступности для растворителей используют определение остаточных блоков, которое впервые было проиллюстрировано в. Структура остаточного блока показана на рисунке ниже. Входной слой сначала подвергается пакетной нормализации, после чего следует нелинейная активация «ReLU». Затем активация проходит через одномерный сверточный слой. Этот промежуточный выходной сигнал одномерного сверточного слоя снова подвергается пакетной нормализации и активируется ReLU, за которым следует еще один одномерный сверточный слой. В конце второй одномерной свертки мы объединяем ее выход с исходным входом. В такой архитектуре входные данные сохраняются в исходном состоянии, а остаточные соединения предохраняются от нелинейных активаций модели.
[00525] Дырчатые/разреженные свертки обеспечивают большие рецептивные поля с новыми обучаемыми параметрами. Дырчатая/разреженная свертка представляет собой свертку, в которой ядро применяется на площади, большей, чем его длина, за счет того, что она пропускает входные значения с определенным шагом, называемым также показателем разрежения или фактором разрежения . Дырчатые/разреженные свертки увеличивают расстояние между элементами свертки фильтра свертки/ядра, в результате чего при осуществлении операции свертки используются соседние входные записи (например, нуклеотиды, аминокислоты) с большими интервалами. Это обеспечивает возможность введения во входные данные контекстуальных зависимостей дальнего действия. Дырчатые (atrous) свертки сохраняют расчет свертки для повторного использования при обработке соседних нуклеотидов.
Новизна нашей модели
[00526] Наш способ отличается от существующих методов предсказания патогенности вариантов по трем аспектам. Во-первых, наш способ использует новую архитектуру глубоких полуконтролируемых сверточных нейронных сетей. Во-вторых, надежные доброкачественные варианты получают из распространенных вариантов человека из gnomAD и вариантов приматов, в то время как высоконадежный патогенный обучающий набор создается с помощью итеративной сбалансированной выборки и обучения, что позволяет избежать циклического обучения и тестирования моделей с использованием идентичных баз данных вариантов, созданных человеком. В-третьих, модели глубокого обучения вторичной структуры и доступности для растворителя интегрированы в архитектуру нашей модели патогенности. Информация, полученная из моделей структуры и растворителя, не ограничивается предсказанием метки для конкретных аминокислотных остатков. Вместо этого, слой считывания удаляется из моделей структуры и растворителя, а предварительно обученные модели объединяются с моделью патогенности. Во время обучения модели патогенности предварительно обученные слои структуры и растворителя также распространяются в обратном направлении, чтобы минимизировать ошибку. Это помогает предварительно обученной модели структуры и растворителя сосредоточиться на проблеме предсказания патогенности.
Обучение моделей вторичной структуры и доступности для растворителя
Подготовка данных
[00527] Мы обучили глубокие сверточные нейронные сети для предсказания вторичной структуры с 3 состояниями и доступности белков для растворителей с 3 состояниями. Аннотации белков из PDB используются для обучения моделей. Согласно одному варианту реализации последовательности с более чем 25% сходством с профилем последовательностей удаляются. Согласно одному варианту реализации в общей сложности 6293 последовательности белка используются для обучения, 392 для валидации и 499 для тестирования.
[00528] Профили консервации матрицы оценок, зависящих от положения (PSSM), для белков генерируются путем запуска PSI-BLAST с пороговым значением E-value 0,001 и 3 итерациями для поиска UniRef90. Любая неизвестная аминокислота определяется как пустая, как и ее вторичная структура. Мы также запускаем PSI-BLAST с аналогичными настройками параметров для всех генов человека, чтобы собрать их профили сохранения PSSM. Эти матрицы используются для интеграции модели структуры для предсказания патогенности. Затем аминокислоты белковых последовательностей преобразуются в векторы быстрого кодирования. А белковые последовательности и матрицы PSSM преобразуются в матрицу Lx20, где L - длина белка. Три предсказанных метки для вторичной структуры включают спираль (H), бета-лист (B) и катушки (C). Три метки для доступа к растворителю включают скрытый (B), промежуточный (I) и открытый (E). Одна метка соответствует одному аминокислотному остатку. Метки кодируются как векторы быстрого кодирования размерности 3.
Архитектура и обучение модели
[00529] Мы обучили две сквозные модели глубокой сверточной нейронной сети для предсказания вторичной структуры с 3 состояниями и доступности белков для растворителей с 3 состояниями, соответственно. Эти две модели имеют схожие конфигурации, включая два входных канала, один для последовательностей белков, а другой - для профилей консервации белков. Каждый входной канал имеет размер L x 20, где L обозначает длину белка.
[00530] Каждый входной канал проходит через одномерный сверточный слой (слои 1a и 1b) с 40 ядрами и линейными активациями. Этот слой используется для повышения дискретизации входных размеров с 20 до 40. Обратите внимание, что во всех остальных слоях модели используется 40 ядер. Активации двух слоев (1a и 1b) объединяются путем суммирования значений по каждому из 40 измерений (то есть, режим объединения = «сумма»). Выход узла объединения проходит через единственный слой 1D-свертки (слой 2) с последующей линейной активацией.
[00531] Активации из слоя 2 проходят через серию из 9 остаточных блоков (слои с 3 по 11), как определено выше. Активация слоя 3 подается на слой 4, а активация слоя 4 подается на слой 5 и так далее. Также существуют соединения с пропуском, которые напрямую суммируют выход каждого третьего остаточного блока (уровни 5, 8 и 11). Затем объединенные активации поступают в две одномерные свертки (слои 12 и 13) с активациями ReLU. Активации из уровня 13 передаются на уровень считывания softmax. Softmax вычисляет вероятности выходов трех классов для данного входа.
[00532] Для получения лучшей модели вторичной структуры одномерные свертки имеют показатель разряжения 1. Для модели доступности для растворителя последние 3 остаточных блока (слои 9, 10 и 11) имеют показатель разряжения 2, чтобы увеличить покрытие ядер. Вторичная структура белка сильно зависит от взаимодействия аминокислот в непосредственной близости. Таким образом, модели с более высоким охватом ядра немного улучшают производительность. С другой стороны, доступность для растворителя зависит от дальнодействующих взаимодействий между аминокислотами. Таким образом, для модели с большим покарытием ядер с использованием разреженных сверток ее точность более чем на 2% выше, чем у моделей с небольшим покрытием.
[00533] В таблице ниже представлены подробности активаций и параметров для каждого уровня модели предсказания вторичной структуры с 3 состояниями согласно одному варианту реализации.
[00534] Подробности модели доступности для растворителя показаны в таблице ниже, в соответствии с одним вариантом реализации.
[00535] Класс вторичной структуры конкретного аминокислотного остатка определяется наибольшими предсказанными вероятностями softmax. Модель обучается с помощью накопленной категориальной функции перекрестной потери энтропии для всей последовательности белка с использованием оптимизатора ADAM для оптимизации обратного распространения.
[00536] Наилучшая точность тестирования для модели предсказания вторичной структуры с 3 состояниями составляет 80,32%, что близко к современной точности, предсказанной моделью DeepCNF на аналогичном наборе обучающих данных.
[00537] Наилучшая точность тестирования для модели предсказания доступности для растворителя с 3 состояниями составляет 64,83%, что близко к текущей наилучшей точности, предсказанной RaptorX на аналогичном наборе обучающих данных.
[00538] Мы интегрировали предварительно обученные модели предсказания вторичной структуры с 3 состояниями и доступности для растворителя в нашу модель предсказания патогенности, как описано ниже.
Обучающие модели для предсказания патогенности вариантов
Входные характеристики для модели предсказания патогенности
[00539] Как обсуждалось выше, для задачи предсказания патогенности существует обучающий набор доброкачественных вариантов и обучающий набор неизвестных вариантов для обучения модели патогенности. Для каждого варианта мы подготовили следующие входные функции для использования в нашей модели.
[00540] Первым входным параметром (признаком) каждого варианта является его фланкирующая аминокислотная последовательность, то есть 25 аминокислот с каждой стороны от варианта, полученного из референсной последовательности hg19, для обеспечения моделей глубокого обучения контекстных последовательностей варианта. В общей сложности эта фланкирующая референсная последовательность имеет длину, равную 51 аминокислоте.
[00541] Вторая характеристика (параметр, признак) - это альтернативная аминокислота, которая определяет вариант. Вместо того, чтобы напрямую предоставлять пару аминокислот референс-альтернативная аминокислота, мы предоставляем альтернативную фланкирующую последовательность для модели. Альтернативная фланкирующая последовательность такая же, как контрольная фланкирующая последовательность в первом параметре, за исключением того, что среднее положение последовательности содержит альтернативную аминокислоту вместо контрольной аминокислоты.
[00542] Обе последовательности затем преобразуются путем кодирования с одним корячим состоянием в векторы длиной 51 × 20, причем каждая аминокислота представлена вектором из 20 нулей или единиц.
[00543] Затем три матрицы весовых коэффициентов положения (PWM) генерируются из множественных выравниваний последовательностей (MSA) 99 позвоночных для варианта, включая одну для 12 приматов, одну для 47 млекопитающих, исключая приматов, и одну для 40 позвоночных, исключая приматов и млекопитающих. Каждая PWM имеет размер L x 20, где L - длина фланкирующих последовательностей вокруг варианта (в нашем случае L представляет 51 аминокислоту). Он включает количество аминокислот, встречающихся в каждой категории видов.
[00544] Мы также генерируем матрицы PSSM для фланкирующих вариант последовательностей из 51 аминокислоты из алгоритма psi blast. Это применяется для интеграции моделей предсказания вторичной структуры с 3 состояниями и доступности для растворителя для предсказания патогенности.
[00545] Мы обучаем модель патогенности с помощью референсной последовательности (input1 (вход 1)), альтернативной последовательности (вход 2), матриц PWM для приматов (вход 3), млекопитающих (вход 4), позвоночных животных (вход 5) и информации из вторичной структуры с 3 состояниями и модели доступности для растворителя.
Обучение (тренировка) модели глубокого обучения
[00546] ФИГ. 19 представляет собой блок-схему, которая дает обзор рабочего процесса моделей глубокого обучения. Обучающиеся модели патогенности включают пять прямых и четыре косвенных входа. Пять функций прямого ввода включают референсную последовательность (1a), альтернативную последовательность (1b), консервацию у приматов (1c), консервацию у млекопитающих (1d) и консервацию у позвоночных (1e). Косвенные входные данные включают вторичную структуру на основе референсной последовательности (1f), вторичную структуру на основе альтернативной последовательности (1g), доступность для растворителя на основе контрольной последовательности (1h) и доступность для растворителя на основе альтернативной последовательности (1i).
[00547] Для косвенных входов 1f и 1g мы загружаем предварительно обученные слои модели предсказания вторичной структуры, исключая слой softmax. Предварительно обученные слои 1f основаны на референсной последовательности человека для вариантов вместе с PSSM, созданным с помощью PSI-BLAST для варианта. Аналогичным образом, для входа 1g предварительно обученные слои моделей предсказания вторичной структуры основаны на альтернативной последовательности человека в качестве входных данных вместе с матрицей PSSM. Входы 1h и 1i соответствуют аналогичным предварительно обученным каналам, содержащим информацию о доступности растворителя для референсной и альтернативной последовательностей варианта соответственно.
[00548] Пять каналов прямого входа пропускаются через сверточный слой с повышающей дискретизацией из 40 ядер с линейными активациями. Слои 1a, 1c, 1d и 1e объединяются со значениями, суммированными по 40 размерам элементов с получением слоя 2a. Другими словами, карта признаков контрольной последовательности объединяется с тремя типами карт параметров консервации. Точно так же 1b, 1c, 1d и 1e объединяются со значениями, суммированными по 40 измерениям признаков с созданием слоя 2b, т.е. признаки альтернативной последовательности объединяются с тремя типами признаков консервации.
[00549] Слои 2a и 2b подвергаются пакетной нормализации с активацией ReLU, и каждый проходит через одномерный сверточный слой с размером фильтра 40 (3a и 3b). Выходы слоев 3a и 3b объединяются с 1f, 1g, 1h и 1i, при этом карты признаков объединяются друг с другом. Другими словами, карты признаков референсной последовательности с профилем консервации и альтернативной последовательности с профилем консервации объединяются с картами признаков вторичной структуры референсной и альтернативной последовательности и картами признаков доступности для растворителя референсной и альтернативной последовательностей (слой 4).
[00550] Выходы уровня 4 проходят через шесть остаточных блоков (слои 5,6,7,8,9,10). Последние три остаточных блока имеют показатель разряжения 2 для одномерных сверток, чтобы обеспечить большее покрытие для ядер. Вход слоя 10 проходит через одномерную свертку с размером фильтра 1 и сигмоидной активацией (слой 11). Выходные данные уровня 11 передаются через глобальное объединение maxpool, которое выбирает одно значение для варианта. Это значение отражает патогенность варианта. Подробности одного варианта реализации модели предсказания патогенности показаны в таблице ниже.
Ансамбли
[00551] В одном варианте реализации для каждого цикла нашего метода мы запускали восемь разных моделей, которые обучаются на одном и том же наборе доброкачественных данных и восьми разных наборах неизвестных данных, и усредняли предсказание наборов данных оценки по восьми моделям. Систематическую ошибку выборки можно уменьшить и хорошо контролировать, если представлять модели несколько случайно выбранных наборов неизвестных вариантов.
[00552] Кроме того, применение подхода ансамблей может улучшить эффективность (показатели работы) нашей модели на нашем наборе данных для оценки. CADD использует ансамбль из 10 моделей и получает среднюю оценку по всем 10 моделям для оценки варианта. Здесь мы попытались использовать аналогичный ансамблевый подход. Мы сравнили результаты, полученные с использованием одного ансамбля, а затем увеличили количество ансамблей, чтобы оценить увеличение эффективности. Обратите внимание, что в каждом ансамбле есть восемь моделей, которые обучаются на одном и том же наборе доброкачественных данных, и восемь разных наборов неизвестных данных. Для разных ансамблей начальные значения генератора случайных чисел различны, так что наборы случайных вариантов отображаются по-разному.
[00553] Подробные результаты согласно одному варианту реализации показаны в таблице ниже.
[00554] По сравнению с одним ансамблем, 5 ансамблей и 10 ансамблей дали более значимые p-значения при оценке с использованием наборов данных DDD. Но увеличение количества ансамблей не приводит к дальнейшему улучшению показателей, что указывает на насыщение ансамблей. Ансамбли уменьшают систематическую ошибку выборки, из-за обусловленную большим количеством неизвестных вариантов. Однако нам также потребовалось сопоставить 192 тринуклеотидных контекста между доброкачественными и патогенными классами, что существенно ограничивает пространство для отбора проб, что приводит к быстрому насыщению. Мы пришли к выводу, что подход ансамбля ансамблей значительно улучшает производительность модели и еще больше обогащает наше понимание моделей.
Раннее прекращение обучения модели патогенности
[00555] Поскольку отсутствуют надежные аннотированные образцы патогенных вариантов, сложно определить критерии остановки для обучения модели. Чтобы избежать использования патогенных вариантов при оценке модели, в одном варианте реализации мы использовали 10 000 доброкачественных вариантов для валидации от ортологичных приматов и 10 000 неизвестных вариантов, соответствующих тринуклеотидному контексту. После обучения каждой эпохи модели мы оценивали доброкачественные варианты валидации и неизвестные варианты валидации. Мы использовали критерий суммы рангов Уилкоксона, чтобы оценить разницу распределений вероятностей обоих наборов вариантов для валидации.
[00556] Р-значение критерия становится более значимым с улучшением способности модели отличать доброкачественные варианты от набора неизвестных вариантов. Мы останавливаем обучение, если не наблюдается улучшения в способности модели различать два распределения в течение любых пяти последовательных эпох обучения модели.
[00557] Ранее мы выделили два отдельных набора из 10 000 отложенных вариантов приматов для обучения, которые мы назвали набором валидации и набором тестирования. Мы использовали набор валидации из 10 000 удерживаемых вариантов приматов и 10 000 неразмеченных вариантов, которые были сопоставлены по тринуклеотидному контексту, для оценки раннего прекращения во время обучения модели. После каждой эпохи обучения мы оценивали способность глубокой нейронной сети различать варианты в маркированном доброкачественном наборе для валидации и неразмеченном подобранном контроле, измеряя разницу в распределениях предсказанных оценок с помощью критерия суммы рангов Вилкоксона. Мы прекращали обучение, как только не наблюдали дальнейшего улучшения после пяти последовательных эпох обучения, чтобы предотвратить переобучение.
Сравнительный анализ эффективности классификатора
[00558] Мы оценили точность классификации двух вариантов сети глубокого обучения, одна обучена только с распространенным человеческими вариантами, а другая обучена с полным размеченным доброкачественным набором данных, включая как распространенные варианты человека, так и варианты приматов, в дополнение к следующим классификаторам: SIFT, PolyPhen-2, CADD, REVEL, M-CAP, LRT, MutationTaster, MutationAssessor, FATHMM, PROVEAN, VEST3, MetaSVM, MetaLR, MutPred, DANN, FATHMM-MKL_coding, Eigen, GenoCanyon,32-48 ++ 13 и GERP. Чтобы получить оценки для каждого из других классификаторов, мы загрузили оценки для всех миссенс-вариантов из dbNSFP 49 (https://sites.google.com/site/jpopgen/dbNSFP) и проверили методы на 10 000 отложенных вариантах приматов, тестовом наборе и вариантах de novo в случаях DDD по сравнению с контролями. Мы выбрали SIFT, PolyPhen-2 и CADD для включения в основную статью, потому что они являются одними из наиболее широко используемых методов, и REVEL, потому что в разных режимах оценки он выделялся как один из лучших из 20 существующих классификаторов, которые мы оценили. Показатели всех оцениваемых нами классификаторов приведены на ФИГ. 28А.
[00559] Для оценки влияния доступного размера обучающих данных на показатели работы сети глубокого обучения мы обучили сети глубокого обучения в каждой точке данных на ФИГ. 6 путем случайной выборки из размеченного доброкачественного обучающего набора из 385 236 вариантов приматов и распространенных человеческих вариантов. Чтобы уменьшить случайный шум в работе классификаторов, мы выполнили эту процедуру обучения пять раз, каждый раз используя случайное отображение начальных весов параметров, и показали среднюю производительность как для 10000 отложенных вариантов приматов, так и для случая DDD по сравнению с контрольным набором данных в ФИГ. 6. Случайно, показатели работы медианного классификатора с полным набором данных из 385 236 размеченных доброкачественных вариантов была немного лучше, чем у того, который мы использовали для остальной части статьи по набору данных DDD (P <10-29 вместо P <10- 28 по критерию суммы рангов Вилкоксона). Чтобы показать, что варианты от каждого отдельного вида приматов вносят свой вклад в точность классификации, тогда как варианты от каждого отдельного вида млекопитающих снижают точность классификации, мы обучили сети глубокого обучения, используя обучающий набор данных, содержащий 83546 человеческих вариантов плюс постоянное количество случайно выбранных вариантов для каждого вида, согласно к одному варианту реализации. Согласно одному варианту реализации, постоянное количество вариантов, которые мы добавили в обучающий набор (23380), представляет собой общее количество вариантов, доступных для вида с наименьшим количеством миссенс-вариантов, то есть бонобо. Чтобы уменьшить шум, мы повторили процедуру обучения еще пять раз и определили среднюю эффективность (показатели работы) классификатора.
Оценка модели
[00560] В одном варианте реализации мы обучили 21 цикл моделей глубокого обучения по итеративной процедуре сбалансированной выборки. Мы применили два типа оценок, чтобы оценить эффективность наших классификаторов. Мы также сравнили наши модели с Polyphen2, SIFT и CADD по двум показателям и оценили потенциал применения наших моделей для клинической аннотации.
Метод 1: Точности для доброкачественного тестового набора
[00561] В одном варианте реализации мы оценили 10 000 доброкачественных вариантов и неизвестных вариантов путем вычисления их предсказанных вероятностей с использованием ансамбля из восьми различных обученных моделей. Мы также получили их предсказанные вероятности, оцененные другими существующими методами, упомянутыми выше.
[00562] Затем мы получили медианное значение предсказанных вероятностей для неизвестных вариантов для тестирования для каждого из методов, использованных при оценке. Используя среднюю оценку, мы нашли количество доброкачественных вариантов, которые получили оценку выше или ниже медианы, в зависимости от аннотации доброкачественных и патогенных вариантов, используемых каждым из методов. SIFT, CADD и наш способ маркируют патогенные варианты как 1, а доброкачественные - как 0. Таким образом, мы подсчитали количество доброкачественных вариантов, получивших оценку ниже медианы. В Polyphen используется противоположная аннотация, и мы подсчитали количество доброкачественных вариантов выше медианы. Отношение количества доброкачественных вариантов, набранных выше / ниже медианы, к общему количеству доброкачественных вариантов, представляет собой точность предсказания доброкачественных вариантов.
Точность для доброкачественных = Общее число доброкачественных вариантов выше (ниже*) медианы ÷ общее число доброкачественных вариантов
[00563] Наши рассуждения, обосновывающие этот метод оценки, основаны на анализе выборочного давления вариантов в gnomAD. Для одиночных вариантов (синглтонов) в gnomAD отношение миссенс-вариантов к синонимичным вариантам составляет ~ 2,26: 1. В то время как для распространенных вариантов (MAF> 0,1%) в gnomAD отношение миссенс/синонимы составляет ~ 1,06: 1. Это указывает на то, что для набора случайных неизвестных вариантов ожидается, что примерно 50% будут очищены естественным отбором и остальные 50% имеют тенденцию быть умеренными и, вероятно, станут распространенным явлением в популяции.
[00564] Как показано в таблице выше, наш способ превосходит второй лучший метод CADD более чем на 8%. Это показывает значительно улучшенную способности нашей модели классифицировать доброкачественные варианты. Хотя такая демонстрация доказывает возможности нашей модели, следующий метод 2 показывает полезность нашей модели на наборах клинических данных для клинической интерпретации.
Метод 2: Оценка набора клинических данных
[00565] В одном из вариантов реализации мы оценили эти методы предсказания патогенности на наборах клинических данных, включая набор данных случай заболевания-контроль для нарушений развития (DDD). Набор данных DDD включает 3821 миссенс-вариант de novo от больных детей и 1231 миссенс-вариант de novo от их здоровых братьев и сестер. Наша гипотеза состоит в том, что варианты de novo от больных детей, как правило, более опасны, чем варианты de novo от их здоровых братьев и сестер.
[00566] Поскольку наборы клинических данных не содержат четкой маркировки патогенных вариантов, мы использовали разделение между двумя наборами вариантов de novo (от больных и здоровых), чтобы оценить эффективность этих методов. Мы применили критерий суммы рангов Вилкоксона, чтобы оценить, насколько хорошо разделяются эти два набора вариантов de novo.
[00567] Согласно приведенной выше таблице, наши полуконтролируемые модели глубокого обучения работают значительно лучше в различении набора вариантов de novo больных пациентов от набора от здоровых пациентов. Это показывает, что наша модель лучше подходит для клинической интерпретации, чем существующие методы. Это также подтверждает, что общий подход к извлечению признаков из последовательностей генома и профилей сохранения превосходит определенные вручную признаки на наборах данных с курированием человеком.
Точность предсказаний доброкачественности на отложенном тестовом наборе из 10000 вариантов приматов
[00568] Мы использовали 10000 отложенных вариантов приматов в наборе тестовых данных для тестирования сети глубокого обучения, а также других 20 классификаторов. Поскольку разные классификаторы имели сильно различающиеся распределения оценок, мы использовали 10000 случайно выбранных неразмеченных вариантов, которые были выровнены с тестовым набором по тринуклеотидному контексту, чтобы определить порог 50-го процентиля для каждого классификатора. Чтобы обеспечить справедливое сравнение методов, мы сравнили каждый классификатор с долей вариантов в наборе из 10 000 отложенных тестовых вариантов приматов, которые были классифицированы как доброкачественные при пороге, равнос 50-му процентилю для этого классификатора.
[00569] Наши рассуждения, обосновывающие применение 50-го процентиля для идентификации доброкачественных вариантов основаны на избирательном давлении, наблюдаемом для миссенс-вариантов в наборе данных ExAC / gnomAD. Для вариантов, встречающихся с частотой одиночного аллеля, отношение миссенс: синонимы составляет ~ 2,2: 1, тогда как для распространенных вариантов (частота аллелей> 0,1%) отношение миссенс: синонимы составляет ~ 1,06: 1. Ожидается, что варианты будут очищены естественным отбором при распространенных частотах аллелей, а оставшиеся 50% являются достаточно мягкими, чтобы они могли стать распространенными в популяции по механизму генетического дрейфа.
[00570] Для каждого из классификаторов показана доля отложенных вариантов приматов для тестирования, предсказанных как доброкачественные с использованием порога, соответствующего 50-му процентилю (см. ФИГ. 28A и Дополнительную таблицу 17 (ФИГ. 34)).
Анализ вариантов de novo по исследованию DDD
[00571] Мы сравнили методы классификации по их способности различать de novo миссенс-варианты у индивидуумов, пораженных DDD, по сравнению с миссенс-вариантами de novo у здоровых родственных братьев. Для каждого классификатора мы определяли p-значение из теста суммы рангов Вилкоксона для разницы между оценками предсказаний для двух распределений (ФИГ. 28B и 28C и Дополнительная таблица 17 (ФИГ. 34)).
[00572] Учитывая, что наши две метрики для анализа показателей работы модели получены из разных источников и методологий, мы проверили, коррелировали ли показатели классификаторов по двум различным метрикам. В самом деле, мы обнаружили, что эти два показателя коррелировали: с коэффициентом Спирмена ρ = 0,57 (P <0,01) между точностью классификации доброкачественности на отложенном тестовом наборе приматов и p-значением суммы рангов Вилкоксона для de novo миссенс-вариантов в случаях DDD по сравнению с контролями. Это показывает, что существует хорошее соответствие между точностью для отложенного тестового набора приматов и p-значениями случаев DDD по сравнению с контролем для сравнительного анализа классификаторов (ФИГ. 30A).
[00573] Кроме того, мы проверили, может ли сеть глубокого обучения помочь в обнаружении генов, связанных с заболеванием. Мы протестировали увеличение количества мутаций de novo в генах, сравнив наблюдаемое количество мутаций de novo с числом, ожидаемым в рамках модели нулевых мутаций.
[00574] Мы исследовали показатели работы сети глубокого обучения, сравнивая результаты всех миссенс-мутаций de novo с результатами миссенс-мутаций с оценкой> 0,803. При тестировании всех миссенс-мутаций использовалась стандартная частота миссенс-мутаций, тогда как при тестировании отфильтрованных миссенс-мутаций использовались коэффициенты миссенс-мутаций, рассчитанные на сайтах с оценками> 0,803. Для каждого гена потребовалось четыре теста: один тест на обогащение вариантами, приводящими к укорочению белка, один тест на обогащение изменяющими белок мутациями de novo, причем оба применялись только для когорты DDD и для более крупного метаанализа когорт тройного секвенирования нервной системы. Обогащение изменяющих белок мутаций de novo было объединено по методу Фишера с тестом кластеризации миссенс-мутаций de novo в кодирующей последовательности (Дополнительные таблицы 20 и 21). Р-значение для каждого гена было взято из минимума из четырех тестов, а значимость для всего генома была определена как P <6,757 x 10-7 (α = 0,05, 18 500 генов с четырьмя тестами).
Характеристики рабочей кривой приемника и точность классификации в пределах 605 DDD генов, связанных с заболеванием
[00575] Чтобы проверить, действительно ли сеть глубокого обучения различает патогенные и доброкачественные варианты в пределах одного и того же гена, а не отдает предпочтение патогенности генов с доминантным типом наследования de novo, мы идентифицировали набор из 605 генов, которые были связаны с развитием нервной системы. заболевание с p-значением <0,05 в когорте DDD (рассчитано с использованием только вариантов de novo, приводящих к укорочению белка) (Дополнительная таблица 18). Мы приводим p-значение суммы рангов Вилкоксона для всех классификаторов для из способности различать распределения вероятностей вариантов в 605 генах в DDD и контрольном наборе данных (ФИГ. 28C и Дополнительная таблица 19 (ФИГ. 35)).
[00576] В этом наборе из 605 генов мы наблюдаем коэффициент обогащения для миссенс-вариантов de novo, который в три раза превышает ожидаемый на основании только степени мутаций. Это указывает на то, что миссенс-варианты de novo у пациентов, пораженных DDD, включают примерно 67% патогенных вариантов и 33% фоновых вариантов, в то время как миссенс-варианты de novo у здоровых контролей состоят в основном из фоновых вариантов, за исключением случаев неполной пенетрантности.
[00577] Чтобы вычислить максимально возможную AUC для классификатора, который отлично различает патогенные и доброкачественные варианты, мы учли, что только 67% de novo миссенс-вариантов у больных (пораженных) индивидуумов в пределах 605 генов были патогенными, а остальные были фоновыми. Чтобы построить кривую рабочих приемника, мы рассматривали классификацию de novo вариантов DDD как патогенных как истинно-положительные определений и рассматривали классификацию вариантов de novo в здоровых контролях как патогенные как ложноположительные определения. Следовательно, идеальный классификатор классифицировал бы 67% вариантов de novo у пациентов с DDD как истинно положительные, 33% вариантов de novo у пациентов с DDD как ложноотрицательные и 100% вариантов de novo в контроле как истинно отрицательные. Визуализация рабочей кривой приемника будет показывать только одну точку с 67% показателем истинноположительных и 0% ложноположительных результатов, соединенную с (0%, 0%) и (100%, 100%) углами графика прямыми линиями, которая дает максимальное значение AUC 0,837 для классификатора с полным различением доброкачественных и патогенных мутаций (ФИГ. 30B и Дополнительная таблица 19 (ФИГ. 35)).
[00578] Мы рассчитали точность классификации сети глубокого обучения для разделения патогенных и доброкачественных вариантов по бинарному порогу путем оценки ожидаемой доли патогенных вариантов в пределах 605 генов в комбинированных наборах данных DDD и здорового контроля. Поскольку набор данных DDD содержал 379 вариантов de novo с превышением на 249 миссенс-вариантов de novo над ожиданием, а контрольный набор данных содержал 65 вариантов de novo, мы ожидали 249 патогенных вариантов из 444 полных вариантов (ФИГ. 22A). Мы выбрали порог для каждого классификатора, который разделил 444 миссенс-варианта de novo на доброкачественные или патогенные категории в соответствии с этой ожидаемой долей, и использовали его как бинарный порог для оценки точности каждого классификатора. Для нашей модели глубокого обучения этот порог был достигнут при пороговом значении ≥ 0,803, с вероятностью истинных положительных результатов 65% и частотой ложных положительных результатов 14%. Для расчета точности классификации, скорректированной с учетом присутствия ~ 33% фоновых вариантов у индивидуумов DDD, мы предположили, что 33% вариантов de novo DDD, которые были фоновыми, будут классифицироваться с той же частотой ложноположительных результатов, которую мы наблюдали у здоровых контролей. Это соответствует 14% × 0,33 = 4,6% истинно положительных классификационных событий в наборе данных DDD, фактически являющихся ложноположительными из фоновых вариантов. По нашим оценкам, скорректированный показатель истинно-положительных результатов для сети глубокого обучения составляет (65% - 4,6%) / 67% = 90%. Мы определяет среднее значение показателя истинно-положительных и показателя истинно-отрицательных, которое составляет 88% для сети глубокого обучения (ФИГ. 30C и Дополнительная таблица 19 (ФИГ. 35)). Эта оценка, вероятно, недооценивает истинную точность классификатора из-за высокой распространенности неполной пенетрантности при нарушениях нервного развития.
Точность классификации ClinVar
[00579] Большинство существующих классификаторов обучаются на ClinVar; даже на классификаторы, которые не обучаются непосредственно на ClinVar, могут повлиять результаты предсказания от классификаторов, обученных на ClinVar. Кроме того, распространенные человеческие варианты высоко обогащены доброкачественными последствиями ClinVar, поскольку частота аллелей является частью критериев для определения доброкачественных последствий для варианта.
[00580] Мы попытались минимизировать зацикливание набора данных ClinVar, чтобы сделать его пригодным для анализа, используя только варианты ClinVar, которые были добавлены в 2017 году, поскольку другие методы классификации были опубликованы в предыдущие годы. Даже среди вариантов ClinVar 2017 года мы исключили все варианты, присутствующие с распространенными частотами аллелей (> 0,1%) в ExAC или присутствующие в HGMD, LSDB или Uniprot. После фильтрации всех таких вариантов и исключения вариантов с неопределенным значением и вариантов с противоречивыми аннотациями в ClinVar осталось 177 вариантов с доброкачественной аннотацией и 969 вариантов с патогенной аннотацией.
[00581] Мы оценили все варианты ClinVar, используя как сеть глубокого обучения, так и существующие методы. Мы выбрали порог для каждого классификатора, который разделил варианты ClinVar на категории доброкачественных или патогенных в соответствии с наблюдаемой долей доброкачественных и патогенных вариантов в этом наборе данных, и использовали его в качестве бинарного порога для оценки точности каждого классификатора. Мы определяет среднее значение показателя истинно положительных определений и показателя истинно отрицательных определений для каждого классификатора (ФИГ. 31A и 31B). Эффективность классификаторов в наборе данных ClinVar не коррелировала значимо с показатели работы классификаторов ни по точности классификации для 10000 отложенных вариантов приматов, ни по p-значению суммы рангов Вилкоксона для случаев DDD по сравнению с контрольным набором данных (ФИГ. 31A и 31B ).
[00582] Мы предполагаем, что существующие классификаторы точно моделируют поведение людей-экспертов, но что человеческие эвристики могут не быть полностью оптимальными для различения патогенных и доброкачественных мутаций в эмпирических данных. Одним из таких примеров является оценка Грэнтэма, которая обеспечивает метрику расстояния для характеристики сходства или несходства аминокислотных замен. Мы вычислили средний балл Грэнтэма (Grantham) для патогенных и доброкачественных вариантов в оценкой полного набора данных ClinVar (~ 42000 вариантов) и сравнили его со средней оценкой Грэнтэма для вариантов de novo у людей, пораженных DDD, и без них в пределах 605 генов. Чтобы скорректировать присутствие ~ 33% фоновых вариантов у лиц, пораженных DDD, мы увеличили разницу в баллах Грэнтэма между случаями DDD и контролями на 50%, что все же было меньше, чем разница между патогенными и доброкачественными вариантами в ClinVar. Одна из возможностей состоит в том, что эксперты-люди придают слишком большое значение мерам, которые легко измерить, таким как расстояние в заменах аминокислот, и в то же время недооценивают такие факторы, как структура белка, которые эксперту-человеку труднее определить количественно.
Интерпретация моделей глубокого обучения
[00583] Часто бывает сложно понять средства, с помощью которых алгоритмы машинного обучения решают проблемы. Мы визуализировали начальные уровни сети глубокого обучения, чтобы понять признаки, которые она научилась извлекать для предсказания патогенности вариантов. Мы рассчитали коэффициенты корреляции для различных аминокислот в пределах первых трех слоев (первый сверточный слой после двух слоев повышающей дискретизации) предварительно обученных моделей предсказания вторичной структуры с 3 состояниями и показали, что веса сверточных слоев обучаются функциям, очень похожим на Матрица BLOSUM62 или расстояние Грантэма.
[00584] Чтобы вычислить коэффициенты корреляции между различными аминокислотами, мы начали с весов первого сверточного слоя, которому предшествовали три уровня повышающей дискретизации (слои 1a, 1b и 1c) в модели вторичной структуры. Мы выполнили матричное умножение между тремя слоями, в результате получилась матрица с размерами (20,5,40), где 20 - количество аминокислот, 5 - размер окна сверточного слоя, а 40 - количество ядер. Мы изменили форму матрицы так, чтобы она имела размерность (20 200), сглаживая последние два измерения, получив матрицу, в которой веса, действующие на каждую из 20 аминокислот, были представлены как вектор длиной 200. Мы рассчитали корреляционную матрицу между 20 аминокислотами. Поскольку каждое измерение представляет каждую аминокислоту, вычисляя матрицу коэффициентов корреляции, мы вычисляем корреляцию между аминокислотами и насколько они похожи на сеть глубокого обучения, основываясь на том, что она извлекла из данных обучения. Визуализация матрицы коэффициентов корреляции представлена на ФИГ. 27 (аминокислоты, отсортированные по матричному порядку BLOSUM62) и показывает два заметных кластера, включающих гидрофобные аминокислоты (метионин, изолейцин, лейцин, валин, фенилаланин, тирозин, триптофан) и гидрофильные аминокислоты (аспарагин, аспарагиновая кислота, глутаминовая кислота). Кислота, глутамин, аргинин и лизин). Выходные данные этих начальных уровней становятся входами для последующих уровней, что позволяет сети глубокого обучения создавать все более сложные иерархические представления данных.
[00585] Чтобы проиллюстрировать окно аминокислотной последовательности, используемое нейронной сетью в ее предсказаниях, мы нарушали (изменяли) каждое положение в приблизительно 5000 случайно выбранных вариантах, чтобы наблюдать ее влияние на предсказанный показатель PrimateAI для варианта (ФИГ. 25B). Мы систематически обнуляли входные данные в каждом соседнем положении аминокислоты (от -25 до +25) вокруг варианта и измеряли изменение прогнозируемой нейронной сетью патогенности варианта и наносили на график среднее абсолютное значение изменения для 5000 вариантов. Аминокислоты, близкие к варианту, имеют наибольший эффект, при примерно симметричном распределении, с постепенным уменьшением по мере увеличения расстояния от варианта. Важно отметить, что модель делает свои предсказания, основываясь не только на аминокислоте в положении варианта, но и на информации из более широкого окна, которая может потребоваться для распознавания белковых мотивов. В соответствии с относительно компактным размером белковых субдоменов, мы эмпирически наблюдали, что увеличение размера окна до более чем 51 аминокислоты не привело к дальнейшему повышению точности.
[00586] Чтобы оценить чувствительность классификатора глубокого обучения к выравниванию, мы исследовали влияние глубины выравнивания на точность классификации вариантов следующим образом. Мы разделили данные на пять интервалов в зависимости от количества видов в выравнивании и оценили точность сети в каждом интервале (ФИГ. 57). Мы обнаружили, что точность сети при отделении набора скрытых доброкачественных мутаций от случайно выбранных мутаций, которые были выровнены с тринуклеотидным контекстом (как в ФИГ. 21D, но выполнялись отдельно для каждого интервала), наиболее высока в трех верхних интервалах и заметно слабее в двух нижних интервалах. Многовидовое выравнивание 99 позвоночных включает 11 приматов, не являющихся людьми, 50 млекопитающих и 38 позвоночных, причем два нижних интервала представляют белки, которые имеют разреженную информацию о выравнивании от других млекопитающих, не являющихся приматами. Сеть глубокого обучения является надежной и точной, когда информация о выравнивании распространяется на приматов и млекопитающих, а информация о консервации от более удаленных позвоночных менее важна.
Определение канонических кодирующих областей
[00587] Для того чтобы определить канонические кодирующие области, из геномного браузера UCSC загружались данные множественных выравниваний 99 геномов позвоночных с геномом человека для областей, содержащих кодирующие последовательности ДНК человека (knownCanonical.exonNuc.fa.gz). Координаты экзонов человека находятся в сборке hg19. Экзоны объединяются, чтобы сформировать ген. Гены на аутосомах и Х-хромосоме остаются без изменений. Негомологичные гены были удалены в соответствии со списком гомологичных генов, загруженным из NCBI (ftp://ftp.ncbi.nih.gov/pub/HomoloGene/current/homologene.data). Для однонуклеотидного полиморфизма с несколькими аннотациями генов в качестве аннотации однонуклеотидного полиморфизма выбирается самый длинный транскрипт.
Данные полиморфизмов человека, обезьяны и млекопитающих
[00588] Мы загружали данные полиморфизмов экзома человека из базы данных агрегации генома (gnomAD), сформированной в результате недавнего крупномасштабного исследования, в которой собраны данные последовательности полного экзома для 123136 человек из 8 субпопуляций по всему миру. Затем извлекали варианты, прошедшие фильтрацию и попавшие в области канонического кодирования.
[00589] Большой проект секвенирования генома обезьяны содержит данные о секвенировании полного генома 24 шимпанзе, 13 бонобо, 27 горилл и 10 орангутанов (включая 5 суматранских орангутанов и 5 калимантанских орангутанов). Результаты исследования шимпанзе и бонобо позволяют дополнить эти данные последовательностями полного генома 25 человекообразных обезьян. Поскольку все данные секвенирования были картированы по hg19, мы загрузили файлы VCF, полученные в результате этих исследований, и непосредственно извлекли варианты в пределах областей канонического кодирования.
[00590] Для сравнения с другими обезьянами и млекопитающими мы также загрузили данные однонуклеотидных полиморфизмов некоторых других видов из базы данных однонуклеотидных полиморфизмов, в том числе макака-резус, мартышки, свиньи, коровы, козы, мыши и курицы. Мы исключили другие виды, такие как собака, кошка или овца, так как базы данных однонуклеотидных полиморфизмов содержат ограниченное число вариантов для этих видов. Сначала мы перенесли однонуклеотидные полиморфизмы каждого вида на hg19. Оказалось, что около 20% вариантов соответствуют областям псевдогенов. Затем для каждого вида мы получили координаты экзонов из файла множественных выравниваний для 100 позвоночных из областей канонического кодирования и определили варианты в этих экзонах. Затем указанные определенные однонуклеотидные полиморфизмы были перенесены на hg19. В тех случаях, когда варианты находились на другой сборке генома указанного выравнивания для данного вида, мы предварительно переносили варианты на искомую сборку генома указанного выравнивания.
[00591] Поскольку данные однонуклеотидных полиморфизмов коровы получены из различных исследований, мы загрузили из базы данных однонуклеотидных полиморфизмов все большие пакеты данных вариантов коровы (16 пакетов с файлами VCF размером более 100 МБ) и оценили качество различных пакетов данных однонуклеотидных полиморфизмов коровы по величине миссенс-синонимического отношения для каждого пакета. Медианное значение миссенс-синонимического отношения составляет 0,781, а среднее абсолютное отклонение (MAD) составляет 0,160 (среднее значение - 0,879, стандартное отклонение (SD) - 0,496). Два пакета с выпадающими значениями указанного отношения (SNPBatch_1000_BULL_GENOMES_1059190.gz с отношением 1,391 и SNPBatch_COFACTOR_GENOMICS_1059634.gz с отношением 2,568) были исключены из дальнейшего анализа.
Оценка свойств полиморфизма у обезьян и млекопитающих
[00592] Для того, чтобы продемонстрировать удобство использования однонуклеотидных полиморфизмов человекообразных обезьян, мы рассчитывали показатель обогащения, как отношение числа синглтонов (единичных вариантов) к числу обыкновенных однонуклеотидных полиморфизмов (частота встречаемости аллелей (AF) > 0,1%). Известно, что синонимичные варианты являются доброкачественными и обычно изменяются нейтрально без какого-либо давления отбора. В отличие от синонимичных вариантов, вредные миссенс-варианты постепенно удаляются естественным отбором, поэтому их распределение по частоте встречаемости аллелей, как правило, изобилует редкими вариантами.
[00593] Мы сосредоточились на таких однонуклеотидных полиморфизмах из базы данных gnomAD, которые перекрываются с однонуклеотидными полиморфизмами, наблюдаемыми у приматов, млекопитающих и домашних птиц. Мы подсчитали количество синонимичных и миссенс-вариантов для каждого вида. После этого миссенс-варианты были разделены на два типа: «миссенс-идентичные», для которых соответствующие изменения аминокислот совпадают с другими видами, и «миссенс-различные», для которых соответствующие изменения аминокислот отличаются от других видов. Соответствующие показатели обогащения затем рассчитывались для каждого вида, как отношение числа синглетонов к числу обыкновенных вариантов.
[00594] Кроме того, проводилась проверка гипотезы об однородности по критерию хи-квадрат (χ2) для таблицы сопряженности 2x2c целью сравнения показателей обогащения, рассчитываемых для синонимичных и миссенс-идентичных вариантов для каждого вида. Для всех приматов существенной разницы в показателях обогащения между синонимичными и миссенс-идентичными вариантами обнаружено не было, в то время как значительная разница была обнаружена для коровы, мыши и курицы.
[00595] В результате обнаружено, что те однонуклеотидные полиморфизмы, которые соответствуют одинаковым изменениям аминокислот у человекообразных обезьян, характеризуются показателями обогащения очень близкими, по своим значениям, к синонимичным однонуклеотидным полиморфизмам, и, таким образом, можно предположить, что они не связаны со значительным влиянием на здоровье человека. В то же время, однонуклеотидные полиморфизмы, соответствующие неодинаковым изменениям аминокислот у человекообразных обезьян, характеризуются показателями обогащения, существенно отличающимся, по своим значениям, от синонимичных однонуклеотидных полиморфизмов. Миссенс-полиморфизмы у видов, не являющихся приматами, также имеют распределение по частоте встречаемости аллелей, отличное от синонимичных вариантов. Таким образом, однонуклеотидные полиморфизмы, которые соответствуют одинаковым изменениям аминокислот у человекообразных обезьян, могут быть добавлены в тренировочный набор доброкачественных вариантов.
[00596] Наше предположение состоит в том, что большинство вариантов образовывается независимо и не связанно с идентичностью по происхождению. Таким образом, мы провели анализ обогащения для редких вариантов в идентичных по происхождению однонуклеотидных полиморфизмах, чтобы оценить различное поведение их показателей обогащения. Идентичные по происхождению однонуклеотидные полиморфизмы определяются как такие однонуклеотидные полиморфизмы человека, которые появляются как у человека, так и у двух или более видов человекообразных обезьян, включая шимпанзе, бонобо, гориллу, суматранского орангутана и калимантанского орангутана. Затем показатели обогащения, определяемые как отношение числа синглетонов к числу обыкновенных вариантов (с частотой встречаемости аллелей AF> 0,1%), рассчитываются отдельно для миссенс-вариантов и рассматриваемых в качестве нейтральной основы для сравнения синонимичных вариантов.
Фиксированные замены среди млекопитающих
Анализ обогащения фиксированных замен
[00597] Мы также проанализировали обогащение межвидовых замен редкими вариантами. Мы загрузили филогенетическое дерево для 100 видов позвоночных из геномного браузера UCSC (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/multiz100way/hg19.100way.commonNames.nh). Затем мы вычисляли филогенетические расстояния между парами и отбирали пары близкородственных видов (расстояние <0,3). Чтобы получить пары для приматов, мы загрузили из геномного браузера UCSC данные выравнивания (hg38) 19 геномов млекопитающих (16 приматов) с геномом человека для областей, содержащих кодирующие последовательности. Четыре пары приматов были добавлены к 13 парам позвоночных. В следующей таблице приведены генетические расстояния для нескольких пар близкородственных видов в соответствии с одним из вариантов реализации.
00598] Мы использовали данные множественных выравниваний 19 геномов млекопитающих или 99 позвоночных с геномом человека для канонических кодирующих областей с целью получения данных о заменах нуклеотидов для каждой выбранной пары позвоночных. Было произведено картирование указанных замен с однонуклеотидными полиморфизмами экзома человека из базы данных gnomAD с учетом идентичных изменений кодонов между парой видов и вариантами человека. Мы разделили варианты на три типа: синонимичные варианты; миссенс-варианты, для которых соответствующие изменения аминокислот совпадают с другими видами; миссенс-варианты, для которых соответствующие изменения аминокислот не совпадают с другими видами. Показатели обогащения были рассчитаны для каждого типа относительно пар видов.
Сравнение внутривидовых и межвидовых полиморфизмов
[00599] Были отобраны шесть видов для проведения сравнения внутривидовых и межвидовых полиморфизмов, включая шимпанзе, макака-резус, мартышку, козу, мышь и курицу, поскольку для этих видов доступны данные вариантов, как внутривидовых, так и межвидовых. Сравнение показателей обогащения внутривидовых и межвидовых вариантов аналогично сравнению коэффициентов несогласия, рассчитываемых для двух таблиц сопряженности 2x2. Для оценки однородности коэффициентов несогласия, рассчитываемых для таблиц сопряженности, обычно применяется тест Вульфа. Таким образом, мы использовали тест Вульфа для оценки различия между показателями обогащения для внутривидовых и межвидовых полиморфизмов.
Анализ обогащения в расчете на ген
[00600] На Фиг. 64 представлен один из вариантов реализации анализа обогащения на ген. В одном из вариантов реализации для анализа обогащения на ген используется классификатор патогенности варианта, разработанный на основе сверточной нейронной сети глубокого обучения, который подтверждает патогенность тех вариантов, патогенность которых была установлена. Для одного конкретного гена, взятого из группы индивидуумов с генетическим заболеванием, анализ обогащения на ген включает в себя: идентификацию патогенных вариантов-кандидатов в указанном конкретном гене с использованием классификатора патогенности вариантов на основе сверточной нейронной сети глубокого обучения; определение фонового числа мутаций для указанного конкретного гена с помощью суммирования наблюдаемых тринуклеотидных мутаций вариантов-кандидатов с последующим умножением полученной суммы на количество трансмиссий и размер когорты; идентификацию полученных de novo патогенных миссенс-вариантов в конкретном гене с использованием классификатора патогенности вариантов на основе сверточной нейронной сети глубокого обучения; сравнение указанного фонового числа мутаций с количеством полученных de novo миссенс-вариантов. На основании указанного сравнения анализ обогащения на ген подтверждает, что конкретный ген связан с генетическим нарушением и что полученные de novo миссенс-варианты являются патогенными. В некоторых вариантах реализации в качестве генетического расстройства рассматривается расстройство аутистического спектра (сокращенно ASD). В других вариантах реализации В других вариантах реализации в качестве генетического расстройства рассматривается расстройство задержки в развитии (сокращенно DDD).
[00601] В примере, представленном на Фиг. 64, пять вариантов-кандидатов в конкретном гене были классифицированы как патогенные с помощью классификатора патогенности вариантов на основе сверточной нейронной сети глубокого обучения. Этим пяти вариантам-кандидатам соответствуют значения наблюдаемых частот мутаций тринуклеотидов равные 10-8, 10-2, 10-1, 105, и 101. Фоновое число мутаций для конкретного гена определено равным 10-5 в результате суммирования соответствующих наблюдаемых частот мутаций тринуклеотидов в пяти вариантах-кандидатах с последующим умножением полученной суммы на количество трансмиссий / хромосом (2) и размер когорты (1000), после чего это значение сравнивается с количеством полученных de novo вариантов (3).
[00602] В некоторых вариантах реализации классификатор патогенности вариантов на основе сверточной нейронной сети глубокого обучения сформирован таким образом, чтобы также проводить указанное сравнение с использованием какого-либо статистического теста, который в качестве выходного значения выдает p-значение уровня значимости.
[00603] В других вариантах реализации классификатор патогенности вариантов на основе сверточной нейронной сети глубокого обучения сформирован таким образом, чтобы также проводить указанное сравнение фонового числа мутаций с количеством полученных de novo миссенс-вариантов, и на основании результатов такого сравнения получать подтверждение того, что конкретный ген не связан с генетическим заболеванием и что полученные de novo миссенс-варианты являются доброкачественными.
Анализ обогащения полного генома
[00604] На Фиг. 65 показан один из вариантов реализации анализа обогащения полного генома. В другом варианте реализации классификатор патогенности вариантов на основе сверточной нейронной сети глубинного обучения сформирован таким образом, чтобы также проводить анализ обогащения полного генома для получения подтверждения патогенности тех вариантов, патогенность которых была установлена. Анализ обогащения полного генома включает в себя: идентификацию первого набора полученных de novo миссенс-вариантов, являющихся патогенными для некоторого количества образцов генов, взятых у группы здоровых индивидуумов, с использованием классификатора патогенности вариантов на основе сверточной нейронной сети глубинного обучения; идентификацию второго набора полученных de novo миссенс-вариантов, являющихся патогенными для некоторого количества образцов генов, взятых у группы индивидуумов с генетическими расстройствами, с использованием классификатора патогенности вариантов на основе сверточной нейронной сети глубинного обучения и последующим сравнением значений, полученных в первом и втором наборах; подтверждение на основании указанного сравнения того, что второй набор полученных de novo миссенс-вариантов, соответствующий группе индивидуумов с генетическими расстройствами, является обогащенным и, следовательно, является патогенным. В некоторых вариантах реализации в качестве генетического расстройства рассматривается расстройство аутистического спектра (сокращенно ASD). В других вариантах реализации в качестве генетического расстройства рассматривается расстройство задержки в развитии (сокращенно DDD).
[00605] В некоторых вариантах реализации классификатор патогенности вариантов на основе сверточной нейронной сети глубинного обучения сформирован таким образом, чтобы также проводить указанное сравнение с использованием какого-либо статистического теста, который в качестве выходного значения выдает p-значение уровня значимости. В одном из вариантов реализации указанное сравнение параметризировано в соответствии с размерами групп (когорт).
[00606] В некоторых вариантах реализации классификатор патогенности вариантов на основе сверточной нейронной сети глубинного обучения сформирован таким образом, чтобы также проводить указанное сравнение соответствующих значений в первом и втором наборах и на основании результатов такого сравнения получать подтверждение того, что второй набор полученных de novo миссенс-вариантов, соответствующий группе индивидуумов с генетическими расстройствами, не является обогащенным и, следовательно, является доброкачественным.
[00608] На Фиг.65 для конкретного случая представлены частота появления мутаций в группе здоровых индивидуумов (0,001) и частота появления мутаций в группе больных индивидуумов (0,004) вместе с соотношением мутаций, приходящихся на одного индивида (4).
Частные варианты реализации
[00608] Мы описываем системы, способы и изделия для конструирования классификатора патогенности вариантов. Один или более признаков одного из вариантов реализации могут быть объединены с базовым вариантом реализации. Подразумевается, что варианты, которые не являются взаимоисключающими, совместимы. Один или более признаков варианта реализации можно комбинировать с другими вариантами реализации. Настоящее описание периодически напоминает пользователю об этих опциях. То, что в некоторых вариантах реализации пропущено упоминание об этих опциях, не следует воспринимать как ограничение комбинаций, описанных в предыдущих разделах - в настоящем тексте такое описание включается посредством ссылки в каждый их последующих вариантов реализации.
[00609] Один вариант реализации системы в соответствии с раскрытой технологией включает один или более процессоров, соединенных с памятью. В память загружены компьютерные команды для обучения детектора сайтов сплайсинга, который идентифицирует сайты сплайсинга в геномных последовательностях (например, нуклеотидных последовательностях).
[00610] Как показано на ФИГ. 48 и 19, система обучает классификатор патогенности вариантов на основе сверточной нейронной сети, который реализуется на множестве процессоров, связанных с памятью. Эта система использует доброкачественные обучающие примеры и патогенные обучающие примеры пар последовательностей белков, сгенерированных по доброкачественным вариантам и патогенным вариантам. Указанные доброкачественные варианты включают распространенные (обычные) миссенс-варианты человека и миссенс-варианты примата, отличного от человека, встречающиеся в альтернативных последовательностях кодонов примата, отличного от человека, которые обладают совпадающими референсными последовательностями кодонов с людьми. Фраза «пары белковых последовательностей» относится к референсной белковой последовательности и альтернативной белковой последовательности, где референсная протеиновая последовательность включает референсные аминокислоты, образованные референсными триплетами нуклеотидных оснований (референсными кодонами), а альтернативная белковая последовательность включает альтернативные аминокислоты, образованные альтернативными триплетами нуклеотидных оснований (альтернативными кодонами), так что альтернативная последовательность белка образуется в результате варианта, встречающегося в контрольных триплетных нуклеотидных основаниях (контрольных кодонах), образующих контрольные аминокислоты последовательности контрольного белка. Вариант может представлять собой SNP, вставку или делецию.
[00611] Этот вариант реализации системы и другие раскрытые системы необязательно включают один или более из следующих признаков. Система может также включать признаки, описанные в связи с раскрытыми способами. Для краткости альтернативные комбинации признаков системы не приводятся по отдельности. Признаки, связанные с системами, способами и изделиями, не повторяются для каждого заявленного набора классов основных признаков. Читатель поймет, как признаки, идентифицированные в этом разделе, можно легко объединять с основными признаками в других заявленных классах.
[00612] Как показано на ФИГ. 44, распространенные (обычные) миссенс-варианты человека, имеющие частоту минорного аллеля (сокращенно MAF) выше чем 0.1% в наборе данных по вариантам в человеческой популяции, полученным по образцам от меньшей мере 100000 человек.
[00613] Как показано на ФИГ. 44, люди, у которых взяты образцы принадлежат к различным субпопуляциям людей, и распространенные (обычные) миссенс-варианты человека имеют MAF выше, чем 0.1% в соответствующих наборах данных по вариантам в соответствующей популяции людей.
[00614] Указанные субпопуляции людей включают африканцев/афро-американцев (сокращенно AFR), американцев (сокращенно AMR), евреев - ашкеназов (сокращенно ASJ), восточных азиатов (сокращенно EAS), финнов (сокращенно FIN), европейцев, отличных от финнов (сокращенно NFE), южных азиатов (сокращенно SAS), и других (сокращенно OTH).
[00615] Как показано на ФИГ. 43 и 44, миссенс-варианты примата, отличного от человека, включают миссенс-варианты из множества видов приматов, отличных от человека, включая шимпанзе, бонобо, гориллу, калимантанского орангутана, суматранского орангутана, Резусов и игрунок.
[00616] Как показано на ФИГ. 45 и 46, на основании анализа обогащения, система принимает конкретный вид приматов, отличный от человека, для включения миссенс-вариантов указанного конкретного вида приматов, отличных от человека, в доброкачественные варианты. Анализ обогащения включает, для указанного конкретного вида приматов, отличного от человека, сравнение первого показателя обогащения синонимичных вариантов указанного конкретного вида приматов, отличного от человека, со вторым показателем обогащения идентичных миссенс-вариантов указанного конкретного вида приматов, отличного от человека.
[00617] ФИГ. 45 демонстрирует один вариант реализации человеческих ортологичных миссенс- SNP. Миссенс-SNP в виде, отличным от человека, который имеет соответствующие референсный и альтернативный кодоны у людей. Как показано на ФИГ. 45, указанные идентичные миссенс-варианты представляют собой миссенс-варианты, обладающие совпадающими общими референсной и альтернативной последовательностями кодонов с людьми.
[00618] Как показано на ФИГ. 46 и 47, указанный первый показатель обогащения получают путем определения отношения редких синонимичных вариантов с MAF ниже 0.1% к обычным синонимичным вариантам с MAF выше 0.1%. Указанный второй показатель обогащения получают путем определения отношения редких идентичных миссенс-вариантов с MAF ниже 0.1% к обычным идентичным миссенс-вариантам с MAF выше 0.1%. Редкие варианты включают одноточечные варианты.
[00619] Как показано на ФИГ. 46 и 47, разница между указанным первым показателем обогащения и указанным вторым показателем обогащения укладывается в заранее определенный диапазон, дополнительно включающий прием указанного конкретного вида примата, отличного от человека, для включения миссенс-вариантов указанного конкретного примата, отличного от человека, в доброкачественные варианты. Эта разница, укладывающаяся в заранее определенный диапазон, указывает на то, что указанные идентичные миссенс-варианты подвергаются естественному отбору в той же степени, что и синонимичные варианты, и, следовательно, являются такими же доброкачественными, как синонимичные варианты.
[00620] Как показано на ФИГ. 48, система повторяет применение анализа обогащения приемом множества видов приматов, отличных от человека, для включения миссенс-вариантов указанных видов приматов, отличны от человека, в число доброкачественных вариантов. Система дополнительно включает критерий хи-квадрат для гомогенности для сравнения первого показателя обогащения синонимичных вариантов и второго показателя обогащения идентичных миссенс-вариантов для каждого из указанных видов приматов, отличных от человека
[00621] Как показано на ФИГ. 48, подсчитанное число миссенс-вариантов примата, отличного от человека, составляет по меньшей мере 100000. Число миссенс-вариантов примата, отличного от человека, составляет 385236. Число обычных миссенс-вариантов человека составляет по меньшей мере 50000. Число обычных миссенс-вариантов человека составляет 83546.
[00622] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению системой описанных выше действий. Еще один вариант реализации может включать выполнение описанных выше действий системы.
[00623] Другой вариант реализации системы согласно раскрытой технологии включает построение классификатора однонуклеотидных полиморфизмов (сокращенно SNP). Система обучает (тренирует) классификатор патогенности SNP на основе сверточной нейронной сети, реализованной на множестве процессоров, связанных с памятью, с применением доброкачественных обучающих примеров и патогенных обучающих примеров аминокислотных последовательностей, экспрессируемых доброкачественными SNP и патогенными SNP. Доброкачественные обучающие примеры включают первый и второй наборы нуклеотидных последовательностей, экспрессируемых в виде пары аминокислотных последовательностей, причем каждая аминокислотная последовательность содержит центральную аминокислоту, фланкированную вышележащими и нижележащими аминокислотами. Каждая пара аминокислотных последовательностей включает референсную последовательность аминокислот, экспрессируемую референсной нуклеотидной последовательностью, и альтернативную последовательность аминокислот, экспрессируемую альтернативной нуклеотидной последовательностью, содержащей SNP.
[00624] Как показано на ФИГ. 9, указанный первый набор содержит пары нуклеотидных последовательностей человека, причем каждая пара включает альтернативную нуклеотидную последовательность человека, содержащую SNP, и имеет частоту минорного аллеля (сокращенно MAF), которая считается распространенной в человеческой популяции. Второй набор содержит референсную нуклеотидную последовательность примата, отличного от человека, в паре с альтернативной нуклеотидной последовательностью примата, отличного от человека. Указанная референсная нуклеотидная последовательность примата, отличного от человека, имеет ортологичную референсную нуклеотидную последовательность человека. Указанная альтернативная нуклеотидная последовательность примата, отличного от человека, содержит SNP.
[00625] Каждый признак, обсуждаемый в этом разделе, относящемся к конкретному варианту реализации, применительно к первому варианту реализации системы, равно применим к этому варианту реализации системы. Как указано выше, здесь не повторяются все признаки системы, подразумевается, что они повторяются посредством ссылки.
[00626] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению системой описанных выше действий. Еще один вариант реализации может включать выполнение описанных выше действий системы.
[00627] Как показано на ФИГ. 48 и 19, первый вариант реализации способа согласно раскрытой технологии включает построение классификатора патогенности вариантов, причем способ включает. Способ дополнительно включает обучение классификатора патогенности вариантов на основе сверточной нейронной сети, который работает на множестве процессоров, связанных с памятью, с применением доброкачественных обучающих примеров и патогенных обучающих примеров пар последовательностей белков, сгенерированных по доброкачественным вариантам и патогенным вариантам. Указанные доброкачественные варианты включают распространенные (обычные) миссенс-варианты человека м миссенс-варианты примата, отличного от человека, встречающиеся в альтернативных последовательностях кодонов примата, отличного от человека, которые обладают совпадающими референсными последовательностями кодонов с людьми.
[00628] Каждый из признаков, обсуждаемых в разделе, относящемся к этому конкретному варианту реализации, для первого варианта реализации системы, равно относится к этому варианту реализации способа. Как указано выше, здесь не повторяются все признаки системы, подразумевается, что они повторяются посредством ссылки.
[00629] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению описанного выше способа. Еще один вариант реализации может включать систему, включающую память и один или более процессоров, выполненных с возможностью исполнять инструкции, хранящиеся в памяти, с реализацией описанного выше способа.
[00630] Как показано на ФИГ. 48 и 19, второй вариант реализации способа согласно раскрытой технологии включает построение классификатор однонуклеотидных полиморфизмов (сокращенно SNP). Способ дополнительно включает обучение классификатора патогенности SNP на основе сверточной нейронной сети, который работает на множестве процессоров, связанных с памятью, с применением доброкачественных обучающих примеров и патогенных обучающих примеров аминокислотных последовательностей, экспрессируемых доброкачественными SNP и патогенными SNP. Доброкачественные обучающие примеры включают первый и второй наборы нуклеотидных последовательностей, экспрессируемых в виде пары аминокислотных последовательностей, причем каждая аминокислотная последовательность содержит центральную аминокислоту, фланкированную вышележащими и нижележащими аминокислотами, и каждая пара аминокислотных последовательностей включает референсную последовательность аминокислот, экспрессируемую референсной нуклеотидной последовательностью, и альтернативную последовательность аминокислот, экспрессируемую альтернативной нуклеотидной последовательностью, содержащей SNP. Указанный первый набор содержит пары нуклеотидных последовательностей человека, причем каждая пара включает альтернативную нуклеотидную последовательность человека, содержащую SNP, и имеет частоту минорного аллеля (сокращенно MAF), которая считается распространенной в человеческой популяции. Второй набор содержит референсную нуклеотидную последовательность примата, отличного от человека, в паре с альтернативной нуклеотидной последовательностью примата, отличного от человека. Указанная референсная нуклеотидная последовательность примата, отличного от человека, имеет ортологичную референсную нуклеотидную последовательность человека and указанная альтернативная нуклеотидная последовательность примата, отличного от человека, содержит SNP.
[00631] Каждый из признаков, обсуждаемых в этом разделе этого конкретного варианта реализации для второго варианта реализации систему равно относится к этому варианту реализации способа. Как указано выше, здесь не повторяются все признаки системы, подразумевается, что они повторяются посредством ссылки.
[00632] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению описанного выше способа. Еще один вариант реализации может включать систему, включающую память и один или более процессоров, выполненных с возможностью исполнять инструкции, хранящиеся в памяти, с реализацией описанного выше способа.
[00633] Мы описываем системы, способы и изделия для применения классификатора патогенности вариантов на основе глубокой сверточной нейронной сети с классификаторами вторичной структуры и доступности для растворителя. Один или более признаков одного из вариантов реализации могут быть объединены с базовым вариантом реализации. Подразумевается, что варианты, которые не являются взаимоисключающими, совместимы. Один или более признаков варианта реализации можно комбинировать с другими вариантами реализации. Настоящее описание периодически напоминает пользователю об этих опциях. То, что в некоторых вариантах реализации пропущено упоминание об этих опциях, не следует воспринимать как ограничение комбинаций, описанных в предыдущих разделах - в настоящем тексте такое описание включается посредством ссылки в каждый их последующих вариантов реализации.
[00634] Один вариант реализации системы в соответствии с раскрытой технологией включает один или более процессоров, соединенных с памятью. В память загружены компьютерные команды по запуску классификатора патогенности вариантов на основе глубокой сверточной нейронной сети с классификаторами вторичной структуры и доступности для растворителя.
[00635] Система содержит первую подсеть вторичной структуры, реализованную на множестве процессоров, связанных с памятью, обученную предсказывать вторичную структуру с тремя состояниями для положений аминокислот в белковой последовательности. Система дополнительно включает вторую подсеть доступности для растворителя, реализованную на множестве процессоров, связанных с памятью, обученную предсказывать доступность для растворителя с тремя состояниями для положений аминокислот в белковой последовательности.
[00636] Вторичная структура с тремя состояниями относится к одному из множества состояний вторичной структуры ДНК: альфа-спираль (H), бета-слой (B) и скрученную спираль (coil, C).
[00637] Доступность для растворителя с тремя состояниями относится к одному из множества состояний доступности для растворителя: заглубленное (B), промежуточное (I) и экспонированное (E).
[00638] Генератор частотной матрицы положений (сокращенно PFM), реализованный на по меньшей мере одном из указанного множества процессоров, применяется к трем группам последовательностей: приматов, млекопитающих и позвоночных за исключением приматов и млекопитающих для генерации PFM приматов, PFM млекопитающих и PFM позвоночных.
[00639] Другими словами, это включает применение генератора PFM к данным последовательностей приматов для генерации PFM приматов, применение генератора PFM к данным последовательностей млекопитающих для генерации PFM млекопитающих и применение генератора PFM к данным последовательностей позвоночных, за исключением приматов и млекопитающих, для создания PFM позвоночных.
[00640] Входной процессор, который принимает вариантную аминокислотную последовательность с целевой вариантной аминокислотой, фланкированной в направлении 5 'и в направлении 3' по меньшей мере 25 аминокислотами в каждом направлении, причем вариант с одним нуклеотидом порождает вариант целевой аминокислоты. Распределитель дополнительных данных, реализованный по меньшей мере на одном из указанного множества процессоров, который сопоставляет референсную аминокислотную последовательность с референсной целевой аминокислотой, фланкированной в направлении 5 'и в направлении 3' по меньшей мере 25 аминокислотами в каждом направлении, выровненную с вариантной аминокислотной последовательностью. После этого он распределяет (присваивает) классификации референсных состояний, созданные первой и второй подсетями для референсной аминокислотной последовательности. После этого распределитель дополнительных данных распределяет классификации состояний вариантов, полученные в первой и второй подсетях для вариантной аминокислотной последовательности. Наконец, он распределяет (присваивает) PFM приматов, млекопитающих и позвоночных, выровненные с эталонной аминокислотной последовательностью.
[00641] В контексте данной заявки фраза «выровненная с» относится к определению по положениям PFM приматов, млекопитающих и позвоночных для каждого аминокислотного положения в референсной аминокислотной последовательности или альтернативной аминокислотной последовательности, а также к кодированию и хранению результатов определения на основе положения или порядкового положения в том же порядке, в котором положения аминокислот встречаются в референсной аминокислотной последовательности или альтернативной аминокислотной последовательности.
[00642] Система также включает глубокую сверточную нейронную сеть, реализуемую на указанном множестве процессов, обученную для классификации вариантной аминокислотной последовательности как доброкачественной или патогенной на основе обработки вариантной аминокислотной последовательности, присвоенной референсной аминокислотной последовательности, присвоенной референсной последовательности. и классификации состояний вариантов и присвоенных PFM. Система включает в себя процессор вывода, который по меньшей мере выдает оценку патогенности для варианта аминокислотной последовательности.
[00643] Этот вариант реализации системы и другие раскрытые системы необязательно включают один или более из следующих признаков. Система может также включать признаки, описанные в связи с раскрытыми способами. Для краткости альтернативные комбинации признаков системы не приводятся по отдельности. Признаки, связанные с системами, способами и изделиями, не повторяются для каждого заявленного набора классов основных признаков. Читатель поймет, как признаки, идентифицированные в этом разделе, можно легко объединять с основными признаками в других заявленных классах.
[00644] Система, содержащая классификатор патогенности вариантов на основе глубокой сверточной нейронной сети, дополнительно сконфигурированная для классификации однонуклеотидного варианта как доброкачественного или патогенного на основе оценки патогенности.
[00645] Система содержит классификатор патогенности вариантов на основе глубокой сверточной нейронной сети, в котором глубокая сверточная нейронная сеть принимает, параллельно, в качестве входных данных, по меньшей мере вариантную аминокислотную последовательность, классификацию присвоенных вторичных состояний варианта, классификацию присвоенных вторичных состояний референса, классификацию присвоенной доступности для растворителя варианта, классификацию присвоенной доступности для растворителя референса, присвоенную PFM приматов, присвоенную PFM млекопитающих и присвоенную PFM позвоночных.
[00646] Система сконфигурирована для использования слоев пакетной нормализации, слоев нелинейности ReLU и меняющих размерность слоев для предварительной обработки вариантной аминокислотной последовательности, присвоенной референсной аминокислотной последовательности, присвоенной PFM приматов, присвоенной PFM млекопитающих и присвоенной PFM позвоночных. Система дополнительно сконфигурирована для суммирования предварительно обработанных характеристик и объединения сумм с присвоенных классификаций состояния вторичной структуры варианта, присвоенных классификаций состояния вторичной структуры референса, присвоенных классификаций доступности для растворителя варианта и присвоенных классификаций доступности для растворителя референса для генерации объединенного (конкатинированного) входа. Система обрабатывает объединенные входные данные через слой изменения размерности и принимает обработанные объединенные входные данные для инициации остаточных блоков глубокой сверточной нейронной сети.
[00647] Указанная глубокая сверточная нейронная сеть содержит группы остаточных блоков, организованных в последовательность от низшего к высшему. Указанная глубокая сверточная нейронная сеть параметризуется числом остаточных блоков, числом соединений с пропуском и числом остаточных связей в нелинейными активациями. Указанная глубокая сверточная нейронная сеть содержит меняющие размерность слои которые изменяют пространственные размерности и размерности признаков предшествующего входа.
[00648] Система дополнительно сконфигурирована для того чтобы классифицировать как патогенный однонуклеотидный вариант, который порождает вариантную целевую аминокислоту, отличную от референсной целевой аминокислоты, и который сохраняется в выровненных последовательностях аминокислот у приматов, млекопитающих и позвоночных.
[00649] Консервация (сохранение) представляет функциональную значимость референсной целевой аминокислоты и определяется по PFW. Система дополнительно сконфигурирована для того чтобы классифицировать как патогенный однонуклеотидный вариант, который приводит к появлению вторичных структур, различающихся между вариантной аминокислотной последовательностью и референсной аминокислотной последовательностью варианта.
[00650] Система дополнительно сконфигурирована для того чтобы классифицировать как патогенный однонуклеотидный вариант, который приводит к появлению различных состояний доступности для растворителя между вариантной аминокислотной последовательностью и референсной аминокислотной последовательностью.
[00651] PFM представляет консервацию аминокислот в последовательности белка человека в среди выровненных последовательностях белка других видов путем определения, на основе положения, частоты встречаемости аминокислоты в последовательности белка человека в выровненных последовательностях белка другого вида.
[00652] Три состояния вторичной структуры - это спираль, лист и скрученная спираль (coil). Первая подсеть вторичной структуры обучена принимать входную последовательность белка и PFM приматов, млекопитающих и позвоночных, выровненные с положениями аминокислот во входной последовательности белка, и предсказывать вторичную структуру с тремя состояниями в каждом из положений аминокислот. Три состояния доступности растворителя: открытое (доступное), скрытое (заглубленное) и промежуточное.
[00653] Вторая подсеть доступности растворителя обучена принимать входную последовательность белка и PFM приматов, млекопитающих и позвоночных, выровненные с положениями аминокислот во входной последовательности белка, и предсказывать доступность растворителя с тремя состояниями в каждом из положений аминокислот. Входная последовательность белка представляет собой референсную последовательность белка. Входная последовательность белка представляет собой альтернативную последовательность белка. Входная последовательность белка представляет собой референсную последовательность белка. Входная последовательность белка представляет собой альтернативную последовательность белка в последовательность от низшего к высшему. Первая подсеть вторичной структуры параметризуется числом остаточных блоков, числом соединений с пропуском и числом остаточных без нелинейных активаций.
[00654] Первая подсеть вторичной структуры содержит меняющие размерность слои которые изменяют пространственные размерности и размерности признаков предшествующего входа. Вторая подсеть доступности для растворителя содержит группы остаточных блоков, организованных в последовательность от низшего к высшему. Вторая подсеть доступности для растворителя параметризуется числом остаточных блоков, числом соединений с пропуском и числом остаточных связей без нелинейных активаций. Вторая подсеть доступности для растворителя содержит меняющие размерность слои которые изменяют пространственные размерности и размерности признаков предшествующего входа.
[00655] Каждый остаточный блок содержит по меньшей мере один слой пакетной нормализации, по меньшей мере один слой блоков линейной ректификации (сокращенно ReLU), по меньшей мере один меняющий размерность слой и по меньшей мере одну остаточную связь. Каждый остаточный блок содержит два слоя пакетной нормализации, два нелинейных слоя ReLU, два меняющих размерность слоя и одно остаточное соединение.
[00656] Глубокая сверточная нейронная сеть, первая подсеть вторичной структуры и вторая подсеть доступности для растворителя составляют каждый конечный слой классификации. Конечный слой классификации- это сигмоидальный слой. Конечный уровень классификации - это слой на основе softmax.
[00657] Система дополнительно сконфигурирована для удаления конечных уровней классификации первой подсети вторичной структуры и второй подсети доступности для растворителя для сотрудничества с глубокой сверточной нейронной сетью.
[00658] Система дополнительно сконфигурирована так, чтобы во время обучения глубокой сверточной нейронной сети дополнительно обучать первую подсеть вторичной структуры и вторую подсеть доступности для растворителя классификации патогенности, включая распространение ошибок в подсетях и обновление весов подсетей.
[00659] Вторая подсеть доступности для растворителя включает по меньшей мере один разреженный сверточный слой. Система дополнительно сконфигурирована для классификации вариантов, вызывающих расстройство с задержкой развития (сокращенно DDD), как патогенное. Вариант аминокислотной последовательности и референсная аминокислотная последовательность имеют общие фланкирующие аминокислоты. Система дополнительно сконфигурирована для использования «горячего» кодирования (с одним активным состоянием) для кодирования входных данных в глубокую сверточную нейронную сеть.
[00660] На ФИГ. 1Q показан пример вычислительной среды, в которой может быть реализована раскрытая технология. Глубокая сверточная нейронная сеть, первая подсеть вторичной структуры и вторая подсеть доступности для растворителя обучаются на одном или большем числе тренировочных серверов. Обученная глубокая сверточная нейронная сеть, обученная первая подсеть вторичной структуры и обученная вторая подсеть доступности для растворителя размещены на одном или более рабочих серверах, которые получают входные последовательности от запрашивающих клиентов. Рабочие серверы обрабатывают входные последовательности по меньшей мере одной глубокой сверточной нейронной сетью, первой подсетью вторичной структуры и второй подсетью доступности для растворителя с получением выходных данных, которые передаются указанным клиентам.
[00661] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению системой описанных выше действий. Еще один вариант реализации может включать выполнение описанных выше действий системы.
[00662] Другой вариант реализации системы согласно раскрытой технологии включает классификатор патогенности вариантов на основе глубокой сверточной нейронной сети, реализованный на множестве процессоров, связанных с памятью. Система включает генератор частотной матрицы положений (сокращенно PFM), работающий на по меньшей мере одном из указанного множества процессоров, применяемый к двум группам последовательностей приматов и млекопитающих для генерации PFM и PFM млекопитающих. Система включает входной процессор, который принимает вариантную аминокислотную последовательность с целевой вариантной аминокислотой, фланкированной в направлении 5’ и в направлении 3’ по меньшей мере 25 аминокислотами в каждом направлении, причем однонуклеотидный вариант дает вариантную аминокислоту. Система также включает распределитель (allocator) дополнительных данных, реализуемый на по меньшей мере одном из указанного множества процессоров, который ставит в соответствие референсной аминокислоту целевую референсную аминокислоту, фланкированную в направлении 5’ и в направлении 3’ по меньшей мере 25 аминокислотами в каждом направлении, выровненную с вариантной аминокислотой последовательностью. От также распределяет PFM приматов и млекопитающих, выровненные с референсными аминокислотными последовательностями. Система дополнительно включает глубокую сверточную нейронную сеть, работающую на указанном множестве процессоров, обученную классифицировать вариантные последовательности аминокислот как доброкачественные или патогенные на основании обработки вариантной аминокислотной последовательности, распределенную (присвоенную) аминокислотную последовательность и распределённые PFM. Наконец, система включает выходной процессор, который выдает по меньшей мере оценку патогенности для вариантной аминокислотной последовательности.
[00663] Этот вариант реализации системы и другие раскрытые системы необязательно включают один или более из следующих признаков. Система может также включать признаки, описанные в связи с раскрытыми способами. Для краткости альтернативные комбинации признаков системы не приводятся по отдельности. Признаки, связанные с системами, способами и изделиями, не повторяются для каждого заявленного набора классов основных признаков. Читатель поймет, как признаки, идентифицированные в этом разделе, можно легко объединять с основными признаками в других заявленных классах.
[00664] Система дополнительно сконфигурирована для того чтобы классифицировать однонуклеотидные варианты как доброкачественные или патогенные на основании оценки патогенности. Глубокая сверточная нейронная сеть параллельно принимает и обрабатывает вариантную аминокислотную последовательность, распределенную референсную аминокислотную последовательность, распределенную PFM приматов и распределенную PFM млекопитающих. Система дополнительно сконфигурирована для того чтобы классифицировать как патогенный однонуклеотидный вариант, который порождает вариантную целевую аминокислоту из референсной целевой аминокислоты, которая сохраняется в рефересных аминокислотных последовательностей у приматов и млекопитающих. Консервация (сохранение) представляет функциональную значимость референсной целевой аминокислоты и определяется по PFW.
[00665] Каждый признак, обсуждаемый в этом разделе, относящемся к конкретному варианту реализации, применительно к первому варианту реализации системы, равно применим к этому варианту реализации системы. Как указано выше, здесь не повторяются все признаки системы, подразумевается, что они повторяются посредством ссылки.
[00666] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению системой описанных выше действий. Еще один вариант реализации может включать выполнение описанных выше действий системы.
[00667] Первый вариант реализации способа согласно раскрытой технологии включает запуск первой подсети вторичной структуры на множестве процессоров, связанных с памятью, обученную предсказывать вторичную структуру с тремя состояниями для положений аминокислот в белковой последовательности. Запуск второй подсети доступности для растворителя на множестве процессоров, связанных с памятью, обученную предсказывать доступность для растворителя с тремя состояниями для положений аминокислот в белковой последовательности. Запуск на по меньшей мере одном из указанного множества процессоров генератора частотной матрицы положений (сокращенно PFM) применяемого трем группам последовательностей: приматов, млекопитающих, и позвоночных за исключением приматов и позвоночных, для генерации PFM приматов, PFM млекопитающих и PFM позвоночных. Прием вариантной аминокислотной последовательности входным процессором с вариантной целевой аминокислотой, фланкированной d в направлении 5’ и в направлении 3’ по меньшей мере 25 аминокислотами в каждом направлении. Однонуклеотидный вариант порождает вариантную целевую аминокислоту. Запуск на по меньшей мере одном из указанного множества процессоров, распределителя дополнительных данных, который сопоставляет (распределяет) референсную аминокислотную последовательность с целевой референсной аминокислотной последовательности, фланкированной в направлении 5’ и в направлении 3’ по меньшей мере 25 аминокислотами в каждом направлении, выровненной с вариантной аминокислотной последовательностью. Он также распределяет классификации состояний, выданные первой и второй подсетями для референсной аминокислотной последовательности. Он также распределяет классификации, выданные первой и второй подсетями для вариантной аминокислотной последовательностью. От распределяет PFM приматов, млекопитающих и позвоночных, выровненные с референсной аминокислотной последовательностью. Запуск на множестве процессоров, глубокой сверточной нейронной сети, обученной классифицировать вариантную аминокислотную последовательность как доброкачественную или патогенную на основании обработки вариантной аминокислотной последовательности, распределенной аминокислотной последовательности, распределенных классификаций состояний референса и варианта, и распределенных PFM. Вывод по меньшей мере оценки патогенности для вариантной аминокислотной последовательности выходным процессором.
[00668] Каждый из признаков, обсуждаемых в разделе, относящемся к этому конкретному варианту реализации, для первого варианта реализации системы, равно относится к этому варианту реализации способа. Как указано выше, здесь не повторяются все признаки системы, подразумевается, что они повторяются посредством ссылки.
[00669] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению описанного выше способа. Еще один вариант реализации может включать систему, включающую память и один или более процессоров, выполненных с возможностью исполнять инструкции, хранящиеся в памяти, с реализацией описанного выше способа.
[00670] Второй вариант реализации способа согласно раскрытой технологии включает реализованный на множестве процессоров, связанных с памятью, классификатор патогенности вариантов на основе глубокой сверточной нейронной сети. Запуск генератора частотных матриц положений (сокращенно PFM) на по меньшей мере одном из указанного множества процессоров, применяемого к двум группам последовательностей: приматов и млекопитающих для генерации PFM приматов и PFM млекопитающий. Прием во входной процессор вариантной аминокислотой последовательности с вариантной целевой аминокислотной последовательностью, фланкированной в направлении 5’ и в направлении 3’ по меньшей мере 25 аминокислотами в каждом направлении. Однонуклеотидный вариант порождает вариантную целевую аминокислоту. Запуск на по меньшей мере одном из указанного множества процессоров, распределителя дополнительных данных, который сопоставляет (распределяет) референсную аминокислотную последовательность с целевой референсной аминокислотной последовательности, фланкированной в направлении 5’ и в направлении 3’ по меньшей мере 25 аминокислотами в каждом направлении, выровненной с вариантной аминокислотной последовательность, и PFM приматов и млекопитающих, выровненные с референсной аминокислотной последовательностью. Запуск на множестве процессоров, глубокой сверточной нейронной сети, обученной классифицировать вариантную аминокислотную последовательность как доброкачественную или патогенную на основании обработки вариантной аминокислотной последовательности, распределенной референсной аминокислотной последовательности и распределенных PFM. Вывод по меньшей мере оценки патогенности для вариантной аминокислотной последовательности выходным процессором.
[00671] Каждый из признаков, обсуждаемых в этом разделе этого конкретного варианта реализации для второго варианта реализации системы равно относится к этому варианту реализации способа. Как указано выше, здесь не повторяются все признаки системы, подразумевается, что они повторяются посредством ссылки.
[00672] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению описанного выше способа. Еще один вариант реализации может включать систему, включающую память и один или более процессоров, выполненных с возможностью исполнять инструкции, хранящиеся в памяти, с реализацией описанного выше способа.
[00673] Еще один вариант реализации системы согласно раскрытой технологии включает систему, которая генерирует крупномасштабные обучающие данные для обучения классификатора однонуклеотидных полиморфизмов (сокращенно SNP).
[00674] Как показано на ФИГ. 19 система обучает классификатор патогенности SNP, который реализуется на множестве процессоров, связанных с памятью, с использованием обучающего набора доброкачественных SNP и обучающего набора элитных предсказанных патогенных SNP, которые отбираются из синтетического набора комбинаторно-сгенерированных SNP. В контексте этой заявки элитные (отобранные) предсказанные патогенные SNP - это те SNP, которые производятся / отбираются в конце каждого цикла на основе их средней или максимальной оценки патогенности, выводимой ансамблем. Термин «элита» заимствован из словаря генетических алгоритмов, и предполагается, что он имеет значение, обычно придаваемое ему в публикациях, описывающих генетические алгоритмы.
[00675] Как показано на ФИГ. 37, 38, 39, 40, 41 и 42, система строит элитный набор итеративно в циклах, начиная с отсутствия предсказанных SNP, накапливая полный набор предсказанных SNP путем отбраковки выбивающихся SNP из синтетического набора. Синтетический набор включает псевдопатогенные SNP, которые являются комбинаторно сгенерированными SNP, не присутствующими в доброкачественном наборе, и их количество в наборе снижается, поскольку выбивающиеся SNP итеративно отбираются из синтетического набора для включения в элитный набор. В контексте этой заявки термин «отбраковка» означает фильтрацию, замену, обновление или выбор предыдущей совокупности новой совокупностью. Термин «отбраковка» заимствован из словаря генетических алгоритмов, и предполагается, что он имеет значение, обычно придаваемое ему в публикациях, описывающих генетические алгоритмы.
[00676] Как показано на ФИГ. 37, 38, 39, 40, 41 и 42 система обучает и применяет ансамбль классификаторов патогенности SNP, чтобы итеративно циклически отбирать выбивающиеся SNP из синтетического набора. Это включает обучение ансамбля с использованием общего обучающего набора доброкачественных SNP, общего обучающего набора элитных предсказанных патогенных SNP и отдельных обучающих наборов псевдопатогенных SNP, выбранных из синтетического набора без замены. Это также включает применение обученного ансамбля для отбраковки выбивающихся SNP из синтетического набора и накопления отбракованных выбивающихся SNP в общем элитном наборе путем применения обученного ансамбля для оценки по меньшей мере некоторых SNP из синтетического набора, которые не использовались для обучения ансамбля в текущем цикле и с использованием оценок для выбора из оцененных SNP, SNP, выбивающихся в текущем цикле, для накопления в общем элитном наборе.
[00677] В контексте данной заявки «псевдопатогенные SNP» - это те SNP, которые помечены как патогенные для целей обучения и взяты из синтетически созданных вариантов без замены во время обучения.
[00678] Кроме того, обучающий набор элитных предсказанных патогенных SNP строится итеративно в течение нескольких циклов.
[00679] Как показано на ФИГ. 37, 38, 39, 40, 41 и 42, система затем сохраняет в памяти параметры классификатора, полученные в результате обучения, общий элитный набор, образованный в течение циклов и в пределах заранее определенного диапазона общего доброкачественного набора, и общий доброкачественный набор для обучения классификатора патогенности SNP.
[00680] Как показано на ФИГ. 37, 38, 39, 40, 41 и 42, элитные предсказанные патогенные SNP, составляют верхние 5% SNP, предсказанных ансамблем. В некоторых вариантах реализации они представляют собой фиксированное количество SNP с наивысшей оценкой, например 20000.
[00681] Классификатор патогенности SNP и ансамбль классификаторов патогенности SNP представляют собой глубокие сверточные нейронные сети (сокращенно DCNN). Ансамбль включает от 4 до 16 DCNN. Как показано на ФИГ. 37, 38, 39, 40, 41 и 42 ансамбль включает 8 DCNN.
[00682] Как показано на ФИГ. 37, 38, 39, 40, 41 и 42 система обучает ансамбль DCCN в эпохах в течение циклов, завершая обучение для определенного цикла, когда предсказания на проверочной выборке формируют дискретные кластеры распределения вероятностей доброкачественных и патогенных предсказаний.
[00683] Как показано на ФИГ. 37, 38, 39, 40, 41 и 42 система использует оценки для выбора выбивающихся SNP из текущего цикла путем суммирования оценок из ансамбля DCCN.
[00684] Как показано на ФИГ. 37, 38, 39, 40, 41 и 42, система использует оценки для выбора выбивающихся SNP из текущего цикла, принимая максимальное среднее значение для каждого из SNP, оцененных ансамблем DCNN.
[00685] Как показано на ФИГ. 37, 38, 39, 40, 41 и 42, выборка без замены во время текущего цикла приводит к несвязанным отдельным обучающим наборам псевдопатогенных SNP в текущем цикле.
[00686] Система продолжает циклы до тех пор, пока не будет достигнуто условие завершения. Условием завершения может быть заданное количество циклов. Как показано на ФИГ. 37, 38, 39, 40, 41 и 42 заданное количество циклов равно 21.
[00687] Как показано на ФИГ. 37, 38, 39, 40, 41 и 42, условие завершения - это когда размер предсказанного элитного патогенного набора находится в пределах заранее определенного диапазона размера доброкачественного набора.
[00688] Параметры классификатора могут быть по меньшей мере весовыми коэффициентами сверточного фильтра и показателем обучения.
[00689] Система может выбрать один из классификаторов патогенности SNP в ансамбле в качестве классификатора патогенности SNP. Выбранный классификатор патогенности SNP может быть тем классификатором, который превосходит другие классификаторы патогенности SNP в ансамбле на проверочной выборке, оцениваемой в конечном цикле.
[00690] Как показано на ФИГ. 37, 38, 39, 40, 41 и 42, общий элитный набор, завершенный в течение циклов, может иметь по меньшей мере 400000 предсказанных элитных патогенных SNP.
[00691] Как показано на ФИГ. 37, 38, 39, 40, 41 и 42, система в каждом цикле может сопоставлять тринуклеотидный контекст между доброкачественными SNP и выбранными псевдопатогенными SNP для предотвращения смещения частоты мутаций в предсказанных элитных патогенных SNP.
[00692] Как показано на ФИГ. 37, 38, 39, 40, 41 и 42, выборка псевдопатогенных SNP из синтетического набора может уменьшаться на 5% в каждом последующем цикле.
[00693] Как показано на ФИГ. 37, 38, 39, 40, 41 и 42, система может фильтровать синтетические SNP, набранные в текущем цикле, по псевдопатогенным SNP, выбранным в текущем цикле для обучения, элитные предсказанные патогенные SNP и доброкачественные SNP, используемым в текущем цикле обучения.
[00694] Каждый признак, обсуждаемый в этом разделе, относящемся к конкретному варианту реализации, применительно к первому варианту реализации системы, равно применим к этому варианту реализации системы. Как указано выше, здесь не повторяются все признаки системы, подразумевается, что они повторяются посредством ссылки.
[00695] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению системой описанных выше действий. Еще один вариант реализации может включать систему, включающую память и один или более процессоров, которые могут исполнять инструкции, хранящиеся в памяти, по выполнению системой описанных выше действий.
[00696] Еще один вариант реализации раскрытой технологии включает полуконтролируемого ученика (обучаемый блок) на основе сверточной нейронной сети (сокращенно CNN), как показано на ФИГ. 36.
[00697] Как показано на ФИГ. 36, полуконтролируемый обучаемый блок может включать ансамбль CNN-сетей, реализованный на множестве процессоров, связанных с памятью, который итерационно обучается на доброкачественном обучающем наборе и патогенном обучающем наборе.
[00698] Как показано на ФИГ. 36, полуконтррлируемый обучаемый блок может включать блок дополнения набора, реализуемый на по меньшей мере одном из процессоров, который прогрессивно увеличивает размер набора патогенного набора на основе оценок синтетического набора обученным ансамблем;
[00699] На каждой итерации оценка выдает избранный предсказанный набор, который блок дополнения набора добавляет к патогенному обучающему набору.
[00700] Полуконтролируемый обучаемый блок может включать средство сборки (builder), которое принимает по меньшей мере одну из CNN, дополненный патогенный обучающий набор и доброкачественный обучающий набор и конструирует и обучает классификатор однонуклеотидных полиморфизмов (сокращенно SNP).
[00701] Каждый признак, обсуждаемый в этом разделе, относящемся к конкретному варианту реализации, применительно к первому варианту реализации системы, равно применим к этому варианту реализации системы. Как указано выше, здесь не повторяются все признаки системы, подразумевается, что они повторяются посредством ссылки.
[00702] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению системой описанных выше действий. Еще один вариант реализации может включать систему, включающую память и один или более процессоров, которые могут исполнять инструкции, хранящиеся в памяти, по выполнению системой описанных выше действий.
[00703] Предшествующее описание приведено для того, чтобы сделать возможными реализацию и применение раскрытой технологии. Различные модификации раскрытых вариантов реализации будут очевидны, и общие принципы, определенные в данном документе, могут быть применены к другим вариантами реализации и приложениям без отступления от сущности и объема раскрытой технологии. Таким образом, не предполагается, что раскрытая технология ограничена показанными вариантами, но соответствует самому широкому объему, согласующемуся с принципами и признаками, раскрытыми в данном документе. Объем раскрытой технологии определяется прилагаемой формулой изобретения.
Компьютерная система
[00704] ФИГ. 66 представляет собой упрощенную блок-схему компьютерной системы, которую можно применять для реализации раскрытой технологии. Компьютерная система обычно включает в себя по меньшей мере один процессор, который связывается с рядом периферийных устройств через подсистему шин. Эти периферийные устройства могут включать в себя подсистему хранения, включая, например, запоминающее устройство и подсистему хранения файлов, устройства ввода пользовательского интерфейса, устройства вывода пользовательского интерфейса и подсистему сетевого интерфейса. Устройства ввода и вывода позволяют пользователю взаимодействовать с компьютерной системой. Подсистема сетевого интерфейса обеспечивает интерфейс с внешними сетями, включая интерфейс с соответствующими интерфейсными устройствами в других компьютерных системах.
[00705] В одном варианте реализации нейронная сеть, такая как генератор доброкачественного набора данных, классификатор патогенности вариантов, классификатор доступности для растворителя и полуконтролируемый обучаемый блок, связаны с возможностью обмена информацией с подсистемой хранения и устройствами ввода пользовательского интерфейса.
[00706] Устройства ввода пользовательского интерфейса могут включать клавиатуру; указывающие устройства, такие как мышь, трекбол, тачпад или графический планшет; сканер; сенсорный экран (тач-скрин), встроенный в дисплей; устройства звукового ввода, такие как системы распознавания голоса и микрофоны; и другие типы устройств ввода. В общем, использование термина «устройство ввода» предназначено для включения всех возможных типов устройств и способов ввода информации в компьютерную систему.
[00707] Устройства вывода пользовательского интерфейса могут включать подсистему дисплея, принтер, факсимильный аппарат или невизуальные дисплеи, такие как устройства звукового вывода. Подсистема дисплея может включать в себя электронно-лучевую трубку (ЭЛТ), устройство с плоской панелью, такое как жидкокристаллический дисплей (ЖКД), проекционное устройство или какой-либо другой механизм для создания видимого изображения. Подсистема дисплея может также обеспечивать невизуальный дисплей, такой как устройства звукового вывода. В целом, использование термина «устройство вывода» предназначено для включения всех возможных типов устройств и способов вывода информации из компьютерной системы пользователю, другой машине или компьютерной системе.
[00708] Подсистема хранения хранит программы и конструкции данных, которые обеспечивают функциональные возможности некоторых или всех модулей и методов, описанных в данном документе. Эти программные модули обычно исполняются одним процессором или в сочетании с другими процессорами.
[00709] Память, используемая в подсистеме хранения, может включать ряд запоминающих устройств, включая основную память с произвольным доступом (RAM) для хранения инструкций и данных во время выполнения программы, и постоянную память (ROM), в которой хранятся фиксированные инструкции. Подсистема хранения файлов может обеспечивать постоянное хранилище для файлов программ и данных и может включать жесткий диск, дисковод гибких дисков вместе со связанным съемным носителем, дисковод компакт-дисков, оптический дисковод или съемные картриджи. Модули, реализующие функциональные возможности определенных реализаций, могут храниться в подсистеме хранения файлов в подсистеме хранения или на других машинах, доступных процессору.
[007710] Подсистема шины обеспечивает механизм, позволяющий различным компонентам и подсистемам компьютерной системы определенным образом связываться друг с другом. Хотя подсистема шины схематично показана как одна шина, альтернативные реализации подсистемы шины могут использовать множество шин.
[00711] Сама компьютерная система может быть различных типов, включая персональный компьютер, портативный компьютер, рабочую станцию, компьютерный терминал, сетевой компьютер, телевизор, мэйнфрейм, серверную ферму, распределенный набор слабо связанных в сеть компьютеров или любую другую систему обработки данных или пользовательское устройство. В связи с изменчивой природой компьютеров и сетей описание компьютерной системы, изображенной на ФИГ. 66 , приведено только в качестве конкретного примера с целью иллюстрации раскрытой технологии. Возможны многие другие конфигурации компьютерной системы, имеющие больше или меньше компонентов, чем компьютерная система, изображенная на ФИГ. 66.
[00712] Процессоры глубокого обучения могут быть графическими процессорами или FPGA и могут размещаться на облачных платформах глубокого обучения, таких как Google Cloud Platform, Xilinx и Cirrascale. Примеры процессоров глубокого обучения включают Tensor Processing Unit (TPU) от Google, стоечные решения, такие как серия GX4 Rackmount, серия GX8 Rackmount, NVIDIA DGX-1, Microsoft Stratix V FPGA, интеллектуальный процессор Graphcore (IPU), платформа Qualcomm Zeroth с процессорами Snapdragon , NVIDIA Volta, NVIDIA DRIVE PX, NVIDIA JETSON TX1 / TX2 MODULE, Nirvana от Intel, Movidius VPU, Fujitsu DPI, DynamicIQ от ARM, IBM TrueNorth и другие.
ПРИЛОЖЕНИЕ
Ниже приведена библиография потенциально релевантных ссылок, перечисленных в статье, написанной авторами настоящего изобретения. Объект этой статьи покрывают предварительные заявки США, на основании которых испрашивается приоритет по этой заявке. Доступ к этим заявкам можно получить по запросу у юрисконсульта или через систему Global Dossier. Эта статья приведена в списке источников первой.
1. Laksshman Sundaram, Hong Gao, Samskruthi Reddy Padigepati, Jeremy F. McRae, Yanjun Li, Jack A. Kosmicki, Nondas Fritzilas, Jörg Hakenberg, Anindita Dutta, John Shon, Jinbo Xu, Serafim Batzloglou, Xiaolin Li & Kyle Kai-How Farh. Predicting the clinical impact of human mutation with deep neural networks. Nature Genetics volume 50, pages1161-1170 (2018). Accessible at https://www.nature.com/articles/s41588-018-0167-z.
2. MacArthur, D. G. et al. Guidelines for investigating causality of sequence variants in human disease. Nature 508, 469-476, doi:10.1038/nature13127 (2014).
3. Rehm, H. L., J. S. Berg, L. D. Brooks, C. D. Bustamante, J. P. Evans, M. J. Landrum, D. H. Ledbetter, D. R. Maglott, C. L. Martin, R. L. Nussbaum, S. E. Plon, E. M. Ramos, S. T. Sherry, M. S. Watson. ClinGen--the Clinical Genome Resource. N. Engl. J. Med. 372, 2235-2242 (2015).
4. Bamshad, M. J., S. B. Ng, A. W. Bigham, H. K. Tabor, M. J. Emond, D. A. Nickerson, J. Shendure. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 12, 745-755 (2011).
5. Rehm, H. L. Evolving health care through personal genomics. Nature Reviews Genetics 18, 259-267 (2017).
6. Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405-424, doi:10.1038/gim.2015.30 (2015).
7. Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285-291, doi:10.1038/nature19057 (2016).
8. Mallick, S. et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538, 201-206, doi:10.1038/nature18964 (2016).
9. Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68-74, doi:10.1038/nature15393 (2015).
10. Liu, X., X. Jian, E. Boerwinkle. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Human Mutation 32, 894-899 (2011).
11. Chimpanzee Sequencing Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69-87, doi:10.1038/nature04072 (2005).
12. Takahata, N. Allelic genealogy and human evolution. Mol Biol Evol 10, 2-22 (1993).
13. Asthana, S., Schmidt, S. & Sunyaev, S. A limited role for balancing selection. Trends Genet 21, 30-32, doi:10.1016/j.tig.2004.11.001 (2005).
14. Leffler, E. M., Z. Gao, S. Pfeifer, L. Ségurel, A. Auton, O. Venn, R. Bowden, R. Bontrop, J.D. Wall, G. Sella, P. Donnelly. Multiple instances of ancient balancing selection shared between humans and chimpanzees. Science 339, 1578-1582 (2013).
15. Samocha, K. E. et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet 46, 944-950, doi:10.1038/ng.3050 (2014).
16. Ohta, T. Slightly deleterious mutant substitutions in evolution. Nature 246, 96-98 (1973).
17. Reich, D. E. & Lander, E. S. On the allelic spectrum of human disease. Trends Genet 17, 502-510 (2001).
18. Whiffin, N., E. Minikel, R. Walsh, A. H. O’Donnell-Luria, K. Karczewski, A. Y. Ing, P. J. Barton, B. Funke, S. A. Cook, D. MacArthur, J. S. Ware. Using high-resolution variant frequencies to empower clinical genome interpretation. Genetics in Medicine 19, 1151-1158 (2017).
19. Prado-Martinez, J. et al. Great ape genome diversity and population history. Nature 499, 471-475 (2013).
20. Klein, J., Satta, Y., O'HUigin, C. & Takahata, N. The molecular descent of the major histocompatibility complex. Annu Rev Immunol 11, 269-295, doi:10.1146/annurev.iy.11.040193.001413 (1993).
21. Kimura, M. The neutral theory of molecular evolution. (Cambridge University Press, 1983).
22. de Manuel, M. et al. Chimpanzee genomic diversity reveals ancient admixture with bonobos. Science 354, 477-481, doi:10.1126/science.aag2602 (2016).
23. Locke, D. P. et al. Comparative and demographic analysis of orang-utan genomes. Nature 469, 529-533 (2011).
24. Rhesus Macaque Genome Sequencing Analysis Consortium et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science 316, 222-234, doi:10.1126/science.1139247 (2007).
25. Worley, K. C., W. C. Warren, J. Rogers, D. Locke, D. M. Muzny, E. R. Mardis, G. M. Weinstock, S. D. Tardif, K. M. Aagaard, N. Archidiacono, N. A. Rayan. The common marmoset genome provides insight into primate biology and evolution. Nature Genetics 46, 850-857 (2014).
26. Sherry, S. T. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29, 308-311 (2001).
27. Schrago, C. G. & Russo, C. A. Timing the origin of New World monkeys. Mol Biol Evol 20, 1620-1625, doi:10.1093/molbev/msg172 (2003).
28. Landrum, M. J. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44, D862-868, doi:10.1093/nar/gkv1222 (2016).
29. Brandon, E. P., Idzerda, R. L. & McKnight, G. S. Targeting the mouse genome: a compendium of knockouts (Part II). Curr Biol 5, 758-765 (1995).
30. Lieschke, J. G., P. D. Currie. Animal models of human disease: zebrafish swim into view. Nature Reviews Genetics 8, 353-367 (2007).
31. Sittig, L. J., P. Carbonetto, K. A. Engel, K. S. Krauss, C. M. Barrios-Camacho, A. A. Palmer. Genetic background limits generalizability of genotype-phenotype relationships. Neuron 91, 1253-1259 (2016).
32. Bazykin, G. A. et al. Extensive parallelism in protein evolution. Biol Direct 2, 20, doi:10.1186/1745-6150-2-20 (2007).
33. Ng, P. C. & Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res 11, 863-874, doi:10.1101/gr.176601 (2001).
34. Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248-249, doi:10.1038/nmeth0410-248 (2010).
35. Chun, S., J. C. Fay. Identification of deleterious mutations within three human genomes. Genome research 19, 1553-1561 (2009).
36. Schwarz, J. M., C. Rödelsperger, M. Schuelke, D. Seelow. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575-576 (2010).
37. Reva, B., Antipin, Y. & Sander, C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 39, e118, doi:10.1093/nar/gkr407 (2011).
38. Dong, C. et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 24, 2125-2137, doi:10.1093/hmg/ddu733 (2015).
39. Carter, H., Douville, C., Stenson, P. D., Cooper, D. N. & Karchin, R. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics 14 Suppl 3, S3, doi:10.1186/1471-2164-14-S3-S3 (2013).
40. Choi, Y., Sims, G. E., Murphy, S., Miller, J. R. & Chan, A. P. Predicting the functional effect of amino acid substitutions and indels. PLoS One 7, e46688, doi:10.1371/journal.pone.0046688 (2012).
41. Gulko, B., Hubisz, M. J., Gronau, I. & Siepel, A. A method for calculating probabilities of fitness consequences for point mutations across the human genome. Nat Genet 47, 276-283, doi:10.1038/ng.3196 (2015).
42. Shihab, H. A. et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 31, 1536-1543, doi:10.1093/bioinformatics/btv009 (2015).
43. Quang, D., Chen, Y. & Xie, X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 31, 761-763, doi:10.1093/bioinformatics/btu703 (2015).
44. Bell, C. J., D. L. Dinwiddie, N. A. Miller, S. L. Hateley, E. E. Ganusova, J. Midge, R. J. Langley, L. Zhang, C. L. Lee, R. D. Schilkey, J. E. Woodward, H. E. Peckham, G. P. Schroth, R. W. Kim, S. F. Kingsmore. Comprehensive carrier testing for severe childhood recessive diseases by next generation sequencing. Sci. Transl. Med. 3, 65ra64 (2011).
45. Kircher, M., D. M. Witten, P. Jain, B. J. O’Roak, G. M. Cooper, J. Shendure. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310-315 (2014).
46. Smedley, D. et al. A Whole-Genome Analysis Framework for Effective Identification of Pathogenic Regulatory Variants in Mendelian Disease. Am J Hum Genet 99, 595-606, doi:10.1016/j.ajhg.2016.07.005 (2016).
47. Ioannidis, N. M. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 99, 877-885, doi:10.1016/j.ajhg.2016.08.016 (2016).
48. Jagadeesh, K. A., A. M. Wenger, M. J. Berger, H. Guturu, P. D. Stenson, D. N. Cooper, J. A. Bernstein, G. Bejerano. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nature genetics 48, 1581-1586 (2016).
49. Grimm, D. G. The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Human mutation 36, 513-523 (2015).
50. He, K., X. Zhang, S. Ren, J. Sun. in Proceedings of the IEEE conference on computer vision and pattern recognition. 770-778.
51. Heffernan, R. et al. Improving prediction of secondary structure, local backbone angles, and solvent accessible surface area of proteins by iterative deep learning. Sci Rep 5, 11476, doi:10.1038/srep11476 (2015).
52. Wang, S., J. Peng, J. Ma, J. Xu. Protein secondary structure prediction using deep convolutional neural fields. Scientific reports 6, 18962-18962 (2016).
53. Harpak, A., A. Bhaskar, J. K. Pritchard. Mutation Rate Variation is a Primary Determinant of the Distribution of Allele Frequencies in Humans. PLoS Genetics 12 (2016).
54. Payandeh, J., Scheuer, T., Zheng, N. & Catterall, W. A. The crystal structure of a voltage-gated sodium channel. Nature 475, 353-358 (2011).
55. Shen, H. et al. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 355, eaal4326, doi:10.1126/science.aal4326 (2017).
56. Nakamura, K. et al. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology 81, 992-998, doi:10.1212/WNL.0b013e3182a43e57 (2013).
57. Henikoff, S. & Henikoff, J. G. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A 89, 10915-10919 (1992).
58. Li, W. H., C. I. Wu, C. C. Luo. Nonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications. Journal of Molecular Evolution 21, 58-71 (1984).
59. Grantham, R. Amino acid difference formula to help explain protein evolution. Science 185, 862-864 (1974).
60. LeCun, Y., L. Bottou, Y. Bengio, P. Haffner. in Proceedings of the IEEE 2278-2324.
61. Vissers, L. E., Gilissen, C. & Veltman, J. A. Genetic studies in intellectual disability and related disorders. Nat Rev Genet 17, 9-18, doi:10.1038/nrg3999 (2016).
62. Neale, B. M. et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242-245, doi:10.1038/nature11011 (2012).
63. Sanders, S. J. et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237-241, doi:10.1038/nature10945 (2012).
64. De Rubeis, S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209-215, doi:10.1038/nature13772 (2014).
65. Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature 519, 223-228, doi:10.1038/nature14135 (2015).
66. Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433-438, doi:10.1038/nature21062 (2017).
67. Iossifov, I. et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216-221, doi:10.1038/nature13908 (2014).
68. Zhu, X., Need, A. C., Petrovski, S. & Goldstein, D. B. One gene, many neuropsychiatric disorders: lessons from Mendelian diseases. Nat Neurosci 17, 773-781, doi:10.1038/nn.3713 (2014).
69. Leffler, E. M., K. Bullaughey, D. R. Matute, W. K. Meyer, L. Ségurel, A. Venkat, P. Andolfatto, M. Przeworski. Revisiting an old riddle: what determines genetic diversity levels within species? PLoS biology 10, e1001388 (2012).
70. Estrada, A. et al. Impending extinction crisis of the world’s primates: Why primates matter. Science advances 3, e1600946 (2017).
71. Kent, W. J., C. W. Sugnet, T. S. Furey, K. M. Roskin, T. H. Pringle, A.M. Zahler, D. Haussler. The human genome browser at UCSC. Genome Res. 12, 996-1006 (2002).
72. Tyner, C. et al. The UCSC Genome Browser database: 2017 update. Nucleic Acids Res 45, D626-D634, doi:10.1093/nar/gkw1134 (2017).
73. Kabsch, W. & Sander, C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577-2637, doi:10.1002/bip.360221211 (1983).
74. Joosten, R. P. et al. A series of PDB related databases for everyday needs. Nucleic Acids Res 39, D411-419, doi:10.1093/nar/gkq1105 (2011).
75. He, K., Zhang, X., Ren, S. & Sun, J. in European Conference on Computer Vision. 630-645 (Springer).
76. Ionita-Laza, I., McCallum, K., Xu, B. & Buxbaum, J. D. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet 48, 214-220, doi:10.1038/ng.3477 (2016).
77. Li, B. et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 25, 2744-2750, doi:10.1093/bioinformatics/btp528 (2009).
78. Lu, Q. et al. A statistical framework to predict functional non-coding regions in the human genome through integrated analysis of annotation data. Sci Rep 5, 10576, doi:10.1038/srep10576 (2015).
79. Shihab, H. A. et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat 34, 57-65, doi:10.1002/humu.22225 (2013).
80. Davydov, E. V. et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 6, e1001025, doi:10.1371/journal.pcbi.1001025 (2010).
81. Liu, X., Wu, C., Li, C. & Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum Mutat 37, 235-241, doi:10.1002/humu.22932 (2016).
82. Jain, S., White, M. & Radivojac, P. in Proceedings of the Thirty-First AAAI Conference on Artificial Intelligence. 2066-2072.
83. de Ligt, J. et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 367, 1921-1929, doi:10.1056/NEJMoa1206524 (2012).
84. Iossifov, I. et al. De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285-299, doi:10.1016/j.neuron.2012.04.009 (2012).
85. O'Roak, B. J. et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246-250, doi:10.1038/nature10989 (2012).
86. Rauch, A. et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380, 1674-1682, doi:10.1016/S0140-6736(12)61480-9 (2012).
87. Epi, K. C. et al. De novo mutations in epileptic encephalopathies. Nature 501, 217-221, doi:10.1038/nature12439 (2013).
88. Euro, E.-R. E. S. C., Epilepsy Phenome/Genome, P. & Epi, K. C. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet 95, 360-370, doi:10.1016/j.ajhg.2014.08.013 (2014).
89. Gilissen, C. et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 511, 344-347, doi:10.1038/nature13394 (2014).
Lelieveld, S. H. et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci 19, 1194-1196, doi:10.1038/nn.4352 (2016).
90. Famiglietti, M. L. et al. Genetic variations and diseases in UniProtKB/Swiss-Prot: the ins and outs of expert manual curation. Hum Mutat 35, 927-935, doi:10.1002/humu.22594 (2014).
91. Horaitis, O., Talbot, C. C., Jr., Phommarinh, M., Phillips, K. M. & Cotton, R. G. A database of locus-specific databases. Nat Genet 39, 425, doi:10.1038/ng0407-425 (2007).
93. Stenson, P. D. et al. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 133, 1-9, doi:10.1007/s00439-013-1358-4 (2014).
| название | год | авторы | номер документа |
|---|---|---|---|
| КЛАССИФИКАЦИЯ САЙТОВ СПЛАЙСИНГА НА ОСНОВЕ ГЛУБОКОГО ОБУЧЕНИЯ | 2018 |
|
RU2780442C2 |
| ФРЕЙМВОРК НА ОСНОВЕ ГЛУБОКОГО ОБУЧЕНИЯ ДЛЯ ИДЕНТИФИКАЦИИ ПАТТЕРНОВ ПОСЛЕДОВАТЕЛЬНОСТИ, КОТОРЫЕ ВЫЗЫВАЮТ ПОСЛЕДОВАТЕЛЬНОСТЬ-СПЕЦИФИЧНЫЕ ОШИБКИ (SSE) | 2019 |
|
RU2745733C1 |
| GAN-CNN ДЛЯ ПРОГНОЗИРОВАНИЯ СВЯЗЫВАНИЯ МНС-ПЕПТИД | 2019 |
|
RU2777926C2 |
| GAN-CNN ДЛЯ ПРОГНОЗИРОВАНИЯ СВЯЗЫВАНИЯ MHC-ПЕПТИД | 2019 |
|
RU2836823C2 |
| МЕТОД ДЛЯ ВЫДЕЛЕНИЯ И КЛАССИФИКАЦИИ ТИПОВ КЛЕТОК КРОВИ С ПОМОЩЬЮ ГЛУБОКИХ СВЕРТОЧНЫХ НЕЙРОННЫХ СЕТЕЙ | 2019 |
|
RU2732895C1 |
| СПОСОБ ОБУЧЕНИЯ ГЛУБОКИХ НЕЙРОННЫХ СЕТЕЙ НА ОСНОВЕ РАСПРЕДЕЛЕНИЙ ПОПАРНЫХ МЕР СХОЖЕСТИ | 2016 |
|
RU2641447C1 |
| СПОСОБ И СИСТЕМЫ ДЛЯ ПРОГНОЗИРОВАНИЯ СПЕЦИФИЧЕСКИХ ДЛЯ HLA КЛАССА II ЭПИТОПОВ И ОХАРАКТЕРИЗАЦИИ CD4+ T-КЛЕТОК | 2019 |
|
RU2826261C2 |
| Компьютерная система и способ для обнаружения вредоносных программ с использованием машинного обучения | 2021 |
|
RU2802860C1 |
| СИСТЕМА И СПОСОБ ОБРАБОТКИ ИЗОБРАЖЕНИЙ С ИСПОЛЬЗОВАНИЕМ ГЛУБИННЫХ НЕЙРОННЫХ СЕТЕЙ | 2018 |
|
RU2743931C1 |
| ОБРАБОТКА ОККЛЮЗИЙ ДЛЯ FRC C ПОМОЩЬЮ ГЛУБОКОГО ОБУЧЕНИЯ | 2020 |
|
RU2747965C1 |
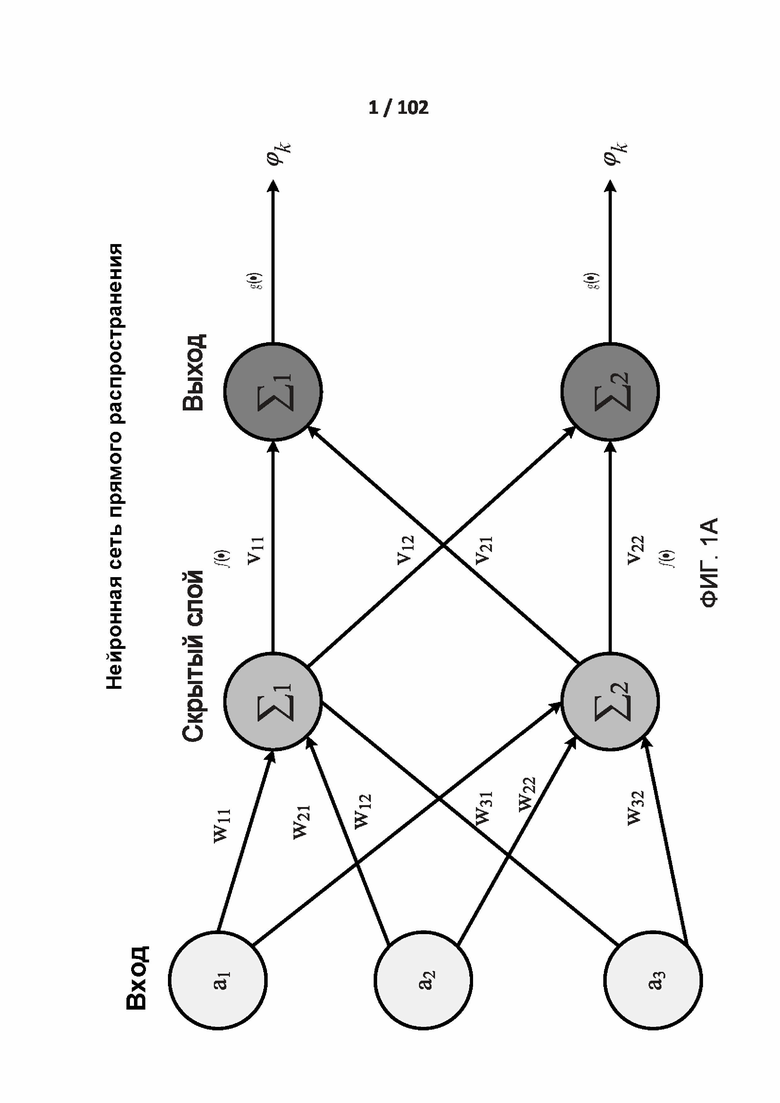

