Область изобретения
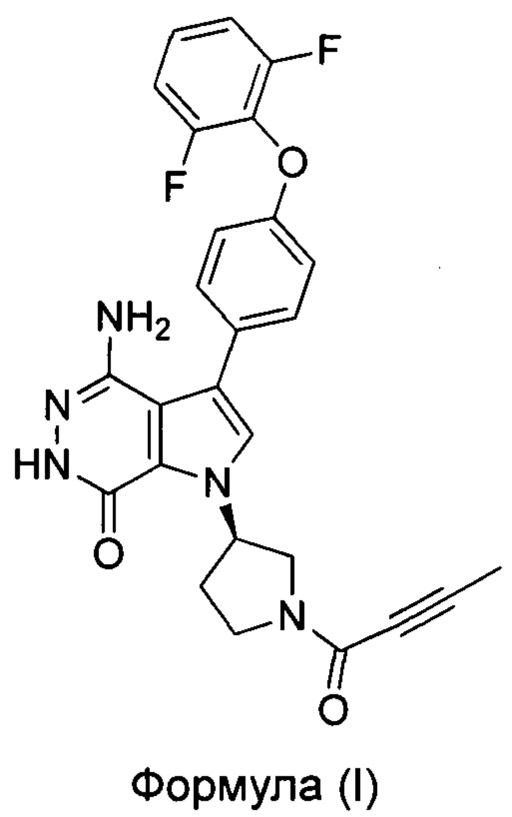
Настоящее изобретение относится к кристаллической форме ингибитора тирозинкиназы Брутона и способу ее получения. В частности, настоящее изобретение относится к кристаллической форме (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1Н-пирроло[2,3-d]пиридазин-7(6Н)-она и способу ее получения. Соединение формулы (I), полученное в соответствии с методом настоящего изобретения, может быть использовано в лечении В-клеточных злокачественных новообразований и аутоиммунных заболеваний.
Уровень техники изобретения
Иммунные клетки, как правило, можно разделить на Т-клетки и В-клетки, при этом основная функция В-клеток заключается в секреции различных антител, цель которых защитить организм от всех видов инвазии извне. Тирозинкиназа Брутона (ВТК - от англ. Bruton tyrosine protein kinase) входит в подсемейство тирозин протеинкиназ, и принадлежит Tec-семейству киназ. В основном она экспрессируется в В-клетках и распределяется по лимфатической, кроветворной и гематологической системам. В-клеточный рецептор (ВКР или BCR - от англ. В-cell receptor) играет решающую роль в регуляции пролиферации и выживаемости различных лимфом, выбранных из подвидов хронического лимфолейкоза (ХЛЛ или CLL - от англ. chronic lymphocytic leukemia) и неходжкинская лимфома (НХЛ или NHL - от англ. non-Hodgkin lymphoma), мантийноклеточная лимформа (МКЛ или MCL - от англ. mantle cell lymphoma) и диффузная В-крупноклеточная лимфома (ДВККЛ или DLBCL - от англ. diffuse large B-cell lymphoma). Кроме того, в клинической практике доказано влияние В-клеток на патогенез ревматоидного артрита, системной красной волчанки, рассеянного склероза и других иммунных заболеваний. Тирозинкиназа Брутона (ВТК) является ключевой протеинкиназой в сигнальном пути В-клетончого рецептора. Она способна регулировать созревание и дифференцирование нормальных В-клеток, а также тесно связана с различными заболеваниями, связанными с нарушениями В-клеточной лимфоидной ткани. Таким образом, низкомолекулярный ингибитор нацеленный на ВТК может быть предпочтителен для лечения злокачественных опухолей В-клеток и аутоиммунных заболеваний.
Ибрутиниб - низкомолекулярный ингибитор ВТК первого поколения, совместно разработанный компаниями Pharmacyclics и Janssen. Он был первым одобрен FDA (от англ. Food and Drug Administration - Управление по санитарному надзору за качеством пищевых продуктов и медикаментов) для лечения мантийноклеточной лимформы (МКЛ) в ноябре 2013 и впоследствии был одобрен для лечения хронического лимфоцитарного лейкоза (ХЛЛ) в феврале 2014. Ибрутиниб необратимо связывается с цистеином 481 в АТФ-связывающем домене тироксиназы Брутона через рецептор Майкла, тем самым ингибируя передачу нисходящего сигнала ВТК, и эффективно контролируя рост опухолевых клеток.
PCT/US14/61393 относится к соединению формулы (I), то есть к (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1Н-пирроло[2,3-d]пиридазин-7(6Н)-ону. Это соединение является новым ингибитором тирозинкиназы Бутона. Оно было улучшено с точки зрения киназной селективности, клинической эффективности или показаний к применению и безопасности. Тем не менее, на кристаллической форме соединения согласно данной заявке на патент, исследований не проводилось.
Кристаллическая структура фармацевтически активного компонента часто влияет на химическую стабильность препарата. Различные условия кристаллизации и условия хранения могут привести к изменениям в кристаллической структуре соединения, а иногда приводить к образованию других кристаллических форм. Обычно аморфный препарат не имеет регулярной кристаллической структуры, и часто имеет другие дефекты, такие как плохая устойчивость продукта, тонкая кристаллизация, сложная фильтрация, легкая агломерация и плохая ликвидность. Поэтому необходимо улучшать различные свойства вышеуказанного продукта. Существует необходимость в поиске новой кристаллической формы с высокой чистотой и хорошей химической устойчивостью.
Описание изобретения
Целью настоящего изобретения является обеспечение кристаллической формы I (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1Н-пирроло[2,3-d]пиридазин-7(6Н)-она и способ ее получения.
