Область изобретения
Данное изобретение относится к органическим соединениям, применимым для терапии или профилактики у млекопитающих и, в частности, к ингибиторам моноацилглицеринлипазы (MAGL, англ. «monoacylglycerol lipase») для лечения или профилактики нейровоспаления, нейродегенеративных заболеваний, боли, онкологических заболеваний, психических расстройств, рассеянного склероза, болезни Альцгеймера, болезни Паркинсона, амиотрофического бокового склероза, травматического повреждения головного мозга, нейротоксичности, инсульта, эпилепсии, тревожности, мигрени и/или депрессии у млекопитающего.
Уровень техники
Эндоканнабиноиды (ЭК) представляют собой сигнальные липиды, биологическое действие которых осуществляется посредством взаимодействия с каннабиноидными рецепторами (CBR, англ. «cannabinoid receptors») СВ1 и СВ2. Они модулируют многие физиологические процессы, включая нейровоспаление, нейродегенерацию и регенерацию тканей (Iannotti, F.A., et al., Progress in lipid research 2016, 62, 107-28.). В головном мозге основной эндоканнабиноид, 2-арахидоноилглицерин (2-AG), вырабатывается диациглицеринлипазами (DAGL, англ. «diacyglycerol lipase») и гидролизуется моноацилглицеринлипазой, MAGL. MAGL гидролизует 85% 2-AG; оставшиеся 15% гидролизуются ABHD6 и ABDH12 (Nomura, D.K., et al., Science 2011, 334, 809.). MAGL экспрессируется по всему головному мозгу и в большинстве клеток головного мозга, включая нейроны, астроциты, олигодендроциты и клетки микроглии (Chanda, Р.K., et al., Molecular pharmacology 2010, 78, 996; Viader, A., et al., Cell reports 2015, 12, 798.). Гидролиз 2-AG приводит к образованию арахидоновой кислоты (АА), предшественницы простогландинов (PG) и лейкотриенов (LT). Окислительный метаболизм АА повышен в воспаленных тканях. Существует два основных ферментативных пути оксигенации арахидоновой кислоты, вовлеченных в воспалительные процессы циклооксигеназа, продуктом которой являются PG, и 5-липоксигеназа, продуктом которой являются LT. Одним из наиболее важных продуктов циклооксигеназы, образуемых во время воспаления, является PGE2. Эти продукты были обнаружены во всех местах воспаления, например, в цереброспинальной жидкости пациентов, страдающих нейродегенеративными расстройствами, и считается, что они участвуют в воспалительном ответе и прогрессировании заболевания. Мыши с отсутствием MAGL (Mgll-/-) демонстрируют сильно сниженную активность 2-AG-гидролазы и повышенные уровни 2-AG в нервной системе, тогда как другие арахидоноил-содержащие виды фосфолипидов и нейтральных липидов, включая анандамид (АЕА), а также другие свободные жирные кислоты, остаются неизменными. И наоборот, уровни АА и полученных из АА простагландинов и других эйкозаноидов, включая простагландин Е2 (PGE2), D2 (PGD2), F2 (PGF2) и тромбоксан В2 (ТХВ2), сильно снижены. Ферменты фосфолипазы А2 (PLA2) рассматривали как основной источник АА, но мыши с дефицитом cPLA2 имеют не измененные уровни АА в головном мозге, подтверждая ключевую роль MAGL в головном мозге для выработки АА и регуляции воспалительных процессов в головном мозге.
Нейровоспаление является распространенной характеристикой патологических изменений при заболеваниях головного мозга, включая, но не ограничиваясь этим, нейродегенеративные заболевания (например, рассеянный склероз, болезнь Альцгеймера, болезнь Паркинсона, амиотрофический боковой склероз, травматическое повреждение головного мозга, нейротоксичность, инсульт, эпилепсия и психические расстройства, такие как тревожность и мигрень). В головном мозге выработки эйкозаноидов и простагландинов регулирует процесс нейровоспаления. Провоспалительный агент липополисахарид (ЛПС) приводит к резкому, зависимому от времени повышению эйкозаноидов в головном мозге, которые существенно снижены у мышей Mgll-/-. Обработка ЛПС также индуцирует повсеместное повышение уровней провоспалительных цитокинов, включая интерлейкин-1-а (IL-1-a), IL-1b, IL-6 и фактор некроза опухоли a (TNF-a), которые ограничены у мышей Mgll-/-.
Нейровоспаление характеризуется активацией клеток врожденного иммунитета центральной нервной системы, микроглии и астроцитов. Сообщалось, что противовоспалительные лекарственные препараты могут подавлять в доклинических моделях активацию клеток глии и прогрессирование заболеваний, включая болезнь Альцгеймера и рассеянный склероз (Lleo A., Cell Mol Life Sci. 2007, 64, 1403.). Также важно, что генетическое и/или фармакологическое нарушение активности MAGL также блокирует индуцированную ЛПС активацию клеток микроглии в головном мозге (Nomura, D.K., et al., Science 2011, 334, 809.).
Кроме того, было показано, что генетическое и/или фармакологическое нарушение активности MAGL продемонстрировало защитные свойства в нескольких животных моделях нейродегенерации, включая, но не ограничиваясь этим, болезнь Альцгеймера, болезнь Паркинсона и рассеянный склероз. Например, необратимый ингибитор MAGL широко использовали в доклинических моделях нейровоспаления и нейродегенерации (Long, J.Z., et al., Nature chemical biology 2009, 5, 37.). Системная инъекция такого ингибитора воспроизводит фенотип мышей Mgll-/- в головном мозге, включая повышение уровней 2-AG, снижение уровней АА и связанную с этим выработку эйкозаноидов, а также предотвращение выработки цитокинов и активации микроглии после ЛПС-индуцированного нейровоспаления (Nomura, D.K., et al., Science 2011, 334, 809.), что вместе подтверждает, что MAGL является поддающейся воздействию лекарственных препаратов мишенью.
Вследствие генетического и/или фармакологического нарушения активности MAGL эндогенные уровни естественного субстрата MAGL в головном мозге, 2-AG, повышаются. Сообщалось, что 2-AG демонстрировал благоприятное воздействие при боли наряду, например, с антиноцицептивным воздействием у мышей (Ignatowska-Jankowska В. et al., J. Pharmacol. Exp.Ther. 2015, 353, 424.) и при психических расстройствах, таких как депрессия, в моделях хронического стресса (Zhong P. et al., Neuropsychopharmacology 2014, 39, 1763.).
Кроме того, олигодендроциты (OL), миелинизирующие клетки центральной нервной системы, и их предшественники (ОРС) экспрессируют на своих мембранах каннабиноидный рецептор 2 (СВ2). 2-AG является эндогенным лигандом рецепторов СВ1 и СВ2. Сообщалось, что как каннабиноиды, так и фармакологическое ингибирование MAGL ослабляют восприимчивость OL и ОРС к эксайтотоксическим стрессам и, следовательно, могут иметь нейропротекторные свойства (Bernal-Chico, A., et al., Glia 2015, 63, 163.). Кроме того, фармакологическое ингибирование MAGL повышает количество миелинизирующих OL в головном мозге мышей, что позволяет предположить, что ингибирование MAGL может стимулировать дифференцировку ОРС в миелинизирующих OL in vivo (Alpar, A., et al., Nature communications 2014, 5, 4421.). Также было показано, что ингибирование MAGL также стимулирует ремиелинизацию и функциональное восстановление в мышиной модели прогрессирующего рассеянного склероза (Feliu A. et al., Journal of Neuroscience 2017, 37 (35), 8385.).
И наконец, в последние годы говорят о большой важности метаболизма в исследованиях онкологических заболеваний, в особенности липидного метаболизма. Исследователи полагают, что de novo синтез жирных кислот играет важную роль в развитии опухолей. Многие исследования продемонстрировали, что эндоканнабиноиды обладают антионкогенным действием, включая антипролиферативные, апоптоз-индуцирующие и антиметастатические эффекты. MAGL как важный разлагающий фермент как для липидного метаболизма, так и для эндоканнабиноидной системы, и дополнительно как часть сигнатуры генной экспрессии, влияет на разные аспекты онкогенеза (Qin, Н., et al., Cell Biochem. Biophys. 2014, 70, 33; Nomura DK et al., Cell 2009, 140(1), 49-61; Nomura DK et al., Chem. Biol. 2011, 18(7), 846-856).
И напоследок, подавление действия и/или активации MAGL является новой перспективной терапевтической стратегией для лечения или предотвращения нейровоспаления, нейродегенеративных заболеваний, боли, онкологических заболеваний и психических расстройств. Помимо этого, подавление действия и/или активации MAGL является новой перспективной терапевтической стратегией для обеспечения нейропротекции и восстановления миелина. Соответственно, существует значительная неудовлетворенная медицинская потребность в новых ингибиторах MAGL.
Сущность изобретения
В первом аспекте в данном изобретении предложено соединение формулы (I)
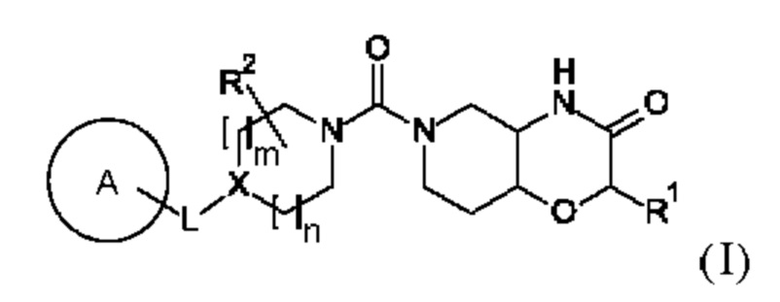
или его фармацевтически приемлемая соль, где A, L, X, m, n, R1 и R2 соответствуют описанию в данном документе.
В одном аспекте в данном изобретении предложен способ производства соединений мочевины формулы (I), описанных в данном документе, включающий: проведение реакции первого амина формулы 1, где R1 соответствует описанию в данном документе, предпочтительно где R1 представляет собой водород,

со вторым амином 2, где A, L, m, n, X и R2 соответствуют описанию в данном документе,

в присутствии основания и образующего мочевину реагента, с образованием указанного соединения формулы (I).
В дополнительном аспекте в данном изобретении предложено соединение формулы (I), описанное в данном документе, произведенное в соответствии со способом, описанным в данном документе.
В дополнительном аспекте в данном изобретении предложено соединение формулы (I), описанное в данном документе, для применения в качестве терапевтически активного вещества.
В дополнительном аспекте в данном изобретении предложена фармацевтическая композиция, содержащая соединение формулы (I), описанное в данном документе, и терапевтически инертный носитель.
В дополнительном аспекте в данном изобретении предложено применение соединения формулы (I), описанного в данном документе, или фармацевтической композиции, описанной в данном документе, для ингибирования моноацилглицеринлипазы (MAGL) у млекопитающего.
В дополнительном аспекте в данном изобретении предложено применение соединения формулы (I), описанного в данном документе, или фармацевтической композиции, описанной в данном документе, для лечения или профилактики нейровоспаления, нейродегенеративных заболеваний, боли, онкологических заболеваний и/или психических расстройств у млекопитающего.
В дополнительном аспекте в данном изобретении предложено применение соединения формулы (I), описанного в данном документе, или фармацевтической композиции, описанной в данном документе, для лечения или профилактики рассеянного склероза, болезни Альцгеймера, болезни Паркинсона, амиотрофического бокового склероза, травматического повреждения головного мозга, нейротоксичности, инсульта, эпилепсии, тревожности, мигрени, депрессии, гепатоцеллюлярной карциномы, канцерогенеза толстой кишки, рака яичника, нейропатической боли, индуцированной химиотерапией нейропатии, острой боли, хронической боли и/или спастичности, связанной с болью, у млекопитающего.
В дополнительном аспекте в данном изобретении предложено соединение формулы (I), описанное в данном документе, или фармацевтическая композиция, описанная в данном документе, для применения в способе ингибирования моноацилглицеринлипазы у млекопитающего.
В дополнительном аспекте в данном изобретении предложено соединение формулы (I), описанное в данном документе, или фармацевтическая композиция, описанная в данном документе, для лечения или профилактики нейровоспаления, нейродегенеративных заболеваний, боли, онкологических заболеваний и/или психических расстройств у млекопитающего.
В дополнительном аспекте в данном изобретении предложено соединение формулы (I), описанное в данном документе, или фармацевтическая композиция, описанная в данном документе, для лечения или профилактики рассеянного склероза, болезни Альцгеймера, болезни Паркинсона, амиотрофического бокового склероза, травматического повреждения головного мозга, нейротоксичности, инсульта, эпилепсии, тревожности, мигрени, депрессии, гепатоцеллюлярной карциномы, канцерогенеза толстой кишки, рака яичника, нейропатической боли, индуцированной химиотерапией нейропатии, острой боли, хронической боли и/или спастичности, связанной с болью, у млекопитающего.
В дополнительном аспекте в данном изобретении предложено применение соединения формулы (I), описанного в данном документе, для изготовления лекарственного средства для ингибирования моноацилглицеринлипазы у млекопитающего.
В дополнительном аспекте в данном изобретении предложено применение соединения формулы (I), описанного в данном документе, для изготовления лекарственного средства для лечения или профилактики нейровоспаления, нейродегенеративных заболеваний, боли, онкологических заболеваний и/или психических расстройств у млекопитающего.
В дополнительном аспекте в данном изобретении предложено применение соединения формулы (I), описанного в данном документе, для изготовления лекарственного средства для лечения или профилактики рассеянного склероза, болезни Альцгеймера, болезни Паркинсона, амиотрофического бокового склероза, травматического повреждения головного мозга, нейротоксичности, инсульта, эпилепсии, тревожности, мигрени, депрессии, гепатоцеллюлярной карциномы, канцерогенеза толстой кишки, рака яичника, нейропатической боли, индуцированной химиотерапией нейропатии, острой боли, хронической боли и/или спастичности, связанной с болью, у млекопитающего.
В дополнительном аспекте в данном изобретении предложен способ ингибирования моноацилглицеринлипазы у млекопитающего, который включает введение млекопитающему эффективного количества соединения формулы (I), описанного в данном документе, или фармацевтической композиции, описанной в данном документе.
В дополнительном аспекте в данном изобретении предложен способ лечения или профилактики нейровоспаления, нейродегенеративных заболеваний, боли, онкологических заболеваний и/или психических расстройств у млекопитающего, который включает введение млекопитающему эффективного количества соединения формулы (I), описанного в данном документе, или фармацевтической композиции, описанной в данном документе.
В дополнительном аспекте в данном изобретении предложен способ лечения или профилактики рассеянного склероза, болезни Альцгеймера, болезни Паркинсона, амиотрофического бокового склероза, травматического повреждения головного мозга, нейротоксичности, инсульта, эпилепсии, тревожности, мигрени, депрессии, гепатоцеллюлярной карциномы, канцерогенеза толстой кишки, рака яичника, нейропатической боли, индуцированной химиотерапией нейропатии, острой боли, хронической боли и/или спастичности, связанной с болью, у млекопитающего, который включает введение млекопитающему эффективного количества соединения формулы (I), описанного в данном документе, или фармацевтической композиции, описанной в данном документе.
Подробное описание сущности изобретения
Определения
Признаки, целые числа, характеристики, соединения, химические фрагменты или группы, описанные в связи с конкретным аспектом, вариантом осуществления или примером данного изобретения, следует понимать как применимые к любому другому аспекту, варианту осуществления или примеру, описанным в данном документе, если они не являются несовместимыми с ними. Все признаки, раскрытые в данном описании (включая прилагаемые формулу изобретения, реферат и графические материалы), и/или все этапы любого способа или процесса, раскрытых таким образом, можно комбинировать в любой комбинации, за исключением комбинаций, в которых по меньшей мере некоторые из таких признаков и/или этапов являются взаимно исключающими. Данное изобретение не ограничено деталями любого из вышеприведенных вариантов осуществления. Данное изобретение распространяется на любой новый признак или любую новую комбинацию признаков, раскрытых в данном описании (включая прилагаемые формулу изобретения, реферат и графические материалы), и/или на любой новый этап или любую новую комбинацию этапов любых способа или процесса, раскрытых таким образом.
Термин «алкил» относится к одно- или многовалентной, например, одно- или двухвалентной, неразветвленной или разветвленной насыщенной углеводородной группе из 1-12 атомов углерода. В некоторых предпочтительных вариантах осуществления алкильная группа содержит 1-6 атомов углерода («C1-6-алкил»), например, 1, 2, 3, 4, 5 или 6 атомов углерода. В других вариантах осуществления алкильная группа содержит 1-3 атома углерода, например, 1, 2 или 3 атома углерода. Некоторые неограничивающие примеры алкилов включают метил, этил, пропил, 2-пропил (изопропил), н-бутил, изобутил, втор-бутил, трет-бутил и 2,2-диметилпропил. Особенно предпочтительными, но не ограничивающими примерами алкила являются метил и трет-бутил.
Термин «алкокси» относится к алкильной группе по определению выше, присоединенной к исходному молекулярному фрагменту посредством атома кислорода. Если не указано иное, алкокси-группа содержит 1-12 атомов углерода. В некоторых предпочтительных вариантах осуществления алкокси-группа содержит 1-6 атомов углерода («С1-6-алкокси»). В других вариантах осуществления алкокси-группа содержит 1-4 атома углерода. В других вариантах осуществления алкокси-группа содержит 1-3 атома углерода. Некоторые неограничивающие примеры алкокси-групп включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси и трет-бутокси. В особенности предпочтительным, но не ограничивающим примером алкокси является метокси.
Термин «галоген» или «гало» относится к фтору (F), хлору (С1), брому (Br) или иоду (I). Предпочтительно термин «галоген» или «гало» относится к фтору (F), хлору (Cl) или брому (Br). В особенности предпочтительными, но не ограничивающими примерами «галогена» или «гало» являются фтор (F) и хлор (Cl).
В контексте данного документа термин «циклоалкил» относится к насыщенной или частично ненасыщенной моноциклической или бициклической углеводородной группе из 3-10 кольцевых атомов углерода. В некоторых предпочтительных вариантах осуществления циклоалкильная группа представляет собой насыщенную моноциклическую углеводородную группу из 3-8 кольцевых атомов углерода. «Бициклический циклоалкил» относится к циклоалкильным фрагментам, состоящим из двух насыщенных карбоциклов, имеющих два общих атома углерода, т.е. мостик, разделяющий два кольца, представляет собой одинарную связь или цепочку из одного или двух кольцевых атомов, и к спироциклическим фрагментам, т.е. в которых два кольца соединены посредством общего кольцевого атома. Предпочтительно циклоалкильная группа представляет собой насыщенную моноциклическую углеводородную группу из 3-6 кольцевых атомов углерода, например, 3, 4, 5 или 6 атомов углерода. Некоторые неограничивающие примеры циклоалкилов включают циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
Термины «гетероциклил» и «гетероциклоалкил» взаимозаменяемо используются в данном документе и относятся к насыщенной или частично ненасыщенной моно- или бициклической, предпочтительно моноциклической кольцевой системе из 3-10 кольцевых атомов, предпочтительно 3-8 кольцевых атомов, при этом 1, 2 или 3 из указанных кольцевых атомов представляют собой гетероатомы, выбранные из N, О и S, а остальные кольцевые атомы представляют собой углерод. Предпочтительно, 1-2 из указанных кольцевых атомов выбраны из N и О, а остальные кольцевые атомы представляют собой углерод. «Бициклический гетероциклил» относится к гетероциклическим фрагментам, состоящим из двух циклов, имеющих два общих кольцевых атома, т.е. мостик, разделяющий два кольца, представляет собой одинарную связь или цепочку из одного или двух кольцевых атомов, и к спироциклическим фрагментам, т.е. в которых два кольца соединены посредством общего кольцевого атома. Некоторые неограничивающие примеры моноциклических гетероциклильных групп включают азетидин-3-ил, азетидин-2-ил, оксетан-3-ил, оксетан-2-ил, 1-пиперидил, 2-пиперидил, 3-пиперидил, 4-пиперидил, 2-оксопирролидин-1-ил, 2-оксопирролидин-3-ил, 5-оксопирролидин-2-ил, 5-оксопирролидин-3-ил, 2-оксо-1-пиперидил, 2-оксо-3-пиперидил, 2-оксо-4-пиперидил, 6-оксо-2-пиперидил, 6-оксо-3-пиперидил, морфолино, морфолин-2-ил и морфолин-3-ил.
Термин «арил» относится к моноциклической, бициклической или трициклической карбоциклической кольцевой системе, имеющей в целом 6-14 кольцевых членов, предпочтительно 6-12 кольцевых членов и более предпочтительно 6-10 кольцевых членов, и при этом по меньшей мере одно кольцо в системе является ароматическим. Некоторые неограничивающие примеры арилов включают фенил и 9Н-фторфенил (например, 9Н-фтор-9-ил). В особенности предпочтительным, но не ограничивающим примером арила является фенил.
Термин «гетероарил» относится к одно- или многовалентной, моноциклической или бициклической кольцевой системе, имеющей в целом 5-14 кольцевых членов, предпочтительно 5-12 кольцевых членов и более предпочтительно 5-10 кольцевых членов, при этом по меньшей мере одно кольцо в системе является ароматическим и по меньшей мере одно кольцо в системе содержит один или более гетероатомов. Предпочтительно, «гетероарил» относится к 5-10-членному гетероарилу, содержащему 1, 2, 3 или 4 гетероатома, независимо выбранных из О, S и N. Наиболее предпочтительно, «гетероарил» относится к 5-10-членному гетероарилу, содержащему 1-2 гетероатома. независимо выбран из О, S и N. Некоторые предпочтительные, но не ограничивающие примеры гетероарила включают тиазолил (например, тиазол-2-ил); оксазолил (например, оксазол-2-ил); 5,6-дигидро-4Н-циклопента[d]тиазол-2-ил; 1,2,4-оксадиазол-5-ил; пиридил (например, 2-пиридил); пиразолил (например, пиразол-1-ил); имидазолил (например, имидазол-1-ил); бензоксазолил (например, бензоксазол-2-ил) и оксазоло[5,4-с]пиридин-2-ил.
Термин «гидрокси» относится к группе ОН.
Термин «циано» относится к -CN (нитрил) группе.
Термин «галогеналкил» относится к любой алкильной группе, в которой по меньшей мере один из атомов водорода алкильной группы был замещен атомом галогена, предпочтительно фтором. Предпочтительно «галогеналкил» относится к алкильной группе, в которой 1, 2 или 3 атома водорода алкильной группы были замещены атомом галогена, предпочтительно фтором. В особенности предпочтительными, но не ограничивающими примерами галогеналкилов являются трифторметил (CF3) и трифторэтил (например, 2,2,2-трифторэтил).
Термин «галогеналкокси» относится к любой алкокси-группе, в которой по меньшей мере один из атомов водорода алкокси-группы был замещен атомом галогена, предпочтительно фтором. Предпочтительно «галогеналкокси» относится к алкокси-группе, в которой 1, 2 или 3 атома водорода алкокси-группы были замещены атомом галогена, предпочтительно фтором. В особенности предпочтительным, но не ограничивающим примером галогеналкокси является трифторметокси (-OCF3).
Термин «гидроксиалкил» относится к любой алкильной группе, в которой по меньшей мере один из атомов водорода алкильной группы был замещен гидрокси-группой. Предпочтительно «гидроксиалкил» относится к алкильной группе, в которой 1, 2 или 3 атома водорода, наиболее предпочтительно 1 атом водорода алкильной группы были замещены гидрокси-группой. Предпочтительными, но не ограничивающими примерами гидроксиалкилов являются гидроксиметил и гидроксиэтил (например, 2-гидроксиэтил). В особенности предпочтительным, но не ограничивающим примером гидроксиалкила является гидроксиметил.
Термин «галогенарил» относится к любой арильной группе, в которой по меньшей мере один из атомов водорода арильной группы был замещен атомом галогена. Предпочтительно «галогенарил» относится к арильной группе, в которой 1, 2 или 3 атома водорода, более предпочтительно 1-2 атома водорода, наиболее предпочтительно 1 атом арильной группы были замещены атомом галогена. В особенности предпочтительным, но не ограничивающим примером галогенарила является хлорфенил, в частности, 4-хлорфенил.
Термин «арилокси» относится к арильной группе по определению выше, присоединенной к исходному молекулярному фрагменту посредством атома кислорода. Предпочтительным, но не ограничивающим примером арилокси является фенокси.
Термин «галогенарилокси» относится к галогенарильной группе по определению выше, присоединенной к исходному молекулярному фрагменту посредством атома кислорода. Предпочтительным, но не ограничивающим примером галогенарилокси является 4-фторфенокси.
Термин «фармацевтически приемлемая соль» относится к тем солям, которые сохраняют биологическую эффективность и свойства свободных оснований или свободных кислот, которые не являются биологически или в других отношениях нежелательными. Соли образуются с такими неорганическими кислотами, как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., в частности хлористоводородная кислота, и такими органическими кислотами, как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота, N-ацетилцистеин и т.п. Кроме того, эти соли могут быть получены добавлением неорганического основания или органического основания к свободной кислоте. Соли, полученные из неорганического основания, включают, но не ограничиваются этим, соли натрия, калия, лития, аммония, кальция, магния и т.п. Соли, полученные из органических оснований, включают, но не ограничиваются этим, соли первичных, вторичных и третичных аминов, замещенных аминов, в том числе встречающихся в природе замещенных аминов, циклических аминов и основных ионообменных смол, например смол изопропиламина, триметиламина, диэтиламина, триэтиламина, трипропиламина, этаноламина, лизина, аргинина, N-этилпиперидина, пиперидина, полиимина и т.п. Конкретными фармацевтически приемлемыми солями соединений формулы (I) являются хлористоводородные соли.
Термин «фармацевтически приемлемый сложный эфир» относится к сложным эфирам, которые гидролизуются in vivo и включают те, которые легко разрушаются в организме человека с высвобождением исходного соединения или его соли. Подходящие сложноэфирные группы включают, например, полученные из фармацевтически приемлемых алифатических карбоновых кислот, в частности, алкановой, алкеновой, циклоалкановой и циклоалкеновой кислот, в которых алкильный или алкенильный фрагмент преимущественно имеет не более 6 атомов углерода. Типовые примеры конкретных сложных эфиров включают, но не ограничиваются этим, формиаты, ацетаты, пропионаты, бутираты, акрилаты и этилсукцинаты. Примеры фармацевтически приемлемых типов пролекарств описаны в Higuchi and Stella, Pro-drugs as Novel Delivery Systems, Vol.14 of the A.C.S. Symposium Series, and in Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987.
Термин «защитная группа» (ЗГ) обозначает группу, которая избирательно блокирует реакционноспособный сайт в многофункциональном соединении, так чтобы можно было избирательно проводить химическую реакцию в другом незащищенном реакционноспособном сайте в понимании, традиционно связанном с этим в области синтетической химии. Защитные группы можно удалять в подходящий момент времени. Типовыми защитными группами являются амино-защитные группы, карбокси-защитные группы или гидрокси-защитные группы. Конкретными защитными группами являются трет-бутоксикарбонил (Boc), бензилоксикарбонил (Cbz), флуоренилметоксикарбонил (Fmoc) и бензил-(Bn). Дополнительными конкретными защитными группами являются трет-бутоксикарбонил (Boc) и флуоренилметоксикарбонил (Fmoc). Более конкретной защитной группой является трет-бутоксикарбонил (Boc). Типовые защитные группы и их применение в органическом синтезе описаны, например, в "Protective Groups in Organic Chemistry" by Т. W. Greene and P. G. M. Wutts, 5th Ed., 2014, John Wiley & Sons, N.Y.
Термин «образующий мочевину реагент» относится к химическому соединению, которое способно отдавать первый амин, который будет вступать в реакцию со вторым амином с образованием, таким образом, производного мочевины. Неограничивающие примеры образующих мочевину реагентов включают бис(трихлорметил)карбонат, фосген, трихлорметилхлорформиат, (4-нитрофенил)карбонат и 1,1'-карбонилдиимидазол. Образующие мочевину реагенты, описанные в G. Sartori et al., Green Chemistry 2000, 2, 140, включены в данный документ посредством ссылки.
Соединения формулы (I) могут содержать несколько асимметрических центров и могут находиться в форме оптически чистых энантиомеров, смесей энантиомеров, таких как, например, рацематы, оптически чистых диастереоизомеров, смесей диастереоизомеров, диастереоизомерных рацематов или смесей диастереоизомерных рацематов. В предпочтительном варианте осуществления соединение формулы (I) согласно данному изобретению представляет собой цис-энантиомер формулы (Ia) или (Ib), соответственно, как описано в данном документе.
В соответствии с правилом Кана - Ингольда - Прелога асимметрический атом углерода может находиться в «R»- или «S»-конфигурации.
Аббревиатура «MAGL» относится к ферменту моноацилглицеринлипазе. Термины «MAGL» и «моноацилглицеринлипаза» взаимозаменяемо используются в данном документе.
В контексте данного документа термин «лечение» включает: (1) ингибирование состояния, расстройства или патологического состояния (например, прекращение, уменьшение или замедление развития заболевания или, в случае поддерживающего лечения, повторного появления по меньшей мере одного его клинического или субклинического симптома); и/или (2) облегчение патологического состояния (т.е. достижение регрессии состояния, расстройства или патологического состояния или по меньшей мере одного его клинического или субклинического симптома). Польза для подлежащего лечению пациента является статистически значимой или, по меньшей мере, ощутимой для пациента или лечащего врача. Однако следует понимать, что когда лекарственное средство вводят пациенту для лечения заболевания, результатом не всегда может быть эффективное лечение.
В контексте данного документа термин «профилактика» включает: предотвращение или замедление появления клинических симптомов состояния, расстройства или патологического состояния, развивающегося у млекопитающего и, в особенности у человека, который может быть подвержен или предрасположен к состоянию, расстройству или патологическому состоянию, но еще не чувствует или не демонстрирует клинических или субклинических симптомов состояния, расстройства или патологического состояния.
В контексте данного документа термин «нейровоспаление» относится к острому и хроническому воспалению нервной ткани, которая является основным тканевым компонентом двух частей нервной системы; головного мозга и спинного мозга центральной нервной системы (ЦНС) и разветвленных периферических нервов периферической нервной системы (ПНС). Хроническое нейровоспаление связано с нейродегенеративными заболеваниями, такими как болезнь Альцгеймера, болезнь Паркинсона и рассеянный склероз. Острое нейровоспаление обычно следует незамедлительно за повреждением центральной нервной системы, например, в результате травматического повреждения головного мозга (ТПГМ).
Термин «травматическое повреждение головного мозга» («ТПГМ», также известное как «внутричерепная травма») относится к повреждению головного мозга в результате действия внешней механической силы, такой как резкое ускорение или торможение, удар, ударные волны или проникновение пули.
Термин «нейродегенеративные заболевания» относится к заболеваниям, связанным с прогрессирующей потерей структуры или функции нейронов, включая гибель нейронов. Примеры нейродегенеративных заболеваний, включают, но не ограничиваются этим, рассеянный склероз, болезнь Альцгеймера, болезнь Паркинсона и амиотрофический боковой склероз.
Термин «психические расстройства» (также называемые психическими заболеваниями или психиатрическими расстройствами) относится к поведенческим или психическим моделям, которые могут приводить к страданиям или недееспособности. Такие признаки могут быть постоянными, появляющимися и исчезающими или появляться в виде единственного эпизода. Примеры психических заболеваний, включают, но не ограничиваются этим, тревожность и депрессию.
Термин «боль» относится к неприятным чувственным и эмоциональным ощущениям, связанным с фактическим или потенциальным повреждением тканей. Примеры боли включают, но не ограничиваются этим, ноцицептивную боль, хроническую боль (включая идиопатическую боль), нейропатическую боль, включая индуцированную химиотерапией нейропатию, фантомную боль и психогенную боль. Конкретным примером боли является нейропатическая боль, вызываемая повреждением или заболеванием, поражающим любую часть нервной системы, участвующую в функции ощущений (т.е. соматосенсорную систему). В одном варианте осуществления «боль» представляет собой нейропатическую боль в результате ампутации или торакотомии. В одном варианте осуществления «боль» представляет собой индуцированную химиотерапией нейропатию.
Термин «нейротоксичность» относится к токсичности в нервной системе. Она возникает, когда воздействие природных или искусственных токсических веществ (нейротоксинов) изменяет нормальную активность нервной системы образом, приводящим к повреждению нервной системы. Примеры нейротоксичности включают, но не ограничиваются этим, нейротоксичность в результате воздействия веществ, используемых в химиотерапии, лучевой терапии, лекарственной терапии, при лекарственной зависимости и при трансплантации органов, а также воздействия тяжелых металлов, определенных видов пищи и пищевых добавок, пестицидов, промышленных и/или чистящих растворителей, косметики и некоторых веществ природного происхождения.
Термин «онкологическое заболевание» («рак») относится к заболеванию, характеризуемому наличием новообразования или опухоли в результате аномального неконтролируемого роста клеток (такие клетки называются «раковыми клетками»). В контексте данного документа термин онкологическое заболевание однозначно включает, но не ограничивается этим, гепатоцеллюлярную карциному, канцерогенез толстой кишки и рак яичника.
В контексте данного документа термин «млекопитающее» включает как людей, так и отличных от людей животных, и включает, но не ограничивается этим, людей, отличных людей приматов, собачьих, кошачьих, мышиных, бычьих, лошадиных и свинообразных. В особенности предпочтительном варианте осуществления термин «млекопитающее» относится к людям.
Соединения по данному изобретению
В первом аспекте в данном изобретении предложено соединение формулы
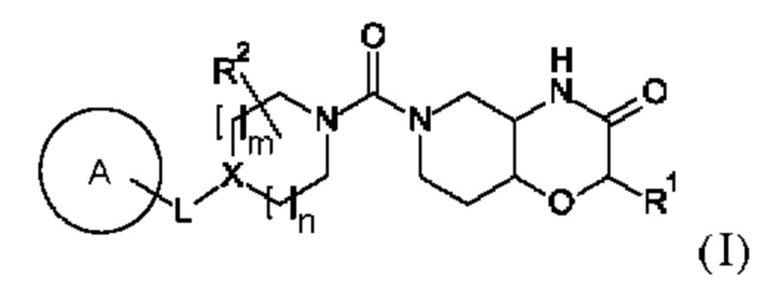
или его фармацевтически приемлемая соль, где:
(i) X представляет собой C-R3; m равно 0 или 1; N выбрано из 0, 1 и 2; и L выбран из -(СН2)р-, -О-, -ОСН2-, -СН2О-, -СН2ОСН2-, -CF2CH2- и -CH2CF2-; или
(ii) X представляет собой N; m равно 1; n равно 1 или 2; и L представляет собой -(СН2)р- или -CF2CH2-;
р выбрано из 1, 2 и 3; А выбран из:
(i) арила, замещенного R4, R5 и R6;
(ii) гетероарила, замещенного R7, R8 и R9; и
(iii) гетероциклоалкила, замещенного R10, R11 и
R1 представляет собой водород или С1-6-алкил; R2 выбран из водорода, С1-6-алкила и гидрокси-С1-6-алкила;
R3 выбран из водорода, галогена, гидрокси, С1-6-алкокси, C1-6-алкила и галоген-С1-6-алкила;
каждый из R4, R5, R6, R7, R8, R9, R10, R11 и R12 независимо выбран из водорода, галогена, циано, гидрокси, C1-6-алкила, галоген-С1-6-алкила, гидрокси-С1-6-алкила, галоген-С1-5-алкила-СН(ОН)-, С1-6-алкокси, галоген-С1-6-алкокси, SF5, CH3SO2, С3-10-циклоалкила, С3-10-циклоалкила, замещенного R13, гетероциклоалкила, гетероциклоалкила, замещенного R14, гетероарила, арила и галогенарила; и
каждый из R13 и R14 независимо представляет собой С1-6-алкил, галоген-С1-6-алкил или гидрокси.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, причем соединение формулы (I) представляет собой соединение формулы (Ia):

где A, L, X, m, n, R1 и R2 соответствуют определениям в данном документе. Предпочтительно указанное соединение формулы (Ia) имеет энантиомерный избыток (ее) равный >80%, более предпочтительно >90%, в частности >99%.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, причем соединение формулы (I) представляет собой соединение формулы (Ib):
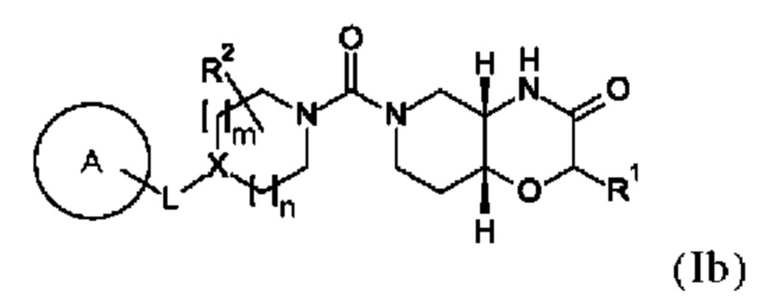
где A, L, X, m, n, R1 и R2 соответствуют определениям в данном документе. Предпочтительно указанное соединение формулы (Ib) имеет энантиомерный избыток (ее) равный >80%, более предпочтительно >90%, в частности >99%.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, причем соединение формулы (I) представляет собой соединение формулы (Ic):

где A, L, X, m, n, R1 и R2 соответствуют определениям в данном документе. Предпочтительно указанное соединение формулы (Ic) имеет энантиомерный избыток (ее) равный >80%, более предпочтительно >90%, в частности >99%.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, причем соединение формулы (I) представляет собой соединение формулы (Id):
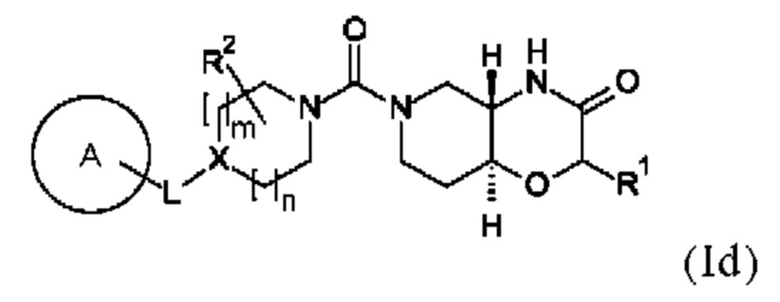
где A, L, X, m, n, R1 и R2 соответствуют определениям в данном документе. Предпочтительно указанное соединение формулы (Id) имеет энантиомерный избыток (ее) равный >80%, более предпочтительно >90%, в частности >99%.
В одном варианте осуществления предложено соединение формулы (I), как описано в данном документе, где, когда X представляет собой C-R3 и R3 представляет собой гидрокси или галоген, L не представляет собой -О- или СН2О.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
(i) X представляет собой C-R3; m равно 0 или 1; n выбрано из 0, 1 и 2; и L выбран из -(СН2)р-, -О-, -ОСН2-, -СН2О- и -СН2ОСН2-; или
(ii) X представляет собой N; m и n оба равны 1; и L представляет собой -(СН2)р-.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
X представляет собой C-R3;
m оба равны 0; или
m оба равны 1; и
L выбран из -(СН2)р -, -О-, -ОСН2- и -СН2О-.
В другом предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
X представляет собой C-R3;
m оба равны 0; или
m оба равны 1;
L выбран из -(СН2)р-, -О-, -ОСН2- и -СН2О-;
R3 выбран из водорода, С1-6-алкила и галогена; и
р равно 1 или 2.
В особенно предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
X представляет собой C-R3;
m оба равны 0; или
m оба равны 1;
L выбран из -(СН2)р-, -О-, -ОСН2- и -СН2О-;
R3 представляет собой водород; и
р равно 1 или 2.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где р равно 1 или 2.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где А выбран из:
(i) арила, замещенного R4, R5 и R6; и
(ii) гетероарила, замещенного R7, R8 и R9.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где А выбран из:
(i) фенила, замещенного R4, R5 и R6; и
(ii) оксазолила, замещенного R7, R8 и R9.
В другом предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где А представляет собой арил, замещенный R4, R5 и R6.
В особенно предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где А представляет собой фенил, замещенный R4, R5 и R6.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
А выбран из:
(i) арила, замещенного R4, R5 и R6; и
(ii) гетероарила, замещенного R7, R8 и R9;
R4 выбран из водорода, галогена, галоген-С1-6-алкокси и галоген-С1-6-алкила;
R5 выбран из водорода, циано, галогена, C1-6-алкила, C1-6-алкокси, гетероциклоалкила, С3-10-никлоалкила, гетероарила или галоарила;
R6 представляет собой водород или галоген;
R7 выбран из водорода, С1-6-алкила, арила и галоген-С1-6-алкила;
R8 представляет собой водород или С1-6-алкил; и
R9 представляет собой водород.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
А выбран из:
(i) арила, замещенного R4, R5 и R6; и
(ii) гетероарила, замещенного R7, R8 и R9;
R4 выбран из галогена, галоген-С1-6-алкокси и галоген-С1-6-алкила;
R5 выбран из водорода, циано, галогена, гетероциклоалкила, С3-10-циклоалкила и галогенарила;
R6 представляет собой водород;
R7 представляет собой C1-6-алкил;
R8 представляет собой водород; и
R9 представляет собой водород.
В особенно предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
А выбран из:
(i) фенила, замещенного R4, R6 и R6; и
(ii) оксазолила, замещенного R7, R8 и R9;
R4 выбран из хлора, OCF3 и CF3;
R5 выбран из водорода, циано, фтора, хлора, пирролидинила, циклопентила, циклопропила и хлорфенила;
R6 представляет собой водород;
R7 представляет собой трет-бутил;
R8 представляет собой водород; и
R9 представляет собой водород.
В следующем конкретно предпочтительном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где А выбран из фенила, 4-трет-бутилтиазол-2-ила, 4-трет-бутилоксазол-2-ила, 2-хлор-4-фтор-фенила, 4-(трифторметил)фенила, 4-(трифторметокси)фенила, 4-хлорфенила, 5,6-дигидро-4Н-циклопента[d]тиазол-2-ила, 3-фенил-1,2,4-оксадиазол-5-ила, 5-(трифторметил)-2-пиридила, 4-(трифторметил)пиразол-1-ила, 2-фтор-4-(трифторметил)фенила, 2,4-дифторфенила, 4-хлор-3-фтор-фенила, 4-цианофенила, 4,4-дифтор-1-пиперидила, 5-трет-бутилоксазол-2-ила, 4-метокси-2-фтор-фенила, 2-хлор-4(трифторметил)фенила, 6-(трифторметил)-3-пиридила, 3-(трифторметил)фенила, 2-хлор-4-(трифторметокси)фенила, 4-хлор-2-фтор-фенила, 4-фтор-2-(трифторметил)фенила, 2-пирролидин-1-ил-4-(трифторметил)фенила, 4-фтор-2-циано-фенила, 2-циклопентил-4-(трифторметил)фенила, 2-хлор-4-цианофенила, 4-(трифторметил)имидазол-1-ила, 4-фтор-2-метил-фенила, 4-трет-бутилпиразол-1-ила, 1,3-бензоксазол-2-ила, 4-хлор-3-(4-хлорфенил)фенила, 2-(1Н-пиразол-4-ил)-4-(трифторметил)фенила, 2,4-дихлорфенила, 3-метокси-4-(трифторметил)фенила, 5-метил-6-(трифторметил)-3-пиридила, 3-хлорфенила, 2-хлорфенила, 2-циклопропил-4-(трифторметил)фенила, 2-метил-4-(трифторметил)фенила, 3-фтор-5-(трифторметил)фенила, 2-фтор-6-(трифторметил)фенила, 3-хлор-4-(трифторметил)фенила, 2,4-дифтор-5-(трифторметил)фенила, 2-фтор-5-(трифторметил)фенила, 2-метокси-4-(трифторметил)фенила, 4-хлор-2-(трифторметил)фенила, 4-хлор-3-(трифторметил)фенила, 4-хлор-3-морфолино-фенила, 2-циано-4-(трифторметил)фенила, оксазоло[5,4-с]пиридин-2-ила, 4-метил-3-(трифторметил)фенила, 3-циклопропил-4-(трифторметил)фенила, 2-фтор-4-метил-фенила, 4-метокси-2-(трифторметил)фенила, 4-метил-2-(трифторметил)фенила, 3,4-дихлорфенила, 2,5-дихлорфенила, 4,5-бис(трифторметил)-2-пиридила, 2-фтор-4-(трифторметил)-фенила, 2-фтор-4-(пентафтор-лямбда6-сульфанил)фенила, 2,4-бис(трифторметил)фенила, 2-метил-3-(трифторметил)фенила, 2-метил-4-(трифторметокси)фенила, 3-хлор-4-(трифторметил)фенила, 3-циклопропил-4-хлор-фенила, 2-хлор-3-(трифторметил)фенила, 2-хлор-3-циклопропил-фенила, 3-(2-азаспиро[3.3]гептан-2-ил)-4-(трифторметил)фенила, 2-хлор-3-(2-азаспиро[3.3]гептан-2-ил)-фенила, 2-хлор-3-(5-окса-2-азаспиро[3.5]нонан-2-ил)фенила, 2-фтор-4-(трифторметил)фенила, 2-фтор-4-метил-фенила, 2-фтор-6-(трифторметил)фенила и 2-(трифторметил)-4-метил-фенила.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R1 представляет собой водород или метил.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R1 представляет собой водород.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R2 представляет собой водород или С1-6-алкил.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R2 представляет собой водород или метил.
В одном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R3 выбран из водорода, галогена, С1-6-алкила и галоген-С1-6-алкила.
В предпочтительном варианте осуществления в настоящем изобретении предложено соединение формулы(I) как описано в данном документе, или его фармацевтически приемлемая соль, где R3 выбран из водорода, галогена и С1-6-алкила.
В конкретно предпочтительном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R3 выбран из водорода, фтора и метила.
В одном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R3 выбран из водорода, метила, фтора и трифторметила.
В одном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R4 выбран из водорода, галогена, галоген-С1-6-алкокси и галоген-С1-6-алкила.
В предпочтительном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R4 выбран из галогена, галоген-С1-6-алкокси и галоген-С1-6-алкила.
В конкретно предпочтительном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R4 выбран из хлора, OCF3 и CF3.
В одном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R4 выбран из водорода, хлора, фтора, OCF3 и CF3.
В одном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R5 выбран из водорода, циано, галогена, C1-6-алкила, С1-6-алкокси, гетероциклоалкила, С3-10-циклоалкила, гетероарила или галоарила.
В предпочтительном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R5 выбран из водорода, циано, галогена, гетероциклоалкила, С3-10-циклоалкила и галогенарила.
В конкретно предпочтительном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R5 выбран из водорода, циано, фтора, хлора, пирролидинила, циклопентила, циклопропила и хлорфенила.
В одном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R5 выбран из водорода, метила, метокси, циано, фтора, хлора, пиролидинила, морфолинила, пиразолила, циклопентила, циклопропила и 4-хлорфенила.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R6 представляет собой водород или галоген.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R6 представляет собой водород или фтор.
В особенно предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R6 представляет собой водород.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R7 выбран из водорода, С1-6-алкила, арила и галоген-С1-6-алкила.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R7 представляет собой С1-6-алкил.
В особенно предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R7 представляет собой трет-бутил.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R7 выбран из водорода, трет-бутил, фенила и CF3.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R8 представляет собой водород или С1-6-алкил.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R8 представляет собой водород или метил.
В особенно предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R8 представляет собой водород.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R9 представляет собой водород.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R10 представляет собой галоген.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R10 представляет собой фтор.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R11 представляет собой галоген.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R11 представляет собой фтор.
В конкретно предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R10 и R11 оба представляют собой фтор.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R12 представляет собой водород.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
(i) X представляет собой C-R3; m равно 0 или 1; n выбрано из 0, 1 и 2; и L выбран из -(СН2)р-, -О-, -ОСН2-, -СН2О- и -СН2ОСН2-; или
(ii) X представляет собой N; m и n оба равны 1; и L представляет собой -(СН2)р-;
р равно 1 или 2;
А выбран из:
(i) арила, замещенного R4, R5 и R6;
(ii) гетероарила, замещенного R7, R8 и R9; и
(iii) гетероциклоалкила, замещенного R10, R11 и
R1 представляет собой водород или С1-6-алкил;
R2 выбран из водорода или С1-6-алкила;
R3 выбран из водорода, галогена, С1-6-алкила и галоген-С1-6-алкила;
R4 выбран из водорода, галогена, галоген-С1-6-алкокси и галоген-С1-6-алкила;
R5 выбран из водорода, циано, галогена, С1-6-алкила, С1-6-алкокси, гетероциклоалкила, С3-10-пиклоалкила, гетероарила или галоарила;
R6 представляет собой водород или галоген;
R7 выбран из водорода, C1-6-алкила, арила и галоген-С1-6-алкила;
R8 представляет собой водород или С1-6-алкил;
R9 представляет собой водород;
R10 представляет собой галоген;
R11 представляет собой галоген, и
R12 представляет собой водород.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
X представляет собой C-R3;
m оба равны 0; или
m оба равны 1;
L выбран из -(СН2)р-, -О-, -ОСН2- и -СН2О-;
р равно 1 или 2;
А выбран из:
(i) арила, замещенного R4, R5 и R6; и
(ii) гетероарила, замещенного R7, R8 и R9;
R1 представляет собой водород;
R2 выбран из водорода или С1-6-алкила;
R3 выбран из водорода, галогена и C1-6-алкила;
R4 выбран из галогена, галоген-С1-6-алкокси и галоген-С1-6-алкила;
R5 выбран из водорода, циано, галогена, гетероциклоалкила, С3-10-циклоалкила и галогенарила;
R6 представляет собой водород;
R7 представляет собой С1-6-алкил;
R8 представляет собой водород; и
R9 представляет собой водород.
В особенно предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
X представляет собой C-R3;
m оба равны 0; или
m оба равны 1;
L выбран из -(СН2)р-, -О-, -ОСН2- и -СН2О-;
р равно 1 или 2;
А выбран из:
(i) фенила, замещенного R4, R5 и R6; и
(ii) оксазолила, замещенного R7, R8 и R9;
R1 представляет собой водород;
R2 выбран из водорода или метила;
R3 выбран из водорода, фтора и метила;
R4 выбран из хлора, OCF3 и CF3;
R5 выбран из водорода, циано, фтора, хлора, пирролидинила, циклопентила, циклопропила и хлорфенила;
R6 представляет собой водород;
R7 представляет собой трет-бутил;
R8 представляет собой водород; и
R9 представляет собой водород.
В одном аспекте в данном изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль, где:
(i) X представляет собой C-R3; m равно 0 или 1; n выбрано из 0, 1 и 2; и L выбран из -(СН2)р-, -О-, -ОСН2-, -СН2ОСН2-, -CF2CH2-, -СН2=СН2-, -(CR16R17)q-CH2O- и -CH2CF2-; или
(ii) X представляет собой N; m равно 1; n равно 1 или 2; и L представляет собой -(СН2)р- или -CF2CH2-;
р выбрано из 1, 2 и 3; q равно 0 или 1; А выбран из:
(i) С6-С14-арила, замещенного R4, R5 и R6;
(ii) 5-14-членного гетероарила, замещенного R7, R8 и R9; и
(iii) 3-14-членного гетероциклоалкила, замещенного R10, R11 и R12;
R1 представляет собой водород или С1-6-алкил;
R2 выбран из водорода, C1-6-алкила и гидрокси-С1-6-алкила;
R3 выбран из водорода, галогена, гидрокси, С1-6-алкокси, C1-6-алкила и галоген-С1-6-алкила;
каждый из R4, R5, R6, R7, R8, R9, R10, R11 и R12 независимо выбран из водорода, галогена, циано, гидрокси, С1-6-алкила, галоген-С1-6-алкила, гидрокси-С1-6-алкила, С1-6-алканоила, галоген-С1-5-алкил-СН(ОН)-, C1-6-алкокси, галоген-С1-6-алкокси, SF5, CH3SO2, С3-10-циклоалкила, С3-10-циклоалкила, замещенного R13, 3-14-членного гетероциклоалкила, 3-14-членного гетероциклоалкила, замещенного R14 и R15, 5-14-членного гетероарила, С6-С14-арила, С6-С14-арилокси, галоген-С6-С14-арила и галоген-С6-С14-арилокси;
каждый из R13, R14 и R15 независимо выбран из С1-6-алкила, С1-6-алкокси, галоген-С1-6-алкила, галоген-С1-6-алкокси, галогена и гидрокси; и
R16 и R17 вместе с атомом углерода, к которому они присоединены, образуют С3-10-циклоалкил.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
(i) X представляет собой C-R3; m равно 0 или 1; n выбрано из 0, 1 и 2; и L выбран из -(СН2)р-, -О-, -ОСН2-, -CF2CH2-, -СН2=СН2-, -(CR16R17)q-CH2O- и -СН2ОСН2-; или
(ii) X представляет собой N; m и n оба равны 1; и L представляет собой -(СН2)р-
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где р равно 2.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где q равно 0 или 1.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где q равно 0.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где А выбран из:
(i) С6-С14-арила, замещенного R4, R5 и R6; и
(ii) 5-14-членного гетероарила, замещенного R7, R8 и R9.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где А выбран из:
(i) фенила, замещенного R4, R5 и R6;
(ii) оксазолила, замещенного R7, R8 и R9; и
(iii) пиридила, замещенного R7, R8 и R9.
В одном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R4 выбран из водорода, галогена, гидрокси, циано, C1-6-алкила, C1-6-алканоила, SF5, C1-6-алкокси, галоген-С1-6-алкокси, галоген-С1-6-алкила, С3-10-циклоалкила, 3-14-членного гетероциклила, 3-14-членного гетероциклоалкила, замещенного R14 и R15, 5-14-членного гетероарила, С6-С14-арилокси и галоген-С6-С14-арила.
В предпочтительном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R4 выбран из галогена, SF5, C1-6-алкила, C1-6-алкокси, галоген-С1-6-алкокси, галоген-С1-6-алкила, С3-10-циклоалкила и 3-14-членного гетероциклоалкила.
В конкретно предпочтительном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R4 выбран из хлора, SF5, метила, метокси, OCF3, CF3, циклопропила и 2-азаспиро[3.3]гептан-2-ила.
В одном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R5 выбран из водорода, циано, галогена, C1-6-алкила, галоген-С1-6-алкила, C1-6-алкокси, 3-14-членного гетероциклоалкила, С3-10-циклоалкила, 5-14-членного гетероарила и галоген-С6-С14-арила.
В предпочтительном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R5 выбран из водорода, циано, галогена, C1-6-алкила, галоген-С1-6-алкила, 3-14-членного гетероциклоалкила, С3-10-циклоалкила и галоген-С6-С14-арила.
В конкретно предпочтительном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R5 выбран из водорода, циано, фтора, хлора, метила, CF3, пирролидинила, циклопентила, циклопропила и хлорфенила.
В одном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R7 выбран из водорода, С1-6-алкила, С6-С14-арила, галоген-С6-С14-арила, галоген-С6-С14-арилокси и галоген-С1-6-алкила.
В предпочтительном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R7 представляет собой С1-6-алкил или галоген-С1-6-алкил.
В конкретно предпочтительном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R7 представляет собой трет-бутил или CF3.
В одном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R8 выбран из водорода, галогена, C1-6-алкила и галоген-C1-6-алкила.
В предпочтительном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R8 представляет собой водород или галоген-С1-6-алкил.
В особенно предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R8 представляет собой водород или CF3.
В одном варианте осуществления в настоящем изобретении предложено соединение формулы (I), как описано в данном документе, или его фармацевтически приемлемая соль, где R14 выбран из С1-6-алкила, С1-6-алкокси и галогена.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где R15 представляет собой водород или галоген.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
(i) X представляет собой C-R3; m равно 0 или 1; n выбрано из 0, 1 и 2; и L выбран из -(СН2)р-, -О-, -ОСН2-, (CR66R17)q-CH2O-, -СН2ОСН2-, -CF2CH2- и -СН2=СН2-; или
(ii) X представляет собой N; m и n оба равны 1; и L представляет собой -(СН2)р-;
р равно 1 или 2;
q равно 0 или 1;
А выбран из:
(i) С6-С14-арила, замещенного R4, R5 и R6;
(ii) 5-14-членного гетероарила, замещенного R7, R8 и R9; и
(iii) 3-14-членного гетероциклоалкила, замещенного R10, R11 и R12;
R1 представляет собой водород или С1-6-алкил;
R2 представляет собой водород или C1-6-алкил;
R3 выбран из водорода, галогена, С1-6-алкила и галоген-С1-6-алкила;
R4 выбран из водорода, галогена, циано, SF5, С1-6-алкила, С1-6-алканоила, C1-6-алкокси, галоген-С1-6-алкокси, галоген-С1-6-алкила, С3-10-циклоалкила, 3-14-членного гетероциклоалкила, 3-14-членного гетероциклоалкила, замещенного R14 и R15, 5-14-членного гетероарила, галоген-С6-С14-арила и С6-С14-арилокси;
R5 выбран из водорода, циано, галогена, C1-6-алкила, галоген-С1-6-алкила, С1-6-алкокси, 3-14-членного гетероциклоалкила, С3-10-циклоалкила, 5-14-членного гетероарила и галоген-С6-С14-арила;
R6 представляет собой водород или галоген;
R7 выбран из водорода, С1-6-алкила, С6-С14-арила, галоген-С6-С14-арила, галоген-С6-С14-арилокси и галоген-С1-6-алкила;
R8 выбран из водорода, галогена, C1-6-алкила и галоген-С1-6-алкила;
R9 представляет собой водород;
R10 представляет собой галоген;
R11 представляет собой галоген;
R12 представляет собой водород;
R14 выбран из галогена, С1-6-алкила и С1-6-алкокси;
R15 представляет собой водород или галоген; и
R16 и R17 вместе с атомом углерода, к которому они присоединены, образуют С3-10-циклоалкил.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
X представляет собой C-R3;
m и n оба равны 0; или
m и n оба равны 1;
L выбран из -(СН2)р-, -О-, -ОСН2- и -CH2O-;
р равно 1 или 2;
А выбран из:
(i) С6-С14-арила, замещенного R4, R5 и R6; и
(ii) 5-14-членного гетероарила, замещенного R7, R8 и R9;
R1 представляет собой водород;
R2 представляет собой водород или C1-6-алкил;
R3 выбран из водорода, галогена и С1-6-алкила;
R4 выбран из галогена, SF5, С1-6-алкила, C1-6-алкокси, галоген-С1-6-алкокси, галоген-С1-6-алкила, С3-10-циклоалкила и 3-14-членного гетероциклоалкила;
R5 выбран из водорода, циано, галогена, C1-6-алкила, галоген-С1-6-алкила, 3-14-членного гетероциклоалкила, С3-10-циклоалкила и галоген-С6-С14-арила;
R6 представляет собой водород;
R7 представляет собой С1-6-алкил и галоген-С1-6-алкил;
R8 представляет собой водород или галоген-С1-6-алкил; и
R9 представляет собой водород.
В особенно предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
X представляет собой C-R3;
m и n оба равны 0; или
m и n оба равны 1;
L выбран из -(СН2)p-, -О-, -ОСН2- и -CH2O-;
р равно 1 или 2;
А выбран из:
(i) фенила, замещенного R4, R5 и R6;
(ii) оксазолила, замещенного R7, R8 и R9; и
(iii) пиридила, замещенного R7, R8 и R9;
R1 представляет собой водород;
R2 представляет собой водород или метил;
R3 выбран из водорода, фтора и метила;
R4 выбран из хлора, SF5, метила, метокси, OCF3, CF3, циклопропила и 2-азаспиро[3.3]гептан-2-ила;
R5 выбран из водорода, циано, фтора, хлора, метила, CF3, пирролидинила, циклопентила, циклопропила и хлорфенила;
R6 представляет собой водород;
R7 выбран из трет-бутила, метила и CF3;
R8 представляет собой водород или CF3; и
R9 представляет собой водород.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
X представляет собой C-R3; и
R3 выбран из водорода, галогена и C1-6-алкила.
В особенно предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
X представляет собой C-R3; и
R3 выбран из водорода, фтора и метила.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
(i) X представляет собой C-R3; m равно 0 или 1; n выбрано из 0, 1 и 2; и L выбран из -(СН2)p-, -О-, -ОСН2-, -(CR16R17)q-CH2O-, -СН2ОСН2-, -CF2CH2- и -СН2=СН2-; или
(ii) X представляет собой N; m и n оба равны 1; и L представляет собой -(СН2)р-;
р равно 1 или 2;
q равно 0 или 1;
R3 выбран из водорода, галогена и С1-6-алкила; и
R16 и R17 вместе с атомом углерода, к которому они присоединены, образуют С3-10-циклоалкил.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
X представляет собой C-R3;
m и n оба равны 0; или
m и n оба равны 1;
L выбран из -(СН2)р-, -О- -ОСН2- и -CH2O-;
р равно 1 или 2; и
R3 выбран из водорода, галогена и С1-6-алкила.
В особенно предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
X представляет собой C-R3;
m и n оба равны 0; или
m и n оба равны 1;
L выбран из -(CH2)p-, -O-, -OCH2- и -CH2O-;
p равно 1 или 2; и
R3 выбран из водорода, фтора и метила.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
А выбран из:
(i) С6-С14-арила, замещенного R4, R5 и R6;
(ii) 5-14-членного гетероарила, замещенного R7, R8 и R9; и
(iii) 3 14-членного гетероциклоалкила, замещенного R10, R11 и R12;
R4 выбран из водорода, галогена, циано, SF5, С1-6-алкила, С1-6-алканоила, С1-6-алкокси, галоген-С1-6-алкокси, галоген-С1-6-алкила, С3-10-циклоалкила, 3-14-членного гетероциклоалкила, 3-14-членного гетероциклоалкила, замещенного R14 и R15, 5-14-членного гетероарила, галоген-С6-С14-арила и С6-С14-арилокси;
R5 выбран из водорода, циано, галогена, С1-6-алкила, галоген-С1-6-алкила, С1-6-алкокси, 3-14-членного гетероциклоалкила, С3-10-циклоалкила, 5-14-членного гетероарила и галоген-С6-С14-арила;
R6 представляет собой водород или галоген;
R7 выбран из водорода, С1-6-алкила, С6-С14-арила, галоген-С6-С14-арила, галоген-С6-С14-арилокси и галоген-С1-6-алкила;
R8 выбран из водорода, галогена, С1-6-алкила и галоген-С1-6-алкила;
R9 представляет собой водород;
R10 представляет собой галоген;
R11 представляет собой галоген;
R12 представляет собой водород;
R14 выбран из галогена, C1-6-алкила и C1-6-алкокси; и
R15 представляет собой водород или галоген.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где: А выбран из:
(i) С6-С14-арила, замещенного R4, R5 и R6; и
(ii) 5-14-членного гетероарила, замещенного R7, R8 и R9;
R4 выбран из галогена, SF5, С1-6-алкила, С1-6-алкокси, галоген-С1-6-алкокси, галоген-С1-6-алкила, С3-10-циклоалкила и 3-14-членного гетероциклоалкила;
R5 выбран из водорода, циано, галогена, С1-6-алкила, галоген-С1-6-алкила, 3-14-членного гетероциклоалкила, С3-10-циклоалкила и галоген-С6-С14-арила;
R6 представляет собой водород;
R7 представляет собой С1-6-алкил и галоген-С1-6-алкил;
R8 представляет собой водород или галоген-С1-6-алкил; и
R9 представляет собой водород.
В особенно предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
А выбран из:
(i) фенила, замещенного R4, R5 и R6;
(ii) оксазолила, замещенного R7, R8 и R9; и
(iii) пиридила, замещенного R7, R8 и R9;
R4 выбран из хлора, SF5, метила, метокси, OCF3, CF3, циклопропила и 2-азаспиро[3.3]гептан-2-ила;
R5 выбран из водорода, циано, фтора, хлора, метила, CF3, пирролидинила, циклопентила, циклопропила и хлорфенила;
R6 представляет собой водород;
R7 представляет собой трет-бутил, метил и CF3;
R8 представляет собой водород или CF3; и
R9 представляет собой водород.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
X представляет собой С-Н;
m и n оба равны 0;
L представляет собой -CH2O-;
А представляет собой С6-С14-арил, замещенный R4, R5 и R6;
R1, R2 и R6 все представляют собой водород;
R4 представляет собой галоген-С1-6-алкил; и
R5 представляет собой галоген или С1-6-алкил.
В особенно предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
X представляет собой С-Н;
m и n оба равны 0;
L представляет собой -CH2O-;
А представляет собой С6-С14-арил, замещенный R4, R5 и R6;
R1, R2 и R6 все представляют собой водород;
R4 представляет собой галоген-С1-6-алкил; и
R5 представляет собой галоген.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
X представляет собой С-Н;
m и n оба равны 0;
L представляет собой -CH2O-;
А представляет собой фенил, замещенный R4, R5 и R6;
R1, R2 и R6 все представляют собой водород;
R4 представляет собой CF3; и
R5 представляет собой фтор или метил.
В особенно предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, где:
X представляет собой C-H;
m и n оба равны 0;
L представляет собой -СН2О-;
А представляет собой фенил, замещенный R4, R5 и R6;
R1, R2 и R6 все представляют собой водород;
R4 представляет собой CF3; и
R5 представляет собой фтор.
В одном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, выбранное из соединений, описанных в Таблице 1.
В предпочтительном варианте осуществления в данном изобретении предложено соединение формулы (I), описанное в данном документе, или его фармацевтически приемлемая соль, выбранное из:
(+)-(4aR,8aS)-6-(4-((4-(трет-бутил)оксазол-2-ил)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)-(4aR,8aS)-6-[4-[[4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(+)-(4aR,8aS)-6-(4-((2-хлор-4-фторфенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
рац-(4aR,8aS)-6-(3-((2-фтор-4-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(3-((2-хлор-4-фторбензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(3-((2-фтор-4-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-(4-(трифторметокси)бензил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-(4-хлор-3-фторбензил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-(2-хлор-4-(трифторметил)фенокси)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-(3-(трифторметил)фенокси)пиперидин-1-карбонил)гексагидро-2H-пиридо[4,3-b][1,4]оксазин-(4H)-она;
(+)- или (-)-(4aR,8aS)-6-[4-[[2-хлор-4-(трифторметокси)фенокси]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(+)- или (-)-(4aR,8aS)-6-(3-((2-фтор-4-(трифторметил)фенокси)метил)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-((4-хлор-2-фторфенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-((4-фтор-2-(трифторметил)фенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-((2-фтор-4-(трифторметил)фенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-(2-(пирролидин-1-ил)-4-(трифторметил)бензил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-((2-хлор-4-(трифторметил)фенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-[4-[[2-циклопентил-4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(+)- или (-)-3-хлор-4-((1-((4aR,8aS)-3-оксооктагидро-2H-пиридо[4,3-b][1,4]оксазин-6-карбонил)пиперидин-4-ил)метокси)бензонитрила;
(+)- или (-)-(4aR,8aS)-6-(3-((2-хлор-4-(трифторметил)фенокси)метил)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-((2-хлор-4-фторфенокси)метил)-4-фторпиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-((4',6-дихлор-[1,1'-бифенил]-3-ил)окси)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-цис-4-((2-хлор-4-фторфенокси)метил)-3-метилпиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(3-((2-хлор-4-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aR)-6-(3-((2-фтор-4-(трифторметокси)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-((2-хлор-4-фторфенокси)метил)-4-метилпиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(4aR,8aS)-6-[3-[(2,4-дихлорфенил)метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[4-[[2-фтор-4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[4-[[2-циклопропил-4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[[3-хлор-4-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[[2-фтор-5-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[2-[2-фтор-6-(трифторметил)фенил]этил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-(3-(2-фтор-4-(трифторметил)фенэтил)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
6-(3-((2,4-бис(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(4aR,8aS)-6-[4-[3-хлор-4-(трифторметил)фенокси]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-(3-метил-4-(((5-метил-6-(трифторметил)пиридин-3-ил)окси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(4aR,8aS)-6-(3-((3,4-дихлорбензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(4aR,8aS)-6-(3-((2,5-дихлорбензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
рац-(4aR,8aS)-6-(2-метил-3-((4-метил-3-(трифторметокси)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(4aR,8aS)-6-(3-(((4,5-бис(трифторметил)пиридин-2-ил)окси)метил)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
рац-(4aR,8aS)-6-(3-((2-фтор-4-(трифторметил)бензил)окси)-2-метилазетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(4aR,8aS)-6-(3-((2-фтор-4-(пентафтор-6-сульфанеил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(4aR,8aS)-6-(3-((4-метил-2-(трифторметокси)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(4aR,8aS)-6-[4-[3-циклопропил-4-(трифторметил)фенокси]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[2-(2-фтор-4-метилфенил)этил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[2-[4-метокси-2-(трифторметил)фенил]этил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[3-(2-азаспиро[3.3]гептан-2-ил)-4-(трифторметил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[2-[4-метил-2-(трифторметил)фенил]этил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-(3-((4-метил-3-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2H-пиридо[4,3-b][1,4]оксазин-3(4H)-она;
(4aR,8aS)-6-(3-((2-метил-3-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
рац-(4aR,8aS)-6-[2-метил-3-[[2-метил-4-(трифторметокси)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
рац-(4aR,8aS)-6-[2-метил-3-[[2-метил-3-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-(4-хлор-3-циклопропилфенокси)азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[4-[2-хлор-3-(трифторметил)фенокси]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-(2-хлор-3-циклопропилфенокси)азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[3-(2-азаспиро[3.3]гептан-2-ил)-2-хлорфенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[2-хлор-3-(5-окса-2-азаспиро[3.5]нонан-2-ил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[(Е)-2-(2-фтор-4-метилфенил)винил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-(3-((Е)-2-фтор-6-(трифторметил)стирил)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она; и
(4aR,8aS)-6-(3-((4-метил-3-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она.
В одном варианте осуществления в данном изобретении предложены фармацевтически приемлемые соли или сложные эфиры соединений формулы (I), описанных в данном документе. В конкретном варианте осуществления в данном изобретении предложены фармацевтически приемлемые соли соединений в соответствии с формулой (I), описанных в данном документе, в частности, гидрохлоридные соли. В дополнительном конкретном варианте осуществления в данном изобретении предложены фармацевтически приемлемые сложные эфиры соединений в соответствии с формулой (I), описанных в данном документе. В дополнительном конкретном варианте осуществления в данном изобретении предложены соединения в соответствии с формулой (I), описанные в данном документе.
В некоторых вариантах осуществления соединения формулы (I) являются изотопно-мечеными за счет наличия одного или более атомов, замещенных атомами, имеющими отличные атомную массу или массовое число. Такие изотопно-меченые (т.е. радиоактивно-меченые) соединения формулы (I) считаются входящими в объем данного описания. Примеры изотопов, которые могут быть включены в соединения формулы (I), включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, хлора и иода, такие как, но не ограничиваясь этим, 2Н, 3Н, 11С, 13С, 14С, 13N, 15N, 15O, 17O, 18O, 31Р, 32Р, 35S, 18F, 36Cl, 123I и 125I, соответственно. Определенные изотопно-меченые соединения формулы (I), например, те, в которые включен радиоактивный изотоп, применимы в исследования тканевого распределения лекарственного препарата и/или субстрата. Радиоактивные изотопы трития, т.е. 3Н, и углерода-14, т.е. 14С, в особенности применимы в этих целях ввиду простоты их включения и наличия готовых средств обнаружения. Например, соединение формулы (I) можно обогащать 1, 2, 5, 10, 25, 50, 75, 90, 95 или даже 99 процентами заданного изотопа.
Замещение более тяжелыми изотопами, таким как дейтерий, т.е. 2Н, может обеспечить определенные терапевтические преимущества в результате большей метаболической стабильности, например, повышение in vivo времени полужизни или снижение необходимых дозировок.
Замещение испускающими позитроны изотопами, таким как 11С, 18F, 15O и 13N, может быть пригодно в исследованиях методом позитронно-эмиссионной томографии (ПЭТ) для изучения занятости рецепторов субстрата. Изотопно-меченые соединения формулы (I) в общем случае можно получать с помощью традиционных методик, известных специалистам в данной области техники, или методами, аналогичными описанным в примерах, приведенных ниже, с использованием соответствующего изотопно меченого реагента вместо применяемого ранее не меченого реагента.
Способы производства
Получение соединений формулы (I) по данному изобретению можно осуществлять путем последовательного или конвергентного синтеза. Способы синтеза по изобретению проиллюстрированы на следующих общих схемах. Навыки, необходимые для проведения реакции и очистки получаемых в результате продуктов, известны специалиста в данной области техники. Заместители и индексы, используемые в нижеприведенном описании способов, имеют значение, указанное в данном документе, если не указано иное.
Если одно из исходных веществ, промежуточных соединений или соединений формулы (I) содержит одну или более функциональных групп, которые являются нестабильными или реакционноспособными в реакционных условиях одного или более этапов реакции, до ключевого этапа можно вводить соответствующие защитные группы (как описано, например, в "Protective Groups in Organic Chemistry" by T.W. Greene and P.G.M. Wutts, 5th Ed., 2014, John Wiley & Sons, N.Y.), используя способы, хорошо известные в данной области техники. Такие защитные группы можно удалять на более поздней стадии синтеза, используя стандартные способы, описанные в литературе.
Если исходные вещества или промежуточные соединения содержат стереогенные центры, соединения формулы (I) можно получать в виде смесей диастереомеров или энантиомеров, которые можно разделять способами, хорошо известными в данной области техники, например, хиральная ВЭЖХ, хиральная СЖХ или хиральная кристаллизация. Рацемические соединения можно, например, разделять на антиподы с помощью диастереомерных солей путем кристаллизации с оптически чистыми кислотами или путем разделения антиподов специальными хроматографическими методами с использованием хирального адсорбента или хирального элюента. Одинаково возможно разделять исходные вещества и промежуточные соединения, содержащие стереогенные центры, с получением диастереомерно/энантиомерно обогащенных исходных материалов и промежуточных соединений. Применение таких диастереомерно/энантиомерно обогащенных исходных веществ и промежуточных соединений в синтезе соединений формулы (I), как правило, приводит к получению соответствующих диастереомерно/энантиомерно обогащенных соединений формулы (I).
Специалисту в данной области техники будет понятно, что при синтезе соединений формулы (I), за исключением случаев, когда необходимо иное, будет применяться «стратегия ортогональных защитных групп», которая позволяет проводить отщепление нескольких защитных групп за раз, не влияя при этом на другие защитные группы в молекуле. Принцип ортогональной защиты хорошо известен в данной области техники и также был описан в литературе (например, Barany and R.В. Merrifield, J. Am. Chem. Soc. 1977, 99, 7363; H. Waldmann et al., Angew. Chem. Int. Ed. Engl. 1996, 35, 2056).
Специалисту в данной области техники будет понятно, что последовательность реакций можно менять в зависимости от реакционной способности и природы промежуточных соединений.
Конкретнее, соединения формулы (I) можно получать способами, приведенными ниже, способами, приведенными в примерах, или аналогичными способами. Соответствующие условия реакций для отдельных этапов реакций известны специалисту в данной области техники. Также, информацию в отношении условий реакций, описанную в литературе, касающейся описанных реакций, можно найти, например, в: Comprehensive Organic Transformations: A Guide to Functional Group Preparations, 2nd Edition, Richard C. Larock. John Wiley & Sons, New York, NY. 1999). Проведение реакций в присутствии или отсутствие растворителя было найдено удобным. Не существует конкретных ограничений по природе предполагаемого растворителя, при условии, что он не оказывает негативное влияние на реакцию или на участвующие в ней реагенты и что он может растворять реагенты, по меньшей мере в некоторой степени. Описанные реакции могут проходить в широком диапазоне температур, а точная температура реакции не является критической для изобретения. Удобно проводить описанные реакции в температурном диапазоне от -78°С до кипения. Время, необходимое для реакции, также может сильно варьироваться в зависимости от многих факторов, а именно температуры реакции и природы реагентов. При этом периода от 0,5 часа до нескольких суток обычно достаточно, чтобы получить описанные промежуточные соединения и соединения. Последовательность реакции не ограничена приведенной на схемах, однако в зависимости от исходных веществ и их соответствующей реакционной способности, последовательность этапов реакции можно свободно менять.
Если исходные материалы или промежуточные соединения коммерчески не доступны или их синтез не описан в литературе, их можно готовить по аналогии с существующими процедурами для близких аналогов или как описано в экспериментальном разделе.
В данном тексте используются следующие сокращения:
АсОН = уксусная кислота, АЦН = ацетонитрил, Bn = бензил, Boc = трет-бутилоксикарбонил, CAS RN = номер по системе кодирования в реферативном журнале «Chemical Abstracts)), Cbz = бензилоксикарбонил, Cs2CO3 = карбонат цезия, СО = монооксид углерода, CuCl = хлорид меди (I), CuCN = цианид меди (I), CuI = иодид меди (I), ДАСТ = (диэтиламино)серы трифторид, ДБУ = 1,8-диазабицикло[5,4,0]ундек-7-ен, ДЭАД = диэтилазодикарбоксилат, ДИАД = диизопропилазодикарбоксилат, ДМАП = 4-диметиламинопиридин, ДМЭ = диметоксиэтан, ДМЭДА = N,N'-диметилэтилендиамин, ДМФА = N,N-диметилформамид, ДИПЭА = N,N-диизопропилэтиламин, dppf = 1,1 бис(дифенилфосфино)ферроцен, ЭДК.HCl = N-(3-диметиламинопропил)-N'-этилкарбодиимидгидрохлорид, ЭУ = электронный удар, ИЭР = ионизация электрораспылением, EtOAc = этилацетат, EtOH = этанол, ч = час(ы), МК = муравьиная кислота, H2O = вода, H2SO4 = серная кислота, HATU = 1-[бис(диметиламино)метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиний-3-оксид гексафторфосфат, HBTU = O-бензотриазол-N,N,N',N'-тетраметилуроний-гексафторфосфат, HCl = хлористый водород, HOBt = 1-гидрокси-1Н-бензотриазол; ВЭЖХ = высокоэффективная жидкостная хроматография, iPrMgCl = изопропилмагнийхлорид, I2 = иод, ИПС = 2-пропанол, ПИР = ионное распыление в положительном (режиме), ОИР = отрицательное ионное распыление в отрицательном (режиме), K2CO3 = карбонат калия, KHCO3 = гидрокарбонат калия, KI = иодид калия, KOH = гидроксид калия, K3PO4 = трехосновный фосфат калия, LiAlH4 или LAH = алюмогидрид лития, LiHMDS = бис(триметилсилил)амид лития, LiOH = гидроксид лития, м-ХПБК = мета-хлорпероксибензойная кислота, MgSO4 = сульфат магния, мин = минута(ы), мл = миллилитр, ЖХСД = жидкостная хроматография среднего давления, МС = масс-спектр, nBuLi = н-бутиллитий, NaBH3CN = цианоборгидрид натрия, NaH = гидрид натрия, NaHCO3 = гидрокарбонат натрия, NaNO2 = нитрит натрия, NaBH(OAc)3 = триацетоксиборгидрид натрия, NaOH = гидроксид натрия, Na2CO3 = карбонат натрия, Na2SO4 = сульфат натрия, Na2S2O3 = тиосульфат натрия, NBS = N-бромсукцинимид, nBuLi = н-бутиллитий, NEt3 = триэтиламин (ТЭА), NH4Cl = хлорид аммония, NMP = N-метил-2-пирролидон, ОАс = ацетокси, Т3Р = пропилфосфоновый ангидрид, ПЭ = петролейный эфир, ЗГ = защитная группа, Pd-С = палладий на активированном угле, PdCl2(dppf)-CH2Cl2 = комплекс дихлорида 1,1'-бис(дифенилфосфино)ферроцен-палладия (II) с дихлорметаном, Pd2(dba)3 = трис(дибензилиденацетон)дипалладий (0), Pd(OAc)2 = ацетат палладия (II), Pd(OH)2 = гидроксид палладия, Pd(PPh3)4 = тетракис(трифенилфосфин)палладий (0), ПТСК = п-толуолсульфоновая кислота, R = любая группа, КТ = комнатная температура, СЖХ = сверхкритическая жидкостная хроматография, S-PHOS = 2-дициклогексилфосфино-2',6'-диметоксибифенил, ТБАЙ = тетра-бутиламмоний-иод, ТЭА = триэтиламин, ТФУ = трифторуксусная кислота, ТГФ = тетрагидрофуран, ТМЭДА = М,М,М',М'-тетраметилэтилендиамин, ZnCl2 = хлорид цинка, Hal = галоген.
Соединения формулы I, где A, L, X, n, n, R1 и R2 соответствуют описанию в данном документе, можно синтезировать по аналогии с описанными в литературе процедурами и/иди как проиллюстрировано, например, на Схеме 1.
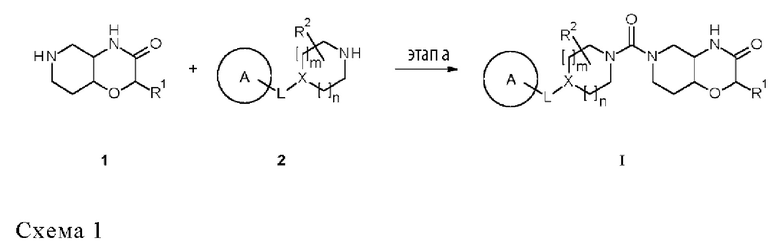
Соответственно, 4а,5,6,7,8,8а-гексагидро-4Н-пиридо[4,3-b][1,4]оксазин-3-оны 1 приводят в реакцию с промежуточными соединениями 2 в присутствии образующего мочевину реагента, такого как бис(трихлорметил)карбонат, используя подходящие основание и растворитель, например, гидрокарбонат натрия в ДХМ, чтобы получить соединения формулы I (этап а). Дополнительные образующие мочевину реагенты включают, но не ограничиваются этим, фосген, трихлорметилхлорформиат, (4-нитрофенил)карбонат или 1,1'-карбонилдиимидазол. Реакции этого типа и применение этих реагентов широко описаны в литературе (например, G. Sartori et al., Green Chemistry 2000, 2, 140). Специалисту в данной области техники будет понятно, что порядок добавления реагентов может быть важен в этом типе реакций в связи с реактивностью и стабильностью образуемых промежуточных карбамоилхлоридов, а также во избежание образования нежелательных симметрических побочных продуктов мочевины.
Промежуточные соединения 1 можно синтезировать, как проиллюстрировано на схеме 2, и/или по аналогии со способами, описанными в литературе.

