ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка является международной заявкой, по которой испрашивается приоритет и преимущество по предварительной заявки на патент США № 62/780,703, поданной 17 декабря 2018 г., содержание которой включено в настоящее описание во всей своей полноте посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001] Настоящее изобретение относится к конкретным пролекарствам и аналогам (3α,5α)-3-гидрокси-21-(1H-имидазол-1-ил)-3-метоксиметил)-прегнан-20-она, находящимся в свободном виде или в виде фармацевтически приемлемой соли и/или по существу чистом виде, как раскрыто в настоящем описании, фармацевтическим композициям на их основе и способам их применения в качестве седативных средств, снотворных средств, анксиолитиков и/или анестетиков и способам лечения депрессии, тревожного расстройства, бессонницы, эпилепсии и других расстройств центральной нервной системы, а также к их комбинациям с другими агентами.
УРОВЕНЬ ТЕХНИКИ
[0002] Возбудимость мозга определяют как уровень возбуждения животного, некий континуум, который варьирует от комы до судорог и регулируется различными нейротрансмиттерами. Как правило, нейротрансмиттеры отвечают за регулирование проводимости ионов через нейронные мембраны.
[0003] Нейромедиатор гамма-аминомасляная кислота (ГАМК) оказывает сильное влияние на общую возбудимость головного мозга, поскольку до 40% нейронов в головном мозге используют ГАМК в качестве нейромедиатора. ГАМК взаимодействует с комплексом рецепторов ГАМК (GRC), опосредуя свое воздействие на нервные клетки во всей нервной системе, включая мозг. ГАМК регулирует возбудимость отдельных нейронов путем регуляции проводимости хлорид-ионов через нейронную мембрану. ГАМК взаимодействует со своим участком узнавания на GRC, облегчая прохождение хлорид-ионов по электрохимическому градиенту GRC в клетку. Внутриклеточное повышение уровней этого аниона вызывает гиперполяризацию трансмембранного потенциала, уменьшая чувствительность нейрона к факторам возбуждения, т.е. снижает возбудимость нейрона. Другими словами, чем выше концентрация хлорид-ионов в нейроне, тем ниже возбудимость головного мозга и уровень возбуждения.
[0004] GRC является ключевым посредником в развитии тревожности, эпилептической активности, депрессии и седативного эффекта. В результате ГАМК и препараты, действующие как ГАМК, облегчающие эффекты ГАМК (например, терапевтически полезные барбитураты и бензодиазепины (BZ), такие как валиум®), оказывают терапевтически полезные эффекты через взаимодействие со специфическими регуляторными участками на GRC. Накопленные к настоящему времени данные указывают на то, что кроме участка связывания бензодиазепинов и барбитуратов, GRC содержит отдельный участок для нейроактивных стероидов.
[0005] Нейроактивные стероиды являются эндогенными. Наиболее эффективными эндогенными нейроактивными стероидами являются 3α-гидрокси-5-восстановленный прегнан-20-он и 3α,21-дигидрокси-5-восстановленный прегнан-20-он, метаболиты гормональных стероидов прогестерона и дезоксикортикостерона, соответственно. Как обсуждается в US 2017/0240589, полностью включенном в настоящее описание в виде ссылки, несколько недавних клинических наблюдений свидетельствуют о решающей роли прогестерона и дезоксикортикостерона и их метаболитов в гомеостатической регуляции возбудимости головного мозга. Это проявляется, например, в увеличении эпилептической активности или симптомов, связанных с менструальной эпилепсией, ПМС и ПРД, а также в корреляции между сниженными уровнями прогестерона и симптомами, связанными с ПМС, ПРД и менструальной эпилепсией. Однако прогестерон не всегда эффективен при лечении вышеупомянутых синдромов. Природные нейроактивные метаболиты прогестерона включают прегнанолон и аллопрегнанолон, и эти метаболиты могут опосредовать по меньшей мере некоторые эффекты прогестерона и дезоксикортикостерона.
[0006] Встречающиеся в природе нейроактивные стероиды обычно непригодны в качестве фармакологических агентов из-за короткого периода полувыведения и плохой биодоступности при пероральном введении, предположительно по причине быстрого метаболизма. Одним из таких стероидов является аллопрегнанолон,
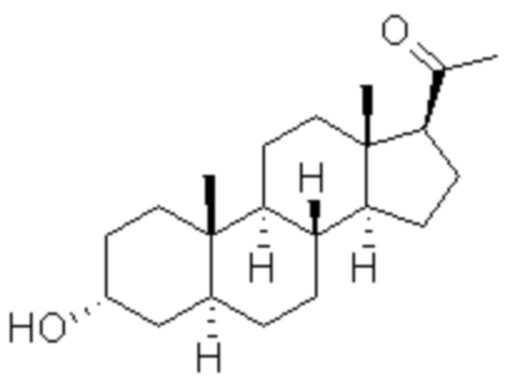 .
.
Он является эндогенным нейроактивным стероидом, демонстрирующим многообещающую фармакологическую активность, но имеет недостатки, такие как низкая биодоступность при приеме внутрь и короткий период полувыведения. Тем не менее, аллопрегнанолон используется для внутривенного лечения эпилепсии, депрессии и других заболеваний ЦНС.
[0007] Необходимы новые и улучшенные нейроактивные стероиды, действующие как агенты, модулирующие возбудимость мозга, такие как седативные средства, снотворные средства и анксиолитики, а также средства для профилактики и лечения заболеваний, связанных с ЦНС.
[0008] В данной области известны синтетические и полусинтетические нейроактивные стероиды, которые были изучены в качестве потенциальных лекарственных веществ для ЦНС.
[0009] Было показано, что добавление 3β-замены приводит к получению нейроактивных стероидов с сильной пероральной активностью, но с нежелательно долгим периодом полувыведения. Например, седативные/снотворные средства предпочтительно должны иметь период полувыведения у людей менее 5 часов, что позволяет избежать развития остаточных эффектов на следующий день и накопления при продолжении дозирования ночью. Ранее было обнаружено, что у 3β-метоксиметил-замещенных стероидов сохраняется требуемая пероральная активность других 3β-замещенных нейроактивных стероидов, но с продолжительностью действия, которая обеспечивает возможность их применения в качестве седативных/снотворных средств и анестетиков. Такие соединения раскрыты, например, в патентах США 5939545 и 6277838.
[00010] 3α-Гидрокси-3β-метоксиметил-21-(1H-имидазол-1-ил)-5α-прегнан-20-он представляет собой синтетический нейроактивный стероид. Его первичной молекулярной мишенью является рецептор γ-аминомасляной кислоты типа A (ГАМКА, GABAA), где он действует как положительный аллостерический модулятор функции канала GRC. Подобно другим классам модуляторов ГАМКА, таких как бензодиазепины и другие лиганды бензодиазепиновых участков, нейроактивные стероиды имеют ряд потенциальных показаний, например, для лечения расстройств сна, тревожного расстройства, депрессии и эпилепсии. Это соединение раскрыто, например, в публикациях США 2004/0034002 и 2009/0131383, содержание которых включено в настоящее описание во всей своей полноте в виде ссылки.
[00011] Клинические исследования подтверждают, что 3α-гидрокси-3β-метоксиметил-21-(1H-имидазол-1-ил)-5α-прегнан-20-он имеет следующие фармакокинетические свойства у людей после перорального приема: (1) быстрое всасывание с Tmax от примерно 1 до примерно 3 часов; (2) уровни Cmax, изменяющиеся между субъектами; (3) более высокие дозозависимые значения Cmax; и (4) значения T1/2, которые в среднем составляли приблизительно 12 часов в пяти различных группах дозирования. См. таблицу 1 ниже. Для фармакокинетических параметров, таких как AUC, Cmax и tmax, приведены средние значения. Значения, указанные в скобках, соответствуют стандартным отклонениям.
Таблица I
[00012] Однако было бы полезно разработать производные 3α-гидрокси-3β-метоксиметил-21-(1H-имидазол-1-ил)-5α-прегнан-20-она, обладающие улучшенными фармакокинетическими свойствами, такими как более высокая устойчивость к метаболическим нарушениям или улучшенное распределение и/или биодоступность.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
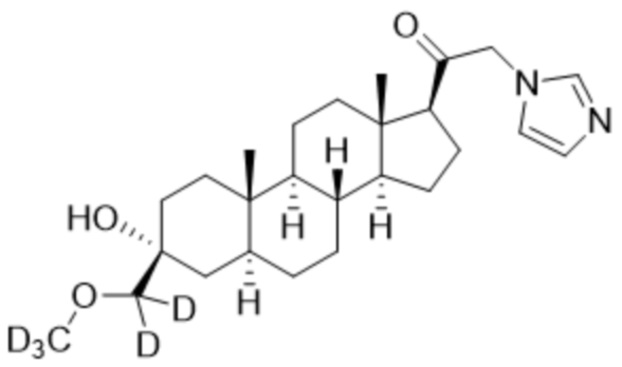
[00013] Соединение формулы A, имеющее химическое название 2-(1H-имидазол-1-ил)-1-((3S,5S,8R,9S,10S,13S,14S,17S)-3-(метоксиметил)-10,13-диметилгексадекагидро-1H-циклопент[a]фенантрен-17-ил)этан-1-он, и общепринятое название 3α-гидрокси-3β-метоксиметил-21-(1H-имидазол-1-ил)-5α-прегнан-20-он, показанное ниже, представляет собой мощный положительный аллостерический модулятор рецептора ГАМКА. Это соединение также может взаимодействовать с ацетилхолиновыми рецепторами и 5-HT3 серотониновыми рецепторами.
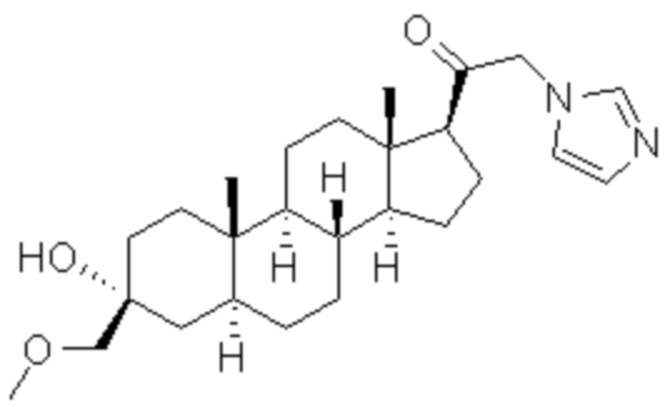
Формула A.
Соединение формулы A полезно в качестве седативного/снотворного средства или анестетика, а также для лечения или профилактики расстройств центральной нервной системы, но в данной области техники существует потребность в аналогах соединения формулы А, таких как изотопные аналоги и пролекарства, которые при введении пациенту могут обеспечивать улучшенные терапевтические концентрации или улучшенное фармакокинетическое распределение или динамику. Настоящее изобретение удовлетворяет эту потребность путем предоставления соединений формулы I et seq., которые являются дейтерированными аналогами и/или пролекарствами соединения формулы A. Благодаря полезному метаболическому и фармакокинетическому профилю соединения по настоящему изобретению могут быть особенно подходящими для приготовления лекарственных форм длительного действия или с пролонгированным высвобождением, которые при введении пациенту могут обеспечивать улучшенные терапевтические количества, концентрации соединения А и его аналогов в течение продолжительного периода времени.
[00014] В первом аспекте настоящее изобретение относится к соединению (Соединение 1) формулы I:
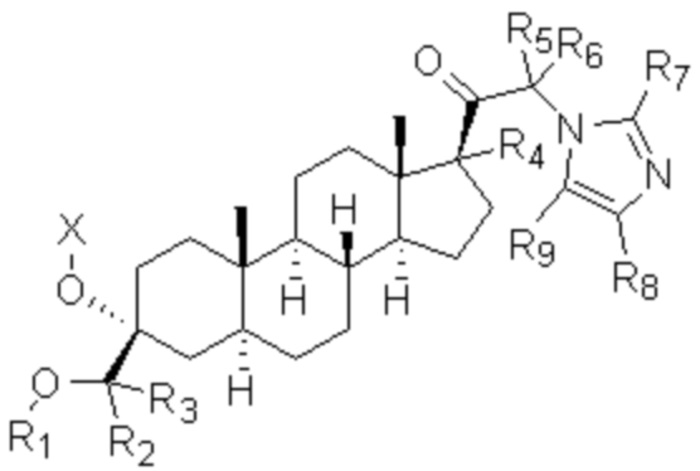 ,
,
формула I
где:
X выбирают из H, -(C=O)-Ra, -CH2-(C=O)-O-Ra и -CH2-(C=O)-N(Ra)(Rb);
R1 выбирают из CH3, CDH2, CD2H и CD3;
каждый из R2-R9 независимо выбирают из H и D;
Ra и Rb независимо выбирают из H, C1-20алкила (например, метила) и C1-4алкил-арила (например, бензила),
в свободном виде или в виде соли (например, в виде фармацевтически приемлемой соли), например, в выделенном или очищенном свободном виде или в виде соли,
при условии, что если R1 представляет собой CH3 и все R2-R9 представляют собой H, то X выбирают из -(C=O)-Ra, -CH2-(C=O)-O-Ra и -CH2-(C=O)-N(Ra)(Rb).
[00015] Во втором аспекте настоящее изобретение относится к фармацевтическим композициям, содержащим соединения формулы I et seq. в комбинации с фармацевтически приемлемым разбавителем или носителем.
[00016] В третьем аспекте настоящее изобретение относится к способам лечения или профилактики расстройств центральной нервной системы, поддающихся облегчению при использовании модулятора рецептора ГАМКА (например, положительного аллостерического модулятора рецептора ГАМКА), при этом способы включают введение пациенту при необходимости соединения формулы I или его фармацевтической композиции.
[00017] В четвертом аспекте настоящее изобретение относится к способам индукции седации или анестезии у нуждающегося в этом пациента, при этом способы включают введение соединения формулы I или его фармацевтической композиции.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[00018] В первом аспекте настоящее изобретение относится к соединению (Соединению 1) формулы I:
,
Формула I
где:
X выбирают из H, -(C=O)-Ra, -CH2-(C=O)-O-Ra и -CH2-(C=O)-N(Ra)(Rb);
R1 выбирают из CH3, CDH2, CD2H и CD3;
каждый из R2-R9 независимо выбирают из H и D;
Ra и Rb независимо выбирают из H, C1-20алкила (например, метила) и C1-4алкил-арила (например, бензила),
в свободном виде или в виде соли (например, в виде фармацевтически приемлемой соли), например, в выделенном или очищенном свободном виде или в виде соли,
при условии, что если R1 представляет собой CH3 и все R2-R9 представляют собой H, то X выбирают из -(C=O)-Ra, -CH2-(C=O)-O-Ra и -CH2-(C=O)-N(Ra)(Rb).