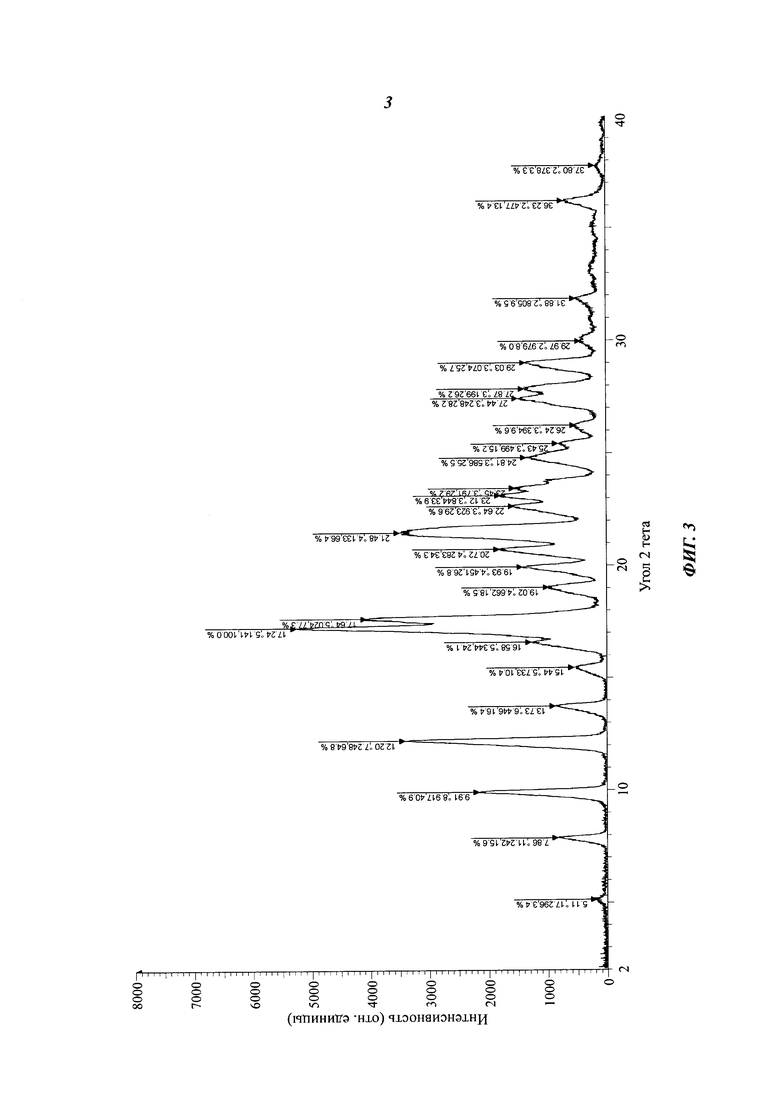
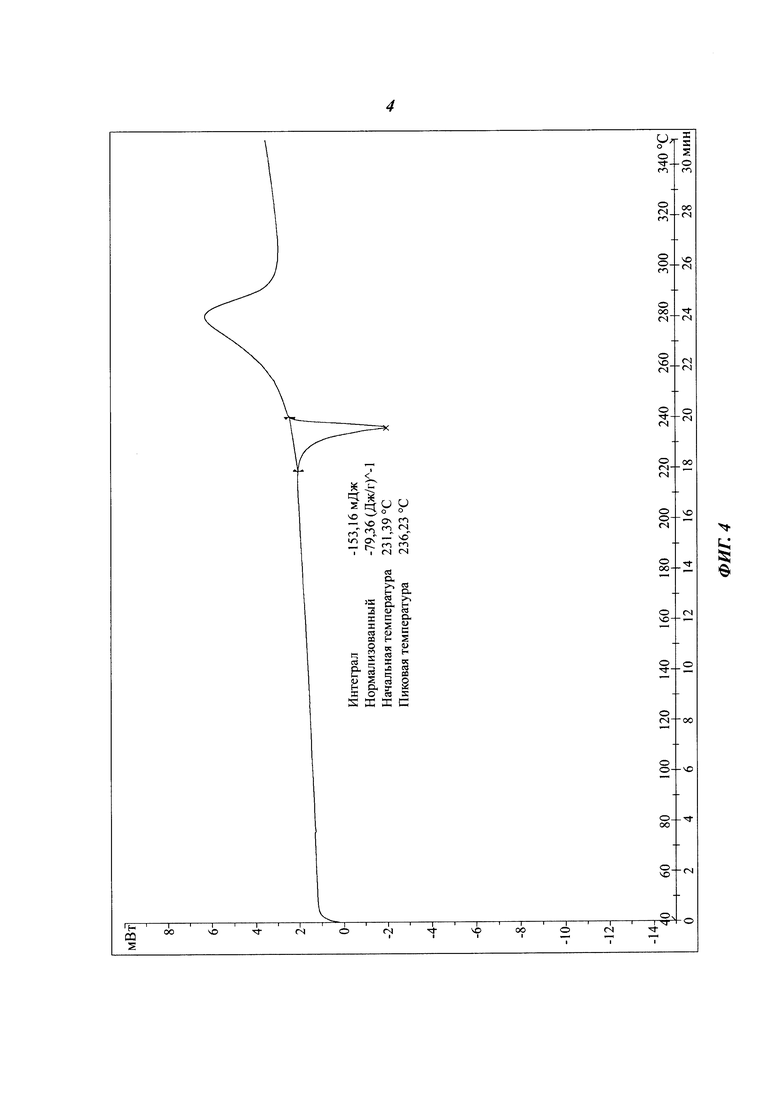
Заявитель исследовал ряд продуктов кристаллизации соединения формулы (I), полученных в различных условиях кристаллизации. Были проведены рентген-дифракционный анализ и дифференциальная сканирующая калориметрия (ДСК) полученных продуктов кристаллизации. Было установлено, что стабильная кристаллическая форма, которая называется кристаллическая форма I, может быть получена в условиях кристаллизации согласно настоящему изобретению. Спектр ДСК кристаллической формы I, согласно настоящей заявке, имеет эндотермический пик плавления при 236,23°С. Спектр рентгеновской порошковой дифрактометрии, представленный углом 2θ и межплоскостным расстоянием, получен с помощью излучения Cu-Кα, который имеет характеристические пики при 2θ±0,2: 9,91, 12,20, 17,24, 17,64 и 21,48.