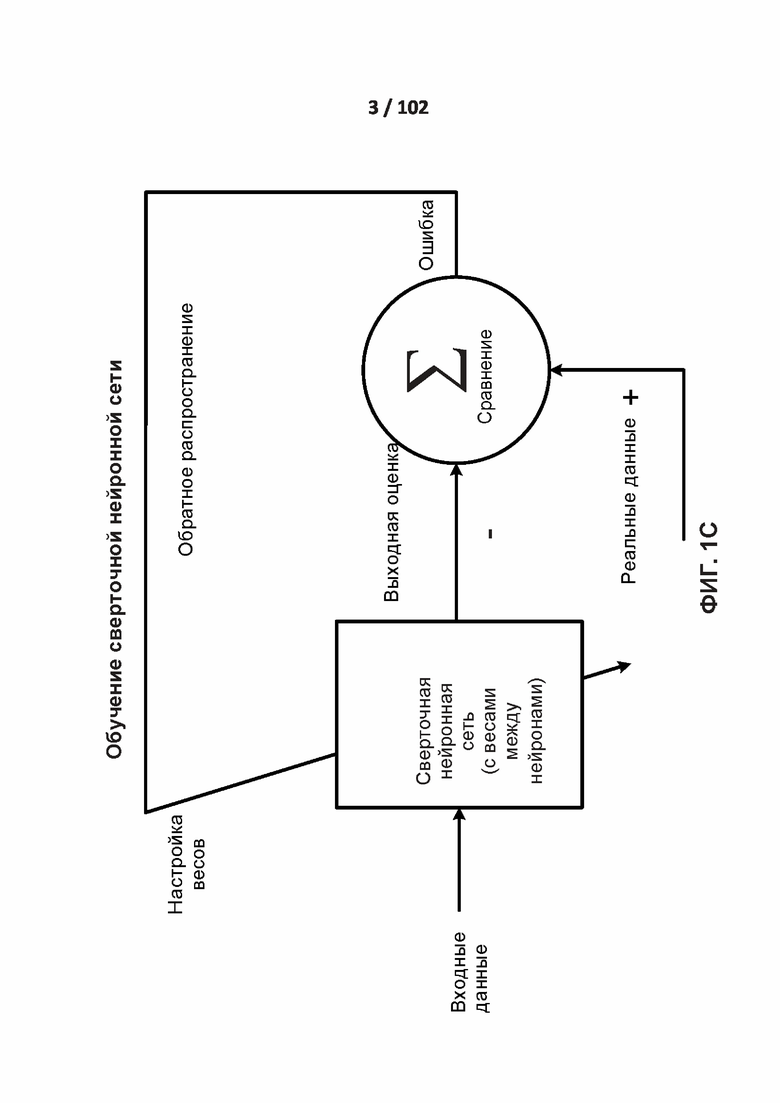
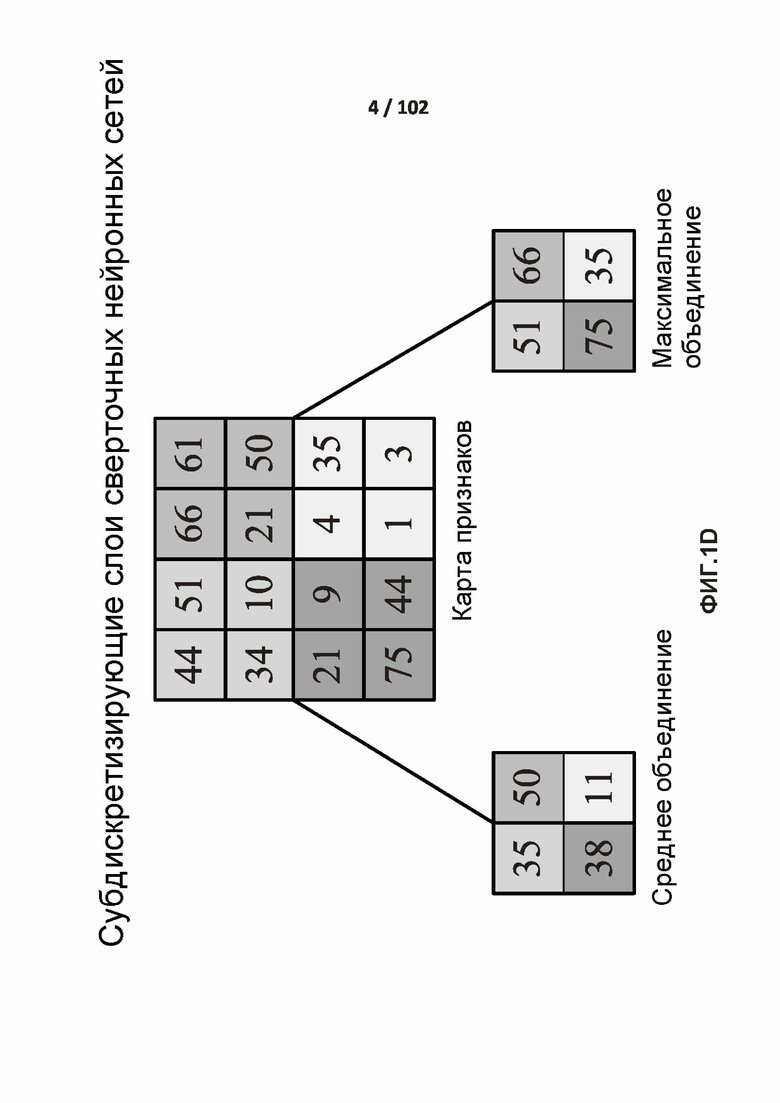