[00019] В настоящем описании представлены дополнительные иллюстративные варианты осуществления соединения формулы I в свободном виде или в виде соли, например, в выделенном или очищенном свободном виде или в виде соли, включая:
1.1 соединение I, в котором X представляет собой H;
1.2 соединение I, в котором X выбирают из -(C=O)-Ra, -CH2-(C=O)-O-Ra и -CH2-(C=O)-N(Ra)(Rb);
1.3 соединение I, в котором X представляет собой -(C=O)-Ra;
1.4 соединение I, в котором X представляет собой -CH2-(C=O)-O-Ra;
1.5 соединение I, в котором X представляет собой CH2-(C=O)-N(Ra)(Rb);
1.6 соединение I или любое из 1.1-1.5, в котором Ra представляет собой H;
1.7 соединение I или любое из 1.1-1.5, в котором Ra представляет собой C1-20алкил (например, метил) или C1-4алкил-арил (например, бензил);
1.8 соединение I или любое из 1.1-1.5, в котором Ra представляет собой C1-20алкил (например, метил);
1.9 соединение I или любое из 1.1-1.5, в котором Ra представляет собой C1-4алкил-арил (например, бензил);
1.10 соединение I или любое из 1.1-1.5, в котором Ra представляет собой метил;
1.11 соединение I или любое из 1.1-1.5, в котором Ra представляет собой бензил;
1.12 соединение I или любое из 1.1-1.11, в котором X представляет собой CH2-(C=O)-N(Ra)(Rb), и Rb представляет собой H;
1.13 соединение I или любое из 1.1-1.12, в котором R1 представляет собой CH3;
1.14 соединение I или любое из 1.1-1.12, в котором R1 представляет собой CDH2, CD2H или CD3;
1.15 соединение I или любое из 1.1-1.12, в котором R1 представляет собой CD3;
1.16 любое из соединений 1.1-1.15, в котором все R2-R9 представляют собой H;
1.17 любое из соединений 1.1-1.15, в котором любой из R2-R9 представляет собой D;
1.18 любое из соединений 1.1-1.15, в котором любые два из R2-R9 представляют собой D;
1.19 любое из соединений 1.1-1.15, в котором любые три из R2-R9 представляют собой D;
1.20 любое из соединений 1.1-1.15, в котором любые четыре из R2-R9 представляют собой D;
1.21 любое из соединений 1.1-1.20, в котором R2 и R3 представляют собой D;
1.22 любое из соединений 1.1-1.21, в котором R5-R6 представляют собой D;
1.23 любое из соединений 1.1-1.22, в котором любое одно, два или три из R7-R9 представляют собой D;
1.24 соединение I или любое из 1.1-1.23, где соединение выбирают из группы, состоящей из:
 ,
, 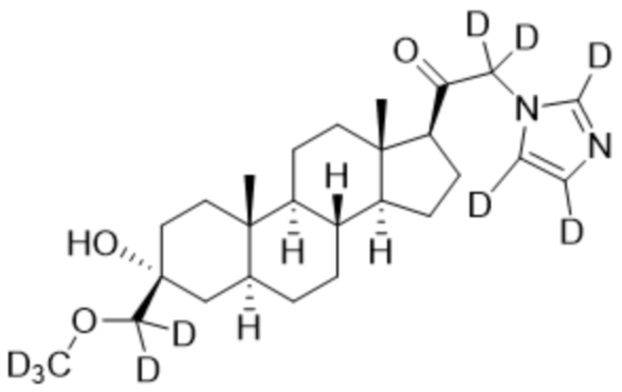 ,
,
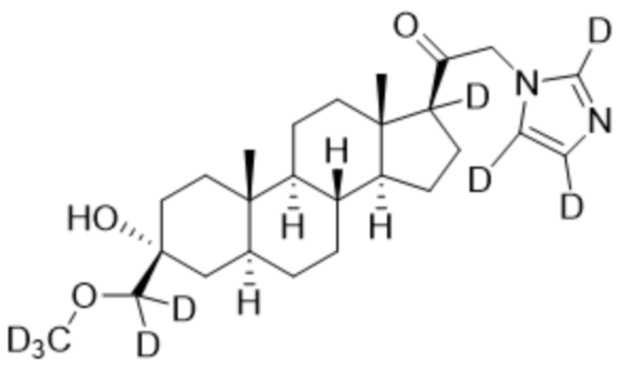 ,
, 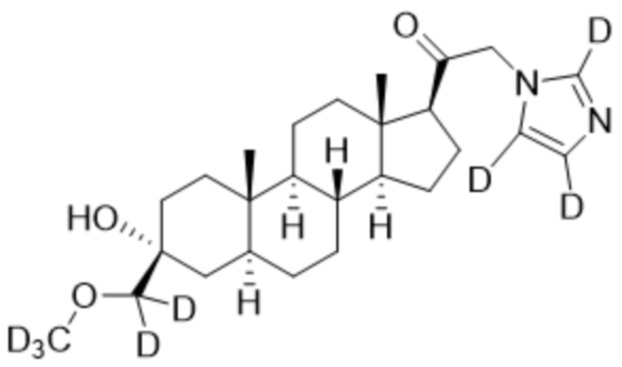 ,
,
 ,
, 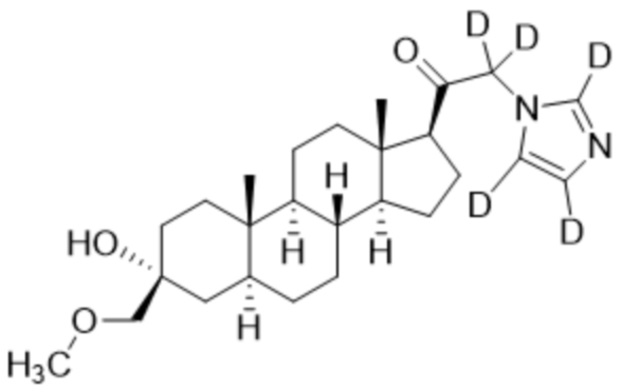 ,
, 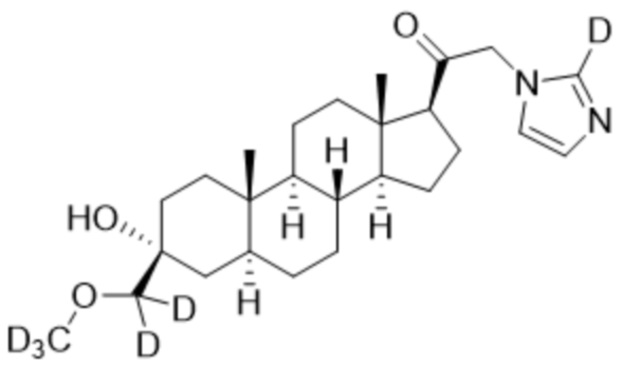 ,
, 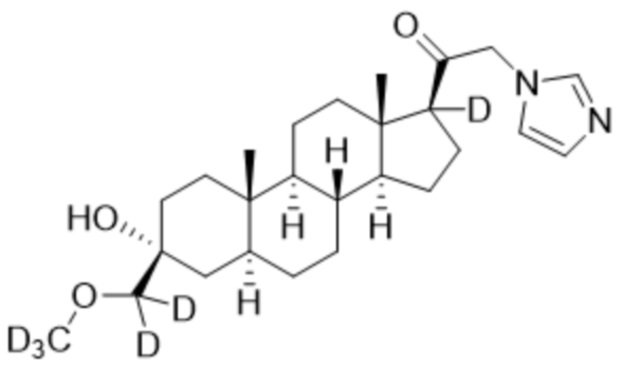 ,
, 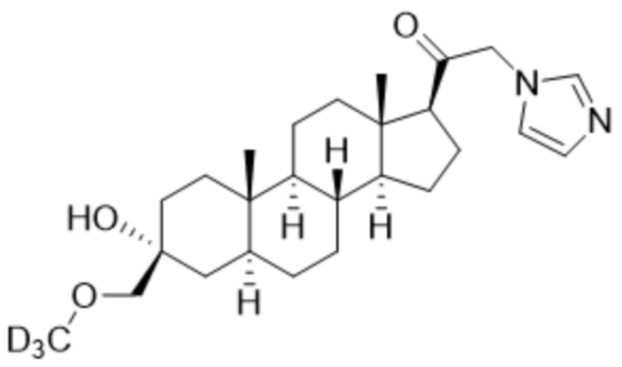 ,
, 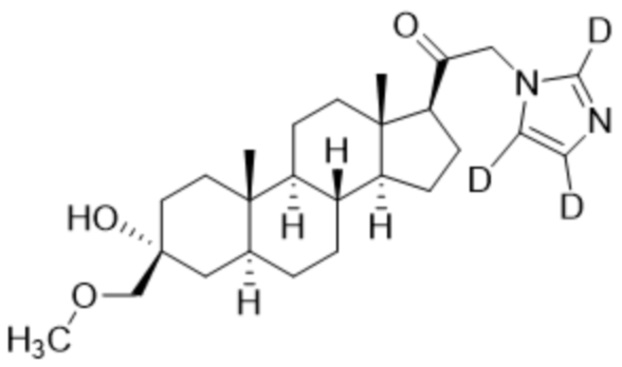 ,
, 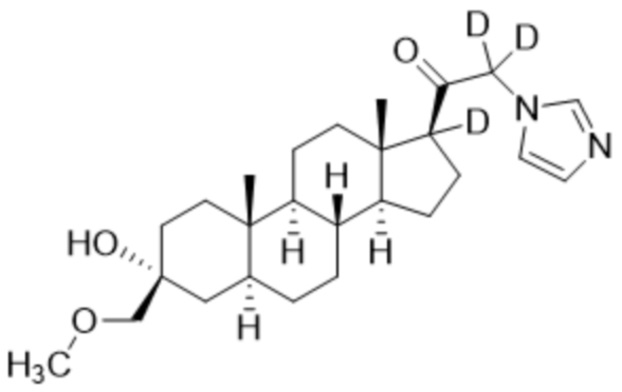 ,
, 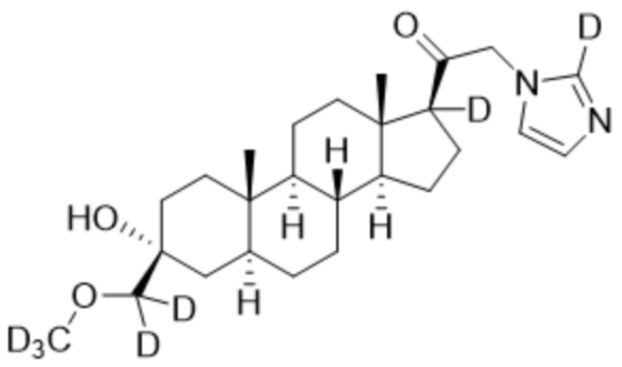 ,
,
 ,
, 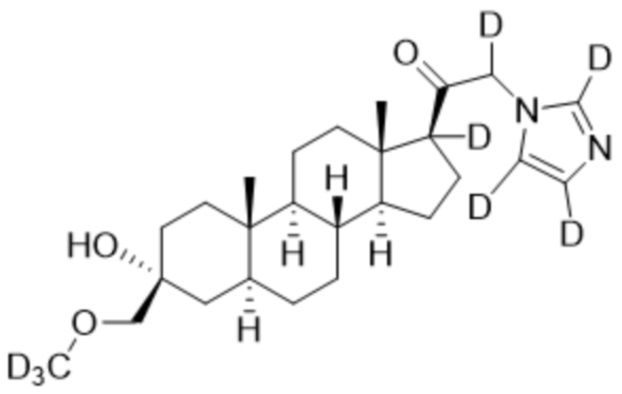 ,
,
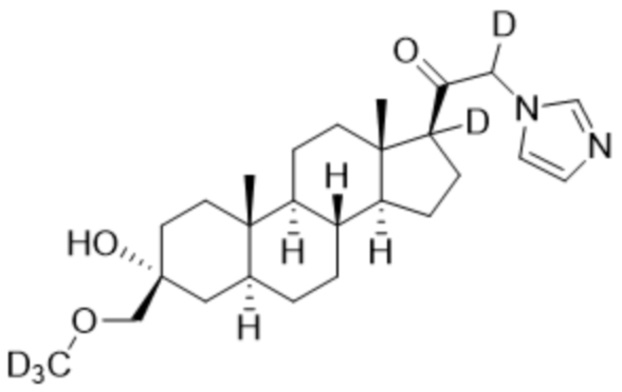 ,
, 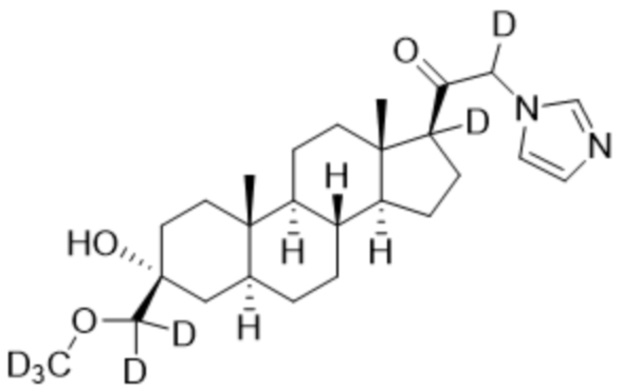 ,
,
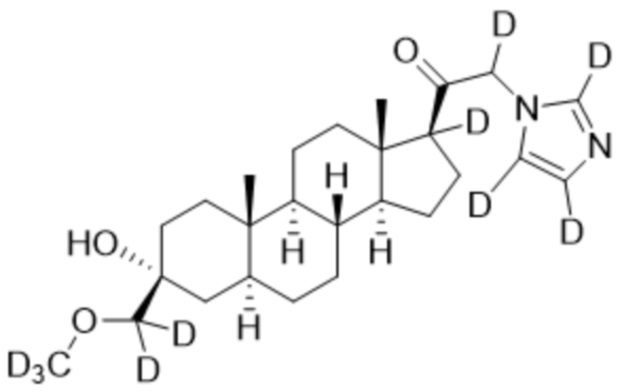 и
и 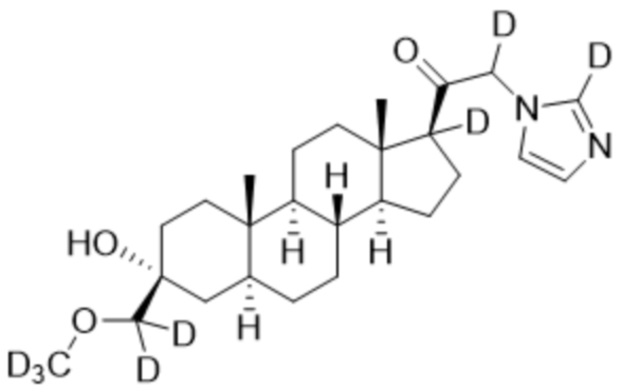 ;
;
1.25 соединение I или любое из 1.1-1.24 в свободном виде (в виде свободного основания);
1.26 соединение I или любое из 1.1-1.24 в виде соли, например, в виде фармацевтически приемлемой соли (например, гидрохлорида);
1.27 соединение I или любое из 1.1-1.24 в виде твердого вещества;
1.28 соединение I или любое из 1.1-1.27 по существу в виде чистого диастереомера (т.е., по существу в виде, не содержащем другие диастереомеры);
1.29 соединение I или любое из 1.1-1.28, имеющее диастереомерный избыток более чем 70%, предпочтительно более чем 80%, более предпочтительно более чем 90% и наиболее предпочтительно более чем 95%;
1.30 соединение I или любое из 1.1-1.28, имеющее более чем 50% введение дейтерия в одном или более из указанных положений структуры (т.е., более чем 50 атом.% D), например, более чем 60% или более чем 70%, или более чем 80%, или более чем 90% или более чем 95%, или более чем 96%, или более чем 97%, или более чем 98%, или более чем 99%.
1.31 соединение I или любое из 1.1-1.30 в выделенном или очищенном виде.
[00020] Во втором аспекте настоящее изобретение относится к фармацевтической композиции (фармацевтическая композиция 2), содержащей соединение формулы I, например соединение 1 или любое из 1.1-1.31, например, в смеси с фармацевтически приемлемым разбавителем или носителем. Настоящее изобретение относится к дополнительным иллюстративным вариантам осуществления фармацевтической композиции 2, включая:
2.1 фармацевтическую композицию 2, в которой соединение формулы I et seq. находится в твердой форме;
2.2 фармацевтическую композицию 2 или 2.1, где композиция представляет собой композицию с немедленным высвобождением;
2.3 фармацевтическую композицию 2 или 2.1, где композиция представляет собой композицию с задержанным высвобождением;
2.4 фармацевтическую композицию 2 или 2.1, где композиция представляет собой композицию с замедленным высвобождением;
2.5 фармацевтическую композицию 2 или любую из 2.1-2.4, где композиция представляет собой пероральную лекарственную форму (например, таблетку или капсулу);
2.6 фармацевтическую композицию 2.5, где композиция представляет собой сублингвальную, буккальную, и/или растворяющуюся во рту таблетку;
2.7 фармацевтическую композицию 2 или любую из 2.1-2.4, где композиция представляет собой инъекционную композицию (например, приготовленную в виде лекарственной формы для внутривенной, подкожной или внутримышечной инъекции);
2.8 фармацевтическую композицию 2.7, где композиция представляет собой инъекционную композицию с замедленным высвобождением (например, состав депо), например, приготовленный в виде инъекционного состава длительного действия для внутримышечной или подкожной инъекции;
2.9 фармацевтическую композицию 2 или любую из 2.1-2.4, где композиция представляет собой трансмукозальную композицию, например, буккальную, сублингвальную, интраназальную композицию или композицию в виде легочного аэрозоля;
2.10 фармацевтическую композицию 2 или любую из 2.1-2.4, где композиция представляет собой офтальмологическую композицию, например, топическую композицию или композицию для внутриглазной инъекции;
2.11 фармацевтическую композицию 2 или любую из 2.1-2.10, где соединение формулы I et seq. находится в полимерной матрице.
[00021] Фармацевтические композиции 2 et seq. включают все композиции, в которых соединения по настоящему изобретению содержатся в количестве, эффективном для достижения их назначения. Несмотря на изменчивость индивидуальных потребностей, определение оптимальных диапазонов эффективных количеств каждого компонента находится в пределах компетенции специалиста в данной области. Обычно соединения можно вводить млекопитающим, например людям, получающим лечение от бессонницы, перорально в суточной дозе от 0,0025 до 50 мг/кг массы тела млекопитающего или его фармацевтически приемлемую соль в эквивалентном количестве. Для внутримышечной инъекции доза обычно составляет примерно половину пероральной дозы.
[00022] Единичная пероральная доза может содержать от примерно 0,01 до примерно 50 мг, предпочтительно от примерно 0,1 до примерно 10 мг соединения. Единичную дозу можно вводить один или более раз в сутки в виде одной или более таблеток, каждая из которых содержит от примерно 0,1 до примерно 10, обычно от примерно 0,25 до 50 мг соединения или его соли.
[00023] Фармацевтические композиции по настоящему изобретению можно вводить любыми способами, обеспечивающими достижение предполагаемой цели. Например, введение может осуществляться парентеральным, сублингвальным, подкожным, внутривенным, внутримышечным, внутрибрюшинным, интраназальным, трансдермальным или трансбуккальным путями. Альтернативно или наряду с этим введение может осуществляться пероральным путем. Вводимая доза будет зависеть от возраста, состояния здоровья и массы реципиента, вида сопутствующего лечения, если таковое имеется, частоты лечения и характера требуемого эффекта.
[00024] Терапевтические уровни соединения 1 в плазме et seq. могут меняться от примерно 5 нг/мл до примерно 500 нг/мл. Другие эффективные терапевтические диапазоны включают от примерно 50 нг/мл до примерно 500 нг/мл, от примерно 50 нг/мл до примерно 400 нг/мл, от примерно 50 нг/мл до примерно 325 нг/мл, от примерно 50 нг/мл до примерно 250 нг/мл, от примерно 50 нг/мл до примерно 100 нг/мл и от примерно 100 нг/мл до примерно 250 нг/мл.
[00025] В некоторых вариантах осуществления фармацевтические композиции по настоящему изобретению представляют собой композиции для инъекций с замедленным или отсроченным высвобождением, например, составы депо.
[00026] В некоторых вариантах осуществления фармацевтические композиции содержат соединение формулы 1 et seq. в полимерной матрице. В одном из вариантов осуществления соединение по настоящему изобретению диспергировано или растворено в полимерной матрице. В другом варианте осуществления полимерная матрица содержит стандартные полимеры, используемые в составах депо, такие как полимеры, выбранные из сложного полиэфира жирной кислоты и ее производных или полимера на основе алкил-альфа-цианоакрилата, полиалкиленоксалата, сложного полиортоэфира, поликарбоната, полиортокарбоната, полиаминокислоты, сложного эфира гиалуроновой кислоты и их смесей. В другом варианте осуществления полимер выбирают из группы, состоящей из полилактида, поли-d, l-лактида, полигликолида, полимера PLGA 50:50, PLGA 65:35, PLGA 75:25, PLGA 85:15 и PLGA 90:10. В другом варианте осуществления полимер выбирают из поли(гликолевой кислоты), поли-D, L-молочной кислоты, поли-L-молочной кислоты, сополимеров вышеперечисленного, поли(алифатических карбоновых кислот), сополимеров на основе оксалатов, поликапролактона, полидиоксанона, поли(орто-карбонатов), поли(ацеталей), поли(молочной кислоты-капролактона), полиортоэфиров, поли(гликолевой кислоты-капролактона), полиангидридов и природных полимеров, включая альбумин, казеин и воски, такие как моно- и дистеарат глицерина и т.п. В предпочтительном варианте осуществления полимерная матрица содержит сополимер поли(d,l-лактид-гликолида).