Так же кристаллическая форма I имеет характеристические пики при 2θ±0,2: 7,86, 9,91, 12,20, 13,73, 17,24, 17,64, 19,02, 19,93, 20,72, 21,48, 22,64, 24,81, 27,44 и 27,87.
Спектр рентгеновской порошковой дифрактометрии кристаллической формы I показан на фиг.3. На нем имеются характеристические пики при 2θ±0,2: 5,11 (17,30), 7,86 (11,24), 9,91 (8,92), 12,20 (7,25), 13,73 (6,45), 15,44 (5,73), 17,24 (5,14), 17,64 (5,02), 19,02 (4,66), 19,93 (4,45), 20,72 (4,28), 21,48 (4,13), 22,64 (3,92), 23,12 (3,84), 24,81 (3,59), 25,43 (3,50), 26,24 (3,39), 27,44 (3,25), 27,87 (3,20) и 29,03 (3,07).
В настоящем изобретении также предложен способ получения кристаллической формы I соединения формулы (I). В частности, способ включает следующие стадии:
1) растворение твердого (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1Н-пирроло[2,3-d]пиридазин-7(6Н)-она находящегося в любой кристаллической форме или аморфном состоянии в соответствующем количестве органического растворителя при нагревании с последующим охлаждением раствора с получением кристаллического осадка; и
2) фильтрация кристаллов с последующей промывкой и сушкой.
На стадии 1) растворитель выбирается из любого одного или более спиртов, кетонов, нитрилов, простых и сложных эфиров, каждый из которых имеет 4 или меньше атомов углерода, или смешанного растворителя из одного или более растворителей, упомянутых выше, и воды. Предпочтительный растворитель - метанол, этанол, изопропанол, ацетон, этилацетат, ацетонитрил, тетрагидрофуран или этанол/вода, N,N-диметилформамид/вода, или 1,4-диоксан/вода. Для кристаллизации может быть использован простой растворитель или смешанный растворитель из вышеупомянутых органических растворителей.
При этом, наиболее предпочтительным простым растворителем является этанол.
Метод рекристаллизации не ограничен конкретным образом и может осуществляться путем обычного процесса рекристаллизации. Например, вещество, то есть соединение формулы (I), может быть растворено в органическом растворителе при нагревании, затем раствор медленно охлаждают для осаждения кристаллов при перемешивании. После завершения кристаллизации, желаемое кристаллическое вещество можно получить посредством фильтрации и сушки. Так, например, кристаллическое вещество, полученное путем фильтрации, сушат в вакууме при пониженном давлении, при температуре от 30 до 100°С, предпочтительно 40-60°С, для удаления растворителя перекристаллизации.
Полученная кристаллическая форма исследуется путем дифференциальной сканирующей калориметрии (ДСК) и рентген-дифракционного анализа. При этом также определяется наличие остаточного растворителя в полученном кристалле.
Кристалл соединения формулы (I), полученный согласно способу настоящего изобретения, не содержит или содержит относительно низкое количество остаточного растворителя, что соответствует требованию Национальной Фармакопеи в отношении ограничения остаточного растворителя в лекарственных препаратах. Таким образом, кристаллическая форма, согласно настоящему изобретению, может быть успешно использована в качестве фармацевтического активного ингредиента.
Результаты эксперимента показывают, что в условиях освещения, высокой температуры и высокой влажности, устойчивость кристаллической формы I соединения формулы (I), которая изготовлена в соответствии с настоящим изобретением, значительно лучше, чем у аморфного образца. Кристаллическая форма I также стабильна в условиях измельчения, давления и нагрева, что отвечает медицинским требованиям производства, транспортировки и хранения. Процесс ее получения стабилен, повторяем и управляем, что предпочтительно для промышленного производства.
Описание графических материалов
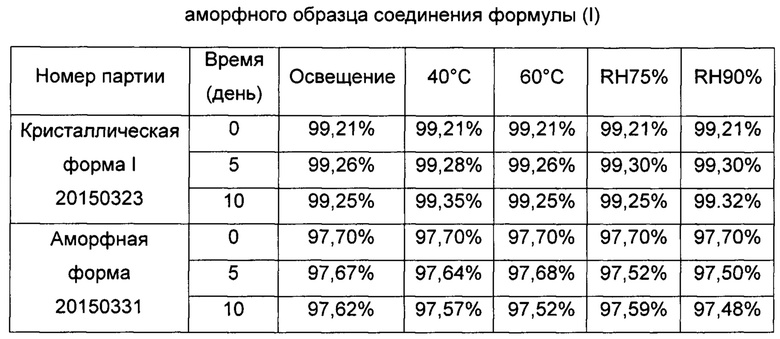
На фиг. 1 представлен спектр рентгеновской порошковой дифрактометрии твердой аморфной формы соединения формулы (I).
