Область техники
Настоящее изобретение относится к соединениям бензимидазола, которые полезны для ингибирования циклинзависимой киназы. Более конкретно, согласно изобретению предложены соединения и содержащие их фармацевтические композиции, которые полезны в качестве ингибиторов CDK4/6, и способы лечения заболеваний, опосредованных CDK4/6, таких как рак.
Предшествующий уровень техники
Циклинзависимые киназы (CDK) опосредуют прохождение клеточного цикла, регулируя переход из G1 в S-фазу и из G2 в М-фазу. Активность CDK строго контролируется на протяжении всего клеточного цикла посттрансляционными модификациями, а также экспрессией циклинов и ингибиторов CDK. Существует четыре пролиферативных CDK: CDK1, которая регулирует преимущественно переход из G2 в М-фазу, и CDK2/4/6, которые регулируют переход из G1 в S-фазу.
Прохождение клеточного цикла является строго регулируемым процессом. В отсутствие соответствующих сигналов роста семейство карманных белков, включающее белок ретинобластомы (pRb), препятствует вхождению клеток в фазу репликации ДНК (S-фазу). Цикл репликации начинается, когда митогены запускают пути передачи сигналов, вызывающих увеличение клеточных уровней D-циклинов. D-циклины, в свою очередь, активируют циклинзависимые киназы 4/6 (CDK4/6), которые фосфорилируют и инактивируют pRb.
Неконтролируемая пролиферация клеток является одним из признаков рака, и инактивация pRb является ключевым событием, которое делает возможным прохождение клеток опухоли по неконтролируемому клеточному циклу. Хотя некоторые опухоли удаляют сам ген pRb, большинство сохраняет функциональный pRb и взамен активирует активность киназ CDK4/6. Абляция активности киназ CDK4/6 приводила к полному ингибированию роста опухоли при многих типах рака, таких как HR+ (рецептор гормона - положительный) рак молочной железы, мантийноклеточная лимфома, глиобластома и плоскоклеточный рак легкого. Кроме того, было показано, что нормальные клетки фибробласты преодолевают отсутствие CDK4/6 за счет компенсации киназой CDK1, чье отсутствие не допускается. В совокупности, это доказательство свидетельствует о том, что селективный ингибитор CDK4/6 может иметь более широкое терапевтическое окно, чем ингибиторы пан-CDK.
Обнаружено, что помимо прямых антинеопластических эффектов, ингибиторы CDK4/6 лечат воспалительные заболевания, костные заболевания, метаболические заболевания, неврологические и нейродегенеративные заболевания, сердечно-сосудистые заболевания, аллергии и астму, болезнь Альцгеймера и гормональные заболевания. Соответственно, в медицинской химии были предприняты значительные усилия, направленные на обнаружение ингибиторов CDK4/6, которые являются эффективными в качестве терапевтических агентов.
CDK1 является ключевым фактором митотической прогрессии, и это единственная CDK, которая может инициировать начало митоза. Эксперименты на мышах с нокаутированным геном показали, что CDK1 требуется для пролиферации клеток млекопитающих[1]. Поскольку CDK1 является ключевой для пролиферации клеток, токсичность, вызываемая ингибированием CDK1, будет ограничивать возможность достижения терапевтических уровней, поэтому необходимо сохранять селективность CDK1 относительно CDK4/6 лекарственной мишени.
CDK2 структурно и функционально родственна CDK1; она имеет значительно более широкий субстратный профиль, чем CDK4 и CDK6, и фосфорилирует большое количество белков, вовлеченных в прохождение клеточного цикла (например, p27KIP1 и RB), репликацию ДНК (например, репликацию факторов А и С), синтез гистонов (например, NPAT), дупликацию центросом (например, нуклеофосмин (NPM)), среди прочих процессов. В отличие от CDK4 и CDK6, CDK2 регулируется не белками INK4, а ингибиторами CDK семейства CDK-взаимодействующих белков/киназа-ингибирующих белков (CIP/KIP), которые связываются с CDK2-циклиновыми комплексами и делают их неактивными [1]. При создании ингибитора CDK4/6 в качестве лекарственного средства против рака лучше сохранять селективность относительно CDK2.
Помимо CDK, которые напрямую стимулируют прохождение клеточного цикла (например, CDK4, CDK6, CDK2 и CDK1), было идентифицировано дополнительное семейство CDK, которые регулируют транскрипцию и которые включают CDK7, CDK8 и CDK9. CDK7 играет общую роль в фосфорилировании карбоксиконцевого домена РНК-полимеразы II, что способствует инициации транскрипции, и CDK9 также фосфорилирует РНК-полимеразу II, стимулируя тем самым элонгацию транскрипции [1]. Ингибиторы CDK первого поколения представляли собой ингибиторы пан-CDK и не имели успеха из-за не поддающейся контролю токсичности. Например, флавопиридол до сих пор является наиболее интенсивно исследуемым ингибитором CDK. Хотя флавопиридол может индуцировать остановку клеточного цикла в фазах G1 и G2, в некоторых обстоятельствах он также индуцирует цитотоксический ответ, вероятно в результате ингибирования CDK7 и CDK9, что приводит к супрессии транскрипции[2]. Поэтому необходимо избегать CDK7/9 при создании лекарственных средств, нацеленных на CDK4/6.
До настоящего времени ряд ингибиторов CDK проходят предклиническую и клиническую оценку. Принимая во внимание сведения, изложенные выше, многие исследовательские группы встали на путь открытия селективного ингибитора CDK4/6, причем хорошо задокументированы палбоциклиб (Palbociclib) (PD-0332991), рибоциклиб (Ribociclib) (LEE-011) и абемациклиб (Abemaciclib) (LY2835219). Тем не менее, остается потребность в создании более сильнодействующих, селективных и безопасных ингибиторов CDK4/6, которые можно применять в лечении клеточных пролиферативных заболеваний, таких как рак.
Источники информации
[1]. Uzma Asghar, Agnieszka K. Witkiewicz, Nicholas С. Turner, Et al. Nat Rev Drug Discov. 2015, 14(2): 130-146.
[2]. Prithviraj Bose, Gary L Simmons, Steven Grant. Expert Opin Investig Drugs. 2013, 22(6): 723-738.
Краткое изложение сущности изобретения
Настоящее изобретение относится к соединениям бензимидазола, которые полезны в качестве ингибиторов CDK4/6 и для лечения заболеваний, опосредованных CDK4/6. Соединения по изобретению имеют общую структурную формулу I. Соединение формулы I или его стереоизомер, таутомер, полиморф, сольват, фармацевтически приемлемая соль или пролекарство