[00027] Эти композиции могут быть приготовлены в виде составов для контролируемого и/или замедленного высвобождения соединений по настоящему изобретению (например, в виде композиции депо) в течение периода до 180 дней, например, от примерно 14-30 до примерно 180 дней. Например, полимерная матрица может разлагаться и высвобождать соединения по настоящему изобретению в течение примерно 30, примерно 60 или примерно 90 дней. В другом примере полимерная матрица может разлагаться и высвобождать соединения по настоящему изобретению в течение примерно 120 или примерно 180 дней.
[00028] В еще одном варианте осуществления фармацевтические композиции по настоящему изобретению, например композиция депо по настоящему изобретению, приготовлены в виде составов для инъекции.
[00029] В третьем аспекте настоящее изобретение относится к способу (способ 3) лечения или профилактики подлежащего облегчению расстройства центральной нервной системы с помощью модулятора рецептора ГАМКА (положительного аллостерического модулятора рецептора ГАМКА), где способы включают введение нуждающемуся в этом пациенту соединения формулы I, например, соединения 1 или любого из 1.1-1.31, или его фармацевтической композиции, например композиции 2 или любой из 2.1-2.11. В дополнительных вариантах осуществления настоящее изобретение относится к:
3.1 Способу 3, включающему введение соединения 1 или любого из 1.1-1.31 в свободном виде или в виде фармацевтически приемлемой соли;
3.2 Способу 3, включающему введение фармацевтической композиции 2 или любой из 2.1-2.11;
3.3 Способу 3 или любому из способов 3.1-3.2, где заболевание центральной нервной системы поддается лечению с помощью положительного аллостерического модулятора рецептора ГАМКА;
3.4 Способу 3.3, при котором расстройство центральной нервной системы выбирают из группы, состоящей из расстройств сна (например, бессонницы), нарушений циркадного ритма, нарушений фазового сдвига (например, смены часовых поясов), тревожного расстройства (включая общую тревожность, социальную тревожность и панические расстройства), посттравматического стрессового расстройства, депрессии (например, рефрактерных форм депрессии, большого депрессивного расстройства, биполярной депрессии, послеродовой депрессии, сезонного аффективного расстройства, дистимии, терапевтически резистентной депрессии, суицидальных мыслей или суицидального поведения и предменструального дисфорического расстройства), компульсивных расстройств (например, обсессивно-компульсивного расстройства), шизофрении, шизоаффективного расстройства, расстройства внимания (например, синдрома дефицита внимания (ADD), синдрома дефицита внимания с гиперактивностью (ADHD)), судорожных расстройств (например, припадочных расстройств, эпилепсии или эпилептического статуса, включая ранний эпилептический статус, установленный эпилептический статус, рефрактерный эпилептический статус, супер-рефрактерный эпилептический статус и неконвульсивный эпилептический статус, такой как генерализованный эпилептический статус и сложный парциальный эпилептический статус), расстройств, сопровождающихся агрессией (например, острой или хронической агрессии), расстройств, сопровождающихся возбуждением (например, острого или хронического возбуждения), расстройств памяти и/или когнитивных функций (таких как нейродегенеративные расстройства, болезнь Альцгеймера, старость, деменция с тельцами Леви, сосудистая деменция), двигательных расстройств (например, болезни Паркинсона, болезни Хантингтона, тремор), аутизма и расстройств аутистического спектра (например, синдрома Аспергера), болевых расстройств (например, невропатической боли, острой боли, хронической боли), расстройств личности (например, антисоциального расстройства личности, депрессивного расстройства личности), сосудистых расстройств (например, инсульта, ишемии, сосудистых мальформаций), расстройств пищевого поведения (например, булимии, анорексии, переедания, кахексии), черепно-мозговой травмы, расстройств, связанных со злоупотреблением психоактивных веществ, расстройств, вызванных употреблением психоактивных веществ, синдрома отмены психоактивных веществ, синдрома Ретта, синдрома ломкой Х-хромосомы, синдрома Ангельмана и шума в ушах, а также нейродегенеративных заболеваний (например, болезни Альцгеймера, бокового амиотрофического склероза, комы, деменции, болезни Паркинсона, болезни Хантингтона, дискинезии, дистонии); и любых расстройств, для эффективного лечения которых требуется седация или анестезия;
3.5 Способу 3.4, где расстройство центральной нервной системы выбирают из группы, состоящей из расстройств сна (например, бессонницы), тревожности (включая общую тревожность, социальную тревожность и панические расстройства), посттравматического стрессового расстройства, депрессии (например, рефрактерных форм депрессии и большого депрессивного расстройства, биполярной депрессии, послеродовой депрессии) и судорожных расстройств (например, припадочных расстройств, эпилепсии или эпилептического статуса);
3.6 Способу 3.5, где расстройство центральной нервной системы выбирают из группы, состоящей из расстройств сна (например, бессонницы), тревожности (включая общую тревожность, социальную тревожность и панические расстройства), депрессии (например, рефрактерной депрессии, большого депрессивного расстройства, биполярной депрессии, послеродовой депрессии) и судорожных расстройств (например, припадочных расстройств, эпилепсии или эпилептического статуса);
3.7 Способу 3 или любому из 3.1-3.6, где соединение или композицию вводят перорально;
3.8 Способу 3.7, где вводимая композиция представляет собой твердую пероральную лекарственную форму (например, таблетку или капсулу);
3.9 Способу 3.7, где твердая пероральная лекарственная форма представляет собой сублингвальную или буккальную таблетку, растворяющуюся во рту;
3.10 Способу 3 или любому из 3.1-3.6, где соединение или композицию вводят интраназально или путем ингаляции легких, например, в виде аэрозоля;
3.11 Способу 3 или любому из 3.1-3.6, где соединение или композицию вводят офтальмологически, например, в виде местного офтальмологического средства;
3.12 Любому из способов 3.7-3.11, где соединение или композицию вводят три раза в сутки или два раза в сутки, или один раз в сутки;
3.13 Способу 3 или любому из 3.1-3.6, где соединение или композицию вводят путем инъекции, например, путем внутривенной, подкожной, внутриглазной, внутрибрюшинной или внутримышечной инъекции;
3.14 Способу 3.13, где соединение или композицию вводят в виде инъекционной композиции длительного действия, например, композиции депо;
3.15 Способу 3.13 или 3.14, где инъекцию вводят один раз в сутки или один раз в двое суток, или один раз каждые три-семь дней, или один раз в неделю, или один раз в одну-четыре недели, или один раз в месяц.
[00030] Другие заболевания и расстройства, поддающиеся лечению с помощью модуляторов ГАМКА по настоящему изобретению, включают заболевания и расстройства, раскрытые и описанные в US 2017/0240589, содержание которого включено в настоящее описание во всей своей полноте в виде ссылки.
[00031] Соединения по настоящему изобретению, представленные в настоящем описании, как правило предназначены для модуляции функции ГАМК и, следовательно, действуют как нейроактивные стероиды для лечения и профилактики связанных с ЦНС состояний у субъекта. В контексте настоящего описания модуляция относится к ингибированию или усилению функции рецептора ГАМК и, в частности, к положительной аллостерической модуляции (усилению) функции рецептора ГАМКА. Соответственно, соединения и фармацевтические композиции, представленные в настоящем описании, находят применение в качестве терапевтических средств для профилактики и/или лечения состояний ЦНС у млекопитающих, включая человека и млекопитающих, не являющихся человеком. Таким образом, и как указывалось ранее, в объем настоящего изобретения включены, и оно охватывает перечисленные способы лечения, а также на соединения для таких способов и применение таких соединений для получения лекарственных средств, полезных для указанных способов.
[00032] В другом варианте осуществления настоящее изобретение относится к любому из способов 3.1-3.15, где способ включает введение фармацевтической композиции 2 или любой из 2.1-2.11, которая приготовлена в виде состава с отсроченным высвобождением и/или замедленным высвобождением соединений по изобретению в течение от примерно 14 дней, примерно 30 до примерно 180 дней, предпочтительно в течение примерно 30, примерно 60 или примерно 90 дней. Отсроченное и/или замедленное высвобождение особенно полезно для предотвращения преждевременного прекращения терапии, особенно для лекарственной терапии, при которой нарушение или несоблюдение схемы лечения является обычным явлением.
[00033] В еще одном варианте осуществления изобретение относится к любому описанному выше способу 3 или 3.1-3.15, где способ включает введение фармацевтической композиции, которая представляет собой композицию депо по настоящему изобретению, вводимую для контролируемого и/или замедленного высвобождения соединения по изобретению в течение некоторого периода времени.
[00034] В четвертом аспекте настоящее изобретение относится к способу (способ 4) индукции седации или анестезии у нуждающегося в этом пациента, где способ включает введение нуждающемуся в этом пациенту соединения формулы I, например соединения 1 или любого из 1.1-1.31 или его фармацевтической композиции, например композиции 2 или любой из 2.1-2.11. В дополнительных вариантах осуществления четвертый аспект относится к:
4.1 Способу 4, при котором вводимая доза является эффективной для генерации седации и/или анестезии у пациента в течение 2 часов после введения, например в течение 1 часа после введения;
4.2 Способу 4, при котором вводимая доза является эффективной для генерации седации и/или анестезии у пациента в течение 45 минут после введения, например в течение 30 минут или в течение 20 минут, или в течение 10 минут после введения;
4.3 Способу 4, при котором вводимая доза является эффективной для генерации седации и/или анестезии у пациента в течение 5 минут после введения, например в течение 3 минут или в течение 2 минут, или в течение 1 минуты после введения;
4.4 Способу 4 или любому из 4.1-4.3, при котором седация и/или анестезия, обеспечиваемые однократной дозой, имеют продолжительность от 1 часа до 24 часов, например от 1 часа до 12 часов или от 1 часа до 6 часов, или от 1 часа до 4 часа или от 1 часа до 2 часов;
4.5 Способу 4 или любому из 4.1-4.4, где соединение или композицию вводят перорально;
4.6 Способу 4.5, где вводимая композиция представляет собой твердую пероральную лекарственную форму (например, таблетку или капсулу);
4.7 Способу 4.6, где твердая пероральная лекарственная форма представляет собой сублингвальную или буккальную таблетку, растворяющуюся во рту;
4.8 Способу 4 или любому из 4.1-4.4, где соединение или композицию вводят интраназально или путем ингаляции в легкие, например в виде аэрозоля;
4.9 Способу 4 или любому из 4.1-4.4, где соединение или композицию вводят путем инъекции, например, путем внутривенной, подкожной, внутрибрюшинной или внутримышечной инъекции.
[00035] Соединения по настоящему изобретению, фармацевтические композиции по настоящему изобретению или композиции депо по настоящему изобретению можно использовать в комбинации со вторым терапевтическим агентом, в частности, в более низких дозах, чем при введении агентов по отдельности в качестве монотерапии, для усиления терапевтической активности комбинированных агентов, не вызывая при этом нежелательных побочных эффектов, обычно возникающих при традиционной монотерапии. Следовательно, соединения по настоящему изобретению можно вводить одновременно, последовательно или параллельно с одним или более антидепрессантами, антипсихотическими средствами, седативными/снотворными средствами (например, бензодиазепинами, противосудорожными средствами, средствами для лечения наркотической зависимости (например, метадон, налоксон) и/или агентами, применяемыми для лечения болезни Паркинсона или расстройств настроения. В другом примере побочные эффекты могут быть уменьшены или минимизированы путем введения соединения по настоящему изобретению в комбинации с одним или более вторыми терапевтическими агентами в свободном виде или в виде соли, причем дозировки (i) второго терапевтического агента(-ов) или (ii) соединения по настоящему изобретению и вторых терапевтических агентов ниже, чем если бы эти агенты/соединения вводили в виде монотерапии.
[00036] Таким образом, в некоторых вариантах осуществления настоящего изобретения предлагается способ 3 или любой из способов 3.1-3.15 или способ 4 или любой из 4.1-4.9, дополнительно включающий введение пациенту одного или более терапевтических средств, при этом одно или более терапевтических средств выбирают из соединений, которые модулируют активность ГАМК (например, усиливают активность и облегчают передачу ГАМК), агониста ГАМК-В, модулятора рецептора 5-HT (например, агониста 5-HT1A, антагониста 5-HT2A, обратного агониста 5-HT2A, ингибитора обратного захвата серотонина и т.д.), агониста рецептора мелатонина, модулятора ионного канала (например, блокатора), антагониста рецептора/ингибитора обратного захвата серотонина-2 (соединения, обладающего как 5-HT2 антагонизмом, так и являющееся ингибитором обратного захвата серотонина, т.е. SARI), антагониста рецептора орексина, агониста или антагониста H3, норадренергического агониста или антагониста, агониста галанина, антагониста CRH, гормона роста человека, агониста гормона роста, эстрогена, агониста эстрогена, других нейроактивных стероидов, прогестерона или метаболитов прогестерона, лекарственного средства нейрокинина-1, антидепрессанта и опиоидных агонистов и/или частичных опиоидных агонистов (например, агониста или частичного агониста мю-, каппа- или дельта-опиоидных рецепторов), агониста ноцицептина, ингибитора метаболизма лекарственных веществ и антипсихотического агента, например атипичного антипсихотического агента, в свободном виде или в виде фармацевтически приемлемой соли. В некоторых вариантах осуществления такие агенты включают ингибиторов метаболизма лекарственных веществ, таких как ингибиторы редуктазы, ингибиторы оксидоредуктазы или ингибиторы цитохромоксидазы (фермент CYP), которые могут служить для снижения скорости метаболизма вводимого соединения по настоящему изобретению. Например, такие агенты могут включать ингибиторы кетонредуктаз и стероидных гидрогеназ (например, 20α-гидроксистероидной гидрогеназы или 20β-гидроксистероидной гидрогеназы). Помимо ингибиторов таких редуктаз, оксидоредуктаз и гидрогеназ, такие ингибиторы метаболизма лекарственных веществ также могут включать конкурирующие субстраты для этих ферментов.
[00037] В некоторых вариантах осуществления комбинация соединения по настоящему изобретению с одним или более описанными выше вторыми терапевтическими средствами может быть введена пациенту в виде описанных выше фармацевтической композиции или композиции депо. Комбинированные композиции могут включать смеси комбинированных лекарственных веществ, а также две или более отдельных композиций лекарственных веществ, причем возможно, например, совместное введение пациенту указанных отдельных композиций.
[00038] В пятом аспекте настоящее изобретение относится к применению соединения формулы I, например соединения 1 или любого из 1-1.31, или его фармацевтической композиции, например композиции 2 или любой из 2.1-2.11, при производстве лекарственного средства для лечения или профилактики одного или более указанных выше расстройств, например, в любом из способа 3 или любых из способов 3.1-3.15, или для индукции седации или анестезии, например, в любом из способа 4 или любого из 4.1-4.9 или любых других вариантов осуществления способа, представленных в настоящем описании.
[00039] В шестом аспекте настоящее изобретение относится к применению соединения Формулы I, например соединения 1 или любого из 1-1.31, или его фармацевтической композиции, например композиции 2 или любой из 2.1-2.11, при лечении или профилактике одного или более описанных выше расстройств, например, в любом из способа 3 или любом из способов 3.1-3.15, или для индукции седации или анестезии, например, в любом из способа 4 или любого из 4.1-4.9 или любых других вариантов осуществления способа, представленных в настоящем описании.
[00040] Не ограничиваясь какой-либо теорией, настоящее изобретение относится к соединениям, которые специфически ограничивают, замедляют, изменяют и/или предотвращают метаболизм, который, как было обнаружено, имеет место у животных и/или людей, получающих лечение соединением формулы A:
формула A.
[00041] Поскольку атомы дейтерия (2H) очень схожи с атомами водорода (1H) по своим химическим и физическим свойствам, например атомному заряду, атомному объему, полярности, валентности и т.д., считается, что лекарственные соединения, в которых водород замещен дейтерием, имеют биологическую активность, аналогичную недейтерированному аналогу, но потенциально улучшенные фармакокинетические свойства. Особенно важным является то, что, хотя атомная масса атомов дейтерия почти в два раза больше, чем у атомов протия, они имеют аналогичные объем и распределение заряда, причем последние факторы имеют решающее значение для связывания с биологическими молекулами. Улучшенные фармакокинетические свойства являются результатом значительно более прочной связи C-D по сравнению с H-D связью и, следовательно, более высокого энергетического барьера для разрушения D/H во время ферментативной (метаболической) реакции (кинетический изотопный эффект). Степень, при которой такая замена приведет к улучшению фармакокинетических свойств без слишком серьезной потери фармакологической активности, варьирует. Таким образом, в некоторых обстоятельствах полученное дейтерированное соединение обеспечивает только умеренное повышение фармакокинетической стабильности, тогда как в других обстоятельствах полученное дейтерированное соединение может обладать значительно улучшенной метаболической стабильностью. К тому же, трудно с уверенностью предсказать эффекты, возникающие при одновременных замещениях дейтерием, поскольку они могут либо привести, либо не привести к аддитивному (синергетическому) улучшению метаболической стабильности.
[00042] Хотя к настоящему времени было предложено и исследовано множество дейтерированных фармацевтических соединений, Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США было одобрено только одно дейтерированное фармацевтическое соединение - дейтерированный препарат тетрабеназин, применяемый при болезни Гентингтона (Teva Pharmaceuticals, апрель 2017 г.), период полувыведения которого с терапевтической точки зрения является более длительным, чем у его недейтерированного аналога.
[00043] В некоторых вариантах осуществления настоящее изобретение относится к соединениям, содержащим атомы дейтерия в конкретных выбранных положениях структуры соединения формулы A. Ожидается, что эти конкретные дейтерирования окажут влияние на метаболическое разложение и клиренс указанных соединений, поскольку их связь с ферментативными путями, определенными авторами изобретения, по всей видимости влияет на эти соединения. Следовательно, ожидается, что эти новые соединения по существу будут иметь такую же фармакологическую активность, что и соединение формулы A, и непредвиденную улучшенную метаболическую стабильность и фармакокинетические свойства.
