ПРИЛОЖЕНИЕ
[0001] Приложение включает библиографию потенциально релевантных источников, перечисленных в статье, написанной авторами настоящего изобретения. Объект этой статьи покрывают предварительные заявки США, на основании которых испрашивается приоритет по этой заявке. Доступ к этим заявкам можно получить по запросу у юрисконсульта или через систему Global Dossier.
ПРИОРИТЕТНЫЕ ЗАЯВКИ
[0002] Настоящая заявка испрашивает приоритет или преимущество предварительной заявки на патент США No. 62/573,125, озаглавленной "Deep Learning-Based Splice Site Classification"(''Классификация сайтов сплайсинга на основе глубокого обучения"), на имя Kishore Jaganathan, Kai-How Farh, Sofia Kyriazopoulou Panagiotopoulou и Jeremy Francis McRae, поданной 16 октября 2017 г. (№ дела поверенного ILLM 1001- l/IP-1610-PRV), предварительной заявки на патент США №62/573,131, озаглавленной "Deep Learning-Based Aberrant Splicing Detection" ("определение аберрантных сайтов сплайсинга"), на имя Kishore Jaganathan, Kai-How Farh, Sofia Kyriazopoulou Panagiotopoulou и Jeremy Francis McRae, поданной 16 октября 2017 г. (№ дела поверенного ILLM 1001-2/IP-1614-PRV), предварительной заявки на патент США №62/573,135, озаглавленной "Aberrant Splicing Detection Using Convolutional Neural Networks (CNNs)" («Детектирование аберрантного сплайсинга с применением сверточных нейронных сетей (CNN, СНС)») («Детектирование аберрантного сплайсинга с применением сверточных нейронных сетей (CNN, СНС)»), на имя Kishore Jaganathan, Kai-How Farh, Sofia Kyriazopoulou Panagiotopoulou и Jeremy Francis McRae, поданной 16 октября 2017 г. (№ дела поверенного ILLM 1001-3/IP-1615-PRV); и предварительной заявки на патент США № 62/726,158, озаглавленной "Predicting Splicing from Primary Sequence with Deep Learning"("Предсказание сплайсинга по первичной последовательности при помощи глубокого обучения"), на имя Kishore Jaganathan, Kai-How Farh, Sofia Kyriazopoulou Panagiotopoulou и Jeremy Francis McRae, поданной 31 августа 2018 г. (№ дела поверенного ILLM 1001-10/IP-1749-PRV). Указанные предварительные заявки включены в настоящий документ посредством ссылки для всех целей.
ВКЛЮЧЕНИЕ
[0003] Следующие документы полностью включены в настоящий текст посредством ссылки так как если бы они были приведены здесь полностью:
[0004] Патентная заявка РСТ №PCT/US18/ озаглавленная "Deep Learning-Based Aberrant Splicing Detection" ("определение аберрантных сайтов сплайсинга"), на имя Kishore Jaganathan, Kai-How Farh, Sofia Kyriazopoulou Panagiotopoulou и Jeremy Francis McRae, поданная одновременно 15 октября 2018 г. (№ дела поверенного ILLM 1001-8ЛР-1614-РСТ), позже опубликованная как публикация РСТ № WO
озаглавленная "Deep Learning-Based Aberrant Splicing Detection" ("определение аберрантных сайтов сплайсинга"), на имя Kishore Jaganathan, Kai-How Farh, Sofia Kyriazopoulou Panagiotopoulou и Jeremy Francis McRae, поданная одновременно 15 октября 2018 г. (№ дела поверенного ILLM 1001-8ЛР-1614-РСТ), позже опубликованная как публикация РСТ № WO
[0005] Патентная заявка РСТ № PCT/US18/ озаглавленная "Aberrant Splicing
озаглавленная "Aberrant Splicing
Detection Using Convolutional Neural Networks (CNNs)" («Детектирование аберрантного сплайсинга с применением сверточных нейронных сетей (CNN, СНС)»), на имя Kishore Jaganathan, Kai-How Farh, Sofia Kyriazopoulou Panagiotopoulou и Jeremy Francis McRae, поданная одновременно 15 октября 2018 г. (№дела поверенного ILLM 1001-9/ТР-1615-РСТ), позже опубликованная как публикация РСТ №WO
[0006] Непредварительная патентная заявка на патент США, озаглавленная "Deep Learning-Based Splice Site Classification"("Классификация сайтов сплайсинга на основе глубокого обучения"), на имя Kishore Jaganathan, Kai-How Farh, Sofia Kyriazopoulou Panagiotopoulou и Jeremy Francis McRae (№ дела поверенного ILLM 1001-4/IP-1610-US), поданная одновременно.
[0007] Непредварительная патентная заявка на патент США, озаглавленная "Deep Learning-Based Aberrant Splicing Detection" ("Определение аберрантных сайтов сплайсинга"), на имя Kishore Jaganathan, Kai-How Farh, Sofia Kyriazopoulou Panagiotopoulou и Jeremy Francis McRae, (№дела поверенного ILLM 1001-5/IP-1614-US), поданная одновременно.
[0008] Непредварительная патентная заявка на патент США, озаглавленная "Aberrant Splicing Detection Using Convolutional Neural Networks (CNNs)" («Детектирование аберрантного сплайсинга с применением сверточных нейронных сетей (CNN, СНС)»), на имя Kishore Jaganathan, Kai-How Farh, Sofia Kyriazopoulou Panagiotopoulou и Jeremy Francis McRae, (№ дела поверенного ILLM 1001-6/IP-1615-US), поданная одновременно.
[0009] Документ 1 - S. Dieleman, Η. Zen, К. Simonyan, О. Vinyals, A. Graves, N. Kalchbrenner, Α. Senior, and К. Kavukcuoglu, "WAVENET: A GENERATIVE MODEL FOR RAW AUDIO", arXiv: 1609.03499, 2016;
[0010] Документ 2 - S.  M. Chrzanowski, A. Coates, G. Diamos, A. Gibiansky, Y. Kang, X. Li, J. Miller, A. Ng, J. Raiman, S. Sengupta, M. Shoeybi, "DEEP VOICE: REAL-TIME NEURAL TEXT-TO-SPEECH", arXiv: 1702.07825, 2017;
M. Chrzanowski, A. Coates, G. Diamos, A. Gibiansky, Y. Kang, X. Li, J. Miller, A. Ng, J. Raiman, S. Sengupta, M. Shoeybi, "DEEP VOICE: REAL-TIME NEURAL TEXT-TO-SPEECH", arXiv: 1702.07825, 2017;
[0011] Документ 3 - F. Yu, V. Koltun, "MULTI-SCALE CONTEXT AGGREGATION BY DILATED CONVOLUTIONS", arXiv: 1511.07122, 2016;
[0012] Документ 4 - K. He, X. Zhang, S. Ren, J. Sun, "DEEP RESIDUAL LEARNING FOR IMAGE RECOGNITION", arXiv: 1512.03385, 2015;
[0013] Документ 5 - R. K. Srivastava, K. Greff, J. Schmidhuber, "HIGHWAY NETWORKS", arXiv: 1505.00387, 2015;
[0014] Документ 6 G. Huang, Z. Liu, L. van der Maaten, K. Q. Weinberger, "DENSELY CONNECTED CONVOLUTIONAL NETWORKS", arXiv: 1608.06993, 2017;
[0015] Документ 7 - С. Szegedy, W. Liu, Y. Jia, P. Sermanet, S. Reed, D. Anguelov, D. Erhan, V. Vanhoucke, A. Rabinovich, "GOING DEEPER WITH CONVOLUTIONS", arXiv: 1409.4842, 2014;
[0016] Документ 8 - S. Ioffe, C. Szegedy, 'ΈATCH NORMALIZATION: ACCELERATING DEEP NETWORK TRAINING BY REDUCING INTERNAL COVARIATE SHIFT", arXiv: 1502.03167, 2015;
[0017] Документ 9 - J. Μ. Wolterink, Τ. Leiner, Μ. Α. Viergever, I. Isgum, "DILATED CONVOLUTIONAL NEURAL NETWORKS FOR CARDIOVASCULAR MR SEGMENTATION IN CONGENITAL HEART DISEASE", arXiv: 1704.03669, 2017;
[0018] Документ 10 - L. C. Piqueras, "AUTOREGRESSIVE MODEL BASED ON A DEEP CONVOLUTIONAL NEURAL NETWORK FOR AUDIO GENERATION", Tampere University of Technology (Технологический университет Тампере), 2016;
[0019] Документ 11 - J. Wu, 'Introduction to Convolutional Neural Networks", Nanjing University (Нанкинский университет), 2017;
[0020] Документ 12 - I. J. Goodfellow, D. Warde-Farley, M. Mirza, A. Courville, and Y. Bengio, "CONVOLUTIONAL NETWORKS", Deep Learning, MIT Press, 2016; and
[0021] Документ 13 - J. Gu, Z. Wang, J. Kuen, L. Ma, A Shahroudy, B. Shuai, T. Liu, X. Wang, G. Wang, "RECENT ADVANCES IN CONVOLUTIONAL NEURAL NETWORKS", arXiv: 1512.07108, 2017.
[0022] В документе 1 описаны архитектуры глубоких сверточных нейронных сетей, в которых используются группы остаточных блоков с фильтрами свертки, имеющими одинаковый размер окна свертки, слои пакетной нормализации, слои блоков линейной ректификации (сокращенно ReLU), меняющие размерность слои, слои разреженной (дырчатой, atrous) свертки с экспоненциально растущими показателями разрежения свертки, соединения с пропуском и слой классификации на основе функции softmax (многопеременной логистической функции), которые принимают входные последовательности и выдают выходные последовательности, которые присваивают оценки записям во входной последовательности. В предложенном способе применяются компоненты нейронной сети и параметры, раскрытые в Документе 1. В одном варианте реализации раскрытая технология модифицирует параметры компонентов нейронной сети, описанных в Документе 1. Например, в отличие от Документа 1, показатель разряжения свертки в раскрытой технологии растет неэкспоненциально от нижней группы остаточных блоков к более высокой группе остаточных блоков. В другом примере в отличие от Документа 1, размер окна свертки в раскрытой технологии в разных группах остаточных блоков различаются.
[0023] Документ 2 описывает детали вариантов архитектуры глубоких сверточных нейронных сетей, описанных в Документе 1.
[0024] Документ 3 описывает разреженные свертки, используемые в раскрытой технологии. В настоящем документе свертки atrous ("дырчатые") называются также "разреженными свертками". Atrous-/разреженные свертки обеспечивают крупные рецептивные поля при меньшем количестве обучающихся параметров. Atrous-/разреженная свертка представляет собой свертку, в которой ядро применяется на площади, большей, чем его длина, за счет того, что она пропускает входные значения с определенным шагом, называемым также показателем разрежения или фактором разрежения. Atrous/разреженные свертки увеличивают расстояние между элементами свертки фильтра свертки/ядра, в результате чего при осуществлении операции свертки используются соседние входные записи (например, нуклеотиды, аминокислоты) с большими интервалами. Это обеспечивает возможность введения во входные данные контекстуальных зависимостей дальнего действия. Дырчатые (atrous) свертки сохраняют расчет свертки для повторного использования при обработке соседних нуклеотидов.
[0025] Документ 4 описывает остаточные блоки и остаточные соединения (связи), применяемые в раскрытой технологии.
[0026] Документ 5 описывает соединения с пропуском, применяемые раскрытой технологией. В настоящем документе соединения с пропуском также называются "скоростными сетями".
[0027] Документ 6 описана архитектура плотно соединенной (связанной) сверточной сети, применяемой в раскрытой технологии.
[0028] Документ 7 описаны меняющие размерность сверточные слои и модульные пайплайны (конвейеры) обработки, применяемые в раскрытой технологии. Одним из примеров свертки с изменением размерности является свертка 1x1.
[0029] Документ 8 описаны слои пакетной нормализации, применяемые в раскрытой технологии.
[0030] Документ 9 также описаны дырчатые (Atrous)/разреженные свертки, применяемые в раскрытой технологии.
[0031] Документ 10 описаны различные архитектуры глубоких нейронных сетей, которые могут применяться в раскрытой технологии, включая сверточные нейронные сети, глубокие сверточные нейронные сети с дырчатыми/разреженными свертками.
[0032] Документ 11 описывает детали сверточной нейронной сети, которая может применяться в раскрытой технологии, включая алгоритмы для обучения (тренировки) сверточной нейронной сети с субдисткретизирующими слоями (слоями подвыборки) (например, объединения) и полностью связанными слоями.
[0033] Документ 12 описывает детали различных операций свертки, которые могут применяться в раскрытой технологии.
[0034] Документ 13 описывает различные архитектуры сверточных нейронных сетей, которые могут применяться в раскрытой технологии.
ВКЛЮЧЕНИЕ ПУТЕМ ССЫЛКИ ТАБЛИЦ, ПОДАННЫХ В ЭЛЕКТРОННОМ ВИДЕ С НАСТОЯЩЕЙ ЗАЯВКОЙ
[0035] С настоящей заявкой поданы три таблицы в текстовом формате ASCII, которые включены в настоящий документ посредством ссылки. Файлы имеют следующие имена, даты создания и размеры:
[0036] table_S4_mutation_rates.txt 31 августа 2018 г. 2,452 KB
[0037] table_S5_gene enrichment.txt 31 августа 2018 г. 362 KB
[0038] table_S6_validation.txt 31 августа 2018 г. 362 KB
ОБЛАСТЬ ТЕХНИКИ
[0039] Раскрытая технология относится к компьютерам и цифровым системам обработки данных, относящихся к типу искусственного интеллекта, и соответствующим способам обработки данных и продуктам для эмуляции интеллекта (т.е. системам, основанным на знаниях, системам построения рассуждений и системам приобретения знаний); включая системы для логических рассуждений в условиях неопределенности (например, системы нечеткой логики), адаптивным системам, системам машинного обучения и искусственным нейронным сетям. В частности, Раскрытая технология относится к применению технологий глубокого обучения для обучения (тренировки) глубоких сверточных нейронных сетей.
УРОВЕНЬ ТЕХНИКИ
[0040] Не следует полагать, что аспекты, обсуждаемые в этом разделе, составляют часть уровня техники только потому, что они упоминаются в этом разделе. Аналогичным образом, не следует полагать, что задача, упоминаемая в этом разделе или связанная с объектом, указанным в качестве предпосылки, является признанным уровнем техники. Предмет этого раздела лишь представляет различные подходы, которые сами по себе также могут соответствовать вариантам реализации заявленной технологии.
Машинное обучение
[0041] В машинном обучении входные переменные используются для предсказания выходных переменных. Входные переменные часто называют признаками и обозначают как X={Х1, Х2, …, Xk), где каждый Xi, i ∈ 1, k представляет собой признак. Выходная переменная часто называется ответом или зависимой переменной и обозначается переменной Yi. Отношение между Υ и соответствующей X можно зависать в общем виде:
Y = ƒ (Х)+е
[0042] В приведенном выше уравнении ƒ представляет собой функцию признаков (Х1, Х2, …, Xk), а ∈ представляет собой показатель случайной ошибки. Указанный показатель случайно ошибки не зависит от Х и имеет среднее значение, равное нулю.
[0043] На практике признаки X доступны в отсутствие Υ или без знания точного отношения X и Υ. Поскольку среднее значение показателя ошибки равно нулю, задача заключается в том, чтобы оценить ƒ.
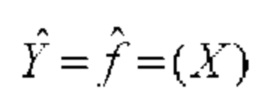
[0044] В приведенном выше уравнении 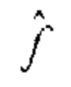 является оценкой ∈, которая часто считается черным ящиком, что означает, что известно только отношение (связь) между входом и выходом
является оценкой ∈, которая часто считается черным ящиком, что означает, что известно только отношение (связь) между входом и выходом 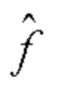 , а ответа на вопрос, как это работает, нет.
, а ответа на вопрос, как это работает, нет.
[0045] Функцию 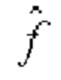 находят при помощи обучения. В машинном обучении применяются два подхода: обучение с учителем и обучение без учителя. В обучении с учителем для обучения (тренировки) используются размеченные данные. Демонстрируя входы и соответствующие выходы (=метки), функцию
находят при помощи обучения. В машинном обучении применяются два подхода: обучение с учителем и обучение без учителя. В обучении с учителем для обучения (тренировки) используются размеченные данные. Демонстрируя входы и соответствующие выходы (=метки), функцию 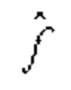 оптимизируют таким образом чтобы она аппроксимировала выход. В обучении без учителя целью является выявить скрытую структуру по неразмеченным данным. Этот алгоритм не имеет меры точности на входных данных, что отличает его от обучения с учителем.
оптимизируют таким образом чтобы она аппроксимировала выход. В обучении без учителя целью является выявить скрытую структуру по неразмеченным данным. Этот алгоритм не имеет меры точности на входных данных, что отличает его от обучения с учителем.
Нейронные сети
[0046] Однослойный перцептрон (SLP) представляет собой простейшую модель нейронной сети. Он содержит один входной слой и одну функцию активацию как показано на ФИГ. 1. Входы прогоняют через взвешенный граф. Функция/использует сумму входов как аргумент и сравнивает ее с порогом θ.
[0047] ФИГ. 2 демонстрирует один из вариантов реализации полностью соединенной (связанной) нейронной сети с несколькими слоями. Нейронная сеть представляет собой систему взаимосвязанных искусственных нейронов (например, a1, а2, а3), которые обмениваются друг с другом сообщениями. Показанная нейронная сеть имеет три входа, два нейрона в скрытом слое и два нейрона в выходном слое. Скрытый слой имеет функцию активации ƒ(•), а выходной слой имеет функцию активации g(•). Связи имеют численные веса (например, w11, w21, w12, w31, w22, w32, v11, v22), которые подстраиваются в процессе обучения (тренировки) таким образом, то обученная приемлемым образом сеть отвечает правильно при предъявлении образа для распознавания. Входной слой обрабатывает необработанные входные данные, скрытый слой обрабатывает данные, полученные на выходе входного слоя на основании весов связей между входным слоем и скрытым слоем. Выходной слой берет выход (выходные данные) скрытого слоя и обрабатывает на основании весов связей между скрытым слоем и выходным слоем. Сеть включает несколько слоев нейронов, детектирующих признаки. Каждый слой содержит много нейронов, которые отвечают на различные комбинации входов от предыдущих слоев. Слои сконструированы таким образом, что первый слой детектирует набор примитивных паттернов в данных входного образа, второй слой детектирует паттерны паттернов, а третий слой детектирует паттерны этих паттернов.
[0048] Обзор применения глубокого обучения в геномике можно найти в следующих публикациях:
• Т. Ching et al., Opportunities And Obstacles For Deep Learning In Biology And Medicine, www.biorxiv.org: 142760, 2017;
• Angermueller C, Parnamaa T, Parts L, Stegle O. Deep Learning For Computational Biology. Mol SystBiol. 2016; 12:878;
• Park Y, Kellis M. 2015 Deep Learning For Regulatory Genomics. Nat. Biotechnol. 33, 825-826. (doi:10.1038/nbt.3313);
• Min, S., Lee, B. & Yoon, S. Deep Learning In Bioinformatics. Brief. Bioinform. bbw068 (2016);
• Leung MK, Delong A, Alipanahi В et al. Machine Learning In Genomic Medicine: A Review of Computational Problems and Data Sets 2016; и
• Libbrecht MW, Noble WS. Machine Learning Applications In Genetics and Genomics. Nature Reviews Genetics 2015; 16(6):321-32.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0049] На чертежах одинаковые ссылочные позиции обычно относятся к одинаковым деталям на разных видах. Кроме того, чертежи не обязательно выполнены в масштабе, вместо этого, как правило, делается акцент на иллюстрации принципов раскрытой технологии. В последующем описании различные реализации раскрытой технологии описаны со ссылкой на следующие чертежи, на которых:
[0050] ФИГ. 1 демонстрирует однослойный перцептрон (SLP).
[0051] ФИГ. 2 демонстрирует один из вариантов реализации многослойной нейронной сети прямого распространения.
[0052] ФИГ. 3 показывает один вариант реализации работы сверточной нейронной сети.
[0053] ФИГ. 4 показывает блок-схему тренировки сверточной нейронной сети в соответствии с одним из вариантов реализации раскрытой технологии.
[0054] ФИГ. 5 показан один из вариантов реализации нелинейного слоя ReLU (с блоками линейной ректификации) в соответствии с одним вариантом реализации раскрытой технологии.
[0055] ФИГ. 6 иллюстрирует разреженные свертки.
[0056] ФИГ. 7 представляет собой один вариант реализации слоев субдискретизации (подвыборки) (максимальное/среднее объединения (пулинг)) в соответствии с одним из вариантов реализации раскрытой технологии.
[0057] ФИГ. 8 показывает один вариант осуществления двухслойной свертки сверточных слоев.
[0058] ФИГ. 9 показывает остаточную связь, которая снова вводит предварительную информацию ниже путем добавления карты признаков.
[0059] ФИГ. 10 показывает один вариант реализации остаточных блоков и связей с пропусками.
[0060] ФИГ. 11 демонстрирует один вариант реализации пакетных разреженных сверток.
[0061] ФИГ. 12 демонстрирует прямой проход пакетной нормализации.
[0062] ФИГ. 13 иллюстрирует преобразование пакетной нормализации в момент теста.
[0063] ФИГ. 14 демонстрирует обратный проход пакетной нормализации.
[0064] ФИГ. 15 показывает применение слоя нормализации слоя со сверточным или плотно связанным слоем.
[0065] ФИГ. 16 демонстрирует один вариант реализации ID-свертки.
[0066] ФИГ. 17 иллюстрирует работу глобального среднего объединения (GAP).
[0067] ФИГ. 18 иллюстрирует один вариант реализации вычислительной среды с обучающими серверами и рабочими серверами, который можно применять для реализации раскрытой технологии.
[0068] ФИГ. 19 показывает один вариант реализации архитектуры разреженной сверточной нейронной сети (сокращенно ACNN), называемой в настоящем документе "SpliceNet".
[0069] ФИГ. 20 демонстрирует один вариант реализации, который может применяться сетью типа ACNN и сверточной нейронной сетью (сокращенно CNN).
[0070] ФИГ. 21 показывает другой вариант реализации архитектуры ACNN (разреженной сверточной нейронной сети), называемой в настоящем документе "SpliceNet80".
[0071] ФИГ. 22 показывает еще один вариант реализации архитектуры ACNN (разреженной сверточной нейронной сети), называемой в настоящем документе "SpliceNet400".
[0072] ФИГ. 23 показывает еще один вариант реализации архитектуры ACNN (разреженной сверточной нейронной сети), называемой в настоящем документе "SpliceNet2000".
[0073] ФИГ. 24 показывает еще один вариант реализации архитектуры ACNN (разреженной сверточной нейронной сети), называемой в настоящем документе "SpliceNet10000".
[0074] ФИГ. 25, 26 и 27 показывают различные типа входов, обрабатываемые сетями ACNN и CNN.
[0075] ФИГ. 28 показывает, что ACNN можно тренировать на по меньшей мере 800 миллионах несплайсирующихся сайтов, a CNN можно тренировать на по меньшей мере 1 миллионе несплайсирущихся сайтов.
[0076] ФИГ. 29 иллюстрирует энкодер с одним горячим состоянием.
[0077] ФИГ. 30 показывает обучение ACNN.
[0078] ФИГ. 31 показывает CNN.
[0079] ФИГ. 32 обучение, валидацию и тестирование ACNN и CNN.
[0080] ФИГ. 33 показывает референсную последовательность и альтернативную последовательность.
[0081] ФИГ. 34 иллюстрирует детектирование (определение) аберрантного сплайсинга.
[0082] ФИГ. 35 иллюстрирует обработку пирамиды сети SpliceNet10000 для классификации сайтов сплайсинга.
[0083] ФИГ. 36 иллюстрирует обработку пирамиды сети SpliceNet10000 для детекции аберрантного сплайсинга.
[0084] ФИГ. 37А, 37В, 37С, 37D, 37Е, 37F, 37G и 37Н иллюстрирует один вариант реализации предсказания сплайсинга по первичной последовательности при помощи глубокого обучения.
[0085] ФИГ. 38А, 38В, 38С, 38D, 38Е, 38F и 38G показывают один вариант реализации валидации редких критических сплайс-мутаций в данных секвенирования РНК.
[0086] ФИГ. 39А, 39В и 39С показывают один вариант реализации криптических сплайс-вариантов (вариантов криптического сплайсинга), которые обуславливают ткань-специфический альтернативный сплайсинг.
[0087] ФИГ. 40А, 40В, 40С, 40D и 40Е показывают один вариант реализации предсказанных сплайс-вариантов, сильно вредоносных для человеческих популяций.
[0088] ФИГ. 41А, 41В, 41С, 41D, 41Е и 41F показывают один вариант реализации критических de novo сплайс-мутаций у пациентов с редкими генетическими заболеваниями.
[0089] ФИГ. 42А и 42В показывают оценку различных алгоритмов предсказания сплайсинга на длинных промежуточных некодирующих РНК.
[0090] ФИГ. 43А и 43В иллюстрируют зависящие от положения эффекты мотивов точки ветвления ТАСТААС и энхансера сплайсинга экзонов GAAGAA.
[0091] ФИГ. 44А и 44В показывают эффекты позиционирования нуклеосом при сплайсинге.
[0092] ФИГ. 45 иллюстрирует пример расчета величины эффекты для нарушающего сплайсинг варианта при сложных эффектах.
[0093] ФИГ. 46А, 46В и 46С показывает оценку модели SpliceNet-10k на единичных и частых вариантах.
[0094] ФИГ. 47А и 47В демонстрируют показатель валидации и величину эффектов вариантов, создающих сайты сплайсинга, разбитые по расположению варианта.
[0095] ФИГ. 48А, 48В, 48С и 48D показывают оценку модели SpliceNet-10k на тренировочных и тестовых хромосомах.
[0096] ФИГ. 49А, 49В и 49С иллюстрируют критические de novo сплайс-мутации (de novo мутации критического сплайсинга) у пациентов с редкими генетическими заболеваниями, только по сайтам синонимичных, интронных или нетранслируемых областей.
[0097] ФИГ. 50А и 50В показывают критические de novo сплайс-мутации (мутации сплайсинга) при РАС как долю всех de novo мутаций.
[0098] ФИГ. 51А, 51В, 51С, 51D, 51Е, 51F, 51G, 51Н, 51I и 51J показывают валидацию по РНК-последовательностям предсказанных критических de novo сплайс-мутаций у пациентов с РАС.
[0099] ФИГ. 52А и 52В демонстрируют показатель валидации и чувствительность на РНК-последовательностях модели, обученной исключительно на канонических транскриптах.
[00100] ФИГ. 53А, 53В и 53С иллюстрирует, что комплексное моделирование улучшает показатели работы SpliceNet-10k.
[00101] ФИГ. 54А и 54В демонстрируют оценку SpliceNet-10k в областях с варьирующей плотностью экзонов.
[00102] ФИГ. 55 представляет собой Таблицу S1, где показан один вариант реализации образцов GTEx, применяемый для демонстрации расчета величины эффекта и ткань-специфического сплайсинга.
[00103] ФИГ. 56 представляет собой Таблицу S2, где показан один вариант реализации остановок, применяемый для оценки параметра валидации и чувствительности различных алгоритмов.
[00104] ФИГ. 57 демонстрирует один вариант реализации анализа обогащения по генам.
[00105] ФИГ. 58 демонстрирует один вариант реализации полногеномного анализа обогащения.
[00106] ФИГ. 59 представляет собой упрощенную блок-схему компьютерной системы, которую можно применять для реализации раскрытой технологии.
ПОДРОБНОЕ ОПИСАНИЕ
[00107] Приведенное ниже описание представлено для того, чтобы любой специалист в данной области техники мог осуществить и применить раскрытую технологию, и представлено в контексте конкретного случая применения и ее требований. Различные модификации раскрытых вариантов осуществления будут очевидны для специалиста в данной области техники, а общие принципы, раскрытые в настоящем документе, могут быть применены к другим вариантам осуществления и областям применения без выхода за пределы идеи и объема раскрытой технологии. Таким образом, раскрытая технология не ограничена представленными вариантами осуществления, и она должна рассматриваться в соответствии с наиболее широким объемом, соответствующим принципам и признакам, раскрытым в настоящем документе. Введение
Сверточные нейронные сети
[00108] Сверточная нейронная сеть представляет собой особый тип нейронной сети. Фундаментальная разница между плотно соединенным (связанным) слоем и сверточным слоем заключается в следующем: Соединенные слои изучают глобальные паттерны в своем пространстве входных признаков, в то время как сверточные слои изучают локальные паттерны: в случае образов паттерны находятся в малых двумерных окнах входных данных. Эта ключевая характеристика придает сверточным нейронным сетям две интересные особенности: (1) паттерны, которые они изучают, являются инвариантными относительно сдвига и (2) они могут изучать пространственные иерархии паттернов.
[00109] В отношении первого можно отметить, что после изучения конкретного паттерна в правом нижнем углу картинки сверточным слой может распознать его где угодно: например, в верхнем левом углу. Плотно соединенным нейронным сетям пришлось бы обучаться паттерну снова, если бы он появился в новом месте. Это делает сверточные нейронные сети эффективными в отношении данных, так как им требуется меньшее количество тренировочных образцов для обучения способу задания функций в связи с тем, что они способны к обобщению.
[00110] В отношении второго можно отметить, что первый сверточный слой может изучать малые локальные паттерны, такие как края, второй сверточный слой будет изучать паттерны большего размера, выполненные из признаков первых слоев, и т.д. Это обеспечивает сверточным нейронным сетям возможность эффективного обучения существенно более сложным и абстрактным визуальным концептам.
[00111] Сверточная нейронная сеть обучена преобразованиям с высокой нелинейностью посредством взаимно соединенных слоев искусственных нейронов, расположенных во множестве различных слоев с функциями активации, которые делают слои зависимыми. Она содержит один или более сверточных слоев, перемежающихся с одним или более субдискретизирующих слоев, за которыми обычно следуют один или более плотно соединенные слоев. Каждый элемент сверточной нейронной сети принимает входные данные из совокупности признаков в предыдущем слое. Сверточная нейронная сеть обучена параллельно, так как нейроны в одной и той же карте признаков имеют идентичные весовые значения. Эти локальные общие весовые коэффициенты снижают сложность сети таким образом, что когда многомерные входные данные попадают в сеть, сверточная нейронная сеть избегает сложностей, связанных с реконструкцией данных при извлечении признаков и процессе регрессии или классификации.
[00112] Свертки осуществляют операции над трехмерными тензорами, называемыми картами признаков, с двумя пространственными осями (высота и ширина), а также с осью глубины (также называемой канальной осью). Для изображения RGB размер оси глубины составляет 3, так как изображение имеет три цветовых канала; красный, зеленый и синий. Для черно-белых картинок глубина составляет 1 (уровни серого). Операция свертки извлекает вставки из карты ее входных признаков и применяет то же преобразование ко всем вставкам с получением карты выходных признаков. Эта карта выходных признаков все еще является трехмерным тензором: она имеет ширину и высоту. Ее глубина может быть произвольной, так как глубина выходных данных является параметром слоя, а различные каналы по указанной оси глубины больше не соответствуют конкретным цветам во входных данных RGB, наоборот, они соответствуют фильтрам. Фильтры кодируют определенные аспекты входных данных: например, на уровне высоты один фильтр может кодировать концепцию «наличие лица во входных данных».
[00113] Например, первый сверточный слой берет карту признаков размером (28, 28, 1) и выдает карту признаков размером (26, 26, 32): он вычисляет 32 фильтра по своим входным данным. Каждый из указанных 32 выходных каналов содержит сетку значений размером 26 × 26, которая представляет собой карту ответов фильтра на входные данные, указывающую ответ паттерна указанного фильтра в различных местах во входных данных. Иными словами, термин «карта признаков» обозначает следующее: каждая координата по оси глубины является признаков (или фильтром), а двумерный тензор выходных данных [:, :, n] представляет собой двумерную пространственную карту ответов указанного фильтра по входным данным.
[00114] Свертки заданы двумя ключевыми параметрами: (1) размер вставок, извлеченных из входных данных - они обычно составляют 1 x 1, 3 x 3 или 5 x 5, и (2) глубина карты выходных признаков - количество фильтров, вычисленных посредством свертки. Зачастую начинают с глубины 32, продолжают с глубиной 64 и заканчивают с глубиной 128 или 256.
[00115] Свертка работает посредством перемещения указанных окон размером 3 x 3 или 5 x 5 по трехмерной карте входных признаков с остановкой в каждом месте и извлечением трехмерной вставки окружающих признаков (shape (window_height, window_width, input_depth); форма (окно_высота, окно_ширина, входные данные_глубина)). Каждую такую трехмерную вставку затем преобразуют (посредством тензорного произведения с весовой матрицей, обученной таким же образом, называемой ядром свертки) в одномерный вектор формы (output_depth; выходные данные_глубина). Все из указанных векторов затем подвергают пространственной обратной сборке в трехмерную карту выходных данных формы (height, width, output_depth; высота, ширина, выходные данные_глубина). Каждое пространственное положение на карте выходных признаков соответствует тому же положению на карте входных признаков (например, нижний правый угол выходных данных содержит информацию о нижнем правом угле входных данных). Например, в случае окон 3 x 3, векторные выходные данные [i, j, :] происходят из входных данных [i-1: i+l, j-1:J+1, :] трехмерной вставки. Полностью процесс подробно показан на ФИГ. 3.
[00116] Сверточная нейронная сеть содержит сверточные слои, которые выполняют операцию свертки между входными значениями и сверточными фильтрами (весовой матрицей), которые обучены на множестве итераций градиентного изменения во время обучения. Пусть (m, n) будет размером фильтр a, a W- весов ой матрицей, тогда сверточный слой выполняет свертку W с входными данными Χ посредством вычисления скалярного произведения W • х+b, где x представляет собой элемент из X, a b представляет собой смещение. Размер шага, на который сверточные фильтры перемещаются по входным данным, называют сдвигом, а область фильтрации (m × n) называют рецептивным полем Один и тот же сверточный фильтр применяют к различным положениям входных данных, что позволяет снизить количество изученных весов. Это также обеспечивает возможность обучения с инвариантностью положений, т.е. если важный паттерн присутствует во входных данных, сверточные фильтры изучают его вне зависимости от его положения в последовательности Обучение сверточной натронной сети
[00117] На ФИГ. 4 показана блок-схема обучения сверточной нейронной сети в соответствии с одним из вариантов реализации раскрытой технологии. Сверточная нейронная сеть настроена или обучена таким образом, что входные данные ведут к особой выходной оценке. Сверточную нейронную сеть настраивают с использованием обратного распространения на основе сравнения выходной оценки и реальных данных до тех пор, пока выходная оценка прогрессивно совпадет или приблизится к реальным данным
[00118] Сверточную нейронную сеть обучают посредством регулировки весов между нейронами на основании разницы между реальными данными и действительными выходными данными. Математически это можно описать следующим образом:
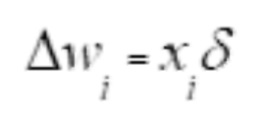
где δ - (реальные данные)-(фактический выход)
[00119] В одном варианте осуществления обучающее правило определено как:
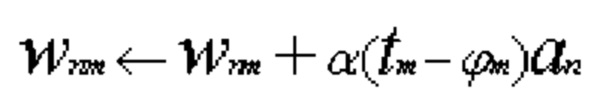
[00120] В представленном выше уравнении: стрелка указывает на изменение значения; tm представляет собой целевое значение нейрона m; ϕm представляет собой вычисленное текущее выходные данные нейрона m; an представляет собой входные данные n; а α представляет собой скорость обучения.
[00121] Промежуточный этап обучения включает выработку вектора признаков из входных данных с использованием сверточных слоев. Вычисляют градиент в отношении весов в каждом слое, начиная с выходных данных. Это называют обратным проходом или прохождением в обратном направлении. Веса в сети изменяют с использованием комбинации отрицательного градиента и предыдущих весов.
[00122] В одном варианте осуществления сверточная нейронная сеть использует алгоритм изменения со стохастическим градиентом (такой как ADAM), который выполняет обратное распространение ошибок посредством градиентного спуска. Один пример алгоритма обратного распространения на основе сигмоидной функции описан ниже:
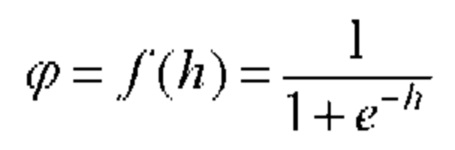
[00123] В приведенной выше сигмоидной функции, h представляет собой взвешенную сумму, вычисленную нейроном. Сигмоидная функция имеет следующую производную:
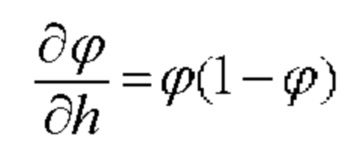
[00124] Алгоритм включает вычисление активации нейронов в сети, вырабатывая выходные данные для прямого прохода. Активация нейрона m в скрытых слоях можно описать как:
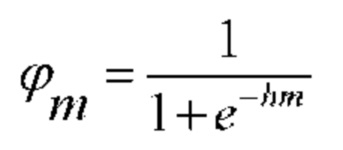
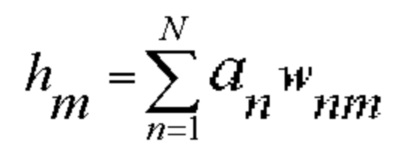
[00125] Это выполняется для всех скрытых слоев для получения активаций, описанных следующим образом:
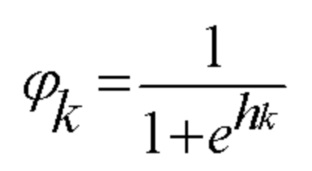
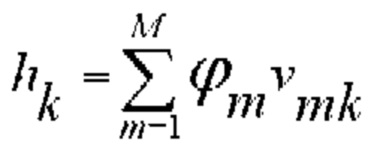
[00126] Затем для каждого слоя вычисляют ошибку и корректировочные веса. Ошибку в выходных данных вычисляют следующим образом:
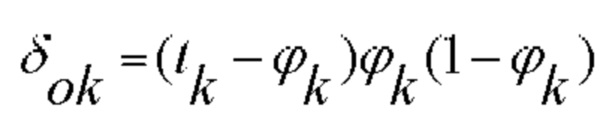
[00127] Ошибку в скрытых слоях вычисляют следующим образом:
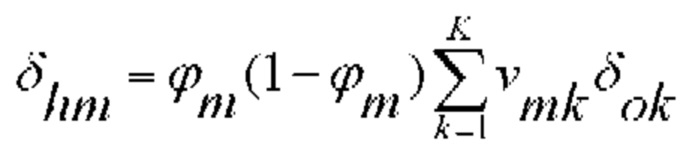
[00128] Веса выходного слоя изменяют следующим образом:
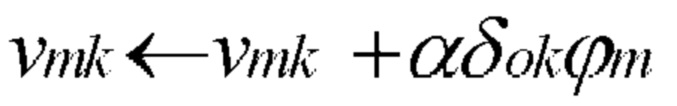
[00129] Веса скрытого слоя изменяют с использованием скорости обучения α следующим образом:
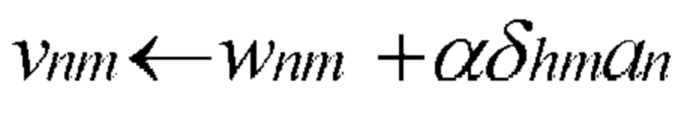
[00130] В одном варианте осуществления сверточная нейронная сеть использует оптимизацию с градиентным спуском для вычисления ошибки по всем слоям. При такой оптимизации для вектора л; входных признаков и спрогнозированных выходных данных 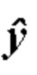 функция потерь определена как
функция потерь определена как 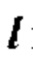 в целях прогнозирования
в целях прогнозирования 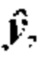 когда целью является у, т.е.
когда целью является у, т.е. 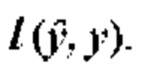 Спрогнозированные выходные данные
Спрогнозированные выходные данные 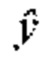 преобразуют из вектора x входных признаков с использованием функции ƒ. Функция ƒ параметризуется весами сверточной нейронной сети, т.е.
преобразуют из вектора x входных признаков с использованием функции ƒ. Функция ƒ параметризуется весами сверточной нейронной сети, т.е. 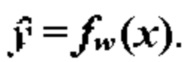 Функция потерь описана как
Функция потерь описана как 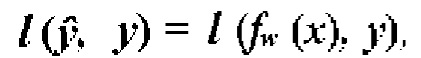 или
или 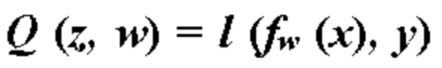 где z представляет собой пару (х, у) входных данных и выходных данных. Оптимизацию с градиентным спуском выполняют путем изменения весов в соответствии с:
где z представляет собой пару (х, у) входных данных и выходных данных. Оптимизацию с градиентным спуском выполняют путем изменения весов в соответствии с:
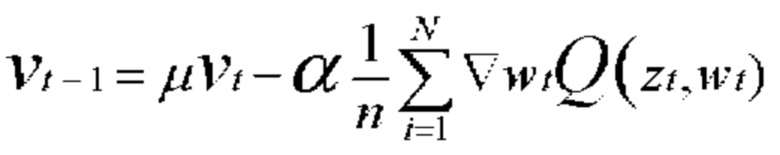
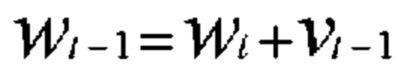
[00131] В приведенном выше уравнении α представляет собой скорость обучения. Кроме того, потери вычисляют как среднее по совокупности n пар данных. Вычисление останавливают, когда скорость обучения α достаточно мала при линейном схождении. В других вариантах осуществления градиент вычисляют с использованием только выбранных пар данных, подаваемых в ускоренный градиент Нестерова и адаптивный градиент для обеспечения эффективности вычисления.
[00132] В одном варианте осуществления сверточная нейронная сеть использует стохастический градиентный спуск (SGD) для вычисления функции потерь (функции стоимости). SGD аппроксимирует градиент в отношении весов в функции потерь посредством его вычисления на основании только одной, выбранной в случайном порядке, пары данных, Zt, что можно описать как:
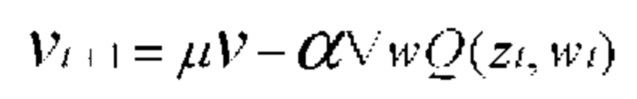
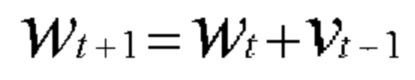
[00133] В приведенном выше уравнении: α представляет собой скорость обучения; μ представляет собой момент; a t представляет собой весовое значение перед изменением. Скорость схождения SGD составляет приблизительно O(1/t), когда скорость обучения α снижается достаточно быстро и достаточно медленно. В другом варианте осуществления сверточная нейронная сеть использует различные функции потерь, такие как евклидова функция потерь и softmax (многопеременная логистическая) функция потерь. В другом варианте осуществления сверточная нейронная сеть использует стохастический оптимизатор Adam.
Сверточные слои
[00134] Сверточные слои сверточной нейронной сети служат в качестве экстракторов признаков. Сверточные слои функционируют как адаптивные экстракторы признаков, способные к обучению и декомпозиции входных данных на иерархические признаки. В одном варианте осуществления сверточные слои берут два изображения в качестве входных данных и выдают третье изображение в качестве выходных данных. В таком варианте осуществления свертка выполняется над двумя изображениями в двух измерениях (2D), причем одно изображение представляет собой входное изображение, а другое изображение, называемое «ядром» и применяемое в качестве фильтра к входному изображению, обеспечивает получение выходного изображения. Таким образом, для входного вектора ƒ длиной n и ядра g длиной m, свертка ƒ* g для ƒ и g определяется как:

[00135] Операция свертки включает перемещение ядра по входному изображению. Для каждого положения ядра перекрывающиеся значения ядра и входного изображения умножаются и результаты складываются. Сумма произведений представляет собой значение выходного изображения в точке на входном изображении, в которой отцентровано ядро. Полученные в результате различные выходные данные от множества ядер называют картами признаков.
[00136] После того как сверточные слои обучены, их применяют для выполнения задач по распознаванию над новыми рассматриваемыми данными. Так как сверточные слои обучаются на тренировочных данных, они избегают извлечения признаков в явном виде и неявно обучаются на тренировочных данных. Сверточные слои используют сверточные веса ядра фильтрации, которые определяются и изменяются как часть процесса обучения. Сверточные слои извлекают различные признаки из входных данных, которые комбинируются на верхних слоях. Сверточная нейронная сеть использует различное количество сверточных слоев, каждый из которых имеет различные параметры свертки, такие как размер ядра, сдвиги, заполнение, количество карт признаков и веса.
Нелинейные слои
[00137] На фиг. 5 показан один вариант осуществления нелинейных слоев в соответствии с одним вариантом осуществления раскрытой технологии. Нелинейные слои используют различные пусковые функции для указания на явную идентификацию или наиболее вероятные признаки на каждом скрытом слое. Нелинейные слои используют множество особых функций для осуществления нелинейного запуска, включая блоки линейной ректификации (ReLU), гиперболический тангенс, абсолютную величину гиперболического тангенса, сигмоидную и непрерывную пусковые (нелинейные) функции. В одном варианте осуществления активация ReLU осуществляет функцию у = max(х, 0) и сохраняет размеры входных и выходных данных одинаковыми. Преимущество использования ReLU заключается в том, что сверточная нейронная сеть обучена во много раз быстрее. ReLU не является непрерывной, насыщающей функцией активации, которая является линейной относительно входных данных, если входные значения больше, чем ноль, и равна нулю в противном случае. С математической точки зрения функцию активации ReLU можно описать как:
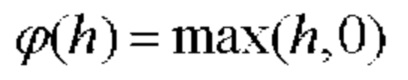
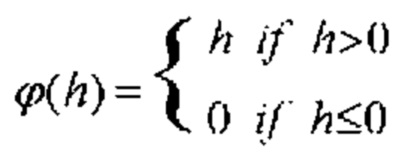
[00138] В других вариантах осуществления сверточная нейронная сеть использует функцию активации со степенным блоком, которая представляет собой непрерывную ненасыщающую функцию, описываемую как:
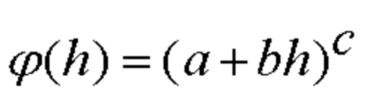
[00139] В приведенном выше уравнении а, b и с представляют собой параметры, управляющие смещением, масштабом и мощностью соответственно. Степенная функция активации может обеспечивать х и y - антисимметричную активацию, если с имеет нечетное значение, и y - симметричную активацию, если с имеет четное значение. В некоторых вариантах осуществления указанный блок обеспечивает невыпрямленную линейную активацию.
[00140] В других вариантах осуществления сверточная нейронная сеть использует функцию активации с сигмоидным блоком, которая представляет собой непрерывную ненасыщающую функцию, описываемую следующей логистической функцией:
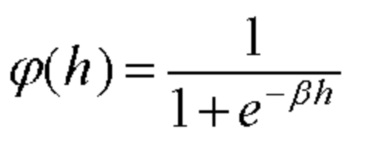
[00141] В приведенном выше уравнении β = 1. Функция активации с сигмоидным блоком не обеспечивает отрицательную активацию и является только асимметричной по отношению к y-оси.
Разреженные свертки
[00142] На фиг.6 показаны разреженные свертки. Разреженные свертки, иногда называемые дырчатыми (atrous) свертками, что буквально означает «с отверстиями». Данное название происходит из алгоритма «а trous» (франц.), который вычисляет быстрое двойное вейвлет преобразование. В сверточных слоях такого типа входные данные, соответствующие рецептивному полю фильтров, не являются соседними точками. Это показано на фиг. 6. Расстояние между входными данными зависит от коэффициента разрежения.
Субдискретизирующие слои (слои подвыборок)
[00143] На фиг. 7 показан один вариант осуществления субдискретизирующих слоев в соответствии с одним вариантом осуществления раскрытой технологии. Субдискретизирующие слои снижают разрешение признаков, извлеченных сверточными слоями, чтобы сделать извлеченные признаки или карты признаков устойчивыми к шуму и искажению. В одном варианте осуществления субдискретизирующие слои используют два типа объединяющих операций, среднее объединение и максимальное объединение. Объединяющие операции разделяют входные данные на неперекрывающиеся двумерные пространства. Для среднего объединения вычисляют среднее для четырех значений в области. Для максимального объединения выбирают максимальное значение из четырех значений.
[00144] В одном варианте осуществления субдискретизирующие слои включают объединяющие операции на совокупности нейронов в предыдущем слое посредством преобразования его выходных данных только до одних из входных данных при максимальном объединении и посредством преобразования его выходных данных до среднего из входных данных про среднем объединении. При максимальном объединении выходные данные объединяющего нейрона представляют собой максимальное значение, которое имеется во входных данных, что описано так:
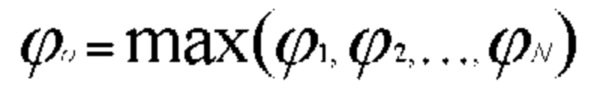
[00145] В приведенном выше уравнении N представляет собой общее количество элементов в совокупности нейронов.
[00146] При среднем объединении выходные данные объединяющего нейрона представляют собой среднее значение входных данных, которое имеется во входной совокупности нейронов, что описано так:
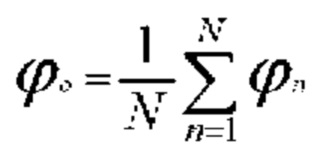
[00147] В приведенном выше уравнении N представляет собой общее количество элементов во входной совокупности нейронов.
[00148] На фиг. 7 входной размер составляет 4 x 4. Для субдискретизации 2 x 2 изображение 4 x 4 разделяют на четыре неперекрывающиеся матрицы размером 2 x 2. Для среднего объединения среднее для четырех значений является полностью целочисленными выходными данными. Для максимального объединения максимальное значение для четырех значений в матрице 2 x 2 является полностью целочисленными выходными данными.
Примеры свертки
[00149] На фиг. 8 показан один вариант осуществления двухслойной свертки сверточных слоев. На фиг. 8 сворачивают входные данные размерностью 2048 измерений. При свертке 1 входные данные сворачивают посредством сверточного слоя, содержащего два канала с шестнадцатью ядрами размером 3 x 3. Полученные в результате шестнадцать карт признаков затем ректифицируют посредством функции активации ReLU при ReLU1, а затем объединяют в Pool 1 посредством среднего объединения с использованием объединяющего слоя с шестнадцатью каналами с ядрами размером 3 x 3. При свертке 2 входные данные из Pool 1 затем сворачивают посредством другого сверточного слоя, содержащего шестнадцать каналов с тридцатью ядрами размером 3 x 3. За этим следует другой ReLU2 и среднее объединение в Pool 2 с ядром размером 2 x 2. Сверточные слои используют переменное количество сдвигов и заполнений, например, ноль, два и три. Полученный в результате вектор признаков имеет пятьсот двенадцать (512) измерений в соответствии с одним вариантом осуществления.
[00150] В другом вариантах осуществления сверточная нейронная сеть использует различное количество сверточных слоев, субдискретизирующих слоев, нелинейных слоев и плотно соединенных слоев. В одном варианте осуществления сверточная нейронная сеть представляет собой неглубокую сеть с меньшим количеством слоев и большим количеством нейронов в каждом слое, например, с одним, двумя или тремя плотно соединенными (плотно связанными) слоями, содержащими сто (100) - двести (200) нейронов на слой. В другом варианте осуществления сверточная нейронная сеть представляет собой глубокую сеть с большим количеством слоев и меньшим количеством нейронов в каждом слое, например, с пятью (5), шестью (6) или восемью (8) Плотно соединенными слоями, содержащими тридцать (30) - пятьдесят (50) нейронов на слой.
Прямой проход
[00151] Выходные данные нейрона в ряду х, столбце у в сверочном слое 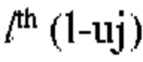 и карте kth (к-го) признаков для количества ƒ ядер свертки на карте признаков определяют с помощью следующего уравнения:
и карте kth (к-го) признаков для количества ƒ ядер свертки на карте признаков определяют с помощью следующего уравнения:

[00152] Выходные данные нейрона в ряду х, столбце у в субдискретизирующем слое 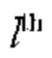 и карте kth признаков определяют с помощью следующего уравнения:
и карте kth признаков определяют с помощью следующего уравнения:
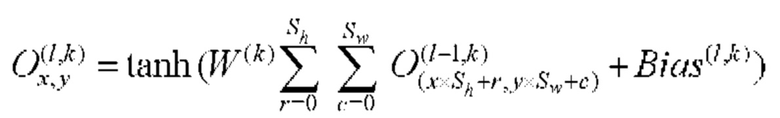
[00153] Выходные данные нейрона ith в выходном слое 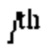 определяют с помощью следующего уравнения:
определяют с помощью следующего уравнения:
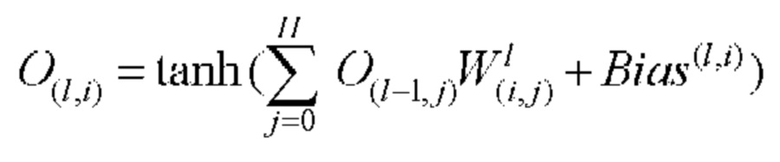
Обратное распространение
[00154] Выходное отклонение kth нейрона в выходном слое определяют с помощью следующего уравнения:
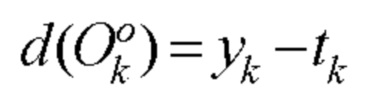
[00155] Входное отклонение kth нейрона в выходном слое определяют с помощью следующего уравнения:
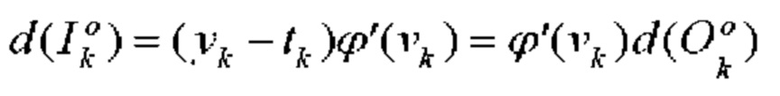
[00156] Вес и изменение смещения kth нейрона в выходном слое определяют с помощью следующего уравнения:
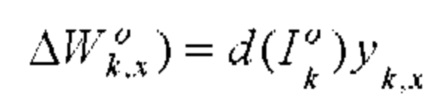
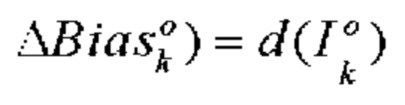
[00157] Выходное смещение kth нейрона в скрытом слое определяют с помощью следующего уравнения:
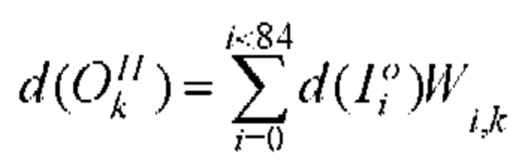
[00158] Входное смещение kth нейрона в скрытом слое определяют с помощью следующего уравнения:
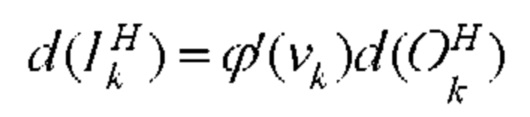
[00159] Вес и изменение смещения в ряду х, столбце у в mth карте признаков первичного слоя, принимающего входные данные от k нейронов в скрытом слое определяют с помощью следующего уравнения:
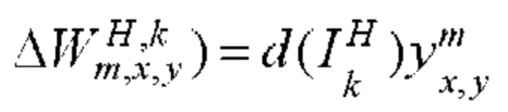
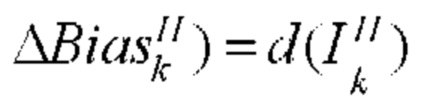
[00160] Выходное смещение в ряду х, столбце у в mth карте признаков субдискретизирующего слоя S определяют с помощью следующего уравнения:
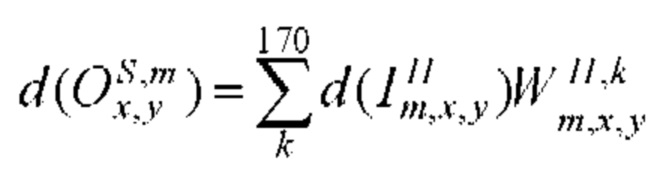
[00161] Входное смещение в ряду х, столбце у в mth карте признаков субдискретизирующего слоя S определяют с помощью следующего уравнения: 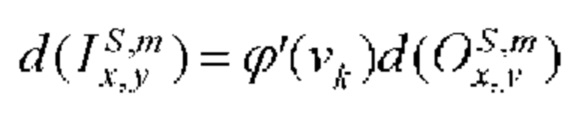
[00162] Вес и изменение смещения в ряду х, столбце у в mth карте признаков субдискретизирующего слоя S и сверточного слоя С определяют с помощью следующего уравнения:
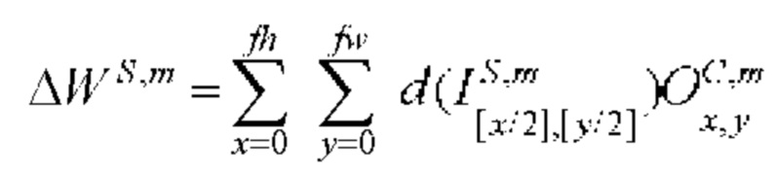
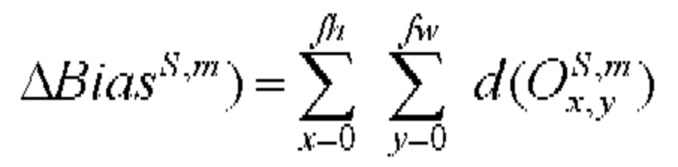
[00163] Выходное смещение в ряду х, столбце у в kth карте признаков сверточного слоя С определяют с помощью следующего уравнения:
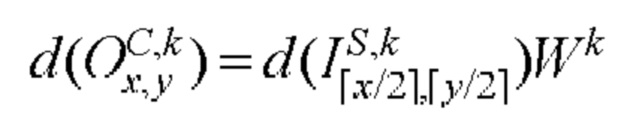
[00164] Входное смещение в ряду х, столбце у в kth карте признаков сверточного слоя С определяют с помощью следующего уравнения:
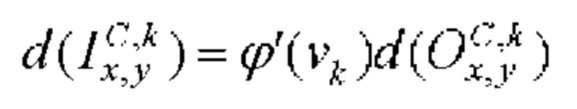
[00165] Вес и изменение смещения в ряду r, столбце с в mth ядре свертки kth карты признаков  сверточного слоя С:
сверточного слоя С:
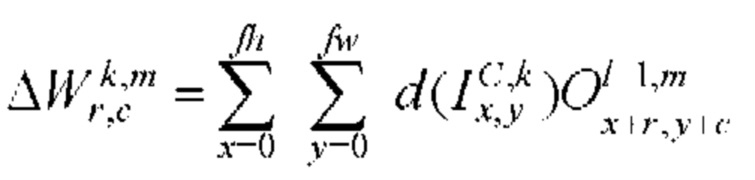
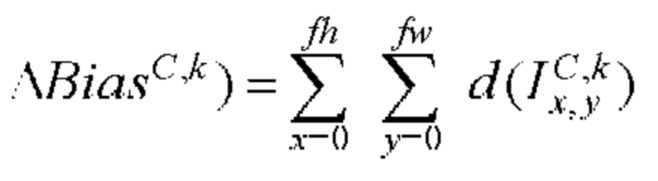
Остаточные соединения
[00166] На фиг. 9 показано остаточное соединение, которое повторно подает первичную информацию ниже по ходу потока посредством добавления карты признаков. Остаточное соединение включает повторную подачу предыдущих представлений в дальнейший поток данных посредством добавления тензора более ранних выходных данных к тензору более поздних выходных данных, что помогает предотвратить потери информации по ходу потока обработки данных. Остаточные соединения обладают двумя общими проблемами, которые наносят вред любой крупномасштабной модели глубокого обучения: исчезающие градиенты и узкие места, связанные со способом задания функций. В целом, добавление остаточных соединений в любую модель, имеющую более 10 слоев, наиболее вероятно обеспечит преимущество. Как описано выше, остаточное соединение включает обеспечение доступности выходных данных более раннего слоя в качестве входных данных более позднего слоя, что с фактически создает короткий путь в последовательной сети. Вместо того, чтобы быть конкатенированными к более поздним активациям, более ранние выходные данные суммируют с более поздними активациями, что предполагает, что обе активации имеют одинаковый размер. Если они имеют различные размеры для изменения формы более ранней активации до целевой формы может быть использовано линейное преобразование.
Остаточное обучение и соединения с пропуском
[00167] На фиг. 10 показан один вариант осуществления остаточных блоков и соединений с пропуском. Основная идея остаточного обучения заключается в том, что остаточное преобразование является более легким для обучения, чем первоначальное преобразование. Остаточная сеть собирает в стек некоторое количество остаточных блоков для того, чтобы уменьшить ухудшение точности обучения. Остаточные блоки используют особые дополнительные соединения с пропуском для борьбы с исчезающими градиентами в глубоких нейронных сетях. В начале остаточного блока поток данных разделяют на два потока: первый переносит неизменные входные данные блока, а второй применяет веса и нелинейности. В конце блока два потока сливаются с использованием поэлементного суммирования. Основным преимуществом таких структур является обеспечение более легкого протекания градиента через сеть.
[00168] Пользуясь преимуществом остаточной сети глубокие сверточные нейронные сети (CNN) могут быть легко обучены и можно достичь повышенной точности для классификации изображений и обнаружения объектов. Сверточные сети с прямой связью соединяют выходные данные 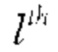 слоя в качестве входных даны
слоя в качестве входных даны 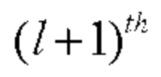 слоя, что обеспечивает следующий переход между слоями:
слоя, что обеспечивает следующий переход между слоями: 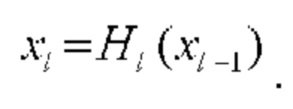 Остаточные блоки добавляют соединения с пропусками, которые обходят нелинейные преобразования с функцией идентификации:
Остаточные блоки добавляют соединения с пропусками, которые обходят нелинейные преобразования с функцией идентификации: 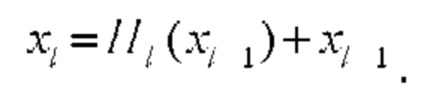 Преимущество остаточных блоков заключается в том, что градиент может проходить непосредственно через функцию идентификации от более поздних слоев к более ранним слоям. Однако функция идентификации и выходные данные
Преимущество остаточных блоков заключается в том, что градиент может проходить непосредственно через функцию идентификации от более поздних слоев к более ранним слоям. Однако функция идентификации и выходные данные 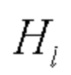 объединяют посредством суммирования, что может препятствовать потоку информации в сети.
объединяют посредством суммирования, что может препятствовать потоку информации в сети.
WaveNet
[00169] WaveNet (WN) представляет собой глубокую нейронную сеть для выработки исходных звуковых сигналов. WaveNet отличается от других сверточных сетей, так как она способна обрабатывать относительно сравнительно большие «визуальные области» малыми ресурсами. Более того, она способна приводить сигналы к требуемым условиям локально и глобально, что позволяет использовать WaveNet в качестве движка перевода текста в речь (TTS) со множеством голосов, при этом TTS дает локальное приведение к требуемым условиям, а конкретный голос -глобальное приведение к требуемым условиям.
[00170] Основные строительные блоки WaveNet представляют собой каузальные разреженные свертки. В качестве разрежения каузальных разреженных сверток WaveNet также позволяет собирать в стеки указанные сборки, как показано на фиг. 11. Для получения такой же рецептивной области с разреженными свертками на данной фигуре необходим другой расширяющий слой. Стеки представляют собой повторение разреженных сверток, что обеспечивает соединение выходных данных разреженного сверточного слоя в одни выходные данные. Это обеспечивает получение WaveNet большой «визуальной» области одного выходного узла с использованием сравнительно малых вычислительных ресурсов. Для сравнения, для получения визуальной области с 512 входными данными полностью сверточной сети (FCN) понадобится 511 слоев. В случае разреженной сверточной сети нам понадобится восемь слоев. Собранные в стек разреженные свертки потребуют только семь слоев с двумя стеками или шесть слоев с четырьмя стеками. Для получения представления о различиях в потребляемых вычислительных ресурсах, требуемых для покрытия одной и той же визуальной области, в приведенной ниже таблице показано количество весов, требуемое в сети с условием одного фильтра на слой и шириной фильтра равной двум. Кроме того, принято, что сеть использует двоичное восьмибитное кодирование.

[00171] WaveNet добавляет соединение с пропуском перед тем как выполнено остаточное соединение, что обеспечивает обход всех последующих остаточных блоков. Каждое из указанных соединений с пропусками суммируют перед их проходом через последовательности функций активации и сверток. Интуитивно это представляет собой сумму информации, извлеченной в каждом слое.
[00172]
Пакетная нормализация (Batch normalization)
[00173] Пакетная нормализация представляет собой способ ускорения обучения глубоких сетей посредством того, что стандартизацию данных делают неотъемлемой частью архитектуры сети. Пакетная нормализация может адаптивным образом нормализовывать данные даже при изменении среднего и дисперсии со временем в процессе обучения. Это работает посредством внутреннего поддержания экспоненциально изменяющегося среднего значения среднего и дисперсии данных для каждого пакета данных, наблюдаемых во время обучения. Основной эффект нормализации пакетов данных заключается в том, что она помогает распространению градиента, наподобие остаточным соединениям, и тем самым обеспечивает получение глубоких сетей. Некоторые очень глубокие сети могут быть обучены, если они включают множество слоев с пакетной нормализации.
[00174] Пакетная нормализация может выглядеть как еще один слой, который может быть вставлен в архитектуру модели, как плотно соединенный или сверточный слой. Слой с пакетной нормализацией (BatchNormalization layer) обычно используют после сверточного или плотно соединенного слоя. Также его могут использовать перед сверточным или Плотно соединенным слоем. Оба варианта осуществления могут быть использованы в раскрытой технологии и показаны на фиг. 15. Пакетная нормализация берет аргумент оси, который определяет ось признаков, которую необходимо нормализовать. Этот аргумент по умолчанию имеет значение -1, последняя ось во входном тензоре. Это корректное значение при использовании слоев Dense, слоев Conv1D, слоев RNN и слоев Conv2D с data format (данные формат), установленным на «channels last» (каналы последний). Однако при нишевом использовании слоев Conv2D с dataformat установленным на "channels first" (каналы последний), признаки оси представляют собой axis 1 (ось 1); аргумент оси при BatchNormalization может быть установлен на 1.
[00175] Пакетная нормализация обеспечивает определение для прямой подачи входных данных и вычисления градиентов относительно параметров и их собственных входных данных посредством обратного прохода. На практике, слои с пакетной нормализацией вставлены после сверточного или плотно соединенного слоя, но перед подачей выходных данных в функцию активации. Для сверточных слоев различные элементы одной карты признаков, т.е. активации, в различных положениях нормализованы одинаковым образом для того, чтобы подчиняться свойству свертки. Таким образом, все активации в малом пакете данных (mini-batch) нормализованы по всем положениям, а не на каждую активацию.
[00176] Внутреннее ковариантное смещение представляет собой главную причину, почему глубокие архитектуры, как хорошо известно, нужно было долго обучать. Это вызвано тем фактом, что глубокие сети не только должны обучаться новому способу задания функций на каждом слое, но также должны учитывать изменение их распределения.
[00177] Ковариантное смещение в целом является известной проблемой в области глубокого обучения и часто встречается в проблемах в реальном мире. Известной проблемой ковариантного смещения является разница в распределении обучающего и тестового набора, что может привести к неоптимальной эффективности обобщения. Эту проблему обычно решают этапом стандартизации или предобработки выбеливанием. Однако особенно операция выбеливания является ресурсозатратной и, таким образом, непрактичной в онлайн системах, особенно, если ковариантное смещение происходит в различных слоях.
[00178] Внутреннее ковариантное смещение представляет собой феномен, при котором распределение активаций сети изменяется в слоях вследствие изменения параметров сети во время обучения. В идеале, каждый слой должен быть преобразован в пространство, в котором они имеют одинаковое распределение, но функциональное взаимодействие остается тем же. Для того, чтобы избежать ресурсозатратных вычислений ковариантных матриц для того, чтобы декореллировать и выбелить данные на каждом слое и этапе, авторы изобретения нормализовали распределение каждого входного признака в каждом слое в каждом малом пакете данных для того, чтобы иметь его нулевое среднее и стандартное отклонение.
Прямой проход
[00179] Во время прямого прохода вычисляют среднее и дисперсию малого пакета данных. С такими статистическими показателями малого пакета данных данные нормализуют путем вычитания среднего и деления на стандартное отклонение. Наконец, данные масштабируют и смещают с изученными параметрами масштабирования и смещения. Прямой проход ƒBN нормализации пакетов данных показан на фиг. 12.
[00180] На фиг. 12 μβ представляет собой среднее пакета данных, а 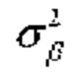 представляет собой дисперсию пакета данных соответственно. Изученные параметры масштабирования и смещения обозначены как γ и β соответственно. Для ясности, процедура нормализации пакетов данных описана в настоящем документе для активации и опускает соответствующие показатели.
представляет собой дисперсию пакета данных соответственно. Изученные параметры масштабирования и смещения обозначены как γ и β соответственно. Для ясности, процедура нормализации пакетов данных описана в настоящем документе для активации и опускает соответствующие показатели.
[00181] Так как нормализация является дифференцируемым преобразованием, ошибки распространяются в указанные изученные параметры и тем самым способны восстановить репрезентативную способность сути путем обучения тождественному преобразованию. В отличие от этого, путем изучения параметров масштабирования и смещения, которые идентичны соответствующим статистическим показателям пакета данных, преобразование с пакетной нормализацией не имело бы эффекта на сеть, если это было бы оптимальной операцией к выполнению. Во время тестирования среднее и дисперсия пакета данных заменены соответствующими статистическими показателями выборки, так как входные данные не зависят от других образцов из малого пакета данных. Другой способ заключается в удержании скользящих средних значений статистических показателей пакета данных во время обучения и в использовании их для вычисления выходных данных сети во время тестирования. Во время тестирования преобразование с пакетной нормализацией может быть выражено как показано на фиг. 13. На фиг. 13 μD и 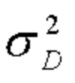 обозначают среднее и дисперсию выборки, а не статистические показатели пакета данных, соответственно.
обозначают среднее и дисперсию выборки, а не статистические показатели пакета данных, соответственно.
Обратный проход
[00182] Так как нормализация является дифференцируемой операцией, обратный проход может быть вычислен как показано на фиг. 14.
Одномерная (1D) свертка
[00183] Одномерные свертки извлекают локальные одномерные вставки или частичные последовательности из последовательностей, как показано на фиг. 16. Одномерная свертка получает каждый выходной шаг по времени из временной вставки во входной последовательности. Одномерные сверточные слои распознают локальные паттерны в последовательности. Так как та же самая входная информация выполняется над каждой вставкой, вставка, изученная в определенном положении во входной последовательности позже может быть распознана в другом положении, что делает одномерные сверточные слои инвариантными в перемещению для временных перемещений. Например, одномерный сверточный слой, обрабатывающий последовательности оснований с использованием окон свертки размером 5, должен быть способен изучать основания или последовательности оснований длиной 5 или менее, и он должен быть способен распознавать основные мотивы в любом контексте во входной последовательности. Одномерная свертка основного уровня таким образом способна учиться в отношении морфологии оснований.
Глобальное среднее объединение
[00184] На фиг. 17 показано, как работает глобальное среднее объединение (GAP). Глобальное среднее объединение может быть использовано для замены плотно соединенных (FC) слоев для классификации посредством взятия средних значений в последнем слое оценки. Это позволяет сократить обучающую нагрузку и обойти проблемы с переподгонкой. Глобальное среднее объединение применяет структурные априорные данные к модели и это эквивалентно линейному преобразованию с заданными весами. Глобальное среднее объединение уменьшает количество параметров и устраняет плотно соединенные слои. Плотно соединенные слои обычно являются наиболее загруженными с точки зрения параметров и соединений слоями, а глобальное среднее объединение обеспечивает значительно менее ресурсозатратный подход к достижению аналогичных результатов. Основная идея глобального среднего объединения заключается в создании среднего значения из каждой последней карты признаков слоя как коэффициента достоверности для оценки, подаваемого непосредственно в softmax слой.
[00185] Глобальное среднее объединение имеет три преимущества: (1) отсутствуют дополнительные параметры в слоях с глобальным средним объединением, тем самым позволяя избежать переподгонки в указанных слоях; (2) так как выходные данные глобального среднего объединения является среднее всей карты признаков, то глобальное среднее объединение будет более устойчивым к пространственным перемещениям; и (3) вследствие огромного числа параметров в плотно соединенных слоях, которое обычно занимает более 50% всех параметров всей сети, их замена слоями с глобальным средним объединением может значительно уменьшить размер модели, и это делает глобальное среднее объединение очень полезным при сжатии моделей.
[00186] Глобальное среднее объединение является целесообразным, так как ожидается, что более эффективные признаки в последнем слое будут иметь большее среднее значение. В некоторых вариантах осуществления глобальное среднее объединение может быть использовано в качестве посредника для оценки классификации. Карты признаков при глобальном среднем объединении могут быть интерпретированы как карты достоверности, и обеспечивают соответствие между картами признаков и категориями. Глобальное среднее объединение может быть особенно эффективным, если признаки последнего слоя достаточно абстрактны для прямой классификации, глобального среднего объединения в отдельности не достаточно, если многоуровневые признаки должны быть комбинированы в группы наподобие частичных моделей, что наилучшим образом выполняется путем добавления простого плотно соединенного слоя или другого классификатора после Глобального среднего объединения.
Терминология
[00187] Все литературные источники и аналогичный материал, цитируемый в настоящей заявке, в том числе, но не ограничиваясь перечисленным, патенты, патентные заявки, статьи, книги, научные работы и веб-страницы, независимо от формата таких литературных источников и аналогичных материалов, явным образом и полностью включены в настоящий документ посредством ссылок. В тех случаях, когда один или более из включенных литературных источников и аналогичных материалов отличается от настоящей заявки или противоречит ей, в том числе, но не ограничиваясь перечисленным, определяемые термины, силу будет иметь настоящая заявка.
[00188] В настоящем документе следующие термины имеют указанные значения.
