Область техники
[0001]
Настоящее изобретение относится к новым содержащим азот 6-членным циклическим соединениям и лекарственным средствам, применяя их в качестве активного ингредиента.
Уровень техники
[0002]
Перелом кости представляет собой состояние, при котором кость частично или полностью разломана или деформирована под действием внешней силы, возникающей в результате несчастного случая или падения. Переломы костей подразделяют на полный перелом (перелом) и неполный перелом (трещина), простой перелом (имеется одна линия перелома) и раздробленный перелом (сложный перелом кости), закрытый перелом (переломанная часть не выходит из тела) и открытый перелом (переломанная часть выходит из тела) или подобные. Переломы костей создают серьезные проблемы для повседневной жизнедеятельности пациентов, и их заживление занимает значительный длительный период времени, хотя это зависит от переломанной части и наличия или отсутствия смещения (вывиха) кости. Состояние сращивания кости без коррекции вывиха называют "неправильно сросшимся переломом". В качестве условий перелома кости, хотя она зависят от места и типа перелома кости, могут возникать такие состояния, как «замедленное срастание», при котором срастание не достигают даже через 3-9 месяцев после повреждения из-за различных факторов, таких как старение, диабет и курение, и "несрастание", при котором срастание не достигают даже через 9 месяцев после повреждения, и подозревается остановка процесса заживления перелома. В таких случаях, как неправильно сросшийся перелом, замедленное срастание и несрастание, возникают боль или дискомфорт, нормальное функциональное заживление переломанных частей не достигается и, следовательно, качество жизни пациентов с переломом заметно ухудшается.
[0003]
Особенно серьезными проблемами при лечении переломов являются переломы, сопровождающие остеопороз. Переломы, сопровождающие остеопороз, часто возникают при метафизах аппендикулярных костей и позвоночника, в частности перелом шейки бедра, компрессионный перелом позвонка, перелом дистального конца лучевой кости и перелом проксимального конца плечевой кости рассматривают как четыре основных перелома, наблюдаемые при остеопорозе. Переломы, сопровождающие остеопороз, имеют проблему, заключающуюся в том, что их восстановление затруднено из-за хрупкости кости, и даже если протекает остеосинтез, едва ли можно получить достаточную стабильность, а неправильная фиксация вызывает неправильный сросшийся перелом, замедленное срастание, а также несрастание. Кроме того, поскольку ежедневная активность ограничена в период лечения переломов, возникает отрицательная спираль, которая препятствует сокращению костной массы и прогрессирует мышечная атрофия, и они вновь вызывают падение и перелом. В частности, задержка нормального заживления перелома тела позвонка или перелома шейки бедра вынуждает пациентов быть прикованными к постели. Частота возникновения различных и критических осложнений, таких как мышечная слабость, контрактура суставов, пролежневая язва, деменция, инфекция мочевыводящих путей и сердечно-легочная гипофункция, сопровождающие системное бездействие у данных пациентов, является чрезвычайно высокой, и сообщается о значительном снижении выживаемости после повреждения (непатентный документ 1).
[0004]
Как описано выше, переломы, особенно переломы, сопровождающие остеопороз, вызывают ухудшение качества жизни, тяжелые осложнения, а также значительное влияние на прогноз жизненно важных функций, и поэтому они создают чрезвычайно серьезные социальные проблемы, такие как повышение стоимости медицинского обслуживания и расходы на уход. Лечение переломов в настоящее время выполняется путем возврата состояния кости в анатомически нормальное положение и выполнения фиксации с целью достижения заживления до функционального уровня до повреждения, насколько это возможно, с помощью механизма нормального восстановления кости с предотвращением осложнений, таких как неправильно сросшийся перелом, замедленное срастание и несрастание.
[0005]
В качестве способов лечения, положительно способствующих заживлению переломов, применяют ультразвуковые аппараты для лечения переломов, а в качестве терапевтических препаратов применяют или предпринимают попытки клинического применения препаратов костного морфогенетического белка (BMP), препаратов паратиреоидного гормона, препаратов фактора роста фибробластов (FGF) и подобных. Однако, несмотря на применение или попытки применения данного разнообразия лекарственных средств, как упомянуто выше, количество пациентов с заболеваниями костей, такими как перелом, с каждым годом увеличивается, например, количество пациентов с переломом шейки бедренной кости, как предполагалось, составляло 1700000 во всем мире в 1990 году и прогнозируется увеличение до 6300000 в 2050 году. В связи с этим, желательна разработка новых инновационных препаратов, обладающих профилактическим и/или терапевтическим эффектом при заболеваниях костей, таких как переломы.
[0006]
Известно, что простагландин E2 (далее сокращенно обозначаемый как PGE2) выполняет различные физиологические функции, такие как болеутоляющее действие и окситоцическое действие, и также хорошо известно, что он играет важную роль в метаболизме костей. Когда PGE2 добавляют в систему культивирования клеток костного мозга, активность щелочной фосфатазы, которая является маркером образования кальцифицированного костного узла и дифференцировки остеобластов, увеличивается. Также было обнаружено, что когда PGE2 реально вводится лабораторным животным, таким как крысы, или людям, остеогенез увеличивается, и костная масса увеличивается. Кроме того, когда PGE2 местно вводят в кость в виде препарата с замедленным высвобождением, в месте введения усиливается остеогенез, и, следовательно, для PGE2 можно ожидать системного или местного эффекта положительного стимулирования остеогенеза.
[0007]
Однако поскольку PGE2 проявляет побочные реакции, такие как вызывающее боль действие и окситоцическое действие, как описано выше, которые следует избегать при длительном введении, существует необходимость в селективном производном PGE2, которое безопасно и эффективно действует на костные ткани. Например, в качестве рецепторов PGE2, для мыши, крысы, собаки, человека и подобных до сих пор были описаны четыре их вида, рецепторы EP1, EP2, EP3 и EP4, и поскольку сайты их экспрессии и системы внутриклеточной передачи сигнала для активации являются разными, были созданы соединения, селективные для каждого подтипа.
[0008]
Предполагается, что среди четырех типов рецепторов, на которые действует PGE2, рецепторы EP2 и EP4 играют важную роль в метаболизме костей в клетках и у животных, и оба сопряжены с Gs белком, увеличивая цАМФ в остеобластах. До настоящего времени были разработаны EP2-селективные агонисты, EP4-селективные агонисты и EP2/EP4-агонисты, и на животных моделях был продемонстрирован значительный эффект на остеогенез или эффект, способствующий заживлению переломов, системным или местным введением. В качестве соединений, которые действуют на PGE рецепторы, например, соединения, описанные в патентных документах 1-8, являются известными.
Ссылки предшествующего уровня техники
Патентные документы
[0009]
Патентный документ 1: международная патентная публикация WO02/24647
Патентный документ 2: международная патентная публикация WO02/42268
Патентный документ 3: международная патентная публикация WO03/007941
Патентный документ 4: международная патентная публикация WO03/035064
Патентный документ 5: международная патентная публикация WO2004/063158
Патентный документ 6: международная патентная публикация WO2004/085430
Патентный документ 7: патент США No. 6747037
Патентный документ 8: Международная патентная публикация WO2006/080323
Непатентные документы
[0010]
Непатентный документ 1: C. Cooper et al., Am. J. Epidemiol., 137, 1001-1005, 1993
Сущность настоящего изобретения
Цель, которую достигают настоящим изобретением
[0011]
Цель, которую достигает настоящее изобретение, заключается в обеспечении нового соединения, обладающего превосходной агонистической активностью по отношению к EP4 рецептору. Предпочтительно, цель настоящего изобретения заключается в обеспечении нового соединения, обладающего превосходной селективной агонистической активностью по отношению к EP4 рецептору. Другая цель настоящего изобретения заключается в обеспечении нового соединения, пригодного в качестве активного ингредиента лекарственного средства для профилактического и/или терапевтического лечения заболевания, связанного с агонизмом EP4 рецептора, например, нового соединения, пригодного в качестве активного ингредиента лекарственного средства для терапевтического лечения перелома или стимулирования заживления перелома. Еще другая цель настоящего изобретения заключается в обеспечении лекарственного средства, содержащего данное соединение.
Способы достижения цели
[0012]
Для достижения вышеупомянутых целей, изобретатели настоящего изобретения провели различные исследования. Как результат, они обнаружили, что соединения настоящего изобретения, представленные следующей формулой (1), обладают превосходной агонистической активностью относительно EP4, в частности, соединения согласно одному варианту осуществления настоящего изобретения обладают превосходной селективной агонистической активностью относительно EP4 рецептора, и данные соединения являются пригодными для профилактического и/или терапевтического лечения заболевания, связанного с агонизмом EP4 рецептора, например, терапевтического лечения и/или стимулирования заживления перелома, и пришли к завершению настоящего изобретения. Считают, что предпочтительно обеспечивать соединение, обладающее селективной агонистической активностью относительно EP4 рецептора по следующим причинам. А именно, тогда как EP4 рецептор наблюдают в остеобластах и остеокластах в человеческих культивируемых остеобластах и костных тканях, экспрессия EP2 рецептора не обнаружена (P. Sarrazin, G et al. Prostaglandins Leukot. Essent. Fatty Acids, 64, 203-210, 2001; I. Fortier et al., Prostaglandins Leukot. Essent. Fatty Acids, 70, 431-439, 2004) и, следовательно, считают, что самой важной мишенью PGE2 для действия в костных тканях является EP4 рецептор, и селективный агонист EP4 рецептора может быть безопасным и эффективным лекарственным средством для регенерации костной ткани. Конечно, соединения, обладающие агонистической активностью относительно EP2, не исключают из соединений настоящего изобретения.
[0013]
Таким образом, настоящее изобретение относится к следующему.
[1] Соединение, представленное следующей общей формулой (1):
[формула 1]
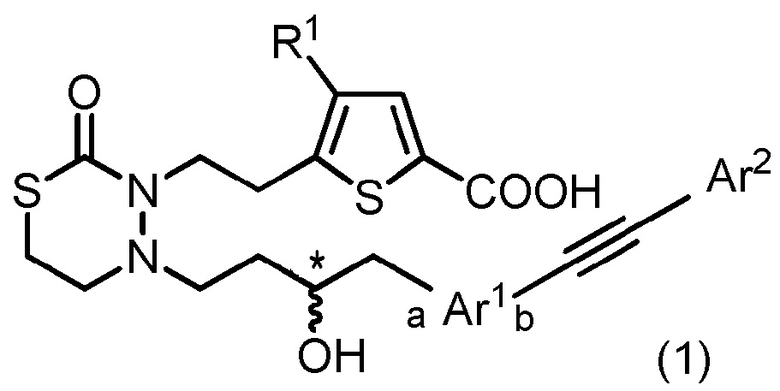
[где в формуле (1),
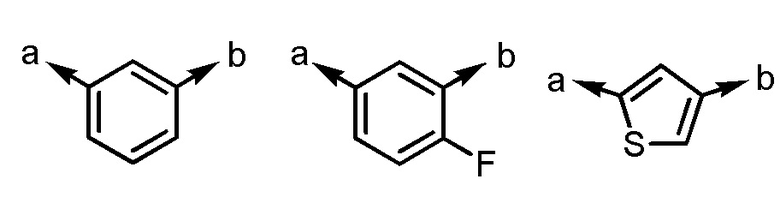
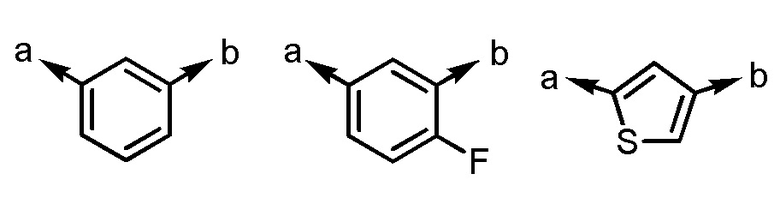
R1 представляет собой -H или галоген;
Ar1 представляет собой любой заместитель, выбранный из группы G1, который может быть замещен 1-3 одинаковыми или различными заместителями, выбранными из группы, состоящей из -F и метила (при условии, что
[формула 2]

исключены),
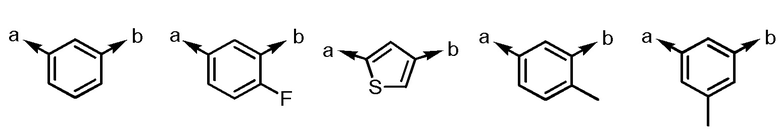
в которых группа G1 представляет собой группу, состоящую из
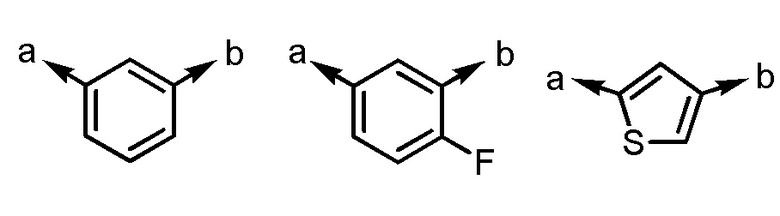
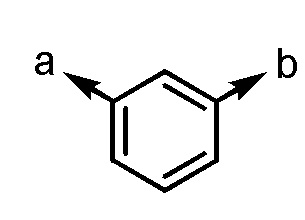
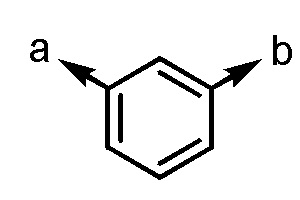
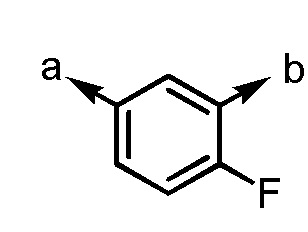
[формула 3]
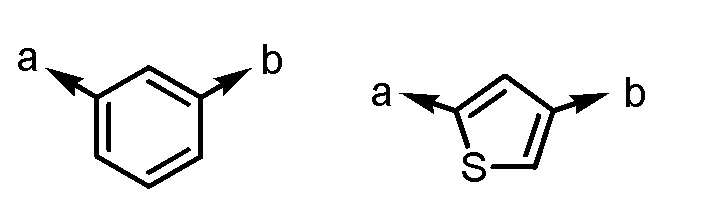
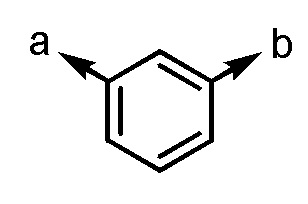
(a и b представляют собой направление присоединения);
Ar2 представляет собой любой заместитель, выбранный из группы G2, который может быть замещен 1-3 одинаковыми или различными заместителями, выбранными из группы, состоящей из циано, -Cl, метила, метокси и фенила (при условии, что
[формула 4]
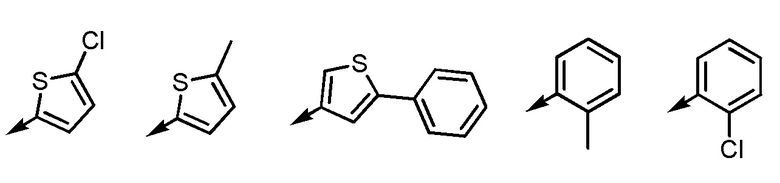
исключены),
в которых группа G2 представляет собой группу, состоящую из фенила, тиенила, фурила и тиазолила; и
* представляет собой асимметрический углерод],
или его соль.
[0014]
[2] Соединение или его соль согласно [1], приведенному выше, в котором R1 представляет собой -H, -Cl или -Br.
[3] Соединение или его соль согласно [2], приведенному выше, в котором Ar1 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 5]
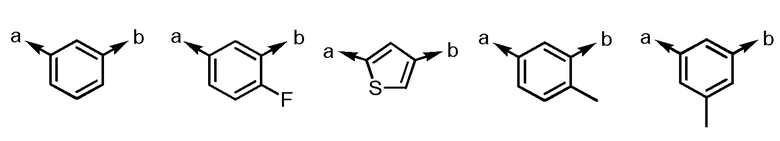 .
.
[0015]
[3-2] Соединение или его соль согласно [1] или [2], приведенным выше, в котором Ar1 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 6]
 .
.
[0016]
[3-3] Соединение или его соль согласно [1] или [2], приведенным выше, в котором Ar1 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 7]
 .
.
[0017]
[4] Соединение или его соль согласно [2], приведенному выше, в котором Ar1 представляет собой
[формула 8]
 .
.
[0018]
[4-2] Соединение или его соль согласно [1] или [2], приведенным выше, в котором Ar1 представляет собой
[формула 9]
 .
.
[0019]
[4-3] Соединение или его соль согласно [1] или [2], приведенным выше, в котором Ar1 представляет собой
[формула 10]
 .
.
[4-4] Соединение или его соль согласно [1] или [2], приведенным выше, в котором Ar1 представляет собой
[формула 11]
 .
.
[0020]
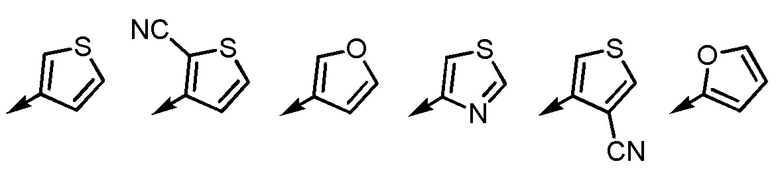
[5] Соединение или его соль согласно [3] или [4], приведенным выше, в котором Ar2 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 12]
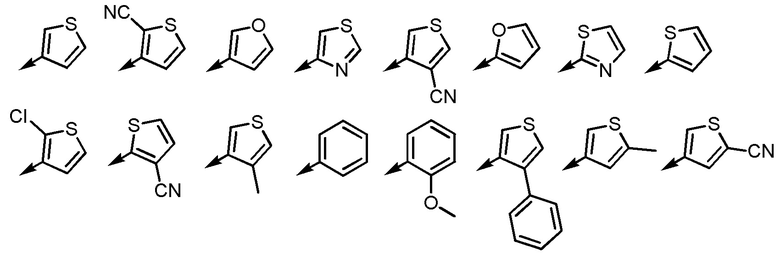 .
.
[0021]
[5-2] Соединение или его соль по любому из [1]-[4-4], приведенным выше, в котором Ar2 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 13]
 .
.
[0022]
Когда номера цитируемых пунктов обозначены таким диапазоном, как [1]-[4-4], приведенном выше, и данный диапазон включает пункт, указанный номером, имеющим дополнительный номер, такой как [3-2], это означает, что также цитируют пункт, указанный номером, имеющим дополнительный номер, такой как [3-2]. То же самое относится к следующим определениям.
[0023]
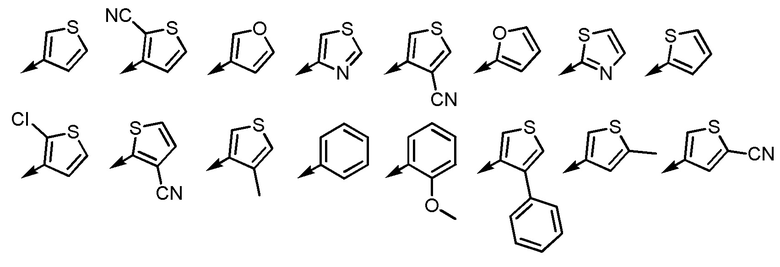
[6] Соединение или его соль согласно [3] или [4], приведенным выше, в котором Ar2 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 14]
 .
.
[0024]
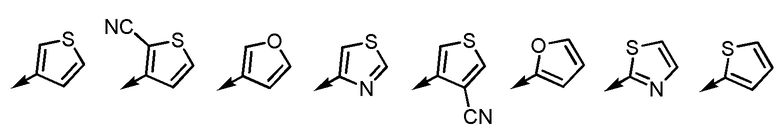
[6-2] Соединение или его соль по любому из [1]-[4-4], приведенным выше, в котором Ar2 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 15]
 .
.
[0025]
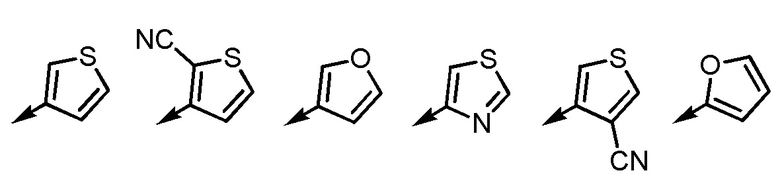
[7] Соединение или его соль согласно [3] или [4], приведенным выше, в котором Ar2 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 16]
 .
.
[0026]
[7-2] Соединение или его соль по любому из [1]-[4-4], приведенным выше, в котором Ar2 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 17]
 .
.
[0027]
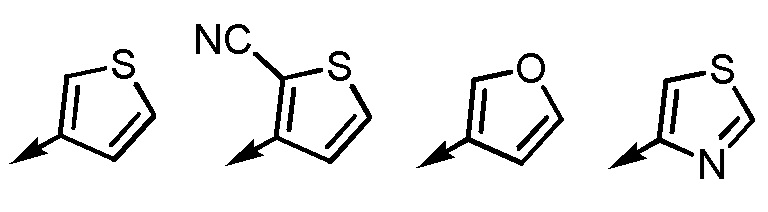
[7-3] Соединение или его соль по любому из [1]-[4-4], приведенным выше, в котором Ar2 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 18]
 .
.
[0028]
[7-4] Соединение или его соль по любому из [1]-[4-4], приведенным выше, в котором Ar2 представляет собой
[формула 19]
 .
.
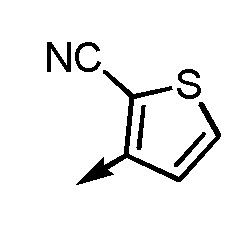
[7-5] Соединение или его соль по любому из [1]-[4-4], приведенным выше, в котором Ar2 представляет собой
[формула 20]
 .
.
[0029]
[7-6] Соединение или его соль по любому из [1]-[4-4], приведенным выше, в котором Ar2 представляет собой
[формула 21]
 .
.
[7-7] Соединение или его соль по любому из [1]-[4-4], приведенным выше, в котором Ar2 представляет собой [формула 22]
 .
.
[0030]
[8] Соединение или его соль согласно [7], приведенному выше, в котором R1 представляет собой -H.
[8-2] Соединение или его соль по любому из [1]-[7-7], приведенным выше, в котором R1 представляет собой -H.
[0031]
[9] Соединение или его соль согласно [7], приведенному выше, в котором R1 представляет собой -Cl.
[9-2] Соединение или его соль по любому из [1]-[7-7], приведенным выше, в котором R1 представляет собой -Cl.
[0032]
[10] Соединение или его соль согласно [7], приведенному выше, в котором R1 представляет собой -Br.
[10-2] Соединение или его соль по любому из [1]-[7-7], приведенным выше, в котором R1 представляет собой -Br.
[0033]
[11] Соединение или его соль согласно [1], приведенному выше, в котором R1 представляет собой -H, -Cl или -Br;
Ar1 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 23]
 ; и
; и
Ar2 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 24]
 .
.
[0034]
[11-2] Соединение или его соль согласно [1], приведенному выше, в котором R1 представляет собой -H, -Cl или -Br;
Ar1 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 25]
 ; и
; и
Ar2 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 26]
 .
.
[0035]
[11-3] Соединение или его соль согласно [1], приведенному выше, в котором R1 представляет собой -H, -Cl или -Br;
Ar1 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 27]
 ; и
; и
Ar2 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 28]
 .
.
[0036]
[11-4] Соединение или его соль согласно [1], приведенному выше, в котором R1 представляет собой -H, -Cl или -Br;
Ar1 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 29]
 ; и
; и
Ar2 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 30]
 .
.
[0037]
[12] Любое соединение, выбранное из следующей группы, или его соль;
[формула 31]
 .
.
[0038]
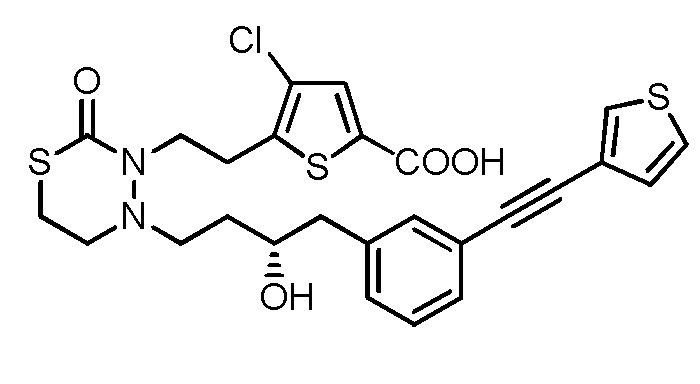
[13] Соединение, приведенное ниже, или его соль;
[формула 32]
 .
.
[0039]
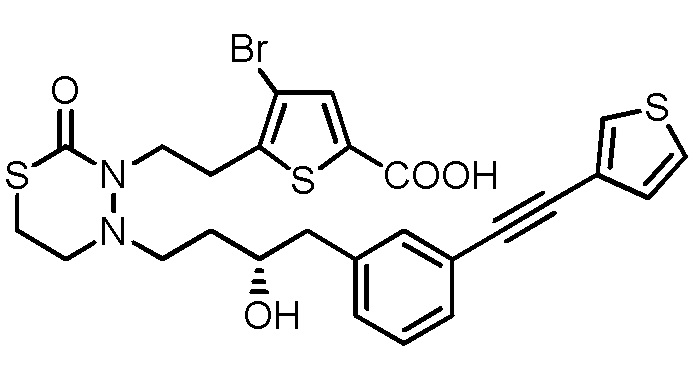
[14] Соединение, приведенное ниже, или его соль;
[формула 33]
 .
.
[0040]
[15] Соединение, приведенное ниже, или его соль;
[формула 34]
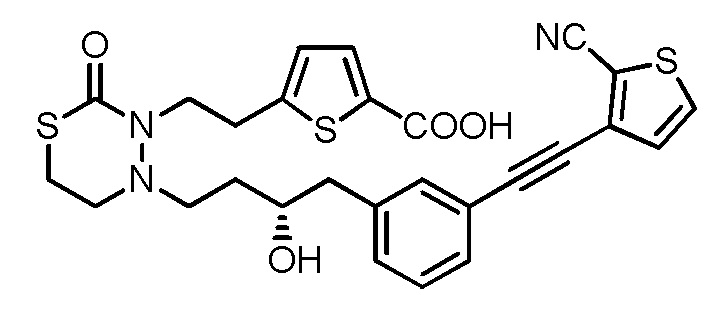 .
.
[0041]
[16] Соединение, приведенное ниже, или его соль;
[формула 35]
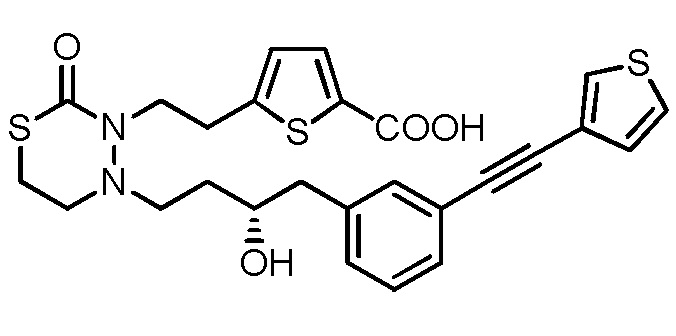 .
.
[0042]
[17] Соединение, приведенное ниже, или его соль;
[формула 36]
 .
.
[0043]
[18] Соединение, приведенное ниже, или его соль;
[формула 37]
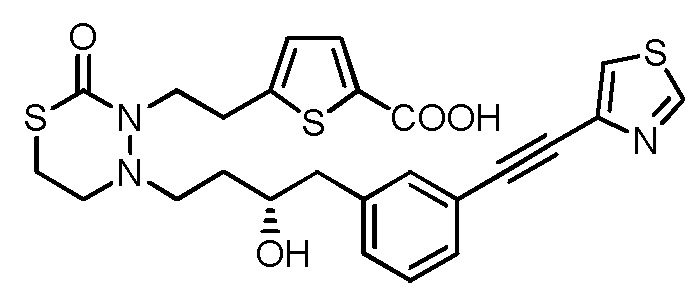 .
.
[0044]
[19] Соединение, приведенное ниже, или его соль;
[формула 38]
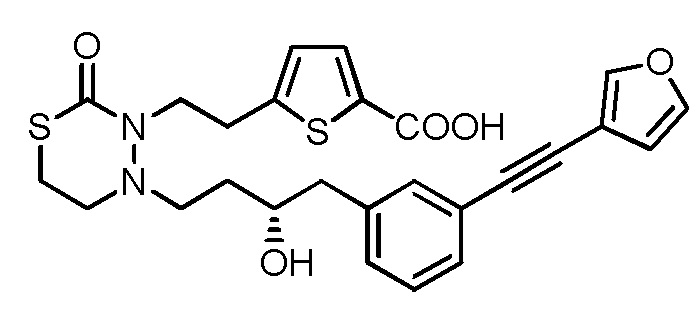 .
.
[0045]
[20] Лекарственное средство, содержащее соединение по любому из [1]-[19], приведенным выше, или его фармацевтически приемлемую соль в качестве активного ингредиента.
[21] Лекарственное средство согласно [20], приведенному выше, которое предназначено для профилактического и/или терапевтического лечения заболевания, связанного с EP4 рецепторным агонизмом.
[0046]
[22] Лекарственное средство согласно [20], приведенному выше, которое предназначено для стимуляции остеогенеза.
[23] Лекарственное средство согласно [20], приведенному выше, которое предназначено для терапевтического лечения и/или стимулирования заживления перелома.
[24] Лекарственное средство согласно [20], приведенному выше, которое предназначено для терапевтического лечения и/или стимулирования заживления костного дефекта.
[0047]
[25] Лекарственное средство согласно [20], приведенному выше, которое предназначено для стимуляции сращивания кости.
[25-2] Лекарственное средство согласно [25], приведенному выше, которое предназначено для стимуляции сращивания кости при фиксации позвоночника.
[26] EP4 агонист, содержащий соединение по любому из [1]-[19], приведенному выше, или его фармацевтически приемлемую соль в качестве активного ингредиента.
[27] Фармацевтическая композиция для терапевтического лечения перелома, которая содержит соединение по любому из [1]-[19], приведенным выше, или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель.
[28] Получение микросфер, содержащих соединение по любому из [1]-[19], приведенных выше, или его фармацевтически приемлемую соль, и сополимер молочной/гликолевой кислоты.
[0048]
[29] Соединение по любому из [1]-[19], приведенному выше, или его фармацевтически приемлемая соль, которую применяют для терапевтического лечения перелома.
[30] Способ терапевтического лечения перелома у млекопитающего, который включает стадию введения эффективного количества соединения по любому из [1]-[19], приведенных выше, или его фармацевтически приемлемой соли, млекопитающему.
Эффект настоящего изобретения
[0049]
"Соединения, представленные формулой (1), и их соли" (далее также называют просто "соединения настоящего изобретения") обладают превосходной агонистической активностью относительно EP4 рецептора. Соединения настоящего изобретения можно применять в качестве активного ингредиента лекарственного средства для профилактического и/или терапевтического лечения заболевания, связанного с EP4 рецепторным агонизмом, например, терапевтического лечения и/или стимулирования заживления перелома. В качестве другого варианта осуществления, соединения настоящего изобретения можно применять в качестве реагента, обладающего агонистической активностью относительно EP4 рецептора.
Варианты осуществления настоящего изобретения
[0050]
В настоящем изобретении далее, настоящее изобретение будет объяснено конкретно.
В настоящем изобретении, атом углерода может быть просто представлен как “C”, атом водорода как H”, атом кислорода как “O”, атом серы как “S” и атом азота как N”. Кроме того, карбонильную группу можно просто представить как “-C(O)-”, карбоксильную группу как “-COO-”, сульфинильную группу как “-S(O)-”, сульфонильную группу как “-S(O)2-”, эфирную связь как “-O-” и тиоэфирную связь как “-S-” (каждый “-” в данных группах обозначает связь).
[0051]
В настоящем изобретении, алкил, содержащий 1-4 атома углерода, обозначает метил, этил, пропил, бутил или его изомер [нормальный (н), изо, вторичный (втор-), третичный (трет) и подобные].
[0052]
В настоящем изобретении ацил, содержащий 2-6 атомов углерода, обозначает ацетил, пропаноил, бутаноил, пентаноил, гексаноил или их изомер.
[0053]
В настоящем изобретении алкокси, содержащий 1-4 атома углерода, обозначает метокси, этокси, пропокси, бутокси или их изомер.
В настоящем изобретении, галоген обозначает фтор (-F), хлор (-Cl), бром (-Br) или йод (-I).
[0054]
В настоящем изобретении, включены все изомеры, если не указано специально. Например, алкил, алкенил, алкинил, алкокси, алкилтио, алкилен, алкенилен и алкинилен включают нормальные и разветвленные группы. Кроме того, любой из изомеров, исходя из двойной связи, кольца или конденсированного кольца (E- или Z-изомеры, или цис- или транс-изомеры), изомеры, исходя из наличия асимметрического углерода и подобных (R- или S-изомер, изомер, исходя из α- или β-конфигурации, энантиомеры, диастереомеры и подобные), оптически активные вещества, проявляющие оптическое вращение (D- или L-изомеры или d- или l-изомеры), изомеры, исходя из полярности при хроматографическом разделении (изомеры высокой полярности или изомеры низкой полярности), уравновешенные соединения, вращательные изомеры, смеси данных изомеров в произвольных соотношениях и рацематы включены в объем настоящего изобретения.
[0055]
В настоящем изобретении, как ясно специалисту в данной области техники, символ:
[формула 39]

показывает, что связь находится за плоскостью (т.е., α-конфигурация), символ:
[формула 40]

показывает, что связь находится перед плоскостью (т.е., β-конфигурация), символ:
[формула 41]

обозначает α-конфигурацию или β-конфигурацию, или их смесь, и символ:
[формула 42]