[00044] В других вариантах осуществления настоящее изобретение относится к соединениям, которые являются аналогами соединения формулы A, имеющими биологически лабильные функциональные группы, расположенные внутри соединений, которые удаляются в результате естественной метаболической активности, обеспечивая in vivo соединение формулы A. Таким образом, введение нуждающемуся в этом пациенту некоторых соединений по настоящему изобретению приводит как к немедленному, так и к отсроченному высвобождению в ткани указанного человека соединения формулы A. Ожидается, что такие соединения по настоящему изобретению сами по себе не обладают значительной фармакологической активностью, но могут служить резервуаром для фармакологически активного соединения формулы A. Таким образом, соединения по настоящему изобретению являются особенно подходящими для изготовления инъекционных препаратов длительного действия (LAI) или фармацевтических композиций «депо». Не ограничиваясь какой-либо теорией, инъекционное «депо», содержащее соединение по настоящему изобретению, будет постепенно высвобождать в ткани организма указанное соединение, при этом указанное соединение будет постепенно подвергаться метаболизму с получением соединения формулы А. Такие составы депо можно дополнительно модифицировать путем выбора подходящих компонентов для контроля скорости растворения и высвобождения соединений по настоящему изобретению.
[00045] В контексте настоящего описания «алкил» означает насыщенный или ненасыщенный углеводородный фрагмент, например, если не указано иное, длиной от одного до двадцати одного атома углерода; причем любой такой алкил может быть линейным или разветвленным (например, н-бутил или трет-бутил), предпочтительно линейным, если не указано иное. Например, «C1-21 алкил» означает алкил, содержащий от 1 до 21 атома углерода. В одном из вариантов осуществления алкил необязательно замещен одной или более гидрокси- или C1-22алкокси (например, этокси) группами. В другом варианте осуществления алкил содержит от 1 до 21 атома углерода, предпочтительно с прямой и необязательно насыщенной или ненасыщенной цепью, например, в некоторых вариантах осуществления, где R1 представляет собой алкильную цепь, содержащую от 1 до 21 атома углерода, предпочтительно 6-15 атомов углерода, 16-21 атома углерода, например, такой, что вместе с -C(O)-, к которому он присоединен, например, при отщеплении от соединения формулы I, образует остаток природной или неприродной насыщенной или ненасыщенной жирной кислоты.
[00046] Термин «D» или «дейтерий» относится к 2H-изотопу атома водорода. Распространенность в природе двух стабильных изотопов водорода составляет примерно 99,98% протия (1H) и 0,02% дейтерия (2H). Таким образом, в среднем любой атом водорода в молекуле, синтезированной с помощью обычных реагентов, будет иметь приблизительно 0,02% дейтерия в каждом положении атома водорода. Следовательно, квалифицированный специалист поймет, что в случае ссылки на химическую структуру, имеющую связь CD или атом «D», в контексте настоящего описания, означает, что указанное положение молекулы является обогащенным до уровня, превышающего природную распространенность дейтерия, 0,02%. Таким образом, символ «D» в молекуле означает, например, по меньшей мере 0,1% дейтерия или по меньшей мере 1% дейтерия, или по меньшей мере 10% дейтерия. Предпочтительно, любое соединение по настоящему изобретению имеет более 50% включения дейтерия в каждом конкретном положении атома «D» в структуре соединения (т.е. более 50 атом.% D), например, более 60% или более 70% или более 80%, или более 90%, или более 95%, или более 96%, или более 97%, или более 98%, или более 99%.
[00047] Термин «фармацевтически приемлемый разбавитель или носитель» означает разбавители и носители, которые используются в фармацевтических препаратах и не содержат веществ, которые являются аллергенными, пирогенными или патогенными и которые, как известно, потенциально могут вызывать или способствовать заболеванию. Таким образом, фармацевтически приемлемые разбавители или носители не включают жидкости организма, такие как кровь, моча, спинномозговая жидкость, слюна и т.п., а также составляющие их компоненты, такие как клетки крови и циркулирующие белки. Подходящие фармацевтически приемлемые разбавители и носители можно найти в любом из нескольких хорошо известных руководств по фармацевтическим составам, например Anderson, Philip O.; Knoben, James E.; Troutman, William G, eds., Handbook of Clinical Drug Data, Tenth Edition, McGraw-Hill, 2002; Pratt and Taylor, eds., Principles of Drug Action, Third Edition, Churchill Livingston, New York, 1990; Katzung, ed., Basic and Clinical Pharmacology, Ninth Edition, McGraw Hill, 20037ybg; Goodman and Gilman, eds., The Pharmacological Basis of Therapeutics, Tenth Edition, McGraw Hill, 2001; Remington’s Pharmaceutical Sciences, 20th Ed., Lippincott Williams & Wilkins., 2000; and Martindale, The Extra Pharmacopoeia, Thirty-Second Edition (The Pharmaceutical Press, London, 1999); все из которых включены в настоящее описание во всей своей полноте в виде ссылки.
[00048] Термины «очищенный», «в очищенном виде» или «в выделенном и очищенном виде» в отношении соединения относятся к физическому состоянию указанного соединения после его выделения в процессе синтеза (например, из реакционной смеси) или из природного источника или их комбинации. Таким образом, термин «очищенный», «в очищенном виде» или «в выделенном и очищенном виде» в отношении соединения относится к физическому состоянию указанного соединения после его получения в процессе или процессах очистки, указанных в настоящем описании или хорошо известных специалисту в данной области (например, хроматографии, перекристаллизации, методов ЖХ-МС и ЖХ-МС/МС и т.п.) с достаточной чистотой, позволяющей охарактеризовать указанное соединение стандартными аналитическими методами, раскрытыми в настоящем описании или хорошо известными специалисту в данной области.
[00049] Если не указано иное, соединения по настоящему изобретению, например соединение I или 1.1-1.31, могут существовать в свободном виде или в виде соли, например, в виде солей, образованных в результате реакции присоединения кислоты. Такая соль образуется соединением по изобретению, которое является в достаточной степени основным для реакции с кислотой, например в результате реакции присоединения кислоты, например неорганической или органической кислоты, например соляной кислоты, и т.п.
[00050] Соединения по настоящему изобретению предназначены для применения в качестве фармацевтических агентов, поэтому фармацевтически приемлемые соли являются предпочтительными. Соли, непригодные для фармацевтического применения, могут быть полезными, например, для выделения или очистки свободных соединений по изобретению, и поэтому они также включены в объем соединений по настоящему изобретению.
[00051] Соединения по настоящему изобретению могут содержать один или более хиральных атомов углерода. Таким образом, соединения существуют в виде отдельных изомерных, например энантиомерных или диастереомерных, форм или в виде смесей отдельных форм, например, в рацемических/диастереомерных смесях. Возможен любой изомер, в котором асимметричный центр находится в (R)-, (S)- или (R, S)-конфигурации. Следует понимать, что изобретение охватывает как отдельные оптически активные изомеры, так и их смеси (например, рацемические/диастереомерные смеси). Соответственно, соединения по изобретению могут находиться в виде рацемической смеси, или они могут быть преимущественно, например, в виде чистого или по существу чистого изомера, например, с более 70% энантиомерным/диастереомерным избытком («ее»), предпочтительно более 80% ее, более предпочтительно более 90% ее, наиболее предпочтительно более 95% ее. Очистка указанных изомеров и разделение указанных смесей изомеров могут быть выполнены стандартными методами, известными в данной области (например, колоночной хроматографией, препаративной ТСХ, препаративной ВЭЖХ, псевдодвижущимся слоем и т.п.).
[00052] Геометрические изомеры по природе заместителей около двойной связи или кольца могут присутствовать в цис (Z) или транс (E) форме, и обе изомерные формы входят в объем настоящего изобретения.
[00053] Также предполагается, что соединения по настоящему изобретению включают стабильные и нестабильные изотопы. Стабильные изотопы представляют собой нерадиоактивные изотопы, которые содержат один дополнительный нейтрон по сравнению с распространенными нуклидами того же вида (т.е. элемента). Ожидается, что активность соединений, содержащих такие изотопы, будет сохраняться, и такое соединение также будет полезным для измерения фармакокинетики неизотопных аналогов. Например, атом водорода в определенном положении в соединениях по изобретению может быть заменен дейтерием (стабильный изотоп, который не является радиоактивным). Примеры известных стабильных изотопов включают, без ограничения, дейтерий, 13C, 15N, 18O. В качестве альтернативы соответствующие распространенные виды I, C и F могут быть заменены нестабильными изотопами, которые представляют собой радиоактивные изотопы, содержащие дополнительные нейтроны по сравнению с распространенными нуклидами того же вида (т.е. элемента), например 123I, 131I, 125I, 11C, 18F. Другим примером полезного изотопа соединения по изобретению является изотоп 11C. Эти радиоизотопы полезны для получения радиоизображения и/или для фармакокинетических исследований соединений по настоящему изобретению.
[00054] Таким образом, в дополнение к дейтерированию, специально предусмотренному объемом соединений формулы I, настоящее изобретение также относится к соединениям формулы I, в которых один или более атомов углерода, атомов азота или атомов кислорода заменены стабильным или нестабильным изотопным вариантом (например, 11C, 13C, 15N, 18O), и, кроме того, в котором один или более атомов водорода заменены тритием (3H). Эти соединения полезны, например, для определений структуры (например, с помощью ядерного магнитного резонанса или масс-спектрального анализа) и для радиовизуализационных исследований для выявления метаболических и экскреторных путей и для измерения клиренса потенциальных лекарственных препаратов-кандидатов.
[00055] Полимеры, полезные для полимерной матрицы в композиции по настоящему изобретению (например, композиции депо по настоящему изобретению), могут включать сложный полиэфир гидроксижирной кислоты и ее производных или других агентов, таких как полимолочная кислота, полигликолевая кислота, полилимонная кислота, полияблочная кислота, поли-бета-гидроксимасляная кислота, полимер с раскрытием кольца эпсилон-капролактона, сополимер молочной кислоты и гликолевой кислоты, сополимер 2-гидроксимасляной кислоты и гликолевой кислоты, сополимер полимолочной кислоты и полиэтиленгликоля или сополимер полигликолевой кислоты и полиэтиленгликоля), полимер алкил-альфа-цианоакрилата (например, поли(бутил-2-цианоакрилат)), полиалкиленоксалат (например, политриметиленоксалат или политетраметиленоксалат), сложный полиортоэфир, поликарбонат (например, полиэтиленкарбонат или полиэтиленпропиленкарбонат), полиортокарбонат, полиаминокислоту (например, поли-гамма-L-аланин, поли-гамма-бензил-L-глутаминовую кислоту или поли-γ-метил-L-глутаминовую кислоту), сложный эфир гиалуроновой кислоты и т.п., и можно использовать один или более из этих полимеров.
[00056] В предпочтительном варианте осуществления полимерная матрица по изобретению представляет собой биосовместимый и биоразлагаемый полимерный материал. Термин «биосовместимый» определяется как полимерный материал, который не является токсичным, канцерогенным и не вызывает значительного воспаления в тканях организма. Материал матрицы должен быть биоразлагаемым, при этом полимерный материал должен разлагаться под действием процессов, происходящих в организме, до продуктов, легко удаляемых из организма, и не должен накапливаться в организме. Продукты биодеградации также должны быть биосовместимы с организмом, поскольку полимерная матрица является биосовместимой с организмом.
[00057] Термины «заболевание», «расстройство» и «состояние» используются взаимозаменяемо, и их не следует интерпретировать, как имеющие какие-либо различия.
[00058] «Терапевтически эффективное количество» представляет собой любое количество соединения по изобретению (например, содержащееся в фармацевтическом депо), которое при введении субъекту, страдающему заболеванием или расстройством, эффективно вызывает уменьшение, ремиссию или регресс заболевания или расстройства в течение периода времени, предназначенного для лечения.
[00059] Дозировки, применяемые при практической реализации настоящего изобретения, конечно, будут меняться в зависимости, например, от конкретного заболевания или состояния, подлежащего лечению, конкретного используемого соединения по изобретению, способа введения и желаемой терапии. Если не указано иное, количество соединения по изобретению, предназначенного для введения (вводимого в виде свободного основания или в виде соли), относится или основано на количестве соединения по изобретению в виде свободного основания (т.е. количество рассчитывается, исходя из количества свободного основания).
[00060] Соединения по настоящему изобретению можно вводить любым подходящим способом, включая пероральный, сублингвальный, парентеральный (внутривенный, внутримышечный, интраназальный или подкожный) или чрескожный, предпочтительным является пероральное введение. В некоторых вариантах осуществления соединения по изобретению, например в составе депо, предпочтительно вводят парентерально, например, путем инъекции.
[00061] Фармацевтически приемлемые соли соединений по настоящему изобретению могут быть синтезированы из исходного соединения, которое содержит основную или кислотную составляющую, обычными химическими методами. Обычно такие соли могут быть получены в результате взаимодействия этих соединений в виде свободного основания со стехиометрическим количеством подходящей кислоты в воде или в органическом растворителе, или в их смеси; обычно предпочтительными являются неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил.
[00062] Фармацевтические композиции, содержащие соединения по настоящему изобретению, могут быть приготовлены с использованием обычных разбавителей или наполнителей (примеры включают, без ограничения, кунжутное масло) и методов, известных в области производства лекарственных форм. Таким образом, пероральные лекарственные формы могут включать таблетки, капсулы, растворы, суспензии и т.п.
[00063] Способы синтеза соединения формулы A, включая его промежуточные соединения раскрыты в публикациях США 2004/0034002 и 2009/0131383, содержание которых включено в настоящее описание во всей своей полноте в виде ссылки.
[00064] Основное ядро других соединений по настоящему изобретению получали согласно аналогичным процедурам, раскрытым в указанных выше публикациях и известных специалистам в данной области. Конкретные дейтерированные соединения по настоящему изобретению как правило могут быть получены аналогичными способами путем замены недейтерированных реагентов коммерчески доступными дейтерированными реагентами, если такие дейтерированные реагенты являются доступными.
[00065] Выделение или очистка диастереомеров соединений по изобретению может быть выполнена обычными методами, известными в данной области техники, например, путем очистки на колонке, с помощью препаративной тонкослойной хроматографии, препаративной ВЭЖХ, кристаллизации, растирания, псевдодвижущихся слоев и т.п.
[00066] Диастереомеры полученных соединений можно разделить, например, с помощью ВЭЖХ, используя CHIRALPAK® AY-H, 5 мкм, 30×250 мм при комнатной температуре путем элюирования смесью 10% этанол/90% гексан/0,1% диметилэтиламин. Пики могут быть обнаружены при 230 нм с получением 98-99,9% ее диастереомера.
[00067] 3α-Гидрокси-3β-метоксиметил-5α-прегнан-20-он может быть получен из (3R)-спиро[оксиран-2α,5α-прегнан]-20-она и метоксида натрия, как описано Hogenkamp, et al., «Synthesis and in Vitro Activity of 3β-Substituted-3α-hydroxypregnan-20-ones: Allosteric Modulators of the GABAA Receptor», J. Med. Chem. 40:61-72 (1997). 21-Замещенные стероиды могут быть получены из соответствующих 21-бромстероидов, которые синтезируют из 20-кетостероидов с использованием Br2 в MeOH с каталитическим HBr. Другие источники полезных синтетических методов включают: Botella et al., J. Med Chem., 48:3500-3511 (2015); Botella et al., J. Med Chem., 60:7810-7819 (2017); Wong et al., Steroids 71:77-82 (2006); Botella et al., WO 2016/061527; Hogenkamp, Derek L., WO 2000/66614; Goliber et al., US 2006/0074059; Chang et al., WO 2005/105822; и Woodward, Richard M., WO 2006/131392; содержание каждого из которых включено в настоящее описание во всей своей полноте в виде ссылки. В общем случае, соединения по настоящему изобретению могут быть получены способами, известными в данной области, начиная с хорошо известного соединения 3α-гидрокси-5α-прегнан-2-она и его диастереомеров.
[00068] Например, это соединение может быть получено согласно приведенной ниже схеме, описанной Wong et al.:
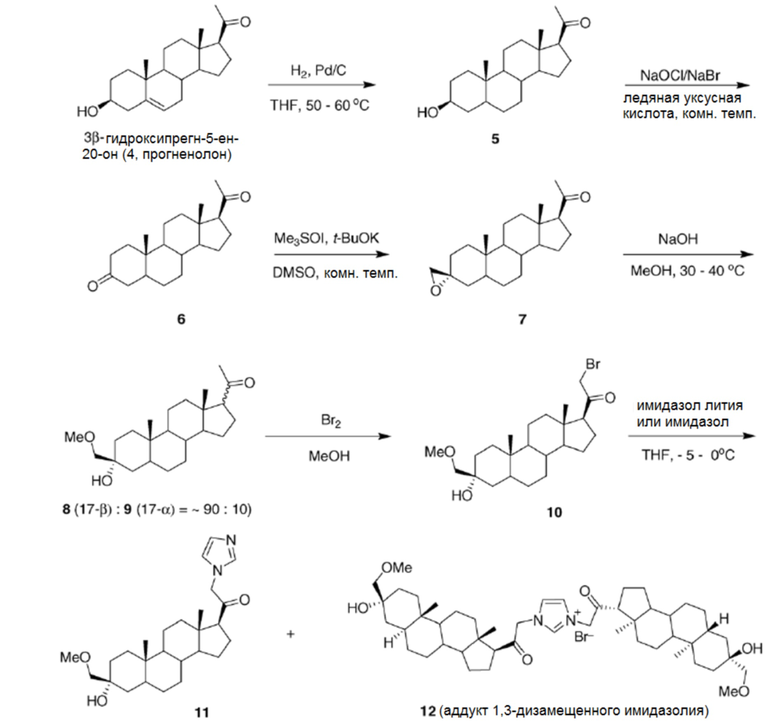
Пример 1: Синтез 3α-гидрокси-21-(1′-имидазолил)-3β-метоксиметил-5α-прегнан-20-она (соединение формулы A)
Этап 1: 21-бром-3α-гидрокси-3β-метоксиметил-5α-прегнан-20-она.