[00189] Основание относится к нуклеотидному основанию или нуклеотиду, А (аденину), С (цитозину), Τ (тимину) или G (гуанину).
[00190] В настоящей заявке взаимозаменяемо используются термины «белок» и «транслируемая последовательность».
[00191] В настоящей заявке взаимозаменяемо используются термины «кодон» и «триплет оснований».
[00192] В настоящей заявке взаимозаменяемо используются термины «аминокислота» и «транслируемая единица».
[00193] В настоящей заявке взаимозаменяемо используются выражения «классификатор патогенное™ вариантов», «классификатор на основе сверточной нейронной сети для классификации вариантов» и «классификатор на основе глубокой сверточной нейронной сети для классификации вариантов».
[00194] Термин «хромосома» относится к носителю генов, передающих наследственные признаки, в живой клетке, происходящему из нитей хроматина, содержащих ДНК и белковые компоненты (в частности, гистоны). В настоящем документе используется стандартная международно признанная система нумерации индивидуальных хромосом генома человека.
[00195] Термин «сайт» относится к уникальному положению (например, идентификатору хромосомы, положению и ориентации хромосомы) на референсном геноме. В некоторых вариантах реализации сайт может представлять собой остаток, метку последовательности или положение сегмента на последовательности. Термин «локус» может применяться для обозначения специфической локализации последовательности нуклеиновой кислоты или полиморфизма на референсной хромосоме.
[00196] Термин «образец» в настоящем документе относится к образцу, как правило, происходящему из биологической жидкости, клетки, ткани, органа или организма, содержащего нуклеиновую кислоту или смесь нуклеиновых кислот, содержащую по меньшей мере одну последовательность нуклеиновой кислоты, подлежащую секвенированию и/или фазированию. Такие образцы включают, не ограничиваясь перечисленными, образцы мокроты/жидкости ротовой полости, амниотической жидкости, крови, фракции крови, тонкоигольной биопсии (например, хирургической биопсии, тонкоигольной биопсии и т.п.), мочи, жидкости брюшной полости, плевральной жидкости, эксплантата ткани, культуры органа и любого другого препарата ткани или клеток, или его фракции или производного, или выделенные из них образцы. Хотя образец часто получают от субъекта-человека (например, пациента), образцы могут быть взяты из любого организма, имеющего хромосомы, в том числе, но не ограничиваясь перечисленными, организма собак, кошек, лошадей, коз, овец, крупного рогатого скота, свиней и т.п. Образец может применяться непосредственно в полученном из биологического источника виде или после предварительной обработки для модификации характера образца. Например, такая предварительная обработка может включать получение плазмы из крови, разведение вязких текучих сред и т.д. Методы предварительной обработки могут также включать, не ограничиваясь перечисленными, фильтрацию, осаждение, разведение, дистилляцию, смешивание, центрифугирование, замораживание, лиофилизацию, концентрацию, амплификацию, фрагментацию нуклеиновых кислот, инактивацию мешающих компонентов, добавление реагентов, лизис и т.п.
[00197] Термин «последовательность» включает или обозначает цепь взаимно сопряженных нуклеотидов. Нуклеотиды могут быть основаны на ДНК или РНК. Следует понимать, что одна последовательность может включать несколько субпоследовательностей. Например, одна последовательность (например, ПЦР-ампликона) может содержать 350 нуклеотидов. Рид образца может включать несколько субпоследовательностей в пределах указанных 350 нуклеотидов. Например, рид образца может включать первую и вторую фланкирующие субпоследовательности, содержащие, например, 20 50 нуклеотидов. Указанные первая и вторая фланкирующие субпоследовательности могут быть локализованы на любой стороне повторяющегося сегмента, содержащего соответствующую субпоследовательность (например, 40 100 нуклеотидов). Каждая из фланкирующих субпоследовательностей может включать (или включать частично) субпоследовательность праймера (например, 10-30 нуклеотидов). Для простоты чтения вместо термина «субпоследовательность» используют «последовательность», но следует понимать, что две последовательности не обязательно отделены одна от другой на общей цепи. Для различения различных последовательностей, описанных в настоящем документе, в указанные последовательности могут быть включены разные метки (например, целевая последовательность, праймерная последовательность, фланкирующая последовательность, референсная последовательность и т.п.). В другие объекты, такие как описываемые термином «аллель», могут быть включены разные метки для дифференциации сходных объектов.
[00198] Термин «парно-концевое секвенирование» относится к способам секвенирования с секвенированием обоих концов целевого фрагмента. Парно-концевое секвенирование может облегчать детекцию геномных перестановок и повторяющихся сегментов, а также слитых генов и новых транскриптов. Методология парно-концевого секвенирования описана в РСТ-публикации WO 07010252, РСТ-публикации сер. № PCTGB2007/003798 и опубликованной заявке на патент США US 2009/0088327, каждая из которых включена посредством ссылки в настоящий документ. Согласно одному примеру может быть выполнен следующий ряд операций; (а) генерация кластеров нуклеиновых кислот; (b) линеаризация указанных нуклеиновых кислот; (с) гибридизация первого праймера для секвенирования и проведение многократных циклов удлинения, сканирования и деблокирования согласно описанию выше; (d) "инверсия» целевых нуклеиновых кислот на поверхности проточной ячейки путем синтеза комплементарной копии; (е) линеаризация ресинтезированной цепи; и (f) гибридизация второго праймера для секвенирования и проведения многократных циклов удлинения, сканирования и деблокирования согласно описанию выше. Операция инверсии может быть проведена с доставкой реагентов согласно описанию выше для одного цикла мостиковой амплификации.
[00199] Термин «референсный геном» или «референсная последовательность» относится к любой конкретной известной последовательности генома, частичной или полной, любого организма, которая может быть использована в качестве референсной для идентифицированных последовательностей субъекта. Например, референсный геном, используемый для субъектов-людей, а также многих других организмов можно найти по ссылке ncbi.nlm.nih.gov от Национального центра биотехнологической информации. "Геном» относится к полной генетической информации организма или вируса, представленной в виде последовательностей нуклеиновых кислот. Геном включает как гены, так и некодирующие последовательности ДНК. Референсная последовательность может быть длиннее ридов, которые на нее выравнивают. Например, она может быть по меньшей мере приблизительно в 100 раз длиннее, или по меньшей мере приблизительно в 1000 раз длиннее, или по меньшей мере приблизительно в 10000 раз длиннее, или по меньшей мере приблизительно в 105 раз длиннее, или по меньшей мере приблизительно 106 раз длиннее, или по меньшей мере приблизительно в 107 раз длиннее. В одном примере референсная последовательность генома представляет собой последовательность полноразмерного генома человека. В другом примере референсная последовательность генома ограничена специфической хромосомой человека, такой как хромосома 13. В некоторых вариантах реализации референсная хромосома представляет собой последовательность хромосомы из генома человек версии hg19. Такие последовательности могут называться референсными последовательностями хромосомы, хотя предполагается, что термин «референсный геном» охватывает такие последовательности. Другие примеры референсных последовательностей включают геномы других видов, а также хромосом, субхромосомных областей (например, цепей) и т.п., любых видов. В различных вариантах реализации референсный геном представляет собой консенсусную последовательность или другую комбинацию, полученную от нескольких индивидуумов. Однако в определенных вариантах применения референсная последовательность может быть получена от конкретного индивидуума.
[00200] Термин «рид» относится к совокупности данных о последовательности, описывающих фрагмент нуклеотидного образца или референсной последовательности. Термин «рид» может относиться к риду образца и/или референсному риду. Обычно, хотя не обязательно, рид представлен короткой последовательностью непрерывно расположенных пар оснований в образце или референсной последовательности. Рид может быть символически представлен последовательностью пар оснований (ATCG) образца или референсного фрагмента. Он может храниться в запоминающем устройстве и обрабатываться подходящим образом для определения того, совпадает ли рид с референсной последовательностью или отвечает ли другим критериям. Рид может быть получен непосредственно из аппарата для секвенирования или непрямо, из сохраненной информации о последовательности, касающейся указанного образца. В некоторых случаях рид представляет собой последовательность ДНК достаточной длины (например, по меньшей мере приблизительно 25 п. о.) которые могут применяться для идентификации последовательности или области большей длины, например, например, которая может быть выравнена и специфическим образом соотнесена с хромосомой, или геномной областью, или генов.
[00201] Методы секвенирования следующего поколения включают, например, технологию секвенирования путем синтеза (Illumina), пиросеквенирование (454), технологию ионного полупроводникового секвенирования (секвенирование Ion Torrent), одномолекулярное секвенирование в реальном времени (Pacific Biosciences) и секвенирование путем лигирования (секвенирование SOLiD). В зависимости от методов секвенирования длина каждого рида может варьировать от приблизительно 30 п.о. до более 10000 п.о. Например, метод секвенирования Illumina с использованием секвенатора SOLiD генерирует риды нуклеиновых кислот длиной приблизительно 50 п. о. В другом примере секвенирование Ion Torrent генерирует риды нуклеиновых кислот длиной до 400 п.о., а пиросеквенирование 454 генерирует риды нуклеиновых кислот длиной приблизительно 700 п.о. В еще одном примере способы одномолекулярного секвенирования в реальном времени могут генерировать риды длиной от 10000 п.о. до 15000 п.о. Соответственно, в определенных вариантах реализации риды последовательностей нуклеиновых кислот имеют длину 30-100 п.о., 50-200 п.о. или 50-400 п.о.
[00202] Термины "рид образца», «последовательность образца» или «фрагмент образца» относятся к данным представляющей интерес геномной последовательности из образца. Например, рид образца содержит данные о последовательности из ПЦР-ампликона, содержащего последовательности прямого и обратного праймера. Данные о последовательности могут быть получены с применением любого выбранного метода секвенирования. Рид образца может быть получен, например, в результате реакции секвенирования путем синтеза (SBS), реакции секвенирования путем лигирования или любого другого подходящего метода секвенирования, для которого требуется определение длины и/или идентичности повторяющегося элемента. Рид образца может представлять собой консенсусную (например, усредненную или взвешенную) последовательность, полученную из нескольких ридов образца. В некоторых вариантах реализации получение референсной последовательности включает идентификацию представляющего интерес локуса на основании последовательности праймера из ПЦР-ампликона.
[00203] Термин "необработанный фрагмент» относится к данным о последовательности части представляющей интерес геномной последовательности, которая по меньшей мере частично перекрывает заданное положение или представляющее интерес вторичное положение в риде образца или фрагменте образца. Неограничивающие примеры необработанных фрагментов включают дуплексный фрагмент со сшивкой, симплексный фрагмент со сшивкой, дуплексный фрагмент без сшивки и симплексный фрагмент без сшивки. Термин "необработанный» используют, чтобы показать, что необработанный фрагмент включает данные о последовательности, определенным образом связанные с данными о последовательности в риде образца, независимо от того, демонстрирует ли необработанный фрагмент подтверждающий вариант, который соответствует и удостоверяет или подтверждает потенциальный вариант в риде образца. Термин "необработанный фрагмент» не указывает на то, что указанный фрагмент обязательно включает подтверждающий вариант, валидирующий распознанный вариант в риде образца. Например, если приложением для распознавания вариантов определено, что рид образца демонстрирует первый вариант, указанное приложение для распознавания вариантов может определить, что в одном или более необработанных фрагментах отсутствует соответствующий тип «подтверждающего» варианта, наличие которого в ином случае можно ожидать на основании варианта в риде образца.
[00204] Термины "картирование», «выравненный», «выравнивание» относятся к процессу сравнения рида или метки с референсной последовательностью, с определением таким образом того, содержит ли указанная референсная последовательность содержит последовательность рида. Если референсная последовательность содержит рид, указанный рид может быть картирован на указанную референсную последовательность или, в определенных вариантах реализации, на конкретное место в референсной последовательности. В некоторых случаях выравнивание просто показывает, входит ли рид в состав конкретной референсной последовательности (т.е. присутствует или отсутствует указанный рид в референсной последовательности). Например, выравнивание рида на референсную последовательность хромосомы 13 человека показывает, присутствует ли указанный рид в указанной референсной последовательности хромосомы 13. Инструмент, который обеспечивает получение указанной информации, может называться тестировщиком принадлежности множеству. В некоторых случаях выравнивание, кроме того, указывает на место в референсной последовательности, куда картируется рид или метка. Например, если референсная последовательность представляет собой полную последовательность генома человека, выравнивание может показать, что рид присутствует на хромосоме 13, и может дополнительно показать, что рид располагается в конкретной цепи и/или сайте хромосомы 13.
[00205] Термин "индел" относится к инсерции и/или делеции оснований в ДНК организма. Микроиндел представляет собой индел, который приводит к чистому изменению 1-50 нуклеотидов. В кодирующих областях генома, за исключением случаев, когда длина индела кратна 3, он дает мутацию со сдвигом рамки. Инделы могут быть противопоставлены точечным мутациям. Индел инсертирует и делетирует нуклеотиды в последовательности, тогда как точечная мутация представляет собой форму замены, при которой один из нуклеотидов заменяют без изменения общего числа в ДНК. Инделы могут также быть противопоставлены тандемной мутации оснований (ТВМ), которая может быть определена как замена нуклеотидов в смежных положениях ("вариант" относится к последовательности нуклеиновой кислоты, отличающейся от референсной нуклеиновой кислоты).
[00206] Термин "вариант" относится к последовательности нуклеиновой кислоты, отличающейся от референсной нуклеиновой кислоты. Типичный вариант последовательности нуклеиновой кислоты включает, без ограничения, однонуклеотидный полиморфизм (SNP), короткие делеционные и инсерционные полиморфизмы (индел), вариацию числа копий (CNV), микросателлитные маркеры или короткие тандемные повторы, и структурную вариацию. Распознавание соматических вариантов представляет собой попытку идентификации вариантов, присутствующих в образце ДНК с низкой частотой. Распознавание соматических вариантов представляет интерес в контексте лечения рака. Образец ДНК из опухоли обычно являются гетерогенным и включает некоторое число нормальных клеток, некоторое число клеток ранней стадии прогрессирования рака (с меньшим количеством мутаций) и некоторое число клеток поздней стадии (с большим количеством мутаций). Из-за указанной гетерогенности при секвенировании опухоли (например, из фиксированного формалином и залитого в парафин (FFPE) образца) соматические мутации часто появляется с низкой частотой. Например, однонуклеотидная вариация (SNV) наблюдается только в 10% ридов, захватывающих заданное основание. Вариант, который подлежит классификации как относящийся к соматической или зародышевой линии классификатором вариантов, также называется в настоящем документе «тестируемым вариантом».
[00207] Термин «шум» относится к ошибочно распознанному варианту, полученному в результате одной или более ошибок в процессе секвенирования и/или в приложении для распознавания вариантов.
[00208] Термин «частота варианта» относится к относительной частоте аллеля (варианта гена) в конкретном локусе в популяции, выраженной в виде доли или процента. Например, указанные доля или процент могут быть представлены долей всех хромосом в популяции, несущих указанный аллель. Например, частота варианта в образце представляет собой относительную частоту аллеля/варианта в конкретном локусе/положении вдоль представляющей интерес геномной последовательности в «популяции», соответствующей числу ридов и/или образцов, полученных для указанной представляющей интерес геномной последовательности от индивидуума. В другом примере исходная частота варианта представляет собой относительную частоту аллеля/варианта в конкретном локусе/положении вдоль одной или более исходных геномных последовательностей, где «популяция» соответствует числу ридов и/или образцов, полученных для одной или более исходных геномных последовательностей из популяции здоровых индивидуумов.
[00209] Термин «частота варианта аллеля (VAF)» относится к наблюдаемому проценту секвенированных ридов, совпадающих с указанным вариантом, разделенному на общее покрытие в целевом положении. VAF представляет собой показатель пропорции секвенированных ридов, несущих указанный вариант.
[00210] Термины «положение», «заданное положение» и «локус» относятся к месту или координатам одного или более нуклеотидов в составе последовательности нуклеотидов. Термины «положение», «заданное положение» и «локус» также относятся к месту или координатам одной или более пар оснований в последовательности нуклеотидов.
[00211] Термин «гаплотип» относится к комбинации аллелей в смежных сайтах на хромосоме, наследуемых вместе. Гаплотип может быть представлен одним локусом, несколькими локусами или всей хромосомой в зависимости от числа событий рекомбинации, произошедших между локусами в определенном наборе локусов, если они вообще происходили.
[00212] Термин «порог» в настоящем документе относится к численному или не-численному значению, которое применяют в качестве значения отсечения для характеризации образца, нуклеиновой кислоты или их части (например, рида). Порог может варьировать на основании результатов эмпирического анализа. Порог можно сравнивать с измеренным или рассчитанным значением для определения того, должен ли источник таких предполагаемых значений быть классифицирован конкретным образом. Выбор порога зависит от уровня доверительности, с которым пользователь желает получить при осуществлении классификации. Порог может быть выбран с конкретной целью (например, для достижения баланса чувствительности и селективности). В настоящем документе порог» указывает на точку, в которой ход анализа может быть изменен, и/или точку, в которой может быть запущено действие. Порог не обязательно должен представлять собой заранее заданное число. Вместо этого порог может представлять собой, например, функцию, основанную на множестве факторов. Порог может быть адаптивно регулируемым с учетом обстоятельств. Кроме того, порог может задавать верхний предел, нижний предел или диапазон между пределами.
[00213] В некоторых вариантах реализации меру или оценку (балл, score), основанная(ый) на данных секвенирования, можно сравнивать с порогом. В настоящем документе термины «мера» или «оценка» могут включать значения или результаты, определенные исходя из данных секвенирования, или могут включать функции, основанные на значениях или результатах, определенных исходя из данных секвенирования. Как и порог, мера или оценка могут быть адаптивно регулироваться с учетом обстоятельств. Например, метрика или оценка может представлять собой нормированное значение. В качестве примера оценки или меры один или более вариантов реализации может задействовать показатели подсчитанных количеств при анализе данных, оценка подсчитанного количества может быть основан на числе ридов образца. Счетная оценка подсчитанного количества может быть основана на числе ридов образца. Риды образца могут быть подвергнуты одной или более стадий фильтрации, таким образом, чтобы они обладали по меньшей мере одной общей характеристикой или одним общим качеством. Например, каждый из ридов образца, который используют для определения оценки подсчитанного количества, может быть выравнен по референсной последовательности или может быть определен как потенциальный аллель. Может быть подсчитано число ридов образца, обладающих общей характеристикой, для определения подсчитанного количества ридов. Счетные оценки могут быть основаны на подсчитанном количестве ридов. В некоторых вариантах реализации счетная оценка может представлять собой значение, равное подсчитанному количеству ридов. Согласно другим вариантам реализации счетная оценка может быть основана на подсчитанном количестве ридов и другой информации. Например, счетная оценка может быть основана на подсчитанном количестве ридов для конкретного аллеля генетического локуса и общего числа ридов для генетического локуса. В некоторых вариантах реализации счетные оценки могут быть основаны на подсчитанном количестве ридов и ранее полученных данных для генетического локуса. В некоторых вариантах реализации счетные оценки могут представлять собой нормированные показатели между заранее заданными значениями. Счетная оценка может также представлять собой функцию от подсчитанных количеств ридов из других локусов образца или функцию от подсчитанных количеств ридов из других образцов, которые анализировали одновременно с представляющим интерес образцом. Например, счетная оценка может представлять собой функцию от подсчитанного количества ридов конкретного аллеля и подсчитанных количеств ридов других локусов в образце, и/или подсчитанных количества ридов из других образцов. В одном примере подсчитанные количества ридов из других локусов и/или подсчитанные количества ридов из других образцов могут быть использованы для нормирования оценки подсчитанного количества для конкретного аллеля.
[00214] Термины «покрытие» или «покрытие фрагмента» относятся к подсчитанному количеству или другой мере ряда ридов образца для одного и того же фрагмента последовательности. Подсчитанное количество ридов может представлять собой подсчитанное количество ридов, покрывающих соответствующий фрагмент. Как вариант, покрытие может быть определено путем умножения подсчитанного количества ридов на заданный коэффициент, основанный на ретроспективной информации, информации об образце, информации о локусе и т.п.
[00215] Термин «глубина считывания» (обычно в виде числа с последующим символом «×») относится к числу секвенированных ридов, перекрывающихся при выравнивании в целевом положении. Его часто выражают через среднее значение или процент, превышающий значение отсечения на протяжении множества интервалов (таких как экзоны, гены или панели). Например, в клиническом заключении может быть сказано, что среднее покрытие панели составляет 1,105× при 98% покрытии целевых оснований >100×.
[00216] Термины «оценка качества распознавания оснований» или «оценка Q» относятся к вероятности по шкале PHRED в диапазоне от 0 20, обратно пропорциональной вероятности того, что отдельное секвенированное основание является корректным. Например, распознанное основание Т с Q, равным 20, считают вероятно корректным с достоверностью, соответствующей Р-значению 0,01. Любые распознанные основания с Q<20 должны считаться результатами низкого качества, и любой идентифицированный вариант с существенной пропорцией имеющих низкое качество секвенированных ридов, подтверждающих указанный вариант, должен считаться потенциально ложноположительным.
[00217] Термины «риды вариантов» или «число ридов вариантов» относятся к числу секвенированных ридов, свидетельствующих о присутствии указанного варианта.
Процесс секвенирования
[00218] Варианты реализации, представленные в данном документе, могут быть применимы к анализу последовательностей нуклеиновых кислот для идентификации вариаций последовательностей. Варианты реализации могут применяться для анализа потенциальных вариантов / аллелей генетического положения / локуса и определения генотипа генетического локуса или, другими словами, обеспечения распознавания генотипа для локуса. В качестве примера, последовательности нуклеиновой кислоты могут быть проанализированы в соответствии со способами и системами, описанными в публикации заявки на патент США №2016/0085910 и публикации заявки на патент США №2013/0296175, полное содержащие которых в явном виде включено в настоящий документ в полном объеме посредством ссылки.
[00219] В одном варианте реализации процесс секвенирования включает получение образца, который содержит или предположительно содержит нуклеиновые кислоты, такие как ДНК. Образец может быть из известного или неизвестного источника, такого как животное (например, человек), растение, бактерии или гриб. Образец может быть взят непосредственно из источника. Например, кровь или слюна могут быть взяты непосредственно от индивидуума. Как вариант, образец может не быть получен непосредственно из источника. Затем один или более процессоров дают системе команду на подготовку образца к секвенированию. Подготовка может включать удаление постороннего материала и/или выделение определенного материала (например, ДНК). Биологический образец может быть подготовлен для включения признаков для конкретного анализа. Например, биологический образец может быть подготовлен для секвенирования путем синтеза (SBS). В некоторых вариантах реализации подготовка может включать амплификацию определенных областей генома. Например, подготовка может включать амплификацию заранее определенных генетических локусов, которые, как известно, включают STR (короткие тандемные повторы)и/или SNP (однонуклеотидные полиморфизмы). Генетические локусы могут быть амплифицированы с использованием предварительно определенных последовательностей праймеров.
[00220] Затем, указанные один или более процессоров передают системе инструкцию секвенировать образец. Секвенирование может осуществляться в соответствии с различными известными протоколами секвенирования. В частных вариантах реализации секвенирование включает SBS. В SBS множество флуоресцентно меченых нуклеотидов используется для последовательности множества кластеров амплифицированной ДНК (возможно, миллионов кластеров), присутствующих на поверхности оптического субстрата (например, поверхности, которая по меньшей мере частично ограничивает канал в проточной ячейке). Проточные ячейки могут содержать образцы нуклеиновых кислот для секвенирования, причем проточные ячейки размещены в соответствующих держателях проточных ячеек.
[00221] Нуклеиновые кислоты могут быть подготовлены таким образом, чтобы они содержали известную последовательность праймера, которая соседствует с неизвестной целевой последовательностью. Чтобы инициировать первый цикл секвенирования SBS, один или несколько нуклеотидов, меченных различным образом, ДНК-полимеразу и т.д., можно подать в проточную ячейку или через нее посредством подсистемы потока жидкости. Можно добавлять либо по одному типу нуклеотида, либо нуклеотиды, используемые в процедуре секвенирования, могут быть специально сконструированы так, чтобы обладать свойством обратимой терминации, что дает возможность одновременного проведения каждого цикла реакции секвенирования в присутствии нескольких типов меченых нуклеотидов (например, А, С, Т, G). Нуклеотиды могут включать обнаруживаемые фрагменты-метки, такие как флуорофоры. Когда четыре нуклеотида смешаны вместе, полимераза может выбрать правильное основание для включения, и каждая последовательность удлиняется на одно основание. Невключенные нуклеотиды можно отмывать потоком промывочного раствора через проточную ячейку. Один или несколько лазеров могут возбуждать нуклеиновые кислоты и вызывать флуоресценцию. Флуоресценция, испускаемая нуклеиновыми кислотами, основана на флуорофорах включенного основания, и разные флуорофоры могут излучать света с разными длинами волн. Деблокирующий реагент может быть добавлен в проточную ячейку для удаления обратимых терминаторных групп из удлинненных и детектированных цепей ДНК. Деблокирующий реагент затем можно отмыть, пропуская промывочный раствор через проточную ячейку. После этого проточная ячейка готова к следующему циклу секвенирования, начиная с введения меченого нуклеотида, как описано выше. Операции с текучей средой и обнаружением могут повторяться несколько раз для завершения последовательности операций. Примеры способов секвенирования описаны, например, в Bentley et al., Nature 456: 53-59 (2008), международной публикации № WO 04/018497, патенте США №7,057,026, международной публикации № WO 91/06678, международной публикации № WO 07/123744, патенте США №7,329,492, патенте США №7,211,414, патенте США №7,315,019, патенте США №7,405,281 и публикации заявки на патент США №2008/0108082, каждый(ая) из которых включен(а) в настоящий документ посредством ссылки.
[00222] В некоторых проявлениях реализации нуклеиновые кислоты могут быть присоединены к поверхности и амплифицированы до или во время секвенирования. Например, амплификация может быть проведена с использованием мостиковой амплификации с образованием кластеров нуклеиновых кислот на поверхности. Применимые методы амплификации описаны, например, в Патенте США №5,641,658, патентной публикации США №2002/0055100, патенте США №7,115,400, патентной публикации США №2004/0096853, патентной публикации США №2004/0002090, патентной публикации США №2007/0128624 и публикации заявки на патент США №2008/0009420, каждый из этих документов полностью включен в настоящую заявку посредством ссылки. Другим полезным способом амплификации нуклеиновых кислот на поверхности является амплификация по типу катящегося кольца (RCA), например, как описано в Lizardi et al., Nat. Genet. 19:225-232 (1998) and U.S. Patent Application Publication No. 2007/0099208 A1, каждый(ая) из которых включен(а) в настоящий документ посредством ссылки.
[00223] Один пример протокола SBS использует модифицированные нуклеотиды, имеющие удаляемые 3'-блоки, например, как описано в международной публикации № WO 04/018497, публикации заявки на патент США №2007/0166705 А1 и патенте США №7057026, каждый(ая) из которых включен(а) в настоящий документ посредством ссылки. Например, реагенты SBS могут доставляться повторяющимися циклами в проточную ячейку, к которой присоединены целевые нуклеиновые кислоты, например, по протоколу мостиковой амплификации. Кластеры нуклеиновых кислот могут быть преобразованы в одноцепочечную форму с использованием линеаризирующего раствора. Линеаризирующий раствор может содержать, например, эндонуклеазу рестрикции, способную расщеплять одну цепь каждого кластера. Линеаризирующий раствор может содержать, например, эндонуклеазу рестрикции, способную расщеплять одну цепь каждого кластера (например, расщепление диольной связи периодатом), расщепление сайтов без оснований путем расщепления эндонуклеазой (например, «USER», которая поставляется компанией NEB, Ипсвич, штат Массачусетс, США, номер компонента (M5505S), (путем воздействия тепла или щелочи, расщепления рибонуклеотидов, включенных в продукты амплификации, в остальном состоящих из дезоксирибонуклеотидов, фотохимического расщепления или расщепления пептидного линкера. После операции линеаризации праймер для секвенирования может быть подан в проточную ячейку в условиях гибридизации праймера для секвенирования с целевыми нуклеиновыми кислотами, которые должны быть секвенированы.
[00224] Затем проточную клетку можно привести в контакт с реагентом-удлинителем SBS, имеющим модифицированные нуклеотиды с удаляемыми 3'-блоками и флуоресцентными метками в условиях, позволяющих удлинить праймер, гибридизованный с каждой целевой нуклеиновой кислотой путем добавления одного нуклеотида. К каждому праймеру добавляется только один нуклеотид, поскольку включение модицифированного нуклеотида в растущую полинуклеотидную, комплементарную секвенируемой области матрицы, обуславливает отсутствие свободной группы 3'-ОН, доступной для направления дальнейшего удлинения последовательности и, следовательно, полимераза, не может добавить дополнительные нуклеотиды. Удлиняющий реагент SBS можно удалить и заменить сканирующим реагентом, содержащим компоненты, которые защищают образец при возбуждении излучением. Примеры компонентов сканирующего реагента описаны в публикации заявки на патент США №2008/0280773 А1 и заявке на патент США №13/018,255, каждый(ая) из которых включен(а) в настоящий документ посредством ссылки. Затем удлиненные нуклеиновые кислоты могут быть детектированы флуоресцентно в присутствии сканирующего реагента. После детектирования флуоресценции 3'-блок может быть удален с использованием деблокирующего реагента, который соответствует используемой блокирующей группе. Примеры деблокирующих реагентов, которые можно применять для соответствующих блокирующих групп, описаны в WO 004018497, US 2007/0166705 А1 и патенте США №7057026, каждый(ая) из которых включен(а) в настоящий документ посредством ссылки. Деблокирующий реагент можно смыть, оставляя целевые нуклеиновые кислоты гибридизованными с удлиненными праймерами, имеющими 3'-OH-группы, к которым теперь можно присоединять другие нуклеотиды. Соответственно, циклы добавления удлиняющего реагента, сканирующего реагента и деблокирующего реагента с необязательными промываниями между одной или несколькими операциями могут повторяться до тех пор, пока не будет получена необходимая последовательность. Вышеуказанные циклы могут быть выполнены с использованием одной операции доставки удлиняющего реагента на цикл, когда к каждому из модифицированных нуклеотидов прикреплена отличная от других метка, о которой известно, что она соответствует конкретному основанию. Различные метки облегчают различение нуклеотидов, добавляемых во время каждой операции включения. В качестве альтернативы, каждый цикл может включать в себя отдельные операции доставки удлиняющего реагента, за которыми следуют отдельные операции доставки и детектирования сканирующего реагента, и в этом случае два или более нуклеотида могут иметь одинаковую метку и могут различаться на основании известного порядка доставки.
[00225] Хотя операция секвенирования обсуждалась выше в отношении конкретного протокола SBS, следует понимать, что при желании могут выполняться другие протоколы для секвенирования любого из множества других молекулярных анализов.
[00226] Затем указанные один или более процессоров системы получают данные секвенирования для последующего анализа. Данные секвенирования могут быть отформатированы различными способами, например, в файле. ВАМ. Данные секвенирования могут включать в себя, например, несколько ридов образцов. Данные секвенирования могут включать в себя множество ридов образцов, которые имеют соответствующие нуклеотидные последовательности образцов. Хотя обсуждается только один рид образца, следует понимать, что данные последовательности могут включать, например, сотни, тысячи, сотни тысяч или миллионы ридов образцов. Различные риды образцов могут содержать различное число нуклеотидов. Например, риды образцов может варьировать от 10 нуклеотидов до 500 нуклеотидов или более. Риды образцов могут охватывать весь геном источника (ов). В качестве одного примера, риды образцов направлены на заранее определенные генетические локусы, такие как генетические локусы, которые имеют подозрительные STR или предполагаемые SNP.
[00227] Каждый рид образца может включать последовательность нуклеотидов, которая может называться последовательностью образца, фрагментом образца или целевой последовательностью. Последовательность образца может включать, например, последовательности праймеров, фланкирующие последовательности и целевую последовательность. В некоторых вариантах реализации один или более ридов образцов (или последовательностей образцов) включают по меньшей мере 150 нуклеотидов, 200 нуклеотидов, 300 нуклеотидов, 400 нуклеотидов, 500 нуклеотидов или более. В некоторых вариантах реализации риды образцов могут включать более 1000 нуклеотидов, 2000 нуклеотидов или более. Риды образцов (или последовательности образцов) могут включать последовательности праймеров на одном или обоих концах.
[00228] Затем, указанные один или более процессоров анализируют данные секвенирования, чтобы получить потенциальные распознавание (ия) варианта (ов) образца и частоту варианта образца для указанных распознавания (ий) варианта (ов) образца. Эта операция также может называться приложением распознавания вариантов или распознавателем (определителем) вариантом. Таким образом, распознаватель вариантов идентифицирует или обнаруживает варианты, а классификатор вариантов классифицирует обнаруженные варианты как соматические или зародышевые. Могут применяться альтернативные распознаватели вариантов в соответствии с приведенным в настоящем документе вариантами реализации, причем могут применяться различные распознаватели вариантов в зависимости от типа выполняемой операции упорядочения, на основе характеристик образца, которые представляют интерес, и т.п. Одним из неограничивающих вариантов такого приложения для распознавания вариантов является приложение Pisces™ от компании Illumina Inc., (San Diego, CA, США), размещенное по адресу и https://github.com/Illumina/Pisces и описанное в статье Dunn, Tamsen & Berry, Gwenn & Emig-Agius, Dorothea & Jiang, Yu & Iyer, Anita & Udar, Nitin & Stromberg, Michael. (2017). Pisces: An Accurate and Versatile Single Sample Somatic and Germline Variant Caller. 595-595. 10.1145/3107411.3108203, полное содержание которой в явном виде полностью включено в настоящий документ посредством ссылки.
[00229] Такое приложение для распознавание вариантов содержит четыре выполняемых последовательно модуля:
[00230] ((1) Pisces Read Stitcher (сшиватель ридов Pisces, PRS): снижает шум путем сшивания парных ридов в ВАМ (рида один и рида два одной молекулы) в консенсусные. На выходе сшитый ВАМ.
[00231] (2) Pisces Variant Caller (определитель вариантов Pisces, PVC): определяет небольшие SNV, вставки (инсерции) и делеции. Pisces включают в себя алгоритм свертки вариантов для объединения вариантов, разбитых по границам ридов, основные алгоритмы фильтрации и простой алгоритм оценки достоверности вариантов на основе пуассоновского процесса. На выходе - VCF.
[00232] (3) Pisces Variant Quality Recalibrator (Рекалибратор качества вариантов Pisces, VQR): В случае, если определения (вызовы) вариантов в подавляющем большинстве случаев следуют некоторому паттерну, связанному с термическим повреждением или дезаминированием FFPE, шаг VQR будет понижать оценку Q варианта для подозрительных определений (вызовов). На выходе-откорректированный VCF.
[00233] (4) Pisces Variant Phaser (Фазировщик фариантов Pisces -Scylla): использует жадный метод кластеризации на основе ридов для сборки небольших вариантов в сложные аллели из клональных субпопуляций. Это позволяет более точно определять функциональные последствия последующими инструментами. На выходе- откорректированный VCF.
[00234] В качестве дополнения или альтернативы для этой операции можно применять приложение для определения вариантов Strelka™, от компании Illumina Inc., размещенное по адресу http://github.com/Illumina/strelka и описанное в статье Т Saunders, Christopher & Wong, Wendy & Swamy, Sajani & Becq, Jennifer & J Murray, Lisa & Cheetham, Keira. (2012). Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics (Oxford, Англия). 28. 1811-7. 10.1093/bioinformatics/bts271, полное содержание которой в явном виде полностью включено в настоящий документ посредством ссылки. Далее, в качестве дополнения или альтернативы, для этой операции можно применять приложение Strelka2™ от компании Illumina Inc., размещенное по адресу https://github.com/Illumina/strelka и описанное в статье Kim, S., Scheffler, K., Halpern, A.L., Bekritsky, М.А., Noh, E., Källberg, M., Chen, X., Beyter, D., Krusche, P., and Saunders, C.T. (2017). Strelka 2: Fast and accurate variant calling for clinical sequencing applications, полное содержание которой в явном виде полностью включено в настоящий документ посредством ссылки. Более того, в качестве дополнения или альтернативы, для этой операции можно применять инструмент для аннотации/определения вариантов, такой как Nirvana™, от компании Illumina Inc., размещенное по адресу https://github.com/Illumina/Nirvana/wiki и описанная в статье Stromberg, Michael & Roy, Rajat & Lajugie, Julien & Jiang, Yu & Li, Haochen & Margulies, Elliott. (2017). Nirvana: Clinical Grade Variant Annotator. 596-596. 10.1145/3107411.3108204, полное содержание которой в явном виде полностью включено в настоящий документ посредством ссылки.
[00235] Такой инструмент для аннотации/определения вариантов может применять различные алгоритмические методики, такие как описанные у Nirvana:
[00236] а. Идентификация всех перекрывающихся транскриптов с помощью массива интервалов: для функциональной аннотации мы можем идентифицировать все транскрипты, перекрывающие вариант, и можно применять дерево интервалов. Однако, поскольку набор (множество) интервалов может быть статическим, мы смогли дополнительно оптимизировать его в Массив Интервалов. Дерево интервалов возвращает все перекрывающиеся транскрипты за время О (min (n, k lg n)), где где n - количество интервалов в дереве, а k - количество перекрывающихся интервалов. На практике, поскольку k на самом деле мало по сравнению с n для большинства вариантов, эффективное время выполнения на дереве интервалов будет О (k lg n). Мы улучшили до О (lg n+k) за счет создания массива интервалов, в котором все интервалы хранятся в отсортированном массиве, так что нам нужно только найти первый перекрывающийся интервал, а затем пронумеровать оставшиеся (k-1).
[00237] b. CNV/SV (Yu): могут быть предоставлены аннотации для вариаций количества копий (CNV) и структурных вариантов (SV). Аналогично аннотациям небольших вариантов, транскрипты, перекрывающиеся с SV, а также ранее определенные структурные варианты могут быть аннотированы в онлайн-базах данных. В отличие от небольших вариантов, не обязательно все перекрывающиеся транскрипты аннотировать, так как слишком много транскриптов будут перекрываться с большими SV. Вместо этого могут быть аннотированы все перекрывающиеся транскрипты, относящиеся к частичному перекрывающемуся гену. В частности, для этих транскриптов могут выявляться (включаться в отчет) затронутые интроны, экзоны и последствия, обусловленные структурными вариантами. Доступна опция, позволяющая выводить все перекрывающиеся транскрипты, но может быть представлена основная информация для этих транскриптов, такая как символ гена, отметка, является ли это каноническим перекрыванием или частичным перекрыванием с транскриптами. Для каждого SV/CNV также интересно знать, были ли изучены эти варианты и их частота в разных популяциях. Соответственно, мы регистрировали перекрывающиеся SV во внешних базах данных, таких как "1000 геномов", DGV и ClinGen. Чтобы избежать применения произвольного отсечения для определения того, какой SV перекрывается, вместо этого можно применять все перекрывающиеся транскрипты и вычислять взаимное перекрывание, вместо этого можно применять все перекрывающиеся транскрипты и вычислять взаимное то есть длину перекрывания, деленную на минимум длины этих двух SV.
[00238] с. Регистрация дополнительных аннотаций: Дополнительные аннотации бывают двух типов: малые и структурные варианты (SV). SV можно моделировать как интервалы и использовать массив интервалов, описанный выше, для идентификации перекрывающихся SV. Небольшие варианты моделируются в виде точек и сопоставляются по положению и (необязательно) аллелю. Соответственно, их ищут с применением алгоритма, подобного бинарному поиску. Поскольку база данных дополнительных аннотаций может быть довольно большой, создают гораздо меньший индекс для картирования хромосомных положений на местоположения файлов, в которых находится дополнительная аннотация. Индекс - это отсортированный массив объектов (состоящих из хромосомного положения и расположения файла), по которым можно выполнять двоичный поиск с использованием положения. Чтобы размер индекса оставался небольшим, множество положений (до определенного максимального числа) сжимают в один объект, который хранит значения для первого положения и только дельты для последующих положений. Поскольку мы используем двоичный поиск, время выполнения - О (lg n), где n - количество элементов в базе данных.
[00239] d. Кэш-файлы VEP
[00240] е. База данных транскриптов: Файлы Transcript Cache (кэш транскриптов, кэш) и Supplementary database (дополнительная база данных, SAdb) представляют собой упорядоченное хранилище объектов данных, таких как транскрипты и дополнительные аннотации. Мы применяем кэш Ensembl VEP cache в качестве источника данных для кэша. Для создания кэша все транскрипты помещают в массив интервалов, а конечное состояние массива сохраняется в файлах кэша. Таким образом, в процессе аннотации нам нужно только загрузить предварительно вычисленный массив интервалов и выполнить поиск по нему. Поскольку кэш загружается в память, а поиск выполняется очень быстро (описано выше), поиск перекрывающихся транскриптов согласно Nirvana выполняется очень быстро (профилировано менее 1% от общего времени выполнения?).
[00241] f. Дополнительная база данных: источники данных для SAdb перечислены в дополнительных материалах. База данных SAdb для небольших вариантов создается путем k-направленного объединения всех источников данных, так что каждый объект в базе данных (идентифицируемый ссылочным именем и положением) содержит все соответствующие дополнительные аннотации. Проблемы, возникающие при парсировании файлов - источников данных, подробно описаны на домашней странице Nirvana. Чтобы ограничить использование памяти, в память загружается только индекс SA. Этот индекс позволяет осуществить быстрый поиск положения файла для дополнительной аннотации. Однако, поскольку данные должны быть извлечены с диска, добавление дополнительных аннотаций было определено как самое узкое место Nirvana (профилируется примерно как 30% от общего времени выполнения).
[00242] g. Последствия и онтология последовательности: Последствия и онтология последовательности. Иногда у нас была возможность выявить проблемы в текущей SO и сотрудничать с командой SO, чтобы улучшить состояние аннотации.
[00243] Такой инструмент вариантов аннотации может включать предварительную обработку. Например, Nirvana включала большое количество аннотаций из внешних источников данных, таких как ЕхАС, EVS, проект "1000 геномов", dbSNP, ClinVar, Cosmic, DGV и ClinGen. Чтобы в полной мере использовать эти базы данных, мы должны очистить информацию из них. Мы реализовали разные стратегии для решения разных конфликтов, обусловленных разными источниками данных. Например, в случае нескольких записей dbSNP для одного и того же положения и другого аллеля, мы объединяем все идентификаторы в список идентификаторов, разделенных запятыми; если есть несколько записей с разными значениями CAF для одного и того же аллеля, мы используем первое значение CAF. Для конфликтующих записей ЕхАС и EVS мы учитываем количество образцов и используем запись с большим количеством образцов. В проекте "1000 геномов" мы удаляли частоту аллеля конфликтующего аллеля. Другая проблема - неточная информация. В основном мы брали информацию о частотах аллелей из проекта "1000 геномов", однако мы заметили, что для GRCh38 частота аллелей, указанная в информационном поле, не исключала образцы с недоступным генотипом, что приводило к повышенным частотам для вариантов, которые доступны не для всех образцов. Чтобы гарантировать точность нашей аннотации, мы используем все генотипы индивидуального уровня для вычисления истинных частот аллелей. Как мы знаем, одни и те же варианты могут иметь разные представления на основе разных выравниваний. Чтобы быть уверенным, что мы можем точно получить (вывести) информацию об уже идентифицированных вариантах, мы должны предварительно обработать варианты из разных ресурсов, чтобы они имели единообразное представление. Для всех внешних источников данных мы удалили аллели, чтобы удалить дублированные нуклеотиды как в референсном аллеле, так и в альтернативном аллелях. Для ClinVar мы непосредственно парсировали xml-файл и выполнили пятизначное выравнивание для всех вариантов, которое часто используется в vcf-файле. Различные базы данных могут содержать одинаковый набор информации. Чтобы избежать ненужного дублирования, мы удалили часть повторяющейся информации Чтобы избежать ненужного дублирования, мы удалили часть повторяющейся информации.
[00244] В соответствии с по меньшей мере некоторыми вариантами реализации, указанное приложение для определения вариантов выдает варианты с низкой частотой, определение зародышевой линии и т.п. В качестве неограничивающего примера, указанное приложение для определения вариантов может работать только с опухолевыми образцами и/или с парными образцами опухоль-норма. Приложение для определения вариантов может искать однонуклеотидные варианты(SNV), много нуклеотидные варианты (MNV), инделы и т.п. ТПриложение определения вариантов идентифицирует варианты, одновременно фильтруя несоответствия из-за ошибок секвенирования или подготовки образца. Для каждого варианта определитель вариантов идентифицирует референсную последовательность, положение варианта и потенциальную последовательность (и) варианта (например, SNV от А до С или делеция из AG в А). Приложение определения вариантов идентифицирует последовательность образца (или фрагмент образца), референсную последовательность / фрагмент и определение варианта как показатель присутствия варианта. Приложение определения вариантов может идентифицировать необработанные фрагменты и выводить обозначение исходных фрагментов, подсчет числа необработанных фрагментов, которые верифицируют возможное определение варианта, положение в исходном фрагменте, в котором присутствует подтверждающий вариант, и другую важную информацию. Неограничивающие примеры необработанных фрагментов включают дуплексных сшитый фрагмент, симплекснный сшитый фрагмент, дуплексный несшитый фрагмент и симплексный несшитый фрагмент.
[00245] Приложение для определения вариантов может выводить определения (вызовы) в различных форматах, например, в файл .VCF или .GVCF. Только в качестве примера указанное приложение для определения вариантов может быть включено в пайплайн MiSeqReporter (например, когда оно реализовано в секвенаторе MiSeq®). При желании приложение может быть реализовано с различными рабочими процессами. Анализ может включать единый протокол или комбинацию протоколов, которые анализируют риды образца определенным образом для получения желаемой информации.
[00246] Затем указанные один или более процессоров осуществляют операцию валидации применительно к определению потенциальных вариантов. Операция валидации может быть основана на оценке качества и/или иерархии многоуровневых тестов, как объясняется ниже. Когда операция валидации (проверки) аутентифицирует или проверяет наличие потенциального определения варианта, операция проверки передает информацию об определенном варианте (из указанного приложения для определения вариантов) в генератор отчетов по образцам. В качестве альтернативы, когда операция проверки делает недействительным или дисквалифицирует потенциальное определение варианта, операция проверки передает соответствующий индикатор (например, отрицательный индикатор, индикатор отсутствия определения, индикатор недействительного определения) генератору отчетов по образцам. Операция проверки также может передавать оценку достоверности, связанную со степенью уверенности в том, что конкретное определение варианта правильно или определение варианта правильно обозначено как недействительное (невалидное).
[00247] Затем, указанные один или более процессоров генерируют и сохраняют отчет по образцу. Отчет по образцу может включать, например, информацию о множестве генетических локусов по отношению к образцу. Например, для каждого генетического локуса заранее определенного набора генетических локусов отчет по образцу может по меньшей мере одно из: определить генотип; указывать, что определение генотипа невозможно; предоставить оценку достоверности определения генотипа; или указать потенциальные проблемы с анализом в отношении одного или нескольких генетических локусов. В отчете по образцу также может быть указан пол человека, предоставившего образец, и/или указано, что образец включает несколько источников. В настоящем документе «отчет по образцу» («отчет об образце») может включать цифровые данные (например, файл данных) генетического локуса или заранее определенного набора генетических локусов и/или печатный отчет о генетическом локусе или наборе генетических локусов. Таким образом, создание или предоставление может включать в себя создание файла данных и/или печать отчета по образцу, или отображение отчета по образцу.
[00248] Отчет по образцу может указывать на то, что определение варианта было установлено, но не было подтверждено. Когда определение варианта определяется как недопустимое, отчет по образцу может указывать дополнительную информацию, касающуюся основания для решения не подтверждать определение варианта. Например, дополнительная информация в отчете может включать описание исходных фрагментов и степень (например, количество), в которой исходные фрагменты поддерживают определение варианта или противоречат ему. Дополнительно или в качестве альтернативы, дополнительная информация в отчете может включать оценку качества, полученную в соответствии с вариантами реализации, описанными в данном документе.
Применение определения вариантов
[00249] Варианты реализации, раскрытые в настоящем документе, включают анализ секвенированных данных для определения потенциальных вариаций. Распознавание вариантов может проводиться над сохраненными данными для выполненной ранее операции секвенирования. В качестве дополнения или альтернативы, его можно проводить в режиме реального времени одновременно с выполнением операции секвенирования. Каждый из ридов образцов ставится в соответствие соответствующим генетическим локусам. Риды образца могут быть поставлены в соответствие определенным генетическим локусам на основании последовательности нуклеотидов рида образца, или, другими словами, порядку нуклеотидов, входящих в рид (например, А, С, G, Т). На основании этого анализа рид образца может быть охарактеризован как включающий возможную вариацию/аллель определенного генетического локуса. Рид образца можно собирать (или агрегировать или группировать) вместе с другими ридами образца, охарактеризованными как включающие возможную вариацию/аллель генетического локуса. Под операцией определения соответствия можно также понимать операцию распознавания, в которой рид образца определяется как возможно ассоциированный с определенным генетическим положением/локусом. Риды образцов можно анализировать с целью локализовать идентифицирующие последовательности (например, последовательности праймеров) нуклеотидов, которые отличают данный рид образца от других ридов образца. Более конкретно, идентифицирующая последовательности и) может идентифицировать рид образца среди других ридов образцов как ассоциированный с определенным генетическим локусом.
[00250] Операция определения соответствия (присваивания) может включать анализ серии n нуклеотидов идентифицирующей последовательности для определения, соответствует ли серия n нуклеотидов, идентифицирующих последовательности, одной или более выбранным последовательностям. В частных вариантах реализации операция определения соответствия (присваивания) может включать анализ первых n нуклеотидов последовательности образца для определения, соответствуют ли первые n нуклеотидов последовательности образца одной или более выбранным последовательностям. Число n может принимать разнообразные значения, которые могут быть заложены в программу протокола или вводиться пользователем. Например, число n может быть определено как число нуклеотидов самой короткой выбранной последовательности в базе данных. Это заранее предопределенное число может составлять, например, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 или 30 нуклеотидов. Однако, в других вариантах реализации может применяться меньшее или большее число нуклеотидов. Число n может также быть выбрано человеком, например пользователем системы. Выбор числа n может быть основан на одном или более условиях. Например, число n может быть определено как число нуклеотидов самой короткой последовательности праймера в базе данных, или определенное число, смотря какое из них меньше. В некоторых вариантах реализации а для n может быть использовано минимальное значение, такое как 15, такое что любую последовательность праймера короче 15 нуклеотидов можно считать исключением.
[00251] В некоторых случаях, серия n нуклеотидов идентифицирующей последовательности может не соответствовать точно нуклеотидам последовательности выборки. Тем не менее, идентифицирующая последовательность может эффективно соответствовать последовательности выборки, если идентифицирующая последовательность почти идентична последовательности выборки. Например, рид образца может быть определен для генетического локуса, если серия n нуклеотидов (например, первые n нуклеотидов) идентифицирующей последовательности совпадают с последовательностью выборки с не более чем установленным числом несоответствий (например, 3) и/или установленным числом сдвигов (например, 2). Правила можно установить так, что каждое несоответствие или сдвиг может считаться как различие между ридом образца и последовательностью праймера. Если число различий меньше установленного значения, операция присваивания может быть применена к риду образца для соответствующего генетического локуса (то есть, рид присвоен соответствующему локусу). В некоторых вариантах реализации, вводится оценка совпадения, которая основана на количестве различий между идентифицирующей последовательностью рида образца и последовательностью выборки, ассоциированной с генетическим локусом. Если оценка совпадения превосходит установленный порог совпадения, генетический локус, соответствующий выбранной последовательности, можно считать потенциальным локусом рида образца. В некоторых вариантах реализации, может проводиться последующий анализ с целью определить, действительно ли рид образца соответствует генетическому локусу.
[00252] Если рид образца эффективно совпадает с одной из выбранных последовательностей в базе данных (т.е., в точности совпадает или совпадает в пределах критериев, описанных выше), то риду образца назначают или ставят в соответствие генетический локус, который коррелирует с выбранной последовательностью. Это можно назвать определением локуса или предварительным определением локуса, где рид образца определен для генетического локуса, который коррелирует с выбранной последовательностью. Однако, как описано выше, рид образца может быть определен для более одного генетического локуса. В таких вариантах осуществления, может проводиться последующий анализ для определения или присваивания рида образца только одному из потенциальных генетических локусов. В некоторых вариантах реализации рид образца, который сравнивают с базой данных референсных последовательностей, представляет собой первый рид из секвенирования спаренных концов. При осуществлении секвенирования спаренных концов, получают второй рид (представляющий фрагмент необработанных данных) который коррелирует с ридом образца. После присваивания, последующий анализ, который проводится с присвоенными ридами, может быть основан на типе генетического локуса, который был определен для этого рида.
[00253] Затем, риды образца анализируют для идентификации потенциальных вариантов. Среди прочего, результаты этого анализа идентифицируют потенциальный вариант, частоту последовательности варианта, референсную последовательность и положение в исследуемой генетической последовательности, в которой встретился вариант. Например, если известно, что генетический локус включает однонуклеотидные полиморфизмы, то присвоенные риды, которые были определены для генетического локуса можно подвергать дополнительному анализу для идентификации однонуклеотидных полиморфизмов присвоенных ридов. Если известно, что генетический локус включает полиморфные повторяющиеся элементы ДНК, то присвоенные риды можно анализировать для того, чтобы идентифицировать или охарактеризовать полиморфные повторяющиеся элементы ДНК в составе ридов образцов. В некоторых вариантах реализации если присвоенный рид эффективно совпадает с STR-локусом и SNP-локусом, риду образца может быть присвоено предупреждение или флаг. Рид образца может быть определен и как STR-локус и как SNP-локус. Анализ может включать выравнивание присвоенных ридов в соответствии с алгоритмом выравнивания с целью определить последовательности и/или длины присвоенных ридов. Протокол выравнивания может включать метод, описанный в Международной Патентной Заявке № PCT/US2013/030867 (№публикации WO 2014/142831), поданной 15 марта 2013, которая в полном объеме включена в данную заявку посредством ссылки.
[00254] Затем один или более процессов анализируют необработанный фрагмент с целью определить, существуют ли поддерживающие варианты в соответствующих положениях необработанных фрагментов. Можно идентифицировать различные типы необработанных фрагментов. Например, определитель вариантов может идентифицировать тип необработанного фрагмента, который имеет вариант, валидирующий (подтверждающий) исходно найденный вариант. Например, тип необработанного фрагмента может представлять двунитевый сшитый фрагмент, однонитевый сшитый фрагмент, двунитевый несшитый фрагмент или однонитевой несшитый фрагмент. Опционально можно идентифицировать другие необработанные фрагменты вместо или в дополнение к приведенным примерам. Вместе с идентификацией каждого типа необработанных фрагментов, пользователь также определяет положение в этом фрагменте, в котором встретился поддерживающий вариант, а также число необработанных фрагментов, в которых этот выявили поддерживающий вариант. Например, определитель вариантов может вывести индикацию того, что 10 ридов необработанных фрагментов идентифицированы как представляющие собой двунитевые сшитые фрагменты, содержащие поддерживающий вариант в определенном положении X. Определитель вариантов может также выводить индикацию того, что пять ридов необработанных фрагментов представляют собой однонитевые несшитые фрагменты, имеющие поддерживающий вариант в определенном положении Y. Определитель вариантов может также выводить число необработанных фрагментов, которые соответствуют референсной последовательности, и таким образом не включают поддерживающий вариант, который в ином случае был бы свидетельством поддерживающим определение потенциального варианта в исследуемой генной последовательности.
[00255] Далее, сохраняется число необработанных фрагментов, которые включают поддерживающие варианты, а также положения, в которых встретились поддерживающие варианты. В качестве дополнения или альтернативы, можно сохранять число необработанных фрагментов, которые не включают поддерживающие варианты в представляющем интерес положении (относительно положения определения потенциального вариантов рида образца или фрагмента образца). В качестве дополнения или альтернативы, может сохраняться число необработанных фрагментов, которые соответствуют референсной последовательности и не удостоверяют/подтверждают определение потенциального варианта. Полученная информация выводится в приложение валидации определения вариантов, включая количество и тип необработанных фрагментов, которые поддерживают определение потенциального варианта, положения поддерживающих вариантов в необработанных фрагментах, число необработанных фрагментов, которые не поддерживают потенциального определение варианта и т.п.
[00256] Когда потенциальный вариант идентифицирован, в выходных данных процесса появляется индикация определения потенциального варианта, последовательность варианта, положение варианта и референсная последовательность, ассоциированная с ним. Вариант обозначается как "потенциальный", поскольку ошибки могут приводить к идентификации ложного варианта. В соответствии с приведенными здесь вариантами осуществления определение потенциального варианта анализируют, чтобы уменьшить или исключить ложные варианты и ложные совпадения. В качестве дополнения или альтернативы, процесс (способ) анализирует один или более необработанных фрагментов, ассоциированных с ридом образца, и дополняет выходные данные соответствующим вариантом, ассоциированным с необработанными фрагментами.
Глубокое Обучение в Геномике
[00257] Генетические вариации могут помочь объяснить многие заболевания. Каждое человеческое существо имеет уникальный генетический код, а в группе индивидуумов встречается множество генетических вариантов. Большинство вредоносных генетических вариантов были исключены из геномов в результате естественного отбора. Важно идентифицировать, генетические варианты, которые вероятно являются патогенными или вредоносными. Это поможет исследователям сосредоточиться на вероятно патогенных вариантах и ускорить диагностику и лечение многих заболеваний.
[00258] Моделирование свойств и функциональных эффектов (например, патогенности) вариантов - это важная, но сложная для ученых область геномики. Несмотря на быстрое развитие функциональных генных технологий секвенирования, Несмотря интерпретация функциональных эффектов вариантов остается крайне непростой задачей из-за сложности специфичных для разных типов клеток систем регуляции транскрипции быстрое развитие функциональных генных технологий секвенирования.
[00259] Применительно к классификаторам патогенности глубокие нейросети это тип искусственных нейронных сетей, которые используют множественные нелинейные и сложные преобразующие слои, чтобы последовательно моделировать высокоуровневые признаки. Глубокие нейросети обеспечивают обратную связь посредством алгоритма обратного распространения, который несет информацию о разнице между наблюдаемыми и ожидаемыми выходными данными, с целью коррекции параметров. Глубокие нейронные сети развивались по мере того, как становились доступны большие объемы данных для обучения, мощности параллельных и распределенных вычислений, и развитые алгоритмы обучения. Глубинные нейросети способствовали существенному развитию в множестве областей, таких как компьютерное зрение, распознавание речи и обработка естественных языков.
[00260] Сверточные нейронные сети (CNN) и рекуррентные нейронные сети (RNN) являются компонентами нейронных сетей глубокого обучения (глубоких нейронных сетей). Сверточные нейронные сети особенно успешно выполняют задачи по распознаванию образов и имеют архитектуру, которая включает слои свертки, нелинейные слои, слои пулинга. Рекуррентные нейронные сети созданы для использования последовательных входных данных с циклическими связями между строительными блоками, перцептронами, единицами долгосрочной и краткосрочной памяти, и управляемые рекуррентные блоки. В дополнение было предложено много других новейших нейросетей глубокого обучения для ограниченных контекстов, например глубокие пространственно-временные нейронные сети, многомерные рекуррентные нейронные сети, и сверточные автоэнкодеры.
[00261] Цель обучения глубоких нейронных сетей заключается в оптимизации веса параметров в каждом слое, который постепенно комбинирует более простые признаки в сложные, что позволяет получить из данных наиболее подходящие иерархические представления. Отдельный цикл процесса оптимизации организован следующим образом. Сначала, на тренировочном (обучающем) наборе данных, прямой проход алгоритма последовательно вычисляет выходные данные в каждом слое, и распространяет сигналы функции вперед по сети. В конечном выходном слое (слове выходных данных), целевая функция потерь измеряет погрешность между выходными данными работы обученной нейронные сети и данными метками. Для минимизации ошибок обучения, при обратном проходе используется правило сложной производной (цепное правило) для обратного распространения сигналов ошибки и вычисления градиентов по всем весам по всей нейронные сети. В конце весовые параметры обновляются посредством алгоритмов оптимизации, основанных на стохастическом градиентном спуске. В то время как градиентный спуск осуществляет обновление параметров для каждого полного набора данных, стохастический градиентный спуск обеспечивает стохастическую аппроксимацию, проводя обновление для каждого небольшого набора семплированных данных (данных в выборке). На принципе стохастического градиентного спуска основаны несколько алгоритмов оптимизации. Например, обучающий алгоритм Адаграда и Адама проводит стохастический градиентный спуск с адаптивным изменением скорости обучения на основе частоты обновления моментов градиентов для каждого параметра, соответственно.
[00262] Другим базовым элементом обучения глубокой нейронной сети является регуляризация, понятие, относящееся к стратегиям, направленным на то, чтобы избежать переобучения нейронные сети, и таким образом добиться хорошей производительности генерализации. Например, сокращение весов добавляет штрафные слагаемые к целевой функции потерь, так что весовые параметры сходятся к меньшим абсолютным значениям. Метод исключения (dropout) случайным образом убирает скрытые узлы из нейронной сети во время обучения, и может рассматриваться как ансамбль возможных подсетей. Чтобы улучшить возможности метода исключения, была предложена новая функция активации, maxout, и определен вариант метода исключения для рекуррентных нейросетей - mnDrop. Кроме того, пакетная нормализация обеспечивает новый метод регуляризации посредством нормализации скалярных признаков для каждой активации внутри мини-пакета с обучением каждого среднего и отклонения в качестве параметров.
[00263] Кроме того, пакетная нормализация обеспечивает новый метод регуляризации посредством нормализации скалярных признаков для каждой активации внутри мини-пакета с обучением каждого среднего и отклонения в качестве параметров. Сверточные нейронные сети адаптированы для решения задач геномики, основанных на последовательностях, таких как обнаружение мотива, идентификация патогенных вариантов и исследование экспрессии генов. Сверточные нейронные сети используют стратегию совместно используемых весов (weight-sharing), которая особенно полезна для изучения ДНК, потому что они могут регистрировать мотивы последовательностей, которые являются короткими, рекуррентными локальными паттернами в ДНК, имеющими, как полагают, важные биологические функции. Характерной чертой сверточных нейросетей является использование сверточных фильтров, отличие от традиционных подходов к классификации, которые основаны на признаках, полученных в результате тщательной ручной работы, сверточные фильтры проводят адаптивное обучение признаков, аналогично процессу картирования необработанных входных данных на информативное представление знаний. В этом смысле, сверточные фильтры служат серией сканеров мотивов, поскольку набор таких фильтров способен опознать релевантные паттерны во входных данных, и адаптироваться в процессе обучения. Рекуррентные нейронные сети могут регистрировать дальномерные зависимости в последовательных данных различной длины, таких как белковые последовательности или ДНК.
[00264] Таким образом, мощная вычислительная модель, предсказывающая патогенность вариантов, может давать огромное преимущество как для фундаментальной, так и для прикладной науки.
[00265] В настоящее время только 25-30% пациентов с редкими заболеваниями проходят молекулярную диагностику на основании исследования белок-кодирующих последователей, что означает, что остальная диагностическая значимость может быть связана с 99% генома, которые являются некодирующими. Здесь мы описываем новую сеть глубокого обучения, точно предсказывает границы сплайсинга по произвольной последовательности пре-мРНК транскрипта, давая возможность точно предсказать эффект альтернативного сплайсинга некодирующих вариантов. Синонимические и интронные мутации с предсказанными изменяющими сплайсинг последствиями часто валидируются в данных РНК-seq (секвенирования РНК) и являются крайне разрушительными для человеческой популяции. Первичные мутации с предсказанными изменяющими сплайсинг последствиями значительно чаще встречаются у пациентов с аутизмом и интеллектуальными отклонениями, по сравнению со здоровыми людьми, и подтверждаются в данных секвенирования РНК у 21 из 28 таких пациентов. Мы оцениваем, что 9-11% патогенных мутаций у пациентов с редкими генетическими заболеваниями вызваны этими ранее недооцениваемыми вариантами.
[00266] Секвенирование экзома существенно изменило клиническую диагностику пациентов и семей с редкими генетическими заболеваниями: при использовании в качестве теста первой линии оно существенно уменьшает время и стоимость постановки диагноза (Monroe et al., 2016; Stark et al., 2016; Tan et al., 2017). Однако, диагностическая значимость секвенирования экзома составляет ~25-30% в случаях редких генетических заболеваний, оставляя большинство пациентов без диагноза даже после комбинированного микроматричного анализа и проверки экзома (Lee et al., 2014; Trujillano et al., 2017; Yang et al., 2014). Некодирующие участки играют существенную роль в регуляции генов и отвечают за 90% локусов, связанных с болезнями, обнаруженных в объективных исследованиях сложных заболеваний человека по всему геному (Ernst et al., 2011; Farh et al., 2015; Maurano et al., 2012), что наводит на мысль, что пенетрантные некодирующие варианты также могут отвечать за значительную часть мутаций, являющихся причиной редких генетических заболеваний. Действительно, пенетрантные некодирующие варианты, которые нарушают нормальный паттерн сплайсинга мРНК несмотря на то, что лежат за пределами необходимых динуклеотидов сплайсинга GT и AG, которые часто называют критическими сплайсинговыми вариантами, уже давно признаны играющими существенную роль в генетических заболеваниях (Cooper et al., 2009; Padgett 2012; Scotti and Swanson, 2016; Wang and Cooper, 2007). Однако, критические сплайсинговые мутации часто упускают из вида в клинической практике из-за нашего неполного понимания кода сплайсинга и обусловленной этим сложность точно идентифицировать меняющие сплайсинг варианты за пределами необходимых GT и AG динуклеотидов (Wang and Burge, 2008).
[00267] В последнее время, секвенирование РНК все активнее рассматривается как многообещающий метод выявления нарушений сплайсинга в Менделевских заболеваниях (Cummings et al., 2017; Kremer et al., 2017), но пока его применение в клинических условиях остается ограниченным небольшим числом случаев, когда релевантный тип клеток известен и доступен для биопсии. Исследование потенциальных изменяющих сплайсинг вариантов методом высокопроизводительного скрининга (Soemedi et al., 2017) расширили описание вариантов сплайсинга, однако они менее практичны для оценки произвольных первичных мутаций в генетических заболеваниях, поскольку пространство генома, где могут появляться меняющие сплайсинг мутации, крайне велико. Общее предсказание сплайсинга по произвольной последовательности пре-мРНК потенциально позволило бы точно предугадывать меняющие сплайсинг последствия некодирующих вариантов, существенно улучшив диагностику у пациентов с генетическими заболеваниями. На сегодняшний день, общая предсказательная модель сплайсинга, основанная на необработанных данных последовательности, которая учитывала бы специфику сплайсосомы, остается недостижимой, несмотря на прогресс в определенных областях применения, таких как моделирование характеристик последовательности коровых сплайсинговых мотивов (Yeo and Burge, 2004), описание энхансеров и сайленсеров экзонных сплайсов (Fairbrother et al., 2002; Wang et al., 2004), и предсказание включений кассетных экзонов (Barash et al., 2010; Jha et al., 2017; Xiong et al., 2015).
[00268] Сплайсинг длинных пре-мРНК в зрелые транскрипты замечателен своей точностью, а также клинической тяжестью меняющих сплайсинг мутаций, в то же время принцип, по которому аппарат клетки определяет его специфичность, понятен не до конца. В данной заявке мы обучаем глубокую нейросеть, которая подходит точности сплайсосомы методом компьютерного моделирования (in silico), идентифицируя границы экзон-интрон по последовательности пре-мРНК с точностью 95%, и предсказывает функциональные криптические мутации сплайсинга с коэффициентом валидации более 80% на данных секвенирования РНК. Некодирующие варианты, которые, как предсказано, меняют сплайсинг, наносят большой вред человеческой популяции, 80% первичных критических мутации сплайсинга испытывают отрицательный отбор, подобно другим классам вариантов, укорачивающих белок. De novo (первичные) мутации критического сплайсинга у пациентов с аутизмом и интеллектуальными отклонениями поражают те же гены, которые рекуррентно мутированы мутациями, укорачивающими белок, что позволяет говорить об обнаружении дополнительных кандидатных генов, связанных с заболеванием. По нашей оценке, до 24% редких генетических заболеваний, вызванных пенетрантными мутациями, у пациентов возникают по причине присутствия этого ранее недооцененного класса связанных с заболеванием вариантов, что подчеркивает необходимость улучшать интерпретацию 99% некодирующего генома при клиническом секвенировании.
[00269] Клиническое секвенирование экзома произвело революцию в диагностике пациентов и семей с редкими генетическими заболеваниями, и, при применении в качестве теста первой линии, существенно уменьшает время и стоимость полной диагностики. Однако, диагностическая значимость секвенирования экзома, по имеющимся данным, находится на уровне 25-30% многочисленных больших когорт пациентов с редкими генетическими заболеваниями и их родителей, то есть большинство пациентов остаются без диагноза даже после комбинированного экзомного и микроматричного анализа. Некодирующий геном крайне активен в регуляции генов, в некодирующие варианты отвечают за ~90% обнаруженных в процессе общегеномного исследования распространенных заболеваний, что дает основания полагать, что редкие варианты в некодирующем геноме также могут отвечать за значительную часть причинных мутаций пенетрантных заболеваний, таких как редкие генетические заболевания и онкология. Однако, сложность интерпретации вариантов в некодирующем геноме означает, что за пределами крупных структурных вариантов некодирующий геном в настоящее время не предлагает существенных улучшений для диагностики в отношении редких пенетрантных вариантов, которые имеют огромное влияние на клиническое ведение пациентов.
[00270] Роль меняющих сплайсинг мутаций за пределами канонических GT и AG сплайсинговых динуклеотидов в вопросе редких заболеваний давно признана. Действительно, эти критические варианты сплайсинга являются самыми распространенными мутациями для некоторых редких генетических заболеваний, таких как гликогеноз XI (гликогеноз II типа) и эритропоэтическая протопорфирия. Расширенные мотивы сплайсинга на 5' и 3' концах интрона в высокой вырождены, и в геноме часто встречаются одинаково хорошие мотивы, что делает попытки предсказать, какой из некодирующих вариантов может вызывать критический сплайсинг, непрактичными в рамках существующих методов.
[00271] Для лучшего понимания того, как сплайсосома хранит свою специфичность, мы обучили глубокую нейросеть предсказывать для каждого нуклеотида в пре-мРНК транскрипте, является ли он акцептором сплайсинга, донором сплайсинга, или ни тем, ни другим, используя в качестве входных данных только последовательности транскрипта (Фиг. 37А). Используя канонические транскрипты четных хромосом в качестве тренировочных данных, и транскрипты нечетных хромосом для тестирования (с исключенными паралогами), глубокая нейросеть отыскивает границы экзон-интрон с 95% точностью. Даже транскрипты, превосходящие 100KB, такие как трансмембранный регулятор муковисцидоза (CFTR) часто реконструируются с точностью до нуклеотида (Фиг. 37В).
[00272] Далее мы хотели понять специфичность детерминант, используемых нейросетью для распознавания границы экзон-интрон с такой поразительной точностью. В отличие от предыдущих классификаторов, которые работали на статистических или разработанных людьми принципах, глубокое обучение непосредственно вырабатывает признаки по данным последовательности в иерархической манере, давая возможность водить в систему дополнительную специфичность из контекста длинномерной последовательности. Действительно, мы установили, что точность нейросети сильно зависит от длины контекста последовательности, окружающей рассматриваемый нуклеотид, которая является входными данными нейросети (Таблица 1), и когда мы обучаем глубокую модель, которая использует только 40-нуклеотидный фрагмент последовательности, производительность лишь умеренно превосходит существующие статистические методы. Это показывает, что глубокое обучение мало добавляет к существующим статистическим методам для распознавания отдельных 9-23 нуклеотидных сплайсинговых мотивов, но более широкий контекст последовательности является ключом к обнаружению функциональных сплайсинговых сайтов на фоне нефункциональных с равносильными мотивами. Если дать нейросети задачу найти экзоны, в области которых последовательность нарушается, оказывается, что разрушение донорного мотива обычно также вызывает исчезновение сигнала акцептора (Фиг. 37С), как это часто наблюдается в случае событий пропуска экзона у пациентов. Это значит, что существенная часть специфичности вносится простым требованием парности между сильными акцепторными и донорными мотивами на приемлемом расстоянии.
[00273] Хотя в большом числе случаев наблюдается, что изменение длины экзонов оказывает сильное влияние на включение экзонов по отношению к пропуску экзонов, это не объясняет, почему точность глубокой нейросети продолжает возрастать после увеличения длины контекста за пределами 1000-нуклеотидов. Чтобы лучше различать локальную специфичность, обусловленную мотивами сплайсинга и длинномерными специфичными детерминантами, мы обучили локальную сеть, которая принимает на вход только 100-нуклеотидный контекст. Используя локальную сеть, чтобы проставить оценки известных границ, мы получаем, что экзоны и интроны имеют оптимальные значения длины (~115 нуклеотидов для экзонов, ~1000 нуклеотидов для интронов), при которой сила мотива минимальна (Фиг. 37D). Эта связь отсутствует в 10000-нуклеотидной глубокой нейросети (Фиг. 37Е), что означает, что длины интронов и экзонов полностью факторизованы в ширококонтекстной глубокой нейросети. Примечательно, что границы интронов и экзонов никогда не подавали на вход ширококонтекстной глубокой обучающей модели, то есть она была способна получить эти длины на основании лишь данных о положениях экзонов и интронов на основании только последовательности.
[00274] Систематический поиск по пространству гексамеров также показал, что глубокая нейросеть использует мотивы в определении экзон-интрон, в частности, мотив точки ветвления ТАСТААС от положения -34 до -14, хорошо изученный энхансер экзонного сплайсинга GAAGAA возле концов экзона, и поли-U мотивы, которые обычно являются частью полипиримидинового тракта, но также, как оказалось, выступают в качестве сайленсеров экзонного сплайсинга (Фигуры. 21, 22, 23, и 24).