Таким образом, производные 3-аминопиперидин-4-ола 3, в которых «ЗГ» обозначает подходящую защитную группу, такую как защитная группа Cbz или Boc, можно ацилировать, например, ацилхлоридами 4, в которых R1 соответствует описанию в данном документе, а «УГ» обозначает подходящую уходящую группу (например, Cl или Br), используя подходящее основание, такое как карбонат натрия или калия, гидроксид натрия или ацетат натрия, в соответствующем растворителе, таком как ТГФ, вода, ацетон или их смеси, чтобы получить промежуточные соединения 5 (этап а). Промежуточные соединения 4 являются коммерчески доступными или же их можно получить в соответствии с описанными в литературе способами в ахиральной (R1=Н) рацемической (R1 не является H) или энантиомерно чистой форме (R1 не является Н).
Промежуточные соединения 5 можно циклизовать до промежуточных соединений 6, используя способы, хорошо известные в данной области техники, например, путем обработки 5 гидридом натрия в ТГФ или трет-бутоксидом калия в ИПС и воде (этап b). Реакции этого типа описаны в литературе (например, Z. Rafinski et al., J. Org. Chem. 2015, 80, 7468; S. Dugar et al., Synthesis 2015, 47(5), 712; WO 2005/066187).
Удаление защитной группы в промежуточных соединениях 6 с применением способов, известных в данной области техники (например, группы Вое, используя ТФУ в ДХМ при температурах от 0°С до комнатной температуры, группы Cbz, используя водород в присутствии подходящего катализатора, такого как Pd или Pd(OH)2 на угле, в подходящем растворителе, таком как МеОН, EtOH, EtOAc или их смеси, и как описано, например, в "Protective Groups in Organic Chemistry" авторства T.W. Greene and P.G.M. Wuts, 4th Ed., 2006, Wiley N.Y.), позволяет получить промежуточные соединения 1 (этап с).
Промежуточные соединения 1 можно получать в виде смесей диастереомеров и энантиомеров, соответственно, или в виде отдельных стереоизомеров в зависимости от того, применяют при их синтезе рацемические смеси или энантиомерно чистые формы производных цис- или транс-3-аминопиперидин-4-ола 3 или промежуточных соединений 4. Промежуточные соединения 3 являются коммерчески доступными, и их синтез также описан в литературе (например, WO 2005/066187; WO 2011/0059118; WO 2016/185279). Оптически чистые промежуточные соединения 1В и 1С в цис-конфигурации можно получать, например, в соответствии со Схемой 3 путем хирального разделения коммерчески доступного рац-(4aR,8aS)-4a,5,6,7,8,8а-гексагидро-4Н-пиридо[4,3-b][1,4]оксазин-3-она (1А) (необязательно в форме соли, такой как, например, гидрохлоридная соль), используя способы, известные в данной области техники, например, с помощью кристаллизации диастереомерной соли или с помощью хиральной хроматографии (этап а).
В некоторых вариантах осуществления изобретения промежуточные соединения 2 представляют собой промежуточные соединения типа В. Промежуточные соединения типа В, в которых A, m, n и R2 соответствуют описанию в данном документе могут быть получены способами, хорошо известными специалистам в данной области, и на примерах общих синтетических методик, изложенных на Схеме 4.

Кетоны 7, как коммерчески доступные, так и полученные способами, известными в данной области, могут быть подвергнуты, например, реакции Виттига с алкилидентрифенилфосфоранами типа 8а в подходящем растворителе, таком как, например, ТГФ, Метил-ТГФ или ДМСО для получения промежуточных соединений 9 (этап а). Фосфораны 8а могут быть образованы обработкой соответствующих фосфониевых солей подходящим основанием, таким как BuLi, NaH или KOtBu, в подходящем растворителе, таком как ТГФ, диоксан или Метил-ТГФ, и могут быть выделены или использованы in situ. Фосфониевые соли, в свою очередь, легко доступны из арил/гетероарил/гетероциклически замещенного алкилгалогенида (галогенидом которого являются Cl, Br и иод) и трифенилфосфина в подходящем растворителе, таком как толуол. Может применяться нагревание для ускорения реакции или доведения реакции до ее завершения (например, Н.J. Cristau, Б. Plénat in PATAI'S Chemistry of Functional Groups, Editor(s): Frank В.. Hartley, 07th August 2006, Series Editor(s): Prof. Saul Patai).
В качестве альтернативы промежуточные соединения 9 могут быть получены с помощью реакции Хорнера-Уодсворта-Эммонса (HWE) с использованием кетонов 7 и фосфонатов 8b, где Ra представляет собой алкил, например метил или этил. Фосфонаты 8b α-металлируют in situ, используя подходящее основание и растворитель, такой как NaH, nBuLi или KOtBu в ТГФ (этап а). Фосфонаты 8b легко получают с использованием, например, реакции Арбузова путем алкилирования арил/гетероарил/гетероциклического галогенида (галогенидом которого являются Cl, Br и иод) коммерчески доступным триалкилфосфитом (например, Chem. Rev. 1984, 84, 577).
Реакции олефинирования обоих типов широко описаны в литературе (например, Current Org. Chem. 2015, 19(9), страница 744; Chem. Rev. 1989, 89(4), 863; Org. React. 1977, 25,73, Liebigs Ann./Recueil 1997, 1283; Acc. Chem. Res. 1983, 16, 411).
Восстановление двойной связи в промежуточных соединениях 9 используя, например, водород в присутствии подходящего катализатора, такого как палладий на угле, в подходящем растворителе или смеси растворителей, такой как EtOAc, МеОН или АсОН, дает соединения 10 (этап b).
Удаление защитной группы из промежуточных соединений 10 с применением способов, известных в данной области техники (например, группы Вое, используя ТФУ в ДХМ или 4 М HCl в диоксане при температурах от 0°С до комнатной температуры, группы Cbz, используя водород в присутствии подходящего катализатора, такого как Pd или Pd(OH)2 на угле, в подходящем растворителе, таком как МеОН, EtOH, EtOAc или их смеси, и как описано, например, в "Protective Groups in Organic Chemistry" T.W. Greene и P.G.M. Wuts, 4th Ed., 2006, Wiley N.Y.)), позволяет получить промежуточные соединения В (этап с).
В некоторых вариантах осуществления изобретения промежуточные соединения 2 представляют собой промежуточные соединения типа С.Промежуточные соединения типа С, в которых A, R2 и р соответствуют описанию в данном документе, r=0, 1 или 2 и (m+n)=2 или 3 могут быть получены способами, хорошо известными в данной области, и на примерах общих синтетических методик, изложенных на Схеме 5.

Алкилирование необязательно монозащищенных производных пиперазина или 1,4-диазепана 11а,b (коммерчески доступны или синтезированы аналогично литературным методам) с производными арил/гетероарил/гетероциклилзамещенного алкила 12, коммерчески доступными или синтезированными согласно литературным методикам, и в которых УГ означает подходящую уходящую группу, такую как хлор, бром, иод, OSO2алкил (например мезилат (метансульфонат), OSO2фторалкил (например, трифлат (трифторметансульфонат) или OSO2арил (например, тозилат (п-толуолсульфонат с использованием подходящего основания в подходящем растворителе (например, гидрид натрия в ДМФА)) при температурах от 0°С до температуры кипения растворителя дает промежуточные соединения 14а,b (этап а).
Альтернативно, соединения 11а,b могут быть подвергнуты реакции восстановительного аминирования с альдегидами типа 13, используя подходящий восстанавливающий агент и растворитель, такой как NaBH3CN в МеОН, АсОН, или их смесь с получением промежуточных соединений 14а,b (этап а).
Удаление защитной группы из промежуточных соединений 14b с применением способов, известных в данной области техники (например, группы Вое, используя ТФУ в ДХМ или 4 М HCl в диоксане при температурах от 0°С до комнатной температуры, группы Cbz, используя водород в присутствии подходящего катализатора, такого как Pd или Pd(OH)2 на угле, в подходящем растворителе, таком как МеОН, EtOH, EtOAc или их смеси, и как описано, например, в "Protective Groups in Organic Chemistry" T.W. Greene и P.G.M. Wuts, 4th Ed., 2006, Wiley N.Y.)), позволяет получить промежуточные соединения С (этап с).
В некоторых вариантах осуществления изобретения соединения формулы I представляют собой соединения типа Ie. Соединения Ie, в которых А, р, R1 и R2 соответствуют определениям в данном документе, и (m+n)=2 или 3 могут быть получены аналогично литературным методикам или методам, описанным на Схеме 6 ниже.

Соединения 1 могут сочетаться с производными пиперазина или 1,4-диазепана 11а, применяя, например, условия, указанные в Схеме 1, этап а, для получения промежуточных соединений 15 (этап а).
Промежуточные соединения 15 могут быть преобразованы в соединения IC по аналогии с методикой, описанной на Схеме 5, этап а (этап b).
Альтернативно, соединения 1 могут сочетаться с моноз ащище иными производными пиперазина или 1,4-диазепана 11b, в которых ЗГ означает подходящую защитную группу, такую как защитная группа Cbz или Boc, применяя, например, условия, указанные на Схеме 1, этап а, с получением промежуточных соединений 16 (этап с).
Удаление защитной группы опубликованными методами или как описано на Схеме 5, этап с, дает промежуточные соединения 15 (этап d).
В некоторых вариантах осуществления изобретения промежуточные соединения 2 представляют собой промежуточные соединения типа D. Промежуточные соединения типа D, в которых A, m, n и R2 соответствуют описанию в данном документе и R3 выбран из водорода, галогена, C1-6-алкокси, C1-6-алкила и галоген-C1-6-алкила, могут быть получены способами, хорошо известными в данной области, и на примерах общих синтетических методик, изложенных на Схеме 7.

Спирты типа 17 могут подвергаться реакции Мицунобу с промежуточными соединениями 18 в которых ЗГ представляет собой подходящую защитную группу, такую как Cbz, Boc или Bn, с использованием подходящего фосфина, такого как трифенилфосфин, и диалкилазодикарбоксилата, такого как DEAD или DIAD, в подходящем растворителе, таком как ТГФ, с получением промежуточных соединений 19 (этап а). Реакции Мицунобу такого типа широко описаны в литературе (например, Org. Chem. Front. 2015, 2, 739; Chem. Rev. 2009, 109 (6), 2551).
Удаление защитной группы из промежуточных соединений 19 с применением способов, описанных в литературе и описанных, например, под Схемой 3, этап с, позволяет получить промежуточные соединения D (этап b).
В качестве альтернативы промежуточные соединения 19 могут быть получены из спиртов 17, которые могут быть алкилированы соединениями 20, в которых УГ является подходящей уходящей группой, такой как хлор, бром, иод, OSO2алкил (например, метансульф онат), OSO2фторалкил (например, трифторметансульфонат) или OSO2арил (например п-толуолсульфонат, используя подходящее основание в подходящем растворителе (например, гидрид натрия в ДМФА) при температурах между 0°С и температуре кипения растворителя (этап с).
Кроме того, промежуточные соединения 19 могут быть синтезированы путем алкилирования спиртов типа 18 соединениями 21 в условиях, описанных на этапе с (этап d).
В другом варианте осуществления промежуточные соединения 2 представляют собой промежуточные соединения типа Е. Промежуточные соединения типа Е, в которых A, m, n, R и R соответствуют описанию в данном документе, могут быть получены способами, хорошо известными в данной области, и на примерах общих синтетических методик, изложенных на Схеме 8. В случае R представляет собой гидроксигруппу, может применяться подходящая стратегия защиты группы, известная специалистам в данной области.
Функциональная группа карбоновой кислоты в производных 22, в которых ЗГ означает подходящую защитную группу, такую как, например, защитная группа Вое, Cbz или Вп, как коммерчески доступную, так и полученную способами, известными в данной области, может быть превращена в хлорангидрид (LG = С1) или амид Вайнреба (LG = NMeOMe) с применением методов, широко описанных в литературе, для получения промежуточных соединений 23 (этап а).
Промежуточные соединения 23 могут быть введены в реакцию с соединениями типа 24, как коммерчески доступными, так и синтезированными методами, известными в данной области и описанными ниже, с получением промежуточных соединений 25 (этап b).
Если соединения 24 не являются коммерчески доступными, их можно получить аналогично литературным методам. Например, депротонирование реакционноспособной метальной группы в необязательно замещенных гетероциклах 27, используя подходящее основание, такое как nBuLi или LiHMDS, в подходящем растворителе, например ТГФ, гексане или их смеси, при температуре от -78°С до комнатной температуры дают промежуточные соединения 24, в которых MX=Li (этап d).
Соединения 24, в которых MX=MgHal, где Hal представляет собой Cl, Br или I (реактивы Гриньяра), могут быть получены реакцией соответствующих замещенных бензилгалогенидов 28 с магнием в подходящем растворителе, таком как ТГФ, необязательно в присутствии каталитических количеств иода при температурах в диапазоне от 0°С до точки кипения растворителя (этап d).
Соединения 25 могут быть далее преобразованы в соединения 26 реакцией дезоксифторирования с использованием подходящего фторирующего агента, такого как DAST, Deoxo-Fluor (трифторид бис(2-метоксиэтил)серы) или тетрафторбораты аминодифторсульфиния (XtalFluor-E®, XtalFluor-M® в присутствии, например тригидрофторида триэтиламина и TEA или DBU) в подходящем растворителе, таком как ДХМ или АЦН (этап d).
Удаление защитной группы из промежуточных соединений 26 с применением способов, описанных в литературе и описанных, например, под Схемой 3, этап с, позволяет получить промежуточные соединения Е (этап е).
В дополнительном варианте осуществления промежуточные соединения 2 представляют собой промежуточные соединения типа F. Промежуточные соединения типа F, в которых A, m, n, R2 и R3 соответствуют описанию в данном документе, могут быть получены способами, хорошо известными в данной области, и на примерах общих синтетических методик, изложенных на Схеме 9. В случае R3 представляет собой гидроксигруппу, может применяться подходящая стратегия защиты группы, известная специалистам в данной области.
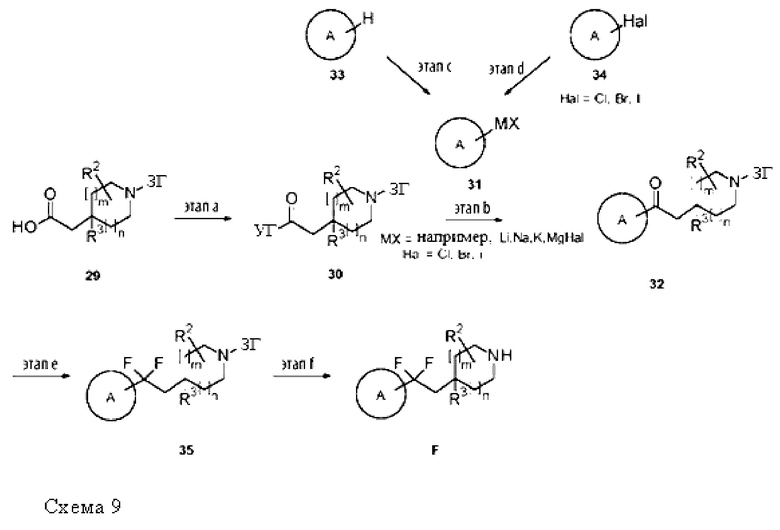
Карбоксильная функциональная группа в промежуточных соединениях 29, как коммерчески доступных, так и полученных методами, известными в данной области, в которых ЗГ означает подходящую защитную группу, такую как, например, защитная группа Boc, Chz или Bn, может быть превращена в хлорангидрид (УГ = Cl) или амид Вайнреба (УГ = NMeOMe) с применением методов, широко описанных в литературе, для получения промежуточных соединений 30 (этап а).
Промежуточные соединения 30 могут быть введены в реакцию с соединениями типа 31, как коммерчески доступными, так и синтезированными методами, известными в данной области и описанными ниже, с получением промежуточных соединений 32 (этап b).
В случае, если соединения 31 не являются коммерчески доступными, их можно получить аналогично литературным методам. Например, депротонирование необязательно замещенных арильных или гетероарильных колец 33, используя подходящее основание, такое как н-BuLi, втор-BuLi, трет-BuLi, LiHMDS, NaH, KH, в подходящем растворителе, таком как ТГФ, н-гексан или их смеси, при температурах от -73°С до комнатной температуры дает промежуточные соединения 31, в которых, в зависимости от используемого основания, MX = Li, Na или K (этап c).
Соединения 31, в которых MX = MgHal, где Hal представляет собой Cl, Br или I (реактивы Гриньяра), могут быть получены реакцией соответствующих необязательно замещенных арил- или гетероарилгалогенидов 34 посредством прямого введения магния (например, магниевые стружки, необязательно в присутствии каталитических количеств иода, порошок в присутствии LiCl или магния Рике, органических галогенидов) или путем галоген-магниевого обмена путем обработки 34, в котором Hal предпочтительно представляет собой бром или иод, алкил-галогенидом магния, таким как iPrMgCl (необязательно в присутствии LiCl) в подходящих растворителях, таких как диэтиловый эфир или ТГФ, при температуре от 0°С до точки кипения растворителя (этап d).
Соединения 32 могут быть далее преобразованы в соединения 35 реакцией дезоксифторирования с использованием подходящего фторирующего агента, такого как DAST, Deoxo-Fluor (трифторид бис(2-метоксиэтил)серы) или тетрафторбораты аминодифторсульфиния (XtalFluor-E®, XtalFluor-M® в присутствии, например тригидрофторида триэтиламина и TEA или DBU) в подходящем растворителе, таком как ДХМ или АЦН (этап е).
Удаление защитной группы из промежуточных соединений 35 с применением способов, описанных в литературе и описанных, например, под Схемой 3, этап с, позволяет получить промежуточные соединения F (этап f).
В некоторых вариантах осуществления изобретения промежуточные соединения 2 представляют собой промежуточные соединения типа G. Промежуточные соединения типа G, в которых A, m, n, R2 соответствуют описанию в данном документе и R3 представляет собой водород, C1-6-алкокси, С1-6-алкил и галоген-С1-6-алкил могут быть получены способами, хорошо известными в данной области и на примерах общих синтетических методик, изложенных на Схеме 10.

Промежуточные соединения 38 можно получать из спиртов 36, в которых ЗГ представляет собой подходящую защитную группу, такую как Boc, Cbz или Bn, которые могут быть алкилированы соединениями 37, в которых УГ представляет собой подходящую уходящую группу, такую как хлор, бром, иод, OSO2алкил (например, метансульфонат), OSO2фторалкил (например, трифторметансульфонат) или OSO2арил (например, п-толуолсульфонат), используя подходящее основание, такое как гидрид натрия, трет-бутоксид калия, в подходящем растворителе (например, в ДМФА или ТГФ) при температурах от 0°С до температуры кипения растворителя (этап а).
Удаление защитной группы из промежуточных соединений 38 с применением способов, описанных в литературе и описанных, например, под Схемой 4, этап с, позволяет получить промежуточные соединения G (этап b).
В некоторых вариантах осуществления изобретения промежуточные соединения 2 представляют собой промежуточные соединения типа Н. Промежуточные соединения типа Н, в которых A, m, n, R2 и R3 соответствуют описанию в данном документе, могут быть получены способами, хорошо известными в данной области, и на примерах общих синтетических методик, изложенных на Схеме 11.

Спирты типа 17 могут подвергаться реакции Мицунобу с промежуточными соединениями 39 в которых ЗГ представляет собой подходящую защитную группу, такую как Cbz, Boc или Bn, с использованием подходящего фосфина, такого как трифенилфосфин, и диалкилазодикарбоксилата, такого как DEAD или DIAD, в подходящем растворителе, таком как ТГФ, с получением промежуточных соединений 41 (этап а). Реакции Мицунобу такого типа широко описаны в литературе (например, Org. Chem. Front. 2015, 2, 739; Chem. Rev. 2009, 109 (6), 2551).
Удаление защитной группы из промежуточных соединений 41 с применением способов, описанных в литературе и описанных, например, под Схемой 4, этап с, позволяет получить промежуточные соединения Н (этап b).
В качестве альтернативы промежуточные соединения 41 могут быть получены из спиртов 17, которые могут быть алкилированы соединениями 40, в которых УГ является подходящей уходящей группой, такой как хлор, бром, иод, OSO2алкил (например, метансульфонат), OSO2фторалкил (например, трифторметансульфонат) или OSO2арил (например п-толуолсульфонат, используя подходящее основание, такое как Cs2CO3, NaH, в подходящем растворителе, таком как ДМФА, при температурах между 0°С и температуре кипения растворителя (этап с).
Введение промежуточных соединений Н в реакцию с промежуточными соединениями 1, например, используя условия, описанные на Схеме 1, этап а, с получением соединений типа If, где A, R1, R2, R3, m и n соответствуют определениям в данном документе.

В качестве альтернативы соединения типа If могут быть получены по Схеме 12.
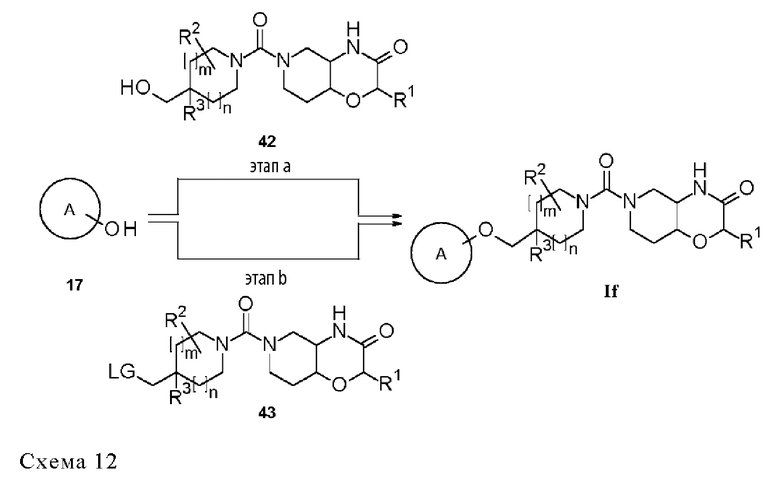
Спирты типа 17 может подвергаться реакции Мицунобу с промежуточными соединениями 42, используя подходящий фосфин, такой как трифенилфосфин, и диалкилазодикарбоксилат, такой как DEAD или DIAD в подходящем растворителе, таком как ТГФ, с получением соединений ID (этап а). Реакции Мицунобу такого типа широко описаны в литературе (например, Org. Chem. Front. 2015, 2, 739; Chem. Rev. 2009, 109 (6), 2551).
В качестве альтернативы соединения ID могут быть получены из спиртов 17, которые непосредственно могут быть алкилированы соединениями 43, в которых УГ является подходящей уходящей группой, такой как хлор, бром, иод, OSO2алкил (например, метансульфонат), OSO2фторалкил (например, трифторметансульфонат) или OSO2арил (например п-толуолсульфонат, используя подходящее основание, такое как Cs2CO3, NaH, в подходящем растворителе, таком как ДМФА, при температурах между 0°С и температуре кипения растворителя (этап b).
В некоторых вариантах осуществления изобретения промежуточные соединения 2 представляют собой промежуточные соединения типа J. Промежуточные соединения типа J, в которых А, m, n, R2 и R3 соответствуют описанию в данном документе, могут быть получены способами, хорошо известными в данной области, и на примерах общих синтетических методик, изложенных на Схеме 13.

Промежуточные соединения 46 могут быть получены из гетероциклоалкилов или гетероарилов 45, которые могут быть алкилированы соединениями 44, в которых УГ является подходящей уходящей группой, такой как хлор, бром, иод, OSO2алкил (например, метансульфонат), OSO2фторалкил (например, трифторметансульфонат) или OSO2арил (например п-толуолсульфонат, используя подходящее основание, такое как Cs2CO3, K2CO3, NaH, в подходящем растворителе, таком как ДМФА, при температурах между 0°С и температуре кипения растворителя (этап а).
Удаление защитной группы из промежуточных соединений 46 с применением способов, описанных в литературе и описанных, например, под Схемой 4, этап с, позволяет получить промежуточные соединения J (этап b).
Введение промежуточных соединений J в реакцию с промежуточными соединениями 1 позволяет получить соединения типа Ig, где А, R1, R2, R3, m и n соответствуют определениям в данном документе.
В качестве альтернативы, соединения Ig непосредственно могут быть получены из гетероциклоалкилов или гетероарилов 45, которые могут быть алкилированы соединениями 47, в которых УГ является подходящей уходящей группой, такой как хлор, бром, иод, OSO2алкил (например, метансульфонат), OSO2фторалкил (например, трифторметансульфонат) или OSO2арил (например п-толуолсульфонат, используя подходящее основание, такое как Cs2CO3, K2CO3, NaH, в подходящем растворителе, таком как ДМФА, при температурах между 0°С и температуре кипения растворителя (Схема 14).

В некоторых вариантах осуществления изобретения промежуточные соединения 2 представляют собой промежуточные соединения типа К. Промежуточные соединения типа K, в которых A, m, n, R2 и R3 соответствуют описанию в данном документе, могут быть получены способами, хорошо известными в данной области, и на примерах общих синтетических методик, изложенных на Схеме 15.
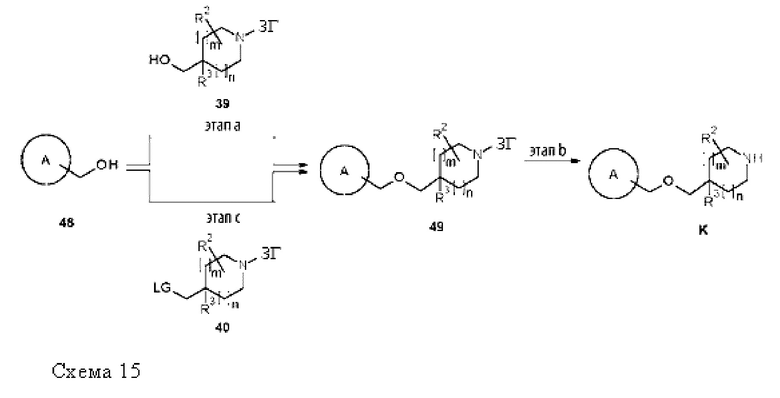
Спирты типа 48 могут подвергаться реакции Мицунобу с промежуточными соединениями 39 в которых ЗГ представляет собой подходящую защитную группу, такую как Cbz, Boc или Bn, с использованием подходящего фосфина, такого как трифенилфосфин, и диалкилазодикарбоксилата, такого как DEAD или DIAD, в подходящем растворителе, таком как ТГФ, с получением промежуточных соединений 49 (этап а). Реакции Мицунобу такого типа широко описаны в литературе (например, Org. Chem. Front. 2015, 2, 739; Chem. Rev. 2009, 109 (6), 2551).
Удаление защитной группы из промежуточных соединений 49 с применением способов, описанных в литературе и описанных, например, под Схемой 4, этап с, позволяет получить промежуточные соединения К (этап b).
В качестве альтернативы промежуточные соединения 49 могут быть получены из спиртов 48, которые могут быть алкилированы соединениями 40, в которых УГ является подходящей уходящей группой, такой как хлор, бром, иод, OSO2алкил (например, метансульфонат), OSO2фторалкил (например, трифторметансульфонат) или OSO2арил (например п-толуолсульфонат, используя подходящее основание, такое как Cs2CO3, NaH, в подходящем растворителе, таком как ДМФА, при температурах между 0°С и температуре кипения растворителя (этап с).
В некоторых вариантах осуществления изобретения промежуточные соединения 2 представляют собой промежуточные соединения типа L. Промежуточные соединения типа L, в которых A, m, n, R2 и R3 соответствуют описанию в данном документе могут быть получены способами, хорошо известными специалистам в данной области, и на примерах общих синтетических методик, изложенных на Схеме 16.

Промежуточные соединения 51 могут быть получены, например, из альдегидов 50, как коммерчески доступных, так и полученных методами, известными в данной области, с использованием реакции Виттига или реакции Хорнера-Уодсворта-Эммонса (HWE), используя алкилидентриф енилфосф ораны типа 8а и фосфонаты 8b, соответственно, как описано на этапе а Схемы 4 (этап а).
Восстановление двойной связи в промежуточных соединениях 51 применяя условия, описанных для этапе b на Схеме 4, дает соединения 52 (этап b).
Удаление защитной группы из промежуточных соединений 52 с применением методов, известных в данной области техники и описанных на этапе с на Схеме 4, дает промежуточные соединения L (этап с).
Удаление защитной группы из промежуточных соединений 51 с применением методов, известных в данной области техники и описанных на этапе с на Схеме 4, дает промежуточные соединения М (этап d).
В одном аспекте в данном изобретении предложен способ производства соединений мочевины формулы (I), описанных в данном документе, включающий: проведение реакции первого амина формулы 1, где R1 соответствует описанию в данном документе, предпочтительно где R1 представляет собой водород,

со вторым амином 2, где A, L, m, n, X и R2 соответствуют описанию в данном документе,

в присутствии основания и образующего мочевину реагента,
с образованием указанного соединения формулы (I).
В одном варианте осуществления предложен способ в соответствии с изобретением, в котором указанное основание представляет собой гидрокарбонат натрия.
В одном варианте осуществления предложен способ в соответствии с изобретением, в котором указанный образующий мочевину реагент выбран из бис(трихлорметил)карбоната, фосгена, трихлорметилхлорформиата, (4-нитрофенил)карбоната и 1,1'-карбонилдиимидазола, предпочтительно, в котором указанный образующий мочевину реагент бис(трихлорметил)карбонат.
В одном аспекте в данном изобретении предложено соединение формулы (I), описанное в данном документе, произведенное в соответствии с любым из способов, описанных в данном документе.
Ингибирующая MAGL активность
Соединения по данному изобретению являются ингибиторами MAGL. Таким образом, в одном аспекте в данном изобретении предложено применение соединений формулы (I), описанных в данном документе, для ингибирования MAGL у млекопитающего.
В дополнительном аспекте в данном изобретении предложены соединения формулы (I), описанные в данном документе, для применения в способе ингибирования MAGL у млекопитающего.
В дополнительном аспекте в данном изобретении предложено применение соединений формулы (I), описанных в данном документе, для изготовления лекарственного средства для ингибирования MAGL у млекопитающего.
В дополнительном аспекте в данном изобретении предложен способ ингибирования MAGL у млекопитающего, который включает введение млекопитающему эффективного количества соединения формулы (I), описанного в данном документе.
Профили соединений в отношении ингибирующей MAGL активности получали путем измерения ферментативной активности MAGL после гидролиза 4-нитрофенилацетата, приводящего к получению 4-нитрофенола, который поглощает на 405-412 нм (G.G. Muccioli, G. Labar, D.M. Lambert, Chem. Bio. Chem. 2008, 9, 2704-2710). Далее по тексту этот анализ сокращенно называется «анализом 4-NPA».
Анализ 4-NPA проводили в 384-луночных аналитических планшетах (черные с прозрачным дном, обеспечивающая несвязывающую поверхность обработка, Corning, реф. 3655) в общем объеме 40 мкл. Разведения соединений делали в 100% ДМСО (VWR Chemicals 23500.297) в полипропиленовом планшете с этапами 3-кратного разведения для получения конечного диапазона концентрации в анализе от 25 мкМ до 1,7 нМ. 1 мкл разведений соединений (100% ДМСО) добавляли в 19 мкл MAGL (рекомбинантный, дикого типа) в аналитическом буфере (50 мМ Трис (GIBCO, 15567-027), 1 мМ ЭДТК (Fluka, 03690-100 мл)). Планшет встряхивали в течение 1 мин при 2000 об/мин (Variomag Teleshake), а затем инкубировали в течение 15 мин при КТ. Чтобы начать реакцию добавляли 20 мкл 4-нитрофенилацетата (Sigma N-8130) в аналитическом буфере с 6% EtOH. Конечные концентрации в анализе составляли 1 нМ MAGL и 300 мкМ 4-нитрофенилацетата. После встряхивания (1 мин, 2000 об/мин) и 5 мин инкубации при КТ первый раз измеряли поглощение на 405 нм (Molecular Devices, SpectraMax Paradigm). Второе измерение проводили после инкубации в течение 80 мин при КТ. По двум измерениям рассчитывали наклон путем вычитания первого измерения из второго.
В альтернативном варианте профили соединений в отношении ингибирующей MAGL активности получали путем определения ферментативной активности, отслеживая гидролиз природного субстрата 2-арахидоноилглицерина, приводящий к получению арахидоновой кислоты, что можно отслеживать с помощью масс-спектрометрии. Далее по тексту этот анализ сокращенно называется «анализом 2-AG».
Анализ 2-AG проводили в 384-луночных аналитических планшетах (РР, Greiner, кат. №784201) в общем объеме 20 мкл. Разведения соединений делали в 100% ДМСО (VWR Chemicals 23500.297) в полипропиленовом планшете с этапами 3-кратного разведения для получения конечного диапазона концентрации в анализе от 12,5 мкМ до 0,8 пМ. 0,25 мкл разведений соединений (100% ДМСО) добавляли к 9 мкл MAGL в аналитическом буфере (50 мМ Трис (GIBCO, 15567-027), 1 мМ ЭДТК (Fluka, 03690-100 мл), 0,01% (об./об.) Tween. После встряхивания планшет инкубировали в течение 15 мин при КТ. Чтобы начать реакцию добавляли 10 мкл 2-арахидоноилглицерина в аналитическом буфере. Конечные концентрации в анализе составляли 50 пМ MAGL и 8 мкМ 2-арахидоноилглицерина. После встряхивания и 30 мин инкубации при КТ реакцию гасили добавлением 40 мкл ацетонитрила, содержащего 4 мкМ с18-арахидоновой кислоты. Количество арахидоновой кислоты отслеживали с помощью онлайн системы SPE (Agilent Rapidfire) в сочетании с тройным квадрупольным масс-спектрометром (Agilent 6460). Картридж С18 SPE (G9205A) использовали с жидкостными настройками ацетонитрил/вода. Масс-спектрометр работал в режиме отрицательного электрораспыления с отслеживанием переноса массы 303,1 → 259,1 для арахидоновой кислоты и 311,1 → 267,0 для d8-арахидоновой кислоты. Активность соединений рассчитывали на основании соотношения интенсивностей [арахидоновая кислота/d8-арахидоновая кислота].
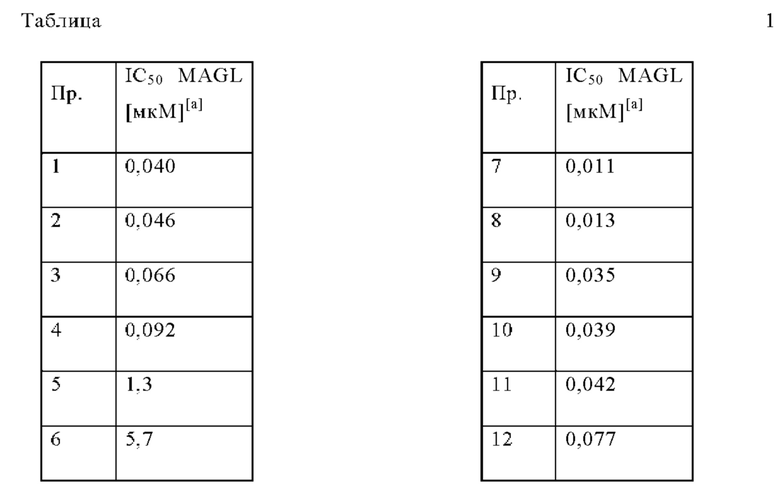
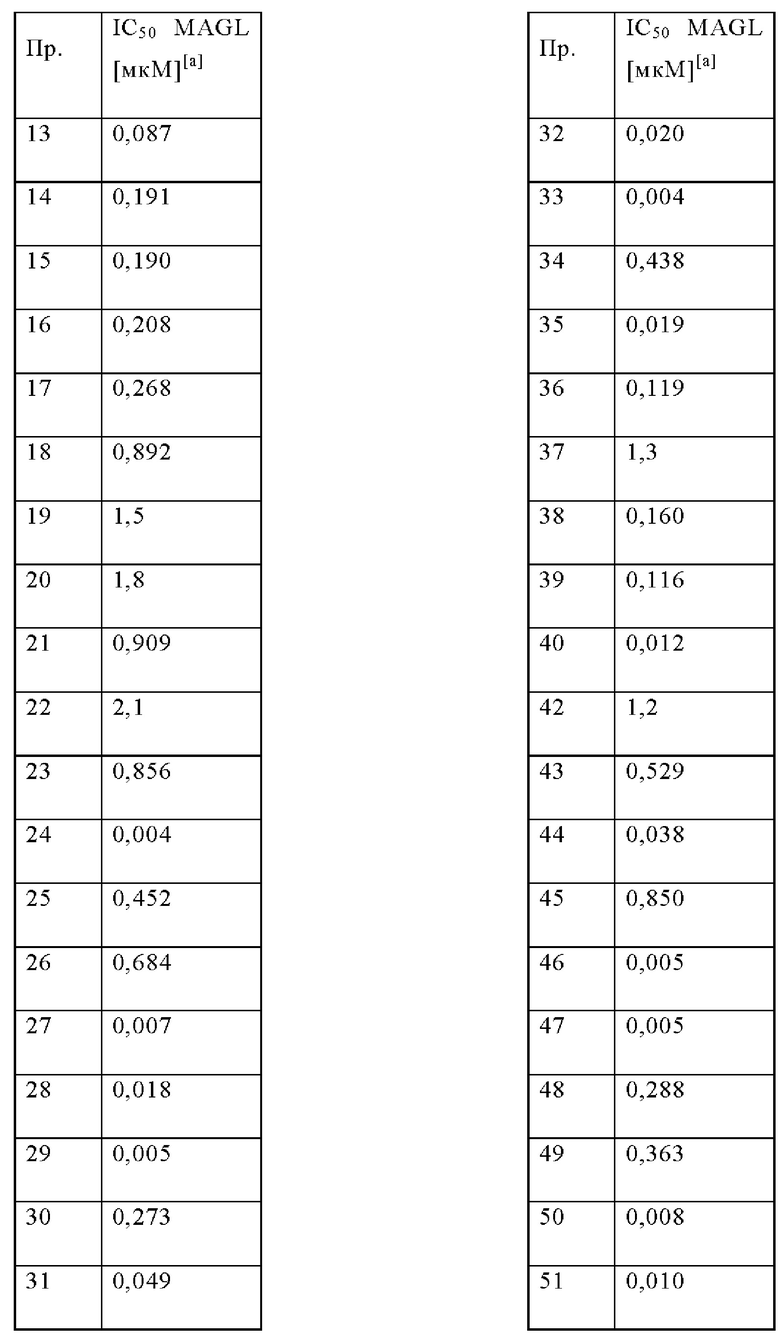
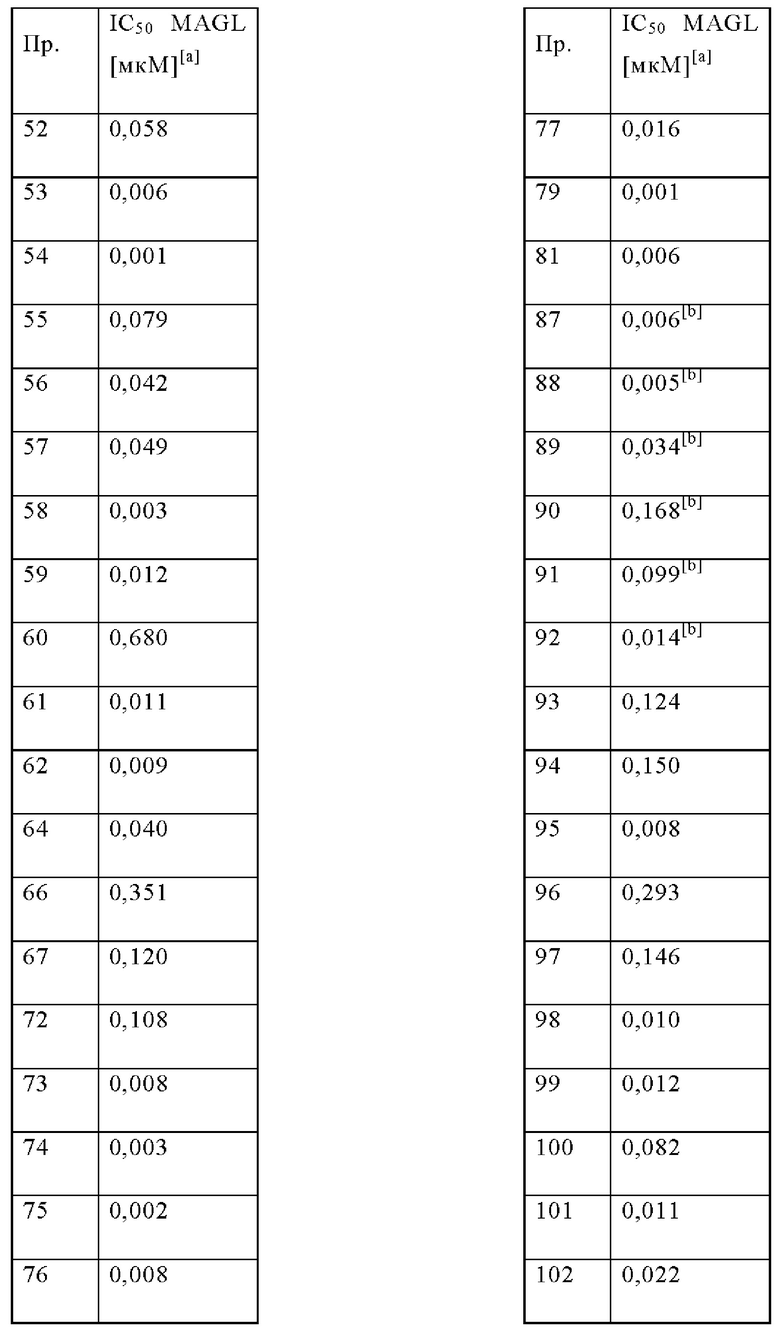



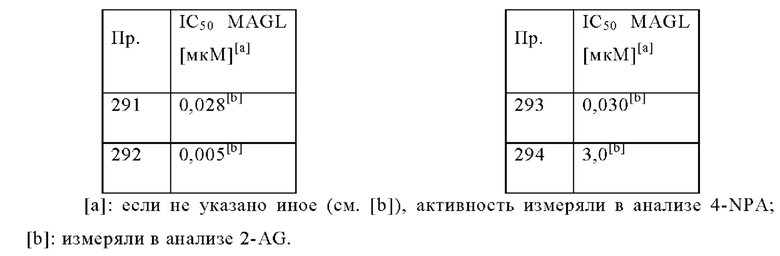
В одном аспекте в данном изобретении предложены соединения формулы (I) и их фармацевтически приемлемые соли или сложные эфиры, описанные в данном документе, причем указанные соединения формулы (I) и их фармацевтически приемлемые соли или сложные эфиры имеют IC50 в отношении ингибирования MAGL менее 25 мкМ, предпочтительно менее 10 мкМ, более предпочтительно менее 5 мкМ согласно измерению в анализе MAGL, описанном в данном документе.
В одном варианте осуществления соединения формулы (I) и их фармацевтически приемлемые соли или сложные эфиры, описанные в данном документе, имеют значения IC50 (ингибирование MAGL) от 0,000001 мкМ до 25 мкМ, конкретные соединения имеют значения IC50 от 0,000005 мкМ до 10 мкМ, дополнительные конкретные соединения имеют значения IC50 от 0,00005 мкМ до 5 мкМ согласно измерению в анализе MAGL, описанном в данном документе.
В одном варианте осуществления в данном изобретении предложены соединения формулы (I) и их фармацевтически приемлемые соли или сложные эфиры, описанные в данном документе, причем указанные соединения формулы (I) и их фармацевтически приемлемые соли или сложные эфиры имеют IC50 в отношении MAGL менее 25 мкМ, предпочтительно менее 10 мкМ, более предпочтительно менее 5 мкМ согласно измерению в анализе, включающем этапы:
а)получение раствора соединения формулы (I) или его фармацевтически приемлемой соли или сложного эфира в ДМСО;
b)получение раствора MAGL (рекомбинантный, дикого типа) в аналитическом буфере (50 мМ трис(гидроксиметил)аминометан; 1 мМ этилендиаминтетрауксусная кислота);
с)добавление 1 мкл раствора соединения с этапа а) в 19 мкл раствора MAGL с этапа b);
с1)встряхивание смеси в течение 1 мин при 2000 об/мин;
е) инкубация в течение 15 мин при КТ;
г)добавление 20 мкл раствора 4-нитрофенилацетата в аналитическом буфере (50 мМ трис(гидроксиметил)аминометан; 1 мМ этилендиаминтетрауксусная кислота, 6% EtOH);
g) в стряхивание смеси в течение 1 мин при 2000 об/мин;
h) инкубация в течение 5 мин при КТ;
i) измерение поглощения смеси на 405 нм первый раз;
j) инкубация в течение дополнительных 80 мин при КТ;
k) измерение поглощения смеси на 405 нм второй раз;
l) вычитание поглощения, измеренного в i), из поглощения, измеренного в к) и расчет наклона поглощения;
где:
i) концентрация соединения формулы (I) или его фармацевтически приемлемой соли или сложного эфира после этапа f) находится в диапазоне от 25 мкМ ло 1,7 нМ;
ii) концентрация MAGL в анализе после этапа f) составляет 1 нМ;
iii) концентрация 4-нитрофенилацетата в анализе после этапа f) составляет 300 мкМ; и
iv) этапы от а) до 1) повторяют по меньшей мере 3 раза, каждый раз с разной концентрацией соединения формулы (I) или его фармацевтически приемлемой соли или сложного эфира.
Применение соединений по изобретению
В одном аспекте в данном изобретении предложены соединения формулы (I), описанные в данном документе, для применения в качестве терапевтически активного вещества.
В дополнительном аспекте в данном изобретении предложено применение соединений формулы (I), описанных в данном документе, для лечения или профилактики нейровоспаления, нейродегенеративных заболеваний, боли, онкологических заболеваний и/или психических расстройств у млекопитающего.
В одном варианте осуществления в данном изобретении предложено применение соединений формулы (I), описанных в данном документе, для лечения или профилактики нейровоспаления и/или нейродегенеративных заболеваний у млекопитающего.
В одном варианте осуществления в данном изобретении предложено применение соединений формулы (I), описанных в данном документе, для лечения или профилактики нейродегенеративных заболеваний у млекопитающего.
В одном варианте осуществления в данном изобретении предложено применение соединений формулы (I), описанных в данном документе, для лечения или профилактики онкологических заболеваний у млекопитающего.
В одном аспекте в данном изобретении предложено применение соединений формулы (I), описанных в данном документе, для лечения или профилактики рассеянного склероза, болезни Альцгеймера, болезни Паркинсона, амиотрофического бокового склероза, травматического повреждения головного мозга, нейротоксичности, инсульта, эпилепсии, тревожности, мигрени, депрессии, гепатоцеллюлярной карциномы, канцерогенеза толстой кишки, рака яичника, нейропатической боли, индуцированной химиотерапией нейропатии, острой боли, хронической боли и/или спастичности, связанной с болью, у млекопитающего.
В предпочтительном варианте осуществления в данном изобретении предложено применение соединений формулы (I), описанных в данном документе, для лечения или профилактики рассеянного склероза, болезни Альцгеймера и/или болезни Паркинсона у млекопитающего.
В особенно предпочтительном варианте осуществления в данном изобретении предложено применение соединений формулы (I), описанных в данном документе, для лечения или профилактики рассеянного склероза у млекопитающего.
В одном аспекте в данном изобретении предложены соединения формулы (I), описанные в данном документе, для применения при лечении или профилактике нейровоспаления, нейродегенеративных заболеваний, боли, онкологических заболеваний и/или психических расстройств у млекопитающего.
В одном варианте осуществления в данном изобретении предложены соединения формулы (I), описанные в данном документе, для применения при лечении или профилактике нейровоспаления и/или нейродегенеративных заболеваний у млекопитающего.
В одном варианте осуществления в данном изобретении предложены соединения формулы (I), описанные в данном документе, для применения при лечении или профилактике онкологических заболеваний у млекопитающего.
В одном варианте осуществления в данном изобретении предложены соединения формулы (I), описанные в данном документе, для применения при лечении или профилактике нейродегенеративных заболеваний у млекопитающего.
В одном аспекте в данном изобретении предложены соединения формулы (I), описанные в данном документе, для применения при лечении или профилактике рассеянного склероза, болезни Альцгеймера, болезни Паркинсона, амиотрофического бокового склероза, травматического повреждения головного мозга, нейротоксичности, инсульта, эпилепсии, тревожности, мигрени, депрессии, гепатоцеллюлярной карциномы, канцерогенеза толстой кишки, рака яичника, нейропатической боли, индуцированной химиотерапией нейропатии, острой боли, хронической боли и/или спастичности, связанной с болью, у млекопитающего.
В предпочтительном варианте осуществления в данном изобретении предложены соединения формулы (I), описанные в данном документе, для применения при лечении или профилактике рассеянного склероза, болезни Альцгеймера и/или болезни Паркинсона у млекопитающего.
В особенно предпочтительном варианте осуществления в данном изобретении предложены соединения формулы (I), описанные в данном документе, для применения при лечении или профилактике рассеянного склероза у млекопитающего.
В одном аспекте в данном изобретении предложено применение соединений формулы (I), описанных в данном документе, для изготовления лекарственного средства для лечения или профилактики нейровоспаления, нейродегенеративных заболеваний, боли, онкологических заболеваний и/или психических расстройств у млекопитающего.
В одном варианте осуществления в данном изобретении предложено применение соединений формулы (I), описанных в данном документе, для изготовления лекарственного средства для лечения или профилактики нейровоспаления и/или нейродегенеративных заболеваний у млекопитающего.
В одном варианте осуществления в данном изобретении предложено применение соединений формулы (I), описанных в данном документе, для изготовления лекарственного средства для лечения или профилактики нейродегенеративных заболеваний у млекопитающего.
В одном варианте осуществления в данном изобретении предложено применение соединений формулы (I), описанных в данном документе, для изготовления лекарственного средства для лечения или профилактики онкологических заболеваний у млекопитающего.
В дополнительном аспекте в данном изобретении предложено применение соединений формулы (I), описанных в данном документе, для изготовления лекарственного средства для лечения или профилактики рассеянного склероза, болезни Альцгеймера, болезни Паркинсона, амиотрофического бокового склероза, травматического повреждения головного мозга, нейротоксичности, инсульта, эпилепсии, тревожности, мигрени, депрессии, гепатоцеллюлярной карциномы, канцерогенеза толстой кишки, рака яичника, нейропатической боли, индуцированной химиотерапией нейропатии, острой боли, хронической боли и/или спастичности, связанной с болью, у млекопитающего.
В предпочтительном варианте осуществления в данном изобретении предложено применение соединений формулы (I), описанных в данном документе, для изготовления лекарственного средства для лечения или профилактики рассеянного склероза, болезни Альцгеймера и/или болезни Паркинсона у млекопитающего.
В особенно предпочтительном варианте осуществления в данном изобретении предложено применение соединений формулы (I), описанных в данном документе, для изготовления лекарственного средства для лечения или профилактики рассеянного склероза у млекопитающего.
В дополнительном аспекте в данном изобретении предложен способ лечения или профилактики нейровоспаления, нейродегенеративных заболеваний, боли, онкологических заболеваний и/или психических расстройств у млекопитающего, который включает введение млекопитающему эффективного количества соединения формулы (I), описанного в данном документе.
В одном варианте осуществления в данном изобретении предложен способ лечения или профилактики нейровоспаления и/или нейродегенеративных заболеваний у млекопитающего, который включает введение млекопитающему эффективного количества соединения формулы (I), описанного в данном документе.
В одном варианте осуществления в данном изобретении предложен способ лечения или профилактики нейродегенеративных заболеваний у млекопитающего, который включает введение млекопитающему эффективного количества соединения формулы (I), описанного в данном документе.
В одном аспекте в данном изобретении предложен способ лечения или профилактики рассеянного склероза, болезни Альцгеймера, болезни Паркинсона, амиотрофического бокового склероза, травматического повреждения головного мозга, нейротоксичности, инсульта, эпилепсии, тревожности, мигрени, депрессии и/или боли у млекопитающего, который включает введение млекопитающему эффективного количества соединения формулы (I), описанного в данном документе.
В предпочтительном варианте осуществления в данном изобретении предложен способ лечения или профилактики рассеянного склероза, болезни Альцгеймера и/или болезни Паркинсона у млекопитающего, который включает введение млекопитающему эффективного количества соединения формулы (I), описанного в данном документе.
В особенно предпочтительном варианте осуществления в данном изобретении предложен способ лечения или профилактики рассеянного склероза у млекопитающего, который включает введение млекопитающему эффективного количества соединения формулы (I), описанного в данном документе.
Фармацевтические композиции и введение
В одном аспекте в данном изобретении предложена фармацевтическая композиция, содержащая соединение формулы (I), описанное в данном документе, и терапевтически инертный носитель.
Соединения формулы (I) и их фармацевтически приемлемые соли и сложные эфиры можно использовать в качестве лекарственных средств (например, в форме фармацевтических препаратов). Фармацевтические препараты можно вводить внутрь, например, перорально (например, в форме таблеток, таблеток с покрытием, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий), назально (например, в форме назальных спреев) или ректально (например, в форме суппозиториев). При этом введение также можно осуществлять парентерально, например, внутримышечно или внутривенно (например, в форме инъекционных растворов).
Соединения формулы (I) и их фармацевтически приемлемые соли и сложные эфиры можно соединять с фармацевтически инертными, неорганическими или органическими адъювантами для получения таблеток, таблеток с покрытием, драже и твердых желатиновых капсул. Например, можно использовать лактозу, кукурузный крахмал или его производные, тальк, стеариновую кислоту или ее соли и т.д. в качестве адъювантов для таблеток, драже и твердых желатиновых капсул.
Подходящими адъювантами для мягких желатиновых капсул являются, например, растительные масла, воски, жиры, полутвердые вещества и жидкие полиолы, и т.д.
Подходящими адъювантами для получения растворов и сиропов являются, например, вода, полиолы, сахароза, инвертный сахар, глюкоза и т.д.
Подходящими адъювантами для инъекционных растворов являются, например, вода, спирты, полиолы, глицерин, растительные масла и т.д.
Подходящими адъювантами для суппозиториев являются, например, натуральные или отвержденные масла, воски, жиры, полутвердые или жидкие полиолы, и т.д.
Кроме того, фармацевтические препараты могут содержать консерванты, солюбилизаторы, вещества, повышающие вязкость, стабилизаторы, смачивающие агенты, эмульгаторы, подсластители, красители, ароматизаторы, соли для изменения осмотического давления, буферы, маскирующие агенты или антиоксиданты. Также они могут содержать другие имеющие терапевтическую ценность вещества.
Дозировка может варьироваться в широком диапазоне и, конечно, будет подбираться под индивидуальные требования в каждом конкретном случае. В целом, в случае перорального введения суточная подходящей будет дозировка, составляющая от около 0,1 мг до 20 мг на кг массы тела, предпочтительно от 0,5 мг до 4 мг на кг массы тела (например, около 300 мг на человека), предпочтительно разделенная на 1-3 отдельные дозы, которые могут составлять, например, одинаковое количество. При этом следует понимать, что приведенный в данном документе верхний предел может быть превышен, если это показано.
В соответствии с изобретением соединения формулы (I) или их фармацевтически приемлемые соли и сложные эфиры можно применять для лечения или профилактики диабета 2 типа, связанных с ним микрососудистых осложнений (таких как, но не ограничиваясь этим, диабетическая ретинопатия, диабетическая нейропатия и диабетическая нефропатия), ишемической болезни сердца, ожирения и первопричинных воспалительных заболеваний, хронических воспалительных и аутоиммунных/воспалительных заболеваний.
Примеры
Данное изобретение будет более понятно со ссылкой на нижеприведенные примеры. При этом формулу изобретения не следует понимать как ограничивающую объем примеров.
В случае, когда препаративные примеры получены в виде смеси энантиомеров, чистые энантиомеры можно разделять способами, описанными в данном документе, или способами, известными специалисту в данной области техники, таким как, например, хиральная хроматография (например, хиральная СЖХ) или кристаллизация.
Все образцы реакций и промежуточные соединения получали в атмосфере аргона, если не указано иное.
Способ А1
Пример 11
рац-(4aR,8aS)-6-[4-[[4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он

В раствор 4-нитрофенил 4-(4-(трифторметил)бензил)пиперидин-1-карбоксилата (100 мг, 245 мкмоль, ВВ2) в ДМФА добавляли (1,5 мл), рац-(4aR,8aS)-гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она дигидрохлорид (45,9 мг, 294 мкмоль, ChemBridge Corporation, ВВ1) и ТЭА (49,6 мг, 68,3 мкл, 490 мкмоль). Полученную в результате реакционную смесь нагревали при 80°С в течение 18 ч. Реакционную смесь разводили EtOAc и три раза промывали Н2О и NaHCO3. Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Неочищенный материал очищали с помощью флэш-хроматографии (силикагель, элюирование градиентом MeOH/EtOAc 0-10%) с получением указанного в заголовке соединения в виде грязно-белого масла (0,045 г; 43,2%). МС (ИЭР): m/z=426,4 [М+Н]+.
Способ А2
Пример 3
рац-(4aR,8aS)-6-[4-[(4-трет-бутилтиазол-2-ил)метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он
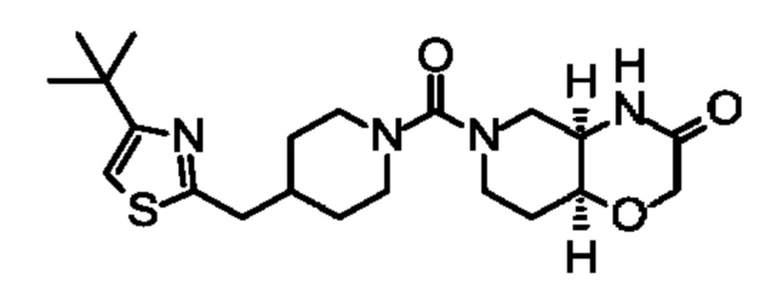
В ледяную суспензию бис(трихлорметил)карбоната (45,3 мг, 153 мкмоль, С AS RN 32315-10-9) и NaHCO3 (73,3 мг, 873 мкмоль) в ДХМ (2 мл) одной частью добавляли 4-трет-бутил-2-(4-пиперидилметил)тиазола гидрохлорид (60 мг, 218 мкмоль, Enamine Ltd) и перемешивали смесь при КТ в течение ночи. Суспензию фильтровали, а фильтрат выпаривали. Остаток растворяли в ДХМ (1 мл) и по каплям добавляли в ледяной раствор рац-(4aR,8aS)-гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она дигидрохлорида (50 мг, 218 мкмоль, ChemBridge Corporation, ВВ1) и ДИПЭА (152 мкл, 870 мкмоль) в ДХМ (1 мл). Суспензию перемешивали при КТ в течение 19 ч, чтобы она стала раствором. Реакционную смесь вливали в Н2О и ДХМ и разделяли слои. Водный слой три раза экстрагировали ДХМ. Органические слои дважды промывали водой, сушили над MgSO4, фильтровали, обрабатывали силикагелем и выпаривали. Соединение очищали с помощью хроматографии на силикагеле на 4 г колонке, используя систему для ЖХСД, с элюированием градиентом ДХМ: МеОН (от 100: от 0 до 90: 10) с получением необходимого соединения в виде бесцветной пены (0,039 г; 42,5%). МС (ИЭР): m/z=421,2 [М+Н]+.
Способ A3
Пример 34
(+)- или (-)-4-[[1-[(4aR,8aS)-3-оксо-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-6-карбонил]-4-пиперидил] метил] бензонитрил

В ледяной раствор бис(трихлорметил)карбоната (39,9 мг, 134 мкмоль, CAS RN 32315-10-9) в ДХМ добавляли NaHCO3 (64,5 мг, 768 мкмоль) и (+)-цис-гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он (30 мг, 192 мкмоль, ВВ1а) и перемешивали смесь при КТ в течение ночи. В суспензию добавляли 4-(пиперидин-4-илметил)бензонитрил (38,5 мг, 192 мкмоль, CAS RN 333987-57-8) и ДИПЭА (99,3 мг, 134 мкл, 768 мкмоль). Суспензию перемешивали при КТ в течение 4,5 ч. Реакционную смесь вливали в Н2О и ДХМ и разделяли слои. Водный слой три раза экстрагировали ДХМ. Органические слои дважды промывали H2O, сушили над MgSO4, фильтровали, обрабатывали силикагелем и выпаривали. Соединение очищали с помощью хроматографии на силикагеле на 4 г колонке, используя систему для ЖХСД, с элюированием градиентом ДХМ: МеОН (от 100: от 0 до 90: 10) с получением необходимого соединения в виде бесцветной смолы (0,023 г; 31,3%). МС (ИЭР): m/z=383,2 [М+Н]+.
Способ А4
Пример 79
(4aR,8aS)-6-(4-((2-хлор-4-фторфенокси)метил)-4-метилпиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b] [1,4]оксазин-3(4Н)-он