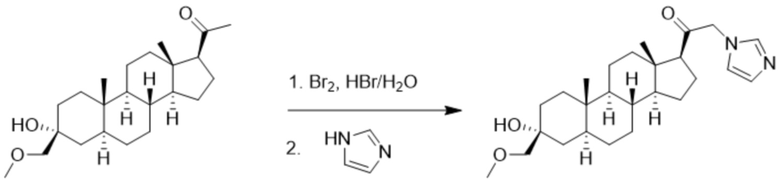
[00069] К раствору 3α-гидрокси-3β-метоксиметил-5α-прегнан-20-она (30,0 г, 82,9 ммоль) в 900 мл метанола, перемешивая при комнатной температуре, добавляют 3 капли 48% водного раствора HBr. Затем по каплям добавляют бром (13,9 г, 87,1 ммоль) в виде раствора в 200 мл метанола в течение 2 часов, в это время реакцию защищают от света. После того как ТСХ (1% ацетон/метиленхлорид) показала, что исходный материал отсутствует и образовался менее полярный продукт, реакционную смесь концентрируют до приблизительно 300 мл. Затем добавляют CH2Cl2 (400 мл), и реакционную смесь выливают в делительную воронку, содержащую 200 мл воды. Фазы разделяют, и водную фазу экстрагируют CH2Cl2 (3×100 мл). Органические фазы объединяют, промывают 200 мл насыщенного водного раствора NaHCO3, сушат над Na2SO4 и концентрируют при пониженном давлении с получением бромида в виде бледно-желтой пены. Никакой дополнительной очистки не требуется.
Этап 2: 3α-Гидрокси-21-(1′-имидазолил)-3β-метоксиметил-5α-прегнан-20-он
[00070] К суспензии полученного выше бромида (36,7 г, 82,9 ммоль) в 800 мл CH3CN добавляют имидазол (28,2 г, 415 ммоль), и реакционную смесь нагревают с обратным холодильником в атмосфере аргона. Реакция завершается через 1 час нагревания с обратным холодильником (ТСХ, 95:4,5:0,5 CH2Cl2:MeOH:триэтиламин (TEA)). Реакционную смесь охлаждают до комнатной температуры и затем концентрируют в вакууме. Полученное масло растворяют в 600 мл CH2Cl2, промывают разбавленным раствором NaHCO3 (4×200 мл), сушат над Na2SO4 и концентрируют в вакууме. Очистка флэш-хроматографией на силикагеле путем элюирования 95:4,5:0,5 CH2Cl2:MeOH:TEA дает 18 г указанного в заголовке соединения в виде белого твердого вещества с температурой плавления 185-187°C (откачанный капилляр). 1H ЯМР (300 МГц, CDCl3) δ7,40 (с, 1H), 7,08 (с, 1H), 6,84 (с, 1H), 4,72 (д, 1H, J=17,7 Гц), 4,64 (д, 1H, J=18 Гц), 3,39 (с, 3H), 3,18 (с, 2H), 2,57 (т, 1H, J=8,7 Гц), 0,76 (с, 3H), 0,66 (с, 3H).
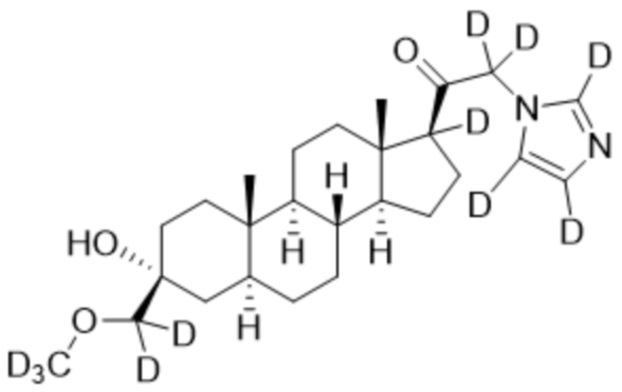
Пример 2: 1-((3R,5S,8R,9S,10S,13S,14S,17S)-3-гидрокси-3-(метоксиметил)-10,13-диметилгексадекагидро-1H-циклопента[a]фенантрен-17-ил)-2-(1H-имидазол-1-ил-d3)этан-1-он
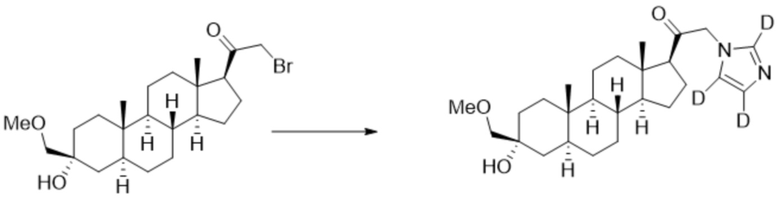
[00071] К раствору имидазола-d4 (0,193 г, 2,72 ммоль, 3,0 экв.) в THF (2 мл) при 0ºC добавляют гидрид лития (0,0237 г, 2,8 ммоль, 3,1 экв.). Раствор перемешивают при 0ºC в течение 2 часов в атмосфере Ar. В реакционную смесь медленно добавляют 21-бром-3α-гидрокси-3β-метоксиметил-5α-прегнан-20-он (0,40 г, 0,906 ммоль, 1,0 экв.) в THF (2,5 мл) при 0ºC в течение 5 мин в атмосфере Ar. После перемешивания при 0ºC в течение 3 часов реакцию гасят метанолом на ледяной бане. Органический слой отделяют и концентрируют при пониженном давлении. Остаток дополнительно очищают с помощью колоночной хроматографии с получением чистого указанного в заголовке соединения (0,184 г, 0,426 ммоль) в виде белого порошка с 50% выходом выделенного соединения. 1H ЯМР (500 МГц, хлороформ-d) δ 4,86-4,60 (м, 2H), 3,41 (с, 3H), 3,20 (с, 2H), 2,60 (т, J=8,9 Гц, 1H), 2,31-2,12 (м, 1H), 2,06-1,90 (м, 1H), 1,84-1,66 (м, 4H), 1,66-1,13 (м, 12H), 1,63-1,51 (м, 3H) 1,00 (м, 1H), 0,87 (м, 1H), 0,78 (с, 3H), 0,68 (с, 3H).
Пример 3: 1-((3R,5S,8R,9S,10S,13S,14S,17S)-3-гидрокси-3-((метокси-d3)метил)-10,13-диметилгексадекагидро-1H-циклопента[a]фенантрен-17-ил)-2-(1H-имидазол-1-ил)этан-1-он
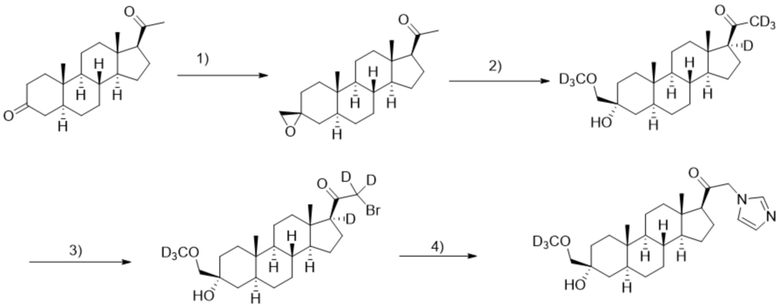
[00072] Этап 1: 1-((3R,5S,8R,9S,10S,13S,14S,17S)-10,13-диметилгексадекагидроспиро[циклопента[a]фенантрен-3,2'-оксиран]-17-ил)этан-1-он.
[00073] Перемешиваемый раствор йодида триметилсульфоксония (2,09 г, 9,5 ммоль) и трет-бутоксида калия (1,134 г, 10,01 ммоль) в ДМСО (30,0 мл) нагревают при 60ºC в течение 1 часа в атмосфере Ar. К реакционной смеси добавляют 5α-прегнанан-3,20-дион (2,0 г, 6,3 ммоль) и перемешивают при комнатной температуре в течение ночи. После завершения реакции реакционную смесь гасят и подвергают преципитации водой (30 мл) на ледяной бане. Полученный преципитат собирают фильтрацией, промывают водой (50 мл ×2). Остаток очищают перекристаллизацией из смеси MeOH/ацетон (4/1), затем твердое вещество сушат в вакууме в течение ночи, получая чистый конечный продукт (1,83 г, 5,54 ммоль) в виде белого порошка с 87% выходом. 1H ЯМР (500 МГц, хлороформ-d) δ 2,64 (д, J=1,4 Гц, 2H), 2,56 (т, J=8,9 Гц, 1H), 2,25-2,15 (м, 1H), 2,14 (с, 3H), 2,11-1,99 (м, 2H), 1,89 (т, J=14,0, 13,1 Гц, 1H), 1,79-1,60 (м, 6H), 1,60-1,51 (м, 1H), 1,50-1,05 (м, 9H), 1,06-0,94 (м, 2H), 0,90 (с, 3H), 0,63 (с, 3H).
[00074] Этап 2: 1-((3R,5S,8R,9S,10S,13S,14S,17S)-3-гидрокси-3-((метокси-d3)метил)-10,13-диметилгексадекагидро-1H-циклопента[a]фенантрен-17-ил-17-d)этан-1-он-2,2,2-d3
[00075] Гидроксид натрия (0,916 г, 22,9 ммоль) растворяют в метаноле-d4 (40 мл) и нагревают с обратным холодильником в течение 30 мин в атмосфере Ar. При комнатной температуре в атмосфере Ar к метанольному раствору медленно добавляют соединение, полученное на этапе 1 (3,8 г, 11,4 ммоль), и раствор нагревают при 40ºC в течение ночи. Реакционную смесь гасят и подвергают преципитиции водой (65 мл) на ледяной бане. Полученный преципитат фильтруют, промывают водой (25 мл ×2). Полученный твердый остаток дополнительно очищают перекристаллизацией из смеси этилацетат/гексаны (1/1) и сушат в вакууме с получением чистого продукта (3,15 г, 8,52 ммоль) в виде белого порошка с 74% выходом. 1H ЯМР (500 МГц, хлороформ-d) δ 3,20 (с, 2H), 2,26-2,13 (м, 1H), 2,04-1,95 (м, 1H), 1,80-1,51 (м, 8H), 1,52-1,12 (м, 12H), 0,98 (м 1H), 0,85 (м, 1H), 0,77 (с, 3H), 0,62 (с, 3H).
[00076] Этап 3: 2-бром-1-((3R,5S,8R,9S,10S,13S,14S,17S)-3-гидрокси-3-((метокси-d3)метил)-10,13-диметилгексадекагидро-1H-циклопента[a]фенантрен-17-ил-17-d)этан-1-он-2,2-d2.
[00077] К раствору соединения, полученного на этапе 2 (3,11 г, 8,41 ммоль) в MeOH (30 мл), добавляют три капли водного раствора HBr (48%). Бром (0,485 мл, 8,88 ммоль) растворяют в MeOH (20 мл) и добавляют по каплям к реакционной смеси в темноте. После завершения реакции реакционную смесь гасят добавлением воды (100 мл). Добавляют этилацетат (30 мл ×3) для экстракции конечного продукта, органическую фазу объединяют и снова промывают водой (20 мл ×2). Выпаривают растворитель, и остаток сушат в вакууме в течение ночи с получением чистого 7 (3,67 г, 8,20 ммоль) в виде белого порошка с 97% выходом. 1H ЯМР (500 МГц, хлороформ-d) δ 3,20 (с, 2H), 2,19 (т, J=10,8 Гц, 0H), 1,92 (ддд, J=11,9, 4,1, 2,9 Гц, 1H), 1,79-1,51 (м, 7H), 1,51-1,15 (м, 12H), 1,06-0,93 (м, 1H), 0,89-0,80 (м, 1H), 0,77 (с, 3H), 0,65 (с, 3H).
[00078] Этап 4: 1-((3R,5S,8R,9S,10S,13S,14S,17S)-3-гидрокси-3-((метокси-d3)метил)-10,13-диметилгексадекагидро-1H-циклопента[a]фенантрен-17-ил)-2-(1H-имидазол-1-ил)этан-1-он
[00079] К раствору имидазола (0,274 г, 4,03 ммоль, 3,0 экв.) в THF (2 мл) добавляют гидрид лития (0,035 г, 4,17 ммоль, 3,1 экв.). Раствор нагревают с обратным холодильником в течение 30 мин в атмосфере Ar. Соединение, полученное на этапе 3 (0,6 г, 1,34 ммоль, 1,0 экв.), в THF (4 мл) медленно добавляют к реакционной смеси при 0ºC в течение 5 мин в атмосфере Ar. После перемешивания при 0ºC в течение 3 часов реакционную смесь гасят метанолом на ледяной бане. Выпаривают растворитель и выполняют очистку колоночной хроматографией с получением указанного в заголовке соединения (0,12 г, 0,277 ммоль) в виде белого порошка с фактическим выходом 21%. 1H ЯМР (500 МГц, хлороформ-d) δ 7,39 (с, 1H), 7,13 (с, 1H), 6,88 (с, 1H), 4,93-4,56 (м, 2H), 3,20 (с, 1H), 2,60 (т, J=8,8 Гц, 1H), 2,33-2,12 (м, 1H), 1,98 (дт, J=11,8, 3,3 Гц, 1H), 1,83-1,66 (м, 4H), 1,63-1,54 (м, 4H), 1,51-1,13 (м, 12H), 1,07-0,94 (м, 1H), 0,91-0,82 (м, 1H), 0,78 (с, 3H), 0,68 (с, 3H).
Пример 4: 1-((3R,5S,8R,9S,10S,13S,14S,17S)-3-гидрокси-3-((метокси-d3)метил)-10,13-диметилгексадекагидро-1H-циклопента[a]фенантрен-17-ил)-2-(1H-имидазол-1-ил-d3)этан-1-он
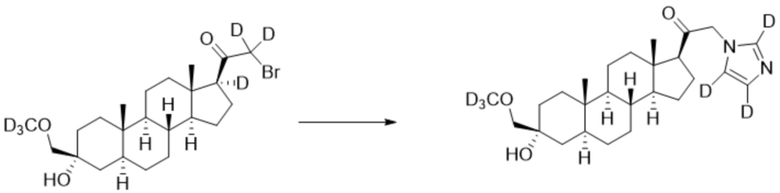
[00080] К раствору имидазола-d4 (0,291 г, 4,03 ммоль, 3,0 экв.) в THF (2 мл) добавляют гидрид лития (0,035 г, 4,17 ммоль, 3,1 экв.). Раствор нагревают с обратным холодильником в течение 30 мин в атмосфере Ar. Соединение, полученное на этапе 3 примера 3 (0,6 г, 1,34 ммоль, 1,0 экв.), в THF (4 мл) медленно добавляют к реакционной смеси при 0ºC в течение 5 мин в атмосфере Ar. После перемешивания при 0ºC в течение 3 часов реакционную смесь гасят метанолом на ледяной бане. Выпаривают растворитель и выполняют очистку колоночной хроматографией с получением указанного в заголовке соединения (0,07 г, 0,016 ммоль) в виде белого порошка с фактическим выходом 12%. 1H ЯМР (500 МГц, хлороформ-d) δ 3,20 (с, 2H), 2,60 (т, J=8,9 Гц, 1H), 2,31-2,15 (м, 1H), 2,08-1,90 (м, 1H), 1,82-1,66 (м, 4H), 1,64-1,14 (м, 15H), 1,08-0,93 (м, 1H), 0,92-0,83 (м, 1H), 0,78 (с, 3H), 0,68 (с, 3H).
Пример 5: 1-((3R,5S,8R,9S,10S,13S,14S,17S)-3-гидрокси-3-((метокси-d3)метил)-10,13-диметилгексадекагидро-1H-циклопента[a]фенантрен-17-ил-17-d)-2-(1H-имидазол-1-ил)этан-1-он-2-d
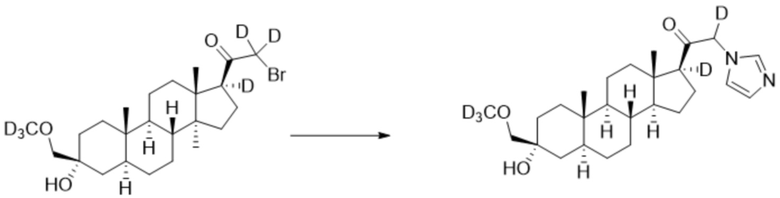
[00081] К раствору имидазола (0,161 г, 2,37 ммоль, 3,0 экв.) в THF (2 мл) добавляют гидрид лития (0,021 г, 2,45 ммоль, 3,1 экв.). Раствор перемешивают при 0ºC в течение 2 часов в атмосфере Ar. Соединение, полученное на этапе 3 примера 3 (0,353 г, 0,79 ммоль, 1,0 экв.), в THF (2,5 мл) медленно добавляют к реакционной смеси при 0ºC в течение 5 мин в атмосфере Ar. После перемешивания при 0ºC в течение 3 часов реакционную смесь гасят D2O (1,5 мл) на ледяной бане. Выпаривают растворитель и выполняют очистку колоночной хроматографией с получением указанного в заголовке соединения (0,097 г, 0,253 ммоль) в виде белого порошка с фактическим выходом 29% выделенного соединения. 1H ЯМР (500 МГц, хлороформ-d) δ 7,56 (с, 1H), 7,14 (с, 1H), 7,04-6,84 (м, 1H), 5,05-4,43 (м, 1H), 3,20 (с, 2H), 2,33-2,14 (м, 1H), 1,97 (дт, J=11,6, 3,4 Гц, 1H), 1,82-1,66 (м, 4H), 1,57 (м, 3H), 1,50-1,15 (м, 12H), 1,06-0,92 (м, 1H), 0,91-0,82 (м, 1H), 0,78 (с, 3H), 0,68 (с, 3H).