[00275] Мы расширяем глубокую нейросеть (нейросеть глубокого обучения) до оценки роли генетических вариантов в изменении сплайсинга за счет предсказаний границ экзон-интрон как в референсной последовательности транскрипта, так и в альтернативной последовательности транскрипта, содержащей вариант и поиска любых изменений границ экзон-интрон. Доступные в данный момент данные экзома 60706 людей позволяют нам оценивать влияние отрицательного отбора на варианты, которые, как предполагается, меняют функцию сплайсинга, исследуя их распределение по спектру частоты аллели. Мы обнаружили, что предсказанные критические варианты сплайсинга подвергаются сильному отрицательному отбору (Фиг. 38А), что подтверждается их относительным убыванием при высокой частоте аллелей в сравнении с ожидаемыми числами, а их амплитуда убывания сравнима с AG или GT вариантами, нарушающими сплайсинг, и вариантами преждевременной терминации (stop-gain). Влияние негативного отбора больше при рассмотрении вариантов критического сплайсинга, которые могут вызвать сдвиги рамки по сравнению с теми, которые вызывают изменения внутри рамки. (Фиг. 38В). Основываясь на истощении вариантов критического сплайсинга, вызывающих сдвиг рамки, по сравнению с другими классами вариантов, вызывающих укорачивание белка, по нашей оценке 88% достоверно предсказанных криптических мутаций сплайсинга являются функциональными.
[00276] Хотя доступно не так много совокупных данных целого генома, как данных экзома, что ограничивает возможность обнаружения влияния естественного отбора в глубоких интронных областях, нам все же удалось вычислить наблюдаемое vs ожидаемое число криптических мутаций сплайсинга вдали от экзонных областей. В общем, мы наблюдаем 60% деплецию (истощение) мутации критического сплайсинга на расстоянии >50 нуклеотидов от границы экзон-интрон (Фиг. 38С). Затухающий сигнал, вероятно, является комбинацией меньшего размера образца с полными данными генома в сравнении с экзомом, и представляет еще большую трудность для предсказания влияния глубоких интронных вариантов.
[00277] Мы также используем данные наблюдаемого против ожидаемого числа вариантов критического сплайсинга для оценки числа вариантов критического сплайсинга в условиях отбора, и как это соотносится с другими классами вариантов, укорачивающих белок. Поскольку критические варианты сплайсинга могут лишь частично подавлять функцию сплайсинга, мы также оценивали данные наблюдаемого против ожидаемого числа вариантов критического сплайсинга при более низких пороговых значениях, и получили, что имеется примерно в 3 раза больше вредоносных редких вариантов критического сплайсинга, чем редких AG или GT нарушающих сплайсинг вариантов в наборе данных ЕхАС (Фиг. 38D). Каждый человек является носителем примерно ~20 редких криптических мутаций сплайсинга, что приблизительно равно числу укорачивающих белок вариантов (Фиг. 38Е), хотя не все эти варианты полностью подавляют функцию сплайсинга.
[00278] Недавний релиз данных GTEx, содержащий данные 148 людей с секвенированием всего генома и PHK-seq из различных участков тканей, позволяет нам искать влияние редких вариантов критического сплайсинга непосредственно в данных секвенирования РНК. Чтобы аппроксимировать сценарий, возникающий при секвенировании редких заболеваний, мы рассматривали только редкие варианты (синглетоны (одиночные)в когорте GTEx, и частоту аллели <1% в проекте «1000 геномов»), и ставили их в соответствие событиям сплайсинга, которые были уникальны для носителя варианта. Хотя различия в экспрессии генов и тканей, а также сложность отклонений сплайсинга от нормы делают оценку чувствительности и специфичности предсказаний глубокой нейросети сложной задачей, мы получили, что при жестких порогах специфичности, более 90% редких мутаций криптического сплайсинга подтверждаются на данных секвенирования РНК (Фиг. 39А). Выяснилось, что большое число аберрантных событий сплайсинга, присутствующих в PHK-seq, ассоциированы с вариантами, которые, как предполагалось, имеют мало выраженное влияние, согласно классификатору глубокого обучения, что говорит о том, что они лишь частично подавляют функцию сплайсинга. При более пониженных порогах чувствительности, приблизительно 75% новых границ, согласно результатам нейросети, будут вызывать аберрации в функции сплайсинга (Фиг. 38В).
[00279] Успех глубокой нейросети в предсказании вариантов криптического сплайсинга, вредоносных для популяции, и высокий процент валидных данных на данных секвенирования РНК, указывают на то, что метод может применяться для идентификации дополнительных диагнозов при секвенировании последовательностей редких заболеваний. Чтобы проверить эту гипотезу, мы изучили de novo варианты в исследованиях с секвенированием экзома при аутизме и заболеваниях неврологического развития, и продемонстрировали, что криптические мутации сплайсинга существенно многочисленнее у пациентов с этими заболеваниями в сравнении с их здоровыми сиблингами (Фиг. 40А). Более того, разнообразие критических мутации сплайсинга слегка меньше, чем разнообразие укорачивающих белок вариантов, что указывает на то, что примерно 90% предсказанных вариантов криптического сплайсинга являются функциональными. Основываясь на этих значениях, примерно ~20% вызывающих заболевание укорачивающих белок вариантов могут быть отнесены к криптическим мутациям сплайсинга в экзонах и нуклеотидах, непосредственно прилегающих к экзонам (Фиг. 40В). Экстраполируя этот паттерн на полногеномные исследования, которые позволяют проверять всю интронную последовательность, мы оцениваем, что 24% причинных мутаций при редких генетических заболевания возникают из-за критических мутации сплайсинга.
[00280] Мы оценили вероятность определения критических мутаций сплайсинга de novo (первичных) для каждого отдельного гена, что позволило оценить разнообразие критических мутаций сплайсинга в генах, которые являются кандидатами на причину болезни, по отношению к вероятности. Первичные криптические мутации сплайсинга были существенно многочисленнее в генах, в которых ранее были обнаружены приводящие к укорочению белка варианты, но не миссенс-вариант (Фиг. 40С), что указывает на то, что они по большей части вызывают заболевание из-за гаплонедостаточности, а не через другие возможные механизмы действия. Добавление критических мутаций сплайсинга к списку укорачивающих белок вариантов позволяет нам идентифицировать 3 дополнительных болезнетворных гена в случае аутизма и 11 дополнительных болезнетворных генов при интеллектуальных отклонениях, по сравнению с использованием исключительно вариантов, укорачивающих белок (Фиг. 40D).
[00281] Для оценки правомерности валидации критических мутаций сплайсинга у пациентов, у которых вероятные пораженные ткани не были доступны (в нашем случае, мозг), мы провели глубинный PHK-seq-анализ у 37 людей с предсказанными первичными критическими мутациями сплайсинга из данных коллекции Simon's Simplex Collection, в которых искали аберрантные события сплайсинга, которые присутствовали у пациента, и отсутствовали у всех остальных пациентов в эксперименте, и у 149 людей из когорты GTEx. Мы обнаружили, что у NN из 37 пациентов имеются уникальные, аберрантные события сплайсинга на данных секвенирования РНК (Фиг. 40Е), объясняемые предсказанным вариантом криптического сплайсинга.
[00282] В целом, мы демонстрируем модель глубокого обучения, которое достоверно предсказывает криптические варианты сплайсинга с достаточной точностью для того, чтобы быть полезной в идентификации мутаций, вызывающих редкие генетические заболевания. По нашей оценке, существенная часть диагностированных редких заболеваний, вызванных критическим сплайсингом, в настоящее время пропускают, рассматривая только области, кодирующие белок, и подчеркиваем необходимость развивать методы интерпретации влияния пенетрантных редких вариаций в некодирующем геноме.
Результаты
Точное предсказание сплайсинга по первичной последовательности с использованием глубокого обучения
[00283] Мы сконструировали глубокую остаточную нейронную сеть (Не et al., 2016а), которая предсказывает, какое положение в пре-мРНК-транскрипте донором сплайсинга, акцептором сплайсинга или не является ни тем, ни другим (ФИГ. 37А и ФИГ. 21, 22, 23 и 24), используя только геномную последовательность пре-мРНК транскрипт. Поскольку доноры сплайсинга и акцепторы сплайсинга могут быть разделены десятками тысяч нуклеотидов, мы использовали новую сетевую архитектуру, состоящую из 32 разреженных сверточных слоев (Yu and Koltun, 2016), которые могут распознавать детерминанты последовательности, охватывающие очень большие геномные расстояния. В отличие от предыдущих методов, которые учитывали только короткие нуклеотидные окна, примыкающие к границам экзон-интрон (Yeo and Burge, 2004), или полагались на функции, созданные человеком (Xing et al, 2015), или экспериментальные данные, такие как экспрессия или связывание фактора сплайсинга (Jha et al., 2017), наша нейронная сеть изучает детерминанты сплайсинга непосредственно по первичной последовательности, оценивая 10000 нуклеотидов фланкирующей контекстной последовательности чтобы предсказать функцию сплайсинга каждом положении в пре-мРНК-транскрипте.
[00284] Мы использовали аннотированные GENCODE последовательности (Harrow et al, 2012) на подмножестве хромосом человека для обучения параметров нейронной сети и транскрипты на остальных хромосомах, исключая паралоги, для проверки прогнозы сети. Для пре-мРНК-транскриптов в тестовом наборе данных сеть предсказывает границы сплайсинга с точностью по к до 95%, что представляет собой долю правильно предсказанных сайтов сплайсинга при пороге, при котором количество предсказанных сайтов равно фактическому количеству сайтов сплайсинга, присутствует в тестовом наборе данных (Boyd et al., 2012; Yeo and Burge, 2004). Даже гены размером более 100 т.п.н., такие как CFTR, часто реконструируются идеально с точностью до нуклеотида (ФИГ. 37 В). Чтобы подтвердить, что сеть не просто полагается на смещения экзонных последовательностей, мы также протестировали сеть на длинных некодирующих РНК. Несмотря на неполноту аннотаций некодирующих транскриптов, которая, как ожидается, снизит нашу точность, сеть предсказывает известные границы сплайсинга в lincRNA (дпнРНК) с точностью до по k 84% (ФИГ. 42А и 42 В), что указывает на то, что она может аппроксимировать поведение сплайсосомы. на произвольных последовательностях, которые свободны от давления отбора кодирования белка.
[00285] Для каждого аннотированного экзона GENCODE в тестовом наборе данных (за исключением первого и последнего экзонов каждого гена) мы также исследовали, коррелируют ли оценки предсказания сети с долей считываний, поддерживающих включение экзона, по сравнению с пропуском экзона, на основании данных РНК- seq из Атласа экспрессии генов и тканей (GTEx) (The GTEx Consortium et al., 2015) (ФИГ. 37C). Экзоны, которые конститутивно включались или исключались при сплайсинге в тканях GTEx, имели предсказательные оценки, близкие к 1 или 0, соответственно, тогда как экзоны, которые в значительной степени подвергались альтернативному слпайсингу (от 10 до 90% включения экзонов, усредненного по образцам), имели тенденцию к промежуточному уровню оценок (корреляция Пирсона=0,78, Р 0).
[00286] Затем мы попытались понять детерминанты последовательности, используемые сетью для достижения ее выдающейся точности. Мы выполняли систематические замены in silico каждого нуклеотида рядом с аннотированными экзонами, измеряя влияние на оценки предсказания сети на прилегающих участках сплайсинга (ФИГ. 37Е). Мы обнаружили, что нарушение последовательности донорного мотива сплайсинга часто приводило к тому, что сеть предсказывала, что вышестоящий акцепторный сайт сплайсинга также будет потерян, как это наблюдается с событиями пропуска экзона in vivo, что указывает на то, что значительную степень специфичности придает определение между парным вышестоящим акцепторным мотивом и нижележащим донорным мотивом, расположенными на оптимальном расстоянии (Berget, 1995). Дополнительные мотивы, которые вносят вклад в сигнал сплайсинга, включают хорошо охарактеризованные связывающие мотивы семейства SR-белков и точки ветвления (ФИГ. 43А и 43 В) (Fairbrother et al., 2002; Reed, Maniatis, 1988). Эффекты этих мотивов сильно зависят от их положения в экзоне, указывая на то, что их роли включают определение точного положения границ интрон-экзон путем различения конкурирующих акцепторных и донорных сайтов.
[00287] Обучение сети с применением различного контекста входных последовательностей влияет на скорость предсказаний (ФИГ. 37Е), что указывает на то, что дальнодействующие детерминанты последовательности на расстоянии до 10 000 нуклеотидов от места сплайсинга имеют важное значение для различения функциональных границ сплайсинга среди большого количества нефункциональных сайтов с почти оптимальными мотивами. Чтобы изучить дальнодействующие или короткодействующие детерминанты специфичности, мы сравнили оценки, присвоенные аннотированным соединениям моделью, обученной на контекстной последовательности длиной 80 нуклеотидов (SpliceNet-80nt), с полной моделью, которая обучена на контексте в 10 000 нуклеотидов (SpliceNet-10k). Сеть, обученная на 80-нуклеотидной контекстной последовательности, присваивает более низкие оценки соединениям, примыкающим к экзонам или интронам типичной длины (150 нуклеотидов для экзонов, ~ 1000 нуклеотидов для нитронов) (ФИГ. 37F), в соответствии с более ранними наблюдениями, что такие сайты имеют тенденцию иметь более слабые мотивы сплайсинга по сравнению с необычно короткими или длинными сайтами сплайсинга экзонов и нитронов, (Amit et al., 2012; Gelfman et al., 2012; Li et al., 2015). Напротив, сеть, обученная на контекстной последовательности из 10 000 нуклеотидов, демонстрирует предпочтение нитронов и экзонов средней длины, несмотря на их более слабые мотивы сплайсинга, поскольку она может учитывать дальнодействующую специфичность, обусловленную длиной экзона или нитрона. Пропуск более слабых мотивов в длинных непрерывных интронах согласуется с более быстрым удлинением РНК-полимеразы II, экспериментально наблюдаемым в отсутствие паузы экзонов, что может оставлять сплайсосоме меньше времени для распознавания субоптимальных мотивов (Close et al., 2012; Jonkers et al., 2014; Veloso et al., 2014). Наши результаты указывают на то, что средняя граница (соединение) сплайсинга обладает благоприятными детерминантами последовательности с большим радиусом действия, которые придают существенную специфичность, объясняя высокую степень вырожденности последовательности, допускаемую для большинства мотивов сплайсинга.
[00288] Поскольку сплайсинг происходит (котранскрипционно) одновременно с транскрипцией (Cramer et al., 1997; Tilgner et al., 2012), взаимодействия между состоянием хроматина и котранскрипционным сплайсингом также могут направлять определение экзона (Luco et al., 2011) и иметь потенциал, который может использоваться сетью в той степени, в которой состояние хроматина можно предсказать по первичной последовательности. В частности, полногеномные исследования позиционирования нуклеосом показали, что занятость нуклеосом выше в экзонах (Andersson et al., 2009; Schwartz et al., 2009; Spies et al., 2009; Tilgner et al., 2009). Чтобы проверить, использует ли сеть детерминанты последовательности позиционирования нуклеосом для предсказания сайта сплайсинга, мы прошлись по паре оптимальных акцепторных и донорных мотивов, разделенных 150 нуклеотидами (примерно размер среднего экзона) в геноме, и попросили сеть предсказать, приведет ли эта пара мотивов к включению экзона в этот локус (ФИГ. 37G). Мы обнаружили, что положения, предсказанные как благоприятные для включения экзона, коррелируют с положениями с высокой занятостью нуклеосом, даже в межгенных областях (корреляция Спирмена=0,36, Р ≈ 0), и этот эффект сохраняется после контроля содержания GC (ФИГ. 44А). Эти результаты указывают на то, что сеть неявно научилась предсказывать положение нуклеосом по первичной последовательности и использует его в качестве детерминанты специфичности при определении экзона. Подобно экзонам и интронам средней длины, экзоны, расположенные над нуклеосомами, имеют более слабые локальные мотивы сплайсинга (ФИГ. 44 В), что согласуется с большей толерантностью к вырожденным мотивам в присутствии компенсаторных факторов (Spies et al., 2009).
[00289] Хотя в многочисленных исследованиях сообщалось о корреляции между экзонами и занятостью нуклеосом, причинная роль позиционирования нуклеосом в определении экзона не была окончательно установлена. Используя данные 149 человек с секвенированием как PHK-seq, так и всего генома из когорты Genotype-Tissue Expression (GTEx) (The GTEx Consortium et al., 2015), мы идентифицировали новые экзоны, которые были специфичными (собственными) для одного человека и соответствовали генетическим мутациям, создающим специфичный сайт сплайсинга. Эти события создания специфичного (собственного) экзона были в значительной степени связаны с существующим положением нуклеосом в клетках К562 и GM12878 (Р=0,006 по тесту перестановки, ФИГ. 37Н), даже несмотря на то, что эти клеточные линии, скорее всего, не имеют соответствующих собственных генетических мутаций. Наши результаты показывают, что генетические варианты с большей вероятностью будут запускать создание нового экзона, если полученный новый экзон будет перекрывать область существующей занятости нуклеосомы, что дополнительно поддерживает предположение о причинной роли позиционирования нуклеосомы в содействии определению экзона.
Проверка предсказанных криптических мутаций сплайсинга в данных секвенирования РНК (RNA-seq)
[00290] Мы расширили сеть глубокого обучения на оценку генетических вариантов для функции изменения сплайсинга за счет предсказания границ экзон-интрон как для референсной последовательности пре-мРНК, так и для альтернативной последовательности транскрипта, содержащей вариант, и учета разницы между оценками (Δ Score) (ФИГ. 38А). Важно отметить, что сеть обучалась только на референсных последовательностях транскриптов и аннотациях границ сплайсинга и никогда не видела данных вариантов во время обучения, что делало предсказание эффектов вариантов сложной проверкой способности сети точно моделировать детерминанты сплайсинга в последовательности.
[00291] Мы искали эффекты вариантов криптического сплайсинга данных секвенирования РНК в когорте GTEx (The GTEx Consortium et al., 2015), состоящей из 149 человек с секвенированием всего генома и секвенированием РНК из нескольких тканей. Чтобы приблизиться к сценарию, встречающемуся при секвенировании редких заболеваний, мы сначала сосредоточились на редких частных мутациях (присутствующих только у одного человека в когорте GTEx). Мы обнаружили, что частные мутации, которые, по прогнозам нейронной сети, имеют функциональные последствия, представлены в больших количествах в частных границах сплайсинга и на границах пропускаемых экзонов в частных событиях пропуска экзонов (ФИГ. 38В), что позволяет предположить, что большая часть этих предсказаний функциональна.
[00292] Чтобы количественно оценить эффекты вариантов, создающих сайт сплайсинга, на относительную продукцию нормальных и аберрантных изоформ сплайсинга, мы измерили количество ридов, поддерживающих новое событие сплайсинга, как долю от общего числа ридов, покрывающих сайт (ФИГ. 38С) (Cummings et al., 2017). Для вариантов, разрушающих сайт сплайсинга, мы наблюдали, что многие экзоны имели низкую базовую скорость пропуска экзонов, и эффект варианта заключался в увеличении доли ридов с пропуском экзонов. Соответственно, мы рассчитали как уменьшение доли ридов, которые сплайсировались на разрушенном соединении, так и увеличение доли ридов, которые пропустили экзон, принимая больший из двух эффектов (ФИГ. 45 и методы STAR).
[00293] Доверительно предсказанные критические варианты сплайсинга (Δ Score (оценки)≥0,5) подтверждаются на данных секвенирования РНК в трех четвертях случаев существенных нарушений сплайсинга GT или AG (ФИГ. 38D). Как уровень валидации, так и величина эффекта для вариантов криптического сплайсинга точно соответствуют их Δ-оценкам (ФИГ. 38D и 38Е), демонстрируя, что показатель предсказания модели является хорошим показателем способности варианта изменять плаайсинг. Валидированные варианты, особенно с более низкими оценками (Δ Score (оценки)<0,5), часто являются неполно пенетрантными и приводят к альтернативному сплайсингу с образованием смеси как аберрантных, так и нормальных транскриптов в данных секвенирования РНК (ФИГ. 38Е). Наши оценки степени валидации и величины эффекта консервативны и, вероятно, занижают истинные значения из-за неучтенных изменений изоформ сплайсинга и нонсенс-опосредованного распада, который разрушает преимущественно аберрантно сплайсированные транскрипты, поскольку они часто вводят преждевременные стоп-кодоны (ФИГ. 38С и ФИГ.45). Об этом свидетельствует средние размеры эффекта вариантов, разрушающих основные динуклеотиды сплайсинга GT и AG, которые составляют менее 50% от ожидаемого для полностью пенетрантных гетерозиготных вариантов.
[00294] Для вариантов криптического сплайсинга, которые продуцируют аберрантные изоформы сплайсинга, по меньшей мере, три десятых наблюдаемых копий транскрипта мРНК, сеть имеет чувствительность 71%, когда вариант находится рядом с экзонами, и 41%, когда вариант присутствует в глубокой интронной последовательности (Δ Score≥0.5, ФИГ. 38F). Эти находки показывают, что варианты глубоких интронов труднее предсказать, возможно, потому, что глубокие интронные области содержат меньше детерминант специфичности, которые были выбраны для присутствия вблизи экзонов.
[00295] Чтобы сравнить производительность нашей сети с существующими методами, мы выбрали три популярных классификатора, которые упоминаются в литературе для диагностики редких генетических заболеваний, GeneSplicer (Pertea et al., 2001), MaxEntScan (Yeo and Burge, 2004) и NNSplice (Reese et al., 1997), и построили график степени валидации и чувствительности на данных секвенирования РНК при различных порогах (ФИГ. 38G). Как и другие специалисты в этой области (Cummings et al., 2017), мы обнаружили, что существующие классификаторы обладают недостаточной специфичностью, учитывая очень большое количество некодирующих вариантов в масштабе всего генома, которые могут повлиять на сплайсинг, предположительно потому, что они сосредоточены на локальных мотивах и в значительной степени не учитывают детерминанты дальнодействующей специфичности.
[00296] Учитывая большой разрыв в производительности по сравнению с существующими методами, мы выполнили дополнительные проверки, чтобы исключить возможность того, что наши результаты на данных секвенирования РНК могут быть искажены из-за переобучения. Во-первых, мы повторили анализ проверки и чувствительности отдельно для частных вариантов и вариантов, присутствующих более чем у одного человека в когорте GTEx (ФИГ. 46А, 46В и 46С). Поскольку ни аппарат сплайсинга, ни модель глубокого обучения не имеют доступа к информации о частотах аллелей, проверка того, что сеть имеет одинаковую производительность в частотном спектре аллелей, является важным элементом контроля. Мы обнаружили, что при одних и тех же пороговых значениях Д-оценки частные и общие варианты криптического сплайсинга не демонстрируют значительных различий степени валидации в последовательности RNA-seq (Р>0,05, точный тест Фишера), что указывает на то, что предсказания сети устойчивы к частоте аллелей.
[00297] Во-вторых, чтобы валидировать предсказания модели для различных типов вариантов критического сплайсинга, которые могут создавать новые границы сплайсинга, мы отдельно оценили варианты, которые порождают новые динуклеотиды GT или AG, те, которые влияют на расширенный акцепторный или донорный мотив, и варианты, которые встречаются в более дистальные области. Мы обнаружили, что критические варианты сплайсинга примерно поровну распределяются между этими тремя группами, и что при одинаковых пороговых значениях Δ Score (оценки) нет значимых различий в степени валидации или величинами эффекта между группами (Р>0,3% χ^2, критерий однородности и Р>0,3 U-критерий Манна-Уитни соответственно, ФИГ. 47А и 47В).
[00298] В-третьих, мы выполнили валидацию и анализ чувствительности по PHK-seq отдельно для вариантов на хромосомах, использованных для обучения, и вариантов на остальных хромосомах (ФИГ. 48А и 48В). Хотя сеть была обучена только на референсной геномной последовательности и аннотациях сплайсинга и не подвергалась воздействию данных вариантов во время обучения, мы хотели исключить возможность ошибок в предсказаниях вариантов, возникающих из-за того, что сеть видела референсную последовательность в тренировочных хромосомах. Мы обнаружили, что сеть одинаково хорошо работает с вариантами из обучающих и тестовых хромосом без существенной разницы в степени валидации или чувствительности (Р>0,05, точный тест Фишера), что указывает на то, что предсказания вариантов сети вряд ли могут быть объяснены переобучением обучающими последовательностями.
[00299] Предсказание вариантов криптического сплайсинга является более сложной задачей, чем предсказание аннотированных границ сплайсинга, что отражается в результатах нашей модели, а также других алгоритмов предсказания сплайсинга (сравните ФИГ. 37Е и ФИГ. 38G). Важной причиной является различие в основном распределении уровней включения экзонов между двумя типами анализов. Подавляющее большинство экзонов, аннотированных GENCODE, имеют сильные детерминанты специфичности, в результате чего показатели конститутивного сплайсинга и предсказания близки к 1 (ФИГ. 37С). Напротив, большинство вариантов криптического сплайсинга являющиеся лишь частично пенетрантными (ФИГ. 38D и 38Е), имеют низкие или промежуточные оценки предсказания и часто приводят к альтернативному сплайсингу с образованием смеси как нормальных, так и аберрантных транскриптов. Это делает последнюю проблему прогнозирования эффектов вариантов криптического сплайсинга существенно более сложной, чем идентификация аннотированных сайтов сплайсинга. Дополнительные факторы, такие как миссенс-распад, неучтенные изоформные изменения и ограничения анализа RNA-seq, дополнительно способствуют снижению степени валидации на данных секвенирования РНК (ФИГ. 38С и ФИГ. 45).
Тканеспецифический альтернативный сплайсинг часто возникает в результате слабых криптических вариантах сплайсинга
[00300] Альтернативный сплайсинг представляет собой основной механизм регуляции генов, который служит для увеличения разнообразия транскриптов в разных тканях и на разных стадиях развития; его дисрегуляция ассоциирована с болезненными процессами (Blencowe, 2006; Irimia et al., 2014; Keren et al., 2010; Licatalosi and Darnell, 2006; Wang et al., 2008). Неожиданно авторы обнаружили, что относительная частота использования новых границ сплайсинга, образующихся при критических мутациях сплайсинга, может существенно варьировать в разных тканях (фиг. 39А). Кроме того, варианты, обуславливающие тканеспецифические различия сплайсинга, воспроизводятся у нескольких индивидуумов (фиг. 39В), что указывает на вероятность того, что в основе указанных различий, вероятно, лежит тканеспецифическая биология, а не стохастические эффекты. Авторы обнаружили, что для 35% вариантов критического сплайсинга со слабыми и промежуточными предсказанными оценками (Δ Оценка 0,35-0,8) наблюдаются значимые различия в доле нормальных и аберрантных транскриптов, образующихся в разных тканях (Р<0,01 с поправкой Бонферрони для критерия χ2, фиг. 39С). Это отличалось от вариантов с высокими предсказанными показателями (ΔПоказатель>0,8), которые со значимо меньшей вероятностью давали тканеспецифические эффекты (Р=0,015). Обнаруженные нами результаты согласуются с более ранним наблюдением, что у альтернативно сплайсированных экзонов наблюдается тенденция к промежуточным предсказанным оценкам (фиг. 37С) в отличие от конститутивно сплайсируемых, с исключением или включением, экзонов, которые имеют показатели, близкие к 1 или 0, соответственно.
[00301] Указанные результаты поддерживают модель, согласно которой тканеспецифические факторы, такие как контекст хроматина и связывание РНК-связывающих белков, могут сдвигать результаты конкуренции двух точек сплайсинга, близких по предпочтительности (Gelfman et al., 2013; Luco et al., 2010; Shukla et al., 2011; Ule et al., 2003). Сильные критические варианты сплайсинга способны полностью смещать сплайсинг от нормальной к аберрантной изоформе независимо от эпигенетического контекста, тогда как более слабые варианты сдвигают выбор границы (точки) сплайсинга ближе к границе решения, что приводит к использованию альтернативных точек в контекстах разных типах тканей и клеток. Это подчеркивает неожиданную роль, которую играют критические мутации сплайсинга в формировании разнообразия новых вариантов альтернативного сплайсинга, поскольку в таком случае при естественном отборе имеется возможность сохранения мутаций, создающих полезные варианты тканеспецифического альтернативного сплайсинга.
Предсказанные криптические варианты сплайсинга оказывают выраженное пагубное действие в популяциях человека
[00302] Хотя предсказанные криптические варианты сплайсинга имеют высокую степень валидации на данных секвенирования РНК (RNA-seq), во многих случаях указанные эффекты не являются полностью пенетрантными, и образуется смесь как нормальных, так и аберрантных изоформ сплайсинга, в результате возможно, что доля указанных критических изменяющих сплайсинг вариантов может не быть функционально значимой. Для изучения особенностей влияния естественного отбора на предсказанные криптические варианты сплайсинга авторы получили оценки для каждого варианта в 60 706 экзомах человека из базы данных Exome Aggregation Consortium (ЕхАС) (Lek et al., 2016), и идентифицировали предсказанные изменяющие границы экзон-интрон варианты.
[00303] Для изменения степени отрицательного отбора, действующего на предсказанные изменяющие сплайсинг варианты, авторы подсчитывали число предсказанных изменяющих сплайсинг вариантов с обнаруживаемыми частотами распространенных аллелей (≥0,1% в популяции человека) и сравнивали с числом предсказанных изменяющих сплайсинг вариантов с частотой единичного аллеля в ЕхАС (т.е. у 1 из 60 706 индивидуумов). Из-за экспоненциального роста популяции человека в последнее время, единичные варианты представляют недавно образовавшиеся мутации, прошедшие минимальную фильтрацию очищающего отбора (Tennessen et al., 2012). И напротив, распространенные (общие) варианты представляют поднабор нейтральных мутаций, прошедших через сито очищающего отбора. Таким образом, истощение (деплеция) по предсказанным изменяющим сплайсинг вариантам в спектре частоты распространенных (общих) аллелей относительно одиночных вариантов обеспечивает расчетную оценку доли предсказанных изменяющих сплайсинг вариантов, которые обладают пагубным действием и, соответственно, функциональны. Чтобы избежать искажающих эффектов на кодирующую белок последовательность, авторы ограничили анализ синонимичными вариантами и интронными вариантами, располагающимися вне необходимых динуклеотидов GT или AG, исключая миссенс-мутации, которые также, как предсказано, имеют изменяющие сплайсинг эффекты.
[00304] При обычных частотах аллелей достоверно предсказанные критические варианты сплайсинга (Δ-Оценка≥0,8) подвергаются выраженному отрицательному отбору, о чем свидетельствует их относительное истощение по сравнению с ожиданием (фиг. 40А). При указанном пороговом значении, когда большинство вариантов, как ожидается, будут близки к полной пенетрантности по данным RNA-seq (фиг. 38D), истощение по предсказанным синонимичным и интронным мутациям скрытого сплайсинга составляет 78% при обычной частоте аллелей, что сопоставимо с 82% истощением по вариантам с мутациями со сдвигом рамки, стоп-мутациями и мутациями, разрушающими необходимые для сплайсинга GT или AG (фиг. 40 В). Влияние отрицательного отбора больше при рассмотрении вариантов скрытого сплайсинга, вызывающих мутации со сдвигом рамки, по сравнению с вариантами, которые вызывают изменения внутри рамки (фиг. 40С). Истощение по вариантам скрытого сплайсинга с последствиями в виде сдвига рамки почти идентично истощению по другим классам вариантов с усечением белка, что указывает на то, что подавляющее большинство достоверно предсказанных мутаций скрытого сплайсинга в около-интронной области (≤50 нуклеотидов от известных экзон-интронных границ) являются функциональными и оказывают выраженные пагубные эффекты в популяции человека.
[00305] Для расширения указанного анализа на глубокие интронные области, >50 нуклеотидов от известных экзон-интронных границ, авторы использовали агрегированные данные полно геномного секвенирования для 15496 людей из когорты базы данных Genome Aggregation Database (gnomAD) (Lek et al., 2016) для вычисления наблюдаемого и ожидаемого количества критических мутаций сплайсинга при обычной частоте аллелей. В общей сложности, авторы наблюдали 56% истощение по обычным мутациям скрытого сплайсинга (Δ Оценка ≥0,8) на расстоянии >50 нуклеотидов от экзон-интронной границы (фиг. 40D), что согласуется с большей сложностью предсказания влияния глубоко интронных вариантов, что авторы наблюдали в данных секвенирования РНК.
[00306] Затем мы попытались получить оценку потенциальной способности мутаций скрытого сплайсинга вносить вклад в пенетрантное генетическое заболевание относительно других типов вариаций кодирования белков, путем измерения числа редких мутаций скрытого сплайсинга у каждого индивидуума в когорте gnomAD. Судя по доле предсказанных мутаций скрытого сплайсинга, находящихся под действием отрицательного отбора (фиг. 40А), средний человек является носителем ~5 редких функциональных мутаций скрытого сплайсинга (частота аллелей <0,1%), и при этом ~11 редких вариантов с усечением белков (фиг. 40Е). Критические варианты сплайсинга численно превосходяп варианты с разрушением необходимых для сплайсинга GT или AG в отношении примерно 2:1. Авторы отмечают, что значительная доля указанных вариантов скрытого сплайсинга может не полностью устранять функцию гена, либо ввиду того, что они вызывают изменения внутри рамки, либо ввиду того, что они не полностью переводят сплайсинг в аберрантную изоформу
De novo криптические мутации сплайсинга представляют собой основную причину редких генетических расстройств
[00307] Исследования с крупномасштабным секвенированием на пациентах с расстройствами аутического спектра и тяжелой умственной отсталостью продемонстрировали центральную роль de novo мутаций кодирования белков (миссенс-мутаций, нонсенс-мутаций, мутаций со сдвигом рамки и мутации динуклеотидов, необходимых для сплайсинга), которые разрушают гены путей развития нервной системы (Fitzgerald et al., 2015; Iossifov et al., 2014; McRae et al., 2017; Neale et al., 2012; De Rubeis et al., 2014; Sanders et al., 2012). Для оценки клинического воздействия некодирующих мутаций, которые действуют за счет изменения сплайсинга, авторы использовали нейронную сеть для предсказания эффектов de novo мутаций у 4293 индивидуумов с умственной отсталостью из когорты проекта Deciphering Developmental Disorders (DDD) (McRae et al., 2017), 3953 индивидуумов с расстройствами аутического спектра (ASD) из проектов Simons Simplex Collection (De Rubeis et al., 2014; Sanders et al., 2012; Turner et al., 2016) и Autism Sequencing Consortium, и 2073 не пораженных указанными расстройствами единокровных братьев и сестер (сиблингов) из Simons Simplex Collection. Для контроля различий при установлении de novo вариантов в разных исследованиях авторы нормировали ожидаемое число de novo вариантов, так что число синонимичных мутаций на каждого индивидуума было одним и тем же во всех когортах.
[00308] 1,51-кратное обогащение по сравнению с здоровыми контрольными индивидуумами по de novo мутациям, которые, как предсказано, способны разрушать сплайсинг, происходит при умственной отсталости (Р=0,000416); и 1,30-кратное - при расстройствах аутического спектра (Р =0,0203) (Δ Оценка ≥0,1, фиг. 41А, фиг. 43А и фиг. 43В). Значимое обогащение по разрушающим сплайсинг мутациям происходит также в случаях расстройств относительно контролей при рассмотрении только синонимичных и интронных мутаций (фиг. 49А, фиг. 49В и фиг. 49С), исключая возможность того, что обогащение может быть объяснено исключительно мутациями с двойным эффектом, в отношении кодирования белков и сплайсинга. Судя по избытку de novo мутаций у пораженных индивидуумов относительно не пораженных, криптические мутации сплайсинга включают, по расчетным оценкам, приблизительно 11% патогенных мутаций при расстройствах аутического спектра и 9% при умственной отсталости (фиг. 41В), после коррекции с учетом ожидаемой доли мутаций в областях, где отсутствовало покрытие секвенирования или установленные варианты, в каждом исследовании. Большинство de novo предсказанных критических мутаций сплайсинга у пораженных индивидуумов имели Δ показатели <0,5 (фиг. 41С, фиг. 50А, и фиг. 50В), и, согласно ожиданиям, должны были давать смесь нормальных и аберрантных транскриптов, судя по вариантам с аналогичными показателями в наборе данных GTEx RNA-seq.
[00309] Для оценки обогащения по криптическим мутациям сплайсинга в кандидатных связанных с заболеванием генах по сравнению со случайным, авторы вычисляли вероятность прогнозирования de novo криптических мутаций сплайсинга для каждого индивидуального гена с использованием тринуклеотидного контекста для коррекции частоты мутаций (Samocha et al., 2014) (таблица S4). Комбинирование криптических мутаций сплайсинга с мутациями кодирования белков при поиске новых генов дает 5 дополнительных кандидатных генов, ассоциированных с умственной отсталостью, и 2 дополнительных гена, ассоциированных с расстройством аутического спектра (фиг. 41D и фиг. 45), которые не достигали бы порога обнаружения (FDR<0,01) при рассмотрении только мутаций кодирования белков (Kosmicki et al., 2017; Sanders et al., 2015).
Экспериментальное подтверждение de novo криптических мутаций сплайсинга у пациентов с аутизмом
[00310] Мы получили клетки лимфобластоидных линий из периферической крови (КЛЛ) от 36 индивидуумов из проекта Simons Simplex Collection, носителей предсказанных de novo криптических мутаций сплайсинга в генах с по меньшей мере минимальным уровнем экспрессии КЛЛ (De Rubeis et al., 2014; Sanders et al., 2012); каждый индивидуум представлял единственный случай аутизма среди близких родственников. Как в случае большинства редких генетических заболеваний, релевантный тип тканей и клеток тип (предположительно, развивающийся мозг) доступен не был. Поэтому авторы проводили глубокое секвенирование мРНК (~350 млн × 150 п. о. одиночных ридов на образец, примерно в 10 раз больше покрытия GTEx) для компенсации слабой экспрессии многих из указанных транскриптов в КЛЛ. Для обеспечения подтверждения репрезентативного набора предсказанных вариантов критического сплайсинга, а не просто ведущих предсказанных вариантов, авторы использовали относительно нестрогие пороги (Δ Оценка >0,1 для вариантов с утратой точки сплайсинга и Δ Оценка>0,5 для вариантов с добавлением точки сплайсинга; методы STAR); и экспериментально подтверждали все de novo варианты, отвечающие указанным критериям.
[00311] После исключения 8 индивидуумов с недостаточным покрытием RNA-seq по представляющему интерес гену мы идентифицировали уникальные события аберрантного сплайсинга, ассоциированные с предсказанной de novo критической мутацией сплайсинга у 21 из 28 пациентов (фиг. 41Е и фиг. 51А, 51В, 51С, 51D, 51Е, 51F, 51G, 51H, 51I и 51J). Указанные события аберрантного сплайсинга отсутствовали у других 35 индивидуумов, для КЛЛ которых проводили глубокое секвенирование РНК, а также у 149 индивидуумов из когорты GTEx. Среди 21 подтвержденных de novo критических мутаций сплайсинга авторы наблюдали 9 случаев образования новых границ (точек) сплайсинга, 8 случаев перепрыгивания экзонов и 4 случая сохранения интронов, а также более сложные аберрации сплайсинга (фиг. 41F, фиг. 46А фиг. 46В, и фиг. 46С). В семи случаях аберрантный сплайсинг в КЛЛ не наблюдался, несмотря на экспрессию соответствующего транскрипта. Хотя такой под набор может давать ложно положительные предсказания, некоторые критические мутации сплайсинга могут приводить к тканеспецифическому альтернативному сплайсингу, который не поддается наблюдению в КЛЛ в указанных экспериментальных условиях.
[00312] Высокая степень валидации предсказанных критических мутаций сплайсинга у пациентов с расстройством аутического спектра (75%), несмотря на ограничения анализа RNA-seq, показывает, что большинство предсказанных вариантов функциональны. Однако обогащение de novo криптическими вариантами сплайсинга в случаях расстройств по сравнению с контролями (1,5-кратное при DDD и 1,3-кратное при ASD, фиг. 41А) составляет только 38% от величины эффекта, наблюдаемого для de novo вариантов с укорочением (усечением) белков (2,5-кратного при DDD и 1,7-кратного при ASD) (Iossifov et al., 2014; McRae et al., 2017; De Rubeis et al., 2014). Это позволило нам количественно определить, что функциональные криптические мутации сплайсинга обеспечивают примерно 50% клинической пенетрантности классических форм мутации с усечением белка (стоп-мутация, мутация со сдвигом рамки и и мутации необходимых для сплайсинга динуклеотидов), вследствие того, что многие из них только частично нарушают образование нормального транскрипта. Так, некоторые из наиболее полно описанных критических мутаций сплайсинга при менделевских заболеваниях, такие как с. 315-48Т>С в FECH (Gouya et al., 2002) и c.-32-13T>G в GAA (Boerkoel et al., 1995), представляют собой гипоморфные аллели, ассоциированные с более мягким фенотипом или более поздним возрастом начала. Расчетную клиническую пенетрантность вычисляют для всех de novo вариантов, отвечающих относительно нестрогому порогу (Δ Оценка≥0,1), и варианты с более сильными предсказанными показателями, как ожидается, будут обладать соответствующей более высокой пенетрантностью.
[00313] На основании избытка de novo мутаций в случаях расстройств относительно контролей во всех когортах ASD и DDD, 250 случаев могут быть объяснены криптическими de novo мутациями сплайсинга, а 909 случаев могут быть объяснены de novo вариантами с усечением белков (фиг. 41В). Эти объяснения согласуются с полученной авторами ранее расчетной оценкой среднего числа редких критических мутаций сплайсинга (~5) по сравнению с редкими вариантами с усечением белков (~11) на человека в общей популяции (фиг. 38А), при учете сниженной пенетрантности критических мутаций сплайсинга. Широкое распределение криптических мутаций сплайсинга в геноме предполагает, что данные о доле случаев, объясняемых мутациями скрытого сплайсинга при нарушениях развития нервной системы (9-11%, фиг. 41В), вероятно, можно распространить и на другие редкие генетические нарушения, при которых первичный механизм заболевания заключается в утрате функционального белка. Для облегчения интерпретации изменяющих сплайсинг мутаций авторы предварительно вычисляли предсказанные Δ Оценки для всех возможных однонуклеотидных замен по всему геному, и предоставляют указанные данные в качестве ресурса научному сообществу. Авторы полагают, что указанный ресурс будет способствовать пониманию указанного ранее недооцененного источника генетической вариативности.
Частные варианты реализации
[00314] Мы описываем системы, способы и изделия для применения обученной разреженной сверточной нейронной сети для детектирования сайтов сплайсинга в геномной последовательности (например, в нуклеотидной последовательности или аминокислотой последовательности). Один или более признаков одного из вариантов реализации могут быть объединены с базовым вариантом реализации. Подразумевается, что варианты, которые не являются взаимоисключающими, совместимы. Один или более признаков варианта реализации можно комбинировать с другими вариантами реализации. Настоящее описание периодически напоминает пользователю об этих опциях. То, что в некоторых вариантах реализации пропущено упоминание об этих опциях, не следует воспринимать как ограничение комбинаций, описанных в предыдущих разделах - в настоящем тексте такое описание включается посредством ссылки в каждый их последующих вариантов реализации.
[00315] В этом раздели термины модуля(ей) и стадии(ий) используются взаимозаменяемо.
[00316] Один вариант реализации системы в соответствии с раскрытой технологией включает один или более процессоров, соединенных с памятью. В память загружены компьютерные команды для обучения детектора сайтов сплайсинга, который идентифицирует сайты сплайсинга в геномных последовательностях (например, нуклеотидных последовательностей).
[00317] Как показано на ФИГ. 30, система обучает разреженную сверточную нейронную сеть (сокращенно ACNN) на по меньшей мере 50000 тренировочных примеров донорных сайтов сплайсинга, по меньшей мере 50000 тренировочных примеров акцепторных сайтов сплайсинга и на по меньшей мере 100000 тренировочных примеров сайтов, не связанных со сплайсингом. Каждый обучающий пример представляет собой целевую нуклеотидную последовательность, имеющую по меньшей мере один целевой нуклеотид, фланкированный по меньшей мере 20 нуклеотидами с каждой стороны.
[00318] ACNN представляет собой сверточную нейронную которая использует дырчатые/разреженные свертки, что дает возможность больших рецептивных полей при небольшом числе обучаемых параметров Atrous/разреженная свертка представляет собой свертку, в которой ядро применяется на площади, большей, чем его длина, за счет того, что она пропускает входные значения с определенным шагом, называемым также показателем разрежения или фактором разрежения. Atrous/разреженные свертки увеличивают расстояние между элементами свертки фильтра свертки/ядра, в результате чего при осуществлении операции свертки используются соседние входные записи (например, нуклеотиды, аминокислоты) с большими интервалами. Это обеспечивает возможность введения во входные данные контекстуальных зависимостей дальнего действия. Дырчатые (atrous) свертки сохраняют расчет свертки для повторного использования при обработке соседних нуклеотидов.
[00319] Как показано на ФИГ. 30, для оценки тренировочного примера с применением указанной ACNN, системя обеспечивает, в качестве входя указанной ACNN, целевую нуклеотидную последовательность, дополнительно фланкированную по меньшей мере 40 расположенными в направлении 5'-конца нуклеотидами и по меньшей мере 40 расположенными в направлении 3'-конца нуклеотидами.
[00320] Как показано на ФИГ. 30, на основании этой оценки, затем ACNN-сеть генерирует, в качестве выхода, тройные оценки для правдоподобия того, что каждый нуклеотид в целевой нуклеотидной последовательности является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом.
[00321] Вариант реализации этой системы и других раскрытых систем, раскрываемых необязательно включает один или более из следующих признаков. Система может дополнительно включать признаки, описанные в связи с раскрытыми способами. Для краткости альтернативные комбинации признаков системы не перечисляются по отдельности. Признаки, применимые к системам, способам и изделиям, не повторяются для каждого предписанного набора множества классов базовых признаков. Читатель поймет, как признаки, идентифицированные в этом разделе, могут быть легко объединены с базовыми признаками других предписанных классов
[00322] Как показано на ФИГ. 25, 26 и 27, вход может включать целевую нуклеотидную последовательность, которая содержит целевой нуклеотид, фланкированный 2500 нуклеотидами с каждой стороны. В каждом таком варианте реализации целевой нуклеотид дополнительно фланкирован 5000 расположенными в направлении 5' контекстными нуклеотидами и 5000 расположенными в направлении 3' контекстными нуклеотидами.
[00323] Входные данные могут включать целевую нуклеотидную последовательность, которая содержит целевой нуклеотид, фланкированный 100 нуклеотидами с каждой стороны. В каждом таком варианте реализации целевой нуклеотид дополнительно фланкирован 200 расположенными в направлении 5' контекстными нуклеотидами и 200 расположенными в направлении 3' контекстными нуклеотидами.
[00324] Входные данные могут включать целевую нуклеотидную последовательность, которая содержит целевой нуклеотид, фланкированный 500 нуклеотидами с каждой стороны. В каждом таком варианте реализации целевой нуклеотид дополнительно фланкирован 5000 расположенных в направлении 5' контекстных нуклеотидов и 5000 расположенных в направлении 3' контекстных нуклеотидов.
[00325] Как показано на ФИГ. 28, система может обучать ACNN-сеть на 150000 тренировочных примеров донорных сайтов сплайсинга, 150000 тренировочных примеров акцепторных сайтов сплайсинга, and 800000000 тренировочных примеров сайтов, не связанных со сплайсингом.
[00326] Как показано на ФИГ. 19, ACNN-сеть может включать группы остаточных блоков, организованных в последовательность от низшего к высшему. Причем каждая группа остаточных блоков параметризируется числом сверточных фильтров в остаточных блоках, размером окна свертки остаточных блоков и показателем разрежения свертки остаточных блоков.
[00327] Как показано на ФИГ. 21, 22, 23 и 24, в указанной ACNN-сети показатель разряжения свертки растет неэкспоненциально от нижней группы остаточных блоков к более высокой группе остаточных блоков.
[00328] Как показано на ФИГ. 21, 22, 23 и 24, в указанной ACNN-сети размер окна свертки в разных группах остаточных блоков различаются.
[00329] Указанная ACNN может быть сконфигурирована для оценки входа, который включает целевую нуклеотидную последовательность, дополнительно фланкированную 40 расположенными в направлении 5' контекстными нуклеотидами и 40 расположенными в направлении 3' контекстными нуклеотидами. В каждом таком варианте реализации ACNN включает одну группу из четырех остаточных блоков и по меньшей мере одно соединение с пропуском. Каждый остаточный блок имеет 32 сверточных фильтра, размер окна свертки 11 и показатель разрежения свертки 1. Этот вариант реализации ACNN называется в настоящем документе "SpliceNet80" и показан на ФИГ. 21.
[00330] Указанная ACNN может быть сконфигурирована для оценки входа, который включает целевую нуклеотидную последовательность, дополнительно фланкированную 200 расположенными в направлении 5' контекстными нуклеотидами и 200 расположенными в направлении 3' контекстными нуклеотидами. В каждом таком варианте реализации ACNN включает по меньшей мере две группы из четырех остаточных блоков и по меньшей мере два соединения с пропуском. Каждый остаточный блок в первой группе имеет 32 сверточных фильтра, размер окна свертки 11 и показатель разрежения свертки 1. Каждый остаточный блок во второй группе имеет 32 сверточных фильтра, размер окна свертки 11 и показатель разрежения свертки 4. Этот вариант реализации ACNN называется в настоящем документе "SpliceNet400" и показан на ФИГ. 22.
[00331] Указанная ACNN может быть сконфигурирована для оценки входа, который включает целевую нуклеотидную последовательность, дополнительно фланкированную 5000 расположенных в направлении 5' контекстных нуклеотидов и 5000 расположенных в направлении 3' контекстных нуклеотидов. В каждом таком варианте реализации ACNN включает по меньшей мере три группы из четырех остаточных блоков и по меньшей мере три соединения с пропуском. Каждый остаточный блок в первой группе имеет 32 сверточных фильтра, размер окна свертки 11 и показатель разрежения свертки 1. Каждый остаточный блок во второй группе имеет 32 сверточных фильтра, размер окна свертки 11 и показатель разрежения свертки 4. Каждый остаточный блок в третьей группе имеет 32 сверточных фильтра, размер окна свертки 21, и показатель разрежения свертки 19. Этот вариант реализации ACNN называется в настоящем документе "SpliceNet2000" и показан на ФИГ. 23.
[00332] Указанная ACNN может быть сконфигурирована для оценки входа, который включает целевую нуклеотидную последовательность, дополнительно фланкированную 5000 расположенными в направлении 5' контекстными нуклеотидами и 5000 расположенными в направлении 3' контекстными нуклеотидами. В каждом таком варианте реализации ACNN включает по меньшей мере четыре группы из четырех остаточных блоков и по меньшей мере четыре соединения с пропуском. Каждый остаточный блок в первой группе имеет 32 сверточных фильтра, размер окна свертки 11 и показатель разрежения свертки 1. Каждый остаточный блок во второй группе имеет 32 сверточных фильтра, размер окна свертки 11 и показатель разрежения свертки 4. Каждый остаточный блок в третьей группе имеет 32 сверточных фильтра, размер окна свертки 21, и показатель разрежения свертки 19. Каждый остаточный блок в четвертой группе имеет 32 сверточных фильтра, размер окна свертки 41 и показатель разрежения свертки 25. Этот вариант реализации ACNN называется в настоящем документе "SpliceNet 10000" и показан на ФИГ. 24.
[00333] Тройные оценки для каждого нуклеотида в целевой нуклеотидной последовательности могут быть экспоненциально нормированы чтобы давать в сумме единицу. В каждом таком варианте реализации система классифицирует каждый нуклеотид в целевом нуклеотиде как донорный сайт сплайсинга, акцепторный сайт сплайсинга или сайт, не связанный со сплайсингом, на основании самой высокой оценки в соответствующих тройных оценках.
[00334] Как показано на ФИГ. 35, размерность входа ACNN-сети может быть определена как (Cu+L+Cd) х 4, где Cu представляет собой число расположенных в 5'-направлении контекстных нуклеотидов, Cd представляет собой число расположенных в 3'-направлении контекстных нуклеотидов, и L представляет собой число нуклеотидов в целевой нуклеотидной последовательности. В одном варианте реализации размерность входа составляет (5000+5000+5000) х 4.
[00335] Как показано на ФИГ. 35, размерность выхода ACNN-сети может быть определена как L х 3. В одном варианте реализации размерность составляет 5000 х 3.
[00336] Как показано на ФИГ. 35, каждая группа остаточных блоков может выдавать промежуточный выход посредством обработки предшествующего входа. Размерность промежуточного выхода может быть определена как (I-[{(W-1) * D} * А]) х N, где I представляет собой размерность предшествующего выхода, W представляет собой размер окна свертки остаточных блоков, D представляет собой показатель разрежения свертки остаточных блоков, А представляет собой число слоев разреженной свертки в указанной группе, и N представляет собой число сверточных фильтров в остаточных блоках.
[00337] Как показано на ФИГ. 32, ACNN пакетно оценивает обучающие примеры на протяжении эпохи. Тренировочные образцы случайным образом группируются в пакеты. Каждый пакет имеет заранее определенный размер пакета. ACNN итерирует оценку обучающих примеров на протяжении нескольких эпох (например, 1-10).
[00338] Входные данные могут включать целевую последовательность, которая содержит два соседних целевых нуклеотида. Два соседних целевых нуклеотида могут представлять собой аденин (сокращенно А) и гуанин (сокращенно G). Два соседних целевых нуклеотида могут представлять собой гуанин (сокращенно G) и урацил (сокращенно U).
[00339] Система включает энкодер с одним горячим состоянием (показан ФИГ. 29), который кодирует с разрежением тренировочные примеры и выдает в качестве выходных данных результаты кодирования с одним горячим состоянием.
[00340] ACNN может быть параметризована числом остаточных блоков, числом соединений с пропуском и числом остаточных связей.
[00341] ACNN может содержать меняющие размерность сверточные слои, которые изменяют пространственные размерности и размерности признаков предшествующего входа.
[00342] Как показано на ФИГ. 20, каждый остаточный блок может включать по меньшей мере один слой пакетной нормализации, по меньшей мере один слой блоков линейной ректификации (сокращенно ReLU), по меньшей мере один слой разреженной свертки и по меньшей мере одну остаточную связь. В каждом таком варианте реализации каждый остаточный блок содержит два слоя пакетной нормализации, два нелинейных слоя ReLU, два слоя разреженной (atrous) свертки и одно остаточное соединение.
[00343] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению системой описанных выше действий. Еще один вариант реализации может включать выполнение описанных выше действий системы.
[00344] Другой системный вариант реализации раскрытой технологии включает обученный предсказатель сайтов сплайсинга, который реализуется на множестве процессоров, работающих параллельно и связанных с памятью. Система обучает (тренирует) разреженную сверточную нейронную сеть (сокращенно ACNN), реализованную на множестве процессоров, на по меньшей мере 50000 тренировочных примеров донорных сайтов сплайсинга, по меньшей мере 50000 тренировочных примеров акцепторных сайтов сплайсинга и по меньшей мере 100000 тренировочных примеров сайтов, не связанных со сплайсингом. Каждый из тренировочных (обучающих) примеров, используемых для обучения, представляет собой нуклеотидную последовательность, которая включает целевой нуклеотид, фланкированный по меньшей мере 400 нуклеотидами с каждой стороны.
[00345] Система включает стадию входа указанной ACNN, которая реализуется на по меньшей мере одном из указанного множества процессоров и обеспечивает подачу входной последовательности из по меньшей мере 801 нуклеотида для оценки целевых нуклеотидов. Каждый целевой нуклеотид фланкирован по меньшей мере 400 нуклеотидами с каждой стороны. В других вариантах реализации система включает модуль ввода указанной ACNN, который реализован на по меньшей мере одном из указанного множества процессоров и обеспечивает подачу входной последовательности из по меньшей мере 801 нуклеотида для оценки целевых нуклеотидов.
[00346] Система включает стадию выхода указанной ACNN, которая реализуется на по меньшей мере одном из указанного множества процессоров и переводит анализ указанной ACNN-сетью в классификационные оценки для правдоподобия того, что каждый из указанных целевых нуклеотидов является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом. В других вариантах реализации система включает модуль вывода указанной ACNN, который реализован на по меньшей мере одном из указанного множества процессоров и переводит анализ указанной ACNN-сетью в классификационные оценки для правдоподобия того, что каждый из указанных целевых нуклеотидов является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом.
[00347] Каждый признак, обсуждаемый в этом разделе, относящемся к конкретному варианту реализации, применительно к первому варианту реализации системы, равно применим к этому варианту реализации системы. Как указано выше, здесь не повторяются все признаки системы, подразумевается, что они повторяются посредством ссылки.
[00348] ACNN-сеть может быть обучена на 150000 тренировочных примеров донорных сайтов сплайсинга, 150000 тренировочных примеров акцепторных сайтов сплайсинга и 800000000 тренировочных примеров сайтов, не связанных со сплайсингом. В другом варианте реализации системы ACNN содержит группы остаточных блоков, организованных в последовательность от низшего к высшему. В еще одном варианте реализации каждая группа остаточных блоков параметризируется числом сверточных фильтров в остаточных блоках, размером окна свертки остаточных блоков и показателем разрежения свертки остаточных блоков.
[00349] ACNN может содержать группы остаточных блоков, организованных в последовательность от низшего к высшему. Причем каждая группа остаточных блоков параметризируется числом сверточных фильтров в остаточных блоках, размером окна свертки остаточных блоков и показателем разрежения свертки остаточных блоков.
[00350] В ACNN показатель разряжения свертки растет неэкспоненциально от нижней группы остаточных блоков к более высокой группе остаточных блоков. Также в указанной ACNN-сети размер окна свертки в разных группах остаточных блоков различаются.
[00351] ACNN-сеть может быть обучена на одном или более обучающих серверов, как показано на ФИГ. 18.
[00352] Обученная сеть ACNN может быть размещена на одном или более рабочих серверах, которые получают входные последовательности от запрашивающих клиентов, как показано ФИГ. 18. В каждом таком варианте реализации указанные рабочие серверы обрабатывают входные последовательности посредством стадий ввода и вывода указанной ACNN с получением выходных данных, которые передаются указанным клиентам, как показано на ФИГ. 18. В других вариантах реализации указанные рабочие серверы обрабатывают входные последовательности посредством входных и выходных модулей сети ACNN с получением выходных данных, которые передаются указанным клиентам, как показано на ФИГ. 18.
[00353] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению системой описанных выше действий. Еще один вариант реализации может включать выполнение описанных выше действий системы.
[00354] Вариант реализации способа согласно раскрытой технологии включает обучение детектора сайтов сплайсинга, который идентифицирует сайты сплайсинга в геномных последовательностях (например, нуклеотидных последовательностях).
[00355] Способ включает ввод в разреженную сверточную нейронную сеть (сокращенно ACNN) входной последовательности из по меньшей мере 801 нуклеотида для оценки целевых нуклеотидов, каждый из которых фланкирован по меньшей мере 400 нуклеотидами с каждой стороны. [00356] ACNN обучается на по меньшей мере 50000 тренировочных примеров донорных сайтов сплайсинга, по меньшей мере 50000 тренировочных примеров акцепторных сайтов сплайсинга и по меньшей мере 100000 тренировочных примеров сайтов, не связанных со сплайсингом. Каждый из тренировочных (обучающих) примеров, используемых для обучения, представляет собой нуклеотидную последовательность, которая включает целевой нуклеотид, фланкированный по меньшей мере 400 нуклеотидами с каждой стороны.
[00357] Способ дополнительно включает перевод анализа указанной ACNN-сетью в классификационные оценки для правдоподобия того, что каждый из указанных целевых нуклеотидов является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом.
[00358] Каждый из признаков, обсуждаемых в разделе, относящемся к этому конкретному варианту реализации, для первого варианта реализации системы, равно относится к этому варианту реализации способа. Как указано выше, здесь не повторяются все признаки системы, подразумевается, что они повторяются посредством ссылки.
[00359] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению описанного выше способа. Еще один вариант реализации может включать систему, включающую память и один или более процессоров, выполненных с возможностью исполнять инструкции, хранящиеся в памяти, с реализацией описанного выше способа.
[00360] Мы описываем системы, способы и изделия для применения обученной разреженной сверточной нейронной сети для выявления аберрантного сплайсинга в геномных последовательностях (например, нуклеотидных последовательностях). Один или более признаков одного из вариантов реализации могут быть объединены с базовым вариантом реализации. Подразумевается, что варианты, которые не являются взаимоисключающими, совместимы. Один или более признаков варианта реализации можно комбинировать с другими вариантами реализации. Настоящее описание периодически напоминает пользователю об этих опциях. То, что в некоторых вариантах реализации пропущено упоминание об этих опциях, не следует воспринимать как ограничение комбинаций, описанных в предыдущих разделах в настоящем тексте такое описание включается посредством ссылки в каждый их последующих вариантов реализации.
[00361] Один вариант реализации системы в соответствии с раскрытой технологией включает один или более процессоров, соединенных с памятью. В память загружены компьютерные команды по реализации детектора аберрантного сплайсинга, реализованного на множестве процессоров, работающих параллельно и связанных с памятью.
[00362] Как показано на ФИГ. 34, система включает обученную разреженную сверточную нейронную сеть (сокращенно ACNN), реализованную на множестве процессоров. ACNN представляет собой сверточную нейронную которая использует дырчатые/разреженные свертки, что дает возможность больших рецептивных полей при небольшом числе обучаемых параметров. Atrous/разреженная свертка представляет собой свертку, в которой ядро применяется на площади, большей, чем его длина, за счет того, что она пропускает входные значения с определенным шагом, называемым также показателем разрежения или фактором разрежения. Atrous/разреженные свертки увеличивают расстояние между элементами свертки фильтра свертки/ядра, в результате чего при осуществлении операции свертки используются соседние входные записи (например, нуклеотиды, аминокислоты) с большими интервалами. Это обеспечивает возможность введения во входные данные контекстуальных зависимостей дальнего действия. Дырчатые (atrous) свертки сохраняют расчет свертки для повторного использования при обработке соседних нуклеотидов.
[00363] Как показано на ФИГ. 34, ACNN классифицирует входные нуклеотиды во входной последовательности и присваивает оценки сайтам сплайсинга для правдоподобия того, что каждый из указанных целевых нуклеотидов является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом. Входная последовательность содержит по меньшей мере 801 нуклеотид, и каждый целевой нуклеотид фланкирован по меньшей мере 400 нуклеотидами с каждой стороны.
[00364] Как показано на ФИГ. 34, система также включает классификатор, реализованный на по меньшей мере одном из указанного множества процессоров, который обрабатывает референсную последовательность и вариантную последовательность посредством ACNN и выдает оценки сайтов сплайсинга, характеризующие правдоподобие того, что каждый целевой в референсной последовательности и в вариантной последовательности является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом. И референсная последовательность, и вариантная последовательность, каждая содержит по меньшей мере 101 целевой нуклеотид, и каждый целевой нуклеотид фланкирован по меньшей мере 400 нуклеотидами с каждой стороны. ФИГ. 33 показывает референсную последовательность и альтернативную/вариантную последовательность.
[00365] Как показано на ФИГ. 34, затем система определяет, по разницам в оценках сайтов сплайсинга целевых нуклеотидов в референсной последовательности и в вариантной последовательности, вызывает ли вариант, порождающий вариантную последовательность, аберрантный сплайсинг, и является ли он, соответственно, патогенным.
[00366] Этот вариант реализации системы и другие раскрытые системы необязательно включают один или более из следующих признаков. Система может также включать признаки, описанные в связи с раскрытыми способами. Для краткости альтернативные комбинации признаков системы не приводятся по отдельности. Признаки, связанные с системами, способами и изделиями, не повторяются для каждого заявленного набора классов основных признаков. Читатель поймет, как признаки, идентифицированные в этом разделе, можно легко объединять с основными признаками в других заявленных классах.
[00367] Как показано на ФИГ. 34, разницы в баллах сайтов сплайсинга можно порциями определять между целевыми нуклеотидами в референсной последовательности и в вариантной последовательности.
[00368] Как показано на ФИГ. 34, для по меньшей мере одного положения целевого нуклеотида, когда глобальный максимум разницы в оценках для сайтов сплайсинга превышает определенный порог, ACNN классифицирует этот вариант как вызывающий аберрантный сплайсинг и, соответственно, патогенный.
[00369] Как показано на ФИГ. 17, для по меньшей мере одного положения целевого нуклеотида, когда глобальный максимум разницы в оценках для сайтов сплайсинга ниже определенного порога, ACNN классифицирует этот вариант как не вызывающий аберрантный сплайсинг и, соответственно, доброкачественный.
[00370] Порог может быть определен из множества кандидатных порогов. Это включает обработку первого набора пар референсная последовательность-вариантная последовательность, порожденных обычными доброкачественными вариантами, с получением первого набора определений аберрантного сплайсинга, обработку второго набора пар референсная последовательность-вариантная последовательность, порожденных патогенными редкими вариантами, с получением второго набора определений аберрантного сплайсинга, и выбор по меньшей мере порога, для применения классификатором, который максимизирует число определений аберрантного сплайсинга во втором наборе и минимизирует число определений аберрантного сплайсинга в первом наборе.
[00371] В одном варианте реализации ACNN идентифицирует варианты, которые вызывают расстройства аутического спектра (сокращенно ASD, РАС). В другом варианте реализации ACNN идентифицирует варианты, которые вызывают расстройства, связанный с задержкой развития (сокращенно DDD).
[00372] Как показано на ФИГ. 36, и референсная, и вариантная последовательность могут каждая иметь по меньшей мере 101 целевых нуклеотидов, и каждый целевой нуклеотид может быть фланкирован по меньшей мере 5000 нуклеотидов с каждой стороны.
[00373] Как показано на ФИГ. 36, оценки сайтов сплайсинга целевых нуклеотидов в референсной последовательности могут кодироваться в первом выходе ACNN, а оценки сайтов сплайсинга целевых нуклеотидов в вариантной последовательности могут кодироваться во втором выходе ACNN. В одном варианте реализации первый выход кодируется в первой матрице 101 х 3, и второй выход кодируется во второй матрице 101 х 3.
[00374] Как показано на ФИГ. 36, В каждом таком варианте реализации каждый ряд в первой матрице 101 х 3 уникальным образом представляет оценки сайтов сплайсинга по правдоподобию того, что целевой нуклеотид в референсной последовательности является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом.
[00375] Как показано на ФИГ. 36, также в каждом таком варианте реализации каждый ряд во второй матрице 101 х 3 уникальным образом представляет оценки сайтов сплайсинга по правдоподобию того, что целевой нуклеотид в вариантой последовательности является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом.
[00376] Как показано на ФИГ. 36, в некоторых вариантах реализации оценки сайтов сплайсинга в каждом ряду первой матрицы 101 х 3 и второй матрицы 101 х 3 могут быть экспоненциально нормированы чтобы давать в сумме единицу.
[00377] Как показано на ФИГ. 36, классификатор может осуществлять построчное сравнение первой матрицы 101 х 3 и второй матрицы 101 х 3 и построчно определять изменения в распределении оценок сайта сплайсинга. По меньшей мере для одного случая построчного сравнения, когда изменение в распределении превышает заранее определенный порог, ACNN классифицирует этот вариант как вызывающий аберрантный сплайсинг и, соответственно, патогенный.
[00378] Система включает энкодер с одним горячим состоянием (показан ФИГ. 29), который кодирует с разряжением референсную последовательность и вариантную последовательность.
[00379] Каждый из признаков, обсуждаемых в этом разделе этого конкретного варианта реализации, для вариантов реализаций систем и способов, в равной степени применим к этому варианту реализации системы. Как указано выше, здесь не повторяются все признаки системы, подразумевается, что они повторяются посредством ссылки.
[00380] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению системой описанных выше действий. Еще один вариант реализации может включать выполнение описанных выше действий системы.
[00381] Вариант реализации способа согласно раскрытой технологии включает обнаружение геномных вариантов, вызывающих аберрантный сплайсинг.
[00382] Способ включает обработку референсной последовательности разреженной сверточной нейронной сетью (сокращенно ACNN), обученной обнаруживать паттерны дифференциального сплайсинга в целевой подпоследовательности входной последовательности путем классификации каждого нуклеотида в целевой подпоследовательности как донорного сайта сплайсинга, акцепторного сайта сплайсинга или сайта, не связанного со сплайсингом.
[00383] Способ включает, на основе обработки, обнаружение первого дифференциального паттерна сплайсинга в референсной целевой подпоследовательности путем классификации каждого нуклеотида в целевой подпоследовательности как донорного сайта сплайсинга, акцепторного сайта сплайсинга или сайта, не связанного со сплайсингом.
[00384] Способ включает в себя обработку вариантной последовательности посредством ACNN. Вариантная последовательность и референсная последовательность различаются по меньшей мере одним вариантным нуклеотидом, расположенным в вариантной целевой подпоследовательности.
[00385] Способ включает, на основе обработки, обнаружение второго паттерна дифференциального сплайсинга в вариантной целевой подпоследовательности путем классификации каждого нуклеотида в вариантной целевой подпоследовательности как донорного сайта сплайсинга, акцепторного сайта сплайсинга или сайта, не связанного со сплайсингом.
[00386] Способ включает определение разницы между первым паттерном дифференциального сплайсинга и вторым паттерном дифференциального сплайсинга путем понуклеотидного сравнения классификаций сайтов сплайсинга референсной целевой подпоследовательности и вариантной целевой подпоследовательности.
[00387] Когда разница превышает заданный порог, способ включает классификацию варианта как вызывающего аберрантный сплайсинг, и, соответственно, патогенного, и сохранение классификации в памяти.
[00388] Каждый из признаков, обсуждаемых в этом разделе этого конкретного варианта реализации для других вариантов реализации систем и способов, равно относится к этому варианту реализации способа. Как указано выше, здесь не повторяются все признаки системы, подразумевается, что они повторяются посредством ссылки.
[00389] Паттерн дифференциального сплайсинга может идентифицировать позиционное распределение возникновения событий сплайсинга в целевой подпоследовательности. Примеры событий сплайсинга включают по меньшей мере одно из критического (скрытого сплайсинга), пропуска экзона, взаимоисключающих экзонов, альтернативного донорного сайта и сохранения интрона.
[00390] Референсная целевая подпоследовательность и вариантная целевая подпоследовательность могут быть выровнены относительно положений нуклеотидов и могут различаться по меньшей мере одним вариантным нуклеотидом.
[00391] Референсная целевая подпоследовательность и вариантная целевая подпоследовательность могут каждая иметь по меньшей мере 40 нуклеотидов и каждая может фланкироваться по меньшей мере 40 нуклеотидами с каждой стороны.
[00392] Референсная целевая подпоследовательность и вариантная целевая подпоследовательность могут каждая иметь по меньшей мере 101 нуклеотид и каждая может быть фланкирована по меньшей мере 5000 нуклеотидов с каждой стороны.
[00393] Вариантная целевая подпоследовательность может включать два варианта.
[00394] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению описанного выше способа. Еще один вариант реализации может включать систему, включающую память и один или более процессоров, выполненных с возможностью исполнять инструкции, хранящиеся в памяти, с реализацией описанного выше способа.
[00395] Мы описываем системы, способы и изделия для применения обученной (тренированной) сверточной нейронной сети для определения сайтов сплайсинга и аберрантного сплайсинга в геномных последовательностях (например, нуклеотидных последовательностях). Один или более признаков одного из вариантов реализации могут быть объединены с базовым вариантом реализации. Подразумевается, что варианты, которые не являются взаимоисключающими, совместимы. Один или более признаков варианта реализации можно комбинировать с другими вариантами реализации. Настоящее описание периодически напоминает пользователю об этих опциях. То, что в некоторых вариантах реализации пропущено упоминание об этих опциях, не следует воспринимать как ограничение комбинаций, описанных в предыдущих разделах - в настоящем тексте такое описание включается посредством ссылки в каждый их последующих вариантов реализации.
[00396] Один вариант реализации системы в соответствии с раскрытой технологией включает один или более процессоров, соединенных с памятью. В память загружены компьютерные команды для обучения детектора сайтов сплайсинга, который идентифицирует сайты сплайсинга в геномных последовательностях (например, нуклеотидных последовательностях).
[00397] Система обучает (тренирует) сверточную нейронную сеть (сокращенно CNN) на по меньшей мере 50000 тренировочных примеров донорных сайтов сплайсинга, по меньшей мере 50000 тренировочных примеров акцепторных сайтов сплайсинга, и по меньшей мере 100000 тренировочных примеров сайтов, не связанных со сплайсингом. Каждый обучающий пример представляет собой целевую нуклеотидную последовательность, имеющую по меньшей мере один целевой нуклеотид, фланкированный по меньшей мере 20 нуклеотидами с каждой стороны.
[00398] Для оценки тренировочного примера с использованием CNN система подает, в качестве входных данных для CNN, целевую нуклеотидную последовательность, дополнительно фланкированную по меньшей мере 40 расположенными в направлении 5'-конца нуклеотидами и по меньшей мере 40 расположенными в направлении 3'-конца нуклеотидами.
[00399] На основании этой оценки, CNN затем генерирует, в качестве выхода, тройных оценок для правдоподобия того, что каждый нуклеотид в целевой нуклеотидной последовательности является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом.
[00400] Этот вариант реализации системы и другие раскрытые системы необязательно включают один или более из следующих признаков. Система может также включать признаки, описанные в связи с раскрытыми способами. Для краткости альтернативные комбинации признаков системы не приводятся по отдельности. Признаки, связанные с системами, способами и изделиями, не повторяются для каждого заявленного набора классов основных признаков. Читатель поймет, как признаки, идентифицированные в этом разделе, можно легко объединять с основными признаками в других заявленных классах.
[00401] Входные данные могут включать целевую нуклеотидную последовательность, которая содержит целевой нуклеотид, фланкированный 100 нуклеотидами с каждой стороны. В каждом таком варианте реализации целевой нуклеотид дополнительно фланкирован 200 расположенными в направлении 5' контекстными нуклеотидами и 200 расположенными в направлении 3' контекстными нуклеотидами.