где
кольцо А представляет собой арил или гетероарил;
Z выбран из группы, состоящей из СН2, NH, О и S;
R1 независимо выбран из группы, состоящей из водорода, галогена, CN, NO2, ОН, NH2, С1-8алкила, С1-8алкокси, C3-8циклоалкила, арила, гетероарила, гетероциклила, группы гетероциклил-(СН2)m-, арил-C1-6алкил-, гетероарил-C1-6алкил-, -NR12R13, -NR12-C1-6алкилен-NR12R13 и гетероциклил-С(О)-, где каждый из С1-8алкила, С1-8алкокси, C3-8циклоалкила, арила, гетероарила, гетероциклила, гетероциклил-(СН2)m-, арил-C1-6алкил-, гетероарил-C1-6алкил- или гетероциклил-С(О)- не замещен или замещен по меньшей мере одним заместителем, выбранным из галогена, гидроксила, С1-8алкила, C3-8циклоалкила, гетероциклила, -NR12R13 или -(СН2)t-ОН;
каждый из R2 и R3 независимо выбран из Н, ОН, CN, NO2, NH2, галогена, С1-8алкила, С1-8алкокси, C3-8циклоалкила, арила, гетероарила, гетероциклила, где каждый из С1-8алкила, С1-8алкокси, C3-8циклоалкила, арила, гетероарила, гетероциклила не замещен или замещен по меньшей мере одним заместителем, выбранным из галогена, гидроксила, С1-8алкила, C3-8циклоалкила или гетероциклила;
каждый из R12 и R13 независимо выбран из Н, С1-8алкила, арила, гетероарила, гетероциклила, C3-8циклоалкила, где каждый из С1-8алкила, арила, гетероарила, гетероциклила или C3-8циклоалкила не замещен или замещен по меньшей мере одним заместителем, выбранным из галогена, гидроксила, С1-8алкила, C3-8циклоалкила или гетероциклила;
m равно 0, 1, 2, 3 или 4;
n равно 0, 1, 2, 3 или 4;
t равно 0, 1, 2, 3 или 4.
В некоторых воплощениях формулы I Z представляет собой СН2.
В некоторых воплощениях формулы I Z представляет собой О.
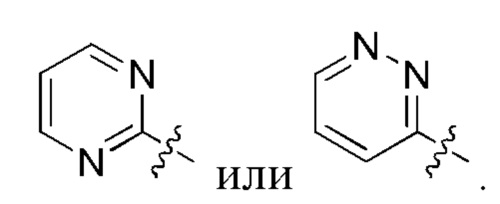
В некоторых воплощениях формулы I кольцо А представляет собой 6-членный гетероарил, содержащий один или два гетероатома N, например пиридил, пиримидинил, пиридазинил и т.п.
В других воплощениях формулы I кольцо А представляет собой 
В некоторых воплощениях формулы I R1 представляет собой гетероциклил-(СН2)m- или гетероциклил-(СН2)m-, замещенный С1-8алкилом, NR12R13, 4-6-членным гетероциклилом, C3-6циклоалкилом или -(СН2)t-ОН.
В других воплощениях формулы I R1 представляет собой 5-6-членный гетероциклил-СН2- или 5-6-членный гетероциклил-СН2-, замещенный C1-3алкилом, -N(CH3)2, -N(CH2CH2OH)CH3, 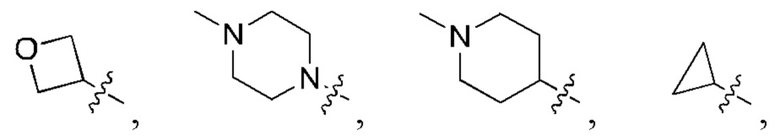 -СН2ОН, -СН2СН2ОН или -ОН.
-СН2ОН, -СН2СН2ОН или -ОН.
В других воплощениях формулы I R1 представляет собой 6-членный гетероциклил-СН2- или 6-членный гетероциклил-СН2-, замещенный метилом или этилом.
В некоторых воплощениях формулы I R1 представляет собой гетероциклил или гетероциклил, замещенный С1-8алкилом, NR12R13, 4-6-гетероциклилом, C3-6циклоалкилом или -(СН2)t-ОН.
В других воплощениях формулы I R1 представляет собой 5-6-членный гетероциклил или 5-6-членный гетероциклил, замещенный C1-3алкилом, -N(СН3)2, -N(CH2CH2OH)CH3,  -СН2ОН, -СН2СН2ОН или ОН.
-СН2ОН, -СН2СН2ОН или ОН.
В других воплощениях формулы I R1 представляет собой 6-членный гетероциклил или 6-членный гетероциклил, замещенный метилом или этилом.
В некоторых воплощениях формулы I R1 представляет собой 6-членный гетероциклил-С(О)- или 6-членный гетероциклил-С(О)-, замещенный C1-3алкилом.
В других воплощениях формулы I R1 представляет собой 6-членный гетероциклил-С(О)-, замещенный метилом.
В некоторых воплощениях формулы I гетероциклил содержит один или два гетероатома N или О в качестве кольцевых атомов.
В некоторых воплощениях формулы I гетероциклил содержит один или два гетероатома N в качестве кольцевых атомов.
В некоторых воплощениях формулы I R1 представляет собой -NR12-C1-3алкилен-NR12R13.
В некоторых воплощениях формулы I каждый из R12 и R13 независимо представляет собой Н, -(СН2)t-ОН или C1-3алкил.
Предпочтительно, каждый из R12 и R13 независимо представляет собой ОН, СН2СН2ОН, метил или этил.
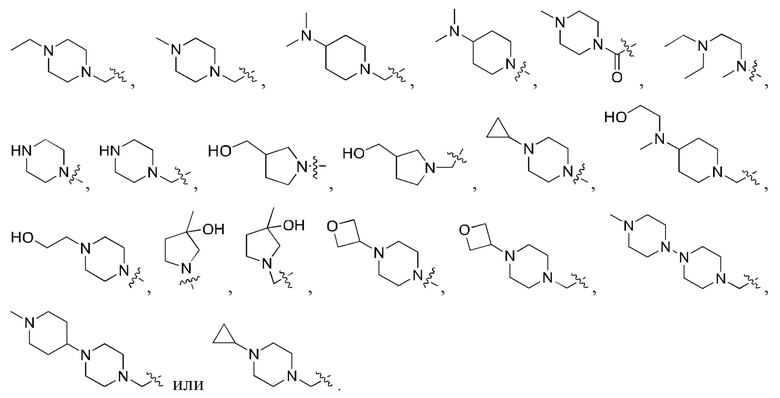
В некоторых воплощениях формулы I R1 представляет собой 
В некоторых воплощениях формулы I m равно 1.
В некоторых воплощениях формулы I, n равно 1.
В некоторых воплощениях формулы I, t равно 0, 1 или 2.
В некоторых воплощениях формулы I каждый из R2 и R3 независимо представляет собой Н, ОН, галоген, C1-6алкил, C1-6алкил, замещенный галогеном, C1-6алкокси, C1-6алкокси, замещенный галогеном.
В других воплощениях формулы I каждый из R2 и R3 независимо представляет собой Н, ОН, F, Cl, СН3, СН2СН3, CF3, -ОСН3 или -OCF3.
В других воплощениях формулы I R2 и R3 оба представляют собой F.
Согласно настоящему изобретению предложены также некоторые предпочтительные технические решения, относящиеся к соединению формулы I, и соединение представляет собой:
1) 4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-а]пиридин-8-ил)-N-(5-(4-метилпиперазин-1-ил)пиридин-2-ил)пиримидин-2-амин;
2) N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-а]пиридин-8-ил)пиримидин-2-амин;
3) 5-фтор-4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-а]пиридин-8-ил)-N-(5-(4-метилпиперазин-1-ил)пиридин-2-ил)пиримидин-2-амин;
4) 5-фтор-4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-а]пиридин-8-ил)-N-(6-((4-метилпиперазин-1-ил)метил)пиридин-3-ил)пиримидин-2-амин;
5) 5-фтор-4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-а]пиридин-8-ил)-N-(5-((4-метилпиперазин-1-ил)метил)пиримидин-2-ил)пиримидин-2-амин;
6) N-(5-((4-(диметиламино)пиперидин-1-ил)метил)пиримидин-2-ил)-5-фтор-4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-а]пиридин-8-ил)пиримидин-2-амин;
7) N-(5-((4-(диметиламино)пиперидин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-а]пиридин-8-ил)пиримидин-2-амин;
8) N-(5-(4-(диметиламино)пиперидин-1-ил)пиридин-2-ил)-5-фтор-4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-а]пиридин-8-ил)пиримидин-2-амин;
9) (2-((5-фтор-4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-а]пиридин-8-ил)пиримидин-2-ил)амино)пиримидин-5-ил)(4-метилпиперазин-1-ил)метанон;
10) (6-((5-фтор-4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-а]пиридин-8-ил)пиримидин-2-ил)амино)пиридин-3-ил)(4-метилпиперазин-1-ил)метанон;
11) N5-(2-(диэтиламино)этил)-N2-(5-фтор-4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-а]пиридин-8-ил)пиримидин-2-ил)-N5-метилпиридин-2,5-диамин;
12) N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-4-(6-фтор-1-метил-1,2,3,4,4а,5-гексагидробензо[4,5]имидазо[1,2-а]пиридин-8-ил)-5-(трифторметил)пиримидин-2-амин;
13) N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-4-(6-фтор-1-метил-1,2,3,4,4а,5-гексагидробензо[4,5]имидазо[1,2-а]пиридин-8-ил)-5-метилпиримидин-2-амин;
14) 5-хлор-N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-а]пиридин-8-ил)пиримидин-2-амин;
15) 5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)-N-(5-(4-метилпиперазин-1-ил)пиридин-2-ил)пиримидин-2-амин;
16) N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)пиримидин-2-амин;
17) N-(5-(4-(диметиламино)пиперидин-1-ил)пиридин-2-ил)-5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)пиримидин-2-амин;
18) N-(5-((4-(диметиламино)пиперидин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)пиримидин-2-амин;
19) 5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)-N-(5-(пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амин;
20) 5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)-N-(5-(пиперазин-1-илметил)пиридин-2-ил)пиримидин-2-амин;
21) N-(5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)пиримидин-2-ил)-6-(4-метилпиперазин-1-ил)пиридазин-3-амин;
22) 6-((4-этилпиперазин-1-ил)метил)-N-(5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)пиримидин-2-ил)пиридазин-3-амин;
23) (1-(6-((5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)пиримидин-2-ил)амино)пиридин-3-ил)пирролидин-3-ил)метанол;
24) (1-((6-((5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)пиримидин-2-ил)амино)пиридин-3-ил)метил)пирролидин-3-ил)метанол;
25) N-(5-(4-циклопропилпиперазин-1-ил)пиридин-2-ил)-5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)пиримидин-2-амин;
26) N-(5-((4-циклопропилпиперазин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)пиримидин-2-амин;
27) 2-((1-((6-((5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)пиримидин-2-ил)амино)пиридин-3-ил)метил)пиперидин-4-ил)(метил)амино)этан-1-ол;
28) 1-(6-((5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)пиримидин-2-ил)амино)пиридин-3-ил)-3-метилпирролидин-3-ол;
29) 1-((6-((5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)пиримидин-2-ил)амино)пиридин-3-ил)метил)-3-метилпирролидин-3-ол;
30) 5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)-N-(5-((4-(оксетан-3-ил)пиперазин-1-ил)метил)пиридин-2-ил)пиримидин-2-амин;
31) 5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)-N-(5-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амин;
32) N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(9-фтор-4-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-а]пиразин-7-ил)пиримидин-2-амин;
33) 5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)-N-(5-((4'-метил-[1,1'-бипиперазин]-4-ил)метил)пиридин-2-ил)пиримидин-2-амин;
34) 5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1Н-бензо[4,5]имидазо[2,1-с][1,4]оксазин-7-ил)-N-(5-((4-(1-метилпиперидин-4-ил)пиперазин-1-ил)метил)пиридин-2-ил)пиримидин-2-амин.
Неожиданно, в высокой степени очищенный (-)-энантиомер соединения формулы I имеет преимущество над (+)-энантиомером в биологической активности. Например, оптически чистый (-)-энантиомер соединения 2 (N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-а]пиридин-8-ил)пиримидин-2-амин) является более сильнодействующим, чем его (+)-энантиомер.
Если не указано иное, "(-)" в настоящем изобретении означает, что оптическое вращение имеет отрицательное значение; и "(+)" означает, что оптическое вращение имеет положительное значение. Соединение по настоящему изобретению, описанное в данном документе, может представлять собой (-)-изомер соединения и/или (+)-изомер соединения.
Согласно настоящему изобретению предложены также фармацевтические композиции, содержащие терапевтически эффективное количество соединения формулы I или его терапевтически приемлемой соли и фармацевтически приемлемый эксципиент.
В некоторых воплощениях массовое отношение указанного соединения к указанному эксципиенту находится в диапазоне от примерно 0,001 до примерно 10.
Согласно настоящему изобретению дополнительно предложено соединение по настоящему изобретению, его фармацевтически приемлемая соль или содержащая его фармацевтическая композиция, упомянутые выше, для приготовления лекарственного средства.
В некоторых воплощениях лекарственное средство применяют для лечения рака, такого как рак ободочной кишки, ректальный рак, мантийноклеточная лимфома, множественная миелома, рак молочной железы, рак предстательной железы, глиобластома, плоскоклеточный рак пищевода, липосаркома, Т-клеточная лимфома, меланома, рак поджелудочной железы, рак головного мозга или рак легкого.
В некоторых воплощениях лекарственное средство применяют в качестве ингибитора CDK, предпочтительно CDK4 и/или CDK6.
Согласно настоящему изобретению предложен способ лечения рака у субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения по настоящему изобретению, его фармацевтически приемлемой соли или вышеупомянутой фармацевтической композиции. В частности, рак выбран из группы, состоящей из рака ободочной кишки, ректального рака, мантийноклеточной лимфомы, множественной миеломы, рака молочной железы, рака предстательной железы, глиобластомы, плоскоклеточного рака пищевода, липосаркомы, Т-клеточной лимфомы, меланомы, рака поджелудочной железы, рака головного мозга или рака легкого.
Согласно данному изобретению предложен также способ лечения заболевания, опосредованного CDK, например CDK4 и/или CDK6, у субъекта, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения по настоящему изобретению, его фармацевтически приемлемой соли или вышеупомянутой фармацевтической композиции.
Общие химические термины, использованные в приведенной выше формуле, имеют их обычные значения. Например, в данном документе "галоген", если не указано иное, означает фтор, хлор, бром или йод. Предпочтительные галогеновые группы включают F, О и Br.
В данном документе, если не указано иное, алкил включает насыщенные одновалентные углеводородные радикалы, имеющие прямые или разветвленные группировки. Например, алкильные радикалы включают метил, этил, пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, 3-(2-метил)бутил, 2-пентил, 2-метилбутил, неопентил, н-гексил, 2-гексил, 2-метилпентил и т.п. Аналогично, С1-8 в С1-8алкиле идентифицирует группу, имеющую 1, 2, 3, 4, 5, 6, 7 или 8 атомов углерода в линейном или разветвленном размещении.
Алкенильные и алкинильные группы включают алкены и алкины с прямой или разветвленной цепью. Аналогично, "C2-8алкенил" и "C2-8алкинил" означают алкенильные или алкинильные радикалы, имеющие 2, 3, 4, 5, 6, 7 или 8 атомов углерода в линейном или разветвленном размещении.
Алкокси представляют собой кислородсодержащие простые эфиры, образованные из ранее описанных алкильных групп с прямой или разветвленной цепью, то есть -О-алкил.
В данном документе термины, указанные в единственном числе, "по меньшей мере один" и "один или более" использованы взаимозаменяемым образом. Так, например, композиция, содержащая фармацевтически приемлемый эксципиент, может быть истолкована как означающая, что композиция содержит "один или более" фармацевтически приемлемых эксципиентов.
Термин "арил" в данном документе, если не указано иное, относится к незамещенной или замещенной моно- или полициклической кольцевой системе, содержащей кольцевые атомы углерода. Предпочтительными арилами являются моноциклические или бициклические 6-10-членные ароматические кольцевые системы Фенил и нафтил являются предпочтительными арилами. Наиболее предпочтительным арилом является фенил.
Термин "гетероциклил" в данном документе, если не указано иное, означает незамещенную или замещенную стабильную трех-восьми-членную моноциклическую насыщенную кольцевую систему, которая состоит из атомов углерода и одного-трех гетероатомов, выбранных из N, О или S, причем гетероатомы азота или серы возможно могут быть окисленными, а гетероатом азота возможно может быть кватернизированным. Гетероциклильная группа может быть присоединена по любому гетероатому или атому углерода, который обеспечивает создание стабильной структуры. Примеры таких гетероциклильных групп включают, без ограничения, азетидинил, пирролидинил, пиперидинил, пиперазинил, оксопиперазинил, оксопиперидинил, тетрагидрофуранил, диоксоланил, тетрагидроимидазолил, тетрагидротиазолил, тетрагидрооксазолил, тетрагидропиранил, морфолинил, тиоморфолинил, тиаморфолинилсульфоксид, тиаморфолинилсульфон и тетрагидрооксадиазолил.
Термин "гетероарил" в данном документе, если не указано иное, означает незамещенную или замещенную стабильную пяти-шести-членную моноциклическую ароматическую кольцевую систему, или незамещенную или замещенную девяти-десяти-членную бензо-конденсированную гетероароматическую кольцевую систему, или бициклическую гетероароматическую кольцевую систему, которая состоит из атомов углерода и одного-четырех гетероатомов, выбранных из N, О или S, причем гетероатомы азота или серы возможно могут быть окисленными, а гетероатом азота возможно может быть кватернизированным. Гетероарильная группа может быть присоединена по любому гетероатому или атому углерода, который обеспечивает создание стабильной структуры. Примеры гетероарильных групп включают, без ограничения тиенил, фуранил, имидазолил, изоксазолил, оксазолил, пиразолил, пирролил, тиазолил, тиадиазолил, триазолил, пиридил, пиридазинил, индолил, азаиндолил, индазолил, бензимидазолил, бензофуранил, бензотиенил, бензизоксазолил, бензоксазолил, бензопиразолил, бензотиазолил, бензотиадиазолил, бензотриазолил, аденинил, хинолинил или изохинолинил.
Термин "циклоалкил" относится к циклической насыщенной алкильной цепи, имеющей от 3 до 12 атомов углерода, например циклопропил, циклобутил, циклопентил или циклогексил.
Термин "замещенный" относится к группе, в которой один или более атомов водорода, каждый независимо, замещены одинаковыми или разными заместителями. Типичные заместители включают, без ограничения, галоген (F, Cl, Br или I), С1-8алкил, C3-12циклоалкил, -OR1, SR1, =O, =S, -С(O)R1, -C(S)R1, =NR1, -C(O)OR1, -C(S)OR1, -NR1R2, -С(O)NR1R2, циано, нитро, -S(O)2R1, -OS(O2)OR1, -OS(O)2R1, -OP(O)(OR1)(OR2), где R1 и R2 независимо выбраны из -H, C1-6алкила, C1-6галогеналкила. В некоторых воплощениях заместитель(и) независимо выбран(ы) из группы, состоящей из -F, -Cl, -Br, -I, -ОН, трифторметокси, этокси, пропилокси, изопропилокси, н-бутилокси, изобутилокси, трет-бутилокси, -SCH3, -SC2H5, формальдегидной группы, -С(ОСН3), циано, нитро, CF3, -OCF3, амино, диметиламино, метила, тио, сульфонила и ацетила.
Примеры замещенных алкильных групп включают, без ограничения, 2-аминоэтил, 2-гидроксиэтил, пентахлорэтил, трифторметил, метоксиметил, пентафторэтил и пиперазинилметил.
Примеры замещенных алкоксигрупп включают, без ограничения, аминометокси, трифторметокси, 2-диэтиламиноэтокси, 2-этоксикарбонилэтокси, 3-гидроксипропокси.
Термин "фармацевтически приемлемые соли" относится к солям, получаемым из фармацевтически приемлемых нетоксичных оснований или кислот. В тех случаях, когда соединение по настоящему изобретению является кислотным, его соответствующая соль легко может быть получена из фармацевтически приемлемых нетоксичных оснований, включая неорганические основания и органические основания. Соли, получаемые из неорганических оснований, включают соли алюминия, аммония, кальция, меди (двухвалентной и одновалентной), трехвалентного железа, двухвалентного железа, лития, магния, марганца (трехвалентного и двухвалентного), калия, натрия, цинка и т.п. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, получаемые из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, а также циклических аминов и замещенных аминов, таких как природные и синтезированные замещенные амины. Другие фармацевтически приемлемые органические нетоксичные основания, из которых могут быть образованы соли, включают ионообменные смолы, такие как, например, аргинин, бетаин, кофеин, холин, N',N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминные смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п.
В тех случаях, когда соединение по настоящему изобретению является основным, его соответствующая соль может быть легко получена из фармацевтически приемлемых нетоксичных кислот, включающих неорганические и органические кислоты. Такие кислоты включают, например, уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, муравьиную, фумаровую, глюконовую, глутаминовую, бромистоводородную, соляную, изетионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, пара-толуолсульфоновую кислоту и т.п. Предпочтительными являются лимонная, бромистоводородная, муравьиная, соляная, малеиновая, фосфорная, серная и винная кислоты; особенно предпочтительными являются муравьиная кислота и соляная кислота. Поскольку соединения формулы I предназначены для фармацевтического применения, их предпочтительно предоставляют в по существу чистой форме, например с чистотой по меньшей мере 60%, лучше с чистотой по меньшей мере 75%, особенно с чистотой по меньшей мере 98% (% являются массовыми).
Соединения по настоящему изобретению могут быть представлены также в форме фармацевтически приемлемых солей. Для применения в медицине соли соединений по данному изобретению относятся к нетоксичным "фармацевтически приемлемым солям". Формы фармацевтически приемлемых солей включают фармацевтически приемлемые кислотные/анионные или основные/катионные соли. Фармацевтически приемлемая кислотная/анионная соль обычно принимает форму, в которой основной азот протонирован неорганической или органической кислотой. Репрезентативные органические или неорганические кислоты включают соляную, бромистоводородную, йодистоводородную, перхлорную, серную, азотную, фосфорную, уксусную, пропионовую, гликолевую, молочную, янтарную, малеиновую, фумаровую, яблочную, винную, лимонную, бензойную, миндальную, метансульфоновую, гидроксиэтансульфоновую, бензолсульфоновую, оксалиновую, памовую, 2-нафталинсульфоновую, пара-толуолсульфоновую, циклогексансульфаминовую, салициловую, сахариновую или трифторуксусную кислоту. Фармацевтически приемлемые основные/катионные соли включают, без ограничения, соли алюминия, кальция, хлорпрокаина, холина, диэтаноламина, этилендиамина, лития, магния, калия, натрия и цинка.
Настоящее изобретение охватывает в своем объеме пролекарства соединений по данному изобретению. Как правило, такие пролекарства представляют собой функциональные производные соединений, которые легко превращаются in vivo в требуемое соединение. Например, любая фармацевтически приемлемая соль, любой сложный эфир, любая соль сложного эфира или другое производное соединения по данному изобретению, которое при введении реципиенту способно обеспечивать образование, либо напрямую, либо опосредованно, соединения по настоящему изобретению или фармацевтически активного метаболита или остатков. Особенно предпочтительными производными или пролекарствами являются те соединения, которые могут увеличивать биодоступность соединения по настоящему изобретению при введении пациенту (например те соединения, которые могут облегчать всасывание в кровь перорально введенного соединения), или которые могут облегчать доставку родительского соединения в биологический организм, или которые доставляются к месту действия (например, головной мозг или лимфатическую систему). Таким образом, в способах лечения по настоящему изобретению термин "введение" охватывает лечение различных расстройств, описанных применительно к соединению, конкретно раскрытому, или к соединению, которое может не быть конкретно раскрыто, но которое превращается в конкретно указанное соединение in vivo после введения субъекту. Стандартные методики выбора и получения подходящих пролекарственных производных описаны, например, в "Design of Prodrugs", ed. H. Bundgaard, Elsevier, 1985.
Предполагается, что определение любого заместителя или любой переменной в конкретном положении в молекуле не зависит от его(ее) определений где-либо еще в этой молекуле. Понятно, что специалист в данной области может выбрать заместители и картины замещения на соединениях по данному изобретению, обеспечивающие получение соединений, которые являются химически стабильными и которые легко могут быть синтезированы способами, известными в данной области, а также способами, изложенными в данном документе.
Настоящее изобретение охватывает описанные в данном документе соединения, которые могут содержать один или более асимметрических центров и, следовательно, могут давать в результате диастереомеры и оптические энантиомеры. Настоящее изобретение охватывает все такие возможные диастереомеры, а также их рацемические смеси, их по существу чистые разделенные энантиомеры, все возможные геометрические энантиомеры и их фармацевтически приемлемые соли.
Теперь обнаружено, что оптически чистый (-)-энантиомер соединения по настоящему изобретению является более сильнодействующим ингибитором CDK4/6. Настоящее изобретение охватывает способы лечения заболевания, опосредованного CDK4/6, у субъекта, включающие введение указанному субъекту (-)-энантиомера, или его фармацевтически приемлемой соли, по существу не содержащего его (+)-энантиомера, в количестве, достаточном для ослабления заболевания, но недостаточном для того, чтобы вызывать неблагоприятные эффекты.
Термин "по существу не содержащий его (+)-энантиомера" в данном документе означает, что композиция содержит большую часть или более высокий процент (-)-энантиомера по сравнению с (+)-энантиомером, причем указанный процент приводится в расчете на общее количество смеси. В одном из воплощений термин "по существу не содержащий его (+)-энантиомера" означает, что композиция содержит по меньшей мере 60 мас.% (-)-энантиомера и 40 мас.% или меньше (+)-энантиомера. В предпочтительном воплощении термин "по существу не содержащий его (+)-энантиомера" означает, что композиция содержит по меньшей мере 70 мас.% (-)-энантиомера и 30 мас.% или меньше (+)-энантиомера. В другом воплощении термин "по существу не содержащий его (+)-энантиомера" означает, что композиция содержит по меньшей мере 80 мас.% (-)-энантиомера и 20 мас.% или меньше (+)-энантиомера. Кроме того, термин "по существу не содержащий его (+)-энантиомера" означает, что композиция содержит по меньшей мере 90 мас.% (-)-энантиомера и 10 мас.% или меньше (+)-энантиомера. Еще термин "по существу не содержащий его (+)-энантиомера" означает, что композиция содержит по меньшей мере 95 мас.% (-)-энантиомера и 5 мас.% или меньше (+)-энантиомера. Более того, термин "по существу не содержащийт его (+)-энантиомера" означает, что композиция содержит по меньшей мере 99 мас.% (-)-энантиомера и 1 мас.% или меньше (+)-энантиомера.
Вышеуказанная формула I показана без определенной стереохимии по некоторым положениям. Настоящее изобретение охватывает все стереоизомеры соединений формулы I и их фармацевтически приемлемые соли. Кроме того, смеси стереоизомеров, а также выделенные конкретные стереоизомеры также охвачены. В ходе осуществления способов синтеза, использованных для получения таких соединений, или при использовании способов рацемизации или эпимеризации, известных специалистам в данной области, продуктами таких способов могут быть смеси стереоизомеров.
Когда существует таутомер соединения формулы I, настоящее изобретение охватывает все возможные таутомеры и их фармацевтически приемлемые соли, и их смеси, за исключением тех случаев, когда конкретно указано иное.
Когда соединение формулы I и его фармацевтически приемлемые соли существуют в форме сольватов или в полиморфных формах, настоящее изобретение охватывает все возможные сольваты и полиморфные формы. Тип растворителя, который образует сольват, конкретно не ограничен при условии, что растворитель является фармакологически приемлемым. Например, можно использовать воду, этанол, пропанол, ацетон или т.п.
Термин "композиция" в данном документе означает продукты, которые содержат конкретно указанные количества конкретно указанных ингредиентов, а также любой продукт, который получен, напрямую или опосредовано, из конкретно указанной комбинации конкретных ингредиентов. Таким образом, фармацевтические композиции, содержащие соединения по настоящему изобретению в качестве активных ингредиентов, и способы получения соединений по изобретению также являются частью данного изобретения. Кроме того, некоторые кристаллические формы соединений могут существовать в виде полиморфов, и такие полиморфы охвачены настоящим изобретением. Кроме того, некоторые соединения могут образовывать сольваты с водой (т.е. гидраты) или с обычными органическими растворителями, и такие сольваты также входят в объем настоящего изобретения.
Фармацевтические композиции по настоящему изобретению содержат соединение, представленное формулой I, (или его фармацевтически приемлемую соль) в качестве активного ингредиента, фармацевтически приемлемый носитель и возможно другие терапевтические ингредиенты или вспомогательные вещества. Композиции включают композиции, подходящие для перорального, ректального, местного и парентерального (включая подкожный, внутримышечный и внутривенный) введения, хотя наиболее подходящий путь в любом данном случае будет зависеть от конкретного хозяина и характера и тяжести состояний, против которых вводят активный ингредиент. Фармацевтические композиции для удобства могут быть представлены в стандартной лекарственной форме и могут быть приготовлены любым способом, известным в области фармации.
На практике соединения, представленные формулой I, или их пролекарство, или метаболит, или фармацевтически приемлемые соли, по данному изобретению могут быть объединены в виде активного ингредиента во внутренней смеси с фармацевтическим носителем согласно общепринятым фармацевтическим методам компаундирования. Носитель может принимать широкое разнообразие форм в зависимости от формы препарата, желаемого для введения, например перорального или парентерального (включая внутривенное). Так, фармацевтические композиции по настоящему изобретению могут быть представлены в виде дискретных единиц, подходящих для перорального введения, таких как капсулы, облатки или таблетки, каждая из которых содержит предопределенное количество активного ингредиента. Кроме того, композиции могут быть представлены в виде порошка, в виде гранул, в виде раствора, в виде суспензии в водной жидкости, в неводной жидкости, в виде эмульсии типа масло-в-воде или в виде жидкой эмульсии типа вода-в-масле. Помимо обычных лекарственных форм, указанных выше, соединение, представленное формулой I или его фармацевтически приемлемую соль можно вводить также методами контролируемого высвобождения и/или посредством устройств для доставки. Композиции могут быть приготовлены любыми способами, известными в фармации. Как правило, такие способы включают стадию приведения активного ингредиента в контакт с носителем, который состоит из одного или более необходимых ингредиентов. Как правило, композиции получают путем равномерного и тщательного смешивания активного ингредиента с жидкими носителями или тонкоизмельченными твердыми носителями, или и теми, и другими. Продукту затем для удобства может быть придана форма желаемой презентации.
Таким образом, фармацевтические композиции по данному изобретению могут содержать фармацевтически приемлемый носитель и соединение формулы I, его стереоизомер, таутомер, полиморф, сольват, фармацевтически приемлемую соль или пролекарство. Соединения формулы I или их фармацевтически приемлемые соли могут быть включены в состав фармацевтической композиции в комбинации с одним или более другими терапевтически активными соединениями.
Используемый фармацевтический носитель может представлять собой, например, твердое вещество, жидкость или газ. Примеры твердых носителей включают такие носители, как лактоза, каолин, сахароза, тальк, желатин, агар, пектин, аравийская камедь, стеарат магния и стеариновая кислота. Примеры жидких носителей включают такие носители, как сахарный сироп, арахисовое масло, оливковое масло и воду. Примеры газообразных носителей включают такие носители, как диоксид углерода и азот. В приготовлении композиций в стандартной лекарственной форме могут быть использованы любые пригодные фармацевтические среды. Например, вода, гликоли, масла, спирты, корригенты, консерванты, красители и т.п. могут быть использованы для создания пероральных жидких препаратов, таких как суспензии, эликсиры и растворы, а такие носители, как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие агенты, смазывающие вещества, связующие, разрыхлители и т.п., могут быть использованы для создания пероральных твердых препаратов, таких как порошки, капсулы и таблетки. Из-за легкости их введения таблетки и капсулы являются предпочтительными пероральными лекарственными единицами, в которых используются твердые фармацевтические носители. Возможно, таблетки могут быть покрыты оболочкой стандартными водными и неводными методами.
Таблетка, содержащая композицию по данному изобретению, может быть изготовлена путем прессования или формования, возможно с одним или более дополнительными ингредиентами или вспомогательными веществами. Прессованные таблетки могут быть изготовлены путем прессования в подходящей машине активного ингредиента в сыпучей форме, например в форме порошка или гранул, возможно смешанных со связывающим веществом, смазывающим веществом, инертным разбавителем, поверхностно-активным веществом или диспергирующим агентом. Формованные таблетки могут быть изготовлены путем формования в подходящей машине смеси порошкообразного соединения, смоченного инертным жидким разбавителем. Каждая таблетка предпочтительно содержит от примерно 0,05 мг до примерно 5 г активного ингредиента, и каждая облатка или капсула предпочтительно содержит от примерно 0,05 мг до примерно 5 г активного ингредиента. Например, композиция для перорального введения людям может содержать от примерно 0,5 мг до примерно 5 г активного агента, смешанного с соответствующим и удобным количеством вещества-носителя, которое может варьировать от примерно 5 до примерно 95 процентов от общего количества композиции. Стандартные лекарственные формы обычно будут содержать от примерно 1 мг до примерно 2 г активного ингредиента, в типичных случаях 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг, 500 мг, 600 мг, 800 мг или 1000 мг.
Фармацевтические композиции по настоящему изобретению, подходящие для парентерального введения, могут быть приготовлены в виде растворов или суспензий активного соединения в воде. В состав композиции может быть включено поверхностно-активное вещество, такое как, например, гидроксипропилцеллюдлоза. Также могут быть приготовлены дисперсии в глицерине, жидких полиэтиленгликолях и их смесях в маслах. Кроме того, в состав может быть включен консервант для предотвращения роста микроорганизмов.
Фармацевтические композиции по настоящему изобретению, подходящие для инъекционного применения, включают стерильные водные растворы или дисперсии. Более того, композиции могут быть в форме стерильных порошков для немедленного приготовления таких стерильных инъекционных растворов или дисперсий. Во всех случаях конечная инъекционная форма должна быть стерильной и должна быть эффективно жидкой для облегчения введения шприцем. Фармацевтические композиции должны быть стабильными в условиях изготовления и хранения, поэтому они должны быть защищены от загрязняющего действия микроорганизмов, таких как бактерии и грибы. Носитель может представлять собой растворитель или дисперсионную среду, содержащую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), растительные масла и подходящие их смеси.
Фармацевтические композиции по настоящему изобретению могут быть в форме, подходящей для местного применения, такой как, например, аэрозоль, крем, мазь, лосьон, присыпка или т.п. Кроме того, композиции могут быть в форме, подходящей для применения в трансдермальных устройствах. Эти композиции могут быть приготовлены с использованием соединения, представленного формулой I, по данному изобретению или его фармацевтически приемлемой соли стандартными методами обработки. В качестве примера, крем или мазь получают путем смешивания гидрофильного вещества и воды вместе с соединением, взятым в количестве от примерно 5 мас.% до примерно 10 мас.%, с получением крема или мази желаемой консистенции.
Фармацевтические композиции по данному изобретению могут быть в форме, подходящей для ректального введения, где носитель представляет собой твердое вещество. Предпочтительно, чтобы смеси были в форме содержащих стандартную дозу суппозиториев. Подходящие носители включают масло какао и другие вещества, обычно используемые в данной области. Суппозитории могут быть легко сформованы сначала смешиванием композиции с размягченным или расплавленным носителем(ями) с последующим охлаждением и формованием в формах.
Помимо вышеупомянутых ингредиентов-носителей фармацевтические композиции, описанные выше, могут содержать, если это целесообразно, один или более дополнительных ингредиентов-носителей, таких как разбавители, буферы, корригенты, связывающие вещества, поверхностно-активные вещества, загустители, смазывающие вещества, консерванты (в том числе антиоксиданты) и т.п. Более того, в состав могут быть включены другие вспомогательные вещества, делающие композицию изотоничной с кровью намеченного реципиента. Композиции, содержащие соединение, описанное формулой I, или его фармацевтически приемлемые соли, могут быть приготовлены также в форме порошка или жидкого концентрата.
Как правило, уровни дозировки порядка от примерно 0,01 мг/кг до примерно 150 мг/кг массы тела в сутки полезны в лечении вышеуказанных состояний, или, альтернативно, от примерно 0,5 мг до примерно 7 г на пациента с сутки. Например, рак ободочной кишки, ректальный рак, мантийноклеточную лимфому, множественную миелому, рак молочной железы, рак предстательной железы, глиобластому, плоскоклеточный рак пищевода, липосаркому, Т-клеточную лимфому, меланому, рак поджелудочной железы, глиобластому или рак легкого можно эффективно лечить путем введения от примерно 0,01 до 50 мг соединения на килограмм массы тела в сутки или, альтернативно, от примерно 0,5 мг до примерно 3,5 г на пациента в сутки.
Понятно, однако, что могут потребоваться дозы ниже или выше, чем дозы перечисленные выше. Конкретная доза и схема лечения для любого конкретного субъекта будет зависеть от целого ряда факторов, включающих активность конкретно используемого соединения, возраст, массу тела, общее состояние здоровья, пол, диету, время введения, путь введения, скорость выведения, комбинацию лекарственных средств, тяжесть и ход конкретного заболевания, подвергаемого лечению, предрасположенность субъекта к заболеванию и суждение лечащего врача.
Эти и другие аспекты станут ясны из нижеследующего описания изобретения.
Примеры
Следует иметь в виду, что вышеприведенное общее описание и следующее далее подробное описание являются только иллюстративными и поясняющими и не ограничивают заявленный предмет изобретения. Все части и проценты массовые, и все температуры приведены в градусах Цельсия, если явно не указано иное. Соединения, описанные в данном документе, могут быть получены из коммерческих источников или могут быть синтезированы стандартными способами, как показано ниже, с использованием коммерчески доступных исходных веществ и реагентов.
В примерах использованы следующие сокращения:
АТР: аденозинтрифосфат;
Вос2О: ди-трет-бутил-дикарбонат;
con-H2SO4: концентрированная серная кислота;
Crk: СТ10 (ретровирус куриной опухоли 10);
DCM: дихлорметан;
DEA: диэтиламин;
DEAD: диэтилазодикарбоксилат;
DIEA: N,N-диизопропилэтиламин;
DMEM: среда Игла, модифицированная по Дульбекко;
DMF: N,N-диметилформамид;
DMA: N,N-диметилацетамид;
DMAP: 4-N,N-диметиламинопиридин;
DMSO: диметилсульфоксид;
DTT: DL-дитиотрейтол;
ЕА: этилацетат;
EDC: 1-этил-3-(3-диметиламинопропил)карбодиимид;
EDTA: этилендиаминтетрауксусная кислота;
EtOH: этиловый спирт;
FBS: фетальная коровья сыворотка;
GSR: глутатион-S-трансфераза;
HATU: гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония;
HEPES: 4-(2-гидроксиэтил)пиперазин-1-этансульфоновая кислота;
Hex: н-гексан;
ч: часы;
IPA: изопропанол
KOAc: ацетат калия;
KTB: трет-бутоксид калия;
МеОН: метанол;
мин: минуты;
MsCl: метилсульфонилхлорид;
MTS: 3-(4,5-диметилтиазол-2-ил)-5-(3-карбоксиметоксифенил)-2-(4-сульфофенил)-2Н-тетразолий;
NaBH4: боргидрид натрия;
NaBH(OAc)3: триацетоксиборгидрид натрия;
Р(Су)3: трициклогексилфосфин;
Pd2(dba)3: трис(дибензилиденацетон)дипалладий
Pd(dppf)Cl2: [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий;
Pd(OAc)2: ацетат палладия;
РЕ: петролейный эфир;
PMS: феназинметосульфат;
POCl3: оксихлорид фосфора;
P/S: раствор пенициллина/стрептомицина;
КТ или к.т.: комнатная температура;
SDS: додецилсульфат натрия;
SDS-PAGE: электрофорез в полиакриламидном геле в присутствии додецилсульфата натрия;
ТВАВ: бромид тетрабутиламмония;
TEA: триэтиламин;
THF: тетрагидрофуран;
ТСХ: тонкослойная хроматография;
Tol: толуол.
Получение 1: 5-(4-метилпиперазин-1-ил)пиридин-2-амин (Промежуточное соединение M1)