В раствор 4-нитрофенил (4aR,8aS)-3-оксогексагидро-2Н-пиридо[4,3-b][1,4]оксазин-6(5Н)-карбоксилата (25 мг, 77,8 мкмоль, ВВ7а) в NMP (1 мл) добавляли ДИПЭА (25,1 мг, 34 мкл, 195 мкмоль) и 4-((2-хлор-4-фторфенокси)метил)-4-метилпиперидин; гидрохлоридную соль (19,5 мг, 66,1 мкмоль, ВВ12). Реакционную пробирку перемешивали при 140°С в течение 45 мин. Неочищенный материал очищали с помощью обращенно-фазовой ВЭЖХ с получение 23,2 мг необходимого продукта. МС (ИЭР): m/z=440,2 [М+Н]+.
Способ А5
Пример 64
(4aR,8aS)-6-(4-((2-хлор-4-фторфенокси)метил)-4-фторпиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b] [1,4]оксазин-3(4Н)-он
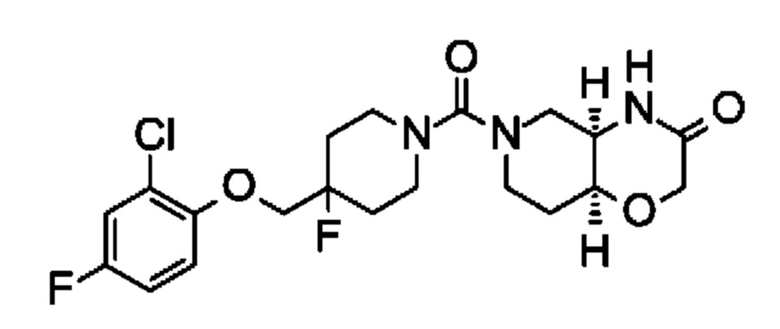
Пробирку для микроволновой обработки сушили феном и наполняли бис(трихлорметил)карбонатом (26,6 мг, 89,6 мкмоль) и гидрокарбонатом натрия (32,3 мг, 384 мкмоль). Пробирку помещали в атмосферу аргона и добавляли ДХМ (1 мл) для получения суспензии. Суспензию охлаждали на ледяной бане и добавляли 4-((2-хлор-4-фторфенокси)метил)-4-фторпиперидин; гидрохлоридную соль (36,1 мг, 121 мкмоль, ВВ15). Смесь перемешивали при 0°С в течение 15 мин и при КТ в течение ночи. Реакционную смесь охлаждали на ледяной бане и добавляли ДХМ (500 мкл) и ДИПЭА (49,7 мг, 67,1 мкл, 384 мкмоль) с последующим добавлением (4aR,8aS)-6-(4-((2-хлор-4-фторфенокси)метил)-4-фторпиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она (21,1 мг, 47,5 мкмоль, ВВ1а). Полученную в результате грязно-белую суспензию перемешивали при комнатной температуре в течение 7 ч. Реакционную смесь вливали в воду, добавляли ДХМ и разделяли слои. Водный слой дважды экстрагировали ДХМ. Объединенные органические слои промывали солевым раствором, сушили над MgSO4, фильтровали и выпаривали с получением желтого масла (58 мг). Неочищенный продукт очищали с помощью обращенно-фазовой ВЭЖХ и лиофилизировали с получением указанного в заголовке соединения в виде белого твердого вещества (21,1 мг, выход 37,1%). МС (ИЭР): m/z=444,2 [М+Н]+.
Способ А6
Пример 39
(4aR,8aS)-6-[4-[(2-фтор-4-метоксифенокси)метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он
В раствор 2-фтор-4-метоксифенола (16,5 мг, 13 мкл, 116 мкмоль), (4aR,8aS)-6-[4-(гидроксиметил)пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она (34,5 мг, 116 мкмоль, ВВ16) и трифенилфосфина (33,5 мг, 128 мкмоль) в ДХМ (580 мкл) по каплям добавляли ДИАД (25,8 мг, 24,8 мкл, 128 мкмоль) и перемешивали реакцию при комнатной температуре в течение 22 ч. Реакционную смесь разводили ДХМ и промывали 1 М водн. NaOH. Фазы разделяли, а водную фазу дважды экстрагировали ДХМ. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали до сухости с получением красного масла (99 мг). Неочищенный продукт очищали с помощью обращенно-фазовой ВЭЖХ и лиофилизировали с получением необходимого соединения (20 мг, выход 40,9%) в виде белого твердого вещества. МС (ИЭР): m/z=422,3 [М+Н]+.
Способ А7
Пример 42 и 43
(4aS,8aR)-6-(4-(((6-(трифторметил)пиридин-3-ил)окси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он (пример 42)
и
(4aR,8aS)-6-(4-(((6-(трифторметил)пиридин-3-ил)окси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он (пример 43)

Этап а) рац-(4aR,8aS)-6-(4-(хлорметил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он

В раствор рац-(4aR,8aS)-6-(4-(гидроксиметил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она (80 мг, 269 мкмоль, ВВ16) в сухом ДМФА (2 мл) добавляли ДИПЭА (52,2 мг, 70,5 мкл, 404 мкмоль), ДМАП (1,64 мг, 13,5 мкмоль) и метансульфонилхлорид (46,2 мг, 404 мкмоль) и перемешивали реакционную смесь при комнатной температуре в течение 2 ч. Добавляли 4,4-дифторпиперидин; гидрохлоридную соль (84,8 мг, 538 мкмоль), ДИПЭА (139 мг, 188 мкл, 1,08 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 2 ч. Затем реакцию перемешивали при 70°С в течение 14 ч. Неочищенную реакционную смесь подвергали очистке методом обращенно-фазовой ВЭЖХ с получением указанного в заголовке соединения в виде побочного продукта (35 мг). МС (ИЭР): m/z=315,1 [М+Н]+.
Этап b) (4aS,8aR)-6-(4-(((6-(трифторметил)пиридин-3-ил)окси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он (пример 42) и (4aR,8aS)-6-(4-(((6-(трифторметил)пиридин-3-ил)окси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он (пример 43)
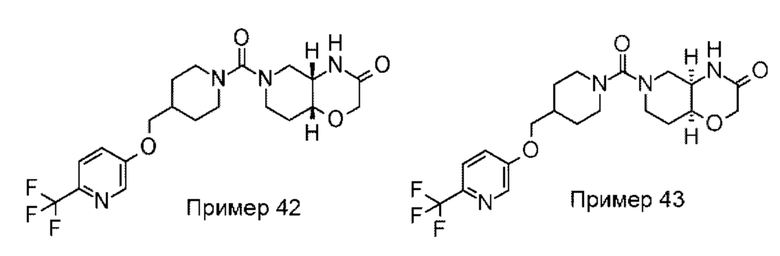
В раствор рац-(4aR,8aS)-6-(4-(хлорметил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она (70 мг, 222 мкмоль) в сухом ДМФА (1 мл) добавляли 6-(трифторметил)пиридин-3-ол (54.2 мг, 332 мкмоль) и Cs2CO3 (108 мг, 332 мкмоль). Реакционную смесь перемешивали при 95°С в течение 18 ч. Нерастворимые вещества удаляли фильтрацией через целит, фильтрат концентрировали до сухости, а неочищенный остаток очищали и разделяли энантиомеры с помощью хиральной СЖХ с получение примера 42 (33,8 мг) и примера 43 (32,5 мг). МС (ИЭР): m/z=443,2 [М+Н]+ для обоих примеров.
Способ А8
Пример 26
(4aS,8aR)-6-(4-((4-(трифторметил)-1H-пиразол-1-ил)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b] [1,4]оксазин-3(4Н)-он

В раствор рац-(4aR,8aS)-6-(4-(гидроксиметил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она (75 мг, 252 мкмоль, ВВ16) в сухом ДМФА (2 мл) добавляли ДИПЭА (39,1 мг, 52,9 мкл, 303 мкмоль), ДМАП (3,08 мг, 25,2 мкмоль) и метансульфонилхлорид (30,3 мг, 265 мкмоль) и перемешивали реакционную смесь при комнатной температуре в течение 2 ч. Добавляли 4-(трифторметил)-1Н-пиразол (68,6 мг, 504 мкмоль) и К2СО3 (87,1 мг, 631 мкмоль) и перемешивали реакционную смесь при 90°С в течение 18 ч. Нерастворимые вещества удаляли фильтрацией через целит, фильтрат концентрировали до сухости в вакууме, а неочищенный остаток непосредственно очищали с помощью флэш-хроматографии с элюированием смесью ДХМ и МеОН (от 0% до 10%) с получением 90 мг необходимого продукта в виде рацемата. Его подвергали хиральному разделению методом СЖХ с получение примера 26 (25 мг) в виде бесцветного масла и энантиомера (31 мг) в виде бесцветного масла. МС (ИЭР): m/z=416,2 [М+Н]+.
Способ А9 Пример 37
(4aR,8aS)-6-(4-((4,4-дифторпиперидин-1-ил)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b] [1,4]оксазин-3(4Н)-он

В раствор (4aR,8aS)-гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она (40 мг, 256 мкмоль, ВВ1а) в сухом ДМФА (2 мл), охлажденный до 0°С, добавляли ДИПЭА (39,7 мг, 53,7 мкл, 307 мкмоль) и 4-нитрофенилкарбонохлоридат (61,9 мг, 307 мкмоль). Реакционную смесь перемешивали при 0°С в течение 20 мин. Контрольная ЖХМС показала образование промежуточного карбамата. Добавляли ДИПЭА (116 мг, 157 мкл, 896 мкмоль) и 4,4-дифтор-1-(пиперидин-4-илметил)пиперидин; дигидрохлоридную соль (89,5 мг, 307 мкмоль, ВВ17) и перемешивали реакционную смесь при комнатной температуре в течение 30 мин, затем перемешивали при 100°С в течение 14 ч. Летучие вещества удаляли в вакууме, а неочищенный остаток непосредственно подвергали очистке СЖХ с получением необходимого соединения (9,5) мг в виде светло-оранжевого масла. МС (ИЭР): m/z=401,3 [М+Н]+.
Способ А10
Пример 125
(+)-(4aR,8aS)-6-[4-[2-(2-хлорфенил)этинил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он
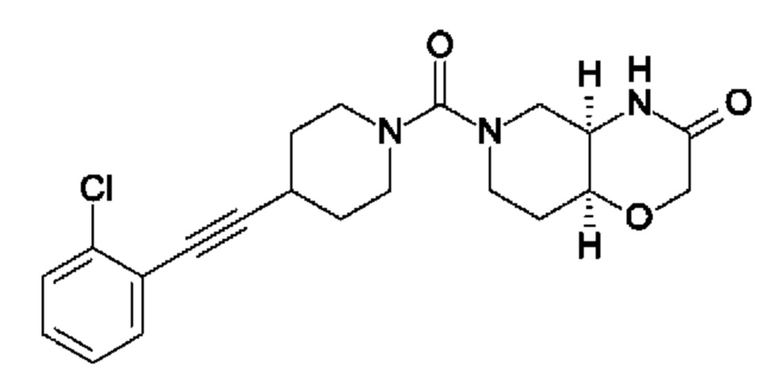
В закрытой пробирке смешивали 4-[2-(2-хлорфенил)этинил]пиперидин (ВВ18, 0,02 г, 0,078 ммоль) и 4-нитрофенил (4aR,8aS)-3-оксогексагидро-2Н-пиридо[4,3-b][1,4]оксазин-6(5Н)-карбоксилат (ВВ7а, 0,025 г, 0,078 ммоль) в АЦН (0,6 мл). Затем добавляли основание Хюнига (0,041 мл, 0,234 ммоль) ДМАП (0,005 г, 0,039 ммоль) и нагревали реакционную смесь до 90°С в течение ночи. Смесь выпаривали до сухости, а неочищенный остаток очищали с помощью обращенно-фазовой ВЭЖХ с получение указанного в заголовке соединения (0,013 г, 41%) в виде бесцветного масла. МС (ИЭР): m/z=402,2 [М+Н]+.
Способ В1
Пример 1
(+)-(4aR,8aS)-6-(4-((4-(трет-бутил)тиазол-2-ил)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b] [1,4]оксазин-3(4Н)-он

Энантиомеры примера 3 разделяли с помощью препаративной хиральной ВЭЖХ (колонка Chiralcel OD), используя изократическую смесь EtOH (содержащего 0,05% NH4OAc):н-гептан (20: 80). Фракции выпаривали с получением необходимого соединения в виде бесцветного твердого вещества (0,012 г; 34,3%). МС (ИЭР): m/z=421,2 [М+Н]+.
Способ В2
Пример 12
(+)-или (-)-(4aR,8aS)-6-(4-(4-(трифторметил)фенокси)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b] [1,4]оксазин-3(4Н)-он
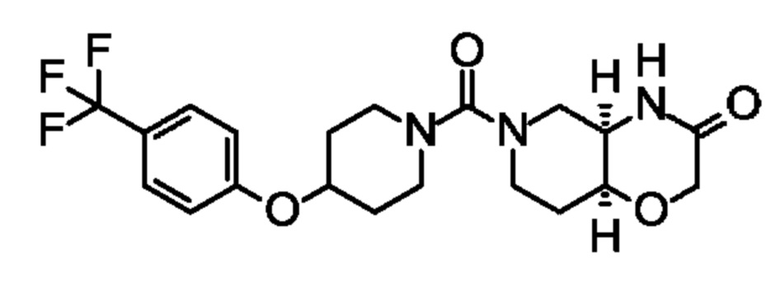
Энантиомеры примера 13 разделяли с помощью препаративной хиральной ВЭЖХ (колонка Chiralpak AD), используя изократическую смесь EtOH (содержащего 0,05% NH4OAc):н-гептан (40: 60). Фракции выпаривали с получением необходимого соединения в виде светло-коричневого масла (0,013 г; 28,4%). МС (ИЭР): m/z=428,2 [М+Н]+.
Способ В3
Примеры 103, 104 и 105
(4aR,8aS)-6-[2-метил-3-[[4-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он (изомер А+В, (изомер С, (изомер D)

Стереоизомеры примера 117 разделяли с помощью препаративной хиральной ВЭЖХ (колонка Reprosil Chiral NR), используя изократическую смесь EtOH (содержащего 0,05% NH4OAC): н-гептан (40: 60) с получением примеров 103 и 104 в виде отдельных изомеров и примера 105 в виде двух стереоизомеров. Фракции выпаривали с получением необходимых соединений в виде бесцветного твердого вещества.
Способ С
Пример 21
рац-(4aR,8aS)-6-(4-(4-(трифторметил)бензил)пиперазин-1-карбонил)гексагидро-2Н-пиридо[4,3-b] [1,4]оксазин-3(4Н)-он

Смесь рац-цис-6-(пиперазин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она (35 мг, 130 мкмоль, ВВ3), 4-(трифторметил)бензальдегида (22,7 мг, 17,4 мкл, 130 мкмоль) и триацетоксиборгидрида натрия (27,6 мг, 130 мкмоль) в ДХМ (1 мл) перемешивали при КТ в течение 15 ч. Реакционную смесь концентрировали, а остаток очищали с помощью препаративной ВЭЖХ с получение необходимого соединения в виде белого твердого вещества (8 мг, 14,4%). МС (ИЭР): m/z=427,4 [М+Н]+.
Если не указано иное, следующие примеры синтезировали из pa4-(4aR,8aS)-гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она дигидрохлорида (ChemBridge Corporation) и подходящих строительных блоков по аналогии с реакционными способами, описанными в данном документе.
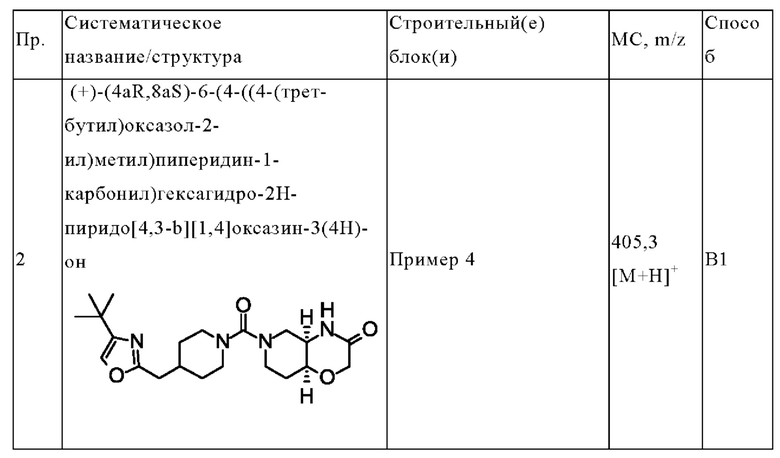
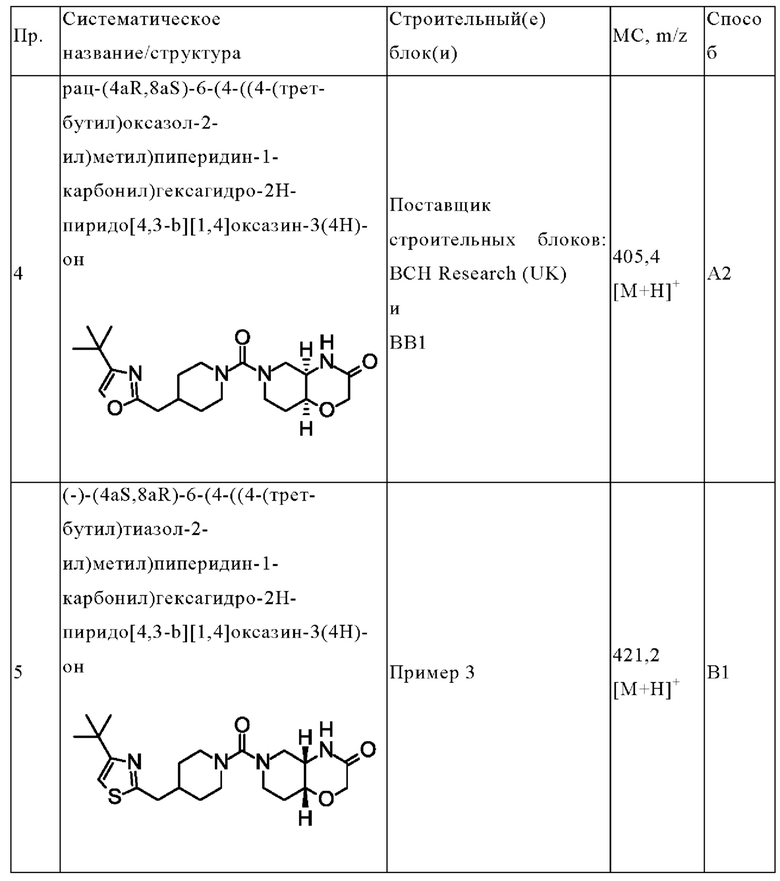
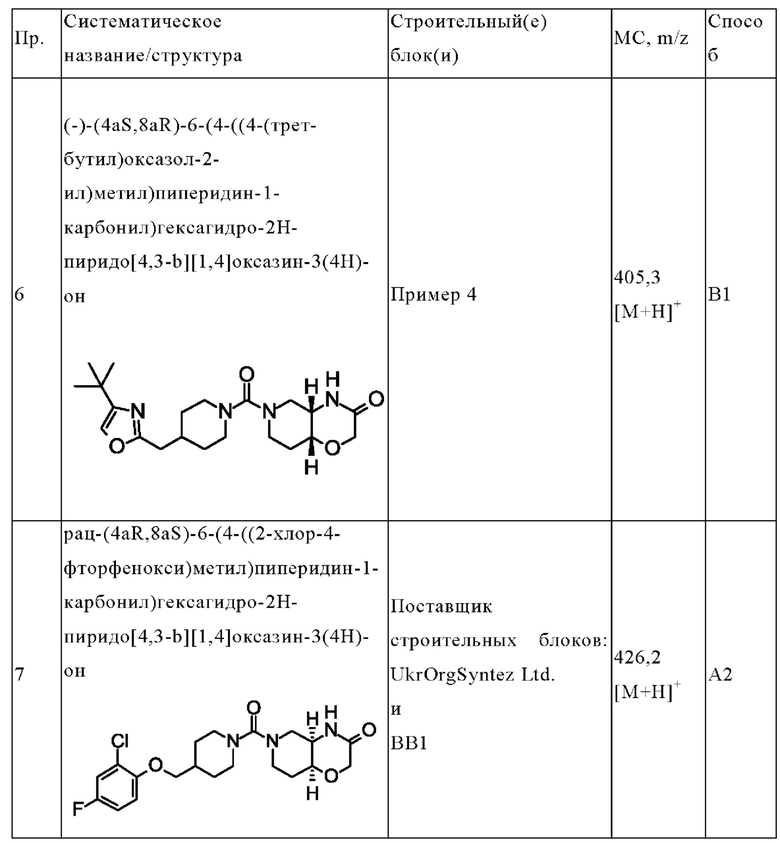
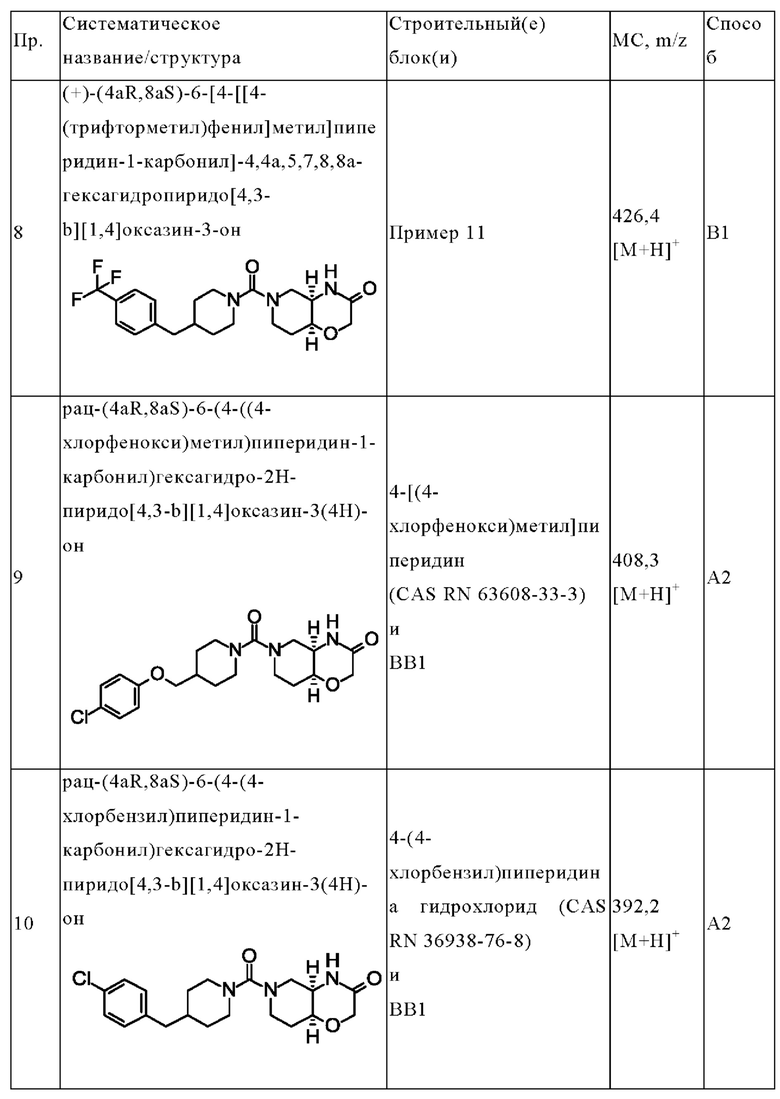
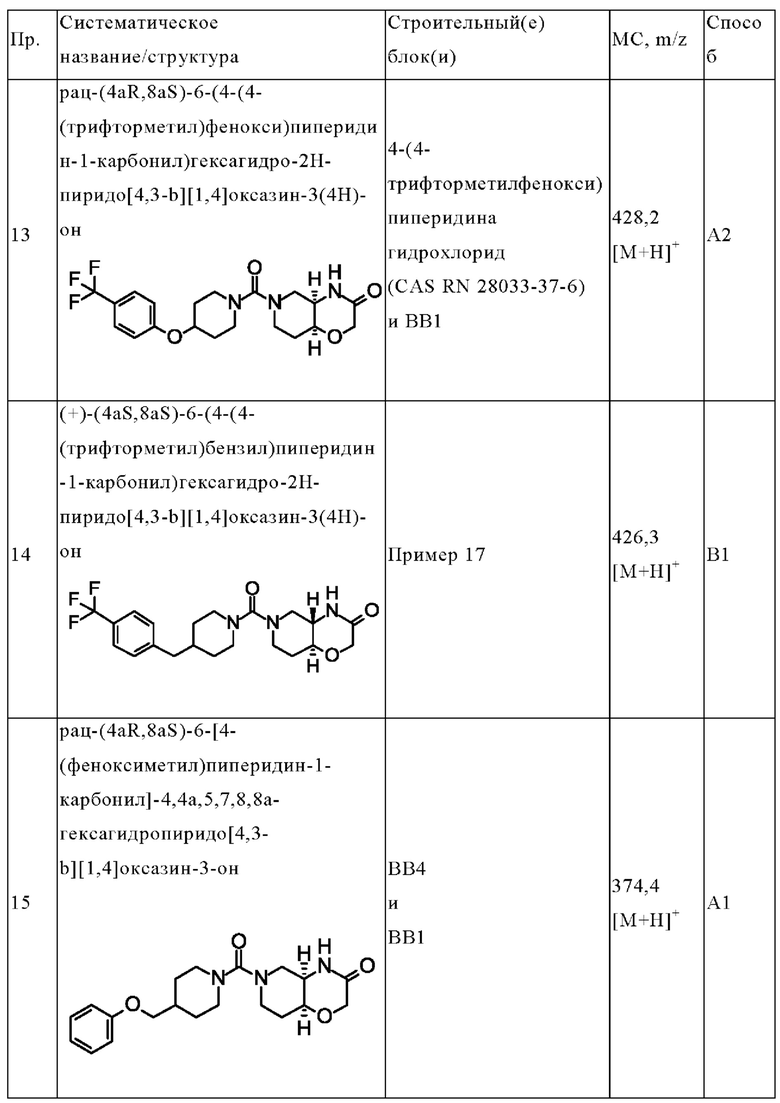


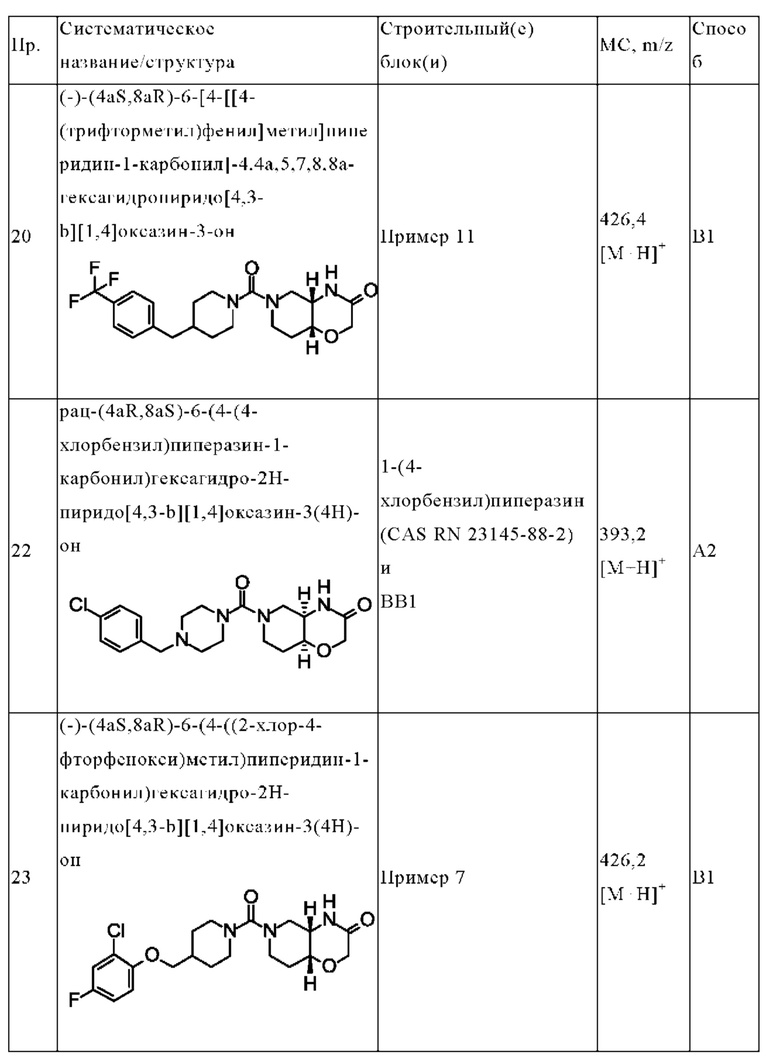
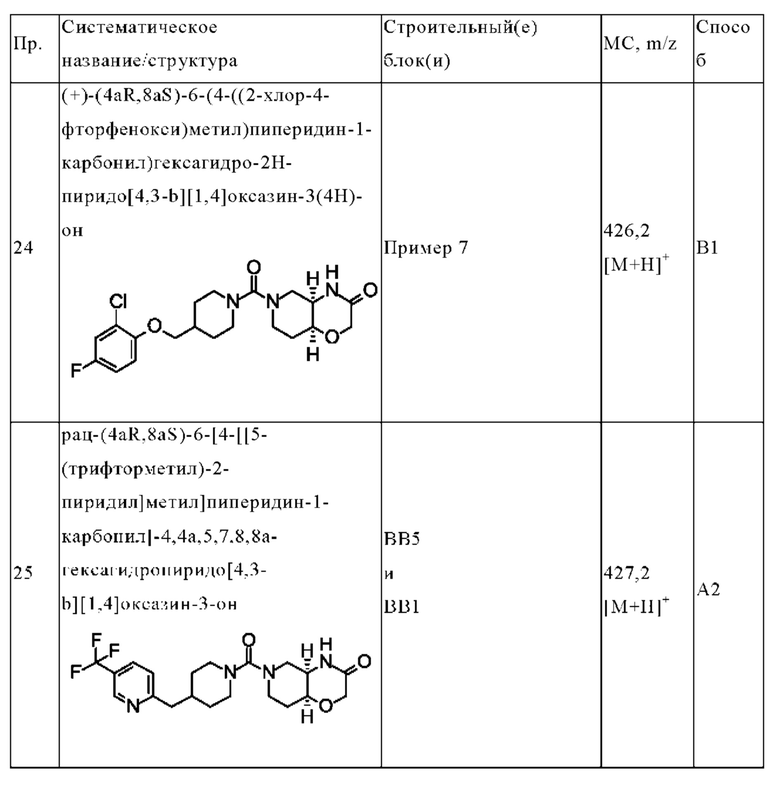

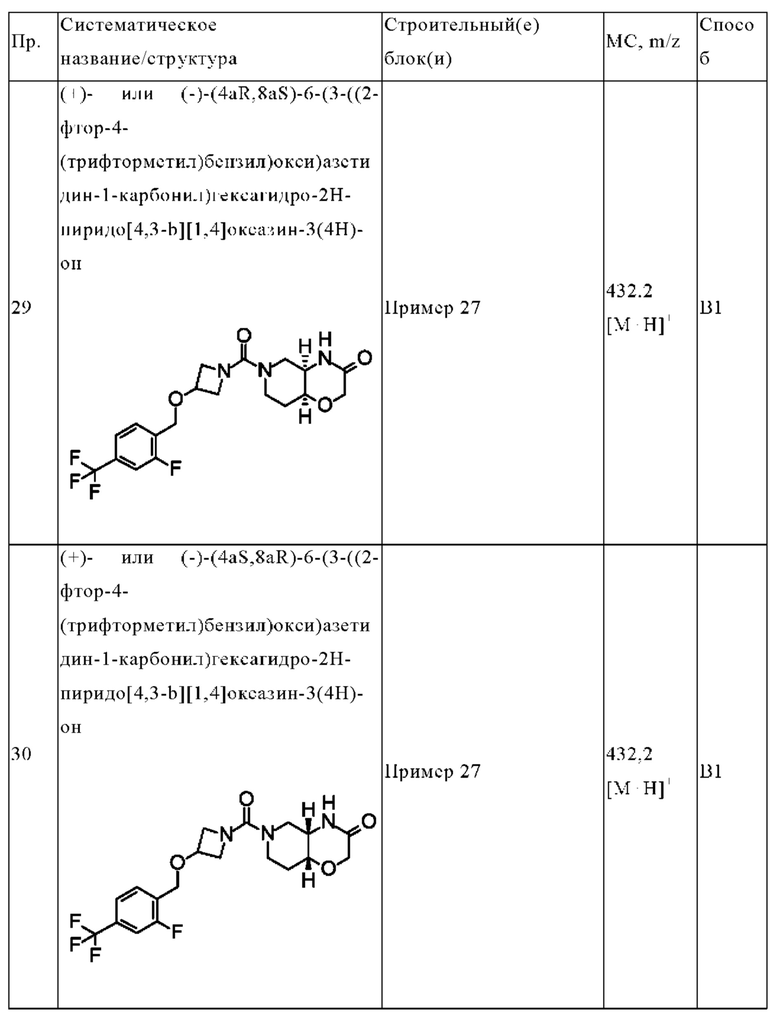


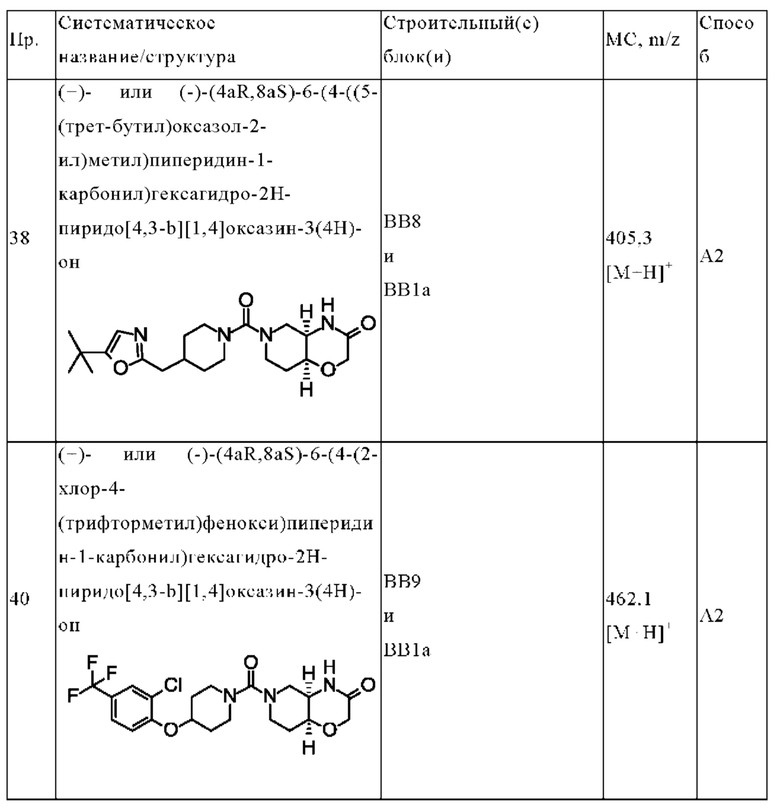
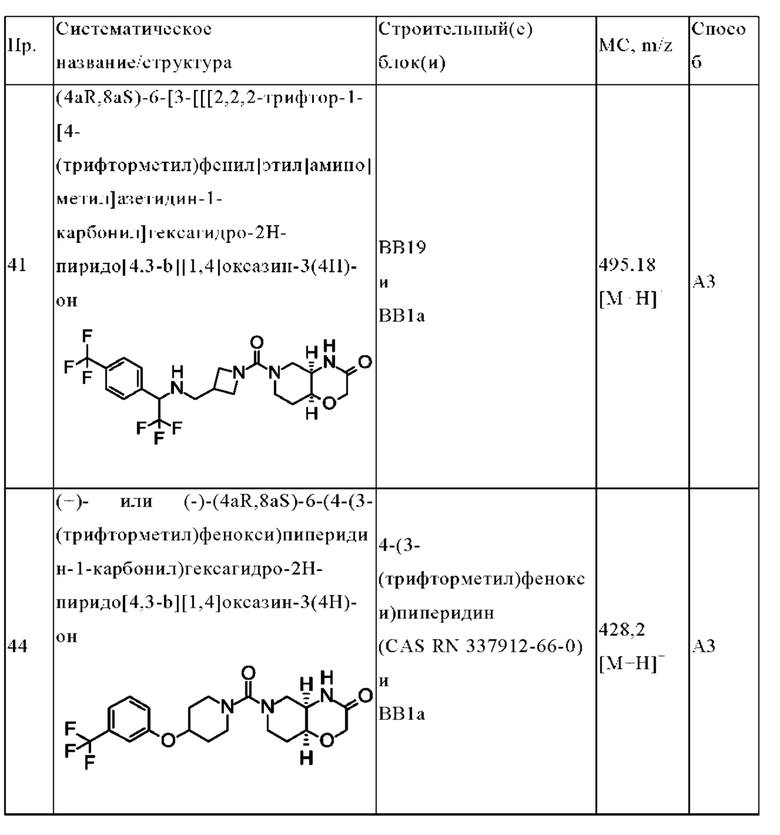
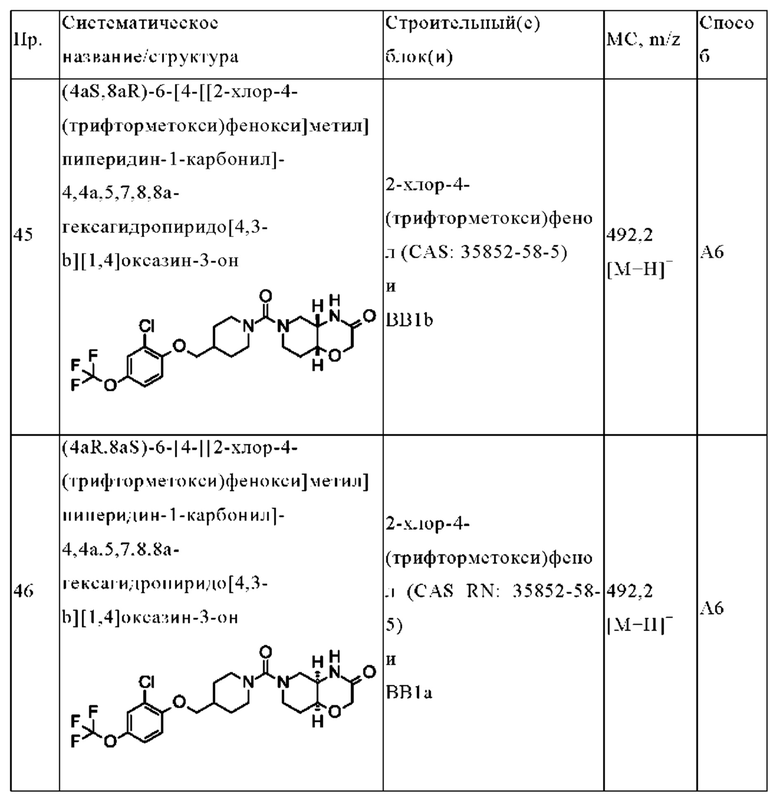




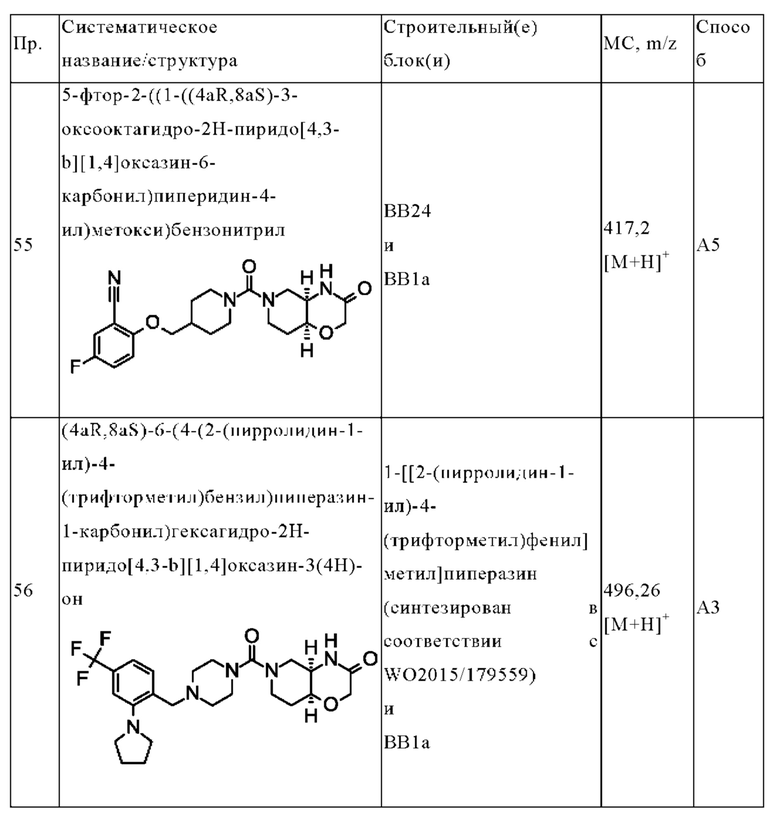

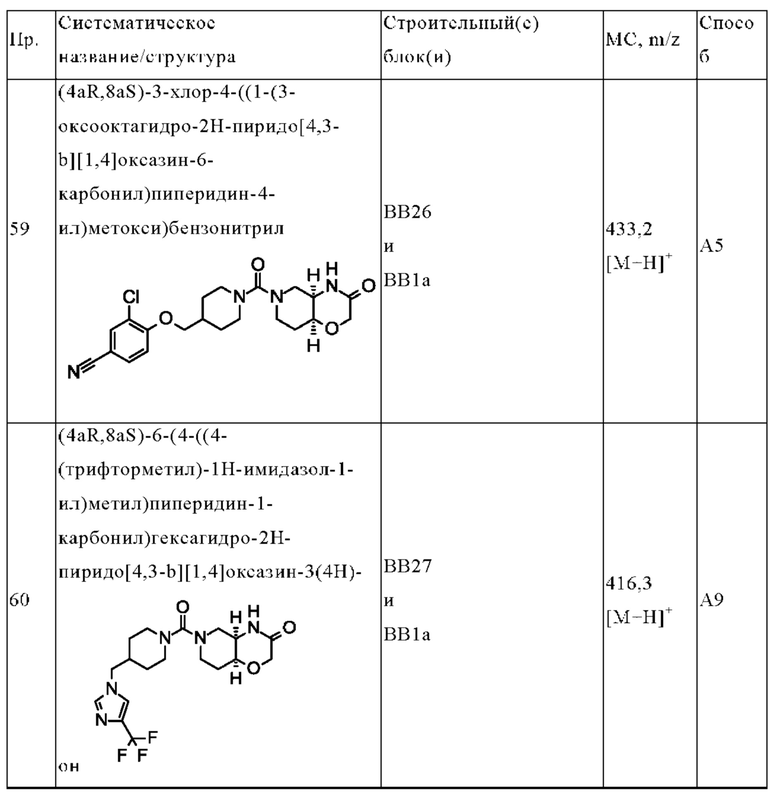

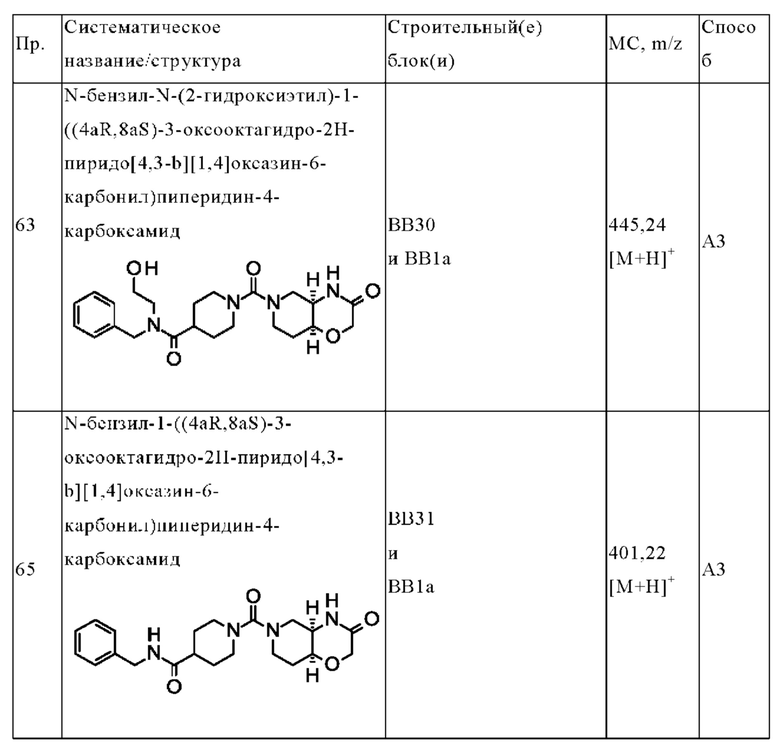
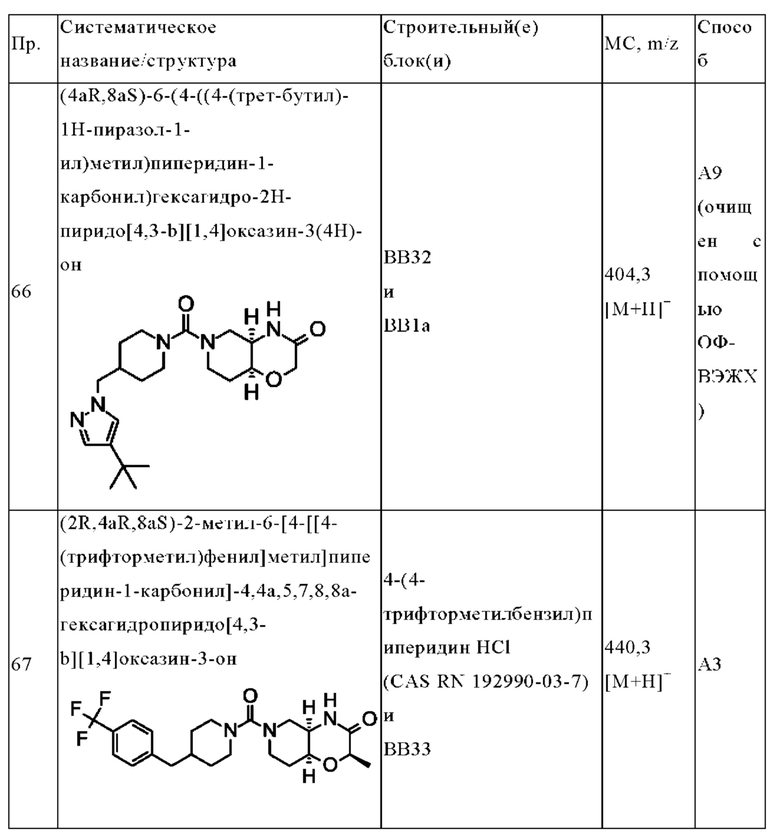

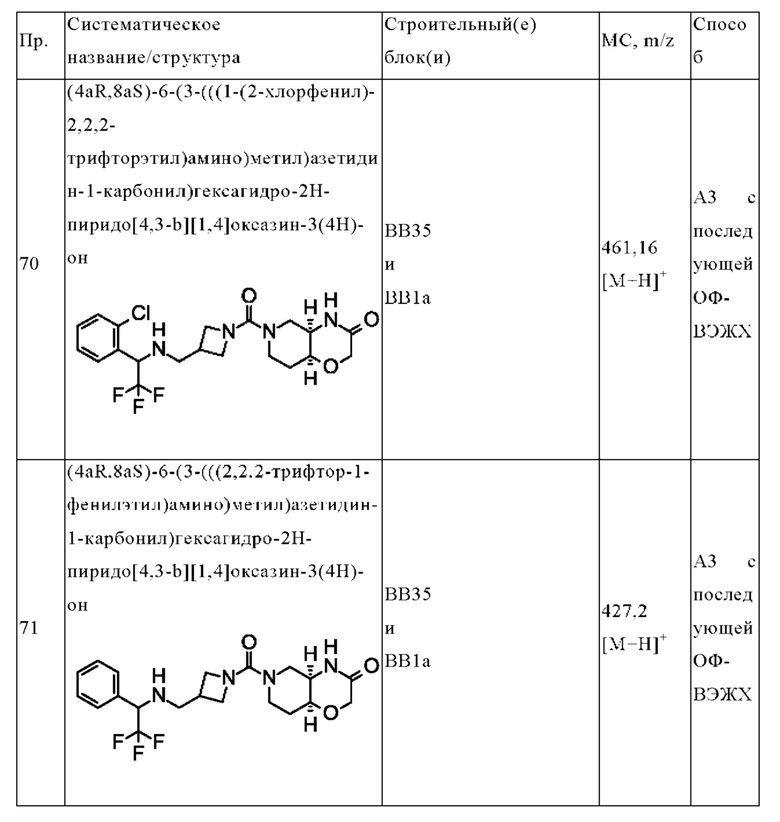


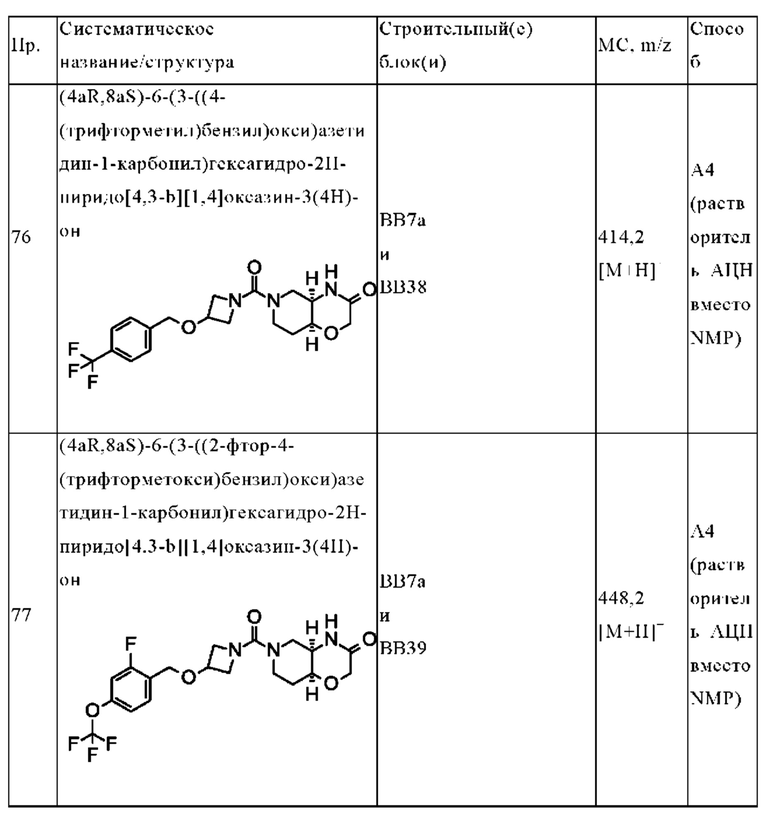






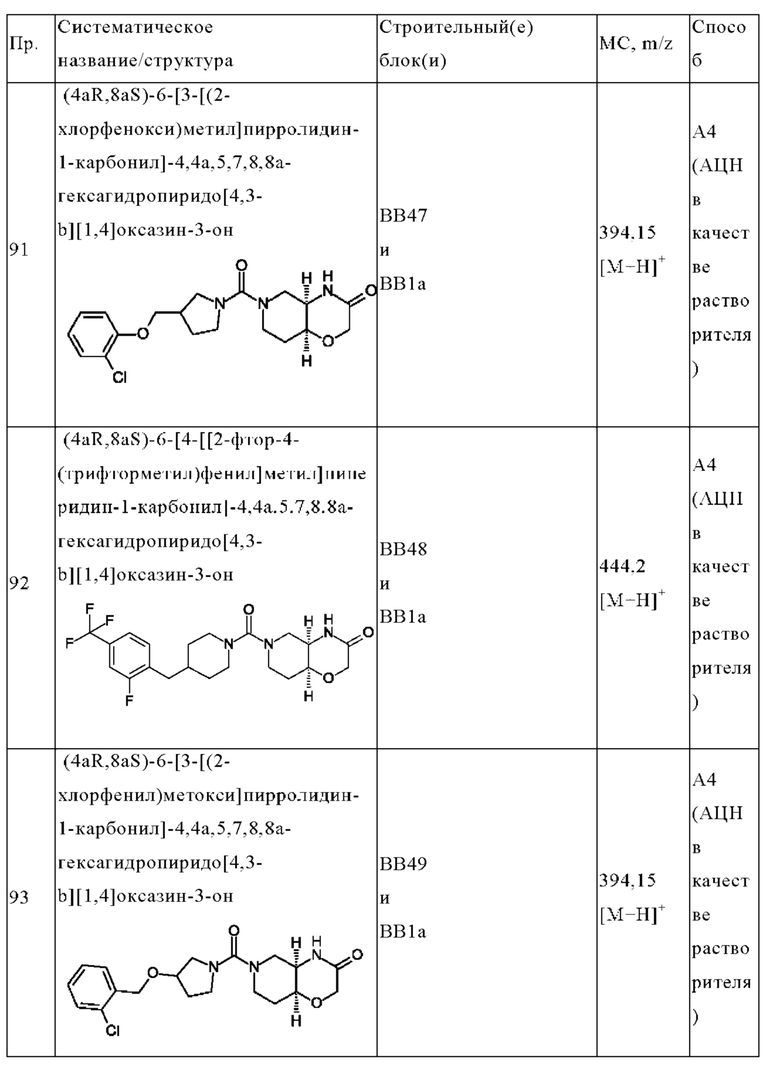



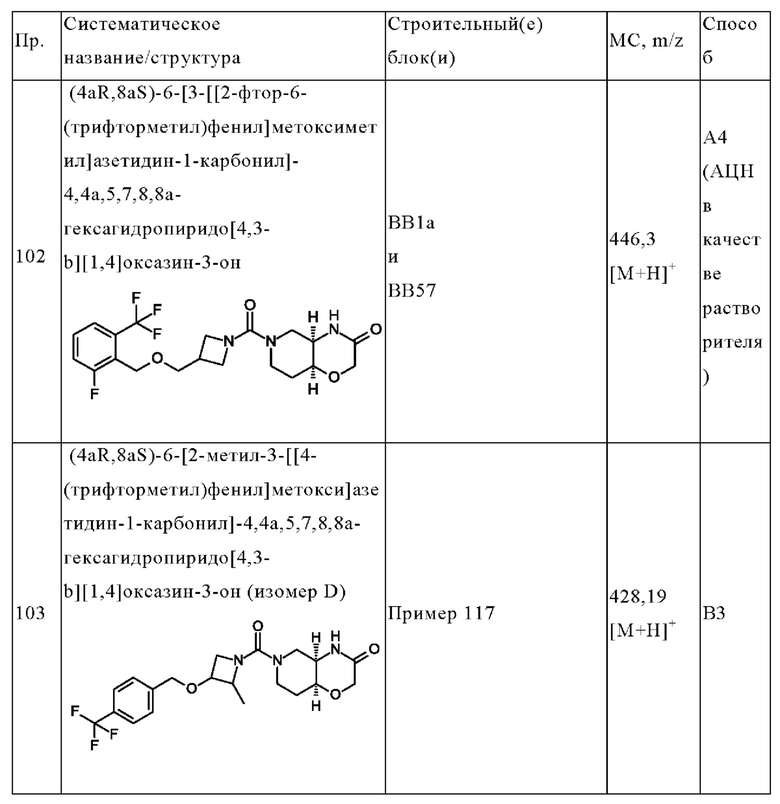

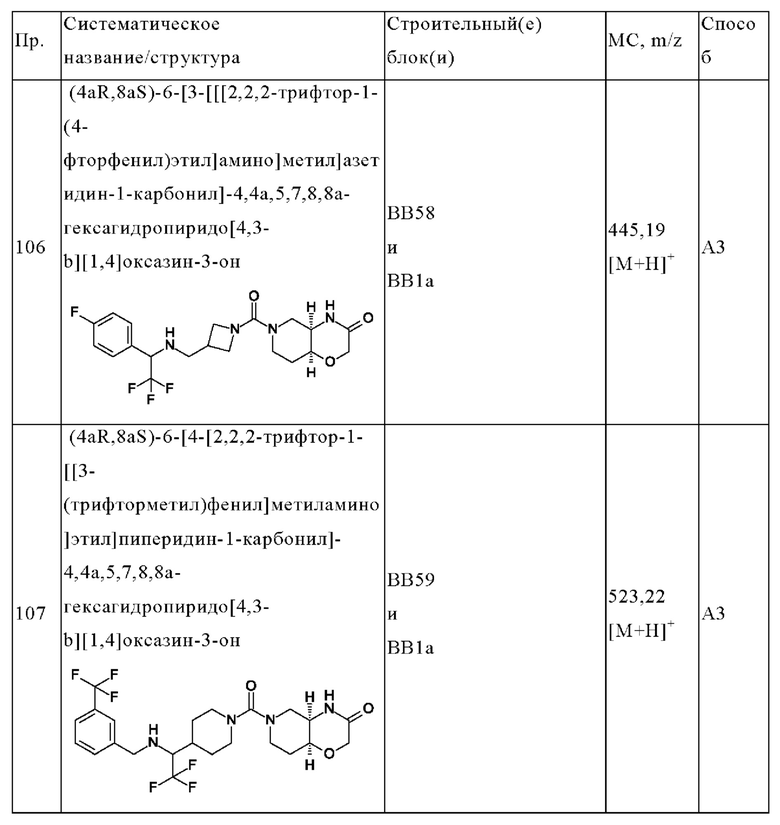

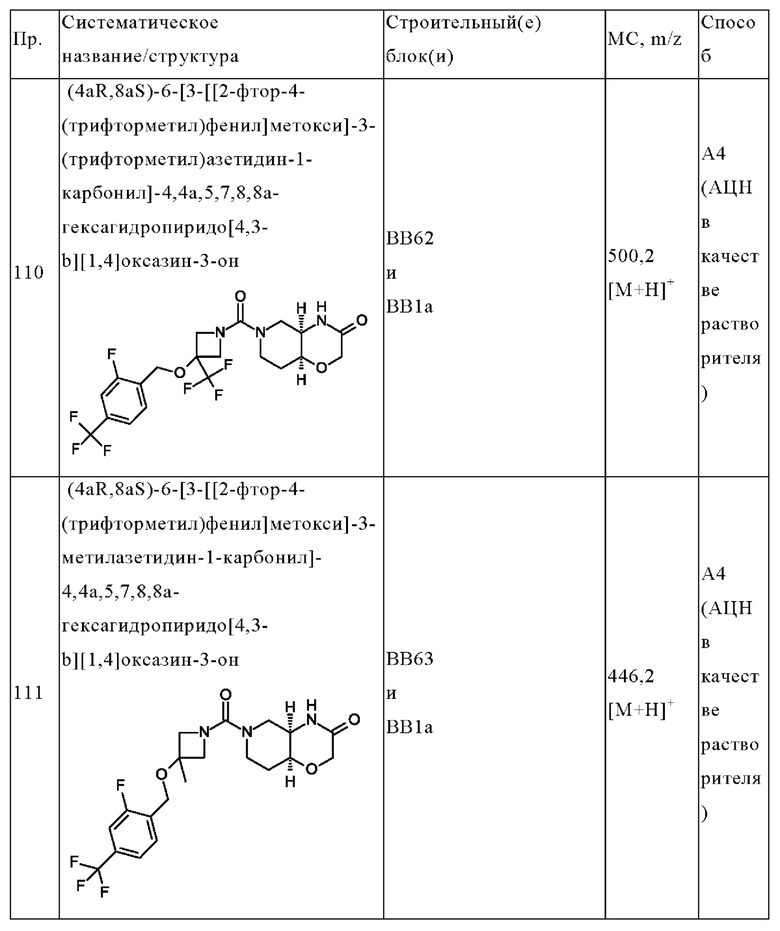
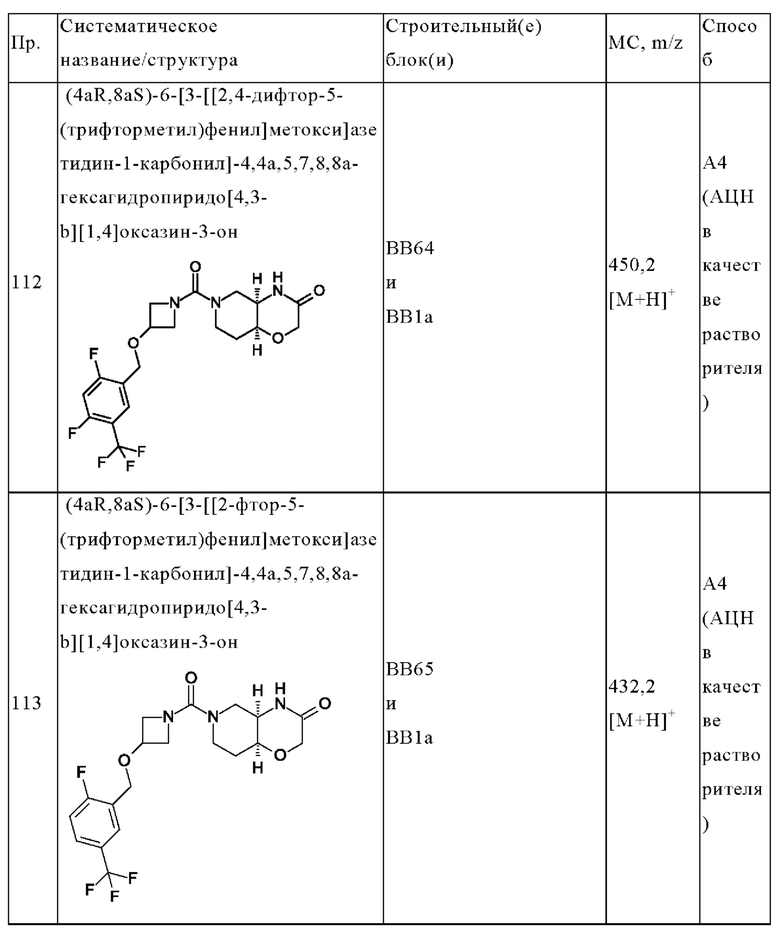

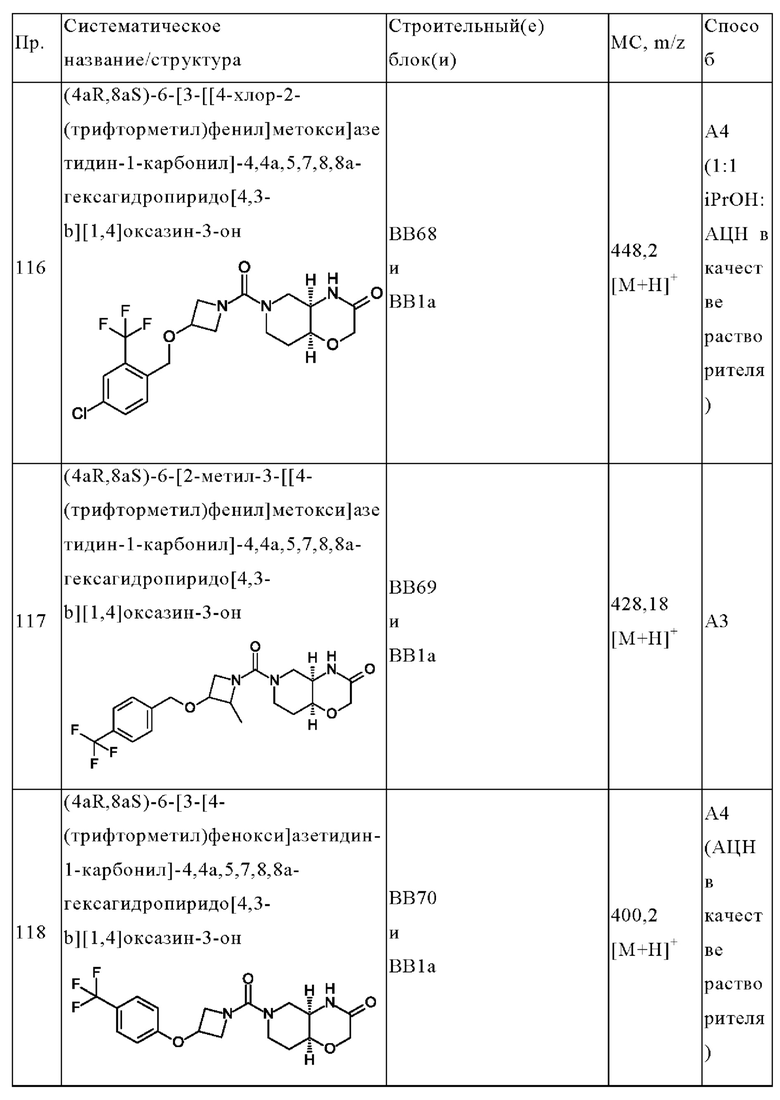


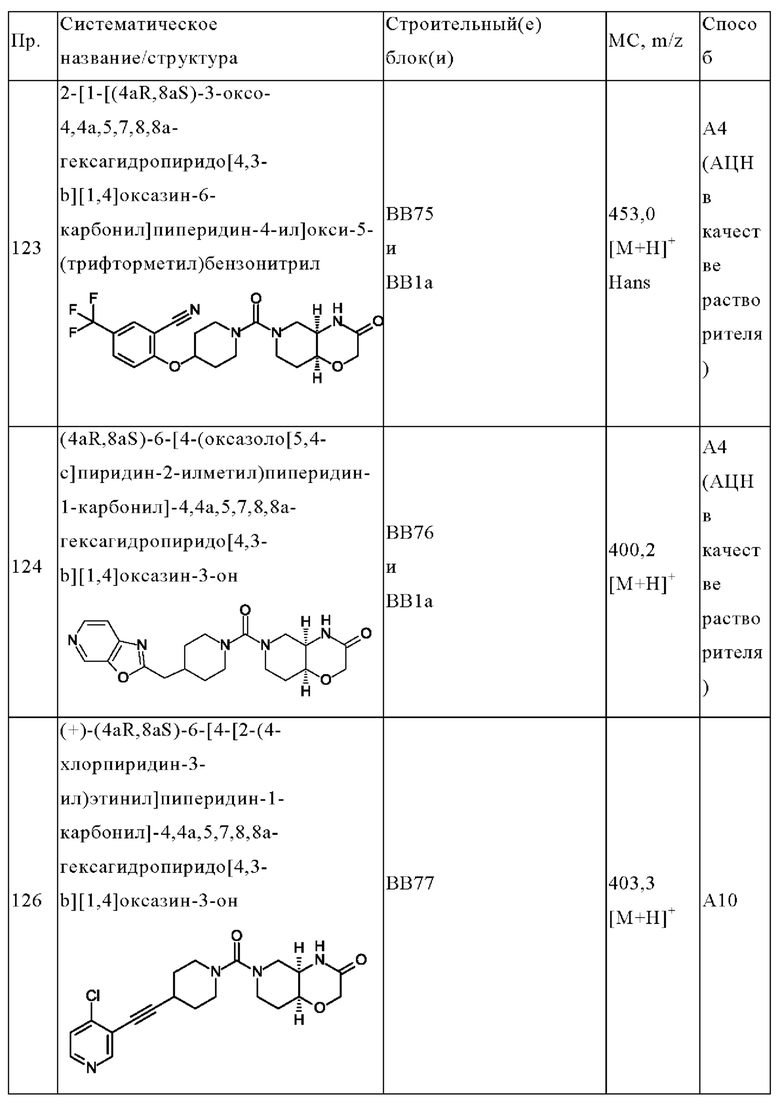
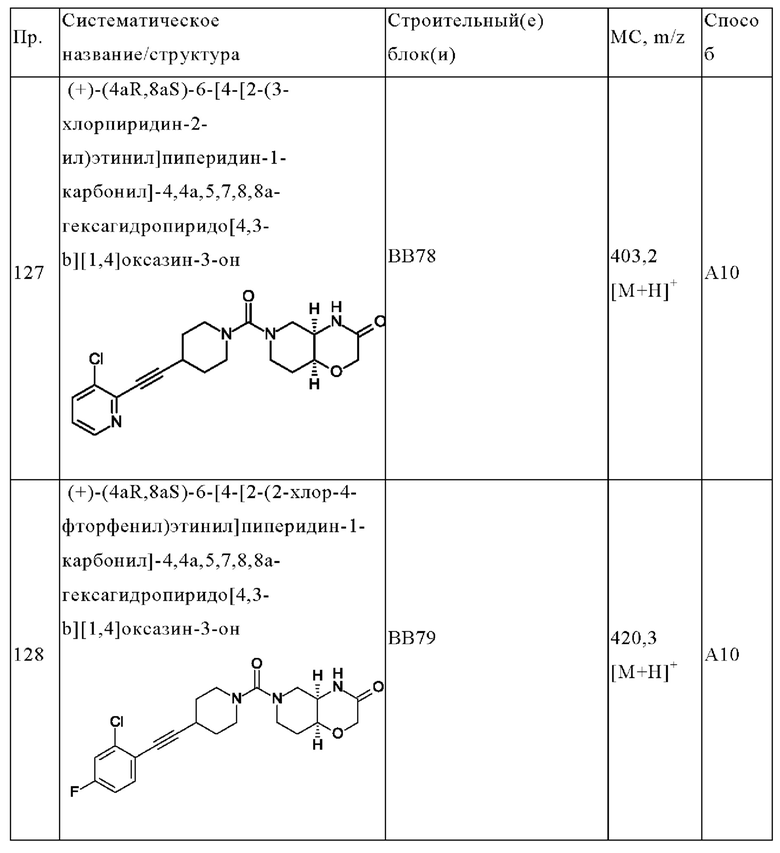