[00402] Как показано на ФИГ. 28, система может обучать (тренировать) CNN на 150000 тренировочных примеров донорных сайтов сплайсинга, 150000 тренировочных примеров акцепторных сайтов сплайсингаи 1000000 тренировочных примеров сайтов, не связанных со сплайсингом.
[00403] Как показано на ФИГ. 31, CNN может быть параметризована числом сверточных слоев, числом сверточных фильтров, и числом субдискретизирующих словев (например, максимальное объединение и среднее объединение).
[00404] Как показано на ФИГ. 31, CNN может включать один или более полностью связанных слоев и конечный слой классификации.
[00405] CNN может включать меняющие размерность сверточные слои которые, меняют пространственные размерности и размерности признаков предшествующего входа.
[00406] Тройные оценки для каждого нуклеотида в целевой нуклеотидной последовательности могут быть экспоненциально нормированы чтобы давать в сумме единицу. В каждом таком варианте реализации система классифицирует каждый нуклеотид в целевом нуклеотиде как донорный сайт сплайсинга, акцепторный сайт сплайсинга или сайт, не связанный со сплайсингом, на основании самой высокой оценки в соответствующих тройных оценках.
[00407] Как показано на ФИГ. 32, CNN пакетно оценивает обучающие примеры на протяжении эпохи. Тренировочные образцы случайным образом группируются в пакеты. Каждый пакет имеет заранее определенный размер пакета. CNN итерирует оценку обучающих примеров на протяжении нескольких эпох (например, 1-10).
[00408] Входные данные могут включать целевую последовательность, которая содержит два соседних целевых нуклеотида. Два соседних целевых нуклеотида могут представлять собой аденин (сокращенно А) и гуанин (сокращенно G). Два соседних целевых нуклеотида могут представлять собой гуанин (сокращенно G) и урацил (сокращенно U).
[00409] Система включает энкодер с одним горячим состоянием (показан на ФИГ. 32) который кодирует с разрежением тренировочные примеры и выдает в качестве выходных данных результаты кодирования с одним горячим состоянием.
[00410] CNN может быть параметризована числом остаточных блоков, числом соединений с пропуском и числом остаточных связей.
[00411] Каждый остаточный блок может включать по меньшей мере один слой пакетной нормализации, по меньшей мере один слой блоков линейной ректификации (сокращенно ReLU), по меньшей мере один меняющий размерность слой и по меньшей мере одну остаточную связь. Каждый остаточный блок может включать два слоя пакетной нормализации, два нелинейных слоя ReLU, два меняющих размерность слоя и одно остаточное соединение.
[00412] Каждый из признаков, обсуждаемых в этом разделе этого конкретного варианта реализации для вариантов реализаций систем и способов, в равной степени применим к этому варианту реализации системы. Как указано выше, здесь не повторяются все признаки системы, подразумевается, что они повторяются посредством ссылки.
[00413] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению системой описанных выше действий. Еще один вариант реализации может включать выполнение описанных выше действий системы.
[00414] Другой системный вариант реализации раскрытой технологии включает обученный предсказатель сайтов сплайсинга, который реализуется на множестве процессоров, работающих параллельно и связанных с памятью. Система обучает (тренирует) сверточную нейронную сеть (сокращенно CNN), реализованную на множестве процессоров, на по меньшей мере 50000 тренировочных примеров донорных сайтов сплайсинга, по меньшей мере 50000 тренировочных примеров акцепторных сайтов сплайсинга и по меньшей мере 100000 тренировочных примеров сайтов, не связанных со сплайсингом. Каждый из тренировочных (обучающих) примеров, используемых для обучения, представляет собой нуклеотидную последовательность, которая включает целевой нуклеотид, фланкированный по меньшей мере 400 нуклеотидами с каждой стороны.
[00415] Система включает стадию ввода CNN, которая реализована на по меньшей мере одном из указанного множества процессоров и обеспечивает подачу входной последовательности из по меньшей мере 801 нуклеотида для оценки целевых нуклеотидов. Каждый целевой нуклеотид фланкирован по меньшей мере 400 нуклеотидами с каждой стороны. В других вариантах реализации система включает модуль ввода CNN, который реализован на по меньшей мере одном из указанного множества процессоров и обеспечивает подачу входной последовательности из по меньшей мере 801 нуклеотида для оценки целевых нуклеотидов.
[00416] Система включает стадию вывода CNN, которая реализована на по меньшей мере одном из указанного множества процессоров и переводит анализ, выполняемый CNN, в баллы классификации для правдоподобия того, что каждый из указанных целевых нуклеотидов является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом. В других вариантах реализации система включает модуль вывода CNN, который реализован на по меньшей мере одном из указанного множества процессоров и переводит анализ, выполняемый CNN, в баллы классификации для правдоподобия того, что каждый из указанных целевых нуклеотидов является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом.
[00417] Каждый из признаков, обсуждаемых в этом разделе этого конкретного варианта реализации для вариантов реализаций систем и способов, в равной степени применим к этому варианту реализации системы. Как указано выше, здесь не повторяются все признаки системы, подразумевается, что они повторяются посредством ссылки.
[00418] CNN может быть обучена на 150000 тренировочных примеров донорных сайтов сплайсинга, 150000 тренировочных примеров акцепторных сайтов сплайсинга, и 800000000 тренировочных примеров сайтов, не связанных со сплайсингом.
[00419] CNN может быть обучена на одном или большем числе тренировочных серверов.
[00420] Обученная CNN может быть размещена на одном или большем числе рабочих серверов, которые получают входные последовательности от запрашивающих клиентов. В каждом таком варианте реализации указанные рабочие серверы обрабатывают входные последовательности посредством стадий ввода и вывода сети CNN с получением выходных данных, которые передаются указанным клиентам. В других вариантах реализации указанные рабочие серверы обрабатывают входные последовательности посредством ввода и вывода сети CNN с получением выходных данных, которые передаются указанным клиентам.
[00421] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению системой описанных выше действий. Еще один вариант реализации может включать выполнение описанных выше действий системы.
[00422] Вариант реализации способа согласно раскрытой технологии включает обучение детектора сайтов сплайсинга, который идентифицирует сайты сплайсинга в геномных последовательностях (например, нуклеотидных последовательностях). Способ включает ввод, в сверточную нейронную сеть (сокращенно CNN), входящей последовательности из по меньшей мере 801 нуклеотидов для оценки целевых нуклеотидов, каждый из которых фланкирован по меньшей мере 400 нуклеотидами с каждой стороны.
[00423] CNN обучается на по меньшей мере 50000 тренировочных примеров донорных сайтов сплайсинга, по меньшей мере 50000 тренировочных примеров акцепторных сайтов сплайсинга, и по меньшей мере 100000 тренировочных примеров сайтов, не связанных со сплайсингом. Каждый из тренировочных (обучающих) примеров, используемых для обучения, представляет собой нуклеотидную последовательность, которая включает целевой нуклеотид, фланкированный по меньшей мере 400 нуклеотидами с каждой стороны.
[00424] Способ дополнительно включает перевод анализа, выполняемого CNN в баллы классификации для правдоподобия того, что каждый из указанных целевых нуклеотидов является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом.
[00425] Каждый из признаков, обсуждаемых в этом разделе этого конкретного варианта реализации для других вариантов реализации систем и способов, равно относится к этому варианту реализации способа. Как указано выше, здесь не повторяются все признаки системы, подразумевается, что они повторяются посредством ссылки.
[00426] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению описанного выше способа. Еще один вариант реализации может включать систему, включающую память и один или более процессоров, выполненных с возможностью исполнять инструкции, хранящиеся в памяти, с реализацией описанного выше способа.
[00427] Еще один вариант реализации системы согласно раскрытой технологии включает один или более процессоров, соединенных с памятью. В память загружены компьютерные команды по реализации детектора аберрантного сплайсинга, реализованного на множестве процессоров, работающих параллельно и связанных с памятью.
[00428] Система включает обученную сверточную нейронную сеть (сокращенно CNN), реализованную на множестве процессоров.
[00429] Как показано на ФИГ. 34, CNN классифицирует целевые нуклеотиды во входной последовательности и присваивает оценки сайтов сплайсинга для правдоподобия того, что каждый из указанных целевых нуклеотидов является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом. Входная последовательность содержит по меньшей мере 801 нуклеотид, и каждый целевой нуклеотид фланкирован по меньшей мере 400 нуклеотидами с каждой стороны.
[00430] Как показано на ФИГ. 34, система также включает классификатор, реализованный на по меньшей мере одном из указанного множества процессоров, который обрабатывает референсную последовательность и вариантную последовательность посредством CNN и выдает оценки сайтов сплайсинга, отражающие правдоподобие того, что каждый целевой нуклеотид в референсной последовательности и в вариантной последовательности является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом. И референсная последовательность, и вариантная последовательность, каждая содержит по меньшей мере 101 целевой нуклеотид, и каждый целевой нуклеотид фланкирован по меньшей мере 400 нуклеотидами с каждой стороны.
[00431] Как показано на ФИГ. 34, затем система определяет, по разницам в оценках сайтов сплайсинга целевых нуклеотидов в референсной последовательности и в вариантной последовательности, вызывает ли вариант, порождающий вариантную последовательность, аберрантный сплайсинг, и является ли он, соответственно, патогенным.
[00432] Каждый из признаков, обсуждаемых в этом разделе этого конкретного варианта реализации для вариантов реализаций систем и способов, в равной степени применим к этому варианту реализации системы. Как указано выше, здесь не повторяются все признаки системы, подразумевается, что они повторяются посредством ссылки.
[00433] Разницы в оценках сайтов сплайсинга можно определить по положениям между целевыми нуклеотидами в референсной последовательности и в вариантной последовательности.
[00434] Для по меньшей мере одного положения целевого нуклеотида, когда глобальный максимум разницы в оценках для сайтов сплайсинга превышает определенный порог, CNN классифицирует этот вариант как вызывающий аберрантный сплайсинг и, соответственно, патогенный.
[00435] Для по меньшей мере одного положения целевого нуклеотида, когда глобальный максимум разницы в оценках для сайтов сплайсинга ниже определенного порога, CNN классифицирует этот варианта как не вызывающий аберрантный сплайсинг и, соответственно, доброкачественный.
[00436] Порог может быть определен из множества кандидатных порогов. Это включает обработку первого набора пар референсная последовательность-вариантная последовательность, порожденных обычными доброкачественными вариантами, с получением первого набора определений аберрантного сплайсинга, обработку второго набора пар референсная последовательность-вариантная последовательность, порожденных патогенными редкими вариантами, с получением второго набора определений аберрантного сплайсинга, и выбор по меньшей мере порога, для применения классификатором, который максимизирует число определений аберрантного сплайсинга во втором наборе и минимизирует число определений аберрантного сплайсинга в первом наборе.
[00437] В одном варианте реализации CNN идентифицирует варианты, которые вызывают расстройств аутического спектра (сокращенно ASD). В другом варианте реализации CNN идентифицирует варианты, которые вызывают расстройства, связанные с нарушением развития (сокращенно DDD).
[00438] Референсная последовательность и вариантная последовательность могут каждая иметь по меньшей мере 101 целевой нуклеотид, и каждый целевой нуклеотид может быть фланкирован по меньшей мере 1000 нуклеотидов с каждой стороны.
[00439] Оценки сайтов сплайсинга для целевых нуклеотидов в референсной последовательности могут кодироваться в первом выводе CNN, а оценки сайтов сплайсинга целевых нуклеотидов в вариантной последовательности могут кодироваться во втором выводе CNN. В одном варианте реализации первый выход кодируется в первой матрице 101 x 3, и второй выход кодируется во второй матрице 101 x 3.
[00440] В каждом таком варианте реализации каждый ряд в первой матрице 101 x 3 уникальным образом представляет оценки сайтов сплайсинга по правдоподобию того, что целевой нуклеотид в референсной последовательности является до норным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом.
[00441] Также в каждом таком варианте реализации каждый ряд во второй матрице 101 x 3 уникальным образом представляет оценки сайтов сплайсинга по правдоподобию того, что целевой нуклеотид в вариантой последовательности является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом.
[00442] В некоторых вариантах реализации оценки сайтов сплайсинга в каждом ряду первой матрицы 101 x 3 и второй матрицы 101 x 3 могут быть экспоненциально нормированы чтобы давать в сумме единицу.
[00443] Классификатор может осуществлять построчное сравнение первой матрицы 101 x 3 и второй матрицы 101 x 3 и построчно определять изменения в распределении оценок сайта сплайсинга. По меньшей мере для одного случая построчного сравнения, когда изменение в распределении превышает заранее определенный порог, CNN классифицирует этот вариант как вызывающий аберрантный сплайсинг и, соответственно, патогенный.
[00444] Система включает энкодер с одним горячим состоянием (показан на ФИГ. 29), который кодирует с разряжением референсную последовательность и вариантную последовательность.
[00445] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению системой описанных выше действий. Еще один вариант реализации может включать выполнение описанных выше действий системы.
[00446] Вариант реализации способа согласно раскрытой технологии включает обнаружение геномных вариантов, вызывающих аберрантный сплайсинг.
[00447] Способ включает обработку референсной последовательности разреженной сверточной нейронной сетью (сокращенно CNN), обученной определять дифференциальные паттерны сплайсинга в целевой подпоследовательности входной последовательности путем классификации каждого нуклеотида в целевой подпоследовательности как донорного сайта сплайсинга, акцепторного сайта сплайсинга или сайта, не связанного со сплайсингом.
[00448] Способ включает, на основе обработки, обнаружение первого дифференциального паттерна сплайсинга в референсной целевой подпоследовательности путем классификации каждого нуклеотида в целевой подпоследовательности как донорного сайта сплайсинга, акцепторного сайта сплайсинга или сайта, не связанного со сплайсингом.
[00449] Способ включает в себя обработку вариантной последовательности посредством CNN. Вариантная последовательность и референсная последовательность различаются по меньшей мере одним вариантным нуклеотидом, расположенным в вариантной целевой подпоследовательности.
[00450] Способ включает, на основе обработки, обнаружение второго паттерна дифференциального сплайсинга в вариантной целевой подпоследовательности путем классификации каждого нуклеотида в вариантной целевой подпоследовательности как донорного сайта сплайсинга, акцепторного сайта сплайсинга или сайта, не связанного со сплайсингом.
[00451] Способ включает определение разницы между первым паттерном дифференциального сплайсинга и вторым паттерном дифференциального сплайсинга путем понуклеотидного сравнения классификаций сайтов сплайсинга референсной целевой подпоследовательности и вариантной целевой подпоследовательности.
[00452] Когда разница превышает заданный порог, способ включает классификацию варианта как вызывающего аберрантный сплайсинг, и, соответственно, патогенного, и сохранение классификации в памяти.
[00453] Каждый из признаков, обсуждаемых в этом разделе этого конкретного варианта реализации для других вариантов реализации систем и способов, равно относится к этому варианту реализации способа. Как указано выше, здесь не повторяются все признаки системы, подразумевается, что они повторяются посредством ссылки.
[00454] Паттерн дифференциального сплайсинга может идентифицировать позиционное распределение возникновения событий сплайсинга в целевой подпоследовательности. Примеры событий сплайсинга включают по меньшей мере одно из критического (скрытого сплайсинга), пропуска экзона, взаимоисключающих экзонов, альтернативного донорного сайта и сохранения интрона.
[00455] Референсная целевая подпоследовательность и вариантная целевая подпоследовательность могут быть выровнены относительно положений нуклеотидов и могут различаться по меньшей мере одним вариантным нуклеотидом.
[00456] Референсная целевая подпоследовательность и вариантная целевая подпоследовательность могут каждая иметь по меньшей мере 40 нуклеотидов и каждая может фланкироваться по меньшей мере 40 нуклеотидами с каждой стороны.
[00457] Референсная целевая подпоследовательность и вариантная целевая подпоследовательность могут каждая иметь по меньшей мере 101 нуклеотидов и могут быть фланкированы по меньшей мере 1000 нуклеотидов с каждой стороны.
[00458] Вариантная целевая подпоследовательность может включать два варианта.
[00459] Другие варианты осуществления могут включать компьютерочитаемый носитель долговременного хранения информации, на котором хранятся исполняемые процессором команды по выполнению описанного выше способа. Еще один вариант реализации может включать систему, включающую память и один или более процессоров, выполненных с возможностью исполнять инструкции, хранящиеся в памяти, с реализацией описанного выше способа.
[00460] раскрытой технологии. Различные модификации раскрытых вариантов реализации будут очевидны, и общие принципы, определенные в данном документе, могут быть применены к другим вариантом реализации и приложениям без отступления от сущности и объема раскрытой технологии. Таким образом, раскрытая технология ограничивается показанными вариантами реализации, но должна соответствовать самому широкому объему, согласующемуся с принципами и признаками, раскрытыми в данном документе. Объем раскрытой технологии определяется прилагаемой формулой изобретения.
Анализ обогащения по генам
[00461] ФИГ. 57 демонстрирует один вариант реализации анализа обогащения по генам. В одном варианте реализации детектор аберрантного сплайсинга дополнительно сконфигурирован для реализации анализа обогащения по генам, который определяет патогенность вариантов, которые, как было установлено, вызывают аберрантный сплайсинг. Для конкретного гена, отобранного из группы людей с генетическим заболеванием, анализ обогащения каждого гена включает применение обученной ACNN для идентификации вариантов-кандидатов в конкретном гене, которые вызывают аберрантное сплайсинг, определение фонового количества мутаций для конкретного гена на основе суммирования наблюдаемых показателей тринуклеотидных мутаций вариантов-кандидатов и умножении суммы на количество передач и размер когорты, применение обученной ACNN для идентификации de novo вариантов в конкретном гене, вызывающих аберрантный сплайсинг, и сравнение фонового количества мутаций с количеством вариантов de novo. На основе результатов сравнения анализ обогащения каждого гена определяет, что конкретный ген связан с генетическим заболеванием и что варианты de novo являются патогенными. В некоторых вариантах реализации генетическим нарушением является расстройство аутического спектра (сокращенно ASD). В других вариантах реализации генетическим расстройством является нарушение, связанное с задержкой развития (сокращенно DDD).
[00462] В примере, показанном на ФИГ. 57, пять вариантов-кандидатов в конкретном гене были классифицированы детектором аберрантного сплайсинга как вызывающие аберрантный сплайсинг. Эти пять вариантов-кандидатов имеют соответствующие наблюдаемые показатели тринуклеотидных мутаций: 10-8, 10-2, 10-1, 105 и 101. Фоновое количество мутаций для конкретного гена определено как 10-5 на основе суммирования соответствующих наблюдаемых частот тринуклеотидных мутаций пяти вариантов-кандидатов и умножения суммы на количество передач / хромосом (2) и размер когорты (1000). Затем его сравнивают с числом вариантов de novo (3).
[00463] В некоторых вариантах реализации детектор аберрантного сплайсинга дополнительно сконфигурирован для выполнения сравнения с использованием статистического критерия, который выдает р-значение на выходе.
[00464] В других вариантах реализации детектор аберрантного сплайсинга дополнительно сконфигурирован для сравнения фонового количества мутаций с количеством вариантов de novo и определения, на основе результатов сравнения, что конкретный ген не связан с генетическим заболеванием, и что варианты de novo безвредны (являются доброкачественными).
[00465] В одном варианте реализации по меньшей мере некоторые из вариантов-кандидатов являются вариантами, укорачивающими белок.
[00466] Другим вариантом реализации по меньшей мере некоторые из вариантов-кандидатов являются миссенс-вариантами.
Полногеномный анализ обогащения
[00467] ФИГ. 58 демонстрирует один вариант реализации полногеномного анализа обогащения. В другом варианте реализации детектор аберрантного сплайсинга дополнительно сконфигурирован для реализации полногеномного анализа обогащения, который определяет патогенность вариантов, которые, как было установлено, вызывают аберрантный сплайсинг. Полногеномный анализ обогащения включает применение обученной ACNN для идентификации первого набора вариантов de novo, которые вызывают аберрантный сплайсинг во множестве генов, взятых из когорты здоровых людей, применение обученной ACNN для идентификации второго набора вариантов de novo, которые вызывают аберрантный сплайсинг во множестве генов, отобранных из группы людей с генетическим заболеванием, и сравнение соответствующих значений первого и второго наборов, и, на основе результатов сравнения, определение, что второй набор вариантов de novo обогащен в когорте людей с генетическим заболеванием и, следовательно, является патогенным. В некоторых вариантах реализации генетическое нарушение является расстройство аутического спектра (сокращенно ASD). В других вариантах реализации генетическим нарушением является нарушение, связанное с задержкой развития (сокращенно DDD).
[00468] В некоторых вариантах реализации детектор аберрантного сплайсинга дополнительно сконфигурирован для выполнения сравнения с использованием статистического критерия, который выдает р-значение на выходе. В одном варианте реализации сравнение может быть дополнительно параметризовано соответствующими размерами когорт.
[00469] В некоторых вариантах реализации детектор аберрантного сплайсинга дополнительно сконфигурирован для сравнения соответствующих счетов первого и второго наборов, и, на основе выходных данных сравнения, определения, что второй набор вариантов de novo не обогащен в когорте люди с генетическим заболеванием и, следовательно, является безвредным (доброкачественным).
[00470] В примере, показанном на ФИГ. 58 показана частота мутаций в когорте здоровых (0,001) и частота мутаций в когорте пораженных (0,004), а также соотношение мутаций на индивидуума (4).
Обсуждение
[00471] Несмотря на ограниченную диагностическую ценность секвенирования экзома у пациентов с тяжелыми генетическими нарушениями, клиническое секвенирование сосредоточено на редких кодирующих мутациях, в значительной степени игнорируя вариации в некодирующем геноме из-за сложности интерпретации. Здесь мы представляем сеть глубокого обучения, которая точно предсказывает сплайсинг на основе первичной нуклеотидной последовательности, тем самым выявляя некодирующие мутации, которые нарушают нормальное формирование паттерна экзонов и интронов с серьезными последствиями для образующегося белка. Мы показываем, что предсказанные критические мутации сплайсинга имеют высокие показатели валидации по РНК последовательности, в большой степени вредоносны для человеческой популяции и являются основной причиной редких генетических заболеваний.
[00472] Используя сеть глубокого обучения в качестве модели сплайсосомы in silico, мы смогли реконструировать детерминанты специфичности, которые позволяют сплайсосомам достичь своей замечательной точности in vivo. Мы подтверждаем многие открытия, которые были сделаны за последние четыре десятилетия исследований механизмов сплайсинга, и показываем, что сплайсосома объединяет в своих решениях большое количество детерминант специфичности короткого и дальнего действия. В частности, мы обнаружили, что предполагаемая вырожденность большинства мотивов сплайсинга объясняется присутствием дальнодействующих детерминант, таких как длина экзона / интрона и расположение нуклеосом, которые с избытком компенсируют и делают ненужной дополнительную специфичность на уровне мотива. Наши результаты демонстрируют перспективность моделей глубокого обучения для получения биологической информации, а не просто служат классификаторами типа «черный ящик».
[00473] Глубокое обучение - это относительно новый метод в биологии, и он не лишен возможных недостатков. Обучаясь автоматическому извлечению признаков из последовательности, модели глубокого обучения могут использовать новые детерминанты последовательности, недостаточно хорошо описанные экспертами-людьми, но также существует риск того, что модель может включать в себя функции, которые не отражают истинное поведение сплайсосомы. Эти нерелевантные особенности могут повысить очевидную точность предсказания аннотированных границ экзон-интрон, но снизят точность предсказания эффектов сплайсинга произвольных изменений последовательности, вызванных генетической изменчивостью. Поскольку точное предсказание вариантов предоставляет наиболее убедительные доказательства того, что модель может быть обобщена для истинной биологии, мы обеспечиваем валидацию предсказанных вариантов, изменяющих сплайсинг, используя три полностью ортогональных метода: последовательность РНК, естественный отбор в человеческих популяциях и обогащение вариантов de novo в отдельных случаях в сравнении с контрольными когортами. Хотя это не полностью исключает включение в модель нерелевантных функций, полученная модель оказалась достаточно точной для истинной биологии сплайсинга, чтобы иметь значительную ценность для практических приложений, таких как идентификация криптических мутаций сплайсинга у пациентов с генетическими заболеваниями.
[00474] По сравнению с другими классами мутаций, укорачивающих белок, особенно интересным аспектом криптических мутаций сплайсинга является широко распространенный феномен альтернативного сплайсинга из-за не полностью пенетрантных изменяющих сплайсинг вариантов, которые имеют тенденцию ослаблять канонические сайты сплайсинга по сравнению с альтернативными сайтами сплайсинга, приводя к образованию смеси как аберрантных, так и нормальных транскриптов в данных РНК-секвенирования. Наблюдение, что эти варианты часто приводят к тканеспецифическому альтернативному сплайсингу, подчеркивает неожиданную роль, которую играют критические мутации сплайсинга в формировании нового разнообразия альтернативного сплайсинга. Потенциальным будущим направлением могло бы стать обучение моделей глубокого обучения на аннотациях границ сплайсинга по последовательности РНК соответствующей ткани, что позволило бы получать тканеспецифичные модели альтернативного сплайсинга. Обучение сети аннотациям, полученным непосредственно из данных РНК-секвенирования, также помогает заполнить пробелы в аннотациях GENCODE, что улучшает производительность модели в предсказании вариантов (ФИГ. 52А и 52В).
[00475] Наше понимание того, как мутации в некодирующем геноме приводят к заболеваниям человека, остается далеко не полным. Открытие пенетрантных критических сплайс-мутаций de novo при нарушениях неврологического развития у детей демонстрирует, что улучшенная интерпретация некодирующего генома может принести прямую пользу пациентам с тяжелыми генетическими нарушениями. Скрытые мутации сплайсинга также играют важную роль в развитии рака (Jung et al., 2015; Sanz et al., 2010; Supek et al., 2014), а повторяющиеся соматические мутации в факторах сплайсинга, как было показано, вызывают широко распространенные изменения в специфичности сплайсинга (Graubert et al., 2012; Shirai et al., 2015; Yoshida et al., 2011). Еще предстоит проделать большую работу, чтобы понять регуляцию сплайсинга в различных тканях и клетках, особенно в случае мутаций, которые непосредственно влияют на белки в сплайсосоме. В свете недавних достижений в терапии олигонуклеотидами, которая потенциально может воздействовать на дефекты сплайсинга специфическим для последовательности образом (Finkel et al., 2017), более глубокое понимание регуляторных механизмов, управляющих этим замечательным процессом, может проложить путь для новых кандидатов для терапевтического вмешательства.
[00476] ФИГ. 37А, 37В, 37С, 37D, 37Е, 37F, 37G и 37Н иллюстрируют один вариант реализации предсказания сплайсинга по первичной последовательности при помощи глубокого обучения.
[00477] В отношении ФИГ. 37А, для каждого положения в транскрипте пре-мРНК, SpliceNet-10k использует 10000 нуклеотидов фланкирующей последовательности в качестве входных данных и предсказывает, является ли это положение акцептором сплайсинга, донором сплайсинга, или ни тем, ни другим.
[00478] В отношении ФИГ. 37В показан полный пре-мРНК-транскрипт для гена CFTR, оцененный с помощью MaxEntScan (вверху) и SpliceNet-10k (внизу), вместе с предсказанными акцепторными (красные стрелки) и донорными (зеленые стрелки) сайтами и фактическими положениями экзонов. (черные прямоугольники). Для каждого метода мы применили порог, при котором количество предсказанных сайтов равнялось общему количеству реальных сайтов.
[00479] В отношении ФИГ. 37С, для каждого экзона мы измерили степень включения экзона в РНК-последовательность и показали распределение оценок SpliceNet-10k для экзонов при различных уровнях включения. Показаны максимальные оценки акцептора и донора экзона.
[00480] В отношении ФИГ. 37D, влияние in silico мутирования каждого нуклеотида вокруг экзона 9 в гене U2SURP. Вертикальный размер каждого нуклеотида показывает уменьшение предсказанной силы акцепторного сайта (черная стрелка), когда этот нуклеотид мутирован (Δ Score (Оценка)).
[00481] В отношении ФИГ. 37Е, влияние размера контекста входной последовательности на точность сети. Точность Тор-k - это доля правильно предсказанных сайтов сплайсинга при пороге, при котором количество предсказанных сайтов равно фактическому количеству присутствующих сайтов. PR-AUC - это площадь под кривой точность-отзыв. Мы также показываем точность top-k и PR-AUC для трех других алгоритмов обнаружения сайта сплайсинга.
[00482] В отношении ФИГ. 37F, взаимосвязь между длиной экзона / интрона и силой соседних сайтов сплайсинга, как было предсказано с помощью SpliceNet-80nt (оценка локальных мотивов) и SpliceNet-10k. Распределение длины экзона (желтый) и длины интрона (розовый) по всему геному показано на заднем плане. Ось x представлена в логарифмическом масштабе.
[00483] В отношении ФИГ. 37G, пару акцепторных и донорных мотивов сплайсинга, разнесенных на 150 нуклеотидов, прогоняют по гену HMGCR. Показаны в каждом положении сигнал нуклеосомы K562 и вероятность того, что пара образует экзон в этом положении, в соответствии с предсказанием SpliceNet-10k.
[00484] В отношении ФИГ. 37Н, средний нуклеосомный сигнал K562 и GM12878 рядом с частными мутациями, которые, по прогнозам модели SpliceNet-10k, создают новые экзоны в когорте GTEx. Показано р-значение по критерию перестановки.
[00485] ФИГ. 38А, 38В, 38С, 38D, 38Е, 38F, и 38G показывают один вариант реализации валидации редких критических сплайс-мутаций в данных секвенирования РНК.
[00486] В отношении ФИГ. 38А, чтобы оценить влияние мутации на изменение сплайсинга, SpliceNet-10k предсказывает оценки акцепторов и доноров в каждом положении в последовательности пре-мРНК гена с мутацией и без мутации, как показано здесь для rs397515893, патогенного критического варианта сплайсинга, в интроне MYBPC3, связанного с кардиомиопатией. Значение Δ Score (Оценка) для мутации - это наибольшее изменение в оценках предсказания сплайсинга в пределах 50 нуклеотидов от варианта.
[00487] В отношении ФИГ. 38В, мы оценили частные генетические варианты (наблюдаемые только у одного из 149 человек в когорте GTEx) с помощью модели SpliceNet-10k. Показано обогащение частных вариантов, которые, согласно предсказанию, изменяют сплайсинг (Δ Score >0,2, синий) или не влияют на сплайсинг (Δ Score <0,01, красный) в непосредственной близости от мест пропуска частных экзонов (вверху) или частных акцепторов и доноров, сайты (внизу). Ось Y показывает, сколько раз частное событие сплайсинга и ближайший частный генетический вариант совпадают у одного и того же человека, по сравнению с ожидаемыми числами, полученными путем перестановок.
[00488] В отношении ФИГ. 38С, пример гетерозиготного синонимичного варианта в PYGB, который создает новый донорный сайт с неполной пенетрантностью. Покрытие РНК-последовательности, количество ридов границ и положения границ (синие и серые стрелки) показаны для индивидуума с вариантом и контрольного индивидуума. Величина эффекта рассчитывается как разница в использовании новой границы (АС) между людьми с вариантом и людьми без варианта. На гистограмме с накоплением ниже мы показываем количество ридов с референсным или альтернативным аллелем, в котором использовалось аннотированная или новая граница («без сплайсинга» и «новая граница» соответственно). Общее количество референсных ридов значительно отличалось от общего количества альтернативных ридов (Р=0,018, биномиальный тест), что свидетельствует о том, что 60% транскриптов, сплайсированных по новой границе, отсутствуют в данных РНК-секвенирования, предположительно из-за обусловленного миссенс-мутацией распада. (NMD).
[00489] В отношении ФИГ. 38D, фракция критических мутаций сплайсинга, предсказанная моделью SpliceNet-10k, подтвержденная данными РНК-последовательностей GTEx. Уровень валидации нарушений основных акцепторных или донорных динуклеотидов (пунктирная линия) составляет менее 100% из-за покрытия и нонсенс-опосредованного распада.
[00490] В отношении ФИГ. 38Е, распределение размеров эффекта для подтвержденных предсказаний криптического сплайсинга. Пунктирная линия (50%) соответствует ожидаемой величине эффекта полностью пенетрантных гетерозиготных вариантов. Измеренная величина эффекта разрушений основных акцепторных или донорных динуклеотидов составляет менее 50% из-за нонсенс-опосредованного распада или неучтенных изменений изоформ.
[00491] В отношении ФИГ. 38F, чувствительность SpliceNet-10k при обнаружении изменяющих сплайсинг частных вариантов в когорте GTEx при различных пороговых значениях Δ Score. Варианты делятся на варианты с глубоким интроном (>50 нуклеотидов от экзонов) и варианты около экзонов (перекрывающиеся экзоны или >50 нуклеотидов от границ экзон-интрон).
[00492] В отношении ФИГ. 38G, уровень валидации и чувствительность SpliceNet-10k и трех других методов для предсказания сайта сплайсинга при различных порогах достоверности. Три точки на кривой SpliceNet-10k показывают производительность SpliceNet-10k при пороговых значениях Δ Score 0,2, 0,5 и 0,8. Для трех других алгоритмов три точки на кривой указывают их производительность при порогах, где они предсказывают такое же количество вариантов криптического сплайсинга, что и SpliceNet-10k при пороговых значениях Δ Score 0,2, 0,5 и 0,8.
[00493] ФИГ. 39А, 39В и 39С показывают один вариант реализации криптических сплайс-вариантов, которые обуславливают ткань-специфический альтернативный сплайсинг.
[00494] В отношении ФИГ. 39А, пример гетерозиготного экзонного варианта в CDC25B, который создает новый донорный сайт. Вариант является частным для одного человека в когорте GTEx и демонстрирует тканеспецифический альтернативный сплайсинг, который способствует большей доле новой изоформы сплайсинга в мышцах по сравнению с фибробластами (Р=0,006 по точному критерию Фишера). Покрытие РНК-последовательности, количество ридов границы и положения границ (синие и серые стрелки) показаны для индивидуума с вариантом и контрольного индивидуума как в мышцах, так и в фибробластах.
[00495] В отношении ФИГ. 39В, пример гетерозиготного варианта, создающего экзонный акцептор в FAM229B, который проявляет согласованные тканеспецифические эффекты у всех трех индивидуумов в когорте GTEx, которые несут этот вариант. РНК-последовательности для артерии и легкого показаны для трех индивидуумов с вариантом и контрольного индивидуума.
[00496] В отношении ФИГ. 39С, доля вариантов, создающих сайт сплайсинга, в когорте GTEx, которые связаны со значительно неравномерным использованием новой границы в экспрессирующих тканях, оцениваемая на гомогенность с помощью критерия хи-квадрат. Подтвержденные варианты критического сплайсинга с низкими и средними значениями Δ Score с большей вероятностью приведут к тканеспецифическому альтернативному сплайсингу (Р=0,015, точный критерий Фишера).
[00497] ФИГ. 40А, 40В, 40С, 40D и 40Е показывают один вариант реализации предсказанных сплайс-вариантов, сильно вредоносных для человеческих популяций.
[00498] В отношении ФИГ. 40А, синонимичные и интронные варианты (≤50 нуклеотидов от известных границ экзон-интрон и исключая основные динуклеотиды GT или AG) с достоверно предсказанными эффектами изменения сплайсинга (Δ-оценка 0,8) сильно истощены при общих частотах аллелей (0,1%) в человеческой популяции относительно редких вариантов наблюдается только один раз у 60 706 особей. Отношение шансов 4,58 (Р <10-127 по критерию хи-квадрат) указывает на то, что 78% недавно возникших предсказанных вариантов критического сплайсинга достаточно опасны, чтобы их можно было удалить естественным отбором.
[00499] отношении ФИГ. 40В, доля вариантов, укорачивающих белок, и предсказанные синонимичные и интронные варианты критического сплайсинга в наборе данных ЕхАС, которые являются вредными, рассчитаны как в (А).
[00500] В отношении ФИГ. 40С, доля синонимичных и интронных вариантов усиления критического сплайсинга в наборе данных ЕхАС, которые являются вредными (Δ Score 0,8), разделены в зависимости от того, вызовет ли вариант сдвиг рамки или нет.
[00501] В отношении ФИГ. 40D, фракция вариантов, укорачивающих белок, и предсказанные варианты критического сплайсинга (>50 нуклеотидов от известных границ экзон-интрон) в наборе данных gnomAD, которые являются вредными.
[00502] В отношении ФИГ. 40Е, среднее количество редких (частота аллелей <0,1%) вариантов укорочения белка и редких функциональных вариантов криптического сплайсинга на индивидуальный геном человека. Количество криптических мутаций сплайсинга, которые, как ожидается, будут функциональными, оценивается на основе доли предсказаний, которые являются вредными. Общее количество предсказаний выше.
[00503] ФИГ. 41А, 41В, 41С, 41D, 41Е и 41F показывают один вариант реализации криптических de novo сплайс-мутаций у пациентов с редкими генетическими заболеваниями.
[00504] В отношении ФИГ. 41А предсказали критические de novo мутации сплайсинга на человека для пациентов из когорты «Расшифровка нарушений развития» (DDD), людей с расстройствами аутического спектра (ASD) из коллекции Simons Simplex и Консорциума по секвенированию аутизма, а также для здоровых людей. Показано обогащение когорт DDD и ASD относительно здоровых контрольных группам с поправкой на выявление вариантов между когортами. Планки погрешностей показывают 95% доверительные интервалы.
[00505] В отношении ФИГ. 41В, расчетная доля патогенных мутаций de novo по функциональным категориям для когорт DDD и ASD, основанная на обогащении каждой категории по сравнению со здоровыми контролями.
[00506] В отношении ФИГ. 41С, обогащение и избыток критических de novo мутаций сплайсинга в когортах DDD и ASD по сравнению со здоровыми контролями при различных порогах Δ Score.
[00507] В отношении ФИГ. 41D, список новых генов-кандидатов для заболеваний, обогащенных мутациями de novo в когортах DDD и ASD (FDR <0,01) при включении предсказанных критических мутаций сплайсинга вместе с мутациями, кодирующими белок, в анализ обогащения. Показаны фенотипы, которые присутствовали у нескольких человек.
[00508] В отношении ФИГ. 41Е, три примера предсказанных de novo критических мутаций сплайсинга у пациентов с аутизмом, которые подтверждаются с помощью данными РНК-секвенирования, приводящее к сохранению интрона, пропуску экзона и удлинению экзона, соответственно. Для каждого примера покрытие РНК-последовательностей и количество границ для пораженного человека показаны вверху, а контрольный человек без мутации показан внизу. На смысловой цепи показаны последовательности относительно транскрипции гена. Синие и серые стрелки разграничивают положения границ у индивидуума с вариантом и контрольного индивидуума соответственно.
[00509] В отношении ФИГ. 41F, статус валидации для 36 предсказанных критических сайтов сплайсинга, выбранных для экспериментальной валидации по данным РНК-секвенирования.
ЭКСПЕРИМЕНТАЛЬНАЯ МОДЕЛЬ И СУБЪЕКТЫ
[00510] Детали субъектов для 36 пациентов с аутизмом были опубликованы ранее в работе Iossifov et al, Nature 2014 (таблица S1), и для них возможны перекрестные ссылки с помощью анонимизированных идентификаторов в столбце 1 таблицы S4 в нашей статье.
ДЕТАЛИ Способа
I. Глубокое обучение для прогнозирования сплайсинга Архитектура SpliceNet
[00511] Мы обучили несколько моделей на основе сверхглубоких сверточных нейронных сетей для компьютерного прогнозирования сплайсинга по нуклеотидной последовательности пре-мРНК. Мы разработали четыре архитектуры, а именно SpliceNet-80nt, SpliceNet-400nt, SpliceNet-2k и SpliceNet-10k, которые используют 40, 200, 1000 и 5000 нуклеотидов с каждой стороны от целевого полодения в качестве входных данных, соответственно, и выводят вероятность того, что данное положение является акцептором и донором сплайсинга. Точнее, входом в модели является последовательность кодированных с одним горячим положением нуклеотидов, где А, С, G и T (или, эквивалентно, U) кодируются как [1, 0, 0, 0], [0, 1, 0, 0], [0, 0, 1, 0] и [0, 0, 0, 1] соответственно, и выходные данные моделей состоят из трех оценок, сумма которых равна единице, которые соответствуют вероятности того, что целевое положение является акцептором сплайсинга, донором сплайсинга или не связано со сплайсингом.
[00512] Основной единицей архитектуры SpliceNet является остаточный блок (Не et al., 2016b), который состоит из слоев пакетной нормализации (Ioffe и Szegedy, 2015), ректифицированных линейных блоков (ReLU) и сверточных блоков, организованных определенным образом (ФИГ. 21. 22, 23 и 24). Остаточные блоки обычно используются при проектировании глубоких нейронных сетей. До разработки остаточных блоков глубокие нейронные сети, состоящие из многих сверточных блоков, складывались один за другим, и их было очень сложно обучать из-за проблемы взрыва/снижения градиентов (Glorot and Bengio, 2010), а увеличение глубины таких нейронных сетей часто приводило к более высокой погрешности обучения (Не et al., 2016а). С помощью полного набора вычислительных экспериментов было показано, что архитектуры, состоящие из множества остаточных блоков, расположенных один за другим, позволяют преодолеть эти проблемы (Не et al., 2016а).
[00513] Полные архитектуры SpliceNet представлены на ФИГ. 21, 22, 23 и 24. Архитектуры состоят из K наложенных друг на друга остаточных блоков, соединяющих входной слой с предпоследним слоем, и сверточного блока с активацией softmax, соединяющего предпоследний слой с выходным слоем. Остаточные блоки складываются таким образом, что выход i-го остаточного блока соединяется с входом 1-го остаточного блока. Далее, выход каждого четвертого остаточного блока добавляется ко входу предпоследнего слоя. Такие «соединения с пропуском» обычно используются в глубоких нейронных сетях для увеличения скорости сходимости во время обучения (Oord et al., 2016).
[00514] Каждый остаточный блок имеет три гиперпараметра N, W и D, где N обозначает количество сверточных ядер, W обозначает размер окна, a D обозначает скорость расширения (Yu and Koltun, 2016) каждого сверточного ядра. Поскольку сверточное ядро с размером окна W и показателем разрежения D извлекает признаки, охватывающие (W-1) D соседних положений, остаточный блок с гиперпараметрами N, W и D извлекает признаки, охватывающие 2 (W-1) D соседних положений. Следовательно, общий диапазон соседей архитектур SpliceNet определяется выражением  ,
,  , Wi и Di представляют собой гипер-параметры
, Wi и Di представляют собой гипер-параметры  остаточного блока. Для архитектур SpliceNet-80nt, SpliceNet-400nt, SpliceNet-2k и SpliceNet-10k число остаточных блоков и гиперпараметры для каждого остаточного блока были выбраны таким образом, чтобы S было равно 80, 400, 2000 и 10000, соответственно.
остаточного блока. Для архитектур SpliceNet-80nt, SpliceNet-400nt, SpliceNet-2k и SpliceNet-10k число остаточных блоков и гиперпараметры для каждого остаточного блока были выбраны таким образом, чтобы S было равно 80, 400, 2000 и 10000, соответственно.
[00515] Архитектура SpliceNet имеет только блоки нормализации и нелинейной активации в дополнение к сверточным блокам. Следовательно, модели могут использоваться в режиме от последовательности к последовательности с переменной длиной последовательности (Oord et al., 2016). Например, вход в модели SpliceNet-10k (S=10,000) представляет собой закодированную с одним горячим положением нуклеотидную последовательность длиной 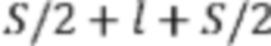 , выход представляет собой матрицу размером
, выход представляет собой матрицу размером 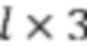 , соответствующую трем оценкам
, соответствующую трем оценкам  центральных положений во входных данных, т. е. положений, оставшихся после исключения первого и последнего нуклеотидов S/2. Эта функция может быть использована для получения огромной экономии вычислительных ресурсов во время обучения, а также тестирования. Это связано с тем, что большинство вычислений для положений, которые близки друг к другу, являются общими, и общие вычисления должны выполняться моделями только один раз, когда они используются в режиме от последовательности к последовательности.
центральных положений во входных данных, т. е. положений, оставшихся после исключения первого и последнего нуклеотидов S/2. Эта функция может быть использована для получения огромной экономии вычислительных ресурсов во время обучения, а также тестирования. Это связано с тем, что большинство вычислений для положений, которые близки друг к другу, являются общими, и общие вычисления должны выполняться моделями только один раз, когда они используются в режиме от последовательности к последовательности.
[00516] В наших моделях используется архитектура остаточных блоков, которая получила широкое распространение благодаря успеху в классификации изображений. Остаточные блоки содержат повторяющиеся сверточные блоки, перемежающиеся соединением с пропуском, что позволяет информации из более ранних уровней пропускать остаточные блоки. В каждом остаточном блоке сначала нормализуется входной слой, а затем слой активации с использованием выпрямленных линейных единиц (ReLU). Затем активация проходит через одномерный сверточный слой. Этот промежуточный выходной сигнал одномерного сверточного слоя снова нормализуется пакетно и активируется ReLU, за которым следует еще один одномерный сверточный слой. В конце второй одномерной свертки мы суммировали ее выход с исходным входом в остаточный блок, который действует как соединение с пропуском, позволяя исходной входной информации обходить остаточный блок. В такой архитектуре, названной ее авторами сетью с глубоким остаточным обучением, входные данные сохраняются в исходном состоянии, а остаточные соединения остаются свободными от нелинейных активаций модели, что позволяет эффективно обучать более глубокие сети.
[00517] После остаточных блоков слой softmax вычисляет вероятности трех состояний для каждой аминокислоты, среди которых наибольшая вероятность softmax определяет состояние аминокислоты. Модель обучается с помощью накопленной категориальной функции кросс-энтропийных потерь для всей последовательности белка с помощью оптимизатора ADAM.
[00518] Atrous/разреженные свертки обеспечивают крупные рецептивные поля при меньшем количестве обучающихся параметров. Atrous/разреженная свертка представляет собой свертку, в которой ядро применяется на площади, большей, чем его длина, за счет того, что она пропускает входные значения с определенным шагом, называемым также показателем разрежения или фактором разрежения. Atrous/разреженные свертки увеличивают расстояние между элементами свертки фильтра свертки/ядра, в результате чего при осуществлении операции свертки используются соседние входные записи (например, нуклеотиды, аминокислоты) с большими интервалами. Это обеспечивает возможность введения во входные данные контекстуальных зависимостей дальнего действия. Дырчатые (atrous) свертки сохраняют расчет свертки для повторного использования при обработке соседних нуклеотидов.
[00519] В показанном примере используются одномерные свертки. В других вариантах реализации модель может использовать различные типы сверток, такие как двумерные свертки, трехмерные свертки, разреженные или дырчатые свертки, транспонированные свертки, разделяемые свертки и разделяемые по глубине свертки. Некоторые слои также используют функцию активации ReLU, которая значительно ускоряет сходимость стохастического градиентного спуска по сравнению с насыщающими нелинейностями, такими как сигмоидальный или гиперболический тангенс. Другие примеры функций активации, которые могут использоваться раскрытой технологией, включают параметрическое ReLU, утечку ReLU и экспоненциальный линейный блок (ELU).
[00520] Некоторые слои также используют пакетную нормализацию (Ioffe и Szegedy, 2015). Что касается пакетной нормализации, распределение каждого слоя в сверточной нейронной сети (CNN) изменяется во время обучения, и оно меняется от одного уровня к другому. Это снижает скорость сходимости алгоритма оптимизации. Пакетная нормализация - это способ решения этой проблемы. Обозначая вход слоя пакетной нормализации через х, а его выход через z, пакетная нормализация применяет следующее преобразование к х:
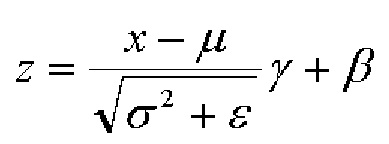
[00521] Пакетная нормализация применяет нормализация среднего отклонения на входе x с использованием μ и σ и линейно масштабирует и сдвигает его, используя γ и β. Параметры нормализации μ и σ вычисляются для текущего слоя по тренировочному (обучающему) набору с использованием метода, называемого экспоненциальным скользящим средним. Другими словами, это не обучаемые параметры. Напротив, γ и β являются обучаемыми параметрами. Значения μ и σ, вычисленные во время обучения, используются при прямом проходе во время вывода.
Тренировка (обучение) и тестирование модели
[00522] Мы загрузили аннотированную таблицу генома GENCODE (Harrow, 2012) V24lift37 из браузера таблиц UCSC и выделили 20287 аннотаций генов, кодирующих белок, при этом в случаях, когда было представлено несколько изоформ, выбирался основной транскрипт. Мы удалили гены, не содержавшие ни одной точки сплайсинга, и разделили оставшиеся гены на тренировочный и тестовый наборы следующим образом: гены, принадлежавшие хромосомам 2, 4, 6, 8, 10-22, X и Y, использовались для тренировки моделей (13384 гена, 130796 пар донор-акцептор). Мы выбрали случайным образом 10% тренировочных генов и использовали их для определения точки ранней остановки в ходе обучения, а оставшиеся использовались нами для тренировки моделей. Для тестирования моделей использовались гены из хромосом 1, 3, 5, 7 и 9, не содержавшие паралогов (1652 гена, 14289 пар донор-акцептор). Для этого мы обращались к списку паралогов генов человека из http://grch37.ensembl.org/biomart/martview.
Для тренировки и тестирования моделей в режиме «последовательность к последовательности» с отрезками длиной 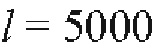 мы использовали следующую процедуру. Последовательность мРНК-транскриптов для каждого гена между каноническими сайтами начала и окончания транскрипции извлекалась из сборки hg19/GRCh37. Входную последовательность мРНК транскриптов представляли в унитарном коде следующим образом: А, С, G, T/U преобразовывали в, соответственно, [1, 0, 0, 0], [0, 1, 0, 0], [0, 0, 1, 0], [0, 0, 0, 1]. Последовательность нуклеотидов в унитарном коде дополнялась нулями до тех пор, пока ее длина не становилась кратной 5000, после чего ее дополняли нулями в начале и в конце фланкирующими последовательностями длиной S/2, где S было равным 80, 400, 2000 и 10000 для моделей SpliceNet-80nt, SpliceNet-400nt, SpliceNet-2k and SpliceNet-10k, соответственно.
мы использовали следующую процедуру. Последовательность мРНК-транскриптов для каждого гена между каноническими сайтами начала и окончания транскрипции извлекалась из сборки hg19/GRCh37. Входную последовательность мРНК транскриптов представляли в унитарном коде следующим образом: А, С, G, T/U преобразовывали в, соответственно, [1, 0, 0, 0], [0, 1, 0, 0], [0, 0, 1, 0], [0, 0, 0, 1]. Последовательность нуклеотидов в унитарном коде дополнялась нулями до тех пор, пока ее длина не становилась кратной 5000, после чего ее дополняли нулями в начале и в конце фланкирующими последовательностями длиной S/2, где S было равным 80, 400, 2000 и 10000 для моделей SpliceNet-80nt, SpliceNet-400nt, SpliceNet-2k and SpliceNet-10k, соответственно.
[00523] Затем дополненную последовательность нуклеотидов разделяли на блоки длиной S/2+5000+S/2 таким образом, чтобы блок с порядковым номером i состоял из нуклеотидов в положениях с 5000(i-1)-S/2+1 по 5000i+S/2. Похожим образом, последовательность выходных меток сплайсинга, представляемая в унитарном коде как: «не сайт сплайсинга», «сайт акцептора сплайсинга» (первый нуклеотид соответствующего экзона), «сайт донора сплайсинга» (последний нуклеотид соответствующего экзона), преобразовывали в [1, 0, 0], [0, 1, 0] и [0, 0, 1], соответственно. Последовательность выходных меток сплайсинга в унитарном коде дополняли нулями до тех пор, пока ее длина не становилась кратной 5000, и разделяли на блоки длиной 5000 таким образом, чтобы блок с порядковым номером i содержал значения в положениях с 5000(i-1)+1 по 5000i. Последовательность нуклеотидов в унитарном коде и соответствующая ей последовательность меток в унитарном коде использовали в качестве входных данных модели и заданных выходных данных модели, соответственно.
[00524] Модель тренировали по 10 эпохам с размером пакета 12 на двух NVIDIA GeForce GTX 1080 Ti GPUs. Значение функции качественного уменьшения перекрестной энтропии между заданными и предсказанными выходными данными в ходе тренировки минимизировали с помощью оптимизатора Адама (Kingma, 2015). Скорость обучения устанавливали равной 0,001 для первых 6 эпох и затем уменьшали в 2 раза в каждой последующей эпохе. Мы повторили процедуру тренировки 5 раз для каждой архитектуры и получили 5 тренированных (обученныз) моделей (Фиг. 53А и 53Б). В процессе тестирования каждый входной набор данных использовался всеми 5 моделями, и средние значения их выходных значений использовали в качестве выходных данных предсказания. Анализ результатов, полученных с использованием этих моделей, представлен на Фиг. 37А и других относящихся к ним фигурах.
[00525] Для анализа, представленного на Фиг. 38А-38Ж, 39А-39В, 40А-40Д и 41А-41Е, в которых задействованы варианты альтернативного сплайсинга, мы дополнили тренировочный набор аннотаций GENCODE новыми границам сплайсинга, часто встречающимися в GTEx группе хромосом 2, 4, 6, 8, 10-22, X, Y (67012 доноров сплайсинга и 62911 акцепторов сплайсинга). Это позволило увеличить число аннотаций границ сплайсинга в тренировочном наборе приблизительно на 50%. Тренировка сети с использованием такого объединенного набора данных привела к повышению чувствительности обнаружения вариантов альтернативного сплайсинга в данных секвенирования РНК по сравнению с тренировкой сети с использованием только GENCODE (Фиг. 52А и 52Б), в частности, при предсказании вариантов альтернативного сплайсинга глубоких интронных мутаций. Мы использовали эту сеть для аналитических исследований, связанных с количественным определением задействованных вариантов (Фиг. 38А-38Ж, 39А-39В, 40А-40Д и 41А-41Е и относящиеся к ним фигуры). Чтобы убедиться в том, что данные из набора GTEx RNA seq не перекрывались при тренировке и при выполнении вычислений, мы включали в тренировочный набор только те точки сплайсинга, которые встречались у 5 и более индивидов, а количественную оценку функциональных характеристик сети проводили для вариантов, которые встречались у 4 и менее индивидов. Новый способ определения точек сплайсинга подробно описан в подразделе «Обнаружение точек сплайсинга», в разделе, посвященном методам анализа GTEx.
Точность Тор-k
[00526] Количественные показатели точности, такие как процент правильно классифицированных положений, являются очень неэффективными, поскольку большинство положений не являются сайтами сплайсинга. Вместо этого, количественную, оценка модели осуществляли с применением двух показателей, являющихся более эффективными в таких обстоятельствах, а именно top-k точности и площади под кривой точности-полноты. Значение top-k точности для конкретного класса определяется следующим образом. Пусть тестовый набор содержит k положений, принадлежащих классу. Мы выбираем такое пороговое значение, при котором ровно k положений контрольного набора отнесены к этому классу в результате предсказания. Значение top-k точности определяется как доля действительно принадлежащих классу положений из числа предсказанных k положений. В действительности, такая величина совпадает с величиной точности, если пороговое значение выбрано таким образом, чтобы величины точности и полноты имели одинаковые значения.
Количественная оценка модели для длинных промежуточных некодирующих РНК (lincRNA, дпнРНК)
[00527] Мы получили список всех транскриптов длинных промежуточных некодирующих РНК (дпнРНК) из аннотаций GENCODE V24lift3. В отличие от кодирующих белки генов, дпнРНК не содержат главного присвоенного транскрипта в аннотациях GENCODE. Для того, чтобы минимизировать избыточные повторения в наборе для валидации, мы идентифицировали транскрипт по самой протяженной последовательности экзонов для дпнРНК каждого гена, и назвали его каноническим транскриптом гена. Так как предполагается, что аннотации дпнРНК являются менее достоверными по сравнению с аннотациями кодирующих белки генов, а ошибочные аннотации могут влиять на наши оценку top-k точности, мы использовали данные GTEx для исключения дпнРНК с потенциально проблемными аннотациями (подробнее об этих данных см. в разделе «Анализ набора данных GTEx»). Для каждого дпнРНК мы подсчитали количество разделенных ридов, картрированных на протяжении всей длины дпнРНК по всем выборкам GTEx (см. подробнее ниже в разделе «Обнаружение точек сплайсинга»). Это позволило получить примерную оценку общего числа покрывающих точки сплайсинга ридов дпнРНК с использованием аннотированных точек сплайсинга или новых точек сплайсинга. Мы также подсчитали количество ридов, покрывающих точки сплайсинга канонического транскрипта. Мы учитывали только дпнРНК, для которых, как минимум, 95% ридов, покрывающих точки сплайсинга по всем выборкам GTEx, соответствовали каноническому транскрипту. Также требовалось, чтобы все точки сплайсинга канонического транскрипта встречались, как минимум, один раз в группе GTEx (за исключением точек, покрывающих интроны длиной менее 10 нуклеотидов). При вычислении top-k точности мы учитывали только точки сплайсинга канонических транскриптов дпнРНК, прошедших указанные выше фильтры (781 транскрипт, 1047 точек сплайсинга).
Идентификация границ сплайсинга по последовательности пре-мРНК
[00528] На Фиг. 37Б показано сравнение функциональных характеристик MaxEntScan и SpliceNet-10k при определении границ канонического экзона гена из его последовательности. В качестве примера мы использовали ген CFTR из нашего контрольного набора, который содержал 26 канонических доноров и акцепторов сплайсинга, и получили значения показателей акцептора и донора для каждого из 188703 положений, начиная с канонического сайта начала транскрипции (chr7:117,120,017) и заканчивая каноническим сайтом конца транскрипции (chr7:117,308,719), с использованием MaxEntScan и SpliceNet-10k. Положение классифицировалось как акцептор или донор сплайсинга, если соответствующий ему показатель был выше порогового значения, выбранного в процессе количественного определения top-k точности. MaxEntScan предсказала 49 акцепторов сплайсинга и 22 донора сплайсинга, из которых 9 и 5 являлись истинными акцепторами и донорами сплайсинга, соответственно. С целью более наглядного представления, показаны предварительно логарифмированные значения показателей MaxEntScan (не превышающие максимального значения, равного 2500). SpliceNet-10k предсказала 26 акцепторов сплайсинга и 26 донора сплайсинга, из которых все оказались верными. Мы провели такой анализ повторно для гена LINC00467.
Оценка включения экзона в GENCODE - аннотированные границы сплайсинга
[00529] Мы вычислили долю включения всех аннотированных в соответствии с GENCODE экзонов из данных РНК-секвенирования GTEx (Фиг. 37В). Для каждого экзона, включая первый и последний экзон каждого гена, доля включения рассчитывалась как:
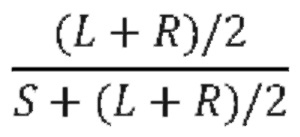
[00530] где L - общее количество ридов границы (точки) сплайсинга, начиная с предыдущего канонического экзона и заканчивая текущим экзоном, во всех выборках GTEx, R - общее количество ридов границы (точки) сплайсинга, начиная с текущего экзона и заканчивая следующим каноническим экзоном, S - общее количество ридов точек пропуска, начиная с предыдущего канонического экзона и заканчивая следующим каноническим экзоном.
Значимость различных нуклеотидов для распознавании сайтов сплайсинга
[00531] На Фиг. 37Г показано, как мы определяем, какие нуклеотиды являются важными для классификации положения акцептора сплайсинга в SpliceNet-10k. Для этого рассмотрим акцептор сплайсинга в chr3:142,740,192 в гене U2SURP, который находится в нашем контрольном наборе. «Показатель значимости» нуклеотида по отношению к акцептору сплайсинга определяется следующим образом. Пусть s_ref обозначает величину показателя рассматриваемого акцептора сплайсинга. Значение показателя акцептора пересчитывается при замене рассматриваемого нуклеотида на А, С, G и Т. Пусть такие показатели обозначаются как sA, sT, s G и T, соответственно. «Показатель значимости нуклеотида» рассчитывается как:

[00532] Эту процедуру часто называют мутагенезом in-silico (Zhou, Troyanskaya, 2015). Мы представили на графике 127 нуклеотидов, начиная с chr3:142,740,137 и заканчивая chr3:142,740,263, таким образом, чтобы высота каждого нуклеотида соответствовала величине его показателя значимости по отношению к акцептору сплайсинга в chr3:142,740,192. При построении графика использовалась функция из программного обеспечения DeepLIFT (Shrikumar, 2017).
Влияние мотивов ТАСТААС и GAAGAA на сплайсинг
С целью изучения влияния положения последовательности точек ветвления на силу акцептора мы сначала получили значения показателя акцептора для 14289 акцепторов сплайсинга из тестового набора с помощью SpliceNet-10k. Пусть y_ref обозначает вектор этих значений. Для каждого значения i в диапазоне от 0 до 100 мы делали следующее. Для каждого акцептора сплайсинга из тестового набора мы заменяли нуклеотиды с i по i-6 позицию перед акцептором сплайсинга на ТАСТААС и повторно вычисляли показатель акцептора с помощью SpliceNet-10k. Вектор, содержащий значения этих показателей, обозначается yalt,i. Мы представили на графике следующую величину, как функцию от i (Фиг. 43А):
mean(yalt,i-yref)
Мы повторили эту процедуру для другого мотива белка SR, GAAGAA. (Фиг. 43Б).
[00533] В этом случае мы также изучили влияние мотива в случае, если он присутствует после акцептора сплайсинга, а также его влияние на силу донора. В результате расширенного поиска в k-мерном пространстве выявлено, что ТАСТААС и GAAGAA. - мотивы, оказывающие наибольшее влияние на силу акцептора и донора.
Роль длины экзона и длины интрона в сплайсинге
[00534] Для изучения влияния длины экзона на сплайсинг, мы отфильтровали экзоны тестового набора, которые были первыми или последними экзонами. В результате такой фильтрации было удалено 16552 из 14289 экзонов. Мы провели сортировку оставшихся экзонов по увеличению длины. Для каждого из них мы рассчитали значение оценки сплайсинга, усредняя значения оценок акцепторов в положении сайта акцептора сплайсинга и значения оценок доноров в положении сайта донора сплайсинга с помощью SpliceNet-80nt. Мы построили график зависимости значений оценки сплайсинга от длины экзона (Фиг. 37Е). Перед построением были проведены следующие процедуры сглаживания. Пусть x обозначает вектор значений длины экзона, а у обозначает вектор советующих значений величины оценки сплайсинга. Мы провели сглаживание x и у с использованием интервала усреднения, равного 2500.
[00535] Мы повторили этот анализ для показателей сплайсинга, рассчитанных с помощью SpliceNet-10k. На заднем фоне показана гистограмма распределения длин 12637 экзонов. участвовавших в этих вычислениях. Мы провели аналогичный анализ для изучения влияния длины нитрона на сплайсинг, с главным отличием отсутствием необходимости исключать первый и последний экзоны. Роль нуклеосом в сплайсинге
[00536] Мы загрузили данные нуклеосом для колонии клеток из геномного браузера UCSC. Мы использовали ген HMGR, присутствовавший в нашем контрольном наборе, в качестве случайного примера, чтобы продемонстрировать влияние положения нуклеосом на показатели SpliceNet-10k. Для каждого положения p в гене мы рассчитывали значение «оценки сеяного сплайсинга» следующим образом:
8 нуклеотидов в положениях р+74 и р+81 заменялись мотивом донора AGGTAAGG.
4 нуклеотида в положениях р-78 и р-75 заменялись мотивом акцептора TAGG.
20 нуклеотидов в положениях р-98 и р-79 заменялись полипиримидиновым трактом CCTCCTTTTTCCTCGCCCTC.
7 нуклеотидов в положениях р-105 и р-99 заменялись последовательностью точки ветвления САСТААС.
Показатель сеяного сплайсинга рассчитывался как среднее значений оценки акцептора в р-75 и оценки донора в р+75, предсказанных с помощью SpliceNet-10k.
[00537] На Фиг. 37G показаны сигналы нуклеосом K562 вместе с оценками сеяного сплайсинга для 5000 положений, начиная с chr5:74,652,154 и заканчивая chr5:74,657,153.
[00538] Для того, чтобы рассчитать коэффициент корреляции Спирмэна между этими двумя величинами по всему геному, мы случайным образом выбрали один миллион межгенных положений, которые находились, как минимум, на расстоянии 100000 нуклеотидов от канонических генов. Для каждого из этих положений мы рассчитали величины оценки сеяного сплайсинга и усредненного сигнала нуклеосом K562 (использовался интервал усреднения, равный 50). Корреляция между этими двумя величинами для одного миллиона положений показана на Фиг. 37G. Далее, мы разделили эти положения по содержанию CG (оценка производилась для нуклеотидов. находящихся между мотивами сеяных акцепторов и доноров) на подклассы с величиной интервала 0,02. Значения коэффициента корреляции Спирмэна по всему геному для каждого из интервалов показаны на Фиг. 44А.
[00539] Для каждого из 14289 акцепторов сплайсинга тестового набора мы извлекали данные нуклеосом в интервале 50 нуклеотидов с каждой стороны и рассчитывали показатель обогащения нуклеосомами, как отношение среднего значения сигнала на стороне экзона к среднему значению сигнала на стороне интрона. Мы провели сортировку акцепторов сплайсинга по обогащению нуклеосомами в порядке возрастания и рассчитали соответствующие им значения показателей акцепторов с помощью SpliceNet-80nt.
[00540] Значения показателей акцепторов на графике зависимости от обогащения нуклеосомами представлены на Фиг. 44Б. Перед построением графиков были проведены процедуры сглаживания, использовавшиеся при построении графиков на Фиг. 37Е. Мы повторили этот анализ, используя SpliceNet-10k, а также для 14289 доноров сплайсинга тестового набора.
Усиление сигнала нуклеосом в новых экзонах
[00541] Мы хотели посмотреть на сигнал нуклеосом возле новых предсказанных экзонов (Фиг. 37Н). Чтобы убедиться в том, что мы рассматривали новые экзоны с высокой степенью доверительности, мы выбирали только частные варианты (варианты, присутствовавшие у одного GTEx индивида), для которых предсказанная приобретенная точка сплайсинга была полностью частным случаем, встречающимся у индивида с таким вариантом. Для того, чтобы дополнительно избежать влияния наложений соседних экзонов, мы рассматривали только варианты интронов, находящихся, как минимум, в 750 нуклеотидах от аннотированного экзона. Мы загрузили сигналы нуклеосом клеточных культур GM12878 и K562 из браузера UCSC и получили сигнал нуклеосом для отрезков длиной 750 нуклеотидов с каждой из сторон нового предсказанного акцептора или донора. Мы усреднили сигнал нуклеосом двух клеточных культур и перевернули сигнальные векторы для вариантов генов на комплементарной цепи. Мы сдвинули сигнал с сайтов акцептора на 70 нуклеотидов вправо, а сигнал с сайтов донора на 70 нуклеотидов влево. После сдвига сигналы нуклеосом с сайтов донора и акцептора были центрированы по отношению к середине идеализированного экзона длиной 140 нуклеотидов, что является медианной длиной экзонов в аннотациях GENCODE v19. Мы усреднили, в качестве последнего шага, все сдвинутые сигналы и провели сглаживание итогового сигнала путем вычисления среднего для 11-нуклеотидного интервала, центрированного относительно каждого положения.
[00542] Для исследования связи мы выбирали случайные одиночные однонуклеотидные вариации, находившиеся, как минимум, на расстоянии 750 нуклеотидов от аннотированных экзонов, и в отношении которых модель предсказывала отсутствие влияния на сплайсинг (Δ Score <0.01). Мы создали 1000 случайных выборок таких однонуклеотидных вариаций, причем каждая выборка содержала такое же число однонуклеотидных вариаций, сколько и набор сайтов появления сайтов сплайсинга, использованных нами для Фиг. 37Н (128 сайтов). Для каждой случайной выборки мы вычисляли сглаженный усредненный сигнал, как это описано выше. Так как случайные однонуклеотидные вариации не были предсказаны при определении новых экзонов, мы центрировали сигналы нуклеосом каждой однонуклеотидной вариации непосредственно по положению однонуклеотидной вариации и случайным образом сдвигали или на 70 нуклеотидов влево или на 70 нуклеотидов вправо. Затем, мы сравнивали сигнал нуклеосом в центральной нижней области (Фиг. 37Н) с сигналами, полученными в результате 1000 имитационных испытаний в этой области. Эмпирическое значение уровня значимости (p-value) было рассчитано как доля наборов, использовавшихся в имитационных испытаниях, для которых значение в центре было больше или равным тому, которое наблюдалось для вариантов появления сайтов сплайсинга.
Устойчивость сети к различиям в плотности экзонов
[00543] Для выяснения того, насколько можно обобщить предсказания сети, мы провели количественную оценку SpliceNet-10k в областях с различной плотностью экзонов. Сначала мы разделили положения тестового набора на 5 категорий в зависимости от процента канонических экзонов в интервале, состоящем из 10000 нуклеотидов (5000 нуклеотидов с каждой стороны) (Фиг. 54). Чтобы удостовериться, что количество экзонов в каждом положении было целочисленным, мы использовали количество начальных положений экзонов в указанном интервале. Для каждой категории мы рассчитали top-k точность и площадь под кривой точности-полноты. Количество положений и значения k различаются в разных категориях (см. подробнее в таблице ниже).
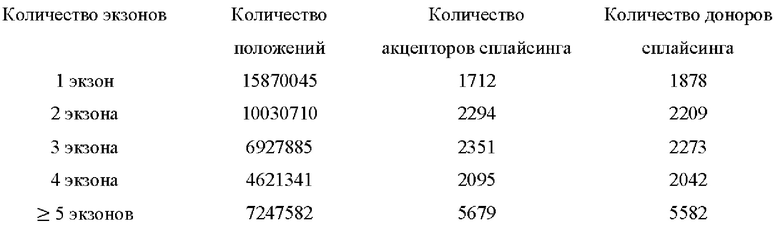
Устойчивость сети для каждой из пяти моделей в ансамбле
[00544] Тренировка нескольких моделей и использование среднего их предсказаний в качестве выходных данных относятся к стратегии ансамблевого обучения, которая обычно используется в машинном обучении для улучшения функциональных характеристик предсказательной способности. На Фиг. 53А показаны значения top-k точности и площади под кривой точности-полноты, полученные для 5 моделей SpliceNet-10k, которые тренировались при создании ансамбля. Полученные результаты свидетельствуют о стабильности тренировочного процесса.
[00545] Мы также рассчитали коэффициенты корреляции Пирсона для их предсказаний. Так как большая часть положений в геноме не являлись сайтами сплайсинга, коэффициенты корреляции между предсказаниями большинства моделей были близки к 1, что лишало дальнейший анализ смысла. Чтобы этого избежать, мы рассматривали только такие положения в тестовом наборе, для которых величина оценки акцептора или величина оценки донора была больше или равной 0,01, хотя бы для одной из моделей. 53272 положений удовлетворяли такому условию (приблизительно равное количество сайтов сплайсинга и сайтов без сплайсинга). Полученные результаты показаны на Фиг. 53В. Наибольший из коэффициентов корреляции Пирсона между предсказаниями моделей в дальнейшем служил показателем устойчивости.
[00546] На Фиг. 53С показано, как число моделей, составляющих ансамбль, влияет на его функциональные характеристики. Полученные результаты свидетельствуют о том, что функциональные характеристики улучшаются с увеличением числа моделей, причем вклад каждой следующей модели в общую эффективность снижается. II. Анализ набора данных РНК-последовательностей GTEx Значение Δ Оценки (Score) однонуклеотидного варианта
[00547] Мы провели количественную оценку изменений в сплайсинге, связанных с однонуклеотидным вариантом следующим образом. Сначала мы использовали референсный нуклеотид и рассчитали значения показателей акцептора и донора для 101 положений возле варианта (50 положений с каждой стороны). Пусть эти показатели обозначаются как векторы и dref, соответственно. Затем мы пересчитали показатели акцептора донора для альтернативного нуклеотида. Пусть эти показатели обозначаются как векторы aalt и dalt, соответственно. Мы вычисляли следующие значения:
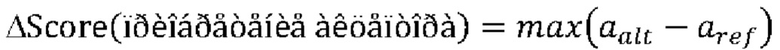
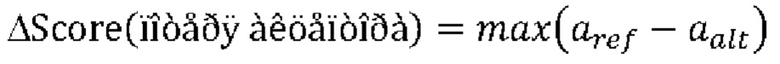
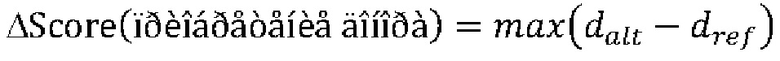
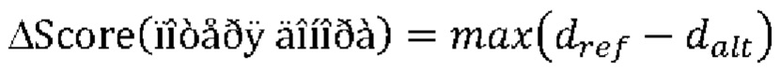
[00548] Максимальное из этих четырех значений определяет величину оценки сплайсинга варианта, Δ оценку (Score).
Критерии, по которым проводились управление качеством и отбор вариантов
Мы загрузили данные GTEx VCF и RNA-seq из dbGaP (доступ для исследований phs000424.v6.p1; https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000424.v6p1)
[00549] Мы оценили функциональные характеристики SpliceNet для аутосомных однонуклеотидных вариаций, встречающихся не более, чем у 4 индивидов в группе GTEx. В частности, рассматривался только такой вариант, который бы удовлетворял следующим критериям для, как минимум, одного индивида А:
1. Вариант не прошел фильтрацию (поле FILTER в VCF определено как PASS).
2. Вариант не был помечен как MULTI_ALLELIC в поле INFO в VCF индивида А и VCF, содержащего одиночную аллель в поле ALT.