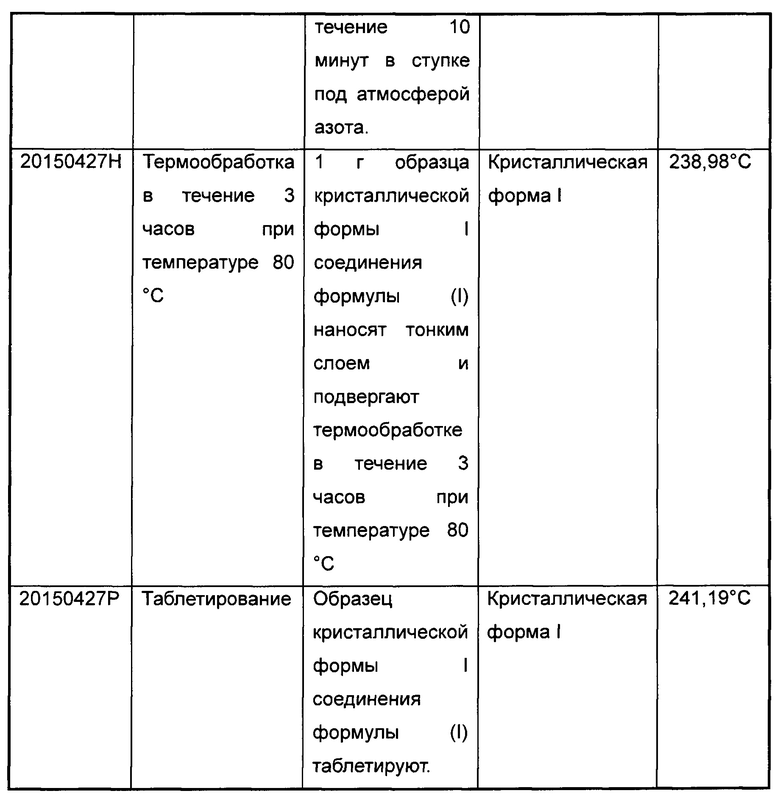
На фиг. 2 представлен ДСК спектр твердой аморфной формы соединения формулы (I).
На фиг. 3 представлен спектр рентгеновской порошковой дифрактометрии кристаллической формы I соединения формулы (I).
На фиг. 4 представлен ДСК спектр кристаллической формы I соединения формулы (I).
Подробное описание изобретения
Настоящее изобретение подробно иллюстрируется следующими примерами. Примеры согласно настоящему изобретению предназначены лишь для описания технического решения настоящего изобретения и не должны рассматриваться как ограничение объема настоящего изобретения.
Испытательные приборы, используемые в экспериментах
1. Спектры ДСК
Тип прибора: Mettler Toledo DSC 1 Staree System
Газ продувки: Азот
Скорость нагрева: 10,0°С/мин
Диапазон температур: 40-350°С
2. Рентген-дифракционный анализ
Тип прибора: Bruker D8 Focus X-Ray порошковый дифрактометр
Луч: монохромный Cu-Ка луч (λ равен 1,5406)
Режим сканирования: θ/28, диапазон сканирования: 2-40°
Напряжение: 40 кВ, Сила тока: 40 мА
Пример 1. Способ получения (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1Н-пирроло[2,3-d]пиридазин-7(6Н)-она, включающий в себя три этапа:
Этап первый: Синтез соединения 1b
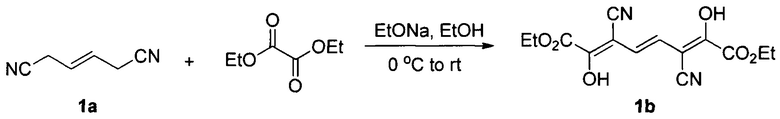
Раствор ацетата натрия в этаноле (160 мл, массовая доля 21%, 0,49 ммоль) добавляют в 110 мл этанола, в ледяную баню добавляют диэтилоксалат (64 мл, 0,47 моль). Смесь перемешивают в течение 30 минут. Затем, добавляют раствор (E)-гекс-3-енонитрила 1а (16 г, 0,15 ммоль) в этаноле (30 мл), и смесь оставляют перемешиваться на ночь при комнатной температуре. После охлаждения в ледяной ванне суспензию отфильтровывают. Осадок отмывают небольшим количеством этанола, а затем растворяют в 380 мл воды. Раствор подкисляют соляной кислотой до рН 4, и осаждают большое количество твердого осадка. Осадок отфильтровывают, промывают водой и сушат, получая 11,9 г 1b в виде желтого твердого порошка.
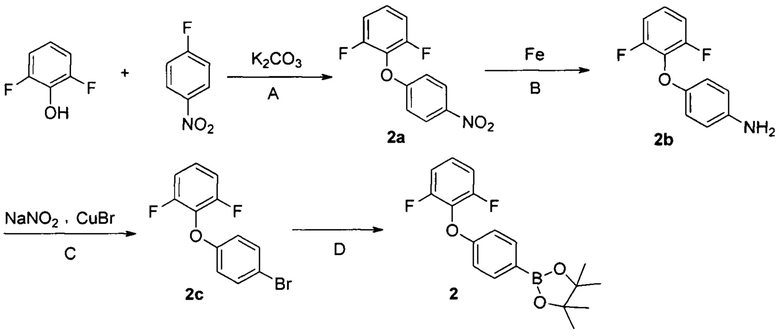
Этап второй: Получение соединения 2
Стадия А
2,6-Дифторфенол (3,0 г, 21,3 ммоль), 1-фтор-4-нитробензол (3,04 г, 23,4 ммоль) и карбонат калия (4,4 г, 32 ммоль) добавляют в 50 мл ацетонитрила, и смесь кипятят с обратным холодильником в течение 16 часов. Далее раствор охлаждают до комнатной температуры и удаляют растворители. Добавляют воду и раствор троекратно экстрагируют этилацетатом. Органические экстракты промывают водой и рассолом, сушат над сульфатом магния, фильтруют и концентрируют, получая 4,9 г соединения 2а в виде масла.
Стадия В
1,3-дифтор-2-(4-нитрофенокси)бензол 2а (4,9 г, 19,5 ммоль), 5 мл насыщенного раствора аммония хлорида и порошка железа (5,5 г, 97,5 ммоль) добавляют в 40 мл метанола, и смесь кипятят с обратным холодильником в течение 3 часов. Смесь фильтруют, добавляют воду к фильтрату и полученный раствор троекратно экстрагируют этилацетатом. Органические экстракты промывают водой и рассолом, сушат над сульфатом магния, фильтруют и концентрируют, получая 4,1 г соединения 2b в виде светло-желтого масла.