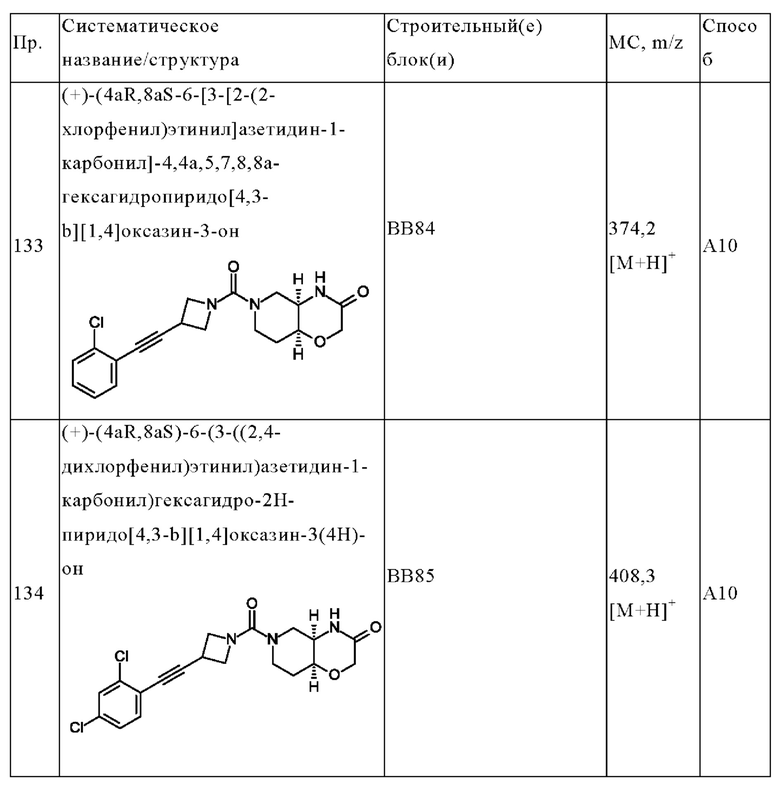
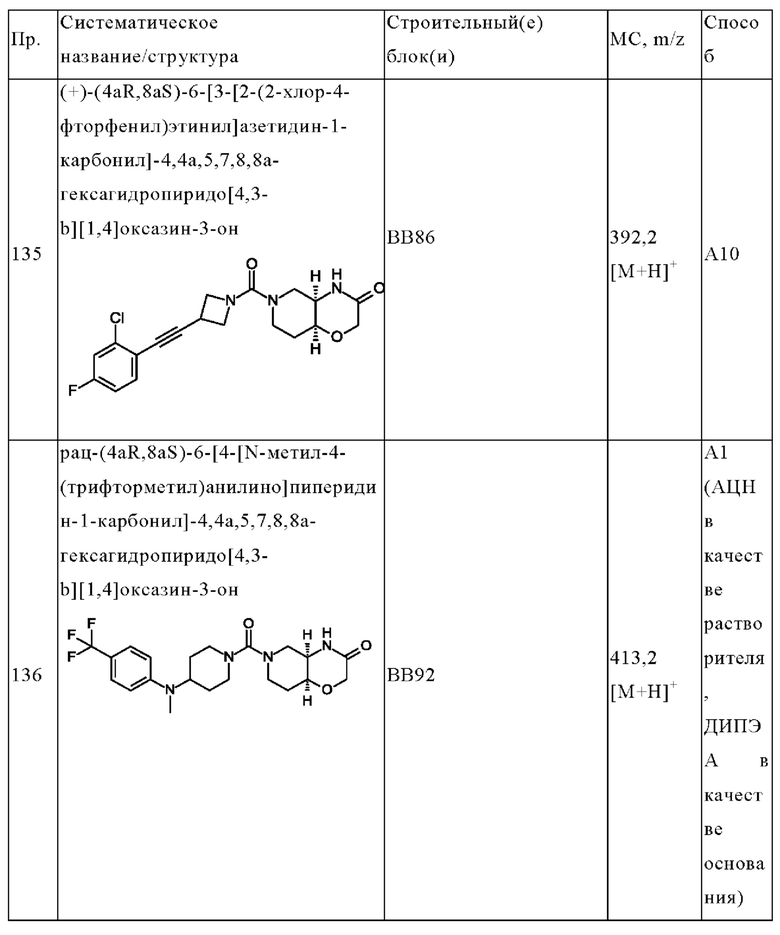
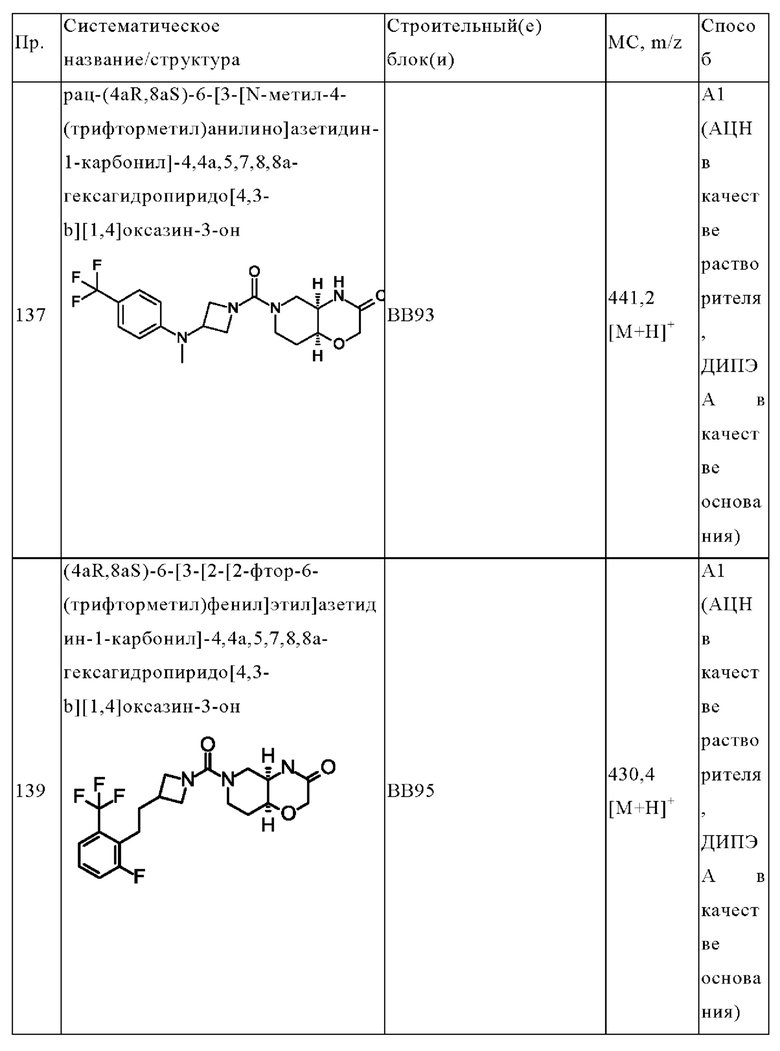

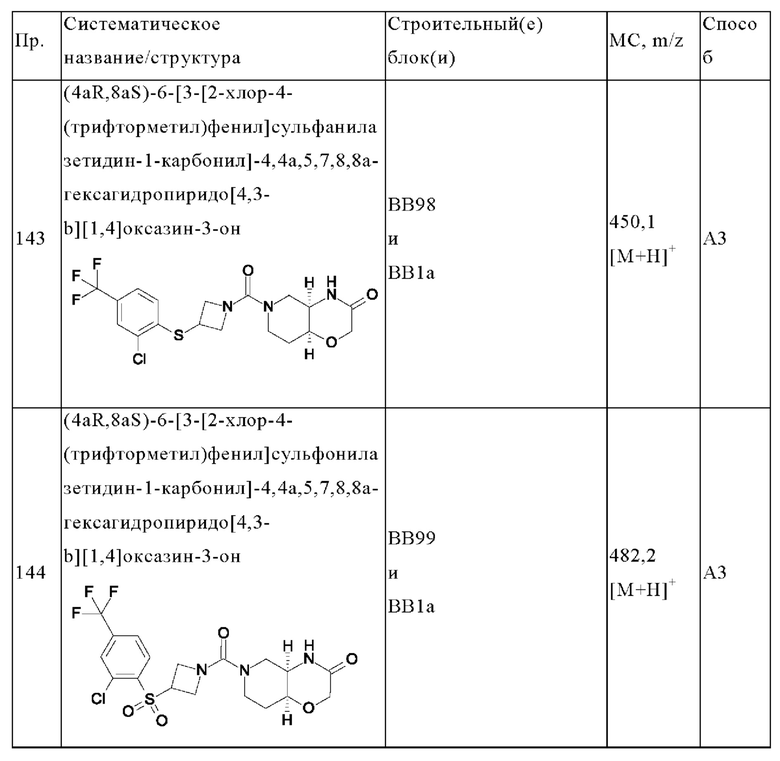
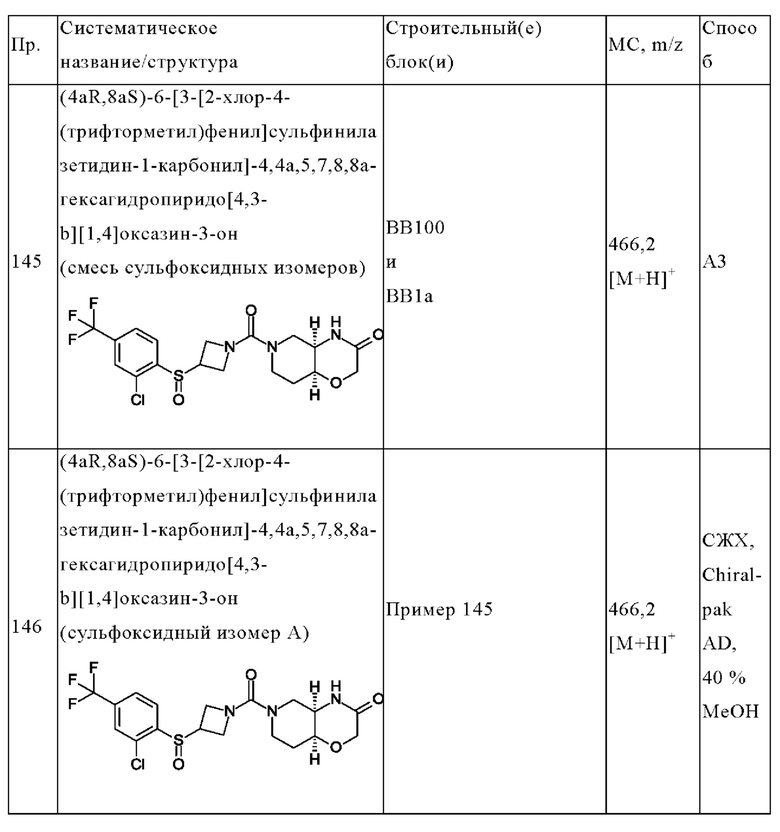
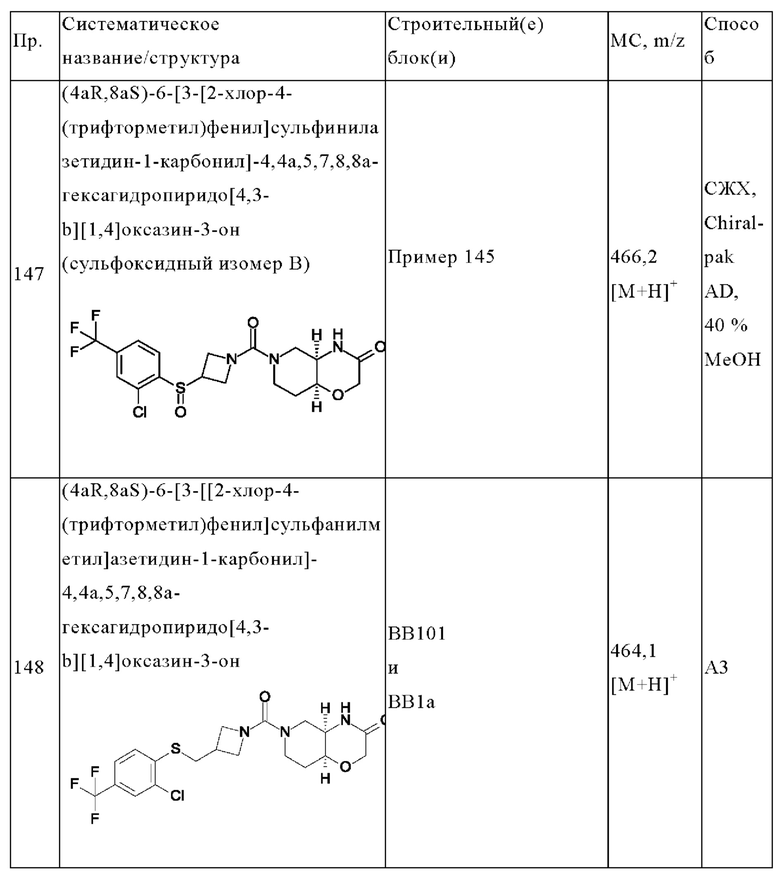




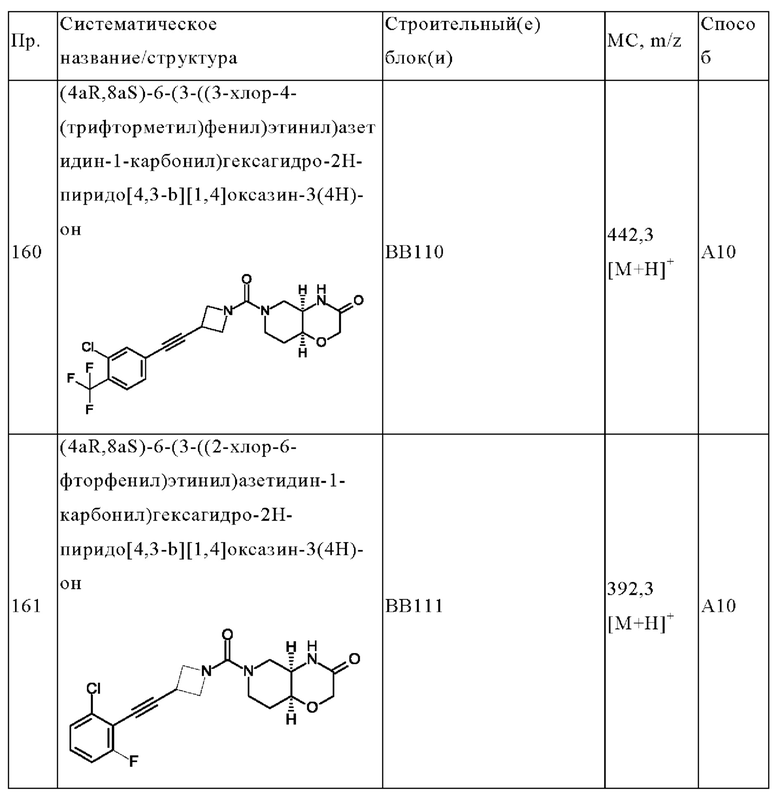
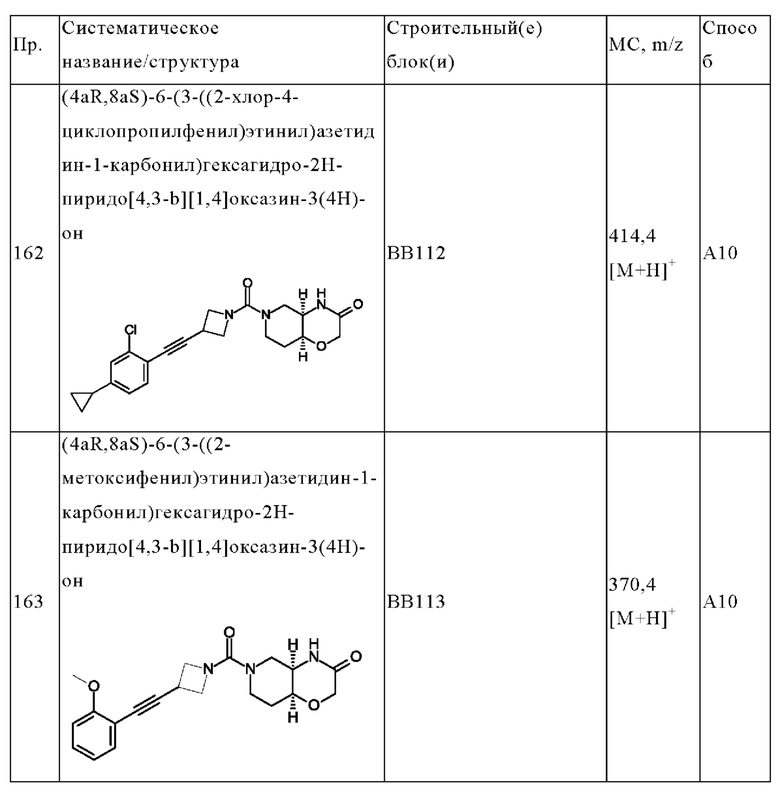




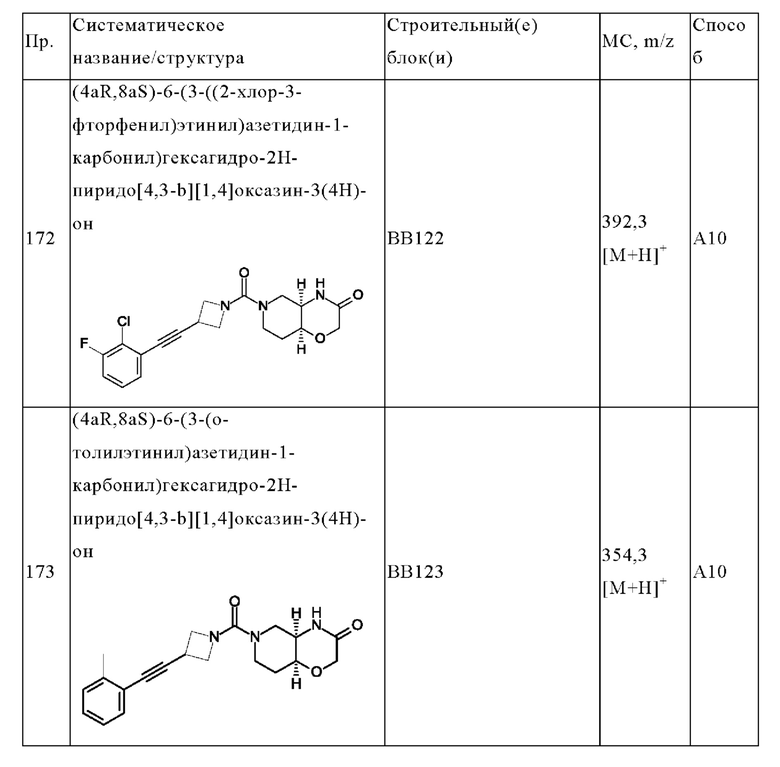
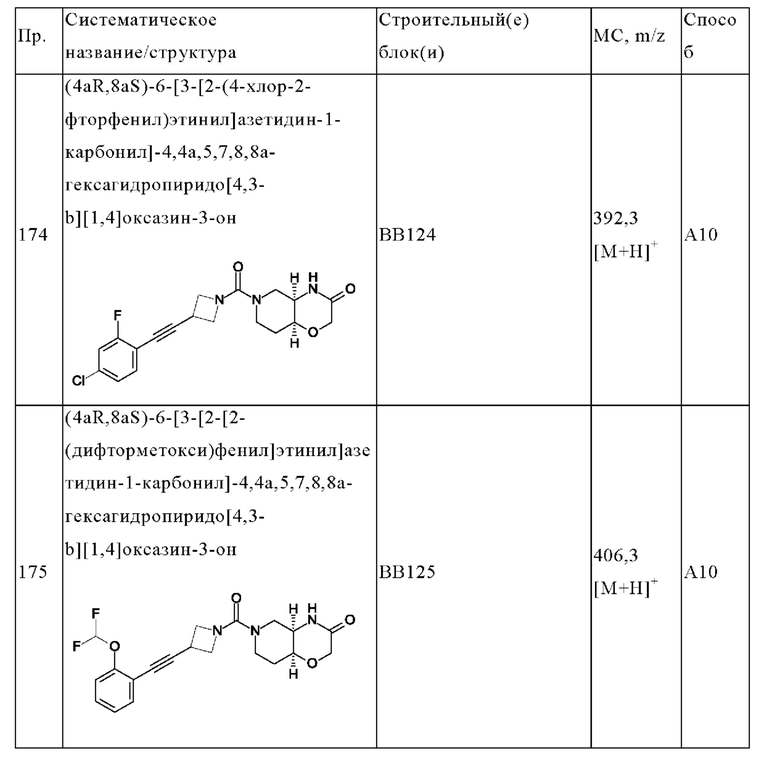


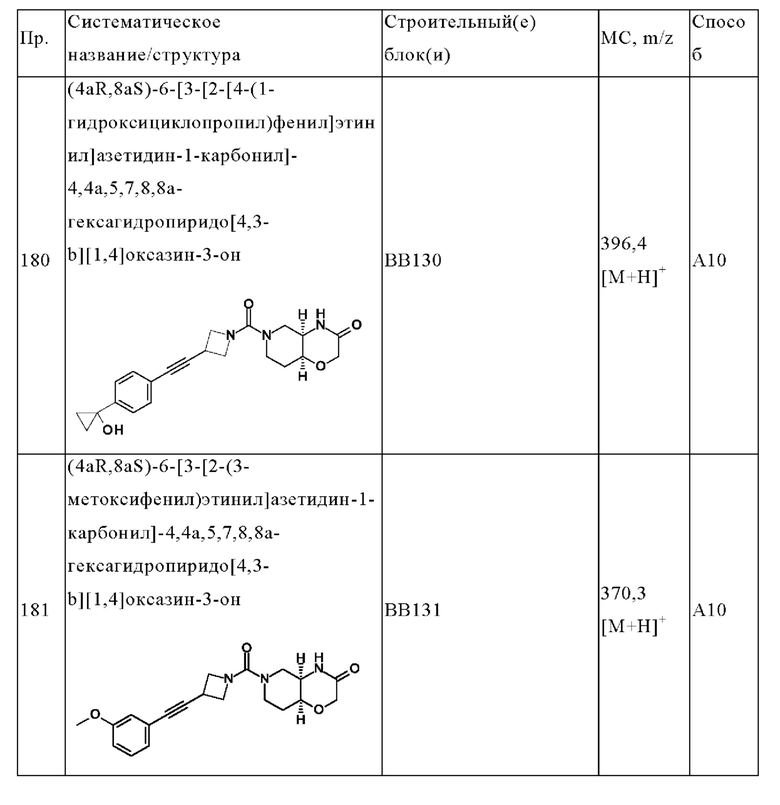

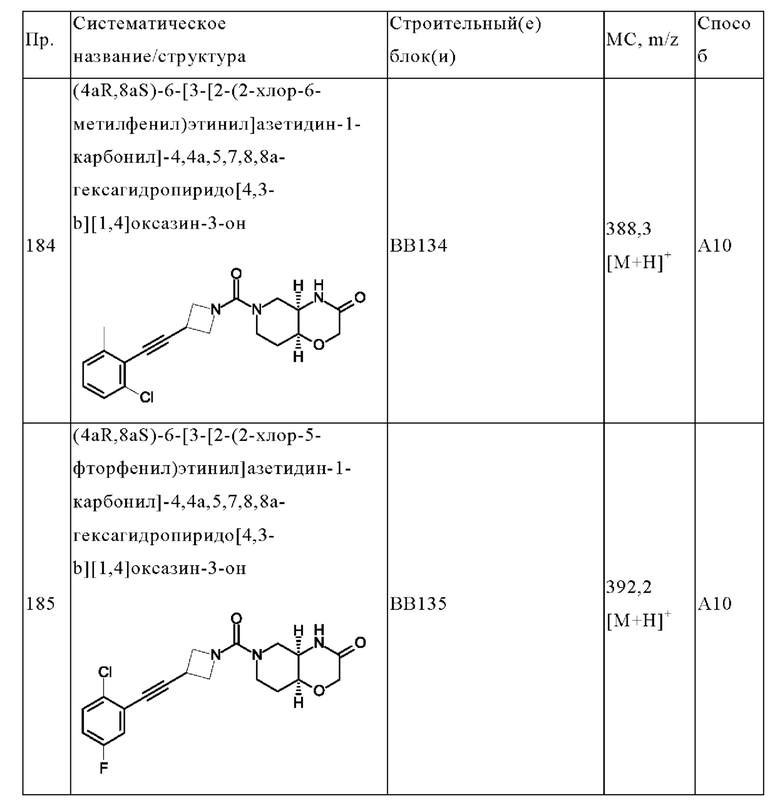

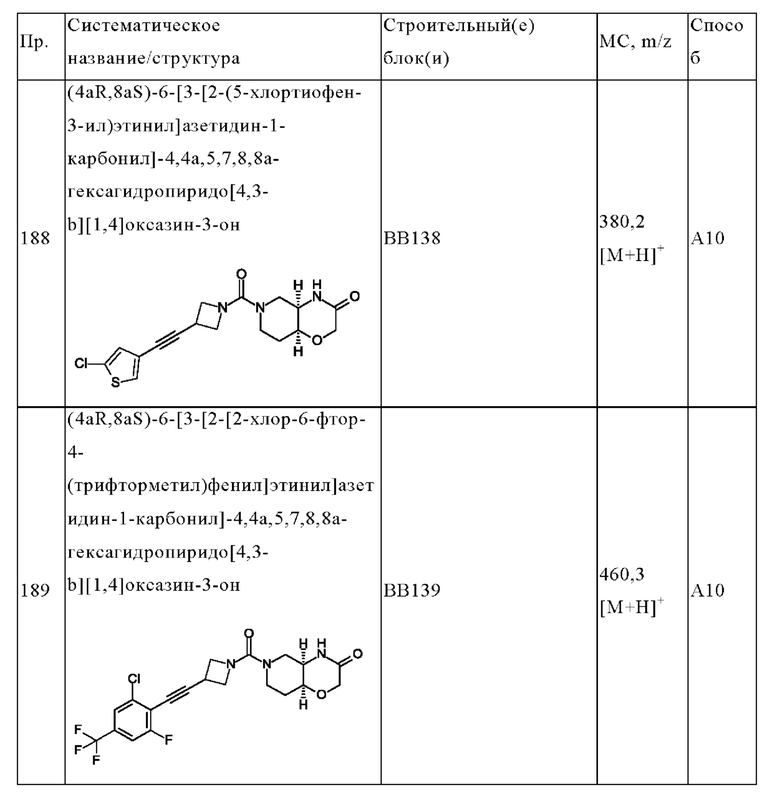
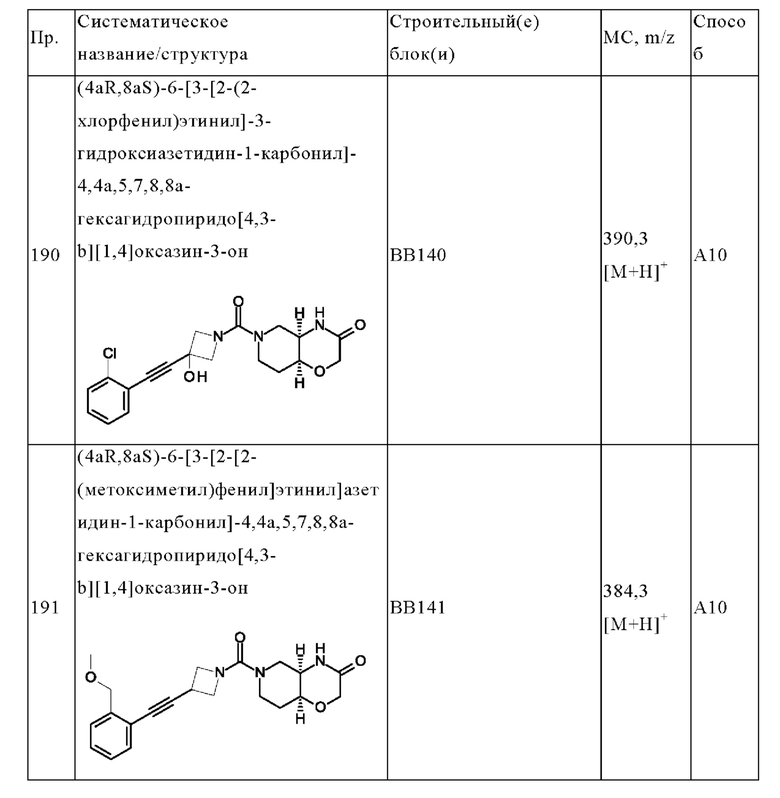
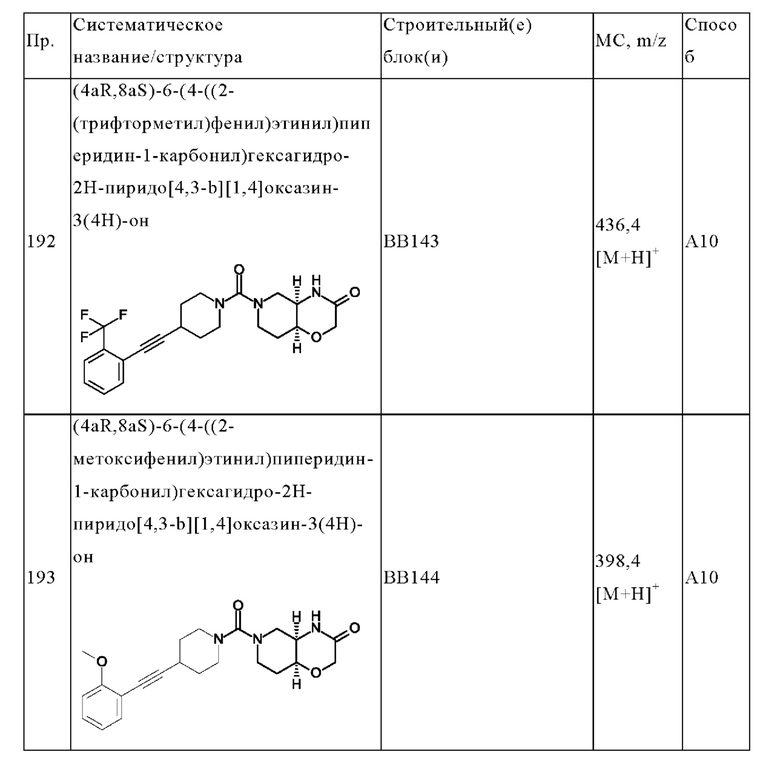
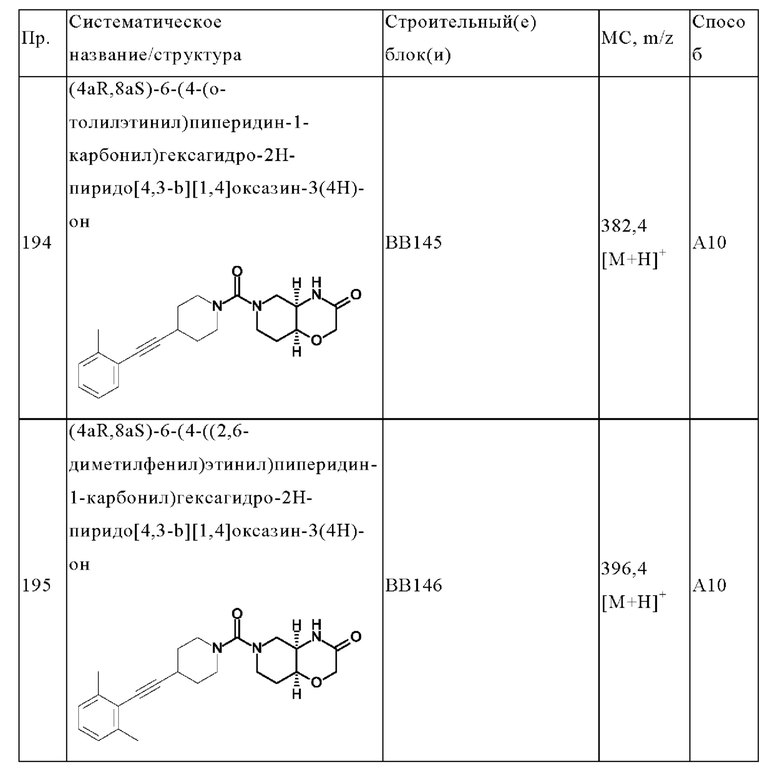

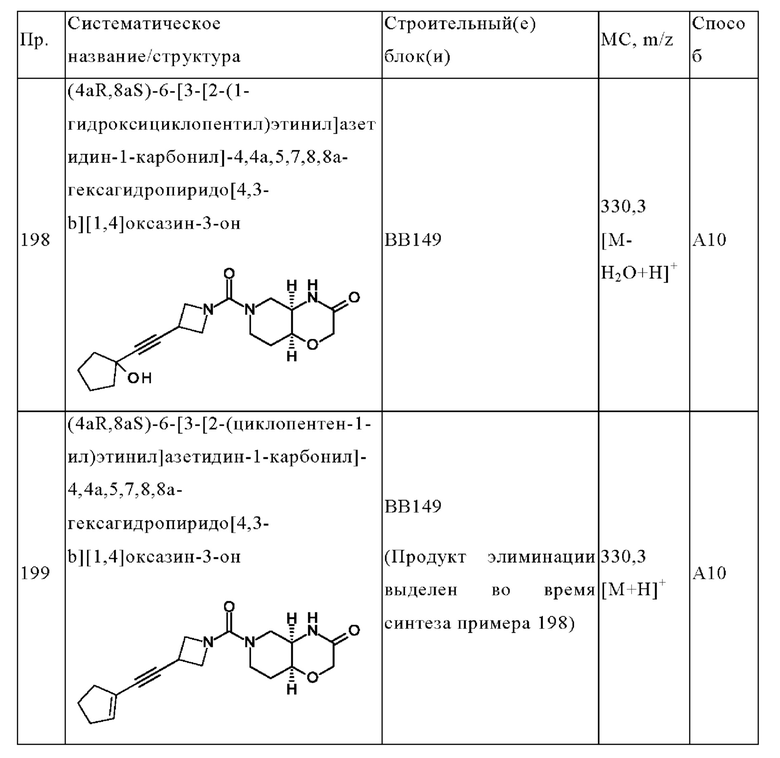


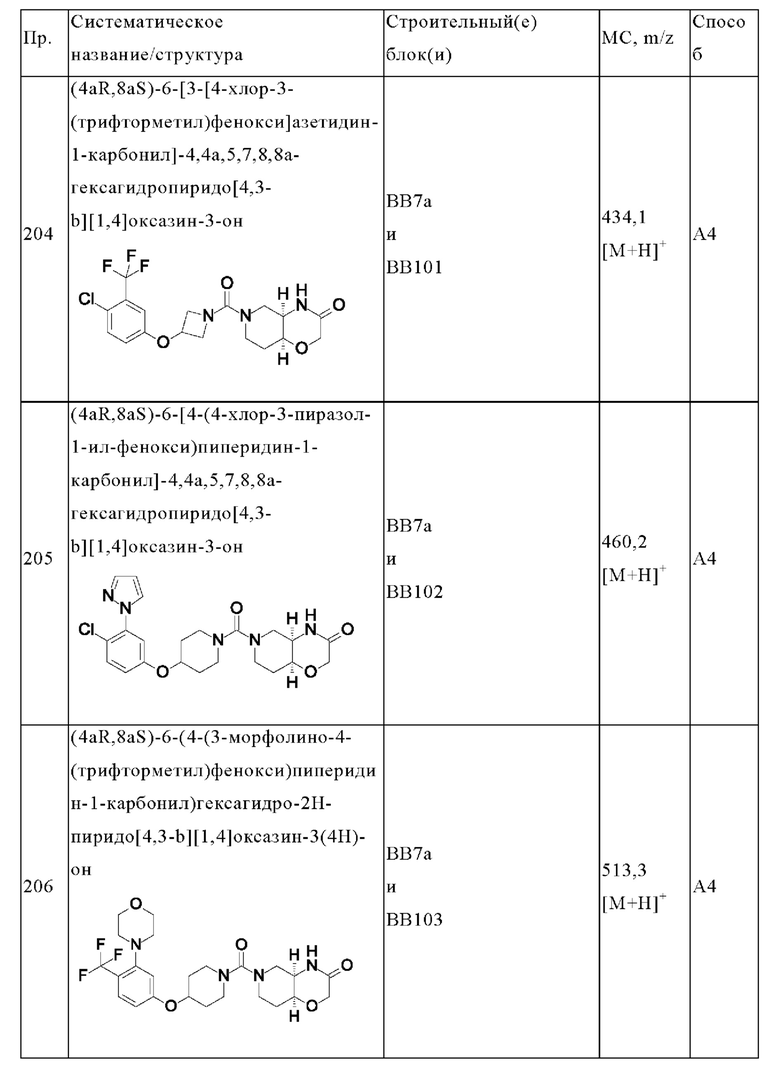

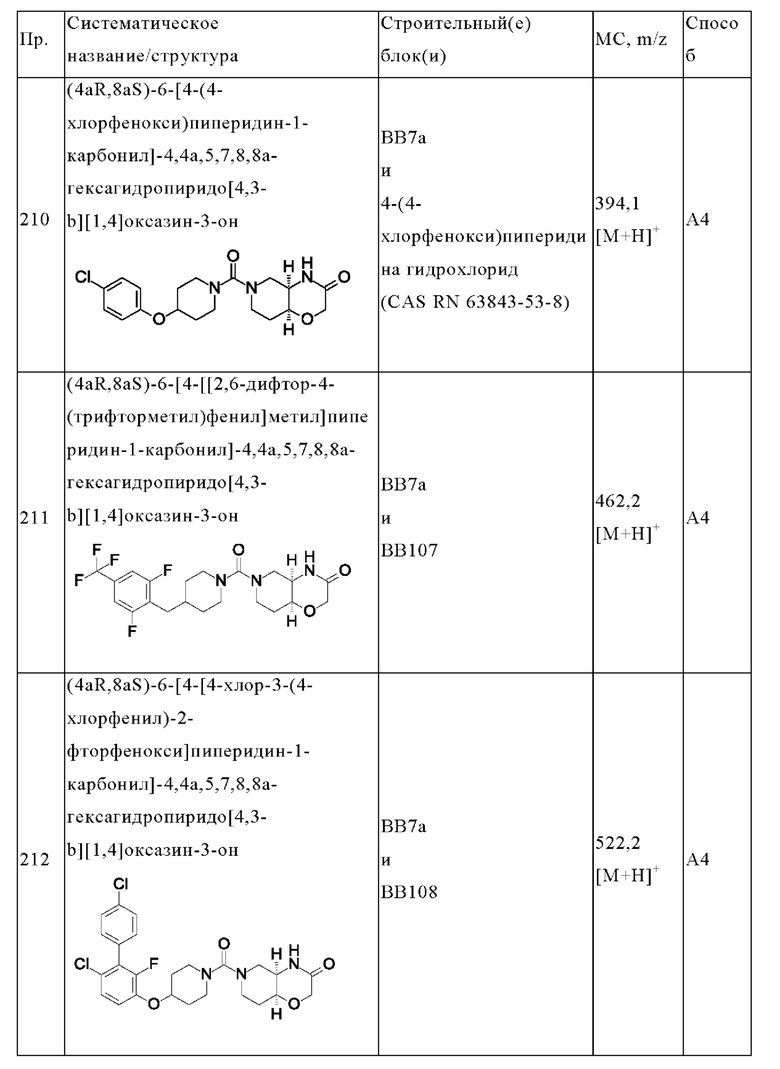

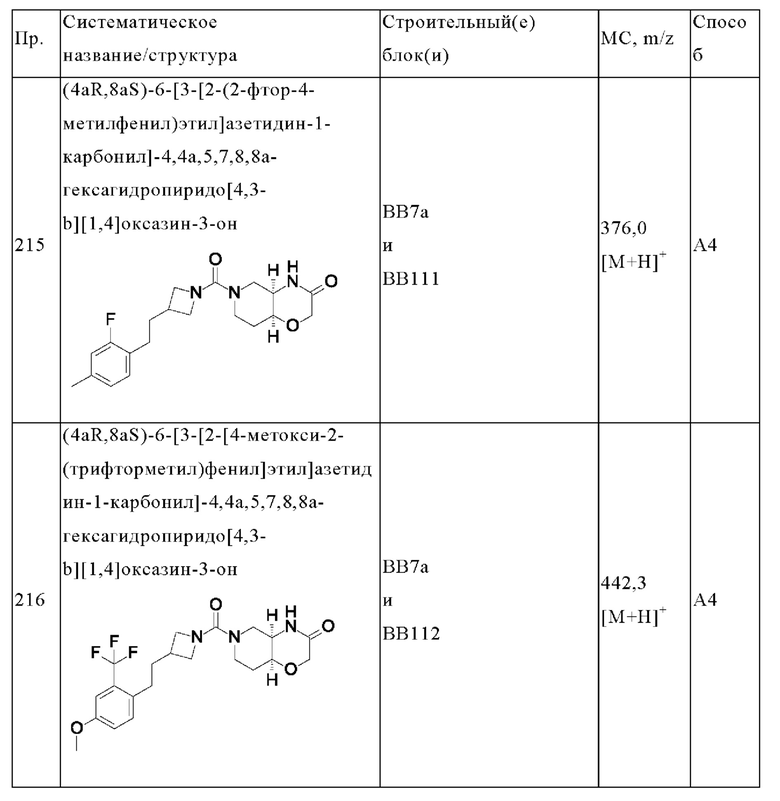

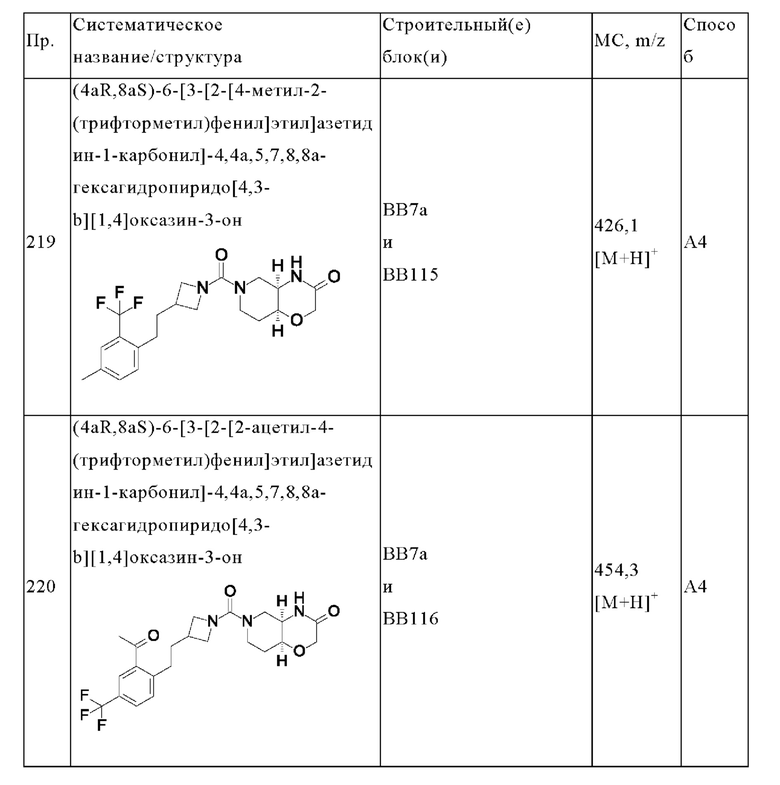





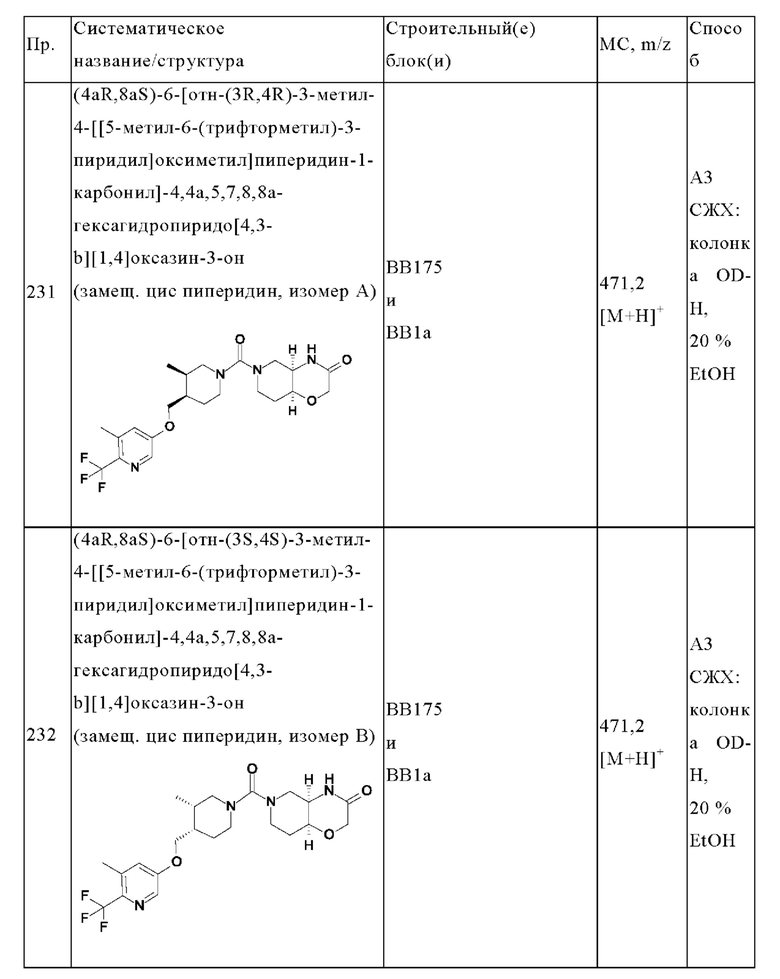

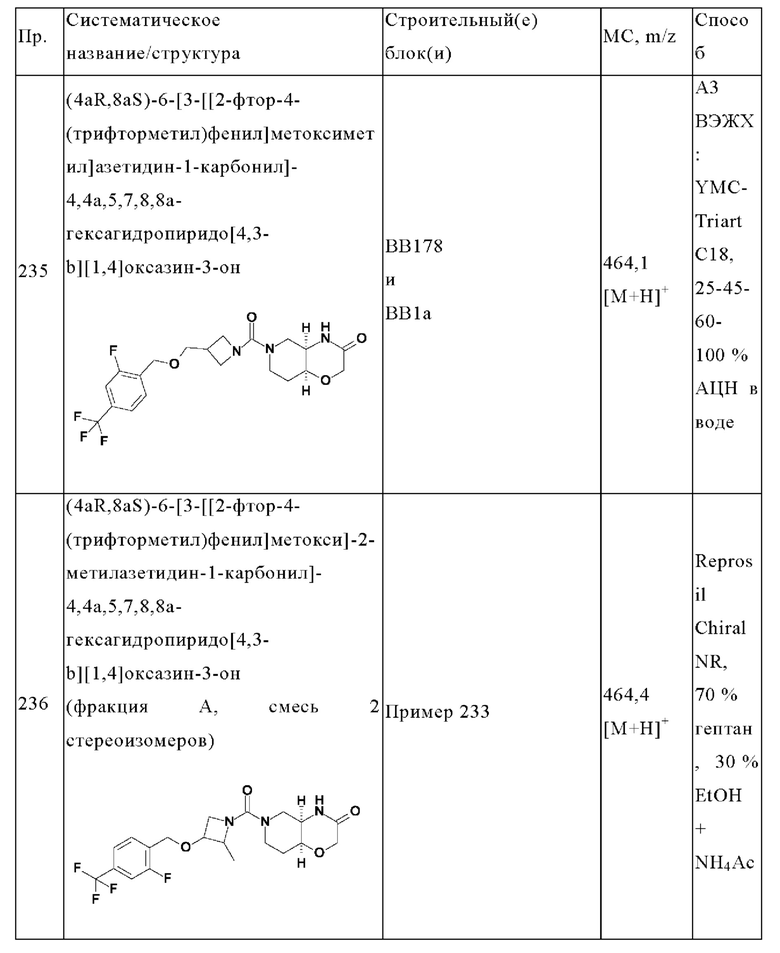





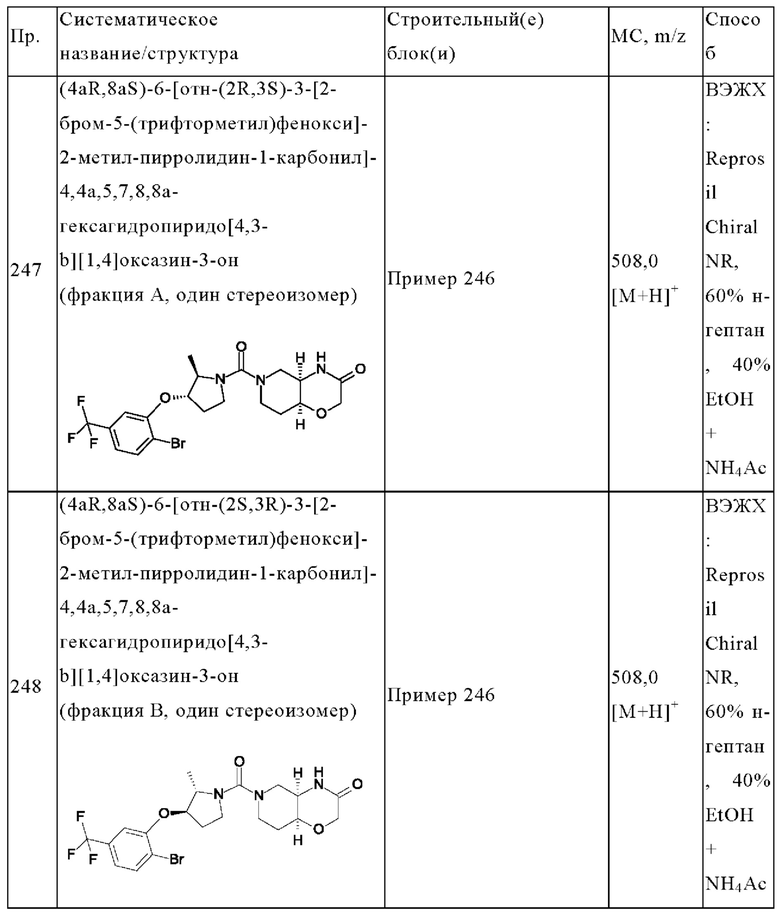



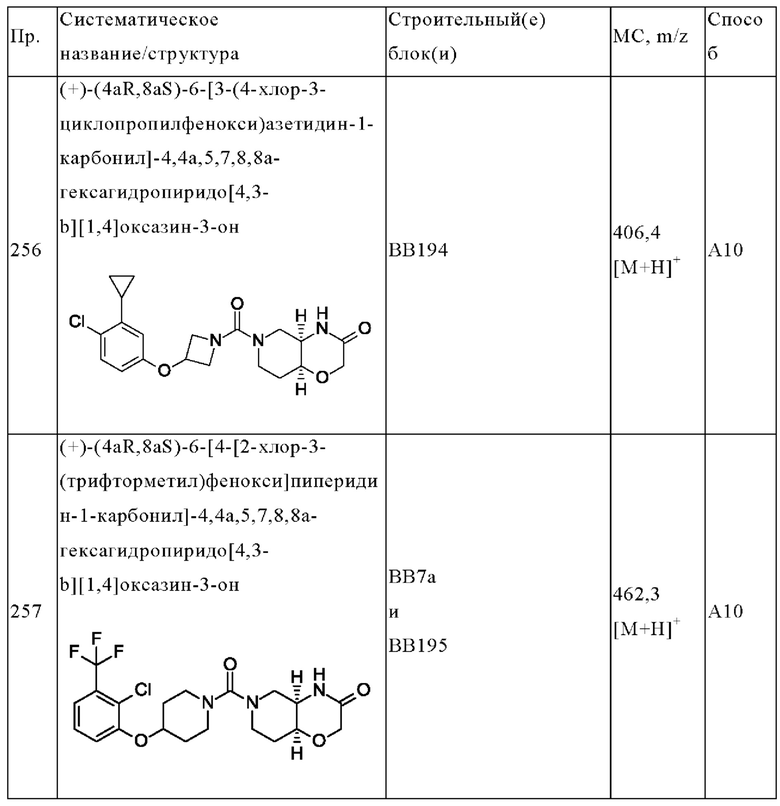
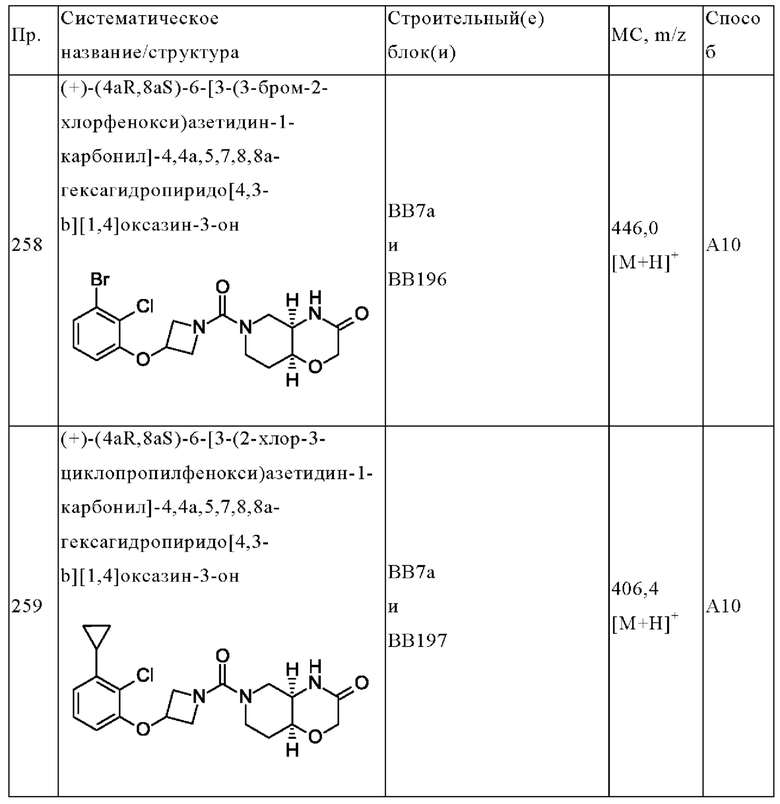
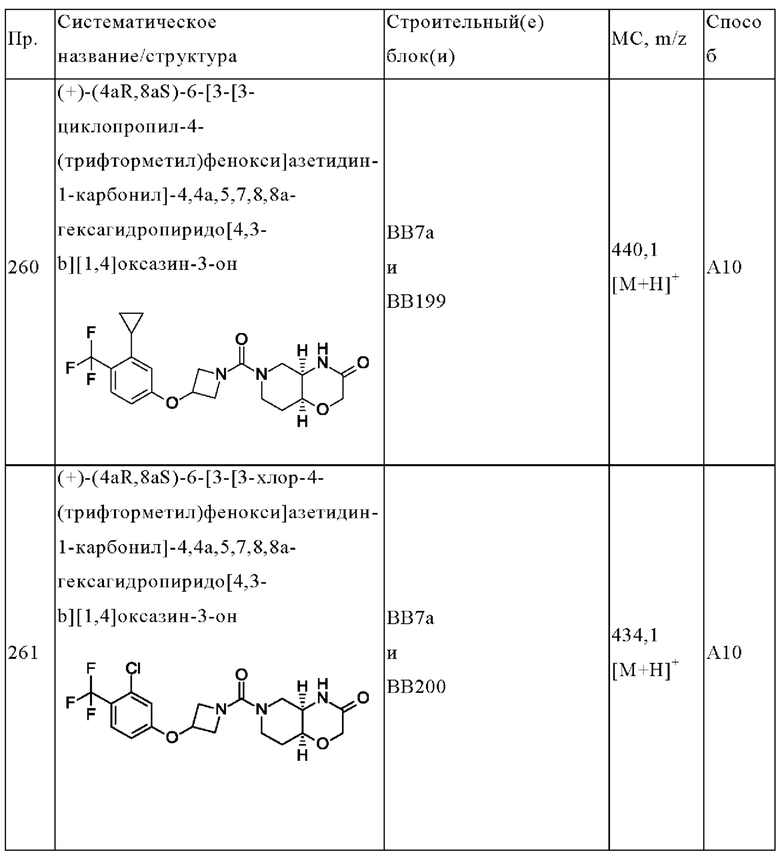

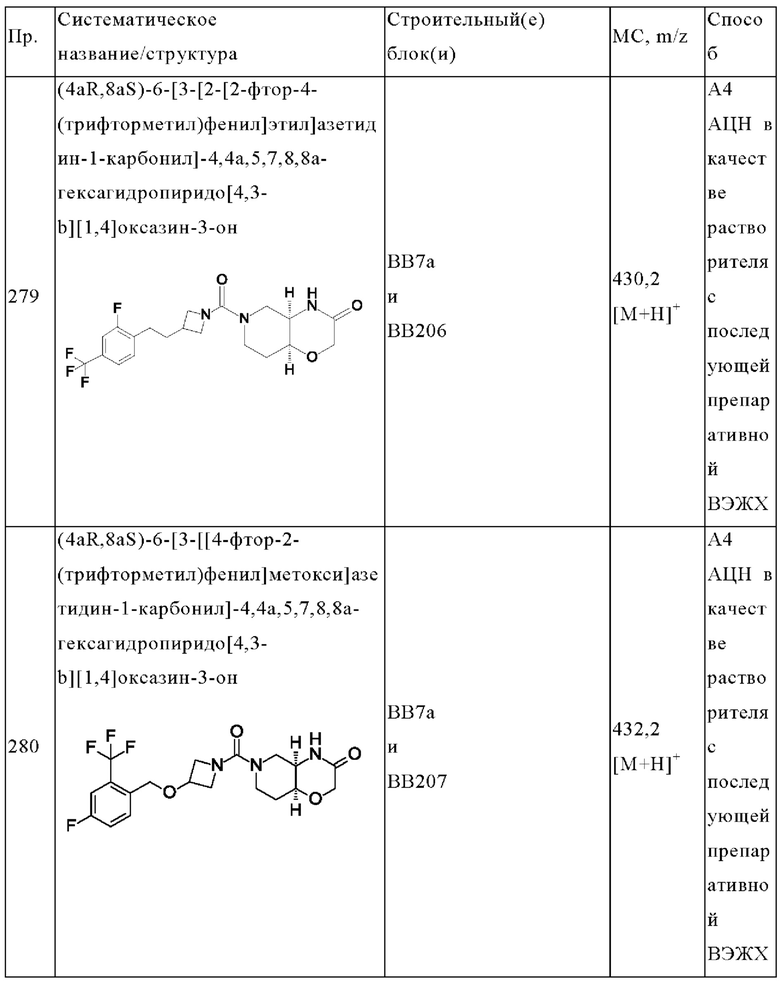








Пример 222
(4aR,8aS)-6-[3-[[6-фтор-4-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он

Этап а) трет-бутил-3-[[6-хлор-4-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбоксилат
В раствор трет-бутил-3-(гидроксиметил)азетидин-1-карбоксилата (CAS Nr, 142253-56-3) (2,60 г, 13,9 ммоль) и 2,6-дихлор-4-(трифторметил)пиридина (CAS Nr, 39890-98-7) (3,00 г, 13,9 ммоль) в ТГФ (60 мл) добавляли NaH (60%, 1,11 г, 27,8 ммоль) и перемешивали смесь в течение 3 ч при 25°С. Раствор вливали в насыщ. води. NH4Cl (50 мл) и экстрагировали EtOAc (2×50 мл). Объединенные органические слои концентрировали под вакуумом с получением неочищенного трет-бутил-3-[[6-хлор-4-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбоксилата (3,00 г, 59%) в виде бесцветного масла, которое непосредственно использовали на следующем этапе. ЖХ-МС (ИЭР): m/z=367,1 [М+Н]+.
Этап b) 2-(азетидин-3-илметокси)-6-хлор-4-(трифторметил)пиридин
Раствор трифторуксусной кислоты (6,3 мл, 81,8 ммоль, 10 экв.) и трет-бутил-3-[[6-хлор-4-(трифторметил)-2-пиридил] оксиметил] азетидин- 1-карбоксилата (3,00 г, 8,18 ммоль) в ДХМ (30 мл) перемешивали при 25°С в течение 4 ч. Раствор концентрировали под вакуумом с получением остатка, который очищали с помощью препаративной ВЭЖХ (условия HCl) с получением 2-(азетидин-3-илметокси)-6-хлор-4-(трифторметил)пиридина (1,00 г, 46%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z=267,0 [М+Н]+.
Этап с) (4aR,8aS)-6-[3-[[6-хлор-4-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо [4,3-b][1,4]оксазин-3-он
Раствор 2-(азетидин-3-илметокси)-6-хлор-4-(трифторметил)пиридина (150 мг, 0,560 ммоль), N,N-диизопропилэтиламина (0,29 мл, 1,69 ммоль) и 4-нитрофенил (4aR,8aS)-3-оксогексагидро-2Н-пиридо[4,3-b][1,4]оксазин-6(5Н)-карбоксилата (ВВ7а) (199 мг, 0,620 ммоль) в АЦН (5 мл) перемешивали при 25°С в течение 16 ч. Раствор концентрировали под вакуумом с получением остатка, который очищали с помощью препаративной ВЭЖХ (условия ТФУ) с получением (4aR,8aS)-6-[3-[[6-хлор-4-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она (100 мг, 40%) в виде бесцветного масла. ЖХ-МС (ИЭР): m/z=449,2 [М+Н]+.
Этап d) (4aR,8aS)-6-[3-[[6-фтор-4-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он
Раствор (4aR,8aS)-6-[3-[[6-хлор-4-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она (75 мг, 0,17 ммоль) и фторида цезия (101 мг, 0,670 ммоль) в ДМСО (3 мл) перемешивали при 80°С в течение 16 ч. Раствор фильтровали и очищали с помощью препаративной ВЭЖХ (условия ТФУ) с получением (4aR,8aS)-6-[3-[[6-фтор-4-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она (22 мг, 28%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z=433,0 [М+Н]+.
Пример 223
(4aR,8aS)-6-[3-[[6-фтор-5-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он