3. Индивид А являлся гетерозиготным по этому варианту.
4. Отношение alt_depth/(alt_depth+ref_depth) находилось между 0,25 и 0,75, где alt_depth и ref_depth - число ридов для альтернативного и референсноо аллелей индивида А, соответственно.
5. Значение общей глубины, alt_depth+ref_depth, было между 20 и 300 в VCF индивида А.
6. Вариант находился в области тела гена. Тело гена определяется как интервал между тем, где транскрипция начинается и тем, где она заканчивается, для канонических транскриптов из GENCODE (V24lift37).
[00550] Если вариант удовлетворял этим критериям для, как минимум, одного индивида, то мы учитывали всех индивидов, у которых присутствовал этот вариант, даже если для них этот вариант не удовлетворял всем критериям. Мы называли вариант, обнаруживаемый у одного индивида, частным вариантом, а варианты, обнаруживаемые у 2-4 индивидов, обычными вариантами. Мы не проводили количественную оценку для вариантов, которые обнаруживались у 5 или более индивидов во избежание перекрывания с тренировочным набором данных.
Выравнивание ридов в РНК-последовательностях
[00551] Мы использовали OLego (Wu, 2013) для картирования ридов выборок GTEx по референсному геному hg19, с расстоянием преобразования между запрашиваемым ридом и референсной последовательностью, не превышающим 4, (параметр -М 4). Отметим, что OLego способен функционировать независимо, без каких-либо аннотаций гена. Так как OLego осуществляет поиск присутствия мотивов сплайсинга на концах разделенных ридов, выравнивание может быть смещенным в области однонуклеотидных вариаций в направлении к референсной последовательности или от референсной последовательности, что может приводить к. соответственно, пропуску или созданию, сайта сплайсинга. Чтобы исключить такие смещения, мы создавали альтернативные референсные последовательности для каждого индивида в GTEx, вставляя в референсный геном hg19 все однонуклеотидные вариации индивида, прошедшие через фильтр PASS. Мы использовали OLego с прежними параметрами для картрирования всех выборок от каждого индивида по указанной альтернативной референсной последовательности. Выравнивание каждой из выборок проводилось по двум наборам выравниваний (по hg19 и альтернативной референсной последовательности индивида) так, что для каждой пары ридов выбиралось лучшее из выравниваний. При выборе лучшего из выравниваний для пары ридов Р мы использовали следующую процедуру:
1. Если оба рида в паре Р не были картрированными в обоих наборах выравниваний, то случайным образом выбиралось любое из выравниваний, hg19 или альтернативное в Р.
2. Если для пары Р в одном из наборов выравниваний содержалось больше картрированых концов, чем в другом (например, оба конца пары Р картрированы по альтернативной последовательности, но только один конец картрирован по hg19), мы выбирали то из выравниваний, при котором оба конца картрированы.
3. Если оба конца пары Р были картрированы в обоих наборах выравниваний, мы выбирали выравнивание с наименьшим общим числом несовпадений, или выбирали случайным образом одно из двух выравниваний, если число несовпадений было одинаковым.
Обнаружение границ сплайсинга в выравненных данных секвенирования РНК (РНК-последовательностей)
[00552] Мы использовали утилиту leafcutter_cluster из библиотеки leafcutter (Li, 2018) для обнаружения и подсчета границ сплайсинга в каждой выборке. Требовалось, чтобы один разделенный рид определял точку сплайсинга, и предполагалось, чтобы максимальная длина интрона была 500кб (параметры -m 1 -l 500000). Чтобы получить набор точек сплайсинга с высокой степенью достоверности для тренировки модели глубинного обучения, мы составляли единый набор всех точек сплайсинга из leafcutter для всех выборок, после чего удаляли границы (точки) сплайсинга, не удовлетворявшие любому из следующих критериев:
1. Один из концов границы сплайсинга перекрывался с областью, соответствующей «черному списку» ENCODE (таблица wgEncodeDacMapabilityConsensusExcludable в hg19 из браузера UCSC) или в простыми повторам (список Простых Повторов в hg19 из браузера UCSC).
2. Оба конца границы (точки) сплайсинга находились в неканонических экзонах (см. канонические транскрипты GENCODE version V24lift37).
3. Концы границы (точки) сплайсинга принадлежали разным генам, или каждый из концов был из некодирующей области.
4. Каждый из концов не содержал необходимое число динуклеотидов GT/AG dinucleotides.
[00553] Границы (точки) сплайсинга, присутствовавшие у 5 или более индивидов, пополняли список аннотированных точек сплайсинга из GENCODE, использовавшийся для предсказания вариантов (Фиг. 38A-G, 39А-С, 40А-Е, и 41A-F). Ссылки на файлы, содержащие список точек сплайсинга для тренировки модели, указаны в Таблице «Key Resources» (Ключевые ресурсы).
[00554] Хотя мы использовали границы (точки) сплайсинга, обнаруженные с помощью leafcutter, для пополнения тренировочного набора данных, мы заметили, что, несмотря на использование нежестких параметров, leafcutter отфильтровал многие границы (точки) сплайсинга с хорошим покрытием в данных секвенирования РНК. Это искусственно снизило показатели валидации. Таким образом, для валидации GTEx RNA-seq (Фиг. 38A-38G и 39А-В), мы повторно вычислили набор границ сплайсинга и число границ сплайсинга непосредственно из данных ридов RNA-seq. Мы посчитали все сдублированные разделенные риды с помощью MAPQ с использованием, как минимум, 10 нуклеотидов, из которых, как минимум, 5 нуклеотидов были выравнены с каждой из сторон границы (точки) сплайсинга. Допускался пропуск ридом более двух экзонов, и, в таком случае, рид относился к каждой точке сплайсинга с, как минимум, 5 нуклеотидами картированной последовательности с каждой из сторон.
Определение частной границы (точки) сплайсинга
[00555] Граница считалась частной для индивида А, если соблюдалось, как минимум, одно из следующих условий:
1. Эта граница имела, как минимум, 3 рида в, как минимум, одной выборке от А, и никогда не встречалась ни у какого другого индивида.
2. Соблюдались оба из следующих условий, как минимум, для двух тканей:
a. Среднее число ридов границы (точки) сплайсинга в выборке индивида А из ткани равнялось, как минимум, 10.
b. Индивид А имел, как минимум, в среднем в два раза больше нормированных ридов из такой ткани, чем любой другой индивид. Здесь, нормированное число ридов границы (точки) сплайсинга в выборке определялось, как число ридов границы (точки) сплайсинга, нормированное по общему числу ридов во всех точках сплайсинга соответствующего гена.
[00556] Ткани с менее, чем 5 выборками, полученными от других индивидов (не А), не использовались в этом тесте.
Обогащение однонуклеотидными вариациями одиночных вариантов в области точек сплайсинга
[00557] Если только один конец частной границы (точки) сплайсинга был аннотирован в соответствии с аннотациям GENCODE, мы рассматривали ее в качестве возможного кандидатного сайта появления акцептора или донора и искали однонуклеотидную вариацию одиночного варианта (однонуклеотидной вариации, возникающей у одного индивида в GTEx), которая являлась бы частной для этого индивида в интервале 150 нуклеотидов начиная с того конца, который не был аннотированным. Если оба конца частной границы (точки) сплайсинга были аннотированными, мы считали это частным событием пропуска экзона, если пропускался, как минимум, один экзон, но не более, чем три экзона того же гена, в соответствии с аннотациями GENCODE. Затем мы искали однонуклеотидную вариацию одиночного варианта в интервале 150 нуклеотидов, начиная с концов каждого пропущенных экзонов. Частная точка сплайсинга с обоими концами, не содержащими аннотаций экзонов из GENCODE, не принималась во внимание, так как существенная часть таких точек связана с ошибками выравнивания.
[00558] Для вычисления обогащения однонуклеотидными вариациями одиночных вариантов в области новых частных акцепторов или доноров (в нижней части Фиг. 38В) мы складывали количества однонуклеотидных вариаций частных вариантов, полученные для каждого положения относительно частной границы (точки) сплайсинга. Если перекрывающийся ген находился в комплементарной цепи, относительные положения переворачивались. Мы разбили однонуклеотидные вариации на две группы: однонуклеотидные вариации, частные для индивида с частной точкой сплайсинга, и однонуклеотидные вариации, частные для другого индивида. Для сглаживания полученных сигналов, мы проводили усреднение полученных значений в интервале длиной 7 нуклеотидов, центрированном относительно каждого из положений. Затем мы вычисляли отношение сглаженных величин для первой группы (частные для одного индивида) к сглаженным величинам для второй группы (частные для другого индивида). Для новых сайтов пропуска экзона (верхняя часть Фиг. 38Б) мы использовали похожую процедуру, складывая количества однонуклеотидных вариаций одиночных вариантов в области концов пропущенных экзонов.
Валидация предсказаний модели в данных РНК-секвенирования (RNA-seq) GTEx
[00559] Как для частных вариантов (обнаруженных у одного индивидуума в группе GTEx), так и для общих вариантов (присутствующая у двух-четырех индивидуумах в группе GTEx) были получены предсказания модели глубокого обучения и для референсной, и для альтернативной аллелей и вычислен Δ Оценка. Также были получены положения, в которых модель предсказала присутствие границы сплайсинга с отклонением от нормы (новой или разрушенной). Затем мы попытались определить, имелось ли в данных РНК-сервенирования (RNA-seq) доказательство, подтверждающее наличие отклонения от нормы у индивидуумов с вариантом в предсказанном положении. Во многих случаях модель может предсказывать множество результатов для одного и того же варианта, т.е. варианта, прерывающего аннотированный донор сплайсинга, может также повысить использование субоптимального донора, как показано на фиг. 45, в таком случае модель может предсказать как потерю донора в аннотированной границе сплайсинга, так и появление донора в субоптимальном месте. Однако для валидации мы рассматривали только эффекты с наибольшей предсказанной Δ Оценкой для каждого варианта. Таким образом, для каждого варианта мы рассматривали предсказанные эффекты, создающие границу сплайсинга и разрушающие границу сплайсинга, по отдельности. Следует отметить, что сайты сплайсинга, присутствующие менее, чем у пяти индивидуумов, исключались во время обучения модели для того, чтобы избежать оценки модели на новых границах сплайсинга, на которых она была обучена.
Валидация предсказанных криптических сплайс-мутаций (мутаций криптического сплайсинга) в частных точках (границах) сплайсинга
[00560] Для каждого частного варианта, для которого предсказано, что он вызовет образование новой границы сплайсинга, мы использовали сеть для предсказывания положения вновь созданных границ сплайсинга с отклонением от нормы и смотрели в данные РНК-секвенирования для валидации того, что такие новые границы сплайсинга появились только у индивидуума с SNV, но не у других индивидуумов GTEx. Аналогичным образом, для варианта, для которого предсказано, что он вызовет потерю сайта сплайсинга, что повлияет на границы сплайсинга экзона X, мы искали новые случаи пропуска экзона, от предыдущего канонического экзона (экзона, расположенного раньше перед X на основании аннотаций GENCODE) до следующего канонического экзона (экзона. расположенного позже X), которые наблюдались только у индивидуумов с вариантом, но не у других индивидуумов GTEx. Мы исключали предсказанные потери, если сайт сплайсинга, для которого модель предсказывала, что он будет потерян, не был аннотирован в GENCODE или никогда не наблюдался у индивидуумов GTEx без вариантов. Мы также исключали предсказанные появления, если сайт сплайсинга, появление которого предсказано, уже был аннотирован в GENCODE. Для распространения данного анализа на общие варианты (присутствующие у двух-четырех индивидуумов) мы также валидировали новые границы сплайсинга, которые присутствовали по меньшей мере у половины индивидуумов с вариантами, но отсутствовали у всех индивидуумов без вариантов.
[00561] Используя требование, что предсказанное событие сплайсинга с отклонением от нормы является частным для индивидуумов с вариантами, мы смогли валидировать 40% предсказанных появлений акцептора и донора с высокой оценкой (Δ Оценка ≥0,5), но только 3,4% из предсказанных потерь с высоким показателем и 5,6% из первичных прерываний GT или AG (при величине ложной валидации <0,2% на основании пермутаций - см. раздел «Оценка величины ложной валидации»). Причиной расхождения в величине валидации получений и потерь является двойственность. Во-первых, в отличии от получений, случаи пропуска экзона редко бывают полностью частными у индивидуумов с вариантом, поскольку экзоны часто пропущены на низких базовых уровнях, что можно наблюдать с достаточно глубокой РНК-последовательности. Во-вторых, потери границ сплайсинга могут иметь другие эффекты помимо повышения пропусков экзонов, например, повышение сохранения интронов или повышение использования альтернативных субоптимальных сайтов сплайсинга. По этой причине мы не полностью полагались на частные новые границы сплайсинга при валидации предсказаний модели, мы также валидировали варианты на основе количественных свидетельств повышения или понижения использования границ сплайсинга, в отношении которых которые, что оно будет оказывать воздействие у индивидуумов с вариантом.
Валидация предсказанных криптических мутаций сплайсинга через количественные критерии
[00562] Для границы j из образца s мы получили нормализованное количество границ cjs:
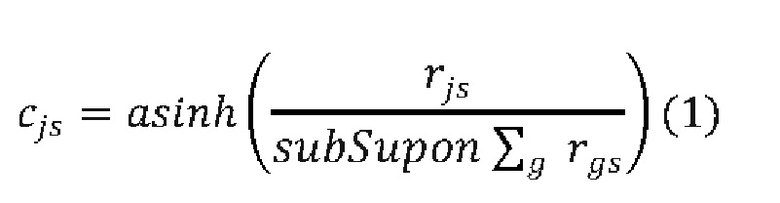
[00563] Здесь, rjs - это необработанное количество границ для границы j в выборке s, а сумма в знаменателе берется по всем другим границам между аннотированными акцепторами и донорами того же гена, что и j (с использованием аннотаций из GENCODE v19). Преобразование asinh определяется как 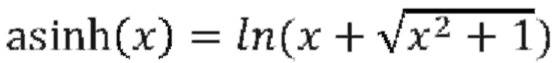 . Это похоже на логарифмическое преобразование, часто используемое для преобразования данных РНК-секвенирования (Lonsdale et al., 2013), однако имеет значение в 0, что устраняет необходимость в псевдосчетах, которые существенно исказили бы значения, поскольку многие границы, особенно новые, имеют низкие значения, близкие к нулю. Преобразование asinh ведет себя как log-преобразование для больших значений, но близко к линейному для малых значений. По этой причине он часто используется в наборах данных (таких как наборы данных секвенирования РНК или ChIP-seq) с большим количеством значений, близких к нулю, чтобы предотвратить доминирование небольшого числа больших значений в сигнале (Azad et al., 2016; Herring et al., 2018; Hoffman et al., 2012; Kasowski et al., 2013; SEQC / MAQC-III Consortium, 2014). Как описано ниже, в разделе «Критерии extnf для валидации» образцы, в которых знаменатель в уравнении (1) был ниже 200, bcrk.xfkbcm из всех анализов валидации, что позволило избежать численных проблем.
. Это похоже на логарифмическое преобразование, часто используемое для преобразования данных РНК-секвенирования (Lonsdale et al., 2013), однако имеет значение в 0, что устраняет необходимость в псевдосчетах, которые существенно исказили бы значения, поскольку многие границы, особенно новые, имеют низкие значения, близкие к нулю. Преобразование asinh ведет себя как log-преобразование для больших значений, но близко к линейному для малых значений. По этой причине он часто используется в наборах данных (таких как наборы данных секвенирования РНК или ChIP-seq) с большим количеством значений, близких к нулю, чтобы предотвратить доминирование небольшого числа больших значений в сигнале (Azad et al., 2016; Herring et al., 2018; Hoffman et al., 2012; Kasowski et al., 2013; SEQC / MAQC-III Consortium, 2014). Как описано ниже, в разделе «Критерии extnf для валидации» образцы, в которых знаменатель в уравнении (1) был ниже 200, bcrk.xfkbcm из всех анализов валидации, что позволило избежать численных проблем.
[00564] Для каждой образованной (новой) или потерянной границы j, которая, согласно предсказаниям, вызвана SNV, появляющимся у набора индивидуумов I, мы вычислили следующую z-оценку для каждой ткани t отдельно:
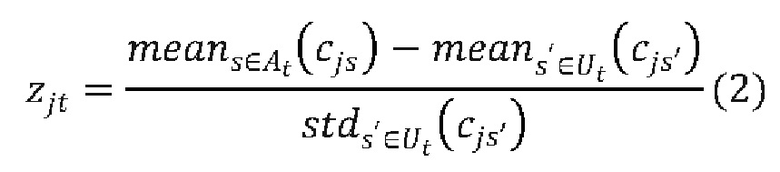
mean=среднее
[00565] где At i это набор образцов от людей в I в ткани t, a Ut набор образцов от всех других людей в ткани t. Следует отметить, что в наборе данных GTEx может быть несколько образцов для одного и того же индивидуума и ткани. Как и раньше, cjs i это количество для границы j в образце s. Для предсказанных потерь мы также вычислили аналогичную z-оценку для границы k, пропускающей предположительно затронутый экзон:
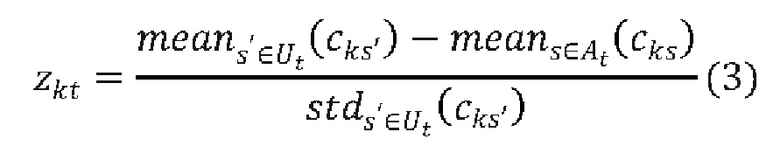
[00566] Обратите внимание, что потеря, которая привела к пропуску, приведет к относительному уменьшению потерянной границы и относительному увеличению пропусков. Это оправдывает обратное изменение разницы в числителях zjt и zkt, поэтому обе эти оценки будут иметь тенденцию быть отрицательными для реальной потери сайта сплайсинга.
[00567] Наконец, мы вычислили медианную z-оценку по всем рассматриваемым тканям. Для потерь мы вычислили медианное значение каждой z-оценки по уравнениям (2) и (3) отдельно. Предсказание потери акцептора или донора считался подтвержденным, если выполнялось одно из следующих условий:
1. Медиана z-значений из уравнения (2), являющаяся количественной оценкой относительной потери границы, была меньше 5-го процентам соответствующего значения в переставленных данных (-1,46), а медианы z-оценок из уравнения (3), являющаяся количественной оценкой относительного изменения пропусков, была неположительной (ноль, отрицательная или отсутствовала, что было бы в случае, если бы приводящая к пропуску граница не наблюдалось ни у одного человека). Другими словами, были убедительные доказательства уменьшения использования затронутой границы и отсутствовали доказательства, свидетельствующие об уменьшении пропусков у затронутого человека.
2. Медиана z-оценок из уравнения (3) была меньше 5-го процентиля соответствующего значения в переставленных данных (-0,74), а медиана z-оценок из уравнения (3) была отрицательной.
3. Медиана z-значений из уравнения (2) была меньше 1-го процентиля соответствующих значений в переставленных данных (-2,54).
4. Медиана z-оценок из уравнения (3) была меньше 1-го процентиля соответствующих значений в переставленных данных (-4,08).
5. Граница, приводящая к пропуску затронутого экзона, наблюдалось по меньшей мере у половины индивидуумов с вариантом и ни у одного из других индивидуумов (как описано выше в разделе «Валидация предсказанных криптических мутаций сплайсинга на основе частных границ сплайсинга»).
[00568] Описание перестановок, используемых для получения указанных выше пороговых значений, приведено в разделе «Оценка показателей ложной валидации».
[00569] Эмпирически путем мы наблюдали, что необходимо применять более строгие критерии валидации для потерь, чем для появления новых границ, поскольку, как объясняется в разделе «Валидация предсказанных криптических мутаций сплайсинга на основе частных границ сплайсинга», потери обычно приводят к более смешанным эффектам, чем появление. Очень маловероятно, что наблюдение новой границы рядом с частным SNV произойдет случайно, поэтому даже незначительного свидетельства границы должно быть достаточно для валидации. И напротив, большинство предсказанныз потерь привели к ослаблению существующей границы, и такое ослабление труднее обнаружить, чем изменение включения-выключения, вызванное усилением и с большей вероятностью связанное с шумом в данных РНК-секвенирования.
Критерии включения для валидационного анализа
[00570] Чтобы избежать вычисления z-оценок при низком количестве или плохом покрытии, мы использовали следующие критерии для фильтрации вариантов для валидационного анализа:
1. Образцы учитывались для вышеуказанного расчета z-оценки, только если они экспрессировали ген (subSupon ∑grgs>200 в уравнении (1)).
2. Ткань не учитывали для расчета z-оценки потери или прироста, если среднее количество потерянных или «референсных» границ, соответственно, у индивидуумов без варианта было меньше 10. «Референсная» граница - это каноническая граница, используемая до усиления новой границы, на основании аннотаций GENCODE (подробности см. в разделе о вычислении величины эффекта). Интуиция подсказывает, что мы не должны пытаться валидировать вариант потери сплайсинга, который влияет на границу, не экспрессируемую у контрольных индивидуумов. Точно так же мы не должны пытаться валидировать вариант с приобретением сплайсинга, если контрольные индивидуумы не экспрессировали в достаточной степени транскрипты, охватывающие затронутый сайт.
3. В случае предсказанной потери сайта сплайсинга образцы от индивидуумов без варианта учитывались только в том случае, если у них было по меньшей мере 10 счетов потерь границы. В случае предсказанного прироста акцептора или донора образцы от контрольных индивидуумов учитывали только в том случае, если они имели по меньшей мере 10 счетов «референсной» границы. Интуитивно, даже в ткани с большим средним уровнем экспрессии затронутой границы (т.е. Прохождение критерия 2) разные образцы могут иметь существенно разную глубину секвенирования, поэтому следует включать только контрольные образцы с достаточной экспрессией.
4. Ткань учитывалась только в том случае, если был по меньшей мере один образец, удовлетворяющий указанным выше критериям от индивидуумов с вариантом, а также по меньшей мере 5 образцов, отвечающих указанным выше критериям, от по меньшей мере 2 отдельных контрольных индивидуумов.
[00571] Варианты, для которых не было тканей, удовлетворяющих вышеуказанным критериям для учета, считались неустановленными и исключались при вычислении уровня валидации. Для вариантов приобретения сплайсинга мы отфильтровали те, которые встречаются на уже существующих сайтах сплайсинга, аннотированных GENCODE. Аналогично, для вариантов с потерей сплайсинга мы рассматривали только те, которые снижают оценки существующих сайтов сплайинга, аннотированных GENCODE. В целом, 55% и 44% предсказанных приобретений и потерь с высокими оценками (Δ Оценка≥0,5), соответственно, считались достоверными и использовались для валидационного анализа.
Оценка уровня ложных валидации
[00572] Чтобы подтвердить, что описанная выше процедура имела разумные показатели истинной валидации, мы сначала рассмотрели SNV, которые появляются у 1-4 индивидуумов GTEx и нарушают основные динуклеотиды GT / AG. Мы утверждали, что такие мутации почти наверняка влияют на сплайсинг, поэтому уровень их валидации должен быть близок к 100%. Среди таких нарушений 39% можно было установить на основе критериев, описанных выше, а среди возможных нарушений уровень валидации составил 81%. Чтобы оценить частоту ложных валидации, мы переставили отдельные метки данных SNV. Для каждого SNV, который появился у k индивидуумов из GTEx, мы выбрали случайную подгруппу из k индивидуумов GTEx и присвоили им SNV. Мы создали 10 таких рандомизированных наборов данных и повторили для них процесс валидации. Уровень валидации в пермутированных наборах данных составлял 1,7-2,1% для приобретений и 4,3-6,9% для потерь, при медиане 1,8% и 5,7% соответственно. Более высокий уровень ложных валидации потерь и относительно низкий уровень валидации существенных нарушений объясняется сложностью валидации потерь сайтов сплайсинга, как указано в разделе «Валидация предсказанных криптических мутаций сплайсинга на основе частных границ сплайсинга».
Расчет величины эффекта вариантов криптического сплайсинга в данных секвенирования РНК
[00573] Мы определили «велицину эффекта» варианта как долю транскриптов затронутого гена, которые изменили паттерны сплайсинга из-за варианта (например, доля, которая переключилась на новый акцептор или донор). В качестве справочного примера для предсказанного варианта усиления в точке рассмотрим вариант в ФИГ. 38С. Для предсказанного полученного донора А мы сначала идентифицировали границу (АС) с ближайшим аннотированным акцептором С.Затем мы идентифицировали «референсный» переход (ВС), где В≠А - аннотированный донор, ближайший к А. Затем в каждом образце s мы вычислили относительное использование новой границы (АС) по сравнению с референсной границей (ВС):
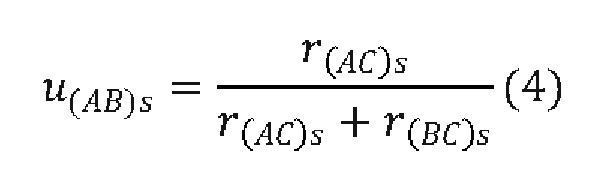
[00574] Здесь r(AC)s - это необработанное количество ридов границы (АС) в образце(выборке) s. Для каждой ткани мы вычислили изменение в использовании границы (АС) между индивидуумами с вариантом и всеми другими индивидуумами:
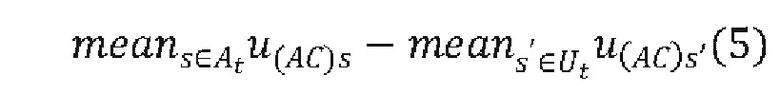
mean=среднее
[00575] где At это набор образцов от индивидуумов с вариантом в ткани t, a Ut набор образцов от других индивидуумов в ткани t Окончательная величина эффекта была рассчитан как среднее значение указанной выше разницы для всех рассматриваемых тканей. Вычисление было аналогичным в случае приобретенного акцептора или в случае, когда вариант, создающий сайт сплайсинга, был интронным. Упрощенный вариант вычисления величины эффекта (предполагающая единственный образец от индивидуумов с вариантом и без него) показан на ФИГ. 38С.
[00576] Для предсказанной потери мы сначала вычислили долю транскриптов, которые пропустили затронутый экзон. Расчет показан на ФИГ. 45. Для предсказанной потери донора С мы идентифицировали границу (СЕ) со следующим аннотированным экзоном ниже по течению, а также границу (АВ) с экзоном выше предположительно затронутого. Мы вычисляли долю транскриптов, которые пропустили пораженный экзон, следующим образом:
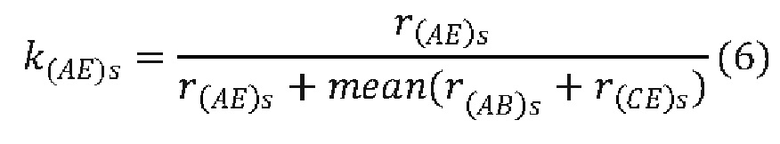
[00577] Что касается приобретения, мы затем вычислили изменение пропущенной доли между образцами от индивидуумов с вариантом и образцами от индивидуумов без варианта:
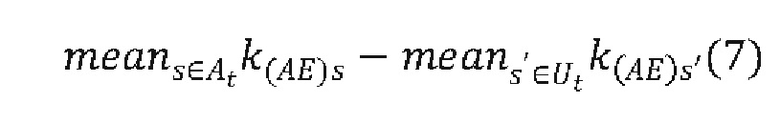
[00578] Доля транскриптов с пропуском, вычисленная выше, не полностью отражает эффекты потери акцептора или донора, поскольку такое нарушение может также приводить к повышенным уровням сохранения интронов или использованию субоптимальных сайтов сплайсинга. Чтобы учесть некоторые из этих эффектов, мы также вычислили использование потерянной границы (СЕ) относительно использования других границ с тем же акцептором Е:
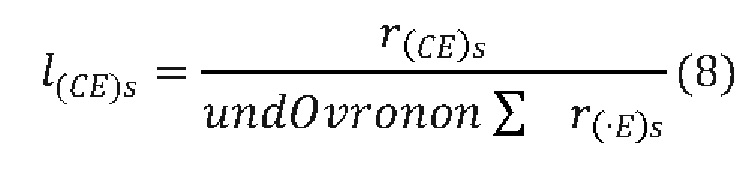
[00579] Здесь  - это сумма всех границ от любого (аннотированного или нового) акцептора к донору Е. Она включает затронутую границу (СЕ), вызывающую пропуск границу (АЕ), а также потенциальные границы от других неоптимальных доноров, которые компенсировали потерю С, как показано в примере на ФИГ. 45. Затем мы вычислили изменение относительного использования затронутой границы:
- это сумма всех границ от любого (аннотированного или нового) акцептора к донору Е. Она включает затронутую границу (СЕ), вызывающую пропуск границу (АЕ), а также потенциальные границы от других неоптимальных доноров, которые компенсировали потерю С, как показано в примере на ФИГ. 45. Затем мы вычислили изменение относительного использования затронутой границы:
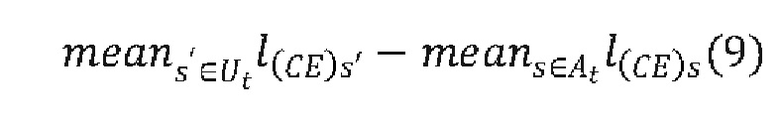
[00580] Обратите внимание, что, в отличие от (5) и (7), которые измеряют увеличение использования приобретенной или пропускающей границы у индивидуумов с вариантом, в (9) мы хотим измерить уменьшение использования потеранной границы, что обуславливает возврат двух частей разницы. Для каждой ткани величина эффекта рассчитывалась как максимум из (7) и (9). Что касается прироста, конечной величиной эффекта для варианта была средняя величина эффекта по тканям.
Критерии включения для анализа величины эффекта
[00581] Вариант учитывали для вычисления размера эффекта только в том случае, если он признавался валидированным на основании критериев, описанных в предыдущем разделе. Чтобы избежать вычисления доли аберрантных транскриптов на очень малых числах, мы рассматривали только образцы, в которых количество аберрантных и референсных границ составляло по меньшей мере 10. Поскольку большинство вариантов критического сплайсинга находились в интроне, величину эффекта нельзя было вычислить непосредственно путем подсчета количества референсных и альтернативных ридов, перекрывающих вариант.Соответственно, величина эффекта потерь рассчитывается косвенно по уменьшению относительного использования нормальной границы. Что касается величины эффекта нового прироста соединения, на аберрантные транскрипты может влиять миссенс-разрушение, ослабляя наблюдаемую величину эффекта. Несмотря на ограничения этих измерений, мы наблюдаем последовательную тенденцию к меньшей величине эффекта для критических вариантов границ с более низкими оценками как при приобретении, так и при потерях.
Ожидаемый размер эффекта полностью пенетрантных гетерозиготных частных SNV
[00582] Для варианта, создающего полностью пенетрантный сайт сплайсинга, который вызывает переключение всех транскриптов с вариантного гаплотипа индивидуумов с вариантом на новую граница, и предполагая, что новая граница не возникает у контрольных индивидуумов, ожидаемая величина эффекта будет 0,5 по уравнению (5).
[00583] Аналогично, если гетерозиготный SNV вызывает новое событие пропуска экзона, и все транскрипты затронутого гаплотипа переключаются на пропускающий сплайсинг, ожидаемая величина эффекта в уравнении (7) составляет 0,5. Если все транскрипты от индивидуумов с вариантом переключились на другую границу (либо пропускающий сплайсинг, либо другой компенсирующий сплайсинг), соотношение в уравнении (8) было бы 0,5 в образцах от индивидуумов с вариантом и 1 в образцах от других индивидуумов, поэтому разница в уравнении (9) будет 0,5. Это предполагает, что у индивидуумов без варианта не было пропуска или других границ в акцептор Е. Это также предполагает, что нарушение сайта сплайсинга не вызывает сохранения интрона. На практике по меньшей мере низкие уровни сохранения интронов часто связаны с нарушениями сайта сплайсинга. Более того, пропуск экзонов широко распространен даже в отсутствие вариантов, изменяющих сплайсинг. Это объясняет, почему измеренная величина эффекта ниже 0,5 даже для вариантов, разрушающих основные динуклеотиды GT / AG
[00584] Ожидаемая величина эффекта 0,5 для полностью пенетрантных гетерозиготных вариантов также предполагает, что вариант не вызывал нонсенс-опосредованного распада (NMD). В присутствии NMD числитель и знаменатель уравнений (4), (6) и (8) будут уменьшаться, уменьшая таким образом наблюдаемую величину эффекта.
Фракция транскриптов, деградированных в результате нонсенс-опосредованного распада (NMD)
[00585] Для ФИГ. 38С, поскольку вариант был экзонным, мы могли подсчитать количество ридов, которые охватывали вариант и имели референсный или альтернативный аллель («Ref (без сплайсинга)» и «Alt (без сплайсинга)» соответственно). Мы также подсчитали количество ридов, которые сплайсировались на новом сайте сплайсинга и предположительно несли альтернативный аллель («Alt (новое соединение)»). На примере ФИГ. 38С и во многих других случаях, которые мы рассмотрели, мы наблюдали, что общее количество ридов, происходящих из гаплотипа с альтернативным аллелем (сумма «Alt (без сплайсинга)» и «Alt (новое соединение)») было меньше, чем количество ридов с референсным аллелем («Ref (без сплайсинга)»). Поскольку мы считаем, что устранили связанные с референсными последовательностями систематические ошибки во время картирования ридов, отображая как эталонный, так и альтернативный гаплотип, и предполагая, что количество ридов пропорционально количеству транскриптов с каждым аллелем, мы ожидали, что референсный аллель будет отвечать за половину ридов в вариантном локусе. Мы предполагаем, что «отсутствующие» риды альтернативных аллелей соответствуют транскриптам из гаплотипа альтернативных аллелей, которые сплайсируются по новой границе и деградировали в результате миссенс-распада (NMD). Мы назвали эту группу «Alt (NMD)».
[00586] Чтобы определить, была ли разница между наблюдаемым числом референсных и альтернативных ридов значимой, мы вычислили вероятность наблюдения Alt (без сплайсинга) + Alt (новая граница) ридов (или меньше) при биномиальном распределении с вероятностью успеха 0,5 и общее количество попыток Alt (без сплайсинга) + Alt (новая граница) + Ref (новая граница). Это консервативное р-значние, поскольку мы недооцениваем общее количество «испытаний», не считая потенциально деградированные транскрипты. Доля транскриптов NMD на ФИГ. 38С вычислялся как количество ридов «Alt (NMD)» по отношению к общему количеству ридов, сплайсированных по новой границе (Alt (NMD) + Alt (новая граница)).
Чувствительность сети при обнаружении криптических границ сплайсинга
[00587] Для оценки чувствительности модели SpliceNet (ФИГ. 38F) мы использовали SNV, которые находились в пределах 20 нуклеотидов от затронутого сайта сплайсинга (т.е. нового или разрушенного акцептора или донора) и не перекрывали основной динуклеотид GT / AG аннотированного экзона и имели предполагаемую величину эффекта по меньшей мере 0,3 (см. раздел «Расчет величины эффекта»). На всех графиках чувствительности SNV определялись как «близкие к экзонам», если они перекрывали аннотированный экзон или находились в пределах 50 нуклеотидов от границ аннотированного экзона. Все остальные SNV считались «глубокими интронными». Используя этот набор данных истинности критических сайтов сплайсинга с сильной поддержкой, мы оценили нашу модель при различных пороговых значениях оценки и зафиксировали долю криптических сайтов сплайсинга в наборе истинных данных, которые предсказываются моделью при этом пороге.
Сравнение с существующими моделями предсказания сплайсинга
[00588] Мы провели прямое сравнение SpliceNet-10k, MaxEntScan (Yeo and Burge, 2004), GeneSplicer (Pertea et al., 2001) и NNSplice (Reese et al., 1997) по различным показателям. Мы загрузили программное обеспечение MaxEntScan и GeneSplicer с http://genes.mit.edu/burgelab/maxent/download/ и http://www.cs.jhu.edu/~genomics/GeneSplicer/ соответственно. NNSplice недоступен в качестве загружаемого программного обеспечения, поэтому мы загрузили наборы для обучения и тестирования с http://www.fruirfly.org/data/seq_tools/datasets/Human/GENIE_96/splicesets/, а также обученные модели с описанными наиболее эффективными архитектурами, в (Reese et al., 1997). В качестве проверки работоспособности мы воспроизвели показатели набора тестов, представленные в (Reese et al., 1997).
Чтобы оценить top - к точности и площадь под кривыми точность-отзыв этих алгоритмов, мы оценили все положения в генах тестового набора и дпнРНК для каждого алгоритма (ФИГ. 37D).
[00589] Выходные данные MaxEntScan и GeneSplicer соответствуют логарифмическим отношениям шансов, тогда как выходные данные NNSplice и SpliceNet-10k соответствуют вероятностям. Чтобы обеспечить максимальные шансы на успех MaxEntScan и GeneSplicer, мы рассчитали Δ Оценки, используя их с выходными данными по умолчанию, а также с преобразованными выходными данными, где мы сначала преобразуем их выходные данные, чтобы они соответствовали вероятностям. Точнее, выходные данные MaxEntScan по умолчанию соответствуют
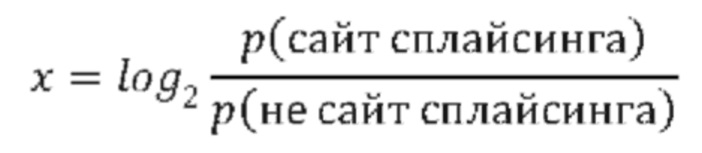
[00590] что после преобразования 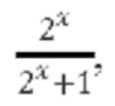 соответствует искомой величине. Мы дважды скомпилировали программное обеспечение GeneSplicer: один раз установив флаг RETURN TRUE PROB на 0 и один раз установив его на 1. Мы выбрали стратегию вывода, которая привела к лучшему уровню валидации данных секвенирования РНК (MaxEntScan: преобразованные выходные данные, GeneSplicer: выходные данные по умолчанию).
соответствует искомой величине. Мы дважды скомпилировали программное обеспечение GeneSplicer: один раз установив флаг RETURN TRUE PROB на 0 и один раз установив его на 1. Мы выбрали стратегию вывода, которая привела к лучшему уровню валидации данных секвенирования РНК (MaxEntScan: преобразованные выходные данные, GeneSplicer: выходные данные по умолчанию).
[00591] Чтобы сравнить скорость проверки и чувствительность различных алгоритмов (ФИГ. 38G), мы нашли граничные значения, при которых все алгоритмы предсказывали одинаковое количество приобретений и потерь для всего генома. То есть для каждого граничного значения на значениях Δ-Оценки SpliceNet-10k мы нашли граничные значения, при которых каждый конкурирующий алгоритм будет делать такое же количество предсказаний приобретения и такое же количество прогнозов потерь, как SpiceNet-10k. Выбранные граничные значения (cutoff) приведены в таблице S2.
Сравнение предсказания вариантов для одиночных и распространенных вариантов
[00592] Мы выполнили валидацию и анализ чувствительности (как описано в разделах «Анализ чувствительности» и «Валидация предсказаний модели») отдельно для одноэлементных SNV и SNV, появляющихся у 2-4 индивидуумов в GTEx (ФИГ. 46А, 46В и 46С). Чтобы проверить, значительно ли различается уровень валидации между одиночными и общими вариантами, мы применили точный критерий Фишера, сравнив уровни валидации в каждой группе Δ-Оценки (0,2 -0,35, 0,35 - 0,5, 0,5 - 0,8, 0,8 - 1) и для каждого предсказанный эффект (приобретение или потеря акцептора или донора). После поправки Бонферрони для учета 16 тестов все Р-значения были больше 0,05. Мы аналогичным образом сравнили чувствительность для обнаружения одиночных или общих вариантов. Мы использовали точный критерий Фишера, чтобы проверить, значимо ли различался уровень валидации между двумя группами вариантов. Мы рассматривали варианты с глубоким интроном и варианты около экзонов отдельно и выполнили коррекцию Бонферрони для двух тестов. Ни одно из Р-значений не было значимым при граничном значении 0,05. Поэтому мы объединили одиночные и обычные варианты GTEx и рассмотрели их вместе для анализа, представленного на ФИГ. 48А, 48В, 48С, 48D, 48Е, 48F, 48G и ФИГ. 39А, 39В и 39С.
Сравнение предсказания вариантов на обучающих и тестовых хромосомах
[00593] Мы сравнили степень валидации на данных секвенирования РНК и чувствительность SpliceNet-10k между вариантами на хромосомах, используемых во время обучения, и вариантами на остальных хромосомах (ФИГ. 48А и 48В). Все Р-значения были больше 0,05 после коррекции Бонферрони. Мы также вычислили долю вредоносных вариантов отдельно для вариантов на обучающей и тестовой хромосомах, как описано в разделе «Доля вредоносных вариантов» ниже (ФИГ. 48С). Для каждой группы Δ-оценки и каждого типа варианта мы использовали точный критерий Фишера, чтобы сравнить количество общих и редких вариантов между обучающими и тестовыми хромосомами. После поправки Бонферрони для 12 тестов все Р-значения были больше 0,05. Наконец, мы вычислили количество вариантов критического сплайсинга de novo на обучающих и тестовых хромосомах (ФИГ. 48D), как описано в разделе «Увеличение количества мутаций de novo на когорту».
Сравнение предсказания вариантов для разных типов вариантов криптического сплайсинга
[00594] Мы разделили предсказанные варианты, создающие сайт сплайсинга, на три группы: варианты, создающие новый динуклеотид сплайсинга GT или AG, варианты, перекрывающие остальную часть мотива сплайсинга (положения вокруг границы экзон-интрон до 3 нуклеотидов в экзон и 8 нуклеотидов в экзон). интрон) и варианты вне сплайсингового мотива (ФИГ. 47А и 47В). Для каждой группы Δ-оценки (0,2 - 0,35, 0,35 - 0,5, 0,5 - 0,8, 0,8 - 1) мы применили критерий χ2 чтобы проверить гипотезу о том, что уровень валидации одинаков для трех типов вариантов, создающих сайты сплайсинга. Все тесты дали Р-значения >0,3 даже до коррекции для множественных гипотез. Чтобы сравнить распределение величины эффекта между тремя типами вариантов, мы использовали U-критерий Манна-Уитни и сравнили все три пары типов вариантов для каждой группы Δ-Оценки (всего 4×3=12 тестов). После поправки Бонферрони для 12 тестов все Р-значения были >0,3.
Обнаружение тканеспецифичных вариантов приобретения сайтов сплайсинга
[00595] За ФИГ. 39С, мы хотели проверить, была ли степень использования новых границ равномерной во всех тканях, экспрессирующих затронутый ген. Мы сосредоточились на SNV, которые создавали новые частные сайты сплайсинга, то есть SNV, приводящие к приобретению сайта сплайсинга, который присутствовал у по меньшей мере половины индивидуумов с вариантом и ни у кого из других индивидуумов. Для каждого такой новой границы j мы вычислили в каждой ткани t общее количество границ по всем образцам от индивидуумов с вариантом в ткани: 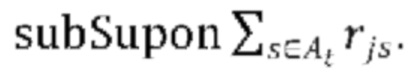 Здесь At набор образцов от индивидуумов с вариантом в ткани t. Точно так же мы вычислили общее количество всех аннотированных границ гена для тех же образцов
Здесь At набор образцов от индивидуумов с вариантом в ткани t. Точно так же мы вычислили общее количество всех аннотированных границ гена для тех же образцов 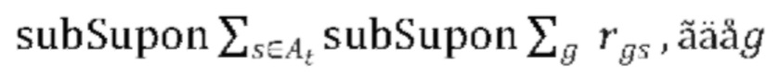 индексирует аннотированные границы гена.
индексирует аннотированные границы гена.
Относительное использование новой границы в ткани t, нормированного относительно фонового количества для гена, затем может быть измерено как:
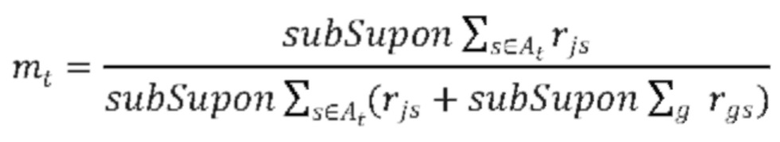
[00596] Мы также вычислили среднее использование границы по тканям:
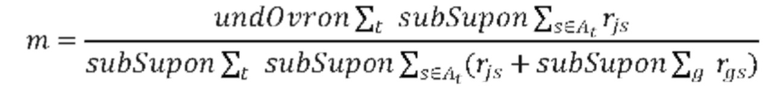
[00597] Мы хотели проверить гипотезу о том, что относительное использование границы равномерно по тканям и равно m. Таким образом, мы применили критерий χ2 для сравнения наблюдаемого количества в ткани 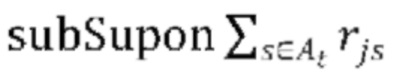 с ожидаемыми количествами при допущении одинакового уровня,
с ожидаемыми количествами при допущении одинакового уровня, 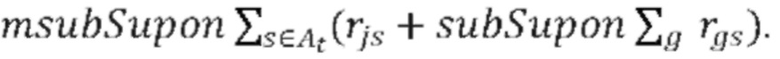 Вариант, создающий сайт сплайсинга считался, тканеспецифичным, если скорректированное по Бонферрони з-значение χ2 было меньше 10-2. Степени свободы для теста Т - 1, где Т - s число рассматриваемых тканей. В тесте использовались только ткани, которые удовлетворяли критериям рассмотрения, описанным в разделе о валидации. Кроме того, чтобы избежать случаев с низким счетом, когда тест на однородность был недостаточно мощным, мы тестировали только варианты однородности с по меньшей мере тремя рассматриваемыми тканями, по меньшей мере одно отклонение от нормы на ткань в среднем (т.е. m>1), и по меньшей мере по мере 15 аберрантных ридов в целом во всех рассматриваемых тканях
Вариант, создающий сайт сплайсинга считался, тканеспецифичным, если скорректированное по Бонферрони з-значение χ2 было меньше 10-2. Степени свободы для теста Т - 1, где Т - s число рассматриваемых тканей. В тесте использовались только ткани, которые удовлетворяли критериям рассмотрения, описанным в разделе о валидации. Кроме того, чтобы избежать случаев с низким счетом, когда тест на однородность был недостаточно мощным, мы тестировали только варианты однородности с по меньшей мере тремя рассматриваемыми тканями, по меньшей мере одно отклонение от нормы на ткань в среднем (т.е. m>1), и по меньшей мере по мере 15 аберрантных ридов в целом во всех рассматриваемых тканях 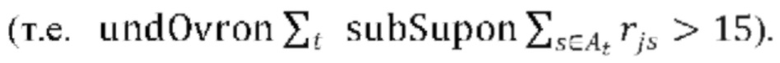 Мы отбрасывали все варианты с оценкой "1-оценкой ниже 0,35, поскольку этот класс вариантов обычно имеет низкую величину эффекта и низкое количество границ. Мы заметили, что доля тканеспецифичных вариантов была очень низкой для этого класса, но мы полагаем, что это произошло из-за проблем, связанных с мощностью.
Мы отбрасывали все варианты с оценкой "1-оценкой ниже 0,35, поскольку этот класс вариантов обычно имеет низкую величину эффекта и низкое количество границ. Мы заметили, что доля тканеспецифичных вариантов была очень низкой для этого класса, но мы полагаем, что это произошло из-за проблем, связанных с мощностью.
III. Анализ наборов данных ЕхАС и gnomAD
Фильтрация вариантов
[00598] Мы загрузили файл Sites VCF, релиз 0.3 (60 706 экзомов) из браузера ЕхАС (Lek et al., 2016) и файл Sites VCF, релиз 2.0.1 (15496 полных геномов) из браузера gnomAD. Мы создали из них отфильтрованный список вариантов, чтобы оценить SpliceNet-10k. В частности, рассматривались варианты, удовлетворяющие следующим критериям:
Поле FILTER (фильтр) - PASS (пройдено).
Вариант представлял собой однонуклеотидный вариант, и был только один альтернативный нуклеотид.
Поле AN (общее количество аллелей в названных генотипах) имело значение по меньшей мере 10000.
Вариант находился между сайтом начала и конца транскрипции канонического транскрипта GENCODE.
[00599] Всего через эти фильтры в наборах данных ЕхАС и gnomAD прошли 7 615 051 и 73 099 995 вариантов соответственно.
Доля вредоносных вариантов
[00600] Для этого анализа мы учитывали только те варианты в отфильтрованных списках ЕхАС и gnomAD, которые были одиночными или общими (частота аллелей (AF)≥0,1%) в когорте. Мы разделили эти варианты на подклассы на основе их геномного положения в соответствии с каноническими аннотациями GENCODE:
Экзонные: эта группа состоит из синонимичных вариантов ЕхАС (676 594 одиночных и 66 524 общих). Миссенс-варианты здесь не рассматривались, чтобы гарантировать, что большая часть вредоносных вариантов в этой группе была вызвана изменениями сплайсинга.
Почти интронные: эта группа состоит из интронных вариантов ЕхАС, которые находятся на расстоянии от 3 до 50 нуклеотидов от границы канонического экзона. Точнее, для анализа вариантов приобретения / потери акцептора и приобретения / потери донора учитывались только те варианты, которые находились на расстоянии 3-50 нуклеотидов от акцептора сплайсинга и донора соответственно (575 636 одиночных и 48 362 общих для приобретений / потерь акцепторов, 567 774 одиночных и 50 614 общих приобретений / потерь доноров).
Глубокий интронные: эта группа состоит из интронных вариантов gnomAD, которые удалены более чем на 50 нуклеотидов от канонической границы экзона (34 150 431 одиночный и 8 215 361 общий).
[00601] Для каждого варианта мы рассчитали его Δ-оценки для четырех типов сплайсинга с помощью SpliceNet-10k. Затем для каждого типа сплайсинга мы построили таблицу сопряженности хи-квадрат 2×2, в которой две строки соответствовали прогнозируемым вариантам с изменением сварки (Δ-Оценка в соответствующем диапазоне для данного типа сплайсинга) по сравнению с предсказанными вариантами без изменения сплайсинга (Δ-Оценка<0,1 для всех типов сплайсинга), и два столбца соответствуют одноэлементным и общим вариантам. Для вариантов приобретения сплайсинга мы отфильтровали те, которые встречаются на уже существующих сайтах сплайсинга, аннотированных GENCODE. Аналогично, для вариантов с потерей границы мы рассматривали только те, которые снижают оценку существующих сайтов сплайсинга, аннотированных GENCODE. Было рассчитано отношение шансов, и доля вредных вариантов была оценена как
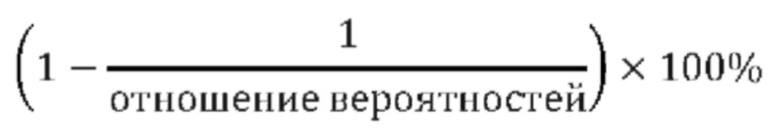
Варианты, укорачивающие белок, в списках фильтров ЕхАС и gnomAD, идентифицировали следующим образом:
Нонсенс: последствием VEP (McLaren et al., 2016) был «stop_gained» (приобретение точки остановаи) (44 046 одиночных и 722 общих в ЕхАС, 20 660 одиночных и 970 общих в gnomAD).
Сдвиг рамки: следствием VEP было «frameshift_variant» (вариант сдвига раски). Критерий однонуклеотидного варианта во время фильтрации вариантов был ослаблен, чтобы создать эту группу (48 265 одиночных и 896 общих в ЕхАС, 30 342 одиночных и 1472 общих в gnomAD).
Потеря существенного акцептора / донора: вариант был в первых или последних двух положениях канонического интрона (29 240 одиночных и 481 общих в ЕхАС, 12 387 одиночных и 746 общих в gnomAD).
[00602] Таблица сопряженности хи-квадрат 2×2 для вариантов, укорачивающих белок, была построена для фильтрованных списков ЕхАС и gnomAD и использовалась для оценки доли вредоносных вариантов. Здесь две строки соответствуют вариантам, укорачивающим белок, и синонимичным вариантам, а два столбца, как и раньше, соответствуют одиночным и общим вариантам.
[00603] Результаты для вариантов ЕхАС (экзонный и почти интронный) и gnomAD (глубокий интронный) представлены в ФИГ. 40 В и 40D соответственно.
Сдвиг рамки и приобретение сплайсинга в рамке
[00604] Для этого анализа мы сосредоточили наше внимание на вариантах ЕхАС, которые были экзонными (только синонимы) или близкими к интронным, и были одиночными или общими (AF≥0,1%) в когорте. Чтобы классифицировать вариант приобретения акцептора как внутрирамочный или со сдвигом рамки, мы измерили расстояние между каноническим акцептором сплайсинга и вновь созданным акцептором сплайсинга и проверили, кратно ли оно 3 или нет. Мы классифицировали варианты приобретения донора аналогичным образом, измерив расстояние между каноническим донором сплайсинга и вновь образованным донором сплайсинга.
[00605] Доля вредоносных вариантов приобретения сплайсинга с сохранением рамки оценивалась по таблице сопряженности хи-квадрат 2×2, где две строки соответствовали предсказанным вариантам приобретения сплайсинга с сохранением рамки (Δ-Оценка≥0,8 для приобретения акцептора или донора) по сравнению предсказанными вариантами, не изменяющими сплайсинг (Δ-Оценка<0,1 для всех типов сплайсинга), и два столбца соответствуют одноэлементным и общим вариантам. Эту процедуру повторяли для вариантов приобретения сплайсинга со сдвигом рамки путем замены первой строки в таблице сопряжения предсказанными вариантами приобретения сплайсинга со сдвигом рамки.
[00606] Для расчета р-значения, указанго на ФИГ. 40С, мы построили таблицу сопряжения хи-квадрат 2×2, используя только предсказанные варианты приобретения сплайсинга. Здесь две строки соответствовали вариантам усиления сплайсинга с сохранением рамки и сдвигом рамки, а два столбца, как и раньше, соответствовали одиночным и общим вариантам. Количество вариантов криптического сплайсинга на человека
[00607] Чтобы оценить количество редких функциональных вариантов криптического сплайсинга на индивидуума (ФИГ. 40Е), мы сначала смоделировали 100 индивидуумов gnomAD, включив каждый вариант gnomAD в каждый аллель с вероятностью, равной частоте его аллеля. Другими словами, каждый вариант отбирался дважды независимо для каждого индивидуума для имитации диплоидии. Мы подсчитали количество редких (AF<0,1%) экзонных (только синонимов), близких к интронным и глубоких интронных вариантов на человека, у которых оценка Δ-оценка была выше или равна 0,2,0,2 и 0,5, соответственно. Это относительно допустимые пороги "1-оценки, которые оптимизируют чувствительность, гарантируя, что по меньшей мере 40% предсказанных вариантов являются вредоносными. При этих порогах мы получили в среднем 7,92 синонимичных / почти интронных и 3,03 глубоких интронных редких критических варианта сплайсинга на человека. Поскольку не все эти варианты являются функциональными, мы умножили количество вариантов на долю вариантов, которые являются вредоносными при этих порогах.
IV. Анализ наборов данных DDD и ASD
De novo мутации криптического сплайсинга
[00608] Мы получили опубликованные de novo мутации (DNM). Среди них 3953 пробанда с расстройством аутистического спектра (Dong et al., 2014; Iossifov et al., 2014; De Rubeis et al., 2014), 4293 пробанда из когорты Deciphering Developmental Disorders (McRae et al., 2017) и 2073 человека, здоровые контроля (Iossifov et al.,2014). DNM низкого качества были исключены из анализа (ASD и здоровые контроля: Confidence (доверительность)=lowConf, DDD: РР (DNM)<0,00781, (McRae et al., 2017)). DNM оценивали с помощью сети, и мы использовали Δ-оценки (см. Методы выше) для классификации мутаций криптического сплайсинга в зависимости от контекста. Мы рассматривали только мутации, аннотированные VEP-последствиями synonymous_variant, splice_region_variant, intron_variant, 5_prime_UTR_variant, 3_prime_UTR_variant или missense_variant. Для ФИГ использовались сайты с Δ-оценками >0,1. 41А, 41В, 41С, 41D, 41Е, 41F и ФИГ. 50А и 50Б, а также сайты с Δ баллами >0,2 для ФИГ. 49А, 49В и 49С.
[00609] На ФИГ. 20, 21, 22, 23 и 24 показано подробное описание архитектур SpliceNet-80nt, SpliceNet-400nt, SpliceNet-2k и SpliceNet-10k. Четыре архитектуры используют фланкирующую нуклеотидную последовательность длиной 40, 200, 1000 и 5000 соответственно с каждой стороны интересующей позиции в качестве входных данных и выводят вероятность того, что положение является акцептором сплайсинга, донором сплайсинга и ни тем, ни другим. Архитектуры в основном состоят из сверточных слоев Conv (N, W, D), где N, W и D - количество сверточных ядер, размер окна и скорость расширения каждого сверточного ядра в слое соответственно.
[00610] Кривые отчность-отзыв различных алгоритмов предсказания сплайсинга при оценке на дпнРНК. На ФИГ. 42В показан полный транскрипт пре-мРНК для гена LINC00467, оцененный с помощью MaxEntScan и SpliceNet-10k, вместе с предсказанными акцепторными (красные стрелки) и донорными (зеленые стрелки) сайтами и фактическими положениями экзонов.
[00611] На ФИГ. 43А и 43В показаны зависящие от положения эффекты мотивов точки ветвления ТАСТААС и энхансера сплайсинга экзонов GAAGAA. Что касается ФИГ. 43А, оптимальная последовательность точки ветвления ТАСТААС была введена на различных расстояниях от каждого из 14 289 тестовых наборов акцепторов сплайсинга, и оценки акцепторов были рассчитаны с использованием SpliceNet-10k. Среднее изменение предсказанной оценки акцептора отображается как функция расстояния от акцептора сплайсинга. Предсказанные оценки увеличиваются, когда расстояние от акцептора сплайсинга составляет от 20 до 45 нуклеотидов; на расстоянии менее 20 нуклеотидов ТАСТААС разрушает полипиримидиновый тракт, что обуславливает очень низкие предсказанные оценки акцепторов.
[00612] Что касается ФИГ. 43В, гексамерный мотив SR-белка GAAGAA был аналогичным образом ввеаналогичным образом вводили ден на различных расстояниях от каждого из 14 289 акцепторов и доноров сплайсинга в тестовом наборе. Среднее изменение предсказанных оценок акцептора и донора SpliceNet-10k наносили на график как функцию расстояния от акцептора и донора сплайсинга соответственно. Предсказанные оценки увеличиваются, когда мотив находится на экзонной стороне и менее чем на расстоянии ~ 50 нуклеотидов от сайта сплайсинга. На больших расстояниях в экзон мотив GAAGAA имеет тенденцию препятствовать использованию рассматриваемого акцепторного или донорного сайта сплайсинга, по-видимому, потому что теперь он предпочтительно поддерживает более проксимальный акцепторный или донорный мотив. Очень низкие оценки акцептора и донора, когда GAAGAA располагается в положениях, очень близких к интрону, обусловлены разрушением удлиненных акцепторных или донорных мотивов сплайсинга.
[00613] На ФИГ. 44А и 44В показаны эффекты позиционирования нуклеосом при сплайсинге. Что касается ФИГ. 44А, в 1 миллион случайно выбранных межгенных положений были введены сильные акцепторные и донорные мотивы, расположенные на расстоянии 150 нуклеотидов друг от друга, и вероятность включения экзона была рассчитана с использованием SpliceNet-10k Чтобы показать, что корреляция между предсказаниями SpliceNet-10k и позиционированием нуклеосом происходит независимо от состава GC, положения объединяли на основе содержания в них GC (рассчитанного с использованием 150 нуклеотидов между введенными сайтами сплайсинга) и корреляции Спирмена между предсказаниями SpliceNet-10k, и показывали на графике сигнал нуклеосомы для каждого интервала.
[00614] Что касается ФИГ. 44В, акцепторные и донорные сайты сплайсинга из тестового набора оценивали с использованием SpliceNet-80nt (называемого баллом локальных мотивов) и SpliceNet-10k, и оценки наносили на график как функцию обогащения нуклеосом. Обогащение нуклеосом рассчитывается как сигнал нуклеосомы, усредненный по 50 нуклеотидам на экзонной стороне сайта сплайсинга, деленный на сигнал нуклеосомы, усредненный по 50 нуклеотидам на интронной стороне сайта сплайсинга. Оценка SpliceNet-80nt, которая является суррогатом силы мотива, отрицательно коррелирует с обогащением нуклеосом, тогда как оценка SpliceNet-10k положительно коррелирует с обогащением нуклеосом. Это указывает на то, что позиционирование нуклеосом является дальнодействующей детерминантой специфичности, которая может компенсировать слабые локальные мотивы сплайсинга.
[00615] На ФИГ. 45 показан пример расчета параметров эффектов для нарушающего сплайсинг варианта при сложных эффектах. Интронный вариант chr9: 386429 А>G разрушает нормальный донорный сайт (С) и активирует ранее подавленный интронный нижележащий донор (D). Показаны покрытие РНК-последовательности и количество ридов границы в цельной крови индивидуума с вариантом и контрольного индивидуума. Донорные сайты у индивидуума с вариантом и контрольного индивидуума отмечены синими и серыми стрелками соответственно. Жирные красные буквы соответствуют конечным точкам границы. Для наглядности длина экзона была увеличена в 4 раза по сравнению с длиной интрона. Чтобы оценить величину эффекта, мы вычисляем как увеличение использования границы с пропуском экзона (АЕ), так и уменьшение использования нарушенной границами (СЕ) по сравнению со всеми другими границы с тем же донором Е. Окончательная величина эффекта является максимальным из двух значений (0,39). В мутированном образце также присутствует повышенное сохранение интронов. Эти переменные эффекты являются общими для событий пропуска экзонов и увеличивают сложность валидации редких вариантов, которые, как предполагается, вызывают потерю акцепторных или донорных сайтов.
[00616] ФИГ. 46А, 46В и 46С показывает оценку модели SpliceNet-10k на одиночных и общих вариантах. Что касается ФИГ. 46А, фракция критических мутаций сплайсинга, предсказанных SpliceNet-10k, которые валидированы на данных секвенирования РНК GTEx. Модель оценивали на всех вариантах, встречающихся не более чем у четырех индивируумов в когорте GTEx. Варианты с предсказанными эффектами изменения сплайсинга валидировали по данным секвенирования РНК. Уровень валидации показан отдельно для вариантов, появляющихся у одного индивидуума GTEx (слева), и вариантов, появляющихся у двух или четырех индивидуумов GTEx (справа). Предсказания сгруппированы по их Δ-оценкам. Мы сравнили уровень валидации между одиночными и общими вариантами для каждого из четырех классов вариантов (приобретения или потеря акцептора или донора) в каждой группе Δ-оценки. Различия незначимы (Р>0,05, точный критерий Фишера с поправкой Бонферрони для 16 тестов).
[00617] Что касается ФИГ. 46В, чувствительность SpliceNet-10k при обнаружении изменяющих соединение вариантов в когорте GTEx при различных пороговых значениях Δ-оценки. Чувствительность модели показана отдельно для одиночного (слева) и общего (справа) вариантов. Различия в чувствительности между одноэлементными и общими вариантами при граничном значении Δ-оценки, равном 0,2, не являются значимыми ни для вариантов рядом с экзонами, ни для вариантов с глубоким интроном (Р>0,05, точный тест Фишера с поправкой Бонферрони для двух тестов).
[00618] Что касается ФИГ. 46С, распределение значений Δ-оценки для проверенных одиночных и общих вариантов. Р-значения предназначены для U-тестов Манна-Уитни, в которых сравниваются оценки одиночных и общих вариантов. Общие варианты имеют значительно более слабые значения Δ-оценки из-за того, что естественный отбор отфильтровывает мутации, нарушающие сплайсинг, с большими эффектами.
[00619] На ФИГ. 47А и 47В показаны уровни валидации и блоков эффектов вариантов, создающих сайты сплайсинга, разбитые по расположению вариантов. Предсказанные варианты, создающие сайт сплайсинга, были сгруппированы в зависимости от того, создавал ли вариант новый существенный динуклеотид сплайсинга GT или AG, перекрывал ли он остальную часть мотива сплайсинга (все позиции вокруг границы экзон-интрон до 3 нуклеотидов в экзон и 8 нуклеотидов в интрон, исключая существенный динуклеотид), или находился ли он вне мотива сплайсинга.
[00620] Что касается ФИГ. 47А, уровень валидации для каждой из трех категорий вариантов, создающих сайта сплайсинга. Общее количество вариантов в каждой категории показано над столбиками. Внутри каждой группы Δ-оценки различия в уровнях валидации между тремя группами вариантов не являются значимыми (Р>0,3, критерий однородности χ2).
[00621] Что касается ФИГ. 47В, распределение величин эффекта для каждой из трех категорий вариантов, создающих сайта сплайсинга. Внутри каждой группы Δ-оценки различия в величине эффекта между тремя группами вариантов не значимы (Р>0,3, U-критерий Манна-Уитни с поправкой Бонферрони).
[00622] На ФИГ. 48А, 48В, 49С и 49D показана оценка модели SpliceNet-10k на тренировочных и тестовых хромосомах. Что касается ФИГ. 48А, фракция криптических мутаций сплайсинга, предсказанная моделью SpliceNet-10k, которая валидирована на данных секвенирования РНК GTEx. Уровень валидации показан отдельно для вариантов хромосом, используемых во время обучения (все хромосомы, кроме chr1, chr3, chr5, chr7 и chr9; слева) и остальных хромосом (справа). Прогнозы сгруппированы по их Δ-оценкам. Мы сравнили уровень валидации между обучающими и тестовыми хромосомами для каждого из четырех классов вариантов (приобретение или потеря акцептора или донора) в каждой группе Δ-оценки. Этим объясняются потенциальные различия в распределении предсказанных значений Δ-оценки между обучающими и тестовыми хромосомами. Различия в показателях валидации незначительны (Р>0,05, точный критерий Фишера с поправкой Бонферрони для 16 тестов).
[00623] Что касается ФИГ. 48В, чувствительность SpliceNet-lOk при обнаружении изменяющих сплайсинг вариантов в когорте GTEx при различных граничныз значениях Δ-оценки. Чувствительность модели показана отдельно для вариантов хромосом, используемых для обучения (слева) и для остальных хромосом (справа). Мы использовали точный тест Фишера, чтобы сравнить чувствительность модели при пороговом значении Δ-оценки 0,2 между обучающими и тестовыми хромосомами. Различия не значимы ни для вариантов около экзонов, ни для вариантов с глубоким интроном (Р>0,05 после поправки Бонферрони для двух тестов).
[00624] Что касается ФИГ. 48С, доля предсказанных синонимичных и интронных вариантов критического сплайсинга в наборе данных ЕхАС, которые являются вредоносными, рассчитывается отдельно для вариантов в хромосомах, используемых для обучения (слева), и для остальных хромосом (справа). Доли и Р-значения вычисляются, как показано на рисунке 4А. Мы сравнили количество общих и редких вариантов между обучающими и тестовыми хромосомами для каждого из четырех классов вариантов (приобретение или потеря акцептора или донора) в каждой группе Δ-оценки. Различия незначимы (Р>0,05, точный критерий Фишера с поправкой Бонферрони для 12 тестов).
[00625] Что касается ФИГ. 48D, предсказанные критические сплайсинговые мутации de novo (DNM) на человека для DDD, ASD и контрольных когорт, показаны отдельно для вариантов в используемых хромосомах.
[00626] для обучения (слева) и остальных хромосом (справа). Планки погрешностей показывают 95% доверительный интервал (ДИ). Количество вариантов критического сплайсинга de novo на человека меньше для тестового набора, потому что он составляет примерно половину размера обучающего набора. Цифры зашумлены из-за небольшого размера выборки.
[00627] ФИГ. 49А, 49В и 49С иллюстрируют криптические de novo сплайс-мутации у пациентов с редкими генетическими заболеваниями, только по сайтам синонимичных, интронных или нетранслируемых областей. Что касается ФИГ. 49А, предсказанные криптические криптические de novo мутации сплайсинга (DNM) с Δ-оценками критического сплайсинга >0,2 на человека для пациентов из когорты Deciphering Developmental Disorders (Расшифровки нарушений развития, DDD), людей с расстройствами аутического спектра (ASD) из Simons Simplex Collection и Консорциума по секвенированию аутизма, а также здоровых контролей. Показано обогащение когорт DDD и ASD относительно здоровых контрольных групп с поправкой на выявление вариантов между когортами. Планки погрешностей показывают 95% доверительный интервал.
[00628] Что касается ФИГ. 49В, расчетная доля патогенных DNM по функциональным категориям для когорт DDD и ASD, основанная на обогащении каждой категории по сравнению со здоровыми контролями. Пропорция скрытого сплайсинга скорректирована с учетом отсутствия миссенс- и более глубоких интронных сайтов.
[00629] Что касается ФИГ. 49С, обогащение и избыток криптических DNM сплайсинга в когортах DDD и ASD по сравнению со здоровыми контролями при различных пороговых значениях Δ-оценки. Избыток криптического сплайсинга корректируется с учетом отсутствия миссенс - и более глубоких интронных сайтов.
[00630] На ФИГ. 50А и 50В показаны новые критические сплайс-мутации при PAC(ASD) как долю всех мутаций de novo. Что касается ФИГ. 50А, обогащение и избыток криптических DNM сплайсинга у пробандов ASD при различных пороговых значениях Δ-оценки для предсказания криптических сайтов сплайсинга.
[00631] Что касается ФИГ. 50В, доля патогенных DNM, относящихся к криптическим сайтам сплайсинга, как фракция всех классов патогенных DNM (включая мутации, кодирующие белок), с использованием различных пороговых значений Δ-оценки для прогнозирования криптических сайтов сплайсинга. Более мягкие пороговые значения Δ-оценки увеличивают количество криптических сайтов сплайсинга, идентифицированных сверх ожидаемого фона, за счет более низкого отношения шансов.
[00632] ФИГ. 51А, 51В, 51С, 51D, 51Е, 51F, 51G, 5H, 51I и 51J показывают валидацию по РНК-последовательностям предсказанных новых криптических сплайс-мутаций у пациентов с РАС. Покрытие и количество границ сплайсинга экспрессии РНК из 36 предсказанных криптических сайтов сплайсинга, выбранных для экспериментальной проверки на данных секвенирования РНК. Для каждого образца покрытие данными секвенирования РНК и количество соединений для пораженного индивидуума показаны вверху, а контрольный индивидуум без мутации показан внизу. Графики сгруппированы по статусу валидации и типу аберрации сплайсинга.
[00633] ФИГ. 52А и 52В демонстрируют показатель валидации на РНК-последовательности для моделей, обученных на канонических транскриптах. Что касается ФИГ. 52А, мы обучили модель SpliceNet-10k, используя только соединения из канонических транскриптов GENCODE, и сравнили производительность этой модели с моделью, обученной как по каноническим границам, так и по границам сплайсинга, присутствующим по меньшей мере у пяти человек в когорте GTEx. Мы сравнили уровни валидации двух моделей для каждого из четырех классов вариантов (приобретение или потеря акцептора или донора) в каждой группе Δ-оценки. Различия в уровнях валидации между двумя моделями незначительны (Р>0,05, точный критерий Фишера с поправкой Бонферрони для 16 тестов).
[00634] Что касается ФИГ. 52В, чувствительность модели, которая была обучена на канонических границах, при определении изменяющих сплайсинг вариантов в когорте GTEx при различных граничных значениях Δ-оценки. Чувствительность этой модели в глубоких интронных областях ниже, чем у модели на рисунке 2 (Р<0,001, точный критерий Фишера с поправкой Бонферрони). Чувствительность около экзонов существенно не отличается.
[00635] ФИГ. 53А, 53В и 53С демонстрируют, что комплексное моделирование улучшает показатели работы SpliceNet-10k. Что касается ФИГ. 53А показаны top-k точности и площадь под кривыми точность-отзыв 5 отдельных моделей SpliceNet-10k. Модели имеют одинаковую архитектуру и были обучены с использованием одного и того же набора данных. Однако они отличаются друг от друга из-за различных случайных аспектов, задействованных в процессе обучения, таких как инициализация параметров, перетасовка данных и т.д.
[00636] Что касается ФИГ. 53В, предсказания 5 отдельных моделей SpliceNet-10k сильно коррелируют. Для этого исследования мы учитывали только те положения в тестовом образце, которым была присвоена оценка акцептора или донора больше или равная 0,01 по меньшей мере одной модели. Подграфик (i, j) строится путем построения графика прогнозов Модели №i относительно предсказаний Модели №j (соответствующая корреляция Пирсона отображается над подграфиком).
[00637] Что касается ФИГ. 53С, производительность улучшается по мере увеличения количества моделей, используемых для построения ансамбля SpliceNet-10k с 1 до 5.
[00638] ФИГ. 54А и 54В демонстрируют оценку SpliceNet-10k в области с различными плотностями экзонов. Что касается ФИГ. 54А, положения тестового набора были разделены на 5 групп в зависимости от количества канонических экзонов, присутствующих в окне из 10000 нуклеотидов. Для каждого интервала мы рассчитали точность top-k и площадь под кривой точность-отзыв для SpliceNet-10k.
[00639] Что касается ФИГ. 54В, мы повторили анализ с MaxEntScan для сравнения. Обратите внимание, что производительность обеих моделей улучшается при более высокой плотности экзонов, что количественно показывает точность top-k и площадь под кривой точность-отзыв, поскольку количество положительных тестовых примеров увеличивается по сравнению с количеством отрицательных тестовых примеров.
Увеличение количества мутаций de novo на когорту
[00640] Кандидатные критические DNM сплайсинга подсчитывали в каждой из трех когорт. Когорта DDD давала интронных DNM на расстоянии >8 нуклеотидов от экзонов, поэтому области >8 нуклеотидов от экзонов исключали из всех когорт для целей анализа обогащения, чтобы обеспечить эквивалентное сравнение между когортами DDD и ASD (ФИГ. 41А). Мы также выполнили отдельный анализ, который исключил мутации с двойным критическим сплайсингом и последствиями функции кодирования белка, чтобы продемонстрировать, что обогащение не связано с обогащением мутаций эффектами кодирования белка в затронутых когортах (ФИГ. 49А, 49В и 49С). Подсчеты масштабировали для дифференцирования подтверждения DNM между когортами путем нормализации количества синонимичных DNM на индивидуума между когортами, используя здоровую контрольную когорту в качестве фонового уровня. Мы сравнили уровень DNM криптического сплайсинга на когорту, используя Е-критерий, чтобы сравнить два Пуассоновских показателя (Krishnamoorthy and Thomson, 2004).