Масс-спектрометрия (ионизация электрораспылением) (МС (ИЭР)):m/z=222.1 [М+Н]+.
Стадия С
4-(2,6-дифторфенокси)анилин 2b (4,1 г, 18,5 ммоль) добавляют в 2М раствор серной кислоты (50 мл) при 0°С, затем добавляют водный раствор (20 мл) нитрита натрия (6,4 г, 92,7 ммоль). Смесь перемешивают в течение 40 минут, затем добавляют бромид меди (5,3 г, 37 ммоль). Полученную смесь кипятят с обратным холодильником 16 часов. Далее раствор охлаждают до комнатной температуры и троекратно экстрагируют этилацетатом. Органические экстракты промывают водой и рассолом, сушат над сульфатом магния, фильтруют и концентрируют, получая 1,6 г соединения 2с в виде бесцветного масла.
Стадия D
2-(4-бромфенокси)-1,3-дифторбензол 2с (1,6 г, 3,6 ммоль), бис(пинаколато)дибор (1,71 г, 6,7 ммоль), ацетат калия (830 мг, 8,4 ммоль) и Pd(PPh3)2Cl2 (126 мг, 0,18 ммоль) добавляют в 40 мл 1,4-диоксана, и смесь перемешивают под атмосферой азота при температуре 80°С в течение 16 часов. Далее раствор охлаждают до комнатной температуры и удаляют растворители. Остаток перегонки очищают хроматографией на силикагеле с получением 1,6 г вещества 2 в виде бесцветного масла.
Этап 3: Способ синтеза (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1Н-пирроло[2,3-d]пиридазин-7(6Н)-она
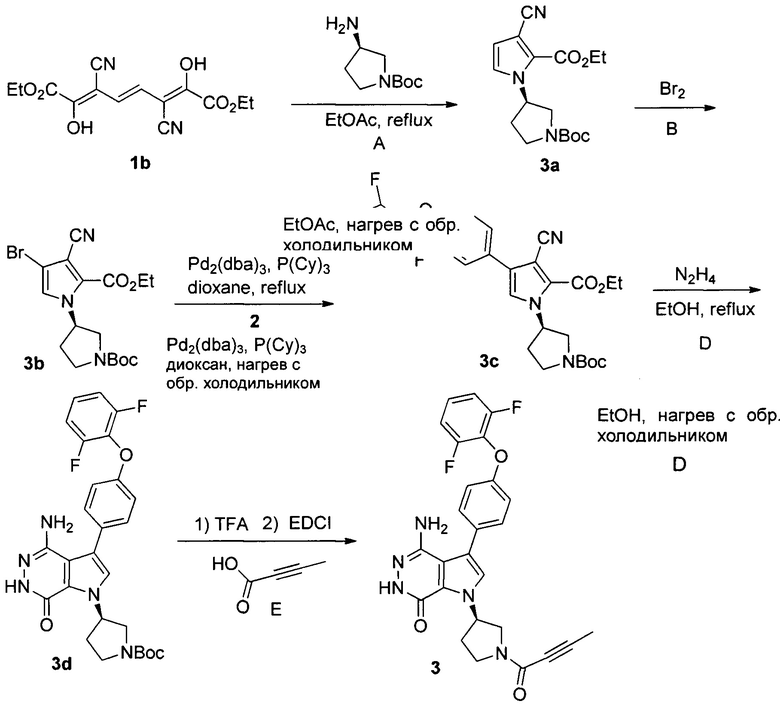
TFA - от англ. trifluoroacetic acid - трифторуксусная кислота,
EDCI - от англ. 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide-1-этил-3-(3-диметиламинопропил)-карбодиимид
Стадия А
1,5 г 1b добавляют в 84 мл этилацетата, и раствор нагревают до 60°С. Затем капельно добавляют раствор (21 мл) (R)-1-трет-бутоксикарбонил-3-аминопирролидина (1,41 г) в этилацетате. Полученную смесь кипятят с обратным холодильником 4 часа. Далее раствор охлаждают до комнатной температуры и удаляют растворители. Остаток очищают хроматографией на силикагеле с получением 0,686 г вещества 3а.
Стадия В
0,686 г 3а добавляют в 120 мл дихлорметана при 0°С, затем медленно добавляют по каплям раствор (5 мл) Br2 (3,7 г) в дихлорметане. Смесь перемешивают в течение 1,5 часов, а затем гасят 10% раствором тиосульфата натрия и насыщенным раствором бикарбоната натрия. Образовавшиеся две фазы разделяют, и водную фазу экстрагируют дихлорметаном. Объединенные органические экстракты обрабатывают избытком ди-трет-бутилдикарбоната (Boc2O), сушат над сульфатом натрия, фильтруют и концентрируют. Остаток перегонки очищают хроматографией на силикагеле с получением 0,342 г вещества 3b.
Стадия С
3b (198 мг, 0,48 ммоль), 2 (160 мг, 0,48 ммоль) и K3PO4⋅3H2O (188 мг, 0,72 ммоль) вводят в раствор 1,4-диоксан/вода (10 мл/1 мл) под атмосферой азота. Затем добавляют трис(дибензилиденацетон)дипалладий(0) (Pd2(dba)3) (22 мг, 0,024 ммоль) и трициклогексилфосфин (Р(Су)3) (14 мг, 0,048 ммоль). Полученную смесь кипятят с обратным холодильником под атмосферой азота 16 часов. После охлаждения до комнатной температуры полученную смесь фильтруют и концентрируют фильтрат. Остаток очищают хроматографией на силикагеле с получением 59 мг вещества 3с в виде белого твердого вещества.
МС (ИЭР): m/z=538 [М+Н]+.