Этап а) трет-бутил-3-[[6-хлор-5-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбоксилат
В раствор трет-бутил-3-(гидроксиметил)азетидин-1-карбоксилата (CAS Nr, 142253-56-3) (1,56 г, 8,33 ммоль) в ТГФ (50 мл) добавляли NaH (60%, 741 мг, 18,5 ммоль), а после - 2,6-дихлор-3-(трифторметил)пиридин (CAS Nr, 55304-75-1) (2,00 г, 9,26 ммоль). Полученную в результате смесь перемешивали при 25°С в течение 3 ч. Раствор вливали в насыщ. водн. NH4Cl (50 мл) и экстрагировали EtOAc (2×30 мл). Объединенные органические слои концентрировали под вакуумом с получением неочищенного остатка, который очищали с помощью колоночной флэш-хроматографии (петролейный эфир : EtOAc = 5:1) с получением трет-бутил-3-[[6-хлор-5-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбоксилата (1,10 г, 32%) в виде бесцветного масла. ЖХ-МС (ИЭР): m/z=311,0 [М-56+Н]+.
Этап b) 6-(азетидин-3-илметокси)-2-хлор-3-(трифторметил)пиридин
Раствор трифторуксусной кислоты (0,37 мл, 4,8 ммоль) и трет-бутил-3-[[6-хлор-5-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбоксилата (1,1 г, 3,0 ммоль) в ДХМ (30 мл) перемешивали при 25°С в течение 4 ч. Раствор концентрировали под вакуумом с получением остатка, который очищали с помощью препаративной ВЭЖХ (условия HCl) с получением 6-(азетидин-3-илметокси)-2-хлор-3-(трифторметил)пиридина (600 мг, 75%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z=267,0 [М+Н]+.
Этап с) (4aR,8aS)-6-[3-[[6-хлор-5-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо [4,3-b][1,4]оксазин-3-он
В раствор 6-(азетидин-3-илметокси)-2-хлор-3-(трифторметил)пиридина (100 мг, 0,380 ммоль) и 4-нитрофенил (4aR,8aS)-3-оксогексагидро-2Н-пиридо[4,3-b][1,4]оксазин-6(5Н)-карбоксилата (ВВ7а) (120 мг, 0,380 ммоль) в АЦН (5 мл) добавляли N,N-диизопропилэтиламин (0,13 мл, 0,75 ммоль) с перемешиванием при 25°С. Раствор перемешивали при 25°С в течение 16 ч. Раствор концентрировали под вакуумом с получением остатка, который очищали с помощью препаративной ВЭЖХ (условия ТФУ) с получением (4aR,8aS)-6-[3-[[6-хлор-5-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она (76 мг, 45%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z=449,1 [М+Н]+.
Этап d) (4aR,8aS)-6-[3-[[6-фтор-5-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он
Раствор (4aR,8aS)-6-[3-[[6-хлор-5-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она (70 мг, 0,16 ммоль) и фторида цезия (95 мг, 0,62 ммоль) в ДМСО (3 мл) перемешивали при 60°С в течение 24 ч. Раствор фильтровали и затем очищали с помощью препаративной ВЭЖХ (условия ТФУ) с получением (4aR,8aS)-6-[3-[[6-фтор-5-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она (38 мг, 49%) в виде белого твердого вещества. ЖХ-МС (ИЭР): m/z=433,3 [М+Н]+.
Синтез строительных блоков ВВ1а и BB1b
(+)-цис-4а,5,6,7,8,8а-гексагидро-4Н-пиридо[4,3-b][1,4]оксазин-3-он и
(-)-цис-4а,5,6,7,8,8а-гексагидро-4Н-пиридо[4,3-b][1,4]оксазин-3-он
Энантиомеры рац-(4aR,8aS)-гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она дигидрохлорида (ВВ1, 500 мг, 2,18 ммоль, ChemBridge Corporation) разделяли с помощью препаративной хиральной ВЭЖХ (колонка ReprosilChiral NR), используя изократическую смесь EtOH (содержащий 0,05% NH4OAc) : н-гептан (30:70).
Первый элюирующийся энантиомер: (+)-цис-4а,5,6,7,8,8а-гексагидро-4Н-пиридо[4,3-b][1,4]оксазин-3-он (ВВ1а). Желтое твердое вещество (0,150 г; 44,0%). МС (ИЭР): m/z=157,1 [М+Н]+.
Второй элюирующийся энантиомер: (-)-цис-4а,5,6,7,8,8а-гексагидро-4Н-пиридо[4,3-b][1,4]оксазин-3-он. (BB1b). Желтое твердое вещество (0,152 г; 44,6%). МС (ИЭР): m/z=157,1 [М+Н]+.
ВВ224
(4-нитрофенил)-4-[[4-(трифторметил)фенил] метил] пиперидин-1-карбоксилат
В раствор 4-(4-(трифторметил)бензил)пиперидина (100 мг, 411 мкмоль, CAS RN 192990-03-7) в ДХМ (1 мл) добавляли ТЭА (83,2 мг, 115 мкл, 822 мкмоль). При охлаждении до 0°С добавляли 4-нитрофенилкарбонохлоридат (91,1 мг, 452 мкмоль, CAS RN 7693-46-1), реакционную смесь оставляли нагреваться до КТ и перемешивали в течение 18 часов. Реакционную смесь разводили ДХМ, а после этого промывали Н2О и насыщ. водным раствором NaHCO3. Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Неочищенный материал очищали с помощью флэш-хроматографии (силикагель, 10 г, элюирование EtOAc/гептан 0-50%) с получением указанного в заголовке соединения в виде светло-желтого твердого вещества. (0,165 г; 98,3%). МС (ИЭР): m/z=409,3 [М+Н]+.
ВВ3
рац-(4aR,8aS)-6-(пиперазин-1-карбонил)-4,4а,5,7,8,8а-гексагидропиридо[4,3-B][1,4]оксазин-3-он
В смесь рац-трет-бутил-4-((4aR,8aS)-3-оксооктагидро-2Н-пиридо[4,3-b][1,4]оксазин-6-карбонил)пиперазин-1-карбоксилата (100 мг, 271 мкмоль) в ДХМ (3 мл) добавляли ТФУ (155 мг, 105 мкл, 1,36 ммоль) и перемешивали смесь при КТ в течение 15 ч в атмосфере аргона. Реакционную смесь промывали насыщенным водным раствором NaHCO3. Слой Н2О концентрировали в вакууме с получение белого твердого вещества, которое растирали с ДХМ в течение 30 мин перед фильтрацией. Фильтрат концентрировали с получением светло-желтой смолы (70 мг, 96,1%). МС (ИЭР): m/z=269,3 [М+Н]+.
Этап а) рац-трет-бутил-4-((4aR,8aS)-3-оксооктагидро-2Н-пиридо[4,3-b][1,4]оксазин-6-карбонил)пиперазин-1-карбоксилат
В смесь трифосгена (1,29 г, 4,36 ммоль) и Na2CO3 (1,98 г, 18,7 ммоль) в ТГФ (3 мл) при 0°С по каплям добавляли раствор трет-бутилпиперазин-1-карбоксилата (1,16 г, 6,23 ммоль, CAS RN 57260-71-6) в ТГФ (90 мл). Реакционную смесь перемешивали в течение 10 мин при 0°С, затем оставляли нагреваться до КТ и продолжали перемешивать при КТ в течение 5 ч. Суспензию отфильтровывали, а фильтрат концентрировали в вакууме. Остаток растворяли в ТГФ (40 мл) и по каплям добавляли в перемешиваемую суспензию рац-(4aR,8aS)-гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она гидрохлорида (1200 мг, 6,23 ммоль, Chembridge Corporation) и ДИПЭА (4,83 г, 6,53 мл, 37,4 ммоль) в ТГФ (40 мл) при 0°С. Через 30 мин при 0°С реакционную смесь оставляли нагреваться до КТ и перемешивали при КТ в течение 15 ч. Смесь фильтровали, а фильтрат концентрировали в вакууме. Остаток разводили ДХМ и промывали водой, насыщ. раствором NaHCO3 и солевым раствором. Органический слой сушили над Na2SO4, фильтровали и концентрировали с получение белого твердого вещества (1,13 г, 58,6%). МС (ИЭР): m/z=313,3 [М+Н]+.
ВВ4
(4-нитрофенил)-4-(феноксиметил)пиперидин- 1-карбоксилат
Это соединение готовили по аналогии с ВВ2, используя 4-(феноксиметил)пиперидин (CAS N63614-86-8), с получением указанного в заголовке соединения в виде белого твердого вещества, которое использовали на следующем этапе без дополнительной очистки.
ВВ5
2-(4-пиперидилметил)-5-(трифторметил)пиридин; гидрохлоридная соль
трет-бутил-4-[[5-(трифторметил)-2-пиридил] метил] пиперидин-1-карбоксилат (320 мг, 0,930 ммоль) растворяли в 4 М HCl в EtOAc (10,0 мл, 40 ммоль) и перемешивали раствор при 20°С в течение 2 ч. Смесь концентрировали с получение необходимого соединения в виде светло-желтого твердого вещества (0,259, 94,8%). МС (ИЭР): m/z=245,0 [М-HCl+Н]+.
Этап а) трет-бутил-4-[[5-(трифторметил)-2-пиридил]метил]пиперидин-1-карбоксилат
2-бром-5-(трифторметил)пиридин (500,0 мг, 2,21 ммоль, CAS RN 1000773-62-5) дегазировали перед добавлением 9-BBN раствора 0,5 М в ТГФ (4,87 мл, 2,43 ммоль, CAS RN 280-64-8). Полученный в результате раствор кипятили с обратным холодильником в течение 1 ч. После охлаждения до КТ раствор добавляли в раствор трет-бутил-4-метиленпиперидин-1-карбоксилата (480,1 мг, 2,43 ммоль, CAS RN 159635-49-1), хлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия (II) (161,89 мг, 0,220 ммоль, CAS RN 72287-26-4) и K2CO3 (611,56 мг, 4,42 ммоль) в ДМФА (5 мл) и воде (0,5 мл). Полученную в результате смесь нагревали при 80°С в течение 4 ч. Смесь охлаждали до КТ и вливали в воду, рН доводили до 11 с помощью 10% водного NaOH и экстрагировали смесь EtOAc. Объединенные органические экстракты промывали солевым раствором, сушили над Na2SO4, фильтровали и выпаривали с получением неочищенного масла, которое очищали с помощью колоночной хроматографии (кремниевый адсорбент; градиент ПЭ : EtOAc от 10:1 потом 5:1) с получением необходимого соединения в виде светло-желтого масла (320 мг, 0,930 ммоль, 42%). МС (ИЭР): m/z=289,0 [М-С4Н8+Н]+.
ВВ6
рац-(4aS,8aS)-гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он
рац-бензил-(4aS,8aS)-3-оксогексагидро-2Н-пиридо[4,3-b][1,4]оксазин-6(5Н)-карбоксилат (125 мг, 431 мкмоль) растворяли в МеОН (5 мл). Реакционный раствор дегазировали в вакууме и снова наполняли аргоном. Pd-C (20 мг, 188 мкмоль) добавляли в атмосфере аргона. Аргон удаляли из реакционной смеси и снова наполняли водородом. Реакционную смесь перемешивали при КТ в течение 15 ч в атмосфере водорода. Реакционную смесь фильтровали через шприцевой фильтр и концентрировали в вакууме с получением необходимого продукта в виде белого твердого вещества (62 мг, 92,2%). МС (ИЭР): m/z=157,098 [М+Н]+.
Этап а) рац-бензил-(3S,4S)-3-(2-хлорацетамидо)-4-гидроксипиперидин-1-карбоксилат
В перемешиваемую суспензию рац-бензил-(3S,4S)-3-амино-4-гидроксипиперидин-1-карбоксилата (317 мг, 1,27 ммоль, синтезированный в соответствии с патентом US 2011/59118 А1) и ацетата натрия (208 мг, 2,53 ммоль, CAS RN 127-09-3) в смеси ацетона (4 мл)/H2O (0,5 мл) по каплям добавляли раствор хлорацетилхлорида (150 мг, 107 мкл, 1,33 ммоль, CAS RN 79-04-9) в ацетоне (3 мл) между 0-5°С. После добавления реакционную смесь перемешивали при КТ в течение 1 ч, а затем выпаривали до сухости с получением желтой смолы. Неочищенный продукт очищали с помощью хроматографии на силикагеле с получением необходимого продукта в виде желтого твердого вещества (385 мг, 93%). МС (ИЭР): m /z=325,2 [М-Н]-.
Этап b) рац-бензил-(4aS,8aS)-3-оксогексагидро-2Н-пиридо[4,3-b][1,4]оксазин-6(5Н)-карбоксилат
В перемешиваемый раствор рац-бензил-(3S,4S)-3-(2-хлорацетамидо)-4-гидроксипиперидин-1-карбоксилата (385 мг, 1,18 ммоль) в сухом ТГФ (4 мл) добавляли NaH (67,9 мг, 1,7 ммоль) при 0°С. Смесь оставляли достигать КТ, а затем перемешивали в течение 90 мин в атмосфере аргона. Добавляли Н2О (5 мл) и продолжали перемешивание в течение 10 мин при КТ. ТГФ из реакционной смеси удаляли под вакуумом. Остаток обрабатывали ДХМ, а органическую фазу промывали Н2О и солевым раствором, сушили над Na2SO4, фильтровали, а затем концентрировали в вакууме. Остаток очищали с помощью флэш-хроматографии (12 г колонка с обращенной фазой, градиент 0-100% АЦН (0,1% МК) в воде (0,1% МК) с получение необходимого продукта в виде белого твердого вещества (133 мг, 38,9%). МС (ИЭР): m/z=291,3 [М+Н]+.
ВВ7а и BB7b
4-нитрофенил (4aR,8aS)-3-оксогексагидро-2Н-пиридо[4,3-b][1,4]оксазин-6(5Н)-карбоксилат (ВВ7а)
и
4-нитрофенил (4aS,8aR)-3-оксогексагидро-2Н-пиридо[4,3-b][1,4]оксазин-6(5Н)-карбоксилат (BB7b)
В суспензию рац-(4aR,8aS)-гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она; дигидрохлоридная соль (4,5 г, 19,6 ммоль, ВВ1) в сухом ДХМ (125 мл) при 0°С добавляли ДИПЭА (6,35 г, 8,58 мл, 49,1 ммоль), а после - 4-нитрофенилкарбонохлоридат (4,35 г, 21,6 ммоль). Реакционную смесь перемешивали при 0°С в течение 10 мин и при КТ в течение 2 ч. Неочищенную реакцию разводили ДХМ и переносили в разделительную воронку для экстракции насыщ. водн. раствором Na2CO3. Органическую фазу собирали, а водную фазу обратно экстрагировали ДХМ. Объединенные органические фазы сушили над Na2SO4 и выпаривали до сухости с получением 6,62 г неочищенного рацемического продукта (ВВ7) в виде желтого твердого вещества. Неочищенный материал непосредственно подвергали хиральному СЖХ-разделению с получением энантиомера BB7b (2,72 г, второй элюирующийся энантиомер) в виде желтого твердого вещества и энантиомера ВВ7а (3,25 г, первый элюирующийся энантиомер) в виде светло-бежевого твердого вещества, но с примесями BB7b. Проводили дополнительное хиральное СЖХ-разделение с получением 2,71 г ВВ7а. МС (ИЭР): m/z=322,2 [М+Н]+ для обоих энантиомеров.
ВВ8
5-трет-бутил-2-(4-пиперидилметил)оксазол; гидрохлоридная соль
Раствор трет-бутил-4-[(5-трет-бутилоксазол-2-ил)метил] пиперидин-1-карбоксилата (167 мг, 518 мкмоль) в HCl 2 M в диэтиловом эфире (2,59 мл, 5,18 ммоль) перемешивали при КТ в течение 5 ч перед добавление дополнительных 1,29 мл (2,59 ммоль) HCl 2 M в диэтиловом эфире. Белую суспензию перемешивали при КТ в течение ночи. Смесь охлаждали на ледяной бане, затем фильтровали и промывали диэтиловым эфиром с получением необходимого соединения в виде бесцветного масла (0,126 г, 94,0%). МС (ИЭР): m/z=223,2 [М+Н]+.
Этап а) (5-трет-бутилоксазол-2-ил)метил-трифенил-фосфония бромид
В раствор 2-(бромметил)-5-(трет-бутил)оксазола (600 мг, 2,75 ммоль, CAS RN 1334492-54-4) в диэтиловом эфире (5 мл) добавляли трифенилфосфин (722 мг, 2,75 ммоль, CAS RN 603-35-0) и перемешивали смесь при КТ в течение 64 ч. Суспензию охлаждали на ледяной бане и затем фильтровали. Фильтрационный осадок промывали небольшим объемом диэтилового эфира с получение необходимого соединения в виде светло-желтого твердого вещества (0,864 г, 65,4%). МС (ИЭР): m/z=400,2 [М-Br+Н]+.
Этап b) трет-бутил-4-[(5-трет-бутилоксазол-2-ил)метилен] пиперидин-1 -карбоксилат
В ледяную смесь 5-трет-бутилоксазол-2-ил)метил-трифенил-фосфония бромида (355 мг, 739 мкмоль) в ТГФ (7 мл) добавляли 1 М раствор трет-бутилата калия в ТГФ (738 мкл, 738 мкмоль) и перемешивали реакцию при этой температуре в течение 15 мин. Затем в непрозрачный оранжевый раствор добавляли трет-бутил-4-оксопиперидин-1-карбоксилат (162 мг, 813 мкмоль, CAS RN 79099-07-3) и продолжали перемешивание при 0°С еще в течение 15 мин, затем при КТ в течение 42 ч. Реакционную смесь вливали в полунасыщенный раствор NH4Cl и EtOAc и разделяли слои. Водный слой дважды экстрагировали EtOAc. Объединенные органические слои сушили над MgSO4, фильтровали, обрабатывали силикагелем и выпаривали. Соединение очищали с помощью хроматографии на силикагеле на 12 г колонке, используя систему для ЖХСД, с элюированием градиентом н-гептан : EtOAc (от 100: от 0 до 50:50) с получением необходимого соединения в виде бесцветного твердого вещества (0,180 мг; 76,0%). МС (ИЭР): m/z=321,3 [М+Н]+.
Этап с) трет-бутил-4-[(5-трет-бутилоксазол-2-ил)метил] пиперидин-1-карбоксилат
В раствор трет-бутил-4-[(5-трет-бутилоксазол-2-ил)метилен]пиперидин-1-карбоксилата (180 мг, 562 мкмоль) в МеОН (1 мл) и EtOAc (1 мл) добавляли Pd/C 10% (17,9 мг, 16,9 мкмоль) и перемешивали суспензию в атмосфере водорода при 1,3 бар в течение 2 ч. Суспензию фильтровали через микрофильтр, а фильтрат выпаривали с получением необходимого соединения в виде бесцветного масла (0,167 г; 92,2%). МС (ИЭР): m/z=323,3 [М+Н]+.
ВВ12
4-[(2-хлор-4-фторфенокси)метил]-4-метилпиперидин; гидрохлоридная соль
В раствор трет-бутил-4-[(2-хлор-4-фторфенокси)метил]-4-метилпиперидин-1-карбоксилата (186 мг, 0,520 ммоль) в EtOAc (1,5 мл) добавляли НСl в EtOAc (4 М, 1,5 мл) при 0°С. Раствор перемешивали при 15°С в течение 3 ч. Раствор концентрировали под вакуумом, затем сушили путем лиофилизации с получением необходимого продукта в виде белого твердого вещества (64,0 мг, 0,220 ммоль, выход 40,3%). МС (ИЭР): m/z=258 [М+Н]+.
Этап а) трет-бутил-4-метил-4-(метилсульфонилоксиметил)пиперидин-1-карбоксилат
В раствор трет-бутил-4-(гидроксиметил)-4-метилпиперидин-1-карбоксилата (500 мг, 2,14 ммоль, CAS RN: 614730-97-1) в ДХМ (5 мл) добавляли NEt3 (0,45 мл, 3,22 ммоль) и метансульфонилхлорид (0,23 мл, 3,0 ммоль) при 0°С. Смесь перемешивали при 0°С в течение 2 ч. Смесь дважды промывали водой (по 3 мл каждый раз) при 0°С и сушили над Na2SO4. Органический слой концентрировали в вакууме с получением необходимого продукта в виде бесцветного масла (766 мг, 2,46 ммоль, 98,5%), которое использовали на следующем этапе без дополнительной очистки. МС (ИЭР): m/z=256 [М-56+Н]+.
Этап b) трет-бутил-4-[(2-хлор-4-фторфенокси)метил]-4-метилпиперидин-1-карбоксилат
В раствор трет-бутил-4-метил-4-(метилсульфонилоксиметил)пиперидин-1-карбоксилата (450 мг, 1,46 ммоль) в ДМФА (5 мл) добавляли CS2CO3 (620 мг, 1,9 ммоль) и 2-хлор-4-фторфенол (0,14 мл, 1,46 ммоль) при 15°С.Смесь нагревали до 90°С и перемешивали в течение 16 ч. Реакционный раствор разводили EtOAc (10 мл), дважды промывали солевым раствором (по 10 мл каждый раз) и сушили над Na2SO4. Органический слой концентрировали под вакуумом с получением неочищенного продукта (0,7 г) в виде светло-желтого масла. Неочищенный продукт очищали с помощью препаративной ВЭЖХ и сушили путем лиофилизации с получением необходимого соединения в виде бесцветного твердого вещества (186 мг, 0,520 ммоль, выход 35,5%). МС (ИЭР): m/z=302 [М-56+Н]+.
ВВ15
4-[(2-хлор-4-фторфенокси)метил]-4-фторпиперидин; гидрохлоридная соль
В раствор трет-бутил-4-[(2-хлор-4-фторфенокси)метил]-4-фторпиперидин-1-карбоксилата (220 мг, 0,610 ммоль) в EtOAc (2 мл) добавляли HCl/EtOAc (0,4 мл, 3,6 ммоль) при 0°С. Раствор перемешивали при 15°С в течение 2,5 ч. Раствор концентрировали в вакууме, затем сушили путем лиофилизации с получением необходимого продукта в виде белого твердого вещества (136,7 мг, 75,4%).
Этап а) трет-бутил-4-фтор-4-(метилсульфонилоксиметил)пиперидин-1-карбоксилат
В раствор трет-бутил-4-фтор-4-(гидроксиметил)пиперидин-1-карбоксилата (500 мг, 2,14 ммоль) в ДХМ (5 мл) добавляли NEt3 (0,45 мл, 3,22 ммоль) и метансульфонилхлорид (0,23 мл, 3 ммоль) при 0°С. Смесь перемешивали при 0°С в течение 2 ч. Смесь дважды промывали Н2О (по 3 мл каждый раз) при 0°С и сушили над Na2SO4. Органический слой концентрировали с получением соединения в виде бесцветного масла (766 мг, 98,5%), которое использовали на следующем этапе без дополнительной очистки. МС (ИЭР: m/z=256 [М-56+Н]+.
Этап b) трет-бутил-4-[(2-хлор-4-фторфенокси)метил]-4-фторпиперидин-1-карбоксилат
В раствор трет-бутил-4-фтор-4-(метилсульфонилоксиметил)пиперидин-1-карбоксилата (383 мг, 1,23 ммоль) в ДМФА (4 мл) добавляли Cs2CO3 (601 мг, 1,85 ммоль), 2-хлор-4-фторфенол (0,13 мл, 1,35 ммоль) и 2-хлор-4-фторфенол (0,13 мл, 1,35 ммоль) при 15°С. Смесь нагревали до 85°С и перемешивали в течение 16 ч. Смесь три раза экстрагировали EtOAc (по 5 мл каждый раз) при 15°С, объединенные органические слои три раза промывали солевым раствором (по 5 мл каждый раз), сушили над Na2SO4, фильтровали и выпаривали. Неочищенный продукт очищали с помощью препаративной ВЭЖХ и сушили путем лиофилизации с получением необходимого соединения в виде светло-желтого масла (275 мг, 0,760 ммоль, 61,5%). МС (ИЭР: m/z=306 [М-56+Н]+.
ВВ16
рац-(4aR,8aS)-6-(4-(гидроксиметил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он
В суспензию рац-(4aR,8aS)-гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она; дигидрохлоридная соль (450 мг, 1,96 ммоль, ВВ1) в сухом ДМФА (9 мл), охлажденную до 0°С в атмосфере инертного газа, добавляли ДИПЭА (787 мг, 1,06 мл, 6,09 ммоль) и 4-нитрофенилкарбонохлоридат (475 мг, 2,36 ммоль). Реакционную смесь перемешивали при 0°С в течение 30 мин. Добавляли пиперидин-4-илметанол (271 мг, 2,36 ммоль, С AS RN 6457-49-4) и ДИПЭА (381 мг, 515 мкл, 2,95 ммоль) и перемешивали реакционную смесь при 100°С в течение 14 ч. Летучие вещества удаляли в вакууме, а неочищенный остаток очищали с помощью флэш-хроматографии с 24 г колонкой с SiO2, используя смесь элюентов из ДХМ и МеОН (от 5% до 25%). Неочищенный продукт подвергали СЖХ-очистке с получением необходимого соединения в виде светло-желтого масла (338 мг). МС (ИЭР): m/z=298,3 [М+Н]+.
ВВ17
4,4-дифтор-1-(пиперидин-4-илметил)пиперидин; дигидрохлоридная соль
В раствор трет-бутил-4-((4,4-дифторпиперидин-1-ил)метил)пиперидин-1-карбоксилата (453 мг, 1,07 ммоль) в диоксане (2,5 мл) добавляли HCl (4,0 М раствор в диоксане) (2,67 мл, 10,7 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 14 ч. Летучие вещества удаляли в вакууме с получением необходимого соединения в виде белого твердого вещества (286 мг), которое использовали на следующем этапе без дополнительной очистки. МС (ИЭР): m/z=219,3 [М+Н]+.
Этап а) трет-бутил-4-((4,4-дифторпиперидин-1-ил)метил)пиперидин-1-карбоксилат
В раствор трет-бутил-4-(бромметил)пиперидин-1-карбоксилата (0,5 г, 1,8 ммоль, CAS RN: 158407-04-6) в сухом ДМФА (4 мл) добавляли 4,4-дифторпиперидин; дигидрохлоридную соль (425 мг, 2,7 ммоль) и Cs2CO3 (1,17 г, 3,59 ммоль). Реакционную смесь перемешивали при 80°C под микроволновым облучением в течение 60 мин. Нерастворимые вещества удаляли путем фильтрации, затем фильтрат концентрировали в вакууме, а полученный неочищенный остаток суспендировали в ДХМ и фильтровали через слой целита с получением неочищенного желтого масла, которое очищали с помощью флэш-хроматографии на колонке с SiO2, используя смесь элюентов из н-гептана и EtOAc (от 10% до 60%) с получением необходимого продукта в виде бесцветного масла (453 мг). Соединение переносили на следующий этап без дополнительной очистки. МС (ИЭР): m/z=319,3 [М+Н]+.
ВВ19
N-(азетидин-3-илметил)-2,2,2-трифтор-1-(4-(трифторметил)фенил)этан-1-амин; бис(трифторацетатная) соль
В раствор трет-бутил-3-(((2,2,2-трифтор-1-(4-(трифторметил)фенил)этил)амино)метил)азетидин-1-карбоксилата (1 г, 2,42 ммоль) в ДХМ (10 мл) добавляли ТФУ (5,53 г, 3,74 мл, 48,5 ммоль). Полученную в результате реакционную смесь перемешивали при КТ в течение 1 ч. Реакционную смесь концентрировали в вакууме с получением необходимого продукта в виде бесцветного масла (1,29 г). МС (ИЭР): m/z=313,5 [М+Н]+.
Этап а) трет-бутил-3-(((2,2,2-трифтор-1-(4-(трифторметил)фенил)этил)амино)метил)азетидин-1-карбоксилат
В сухую колбу с перегородкой в атмосфере азота добавляли трет-бутил-3-(аминометил)азетидин-1-карбоксилат (0,852 г, 4,57 ммоль), триэтиламин (1,39 г, 1,91 мл, 13,7 ммоль), 2,2,2-трифтор-1-(4-(трифторметил)фенил)этан-1-он (1,11 г, 780 мкл, 4,57 ммоль) и сухой ДХМ (28 мл). 1 М тетрахлорид титана в ДХМ (2,29 мл, 2,29 ммоль) с помощью шприца добавляли в ледяную колбу (экзотермически). Реакцию перемешивали в течение ночи при КТ, затем аккуратно гасили раствором NaCNBH3 (862 мг, 13,7 ммоль) в МеОН (8,79 г, 11,1 мл, 274 ммоль) и перемешивали в течение ночи. Реакцию подщелачивали насыщ. раствором NaHCO3. Полученный нерастворимый материал отфильтровывали через целит. Фильтрат экстрагировали ДХМ, органические слои объединяли, промывали солевым раствором, сушили над Na2SO4 и концентрировали. Неочищенный материал очищали с помощью флэш-хроматографии (силикагель, 50 г, от 0% до 50% EtOAc в н-гептане с получением трет-бутил-3-(((2,2,2-трифтор-1-(4-(трифторметил)фенил)этил)амино)метил)азетидин-1-карбоксилата, который использовали на следующем этапе без дополнительной очистки.
ВВ26
3-хлор-4-(4-пиперидилметокси)бензонитрил; гидрохлоридная соль
В раствор трет-бутил-4-[(2-хлор-4-цианофенокси)метил]пиперидин-1-карбоксилата (300 мг, 0,860 ммоль) в EtOAc (3 мл) добавляли HCl в EtOAc (4 М, 2,0 мл) при 0°C. Раствор перемешивали при 15°C в течение 3 ч. Раствор концентрировали в вакууме, затем сушили путем лиофилизации с получением необходимого продукта в виде белого твердого вещества (238 мг, 0,830 ммоль, выход 96%). МС (ИЭР): m/z=251 [М+Н]+.
Этап а) трет-бутил-4-(((метилсульфонил)окси)метил)пиперидин- 1-карбоксилат
В раствор N-Boc-4-пиперидинметанола (10,0 г, 46,5 ммоль, 1 экв.) в ДХМ (200 мл) добавляли NEt3 (7,04 г, 69,7 ммоль), затем добавляли метансульфонилхлорид (3,95 мл, 51,1 ммоль) при 0°C и перемешивали смесь при 0°C в течение 1 ч. Смесь вливали в ледяную воду, водную фазу дважды экстрагировали ДХМ (по 50 мл каждый раз). Объединенные органические слои промывали солевым раствором (50 мл), и концентрировали под вакуумом. Остаток использовали непосредственно без какой-либо очистки. МС (ИЭР): m/z=238,1 [М+Н]+.
Этап b) трет-бутил-4-[(2-хлор-4-цианофенокси)метил]пиперидин-1-карбоксилат
В раствор трет-бутил-4-(метилсульфонилоксиметил)пиперидин-1-карбоксилата (700 мг, 2,39 ммоль) в ДМФА (7 мл) добавляли Cs2CO3 (855 мг, 2,62 ммоль) и 3-хлор-4-гидроксибензонитрил (0,25 мл, 2,39 ммоль) при 15°C. Смесь нагревали до 85°C и перемешивали в течение 16 ч. Реакционный раствор разводили EtOAc (8 мл) при 15°C, три раза промывали солевым раствором (по 8 мл каждый раз), объединенные органические слои сушили над Na2SO4 и выпаривали. Бесцветный остаток (0,75 г) очищали с помощью препаративной ВЭЖХ и сушили путем лиофилизации с получением необходимого продукта в виде белого твердого вещества (531 мг, 1,51 ммоль, 53,4%). МС (ИЭР: m/z=295 [М-56+Н]+.
ВВ27
4-((4-(трифторметил)-1Н-имидазол-1-ил)метил)пиперидин; гидрохлоридная соль
В раствор трет-бутил-4-((4-(трифторметил)-1Н-имидазол-1-ил)метил)пиперидин-1-карбоксилата (430 мг, 1,29 ммоль) в диоксане (3 мл) добавляли HCl (4 М раствор в диоксане; 3,22 мл, 12,9 ммоль) и перемешивали реакционную смесь при КТ в течение 14 ч. Летучие вещества удаляли в вакууме с получением неочищенного продукта (362 мг), который использовали на следующем этапе без дополнительной очистки. МС (ИЭР): m/z=234,2 [М+Н]+.
Этап а) трет-бутил-4-((4-(трифторметил)-1Н-имидазол-1-ил)метил)пиперидин-1-карбоксилат
В раствор трет-бутил-4-(бромметил)пиперидин-1-карбоксилата (0,5 г, 1,8 ммоль, CAS RN: 158407-04-6) в сухом ДМФА (4 мл) добавляли 4-(трифторметил)-1Н-имидазол (293 мг, 2,16 ммоль) и Cs2CO3 (1,17 г, 3,59 ммоль). Затем реакционную смесь перемешивали при 80°C в течение 14 ч. Нерастворимые вещества удаляли путем фильтрации, а фильтрат концентрировали в вакууме. Неочищенный остаток суспендировали в ДХМ и фильтровали через слой целита с получением желтого масла, которое очищали с помощью флэш-хроматографии с колонкой SiO2, используя смесь элюентов из н-гептана и EtOAc (от 10% до 90%). Это позволило получить первую фракцию (301 мг) необходимого продукта в виде бесцветного масла и вторую фракцию (261 мг) смеси необходимого продукта с примесями. Вторую фракцию подвергали СЖХ-очистке, а очищенный продукт объединяли с первой фракцией с получением 430 мг необходимого продукта в виде бесцветного масла. МС (ИЭР): m/z=334,2 [М+Н]+.
ВВ29
3-((2-хлор-4-(трифторметил)фенокси)метил)азетидин
Трифторуксусную кислоту (2 г, 1,35 мл, 17,5 ммоль) добавляли в раствор трет-бутил-3-((2-хлор-4-(трифторметил)фенокси)метил)азетидин-1-карбоксилата (320 мг, 875 мкмоль) в ДХМ (4,37 мл) и перемешивали раствор при КТ в течение 2 ч. Растворитель удаляли при пониженном давлении, а полученное в результате светлое масло (470 мг) разводили EtOAc и промывали насыщ. раствором Na2CO3. Водную фазу экстрагировали три раза EtOAc, а объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении с получением соединения в виде желтого масла (259 мг, 877 мкмоль). МС (ИЭР): m/z=266,1 [М+Н]+.
Этап а) трет-бутил-3-((2-хлор-4-(трифторметил)фенокси)метил)азетидин-1-карбоксилат
В раствор 2-хлор-4-(трифторметил)фенола (525 мг, 357 мкл, 2,67 ммоль), трет-бутил-3-(гидроксиметил)азетидин-1-карбоксилата (500 мг, 2,67 ммоль, CAS RN: 142253-56-3) и трифенилфосфина (770 мг, 2,94 ммоль) в ДХМ (13,4 мл) по каплям добавляли ДИАД (594 мг, 571 мкл, 2,94 ммоль) и перемешивали реакцию при КТ в течение 17 ч. Реакционную смесь гасили путем добавления насыщ. водн. раствора NaHCO3 (20 мл). Фазы разделяли, а водную фазу дважды экстрагировали ДХМ. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали до сухости. Остаток растворяли в EtOH (7 мл) и добавляли гомогенный раствор хлорида цинка (218 мг, 1,6 ммоль) в EtOH (2 мл, 0,5 М). Смесь перемешивали в течение 30 мин, во время чего выпадал белый твердый осадок. Белое твердое вещество отфильтровывали и промывали EtOH. Фильтрат концентрировали с получением желтого масла с белым осадком. Неочищенный материал иммобилизовали на Isolute и очищали с помощью колоночной хроматографии (40 г, от 0 до 30% EtOAc в гептанах) с получением указанного в заголовке соединения в виде белого твердого вещества (764,6 мг, 1,99 ммоль, 74,4%). МС (ИЭР): m/z=310,1 [М-56+Н]+.
BB30
N-бензил-N-(2-гидроксиэтил)пиперидин-4-карбоксамида гидрохлорид
В раствор трет-бутил-4-(бензил(2-гидроксиэтил)карбамоил)пиперидин-1-карбоксилата (0,080 г, 221 мкмоль) в ДХМ (1 мл) добавляли 2 М HCl в диэтиловом эфире (1,1 мл, 2,21 ммоль). Полученную в результате реакционную смесь перемешивали при КТ в течение 1 ч, а затем концентрировали в вакууме при 22°C с получением необходимого соединения в виде бесцветного масла (63 мг) (BB30). МС (ИЭР): m/z=263,18 [М+Н]+.
Этап а) трет-бутил-4-(бензил(2-гидроксиэтил)карбамоил)пиперидин-1-карбоксилат
В 10 мл стеклянной пробирке к 1-(трет-бутоксикарбонил)пиперидин-4-карбоновой кислоте (0,1 г, 436 мкмоль) в ДМФА (2 мл) добавляли 2-(бензиламино)этан-1-ол (72,5 мг, 480 мкмоль), ДИПЭА (169 мг, 229 мкл, 1,31 ммоль) и HATU (182 мг, 480 мкмоль), перемешивали при КТ в течение 1 ч и экстрагировали H2O/ДХМ. Неочищенный материал очищали с помощью флэш-хроматографии (силикагель, 20 г, от 50% до 100% EtOAc в н-гептане) с получением соединения в виде светло-желтого масла (156 мг).
ВВ31
N-бензилпиперидин-4-карбоксамида гидрохлорид
трет-бутил-4-(бензилкарбамоил)пиперидин-1-карбоксилат (0,138 г, 433 мкмоль) растворяли в ДХМ (1 мл) и добавляли 2 М HCl в диэтиловом эфире (2,17 мл, 4,33 ммоль). Реакционную смесь перемешивали в течение 2 ч. Остаток концентрировали в вакууме с получением соединения (108 мг) в виде бесцветного масла. МС (ИЭР): m/z=219,15 [М+Н]+.
Этап а) трет-бутил-4-(бензилкарбамоил)пиперидин-1-карбоксилат
В 10 мл стеклянной пробирке к 1-(трет-бутоксикарбонил)пиперидин-4-карбоновой кислоте (0,1 г, 436 мкмоль) в ДМФА (2 мл) добавляли фенилметанамин (51,4 мг, 52,4 мкл, 480 мкмоль), ДИПЭА (169 мг, 229 мкл, 1,31 ммоль) и HATU (182 мг, 480 мкмоль), перемешивали при КТ в течение 2 ч и экстрагировали Н2О/ДХМ. Неочищенный материал очищали с помощью флэш-хроматографии (силикагель, 20 г, от 50% до 100% EtOAc в н-гептане) с получением соединения в виде бесцветного масла (0,138 г).
ВВ32
4-((4-(трет-бутил)-1Н-пиразол-1-ил)метил)пиперидин; гидрохлоридная соль
В раствор трет-бутил-4-((4-(трет-бутил)-1Н-пиразол-1-ил)метил)пиперидин-1-карбоксилата (100 мг, 311 мкмоль) в диоксане (1 мл) добавляли HCl (4,0 М раствор в диоксане; 1,17 мл, 4,67 ммоль) и перемешивали реакционную смесь при КТ в течение 14 ч. Летучие вещества удаляли в вакууме с получением 84 мг неочищенного продукта, который использовали на следующем этапе без дополнительной очистки. МС (ИЭР): m/z=222,3 [М+Н]+.
Этап а) трет-бутил-4-((4-(трет-бутил)-1Н-пиразол-1-ил)метил)пиперидин-1-карбоксилат
В раствор трет-бутил-4-(бромметил)пиперидин-1-карбоксилат (0,5 г, 1,8 ммоль, CAS RN 158407-04-6) в сухом ДМФА (4 мл) добавляли 4-(трет-бутил)-1Н-пиразол (268 мг, 2,16 ммоль) и NaH (86,3 мг, 2,16 ммоль). Реакционную смесь перемешивали при 80°C в течение 14 ч. Реакцию гасили путем добавления нескольких капель насыщ. водн. раствора NH4Cl и переносили в разделительную воронку для разделения между ДХМ и насыщ. водн. раствором NaHCO3. Органическую фазу собирали, а водную фазу обратно экстрагировали ДХМ. Объединенные органические фазы сушили над Na2SO4 и выпаривали до сухости. Неочищенный материал очищали с помощью флэш-хроматографии на колонке с SiO2 с элюированием смесью н-гептана и EtOAc (от 5% до 60%) с получением необходимого соединения в виде бесцветного масла (102 мг). МС (ИЭР): m/z=322,3 [М+Н]+.
BB33
(2R,4aR,8aS)-2-метил-4а,5,6,7,8,8а-гексагидро-4Н-пиридо[4,3-b][1,4]оксазин-3-он
В раствор 6-бензил-2-метил-5,6,7,8-тетрагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она (изомер А, 1,10 г, 4,26 ммоль) в EtOAc (16 мл) и МеОН (16 мл) в атмосфере аргона добавляли Pd-C (227 мг, 213 мкмоль) и перемешивали суспензию в атмосфере водорода (баллон) при 1 бар в течение 24 ч. Суспензию фильтровали через тонкий стеклянный фильтр и промывали 20 мл EtOAc в атмосфере инертного газа. Фильтрат выпаривали с получением BB33 в виде бесцветного твердого вещества (715 мг). МС (ИЭР): m/z=170,8 [М+Н]+. Примечание: Во время восстановления образовался только один энантиомер.
Этап а) 2-метил-4Н-пиридо[4,3-b][1,4]оксазин-3-он
В раствор 3-аминопиридин-4-ола (2,5 г, 22,7 ммоль) в ДМФА (100 мл) по каплям добавляли 2-хлорпропаноилхлорид (3,03 г, 2,31 мл, 23,8 ммоль) и перемешивали смесь при КТ в течение 30 мин. После добавления K2CO3 (7,84 г, 56,8 ммоль) суспензию нагревали до 100°C (масляная баня) в течение 20 ч. ДМФА удаляли в вакууме, затем добавляли 100 мл EtOAc и перемешивали при КТ в течение 10 мин, промывали 50 мл H2O, 3 раза экстрагировали EtOAc. Органические фазы комбинировали, сушили с MgSO4 и концентрировали в вакууме с получением 3,72 г 2-метил-4Н-пиридо[4,3-b][1,4]оксазин-3-она, который использовали на следующем этапе без дополнительной очистки.
Этап b) 6-бензил-2-метил-3-оксо-3,4-дигидро-2Н-пиридо[4,3-b][1,4]оксазин-6-ий бромид
Суспензию 2-метил-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она (3,72 г, 22,7 ммоль) в ДХМ (32 мл) и МеОН (8 мл) обрабатывали (бромметил)бензолом (4,65 г, 3,23 мл, 27,2 ммоль) и перемешивали смесь при КТ в течение 60 ч. К образованной суспензии, которую охладили до 0°C, добавляли 20 мл н-гексана, после чего фильтровали осадок. Остаток промывали 15 мл холодного ДХМ/н-гексана с получением соединения в виде грязно-белого твердого вещества (5,2 г). МС (ИЭР): m/z=255 [М+Н]+.
Этап с) 6-бензил-2-метил-5,6,7,8-тетрагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он
В суспензию 6-бензил-2-метил-3-оксо-3,4-дигидро-2Н-пиридо[4,3-b][1,4]оксазин-6-ий бромида (5,2 г, 15,5 ммоль) в EtOH (38 мл) частями добавляли NaBH4 (763 мг, 20,2 ммоль) (экзотермически, от 22°C до 30°C, желтая суспензия). После окончания экзотермической реакции смесь перемешивали при комнатной температуре в течение 3 ч, затем при 60°C в течение 1 ч и при 22°C в течение 1 ч. Реакционную смесь выпаривали, разделяли между Н2О и EtOAc и разделяли слои. Водный слой один раз экстрагировали EtOAc. Органические слои дважды промывали Н2О, сушили над MgSO4, фильтровали, обрабатывали силикагелем и выпаривали. Соединение очищали с помощью хроматографии на силикагеле на 120 г колонке, используя систему для ЖХСД, с элюированием градиентом н-гептан: EtOAc (от 50 до 100 за 30 мин) с получением соединения в виде светло-желтого твердого вещества (2,48 г), которое можно было использовать на следующем этапе без дополнительной очистки.
Этап d) 6-бензил-2-метил-5,6,7,8-тетрагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он
Энантиомеры разделяли с помощью препаративной хиральной ВЭЖХ (колонка Chiralcel OD), используя изократическую смесь ETOH (содержащего 0,05% NH4OAc):н-гептан (10:90). Фракции выпаривали с получением необходимых соединений в виде светло-желтых твердых веществ (изомер А, 1,17 г; изомер В, 1,10 г).
ВВ34
N-(азетидин-3-илметил)-2,2,2-трифтор-1-(3-(трифторметил)фенил)этан-1-амин
В 100 мл пробирке с двумя горлышками растворяли бензил-3-(((2,2,2-трифтор-1-(3-(трифторметил)фенил)этил)амино)метил)азетидин-1-карбоксилат (0,913 г, 2,05 ммоль) в смеси ТГФ (5 мл) и МеОН (5 мл). Pd/C 10% (109 мг, 102 мкмоль) добавляли в атмосфере аргона. Колбу продували и обратно заполняли газообразным Н2 (3 раза). Затем реакционную смесь перемешивали при 25°C в течение 4 ч. Суспензию фильтровали через дикалит, концентрировали, а полученное в результате указанное в заголовке соединение (611 мг, бесцветное масло) использовали непосредственно н следующем этапе. МС (ИЭР): m/z=313,4 [М+Н]+.
Этап а) бензил-3-(((2,2,2-трифтор-1-(3-(трифторметил)фенил)этил)амино)метил)азетидин-1-карбоксилат
В сухую колбу с перегородкой добавляли бензил-3-(аминометил)азетидин-1-карбоксилат (0,5 г, 2,27 ммоль), NEt3 (689 мг, 949 мл, 6,81 ммоль), 2,2,2-трифтор-1-(3-(трифторметил)фенил)этан-1-он (554 мг, 391 мкл, 2,27 ммоль) и сухой ДХМ (15 мл). 1 М тетрахлорид титана в ДХМ (1,13 мл, 1,13 ммоль) добавляли с помощью шприца, а колбу охлаждали на ледяной бане (экзотермически). Реакцию перемешивали в течение ночи при КТ, аккуратно гасили раствором NaCNBH3 (428 мг, 6,81 ммоль) в МеОН (4,36 г, 5,51 мл, 136 ммоль) и уксусной кислоты (,01 мл) и перемешивали при КТ в течение ночи. Реакцию подщелачивали насыщ. водн. раствором NaHCO3, а полученный нерастворимый материал отфильтровывали через целит. Фильтрат экстрагировали ДХМ. Органические слои объединяли, промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Неочищенный материал очищали с помощью флэш-хроматографии (силикагель, 50 г, от 0% до 50% EtOAc в н-гептане) с получением необходимого соединения в виде бесцветного масла (913 мг). МС (ИЭР): m/z=447,2 [М+Н]+.
ВВ35
N-(азетидин-3-илметил)-1-(2,4-дихлорфенил)-2,2,2-трифторэтан-1-амин В 100 мл пробирке с двумя горлышками растворяли бензил-3-(((1-(2,4-дихлорфенил)-2,2,2-трифторэтил)амино)метил)азетидин-1-карбоксилат (0,660 г, 1,48 ммоль) в EtOAc (20 мл) с получением бесцветного раствора. Pd/C 10% (78,5 мг, 73,8 мкмоль) добавляли в атмосфере аргона. Колбу продували и обратно заполняли газообразным Н2 (3 раза). Реакционную смесь перемешивали при 25°C в течение 4 ч. ЖХ-МС показала наличие смеси указанного в заголовке продукта N-(азетидин-3-илметил)-1-(2,4-дихлорфенил)-2,2,2-трифторэтан-1-амина вместе с дегалогенизированными побочными продуктами N-(азетидин-3-илметил)-1-(2-хлорфенил)-2,2,2-трифторэтан-1-амином и N-(азетидин-3-илметил)-1-фенил-2,2,2-трифторэтан-1-амином. Реакционную смесь фильтровали через дикалит, концентрировали в вакууме и использовали непосредственно н следующем этапе.
Этап а) бензил-3-[[[1-(2,4-дихлорфенил)-2,2,2-трифторэтилиден]амино]метил]азетидин-1-карбоксилат
В сухую колбу с перегородкой в атмосфере азота добавляли бензил-3-(аминометил)азетидин-1-карбоксилат (0,500 г, 2,27 ммоль, CAS RN 1016731-24-0), NEt3 (689 мг, 949 мкл, 6,81 ммоль), 1-(2,4-дихлорфенил)-2,2,2-трифторэтан-1-он (556 мг, 2,27 ммоль) и сухой ДХМ (16,4 мл). Тетрахлорид титана (1 М раствор в ДХМ; 1,13 мл, 1,13 ммоль) с помощью шприца добавляли в ледяную колбу (экзотермически). Реакцию перемешивали в течение ночи при КТ, аккуратно гасили раствором NaCNBH3 (428 мг, 6,81 ммоль) в МеОН (4,36 г, 5,51 мл, 136 ммоль) и перемешивали в течение 6 ч. ЖХМС показала, что реакция завершилась на имине.
Реакцию подщелачивали насыщ. NaHCO3. Полученный нерастворимый материал фильтровали через целит, а фильтрат экстрагировали ДХМ. Органические слои объединяли, промывали солевым раствором, сушили над Na2SO4 и концентрировали. Неочищенный материал очищали с помощью флэш-хроматографии (силикагель, 50 г, от 0% до 50% EtOAc в н-гептане) с получением необходимого соединения в виде бесцветного масла (1 г).
Этап b) бензил-3-(((1-(2,4-дихлорфенил)-2,2,2-трифторэтил)амино)метил)азетидин-1-карбоксилат
В 25 мл пробирке с двумя горлышками растворяли бензил-3-[[[1-(2,4-дихлорфенил)-2,2,2-трифторэтилиден]амино]метил]азетидин-1-карбоксилат (1 г, 2,25 ммоль) в ТГФ (10 мл) и МеОН (1 мл) с получением бесцветного раствора. Добавляли уксусную кислоту (135 мг, 129 мкл, 2,25 ммоль) и NaCNBH3 (423 мг, 6,74 ммоль). Реакционную смесь перемешивали при 25°C в течение 6 ч. Реакцию подщелачивали насыщ. NaHCO3. Полученный нерастворимый материал фильтровали через целит, а фильтрат экстрагировали ДХМ. Органические слои объединяли, промывали солевым раствором, сушили над Na2SO4 и концентрировали. Неочищенный материал очищали с помощью флэш-хроматографии (силикагель, 50 г, от 0% до 50% EtOAc в гептане) с получением указанного в заголовке соединения в виде бесцветного масла (660 мг), которое использовали на следующем этапе без дополнительной очистки.
ВВ36
цис-4-((2-хлор-4-фторфенокси)метил)-3-метилпиперидин; гидрохлоридная соль
трет-бутил цис-4-((2-хлор-4-фторфенокси)метил)-3-метилпиперидин-1-карбоксилат (115 мг, 321 мкмоль) растворяли в ДХМ (2 мл) и добавляли 2 М HCl в эфире (161 мкл, 321 мкмоль). Реакцию перемешивали при КТ в течение 6 ч, затем в вакууме удаляли растворитель. Неочищенный продукт (94 мг, бесцветная пена) использовали на следующем этапе без очистки. МС (ИЭР): m/z=258,2 [М+Н]+.
Этап а) трет-бутил цис-4-((2-хлор-4-фторфенокси)метил)-3-метилпиперидин-1-карбоксилат
Реакция Мицунобу: В 50 мл колбу для сульфирования с четырьмя горлышками в атмосфере аргона, в которой был растворен трет-бутил цис-4-(гидроксиметил)-3-метилпиперидин-1-карбоксилат (840 мг, 3,66 ммоль) в ТГФ (15 мл), добавляли 2-хлор-4-фторфенол (590 мг, 439 мкл, 4,03 ммоль) и трифенилфосфин (1,06 г, 4,03 ммоль). Прозрачный раствор перемешивали 5 мин при КТ, затем охлаждали до 0-2°C и добавляли ДЭАД (702 мг, 638 мкл, 4,03 ммоль) в течение 10 мин. Реакционную смесь перемешивали при 2-4°C в течение 1 ч, затем перемешивали в течение ночи при КТ. Добавляли 50 мл диэтилэфира и промывали смесь 2×25 мл воды, 3×20 мл 1 Н NaOH, 1×20 мл солевого раствора, органическую фазу сушили с Mg2SO4, после удаления растворителя в вакууме получали 2,7 г желтого масла. Для удаления трифенилфосфиноксида остаток перемешивали в н-гептане/диэтилэфире в течение 30 мин, твердые вещества отфильтровывали, фильтрат концентрировали в вакууме с получением 1,8 г неочищенного продукта, который очищали с помощью флэш-хроматографии (силикагель, 50 г, от 0% до 30% EtOAc в гептане, 40 мин): трет-бутил цис-4-((2-хлор-4-фторфенокси)метил)-3-метилпиперидин-1-карбоксилат, 1,21 г, белое твердое вещество.
ВВ39
3-((2-фтор-4-(трифторметил)бензил)окси)азетидин; трифторацетатная соль
В раствор трет-бутил-3-((2-фтор-4-(трифторметокси)бензил)окси)азетидин-1-карбоксилата (415 мг, 1,14 ммоль) в ДХМ (5 мл) добавляли ТФУ (1,3 г; 875 мкл, 11,4 ммоль) и перемешивали реакционную смесь при КТ в течение 3 ч. Летучие вещества удаляли в вакууме с получением 455 мг светло-желтого масла, которое использовали на следующем этапе без дополнительной очистки. МС (ИЭР): m/z=266,1 [М+Н]+.
Этап а) трет-бутил-3-((2-фтор-4-(трифторметокси)бензил)окси)азетидин-1-карбоксилат
В раствор трет-бутил-3-гидроксиазетидин-1-карбоксилата (200 мг, 1,15 ммоль) в сухом ТГФ (5 мл) добавляли трет-бутоксид калия (1,65 М раствор в ТГФ, 735 мкл, 1,21 ммоль) и перемешивали реакционную смесь при КТ в течение 15 мин с последующим добавлением 1-(бромметил)-2-фтор-4-(трифторметокси)бензола (315 мг, 1,15 ммоль). Затем реакционную смесь перемешивали при комнатной температуре в течение 14 ч. Неочищенную реакцию разводили EtOAc и экстрагировали водн. 1 М раствором NaHCO3, органическую фазу собирали, а водный слой обратно экстрагировали EtOAc. Объединенные органические фазы сушили над NaSO4 и выпаривали до сухости с получением 418 мг неочищенного продукта, который использовали на следующем этапе без дополнительной очистки. МС (ИЭР: m/z=310,1 [М-56+Н]+.
ВВ40
N-(азетидин-3-ил)-2-хлор-4-фторбензамид; трифторацетатная соль
В раствор трет-бутил-3-[(2-хлор-4-фторбензоил)амино]азетидин-1-карбоксилата (346 мг, 1,05 ммоль) в ДХМ (3,5 мл) добавляли ТФУ (0,7 мл) при 0°C. Раствор перемешивали при 0°C в течение 2 ч. Раствор концентрировали в вакууме с получением неочищенного продукта (600 мг) в виде светло-желтого масла. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (0,1% ТФУ в H2O и MeCN) и сушили путем лиофилизации с получением необходимого соединения в виде бесцветного твердого вещества (223 мг, 0,650 ммоль, выход 59,2%). МС (ИЭР): m/z=229 [М+Н]+.
Этап а) трет-бутил-3-[(2-хлор-4-фторбензоил)амино]азетидин-1-карбоксилат
В раствор 2-хлор-4-фторбензойной кислоты (500 мг, 2,86 ммоль), 1-Вос-3-(амино)азетидина (493 мг, 2,86 ммоль) и ДМАП (35,0 мг, 0,290 ммоль) в ТГФ (10 мл) добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид (714 мг, 3,72 ммоль) при 0°C. Смесь нагревали до 30°C и перемешивали в течение 16 ч. Реакцию разводили EtOAc (5 мл), три раза промывали солевым раствором (по 10 мл каждый раз) и сушили над Na2SO4. Органический слой концентрировали в вакууме с получением неочищенного продукта (0,72 г) в виде желтого масла. Неочищенный продукт очищали с помощью препаративной ВЭЖХ и сушили путем лиофилизации с получением необходимого соединения в виде бесцветного твердого вещества (546 мг, 1,66 ммоль, выход 57,9%). МС (ИЭР: m/z=273 [М-56+Н]+.
ВВ41
N-(азетидин-3-илметил)-2,2,2-трифтор-N-метил-1-[4-(трифторметил)фенил)этанамин; трифторацетатная соль
В раствор трет-бутил-3-((метил(2,2,2-трифтор-1-(4-(трифторметил)фенил)этил)амино)метил)азетидин-1-карбоксилата (0,256 г, 600 мкмоль) в ДХМ (5 мл) добавляли ТФУ (1,37 г, 925 мкл, 12 ммоль). Полученную в результате реакционную смесь перемешивали при КТ в течение 1 ч. Реакционную смесь концентрировали в вакууме с получением необходимого продукта в виде бесцветного масла (268 мг). МС (ИЭР): m/z=327,4 [М+Н]+.
Этап а) трет-бутил-3-((метил(2,2,2-трифтор-1-(4-(трифторметил)фенил)этил)амино)метил)азетидин-1-карбоксилат
В сухую колбу с перегородкой и  молекулярными ситами в атмосфере азота добавляли трет-бутил-3-((метиламино)метил)азетидин-1-карбоксилат (0,300 г, 293 мкл, 1,5 ммоль), ТЭА (455 мг, 626 мкл, 4,49 ммоль), 2,2,2-трифтор-1-(4-(трифторметил)фенил)этан-1-он (363 мг, 255 мкл, 1,5 ммоль) и сухой ДХМ (9,86 мл). 1 М тетрахлорид титана в ДХМ (749 мл, 749 мкмоль) с помощью шприца добавляли в ледяную колбу (экзотермически). Реакцию перемешивали в течение ночи при КТ, аккуратно гасили раствором NaCNBH3 (282 мг, 4,49 ммоль) в МеОН (3,64 мл, 89,9 ммоль) и перемешивали при КТ в течение 2 ч. Реакцию подщелачивали насыщ. раствором NaHCO3. Полученный нерастворимый материал фильтровали через целит, а фильтрат экстрагировали ДХМ. Органические слои объединяли, промывали солевым раствором, сушили над Na2SO4 и концентрировали. Неочищенный материал очищали с помощью флэш-хроматографии (силикагель, 50 г, от 0% до 50% EtOAc в н-гептане) и использовали непосредственно на следующем этапе.
молекулярными ситами в атмосфере азота добавляли трет-бутил-3-((метиламино)метил)азетидин-1-карбоксилат (0,300 г, 293 мкл, 1,5 ммоль), ТЭА (455 мг, 626 мкл, 4,49 ммоль), 2,2,2-трифтор-1-(4-(трифторметил)фенил)этан-1-он (363 мг, 255 мкл, 1,5 ммоль) и сухой ДХМ (9,86 мл). 1 М тетрахлорид титана в ДХМ (749 мл, 749 мкмоль) с помощью шприца добавляли в ледяную колбу (экзотермически). Реакцию перемешивали в течение ночи при КТ, аккуратно гасили раствором NaCNBH3 (282 мг, 4,49 ммоль) в МеОН (3,64 мл, 89,9 ммоль) и перемешивали при КТ в течение 2 ч. Реакцию подщелачивали насыщ. раствором NaHCO3. Полученный нерастворимый материал фильтровали через целит, а фильтрат экстрагировали ДХМ. Органические слои объединяли, промывали солевым раствором, сушили над Na2SO4 и концентрировали. Неочищенный материал очищали с помощью флэш-хроматографии (силикагель, 50 г, от 0% до 50% EtOAc в н-гептане) и использовали непосредственно на следующем этапе.
ВВ42
метил-N-(пиперидин-4-ил)-1-(3-(трифторметил)фенил)циклопропан-1-карбоксамид гидрохлорид
В раствор трет-бутил-4-(N-метил-1-(3-(трифторметил)фенил)циклопропан-1-карбоксамидо)пиперидин-1-карбоксилата (0,301 г, 706 мкмоль) в ДХМ (2 мл) добавляли 2 М HCl в диэтиловом эфире (3,53 мл, 7,06 ммоль). Полученную в результате реакционную смесь перемешивали при КТ в течение ночи, а затем концентрировали в вакууме при 22°C с получение 256 мг ВВ42 в виде грязно-белого твердого вещества, МС (ИЭР): m/z=327,2 [М+Н]+
Этап а) трет-бутил-4-(N-метил-1-(3-(трифторметил)фенил)циклопропан-1-карбоксамидо)пиперидин-1-карбоксилат
В 20 мл стеклянной пробирке к 1-(3-(трифторметил)фенил)циклопропан-1-карбоновой кислоте (177 мг, 770 мкмоль) в ДМФА (5 мл) добавляли HATU (293 мг, 770 мкмоль) и ДИПЭА (271 мг, 367 мкл, 2,1 ммоль). Реакционную смесь перемешивали в течение 15 мин, а затем добавляли трет-бутил-4-(метиламино)пиперидин-1-карбоксилат (0,15 г, 700 мкмоль). Реакционную смесь перемешивали при КТ в течение 2 часов. Реакционную смесь экстрагировали водой/ДХМ. Неочищенный материал очищали с помощью флэш-хроматографии (силикагель, 20 г, от 0% до 100% EtOAc в гептане) с получением необходимого соединения в виде светло-желтого масла (301 мг). МС (ИЭР): m/z=371,2 [М-56+Н]+
ВВ43
2-(2-хлор-3-(трифторметил)фенил)-N-метил-N-(пиперидин-4-ил)ацетамид; гидрохлоридная соль
В раствор трет-бутил-4-(2-(2-хлор-3-(трифторметил)фенил)-N-метилацетамидо)пиперидин-1-карбоксилата (0,301 г, 692 мкмоль) в ДХМ (2 мл) добавляли HCl (3,46 мл, 6,92 ммоль). Полученную в результате реакционную смесь перемешивали при КТ в течение 2 суток, а затем концентрировали в вакууме при 22°C с получением 252 мг ВВ43 в виде грязно-белого твердого вещества. МС (ИЭР): m/z=335,1 [М+Н]+.
Этап а) трет-бутил-4-(2-(2-хлор-3-(трифторметил)фенил)-N-метилацетамидо)пиперидин-1-карбоксилат
В 20 мл стеклянной пробирке к 2-(2-хлор-3-(трифторметил)фенил)уксусной кислоте (184 мг, 770 мкмоль) в ДМФА (5 мл) добавляли HATU (293 мг, 770 мкмоль), ДИПЭА (271 мг, 367 мкл, 2,1 ммоль). Реакционную смесь перемешивали в течение 15 мин, а затем добавляли трет-бутил-4-(метиламино)пиперидин-1-карбоксилат (0,150 г, 700 мкмоль). Реакционную смесь перемешивали при КТ в течение 2 часов, а затем экстрагировали водой/ДХМ. Неочищенный материал очищали с помощью флэш-хроматографии (силикагель, 20 г, от 0% до 100% EtOAc в гептане) с получением трет-бутил-4-(2-(2-хлор-3-(трифторметил)фенил)-N-метилацетамидо)пиперидин-1-карбоксилата в виде светло-желтого масла, 301 мг, МС (ИЭР): m/z=379,1 [М-56+Н]+
ВВ44
2-(2-хлор-5-(трифторметил)фенил)-N-метил-N-(пиперидин-4-ил)ацетамид; гидрохлоридная соль
Синтезирован из 2-(2-хлор-5-(трифторметил)фенил)уксусной кислоты и трет-бутил-4-(метиламино)пиперидин-1-карбоксилата. В отношении деталей смотрите синтез ВВ43. МС (ИЭР): m/z=335,1 [М+Н]+.
ВВ46
3-метил-5-(пиперидин-4-илметокси)-2-(трифторметил)пиридин; дигидрохлоридная соль
В 25 мл пробирке трет-бутил-4-(((5-метил-6-(трифторметил)пиридин-3-ил)окси)метил)пиперидин-1-карбоксилат (87 мг, 232 мкмоль) растворяли в ДХМ (2 мл) и затем добавляли 2 М HCl в эфире (697 мкл, 1,39 ммоль), реакционную смесь перемешивали в течение 12 ч при КТ. Смесь концентрировали в вакууме с получением 80 мг ВВ46 в виде белого твердого вещества. МС (ИЭР): m/z=275,2 [М+Н]+.
Этап а) трет-бутил-4-(((5-метил-6-(трифторметил)пиридин-3-ил)окси)метил)пиперидин-1-карбоксилат
В 5 мл пробирке трет-бутил-4-(гидроксиметил)пиперидин-1-карбоксилат (80,7 мг, 375 мкмоль) растворяли в ДМФА (1,5 мл), затем добавляли NaH в масле 60% (18 мг, 450 мкмоль) при комнатной температуре, смесь перемешивали в течение 20 мин, затем добавляли 5-бром-3-метил-2-(трифторметил)пиридин (90 мг, 60 мкл, 375 мкмоль) и перемешивали в течение 2 ч с получением коричневого раствора. Добавляли 10 мл насыщ. NH4Cl, экстрагировали водой/этилацетатом, сушили над MgSO4, растворитель удаляли при 40°C/150 мбар. Неочищенный продукт очищали с помощью флэш-хроматографии (силикагель, 20 г, от 0% до 40% EtOAc в н-гептане за 35 мин) с получением 87 мг трет-бутил-4-(((5-метил-6-(трифторметил)пиридин-3-ил)окси)метил)пиперидин-1-карбоксилата. МС (ИЭР): m/z=319,2 [М-56+Н]+
ВВ58
N-(азетидин-3-илметил)-2,2,2-трифтор-1-(4-фторфенил)этан-1-амин
В 100 мл пробирке с двумя горлышками бензил-3-(((1-(2-хлор-4-фторфенил)-2,2,2-трифторэтил)амино)метил)азетидин-1-карбоксилат (707 мг, 1,64 ммоль) объединяли с ТГФ (5 мл) и МеОН (5 мл) с получением бесцветного раствора. Pd/C 10% (87,3 мг, 82,1 мкмоль) добавляли в атмосфере аргона. Колбу продували и обратно заполняли Н2 (3 раза). Реакционную смесь перемешивали при 25°C в течение 1 ч. Реакционную смесь фильтровали через дикалит, концентрировали и использовали непосредственно н следующем этапе. Бесцветное масло (472 мг). МС (ИЭР): m/z=263,2 [М+Н]+ (орто-хлор был потерян во время гидрогенизации).
Этап а:) бензил-3-(((1-(2-хлор-4-фторфенил)-2,2,2-трифторэтил)амино)метил)азетидин-1-карбоксилат
В сухую колбу в потоке азота добавляли бензил-3-(аминометил)азетидин-1-карбоксилат (0,5 г, 2,27 ммоль), триэтиламин (689 мг, 949 мкл, 6,81 ммоль), 1-(2-хлор-4-фторфенил)-2,2,2-трифторэтанон (519 мг, 2,27 ммоль) и сухой ДХМ (15 мл). 1 М тетрахлорид титана в ДХМ (1,13 мл, 1,13 ммоль) с помощью шприца добавляли в ледяную колбу (экзотермически). Реакцию перемешивали в течение ночи при КТ, аккуратно гасили метанольным раствором цианоборгидрида натрия (428 мг, 6,81 ммоль) в метаноле (4,36 г, 5,51 мл, 136 ммоль) + уксусная кислота (0,1 мл) и перемешивали при КТ в течение ночи. Реакцию подщелачивали насыщ. NaHCO3. Полученный нерастворимый материал отфильтровывали через целит, фильтрат экстрагировали ДХМ, органические слои объединяли, промывали солевым раствором, сушили над Na2SO4 и концентрировали. Очистка: Неочищенный продукт очищали с помощью флэш-хроматографии (силикагель, 50 г, от 0% до 50% EtOAc в гептане) с получением 707 мг бензил-3-(((1-(2-хлор-4-фторфенил)-2,2,2-трифторэтил)амино)метил)азетидин-1-карбоксилата в виде бесцветного масла. МС (ИЭР): m/z=431,2 [М+Н]+.
ВВ59
2,2,2-трифтор-1-(пиперидин-4-ил)-N-(3-(трифторметил)бензил)этан-1-амин; гидрохлоридная соль
В раствор трет-бутил-4-(2,2,2-трифтор-1-((3-(трифторметил)бензил)амино)этил)пиперидин-1-карбоксилата (0,140 г, 318 мкмоль) в ДХМ (2 мл) добавляли 2 М HCl в диэтиловом эфире (1,59 мл, 3,18 ммоль). Полученную в результате реакционную смесь перемешивали при КТ в течение ночи, а затем концентрировали в вакууме при 22°C с получением 119 мг указанного в заголовке соединения в виде грязно-белого твердого вещества. МС (ИЭР): m/z=340,8 [М+Н]+.
Этап а) трет-бутил-4-(2,2,2-трифтор-1-((3-(трифторметил)бензил)амино)этил)пиперидин-1-карбоксилат
Раствор трет-бутил-4-(1-амино-2,2,2-трифторэтил)пиперидин-1-карбоксилата (0,150 г, 531 мкмоль) и 3-(трифторметил)бензальдегида (92,5 мг, 71,1 мкл, 531 мкмоль) в 1,2-ДХЭ (1 мл) перемешивали в течение 1 часа при КТ. Затем добавляли триацетоксиборгидрид натрия (225 мг, 1,06 ммоль) при 0°C, и перемешивали реакционную смесь при КТ в течение ночи. Реакционную смесь вливали в насыщ. NaHCO3 и экстрагировали ДХМ. Органические слои объединяли, промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Неочищенный материал очищали с помощью флэш-хроматографии (силикагель, 20 г, от 0% до 100% EtOAc в гептане) с получением 145 мг необходимого соединения в виде бесцветного масла. МС (ИЭР): m/z=383,1 [М-56+Н]+
ВВ69
2-метил-3-((4-(трифторметил)бензил)окси)азетидин; трифторацетатная соль
В раствор трет-бутил-2-метил-3-((4-(трифторметил)бензил)окси)азетидин-1-карбоксилата (0,36 г, 1,04 ммоль) в ДХМ (4 мл) добавляли трифторуксусную кислоту (1,19 г, 10,4 ммоль). Полученную в результате реакционную смесь перемешивали при КТ в течение 1 часа. Реакционную смесь концентрировали в условиях высокого вакуума с получением ВВ69 в виде светло-желтого масла, 399 мг, смесь всех четырех стереоизомеров. МС (ИЭР): m/z=246,1 [М+Н]+.
Этап а) трет-бутил-2-метил-3-((4-(трифторметил)бензил)окси)азетидин-1-карбоксилат
В 25 мл пробирке с двумя горлышками трет-бутил-3-гидрокси-2-метилазетидин-1-карбоксилат (215 мг, 1,15 ммоль) растворяли в ДМФА (5 мл) с получением бесцветного раствора. При 0°C добавляли гидрид натрия (60% дисперсия в минеральном масле) (41,8 мг, 1,05 ммоль). Реакционную смесь перемешивали при 0°C в течение 15 мин. Затем добавляли 1-(бромметил)-4-(трифторметил)бензол (0,250 г, 1,05 ммоль) при 0°C. Реакционную смесь перемешивали при КТ в течение ночи. Реакционную смесь вливали в 20 мл насыщ. NH4Cl и экстрагировали EtOAc (2×50 мл). Органические слои объединяли, промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Неочищенный продукт очищали с помощью флэш-хроматографии (силикагель, 20 г, от 0% до 70% EtOAc в гептане) с получением 360 мг трет-бутил-2-метил-3-((4-(трифторметил)бензил)окси)азетидин-1-карбоксилата в виде бесцветного масла. МС (ИЭР): m/z=290,1 [М-56+Н]+
ВВ87
3-фтор-5-(трифторметил)бензил 4-метилбензолсульфонат
В раствор (3-фтор-5-(трифторметил)фенил)метанола (100 мг, 72,5 мкл, 515 мкмоль, экв: 1) в ДХМ (2,58 мл) добавляли п-толуолсульфоновый ангидрид (185 мг, 567 мкмоль), ДИПЭА (79,9 мг, 108 мкл, 618 мкмоль) и ДМАП (6,29 мг, 51,5 мкмоль). Реакционную смесь перемешивали в течение 4 ч при 0°C и в течение 2 суток при комнатной температуре. Реакционную смесь вливали в EtOAc и промывали водой и солевым раствором. Органические слои сушили над MgSO4 и концентрировали в вакууме с получением желтого масла (178 мг), которое использовали без дополнительной очистки.
По аналогии с ВВ39 и если не указано иное, промежуточные соединения, приведенные в следующей таблице, получали из коммерчески доступных бензилбромидов или приготовленных тозилатных промежуточных соединений и соответствующих трет-бутил-3-гидроксиазетидин-1-карбоксилатных строительных блоков.



По аналогии с ВВ29 промежуточные соединения ВВ20, ВВ25 и ВВ61 из следующей таблицы получали из коммерчески доступных фенолов. Если указаны трифторацетатные соли, неочищенный продукт, получаемый в результате концентрирования реакционной смеси, использовали непосредственно без дополнительной нейтрализации или очистки.