Добавить 1-метилпиперазин (1,180 г) и K2CO3 (2,720 г) последовательно к раствору 5-бром-2-нитропиридина (2,010 г) в DMSO (20 мл). Перемешивать эту реакционную смесь при 82°С в течение 15 ч в масляной бане. Добавить воду (50 мл), экстрагировать DCM (20 мл × 8), объединенную органическую фазу сушили над безводным Na2SO4, концентрировали при пониженном давлении, очищали колоночной хроматографией (DCM/MeOH=10/1) с получением 1,940 г 1-метил-4-(6-нитропиридин-3-ил)пиперазина.
Добавить Pd/C (0,194 г) к раствору 1-метил-4-(6-нитропиридин-3-ил)пиперазина (1,940 г) в THF (25 мл) в атмосфере водорода в течение 2 ч при КТ. Фильтрат собирали фильтрованием и затем концентрировали с получением 1,480 г 5-(4-метилпиперазин-1-ил)пиридин-2-амина.
МС(ЭРИ+) (масс-спектрометрия с электрораспылительной ионизацией с регистрацией положительных ионов): m/z=193,1 (М+Н)+.
Получить нижеследующие промежуточные соединения (показанные в Таблице 1), в сущности как описано для 5-(4-метилпиперазин-1-ил)пиридин-2-амина (именуемого здесь как Промежуточное соединение M1), с использованием соответствующего производного пиперазина.
Получение 4: 6-((4-метилпиперазин-1-ил)метил)пиридин-3-амин (Промежуточное соединение М4)