обозначает смесь α-конфигурации и β-конфигурации, если не указано специально.
[0056]
В настоящем изобретении далее соединения, представленные формулой (1), или их соли, будут описаны подробно.
R1 представляет собой, например, -H или галоген. Согласно другому варианту осуществления, R1 представляет собой, например, -H, -Cl или -Br. Согласно еще другому варианту осуществления, R1 представляет собой, например, -H.
[0057]
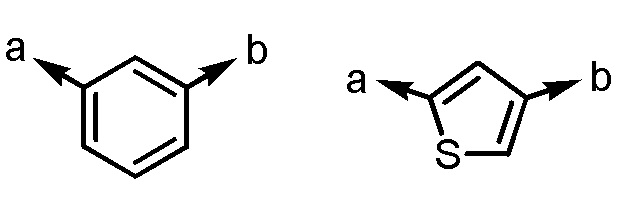
Ar1 представляет собой, например, любой заместитель, выбранный из группы G1, который может быть замещен 1-3 одинаковыми или различными заместителями, выбранными из группы, состоящей из -F и метила (при условии, что
[формула 43]
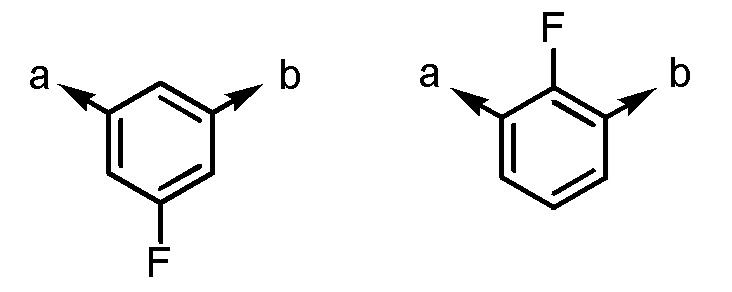
исключены), в которых группа G1 представляет собой группу, состоящую из
[формула 44]
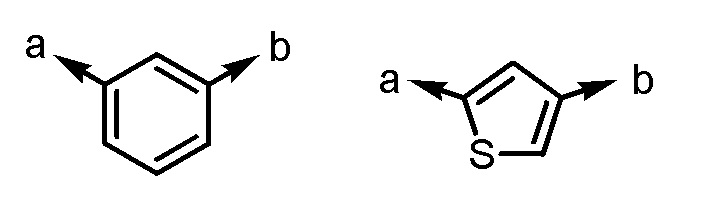
(a и b представляют собой направление присоединения).
[0058]
Согласно другому варианту осуществления данной группы, состоящей из -F и метила, приведенной выше, группа состоит, например, из -F, и согласно еще другому варианту осуществления, группа состоит, например, из метила.
[0059]
Согласно другому варианту осуществления группы G1, группа состоит, например, из
[формула 45]
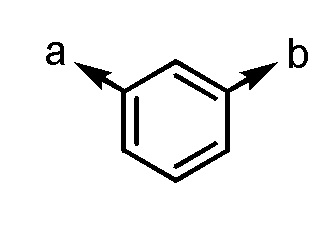 .
.
[0060]
Согласно другому варианту осуществления Ar1, Ar1 представляет собой, например, любой заместитель, выбранный из группы, состоящей из
[формула 46]
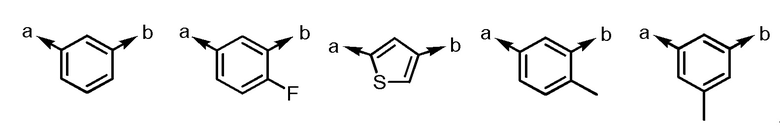 .
.
[0061]
Согласно еще другому варианту осуществления Ar1, Ar1 представляет собой, например, любой заместитель, выбранный из группы, состоящей из
[формула 47]
 .
.
[0062]
Согласно еще другому варианту осуществления Ar1, Ar1 представляет собой, например,
[формула 48]
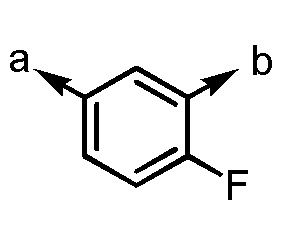
 .
.
[0063]
Согласно еще другому варианту осуществления Ar1, Ar1 представляет собой, например,
[формула 49]
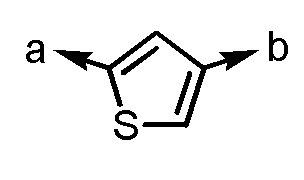
 .
.
[0064]
Согласно еще другому варианту осуществления Ar1, Ar1 представляет собой, например,
[формула 50]
 .
.
[0065]
Когда Ar1 замещена 1-3 одинаковыми или различными заместителями, выбранными из группы, состоящей из -F и метила, согласно другому варианту осуществления, Ar1 замещена, например, 1 или 2 одинаковыми или различными заместителями, выбранными из группы, состоящей из -F и метила, и согласно еще другому варианту осуществления, Ar1 замещена, например, одним из -F или метила. Незамещенная Ar1 также представляет собой один из предпочтительных вариантов осуществления.
[0066]
Ar2 представляет собой, например, любой заместитель, выбранный из группы G2, который может быть замещен 1-3 одинаковыми или различными заместителями, выбранными из группы, состоящей из циано, -Cl, метила, метокси и фенила (при условии, что
[формула 51]
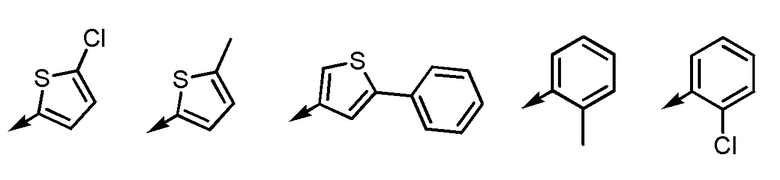
исключены).
[0067]
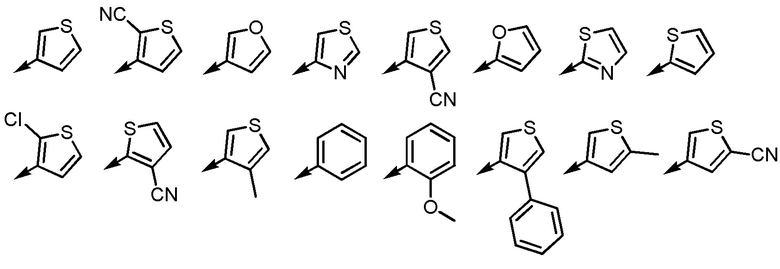
Группа G2, приведенная выше, представляет собой группу, состоящую из фенила, тиенила, фурила и тиазолила.
Согласно другому варианту осуществления данной группы, состоящей из циано, -Cl, метила, метокси и фенила, группа состоит, например, из циано.
Согласно другому варианту осуществления группы G2, G2 представляет собой, например, группу, состоящую из тиенила и фурила.
[0068]
Согласно еще другому варианту осуществления группы G2, группа состоит, например, из тиенила.
Согласно другому варианту осуществления Ar2, Ar2 представляет собой, например, любой заместитель, выбранный из группы, состоящей из
[формула 52]
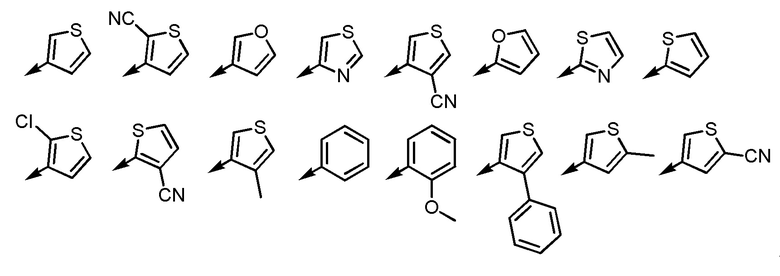 .
.
[0069]
Согласно еще другому варианту осуществления Ar2, Ar2 представляет собой, например, любой заместитель, выбранный из группы, состоящей из
[формула 53]
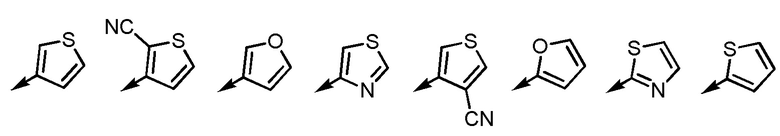 .
.
[0070]
Согласно еще другому варианту осуществления Ar2, Ar2 представляет собой, например, любой заместитель, выбранный из группы, состоящей из
[формула 54]
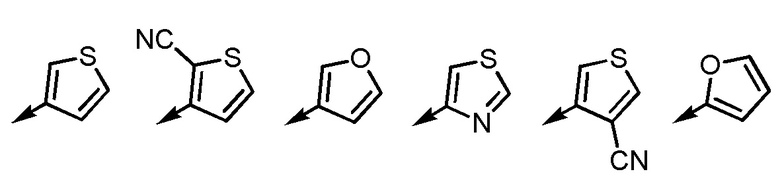 .
.
[0071]
Согласно еще другому варианту осуществления Ar2, Ar2 представляет собой, например, любой заместитель, выбранный из группы, состоящей из
[формула 55]
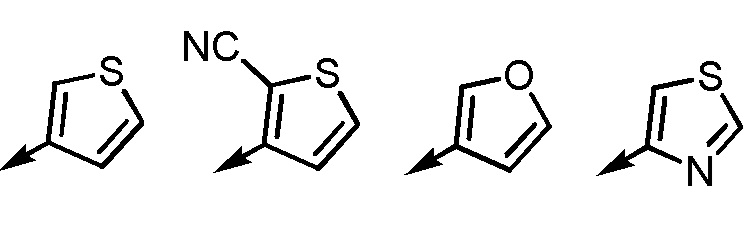 .
.
[0072]
Согласно еще другому варианту осуществления Ar2, Ar2 представляет собой, например,
[формула 56]
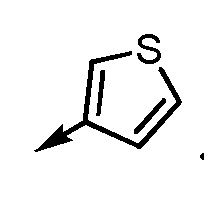 .
.
[0073]
Согласно еще другому варианту осуществления Ar2, Ar2 представляет собой, например,
[формула 57]
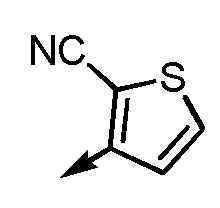 .
.
[0074]
Согласно еще другому варианту осуществления Ar2, Ar2 представляет собой, например,
[формула 58]
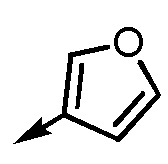 .
.
[0075]
Согласно еще другому варианту осуществления Ar2, Ar2 представляет собой, например,
[формула 59]
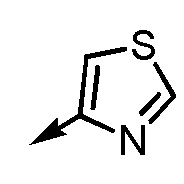 .
.
[0076]
Когда Ar2 замещена 1-3 одинаковыми или различными заместителями, выбранными из группы, состоящей из циано, -Cl, метила, метокси и фенила, согласно другому варианту осуществления, Ar2 замещена, например, 1 или 2 одинаковыми или различными заместителями, выбранными из группы, состоящей из циано, -Cl, метила, метокси и фенила, и согласно еще другому варианту осуществления, Ar2 замещена, например, 1 циано, -Cl, метилом, метокси или фенилом. Незамещенная Ar2 также представляет собой один из предпочтительных вариантов осуществления.
[0077]
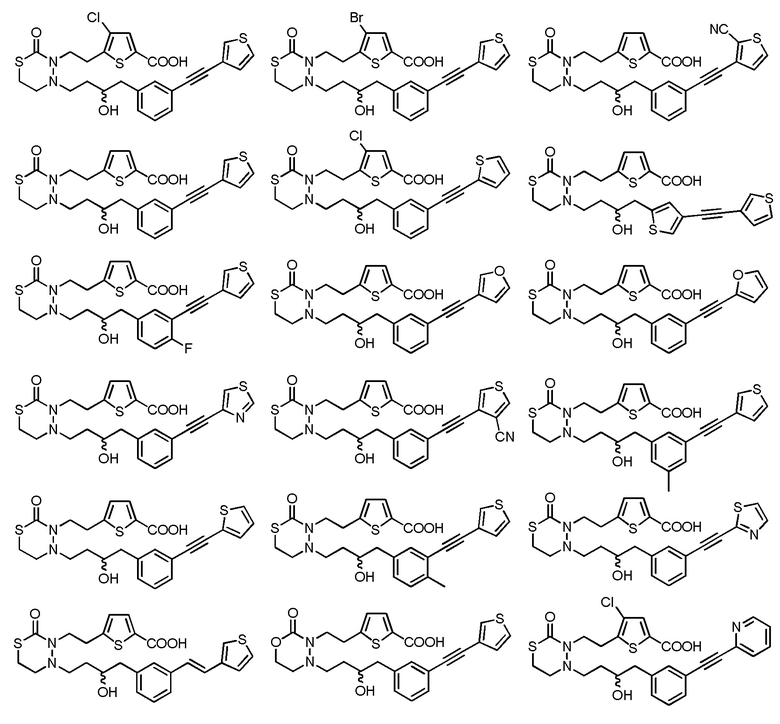
В качестве конкретных соединений, попадающих в пределы объема настоящего изобретения, можно привести в качестве примера следующие соединения.
[формула 60]
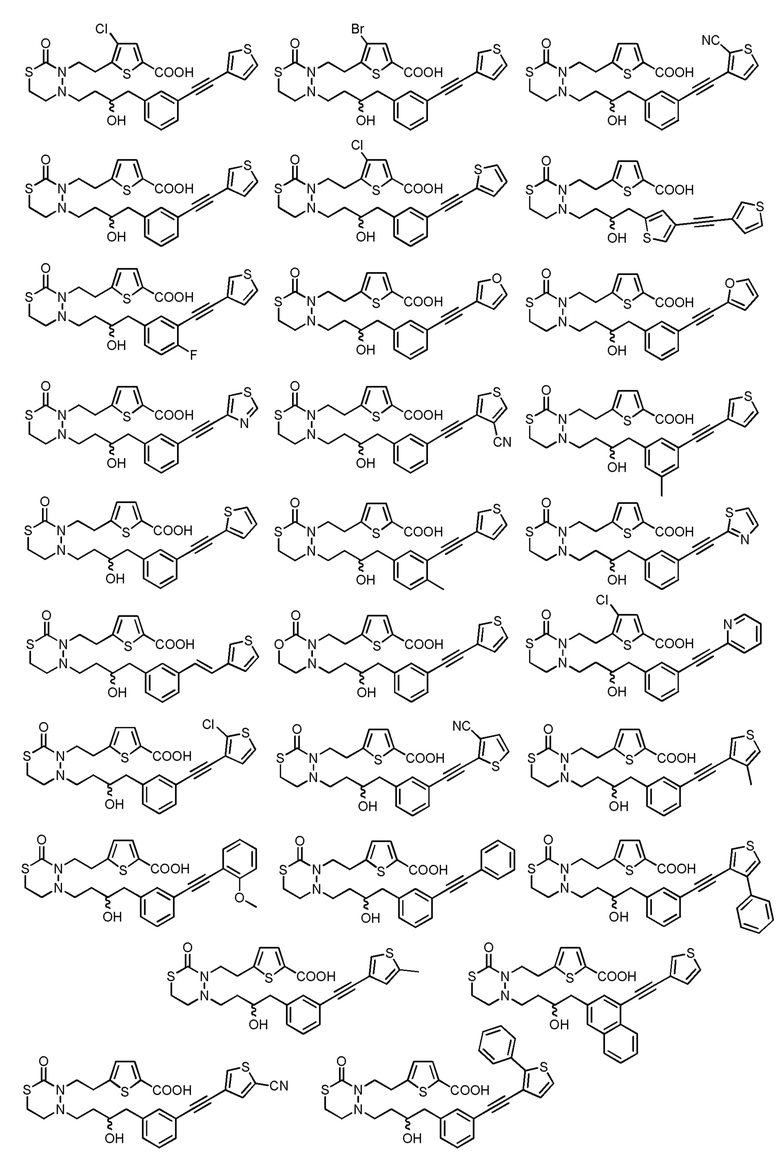
[0078]
В качестве еще других примеров соединений, попадающих в пределы объема настоящего изобретения, можно привести в качестве примера следующие соединения.
[формула 61]
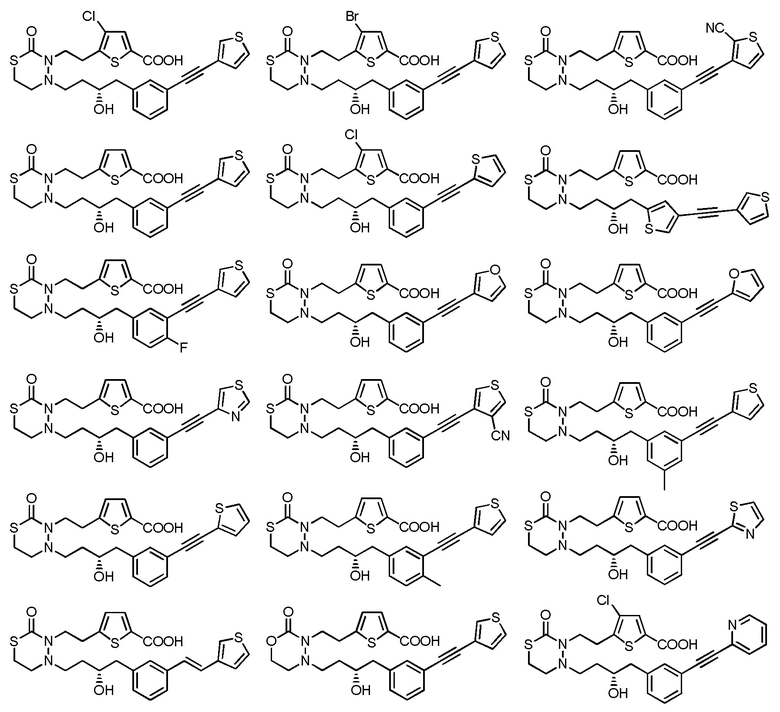
[0079]
В данном описании, "соединения, представленные формулой (1)" обычно подразумевают соединения, представленные формулой (1), в свободной форме. Примеры их солей включают следующие соли.
[0080]
Тип соли соединений, представленных формулой (1), конкретно не ограничен, и она может представлять собой соль присоединения кислоты или соль присоединения основания, и может быть в виде внутримолекулярного противоиона. В частности, когда соль применяют в качестве активного ингредиента лекарственного средства, соль предпочтительно представляет собой фармацевтически приемлемую соль. Когда описание приведено для применения в качестве лекарственного средства в данном описании, соль соединений, представленных формулой (1), обычно понимают как фармацевтически приемлемую соль. Соли присоединения кислоты включают, например, соли присоединения неорганической кислоты, такой как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота и фосфорная кислота и соли присоединения органической кислоты, такой как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, метансульфокислота, этансульфоновая кислота, бензолсульфоновая кислота, лимонная кислота, яблочная кислота, винная кислота, дибензоилтартаровая кислота, миндальная кислота, малеиновая кислота, фумаровая кислота, аспарагиновая кислота и глутаминовая кислота. В качестве солей присоединения оснований можно привести, например, соли присоединения неорганического основания, такого как натрий, калий, магний, кальций и алюминий, соли присоединения органических оснований, таких как метиламин, 2-аминоэтанол, аргинин, лизин и орнитин и подобных. Однако тип соли ими не ограничивается, и, конечно, он может быть соответствующим образом выбран специалистами в данной области техники.
[0081]
Соединения настоящего изобретения могут быть в виде гидрата. Соединения настоящего изобретения могут быть в виде ангидрида
Соединения настоящего изобретения могут быть в виде сольвата. Соединения настоящего изобретения могут также быть в виде, отличном от сольвата.
[0082]
Соединения настоящего изобретения могут быть в виде кристалла. Соединения настоящего изобретения могут также быть в аморфной форме.
Более конкретно, соединения настоящего изобретения включают ангидриды и формы, отличные от сольватов, "соединений, представленных формулой (1)", их гидраты и/или сольваты, и их кристаллы.
[0083]
Соединения настоящего изобретения также включают ангидриды и формы, отличные от сольватов, "солей соединений, представленных формулой (1)", гидраты и/или сольваты солей, и их кристаллы.
[0084]
Соединения настоящего изобретения могут также представлять собой фармацевтически приемлемое пролекарарство "соединений, представленных формулой (1)". Фармацевтически приемлемое пролекарство представляет собой соединение, содержащее группу, которую можно обменять на аминогруппу, гидроксильную группу, карбоксильную группу или подобную сольволизом или при физиологических условиях. Например, в качестве группы, которая образует пролекарство для гидрокси группы или аминогруппы, можно привести в качестве примера, например, ацильную группу и алкоксикарбонильную группу. В качестве группы, которая образует пролекарство для карбоксильной группы, можно привести в качестве примеров, например, метильную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, аминогруппу, метиламиногруппу, этиламиногруппу, диметиламиногруппу и диэтиламиногруппу.
[0085]
Данное пролекарство можно получить, например, подходящим введение группы, которая образует пролекарство в любом из соединений настоящего изобретения по одной или более произвольных групп, выбранных из гидроксильной группы и аминогруппы, применяя реагент, образующий пролекарство, такой как соответствующий галогенид, общепринятым способом, затем, при необходимости, подходящим выделением и очисткой соединения общепринятым способом. Группу, которая образует пролекарство, можно также подходящим образом ввести в соединение настоящего изобретения по карбоксильной группе, применяя такой реагент, образующий пролекарство, как соответствующий спирт или амин общепринятым способом.
[0086]
Соединения настоящего изобретения могут содержать асимметрический углерод. Пространственная конфигурация данного асимметрического углерода конкретно не ограничена, и она может представлять собой S-конфигурацию или R-конфигурацию, или смесь обеих. Любые оптически активные вещества, исходя из данного асимметрического углерода, в чистой форме, в виде стереоизомеров, таких как диастереоизомеры, произвольных смесей стереоизомеров, рацематов и подобных, попадают в пределы объема соединений настоящего изобретения.
[0087]
В частности, пространственная конфигурация асимметрического углерода, указанная "*" в формуле (1), конкретно не ограничена. Однако конфигурация, показанная ниже, является одним из предпочтительных вариантов осуществления.
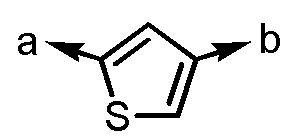
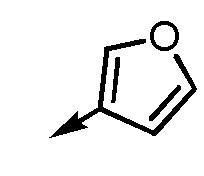
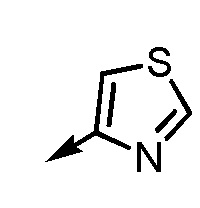
[формула 62]
Когда Ar1 представляет собой бензольное кольцо среди групп группы G1, пространственная конфигурация в формуле выше представляет собой S-конфигурацию, и когда Ar1 представляет собой тиофеновое кольцо среди групп группы G1, пространственная конфигурация в формуле выше представляет собой R-конфигурацию.
[0088]
<Способы получения соединений настоящего изобретения>
Соединения настоящего изобретения представляют собой новые соединения, неописанные в литературе. Хотя соединения настоящего изобретения можно получить, например, следующими способами, способ получения соединений настоящего изобретения не ограничен следующими способами.
[0089]
Хотя продолжительность реакции в каждой из реакций конкретно не ограничена, ход реакций можно легко контролировать способами анализа, описанными ниже, и, следовательно, реакции можно прекращать при достижении максимального выхода целевого вещества. Каждую из реакций можно проводить в атмосфере инертного газа, например, в потоке азота или в потоке аргона, если потребуется. Когда защита защитной группой и последующее деблокирование необходимы в каждой из реакций, реакции можно надлежащим образом проводить, применяя способы, описанные ниже.
[0090]
Примеры защитной группы, применяемой в настоящем изобретении, включают следующие группы: защитные группы для карбоксильной группы (-COOH), защитные группы для гидроксильной группы (-OH), защитные группы для алкинильной группы, защитные группы для амино группы (-NH2) и подобные.
[0091]
Примеры защитной группы для карбоксильной группы включает, например, алкил, содержащий 1-4 атома углерода, алкенил, содержащий 2-4 атома углерода, алкил, содержащий 1-4 атома углерода и замещенный алкокси, содержащий 1-4 атома углерода, алкил, содержащий 1-4 атома углерода и замещенный 1-3 галогенами, и подобными. Конкретные примеры включают метил, этил, трет-бутил, аллил, метоксиэтил, трихлорэтил и подобные.
[0092]
Примеры защитных групп для гидроксильной группы включают, например, алкил, содержащий 1-4 атома углерода, алкенил, содержащий 2-4 атома углерода, алкил, содержащий 1-4 атома углерода и замещенный алкокси, содержащей 1-4 атома углерода, алкил, содержащий 1-4 атома углерода и замещенный 1-3 галогенами, силил, замещенный тремя одинаковыми или различными алкилами, содержащими 1-4 атома углерода, или фенилами, тетрагидропиранил, тетрагидрофурил, пропаргил, триметилсилилэтильную группу и подобные. Конкретные примеры включают метил, этил, трет-бутил, аллил, метоксиметил (MOM), метоксиэтил (MEM), трихлорэтил, фенил, метилфенил, хлорфенил, бензил, метилбензил, хлорбензил, дихлорбензил, фторбензил, трифторметилбензил, нитробензил, метоксифенил, N-метиламинобензил, N, N-диметиламинобензил, фенацил, тритил, 1-этоксиэтил (EE), тетрагидропиранил (THP), тетрагидрофурил, пропаргил, триметилсилил (TMS), триэтилсилил (TES), трет-бутилдиметилсилил (TBDMS), трет-бутилдифенилсилил (TBDPS), ацетил (Ac), пивалоил, бензоил, аллилоксикарбонил (Alloc), 2,2,2-трихлорэтоксикарбонил (Troc) и подобные.
[0093]
Примеры защитных групп для алкинила включают триметилсилил, 2-гидрокси-2-пропил и подобные.
Примеры защитных групп для амино группы включают, например, бензил, метилбензил, хлорбензил, дихлорбензил, фторбензил, трифторметилбензил, нитробензил, метоксифенил, N-метиламинобензил, N, N-диметиламинобензил, фенацил, ацетильная группа, трифторацетил, пивалоил, бензоил, аллилоксикарбонил, 2,2,2-трихлорэтоксикарбонил, бензилоксикарбонил, трет-бутоксикарбонил (Boc), 1-метил-1-(4-бифенил)этоксикарбонил (Bpoc), 9-флуоренилметоксикарбонил, бензилоксиметил (BOM), 2-(триметилсилил)этоксиметил (SEM) и подобные.
[0094]
Удалив данные защитные группы одновременно с получением или постадийно в процессе получения или на конечной стадии, защищенные соединения можно превратить в целевые соединения. Реакции защиты и деблокирования можно проводить согласно известным способам, таким как способы, описанные, например, в Protective groups in Organic Synthesis, опубликованной John Wiley and Sons (2007) и подобных, и их можно осуществлять, например, способами (1)-(6), приведенными ниже, и подобными.
[0095]
(1) Реакцию деблокирования щелочным гидролизом проводят, например, реакцией защищенного соединения с основанием в полярном растворителе. Примеры основания, применяемого в данной реакции, включают, например, основания щелочных металлов, такие как гидроксид натрия, гидроксид калия, гидроксид лития, гидроксид бария, гидроксид кальция, карбонат натрия, карбонат калия, метоксид натрия и трет-бутоксид калия, и органические основания, такие как триэтиламин. Например, их можно обычно применять в количестве 1-20 кратный мольный избыток, предпочтительно 1-10 кратный мольный избыток, относительно реагента, когда применяют основание щелочного металла, или от 1 кратного мольного избытка до большего избыточного количества, когда применяют органическое основание. Что касается растворителя реакции, обычно предпочтительно проводить реакцию в неактивной среде, которая не ингибирует реакцию, предпочтительно полярном растворителе. Примеры полярного растворителя включают воду, метанол, этанол, тетрагидрофуран, диоксан и подобные, и их можно применять в виде смеси, при необходимости. Что касается температуры реакции, выбирают подходящую температуру, например, от -10°C до температуры кипения растворителя. Продолжительность реакции, например, обычно составляет 0,5-72 часа, предпочтительно 1-48 часов, когда применяют основание щелочного металла, или обычно 5 часов-14 дней, когда применяют органическое основание. Однако за ходом реакции можно следить с помощью тонкослойной хроматографии (ТСХ), высокоэффективной жидкостной хроматографии (ВЭЖХ) или подобных, и, соответственно, реакцию можно обычно прекратить, когда получен максимальный выход целевого соединения.
[0096]
(2) Реакцию деблокирования в кислых условиях проводят, например, в органическом растворителе (дихлорметан, хлороформ, диоксан, этилацетат, анизол и подобные) в присутствии органической кислоты (уксусная кислота, трифторуксусная кислота, метансульфокислота, п-толуолсульфокислота и подобные), кислоты Льюиса (трибромид бора, трифторид бора, бромид алюминия, хлорид алюминия и подобные) или неорганической кислоты (хлористоводородная кислота, серная кислота и подобные) или их смеси (бромистоводородная кислота/уксусная кислота и подобные) при температуре -10-100°C. Есть также способ добавления этантиола, 1,2-этандитиола или подобных в качестве добавки.
[0097]
(3) Реакцию деблокирования гидрогенолизом проводят, например, в растворителе [растворителях эфирного типа (тетрагидрофуран, диоксан, диметоксиэтан, диэтиловый эфир и подобные), растворителях спиртового типа (метанол, этанол и подобные), растворителях бензольного типа (бензол, толуол и подобные), растворителях кетонового типа (ацетон, метилэтилкетон и подобные), растворителях нитрильного типа (ацетонитрил и подобные), растворителях амидного типа (диметилформамид и подобные), растворителях сложноэфирного типа (этилацетат и подобные), воде, уксусной кислоте, смеси двух или более типов данных растворителей и подобных] в присутствии катализатора (палладия/угольный порошок, оксид платины (PtO2), активированный никель и подобные) и источника водорода, такого как газообразный водород стандартного давления или под давлением, формиат аммония, или гидразингидрат, при температуре -10-60°C.
[0098]
(4) Реакцию деблокирования силильной группы проводят, например, применяя фторид тетра-н-бутиламмония или подобные, в органическом растворителе, смешивающимся с водой (тетрагидрофуран, ацетонитрил и подобные) при температуре -10-60°C.
[0099]
(5) Реакцию деблокирования, применяя металл, проводят, например, в кислом растворителе (уксусная кислота, буфер pH 4,2-7,2, смеси данного раствора и органического растворителя, такого как тетрагидрофуран) в присутствии цинкового порошка с или без обработки ультразвуком при температуре -10-60°C.
[0100]
(6) Реакцию деблокирования, применяя комплекс металла, проводят, например, в органическом растворителе (дихлорметан, диметилформамид, тетрагидрофуран, этилацетат, ацетонитрил, диоксан, этанол и подобные), воде или их смеси, в присутствии агента, являющего ловушкой (гидрид трибутилолова, триэтилсилан, димедон, морфолин, диэтиламин, пирролидин и подобные), органической кислоты (уксусная кислота, муравьиная кислота, 2-этилгексановая кислота и подобные) и/или соли органической кислоты (2-этилгексаноат натрия, 2-этилгексаноат калия и подобные) в присутствии или отсутствии реагента фосфинового типа (трифенилфосфин и подобные), применяя комплекс металла [тетракистрифенилфосфин палладия (0), дихлорид бис(трифенилфосфин)палладия (II), ацетат палладия(II), хлорид трис(трифенилфосфин)родия(I) и подобные] при температуре -10-60°C.
[0101]
Соединения настоящего изобретения, представленные формулой (1), можно получить, например, согласно следующим реакционным способам. На следующих схемах, "стадия" обозначает каждую стадию, например, "стадия 1-1" обозначает стадию 1-1.
Стадия 1-1
[формула 63]
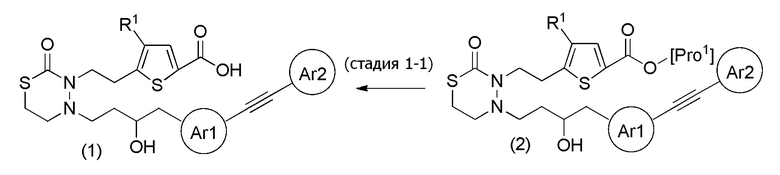
Соединения, представленные формулой (1), можно получить деблокированием соединения, представленного формулой (2) [в формуле (2), "Pro1" представляет собой защитную группу карбоксила в формуле (1)] для защитной группы Pro1. Реакцию деблокирования можно осуществлять согласно известным способам, например, способам, описанным в Protective groups in Organic Synthesis, опубликованной John Wiley and Sons (2007), и подобных.
[0102]
Pro1 конкретно не ограничена при условии, что она представляет собой защитную группу карбоксила, приведенного выше, и ее примеры включают, например, алкил, содержащий 1-4 атома углерода.
[формула 64]
[0103]
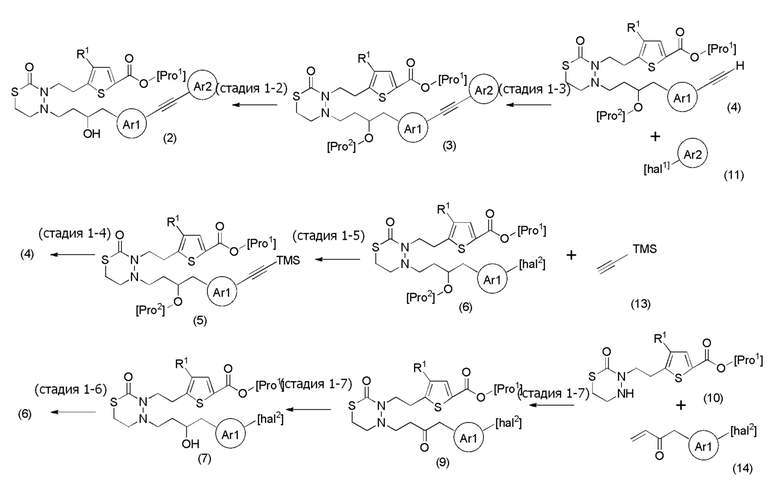
Стадия 1-2
Соединения, представленные формулой (2), можно получить деблокированием соединения, представленного формулой (3) [в формуле (3), "Pro2" представляет собой защитную группу гидроксильной группы в формуле (1), и "Pro1" имеет тоже значение, как определено выше] для защитной группы соединения, представленного формулой (3). Реакцию деблокирования можно осуществлять согласно известным способам, например, способам, описанным в Protective groups in Organic Synthesis, опубликованной John Wiley and Sons (2007), и подобных.
[0104]
Хотя Pro2 конкретно не ограничена при условии, что она представляет собой приведенную выше защитную группу гидроксильной группы, Pro2 предпочтительно представляет собой группу, отличную от TMS для того, чтобы селективно проводить деблокирование Pro2 относительно TMS в формуле (5). Примеры Pro2 включают, например, трет-бутильную группу, MOM группу, MEM группу, THP группу, ацетильную группу и TBDMS группу.
[0105]
Стадия 1-3
Соединения, представленные формулой (3), можно получить конденсацией соединения, представленного формулой (4) [в формуле (4), "Pro1" и "Pro2" имеют те же значения, как определено выше], и соединения, представленного формулой (11) [в формуле (11), "hal1" представляет собой бром или йод] в присутствии основания и палладиевого катализатора. Что касается количества соединения, представленного формулой (11), применяемого в реакции соединения, представленного формулой (4), и соединения, представленного формулой (11), 1/5-20 эквивалентов, предпочтительно 1/2-10 эквивалентов, более предпочтительно 1-5 эквивалентов соединения, представленного формулой (11), можно применять относительно соединения, представленного формулой (4). Однако количество соединения, представленного формулой (11), которое будут применять, можно подходящим образом определить с учетом чистоты, выхода, эффективности очистки и подобных соединения, представленного формулой (4).
[0106]
В качестве основания можно применять, например, карбонат цезия, карбонат натрия, карбонат калия и подобные, и карбонат цезия является предпочтительным. Что касается количества основания, которое будут применять, его можно применять в количестве от эквивалента до избыточного количества, например, 1-10 эквивалентов, предпочтительно 1-5 эквивалентов, относительно соединения, представленного формулой (4), которое служит в качестве исходного соединения.
[0107]
В качестве палладиевого катализатора, например, имеющиеся в продаже катализаторы, такие как тетракис(трифенилфосфин)палладий, тетракис(метилдифенилфосфин)палладий, дихлорбис(трифенилфосфин)палладий, дихлорбис(три-o-толилфосфин)палладий, дихлорбис(трициклогексилфосфин)палладий, дихлорбис(триэтилфосфин)палладий, ацетат палладия, хлорид палладия, хлорид бис(ацетонитрил)палладия, бис(дибензилиденацетон)палладий, трис(дибензилиденацетон)дипалладий и хлорид бис(дифенилфосфиноферроцен)палладия можно добавлять в реакционную систему как есть или в виде катализатора, отдельно полученного и выделенного из ацетата палладия, трис(дибензилиденацетон)дипалладия или подобных и произвольного лиганда. Катализатор, который, как считается, фактически участвует в реакции, можно получить в реакционной системе смешиванием ацетата палладия, трис(дибензилиденацетон)дипалладия или подобных и произвольного лиганда. Валентность палладия может быть 0 или +2. В частности, хлорид бис(ацетонитрил)палладия можно упомянуть в качестве предпочтительного примера.
[0108]
В качестве лиганда, применяемого для получения палладиевого катализатора из произвольного лиганда, можно привести в качестве примера фосфиновые лиганды, такие как трифенилфосфин, три(o-толил)фосфин, три(циклогексил)фосфин, три(трет-бутил)фосфин, дициклогексилфенилфосфин, 1,1'-бис(ди-трет-бутилфосфино)ферроцен, 2-дициклогексилфосфино-2'-диметиламино-1,1'-бифенил, 2-(ди-трет-бутилфосфино)бифенил, 2-(дициклогексилфосфино)бифенил, 2,2'-бис(дифенилфосфино)-1,1'-бинафтил, xantphos, и три(трет-бутил)фосфин. Также можно привести в качестве примера 2-дициклогексилфосфино-2',6'-диметоксибифенил, 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил, 1,2,3,4,5-пентаметил-1'-(ди-трет-бутилфосфино)ферроцен) и подобные, и можно предпочтительно упомянуть 2-дициклогексил-2',4',6'-триизопропилбифенил.
[0109]
Хотя количество в эквиваленте применяемого палладиевого катализатора может быть эквивалентным количеством или каталитическим количеством, оно составляет 0,01 мол% или более, более предпочтительно, в частности от 0,10 до 50,0 мол % в расчете на количество исходного соединения. Примеры растворителя, применяемого для реакции, включают, например, N, N-диметилформамид, N, N-диметилацетамид, ацетонитрил, ксилол, толуол, 1,4-диоксан и тетрагидрофуран, и предпочтительные примеры включают ацетонитрил. Два или более типов данных растворителей можно также применять в виде смеси. Что касается температуры реакции, реакцию можно проводить обычно при -40-100°C, предпочтительно при -20°C-60°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 0,5-48 часов, предпочтительно 1-24 часа.
[0110]
Стадия 1-4
Соединения, представленные формулой (4), можно получить селективным деблокированием TMD соединения, представленного формулой (5) [в формуле (5), "Pro1" и "Pro2" имеют те же значения, как определено выше]. Реакцию деблокирования можно осуществлять согласно известным способам, например, способам, описанным в Protective groups in Organic Synthesis, опубликованной John Wiley and Sons (2007), и подобных.
[0111]
Конкретно, соединения можно получить, например, воздействием неорганического основания на соединение, представленное формулой (5), в органическом растворителе. В качестве неорганического основания можно применять, например, гидроксид натрия, гидроксид калия, карбонат цезия, карбонат натрия, карбонат калия или подобные, и карбонат калия является предпочтительным. Что касается количества основания, которое будут применять, основание можно применять в количестве от эквивалентного количества до избыточного количества относительно соединения, представленного формулой (5), которое служит в качестве исходного соединения, и количество составляет, например, 1-10 эквивалентов, предпочтительно 1-5 эквивалентов. Примеры растворителя, применяемого для реакции, включают метанол и этанол, и предпочтительные примеры включают метанол. Что касается температуры реакции, реакцию можно проводить обычно при -20-60°C, предпочтительно при 0-40°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 0,5-48 часов, предпочтительно 1-24 часа.
[0112]
Стадия 1-5
Соединения, представленные формулой (5), можно получить конденсацией соединения, представленного формулой (6) [в формуле (6), "Pro1" и "Pro2" имеют те же значения, как определено выше, и "hal2" представляет собой бром или йод] с соединением, представленным формулой (13), в органическом растворителе в присутствии неорганического основания. Соединения можно получить тем же способом, как соединение стадии 1-3. В данном получении, соединение, представленное формулой (13), можно применять в количестве 1/5-20 эквивалентов, предпочтительно 1/2-10 эквивалентов, более предпочтительно 1-5 эквивалентов, относительно соединения, представленного формулой (6).
[0113]
Стадия 1-6
Соединения, представленные формулой (6), можно получить защитой гидроксильной группы соединения, представленного формулой (7) [в формуле (7), "Pro1" и "hal2" имеют те же значения, как определено выше]. Реакцию защиты для гидроксильной группы можно осуществлять согласно известным способам, например, способам, описанным в Protective groups in Organic Synthesis, опубликованной John Wiley and Sons (2007), и подобных. Хотя защитная группа гидроксильной группы конкретно не ограничена при условии, что выбрана приведенная выше защитная группа гидроксильной группы, например, можно применять трет-бутильную группу, MOM группу, MEM группу, THP группу, ацетильную группу, TBDMS группу и подобные.
[0114]
Стадия 1-7
Соединения, представленные формулой (7), можно получить воздействием восстанавливающего агента на соединение, представленное формулой (9) [в формуле (9), "Pro1" и "hal2" имеют те же значения, как определено выше] в органическом растворителе. В качестве восстанавливающего агента можно применять, например, боргидрид натрия, боргидрид лития, триацетоксиборгидрид, цианоборгидрид и подобные, и боргидрид натрия является предпочтительным. Что касается количества восстанавливающего агента, которое будут применять, его можно применять в количестве от 1/4 эквивалента до избыточного количества относительно соединения, представленного формулой (9), которое служит в качестве исходного соединения, и количество составляет, например, 1/4-10 эквивалентов, предпочтительно 1-5 эквивалентов. Примеры органического растворителя, применяемого для реакции, включают метанол, этанол, изопропанол и смешанный растворитель их с тетрагидрофураном, и предпочтительные примеры включают метанол. Что касается температуры реакции, реакцию можно проводить обычно при -20-60°C, предпочтительно при 0-40°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 0,5-48 часов, предпочтительно 1-24 часа.
[0115]
Стадия 1-8
Соединения, представленные формулой (9), можно получить воздействием соединения, представленного формулой (14) [в формуле (14), "hal2" имеет тоже значение, как определено выше] на соединение, представленное формулой (10) [в формуле (10), "Pro1" имеет тоже значение, как определено выше]. Что касается количества соединения, представленного формулой (14), которое будут применять, его можно применять в количестве от эквивалентного количества до избыточного количества относительно соединения, представленного формулой (10), которое служит в качестве исходного соединения, и количество составляет, например, эквивалентное количество-10 эквивалентов, предпочтительно 1-5 эквивалентов. Примеры растворителя, применяемого для реакции, включают метанол, этанол, изопропанол, и их смешанный растворитель с водой, и предпочтительные примеры включают этанол. Что касается температуры реакции, реакцию можно проводить обычно при 0-120°C, предпочтительно при 40-100°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 0,5-48 часов, предпочтительно 1-24 часа.
[0116]
Стадия 1-9
[формула 65]
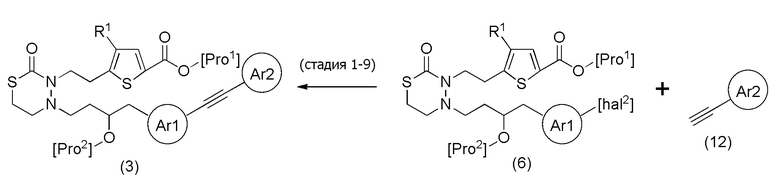
Соединения, представленные формулой (3), можно также получить из соединения, представленного формулой (6) [в формуле (6), "Pro1", "Pro2" и "hal2" имеют те же значения, как определено выше], и соединения, представленного общей формулой (12) тем же способом, как соединение способа стадии 1-3. Что касается количества соединения, представленного формулой (12), применяемого в данном случае, его можно применять в количестве 1/5-20 эквивалентов, предпочтительно 1/2-10 эквивалентов, более предпочтительно 1-5 эквивалентов, относительно соединения, представленного формулой (6).
[0117]
Стадия 1-10
[формула 66]
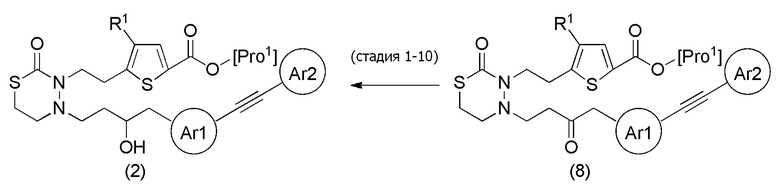
Соединения, представленные формулой (2), можно также получить из соединения, представленного формулой (8) [в формуле (8), "Pro1" имеет тоже значение, как определено выше] тем же способом, как способ стадии 1-7.
[0118]
Стадия 1-11
[формула 67]
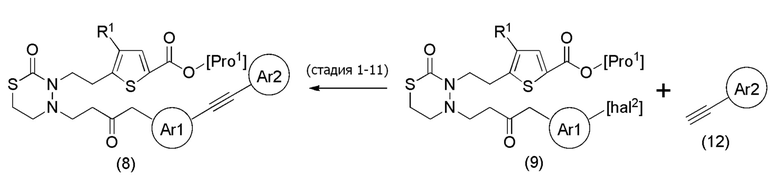
Соединения, представленные формулой (8), получают конденсацией соединения, представленного формулой (9), в котором "hal2" представляет собой атом йода [в формуле (9), "Pro1" имеет тоже значение, как определено выше], и соединения, представленного формулой (12), в присутствии основания, медного катализатора и палладиевого катализатора. Что касается количества соединения, представленного формулой (12), применяемого в реакции соединения, представленного формулой (9), и соединения, представленного формулой (12), его можно применять в количестве 1/5-20 эквивалентов, предпочтительно 1/2-10 эквивалентов, более предпочтительно 1-5 эквивалентов, относительно соединения, представленного формулой (9), но его можно подходящим образом определить с учетом чистоты, выхода, эффективности очистки и подобных соединения, представленного формулой (8).
[0119]
Что касается основания, например, триэтиламин, диэтиламин, диизопропиламин, диизопропилэтиламин, морфолин, пиперидин, пиридин и подобные можно применять, и диэфирные амины являются предпочтительными. Что касается количества основания, которое будут применять, его можно применять в количестве от эквивалентного количества до избыточного количества, например, 1-10 эквивалентов, предпочтительно 1-5 эквивалентов, относительно соединения, представленного формулой (9), которое служит в качестве исходного соединения.
[0120]
Примеры медного катализатора включают, например, йодид меди(I), бромид меди(I), хлорид меди(I) и подобные, и йодид меди (I) является предпочтительным.
Хотя количество в эквивалентах медного катализатора, которое будут применять, может представлять собой эквивалентное количество или каталитическое количество, оно предпочтительно составляет 0,01 моль % или более, особенно предпочтительно 0,10-50,0 моль %, относительно исходного соединения.
[0121]
В качестве палладиевого катализатора, например, имеющиеся в продаже катализаторы, такие как тетракис(трифенилфосфин)палладий, тетракис(метилдифенилфосфин)палладий, дихлорбис(трифенилфосфин)палладий, дихлорбис(три-o-толилфосфин)палладий, дихлорбис(трициклогексилфосфин)палладий, дихлорбис(триэтилфосфин)палладий, ацетат палладия, хлорид палладия, хлорид бис(ацетонитрил)палладия, бис(дибензилиденацетон)палладий, трис(дибензилиденацетон)дипалладий и хлорид бис(дифенилфосфиноферроцен)палладия можно добавлять к реакционной системе, как есть или в виде катализатора, отдельно полученного и выделенного из ацетата палладия, трис(дибензилиденацетон)дипалладия или подобных, и произвольного лиганда. Катализатор, который, как считается, фактически участвует в реакции, можно получить в реакционной системе смешением ацетата палладия, трис(дибензилиденацетон)дипалладия или подобных, и произвольного лиганда. Валентность палладия может быть 0 или +2. В частности, тетракис(трифенилфосфин)палладий можно упомянуть в качестве предпочтительного примера. Когда палладиевый катализатор получают из произвольного лиганды, можно применять те же лиганды, как лиганды, приведенные в качестве примеров для стадии 1-3.
[0122]
Хотя количество в эквивалентах палладиевого катализатора, которое будут применять, может представлять собой эквивалентное количество или каталитическое количество, оно предпочтительно составляет 0,01 моль % или более, более предпочтительно, особенно 0,10-50,0 моль % относительно исходного соединения.
[0123]
Примеры растворителя, применяемого для реакции, включают, например, N, N-диметилформамид, N, N-диметилацетамид, ацетонитрил, ксилол, толуол, 1,4-диоксан, тетрагидрофуран и подобные, или реакцию можно проводить без растворителя. Реакция, проводимая без растворителя, представляет собой предпочтительный пример. Что касается температуры реакции, реакцию можно проводить обычно при -40-100°C, предпочтительно при -20-60°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 0,5-48 часов, предпочтительно 1-24 часа.
[0124]
Стадия 1-12
[формула 68]
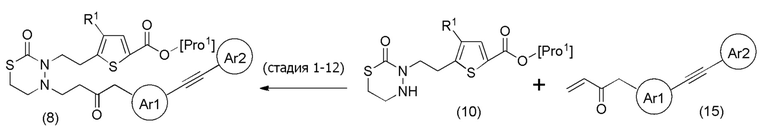
Соединения, представленные формулой (8), можно также получить из соединения, представленного формулой (10) [в формуле (10), "Pro1" имеет тоже значение, как определено выше], и соединения, представленного формулой (15), тем же способом, как способ стадии 1-8.
[формула 69]
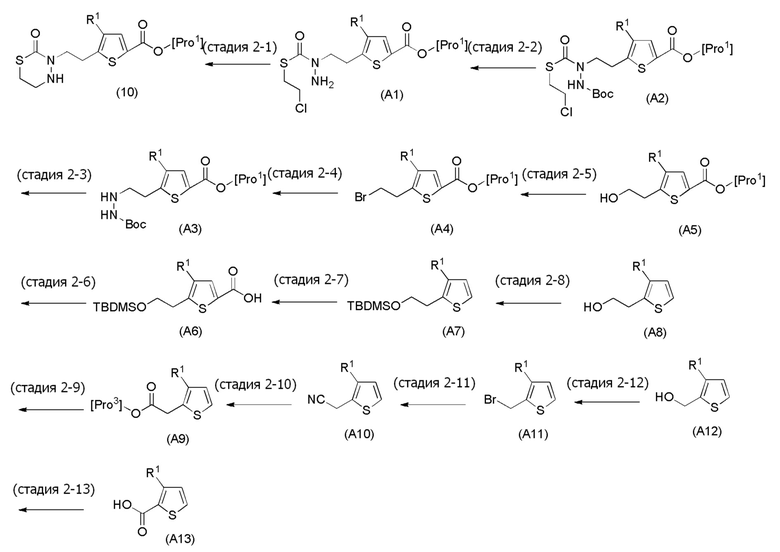
[0125]
Стадия 2-1
Соединения, представленные формулой (10), можно получить действием основания на соединение, представленное формулой (A1) [в формуле (A1), "Pro1" имеет тоже значение, как определено выше] в органическом растворителе. В качестве основания можно применять, например, гидроксид натрия, гидроксид калия, карбонат цезия, карбонат натрия, гидрокарбонат натрия, карбонат калия и подобные, и гидрокарбонат натрия является предпочтительным. Основание можно применять в количестве от эквивалентного количества до избыточного количества, например, 1-20 эквивалентов, предпочтительно 1-10 эквивалентов, относительно соединения, представленного формулой (10), которое служит в качестве исходного соединения. Йодид натрия можно применять в качестве добавки, и можно применять в количестве от эквивалентного количества до избыточного количества, например, 1-10 эквивалентов, предпочтительно 1-5 эквивалентов, относительно соединения, представленного формулой (10), которое служит в качестве исходного соединения. Примеры органического растворителя, применяемого для реакции, включают N, N-диметилформамид, N, N-диметилацетамид, ацетонитрил, толуол, тетрагидрофуран, 1,4-диоксан, диэтиловый эфир и их смешанный растворитель, и предпочтительные примеры включают ацетонитрил. Что касается температуры реакции, реакцию можно проводить обычно при 0-100°C, предпочтительно при 20-60°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 0,5-48 часов, предпочтительно 1-24 часа.
[0126]
Стадия 2-2
Соединения, представленные формулой (A1), можно получить деблокированием соединения, представленного формулой (A2) [в формуле (A2), "Pro1" имеет тоже значение, как определено выше] защитной группы. Реакцию деблокирования можно осуществлять согласно известным способам, например, способам, описанным в Protective groups in Organic Synthesis, опубликованной John Wiley and Sons (2007) и подобных.
[0127]
Стадия 2-3
Соединения, представленные формулой (A2), можно получить воздействием эфира хлортиомуравьиной кислоты, такой как 2-хлорэтилхлортиоформиат, на соединение, представленное формулой (A3) [в формуле (A3), "Pro1" имеет тоже значение, как определено выше] в присутствии основания. Эфир хлортиомуравьиной кислоты можно применять в количестве от эквивалентного количества до избыточного количества, например, 1-5 эквивалентов, предпочтительно 1-2 эквивалентов, относительно соединения, представленного формулой (A2), которое служит в качестве исходного соединения. В качестве основания, которое будут применять, можно применять, например, карбонат натрия, карбонат калия, гидрокарбонат натрия, карбонат цезия, гидроксид натрия, диизопропилэтиламин, триэтиламин и подобные, и гидрокарбонат натрия является предпочтительным. Примеры растворителя, применяемого для реакции, включают дихлорметан, толуол, тетрагидрофуран, 1,4-диоксан, ацетонитрил и подобные, и дихлорметан является предпочтительным. Что касается температуры реакции, реакцию можно проводить при 0-100°C, предпочтительно при 10-30°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 1-24 часов, предпочтительно 2-4 часа.
[0128]
Стадия 2-4
Соединения, представленные формулой (A3), можно получить воздействием трет-бутоксикарбонилгидразина на соединение, представленное формулой (A4) [в формуле (A4), "Pro1" имеет то же значение, как определено выше] в присутствии основания. В качестве основания, которое будут применять, можно применять, например, карбонат натрия, карбонат калия, гидрокарбонат натрия, карбонат цезия, гидроксид натрия, диизопропилэтиламин, триэтиламин и подобные, и гидрокарбонат натрия является предпочтительным. Количество основания, которое будут применять, составляет, например, 1-20 эквивалентов, предпочтительно 3-5 эквивалентов, относительно соединения, представленного формулой (A4), которое служит в качестве исходного соединения. Йодид натрия или подобные можно применять в качестве добавки. Примеры растворителя, применяемого для реакции, включают ацетонитрил, пропионитрил, N, N-диметилформамид, диметилсульфоксид, N-метилпирролидон и подобные, и предпочтительные примеры включают ацетонитрил. Что касается температуры реакции, реакцию можно проводить обычно при комнатной температуре-150°C, предпочтительно при 70°C-100°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, 3-36 часов, предпочтительно 3-18 часов.
[0129]
Стадия 2-5
Соединения, представленные формулой (A4), можно получить замещением атомом брома гидроксильной группы соединения, представленного формулой (A5) [в формуле (A5), "Pro1" имеет тоже значение, как определено выше]. Реакцию замещения на атом брома можно проводить воздействием тетрабромида углерода, N-бромсукцинимида или подобных на соединение в присутствии трифенилфосфина или подобных. Количество трифенилфосфина, которое будут применять, составляет, например, 1-5 эквивалентов, предпочтительно 1-2 эквивалента, относительно соединения, представленного формулой (A5), которое представляет собой исходное соединение. Количество тетрабромида углерода или подобных, которое будут применять, составляет, например, 1-5 эквивалентов, предпочтительно 1-2 эквивалента, относительно соединения, представленного формулой (A5), которое представляет собой исходное соединение. Примеры растворителя, применяемого для реакции, включают дихлорметан, толуол, тетрагидрофуран, 1,4-диоксан, ацетонитрил и подобные, и дихлорметан является предпочтительным. Что касается температуры реакции, реакцию можно проводить обычно при -20-40°C, предпочтительно при -10-10°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 3-36 часов, предпочтительно 12-20 часов.
[0130]
Стадия 2-6
Соединения, представленные формулой (A5), можно получить деблокированием защитной группы гидроксильной группы соединения, представленного формулой (6A). Деблокирование можно осуществлять согласно известным способам, например, способам, описанным в Protective groups in Organic Synthesis, опубликованной John Wiley and Sons (2007) и подобные.
[0131]
Стадия 2-6
Соединения, представленные формулой (A5), можно получить превращением соединения, представленного формулой (A6), являющегося карбоновой кислотой, в эфир, и проведением деблокирования, удаляя защитную группу гидроксильной группы. Реакцию можно проводить в спиртовом растворителе в присутствии кислоты. Примеры кислоты, применяемой для реакции, включают серную кислоту, хлористоводородную кислоту, метансульфокислоту, п-толуолсульфокислоту, трифторуксусную кислоту и подобные, и предпочтительные примеры включают серную кислоту. В качестве растворителя можно применять метанол, этанол или подобные, и предпочтительные примеры включают метанол. Что касается температуры реакции, реакцию можно проводить обычно при комнатной температуре-140°C, предпочтительно при 50-80°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 2-24 часа, предпочтительно 8-16 часов.
[0132]
Стадия 2-7
Соединения, представленные формулой (A6), можно получить воздействием сильное основание, и затем диоксида углерода на соединение, представленное формулой (A7). В качестве сильного основания можно применять амид лития, такой как диизопропиламид лития или гексаметилдисилазид лития. Когда R1 представляет собой водород, можно также применять низший алкиллитий, такой как н-бутиллитий, втор-бутиллитий и н-пропиллитий, и предпочтительно применять диизопропиламид лития. Количество применяемого сильного основания составляет, например, 1-3 эквивалента, предпочтительно 1-2 эквивалента, относительно соединения, представленного формулой (A7), которое представляет собой исходное соединение. Примеры растворителя, применяемого для реакции, включают тетрагидрофуран, диэтиловый эфир, 1,4-диоксан и подобные, и тетрагидрофуран является предпочтительным. Что касается температуры реакции с сильным основанием, реакцию можно проводить обычно при -100--20°C, предпочтительно при -80--60°C. Последующую реакцию с диоксидом углерода или подобными можно обычно проводить при -40-40°C и предпочтительно -20-10°C. Хотя продолжительность реакции с сильным основанием конкретно не ограничена, она составляет, например, 0,2-3 часа, предпочтительно 0,5-1 час. Хотя продолжительность реакции с диоксидом углерода или подобными конкретно не ограничена, она составляет, например, обычно 0,5-24 часа, предпочтительно 0,75-2 часа.
[0133]
Стадия 2-8
Соединения, представленные формулой (A7), можно получить защитой гидроксильной группы соединения, представленного формулой (A8), TBDMS. Защиту гидроксильной группы можно проводить, применяя способ, аналогичный способу стадии 1-6.
[0134]
Соединение формулы (A8), в котором R1 представляет собой H, представляет собой имеющееся в продаже соединение (2-(тиофен-2-ил)этанол, Tokyo Chemicals). Следовательно, когда R1 в формуле (A8) представляет собой H, следующие стадии не требуются.
[0135]
Стадия 2-9
Соединения, представленные формулой (A8), можно получить восстановлением эфирной группы соединения, представленного формулой (A9) [в формуле (A9), "Pro3" представляет собой защитную группу карбоксила соединения формулы (A8)]. ТО есть, в качестве Pro3 можно применять, например, алкил, содержащий 1-4 атома углерода.
[0136]
В качестве восстанавливающего агента можно применять, например, алюмогидрид лития, диизобутилалюмогидрид, гидрид лития-триэтилборан и подобные, и алюмогидрид лития является предпочтительным. Количество восстанавливающего агента, которое будут применять, составляет, например, 0,5-5 моль эквивалентов, предпочтительно 1-2 моль эквивалента, относительно соединения, представленного формулой (A9), которое служит в качестве исходного соединения.
[0137]
Примеры растворителя, применяемого для реакции, включают тетрагидрофуран, диэтиловый эфир, толуол, и их смешанный растворитель, и предпочтительные примеры включают тетрагидрофуран и диэтиловый эфир. Что касается температуры реакции, реакцию можно проводить обычно при -10-20°C, предпочтительно при -5-5°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 0,08-0,5 часа, предпочтительно 0,15-0,3 часа.
[0138]
Стадия 2-10
Соединения, представленные формулой (A9), можно получить, например, проведением сольволиза соединения, представленного формулой (A10), до спирта в присутствии кислоты. В качестве кислоты можно применять серную кислоту, метансульфокислоту, хлористоводородную кислоту и подобные, и серная кислота является предпочтительным. Количество серной кислоты, которое будут применять, составляет, например, 0,0001-0,005 моль эквивалента, предпочтительно 0,0002-0,001 моль эквивалента, относительно соединения, представленного формулой (A10), которое служит в качестве исходного соединения. В качестве спирта, применяемого в качестве растворителя, можно применять, например, этанол, метанол, н-пропанол, н-бутиловый спирт, изобутиловый спирт и подобные. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 6-48 часов, предпочтительно 16-24 часа.
[0139]
Стадия 2-11
Соединения, представленные формулой (A10), можно получить воздействием цианида на соединение, представленное формулой (A11). В качестве цианида можно применять, например, цианид натрия, цианид калия и подобные. Количество цианина, которое будут применять, составляет, например, 1-5 эквивалентов, предпочтительно 1-2 эквивалента, относительно соединения, представленного формулой (A10), которое представляет собой исходное соединение. В качестве растворителя применяемого для реакции можно применять тетрагидрофуран, ацетонитрил, диметилсульфоксид, N, N-диметилацетамид, N, N-диметилформамид и подобные, и предпочтительные примеры включают смешанный растворитель ацетонитрила и диметилсульфоксида. Что касается температуры реакции, реакцию можно проводить обычно при 0-60°C, предпочтительно 10-40°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 0,5-20 часов, предпочтительно 2-6 часов.
[0140]
Стадия 2-12
Соединения, представленные формулой (A11), можно получить превращением гидроксильной группы соединения, представленного формулой (A12), в атом брома. Превращение в атом брома можно осуществлять тем же способом, как способ стадии 2-5.
[0141]
Стадия 2-13
Соединения, представленные формулой (A12), можно получить восстановлением карбоксильной группы имеющегося в продаже соединения, представленного формулой (A13), до гидроксильной группы. В качестве восстанавливающего агента можно применять, например, боран-диметилсульфид, боран-тетрагидрофуран и подобные, и боран-тетрагидрофуран является предпочтительным. Количество восстанавливающего агента, которое будут применять, составляет, например, 1-5 моль эквивалентов, предпочтительно 1-2 моль эквивалента, относительно соединения, представленного формулой (A13), которое служит в качестве исходного соединения.
[0142]
В качестве растворителя, применяемого для реакции, можно применять тетрагидрофуран, диэтиловый эфир и подобные, и предпочтительные примеры включают тетрагидрофуран. Что касается температуры реакции, реакцию можно проводить обычно при 0-60°C, предпочтительно при 10-40°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 4-24 часа, предпочтительно 10-18 часов.
[0143]
Примеры имеющегося в продаже соединения, представленного формулой (A13), включают, например, 3-хлортиофен-2-карбоновую кислоту, 3-бромтиофен-2-карбоновую кислоту и подобные, и их можно купить, например, у Sigma-Aldrich.
[0144]
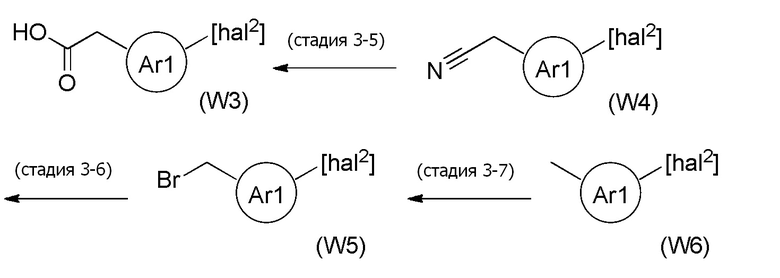
Стадия 3-1
[формула 70]
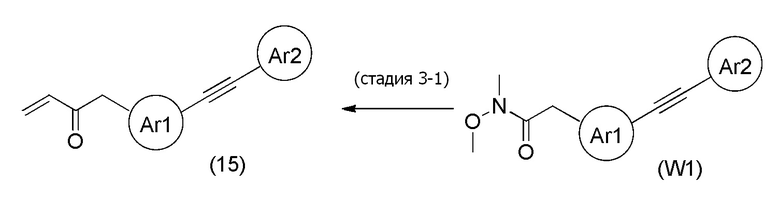
Соединения, представленные формулой (15), можно получить воздействием винилирующего агента на соединение, представленное формулой (W1), в органическом растворителе. В качестве винилирующего агента можно применять, например, винилмагнийбромид, винилмагнийхлорид, виниллитий и подобные, и винилмагнийбромид и винилмагнийхлорид являются предпочтительными. Винилмагний бромид и винилмагний хлорид можно применять в виде раствора в тетрагидрофуране, диэтиловом эфире или толуоле, и тетрагидрофурановый раствор является предпочтительным. Что касается количества винилирующего агента, которое будут применять, агент можно применять в количестве от эквивалентного количества до избыточного количества, например, 1-10 эквивалентов, предпочтительно 1-5 эквивалентов, относительно соединения, представленного формулой (W1), которое служит в качестве исходного соединения. Примеры растворителя, применяемого для реакции, включают толуол, тетрагидрофуран, 1,4-диоксан, диэтиловый эфир, 1,2-диметоксиэтан, и их смешанный растворитель, и предпочтительные примеры включают тетрагидрофуран и 1,2-диметоксиэтан. Что касается температуры реакции, реакцию можно проводить обычно при -78-0°C, предпочтительно при -50-0°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 0,5-24 часа, предпочтительно 1-12 часов.
[0145]
Стадия 3-2
[формула 71]
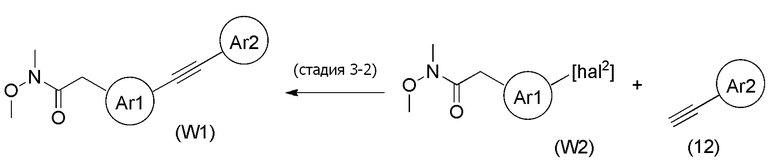
Соединения, представленные формулой (W1), можно получить из соединения, представленного формулой (W2) [в формуле (W2), "hal2" имеет тоже значение, как определено выше] и соединения, представленного формулой (12), тем же способом, как способ стадии 1-3. В данной реакции, соединение, представленное формулой (12), можно применять в количестве 1/5-20 эквивалентов, предпочтительно 1/2-10 эквивалентов, более предпочтительно 1-5 эквивалентов, относительно соединения, представленного формулой (W2).
[0146]
Стадия 3-3
[формула 72]
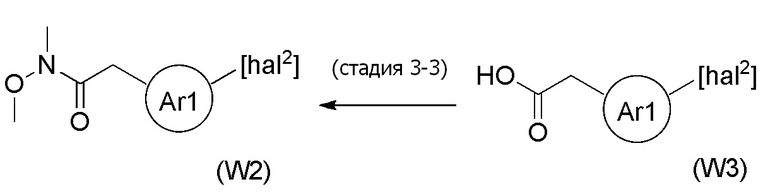
Соединения, представленные формулой (W2), можно получить реакцией соединения, представленного формулой (W3) [в формуле (W3), "hal2" имеет тоже значение, как определено выше] с гидрохлоридом N, O-диметилгидроксиламина в органическом растворителе в присутствии основания и конденсирующего агента, отщепляющего воду. Количество гидрохлорида N, O-диметилгидроксиламина, применяемого в реакции соединения, представленного формулой (W3), и гидрохлорида N, O-диметилгидроксиламина может представлять собой от эквивалентного количества до избыточного количества, например, 1-10 эквивалентов, предпочтительно 1-5 эквивалентов, относительно соединения, представленного формулой (W3), но количество можно подходящим образом определить с учетом чистоты, выхода, эффективности очистки и подобных соединения, представленного формулой (W3).
[0147]
В качестве основания можно применять, например, триэтиламин, диизопропилэтиламин, 1,4-диазабицикло[2,2,2]октан, N, N-диметил-4-аминопиридин и подобные, и диизопропилэтиламин является предпочтительным. Количество основания, которое будут применять, может представлять собой от эквивалентного количества до избыточного количества, например, 1-10 эквивалентов, предпочтительно 1-5 эквивалентов, относительно суммы эквивалентов соединения, представленного формулой (W3), которое служит в качестве исходного соединения, и эквивалентов гидрохлорида N, O-диметилгидроксиламина.
[0148]
В качестве конденсирующего агента, отщепляющего воду, можно применять N, N'-дициклогексилкарбодиимид, 1-этил-3-(3-диметиламинопропил)карбодиимид, N, N'-диизопропилкарбодиимид, N-циклогексил-N'-(2-морфолиноэтил)карбодиимид п-толуолсульфонат, N, N'-карбонилдимидазол, хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния, гексафторфосфат 1H-бензотриазол-1-илокси-трис(диметилфосфония), гексафторфосфат 1H-бензотриазол-1-илокси-трипирролидинофосфония, гексафторфосфат O-(бензотриазол-1-ил)-N, N,N',N'-тетраметилурония, гексафторфосфат O-(7-азабензотриазол-1-ил)-N, N,N',N'-тетраметилурония и подобные, и 1-этил-3-(3-диметиламинопропил)карбодиимид является предпочтительным. Количество конденсирующего агента, отщепляющего воду, которое будут применять, может представлять собой от эквивалентного количества до избыточного количества, например, 1-10 эквивалентов, предпочтительно 1-5 эквивалентов, относительно эквивалента соединения, представленного формулой (W3), которое служит в качестве исходного соединения.
[0149]
В качестве активатора можно добавлять N, N-диметил-4-аминопиридин или подобные. Количество активатора, которое будут применять, может представлять собой от каталитического количества до избыточного количества, например, 0,01-5 эквивалентов, предпочтительно 0,1-1 эквивалент, относительно эквивалента соединения, представленного формулой (W3), которое служит в качестве исходного соединения.
[0150]
Примеры растворителя, применяемого для реакции, включают, например, N, N-диметилформамид, N, N-диметилацетамид, ацетонитрил, ксилол, толуол, 1,4-диоксан, дихлорметан, хлороформ, 1,2-дихлорэтан, тетрагидрофуран и подобные, и предпочтительные примеры включают дихлорметан. Два или более типов данных растворителей можно также применять в виде смеси. Что касается температуры реакции, реакцию можно проводить обычно при 0-100°C, предпочтительно при 20-60°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 0,5-48 часов, предпочтительно 1-24 часа.
[0151]
Стадия 3-4
[формула 73]
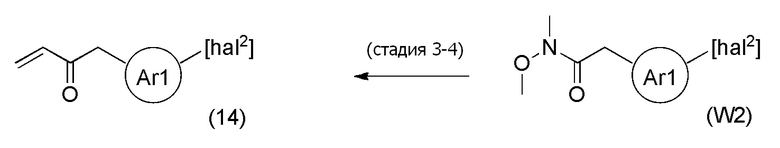
Соединения, представленные формулой (14), можно получить из соединения, представленного формулой (W2) [в формуле (W2), "hal2" имеет тоже значение, как определено выше] тем же способом, как способ стадии 3-1.
[формула 74]
[0152]
Стадия 3-5
Соединения, представленные формулой (W3), можно получить нагреванием соединения, представленного формулой (W4) [в формуле (W4), "hal2" имеет тоже значение, как определено выше] в разбавленной серной кислоте. В качестве разбавленной серной кислоты, применяемой для реакции, можно применять подходящим образом разбавленную концентрированную серную кислоту или разбавленную серную кислоту, и ее концентрация составляет, например, 0,1-15 моль/л, предпочтительно 1-10 моль/л. Количество разбавленной серной кислоты, которое будут применять, может представлять собой избыточное количество для соединения, представленного формулой (W4), и его можно определить с учетом выхода, эффективности очистки и подобных. Что касается температуры реакции, реакцию можно проводить обычно при 20-100°C, предпочтительно при 60-100°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 0,5-48 часов, предпочтительно 1-24 часа.
[0153]
Среди соединений, представленных формулой (W3), 3-бромфенилуксусная кислота, 3-йодфенилуксусная кислота и 3-бром-4-фторфенилуксусная кислота являются имеющимися в продаже соединениями, и их можно получить у Tokyo Chemical Industry. 2-(4-Бромтиофен-2-ил)уксусная кислота представляет собой имеющееся в продаже соединение, и его можно получить у APOLLO.
[0154]
Стадия 3-6
Соединения, представленные формулой (W4), можно получить воздействием цианида на соединение, представленное формулой (W5) [в формуле (W5), "hal2" имеет тоже значение, как определено выше]. В качестве цианида можно применять цианид натрия, цианид калия, цианид меди и подобные, и цианид натрия и цианид калия являются предпочтительными. Количество цианида, которое будут применять, может представлять собой от эквивалентного количества до избыточного количества, например, 1-10 эквивалентов, предпочтительно 1-5 эквивалентов, относительно соединения, представленного формулой (W5), которое служит в качестве исходного соединения. Примеры растворителя, применяемого для реакции, включают, например, метанол, этанол, изопропанол, воду, их смешанный растворитель и подобные, и предпочтительные примеры включают смешанный растворитель этанола и воды при соотношении 2:1. Что касается температуры реакции, реакцию можно проводить обычно при 0-100°C, предпочтительно при 20-100°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 0,5-24 часа, предпочтительно 1-12 часов.
[0155]
Стадия 3-7
Соединения, представленные формулой (W5), можно получить бромированием соединения, представленного формулой (W6) [в формуле (W6), "hal2" имеет тоже значение, как определено выше]. Примеры бромирующего агента включает N-бромсукцинимид и 1,3-дибром-5,5-диметилгидантоин, и N-бромсукцинимид является предпочтительным. Количество бромирующего агента, которое будут применять, может представлять собой от эквивалентного количества до избыточного количества, например, 1-10 эквивалентов, предпочтительно 1-5 эквивалентов, относительно соединения, представленного формулой (W6), которое служит в качестве исходного соединения. Примеры активатора, который добавляют вместе с бромирующим агентом, включают бензоилпероксид, трет-бутилгидропероксид и азабисизобутиронитрил, и бензоилпероксид является предпочтительным. Количество активатора, которое будут применять, может представлять собой от каталитического количества до избыточного количества, например, 0,01-2 эквивалентов, предпочтительно 0,05-1 эквивалент, относительно соединения, представленного формулой (W6), которое служит в качестве исходного соединения. Примеры растворителя, применяемого для реакции, включают, например, тетрахлорид углерода, хлороформ, 1,2-дихлорэтан, их смешанный растворитель и подобные, и предпочтительные примеры включают тетрахлорид углерода. Что касается температуры реакции, реакцию можно проводить обычно при 20-90°C, предпочтительно при 60-90°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 0,5-24 часа, предпочтительно 1-12 часов.
[0156]
Примеры соединения, представленного формулой (W6), включают 2-бром-1,4-диметилбензол, 1-бром-3,5-диметилбензол и подобные, и их можно приобрести как имеющиеся в продаже соединения, например, у Tokyo Chemical Industry.
[0157]
Стадия 3-8
[формула 75]
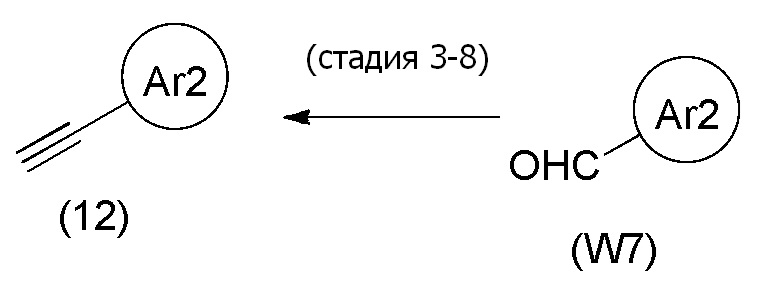
Соединения, представленные формулой (12), можно получить воздействием α-диазофосфатным соединением на соединение, представленное формулой (W7), вместе с неорганическим основанием. Примеры комбинации α-диазофосфатного соединения и неорганического основания включают, например, комбинации диметил(диазометил)фосфата и трет-бутоксида калия, диметил(диазометил)фосфата и трет-бутоксида натрия, диметил(1-диазо-2-оксопропил)фосфата и карбоната калия и диметил(1-диазо-2-оксопропил)фосфата и карбоната натрия, и комбинация диметил(1-диазо-2-оксопропил)фосфата и карбоната калия является предпочтительным. Количество α-диазофосфата, которое будут применять, может представлять собой от эквивалентного количества до избыточного количества, например, 1-10 эквивалентов, предпочтительно 1-5 эквивалентов, относительно соединения, представленного формулой (W7), которое служит в качестве исходного соединения. Количество неорганического основания, которое будут применять, может представлять собой от эквивалентного количества до избыточного количества, например, 1-5 эквивалентов, предпочтительно 1-3 эквивалентов, относительно α-диазофосфата, которое будут применять. Примеры растворителя, применяемого для реакции, включают, например, метанол, этанол, изопропанол, трет-бутанол, их смешанный растворитель и подобные, и предпочтительные примеры включают метанол. Что касается температуры реакции, реакцию можно проводить обычно при -20-80°C, предпочтительно при 0-60°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 0,5- 24 часа, предпочтительно 1-12 часов.
[0158]
Среди соединений, представленных формулой (12), 3-этинилтиофен и 2-этинилтиофен являются имеющимися в продаже соединениями, и их можно получить у Tokyo Chemical Industry.
[0159]
Стадия 3-9
4-Фенилтиофен-3-карбальдегид в качестве соединения, представленного формулой (W7), можно получить реакцией 4-формилтиофен-3-бороновой кислоты и бромбензола в растворителе в присутствии основания и палладиевого катализатора.
[0160]
В качестве палладиевого катализатора, например, имеющиеся в продаже катализаторы, такие как тетракис(трифенилфосфин)палладий, тетракис(метилдифенилфосфин)палладий, дихлорбис(трифенилфосфин)палладий, дихлорбис(три-o-толилфосфин)палладий, дихлорбис(трициклогексилфосфин)палладий, дихлорбис(триэтилфосфин)палладий, ацетат палладия, хлорид палладия, хлорид бис(ацетонитрил)палладия, бис(дибензилиденацетон)палладий, трис(дибензилиденацетон)дипалладий и хлорид бис(дифенилфосфиноферроцен)палладия можно добавлять к реакционной системе, как есть или в виде катализатора, отдельно полученного и выделенного из ацетата палладия, трис(дибензилиденацетон)дипалладия или подобных и произвольного лиганда. Катализатор, который, как считается, фактически участвует в реакции, можно получить в реакционной системе смешением ацетата палладия, трис(дибензилиденацетон)дипалладия или подобных, и произвольного лиганда. Валентность палладия может быть 0 или +2. В частности, трис(дибензилиденацетон)дипалладия(0), ацетат палладия(II) и подобные можно упомянуть в качестве предпочтительных примеров.
[0161]
В качестве лиганда, применяемого в получении палладиевого катализатора из произвольного лиганда, есть примеры фосфиновых лигандов, таких как трифенилфосфин, три(o-толил)фосфин, три(циклогексил)фосфин, три(трет-бутил)фосфин, дициклогексилфенилфосфин, 1,1'-бис(ди-трет-бутилфосфино)ферроцен, 2-дициклогексилфосфино-2'-диметиламино-1,1'-бифенил, 2-(ди-трет-бутилфосфино)бифенил, 2-(дициклогексилфосфино)бифенил, 2,2'-бис(дифенилфосфино)-1,1'-бинафтил, xantphos, три(трет-бутил)фосфин, 2-дициклогексилфосфино-2',6'-диметоксибифенил, 2-дициклогексил-2',4',6'-триизопропилбифенил и 1,2,3,4,5-пентаметил-1'-(ди-трет-бутилфосфино)ферроцен), и предпочтительные примеры включают 2-дициклогексилфосфино-2',6'-диметоксибифенил и подобные.