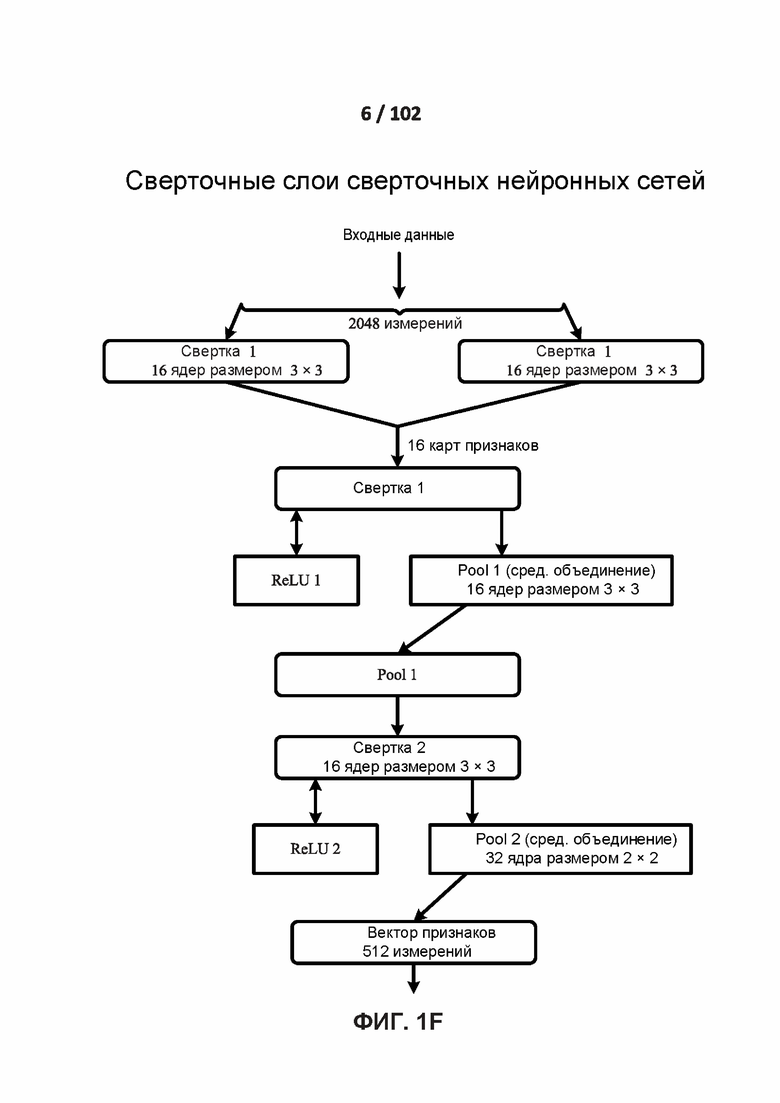
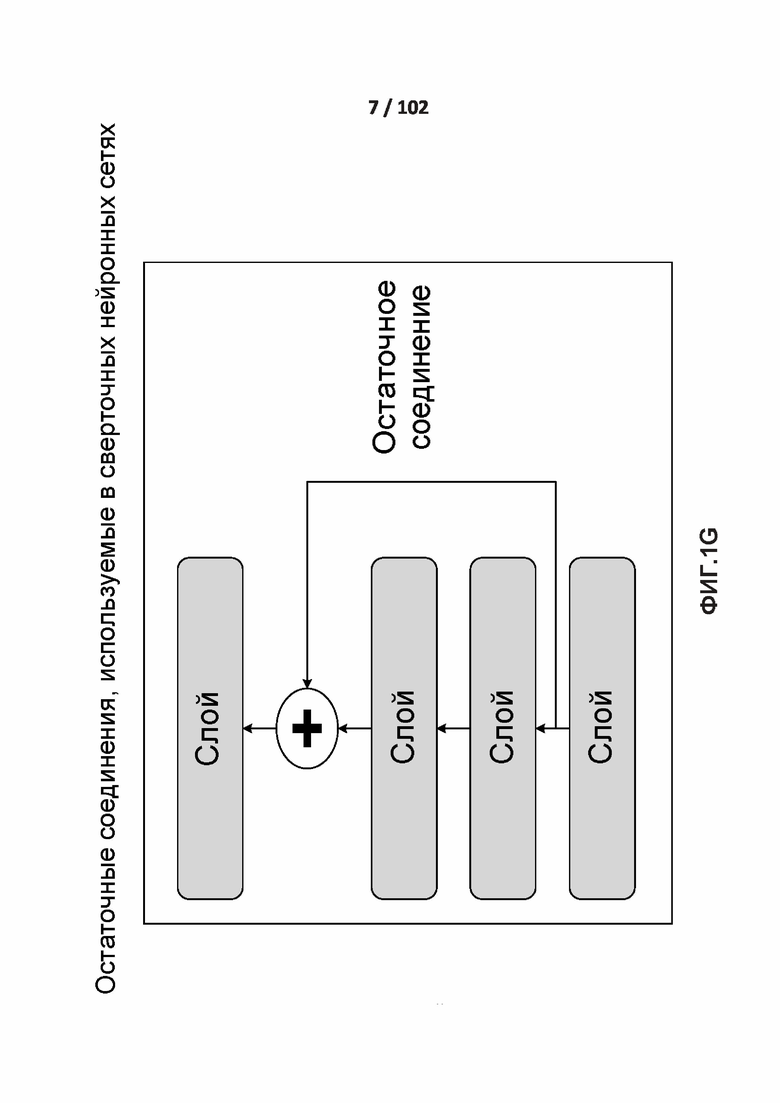
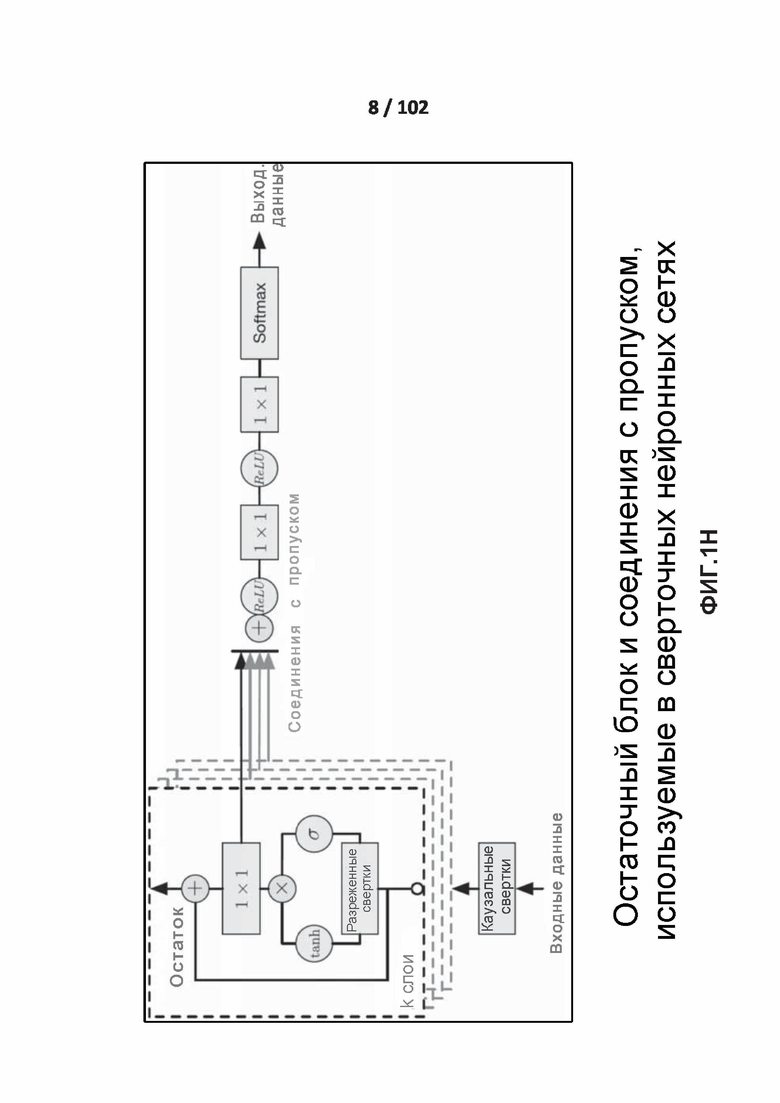
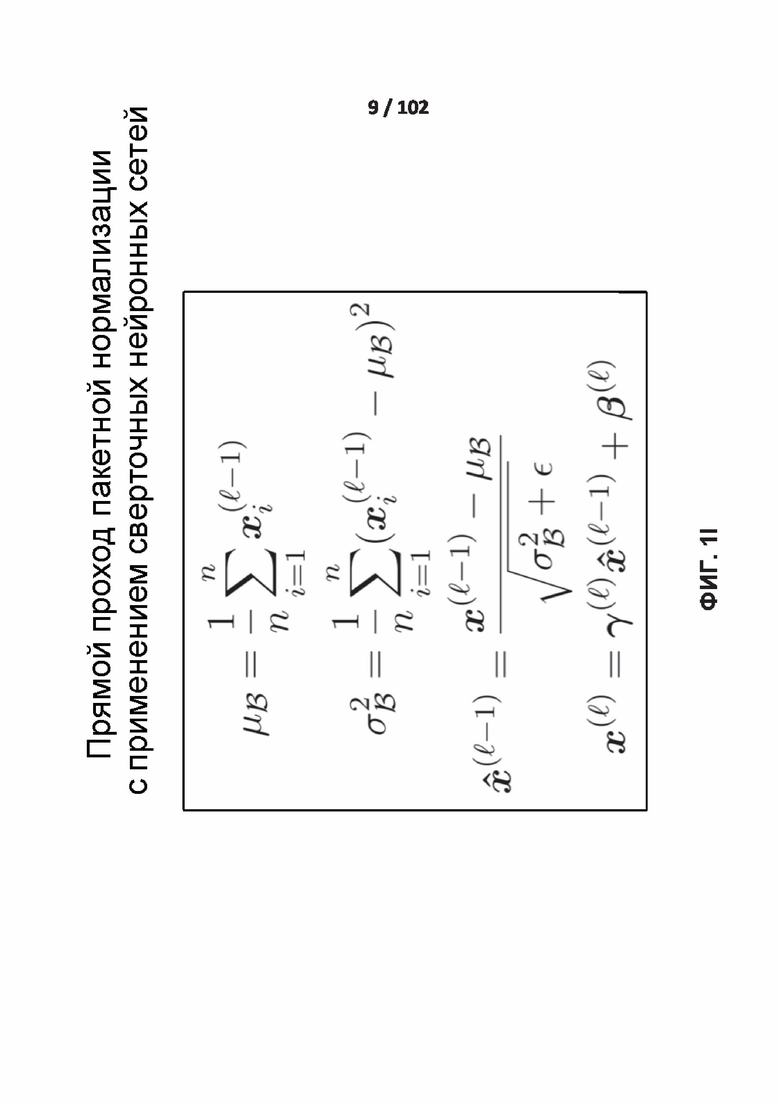