Добавить NaBH4 (1,220 г) к раствору 5-бромпиколинальдегида (2,010 г) в МеОН (30 мл) при 0°С в ледяной бане, после окончания добавления NaBH4 удалить ледяную баню и нагревать до комнатной температуры в естественных условиях. После перемешивания в течение 2 ч при КТ реакционную смесь гасили водой (50 мл) при 0°С. Экстрагировать ЕА (50 мл × 2), объединенную органическую фазу промывали насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали с получением 1,940 г (5-бромпиридин-2-ил)метанола.
Раствор (5-бромпиридин-2-ил)метанола (1,940 г) в THF (20 мл) охлаждают до 0°С в ледяной бане, затем в раствор по каплям добавить метилсульфонилхлорид (1,780 г). После окончания добавления метилсульфонилхлорида удалить ледяную баню, нагревать до комнатной температуры в естественных условиях. После перемешивания в течение 2 ч при КТ реакционную смесь гасили водой (50 мл). Экстрагировать ЕА (50 мл × 2), объединенную органическую фазу промывали насыщенным раствором NaCl (50 мл) и сушили над безводным Na2SO4, концентрировали с получением 2,750 г неочищенного продукта, (5-бромпиридин-2-ил)метилметансульфоната.
Добавить K2CO3 (2,870 г) и 1-метилпиперазин (1,560 г) последовательно к раствору (5-бромпиридин-2-ил)метилметансульфоната (2,750 г) в ацетонитриле (30 мл), нагреть до 50°С в масляной бане и подвергать взаимодействию в течение 2 ч. Затем охладить до комнатной температуры, добавить воду, экстрагировать ЕА (50 мл × 3), объединенную органическую фазу промывали насыщенным раствором NaCl (50 мл) и сушили над безводным Na2SO4, концентрировали и очищали колоночной хроматографией (DCM/MeOH=10/1) с получением 2,010 г 1-((5-бромпиридин-2-ил)метил)-4-метилпиперазина.
Добавить МеОН (20 мл) в герметизируемую пробирку на 100 мл под аммиаком при -78°С, затем последовательно добавить 1-((5-бромпиридин-2-ил)метил)-4-метилпиперазин (1,000 г) и оксид одновалентной меди (0,532 г) до увеличения объема раствора до 30 мл. Удалить внешнюю баню, нагреть до комнатной температуры в естественных условиях, затем нагреть до 70°С и подвергать взаимодействию в течение 12 ч. Фильтрат собирали фильтрованием, концентрировали и очищали колоночной хроматографией (DCM/MeOH=15/1) с получением 0,730 г 6-((4-метилпиперазин-1-ил)метил)пиридин-3-амина.
МС(ЭРИ+): m/z=207,2 (М+Н)+.
Получить нижеследующие промежуточные соединения (показанные в Таблице 2), в сущности как описано для 6-((4-метилпиперазин-1-ил)метил)пиридин-3-амина (именуемого здесь как Промежуточное соединение М4), с использованием соответствующего производного пиперазина.

Получение 7: 5-((4-(диметиламино)пиперидин-1-ил)метил)пиридин-2-амин (Промежуточное соединение М7)
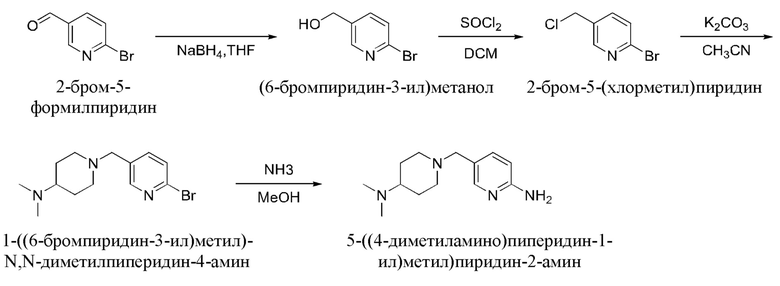
Добавить NaBH4 (1,640 г) к раствору 2-бром-5-формилпиридина (2,010 г) в THF (20 мл) при 0°С в ледяной бане, после окончания добавления NaBH4 удалить ледяную баню, нагревать до комнатной температуры в естественных условиях. После перемешивания в течение 2 ч при КТ реакционную смесь гасили водой (50 мл), экстрагировали ЕА (50 мл × 2), объединенную органическую фазу сушили над безводным Na2SO4, концентрировали и очищали колоночной хроматографией (РЕ/ЕА=5/1) с получением 1,900 г (6-бромпиридин-3-ил)метанола.
Раствор (6-бромпиридин-3-ил)метанола (1,000 г) в DCM (10 мл) охлаждали до 0°С в ледяной бане и по каплям добавляли к тионилхлориду (1,260 г), после окончания добавления удалить ледяную баню, раствор нагревать до комнатной температуры в естественных условиях при перемешивании в течение 2 ч, затем напрямую концентрировали с получением 1,050 г 2-бром-5-(хлорметил)пиридина.
Добавить N,N-диметилпиперидин-4-амин (0,586 г) и K2CO3 (1,160 г) добавляли к раствору 2-бром-5-(хлорметил)пиридина (0,853 г) в ацетонитриле (10 мл). Добавить воду (30 мл), экстрагировать ЕА (50 мл × 3), объединенную органическую фазу сушили над безводным Na2SO4, концентрировали и очищали колоночной хроматографией (DCM/MeOH=10/1) с получением 0,730 г 1-((6-бромпиридин-3-ил)метил)-N,N-диметилпиперидин-4-амина.
Добавить МеОН (20 мл) в герметизируемую пробирку на 100 мл под аммиаком при -78°С, затем добавить последовательно 1-((6-бромпиридин-3-ил)метил)-N,N-диметилпиперидин-4-амин (0,35 мг) и оксид одновалентной меди (0,168 г) до увеличения объема до 30 мл. Удалить внешнюю баню, нагреть до комнатной температуры в естественных условиях, затем нагреть до 70°С и подвергать взаимодействию в течение 12 ч. Фильтрат собирали фильтрованием, концентрировали и очищали колоночной хроматографией (DCM/MeOH=10/1) с получением 0,260 г 5-((4-(диметиламино)пиперидин-1-ил)метил)пиридин-2-амина.
МС(ЭРИ+):m/z=235,2(М+Н)+.
Получить нижеследующие промежуточные соединения (показанные в Таблице 3), в сущности как описано для 5-((4-(диметиламино)пиперидин-1-ил)метил)пиридин-2-амина (именуемого здесь как Промежуточное соединение М7), с использованием соответствующего производного пиперидина.

Получение 11: 5-((4-метилпиперазин-1-ил)метил)пиримидин-2-амина гидрохлорид (Промежуточное соединение М11)

Добавить водный аммиак (25%) (1,200 г) к раствору 2-хлорпиримидин-5-карбальдегида (0,500 г) в THF (50 мл), перемешивая в течение 12 ч. Добавить воду (80 мл), экстрагировать DCM (80 мл × 8), объединенную органическую фазу сушили над безводным Na2SO4 и концентрировали с получением 0,540 г неочищенного продукта, 2-аминопиримидин-5-карбальдегида.
Добавить Boc2O (2,817 г), триэтиламин (1,310 г) и DMAP (0,054 г) последовательно к раствору 2-аминопиримидин-5-карбальдегида (0,540 г) в THF (30 мл) при перемешивании в течение 2 ч. Добавить воду, экстрагировать ЕА (50 мл × 2), объединенную органическую фазу сушили над безводным Na2SO4, концентрировали и очищали колоночной хроматографией (РЕ/ЕА=5/1) с получением 0,514 г соединения МН2-12.
Добавить 1-метилпиперазин (0,109 г) и безводный сульфат магния (0,216 г) последовательно к раствору соединения МН2-12 (0,290 г) в DCM (10 мл) при перемешивании в течение 2 ч, затем подвергать взаимодействию в течение 3 ч при КТ после добавления триацетоксиборгидрида натрия, реакционную смесь гасили водой (20 мл), экстрагировали DCM (20 мл × 3), объединенную органическую фазу сушили над безводным Na2SO4, концентрировали и очищали колоночной хроматографией (DCM/MeOH=10/1) с получением 0,350 г соединения МН2-13.
Раствор соединения МН2-13 (0,350 г) в DCM подвергали взаимодействию в течение 2 ч под газообразным хлористым водородом при КТ, реакционную смесь концентрируют с получением 0,210 г 5-((4-метилпиперазин-1-ил)метил)пиримидин-2-амина гидрохлорида.
МС(ЭРИ+):m/z=244,1 (М+Н)+.
Получить нижеследующее промежуточное соединение (показанное в Таблице 4), в сущности как описано для 5-((4-метилпиперазин-1-ил)метил)пиримидин-2-амина гидрохлорида (именуемого здесь как Промежуточное соединение М11), с использованием соответствующего производного пиперидина вместо производного пиперазина.

Получение 13: (2-аминопиримидин-5-ил)(4-метилпиперазин-1-ил)метанон (Промежуточное соединение М13)

Добавить Оксон (1,810 г) к смеси соединения МН2-12 (0,315 г) в ацетоне (10 мл) и воде (3 мл) при перемешивании в течение 2 ч при КТ. Добавить воду (20 мл), экстрагировать DCM (25 мл × 3), объединенную органическую фазу сушили над безводным Na2SO4 и концентрировали с получением 0,290 г соединения МН8-01.
Добавить HATU (0,488 г) и DIEA (0,221 г) последовательно к раствору соединения МН8-01 (0,290 г) в DCM (10 мл) при перемешивании в течение 1 ч при КТ. Раствор подвергать взаимодействию в течение 2 ч после добавления 1-метилпиперазина (0,105 г) при КТ. Добавить воду (20 мл), экстрагировать DCM (20 мл × 3), объединенную органическую фазу сушили над безводным Na2SO4, концентрировали и очищали колоночной хроматографией (DCM/MeOH=50/1) с получением 0,250 г соединения МН8-02.
Раствор соединения МН8-02 (0,150 г) в DCM (10 мл) подвергают взаимодействию в течение 2 ч под газообразным хлористым водородом при КТ, добавить воду (10 мл), затем довести рН до 8-9 добавлением Na2CO3, полученный водный раствор экстрагировали смешанным растворителем (DCM/MeOH=10/1) (20 мл × 5). Объединенную органическую фазу сушили над безводным Na2SO4 и концентрировали с получением 0,060 г (2-аминопиримидин-5-ил)(4-метилпиперазин-1-ил)метанона.
МС(ЭРИ+):m/z=223,1(М+Н)+.
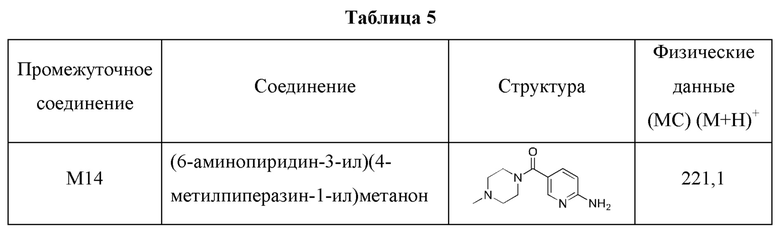
Получить нижеследующее промежуточное соединение (показанное в Таблице 5), в сущности как описано для (2-аминопиримидин-5-ил)-(4-метилпиперазин-1-ил)метанона (именуемого здесь как Промежуточное соединение М13), с использованием соответствующего производного пиридина вместо производного пиримидина.
Получение 15: N5-(2-(диэтиламино)этил)-N5-метилпиридин-2,5-диамин (Промежуточное соединение М15)
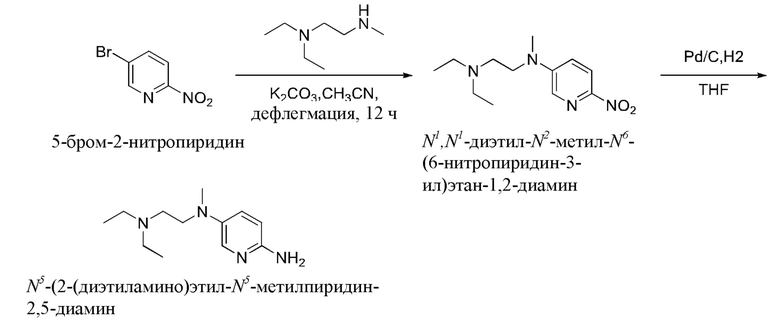
Добавить N,N-диэтил-N'-метилэтилендиамин (0,305 г) и K2CO3 (0,679 г) последовательно к раствору 2-нитро-5-бромпиридина (0,500 г) в ацетонитриле (10 мл). Перемешивать эту реакционную смесь при 82°С в течение 15 ч в масляной бане. Добавить воду (50 мл), экстрагировать DCM (80 мл × 3), объединенную органическую фазу сушили над безводным Na2SO4, концентрировали и очищали колоночной хроматографией (DCM/MeOH=10/1) с получением 0,400 г 1-метил-4-(6-нитропиридин-3-ил)пиперазина.
Добавить Pd/C (0,040 г) к раствору 1-метил-4-(6-нитропиридин-3-ил)пиперазина в THF (15 мл) при перемешивании в течение 2 ч при КТ под газом водородом. Фильтрат собирали фильтрованием и затем концентрировали с получением 0,350 г N5-(2-(диэтиламино)этил)-N5-метилпиридин-2,5-диамина.
МС(ЭРИ+): m/z=223,2(М+Н)+.
Получить нижеследующее промежуточное соединение (показанное в Таблице 6), в сущности как описано для N5-(2-(диэтиламино)этил)-N5-метилпиридин-2,5-диамина (именуемого здесь как Промежуточное соединение M15), с использованием N1,N1-диэтил-N2,N2-диметилэтан-1,2-диамина вместо N,N-диэтил-N'-метилэтилендиамина.