[0162]
Хотя количество эквивалентов палладиевого катализатора, которое будут применять, может представлять собой эквивалентное количество или каталитическое количество, оно предпочтительно составляет 0,01 моль % или более, особенно предпочтительно 0,10-50,0 моль %, относительно исходного соединения. Примеры основания включают, например, трет-бутоксид натрия, карбонат цезия, фосфат калия и подобные, и фосфат калия является предпочтительным. Количество эквивалентов основания, которое будут применять, может представлять собой эквивалентное количество или избыточное количество, например, 1-5 эквивалентов, предпочтительно 1-3 эквивалентов. Примеры растворителя, применяемого для реакции, включают, например, растворители эфирного типа, такие как тетрагидрофуран, 1,4-диоксан, и 1,2-диметоксиэтан, толуол, N, N-диметилформамид, N, N-диметилацетамид, н-бутанол, воду их смешанный растворитель и подобные, и смешанный растворитель н-бутанола и воды при соотношении 5:1 можно упомянуть в качестве предпочтительного примера. Что касается температуры реакции, реакцию можно проводить обычно при -20-120°C, предпочтительно при 0-100°C. Хотя продолжительность реакции конкретно не ограничена, она составляет, например, обычно 0,5-48 часов, предпочтительно 1-24 часа.
[0163]
Способы получения соединений настоящего изобретения не ограничены способами, описанными в настоящем изобретении. Например, соединения настоящего изобретения можно получить модификацией или превращением заместителей соединений в качестве предшественников соединений настоящего изобретения, применяя одну или комбинацию двух или более реакций, описанных в обычных химических статьях и подобных
[0164]
Примеры способа получения соединений настоящего изобретения, которые содержат асимметрический углерод, включают способ получения на основе асимметрического восстановления, способ применение имеющегося в продаже исходного соединения (или исходного соединения, которое можно получить известным способом или способом, аналогичным известному способу), у которого фрагмент, соответствующий асимметрическому углероду, является первоначально оптически активным, и подобные. Также есть способ, в котором соединение настоящего изобретения или его предшественник выделяют в виде активного изомера общепринятым способом. Примеры данного способа включают, например, способ, применяя высокоэффективную жидкостную хроматографию (ВЭЖХ), применяя хиральную колонку, классическую фракционную кристаллизацию для разделения оптически активных веществ, включающую получение соли с активным агентом, разделение фракционной кристаллизацией или подобными, и превращение соли в соединение в свободной форме, причем способ включает конденсацию с оптически активным агентом, получая диастереомер, с последующим разделением, очисткой и разложением полученного диастереомера, и подобные. Когда выделяют предшественник для получения оптически активных веществ, оптически активное соединение настоящего изобретения можно затем получить проведением приведенных выше способов получения.
[0165]
Когда соединение настоящего изобретения содержит кислую функциональную группу, такую как карбоксильная группа, фенольная гидроксильная группа или тетразольное кольцо, соединение можно превратить в фармацевтически приемлемую соль (например, неорганические соли с натрием и подобными, или органические соли с триэтиламином и подобными) известными способами. Например, когда предполагается получить неорганическую соль, можно растворять соединение настоящего изобретения в воде, содержащей, по меньшей мере, 1 эквивалент гидроксида, карбоната, бикарбоната или подобного, соответствующего требуемой неорганической соли. Для реакции можно смешивать смешивающийся с водой неактивный органический растворитель, такой как метанол, этанол, ацетон и диоксан. Например, применяя гидроксид натрия, карбонат натрия, или гидрокарбонат натрия, можно получить раствор натриевой соли.
[0166]
Когда соединение настоящего изобретения содержит аминогруппу, другую основную функциональную группу или ароматическое кольцо, которое само обладает основными свойствами (например, пиридиновое кольцо и подобные), соединение можно также превратить в фармацевтически приемлемую соль (например, соль с неорганической кислотой, такой как хлористоводородная кислота, или соль с органической кислотой, такой как уксусная кислота) известными способами. Например, когда предполагается получить соль с неорганической кислотой, можно растворять соединение настоящего изобретения в воде, содержащей, по меньшей мере, 1 эквивалент требуемой неорганической кислоты. Для реакции можно смешивать смешивающийся с водой неактивный органический растворитель, такой как метанол, этанол, ацетон и диоксан. Например, применяя хлористоводородную кислоту, можно получить раствор гидрохлорида.
[0167]
Если требуется твердая соль, раствор можно упарить или к раствору можно добавить смешивающийся с водой органический растворитель, обладающий полярностью до некоторой степени, такой как н-бутанол или этилметилкетон, для получения твердой соли.
[0168]
Различные соединения, описанные настоящим изобретением, можно очистить известными способами, такими как ряд хроматографических способов (колоночная хроматография, колоночная флэш хроматография, тонкослойная хроматография, высокоэффективная жидкостная хроматография).
[0169]
Соединения настоящего изобретения согласно одному варианту осуществления обладают EP4 агонистической активностью, и их можно применять в качестве EP4 агониста. То есть, соединения настоящего изобретения согласно одному варианту осуществления можно применять в качестве лекарственного средства для профилактического и/или терапевтического лечения заболевания, связанного с EP4 рецепторным агонизмом. Заболевание, связанное с EP4 рецепторным агонизмом, представляет собой, более точно, заболевание, для которого EP4 рецепторный агонизм является эффективным, и более конкретно, оно конкретно не ограничено при условии, что оно представляет собой заболевание, которое можно предотвращать и/или лечить повышением количества синтезируемого цАМФ в остеобластах.
[0170]
EP4 агонистическую активность можно измерить, например, способами, описанными ниже. Таким образом, можно упомянуть способ подтверждения стимулирования продукции цАМФ в клетке, экспрессирующей рецептор EP4 человека. В качестве другого варианта осуществления можно упомянуть способ подтверждения действия, стимулирующего остеогенез, на основе стимулирования продукции цАМФ в присутствии ингибитора циклооксигеназы 2 (СОХ-2) в клетках костного мозга крыс. В качестве еще одного варианта осуществления можно упомянуть способ подтверждения активности связывания с человеческим рецептором EP4. В качестве способа подтверждения действия, стимулирующего остеогенез, основанного на стимулировании продуцирования цАМФ, конкретно можно привести в качестве примера способ, описанный в тестовом примере 1, упомянутом ниже.
[0171]
EP4 агонистическая активность, которую можно подтвердить способом, описанным в тестовом примере 1, составляет, например, 10 нМ или меньше, предпочтительно 1 нМ или меньше, более предпочтительно 0,6 нМ или меньше, еще более предпочтительно 0,3 нМ или меньше, особенно предпочтительно 0,1 нМ или меньше, самое предпочтительное 0,05 нМ или меньше.
[0172]
Соединения настоящего изобретения согласно одному варианту осуществления показывают высокую специфичность (селективность) к EP4. Селективность к EP4 можно оценить, например, проведением измерения агонистической активности и теста на связывание с рецептором, применяя клетки, которые экспрессируют каждый из человеческих EP1, EP2 и EP3 рецепторов, рассчитывая соотношение IC50 величин (концентрация соединения настоящего изобретения, при которой связывание [3H]PGE2 и рецептора подавляется на 50%), или Ki величин. Конкретно, можно привести в качестве примера способ, описанный в тестовом примере 2.
[0173]
Отношение IC50 величины (продолжительность)=IC50 для каждого рецептора/IC50 для EP4
Отношение Ki величины (продолжительность)=константа диссоциации Ki для каждого рецептора/константа диссоциации Ki для EP4
[0174]
Для того чтобы избежать побочных реакций, предпочтительно, чтобы соединения настоящего изобретения согласно одному варианту осуществления проявляли высокую специфичность к EP4. Например, отношение IC50 величины или Ki величины должно быть 10 кратным или более, предпочтительно 100 кратным или более, более предпочтительно 1000 кратным или более, более предпочтительно 3000 кратным или более, особенно предпочтительно 10000 кратным или более.
[0175]
Также предпочтительно, чтобы соединения настоящего изобретения согласно одному варианту осуществления селективно действовали на или связывались с EP4 рецептором, но не действовали на или связывались с EP1 рецептором, EP2 рецептором и EP3 рецептором, а также DP рецептором, FP рецептором, IP рецептором, TP рецептором, PPARα рецептором, PPARδ рецептором, PPARγ рецептором, S1P рецепторами (например, S1P1 рецептором, S1P2 рецептором, S1P3 рецептором и подобными), LTB4 рецепторами (например, BLT1, BLT2 и подобными), LPA рецептором (например, LPA1 рецептором, LPA2 рецептором, LPA3 рецептором и подобными), и каннабиноидными рецепторами (например, CB1 рецептором, CB2 рецептором и подобными), или действовали на или связывались с ними более слабо по сравнению с действием на или связыванием с EP4 рецептором.
Заболевание, связанное с агонизмом EP4 рецептора, конкретно не ограничено при условии, что оно представляет собой заболевание, для которого антагонизм EP4 рецептора является эффективным, и конкретные примеры включают, например, перелом кости и костный дефект.
[0176]
Соединения настоящего изобретения согласно одному варианту осуществления обладают действием, стимулирующим остеогенез, как показано в тестовых примерах, упоминаемых ниже, и являются пригодными в качестве активного ингредиента лекарственного средства. Соединения настоящего изобретения согласно одному варианту осуществления применяют, в частности, для терапевтического лечения и/или стимулирования заживления перелома или костный дефект, и предпочтительно применяют для терапевтического лечения и/или стимулирования заживления перелома. В качестве другого варианта осуществления, их также предпочтительно применять для терапевтического лечения и/или стимулирования заживления костного дефекта.
[0177]
Пригодность лекарственного средства настоящего изобретения согласно одному варианту осуществления для терапевтического лечения и/или стимулирования заживления перелома или костный дефект можно подтвердить, применяя модель закрытого перелома или модель частичного или самого длительного костного дефекта. Конкретно, примером является способ, описанный в тестовом примере 5.
[0178]
Можно ожидать, что лекарственное средство настоящего изобретения согласно одному варианту осуществления будет проявлять системное действие, увеличивающее плотность костей и действие, увеличивающее прочность костей, или местное действие, симулирующее регенерацию/остеоанагенез кости. Действие, стимулирующее остеогенез, соединений настоящего изобретения согласно одному варианту осуществления можно оценить, например, применяя клетки костного мозга, выделенные от экспериментальных животных, таких как крысы, или людей, и культивируемые, и применяя количество сформированных кальцинированных костоподобных узелков, активность щелочной фосфатазы, которая представляет собой маркер дифференцировки остеобластов, или подобных в качестве маркера. Его также можно оценить, применяя патологических модельных животных, таких как модельные крысы с уменьшенной костной массой, подвергнутые резекции седалищного нерва и овариэктомии или подобным, а также плотность или прочность аппендикулярных скелетов или подобных в качестве маркера. Его также можно оценить, применяя крысиную модель закрытого перелома трубчатой кости или модель разреза кости, полученной инвазивной операцией, модель, в которой в произвольной области создается костный дефект или подобные, и остеогенез, скорость сращивания кости, прочность костей восстановленной кости или подобные в качестве маркера.
[0179]
Перелом обозначает состояние, при котором кость частично или полностью разломана или деформирована под действием внешней силы. Кость, которая может страдать от перелома, конкретно не ограничена, при условии, что она относится к пациенту, чья костная ткань повреждена, и примеры включают кости лица (орбитальная кость, скула, нижняя челюсть), кости туловища (ребро, таз, шейный позвонок, грудной позвонок, поясничный позвонок, крестцовая кость, копчиковая кость), кости верхней конечности (лопатка, ключица, плечевая кость, локоть, лучевая кость, локтевая кость, лопаточная кость, хаматум, метакарпус, фаланга), кости нижней конечности (тазобедренный сустав, бедренная кость, большеберцовая кость, малоберцовая кость, голеностопный сустав, пяточная кость, скафоид, плюсневая кость) и подобные, и целевая кость может быть любой частью. Тип повреждения костной ткани конкретно не ограничен, и также включены стимулирование сращивания костей при переломе (полный перелом, неполный перелом, простой перелом, осколочный перелом и подобные) или преднамеренно разрезанной кость при остеотомии или при операции по расширению кости, адаптированной как один из способов хирургического лечения. Перелом шейки бедра, компрессионный перелом позвоночника, перелом дистального отдела лучевой кости, перелом проксимального конца плечевой кости и подобные, для которых причиной заболевания является остеопороз, также включены в объем перелома, приведенный выше.
[0180]
Костный дефект обозначает сами различные заболевания кости, такие как остеонус, остеомиелит, травматическое повреждение, хроническое заболевание суставов, длительное заживление после перелома и слабость искусственного сустава или состояние, при котором дефект кости образуется при хирургическом удалении поражения при лечении данных заболеваний. Его часть конкретно не ограничена, при условии, что он относится к пациенту, который вынужден иметь костный дефект. Примеры включают кости лица (орбитальная кость, скула, нижняя челюсть), кости туловища (ребро, таз, шейный позвонок, грудной позвонок, поясничный позвонок, крестцовая кость, копчиковая кость), кости верхней конечности (лопатка, ключица, плечевая кость, локоть, лучевая кость, локтевая кость, лопаточная кость, хаматум, метакарпус, фаланга), кости нижней конечности (тазобедренный сустав, бедренная кость, большеберцовая кость, малоберцовая кость, голеностопный сустав, пяточная кость, скафоид, плюсневая кость) и подобные, и целевая кость может быть любой частью. Тип костного дефекта конкретно не ограничена, и включены костные дефекты любого типа, как состояние, при котором промежуточная часть кости сильно повреждена, и состояние, при котором кость становится частично дефектной из-за осколочного перелома.
[0181]
Лекарственное средство настоящего изобретения согласно одному варианту осуществления можно применять в качестве агента, стимулирующего сращивание кости, в момент хирургических терапевтических вмешательств. Например, его можно применять при фиксации позвоночника (шейный позвонок, грудной и поясничный позвонки), денатурированной хирургии сколиоза, замене сустава, расширении позвоночного канала, остеотомии, операции по расширению кости, компенсации черепного костного дефекта, краниопластике, фиксации подвздошной кости с костной поддержкой, гетерологичной костной пластике, гомологичной костной пластике, аутологичной костной пластике и терапии с заместителем костного трансплантата, а также восстановлении костей и/или реконструкции костей после хирургического удаления первичной злокачественной опухоли или метастазного поражения кости, которые являются примерами медицинских вмешательств.
[0182]
Лекарственное средство настоящего изобретения согласно одному варианту осуществления предпочтительно применять в качестве агента, стимулирующего остеогенез. Лекарственное средство настоящего изобретения согласно одному варианту осуществления более предпочтительно применять для терапевтического лечения и/или стимулирования заживления перелома или костный дефект. Кроме того, лекарственное средство настоящего изобретения согласно одному варианту осуществления чрезвычайно предпочтительно применять для профилактического и/или терапевтического лечения перелома. Кроме того, специалисту в данной области техники понятно, что лекарственное средство для предотвращения или подавления развития патологического состояния попадает в пределы объема лекарственного средства для профилактического и/или терапевтического лечения, относящегося к настоящему изобретению, в зависимости от обстоятельств.
[0183]
Лекарственное средство настоящего изобретения согласно одному варианту осуществления можно получить в виде лекарственного средства, содержащего соединение, представленное формулой (1) или его фармацевтически приемлемую соль в качестве активного ингредиента, и например, лекарственное средство, содержащее соединение или его фармацевтически приемлемую соль, которое подвергается метаболизму в живом теле, давая соединение, представленное формулой (1), или его фармацевтически приемлемую соль при введении в виде пролекарства, также попадает в пределы объема лекарственного средства настоящего изобретения.
Хотя путь введения лекарственного средства настоящего изобретения согласно одному варианту осуществления конкретно не ограничен, схему введения можно подходящим образом выбрать, например, из перорального введения, подкожного введения, внутрикожного введения, внутримышечного введения, внутривенного введения, перназального введения, интравагинального введения, интраректального введения, местного воздействия на пораженную часть и подобных. Местное введение на пораженную часть является одним из предпочтительных схем введения.
[0184]
В виде лекарственного средства настоящего изобретения можно применять соединение, представленное формулой (1) или его фармацевтически приемлемая соль, саму по себе. Однако предпочтительно добавлять один или более типов фармацевтически приемлемых носителей к соединению, представленному формулой (1), или его фармацевтически приемлемой соли, получая фармацевтическую композицию и вводить композицию. Кроме того, в качестве активного ингредиента лекарственного средства настоящего изобретения можно применять гидрат или сольват соединения, представленного общей формулой (I) или его фармацевтически приемлемую соль.
[0185]
Примеры лекарственной формы, применяемой для получения приведенной выше фармацевтической композиции, включают таблетку, порошок, гранулу, сироп, суспензию, капсулы, средство, применяемое для ингаляции, инъекцию и подобные. Для их получения применяют различные носители, пригодные для данных препаратов. Например, примеры носителя для пероральных препаратов включают вспомогательные вещества, связующие, смазывающие агенты, ускорители текучести и красители. Примеры способа применения композиции в виде средств, применяемых для ингаляции, включают способ ингаляции порошка фармацевтической композиции или жидкой лекарственной формы, полученной растворением или суспендированием фармацевтической композиции в растворителе как есть, способ ингаляции его аэрозоля, применяя распылитель, называемый пульверизатором, или нубелайзер и подобные. Когда композицию формулируют в виде инъекции, в качестве разбавителя обычно можно применять дистиллированную воду для инъекций, физиологический солевой раствор, водный раствор глюкозы, растительное масло для инъекций, пропиленгликоль, полиэтиленгликоль и подобные. Дезинфицирующие, антисептические, стабилизирующие, изотонические, успокаивающие агенты и подобные можно добавлять при необходимости. Клатратное соединение, в котором соединение настоящего изобретения клатрируется в циклодекстрин, можно получить и применять в виде лекарственного средства настоящего изобретения.
[0186]
Когда вводят лекарственное средство настоящего изобретения согласно одному варианту осуществления, подходящую лекарственную форму можно подходящим образом выбрать и вводить подходящим путем. Например, ее можно вводит перорально в виде таблетки, порошка, гранулы, сиропа, суспензии, капсулы или подобных. Лекарственное средство можно также вводить через респираторный тракт в виде средства, применяемого для ингаляции. Кроме того, лекарственное средство можно вводить подкожно, внутрикожно, интраваскулярно, внутримышечно или внутрибрюшинно в форме инъекции, включая капельную инфузию. Кроме того, лекарственное средство можно вводить трансмукозально в виде сублингвальной таблетки, суппозитория или подобных, и можно вводить чрескожно в виде геля, лосьона, мази, крема, спрея или подобных. Кроме того, лекарственное средство можно также вводить в виде лекарственного средства пролонгированного действия, например, инъекции с замедленным высвобождением или композиции для имплантации (например, пленочной композиции и подобных).
[0187]
Когда лекарственное средство настоящего изобретения согласно одному варианту осуществления вводят местно, лекарственное средство можно непосредственно вводить в местный участок, такой как место перелома. В данном случае, соединение можно непосредственно вводить в местный участок вместе с подходящим негидрофильным растворителем, или соединение можно также формулировать в подходящем носителе, таком как биоразлагаемые полимеры, и применять в виде лекарственного средства, формованного в виде стержня, в виде иглы, сферической формы, в виде пленки или подобных, или в виде мази, крема или геля, или препарата с замедленным высвобождением путем трансплантации или введения лекарственного средства в местный участок, такой как переломная часть. Примеры биоразлагаемого высокомолекулярного полимера включают, например, полиэфиры алифатических кислот (полимеры и сополимеры одного или более типов α-гидроксикарбоновых кислот, гидроксидикарбоновых кислот, молочной кислоты/капролактона, валеролактона и подобных, и их смеси), их производные (полимолочные кислоты, полигликолевые кислоты, блочные полимеры полиэтиленгликоля и подобных), эфиры поли-α-цианоакриловой кислоты, поли-β-гидроксимасляные кислоты, полиалкиленоксалаты, полиортоэфиры, полиортокарбонаты, поликарбонаты, полиаминокислоты, эфиры гиалоуроновой кислоты, полистирольные группы, полиметакриловые кислоты, сополимеры акриловой кислоты и метакриловой кислоты, полиаминокислоты, децинстеарат, этилцеллюлозу, ацетилцеллюлозу, нитроцеллюлозу, сополимеры малеинового ангидрида, этиленвинилацетатные сополимеры, поливинилацетаты, полиакриламиды, коллаген, желатин, фибрин, костную муку, костный цемент и подобные.
[0188]
Биоразлагаемый высокомолекулярный полимер может состоять из одного типа вещества или сополимера, комплекса или простой смеси двух или более типов веществ, и схема его полимеризации может быть любой из случайной, блочной и графт-полимеризаций.
Лекарственное средство настоящего изобретения согласно одному варианту осуществления можно также наносить или адсорбировать на искусственной кости (имплантате), состоящей из хорошо биосовместимого материала (металл, кальций, керамические материалы, полимерные материалы и подобные), костных протезах (гидроксиапатит, фосфат β-трикальция и подобные) или подобных вместе с подходящим растворителем или носителем, или имплантировать в них и вводить в местный участок.
[0189]
Период введения лекарственного средства настоящего изобретения согласно одному варианту осуществления конкретно не ограничен. В принципе, лекарственное средство вводят в течение периода, когда установлено, что выражены клинические симптомы заболевания, и обычно продолжают прием от нескольких недель до одного года. Однако также можно продлить период введения в зависимости от патологических состояний или продолжить введение даже после выздоровления от клинических симптомов. Лекарственное средство также можно назначать профилактически по решению врача, даже если какой-либо клинический симптом не выражен. Доза лекарственного средства настоящего изобретения согласно одному варианту осуществления конкретно не ограничена. Когда лекарственное средство настоящего изобретения вводят непосредственно в местный участок, такой как часть перелома, 0,01-1000 мкг активного ингредиента можно вводить взрослому за одно введение. Что касается частоты введения в случае выше, лекарственное средство можно вводить при частоте от каждых 6 месяцев до каждого дня, и лекарственное средство можно предпочтительно вводить от одного раз в 3 месяца до одного раза в месяц, или один раз в неделю.
[0190]
Ежедневная доза и/или однократная доза, период введения и частота введения могут быть соответственно увеличены или уменьшены в зависимости от различных условий, таких как возраст, вес, степень физического здоровья пациента, тип и тяжесть заболевания, подлежащего лечению, путь введения и лекарственная форма (свойство замедленного высвобождения носителя для активного ингредиента и подобные).
[0191]
Когда лекарственное средство настоящего изобретения согласно одному варианту осуществления применяют для терапевтического лечения и/или стимулирования заживления перелома или костного дефекта, или профилактического и/или терапевтического лечения перелома, лекарственное средство настоящего изобретения согласно одному варианту осуществления можно применять вместе с одним или более видами лекарственных средств, выбранными из группы, состоящей из агентов, активирующих кость, агентов, стимулирующих остеогенез, агентов, подавляющих резорбцию кости, агентов, улучшающих метаболизм кости, препаратов половых гормонов и препаратов кальция, одновременно или в различные моменты времени. Кроме того, лекарственное средство настоящего изобретения согласно одному варианту осуществления можно также получить в виде так называемого комбинированного лекарственного средства вместе с лекарственными средствами, приведенными в качестве примеров выше, и затем вводить. Вышеупомянутое комбинированное лекарственное средство может быть в лекарственной форме в виде полной смеси активных ингредиентов, аналогичных типичным композициям данного типа, а также в виде лекарственной формы, набора или упаковки, содержащей несмешанную комбинацию ингредиентов, отдельно вводимых из двух или более контейнеров, каждый из которых содержит каждый активный ингредиент.
[0192]
Примеры агентов, активирующих кости, пригодных в комбинации с лекарственным средством настоящего изобретения, согласно одному варианту осуществления включают, например, витамин D или производные витамина D, такие как кальцитриол, альфакальцидол, OCT, 2MD и ED-71, примеры агентов, стимулирующих остеогенез, включают, например, менатетренон, терипаратид, соматропин, инсулиноподобный фактор роста-I (IGF-I), костные морфогенетические белки (BMP), основной фактор роста фибробластов (bFGF), трансформирующий фактор роста-β (TGF-β), EP2 агонист, LRP5 агонист, анти-SOST антитело, ингибитор GSK-3, ингибитор Dkk1, кальцилитики, агенты, стимулирующие секрецию гормона роста и подобные, примеры агентов, подавляющих резорбцию кости, включают, например, элкатонин, кальцитонин лососевых рыб, этидронат, памидронат, клодронат, алендронат, инкадронат, ризедронат, минодронат, ибандронат, ингибиторы катепсина К, остеопротегерин, антитела против RANKL и подобные, примеры агентов, улучшающих метаболизм кости, включают, например, фтор, ранелат стронция, иприфлавон и подобные, примеры препаратов половых гормонов включают, например, эстриол, эстрадиол, конъюгированный эстроген, прогестерон, медроксипрогестерон, тестостерон, метилтестостерон, местанолон, станозолол, метенолон, нандролон, селективные модуляторы рецептора эстрогена (SERM: ралоксифен, лазофоксифен, базедоксифен, оспемифен, арзоксифен, CHF4227, PSK-3471 и другие), селективные модуляторы рецепторов андрогена (SARM) и подобные, и примеры препаратов кальция включают, например, карбонат кальция, лактат кальция, глюконат кальция, ацетат кальция, хлорид кальция, цитрат кальция, гидрофосфат кальция, L-аспартат кальция и подобные. Его можно также применять вместе с различными видами лекарственных средств от болезней костей, которые будут созданы в будущем. Данные комбинированные лекарственные средства не ограничиваются при условии, что комбинации являются клинически значимыми.
[0193]
Соединения настоящего изобретения согласно одному варианту осуществления включают соединения, показывающие превосходную безопасность (включая различные токсичности и фармакологическую безопасность), фармакокинетические характеристики и подобные, и их применимость в качестве активных ингредиентов лекарственных средств можно подтвердить, например, способами, показанными ниже.
[0194]
Примеры тестов, относящихся к безопасности, включают, например, тесты, перечисленные ниже. Однако они не ограничиваются данными примерами. Примеры включают тесты на цитотоксичность (тесты, применяя HL60 клетки, гепатоциты и подобные), тесты на генотоксичность (тест Эймса, TK-тест мышиной лимфомы, тест на хромосомную аберрацию, микроядерный тест и подобные), тесты на сенсибилизацию кожи (способ Бюлера, способ GPMT, способ APT, тест LLNA и подобные), тесты на фотосенсибилизацию кожи (адъювантный и стрип-способ и подобные), тесты на раздражение глаз (однократное закапывание, кратковременное непрерывное закапывание, повторное закапывание и подобные), фармакологические тесты на безопасность для сердечнососудистой системы (способ телеметрии, способ APD, тес на ингибирование hERG и подобные), фармакологические тесты на безопасность для центральной нервной системы (FOB способ, модифицированная версия способа Ирвина и подобные), фармакологические тесты на безопасность для дыхательной системы (способ измерения, применяя прибор для измерения дыхательной функции, способ измерения, применяя анализатор газов крови, и подобные), общие тесты на токсичность, тесты на репродуктивную и эмбриотоксичность и подобные.
[0195]
Примеры тестов, относящихся к фармакокинетическим параметрам, включают, например, тесты, перечисленные ниже. Однако они не ограничены данными примерами. Примеры включают тесты на ингибирование или индукцию фермента цитохрома P450, тесты на проницаемость клеток (тесты, применяя клети CaCO-2, клети MDCK и подобные), анализ АТФазного транспортера лекарственных средств, тесты пероральной абсорбции, тесты для измерения изменения концентрации в крови, тесты на метаболизм (тест на стабильность, тест на метаболические молекулярные фрагменты, тест на реактивность и подобные), тесты на растворимость (тест на растворимость на основе турбидиметрического способа и подобные) и подобные.
[0196]
Пригодность соединений настоящего изобретения согласно одному варианту осуществления в качестве активного ингредиента лекарственного средства можно подтвердить проведением, например, теста на цитотоксичность. Примеры теста на цитотоксичность включают способы, применяя различные культивируемые клетки, например, HL-60 клетки, которые представляют собой клетки человеческой пролейкемии, первичные выделенные культивируемые клетки гепатоцитов, фракцию нейтрофилов, полученную из периферической крови человека, и подобные. Хотя тест можно осуществлять способом, описанный ниже, способ не ограничивается только следующим описанием. Клетки получают в виде суспензий 105-107 клеток/мл, суспензию добавляют в микропробирки или микропланшеты в объеме от 0,01 до 1 мл. К суспензии добавляют растворяющий соединение раствор в объеме от 1/100 до 1-кратного объема клеточной суспензии, и клетки культивируют в среде для культивирования клеток с конечной концентрацией соединения от 0,001 до 1000 мкМ в течение от 30 минут до нескольких дней при 37° C в атмосфере 5% CO2. После прекращения культивирования доли выживших клеток оценивают, применяя МТТ способ, WST-1 способ (Ishiyama, M., et al., In Vitro Toxicology, 8, стр. 187, 1995) или подобные. Измерив цитотоксичность соединения в клетках, можно подтвердить пригодность соединения в качестве активного ингредиента лекарственного средства.
[0197]
Пригодность соединений настоящего изобретения согласно одному варианту осуществления в качестве активного ингредиента лекарственного средства можно подтвердить проведением, например, теста на генотоксичность. Примеры теста на генотоксичность включают тест Эймса, TK-тест мышиной лимфомы, тест на хромосомную аберрацию, микроядерный тест и подобные. Тест Эймса представляет собой способ определения обратной мутации культивированием бактерий Salmonella или Escherichia bacteria указанного штамма в чашках для культивирования или подобных с добавлением соединения (ссылка на IYAKUSHIN (Notification by the chief of Evaluation and Licensing Division, Pharmaceutical and Medical Safety Bureau, Ministry of Health, Labor and Welfare, Japan), No. 1604, 1999, “Guideline for Genotoxicity Test”, II-1. Genotoxicity Test и подобные). TK-тест мышиной лимфомы представляет собой тест на обнаружение возможности генетических мутаций, направленных на ген тимидинкиназы L5178Y клеток лимфомы мыши (ссылка на IYAKUSHIN No. 1604, 1999, “Guideline for Genotoxicity Test”, II-3. Mouse Lymphoma TK Test; Clive, D. et al., Mutat. Res., 31, стр. 17-29, 1975; Cole, J., et al., Mutat. Res., 111, стр. 371-386, 1983 и подобные). Тест на хромосомную аберрацию представляет собой способ определения активности по вызыванию хромосомной абберации культивированием клеток млекопитающих в присутствии соединения, затем после фиксации клеток, окрашивание и наблюдение хромосом клеток (ссылка на IYAKUSHIN No. 1604, 1999, “Guideline for Genotoxicity Test”, II-2. Chromosomal Aberration Test Utilizing Mammalian Cultured Cells и подобные). Микроядерный тест представляет собой способ оценки способности образовывать микроядра, вызванной хромосомной абберацией, и имеются в наличие способ применения грызунов (in vivo тест) (IYAKUSHIN No. 1604, 1999, “Guideline for Genotoxicity Test”, II-4. Micronucleus Test Using Rodent; Hayashi M. et al., Mutat. Res., 312, стр. 293-304, 1994; Hayashi, M. et al., Environ. Mol. Mutagen., 35, стр. 234-252, 2000), способ применения культивируемых клеток (in vitro тест) (Fenech M., et al., Mutat. Res., 147, стр. 29-36, 1985; Miller, B., et al., Mutat. Res., 392, стр. 45-59, 1997) и подобные. Выяснив генотоксичность соединения, применяя один или несколько из данных способов, можно подтвердить пригодность соединения в качестве активного ингредиента лекарственного средства.
[0198]
Пригодность соединений настоящего изобретения согласно одному варианту осуществления в качестве активного ингредиента лекарственного средства можно подтвердить проведением, например, теста на сенсибилизацию кожи. Тесты на сенсибилизацию кожи включают, в качестве тестов на сенсибилизацию кожи, применяя морских свинок, способ Бюхлера (Buehler, E.V., Arch. Dermatol., 91, стр. 171-177, 1965), GPMT способ (способ максимизации, Magnusson B., et al., J. Invest. Dermatol., 52, стр. 268-276, 1969), APT способ (адьювантный и аппликационный способ (Sato, Y. et al., Contact Dermatitis, 7, стр. 225-237, 1981)) и подобные. Кроме того, в качестве теста на сенсибилизацию кожи, применяя мышей, LLNA (анализ реакции регионарных лимфоузлов) способ (OECD Guideline for the testing of chemicals 429, Skin sensitization 2002; Takeyoshi, M.et al., Toxicol. Lett., 119 (3), стр. 203-8, 2001; Takeyoshi, M. et al., J. Appl. Toxicol., 25 (2), стр. 129-34, 2005) и подобные имеются в наличие. Выяснив способность сенсибилизировать кожу, применяя один или более из данных способов, можно подтвердить пригодность соединения в качестве активного ингредиента лекарственного средства.
[0199]
Пригодность соединений настоящего изобретения согласно одному варианту осуществления в качестве активного ингредиента лекарственного средства можно подтвердить проведением, например, тест на фотосенсибилизацию кожи. Примеры теста на фотосенсибилизацию кожи включают тест на фотосенсибилизацию кожи, применяя морских свинок (ссылка на “Drug Nonclinical Test Guideline Commentary 2002”, Yakuji Nippo, опубликованный в 2002, 1-9: Skin Photosensitization Test и подобные) и подобные, и примеры способа включают способ адьювантной полоски (Ichikawa, H. et al., J. Invest. Dermatol., 76, стр. 498-501, 1981), способ Харбера (Harber, L.C., Arch. Dermatol., 96, стр. 646-653, 1967), способ Хорио (Horio, T., J. Invest. Dermatol., 67, стр. 591-593, 1976), способ Джордана (Jordan, W.P., Contact Dermatitis, 8, стр. 109-116, 1982), способ Кочевера (Kochever, Т.Е. et al., J. Invest. Dermatol., 73, стр. 144-146, 1979), способ Маурера (Maurer, T. et al., Br. J. Dermatol., 63, стр. 593-605, 1980), способ Морикава (Morikawa, F. et al., “Sunlight and Man”, Tokyo Univ. Press, Tokyo, стр. 529-557, 1974), способ Винсона (Vinson, L.J., J. Soc. Cosm. Chem., 17, стр. 123-130, 1966) и подобные. Выяснив способность фотосенсибилизировать кожу соединением, применяя один или более из данных способов, можно подтвердить пригодность соединения в качестве активного ингредиента лекарственного средства.
[0200]
Пригодность соединений настоящего изобретения согласно одному варианту осуществления в качестве активного ингредиента лекарственного средства можно подтвердить проведением, например, теста на раздражение глаз. Примеры теста на раздражение глаз включают способ тестирования с однократным закапыванием, применяя глаза кроликов, глаза обезьян и подобные (закапывание один раз), способ испытания с коротким непрерывным закапыванием (закапывание несколько раз в течение короткого периода времени с равными интервалами), способ испытания с повторяющимся закапыванием (повторяющееся прерывистое закапывание в течение от нескольких дней до 10 дней) и подобные, и имеются в наличие способ оценки симптомов раздражения глаз в течение определенного периода времени после закапывания согласно улучшенной шкале Дрейза (Fukui, N. et al., Gendai no Rinsho, 4 (7), стр. 277-289, 1970) и подобные. Выяснив раздражение глаз соединением, применяя один или более из данных способов, можно подтвердить пригодность соединения в качестве активного ингредиента лекарственного средства.
[0201]
Пригодность соединений настоящего изобретения согласно одному варианту осуществления в качестве активного ингредиента лекарственного средства можно подтвердить проведением, например, фармакологического теста на безопасность для сердечнососудистой системы. Примеры фармакологического теста на безопасность для сердечнососудистой системы включают телеметрический способ (способ измерения влияния введения соединения без анестезии на электрокардиограмму, частоту сердечных сокращений, артериальное давление, кровоток и подобные (Electrocardiogram, Echocardiography, Blood Pressure and Pathological Tests of Animals for Fundamental and Clinical Medicine, edited by Sugano S., Tsubone H., Nakada Y., опубликованный в 2003, Maruzen), APD способ (способ измерения времени удержания потенциала действия клетками сердечной мышцы (Muraki, K. et al., AM. J. Physiol., 269, H524-532, 1995; Ducic, I. et al., J. Cardiovasc. Pharmacol., 30 (1), стр. 42-54, 1997)), способ оценки ингибирования hERG (пэтч-клэмп способ (Chachin, M. et al., Nippon Yakurigaku Zasshi, 119, стр. 345-351, 2002), способ с анализом на связывание (Gilbert, J.D. et al., J. Pharm. Tox. Methods, 50, стр. 187-199, 2004), способ с анализом истечения Rb+ (Cheng, C.S. et al., Drug Develop. Indust. Pharm., 28, стр. 177-191, 2002), способ анализа мембранного потенциала (Dorn, A. et al., J. Biomol. Screen., 10, стр. 339-347, 2005) и подобные. Выяснив влияние на сердечнососудистую систему соединения, применяя один или более из данных способов, можно подтвердить пригодность соединения в качестве активного ингредиента лекарственного средства.
[0202]
Пригодность соединений настоящего изобретения согласно одному варианту осуществления в качестве активного ингредиента лекарственного средства можно подтвердить проведением, например, фармакологического теста на безопасность для центральной нервной системы. Примеры фармакологического теста на безопасность для центральной нервной системы включают способ FOB (батарея стандартных тестов, Mattson, J.L. et al., J. American College of Technology, 15 (3), стр. 239-254, 1996)), модифицированную версию способа Ирвина (способ оценки наблюдения общих симптомов и поведения (Irwin, S., Comprehensive Observational Assessment (Berl.) 13, стр. 222-257, 1968)) и подобные. Выяснив действие на центральную нервную систему соединения, применяя один или более из данных способов, можно подтвердить пригодность соединения в качестве активного ингредиента лекарственного средства.
[0203]
Пригодность соединений настоящего изобретения согласно одному варианту осуществления в качестве активного ингредиента лекарственного средства можно подтвердить проведением, например, фармакологического теста на безопасность для дыхательной системы. Примеры фармакологического теста на безопасность для дыхательной системы включают способ измерения, применяя аппарат для измерения дыхательной функции (способ измерения частоты дыхания, единичного объема легочной вентиляции, минутной вентиляции и подобных, Drorbaugh, J.E. et al., Pediatrics, 16, стр. 81-87, 1955; Epstein, M.A. et al., Respir. Physiol., 32, стр. 105-120, 1978), способ измерения, применяя анализатор газов в крови (способ измерения газов крови, насыщения гемоглобина кислородом и подобных, Matsuo, S., Medicina, 40, стр. 188-, 2003) и подобные. Выяснив действие на дыхательную систему соединения, применяя один или более из данных способов, можно подтвердить пригодность соединения в качестве активного ингредиента лекарственного средства.
[0204]
Пригодность соединений настоящего изобретения согласно одному варианту осуществления в качестве активного ингредиента лекарственного средства можно подтвердить проведением, например, теста на общую токсичность. Тест на общую токсичность представляет собой способ перорального или внутривенного введения соединения, растворенного или суспендированного в подходящем растворителе, однократно или повторно (в течение нескольких дней) грызунам, таким как крысы и мыши или видам, отличным от грызунов, таким как обезьяны и собаки, и оценки на основе наблюдения общего состояния, клинико-химических изменений, патогистологических изменений и подобных у животного, которому осуществляют введение. Выяснив общую токсичность соединения, применяя данные способы, можно подтвердить пригодность соединения в качестве активного ингредиента лекарственного средства.
[0205]
Пригодность соединений настоящего изобретения согласно одному варианту осуществления в качестве активного ингредиента лекарственного средства можно подтвердить проведением, например, теста на репродуктивную токсичность и эмбриотоксичность. Тест на репродуктивную токсичность и эмбриотоксичность представляет собой тест для изучения влияния вредного воздействия соединения на процессы размножения и развития, применяя грызунов, таких как крыса и мышь, или видов, отличных от грызунов, таких как обезьяны и собаки (ссылка на “Drug Nonclinical Test Guideline Commentary 2002”, Yakuji Nippo, published on 2002, 1-6: тест на репродуктивную токсичность и эмбриотоксичность и подобные). Примеры теста на репродуктивную токсичность и эмбриотоксичность включают тесты на фертильность и ранний эмбриогенез вплоть до нидации, тесты на развитие и материнские функции до и после рождения, тесты на эмбриогенез и развитие плода (ссылка на IYAKUSHIN No. 1834, 2000, Appendix, “Guideline for Drug Toxicity Test”, [3] тест на репродуктивную и эмбриотоксичность и подобные) и подобные. Выяснив репродуктивную и эмбриотоксичность соединения, применяя данные способы, можно подтвердить пригодность соединения в качестве активного ингредиента лекарственного средства.
[0206]
Пригодность соединений настоящего изобретения согласно одному варианту осуществления в качестве активного ингредиента лекарственного средства можно подтвердить проведением, например, теста на индукцию и ингибирование фермента цитохром P450 (Gomez-Lechon, M.J. et al., Curr. Drug Metab., 5 (5), стр. 443-462, 2004). Примеры теста на индукцию и ингибирование фермента цитохром P450 включают, например, способ определения in vitro, ингибирует ли соединения активность фермента цитохром P450, применяя фермент цитохром P450 каждого молекулярного вида, очищенный из клеток или полученный, применяя генетическую рекомбинацию, или микросомную систему экспрессии человеческого P450 (Miller, V.P. et al., Ann. N.Y. Acad. Sci., 919, стр. 26-32, 2000), способ измерения изменений экспрессии фермента цитохром P450 каждого молекулярного вида или ферментативной активности, применяя микросомы печени человека или суспензию разрушенных клеток (Hengstler, J.G. et al., Drug Metab. Rev., 32, стр. 81-118, 2000), способ экстракции РНУ из гепатоцитов человека, подверженного воздействию соединения, и сравнения степени экспрессии мРНК со степенью для контроля для изучения способности соединения индуцировать фермент (Kato, M. et al., Drug Metab. Pharmacokinet., 20 (4), стр. 236-243, 2005) и подобные. Выяснив воздействие соединения на ингибирование или индукцию фермента цитохром P450, применяя один или более из данных способов, можно подтвердить пригодность соединения в качестве активного ингредиента лекарственного средства.
[0207]
Пригодность соединений настоящего изобретения согласно одному варианту осуществления в качестве активного ингредиента лекарственного средства можно подтвердить проведением, например, теста на проницаемость клеток. Примеры теста на проницаемость клеток включают, например, способ измерения проницаемости клеточной мембраны для соединения в системе in vitro клеточной культуры, применяя CaCO-2 клетки (Delie, F. et al., Crit. Rev. Ther. Drug Carrier Syst., 14, стр. 221-286, 1997; Yamashita, S. et al., Eur. J. Pham. Sci., 10, стр. 195-204, 2000; Ingels, F.M. et al., J. Pham. Sci., 92, стр. 1545-1558, 2003), способ измерения проницаемости клеточной мембраны для соединения в системе in vitro клеточной культуры, применяя MDCK клетки (Irvine, J.D. et al., J. Pham. Sci., 88, стр. 28-33, 1999) и подобные. Выяснив проницаемость клеток для соединения, применяя один или более из данных способов, можно подтвердить пригодность соединения в качестве активного ингредиента лекарственного средства.
[0208]
Пригодность соединений настоящего изобретения согласно одному варианту осуществления в качестве активного ингредиента лекарственного средства можно подтвердить проведением, например, анализа АТФазного транспортера лекарственных средств для АТФ-связывающего кассетного (ABC) транспортера. Примеры анализа АТФазного транспортера лекарственных средств включают способ изучения, является ли соединение субстратом P-гликопротеина (P-gp), применяя P-gp бакуловирусную систему экспрессии (Germann, U.A., Methods Enzymol., 292, стр. 427-41, 1998) и подобные. Более того, пригодность можно также проверить проведением, например, теста на транспортер, применяя ооциты, полученные у африканских шпорцевых лягушек (Xenopus laevis), в качестве транспортера растворенных веществ (SLC). Тесты на транспортеры включают способ изучения, является ли испытуемое соединение субстратом OATP2, применяя ооциты, экспрессирующие OATP2 (Tamai I. et al., Pharm. Res., 2001 September; 18 (9), 1262-1269) и подобные. Выяснив действие соединения на ABC транспортер или SLC транспортер, применяя данные способы, можно подтвердить пригодность соединения в качестве активного ингредиента лекарственного средства.
[0209]
Пригодность соединений настоящего изобретения согласно одному варианту осуществления в качестве активного ингредиента лекарственного средства можно подтвердить проведением, например, теста на пероральную абсорбцию. Примеры теста на пероральную абсорбцию включают способ перорального введения определенного количества соединения, растворенного или суспендированного в подходящем растворителе, грызуну, обезьяне, собаке или подобному, и измерение концентрации в крови соединения после перорального введения с течением времени, оценивая перемещение в крови соединения пероральным введением, применяя способ LC-MS/MS (“Newest Mass Spectrometry for Life Science”, Kodansha Scientific, 2002, edited by Harada K. et al и подобные) и подобные. Выяснив пероральную абсорбцию соединения, применяя данные способы, можно подтвердить пригодность соединения в качестве активного ингредиента лекарственного средства.
[0210]
Пригодность соединений настоящего изобретения согласно одному варианту осуществления в качестве активного ингредиента лекарственного средства можно подтвердить проведением, например, теста на измерение изменения концентрации в крови. Примеры теста на измерение изменения концентрации в крови включают способ перорального или парентерального (например, внутривенного, внутримышечного, внутрибрюшинного, подкожного, трансдермального, путем инстилляции, трансназального и подобных) введения соединения грызуну, обезьяне, собаке или подобному, и измерения изменения концентрации в крови соединения с течением времени после введения, применяя способ LC-MS/MS (“Newest Mass Spectrometry for Life Science”, Kodansha Scientific, 2002, edited by Harada K. et al и подобные) и подобные. Выяснив изменение концентрации в крови соединения, применяя данные способы, можно подтвердить пригодность соединения в качестве активного ингредиента лекарственного средства. В случае, в частности, местного введения среди парентеральных способов введения, для того чтобы избежать побочных реакций, соединения настоящего изобретения согласно одному варианту осуществления, дающие низкую концентрацию в крови после их введения, могут быть предпочтительными.
[0211]
Пригодность соединений настоящего изобретения согласно одному варианту осуществления в качестве активного ингредиента лекарственного средства можно подтвердить проведением, например, метаболического теста. Примеры метаболического теста включают способ тестирования на стабильность в крови (способ прогнозирования метаболического клиренса in vivo на основе скорости метаболизма соединения в печеночных микросомах человека или других видов животных (ссылка на Shou, W.Z. et al., J. Mass Spectrom., 40 (10) стр. 1347-1356, 2005; Li, C. et al., Drug Metab. Dispos., 34 (6), 901-905, 2006 и подобные), способ тестирования на молекулярные метаболиты, способ тестирования на реакционноспособные метаболиты и подобные. Выяснив метаболический профиль соединения, применяя один или более из данных способов, можно подтвердить пригодность соединения в качестве активного ингредиента лекарственного средства.
[0212]
Пригодность соединений настоящего изобретения согласно одному варианту осуществления в качестве активного ингредиента лекарственного средства можно подтвердить проведением, например, теста на растворимость. В качестве способа оценки растворимости в воде примерами являются подтверждение растворимости в кислотных условиях, нейтральных условиях или основных условиях, и также включено подтверждение изменения растворимости в присутствии или отсутствии желчной кислоты. Примеры теста на растворимость включают тест на растворимость, основанный на турбидиметрическом способе (Lipinski, C.A. et al., Adv. Drug Deliv. Rev., 23, стр. 3-26, 1997; Bevan, C.D. et al., Anal. Chem., 72, стр. 1781-1787, 2000) и подобные. Выяснив растворимость соединения, применяя данные способы, можно подтвердить пригодность соединения в качестве активного ингредиента лекарственного средства.
[0213]
Пригодность соединений настоящего изобретения согласно одному варианту осуществления в качестве активного ингредиента лекарственного средства можно подтвердить осмотром, например, травмы верхних отделов желудочно-кишечного тракта, почечной дисфункции и подобных. В качестве фармакологического теста для верхних отделов желудочно-кишечного тракта, действие на слизистую желудка можно исследовать, применяя модель повреждения слизистой оболочки желудка крыс. Примеры фармакологического теста на функционирования почек включают способ измерения почечного кровотока и скорости клубочковой фильтрации [Physiology, 18th edition, Bunkodo, 1986, глава 17] и подобные. Выяснив действия соединения на верхние отделы желудочно-кишечного тракта и функционирование почек, применяя один или более из данных способов, можно подтвердить пригодность соединения в качестве активного ингредиента лекарственного средства.
Примеры
[0214]
В настоящем изобретении далее, настоящее изобретение будет дополнительно конкретно объяснено со ссылкой на примеры, справочные примеры, примеры получения и примеры испытаний (далее они могут совместно именоваться "примеры и подобные"). Однако объем настоящего изобретения не ограничивается следующими примерами и подобными.
[0215]
В примерах и подобных, для тонкослойной хроматографии (ТСХ) применяли предварительно нанесенный силикагель 60 F254 (полученный Merck, номер продукта 5715-1M)). После прогона со смесью хлороформ:метанол (1:0-1:1), ацетонитрил:уксусная кислота:вода (200:1:1-100:4:4) или этилацетат:гексан (1:0 -0:1), подтверждение проводили УФ облучением (254 нм или 365 нм), или окрашиванием йодным раствором, водным перманганатом калия, фосфомолибденовой кислотой (этанольный раствор) или подобными.
Для сушки органического растворителя применяли безводный сульфат магний или безводный сульфат натрия.
[0216]
Для колоночной хроматографии применяли Multi Prep YFLC, полученный Yamazen Corporation, или 2-ch аппарат для параллельной очистки ”Purif-α2(50F)”, полученный MORITEX Corporation. В случае Multi Prep YFLC, любую из Ultra Pack Si-40A, 40B и 40D, полученную Yamazen Corporation, применяли в качестве колонки, и в случае Purif-α2(50F), PurifPack-Si серию, полученную MORITEX Corporation, применяли в качестве колонки.
Для колоночной флэш хроматографии применяли силикагель 60N (сферическая форма, нейтральный, 40-100 мкм, полученный Kanto Chemicals).
[0217]
Препаративную тонкослойную хроматографию (далее также называемую “ПТСХ”) проводили, применяя одну или несколько платсин LC Plate Silica Gel 60 F254 (20×20 см, толщина слоя 2 мм, включая концентрационные зоны (4 см), полученные Merck, номер продукта 13793-1M) в зависимости от количества образца.
[0218]
Для ВЭЖХ очистки применяли аппарат для хроматографического получения и очистки, полученный Waters Japan, Develosil C30-UG-5 (полученную Nomura Kagaku), или подобные применяли в качестве колонки, и растворитель вода-ацетонитрил, содержащий 0,1% уксусную кислоту, применяли в качестве элюента.
[0219]
Когда очистку проводили ВЭЖХ, целевое соединение получали удалением растворителя лиофилизацией, если не указано конкретно. Для получения спектров ядерного магнитного резонанса (ЯМР), применяли Gemini-300 (FT-NMR, Varian Co., Ltd.) или AL-300 (FT-NMR, полученный JEOL Co., Ltd.). В качестве растворителя применяли дейтерированный хлороформ, если не указано специально, химические сдвиги измеряли, применяя тетраметилсилан (TMS) в качестве внутреннего стандарта, и указывали δ (ppm), и константы связывания указывали J (Гц).
[0220]
Для LCMS, масс-спектр измеряли жидкостным хроматографом-масс-спектрометрией (LC-MS). Если не указано специально, одноквадрупольный масс спектрометр, UPLC/SQD System (полученный Waters) применяли в качестве масс-спектрометра, и измерение проводили способом электроспрей ионизации (ESI). В качестве аппарата для жидкостной хроматографии применяли Acquity Ultra Performance LC систему, полученную Waters. В качестве колонки для разделения применяли ACQUITY UPLC BEH C18 (1×50 мм, 1,7 мкм, полученную Waters).
[0221]
Однако для условий LC FLC-1, приведенных ниже, одноквадрупольный масс-спектрометр, Platform-LC (полученный Waters) применяли в качестве масс-спектрометра, и измерение проводили способом ионизации электроспреем (ESI). В качестве аппарата для жидкостной хроматографии применяли 306 PUMP систему, полученную GILSON. В качестве колонки для разделения применяли Develosil C30-UG-5 (50×4,6 мм, полученную Nomura Kagaku).
[0222]
Когда условия LC упоминают специально в примерах и справочных примерах, это значит, что измерение проводили со следующими условиями растворителей. Символ m/z обозначает данные масс-спектра (MH+, M+NH4+, или MH- также указывают).
[0223]
(LC-1) Измерение проводили в таких условиях, что элюирование проводили при скорости потока 0,6 мл/минута, применяя линейный градиент 5-90% (об/об) раствора B [ацетонитрил, содержащий 0,1% (об/об) уксусную кислоту] в растворе A [вода, содержащая 0,1% (об/об) уксусную кислоту] от 0 минуты до 2,0 минут, и затем линейный градиент 90-98% раствора B в растворе A от 2,0 до 2,5 минут.
[0224]
(LC-6) Измерение проводили в таких условиях, что элюирование проводили при скорости потока 0,6 мл/минута, применяя линейный градиент 70-90% (об/об) раствора B [ацетонитрил, содержащий 0,1% (об/об) уксусную кислоту] в растворе A [вода, содержащая 0,1% (об/об) уксусную кислоту] от 0 до 2,0 минут, и затем линейный градиент 90-98% раствора B в растворе A от 2,0 до 2,5 минут.
[0225]
(NLC-1) Измерение проводили в таких условиях, что элюирование проводили при скорости потока 0,6 мл/минута, применяя линейный градиент 5-90% (об/об) раствора B [ацетонитрил] в растворе A [10 мм водный ацетат аммония] от 0 минуты до 2,0 минут, и затем линейный градиент 90-98% раствора B в растворе A от 2,0 до 2,5 минут.
[0226]
(NLC-6) Измерение проводили в таких условиях, что элюирование проводили при скорости потока 0,6 мл/минута, применяя линейный градиент 70-90% (об/об) раствора B [ацетонитрил] в растворе A [10 мм водный ацетат аммония] от 0 минуты до 2,0 минут, и затем линейный градиент 90-98% раствора B в растворе A от 2,0 до 2,5 минут.
[0227]
(FLC-1) Измерение проводили в таких условиях, что элюирование проводили при скорости потока o2 мл/минута, применяя линейный градиент 5-98% (об/об) раствора B [ацетонитрил, содержащий 0,1% (об/об) уксусную кислоту] в растворе A [вода, содержащая 0,1% (об/об) уксусную кислоту] от 0 до 5 минут, 98% (об/об) раствора B в растворе A от 5 до 6 минут, линейный градиент 98-5% (об/об) раствора B в растворе A от 6 до 6,01 минут, и 5% (об/об) раствора B в растворе A от 6,01 до 7,5 минут.
[0228]
Для хиральной LC время удерживания измеряли высокоэффективной жидкостной хроматографией (ВЭЖХ). Когда условия хиральной LC упоминают конкретно в примерах и справочных примерах, это значит, что измерение проводили со следующими условиями измерения.
[0229]
(Хиральная LC-1) Измерение проводили, применяя CHIRALCEL OD-H (4,6×250 мм, 5, полученную Daicel Corporation) в качестве колонки для разделения в таких условиях, что элюирование проводили при скорости потока 0,6 мл/минута в качестве изократического элюирования, применяя 5% (об/об) раствора B (этанол) в растворе A (н-гексан).
[0230]
(Хиральная LC-2) Измерение проводили, применяя CHIRALCEL OJ-H (4,6×250 мм, 5 мкм, полученную Daicel Corporation) в качестве колонки для разделения в таких условиях, что элюирование проводили при скорости потока 1,0 мл/минута, применяя этанол, содержащий 0,1% (об/об) трифторуксусной кислоты.
[0231]
Изготовители применяемых реагентов можно иногда указывать со следующими сокращениями: Tokyo Chemical Industry Co., Ltd., TCI; Sigma-Aldrich Co. LLC., ALDRICH; Kanto Kagaku Co., Inc., KANTO; Wako Pure Chemical Industries Ltd., WAKO; Maybridge Co., Ltd., MAYBRIDGE; APOLLO Co., Ltd., APOLLO; Combi-Blocks Inc., COMBI-BLOCKS; Takasago International Corporation, TAKASAGO; Johnson Matthey Co., Ltd., JOHNSON; Nippon Chemical Industrial Co., Ltd., Nippon Chemical; и Japan EnviroChemicals, Limited, Japan EnviroChemicals.
[0232]
Сокращения или символы, применяемые в описании, имеют следующие значения: n, нормальный; i, изо; s, вторичный; т, третичный; c, цикло; Me, метил; Et, этил; Pr, пропил; Bu, бутил; Pen, пентил; Hex, гексил; Hep, гептил; Ph, фенил; Bn, бензил; Py, pyridyl; Ac, ацетил; CHO, формил; COOH, карбоксил; NO2, нитро; DMA, диметиламино; NH2, амино; CF3, трифторметил; F, фтор; Cl, хлор; Br, бром; OMe, метокси; OH, гидрокси; TFA, трифторацетил; SO2, сульфонил; CO, карбонил; THF, тетрагидрофуран; DMF, N, N-диметилформамид; DMSO, диметилсульфоксид; и DME, диметоксиэтан.
[0233]
Числа, указанные перед заместителями, представляют положения замещения. Числа, указанные перед сокращениями ароматических колец с дефисами, указывают позиции замещения в ароматических кольцах. Символ (S), упомянутый в именах соединений и структурных формулах, показывает, что соответствующий асимметрический углерод находится в S-конфигурации, и (R) обозначает, что соответствующий асимметрический углерод находится в R-конфигурации. Кроме того, когда (R) или (S) не указано для соединения, содержащего асимметрический углерод, это означает, что соединение состоит из смеси (R)-изомера и (S)-изомера в произвольном соотношении. Такое соединение может представлять собой рацемическую смесь (R)-изомера и (S)-изомера.
[0234]
Когда требуется деблокирование с процессе получения соединения примеров, его проводят согласно известным способам, таким как способы, описанные в Protective groups in Organic Synthesis, опубликованной John Wiley and Sons (2007).
[0235]
Справочный пример A-2: трет-бутилдиметил(2-(тиофен-2-ил)этокси)силан (промежуточное соединение A-2)
[формула 76]
К раствору 2-(тиофен-2-ил)этанола (4 г, TCI) в N, N-диметилформамиде (312 мл) последовательно добавляли имидазол (4,3 г), трет-бутилдиметилхлорсилан (7,05 г) и N, N-диметил-4-аминопиридин (763 мг) при охлаждении на льду, и смесь перемешивали при комнатной температуре в течение 2,75 часов. К реакционной смеси добавляли этилацетат, и органический слой последовательно промывали 1 моль/л хлористоводородной кислотой, насыщенным соляным раствором, насыщенным водным гидрокарбонатом натрия и насыщенным соляным раствором, и затем сушили. Растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (7,32 г).
(промежуточное соединение A-2: Rf (ТСХ)=0,70 (гексан:этилацетат=4:1))
[0236]
Справочный пример A-3: 5-(2-((трет-бутилдиметилсилил)окси)этил)тиофен-2-карбоновая кислота (промежуточное соединение A-3)
[формула 77]
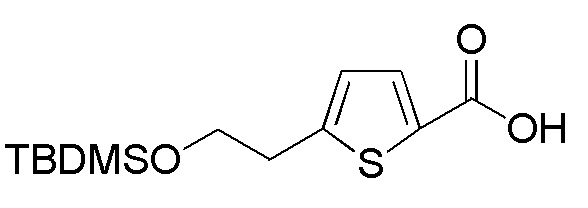
Раствор промежуточного соединения A-2 (7,15 г) в тетрагидрофуране (111 мл) охлаждали до -78°C в атмосфере азота. К реакционной смеси добавляли по каплям н-бутиллитий (2,6 моль/л раствор в гексане, 14,3 мл, KANTO), и смесь перемешивали в течение 0,75 часов как есть. Реакционную смесь нагревали до -5°C, затем добавляли порциями сухой лед (125 г), и после завершения добавления, реакционную смесь дополнительно перемешивали в течение 0,75 часа. К реакционной смеси добавляли насыщенный водный хлорид аммония, и смесь перемешивали при комнатной температуре. Добавляли к реакционной смеси этилацетат для экстракции, и органический слой промывали последовательно насыщенным водным хлоридом аммония и насыщенным соляным раствором, и затем сушили. Растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (9,03 г).
(промежуточное соединение A-3: LCMS m/z 287,0 (MH+), время удерживания 1,35 минут, LC условия NLC-1)
[0237]
Справочный пример A-4: Метил 5-(2-гидроксиэтил)тиофен-2-карбоксилат (промежуточное соединение A-4)
[формула 78]