Пример 6: 1-((3R,5S,8R,9S,10S,13S,14S,17S)-3-гидрокси-3-((метокси-d3)метил)-10,13-диметилгексадекагидро-1H-циклопента[a]фенантрен-17-ил-17-d)-2-(1H-имидазол-1-ил-d3)этан-1-он-2-d
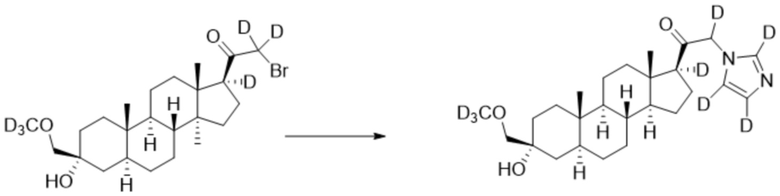
[00082] К раствору имидазола-d4 (0,194 г, 2,69 ммоль, 3,0 экв.) в THF (2 мл) добавляют гидрид лития (0,023 г, 2,78 ммоль, 3,1 экв.). Раствор перемешивают при 0ºC в течение 2 часов в атмосфере Ar. Соединение, полученное на этапе 3 примера 3 (0,4 г, 0,89 ммоль, 1,0 экв.), в THF (2,5 мл) медленно добавляют к реакционной смеси при 0ºC в течение 5 мин в атмосфере Ar. После перемешивания при 0ºC в течение 3 часов реакционную смесь гасят D2O (1,5 мл) на ледяной бане. Выпаривают растворитель и выполняют очистку колоночной хроматографией с получением чистого 11 (0,17 г, 0,389 ммоль) в виде белого порошка с фактическим выходом 44%. 1H ЯМР (500 МГц, хлороформ-d) δ 4,92-4,38 (м, 1H), 3,20 (с, 2H), 2,36-2,06 (м, 1H), 1,97 (дт, J=11,8, 3,4 Гц, 1H), 1,85-1,63 (м, 4H), 1,64-1,14 (м, 11H), 0,99 (м, 1H), 0,86 (м, 1H), 0,78 (с, 3H), 0,68 (с, 3H).
Пример 7: 1-((3R,5S,8R,9S,10S,13S,14S,17S)-3-гидрокси-3-((метокси-d3)метил-d2)-10,13-диметилгексадекагидро-1H-циклопента[a]фенантрен-17-ил)-2-(1H-имидазол-1-ил)этан-1-он
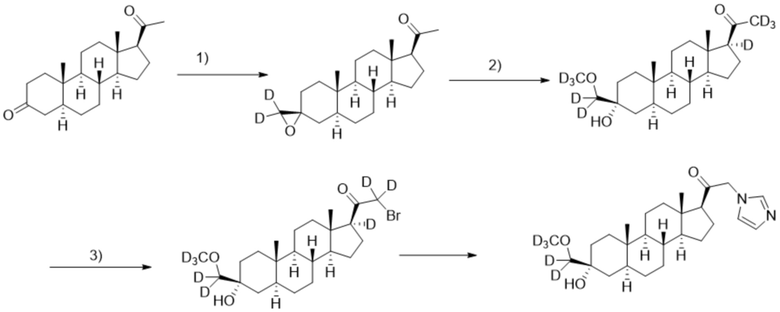
[00083] Этап 1: 1-((3R,5S,8R,9S,10S,13S,14S,17S)-10,13-диметилгексадекагидроспиро[циклопента[a]фенантрен-3,2'-оксиран]-17-ил-3',3'-d2)этан-1-он
[00084] Перемешиваемый раствор иодида триметилсульфоксония-d9 (2,17 г, 9,48 ммоль) и трет-бутоксида калия (1,13 г, 10,1 ммоль) в ДМСО-d6 (26,0 мл) нагревают при 60ºC в течение 1 часа в атмосфере Ar. К реакционной смеси добавляют 5α-прегнан-3,20-дион (2,0 г, 6,3 ммоль) и перемешивают при комнатной температуре в течение ночи. После завершения реакции реакционную смесь гасят подвергают преципитации водой (60 мл) на ледяной бане. Полученный преципитат собирают фильтрацией, промывают водой (50 мл ×2). Остаток дополнительно очищают перекристаллизацией из смеси MeOH/ацетон (4/1) и сушат под вакуумом с получением чистого конечного продукта (2,08 г, 6,2 ммоль) в виде белого порошка с 99% выходом и 91,5% изотопной чистоты. 1H ЯМР (500 МГц, хлороформ-d) δ 2,56 (т, J=9,0 Гц, 1H), 2,28-2,09 (м, 3H), 2,08-1,98 (м, 2H), 1,89 (т, J=13,5 Гц, 1H), 1,80-1,62 (м, 5H), 1,57 (м, 2H), 1,50-1,06 (м, 10H), 0,97 (м, 1H), 0,92-0,73 (м, 5H), 0,63 (с, 3H).
[00085] Этап 2: 1-((3R,5S,8R,9S,10S,13S,14S,17S)-3-гидрокси-3-((метокси-d3)метил-d2)-10,13-диметилгексадекагидро-1H-циклопента[a]фенантрен-17-ил-17-d)этан-1-он-2,2,2-d3
[00086] Гидроксид натрия (0,49 г, 12,2 ммоль) растворяют в метаноле-d4 (20 мл) и нагревают с обратным холодильником в течение 30 мин в атмосфере Ar. Соединение, полученное на этапе 1 (2,04 г, 6,12 ммоль), медленно добавляют к метанольному раствору при комнатной температуре в атмосфере Ar, и раствор нагревают при 40ºC в течение ночи. Реакционную смесь гасят и подвергают преципитации водой (60 мл) на ледяной бане. Полученный преципитат фильтруют, промывают водой (25 мл ×2). Твердый осадок дополнительно очищают перекристаллизацией из смеси этилацетат/гексаны (1/1) и сушат под вакуумом с получением чистого продукта (1,43 г, 3,85 ммоль) в виде белого порошка с 63% выходом. 1H ЯМР (500 МГц, хлороформ-d) δ 2,23-2,12 (м, 1H), 2,01 (дт, J=12,1, 3,4 Гц, 1H), 1,80-1,51 (м, 7H), 1,49-1,12 (м, 11H), 1,06-0,92 (м, 1H), 0,89-0,79 (м, 1H), 0,77 (с, 3H), 0,62 (с, 3H).
[00087] Этап 3: 2-бром-1-((3R,5S,8R,9S,10S,13S,14S,17S)-3-гидрокси-3-((метокси-d3)метил-d2)-10,13-диметилгексадекагидро-1H-циклопента[a]фенантрен-17-ил-17-d)этан-1-он-2,2-d2
[00088] К раствору соединения, полученного на этапе 2 (1,43 г, 3,85 ммоль) в MeOH (12 мл) добавляют три капли водного раствора HBr (48%). Бром (0,209 мл, 4,04 ммоль) растворяют в MeOH (8 мл) и по каплям добавляют к реакционному раствору в темноте. После завершения реакции реакционную смесь гасят добавлением воды (60 мл). Добавляют этилацетат (40 мл x2) для экстракции конечного продукта, органическую фазу объединяют и снова промывают водой (30 мл ×2). Растворитель выпаривают, и остаток сушат под вакуумом в течение ночи с получением чистого продукта (1,70 г, 3,78 ммоль) в виде белого порошка с 98% выходом. 1H ЯМР (500 МГц, хлороформ-d) δ 2,24-2,12 (м, 1H), 1,92 (дт, J=11,8, 3,5 Гц, 1H), 1,79-1,64 (м, 4H), 1,62-1,50 (м, 3H), 1,50-1,15 (м, 11H), 1,04-0,92 (м, 1H), 0,90-0,80 (м, 1H), 0,77 (с, 3H), 0,65 (с, 3H).
[00089] Этап 4: 1-((3R,5S,8R,9S,10S,13S,14S,17S)-3-гидрокси-3-((метокси-d3)метил-d2)-10,13-диметилгексадекагидро-1H-циклопента[a]фенантрен-17-ил)-2-(1H-имидазол-1-ил)этан-1-он
[00090] К раствору имидазола (0,182 г, 2,7 ммоль, 3,0 экв.) в THF (1,5 мл) добавляют гидрид лития (0,023 г, 2,76 ммоль, 3,1 экв.). Раствор перемешивают при 0ºC в течение 2 часов в атмосфере Ar. Соединение, полученное на этапе 3 (0,4 г, 0,89 ммоль, 1,0 экв.), в THF (3,0 мл) медленно добавляют к реакционной смеси при 0ºC в течение 5 мин в атмосфере Ar. После перемешивания при 0ºC в течение 3 часов реакционную смесь гасят метанолом (3,0 мл) на ледяной бане. Растворитель выпаривают, и остаток очищают колоночной флэш-хроматографией на силикагеле с получением соответствующего продукта, затем повторно очищают полупрепаративной ВЭЖХ с получением конечного продукта (0,063 г, 0,145 ммоль) в виде белого порошка с фактическим выходом 16%. 1H ЯМР (500 МГц, хлороформ-d) δ 7,49 (с, 1H), 7,13 (с, 1H), 6,88 (с, 1H), 4,97-4,45 (м, 2H), 2,60 (т, J=9,0 Гц, 1H), 2,27-2,10 (м, 1H), 1,98 (д, J=11,9, 3,4 Гц, 1H), 1,80-1,66 (м, 4H), 1,62-1,52 (м, 3H), 1,51-1,16 (m, 12H), 1,06-0,93 (м, 1H), 0,90-0,81 (м, 1H), 0,78 (с, 3H), 0,68 (с, 3H).
[00091] Пример 8: 1-((3R,5S,8R,9S,10S,13S,14S,17S)-3-гидрокси-3-((метокси-d3)метил-d2)-10,13-диметилгексадекагидро-1H-циклопента[a]фенантрен-17-ил)-2-(1H-имидазол-1-ил-2-d)этан-1-он
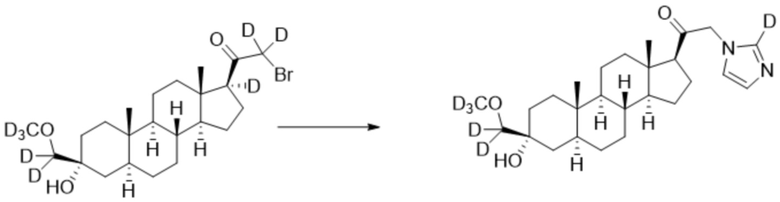
[00092] К раствору имидазола-d1 (0,184 г, 2,67 ммоль, 3,0 экв.) в THF (2,0 мл) добавляют гидрид лития (0,023 г, 2,76 ммоль, 3,1 экв.). Раствор перемешивают при 0ºC в течение 2 часов в атмосфере Ar. Соединение, полученное на этапе 3 примера 7 (0,4 г, 0,89 ммоль, 1,0 экв.), в THF (3,0 мл) медленно добавляют к реакционной смеси при 0ºC в течение 5 мин в атмосфере Ar. После перемешивания при 0ºC в течение 3 часов реакционную смесь гасят метанолом (3,0 мл) на ледяной бане. Растворитель выпаривают, и остаток очищают колоночной флэш-хроматографией на силикагеле с получением соответствующего продукта, затем повторно очищают полупрепаративной ВЭЖХ с получением конечного продукта (0,163 г, 0,375 ммоль) в виде белого порошка с фактическим выходом 42%. 1H ЯМР (500 МГц, хлороформ-d) δ 7,28 (с, 1H), 6,88 (с, 1H), 5,06-4,50 (м, 2H), 2,60 (т, J=8,9 Гц, 1H), 2,34-2,12 (м, 1H), 1,98 (дт, J=11,8, 3,4 Гц, 1H), 1,80-1,67 (м, 4H), 1,64-1,51 (м, 3H), 1,51-1,15 (м, 11H), 1,07-0,94 (м, 1H), 0,91-0,82 (м, 1H), 0,78 (с, 3H), 0,68 (с, 3H).
Пример 9: 1-((3R,5S,8R,9S,10S,13S,14S,17S)-3-гидрокси-3-((метокси-d3)метил-d2)-10,13-диметилгексадекагидро-1H-циклопента[a]фенантрен-17-ил)-2-(1H-имидазол-1-ил-d3)этан-1-он
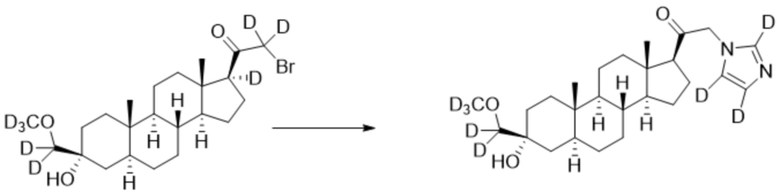
[00093] К раствору имидазола-d4 (0,144 г, 2,00 ммоль, 3,0 экв.) в THF (2,0 мл) добавляют гидрид лития (0,0174 г, 2,07 ммоль, 3,1 экв.). Раствор перемешивают при 0ºС в течение 2 часов в атмосфере Ar. Соединение, полученное на этапе 3 примера 7 (0,3 г, 0,667 ммоль, 1,0 экв.), в THF (3,0 мл) медленно добавляют к реакционной смеси при 0ºС в течение 5 мин в атмосфере Ar. После перемешивания при 0ºС в течение 3 часов реакционную смесь гасят метанолом (3,0 мл) на ледяной бане. Растворитель выпаривают, и остаток очищают колоночной флэш-хроматографией на силикагеле с получением соответствующего продукта, затем повторно очищают полупрепаративной ВЭЖХ с получением конечного продукта (0,147 г, 0,337 ммоль) в виде белого порошка с фактическим выходом 51%. 1H ЯМР (500 МГц, хлороформ-d) δ 4,87-4,36 (м, 2H), 2,60 (т, J=8,9 Гц, 0H), 2,34-2,07 (м, 2H), 1,98 (дт, J=11,7, 3,4 Гц, 0H), 1,80-1,65 (м, 2H), 1,64-1,52 (м, 1H), 1,51-1,15 (м, 11H), 1,06-0,94 (м, 1H), 0,90-0,82 (м, 1H), 0,78 (с, 3H), 0,68 (с, 3H).
Пример 10: 1-((3R,5S,8R,9S,10S,13S,14S,17S)-3-гидрокси-3-((метокси-d3)метил-d2)-10,13-диметилгексадекагидро-1H-циклопента[a]фенантрен-17-ил-17-d)-2-(1H-имидазол-1-ил)этан-1-он
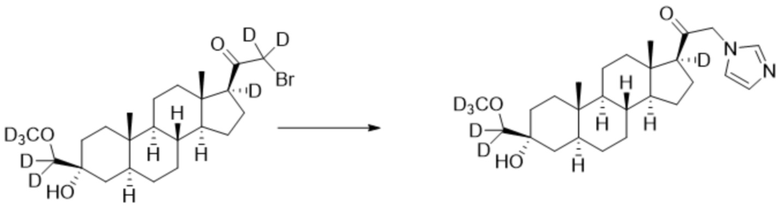
[00094] К раствору имидазола (0,136 г, 2,00 ммоль, 3,0 экв.) в THF (2,0 мл) добавляют гидрид лития (0,0174 г, 2,07 ммоль, 3,1 экв.). Раствор перемешивают при 0ºC в течение 2 часов в атмосфере Ar. Соединение, полученное на этапе 3 примера 7 (0,3 г, 0,667 ммоль, 1,0 экв.), в THF (2,5 мл) медленно добавляют к реакционной смеси при 0ºC в течение 5 мин в атмосфере Ar. После перемешивания при 0ºC в течение 3 часов реакционную смесь гасят D2O (1,50 мл) на ледяной бане. Растворитель выпаривают, и остаток очищают колоночной флэш-хроматографией на силикагеле с получением соответствующего продукта, затем повторно очищают полупрепаративной ВЭЖХ с получением конечного продукта (0,140 г, 0,322 ммоль) в виде белого порошка с фактическим выходом 48%. 1H ЯМР (500 МГц, хлороформ-d) δ 7,60 (с, 1H), 7,15 (с, 1H), 6,89 (с, 1H), 5,20-4,42 (м, 2H), 2,41-2,11 (м, 1H), 2,05-1,87 (м, 1H), 1,80-1,65 (м, 4H), 1,63-1,52 (м, 3H), 1,50-1,15 (м, 13H), 1,06-0,93 (м, 1H), 0,91-0,83 (м, 1H), 0,78 (с, 3H), 0,68 (с, 3H).
Пример 11: 1-((3R,5S,8R,9S,10S,13S,14S,17S)-3-гидрокси-3-((метокси-d3)метил-d2)-10,13-диметилгексадекагидро-1H-циклопента[a]фенантрен-17-ил-17-d)-2-(1H-имидазол-1-ил-2-d)этан-1-он
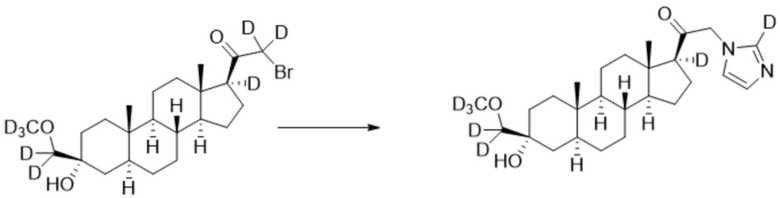
[00095] К раствору имидазола-d1 (0,102 г, 1,48 ммоль, 3,0 экв.) в THF (2,0 мл) добавляют гидрид лития (0,0128 г, 1,52 ммоль, 3,1 экв.). Раствор перемешивают при 0ºC в течение 2 часов в атмосфере Ar. Соединение, полученное на этапе 3 примера 7 (0,221 г, 0,492 ммоль, 1,0 экв.), в THF (3,0 мл) медленно добавляют к реакционной смеси при 0ºC в течение 5 мин в атмосфере Ar. После перемешивания при 0ºC в течение 3 часов реакционную смесь гасят D2O (1,50 мл) на ледяной бане. Растворитель выпаривают, и остаток очищают колоночной флэш-хроматографией на силикагеле с получением соответствующего продукта, затем повторно очищают полупрепаративной ВЭЖХ с получением конечного продукта (0,156 г, 0,358 ммоль) в виде белого порошка с фактическим выходом 73%. 1H ЯМР (500 МГц, хлороформ-d) δ 7,51-7,30 (м, 1H), 7,13 (д, J=2,5 Гц, 1H), 5,76-5,03 (м, 2H), 2,26-2,14 (м, 1H), 2,10 (д, J=12,2 Гц, 1H), 1,90-1,80 (м, 1H), 1,79-1,73 (м, 1H), 1,73-1,64 (м, 2H), 1,65-1,46 (м, 5H), 1,44-1,15 (м, 10H), 1,07-0,94 (м, 1H), 0,91-0,83 (м, 1H), 0,77 (с, 3H), 0,70 (с, 3H).