По аналогии с ВВ26 промежуточные соединения ВВ21-ВВ24 и ВВ28 из следующей таблицы получали из коммерчески доступных фенолов.
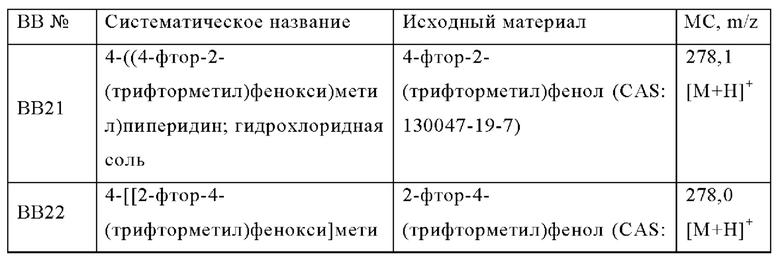
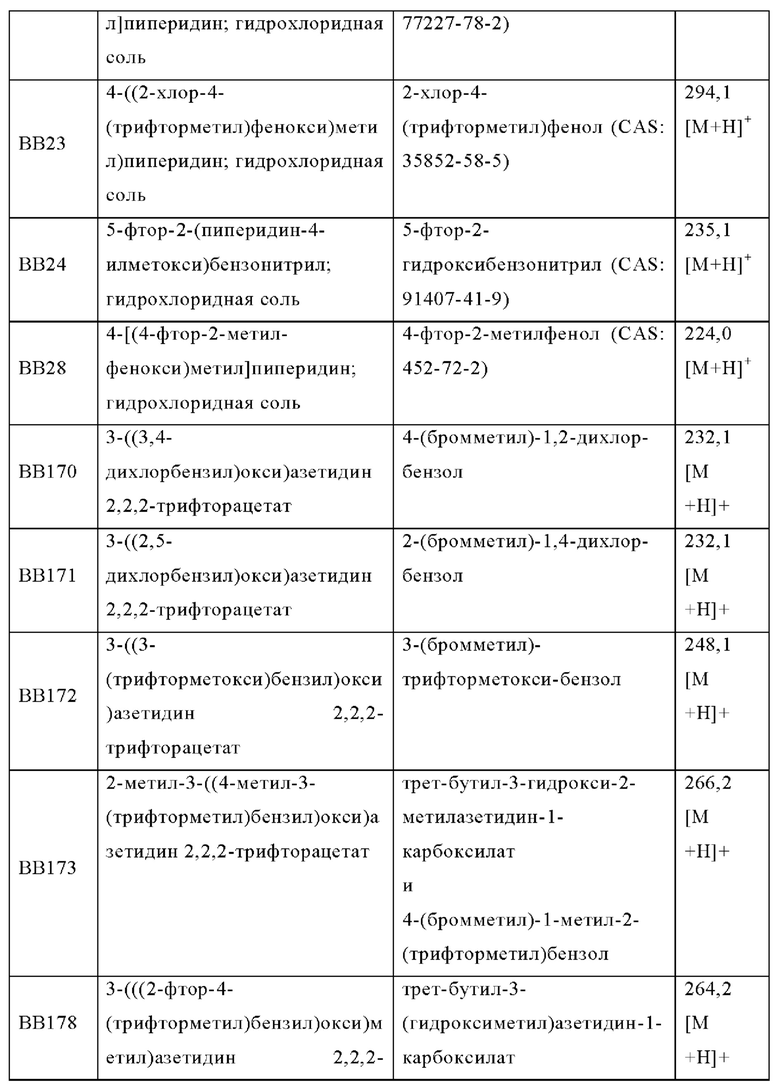

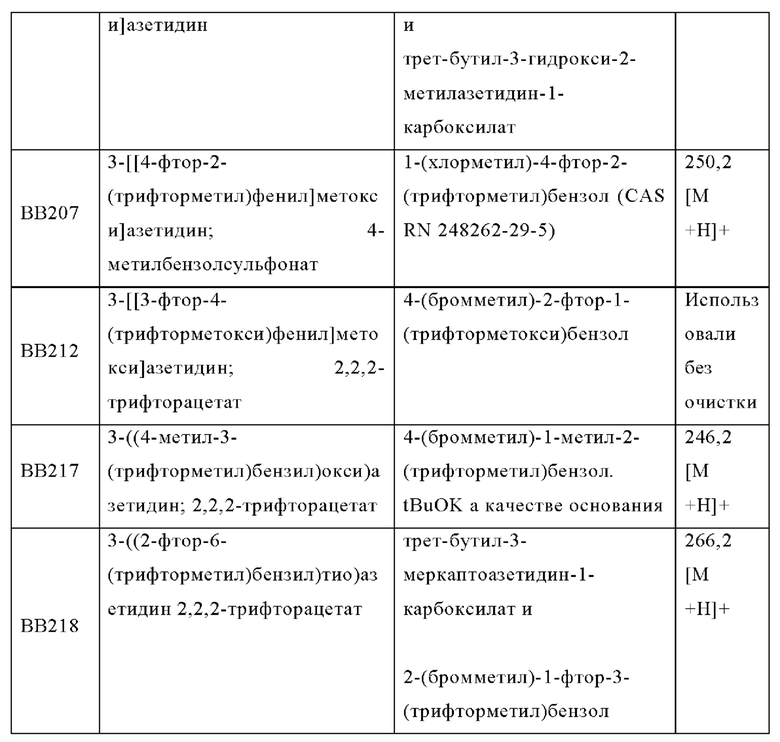
Способ D1
ВВ9
4-[2-Хлор-4-(трифторметил)фенокси]пиперидин; трифторацетатная соль
Смесь трет-бутил-4-[2-хлор-4-(трифторметил)фенокси]пиперидин-1-карбоксилата (750,0 мг, 1,97 ммоль) в ДХМ (20 мл) и ТФУ (0,76 мл) перемешивали при 20°C в течение 12 ч. Смесь концентрировали. Остаток растворяли в H2O (20 мл) и дважды промывали ПЭ:ЭА=10:1 (по 20 мл каждый раз). Водный слой лиофилизировали с получением необходимого продукта (716 мг, 1,82 ммоль, 87,8%). МС (ИЭР): m/z=280,1 [М+Н]+.
Этап а) трет-бутил-4-[2-хлор-4-(трифторметил)фенокси]пиперидин-1-карбоксилат
Смесь 2-хлор-4-(трифторметил)фенола (500 мг, 2,54 ммоль), 1-Вос-4-гидроксипиперидина (768 мг, 3,82 ммоль) и трифенилфосфина (1334 мг, 5,09 ммоль) в ТГФ (10 мл) перемешивали при 0°C до полного растворения. Медленно по каплям добавляли ДИАД (1542 мг, 7,63 ммоль) при 0°C. Смесь перемешивали при 20°C в течение 3 ч, а затем концентрировали в вакууме. Остаток очищали с помощью препаративной ВЭЖХ с получением необходимого соединения в виде светло-желтого твердого вещества (760 мг, 2 ммоль, выход 78,7%). МС (ИЭР: m/z=324,0 [М-56+Н]+.
ВВ57
3-(((2-фтор-6-(трифторметил)бензил)окси)метил)азетидин; трифторацетатная соль
В раствор трет-бутил-3-(((2-фтор-6-(трифторметил)бензил)окси)метил)азетидин-1-карбоксилата (158 мг, 435 мкмоль) в ДХМ (1,74 мл) добавляли ТФУ (793 мг, 536 мкл, 6,96 ммоль) и перемешивали реакцию при комнатной температуре в течение 3 ч. Реакционную смесь концентрировали с получением 3-(((2-фтор-6-(трифторметил)бензил)окси)метил)азетидина; трифторацетатной соли (202 мг, 434 мкмоль, выход 99,7%) в виде бесцветного масла. Неочищенный материал использовали без дополнительной очистки. МС (ИЭР): m/z=264,1 [М+Н]+.
Этап а) трет-бутил-3-(((2-фтор-4-(трифторметил)бензил)окси)метил)азетидин-1-карбоксилат
В раствор трет-бутил-3-(гидроксиметил)азетидин-1-карбоксилата (100 мг, 534 мкмоль) в сухом ТГФ (2,67 мл) добавляли 1,65 М раствор трет-бутоксида калия в ТГФ (340 мкл, 561 ммоль) и перемешивали непрозрачную реакционную смесь при КТ в течение 15 мин с последующим добавлением 1-(бромметил)-2-фтор-6-(трифторметил)бензола (137 мг, 534 мкмоль). Затем реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Неочищенную реакцию разводили этилацетатом и экстрагировали насыщ. води, раствором NaHCO3, органическую фазу собирали, а водный слой обратно экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом натрия и выпаривали до сухости с получением прозрачного масла. Неочищенный материал иммобилизовали на Isolute и очищали с помощью колоночной хроматографии с элюированием от 0 до 30% EtOAc в гептанах с получением трет-бутил-3-(((2-фтор-6-(трифторметил)бензил)окси)метил)азетидин-1-карбоксилата (158 мг, 413 мкмоль, выход 77,3%) в виде бесцветного масла. МС (ИЭР): m/z=308,1 [М-56+Н]+
Способ D2
ВВ10
4-[[2-циклопентил-4-(трифторметил)фенил] метил] пиперидин; соль муравьиной кислоты
Смесь трет-бутил-4-[[2-циклопентил-4-(трифторметил)фенил]метил]пиперидин-1-карбоксилата (440 мг, 0,610 ммоль) и 5,0 мл 4 М HCl в EtOAc в EtOAc (10 мл) перемешивали при 20°C в течение 12 ч. Смесь концентрировали в вакууме. Остаток перерастворяли в H2O (5 мл) и дважды промывали ПЭ:ЭА=(3:1; по 10 мл каждый раз) и разделяли слои. Остаток очищали с помощью препаративной ВЭЖХ с получением необходимого соединения в виде светло-желтого твердого вещества (124 мг, 0,350 ммоль, выход 65,3%). МС (ИЭР): m/z=312,2 [М+Н]+.
Этап а) трет-бутил-4-[[2-циклопентил-4-(трифторметил)фенил]метилен] пиперидин-1-карбоксилат
Раствор трет-бутил-4-[[2-бром-4-(трифторметил)фенил]метилен]пиперидин-1-карбоксилата (500 мг, 1,19 ммоль), циклопентилбромида (266 мг, 1,78 ммоль), Ir(dF(CF3)ppy)2(dtbbpy)PF6 (13,4 мг, 0,010 ммоль, CAS RN 870987-63-6), NiCl2.glyme (0,77 мг, 0,060 ммоль), dtbbpy (19,2 мг, 0,070 ммоль, CAS RN 72914-19-3), TTMSS (296 мг, 1,19 ммоль, CAS RN 1873-77-4) и Na2CO3 (252 мг, 2,38 ммоль) в ДМФА (20 мл) дегазировали путем барботирования потоком аргона в течение 20 мин. Реакционную смесь облучали синим светодиодом (4x1) при 25°C в течение 16 ч. Смесь разводили Н2О, а затем экстрагировали три раза EtOAc (по 100 мл каждый раз). Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью препаративной ВЭЖХ с получением соединения в виде бесцветного масла (460 мг, 1,12 ммоль, выход 53,8%). МС (ИЭР: m/z=354,1 [М-56+Н]+.
Этап b) трет-бутил-4-[[2-циклопентил-4-(трифторметил)фенил]метил] пиперидин-1-карбоксилат
В смесь трет-бутил-4-[[2-циклопентил-4-(трифторметил)фенил]метилен]пиперидин-1-карбоксилата (460 мг, 0,640 ммоль) в EtOAc (10 мл) добавляли влажный Pd/C (40 мг), а затем смесь перемешивали при 20°C в течение 12 ч в атмосфере Н2 (1520 мм рт. ст.). Смесь фильтровали, а фильтрат концентрировали с получением соединения в виде бесцветного масла (460 мг, 1,12 ммоль, выход 99,5%). МС (ИЭР): m/z=356,1 [М+Н-56]+.
ВВП
2-(4-пиперидилметил)-1,3-бензоксазол; соль муравьиной кислоты
Раствор 2-аминофенола (1,0 г, 9,16 ммоль) и 1-Boc-4-пиперидилуксусной кислоты (2,68 г, 11 ммоль) в полифосфорной кислоте (2,2 г) перемешивали при 180°C в течение 2 ч. Смесь разводили смесью 12 М водного раствора NH4OH и льда до достижения рН>7, а затем три раза экстрагировали EtOAc (по 10 мл каждый раз). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали, а остаток очищали с помощью препаративной ВЭЖХ с получением необходимого соединения в виде коричневого масла (251 мг, 0,960 ммоль, 9,7%). МС (ИЭР): m/z=217,2 [М+Н]+.
Способ D3
ВВ13
4-[4-хлор-3-(4-хлорфенил)фенокси]пиперидин; гидрохлоридная соль
Раствор трет-бутил-4-[4-хлор-3-(4-хлорфенил)фенокси] пиперидин-1-карбоксилата (1000 мг, 2,37 ммоль) в 4 М растворе HCl в диоксане (50 мл) перемешивали при 20°C в течение 12 ч. Смесь концентрировали с получением указанного в заголовке соединения в виде белого твердого вещества (845 мг, 2,35 ммоль, 96,2%). МС (ИЭР): m/z=322,0 [М+Н]+.
Этап а) трет-бутил-4-(3-бром-4-хлорфенокси)пиперидин-1-карбоксилат
Смесь 3-бром-4-хлорфенола (1000 мг, 4,82 ммоль), 1-Вос-4-гидроксипиперидина (1164 мг, 5,78 ммоль) и трифенилфосфина (2529 мг, 9,64 ммоль) перемешивали в ТГФ (10 мл) до полного растворения. Затем медленно по каплям добавляли ДИАД (1948 мг, 9,64 ммоль) при 0°C. Смесь перемешивали при 20°C в течение 12 ч, концентрировали, а остаток очищали с помощью обращенной флэш-хроматографии с получением соединения в виде желтого масла (1300 мг, 3,33 ммоль, 69,0%). МС (ИЭР: m/z=336,0 [М-56+Н]+.
Этап b) трет-бутил-4-[4-хлор-3-(4-хлорфенил)фенокси]пиперидин-1-карбоксилат
В раствор трет-бутил-4-(3-бром-4-хлорфенокси)пиперидин-1-карбоксилата (1150 мг, 2,94 ммоль) и 4-хлорфенилбороновой кислоты (506 мг, 3,24 ммоль), Na2CO3 (1248 мг, 11,8 ммоль) в 1,4-диоксане (20 мл) и H2O (5 мл) добавляли тетракис(трифенилфосфин)палладий (0) (340 мг, 0,290 ммоль, CAS RN 14221-01-3), и перемешивали смесь при 110°C в атмосфере N2 в течение 12 ч. Смесь фильтровали, фильтрат концентрировали, а остаток очищали с помощью колоночной хроматографии на силикагеле с элюированием градиентом 5-20% EtOAc - ПЭ с получением необходимого соединения в виде светло-желтого масла (1100 мг, 2,6 ммоль, 88,5%). МС (ИЭР: m/z=366,1 [М-56+Н]+.
ВВ14
4-[[2-(1Н-пиразол-4-ил)-4-(трифторметил)фенил]метил]пиперидин; трифторацетатная соль
В раствор трет-бутил-4-[[2-(1-трет-бутоксикарбонилпиразол-4-ил)-4-(трифторметил)фенил]метил]пиперидин-1-карбоксилата (150,0 мг, 0,290 ммоль) в ДХМ (5 мл) добавляли ТФУ (1,0 мл). Смесь перемешивали при 20°C в течение 15 ч. Смесь концентрировали в вакууме, а затем лиофилизировали с получением указанного в заголовке соединения в виде светло-желтой смолы (149 мг, 0,280 ммоль, выход 85,1%). МС (ИЭР): m/z=310,0 [М+Н]+.
Этап а) трет-бутил-4-[[2-(1-трет-бутоксикарбонилпиразол-4-ил)-4-(трифторметил)фенил]метилен] пиперидин-1-карбоксилат
Смесь трет-бутил-4-[[2-бром-4-(трифторметил)фенил] метилен] пиперидин-1-карбоксилата (600 мг, 1,43 ммоль), трет-бутил-4-(4,4,5,5-тетраметил-1,3-диоксолан-2-ил)пиразол-1-карбоксилата (846 мг, 2,86 ммоль) и K2CO3 (592 мг, 4,28 ммоль) в ДМФА (10 мл) и Н2О (0,5 мл) перемешивали при 80°C в течение 12 ч. Смесь вливали в Н2О (30 мл) и дважды экстрагировали EtOAc (по 50 мл каждый раз). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме с получением соединения в виде светло-желтого твердого масла (520 мг, 1,02 ммоль, выход 71,8%). МС (ИЭР): m/z=308,1 [М+Н]+.
Этап b) трет-бутил-4-[[2-(1-трет-бутоксикарбонилпиразол-4-ил)-4-(трифторметил)фенил]метил] пиперидин-1-карбоксилат
Смесь трет-бутил-4-[[2-(1-трет-бутоксикарбонилпиразол-4-ил)-4-(трифторметил)фенил]метилен]пиперидин-1-карбоксилата (180 мг, 0,350 ммоль) и влажного Pd/C (18 мг) в EtOAc (10 мл) перемешивали при 30°C в течение 24 ч в атмосфере Н2 (~1520 мм рт.ст.). Смесь фильтровали и концентрировали в вакууме с получением соединения в виде коричневого масла (150 мг, 0,290 ммоль, выход 83%). МС (ИЭР: m/z=354,1 [М-56-100+Н]+.
ВВ18
4-[2-(2-хлорфенил)этинил] пиперидин
В суспензию трет-бутил-4-((2-хлорфенил)этинил]пиперидин-1-карбоксилата (0,05 г, 0,156 ммоль) в МеОН (3 мл) добавляли 4 М HCl в диоксане (0,391 мл, 1,56 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 2 ч. Смесь выпаривали до сухости, а остаток растирали в диизопропиловом эфире, отфильтровывали и дополнительно сушили в условиях высокого вакуума с получением указанного в заголовке соединения в виде белого твердого вещества в виде гидрохлоридной соли (0,02 г, 50%). МС (ИЭР): m/z=220,1 [М+Н]+.
Этап а) трет-бутил-4-[2-(2-хлорфенил)этинил]пиперидин-1-карбоксилат
В закрытой пробирке смесь трет-бутил-4-этинилпиперидин-1-карбоксилата (0,1 г, 0,478 ммоль, CAS RN 287192-97-6,), 1-бром-2-хлорбензола (0,084 мл, 0,717 ммоль), иодида меди (I) (0,002 г, 0,009 ммоль), ТЭА (0,666 мл, 4,78 ммоль) и хлорида бис(трифенилфосфин)палладия (II) (0,027 г, 0,038) в ТГФ (2,8 мл) дегазировали в течение 5 мин в атмосфере аргона. Затем реакционную смесь нагревали до 70°C и перемешивали в течение 4 ч. Смесь отфильтровывали через слой дикалита, промывали EtOAc и выпаривали до сухости маточные растворы. Остаток очищали с помощью флэш-хроматографии на силикагеле с элюированием градиентом 0-50% EtOAc/n-гептан с получением указанного в заголовке соединения в виде белого твердого вещества (0,05 г, 33%). МС (ИЭР: m/z=264,1 [М-56+Н]+.
ВВ48а
трет-бутил-4-[[2-фтор-4-(трифторметил)фенил]метил]пиперидин-1-карбоксилат
Дегазированный раствор трет-бутил-4-метилпиперидин-1-карбоксилата (4465 мг, 22,6 ммоль, CAS RN 159635-49-1) в 9-BBN (45,3 мл, 22,6 ммоль) кипятили с обратным холодильником в течение 1 ч. После охлаждения до комнатной температуры раствор добавляли в раствор 4-бром-3-фторбензотрифторида (5,0 г, 20,6 ммоль, CAS RN 40161-54-4), Pd(dppf)Cl2 (1514 мг, 2,06 ммоль) и K2CO3 (5687 мг, 41,1 ммоль) в ДМФА (50 мл) и воде (5 мл). Полученную в результате смесь нагревали при 80°C в течение 5 ч. После этого смесь охлаждали до комнатной температуры и вливали в воду, рН доводили до 11 с помощью 10% водного NaOH и экстрагировали смесь EtOAc. Объединенные органические экстракты промывали солевым раствором, сушили над Na2SO4, фильтровали и выпаривали с получением остатка, который дополнительно очищали с помощью колоночной хроматографии (силикагель, ПЭ: EtOAc=10 от 1 до 5:1) с получением соединения в виде светло-желтого твердого вещества (240 мг; 3,2%). МС (ИЭР): m/z=306 [М+Н-56]+.
ВВ51а
Смесь трет-бутил-4-[[2-циклопропил-4-(трифторметил)фенил]метилен]пиперидин-1-карбоксилата (1000 мг, 2,62 ммоль) и PtO2 (100 мг, 0,440 ммоль) в EtOAc (20 мл) перемешивали при 20°C в течение 12 ч в атмосфере Н2 (1520 мм рт.ст.). Смесь фильтровали, а фильтрат концентрировали с получением соединения в виде светло-желтого твердого вещества (940 мг, 93,5%). МС (ИЭР): m/z=328,2 [М+Н]+.
Этап а) 2-бром-1-(бромметил)-4-(трифторметил)бензол
Смесь 2-бром-1-метил-4-(трифторметил)бензола (5,5 г, 23,0 ммоль, CAS RN 128-08-5), бензоилпероксида (835 мг, 3,45 ммоль) и NBS (4,07 г, 23,01 ммоль) в ССЦ (50,0 мл, 23,0 ммоль) перемешивали при 70°C в течение 5 ч. Смесь вливали в воду (20 мл) и дважды экстрагировали ДХМ (по 20 мл каждый раз). Объединенный органический слой промывали солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением необходимого соединения в виде светло-желтого масла, которое использовали на следующем этапе без дополнительной очистки (7,1 г, 97%).
Этап b) 2-бром-1-(диэтоксифосфорилметил)-4-(трифторметил)бензол
Смесь 2-бром-1-(бромметил)-4-(трифторметил)бензола (7,1 г, 22,3 ммоль) и триэтилфосфита (30 мл) перемешивали при 155°C в течение 5 ч. Смесь концентрировали в вакууме для удаления триэтилфосфита, остаток разводили водой (100 мл) и три раза экстрагировали EtOAc (по 100 мл каждый раз). Объединенные органические слои промывали солевым раствором (100 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии (силикагель, ПЭ: EtOAc=100:1 до 10:1) с получением соединения в виде светло-желтого масла, которое использовали на следующем этапе без дополнительной очистки (8 г, 95,5%).
Этап с) трет-бутил-4-(2-бром-4-(трифторметил)бензилиден)пиперидин-1-карбоксилат
В смесь 2-бром-1-(диэтоксифосфорилметил)-4-(трифторметил)бензола (6,9 г, 18,4 ммоль) в ТГФ (100 мл) добавляли гидрид натрия (2,21 г, 55,2 ммоль) при 0°С. Смесь перемешивали при 0°С в течение 1 ч, затем добавляли 1-Boc-4-пиперидон (7,33 г, 36,79 ммоль, CAS RN 79099-07-3) и перемешивали смесь при 20°С в течение 12 ч. Смесь вливали в воду (100 мл) и три раза экстрагировали EtOAc (по 100 мл каждый раз). Объединенные органические слои промывали солевым раствором (100 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии (силикагель, ПЭ: ЭА = 100: от 1 до 50: 1) с получением необходимого соединения в виде грязно-белого твердого вещества (4 г; 51,7%). МС (ИЭР): m/z = 365,9 [М-56+Н]+.
Этап d) трет-бутил-4-[[2-циклопропил-4-(трифторметил)фенил]метилен]пиперидин-1-карбоксилат
Смесь трет-бутил-4-[[2-бром-4-(трифторметил)фенил]метилен]пиперидин-1-карбоксилата (2,0 г, 4,76 ммоль), циклопропилбороновой кислоты (818 мг, 9,52 ммоль, CAS RN 411235-57-9) и карбоната калия (1973 мг, 14,3 ммоль) в ДМФА (10 мл) и воде (0,5 мл) перемешивали при 80°С в атмосфере азота в течение 12 ч. Смесь вливали в воду (50 мл), три раза экстрагировали EtOAc (по 50 мл каждый раз). Объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью препаративной ВЭЖХ с получением соединения в виде светло-желтого масла (1020 мг, выход 56,2%). МС (ИЭР: m/z = 326,0 [М-56+Н]+.
ВВ53а
трет-бутил-3-[(4-хлорфенил)метокси]пирролидин-1-карбоксилат
В раствор N-Boc-3-гидроксипирролидина (1,0 г, 5,34 ммоль) и 4-хлорбензилбромида (1,32 г, 6,41 ммоль) в АЦН (10 мл) добавляли карбонат калия (1,48 г, 10,68 ммоль). Смесь перемешивали при 80°С в течение 15 ч. Затем смесь концентрировали, разводили водой и три раза экстрагировали EtOAc (по 10 мл каждый раз). Объединенные органические слои концентрировали с получением необходимого соединения в виде бесцветного масла (326 мг, выход 19,6%) МС (ИЭР): m/z = 256,0 [М-56+Н]+.
Способ D4
ВВ70
3-[4-(трифторметил)фенокси]азетидин
В раствор трет-бутил-3-[4-(трифторметил)фенокси]азетидин-1-карбоксилата (500 мг, 1,58 ммоль, ВВ70а) в ДХМ (3 мл) добавляли ТФУ (1,0 мл, 0,950 ммоль) при 25°С, реакцию перемешивали при этой температуре в течение 12 ч. Смесь концентрировали, а остаток очищали с помощью препаративной ВЭЖХ с получением соединения в виде бесцветного твердого вещества (150 мг, 0,690 ммоль, 43,8%). МС (ИЭР): m/z = 218,1 [М+Н]+.
ВВ72а
трет-бутил-4-(4-хлор-3-циклопропилфенокси)пиперидин-1-карбоксилат
В раствор трет-бутил-4-(3-бром-4-хлорфенокси)пиперидин-1-карбоксилата (500 мг, 1,28 ммоль, ВВ90), карбоната калия (354 мг, 2,56 ммоль) и циклопропилбороновой кислоты (121 мг, 1,41 ммоль) в 1,4-диоксане (5 мл) и воде (1 мл) добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлор палладий (II) (187,28 мг, 0,260 ммоль). Смесь перемешивали при 100°С в атмосфере азота в течение 12 ч. Реакционную смесь фильтровали, а фильтрат разводили EtOAc (30 мл), промывали водой и затем солевым раствором, органическую фазу сушили над Na2SO4, концентрировали. Остаток очищали с помощью хроматографии на силикагеле (элюирование градиентом 5% - 10% EtOAc-ПЭ) с получением соединения в виде светло-желтого масла (220 мг, 48,9%). МС (ИЭР: m/z = 296,1 [М-56+Н]+.
ВВ73а
трет-бутил-4-(4-хлор-3-морфолинофенокси)пиперидин-1-карбоксилат
В раствор трет-бутил-4-(3-бром-4-хлорфенокси)пиперидин-1-карбоксилата (500 мг, 1,28 ммоль, ВВ90), карбоната цезия (834 мг, 2,56 ммоль), (R)-(+)-2,2'-бис(дифенилфосфино)-1,1'-бинафталина (159 мг, 0,260 ммоль) и морфолина (112 мг, 1,28 ммоль) в ДМФА (10 мл) добавляли трис(дибензилиденацетон)дипалладий (0) (187 мг, 0,260 ммоль) и перемешивали смесь при 110°С в атмосфере азота в течение 12 ч. Реакционную смесь фильтровали, фильтрат разводили EtOAc (30 мл), промывали водой и затем солевым раствором, органическую фазу сушили над Na2SO4 и концентрировали. Остаток очищали с помощью силикагелевой колонки (элюирование градиентом 5% - 10% EtOAc-ПЭ) с получением необходимого соединения в (360 мг, 70,9%) виде светло-желтого масла. МС (ИЭР): m/z = 397,1 [М+H]+.
ВВ74а
трет-бутил-4-[2-метил-4-(трифторметил)фенокси]пиперидин-1-карбоксилат
В раствор трет-бутил-4-[2-бром-4-(трифторметил)фенокси]пиперидин-1-карбоксилата (2,0 г, 4,71 ммоль, BB74b) в ТГФ (40 мл) по каплям добавляли метид лития (11,8 мл, 18,9 ммоль) при -70°С. Смесь перемешивали при -70°С в течение 1 ч, а затем перемешивали при 20°С в течение 12 ч. Смесь вливали в ледяную воду (100 мл) и три раза экстрагировали EtOAc (по 50 мл каждый раз). Объединенный органический слой промывали солевым раствором (100 мл), сушили над Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме с получением соединения в виде светло-желтого твердого вещества (780 мг, 46%). МС (ИЭР: m/z = 260,1 [М-100+H]+.
ВВ75а
трет-бутил-4-[2-циано-4-(трифторметил)фенокси]пиперидин-1-карбоксилат
В раствор цианида цинка (2214 мг, 18,9 ммоль) и терт-бутил-4-[2-бром-4-(трифторметил)фенокси]пиперидин-1-карбоксилата (1600 мг, 3,77 ммоль, BB74b) в ДМА (30 мл) добавляли dppf (627 мг, 1,13 ммоль), N,N-диизопропилэтиламин (1,97 мл, 11,3 ммоль), цинковую пыль (245 мг, 3,77 ммоль) и Pd2(dba)3 (1036 мг, 1,13 ммоль) при 20°С, затем смесь перемешивали при 140°С в атмосфере азота в течение 4 ч. Смесь фильтровали. Фильтрат вливали в воду (100 мл) и три раза экстрагировали EtOAc (по 50 мл каждый раз). Объединенный органический слой промывали солевым раствором (50 мл), сушили над Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме с получением указанного в заголовке соединения в виде светло-коричневого твердого вещества (2,3 г; неочищенное). МС (ИЭР: m/z = 315,0 [M-56+H]+.
ВВ76а
трет-бутил-4-(оксазоло[5,4-с]пиридин-2-илметил)пиперидин-1-карбоксилат
В раствор гексахлорэтана (2,47 г, 10,4 ммоль) в толуоле (20 мл) добавляли трифенилфосфин (3,28 г, 12,5 ммоль) и NEt3 (4,65 мл, 33,4 ммоль). Смесь перемешивали при 80°С в течение 5 мин, затем добавляли трет-бутил-4-[2-[(3-гидрокси-4-пиридил)амино]-2-оксоэтил]пиперидин-1-карбоксилат (1,4 г, 4,17 ммоль) и перемешивали при 80°С в течение 12 ч. Смесь концентрировали, чтобы удалить толуол, затем разводили водой (100 мл) и три раза экстрагировали EtOAc (по 50 мл каждый раз). Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенное вещество очищали с помощью хроматографии на силикагеле (ПЭ: EtOAc - 10: от 1 до 1: 0) с получением соединения в виде желтого масла (814 мг; 21%). МС (ИЭР): m/z = 318,1 [М+Н]+.
Этап а) трет-бутил-4-[2-[(3-гидрокси-4-пиридил)амино]-2-оксоэтил]пиперидин-1-карбоксилат
В раствор 4-аминопиридин-3-ола (3,0 г, 27,3 ммоль) и 1-Boc-4-пиперидилуксусной кислоты (7,95 г, 32,7 ммоль) в ДМФА (30 мл) добавляли HOBt (6,26 г, 40,9 ммоль), ЭДКИ (6,34 г, 40,87 ммоль) и NEt3 (11,39 мл, 81,74 ммоль). Смесь перемешивали при 20°С в течение 15 ч. Затем смесь концентрировали, остаток вливали в воду (100 мл), а затем три раза экстрагировали EtOAc (по 20 мл каждый раз). Органическую фазу промывали солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали с помощью обращенно-фазовой хроматографии и лиофилизировали с получением двух партий необходимого соединения. Партия 1 в виде бесцветного твердого вещества (1,2 г, чистота 85%, 11,1%), и партия 2 в виде бесцветного твердого вещества (520 мг, чистота 76,7%, выход 4,4%). МС (ИЭР): m/z = 336,1 [М+Н]+ для обеих партий.
ВВ77
4-хлор-3-(2-пиперидин-4-илэтинил)пиридин
Промежуточное соединение ВВ77 получали по аналогии с ВВ18, но используя 3-бром-4-хлорпиридин на этапе а), с получением указанного в заголовке соединения в виде оранжевого твердого вещества. МС (ИЭР): m/z = 221,1 [М+Н]+.
ВВ78
3-хлор-2-(2-пиперидин-4-илэтинил)пиридин
Промежуточное соединение ВВ78 получали по аналогии с ВВ18, но используя 2-бром-3-хлорпиридин на этапе а), с получением указанного в заголовке соединения в виде желтого твердого вещества. МС (ИЭР): m/z = 221,1 [М+Н]+.
ВВ79
4-[2-(2-хлор-4-фторфенил)этинил]пиперидин
Промежуточное соединение ВВ79 получали по аналогии с ВВ18, но используя 1-бром-2-хлор-4-фторбензол на этапе а), с получением указанного в заголовке соединения в виде белого твердого вещества. МС (ИЭР): m/z = 238,1 [M+H]+.
ВВ80
4-[2-(3-хлорфенил)этинил]пиперидин
Промежуточное соединение ВВ80 получали по аналогии с ВВ18, но используя 1-бром-3-хлорбензол на этапе а), с получением указанного в заголовке соединения в виде бесцветного аморфного твердого вещества. МС (ИЭР): m/z = 220,2 [М+Н]+.
ВВ81
4-[2-(4-хлорфенил)этинил]пиперидин
Промежуточное соединение ВВ81 получали по аналогии с ВВ18, но используя 1-бром-4-хлорбензол на этапе а), с получением указанного в заголовке соединения в виде желтого аморфного твердого вещества. МС (ИЭР): m/z = 220,2 [М+Н]+.
ВВ82
4-[2-(2-хлор-4-хлорфенил)этинил]пиперидин
Промежуточное соединение ВВ82 получали по аналогии с ВВ18, но используя 1-бром-2,4-дихлорбензол на этапе а), с получением указанного в заголовке соединения в виде светло-желтого аморфного твердого вещества. МС (ИЭР): m/z = 254,1 [М+Н]+.
ВВ83
4-[2-(2-хлорфенил)этинил]пиперидин-4-ол
Промежуточное соединение ВВ83 получали по аналогии с ВВ18, но используя трет-бутил-4-этинил-4-гидроксипиперидин-1-карбоксилат (CAS RN 275387-83-2) на этапе а), с получением указанного в заголовке соединения в виде желтого аморфного твердого вещества. МС (ИЭР): m/z = 218,1[M-H2O+H]+.
ВВ84
3-[2-(2-хлорфенил)этинил]азетидин
В раствор трет-бутил-3-[2-(2-хлорфенил)этинил]азетидин-1-карбоксилата (0,035 г, 0,120 ммоль) в ДХМ (0,6 мл) добавляли ТФУ (0,924 мл, 1,2 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 2 ч. Смесь разводили ДХМ, вливали в насыщенный води. раствор NaHCO3 и экстрагировали ДХМ. Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали, выпаривали и дополнительно сушили в условиях высокого вакуума с получением неочищенного указанного в заголовке соединения (0,02 г, 87%) в виде светло-желтого масла. МС (ИЭР): m/z = 192.0 [М+Н]+.
Этап а) трет-бутил-3-[2-(2-хлорфенил)этинил]азетидин-1-карбоксилат
Это соединение получали по аналогии с промежуточным соединением ВВ18, но используя трет-бутил-3-этинилазетидин-1-карбоксилат (CAS RN 287193-01-5) на этапе а), с получением указанного в заголовке соединения в виде белого твердого вещества. МС (ИЭР: m/z = 236,1 [М-56+Н]+.
ВВ85
3-[2-(2,4-дихлорфенил)этинил]азетидин
Промежуточное соединение ВВ85 получали по аналогии с промежуточным соединением ВВ84, но используя 1-бром-2,4-дихлорбензол на этапе а), с получением указанного в заголовке соединения в виде светло-желтого масла. МС (ИЭР): m/z = 226,1 [M+H]+.
ВВ86
3-[2-(2-хлор-4-фторфенил)этинил]азетидин
Промежуточное соединение ВВ86 получали по аналогии с промежуточным соединением ВВ84, но используя 1-бром-2-хлор-4-фторбензол на этапе а), с получением указанного в заголовке соединения в виде желтого масла. МС (ИЭР): m/z = 210,1 [М+Н]+.
По аналогии с ВВ9а следующие составляющие компоненты получали из соответствующих строительных блоков.


По аналогии с ВВ15а следующие составляющие компоненты получали из соответствующих строительных блоков.

По аналогии с ВВ9, этап а., следующие составляющие компоненты получали из соответствующих исходных материалов.
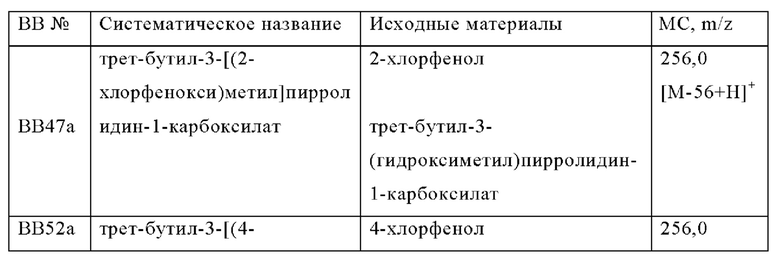

Способ D5
ВВ51
4-[[2-циклопропил-4-(трифторметил)фенил]метил]пиперидин; соль муравьиной кислоты
В смесь трет-бутил-4-[[2-циклопропил-4-(трифторметил)фенил]метил]пиперидин-1-карбоксилата (940 мг, 2,45 ммоль, ВВ51а) в ДХМ (10 мл) добавляли ТФУ (2,0 мл, 2,45 ммоль). Смесь перемешивали при 20°С в течение 12 ч. Смесь концентрировали в вакууме. Остаток дважды очищали с помощью препаративной ВЭЖХ с получением необходимого соединения в виде светло-желтой смолы (111 мг, 12,4%). МС (ИЭР): m/z = 284,2 [М+Н]+.
Этап а) 2-бром-1-(бромметил)-4-(трифторметил)бензол
Смесь 2-бром-1-метил-4-(трифторметил)бензола (5,5 г, 23,0 ммоль, CAS RN 128-08-5), бензоилпероксида (835 мг, 3,45 ммоль) и NBS (4,07 г, 23,0 ммоль) в CCl4 (50,0 мл, 23,0 ммоль) перемешивали при 70°С в течение 5 ч. Смесь вливали в воду (20 мл) и дважды экстрагировали ДХМ (по 20 мл каждый раз). Объединенные органические слои промывали солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением соединения в виде светло-желтого масла (7,1 г, 97%), которое использовали на следующем этапе без дополнительной очистки.
Этап b) 2-бром-1-(диэтоксифосфорилметил)-4-(трифторметил)бензол
Смесь 2-бром-1-(бромметил)-4-(трифторметил)бензола (7,1 г, 22,3 ммоль) и триэтилфосфита (30,0 мл) перемешивали при 155°С в течение 5 ч. Смесь концентрировали в вакууме для удаления триэтилфосфита. Остаток разводили водой (100 мл) и три раза экстрагировали EtOAc (по 100 мл каждый раз). Объединенные органические слои промывали солевым раствором (100 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии (силикагель, ПЭ: ЭА = 100: от 1 до 10: 1) с получением указанного в заголовке соединения в виде светло-желтого масла (8 г, 21,3 ммоль, 95,5%), которое использовали на следующем этапе без дополнительной очистки.
Этап с) трет-бутил-4-(2-бром-4-(трифторметил)бензилиден)пиперидин-1-карбоксилат
В смесь 2-бром-1-(диэтоксифосфорилметил)-4-(трифторметил)бензол (6,9 г, 18,4 ммоль) в ТГФ (100 мл) добавляли NaH (2,21 г, 55,2 ммоль) при 0°С. Смесь перемешивали при 0°С в течение 1 ч, затем добавляли 1-Boc-4-пиперидон (7,33 г, 36,8 ммоль, CAS RN 79099-07-3) и перемешивали смесь при 20°С в течение 12 ч. Смесь вливали в воду (100 мл) и три раза экстрагировали EtOAc (по 100 мл каждый раз). Объединенный органический слой промывали солевым раствором (100 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии (силикагель, ПЭ: ЭА = 100: от 1 до 50: 1) с получением необходимого соединения в виде грязно-белого твердого вещества (4 г, 9,52 ммоль, 51,7%). МС (ИЭР): m/z = 365,9 [M-56+H]+.
Этап d) трет-бутил-4-[[2-циклопропил-4-(трифторметил)фенил]метилен]пиперидин-1-карбоксилат
Смесь трет-бутил-4-[[2-бром-4-(трифторметил)фенил]метилен]пиперидин-1-карбоксилата (2,0 г, 4,76 ммоль), циклопропилбороновой кислоты (818 мг, 9,52 ммоль, CAS RN 411235-57-9) и карбоната калия (1973 мг, 14,3 ммоль) в ДМФА (10 мл) и воде (0,5 мл) перемешивали при 80°С в атмосфере азота в течение 12 ч. Смесь вливали в воду (50 мл) и три раза экстрагировали EtOAc (по 50 мл каждый раз). Объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью препаративной ВЭЖХ с получением соединения в виде светло-желтого масла (1020 мг, выход 56,2%) МС (ИЭР): m/z = 326,0 [М-56+Н]+.
Этап е) трет-бутил-4-[[2-циклопропил-4-(трифторметил)фенил]метил]пиперидин-1-карбоксилат
Смесь трет-бутил-4-[[2-циклопропил-4-(трифторметил)фенил]метилен]пиперидин-1-карбоксилата (1000 мг, 2,62 ммоль) и PtO2 (100 мг, 0,440 ммоль) в EtOAc (20 мл) перемешивали при 20°С в течение 12 ч в атмосфере водорода (1520 мм рт. ст.). Затем смесь фильтровали, а фильтрат концентрировали с получением соединения в виде светло-желтого твердого вещества (940 мг, выход 93,5%). МС (ИЭР): m/z = 328,2 [М+Н]+.
Способ D6
ВВ92
N-метил-N-[4-(трифторметил)фенил]пиперидин-4-амин; трифторац статная соль
В раствор трет-бутил-4-[N-метил-4-(трифторметил)анилино]пиперидин-1-карбоксилата (150 мг, 0,420 ммоль) в ДХМ (1 мл) добавляли ТФУ (0,1 мл) при 0°С. Смесь перемешивали при 25°С в течение 12 ч. Реакционную смесь концентрировали в вакууме. Остаток очищали с помощью препаративной ВЭЖХ (в присутствии ТФА) с получением необходимого продукта в виде желтого твердого вещества (120 мг, 77,0%). МС (ИЭР): m/z = 259,2 [М+Н]+.
Этап а) трет-бутил-4-[4-(трифторметил)анилино]пиперидин-1-карбоксилат
В раствор п-трифторметиланилина (1,17 мл, 9,31 ммоль, CAS RN 455-14-1) в ДХМ (30 мл) добавляли АсОН (0,560 г, 9,31 ммоль) и 1-BOC-4-пиперидон (2,78 г, 14,0 ммоль, CAS RN 79099-07-3). Затем аккуратно добавляли 1 М раствор ВН3/ТГФ (27,9 мл, 27,9 ммоль) при 0°С в атмосфере азота. Реакционную смесь перемешивали при 25°С в течение 12 ч. Смесь вливали в насыщенный водный раствор NH4Cl (30 мл) и три раза экстрагировали EtOAc. Объединенные органические слои дважды промывали водой H2O, а затем солевым раствором, сушили над Na2SO4 и концентрировали в вакууме с получением желтого остатка, который очищали с помощью силикагелевой колонки с элюированием градиентом ПЭ: EtOAc (от 20: от 1 до 5: 1) с получением необходимого продукта в виде белого твердого вещества (2,0 г; 62,4%). МС (ИЭР: m/z = 289,1 [М-56+Н]+.
Этап b) трет-бутил-4-[N-метил-4-(трифторметил)анилино]пиперидин-1-карбоксилат
В раствор NaH (52,3 мг, 60,0% масс. %, 1,31 ммоль) в ДМФА (5 мл) добавляли трет-бутил-4-[4-(трифторметил)анилино]пиперидин-1-карбоксилат (300 мг, 0,870 ммоль) при 0°С в атмосфере азота. Смесь перемешивали при 0°С в течение 15 мин, затем добавляли иодметан (371 мг, 2,61 ммоль). Реакционную смесь перемешивали при 80°С в течение 12 ч. Реакционную смесь вливали в воду (20 мл) и три раза экстрагировали EtOAc, объединенные органические слои дважды промывали водой и солевым раствором, сушили над сульфатом натрия и концентрировали в вакууме с получением светло-желтого остатка, который очищали с помощью силикагелевой колонки с элюированием градиентом ПЭ: EtOAc (от 20: от 1 до 5: 1) с получением необходимого продукта в виде белого твердого вещества (160 мг, 51,3%). МС (ИЭР: m/z = 303,1 [М-56+Н]+
ВВ93
N-метил-N-(4-(трифторметил)фенил)азетидин-3-амин (соль трифторуксусной кислоты)
Указанное в заголовке соединение получали по аналогии со способом D6 из трет-бутил-3-[N-метил-4-(трифторметил)анилино]азетидин-1-карбоксилата (48%). МС (ИЭР): m/z = 231,1 [М+Н]+.
Этап а) трет-бутил-3-[4-(трифторметил)анилино]азетидин-1-карбоксилат
В раствор п-трифторметиланилина (0,780 мл, 6,21 ммоль, CAS RN 455-14-1), АсОН (1,86 г, 31,0 ммоль) и 1-ВОС-3-азетидинона (2,13 г, 12,4 ммоль, CAS RN 398489-26-4) в EtOH (10 мл) добавляли NaBH3CN (1,95 г, 31,0 ммоль) при 25°С. Смесь перемешивали при 25°С в течение 12 ч. Реакционную смесь вливали в насыщенный водный раствор NH4Cl (20 мл) и дважды экстрагировали EtOAc. Объединенные органические слои дважды промывали H2O и солевым раствором, сушили над сульфатом натрия и концентрировали в вакууме с получением желтого остатка, который очищали с помощью силикагелевой колонки с элюированием градиентом ПЭ: EtOAc (от 10: от 1 до 5: 1) с получением необходимого продукта в виде белого твердого вещества (340 мг, 17,3%). МС (ИЭР: m/z = 261,1 [М-56+Н]+.
Этап b) трет-бутил-3-[N-метил-4-(трифторметил)анилино]азетидин-1-карбоксилат
В раствор трет-бутил-3-[4-(трифторметил)анилино]азетидин-1-карбоксилата (300 мг, 0,950 ммоль) в ДМФА (5 мл) добавляли NaH (45,5 мг, 60% масс. %, 1,14 ммоль) при 0°С. Смесь перемешивали в течение 15 мин, а затем добавляли иодметан (404 мг, 2,85 ммоль). Реакционную смесь перемешивали при 25°С в течение 12 ч. Реакционную смесь вливали в H2O (20 мл) и дважды экстрагировали EtOAc. Объединенные органические слои три раза промывали Н2О и солевым раствором, сушили над Na2SO4 и концентрировали в вакууме с получением желтого остатка. Остаток очищали с помощью силикагелевой колонки с элюированием градиентом ПЭ: EtOAc (от 10: от 1 до 5: 1) с получением необходимого продукта в виде белого твердого вещества (310 мг, 98,9%). МС (ИЭР): m/z = 275,2 [М-56+Н]+.
Способ D7
ВВ94
N-метил-N-(пиперидин-4-ил)-2-(3-(трифторметил)фенил)ацетамида гидрохлорид
В раствор трет-бутил-4-[метил-[2-[3-(трифторметил)фенил)ацетил]амино]пиперидин-1-карбоксилата (0,080 г, 200 мкмоль) в ДХМ (1 мл) добавляли 2 М раствор HCl в диэтиловом эфире (999 мкл, 2 ммоль). Реакционную смесь перемешивали при КТ в течение ночи, а затем концентрировали в вакууме с получением указанного в заголовке соединения (67 мг, 199 мкмоль) в виде грязно-белого твердого вещества. МС (ИЭР): m/z = 301,2 [M+H]+.
Этап а) трет-бутил-4-[метил-[2-[3-(трифторметил)фенил)ацетил]амино]пиперидин-1-карбоксилат
В перемешиваемую смесь 2-(3-(трифторметил)фенил)уксусной кислоты (105 мг, 513 мкмоль, CAS RN 351-35-9) в ДМФА (5 мл) добавляли HATU (195 мг, 513 мкмоль) и ДИПЭА (181 мг, 244 мкл, 1,4 ммоль). Через 15 мин перемешивания добавляли трет-бутил-4-(метиламино)пиперидин-1-карбоксилат (0,100 г, 467 мкмоль, CAS RN 147539-41-1) и перемешивали реакционную смесь при КТ в течение 2 ч. Реакционную смесь разводили ДХМ и промывали Н2О. Органическую фазу концентрировали с получением неочищенного продукта, который очищали с помощью флэш-хроматографии на 20 г колонке с SiO2, использую смесь элюентов из н-гептана и EtOAc (от 0% до 100%) с получением необходимого соединения в виде светло-желтого масла (85 мг, 213 мкмоль). МС (ИЭР): m/z = 459,259 [М+CH3CN+NH4]+.
Способ D8
ВВ194
3-(4-хлор-3-циклопропилфенокси)азетидин
В раствор трет-бутил-3-(4-хлор-3-циклопропилфенокси)азетидин-1-карбоксилата (0,023 г, 0,057 ммоль) в ДХМ (1 мл) добавляли ТФУ (0,088 мл, 1,14 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 18 часов. Смесь разводили ДХМ, вливали в насыщенный водн. раствор NaHCO3 и экстрагировали ДХМ. Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали и выпаривали до сухости с получением неочищенного указанного в заголовке соединения (0,007 г, 35%) в виде бесцветного масла. МС (ИЭР): m/z = 224,1 [М+Н]+.
Этап а) трет-бутил-3-(3-бром-4-хлорфенокси)азетидин-1-карбоксилат
В закрытой пробирке 3-бром-4-хлорфенол (0,1 мг, 0,482 ммоль) и трет-бутил-3-гидроксиазетидин-1-карбоксилат (0,083 г, 0,482 ммоль) растворяли в толуоле (1,5 мл). Пробирку дегазировали аргоном, затем добавляли (трибутилфосфоранилиден)ацетонитрил (CAS RN 157141-27-0, 0,195 мл, 0,723 ммоль) и нагревали реакционную смесь до 100°С в течение 30 мин. Смесь разводили EtOAc, вливали в насыщ. водн. раствор NaHCO3 и экстрагировали водный слой EtOAc. Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью флэш-хроматографии на силикагеле с элюированием градиентом от 0 до 20% EtOAc/гептан с получением указанного в заголовке соединения (0,116 г, 53%) в виде желтого масла. МС (ИЭР: m/z = 308,1 [М-56+Н]+.
Этап b) трет-бутил-3-(4-хлор-3-циклопропилфенокси)азетидин-1-карбоксилат
В пробирке для микроволновой обработки трет-бутил-3-(3-бром-4-хлорфенокси)азетидин-1-карбоксилат (0,075 г, 0,165 ммоль), циклопропилбороновую кислоту (0,021 г, 0,248 ммоль) и K2CO3 (0,046 г, 0,331 ммоль) смешивали в диоксане (1,6 мл). Затем добавляли воду (0,4 мл) и после этого - хлорид бис(трифенилфосфин)палладия (II) (0,012 г, 0,016 ммоль), а реакционную смесь нагревали при 130°С микроволновым облучением в течение 1 часа. Реакционную смесь разводили EtOAc, вливали в воду и экстрагировали EtOAc. Органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью флэш-хроматографии на силикагеле с элюированием градиентом от 0 до 10% EtOAc/гептан с получением указанного в заголовке соединения (0,023 г, 43%) в виде бесцветного масла. МС (ИЭР: m/z = 268,2 [М-56+Н]+.
Способ D9
ВВ197
3-(2-хлор-3-циклопропилфенокси)азетидин, три фтор ацетатная соль
В раствор трет-бутил-3-(2-хлор-3-циклопропилфенокси)азетидин-1-карбоксилата (0,1 г, 0,310 ммоль) в ДХМ (2,5 мл) добавляли ТФУ (0,25 мл) и перемешивали реакционную смесь при комнатной температуре в течение 2 часов. Смесь концентрировали в вакууме с получением неочищенного указанного в заголовке соединения (0,083 г, выход 80%) в виде темно-коричневого масла. МС (ИЭР): m/z = 224,6 [М+H]+.
Этап а) трет-бутил-3-(3-бром-2-хлорфенокси)азетидин-1-карбоксилат
В раствор трет-бутил-3-гидроксиазетидин-1-карбоксилата (0,5 г, 2,89 ммоль) и 3-бром-2-хлорфенола (0,5 г, 2,41 ммоль) в ТГФ (10 мл) добавляли PPh3 (0,948 г, 3,62 ммоль) с последующим добавлением диэтилазодикарбоксилата (0,47 мл, 3,62 ммоль), и перемешивали реакционную смесь при комнатной температуре в течение 12 часов. Смесь очищали с помощью обращенно-фазовой ВЭЖХ с получением указанного в заголовке соединения (0,4 г, 46%) в виде светло-желтого масла. МС (ИЭР): m/z = 308,3 [М-56+H]+.
Этап b) трет-бутил-3-(2-хлор-3-циклопропилфенокси)азетидин-1-карбоксилат
В закрытой пробирке циклопропилбороновую кислоту (0,071 г, 0,830 ммоль,), трет-бутил-3-(3-бром-2-хлорфенокси)азетидин-1-карбоксилат (0,2 г, 0,550 ммоль) и Na2CO3 (0,117 г, 1,1 ммоль) смешивали в 1,4-диоксане (5 мл) и воде (1 мл). Затем добавляли Pd(dppf)Cl2 (0,040 г, 0,060 ммоль) и перемешивали смесь при 110°С в течение 12 часов. Смесь очищали с помощью обращенно-фазовой ВЭЖХ с получением указанного в заголовке соединения (0,12 г, 67%) в виде светло-желтого масла. МС (ИЭР): m/z = 268,1 [М-56+Н]+.
Способ D10
ВВ202
5-(4-пиперидилокси)-2-(трифторметил)бензонитрил, трифторацетат
В раствор трет-бутил-4-[3-циано-4-(трифторметил)фенокси]пиперидин-1-карбоксилата (0,05 г, 0,140 ммоль) в ДХМ (1,5 мл) добавляли ТФУ (0,2 мл) и перемешивали реакционную смесь при комнатной температуре в течение 12 часов. Смесь концентрировали в вакууме с получением неочищенного указанного в заголовке соединения (0,051 г, 98%) в виде светло-коричневого масла. МС (ИЭР): m/z = 271,6 [М+Н]+.
Этап а) трет-бутил-4-[3-бром-4-(трифторметил)фенокси]пиперидин-1-карбоксилат
В раствор 3-бром-4-(трифторметил)фенола (0,5 г, 2,54 ммоль) и 1-Boc-4-гидроксипиперидина (0,512 г, 2,54 ммоль) в ТГФ (8,5 мл) добавляли PPh3 (1 г, 3,82 ммоль) с последующим добавлением диэтилазодикарбоксилата (0,665 мл, 3,82 ммоль), и перемешивали реакционную смесь при комнатной температуре в течение 12 часов. Смесь с помощью флэш-хроматографин на силикагеле с элюированием ПЭ:EtOAc 5:1 с получением указанного в заголовке соединения (0,5 г, 47%) в виде светло-желтого масла. МС (ИЭР): m/z = 370,2 [М-56+H]+.
Этап b) трет-бутил-4-[3-циано-4-(трифторметил)фенокси]пиперидин-1-карбоксилат
В закрытой пробирке трет-бутил-4-[3-бром-4-(трифторметил)фенокси]пиперидин-1-карбоксилат (0,2 г, 0,470 ммоль), Zn(CN)2 (0,111 г, 0,940 ммоль), CuI (0,09 г, 0,470 ммоль) смешивали в ДМФА (10 мл). Затем добавляли Pd(PPh3)4 (0,109 г, 0,090 ммоль) и перемешивали реакционную смесь при 130°С в течение 16 часов. Смесь очищали с помощью обращенно-фазовой ВЭЖХ с получением указанного в заголовке соединения (0,05 г, 29%) в виде бесцветного масла. МС (ИЭР: m/z = 315,5 [М-56+Н]+.
Способ Е
Пример 263
(+)-5-[1-[(4aR,8aS)-3-оксо-4,4a,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-6-карбонил]азетидин-3-ил]окси-2-(трифторметил)бензонитрил

В закрытой пробирке (+)-(4aR,8aS)-6-[3-[3-6ром-4-(трифторметил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он (ВВ 205, 0,2 г, 0,420 ммоль), Zn(CN)2 (0,098 г, 0,840 ммоль), Zn (0,027 г, 0,420 ммоль), dppf (0,232 г, 0,420 ммоль), основание Хюнига (0,108 г, 0,840 ммоль) смешивали в ДМА (10 мл) и дегазировали смесь. Затем добавляли Pd2(dba)3 (76,59 мг, 0,080 ммоль) и перемешивали реакционную смесь при 130°С в течение 16 ч. Смесь очищали с помощью обращенно-фазовой ВЭЖХ с получением указанного в заголовке соединения (0,055 г, 30%) в виде светло-желтого твердого вещества. МС (ИЭР): m/z = 425,3 [М+Н]+.
Способ F
Пример 265
(+)-(4aR,8aS)-6-[3-[3-(2-азаспиро[3.3]гептан-2-ил)-4-(трифторметил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он
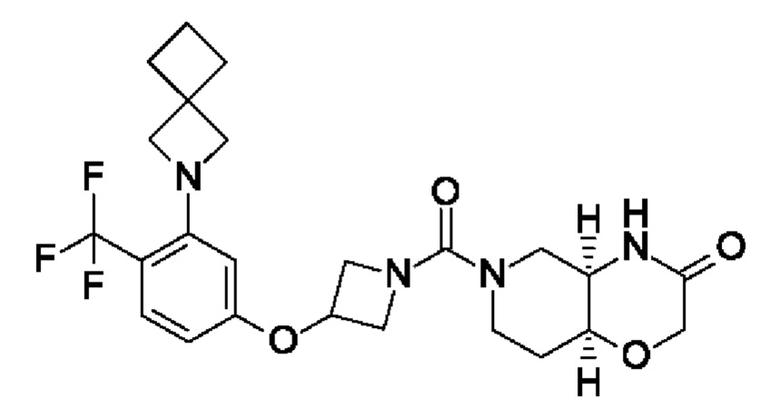
В закрытой пробирке (+)-(4aR,8aS)-6-[3-[3-6ром-4-(трифторметил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он (ВВ203, 0,2 г, 0,420 ммоль), 2-азаспиро[3.3]гептан (CAS RN 665-04-03, 0,117 г, 0,630 ммоль), BINAP (0,052 г, 0,080 ммоль) и K2CO3 (0,173 г, 1,25 ммоль) смешивали в ДМФА (10 мл) и дегазировали смесь. Затем добавляли Pd2(dba)3 (76,59 г, 0,080 ммоль) и перемешивали реакционную смесь при 110°С в течение 16 часов. Реакционную смесь отфильтровывали, фильтрат разводили водой (50 мл) и экстрагировали EtOAc (3 х 20 мл). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью обращенно-фазовой ВЭЖХ с получением указанного в заголовке соединения (0,06 г, 29%) в виде белого твердого вещества. МС (ИЭР): m/z = 495,1 [М+Н]+.
Способ G
Пример 293
(4aR,8aS)-6-(3-(4-гидрокси-2-(трифторметил)фенэтил)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он

Трибромид бора (11,3 мг, 4,29 мкл, 45,3 мкмоль) добавляли в ледяной раствор (4aR,8aS)-6-(3-(4-метокси-2-(трифторметил)фенэтил)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она (пример 216, 20 мг, 45,3 мкмоль) в ДХМ (0,5 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 3 ч. Добавляли насыщенный водный раствор NaHCO3 и экстрагировали смесь AcOEt. Слои разделяли, органический слой сушили над Na2SO4, фильтровали и удаляли растворитель при пониженном давлении. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением указанного в заголовке соединения (19%) в виде бесцветного твердого вещества. МС (ИЭР): m/z = 427,2 [М+Н]+.
Следующие примеры, перечисленные в нижеприведенной таблице, получали по аналогии с процедурой, описанной для получения примера 265, используя указанные промежуточные соединения и/или коммерчески доступные соединения и используя упомянутый способ очистки, такой как обращение-фазовая ВЭЖХ или флэш-хроматография на силикагеле.





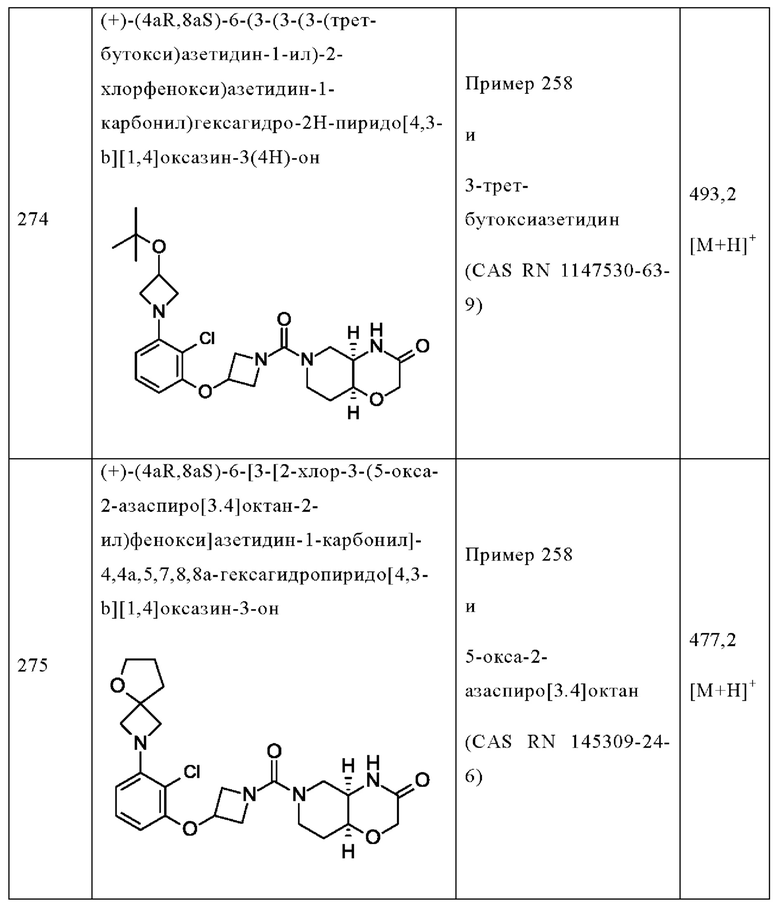


По аналогии со способами, описанными выше в данном документе, нижеприведенные составляющие компоненты получали из соответствующих исходных материалов, указанных в таблице ниже.





ВВ91
4-[[2-пирролидин-1-ил-4-(трифторметил)фенил]метил]пиперидин; соль муравьиной кислоты
Раствор трет-бутил-4-[[2-пирролидин-1-ил-4-(трифторметил)фенил]метил]пиперидин-1-карбоксилата (500 мг, 1,21 ммоль) в 6 М растворе HCl в МеОН (10,0 мл) перемешивали при 20°С в течение 1 ч. Смесь концентрировали в вакууме, очищали в помощью обращенно-фазовой колонки с получением указанного в заголовке соединения в виде оранжевого масла (84,4 мг, выход 21.8%). МС (ИЭР): m/z = 313,2 [М+Н]+.
Этап а) трет-бутил-4-[[2-пирролидин-1-ил-4-(трифторметил)фенил]метилен]пиперидин-1-карбоксилат
В раствор трет-бутил-4-[[2-бром-4-(трифторметил)фенил]метилен]пиперидин-1-карбоксилата (800 мг, 1,90 ммоль; ВВ51, этап с), пирролидина (163 мг, 2,28 ммоль), Ruphos (4,25 мг, 0,010 ммоль) и трет-бутоксида калия (320 мг, 2,86 ммоль) в толуоле (15 мл) добавляли ацетат палладия (II) (1,28 мг, 0,010 ммоль). Смесь перемешивали при 80°С в течение 15 ч в атмосфере N2. Смесь фильтровали и концентрировали в вакууме для удаления толуола. Смесь разводили H2O (40 мл) и три раза экстрагировали EtOAc (по 40 мл каждый раз). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (ПЭ/EtOAc = от 1:0 до 8:1) с получением соединения в виде светло-желтого масла (552 мг, 1,34 ммоль, 36,7%) МС (ИЭР): m/z = 411,1 [M+H]+.
Этап b) трет-бутил-4-[[2-пирролидин-1-ил-4-(трифторметил)фенил]метил]пиперидин-1-карбоксилат
В раствор трет-бутил-4-[[2-пирролидин-1-ил-4-(трифторметил)фенил]метилен]пиперидин-1-карбоксилата (525 мг, 0,660 ммоль) в МеОН (20 мл) добавляли влажный Pd/C (~52 мг) и перемешивали смесь при 20°С в атмосфере Н2 (баллон) в течение 1 ч. Смесь фильтровали и концентрировали в вакууме с получением необходимого соединения в виде бесцветного масла (500 мг), которое использовали на следующем этапе без дополнительной очистки.
ВВ95
3-[2-[2-фтор-6-(трифторметил)фенил]этил]азетидин 4-метилбензолсульфонат
В раствор 3-[2-[2-фтор-6-(трифторметил)фенил]этил]азетидин-1-карбоксилата (50 мг, 144 мкмоль, экв.: 1) в EtOAc (0,8 мл) добавляли 4-метилбензолсульфоновой кислоты моногидрат (29,7 мг, 173 мкмоль, экв.: 1,2) и нагревали смесь с рефлюксом в течение 1,5 часа. Прозрачный, бесцветный раствор оставляли охлаждаться до КТ. Осадок не выпадал. Раствор выпаривали с получение необходимого продукта в виде бесцветной пены. МС (ИЭР): m/z = 248,1 [M-TsOH+H]+.
Этап а) трет-бутил-3-[(Е)-2-[2-фтор-6-(трифторметил)фенил]этенил]азетидин-1-карбоксилат
В ледяной раствор диэтил(2-фтор-4-(трифторметил)бензил)фосфоната (300 мг, 955 мкмоль) в ТГФ (2 мл) добавляли гидрид натрия, 55% в минеральном масле (41,7 мг, 955 мкмоль), и перемешивали смесь при этой температуре в течение 30 минут. К светло-коричневой смеси по каплям добавляли раствор трет-бутил-3-формилазетидин-1-карбоксилата (177 мг, 955 мкмоль) в ТГФ (1 мл). Это приводило к незамедлительному обесцвечиванию реакционной смеси. Перемешивание продолжали в течение 1 часа при температуре ледяной бани, за ним следовало перемешивание при КТ в течение 1,5 часа. Реакционную смесь вливали в воду и этилацетат и разделяли слои. Водный слой дважды экстрагировали этилацетатом. Органические слои один раз промывали солевым раствором, сушили над MgSO4, фильтровали, обрабатывали силикагелем и выпаривали. Соединение очищали с помощью хроматографии на силикагеле на 12 г колонке, используя систему для ЖХСД, с элюированием градиентом н-гептан:этилацетат (от 100: от 0 до 25: 75) с получением необходимого соединения в виде бесцветного твердого вещества (0,108 г; 32,8%). МС (ИЭР): m/z = 290,2 [М-56+Н]+.
Этап b) трет-бутил-3-[2-[2-фтор-6-(трифторметил)фенил]этил]азетидин-1-карбоксилат
В раствор трет-бутил-(Е)-3-(2-фтор-4-(трифторметил)стирил)азетидин-1-карбоксилата (105 мг, 304 мкмоль) в МеОН (1 мл) и этилацетате (1 мл) добавляли Pd/C 10% (11 мг, 304 мкмоль) и перемешивали смесь в атмосфере водорода при 1,7 бар и КТ в течение 30 минут. Суспензию фильтровали. Фильтрат выпаривали с получением необходимого соединения в виде бесцветного масла (0,104 г; 98,5%). МС (ИЭР): m/z = 292,2 [М-56+Н]+.
ВВ96
4-((2-хлор-4-фторфенокси)метил)азепан гидрохлорид
В раствор трет-бутил-4-((2-хлор-4-фторфенокси)метил)азепан-1-карбоксилата (620 мг, 1,73 ммоль) в ДХМ (7,5 мл) добавляли 2 М HCl в эфире (10 мл, 20 ммоль) и перемешивали реакционную смесь в течение ночи при КТ. Смесь концентрировали в вакууме, неочищенный материал собирали в виде белого твердого вещества (490 мг, 1,67 ммоль, 96,1%) и непосредственно использовали на следующем этапе. ЖХ-МС (ИЭР): m/z: 258,2 [М+Н]+.
Этап а) трет-бутил-4-((2-хлор-4-фторфенокси)метил)азепан-1-карбоксилат
В 25 мл пробирке для сульфирования с четырьмя горлышками в атмосфере аргона трет-бутил-4-(гидроксиметил)азепан-1-карбоксилат (480 мг, 2,09 ммоль) растворяли в ТГФ (10 мл). После этого добавляли 2-хлор-4-фторфенол (337 мг, 251 мкд, 2,3 ммоль) и трифенилфосфин (604 мг, 2,3 ммоль) и перемешивали прозрачный раствор в течение 5 мин при КТ. Смесь охлаждали до 0°С и частями добавляли ДЭАД (401 мг, 365 мкл, 2,3 ммоль) в течение 10 мин. Смесь перемешивали в течение 1 ч при 0°С, затем в течение ночи при КТ. Смесь вливали в EtOAc (50 мл), промывали водой (2х25 мл), органическую фазу промывали 1 М NaOH (3х25 мл), солевым раствором (20 мл), сушили с Na2SO4, фильтровали и концентрировали в вакууме. Остаток растворяли в н-гептане/диэтилэфире и перемешивали смесь в течение 30 мин, фильтровали осадок ТРРО и концентрировали неочищенное вещество в вакууме. Неочищенный материал адсорбировали на Isolate® и очищали с помощью колоночной флэш-хроматографии (0 - 30% EtOAc/гептан) на силикагеле (50 г) с получением необходимого продукта (630 мг, 1,76 ммоль, 84,1%) в виде желтого масла. ЖХ-МС (ИЭР): m/z: 302,1 [М-56+Н]+
ВВ97
4-[[4-(трифторметил)фенил]метил]азепан гидрохлорид
В раствор трет-бутил-4-(4-(трифторметил)бензил)азепан-1-карбоксилата (88 мг, 246 мкмоль, экв.: 1) в ДХМ (1,5 мл) добавляли 2 М HCl в эфире (3,08 мл, 6,16 ммоль) и перемешивали реакционную смесь в течение ночи при комнатной температуре. Смесь концентрировали в вакууме, неочищенный материал собирали в виде белого твердого вещества (71 мг, 0,24 ммоль, 98,2%) и непосредственно использовали на следующем этапе. ЖХ-МС (ИЭР): m/z: 258,2 [М+Н]+
Этап а: Трифенил(4-(трифторметил)бензил)фосфоний бромид
Трифенилфосфин (1,84 г, 7 ммоль) и 1-(бромметил)-4-(трифторметил)бензол (1,61 г, 6,74 ммоль) растворяли в ксилоле (35 мл). Реакционную смесь нагревали до рефлюкса при 155°С в течение 3,5 ч, а затем охлаждали до комнатной температуры. Выпавшее в осадок белое кристаллическое твердое вещество собирали путем фильтрации, промывали диэтиловым эфиром и сушили в вакууме. Конечное соединение (3,30 г, 6,58 ммоль, выход 97,7%) получали в виде белого
порошка и непосредственно использовали на следующем этапе. ЖХ-МС (ИЭР): m/z: 421,2 [М+Н]+
Этап b: трет-бутил-(Е)-4-(4-(трифторметил)бензилиден)азепан-1-карбоксилат
Суспензию гидрида натрия (88,6 мг, 2,22 ммоль) в ДМФА (7,5 мл) охлаждали на ледяной бане, затем добавляли трифенил(4-(трифторметил)бензил)фосфоний бромид (1,11 г, 2,22 ммоль). Суспензию перемешивали при 0°С в течение 5 мин, затем в течение 25 мин при КТ. Добавляли трет-бутил-4-оксоазепан-1-карбоксилат (315 мг, 1,48 ммоль), а полученную в результате смесь перемешивали при 80°С в течение 28 ч. Смесь концентрировали в вакууме, разводили водой (50 мл) и EtOAc (40 мл) и экстрагировали EtOAc (3×30 мл). Объединенные органические фракции промывали водой, 10% раствором LiCl, сушили над Na2SO4 и концентрировали в вакууме. Остаточное масло обрабатывали Et2O, чтобы осадить трифенилфосфоксид, который отфильтровывали. Раствор концентрировали в вакууме, а остаток очищали с помощью колоночной флэш-хроматографии (0-35% EtOAc/гептан) на силикагеле (50 г) с получением необходимого продукта (92 мг, 259 мкмоль, выход 17,5%) в виде желтого масла. ЖХ-МС (ИЭР): m/z: 300,2 [М-56+Н]+
Этап с: трет-бутил-4-(4-(трифторметил)бензил)азепан-1-карбоксилат
Раствор трет-бутил-(Е)-4-(4-(трифторметил)бензилиден)азепан-1-карбоксилата (90 мг, 253 мкмоль) растворяли в МеОН (2,5 мл). Реакционный сосуд опустошали и снова наполняли аргоном 5 раз. В атмосфере аргона добавляли Pd-C (13,5 мг, 12,7 мкмоль) и три раза меняли атмосферу на водородную. Реакцию перемешивали в атмосфере водорода при 1 бар в течение 24 ч. Атмосферу меняли на аргоновую и фильтровали реакционную смесь через слой дикалита. Фильтрационный осадок промывали метанолом. Фильтрат концентрировали в вакууме с получением необходимого продукта (89 мг, 249 мкмоль, выход 98,3%) в виде бесцветного масла, которое использовали без дополнительной очистки. ЖХ-МС (ИЭР): m/z: 302,2 [М-56+Н]+
ВВ98
3-((2-хлор-4-(трифторметил)фенил)тио)азетидин 2,2,2-трифторацетат
трет-бутил-3-((2-хлор-4-(трифторметил)фенил)тио)азетидин-1-карбоксилат (110 мг, 299 мкмоль) растворяли в ДХМ (2 мл) и добавляли ТФУ (273 мг, 184 мкл, 2,39 ммоль). Реакционную смесь перемешивали при КТ в течение 3 ч. Летучие вещества удаляли в вакууме с получением 110 мг светло-желтого твердого вещества (96%). МС (ИЭР): m/z = 268,1 [M+H]+.
Этап а) трет-бутил-3-((2-хлор-4-(трифторметил)фенил)тио)азетидин-1-карбоксилат
В 20 мл стеклянной пробирке в раствор 2-хлор-4-(трифторметил)бензолтиола (440 мг, 2,07 ммоль) в сухом ТГФ (6 мл) добавляли 1 М раствор трет-бутоксида калия в ТГФ (2,17 мл, 2,17 ммоль) и перемешивали желтую реакционную смесь при КТ в течение 15 мин с последующим добавлением трет-бутил-3-бромазетидин-1-карбоксилата (489 мг, 2,07 ммоль). Затем реакционную смесь перемешивали при КТ в течение 5 ч и в течение ночи при 70°С. Неочищенную реакцию разводили EtOAc и экстрагировали H2O, органическую фазу собирали, а водную фазу обратно экстрагировали EtOAc. Объединенные органические фазы сушили над Na2SO4 и выпаривали до сухости. Остаток очищали с помощью хроматографии (SiO2, н-гептан/EtOAc (от 0 до 40% за 40 мин) с получением продукта в виде вязкого масла (467 мг, 61%). МС (ИЭР): m/z = 312,1 [М-56]+.
ВВ99
3-((2-хлор-4-(трифторметил)фенил)сульфонил)азетидин 2,2,2-трифторацетат
трет-бутил-3-((2-хлор-4-(трифторметил)фенил)сульфонил)азетидин-1-карбоксилат (100 мг, 250 мкмоль) растворяли в ДХМ и добавляли ТФУ (228 мг, 154 мкл, 2 ммоль). Реакционную смесь перемешивали при КТ в течение 8 ч. Летучие вещества удаляли в вакууме с получением необходимого соединения в виде светло-желтого твердого вещества (102, мг, 98%). МС (ИЭР): m/z = 300,0 [M+H]+.
Этап а) трет-бутил-3-((2-хлор-4-(трифторметил)фенил)тио)азетидин-1-карбоксилат
В 20 мл стеклянной пробирке в раствор 2-хлор-4-(трифторметил)бензолтиола (440 мг, 2,07 ммоль) в сухом ТГФ (6 мл) добавляли 1 М раствор трет-бутоксида калия в ТГФ (2,17 мл, 2,17 ммоль) и перемешивали желтую реакционную смесь при КТ в течение 15 мин с последующим добавлением трет-бутил-3-бромазетидин-1-карбоксилата (489 мг, 2,07 ммоль). Затем реакционную смесь перемешивали при КТ в течение 5 ч и в течение ночи при 70°С. Неочищенную реакцию разводили EtOAc и экстрагировали H2O, органическую фазу собирали, а водную фазу обратно экстрагировали EtOAc. Объединенные органические фазы сушили над Na2SO4 и выпаривали до сухости. Остаток очищали с помощью хроматографии (SiO2, н-гептан/EtOAc (от 0 до 40% за 40 мин) с получением необходимого продукта в виде вязкого масла (467 мг, 61%). МС (ИЭР: m/z = 312,1 [М-56+Н]+.
Этап b) трет-бутил-3-((2-хлор-4-(трифторметил)фенил)сульфонил)азетидин-1-карбоксилат
м-ХПБК (347 мг, 1,41 ммоль) одной частью добавляли в перемешиваемый раствор трет-бутил-3-((2-хлор-4-(трифторметил)фенил)тио)азетидин-1-карбоксилата (345 мг, 938 мкмоль) в ДХМ (6 мл) на ледяной бане. Реакцию перемешивали при КТ в течение 20 мин. Реакционную смесь вливали в 5 мл насыщенного раствора Na2CO3 и дважды экстрагировали ДХМ (по 20 мл каждый раз). Органические слои объединяли, промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Неочищенный материал очищали с помощью препаративной ВЭЖХ (YMC-Triart С 18, АЦН/H2O+0,1% НСООН) с получением продукта в виде белого порошка (253 мг, 67,5%) МС (ИЭР): m/z = 344,0 [M-56+H]+.
BB100
3-((2-хлор-4-(трифторметил)фенил)сульфинил)азетидин 2,2,2-трифторацетат
трет-бутил-3-((2-хлор-4-(трифторметил)фенил)сульфинил)азетидин-1-карбоксилат (50 мг, 130 мкмоль) растворяли в ДХМ (1,5 мл), добавляли ТФУ (149 мг, 100 мкл, 1,3 ммоль) и перемешивали реакционную смесь при КТ в течение 8 ч.
Летучие вещества удаляли в вакууме с получением соединения в виде белого твердого вещества (51 мг, 98%). МС (ИЭР): m/z = 284,1 [M+H]+.
Этап а) трет-бутил-3-((2-хлор-4-(трифторметил)фенил)тио)азетидин-1-карбоксилат
Это соединение готовили по аналогии с ВВ99, этап а, и использовали на следующем этапе без дополнительной очистки.
Этап b) трет-бутил-3-((2-хлор-4-(трифторметил)фенил)сульфинил)азетидин-1-карбоксилат
Промежуточный сульфоксид выделяли из реакции синтеза соответствующего сульфонового составляющего компонента ВВ99, этап b. Продукт получали в виде белого лиофилизированного порошка (50 мг, 13,9%) МС (ИЭР): m/z = 328,1 [М-56+H]+.
ВВ101
3-(((2-хлор-4-(трифторметил)фенил)тио)метил)азетидин 2,2,2-трифторацетат
В раствор трет-бутил-3-(((2-хлор-4-(трифторметил)фенил)тио)метил)азетидин-1-карбоксилата (0,200 г, 524 мкмоль) в ДХМ (3 мл) добавляли ТФУ (478 мг; 323 мкл, 4,19 ммоль) и перемешивали реакционную смесь при КТ в течение 3 ч. Летучие вещества удаляли в вакууме с получением соединения в виде светло-желтого масла, которое использовали на следующем этапе без дополнительной очистки (267 мг). МС (ИЭР): m/z = 282,2 [M+H]+.
Этап а) трет-бутил-3-[[2-хлор-4-(трифторметил)фенил]сульфанилметил]азетидин-1-карбоксилат
В раствор 2-хлор-4-(трифторметил)бензолтиола (0,400 г, 1,88 ммоль) в сухом ТГФ (6 мл) добавляли 1 М раствор трет-бутоксида калия в ТГФ (1,98 мл, 1,98 ммоль) и перемешивали непрозрачную реакционную смесь при КТ в течение 15 мин с последующим добавлением трет-бутил-3-(бромметил)азетидин-1-карбоксилата (471 мг, 1,88 ммоль). Затем реакционную смесь перемешивали при КТ в течение 19 ч. Неочищенную реакцию разводили EtOAc и экстрагировали водн. 1 М раствором NaHCO3, органическую фазу собирали, а водный слой обратно экстрагировали EtOAc. Объединенные органические фазы сушили над Na2SO4 и выпаривали до сухости с получением неочищенного продукта, который использовали на следующем этапе без дополнительной очистки (762 мг). МС (ИЭР: m/z = 326,1 [М-56+Н]+.
ВВ102
3-((2-фтор-6-(трифторметил)бензил)сульфонил)азетидин 2,2,2-трифторацетат
трет-бутил-3-((2-фтор-6-(трифторметил)фенил)метилсульфонил)азетидин-1-карбоксилат (0,047 г, 118 мкмоль) растворяли в ДХМ (0,5 мл) и добавляли ТФУ (108 мг, 72,9 мкл, 946 мкмоль). Реакционную смесь перемешивали при КТ в течение 2 ч. Летучие вещества удаляли в вакууме с получением соединения в виде желтого масла (56 мг), которое использовали на следующем этапе без дополнительной очистки. МС (ИЭР): m/z = 298,2 [М+Н]+.
Этап а) трет-бутил-3-((2-фтор-6-(трифторметил)бензил)тио)азетидин-1-карбоксилат
В раствор трет-бутил-3-меркаптоазетидин-1-карбоксилата (0,400 г, 2,11 ммоль) в сухом ТГФ (5 мл) добавляли 1 М раствор трет-бутоксида калия в ТГФ (2,22 мл, 2,22 ммоль) и перемешивали реакционную смесь при КТ в течение 15 мин с последующим добавлением 2-(бромметил)-1-фтор-3-(трифторметил)бензола (CAS RN 239087-08-2). Затем реакционную смесь перемешивали при КТ в течение 14 ч. Неочищенную реакцию разводили EtOAc и экстрагировали водн. 1 М раствором NaHCO3, органическую фазу собирали, а водный слой обратно экстрагировали EtOAc. Объединенные органические фазы сушили над NaSO4 и выпаривали до сухости с получением неочищенного продукта (805 мг), который использовали на следующем этапе без дополнительной очистки. МС (ИЭР: m/z = 310,2 [M-56+H]+.
Этап b) трет-бутил-3-[[2-фтор-6-(трифторметил)фенил]метилсульфонил]азетидин-1-карбоксилат
3-хлорпероксибензойную кислоту (283 мг, 1,64 ммоль) одной частью добавляли в перемешиваемый раствор трет-бутил-3-((2-фтор-6-(трифторметил)бензил)тио)азетидин-1-карбоксилата (0,300 г, 821 мкмоль) в ДХМ (5 мл) на ледяной бане. Реакционную смесь перемешивали при КТ в течение 15 мин и вливали в 5 мл насыщенного водного раствора NaHCO3 и дважды экстрагировали ДХМ (по 10 мл каждый раз). Органические слои объединяли, промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Неочищенный материал очищали с помощью колоночной флэш-хроматографии (силикагель, 20 г, от 0% до 100% EtOAc в н-гептане) с получением необходимого продукта в виде бесцветного масла (47 мг, 15%). МС (ИЭР): m/z = 415,1 [M+NH4]+.
ВВ103
3-((2-фтор-6-(трифторметил)бензил)сульфинил)азетидин 2,2,2-трифторацетат
трет-бутил-3-[[2-фтор-6-(трифторметил)фенил]метилсульфинил]азетидин-1-карбоксилат (0,086 г, 225 мкмоль) растворяли в ДХМ (1 мл) и добавляли ТФУ (206 мг, 139 мкл, 1,8 ммоль). Реакционную смесь перемешивали при КТ в течение 2 ч. Летучие вещества удаляли в вакууме с получением соединения в виде желтого масла (93 мг), которое использовали на следующем этапе без дополнительной очистки. МС (ИЭР): m/z = 282,2 [М+Н]+.
Этап а) трет-бутил-3-[[2-фтор-6-(трифторметил)фенил]метилсульфинил]азетидин-1-карбоксилат
Промежуточный сульфоксид выделяли из реакции синтеза ВВ102, этап b, в виде бесцветного масла (86 мг, 28%). МС (ИЭР): m/z = 404,1 [M+Na]+.
ВВ104
3-(((2-хлор-4-(трифторметил)фенил)сульфонил)метил)азетидин 2,2,2-трифторацетат
трет-бутил-3-(((2-хлор-4-(трифторметил)фенил)сульфонил)метил)азетидин-1-карбоксилат (0,145 г, 350 мкмоль) растворяли в ДХМ (2 мл) и добавляли ТФУ (320 мг, 216 мкл, 2,8 ммоль). Реакционную смесь перемешивали при КТ в течение 2 ч. Летучие вещества удаляли в вакууме с получением соединения в виде желтого масла (181 мг), которое использовали на следующем этапе без дополнительной очистки. МС (ИЭР): m/z = 314,1 [М+Н]+.
Этап а) трет-бутил-3-(((2-хлор-4-(трифторметил)фенил)сульфонил)метил)азетидин-1-карбоксилат
3-хлорпероксибензойную кислоту (352 мг, 1,57 ммоль) частями добавляли в перемешиваемый раствор трет-бутил-3-(((2-хлор-4-(трифторметил)фенил)тио)метил)азетидин-1-карбоксилата (ВВ101, этап а) (0,300 г, 786 мкмоль) в ДХМ (5 мл) на ледяной бане. Реакционную смесь перемешивали при КТ в течение 15 мин и вливали в 5 мл насыщенного водного раствора NaHCO3 и дважды экстрагировали ДХМ (по 10 мл каждый раз). Органические слои объединяли, промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Неочищенный материал очищали с помощью флэш-хроматографии (силикагель, 20 г, от 0% до 100% EtOAc в н-гептане) с получением необходимого продукта в виде бесцветного масла (145 мг, 45%). МС (ИЭР: m/z = 314,0 [M-56+H]+.
ВВ105
3-(((2-хлор-4-(трифторметил)фенил)сульфинил)метил)азетидин 2,2,2-трифторацетат
трет-бутил-3-(((2-хлор-4-(трифторметил)фенил)сульфинил)метил)азетидин-1-карбоксилат (0,086 г, 216 мкмоль) растворяли в ДХМ (1 мл) и добавляли ТФУ (197 мг, 133 мкл, 1,73 ммоль). Реакционную смесь перемешивали при КТ в течение 2 ч. Летучие вещества удаляли в вакууме с получением соединения в виде желтого масла (99 мг), которое использовали на следующем этапе без дополнительной очистки. МС (ИЭР): m/z = 298,1 [М+Н]+.
Этап а) трет-бутил-3-(((2-хлор-4-(трифторметил)фенил)сульфинил)метил)азетидин-1-карбоксилат
Промежуточный сульфоксид выделяли из реакции синтеза ВВ104, этап а. Необходимый продукт получали в виде желтого масла (80 мг, 25,6%). МС (ИЭР): m/z = 398,1 [М+Н]+.
ВВ106
3-((2-фтор-4-(трифторметил)бензил)тио)азетидин 2,2,2-трифторацетат
В раствор трет-бутил-3-((2-фтор-4-(трифторметил)бензил)тио)метил)азетидин-1-карбоксилата (0,282 г, 772 мкмоль) в ДХМ (3 мл) добавляли ТФУ (880 мг; 595 мкл, 7,72 ммоль) и перемешивали реакционную смесь при КТ в течение 3 ч. Летучие вещества удаляли в вакууме с получением необходимого соединения в виде бесцветного масла (302 мг), которое использовали на следующем этапе без дополнительной очистки. МС (ИЭР): m/z =266,2 [М+H]+.
Этап а) трет-бутил-3-[[2-фтор-4-(трифторметил)фенил]метилсульфанил]азетидин-1-карбоксилат
В раствор трет-бутил-3-меркаптоазетидин-1-карбоксилата (0,200 г, 1,06 ммоль) в сухом ТГФ (2 мл) добавляли 1 М раствор трет-бутоксида калия в ТГФ (1,11 мл, 1,11 ммоль) и перемешивали реакционную смесь при КТ в течение 15 мин с последующим добавлением 1-(бромметил)-2-фтор-4-(трифторметил)бензола (272 мг, 1,06 ммоль, CAS RN 239087-07-1). Затем реакционную смесь перемешивали при КТ в течение 14 ч. Неочищенную реакцию разводили EtOAc и экстрагировали водн. 1 М раствором NaHCO3, органическую фазу собирали, а водный слой обратно экстрагировали EtOAc. Объединенные органические фазы сушили над NaSO4, выпаривали до сухости и очищали с помощью флэш-хроматографии (силикагель, 20 г, от 0% до 80% EtOAc в н-гептане) с получением необходимого продукта в виде бесцветного масла (288 мг, 75%). МС (ИЭР: m/z = 310,2 [М-56+Н]+.
По аналогии с ВВ84 следующие промежуточные соединения получали из соответствующих коммерчески доступных исходных материалов.