Раствор промежуточного соединения A-3 (9,03 г) в метаноле (64 мл) охлаждали до 0°C, добавляли порциями к раствору концентрированную серную кислоту (3,2 мл), и смесь перемешивали как есть в течение 5 минут. Реакционную смесь нагревали до 70°C, перемешивали в течение 24 часов, и затем охлаждали до 0°C, и добавляли порциями насыщенный водный гидрокарбонат натрия до того, как реакционная смесь становилась нейтральной. Этилацетат добавляли к реакционной смеси для экстракции, и органический слой промывали последовательно насыщенным водным гидрокарбонатом натрия и насыщенным соляным раствором, и затем сушили. Растворитель упаривали при пониженном давлении, и остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (4,61 г).
(промежуточное соединение A-4: Rf (ТСХ)=0,33 (гексан:этилацетат=1:1))
[0238]
Справочный пример A-5: Метил 5-(2-бромэтил)тиофен-2-карбоксилат (промежуточное соединение A-5)
[формула 79]

К раствору промежуточного соединения A-4 (4,61 г) в дихлорметане (200 мл) добавляли трифенилфосфин (9,8 г), и затем смесь охлаждали до 0°C. К реакционной смеси добавляли порциями тетрабромид углерода (12,3 г), и смесь нагревали до комнатной температуры, и затем перемешивали в течение 13,5 часов. Реакционная смесь подвергали декомпрессии, упаривая растворитель, и затем остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (5,4 г).
(промежуточное соединение A-5: Rf (ТСХ)=0,70 (гексан:этилацетат=1:1))
[0239]
Справочный пример A-6: трет-бутил 2-(2-(5-(метоксикарбонил)тиофен-2-ил)этил)гидразинкарбоксилат (промежуточное соединение A-6)
[формула 80]
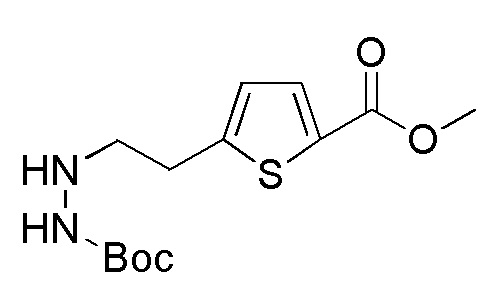
К раствору промежуточного соединения A-5 (6,2 г) в ацетонитриле (125 мл) последовательно добавляли трет-бутилкарбазат (16,5 г, TCI), гидрокарбонат натрия (10,5 г) и йодид натрия (700 мг), и смесь перемешивали при 90°C в течение 13 часов. Реакционную смесь охлаждали до 0°C, добавляли к реакционной смеси этилацетат (155 мл), и органический слой последовательно промывали 1 моль/л хлористоводородной кислотой, насыщенным водным гидрокарбонатом натрия и насыщенным соляным раствором, и затем сушили. Растворитель упаривали при пониженном давлении, и остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (5,1 г).
(промежуточное соединение A-6: LCMS m/z 301,1 (MH+), время удерживания 1,42 минут, LC условия NLC-1)
1H-NMR (CDCl3): δ (ppm) 7,64 (1H, д, J=4,0Гц), 6,87 (1H, д, J=4,0Гц), 3,86 (3H, с), 3,18 (2H, т, J=7,2Гц), 3,02 (2H, т, J=7,2Гц), 2,60-1,90 (2H, уш), 1,64 (9H, с)
[0240]
Справочный пример B-3: S-(2-хлорэтил)карбохлоридтиоат (промежуточное соединение B-3)
[формула 81]
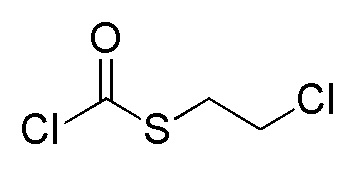
Смешанный раствор этиленсульфида (320 г, TCI) и пиридина (4,3 мл) охлаждали на бане со льдом в атмосфере аргона, добавляли порциями к реакционной смеси трифосген (474 г, TCI), и смесь перемешивали как есть в течение 4 часов. Реакционную смесь перегоняли при пониженном давлении (0,7-0,8 кПа, 50-52°C) для очистки, получая заявленное в заголовке соединение (281 г).
1H-NMR (CDCl3): δ (ppm) 3,72 (2H, т, J=7,0Гц), 3,30 (2H, т, J=7,0Hz)
[0241]
Справочный пример Z-1: трет-бутил 2-(((2-хлорэтил)тио)карбонил)-2-(2-(5-(метоксикарбонил)тиофен-2-ил)этил)гидразинкарбоксилат (промежуточное соединение Z-1)
[формула 82]
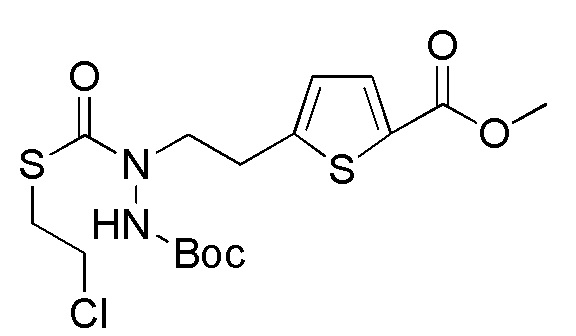
К раствору промежуточного соединения A-6 (140,3 г) в дихлорметане (660 мл) добавляли воду (330 мл) и гидрокарбонат натрия (78,09 г), смесь перемешивали в течение 10 минут, и затем промежуточное соединение B-3 (81,71 г) добавляли порциями к реакционной смеси, в то время как температуру внутри колбы реакционной смеси поддерживали равной 20-25°C. Смесь перемешивали как есть в течение 1 часа, и затем органический слой промывали насыщенным соляным раствором и сушили. Растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (199,5 г).
(промежуточное соединение Z-1: Rf (ТСХ)=0,43 (гексан:этилацетат=2:1))
[0242]
Справочный пример Z-2: Метил 5-(2-(1-(((2-хлорэтил)тио)карбонил)гидразинил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-2)
[формула 83]
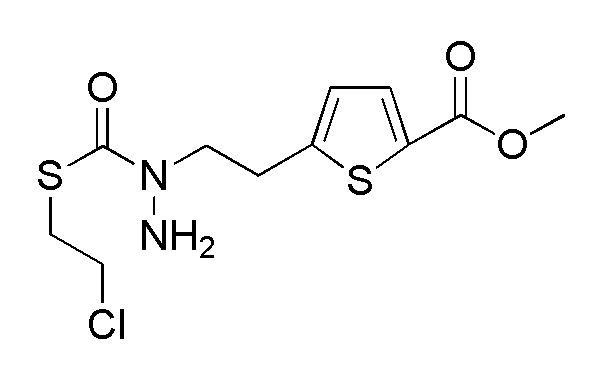
К промежуточному соединению Z-1 (199 г) добавляли 4 моль/л раствор хлористоводородной кислоты в диоксане (800 мл), и смесь перемешивали при комнатной температуре в течение 18 часов. Растворитель упаривали при пониженном давлении, и затем дихлорметан (6 л) и насыщенный водный гидрокарбонат натрия (2 л) добавляли к остатку для экстракции. Органический слой промывали насыщенным соляным раствором (2 л) и сушили, и затем растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (172 г).
(промежуточное соединение A-4: Rf (ТСХ)=0,49 (гептан:этилацетат=1:1))
[0243]
Справочный пример Z-3: Метил 5-(2-(2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-3)
[формула 84]
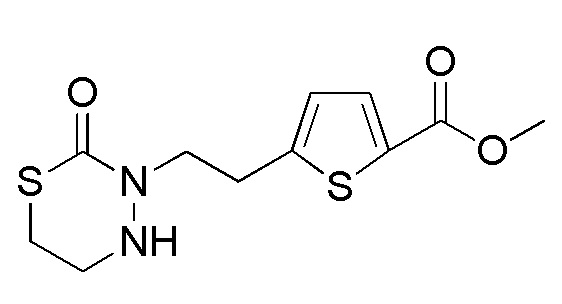
К раствору промежуточного соединения Z-2 (172 г) в ацетонитриле (3,4 л) последовательно добавляли гидрокарбонат натрия (223 г) и йодид натрия (397 г), и смесь перемешивали при 75°C в течение 15 часов. Смесь дополнительно перемешивали при 83°C в течение 15 часов, и затем охлаждали до комнатной температуры. Реакционную смесь фильтровали, применяя фильтровальную бумагу, и остаток, оставшийся на фильтровальной бумаге, промывали ацетонитрилом (1 л), смешивали с фильтратом, и концентрировали при пониженном давлении. К остатку, полученному после концентрирования, добавляли дихлорметан (3 л), и затем смесь фильтровали, применяя фильтровальную бумагу, остаток, оставшийся на фильтровальной бумаге, промывали дихлорметаном (1 л), смешивали с фильтратом, и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), и затем концентрировали при пониженном давлении. Остаток растворяли в этилацетате (1 л) нагреванием, затем добавляли к раствору гептан, и смесь охлаждали на льду. Выпавший осадок собирали фильтрованием, получая заявленное в заголовке соединение (97 г).
1H-NMR (CDCl3): δ (ppm) 7,64 (1H, д, J=3,8Гц), 6,88 (1H, д, J=3,8Гц), 3,86 (3H, с), 3,85 (2H, т, 7,0Гц), 3,30 (2H, т, J=7,0Гц), 3,25-3,17 (4H, м), 3,16 (2H, т, J=7,0 Гц)
[0244]
Справочный пример C-2: 2-(3-Бромфенил)-N-метокси-N-метилацетамид (промежуточное соединение C-2)
[формула 85]
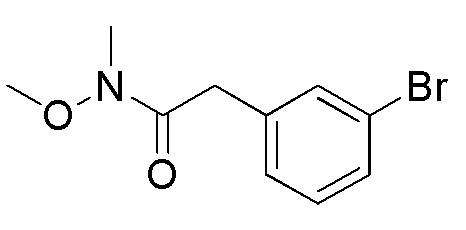
Раствор диизопропилэтиламина (800 мл) в дихлорметане (1,8 л) охлаждали на льду, последовательно добавляли 3-бромфенилуксусную кислоту (313 г, TCI), гидрохлорид N, O-диметилгидроксиламина (284 г), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (334 г) и N, N-диметил-4-аминопиридин (18 г), и смесь перемешивали при комнатной температуре в течение 12,5 часов. К реакционной смеси добавляли воду (630 мл) и дихлорметан (630 мл), и затем органический слой последовательно промывали дважды 2 моль/л хлористоводородной кислотой (630 мл), и по одному разу насыщенным водным гидрокарбонатом натрия (630 мл) и насыщенным соляным раствором (630 мл). Органический слой сушили, и затем растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (372 г).
(промежуточное соединение C-2: LCMS m/z 257,9 (MH+), время удерживания 1,37 минут, LC условия NLC-1)
1H-NMR (CDCl3): δ (ppm) 7,46-7,44 (1H, м), 7,39-7,36 (1H, м), 7,25-7,15 (2H, м), 3,74 (2H, с), 3,64 (3H, с), 3,20 (3H, с)
[0245]
Справочный пример C-3: 1-(3-Бромфенил)бут-3-ен-2-он (промежуточное соединение C-3)
[формула 86]
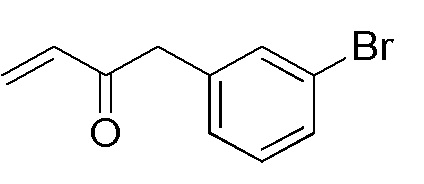
Раствор промежуточного соединения C-2 (104,2 г) в тетрагидрофуране (2,1 л) охлаждали до -45°C в атмосфере азота. К реакционной смеси добавляли винилмагнийбромид (1 моль/л раствор в тетрагидрофуране, 605 мл, Aldrich) в течение 30 минут, и смесь нагревали до 0°C, и затем перемешивали в течение 1,5 часов. Реакционную смесь добавляли к смеси ледяной воды (1 л) и 2 моль/л хлористоводородной кислотой (1 л), и смесь перемешивали в течение 1 минуты. Изопропиловый эфир (2 л) добавляли для экстракции, и органический слой последовательно промывали 1 моль/л хлористоводородной кислотой (1 л), водой (1 л) и насыщенным соляным раствором (1 л) и сушили. Растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (92,1 г).
(промежуточное соединение C-3: Rf (ТСХ)=0,74 (гептан:этилацетат=2:1))
[0246]
Справочный пример C-2-2: 2-(3-Йодфенил)-N-метокси-N-метилацетамид (промежуточное соединение C-2-2)
[формула 87]
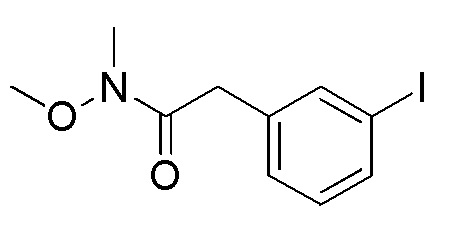
Промежуточное соединение C-2-2 получали согласно способу, описанному в справочном примере C-2, применяя 3-йодфенилуксусную кислоту (2,85 г) вместо 3-бромфенилуксусной кислоты, и таким образом получали заявленное в заголовке соединение (3,07 г).
(промежуточное соединение C-2-2: Rf (ТСХ)=0,42 (гексан:этилацетат=1:2))
Когда соединение получали согласно способу, описанному выше, количество реагентов, количество растворителя, продолжительность реакции и подобные можно подходящим образом изменять в зависимости от эквивалентного количества исходного соединения, которое будут применять в свете известного уровня техники для специалистов в данной области техники. То же самое применимо к следующим примерам.
[0247]
Справочный пример C-3-2: 1-(3-Йодфенил)бут-3-ен-2-он (промежуточное соединение C-3-2)
[формула 88]

Промежуточное соединение C-3-2 получали согласно способу, описанному в справочном примере C-3, применяя промежуточное соединение C-2-2 (100 мг) вместо промежуточного соединения C-2, и таким образом получали заявленное в заголовке соединение (58,7 мг).
(промежуточное соединение C-3-2: Rf (ТСХ)=0,60 (гексан:этилацетат=1:2))
[0248]
Справочный пример Z-4: Метил 5-(2-(4-(4-(3-бромфенил)-3-оксобутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-4)
[формула 89]
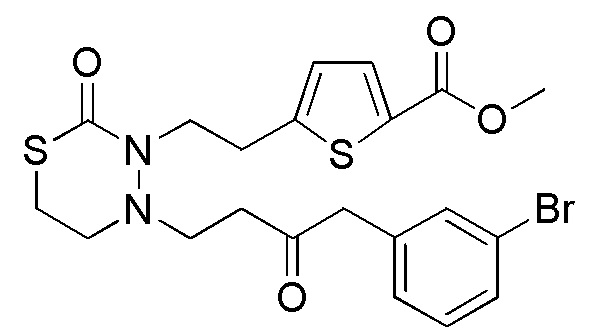
К раствору промежуточного соединения Z-3 (44,4 г) в этаноле (444 мл) добавляли промежуточное соединение C-3 (92,1 г), и смесь перемешивали при 110°C в течение 40 часов. Реакционную смесь подвергали декомпрессии, упаривая растворитель. Полученный в результате остаток очищали колоночной хроматографией (толуол/этилацетат), получая заявленное в заголовке соединение (75,9 г).
(промежуточное соединение Z-4: LCMS m/z 511,2 (MH+), время удерживания 1,75 минут, LC условия NLC-1)
[0249]
Справочный пример Z-4-2: Метил 5-(2-(4-(4-(3-йодфенил)-3-оксобутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-4-2)
[формула 90]
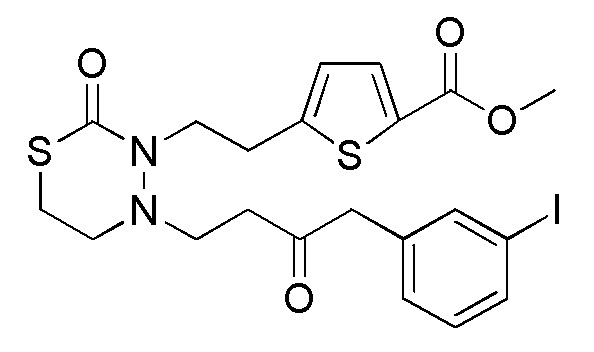
Промежуточное соединение Z-4-2 получали согласно способу, описанному в справочном примере Z-4, применяя промежуточное соединение C-3-2 (680,2 мг) вместо промежуточного соединения C-3, и таким образом получали заявленное в заголовке соединение (120,1 мг).
(промежуточное соединение Z-4-2: Rf (ТСХ)=0,50 (гексан:этилацетат=1:2), LCMS m/z 559,0 (MH+), время удерживания 1,84 минут, LC условия LC-1)
[0250]
Справочный пример Z-5: Метил 5-(2-(4-(4-(3-бромфенил)-3-гидроксибутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-5)
[формула 91]
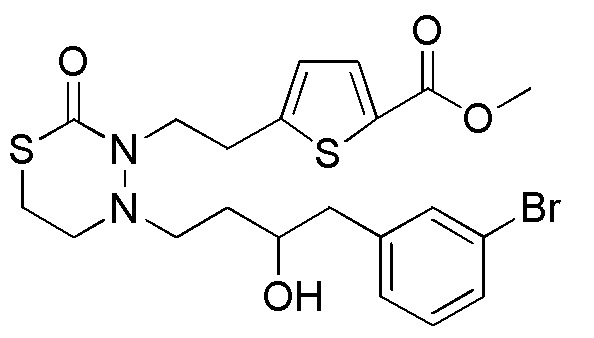
Раствор промежуточного соединения Z-4 (75,7 г) в метаноле (1,14 л) охлаждали до 0°C и добавляли порциями к раствору боргидрид натрия (7,47 г). Смесь перемешивали при 0°C в течение 1 часа, и затем добавляли порциями разбавленную хлористоводородную кислоту к реакционной смеси до того, как реакционная смесь становилась нейтральной. Органический растворитель упаривали при пониженном давлении, затем добавляли к остатку этилацетат (2 л), смесь охлаждали до 0°C, добавляли к смеси порциями насыщенный водный гидрокарбонат натрия (1 л), и полученную в результате смесь перемешивали в течение 5 минут. Органический слой экстрагировали, и сушили, и затем растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (71,0 г).
(промежуточное соединение Z-5: LCMS m/z 513,15 (MH+), время удерживания 1,70 минут, LC условия LC-1)
[0251]
Справочный пример Z-14: Метил 5-(2-(2-оксо-4-(3-оксо-4-(3-(тиофен-3-илэтинил)фенил)бутил)-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-14)
[формула 92]
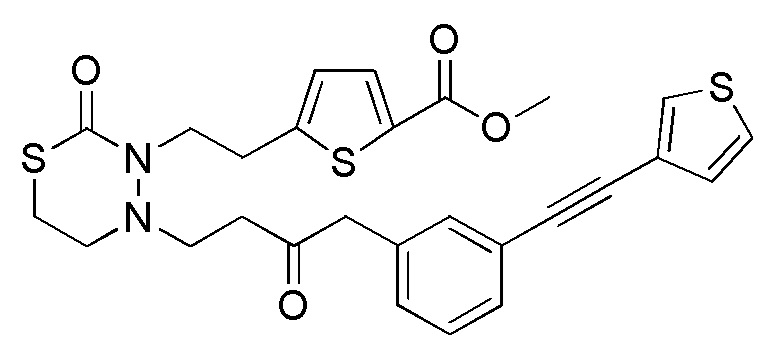
К промежуточному соединению Z-4-2 (86,6 мг) последовательно добавляли диэтиламин (78 мкл), 3-этинилтиофен (21 мкл), йодид меди (I) (0,8 мг) и тетракис(трифенилфосфин)палладия (1,1 мг), и смесь перемешивали при комнатной температуре в течение 2 часов. Кроме того, последовательно добавляли диэтиламин (600 мкл), йодид меди (I) (1,5 мг) и тетракис(трифенилфосфин)палладия (2,0 мг), и смесь перемешивали при комнатной температуре в течение 12 часов. К реакционной смеси добавляли диэтиловый эфир и 1 моль/л хлористоводородную кислоту (0,5 мл), и органический слой последовательно промывали 5 раз 1 моль/л хлористоводородной кислотой (1 мл), и один раз насыщенным водным гидрокарбонатом натрия (0,5 мл) и сушили. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (31,7 мг).
(промежуточное соединение Z-14: Rf (ТСХ)=0,12 (гексан:этилацетат=1:2), LCMS m/z 539,1 (MH+), время удерживания 1,95 минут, LC условия LC-1)
[0252]
Справочный пример Z-17: Метил 5-(2-(4-(3-гидрокси-4-(3-(тиофен-3-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-17)
[формула 93]

Промежуточное соединение Z-17 получали согласно способу, описанному в справочном примере Z-5, применяя промежуточное соединение Z-14 (31,7 мг) вместо промежуточного соединения Z-4, и таким образом получали заявленное в заголовке соединение (31,8 мг),.
(промежуточное соединение Z-17: LCMS m/z 541,1 (MH+), время удерживания 1,90 минут, LC условия LC-1)
[0253]
Пример 1: 5-(2-(4-(3-Гидрокси-4-(3-(тиофен-3-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновую кислоту
[формула 94]
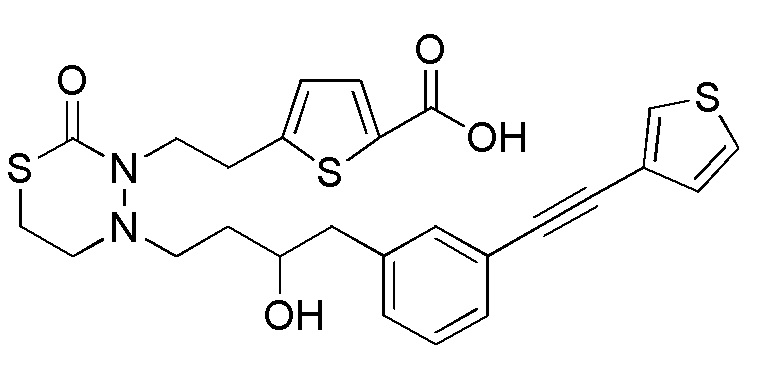
К раствору промежуточного соединения Z-17 (31,8 мг) в тетрагидрофуране (884 мкл) добавляли воду (221 мкл) и 2 моль/л водный гидроксид лития (442 мкл), и смесь перемешивали при 50°C в течение 17,5 часов. Реакционную смесь охлаждали до 0°C, добавляли к смеси 2 моль/л хлористоводородную кислоту (660 мкл), и затем полученную в результате смесь экстрагировали этилацетатом. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (22,7 мг).
(LCMS m/z 527,2 (MH+), время удерживания 1,68 минут, LC условия LC-1)
[0254]
Справочный пример A-10: (3-Бромтиофен-2-ил)метанол (промежуточное соединение A-10)
[формула 95]
Раствор 3-бромтиофен-2-карбоновой кислоты (3,0 г, Aldrich) в тетрагидрофуране (46 мл) охлаждали до 0°C в атмосфере газообразного азота, добавляли по каплям к раствору в течение 15 минут 1 моль/л раствор боран-тетрагидрофуранового комплекса в тетрагидрофуране (26,1 мл), и затем смесь перемешивали при комнатной температуре в течение 21,5 часов. Реакционную смесь охлаждали до 0°C, добавляли к смеси ледяную воду, 1 моль/л хлористоводородную кислоту и этилацетат, и полученную в результате смесь перемешивали. Органический растворитель упаривали при пониженном давлении, затем добавляли к остатку этилацетат, и органический слой последовательно промывали 1 моль/л хлористоводородной кислотой, насыщенным водным гидрокарбонатом натрия и насыщенным соляным раствором, и сушили. Растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (2,87 г).
(промежуточное соединение A-10: Rf (ТСХ)=0,42 (гексан:этилацетат=2:1))
[0255]
Справочный пример A-11: 3-Бром-2-(бромметил)тиофен (промежуточное соединение A-11)
[формула 96]
Раствор промежуточного соединения A-10 (7,14 г) в дихлорметане (169 мл) охлаждали до 0°C, добавляли к раствору трифенилфосфин (13,3 г) и тетрабромид углерода (13,45 г), и смесь перемешивали при комнатной температуре в течение 2,75 часов. К реакционной смеси добавляли насыщенный водный гидрокарбонат натрия, и затем органический растворитель упаривали при пониженном давлении. Добавляли к остатку этилацетат, и затем органический слой последовательно промывали насыщенным водным гидрокарбонатом натрия и насыщенным соляным раствором, и сушили. Органический растворитель упаривали при пониженном давлении, затем смешанный растворитель гексана и этилацетата (8:1) добавляли к полученному в результате остатку, получая суспензию, и суспензию фильтровали через фильтровальную бумагу, покрытую силикагелем. Растворитель фильтрата упаривали при пониженном давлении, получая заявленное в заголовке соединение (9,70 г).
(промежуточное соединение A-11: Rf (ТСХ)=0,64 (гексан:этилацетат=8:1))
[0256]
Справочный пример A-12: 2-(3-Бромтиофен-2-ил)ацетонитрил (промежуточное соединение A-12)
[формула 97]
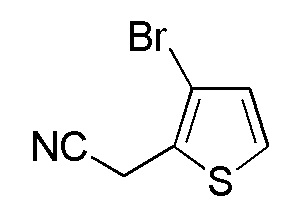
К промежуточному соединению A-11 (9,70 г) добавляли диметилсульфоксид (28 мл) и ацетонитрил (140 мл), смесь охлаждали до 0°C, затем добавляли к смеси цианид натрия (2,15 г), и полученную в результате смесь перемешивали при комнатной температуре в течение 16 часов. К реакционной смеси добавляли насыщенный водный гидрокарбонат натрия, и смесь перемешивали, и затем концентрировали при пониженном давлении. Реакционную смесь фильтровали через фильтровальную бумагу, покрытую целитом, и остаток, оставшийся на целите, промывали этилацетатом. Фильтрат и промывочную жидкость смешивали, и органический слой последовательно промывали насыщенным водным гидрокарбонатом натрия и насыщенным соляным раствором, и сушили. Органический растворитель упаривали при пониженном давлении, и затем полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (3,89 г).
(промежуточное соединение A-12: Rf (ТСХ)=0,18 (гексан:этилацетат=8:1))
[0257]
Справочный пример A-13: Этил 2-(3-бромтиофен-2-ил)ацетат (промежуточное соединение A-13)
[формула 98]
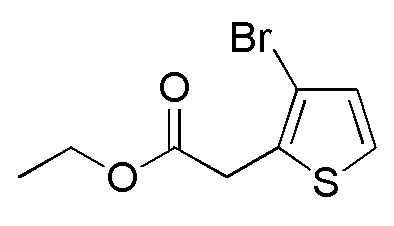
К раствору промежуточного соединения A-12 (3,89 г) в этаноле (32,3 мл) добавляли воду (0,4 мл), смесь охлаждали до 0°C, и затем добавляли к смеси порциями концентрированную серную кислоту (5,63 мл). Реакционную смесь перемешивали при 85°C в течение 115 часов, и затем охлаждали до 0°C, и добавляли насыщенный водный гидрокарбонат натрия до того, как реакционная смесь становилась нейтральной. Этилацетат добавляли к реакционной смеси, и полученную в результате смесь перемешивали, и затем концентрировали при пониженном давлении. Этилацетат добавляли к реакционной смеси, и органический слой последовательно промывали насыщенным водным гидрокарбонатом натрия и насыщенным соляным раствором, и сушили. Растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (4,40 г).
(промежуточное соединение A-13: Rf (ТСХ)=0,33 (гексан:этилацетат=8:1))
[0258]
Справочный пример A-14: 2-(3-Бромтиофен-2-ил)этанол (промежуточное соединение A-14)
[формула 99]
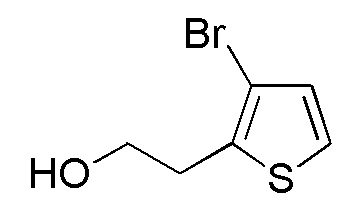
Раствор промежуточного соединения A-13 (4,40 г) в тетрагидрофуране (88,5 мл) охлаждали до 0°C в атмосфере газообразного азота, добавляли к раствору алюмогидрид лития (672 мг), и смесь перемешивали в течение 0,6 часа. К реакционной смеси, добавляли ледяную воду, 1 моль/л хлористоводородную кислоту и этилацетат, и полученную в результате смесь перемешивали, и затем органический слой последовательно промывали 1 моль/л хлористоводородной кислотой, насыщенным водным гидрокарбонатом натрия и насыщенным соляным раствором, и сушили. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (2,69 г).
(промежуточное соединение A-14: Rf (ТСХ)=0,23 (гексан:этилацетат=4:1))
[0259]
Справочный пример A-2-2: (2-(3-Бромтиофен-2-ил)этокси)(трет-бутил)диметилсилан (промежуточное соединение A-2-2)
[формула 100]
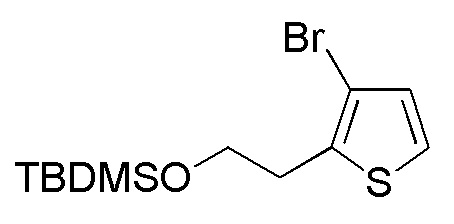
Промежуточное соединение A-2-2 получали согласно способу, описанному в справочном примере A-2, применяя промежуточное соединение A-14 (2,69 г) вместо 2-(тиофен-2-ил)этанола, и таким образом получали заявленное в заголовке соединение (3,93 г).
(промежуточное соединение A-2-2: Rf (ТСХ)=0,76 (гексан:этилацетат=4:1))
[0260]
Справочный пример A-3-2: 4-Бром-5-(2-((трет-бутилдиметилсилил)окси)этил)тиофен-2-карбоновая кислота (промежуточное соединение A-3-2)
[формула 101]
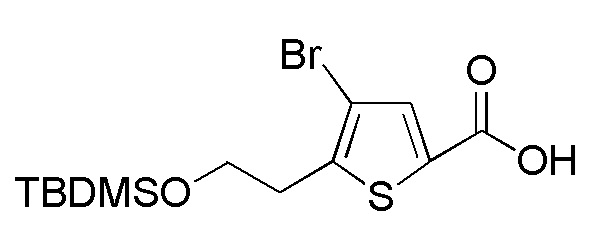
Промежуточное соединение A-3-2 получали согласно способу, описанному в справочном примере A-3, применяя промежуточное соединение A-2-2 (293 мг) вместо промежуточного соединения A-2, и диизопропиламид лития (1,09 моль/л раствор в гексане/тетрагидрофуран, 928 мкл, KANTO) вместо н-бутиллития (2,6 моль/л раствор в гексане, KANTO), и таким образом получали заявленное в заголовке соединение (339 мг).
(промежуточное соединение A-3-2: Rf (ТСХ)=0,12 (гексан:этилацетат=1:1))
[0261]
Справочный пример A-4-2: Метил 4-бром-5-(2-гидроксиэтил)тиофен-2-карбоксилат (промежуточное соединение A-4-2)
[формула 102]
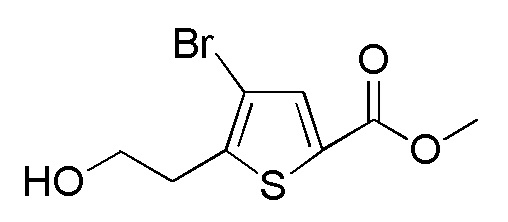
Промежуточное соединение A-4-2 получали согласно способу, описанному в справочном примере A-4, применяя промежуточное соединение A-3-2 (377 мг) вместо промежуточного соединения A-3, и таким образом получали заявленное в заголовке соединение (217 мг).
(промежуточное соединение A-4-2: Rf (ТСХ)=0,53 (гексан:этилацетат=1:1))
[0262]
Справочный пример A-5-2: Метил 4-бром-5-(2-бромэтил)тиофен-2-карбоксилат (промежуточное соединение A-5-2)
[формула 103]
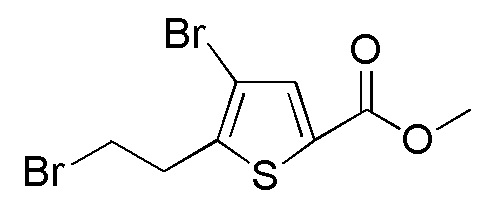
Промежуточное соединение A-5-2 получали согласно способу, описанному в справочном примере A-5, применяя промежуточное соединение A-4-2 (217 мг) вместо промежуточного соединения A-4, и таким образом получали заявленное в заголовке соединение (315 мг).
(промежуточное соединение A-5-2: Rf (ТСХ)=0,44 (гексан:этилацетат=8:1))
[0263]
Справочный пример A-6-2: трет-бутил 2-(2-(3-бром-5-(метоксикарбонил)тиофен-2-ил)этил)гидразинкарбоксилат (промежуточное соединение A-6-2)
[формула 104]