Стадия D
3с (72 мг, 0,13 ммоль) и 1 мл N2H4⋅H2O добавляют в 5 мл этанола, и смесь кипятят с обратным холодильником в течение 16 часов. Далее раствор охлаждают до комнатной температуры и удаляют растворители. Остаток очищают хроматографией на силикагеле с получением 24 мг вещества 3d в виде белого твердого вещества.
МС (ИЭР): m/z=524 [М+Н]+.
Стадия Е
3d (40 мг, 0,08 ммоль) добавляют в 5 мл дихлорметана, затем добавляют 1 мл трифторуксусной кислоты. Смесь перемешивают при комнатной температуре в течение 3 часов, затем концентрируют с получением 49 мг 3е в виде масла. Тетроловую кислоту (13 мг, 0,16 ммоль), карбодиимид (31 мг, 0,16 ммоль) и ангидрид трифторуксусной кислоты (17 мг, 0,16 ммоль) добавляют в раствор (5 мл) 3е (49 мг) в дихлорметане. Полученную смесь перемешивают при комнатной температуре в течение 18 часов и затем концентрируют. Остаток очищают хроматографией на силикагеле с получением 20 мг указанного вещества 3 в виде белого твердого вещества. Рентген-дифракционный спектр твердого образца показан на фиг. 1, на котором отсутствуют характеристические пики поглощения кристаллического вещества. Спектр ДСК твердого образца показан на фиг. 2. В спектре отсутствует эндотермический пик плавления ниже 350°С. Таким образом, продукт представляет собой аморфное твердое тело.
МС (ИЭР): m/z=490 [М+Н]+.
Пример 2
Соединение формулы (I) (1,0 г, 2,04 ммоль) (полученное согласно Примеру 1) вносят в одногорлую колбу объемом 50 мл с последующим добавлением 15 мл безводного этанола. Смесь кипятят с обратным холодильником, пока раствор не станет прозрачным. Раствор охлаждают и осаждают большое количество твердого вещества. Смесь фильтруют и сушат с получением твердого вещества (805 мг, выход: 80,5%). Спектр рентгеновской порошковой дифрактометрии кристаллического образца приведен на фиг. 3. Спектр имеет характеристические пики при значениях около 5,11 (17,30), 7,86 (11,24), 9,91 (8,92), 12,20 (7,25), 13,73 (6,45), 15,44 (5,73), 17,24 (5,14), 17,64 (5,02), 19,02 (4,66), 19,93 (4,45), 20,72 (4,28), 21,48 (4,13), 22,64 (3,92), 23,12 (3,84), 24,81 (3,59), 25,43 (3,50), 26,24 (3,39), 27,44 (3,25), 27,87 (3,20) и 29,03 (3,07). ДСК спектр имеет резкий эндотермический пик плавления при 236,23°С, что видно на фиг. 4. Кристаллическая форма определена как кристаллическая форма I.
Пример 3
Соединение формулы (I) (1,0 г, 2,04 ммоль) (полученное согласно Примеру 1) вносят в одногорлую колбу объемом 50 мл с последующим добавлением 15 мл безводного метанола. Смесь кипятят с обратным холодильником, пока раствор не станет прозрачным. Раствор охлаждают и осаждают большое количество твердого вещества. Смесь фильтруют и сушат с получением твердого вещества (765 мг, выход: 76,5%). После изучения и сравнения рентген-дифракционного спектра и ДСК спектра продукт идентифицируется как кристаллическая форма I.
Пример 4
Соединение формулы (I) (1,0 г, 2,04 ммоль) (полученное согласно Примеру 1) вносят в одногорлую колбу объемом 50 мл с последующим добавлением 15 мл изопропанола. Смесь кипятят с обратным холодильником, пока раствор не станет прозрачным. Раствор охлаждают и осаждают большое количество твердого вещества. Смесь фильтруют и сушат с получением твердого вещества (745 мг, выход: 74,5%). После изучения и сравнения рентген-дифракционного спектра и ДСК спектра продукт идентифицируется как кристаллическая форма I.
Пример 5
Соединение формулы (I) (1,0 г, 2,04 ммоль) (полученное согласно Примеру 1) вносят в одногорлую колбу объемом 50 мл с последующим добавлением 15 мл этилацетата. Смесь кипятят с обратным холодильником, пока раствор не станет прозрачным. Раствор охлаждают и осаждают большое количество твердого вещества. Смесь фильтруют и сушат с получением твердого вещества (690 мг, выход: 69,0%). После изучения и сравнения рентген-дифракционного спектра и ДСК спектра продукт идентифицируется как кристаллическая форма I.
Пример 6
Соединение формулы (I) (1,0 г, 2,04 ммоль) (полученное согласно Примеру 1) вносят в одногорлую колбу объемом 50 мл с последующим добавлением 10 мл ацетона. Смесь кипятят с обратным холодильником, пока раствор не станет прозрачным. Раствор охлаждают и осаждают большое количество твердого вещества. Смесь фильтруют и сушат с получением твердого вещества (660 мг, выход: 66,0%). После изучения и сравнения рентген-дифракционного спектра и ДСК спектра продукт идентифицируется как кристаллическая форма I.
Пример 7
Соединение формулы (I) (1,0 г, 2,04 ммоль) (полученное согласно Примеру 1) вносят в одногорлую колбу объемом 50 мл с последующим добавлением 10 мл ацетонитрила. Смесь кипятят с обратным холодильником, пока раствор не станет прозрачным. Раствор охлаждают и осаждают большое количество твердого вещества. Смесь фильтруют и сушат с получением твердого вещества (810 мг, выход: 81,0%). После изучения и сравнения рентген-дифракционного спектра и ДСК спектра продукт идентифицируется как кристаллическая форма I.