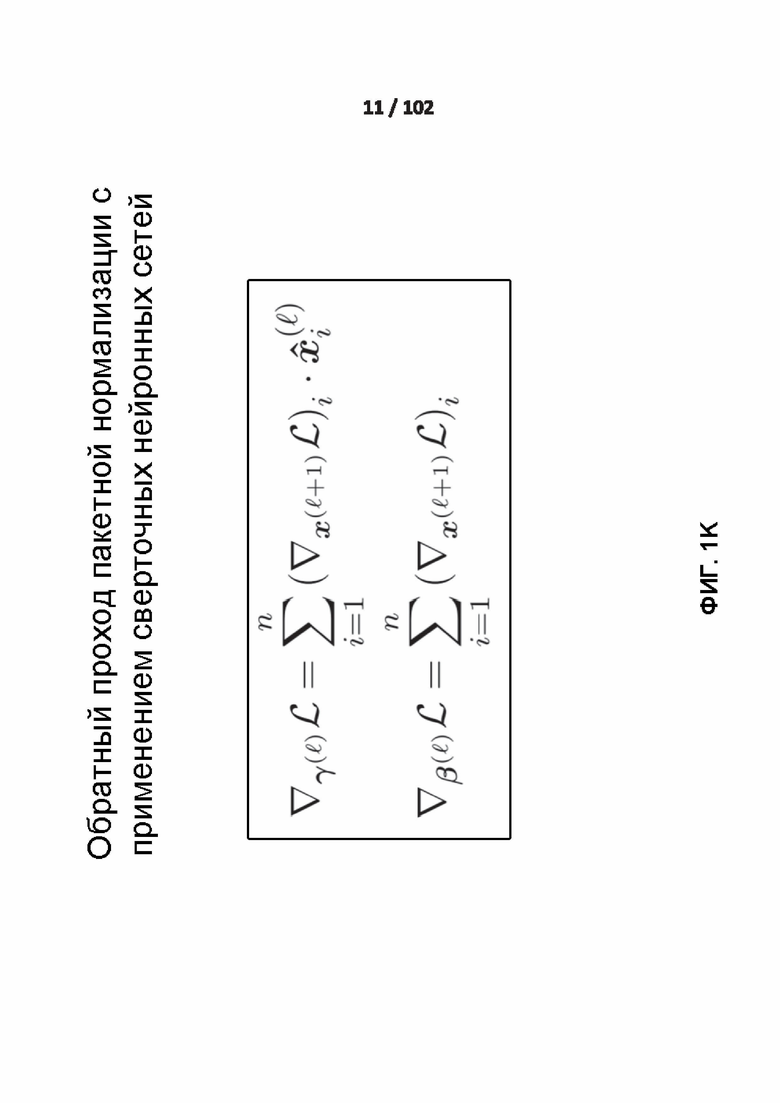

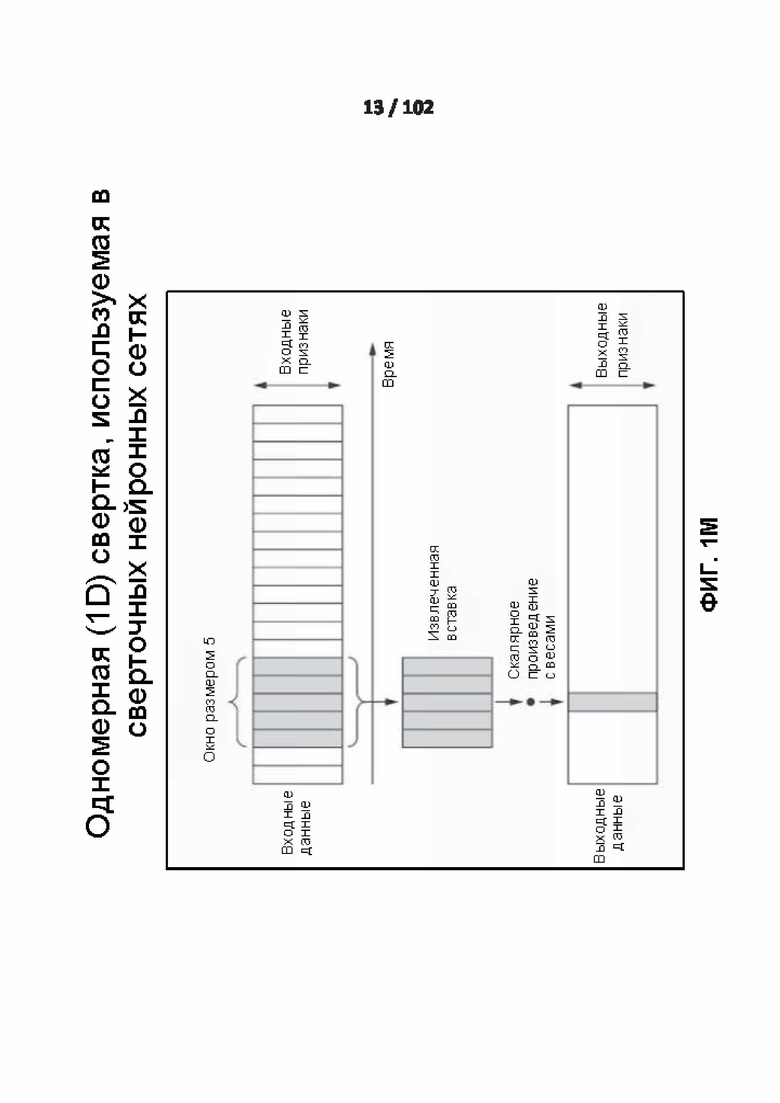
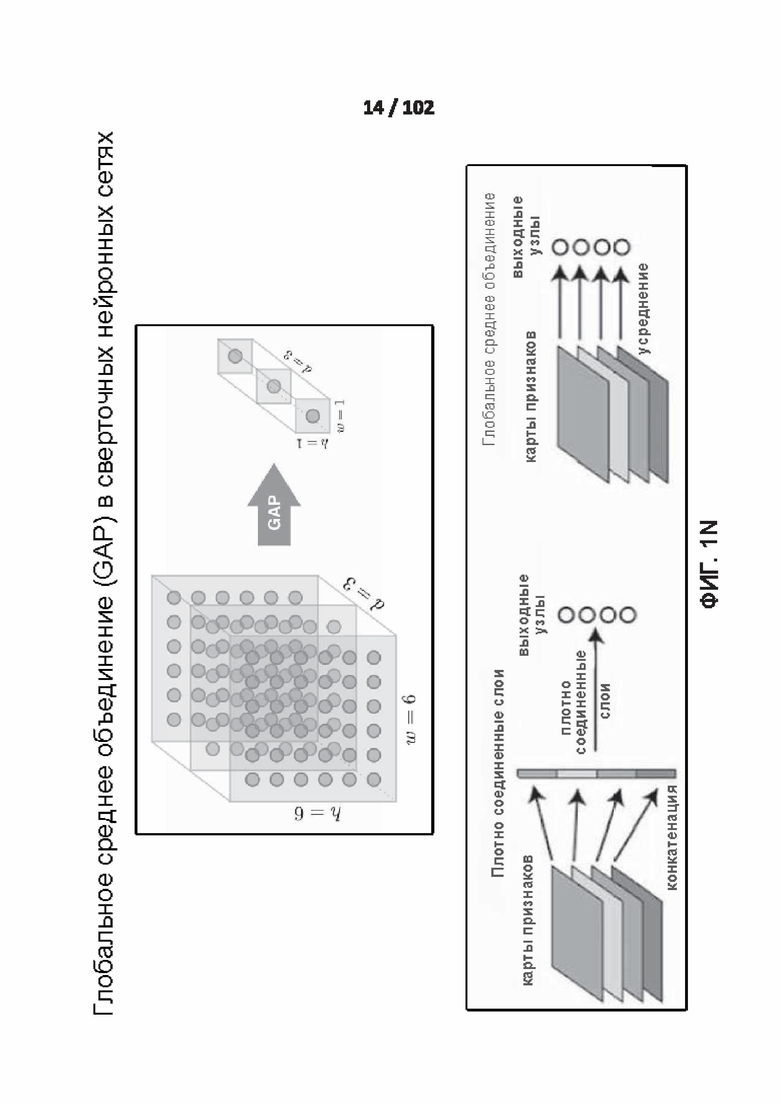
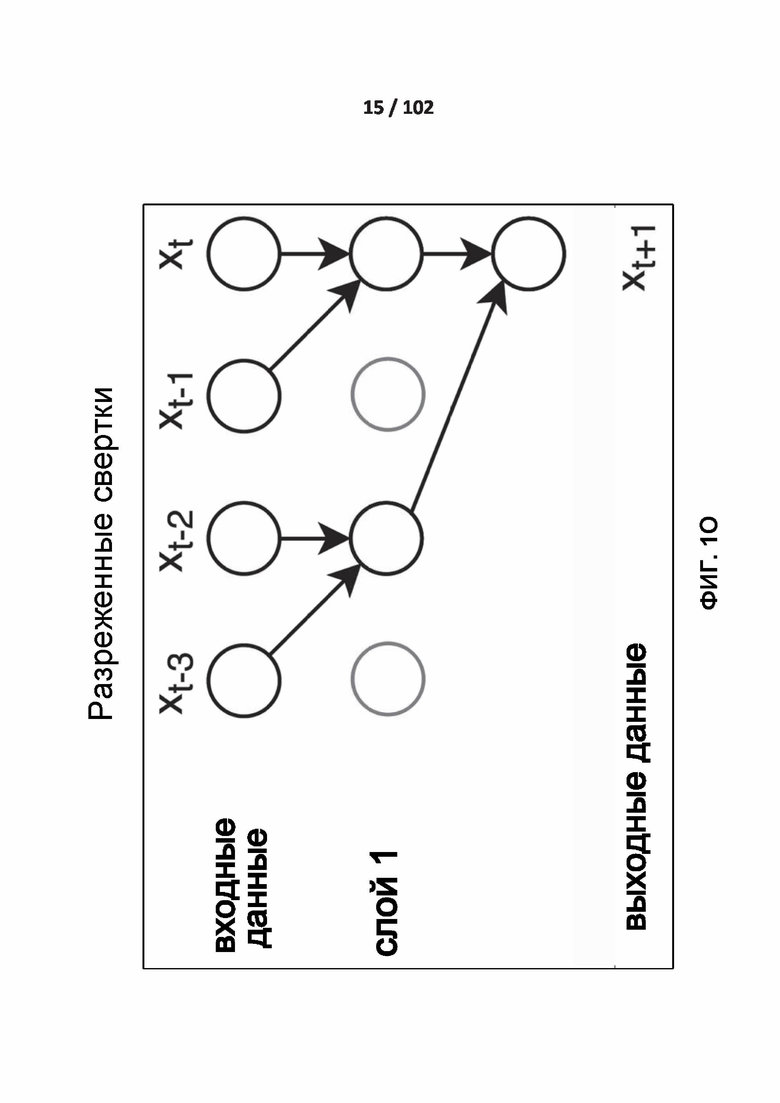

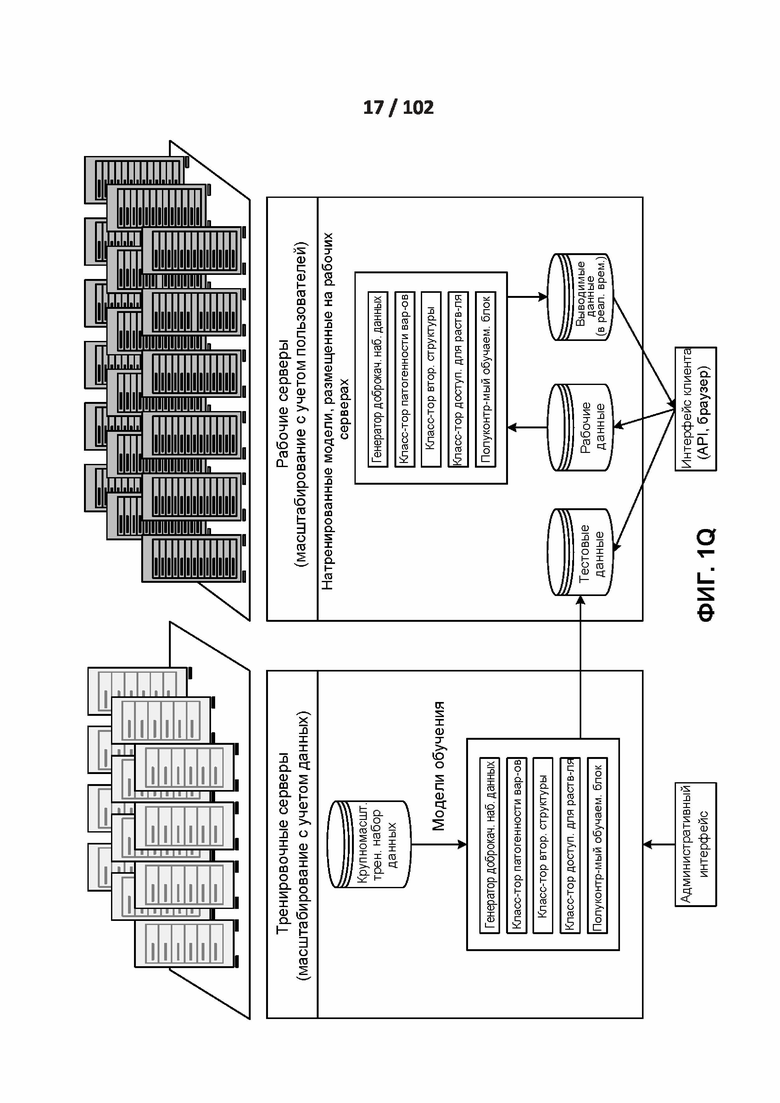

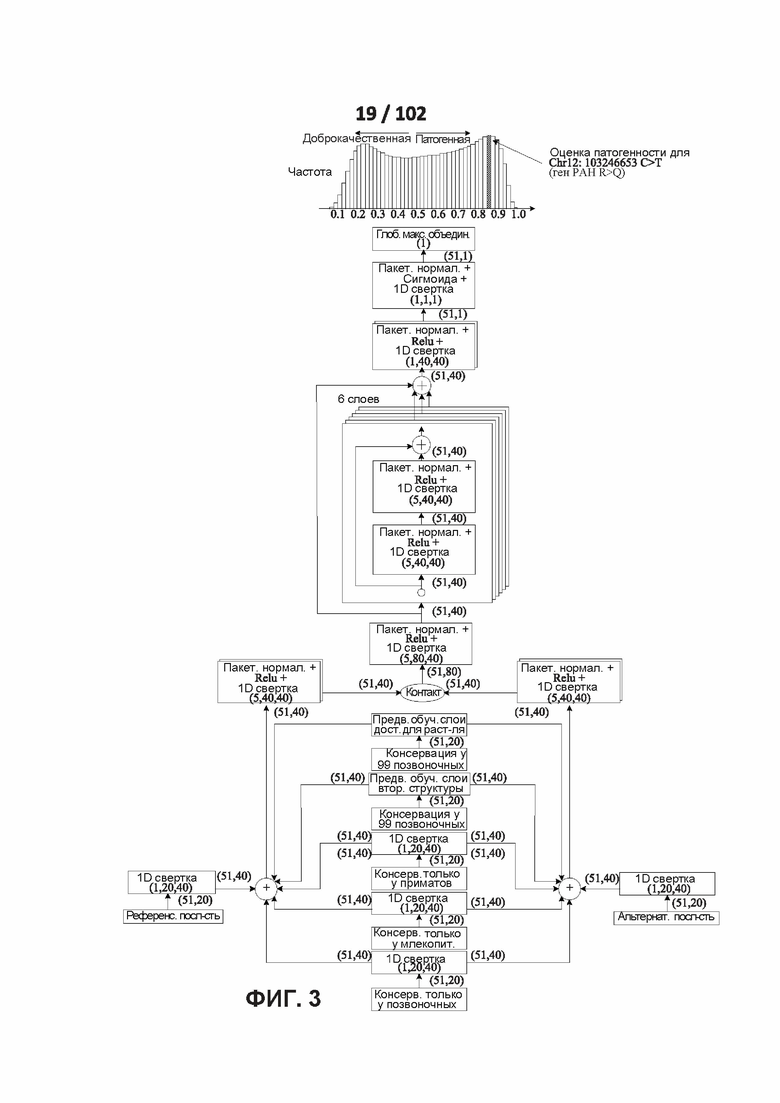
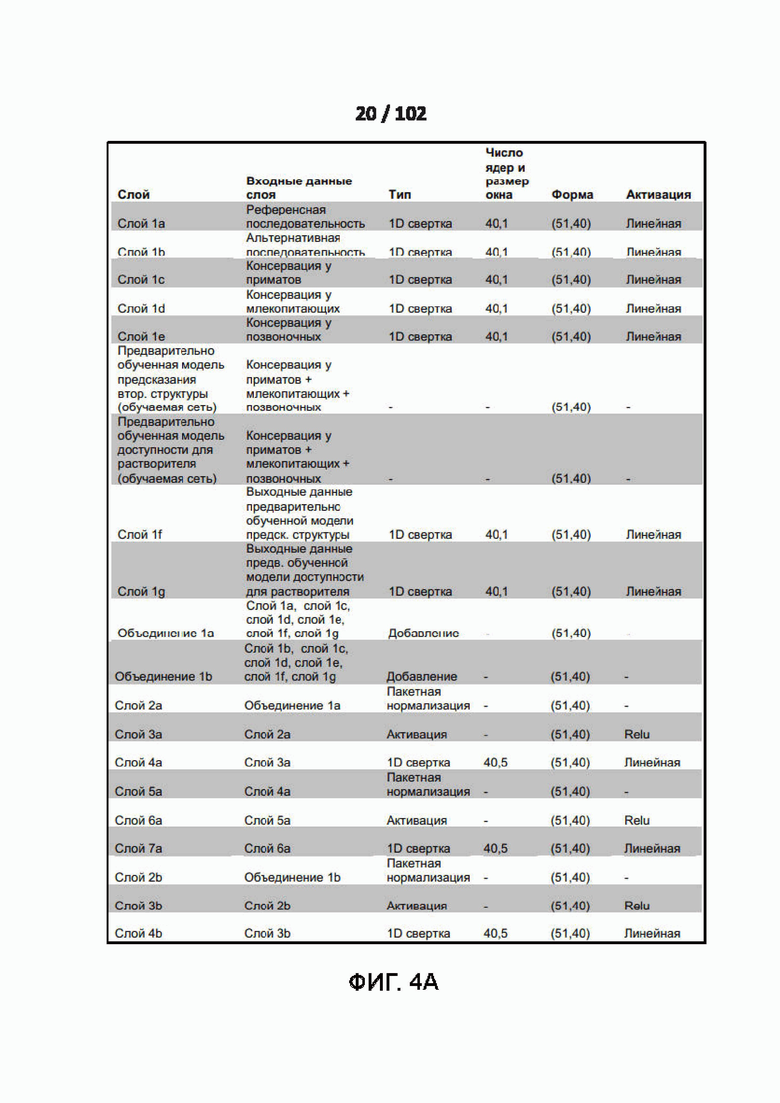
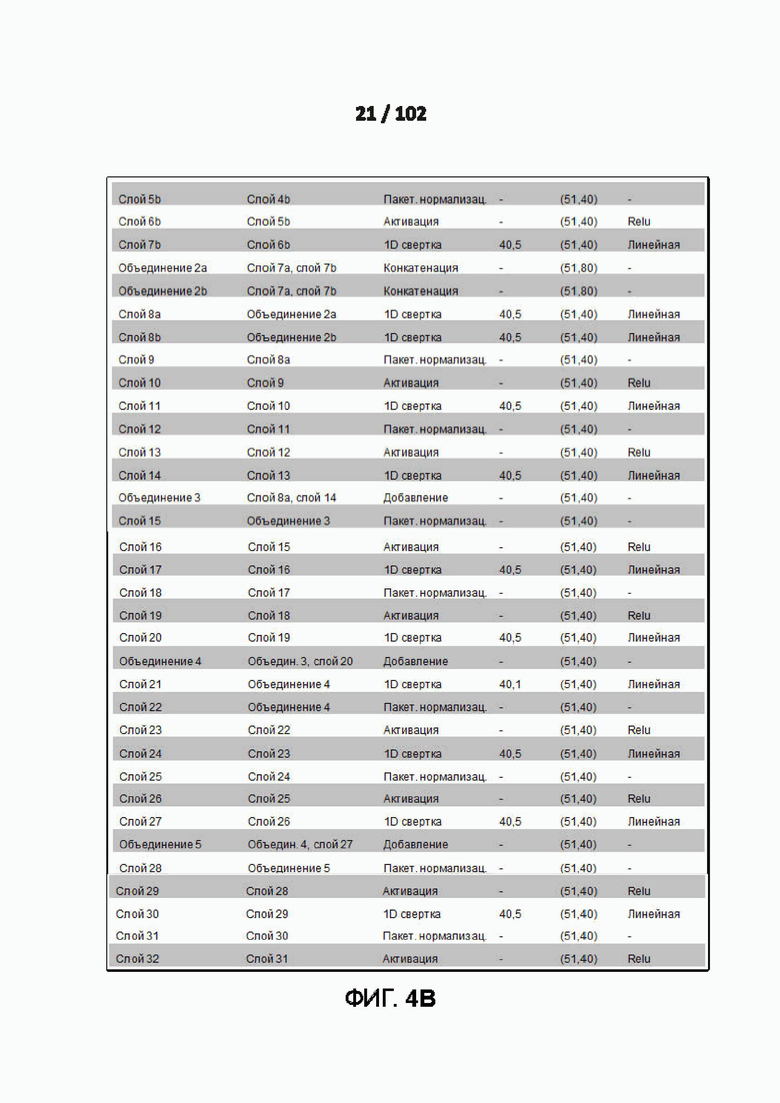