Промежуточное соединение A-6-2 получали согласно способу, описанному в справочном примере A-6, применяя промежуточное соединение A-5-2 (315 мг) вместо промежуточного соединения A-5, и таким образом получали заявленное в заголовке соединение (168 мг).
(промежуточное соединение A-5-2: Rf (ТСХ)=0,48 (гексан:этилацетат=1:1))
[0264]
Справочный пример Z-1-2: трет-бутил 2-(2-(3-бром-5-(метоксикарбонил)тиофен-2-ил)этил)-2-(((2-хлорэтил)тио)карбонил)гидразинкарбоксилат (промежуточное соединение Z-1-2)
[формула 105]
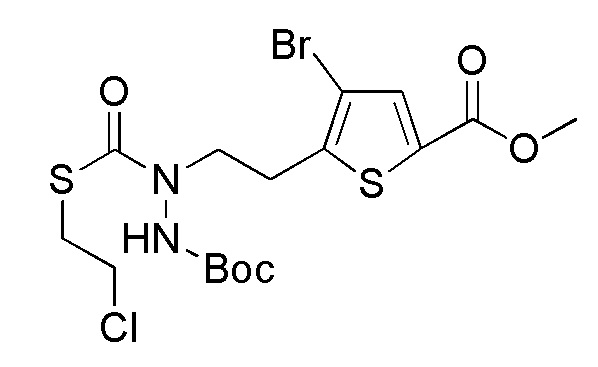
К раствору промежуточного соединения A-6-2 (1,98 г) в дихлорметане (13,1 мл) добавляли гидрокарбонат натрия (880 мг), смесь охлаждали до 0°C, и затем добавляли порциями к смеси промежуточное соединение B-3 (993 мг). Полученную в результате смесь перемешивали при комнатной температуре в течение 0,5 часа, затем добавляли к реакционной смеси воду и этилацетат, и органический слой последовательно промывали насыщенным водным гидрокарбонатом натрия и насыщенным соляным раствором, и сушили. Растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (796 мг).
(промежуточное соединение Z-1-2: Rf (ТСХ)=0,53 (толуол:этилацетат=8:1))
[0265]
Справочный пример Z-2-2: Метил 4-бром-5-(2-(1-(((2-хлорэтил)тио)карбонил)гидразинил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-2-2)
[формула 106]
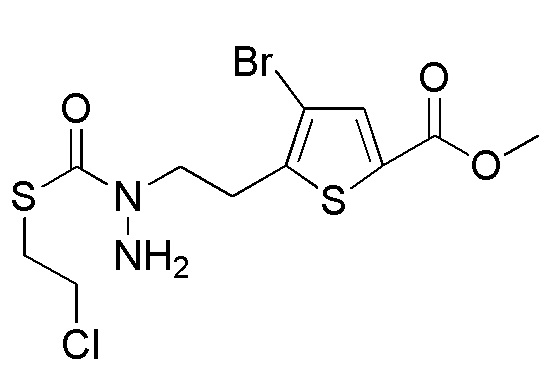
К промежуточному соединению Z-1-2 (796 мг) добавляли 4 моль/л раствор хлористоводородной кислоты в диоксане (7,5 мл), и смесь перемешивали при комнатной температуре в течение 17,6 часов. К реакционной смеси добавляли этилацетат и 5 моль/л водный гидроксид натрия, и затем добавляли насыщенный водный гидрокарбонат натрия к смеси до того, как смесь становилась основной. Органический слой последовательно промывали насыщенным водным гидрокарбонатом натрия и насыщенным соляным раствором, и сушили, и затем растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (594 мг).
(промежуточное соединение Z-2-2: Rf (ТСХ)=0,42 (толуол:этилацетат=8:1))
[0266]
Справочный пример Z-3-2: Метил 4-бром-5-(2-(2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-3-2)
[формула 107]
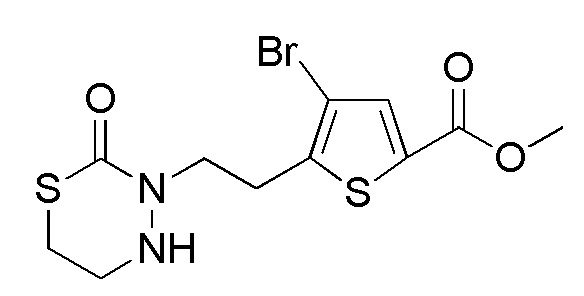
К раствору промежуточного соединения Z-2-2 (594 мг) в ацетонитриле (14,9 мл) последовательно добавляли гидрокарбонат натрия (626 мг) и йодид натрия (1,12 г), и смесь перемешивали при 85°C в течение 120 часов. Реакционную смесь охлаждали до комнатной температуры, и затем добавляли к смеси этилацетат и воду для экстракции. Органический слой последовательно промывали насыщенным водным гидрокарбонатом натрия и насыщенным соляным раствором, и сушили, затем растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (374 мг).
(промежуточное соединение Z-3-2: Rf (ТСХ)=0,13 (гексан:этилацетат=2:1))
[0267]
Справочный пример C-4: N-Метокси-N-метил-2-(3-(тиофен-3-илэтинил)фенил)ацетамид (промежуточное соединение C-4)
[формула 108]
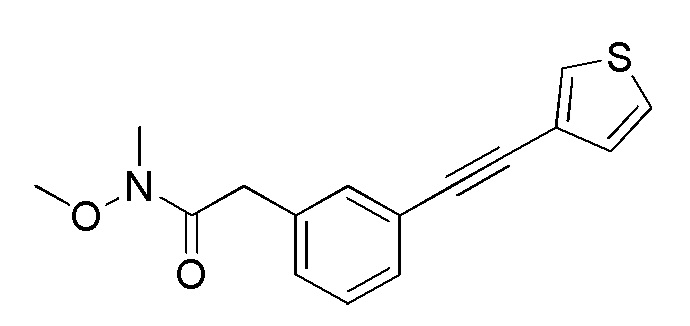
К раствору промежуточного соединения C-2-2 (1,0 г) в ацетонитриле (26 мл) последовательно добавляли хлорид бис(ацетонитрил)палладия (43 мг), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (243 мг), карбонат цезия (2,1 г) и 3-этинилтиофен (650 мкл), и смесь перемешивали при 60°C в течение 14 часов в атмосфере газообразного азота. Реакционную смесь фильтровали через фильтровальную бумагу, покрытую целитом, и остаток, оставшийся на целите, промывали этилацетатом. Фильтрат и промывочную жидкость смешивали, органический растворитель упаривали при пониженном давлении, и затем полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (850 мг).
(промежуточное соединение C-4: Rf (ТСХ)=0,40 (гексан:этилацетат=1:1), LCMS m/z 286,13 (MH+), время удерживания 1,70 минут, LC условия LC-1)
[0268]
Справочный пример Z-14-2: Метил 4-бром-5-(2-(2-оксо-4-(3-оксо-4-(3-(тиофен-3-илэтинил)фенил)бутил)-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-14-2)
[формула 109]
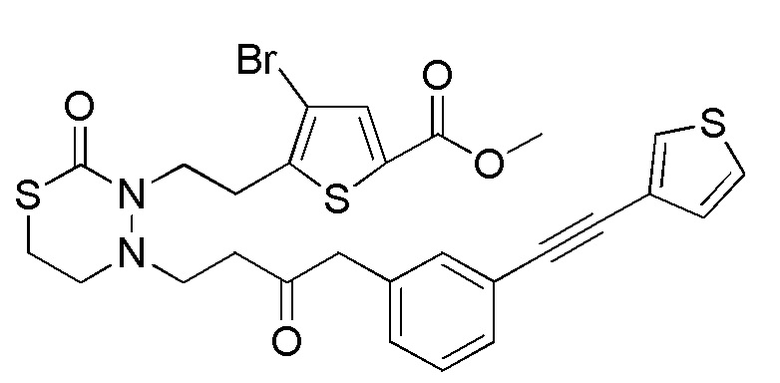
Раствор промежуточного соединения C-4 (117 мг) в 1,2-диметоксиэтане (2,3 мл) охлаждали до 0°C в атмосфере азота. К реакционной смеси добавляли винилмагнийбромид (1 моль/л раствор в тетрагидрофуране, 620 мкл, Aldrich), и полученную в результате смесь перемешивали в течение 3 часов. К реакционной смеси добавляли 2 моль/л хлористоводородную кислоту, и полученную в результате смесь перемешивали в течение 1 минуты. Этилацетат добавляли к реакционной смеси для экстракции, органический слой сушили, и затем растворитель упаривали при пониженном давлении. К полученному в результате остатку добавляли этанол (3 мл), воду (3 мл) и промежуточное соединение Z-3-2 (100 мг), и смесь перемешивали при 110°C в течение 18 часов. К реакционной смеси добавляли насыщенный соляной раствор и хлороформ для экстракции, органический слой сушили, и затем растворитель упаривали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (156,1 мг).
(промежуточное соединение Z-14-2: LCMS m/z 617,2 (MH+), время удерживания 2,08 минут, LC условия LC-1)
[0269]
Пример 2: 4-Бром-5-(2-(4-(3-гидрокси-4-(3-(тиофен-3-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 110]
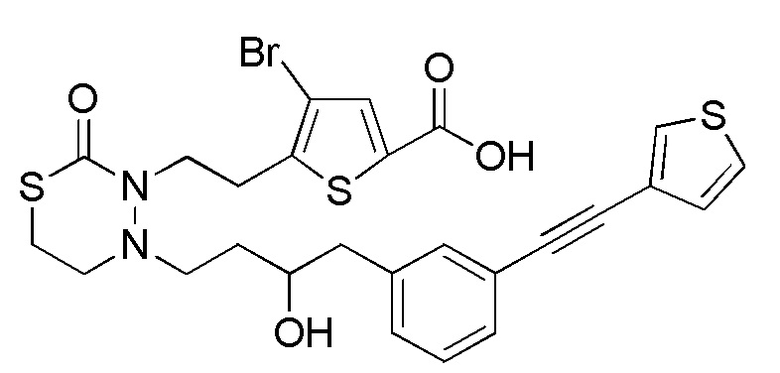
Раствор промежуточного соединения Z-14-2 (156,1 мг) в метаноле (3 мл) охлаждали до 0°C и добавляли порциями к раствору боргидрид натрия (17,3 мг). Полученную в результате смесь перемешивали при 0°C в течение 1 часа, и затем разбавленную хлористоводородную кислоту добавляли порциями к смеси до того, как реакционная смесь становилась нейтральной. Органический растворитель упаривали при пониженном давлении, затем добавляли к остатку этилацетат (2 л), смесь охлаждали до 0°C, добавляли порциями к смеси насыщенный водный гидрокарбонат натрия (1 л), и полученную в результате смесь перемешивали в течение 5 минут. Органический слой экстрагировали и сушили, и затем растворитель упаривали при пониженном давлении. К полученному в результате остатку добавляли тетрагидрофуран (4,6 мл), метанол (4,6 мл) и 1 моль/л водный гидроксид натрия (4,6 мл), и смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь охлаждали до 0°C, добавляли к реакционной смеси 2 моль/л хлористоводородную кислоту, затем полученную в результате смесь экстрагировали этилацетатом, и органический слой сушили. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (хлороформ/метанол), получая заявленное в заголовке соединение (140 мг).
(LCMS m/z 605,1 (MH+), время удерживания 1,78 минут, LC условия LC-1)
[0270]
Справочный пример Z-14-3: Метил 5-(2-(2-оксо-4-(3-оксо-4-(3-(тиофен-2-илэтинил)фенил)бутил)-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-14-3)
[формула 111]
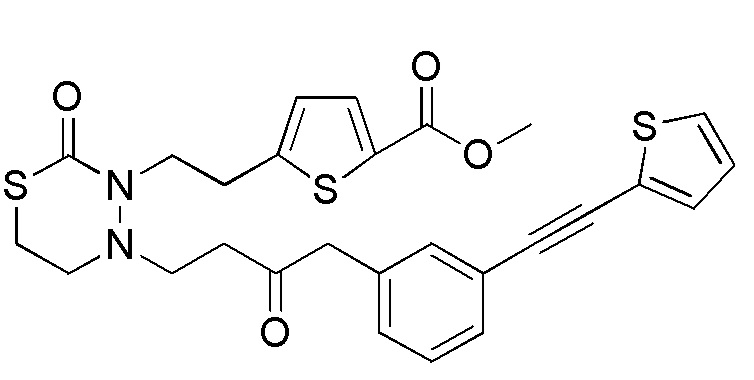
Промежуточное соединение Z-14-3 получали согласно способу, описанному в справочном примере Z-14, применяя 2-этинилтиофен (33,6 мг, MAYBRIDGE) вместо 3-этинилтиофена, и таким образом получали заявленное в заголовке соединение (32,9 мг).
(промежуточное соединение Z-14-3: LCMS m/z 539,0 (MH+), время удерживания 2,00 минут, LC условия LC-1)
[0271]
Пример 3: 5-(2-(4-(3-Гидрокси-4-(3-(тиофен-2-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновую кислоту
[формула 112]

Раствор промежуточного соединения Z-14-3 (32,9 мг) в метаноле (0,6 мл) охлаждали до 0°C, и добавляли порциями к раствору боргидрид натрия (3,6 мг). Смесь перемешивали при 0°C в течение 1,5 часов, и затем разбавленную этилацетатом, и добавляли к смеси разбавленную хлористоводородную кислоту (1,5 мл). Насыщенный водный гидрокарбонат натрия добавляли к смеси до того, как смесь становилась нейтральной. Полученную в результате смесь экстрагировали этилацетатом, органический слой сушили, и затем растворитель упаривали при пониженном давлении. К полученному в результате остатку добавляли тетрагидрофуран (920 мкл), воду (230 мкл) и 2 моль/л водный гидроксид натрия (460 мкл), и полученную в результате смесь перемешивали при 50°C в течение 14 часов. Реакционную смесь охлаждали до 0°C, и оставляли на 5,5 часов, затем добавляли к смеси 2 моль/л хлористоводородную кислоту, смесь экстрагировали этилацетатом, и органический слой сушили. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали жидкостной хроматографией (ацетонитрил/вода), получая заявленное в заголовке соединение (3,6 мг).
(LCMS m/z 527,0 (MH+), время удерживания 1,79 минут, LC условия LC-1)
[0272]
Справочный пример Z-6: Метил 5-(2-(4-(4-(3-бромфенил)-3-((трет-бутилдиметилсилил)окси)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-6)
[формула 113]
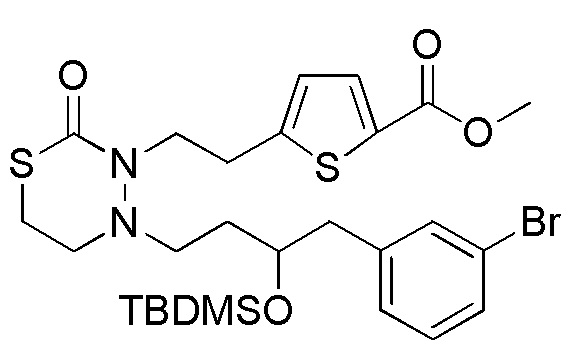
К раствору промежуточного соединения Z-5 (1,0 г) в N, N-диметилформамиде (19,5 мл) добавляли имидазол (265 мг) и трет-бутилдиметилхлорсилан (596 мг), и полученную в результате смесь перемешивали при 30°C в течение 15 часов. К реакционной смеси добавляли насыщенный водный гидрокарбонат натрия, и полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали насыщенным соляным раствором, и затем сушили, растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (1,16 г).
(промежуточное соединение Z-6: LCMS m/z 627,0 (MH+), время удерживания 2,53 минут, LC условия LC-1)
[0273]
Справочный пример Z-21: Метил 5-(2-(4-(3-((трет-бутилдиметилсилил)окси)-4-(3-((триметилсилил)этинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-21)
[формула 114]
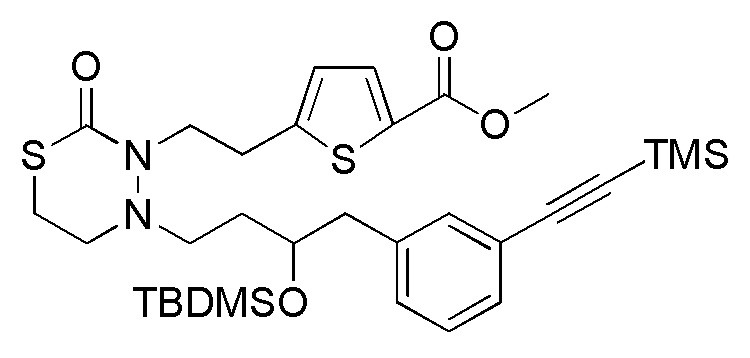
К раствору промежуточного соединения Z-6 (300 мг) в ацетонитриле (15,3 мл) последовательно добавляли хлорид бис(ацетонитрил)палладия (12,4 мг), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (68,3 мг), карбонат цезия (311 мг) и этинилтриметилсилан (331 мкл), и полученную в результате смесь перемешивали при 60°C в течение 19 часов в атмосфере газообразного азота. К реакционной смеси добавляли этинилтриметилсилан (199 мкл), хлорид бис(ацетонитрил)палладия (6,2 мг), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (34,1 мг) и карбонат цезия (187 мг), и полученную в результате смесь перемешивали при 60°C в течение 3,75 часов. Растворитель реакционной смеси упаривали при пониженном давлении, и затем полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (245 мг).
(промежуточное соединение Z-21: LCMS m/z 645,4 (MH+), время удерживания 2,35 минут, LC условия LC-6)
[0274]
Справочный пример Z-22: Метил 5-(2-(4-(3-((трет-бутилдиметилсилил)окси)-4-(3-этинилфенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-22)
[формула 115]
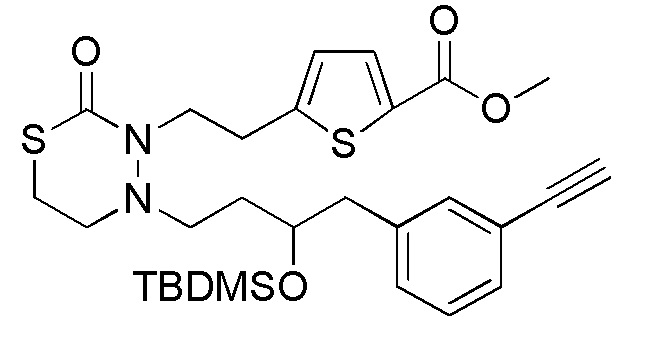
К раствору промежуточного соединения Z-21 (225 мг) в метаноле (3,6 мл) добавляли карбонат калия (50 мг), и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь фильтровали, и фильтрат смешивали с метанольной промывкой, применяемой для промывки остатка, оставшегося на фильтровальной бумаге, и концентрировали. Полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (198 мг).
(промежуточное соединение Z-22: LCMS m/z 573,3 (MH+), время удерживания 1,37 минут, LC условия LC-6)
[0275]
Пример 4: 5-(2-(4-(3-Гидрокси-4-(3-((4-метилтиофен-3-ил)этинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 116]
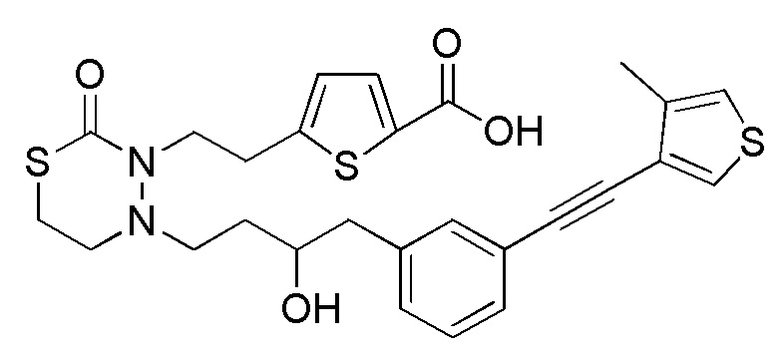
[Стадия a]
Метил 5-(2-(4-(3-((трет-бутилдиметилсилил)окси)-4-(3-((4-метилтиофен-3-ил)этинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-7-4)
[формула 117]
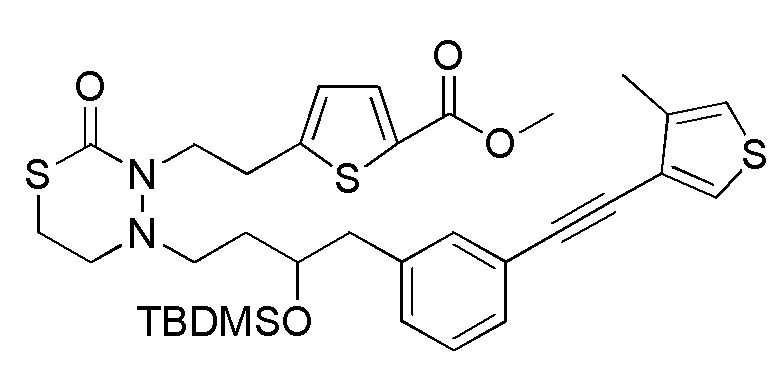
К раствору промежуточного соединения Z-22 (10 мг) в ацетонитриле (280 мкл) последовательно добавляли хлорид бис(ацетонитрил)палладия (0,5 мг), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (2,5 мг), карбонат цезия (6,8 мг), и 3-бром-4-метилтиофен (9,3 мг, TCI), и полученную в результате смесь перемешивали при 60°C в течение 4 часов в атмосфере газообразного азота. Реакционную смесь фильтровали через фильтровальную бумагу, покрытую целитом, и остаток, оставшийся на целите, промывали этилацетатом. Фильтрат и промывочную жидкость смешивали, и органический слой последовательно промывали водой и насыщенным соляным раствором, и затем сушили. Растворитель упаривали при пониженном давлении, и затем полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (7,4 мг).
(промежуточное соединение Z-7-4: LCMS m/z 699,4 (MH+), время удерживания 2,18 минут, LC условия LC-6)
[0276]
[Стадия b]
5-(2-(4-(3-Гидрокси-4-(3-((4-метилтиофен-3-ил)этинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
Раствор промежуточного соединения Z-7-4 (7,4 мг) в тетрагидрофуране (390 мкл) охлаждали до 0°C, добавляли к раствору фторид тетрабутиламмония (1 моль/л раствор в тетрагидрофуране, 33 мкл), и полученную в результате смесь перемешивали при комнатной температуре в течение 2,5 часов. К реакционной смеси добавляли метанол (390 мкл) и 1 моль/л водный гидроксид натрия (390 мкл), и полученную в результате смесь перемешивали при комнатной температуре в течение 2 часов. К реакционной смеси добавляли 2 моль/л хлористоводородную кислоту (100 мкл) и воду (400 мкл), смесь экстрагировали 5 раз этилацетатом (1 мл), и затем органический слой сушили. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (хлороформ/метанол), получая заявленное в заголовке соединение (4,9 мг).
(LCMS m/z 541,2 (MH+), время удерживания 1,74 минут, LC условия LC-1)
[0277]
Пример 5: 5-(2-(4-(4-(3-((2-Хлортиофен-3-ил)этинил)фенил)-3-гидроксибутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 118]
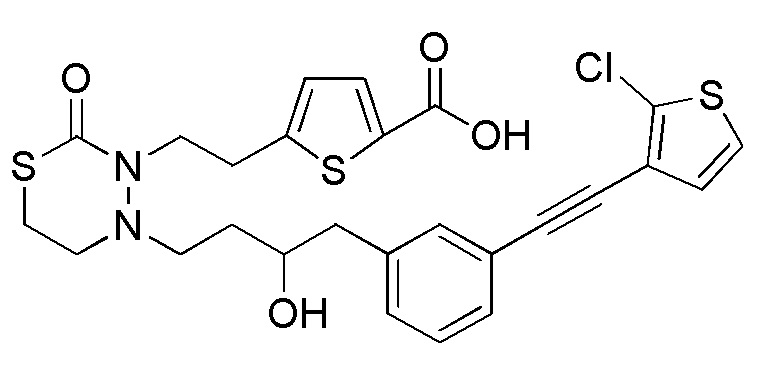
Согласно способу, описанному в примере 4, получение проводили, применяя 3-бром-2-хлортиофен (20,7 мг, TCI) вместо 3-бром-4-метилтиофена, получая заявленное в заголовке соединение (12,9 мг).
(LCMS m/z 561,1 (MH+), время удерживания 1,77 минут, LC условия LC-1)
[0278]
Пример 6: 5-(2-(4-(3-Гидрокси-4-(3-((5-метилтиофен-3-ил)этинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 119]
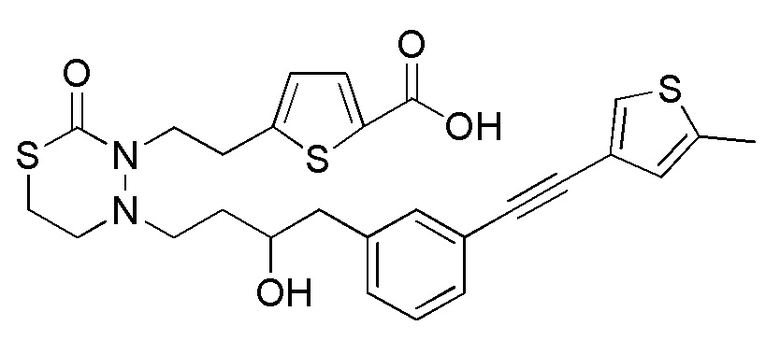
[Стадия a]
Метил 5-(2-(4-(3-((трет-бутилдиметилсилил)окси)-4-(3-((5-метилтиофен-3-ил)этинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-7-6)
[формула 120]
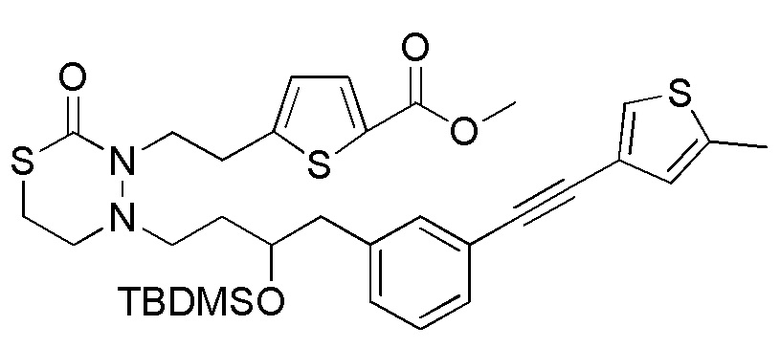
К раствору промежуточного соединения Z-22 (20 мг) в ацетонитриле (1120 мкл) последовательно добавляли хлорид бис(ацетонитрил)палладия (0,9 мг), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (5,0 мг), карбонат цезия (13,7 мг) и 3-бром-5-метилтиофен (18,5 мг, TCI), и полученную в результате смесь перемешивали при 60°C в течение 4 часов в атмосфере газообразного азота. Реакционную смесь фильтровали через фильтровальную бумагу, покрытую целитом, и остаток, оставшийся на целите, промывали смешанным растворителем хлороформа и метанола (9:1). Фильтрат и промывочную жидкость смешивали, растворитель упаривали при пониженном давлении, и затем полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (16,2 мг).
(промежуточное соединение Z-7-6: LCMS m/z 699,4 (MH+), время удерживания 2,20 минут, LC условия LC-6)
[0279]
[Стадия b]
5-(2-(4-(3-Гидрокси-4-(3-((5-метилтиофен-3-ил)этинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
Раствор промежуточного соединения Z-7-6 (16,2 мг) в тетрагидрофуране (850 мкл) охлаждали до 0°C, добавляли фторид тетрабутиламмония (1 моль/л раствор в тетрагидрофуране, 73 мкл), и смесь перемешивали при комнатной температуре в течение 2,5 часов. К реакционной смеси добавляли 1 моль/л водный гидроксид натрия (66 мкл), и полученную в результате смесь перемешивали при комнатной температуре в течение 5 часов. К реакционной смеси добавляли 1 моль/л хлористоводородную кислоту (500 мкл), полученную в результате смесь экстрагировали 5 раз этилацетатом (1 мл), и затем органический слой промывали насыщенным соляным раствором (500 мкл), и сушили. Растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (22,1 мг).
(LCMS m/z 541,2 (MH+), время удерживания 1,76 минут, LC условия LC-1)
[0280]
Пример 7: 5-(2-(4-(4-(3-((4-Цианотиофен-3-ил)этинил)фенил)-3-гидроксибутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 121]

Получение проводили согласно способу, описанному в примере 6, применяя 4-бромтиофен-3-карбонитрил (18,9 мг, COMBI-BLOCKS) вместо 3-бром-5-метилтиофена, получая заявленное в заголовке соединение (3,8 мг).
(LCMS m/z 552,1 (MH+), время удерживания 1,52 минут, LC условия LC-1)
[0281]
Пример 8: 5-(2-(4-(4-(3-((2-Цианотиофен-3-ил)этинил)фенил)-3-гидроксибутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 122]
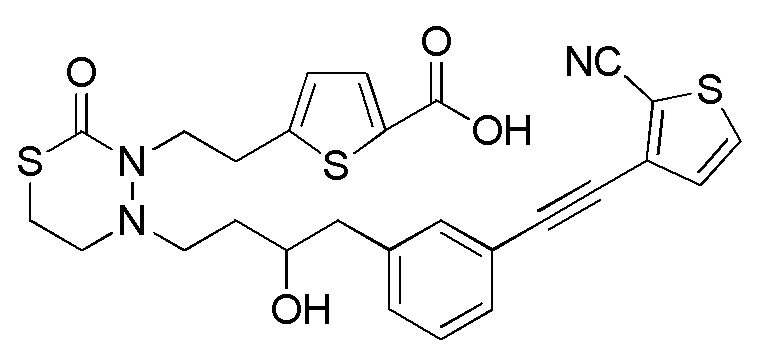
Получение проводили согласно способу, описанному в примере 6, применяя 3-бромтиофен-2-карбонитрил (18,9 мг, APOLLO) вместо 3-бром-5-метилтиофена, получая заявленное в заголовке соединение (5,5 мг).
(LCMS m/z 552,2 (MH+), время удерживания 1,56 минут, LC условия LC-1)
[0282]
Пример 9: 5-(2-(4-(3-Гидрокси-4-(3-(тиазол-4-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 123]

Получение проводили согласно способу, описанному в примере 6, применяя 4-бромтиазол (17,2 мг, ALDRICH) вместо 3-бром-5-метилтиофена, получая заявленное в заголовке соединение (14,3 мг).
(LCMS m/z 528,2 (MH+), время удерживания 1,38 минут, LC условия LC-1)
[0283]
Пример 10: 5-(2-(4-(4-(3-(фуран-3-илэтинил)фенил)-3-гидроксибутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 124]
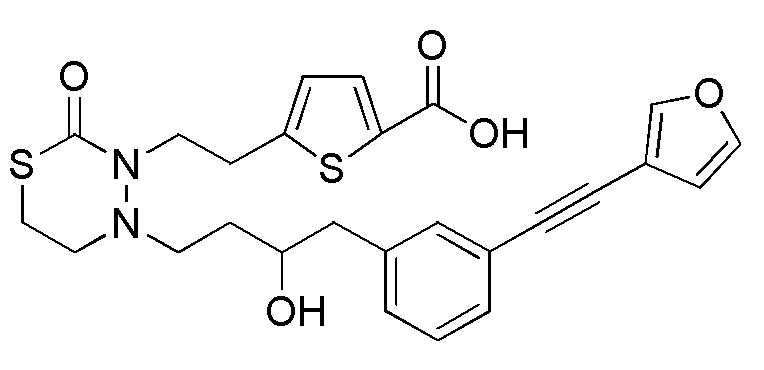
Получение проводили согласно способу, описанному в примере 6, применяя 3-бромфуран (15,4 мг, TCI) вместо 3-бром-5-метилтиофена, получая заявленное в заголовке соединение (13,2 мг).
(LCMS m/z 511,2 (MH+), время удерживания 1,57 минут, LC условия LC-1)
[0284]
Пример 11: 5-(2-(4-(4-(3-(фуран-2-илэтинил)фенил)-3-гидроксибутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 125]
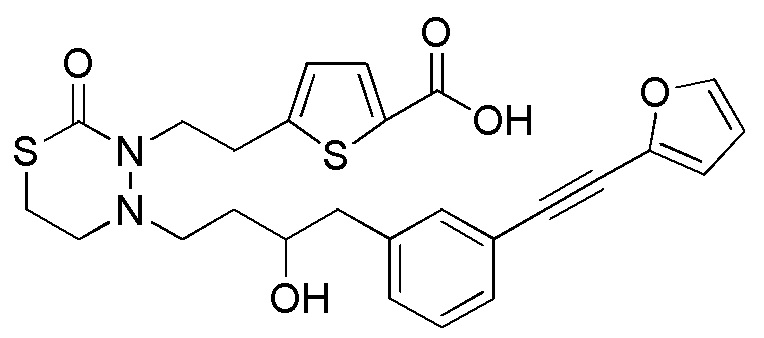
Получение проводили согласно способу, описанному в примере 6, применяя 2-бромфуран (14,8 мг, ALDRICH) вместо 3-бром-5-метилтиофена, получая заявленное в заголовке соединение (4,5 мг).
(LCMS m/z 511,2 (MH+), время удерживания 1,58 минут, LC условия LC-1)
[0285]
Пример 12: 5-(2-(4-(4-(3-((5-Цианотиофен-3-ил)этинил)фенил)-3-гидроксибутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 126]
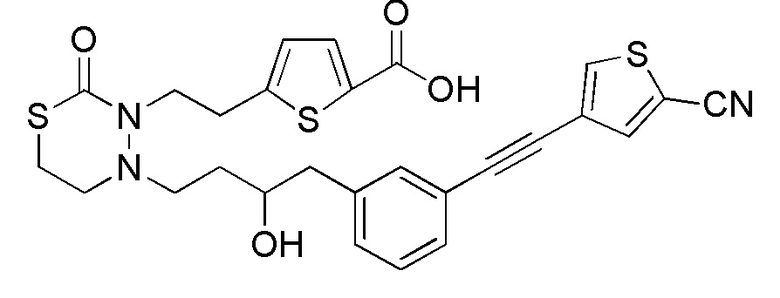
[Стадия a]
Метил 5-(2-(4-(3-((трет-бутилдиметилсилил)окси)-4-(3-((5-цианотиофен-3-ил)этинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-7-12)
[формула 127]
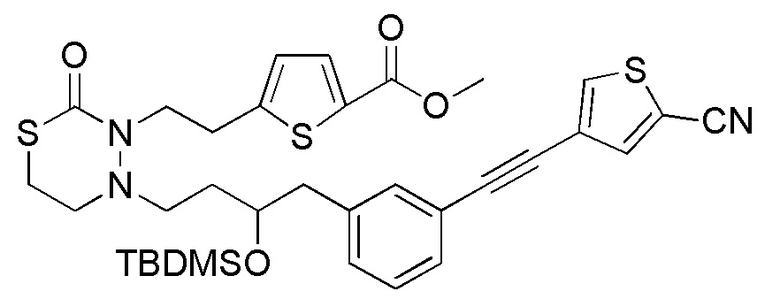
К раствору промежуточного соединения Z-22 (14 мг) в ацетонитриле (400 мкл) последовательно добавляли хлорид бис(ацетонитрил)палладия (0,7 мг), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (3,6 мг), карбонат цезия (12,3 мг) и 4-бромтиофен-2-карбонитрил (18,9 мг, COMBI-BLOCKS), и полученную в результате смесь перемешивали при 60°C в течение 6 часов в атмосфере газообразного азота. Растворитель упаривали при пониженном давлении, и затем полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (6,4 мг).
(промежуточное соединение Z-7-12: LCMS m/z 680,4 (MH+), время удерживания 1,71 минут, LC условия LC-6)
[0286]
[Стадия b]
5-(2-(4-(4-(3-((5-Цианотиофен-3-ил)этинил)фенил)-3-гидроксибутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновую кислоту
Раствор промежуточного соединения Z-7-12 (6,4 мг) в тетрагидрофуране (330 мкл) охлаждали до 0°C, добавляли к раствору фторид тетрабутиламмония (1 моль/л раствор в тетрагидрофуране, 28 мкл), и полученную в результате смесь перемешивали при комнатной температуре в течение 4 часов. К реакционной смеси добавляли 1 моль/л водный гидроксид натрия (30 мкл), и смесь перемешивали при комнатной температуре в течение 2 часов. К реакционной смеси добавляли 1 моль/л хлористоводородную кислоту, смесь экстрагировали этилацетатом, и затем органический слой сушили. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (хлороформ/метанол), получая заявленное в заголовке соединение (0,7 мг).
(LCMS m/z 552,2 (MH+), время удерживания 1,60 минут, LC условия LC-1)
[0287]
Пример 13: 5-(2-(4-(3-Гидрокси-4-(3-(тиазол-2-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 128]
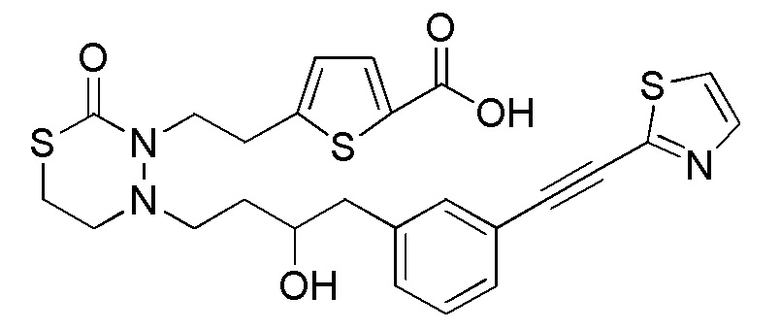
Получение проводили согласно способу, описанному в примере 6, применяя 2-бромтиазол (16,5 мг, TCI) вместо 4-бромтиофен-2-карбонитрила, получая заявленное в заголовке соединение (0,4 мг).
(LCMS m/z 528,2 (MH+), время удерживания 1,43 минут, LC условия LC-1)
[0288]
Пример 14: 5-(2-(4-(4-(3-((3-Цианотиофен-2-ил)этинил)фенил)-3-гидроксибутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 129]