Пример 8
Соединение формулы (I) (1,0 г, 2,04 ммоль) (полученное согласно Примеру 1) вносят в одногорлую колбу объемом 25 мл с последующим добавлением 3 мл тетрагидрофурана. Смесь кипятят с обратным холодильником, пока раствор не станет прозрачным. Раствор охлаждают и осаждают большое количество твердого вещества. Смесь фильтруют и сушат с получением твердого вещества (587 мг, выход: 58,7%). После изучения и сравнения рентген-дифракционного спектра и ДСК спектра продукт идентифицируется как кристаллическая форма I.
Пример 9
Соединение формулы (I) (1,0 г, 2,04 ммоль) (полученное согласно Примеру 1) вносят в одногорлую колбу объемом 25 мл с последующим добавлением 7 мл раствора этанол/вода (V:V составляет 1:1). Смесь кипятят с обратным холодильником, пока раствор не станет прозрачным. Раствор охлаждают и осаждают большое количество твердого вещества. Смесь фильтруют и сушат с получением твердого вещества (657 мг, выход: 65,7%). После изучения и сравнения рентген-дифракционного спектра и ДСК спектра продукт идентифицируется как кристаллическая форма I.
Пример 10
Соединение формулы (I) (1,0 г, 2,04 ммоль) (полученное согласно Примеру 1) вносят в одногорлую колбу объемом 25 мл с последующим добавлением 7 мл раствора N,N-диметилформамид/вода (V:V составляет 1:1). Смесь кипятят с обратным холодильником, пока раствор не станет прозрачным. Раствор охлаждают и осаждают большое количество твердого вещества. Смесь фильтруют и сушат с получением твердого вещества (600 мг, выход: 60,0%). После изучения и сравнения рентген-дифракционного спектра и ДСК спектра продукт идентифицируется как кристаллическая форма I.
Пример 11
Соединение формулы (I) (1,0 г, 2,04 ммоль) (полученное согласно Примеру 1) вносят в одногорлую колбу объемом 25 мл с последующим добавлением 10 мл раствора 1,4-диоксан/вода (V:V составляет 1:2). Смесь кипятят с обратным холодильником, пока раствор не станет прозрачным. Раствор охлаждают и осаждают большое количество твердого вещества. Смесь фильтруют и сушат с получением твердого вещества (793 мг, выход: 79,3%). После изучения и сравнения рентген-дифракционного спектра и ДСК спектра продукт идентифицируется как кристаллическая форма I.
Пример 12
Аморфный образец, изготовленный по Примеру 1, и образец кристаллической формы I, изготовленный согласно Примеру 2, были нанесены тонким слоем на поверхность в атмосфере воздуха, чтобы проверить и сравнить их стабильность в условиях освещения (4500 лк), нагрева (до 40°С, 60°С) и высокой влажности (Относительная влажность (ОВ) 75%, ОВ 90%) соответственно. Отборы проб проводят на пятый и десятый день. Чистоту проб определяют с помощью ВЭЖХ. Результаты эксперимента приведены в Таблице 1.

В дальнейшем кристаллическая форма I и аморфный образец были нанесены тонким слоем на поверхность в атмосфере воздуха, чтобы проверить и сравнить их стабильность в условиях освещения, высокой температуры, и высокой влажности, результаты исследования стабильности показали, что в условиях освещения, высокой температуры и высокой влажности стабильность кристаллической формы значительно лучше, чем у аморфного образца.
Пример 13
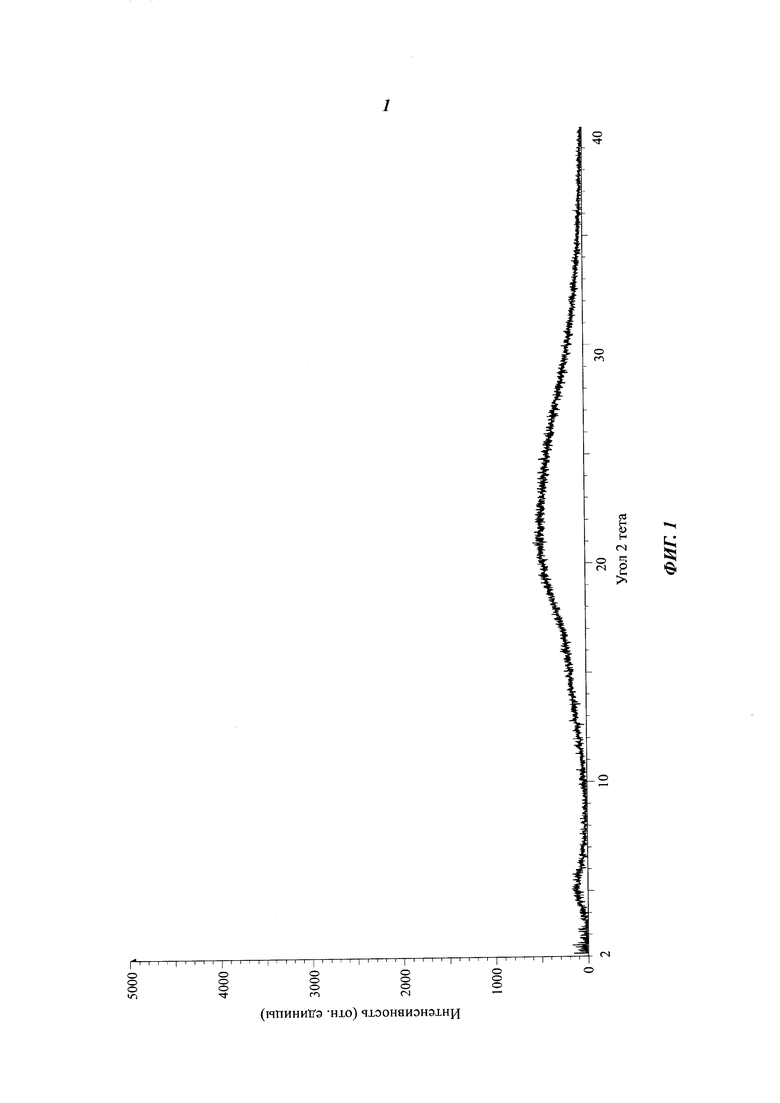
Кристаллическую форму I соединения формулы (I) синтезируют по методике Примера 2, измельчают, нагревают и таблетируют. Результаты показали, что кристаллическая форма является стабильной. Подробные экспериментальные данные приведены ниже в таблице 2.