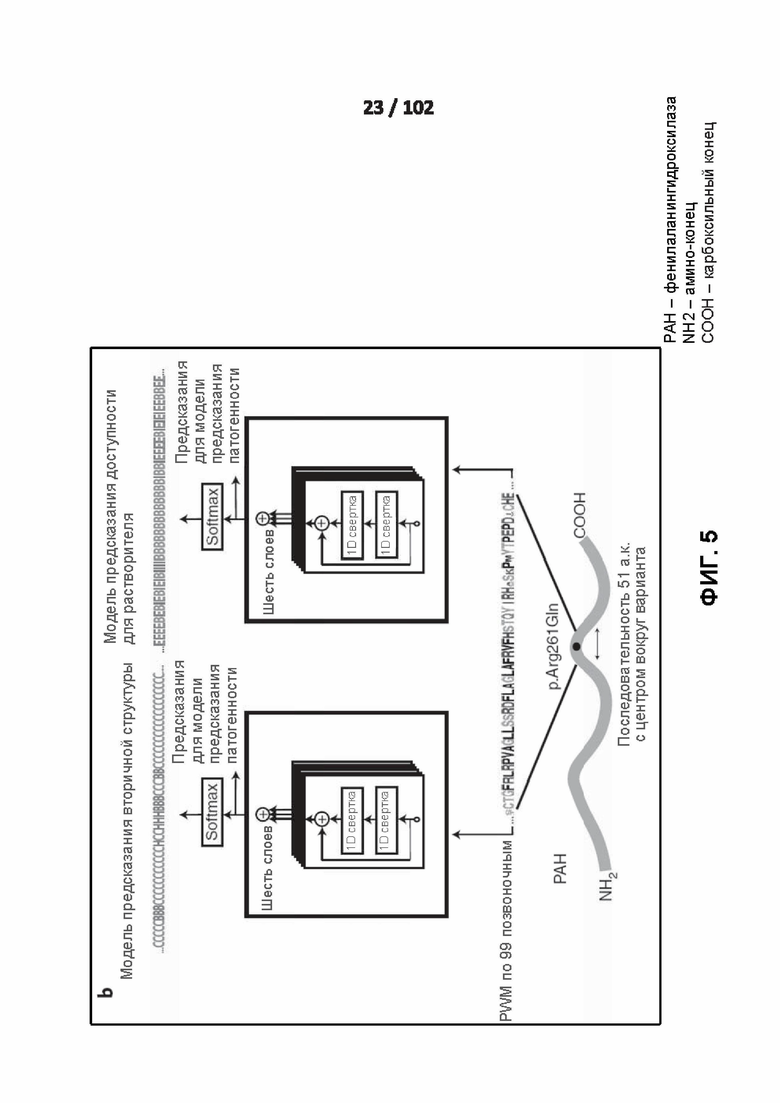

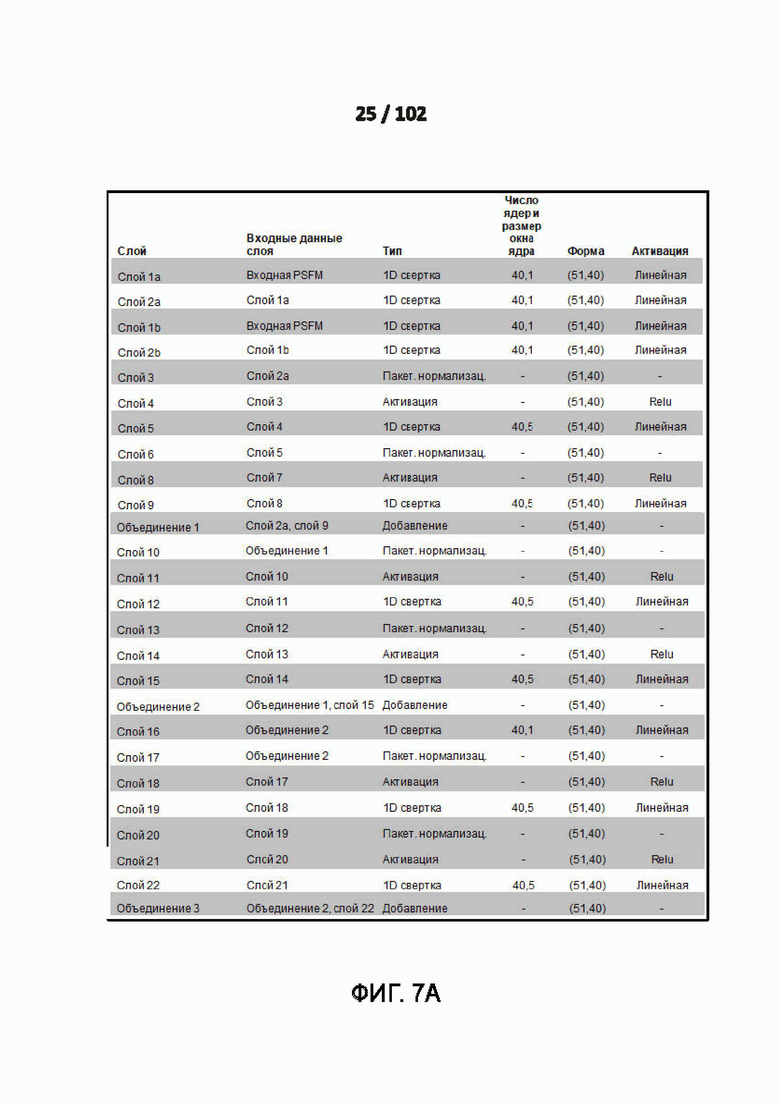
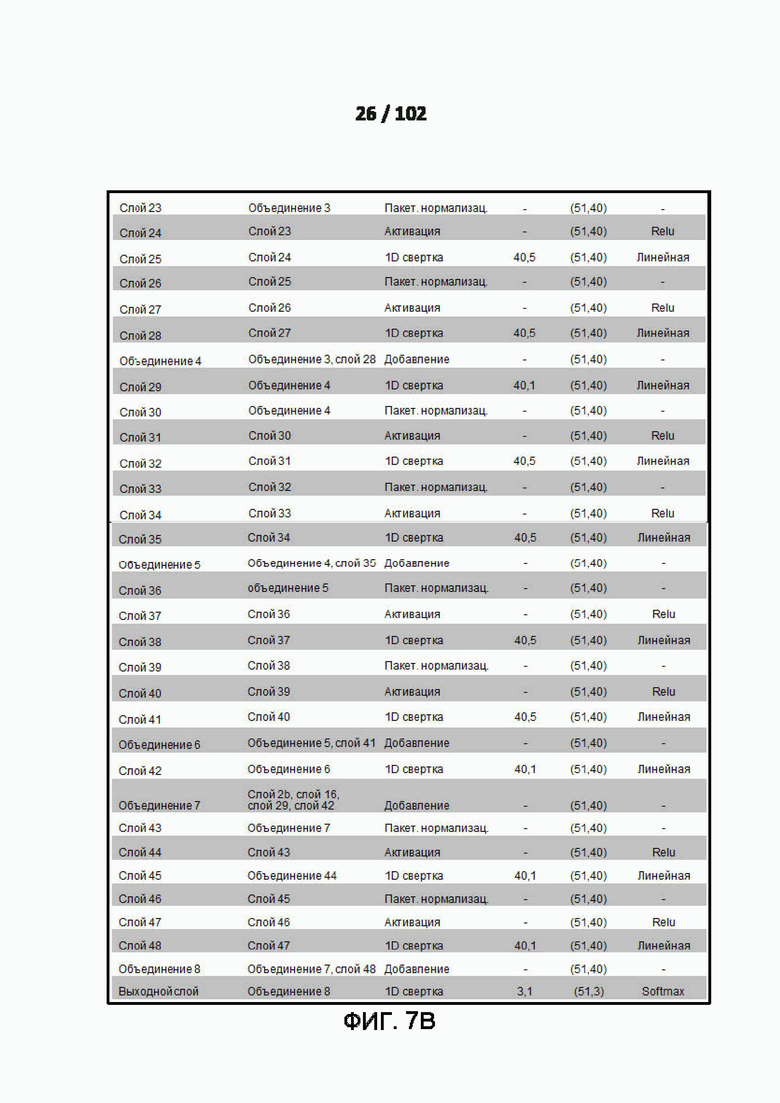
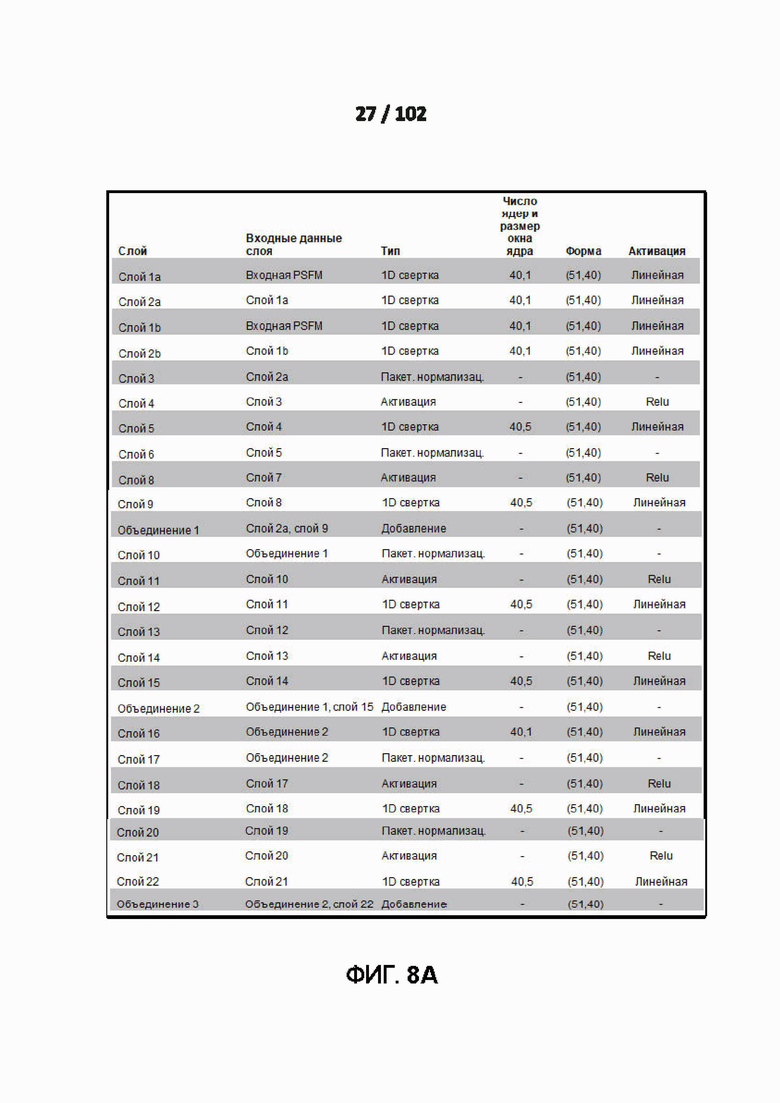

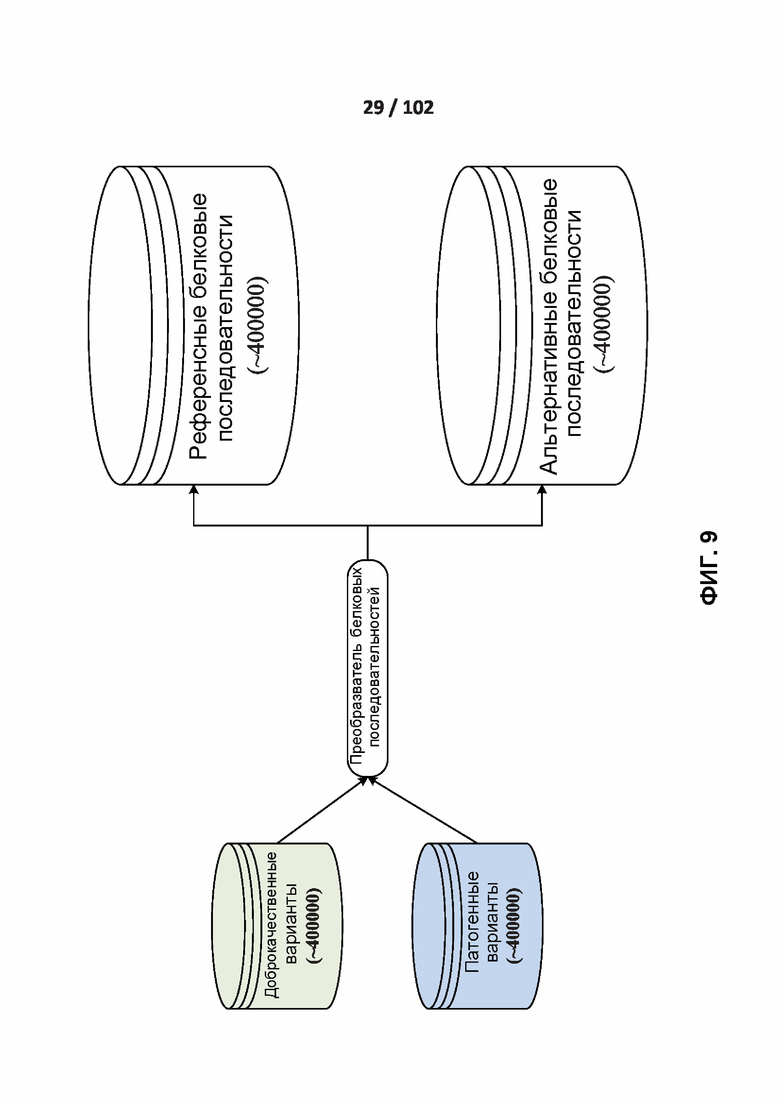
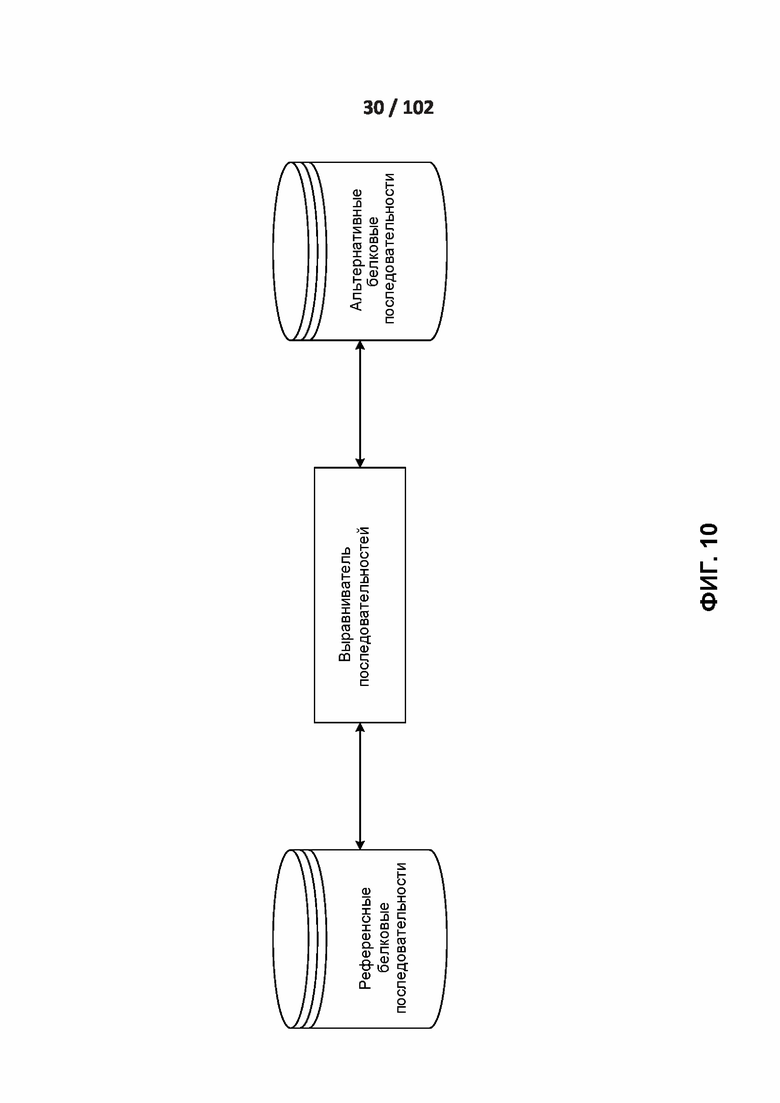

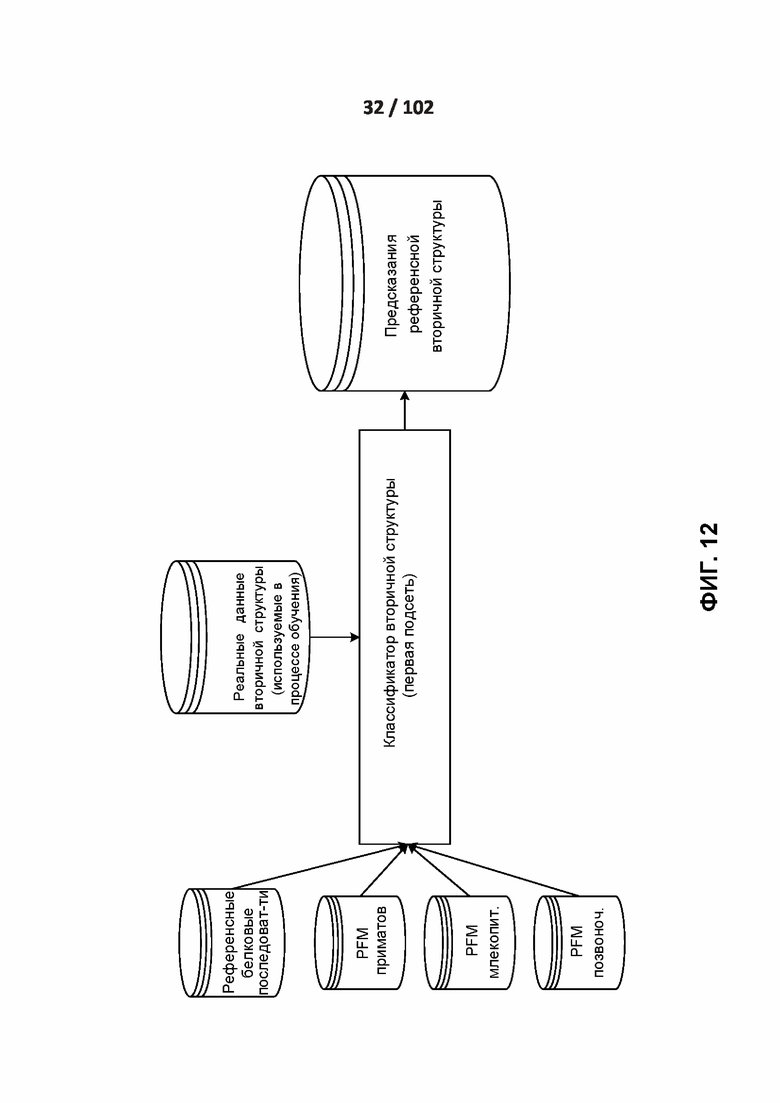
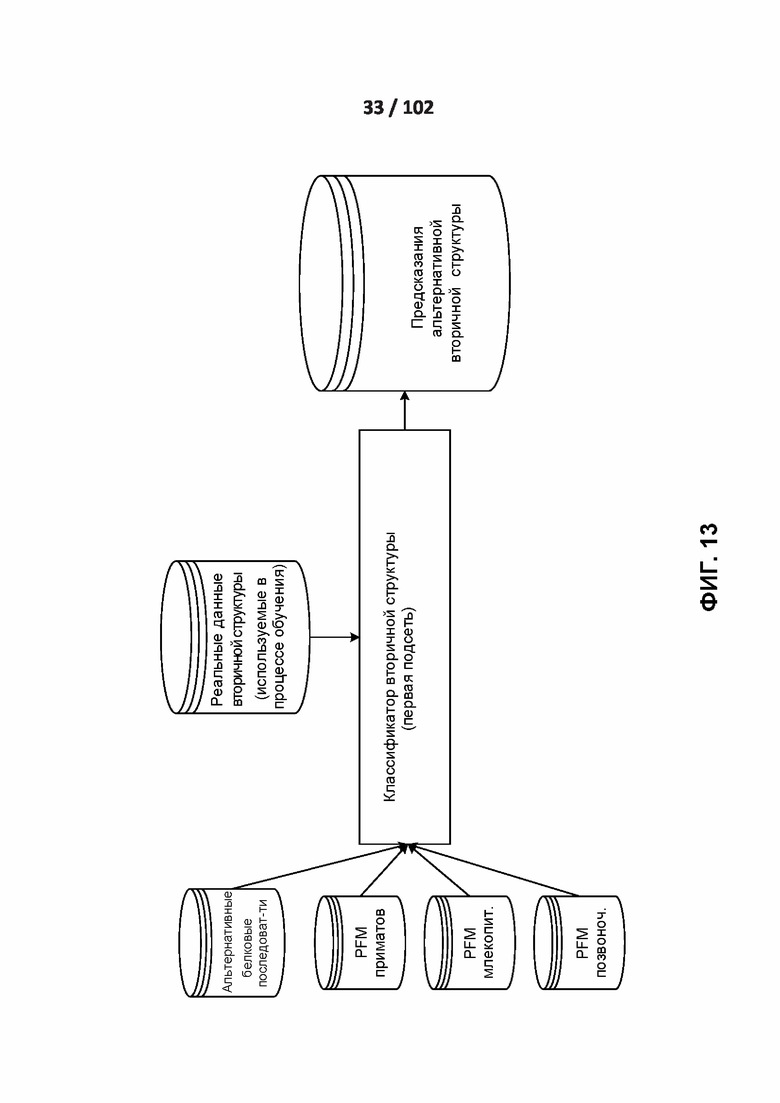
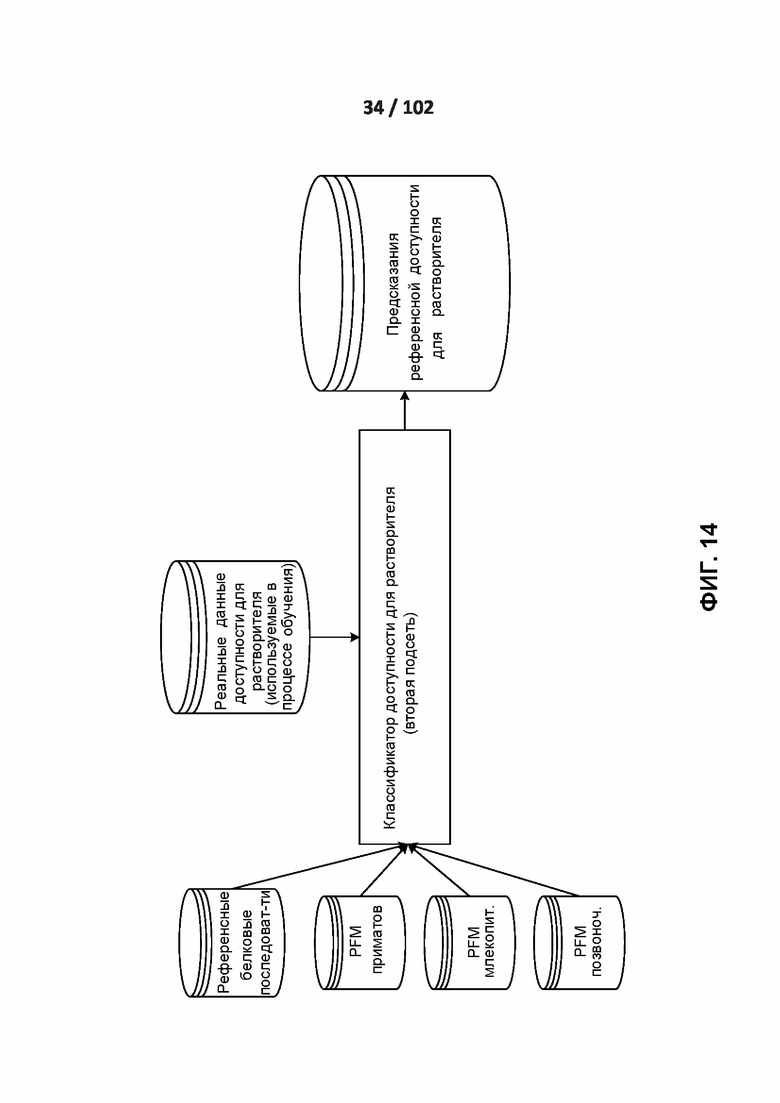
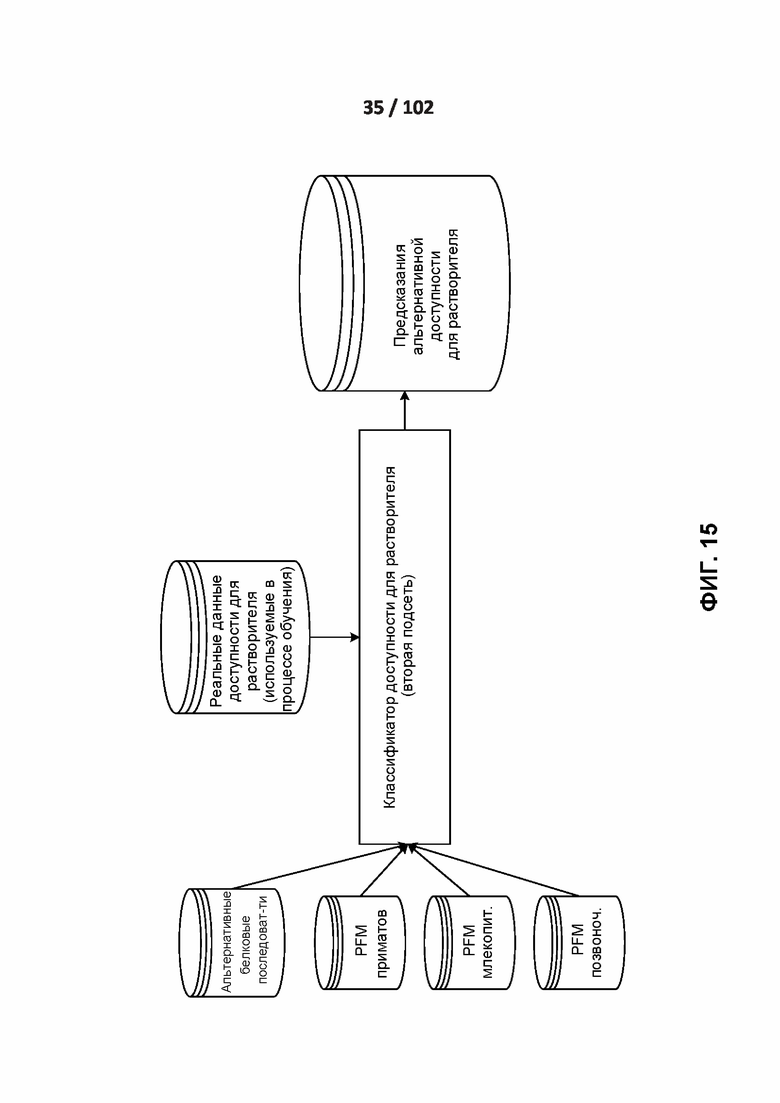
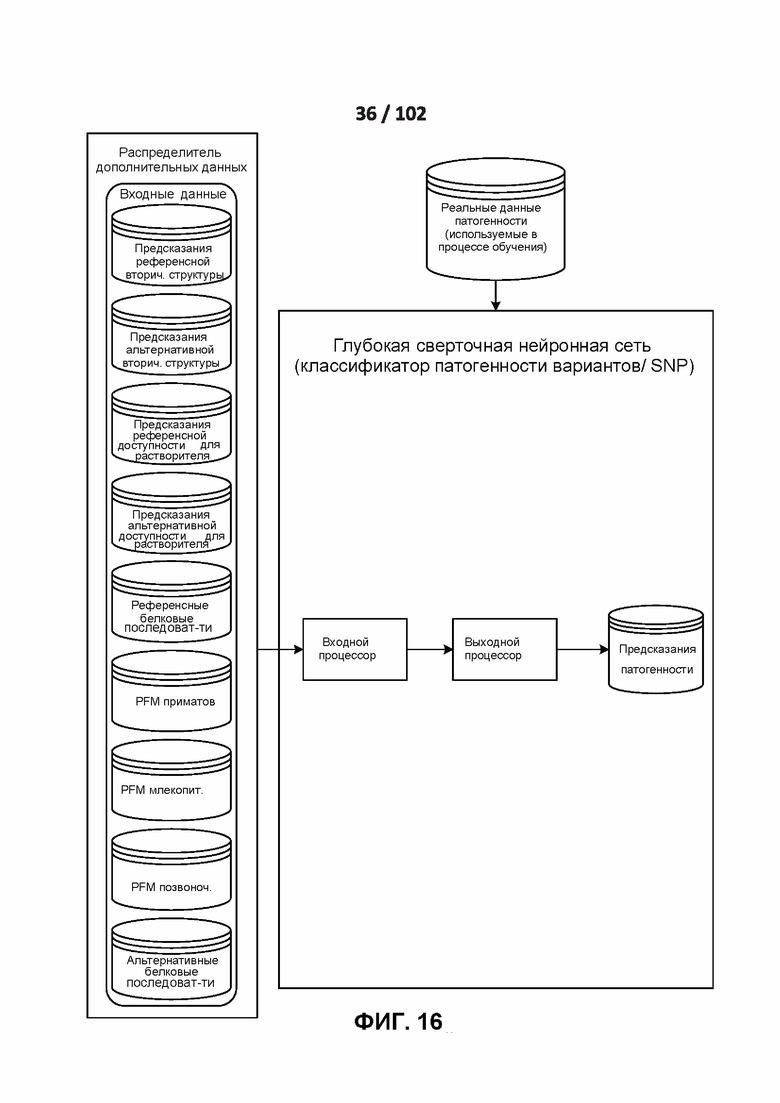

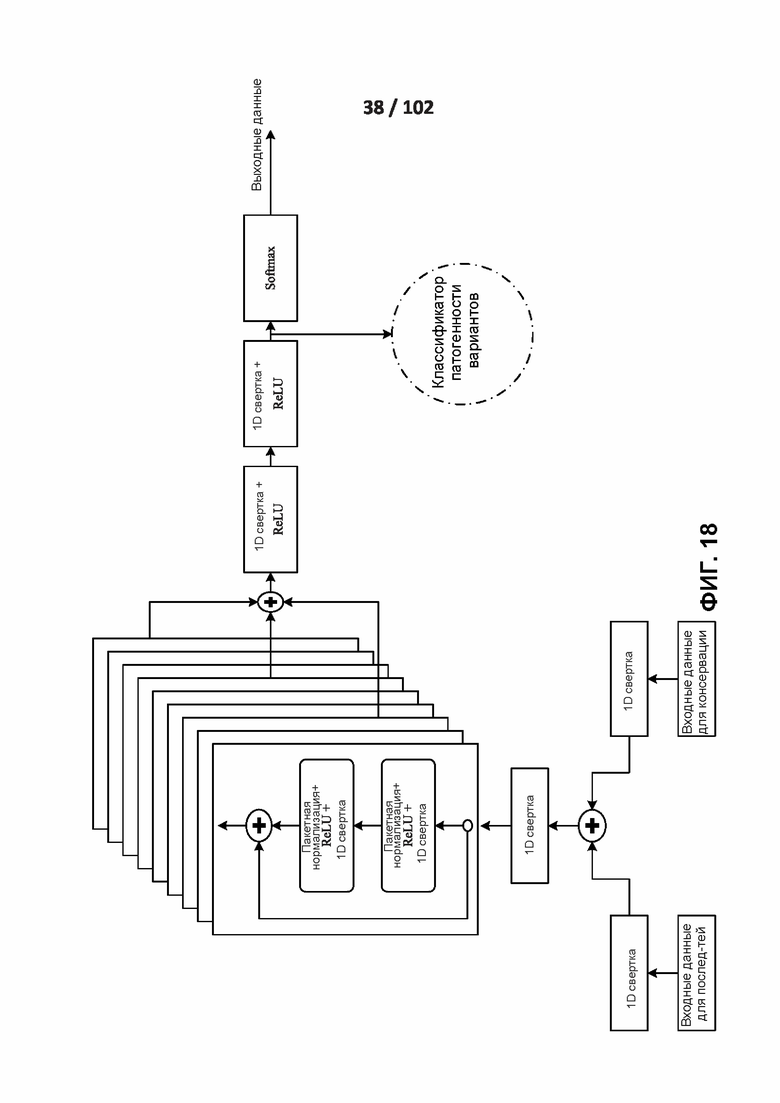
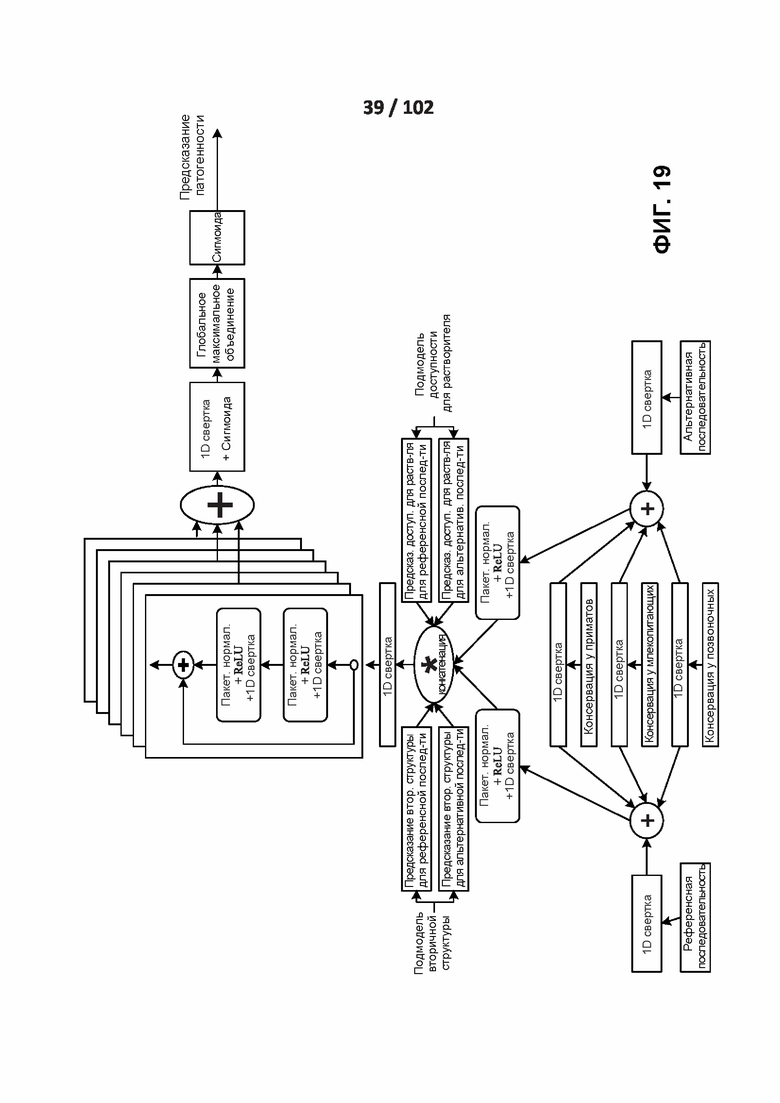