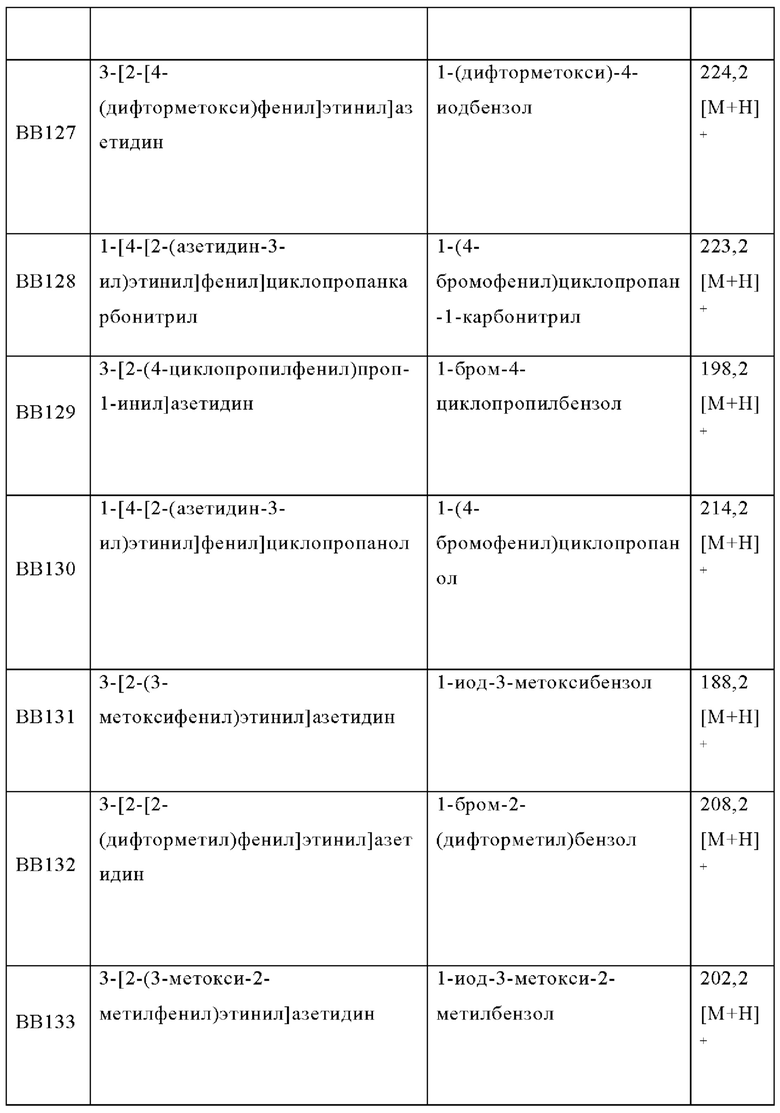
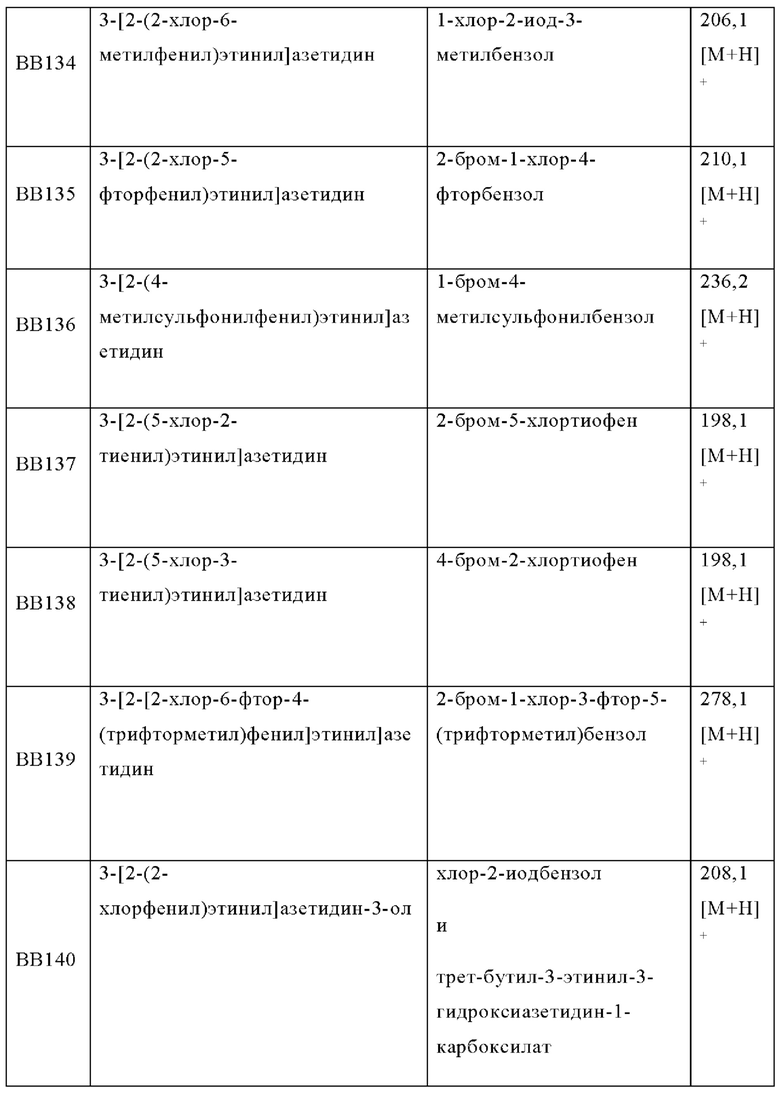

По аналогии с ВВ18 следующие промежуточные соединения получали из соответствующих коммерчески доступных исходных материалов.

ВВ149
1-[2-(азетидин-3-ил)этинил]циклопентанол гидрохлорид
В раствор трет-бутил-3-[2-(1-гидроксициклопентил)этинил]азетидин-1-карбоксилата (0,02 г, 0,075 ммоль) в диоксане (0,5 мл) добавляли 4 М HCl в диоксане (0,094 мл, 0,377 ммоль) и перемешивали реакционную смесь при КТ в течение 18 ч. Смесь выпаривали до сухости, а остаток растирали в диизопропиловом эфире, отфильтровывали и дополнительно сушили в условиях высокого вакуума с получением указанного в заголовке соединения в виде белого твердого вещества в виде гидрохлоридной соли (0,013 г, 87%). МС (ИЭР): m/z =166,1 [М+Н]+.
Этап а) трет-бутил-3-[2-(1-гидроксициклопентил)этинил]азетидин-1-карбоксилат
В раствор трет-бутил-3-этинилазетидин-1-карбоксилата (0,2 г, 1,1 ммоль) в ТГФ (6,5 мл) при -78°С по каплям добавляли nBuLi (0,759 мл, 1,21 ммоль) и перемешивали реакционную смесь при этой температуре в течение 1 ч. Затем по каплям добавляли циклопентанон (0,107 мл, 1,21 ммоль) в ТГФ (3 мл) и перемешивали смесь при -78°С в течение 2 ч. Смесь оставляли нагреваться до 0°С, вливали в насыщ. водный раствор NH4OH и экстрагировали EtOAc. Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали и выпаривали. Остаток очищали с помощью флэш-хроматографии на силикагеле с элюированием градиентом EtOAc:n-гептан (от 0 до 100%) с получением указанного в заголовке соединения в виде светло-желтого масла (0,020 г, 7%). МС (ИЭР: m/z = 192,2 [M-56-18+H]+.
ВВ150
4-[3-пиразол-1-ил-5-(трифторметил)фенокси]пиперидин формиат
Смесь трет-бутил-4-[3-пиразол-1-ил-5-(трифторметил)фенокси]пиперидин-1-карбоксилата (400,0 мг, 0,970 ммоль) и ТФУ (1,0 мл, 0,970 ммоль) в ДХМ (10 мл) перемешивали при 20°С в течение 12 ч. Смесь очищали с помощью препаративной ВЭЖХ (АЦН и вода с содержанием 0,225% об./об. МК) с получением необходимого продукта (300 мг, 94,4%) в виде бесцветной смолы. МС (ИЭР): m/z = 312,1 [М+Н]+.
Этап а: трет-бутил-4-[3-бром-5-(трифторметил)фенокси]пиперидин-1-карбоксилат
В раствор 3-бром-5-(трифторметил)фенола (2,0 г, 8,3 ммоль), 1-ВОС-4-гидроксипиперидина (1,84 г, 9,13 ммоль, CAS RN 106-52-5) и PPh3 (2,61 г, 9,96 ммоль) в ТГФ (32,6 мл) добавляли диизопропилазодикарбоксилат (1,96 мл, 9,96 ммоль) и перемешивали смесь при 20°С в течение 15 ч. Смесь концентрировали в вакууме. Остаток очищали с помощью препаративной ВЭЖХ (АЦН и вода с содержанием 0,225% об./об. МК) и концентрировали в вакууме с получением необходимого продукта (2,6 г, выход 73,9%) в виде светло-желтого масла. МС (ИЭР: m/z = 367.9 [М-56+Н]+.
Этап b) трет-бутил-4-[3-пиразол-1-ил-5-(трифторметил)фенокси]пиперидин-1-карбоксилат
Смесь трет-бутил-4-[3-бром-5-(трифторметил)фенокси]пиперидин-1-карбоксилата (500,0 мг, 1,18 ммоль), пиразола (160,47 мг, 2,36 ммоль), CuI (22,37 мг, 0,120 ммоль), карбоната цезия (1152 мг, 3,54 ммоль) и N,N'-диметилэтилендиамина (519,15 мг, 5,89 ммоль) в ДМФА (5 мл) перемешивали при 110°С в течение 12 ч. Смесь вливали в H2O воду (30 мл) и три раза экстрагировали EtOAc (50 мл). Объединенный органический слой промывали раствором аммиака (10 мл), солевым раствором (50 мл), сушили над Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме с получением необходимого продукта (400 мг. выход 82,5%) в виде светло-желтого масла. МС (ИЭР: m/z = 356,2 [М-56+Н]+.
ВВ151
4-[[2-(2,2,2-трифторэтокси)-4-(трифторметил)фенил]метил]пиперидин
Смесь 4-[[2-(2,2,2-трифторэтокси)-4-(трифторметил)фенил]метилен]пиперидина (250,0 мг, 0,740 ммоль) и Pd/C (50,0 мг, 10 масс. %) в ТГФ (10 мл) перемешивали при 20°С в течение 12 ч в атмосфере H2 (1520 мм рт. ст.). Смесь фильтровали и концентрировали в вакууме с получением необходимого соединения (240 мг, 95,4%) в виде светло-коричневой смолы. МС (ИЭР): m/z = 342.1 [М+Н]+.
Этап а) трет-бутил-4-(п-толилсульфонилгидразоно)пиперидин-1-карбоксилат
В раствор 4-метилбензолсульфонилгидразина (9,35 г, 50,19 ммоль, CAS RN 1576-35-8) в МеОН (100 мл) добавляли 1-BOC-4-пиперидон (10,0 г, 50,19 ммоль, CAS RN 17502-28-8) и перемешивали смесь при 25°С в течение 12 ч. Смесь концентрировали с получением необходимого продукта (18,4 г, 99,8%) в виде грязно-белого твердого вещества. МС (ИЭР): m/z = 368,2 [М+Н]+.
Этап b) 2-(2,2,2-трифторэтокси)-4-(трифторметил)бензальдегид
Смесь NaH (187,39 мг, 60% дисперсия в минеральном масле, 4,68 ммоль) в 2,2,2-трифторэтаноле (16,67 мл, 228,74 ммоль, CAS RN75-89-8) перемешивали при 0°С. Охлаждающую баню удаляли и перемешивали смесь при 20°С в течение 2 ч, а затем добавляли 2-фтор-4-(трифторметил)бензальдегид (1,0 г, 5,21 ммоль, CAS RN 763-93-9) и перемешивали смесь при 20°С в течение 12 ч. Смесь вливали в H2O (30 мл) и дважды экстрагировали EtOAc (по 30 мл каждый раз). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме с получением необходимого продукта (1,2 г, выход 84,7%) в виде светло-желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6) δ 10,44-10,34 (м, 1Н), 7,93 (д, J=8,1 Гц, 1Н), 7,75 (с, 1Н), 7,55 (д, J = 8,1 Гц, 1Н), 5,11 (кв, J = 8,7 Гц, 2Н).
Этап с) трет-бутил-4-[2-(2,2,2-трифторэтокси)-4-(трифторметил)бензоил]пиперидин-1-карбоксилат
Смесь 2-(2,2,2-трифторэтокси)-4-(трифторметил)бензальдегида (1000,0 мг, 3,67 ммоль), трет-бутил-4-(п-толилсульфонилгидразоно)пиперидин-1-карбоксилата (1350,3 мг, 3,67 ммоль) и карбоната цезия (1795,9 мг, 5,51 ммоль) в 1,4-диоксане (30 мл) перемешивали при 110°С в течение 12 ч в атмосфере N2. Смесь вливали в H2O (50 мл) и три раза экстрагировали EtOAc (по 50 мл каждый раз). Объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме, а остаток очищали с помощью препаративной ВЭЖХ (MeCN и вода с содержанием 0,225% об./об. МК) с получением необходимого продукта (980 мг, выход 58,6%) в виде светло-желтой смолы. МС (ИЭР: m/z = 400,1 [М-56+Н]+.
Этап d) трет-бутил-4-[гидрокси-[2-(2,2,2-трифторэтокси)-4-(трифторметил)фенил]метил]пиперидин-1-карбоксилат
В раствор трет-бутил-4-[2-(2,2,2-трифторэтокси)-4-(трифторметил)бензоил]пиперидин-1-карбоксилата (900,0 мг, 1,98 ммоль) в МеОН (45 мл) добавляли NaBH4 (149,54 мг, 3,95 ммоль) при 0°С и перемешивали смесь при 20°С в течение 12 ч. Смесь очищали с помощью препаративной ВЭЖХ (MeCN и вода с содержанием 0,225% об./об. МК) (650 мг, 71,9%) в виде светло-желтого масла. МС (ИЭР: m/z = 384,0 [М-56-ОН+Н]+.
Этап е) 4-[[2-(2,2,2-трифторэтокси)-4-(трифторметил)фенил]метилен]пиперидин
Смесь трет-бутил-4-[гидрокси-[2-(2,2,2-трифторэтокси)-4-(трифторметил)фенил]метил]пиперидин-1-карбоксилата (400,0 мг, 0,870 ммоль) и MsOH (840,43 мг, 8,74 ммоль) в ДХМ (4 мл) перемешивали при 40°С в течение 24 ч. Смесь вливали в насыщенный водный раствор Na2CO3 (5 мл) и три раза экстрагировали EtOAc (по 10 мл каждый раз). Объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме с получением необходимого соединения в виде светло-желтого масла (260 мг, 76,2%). МС (ИЭР): m/z = 340,1 [М+Н]+.
ВВ152
4-[3-(1,2,4-триазол-1-ил)-5-(трифторметил)фенокси]пиперидин трифторацетат
В смесь трет-бутил-4-[3-(1,2,4-триазол-1-ил)-5-(трифторметил)фенокси]пиперидин-1-карбоксилата (240,0 мг, 0,580 ммоль) в ДХМ (10 мл) добавляли ТФУ (1,0 мл). Смесь перемешивали при 20°С в течение 12 ч, а затем концентрировали в вакууме с получением соли 4-[3-(1,2,4-триазол-1-ил)-5-(трифторметил)фенокси]пиперидин 2,2,2-трифторуксусной кислоты (240 мг, 96,7%) в виде светло-желтой смолы. МС (ИЭР): m/z = 313,1 [М+Н]+.
Этап а) трет-бутил-4-[3-(1,2,4-триазол-1-ил)-5-(трифторметил)фенокси]пиперидин-1-карбоксилат
Смесь трет-бутил-4-[3-бром-5-(трифторметил)фенокси]пиперидин-1-карбоксилата (500,0 мг, 1,18 ммоль, ВВ98, промежуточное соединение а), 1,2,4-триазола (162,8 мг, 2,36 ммоль) и CuI (22,37 мг, 0,120 ммоль) в ДМФА (5 мл) перемешивали при 110°С в течение 12 ч. Смесь вливали в H2O (20 мл) и три раза экстрагировали EtOAc (по 30 мл каждый раз). Объединенные органические слои промывали раствором аммиака (20 мл), солевым раствором (20 мл, три раза), сушили над Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме, а остаток очищали с помощью колоночной хроматографии (ПЭ: ЭА = 50: 1 ~ 3: 1) с получением необходимого продукта (240 мг, 49,4%) в виде светло-желтого твердого вещества. МС (ИЭР): m/z = 357,1 [М-56+Н]+.
ВВ153
3-[4-хлор-3-(трифторметил)фенокси]азетидин трифторацетат
В раствор трет-бутил-3-[4-хлор-3-(трифторметил)фенокси]азетидин-1-карбоксилата (300,0 мг, 0,530 ммоль) в ДХМ (7,5 мл) добавляли ТФУ (1,04 мл) при 0°С и перемешивали смесь при 20°С в течение 2 ч. Смесь концентрировали с получением указанного в заголовке соединения в виде желтого масла (280 мг, 97%). МС (ИЭР): m/z =252,0 [М+Н]+.
Этап а) трет-бутил-3-[4-хлор-3-(трифторметил)фенокси]азетидин-1-карбоксилат
В раствор 2-хлор-5-гидроксибензотрифторида (1 г, 5,1 ммоль CAS RN 6294-93-5), трет-бутил-3-гидроксиазетидин-1-карбоксилата (0,97 г, 5,6 ммоль CAS RN 141699-55-0) и трифенилфосфина (1,6 г, 6,11 ммоль) в ТГФ (20 мл) добавляли диизопропилазодикарбоксилат (1,2 мл, 6,11 ммоль) и перемешивали смесь при 20°С в течение 15 ч. Смесь концентрировали и очищали с помощью обращенно-фазовой хроматографии (MeCN и вода с содержанием 0,225% об./об. МК) с получением указанного в заголовке соединения (820 мг, 28,7%) в виде коричневого твердого вещества. МС (ИЭР: m/z = 295,9 [М-56+Н]+.
ВВ154
4-(4-хлор-3-пиразол-1-ил-фенокси)пиперидин трифторацетат
В раствор трет-бутил-4-(4-хлор-3-пиразол-1-ил-фенокси)пиперидин-1-карбоксилата (260,0 мг, 0,690 ммоль) в ДХМ (5,38 мл) добавляли ТФУ (1,34 мл, 17,46 ммоль) при 0°С и перемешивали смесь при 20°С в течение 1 ч. Смесь концентрировали с получением указанного в заголовке соединения в виде оранжевого масла (250 мг, выход 92,7). МС (ИЭР): m/z =278,1 [М+Н]+.
Этап а) трет-бутил-4-(3-бром-4-хлорфенокси)пиперидин-1-карбоксилат
В раствор 1-BOC-4-гидроксипиперидина (2,04 г, 10,12 ммоль, CAS RN 106-52-5), 3-бром-4-хлорфенола (2,0 г, 9,64, ммоль CAS RN 2402-82-6) и трифенилфосфина (3,03 г, 11,57 ммоль) в ТГФ (50 мл) добавляли диизопропилазодикарбоксилат (2,28 мл, 11,57 ммоль) и перемешивали смесь при 20°С в течение 15 ч. Смесь концентрировали, а остаток очищали с помощью обращенной флэш-хроматографии (MeCN и вода с содержанием 0,1% об./об. МК) с получением необходимого продукта (2,8 г, 74,3%) в виде светло-желтого масла. МС (ИЭР: m/z = 335,9 [М-56+Н]+.
Этап b) трет-бутил-4-(4-хлор-3-пиразол-1-ил-фенокси)пиперидин-1-карбоксилат
В смесь трет-бутил-4-(3-бром-4-хлорфенокси)пиперидин-1-карбоксилата (1,0 г, 2,56 ммоль), пиразола (139,4 мг, 2,05 ммоль), карбоната цезия (2501,8 мг, 7,68 ммоль) и 1,10-фенантролина (225,49 мг, 2,56 ммоль) в ДМФА (20 мл) добавляли CuI (48,59 мг, 0,260 ммоль) и перемешивали смесь при 110°С в течение 12 ч в атмосфере N2. Смесь концентрировали, разводили H2O (20 мл) и три раза экстрагировали EtOAc (10 мл). Объединенные органические слои концентрировали, а остаток очищали с помощью обращенно-фазовой хроматографии (АЦН и вода с содержанием 0,1% об./об. МК) с получением необходимого продукта (265 мг, 22,5%, чистота 82%) в виде желтого масла. МС (ИЭР): m/z = 378,1 [М+Н]+.
ВВ155
4-[5-(4-пиперидилокси)-2-(трифторметил)фенил]морфолин трифторацетат
В раствор трет-бутил-4-[3-морфолино-4-(трифторметил)фенокси]пиперидин-1-карбоксилата (400,0 мг, 0,93 ммоль) в ДХМ (3 мл) добавляли ТФУ (1,0 мл) и перемешивали реакционную смесь при 25°С в течение 12 ч. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта (300 мг) в виде желтого масла, которое использовали на следующем этапе без дополнительной очистки. МС (ИЭР): m/z = 331.2 [M+H]+.
Этап а) трет-бутил-4-(3-бром-4-(трифторметил)фенокси)пиперидин-1-карбоксилат
В раствор 3-бром-4-(трифторметил)фенола (500,0 мг, 2,54 ммоль, CAS RN1214385-56-4) и 1-BOC-4-гидроксипиперидина (512 мг, 2,54 ммоль, CAS RN 106-52-5) в ТГФ (8,5 мл) добавляли PPh3 (1000,9 мг, 3,82 ммоль) и диэтилазодикарбоксилат (664,53 мг, 3,82 ммоль) и перемешивали смесь при 25°С в течение 12 ч. Смесь очищали с помощью хроматографии на силикагеле, используя ПЭ: ЭА = 5: 1 в качестве элюента, с получением необходимого продукта (503 мг, выход 46,6%) в виде светло-желтого масла. МС (ИЭР: m/z = 369,2 [М-56+Н]+.
Этап b) трет-бутил-4-(3-морфолино-4-(трифторметил)фенокси)пиперидин-1-карбоксилат
Смесь трет-бутил-4-[3-бром-4-(трифторметил)фенокси]пиперидин-1-карбоксилата (500,0 мг, 1,18 ммоль), морфолина (154 мг, 1,77 ммоль, CAS RN 110-91-8), (R)-(+)-2,2'-бис(дифенилфосфино)-1,1'-бинафталина (146,77 мг, 0,24 ммоль, CAS RN 76189-55-4), карбоната цезия (1,15 г, 3,54 ммоль) и трис(дибензилиденацетон)дипалладия (0) (172,47 мг, 0,240 ммоль, CAS RN 76971-72-7) в ДМФА (10 мл) перемешивали при 110°С в течение 12 ч. Смесь вливали в H2O и три раза экстрагировали EtOAc. Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме, а остаток очищали с помощью колоночной хроматографии (градиент EtOAc в ПЭ от 5% до 33%) с получением необходимого продукта (480 мг, выход 94,6%) в виде светло-желтого твердого вещества. МС (ИЭР): m/z = 431,1 [M+H]+.
ВВ156
4-(4-хлор-3-(1,2,4-триазол-1-ил)фенокси)пиперидин трифторацетат
В раствор трет-бутил-4-[4-хлор-3-(1,2,4-триазол-1-ил)фенокси]пиперидин-1-карбоксилата (196,0 мг, 0,520 ммоль) в ДХМ (5 мл) добавляли ТФУ (1,01 мл, 13,13 ммоль) при 0°С и перемешивали смесь при 20°С в течение 1 ч. Смесь концентрировали с получением указанного в заголовке соединения (178 мг, 87,6%) в виде коричневого масла. МС (ИЭР): m/z = 279,1 [М+Н]+.
Этап а) трет-бутил-4-[4-хлор-3-(1,2,4-триазол-1-ил)фенокси]пиперидин-1-карбоксилат
Смесь трет-бутил-4-(3-бром-4-хлорфенокси)пиперидин-1-карбоксилата (500,0 мг, 1,28 ммоль, ВВ102, промежуточное соединение а), 1,2,4-триазола (176,8 мг, 2,56 ммоль) и CuI (24,3 мг, 0,130 ммоль), карбоната цезия (1250,9 мг, 3,84 ммоль) и диметилглицина (1,0 мл, 1,28 ммоль) в ДМФА (10 мл) перемешивали при 120°С в течение 12 ч. Смесь концентрировали для удаления ДМФА, разводили H2O (50 мл) и три раза экстрагировали EtOAc (по 20 мл каждый раз). Объединенные органические слои выпаривали, а остаток очищали с помощью обращенно-фазовой флэш-хроматографии (АЦН и вода с содержанием 0,1% об./об. МК) с получением указанного в заголовке соединения (196 мг, 37,1%) в виде бесцветного масла. МС (ИЭР: m/z = 323,0 [M-56+H]+.
ВВ157
4-[3-циклопропил-4-(трифторметил)фенокси]пиперидин трифторацетат
В смесь трет-бутил-4-[3-циклопропил-4-(трифторметил)фенокси]пиперидин-1-карбоксилата (360,0 мг, 0,930 ммоль) в ДХМ (18 мл) добавляли ТФУ (1,8 мл). Смесь перемешивали при 25°С в течение 12 ч. Смесь концентрировали в вакууме с получением необходимого соединения в виде светло-желтой смолы (370 мг, 99,2%). МС (ИЭР): m/z =286,2 [М+Н]+.
Этап а) трет-бутил-4-[3-циклопропил-4-(трифторметил)фенокси]пиперидин-1-карбоксилат
Смесь трет-бутил-4-[3-бром-4-(трифторметил)фенокси]пиперидин-1-карбоксилата (500,0 мг, 1,18 ммоль, ВВ103, промежуточное соединение b), циклопропилбороновой кислоты (151,86 мг, 1,77 ммоль), Na2CO3 (374,74 мг, 3,54 ммоль) и Pd(PPh3)4 (13,6 мг, 0,010 ммоль) в 1,4-диоксане (10 мл) и H2O (1 мл) перемешивали при 95°С в течение 12 ч. Смесь вливали в H2O (50 мл) и три раза экстрагировали EtOAc (по 50 мл каждый раз). Объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме и очищали с помощью колоночной хроматографии (ПЭ: EtOAc = 20: 1 ~ 5: 1) с получением необходимого продукта (380 мг, 83,7%) в виде бесцветной смолы. МС (ИЭР: m/z = 330,1 [М-56+Н]+.
ВВ158
4-[3-пиразол-1-ил-4-(трифторметил)фенокси]пиперидин трифторацетат
В раствор трет-бутил-4-[3-пиразол-1-ил-4-(трифторметил)фенокси]пиперидин-1-карбоксилата (180,0 мг, 0,440 ммоль) в ДХМ (5 мл) добавляли ТФУ (0,5 мл). Смесь перемешивали при 25°С в течение 12 ч, а затем концентрировали в вакууме с получением необходимого соединения (180 мг, 96,7%) в виде светло-желтой смолы. МС (ИЭР): m/z = 312,1 [М+Н]+.
Этап а) трет-бутил-4-[3-пиразол-1-ил-4-(трифторметил)фенокси]пиперидин-1-карбоксилат
Смесь трет-бутил-4-[3-бром-4-(трифторметил)фенокси]пиперидин-1-карбоксилата (500,0 мг, 1,18 ммоль, ВВ103, промежуточное соединение b), пиразола (120,35 мг, 1,77 ммоль), CuI (22,37 мг, 0,120 ммоль), N,N'-диметилэтилендиамина (519,45 мг, 5,89 ммоль) и Cs2CO3 (767,99 мг, 2,36 ммоль) в ДМФА (10 мл) перемешивали при 110°С в течение 12 ч. Смесь вливали в H2O (30 мл) и три раза экстрагировали EtOAc (по 50 мл каждый раз). Объединенные органические слои промывали раствором аммиака (20 мл), солевым раствором (50 мл), сушили над Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме, а неочищенное вещество очищали с помощью препаративной ТСХ (ПЭ: ЭА = 5: 1) с получением необходимого продукта (190 мг, 39,2%) в виде бесцветной смолы. МС (ИЭР: m/z = 356,1 [M-56+H]+.
ВВ159
4-[[2,6-дифтор-4-(трифторметил)фенил]метил]пиперидин трифторацетат
В раствор трет-бутил-4-[[2,6-дифтор-4-(трифторметил)фенил]метил]пиперидин-1-карбоксилата (70,0 мг, 0,180 ммоль) в ДХМ (1 мл) добавляли ТФУ (0,2 мл) и перемешивали смесь при 20°С в течение 1 ч. Смесь концентрировали с получением указанного в заголовке соединения (50 мг, 68,9%) в виде коричневого масла. МС (ИЭР):
m/z = 280,1 [M+H]+.
Этап а) 2-(диэтоксифосфорилметил)-1,3-дифтор-5-(трифторметил)бензол
Раствор 2-(бромметил)-1,3-дифтор-5-(трифторметил)бензола (1,29 мл, 3,27 ммоль, CAS RN 493038-91-8) в триэтилфосфите (5,44 г, 32,73 ммоль) перемешивали при 160°С в течение 5 ч. Смесь концентрировали в вакууме с получением указанного в заголовке соединения (600 мг, 55,2%; бесцветное масло), которое использовали на следующем этапе без дополнительной очистки.
Этап b) трет-бутил-4-[[2,6-дифтор-4-(трифторметил)фенил]метилен]пиперидин-1-карбоксилат
Смесь 2-(диэтоксифосфорилметил)-1,3-дифтор-5-(трифторметил)бензола (400,0 мг, 1,2 ммоль) в ТГФ (4 мл) добавляли в гидрид натрия (144,49 мг, 3,61 ммоль) в ТГФ (4 мл) при 0°С. Смесь перемешивали при 0°С в течение 1 ч, а затем в вышеуказанную смесь добавляли 1-BOC-4-пиперидон (479,83 мг, 2,41 ммоль, CAS RN 79099-07-3). Смесь перемешивали при 20°С в течение 12 ч. Смесь вливали в H2O (50 мл) и три раза экстрагировали EtOAc (по 20 мл каждый раз). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии (силикагель, ПЭ: ЭА = 1: от 0 до 2: 1) с получением указанного в заголовке соединения (100 мг, 22,0%) в виде бесцветного масла. МС (ИЭР: m/z = 322,0 [М-56+Н]+.
Этап с) трет-бутил-4-[[2,6-дифтор-4-(трифторметил)фенил] метил]пиперидин-1-карбоксилат
В раствор трет-бутил-4-[[2,6-дифтор-4-(трифторметил)фенил] метилен]пиперидин-1-карбоксилата (100,0 мг, 0,270 ммоль) в МеОН (8 мл) добавляли Pd/C (10,0 мг, 10 масс. %). Смесь перемешивали при 20°С в течение 1 ч в атмосфере Н2, затем фильтровали и концентрировали с получением указанного в заголовке соединения в виде бесцветного масла (70 мг, 69,6%). МС (ИЭР): m/z = 324,1 [M-56+H]+.
ВВ160
4-[4-хлор-3-(4-хлорфенил)-2-фторфенокси]пиперидин трифторацетат
В смесь трет-бутил-4-[4-хлор-3-(4-хлорфенил)-2-фторфенокси]пиперидин-1-карбоксилата (145,0 мг, 0,330 ммоль) в ДХМ (10 мл) добавляли ТФУ (1,0 мл). Смесь перемешивали при 20°С в течение 5 ч. Смесь концентрировали в вакууме с получением необходимого продукта (149 мг, 99,6%) в виде светло-коричневой смолы. МС (ИЭР): m/z = 340,1 [М+Н]+.
Этап а) 1-хлор-2-(4-хлорфенил)-3-фтор-4-метоксибензол
Смесь 4-бромхлорбензола (1,41 г, 7,34 ммоль, CAS RN 106-39-8), (6-хлор-2-фтор-3-метоксифенил) борной кислоты (1,0 г, 4,89 ммоль, CAS RN 867333-04-8) и K2CO3 (2,03 г, 14,68 ммоль) в 1,4-диоксане (15 мл) и H2O (1,5 мл) перемешивали в атмосфере N2 при 110°С в течение 1 ч в микроволновой печи. Смесь вливали в H2O (20 мл) и три раза экстрагировали EtOAc (по 20 мл каждый раз). Объединенные органические слои промывали солевым раствором (20 мл), сушили над Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме, а остаток очищали с помощью колоночной хроматографии, используя ПЭ в качестве элюента, с получением необходимого продукта (110 мг, 8,3%) в виде бесцветного масла, которое использовали на следующем этапе без дополнительной очистки.
Этап b) 4-хлор-3-(4-хлорфенил)-2-фторфенол
В смесь 1-хлор-2-(4-хлорфенил)-3-фтор-4-метоксибензола (215,0 мг, 0,790 ммоль) в ДХМ (7 мл) по каплям добавляли раствор BBr3 (993,36 мг, 3,97 ммоль) в ДХМ (7 мл) при -78°С. Смесь перемешивали при 20°С в течение 12 ч. Реакцию гасили добавлением МеОН (1 мл) и после него воды (10 мл), и три раза экстрагировали смесь ДХМ (по 10 мл каждый раз). Объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме с получением необходимого продукта (120 мг, 57,5%) в виде светло-коричневого твердого вещества, которое использовали на следующем этапе без дополнительной очистки.
Этап с) трет-бутил-4-[4-хлор-3-(4-хлорфенил)-2-фторфенокси]пиперидин-1-карбоксилат
Смесь 4-хлор-3-(4-хлорфенил)-2-фторфенола (120,0 мг, 0,470 ммоль), 1-ВОС-4-гидроксипиперидина (187,88 мг, 0,930 ммоль, CAS RN 106-52-5), PPh3 (244,85 мг, 0,930 ммоль) и ДИАД (0,18 мл, 0,930 ммоль) в ТГФ (12 мл) перемешивали при 20°С в течение 12 ч. Смесь вливали в H2O и три раза экстрагировали EtOAc. Объединенный органический слой промывали солевым раствором, сушили над Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме, а остаток очищали с помощью колоночной хроматографии (ПЭ: ЭА = 1:0 ~ 20: 1) с получением необходимого продукта в виде светло-желтой смолы (150 мг, 73%). МС (ИЭР: m/z = 384,0 [M-56+H]+.
ВВ161
3-[2-хлор-4-(трифторметил)фенокси]азетидин трифторацетат
В раствор трет-бутил-3-[2-хлор-4-(трифторметил)фенокси]азетидин-1-карбоксилата (400,0 мг, 1,14 ммоль) в ДХМ (10 мл) добавляли ТФУ (2,0 мл) при 20°С. После перемешивания в течение 2 ч смесь концентрировали с получением неочищенного продукта (410 мг, 98,6%) в виде светло-желтого масла, которое использовали на следующем этапе без дополнительной очистки.
Этап а) трет-бутил-3-[2-хлор-4-(трифторметил)фенокси]азетидин-1-карбоксилат
В раствор 2-хлор-4-(трифторметил)фенола (1000,0 мг, 5,09 ммоль, CAS RN 35852-58-5) и трет-бутил-3-гидроксиазетидин-1-карбоксилата (1057,5 мг, 6,11 ммоль, CAS RN 141699-55-0) в ТГФ (20 мл) добавляли PPh3 (1999,49 мг, 7,63 ммоль) и диэтилазодикарбоксилат (1329,05 мг, 7,63 ммоль), смесь перемешивали при 25°С в течение 12 ч. Реакционный раствор выпаривали в вакууме, остаток очищали с помощью обращенно-фазовой флэш-хроматографии (0,1% об./об. МК) с получением необходимого продукта (800 мг, 2,27 ммоль, выход 44,7%) в виде светло-желтого масла. МС (ИЭР: m/z = 296,0 [М-56+Н]+.
ВВ162
3-((2-фтор-6-(трифторметил)бензил)окси)азетидин трифторацетат
В раствор трет-бутил-3-[[2-фтор-6-(трифторметил)фенил]метокси]азетидин-1-карбоксилата (400,0 мг, 1,15 ммоль) в сухом ДХМ (10 мл) добавляли ТФУ (2,0 мл) при 25°С и перемешивали смесь при 25°С в течение 12 ч. Растворитель удаляли, а остаток сушили в вакууме с получением необходимого соединения в виде желтого масла (300 мг, 22%). МС (ИЭР): m/z = 250,0 [М+Н]+.
Этап а) трет-бутил-3-[[2-фтор-6-(трифторметил)фенил]метокси]азетидин-1-карбоксилат
В раствор 2-фтор-6-(трифторметил)бензилбромида (1000,0 мг, 3,89 ммоль, CAS RN 239087-08-2) и трет-бутил-3-гидроксиазетидин-1-карбоксилата (673,92 мг, 3,89 ммоль, CAS RN 141699-55-0) в сухом ТГФ (10 мл) при 25°С, добавляли t-BuOK (5,84 мл, 5,84 ммоль; 1,0 М в сухом ТГФ) и перемешивали смесь при 25°С в течение 12 ч. Смесь вливали в H2O (10 мл) и три раза экстрагировали ЭА (по 20 мл каждый раз). Объединенные органические слои, сушили над безводным Na2SO4 и фильтровали. Фильтрат концентрировали при пониженном давлении, очищали с помощью флэш-хроматографии на силикагеле (градиент ПЭ: ЭА = 10: от 1 до 2: 8) с получением указанного в заголовке соединения в виде бесцветного масла (1100 мг, 80,9%). МС (ИЭР: m/z = 294,0 [М-56+Н]+.
ВВ163
3-[2-(2-фтор-4-метилфенил]этил]азетидин трифторацетат
В раствор трет-бутил-3-[2-(2-фтор-4-метилфенил]этил]азетидин-1-карбоксилата (350,0 мг, 1,19 ммоль) в сухом ДХМ (10 мл) при 25°С добавляли ТФУ (1,0 мл, 1,19 ммоль) и перемешивали смесь при 25°С в течение 12 ч.
Реакционную смесь концентрировали при пониженном давлении, а остаток сушили в вакууме с получением необходимого соединения в виде бесцветного масла (260 мг, 70,9%). МС (ИЭР): m/z = 194,0 [М+Н]+.
Этап а) трет-бутил-3-(2-триметилсилилэтинил)азетидин-1-карбоксилат
В раствор триметилсилилацетилена (9,97 г, 101,55 ммоль, CAS RN 1066-54-2) в сухом ТГФ (200 мл) при 25°С добавляли i-PrMgCl (48,57 мл, 97,14 ммоль; 1,0 М в сухом ТГФ) и перемешивали смесь при 25°С в течение 15 мин. Затем добавляли раствор 1-ВОС-3-иодазетидина (25,0 г, 88,3 ммоль, CAS RN 254454-54-1), с последующим добавлением FeCl2 (0,34 г, 2,65 ммоль) в сухом ДМЭ (606 мл), и перемешивали смесь при 25°С в течение 12 ч. Смесь вливали в насыщенный водн. раствор NH4Cl (200 мл) и три раза экстрагировали EtOAc (по 150 мл каждый раз). Органические слои объединяли, сушили над безводным Na2SO4, фильтровали, а фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью флэш-хроматографии на силикагеле (ПЭ: ЭА = 20: от 1 до 10: 1) с получением необходимого продукта в виде черного масла (18 г, 80,4%).
1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ=4,11 (т, J = 8,4 Гц, 2Н), 3,92 (дд, J = 6,5, 8,1 Гц, 2Н), 3,51-3,17 (м, 1Н), 1,44 (с, 10Н), 0,16 (с, 9Н).
Этап b) трет-бутил-3-этинилазетидин-1-карбоксилат
В раствор трет-бутил-3-(2-триметилсилилэтинил)азетидин-1-карбоксилата (6243 мг, 24,64 ммоль) в сухом МеОН (40 мл) добавляли карбонат калия (1700 мг, 12,32 ммоль) при 25°С и перемешивали реакционную смесь при 25°С в течение 2 ч. Смесь фильтровали, фильтрат вливали в насыщенный водн. раствор NH4Cl (100 мл) и экстрагировали ЭА (100 мл, три раза). Объединенные органические слои сушили над безводным Na2SO4, фильтровали, а фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью флэш-хроматографии на силикагеле (ПЭ: ЭА = 50: от 1 до 15: 1) с получением указанного в заголовке соединения в виде светло-желтого масла (4100 мг, 91,8%). 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ=4,16-4,11 (м, 2Н), 3,93 (дд, J = 6,5, 8,2 Гц, 2Н), 3,37-3,20 (м, 1Н), 2,28 (д, J = 2,4 Гц, 1Н), 1,43 (с, 9Н).
Этап с) трет-бутил-3-[2-(2-фтор-4-метилфенил)этинил]азетидин-1-карбоксилат
В раствор трет-бутил-3-этинилазетидин-1-карбоксилата (1000,0 мг, 5,52 ммоль) и 4-бром-3-фтортолуола (1251,58 мг, 6,62 ммоль, CAS RN 452-74-4) в сухом ТГФ (20 мл) добавляли Pd(PPh3)4 (530,63 мг.0,460 ммоль). CuI (87.83 мг, 0,460 ммоль) и ТЭА (4644,2 мг, 46,0 ммоль) при 25°С.Смесь продували N2 в течение 1 мин. а затем перемешивали при 60°С в атмосфере N2 в течение 12 ч. Смесь вливали в насыщенный водн. раствор NH4Cl (50 мл) и три раза экстрагировали EtOAc (по 30 мл каждый раз). Объединенные органические слои сушили над безводным Na2SO4, фильтровали, фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью флэш-хроматографии на силикагеле (ПЭ: ЭА = 20: от 1 до 10: 1) с получением необходимого соединения в виде бесцветного масла (650 мг; 40,7%). 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ=7,33-7,28 (м, 1Н), 6,94-6,85 (м, 2Н), 4,26-4,19 (м, 2Н), 4,05 (дд, J=6,4, 8,1 Гц, 2Н), 3,66-3,49 (м, 1Н), 2,36 (с, 3Н), 1,46 (с, 9Н).
Этап d) трет-бутил-3-[2-(2-фтор-4-метилфенил)этинил]азетидин-1-карбоксилат
Партия а: В раствор трет-бутил-3-[2-(2-фтор-4-метилфенил)этинил]азетидин-1-карбоксилата (50,0 мг, 0,170 ммоль, 1 экв.) в EtOAc (5 мл) добавляли Pd /С (50,0 мг, 10 масс. %) при 25°С. Смесь перемешивали при 40°С в условиях баллонного водорода в течение 12 ч. ЖХМС-анализ показал 79,8% необходимого продукта.
Партия b: В раствор трет-бутил-3-[2-(2-фтор-4-метилфенил)этинил]азетидин-1-карбоксилата (500,0 мг, 1,73 ммоль) в EtOAc (10 мл) добавляли Pd /С (250,0 мг, 10 масс. %) при 25°С и перемешивали смесь при 40°С в условиях баллонного водорода в течение 6 ч. ЖХМС-анализ показал 80,4% необходимого продукта. Партии а и b объединяли, реакционную смесь фильтровали через слой целита, фильтрат концентрировали при пониженном давлении, а остаток сушили в вакууме с получением соединения в виде бесцветного масла (350 мг, 69,0%). МС (ИЭР: m/z = 238,1 [M-56+H]+.
ВВ164
3-[2-[4-метокси-2-(трифторметил)фенил]этил]азетидин трифторацетат
В раствор трет-бутил-3-[2-(2-фтор-4-метилфенил)этил]азетидин-1-карбоксилата (180,0 мг, 0,5 ммоль) в сухом ДХМ (10 мл) добавляли ТФУ (1,0 мл, 1,19 ммоль) при 25°С и перемешивали смесь при 25°С в течение 12 ч. Реакционную смесь концентрировали при пониженном давлении, а остаток сушили в вакууме с получением указанного в заголовке соединения (150 мг, 80,2%) в виде бесцветного масла. МС (ИЭР): m/z = 260,1 [М+Н]+.
Этап а) трет-бутил-3-[2-[4-метокси-2-(трифторметил)фенил]этинил]азетидин-1-карбоксилат
В раствор трет-бутил-3-этинилазетидин-1-карбоксилата (800,0 мг, 4,41 ммоль, ВВ111, промежуточное соединение b) и 3-трифторметил-4-броманизола (1350,9 мг, 5,3 ммоль, CAS RN 400-72-6) в сухом ТГФ (30 мл) при 25°С добавляли Pd(PPh3)4 (509,41 мг, 0,440 ммоль), CuI (84,31 мг, 0,440 ммоль) и ТЭА (4458,42 мг, 44,14 ммоль). Смесь продували N2 в течение 1 мин, а затем перемешивали при 60°С в атмосфере N2 в течение 12 ч. Смесь вливали в насыщенный водн. раствор NH4Cl (100 мл) и три раза экстрагировали EtOAc (по 50 мл каждый раз).
Органические слои объединяли, сушили над безводным Na2SO4, фильтровали, а фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью флэш-хроматографии на силикагеле (ПЭ: ЭА = 20: от 1 до 10: 1) с получением продукта в виде бесцветного масла (160 мг; 8,2%). МС (ИЭР: m/z = 300,1 [M-56+H]+.
Этап b) трет-бутил-3-[2-[4-метокси-2-(трифторметил)фенил]этил]азетидин-1-карбоксилат
В раствор трет-бутил-3-[2-(2-фтор-4-метилфенил)этинил]азетидин-1-карбоксилата (230,0 мг, 0,65 ммоль) в EtOAc (10 мл) при 25°С добавляли Pd/C (150,0 мг, 10 масс. %), смесь перемешивали при 40°С в условиях баллонного Н2 (около 15 фунт/кв. дюйм) в течение 12 ч. Реакционную смесь фильтровали через слой целита, а фильтрат концентрировали при пониженном давлении. Остаток сушили в вакууме с получением необходимого соединения в виде бесцветного масла (180 мг, 77,4%). МС (ИЭР: m/z = 304,1 [М-56+Н]+.
ВВ165
3-[[4-метил-2-(трифторметил)фенил]метокси]азетидин трифторацетат
В раствор трет-бутил-3-[[4-метил-2-(трифторметил)фенил]метокси]азетидин-1-карбоксилата (130,0 мг, 0,380 ммоль) в ДХМ (6,5 мл) добавляли ТФУ (1,3 мл, 16,87 ммоль) и перемешивали реакцию при 20°С, Через 12 ч смесь выпаривали с получением необходимого неочищенного продукта в виде светло-коричневого масла (130 мг, 96,1%). МС (ИЭР): m/z = 246,5 [М+Н]+.
Этап а) 4-бром-1-(бромметил)-2-(трифторметил)бензол
Раствор 5-бром-2-метилбензотрифторида (2000 мг, 8,37 ммоль, CAS RN 86845-27-4), N-бромсукцинимида (1489 мг, 8,37 ммоль, CAS RN 128-08-5) и бензоилпероксида (101,34 мг, 0,420 ммоль, CAS RN 2685-64-5) в тетрахлориде углерода (30 мл) перемешивали при 90°С в течение 12 ч. Смесь выпаривали, а остаток очищали с помощью колоночной хроматографии на силикагеле (100% ПЭ) с получением необходимого продукта в виде светло-коричневого масла (690 мг, 25,9%), которое использовали на следующем этапе без дополнительной очистки.
Этап b) трет-бутил-3-[[4-бром-2-(трифторметил)фенил]метокси]азетидин-1-карбоксилат
В раствор трет-бутил-3-гидроксиазетидин-1-карбоксилата (337,5 мг, 1,95 ммоль, CAS RN 22214-30-8) в ТГФ (9 мл) добавляли t-BuOK (1,95 мл, 1,95 ммоль), затем добавляли 4-бром-1-(бромметил)-2-(трифторметил)бензол (590,0 мг, 1,86 ммоль) и перемешивали смесь при 20°С в течение 12 ч. Смесь вливали в водн. раствор NH4Cl (200 мл) и три раза экстрагировали EtOAc (50 мл). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали с помощью препаративной ВЭЖХ (АЦН и вода с содержанием 0,225% об./об. МК) с получением необходимого продукта в виде светло-коричневого масла (300 мг, 39,4%). МС (ИЭР: m/z = 356,3 [М-56+Н]+.
Этап с) трет-бутил-3-[[4-метил-2-(трифторметил)фенил]метокси]азетидин-1-карбоксилат
В раствор трет-бутил-3-[[4-бром-2-(трифторметил)фенил]метокси]азетидин-1-карбоксилата (250,0 мг, 0,610 ммоль), триметилбороксина (114,8 мг, 0,910 ммоль), K2CO3 (168,5 мг, 1,22 ммоль) в 1,4-диоксане (10 мл) и H2O (2,5 мл) добавляли Pd(dppf)Cl2 (89,18 мг, 0,120 ммоль). Реакцию перемешивали при 100°С в течение 12 ч. Смесь фильтровали, концентрировали, а остаток очищали с помощью обращенной флэш-хроматографии (АЦН и вода с содержанием 0,1% об./об. МК) с получением необходимого продукта в виде светло-коричневого масла (146 мг, 69,4%). МС (ИЭР: m/z = 290,4 [M-56+H]+.
ВВ166
3-[2-[2-метокси-6-(трифторметил)фенил]этил]азетидин трифторацетат
В раствор трет-бутил-3-[2-[2-метокси-6-(трифторметил)фенил]этил]азетидин-1-карбоксилата (300,0 мг, 0,830 ммоль) в ДХМ (5 мл) добавляли ТФУ (1,0 мл) и перемешивали смесь при 25°С в течение 1 ч. Реакционную смесь выпаривали при пониженном давлении с получением необходимого продукта (300 мг, 96,3%) в виде бесцветного масла. МС (ИЭР): m/z = 260,1 [М+Н]+.
Этап а) трет-бутил-3-[2-[2-метокси-6-(трифторметил)фенил]этинил]азетидин-1-карбоксилат
В раствор трет-бутил-3-этинилазетидин-1-карбоксилата (710,6 мг, 3,92 ммоль, B111, промежуточное соединение b) и 2-бром-1-метокси-3-(трифторметил)бензола (500,0 мг, 1,96 ммоль) в сухом ДМСО (17, 5 мл) при 25°С добавляли Pd(PPh3)2Cl2 (137,6 мг, 0,200 ммоль) и Cs2CO3 (1278 мг, 3,92 ммоль). Смесь продували N2 в течение 1 мин, а затем перемешивали при 110°С в атмосфере N2 в течение 12 ч. Смесь фильтровали, фильтрат концентрировали, а остаток очищали с помощью силикагеля (ПЭ: EtOAc = 20: 1) с получением необходимого продукта в виде светло-желтого масла (600 мг, 86,1%), которое использовали на следующем этапе без дополнительной очистки.
Этап b) трет-бутил-3-[2-[2-метокси-6-(трифторметил)фенил]этинил]азетидин-1-карбоксилат
В раствор трет-бутил-3-[2-[2-метокси-6-(трифторметил)фенил]этинил]азетидин-1-карбоксилата (400,0 мг, 1,13 ммоль) в EtOAc (20 мл) добавляли влажный Pd/C (50 мг, 10 масс. %). Смесь 3 раза продували H2, а затем перемешивали при 40°С в атмосфере H2 (баллон) в течение 12 ч. Смесь фильтровали, а фильтрат концентрировали с получением необходимого продукта в виде светло-желтого масла (300 мг, 74,2%), которое использовали на следующем этапе без дополнительной очистки.
ВВ167
3-[2-[4-метил-2-(трифторметил)фенил]этил]азетидин трифторацетат
В раствор трет-бутил-3-[2-[4-метил-2-(трифторметил)фенил]этил]азетидин-1-карбоксилата (100,0 мг, 0,290 ммоль) в ДХМ (4 мл) добавляли ТФУ (0,5 мл) и перемешивали смесь при 20°С в течение 12 ч. Реакционную смесь выпаривали при пониженном давлении с получением необходимого продукта в виде желтого масла (98 мг, 94,2%). МС (ИЭР): m/z = 244,1 [М+Н]+.
Этап а) трет-бутил-3-[2-[4-метил-2-(трифторметил)фенил]этинил]азетидин-1-карбоксилат
В раствор трет-бутил-3-этинилазетидин-1-карбоксилата (606,6 мг, 3,35 ммоль) и 2-бром-5-метилбензотрифторида (400,0 мг, 1,67 ммоль) в сухом ДМСО (14,9 мл) при 25°С добавляли Pd(PPh3)2Cl2 (117,46 мг, 0,170 ммоль) и Cs2CO3 (1091 мг, 3,35 ммоль). Смесь продували N2 в течение 1 мин, а затем перемешивали при 110°С в
атмосфере N2 в течение 12 ч. Реакционную смесь вливали в Н2O и экстрагировали EtOAc. Органический слой выпаривали, а остаток очищали с помощью колоночной хроматографии на силикагеле (ПЭ: ЕtOАс=20: 1) с получением необходимого соединения в виде желтого масла (390 мг; 68,7%). МС (ИЭР: m/z=284,1 [М-56+Н]+.
Этап b) трет-бутил-3-[2-[4-метил-2-(трифторметил)фенил]этил]азетидин-1-карбоксилат
В раствор трет-бутил-3-[2-[4-метил-2-(трифторметил)фенил]этинил]азетидин-1-карбоксилата (390,0 мг, 1,15 ммоль) в ЕtOАс (19,5 мл) добавляли влажный Pd/C (150 мг, 10 масс. %), смесь 3 раза продували Н2 и перемешивали при 40°С в атмосфере Н2 (баллон) в течение 12 ч. Смесь фильтровали, а фильтрат концентрировали с получением необходимого продукта в виде светло-желтого масла (295 мг, выход 72,9%). МС (ИЭР: m/z=288,1 [М-56+Н]+.
ВВ168
1-[2-[2-(азетидин-3-ил)этил]-5-(трифторметил)фенил]этанон трифторацетат
В раствор трет-бутил-3-[2-[2-ацетил-4-(трифторметил)фенил]этил]азетидин-1-карбоксилата (50,0 мг, 0,130 ммоль) в ДХМ (1 мл) добавляли ТФУ (0,2 мл) и перемешивали раствор при 20°С в течение 12 ч. Смесь концентрировали с получением необходимого продукта в виде светло-коричневого масла (50 мг, 96,4%). МС (ИЭР): m/z=272,1 [М+Н]+.
Этап а) 2-бром-1-(бромметил)-4-(трифторметил)бензол
В раствор [2-бром-4-(трифторметил)фенил]метанола (500,0 мг, 1,96 ммоль, СAS RN 497959-33-8) и PPh3 (770,5 мг, 2,94 ммоль) в ТГФ (10 мл) добавляли тетрабромид углерода (975,3 мг, 2,94 ммоль) и перемешивали смесь при 25°С в течение 12 ч. Реакционную смесь концентрировали в вакууме, а остаток очищали с помощью колоночной хроматографии на силикагеле (ПЭ: ЭА=0: 1 ~ 20: 1) с получением необходимого продукта в виде бесцветного масла (600 мг; 96,3%). 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ=7,78 (с, 1Н), 7,55-7,46 (м, 2Н), 4,53 (с, 2Н).
Этап b) 2-бром-1-(диэтоксифосфорилметил)-4-(трифторметил)бензол
Раствор 2-бром-1-(бромметил)-4-(трифторметил)бензола (600,0 мг, 1,89 ммоль) в триэтилфосфите (3136 мг, 18,87 ммоль) перемешивали при 160°С в течение 5 ч. Смесь концентрировали при 100°С при пониженном давлении для удаления большей части триэтилфосфита с получением неочищенного продукта (700 мг) в виде светло-желтого масла. МС (ИЭР): m/z=375,2 [М+Н]+.
Этап с) трет-бутил-3-[(Е)-2-[2-бром-4-(трифторметил)фенил]винил]азетидин-1-карбоксилат
Смесь 2-бром-1-(диэтоксифосфорилметил)-4-(трифторметил)бензола (600,0 мг, 1,6 ммоль) в ТГФ (10 мл) добавляли к другой суспензии NaH (191,9 мг, 4,8 ммоль, 60% дисперсия в минеральном масле) в ТГФ (10 мл) при 0°С. Смесь перемешивали при 0°С в течение 1 ч. Затем добавляли трет-бутил-3-формилазетидин-1-карбоксилат (296,3 мг, 1,6 ммоль) и перемешивали смесь при 20°С в течение 11 ч. Реакционную смесь вливали в водн. раствор NH4Cl (100 мл) и три раза экстрагировали EtOAc (по 50 мл каждый раз). Объединенные органические слои промывали солевым раствором (100 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (ПЭ: ЕtOАс=20: 1) с получением необходимого продукта в виде светло-желтого масла (450 мг, 69,3%). МС (ИЭР: m/z=352,0 [М56+Н]+. 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ=7,74 (д, J=0,8 Гц, 1Н), 7,58-7,51 (м, 1Н), 7,49-7,41 (м, 1Н), 6,71 (д, J=15,8 Гц, 1Н), 6,36 (дд, J=8,4, 15,8 Гц, 1Н), 4,13 (т, J=8,5 Гц, 2Н), 3,78 (дд, J=5,8, 8,6 Гц, 2Н), 3,44-3,31 (м, 1H), 1,39 (с, 9Н).
Этап d) трет-бутил-3-[(Е)-2-[2-ацетил-4-(трифторметил)фенил]винил]азетидин-1-карбоксилат
Раствор трибутил(1-этоксивинил)олова (426,7 мг, 1,18 ммоль), трет-бутил-3-[(Е)-2-[2-бром-4-(трифторметил)фенил]винил]азетидин-1-карбоксилата (400,0 мг, 0,980 ммоль) и Pd(Ph3P)2Cl2 (138,2 мг, 0,200 ммоль) в ТГФ (16 мл) перемешивали при 80°С в атмосфере N2 в течение 4 ч. Смесь охлаждали до комнатной температуры и добавляли водн. раствор KF (10 мл). Смесь перемешивали в течение 10 мин, три раза экстрагировали ЕtOАс (по 20 мл каждый раз) и концентрировали объединенные органические слои. Остаток растворяли в (20 мл) и добавляли водн. НСl (0,6 Н, 20 мл). Смесь перемешивали при 20°С в течение 0,5 ч, три раза экстрагировали ЕtOАс (по 20 мл каждый раз) и концентрировали объединенные органические слои. Остаток очищали с помощью колоночной хроматографии на силикагеле (ПЭ: ЕtOАс=20: 1) с получением необходимого продукта (280 мг, 77%) в виде светло-желтого масла. МС (ИЭР: m/z=314,1 [М-56+Н]+.
Этап е) трет-бутил-3-[2-[2-ацетил-4-(трифторметил)фенил]этил]азетидин-1-карбоксилат
В раствор трет-бутил-3-[(Е)-2-[2-ацетил-4-(трифторметил)фенил]винил]азетидин-1-карбоксилата (50,0 мг, 0,140 ммоль) в ЕtOАс (5 мл) добавляли влажный Pd/C (20,0 мг, 10 масс. %) и перемешивали смесь при 20°С в атмосфере Н2 (баллон) в течение 12 ч. Затем реакционную смесь нагревали до 50°С и перемешивали еще 12 ч. Смесь фильтровали, а фильтрат концентрировали с получением необходимого продукта (50 мг, 99,5%) в виде светло-желтого масла. МС (ИЭР: m/z=316,2 [М-56+Н]+.
ВВ169
3-[2-[2-бром-4-(трифторметил)фенил]этил]азетидин трифторацетат
В раствор трет-бутил-3-[2-[2-бром-4-(трифторметил)фенил]этил]азетидин-1-карбоксилата (400,0 мг, 0,980 ммоль) в ДХМ (10 мл) добавляли ТФУ (1,0 мл) и перемешивали смесь при 20°С в течение 12 ч. Реакционную смесь выпаривали при пониженном давлении с получением необходимого продукта (413 мг, выход 99,8%) в виде желтого масла. МС (ИЭР): m/z=308,1 [М+Н]+.
Этап а) трет-бутил-3-[2-[2-бром-4-(трифторметил)фенил]этил]азетидин-1-карбоксилат
В суспензию трет-бутил-3-[(Е)-2-[2-бром-4-(трифторметил)фенил]винил]азетидин-1-карбоксилата (600,0 мг, 1,48 ммоль, ВВ116, промежуточное соединение с) и MgO (118,1 мг, 2,95 ммоль) в ЕtOАс (20 мл) добавляли Pd/C (300,0 мг, 10 масс. %), смесь перемешивали при 25°С в атмосфере Н2 (баллон) в течение 1 ч. Реакционную смесь фильтровали, а фильтрат выпаривали при пониженном давлении с получением необходимого продукта (500 мг, 82,9%) в виде светло-желтого масла. МС (ИЭР: m/z=352,0 [М-56+Н]+.
ВВ174
2-(азетидин-3-илметокси)-5-(трифторметил)пиридин 2,2,2-трифторацетат
Синтез ВВ174 проводили по аналогии с ВВ57, начиная с трет-бутил-3-(гидроксиметил)азетидин-1-карбоксилата и 2-бром-5-(трифторметил)пиридина. МС (ИЭР): m/z=233,1 [М+Н]+.
ВВ175
3-метил-5-[[рац-(3R,4R)-3-метил-4-пиперидил]метокси]-2-(трифторметил)пиридин дигидрохлорид
трет-бутил-(рац-3R,4R)-3-метил-4-(((5-метил-6-(трифторметил)пиридин-3-ил)окси)метил)пиперидин-1-карбоксилат (198 мг, 510 мкмоль) растворяли в ДХМ (2 мл) и добавляли 2 М НСl в эфире (1,53 мл, 3,06 ммоль). Реакционную смесь перемешивали при КТ в течение 8 ч. Реакционную смесь концентрировали в вакууме с получением 180 мг необходимого продукта в виде белого твердого вещества (98%) МС (ИЭР): m/z=289,3 [М+Н]+.
а) трет-бутил-(рац-3R,4R)-4-(гидроксиметил)-3-метилпиперидин-1-карбоксилат
В перемешиваемый раствор сложного метилового эфира цис-N-ВОС-3-метилпиперидин-4-карбоновой кислоты (2 г, 7,77 ммоль) в ТГФ (10 мл) добавляли борогидрид лития (5,83 мл, 11,7 ммоль) при 2-5°С. Затем реакционную смесь нагревали с рефлюксом в течение 3 ч, а после этого охлаждали до 2-5°С. Добавляли воду и дважды экстрагировали водный слой ЕtOАс (по 30 мл каждый раз). Органический слой промывали водой, NaHCO3 и солевым раствором, слои разделяли, органические сушили над Na2SO4 и концентрировали в вакууме.
Очистка с помощью флэш-хроматографии (градиент ЕtOАс в н-гептане от 0 до 65%) позволила получить продукт в виде бесцветного масла (930 мг, 50%). МС (ИЭР: m/z=174,1 [М-56+Н]+.
b) трет-бутил-(рац-3R,4R)-3-метил-4-(((5-метил-6-(трифторметил)пиридин-3-ил)окси)метил)пиперидин-1-карбоксилат
трет-бутил-(3R,4R)-4-(гидроксиметил)-3-метилпиперидин-1-карбоксилат (239 мг, 1,04 ммоль) растворяли в ДМФА (4,17 мл) и добавляли NaH в минеральном масле (60%, 45,8 мг, 1,15 ммоль) при КТ. Реакцию перемешивали в течение 20 мин, затем добавляли 5-бром-3-метил-2-(трифторметил)пиридин (250 мг, 167 мкл, 1,04 ммоль) и продолжали перемешивание в течение 12 ч при КТ. Реакцию гасили 10 мл насыщ. раствора NH4Cl и три раза экстрагировали водой/ЕtOАс. Органические фазы объединяли и сушили над MgSO4, а растворитель удаляли в вакууме. Флэш-хроматография (градиент ЕtOАс в н-гептане от 0 до 50%) позволила получить продукт в виде белого твердого вещества (148 мг, 49%). МС (ИЭР: m/z=333,2 [М-56+Н]+.
ВВ176
3-((2-фтор-4-(трифторметил)бензил)окси)-2-метилазетидин 2,2,2-трифторацетат
В раствор трет-бутил-3-((2-фтор-4-(трифторметил)бензил)окси)-2-метилазетидин-1-карбоксилата (0,265 г, 729 мкмоль) в ДХМ (4 мл) добавляли ТФУ (832 мг, 562 мкл, 7,29 ммоль). Полученную в результате реакционную смесь перемешивали при КТ в течение 1 ч. Реакционную смесь концентрировали с получением указанного в заголовке соединения в виде бесцветного масла. Неочищенный продукт использовали без дополнительной очистки. МС (ИЭР): m/z=264,2 [М+Н]+.
Этап а) [2-фтор-4-(трифторметил)фенил]метилметансульфонат
В ледяной раствор (2-фтор-4-(трифторметил)фенил)метанола (840 мг, 4,33 ммоль) и триэтиламина (1,31 g, 1,81 мл, 13 ммоль) в ДХМ (8 мл) по каплям добавляли метансульфонилхлорид (496 мг, 337 мкл, 4,33 ммоль) и перемешивали смесь при 0°С в течение 1 ч. Реакционную смесь вливали в насыщенный водный раствор NaHCO3 (10 мл) и ДХМ (20 мл) и разделяли слои. Водный слой один раз экстрагировали ДХМ (20 мл). Органические слои промывали один раз солевым раствором, сушили над MgSO4, фильтровали и выпаривали с получением необходимого соединения в виде желтого масла (1,13 г, 96%).
Этап b) трет-бутил-3-[[2-фтор-4-(трифторметил)фенил]метокси]-2-метилазетидин- 1-карбоксилат
В ледяной раствор трет-бутил-3-гидрокси-2-метилазетидин-1-карбоксилата (250 мг, 1,34 ммоль) в ДМФА (3 мл) частями добавляли NaH (60% в минеральном масле, 58,7 мг, 1,47 ммоль) и перемешивали смесь при температуре ледяной бани в течение 5 мин с последующим перемешиванием при КТ в течение 40 мин. В смесь по каплям добавляли раствор 2-фтор-4-(трифторметил)бензилметансульфоната (436 мг, 1,6 ммоль) в ДМФА (1 мл) при КТ. Перемешивание суспензии продолжали при КТ в течение 16 ч. Реакционную смесь вливали в насыщенный водный раствор NH4Cl (10 мл) и ЕtOАс (20 мл) и разделяли слои. Водный слой один раз экстрагировали EtOAc (50 мл). Органические слои дважды промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Неочищенное соединение очищали с помощью хроматографии на силикагеле (градиент н-гептана: ЕtOАс от 100: от 0 до 0: 100) с получением трет-бутил-3-[[2-фтор-4-(трифторметил)фенил]метокси]-2-метилазетидин-1-карбоксилата в виде бесцветного масла (0,265 г, выход 54,6%). МС (ИЭР: m/z=308,2 [М-56+Н]+.
ВВ 177
2-(азетидин-3-илметокси)-4,5-бис(трифторметил)пиридин 2,2,2- трифторацетат
Синтез ВВ177 проводили по аналогии с ВВ57, начиная с трет-бутил-3-(гидроксиметил)азетидин-1-карбоксилата и 2-хлор-4,5-бис(трифторметил)пиридина. МС (ИЭР): m/z=301,2 [М+Н] +.
ВВ179
3-((4-хлор-2-феноксибензил)окси)азетидин 2,2,2-трифторацетат
Синтез ВВ179 проводили по аналогии с ВВ39, начиная с трет-бутил-3-гидроксиазетидин-1-карбоксилата и 1-(бромметил)-4-хлор-2-феноксибензола (синтез описан ниже). МС (ИЭР): m/z=290,2 [М+Н]+.
1-(бромметил)-4-хлор-2-феноксибензол
i) В 10 мл пробирке с круглым дном разводили метил 4-хлор-2- феноксибензоат (547 мг, 2,08 ммоль) в толуоле (3,82 мл) и охлаждали реакционную смесь на ледяной бане. Медленно, по каплям добавляли бис(2- метоксиэтокси)алюминийгидрид натрия, 70% в толуоле (649 мг, 637 мкл, 2,25 ммоль), мак. при 15°С с получением светло-желтого раствора. Реакционную смесь перемешивали при КТ в течение 30 мин. Неочищенную реакционную смесь, содержащую продукт (4-хлор-2-феноксифенил)метанол, непосредственно использовали на следующем этапе.
ii) В 25 пробирке с круглым дном бромистоводородную кислоту, 48% в Н2О (6,49 г, 4,35 мл, 38,5 ммоль), охлаждали на ледяной бане. Затем медленно, по каплям добавляли (4-хлор-2-феноксифенил)метанол (неочищенный, 488 мг, 2,08 ммоль) и перемешивали смесь при 50°С в течение 2 ч. Добавляли бромистоводородную кислоту, 48% в Н2О (6,25 г, 2,18 мл, 19,25 ммоль) и перемешивали смесь при 60°С в течение 1 ч, затем охлаждали до КТ. Водную фазу отделяли, органическую фазу четыре раза промывали Н2О и выпаривали. Неочищенный материал очищали с помощью колоночной флэш-хроматографии (градиент от 0% до 25% ЕtOАс в гексанах) и использовали на следующем этапе без дополнительной очистки. Выход: 85%.
ВВ 181
3-((1-(2,4-дихлорфенил)циклопропилметокси)азетидин 2,2,2-трифторацетат
В раствор трет-бутил-3-((1-(2,4-дихлорфенил)циклопропил)метокси)азетидин-1-карбоксилата (165 мг, 443 мкмоль) в ДХМ (2 мл) добавляли ТФУ (202 мг, 137 мкл, 1,77 ммоль) и перемешивали реакцию при КТ в течение 8 ч. Смесь концентрировали в вакууме (азеотроп с толуолом, ЕtOАс и н-гептан) с получением соединения в виде бесцветного масла (170 мг, 99%). МС (ИЭР): m/z=272,2 [М+Н] +.
Этап а) 1-(2,4-дихлорфенил)циклопропил)метанол
В 50 мл колбе с тремя горлышками 1-(2,4-дихлорфенил)циклопропан-1-карбоновую кислоту (1 г, 4,33 ммоль) объединяли с ТГФ (20 мл) с получением бесцветного раствора. При 0°С по каплям добавляли 1,0 М раствор комплекса боран-тетрагидрофуран в ТГФ (6,49 мл, 6,49 ммоль) в течение периода 15 мин. Реакцию перемешивали при КТ в течение 2 ч. По каплям добавляли МеОН (2 мл), а после - 1 М водн. раствор НСl, и перемешивали в течение 30 мин. Реакционную смесь дважды экстрагировали ЕtOАс (по 40 мл каждый раз), а органические слои промывали 10% водн. раствором Nа2СО3 (40 мл) и с последующим промыванием солевым раствором (40 мл). Органические фракции объединяли, сушили над Na2SO4 и концентрировали в вакууме. Неочищенный материал очищали с помощью колоночной флэш-хроматографии (градиент ЕtOАс в н-гептане, от 0% до 30%) с получением соединения в виде бесцветного масла (90%) МС (ИЭР): m/z=201,0 [М-16+Н] +.
Этап b) 1-(2,4-дихлорфенил)циклопропил]метилметансульфонат
В ледяной раствор (1-(2,4-дихлорфенил)циклопропил)метанола (350 мг, 1,61 ммоль) и ТЭА (326 мг, 449 мкл, 3,22 ммоль) в ДХМ (6 мл) по каплям добавляли метансульфонилхлорид (185 мг, 126 мкл, 1,61 ммоль) и перемешивали смесь при 0°С в течение 1 ч, затем при КТ в течение ночи. Реакционную смесь вливали в насыщенный водный раствор NаНСО3 (10 мл) и ДХМ (10 мл) и разделяли слои. Водный слой один раз экстрагировали ДХМ (10 мл). Органические слои промывали солевым раствором, сушили над MgSO4, фильтровали и выпаривали с получением необходимого промежуточного соединения мезилата в виде желтого масла (435 мг, 91%). МС (ИЭР): m/z=201,0 [M-mesyl+H] +.
Этап с) трет-бутил-3-((1-(2,4-дихлорфенил)циклопропил)метокси)азетидин-1-карбоксилат
В ледяной раствор трет-бутил-3-гидроксиазетидин-1-карбоксилата (220 мг, 1,27 ммоль) в ДМФА (4 мл) частями добавляли гидрид натрия в минеральном масле (60%, 61 мг, 1,52 ммоль) и перемешивали смесь при температуре ледяной бани в течение 5 мин с последующим перемешиванием при КТ в течение 40 мин. В смесь по каплям добавляли раствор 1-(2,4-дихлорфенил)циклопропил)метилметансульфоната (431 мг, 1,46 ммоль) в ДМФА (1 мл) при КТ. Перемешивание суспензии продолжали при КТ в течение 16 ч, затем при 55°С в течение 2,5 ч. Реакционную смесь вливали в насыщенный водный раствор NH4Cl (10 мл) и ЕtOАс (20 мл) и разделяли слои. Водный слой один раз экстрагировали ЕtOАс (50 мл). Органические слои дважды промывали водой, сушили над MgSO4, фильтровали и концентрировали. Флэш-хроматография (градиент ЕtOАс в н-гептане от 0 до 40%) позволила получить продукт в виде бесцветного масла (165 мг, 35%) МС (ИЭР): m/z=316,2 [М-56+Н]+.
ВВ182
2-((азетидин-3-илокси)метил)-6-(4-фторфенокси)-4-(трифторметил)пиридин 4-метилбензолсульфонат
Трет-бутил-3-((6-(4-фторфенокси)-4-(трифторметил)пиридин-2-ил)метокси)азетидин-1-карбоксилат (150 мг, 339 мкмоль) растворяли в атмосфере аргона в ЕtOАс (2 мл), добавляли моногидрат п-толуолсульфоновой кислоты (77,4 мг, 407 мкмоль) и перемешивали смесь при КТ в течение 5 мин, затем при 80°С в течение 3 ч и при КТ в течение ночи. Реакционную смесь выпаривали с получением соединения в виде 180 мг желтого масла, которое использовали на следующем этапе без дополнительной очистки. МС (ИЭР): m/z=343,2 [М+Н]+.
Этап а) трет-бутил-3-((6-бром-4-(трифторметил)пиридин-2-ил)метокси)азетидин-1 -карбоксилат
В раствор трет-бутил-3-гидроксиазетидин-1-карбоксилата (272 мг, 1,57 ммоль) в сухом ТГФ (8 мл) добавляли 1 М трет-бутоксид калия в ТГФ (1,57 мл, 1,57 ммоль) и перемешивали непрозрачную реакционную смесь при КТ в течение 30 мин. Добавляли 2-бром-6-(бромметил)-4-(трифторметил)пиридин (500 мг, 1,57 ммоль) при 0-2°С и перемешивали реакцию при 0-2°С в течение 20 мин. Затем реакционную смесь перемешивали при КТ в течение 16 ч. Реакционную смесь разводили ЕtOАс, экстрагировали водой, органическую фазу собирали, а водную фазу обратно экстрагировали ЕtOАс. Объединенные органические слои сушили над сульфатом натрия и выпаривали до сухости. Неочищенный материал очищали с помощью колоночной флэш-хроматографии (градиент ЕtOАс в н-гептане, от 0% до 40%) с получением продукта в виде светло-желтого масла (41%) МС (ИЭР): m/z=355,1 [М-56+Н] +.
Этап b) трет-бутил-3-[[6-(4-фторфенокси)-4-(трифторметил)-2-пиридил]метокси]азетидин-1-карбоксилат
Трет-бутил-3-((6-бром-4-(трифторметил)пиридин-2-ил)метокси)азетидин-1-карбоксилат (260 мг, 632 мкмоль) и 4-фторфенол (70,9 мг, 632 мкмоль) растворяли в ДМФА (2 мл), затем добавляли K2СО3 (131 мг, 948 мкмоль) и перемешивали смесь при 80° в течение 30 ч. Реакционную смесь выпаривали в вакууме, а остаток растворяли в EtOAc и экстрагировали водой и солевым раствором. Органические слои сушили над MgSO4, фильтровали, а растворитель удаляли в вакууме. Остаток очищали с помощью флэш-хроматографии (градиент ЕtOАс в n-гептане, от 0 до 30%) с получением продукта в виде светло-желтого масла (93%). МС (ИЭР): m/z=443,4 [М+Н] +.
ВВ183
6-((азетидин-3-илокси)метил)-2-(4-фторфенокси)-3-(трифторметил)пиридин 4-метилбензолсульфонат
трет-бутил-3-((6-(4-фторфенокси)-5-(трифторметил)пиридин-2-ил)метокси)азетидин-1-карбоксилат (170 мг, 384 мкмоль) растворяли в атмосфере аргона в ЕtOАс (2,27 мл) и добавляли моногидрат п-толуолсульфоновой кислоты (87,7 мг, 461 мкмоль). Реакцию перемешивали при КТ в течение 5 мин, затем при 80°С в течение 3 ч и перемешивали при КТ в течение ночи. Реакционную смесь выпаривали при пониженном давлении до сухости с получением необходимого продукта в виде светло-желтого масла (89%) МС (ИЭР): m/z=343,2 [М+Н] +.
Этап а) метил 6-(4-фторфенокси)-5-(трифторметил)пиколинат
Метил 6-хлор-5-(трифторметил)пиколинат (800 мг, 3,34 ммоль), 4-фторфенол (412 мг, 3,67 ммоль) и K2СО3 (692 мг, 5,01 ммоль) растворяли в ДМФА (6 мл) и перемешивали при 80°С в течение 6 ч. Реакционную смесь охлаждали до КТ и три раза экстрагировали водой (по 20 мл каждый раз), дважды ЕtOАс (по 30 мл каждый раз), солевым раствором (20 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме. Остаток очищали с помощью колоночной флэш-хроматографии (градиент ЕtOАс в n-гептане, от 0 до 50%) с получением продукта в виде белого твердого вещества (67%). МС (ИЭР): m/z=316,1 [М+Н] +.
Этап b) (6-(4-фторфенокси)-5-(трифторметил)пиридин-2-ил)метанол
В перемешиваемый раствор метил 6-(4-фторфенокси)-5-(трифторметил)пиколината (705 мг, 2,24 ммоль) в ТГФ (8 мл) добавляли 2 М борогидрид лития в ТГФ (1,34 мл, 2,68 ммоль) при 2-5°С. Реакционную смесь перемешивали при КТ в течение 3 ч, а затем охлаждали до 2-4°С и гасили 10 мл воды (медленно добавляемой). Водный слой дважды экстрагировали ЕtOАс (по 30 мл каждый раз), а объединенные органические слои промывали водой, 10 мл раствора NaHCO3 и 10 мл солевого раствора. Органический слой сушили над Na2SO4 и концентрировали в вакууме. Очистка с помощью колоночной флэш-хроматографии (градиент ЕtOАс в н-гептане от 0 до 50%) позволила получить продукт в виде бесцветного твердого вещества (95%). МС (ИЭР): m/z=288,2 [М+Н] +.
Этап с) 6-(бромметил)-2-(4-фторфенокси)-3-(трифторметил)пиридин
В раствор (6-(4-фторфенокси)-5-(трифторметил)пиридин-2-ил)метанола (330 мг, 1,15 ммоль) в сухом ДХМ (5 мл) добавляли тетрабромметан (457 мг, 1,38 ммоль). Смесь охлаждали до 0-3°С и добавляли трифенилфосфин (392 мг, 1,49 ммоль) в 1 мл сухого ДХМ за 10 мин. Смесь перемешивали 1 ч при 2-4°С, затем добавляли 20 мл ДХМ и силикагеля. Растворитель удаляли в вакууме, а остаток подвергали колоночной флэш-хроматографии (градиент ЕtOАс в н-гептане от 0 до 40%) с получением необходимого продукта в виде бесцветного масла (94%). МС (ИЭР): m/z=350,0 [М+Н] +.
Этап d) трет-бутил-3-((6-(4-фторфенокси)-5-(трифторметил)пиридин-2-ил)метокси)азетидин-1-карбоксилат
В раствор трет-бутил-3-гидроксиазетидин-1-карбоксилата (183 мг 1,06 ммоль) в сухом ТГФ (5 мл) добавляли 1 М трет-бутоксид калия в ТГФ (1,11 мл, 1,11 ммоль) и перемешивали реакционную смесь при КТ в течение 15 мин. Затем добавляли 6-(бромметил)-2-(4-фторфенокси)-3-(трифторметил)пиридин (370 мг, 1,06 ммоль). Реакционную смесь перемешивали при КТ в течение 1 ч, а затем разводили ЕtOАс и экстрагировали 1 М водн. раствором NaHCO3. Органическую фазу собирали, а водную фазу обратно экстрагировали ЕtOАс. Объединенные органические фазы сушили над сульфатом натрия и выпаривали до сухости. Остаток очищали с помощью колоночной флэш-хроматографии (градиент ЕtOАс в n-гептане, от 0 до 30%) с получением продукта в виде бесцветного масла (34%). МС (ИЭР: m/z=387,2 [М-56+Н] +.
ВВ184
2-((азетидин-3-илокси)метил)-4-(4-фторфенил)тиазол 2,2,2-трифторацетат
В раствор трет-бутил-3-((4-(4-фторфенил)тиазол-2-ил)метокси)азетидин-1-карбоксилата (150 мг, 412 мкмоль) в сухом ДХМ (1,5 мл) в атмосфере аргона добавляли ТФУ (282 мг, 190 мкл, 2,47 ммоль) и перемешивали раствор при КТ в течение 8 ч. Реакционную смесь концентрировали в вакууме (азеотроп с толуолом, EtOAc и гептан) с получением необходимого продукта в виде желтого твердого вещества (98%). МС (ИЭР): m/z=265,2 [М+Н] +.
Этап а) (4-(4-фторфенил)тиазол-2-ил)метанол
В перемешиваемый раствор этил 4-(4-фторфенил)тиазол-2-карбоксилата (835 мг, 3,32 ммоль) в сухом ТГФ (10 мл) добавляли 2 М борогидрид лития в ТГФ (1,99 мл, 3,99 ммоль) при 2-5°С. Реакционную смесь перемешивали при КТ в течение 3 ч, а затем охлаждали до 2-4°С и гасили водой (10 мл, медленно добавляемой). Водный слой дважды экстрагировали ЕtOАс (по 30 мл каждый раз), а органические слои промывали водой, 10 мл раствора NaHCO3 и 10 мл солевого раствора. Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали с помощью колоночной флэш-хроматографии (градиент ЕtOАс в n-гептане, от 0 до 60%) с получением необходимого продукта в виде белого твердого вещества (94%) МС (ИЭР): m/z=210,1 [М+Н] +.
Этап b) 2-(бромметил)-4-(4-фторфенил)тиазол
В раствор (4-(4-фторфенил)тиазол-2-ил)метанола (400 мг, 1,91 ммоль) в сухом ДХМ (7 мл) добавляли тетрабромметан (761 мг, 2,29 ммоль), раствор охлаждали до 0-3°С и добавляли трифенилфосфин (652 мг, 2,49 ммоль) в 1 мл сухого ДХМ за 10 мин. Смесь перемешивали при 2-4°С в течение 1 ч, затем добавляли 20 мл ДХМ. Реакционную смесь экстрагировали водой, насыщенным раствором NH4Cl и солевым раствором. Органическую фазу сушили над MgSO4, фильтровали и выпаривали. Остаток очищали с помощью флэш-хроматографии (градиент EtOAc в n-гептане, от 0 до 40%) с получением 480 мг указанного в заголовке соединения в виде светло-желтого масла (83%). МС (ИЭР): m/z=273,9 [М+Н] +
Этап с) трет-бутил-3-((4-(4-фторфенил)тиазол-2-ил)метокси)азетидин-1-карбоксилат
В раствор трет-бутил-3-гидроксиазетидин-1-карбоксилата (293 мг 1,69 ммоль) в сухом ТГФ (6 мл) добавляли 1 М трет-бутоксид калия в ТГФ (1,77 мл, 1,77 ммоль) и перемешивали реакционную смесь при КТ в течение 15 мин. После охлаждения до 2-4°С добавляли 2-(бромметил)-4-(4-фторфенил)тиазол (460 мг, 1,69 ммоль) в 1 мл ТГФ. Реакционную смесь перемешивали при КТ в течение 1 ч, разводили ЕtOАс и экстрагировали 1 М водн. раствором NaHCO3. Органическую фазу собирали, а водную фазу обратно экстрагировали ЕtOАс. Объединенные органические фазы сушили над Na2SO4 и выпаривали до сухости. Остаток очищали с помощью колоночной флэш-хроматографии (градиент ЕtOАс в n-гептане, от 0 до 40%) с получением необходимого продукта в виде желтого твердого вещества (89%). МС (ИЭР): m/z=365,2 [М+Н] +.
ВВ186
рац-(2R,3S)-3-(2-бром-5-(трифторметил)фенокси)-2-метилпирролидин 2,2,2-трифторацетат
В раствор рац-трет-бутил-(2R,3S)-3-(2-бром-5-(трифторметил)фенокси)-2-метилпирролидин-1-карбоксилата (225 мг, 530 мкмоль) в сухом ДХМ (2 мл) в атмосфере аргона добавляли ТФУ (242 мг, 163 мкл, 2,12 ммоль) и перемешивали раствор при КТ в течение ночи. Реакционную смесь концентрировали в вакууме до сухости (азеотроп с н-гептаном) с получением 233 мг указанного в заголовке соединения в виде бесцветного масла (97%). МС (ИЭР): m/z=324,1 [М+Н] +.
Этап а) рац-трет-бутил-(2R,3S)-3-(2-бром-5-(трифторметил)фенокси)-2-метилпирролидин-1-карбоксилат
В раствор рац-трет-бутил-(2R,3S)-3-гидрокси-2-метилпирролидин-1-карбоксилата (CAS:1807941-04-3, 150 мг, 745 мкмоль) в сухом ТГФ (4 мл) в атмосфере аргона добавляли 1 М трет-бутоксид калия в ТГФ (783 мкл, 783 мкмоль). Смесь перемешивали при КТ в течение 15 мин, затем охлаждали до 2-4°С и медленно добавляли раствор 1-бром-2-фтор-4-(трифторметил)бензола (181 мг, 745 мкмоль) в 0,5 мл сухого ТГФ. Смесь перемешивали при КТ в течение 2 ч, затем экстрагировали EtOAc и водным 5% раствором NaHCO3, а после - водой и солевым раствором. Органическую фазу сушили над MgSO4, фильтровали и выпаривали до сухости. Остаток очищали с помощью колоночной флэш-хроматографии (градиент EtOAc в n-гептане, от 0 до 40%) с получением продукта в виде светло-желтого масла (71%). МС (ИЭР: m/z=368 [М-56+Н] +.
Следующие промежуточные соединения синтезировали из 4-нитрофенил (4аR,8аS)-3-оксогексагидро-2Н-пиридо[4,3-b] [1,4]оксазин-6(5Н)-карбоксилата (ВВ7а) и подходящих строительных блоков по аналогии с реакционными способами, описанными в данном документе.