| название | год | авторы | номер документа |
|---|---|---|---|
| НАТРИЕВАЯ СОЛЬ ИНГИБИТОРА ТРАНСПОРТЕРА МОЧЕВОЙ КИСЛОТЫ И ЕГО КРИСТАЛЛИЧЕСКАЯ ФОРМА | 2016 |
|
RU2719484C2 |
| П-ТОЛУОЛСУЛЬФОНАТ ДЛЯ ИНГИБИТОРА МЕК-КИНАЗЫ И ЕГО КРИСТАЛЛИЧЕСКАЯ ФОРМА, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2016 |
|
RU2704251C2 |
| ИНГИБИТОР JAK И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2019 |
|
RU2812575C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2018 |
|
RU2767872C2 |
| СОЕДИНЕНИЯ ФЕНИЛПИРИДАЗИНА И СОДЕРЖАЩИЕ ИХ ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 2001 |
|
RU2269519C2 |
| ЗАМЕЩЕННЫЕ ПИРРОЛО[2,3-D]ПИРИДАЗИН-4-ОНЫ И ПИРАЗОЛО[3,4-D]ПИРИДАЗИН-4-ОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2017 |
|
RU2749038C2 |
| СТАБИЛЬНЫЙ КРИСТАЛЛ 4-ОКСОХИНОЛИНОВОГО СОЕДИНЕНИЯ | 2005 |
|
RU2330845C1 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА И СОЛЕВАЯ ФОРМА ИНГИБИТОРА TGF-βRI И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2018 |
|
RU2750702C1 |
| АМИНОПИРИДАЗИНОНОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2014 |
|
RU2674701C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА БИСУЛЬФАТА ИНГИБИТОРА JAK И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2015 |
|
RU2704795C2 |
Изобретение относится к кристаллической форме I (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1Н-пирроло[2,3-d]пиридазин-7(6Н)-она, обладающей спектром рентгеновской порошковой дифрактометрии, полученным с помощью излучения Cu-Ка и представленным углом 2θ и межплоскостным расстоянием, который имеет характеристические пики при 2θ±0,2: 9,91, 12,20, 17,24, 17,64 и 21,48. Изобретение также относится к способу получения кристаллической формы I, к фармацевтической композиции, к применению. Технический результат: получена стабильная кристаллическая форма I (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1Н-пирроло[2,3-d]пиридазин-7(6Н)-она, обладающая свойствами ингибитора тирозинкиназы Брутона. 4 н. и 3 з.п. ф-лы, 4 ил., 2 табл., 13 пр.
1. Кристаллическая форма I (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1Н-пирроло[2,3-d]пиридазин-7(6Н)-она, отличающаяся тем, что указанное кристаллическое вещество обладает спектром рентгеновской порошковой дифрактометрии, полученным с помощью излучения Cu-Ка и представленным углом 2θ и межплоскостным расстоянием, который имеет характеристические пики при 2θ±0,2: 9,91, 12,20, 17,24, 17,64 и 21,48.
2. Кристаллическая форма I по п. 1, отличающаяся тем, что кристаллическая форма I имеет характеристические пики при 2θ±0,2: 7,86, 9,91, 12,20, 13,73, 17,24, 17,64, 19,02, 19,93, 20,72, 21,48, 22,64, 24,81, 27,44 и 27,87.
3. Кристаллическая форма I по п.1, отличающаяся тем, что указанная кристаллическая форма I обладает спектром рентгеновской порошковой дифрактометрии, как показано на фиг. 3, который имеет характеристические пики при 2θ±0,2: около 5,11, 7,86, 9,91, 12,20, 13,73, 15,44, 17,24, 17,64, 19,02, 19,93, 20,72, 21,48, 22,64, 23,12, 24,81, 25,43, 26,24, 27,44, 27,87, и 29,03.
4. Способ получения кристаллической формы I (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1Н-пирроло[2,3-d]пиридазин-7(6Н)-она по любому из пп.1-3, включающий следующие стадии:
1) растворение твердого (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1Н-пирроло[2,3-d]пиридазин-7(6Н)-она в соответствующем количестве органического растворителя при нагревании до кипения с обратным холодильником с последующим охлаждением с выделением кристаллического осадка, причем растворитель представляет собой метанол, этанол, изопропанол, ацетон, этилацетат, ацетонитрил или тетрагидрофуран, или этанол/вода, N,N-диметилформамид/вода, или 1,4-диоксан/вода; и
2) фильтрация кристаллов с последующей промывкой и сушкой.
5. Способ по п. 4, отличающийся тем, что одиночный растворитель предпочтительно представляет собой этанол.
6. Фармацевтическая композиция для лечения заболевания, связанного с тирозинкиназой Брутона, включающая кристаллическую форму I (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1Н-пирроло[2,3-d]пиридазин-7(6Н)-она по любому из пп.1-3 и фармацевтически приемлемый носитель.
7. Применение кристаллической формы I по любому из пп.1-3 или фармацевтической композиции по п.6 в приготовлении лекарственного средства для лечения заболевания, связанного с тирозинкиназой Брутона.
| АМИНОПИРИДАЗИНОНОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2014 |
|
RU2674701C2 |
| RU 2014106020 A, 27.08.2015 | |||
| Шлифовальный станок | 1929 |
|
SU21715A1 |
| WO 2007138355 A1, 06.12.2007 | |||
| US 8673925 B1, 18.03.2014. | |||