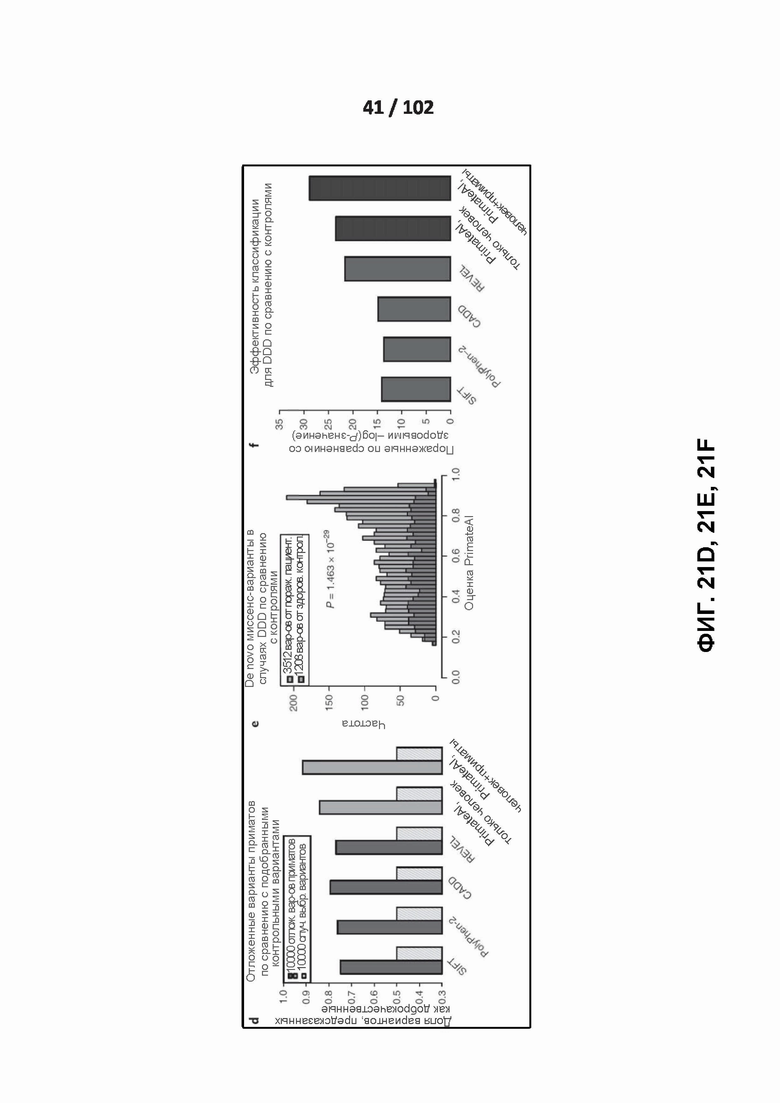

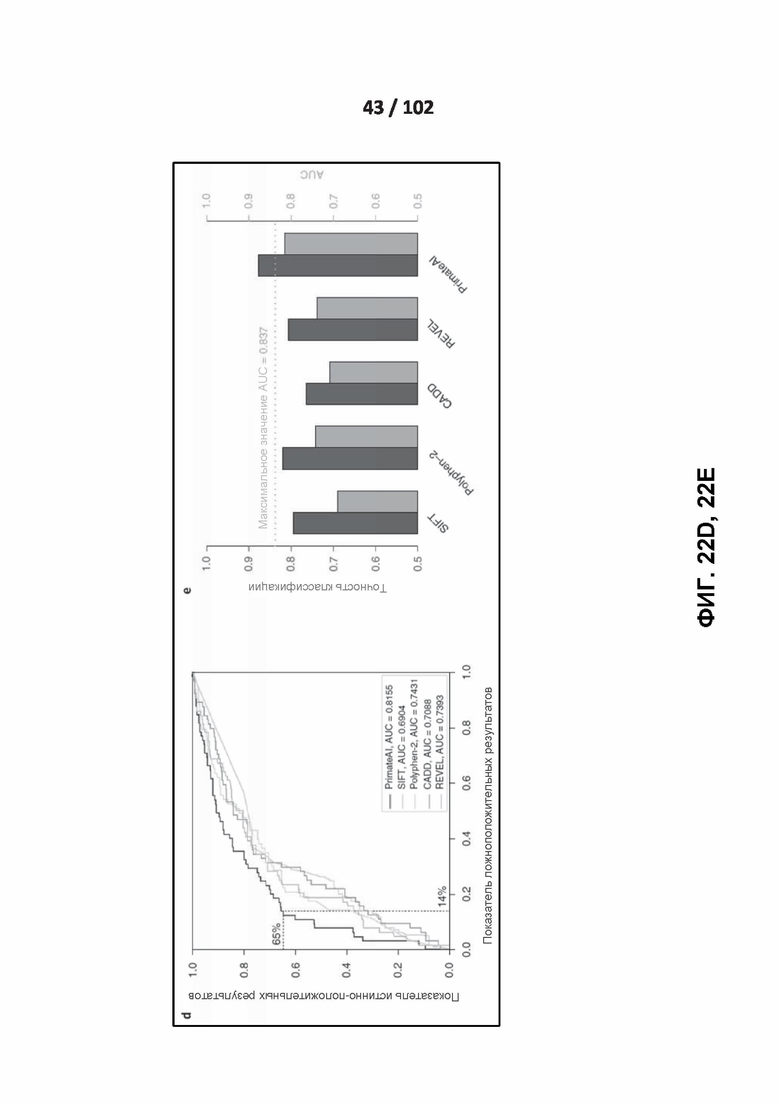
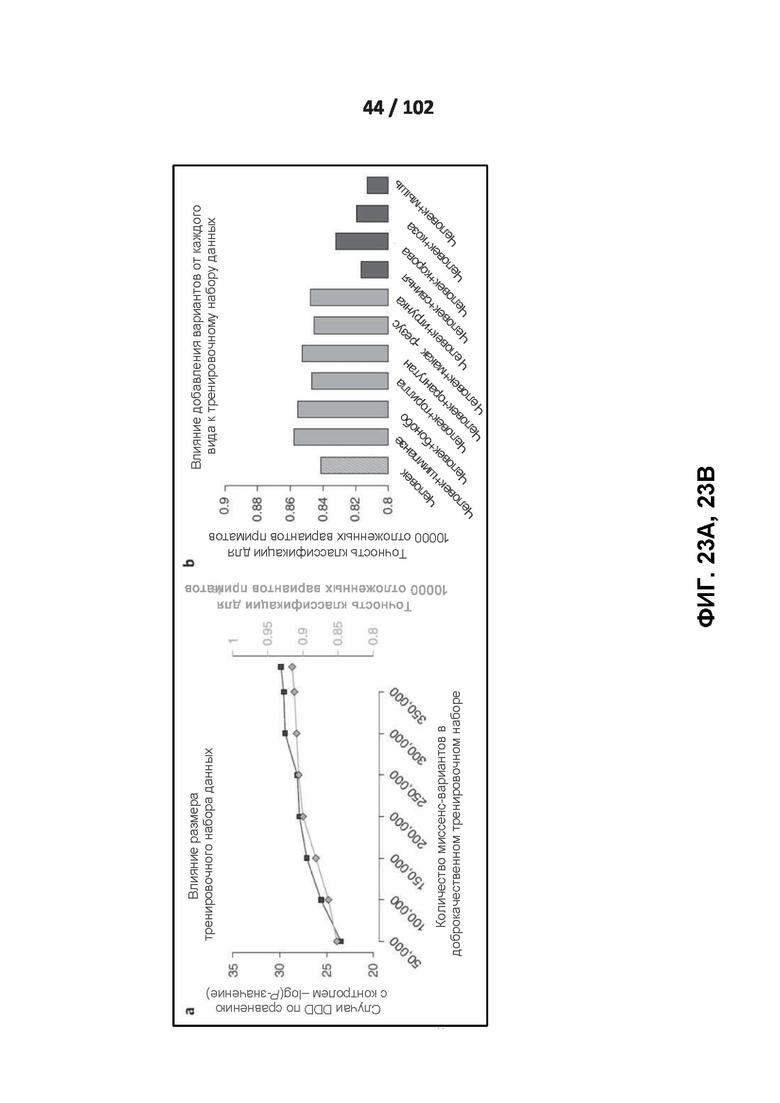
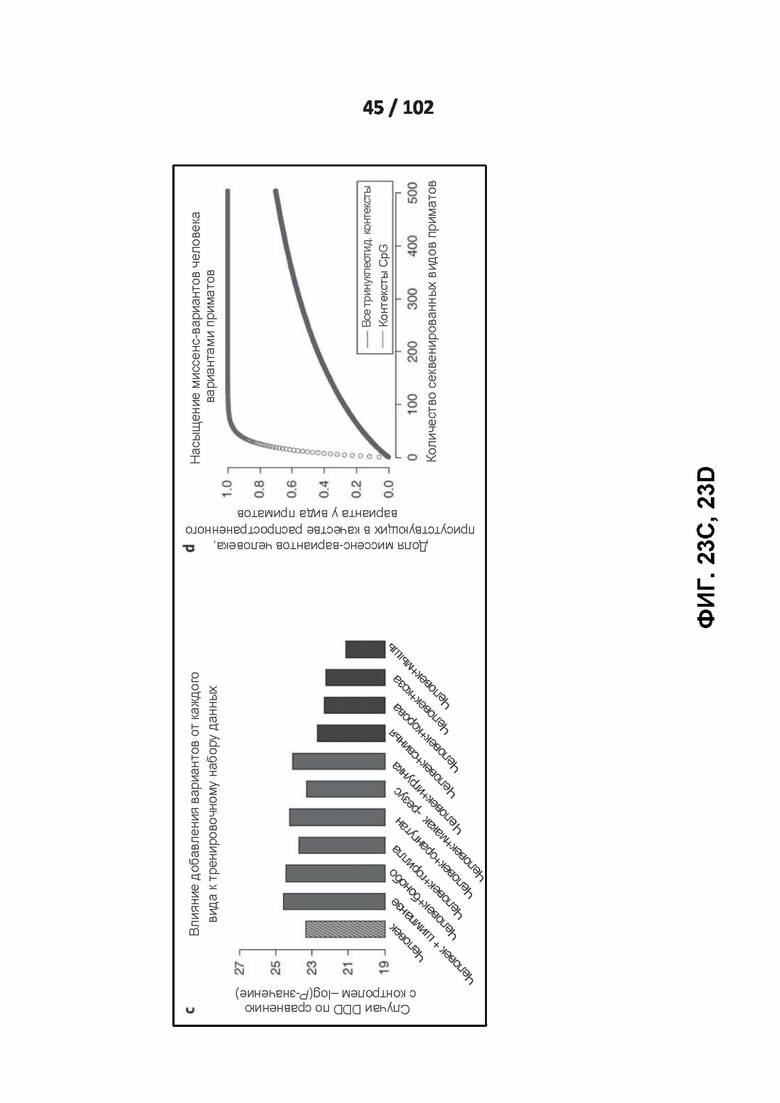
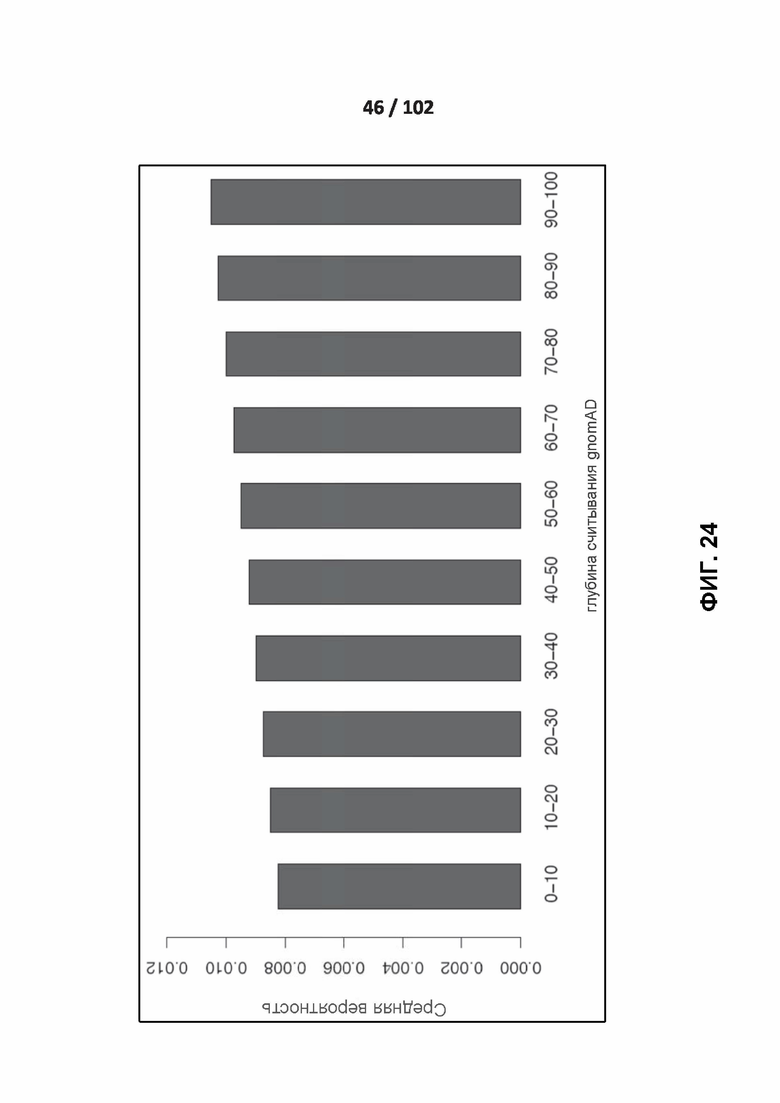
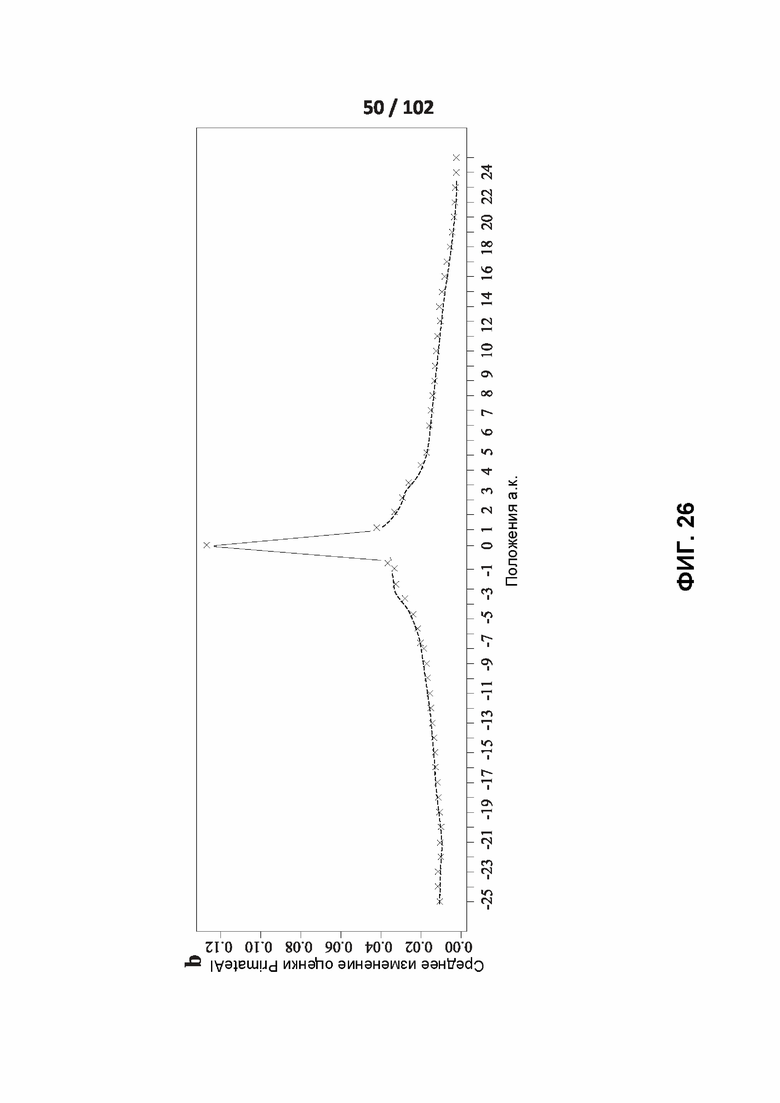

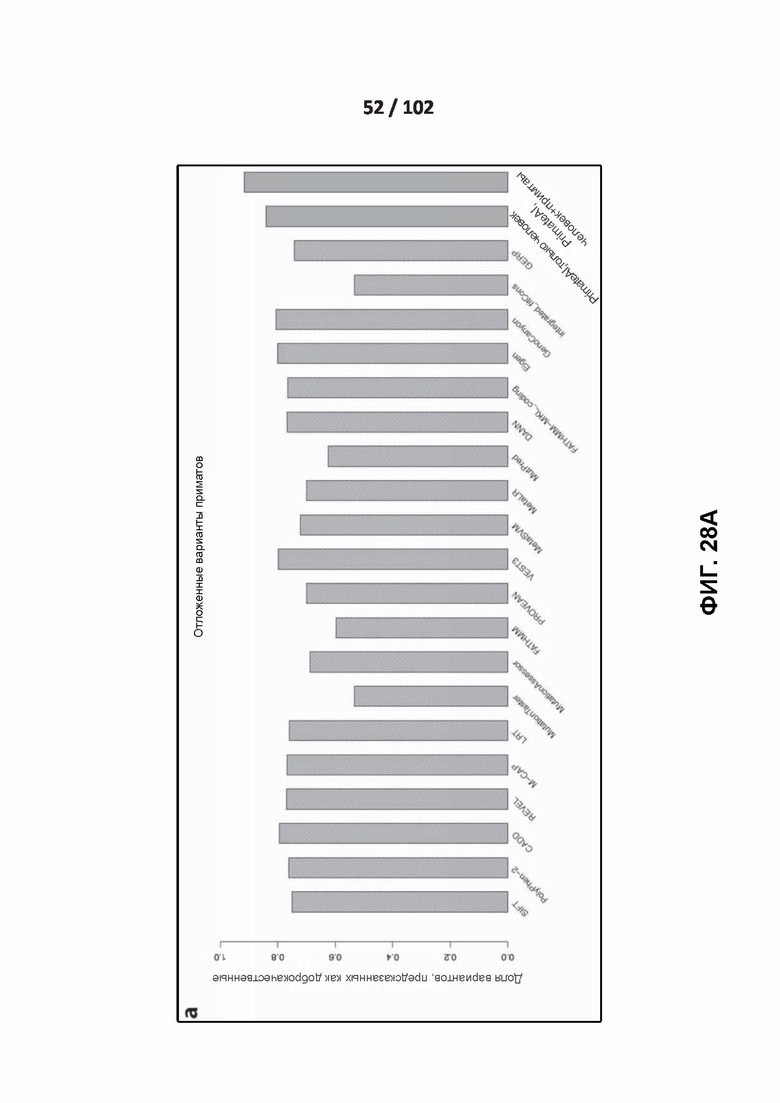
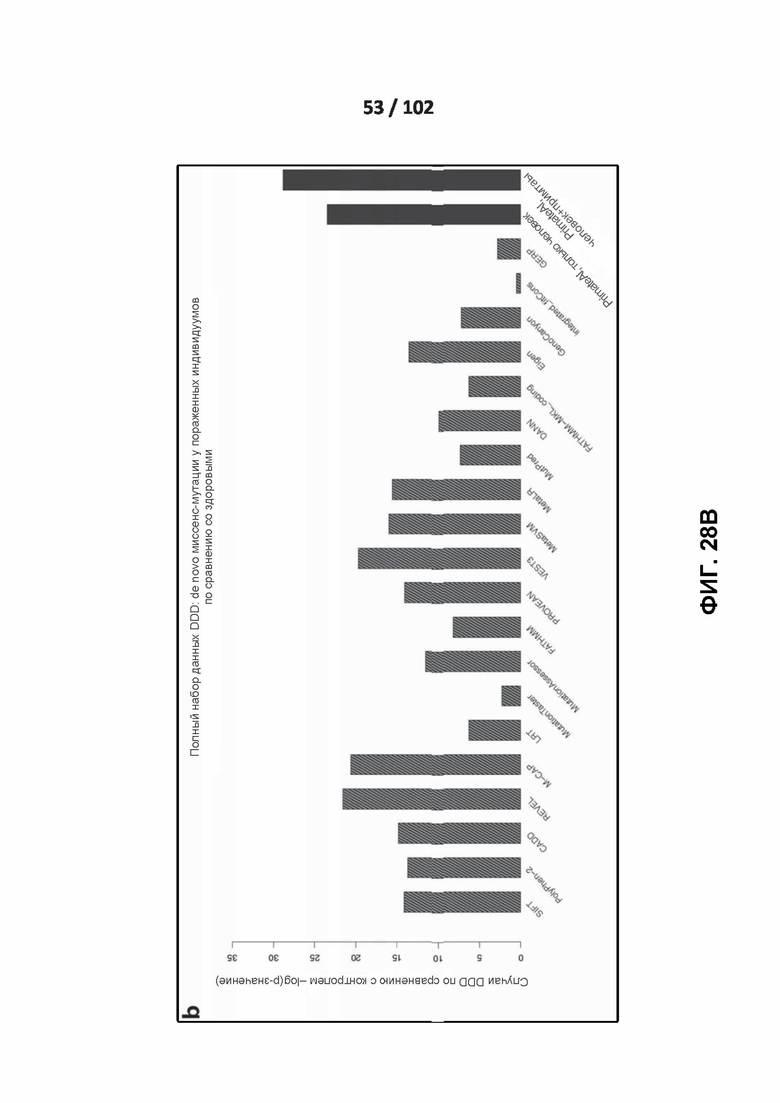


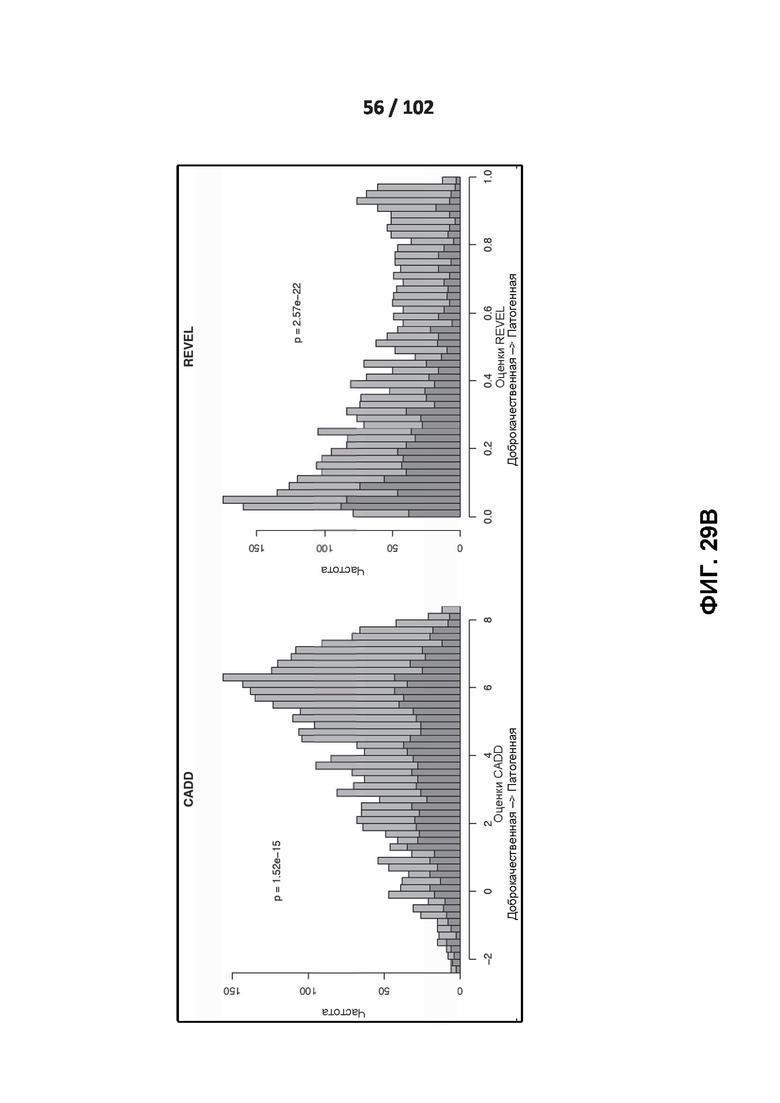
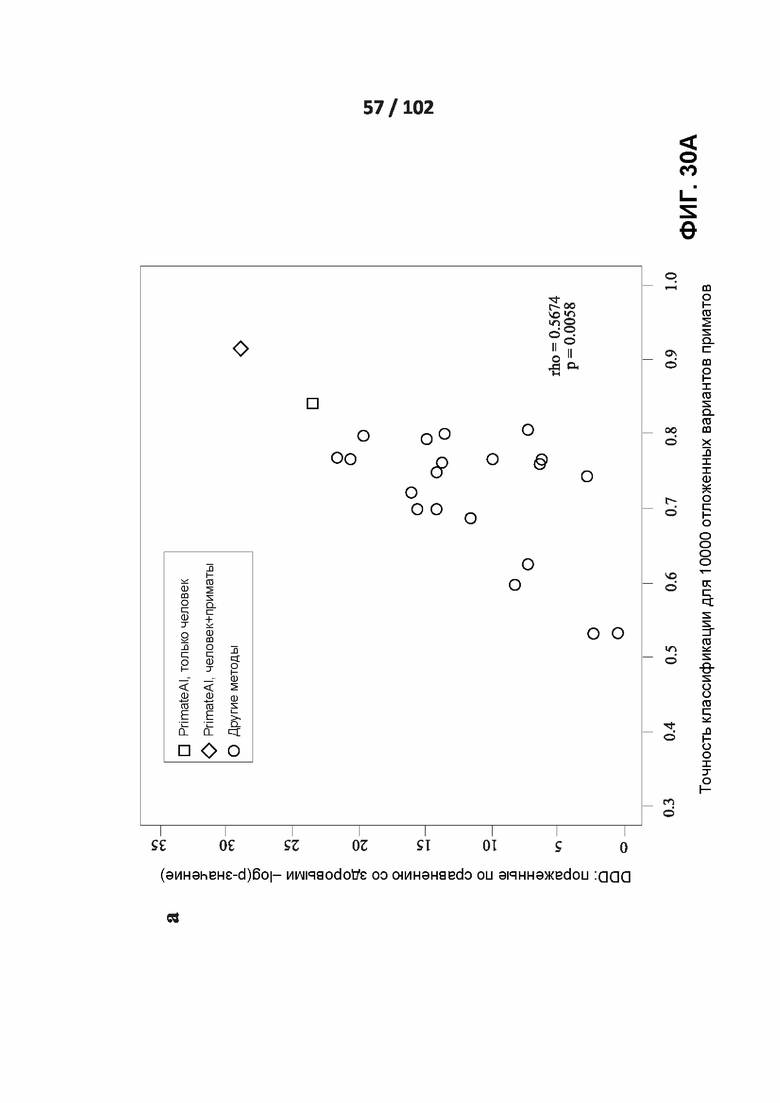
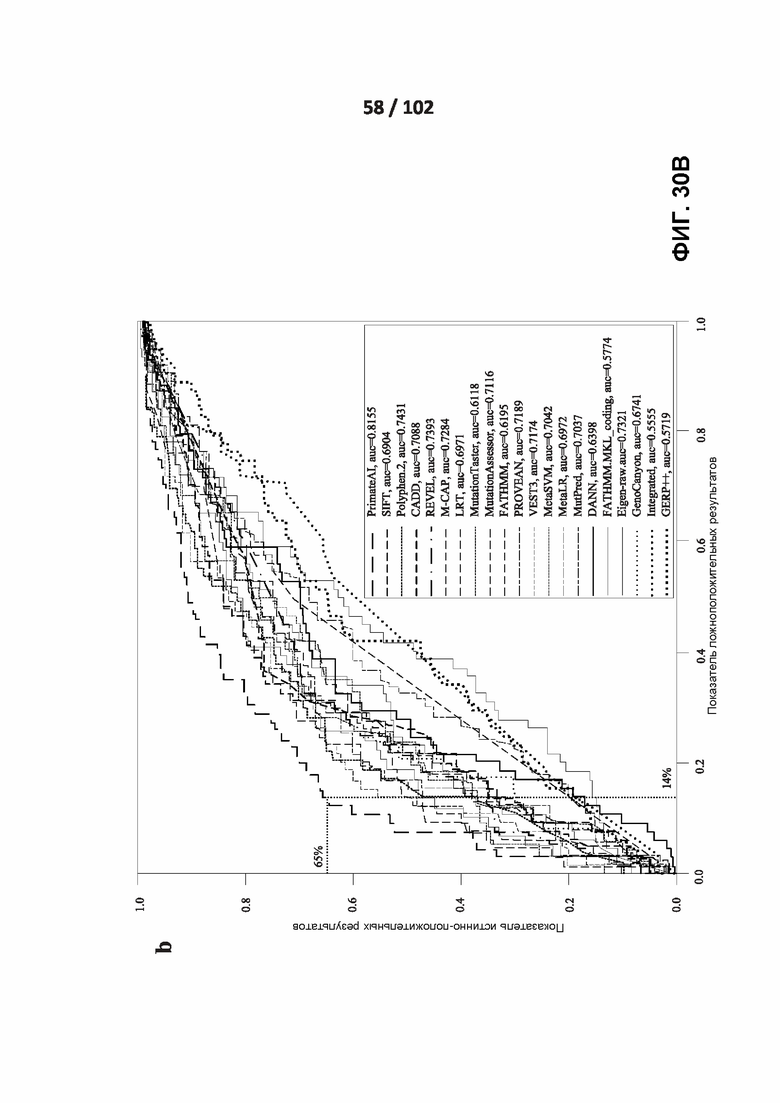
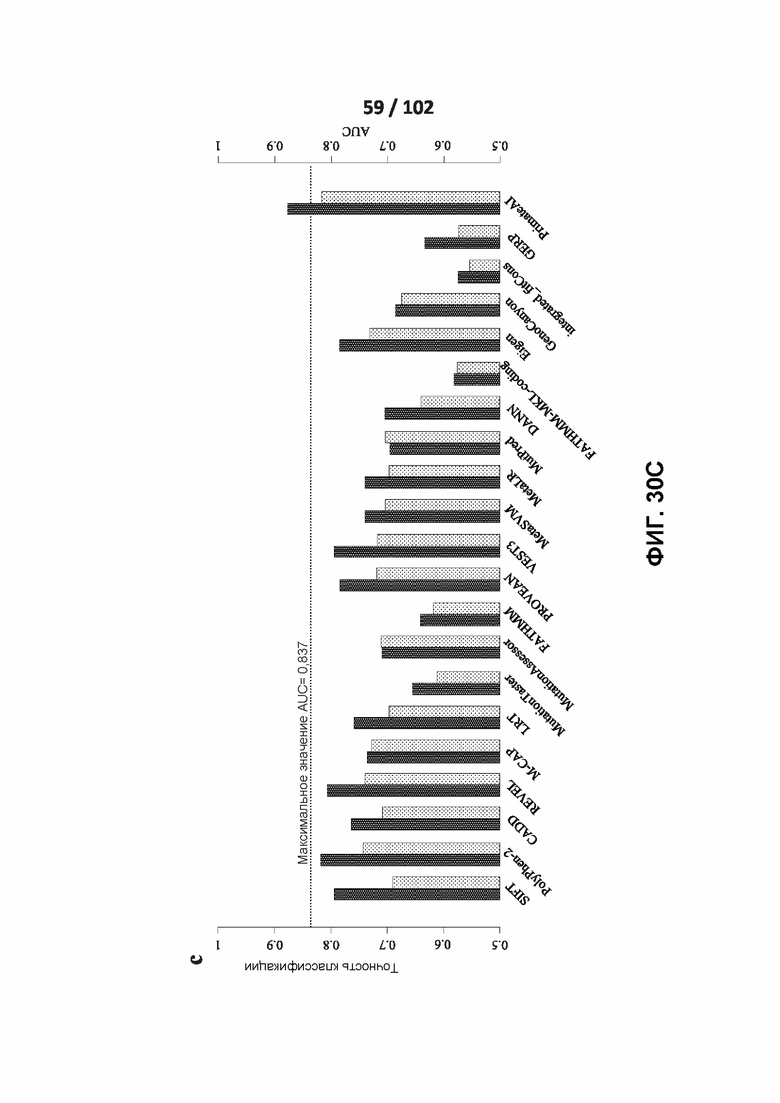
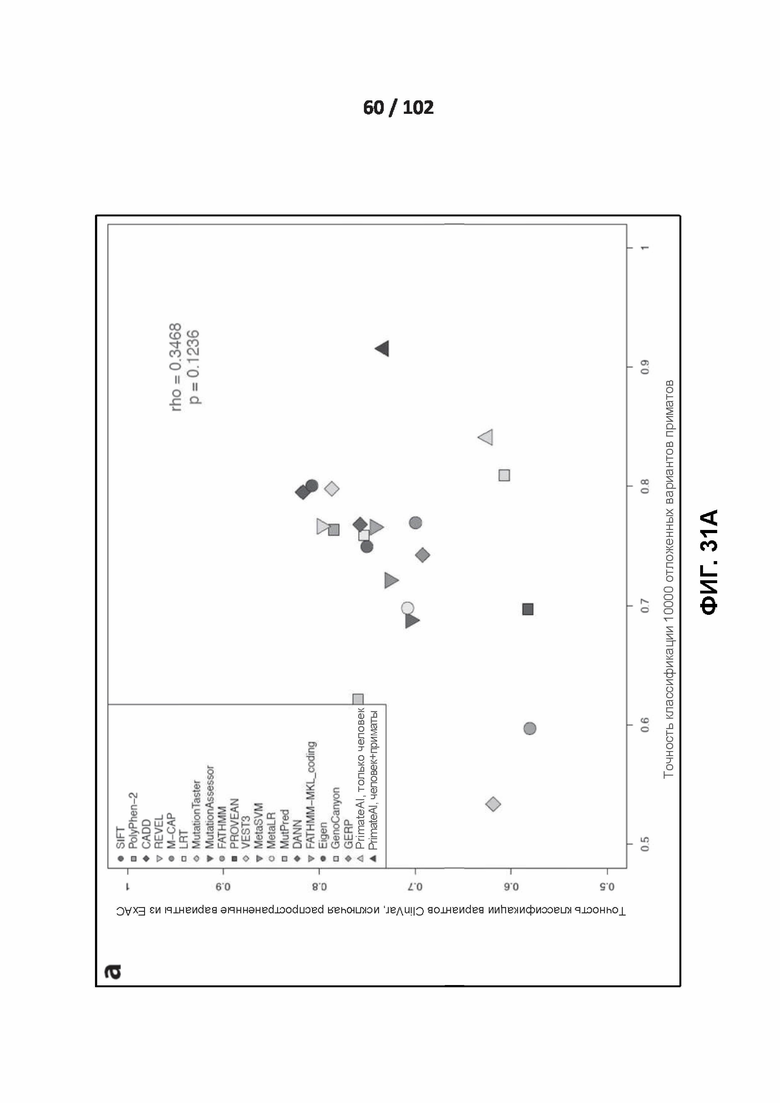
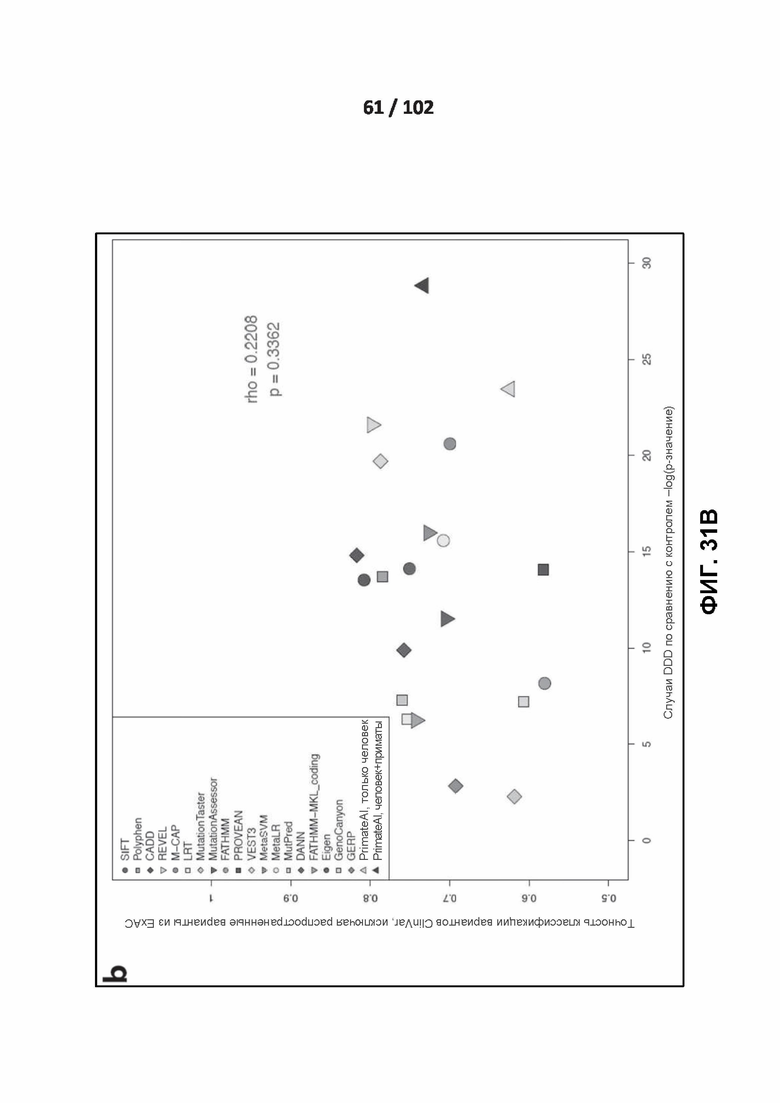