Получение 17: 6-((4-(диметиламино)пиперидин-1-ил)метил)пиридазин-3-амин (Промежуточное соединение М17)
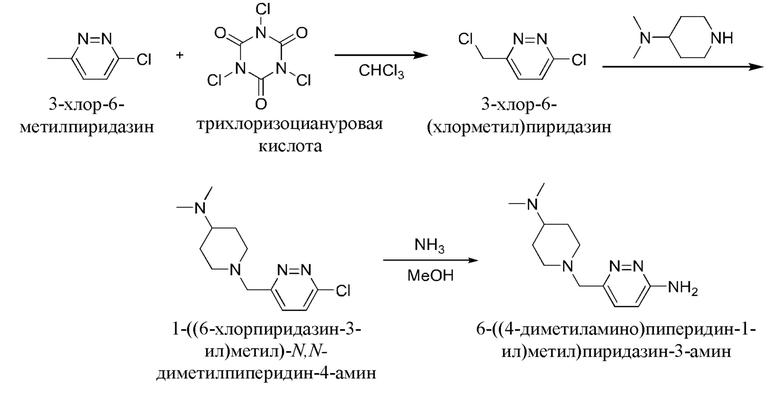
Добавить трихлоризоциануровую кислоту (0,189 г) к раствору 3-хлор-6-метилпиридазина (0,208 г) в CHCl3 (10 мл) и нагревать до 60°С в течение 12 ч в масляной бане. Охладить до комнатной температуры, фильтрат собирали фильтрованием, концентрировали и очищали колоночной хроматографией (РЕ/ЕА=10/1) с получением 0,201 г 3-хлор-6-(хлорметил)пиридазина.
Добавить K2CO3 (0,578 г), KI (0,070 г) и N,N-диметилпиперидин-4-амин (0,322 г) последовательно к раствору 3-хлор-6-(хлорметил)пиридазина (0,340 г) в DMF (15 мл), нагревать до 50°С в течение 1 ч в масляной бане. Охладить до комнатной температуры, добавить DCM (50 мл), промыть объединенные органические слои насыщенным раствором NaCl, и сушили над безводным Na2SO4, концентрировали и очищали колоночной хроматографией (DCM/MeOH=10/1) с получением 0,370 г 1-((6-хлорпиридазин-3-ил)метил)-N,N-диметилпиперидин-4-амина.
Добавить МеОН (20 мл) в герметизируемую пробирку на 100 мл под аммиаком при -78°С, затем добавить последовательно 1-((6-хлорпиридазин-3-ил)метил)-N,N-диметилпиперидин-4-амин (0,370 г) и оксид одновалентной меди (0,532 г) до увеличения объема до 30 мл. Удалить внешнюю баню, нагреть до комнатной температуры в естественных условиях, затем нагреть до 70°С и подвергать взаимодействию в течение 12 ч. Фильтрат собирали фильтрованием, концентрировали и очищали колоночной хроматографией (DCM/MeOH=15/1) с получением 0,230 г 6-((4-(диметиламино)пиперидин-1-ил)метил)пиридазин-3-амина. МС(ЭРИ+):m/z=236,2 (М+Н)+.
Получить нижеследующее промежуточное соединение (показанное в Таблице 7), в сущности как описано для 6-((4-(диметиламино)пиперидин-1-ил)метил)пиридазин-3-амина (именуемого здесь как Промежуточное соединение M17), с использованием соответствующего производного пиперазина вместо производного пиперидина.
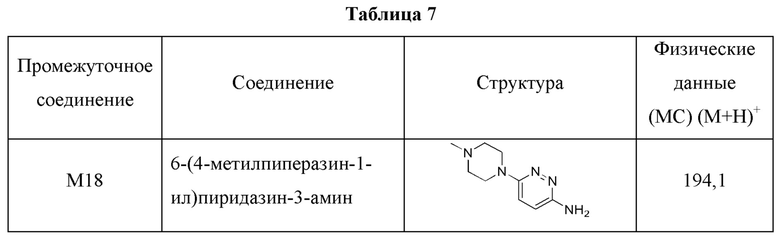
Пример 1: Синтез соединения 1
1. Соединение 1-01
Смесь 2-метилциклопентанона (5,200 г), гидроксиламина гидрохлорида (9,200 г) и триэтиламина (16,080 г) в безводном этаноле (70 мл) перемешивали при 85°С в течение ночи в масляной бане. Затем реакционный раствор концентрировали, остаток промывали ЕА. Фильтрат собирали фильтрованием и затем концентрировали с получением 5,820 г неочищенного соединения 1-01.
2. Соединение 1-02
Неочищенное соединение 1-01 (5,820 г) растворяли в растворе серной кислоты (con-H2SO4 : Н2О = 20 мл : 5 мл), полученную смесь перемешивали при 90°С в масляной бане в течение 90 мин, добавляли воду (10 мл), затем доводили рН до 8-9 добавлением Na2CO3. Полученный водный раствор экстрагировали DCM (20 мл × 5). Объединенную органическую фазу сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 4,110 г неочищенного соединения 1-02.
3. Соединение 1-03
К смеси неочищенного Соединения 1-02 (4,110 г) и 4-бром-2,6-дифторанилина (3,780 г) в метилбензоле (40 мл) добавляли POCl3 (4,180 г) и нагревали в масляной бане. Когда температура поднялась до 110°С, добавляли TEA (2,770 г), полученную смесь подвергали взаимодействию при 110°С в течение 20 мин. Часть метилбензола удаляли, затем доводили рН до 8-9 добавлением Na2CO3, экстрагировали ЕА, объединенную органическую фазу промывали насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 6,550 г неочищенного Соединения 1-03.
4. Соединение 1-04
Смесь неочищенного Соединения 1-03 (6,100 г) и трет-бутоксида калия (4,520 г) в DMF (60 мл) перемешивали при 100°С в течение 20 мин в масляной бане и затем экстрагировали 300 мл ЕА, объединенную органическую фазу промывали насыщенным раствором NaCl (120 мл × 3), сушили над безводным Na2SO4, концентрировали и затем очищали колоночной хроматографией (РЕ/ЕА=1/5) с получением 0,705 г соединения 1-04-А и 1,500 г неочищенного соединения 1-04-С.
5. Соединение 1-05
Смесь неочищенного соединения 1-04-С (0,957 г), бис(пинаколато)дибора (1,290 г), трициклогексилфосфина (0,047 г), ацетата палладия (0,038 г) в DMSO (20 мл) перемешивали под азотом в течение 1 ч при 90°С в масляной бане. Полученную смесь экстрагировали ЕА (60 мл), объединенную органическую фазу промывали насыщенным раствором NaCl (30 мл × 3), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 2,290 г неочищенного Соединения 1-05.
6. Соединение 1-06
Смесь неочищенного Соединения 1-05 (2,290 г), 2,4-дихлорпиримидина (0,755 г), K2CO3 (1,400 г) и Pd(dppf)Cl2⋅DCM (0,138 г) в 1,4-диоксане (30 мл) и воде (3 мл) перемешивали под азотом в течение 2 ч при 60°С в масляной бане. Полученную смесь экстрагировали ЕА (30 мл × 2), объединенную органическую фазу промывали насыщенным раствором NaCl (30 мл × 1), сушили над безводным Na2SO4, концентрировали и затем очищали колоночной хроматографией (РЕ/ЕА=1/1) с получением 0,927 г Соединения 1-06.
7. Соединение 1
Смесь соединения 1-06 (0,400 г), промежуточного соединения M1 (0,291 г), Cs2CO3 (0.822 г), Xanphos (0,017 г) и Pd2(dba)3 (0,027 г) в 1,4-диоксане (12 мл) перемешивали в атмосфере газа азота в течение 1 ч при 110°С в масляной бане, взаимодействие продолжали под воздействием микроволнового излучения в течение 0,5 ч при 110°С. В полученную смесь добавляли воду (10 мл), затем экстрагировали DCM (20 мл × 3), объединенную органическую фазу промывали насыщенным раствором NaCl (30 мл × 1), сушили над безводным Na2SO4, концентрировали и затем очищали колоночной хроматографией (DCM/MeOH=20/1). Твердое вещество промывали метил-трет-бутиловым эфиром (10 мл) и н-гексаном (10 мл) с получением 290 мг Соединения 1.
МС(ЭРИ+):m/z=473,2(М+Н)+.
Н-ЯМР(CDCl3): δ 8.498-8.511 (d, 1H, CH), 8.373-8.396 (d, 1H, CH), 8.167 (s, 1H, CH), 8.040-8.047 (d, 1H, CH), 7.963 (s, 1H, CH), 7.631-7.660 (d, 1H, CH), 7.346-7.660 (dd, 1H, CH), 7.177-7.190 (d, 1H, CH), 4.687-4.719 (m, 1H, CH), 3.190-3.10 (m, 4H, CH2), 2.651-2.675 (m, 4H, CH2), 2.999-3.024 (m, 1H, CH2), 2.403 (s, 3H, CH3), 2.337-2.356 (m, 1H, CH2), 2.267-2.357 (m, 1H, CH2), 2.219-2.248 (m, 1H, CH2), 2.010-2.056 (m, 2H, CH2), 1.602-1.618 (d, 3H, CH3).
Пример 1-1. Хиральное разделение Соединения 1-06
Методы, используемые для разделения изомеров, например энантиомеров, находятся в компетенции специалистов в данной области и описаны в Eliel, E.L.; Wilen, S.H.; Mander, L.N. Stereochemistry of Organic Compounds, Wiley Interscience, NY, 1994. Например, соединение 1, 2 или 15 может быть разделено с достижением высокого энантиомерного избытка (например, 60%, 70%, 80%, 90%, 95%, 99% или больше) посредством высокоэффективной жидкостной хроматографии с использованием хиральной колонки. В некоторых воплощениях неочищенное соединение 1-06 из Примера 1 очищают напрямую на хиральной колонке с получением энантиомерно обогащенного соединения.
Условия хиральной ВЭЖХ:

Пример 1-2. Синтез Соединения 1а и Соединения 1b
Неочищенное Соединение 1-06 очищают на хиральной колонке в вышеуказанных условиях с получением Соединения 1-06-А и Соединения 1-06-В.
Получить Соединение 1а и Соединение 1b, в сущности как описано для стадии 7 Примера 1, с использованием Соединения 1-06-А и Соединения 1-06-В соответственно.
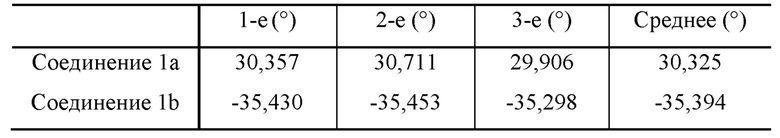
Кроме того, оптическое вращение измеряли 3 раза для каждого соединения, как показано ниже, на поляриметре Rudolf.
Условия:
Результаты:
Пример 2: Синтез соединения 2
1. Соединение 1-01
Смесь 2-метилциклопентанона (5,200 г), гидроксиламина гидрохлорида (9,200 г) и триэтиламина (16,080 г) в безводном этаноле (70 мл) перемешивали при 85°С в течение ночи в масляной бане. Затем реакционный раствор концентрировали; остаток промывали ЕА. Фильтрат собирали фильтрованием и затем концентрировали с получением 5,820 г неочищенного соединения 1-01.
2. Соединение 1-02
Неочищенное соединение 1-01 (5,820 г) растворяли в растворе серной кислоты (con-H2SO4 : Н2О = 20 мл : 5 мл), полученную смесь перемешивали при 90°С в масляной бане в течение 90 мин. Добавляли воду (10 мл), затем доводили рН до 8-9 добавлением Na2CO3, полученный водный раствор экстрагировали DCM (20 мл × 5), объединенную органическую фазу сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 4,110 г неочищенного соединения 1-02.
3. Соединение 1-03
К смеси неочищенного Соединения 1-02 (4,110 г) и 4-бром-2,6-дифторанилина (3,780 г) в метилбензоле (40 мл) добавляли POCl3 (4,180 г) и нагревали в масляной бане. Когда температура поднялась до 110°С, добавляли TEA (2,770 г), полученную смесь подвергали взаимодействию при 110°С в течение 20 мин. Часть метилбензола удаляли, затем доводили рН до 8-9 добавлением Na2CO3, экстрагировали ЕА, объединенную органическую фазу промывали насыщенным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 6,550 г неочищенного Соединения 1-03.
4. Соединение 1-04
Смесь неочищенного Соединения 1-03 (6,100 г) и трет-бутоксида калия (4,520 г) в DMF (60 мл) перемешивали при 100°С в течение 20 мин в масляной бане и затем экстрагировали 300 мл ЕА, объединенную органическую фазу промывали насыщенным раствором NaCl (120 мл × 3), сушили над безводным Na2SO4, концентрировали и затем очищали колоночной хроматографией (РЕ/ЕА=1/5) с получением 0,705 г соединения 1-04-А и 1,500 г неочищенного соединения 1-04-С.
5. Соединение 1-05
Смесь неочищенного соединения 1-04-С (0,200 г), бис(пинаколато)дибора (0,270 г), трициклогексилфосфина (0,039 г), ацетата палладия (0,031 г) в DMSO (5 мл) перемешивали под азотом в течение 1 ч при 90°С в масляной бане. Полученную смесь экстрагировали ЕА (50 мл), объединенную органическую фазу промывали насыщенным раствором NaCl (20 мл × 3), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 0,398 г неочищенного Соединения 1-05.
6. Соединение 2-01
Смесь неочищенного соединения 1-05 (0,398 г), 2,4-дихлорпиримидина (0,304 г), K2CO3 (0,502 г) и Pd(dppf)Cl2⋅DCM (0,050 г) в 1,4-диоксане (10 мл) и воде (1 мл) перемешивали под азотом в течение 80 мин при 60°С в масляной бане. В полученную смесь добавляли воду (10 мл) и затем экстрагировали ЕА (20 мл × 2), объединенную органическую фазу промывали насыщенным раствором NaCl (20 мл × 1), сушили над безводным Na2SO4, концентрировали и затем очищали колоночной хроматографией (РЕ/ЕА=1/1) с получением 0,165 г Соединения 2-01.
7. Соединение 2
Смесь соединения 2-01 (0,030 г), Промежуточного соединения М5 (0,025 г), Cs2CO3 (0,067 г), Xanphos (0,005 г) и Pd2(dba)3 (0,005 г) в 1,4-диоксане (2 мл) перемешивали под азотом в течение 3,5 ч при 110°С в масляной бане. В полученную смесь добавляли 20 мл смеси растворителей (DCM/MeOH=10/1), затем фильтровали, концентрировали и очищали колоночной хроматографией (DCM/MeOH=8/1). Твердое вещество промывали 1,4-диоксаном (5 мл) с получением 0,024 г Соединения 2.
МС(ЭРИ+):m/z=519,3(М+Н)+.
Н-ЯМР (CDCl3): δ 8.587 (s, 1H, NH), 8.448-8.457 (d, 1H, CH), 8.383-8.405 (d, 1H, CH), 8.280-8.286 (d, 1H, CH), 8.009-8.013 (d, 1H, CH), 7.804-7.837 (dd, 1H, CH), 7.6867.714 (dd, 1H, CH), 4.681-4.728 (m, 1H, CH), 3.532 (s, 2H, CH2), 3.189-3.256 (m, 1H, CH2), 3.008-3.091 (m, 1H, CH2), 2.654 (s, 8H, CH2), 2.571-2.608 (m, 2H, CH2), 2.226-2.299 (m, 1H, CH2), 2.111-2.15 (m, 1H, CH2), 2.012-2.053 (m, 2H, CH2), 1.596-1.612 (d, 3H, CH3), 1.174-1.210 (t, 3H, CH3).
Пример 2-1. Хиральное разделение Соединения 2-01
В этом воплощении неочищенное Соединение 2-01 из Примера 2 очищали напрямую на хиральной колонке с получением соединения 2-01-А и соединения 2-01-В в нижеследующих условиях.
Условия хиральной ВЭЖХ:

Пример 2-2. Синтез Соединения 2а и Соединения 2b
Неочищенное Соединение 2-01 очищают на хиральной колонке в вышеуказанных условиях с получением Соединения 2-01-А и Соединения 2-01-В.
Получить Соединение 2а и Соединение 2b, в сущности как описано для стадии 7 Примера 2, с использованием Соединения 2-01-А и Соединения 2-01-В соответственно.
Кроме того, оптическое вращение измеряли 3 раза для каждого соединения, как показано ниже, на поляриметре Rudolf.
Условия:
Результаты:

Получить соединения нижеследующих примеров (показаны в Таблице 8), в сущности как описано для Примера 2, с использованием соответствующих промежуточных соединений. При этом два энантиомера каждого соединения разделяют на хиральной колонке, затем тестируют их оптическое вращение соответственно, в сущности как описано в Примере 2-2.



Пример 15: Синтез Соединения 15b

1. Соединение 15-01
К смеси (-)-энантиомера 5-метилморфолин-3-она (0,200 г) и 4-бром-2,6-дифторанилина (0,210 г) в метилбензоле (20 мл) добавляли POCl3 (0,510 г) и нагревали в масляной бане. Когда температура поднялась до 110°С, добавляли TEA (0,210 г), полученную смесь подвергали взаимодействию при 110°С в течение 40 мин. Часть метилбензола удаляли, доводили рН до 8-9 добавлением Na2CO3 после добавления воды (10 мл). Затем экстрагировали ЕА (20 мл × 2), объединенную органическую фазу промывали насыщенным раствором NaCl (20 мл × 1), сушили над безводным Na2SO4 и концентрировали с получением 0,280 г Соединения 15-01.
2. Соединение 15-02
К смеси Соединения 15-01 (0,275 г) в DMF (5 мл) добавляли трет-бутоксид калия (0,482 г), затем перемешивали в течение 30 мин при 70°С в масляной бане. Полученную смесь экстрагировали ЕА (20 мл × 3), объединенную органическую фазу промывали насыщенным раствором NaCl (30 мл × 4), сушили над безводным Na2SO4, концентрировали и очищали колоночной хроматографией (РЕ/ЕА=5/1) с получением 0,260 г Соединения 15-02.
3. Соединение 15-03
Смесь Соединения 15-02 (0,260 г), бис(пинаколато)дибора (0,320 г), трициклогексилфосфина (0,050 г), ацетата палладия (0,040 г) в DMSO (6 мл) перемешивали под азотом в течение 50 мин при 90°С в масляной бане. Полученную смесь экстрагировали ЕА (20 мл × 2), объединенную органическую фазу промывали насыщенным раствором NaCl (10 мл × 3), сушили над безводным Na2SO4 и концентрировали с получением 0,258 г Соединения 15-03.
4. Соединение 15-04
Смесь Соединения 15-03 (0,120 г), 2,4-дихлор-5-фторпиримидина (0,060 г), K2CO3 (0,099 г) и Pd(dppf)Cl2⋅DCM (0,012 г) в 1,4-диоксане (10 мл) и воде (1 мл) перемешивали под азотом в течение 1 ч при 60°С в масляной бане. Полученную смесь экстрагировали ЕА (20 мл × 2), объединенную органическую фазу промывали насыщенным раствором NaCl (20 мл × 1), сушили над безводным Na2SO4, концентрировали и затем очищали препаративной ВЭЖХ (DCM/MeOH=30/1) с получением 0,052 г (-)-энантиомера Соединения 15-04.
5. Соединение 15b
(-)-Энантиомер Соединения 15-04 (20 мг), промежуточное соединение M1 (0,017 г), Cs2CO3 (0,056 г), Xanphos (0,005 г) и Pd2(dba)3 (0,005 г) в 1,4-диоксане (2 мл) подвергали взаимодействию под воздействием микроволнового излучения при 110°С в течение 1,5 ч в потоке азота. В полученную смесь добавляли 20 мл смеси растворителей (DCM/MeOH=10/1), фильтрат собирали фильтрованием, затем концентрировали, очищали препаративной ВЭЖХ (DCM/MeOH=20/1), полученное твердое вещество промывали н-гексаном (10 мл) с получением 0,014 г Соединения 15b.
МС(ЭРИ+):m/z=493,2(М+Н)+.
Н-ЯМР (CDCl3): δ 8.409-8.418 (d, 1H, CH), 8.211-8.288 (m, 2H, CH), 8.070-8.077 (d, 1H, CH), 8.019-8.023 (d, 1H, CH), 7.833-7.868 (m, 1H, CH), 5.118-5.158 (d, 1H, CH2), 4.912-5.002 (d, 1H, CH2), 4.522-4.584 (m, 1H, CH), 4.111-4.168 (m, 1H, CH2), 4.039-4.075 (m, 1H, CH2), 3.249-3.275 (m, 4H, CH2), 2.724-2.748 (m, 4H, CH2), 2.462 (s, 3H, CH3), 1.662-1.678 (d, 3H, CH3).
Пример 15-1. Синтез Соединения 15а и Соединения 15
Соединение 15а и Соединение 15b являются энантиомерами. Получить Соединение 15а получают, в сущности как описано для соединения Примера 15, с использованием (+)-энантиомера 5-метилморфолин-3-она в качестве исходного вещества.
В другом воплощении в качестве исходного вещества используют неочищенный 5-метилморфолин-3-он; неочищенное соединение 15, включающее в себя Соединение 15а и Соединение 15b, будет получено в конце.
Кроме того, оптическое вращение измеряли 3 раза для каждого соединения, как показано ниже, на поляриметре Rudolf.
Условия:

Результаты:
Получить соединения нижеследующих примеров (показаны в Таблице 9), в сущности как описано для соединения Примера 15, с использованием соответствующих промежуточных соединений и с использованием (+) и/или (-)-энантиомера 5-метилморфолин-3-она в качестве исходного вещества. Их оптическое вращение тестировали, в сущности как описано для соединения Примера 15-1.





ПРИМЕРЫ ДЛЯ СРАВНЕНИЯ
Получить соединения нижеследующих примеров сравнения, в сущности как описано для соединения Примера 1, 2 или 15, с использованием соответствующих промежуточных соединений или исходных веществ. Например, получить соединения нижеследующих примеров сравнения 8, 9 и 10 (показаны в Таблице 10), в сущности как описано для соединения Примера 2, с использованием  вместо
вместо  . И получить соединение нижеследующего примера сравнения 7, в сущности как описано для соединения Примера 1, с использованием
. И получить соединение нижеследующего примера сравнения 7, в сущности как описано для соединения Примера 1, с использованием  вместо
вместо 


В приведенной выше таблице соединения Пр. сравн. 3b и 4b синтезируют, в сущности как описано для соединения Примера 15, и затем их оптическое вращение протестируют соответственно, в сущности как описано для соединения Примера 15-1. Соединения Пр. сравн. 3b и 4b оба показывают отрицательное оптическое вращение.
ФАРМАКОЛОГИЧЕСКОЕ ТЕСТИРОВАНИЕ
Результаты нижеследующих анализов демонстрируют доказательство, что соединения, проиллюстрированные примерами в данном документе, полезны в качестве специфических ингибиторов CDK4/6 и в качестве противораковых агентов. В данном документе "IC50" относится к концентрации агента, который продуцирует 50% от максимального ингибирующего ответа, возможного для агента.
Для наглядной иллюстрации общая структура показана ниже. Неожиданно, авторы изобретения обнаружили, что "R" оказывает решающее влияние на биологическую активность, селективность и безопасность.
Тест 1. Сравнение разных заместителей посредством анализа пролиферации клеток
Воздействие тестируемых соединений на пролиферацию in vitro определяли посредством анализа жизнеспособности клеток с использованием MTS (3-(4,5-диметилтиазол-2-ил)-5-(3-карбоксиметоксифенил)-2-(4-сульфофенил)-2Н-тетразолий).
Культура клеток
Клетки колоректального рака человека (colo-205) размножают в культуре (colo-205 выращивают в среде DMEM с 12% FBS, 1% P/S и 1% L-глутамина).
Анализ жизнеспособности клеток с использованием MTS:
1. Произвести посев клеток при плотности 4×103 клеток на лунку в 96-луночные планшеты, выращивать в течение 24 ч.
2. Добавить к клеткам тестируемые соединения в различных концентрациях.
3. Инкубировать в течение 7 суток воздействия.
4. Приготовить реагенты, следуя инструкциям в наборе для анализа пролиферации клеток (Promega).
5. Заменить на среду, не содержащую сыворотку, с конечным объемом 100 мкл на лунку. Приготовить комплект лунок только со средой для вычитания фона.
6. Добавить в каждую лунку 20 мкл раствора MTS, содержащего PMS (конечная концентрация MTS будет составлять 0,33 мг/мл).
7. Инкубировать от 1 до 4 ч при 37°С во влажной атмосфере с 5% CO2.
8. Зарегистрировать поглощение при 490 нм с использованием планшет-ридера VICTOR™X5 (PerkinElmer).
Все экспериментальные точки создавали в трех лунках, и все эксперименты повторяли по меньшей мере три раза. Значение IC50 определяли из кривой доза-ответ, используя программное обеспечение (Graphpad prism 6), и результаты показаны в Таблице 11.