[00641] График показателей обогащения сверх ожидаемого (ФИГ. 41С) был скорректирован с учетом отсутствия DNM >8 нуклеотидов от экзонов путем масштабирования в сторону увеличения на долю всех DNM криптического сплайсинга, которые, как ожидается, возникнут на расстоянии 9-50 нуклеотидов от экзонов, с использованием контекстной модели тринуклеотидной последовательности (см. ниже, «Обогащение de novo мутаций на ген»). Доля криптических диагностических сайтов и избыток криптических сайтов (ФИГ. 49В и 49С) также были скорректированы с учетом отсутствия миссенс-сайтов путем масштабирования критического количества на долю криптических сайтов сплайсинга, которые, как ожидается, будут встречаться на миссенс-сайтах, по сравнению с синонимичными сайтами. Влияние порогового значения Δ-оценки на обогащение оценивали путем расчета обогащения DNM криптического сплайсинга в когорте DDD по диапазону граничных значений. Для каждого из них вычисляли отношение шансов наблюдаемое: ожидаемое, а также избыток DNM криптического сплайсинга.
Доля патогенных DNM
[00642] Избыток DNM по сравнению с фоновой частотой мутаций можно рассматривать как патогенный выход в когорте. Мы оценили избыток DNM по функциональному типу в когортах ASD и DDD на фоне здоровой контрольной когорты (ФИГ. 41Б). Подсчеты DNM были нормированы по количеству синонимичных DNM на человека, как описано выше. Подсчет DDD криптического сплайсинга был скорректирован с учетом отсутствия DNM на расстоянии 9-50 нуклеотидов от интронов, как описано выше. Для когорт как ASD, так и DDD мы также провели коррекцию по недостающему определению глубоких интронных вариантов на расстоянии >50 нт от экзонов, используя соотношение почти интронных (<50 н.) и глубоких интронных (>50 н.) вариантов криптического сплайсинга из анализа отрицательного отбора (ФИГ. 38Г).
Обогащение de novo мутаций на ген
[00643] Мы определили частоту нулевых мутаций для каждого варианта в геноме, используя контекстную модель тринуклеотидной последовательности (Samocha et al., 2014). Мы использовали сеть для прогнозирования Δ-оценки для всех возможных однонуклеотидных замен в экзонах и до 8 нуклеотидов в интроне. На основе модели частоты нулевых мутаций мы получили ожидаемое количество мутаций криптического сплайсинга de novo на ген (используя Δ-оценку>0,2 в качестве граничного значения).
[00644] В исследовании DDD (McRae et al., 2017) гены оценивали на предмет обогащения DNM по сравнению со случайностью в двух моделях: одна учитывала только DNM, укорачивающие белок (PTV), а другая - все DNM, изменяющие белок. (PTV, миссенс-мутации, пропуски и вставки с сохранением рамки). Для каждого гена мы выбрали наиболее значимую модель и скорректировали Р-значение для проверки нескольких гипотез. Эти тесты проводили один раз, когда мы не учитывали скрытые DNM сплайсинга или показатели криптического сплайсинга (тест по умолчанию, использованный в исходном исследовании DDD), и один раз, когда мы также подсчитывали скрытые DNM сплайсинга и частоту мутаций для них. Мы вычвили дополнительные кандидатные гены, которые были идентифицированы как гены со скорректированным по FDR Р-значением<0,01 при включении DNM криптического сплайсинга, со скорректированным по FDR Р-значением>0,01, в случае, когда не включали DNM критического сплайсинга (тест по умолчанию). Аналогичные тесты обогащения были проведены для когорты ASD.
Валидации предсказанных криптических сайтов слпайсинга
[00645] Мы отобрали de novo мутации с высокой степенью достоверности из затронутых пробандов в коллекции Simons Simplex, с экспрессией RNA-seq с по меньшей мере RPKM>1 в линиях лимфобластоидных клеток. Мы выбрали de novo варианты критического сплайсинга для валидации на основе порогового значения Δ-оценка>0,1 для вариантов потерь при сварке и порогового значения Δ-оценки>0,5 для вариантов приобретения сплайсинга. Поскольку клеточные линии необходимо было закупить заранее, эти пороговые значения отражают более раннюю итерацию наших методов по сравнению с пороговыми значениями, которые мы использовали в других местах статьи (ФИГ. 38G и ФИГ. 41А, 41В, 41С и 41D), и сеть не включала новые границы сплайсинга GTEx для обучения модели.
[00646] Линии лимфобластоидных клеток были получены из SSC для этих пробандов. Клетки культивировали в культуральной среде (RPMI 1640, 2 мМ L-глутамин, 15% фетальная бычья сыворотка) до максимальной плотности клеток 1×106 клеток / мл. Когда клетки достигли максимальной плотности, их пассировали путем диссоциации клеток путем пипетирования вверх и вниз 4 или 5 раз и посева до плотности 200000-500000 жизнеспособных клеток / мл. Клетки выращивали при 37°С, 5% CO2 в течение 10 дней. Затем отделяли приблизительно 5×105 клеток и центрифугировали при 300×g в течение 5 минут при 4°С. РНК экстрагировали с помощью набора RNeasy® Plus Micro Kit (QIAGEN) в соответствии с протоколом производителя. Качество РНК оценивали с помощью набора Agilent RNA 6000 Nano Kit (Agilent Technologies) и запускали на Bioanalyzer 2100 (Agilent Technologies). Библиотеки PHK-seq были созданы с помощью набора для подготовки библиотеки TruSeq® Stranded Total RNA Library с набором A Ribo-Zero Gold (Illumina). Библиотеки секвенировали на приборах HiSeq 4000 в Центре передовых технологий (UCSF) с использованием 150-нуклеотидного секвенирования за одно считывание с охватом 270-388 миллионов ридов (в среднем 358 миллионов ридов).
[00647] Риды секвенирования для каждого пациента выравнивали при помощи OLego (Wu et al., 2013) относительно референса, созданного из hg19, путем замены de novo вариантов пациента (Iossifov et al., 2014) на соответствующий альтернативный аллель. Покрытие секвенированием, использование границ сплайсинга и положения транскриптов были нанесены на график типа «сашими» из MISO (Katz et al., 2010). Мы оценили предсказанные криптические сайты сплайсинга, как описано выше в разделе валидации предсказаний модели. Было подтверждено 13 новых сайтов сплайсинга (9 новых границ, 4 пропуска экзонов), поскольку они наблюдались только в образце, содержащем криптическиц сайт сплайсинга, и не наблюдались ни в одном из 149 образцов GTEx или в других 35 секвенированных образцах. Для 4 дополнительных событий пропуска экзонов в GTEx часто наблюдались низкие уровни пропуска экзонов. В этих случаях мы вычислили долю ридов, в которых использовалась пропускающая граница, и подтвердили, что эта доля была самой высокой в образце, содержащим критический сайт сплайсинга, по сравнению с другими образцами. 4 дополнительных случая были подтверждены на основе заметного сохранения интронов, которое отсутствовало или было намного ниже в других образцах. Умеренное сохранение нитронов в контрольных образцах не позволило нам разрешить события в DDX11 и WDR4. Два события (в CSAD и GSAP) были классифицированы как не прошедшие валидацию, потому что вариант не присутствовал в ридах последовательности.
ДОСТУПНОСТЬ ДАННЫХ И ПРОГРАММНОГО ОБЕСПЕЧЕНИЯ
[00648] Тренировочные (обучающие) и исследуемые данные, оценки предсказаний для всех однонуклеотидных замен в референсном геноме, результаты валидации на данных секвенирования РНК, границы сплайсинга RNA-seq и исходный код размещены для общественного доступа на:
[0001] https://basespace.illumina.com/s/5u6ThOblecrh
[00649] Данные RNA-seq для 36 линий лимфобластоидных клеток, депонируются в базе данных ArrayExpress на EMBL-EBI (www.ebi.ac.uk/arrayexpress) под номером доступа Е-МТАВ-хххх.
[00650] Оценки предсказаний и исходный код сделаны общедоступными в соответствии с модифицированной лицензией Apache License v2.0, и их можно бесплатно использовать для академических и некоммерческих программных приложений. Для снижения проблемы с зацикливанием, которая вызывает беспокойства в данной области, авторы в явном виде требуют, что оценки предсказаний, полученные этим способом, не должны включаться в качестве компонентов в другие классификаторы; вместо этого авторы просят заинтересованные стороны применять предоставленный исходный код и данные для прямого обучения и улучшения собственных моделей глубокого обучения.


НАЗВАНИЯ ДОПОЛНИТЕЛЬНЫХ ТАБЛИЦ
[00651] В таблице S1 показаны образцы GTEx, использованные для демонстрации вычислений величины эффекта и тканеспецифических эффектов в отношении сплайсинга на ФИГ. 38А, 38В, 38С, 38D, 38Е, 38F, и 38G, ФИГ. 39А, ФИГ. 39В и ФИГ. 45
[00652] В таблице S2 соответствующие граничные значения достоверности для SpliceNet-10k, GeneSplicer, MaxEntScan и NNSplice, при которых все алгоритмы предсказывают одно и то же число приобретений и потеть по геному. Связана с ФИГ. 38G.
[00653] В Таблице S3 показаны количестве предсказанных DNM криптического сплайсинга в каждой когорте. Связана с ФИГ. 41А, 41В, 41С, 41D, 41Е и 41, и представлена ниже:

[00654] В Таблице S4 показаны ожидаемые уровни de novo мутаций на ген для каждой категории мутаций. Связана с ФИГ. 41А, 41В, 41С, 41D, 41Е и 41F.
[00655] В Таблице S5 показаны р-значения для обогащения генов в DDD и ASD. Связана с ФИГ. 41А, 41В, 41С, 41D, 41Е и 41F.
[00656] В Таблице S6 показаны результаты валидации 36 предсказанных DNM критических сайтов сплайсинга у пациентов с аутизмом. Связана с ФИГ. 41А, 41В, 41С, 41D, 41Е и 41F.
Компьютерная система
[00657] ФИГ. 59 представляет собой упрощенную блок-схему компьютерной системы, которую можно применять для реализации раскрытой технологии. Компьютерная система обычно включает в себя по меньшей мере один процессор, который связывается с рядом периферийных устройств через подсистему шин. Эти периферийные устройства могут включать в себя подсистему хранения, включая, например, запоминающее устройство и подсистему хранения файлов, устройства ввода пользовательского интерфейса, устройства вывода пользовательского интерфейса и подсистему сетевого интерфейса. Устройства ввода и вывода позволяют пользователю взаимодействовать с компьютерной системой. Подсистема сетевого интерфейса обеспечивает интерфейс с внешними сетями, включая интерфейс с соответствующими интерфейсными устройствами в других компьютерных системах.
[00658] В одном варианте реализации нейронные сети, такие как ACNN и CNN, связаны с возможностью обмена данными с подсистемой хранения данных и устройствами ввода пользовательского интерфейса.
[00659] Устройства ввода пользовательского интерфейса могут включать клавиатуру; указывающие устройства, такие как мышь, трекбол, тачпад или графический планшет; сканер; сенсорный экран (тач-скрин), встроенный в дисплей; устройства звукового ввода, такие как системы распознавания голоса и микрофоны; и другие типы устройств ввода. В общем, использование термина «устройство ввода» предназначено для включения всех возможных типов устройств и способов ввода информации в компьютерную систему.
[00660] Устройства вывода пользовательского интерфейса могут включать подсистему дисплея, принтер, факсимильный аппарат или невизуальные дисплеи, такие как устройства звукового вывода. Подсистема дисплея может включать в себя электронно-лучевую трубку (ЭЛТ), устройство с плоской панелью, такое как жидкокристаллический дисплей (ЖКД), проекционное устройство или какой-либо другой механизм для создания видимого изображения. Подсистема дисплея может также обеспечивать невизуальный дисплей, такой как устройства звукового вывода. В общем, использование термина «устройство вывода» предназначено для включения всех возможных типов устройств и способов вывода информации из компьютерной системы пользователю, другой машине или компьютерной системе.
[00661] Подсистема хранения хранит программы и конструкции данных, которые обеспечивают функциональные возможности некоторых или всех модулей и методов, описанных в данном документе. Эти программные модули обычно исполняются одним процессором или в сочетании с другими процессорами.