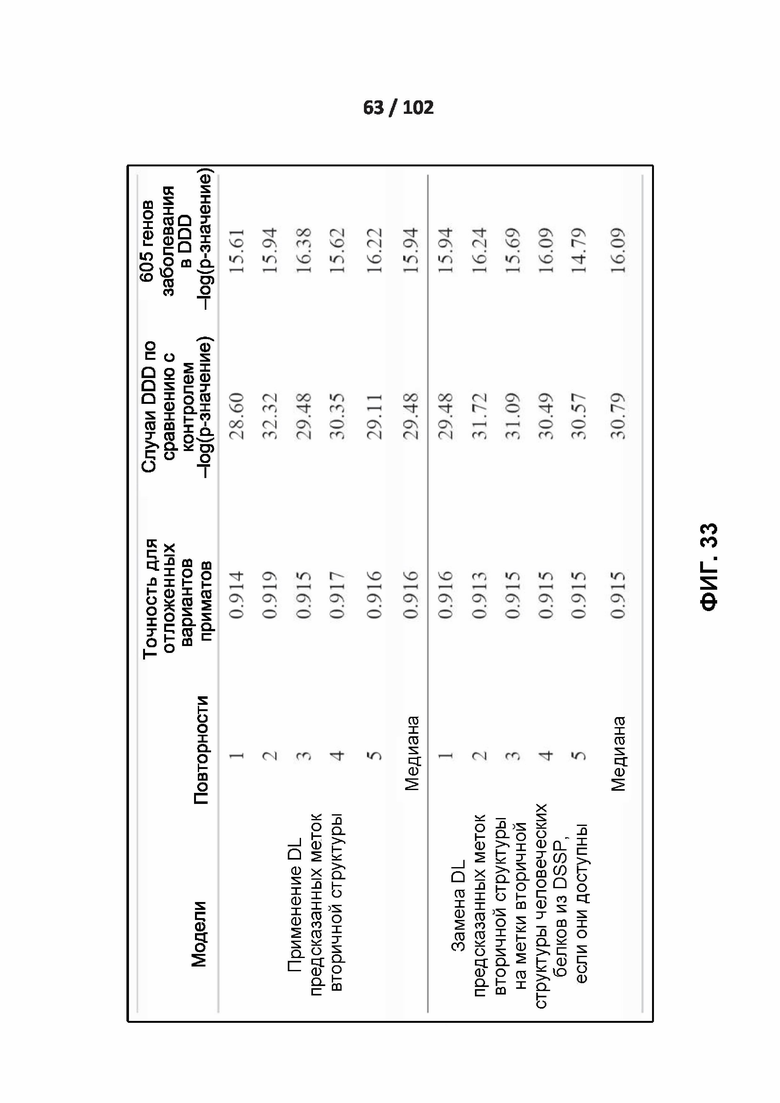


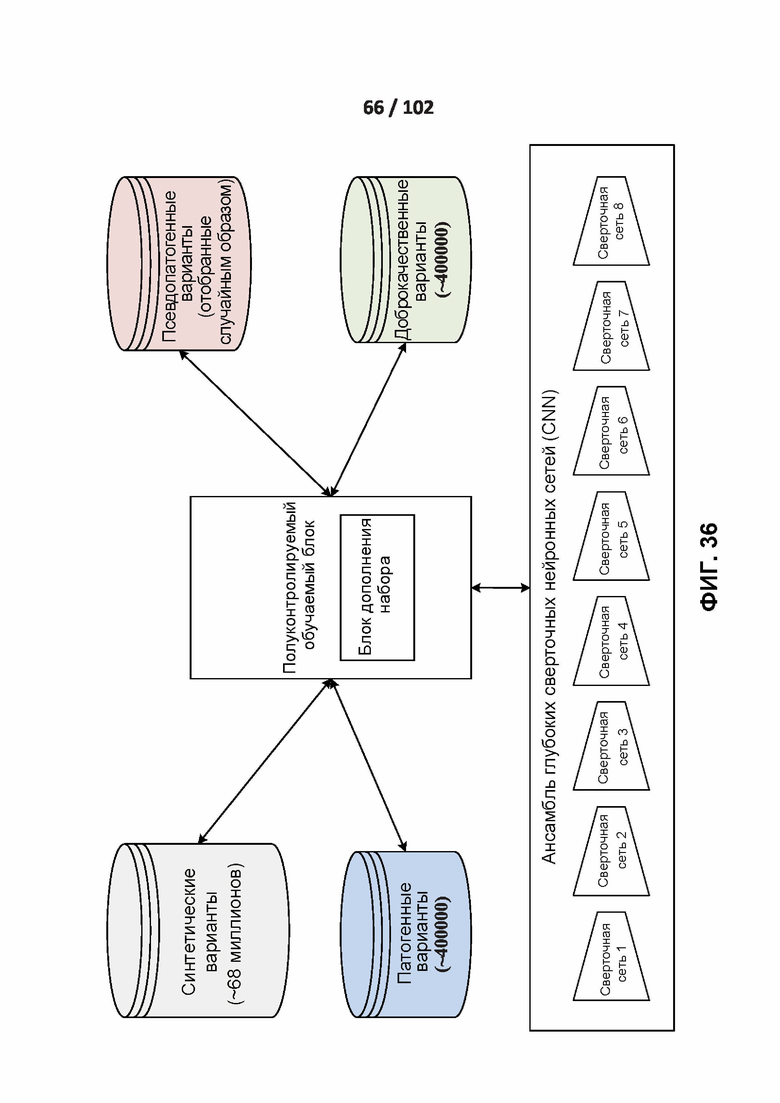


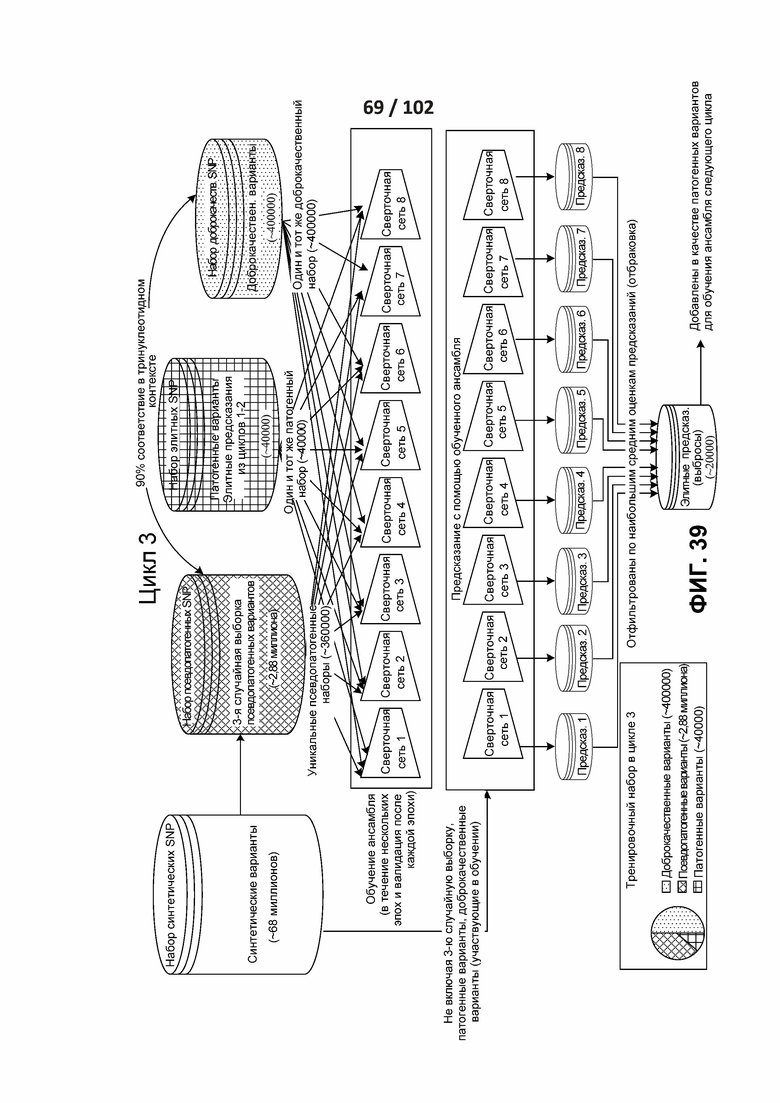
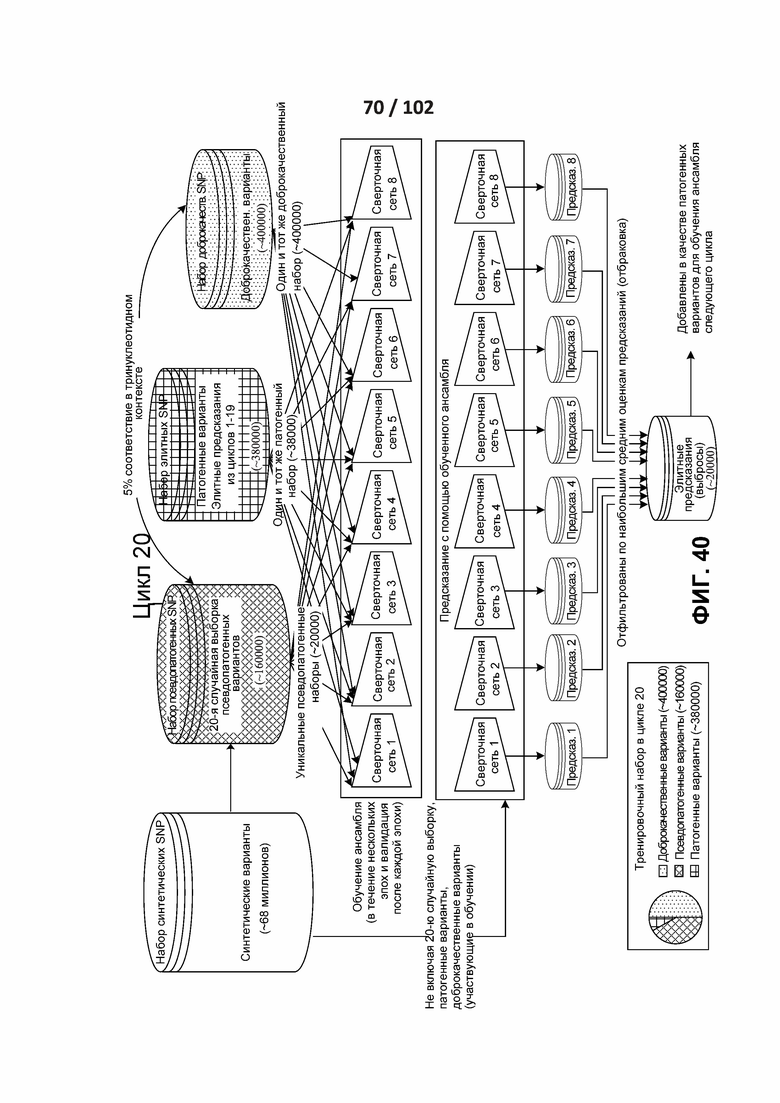
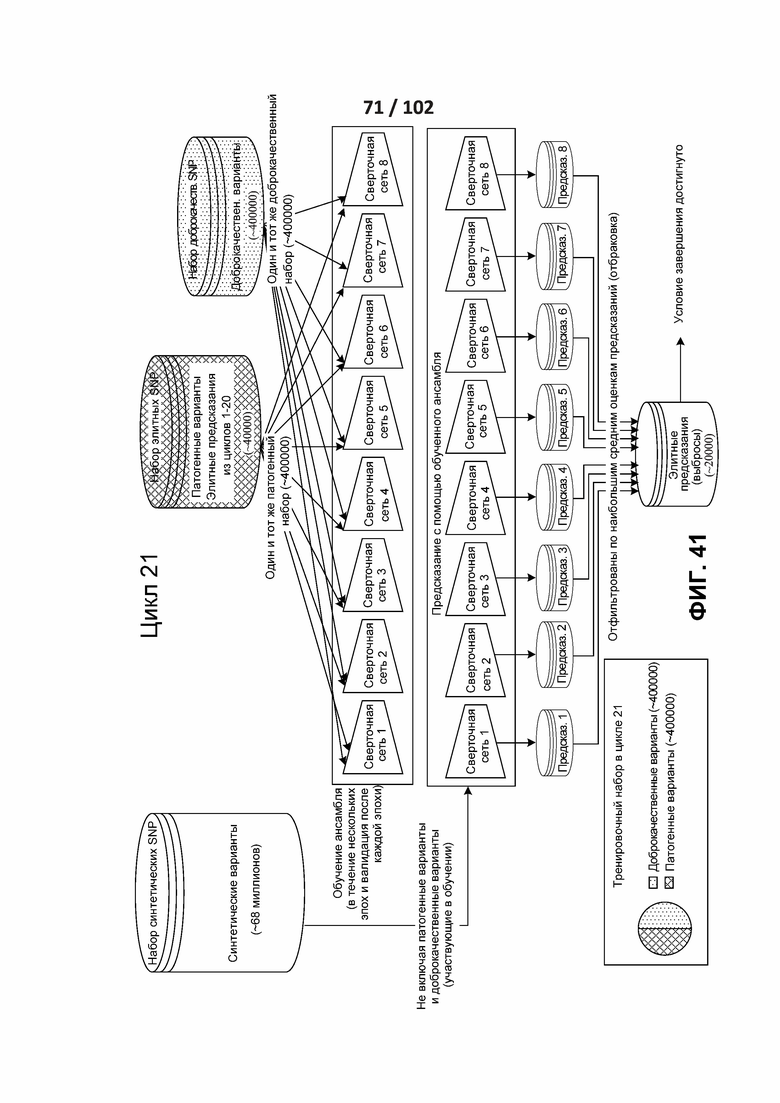
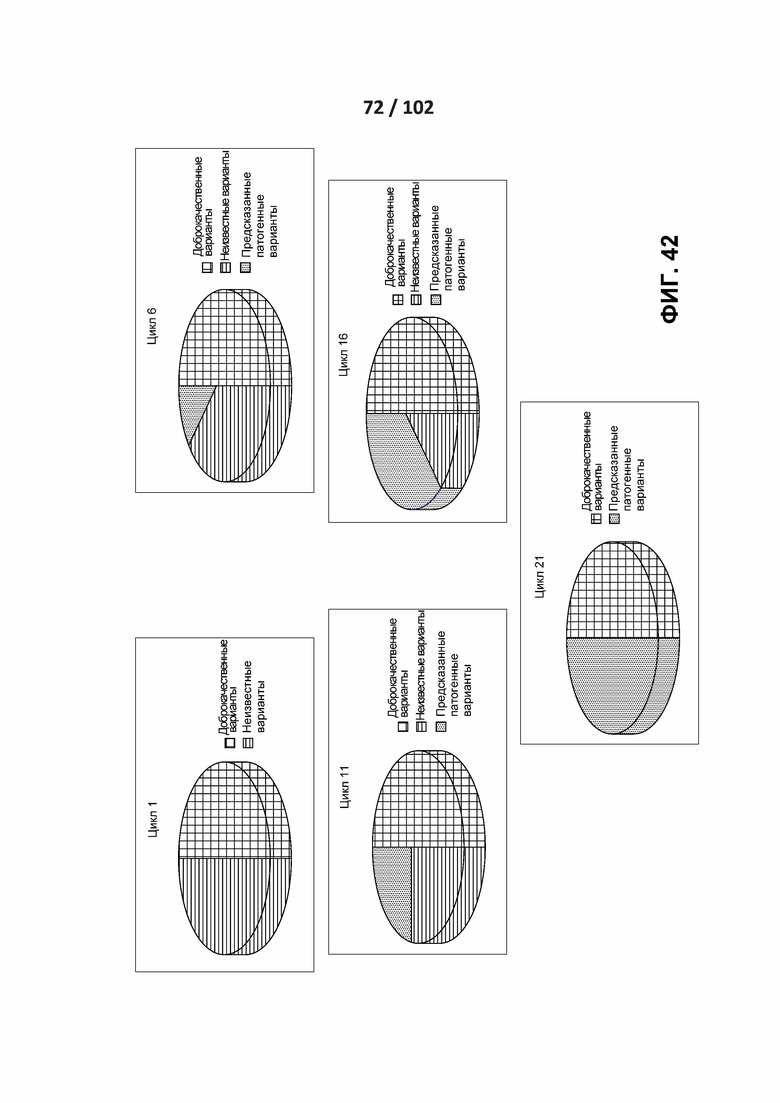
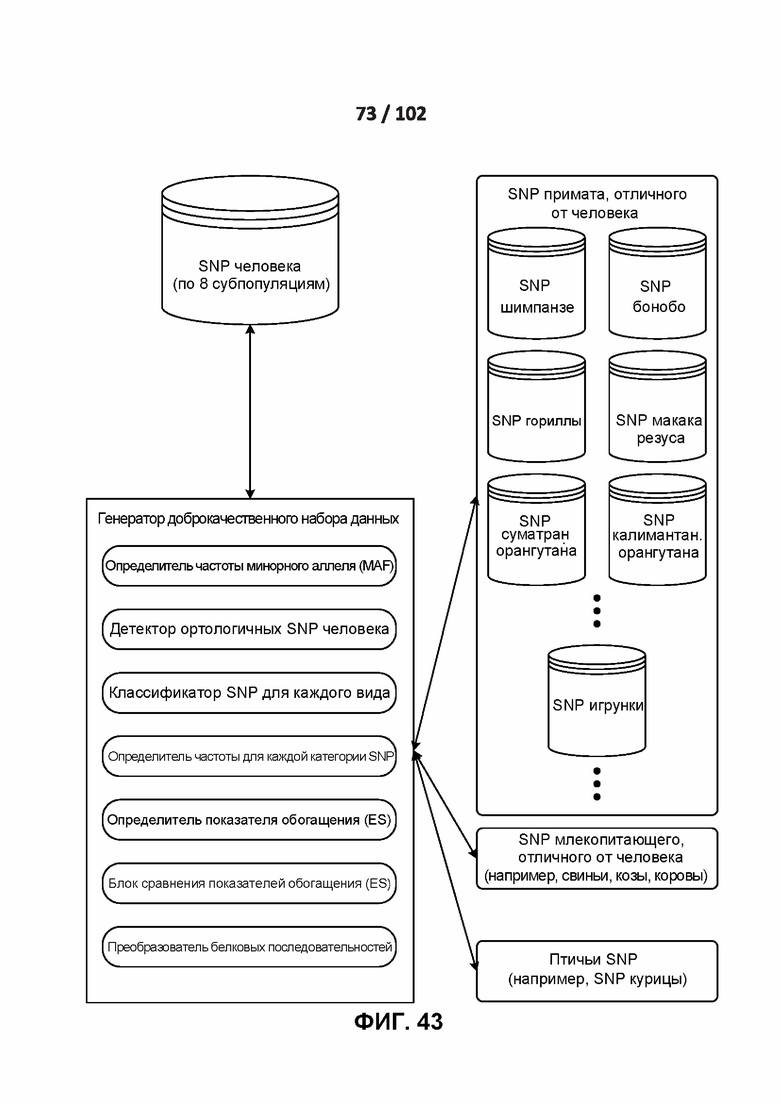
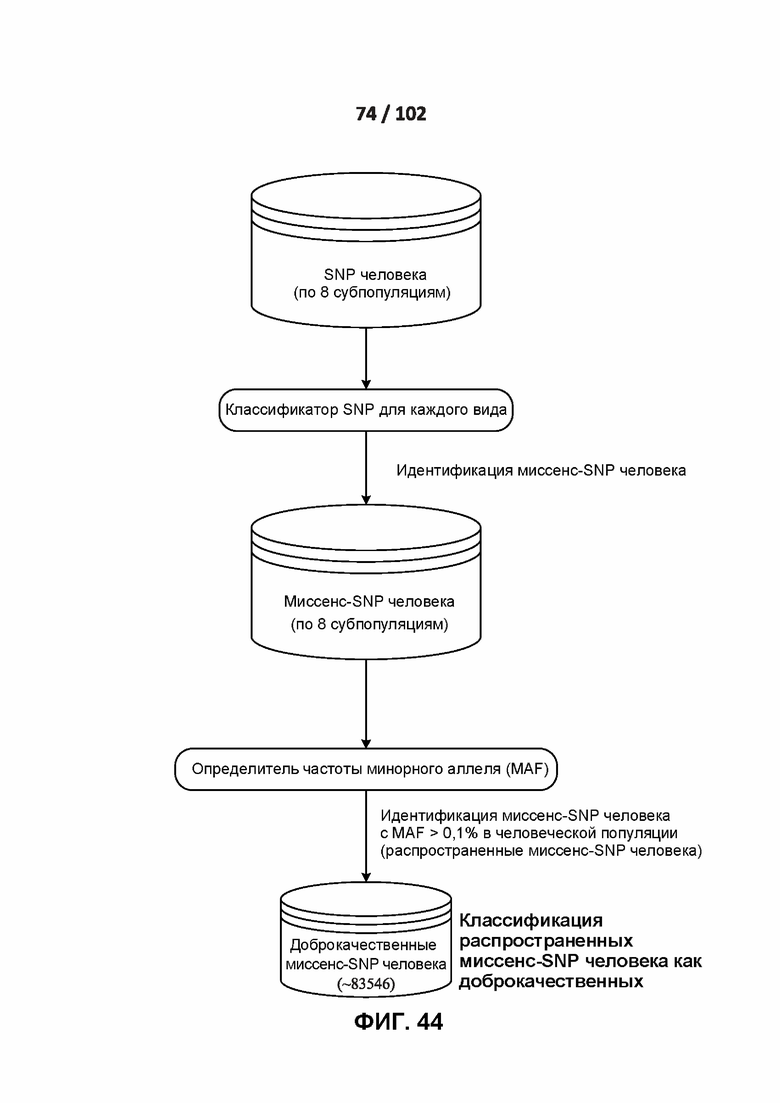

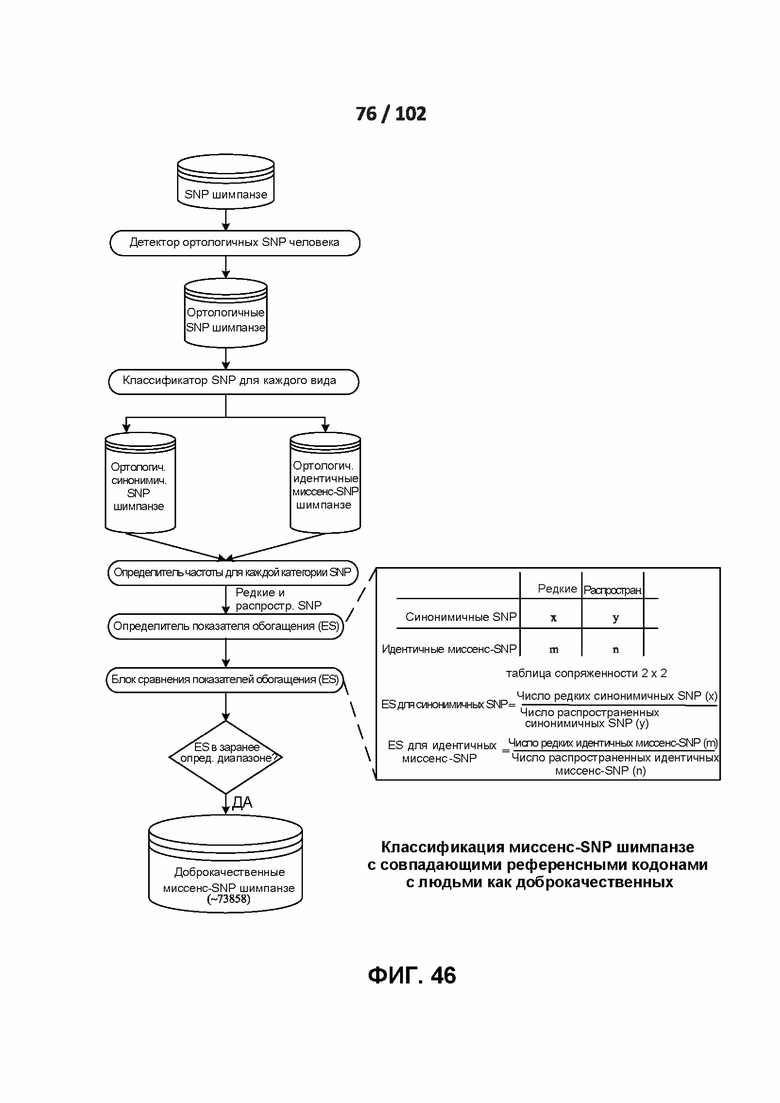

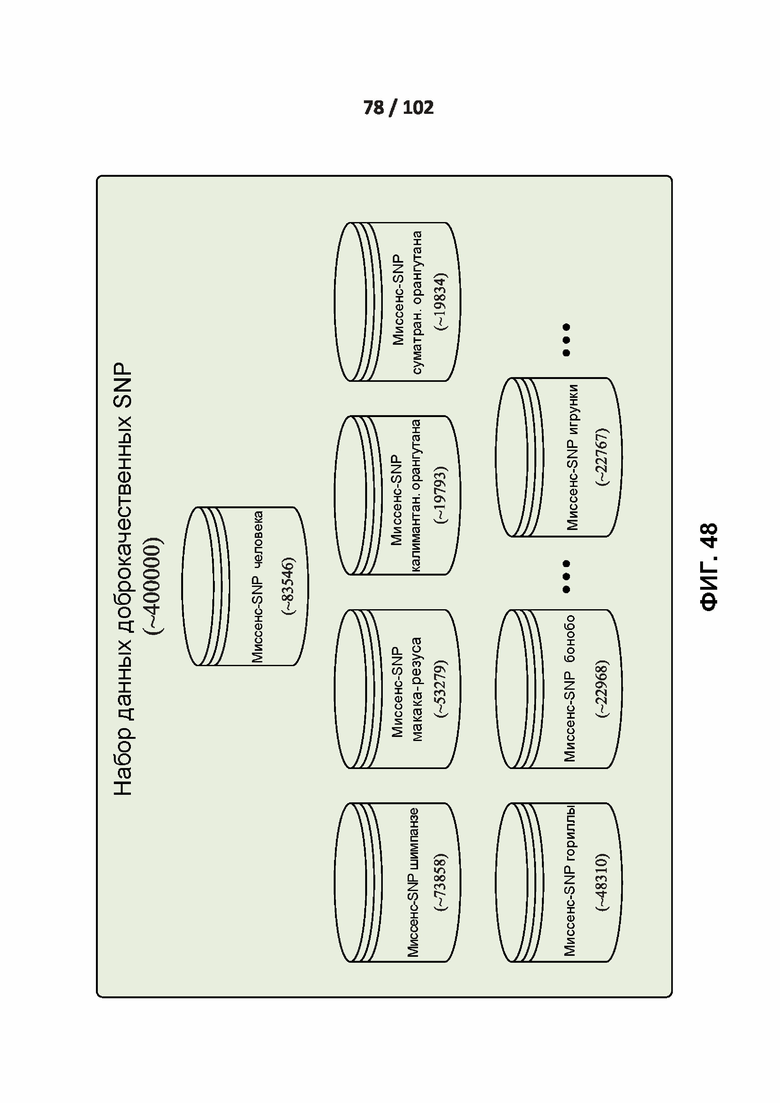
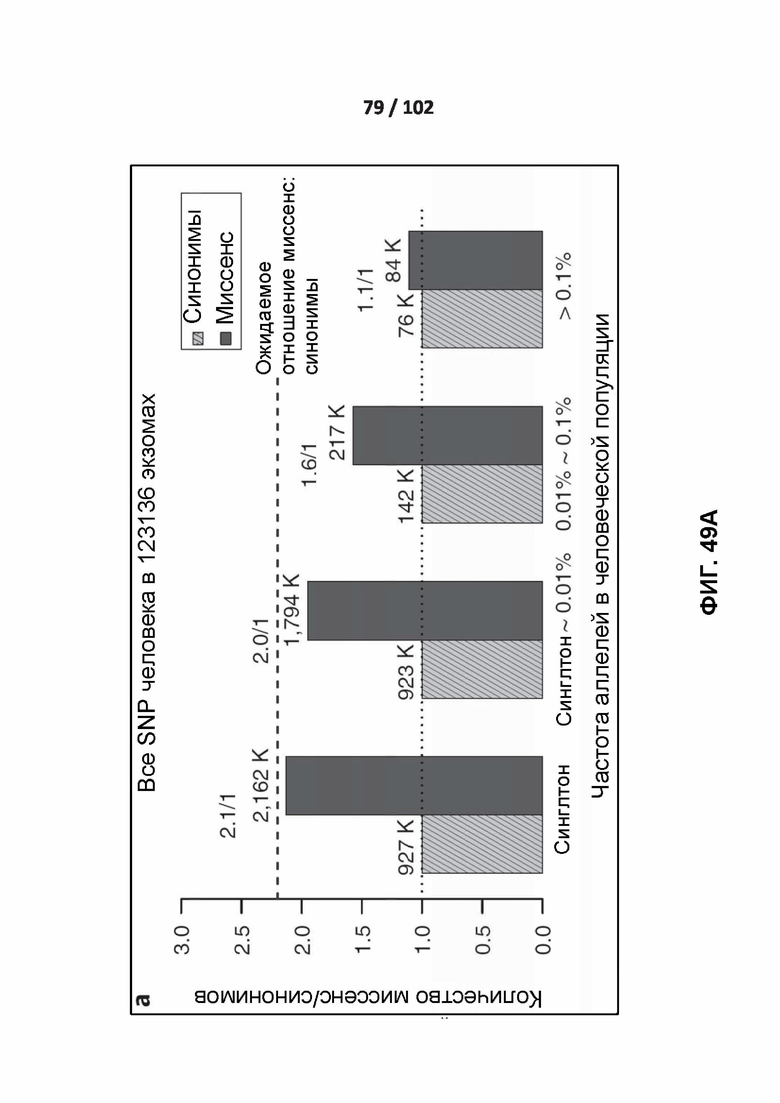

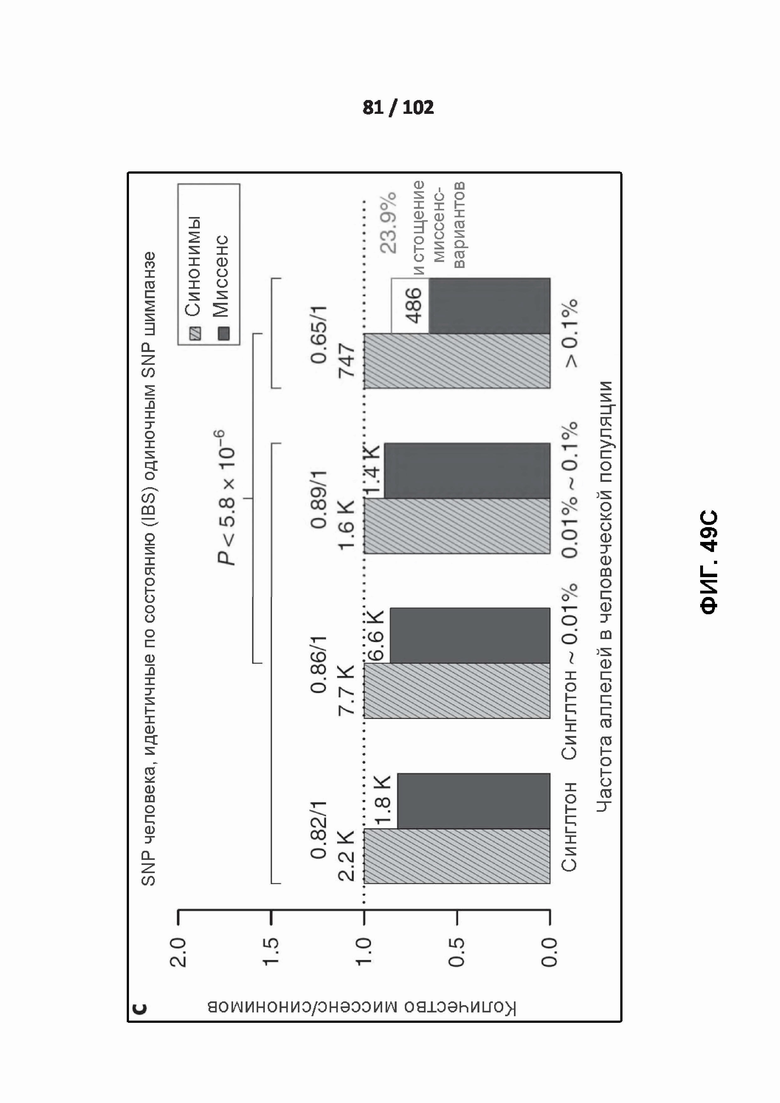
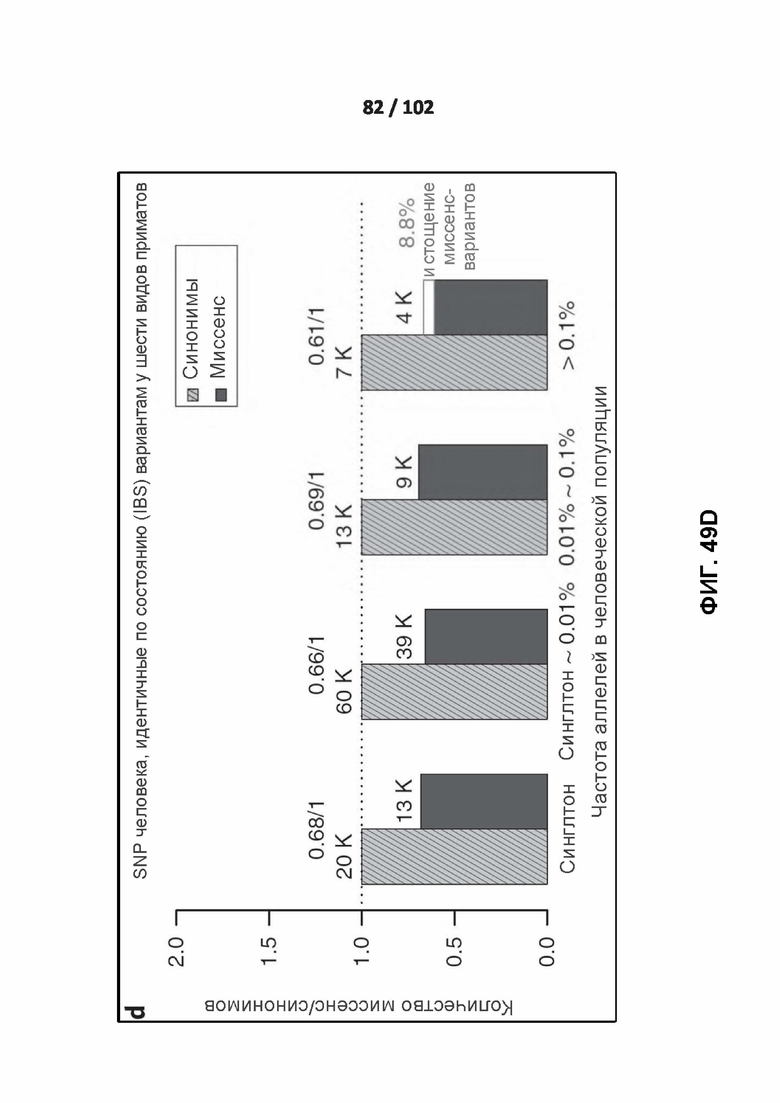
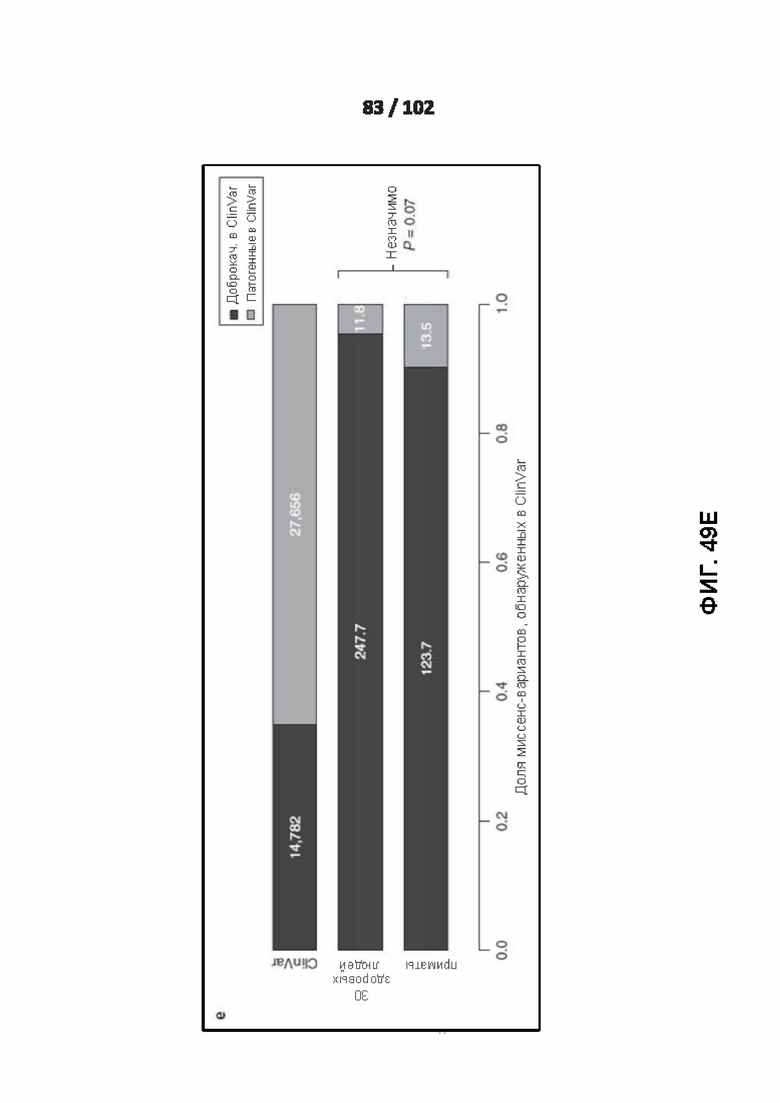
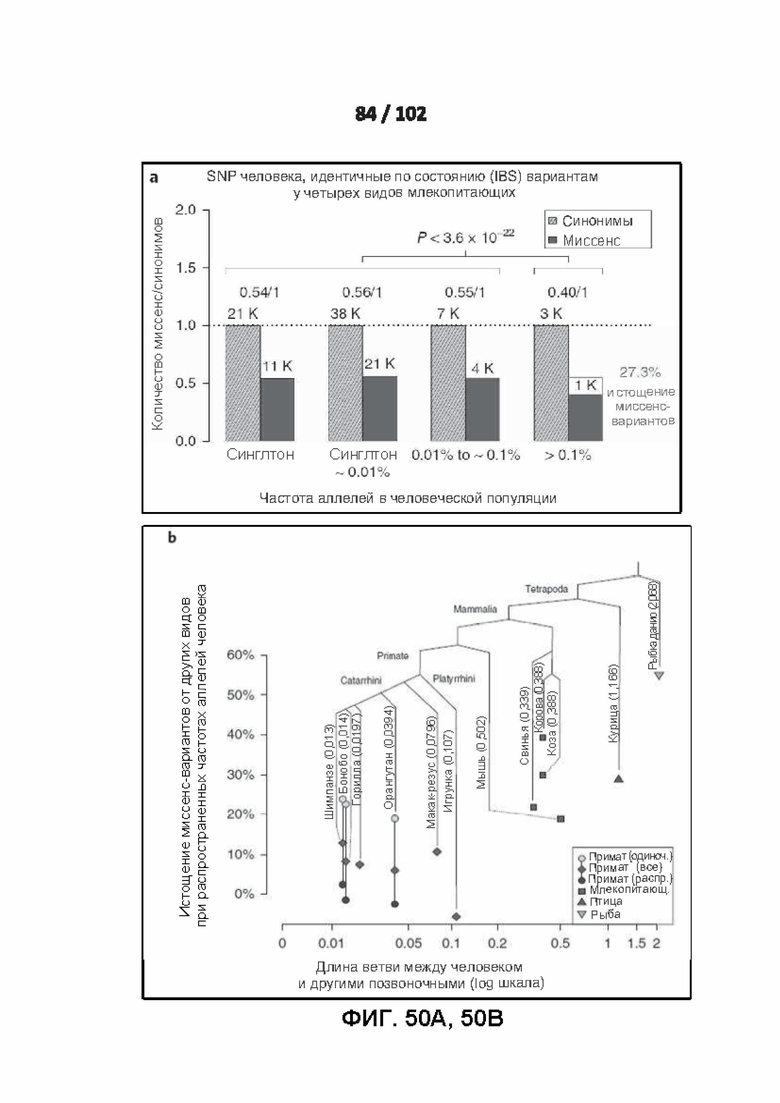
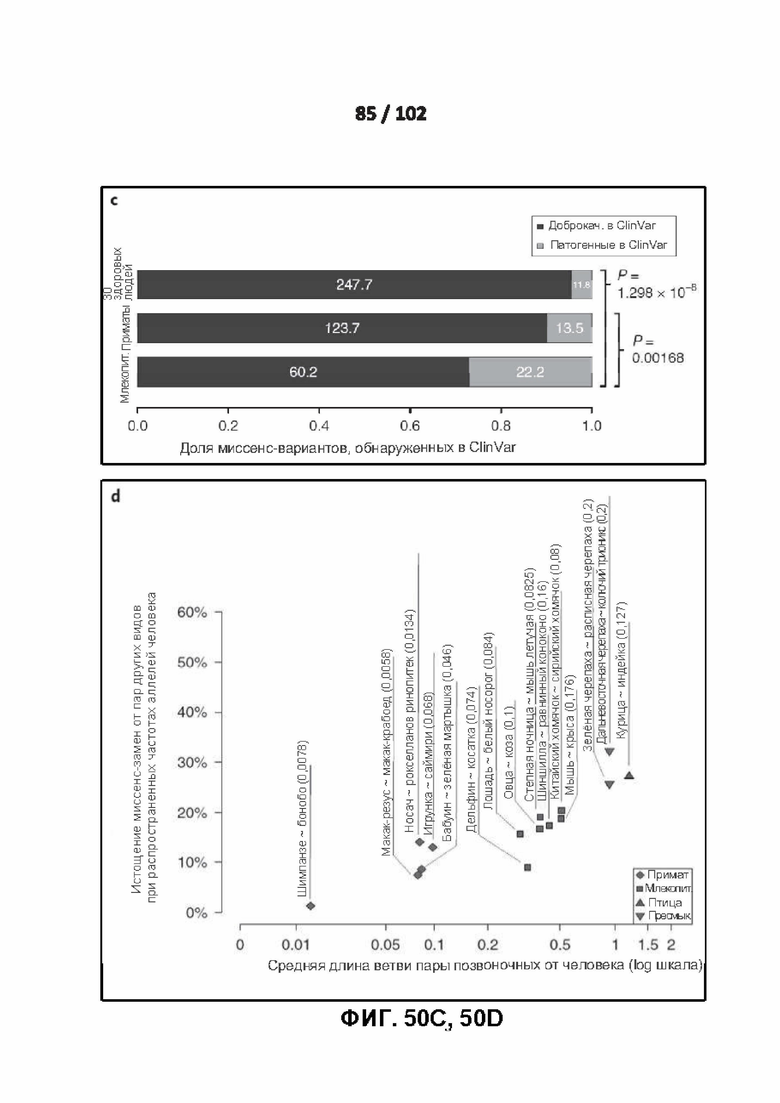