ВВ206
3-[2-[2-фтор-4-(трифторметил)фенил]этил]азетидин; 4-метилбензолсульфоновая кислота
Это соединение получали по аналогии с ВВ95 из трет-бутил-3-(2-фтор-4-(трифторметил)фенэтил)азетидин-1-карбоксилата и моногидрата 4-метилбензолсульфоновой кислоты. После охлаждения образовывалась суспензия, которую фильтровали. Фильтрационный осадок промывали небольшим объемом ЕtOАс с получением необходимого продукта в виде бесцветного твердого вещества (71,6%). МС (ИЭР): m/z=248,2 [М+Н] +.
Этап а) диэтил (2-фтор-4-(трифторметил)бензил)фосфонат
Это соединение получали по аналогии с ВВ159, этап а, из 1-(бромметил)-2-фтор-4-(трифторметил)бензола и триэтилфосфина. Бесцветное масло (83,4%). МС (ИЭР): m/z=315,2 [М+Н]+.
Этап b) трет-бутил-3-[(Е)-2-[2-фтор-4-(трифторметил)фенил]винил]азетидин-1-карбоксилат
Это соединение получали по аналогии с ВВ95, этап а, из диэтил (2-фтор-4-(трифторметил)бензил)фосфоната и трет-бутил-3-формилазетидин-1-карбоксилата с получением соединения в виде бесцветного масла (69,9%). МС (ИЭР: m/z=290,1 [М-56+Н] +.
Этап с) трет-бутил-3-[2-[2-фтор-4-(трифторметил)фенил]этил]азетидин-1-карбоксилат
Это соединение получали по аналогии с ВВ95, этап b, из трет-бутил-3-[(Е)-2-[2-фтор-4-(трифторметил)фенил]винил]азетидин-1-карбоксилата. Бесцветное масло (92,0%). МС (ИЭР: m/z=292,2 [М-56+Н]+.
ВВ208
3-[2,2-дифтор-2-[4-(трифторметил)фенил]этил]азетидин; 4-метилбензолсульфоновая кислота
Это соединение получали по аналогии с ВВ95 из трет-бутил-3-(2,2-дифтор-2-(4-(трифторметил)фенил)этил)азетидин-1-карбоксилата и моногидрата 4-метилбензолсульфоновой кислоты и с использованием материала, выделенного после выпаривания, который использовали без дополнительной очистки (30%). МС (ИЭР): m/z=266,2 [М+Н] +.
Этап а) трет-бутил-3-[2-[метокси(метил)амино]-2-оксоэтил]азетидин-1-карбоксилат
В суспензию 2-(1-(трет-бутоксикарбонил)азетидин-3-ил)уксусной кислоты (2 г, 9,29 ммоль) и HATU (3,89 г, 10,2 ммоль) в ДХМ (65 мл) добавляли ДИПЭА (2,64 г, 3,57 мл, 20,4 ммоль) и перемешивали смесь при КТ в течение 30 мин перед добавлением N,O-диметилгидроксиламин гидрохлорида (906 мг, 9,29 ммоль). Перемешивание продолжали в течение ночи при КТ. Реакционную смесь вливали в насыщенный водный раствор NH4Cl и ЕtOАс и разделяли слои. Водный слой дважды экстрагировали EtOAc. Органические слои дважды промывали водой, сушили над MgSO4, фильтровали, обрабатывали силикагелем и выпаривали. Соединение очищали с помощью хроматографии на силикагеле на 25 г колонке, используя систему для ЖХСД, с элюированием градиентом н-гептан: ЕtOАс (от 100: от 0 до 0: 100) с получением необходимого соединения в виде бесцветного масла (100%), которое использовали на следующем этапе без дополнительной очистки. МС (ИЭР: m/z=203,2 [М-56+Н]+.
Этап b) трет-бутил-3-[2-оксо-2-[4-(трифторметил)фенил]этил]азетидин-1-карбоксилат
В ледяной раствор трет-бутил-3-[2-[метокси(метил)амино]-2-оксоэтил]азетидин-1-карбоксилата (0,8 г, 3,1 ммоль) в ТГФ (5 мл) в продутой аргоном и термически высушенной колбе с 2 горлышками по каплям добавляли непрозрачный 2,22 М раствор (4-(трифторметил)фенил)магния бромида в ТГФ (1,95 мл, 4,34 ммоль). Коричневый раствор перемешивали на ледяной бане в течение 2,5 ч, позволяя температуре подняться до КТ. Реакционную смесь вливали в насыщенный водный раствор NH4Cl и EtOAc и разделяли слои. Водный слой дважды экстрагировали ЕtOАс. Органические слои сушили над MgSO4, фильтровали, обрабатывали силикагелем и выпаривали. Соединение очищали с помощью хроматографии на силикагеле на 25 г колонке, используя систему для ЖХСД, с элюированием градиентом н-гептан: EtOAc (от 100: от 0 до 0: 100) с получением необходимого соединения в виде бесцветного твердого вещества (25,9%). МС (ИЭР): m/z=342,3 [М-Н]-
Этап с) трет-бутил-3-[2,2-дифтор-2-[4-(трифторметил)фенил]этил]азетидин-1-карбоксилат
В раствор трет-бутил-3-[2-оксо-2-[4-(трифторметил)фенил]этил]азетидин-1-карбоксилата (50 мг, 146 мкмоль) в толуоле (0,3 мл) в атмосфере аргона добавляли бис(2-метоксиэтил)аминосерный трифторид (50% раствор в ТГФ, 387 мг, 379 мкл, 874 мкмоль) и перемешивали смесь при 80°С в течение 19 ч. Темную смесь оставляли охлаждаться и добавляли другую партию бис(2-метоксиэтил)аминосерного трифторида (50% раствор в ТГФ, 387 мг, 379 мкл, 874 мкмоль). Нагревание продолжали до 80°С еще в течение 4 ч. Реакционную смесь вливали в насыщенный водный раствор NaHCO3 и EtOAc и разделяли слои. Водный слой дважды экстрагировали ЕtOАс. Органические слои сушили над MgSO4, фильтровали, обрабатывали силикагелем и выпаривали. Соединение очищали с помощью хроматографии на силикагеле на 4 г колонке, используя систему для ЖХСД, с элюированием градиентом н-гептан: EtOAc (от 100: от 0 до 50: 50) с получением необходимого соединения в виде светло-коричневого масла (45,1%). МС (ИЭР): m/z=266,1 [М+Н]+.
ВВ209
3-[2-Фтор-5-(трифторметил)фенокси]пирролидин; 4-метилбензолсульфоновая кислота
Это соединение получали по аналогии с ВВ95 из трет-бутил-3-[2-фтор-5-(трифторметил)фенокси]пирролидин-1-карбоксилата. Бесцветное масло использовали на следующем этапе без дополнительной очистки. МС (ИЭР): m/z=250,1 [М+Н]+.
Этап а) трет-бутил-3-[2-фтор-5-(трифторметил)фенокси]пирролидин-1-карбоксилат
В раствор 2-фтор-5-(трифторметил)фенола (321 мг, 1,78 ммоль), трет-бутил-3-гидроксипирролидин-1-карбоксилата (334 мг, 1,78 ммоль, CAS RN: 103057-44-9) и трифенилфосфина (467 мг, 1,78 ммоль) в ТГФ (5 мл) частями добавляли (Е)-диазен-1,2-диилбис(пиперидин-1-илметанон) (450 мг, 1,78 ммоль, CAS RN 10465-81-3) и перемешивали смесь при КТ в течение 40 ч. В суспензию добавляли силикагель и выпаривали. Соединение очищали с помощью хроматографии на силикагеле на 24 г колонке, используя систему для ЖХСД (ISCO), с элюированием градиентом н-гептан: ЕtOАс (от 100: от 0 до 75: 25) с получением необходимого соединения в виде бесцветного масла (8,3%), которое использовали на следующем этапе без дополнительной очистки. МС (ИЭР: m/z=294,1 [М-56+Н]+.
ВВ210
3-[2-хлор-5-(трифторметил)фенокси] пирролидин; 4-метилбензолсульфоновая кислота
Это соединение получали по аналогии с ВВ95 из трет-бутил-3-[2-хлор-5-(трифторметил)фенокси] пирролидин-1-карбоксилата. Бесцветное масло. МС (ИЭР): m/z=266,1 [М+Н]+.
Этап а) трет-бутил-3-[2-хлор-5-(трифторметил)фенокси] пирролидин-1-карбоксилат
Это соединение получали по аналогии с ВВ209, этап а, из 2-хлор-5-(трифторметил)фенола и трет-бутил-3-гидроксипирролидин-1-карбоксилата. Бесцветное твердое вещество использовали после хроматографии без дополнительной очистки. МС (ИЭР: m/z=310,1 [М-56+Н]+.
ВВ211
3-[(Е)-2-(2-фтор-4-метилфенил)винил]азетидин; 4-метилбензолсульфоновая кислота
Это соединение получали по аналогии с ВВ95 из трет-бутил-3-[(Е)-2-(2-фтор-4-метилфенил)винил]азетидин-1-карбоксилата и моногидрата 4-метилбензолсульфоновой кислоты. Бесцветное твердое вещество (87%). МС (ИЭР): m/z=192,2 [М+Н]+.
Этап а) 1-(диэтоксифосфорилметил)-2-фтор-4-метилбензол
Это соединение получали по аналогии с ВВ206, этап а, из 1-(бромметил)-2-фтор-4-метилбензола и триэтилфосфина с последующей хроматографией на силикагеле на 40 г колонке с использованием системы для ЖХСД (ISCO), с элюированием градиентом н-гептан: EtOAc (от 100: от 0 до 0: 100). Бесцветная жидкость (85%). МС (ИЭР): m/z=261,1 [М+Н]+.
Этап b) трет-бутил-3-[(Е)-2-(2-фтор-4-метилфенил)винил]азетидин-1-карбоксилат
Это соединение получали по аналогии с примером ВВ206, этап Ь, из трет-бутил-3-формилазетидин-1-карбоксилата и 1-(диэтоксифосфорилметил)-2-фтор-4-метилбензола. Бесцветное масло (7%). МС (ИЭР: m/z=236,2 [М-56+Н]+.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ МОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ | 2019 |
|
RU2801190C2 |
| ИНГИБИТОРЫ ОКСАЗИНМОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ (MAGL) | 2019 |
|
RU2794334C2 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2019 |
|
RU2809257C2 |
| ОКТАГИДРО КОНДЕНСИРОВАННЫЕ АЗАДЕКАЛИНОВЫЕ МОДУЛЯТОРЫ ГЛЮКОКОРТИКОИДНОГО РЕЦЕПТОРА | 2014 |
|
RU2674983C1 |
| ИНГИБИТОРЫ ИНТЕГРИНА AVB6 | 2018 |
|
RU2769702C2 |
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ FASN | 2014 |
|
RU2737434C2 |
| КОМПОЗИЦИИ ТЕТРАГИДРОХИНОЛИНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ БРОМОДОМЕНА ВЕТ | 2014 |
|
RU2727169C2 |
| Производные хинолона как ингибиторы рецептора фактора роста фибробластов | 2015 |
|
RU2721723C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПРОСТОГО ЭФИРА ИЗОКСАЗОЛИЛА В КАЧЕСТВЕ ПАМ ГАМК A АЛЬФА5 | 2019 |
|
RU2800160C2 |
| ЗАМЕЩЕННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ КОНДЕНСИРОВАННОЕ ЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2815814C1 |
Изобретение относится к соединению формулы (I), где (i) X представляет собой C-R3; m равно 0 или 1; n выбрано из 0, 1 и 2; и L выбран из -(СН2)Р-, -О-, -ОСН2-, -СН2ОСН2-, -CF2CH2-, -СН2=СН2- и -(CR16R17)q-CH2O-; или (ii) X представляет собой N; m равно 1; n равно 1; и L представляет собой -(СН2)Р-; р выбрано из 1 и 2; q равно 0 или 1; А выбран из: (i) фенила, замещенного R4, R5 и R6; (ii) 5-9-членного гетероарила, содержащего 1, 2 или 3 гетероатома, независимо выбранных из О, S и N, замещенного R7, R8 и R9; и (iii) 6-членного гетероциклоалкила, содержащего 1 гетероатом, представляющий собой N, замещенного R10, R11 и R12, где один из R10, R11 и R12 представляет собой водород, а два других представляют собой галоген; R1 представляет собой водород или С1-6-алкил; R2 выбран из водорода и С1-6-алкила; R3 выбран из водорода, галогена, С1-6-алкила и галоген-С1.6-алкила; каждый из R4, R5, R6, R7, R8 и R9 независимо выбран из водорода, галогена, циано, гидрокси, С1-6-алкила, галоген-С1-6-алкила, С1-6-алканоила, С1-6-алкокси, галоген-С1-6-алкокси, SF5, С3-10-циклоалкила, 5-9-членного гетероциклоалкила, где 1 или 2 из указанных кольцевых атомов представляют собой гетероатомы, выбранные из N и О, 4-7-членного гетероциклоалкила, где 1 из указанных кольцевых атомов представляет собой гетероатом, выбранный из N, замещенного R14 и R15, 5-членного гетероарила, содержащего 2 или 3 гетероатома, выбранных из N, фенила, фенокси, галоген-фенила и галоген-фенокси; каждый из R14 и R15 независимо выбран из С1-6-алкила, С1-6-алкокси и галогена; и R16 и R17 вместе с атомом углерода, к которому они присоединены, образуют С3-10-циклоалкил. Изобретение также относится к способу получения указанных соединений формулы (I). Технический результат – получены новые соединения, которые могут найти применение в медицине для получения лекарственного средства для ингибирования MAGL. 6 н. и 35 з.п. ф-лы, 13 табл., 278 пр.
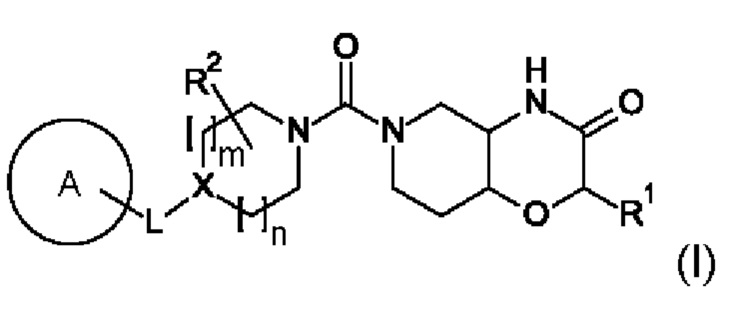
1. Соединение формулы (I)

где:
(i) X представляет собой C-R3; m равно 0 или 1; n выбрано из 0, 1 и 2; и L выбран из -(СН2)Р-, -О-, -ОСН2-, -СН2ОСН2-, -CF2CH2-, -СН2=СН2- и -(CR16R17)q-CH2O-; или
(ii) X представляет собой N; m равно 1; n равно 1; и L представляет собой -(СН2)Р-;
р выбрано из 1 и 2;
q равно 0 или 1;
А выбран из:
(i) фенила, замещенного R4, R5 и R6;
(ii) 5-9-членного гетероарила, содержащего 1, 2 или 3 гетероатома, независимо выбранных из О, S и N, замещенного R7, R8 и R9; и
(iii) 6-членного гетероциклоалкила, содержащего 1 гетероатом, представляющий собой N, замещенного R10, R11 и R12, где один из R10, R11 и R12 представляет собой водород, а два других представляют собой галоген;
R1 представляет собой водород или С1-6-алкил;
R2 выбран из водорода и С1-6-алкила;
R3 выбран из водорода, галогена, С1-6-алкила и галоген-С1.6-алкила;
каждый из R4, R5, R6, R7, R8 и R9 независимо выбран из водорода, галогена, циано, гидрокси, С1-6-алкила, галоген-С1-6-алкила, С1-6-алканоила, С1-6-алкокси, галоген-С1-6-алкокси, SF5, С3-10-циклоалкила, 5-9-членного гетероциклоалкила, где 1 или 2 из указанных кольцевых атомов представляют собой гетероатомы, выбранные из N и О, 4-7-членного гетероциклоалкила, где 1 из указанных кольцевых атомов представляет собой гетероатом, выбранный из N, замещенного R14 и R15, 5-членного гетероарила, содержащего 2 или 3 гетероатома, выбранных из N, фенила, фенокси, галоген-фенила и галоген-фенокси;
каждый из R14 и R15 независимо выбран из С1-6-алкила, С1-6-алкокси и галогена; и
R16 и R17 вместе с атомом углерода, к которому они присоединены, образуют С3-10-циклоалкил.
2. Соединение формулы (I) по п. 1, где соединение формулы (I) представляет собой соединение формулы (Ia):

где A, L, X, m, n, R1 и R2 такие, как определено в п. 1 формулы.
3. Соединение формулы (I) по п. 1, где соединение формулы (I) представляет собой соединение формулы (Ib):

где A, L, X, m, n, R1 и R2 такие, как определено в п. 1 формулы.
4. Соединение формулы (I) по любому из пп. 1-3, где:
(i) X представляет собой C-R3; m равно 0 или 1; n выбрано из 0, 1 и 2; и L выбран из -(СН2)Р-, -О-, -ОСН2-, -CF2CH2-, -СН2=СН2- и -(CR16R17)q-CH2O-; или
(ii) X представляет собой N; m и n оба равны 1; и L представляет собой -(СН2)p-.
5. Соединение формулы (I) по любому из пп. 1-4, где:
X представляет собой C-R3;
m и n оба равны 0; или
m и n оба равны 1; и
L выбран из -(СН2)Р-, -О- и -ОСН2-.
6. Соединение формулы (I) по любому из пп. 1-5, где р равно 2.
7. Соединение формулы (I) по любому из пп. 1-6, где q равно 0.
8. Соединение формулы (I) по любому из пп. 1-7, где А выбран из:
(i) фенила, замещенного R4, R5 и R6; и
(ii) 5-9-членного гетероарила, содержащего 1, 2 или 3 гетероатома, независимо выбранных из О, S и N, замещенного R7, R8 и R9.
9. Соединение формулы (I) по любому из пп. 1-7, где А выбран из:
(i) фенила, замещенного R4, R5 и R6;
(ii) оксазолила, замещенного R7, R8 и R9; и
(iii) пиридила, замещенного R7, R8 и R9.
10. Соединение формулы (I) по любому из пп. 1-9, где R1 представляет собой водород.
11. Соединение формулы (I) по любому из пп. 1-10, где R2 представляет собой водород или метил.
12. Соединение формулы (I) по любому из пп. 1-11, где R3 выбран из водорода, галогена или С1-6-алкила.
13. Соединение формулы (I) по любому из пп. 1-11, где R3 выбран из водорода, фтора и метила.
14. Соединение формулы (I) по любому из пп. 1-13, где R4 выбран из водорода, галогена, гидрокси, циано, С1-6-алкила, С1-6-алканоила, SF5, С1-6-алкокси, галоген-С1-6-алкокси, галоген-С1.6-алкила, С3-10-циклоалкила, 5-9-членного гетероциклила, где 1 или 2 из указанных кольцевых атомов представляют собой гетероатомы, выбранные из N и О, 4-7-членного гетероциклоалкила, где 1 из указанных кольцевых атомов представляет собой гетероатом, выбранный из N, замещенного R14 и R15, 5-членного гетероарила, содержащего 2 или 3 гетероатома, выбранных из N, фенокси и галоген-фенила.
15. Соединение формулы (I) по любому из пп. 1-13, где R4 выбран из галогена, SF5, С1-6-алкила, С1-6-алкокси, галоген-С1-6-алкокси, галоген-С1-6-алкила, С3-10-циклоалкила и 5-9-членного гетероциклоалкила, где 1 или 2 из указанных кольцевых атомов представляют собой гетероатомы, выбранные из N и О.
16. Соединение формулы (I) по любому из пп. 1-13, где R4 выбран из хлора, SF5, метила, метокси, OCF3, CF3, циклопропила и 2-азаспиро[3.3]гептан-2-ила.
17. Соединение формулы (I) по любому из пп. 1-16, где R5 выбран из водорода, циано, галогена, С1-6-алкила, галоген-С1-6-алкила, С1-6-алкокси, 5-9-членного гетероциклоалкила, где 1 или 2 из указанных кольцевых атомов представляют собой гетероатомы, выбранные из N и О, С3-10-циклоалкила, 5-членного гетероарила, содержащего 2 или 3 гетероатома, выбранных из N, и галоген-фенила.
18. Соединение формулы (I) по любому из пп. 1-16, где R5 выбран из водорода, циано, галогена, С1-6-алкила, галоген-С1-6-алкила, 5-9-членного гетероциклоалкила, где 1 или 2 из указанных кольцевых атомов представляют собой гетероатомы, выбранные из N и О, С3-10-циклоалкила и галоген-фенила.
19. Соединение формулы (I) по любому из пп. 1-16, где R5 выбран из водорода, циано, фтора, хлора, метила, CF3, пирролидинила, циклопентила, циклопропила и хлорфенила.
20. Соединение формулы (I) по любому из пп. 1-19, где R6 представляет собой водород или галоген.
21. Соединение формулы (I) по любому из пп. 1-19, где R6 представляет собой водород.
22. Соединение формулы (I) по любому из пп. 1-21, где R7 выбран из водорода, С1-6-алкила, фенила, галоген-фенила, галоген-фенокси и галоген-С1-6-алкила.
23. Соединение формулы (I) по любому из пп. 1-21, где R7 представляет собой С1-6-алкил или галоген-С1-6-алкил.
24. Соединение формулы (I) по любому из пп. 1-21, где R7 представляет собой трет-бутил или CF3.
25. Соединение формулы (I) по любому из пп. 1-24, где R8 выбран из водорода, галогена, С1-6-алкила или галоген-С1-6-алкила.
26. Соединение формулы (I) по любому из пп. 1-24, где R8 представляет собой водород или галоген-С1-6-алкил.
27. Соединение формулы (I) по любому из пп. 1-24, где R8 представляет собой водород или CF3.
28. Соединение формулы (I) по любому из пп. 1-27, где R9 представляет собой водород.
29. Соединение формулы (I) по любому из пп. 1-28, где R10 представляет собой галоген.
30. Соединение формулы (I) по любому из пп. 1-29, где R11 представляет собой галоген.
31. Соединение формулы (I) по любому из пп. 1-30, где R12 представляет собой водород.
32. Соединение формулы (I) по любому из пп. 1-31, где R15 представляет собой галоген.
33. Соединение формулы (I) по любому из пп. 1-3, где:
(i) X представляет собой C-R3; m равно 0 или 1; n выбрано из 0, 1 и 2; и L выбран из -(СН2)Р-, -О-, -ОСН2-, -(CR16R17)q-CH2O-, -СН2ОСН2-, -CF2CH2- и -СН2=СН2-; или
(ii) X представляет собой N; m и n оба равны 1; и L представляет собой -(СН2)p-;
р равно 1 или 2;
q равно 0 или 1;
А выбран из:
(i) фенила, замещенного R4, R5 и R6;
(ii) 5-9-членного гетероарила, содержащего 1, 2 или 3 гетероатома, независимо выбранных из О, S и N, замещенного R7, R8 и R9; и
(iii) 6-членного гетероциклоалкила, содержащего 1 гетероатом, представляющий собой N, замещенного R10, R11 и R12;
R1 представляет собой водород или С1-6-алкил;
R2 представляет собой водород или С1-6-алкил;
R3 выбран из водорода, галогена, С1-6-алкила и галоген-С1-6-алкила;
R4 выбран из водорода, галогена, циано, SF5, С1-6-алкила, С1.6-алканоила, С1-6-алкокси, галоген-С1.6-алкокси, галоген-С1-6-алкила, С3.10-циклоалкила, 5-9-членного гетероциклоалкила, где 1 или 2 из указанных кольцевых атомов представляют собой гетероатомы, выбранные из N и О, 4-7-членного гетероциклоалкила, где 1 из указанных кольцевых атомов представляет собой гетероатом, выбранный из N, замещенного R14 и R15, 5-членного гетероарила, содержащего 2 или 3 гетероатома, выбранных из N, галоген-фенила и фенокси;
R5 выбран из водорода, циано, галогена, С1-6-алкила, галоген-С1-6-алкила, C1-6-алкокси, 5-9-членного гетероциклоалкила, где 1 или 2 из указанных кольцевых атомов представляют собой гетероатомы, выбранные из N и О, С3-10-циклоалкила, 5-членного гетероарила, содержащего 2 или 3 гетероатома, выбранных из N, и галоген-фенила;
R6 представляет собой водород или галоген;
R7 выбран из водорода, С1-6-алкила, фенила, галоген-фенила, галоген-фенокси и галоген-С1.6-алкила;
R8 выбран из водорода, галогена, С1-6-алкила и галоген-С1.6-алкила;
R9 представляет собой водород;
R10 представляет собой галоген;
R11 представляет собой галоген;
R12 представляет собой водород;
R14 выбран из галогена, С1-6-алкила и С1-6-алкокси;
R15 представляет собой галоген; и
R16 и R17 вместе с атомом углерода, к которому они присоединены, образуют С3-10-циклоалкил.
34. Соединение формулы (I) по любому из пп. 1-3, где:
X представляет собой C-R3;
m и n оба равны 0; или
m и n оба равны 1;
L выбран из -(СН2)Р-, -О- и -ОСН2-;
р равно 1 или 2;
А выбран из:
(i) фенила, замещенного R4, R5 и R6; и
(ii) 5-9-членного гетероарила, содержащего 1, 2 или 3 гетероатома, независимо выбранных из О, S и N, замещенного R7, R8 и R9;
R1 представляет собой водород;
R2 представляет собой водород или С1-6-алкил;
R3 выбран из водорода, галогена и С1-6-алкила;
R4 выбран из галогена, SF5, С1-6-алкила, С1-6-алкокси, галоген-С1.6-алкокси, галоген-С1.6-алкила, С3-10-циклоалкила и 5-9-членного гетероциклоалкила, где 1 или 2 из указанных кольцевых атомов представляют собой гетероатомы, выбранные из N и О;
R5 выбран из водорода, циано, галогена, С1-6-алкила, галоген-С1-6-алкила, 5-9-членного гетероциклоалкила, где 1 или 2 из указанных кольцевых атомов представляют собой гетероатомы, выбранные из N и О, С3-10-циклоалкила и галоген-фенила;
R6 представляет собой водород;
R7 представляет собой С1-6-алкил и галоген-С1-6-алкил;
R8 представляет собой водород или галоген-С1-6-алкил; и
R9 представляет собой водород.
35. Соединение формулы (I) по любому из пп. 1-3, где:
X представляет собой C-R3;
m и n оба равны 0; или
m и n оба равны 1;
L выбран из -(СН2)Р-, -О- и -ОСН2-;
р равно 1 или 2;
А выбран из:
(i) фенила, замещенного R4, R5 и R6;
(ii) оксазолила, замещенного R7, R8 и R9; и
(iii) пиридила, замещенного R7, R8 и R9;
R1 представляет собой водород;
R2 представляет собой водород или метил;
R3 выбран из водорода, фтора и метила;
R4 выбран из хлора, SF5, метила, метокси, OCF3, CF3, циклопропила и 2-азаспиро[3.3]гептан-2-ила;
R5 выбран из водорода, циано, фтора, хлора, метила, CF3, пирролидинила, циклопентила, циклопропила и хлорфенила;
R6 представляет собой водород;
R7 выбран из трет-бутила, метила и CF3;
R8 представляет собой водород или CF3; и
R9 представляет собой водород.
36. Соединение формулы (I) по любому из пп. 1-35, выбранное из соединений:
(+)-(4aR,8aS)-6-(4-((4-(трет-бутил)тиазол-2-ил)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)-(4aR,8aS)-6-(4-((4-(трет-бутил)оксазол-2-ил)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
рац-(4aR,8aS)-6-[4-[(4-трет-бутилтиазол-2-ил)метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
рац-(4aR,8aS)-6-(4-((4-(трет-бутил)оксазол-2-ил)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(-)-(4aS,8aR)-6-(4-((4-(трет-бутил)тиазол-2-ил)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(-)-(4aS,8aR)-6-(4-((4-(трет-бутил)оксазол-2-ил)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
рац-(4aR,8aS)-6-(4-((2-хлор-4-фторфенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)-(4aR,8aS)-6-[4-[[4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
рац-(4aR,8aS)-6-(4-((4-хлорфенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
рац-(4aR,8aS)-6-(4-(4-хлорбензил)пиперидин-1-карбонил)гексагидро-2H-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
рац-(4aR,8aS)-6-[4-[[4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)- или (-)-(4aR,8aS)-6-(4-(4-(трифторметил)фенокси)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
рац-(4aR,8aS)-6-(4-(4-(трифторметил)фенокси)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)-(4aS,8aS)-6-(4-(4-(трифторметил)бензил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
рац-(4aR,8aS)-6-[4-(феноксиметил)пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
рац-(4aR,8aS)-6-(4-((5,6-дигидро-4Н-циклопента[с1]тиазол-2-ил)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
рац-(4aS,8aS)-6-(4-(4-(трифторметил)бензил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
рац-(4aR,8aS)-6-(4-((3-фенил-1,2,4-оксадиазол-5-ил)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(-)-(4aR,8aR)-6-(4-(4-(трифторметил)бензил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(-)-(4aS,8aR)-6-[4-[[4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
рац-(4aR,8aS)-6-(4-(4-(трифторметил)бензил)пиперазин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
рац-(4aR,8aS)-6-(4-(4-хлорбензил)пиперазин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(-)-(4aS,8aR)-6-(4-((2-хлор-4-фторфенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)-(4aR,8aS)-6-(4-((2-хлор-4-фторфенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
рац-(4aR,8aS)-6-[4-[[5-(трифторметил)-2-пиридил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aS,8aR)-6-(4-((4-(трифторметил)-1Н-пиразол-1-ил)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
рац-(4aR,8aS)-6-(3-((2-фтор-4-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)- или (-)-(4aR,8aS)-6-(3-((2-хлор-4-фторбензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)- или (-)-(4aR,8aS)-6-(3-((2-фтор-4-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)- или (-)-(4aS,8aR)-6-(3-((2-фтор-4-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)- или (-)-(4aR,8aS)-6-(4-(4-(трифторметокси)бензил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)- или (-)-(4aR,8aS)-6-(4-((2,4-дифторфенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)- или (-)-(4aR,8aS)-6-(4-(4-хлор-3-фторбензил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)- или (-)-4-[[1-[(4aR,8aS)-3-оксо-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-6-карбонил]-4-пиперидил]метил]бензонитрил,
(+)- или (-)-(4aR,8aS)-6-(4-(4-хлорбензил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)- или (-)-(4aR,8aS)-6-[3-[[4-(трифторметил)фенил]метил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-(4-((4,4-дифторпиперидин-1-ил)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)- или (-)-(4aR,8aS)-6-(4-((5-(трет-бутил)оксазол-2-ил)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-[4-[(2-фтор-4-метоксифенокси)метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)- или (-)-(4aR,8aS)-6-(4-(2-хлор-4-(трифторметил)фенокси)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aS,8aR)-6-(4-(((6-(трифторметил)пиридин-3-ил)окси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(4-(((6-(трифторметил)пиридин-3-ил)окси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)- или (-)-(4aR,8aS)-6-(4-(3-(трифторметил)фенокси)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aS,8aR)-6-[4-[[2-хлор-4-(трифторметокси)фенокси]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-[[2-хлор-4-(трифторметокси)фенокси]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-(3-((2-фтор-4-(трифторметил)фенокси)метил)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aS,8aR)-6-[4-[(2-хлор-4-фторфенокси)метил]-4-метилпиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aS,8aR)-6-(4-((2,4-дифторфенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(4-((4-хлор-2-фторфенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(4-((4-фтор-2-(трифторметил)фенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(4-((2-фтор-4-(трифторметил)фенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)- или (-)-(4aR,8aS)-6-(4-(2-(пирролидин-1-ил)-4-(трифторметил)бензил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(4-((2-хлор-4-(трифторметил)фенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
5-фтор-2-((1-((4aR,8aS)-3-оксооктагидро-2Н-пиридо[4,3-b][1,4]оксазин-6-карбонил)пиперидин-4-ил)метокси)бензонитрил,
(4aR,8aS)-6-(4-(2-(пирролидин-1-ил)-4-(трифторметил)бензил)пиперазин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(3-((2-хлор-4-фторфенокси)метил)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)- или (-)-(4aR,8aS)-6-[4-[[2-цикпопентил-4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-3-хлор-4-((1-(3-оксооктагидро-2Н-пиридо[4,3-b][1,4]оксазин-6-карбонил)пиперидин-4-ил)метокси)бензонитрил,
(4aR,8aS)-6-(4-((4-(трифторметил)-1Н-имидазол-1-ил)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(4-((4-фтор-2-метилфенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(3-((2-хлор-4-(трифторметил)фенокси)метил)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(4-((2-хлор-4-фторфенокси)метил)-4-фторпиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(4-((4-(трет-бутил)-1Н-пиразол-1-ил)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(2R,4aR,8aS)-2-метил-6-[4-[[4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)- или (-)-(4aR,8aS)-6-(4-(бензо[d]оксазол-2-илметил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)- или (-)-(4aR,8aS)-6-(4-((4',6-дихлор-[1,1'-бифенил]-3-ил)окси)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-цис-4-((2-хлор-4-фторфенокси)метил)-3-метилпиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(3-((2-хлор-4-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(3-((4-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(3-((2-фтор-4-(трифторметокси)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(4-((2-хлор-4-фторфенокси)метил)-4-метилпиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)- или (-)-(4aR,8aS)-6-(4-(2-(1H-пиразол-4-ил)-4-(трифторметил)бензил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-[3-[(2,4-дихлорфенил)метокси]азетидин-1-карбонил]-4,4а,5,7,8,8a-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[3-метокси-4-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-[[5-метил-6-(трифторметил)пиридин-3-ил]оксиметил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[(3-хлорфенокси)метил]пирролидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[(2-хлорфенокси)метил]пирролидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-[[2-фтор-4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[(2-хлорфенил)метокси]пирролидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[(3-хлорфенил)метокси]пирролидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-[[2-циклопропил-4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[(4-хлорфенокси)метил]пирролидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[(4-хлорфенил)метокси]пирролидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-[[2-метил-4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-[[2-хлор-4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[4-(трифторметил)фенил]метил]пирролидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[3-фтор-5-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[2-фтор-6-(трифторметил)фенил]метоксиметил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[2-метил-3-[[4-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он (изомер D),
(4aR,8aS)-6-[2-метил-3-[[4-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он (изомер С),
(4aR,8aS)-6-[2-метил-3-[[4-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он (изомеры А и D),
(4aR,8aS)-6-[3-[[3-хлор-4-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[(2-хлор-4-фторфенокси)метил]-3-фторазетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[2-фтор-4-(трифторметил)фенил]метокси]-3-(трифторметил)азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[2-фтор-4-(трифторметил)фенил]метокси]-3-метилазетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[2,4-дифтор-5-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[2-фтор-5-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[3-фтор-4-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[2-метокси-4-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[4-хлор-2-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[2-метил-3-[[4-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[4-(трифторметил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-[4-хлор-3-(трифторметил)фенокси]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-(4-хлор-3-циклопропилфенокси)пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-(4-хлор-3-морфолин-4-илфенокси)пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-[2-метил-4-(трифторметил)фенокси]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
2-[1-[(4aR,8aS)-3-оксо-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-6-карбонил]пиперидин-4-ил]окси-5-(трифторметил)бензонитрил,
(4aR,8aS)-6-[4-(оксазоло[5,4-с]пиридин-2-илметил)пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[2-[2-фтор-6-(трифторметил)фенил]этил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-[(2-хлор-4-фторфенокси)метил]азепан-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он (эпимер А),
(4aR,8aS)-6-[4-[(2-хлор-4-фторфенокси)метил]азепан-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он (эпимер В),
(4aR,8aS)-6-[4-[[4-(трифторметил)фенил]метил]азепан-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-[3-пиразол-1-ил-5-(трифторметил)фенокси]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-[[2-(2,2,2-трифторэтокси)-4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-[3-(1,2,4-триазол-1-ил)-5-(трифторметил)фенокси]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[4-хлор-3-(трифторметил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-(4-хлор-3-пиразол-1-ил-фенокси)пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-(4-(3-морфолино-4-(трифторметил)фенокси)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-[4-[4-xлop-3-(1,2,4-триазол-1-ил)фенокси]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-[3-циклопропил-4-(трифторметил)фенокси]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-[3-пиразол-1-ил-4-(трифторметил)фенокси]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-(4-хлорфенокси)пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-[[2,6-дифтор-4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[4-[4-хлор-3-(4-хлорфенил)-2-фторфенокси]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[2-хлор-4-(трифторметил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[2-фтор-6-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[2-(2-фтор-4-метилфенил)этил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[2-[4-метокси-2-(трифторметил)фенил]этил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[4-метил-2-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[2-[2-метокси-6-(трифторметил)фенил]этил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[2-[4-метил-2-(трифторметил)фенил]этил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[2-[2-ацетил-4-(трифторметил)фенил]этил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[2-[2-бром-4-(трифторметил)фенил]этил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[6-фтор-4-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[6-фтор-5-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[(3,4-дихлорфенил)метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[(2,5-дихлорфенил)метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[3-(трифторметокси)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[2-метил-3-[[4-метил-3-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он (фракция А, смесь 2 изомеров),
(4aR,8aS)-6-[2-метил-3-[[4-метил-3-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он (фракция В, один изомер),
(4aR,8aS)-6-[2-метил-3-[[4-метил-3-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он (фракция С, один изомер),
(4aR,8aS)-6-[3-[[5-(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[отн-(3R,4R)-3-метил-4-[[5-метил-6-(трифторметил)-3-пиридил]оксиметил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[отн-(3S,4S)-3-метил-4-[[5-метил-6-(трифторметил)-3-пиридил]оксиметил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[2-фтор-4-(трифторметил)фенил]метокси]-2-метилазетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[4,5-бис(трифторметил)-2-пиридил]оксиметил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[2-фтор-4-(трифторметил)фенил]метоксиметил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[2-фтор-4-(трифторметил)фенил]метокси]-2-метилазетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он (фракция А, смесь 2 стереоизомеров),
(4aR,8aS)-6-[3-[[2-фтор-4-(трифторметил)фенил]метокси]-2-метилазетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он (фракция В, один стереоизомер),
(4aR,8aS)-6-[3-[[2-фтор-4-(трифторметил)фенил]метокси]-2-метилазетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он (фракция С, один стереоизомер),
(4aR,8aS)-6-[3-[(4-хлор-2-феноксифенил)метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[2-фтор-4-(пентафтор-лямда6-сульфанил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[1-(2,4-дихлорфенил)циклопропил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[6-(4-фторфенокси)-4-(трифторметил)-2-пиридил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[6-(4-фторфенокси)-5-(трифторметил)-2-пиридил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[4-(4-фторфенил)тиазол-2-ил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[2-фтор-4-(трифторметил)фенил]метокси]-3-(трифторметил)пирролидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[рац-(2R,3S)-3-[2-бром-5-(трифторметил)фенокси]-2-метил-пирролидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[отн-(2R,3S)-3-[2-бром-5-(трифторметил)фенокси]-2-метил-пирролидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[отн-(2S,3R)-3-[2-бром-5-(трифторметил)фенокси]-2-метил-пирролидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[3-[[2,4-бис(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-(3-((2-метил-3-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)-(4aR,8aS)-6-(3-((4-метил-2-(трифторметокси)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-[2-метил-3-[[2-метил-4-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[2-метил-3-[[2-метил-3-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[4-[2-фтор-4-(трифторметил)фенокси]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[4-[3-хлор-4-(трифторметил)фенокси]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[3-(4-хлор-3-циклопропилфенокси)азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[4-[2-хлор-3-(трифторметил)фенокси]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[3-(3-бром-2-хлорфенокси)азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[3-(2-хлор-3-циклопропилфенокси)азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[3-[3-циклопропил-4-(трифторметил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[3-[3-хлор-4-(трифторметил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-5-[[1-[(4aR,8aS)-3-оксо-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-6-карбонил]-4-пиперидил]окси]-2-(трифторметил)бензонитрил,
(+)-5-[1-[(4aR,8aS)-3-оксо-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-6-карбонил]азетидин-3-ил]окси-2-(трифторметил)бензонитрил,
(+)-(4aR,8aS)-6-[3-(3-бром-4-хлорфенокси)азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[3-[3-(2-азаспиро[3.3]гептан-2-ил)-4-(трифторметил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[3-[3-(3-метилазетидин-1-ил)-4-(трифторметил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[3-[3-(3,3-дифторазетидин-1-ил)-4-(трифторметил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[3-[3-(3-фтор-3-метилазетидин-1-ил)-4-(трифторметил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[3-[3-(6,6-дифтор-2-азаспиро[3.3]гептан-2-ил)-4-(трифторметил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[3-[3-(5-окса-2-азаспиро[3.5]нонан-2-ил)-4-(трифторметил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[3-[3-(2-азаспиро[3.3]гептан-2-ил)-2-хлорфенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[3-[2-хлор-3-(3-метилазетидин-1-ил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[3-[2-хлор-3-(3-фтор-3-метилазетидин-1-ил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-(3-(3-(3-(трет-бутокси)азетидин-1-ил)-2-хлорфенокси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(+)-(4aR,8aS)-6-[3-[2-хлор-3-(5-окса-2-азаспиро[3.4]октан-2-ил)фенокси]азетидин-1-карбонил]-4,4а, 5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[3-[2-хлор-3-(5-окса-2-азаспиро[3.5]нонан-2-ил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[3-[3-(2-азаспиро[3.3]гептан-2-ил)-5-хлорфенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(+)-(4aR,8aS)-6-[3-(3-хлор-5-пирродидин-1-илфенокси)азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[2-[2-фтор-4-(трифторметил)фенил]этил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[4-фтор-2-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[2,2-дифтор-2-[4-(трифторметил)фенил]этил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[[3-(трифторметокси)фенил]метил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[2-фтор-5-(трифторметил)фенокси]пирролидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[2-хлор-5-(трифторметил)фенокси]пирролидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[(3R или 3S)-3-[2-фтор-5-(трифторметил)фенокси]пирролидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[(3S или 3R)-3-[2-фтор-5-(трифторметил)фенокси]пирролидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-[3-[(Е)-2-(2-фтор-4-метилфенил)винил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он,
(4aR,8aS)-6-(3-((3-фтор-4-(трифторметокси)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(3-((Е)-2-фтор-6-(трифторметил)стирил)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(3-((2,3-диметилбензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(3-((2,4-диметилбензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(3-((2-метил-4-(трифторметокси)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-(3-(4-гидрокси-2-(трифторметил)фенэтил)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-он,
(4aR,8aS)-6-[3-[[4-метил-3-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-он.
37. Соединение, выбранное из:
(+)-(4aR,8aS)-6-(4-((4-(трет-бутил)оксазол-2-ил)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)-(4aR,8aS)-6-[4-[[4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(+)-(4aR,8aS)-6-(4-((2-хлор-4-фторфенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
рац-(4aR,8aS)-6-(3-((2-фтор-4-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(3-((2-хлор-4-фторбензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(3-((2-фтор-4-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-(4-(трифторметокси)бензил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-(4-хлор-3-фторбензил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-(2-хлор-4-(трифторметил)фенокси)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-(3-(трифторметил)фенокси)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-[4-[[2-хлор-4-(трифторметокси)фенокси]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(+)- или (-)-(4aR,8aS)-6-(3-((2-фтор-4-(трифторметил)фенокси)метил)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-((4-хлор-2-фторфенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-((4-фтор-2-(трифторметил)фенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-((2-фтор-4-(трифторметил)фенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-(2-(пирролидин-1-ил)-4-(трифторметил)бензил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-((2-хлор-4-(трифторметил)фенокси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-[4-[[2-циклопентил-4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(+)- или (-)-3-хлор-4-((1-((4aR,8aS)-3-оксооктагидро-2Н-пиридо[4,3-b][1,4]оксазин-6-карбонил)пиперидин-4-ил)метокси)бензонитрила;
(+)- или (-)-(4aR,8aS)-6-(3-((2-хлор-4-(трифторметил)фенокси)метил)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-((2-хлор-4-фторфенокси)метил)-4-фторпиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-((4',6-дихлор-[1,1'-бифенил]-3-ил)окси)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-цис-4-((2-хлор-4-фторфенокси)метил)-3-метилпиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(3-((2-хлор-4-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(3-((2-фтор-4-(трифторметокси)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(+)- или (-)-(4aR,8aS)-6-(4-((2-хлор-4-фторфенокси)метил)-4-метилпиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(4aR,8aS)-6-[3-[(2,4-дихлорфенил)метокси]азетидин-1-карбонил]-4,4а,5,7,8,8a-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[4-[[2-фтор-4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[4-[[2-циклопропил-4-(трифторметил)фенил]метил]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[[3-хлор-4-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[[2-фтор-5-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[2-[2-фтор-6-(трифторметил)фенил]этил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-(3-(2-фтор-4-(трифторметил)фенэтил)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
6-(3-((2,4-бис(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(4aR,8aS)-6-[4-[3-хлор-4-(трифторметил)фенокси]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-(3-метил-4-(((5-метил-6-(трифторметил)пиридин-3-ил)окси)метил)пиперидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(4aR,8aS)-6-(3-((3,4-дихлорбензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(4aR,8aS)-6-(3-((2,5-дихлорбензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
рац-(4aR,8aS)-6-(2-метил-3-((4-метил-3-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(4aR,8aS)-6-(3-(((4,5-бис(трифторметил)пиридин-2-ил)окси)метил)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
рац-(4aR,8aS)-6-(3-((2-фтор-4-(трифторметил)бензил)окси)-2-метилазетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(4aR,8aS)-6-(3-((2-фтор-4-(пентафтор-16-сульфанеил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(4aR,8aS)-6-(3-((4-метил-2-(трифторметокси)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(4aR,8aS)-6-[4-[3-циклопропил-4-(трифторметил)фенокси]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[2-(2-фтор-4-метилфенил)этил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[2-[4-метокси-2-(трифторметил)фенил]этил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[3-(2-азаспиро[3.3]гептан-2-ил)-4-(трифторметил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[2-[4-метил-2-(трифторметил)фенил]этил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-(3-((4-метил-3-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
(4aR,8aS)-6-(3-((2-метил-3-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она;
рац-(4aR,8aS)-6-[2-метил-3-[[2-метил-4-(трифторметокси)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
рац-(4aR,8aS)-6-[2-метил-3-[[2-метил-3-(трифторметил)фенил]метокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-(4-хлор-3-циклопропилфенокси)азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[4-[2-хлор-3-(трифторметил)фенокси]пиперидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-(2-хлор-3-циклопропилфенокси)азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[3-(2-азаспиро[3.3]гептан-2-ил)-2-хлорфенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[2-хлор-3-(5-окса-2-азаспиро[3.5]нонан-2-ил)фенокси]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-[3-[(Е)-2-(2-фтор-4-метилфенил)винил]азетидин-1-карбонил]-4,4а,5,7,8,8а-гексагидропиридо[4,3-b][1,4]оксазин-3-она;
(4aR,8aS)-6-(3-((Е)-2-фтор-6-(трифторметил)стирил)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она; и
(4aR,8aS)-6-(3-((4-метил-3-(трифторметил)бензил)окси)азетидин-1-карбонил)гексагидро-2Н-пиридо[4,3-b][1,4]оксазин-3(4Н)-она.
38. Способ получения соединений формулы (I) по любому из пп. 1-37, включающий:
проведение реакции первого амина формулы 1, где R1 такой, как описано в любом из пп. 1-37,

со вторым амином формулы 2, где A, L, m, n, X и R2 такие, как описано в любом из пп. 1-37,

в присутствии основания и образующего мочевину реагента, с образованием указанного соединения формулы (I).
39. Применение соединения формулы (I) по любому из пп. 1-37 для ингибирования моноацилглицеринлипазы (MAGL) у млекопитающего.
40. Применение соединения формулы (I) по любому из пп. 1-37 для получения лекарственного средства для ингибирования MAGL у млекопитающего.
41. Способ ингибирования MAGL у млекопитающего, который включает введение млекопитающему эффективного количества соединения формулы (I) по любому из пп. 1-37.
| EP 3279191 А1, 07.02.2018 | |||
| WO 2011058766 A1, 19.05.2011 | |||
| PATEL JAYENDRA Z ET AL, Bioorganic & Medicinal Chemistry Letters, vol.25, no | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| 1436-1442 | |||
| JAE WON CHANG ET AL, Chemistry & Biology, vol.19, no | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Предохранительный прибор от вылета челнока на ткацких станках | 1924 |
|
SU579A1 |
| ЦЕНТРОБЕЖНЫЙ УВЛАЖНИТЕЛЬ ВОЗДУХА | 1928 |
|
SU9645A1 |
| 1-[ПИРИДО(3,4-B) -1,4-ОКСАЗИНИЛ -4-ИЛ] -1Н-ИНДОЛЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОИЗВОДНОЕ N-(4-ПИРИДИНИЛ)-1Н-ИНДОЛ-1-АМИНА | 1992 |
|
RU2042680C1 |