[00662] Память, используемая в подсистеме хранения, может включать ряд запоминающих устройств, включая основную память с произвольным доступом (RAM) для хранения инструкций и данных во время выполнения программы, и постоянную память (ROM), в которой хранятся фиксированные инструкции. Подсистема хранения файлов может обеспечивать постоянное хранилище для файлов программ и данных и может включать жесткий диск, дисковод гибких дисков вместе со связанным съемным носителем, дисковод компакт-дисков, оптический дисковод или съемные картриджи. Модули, реализующие функциональные возможности определенных реализаций, могут храниться в подсистеме хранения файлов в подсистеме хранения или на других машинах, доступных процессору.
[00663] Подсистема шины обеспечивает механизм, позволяющий различным компонентам и подсистемам компьютерной системы определенным образом связываться друг с другом. Хотя подсистема шины схематично показана как одна шина, альтернативные реализации подсистемы шины могут использовать множество шин.
[00664] Сама компьютерная система может быть различных типов, включая персональный компьютер, портативный компьютер, рабочую станцию, компьютерный терминал, сетевой компьютер, телевизор, мэйнфрейм, серверную ферму, распределенный набор слабо связанных в сеть компьютеров или любую другую систему обработки данных или пользовательское устройство. В связи с изменчивой природой компьютеров и сетей описание компьютерной системы, изображенной на ФИГ. 59, приведено только в качестве конкретного примера с целью иллюстрации раскрытой технологии. Возможны многие другие конфигурации компьютерной системы, имеющие больше или меньше компонентов, чем компьютерная система, изображенная на ФИГ. 59.
[00665] Процессоры глубокого обучения могут быть графическими процессорами или FPGA и могут размещаться на облачных платформах глубокого обучения, таких как Google Cloud Platform, Xilinx и Cirrascale. Примеры процессоров глубокого обучения включают Tensor Processing Unit (TPU) от Google, стоечные решения, такие как серия GX4 Rackmount, серия GX8 Rackmount, NVIDIA DGX-1, Microsoft Stratix V FPGA, интеллектуальный процессор Graphcore (IPU), платформа Qualcomm Zeroth с процессорами Snapdragon, NVIDIA Volta, NVIDIA DRIVE PX, NVIDIA JETSON TX1 / TX2 MODULE, Nirvana от Intel, Movidius VPU, Fujitsu DPI, DynamicIQ от ARM IBM TrueNorth и другие.
[00666] Предшествующее описание приведено для того, чтобы сделать возможным создание и применение раскрытой технологии. Различные модификации раскрытых вариантов реализации будут очевидны, и общие принципы, определенные в данном документе, могут быть применены к другим вариантам реализации и приложениям без отступления от сущности и объема раскрытой технологии. Таким образом, не предполагается, что раскрытая технология ограничена показанными вариантами реализации, она должна соответствовать самому широкому объему, согласующемуся с принципами и признаками, раскрытыми в данном документе. Объем раскрытой технологии определяется прилагаемой формулой изобретения.
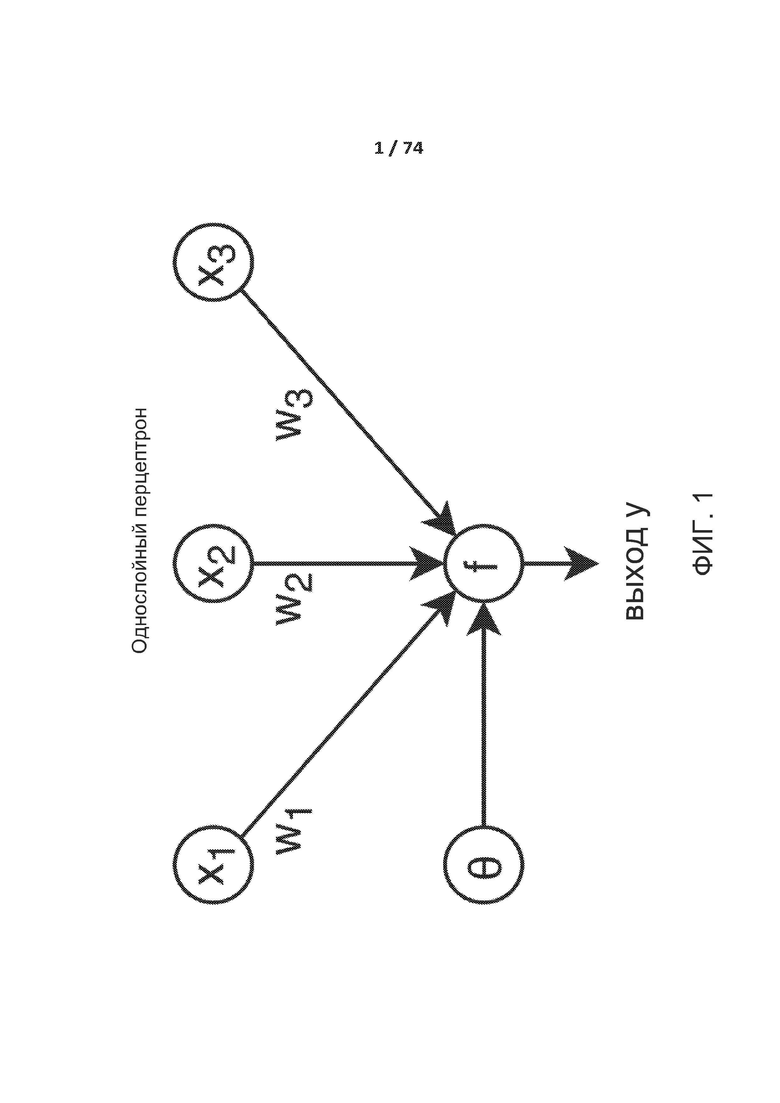
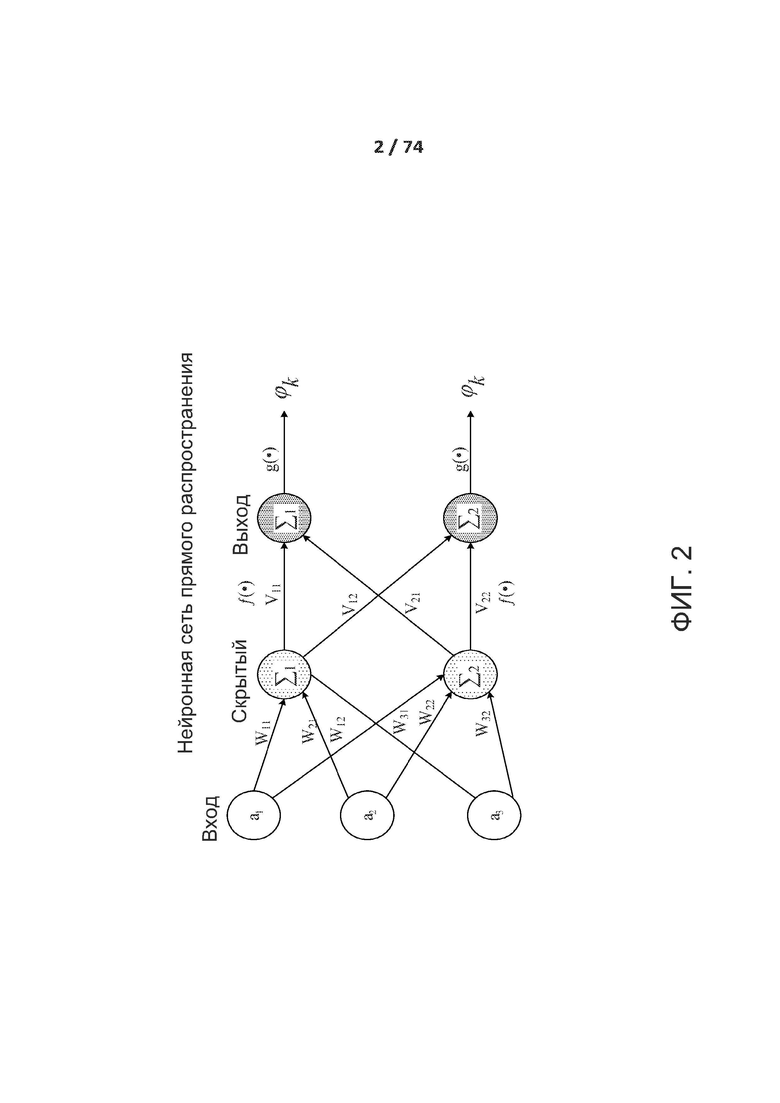
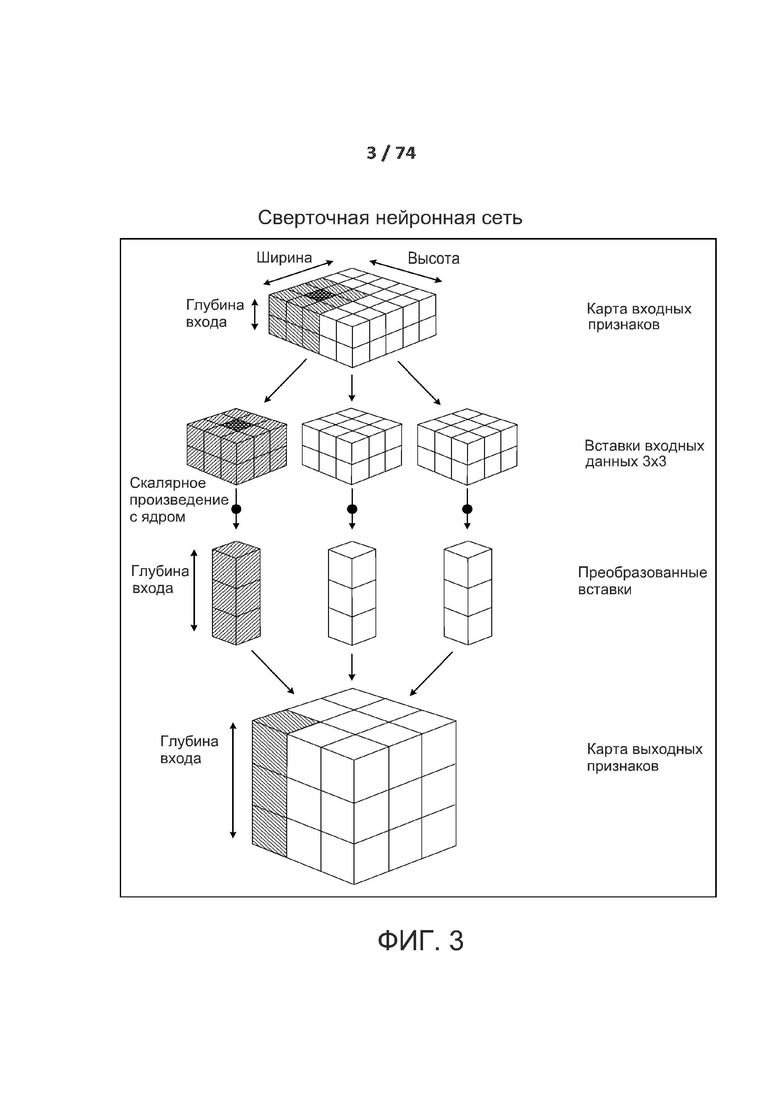
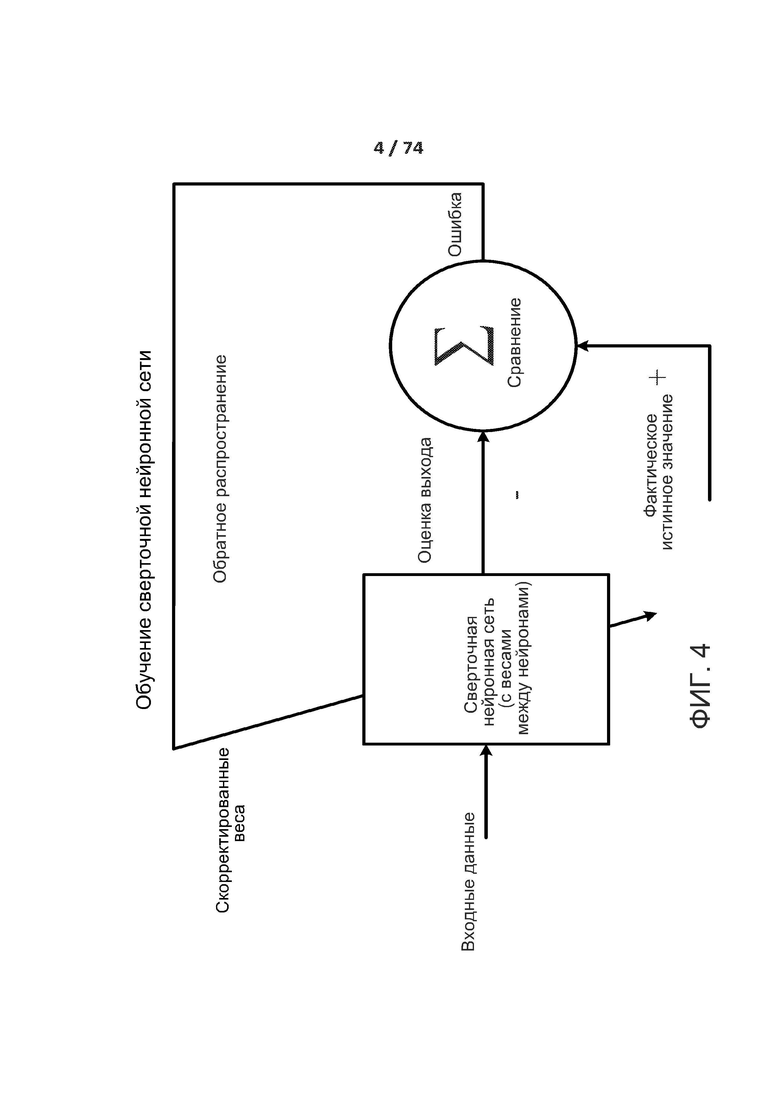
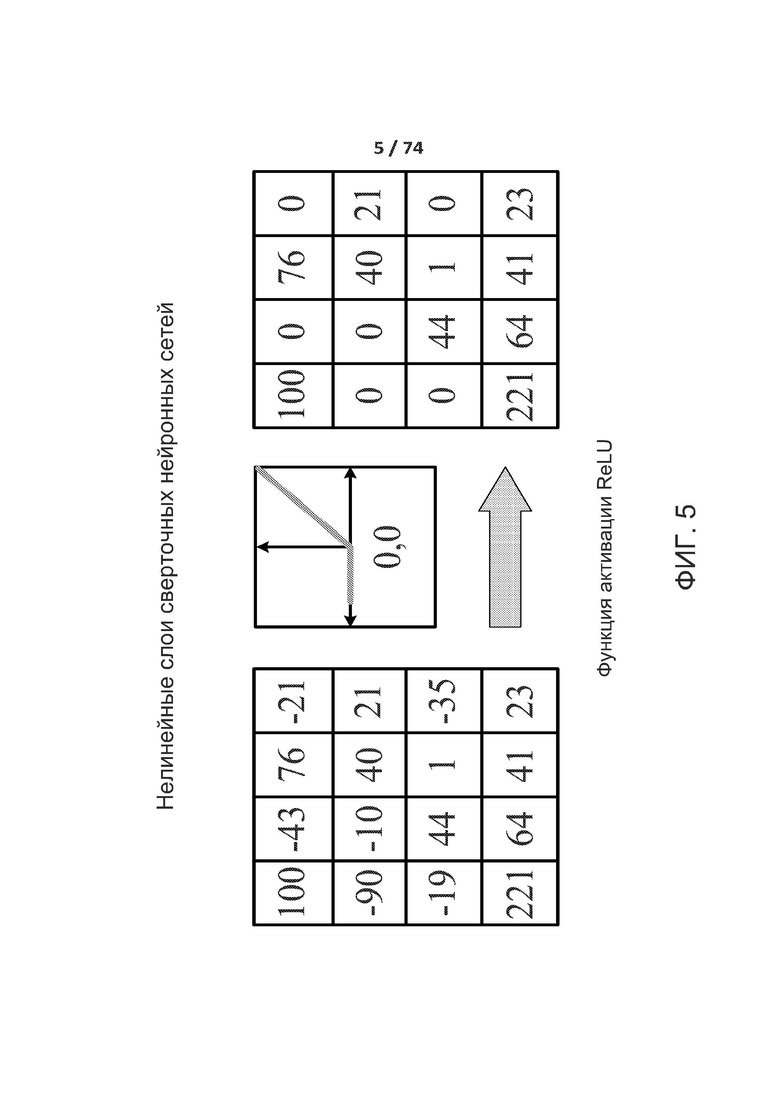
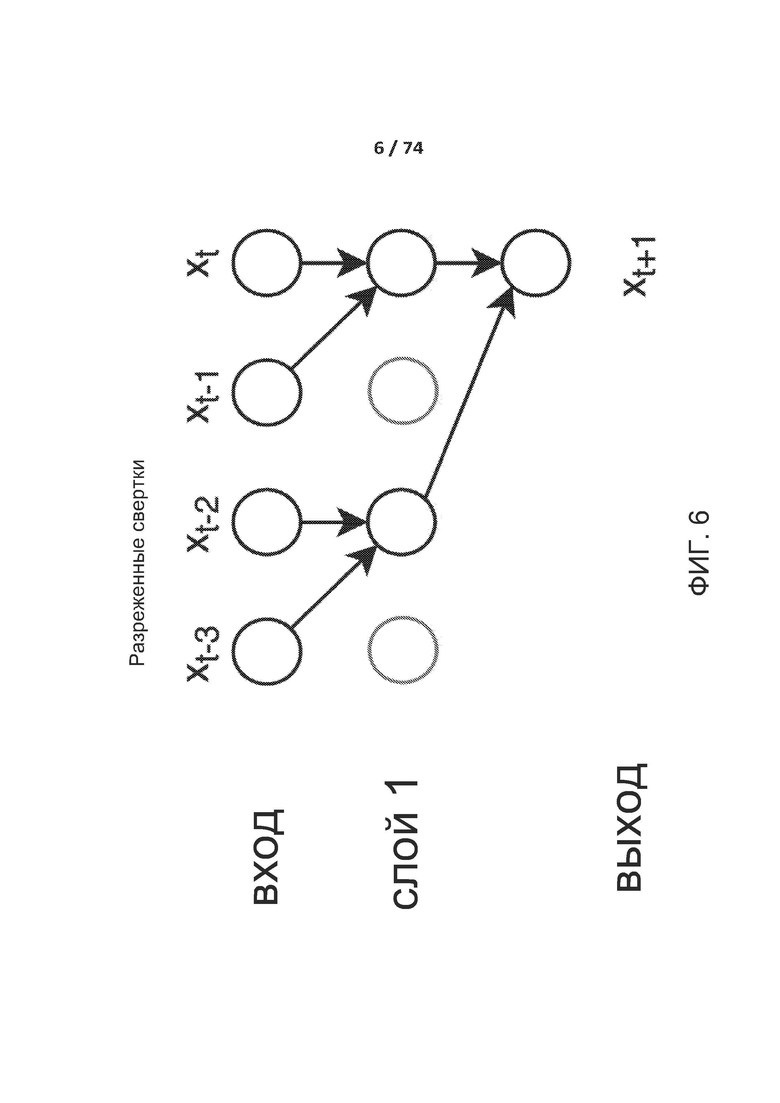
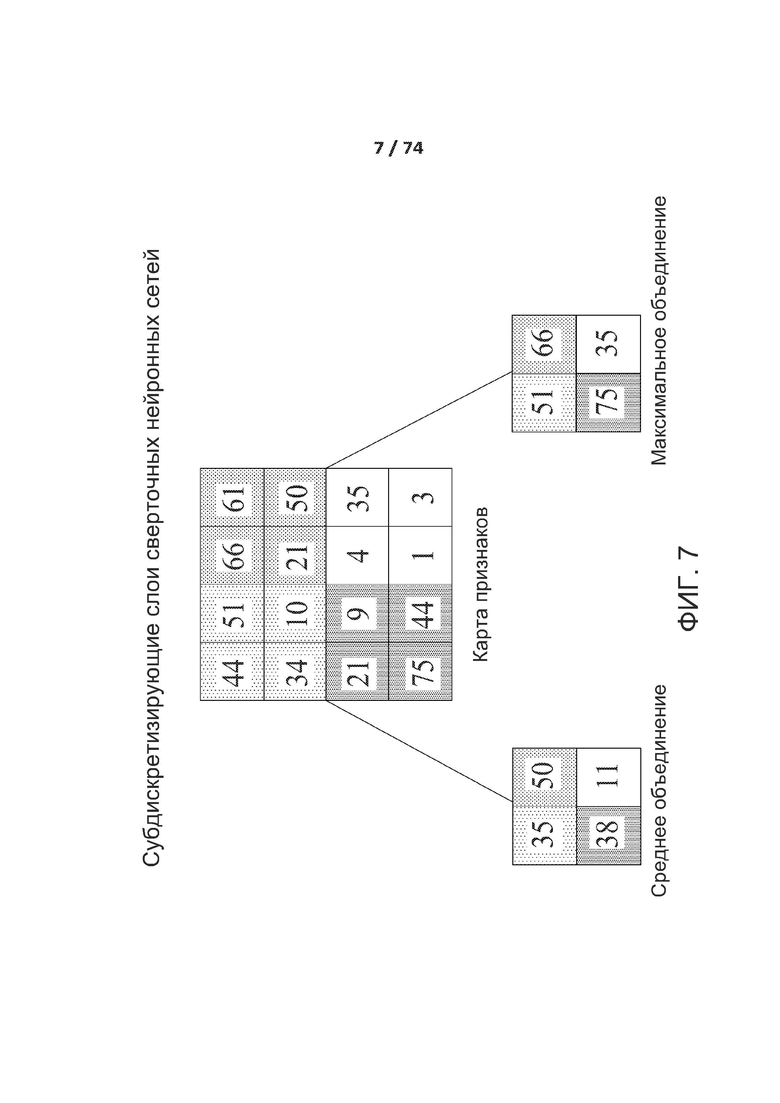
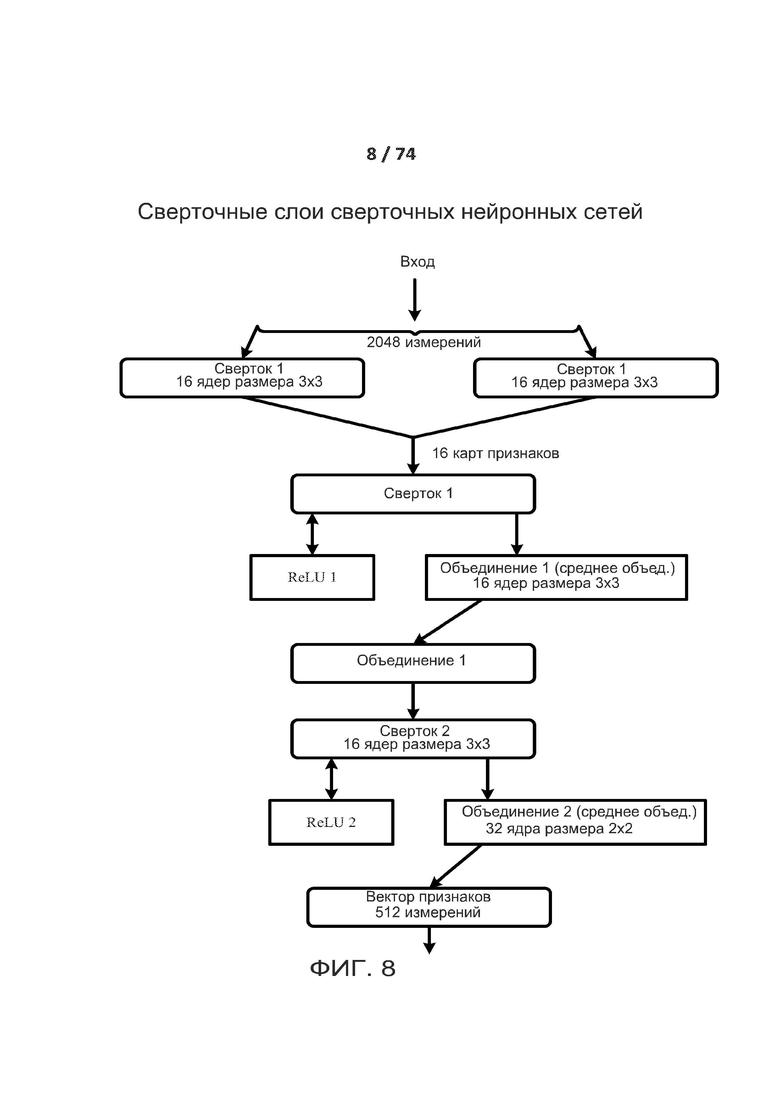
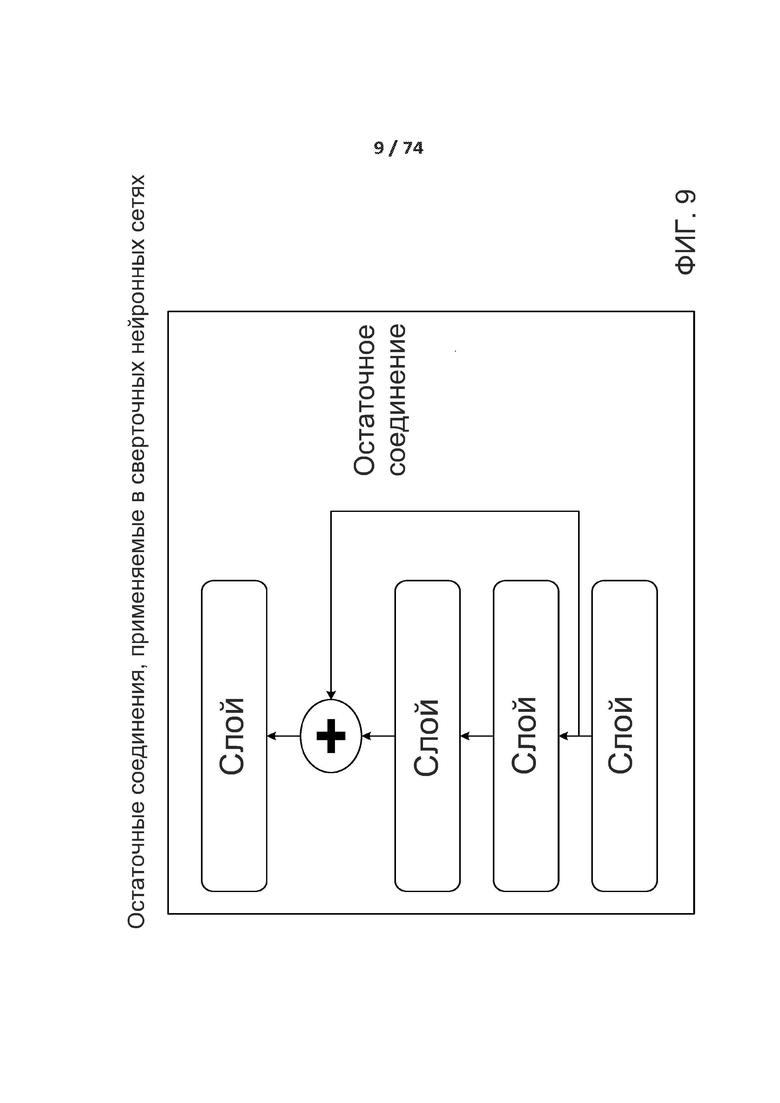
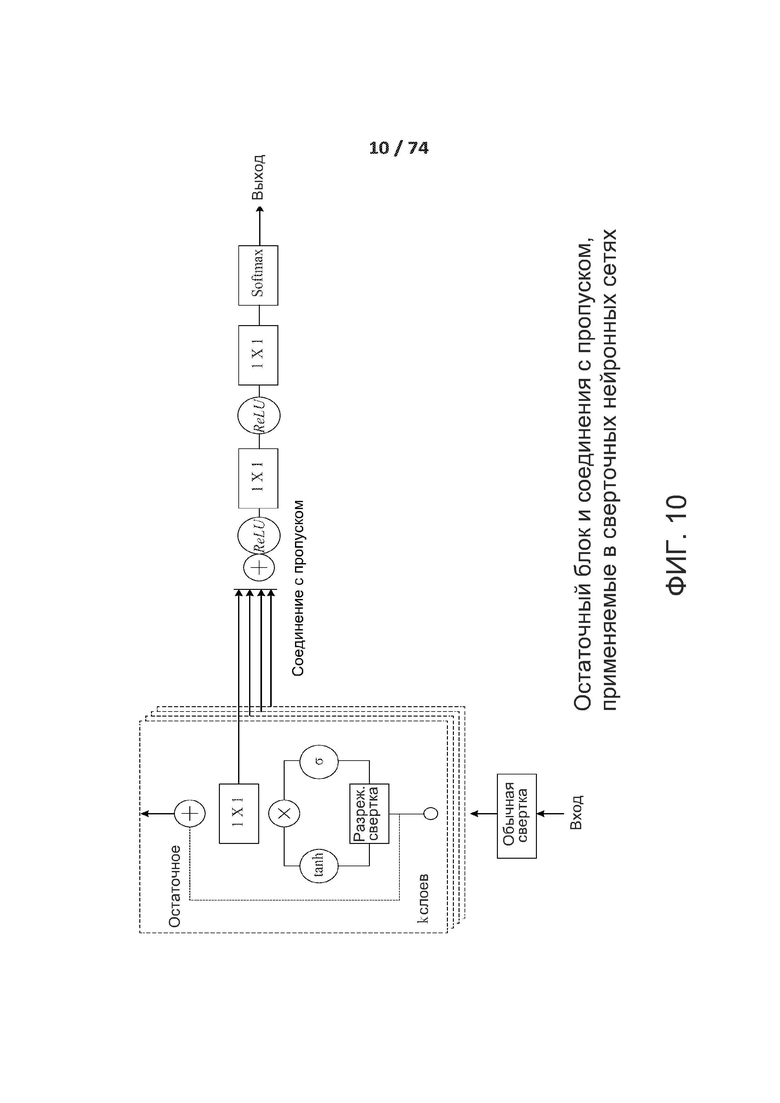
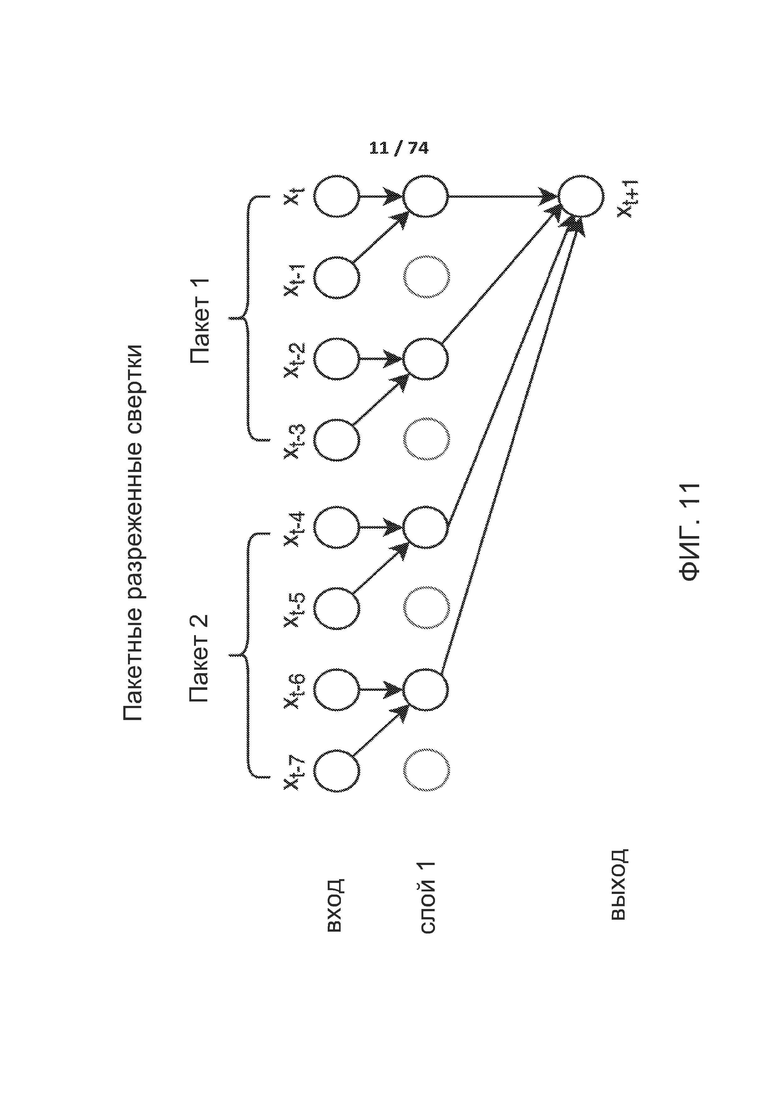
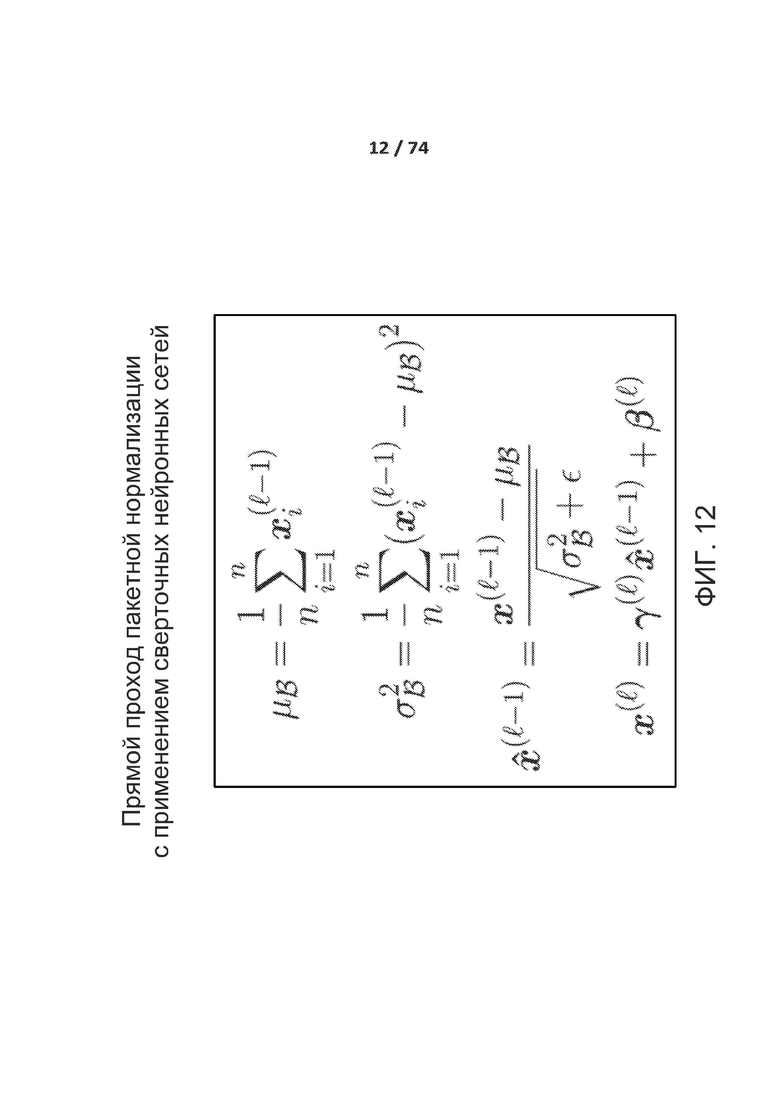

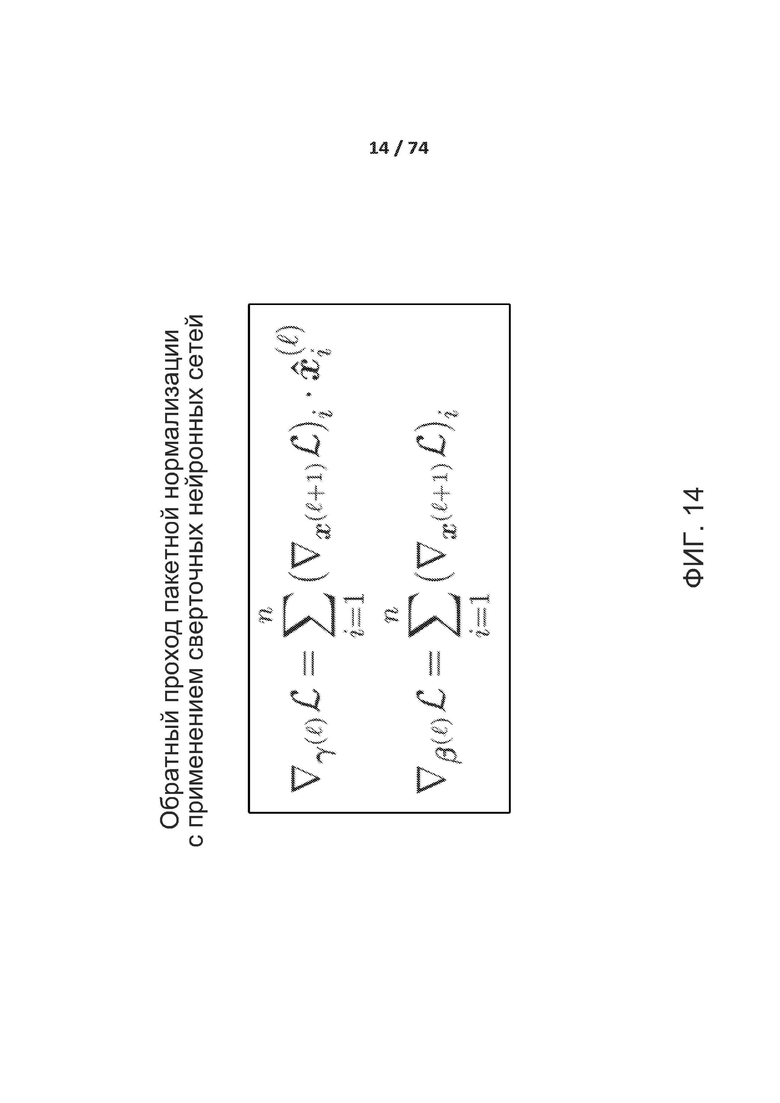
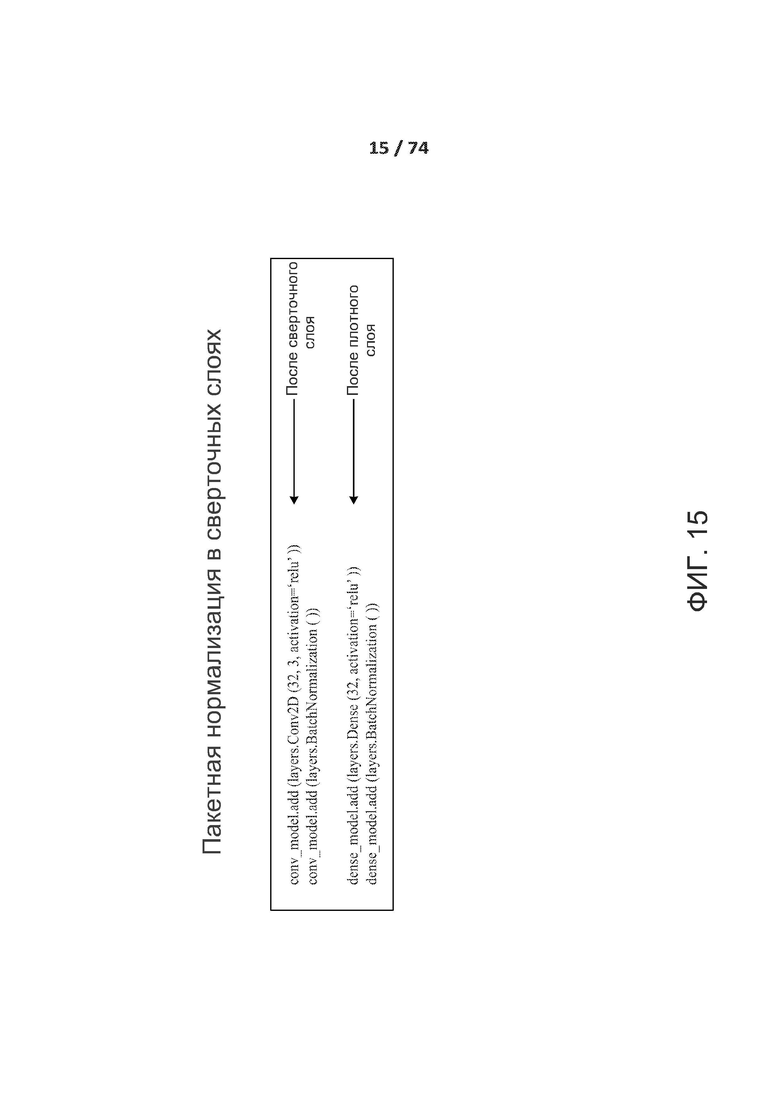
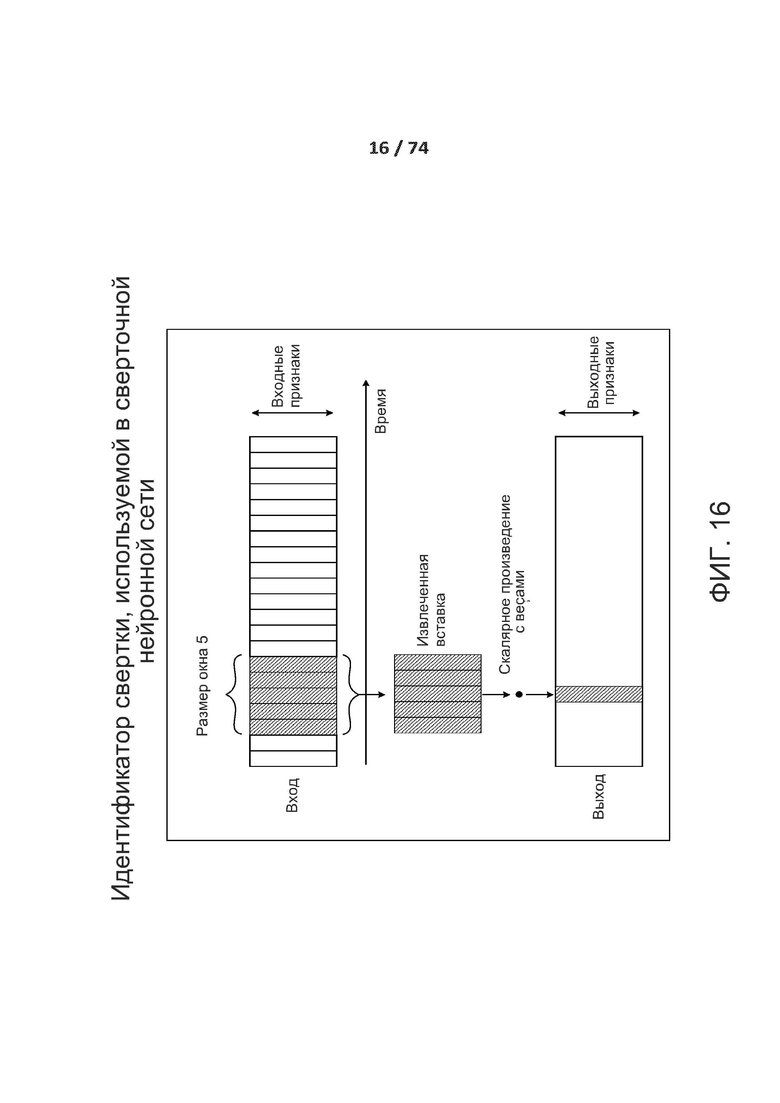
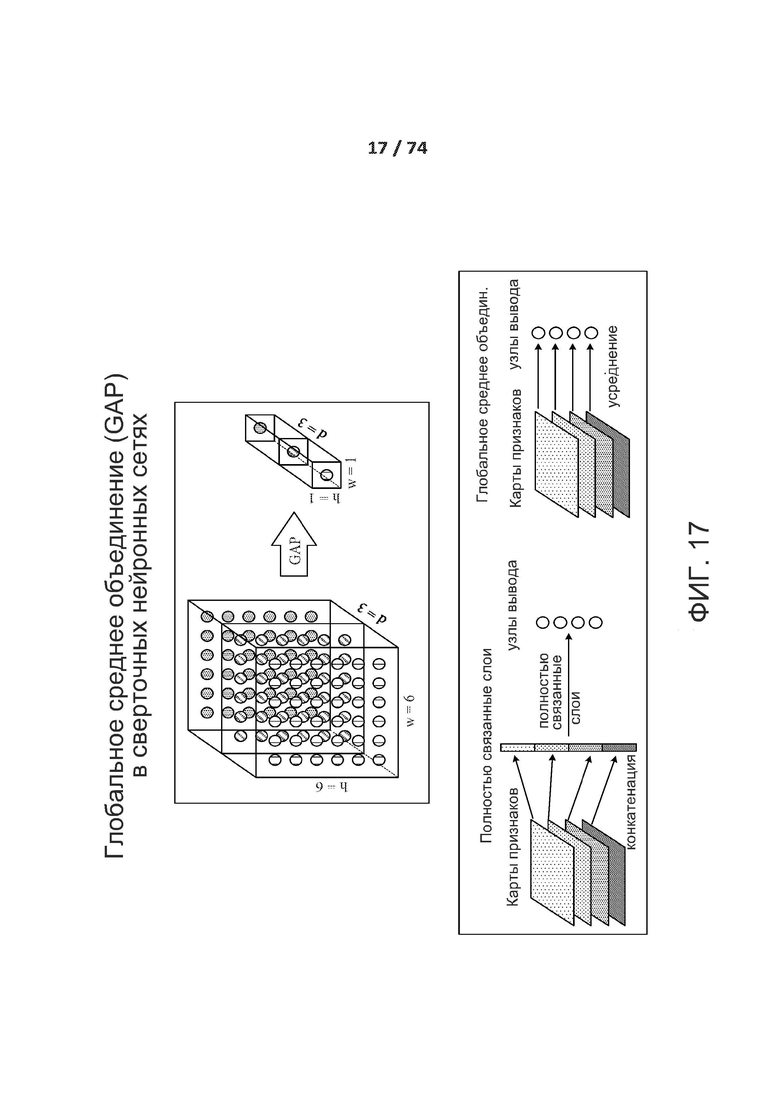
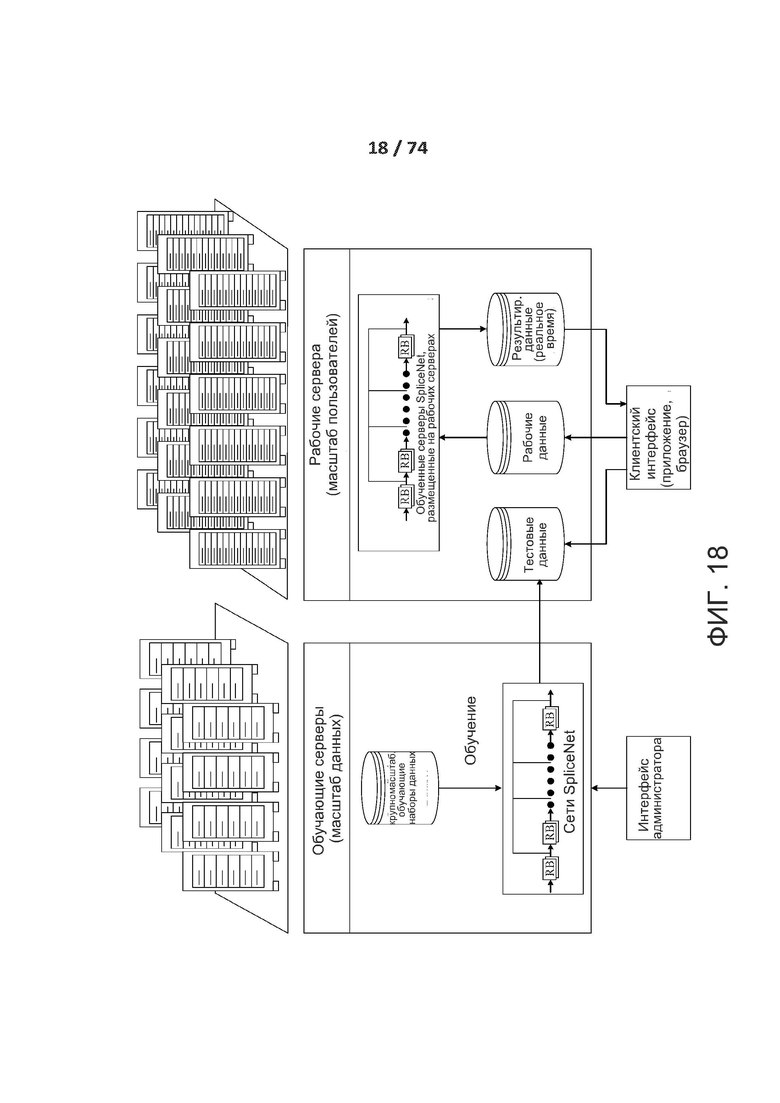
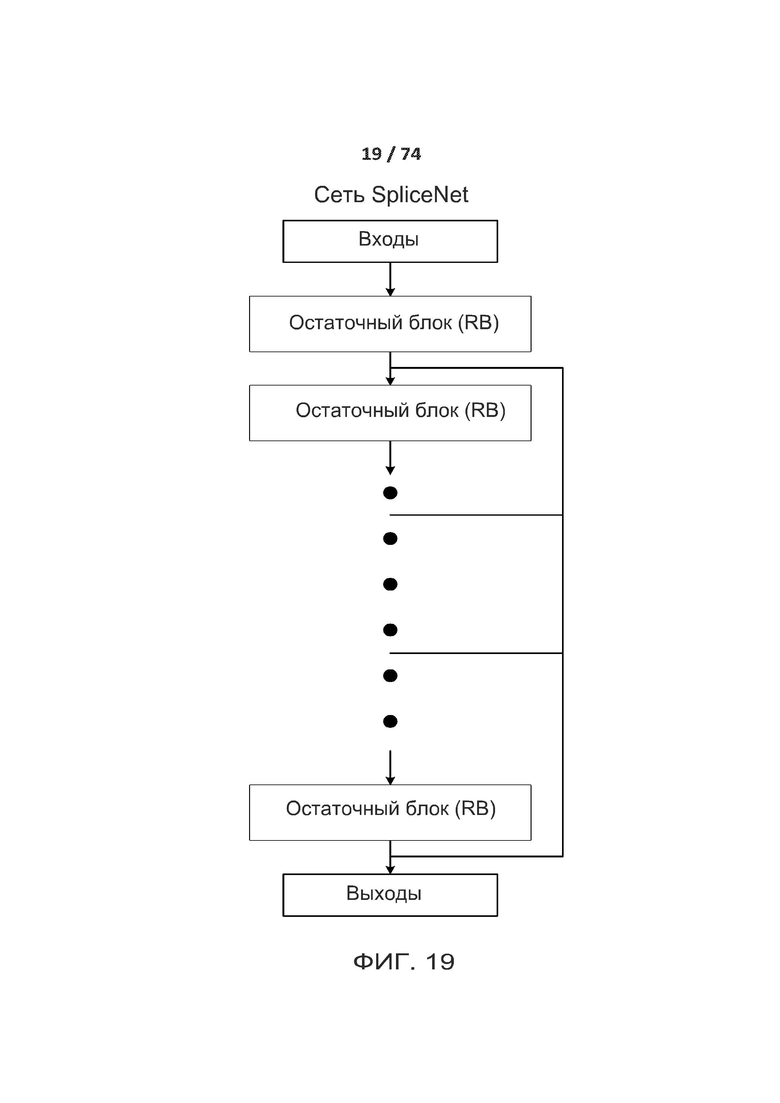
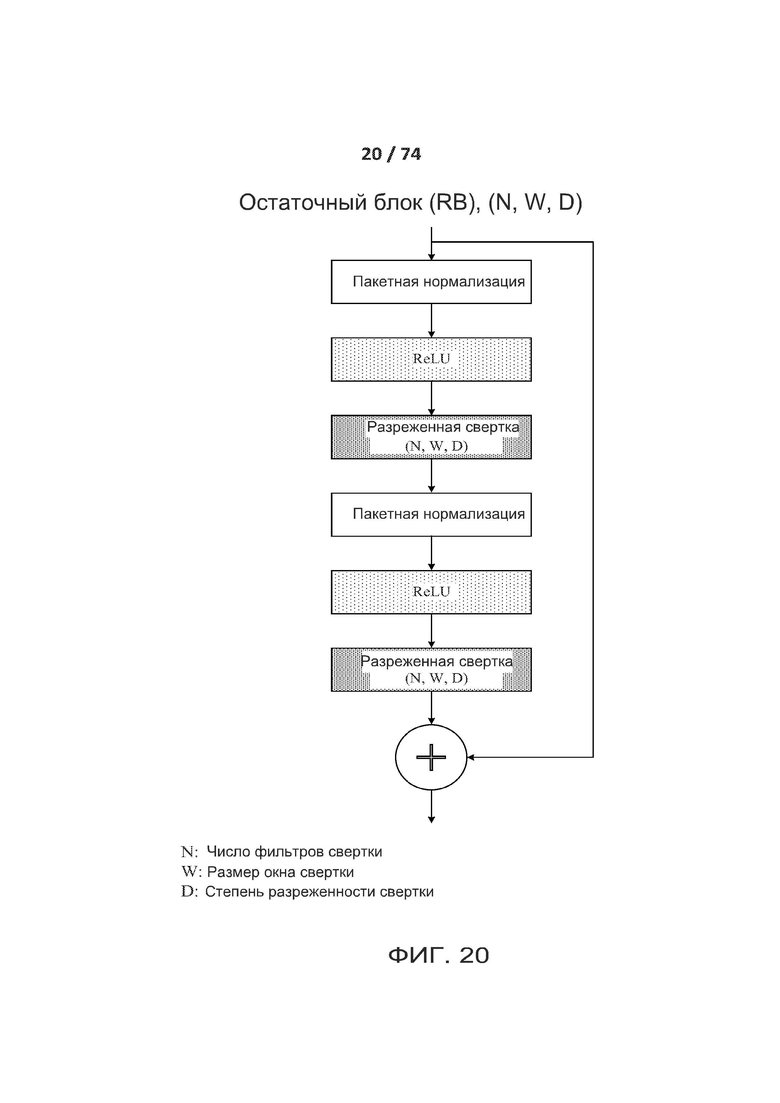
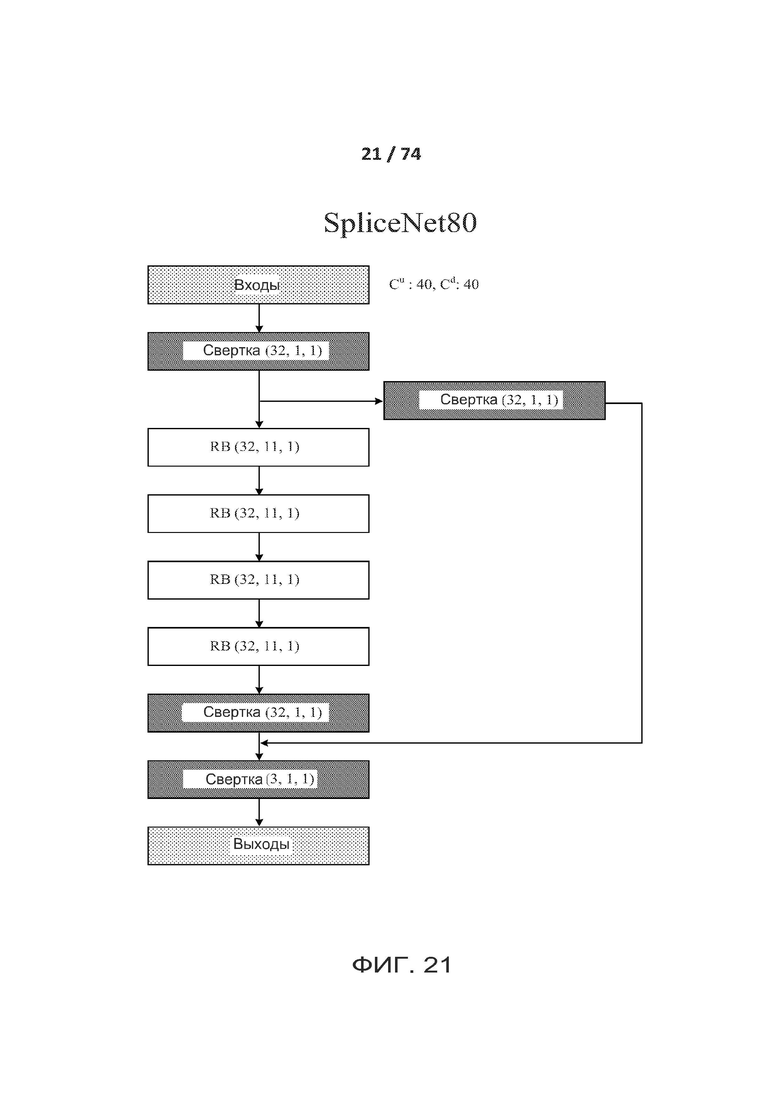
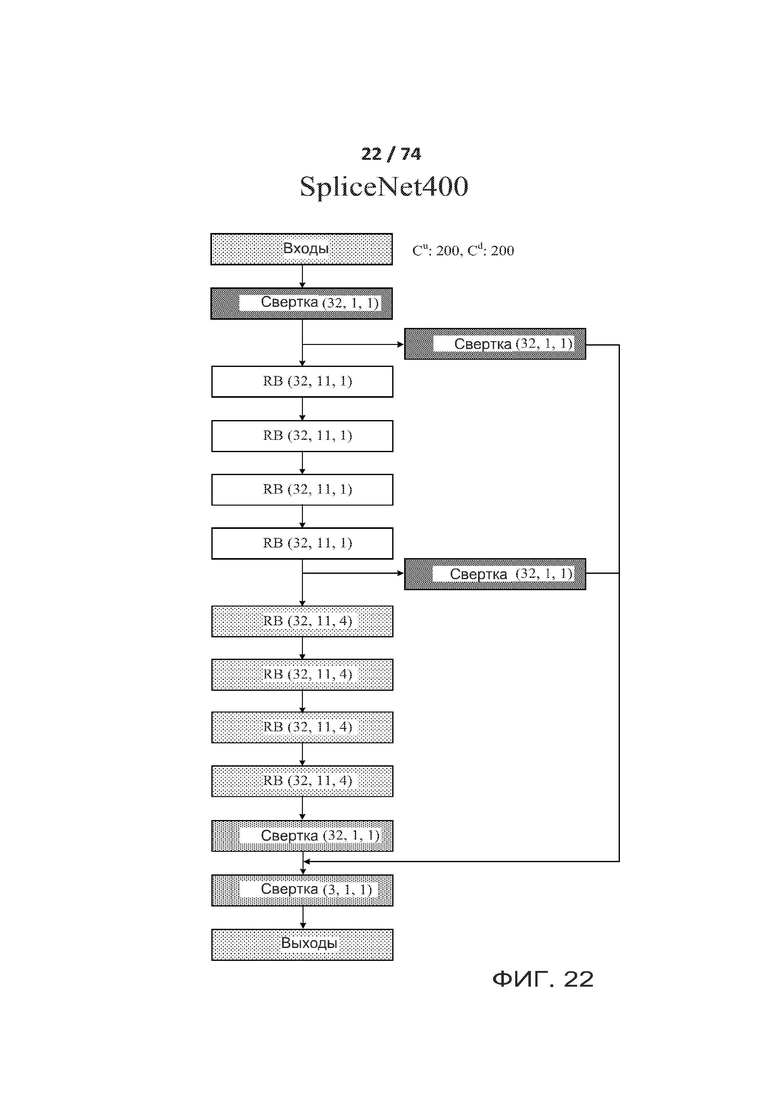
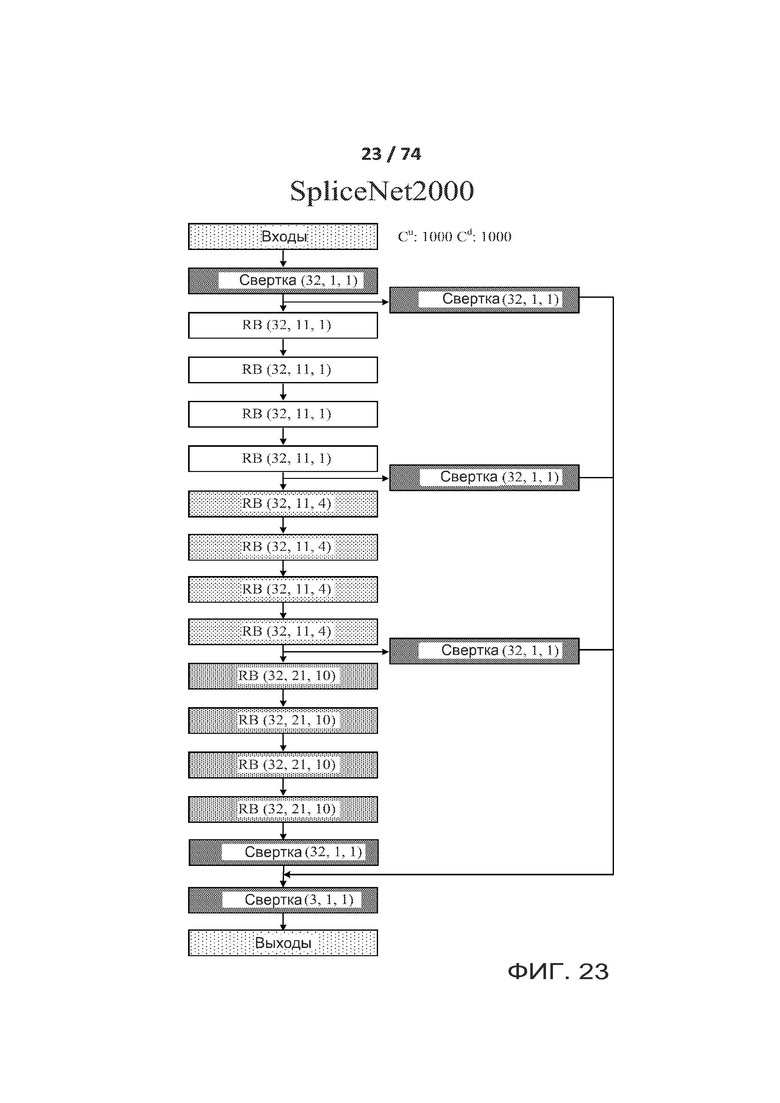
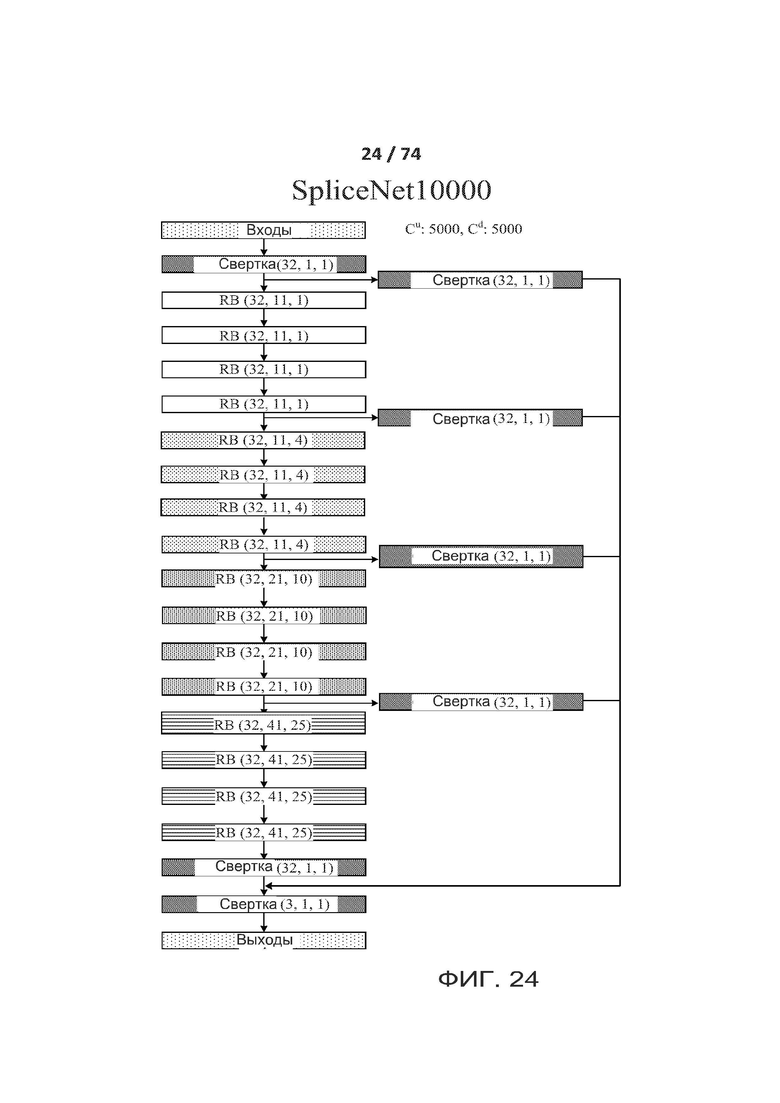
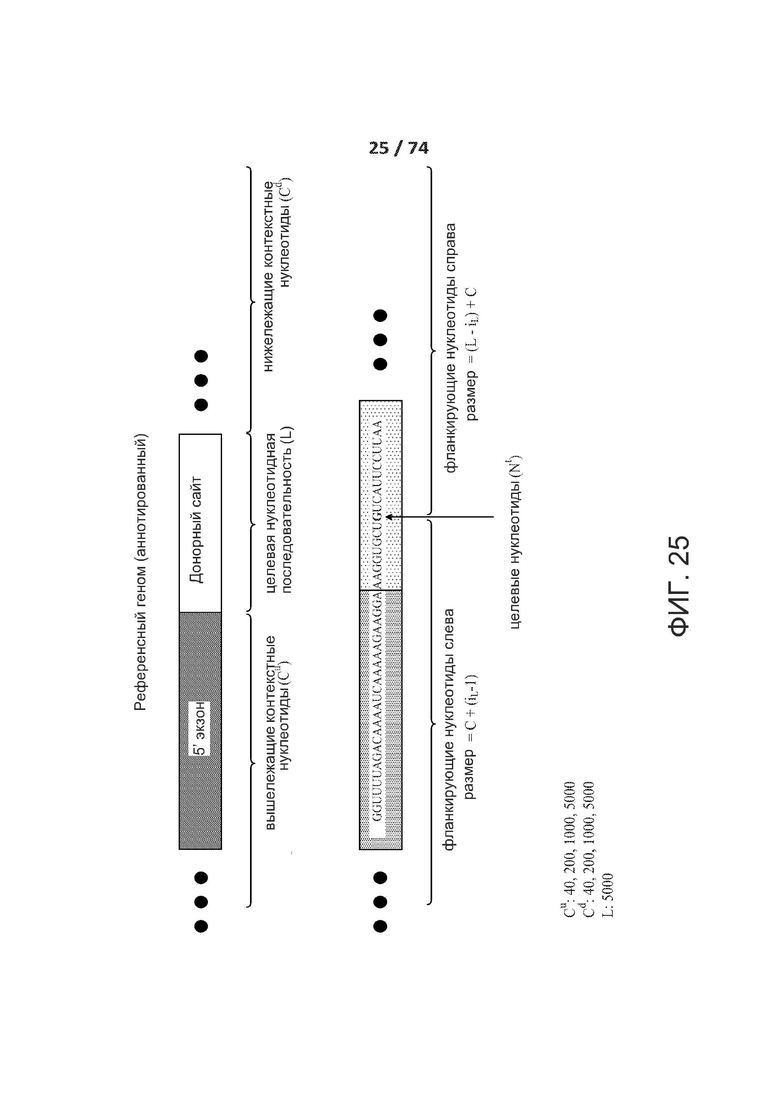
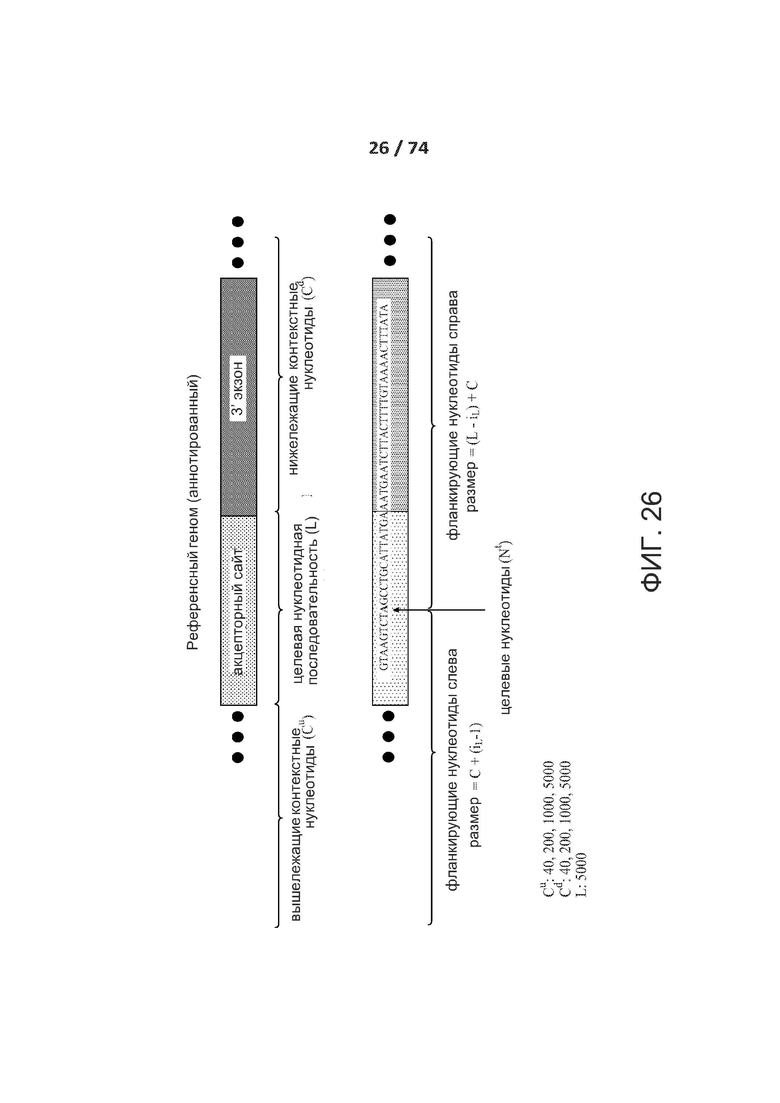
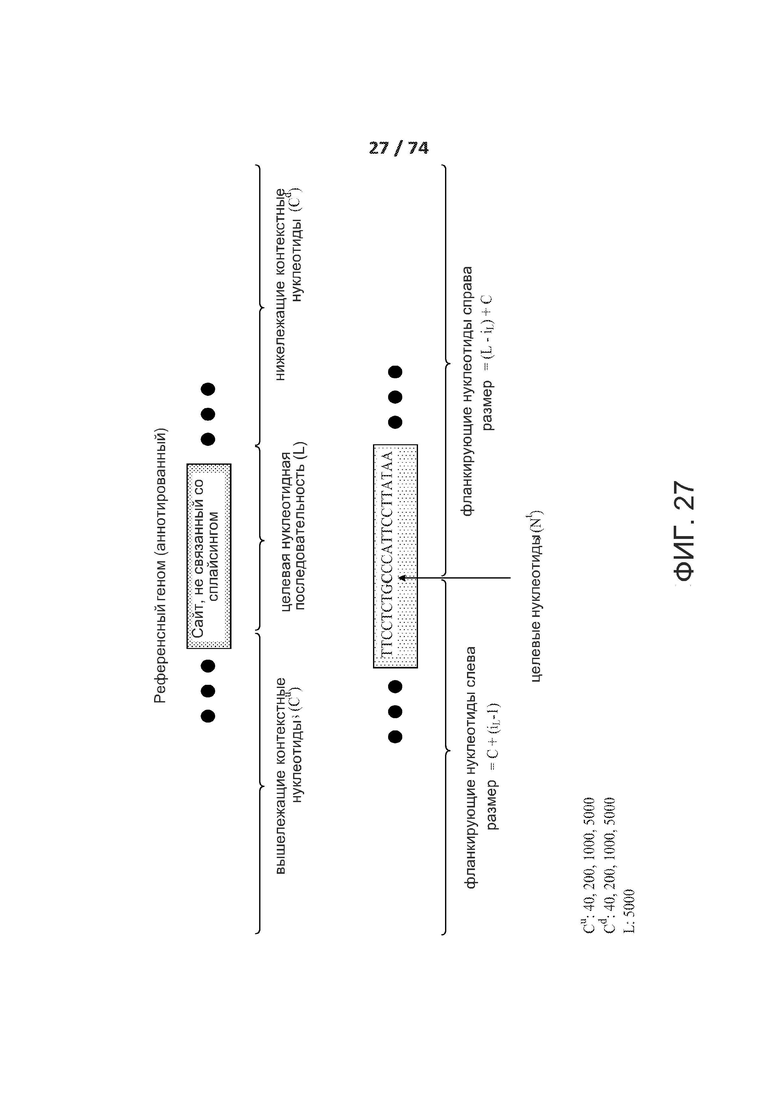
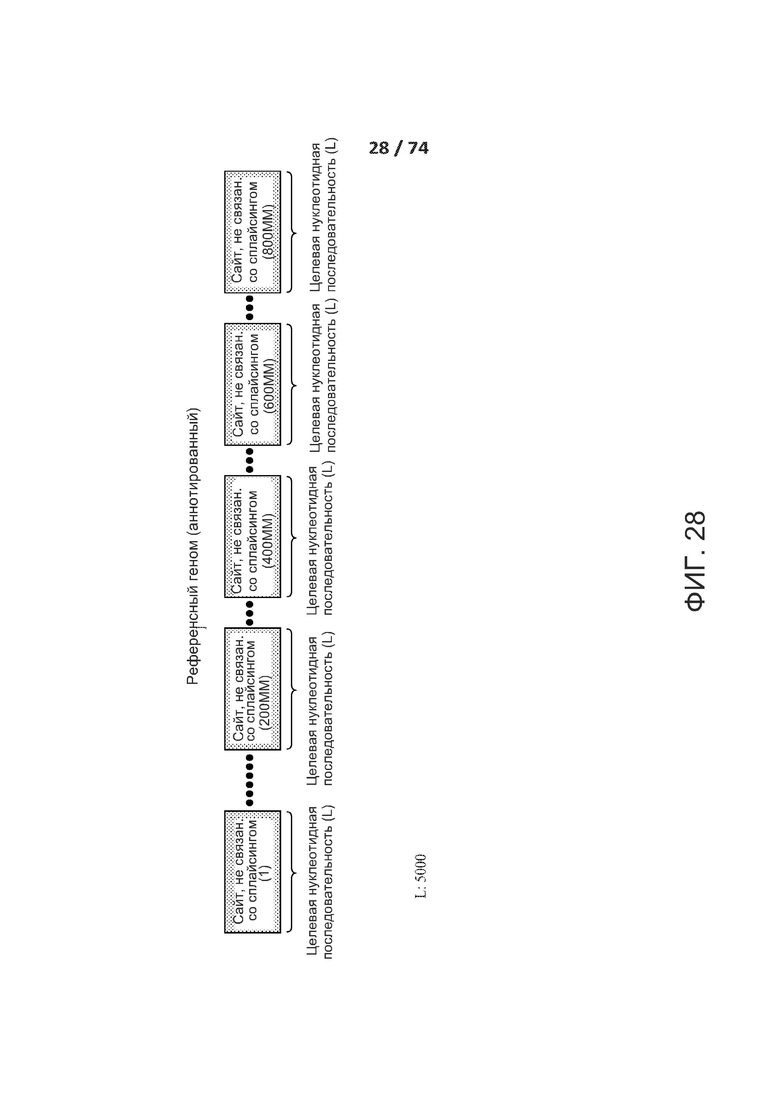
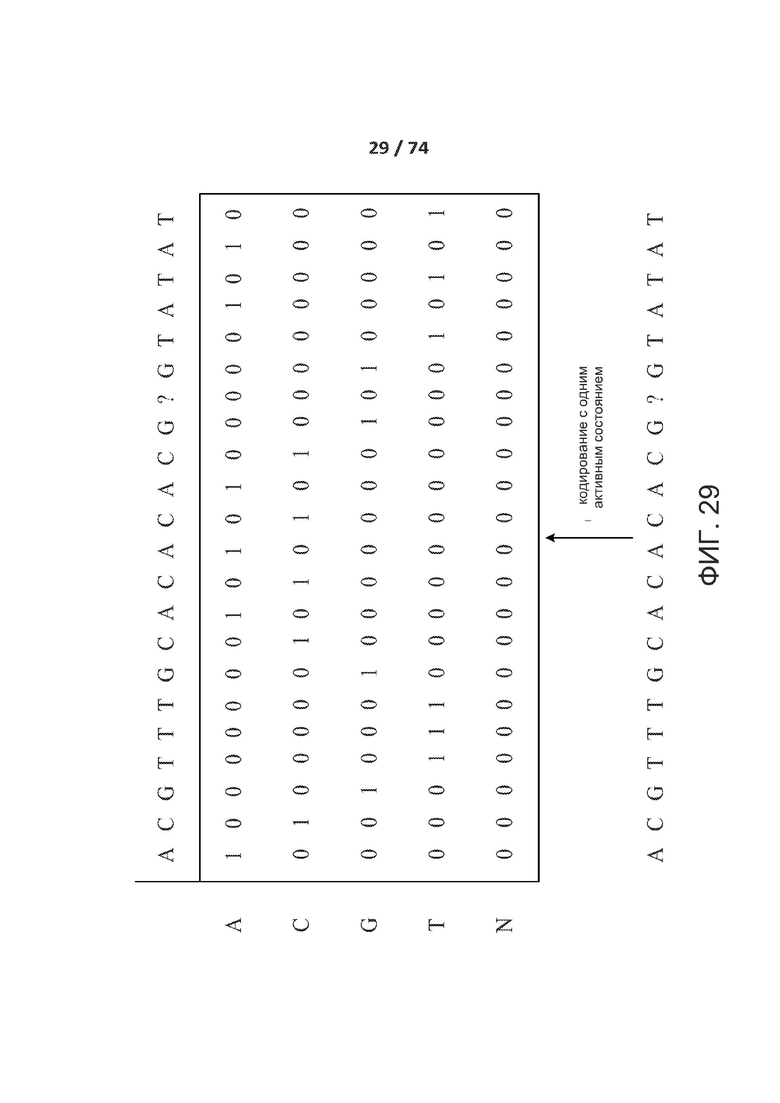
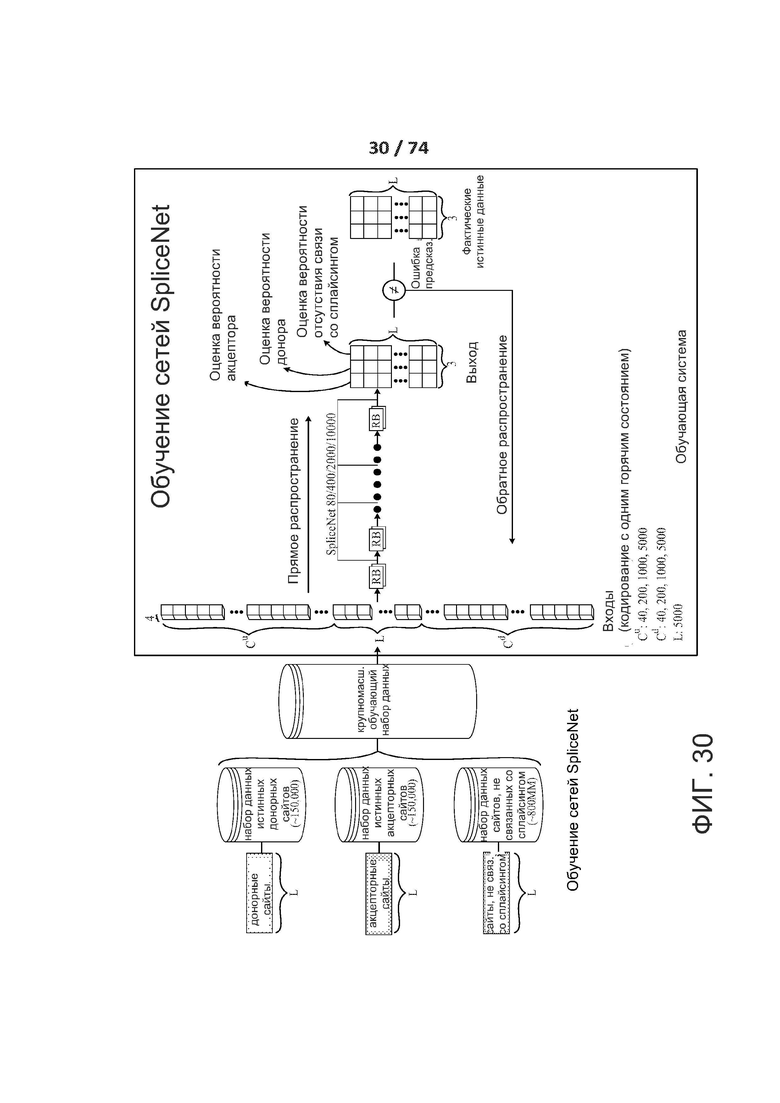
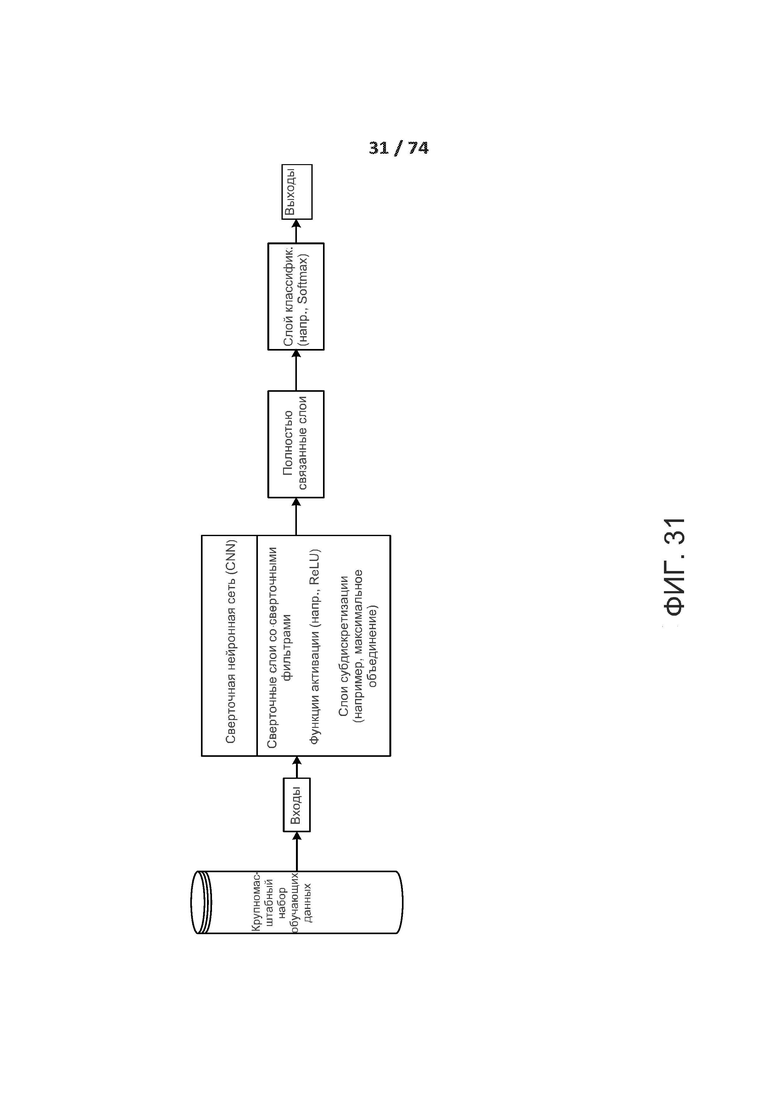

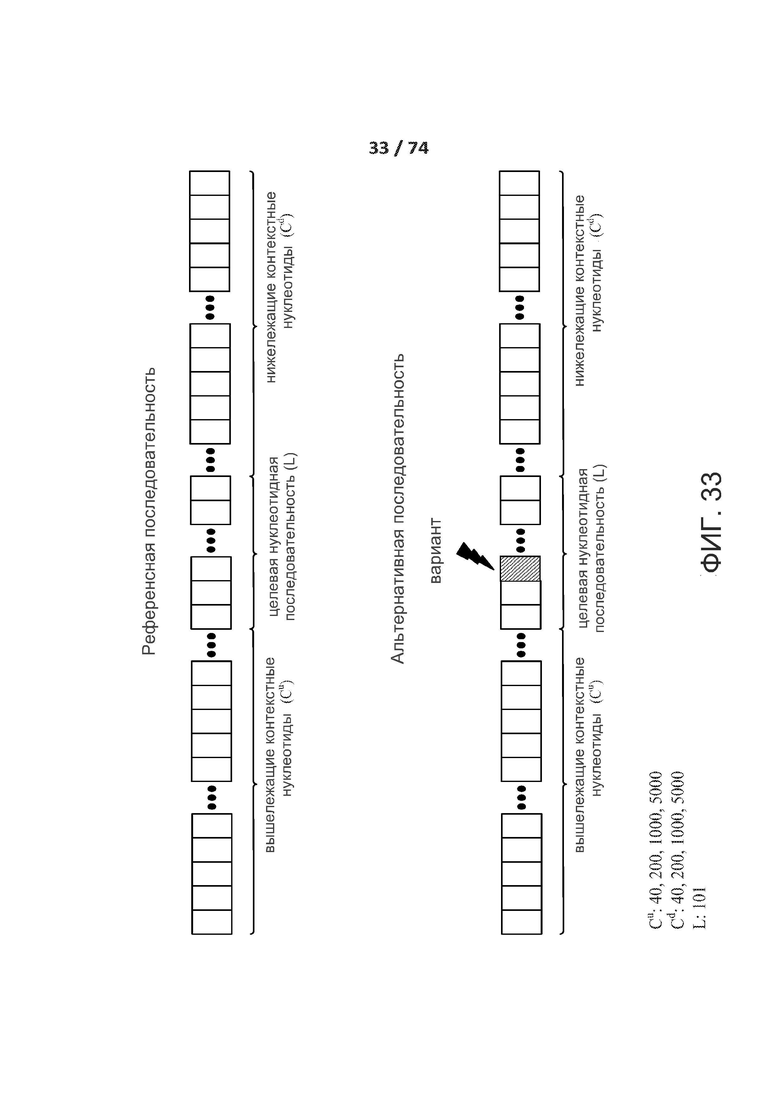
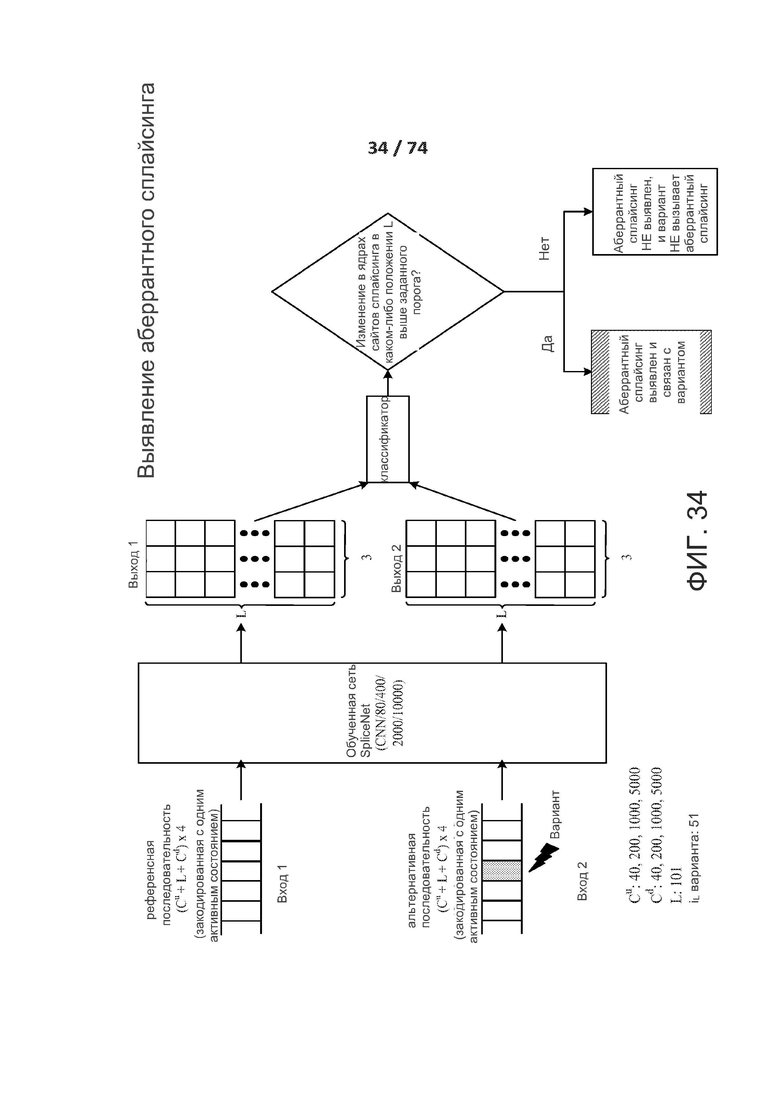
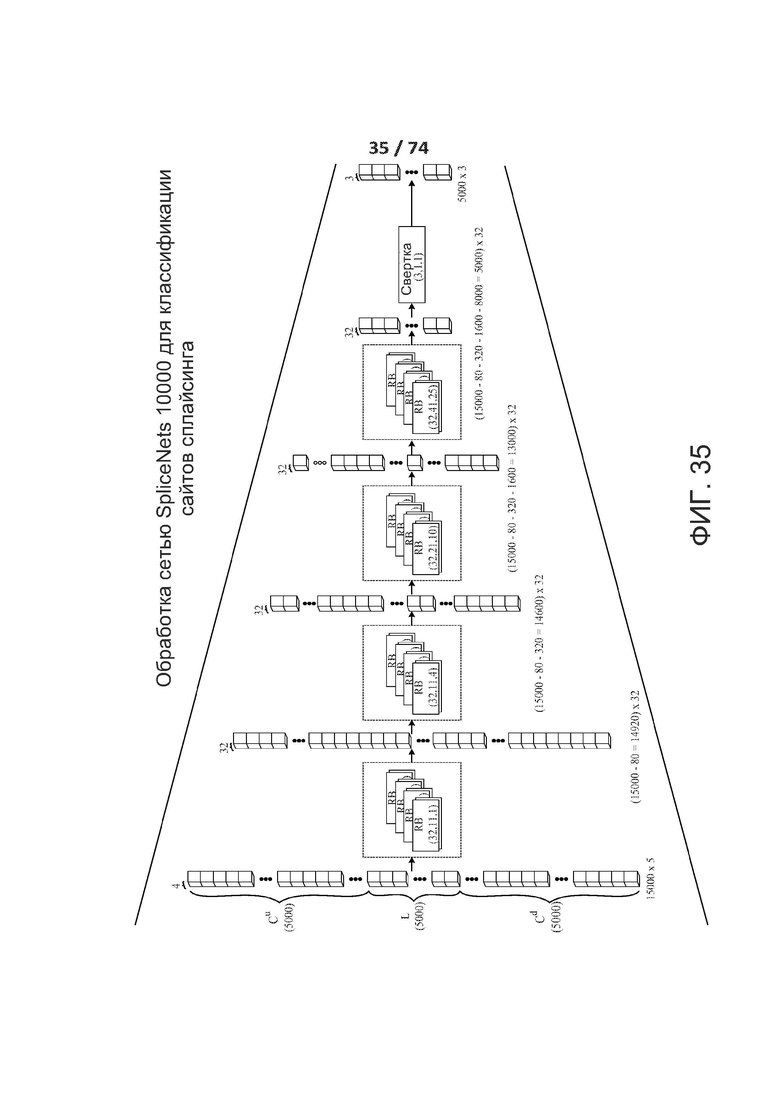
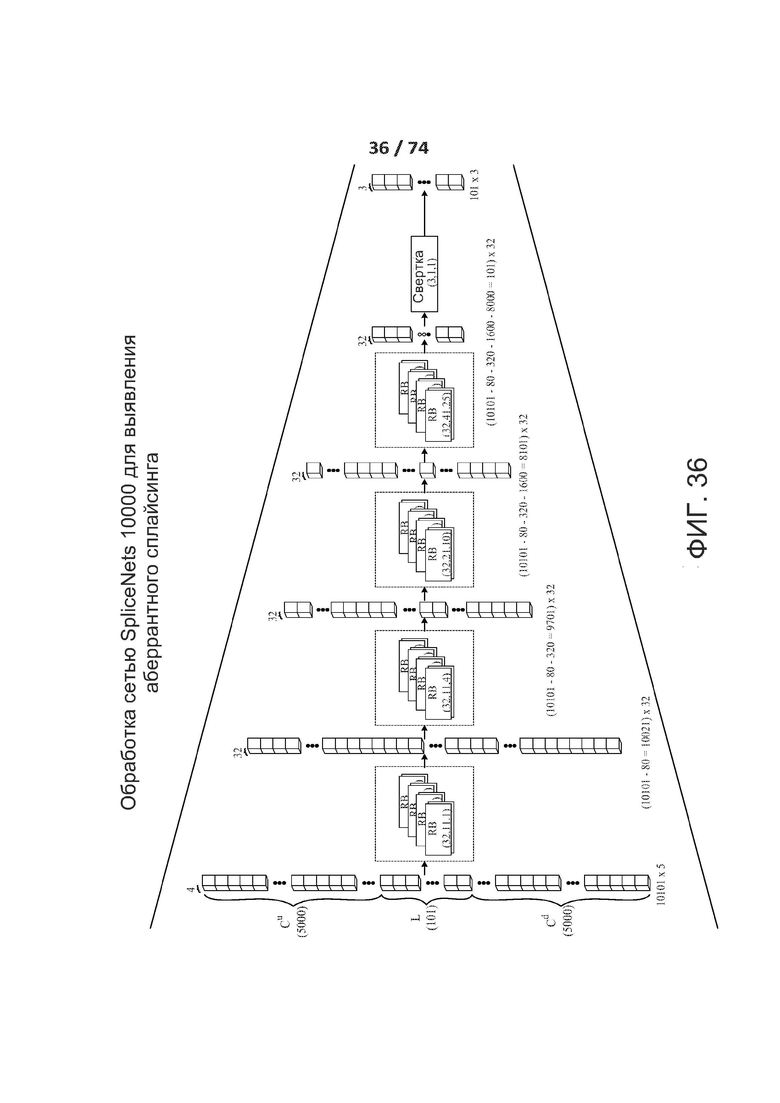
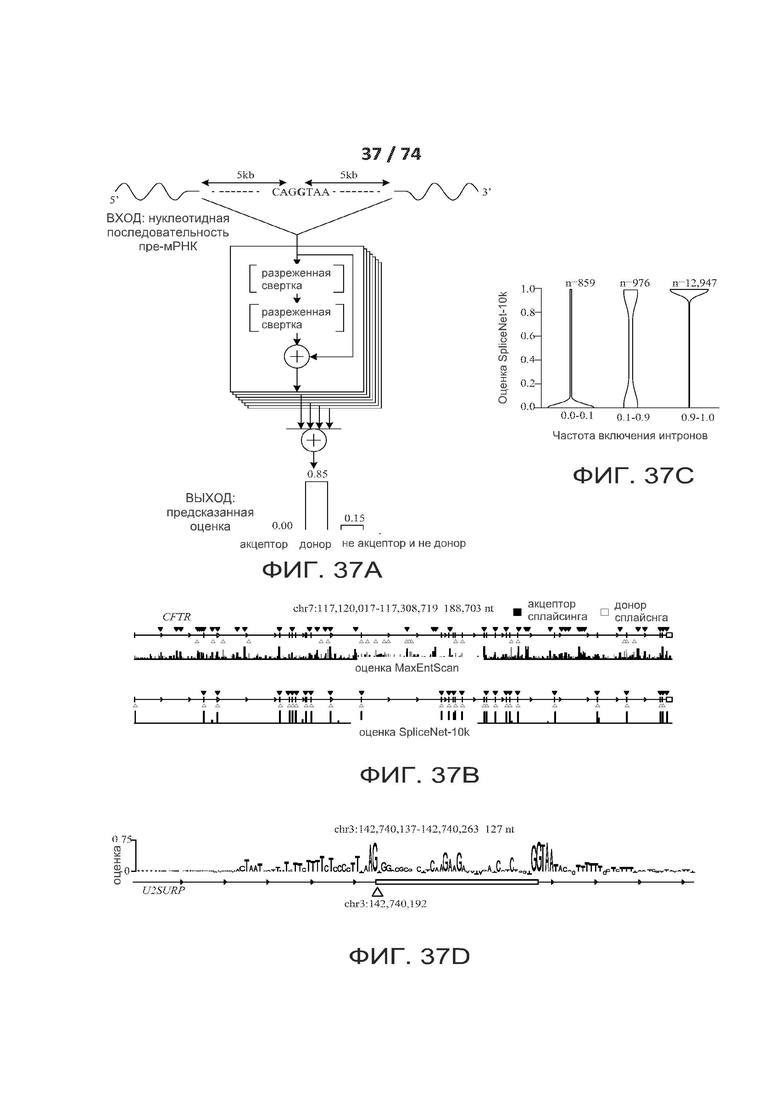
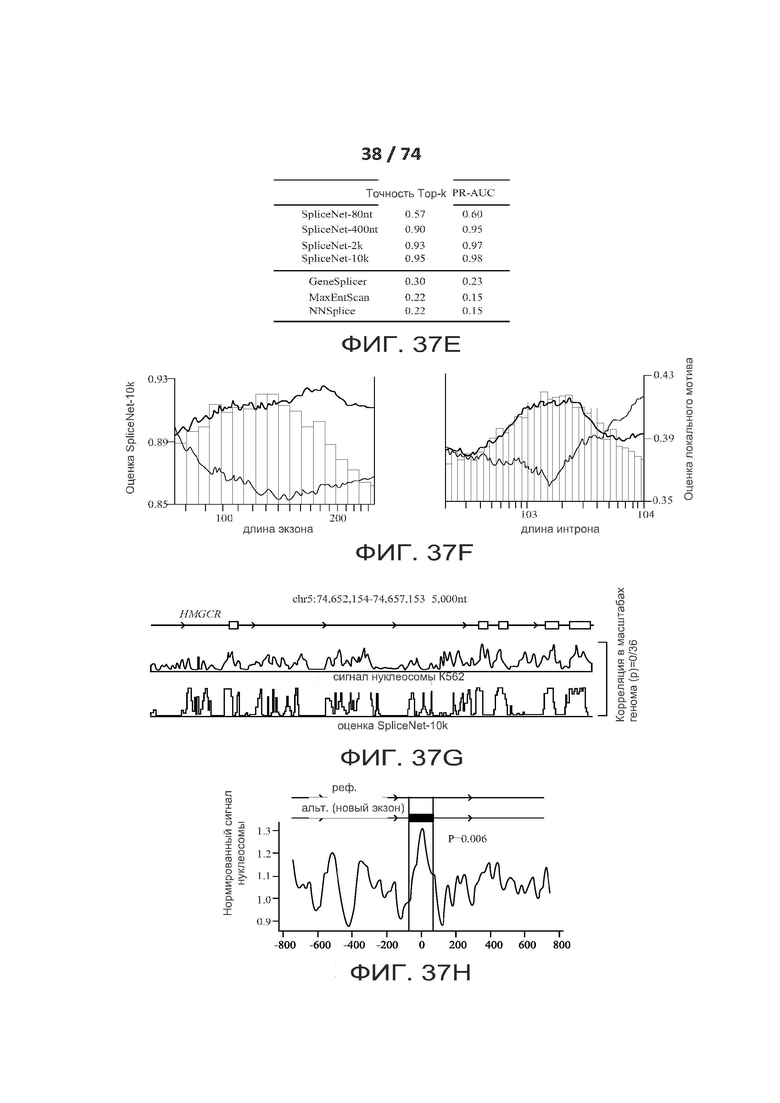

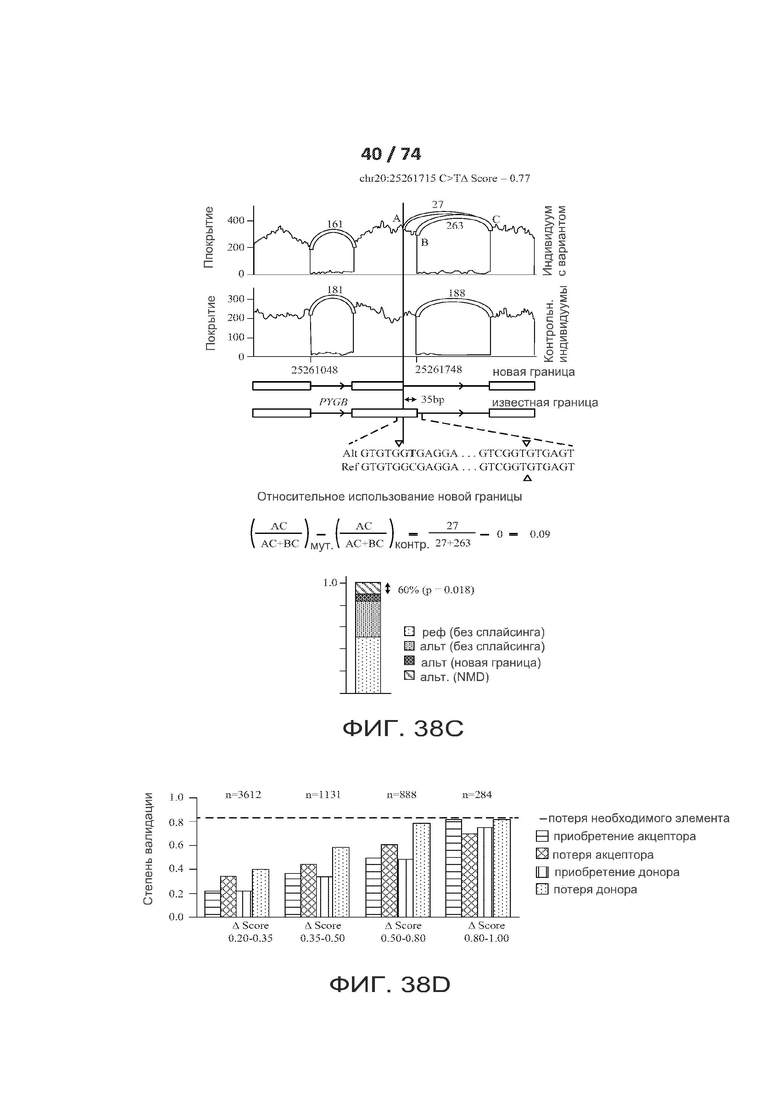
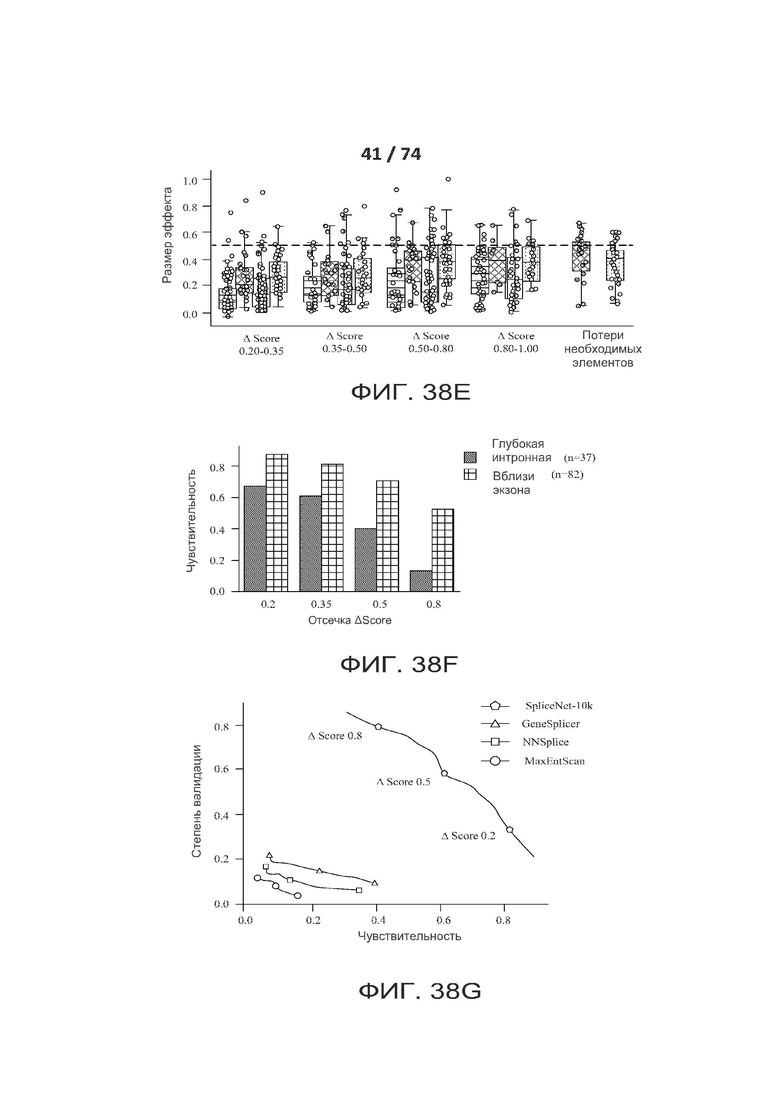
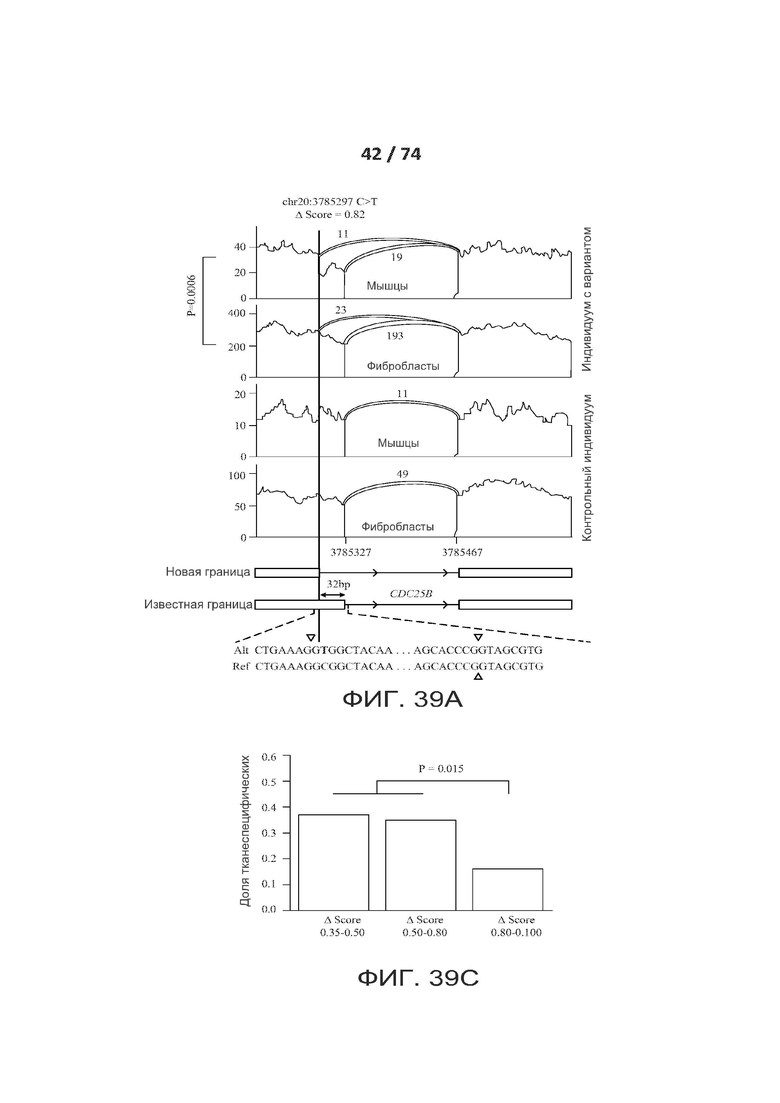
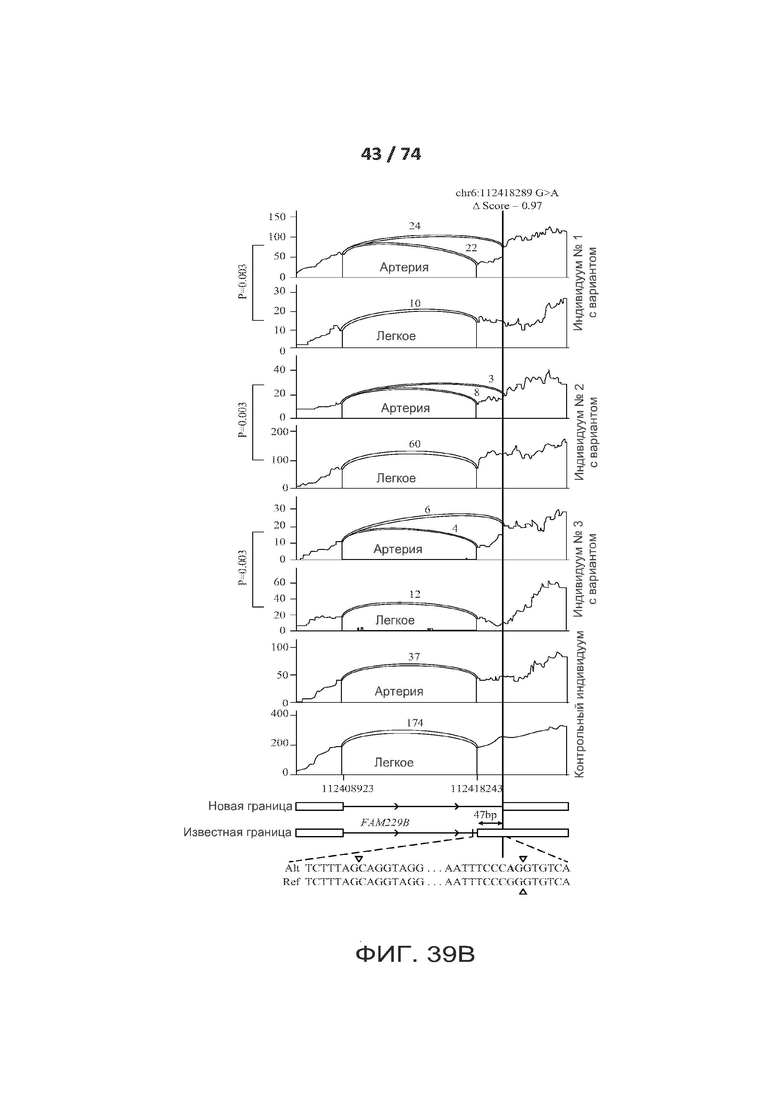
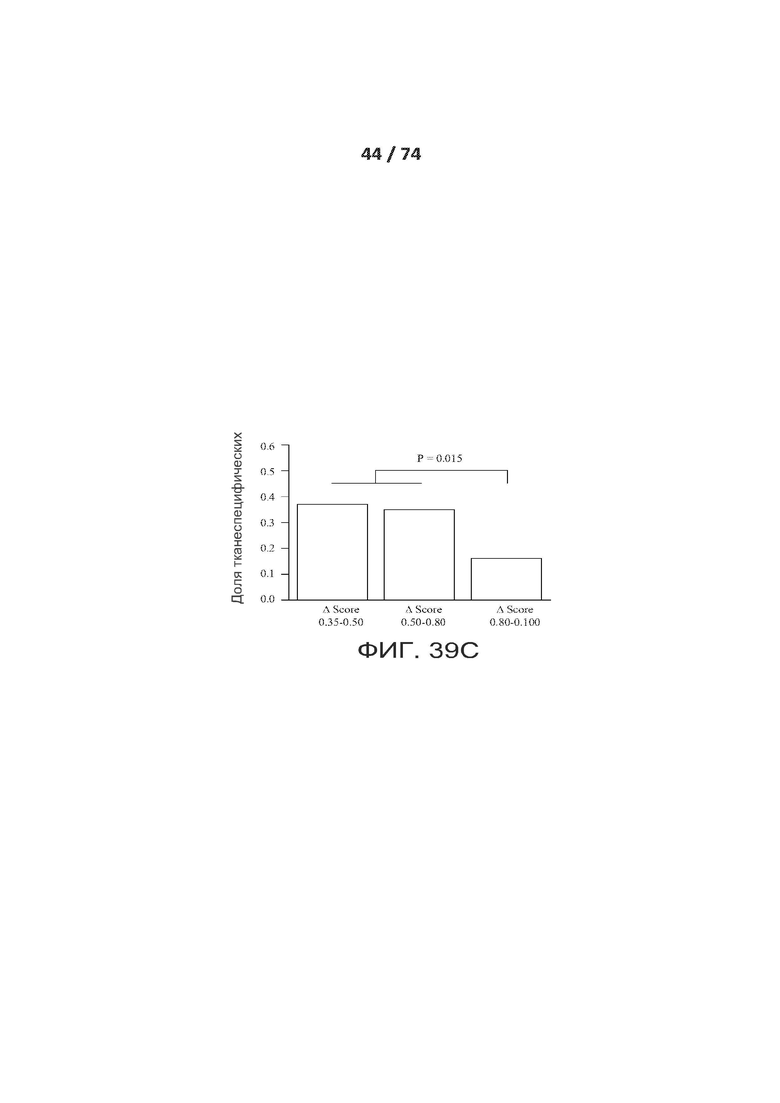
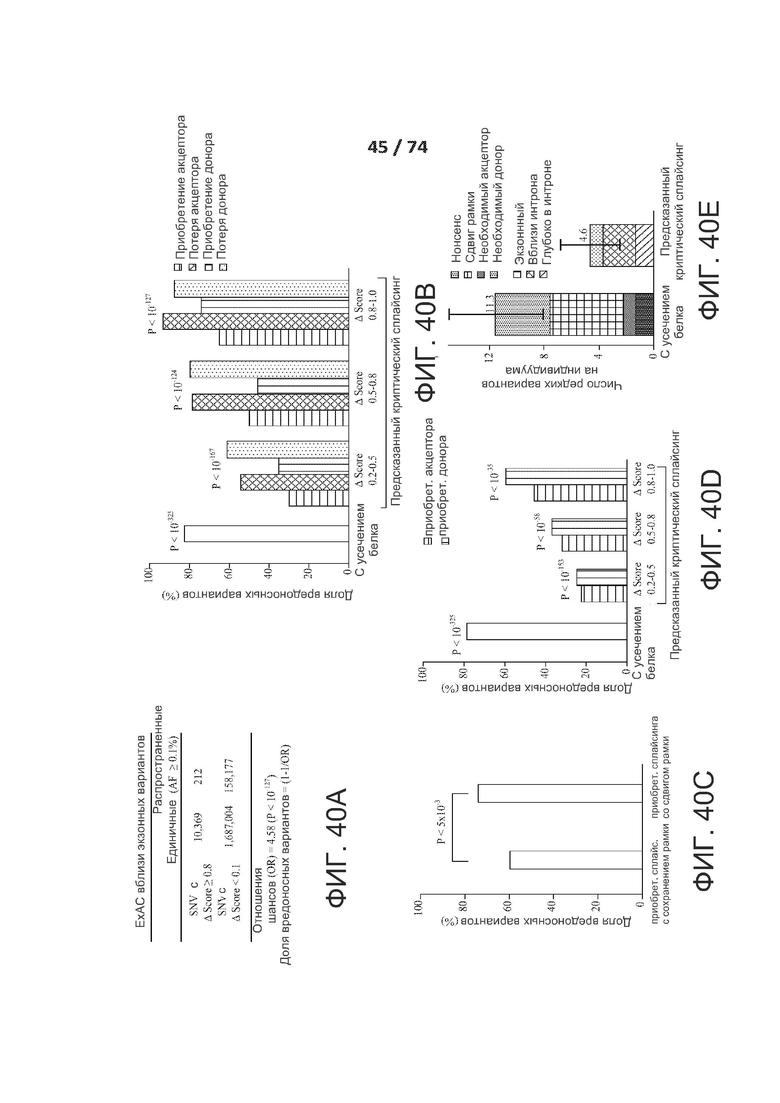
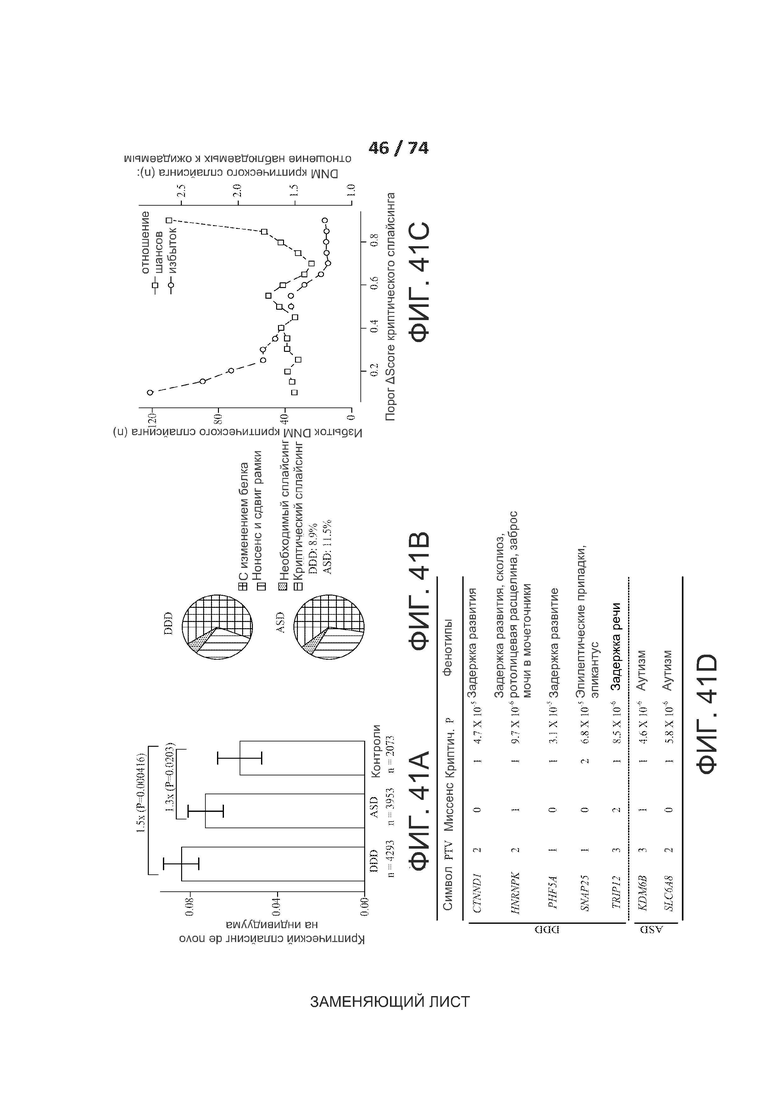
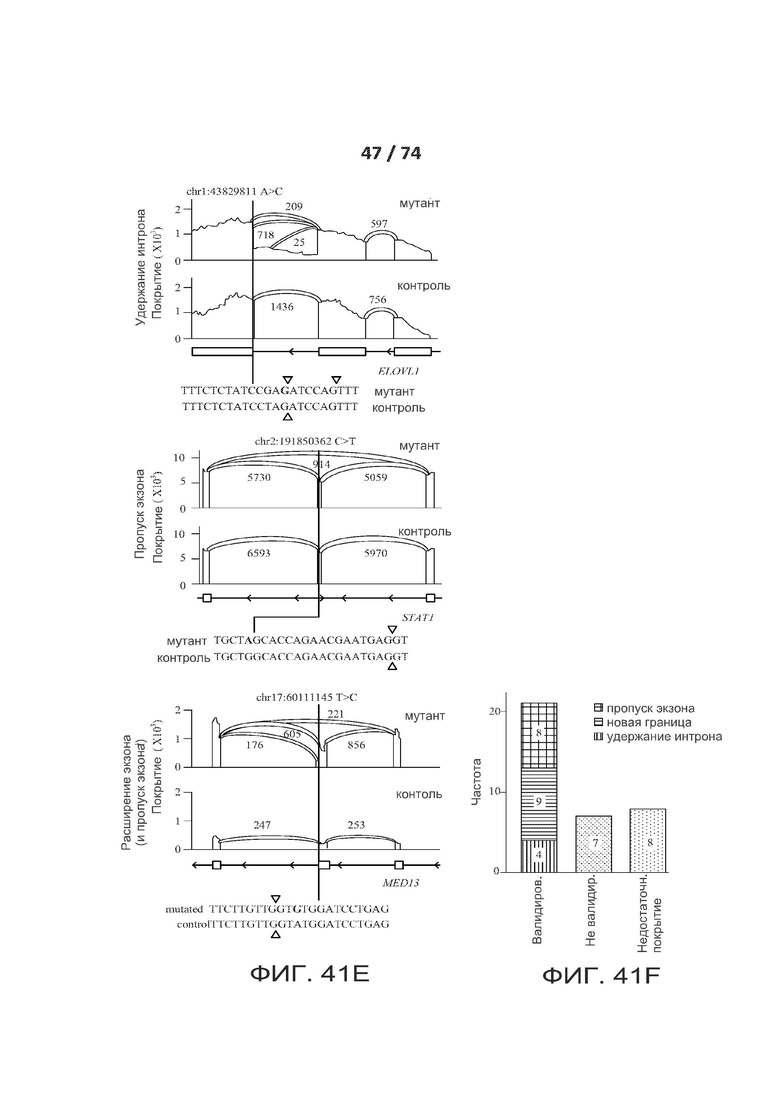
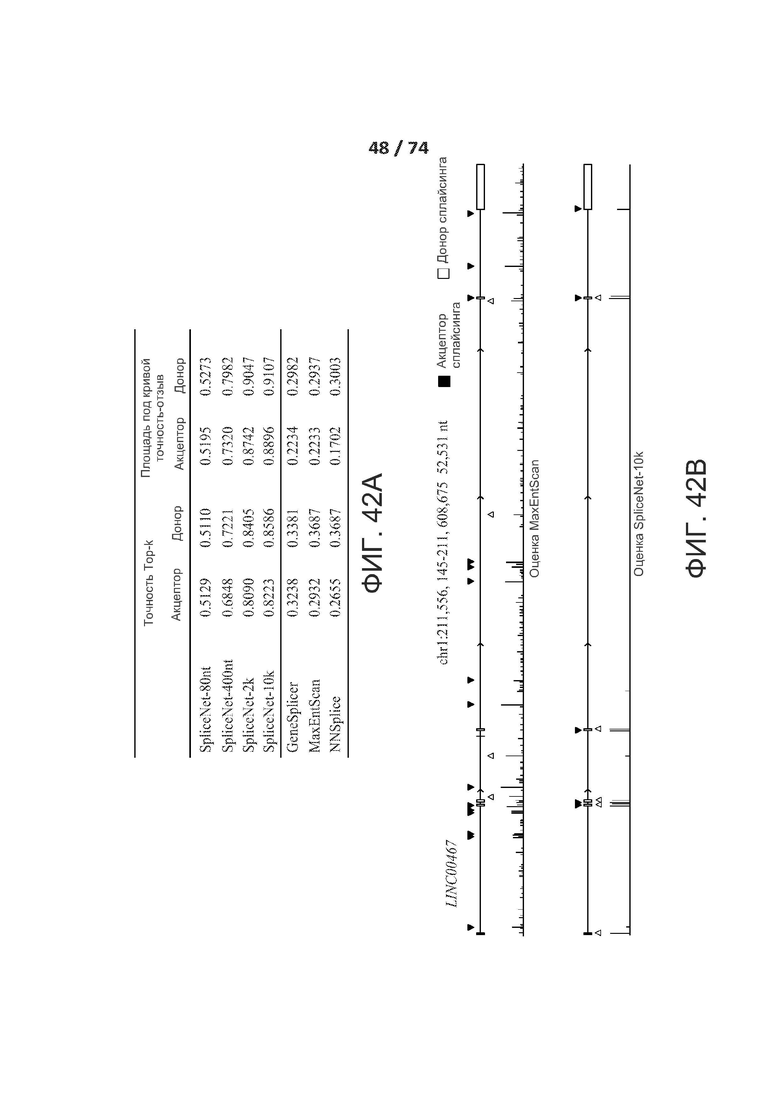
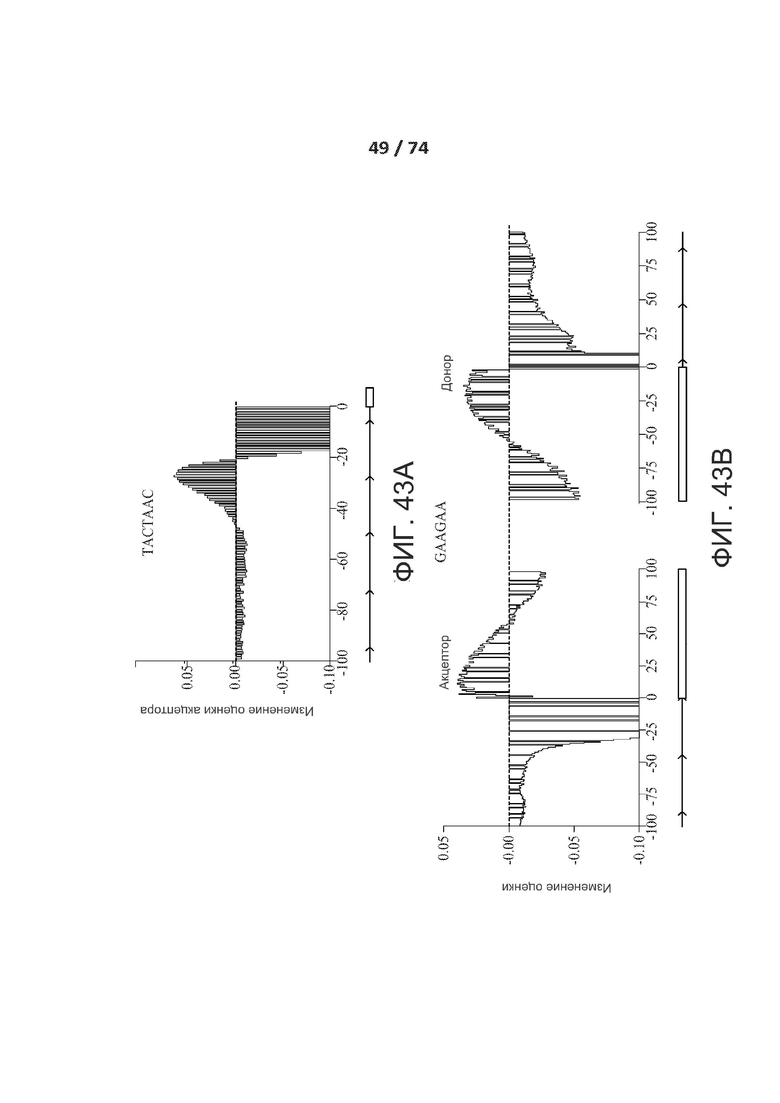
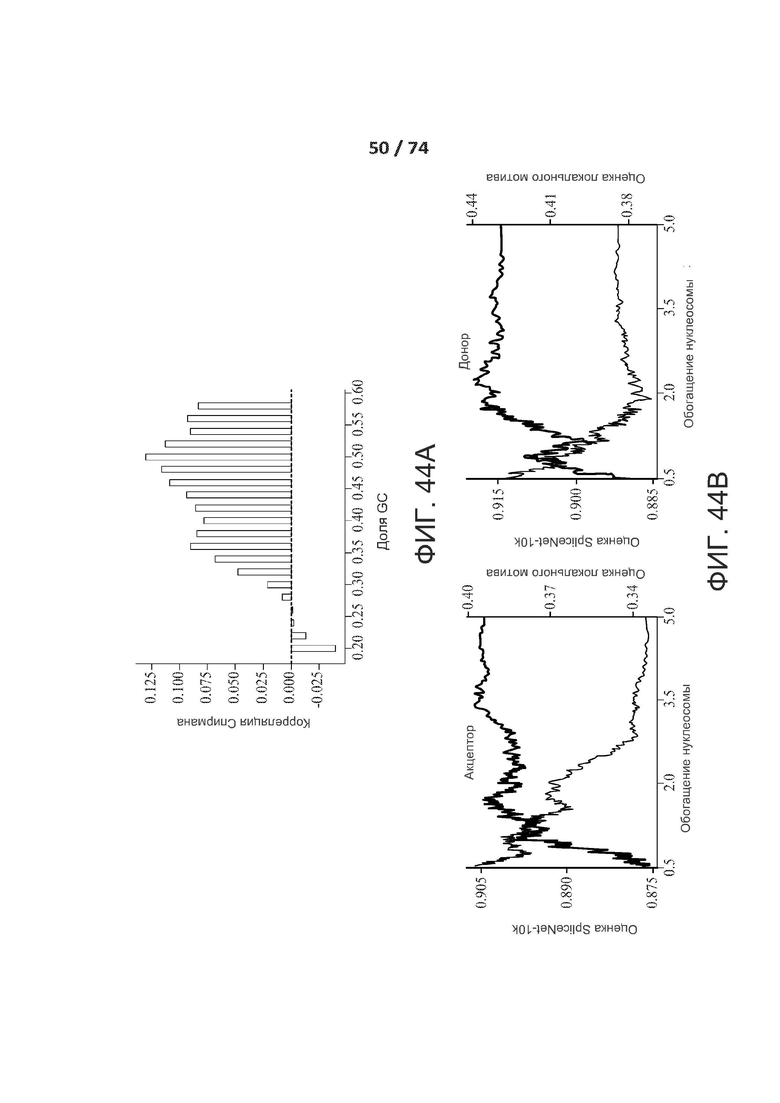
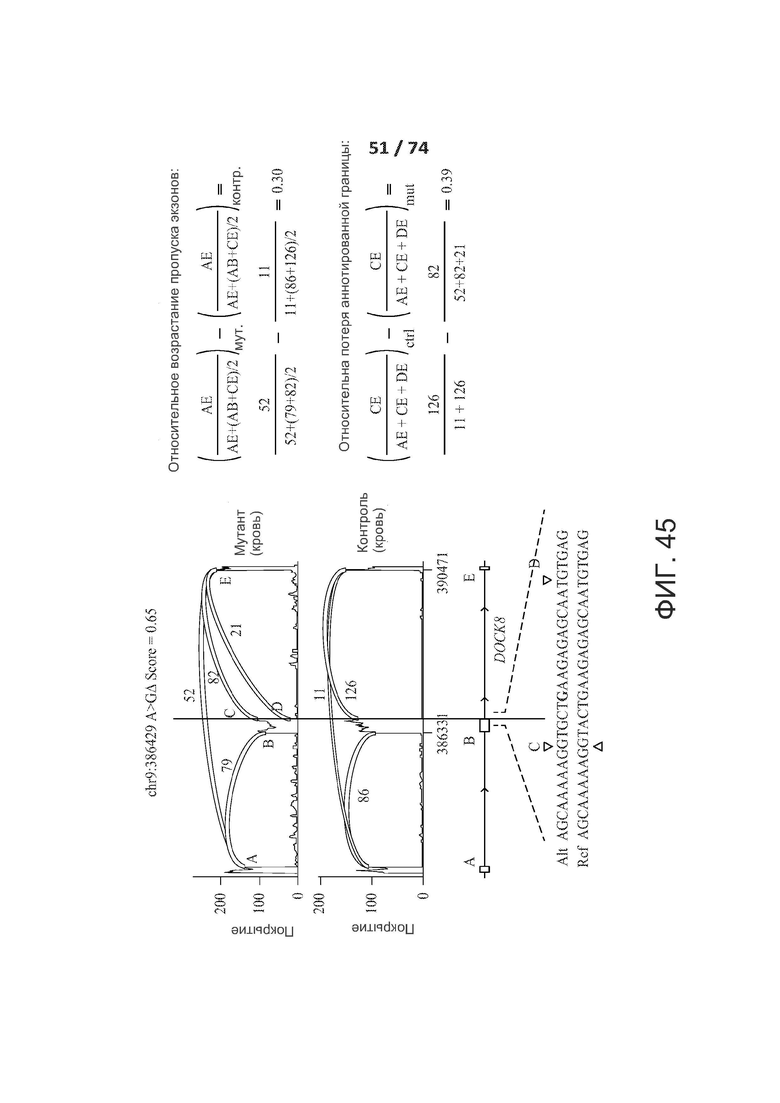
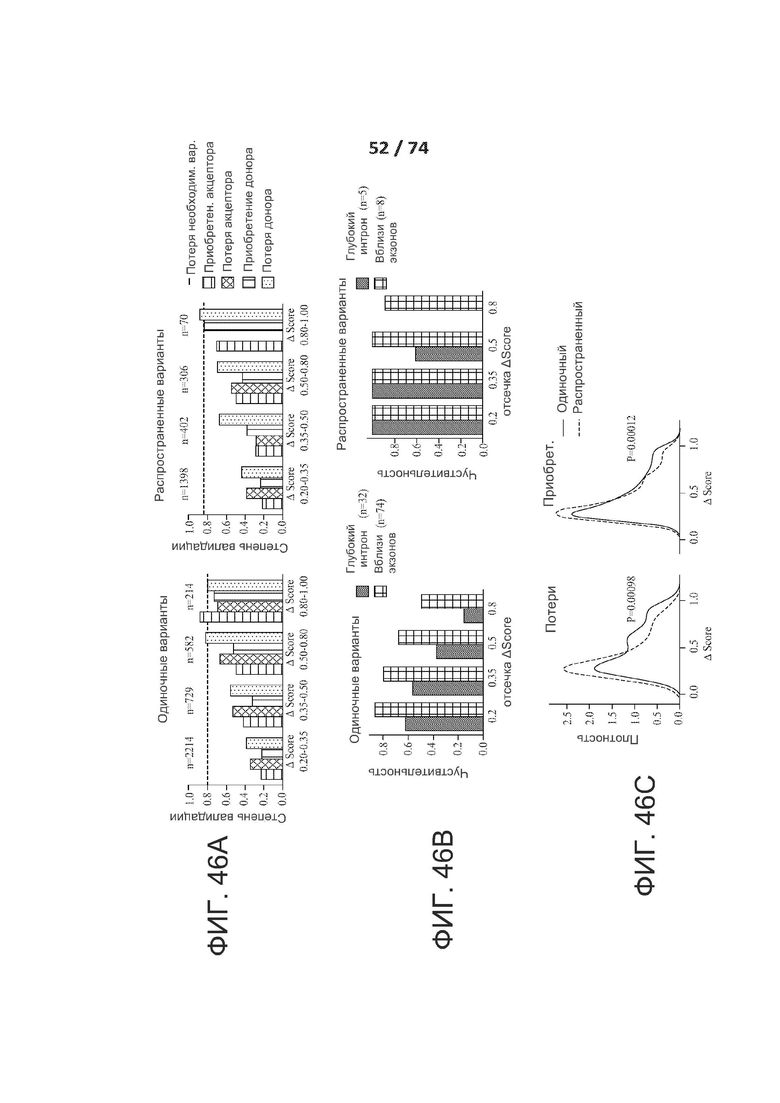
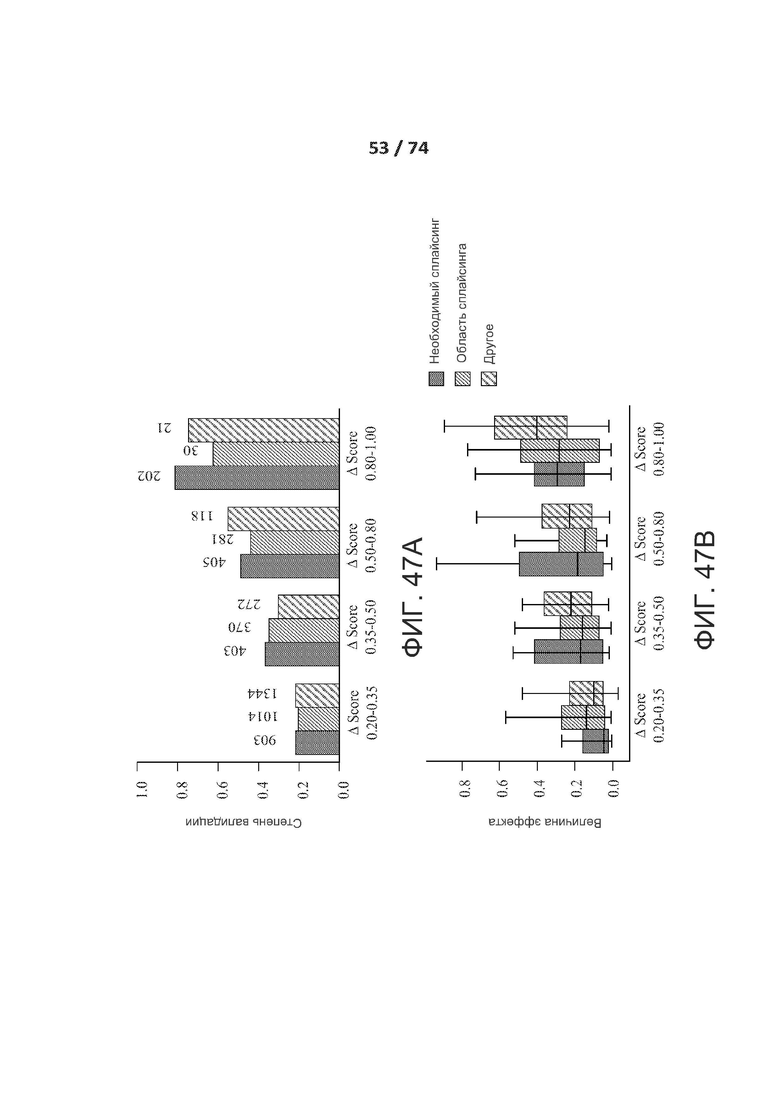
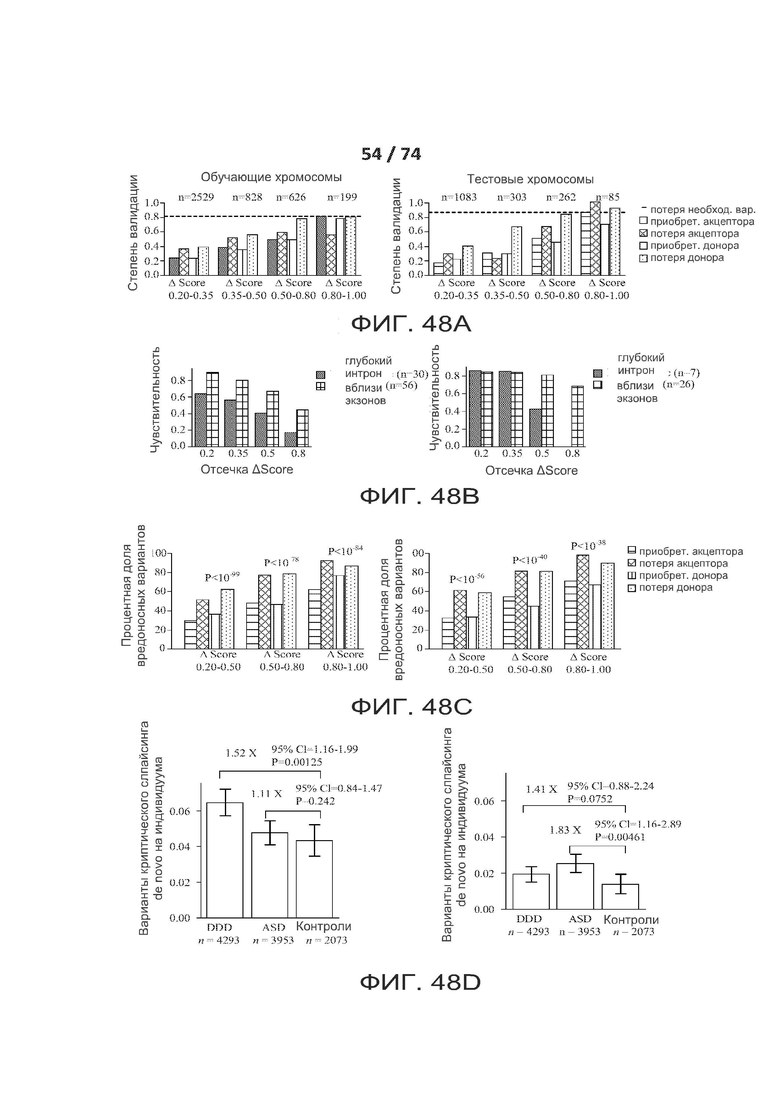
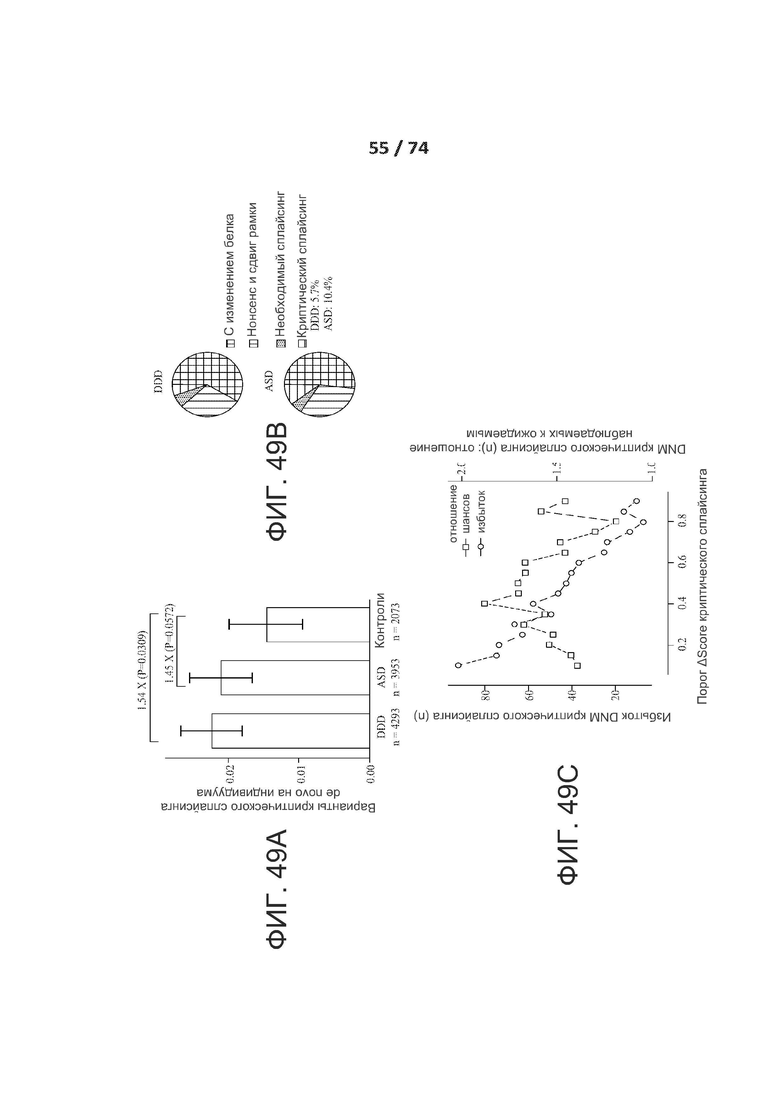
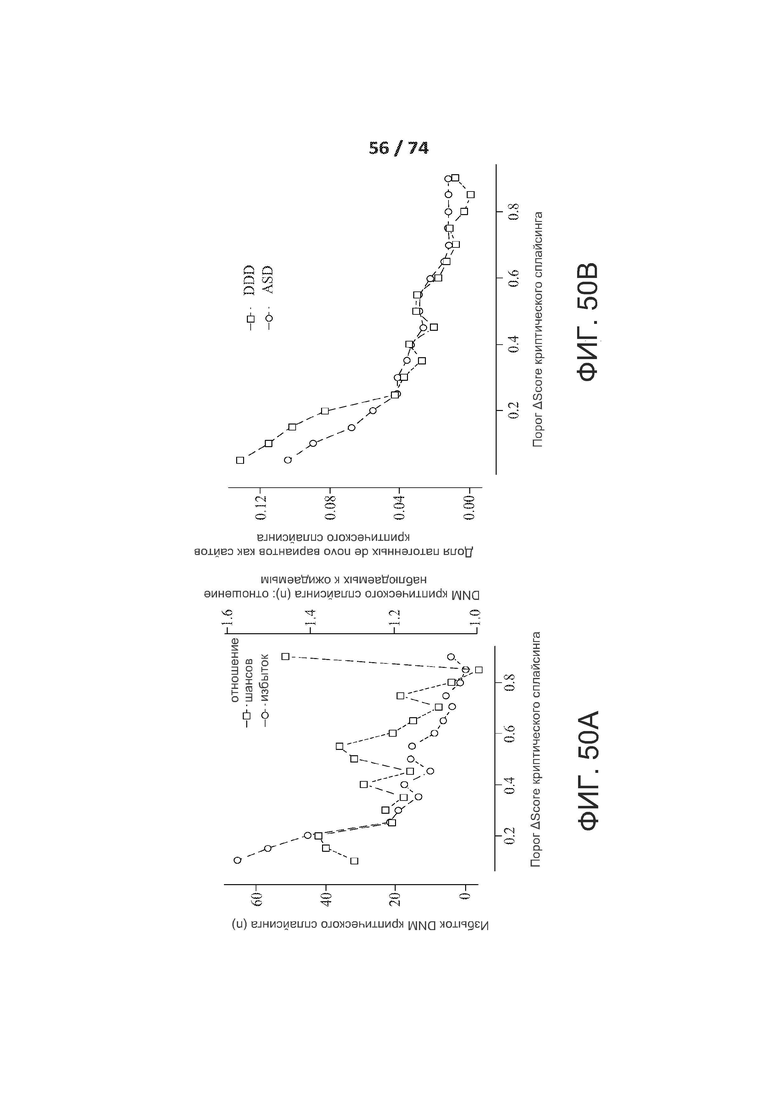
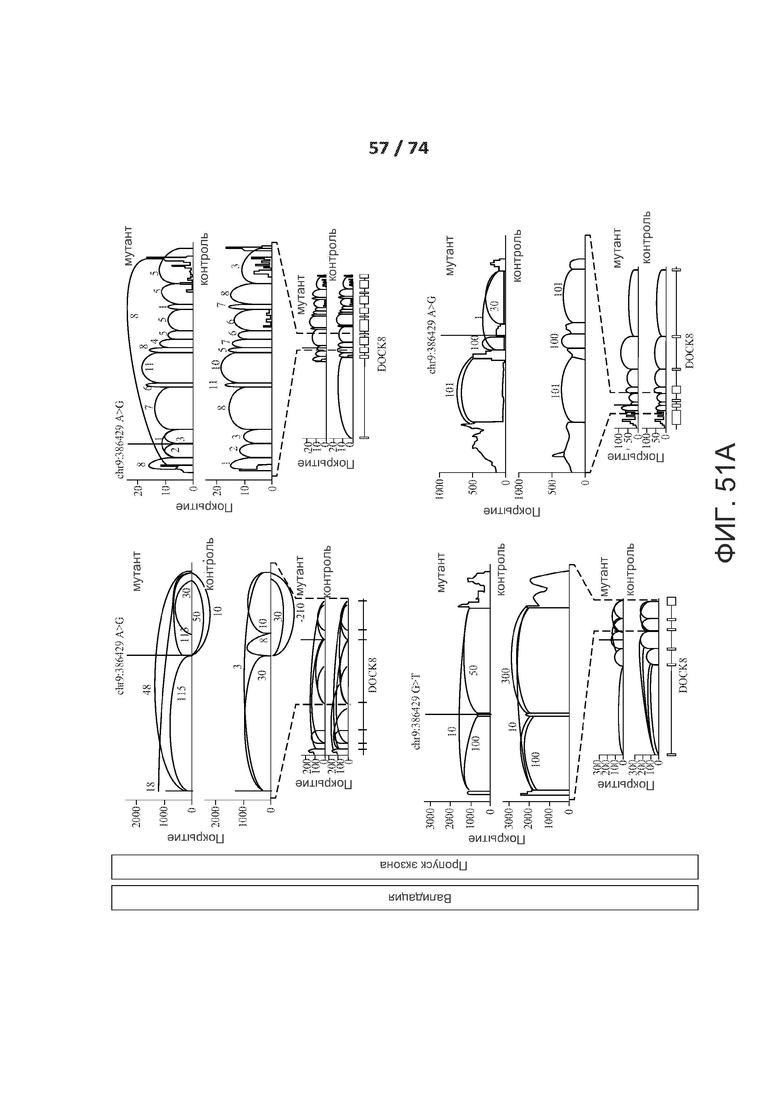
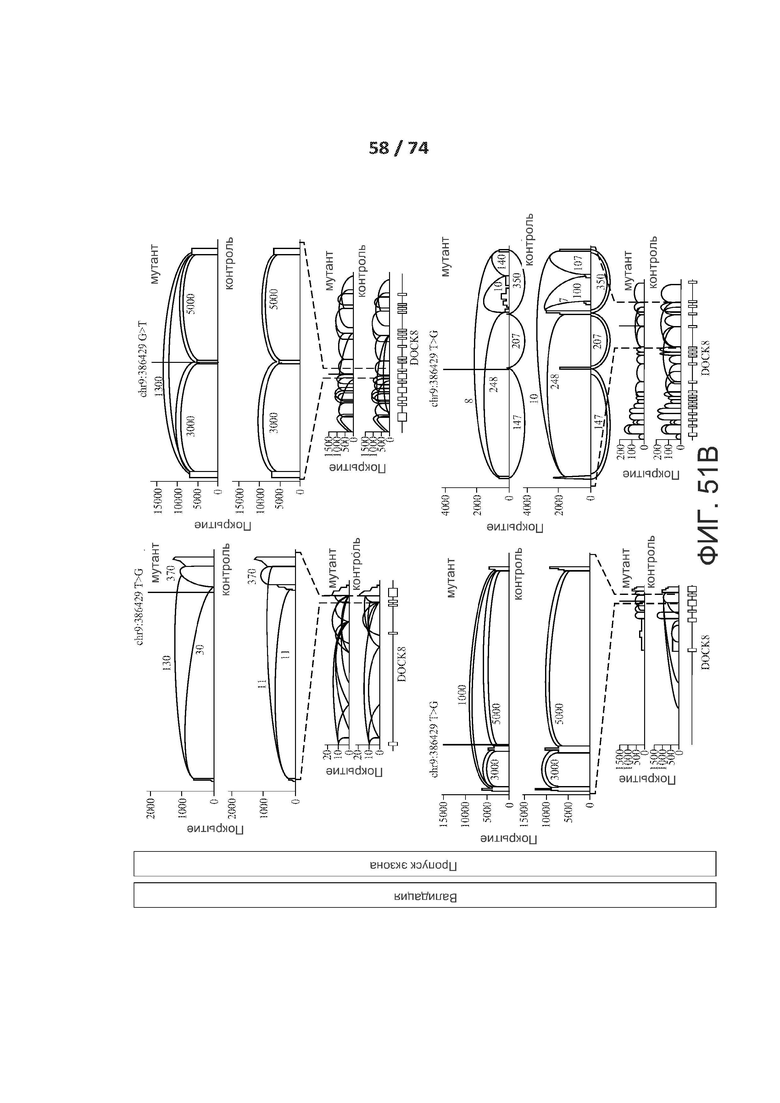
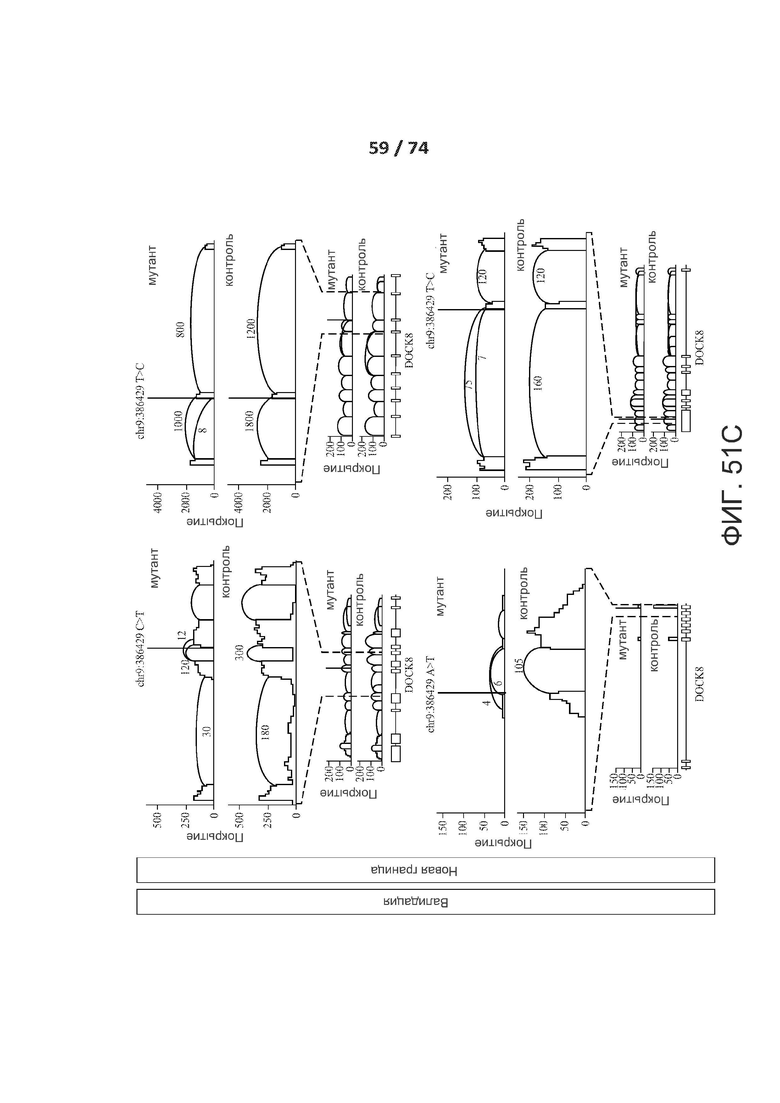
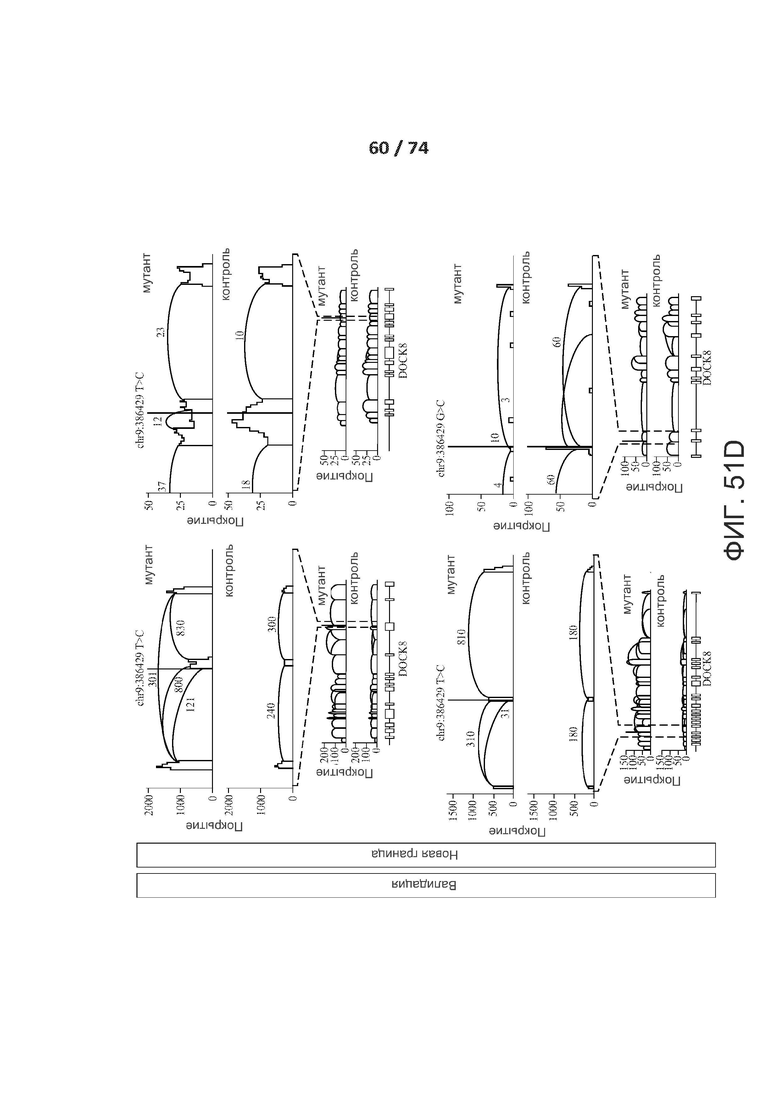
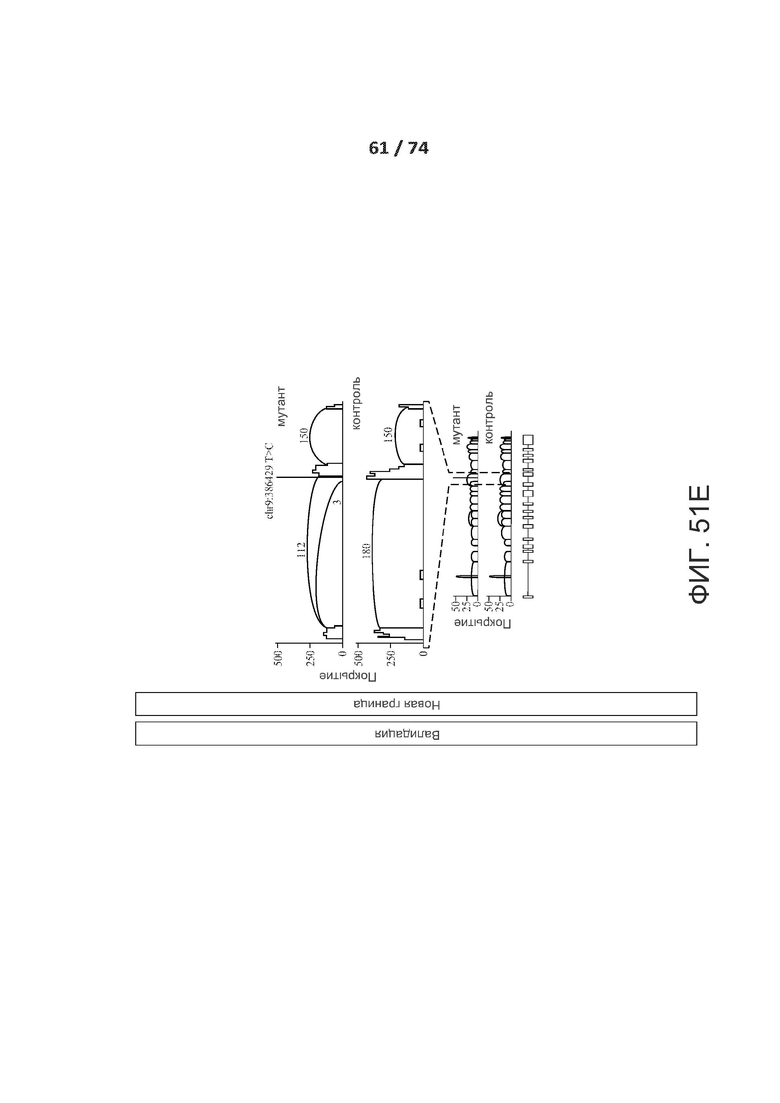
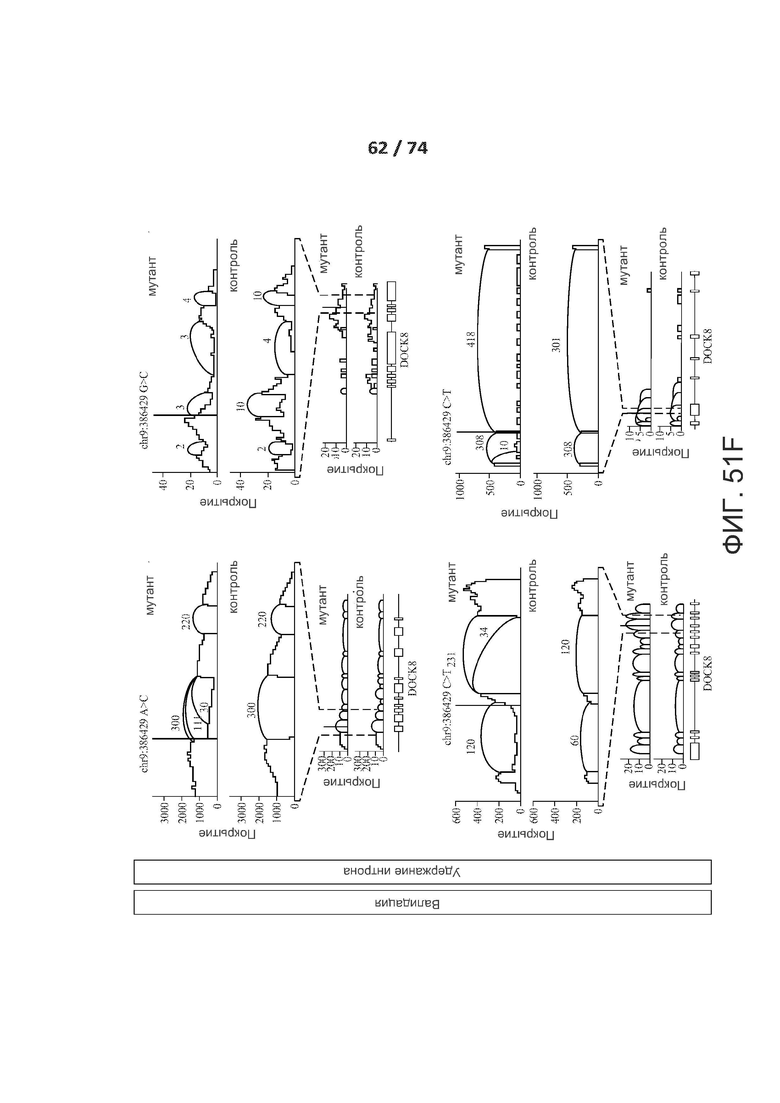
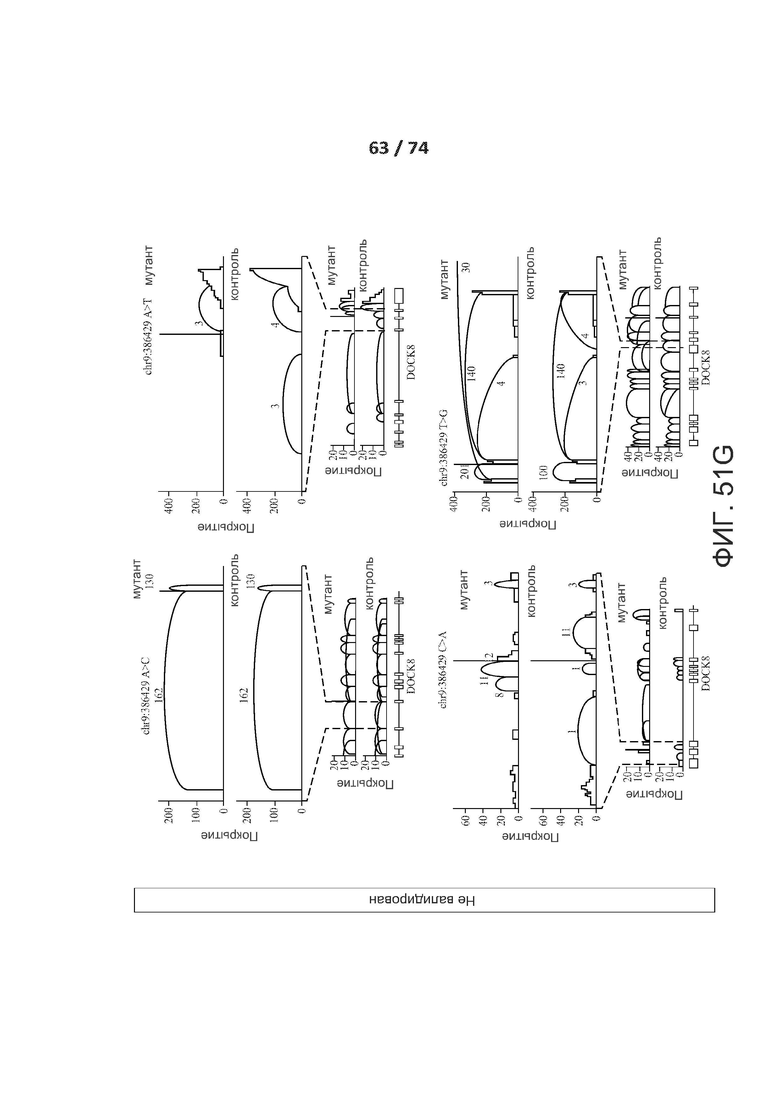
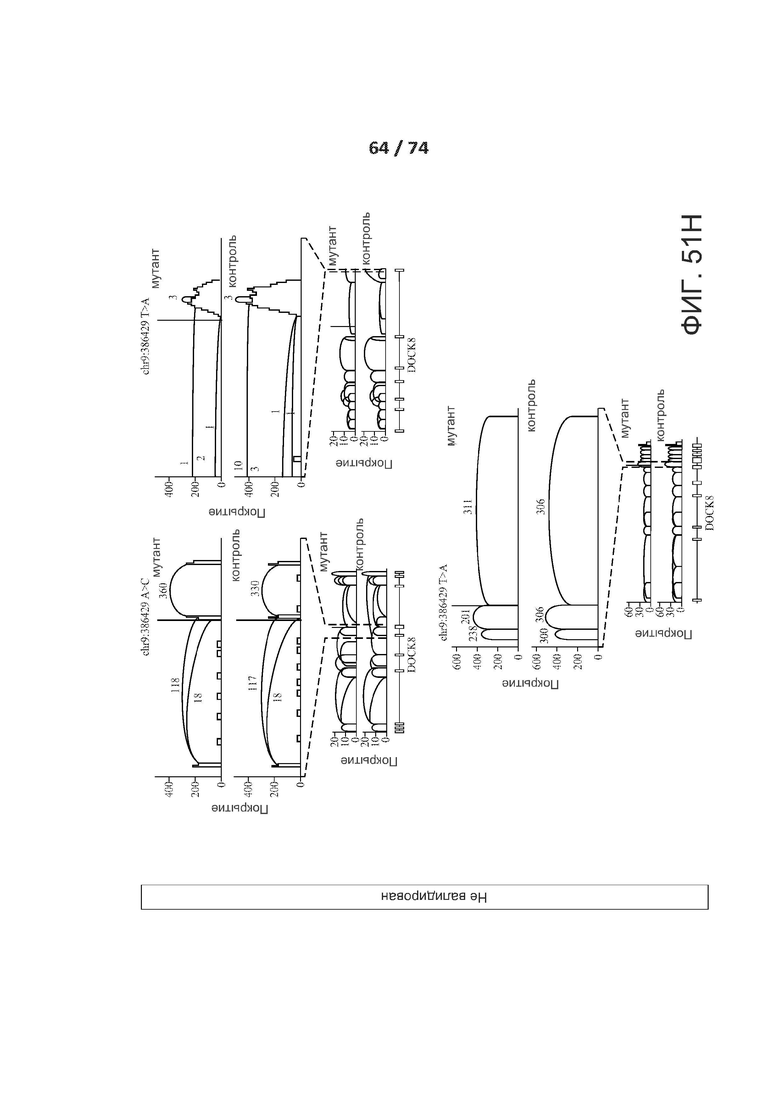
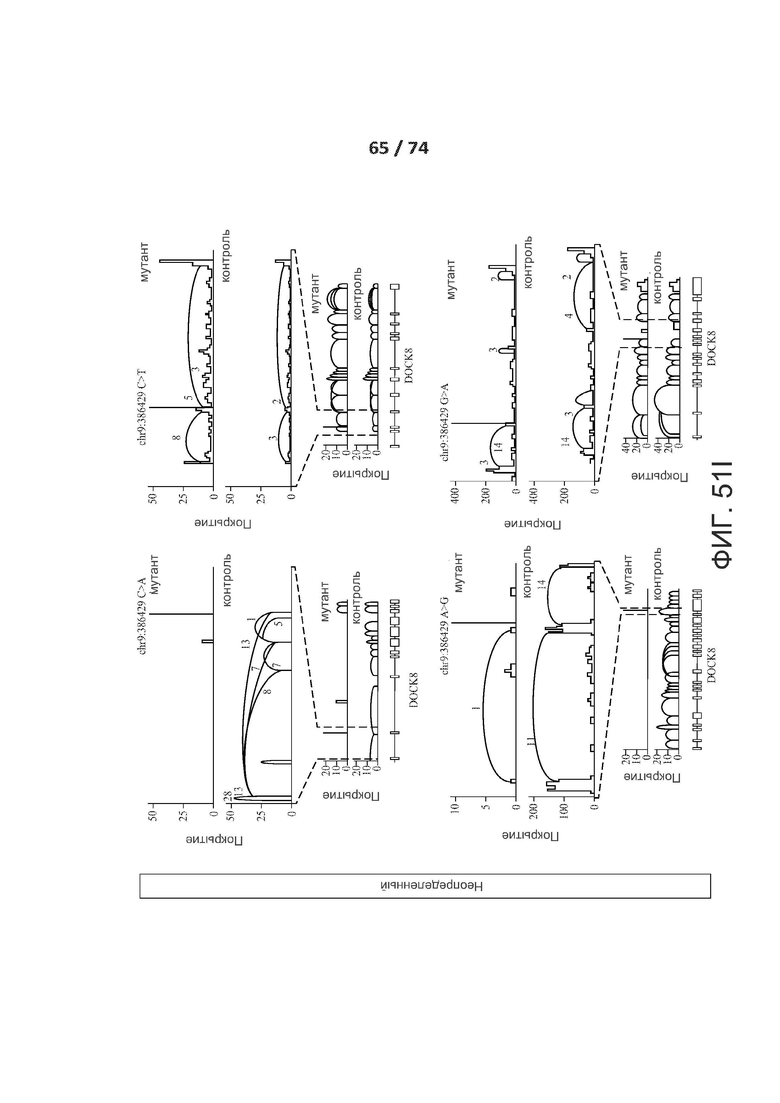
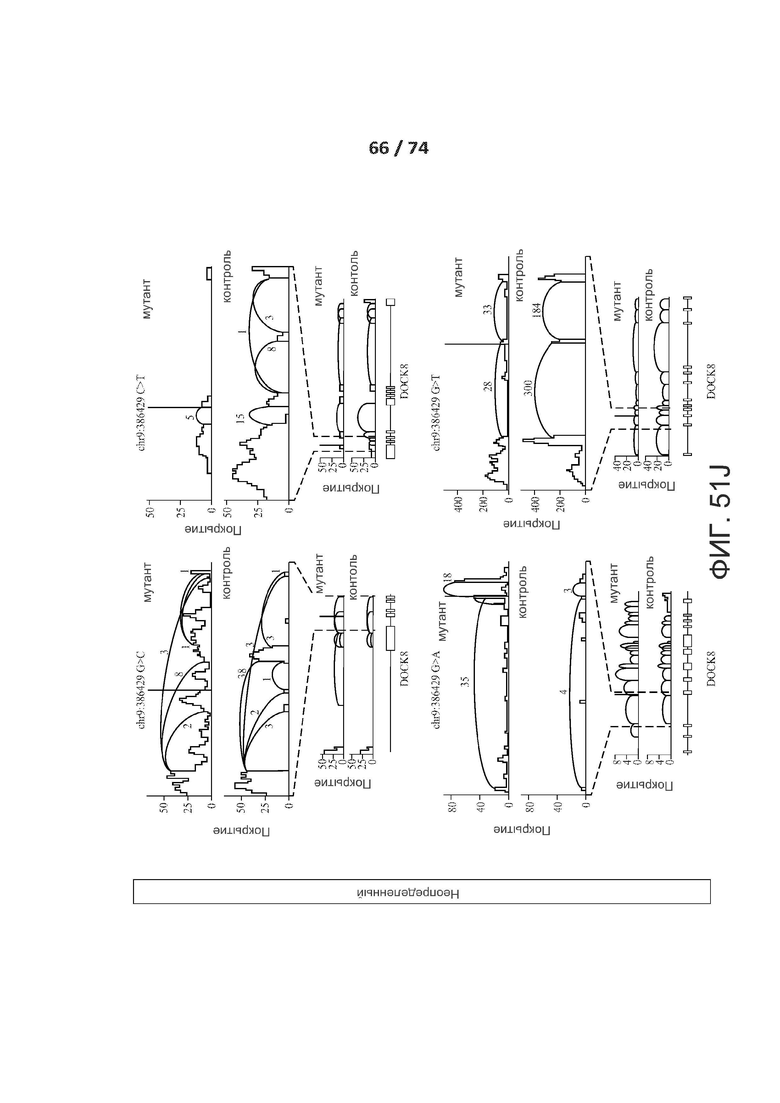
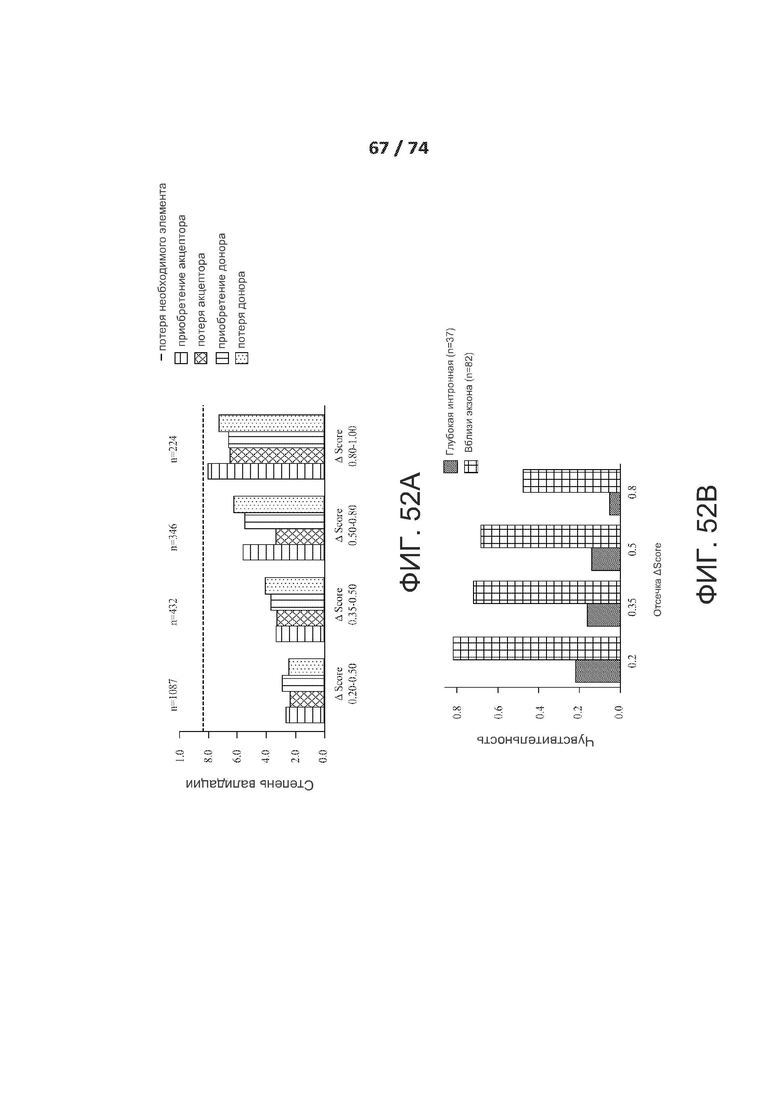
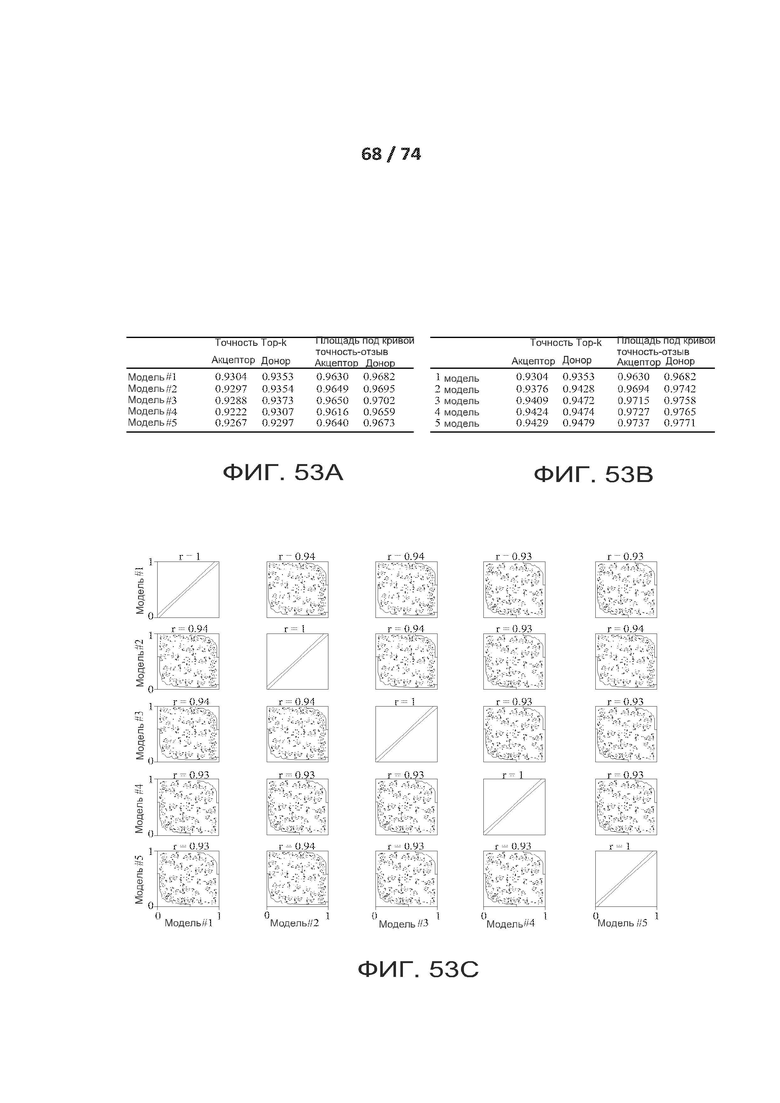

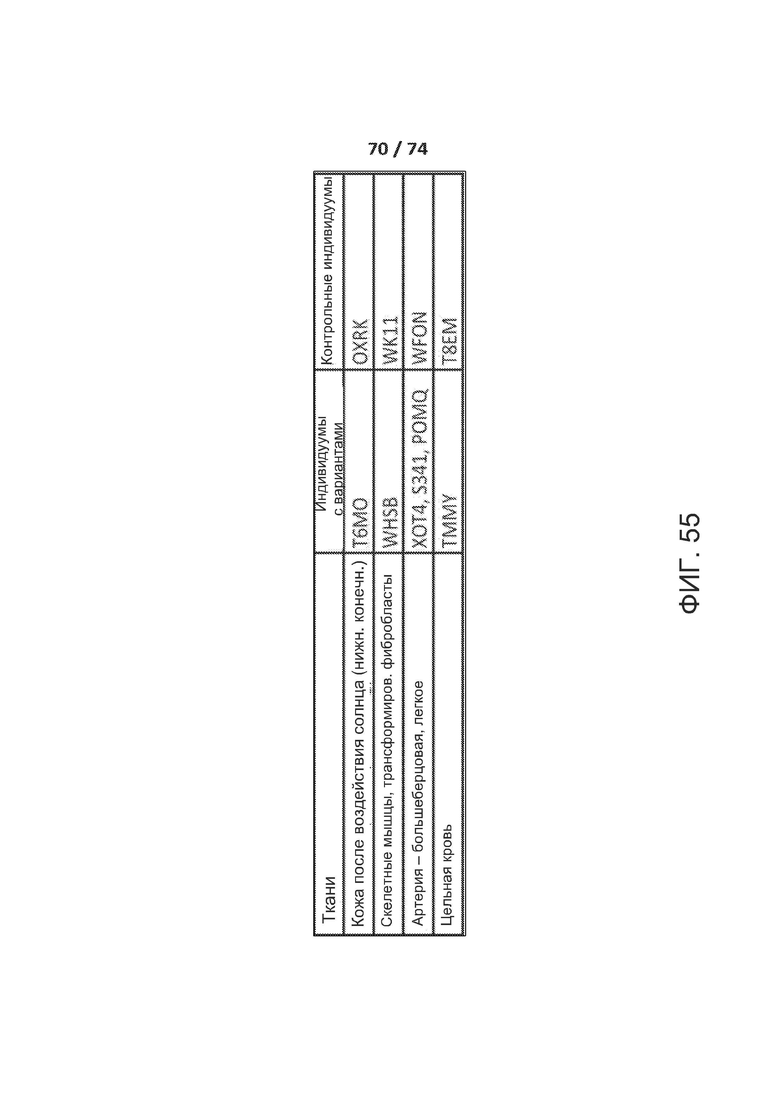
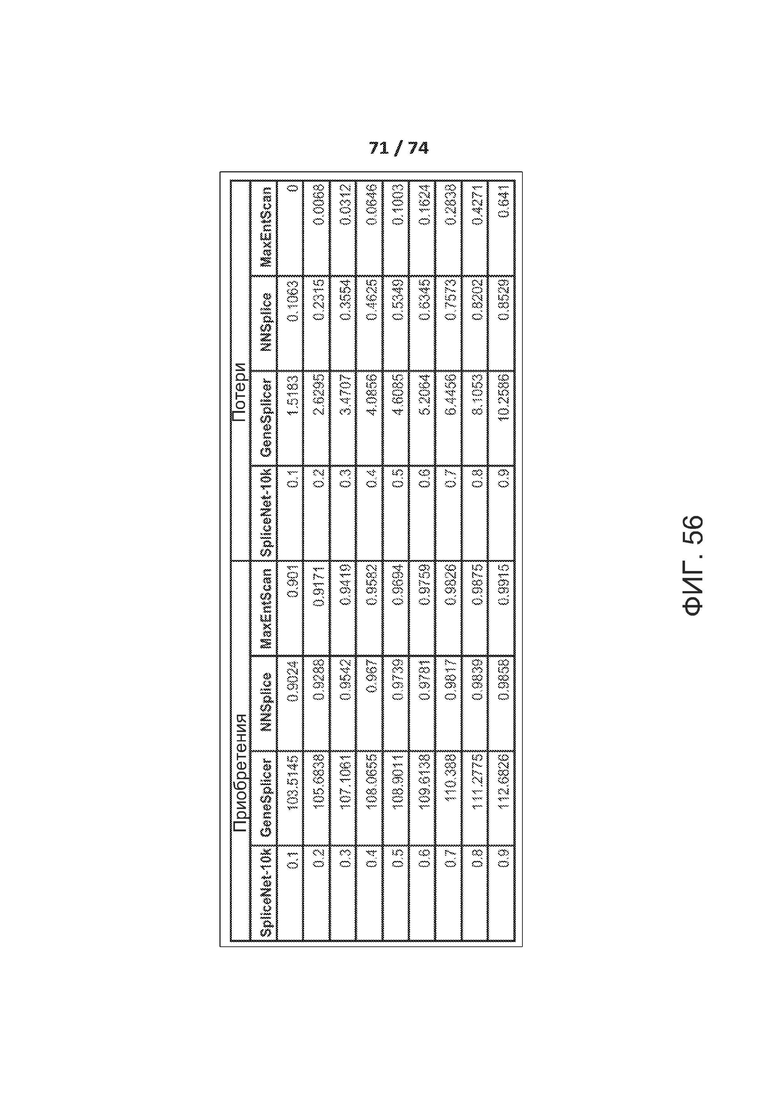
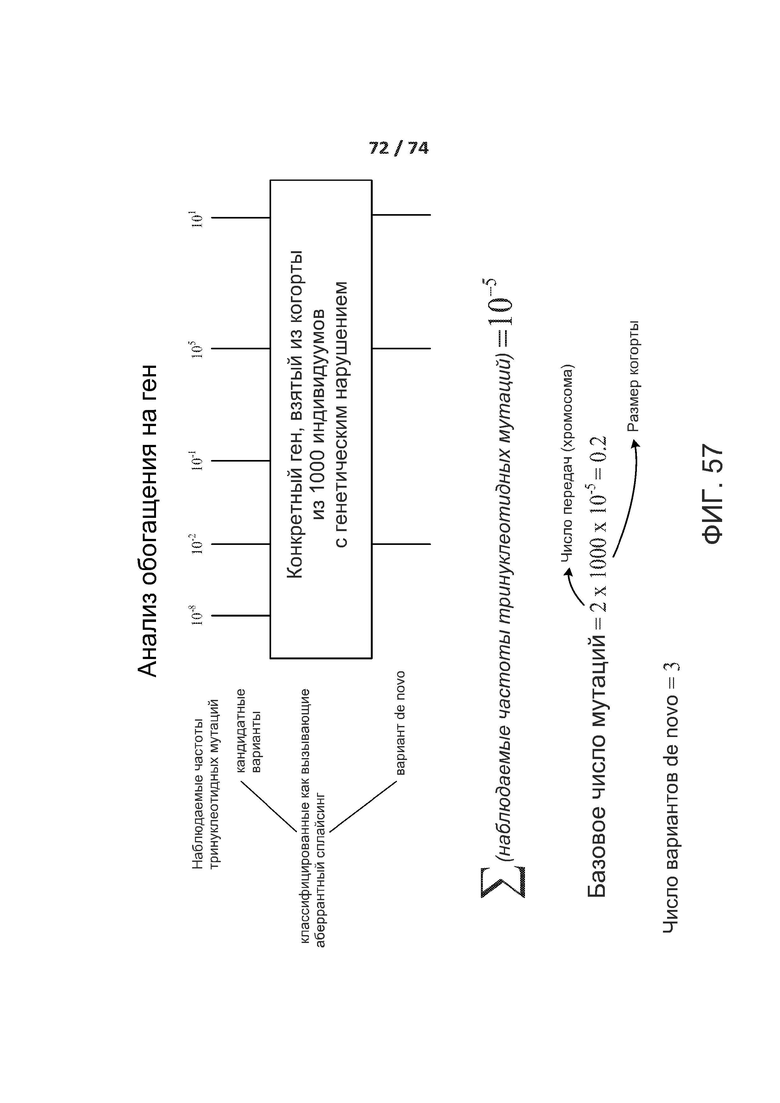
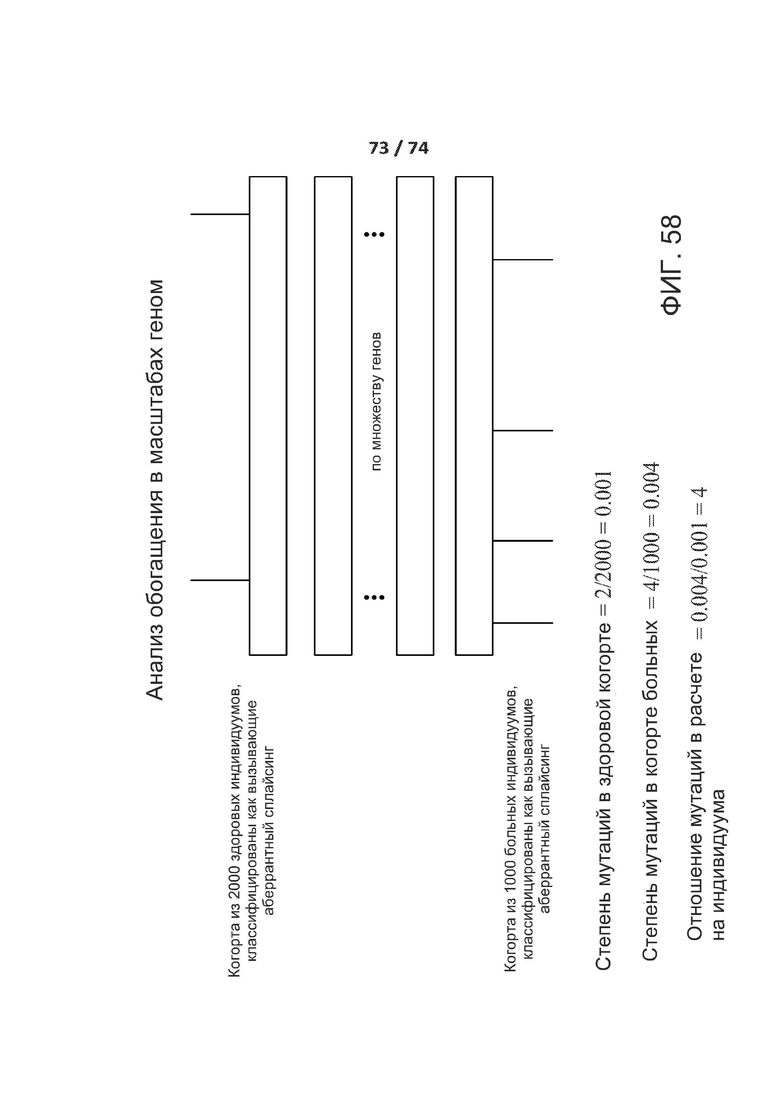
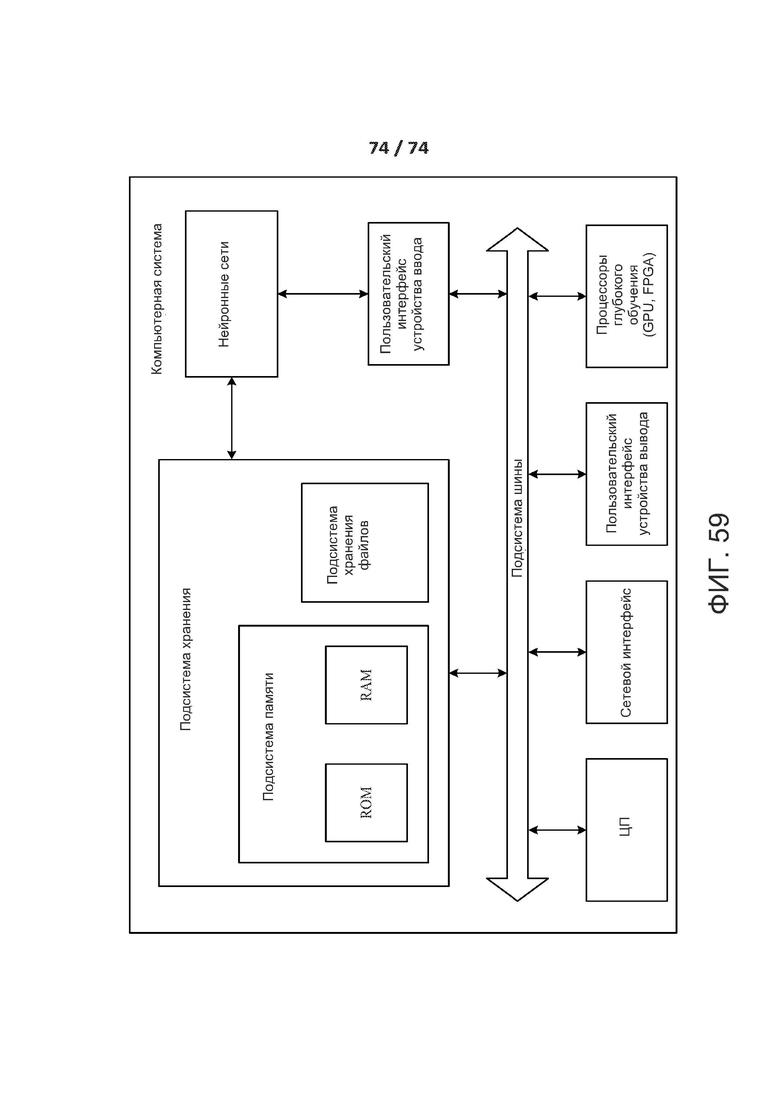
Изобретение относится к биотехнологии. Описан реализуемый с применением компьютера способ предсказания правдоподобия сайтов сплайсинга в пре-мРНК геномных последовательностях. Способ включает: получение пре-мРНК геномных последовательностей путем секвенирования пре-мРНК транскриптов и обучение разреженной сверточной нейронной сети, ACNN, на обучающих примерах пре-мРНК нуклеотидных последовательностей, включая по меньшей мере 50000 обучающих примеров донорных сайтов сплайсинга, по меньшей мере 50000 обучающих примеров акцепторных сайтов сплайсинга и по меньшей мере 100000 обучающих примеров сайтов, не связанных со сплайсингом, и обученная ACNN генерирует тройные оценки для оценки правдоподобия того, что каждый нуклеотид в целевых нуклеотидах является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом. При этом указанное обучение включает: ввод закодированных кодированием с одним активным состоянием обучающих примеров нуклеотидных последовательностей, причем каждая нуклеотидная последовательность содержит по меньшей мере 401 нуклеотид, с по меньшей мере одним целевым нуклеотидом и контекстом из по меньшей мере 200 фланкирующих нуклеотидов с каждой стороны, в направлении 5’ и в направлении 3’ от целевого нуклеотида; и корректировку, путем обратного распространения, параметров фильтров в указанной ACNN для предсказания оценок правдоподобия того, что каждый целевой нуклеотид в указанной нуклеотидной последовательности является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом; причем обученная ACNN получает в качестве входа пре-мРНК нуклеотидную последовательность из по меньшей мере 401 нуклеотида, которая закодирована кодированием с одним активным состоянием и которая включает по меньшей мере один целевой нуклеотид и контекст из по меньшей мере 200 фланкирующих нуклеотидов с каждой стороны. Также описана система для предсказания правдоподобия сайтов сплайсинга в пре-мРНК геномных последовательностях, включающая один или более процессоров, связанных с памятью, причем в память загружены компьютерные команды, которые при исполнении на указанных процессорах реализуют действия, включающие: обучение разреженной сверточной нейронной сети, ACNN, на обучающих примерах пре-мРНК нуклеотидных последовательностей, включая по меньшей мере 50000 тренировочных примеров донорных сайтов сплайсинга, по меньшей мере 50000 тренировочных примеров акцепторных сайтов сплайсинга и по меньшей мере 100000 тренировочных примеров сайтов, не связанных со сплайсингом, и обученная ACNN генерирует тройные оценки для оценки правдоподобия того, что каждый нуклеотид в целевых нуклеотидах является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом. Изобретение расширяет ассортимент средств для обучения (тренировки) глубоких сверточных нейронных сетей. 2 н. и 39 з.п. ф-лы, 59 ил., 3 табл.
1. Реализуемый с применением компьютера способ предсказания правдоподобия сайтов сплайсинга в пре-мРНК геномных последовательностях, где указанный способ включает:
- получение пре-мРНК геномных последовательностей путем секвенирования пре-мРНК транскриптов и
- обучение разреженной сверточной нейронной сети, ACNN, на обучающих примерах пре-мРНК нуклеотидных последовательностей, включая по меньшей мере 50000 обучающих примеров донорных сайтов сплайсинга, по меньшей мере 50000 обучающих примеров акцепторных сайтов сплайсинга и по меньшей мере 100000 обучающих примеров сайтов, не связанных со сплайсингом, где указанное обучение включает:
ввод закодированных кодированием с одним активным состоянием обучающих примеров нуклеотидных последовательностей, причем каждая нуклеотидная последовательность содержит по меньшей мере 401 нуклеотид, с по меньшей мере одним целевым нуклеотидом и контекстом из по меньшей мере 200 фланкирующих нуклеотидов с каждой стороны, в направлении 5’ и в направлении 3’ от целевого нуклеотида; и
корректировку, путем обратного распространения, параметров фильтров в указанной ACNN для предсказания оценок правдоподобия того, что каждый целевой нуклеотид в указанной нуклеотидной последовательности является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом;
причем обученная ACNN получает в качестве входа пре-мРНК нуклеотидную последовательность из по меньшей мере 401 нуклеотида, которая закодирована кодированием с одним активным состоянием и которая включает по меньшей мере один целевой нуклеотид и контекст из по меньшей мере 200 фланкирующих нуклеотидов с каждой стороны; и
обученная ACNN генерирует тройные оценки для оценки правдоподобия того, что каждый нуклеотид в целевых нуклеотидах является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом.
2. Реализуемый с применением компьютера способ по п. 1, где каждый из обучающих примеров пре-мРНК нуклеотидных последовательностей и указанной входной пре-мРНК нуклеотидной последовательности включает 2500 фланкирующих нуклеотидов с каждой стороны от целевого нуклеотида, за счет чего обученная ACNN конфигурируется для приема в качестве входа пре-мРНК нуклеотидной последовательности из по меньшей мере 5001 нуклеотида.
3. Реализуемый с применением компьютера способ по п. 1, где каждый из обучающих примеров пре-мРНК нуклеотидных последовательностей и указанной входной пре-мРНК нуклеотидной последовательности фланкирован 5000 расположенными в направлении 5’ контекстными нуклеотидами и 5000 расположенными в направлении 3’ контекстными нуклеотидами целевого нуклеотида, за счет чего обученная ACNN конфигурируется для приема в качестве входа пре-мРНК нуклеотидной последовательности из по меньшей мере 10001 нуклеотида.
4. Реализуемый с применением компьютера способ по п. 1, где каждый из указанных обучающих примеров пре-мРНК нуклеотидных последовательностей и указанной входной пре-мРНК нуклеотидной последовательности содержит целевой нуклеотид, фланкированный 500 нуклеотидами с каждой стороны от целевого нуклеотида.