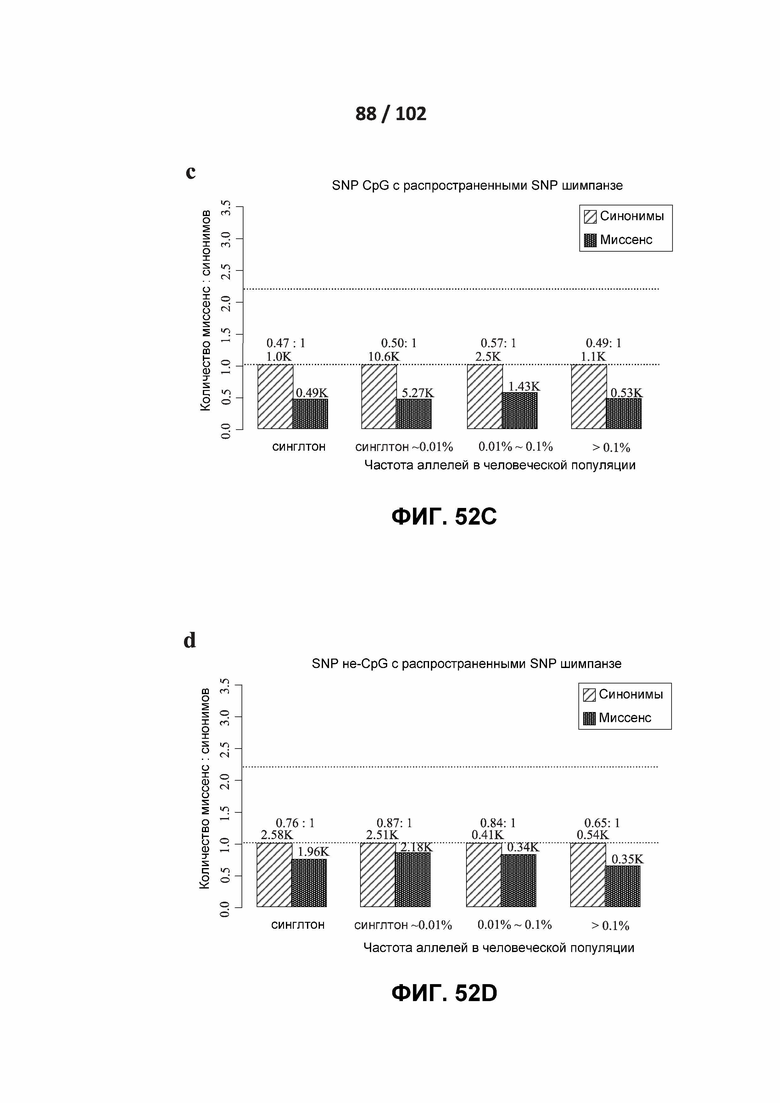
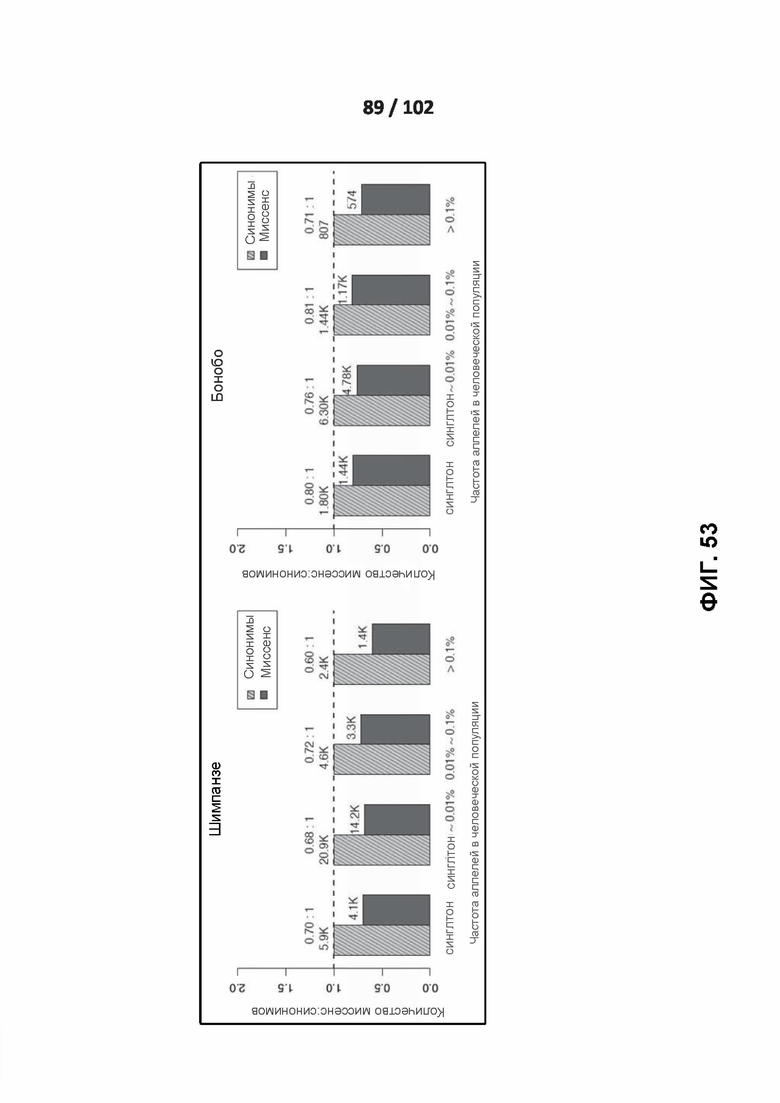
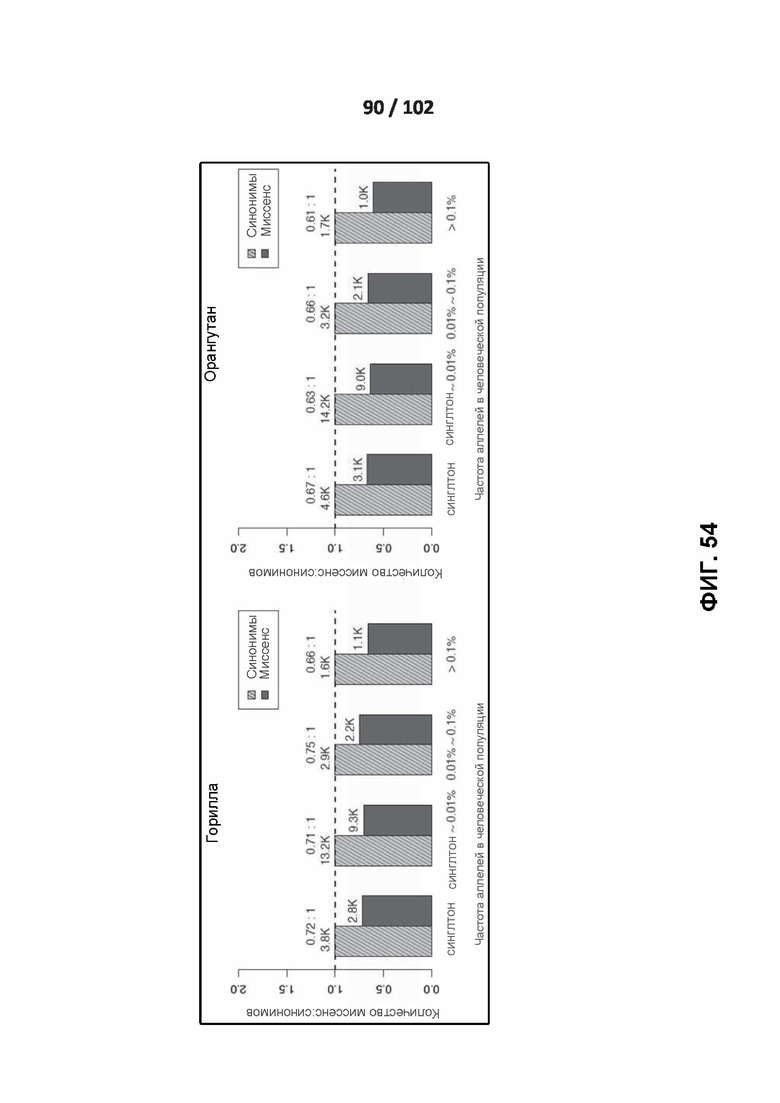
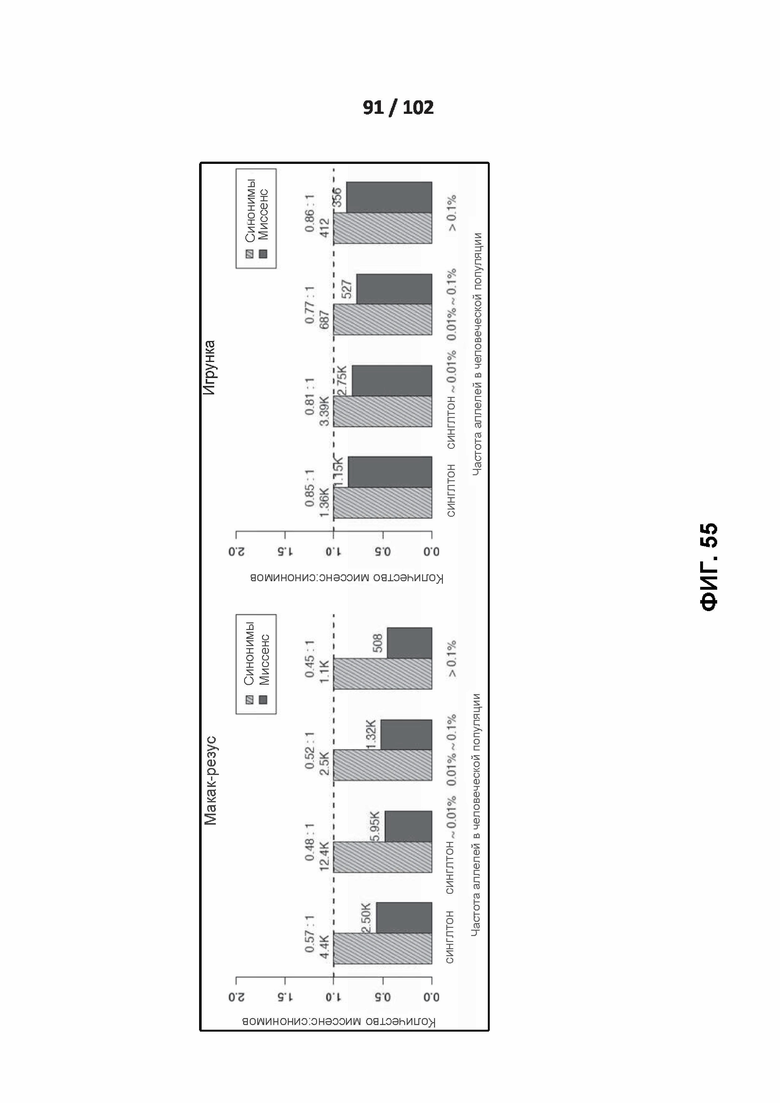
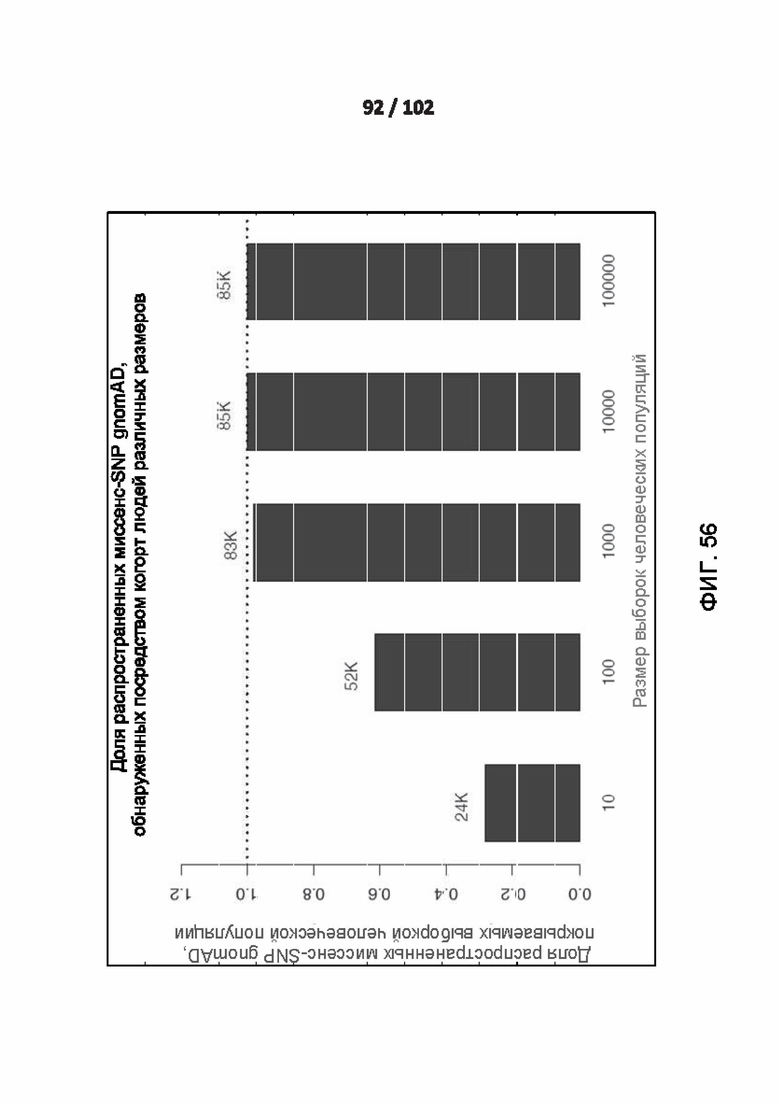
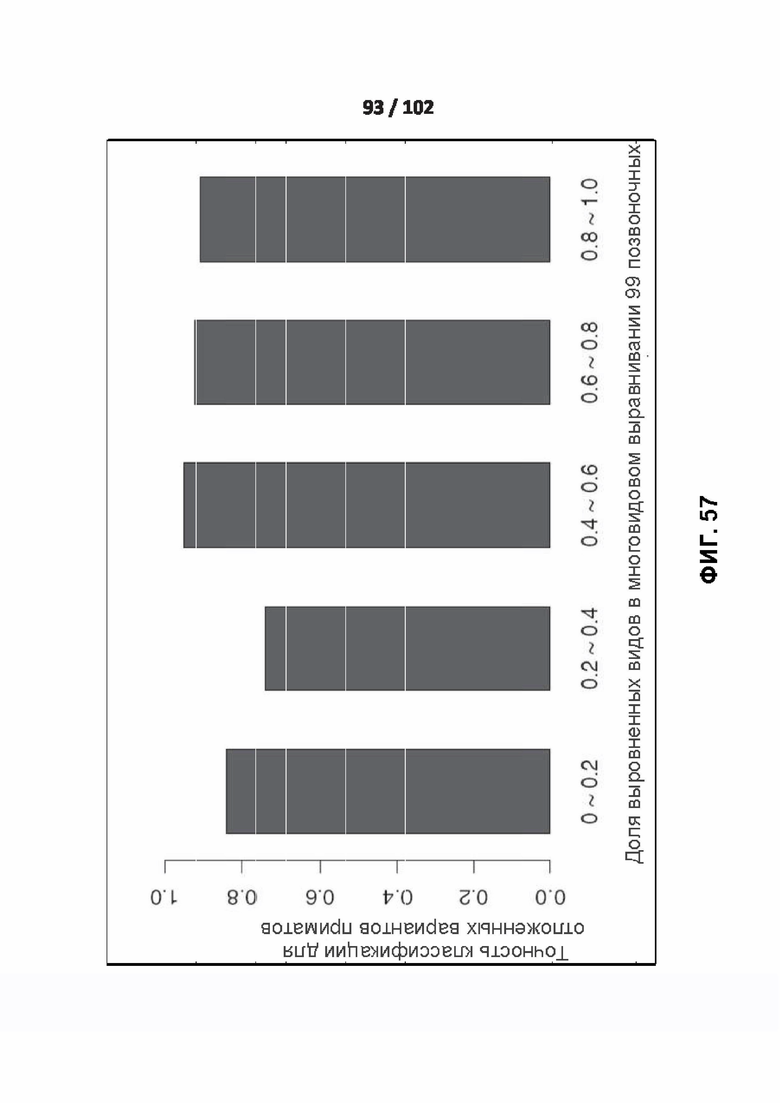
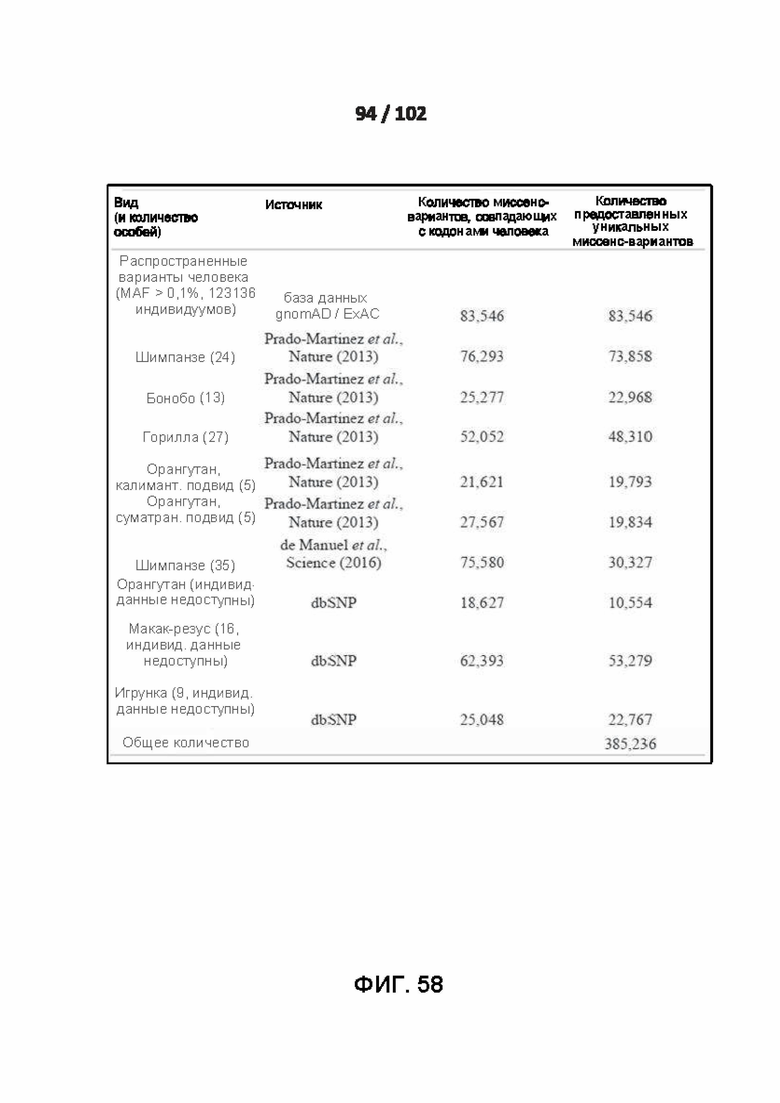
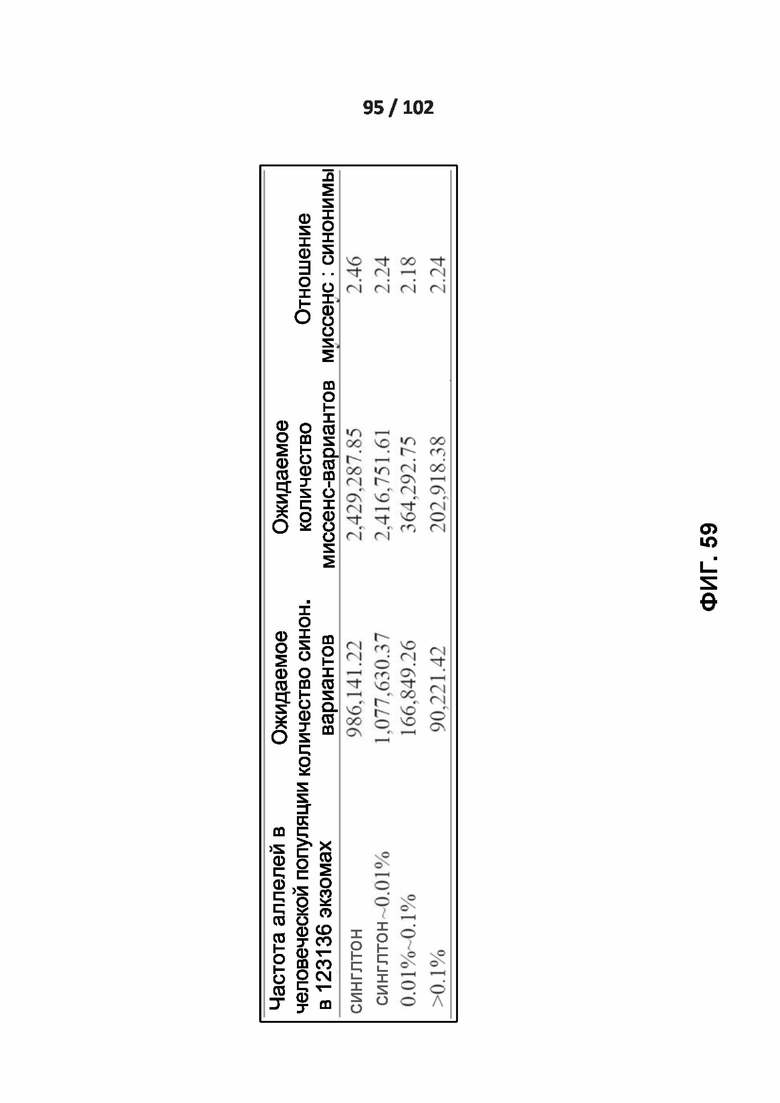



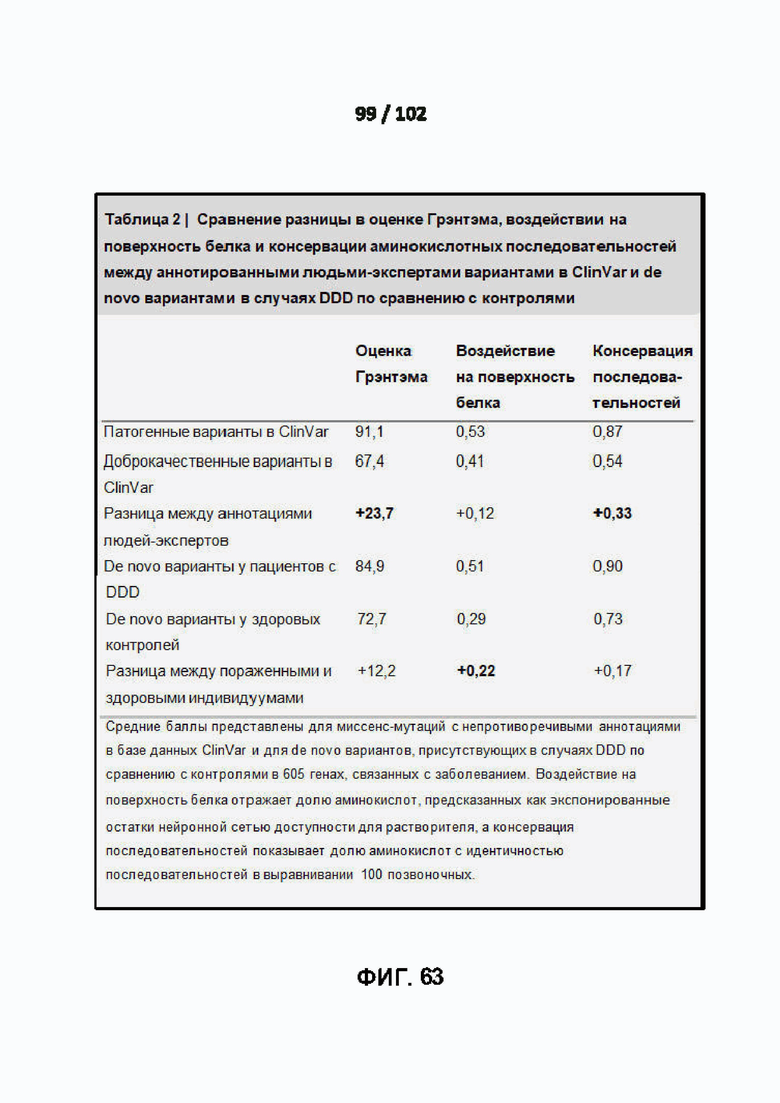
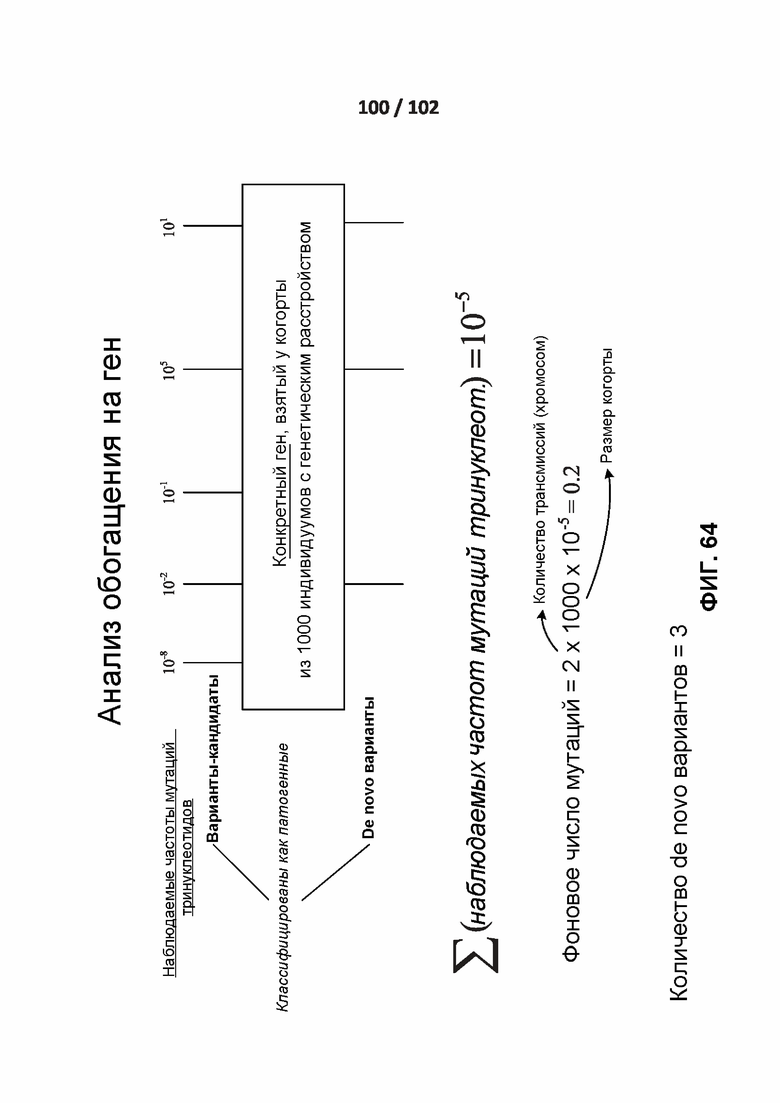
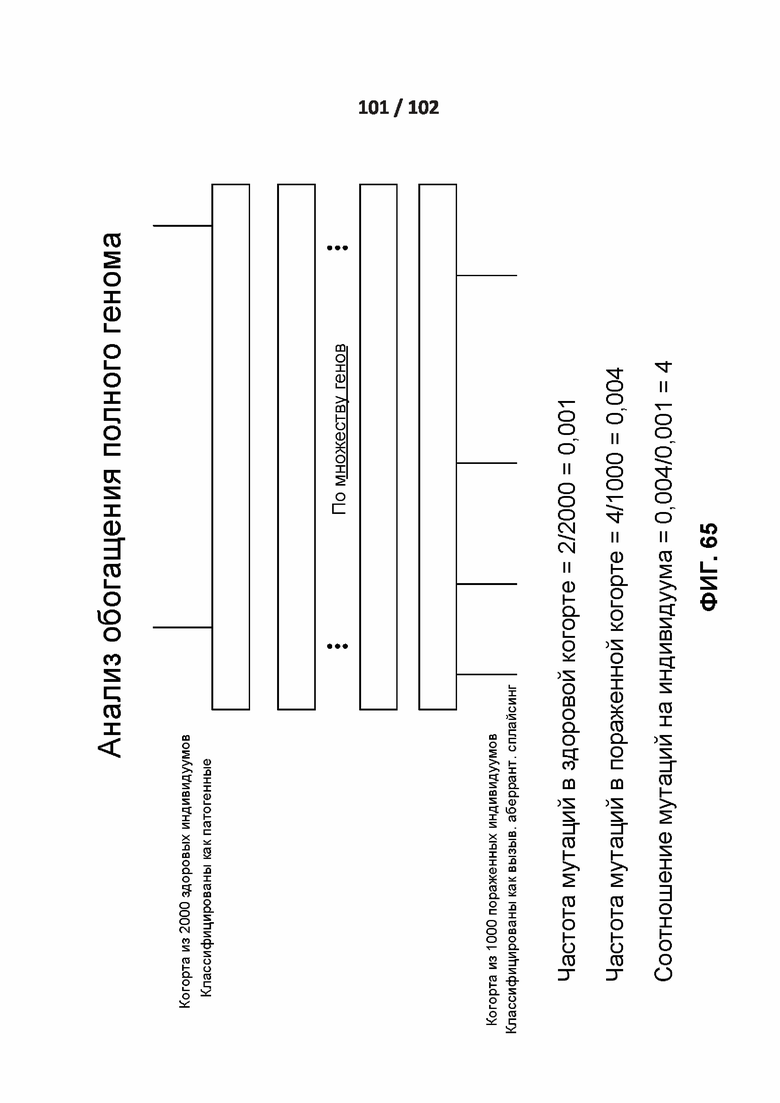

Изобретение относится к способу построения классификатора патогенности вариантов. А также к способу построения классификатора на основе сверточной нейронной сети для классификации вариантов, реализуемому при помощи компьютера, компьютерочитаемым носителям долговременного хранения информации и системам, включающим один или несколько процессоров, связанных с памятью. 6 н. и 17 з.п. ф-лы, 1 пр., 66 ил., 8 табл.
1. Способ построения классификатора патогенности вариантов, включающий
обучение классификатора патогенности вариантов на основе сверточной нейронной сети, который работает на множестве процессоров, связанных с памятью, с применением в качестве входа пар доброкачественных обучающих примеров и пар патогенных обучающих примеров референсных последовательностей белков и альтернативных последовательностей белков, причем альтернативные последовательности белков сгенерированы по доброкачественным вариантам и патогенным вариантам; и
при этом указанные доброкачественные варианты включают распространенные миссенс-варианты человека и миссенс-варианты примата, отличного от человека, встречающиеся в альтернативных последовательностях кодонов примата, отличного от человека, которые обладают совпадающими референсными последовательностями кодонов с людьми.
2. Способ по п. 1, где указанные распространенные миссенс-варианты человека имеют частоту минорного аллеля (MAF) выше 0.1% в наборе данных по вариантам в человеческой популяции, полученным по образцам от по меньшей мере 100000 человек.
3. Способ по п. 2, где люди, у которых взяты образцы, принадлежат к различным субпопуляциям людей и указанные распространенные миссенс-варианты человека имеют MAF выше 0.1% в соответствующих наборах данных по вариантам в соответствующей субпопуляции людей.
4. Способ по п. 3, где указанные субпопуляции людей включают африканцев/афро-американцев (AFR), американцев (AMR), евреев - ашкеназов (ASJ), восточных азиатов (EAS), финнов (FIN), европейцев, отличных от финнов (NFE), южных азиатов (SAS) и других (OTH).
5. Способ по п. 1, где указанные миссенс-варианты примата, отличного от человека, включают миссенс-варианты из множества видов приматов, отличных от человека, включая шимпанзе, бонобо, гориллу, калимантанского орангутана, суматранского орангутана, резусов и игрунок.
6. Способ по п. 1, дополнительно включающий, на основе анализа обогащения, прием конкретного вида приматов, отличного от человека, для включения миссенс-вариантов указанного конкретного вида приматов, отличных от человека, в доброкачественные варианты, причем указанный анализ обогащения включает, для указанного конкретного вида приматов, отличного от человека, сравнение первого показателя обогащения синонимичных вариантов указанного конкретного вида приматов, отличного от человека, со вторым показателем обогащения идентичных миссенс-вариантов указанного конкретного вида приматов, отличного от человека,
где идентичные миссенс-варианты представляют собой миссенс-варианты, обладающие совпадающими общими референсной и альтернативной последовательностями кодонов с людьми;
причем указанный первый показатель обогащения получают путем определения отношения редких синонимичных вариантов с MAF ниже 0.1% к распространенным синонимичным вариантам с MAF выше 0.1%; и
при этом указанный второй показатель обогащения получают путем определения отношения редких идентичных миссенс-вариантов с MAF ниже 0.1% к распространенным идентичным миссенс-вариантам с MAF выше 0.1%.
7. Способ по п. 6, где редкие синонимичные варианты включают единичные варианты.
8. Способ по п. 6, где разница между указанным первым показателем обогащения и указанным вторым показателем обогащения укладывается в заранее определенный диапазон, дополнительно включающий прием указанного конкретного вида примата, отличного от человека, для включения миссенс-вариантов указанного конкретного примата, отличного от человека, в доброкачественные варианты.
9. Способ по п. 6, где разница, укладывающаяся в заранее определенный диапазон, указывает на то, что указанные идентичные миссенс-варианты подвергаются естественному отбору в той же степени, что и синонимичные варианты, и, следовательно, являются такими же доброкачественными, как синонимичные варианты.
10. Способ по п. 6, дополнительно включающий повторное применение анализа обогащения с приемом множества видов приматов, отличных от человека, для включения миссенс-вариантов указанных видов приматов, отличных от человека, в число доброкачественных вариантов.
11. Способ по п. 1, дополнительно включающий применение критерия однородности хи-квадрат для сравнения первого показателя обогащения синонимичных вариантов и второго показателя обогащения идентичных миссенс-вариантов для каждого из указанных видов приматов, отличных от человека.
12. Способ по п. 1, где число миссенс-вариантов примата, отличного от человека, составляет по меньшей мере 100000.
13. Способ по п. 12, где число миссенс-вариантов примата, отличного от человека, составляет 385236.
14. Способ по п. 1, где число распространенных миссенс-вариантов человека составляет по меньшей мере 50000.
15. Способ по п. 14, где число распространенных миссенс-вариантов человека составляет 83546.
16. Компьютерочитаемый носитель долговременного хранения информации, на который нанесены компьютерные программные инструкции по построению классификатора патогенности вариантов, которые при исполнении процессором реализуют способ, включающий
обучение классификатора патогенности вариантов на основе сверточной нейронной сети, который работает на множестве процессоров, связанных с памятью, с применением в качестве входа пар доброкачественных обучающих примеров и пар патогенных обучающих примеров референсных последовательностей белков и альтернативных последовательностей белков, причем альтернативные последовательности белков сгенерированы по доброкачественным вариантам и патогенным вариантам; и
причем указанные доброкачественные варианты включают распространенные миссенс-варианты человека и миссенс-варианты примата, отличного от человека, встречающиеся в альтернативных последовательностях кодонов примата, отличного от человека, которые обладают совпадающими референсными последовательностями кодонов с людьми.
17. Компьютерочитаемый носитель долговременного хранения информации по п. 16, реализующий способ, дополнительно включающий, на основе анализа обогащения, прием конкретного вида приматов, отличного от человека, для включения миссенс-вариантов указанного конкретного вида приматов, отличных от человека, в доброкачественные варианты, причем указанный анализ обогащения включает, для указанного конкретного вида приматов, отличного от человека, сравнение первого показателя обогащения синонимичных вариантов указанного конкретного вида приматов, отличного от человека, со вторым показателем обогащения идентичных миссенс-вариантов указанного конкретного вида приматов, отличного от человека,
где идентичные миссенс-варианты представляют собой миссенс-варианты, обладающие совпадающими общими референсной и альтернативной последовательностями кодонов с людьми;
причем указанный первый показатель обогащения получают путем определения отношения редких синонимичных вариантов с MAF ниже 0.1% к распространенным синонимичным вариантам с MAF выше 0.1%; и
при этом указанный второй показатель обогащения получают путем определения отношения редких идентичных миссенс-вариантов с MAF ниже 0.1% к распространенным идентичным миссенс-вариантам с MAF выше 0.1%.
18. Компьютерочитаемый носитель долговременного хранения информации по п. 16, реализующий способ, дополнительно включающий применение критерия однородности хи-квадрат для сравнения первого показателя обогащения синонимичных вариантов и второго показателя обогащения идентичных миссенс-вариантов для каждого из указанных видов приматов, отличных от человека.
19. Система, включающая один или несколько процессоров, связанных с памятью, где в указанную память загружены компьютерные инструкции по построению классификатора патогенности вариантов, причем указанные инструкции при исполнении указанными процессорами реализуют операции, включающие:
обучение классификатора патогенности вариантов на основе сверточной нейронной сети, который работает на множестве процессоров, связанных с памятью, с применением в качестве входа пар доброкачественных обучающих примеров и пар патогенных обучающих примеров референсных последовательностей белков и альтернативных последовательностей белков, причем альтернативные последовательности белков сгенерированы по доброкачественным вариантам и патогенным вариантам; и
при этом указанные доброкачественные варианты включают распространенные миссенс-варианты человека и миссенс-варианты примата, отличного от человека, встречающиеся в альтернативных последовательностях кодонов примата, отличного от человека, которые обладают совпадающими референсными последовательностями кодонов с людьми.
20. Система по п. 19, дополнительно реализующая следующие действия: на основе анализа обогащения, прием конкретного вида приматов, отличного от человека, для включения миссенс-вариантов указанного конкретного вида приматов, отличных от человека, в доброкачественные варианты, причем указанный анализ обогащения включает, для указанного конкретного вида приматов, отличного от человека, сравнение первого показателя обогащения синонимичных вариантов указанного конкретного вида приматов, отличного от человека, со вторым показателем обогащения идентичных миссенс-вариантов указанного конкретного вида приматов, отличного от человека,
где идентичные миссенс-варианты представляют собой миссенс-варианты, обладающие совпадающими общими референсной и альтернативной последовательностями кодонов с людьми;
причем указанный первый показатель обогащения получают путем определения отношения редких синонимичных вариантов с MAF ниже 0.1% к распространенным синонимичным вариантам с MAF выше 0.1%; и
при этом указанный второй показатель обогащения получают путем определения отношения редких идентичных миссенс-вариантов с MAF ниже 0.1% к распространенным идентичным миссенс-вариантам с MAF выше 0.1%.
21. Реализуемый при помощи компьютера способ построения классификатора на основе сверточной нейронной сети для классификации вариантов, включающий
обучение классификатора на основе сверточной нейронной сети, который работает на множестве процессоров, связанных с памятью, на обучающих данных с применением технологии градиентного обновления на основе обратного распространения, которая последовательно сопоставляет выходные данные классификатора на основе сверточной нейронной сети с соответствующими метками истинных значений;
причем указанный классификатор на основе сверточной нейронной сети содержит группы остаточных блоков,
где каждая группа остаточных блоков параметризируется числом сверточных фильтров в остаточных блоках, размером окна свертки остаточных блоков и показателем разрежения свертки остаточных блоков;
причем указанные размеры окна свертки в разных группах остаточных блоков различаются;
причем показатели разрежения свертки в разных группах остаточных блоков различаются;
при этом указанные обучающие данные включают пары транслируемых последовательностей, сгенерированные по доброкачественным вариантам, и применяются в качестве доброкачественных обучающих примеров и в качестве патогенных обучающих примеров; и
при этом доброкачественные варианты включают распространенные миссенс-варианты человека и миссенс-варианты примата, отличного от человека, встречающиеся в альтернативных последовательностях триплетов оснований, которые обладают совпадающими референсными последовательностями типлетов оснований с людьми.
22. Компьютерочитаемый носитель долговременного хранения информации, на который нанесены компьютерные программные инструкции по построению классификатора на основе сверточной нейронной сети для классификации вариантов, причем указанные инструкции при исполнении процессором реализуют способ, включающий
обучение классификатора на основе сверточной нейронной сети, который работает на множестве процессоров, связанных с памятью, на обучающих данных с применением технологии градиентного обновления на основе обратного распространения, которая последовательно сопоставляет выходные данные классификатора на основе сверточной нейронной сети с соответствующими метками истинных значений;
причем указанный классификатор на основе сверточной нейронной сети содержит группы остаточных блоков,
где каждая группа остаточных блоков параметризируется числом сверточных фильтров в остаточных блоках, размером окна свертки остаточных блоков и показателем разрежения свертки остаточных блоков;
причем указанные размеры окна свертки в разных группах остаточных блоков различаются;
причем показатели разрежения свертки в разных группах остаточных блоков различаются;
при этом указанные обучающие данные включают пары транслируемых последовательностей, сгенерированные по доброкачественным вариантам и патогенным вариантам, и применяются в качестве доброкачественных обучающих примеров и в качестве патогенных обучающих примеров; и
при этом доброкачественные варианты включают распространенные миссенс-варианты человека и миссенс-варианты примата, отличного от человека, встречающиеся в альтернативных последовательностях триплетов, которые обладают совпадающими референсными последовательностями типлетов оснований с людьми.
23. Система, включающая один или несколько процессоров, связанных с памятью, где в указанную память загружены компьютерные инструкции по построению классификатора на основе сверточной нейронной сети для классификации вариантов, причем указанные инструкции при исполнении указанными процессорами реализуют операции, включающие
обучение классификатора на основе сверточной нейронной сети, который работает на множестве процессоров, связанных с памятью, на обучающих данных с применением технологии градиентного обновления на основе обратного распространения, которая последовательно сопоставляет выходные данные классификатора на основе сверточной нейронной сети с соответствующими метками истинных значений;
причем указанный классификатор на основе сверточной нейронной сети содержит группы остаточных блоков,
где каждая группа остаточных блоков параметризируется числом сверточных фильтров в остаточных блоках, размером окна свертки остаточных блоков и показателем разрежения свертки остаточных блоков;
причем указанные размеры окна свертки в разных группах остаточных блоков различаются;
причем показатели разрежения свертки в разных группах остаточных блоков различаются;
при этом указанные обучающие данные включают пары транслируемых последовательностей, сгенерированные по доброкачественным вариантам и патогенным вариантам, и применяются в качестве доброкачественных обучающих примеров и в качестве патогенных обучающих примеров; и
при этом доброкачественные варианты включают распространенные миссенс-варианты человека и миссенс-варианты примата, отличного от человека, встречающиеся в альтернативных последовательностях триплетов, которые обладают совпадающими референсными последовательностями типлетов оснований с людьми.
| CA 2894317 A1, 15.12.2016 | |||
| WEI Q | |||
| et al., The Role of Balanced Training and Testing Data Sets for Binary Classifiers in Bioinformatics // PLOS ONE, 2013, vol.8, no.7, p.e67863 | |||
| IOANNIDIS N.M | |||
| et al., REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants // AMERICAN JOURNAL OF HUMAN GENETICS, AMERICAN SOCIETY OF |