[Стадия a]
Метил 5-(2-(4-(3-((трет-бутилдиметилсилил)окси)-4-(3-((3-цианотиофен-2-ил)этинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-7-14)
[формула 130]
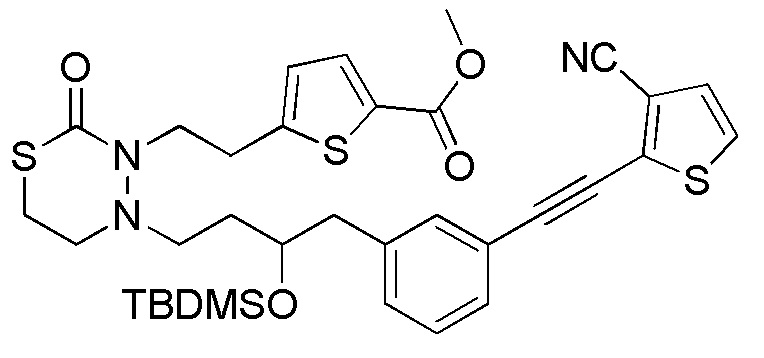
К раствору промежуточного соединения Z-22 (16 мг) в ацетонитриле (1 мл) последовательно добавляли хлорид бис(ацетонитрил)палладия (0,7 мг), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (4,0 мг), карбонат цезия (10,9 мг) и 2-бромтиофен-3-карбонитрил (15,7 мг, MAYBRIDGE), и полученную в результате смесь перемешивали при 60°C в течение 18 часов в атмосфере газообразного азота. Реакционную смесь фильтровали через фильтровальную бумагу, покрытую целитом, остаток, оставшийся на целите, промывали смешанным растворителем хлороформа и метанола (9:1). Фильтрат и промывочную жидкость смешивали, растворитель упаривали при пониженном давлении, и затем полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (7,9 мг).
(промежуточное соединение Z-7-14: LCMS m/z 680,5 (MH+), время удерживания 2,52 минут, LC условия LC-1)
[0289]
[Стадия b]
5-(2-(4-(4-(3-((3-Цианотиофен-2-ил)этинил)фенил)-3-гидроксибутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
Раствор промежуточного соединения Z-7-14 (7,9 мг) в тетрагидрофуране (1 мл) охлаждали до 0°C, добавляли к раствору фторид тетрабутиламмония (1 моль/л раствор в тетрагидрофуране, 0,5 мл), и полученную в результате смесь перемешивали при комнатной температуре в течение 18 часов. К реакционной смеси добавляли метанол (0,5 мл) и 1 моль/л водный гидроксид натрия (0,5 мл), и полученную в результате смесь перемешивали при комнатной температуре в течение 2,5 часов. К реакционной смеси добавляли 1 моль/л хлористоводородную кислоту, смесь экстрагировали этилацетатом, и органический слой сушили. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (хлороформ/метанол), получая заявленное в заголовке соединение (3,0 мг).
(LCMS m/z 552,0 (MH+), время удерживания 1,57 минут, LC условия LC-1)
[0290]
Пример 15: 5-(2-(4-(3-Гидрокси-4-(3-(фенилэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 131]
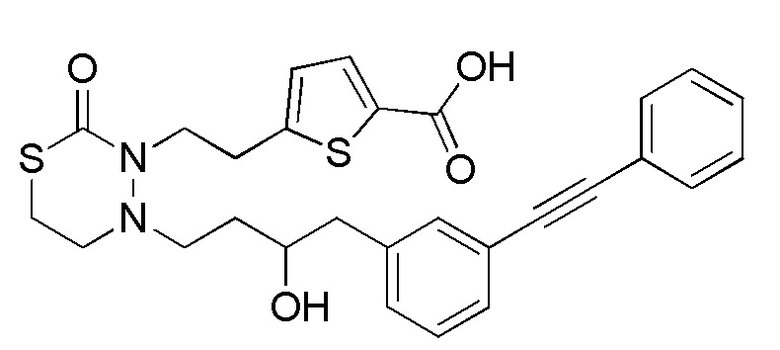
Получение проводили согласно способу, описанному в примере 14, применяя бромбензол (12,3 мг, TCI) вместо 2-бромтиофен-2-карбонитрила, получая заявленное в заголовке соединение (2,4 мг).
(LCMS m/z 521,0 (MH+), время удерживания 1,71 минут, LC условия LC-1)
[0291]
Пример 16: 5-(2-(4-(3-Гидрокси-4-(3-((2-метоксифенил)этинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 132]
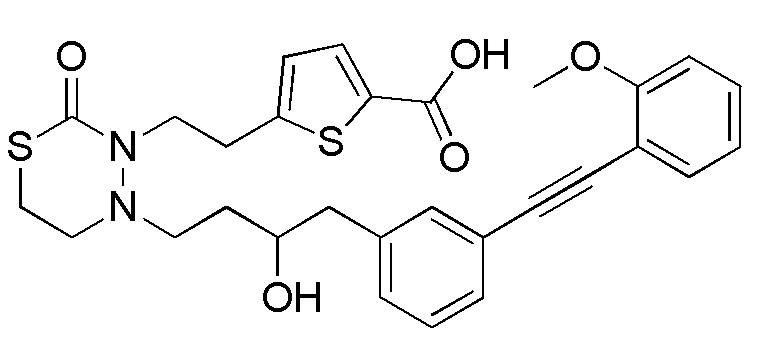
Получение проводили согласно способу, описанному в примере 14, применяя 1-бром-2-метоксибензол (14,7 мг, WAKO) вместо 2-бромтиофен-2-карбонитрила, получая заявленное в заголовке соединение (1,6 мг).
(LCMS m/z 551,3 (MH+), время удерживания 1,64 минут, LC условия LC-1)
[0292]
Справочный пример A-10-2: (3-Хлортиофен-2-ил)метанол (промежуточное соединение A-10-2)
[формула 133]
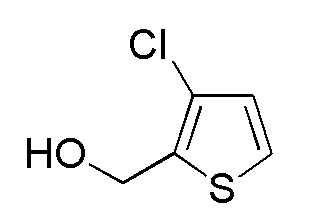
Раствор 3-хлортиофен-2-карбоновой кислоты(4,47 г, ALDRICH) в тетрагидрофуране (88,1 мл) охлаждали до 0°C в атмосфере газообразного азота, добавляли по каплям к раствору 1 моль/л раствора боран-тетрагидрофуранового комплекса в тетрагидрофуране (49,7 мл), и полученную в результате смесь перемешивали при комнатной температуре в течение 22 часов. Реакционную смесь охлаждали до 0°C, метанол, добавляли к смеси воду и этилацетат, и полученную в результате смесь перемешивали. Органический растворитель упаривали при пониженном давлении, затем добавляли к остатку этилацетат, и органический слой последовательно промывали насыщенным водным гидрокарбонатом натрия и насыщенным соляным раствором, и сушили. Растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (4,62 г).
(промежуточное соединение A-10-2: Rf (ТСХ)=0,40 (гексан:этилацетат=2:1))
[0293]
Справочный пример A-11-2: 3-Хлор-2-(бромметил)тиофен (промежуточное соединение A-11-2)
[формула 134]
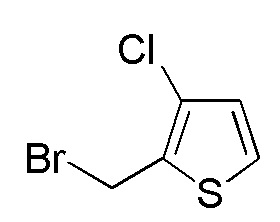
Раствор промежуточного соединения A-10-2 (4,62 г) в дихлорметане (110 мл) охлаждали до 0°C, добавляли к раствору трифенилфосфин (10,8 г) и тетрабромид углерода (10,9 г), и полученную в результате смесь перемешивали при комнатной температуре в течение 1 часа. К реакционной смеси добавляли воду, насыщенный соляной раствор и этилацетат, смесь перемешивали, и затем органический растворитель упаривали при пониженном давлении. Этилацетат добавляли к остатку, и органический слой последовательно промывали насыщенным соляным раствором, и сушили. Органический растворитель упаривали при пониженном давлении, и затем добавляли к полученному в результате остатку смешанный растворитель гексана и этилацетата (9:1), получая суспензию, и суспензию фильтровали через фильтровальную бумагу, покрытую силикагелем. Растворитель фильтрата упаривали при пониженном давлении, получая заявленное в заголовке соединение (9,09 г).
(промежуточное соединение A-11-2: Rf (ТСХ)=0,56 (гексан:этилацетат=8:1))
[0294]
Справочный пример A-12-2: 2-(3-Хлортиофен-2-ил)ацетонитрил (промежуточное соединение A-12-2)
[формула 135]
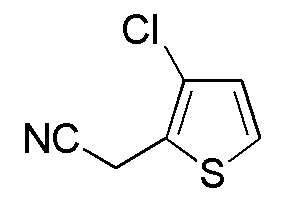
К промежуточному соединению A-11-2 (9,09 г) добавляли диметилсульфоксид (42 мл) и ацетонитрил (126 мл), смесь охлаждали до 0°C, затем добавляли к смеси цианид натрия (2,46 г), и полученную в результате смесь перемешивали при комнатной температуре в течение 2 часов. К реакционной смеси добавляли воду, насыщенный соляной раствор и этилацетат, полученную в результате смесь перемешивали, и затем органический слой промывали насыщенным соляным раствором, и сушили. Растворитель упаривали при пониженном давлении, и затем полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (3,41 г).
(промежуточное соединение A-12-2: Rf (ТСХ)=0,20 (гексан:этилацетат=8:1))
[0295]
Справочный пример A-13-2: Этил 2-(3-хлортиофен-2-ил)ацетат (промежуточное соединение A-13-2)
[формула 136]
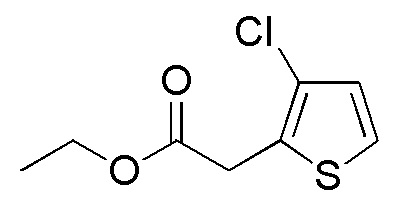
К раствору промежуточного соединения A-12-2 (3,41 г) в этаноле (36 мл) добавляли воду (0,46 мл), смесь охлаждали до 0°C, и затем добавляли порциями к смеси концентрированную серную кислоту (6,3 мл). Реакционную смесь перемешивали при 85°C в течение 88 часов, и затем охлаждали до 0°C, и добавляли к смеси насыщенный водный гидрокарбонат натрия до того, как смесь становилась нейтральной. Добавляли к смеси этилацетат, и полученную в результате смесь перемешивали, и затем концентрировали при пониженном давлении. Добавляли к смеси этилацетат, и органический слой последовательно промывали насыщенным водным гидрокарбонатом натрия и насыщенным соляным раствором, и сушили. Растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (4,56 г).
(промежуточное соединение A-13-2: Rf (ТСХ)=0,31 (гексан:этилацетат=8:1))
[0296]
Справочный пример A-14-2: 2-(3-Хлортиофен-2-ил)этанол (промежуточное соединение A-14-2)
[формула 137]
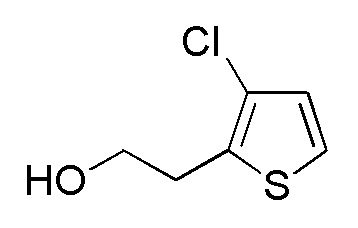
Раствор промежуточного соединения A-13-2 (4,56 г) в тетрагидрофуране (108 мл) охлаждали до 0°C в атмосфере газообразного азота, добавляли к раствору алюмогидрид лития (1,48 г), и смесь перемешивали в течение 0,7 часа. К реакционной смеси добавляли воду, диэтиловый эфир и 1 моль/л хлористоводородную кислоту, полученную в результате смесь перемешивали, и затем органический слой последовательно промывали 1 моль/л хлористоводородной кислотой, насыщенным водным гидрокарбонатом натрия и насыщенным соляным раствором, и сушили. Растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (3,67 г).
(промежуточное соединение A-14-2: Rf (ТСХ)=0,13 (гексан:этилацетат=4:1))
[0297]
Справочный пример A-2-3: (2-(3-Хлортиофен-2-ил)этокси)(трет-бутил)диметилсилан (промежуточное соединение A-2-3)
[формула 138]
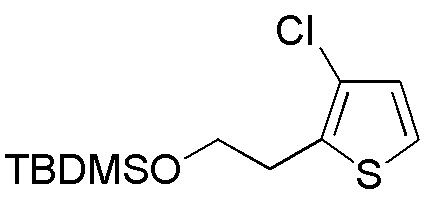
Промежуточное соединение A-2-3 получали согласно способу, описанному в справочном примере A-2, применяя промежуточное соединение A-14-2 (3,67 г) вместо 2-(тиофен-2-ил)этанола, и таким образом получали заявленное в заголовке соединение (4,29 г).
(промежуточное соединение A-2-3: Rf (ТСХ)=0,61 (гексан:этилацетат=4:1))
[0298]
Справочный пример A-3-3: 4-Хлор-5-(2-((трет-бутилдиметилсилил)окси)этил)тиофен-2-карбоновая кислота (промежуточное соединение A-3-3)
[формула 139]
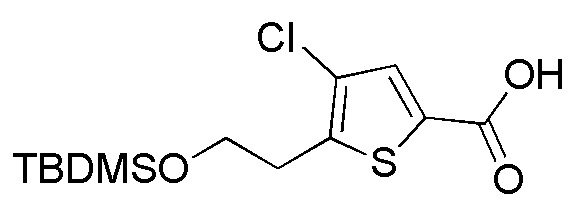
Промежуточное соединение A-3-3 получали согласно способу, описанному в справочном примере A-3, применяя промежуточное соединение A-2-3 (3,03 г) вместо промежуточного соединения A-2, и таким образом получали заявленное в заголовке соединение (3,41 г).
(промежуточное соединение A-3-3: Rf (ТСХ)=0,11 (гексан:этилацетат=4:1))
[0299]
Справочный пример A-4-3: Метил 4-хлор-5-(2-гидроксиэтил)тиофен-2-карбоксилат (промежуточное соединение A-4-3)
[формула 140]
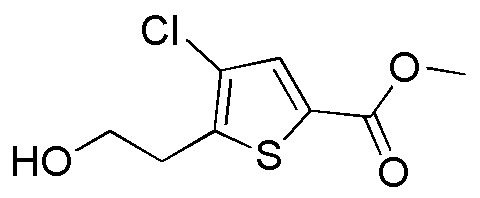
Промежуточное соединение A-4-3 получали согласно способу, описанному в справочном примере A-4, применяя промежуточное соединение A-3-3 (4,35 г) вместо промежуточного соединения A-3, и таким образом получали заявленное в заголовке соединение (2,23 г).
(промежуточное соединение A-4-3: Rf (ТСХ)=0,38 (гексан:этилацетат=1:1))
[0300]
Справочный пример A-5-3: Метил 4-хлор-5-(2-бромэтил)тиофен-2-карбоксилат (промежуточное соединение A-5-3)
[формула 141]
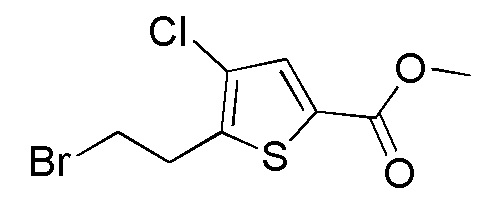
Промежуточное соединение A-5-3 получали согласно способу, описанному в справочном примере A-5, применяя промежуточное соединение A-4-3 (2,23 г) вместо промежуточного соединения A-4, и таким образом получали заявленное в заголовке соединение (3,18 г).
(промежуточное соединение A-5-3: Rf (ТСХ)=0,52 (гексан:этилацетат=2:1))
[0301]
Справочный пример A-6-3: трет-бутил 2-(2-(3-хлор-5-(метоксикарбонил)тиофен-2-ил)этил)гидразинкарбоксилат (промежуточное соединение A-6-3)
[формула 142]
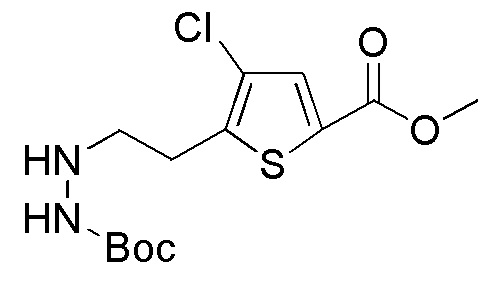
Промежуточное соединение A-6-3 получали согласно способу, описанному в справочном примере A-6, применяя промежуточное соединение A-5-3 (3,18 г) вместо промежуточного соединения A-5, и таким образом получали заявленное в заголовке соединение (3,29 г).
(промежуточное соединение A-5-3: Rf (ТСХ)=0,24 (гексан:этилацетат=2:1))
[0302]
Справочный пример Z-1-3: трет-бутил 2-(2-(3-хлор-5-(метоксикарбонил)тиофен-2-ил)этил)-2-(((2-хлорэтил)тио)карбонил)гидразинкарбоксилат (промежуточное соединение Z-1-3)
[формула 143]

Промежуточное соединение Z-1-3 получали согласно способу, описанному в справочном примере Z-1-2, применяя промежуточное соединение A-6-3 (1,79 г) вместо промежуточного соединения A-6-2, и таким образом получали заявленное в заголовке соединение (2,36 г).
(промежуточное соединение Z-1-3: Rf (ТСХ)=0,24 (гексан:этилацетат=4:1))
[0303]
Справочный пример Z-2-3: Метил 4-хлор-5-(2-(1-(((2-хлорэтил)тио)карбонил)гидразинил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-2-3)
[формула 144]
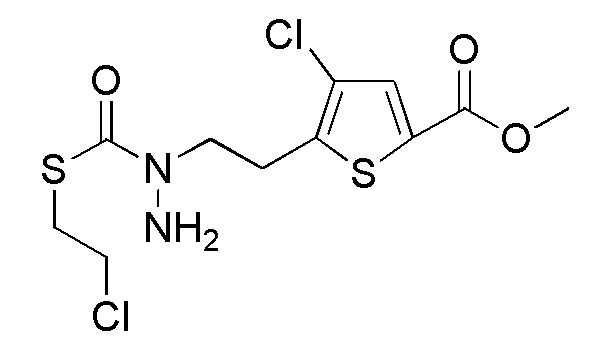
Промежуточное соединение Z-2-3 получали согласно способу, описанному в справочном примере Z-2-2, применяя промежуточное соединение Z-1-3 (2,36 г) вместо промежуточного соединения Z-1-2, и таким образом получали заявленное в заголовке соединение (1,73 г).
(промежуточное соединение Z-2-3: Rf (ТСХ)=0,69 (гексан:этилацетат=1:1))
[0304]
Справочный пример Z-3-3: Метил 4-хлор-5-(2-(2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-3-3)
[формула 145]
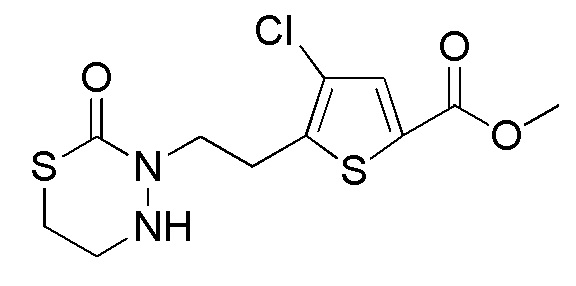
Промежуточное соединение Z-3-3 получали согласно способу, описанному в справочном примере Z-3-2, применяя промежуточного соединения Z-2-3 (1,73 г) вместо промежуточного соединения Z-2-2, и таким образом получали заявленное в заголовке соединение (1,04 г).
(промежуточное соединение Z-3-3: Rf (ТСХ)=0,23 (гексан:этилацетат=1:1))
[0305]
Справочный пример Z-4-3: Метил 4-хлор-5-(2-(4-(4-(3-йодфенил)-3-оксобутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-4-3)
[формула 146]
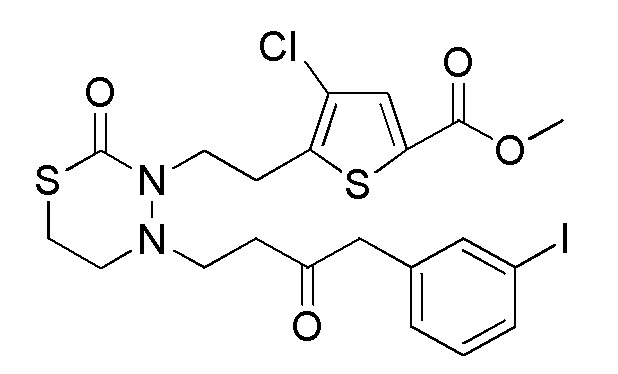
Промежуточное соединение Z-4-3 получали согласно способу, описанному в справочном примере Z-4, применяя промежуточное соединение Z-3-3 (0,30 г) вместо промежуточного соединения Z-3, и промежуточное соединение C-3-2 (суммарное количество промежуточного соединения C-3-2, синтезированного и полученного согласно способу, описанному в справочном примере C-3-2, применяя 2,56 г исходного соединения, промежуточного соединения C-2-2) вместо промежуточного соединения C-3, и таким образом получали заявленное в заголовке соединение (0,64 г).
(промежуточное соединение Z-4-3: Rf (ТСХ)=0,31 (гексан:этилацетат=1:1))
[0306]
Справочный пример Z-14-4: Метил 4-хлор-5-(2-(2-оксо-4-(3-оксо-4-(3-(тиофен-3-илэтинил)фенил)бутил)-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-14-4)
[формула 147]
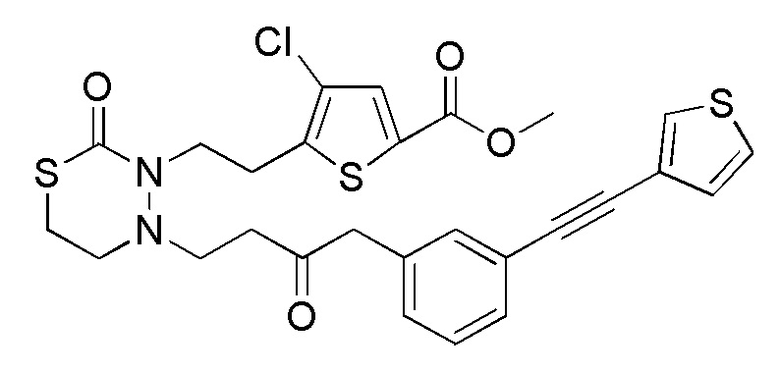
К промежуточному соединению Z-4-3 (429 мг) последовательно добавляли диэтиламин (3,62 мл), 3-этинилтиофен (93 мкл), йодид меди(I) (13,8 мг), и тетракис(трифенилфосфин)палладия (41,8 мг), и смесь перемешивали при комнатной температуре в течение 3,5 часов в атмосфере газообразного азота. К реакционной смеси добавляли 1 моль/л хлористоводородную кислоту, и смесь экстрагировали дважды этилацетатом. Органический слой последовательно промывали насыщенным соляным раствором, насыщенным водным гидрокарбонатом натрия и насыщенным соляным раствором, и сушили. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (хлороформ/метанол), получая заявленное в заголовке соединение (290,4 мг).
(промежуточное соединение Z-14-4: LCMS m/z 573,2 (MH+), время удерживания 2,03 минут, LC условия LC-1)
[0307]
Справочный пример Z-17-17: Метил 4-хлор-5-(2-(4-(3-гидрокси-4-(3-(тиофен-3-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-17-17)
[формула 148]
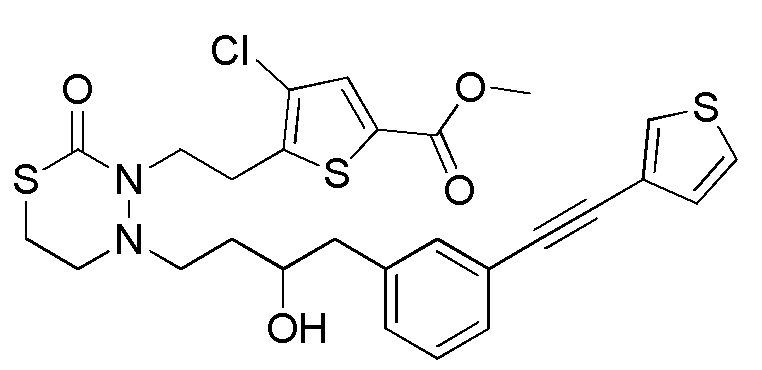
К раствору промежуточного соединения Z-14-4 (290 мг) в метаноле (5 мл) добавляли тетрагидрофуран (1 мл), смесь охлаждали до 0°C, и добавляли порциями к смеси боргидрид натрия (28,8 мг). Смесь перемешивали при комнатной температуре в течение 1,6 часов, затем добавляли к смеси воду, и 1 N хлористоводородной кислотой дополнительно добавляли порциями к реакционной смеси до того, как смесь становилась слабокислой. Смесь экстрагировали дважды этилацетатом, и затем органический слой последовательно промывали насыщенным водным гидрокарбонатом натрия и насыщенным соляным раствором, и сушили. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (хлороформ/метанол), получая заявленное в заголовке соединение (183 мг).
(промежуточное соединение Z-17-17: LCMS m/z 575,2 (MH+), время удерживания 1,98 минут, LC условия LC-1)
[0308]
Пример 17: 4-Хлор-5-(2-(4-(3-гидрокси-4-(3-(тиофен-3-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 149]
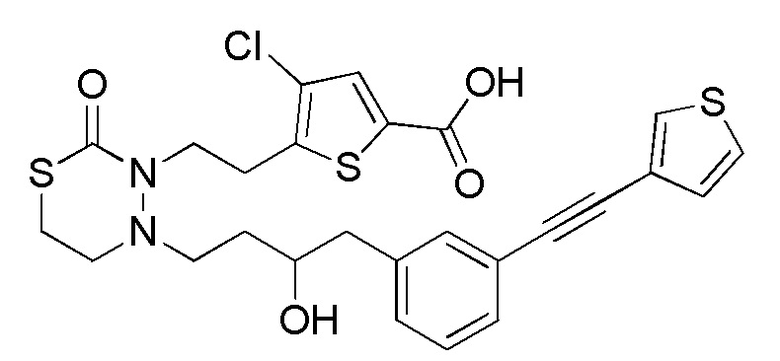
К раствору промежуточного соединения Z-17-17 (183 мг) в тетрагидрофуране (2 мл) добавляли метанол (2 мл), смесь охлаждали до 0°C, и добавляли к смеси воду (2,38 мл) и 4 моль/л водный гидроксид лития (2,38 мл). Полученную в результате смесь перемешивали при комнатной температуре в течение 1 часа, затем добавляли к реакционной смеси воду, и дополнительно 2 N хлористоводородную кислоту добавляли порциями к реакционной смеси до того, как смесь становилась слабокислой. Реакционную смесь экстрагировали дважды этилацетатом, и затем органический слой последовательно промывали насыщенным водным гидрокарбонатом натрия и насыщенным соляным раствором, и сушили. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (хлороформ/метанол), получая заявленное в заголовке соединение (129 мг).
(LCMS m/z 561,25 (MH+), время удерживания 1,72 минут, LC условия LC-1)
[0309]
Справочный пример C-1: 2-(3-Бром-4-метилфенил)ацетонитрил (промежуточное соединение C-1)
[формула 150]
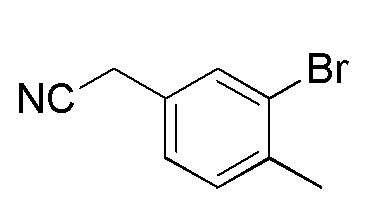
К раствору 2-бром-1,4-диметилбензола (2 г, TCI) в тетрахлориде углерода (21,6 мл) добавляли N-бромсукцинимид (1,06 г) и бензоилпероксид (56,7 мг), и смесь перемешивали при 85°C в течение 1,5 часов. К реакционной смеси добавляли N-бромсукцинимид (1,06 г) и бензоилпероксид (56,7 мг), и полученную в результате смесь дополнительно перемешивали при 85°C в течение 4,5 часов. Реакционную смесь охлаждали до комнатной температуры, и затем фильтровали через фильтровальную бумагу, и остаток, оставшийся на фильтровальной бумаге, промывали дихлорметаном. Фильтрат и промывочную жидкость смешивали, и растворитель упаривали при пониженном давлении. К полученному в результате остатку добавляли этанол (10,8 мл) воду (5,4 мл) и цианид калия (2,1 г), и смесь перемешивали при 100°C в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры, и экстрагировали этилацетатом, и затем органический слой сушили. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (576 мг).
(промежуточное соединение C-1: Rf (ТСХ)=0,58 (гексан:этилацетат=2:1))
1H-NMR (CDCl3): δ (ppm) 7,51 (1H, с), 7,24 (1H, д, J=7,5Гц), 7,18 (1H, д, J=7,5Гц), 3,70 (2H, с), 2,40 (3H, с)
[0310]
Справочный пример C-2-4: 2-(3-Бром-4-метилфенил)-N-метокси-N-метилацетамид (промежуточное соединение C-2-4)
[формула 151]

К промежуточному соединению C-1 (300 мг) добавляли воду (7,1 мл), смесь охлаждали до 0°C, добавляли порциями к смеси концентрированную серную кислоту (5,7 мл), и затем полученную в результате смесь перемешивали при 105°C в течение 15 часов. Реакционную смесь охлаждали до комнатной температуры, и затем этилацетат добавляли для экстракции. Гексан добавляли к водному слою для экстракции, и затем водный слой дополнительно экстрагировали диэтиловым эфиром. Полученные в результате органические слои смешивали, и промывали насыщенным водным гидрокарбонатом натрия, и сушили. Растворитель упаривали при пониженном давлении, и к полученному в результате остатку последовательно добавляли N, N-диметилформамид (14,3 мл), гидрохлорид N, O-диметилгидроксиламина (557 мг), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (821 мг), N, N-диметил-4-аминопиридин (17 мг) и диизопропилэтиламин (1,2 мл), и полученную в результате смесь перемешивали при комнатной температуре в течение 17 часов. К реакционной смеси добавляли диэтиловый эфир, и затем органический слой промывали 3 раз 1 моль/л хлористоводородной кислотой, и один раз насыщенным соляным раствором, и сушили. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (221 мг).
(промежуточное соединение C-2-4: LCMS m/z 272,3 (MH+), время удерживания 1,57 минут, LC условия LC-1)
[0311]
Справочный пример Z-4-4: Метил 5-(2-(4-(4-(3-бром-4-метилфенил)-3-оксобутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-4-4)
[формула 152]
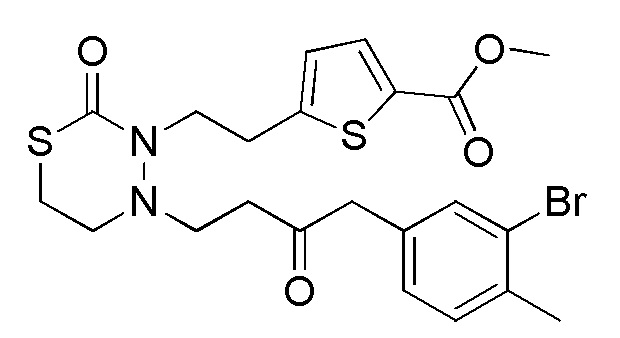
Раствор промежуточного соединения C-2-4 (142,6 мг) в диметоксиэтане (2,85 мл) охлаждали до 0°C в атмосфере азота. К реакционной смеси добавляли винилмагний бромид (1 моль/л раствор в тетрагидрофуране, 790 мкл, ALDRICH), и смесь перемешивали в течение 4 часов. К реакционной смеси добавляли 2 моль/л хлористоводородную кислоту, и смесь перемешивали в течение 1 минуты. Этилацетат добавляли к смеси для экстракции, и органический слой сушили. Растворитель упаривали при пониженном давлении, добавляли к полученному в результате остатку этанол (3 мл), воду (3 мл) и промежуточное соединение Z-3 (100 мг), и смесь перемешивали в течение ночи при 110°C. К реакционной смеси добавляли насыщенный соляной раствор, и смесь экстрагировали хлороформом. Органический слой сушили, и растворитель упаривали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (151,9 мг).
(промежуточное соединение Z-4-4: LCMS m/z 525,1 (MH+), время удерживания 1,87 минут, LC условия LC-1)
[0312]
Справочный пример Z-6-4: Метил 5-(2-(4-(4-(3-бром-4-метилфенил)-3-((трет-бутилдиметилсилил)окси)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-6-4)
[формула 153]
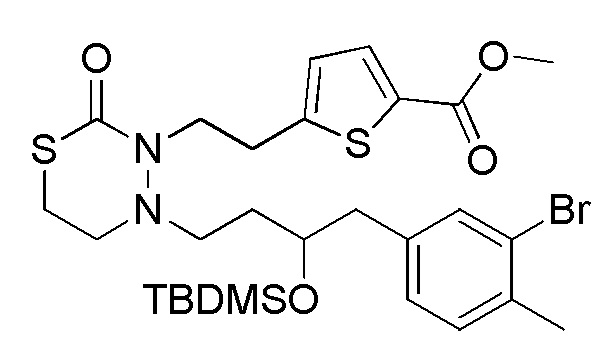
К раствору промежуточного соединения Z-4-4 (152 мг) в метаноле (2,9 мл) добавляли тетрагидрофуран (5 мл), смесь охлаждали до 0°C, и добавляли к смеси боргидрид натрия (16,4 мг). Смесь перемешивали при 0°C в течение 1 часа, и затем добавляли порциями к реакционной смеси разбавленную хлористоводородную кислоту до того, как смесь становилась слабокислой. Органический растворитель упаривали при пониженном давлении, затем этилацетат добавляли к остатку для экстракции, органический слой сушили, и затем растворитель упаривали при пониженном давлении. К полученному в результате остатку последовательно добавляли N, N-диметилформамид (1,4 мл), имидазол (98 мг) и трет-бутилдиметилхлорсилан (131 мг), и полученную в результате смесь перемешивали в течение ночи при комнатной температуре. К реакционной смеси добавляли 2 моль/л хлористоводородную кислоту, и смесь экстрагировали этилацетатом. Органический слой последовательно промывали водой и насыщенным соляным раствором, и затем сушили. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (165,7 мг).
(промежуточное соединение Z-6-4: LCMS m/z 641,2 (MH+), время удерживания 2,02 минут, LC условия LC-6)
[0313]
Справочный пример Z-7-18: Метил 5-(2-(4-(3-((трет-бутилдиметилсилил)окси)-4-(4-метил-3-(тиофен-3-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-7-18)
[формула 154]
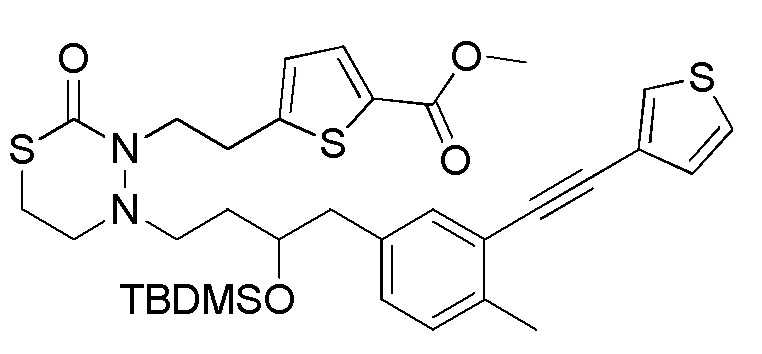
Промежуточное соединение Z-7-18 получали согласно способу, описанному в справочном примере C-4, применяя промежуточное соединение Z-6-4 (20,0 мг) вместо промежуточного соединения C-2-2, и таким образом получали заявленное в заголовке соединение (23,4 мг),.
(промежуточное соединение Z-7-18: LCMS m/z 669,3 (MH+), время удерживания 2,25 минут, LC условия LC-6)
[0314]
Пример 18: 5-(2-(4-(3-Гидрокси-4-(4-метил-3-(тиофен-3-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 155]
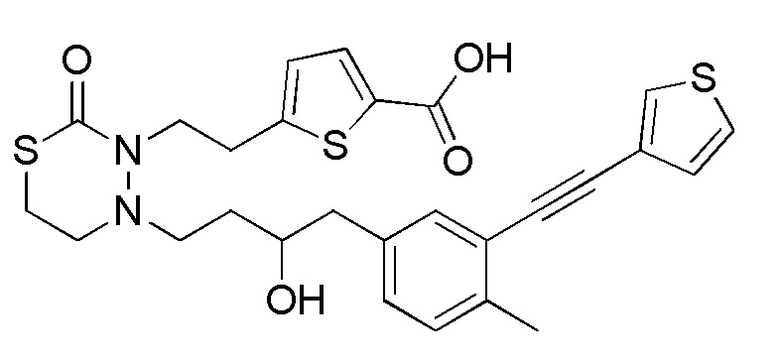
Раствор промежуточного соединения Z-7-18 (23,4 мг) в тетрагидрофуране (0,93 мл) охлаждали до 0°C, добавляли к раствору фторид тетрабутиламмония (1 моль/л раствор в тетрагидрофуране, 93 мкл), и смесь перемешивали при комнатной температуре в течение 1,5 часов. К реакционной смеси добавляли фторид тетрабутиламмония (1 моль/л раствор в тетрагидрофуране, 93 мкл), и смесь перемешивали при комнатной температуре в течение следующих 1,5 часов. К реакционной смеси добавляли метанол (0,93 мл) и 1 моль/л водный гидроксид натрия (0,93 мл), и смесь перемешивали в течение ночи при комнатной температуре. К реакционной смеси добавляли 1 моль/л хлористоводородную кислоту, смесь экстрагировали этилацетатом, и органический слой сушили. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (хлороформ/метанол), получая заявленное в заголовке соединение (17,3 мг).
(LCMS m/z 541,2 (MH+), время удерживания 1,74 минут, LC условия LC-1)
[0315]
Справочный пример C-1-2: 2-(3-Бром-5-метилфенил)ацетонитрил (промежуточное соединение C-1-2)
[формула 156]
Промежуточное соединение C-1-2 получали согласно способу, описанному в справочном примере C-1, применяя 1-бром-3,5-диметилбензол (4,00 г, TCI) вместо 2-бром-1,4-диметилбензола, и таким образом получали заявленное в заголовке соединение (2,34 г).
(промежуточное соединение C-1-2: Rf (ТСХ)=0,64 (гексан:этилацетат=2:1))
1H-NMR (CDCl3): δ (ppm) 7,31 (1H, м), 7,28 (1H, м), 7,09 (1H, м), 3,69 (2H, с), 2,35 (3H, с)
[0316]
Справочный пример C-2-5: 2-(3-Бром-5-метилфенил)-N-метокси-N-метилацетамид (промежуточное соединение C-2-5)
[формула 157]
Промежуточное соединение C-2-5 получали согласно способу, описанному в справочном примере C-2-4, применяя промежуточное соединение C-1-2 (500 мг) вместо промежуточного соединения C-1, и таким образом получали заявленное в заголовке соединение (553 мг).
(промежуточное соединение C-2-5: LCMS m/z 272,3 (MH+), время удерживания 1,57 минут, LC условия LC-1)
[0317]
Справочный пример Z-4-5: Метил 5-(2-(4-(4-(3-бром-5-метилфенил)-3-оксобутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-4-5)
[формула 158]

Промежуточное соединение Z-4-5 получали согласно способу, описанному в справочном примере Z-4-4, применяя промежуточное соединение C-2-5 (142,6 мг) вместо промежуточного соединения C-2-4, и таким образом получали заявленное в заголовке соединение (149,9 мг).
(промежуточное соединение Z-4-5: LCMS m/z 525,1 (MH+), время удерживания 1,88 минут, LC условия LC-1)
[0318]
Справочный пример Z-6-5: Метил 5-(2-(4-(4-(3-бром-5-метилфенил)-3-((трет-бутилдиметилсилил)окси)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-6-5)
[формула 159]

Промежуточное соединение Z-6-5 получали согласно способу, описанному в справочном примере Z-6-4, применяя промежуточное соединение Z-4-5 (149,9 мг) вместо промежуточного соединения Z-4-4, и таким образом получали заявленное в заголовке соединение (163,0 мг).
(промежуточное соединение Z-6-5: LCMS m/z 641,3 (MH+), время удерживания 2,00 минут, LC условия LC-6)
[0319]
Справочный пример Z-7-19: Метил 5-(2-(4-(3-((трет-бутилдиметилсилил)окси)-4-(3-метил-5-(тиофен-3-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-7-19)
[формула 160]
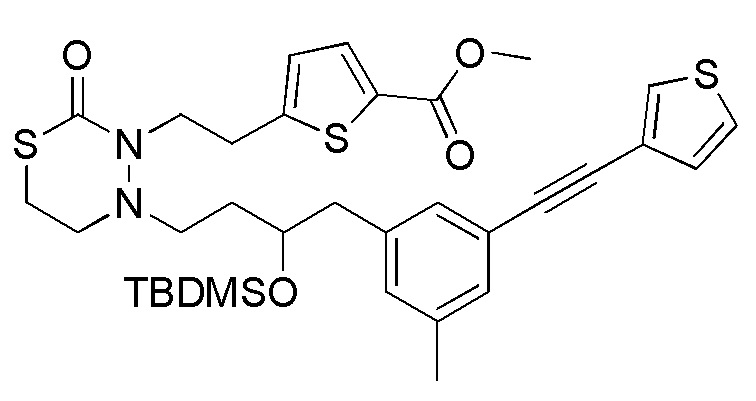
Промежуточное соединение Z-7-19 получали согласно способу, описанному в справочном примере C-4, применяя промежуточное соединение Z-6-5 (20 мг) вместо промежуточного соединения C-2-2, и таким образом получали заявленное в заголовке соединение (19,2 мг).
(промежуточное соединение Z-7-19: LCMS m/z 669,3 (MH+), время удерживания 2,25 минут, LC условия LC-6)
[0320]
Пример 19: 5-(2-(4-(3-Гидрокси-4-(3-метил-5-(тиофен-3-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 161]
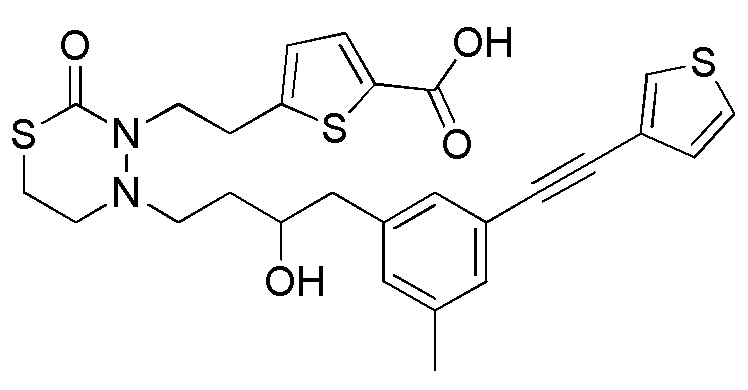
Получение проводили согласно способу, описанному в примере 18, применяя промежуточное соединение Z-7-19 (19,2 мг) вместо промежуточного соединения Z-7-18, получая заявленное в заголовке соединение (13,5 мг).
(LCMS m/z 541,2 (MH+), время удерживания 1,74 минут, LC условия LC-1)
[0321]
Справочный пример C-2-6: 2-(3-Бром-4-фторфенил)-N-метокси-N-метилацетамид (промежуточное соединение C-2-6)
[формула 162]

К раствору 2-(3-бром-4-фторфенил)уксусной кислоты (500 мг) в дихлорметане (43 мл) последовательно добавляли гидрохлорид N, O-диметилгидроксиламина (419 мг), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (494 мг), N, N-диметил-4-аминопиридин (26 мг) и диизопропилэтиламин (1,2 мл), и полученную в результате смесь перемешивали в течение ночи при комнатной температуре. Растворитель реакционной смеси упаривали при пониженном давлении, добавляли к остатку этилацетат, и затем органический слой последовательно промывали 1 моль/л хлористоводородной кислотой и насыщенным соляным раствором, и сушили. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (449 мг).
(промежуточное соединение C-2-6: LCMS m/z 276,2 (MH+), время удерживания 1,37 минут, LC условия LC-1)
[0322]
Справочный пример Z-4-6: Метил 5-(2-(4-(4-(3-бром-4-фторфенил)-3-оксобутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-4-6)
[формула 163]
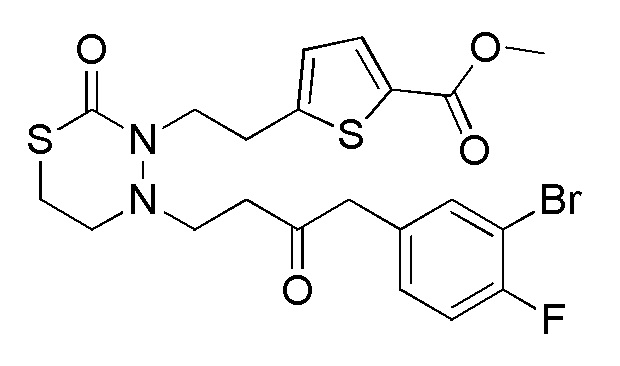
Промежуточное соединение Z-4-6 получали согласно способу, описанному в справочном примере Z-4-4, применяя промежуточное соединение C-2-6 (450,5 мг) вместо промежуточного соединения C-2-4, и таким образом получали заявленное в заголовке соединение (562,8 мг).
(промежуточное соединение Z-4-6: LCMS m/z 529,1 (MH+), время удерживания 1,78 минут, LC условия LC-1)
[0323]
Справочный пример Z-6-6: Метил 5-(2-(4-(4-(3-бром-4-фторфенил)-3-((трет-бутилдиметилсилил)окси)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-6-6)
[формула 164]
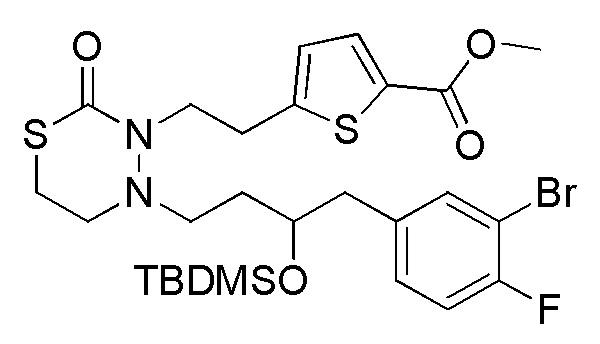
Промежуточное соединение Z-6-6 получали согласно способу, описанному в справочном примере Z-6-4, применяя промежуточное соединение Z-4-6 (562,8 мг) вместо промежуточного соединения Z-4-4, и таким образом получали заявленное в заголовке соединение (719,7 мг).
(промежуточное соединение Z-6-6: LCMS m/z 645,3 (MH+), время удерживания 1,64 минут, LC условия LC-6)
[0324]
Справочный пример Z-7-20: Метил 5-(2-(4-(3-((трет-бутилдиметилсилил)окси)-4-(4-фтор-3-(тиофен-3-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-7-20)
[формула 165]
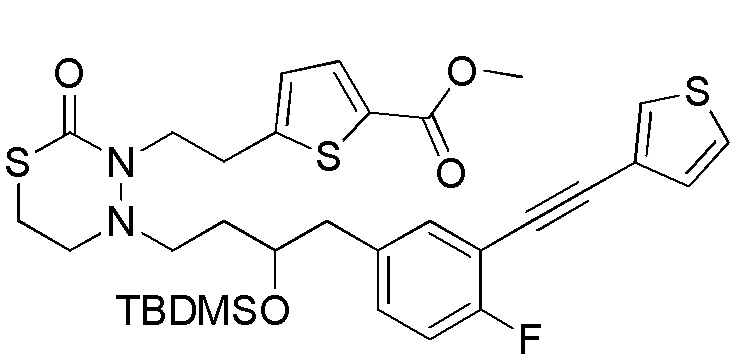
Промежуточное соединение Z-7-20 получали согласно способу, описанному в справочном примере C-4, применяя промежуточное соединение Z-6-6 (15,0 мг) вместо промежуточного соединения C-2-2, и таким образом получали заявленное в заголовке соединение (15,3 мг).
(промежуточное соединение Z-7-20: LCMS m/z 673,4 (MH+), время удерживания 1,87 минут, LC условия LC-6)
[0325]
Пример 20: 5-(2-(4-(4-(4-Фтор-3-(тиофен-3-илэтинил)фенил)-3-гидроксибутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 166]
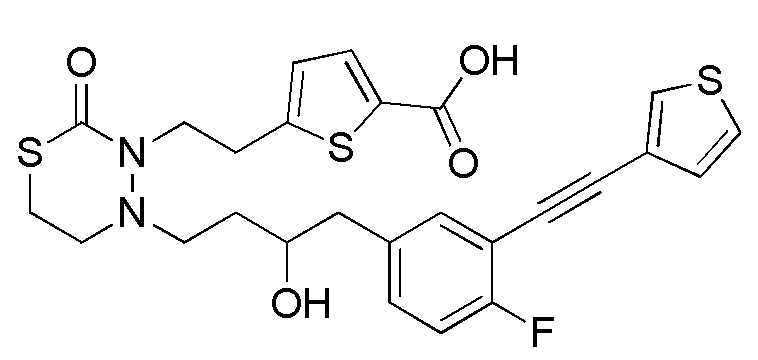
Раствор промежуточного соединения Z-7-20 (15,3 мг) в тетрагидрофуране (345 мкл) охлаждали до 0°C, добавляли к раствору фторид тетрабутиламмония (1 моль/л раствор в тетрагидрофуране, 69 мкл), и смесь перемешивали при комнатной температуре в течение 4 часов. К реакционной смеси добавляли фторид тетрабутиламмония (1 моль/л раствор в тетрагидрофуране, 70 мкл) и тетрагидрофуран (350 мкл), и смесь перемешивали при комнатной температуре в течение следующих 2,5 часов. К реакционной смеси добавляли метанол (345 мкл) и 1 моль/л водный гидроксид натрия (345 мкл), и смесь перемешивали в течение ночи при комнатной температуре. К реакционной смеси добавляли 1 моль/л хлористоводородную кислоту и этилацетат для экстракции, и затем органический слой промывали 1 моль/л хлористоводородной кислотой, и сушили. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали анионообменной смолой, получая заявленное в заголовке соединение (15,3 мг).
(LCMS m/z 545,2 (MH+), время удерживания 1,67 минут, LC условия LC-1)
[0326]
Справочный пример X-1: 4-Фенилтиофен-3-карбальдегид (промежуточное соединение X-1)
[формула 167]
К раствору (4-формилтиофен-3-ил)бороновой кислоты (0,5 г, COMBI-BLOCKS) в н-бутаноле (32 мл) последовательно добавляли бромтиофен (1,0 мл, TCI), воду (6,4 мл), ацетат палладия (36 мг), 2-дициклогексилфосфино-2',6'-диметоксибифенил (132 мг) и фосфат калия (1,36 г), и полученную в результате смесь перемешивали в течение ночи при 95°C в атмосфере газообразного азота. К реакционной смеси, добавляли диэтиловый эфир, и затем органический слой промывали водой, и сушили. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (445 мг).
(промежуточное соединение X-1: LCMS m/z 189,0 (MH+), время удерживания 1,54 минут, LC условия LC-1)
[0327]
Справочный пример X-2: 3-Этинил-4-фенилтиофен (промежуточное соединение X-2)
[формула 168]
К раствору диметил (1-диазо-2-оксопропил)фосфата (727 мг, TCI) в метаноле (14,9 мл) добавляли промежуточное соединение X-1 (445 мг), и смесь охлаждали до 0°C. К реакционной смеси добавляли порциями карбонат калия (686 мг), и смесь перемешивали в течение ночи при комнатной температуре. К реакционной смеси добавляли насыщенный водный хлорид аммония, и смесь экстрагировали диэтиловым эфиром. Органический слой сушили, затем растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (384 мг).
(промежуточное соединение X-2: LCMS m/z 185,1 (MH+), время удерживания 1,81 минут, LC условия LC-1)
[0328]
Справочный пример Z-7-21: Метил 5-(2-(4-(3-((трет-бутилдиметилсилил)окси)-4-(3-((4-фенилтиофен-3-ил)этинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-7-21)
[формула 169]
Промежуточное соединение Z-7-21 получали согласно способу, описанному в справочном примере C-4, применяя промежуточное соединение Z-6 (15,0 мг) вместо промежуточного соединения C-2-2, и промежуточное соединение X-2 (9,4 мг) вместо 3-этинилтиофена, и таким образом получали заявленное в заголовке соединение (15,2 мг).
(промежуточное соединение Z-7-21: LCMS m/z 731,21 (MH+), время удерживания 2,41 минут, LC условия LC-6)
[0329]
Пример 21: 5-(2-(4-(3-Гидрокси-4-(3-((4-фенилтиофен-3-ил)этинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 170]
Получение проводили согласно способу, описанному в примере 18, применяя промежуточное соединение Z-7-21 (15,2 мг) вместо промежуточного соединения Z-7-18, получая заявленное в заголовке соединение (5,7 мг).
(LCMS m/z 603,0 (MH+), время удерживания 1,87 минут, LC условия LC-1)
[0330]
Справочный пример T-2: 2-(4-Бромтиофен-2-ил)-N-метокси-N-метилацетамид (промежуточное соединение T-2)
[формула 171]

Раствор 2-(4-бромтиофен-2-ил)уксусной кислоты (1,0 г) в дихлорметане (9 мл) охлаждали до 0°C, добавляли к раствору гидрохлорид N, O-диметилгидроксиламина (882 мг), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (1,04 г), N, N-диметил-4-аминопиридин (55 мг) и диизопропилэтиламин (3,38 мл), затем дополнительно добавляли диизопропилэтиламин (0,35 мл), и смесь перемешивали при комнатной температуре в течение 41 часов. Реакционную смесь концентрировали, и затем добавляли этилацетат и воду, 1 моль/л хлористоводородную кислоту добавляли для распределения, и дополнительно проводили экстракцию дважды этилацетатом. Органический слой последовательно промывали 1 моль/л хлористоводородной кислотой, водой, насыщенным водным гидрокарбонатом натрия, водой и насыщенным соляным раствором, сушили над сульфатом магния и концентрировали при пониженном давлении, получая заявленное в заголовке соединение (891 мг).
(промежуточное соединение T-2: LCMS m/z 264,2, 266 (MH+), время удерживания 1,36 минут, LC условия LC-1)
[0331]
Справочный пример T-3: 1-(4-Бромтиофен-2-ил)-4-(метокси(метил)амино)бутан-2-он (промежуточное соединение T-3)
[формула 172]
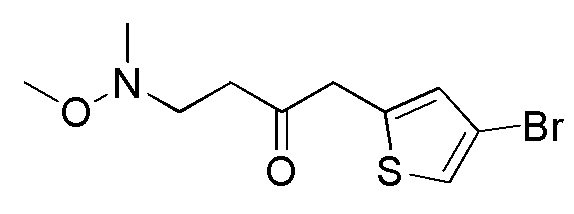
Раствор промежуточного соединения T-2 (200 мг) в 1,2-диметоксиэтане (7 мл) охлаждали до 0°C в атмосфере азота. К реакционной смеси добавляли винилмагнийбромид (1 моль/л раствор в тетрагидрофуране, 1,1 мл, ALDRICH), и смесь перемешивали при той же температуре в течение 2 часов и 50 минут. К реакционной смеси добавляли 1 моль/л хлористоводородную кислоту, делая смесь кислой все еще при 0°C, и затем смесь экстрагировали 3 раз этилацетатом. Органический слой промывали водой, и насыщенным соляным раствором, сушили над сульфатом магния, и затем концентрировали при пониженном давлении, получая заявленное в заголовке соединение (172,8 мг).
(промежуточное соединение T-3: LCMS m/z 292,1, 294 (MH+), время удерживания 1,55 минут, LC условия LC-1)
[0332]
Справочный пример T-4: Метил 5-(2-(4-(4-(4-бромтиофен-2-ил)-3-оксобутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение T-4)
[формула 173]
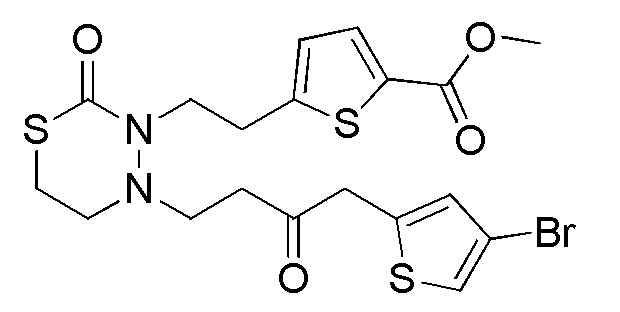
Промежуточное соединение T-3 (172,8 мг), и промежуточное соединение Z-3 (181 мг) растворяли в этаноле (5 мл), добавляли к раствору воду (5 мл), и смесь перемешивали при 105°C в течение 16,5 часов. Реакционную смесь выливали в насыщенный соляной раствор, разбавленный водой, и смесь экстрагировали 3 раз хлороформом. Органический слой промывали насыщенным соляным раствором и сушили над сульфатом магния, и затем растворитель упаривали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией (н-гексан/этилацетат), получая заявленное в заголовке соединение (112,0 мг).
(промежуточное соединение T-4: LCMS m/z 517,0, 519,1 (MH+), время удерживания 1,76 минут, LC условия LC-1)
[0333]
Справочный пример T-5: Метил 5-(2-(4-(4-(4-бромтиофен-2-ил)-3-гидроксибутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение T-5)
[формула 174]
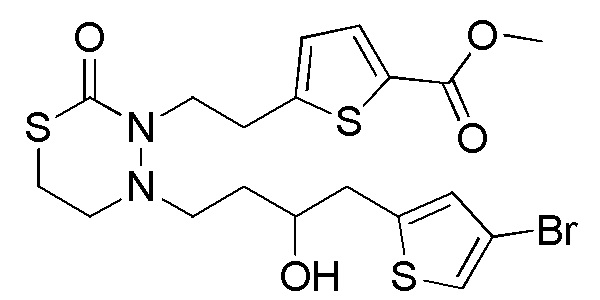
Раствор промежуточного соединения T-4 (112 мг) в метаноле (2 мл) охлаждали до 0°C, добавляли порциями к раствору боргидрид натрия (12,3 мг). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, и затем выливали в воду, и смесь экстрагировали 3 раз этилацетатом. Органический слой промывали водой, и затем насыщенным соляным раствором, и сушили над сульфатом магния, и затем растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (99,6 мг).
(промежуточное соединение T-5: LCMS m/z 519,08, 521,08 (MH+), время удерживания 1,70 минут, LC условия LC-1)
[0334]
Справочный пример T-6: Метил 5-(2-(4-(3-ацетокси-4-(4-бромтиофен-2-ил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение T-6)
[формула 175]
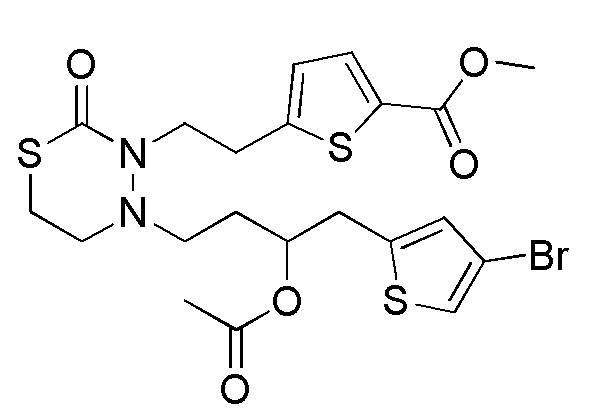
Раствор промежуточного соединения T-5 (95,7 мг) в дихлорметане (1,8 мл) охлаждали до 0°C, добавляли к раствору уксусный ангидрид (348 мкл) и пиридин (741 мкл), смесь перемешивали при комнатной температуре в течение 17 часов, затем добавляли к смеси уксусный ангидрид (348 мкл), и полученную в результате смесь перемешивали при комнатной температуре в течение следующих 4 часов. Добавляли к реакционной смеси воду, и затем смесь экстрагировали 3 раз хлороформом. Органический слой промывали насыщенным водным гидрокарбонатом натрия, и затем насыщенным соляным раствором, и сушили над сульфатом магния, и затем растворитель упаривали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией (н-гексан/этилацетат), получая заявленное в заголовке соединение.
(промежуточное соединение T-6: LCMS m/z 561,1, 563,1 (MH+), время удерживания 1,89 минут, LC условия LC-1)
[0335]
Пример 22: 5-(2-(4-(3-Гидрокси-4-(4-(тиофен-3-илэтинил)тиофен-2-ил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 176]