5. Реализуемый с применением компьютера способ по п. 1, где каждый из указанных обучающих примеров пре-мРНК нуклеотидных последовательностей и указанной входной пре-мРНК нуклеотидной последовательности содержит целевой нуклеотид, фланкированный 1000 расположенными в направлении 5’ контекстными нуклеотидами и 1000 расположенными в направлении 3’ контекстными нуклеотидами.
6. Реализуемый с применением компьютера способ по любому из пп. 1-5, дополнительно включающий обучение указанной ACNN на по меньшей мере 150000 тренировочных примерах донорных сайтов сплайсинга, 150000 тренировочных примерах акцепторных сайтов сплайсинга и 800000000 тренировочных примерах сайтов, не связанных со сплайсингом.
7. Реализуемый с применением компьютера способ по любому из пп. 1-6, где указанная ACNN содержит группы остаточных блоков.
8. Реализуемый с применением компьютера способ по п. 7, где каждая группа остаточных блоков параметризируется числом сверточных фильтров в остаточных блоках, размером окна свертки остаточных блоков и фактором разрежения остаточных блоков.
9. Реализуемый с применением компьютера способ по любому из пп. 7, 8, где фактор разрежения изменяется неэкспоненциально между группами остаточных блоков.
10. Реализуемый с применением компьютера способ по любому из пп. 8, 9, где размеры окна свертки в разных группах остаточных блоков различаются.
11. Реализуемый с применением компьютера способ по любому из пп. 1-9, где указанная ACNN дополнительно включает по меньшей мере одну группу из четырех остаточных блоков и по меньшей мере одно соединение с пропуском, причем каждый остаточный блок содержит 32 сверточных фильтра, размер окна свертки, составляющий 11, и фактор разрежения, составляющий 1.
12. Реализуемый с применением компьютера способ по п. 1, где указанная ACNN обучена на и сконфигурирована для оценки входов, включающих целевой нуклеотид, фланкированный 500 расположенными в направлении 5’ контекстными нуклеотидами и 500 расположенными в направлении 3’ контекстными нуклеотидами, дополнительно включающий:
по меньшей две группы из четырех остаточных блоков и по меньшей мере два соединения с пропусками, причем каждый остаточный блок в первой группе имеет 32 сверточных фильтра, размер окна свертки, составляющий 11, и фактор разрежения, составляющий 1, и каждый остаточный блок во второй группе имеет 32 сверточных фильтра, размер окна свертки, составляющий 11, и фактор разрежения, составляющий 4.
13. Реализуемый с применением компьютера способ по п. 1, где указанная ACNN обучена на и сконфигурирована для оценки входов, включающих целевой нуклеотид, фланкированный 1000 расположенными в направлении 5’ контекстными нуклеотидами и 1000 расположенными в направлении 3’ контекстными нуклеотидами, дополнительно включающий:
по меньшей мере три группы из четырех остаточных блоков и по меньшей мере три соединения с пропуском, где каждый остаточный блок в первой группе имеет 32 сверточных фильтра, размер окна свертки, составляющий 11, и фактор разрежения, составляющий 1, каждый остаточный блок во второй группе имеет 32 сверточных фильтра, размер окна свертки, составляющий 11, и фактор разрежения, составляющий 4, и каждый остаточный блок в третьей группе имеет 32 сверточных фильтра, размер окна свертки, составляющий 21, и фактор разрежения, составляющий 19.
14. Реализуемый с применением компьютера способ по п. 1, где указанная ACNN обучена на и сконфигурирована для оценки входов, включающих целевой нуклеотид, фланкированный 5000 расположенными в направлении 5’ контекстными нуклеотидами и 5000 расположенными в направлении 3’ контекстными нуклеотидами, дополнительно включающий:
по меньшей мере четыре группы из четырех остаточных блоков и по меньшей мере четыре соединения с пропуском, где каждый остаточный блок в первой группе имеет 32 сверточных фильтра, размер окна свертки, составляющий 11, и фактор разрежения, составляющий 1, каждый остаточный блок во второй группе имеет 32 сверточных фильтра, размер окна свертки, составляющий 11, и фактор разрежения, составляющий 4, каждый остаточный блок в третьей группе имеет 32 сверточных фильтра, размер окна свертки, составляющий 21, и фактор разрежения, составляющий 19, и каждый остаточный блок в четвертой группе имеет 32 сверточных фильтра, размер окна свертки, составляющий 41, и фактор разрежения, составляющий 25.
15. Реализуемый с применением компьютера способ по любому из пп. 1-14, где указанные оценки для каждого целевого нуклеотида экспоненциально нормированы и в сумме равны единице.
16. Реализуемый с применением компьютера способ по любому из пп. 1-15, дополнительно включающий классификацию каждого целевого нуклеотида как донорного сайта сплайсинга, акцепторного сайта сплайсинга или сайта, не связанного со сплайсингом, на основании наивысшей оценки для данного целевого нуклеотида.
17. Реализуемый с применением компьютера способ по любому из пп. 1-16, где размерность входа составляет (Cu + L + Cd) x 4, где:
Cu представляет собой число расположенных в направлении 5’ контекстных нуклеотидов;
Cd представляет собой число расположенных в направлении 3’ контекстных нуклеотидов; и
L представляет собой число нуклеотидов в целевой нуклеотидной последовательности.
18. Реализуемый с применением компьютера способ по п. 17, где размерность выхода составляет L x 3.
19. Реализуемый с применением компьютера способ по п. 17, где размерность входа составляет (5000 + 5000 + 5000) x 4.
20. Реализуемый с применением компьютера способ по п. 19, где размерность выхода составляет 5000 x 3.
21. Реализуемый с применением компьютера способ по любому из пп. 7-14, где каждая группа остаточных блоков выдает промежуточный выход посредством обработки предшествующего входа, причем размерность указанного промежуточного выхода составляет (I-[{(W-1) * D} * A]) x N, где:
I представляет собой размерность предшествующего входа;
W представляет собой размер окна свертки остаточных блоков;
D представляет собой показатель разрежения свертки остаточных блоков;
A представляет собой число слоев разреженной свертки в указанной группе; и
N представляет собой число сверточных фильтров в остаточных блоках.
22. Реализуемый с применением компьютера способ по любому из пп. 1-21, где указанная ACNN пакетно оценивает обучающие примеры на протяжении эпохи.
23. Реализуемый с применением компьютера способ по любому из пп. 1-22, где обучающие примеры случайным образом группируют в пакеты, причем каждый пакет имеет заранее определенный размер пакета.
24. Реализуемый с применением компьютера способ по любому из пп. 1-23, где указанная ACNN итерирует оценку обучающих примеров на протяжении по меньшей мере десяти эпох.
25. Реализуемый с применением компьютера способ по п. 24, где указанный вход включает целевую нуклеотидную последовательность, имеющую два соседних целевых нуклеотида.
26. Реализуемый с применением компьютера способ по п. 25, где указанные два соседних целевых нуклеотида представляют собой аденин, сокращенно A, и гуанин, сокращенно G.
27. Реализуемый с применением компьютера способ по п. 25, где указанные два соседних целевых нуклеотида представляют собой гуанин, сокращенно G, и урацил, сокращенно D.
28. Реализуемый с применением компьютера способ по любому из пп. 1-6 и 15-27, где указанная ACNN параметризуется числом остаточных блоков, числом соединений с пропуском и числом остаточных связей.
29. Реализуемый с применением компьютера способ по любому из пп. 1-28, где разреженные свертки сохраняют частичные сверточные вычисления для повторного использования по мере обработки соседних нуклеотидов.
30. Реализуемый с применением компьютера способ по любому из пп. 1-29, где указанная ACNN включает сверточные меняющие размерность слои, которые изменяют пространственные размерности и размерности признаков предшествующего входа.
31. Реализуемый с применением компьютера способ по любому из пп. 7-14 и 28-30, где каждый остаточный блок содержит по меньшей мере один слой пакетной нормализации, по меньшей мере один слой блоков линейной ректификации (сокращенно ReLU), по меньшей мере один слой разреженной свертки и по меньшей мере одно остаточное соединение.
32. Реализуемый с применением компьютера способ по любому из пп. 7-14 и 28-30, где каждый остаточный блок содержит два слоя пакетной нормализации, два нелинейных слоя ReLU, два слоя разреженной свертки и одно остаточное соединение.
33. Система для предсказания правдоподобия сайтов сплайсинга в пре-мРНК геномных последовательностях, включающая один или более процессоров, связанных с памятью, причем в память загружены компьютерные команды, которые при исполнении на указанных процессорах реализуют действия, включающие:
обучение разреженной сверточной нейронной сети, ACNN, на обучающих примерах пре-мРНК нуклеотидных последовательностей, включая по меньшей мере 50000 тренировочных примеров донорных сайтов сплайсинга, по меньшей мере 50000 тренировочных примеров акцепторных сайтов сплайсинга и по меньшей мере 100000 тренировочных примеров сайтов, не связанных со сплайсингом, где указанное обучение включает:
ввод закодированных кодированием с одним активным состоянием примеров нуклеотидных последовательностей, где каждая нуклеотидная последовательность содержит по меньшей мере 401 нуклеотид, с по меньшей мере одним целевым нуклеотидом и контекстом из по меньшей мере 200 фланкирующих нуклеотидов с каждой стороны, в направлении 5’ и в направлении 3’; и
корректировку, путем обратного распространения, параметров фильтров в указанной ACNN для предсказания оценок правдоподобия того, что каждый целевой нуклеотид в указанной нуклеотидной последовательности является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом;
причем обученная ACNN получает в качестве входа пре-мРНК нуклеотидную последовательность из по меньшей мере 401 нуклеотида, которая закодирована кодированием с одним активным состоянием и которая включает по меньшей мере один целевой нуклеотид и контекст из по меньшей мере 200 фланкирующих нуклеотидов с каждой стороны, причем указанная последовательность получена путем секвенирования пре-мРНК транскрипта; и
обученная ACNN генерирует тройные оценки правдоподобия того, что каждый нуклеотид в целевых нуклеотидах является донорным сайтом сплайсинга, акцепторным сайтом сплайсинга или сайтом, не связанным со сплайсингом.
34. Система по п. 33, где указанная ACNN обучена на 150000 тренировочных примерах донорных сайтов сплайсинга, 150000 тренировочных примерах акцепторных сайтов сплайсинга и 800000000 тренировочных примерах сайтов, не связанных со сплайсингом.
35. Система по любому из пп. 33 или 34, где ACNN содержит группы остаточных блоков.
36. Система по п. 35, где каждая группа остаточных блоков параметризируется числом сверточных фильтров в остаточных блоках, размером окна свертки остаточных блоков и фактором разрежения остаточных блоков.
37. Система по п. 36, где фактор разрежения неэкспоненциально меняется между группами остаточных блоков.
38. Система по п. 37, где размеры окна свертки в разных группах остаточных блоков различаются.
39. Система по любому из пп. 33-38, где указанная ACNN обучена на одном или более обучающих серверах.
40. Система по любому из пп. 33-39, где указанная обученная ACNN размещена на одном или большем числе рабочих серверов, которые получают входные последовательности от запрашивающих клиентов.
41. Система по любому из пп. 33-40, где указанные рабочие серверы обрабатывают входные последовательности посредством стадий ввода и вывода указанной ACNN с генерацией выходных данных, которые передаются указанным клиентам.
| ОБНАРУЖЕНИЕ СКРЫТОГО ОБЪЕКТА | 2004 |
|
RU2371735C2 |
| WO 2016201564 А1, 22.12.2016 | |||
| ДИНАМИЧЕСКОЕ КОНФИГУРИРОВАНИЕ, ВЫДЕЛЕНИЕ И РАЗВЕРТЫВАНИЕ ВЫЧИСЛИТЕЛЬНЫХ СИСТЕМ | 2007 |
|
RU2429529C2 |