Пример 12: 1-((3R,5S,8R,9S,10S,13S,14S,17S)-3-гидрокси-3-((метокси-d3)метил-d2)-10,13-диметилгексадекагидро-1H-циклопента[a]фенантрен-17-ил-17-d)-2-(1H-имидазол-1-ил-d3)этан-1-он
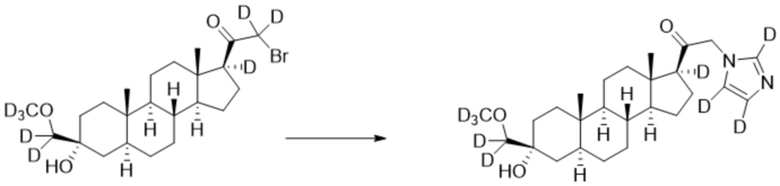
[00096] К раствору имидазола-d4 (0,144 г, 2,00 ммоль, 3,0 экв.) в THF (2,0 мл) добавляют гидрид лития (0,0174 г, 2,07 ммоль, 3,1 экв.). Раствор перемешивают при 0ºC в течение 2 часов в атмосфере Ar. Соединение, полученное на этапе 3 примера 7 (0,300 г, 0,667 ммоль, 1,0 экв.), в THF (3,0 мл) медленно добавляют к реакционной смеси при 0ºC в течение 5 мин в атмосфере Ar. После перемешивания при 0ºC в течение 3 часов реакционную смесь гасят D2O (1,50 мл) на ледяной бане. Растворитель выпаривают, и остаток очищают колоночной флэш-хроматографией на силикагеле с получением соответствующего продукта, затем повторно очищают полупрепаративной ВЭЖХ с получением конечного продукта (0,160 г, 0,366 ммоль) в виде белого порошка с фактическим выходом 55%. 1H ЯМР (500 МГц, хлороформ-d) δ 5,04-4,56 (м, 2H), 2,30-2,13 (м, 1H), 1,98 (дт, J=11,8, 3,4 Гц, 1H), 1,82-1,66 (м, 4H), 1,63-1,52 (м, 3H), 1,51-1,14 (м, 11H), 1,07-0,93 (м, 1H), 0,93-0,83 (м, 1H), 0,78 (м, 3H), 0,68 (с, 3H).
Пример 13: Другие аналоги 3α-гидрокси-21-(1′-имидазолил)-3β-метоксиметил-5α-прегнана-20-она
[00097] Другие дейтерированные аналоги соединений в рамках настоящего изобретения могут быть получены соответствующими способами, известными специалистам в данной области техники, с использованием дейтерированных реагентов вместо обычных реагентов, следуя указанным выше процедурам предшествующего уровня техники, на которые приведены ссылки в настоящем описании. Например:
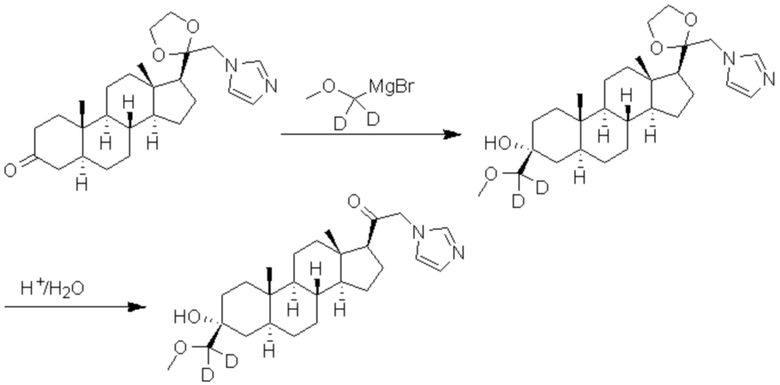
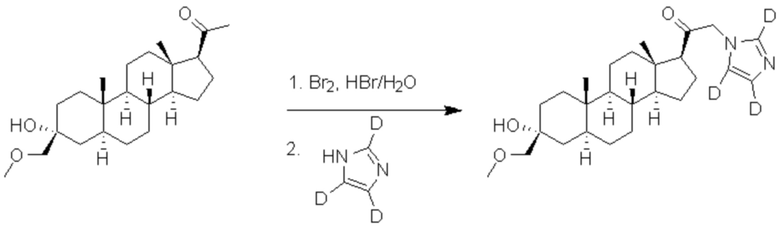
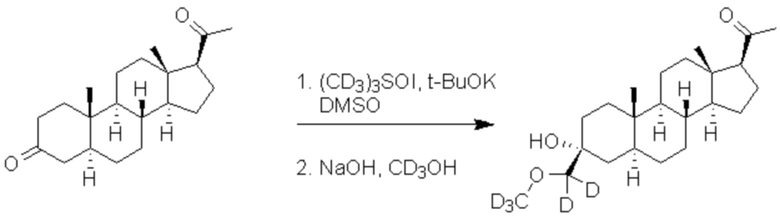
Пример 14: Фармакологическая активность 3α-гидрокси-21-(1′-имидазолил)-3β-метоксиметил-5α-прегнана-20-она
[00098] Определяли эффективность in vitro [способность ингибировать связывание [35S]-трет-бутилбициклофосфоротионата (TBPS)], TD50 в тесте ротород (доза, при которой половина испытуемых животных не может удержаться на вращающемся стержне в течение 1 минуты) и промежуток времени до момента, когда тестируемые животные, получившие 3α-гидрокси-21-(1′-имидазолил)-3β-метоксиметил-5α-прегнана-20-он, были способны пройти тест ротород (продолжительность действия). Эти способы измерения активности соединений по изобретению in vitro и in vivo полностью описаны в патенте США № 5,232,917, включенном в настоящее описание во всей своей полноте в виде ссылки. Анализ TBPS позволяет определить активность соединений in vitro, тогда как анализ с вращающимся стержнем позволяет оценить седативную/снотворную активность соединений. Поскольку продолжительность действия соединения зависит от дозы и будет увеличиваться при более высоких дозах, продолжительность действия измеряют при самой низкой дозе, при которой все животные не прошли испытание на вращающемся стержне. IC50 представляет собой дозу стероида, ингибирующую 50% специфического связывания [35S]-трет-бутилбициклофоротионата (TBPS). TD50 в тесте ротород представляет собой дозу, при которой половина животных не способна пройти тест ротород. Продолжительность действия, измеренная при наименьшей дозе, при которой все животные не прошли тест ротород, представляет собой время, необходимое всем испытуемым животным для повторного прохождения теста ротород.
[00099] Результаты показывают, что соединение формулы A (пример 1), 3α-гидрокси-21-(1'-имидазолил)-3β-метоксиметил-5α-прегнан-20-он, имеет TBPS IC50 138 нМ, TD50 (пероральная) в тесте ротород 28 мг/кг и продолжительность действия 140 минут.
Пример 15: In vivo фармакокинетика дейтерированных соединений у мышей
[000100] В первом исследовании соединения примеров 2-6 сравнивают с соединением формулы A (пример 1) у мышей, используя стандартные процедуры. В каждом тесте выполняют совместное дозирование двух соединений одному животному и определяют относительную фармакокинетику в плазме и мозге. Каждое тестируемое соединение растворяют в носителе, полиэтиленгликоле 400, и вводят перорально в дозе 10 мг/кг. После перорального совместного введения однократной дозы тестируемых соединений измеряют уровни в плазме и мозге через 0,25, 0,5, 1, 2, 4 и 6 часов после введения дозы. Определяют средние значения максимальной концентрации, времени достижения максимальной концентрации и площади под кривой (AUC) для обоих соединений. Результаты представлены в приведенных ниже таблицах для различных пар экспериментов совместных дозирований:
(нг/мл)
(нг/мл)
(нг/мл)
[000101] Эти результаты неожиданно показали, что каждое из дейтерированных соединений примеров 2, 4 и 6 оказывает гораздо более высокое воздействие на мозг в качестве лекарственного вещества по сравнению с недейтерированным соединением примера 1 и, таким образом, имеет гораздо более высокое соотношение мозг/плазма AUC. Напротив, дейтерированное соединение примера 3 имеет такие же значения AUC в плазме и головном мозге и соотношение B/P, что и соединение примера 1. Эти данные позволяют предположить, что дейтерирование в имидазольном кольце или рядом с ним оказывает благоприятное влияние на распределение соединения между мозгом и плазмой и, следовательно, приводит к более высокому воздействию лекарственного вещества на центральную нервную систему. Это особенно важно, поскольку эти соединения представляют собой нейроактивные стероиды с предполагаемым связыванием с рецепторами ГАМК центральной нервной системы.
Пример 16: In vitro фармакокинетика дейтерированных соединений в гепатоцитах человека
[000102] Соединения примеров 1, 2, 5 и 6 сравнивали в стандартном анализе стабильности гепатоцитов человека in vitro, проводимом в двух повторах. Соединения находились в виде 1 мкМ растворов в ДМСО, и концентрацию тестируемого соединения определяли через 0,5, 1,0 и 4,0 часа после добавления тестируемого соединения. Результаты показаны ниже в процентах от начальной концентрации.
[000103] Эти результаты позволяют предположить, что увеличение дейтерирования имидазола может увеличивать метаболическую стабильность этих соединений в гепатоцитах (ср. пример 1 с примером 2), в то время как дейтерирование метокси-группы или атомов углерода, соседних с карбонильной группой, дает неясные эффекты.
| название | год | авторы | номер документа |
|---|---|---|---|
| РЕГУЛЯТОРЫ ПРОИЗВОДНЫХ СТЕРОИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2803499C1 |
| РЕГУЛЯТОРЫ-ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2797408C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ ЦНС | 2015 |
|
RU2733756C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2830163C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ | 2018 |
|
RU2810331C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ УСИЛЕНИЯ ДЕГРАДАЦИИ БЕЛКОВ-МИШЕНЕЙ И ДРУГИХ ПОЛИПЕПТИДОВ С ПОМОЩЬЮ Е3 УБИКВИТИН ЛИГАЗЫ | 2013 |
|
RU2666530C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ УСИЛЕНИЯ ДЕГРАДАЦИИ БЕЛКОВ-МИШЕНЕЙ И ДРУГИХ ПОЛИПЕПТИДОВ С ПОМОЩЬЮ E3 УБИКВИТИН ЛИГАЗЫ | 2013 |
|
RU2781452C2 |
| НОВЫЕ СТЕРОИДНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ ПОВЫШЕННОЙ РАСТВОРИМОСТЬЮ В ВОДЕ И УСТОЙЧИВОСТЬЮ К МЕТАБОЛИЗМУ, И СПОСОБЫ ИХ ПРИГОТОВЛЕНИЯ | 2007 |
|
RU2458065C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ, ЗАМЕЩЕННЫЕ В ПОЛОЖЕНИИ 10 ЦИКЛИЧЕСКОЙ ГРУППОЙ, ДЛЯ ИСПОЛЬЗОВАНИЯ ПРИ ЛЕЧЕНИИ РАССТРОЙСТВ ЦНС | 2019 |
|
RU2796006C2 |
| МОДУЛЯТОРЫ SHIP1 И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2014 |
|
RU2679805C2 |
Группа изобретений относится к области органической химии и фармакологии и направлена на лечение или профилактику расстройства центральной нервной системы, поддающегося облегчению с помощью модулятора рецептора ГАМКА. Раскрывается соединение формулы I, где X выбирают из H; R1 выбирают из CH3 и CD3; каждый из R2-R9 независимо выбирают из H и D; в свободном виде или в виде фармацевтически приемлемой соли, при условии, что по меньшей мере один из R4-R9 представляет собой D. Кроме того, раскрываются фармацевтическая композиция для лечения или профилактики расстройства центральной нервной системы, поддающегося облегчению с помощью модулятора рецептора ГАМКА, содержащая терапевтически эффективное количество указанного выше соединения, способ для лечения или профилактики расстройства центральной нервной системы, поддающегося облегчению с помощью модулятора рецептора ГАМКА, включающий введение нуждающемуся в этом пациенту соединения, способ индукции седации или анестезии у нуждающегося в этом пациента, где способ включает введение нуждающемуся в этом пациенту соединения формулы I, а также применение этого соединения при производстве лекарственного средства. Группа изобретений обеспечивает эффективное лечение расстройств центральной нервной системы, ассоциированных с рецептором ГАМКА. 5 н. и 24 з.п. ф-лы, 3 табл., 16 пр.
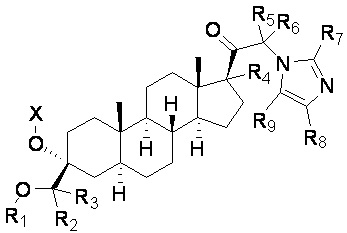
1. Соединение формулы I
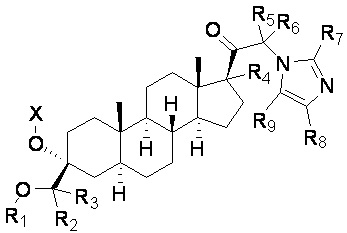
формула I,
где X выбирают из H;
R1 выбирают из CH3 и CD3;
каждый из R2-R9 независимо выбирают из H и D;
в свободном виде или в виде фармацевтически приемлемой соли,
при условии, что по меньшей мере один из R4-R9 представляет собой D.
2. Соединение по п.1, где R1 представляет собой CH3.
3. Соединение по п.1, где R1 представляет собой CD3.
4. Соединение по любому из пп.1-3, где любой один, или два, или три из R4, R5 и R6 представляют собой D.
5. Соединение по любому из пп.1-4, где R4 представляет собой D.
6. Соединение по любому из пп.1-5, где R2 и R3 представляют собой D.
7. Соединение по любому из пп.1-6, где R5 или R6 представляет собой D.
8. Соединение по любому из пп.1-6, где R5 и R6 оба представляют собой D.
9. Соединение по любому из пп.1-8, где любой один, или два, или три из R7-R9 представляют собой D.
10. Соединение по любому из пп.1-9, где R7 представляет собой D.
11. Соединение по любому из пп.1-10, где R8 и R9 представляют собой D.
12. Соединение по любому из пп.1-8, где каждый из R7-R9 представляет собой D.