[Стадия a]
Метил 5-(2-(4-(3-ацетокси-4-(4-(тиофен-3-илэтинил)тиофен-2-ил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение T-7)
[формула 177]
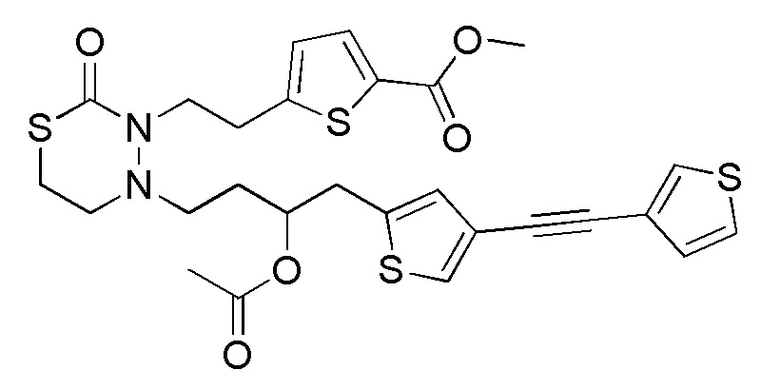
К раствору промежуточного соединения T-6 (17,5 мг) в ацетонитриле (1 мл) последовательно добавляли хлорид бис(ацетонитрил)палладия (1,2 мг), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (6,7 мг), карбонат цезия (13,2 мг) и 3-этинилтиофен (5,6 мкл), и полученную в результате смесь перемешивали при 60°C в течение 17 часов в атмосфере газообразного аргона. Реакционную смесь фильтровали через фильтровальную бумагу, покрытую целитом, и остаток, оставшийся на целите, промывали этилацетатом. Фильтрат и промывочную жидкость смешивали, растворитель упаривали при пониженном давлении, и затем полученный в результате остаток очищали колоночной хроматографией (н-гексан/этилацетат), получая смесь (18,6 мг) промежуточного соединения T-6 и заявленное в заголовке соединение. К раствору полученной в результате смеси в ацетонитриле (2 мл) последовательно добавляли хлорид бис(ацетонитрил)палладия (1,2 мг), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (6,7 мг), карбонат цезия (13,2 мг), и 3-этинилтиофен (6,14 мкл), и полученную в результате смесь перемешивали при 60°C в течение 19 часов в атмосфере газообразного аргона. Реакционную смесь фильтровали через фильтровальную бумагу, покрытую целитом, и остаток, оставшийся на целите, промывали этилацетатом. Фильтрат и промывочную жидкость смешивали, растворитель упаривали при пониженном давлении, и затем полученный в результате остаток очищали колоночной хроматографией (н-гексан/этилацетат), получая заявленное в заголовке соединение (14,2 мг).
(промежуточное соединение T-7: LCMS m/z 589,1 (MH+), время удерживания 2,03 минут, LC условия LC-1)
[0336]
[Стадия b]
5-(2-(4-(3-Гидрокси-4-(4-(тиофен-3-илэтинил)тиофен-2-ил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
К раствору промежуточного соединения T-7 (14,2 мг) в тетрагидрофуране (0,36 мл) добавляли 1 моль/л водный гидроксид лития (0,36 мл), и смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь охлаждали до 0°C и добавляли к смеси 1 моль/л хлористоводородную кислоту (0,36 мл). Смесь разбавляли водой и экстрагировали 3 раз хлороформом, затем органический слой промывали насыщенным соляным раствором, и сушили над сульфатом магния, и растворитель упаривали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией (хлороформ/метанол), получая заявленное в заголовке соединение (3,5 мг).
(LCMS m/z 533,0 (MH+), время удерживания 1,64 минут, LC условия LC-1)
[0337]
Пример 23: (S)-5-(2-(4-(3-Гидрокси-4-(3-(тиофен-3-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 178]

[Стадия a]
(S)-Метил 5-(2-(4-(3-гидрокси-4-(3-(тиофен-3-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-17-S)
[формула 179]
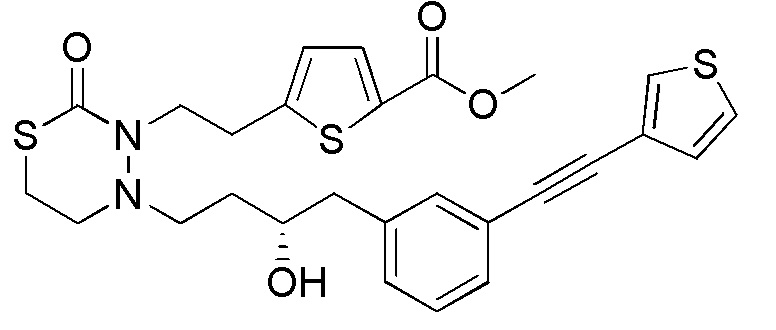
Промежуточное соединение Z-17 (592 мг) подвергали разделению ВЭЖХ, применяя хиральную колонку (ВЭЖХ установка представляла собой установку для препаративной очистки, полученную Japan Waters, хиральная колонка CHIRALCEL AD-H (Daicel Corporation), элюент этанол, скорость потока 0,5 мл/минута, время удерживания 14,91 минут), получая заявленное в заголовке соединение (194,2 мг).
[0338]
[Стадия b]
(S)-5-(2-(4-(3-Гидрокси-4-(3-(тиофен-3-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
Получение проводили согласно способу, описанному в примере 1, применяя промежуточное соединение Z-17-S (41,4 мг) вместо промежуточного соединения Z-17, получая заявленное в заголовке соединение (31,8 мг).
(LCMS m/z 527,2 (MH+), время удерживания 1,68 минут, LC условия LC-1)
[0339]
Справочный пример U: (S)-Метил 5-(2-(4-(4-(3-бромфенил)-3-гидроксибутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение U)
[формула 180]
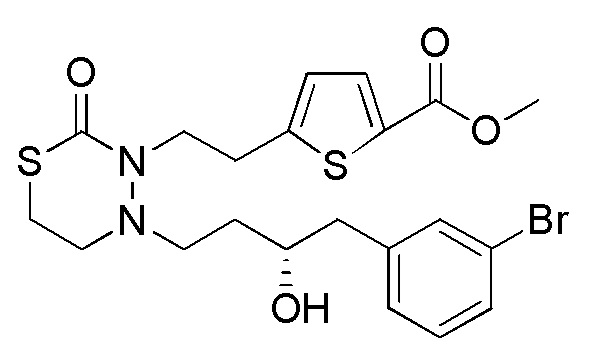
Промежуточное соединение Z-5 (1 г) подвергали разделению ВЭЖХ, применяя хиральную колонку (ВЭЖХ установка представляла собой установку для препаративной очистки, полученную Japan Waters, хиральная колонка CHIRALCEL OJ-H (Daicel Corporation), элюент метанол, скорость потока 0,5 мл/минута, время удерживания 20,72 минут), получая заявленное в заголовке соединение (309 мг).
[0340]
Справочный пример V-1: (S)-Метил 5-(2-(4-(4-(3-бромфенил)-3-((трет-бутилдиметилсилил)окси)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение V-1)
[формула 181]
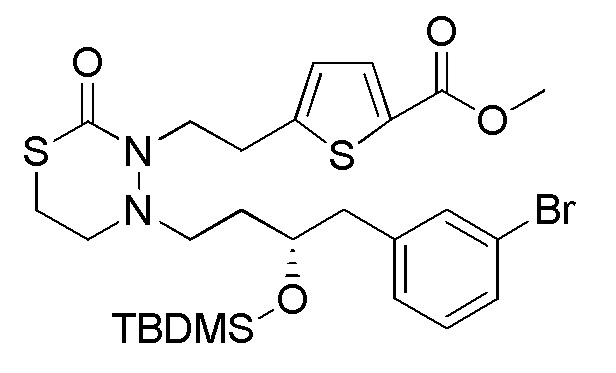
Получение проводили согласно способу, описанному в справочном примере Z-6, применяя промежуточное соединение U (299,3 мг) вместо промежуточного соединения Z-5, получая заявленное в заголовке соединение (333,9 мг).
(промежуточное соединение V-1: LCMS m/z 627,35, 629,35 (MH+), время удерживания 1,79 минут, LC условия NLC-1)
[0341]
Справочный пример V-2: (S)-Метил 5-(2-(4-(3-((трет-бутилдиметилсилил)окси)-4-(3-((триметилсилил)этинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение V-2)
[формула 182]
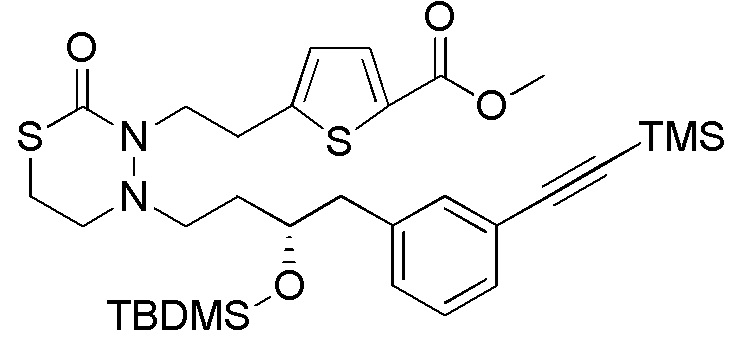
Получение проводили согласно способу, описанному в справочном примере Z-21, применяя промежуточное соединение V-1 (333,9 мг) вместо промежуточного соединения Z-6, получая заявленное в заголовке соединение (91,3 мг).
(промежуточное соединение V-2: LCMS m/z 645,49 (MH+), время удерживания 2,38 минут, LC условия NLC-6)
[0342]
Справочный пример V-3: (S)-Метил 5-(2-(4-(3-((трет-бутилдиметилсилил)окси)-4-(3-этинилфенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение V-3)
[формула 183]
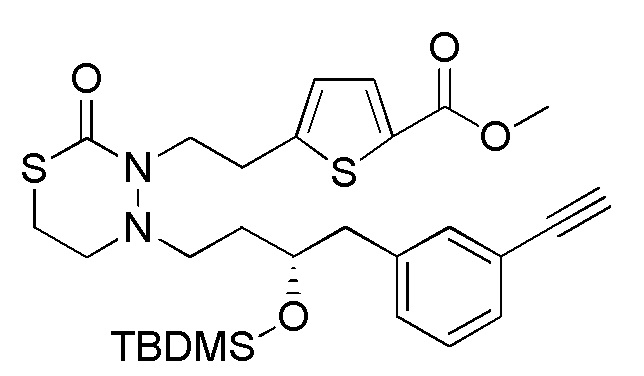
Получение проводили согласно способу, описанному в справочном примере Z-22, применяя промежуточное соединение V-2 (91,3 мг) вместо промежуточного соединения Z-21, получая заявленное в заголовке соединение (91,6 мг).
(промежуточное соединение V-3: LCMS m/z 573,45 (MH+), время удерживания 1,41 минут, LC условия NLC-6)
[0343]
Пример 24: (S)-5-(2-(4-(4-(3-((2-Цианотиофен-3-ил)этинил)фенил)-3-гидроксибутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 184]

[Стадия a]
(S)-Метил 5-(2-(4-(3-((трет-бутилдиметилсилил)окси)-4-(3-((2-цианотиофен-3-ил)этинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-24-1)
[формула 185]
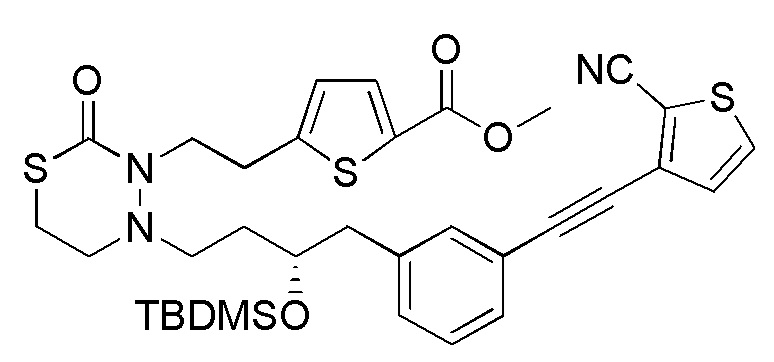
К раствору промежуточного соединения V-3 (50,6 мг) в ацетонитриле (1 мл) последовательно добавляли хлорид бис(ацетонитрил)палладия (2,0 мг), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (11,2 мг), карбонат цезия (51,1 мг) и 3-бромтиофен-2-карбонитрил (73,8 мг), и полученную в результате смесь перемешивали при 60°C в течение 0,75 часа в атмосфере газообразного азота. Реакционную смесь фильтровали через фильтровальную бумагу, покрытую целитом, и остаток, оставшийся на целите, промывали смешанным растворителем хлороформа и метанола (9:1). Фильтрат и промывочную жидкость смешивали, растворитель упаривали при пониженном давлении, и затем полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (10,1 мг).
(промежуточное соединение Z-24-1: LCMS m/z 680,44 (MH+), время удерживания 1,73 минут, LC условия NLC-6)
[0344]
[Стадия b]
(S)-Метил 5-(2-(4-(4-(3-((2-цианотиофен-3-ил)этинил)фенил)-3-гидроксибутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-24-2)
[формула 186]
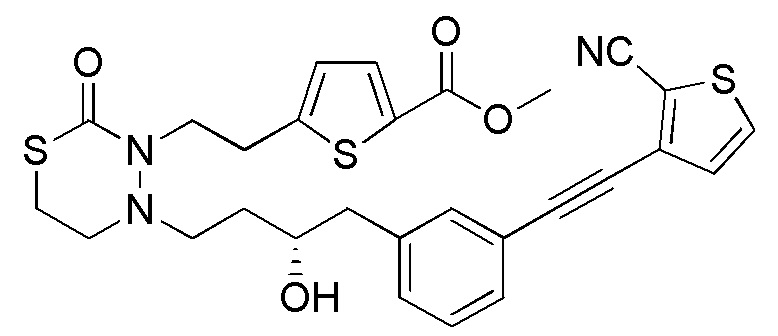
К раствору промежуточного соединения Z-24-1 (10,1 мг) в тетрагидрофуране (0,5 мл) добавляли фторид тетрабутиламмония (1 моль/л раствор в тетрагидрофуране, 44,6 мкл), и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционная смесь очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (9,3 мг).
(промежуточное соединение Z-24-2: LCMS m/z 566,35 (MH+), время удерживания 1,77 минут, LC условия NLC-6)
[0345]
[Стадия c]
(S)-5-(2-(4-(4-(3-((2-Цианотиофен-3-ил)этинил)фенил)-3-гидроксибутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
К раствору промежуточного соединения Z-24-2 (9,3 мг) в тетрагидрофуране (400 мкл) добавляли 1 моль/л водный гидроксид натрия (197 мкл), и смесь перемешивали при комнатной температуре в течение 60 часов. К реакционной смеси добавляли 1 моль/л хлористоводородную кислоту (400 мкл), смесь экстрагировали 3 раз хлороформом, и затем органический слой промывали насыщенным соляным раствором, и сушили. Растворитель упаривали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией (метанол/хлороформ), получая заявленное в заголовке соединение (7,5 мг).
(LCMS m/z 552,30,2 (MH+), время удерживания 1,23 минут, LC условия NLC-1)
[0346]
Пример 25: (S)-5-(2-(4-(3-Гидрокси-4-(3-(тиазол-4-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 187]
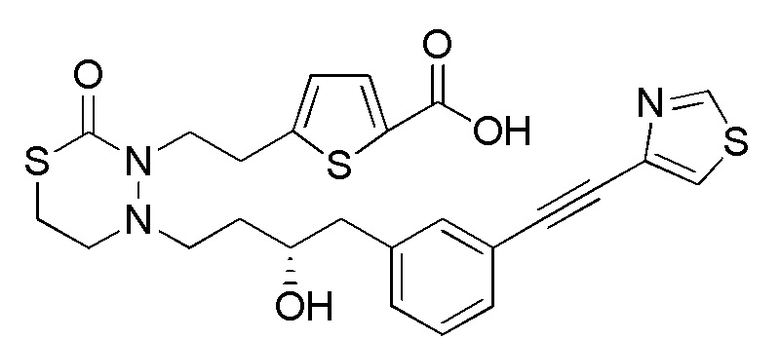
[Стадия a]
(S)-Метил 5-(2-(4-(3-((трет-бутилдиметилсилил)окси)-4-(3-(тиазол-4-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-25-1)
[формула 188]

Получение проводили согласно способу, описанному в примере 24, стадия a, применяя 4-бромтиазол (28,6 мкл) вместо 3-бромтиофен-2-карбонитрила, получая заявленное в заголовке соединение (21,0 мг).
(промежуточное соединение Z-25-1: LCMS m/z 656,43 (MH+), время удерживания 1,37 минут, LC условия NLC-1)
[0347]
[Стадия b]
(S)-5-(2-(4-(3-Гидрокси-4-(3-(тиазол-4-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
К раствору промежуточного соединения Z-25-1 (21,0 мг) в тетрагидрофуране (1 мл) добавляли фторид тетрабутиламмония (1 моль/л раствор в тетрагидрофуране, 92,6 мкл), и смесь перемешивали при комнатной температуре в течение 3 часов. Реакционная смесь очищали колоночной хроматографией (гексан/этилацетат). К раствору полученного в результате промежуточного соединения (12,2 мг) в тетрагидрофуране (540 мкл) и и метаноле (270 мкл) добавляли 1 моль/л водный гидроксид натрия (270 мкл), и смесь перемешивали при комнатной температуре в течение 16 часов. К реакционной смеси добавляли 1 моль/л хлористоводородную кислоту, смесь экстрагировали 5 раз этилацетатом, и затем органический слой промывали насыщенным соляным раствором, и сушили. Растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (12,5 мг).
(LCMS m/z 528,29 (MH+), время удерживания 1,08 минут, LC условия NLC-1)
[0348]
Пример 26: (S)-5-(2-(4-(4-(3-(Furan-3-илэтинил)фенил)-3-гидроксибутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 189]
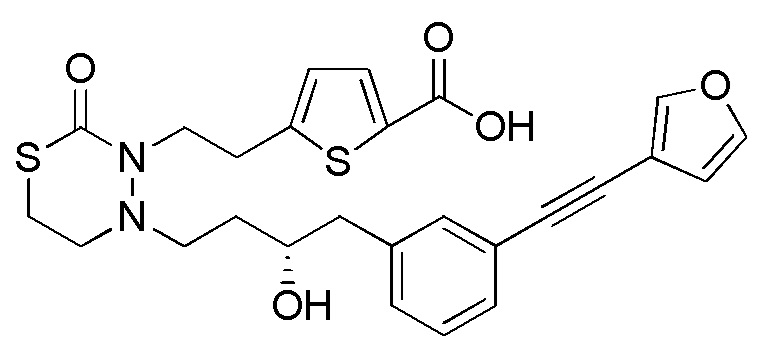
Получение проводили согласно способу, описанному в примере 25, применяя 3-бромфуран (31,2 мг) вместо 4-бромтиазола, и затем полученный в результате остаток очищали колоночной хроматографией (метанол/хлороформ), получая заявленное в заголовке соединение (5,1 мг).
(LCMS m/z 511,35 (MH+), время удерживания 1,23 минут, LC условия NLC-1)
[0349]
Справочный пример Z-27: (E)-Метил 5-(2-(4-(3-((трет-бутилдиметилсилил)окси)-4-(3-(2-(тиофен-3-ил)винил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-27)
[формула 190]
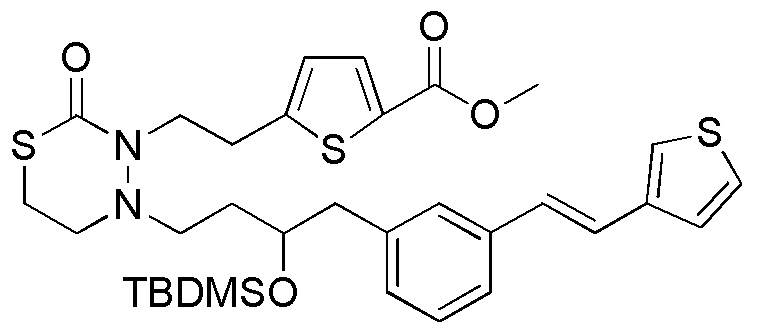
К раствору промежуточного соединения Z-6 (50,0 мг) в 1,4-диоксане (637 мкл) последовательно добавляли (E)-4,4,5,5-тетраметил-2-(2-(тиофен-3-ил)винил)-1,3,2-диоксаборолан (22,6 мг, ALDRICH), хлорид бис(трифенилфосфин)палладия (5,6 мг), карбонат натрия (21,1 мг) и воду (199 мкл), и полученную в результате смесь перемешивали при 85°C в течение 11 часов в атмосфере газообразного азота. К реакционной смеси, добавляли воду, и смесь экстрагировали этилацетатом. Органический слой сушили, затем растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (44,5 мг).
(промежуточное соединение Z-27: LCMS m/z 657,3 (MH+), время удерживания 1,93 минут, LC условия LC-6)
[0350]
Пример 27: (E)-5-(2-(4-(3-Гидрокси-4-(3-(2-(тиофен-3-ил)винил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 191]
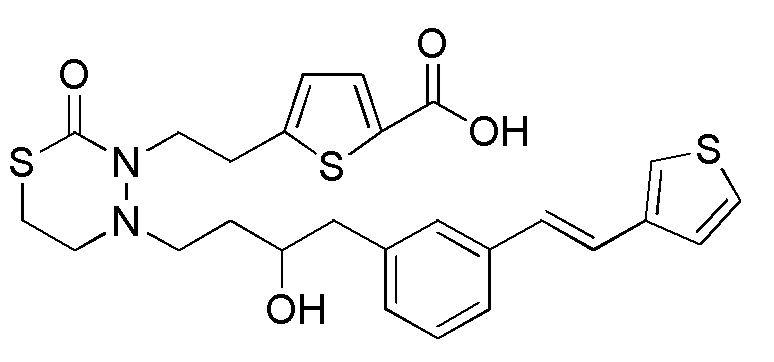
Получение проводили согласно способу, описанному в примере 18, применяя промежуточное соединение Z-27 (24,8 мг) вместо промежуточного соединения Z-7-18, получая заявленное в заголовке соединение (17,8 мг).
(LCMS m/z 529,1 (MH+), время удерживания 1,63 минут, LC условия LC-1)
[0351]
Справочный пример Z-14-5: Метил 4-хлор-5-(2-(2-оксо-4-(3-оксо-4-(3-(пиридин-2-илэтинил)фенил)бутил)-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-14-5)
[формула 192]
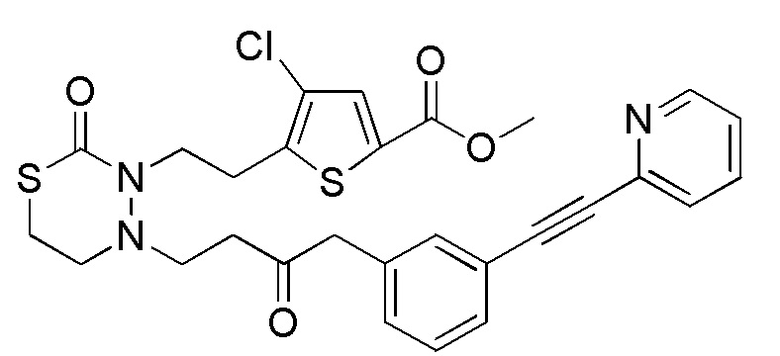
К промежуточному соединению Z-4-3 (100 мг) последовательно добавляли диэтиламин (850 мкл), 2-этинилпиридин (22,8 мкл), йодид меди (I) (2,9 мг) и тетракис(трифенилфосфин)палладия (3,9 мг), и смесь перемешивали при комнатной температуре в течение 15,5 часов в атмосфере газообразного азота. Реакционную смесь разбавляли этилацетатом, затем растворитель упаривали, и полученный в результате остаток очищали колоночной хроматографией (толуол/ацетонитрил), получая заявленное в заголовке соединение (47,6 мг).
(промежуточное соединение Z-14-5: LCMS m/z 570,425 (MH+), время удерживания 4,86 минут, LC условия FLC-1))
[0352]
Пример 28: 4-Хлор-5-(2-(4-(3-гидрокси-4-(3-(пиридин-2-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 193]

К раствору промежуточного соединения Z-14-5 (47,6 мг) в метаноле (840 мкл) добавляли порциями боргидрид натрия (4,8 мг). Смесь перемешивали при комнатной температуре в течение 0,5 часа, и затем добавляли к смеси воду и этилацетат, и органический слой последовательно промывали насыщенным водным гидрокарбонатом натрия и насыщенным соляным раствором, и сушили. Растворитель упаривали при пониженном давлении, и затем добавляли к остатку тетрагидрофуран (640 мкл), метанол (640 мкл) и 2 моль/л водный гидроксид натрия (640 мкл). Реакционную смесь перемешивали при комнатной температуре в течение 1,8 часов, и затем охлаждали до 0°C, и добавляли порциями к смеси 2 моль/л хлористоводородную кислоту (640 мкл). К реакционной смеси добавляли этилацетат, и органический слой промывали насыщенным соляным раствором, и сушили. Растворитель упаривали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией (толуол/этанол/уксусная кислота), получая заявленное в заголовке соединение (15,0 мг).
(LCMS m/z 556,023 (MH+), время удерживания 4,49 минут, LC условия FLC-1)
[0353]
Справочный пример Z-29-1: 2-трет-бутил 1-(2-хлорэтил) 1-(2-(5-(метоксикарбонил)тиофен-2-ил)этил)гидразин-1,2-дикарбоксилат (промежуточное соединение Z-29-1)
[формула 194]
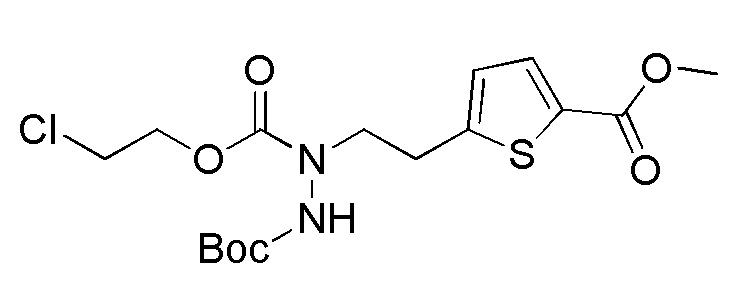
К раствору промежуточного соединения A-6 (5,0 г) в ацетонитриле (165 мл) добавляли карбонат калия (0,46 г), и смесь охлаждали до 0°C. 2-Хлорэтилхлороформиат (2,07 мл) медленно добавляли к смеси, и полученную в результате смесь перемешивали при той же температуре в течение 1 часа. К реакционной смеси добавляли воду, и полученную в результате смесь нагревали до комнатной температуры, и экстрагировали 3 раз этилацетатом. Органический слой промывали водой, и затем насыщенным соляным раствором, и сушили, и затем растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (8,34 г).
(промежуточное соединение Z-29-1: LCMS m/z 307,14, 309,11 (MH+-Boc), время удерживания 1,64 минут, LC условия LC-1)
[0354]
Справочный пример Z-29-2: трет-бутил 3-(2-(5-(метоксикарбонил)тиофен-2-ил)этил)-2-оксо-1,3,4-оксадиазинан-4-карбоксилат (промежуточное соединение Z-29-2)
[формула 195]
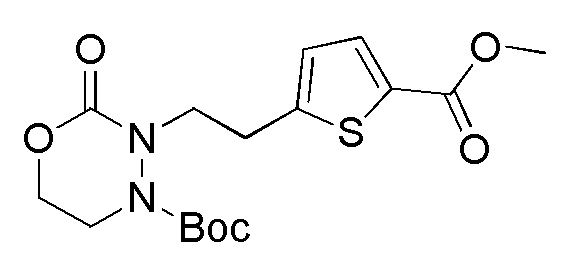
Раствор промежуточного соединения Z-29-1 (8,34 г) в DMF (140 мл) охлаждали до 0°C, добавляли порциями гидрид натрия (55%, 0,87 г) в течение 30 минут, и смесь перемешивали в течение 2 часов. Реакционную смесь нагревали до комнатной температуры, и перемешивали в течение 1 часа, затем добавляли к смеси снова гидрид натрия (55%, 0,1 г) при 0°C, и смесь перемешивали при комнатной температуре в течение 15 часов. Реакционную смесь охлаждали до 0°C, добавляли воду, и полученную в результате смесь экстрагировали 4 раз этилацетатом. Органический слой промывали дважды водой и один раз насыщенным соляным раствором, и сушили, и затем растворитель упаривали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (1,75 г).
(промежуточное соединение Z-29-2: LCMS m/z 371,3 (MH+), время удерживания 1,51 минут, LC условия LC-1)
[0355]
Справочный пример Z-29-3: Метил 5-(2-(2-оксо-1,3,4-оксадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-29-3)
[формула 196]
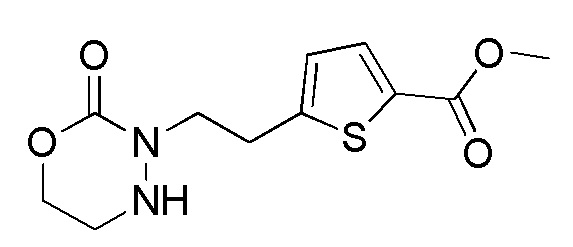
Раствор промежуточного соединения Z-29-2 (1,75 г) в дихлорметане (25 мл) охлаждали до 0°C, добавляли к раствору трифторуксусную кислоту (12,5 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь охлаждали до 0°C, нейтрализовали 5 M водным гидроксидом натрия, и затем экстрагировали 3 раза хлороформом. Органический слой промывали водой, и затем сушили, и растворитель упаривали при пониженном давлении. Остаток очищали короткой колонкой с силикагелем (хлороформ, затем этилацетат), и затем выпавший осадок промывали диэтиловым эфиром, получая заявленное в заголовке соединение (0,90 г).
(промежуточное соединение Z-29-3: LCMS m/z 271,2 (MH+), время удерживания 0,94 минут, LC условия LC-1)
[0356]
Справочный пример Z-29-4: Метил 5-(2-(4-(4-(3-йодфенил)-3-оксобутил)-2-оксо-1,3,4-оксадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-29-4)
[формула 197]

Раствор промежуточного соединения C-2-2 (250 мг) в DME (5 мл) охлаждали до 0°C в потоке азота, добавляли по каплям к раствору винилмагнийбромид (1 M раствор, 1,2 мл), и смесь перемешивали при той же температуре в течение 2 часов. Реакционную смесь подкисляли добавлением 2 M хлористоводородной кислоты, и затем добавляли к смеси воду, и полученную в результате смесь экстрагировали 3 раза этилацетатом. Органический слой промывали дважды водой, и сушили, и затем растворитель упаривали при пониженном давлении.
В колбу, содержащую промежуточное соединение Z-29-3 (110,7 мг) и воду (3,3 мл), добавляли раствор остатка, полученного выше, в этаноле (3,3 мл), и смесь перемешивали при внутренней температуре 110°C в течение 15 часов. Реакционную смесь нагревали до комнатной температуры, и затем выливали в воду, и полученную в результате смесь экстрагировали 3 раза этилацетатом. Органический слой промывали насыщенным соляным раствором, и сушили, и затем растворитель упаривали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией (гексан/этилацетат), получая заявленное в заголовке соединение (126,2 мг).
(промежуточное соединение Z-29-4: LCMS m/z 541,2 (MH+), время удерживания 1,66 минут, LC условия LC-1)
[0357]
Справочный пример Z-29-5: Метил 5-(2-(4-(3-гидрокси-4-(3-йодфенил)бутил)-2-оксо-1,3,4-оксадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-29-5)
[формула 198]
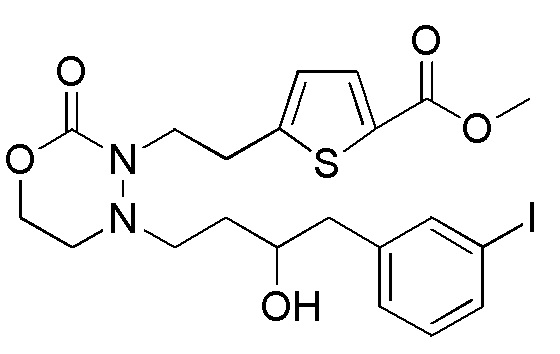
К раствору промежуточного соединения Z-29-4 (58,1 мг) в метаноле (1 мл) добавляли боргидрид натрия (6,1 мг), и смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли к смеси воду, и полученную в результате смесь экстрагировали 3 раза этилацетатом. Органический слой промывали насыщенным соляным раствором, и сушили, и затем растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (61,4 мг).
(промежуточное соединение Z-29-5: LCMS m/z 545,2 (MH+), время удерживания 1,60 минут, LC условия LC-1)
[0358]
Справочный пример Z-29-6: Метил 5-(2-(4-(3-ацетокси-4-(3-йодфенил)бутил)-2-оксо-1,3,4-оксадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-29-6)
[формула 199]
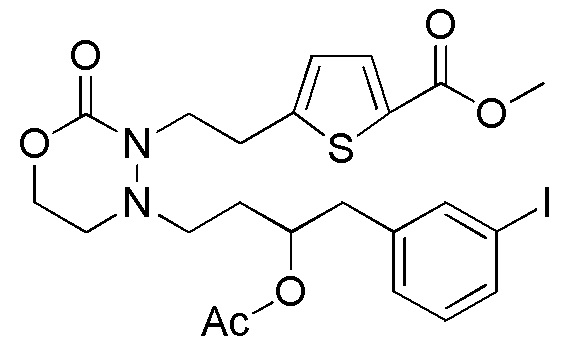
Промежуточное соединение Z-29-5 (61,4 мг) растворяли в дихлорметане (2 мл), добавляли к раствору уксусный ангидрид (0,27 мл) и пиридин (0,23 мл), и смесь перемешивали при комнатной температуре 5,5 часов. К реакционной смеси добавляли воду, и полученную в результате смесь экстрагировали 3 раза хлороформом. Органический слой промывали водой и сушили, и затем растворитель упаривали при пониженном давлении. Остаток очищали короткой колоночной хроматографией, получая заявленное в заголовке соединение (71,8 мг).
(промежуточное соединение Z-29-6: LCMS m/z 587,26 (MH+), время удерживания 1,82 минут, LC условия LC-1)
[0359]
Справочный пример Z-29-7: Метил 5-(2-(4-(3-ацетокси-4-(3-(тиофен-3-илэтинил)фенил)бутил)-2-оксо-1,3,4-оксадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-29-7)
[формула 200]
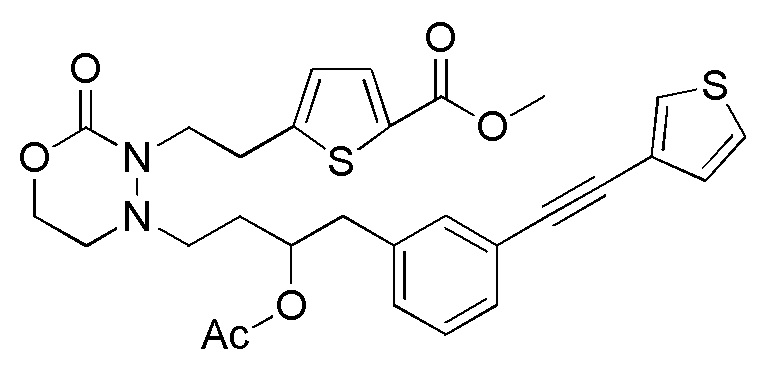
Получение проводили согласно способу, описанному в справочном примере Z-14, применяя промежуточное соединение Z-29-6 (71,8 мг) вместо промежуточного соединения Z-4-2, получая заявленное в заголовке соединение (66,4 мг).
(промежуточное соединение Z-29-7: LCMS m/z 567,36 (MH+), время удерживания 1,94 минут, LC условия LC-1)
[0360]
Пример 29: 5-(2-(4-(3-Гидрокси-4-(3-(тиофен-3-илэтинил)фенил)бутил)-2-оксо-1,3,4-оксадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 201]

Получение проводили согласно способу, описанному в примере 22, стадия b, применяя промежуточное соединение Z-29-7 (66,4 мг) вместо промежуточного соединения T-7, получая заявленное в заголовке соединение (45,6 мг).
(LCMS m/z 509,2 (MH+), время удерживания 1,53 минут, LC условия LC-1)
[0361]
Справочный пример D-2: Метил 4-(3-бромфенил)-3-оксобутаноат (промежуточное соединение D-2)
[формула 202]
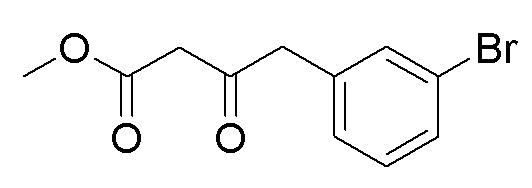
К раствору добавляли монометилмалоната калия (0,885 кг), THF (4,077 кг) и хлорид магния (0,47 кг), и смесь перемешивали при 50°C в течение 10 минут. К данной смеси, реакционную смесь, полученную добавлением раствора карбонилдимидазола (0,801 кг) в DMF (4,025 кг) к раствору 3-бромфенилуксусной кислоты (1,005 кг) в THF (2,023 кг), и перемешивали смесь при комнатной температуре в течение 1 часа. Дополнительно добавляли THF (0,508 кг) к реакционной смеси, и полученную в результате смесь перемешивали при 50°C в течение 30 минут. Реакционную смесь охлаждали до комнатной температуры, добавляли к смеси этилацетат (8,071 кг), органический слой промывали дважды 20% водной лимонной кислотой (6,032 кг), и затем растворитель упаривали при пониженном давлении, получая концентрат (2,358 кг). К данному концентрату добавляли этилацетат (1,011 кг), получая раствор, содержащий промежуточное соединение D-2 (3,369 кг). Раствор, содержащий промежуточное соединение D-2, смешивали с раствором, полученным аналогичным способом. К данной смеси (6,770 кг) добавляли этилацетат (6,063 кг), и дополнительно добавляли 5% водный гидрокарбонат натрия (10,055 кг) хлорид и натрия (0,506 кг), промывая органический слой. Добавляли 5% водный гидрокарбонат натрия (10,054 кг) и хлорид натрия (0,503 кг), дополнительно промывая органический слой. Органический слой дополнительно промывали 20% водным хлоридом натрия (10,064 кг), добавляли этилацетат (1,01 кг), и затем растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (2,52 кг).
(LCMS m/z 268,9, 270,9 (MH-), время удерживания 1,44 минут, LC условия NLC-1)
[0362]
Справочный пример D-3: Метил (S)-4-(3-бромфенил)-3-гидроксибутаноат (промежуточное соединение D-3)
[формула 203]
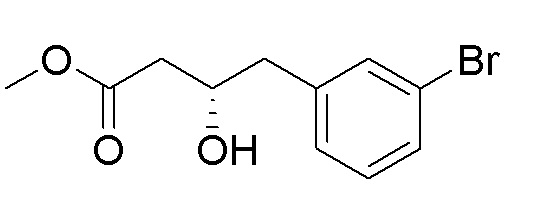
К промежуточному соединению D-2 (2,52 кг) добавляли метанол (14,325 кг), воду (0,237 кг), и [NH2Me2][(RuCl((S)-dm-segphos))2(u-Cl)3] (82,46 г, TAKASAGO), смесь перемешивали при 60°C в течение 6 часов в атмосфере водорода, и затем растворитель упаривали при пониженном давлении. Добавляли к остатку толуол (20,70 кг) и QuadraSil AP (0,60 кг, JOHNSON), смесь перемешивали при 60°C в течение 1 часа, затем реакционную смесь фильтровали, и остаток на фильтре промывали толуолом (2,066 кг). Растворитель фильтрата упаривали при пониженном давлении, получая концентрат (5,68 кг), и затем, к концентрату добавляли по каплям н-гептан (1,22 кг) при 10°C в течение 15 минут, и н-гептан (1,22 кг) дополнительно добавляли по каплям при 5°C в течение 50 минут. Реакционную смесь перемешивали как есть при 5°C в течение 45 минут, и затем фильтровали. Остаток на фильтре промывали смесью н-гептана (0,48 кг) и толуола (0,24 кг), и сушили, получая заявленное в заголовке соединение (1,923 кг).
(LCMS m/z 273,0, 275,1 (MH+), время удерживания 1,36 минут, LC условия NLC-1)
(Хиральная LC: время удерживания 21,1 минут, LC условия Хиральная LC-1)
[0363]
Справочный пример D-35: Метил (S)-3-гидрокси-4-(3-(тиофен-3-илэтинил)фенил)бутаноат (промежуточное соединение D-35)
[формула 204]
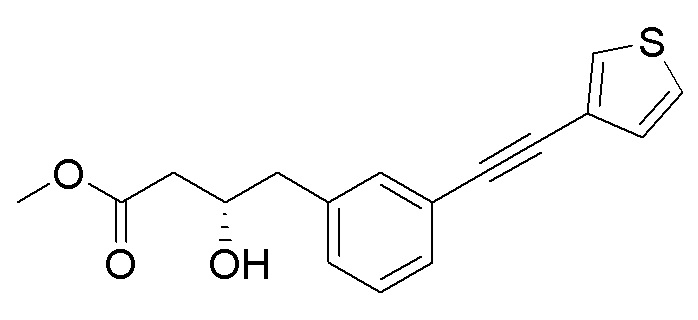
К промежуточному соединению D-3 (558 г) добавляли ацетонитрил (1386 г), карбонат цезия (665,7 г), X-Phos (48,7 г, Nippon Kagaku) и дихлорид бис(ацетонитрил)палладия(II) (13,28 г, TCI), и раствор 3-этинилтиофена (287,3 г) в ацетонитриле (277,2 г) добавляли по каплям в течение 30 минут в атмосфере азота. Реакционную смесь перемешивали при 40°C в течение 30 минут, и затем при 60°C в течение 90 минут. Реакционную смесь охлаждали до комнатной температуры, добавляли к смеси толуол (1208,3 г) и воду (3488,2 г), и полученную в результате смесь перемешивали в течение 20 минут, и затем фильтровали через целит. Остаток на фильтре промывали толуолом (2416,3 г), фильтрат перемешивали в течение 10 минут, и затем оставляли до того, как он не разделялся на органический слой и водный слой, и водный слой удаляли. Органический слой промывали водой (2232,3 г), и затем растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (753,9 г).
(LCMS m/z 301,2 (MH+), время удерживания 1,65 минут, LC условия NLC-1)
[0364]
Справочный пример D-50: Метил (S)-3-((2-метоксиэтокси)метокси)-4-(3-(тиофен-3-илэтинил)фенил)бутаноат (промежуточное соединение D-50)
[формула 205]
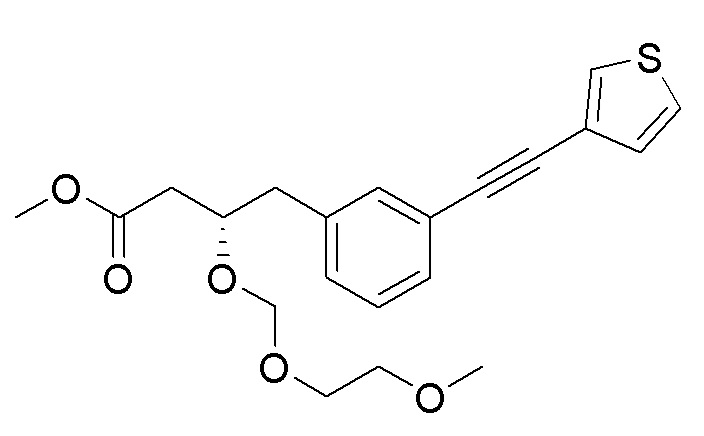
К промежуточному соединению D-35 (753,5 г) добавляли в атмосфере азота толуол (2588,1 г), диизопропилэтиламин (334,3 г), и 2-метоксиэтоксиметилхлорид (322,2 г), и смесь перемешивали при 80°C в течение 150 минут. Реакционную смесь охлаждали, добавляли к смеси воду (3107,7 г), и полученную в результате смесь перемешивали в течение 10 минут, и затем фильтровали. Остаток на фильтре промывали толуолом (1031,6 г), фильтрат оставляли до того, как он не разделялся на органический слой и водный слой, и водный слой удаляли. Органический слой промывали водой (2071,9 г), и затем растворитель упаривали при пониженном давлении, получая маслянистое вещество, содержащее промежуточное соединение D-50 (915,8 г). К данному маслянистому веществу, содержащему промежуточное соединение D-50 (915,4 г) добавляли метанол (2310,7 г) и активированный уголь Shirasagi A (292,3 г, Japan EnviroChemicals), и смесь перемешивали при комнатной температуре в течение 1 часа, и затем фильтровали через фильтровальную бумагу. Остаток на фильтре промывали метанолом (928,8 г), затем фильтрат фильтровали через мембранный фильтр, имеющий диаметр пор 0,5 мкм, и остаток на фильтре промывали метанолом (464,2 г). Фильтрат концентрировали, добавляли к концентрату толуол (2479,1 г) и QuadraSil MTU (349,6 г, JOHNSON), и смесь перемешивали при 40°C в течение 1 часа, и затем фильтровали через фильтровальную бумагу. Остаток на фильтре промывали толуолом (1008,0 г), затем фильтрат фильтровали через мембранный фильтр, имеющий диаметр пор 0,5 мкм, и остаток на фильтре промывали толуолом (503,8 г). Фильтрат концентрировали при пониженном давлении, получая заявленное в заголовке соединение (710,9 г).
(LCMS m/z 406,2 (M+NH4+), время удерживания 1,88 минут, LC условия NLC-1)
[0365]
Справочный пример D-60: (S)-3-((2-Метоксиэтокси)метокси)-4-(3-(тиофен-3-илэтинил)фенил)бутаналь (промежуточное соединение D-60)
[формула 206]