В этой модели соединения, проиллюстрированные приведенными выше примерами, проявляют противоопухолевую активность, как показано в Таблице 11, демонстрируя тем самым, что проиллюстрированные примерами соединения по настоящему изобретению обладают более мощной активностью in vivo по отношению к Rb+ опухолям. По сравнению с известным соединением LY2835219 (абемациклиб) соединение по настоящему изобретению, например соединение Примера 1 или 2, оказывает более сильное ингибирующее действие по отношению к Rb+ опухолям. По сравнению с соединением Примера сравнения 1 (упоминаемого здесь далее как Пр. сравн. 1; R представляет собой спиро) и с соединением Примера сравнения 2 (упоминаемого здесь далее как Пр. сравн. 2; R представляет собой Н) соединение по настоящему изобретению, например соединение Примера 1 или 2 (R представляет собой метил), оказывает намного более сильное ингибирующее действие по отношению к Rb+ опухолям.
Проиллюстрированные приведенными выше примерами соединения также демонстрируют, что соединение, где R представляет собой метил, а не Н или спиро, обладает намного более мощной биологической активностью в этой модели. Из приведенных выше результатов следует, что тип заместителей имеет значительное влияние на ингибирование по отношению к Rb+ опухолям.
Тест 2. Сравнение разных заместителей посредством тестирования на безопасность
Выбранные соединения, полученные, как описано выше, анализировали на безопасность исходя из изменения массы тела и исходя из того, имела ли место смертность, и по методикам, описанным в данном документе. Тестируемое соединение подготавливают в подходящем носителе и вводят мышам BALB/c (22-23 г) перорально через желудочный зонд. Массу тела и смертность принимают за общий показатель токсичности. Потерю массы тела (% измененной массы тела) вычисляют дважды в неделю путем сравнения групп, подвергавшихся лечению, с контрольной группой, получавшей носитель, во время курса лечения. Соединение 2 и Соединение 16 демонстрируют почти несущественную потерю массы тела в этих моделях при введении доз 200 мг/кг (qd (один раз в сутки)). Но в такой же дозировке соединения Примеров сравнения, например Пр. сравн. 3b и Пр. сравн. 4b, способны вызывать намного большую потерю массы тела и даже приводить к высокой смертности с гибелью 4 и 5 мышей в каждой группе (6 мышей) после лечения в течение 2 недель.
Результаты приведены в Таблице 12. "*" означает "потерю массы тела менее 5%"; "**" означает "потерю массы тела более 5% и менее 10%"; "***" означает "потерю массы тела более 10% и менее 30%"; "****" означает "потерю массы тела более 30%". "+" означает "смертность имела место"; "-" означает "смертность отсутствовала".
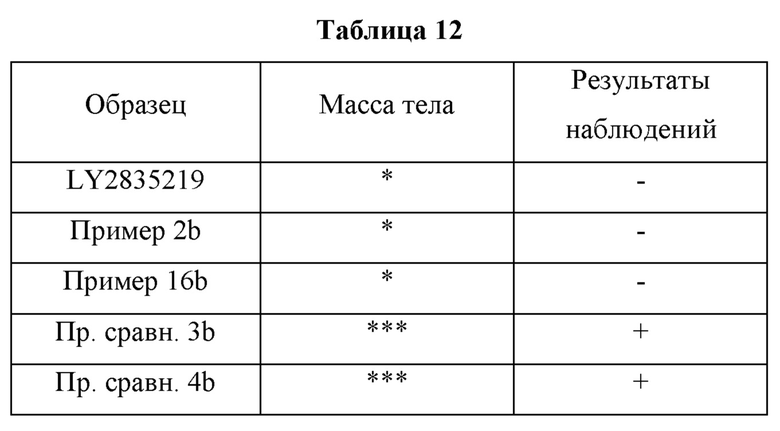
Неожиданно, авторы изобретения обнаружили, что соединение Пр. сравн. 3b (R представляет собой изопропил) или соединение Пр. сравн. 4b (R представляет собой этил) имеют больше побочных эффектов и токсичность. Однако проиллюстрированные примерами соединения по настоящему изобретению, например соединение 2b или 16b (R представляет собой метил), более безопасны, демонстрируя таким образом, что тип заместителей оказывает значительное влияние на безопасность.
Тест 3. Влияние количества заместителей посредством анализов CDK киназ
Чтобы продемонстрировать, что соединения проявляют аффинность к CDK киназам (CDK2/CycA2, CDK4/CycD3, CDK6/cycD3), были выполнены анализы CDK киназ.
Были приготовлены следующие реакционные буферы: киназный базовый буфер для CDK2,6 (50 мМ HEPES, рН 7,5; 0,0015% Brij-35; 10 мМ MgCl2; 2 мМ DTT); киназный базовый буфер для CDK4 (20 мМ HEPES, рН 7,5; 0,01% Triton Х-100; 10 мМ MgCl2; 2 мМ DTT); стоп-буфер (100 мМ HEPES, рН 7,5; 0,015% Brij-35; 0,2% покровного реагента #3; 50 мМ EDTA).
Протокол ферментативной реакции:
1) Разбавить соединение до 50Х конечной желаемой наивысшей концентрации в реакционной смеси 100%-ным DMSO. Перенести 100 мкл этого разбавленного соединения в лунку в 96-луночном планшете. Затем выполнить последовательное разведение соединения путем переноса 30 мкл в 60 мкл 100%-ного DMSO в следующей лунке и так далее с получением в сумме 10 концентраций. Добавить 100 мкл 100%-ного DMSO в две пустые лунки для создания контроля без соединения и контроля без фермента в том же 96-луночном планшете. Маркировать планшет как исходный планшет.
2) Приготовить промежуточный планшет путем переноса 10 мкл соединения из исходного планшета в новый 96-луночный планшет, содержащий 90 мкл киназного буфера, в качестве промежуточного планшета.
3) Перенести 5 мкл соединения из 96-луночного промежуточного планшета в 384-луночный планшет в двух повторах.
4) Добавить 10 мкл 2,5х раствора фермента в каждую лунку 384-луночного планшета для анализа.
5) Инкубировать при комнатной температуре в течение 10 мин.
6) Добавить 10 мкл 2,5х раствора субстрата, приготовленного путем добавления FAM-меченого пептида и АТР в киназном базовом буфере. Реакционные концентрации для ферментов и субстратов приведены в нижеследующей таблице (Таблица 13).

7) Инкубировать при 28°С в течение конкретно указанного периода времени.
8) Добавить 25 мкл стоп-буфера для остановки реакции.
9) Собрать данные на Caliper. Затем преобразовать значения конверсии в значения ингибирования.
Процент ингибирования = (макс-конверсия)/(макс-мин)*100.
Здесь "макс" означает DMSO-контроль, и "мин" означает низший контроль.
10) Произвести выравнивание кривых с использованием процента ингибирования в XLFit excel add-in version 4.3.1 для получения значений IC50. Используемое уравнение: Y = минимум + (максимум - минимум/(1 + (IC50/X) ∧ угловой коэффициент Хилла), где Y означает процент ингибирования (%); X означает концентрацию тестируемого соединения.
Результаты выражены в виде значения IC50, которое показано в Таблице 14.

Как показано в приведенной выше таблице, количество метальных групп оказывает весьма значительное влияние на селективность. Неожиданно, проиллюстрированные примерами соединения по настоящему изобретению демонстрируют IC50 > 0,3 мкМ в вышеуказанном анализе ингибирования киназы CDK2 и IC50 < 0,04 мкМ в вышеуказанном анализе ингибирования киназ CDK4/6, как показано в Таблице 14. Это свидетельствует о том, что проиллюстрированные примерами соединения по настоящему изобретению являются в большей степени селективными ингибиторами активности киназ CDK4/6. Таким образом, показано, что проиллюстрированные примерами соединения (R представляет собой только один метил) являются в большей степени специфическими ингибиторами CDK4/6 по сравнению с известным соединением LY2835219 и соединениями примеров сравнения, например Пр. сравн. 5 или Пр. сравн. 6 (R представляет собой два метила).
Тест 4. Влияние местоположения заместителей
Выбранное соединение, полученное, как описано выше, анализировали согласно биологическим методикам, описанным в данном документе. Результаты показаны в приведенной ниже таблице.
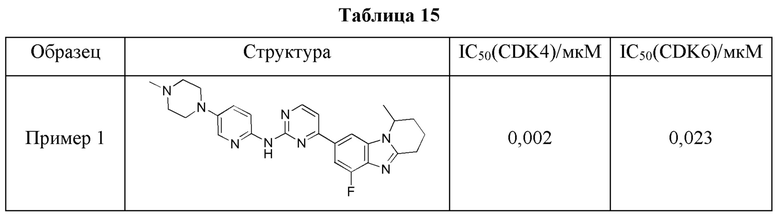

Дополнительно, прямое сравнение соединения Примера 2 и соединения Примера сравнения 8, 9 или 10 показано в нижеследующей таблице. Результаты приведены в Таблице 16. "++++" означает значение "IC50 меньше 0,2 мкМ"; "+++" означает значение "IC50 больше 0,2 мкМ и меньше 1,0 мкМ"; "++" означает значение "IC50 больше 1,0 мкМ и меньше 2,0 мкМ"; "+" означает значение "IC50 больше 2,0 мкМ".
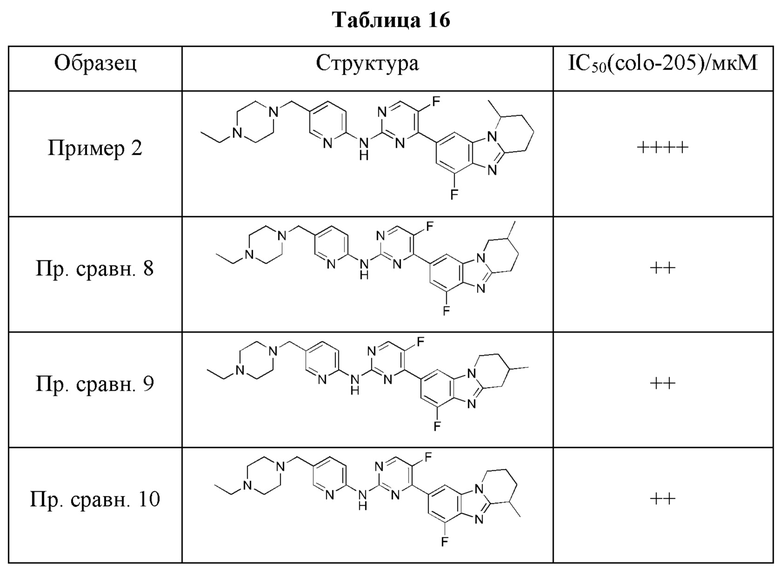
Как показано в приведенных выше таблицах 15 и 16, местоположение заместителей также играет важную роль в биологической активности. Влияние на биологическую активность в связи с изменением местоположения заместителей является в значительной степени неожиданным.
Как видно из результатов, представленных выше, "R" 6-членного гетероциклического кольца оказывает решающее влияние на биологическую активность, селективность и безопасность. Неожиданно, когда имеется один и только один метил на указанном 6-членном гетероциклическом кольце, тогда достигаются по меньшей мере следующие эффекты:
 повышенная биологическая активность;
повышенная биологическая активность;
 хорошая селективность; и
хорошая селективность; и
 низкие побочные эффекты.
низкие побочные эффекты.
Тест 5. Влияние оптической активности
Выбранные соединения, полученные, как описано выше, анализировали согласно биологическим методикам, описанным как Тест 3 (анализы CDK киназ). Результаты показаны в приведенной ниже таблице.

Как показано в приведенной выше таблице, соединения 1b, 2b, 15b, 16b являются более сильнодействующими, чем соединения 1а, 2а, 15а, 16а соответственно, в ингибировании CDK4/6, демонстрируя тем самым, что (-)-энантиомеры соединений по настоящему изобретению имеют преимущество по сравнению с (+)-энантиомерами.
В некоторых случаях вводят раскрытое в данном документе соединение, где один энантиомер [например, (-)-энантиомер или (+)-энантиомер] присутствует в большом энантиомерном избытке. В одном случае энантиомер соединения 1b, имеющий отрицательное оптическое вращение, например -35,394° (с=3,0 мг/мл, EtOH), обладает более высокой активностью по отношению к ферменту CDK4/6, чем энантиомер (соединение 1а), который имеет положительное оптическое вращение +30,325° (с=3,0 мг/мл, EtOH). В другом случае энантиомер соединения 2b, имеющий отрицательное оптическое вращение, например -32,036° (с=4,0 мг/мл, DCM), имеет более высокую активность по отношению к ферменту CDK4/6, чем энантиомер, который имеет положительное оптическое вращение +38,088° (с=4,0 мг/мл, DCM). В других случаях энантиомер соединения 15b, имеющий отрицательное оптическое вращение, например -28,929° (с=4,6 мг/мл, DCM/MeOH=1:1), имеет более высокую активность по отношению к ферменту CDK4/6, чем энантиомер, который имеет положительное оптическое вращение +32,829° (с=4,6 мг/мл, DCM/MeOH=1:1).
Тест 6. Тест на ингибирующую активность и селективность на других подтипах CDK киназ на молекулярном уровне
Репрезентативное соединение 2b по настоящему изобретению использовали в качестве тестируемого соединения и сравнивали с положительным контролем, лекарственным средством LY2835219 (абемациклиб), для сравнения ингибирующей активности по отношению к CDK киназам и селективной специфичности между ними.
Механизм этого метода показан в формуле (II). Киназа катализирует фосфорилирование белкового субстрата с переносом 33Р на 33Р-меченом АТР (γ-33Р-АТР) на белковый субстрат в реакционной системе; реакционную систему наносили на ионообменную мембрану Р81, и эту мембрану тщательно промывали 0,75% фосфатным буфером; фосфорилированный радиоактивным изотопом субстрат оставляли на мембране, и активность киназы определяли, регистрируя интенсивность радиоактивной метки белка-субстрата.

Данные обрабатывали с использованием программного обеспечения Prism4 Software (GraphPad), и формула выравнивания кривой была следующая:
Y = минимум + (максимум - минимум)/(1 + 10 ∧ ((LogIC50-X) * угловой коэффициент Хилла)), где Y означает процент ингибирования (%); X означает логарифм концентрации ингибитора.
Результаты: В результате скрининга различных CDK киназ было обнаружено, что репрезентативные соединения 2, 2а и 2b имеют IC50 больше 0,4 мкМ в ингибировании CDK1/2/7/9, которая в десятки-тысячи раз выше, чем IC50 в ингибировании CDK4/6 (см. Таблицу 18).

Заключение: На молекулярном уровне репрезентативные соединения 2 и 2b по настоящему изобретению показали сильное ингибирующее воздействие на CDK4/6 и слабое ингибирующее воздействие на CDK1/2/7/9, что свидетельствует о том, что соединение 2 и соединение 2b являются ингибиторами киназ CDK4/6 с великолепной селективностью. Соединение 2а показало сильное ингибирующее воздействие на CDK4, слабое ингибирующее воздействие на CDK6 и очень слабое ингибирующее воздействие на CDK1/2/7/9, что свидетельствует о том, что соединение 2а является ингибитором киназы CDK4 с чрезвычайно высокой селективностью и ингибитором киназы CDK6 с хорошей селективностью. Дополнительно, селективность репрезентативных соединений по настоящему изобретению между CDK1/2/9 и CDK4/6 была значительно выше, чем селективность LY2835219 (абемациклиба).
Тест 7. Эффект регрессии опухоли на модели животного с ксенотранстплантатом JeKo-1
Клетки JeKo-1 культивировали в среде RPMI 1640, содержащей 20% фетальной коровьей сыворотки. Экспоненциально растущие клетки JeKo-1 собирали и ресуспендировали в PBS (забуференный фосфатами физиологический раствор) до подходящей концентрации для подкожной инокуляции опухоли мышам NOD/SCID. Семидесяти самкам мышей инокулировали подкожно справа 5×106 клеток JeKo-1, ресуспендированных в PBS и матригеле (1:1). Когда средний объем опухоли достигал 134 мм, мышей произвольно распределяли по группам согласно размерам опухоли и осуществляли введение. Сорок восемь мышей были поделены на экспериментальные группы, а остальные двадцать две мыши не использовались для эксперимента. Объем опухоли вычисляли следующим образом: длинный диаметр х короткий диаметр2/2. Тестирование подразделялось на группу контрольного растворителя, тестируемого лекарственного репрезентативного соединения 2b (10 мг/кг), тестируемого лекарственного репрезентативного соединения 2b (25 мг/кг), тестируемого лекарственного репрезентативного соединения 2b (50 мг/кг), тестируемого лекарственного репрезентативного соединения 2b (100 мг/кг), в сумме 6 групп по 8 мышей в каждой, и введение мышам осуществляли перорально посредством желудочного зонда один раз в сутки, и затем непрерывное введение в течение 19 суток. Эффективность оценивают по относительной скорости ингибирования роста опухоли (TGI).
Формула для вычисления следующая: TGI (%) = (С-Т)/С × 100% (С и Т означают среднюю массу опухоли в группе контрольного растворителя и среднюю массу опухоли в группе лечения соответственно). Чем выше значение TGI (%), тем выше эффективность, и наоборот.
Результаты: Соединение 2b демонстрирует великолепную противоопухолевую активность.

Соответственно, в некоторых случаях для лечения заболевания полезно вводить субъекту соединение 1, 2 или 15, имеющее высокий энантиомерный избыток энантиомера, имеющего отрицательное оптическое вращение. Неожиданно оказалось, что оптически чистый (-)-энантиомер соединения по настоящему изобретению, в том числе, без ограничения, соединение 1b, 2b или 15b, является более эффективным лекарственным средством для лечения заболевания, опосредованного CDK4/6, у субъекта.
Описан целый ряд воплощений изобретения. Тем не менее, следует понимать, что могут быть сделаны различные модификации без отклонения от замысла и объема изобретения. Другие воплощения охвачены формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОР НЕКОТОРЫХ ПРОТЕИНКИНАЗ | 2016 |
|
RU2732952C2 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ с-MET | 2013 |
|
RU2619130C2 |
| ИМИДАЗОПИРИМИДИНЫ КАК ИНГИБИТОРЫ EED И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2836176C2 |
| ЗАМЕЩЕННЫЕ 1,2-ДИГИДРО-3Н-ПИРАЗОЛО[3,4-D]ПИРИМИДИН-3-ОНЫ | 2019 |
|
RU2812726C2 |
| НЕКОТОРЫЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ | 2015 |
|
RU2671494C2 |
| ПРОИЗВОДНЫЕ 6-(ПИРИМИДИНОАМИНОПИРИДИН)БЕНЗОИМИДАЗОЛА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2014 |
|
RU2670762C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОПИРИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ HDM2 | 2013 |
|
RU2690663C2 |
| Макроциклический ингибитор тирозинкиназы и его применение | 2019 |
|
RU2798231C2 |
| БИЦИКЛИЧЕСКИЕ НИТРОИМИДАЗОЛЫ, КОВАЛЕНТНО СОЕДИНЕННЫЕ С ЗАМЕЩЕННЫМИ ФЕНИЛОКСАЗОЛИДИНОНАМИ | 2009 |
|
RU2504547C2 |
| НЕКОТОРЫЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ | 2017 |
|
RU2738837C2 |
Группа изобретений относится к фармацевтической химии и включает соединение формулы (I), его энантиомеры или фармацевтически приемлемые соли, 4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-a]пиридин-8-ил)-N-(5-(4-метилпиперазин-1-ил)пиридин-2-ил)пиримидин-2-амин, N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-a]пиридин-8-ил)пиримидин-2-амин, фармацевтическую композицию на их основе и ее применение, а также способ лечения заболевания, опосредованного CDK4 и/или CDK6 у субъекта. В формуле (I) A выбран из пиридила, пиримидинила и пиридазинила; Z представляет собой NH или O; R1 независимо выбран из группы, состоящей из гетероциклила, группы гетероциклил-(CH2)m-, -NR12R13, -NR12-C1-6алкилен-NR12R13 и гетероциклил-C(O)-, где каждый гетероциклил является 5-6-членным и выбран из пирролидинила, пиперидинила и пиперазинила и может быть замещен 1-2 заместителями, выбранными из гидроксила, C1-8алкила, C3-8циклоалкила, оксетанила,  -NR12R13 или -(CH2)t-OH; каждый R2 и R3 независимо выбран из H, галогена, C1-8алкила, где C1-8алкил может быть замещен 1-3 атомами галогенов; каждый R12 и R13 независимо выбран из C1-8алкила, который может быть замещен одним гидроксилом; m равно 0 или 1; n и t равны 1. Технический результат – производные бензимидазола, проявляющие свойства ингибиторов CDK4 и/или CDK6. 4 н. и 26 з.п. ф-лы, 19 табл., 7 пр.
-NR12R13 или -(CH2)t-OH; каждый R2 и R3 независимо выбран из H, галогена, C1-8алкила, где C1-8алкил может быть замещен 1-3 атомами галогенов; каждый R12 и R13 независимо выбран из C1-8алкила, который может быть замещен одним гидроксилом; m равно 0 или 1; n и t равны 1. Технический результат – производные бензимидазола, проявляющие свойства ингибиторов CDK4 и/или CDK6. 4 н. и 26 з.п. ф-лы, 19 табл., 7 пр.

1. Соединение формулы I или его энантиомер, фармацевтически приемлемая соль
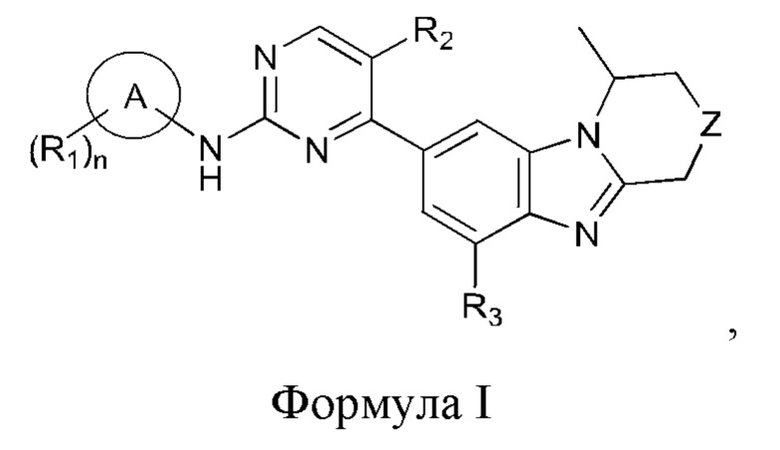
где
кольцо A представляет собой 6-членный гетероарил, который содержит один или два гетероатома N и выбран из пиридила, пиримидинила и пиридазинила;
Z выбран из группы, состоящей из NH и O;
R1 независимо выбран из группы, состоящей из гетероциклила, группы гетероциклил-(CH2)m-, -NR12R13, -NR12-C1-6алкилен-NR12R13 и гетероциклил-C(O)-, где каждый гетероциклил является 5-6-членным и выбран из пирролидинила, пиперидинила и пиперазинила и может быть замещен 1-2 заместителями, выбранными из гидроксила, C1-8алкила, C3-8циклоалкила, оксетанила, 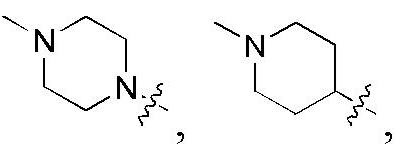 -NR12R13 или -(CH2)t-OH;
-NR12R13 или -(CH2)t-OH;
каждый из R2 и R3 независимо выбран из H, галогена, C1-8алкила, где C1-8алкил может быть замещен 1-3 атомами галогенов;
каждый из R12 и R13 независимо выбран из C1-8алкила, который может быть замещен одним гидроксилом;
m равно 0 или 1;
n и t равны 1; или
4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-a]пиридин-8-ил)-N-(5-(4-метилпиперазин-1-ил)пиридин-2-ил)пиримидин-2-амин, или
N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-a]пиридин-8-ил)пиримидин-2-амин.
2. Соединение по п. 1, где Z представляет собой O.
3. Соединение по п. 1 или 2, где кольцо A представляет собой 
4. Соединение по любому из пп. 1-3, где R1 представляет собой гетероциклил- (CH2)m- или гетероциклил-(CH2)m-, замещенный C1-8алкилом, -NR12R13, оксетанилом, C3-6циклоалкилом или (CH2)t-OH.
5. Соединение по любому из пп. 1-4, где R1 представляет собой 5-6-членный гетероциклил-CH2- или 5-6-членный гетероциклил-CH2-, замещенный C1-3алкилом, - N(CH3)2, -N(CH2CH2OH)CH3,  -CH2OH или OH.
-CH2OH или OH.
6. Соединение по любому из пп. 1-5, где R1 представляет собой 6-членный гетероциклил-CH2- или 6-членный гетероциклил-CH2-, замещенный метилом или этилом.
7. Соединение по любому из пп. 1-3, где R1 представляет собой гетероциклил или гетероциклил, замещенный C1-8алкилом, -NR12R13, оксетанилом, C3-6циклоалкилом или (CH2)t-OH.
8. Соединение по любому из пп. 1-3 или 7, где R1 представляет собой 5-6-членный гетероциклил или 5-6-членный гетероциклил, замещенный C1-3алкилом, - N(CH3)2, -N(CH2CH2OH)CH3, -CH2OH или OH.
9. Соединение по любому из пп. 1-3, 7 или 8, где R1 представляет собой 6-членный гетероциклил или 6-членный гетероциклил, замещенный метилом или этилом.
10. Соединение по любому из пп. 1-3, где R1 представляет собой 6-членный гетероциклил-C(O)- или 6-членный гетероциклил-C(O)-, замещенный C1-3алкилом.
11. Соединение по любому из пп. 1-3 или 10, где R1 представляет собой 6-членный гетероциклил-C(O)-, замещенный метилом.
12. Соединение по любому из пп. 1-3, где R1 представляет собой -NR12-C1- 3алкилен-NR12R13.
13. Соединение по любому из пп. 1-12, где R12 и R13, каждый независимо, представляют собой CH2CH2OH или C1-3алкил.
14. Соединение по любому из пп. 1-13, где R12 и R13, каждый независимо, представляют собой CH2CH2OH, метил или этил.
15. Соединение по п. 1, где R1 представляет собой 

16. Соединение по любому из пп. 1-15, где m равно 1.
17. Соединение по любому из пп. 1-15, где R2 и R3, каждый независимо, представляют собой H, галоген, C1-6алкил, C1-6алкил, замещенный галогеном.
18. Соединение по любому из пп. 1-16, где R2 и R3, каждый независимо, представляют собой H, F, Cl, CH3, CH2CH3, CF3.
19. Соединение по любому из пп. 1-17, где R2 и R3 оба представляют собой F.
20. Соединение по п. 1, представляющее собой:
1) 4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-a]пиридин-8-ил)-N-(5-(4-метилпиперазин-1-ил)пиридин-2-ил)пиримидин-2-амин;
2) N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(6-фтор-1-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-a]пиридин-8-ил)пиримидин-2-амин;
3) 5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1H-бензо[4,5]имидазо[2,1- c][1,4]оксазин-7-ил)-N-(5-(4-метилпиперазин-1-ил)пиридин-2-ил)пиримидин-2-амин;
4) N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(9-фтор-4-метил- 3,4-дигидро-1H-бензо[4,5]имидазо[2,1-c][1,4]оксазин-7-ил)пиримидин-2-амин;
5) N-(5-(4-(диметиламино)пиперидин-1-ил)пиридин-2-ил)-5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1H-бензо[4,5]имидазо[2,1-c][1,4]оксазин-7-ил)пиримидин-2-амин;
6) N-(5-((4-(диметиламино)пиперидин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1H-бензо[4,5]имидазо[2,1-c][1,4]оксазин-7-ил)пиримидин-2-амин;
7) 5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1H-бензо[4,5]имидазо[2,1-c][1,4]оксазин-7-ил)-N-(5-(пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амин;
8) 5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1H-бензо[4,5]имидазо[2,1-c][1,4]оксазин-7-ил)-N-(5-(пиперазин-1-илметил)пиридин-2-ил)пиримидин-2-амин;
9) N-(5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1H-бензо[4,5]имидазо[2,1-c][1,4]оксазин-7-ил)пиримидин-2-ил)-6-(4-метилпиперазин-1-ил)пиридазин-3-амин;
10) 6-((4-этилпиперазин-1-ил)метил)-N-(5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1H-бензо[4,5]имидазо[2,1-c][1,4]оксазин-7-ил)пиримидин-2-ил)пиридазин-3-амин;
11) (1-(6-((5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1H-бензо[4,5]имидазо[2,1-c][1,4]оксазин-7-ил)пиримидин-2-ил)амино)пиридин-3-ил)пирролидин-3-ил)метанол;
12) (1-((6-((5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1H-бензо[4,5]имидазо[2,1-c][1,4]оксазин-7-ил)пиримидин-2-ил)амино)пиридин-3-ил)метил)пирролидин-3-ил)метанол;
13) N-(5-(4-циклопропилпиперазин-1-ил)пиридин-2-ил)-5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1H-бензо[4,5]имидазо[2,1-c][1,4]оксазин-7-ил)пиримидин-2-амин;
14) N-(5-((4-циклопропилпиперазин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1H-бензо[4,5]имидазо[2,1-c][1,4]оксазин-7-ил)пиримидин-2-амин;
15) 2-((1-((6-((5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1H-бензо[4,5]имидазо[2,1-c][1,4]оксазин-7-ил)пиримидин-2-ил)амино)пиридин-3-ил)метил)пиперидин-4-ил)(метил)амино)этан-1-ол;
16) 1-(6-((5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1H-бензо[4,5]имидазо[2,1-c][1,4]оксазин-7-ил)пиримидин-2-ил)амино)пиридин-3-ил)-3-метилпирролидин-3-ол;
17) 1-((6-((5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1H-бензо[4,5]имидазо[2,1-c][1,4]оксазин-7-ил)пиримидин-2-ил)амино)пиридин-3-ил)метил)-3-метилпирролидин-3-ол;
18) 5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1H-бензо[4,5]имидазо[2,1-c][1,4]оксазин-7-ил)-N-(5-((4-(оксетан-3-ил)пиперазин-1-ил)метил)пиридин-2-ил)пиримидин-2-амин;
19) 5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1H-бензо[4,5]имидазо[2,1-c][1,4]оксазин-7-ил)-N-(5-(4-(оксетан-3-ил)пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амин;
20) N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(9-фтор-4-метил-1,2,3,4-тетрагидробензо[4,5]имидазо[1,2-a]пиразин-7-ил)пиримидин-2-амин;
21) 5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1H-бензо[4,5]имидазо[2,1-c][1,4]оксазин-7-ил)-N-(5-((4'-метил-[1,1'-бипиперазин]-4-ил)метил)пиридин-2-ил)пиримидин-2-амин;
22) 5-фтор-4-(9-фтор-4-метил-3,4-дигидро-1H-бензо[4,5]имидазо[2,1-c][1,4]оксазин-7-ил)-N-(5-((4-(1-метилпиперидин-4-ил)пиперазин-1-ил)метил)пиридин-2-ил)пиримидин-2-амин.
21. Соединение по любому из пп. 1-20, где соединение представляет собой (-)-энантиомер соединения.
22. Соединение по любому из пп. 1-20, где соединение представляет собой (+)-энантиомер соединения.
23. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении CDK4 и/или CDK6, содержащая терапевтически эффективное количество соединения по любому из пп. 1-22 и фармацевтически приемлемый эксципиент.
24. Фармацевтическая композиция по п. 23, где указанное соединение в массовом отношении к указанному эксципиенту находится в диапазоне от примерно 0,001 до примерно 10.
25. Применение фармацевтической композиции по п. 23 или 24 или соединения по любому из пп. 1-22 для приготовления лекарственного средства для лечения рака, опосредованного CDK4 и/или CDK6.
26. Применение по п. 25, где рак представляет собой рак ободочной кишки, ректальный рак, мантийноклеточную лимфому, рак молочной железы, глиобластому, липосаркому, T-клеточную лимфому, меланому или рак легкого.
27. Способ лечения заболевания, опосредованного CDK4 и/или CDK6 у субъекта, включающий введение терапевтически эффективного количества соединения по любому из пп. 1-22 или фармацевтической композиции по п. 23 или 24.
28. Способ по п. 27, где заболевание, опосредованное CDK4 и/или CDK6, представляет собой рак.
29. Способ по п. 28, где рак представляет собой рак ободочной кишки, ректальный рак, мантийноклеточную лимфому, рак молочной железы, глиобластому, липосаркому, T-клеточную лимфому, меланому или рак легкого.
30. Способ по любому из пп. 27-29, где субъектом является человек.
| Токарный резец | 1924 |
|
SU2016A1 |
| Токарный резец | 1924 |
|
SU2016A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| CN 105153119 A, 16.12.2005 | |||
| Разрядная трубка | 1929 |
|
SU23263A1 |