13. Соединение по любому из пп.1-12, где соединение выбирают из группы, состоящей из:
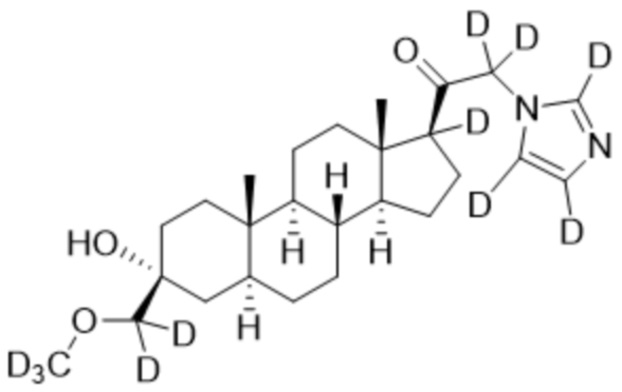 ,
, 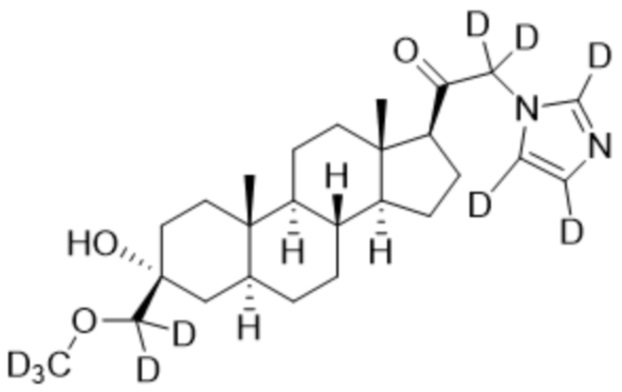 ,
,
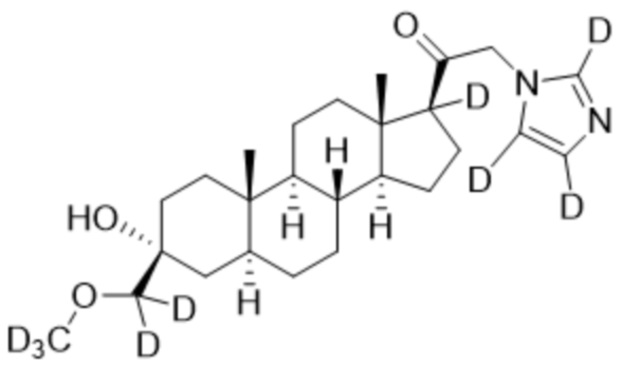 ,
, 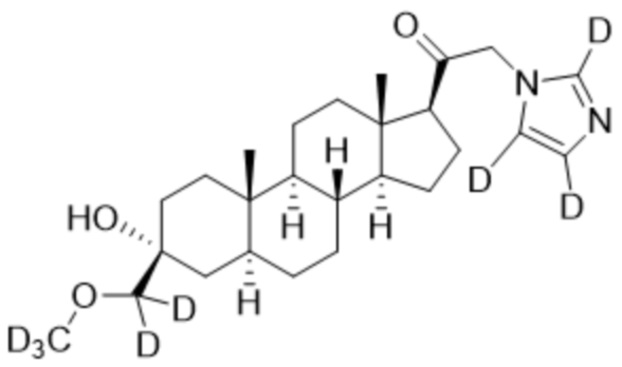 ,
,
, 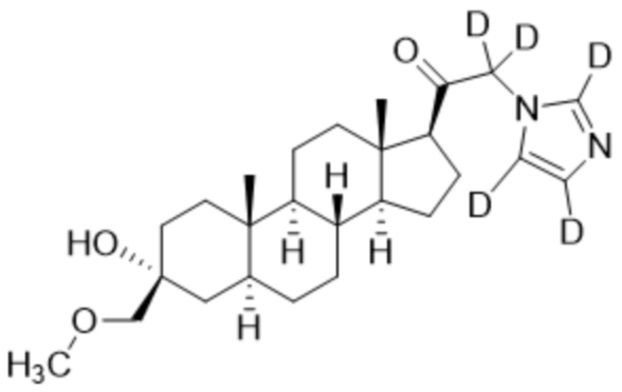 ,
,
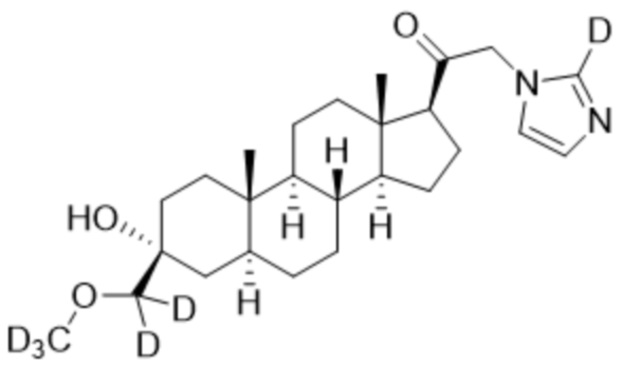 ,
, 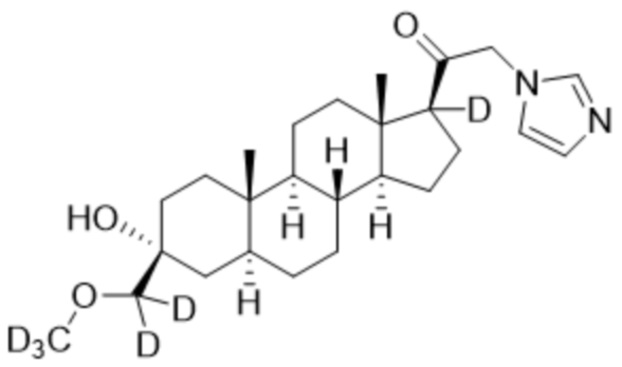 ,
,
, 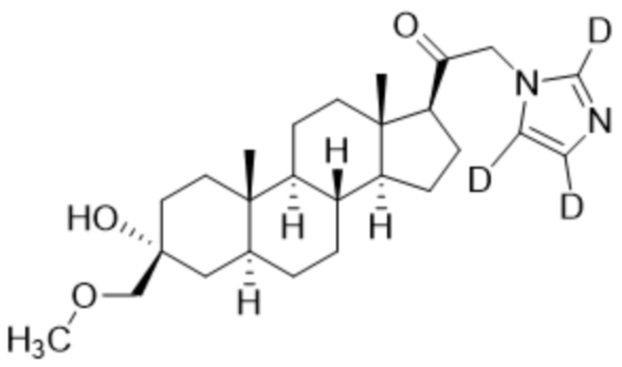 ,
,
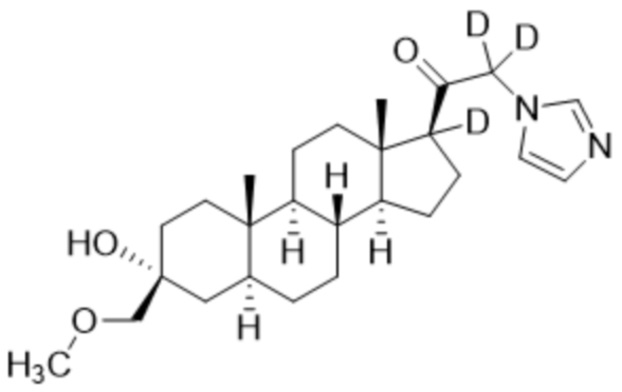 ,
, 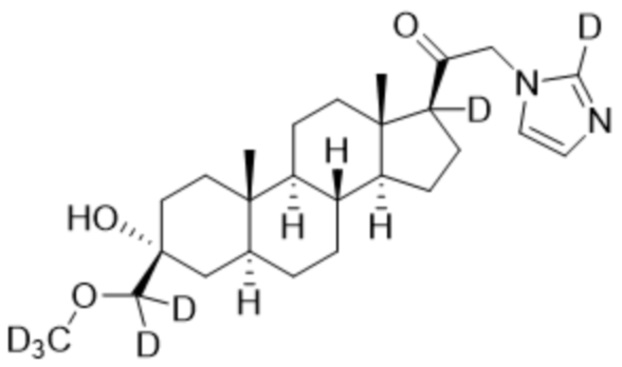 ,
,
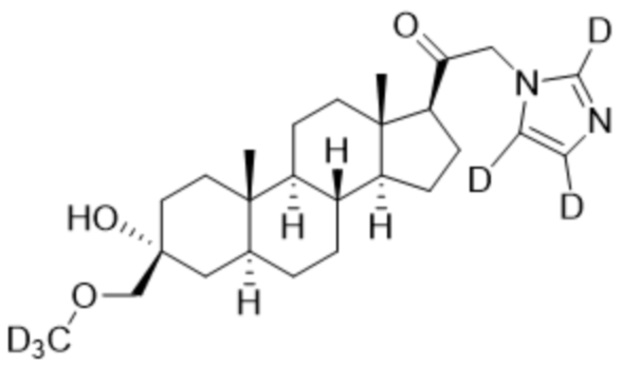 ,
, 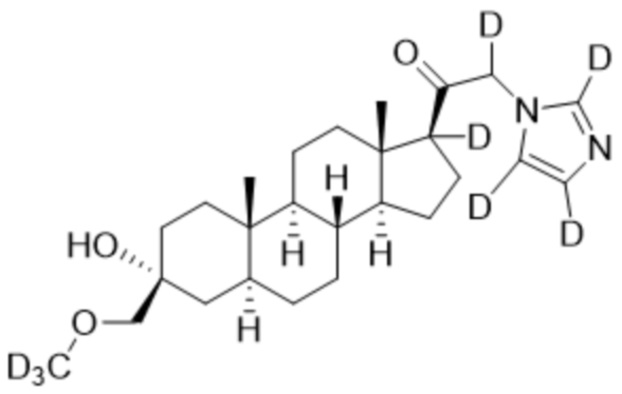 ,
,
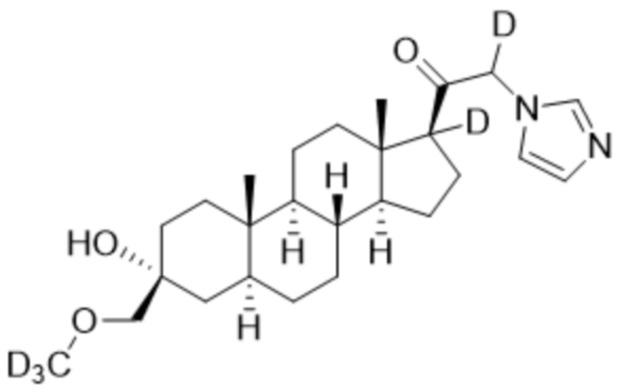 ,
, 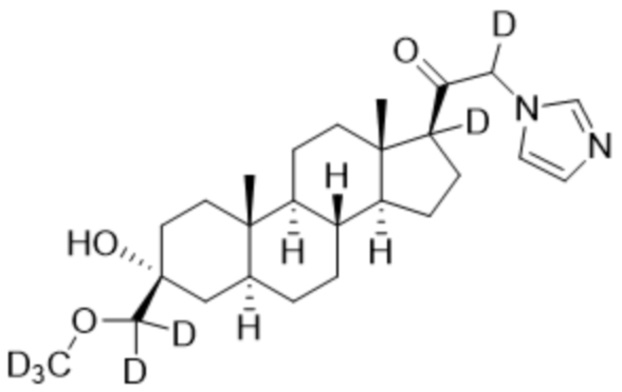 ,
,
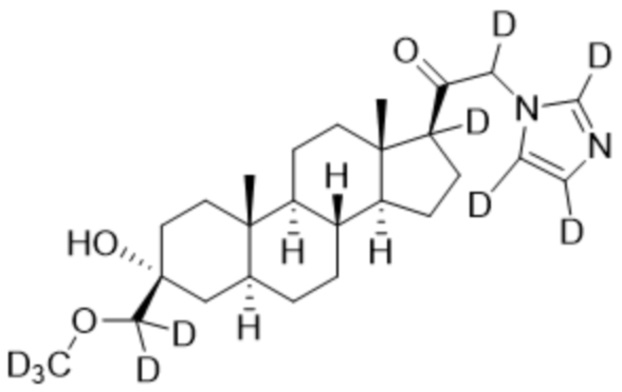 и
и 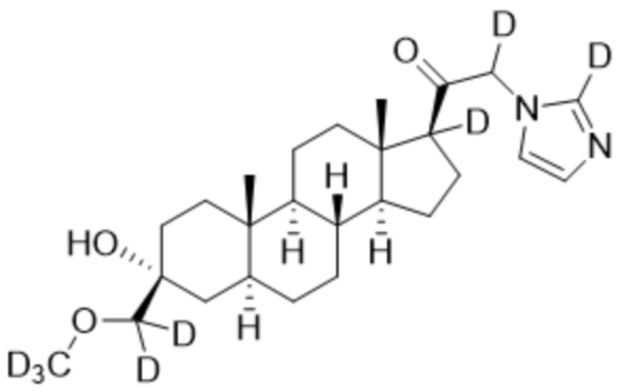 .
.
14. Соединение по любому из пп.1-12, где соединение выбирают из группы, состоящей из:
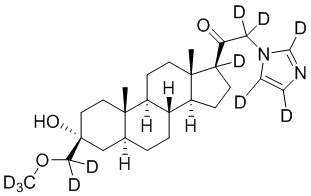 ,
,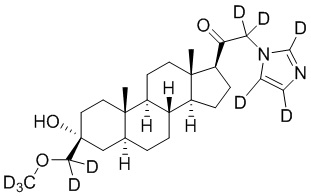 ,
,
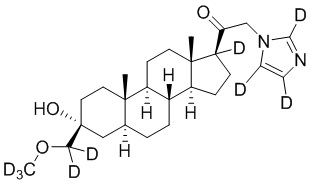 ,
,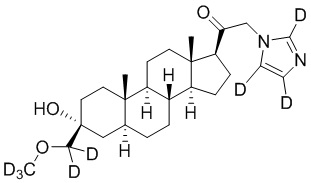 ,
,
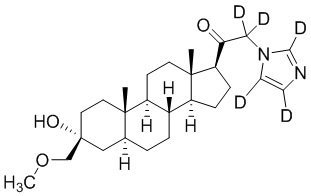 ,
,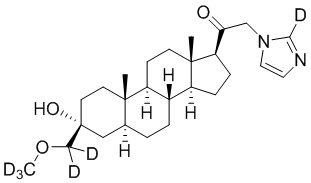 ,
,
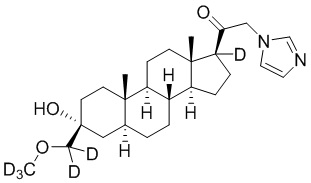 ,
,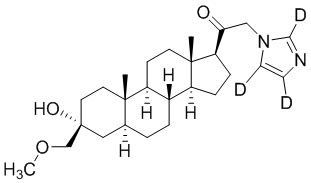 ,
,
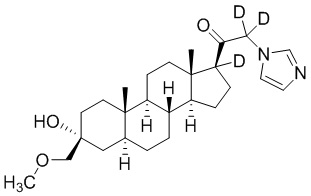 ,
,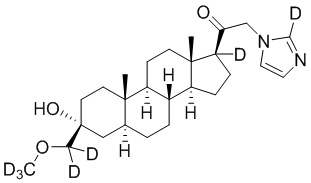 ,
,
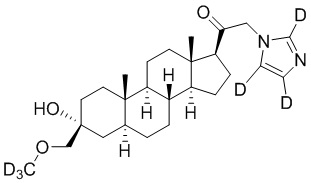 ,
,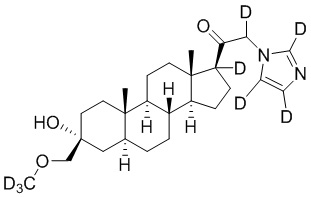 ,
,
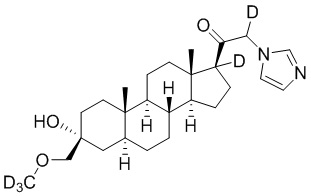 ,
,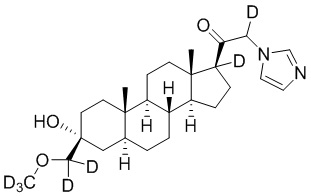 ,
,
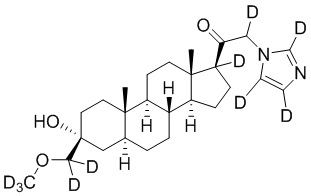 и
и 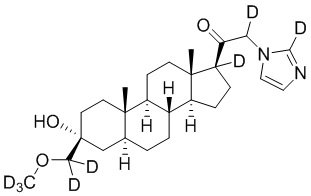 .
.
15. Соединение по любому из пп.1-12, где соединение выбирают из группы, состоящей из:
, ,
и .
16. Соединение по любому из пп.1-12, где соединение выбирают из группы, состоящей из:
и .
17. Соединение по любому из пп.1-16 в виде фармацевтически приемлемой соли.
18. Соединение по любому из пп.1-17, имеющее более 70% включения дейтерия в одном или более из указанных положений структуры.
19. Фармацевтическая композиция для лечения или профилактики расстройства центральной нервной системы, поддающегося облегчению с помощью модулятора рецептора ГАМКА, содержащая терапевтически эффективное количество соединения по любому из пп.1-18 в свободном виде или в виде фармацевтически приемлемой соли в смеси с фармацевтически приемлемым разбавителем или носителем.
20. Фармацевтическая композиция по п.19, где композиция находится в виде пероральной лекарственной формы.
21. Фармацевтическая композиция по п.20, где композиция представляет собой пероральную таблетку или капсулу.
22. Фармацевтическая композиция по п.19, где композиция приготовлена в виде инъекционного состава длительного действия.
23. Фармацевтическая композиция по п.22, где инъекционный состав длительного действия приготовлен в виде мышечной или подкожной инъекции.
24. Способ для лечения или профилактики расстройства центральной нервной системы, поддающегося облегчению с помощью модулятора рецептора ГАМКА, включающий введение нуждающемуся в этом пациенту соединения по любому из пп.1-18 в свободном виде или в виде фармацевтически приемлемой соли или фармацевтической композиции по любому из пп.19-23.
25. Способ по п.24, где указанное расстройство выбирают из группы, состоящей из расстройств сна, нарушений циркадного ритма, нарушений фазового сдвига, тревожного расстройства, посттравматического стрессового расстройства, депрессии, компульсивных расстройств, шизофрении, шизоаффективного расстройства, расстройства внимания, судорожных расстройств, расстройств, сопровождающихся агрессией, расстройств, сопровождающихся возбуждением, расстройств памяти и/или когнитивных расстройств, двигательных расстройств, аутизма и расстройств аутистического спектра, болевых расстройств, расстройств личности, сосудистых расстройств, расстройств пищевого поведения, черепно-мозговой травмы, расстройств, связанных со злоупотреблением психоактивных веществ, расстройств, вызванных употреблением психоактивных веществ, синдрома отмены психоактивных веществ, синдрома Ретта, синдрома ломкой Х-хромосомы, синдрома Ангельмана и шума в ушах и нейродегенеративных заболеваний; и любых расстройств, для эффективного лечения которых требуется седация или анестезия.
26. Способ по п.24, где указанное расстройство выбирают из группы, состоящей из бессонницы, смены часовых поясов, общей тревожности, социальной тревожности, панических расстройств, посттравматического стрессового расстройства, рефрактерных форм депрессии, большого депрессивного расстройства, биполярной депрессии, послеродовой депрессии, сезонного аффективного расстройства, дистимии, терапевтически резистентной депрессии, суицидальных мыслей, суицидального поведения, предменструального дисфорического расстройства, обсессивно-компульсивного расстройства, шизофрении, шизоаффективного расстройства, синдрома дефицита внимания (ADD), синдрома дефицита внимания с гиперактивностью (ADHD), припадочных расстройств, эпилепсии, эпилептического статуса, раннего эпилептического статуса, установленного эпилептического статуса, рефрактерного эпилептического статуса, супер-рефрактерного эпилептического статуса, неконвульсивного эпилептического статуса, генерализованного эпилептического статуса, сложного парциального эпилептического статуса, острой или хронической агрессии, острого или хронического возбуждения, болезни Альцгеймера, старости, деменции с тельцами Леви, сосудистой деменции, болезни Паркинсона, болезни Хантингтона, тремора, синдрома Аспергера, невропатической боли, острой боли, хронической боли, антисоциального расстройства личности, депрессивного расстройства личности, инсульта, ишемии, сосудистых мальформаций, булимии, анорексии, переедания, кахексии, бокового амиотрофического склероза, комы, дискинезии и дистонии.
27. Способ по п.24, где указанное расстройство выбирают из группы, состоящей из бессонницы, общей тревожности, социальной тревожности, панических расстройств, рефрактерных форм депрессии, большого депрессивного расстройства, биполярной депрессии, послеродовой депрессии, терапевтически резистентной депрессии, припадочных расстройств, эпилепсии, эпилептического статуса, раннего эпилептического статуса, установленного эпилептического статуса, рефрактерного эпилептического статуса, супер-рефрактерного эпилептического статуса, неконвульсивного эпилептического статуса, генерализованного эпилептического статуса и сложного парциального эпилептического статуса.
28. Способ индукции седации или анестезии у нуждающегося в этом пациента, где способ включает введение нуждающемуся в этом пациенту соединения по любому из пп.1-18 в свободном виде или в виде фармацевтически приемлемой соли или фармацевтической композиции по любому из пп.19-23.
29. Применение соединения по любому из пп.1-18 в свободном виде или в виде фармацевтически приемлемой соли или фармацевтической композиции по любому из пп.19-23, в свободном виде или в виде фармацевтически приемлемой соли, при производстве лекарственного средства для лечения или профилактики расстройства центральной нервной системы или для индукции седации или анестезии.
| US 20170240589 A1, 24.08.2017 | |||
| US 20120276196 A1, 01.11.2012 | |||
| US 20040034002 A1, 19.02.2004 | |||
| WO 2016134301 A3, 25.08.2016 | |||
| RU 2015149008 A, 22.05.2017. |