К промежуточному соединению D-50 (355,3 г) добавляли в атмосфере азота толуол (2740,1 г), и смесь охлаждали до -83°C. К данной смеси добавляли по каплям 1 моль/л раствор диизобутилалюмогидрида в толуоле (571,6 г) в течение 90 минут. К реакционной смеси добавляли 1 моль/л хлористоводородную кислоту (1884,1 г), смесь перемешивали при комнатной температуре в течение 1 часа, добавляли к смеси толуол (64,3 г), полученную в результате смесь перемешивали в течение 10 минут, и затем оставляли до того, как она не разделялась на органический слой и водный слой, и водный слой удаляли. Органический слой последовательно промывали 1 моль/л хлористоводородной кислотой (1004,9 г), 1% водным гидрокарбонатом натрия (1997,8 г) и водой (1978,1 г), и затем фильтровали через мембранный фильтр, имеющий диаметр пор 0,5 мкм. Остаток на фильтре промывали толуолом (214,3 г), получая толуольный раствор (4061,0 г), содержащий заявленное в заголовке соединение (248,5 г).
(LCMS m/z 376,2 (M+NH4+), время удерживания 1,80 минут, LC условия NLC-1)
[0366]
Справочный пример Z-70: Метил (S)-5-(2-(4-(3-((2-метоксиэтокси)метокси)-4-(3-(тиофен-3-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-70)
[формула 207]
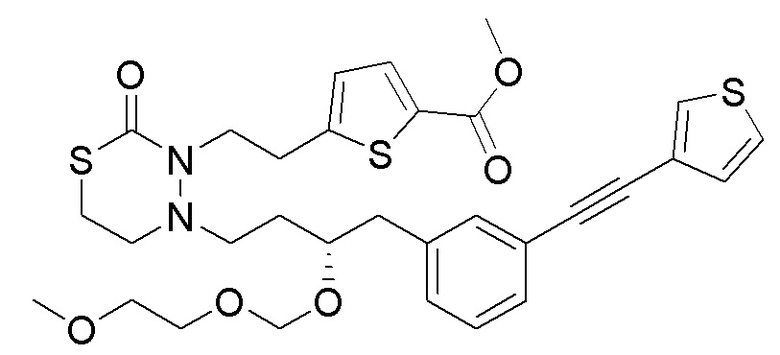
К промежуточному соединению Z-3 (286,3 г) добавляли в атмосфере азота толуол (1959,6 г), уксусную кислоту (474,5 г) и триацетоксиборгидрид натрия (498,5 г). Растворитель раствора в толуоле (7927,0 г), содержащий промежуточное соединение D-60 (460,28 г), упаривали при пониженном давлении, получая концентрированный раствор промежуточного соединения D-60 (1503,9 г), и данный раствор добавляли к реакционной смеси при комнатной температуре в течение 50 минут. Добавляли к реакционной смеси толуол (245 г), и полученную в результате смесь перемешивали в течение 2 часов, и затем последовательно промывали 5% водным гидрокарбонатом натрия (7326,6 г), 5% водным гидрокарбонатом натрия (7327,2 г) и водой (7071,4 г). Растворитель упаривали при пониженном давлении, получая заявленное в заголовке соединение (903,8 г).
(LCMS m/z 629,29 (MH+), время удерживания 2,05 минут, LC условия NLC-1)
[0367]
Справочный пример Z-17-S: Метил (S)-5-(2-(4-(3-гидрокси-4-(3-(тиофен-3-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоксилат (промежуточное соединение Z-17-S)
[формула 208]
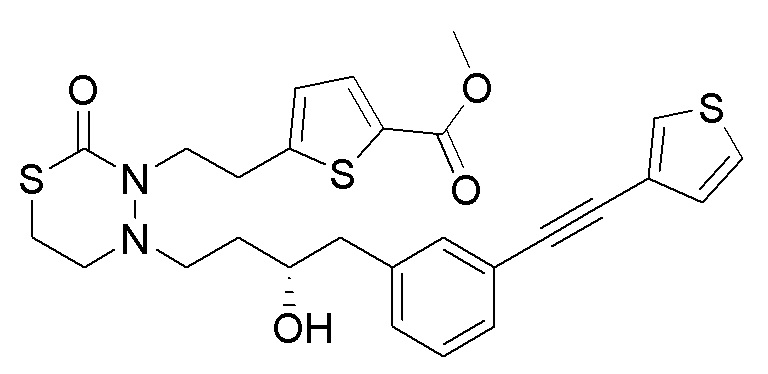
К промежуточному соединению Z-70 (903,5 г) добавляли метанол (4293,5 г) и активированный уголь Shirasagi A (90,35 г, Japan EnviroChemicals), реакционную смесь перемешивали при 40°C в течение 1 часа, и затем фильтровали, и остаток на фильтре промывали метанолом (715,1 г). К фильтрату добавляли 36% хлористоводородную кислоту (588,6 г), смесь перемешивали при 40°C в течение 6 часов, затем добавляли к смеси толуол (2406,8 г), и полученную в результате смесь охлаждали до комнатной температуры. Реакционная смесь последовательно промывали 5% водным гидрокарбонатом натрия (9765,4 г) и водой (1685,1 г), и затем растворитель упаривали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией (н-гептан/этилацетат), получая заявленное в заголовке соединение (471,6 г).
(LCMS m/z 541,19 (MH+), время удерживания 1,87 минут, LC условия NLC-1)
[0368]
Пример 30: (S)-5-(2-(4-(3-Гидрокси-4-(3-(тиофен-3-илэтинил)фенил)бутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)тиофен-2-карбоновая кислота
[формула 209]
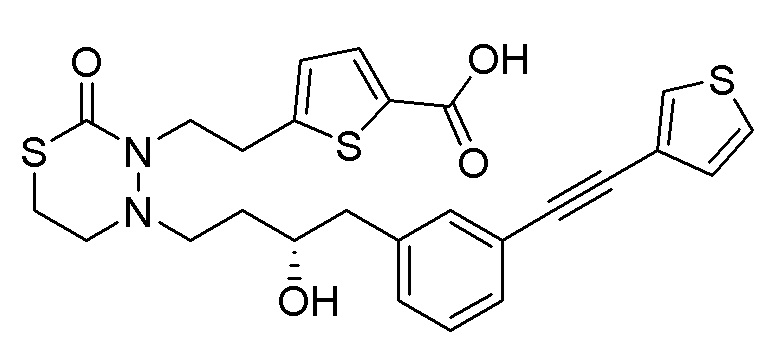
К промежуточному соединению Z-17-S (469,5 г) добавляли THF (2393,3 г) и активированный уголь Shirasagi A (94,0 г, Japan EnviroChemicals), реакционную смесь перемешивали при комнатной температуре в течение 1 часа, и затем фильтровали, и остаток на фильтре промывали THF (1597,1 г). К фильтрату добавляли THF (378,9 г), метанол (1421,8 г) и 1 моль/л водный гидроксид натрия (1728,2 г), и смесь перемешивали в течение 17 часов. К реакционной смеси добавляли толуол (2331,5 г) и воду (1346,5 г), смесь перемешивали, и оставляли до того, как она не разделялась на органический слой и водный слой, и органический слой удаляли. К водному слою добавляли смесь толуола (2332 г), THF (1596 г) и метанола (710 г), полученную в результате смесь перемешивали, и оставляли до того, как она не разделялась на органический слой и водный слой, и органический слой удаляли. Затем, промывку смесью толуола, THF и метанола осуществляли 3 раза аналогичным способом. К водному слою добавляли толуол (4663,3 г), смесь перемешивали, и оставляли до того, как она не разделялась на органический слой и водный слой, и органический слой удаляли. К водному слою добавляли 1 моль/л хлористоводородную кислоту до того, как pH смеси становилась 7,0, затем добавляли этилацетат (2425,9 г), дополнительно добавляли 1 моль/л хлористоводородную кислоту до того, как pH смеси становилась 2,2, смесь перемешивали в течение 10 минут, и затем оставляли до того, как она не разделялась на органический слой и водный слой, и водный слой удаляли. К органическому слою добавляли этилацетат (1347,1 г) и воду (897,5 г), смесь перемешивали в течение 10 минут, и затем оставляли до того, как она не разделялась на органический слой и водный слой, и водный слой удаляли. Растворитель органического слоя упаривали при пониженном давлении, получая остаток, содержащий заявленное в заголовке соединение (358,6 г). К данному остатку, содержащему заявленное в заголовке соединение (358,3 г), добавляли метанол (1860,0 г) и активированный уголь Shirasagi A (47,6 г, Japan EnviroChemicals), смесь перемешивали при комнатной температуре в течение 1 часа, и затем фильтровали через мембранный фильтр, имеющий диаметр пор 0,5 мкм, и остаток на фильтре промывали метанолом (2231,2 г). К фильтрату добавляли активированный уголь Shirasagi A (188,6 г, Japan EnviroChemicals), смесь перемешивали при комнатной температуре в течение 1 часа, и затем фильтровали через фильтровальную бумагу, и остаток на фильтре промывали метанолом (1490,5 г). Фильтрат фильтровали через мембранный фильтр, имеющий диаметр пор 0,2 мкм, и остаток на фильтре промывали метанолом (743,7 г). К фильтрату добавляли активированный уголь Shirasagi A (94,0 г, Japan EnviroChemicals), смесь перемешивали при комнатной температуре в течение 1 часа, и затем фильтровали через мембранный фильтр, имеющий диаметр пор 0,2 мкм, и остаток на фильтре промывали метанолом (2231,2 г). К фильтрату добавляли активированный уголь Shirasagi A (94,1 г, Japan EnviroChemicals), смесь перемешивали при комнатной температуре в течение 1 часа, и затем фильтровали через мембранный фильтр, имеющий диаметр пор 0,2 мкм, и остаток на фильтре промывали метанолом (742,9 г). Растворитель фильтрата упаривали при пониженном давлении, получая заявленное в заголовке соединение (160,1 г).
(LCMS m/z 527,2 (MH+), время удерживания 1,26 минут, LC условия NLC-1)
(Хиральная LC: время удерживания: 21,3 минут, LC условия хиральная LC-2)
Соединение примера 30 является таким же, как соединение примера 23.
[0369]
Сравнительный пример 1: 4-(2-(4-(4-(3-Бромфенил)-3-гидроксибутил)-2-оксо-1,3,4-тиадиазинан-3-ил)этил)бензойная кислота
[формула 210]
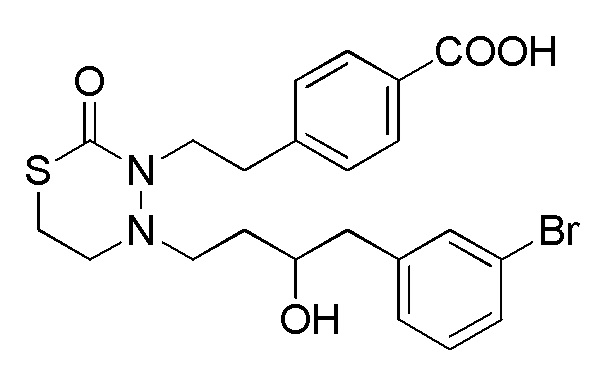
Заявленное в заголовке соединение можно получить способом получения, описанным в международной патентной публикации WO2006/080323 (патентный документ 8), Пример IAH-H072.
[0370]
Пример получения 1
Дихлорметан (20 мл) добавляли к поли(молочоной-со-гликолевой кислоте)) (RESOMER RG504, полученной Evonik Industries, 2,0 г), которую растворяли, применяя машину для ультразвуковой очистки, и соединение примера 23 (1,6 мг), дополнительно добавляли к раствору, и растворяли. Данный раствор добавляли порциями к 0,1% водному раствору поливинилового спирта (300 мл), перемешиваемому при 3000 об/мин, применяя гомогенизатор (MARK II, полученный PRIMIX Corporation), и смесь перемешивали при комнатной температуре в течение 10 минут, получая эмульсию масла в воде. Данную эмульсию масла в воде перемешивали при комнатной температуре в течение 16 часов, дихлорметан упаривали для отверждения масляной фазы, и полученный в результате остаток центрифугировали (3000 об/мин, 20°C, 15 минут), применяя центрифугу. После удаления надосадочной жидкости, осадок диспергировали в 0,1% (вес/об) растворе Tween 80, дисперсию пропускали через сита 53 мкм и 20 мкм, и 20 мкм образец, оставшийся на ситах, центрифугировали (3000 об/мин, 20°C, 15 минут). Надосадочную жидкость удаляли, добавляли к остатку очищенную воду, смесь снова центрифугировали (3000 об/мин, 20°C, 15 минут), и надосадочную жидкость удаляли. Осадок замораживали при -80°C, и сушили при пониженном давлении (48 часов), получая микросферы, содержащие лекарственное средство (1,2 г) имеющие долю захваченного лекарственного средства 0,06%.
[0371]
Пример получения 2
Дихлорметан (20 мл) добавляли к поли(молочоной-со-гликолевой кислоте)) (RESOMER RG504, полученной Evonik Industries, 2,0 г), которую растворяли, применяя машину для ультразвуковой очистки, и соединение примера 23 (20 мг) дополнительно добавляли к раствору, и растворяли. Данный раствор добавляли порциями к 0,1% водному раствору поливинилового спирта (300 мл), перемешиваемому при 3000 об/мин, применяя гомогенизатор (MARK II, полученный PRIMIX Corporation), и смесь перемешивали при комнатной температуре в течение 10 минут, получая эмульсию масла в воде. Данную эмульсию масла в воде перемешивали при комнатной температуре в течение 16 часов, дихлорметан упаривали для отверждения масляной фазы, и полученный в результате остаток центрифугировали (3000 об/мин, 20°C, 15 минут), применяя центрифугу. После удаления надосадочной жидкости, осадок диспергировали в 0,1% (вес/об) растворе Tween 80, дисперсию пропускали через сита 53 мкм и 20 мкм, и 20 мкм образец, оставшийся на ситах, центрифугировали (3000 об/мин, 20°C, 15 минут). Надосадочную жидкость удаляли, затем добавляли к остатку очищенную воду, смесь снова центрифугировали (3000 об/мин, 20°C, 15 минут), и надосадочную жидкость удаляли. Осадок замораживали при -80°C и сушили при пониженном давлении (48 часов), получая микросферы, содержащие лекарственное средство (1,3 г), имеющие долю захваченного лекарственного средства 0,8%.
[0372]
Пример получения 3
Дихлорметан (20 мл) добавляли к поли(молочной-со-гликолевой кислоте)) (RESOMER RG504, полученной Evonik Industries, 2,0 г), которую растворяли, применяя машину для ультразвуковой очистки, и соединение примера 23 (124 мг), дополнительно добавляли к раствору, и растворяли. Данный раствор добавляли порциями к 0,1% водному раствору поливинилового спирта (300 мл), перемешиваемому при 3000 об/мин, применяя гомогенизатор (MARK II, полученную PRIMIX Corporation), и смесь перемешивали при комнатной температуре в течение 10 минут, получая эмульсию масла в воде. Данную эмульсию масла в воде перемешивали при комнатной температуре в течение 16 часов, дихлорметан упаривали для отверждения масляной фазы, и полученный в результате остаток центрифугировали (3000 об/мин, 20°C, 15 минут), применяя центрифугу. После удаления надосадочной жидкости, осадок диспергировали в 0,1% (вес/об) растворе Tween 80, дисперсию пропускали через сита 53 мкм и 20 мкм, и 20 мкм образец, оставшийся на ситах, центрифугировали (3000 об/мин, 20°C, 15 минут). Надосадочную жидкость удаляли, затем добавляли к остатку очищенную воду, смесь снова центрифугировали (3000 об/мин, 20°C, 15 минут), и надосадочную жидкость удаляли. Осадок замораживали при -80°C и сушили при пониженном давлении (48 часов), получая микросферы, содержащие лекарственное средство (1,1 г), имеющие долю захваченного лекарственного средства 3,7%.
[0373]
<Пример испытаний 1: измерение EP4 агонистической активности>
Для того чтобы исследовать EP4 рецепторную агонистическую активность соединений настоящего изобретения, синтез цАМФ измеряли в HEK293 клетках, которые делали стабильно экспрессирующими человеческий EP4 рецептор.
(1) Способ измерения
Применяя базу данных Refseq, проводили поиск рецептора простагландина E. Как результат, получали информацию о гене человеческого EP4 (NM_000958) рецептора. На основе данной информации о последовательности, ген человеческого EP4 рецептора клонировали ПЦР общепринятым способом, применяя человеческую кДНК в качестве матрицы, и получали HEK293 клетки, которые делали стабильно экспрессирующими человеческий EP4 рецептор. Когда криоконсервированные клетки данного штамма размораживали и применяли, применяли клетки, суспендированные три раз или более в течение некоторого периода времени (приблизительно 1-2 недели), применяя среду Игла в модификации Дульбекко (среда Игла в модификации Дульбекко можно сократить далее до DMEM), содержащую 10% FBS, 50 единиц каждого из пенициллина и стрептомицина. Субкультивированные клетки инокулировали в лунках 96-луночного планшета, покрытых поли-D-лизином, при плотности 2×104-2,5×104 клеток/лунка, и культивировали в течение одного дня. Среду в лунках удаляли отсасыванием, затем DMEM (80 мкл) добавляли к каждой лунке, и инкубирование проводили при 37°C в течение 15 минут. Затем, 20 мкл среды для анализа (DMEM, содержащая 100 мМ HEPES и 1 мМ IBMX), содержащей PGE2 или испытуемое соединение (при концентрации в 5 раз большей, чем конечная концентрация) добавляли к каждой лунке, начиная реакцию. Реакцию проводили при 37°C в течение 30 минут, затем среду удаляли отсасыванием. Буфер для анализа/лизиса(100 мкл), содержащийся в цАМФ скрининговом наборе (полученным Applied Biosystems), добавляли для прекращения реакции. Затем, реакционная смесь инкубировали при 37°C в течение 30 минут, получая образец для количественного определения цАМФ, и количество цФМФ в образце определяли согласно способу, указанному в цАМФ скрининговом наборе. Нелинейной регрессией концентрации соединения и количества цАМФ рассчитывали концентрацию соединения, необходимую для увеличения цАМФ до 50% от максимального увеличения (значение ЕС50), применяя the Kaleida Graph.
[0374]
(2) Результаты измерения
Как показано в таблице 1, соединения настоящего изобретения показали превосходную EP4 агонистическую активность. В частности, они показали превосходную EP4 агонистическая активность даже по сравнению с агонистической активностью соединения сравнительного примера 1, которое представляет собой известное соединение, аналогичное соединениям настоящего изобретения.
Для соединений, для которых EP4 агонистическую активность измеряли два или более раз, показаны средние величины, при необходимости. Пример No., приведенный в таблице, обозначает номер примера.
[0375]
[Таблица 1]
[0376]
<Пример испытаний 2: Тест на связывание рецептора, применяя клетки, экспрессирующие человеческий EP рецептор>
Для того чтобы оценить селективность к каждому подтипу EP рецептора, измеряли активности по ингибированию связывания [3H]PGE2 испытуемых соединений для клеточных мембран, на которых стабильно экспрессируются человеческий EP2, человеческий EP3 и человеческий EP4 рецепторы.
(1) Способ измерения
В качестве мембранных фракций рецепторов простагландина E EP2, EP3 и EP4, применяли 10,0 мкг белка/пробирка каждого из HTS185M, HTS092M и HTS142M Merck Millipore, соответственно. Каждую мембранную фракцию инкубировали с реакционной смесью (250 мкл/пробирка), содержащей испытуемое соединение и [3H]PGE2 при 25°C в течение 60 минут. Конечные концентрации [3H]PGE2 были 2,56 нмоль/л в EP2 системе измерения, 1,54 нмоль/л в EP3 системе измерения и 1,24 нмоль/л в EP4 системе измерения. После реакции, мембранную фракцию собирали на фильтровальной бумаге, применяя коллектор клеток, фильтровальную бумагу переносили в пробирку для измерения, и измерение проводили на жидкостном сцинтилляционном счетчике.
[0377]
Неспецифическое связывание определяли в виде связывания, наблюдаемого в присутствии избыточного количества (10 мкм) немеченого PGE2. Измерение активности по ингибированию связывания [3H]PGE2 испытуемого соединения проводили добавлением испытуемого соединения при различных концентрациях. Следующие буферы применяли для реакции.
Буфер для EP2: 50 ммоль/л HEPES-NaOH (pH 7,4), содержащий 5 ммоль/л MgCl2, 1 ммоль/л CaCl2 и 0,2% BSA
Буфер для EP3: 50 ммоль/л Tris-HCl буфер, содержащий 10 ммоль/л MgCl2 и 1 ммоль/л EDTA
Буфер для EP4: 50 ммоль/л HEPES-NaOH (pH7,4), содержащий 5 ммоль/л MgCl2, 1 ммоль/л CaCl2 и 0,2% BSA
Кривая доза-эффект для активности по ингибированию связывания [3H]PGE2 была построена для каждого испытуемого соединения, и рассчитывали концентрацию испытуемого соединения, которое ингибирует 50% связывания PGE2 и рецептора (значение IC50).
[0378]
(2) Результаты измерения
Как показано в таблице 2, соединения настоящего изобретения показали превосходную EP4 селективность. Пример No., приведенный в таблице, обозначает номер примера.
[0379]
[Таблица 2]
[0380]
<Пример испытаний 3: Неоостеогенезное действие на бедренную кость крысы>
Для того чтобы исследовать действие, стимулирующее остеогенез, соединений настоящего изобретения, соединения действовали на бедренные кости крыс, и оценивали образовавшуюся новую кость.
(1) Способ измерения
Восьминедельных женских особей SD крыс (Charles River Japan) фиксировали в положении лежа на боку под тройной анестезией (гидрохлорид медетомидина, мидазолам и тартрат буторфанола). После того, как волосы левой бедренной области выбривали с помощью машинки для стрижки волос и проводили дезинфекционную обработку 70% этанолом, отвержденные in situ гелевые растворы испытуемого соединения, конкретно, испытуемого соединения, растворенного в блочном сополимере поли(молочной-со-гликолевой кислоты) (RESOMER RG502H, полученной Evonik Industries)/поли(молочной-со-гликолевой кислоты)-полиэтиленгликоля (5050 DLG mPEG 5000, полученной Lakeshore Biomaterials)/N-метил-2-пирролидона (полученный Wako) (весовое соотношение 47%/3%/50%), заполняли в 1-мл шприц, иглу для инъекций 21G прикрепляли к шприцу, и иглу трансдермально вводили ударом от четырехглавой мышцы к надкостнице вблизи центра бедренного диафиза. Затем, 100 мкг в виде количества испытуемого соединения или 50 мкл в виде объема для введения раствора вводили между четырехглавой мышцей и надкостницей, и иглу для инъекции вытягивали. Крысам контрольной группы вводили только вышеупомянутый раствор отвержденного in situ гелевого раствора. Через одну неделю после введения раствора лекарственного средства, животным давали тройную анестезию, и их фиксировали в положении лежа на спине и усыпляли кровопусканием. Извлекали левое бедро, удаляли периферические ткани, такие как мышцы, измеряли содержание минеральных веществ в костях всей бедренной кости, применяли анализатор минералов кости DCS-600EX (производства ALOKA), затем его разделяли на три части вдоль длинной оси, и оценивали содержание минеральных веществ кости центральной части (диафиз). Тест проводили с группами, каждая из которых состояла из 6 животных
[0381]
(2) Результаты измерения
В группах, которым вводили типичные соединения настоящего изобретения, содержание минеральных веществ в костях диафиза левой бедренной кости увеличилось по сравнению с контрольной группой (таблицы 3-5). С другой стороны, содержание минеральных веществ в костях диафиза правой бедренной кости, в которые не вводили раствор препарата, не изменялось. На основании данных результатов было подтверждено, что соединения настоящего изобретения являются пригодными в качестве агента, способствующего образованию костной ткани, применяемого местным введением. Во всех группах введения соединения гибель животных не наблюдали, побочных реакций, наблюдаемых при введении PGE2, также не наблюдали, и, таким образом, было продемонстрировано, что соединения настоящего изобретения можно вводить безопасно.
Результаты тестирования приводятся для каждого теста.
[0382]
[таблица 3]
[0383]
[таблица 4]
[0384]
[таблица 5]
[0385]
<Пример испытаний 4: Неоостеогенезное действие на бедренную кость собаки>
Для того чтобы исследовать действие, стимулирующее остеогенез, соединений настоящего изобретения, при введении микросфер, содержащих испытуемое соединение, вблизи бедра собаки, оценивали действие, стимулирующее остеогенез, измерением новой кости, образовавшейся после введения.
(1) Способ измерения
Женских особей собак породы бигля в возрасте от девяти до одиннадцати месяцев (KITAYAMA LABES) анестезировали введением 1:1 смеси гидрохлорида кетамина (Кеталар 500 mg, Daiichi Sankyo Propharma Co., Ltd.) и ксилазина (2% инъекция селактара, Bayer Yakuhin, Ltd.) при дозе приблизительно 0,5 мл/кг, и изофлуран, перечисленный в японской фармакопее (Elucaine, Mylan Pharmaceutical), применяли для поддержания анестезии, который вводили ингалятором IMPAC6 (VetEquip Inc.). После того, как волосы бедренной области правой задней ноги сбривали и проводили дезинфекционную обработку, 350 мкл микросферной суспензии, полученной суспендированием микросфер (приготовленных согласно способу получения 1 или 2), содержащих испытуемое соединение (соединение примера 23) в растворе КМЦ, трансдермально вводили вокруг надкостницы диафиза бедренной кости, применяя инъекционный шприц объемом 1 мл и инъекционную иглу 21G. Дозы испытуемого соединения составляли 0,01, 0,1, 1,0, 10 и 100 мкг/место, и вышеупомянутые микросферы применяли в количестве, соответствующем каждой дозе. В качестве контрольной группы вводили лекарственную жидкость, полученную суспендированием микросфер, не содержащих испытуемое соединение, только в 350 мкл раствора КМЦ. Через четыре недели после введения лекарственной жидкости, животных умерщвляли кровопусканием под анестезией пентобарбиталом натрия (сомнопентил). Бедренные кости обеих сторон отделяли, погружали в 10% нейтральный забуференный формалиновый раствор и хранили. Содержание минеральных веществ в бедренной кости измеряли, применяя рентгеновский анализатор Discovery для определения плотности костей (производства Toyo Medic). Тест проводили с группами, каждая из которых состояла из 4 собак.
[0386]
(2) Результаты измерения
Через четыре недели после введения вышеупомянутой микросферной суспензии в бедренные диафизы собак, содержание минеральных веществ в бедрах увеличивалось с дозами 1,0, 10 и 100 мкг в виде введенного количества испытуемого соединения дозозависимым способом по сравнению с контрольной группой (таблица 6). На основании данных результатов было подтверждено, что соединения настоящего изобретения являются пригодными в качестве агента, стимулирующего остеогенез, применяемого местным введением. Для всех групп введения соединения гибель животных не наблюдали, побочные реакции, обычно наблюдаемые при введении PGE2, также не наблюдали, и, таким образом, было продемонстрировано, что вышеупомянутый препарат микросфер, содержащий соединение настоящего изобретения, можно вводить безопасно.
[0387]
[таблица 6]
[0388]
<Пример испытаний 5: действие, способствующее заживлению перелома, в модели закрытого перелома бедренной кости крыс>
Для того чтобы исследовать действие, способствующее заживлению перелома, соединений настоящего изобретения, подтверждали влияние инъекции микросфер, содержащих испытуемое соединение, в место перелома в модели закрытого перелома бедренной кости крыс.
(1) Способ измерения
Тринадцатинедельных женских особей SD крыс (Japan SLC) фиксировали в положении лежа на боку под тройной анестезией (гидрохлорид медетомидина, мидазолам и тартрат буторфанола). После того, как выбривали волосы области от левого колена до бедренной области с помощью машинки для стрижки волос, и проводили дезинфекционную обработку повидоном-йодом, указанным в японской фармакопее (раствор изодина для животных, 20 мг повидона-йода, перечисленного в японской фармакопее в 1 мл, Meiji Seika Pharma), кожу коленной части и медиальной большой мышцы боковой части надколенника удаляли, и надколенник смещали от головки бедра. Сверло, прикрепленное к трепану, помещали на межмыщелковую ямку открытой головки бедренной кости и вращали вручную, делая отверстие. Проволоку Киршнера (Mizuho Co., Ltd.), имеющую диаметр 1,2 мм, предварительно разрезанную на длину 31 мм, вводили в медуллярную полость бедренной кости через отверстие. Затем, левую бедренную область фиксировали на фиксаторе для испытания на трехточечный изгиб небольшой настольной универсальной испытательной машине (EZ Test, Shimadzu Corp.), а закрытый перелом диафиза бедренной кости вызывали динамической нагрузкой. Был ли перелом успешно создан или нет, подтверждали подтверждением генерации полного поперечного перелома бедренного диафиза на рентгеновском изображении, полученном с помощью мягкого рентгеновского генератора (M-100W, Softex) и цифрового рентгеновского датчика (NX-04, RF Co., Ltd.). Микросферную суспензию, полученную суспендированием микросфер (полученных согласно способу получения 3), содержащую испытуемое соединение (соединение примера 23) в растворе КМЦ, вводили в часть перелома в объеме 100 мкл в качестве объема для введения (содержащего 100 или 300 мкг испытуемого соединения). В качестве контрольной группы вводили лекарственную жидкость, полученную суспендированием микросфер, не содержащих вышеупомянутое испытуемое соединение, только в растворе КМЦ (100 мкл). Через одну, две и три недели после генерации перелома мягкие рентгеновские снимки делали под изофлурановой анестезией, и площади каллусной части на рентгеновских снимках определяли количественно, применяя Image J. Через четыре недели после генерации перелома мягкие рентгеновские снимки делали под тройной анестезией, животных фиксировали в положении лежа на спине, усыпляли кровопусканием и извлекали левые бедренные кости. По подтверждению наличия или отсутствия непрерывности каллуса слепым способом по мягким рентгеновским снимкам, полученным через 4 недели после генерации перелома, определяли сращивание костей. Образцы бедренной кости подвергали криоконсервации до проведения испытания на прочность кости, и прочность на кручение измеряли в день испытания, применяя прибор для измерения прочности костей (MZ-500S, Maruto Instrument Co., Ltd.).
Данный тест можно также проводить, применяя собак вместо крыс.
[0389]
(2) Результаты измерения
В группе, которой вводили испытуемое соединение, через две недели после генерации перелома и далее наблюдали увеличение площади каллуса (таблица 7), и степень сращивания кости, определенная на основе рентгеновских изображений, полученных через 25 дней после генерации перелома, было явно увеличена (таблица 8) по сравнению с контрольной группой. Для бедер, выделенных у животных из группы введения испытуемого соединения через четыре недели после генерации перелома, наблюдали увеличение прочности костей, определяемой тестом на кручение (максимальный крутящий момент) по сравнению с контрольной группой (таблица 8). На основании данных результатов подтверждали, что соединения настоящего изобретения являются пригодными в качестве агента, способствующего сращиванию костей, в процессе заживления перелома. Для всех групп введения соединения гибель животных не наблюдали, побочные реакции, обычно наблюдаемые при введении PGE2, также не наблюдали, и, таким образом, было продемонстрировано, что вышеупомянутый препарат микросфер, содержащий соединение настоящего изобретения, можно вводить безопасно.
[0390]
[таблица 7]
[0391]
[таблица 8]
[0392]
<Пример испытаний 6: Действие, стимулирующее сращивание кости, в модели постолатеральной фиксации поясничных позвонков у собак>
Для того чтобы исследовать действие, стимулирующее сращивание кости, соединений настоящего изобретения, влияние добавления микросфер, содержащих испытуемое соединение, во время аутотрансплантации кости подтверждали, применяя модель постолатеральной фиксации поясничных позвонков.
(1) Способ измерения
Мужских особей собак породы бигль в возрасте двенадцать-тринадцать месяцев (KITAYAMA LABES) анестезировали введением 1:1 смеси гидрохлорида кетамина (Кеталар 500 мг, Daiichi Sankyo Propharma Co., Ltd.) и ксилазина (2% инъекция селактара, Bayer Yakuhin, Ltd.) при дозе приблизительно 0,5 мл/кг, и изофлуран, перечисленный в японской фармакопее (элюкаин, полученный Mylan Pharmaceutical), ингалировали ингалятором IMPAC6 (VetEquip Inc.) для поддержания анестезии. Животных фиксировали в положении лежа, волосы на большой площади над правой и левой задней шейкой подвздошной кости, часть вокруг гребня подвздошной кости и нижней части спины выбривали и проводили дезинфекционную обработку повидоном-йодом, указанным в японской фармакопее (раствор изодина для животных 20 мг повидона-йода, указанного в японской фармакопее в 1 мл (Meiji Seika Pharma) и этанолом для дезинфекции (Wako Pure Chemical Industries). Кожу и мягкие ткани разрезали и вскрывали от задней верхней подвздошной кости, вдоль гребня подвздошной кости, применяя скальпель, и затем мышцы, покрывающие гребень подвздошной кости, отделяли под надкостницей для обнажения гребня подвздошной кости. Приблизительно 2 г каждого из правой и левой подвздошных костей отделяли, применяя ножницы для кости, и проводили сжатие для остановки кровотечения. Собранную подвздошную кость, с которой были удалены мягкие ткани, мелко ломали костными ножницами и, таким образом, превращали в обрезки размером 1 мм, получая по 2 г каждого из костных трансплантатов для правой и левой частей. Затем, кожу рассекали вдоль остистого отростка нижней части спины скальпелем, рассекали левую и правую пояснично-крестцовую фасции, и затем обнажали поперечные отростки четвертого и пятого поясничных позвонков, в то время как мультифидусную мышцу и длинную мышцу отделяли и рассекали между их мышечной оболочкой. Мягкие ткани, прилипшие к поперечным отросткам, удаляли, и затем декортикацию кортикальной кости на поверхности поперечных отростков проводили электродрелью (OS-40MV2, Osada Electric Co., Ltd.) для подготовки ложа костного трансплантата. Костный трансплантат (2 г), полученный выше, в достаточной степени смешивали с микросферной суспензией, полученной суспендированием микросфер, содержащих испытуемое соединение (соединение примера 23) в количестве, соответствующем 10, 30 или 100 мкг испытуемого соединения (полученного согласно способу примера получение 1) в 800 мкл раствора КМЦ, внедряли и прививали на поперечные отростки четвертого и пятого поясничных позвонков, и ложе костного трансплантата между поперечными отростками. После аутологичной костной пластики зашивали пояснично-крестцовую фасцию, подкожные ткани и кожу, и участок операции дезинфицировали. Через двенадцать недель после операции животных умерщвляли передозировкой (30 мг/кг) пентобарбитурата натрия (сомнопентил, Kyoritsuseiyaku Corporation), и затем выделяли поясничные позвонки. О сращивании кости судили по подтверждению руками подвижности четвертого и пятого поясничных позвонков (ручная пальпация) слепым способом, и мягкие рентгеновские изображения получали в одном направлении, применяя Softex M-60 (Softex). Каждое мягкое рентгеновское изображение оценивал один человек слепым способом в соответствии с критериями оценки, приведенными в таблице 9. Тест проводили с группами, каждая из которых состояла из 5 животных.
[0393]
[таблица 9]
[0394]
(2) Результаты измерения
Когда образцы поясничного позвонка отделяли, и остеогенез оценивали на основе степени кальцификации, применяя мягкие рентгеновские изображения, в контрольной группе, где микросферы, не содержащие испытуемое соединение, добавляли к подвздошной кости, подвижность наблюдали между 4-м и 5-м поясничным позвонок, но кальцификация между поперечными отростками не наблюдали на мягких рентгеновских снимках во всех пяти примерах. С другой стороны, в группе, в которой вводили микросферы, содержащие испытуемое соединение, в каждой из доз 10, 30 или 100 мкг, было установлено, что подвижность наблюдалась между 4-м и 5-м поясничными позвонками в одном примере из пяти примеров, но подвижность не наблюдали в четырех других примерах. На мягких рентгеновских снимках наблюдали продвижение остеогенов и костной непрерывности между поперечными отростками 4-го и 5-го поясничного позвонка, а именно, показатель непрерывности был 2 или выше при всех дозах. Кроме того, показатель увеличивался в зависимости от дозы (таблица 10, 2,4±0,5, 2,6±0,5 и 2,8±0,4, соответственно).
На основании данных результатов подтверждали, что соединения настоящего изобретения являются пригодными в качестве агента для стимулирования сращивания костей при фиксации позвоночника на основе аутологичной костной пластики. Во всех группах введения соединения гибель животных не наблюдали, побочных реакций, обычно наблюдаемых при введении PGE2, также не наблюдали, и, таким образом, было продемонстрировано, что вышеупомянутые микросферы, содержащие соединение настоящего изобретения, можно безопасно вводить при фиксации позвоночника.
[0395]
[таблица 10]
(среднее±SD)
| название | год | авторы | номер документа |
|---|---|---|---|
| СПЕЦИФИЧЕСКОЕ ГЛЮКОКОРТИКОСТЕРОИДНОЕ СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 2004 |
|
RU2348645C2 |
| ПРОИЗВОДНЫЕ 5-ОКСА-2-АЗАСПИРО[3.4]ОКТАНА В КАЧЕСТВЕ АГОНИСТОВ M4 | 2020 |
|
RU2820477C1 |
| АГЕНТЫ, ИНГИБИРУЮЩИЕ Р38 КИНАЗУ | 2010 |
|
RU2532376C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛО[1,5-А]ПИРАЗИН-4-ИЛА В КАЧЕСТВЕ JAK-ИНГИБИТОРОВ | 2017 |
|
RU2718902C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDK1 | 2010 |
|
RU2615130C2 |
| ПРОИЗВОДНЫЕ СЛОЖНОГО АМИНОЭФИРА АЛКАЛОИДА И ИХ ЛЕКАРСТВЕННЫЕ КОМПОЗИЦИИ | 2011 |
|
RU2567548C2 |
| СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ KDM5, И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2840256C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ 1β-ДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2006 |
|
RU2415855C2 |
| ПРОИЗВОДНЫЕ ПИКОЛИНАМИДА, ПРИГОДНЫЕ В КАЧЕСТВЕ СЕЛЬСКОХОЗЯЙСТВЕННЫХ ФУНГИЦИДОВ | 2020 |
|
RU2826642C2 |
| БИЦИКЛИЧЕСКИЕ АРИЛЬНЫЕ МОНОБАКТАМОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2017 |
|
RU2733402C2 |
Изобретение относится к соединению, представленному общей формулой (1), где R1 представляет собой -H или галоген; Ar1 представляет собой любой заместитель, выбранный из группы G1, который может быть замещен 1-3 одинаковыми или различными заместителями, выбранными из группы, состоящей из -F и метила (при условии, что структуры, представленные формулой 2 исключены), в которых группа G1 представляет собой группу, состоящую из структур формулы 3 (a и b представляют собой направление присоединения); Ar2 представляет собой любой заместитель, выбранный из группы G2, который может быть замещен 1-3 одинаковыми или различными заместителями, выбранными из группы, состоящей из циано, -Cl, метила, метокси и фенила (при условии, что структуры формулы 4 исключены), в которых группа G2 представляет собой группу, состоящую из фенила, тиенила, фурила и тиазолила; и * представляет собой асимметрический углерод, или его фармацевтически приемлемой соли. Также изобретение относится к фармацевтической композиции, обладающей превосходной агонистической активностью по отношению к EP4 рецептору, содержащей эффективное количество соединения по изобретению или его фармацевтически приемлемой соли, в качестве активного ингредиента, и фармацевтически приемлемый носитель. Также изобретение относится к фармацевтической композиции для терапевтического лечения и/или стимулирования заживления перелома, для стимуляции сращивания кости при операциях на позвоночнике, содержащей эффективное количество соединения по изобретению или его фармацевтически приемлемой соли, в качестве активного ингредиента, и фармацевтически приемлемый носитель. Технический результат - соединения, обладающие превосходной агонистической активностью по отношению к EP4 рецептору. 11 н. и 10 з.п. ф-лы, 10 табл., 39 пр.
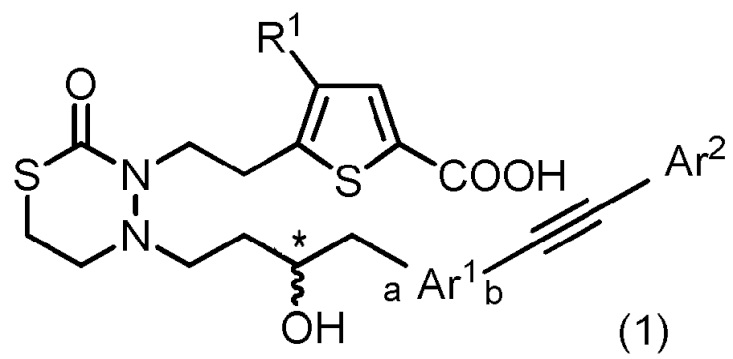

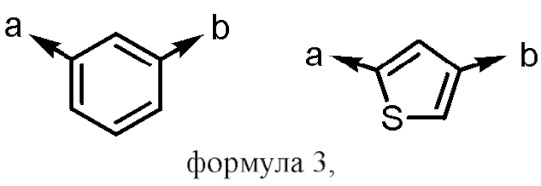

1. Соединение, представленное следующей общей формулой (1):
[формула 1]
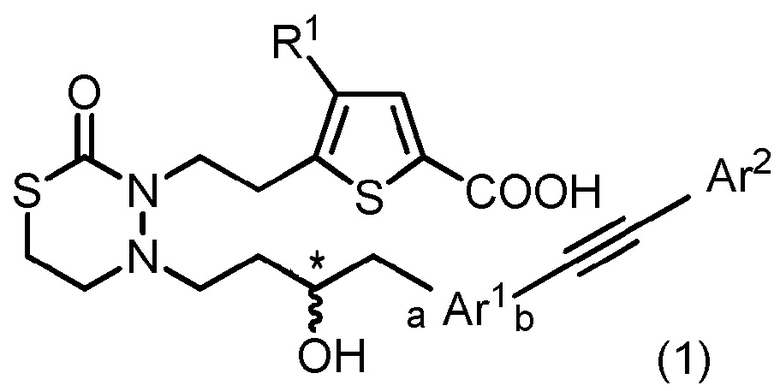
[где в формуле (1)
R1 представляет собой -H или галоген;
Ar1 представляет собой любой заместитель, выбранный из группы G1, который может быть замещен 1-3 одинаковыми или различными заместителями, выбранными из группы, состоящей из -F и метила (при условии, что
[формула 2]

исключены),
где группа G1 представляет собой группу, состоящую из
[формула 3]
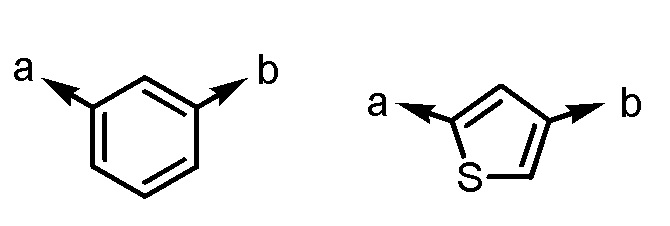
(a и b представляют собой направление присоединения);
Ar2 представляет собой любой заместитель, выбранный из группы G2, который может быть замещен 1-3 одинаковыми или различными заместителями, выбранными из группы, состоящей из циано, -Cl, метила, метокси и фенила (при условии, что
[формула 4]
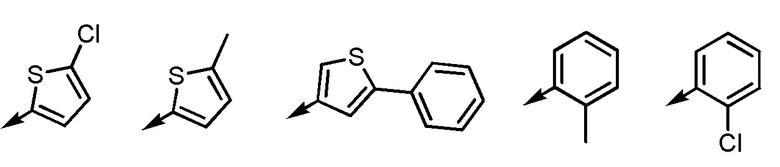
исключены),
где группа G2 представляет собой группу, состоящую из фенила, тиенила, фурила и тиазолила; и
* представляет собой асимметрический углерод],
или его фармацевтически приемлемая соль.
2. Соединение или его фармацевтически приемлемая соль по п. 1, в котором R1 представляет собой -H, -Cl или -Br.
3. Соединение или его фармацевтически приемлемая соль по п. 2, в котором Ar1 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 5]
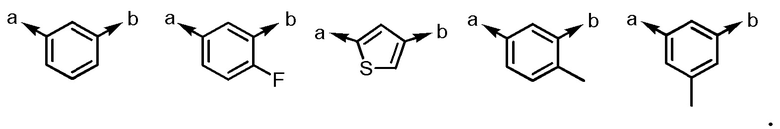
4. Соединение или его фармацевтически приемлемая соль по п. 2, в котором Ar1 представляет собой
[формула 6]
5. Соединение или его фармацевтически приемлемая соль по п. 3 или 4, в котором Ar2 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 7]
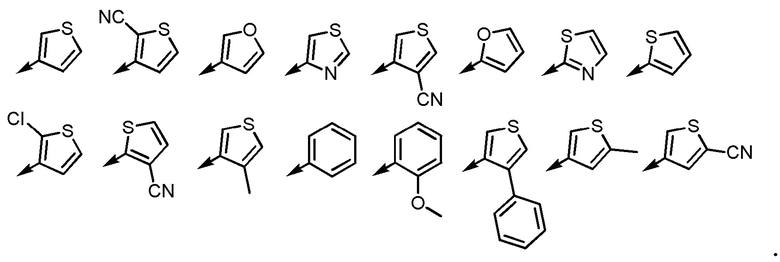
6. Соединение или его фармацевтически приемлемая соль по п. 3 или 4, в котором Ar2 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 8]

7. Соединение или его фармацевтически приемлемая соль по п. 3 или 4, в котором Ar2 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 9]

8. Соединение или его фармацевтически приемлемая соль по п. 7, в котором R1 представляет собой -H.
9. Соединение или его фармацевтически приемлемая соль по п. 7, в котором R1 представляет собой -Cl.
10. Соединение или его фармацевтически приемлемая соль по п. 7, в котором R1 представляет собой -Br.
11. Соединение или его фармацевтически приемлемая соль по п. 1, в котором R1 представляет собой -H, -Cl или -Br;
Ar1 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 10]

Ar2 представляет собой любой заместитель, выбранный из группы, состоящей из
[формула 11]
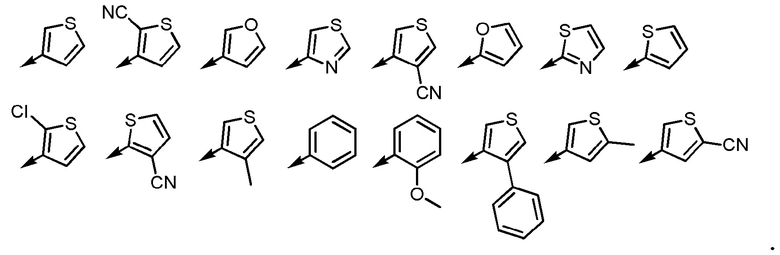
12. Любое соединение, выбранное из следующей группы, или его фармацевтически приемлемая соль:
[формула 12]
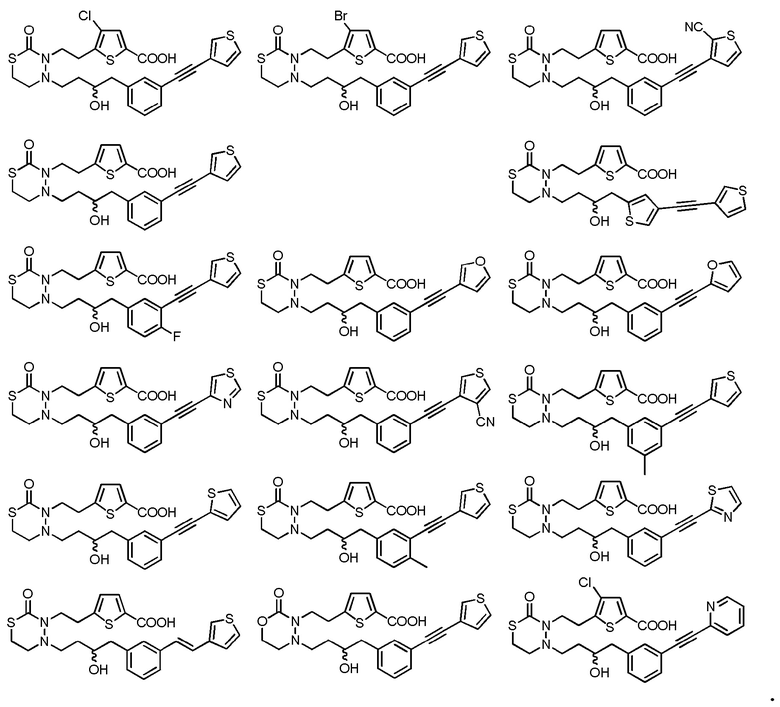
13. Соединение, приведенное ниже, или его фармацевтически приемлемая соль:
[формула 13]
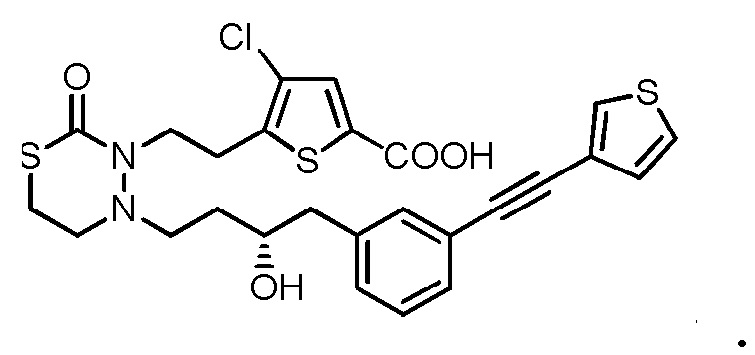
14. Соединение, приведенное ниже, или его фармацевтически приемлемая соль:
[формула 14]

15. Соединение, приведенное ниже, или его фармацевтически приемлемая соль:
[формула 15]
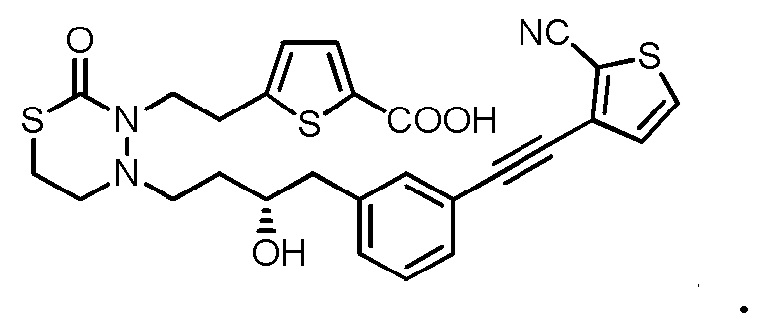
16. Соединение, приведенное ниже, или его фармацевтически приемлемая соль:
[формула 16]
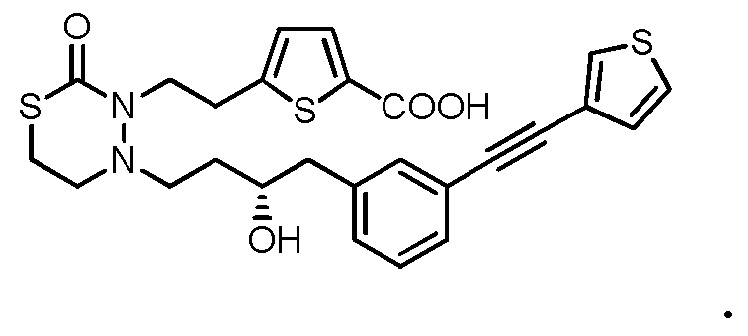
17. Соединение, приведенное ниже, или его фармацевтически приемлемая соль:
[формула 17]
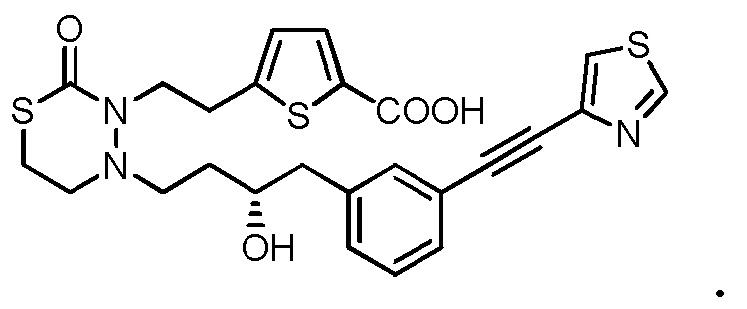
18. Соединение, приведенное ниже, или его соль:
[формула 18]
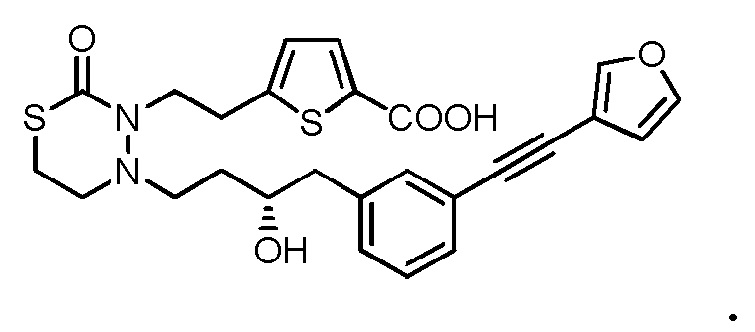
19. Фармацевтическая композиция, обладающая превосходной агонистической активностью по отношению к EP4 рецептору, содержащая эффективное количество соединения по любому из пп. 13-18 или его фармацевтически приемлемой соли, в качестве активного ингредиента, и фармацевтически приемлемый носитель.
20. Фармацевтическая композиция для терапевтического лечения и/или стимулирования заживления перелома, содержащая эффективное количество соединения по любому из пп. 13-18 или его фармацевтически приемлемой соли, в качестве активного ингредиента, и фармацевтически приемлемый носитель.
21. Фармацевтическая композиция для стимуляции сращивания кости при операциях на позвоночнике, содержащая эффективное количество соединения по любому из пп. 13-18 или его фармацевтически приемлемой соли, в качестве активного ингредиента, и фармацевтически приемлемый носитель.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| US 20160060216 A1, 03.03.2016 | |||
| Zhong Zhao; Gian Luca Araldi; Yufang Xiao; Adulla P | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |

