Область техники
Настоящее изобретение принадлежит к технической области лекарственных средств. В частности, настоящее изобретение относится к ингибитору CDK4/6 киназы или его фармацевтически приемлемой соли, сложному эфиру или сольвату или их стереоизомерам; фармацевтическому составу, фармацевтической композиции и набору, содержащим указанный ингибитор CDK4/6 киназы или его фармацевтически приемлемую соль, сложный эфир или сольват или их стереоизомеры; и применению указанного ингибитора CDK4/6 киназы и его фармацевтически приемлемой соли, сложного эфира или сольвата или их стереоизомеров. Например, соединения настоящего изобретения полезны для снижения или ингибирования активности CDK4/6 киназы в клетке и/или при лечении и/или предотвращении связанных с раком заболеваний, опосредованных CDK4/6 киназой.
Уровень техники
Образование опухолей связано с дисбалансом онкогенов и антионкогенов. Почти для всех онкогенов или антионкогенов, их функции и эффекты в конце устремляются к клеточному циклу. Таким образом, опухоли могут быть захвачены в виде клеточного цикла заболевания (CCD), и это является одним из путей лечения опухоли, регулируя или блокируя клеточный цикл. В настоящее время выяснилось, что существует много молекул, связанных с регуляцией клеточного цикла, среди которых циклин-зависимые киназы (CDK) являются основной молекулой клеточного цикла регуляторной сети. CDK, в качестве катализаторов субъединиц, представляют собой класс Ser/Thr киназ, которые участвуют в различных стадиях клеточного цикла в качестве важных сигнальных молекул в клетках. Исследования показывают, что в клетке цикла регуляторной сети с CDK в виде ядра, любые патологии приводят в результате к аномальному клеточному циклу и, в конечном результате, приводят к онкогенезу. Семейство CDK теперь имеет 21 изоформу, которые работают путем связывания с их субъединицей регуляторных циклинов. Кроме того, играя роль в регуляции клеточного цикла, изоформы CDK также участвуют в регулировании транскрипции, репарации ДНК, дифференциации и программировании клеточной смерти. Основываясь на ключевой роли CDK в регулировании пролиферации и гибели опухолевых клеток, семейство CDK предоставляет шанс и новое поле для обнаружения и усовершенствования противоопухолевых лекарственных средств.
В усовершенствовании лекарственных средств первое поколение ингибиторов CDK, представленное флавопиридолом, UCN-01 и тому подобное, обозначается как «pan-CDK» ингибиторы, которые блокируют все изоформы семейства CDK, эквивалентно и сравнительно проявляют высокую токсичность в клинических испытаниях, и некоторые из них не могут быть введены в терапевтически эффективном количестве. Таким образом, человечество начинает развивать селективные ингибиторы CDK для повышения селективности терапии и предохранения нормальных клеток от повреждений некоторыми побочными эффектами.
Среди изоформ CDK, участвующих в клеточном цикле, несомненную роль играет CDK4/6. Мутации, вязанные с раком клеточного цикла, главным образом присутствуют в фазе G1 и G1/S транзиции. Комплекс, образованный CDK4/6 и циклином D, относится к транскрипционному фактору E2F, связанному фосфорилированием (pRb) продукта антионкогена Rb, и вызывает транскрипцию генов, связанных с S-фазой, способствуя, тем самым, прохождению клетками контрольной точки клеточного цикла и транзита от фазы G1 в фазу S. Около 80% опухолей человека являются аномальными в пути циклина D-CDK4/6-INK4-Rb. Из-за изменения пути фаза G1 ускоряется так, что опухолевые клетки имеют ускоренную пролиферацию и, таким образом, приобретают преимущество выживания. Таким образом, интерференция пути стала стратегией лечения, и CDK4/6 стали новой мишенью против опухоли. CDK4/6 в качестве противоопухолевой мишени обладают следующими преимуществами: (1) для большинства пролиферативных клеток, их пролиферация является CDK2 или CDK4/6-зависимой, однако, ингибиторы CDK4/6 не проявляют цитотоксичности ингибиторов “pan-CDK”, такой как подавление деятельности костного мозга и кишечные реакции; и (2) преклинические испытания показывают, что если уровень циклина D увеличен или P16INK4a инактивирован в клетках, чувствительность клеток к лекарственным средствам может быть увеличена; поскольку опухолевые клетки имеют отношение к указанному феномену нормальных клеток, нацеливающие свойства лекарственных средств до некоторой степени увеличивается.
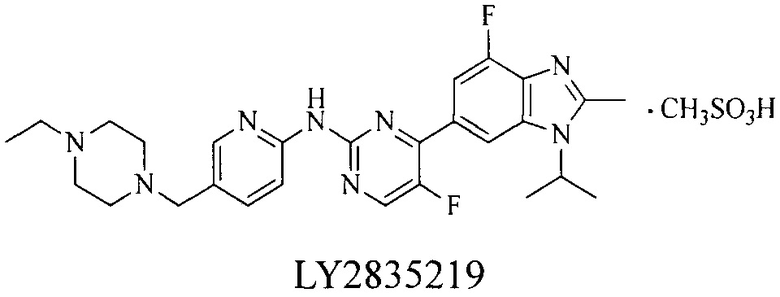
До настоящего времени, нет лекарственных средств, применяемых в качестве ингибиторов CDK, утвержденных для коммерческого маркетинга. Серия CDK4/6 ингибиторов с хорошей избирательностью, о которых сообщалось некоторыми фармацевтическими компаниями, включая Пфайзер, Эли Лилли и Новартис (Pfizer, Eli Lilly и Novartis), находятся на клинических испытаниях. Среди них, особое значение представляют PD0332991 (палбоциклиб), разработанный Пфайзер, LY2835219 (фаза III), разработанный Эли Лилли, и LEE-011 (фаза III), разработанный Новартис:
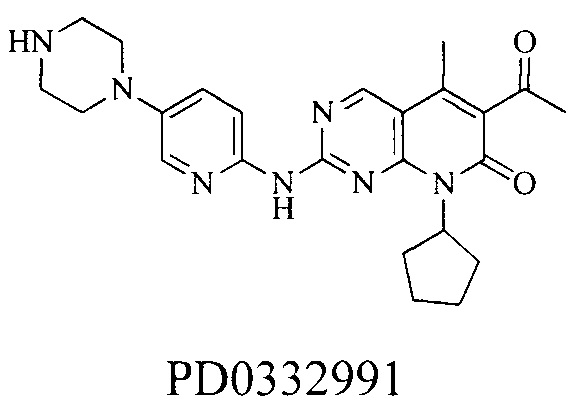
В апреле 2013 разработанный фирмой Пфайзер PD0332991 получил статус принципиально нового средства лечения от Управления по контролю за продуктами и лекарственными средствами (FDA); и в августе 2014 фирма Пфайзер представила FDA совместную заявку на одобрение нового лекарственного средства (NDA), предназначенный для утверждения PD0332991 (палбоциклиб) в комбинации с летрозолом в качестве средства для лечения женщин после менопаузы с местным или метастазирующим раком груди, у которых эстрогеновый рецептор является положительным (ER+), и человеческий эпидермальный фактор роста 2 является отрицательным (HER2-), и ранее не получавших системного лечения. Он обладает весьма положительным эффектом на развитие ингибиторов CDK4/6.
Для достижения лучшего терапевтического воздействия на опухоль и улучшения обеспечения рыночного спроса, авторы настоящего изобретения надеются разработать новое поколение ингибиторов CDK4/6 с высокой эффективностью и низкой токсичностью. Настоящее изобретение предоставляет селективные ингибиторы CDK4/6 с новой структурой, и констатирует, что соединения с такой структурой обладают хорошей эффективностью и могут эффективно проходить через гематоэнцефалический барьер, что делает возможным создание ингибиторов CDK в качестве средств для лечения рака мозга.
Содержание изобретения
В аспекте настоящее изобретение относится к ингибитору/соединению, нацеленному на CDK4/6 киназу. В частности, примерами служат технические решения настоящего изобретения, являющиеся следующими:
1. Соединение формулы (I') или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры:
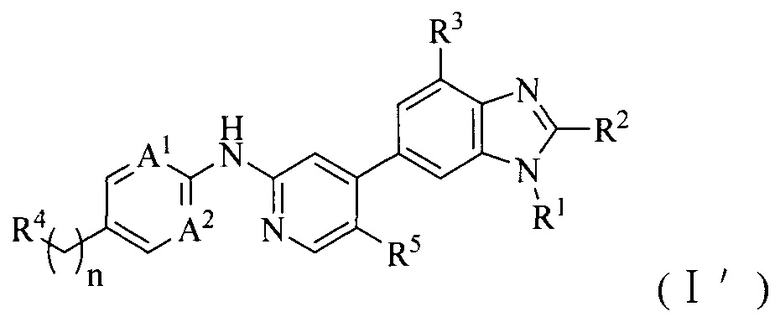
где:
A1 и A2, каждый независимо, выбран из азота;
R1 выбран из C1-6алкила, C1-6алкокси или 3-8-членного циклоалкила, необязательно замещенного Q1, где Q1 выбран из C1-6алкила или C1-6алкокси;
R2 выбран из C1-6алкила, C1-6алкокси, циано, карбамоила или C1-6алкилкарбониламино;
R3 и R5, каждый независимо, выбран из галогена или водорода, и по меньшей мере один из R3 и R5 представляет собой галоген;
R4 выбран из 3-8-членного гетероциклила, 6-14-членного конденсированного гетероциклила, 5-8-членного гетероарила, 6-14-членного конденсированного гетероарила, фенила, нафтила, 6-12-членного мостикового гетероциклила или 6-12-членного спирогетероциклила, каждый из которых необязательно замещен Q2;
Q2 выбран из амино, гидроксила, галогена, трифторметила, циано, C1-6алкокси, C1-6алкилсульфонила, C1-6алкилсульфониламино или ди-C1-6алкиламино; или C1-6алкила, 3-8-членного циклоалкила, 3-8-членного гетероциклила или 6-9-членного мостикового гетероциклила, каждый из которых необязательно замещен заместителем, где данный заместитель выбран из амино, гидроксила, галогена, трифторметила, циано, C1-6алкила, C1-6алкокси, C1-6алкиламино, ди-C1-6алкиламино, C1-6алкилсульфонила, 3-8-членного гетероциклила или 3-8-членного циклоалкила;
n выбран из 0, 1, 2, 3, 4 или 5.
2. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 1, где
A1 и A2, каждый независимо, выбран из азота;
R1 выбран из C1-4алкила или C1-4алкокси;
R2 выбран из C1-4алкила, C1-4алкокси, циано, карбамоила или C1-4алкилкарбониламино;
R3 и R5, каждый независимо, выбран из галогена;
R4 выбран из азотсодержащего 5-6-членного гетероциклила, необязательно замещенного Q2; где “азотсодержащий 5-6-членный гетероциклил” предпочтительно представляет собой “азотсодержащий 6-членный гетероциклил”;
Q2 выбран из амино, гидроксила, галогена, трифторметила, циано, C1-4алкокси или ди-C1-4алкиламино; или C1-4алкила, 3-6-членного циклоалкила или 3-6-членного гетероциклила, каждый из которых необязательно замещен заместителем, где данный заместитель выбран из амино, гидроксила, галогена, трифторметила, C1-4алкила, C1-4алкокси, C1-4алкиламино, ди-C1-4алкиламино или 3-6-членного циклоалкила;
n выбран из 0.
3. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 2, где данное соединение выбрано из:
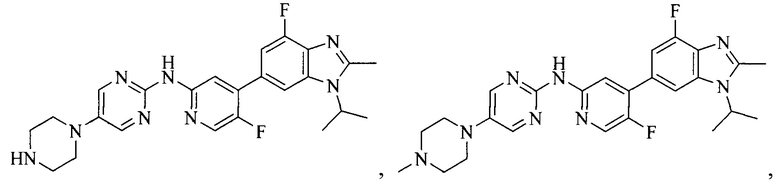
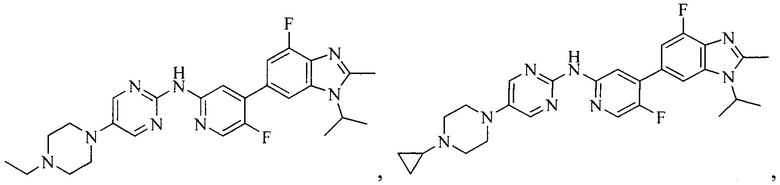
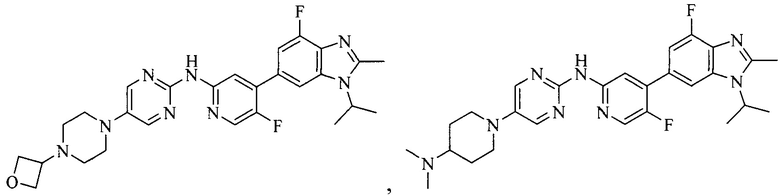 или
или 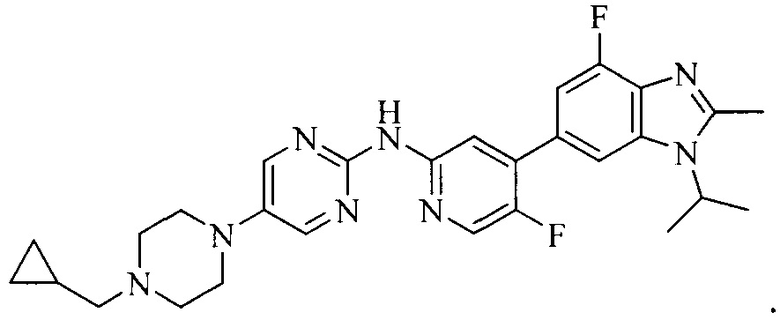 .
.
4. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 1, где данное соединение имеет структуру формулы (I):
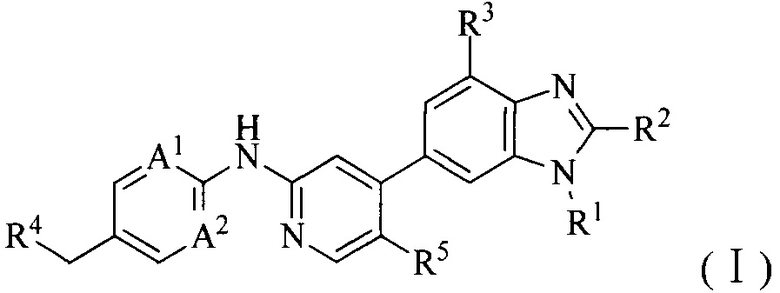
где:
A1 и A2, каждый независимо, выбран из азота;
R1 выбран из C1-6алкила, C1-6алкокси или 3-8-членного циклоалкила, необязательно замещенного Q1, где Q1 выбран из C1-6алкила или C1-6алкокси;
R2 выбран из C1-6алкила, C1-6алкокси, циано, карбамоила или C1-6алкилкарбониламино;
R3 и R5, каждый независимо, выбран из галогена или водорода, и по меньшей мере один из R3 и R5 представляет собой галоген;
R4 выбран из 3-8-членного гетероциклила, 6-14-членного конденсированного гетероциклила, 5-8-членного гетероарила, 6-14-членного конденсированного гетероарила, фенила, нафтила, 6-12-членного мостикового гетероциклила или 6-12-членного спирогетероциклила, каждый из которых необязательно замещен Q2; где Q2 выбран из амино, гидроксила, галогена, трифторметила, циано, C1-6алкила, C1-6алкокси, 3-8-членного гетероциклила или 6-9-членного мостикового гетероциклила.
5. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 4, где
A1 и A2, каждый независимо, выбран из азота;
R1 выбран из C1-4алкила или C1-4алкокси;
R2 выбран из C1-4алкила, C1-4алкокси, циано, карбамоила или C1-4алкилкарбониламино;
R3 и R5, каждый независимо, выбран из галогена;
R4 выбран из 5-7-членного гетероциклила, 6-11-членного конденсированного гетероциклила, 6-11-членного мостикового гетероциклила или 6-11-членного спирогетероциклила, каждый из которых необязательно замещен Q2; где Q2 выбран из амино, гидроксила, трифторметила, циано, C1-4алкила, C1-4алкокси, 5-6-членного гетероциклила или 7-9-членного мостикового гетероциклила.
6. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 5, где
A1 и A2, каждый независимо, выбран из азота;
R1 представляет собой изопропил;
R2 выбран из метила, метокси, циано, карбамоила или ацетиламино;
R3 и R5, каждый независимо, представляет собой F;
R4 выбран из 5-6-членного гетероциклила, необязательно замещенного Q2; где Q2 выбран из амино, гидроксила, трифторметила, циано, C1-4алкила, C1-4алкокси, 6-членного гетероциклила или 8-членного мостикового гетероциклила.
7. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 6, где
R2 представляет собой метил;
R4 выбран из азотсодержащего 5-6-членного гетероциклила, необязательно замещенного Q2; где азотсодержащий 5-6-членный гетероциклил связан с метиленом в формуле (I) через атом азота, где Q2 выбран из амино, гидроксила, трифторметила, циано, C1-4алкила, C1-4алкокси или азотсодержащего 8-членного мостикового гетероциклила;
где азотсодержащий 5-6-членный гетероциклил предпочтительно представляет собой азотсодержащий 5-6-членный гетероциклил, содержащий 1-2 атома азота.
8. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 7, где
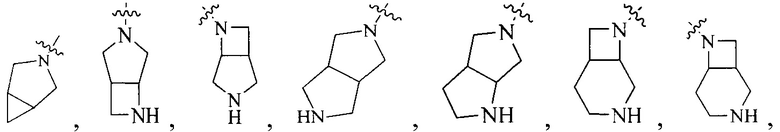
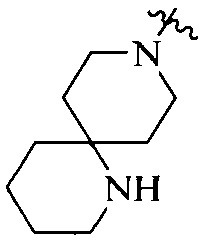
R4 выбран из 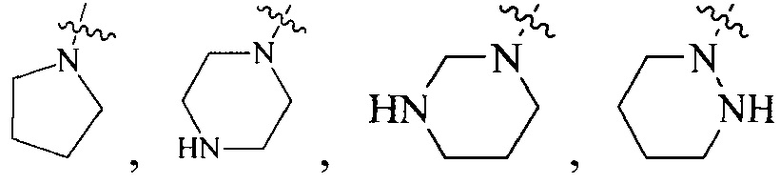 или
или  , каждый из которых необязательно замещен Q2, где Q2 выбран из C1-4алкила или азотсодержащего 8-членного мостикового гетероциклила.
, каждый из которых необязательно замещен Q2, где Q2 выбран из C1-4алкила или азотсодержащего 8-членного мостикового гетероциклила.
9. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 5, где
A1 и A2, каждый независимо, выбран из азота;
R1 представляет собой изопропил;
R2 выбран из метила, метокси, циано, карбамоила или ацетиламино;
R3 и R5, каждый независимо, представляет собой F;
R4 выбран из 7-9-членного мостикового гетероциклила, необязательно замещенного Q2; где Q2 выбран из амино, гидроксила, трифторметила, циано, C1-4алкила, 6-членного гетероциклила или 8-членного мостикового гетероциклила.
10. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 9, где
R2 представляет собой метил;
R4 выбран из азотсодержащего 7-9-членного мостикового гетероциклила, необязательно замещенного Q2; где азотсодержащий 7-9-членный мостиковый гетероциклил связан с метиленом в формуле (I) через атом азота, где Q2 выбран из амино, гидроксила, трифторметила, циано, C1-4алкила или азотсодержащего 6-членного гетероциклила;
где азотсодержащий 7-9-членный мостиковый гетероциклил предпочтительно представляет собой азотсодержащий 7-9-членный мостиковый гетероциклил, содержащий 1-2 атома азота.
11. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 10, где
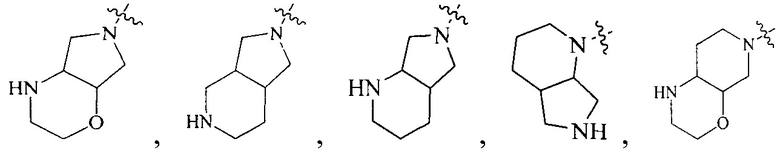
R4 выбран из 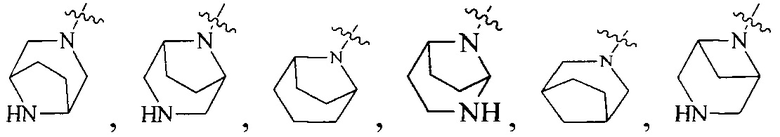 или
или  , каждый из которых необязательно замещен Q2, где Q2 выбран из C1-4 алкила или азотсодержащего 6-членного гетероциклила.
, каждый из которых необязательно замещен Q2, где Q2 выбран из C1-4 алкила или азотсодержащего 6-членного гетероциклила.
12. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 5, где
A1 и A2, каждый независимо, выбран из азота;
R1 представляет собой изопропил;
R2 выбран из метила, метокси, циано, карбамоила или ацетиламино;
R3 и R5, каждый представляет собой F;
R4 выбран из 6-10-членного конденсированного гетероциклила, необязательно замещенного Q2; где Q2 выбран из амино, гидроксила, трифторметила, циано, C1-4алкила, 6-членного гетероциклила или 8-членного мостикового гетероциклила.
13. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 12, где
R2 представляет собой метил;
R4 выбран из азотсодержащего 6-10-членного конденсированного гетероциклила, который содержит 1, 2 или 3 одинаковых или различных гетероатома и необязательно замещен Q2; где данные гетероатомы предпочтительно выбраны из атома азота и атома кислорода, и содержит по меньшей мере один атом азота, и 6-10-членный конденсированный гетероциклил связан с метиленом в формуле (I) через атом азота, где Q2 выбран из амино, гидроксила, трифторметила, циано или C1-4алкила.
14. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 13, где
R4 выбран из
 или
или  , каждый из которых необязательно замещен Q2, где Q2 выбран из амино или C1-4алкила.
, каждый из которых необязательно замещен Q2, где Q2 выбран из амино или C1-4алкила.
15. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 5, где
A1 и A2, каждый независимо, выбран из азота;
R1 представляет собой изопропил;
R2 выбран из метила, метокси, циано, карбамоила или ацетиламино;
R3 и R5, каждый представляет собой F;
R4 выбран из 7-11-членного спирогетероциклила, необязательно замещенного Q2; где Q2 выбран из амино, гидроксила, трифторметила, циано, C1-4алкила, 6-членного гетероциклила или 8-членного мостикового гетероциклила.
16. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 15, где
R2 выбран из метила;
R4 выбран из азотсодержащего 7-11-членного спирогетероциклила, необязательно замещенного Q2; где азотсодержащий 7-11-членный спирогетероциклил связан с метиленом в формуле (I) через атом азота, где Q2 выбран из амино, гидроксила, трифторметила, циано и C1-4алкила;
где азотсодержащий 7-11-членный спирогетероциклил предпочтительно представляет собой азотсодержащий 7-11-членный спирогетероциклил, содержащий 1-2 атома азота.
17. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 16, где
R4 выбран из
 или
или 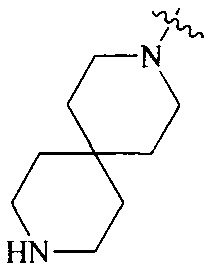 , каждый из которых необязательно замещен Q2, где Q2 выбран из C1-4алкила.
, каждый из которых необязательно замещен Q2, где Q2 выбран из C1-4алкила.
18. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 1, где данное соединение выбрано из соединений, указанных в таблице A.
Часть соединений по изобретению
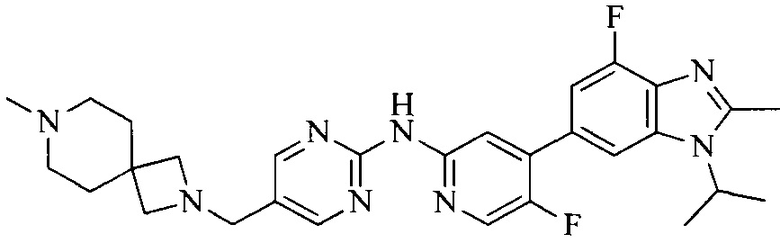
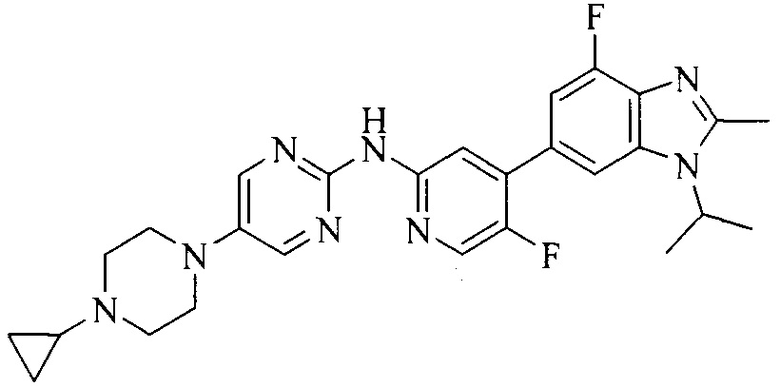
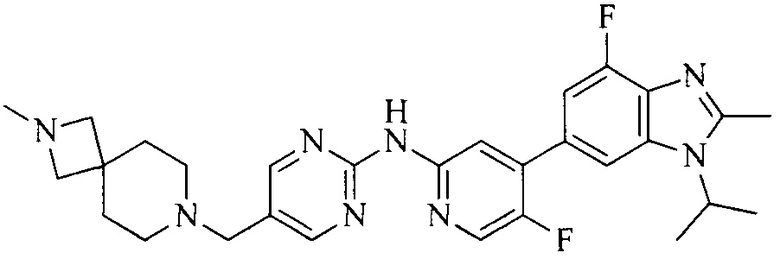
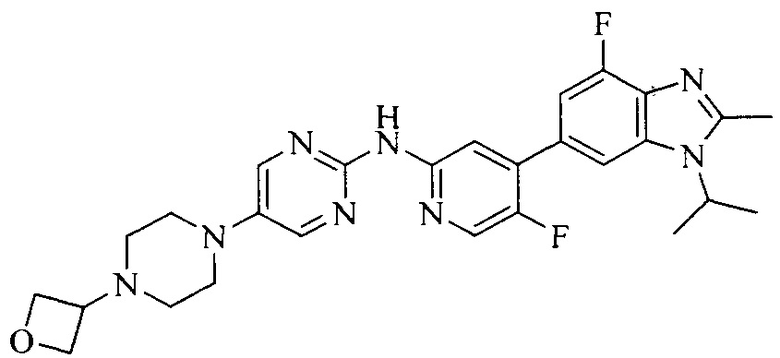
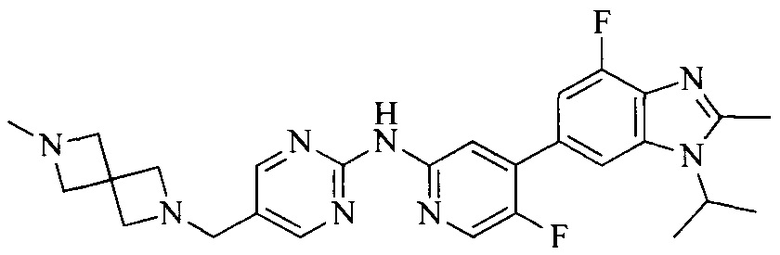
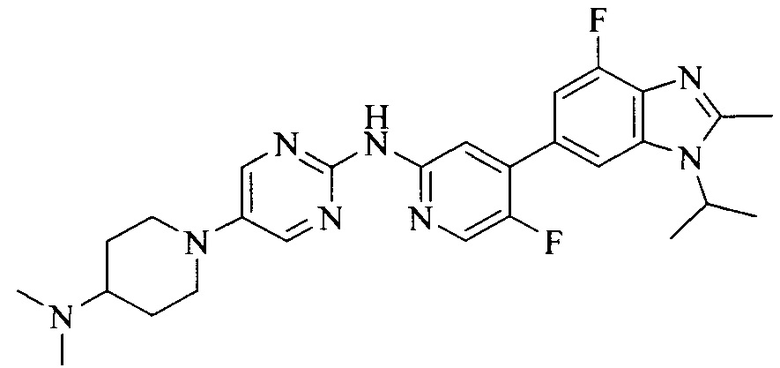
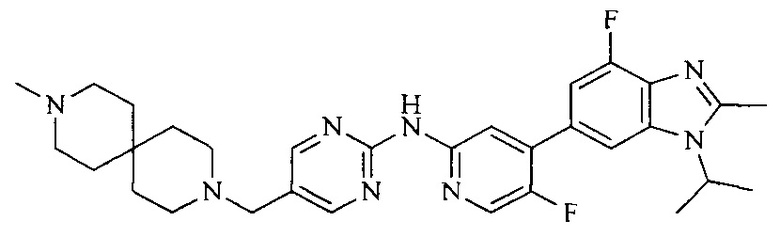
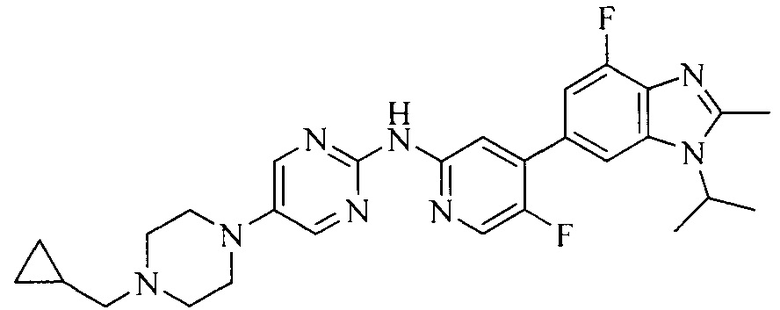
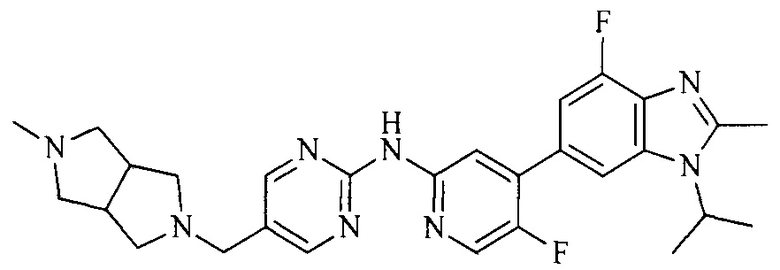
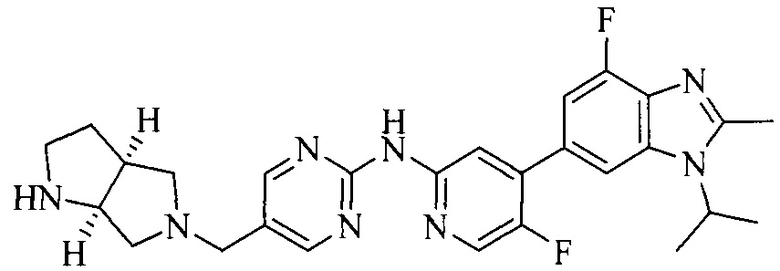
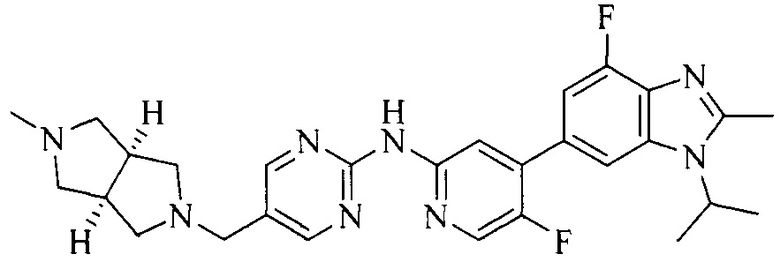
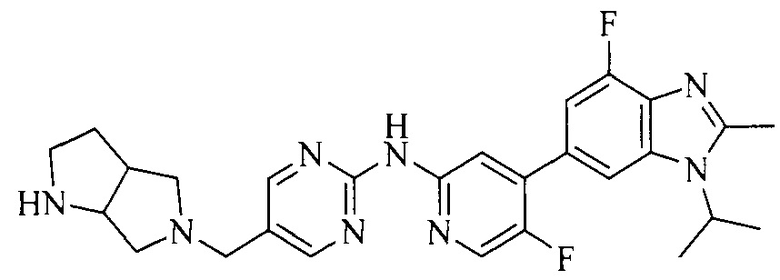
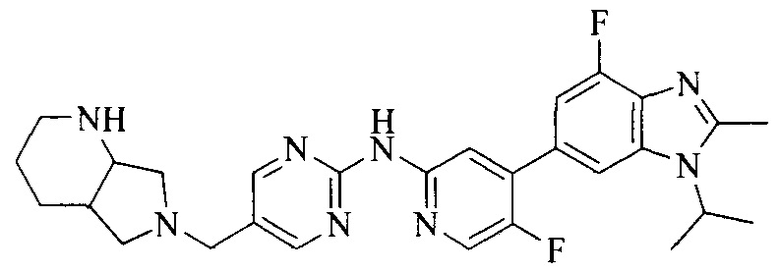
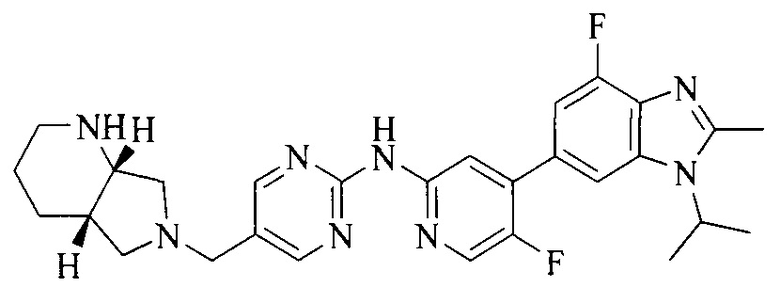
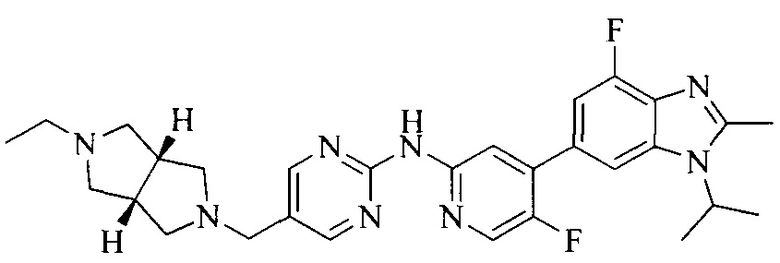
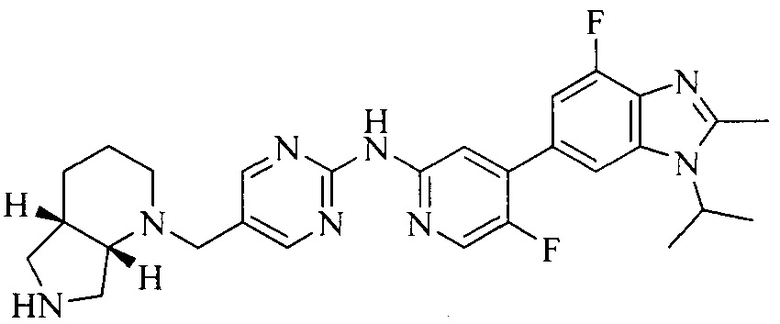
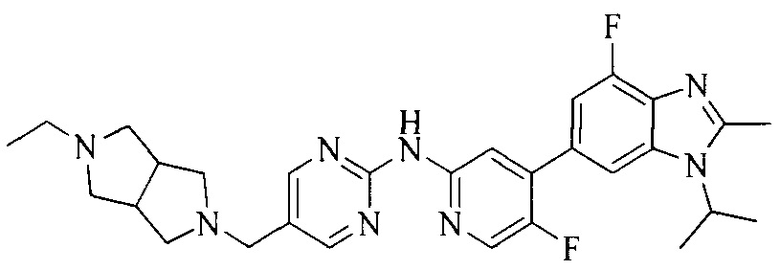
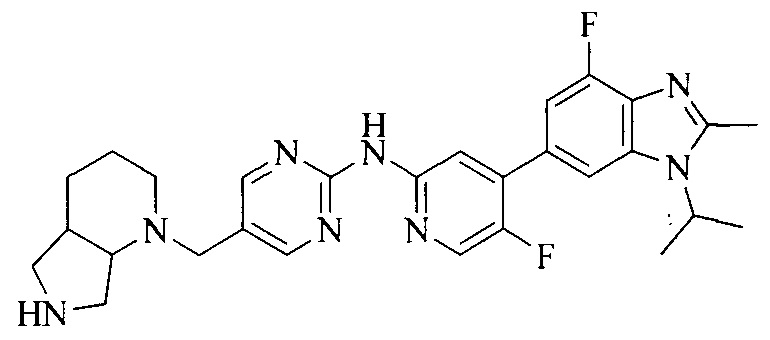
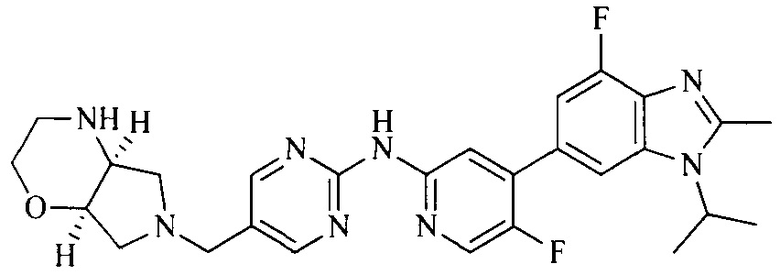
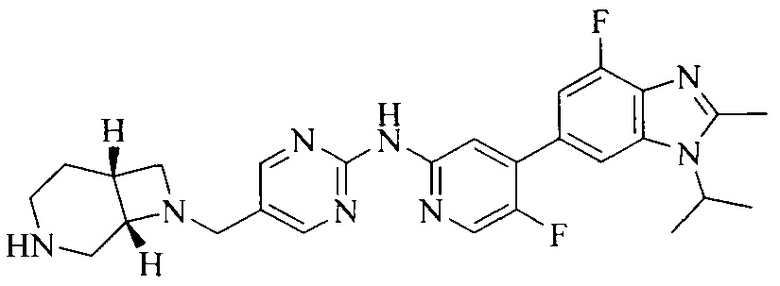
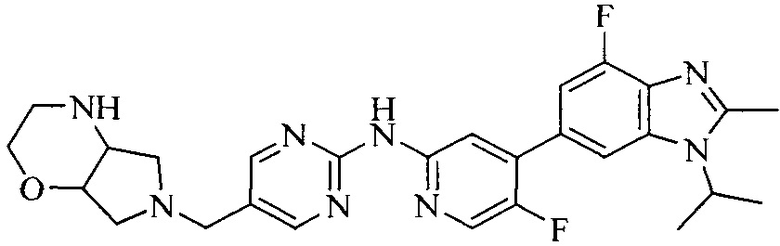
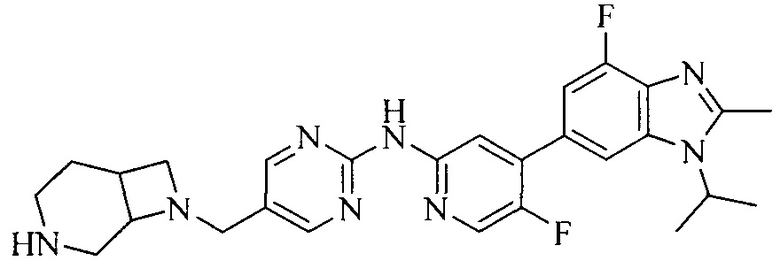
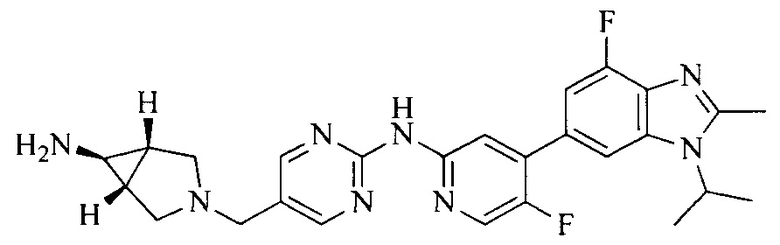
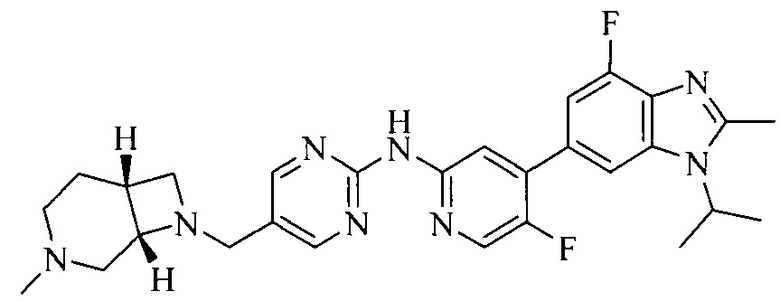
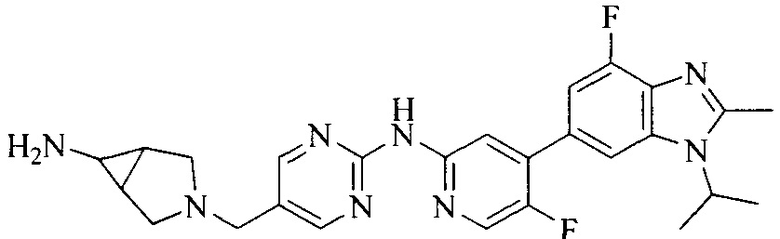
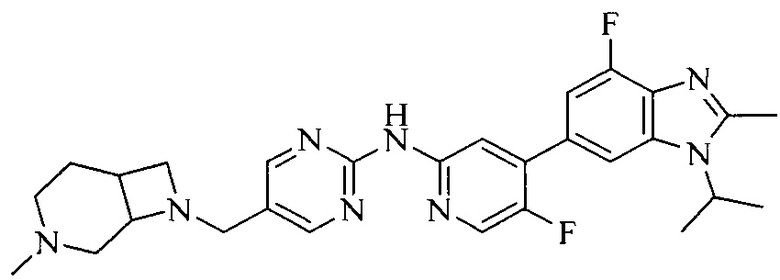
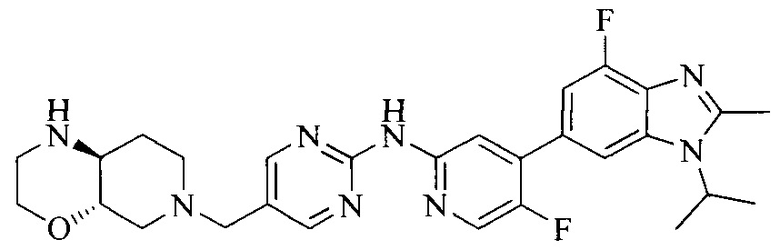
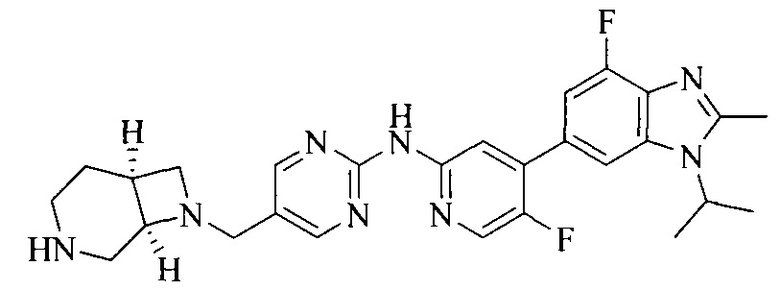
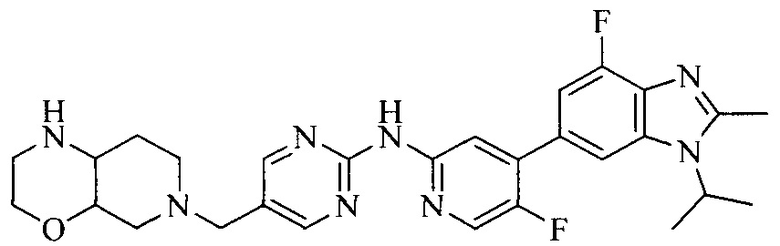
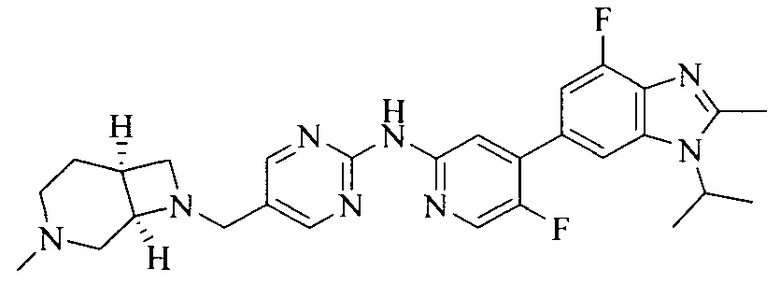
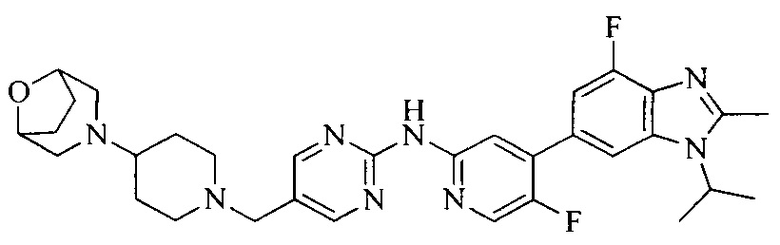
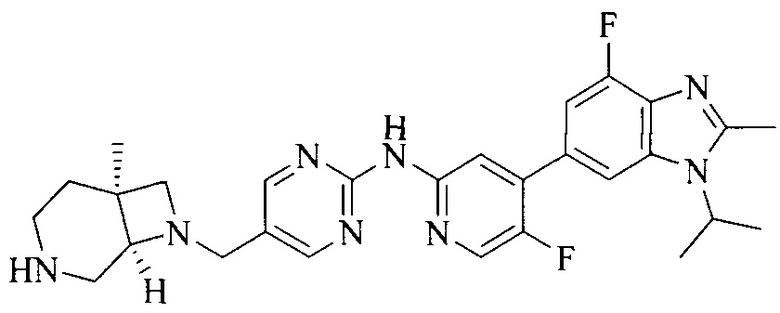
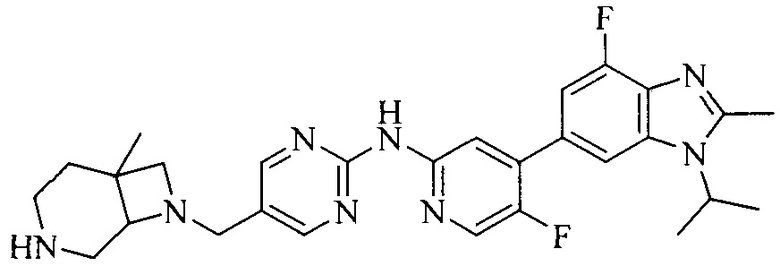
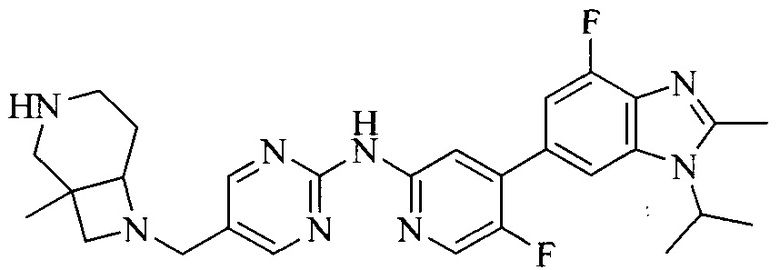
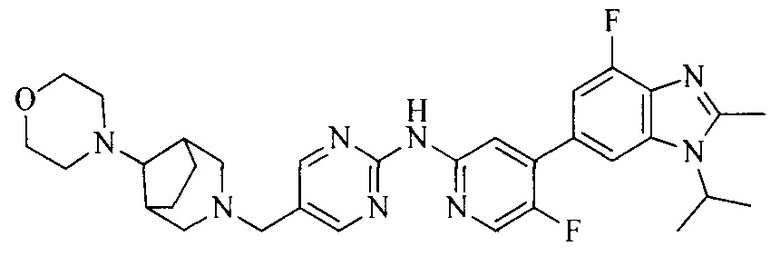
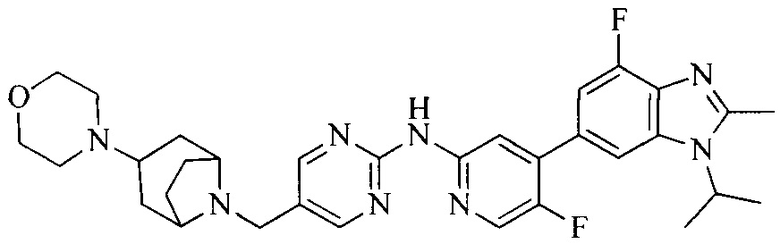
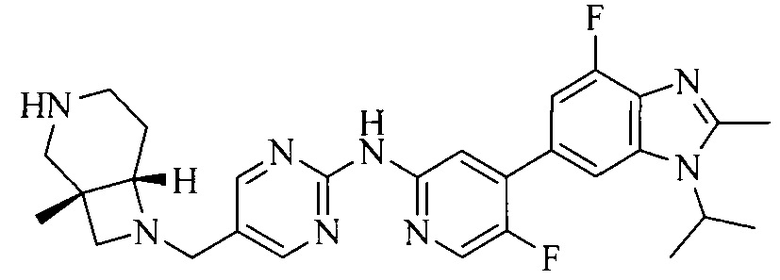
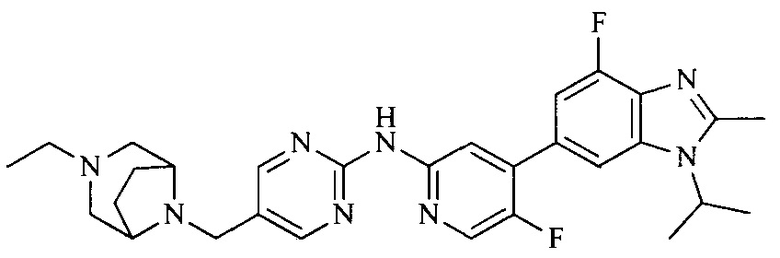
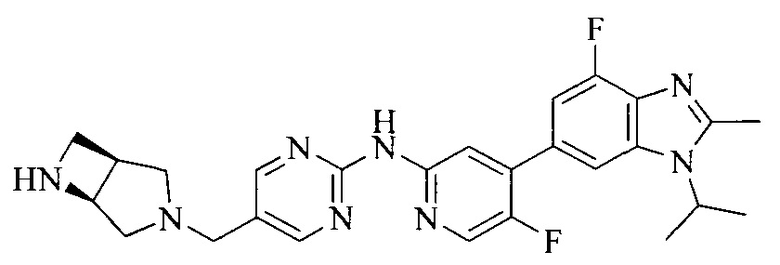
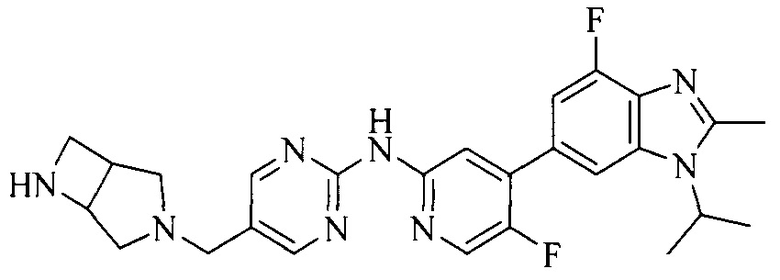
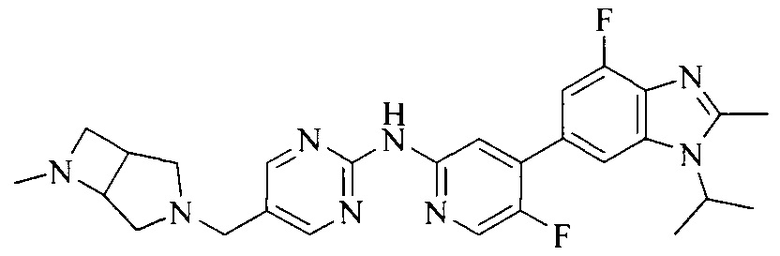
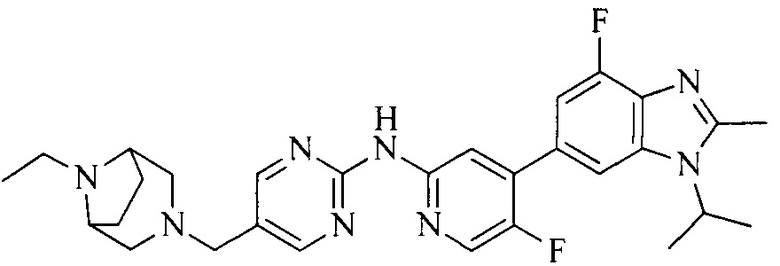
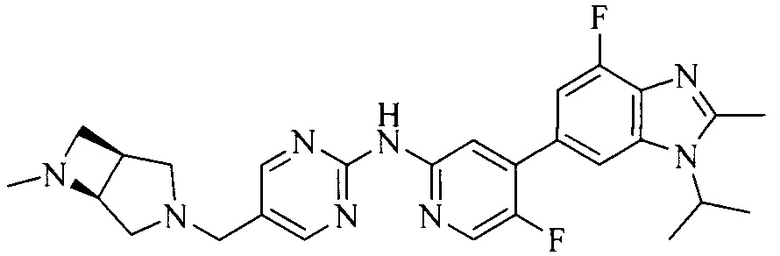
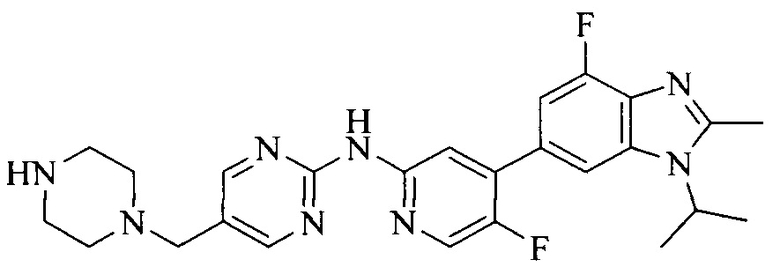
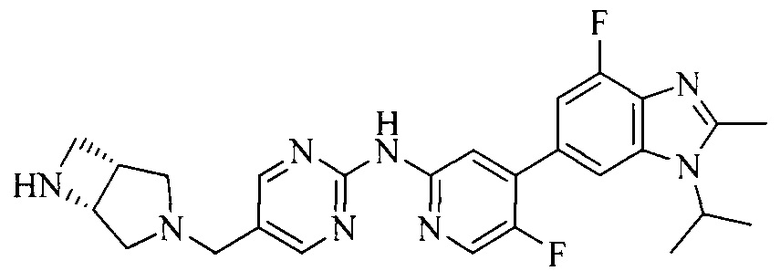
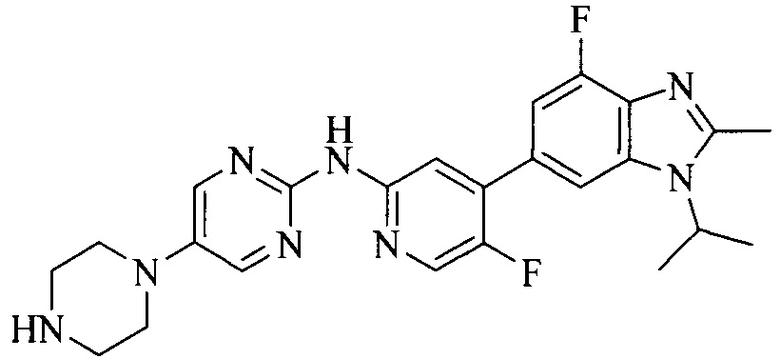
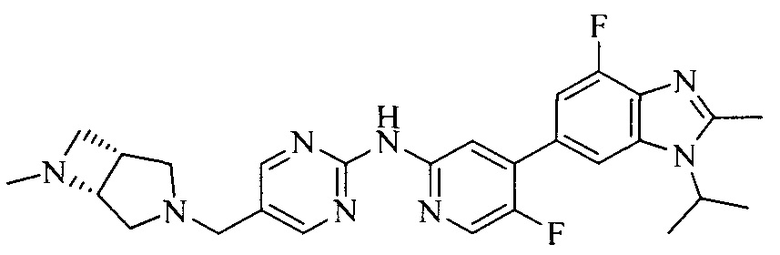
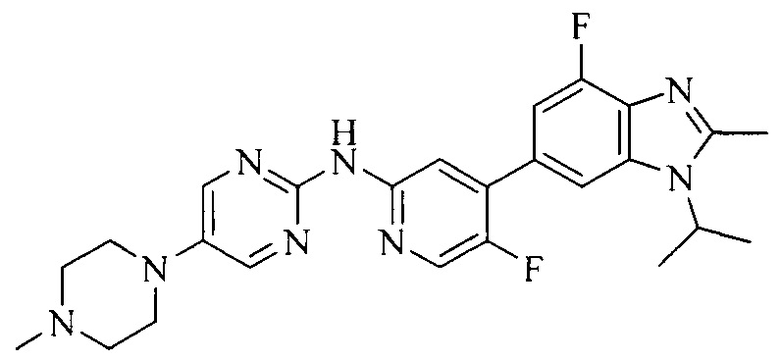
Настоящее изобретение также относится к применению описываемых соединений. Поэтому, настоящее изобретение также относится к следующим примерам технических решений:
19. Фармацевтическая композиция, содержащая соединение или его фармацевтически приемлемую соль, сложный эфир или сольват или их стереоизомеры по любому из решений 1-18 и необязательно один или более фармацевтически приемлемых носителей.
20. Фармацевтическая композиция по решению 19, дополнительно содержащая один или более дополнительных противоопухолевых средств и/или иммуносуппрессоров.
21. Фармацевтическая композиция по решению 20, где дополнительные противоопухолевые средства и/или иммуносуппрессоры выбраны из одного или более: метотрексата, капецитабина, гемцитабина, доксифлуридина, пеметрекседа динатрия, пазопаниба, иматиниба, эрлотиниба, лапатиниба, гефитиниба, вандетаниба, герцептина, бевацизумаба, ритуксимаба, трастузумаба, паклитаксела, винорельбина, доцетаксела, доксорубицина, гидроксикамптотецина, митомицина, эпирубицина, пирарубицина, блеомицина, летрозола, тамоксифена, фульвестранта, трипторелина, флутамида, лейпрорелина, анастрозола, ифосфамида, бусульфана, циклофосфамида, кармустина, нимустина, семустина, мехлорэтамина, мелфалана, хлорамбуцила, карбоплатина, цисплатина, оксалиплатина, лобаплатина, топотекана, камптотецина, топотекана, эверолимуса, сиролимуса, темсиролимуса, 6-меркаптопурина, 6-тиогуанина, азатиоприна, актиномицина D, даунорубицина, адриамицина, митоксантрона, блеомицина, митрамицина и аминоглутетимида.
22. Применение соединения или его фармацевтически приемлемой соли, сложного эфира или сольвата или их стереоизомеров по любому из решений 1-18 при производстве лекарственного средства для лечения и/или предотвращения связанных с раком заболеваний, опосредованных CDK4/6 киназой, у субъекта.
23. Применение по решению 22, где связанное с раком заболевание выбрано из опухоли головного мозга, рака легких, плоскоклеточной карциномы, карциномы мочевого пузыря, рака желудка, рака яичников, перитонеального рака, рака поджелудочной железы, рака молочной железы, рака головы и шеи, рака шейки матки, рака эндометрия, рака прямой кишки, рака печени, почечной карциномы, аденокарциномы пищевода, плоскоклеточного рака пищевода, рака предстательной железы, рака женского репродуктивного протока, рака in situ, лимфомы, нейрофибромы, карциномы щитовидной железы, остеокарциномы, рака кожи, рака мозга, рака толстой кишки, рака мужских половых органов, стромальной опухоли желудочно-кишечного тракта, новообразований предстательной железы, опухоли тучных клеток, множественной миеломы, меланомы, глиомы или саркомы.
24. Применение по решению 22 или 23, где субъектом является млекопитающее, такое как корова, лошадь, коза, свинья, собака, кошка, грызуны и приматы; где особенно предпочтительным субъектом является человек.
25. Применение по любому из решений 22-24, где средство для лечения дополнительно включает одно или более дополнительных противоопухолевых средств и/или иммуносуппрессоров; предпочтительно, дополнительные противоопухолевые средства и/или иммуносуппрессоры выбраны из одного или более: метотрексата, капецитабина, гемцитабина, доксифлуридина, пеметрекседа динатрия, пазопаниба, иматиниба, эрлотиниба, лапатиниба, гефитиниба, вандетаниба, герцептина, бевацизумаба, ритуксимаба, трастузумаба, паклитаксела, винорельбина, доцетаксела, доксорубицина, гидроксикамптотецина, митомицина, эпирубицина, пирарубицина, блеомицина, летрозола, тамоксифена, фульвестранта, трипторелина, флутамида, лейпрорелина, анастрозола, ифосфамида, бусульфана, циклофосфамида, кармустина, нимустина, семустина, мехлорэтамина, мелфалана, хлорамбуцила, карбоплатина, цисплатина, оксалиплатина, лобаплатина, топотекана, камптотецина, топотекана, эверолимуса, сиролимуса, темсиролимуса, 6-меркаптопурина, 6-тиогуанина, азатиоприна, актиномицина D, даунорубицина, адриамицина, митоксантрона, блеомицина, митрамицина и аминоглутетимида.
26. Способ лечения и/или предотвращения связанных с раком заболеваний, опосредованных CDK4/6 киназой, включающий введение субъекту, нуждающемуся в этом, терапевтически и/или профилактически эффективного количества соединения или его фармацевтически приемлемой соли, сложного эфира или сольвата или их стереоизомеров по любому из решений 1-18 или фармацевтической композиции по любому из решений 19-21.
27. Способ по решению 26, где связанное с раком заболевание выбрано из опухоли головного мозга, рака легких, плоскоклеточной карциномы, карциномы мочевого пузыря, рака желудка, рака яичников, перитонеального рака, рака поджелудочной железы, рака молочной железы, рака головы и шеи, рака шейки матки, рака эндометрия, рака прямой кишки, рака печени, почечной карциномы, аденокарциномы пищевода, плоскоклеточного рака пищевода, рака предстательной железы, рака женского репродуктивного протока, рака in situ, лимфомы, нейрофибромы, карциномы щитовидной железы, остеокарциномы, рака кожи, рака мозга, рака толстой кишки, рака мужских половых органов, стромальной опухоли желудочно-кишечного тракта, новообразований предстательной железы, опухоли тучных клеток, множественной миеломы, меланомы, глиомы или саркомы.
28. Способ по решению 26 или 27, где субъектом является млекопитающее, такое как корова, лошадь, коза, свинья, собака, кошка, грызуны и приматы; где особенно предпочтительным субъектом является человек.
29. Способ по любому из решений 25-28, где способ дополнительно включает введение данному субъекту одного или более дополнительных противоопухолевых средств и/или иммуносуппрессоров; предпочтительно, дополнительные противоопухолевые средства и/или иммуносуппрессоры выбраны из одного или более: метотрексата, капецитабина, гемцитабина, доксифлуридина, пеметрекседа динатрия, пазопаниба, иматиниба, эрлотиниба, лапатиниба, гефитиниба, вандетаниба, герцептина, бевацизумаба, ритуксимаба, трастузумаба, паклитаксела, винорельбина, доцетаксела, доксорубицина, гидроксикамптотецина, митомицина, эпирубицина, пирарубицина, блеомицина, летрозола, тамоксифена, фульвестранта, трипторелина, флутамида, лейпрорелина, анастрозола, ифосфамида, бусульфана, циклофосфамида, кармустина, нимустина, семустина, мехлорэтамина, мелфалана, хлорамбуцила, карбоплатина, цисплатина, оксалиплатина, лобаплатина, топотекана, камптотецина, топотекана, эверолимуса, сиролимуса, темсиролимуса, 6-меркаптопурина, 6-тиогуанина, азатиоприна, актиномицина D, даунорубицина, адриамицина, митоксантрона, блеомицина, митрамицина и аминоглутетимида.
30. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по любому из решений 1-18, для применения при лечении и/или предупреждении связанных с раком заболеваний, опосредованных CDK4/6 киназой, у субъекта.
31. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 30, где связанное с раком заболевание выбрано из опухоли головного мозга, рака легких, плоскоклеточной карциномы, карциномы мочевого пузыря, рака желудка, рака яичников, перитонеального рака, рака поджелудочной железы, рака молочной железы, рака головы и шеи, рака шейки матки, рака эндометрия, рака прямой кишки, рака печени, почечной карциномы, аденокарциномы пищевода, плоскоклеточного рака пищевода, рака предстательной железы, рака женского репродуктивного протока, рака in situ, лимфомы, нейрофибромы, карциномы щитовидной железы, остеокарциномы, рака кожи, рака мозга, рака толстой кишки, рака мужских половых органов, стромальной опухоли желудочно-кишечного тракта, новообразований предстательной железы, опухоли тучных клеток, множественной миеломы, меланомы, глиомы или саркомы.
32. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 30 или 31, где субъектом является млекопитающее, такое как корова, лошадь, коза, свинья, собака, кошка, грызуны и приматы; где особенно предпочтительным субъектом является человек.
33. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по любому из решений 30-32, которое применяют в комбинации с одним или более дополнительных противоопухолевых средств и/или иммуносуппрессоров; предпочтительно, дополнительные противоопухолевые средства и/или иммуносуппрессоры выбраны из одного или более: метотрексата, капецитабина, гемцитабина, доксифлуридина, пеметрекседа динатрия, пазопаниба, иматиниба, эрлотиниба, лапатиниба, гефитиниба, вандетаниба, герцептина, бевацизумаба, ритуксимаба, трастузумаба, паклитаксела, винорельбина, доцетаксела, доксорубицина, гидроксикамптотецина, митомицина, эпирубицина, пирарубицина, блеомицина, летрозола, тамоксифена, фульвестранта, трипторелина, флутамида, лейпрорелина, анастрозола, ифосфамида, бусульфана, циклофосфамида, кармустина, нимустина, семустина, мехлорэтамина, мелфалана, хлорамбуцила, карбоплатина, цисплатина, оксалиплатина, лобаплатина, топотекана, камптотецина, топотекана, эверолимуса, сиролимуса, темсиролимуса, 6-меркаптопурина, 6-тиогуанина, азатиоприна, актиномицина D, даунорубицина, адриамицина, митоксантрона, блеомицина, митрамицина и аминоглутетимида.
34. Применение соединения или его фармацевтически приемлемой соли, сложного эфира или сольвата или их стереоизомеров по любому из решений 1-18 при производстве лекарственных составов для снижения и/или ингибирования активности CDK4 и/или CDK6 киназ в клетке.
35. Применение по решению 34, где данный лекарственный состав вводят in vivo или in vitro; например, данный лекарственный состав вводят субъекту (например, млекопитающему, такому как корова, лошадь, коза, свинья, собака, кошка, грызуны и приматы; например, человеку), для снижения и/или ингибирования активности CDK4 и/или CDK6 киназ в клетке субъекта; или данный лекарственный состав вводят в in vitro клетку (например, линию клеток или клетку от субъекта, такую как раковая клетка), для снижения и/или ингибирования активности CDK4 и/или CDK6 киназ в данной in vitro клетке.
36. Применение по решению 34 или 35, где данная клетка выбрана из клеток опухоли головного мозга, клеток рака легких, клеток плоскоклеточной карциномы, клеток карциномы мочевого пузыря, клеток рака желудка, клеток рака яичников, клеток перитонеального рака, клеток рака поджелудочной железы, клеток рака молочной железы, клеток рака головы и шеи, клеток рака шейки матки, клеток рака эндометрия, клеток рака прямой кишки, клеток рака печени, клеток почечной карциномы, клеток аденокарциномы пищевода, клеток плоскоклеточного рака пищевода, клеток рака предстательной железы, клеток рака женского репродуктивного протока, клеток рака in situ, клеток лимфомы, клеток нейрофибромы, клеток карциномы щитовидной железы, клеток остеокарциномы, клеток рака кожи, клеток рака мозга, клеток рака толстой кишки, клеток рака мужских половых органов, клеток стромальной опухоли желудочно-кишечного тракта, клеток новообразований предстательной железы, клеток опухоли тучных клеток, клеток множественной миеломы, клеток меланомы, клеток глиомы или клеток саркомы.
37. Способ снижения и/или ингибирования активности CDK4 и/или CDK6 киназ в клетке, включающий введение в данную клетку эффективного количества соединения или его фармацевтически приемлемой соли, сложного эфира или сольвата или их стереоизомеров по любому из решений 1-18.
38. Способ по решению 37, где данный способ осуществляют in vivo, например, данная клетка является клеткой субъекта (например, млекопитающего, такого как корова, лошадь, коза, свинья, собака, кошка, грызуны и приматы; например, человек); или данный способ осуществляют in vitro, например, данная клетка является in vitro клеткой (например, линией клеток или клеткой от субъекта, такой как раковая клетка).
39. Способ по решению 37 или 38, где данная клетка выбрана из клеток опухоли головного мозга, клеток рака легких, клеток плоскоклеточной карциномы, клеток карциномы мочевого пузыря, клеток рака желудка, клеток рака яичников, клеток перитонеального рака, клеток рака поджелудочной железы, клеток рака молочной железы, клеток рака головы и шеи, клеток рака шейки матки, клеток рака эндометрия, клеток рака прямой кишки, клеток рака печени, клеток почечной карциномы, клеток аденокарциномы пищевода, клеток плоскоклеточного рака пищевода, клеток рака предстательной железы, клеток рака женского репродуктивного протока, клеток рака in situ, клеток лимфомы, клеток нейрофибромы, клеток карциномы щитовидной железы, клеток остеокарциномы, клеток рака кожи, клеток рака мозга, клеток рака толстой кишки, клеток рака мужских половых органов, клеток стромальной опухоли желудочно-кишечного тракта, клеток новообразований предстательной железы, клеток опухоли тучных клеток, клеток множественной миеломы, клеток меланомы, клеток глиомы или клеток саркомы.
40. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по любому из решений 1-18, для применения в снижении или ингибировании активности CDK4 и/или CDK6 киназ в клетке.
41. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их изомер по решению 40, применяемое для in vivo или in vitro введения; например, данный лекарственный состав вводят субъекту (например, млекопитающему, такому как корова, лошадь, коза, свинья, собака, кошка, грызуны и приматы; например, человеку), для снижения и/или ингибирования активности CDK4 и/или CDK6 киназ в клетке субъекта; или данный лекарственный состав вводят в in vitro клетку (например, линию клеток или клетку от субъекта, такую как раковая клетка), для снижения и/или ингибирования активности CDK4 и/или CDK6 киназ в in vitro клетке.
42. Соединение или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по решению 40 или 41, где данная клетка выбрана из клеток опухоли головного мозга, клеток рака легких, клеток плоскоклеточной карциномы, клеток карциномы мочевого пузыря, клеток рака желудка, клеток рака яичников, клеток перитонеального рака, клеток рака поджелудочной железы, клеток рака молочной железы, клеток рака головы и шеи, клеток рака шейки матки, клеток рака эндометрия, клеток рака прямой кишки, клеток рака печени, клеток почечной карциномы, клеток аденокарциномы пищевода, клеток плоскоклеточного рака пищевода, клеток рака предстательной железы, клеток рака женского репродуктивного протока, клеток рака in situ, клеток лимфомы, клеток нейрофибромы, клеток карциномы щитовидной железы, клеток остеокарциномы, рака кожи, клеток рака мозга, клеток рака толстой кишки, клеток рака мужских половых органов, клеток стромальной опухоли желудочно-кишечного тракта, клеток новообразования простаты, клеток опухоли тучных клеток, клеток множественной миеломы, клеток меланомы, клеток глиомы или клеток саркомы.
43. Набор для снижения или ингибирования активности CDK4 и/или CDK6 киназ в клетке, содержащий соединение или его фармацевтически приемлемую соль, сложный эфир или сольват или их стереоизомеры по любому из решений 1-18 и необязательно инструкции.
Подробное описание изобретения
В данном описании и формуле изобретения настоящего описания данные соединения названы согласно их формулам, и если название и формула для некоторых соединений не согласуются друг с другом, формула будет иметь преимущественную силу.
В настоящем описании, если специально не указано иное, использованные специфические и технические термины имеют значения, как правило, понятные специалисту в данной области техники. Однако для лучшего понимания настоящего изобретения, определения и пояснения предоставлены частью терминов. Кроме того, если определения и объяснения терминов, предоставленных в настоящем описании, отличаются от значений, обычно допускаемых специалистом в данной области техники, определения и объяснения терминов, предоставленные в настоящем описании, будут иметь преимущественную силу.
Термин “Me”, используемый в данном описании, означает метил.
В настоящем изобретении волнистая линия “ ” в заместителе означает, что данный радикал данного заместителя присоединен к данному радикалу основной цепи (такому как фенольное кольцо) через химическую связь в положении волнистой линии.
” в заместителе означает, что данный радикал данного заместителя присоединен к данному радикалу основной цепи (такому как фенольное кольцо) через химическую связь в положении волнистой линии.
Термин “галоген”, используемый в данном описании, относится к атому F, Cl, Br и I.
Термин “C1-6алкил”, используемый в данном описании, относится к линейному или разветвленному алкилу, включая "C1-4алкил", "C1-3алкил" и тому подобное, где его примеры включают, но, не ограничиваясь ими, метил, этил, н-пропил, изопропил, н-бутил, 2-метилпропил, 1-метилпропил, 1,1-диметилэтил, н-пентил, 3-метилбутил, 2-метилбутил, 1-метилбутил, 1-этилпропил, н-гексил, 4-метилпентил, 3-метилпентил, 2-метилпентил, 1-метилпентил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил, 2-этилбутил, 1,2-диметилпропил и т.д.
Термины “C1-6алкокси, C1-6алкилкарбониламино, C1-6алкилсульфонил, C1-6алкилсульфониламино, C1-6алкиламино, ди-C1-6алкиламино”, используемые в данном описании, относятся к группам, образованным в виде C1-6алкил-O-, C1-6алкил-C(O)NH-, C1-6алкил-SO2-, C1-6алкил-SO2NH-, C1-6алкил-NH-, (C1-6алкил)2-N-, где термин “C1-6алкил” имеет такие же значения, как определено выше.
Термины “C1-4алкокси, C1-4алкилкарбониламино, C1-4алкилсульфонил, C1-4алкилсульфониламино, C1-4алкиламино, ди-C1-4алкиламино”, используемые в данном описании, относятся к группам, образованным в виде C1-4алкил-O-, C1-4алкил-C(O)NH-, C1-4алкил-SO2-, C1-4алкил-SO2NH-, C1-4алкил-NH-, (C1-4алкил)2-N-, где термин “C1-4алкил” имеет такие же значения, как определено выше.
Термин “3-8-членный циклоалкил”, используемый в данном описании, относится к циклоалкилу, образованному при удалении одного атома водорода из циклоалкана, имеющего 3-8 атомов углерода, включая, например, “3-6-членный циклоалкил” и “4-6-членный циклоалкил”, и т.д. Их примеры включают, но, не ограничиваясь ими: циклопропил, циклобутил, циклопентил и циклогексил, циклогептил, циклооктил и т.д.
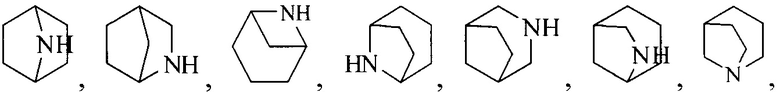
Термин “3-8-членный гетероциклил”, используемый в данном описании, включает, например “3-7-членный гетероциклил”, “3-6-членный гетероциклил”, “4-7-членный гетероциклил”, “4-6-членный гетероциклил”, “5-7-членный гетероциклил”, “5-6-членный гетероциклил”, “5-6-членный азотсодержащий гетероциклил”, “6-членный гетероциклил”, “6-членный азотсодержащий гетероциклил” и т.д. Их примеры включают, но, не ограничиваясь ими: азиридинил, 2H-азиридинил, диазациклопропил, 3H-диазациклопропенил, азетидинил, 1,4-диоксанил, 1,3-диоксанил, 1,3-диоксациклопентил, 1,4-диоксациклогексадиенил, тетрагидрофурил, дигидропирролил, пирролидинилил, имидазолидинил, 4,5-дигидроимидазолил, пиразолидинил, 4,5-дигидропиразолил, 2,5-дигидротиенил, тетрагидротиенил, 4,5-дигидротиазолил, пиперидил, пиперазинил, морфолинил, гексагидропиримидинил, гексагидропиридазинил, 4,5-дигидрооксазолил, 4,5-дигидроизоксазолил, 2,3-дигидроизоксазолил, 2H-1,2-оксазинил, 6H-1,3-оксазинил, 4H-1,3-тиазинил, 6H-1,3-тиазинил, 2H-пиранил, 2H-пиран-2-он, 3,4-дигидро-2H-пиранил и т.д., предпочтительно “5-6-членный азотсодержащий гетероциклил”.
Термин “связанный с метиленом в формуле (I) через атом азота”, используемый в данном описании, относится к азотсодержащей группе (например, азотсодержащему гетероциклилу, такому как “5-6-членный азотсодержащий гетероциклил” или “6-членный азотсодержащий гетероциклил”; азотсодержащему конденсированному гетероциклилу, такому как “6-10-членный азотсодержащий конденсированный гетероциклил”; азотсодержащему мостиковому гетероциклилу, такому как “7-9-членный азотсодержащий мостиковый гетероциклил” или “8-членный азотсодержащий мостиковый гетероциклил”; азотсодержащему спирогетероциклилу, такому как “7-11-членный азотсодержащий спирогетероциклил”), присоединенной к метилену в формуле (I) через атом азота.
В соответствии с правилами номенклатуры IUPAC, конденсированное кольцо по настоящему изобретению относится к конденсированной кольцевой структуре, образованной двумя или более кольцевыми структурами, которые разделены двумя смежными атомами (т.е. разделены одной связью). Мостиковые кольца по настоящему изобретению относятся к мостиковой кольцевой структуре, образованной двумя или более кольцевыми структурами, которые разделены двумя несмежными атомами углерода. Спирокольцо по настоящему изобретению относится к спирокольцевой структуре, образованной двумя или более кольцевыми структурами, которые разделены одним атомом углерода.
Термин “6-14-членный конденсированный гетероциклил”, используемый в данном описании, относится к 6-14-членной конденсированной кольцевой структуре, содержащей по меньшей мере один гетероатом, образованной двумя или более кольцевыми структурами, которые разделены двумя смежными атомами (т.е. разделены одной связью), включая, например, “6-11-членный конденсированный гетероциклил”, “6-10-членный конденсированный гетероциклил”, “7-10-членный конденсированный гетероциклил”, “9-10-членный конденсированный гетероциклил”, “6-10-членный азотсодержащий конденсированный гетероциклил” и т.д. Их примеры включают, но, не ограничиваясь ими: 3-азабицикло[3.1.0]гексан, 3,6-диазабицикло[3.2.0]гептан, 3,8-диазабицикло[4.2.0]октан, 3,7-диазабицикло[4.2.0]октан, октагидропирроло[3,4-c]пиррол, октагидропирроло[3,4-b]пиррол, октагидропирроло[3,4-b][1,4]оксазин, октагидро-1H-пирроло[3,4-c]пиридин, октагидро-1H-пирроло[3,4-b]пиридин, октагидро-1H-пиридо[3,4-b][1,4]оксазин, декагидро-2,6-нафталин, тетрагидроимидазо[4,5-c]пиридинил, 3,4-дигидрохиназолинил, 1,2-дигидрохиноксалинил, бензо[d][1,3]диоксациклопентенил, 1,3-дигидроизобензофурил, 2H-хроменил, 2H-хромен-2-он, 4H-хроменил, 4H-хромен-4-он, хроманил, 4H-1,3-бензоксазинил, 4,6-дигидро-1H-фуро[3,4-d]имидазолил, 3a,4,6,6a-тетрагидро-1H-фуро[3,4-d]имидазолил, 4,6-дигидро-1H-тиено[3,4-d]имидазолил, 4,6-дигидро-1H-пирроло[3,4-d]имидазолил, 4,5,6,7-тетрагидро-1H-бензо[d]имидазолил и т.д.
Термин “5-8-членный гетероарил”, используемый в данном описании, включает, например, “5-7-членный гетероарил”, “5-6-членный гетероарил” и т.д. Их примеры включают, но, не ограничиваясь ими: фурил, тиенил, пирролил, тиазолил, изотиазолил, тиадиазолил, оксазолил, изоксазолил, оксадиазолил, имидазолил, пиразолил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, пиридил, 2-пиридон, 4-пиридон, пиримидинил, 1,4-диоксоциклогексадиенил, 2H-1,2-оксазинил, 4H-1,2-оксазинил, 6H-1,2-оксазинил, 4H-1,3-оксазинил, 6H-1,3-оксазинил, 4H-1,4-оксазинил, пиридазинил, пиразинил, 1,2,3-триазинил, 1,3,5-триазинил, 1,2,4,5-тетразинил, азепинил, 1,3-диазепинил, азоцинил и т.д., предпочтительно “5~6-членный гетероарил”.
Термин “6-14-членный конденсированный гетероарил”, используемый в данном описании, относится к ароматической 6-14-членной конденсированной кольцевой структуре, содержащей по меньшей мере один гетероатом, образованной двумя или более кольцевыми структурами, которые разделены двумя смежными атомами (т.е. разделены одной связью), включая, например, “6-10-членный конденсированный гетероарил”, “7-10-членный конденсированный гетероарил”, “9-10-членный конденсированный гетероарил” и т.д. Их примеры включают, но, не ограничиваясь ими: бензофуранил, изобензофуранил, бензотиенил, индолил, изоиндолил, бензоксазолил, бензоимидазолил, индазолил, бензотриазолил, хинолинил, хинолин-2-он, хинолин-4-он, изохинолин-1-он, изохинолинил, акридинил, фенантридинил, бензопиридазинил, фталазинил, хиназолинил, хиноксалинил, феназинил, птеридинил, пуринил, нафтиридинил, феназин, фенотиазин и т.д.
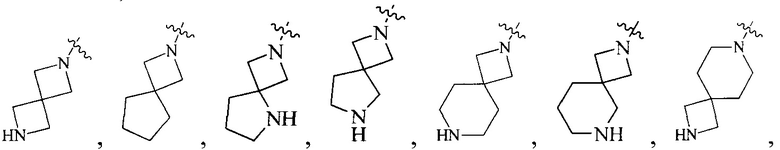
Термин “6-12-членный мостиковый гетероциклил”, используемый в данном описании, относится к 6-12-членной мостиковой кольцевой структуре, содержащей по меньшей мере один гетероатом, образованной любыми двумя кольцами, которые разделены двумя несмежными атомами, где гетероатом выбран из N, S, O, CO, SO и/или SO2 и т.д. Он включает, например, “6-11-членный мостиковый гетероциклил”, “6-9-членный мостиковый гетероциклил”, “7-10-членный мостиковый гетероциклил”, “7-9-членный мостиковый гетероциклил”, “7-9-членный азотсодержащий мостиковый гетероциклил”, “7-8-членный мостиковый гетероциклил”, “8-членный мостиковый гетероциклил”, “8-членный азотсодержащий мостиковый гетероциклил” и т.д. Их примеры включают, но, не ограничиваясь ими:

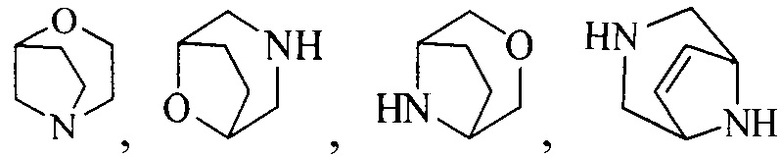 и т.д.
и т.д.
Термин “6-12-членный спирогетероциклил”, используемый в данном описании, относится к 6-12-членной спирокольцевой структуре, содержащей по меньшей мере один гетероатом, образованной по меньшей мере двумя кольцами, которые разделены одним атомом, где гетероатом выбран из N, S, O, CO, SO и/или SO2 и т.д. Он включает, например, “6-11-членный спирогетероциклил”, “7-11-членный спирогетероциклил”, “7-11-членный азотсодержащий спирогетероциклил”, “7-10-членный спирогетероциклил”, “7-9-членный спирогетероциклил”, “7-8-членный спирогетероциклил” и т.д. Их примеры включают, но, не ограничиваясь ими:

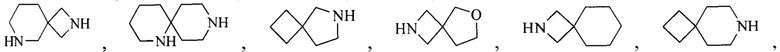

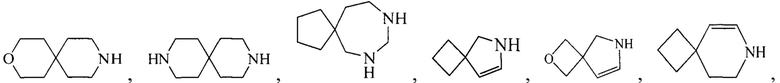 и т.д.
и т.д.
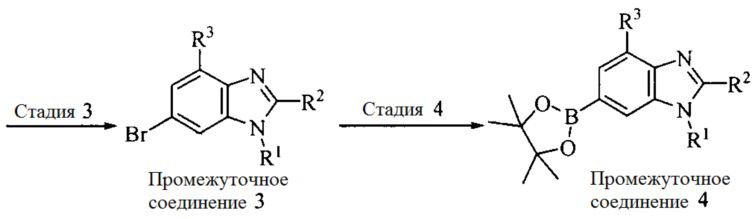
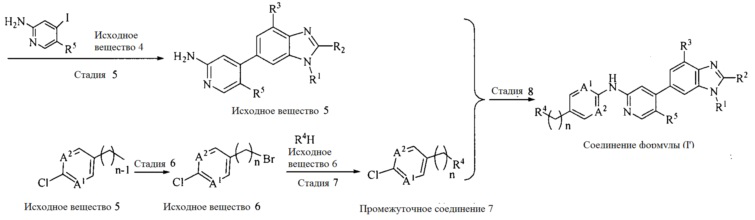
Настоящее изобретение также предоставляет способ получения соединения формулы (I'), включая, но без ограничения, следующую схему (где значения аббревиатур описаны следующим образом: ДХМ: дихлорметан; DIPEA: N,N-диизопропилэтиламин; ДМФА: N,N-диметилформамид; ДМСО: диметилсульфоксид; EA: этилацетат; HATU: гексафторфосфат 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония; MeOH: метанол; NBS: N-бромбутанимид; PE: петролейный эфир; ТГФ: тетрагидрофуран; Xant-phos: 4,5-бис(дифенилфосфино)-9,9-диметилксантен; x-phos: 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил):
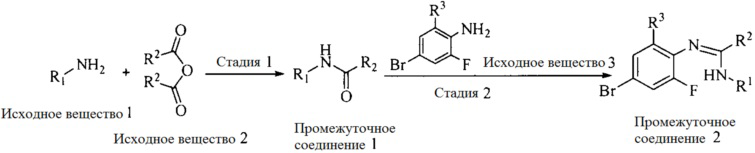
где R1, R2, R3, R4, R5, n, A1, A2 имеют такие же значения, как определено выше, X представляет собой галоген, выбранный из F, Cl, Br и I; и галогенирующий агент выбран из I2 и Br2.
Иллюстративными стадиями являются следующие.
1. Получение промежуточного соединения 1
Исходное вещество 1 и органическое основание растворяют в органическом растворителе, и исходное вещество 2 добавляют медленно по каплям при низкой температуре. Взаимодействие осуществляют при перемешивании. После взаимодействия, полученную реакционную смесь экстрагируют; органическую фазу сушат и концентрируют, с получением промежуточного соединения 1, где органическим растворителем предпочтительно является ДХМ или 1,4-диоксан, и органической фазой предпочтительно является триэтиламин.
2. Получение промежуточного соединения 2
Промежуточное соединение 1, исходное вещество 3 и органическое основание растворяют в органическом растворителе, и по каплям добавляют оксихлорид фосфора. После взаимодействия, добавляют основание для доведения pH полученной реакционной смеси до нейтрального. Полученную в результате смесь экстрагируют, и отделенную органическую фазу сушат и концентрируют, с получением промежуточного соединения 2, где органическим растворителем предпочтительно является ДХМ или 1,2-дихлорэтан, и органической фазой предпочтительно является триэтиламин.
3. Получение промежуточного соединения 3
Промежуточное соединение 2 растворяют в органическом растворителе, и добавляют трет-бутоксид калия. Полученную в результате смесь нагревают до 100°C и подвергают взаимодействию в течение 2 часов. После взаимодействия, добавляют воду для гашения реакции. Полученную реакционную смесь экстрагируют, и органическую фазу сушат и концентрируют. Полученный в результате остаток подвергают колоночной хроматографии, с получением промежуточного соединения 3, где органическим растворителем предпочтительно является ДХМ.
4. Получение промежуточного соединения 4
Промежуточное соединение 3 и пинаколборат растворяют в органическом растворителе; и добавляют диацетат палладия, трициклогексил фосфора и ацетат калия. В защитной атмосфере газообразного азота данное взаимодействие осуществляют при нагревании. После взаимодействия, добавляют воду и органический растворитель для экстракции полученной реакционной смеси. Органическую фазу сушат, концентрируют и разделяют колоночной хроматографией, с получением промежуточного соединения 4, где органическим растворителем предпочтительно является ДМФА или 1,4-диоксан.
5. Получение промежуточного соединения 5
Исходное вещество 4 и промежуточное соединение 4 растворяют в органическом растворителе, и добавляют неорганическое основание и тетракис(трифенилфосфин)палладий. В защитной атмосфере газообразного азота данное взаимодействие осуществляют при нагревании. После взаимодействия, добавляют воду. Полученную реакционную смесь экстрагируют, и органическую фазу сушат и концентрируют. Полученный в результате остаток отделяют колоночной хроматографией, с получением промежуточного соединения 5, где органическим растворителем предпочтительно является 1,4-диоксан или ДМФА.
6. Получение промежуточного соединения 6
Исходное вещество 5 растворяют в органическом растворителе, и добавляют бензоилпероксид и NBS. Данное взаимодействие осуществляют при нагревании. После взаимодействия, полученную реакционную смесь фильтруют, и фильтрат концентрируют. Полученный в результате остаток разделяют колоночной хроматографией, с получением промежуточного соединения 6, где органическим растворителем предпочтительно является четыреххлористый углерод.
7. Получение промежуточного соединения 7
Промежуточное соединение 6 и исходное вещество 6 растворяют в органическом растворителе, и добавляют неорганическое основание. Данное взаимодействие осуществляют при комнатной температуре. После взаимодействия, полученную реакционную смесь фильтруют, фильтрат концентрируют, и остаток разделяют колоночной хроматографией, с получением промежуточного соединения 7, где органическим растворителем предпочтительно является ацетонитрил.
8. Получение соединения формулы (I')
Промежуточное соединение 5 и промежуточное соединение 7 растворяют в органическом растворителе; и добавляют трис(дибензилиденацетон)дипалладий, 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил и карбонат цезия. В защитной атмосфере газообразного азота данное взаимодействие осуществляют при нагревании. После взаимодействия, добавляют воду. Полученную реакционную смесь экстрагируют, и органическую фазу сушат и концентрируют. Полученный в результате остаток разделяют колоночной хроматографией, с получением соединения формулы (I'), где органическим растворителем предпочтительно является 1,4-оксан или ДМФА.
Исходное вещество 6 представляет собой первичный амин или вторичный амин и т.д.
"Фармацевтически приемлемая соль" соединения формулы (I') или формулы (I) по настоящему изобретению относится к соли, образованной взаимодействием кислотной группы(групп) (например, -COOH, -OH, -SO3H и т.д.) в соединении формулы (I') или формулы (I) с подходящим неорганическим или органическим катионом(и) (основанием), включая соль, образованную с щелочным металлом или щелочноземельным металлом, аммонийную соль и соль, образованную с азотсодержащим органическим основанием; и к соли, образованной взаимодействием основной группы(групп) (например, -NH2 и т.д.) в соединении формулы (I') или формулы (I) с подходящим неорганическим или органическим анионом(и) (кислотой), включая неорганическую кислоту и органическую карбоновую кислоту.
"Сложный эфир" соединения формулы (I') или формулы (I) по настоящему изобретению относится к сложному эфиру, образованному реакцией этерификации соединения формулы (I') или формулы (I) со спиртом, когда соединение формулы (I') или формулы (I) имеет карбоксильную группу; или к сложному эфиру, образованному реакцией этерификации соединения формулы (I') или формулы (I) с органической кислотой, неорганической кислотой или солью органической кислоты и т.д., когда соединение формулы (I') или формулы (I) имеет гидроксильную группу. В присутствии кислоты или основания сложный эфир может быть гидролизован до получения соответствующей кислоты или спирта.
"Сольват" соединения формулы (I') или формулы (I) по настоящему изобретению относится к веществу, образованному путем его ассоциации с молекулой(ами) растворителя. Данным растворителем может быть органический растворитель (например, метанол, этанол, пропанол, ацетонитрил и т.д.) и вода, и т.д. Например, соединение формулы (I') или формулы (I) по настоящему изобретению может образовывать алкоголят с этанолом, или образовывать гидрат с водой.
“Стереоизомеризм” соединения по настоящему изобретению делится на конформационную изомерию и конфигурационную изомерию, где конфигурационная изомерия делится на цис-транс изомерию и оптическую изомерию. Конформационная изомерия представляет собой форму стереоизомеризма, в которой результатом являются ротации или искажения простой связи C-C при различных пространственных механизмах атомов или атомных групп в органической молекуле с определенной конфигурацией, обычно в алкановых и циклоалкановых соединениях, такие как конформеры при конформации в циклогексане в форме кресла и лодки. Термин "стереоизомеры” означает, что соединения по настоящему изобретению имеют один или более асимметрических центров и, таким образом, могут представлять собой рацематы и рацемические смеси, индивидуальные энантиомеры, диастереоизомерные смеси и индивидуальные диастереоизомеры. Соединения по настоящему изобретению имеют асимметрические центры, каждый из которых независимо приводит к двум оптическим изомерам. В объем настоящего изобретения включены все возможные оптические изомеры и диастереоизомерные смеси, а также чистые или частично чистые соединения. Если соединения по настоящему изобретению имеют алкеновую углерод-углеродную двойную связь, если специально не указано иное, соединения по настоящему изобретению включают цис-изомеры и транс-изомеры. Соединения по настоящему изобретению могут присутствовать в форме таутомеров, которые имеют различные сайты водородных связей благодаря одному или более сдвигов двойных связей. Например, кетон и его енол образуют кето-енольные таутомеры. Различные таутомеры и их смеси, все включены в соединения по настоящему изобретению. Все энантиомеры, диастереоизомеры, рацематы, цис-транс-изомеры, таутомеры, геометрические изомеры и эпимеры соединения формулы (I') или формулы (I), и их смеси входят в объем настоящего изобретения.
Настоящее изобретение дополнительно предоставляет фармацевтическую композицию, содержащую соединения формулы (I') или формулы (I) или его фармацевтически приемлемую соль, сложный эфир или сольват или их стереоизомеры и необязательно один или более фармацевтически приемлемых носителей. Фармацевтическая композиция может быть получена в любой фармацевтически приемлемой форме. Фармацевтическая композиция может быть введена пациенту или субъекту, нуждающемуся в этом, любым подходящим путем, таким как пероральный, парентеральный, ректальный или внутрилегочный и т.д. Для перорального введения, фармацевтическая композиция может быть получена в форме общепринятого твердого состава, такого как таблетка, капсула, пилюля и гранула; или может быть получена в форме перорального жидкого состава, такого как пероральный раствор, пероральная суспензия и сироп. Когда фармацевтическую композицию получают в форме перорального состава, могут быть добавлены подходящие наполнители, связующие агенты, дезинтегранты, лубриканты и тому подобное. Для парентерального введения, фармацевтическая композиция может быть получена в форме инъекции, включая инъекционный раствор, стерильный порошок для инъекций и концентрированный раствор для инъекций. При получении фармацевтической композиции в форме инъекции могут быть использованы общепринятые способы в фармацевтической области. При получении инъекций добавки могут не добавляться или подходящие добавки могут быть добавлены в зависимости от свойств лекарственного средства. Для ректального введения, фармацевтическая композиция может быть получена в форме суппозитория и т.д. Для внутрилегочного введения, фармацевтическая композиция может быть получена в форме ингаляции или аэрозоля и т.д.
В дополнение к соединению формулы (I') или формулы (I), или его фармацевтически приемлемой соли, сложному эфиру или сольвату или их стереоизомерам, фармацевтическая композиция по настоящему изобретению может дополнительно содержать один или более дополнительных противоопухолевых средств и/или иммуносуппрессоров. Данные противоопухолевые средства и/или иммуносуппрессоры включают, но, не ограничиваясь ими, метотрексат, капецитабин, гемцитабин, доксифлуридин, пеметрексед динатрия, пазопаниб, иматиниб, эрлотиниб, лапатиниб, гефитиниб, вандетаниб, герцептин, бевацизумаб, ритуксимаб, трастузумаб, паклитаксел, винорельбин, доцетаксел, доксорубицин, гидроксикамптотецин, митомицин, эпирубицин, пирарубицин, блеомицин, летрозол, тамоксифен, фульвестрант, трипторелин, флутамид, лейпрорелин, анастрозол, ифосфамид, бусульфан, циклофосфамид, кармустин, нимустин, семустин, мехлорэтамин, мелфалан, хлорамбуцил, карбоплатин, цисплатин, оксалиплатин, лобаплатин, топотекан, камптотецин, топотекан, эверолимус, сиролимус, темсиролимус, 6-меркаптопурин, 6-тиогуанин, азатиоприн, актиномицин D, даунорубицин, адриамицин, митоксантрон, блеомицин, митрамицин и аминоглутетимид.
Настоящее изобретение дополнительно предоставляет применение соединения формулы (I') или формулы (I) или его фармацевтически приемлемой соли, сложного эфира или сольвата или их стереоизомеров по настоящему изобретению при производстве лекарственного средства для лечения и/или предотвращения связанных с раком заболеваний, опосредованных CDK4/6 киназой, у субъекта. В предпочтительном варианте осуществления связанное с раком заболевание выбрано из опухоли головного мозга, рака легких, плоскоклеточной карциномы, карциномы мочевого пузыря, рака желудка, рака яичников, перитонеального рака, рака поджелудочной железы, рака молочной железы, рака головы и шеи, рака шейки матки, рака эндометрия, рака прямой кишки, рака печени, почечной карциномы, аденокарциномы пищевода, плоскоклеточного рака пищевода, рака предстательной железы, рака женского репродуктивного протока, рака in situ, лимфомы, нейрофибромы, карциномы щитовидной железы, остеокарциномы, рака кожи, рака мозга, рака толстой кишки, рака мужских половых органов, стромальной опухоли желудочно-кишечного тракта, новообразований предстательной железы, опухоли тучных клеток, множественной миеломы, меланомы, глиомы или саркомы.
В настоящем изобретении субъектом или пациентом может быть любое животное, предпочтительно млекопитающее, такое как корова, лошадь, коза, свинья, собака, кошка, грызуны и приматы; где особенно предпочтительным субъектом является человек.
Настоящее изобретение дополнительно предоставляет способ лечения и/или предотвращения связанных с раком заболеваний, опосредованных CDK4/6 киназой, включающий введение субъекту, нуждающемуся в этом, терапевтически и/или профилактически эффективного количества соединения или его фармацевтически приемлемой соли, сложного эфира или сольвата или их стереоизомеров по настоящему изобретению или фармацевтической композиции по настоящему изобретению.
Как использовано в данном описании, термин “эффективное количество” относится к количеству, которое достаточно для достижения или по меньшей мере для частичного достижения желаемого эффекта. Например, эффективное количество для предотвращения заболевания (такого как связанное с раком заболевание, опосредованное CDK4/6 киназой) относится к количеству, которое достаточно для предотвращения, подавления или задержки развития заболевания (такого как связанное с раком заболевание, опосредованное CDK4/6 киназой); терапевтически эффективное количество относится к количеству, которое достаточно для лечения или по меньшей мере частичного подавления заболевания и его осложнений у пациента с данным заболеванием. Возможность определения такого эффективного количества полностью находится в компетенции специалиста в данной области. Например, терапевтически эффективное количество будет зависеть от тяжести заболевания, подвергаемого лечению, общего состояния иммунной системы пациента, общих состояний пациента, таких как возраст, масса тела и пол, пути введения лекарственного средства и другой терапии, используемой в комбинации, и тому подобное.
Как подробно описано выше, соединения или фармацевтические композиции по настоящему изобретению могут быть введены субъекту, нуждающемуся в этом, любым подходящим путем в любой подходящей форме. Например, соединения или фармацевтическая композиция по настоящему изобретению могут быть введены субъекту, нуждающемуся в этом, перорально, парентерально, ректально или внутрилегочно и т.д. Соединения или фармацевтическая композиция по настоящему изобретению могут быть в форме таблеток, капсул, пилюль, гранул, растворов, суспензий, сиропов, инъекций (включая раствор для инъекций, стерильный порошок для инъекций и концентрированный раствор для инъекций), суппозиториев, ингаляций или аэрозолей.
Кроме того, способ по настоящему изобретению может быть применен для любого субъекта, предпочтительно млекопитающего, такого как корова, лошадь, коза, свинья, собака, кошка, грызуны и приматы; где особенно предпочтительным субъектом является человек.
Кроме того, как подробно описано выше, способ по настоящему изобретению полезен при лечении и/или предотвращении различных связанных с раком заболеваний, опосредованных CDK4/6 киназами, включая, но, не ограничиваясь ими, опухоль головного мозга, рак легких, плоскоклеточную карциному, карциному мочевого пузыря, рак желудка, рак яичников, перитонеальный рак, рак поджелудочной железы, рак молочной железы, рак головы и шеи, рак шейки матки, рак эндометрия, рак прямой кишки, рак печени, почечную карциному, аденокарциному пищевода, плоскоклеточный рак пищевода, рак предстательной железы, рак женского репродуктивного протока, рак in situ, лимфому, нейрофиброму, карциному щитовидной железы, остеокарциному, рак кожи, рак мозга, рак толстой кишки, рак мужских половых органов, стромальную опухоль желудочно-кишечного тракта, новообразования предстательной железы, опухоль тучных клеток, множественную миелому, меланому, глиому или саркому.
Более того, в дополнение к соединению формулы (I') или формулы (I) или его фармацевтически приемлемой соли, сложному эфиру или сольвату или их стереоизомерам, способ по настоящему изобретению может дополнительно включать введение субъекту одного или более дополнительных противоопухолевых средств и/или иммуносуппрессоров. Другими словами, в способе по настоящему изобретению соединение по настоящему изобретению может быть использовано в комбинации с одним или более дополнительными противоопухолевыми средствами и/или иммуносуппрессорами.
В одном из предпочтительных вариантах осуществления дополнительные противоопухолевые средства и/или иммуносуппрессоры выбраны из одного или более: метотрексата, капецитабина, гемцитабина, доксифлуридина, пеметрекседа динатрия, пазопаниба, иматиниба, эрлотиниба, лапатиниба, гефитиниба, вандетаниба, герцептина, бевацизумаба, ритуксимаба, трастузумаба, паклитаксела, винорельбина, доцетаксела, доксорубицина, гидроксикамптотецина, митомицина, эпирубицина, пирарубицина, блеомицина, летрозола, тамоксифена, фульвестранта, трипторелина, флутамида, лейпрорелина, анастрозола, ифосфамида, бусульфана, циклофосфамида, кармустина, нимустина, семустина, мехлорэтамина, мелфалана, хлорамбуцила, карбоплатина, цисплатина, оксалиплатина, лобаплатина, топотекана, камптотецина, топотекана, эверолимуса, сиролимуса, темсиролимуса, 6-меркаптопурина, 6-тиогуанина, азатиоприна, актиномицина D, даунорубицина, адриамицина, митоксантрона, блеомицина, митрамицина и аминоглутетимида. В предпочтительном варианте осуществления соединение по настоящему изобретению и дополнительное противоопухолевое средство и/или иммуносуппрессор могут быть введены в любом порядке. Например, дополнительное противоопухолевое средство и/или иммуносуппрессор можно вводить субъекту перед, одновременно или после введения соединения по настоящему изобретению.
Настоящее изобретение дополнительно предоставляет применение соединения формулы (I') или формулы (I) или его фармацевтически приемлемой соли, сложного эфира или сольвата или их стереоизомеров при производстве лекарственных составов для снижения или ингибирования активности CDK4 и/или CDK6 киназ в клетке. В предпочтительном варианте осуществления данный лекарственный состав вводят in vivo или in vitro. Например, данный лекарственный состав вводят субъекту (например, млекопитающему, такому как корова, лошадь, коза, свинья, собака, кошка, грызуны и приматы; например, человеку) для снижения и/или ингибирования активности CDK4 и/или CDK6 киназ в клетке субъекта; или данный лекарственный состав вводят в in vitro клетку (например, линию клеток или клетку от субъекта, такую как раковая клетка), для снижения и/или ингибирования активности CDK4 и/или CDK6 киназ в in vitro клетке. В предпочтительном варианте осуществления данная клетка выбрана из клеток опухоли головного мозга, клеток рака легких, клеток плоскоклеточной карциномы, клеток карциномы мочевого пузыря, клеток рака желудка, клеток рака яичников, клеток перитонеального рака, клеток рака поджелудочной железы, клеток рака молочной железы, клеток рака головы и шеи, клеток рака шейки матки, клеток рака эндометрия, клеток рака прямой кишки, клеток рака печени, клеток почечной карциномы, клеток аденокарциномы пищевода, клеток плоскоклеточного рака пищевода, клеток рака предстательной железы, клеток рака женского репродуктивного протока, клеток рака in situ, клеток лимфомы, клеток нейрофибромы, клеток карциномы щитовидной железы, клеток остеокарциномы, клеток рака кожи, клеток рака мозга, клеток рака толстой кишки, клеток рака мужских половых органов, клеток стромальной опухоли желудочно-кишечного тракта, клеток новообразований предстательной железы, клеток опухоли тучных клеток, клеток множественной миеломы, клеток меланомы, клеток глиомы или клеток саркомы.
Настоящее изобретение дополнительно предоставляет способ снижения и/или ингибирования активности CDK4 и/или CDK6 киназ в клетке, включающий введение в данную клетку эффективного количества соединения или его фармацевтически приемлемой соли, сложного эфира или сольвата или их стереоизомеров по настоящему изобретению. В предпочтительном варианте осуществления данный способ осуществляют in vivo, например, данной клеткой является клетка от субъекта (например, млекопитающего, такого как корова, лошадь, коза, свинья, собака, кошка, грызуны и приматы; например, человек); или данный способ осуществляют in vitro, например, данной клеткой является клетка in vitro (например, линия клеток или клетка от субъекта, такая как раковая клетка). В предпочтительном варианте осуществления данная клетка выбрана из клеток опухоли головного мозга, клеток рака легких, клеток плоскоклеточной карциномы, клеток карциномы мочевого пузыря, клеток рака желудка, клеток рака яичников, клеток перитонеального рака, клеток рака поджелудочной железы, клеток рака молочной железы, клеток рака головы и шеи, клеток рака шейки матки, клеток рака эндометрия, клеток рака прямой кишки, клеток рака печени, клеток почечной карциномы, клеток аденокарциномы пищевода, клеток плоскоклеточного рака пищевода, клеток рака предстательной железы, клеток рака женского репродуктивного протока, клеток рака in situ, клеток лимфомы, клеток нейрофибромы, клеток карциномы щитовидной железы, клеток остеокарциномы, клеток рака кожи, клеток рака мозга, клеток рака толстой кишки, клеток рака мужских половых органов, клеток стромальной опухоли желудочно-кишечного тракта, клеток новообразований предстательной железы, клеток опухоли тучных клеток, клеток множественной миеломы, клеток меланомы, клеток глиомы или клеток саркомы.
Настоящее изобретение дополнительно предоставляет набор для снижения или ингибирования активности CDK4 и/или CDK6 киназ в клетке, содержащий соединение или его фармацевтически приемлемую соль, сложный эфир или сольват или их стереоизомеры по настоящему изобретению и необязательно инструкции.
Технические преимущества настоящего изобретения
При сравнении с предшествующим уровнем техники, технические решения по настоящему изобретению обладают следующими преимуществами:
(1) соединение формулы (I') или формулы (I) или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по настоящему изобретению обладают прекрасной ингибирующей активностью в отношении CDK4/6 киназ;
(2) соединение формулы (I') или формулы (I) или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по настоящему изобретению проявляют хорошую биостабильность, более длительный эффект и высокую биодоступность;
(3) соединение формулы (I') или формулы (I) или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по настоящему изобретению проявляют хорошую проницаемость через гематоэнцефалический барьер, что делает возможным создание ингибиторов CDK в качестве лечебного средства при раке головного мозга;
(4) соединение формулы (I') или формулы (I) или его фармацевтически приемлемая соль, сложный эфир или сольват или их стереоизомеры по настоящему изобретению проявляют низкую токсичность, хорошую лекарственную резистентность и высокую безопасность.
Конкретные способы осуществления настоящего изобретения
Настоящее изобретение далее описано, но без ограничения, следующими вариантами осуществления. Специалист в данной области техники, на основе технических решений по настоящему изобретению, сможет осуществить различные модификации или улучшения, не отходя от существа и объема настоящего изобретения.
Эксперименты
Иллюстративные эксперименты предоставлены для части соединений по настоящему изобретению, чтобы продемонстрировать превосходство в активности и преимущества технического эффекта соединений по настоящему изобретению. Однако следует понимать, что следующие эксперименты предоставлены только в иллюстративных целях, а не ограничения объема настоящего изобретения. Специалист в данной области техники, на основе описанных технических решений по настоящему изобретению, сможет провести различные модификации или улучшения технических решений по настоящему изобретению, не отходя от существа и объема настоящего изобретения.
Экспериментальный пример 1: Биологические испытания соединений по настоящему изобретению на in vitro ферментативную ингибирующую активность
Тестируемое соединения: соединения 1, 13 и 14 по настоящему изобретению, химические названия, структуры и способы получения которых можно найти в примерах их получения.
Контрольные агенты: LY2835219, структуры которого можно найти в предшествующем уровне техники, получен авторами настоящего изобретения (ссылка на патент CN102264725A для способов получения).
Значения аббревиатур в представленных ниже экспериментах описаны следующим образом.
Экспериментальный метод: Измерение активности при ингибировании CDK4/6 киназ методом анализа по сдвигу пятна.
1. Получение 1-кратного буфера для киназ
1) Получение 1-кратного буфера для CDK4 киназы
800 мкл исходного раствора, содержащего 1000 мМ HEPES (pH 7,5), и 40 мкл исходного раствора, содержащего 10% Triton X-100, добавляли к 39160 мкл сверхчистой воды, и полученную в результате смесь смешивали до гомогенности.
2) Получение 1-кратного буфера для CDK6 киназы
50 мл исходного раствора, содержащего 1000 мМ HEPES (pH 7,5), и 50 мкл исходного раствора, содержащего 30% Brij-35, добавляли к 949,95 мл сверхчистой воды, и полученную в результате смесь смешивали до гомогенности.
2. Получение исходного раствора
25 мл исходного раствора, содержащего 4% окрашивающий агент #3 (подаваемый с применением 12-трубчатой пластины, используемой в измерительном устройстве для пятен), 50 мл исходного раствора, содержащего 1000 мМ HEPES (pH 7,5), 50 мл исходного раствора, содержащего 0,5M EDTA, и 0,25 мл исходного раствора, содержащего 30% Brij-35, добавляли к 374,75 мл сверхчистой воды, и полученную в результате смесь смешивали до гомогенности.
3. Получение 2,5-кратного раствора киназ
1) Получение 2,5-кратного раствора CDK4/D3 киназ
7 мкл CDK4/D3 ферментативного раствора и 9 мкл исходного раствора, содержащего 1M DTT, добавляли к 1784 мкл 1-кратного CDK4 киназного буфера, и полученную в результате смесь смешивали до гомогенности.
2) Получение 2,5-кратного раствора CDK6/D3 киназ
18 мкл CDK6/D3 ферментативного раствора и 14 мкл исходного раствора, содержащего 1M DTT, добавляли к 2768 мкл 1-кратного CDK6 киназного буфера, и полученную в результате смесь смешивали до гомогенности.
4. Получение 2,5-кратного полипептидного раствора
1) Получение 2,5-кратного CDK4/D3 полипептидного раствора
10 мкл исходного раствора, содержащего 100 мМ ATP, 45 мкл исходного раствора, содержащего 1M MgCl2, и 45 мкл FAM-меченного полипептида 8, добавляли к 1700 мкл 1-кратного CDK4 киназного буфера, и полученную в результате смесь смешивали до гомогенности.
2) Получение 2,5-кратного CDK6/D3 полипептидного раствора
23 мкл исходного раствора, содержащего 100 мМ ATP, 75 мкл исходного раствора, содержащего 1M MgCl2, и 75 мкл FAM-меченного полипептида 8, добавляли к 2827 мкл 1-кратного CDK6 киназного буфера, и полученную в результате смесь смешивали до гомогенности.
5. Получение 5-кратного раствора тестируемого соединения
ДМСО раствор, содержащий 10 мМ тестируемого соединения, разводили ДМСО до раствора, содержащего 50 мкМ тестируемого соединения (который использовали в качестве исходного раствора). Указанный исходный раствор доводили до 4-кратного ступенчатого разведения ДМСО, с получением растворов, содержащих 12,5 мкМ, 3,125 мкМ, 0,78 мкМ, 0,195 мкМ, 0,0488 мкМ, 12,2 нМ, 3 нМ, 0,76 нМ и 0,19 нМ тестируемого соединения, соответственно, и каждый из них разводили до 10-кратного 1-кратным киназным буфером до получения 5-кратного раствора соединения.
6. CDK4/6 ферментативная реакция
1) 5 мкл 5-кратного раствора тестируемого соединения и 10 мкл 2,5-кратного киназного раствора добавляли в соответствующие лунки 384-луночного планшета и инкубировали при комнатной температуре в течение 10 мин.
2) В соответствующие лунки добавляли 10 мкл 2,5-кратного полипептидного раствора для начала ферментативной реакции, и инкубацию осуществляли при 28°C в течение 5 часов.
7. Ферментативный анализ
В соответствующие лунки добавляли 25 мкл стоп-раствора для остановки реакции.
8. Полученные данные считывали измерительным устройством, скорость ингибирования рассчитывали по следующей формуле, и кривую строили с использованием программного обеспечения GraphPad5.0 с получением значений IC50.
Скорость ингибирования=(максимальное значение-значение образца)/(максимальное значение-минимальное значение)×100,
где максимальное значение: положительный контроль без добавления тестируемого соединения; минимальное значение: отрицательный контроль без добавления фермента.
Результаты эксперимента показаны в таблице 1:
In vitro ферментативное ингибирование активности соединениями по настоящему изобретению
Экспериментальное заключение: в таблице 1 показано, что соединения по настоящему изобретению обладают ингибирующей активностью в отношении CDK4 и CDK6 киназ, которая сравнима с контрольным агентом, т.е. показывают достаточную ингибирующую активность.
Экспериментальный пример 2: Биологические испытания на in vitro клеточную ингибирующую активность соединений по настоящему изобретению
Тестируемые соединения: соединения по настоящему изобретению, химические названия, формулы и способы получения которых можно найти в их примерах получения.
Контрольный агент: LY2835219, формулу которого можно найти в предшествующем уровне техники, получен авторами настоящего изобретения (ссылка на патент CN102264725A для способов получения).
Значения аббревиатур в представленных ниже экспериментах описаны следующим образом.
Экспериментальный метод: анализ на пролиферацию клеток осуществляли методом BrdU (BrdU набор для анализа клеточной пролиферации, от компании Cell Signaling Technology Company).
1. Получение реагентов и соединений
Получение 1-кратной промывной жидкости
20-кратную промывную жидкость в качестве исходного раствора разводили сверхчистой водой с получением 1-кратной жидкости.
Получение 1-кратного детекционного раствора антител
100-кратный BrdU детекционный исходный раствор антител разводили детекционным разбавителем антител с получением 1-кратного детекционного раствора антител.
Получение 1-кратного HRP-меченного вторичного раствора антител
100-кратный исходный раствор HRP-меченных антимышиных IgG антител разводили HRP-меченным разбавителем антител с получением 1-кратного HRP-меченного вторичного раствора антител.
10-кратный BrdU раствор
1000-кратный BrdU исходный раствор разводили соответствующей средой с получением 10-кратного BrdU раствора.
Получение тестируемого соединения
Получение исходного раствора тестируемого соединения: 10 мМ исходного раствора тестируемого соединения получали с использованием 100% ДМСО.
Получение градиентного разведения растворов тестируемых соединений: исходный раствор, содержащий 10 мМ тестируемого соединения, подвергали 4-кратному ступенчатому разведению ДМСО с получением растворов, содержащих 2,5 мМ, 625 мкМ, 156 мкМ, 39 мкМ, 9,8 мкМ и 2,5 мкМ тестируемого соединения, соответственно. 2 мкл ДМСО-разведенного соединения добавляли к 198 мкл культуральной среды, содержащей 10% FBS, с получением 10-кратного тестируемого соединения, где максимальная концентрация тестируемого соединения составляла 100 мкМ, концентрация ДМСО составляла 1%, и присутствовало 7 концентрационных градиентов.
Получение культуральной среды
MDA-MB-435S среда: L-15+10% FBS+0,01 мг/мл инсулина
MCF-7 среда: DMEM+10% FBS+0,01 мг/мл инсулина
U87MG среда: MEM+10% FBS
2. Стадии эксперимента
(1) Клетки, выращенные до 80% слияния (в фазе экспоненциального роста), подвергали ферментативному расщеплению с панкреатином и центрифугировали для собирания клеток. Клетки MDA-MB-435S и U87MG ресуспендировали в свободной от FBS- культуральной среде, подсчитывали и высевали в 96-луночный планшет; клетки MDA-MB-435S высевали при 3000 клеток/лунка/81 мкл; клетки U87MG высевали при 4000 клеток/лунка/81 мкл. Клетки MCF-7 ресуспендировали в культуральной среде, содержащей 1% FBS, и подсчитывали и высевали в 96-луночный планшет при 4000 клеток/лунка/82 мкл. Планшеты помещали в клеточный инкубатор при 37°C.
(2) Спустя 24 часа культивирования, добавляли FBS (9 мкл) в каждую лунку с клетками MDA-MB-435S и U87МГ, и добавляли 8 мкл FBS в каждую лунку с клетками MCF-7 для доведения конечной концентрации FBS до 10%.
(3) В каждую лунку добавляли различную концентрацию 10-кратного тестируемого соединения (10 мкл), получая конечную концентрацию тестируемого соединения 10 мкМ, 2,5 мкМ, 625 нМ, 156 нМ, 39 нМ, 9,8 нМ и 2,5 нМ, соответственно, в 3 повторах клеток/группа, и данные клетки культивировали в течение 72 часов при 37°C.
Контрольный растворитель: 0,1% ДМСО
Слепая контрольная проба: культуральная среда без добавления клеток
Нормальный клеточный контроль: нормальные клетки без какой-либо обработки
(4) В каждую лунку добавляли 10-кратный BrdU раствор (10 мкл), и полученную культуральную среду отбрасывали после инкубации в течение 4 часов в инкубаторе.
(5) В каждую лунку добавляли фиксирующий/денатурирующий раствор (10 мкл), полученный раствор отбрасывали после инкубации в течение 30 мин при комнатной температуре.
(6) В каждую лунку добавляли 1-кратный детекционный раствор антител (100 мкл), полученный раствор отбрасывали после инкубации в течение 1 часа, и лунки промывали 1-кратной промывной жидкостью при 200 мкл/лунка три раза.
(7) В каждую лунку добавляли 1-кратный HRP-меченный вторичный раствор антител (100 мкл), полученный раствор отбрасывали после инкубации в течение 30 мин при комнатной температуре, и лунки промывали 1-кратной промывной жидкостью при 200 мкл/лунка три раза.
(8) В каждую лунку добавляли раствор TMB субстрата (100 мкл), и инкубацию осуществляли при комнатной температуре в течение 30 мин.
(9) В каждую лунку добавляли стоп-раствор (100 мкл), и значение OD при 450 нм измеряли с помощью инструмента ELISA.
3. Обработка данных
1) Выживаемость клеток (%)=(ODтестируемое соединение-ODслепой контроль)/(ODнормальный клеточный контроль-ODслепой контроль)×100%,
где ODслепой контроль: значение слепого контроля; ODнормальный клеточный контроль: значение нормального клеточного контроля;
2) полученные данные обрабатывали с использованием программного обеспечения GraphPad Prism 5 для построения кривой и получения значений IC50.
Результаты экспериментов показаны в таблице 2:
In vitro клеточная ингибирующая активность соединений по настоящему изобретению
Экспериментальное заключение: как показано в таблице 2, соединения по настоящему изобретению обладают высокой in vitro клеточной ингибирующей активностью и явно превосходят LY2835219.
Экспериментальный пример 3: Биологические испытания фармакокинетики соединений по настоящему изобретению на голых мышах
Тестируемые соединения: часть соединений по настоящему изобретению, получены авторами настоящего изобретения; химические названия и способы их получения могут быть найдены в примерах их получения.
Контрольный агент: LY2835219, получен авторами настоящего изобретения (ссылка на патент CN102264725A для способов получения), их формула может быть найдена в предшествующем уровне техники.
Тестируемые животные: самки голых мышей (BALB/c); контрольный агент: 3 мыши/путь введения в каждой временной точке, масса: 22-25 г/мышь; соединение 1: 3 мыши/путь введения в каждой временной точке, масса: 22-25 г/мышь; соединение 6-1: 6 мышей/путь введения/тестируемое соединение, масса: 18-26 г/мышь.
Получение раствора тестируемых соединений
Получение пустого растворителя 1
MC (метилцеллюлоза) (500 мг) и SDS (додецилсульфат натрия) (100 мг) взвешивали и смешивали до гомогенности, и добавляли воду (100 мл); полученную в результате смесь растирали и смешивали до гомогенности при встряхивании с получением 0,5% MC+0,1% SDS раствора.
Получение пустого растворителя 2
Получение pH 5,0 буфера: дигидрофосфатидгидрат натрия (1,56 г) взвешивали и добавляли к очищенной воде, с получением 50 мл раствора, который доводили до pH 5,0 с использованием гидроксида натрия.
Получение пустого растворителя 3
0,1% Tween 80+2% HPC: HPC (гидроксипропилцеллюлоза) (20 г) взвешивали и медленно добавляли к очищенной воде (1000 мл) при перемешивании; затем добавляли Tween 80 (1 мл), и полученную в результате смесь перемешивали до получения прозрачного раствора.
Контрольный агент:
(1) Контрольный агент (12,18 мг) взвешивали, и добавляли нормальный солевой раствор (9,529 мл); полученную в результате смесь растворяли при воздействии ультразвука и смешивали до гомогенности при встряхивании, с получением 1 мг/мл бесцветного прозрачного раствора 1, который использовали в качестве жидкости 1 контрольного агента для ВВ введения голым мышам;
(2) контрольный агент (13,12 мг) взвешивали, и добавляли пустой растворитель 1 (10,265 мл); полученную в результате смесь растирали и смешивали до гомогенности, с получением 1 мг/мл гомогенного раствора 2, который использовали в качестве жидкости 2 контрольного агента для ПО введения голым мышам.
Соединение 1:
(1) Соединение 1 (5,79 мг) взвешивали, и добавляли ДМФА (N,N-диметилформамид) (0,556 мл); полученную в результате смесь растворяли при воздействии ультразвука; затем добавляли нормальный солевой раствор (4,999 мл), и полученную в результате смесь смешивали до гомогенности при встряхивании, с получением прозрачного раствора, который использовали в качестве жидкости 1 соединения 1 для ВВ введения голым мышам;
(2) соединение 1 (12,20 мг) взвешивали, и добавляли пустой растворитель 1 (11,706 мл); полученную в результате смесь растирали и смешивали до гомогенности, с получением 1 мг/мл гомогенного раствора, который использовали в качестве жидкости 2 соединения 1 для ПО введения голым мышам;
(3) соединение 1 (84,63 мг) взвешивали, и добавляли пустой растворитель 1 (8,120 мл); полученную в результате смесь растирали и смешивали до гомогенности, с получением 10 мг/мл гомогенного раствора, который использовали в качестве жидкости 3 соединения 1 для ПО введения голым мышам.
Соединение 6-1:
(1) Соединение 6-1 (2,01 мг) взвешивали, и добавляли пустой растворитель 2 (3,928 мл); полученную в результате смесь растворяли при воздействии ультразвука и смешивали до гомогенности при встряхивании, с получением 0,5 мг/мл гомогенного раствора, который использовали в качестве жидкости 1 соединения 6-1 для ВВ введения голым мышам;
(2) соединение 6-1 (5,11 мг) взвешивали, и добавляли пустой растворитель 3 (4,951 мл); полученную в результате смесь растирали до гомогенности при 1000 об./мин в измельчителе ткани; измельченную текучую среду переносили в пробирки для центрифугирования и смешивали до гомогенности при встряхивании, с получением 1 мг/мл гомогенного раствора, который использовали в качестве жидкости 2 соединения 6-1 для ПО введения голым мышам при 10 мг/кг;
(3) соединение 6-1 (51,10 мг) взвешивали и добавляли пустой растворитель 3 (1,496 мл); полученную в результате смесь растирали до гомогенности при 1000 об./мин в измельчителе ткани; измельченную текучую среду переносили в пробирки для центрифугирования, и добавляли указанный растворитель (1 мл) для промывания измельчителя ткани; после промывания, растворитель переносили в пробирки для центрифугирования; полученную в результате смесь смешивали до гомогенности при встряхивании, с получением 20 мг/мл гомогенного раствора, который использовали в качестве жидкости 3 соединения 6-1 для ПО введения голым мышам при 200 мг/кг.
Экспериментальный метод
Введение:
Тестируемые соединения вводили способами, перечисленными в следующей таблице:
Отбор крови:
Временная точка отбора крови
Контрольный агент/соединение 1, группа ВВ введения: перед введением, 0,083, 0,25, 0,5, 1, 2, 4, 6, 8, 24 и 30 часов после введения; группа ПО введения (10 мг/кг): перед введением, 0,167, 0,5, 1, 2, 4, 6, 8, 24 и 30 часов после введения.
Соединение 1, группа ПО введения (100 мг/кг): 0,167, 0,5, 1, 2, 4, 6, 8, 24, 30 и 48 часов после введения.
Соединение 6-1, группа ВВ введения (2,5 мг/кг) и группа ПО введения (10 мг/кг): 0,083, 0,25, 0,5, 1, 2, 4, 6, 8 и 24 часов после введения; соединение 6-1, группа ПО введения (100 мг/кг): 0,083, 0,25, 0,5, 1, 2, 4, 6, 8, 24, 30, 48 часов после введения.
Контрольный агент/соединение 1: 300 мкл цельной крови отбирали из внутриглазного угла глазной щели в каждой временной точке и центрифугировали при 8000 об./мин в течение 6 мин на сверхскоростной центрифуге для отделения плазмы; плазму сохраняли в холодильнике при -80°C. Соединение 6-1: 60 мкл цельной крови отбирали из внутриглазного угла глазной щели в каждой временной точке и помещали в антикоагуляционную пробирку, содержащую антикоагуляционный агент K2EDTA; образец крови центрифугировали при 8000 об./мин в течение 6 мин, с получением образца плазмы.
Анализ образца плазмы
Образцы плазмы контрольного агента и соединения 1 анализировали путем осаждения белка: к 50 мкл плазмы добавляли 200 мкл внутреннего стандарта (ацетонитрильный раствор, содержащей 500 нг/мл варфарина); полученную в результате смесь перемешивали при встряхивании в течение 3 мин при 1500 об./мин и центрифугировали в течение 5 мин при 13000 об./мин; к 100 мкл супернатанта добавляли 400 мкл воды; полученную в результате смесь смешивали до гомогенности при встряхивании и анализировали посредством ЖХ-МС/МС.
Образец плазмы соединения 6-1 анализировали путем осаждения белка: к подходящему количеству плазмы добавляли данный объем раствора внутреннего стандарта; полученную в результате смесь перемешивали при встряхивании и центрифугировали; супернатант разводили добавлением данного объема воды; полученную в результате смесь смешивали до гомогенности при встряхивании и анализировали посредством ЖХ-МС/МС.
Результаты оценки PK (ВВ) на голых мышах
(мг/кг)
(ч*нг/мл)
(л/ч/кг)
(л/кг)
Результаты оценки PK (ПО) на голых мышах
(мг/кг)
(ч*нг/мл)
(%)
CL представляет коэффициент очищения.
Vss представляет наблюдаемый объем характера распределения устойчивого состояния.
Tmax представляет время максимальной концентрации крови.
Cmax представляет максимальную концентрацию крови.
F% представляет абсолютную биодоступность.
Как видно из экспериментальных результатов, показанных в таблицах 3-1 и 3-2, при сравнении с контрольным агентом при такой же самой дозе, соединения по настоящему изобретению обладают хорошими фармакокинетическими свойствами и превосходят контрольный агент, в частности, что касается воздействия дозы и биодоступности; и группа высокого дозирования обладает биодоступностью, близкой к 100%.
Экспериментальный пример 4: Биологические испытания линейной фармакокинетики соединений по настоящему изобретению на крысах
Тестируемые соединения: соединение 1 по настоящему изобретению, получено авторами настоящего изобретения; химическое название и способ его получения можно найти в примерах получения.
Контрольный агент: LY2835219, получен авторами настоящего изобретения (ссылка на патент CN102264725A для способов получения); его формула может быть найдена в предшествующем уровне техники.
Тестируемые животные: самцы SD крыс, 3 крысы/вводимая доза/тестируемое соединение, масса: 200-240 г/крыса.
Получение раствора тестируемых соединений
Получение буферного раствора pH 4,0: лимонную кислоту (21g) растворяли в воде, и добавляли воду до конечного объема 1000 мл с получением раствора A, который хранили для дальнейшего использования; дигидрофосфат натрия (71,63 г) растворяли в воде, и добавляли воду до конечного объема 1000 мл, с получением раствора B, который хранили для дальнейшего использования. Указанный раствор A (614,5 мл) смешивали с указанным раствором B (385,5 мл) до гомогенности при встряхивании, с получением буферного растворителя с pH 4,0.
Контрольный агент:
(1) буферный растворитель с pH 4,0 использовали в качестве растворителя для получения раствора 1 контрольного агента LY2835219 при концентрации 1,5 мг/мл;
(2) буферный растворитель с pH 4,0 использовали в качестве растворителя для получения раствора 2 контрольного агента LY2835219 при концентрации 4,5 мг/мл;
(3) буферный растворитель с pH 4,0 использовали в качестве растворителя для получения раствора 3 контрольного агента LY2835219 при концентрации 13,5 мг/мл;
указанные растворы 1-3 использовали в качестве текучей среды для ПО введения контрольного агента LY2835219 при 15 мг/кг, 45 мг/кг и 135 мг/кг.
Для соединения 1:
(1) буферный растворитель с pH 4,0 использовали в качестве растворителя для получения раствора 4 соединения 1 при концентрации 13,5 мг/мл;
(2) указанный гомогенный раствор 4 (4 мл) разводили буферным растворителем с pH 4,0 для получения раствора 5 соединения 1 при концентрации 4,5 мг/мл;
(3) указанный гомогенный раствор 4 (1,4 мл) разводили буферным растворителем с pH 4,0 для получения раствора 6 соединения 1 при концентрации 1,5 мг/мл;
указанные растворы 4-6 использовали в качестве текучей среды для ПО введения соединения 1 при 15 мг/кг, 45 мг/кг и 135 мг/кг.
Экспериментальный метод
Введение:
Тестируемые соединения вводили способами, перечисленными в следующей таблице:
Отбор крови
Контрольный агент:
Временная точка отбора крови для группы ПО введения (15 мг/кг), 0,167, 0,5, 1, 2, 4, 6, 8, 24, 30 и 48 часов после введения. Для группы ПО введения (45 мг/кг), 0,167, 0,5, 1, 2, 4, 6, 8, 24, 30, 48, 56 и 72 часов после введения. Для группы ПО введения (135 мг/кг), 0,167, 0,5, 1, 2, 4, 6, 8, 24, 30, 48, 56, 72, 96 и 102 часов после введения.
Для соединения 1:
Временная точка отбора крови для группы ПО введения (15 мг/кг), 0,167, 0,5, 1, 2, 4, 6, 8, 24, 30 и 48 часов после введения. Для группы ПО введения (45 мг/кг), 0,167, 0,5, 1, 2, 4, 6, 8, 24, 30 и 48 часов после введения. Для группы ПО введения (135 мг/кг), 0,167, 0,5, 1, 2, 4, 6, 8, 24, 30, 48, 56, 72 и 96 часов после введения.
100 мкл цельной крови отбирали из хвостовой вены в каждой временной точке, добавляли в антикоагуляционную пробирку, содержащую K2EDTA, и центрифугировали при 8000 об./мин на сверхскоростной центрифуге в течение 6 мин для отделения плазмы; плазму сохраняли в холодильнике при -80°C.
Анализ образца плазмы
Образцы плазмы контрольного агента и соединения 1 анализировали путем осаждения белка: к 30 мкл плазмы добавляли 200 мкл внутреннего стандарта (ацетонитрильный раствор, содержащей 50 нг/мл PD0332991, формулы 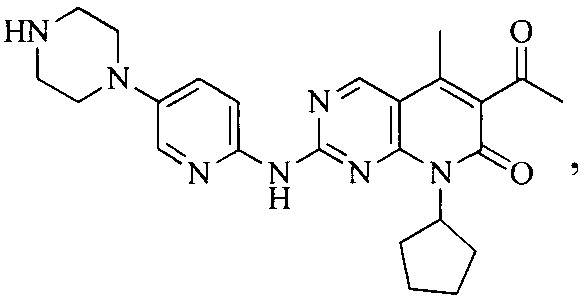 , полученного со ссылкой на способ получения по патенту CN101001857A); полученную в результате смесь перемешивали при встряхивании в течение 10 мин при 1500 об./мин и центрифугировали в течение 20 мин при 4000 об./мин; к 100 мкл супернатанта добавляли 100 мкл воды; полученную в результате смесь смешивали до гомогенности при встряхивании и анализировали посредством ЖХ-МС/МС.
, полученного со ссылкой на способ получения по патенту CN101001857A); полученную в результате смесь перемешивали при встряхивании в течение 10 мин при 1500 об./мин и центрифугировали в течение 20 мин при 4000 об./мин; к 100 мкл супернатанта добавляли 100 мкл воды; полученную в результате смесь смешивали до гомогенности при встряхивании и анализировали посредством ЖХ-МС/МС.
Результаты оценки PK контрольного агента (ПО) на крысах
(мг/кг)
(ч)
(нг/мл)
(ч*нг/мл)
Результаты оценки PK соединения 1 (ПО) на крысах
(мг/кг)
(ч)
(нг/мл)
(ч*нг/мл)
Cmax представляет максимальную концентрацию крови.
AUClast представляет область под кривой концентрация-время в ходе введения 0→t.
Как видно из экспериментальных результатов, показанных в таблицах 4-1 и 4-2, при сравнении с контрольным агентом при такой же самой дозе, соединения по настоящему изобретению превосходят контрольный агент, что касается Cmax и AUClast; и с увеличением дозы AUClast увеличивается линейно; соединение 1 по настоящему изобретению обладает хорошими линейными фармакокинетическими свойствами.
Экспериментальный пример 5: Оценка фармакодинамики и фармацевтической безопасности соединений по настоящему изобретению на модели подкожной опухоли BALB/c голых мышей с клеточным раком толстой кишки Colo-205 человека
Тестируемые соединения: соединение 1 по настоящему изобретению, химическое название и способ получения которого можно найти в примере получения.
Контрольный агент: LY2835219, формулу которого можно найти в предшествующем уровне техники, получен авторами настоящего изобретения (ссылка на патент CN102264725A для способов получения).
Значения аббревиатур в представленных ниже экспериментах описаны следующим образом.
Тестируемые животные: самки голых мышей (BALB/c), 8 мышь/группа дозирования/тестируемое соединение, масса: 19-27 г/мышь.
Получение раствора тестируемых соединений
Получение пустого растворителя, содержащего 0,5% MC и 0,1% SDS: MC (2 г) взвешивали и растворяли добавлением сверхчистой воды (350 мл); затем добавляли SDS (0,4 г), и объем доводили до 400 мл. После тщательного смешивания, полученную в результате смесь фильтровали через 0,22 мкМ фильтр для удаления бактерий, и упаковывали в субпакеты и хранили при 4°C, с получением данного пустого растворителя.
Контрольный агент (32 мг) взвешивали, и добавляли растворитель (2,5 мл); полученную в результате смесь смешивали до гомогенности при встряхивании, с получением гомогенного раствора 1, содержащего контрольный агент при концентрации 10 мг/мл; добавляли растворитель (1,5 мл) к гомогенному раствору 1 (0,5 мл), с получением гомогенного раствора 2, содержащего контрольный агент при концентрации 2,5 мг/мл.
Соединение 1 (6,3 мг) взвешивали, и добавляли растворитель (2,4 мл); полученную в результате смесь смешивали до гомогенности при встряхивании, с получением гомогенного раствора 1, содержащего соединение 1 при концентрации 2,5 мг/мл; соединение 1 (12,6 мг) взвешивали, и добавляли растворитель (2,4 мл); полученную в результате смесь смешивали до гомогенности при встряхивании, с получением гомогенного раствора 2, содержащего соединение 1 при концентрации 5 мг/мл; соединение 1 (25,2 мг) взвешивали, и добавляли растворитель (2,4 мл); полученную в результате смесь смешивали до гомогенности при встряхивании, с получением гомогенного раствора 3, содержащего соединение 1 при концентрации 10 мг/мл. Указанные гомогенные растворы контрольного агента и соединения 1 получали непосредственно перед введением каждый день и хранили в темноте.
Экспериментальный метод
Культура клеток:
Раковые клетки COLO205 культивировали в среде RPMI-1640, содержащей инактивированную 10% фетальную телячью сыворотку, 100 Ед/мл пенициллина, 100 мкг/мл стрептомицина и 2 мМ глутамина, в инкубаторе при 37°C, 5% CO2. Первоначальная концентрация клеточной культуры составляла 5×105 клеток/мл, и данные клетки переносили в другие бутыли каждые 3 или 4 дня и пассировали. Опухолевые клетки в фазе экспоненциального роста использовали для in vivo посева опухоли.
Посев клеток и группировка:
Клетки опухоли COLO205, суспендированные в PBS, высевали подкожно на правой боковой грудной клетки экспериментальных животных при 1×107 клеток/0,1 мл, и животного умерщвляли, когда опухоль дорастала до объема от 800 мм до 1000 мм; в асептических условиях опухоли снимали, и некротические ткани удаляли; отбирали хорошо выросшие опухолевые ткани; опухолевые клетки отделяли и подвергали воздействию на первичную культуру и пролиферацию, и затем подкожно высевали животным. Опухолевые клетки пассировали двум поколениям мышей этим методом, и первичную культуру клеток высевали подкожно на правой боковой грудной клетке экспериментальных животных, при 5×106 клеток/0,1 мл на животное. Когда опухоль вырастала до объема около 100 мм3, животных группировали и осуществляли введение. Для перорального введения вводимый объем составлял 0,1 мл/10 г массы тела животного.
Введение:
Тестируемые соединения вводили способами, перечисленными в следующей таблице:
(мг/мл)
Отбор крови: кровь отбирали из внутриглазного угла глазной щели. После последнего введения (22-ой день) контрольного агента и соединения 1, отбирали кровь/опухоль в тех же самых временных точках, где 8 голых мышей каждой группы дозирования разделяли на группы A и B.
Группа A (№ 1-4): кровь отбирали на 15 минуте, 1 часе и 2 часе, и мышей умерщвляли после отбора крови на 2 часе;
группа B (№ 5-8): кровь отбирали на 4 часе, 8 часе и 24 часе, и мышей умерщвляли после отбора крови на 24 часе;
100 мкл цельной крови в каждой временной точке отбирали и добавляли в антикоагуляционную пробирку, содержащую K2EDTA, и центрифугировали при 4500 об./мин при 4°C на сверхскоростной центрифуге в течение 5 мин для отделения плазмы; плазму сохраняли в холодильнике при -80°C.
Анализ образца плазмы
Образцы плазмы контрольного агента и соединения 1 анализировали путем осаждения белка: к 50 мкл плазмы добавляли 150 мкл внутреннего стандарта (ацетонитрильный раствор, содержащей 50 нг/мл дексаметазона); полученную в результате смесь перемешивали при встряхивании при 1500 об./мин в течение 3 мин и центрифугировали при 4000 об./мин в течение 20 мин; к 100 мкл супернатанта добавляли 100 мкл воды; полученную в результате смесь смешивали до гомогенности при встряхивании и анализировали посредством ЖХ-МС/МС.
Результаты оценки PK контрольного агента на голых мышах
(мг/кг)
(ч*нг/мл)
Результаты оценки PK соединения 1 на голых мышах
(мг/кг)
(h*нг/мл)
Tmax представляет время максимальной концентрации крови.
Cmax представляет максимальную концентрацию крови.
Как видно из экспериментальных результатов, показанных в таблицах 5-1 и 5-2, при сравнении с контрольным агентом, соединение 1 по настоящему изобретению обладает хорошими фармакокинетическими свойствами; соединение 1 превосходит контрольный агент при такой же самой дозе, что касается влияния дозы на AUClast и Cmax.
Экспериментальный пример 6: In vivo биологические испытания по оценке проницаемости через гематоэнцефалический барьер головного мозга соединений по настоящему изобретению на крысах
Тестируемые соединения: соединение 1 по настоящему изобретению, получено авторами настоящего изобретения; химическое название и способ его получения можно найти в примерах получения.
Контрольный агент: LY2835219, формулу которого можно найти в предшествующем уровне техники, получен авторами настоящего изобретения (ссылка на патент CN102264725A для способов получения).
Тестируемые животные: самцы SD крыс, 3 крысы/тестируемое соединение/временная точка, масса: 250-300 г/крыса.
Получение раствора тестируемого соединения
Получение растворителя (0,5% MC (метилцеллюлоза)+0,1% SDS (додецилсульфат натрия) раствор): MC (500 мг) и SDS (100 мг) смешивали до гомогенности, и добавляли воду (100 мл); полученную в результате смесь растирали и смешивали до гомогенности при встряхивании.
Контрольный агент (108,22 мг) взвешивали, и добавляли растворитель (36,07 мл); полученную в результате смесь растирали и суспендировали до гомогенности, с получением гомогенного раствора 1. Полученный раствор использовали в качестве жидкости для ПО введения контрольного агента LY2835219 крысам.
Соединение 1 (111,52 мг) взвешивали, и добавляли растворитель (35,39 мл); полученную в результате смесь растирали и суспендировали до гомогенности, с получением гомогенного раствора 2. Полученный раствор использовали в качестве жидкости для ПО введения соединения 1 крысам.
Экспериментальный метод
Введение:
Тестируемые соединения вводили способами, перечисленными в следующей таблице:
Метод обора:
После раздельного PO введения контрольного агента и соединения 1, отбирали кровь пункцией из сердца во временных точках 2, 4, 24 и 48 часа; отбирали около 8 мл цельной крови и помещали в центрифужную пробирку, содержащую антикоагуляционный агент K2EDTA; спинномозговую жидкость и ткани мозга также собирали в то же самое время, у 3 крыс в каждой временной точке. Собранную цельную кровь центрифугировали при 3000 об./мин при 4°C на сверхскоростной центрифуге в течение 10 мин для отделения плазмы; и плазму сохраняли в холодильнике при -80°C. После сбора образцов, определяли концентрации контрольного агента и соединения 1 в плазме, ткани мозга и спинномозговой жидкости, соответственно.
Анализ образца плазмы
Образцы плазмы контрольного агента и соединения 1 анализировали путем осаждения белка: тестируемые образцы вынимали из холодильника (-80°C), оттаивали при комнатной температуре и затем перемешивали при встряхивании в течение 3 мин; к 30 мкл плазмы добавляли 200 мкл внутреннего стандарта (ацетонитрильный раствор, содержащей 25 нг/мл PD0332991); полученную в результате смесь перемешивали при встряхивании в течение 10 мин при 1500 об./мин и центрифугировали при 4000 об./мин в течение 20 мин; к 100 мкл супернатанта добавляли 100 мкл воды; полученную в результате смесь смешивали до гомогенности при встряхивании и анализировали посредством ЖХ-МС/МС.
Анализ образцов ткани головного мозга спинномозговой жидкости
Образцы ткани головного мозга и спинномозговой жидкости для контрольного агента и соединения 1 анализировали путем осаждения белка: тестируемые образцы вынимали из холодильника (-80°C), оттаивали при комнатной температуре и затем перемешивали при встряхивании в течение 3 мин; к 20 мкл образцу гомогенизированной ткани головного мозга добавляли 200 мкл внутреннего стандарта (раствор метанола и воды при 1:1, содержащий 40 нг/мл декстрометорфана и 50 нг/мл декстрорфана); полученную в результате смесь перемешивали при встряхивании в течение 3 мин при 1500 об./мин и центрифугировали при 4°C, 4000 g в течение 5 мин; к 100 мкл супернатанта добавляли 50 мкл 0,1% смеси муравьиная кислота-вода; полученную в результате смесь смешивали до гомогенности при встряхивании и анализировали посредством ЖХ-МС/МС. Образец спинномозговой жидкости обрабатывали таким же образом, как образец гомогенизированной ткани головного мозга.
Экспериментальные результаты оценки проницаемости гематоэнцефалического барьера головного мозга соединением 1 по настоящему изобретению у крыс
(ч)
(нг/мл)
(нг·ч/мл)
Tmax представляет время максимальной концентрации крови.
Cmax представляет максимальную концентрацию крови.
Как видно из экспериментальных результатов, показанных в таблице 6, при сравнении с контрольным агентом, соединения по настоящему изобретению могут эффективно войти в ткань головного мозга из плазмы при биологическом испытании оценки проницаемости гематоэнцефалического барьера головного мозга, и имеют преимущества перед контрольным агентом LY2835219, что касается влияния дозы на спинномозговую жидкость.
Экспериментальный пример 7: Оценка фармакодинамики и фармацевтической безопасности соединений по настоящему изобретению на модели подкожной опухоли BALB/c голых мышей с клеточной глиомой головного мозга U-87MG человека
Тестируемые соединения: соединения настоящего изобретения, получены авторами настоящего изобретения; химические названия и способы их получения могут быть найдены в примерах их получения.
Контрольный агент: LY2835219, формулу которого можно найти в предшествующем уровне техники, получен авторами настоящего изобретения (ссылка на патент CN102264725A для способов получения).
Значения аббревиатур в представленных ниже экспериментах описаны следующим образом.
Тестируемые животные: самки голых мышей (BALB/c), 10 мышей/группа дозирования/тестируемое соединение, масса: 18-22 г/мышь.
Получение раствора тестируемых соединений
Получение растворителя, содержащего 0,5% MC: подходящее количество MC растворяли в заданном объеме сверхчистой воды; полученную в результате смесь смешивали до гомогенности, с получением растворителя 1, содержащего 0,5% MC.
Получение растворителя, содержащего 0,5% MC и 0,1% SDS: подходящее количество MC и подходящее количество SDS растворяли в заданном объеме сверхчистой воды; полученную в результате смесь смешивали до гомогенности, с получением растворителя 2, содержащего 0,5% MC и 0,1% SDS.
Контрольный агент (508,87 мг) и SDS (39,37 мг) взвешивали, и разбавляли растворителем 1 (39,37 мл); полученную в результате смесь растирали до гомогенности, с получением гомогенного раствора 1, содержащего контрольный агент при концентрации 10,1 мг/мл; брали гомогенный раствор 1 (13,125 мл), содержащей контрольный агент, и добавляли растворитель 2 (13,125 мл); полученную в результате смесь растирали до гомогенности, с получением гомогенного раствора 2, содержащего контрольный агент при концентрации 5,0 мг/мл.
Соединение 1 (931,77 мг) и SDS (45,9 мг) взвешивали и разбавляли растворителем 1 (45,9 мл); полученную в результате смесь растирали до гомогенности, с получением гомогенного раствора 1, содержащего соединение 1 при концентрации 20,0 мг/мл; брали гомогенный раствор 1 (19,69 мл), содержащей соединение 1 (20,0 мг/мл), и добавляли растворитель 2 (19,69 мл); полученную в результате смесь растирали до гомогенности, с получением гомогенного раствора 2, содержащего соединение 1 при концентрации 10,0 мг/мл; брали гомогенный раствор 2 (13,125 мл), содержащей соединение 1 (10,0 мг/мл), и добавляли растворитель 2 (13,125 мл); полученную в результате смесь растирали до гомогенности, с получением гомогенного раствора 3, содержащего соединение 1 при концентрации 5,0 мг/мл.
Экспериментальный метод
Культура клеток:
Клетки U87MG из ATCC культивировали в MEM среде, содержащей 10% фетальную телячью сыворотку; данные клетки подвергали ферментативному расщеплению с трипсином, содержащим EDTA, общепринятыми методами; полученные клетки пассировали двум поколениям каждую неделю и продолжали культивировать в инкубаторе при 37°C, 5% CO2.
Посев клеток и группировка:
Опухолевые клетки в фазе экспоненциального роста собирали; клетки U87MG корректировали с помощью PBS и матригеля в соотношении 1:1 до концентрации 5×107 клеток/мл; 0,1 мл клеточной суспензии высевали подкожно в правой задней части каждой мыши путем инъекции 1 мл шприцем. Объемы опухолей обследовали и измеряли, и средним объемом опухоли U87MG считалось около 200 мм3 через 8 дней после высевания; страдающих опухолью мышей группировали и обрабатывали путем перорального введения тестируемых соединений при дозе 0,1 мл/10 г массы тела.
Введение:
Тестируемые соединения вводили способами, перечисленными в следующей таблице:
Отбор крови: кровь отбирали из внутриглазного угла глазной щели. После последнего введения (день 22) контрольного агента и соединения 1, кровь/опухоль отбирали в тех же самых временных точках, где, как и у 10 голых мышей для каждой группы дозирования, одна была в режиме ожидания, и 9 других мышей были разделены на три группы, т.е. группу A, B и C.
Группа A (№ 1-3): кровь отбирали перед введением и во временной точке 1 час и 2 часа после введения, и мышей умерщвляли после отбора крови в точке 2 часа;
группа B (№ 4-6): кровь отбирали во временной точке 15 мин, 4 часа и 6 часов после введения, и мышей умерщвляли после отбора крови в точке 6 часов;
группа C (№ 7-9): кровь отбирали в точке 30 мин, 10 часов и 24 часа после введения, и мышей умерщвляли после отбора крови в точке 24 часа;
300 мкл цельной крови отбирали в каждой временной точке, добавляли в антикоагуляционную пробирку, содержащую K2EDTA, и центрифугировали при 8000 об./мин на сверхскоростной центрифуге в течение 6 мин для отделения плазмы; плазму сохраняли в холодильнике при -80°C.
Анализ образца плазмы
Образцы плазмы контрольного агента и соединения 1 анализировали путем осаждения белка: 30 мкл плазмы добавляли в 96-луночный глубокий планшет, и добавляли 200 мкл внутреннего стандарта (ацетонитрильный раствор, содержащей 25 нг/мл PD0332991); полученную в результате смесь перемешивали при встряхивании в течение 3 мин при 1500 об./мин и центрифугировали в течение 20 мин при 4000 об./мин; к 100 мкл супернатанта добавляли 100 мкл воды; полученную в результате смесь смешивали до гомогенности при встряхивании и анализировали посредством ЖХ-МС/МС.
Результаты оценки PK контрольного агента на голых мышах
(мг/кг)
(ч*нг/мл)
Результаты оценки PK соединения 1 на голых мышах
(мг/кг)
(ч*нг/мл)
Tmax представляет время максимальной концентрации крови.
Cmax представляет максимальную концентрацию крови.
Как видно из экспериментальных результатов, показанных в таблицах 7-1 и 7-2, при сравнении с контрольным агентом, соединения по настоящему изобретению обладают хорошими фармакокинетическими свойствами; соединение 1 превосходит положительный контрольный агент при такой же самой дозе, что касается влияния дозы и Cmax.
Схемы получения
Иллюстративные схемы получения предоставлены для части соединений по настоящему изобретению. Однако следует понимать, что следующие эксперименты предоставлены только для целей, иллюстрирующих изобретение, не ограничивая объем настоящего изобретения. Специалист в данной области техники на основе технических решений, содержащихся в данном описании, сможет произвести различные модификации или улучшения технических решений по настоящему изобретению, не отходя от существа и объема настоящего изобретения.
Пример получения 1: Получение 5-(бромметил)-2-хлорпиримидина
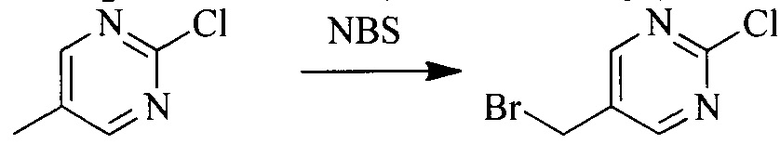
2-хлор-5-метилпиримидин (12,86 г, 0,1 моль) растворяли в четыреххлористом углероде (300 мл), и добавляли N-бромбутанимид (25,72 г, 0,14 моль), и добавляли бензоилпероксид (1,29 г, 5 ммоль) при перемешивании. Полученную в результате смесь нагревали при кипячении с обратным холодильником на масляной бане и охлаждали до комнатной температуры после взаимодействия в течение 8 часов. Полученную смесь фильтровали при отсасывании. Фильтрат концентрировали и затем подвергали колоночной хроматографии на силикагеле (петролейный эфир:этилацетат=1:1), с получением указанного в заголовке соединения (8,7 г, выход: 42%).
Пример получения 2: Получение 2,5-дифтор-4-йодпиридина
Диизопропиламин (17 мл, 122 ммоль) добавляли к тетрагидрофурану (220 мл), и полученную смесь охлаждали до -20°C. В защитной атмосфере азота медленно добавляли н-бутиллитий (49 мл, 122,5 ммоль). После добавления, полученную смесь перемешивали при -20°C в течение 0,5 часа и охлаждали до -78°C. Медленно добавляли 2,5-дифторпиридин (13,3 г, 115 ммоль) в тетрагидрофуране (30 мл) и перемешивали при данной температуре в течение 4 часов. Йод (32 г, 126 ммоль) растворяли в тетрагидрофуране (100 мл) и медленно добавляли к указанному реакционному раствору при -78°C. После добавления, полученную смесь перемешивали в течение 1 часа. После добавления воды (10 мл) и тетрагидрофурана (30 мл), температуру поднимали до комнатной температуры. Добавляли насыщенный раствор тиосульфита натрия. Органическую фазу отделяли, и водную фазу экстрагировали этилацетатом (3×100 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали при отсасывании, и фильтрат концентрировали и затем подвергали колоночной хроматографии на силикагеле (петролейный эфир:этилацетат=50:1), с получением указанного в заголовке соединения (13,5 г, выход: 48%).
Пример получения 3: Получение 5-фтор-4-йодпиридин-2-амина
2,5-дифтор-4-йодпиридин (4,82 г, 20 ммоль) растворяли в ДМСО (40 мл), и добавляли водный аммиак (40 мл) при перемешивании. Полученную смесь перемешивали в течение 12 ч при 90°C в закрытой трубке. Добавляли этилацетат (150 мл) для отделения органической фазы, и органическую фазу сушили над безводным сульфатом натрия, фильтровали при отсасывании. Фильтрат концентрировали и затем подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=30:1), с получением указанного в заголовке соединения (2,38 г, выход: 50%).
Пример получения 4: Получение N-изопропилацетамида
Изопропиламин (5,0 г, 84,7 ммоль) и триэтиламин (12,8 г, 126,7 ммоль) растворяли в дихлорметане (50 мл), и температуру снижали до 0°C. Медленно добавляли уксусный ангидрид (17,3 г, 169,4 ммоль). После добавления, полученную смесь нагревали до комнатной температуры и подвергали взаимодействию в течение 12 ч. Растворитель удаляли перегонкой при пониженном давлении. К полученному остатку добавляли этилацетат (100 мл) и карбонат калия (20 г). После перемешивания в течение 6 часа, полученную смесь фильтровали при отсасывании, и растворитель удаляли перегонкой при пониженном давлении, с получением указанного в заголовке соединения в виде масла (7,65 г, выход: 89,5%).
Пример получения 5: Получение (Z)-N'-(4-бром-2,6-дифторфенил)-N-изопропилацетоамидина
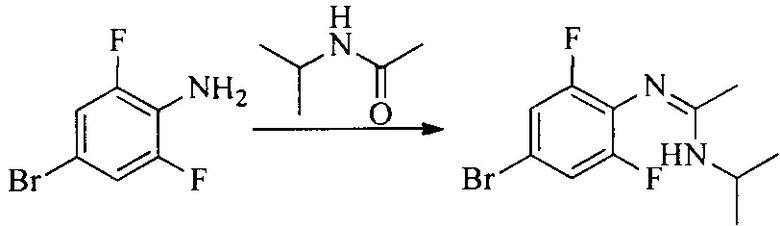
4-бром-2,6-дифторанилин (7,87 г, 37,9 ммоль), N-изопропилацетамид (7,65 г, 75,7 ммоль) и триэтиламин (5,7 г, 56,9 ммоль) растворяли в толуоле (150 мл), и медленно добавляли оксихлорид фосфора (5,8 г, 37,9 ммоль). После добавления, полученную смесь нагревали при кипячении с обратным холодильником в течение 3 часов. Затем полученную реакционную смесь охлаждали до комнатной температуры, растворитель удаляли перегонкой при пониженном давлении, и добавляли дихлорметан (200 мл). Полученную в результате смесь дважды промывали насыщенным раствором гидрокарбоната натрия (2×100 мл), сушили над безводным сульфатом натрия, фильтровали при отсасывании и перегоняли при пониженном давлении, с получением указанного в заголовке соединения (7,3 г, выход: 66,2%).
Пример получения 6: Получение 6-бром-4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазола
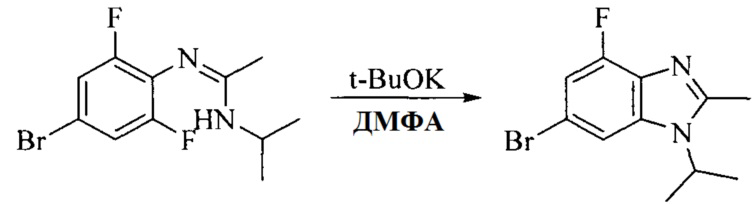
(Z)-N'-(4-бром-2,6-дифторфенил)-N-изопропилацетоамидин (7,0 г, 24,1 ммоль) растворяли в N,N-диметилформамиде (50 мл), и добавляли трет-бутоксид калия (3,2 г, 28,9 ммоль). Полученную в результате смесь нагревали до 100°C и подвергали взаимодействию в течение 2 ч. Полученную реакционную смесь охлаждали до комнатной температуры, добавляли дихлорметан (100 мл) и насыщенную NaCl воду (50 мл) к отделенной органической фазе. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали, с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=2:1), с получением указанного в заголовке соединения (4,0 г, выход: 61,3%).
Пример получения 7: Получение 4-фтор-1-изопропил-2-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-бензо[d]имидазола
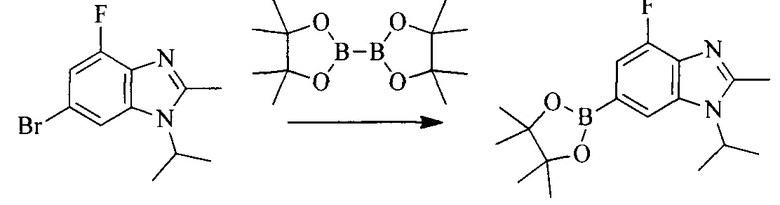
6-бром-4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол (9,0 г, 33,2 ммоль), бис(пинаколато)диборон (12,65 г, 49,8 ммоль), ацетат палладия (840 мг, 3,75 ммоль), трициклогексилфосфин (1,63 г, 5,8 ммоль) и ацетат калия (9,78 г, 99,8 ммоль) добавляли к ДМСО (60 мл). В защитной атмосфере азота полученную смесь нагревали до 80°C и подвергали взаимодействию в течение 6 часов. Воду (200 мл) и этилацетат (200 мл) добавляли к отделенной от водной фазы органической фазе. Водную фазу дважды экстрагировали этилацетатом, и органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали при отсасывании. Фильтрат концентрировали и затем подвергали колоночной хроматографии на силикагеле (петролейный эфир:этилацетат=1:2), с получением указанного в заголовке соединения (6,0 г, выход: 56,8%).
Пример получения 8: Получение 5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амина
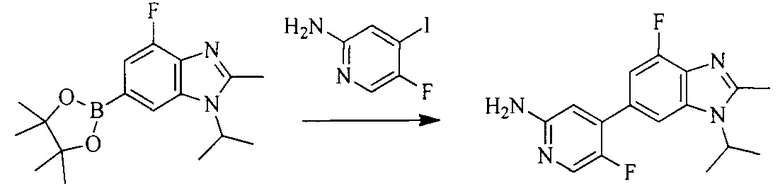
4-фтор-1-изопропил-2-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-бензо[d]имидазол (3,18 г, 10 ммоль), 5-фтор-4-йодпиридин-2-амин (2,38 г, 10 ммоль), карбонат калия (2,76 г, 20 ммоль) и тетракис(трифенилфосфин)палладий(1,15 г, 1 ммоль) добавляли в 35 мл микроволновую пробирку, и добавляли 1,4-диоксан (15 мл) и воду (3 мл). Полученной смеси давали взаимодействовать при 120°C в условиях микроволнового облучения в течение 1 часа. Воду и этилацетат добавляли к отделенной от водной фазы органической фазе. Органическую фазу сушили над безводным сульфатом натрия и фильтровали при отсасывании. Фильтрат концентрировали и затем подвергали колоночной хроматографии на силикагеле (петролейный эфир:этилацетат=1:1), с получением указанного в заголовке соединения (2,1 г, выход: 69,5%).
Пример получения 9: Получение 4-фтор-6-(5-фтор-2-йодпиридин-4-ил)-1-изопропил-2-метил-1H-бензо[d]имидазола
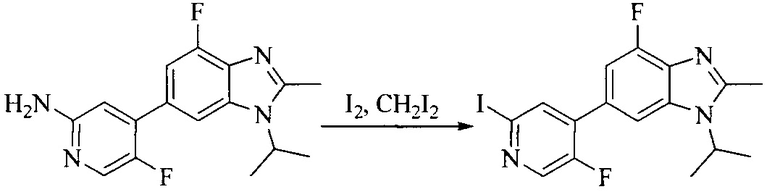
5-Фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (1,0 г, 3,31 ммоль) растворяли в дийодометане (30 мл), и добавляли CuI (0,63 г, 3,32 ммоль), трет-бутилнитрил (0,341 г, 3,31 ммоль) и йод (0,84 г, 3,31 ммоль). Полученную смесь нагревали до 85°C и подвергали взаимодействию в течение 15 мин. Полученную в результате смесь охлаждали и затем непосредственного разделяли колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=200:1~1:1), с получением указанного в заголовке соединения (700 мг, выход: 51,1%).
Пример получения 10: Получение 5-бром-2-аминопиримидина
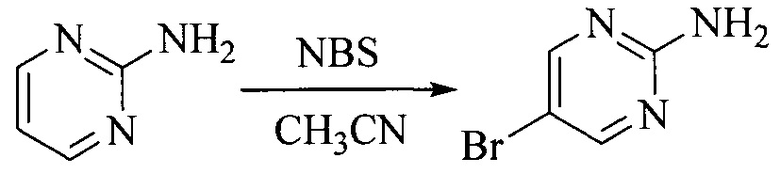
2-аминопиримидин (20,0 г, 0,21 моль) растворяли в ацетонитриле (500 мл), и добавляли N-бромбутанимид (37,0 г, 0,21 моль). Полученную смесь перемешивали при 20°C в течение 4 часов и фильтровали при отсасывании. Осадок на фильтре сушили, с получением продукта (33,0 г, выход: 90,2%).
Пример получения 11: Получение 5-бром-N,N-бис(4-метоксибензил)пиримидин-2-амина
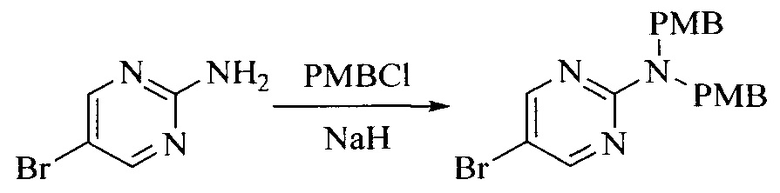
5-Бром-2-аминопиримидин (8,9 г, 51,15 ммоль) растворяли в тетрагидрофуране (150 мл), и медленно добавляли гидрид натрия (4,4 г, 110,0 ммоль, 60%) в условиях охлаждения на ледяной бане. Полученную смесь нагревали до комнатной температуры и перемешивали в течение 30 мин. Добавляли 4-метоксибензилхлорид (20,0 г, 127,71 ммоль), и полученную смесь нагревали до 75°C и подвергали взаимодействию в течение 8 ч. Добавляли ледяную воду для гашения реакции, и реакционный раствор экстрагировали этилацетатом (200 мл). Водную фазу промывали этилацетатом (50 мл), и органические фазы объединяли, сушили, концентрировали и разделяли колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=20:1), с получением указанного в заголовке соединения (10,0 г, выход: 47,2%).
Пример получения 12: Получение трет-бутил 4-(2-(бис(4-метоксибензил)амино)пиримидин-5-ил)пиперазин-1-карбоксилата
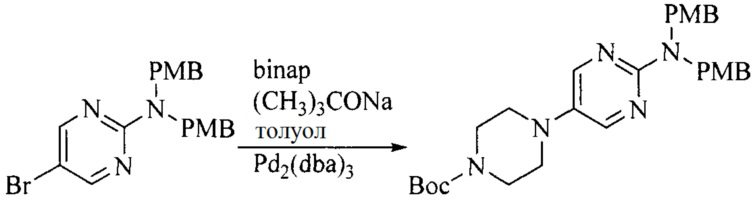
5-бром-N,N-бис(4-метоксибензил)пиримидин-2-амин (10,0 г, 24,1 ммоль), трет-бутилпиперазин-1-карбоксилат (8,97 г, 48,2 ммоль), трет-бутоксид натрия (4,63 г, 48,2 ммоль), трис(дибензилиденацетон)дипалладий (200 мг) и рацемический -2,2'-бис(дифенилфосфино)-1,1'-бинафтил (400 мг) добавляли к толуолу (200 мл). В защитной атмосфере азота полученную смесь нагревали при 80°C на масляной бане в течение 8 часов, и растворитель удаляли отгонкой на роторном испарителе. Полученный остаток очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=1:1), с получением указанного в заголовке соединения (6,2 г, выход: 49,6%).
Пример получения 13: Получение 5-(пиперазин-1-ил)пиримидин-2-амина
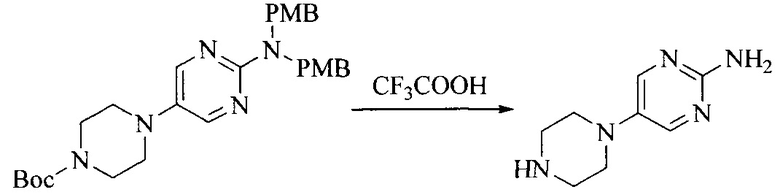
Трет-бутил 4-(2-(бис(4-метоксибензил)амино)пиримидин-5-ил)пиперазин-1-карбоксилат (6,2 г, 11,9 ммоль) растворяли в смешанном растворе дихлорметана (30 мл) и трифторуксусной кислоты (30 мл) и перемешивали при комнатной температуре в течение 30 мин. Растворитель удаляли отгонкой на роторном испарителе. Добавляли небольшое количество дихлорметана, и снова проводили отгонку на роторном испарителе для удаления растворителя, и получали неочищенный продукт (2,1 г). Данный продукт использовали на следующей стадии без очистки.
Пример получения 14: Получение трет-бутил 4-(2-аминопиримидин-5-ил)пиперазин-1-карбоксилата
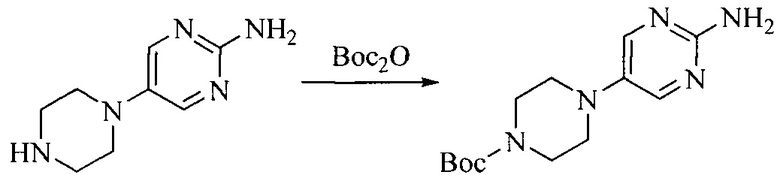
5-(Пиперазин-1-ил)пиримидин-2-амин (2,1 г, 11,7 ммоль) растворяли в дихлорметане (25 мл), и добавляли триэтиламин (1,77 г, 17,5 ммоль). По каплям добавляли ди-трет-бутилдикарбонат (3,05 г, 14,0 ммоль). После добавления, полученную смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли отгонкой на роторном испарителе. Полученный остаток очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=10:1), с получением продукта (1,52 г, выход в две стадии: 45,8%).
Пример получения 15: Получение трет-бутил 4-(2-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)пиперазин-1-карбоксилата
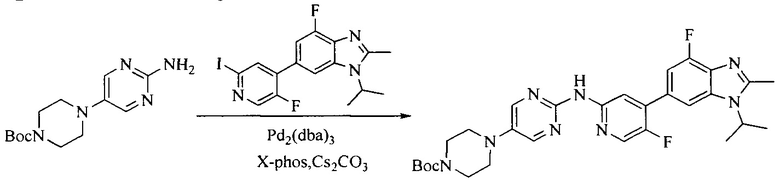
Трет-бутил 4-(2-аминопиримидин-5-ил)пиперазин-1-карбоксилат (475 мг, 1,70 ммоль) и 4-фтор-6-(5-фтор-2-йодпиридин-4-ил)-1-изопропил-2-метил-1H-бензо[d]имидазол (350 мг, 0,85 ммоль) растворяли в 1,4-диоксане (20 мл), и добавляли карбонат цезия (826 мг, 2,54 ммоль), трис(дибензилиденацетон)дипалладий (78 мг, 0,085 ммоль) и 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (81 мг, 0,17 ммоль). В защитной атмосфере азота полученную смесь нагревали до 110°C и подвергали взаимодействию в течение 16 часов. Полученный реакционный раствор фильтровали, и фильтрат концентрировали и разделяли колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=1:1), с получением указанного в заголовке соединения (250 мг, выход: 52,1%).
Пример получения 16: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(пиперазин-2-ил)пиримидин-2-амина
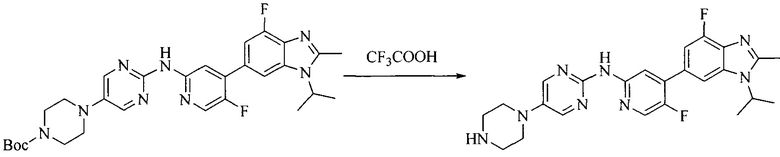
Трет-бутил 4-(2-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)пиперазин-1-карбоксилат (250 мг, 0,44 ммоль) растворяли в смешанном растворе дихлорметана (5 мл) и трифторуксусной кислоты (5 мл), и полученную смесь перемешивали при 20°C в течение 30 мин. Растворитель удаляли отгонкой на роторном испарителе, и добавляли дихлорметан (10 мл). Растворитель удаляли отгонкой на роторном испарителе, и полученный остаток разделяли колоночной хроматографией на силикагеле (дихлорметан:метанол=10:1), с получением указанного в заголовке соединения (180 мг, выход: 88,1%).
Пример 1: Получение 5-((4-этилпиперазин-1-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина (соединение 1)
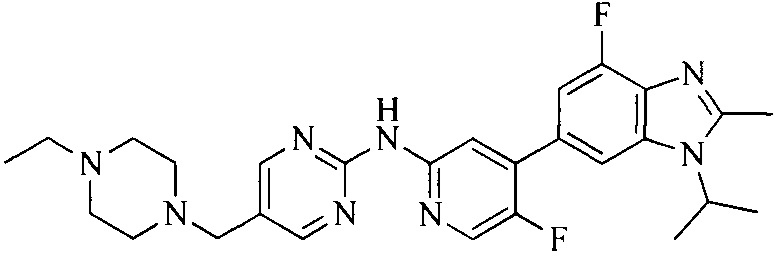
(1) Получение 2-хлор-5-((4-этилпиперазин-1-ил)метил)пиримидина
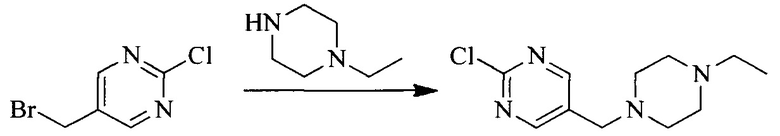
5-(Бромметил)-2-хлорпиримидин (4,14 г, 20 ммоль) растворяли в тетрагидрофуране (70 мл), и добавляли триэтиламин (6,06 г, 60 ммоль) и 1-этилпиперазин (4,56 г, 40 ммоль) при перемешивании. Полученную смесь перемешивали при комнатной температуре в течение 4 часов. После расходования исходных веществ, как установлено ТСХ, полученную смесь фильтровали при отсасывании. Фильтрат концентрировали и затем подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=30:1), с получением указанного в заголовке соединения (3,4 г, выход: 70,8%).
(2) Получение 5-((4-этилпиперазин-1-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина
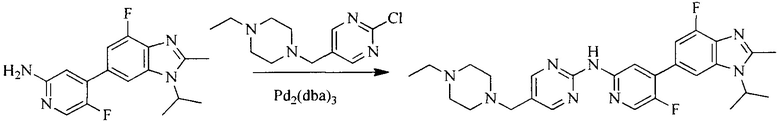
5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (700 мг, 2,32 ммоль), 2-хлор-5-((4-этилпиперазин-1-ил)метил)пиримидин (557 мг, 2,32 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (221 мг, 0,38 ммоль), карбонат цезия (2,26 г, 6,96 ммоль) и трис(дибензилиденацетон)дипалладий (212 мг, 0,23 ммоль) добавляли в 100 мл баклажановидный сосуд, и добавляли 1,4-диоксан (50 мл). В защитной атмосфере азота данное взаимодействие осуществляли при 110°C в течение 4 часов, и полученную в результате смесь фильтровали при отсасывании. Фильтрат концентрировали и затем подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=15:1), с получением указанного в заголовке соединения (500 мг, выход: 42,6%).
Молекулярная формула: C27H32F2N8. Молекулярная масса: 506,3. ЖХ-МС (m/z): 507,4(M+H+).
1H-ЯМР (400 МГц, MeOD-d4): δ: 8,56 (д, J=6,4 Гц, 1H), 8,49 (с, 2H), 8,26 (д, J=2,4 Гц, 1H), 7,79 (с, 1H), 7,27 (д, J=11,6 Гц, 1H), 3,50 (с, 2H), 2,69 (с, 3H), 2,38-2,64 (м, 10H), 1,70 (д, J=7,2 Гц, 6H), 1,11 (т, J=7,2 Гц, 3H).
Пример 2: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-((7-метил-2,7-диазаспиро[3.5]нонан-2-ил)метил)пиримидин-2-амина (соединение 2)
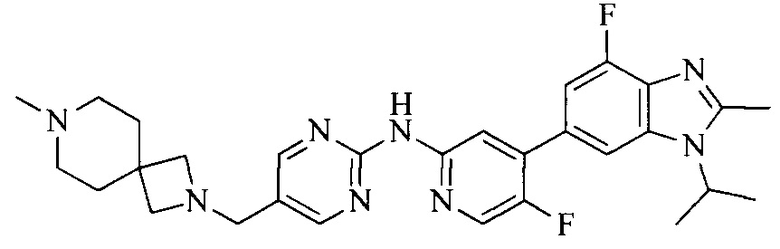
(1) Получение трет-бутил 2-((2-хлорпиримидин-5-ил)метил)-2,7-диазаспиро[3.5]нонан-7-карбоксилата
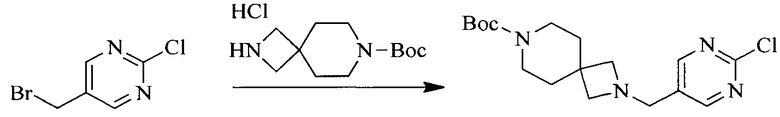
5-Бромметил-2-хлорпиримидин (235 мг, 1,14 ммоль) растворяли в тетрагидрофуране (5 мл), и добавляли гидрохлорид трет-бутил 2,7-диазаспиро[3.5]нонан-7-карбоксилата (300 мг, 1,14 ммоль) и триэтиламин (345 мг, 3,42 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи, и избыток растворителя удаляли перегонкой при пониженном давлении. Полученный остаток разбавляли водой (5 мл) и экстрагировали этилацетатом (3×10 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и сушили на роторном испарителе. Неочищенный продукт очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=20:1), с получением указанного в заголовке соединения (240 мг, выход: 60%).
(2) Получение 2-((2-хлорпиримидин-5-ил)метил)-2,7-диазаспиро[3.5]нонана
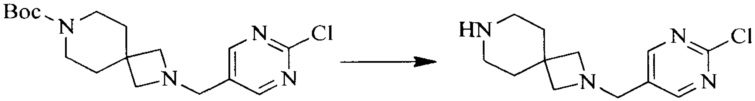
Трет-бутил 2-((2-хлорпиримидин-5-ил)метил)-2,7-диазаспиро[3.5]нонан-7-карбоксилат (240 мг, 0,68 ммоль) растворяли в дихлорметане (2 мл), и добавляли трифторуксусную кислоту (1 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Избыток растворителя удаляли перегонкой при пониженном давлении, с получением указанного в заголовке соединения (156 мг, выход: 91%).
(3) Получение 2-((2-хлорпиримидин-5-ил)метил)-7-метил-2,7-диазаспиро[3.5]нонана
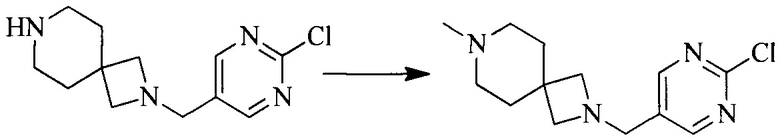
2-((2-Хлорпиримидин-5-ил)метил)-2,7-диазаспиро[3.5]нонан (156 мг, 0,62 ммоль) растворяли в ацетонитриле (5 мл), и добавляли 37% водный раствор формальдегида (251 мг, 3,1 ммоль) и цианоборогидрид натрия (78 мг, 1,24 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Избыток растворителя удаляли перегонкой при пониженном давлении, и неочищенный продукт подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=20:1), с получением указанного в заголовке соединения (100 мг, выход: 61%).
(4) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-((7-метил-2,7-диазаспиро[3.5]нонан-2-ил)метил)пиримидин-2-амина
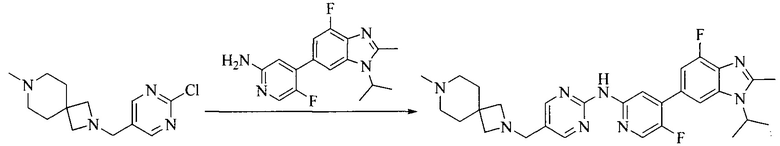
5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (113 мг, 0,37 ммоль) и 2-((2-хлорпиримидин-5-ил)метил)-7-метил-2,7-диазаспиро[3.5]нонан (100 мг, 0,37 ммоль) растворяли в 1,4-диоксане (5 мл), и добавляли трис(дибензилиденацетон)дипалладий (34 мг, 0,037 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (35 мг, 0,074 ммоль) и карбонат цезия (300 мг, 0,93 ммоль). В защитной атмосфере азота полученную смесь перемешивали при кипячении с обратным холодильником в течение ночи и фильтровали. Избыток растворителя удаляли перегонкой при пониженном давлении. Неочищенный продукт подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=10:1), с получением указанного в заголовке соединения (17 мг, выход: 8,6%).
Молекулярная формула: C29H34F2N8. Молекулярная масса: 532,6. ЖХ-МС (m/z): 533(M+H+).
1H-ЯМР (400 МГц, MeOD-d4) δ: 8,53 (д, J=6,0 Гц, 1H), 8,50 (с, 2H), 8,27 (д, J=2,4 Гц, 1H), 7,79 (с, 1H), 7,27 (д, J=11,2 Гц, 1H), 3,68 (с, 2H), 3,21-3,22 (м, 4H), 2,80-3,00 (м, 4H), 2,69 (с, 3H), 2,63 (с, 3H), 1,90-2,00 (м, 4H), 1,70 (д, J=6,8 Гц, 6H).
Пример 3: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-((2-метил-2,7-диазаспиро[3.5]нонан-7-ил)метил)пиримидин-2-амина (соединение 3)
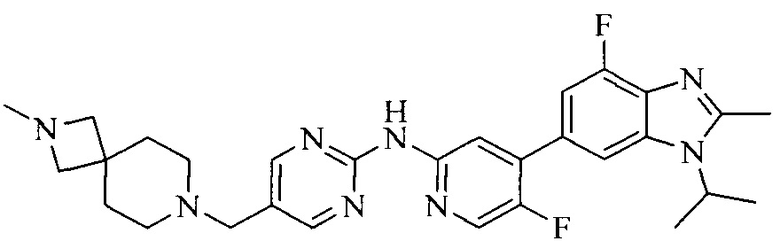
(1) Получение трет-бутил 7-((2-хлорпиримидин-5-ил)метил)-2,7-диазаспиро[3.5]нонан-2-карбоксилата
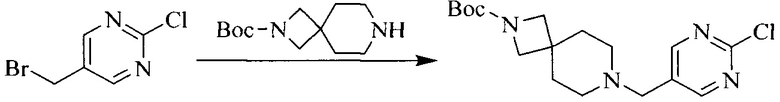
5-(Бромметил)-2-хлорпиримидин (220 мг, 1,06 ммоль) растворяли в тетрагидрофуране (5 мл), и добавляли трет-бутил 2,7-диазаспиро[3.5]нонан-2-карбоксилат (200 мг, 0,88 ммоль) и триэтиламин (268 мг, 2,65 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли перегонкой при пониженном давлении. Полученный остаток разбавляли водой (5 мл) и экстрагировали этилацетатом (3×10 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и сушили на роторном испарителе. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=1:1), с получением указанного в заголовке соединения (190 мг, выход: 60,5%).
(2) Получение 7-((2-хлорпиримидин-5-ил)метил)-2,7-диазаспиро[3.5]нонана
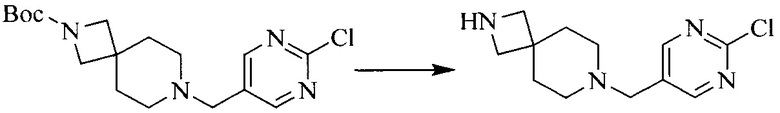
Трет-бутил 7-((2-хлорпиримидин-5-ил)метил)-2,7-диазаспиро[3.5]нонан-2-карбоксилат (190 мг, 0,54 ммоль) растворяли в дихлорметане (2 мл), и добавляли трифторуксусную кислоту (1 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли перегонкой при пониженном давлении, с получением указанного в заголовке соединения (110 мг, выход: 81%).
(3) Получение 7-((2-хлорпиримидин-5-ил)метил)-2-метил-2,7-диазаспиро[3.5]нонана

7-((2-Хлорпиримидин-5-ил)метил)-2,7-диазаспиро[3.5]нонан (110 мг, 0,44 ммоль) растворяли в ацетонитриле (5 мл), и добавляли 37% водный раствор формальдегида (178 мг, 2,2 ммоль) и цианоборогидрид натрия (55 мг, 0,88 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли перегонкой при пониженном давлении, и неочищенный продукт подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=20:1), с получением указанного в заголовке соединения (80 мг, выход: 68%).
(4) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-((2-метил-2,7-диазаспиро[3.5]нонан-7-ил)метил)пиримидин-2-амина
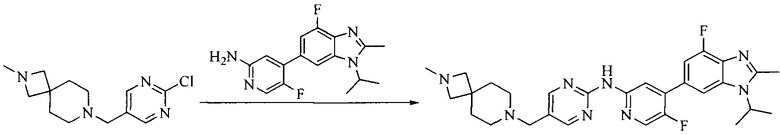
5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (90 мг, 0,3 ммоль) и 7-((2-хлорпиримидин-5-ил)метил)-2-метил-2,7-диазаспиро[3.5]нонан (80 мг, 0,3 ммоль) растворяли в 1,4-диоксане (5 мл), и добавляли трис(дибензилиденацетон)дипалладий (27 мг, 0,03 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (29 мг, 0,06 ммоль) и карбонат цезия (244 мг, 0,75 ммоль). В защитной атмосфере азота полученную смесь перемешивали при кипячении с обратным холодильником в течение ночи и фильтровали. Растворитель удаляли перегонкой при пониженном давлении. Неочищенный продукт подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=10:1), с получением указанного в заголовке соединения (8 мг, выход: 5%).
Молекулярная формула: C29H34F2N8. Молекулярная масса: 532,6. ЖХ-МС (m/z): 533(M+H+).
1H-ЯМР (400 МГц, MeOD-d4) δ: 8,67 (с, 2H), 8,56 (д, J=6,0 Гц, 1H), 8,31 (д, J=2,4 Гц, 1H), 7,77 (с, 1H), 7,23 (д, J=11,6 Гц, 1H), 4,00-4,11 (м, 6H), 3,00-3,19 (м, 4H), 2,96 (с, 3H), 2,69 (с, 3H), 2,11-2,21 (м, 4H), 1,69 (д, J=6,8 Гц, 6H).
Пример 4: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-((6-метил-2,6-диазаспиро[3.3]гептан-2-ил)метил)пиримидин-2-амина (соединение 4)
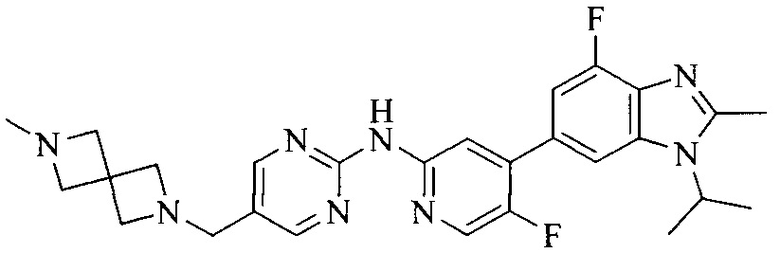
(1) Получение трет-бутил 6-((2-хлорпиримидин-5-ил)метил)-2,6-диазаспиро[3.3]гептан-2-карбоксилата
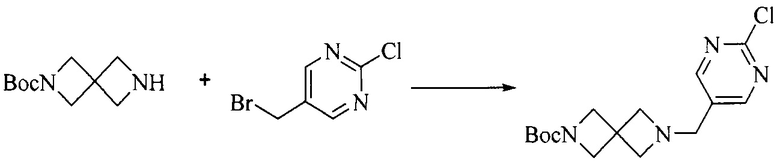
5-(Бромметил)-2-хлорпиримидин (326 мг, 1,57 ммоль) растворяли в ацетонитриле (10 мл), и добавляли карбонат калия (433 мг, 3,14 ммоль) и трет-бутил 2,6-диазаспиро[3.3]гептан-2-карбоксилат (310 мг, 1,57 ммоль). Данное взаимодействие осуществляли при комнатной температуре в течение 16 часов. Полученную смесь фильтровали при отсасывании. Фильтрат концентрировали и очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=20:1), с получением указанного в заголовке соединения (330 мг, выход: 65%).
(2) Получение 2-((2-хлорпиримидин-5-ил)метил)-2,6-диазаспиро[3.3]гептана
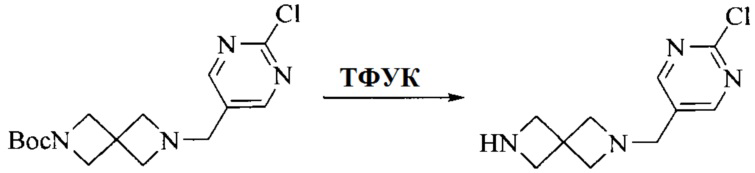
Трет-бутил 6-((2-хлорпиримидин-5-ил)метил)-2,6-диазаспиро[3.3]гептан-2-карбоксилат (330 мг, 1,01 ммоль) растворяли в дихлорметане (10 мл), и добавляли трифторуксусную кислоту (5 мл). Полученную смесь перемешивали при комнатной температуре в течение 1 часа и концентрировали, с получением указанного в заголовке соединения в виде масла (210 мг, выход: 93%).
(3) Получение 2-((2-хлорпиримидин-5-ил)метил)-6-метил-2,6-диазаспиро[3.3]гептана
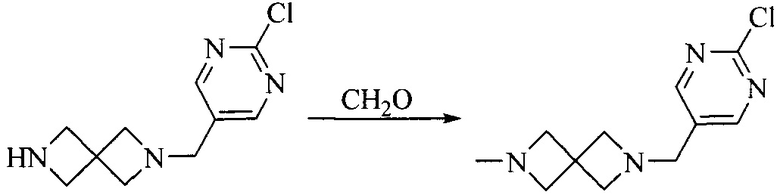
2-((2-Хлорпиримидин-5-ил)метил)-2,6-диазаспиро[3.3]гептан (210 мг, 0,94 ммоль) растворяли в ацетонитриле (5 мл), и добавляли раствор формальдегида (0,5 мл) и цианоборогидрид натрия (65 мг, 1,04 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли перегонкой при пониженном давлении, и неочищенный продукт подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=10:1), с получением указанного в заголовке соединения (150 мг, выход: 67%).
(4) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-((6-метил-2,6-диазаспиро[3.3]гептан-2-ил)метил)пиримидин-2-амина
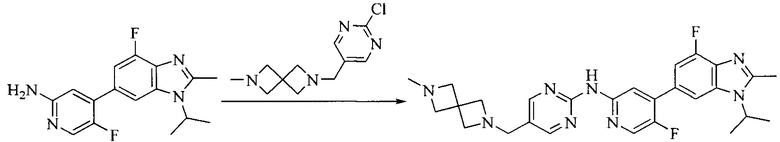
5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (190 мг, 0,63 ммоль) и 2-((2-хлорпиримидин-5-ил)метил)-6-метил-2,6-диазаспиро[3.3]гептан (150 мг, 0,63 ммоль) растворяли в 1,4-диоксане (10 мл), и добавляли 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (60 мг, 0,13 ммоль), карбонат цезия (614 мг, 1,89 ммоль) и трис(дибензилиденацетон)дипалладий (60 мг, 0,06 ммоль). В защитной атмосфере азота полученную смесь нагревали до 110°C и подвергали взаимодействию в течение 16 часов. Полученную в результате смесь фильтровали при отсасывании. Фильтрат концентрировали и очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=5:1), с получением указанного в заголовке соединения в виде твердого вещества (23 мг, выход: 7%).
Молекулярная формула: C27H30F2N8. Молекулярная масса: 504,3. ЖХ-МС (m/z): 505,3(M+H+).
1H-ЯМР (400 МГц, CD3OD-d4) δ: 8,54 (д, J=6,0 Гц, 1H), 8,46 (с, 2H), 8,26 (д, J=2,4 Гц, 1H), 7,79 (с, 1H), 7,27 (д, J=11,2 Гц, 1H), 4,84-4,85 (м, 1H), 3,54 (с, 6H), 3,39 (с, 4H), 2,69 (с, 3H), 2,41 (с, 3H), 1,69 (д, J=6,8 Гц, 6H).
Пример 5: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-((9-метил-3,9-диазаспиро[5.5]ундекан-3-ил)метил)пиримидин-2-амина (соединение 5)
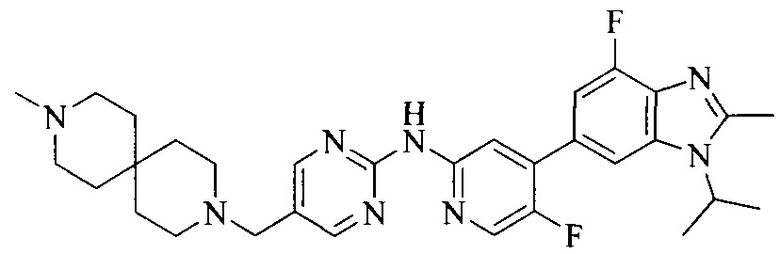
(1) Получение трет-бутил 9-((2-хлорпиримидин-5-ил)метил)-3,9-диазаспиро[5.5]ундекан-3-карбоксилата
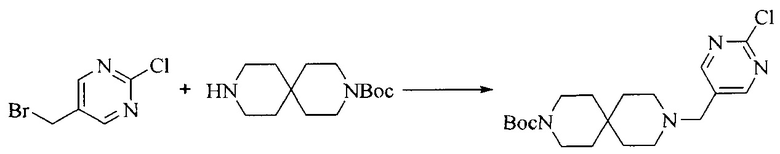
5-(Бромметил)-2-хлорпиримидин (326 мг, 1,57 ммоль) растворяли в тетрагидрофуране (10 мл), и добавляли триэтиламин (158 мг, 1,57 ммоль) и трет-бутил 3,9-диазаспиро[5.5]ундекан-3-карбоксилат (200 мг, 0,79 ммоль) при перемешивании. Полученную реакционную смесь перемешивали при комнатной температуре в течение 4 часов и фильтровали при отсасывании. Фильтрат концентрировали и очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=30:1), с получением указанного в заголовке соединения (210 мг, выход: 70%).
(2) Получение 3-((2-хлорпиримидин-5-ил)метил)-3,9-диазаспиро[5.5]ундекана
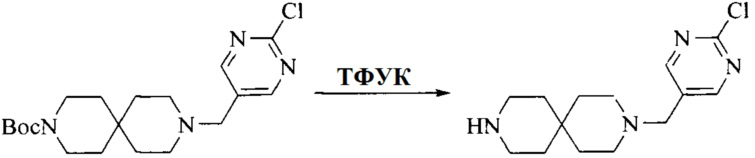
Трет-бутил 9-((2-хлорпиримидин-5-ил)метил)-3,9-диазаспиро[5.5]ундекан-3-карбоксилат (210 мг, 0,55 ммоль) добавляли в дихлорметан (10 мл), и добавляли трифторуксусную кислоту (5 мл). Полученную смесь перемешивали при комнатной температуре в течение 1 часа и концентрировали, с получением указанного в заголовке соединения (145 мг, выход: 94%).
(3) Получение 3-((2-хлорпиримидин-5-ил)метил)-9-метил-3,9-диазаспиро[5.5]ундекана
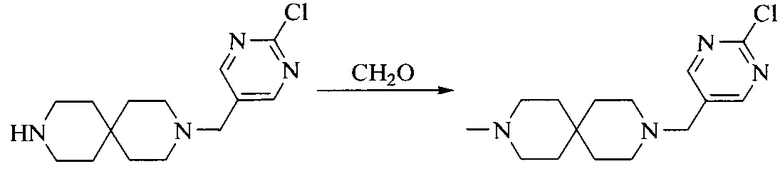
3-((2-Хлорпиримидин-5-ил)метил)-3,9-диазаспиро[5.5]ундекан (145 мг, 0,52 ммоль) растворяли в ацетонитриле (5 мл), и добавляли 37% водный раствор формальдегида (0,5 мл) и цианоборогидрид натрия (65 мг, 1,04 ммоль) при перемешивании. После добавления, полученную смесь перемешивали при комнатной температуре в течение 2 часов. Полученную в результате смесь концентрировали и подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=20:1), с получением указанного в заголовке соединения (65 мг, выход: 42,7%).
(4) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-((9-метил-3,9-диазаспиро[5.5]ундекан-3-ил)метил)пиримидин-2-амина
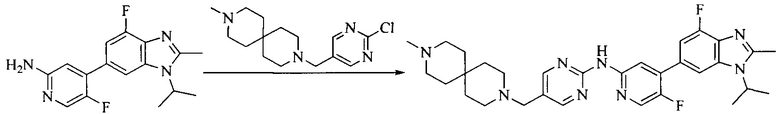
5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (77 мг, 0,26 ммоль), 3-((2-хлорпиримидин-5-ил)метил)-9-метил-3,9-диазаспиро[5.5]ундекан (65 мг, 0,22 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (20 мг, 0,04 ммоль), карбонат цезия (205 мг, 0,63 ммоль) и трис(дибензилиденацетон)дипалладий (20 мг, 0,02 ммоль) добавляли в 1,4-диоксан (10 мл). В защитной атмосфере азота данное взаимодействие осуществляли при 110°C в течение 16 часов, и затем полученную реакционную смесь фильтровали при отсасывании. Фильтрат концентрировали и подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=10:1), с получением указанного в заголовке соединения (10 мг, выход: 8%).
Молекулярная формула: C31H38F2N8. Молекулярная масса: 560,7. ЖХ-МС (m/z): 561,4 (M+H+).
1H-ЯМР (400 МГц, CD3OD-d4) δ: 8,54 (д, J=6,0 Гц, 1H), 8,49 (с, 2H), 8,26 (д, J=2,4 Гц, 1H), 7,79 (с, 1H), 7,28 (д, J=11,6 Гц, 1H), 3,53 (с, 2H), 2,98-3,07 (м, 4H), 2,71-2,68 (м, 6H), 2,50-2,52 (м, 4H), 1,66-1,71 (м, 9H), 1,61-1,65 (м, 5H).
Пример 6: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-((5-метилгексагидропирроло[3,4-c]пиррол-2(1H)-ил)метил)пиримидин-2-амина (соединение 6)
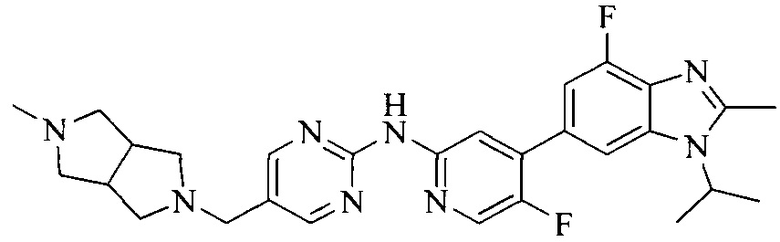
(1) Получение трет-бутил 5-((2-хлорпиримидин-5-ил)метил)гексагидропирроло[3,4-c]пиррол-2(1H)-карбоксилата
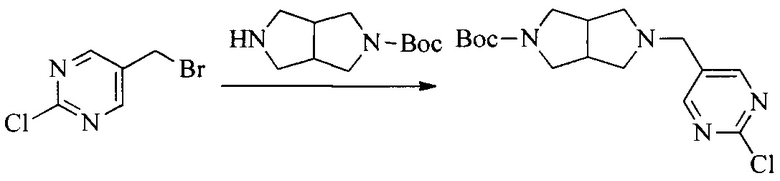
Трет-бутил гексагидропирроло[3,4-c]пиррол-2(1H)-карбоксилат (318 мг, 1,5 ммоль) и 5-(бромметил)-2-хлорпиримидин (310 мг, 1,5 ммоль) растворяли в ацетонитриле (50 мл), и добавляли карбонат калия (621 мг, 4,5 ммоль). Полученной смеси давали взаимодействовать при комнатной температуре в течение 8 часов. Полученную смесь фильтровали. Фильтрат концентрировали и очищали колоночной хроматографией на силикагеле (элюент: смесь дихлорметан:метанол=50:1), с получением указанного в заголовке соединения (430 мг, выход: 85%).
(2) Получение 2-((2-хлорпиримидин-5-ил)метил)октагидропирроло[3,4-c]пиррола
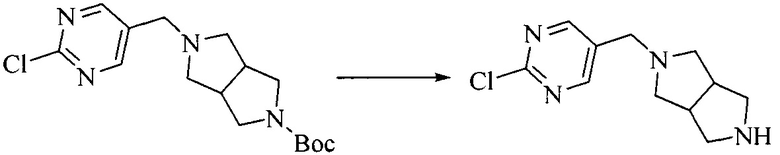
Трет-бутил 5-((2-хлорпиримидин-5-ил)метил)гексагидропирроло[3,4-c]пиррол-2(1H)-карбоксилат (430 мг, 1,27 ммоль) растворяли в дихлорметане (10 мл), и добавляли трифторуксусную кислоту (5 мл). Полученную смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель удаляли перегонкой при пониженном давлении, с получением указанного в заголовке соединения в виде желтого масла (302 мг, неочищенное), которое непосредственно использовали на следующей стадии без очистки.
(3) Получение 2-((2-хлорпиримидин-5-ил)метил)-5-метилоктагидропирроло[3,4-c]пиррола
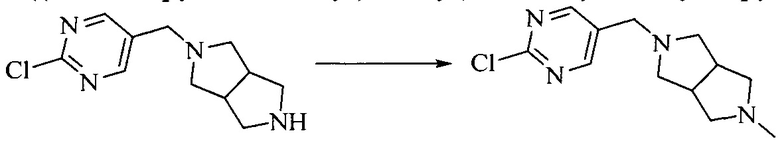
2-((2-Хлорпиримидин-5-ил)метил)гексагидропирроло[3,4-c]пиррол (302 мг, неочищенный продукт) растворяли в метаноле (50 мл). Водный раствор формальдегида (37%, 1 г, 12,3 ммоль) добавляли при комнатной температуре, и данное взаимодействие осуществляли в течение 2 часов. Добавляли цианоборогидрид натрия (0,8 г, 12,7 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 12 часов. После взаимодействия, полученный реакционный раствор концентрировали и очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=20:1), с получением указанного в заголовке соединения (260 мг, выход в две стадии: 81%).
(4) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-((5-метилгексагидропирроло[3,4-c]пиррол-2(1H)-ил)метил)пиримидин-2-амина
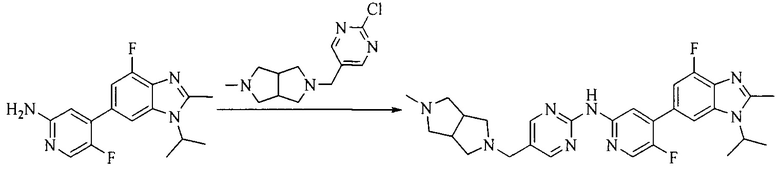
5-Фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (302 мг, 1,0 ммоль) и 2-((2-хлорпиримидин-5-ил)метил)-5-метилоктагидропирроло[3,4-c]пиррол (252 мг, 1,0 ммоль) растворяли в 1,4-диоксане (50 мл), и добавляли трис(дибензилиденацетон)дипалладий (92 мг, 0,1 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (95 мг, 0,2 ммоль) и карбонат цезия (975 мг, 3,0 ммоль). В защитной атмосфере азота полученную смесь нагревали до 110°C и подвергали взаимодействию в течение 8 часов. Полученную смесь охлаждали до комнатной температуры и концентрировали. Воду (100 мл) и этилацетат (150 мл) добавляли к отделенной от водной фазы органической фазе. Водную фазу экстрагировали этилацетатом (150 мл ×2), и органические фазы объединяли и промывали насыщенным раствором NaCl, сушили над безводным сульфатом натрия и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь дихлорметан:метанол=20:1), с получением указанного в заголовке соединения (223 мг, выход: 43%).
Молекулярная формула: C28H32F2N8. Молекулярная масса: 518,6. ЖХ-МС (m/z): 519,3(M+H+).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 10,03 (с, 1H), 8,45 (с, 2H), 8,43 (д, J=6,4 Гц, 1H), 8,36 (с, 1H), 7,75 (с, 1H), 7,23 (д, J=11,6 Гц, 1H), 4,80-4,83 (м, 1H), 3,34-3,49 (м, 6H), 2,66 (шир.с, 4H), 2,61 (с, 3H), 2,35 (шир.с, 5H), 1,57 (д, J=6,8 Гц, 6H).
Пример 6-1: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((цис)-5-метилгексагидропирроло[3,4-c]пиррол-2(1H)-ил)метил)пиримидин-2-амина (соединение 6-1)
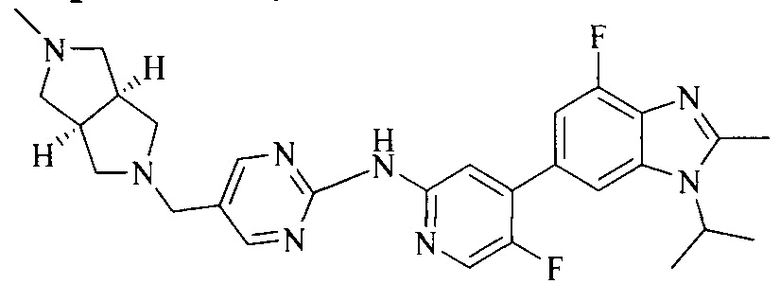
(1) Получение трет-бутил 5-((2-хлорпиримидин-5-ил)метил)-(цис)-гексагидропирроло[3,4-c]пиррол-2(1H)-карбоксилата
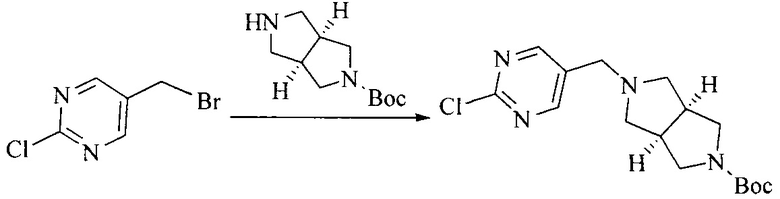
Трет-бутил (цис)-гексагидропирроло[3,4-c]пиррол-2(1H)-карбоксилат (300 мг, 1,41 ммоль) и 5-(бромметил)-2-хлорпиримидин (292,5 мг, 1,41 ммоль) растворяли в ацетонитриле (50 мл), и добавляли карбонат калия (585 мг, 4,23 ммоль). Данное взаимодействие осуществляли при комнатной температуре в течение 4 часов. Полученную смесь фильтровали. Фильтрат сушили дистилляцией и очищали колоночной хроматографией на силикагеле (элюент: смесь дихлорметан:метанол=50:1), с получением указанного в заголовке соединения (402 мг, выход: 82%).
(2) Получение 2-((2-хлорпиримидин-5-ил)метил)-(цис)-октагидропирроло[3,4-c]пиррола
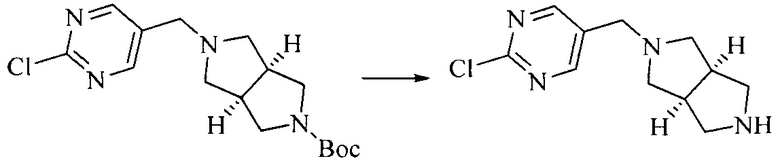
Трет-бутил 5-((2-хлорпиримидин-5-ил)метил)-(цис)-гексагидропирроло[3,4-c]пиррол-2(1H)-карбоксилат (400 мг, 1,18 ммоль) растворяли в дихлорметане (10 мл), и добавляли трифторуксусную кислоту (5 мл). Полученную смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель удаляли перегонкой при пониженном давлении, с получением указанного в заголовке соединения (300 мг, неочищенный продукт), который непосредственно использовали на следующей стадии без очистки.
(3) Получение 2-((2-хлорпиримидин-5-ил)метил)-5-метил-(цис)-октагидропирроло[3,4-c]пиррола
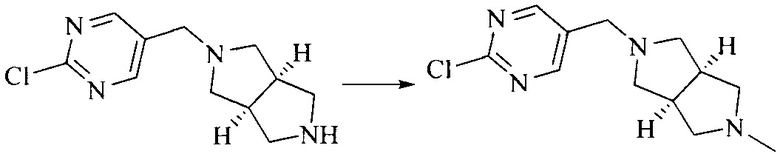
Неочищенный продукт 2-((2-хлорпиримидин-5-ил)метил)-(цис)-октагидропирроло[3,4-c]пиррола (300 мг) растворяли в метаноле (50 мл). Водный раствор формальдегида (37%, 0,96 г, 11,8 ммоль) добавляли при комнатной температуре, и данное взаимодействие осуществляли в течение 2 часов. Добавляли цианоборогидрид натрия (0,74 г, 11,8 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 12 часов. После взаимодействия, растворитель сушили дистилляцией, и полученный остаток подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=20:1), с получением указанного в заголовке соединения (175 мг, выход в две стадии: 58%).
(4) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((цис)-5-метилгексагидропирроло[3,4-c]пиррол-2(1H)-ил)метил)пиримидин-2-амина
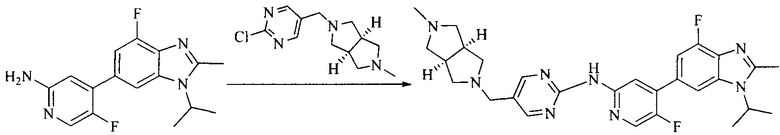
5-Фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (302 мг, 1,0 ммоль) и 2-((2-хлорпиримидин-5-ил)метил)-5-метил-(цис)-октагидропирроло[3,4-c]пиррол (252 мг, 1,0 ммоль) растворяли в 1,4-диоксане (50 мл), и добавляли трис(дибензилиденацетон)дипалладий (92 мг, 0,1 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (95 мг, 0,2 ммоль) и карбонат цезия (975 мг, 3,0 ммоль). В защитной атмосфере азота полученную смесь нагревали до 110°C и подвергали взаимодействию в течение 8 часов. Полученную смесь охлаждали до комнатной температуры и концентрировали. Воду (100 мл) и этилацетат (150 мл) добавляли к отделенной от водной фазы органической фазе. Водную фазу экстрагировали этилацетатом (150 мл ×2), и органические фазы объединяли и промывали насыщенным раствором NaCl, сушили над безводным сульфатом натрия и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь дихлорметан:метанол=20:1), с получением указанного в заголовке соединения (207 мг, выход: 40%).
Молекулярная формула: C28H32F2N8. Молекулярная масса: 518,60. ЖХ-МС (m/z): 519,3(M+H+).
1H-ЯМР (400 МГц, CDCl3) δ: 8,64 (д, J=6,0 Гц, 1H), 8,48-8,47 (м, 3H), 8,29 (д, J=2,4 Гц, 1H), 7,64 (т, J=1,2 Гц, 1H), 7,25 (д, J=9,6 Гц, 1H), 4,70-4,74 (м, 1H), 3,57 (с, 2H), 3,40 (шир.с, 2H), 3,07 (с, 2H), 2,64-2,69 (м, 10H), 2,40-2,48 (м, 2H), 1,69 (д, J=6,8 Гц, 6H).
Пример 6-2: Получение 5-(((цис)-5-этилгексагидропирроло[3,4-c]пиррол-2(1H)-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина (соединение 6-2)
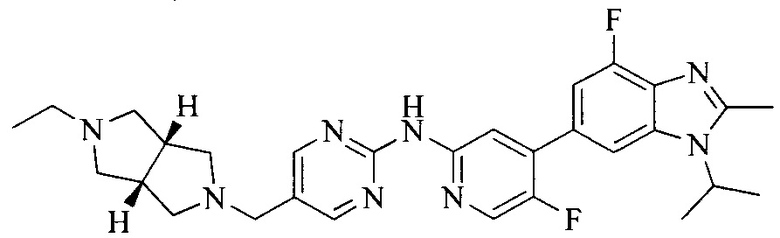
Получение трет-бутил (цис)-5-этилгексагидропирроло[3,4-c]пиррол-2(1H)-карбоксилата
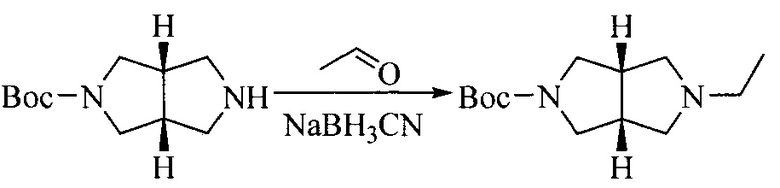
Трет-бутил (цис)-гексагидропирроло[3,4-c]пиррол-2(1H)-карбоксилат (424 мг, 2,0 ммоль) растворяли в метаноле (10 мл), и по каплям добавляли водный раствор ацетальдегида (40%, 2,2 мл, 20 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Медленно добавляли цианоборогидрид натрия (1,26 г, 20 ммоль). После добавления, полученную смесь дополнительно перемешивали при комнатной температуре в течение 30 мин. Растворитель удаляли выпариванием на роторном испарителе, и полученный остаток разделяли колоночной хроматографией на силикагеле (дихлорметан:метанол=10:1), с получением продукта (317 мг, выход: 66,0%).
(2) Получение (цис)-2-этилоктагидропирроло[3,4-c]пиррола
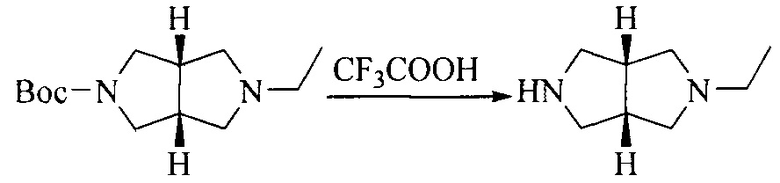
Трет-бутил (цис)-5-этилгексагидропирроло[3,4-c]пиррол-2(1H)-карбоксилат (317 мг, 1,32 ммоль) растворяли в смешанном растворе дихлорметана (3 мл) и трифторуксусной кислоты (3 мл), и перемешивали при комнатной температуре в течение 30 мин. Растворитель удаляли отгонкой на роторном испарителе. Добавляли небольшое количество дихлорметана, и растворитель удаляли отгонкой на роторном испарителе, с получением неочищенного продукта (181 мг). Данный продукт использовали на следующей стадии без очистки.
(3) Получение (цис)-2-((2-хлорпиримидин-5-ил)метил)-5-этилоктагидропирроло[3,4-c]пиррола
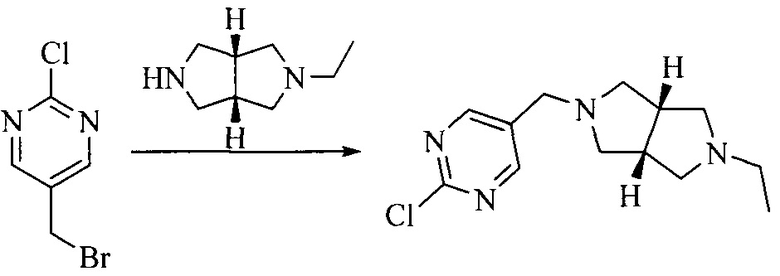
К ацетонитрилу (15 мл) добавляли (цис)-2-этилоктагидропирроло[3,4-c]пиррол (181 мг, 1,29 ммоль), 5-(бромметил)-2-хлорпиримидин (267 мг, 1,29 ммоль) и карбонат калия (178 мг, 1,29 ммоль), и полученную смесь нагревали на масляной бане при 60°C в течение 1 часа. Растворитель удаляли отгонкой на роторном испарителе, и полученный остаток разделяли колоночной хроматографией на силикагеле (дихлорметан:метанол=10:1), с получением продукта (300 мг, выход: 87,0%).
(4) Получение 5-(((цис)-5-этилгексагидропирроло[3,4-c]пиррол-2(1H)-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина
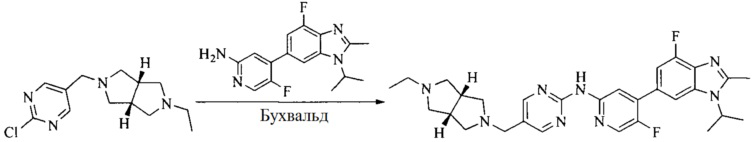
(цис)-2-((2-Хлорпиримидин-5-ил)метил)-5-этилоктагидропирроло[3,4-c]пиррол (300 мг, 1,12 ммоль), 5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (338 мг, 1,12 ммоль), карбонат цезия (730 мг, 2,24 ммоль), трис(дибензилиденацетон)дипалладий (30 мг) и 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (60 мг) добавляли к 1,4-диоксану (20 мл). В защитной атмосфере азота полученную смесь нагревали при 110°C на масляной бане в течение 8 часов. Растворитель удаляли отгонкой на роторном испарителе. Проводили колоночную хроматографию с обратной фазой (вода:метанол=10:1-1:1), с получением указанного в заголовке соединения (176 мг, выход: 29,5%).
Молекулярная формула: C29H34F2N8. Молекулярная масса: 532,6. ЖХ-МС (m/z): 533,3(M+H+).
1H-ЯМР (400 МГц, CDCl3) δ: 8,64 (д, J=5,6 Гц, 2H), 8,44 (с, 2H), 8,29 (д, J=2,0 Гц, 1H), 8,24 (с, 1H), 8,06 (с, 1H), 7,63 (с, 1H), 7,24-7,26 (м, 1H), 4,70-4,73 (м, 1H), 3,55 (с, 2H), 2,90-3,05 (м, 2H), 2,80-2,87 (м, 2H), 2,69 (с, 3H), 2,45-2,60 (м, 6H), 2,25-2,35 (м, 2H), 1,68 (д, J=8,4 Гц, 6H), 1,15-1,18 (м, 3H).
Пример 7: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((цис)-гексагидропирроло[3,4-b][1,4]оксазин-6(2H)-ил)метил)пиримидин-2-амина (соединение 7)
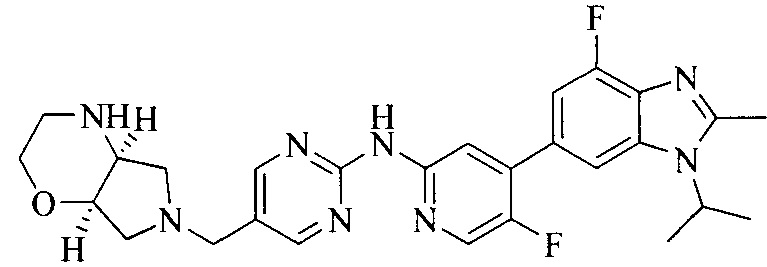
(1) Получение 4-бензил-6-трет-бутил (цис)-гексагидропирроло[3,4-b][1,4]оксазин-4,6-дикарбоксилата
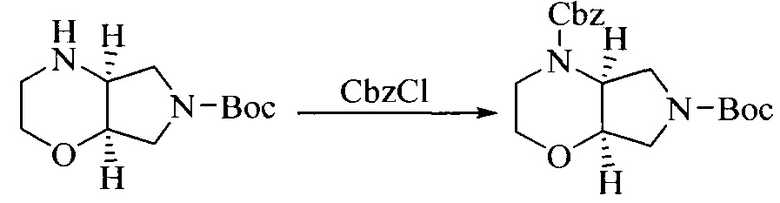
Трет-бутил (цис)-гексагидропирроло[3,4-b][1,4]оксазин-6(2H)-карбоксилат (342 мг, 1,5 ммоль) и N,N-диизопропилэтиламин (581 мг, 4,5 ммоль) растворяли в дихлорметане (30 мл), и по каплям добавляли бензилхлорформиат (358 мг, 2,1 ммоль) в условиях охлаждения на ледяной бане. После добавления, данное взаимодействие осуществляли при комнатной температуре в течение 4 часов. Воду (30 мл) добавляли для отделения органической фазы от водной фазы. Водную фазу экстрагировали дихлорметаном (50 мл ×2). Органические фазы объединяли, промывали насыщенным раствором NaCl, сушили над безводным сульфатом натрия и очищали колоночной хроматографией на силикагеле (элюент: смесь петролейный эфир:этилацетат=5:1), с получением указанного в заголовке соединения (473 мг, выход: 87%).
(2) Получение бензил (цис)-гексагидропирроло[3,4-b][1,4]оксазин-4(4aH)-карбоксилата
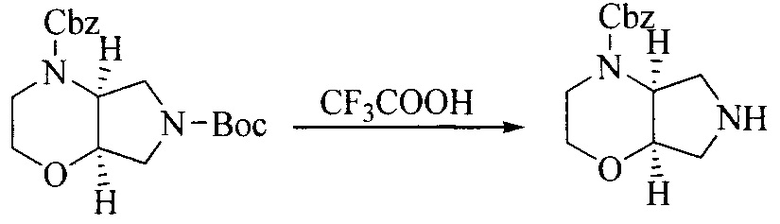
4-Бензил-6-трет-бутил (цис)-гексагидропирроло[3,4-b][1,4]оксазин-4,6-дикарбоксилат (473 мг, 1,3 ммоль) растворяли в дихлорметане (10 мл), и добавляли трифторуксусную кислоту (5 мл). Полученную смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель удаляли перегонкой при пониженном давлении, с получением указанного в заголовке соединения (400 мг, неочищенный продукт), который непосредственно использовали на следующей стадии без очистки.
(3) Получение бензил (цис)-6-((2-хлорпиримидин-5-ил)метил)гексагидропирроло[3,4-b][1,4]оксазин-4(4aH)-карбоксилата
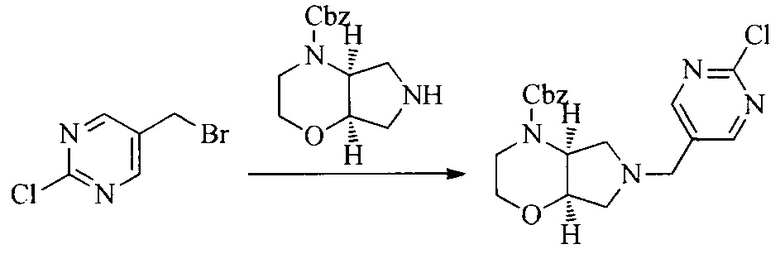
Бензил (цис)-гексагидропирроло[3,4-b][1,4]оксазин-4(4aH)-карбоксилат (400 мг, неочищенный продукт) и 5-(бромметил)-2-хлорпиримидин (269 мг, 1,3 ммоль) растворяли в ацетонитриле (50 мл), и добавляли карбонат калия (538 мг, 3,9 ммоль). Данное взаимодействие осуществляли при комнатной температуре в течение 8 часов. Полученную смесь фильтровали. Фильтрат концентрировали и очищали колоночной хроматографией на силикагеле (элюент: смесь дихлорметан:метанол=50:1), с получением указанного в заголовке соединения (409 мг, выход: 81%).
(4) Получение бензил (цис)-6-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)гексагидропирроло[3,4-b][1,4]оксазин-4(4aH)-карбоксилата
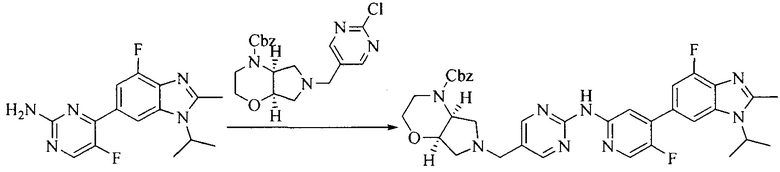
5-Фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (302 мг, 1,0 ммоль) и бензил 6-((2-хлорпиримидин-5-ил)метил)(цис)гексагидропирроло[3,4-b] [1,4]оксазин-4(4aH)-карбоксилат (389 мг, 1,0 ммоль) растворяли в 1,4-диоксане (50 мл), и добавляли трис(дибензилиденацетон)дипалладий (92 мг, 0,1 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (95 мг, 0,2 ммоль) и карбонат цезия (975 мг, 3,0 ммоль). В защитной атмосфере азота полученную смесь нагревали до 110°C и подвергали взаимодействию в течение 8 часов. Полученную смесь охлаждали до комнатной температуры и концентрировали. Воду (100 мл) и этилацетат (150 мл) добавляли к отделенной от водной фазы органической фазе. Водную фазу экстрагировали этилацетатом (150 мл ×2), и органические фазы объединяли, промывали насыщенным раствором NaCl, сушили над безводным сульфатом натрия и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь дихлорметан:метанол=20:1), с получением указанного в заголовке соединения (288 мг, выход: 44%).
(5) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((цис)-гексагидропирроло[3,4-b][1,4]оксазин-6(2H)-ил)метил)пиримидин-2-амин
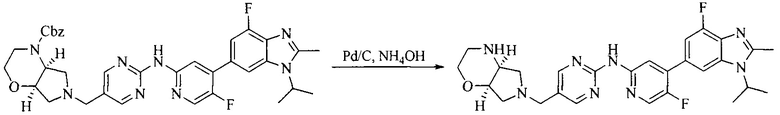
Бензил 6-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)(цис)гексагидропирроло[3,4-b][1,4]оксазин-4(4aH)-карбоксилат (280 мг, 0,43 ммоль) и палладий на углероде (30 мг) суспендировали в метаноле (40 мл) и аммиачной воде (6 мл). Данную систему вакуумировали и заполняли водородом. Данное взаимодействие осуществляли при комнатной температуре в течение 16 часов. Полученную смесь фильтровали через целит, фильтрат концентрировали, и неочищенный продукт очищали колоночной хроматографией на силикагеле (метанол:дихлорметан=1:20, с добавлением 0,2% триэтиламина), с получением указанного в заголовке соединения в виде белого твердого вещества (178 мг, выход: 80%).
Молекулярная формула: C27H30F2N8O. Молекулярная масса: 520,6. ЖХ-МС (m/z): 521,3(M+H+).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 10,02 (с, 1H), 8,44-8,42 (м, 3H), 8,35 (с, 1H), 7,75 (с, 1H), 7,23 (д, J=11,6 Гц, 1H), 4,79-4,83 (м, 1H), 3,79 (с, 1H), 3,16-3,60 (м, 6H), 3,15 (м, 1H), 2,91-2,95 (м, 1H), 2,80-2,85 (м, 1H), 2,73-2,69 (м, 1H), 2,54-2,65 (м, 5H), 1,57 (д, J=6,8 Гц, 6H).
Пример 8: Получение (экзо)-3-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-3-азабицикло[3.1.0]гексан-6-амина (соединение 8)
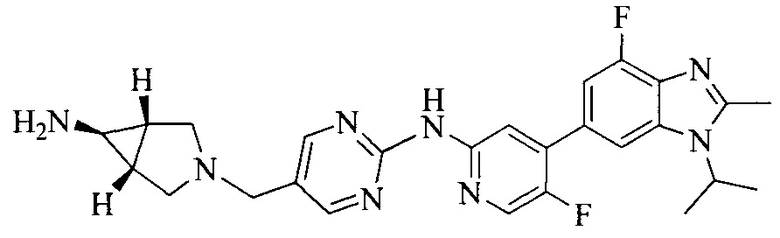
(1) Получение трет-бутил (экзо)-6-((карбобензокси)амино)-3-азабицикло[3.1.0]гексан-3-карбоксилата
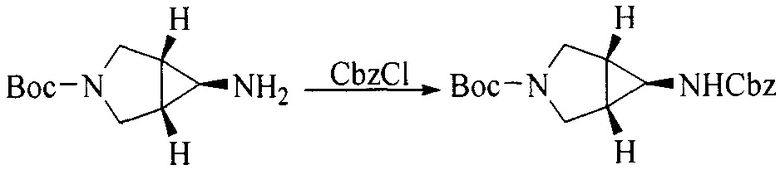
Трет-бутил (экзо)-6-амино-3-азабицикло[3.1.0]гексан-3-карбоксилат (396 мг, 2,0 ммоль) и N,N-диизопропилэтиламин (774 мг, 6,0 ммоль) растворяли в дихлорметане (30 мл), и по каплям добавляли бензилхлорформиат (409 мг, 2,4 ммоль) в условиях охлаждения на ледяной бане. После добавления, данное взаимодействие осуществляли при комнатной температуре в течение 4 часов. Полученный реакционный раствор концентрировали и очищали колоночной хроматографией на силикагеле (элюент: смесь петролейный эфир:этилацетат=10:1), с получением указанного в заголовке соединения (578 мг, выход: 87%).
(2) Получение бензил ((экзо)-3-азабицикло[3.1.0]гексан-6-ил)карбамата
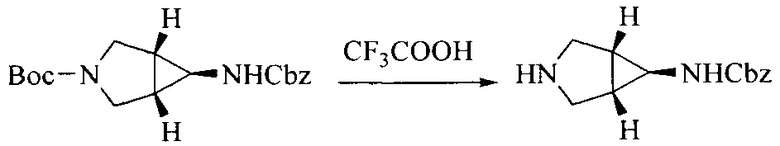
Трет-бутил (экзо)-6-((карбобензокси)амино)-3-азабицикло[3.1.0]гексан-3-карбоксилат (578 мг, 1,74 ммоль) растворяли в дихлорметане (10 мл), и добавляли трифторуксусную кислоту (5 мл). Полученную смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель удаляли перегонкой при пониженном давлении, с получением указанного в заголовке соединения (450 мг, неочищенный продукт), который непосредственно использовали на следующей стадии без очистки.
(3) Получение бензил ((экзо)-3-((2-хлорпиримидин-5-ил)метил)-3-азабицикло[3.1.0] гексан-6-ил)карбамата
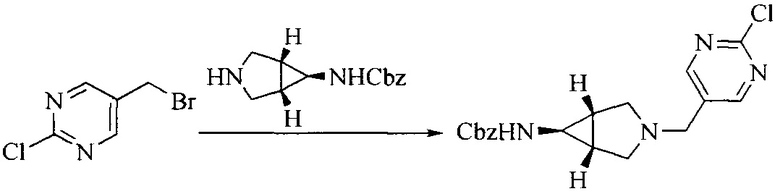
Бензил ((экзо)-3-азабицикло[3.1.0]гексан-6-ил)карбамат (450 мг, неочищенный продукт) и 5-(бромметил)-2-хлорпиримидин (415 мг, 2,0 ммоль) растворяли в ацетонитриле (50 мл), и добавляли карбонат калия (828 мг, 6,0 ммоль). Данное взаимодействие осуществляли при комнатной температуре в течение 4 часов. Полученный реакционный раствор фильтровали. Фильтрат концентрировали и подвергали колоночной хроматографии на силикагеле (элюент: смесь дихлорметан:метанол=50:1), с получением указанного в заголовке соединения (487 мг, выход в две стадии: 78%).
(4) Получение бензил (экзо)-3-((2-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-3-азабицикло[3.1.0]гексан-6-карбамата
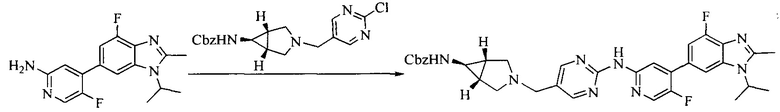
Бензил ((экзо)-3-((2-хлорпиримидин-5-ил)метил)-3-азабицикло[3.1.0]гексан-6-ил)карбамат (480 мг, 1,34 ммоль) и 5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (405 мг, 1,34 ммоль) растворяли в 1,4-диоксане (50 мл), и добавляли трис(дибензилиденацетон)дипалладий (119 мг, 0,13 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (124 мг, 0,26 ммоль) и карбонат цезия (1,3 г, 4,02 ммоль). В защитной атмосфере азота полученную смесь нагревали до 110°C и подвергали взаимодействию в течение 8 часов. Полученную смесь охлаждали до комнатной температуры и концентрировали. Воду (100 мл) и этилацетат (150 мл) добавляли к отделенной от водной фазы органической фазе. Водную фазу экстрагировали этилацетатом (150 мл ×2). Органические фазы объединяли, промывали насыщенным раствором NaCl, сушили над безводным сульфатом натрия и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь дихлорметан:метанол=20:1), с получением указанного в заголовке соединения (527 мг, выход: 63%).
(5) Получение (экзо)-3-((2-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-3-азабицикло[3.1.0]гексан-6-амина
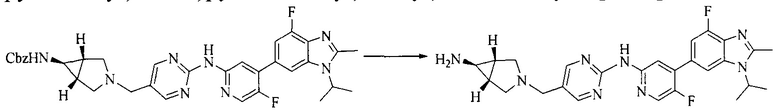
Бензил (экзо)-3-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-3-азабицикло[3.1.0]гексан-6-ил)карбамат (500 мг, 0,80 ммоль) и палладий на углероде (50 мг) суспендировали в метаноле (40 мл), и по каплям добавляли водный аммиак (6 мл). Данную систему вакуумировали и заполняли водородом. После взаимодействия при комнатной температуре в течение 16 часов, полученную реакционную смесь фильтровали. Фильтрат концентрировали и очищали колоночной хроматографией на силикагеле (элюент: смесь дихлорметан:метанол=10:1), с получением указанного в заголовке соединения (192 мг, выход: 49%).
Молекулярная формула: C26H28F2N8. Молекулярная масса: 490,6. ЖХ-МС (m/z): 491,3(M+H+).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 10,04 (с, 1H), 8,43 (д, J=6,0 Гц, 1H), 8,39 (с, 2H), 8,35 (д, J=2,0 Гц, 1H), 7,75 (с, 1H), 7,23 (д, J=11,6 Гц, 1H), 4,78-4,85 (м, 1H), 3,46 (с, 2H), 2,87 (д, J=8,8 Гц, 2H), 2,62 (с, 3H), 2,55 (с, 1H), 2,33 (д, J=8,4 Гц, 2H), 1,57-1,60 (м, 8H).
Пример 9: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((экзо)-октагидро-6H-пиридо[3,4-b][1,4]оксазин-6-ил)метил)пиримидин-2-амина (соединение 9)
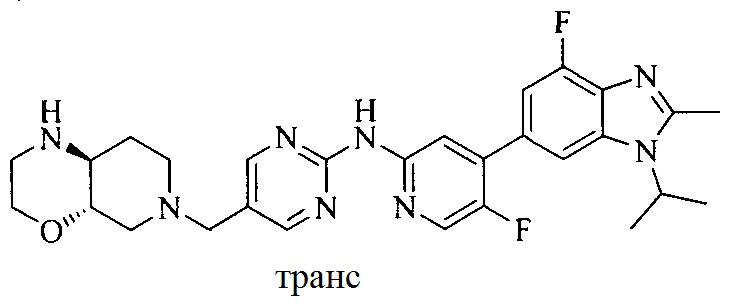
(1) Получение 1-бензил 6-трет-бутил (транс)-гексагидро-1H-пиридо[3,4-b][1,4]оксазин- 1,6(5H)-дикарбоксилата
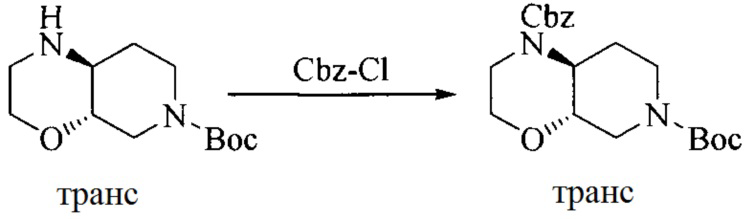
Трет-бутил (транс)-октагидро-6H-пиридо[3,4-b][1,4]оксазин-6-карбоксилат (242 мг, 1,0 ммоль) и триэтиламин (120 мг, 1,2 ммоль) добавляли к дихлорметану (5 мл). По каплям и медленно в условиях охлаждения на ледяной бане добавляли бензилхлорформиат (188 мг, 1,1 ммоль). После добавления, полученную смесь нагревали до комнатной температуры и перемешивали в течение 30 мин. Растворитель отгоняли на роторном испарителе, с получением неочищенного продукта (300 мг, неочищенный продукт), который использовали на следующей стадии без очистки.
(2) Получение бензил (транс)-октагидро-1H-пиридо[3,4-b][1,4]оксазин-1-карбоксилата
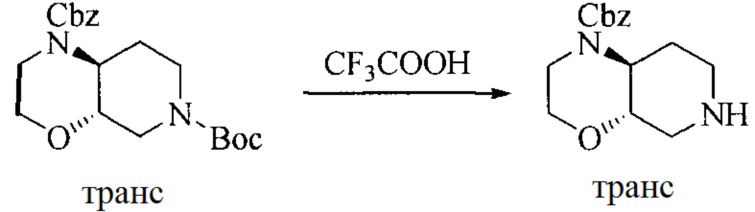
1-бензил 6-трет-бутил (транс)-гексагидро-1H-пиридо[3,4-b][1,4]оксазин-1,6(5H)-дикарбоксилат растворяли в смешанном растворе дихлорметана (3 мл) и трифторуксусной кислоты (3 мл). Полученную смесь перемешивали при комнатной температуре в течение 30 мин. Растворитель отгоняли на роторном испарителе, и добавляли небольшое количество дихлорметана. Растворитель снова отгоняли на роторном испарителе, с получением неочищенного продукта (260 мг), который использовали на следующей стадии без очистки.
(3) Получение бензил (транс)-6-((2-хлорпиримидин-5-ил)метил)октагидро-1H- пиридо[3,4-b][1,4]оксазин-1-карбоксилата
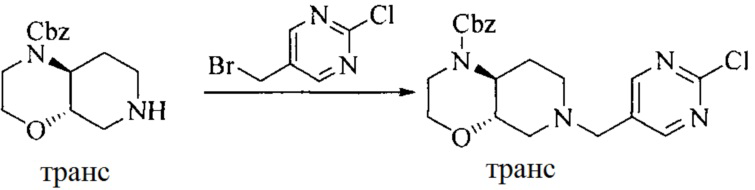
Бензил (транс)-октагидро-1H-пиридо[3,4-b][1,4]оксазин-1-карбоксилат (260 мг, 0,94 ммоль), 5-(бромметил)-2-хлорпиримидин (393 мг, 1,9 ммоль) и карбонат калия (786 мг, 5,7 ммоль) добавляли к ацетонитрилу (15 мл), и полученную смесь нагревали на масляной бане при 60°C в течение 1 часа. Полученный раствор подвергали дистилляции на роторном испарителе, и разделяли препаративной хроматографией (элюент: смесь вода:ацетонитрил=10:1-1:1), с получением продукта (300 мг, выход в три стадии: 75%).
(4) Получение бензил (транс)-6-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)октагидро-1H-пиридо[3,4-b][1,4]оксазин-1-карбоксилата
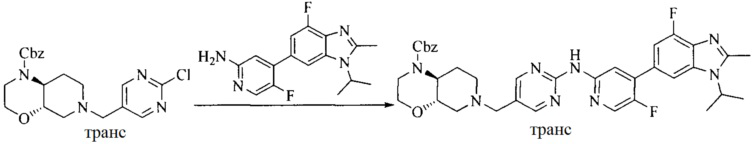
Бензил (транс)-6-((2-хлорпиримидин-5-ил)метил)октагидро-1H-пиридо[3,4-b][1,4]оксазин-1-карбоксилат (300 мг, 0,75 ммоль), 5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (449 мг, 1,5 ммоль), карбонат калия (317 мг, 2,3 ммоль), трис(дибензилиденацетон)дипалладий (30 мг) и 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (60 мг, 0,13 ммоль) добавляли к 20 мл 1,4-диоксана. В защитной атмосфере азота полученную смесь нагревали при 110°C на масляной бане в течение 8 часов, и растворитель удаляли отгонкой на роторном испарителе. Полученный остаток очищали препаративной хроматографией (элюент: смесь вода:ацетонитрил=10:1-1:1), с получением указанного в заголовке соединения (113 мг, выход: 23%).
(5) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((транс)-октагидро-6H-пиридо[3,4-b][1,4]оксазин-6-ил)метил)пиримидин-2-амина
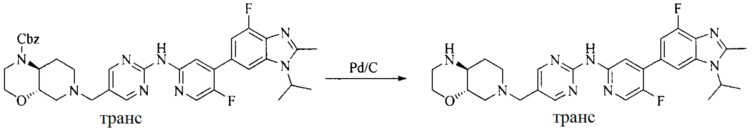
Бензил (транс)-6-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)октагидро-1H-пиридо[3,4-b][1,4]оксазин-1-карбоксилат (113 мг, 0,17 ммоль) растворяли в метаноле (5 мл) и аммиачной воде (0,50 мл). Добавляли катализатор палладий на углероде (30 мг), заполняли водородом и реакцию восстановления осуществляли в течение 2 часов. Полученную смесь фильтровали при отсасывании, и данный раствор подвергали дистилляции на роторном испарителе. Полученный остаток разделяли препаративной хроматографией (элюент: смесь вода:ацетонитрил=10:1-1:1), с получением продукта (30 мг, выход: 33%).
Молекулярная формула: C28H32F2N8O. Молекулярная масса: 534,6. ЖХ-МС (m/z): 535,3(M+H+).
1H-ЯМР (400 МГц, CDCl3) δ: 8,63-8,66 (м, 2H), 8,44 (с, 2H), 8,29 (д, J=2,0 Гц, 1H), 7,62 (с, 1H), 7,26 (д, J=8,0 Гц, 1H), 4,70-4,73 (м, 1H), 3,84 (дд, J1=2,8 Гц, J2=11,2 Гц, 1H), 3,62-3,63 (м, 1H), 3,47-3,48 (м, 2H), 3,20-3,21 (м, 1H), 2,86-3,02 (м, 4H), 2,68 (с, 3H), 2,35-2,40 (м, 1H), 2,11-2,12 (м, 1H), 1,94-2,01 (м, 1H), 1,68 (д, J=10,8 Гц, 6H), 1,66-1,63 (м, 1H), 1,55-1,45 (м, 1H).
Пример 10: Получение 5((4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)пиперидин-1-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина (соединение 10)
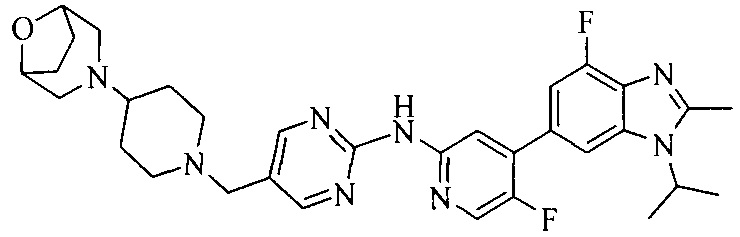
(1) Получение 1-((2-хлорпиримидин-5-ил)метил)пиперидин-4-ола
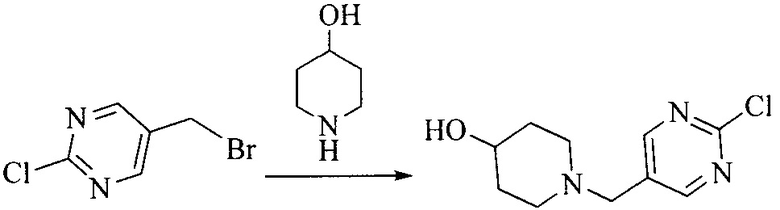
4-Гидроксипиперидин (202 мг, 2 ммоль) и 5-(бромметил)-2-хлорпиримидин (414 мг, 2,0 ммоль) растворяли в ацетонитриле (50 мл), и добавляли карбонат калия (828 мг, 6,0 ммоль). Полученной смеси давали взаимодействовать при комнатной температуре в течение 4 часов. Полученную смесь фильтровали, и растворитель удаляли дистилляцией. Данный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь дихлорметан:метанол=50:1), с получением указанного в заголовке соединения (378 мг, выход: 83%).
(2) Получение 1-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)пиперидин-4-ола
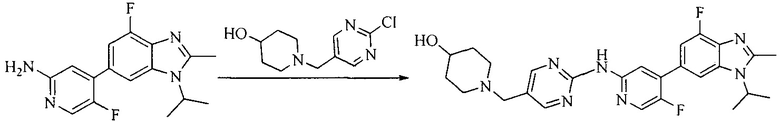
1-((2-Хлорпиримидин-5-ил)метил)пиперидин-4-ол (342 мг, 1,5 ммоль) и 5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (453 мг, 1,5 ммоль) растворяли в 1,4-диоксане (50 мл), и добавляли трис(дибензилиденацетон)дипалладий (137 мг, 0,15 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (143 мг, 0,3 ммоль) и карбонат цезия (1,47 г, 4,5 ммоль). В защитной атмосфере азота полученную смесь нагревали до 110°C и подвергали взаимодействию в течение 8 часов. Полученную смесь охлаждали до комнатной температуры и концентрировали. Воду (100 мл) и этилацетат (150 мл) добавляли к отделенной от водной фазы органической фазе. Водную фазу экстрагировали этилацетатом (150 мл ×2). Органические фазы объединяли, промывали насыщенным раствором NaCl, сушили над безводным сульфатом натрия и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь дихлорметан:метанол=20:1), с получением указанного в заголовке соединения (489 мг, выход: 66%).
(3) Получение 1-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)пиперидин-4-она
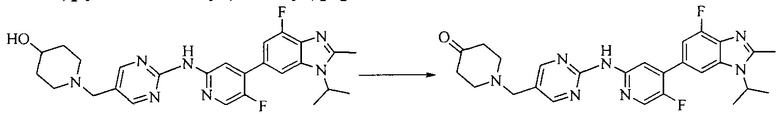
1-((2-((5-Фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)пиперидин-4-ол (450 мг, 0,91 ммоль) растворяли в ДМСО (6 мл) и триэтиламине (4 мл) и добавляли пиридин триоксид серы (723 мг, 4,55 ммоль) при перемешивании. После добавления, данное взаимодействие осуществляли при перемешивании при комнатной температуре в течение 2 часов. Добавляли воду (100 мл). Водную фазу экстрагировали этилацетатом (50 мл ×3). Органические фазы объединяли, промывали насыщенным раствором NaCl, сушили над безводным сульфатом натрия и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь дихлорметан:метанол=20:1), с получением указанного в заголовке соединения (363 мг, выход: 81%).
(4) Получение 5-((4-(8-окса-3-азабицикло[3.2.1]октан-3-ил)пиперидин-1-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина
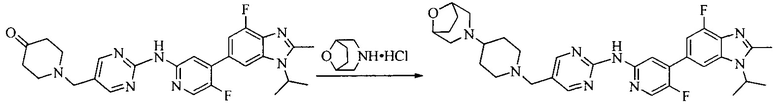
1-((2-((5-Фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)пиперидин-4-он (300 мг, 0,61 ммоль) и гидрохлорид 8-окса-3-азабицикло[3.2.1]октана (91,3 мг, 0,61 ммоль) растворяли в ДМФА (5 мл), и добавляли трихлорид индия (270 мг, 1,22 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 12 часов. Добавляли цианоборогидрид натрия (77 мг, 1,22 ммоль), и полученную смесь дополнительно перемешивали при комнатной температуре в течение 2 часов. Воду (20 мл) и этилацетат (20 мл ×5) добавляли для экстракции. Органические фазы объединяли, концентрировали и сушили над безводным сульфатом натрия. Неочищенный продукт подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=10:1), с получением указанного в заголовке соединения (133 мг, выход: 37%).
Молекулярная формула: C32H38F2N8O. Молекулярная масса: 588,7. ЖХ-МС (m/z): 589,4(M+H+).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 10,01 (с, 1H), 8,43-8,42 (м, 3H), 8,35 (д, J=2,0 Гц, 1H), 7,75 (с, 1H), 7,23 (д, J=11,6 Гц, 1H), 4,79-4,83 (м, 1H), 4,10-4,17 (м, 2H), 3,44-3,49 (м, 2H), 2,81-2,87 (м, 2H), 2,65 (с, 3H), 2,22-2,24 (м, 2H), 1,99-2,06(м, 3H), 1,74-1,76 (м, 2H), 1,67-1,73 (м, 5H), 1,56-1,58 (м, 6H), 1,36-1,38 (м, 2H).
Пример 11: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-((8-морфолино-3-азабицикло[3.2.1]октан-3-ил)метил)пиримидин-2-амина (соединение 11)
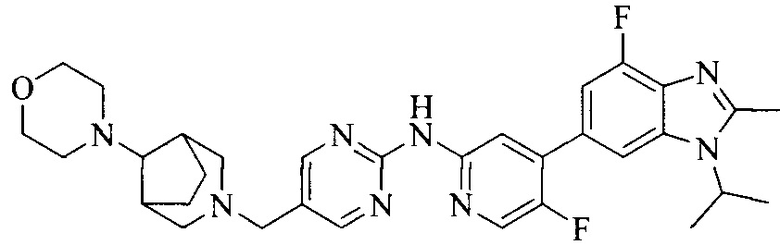
(1) Получение трет-бутил 8-морфолино-3-азабицикло[3.2.1]октан-3-карбоксилата
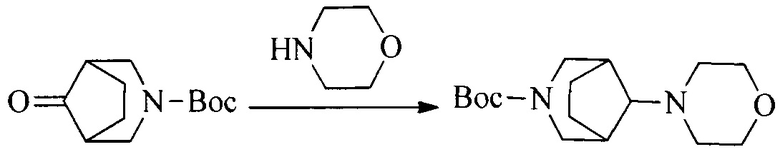
Трет-бутил 8-оксо-3-азабицикло[3.2.1]октан-3-карбоксилат (226 мг, 1,0 ммоль) и морфолин (870 мг, 10 ммоль) растворяли в тетрагидрофуране (5 мл). Добавляли каталитическое количество уксусной кислоты (20 мг). Полученную смесь перемешивали при комнатной температуре в течение 30 мин. Добавляли цианоборогидрид натрия (95 мг, 1,5 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли этилацетат (100 мл). Полученную смесь промывали. Органическую фазу сушили, и подвергали дистилляции на роторном испарителе, с получением неочищенного продукта (300 мг), который использовали на следующей стадии без очистки.
(2) Получение 4-(3-((2-хлорпиримидин-5-ил)метил)-3-азабицикло[3.2.1]октан-8-ил) морфолина
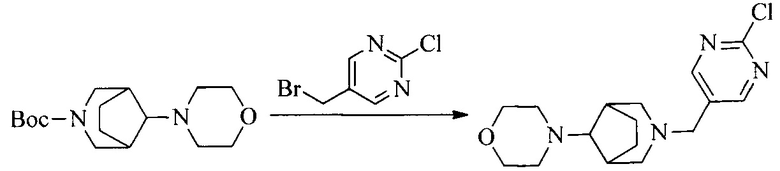
Трет-бутил 8-морфолино-3-азабицикло[3.2.1]октан-3-карбоксилат (300 мг, 1,0 ммоль) растворяли в смешанном растворе дихлорметана (5 мл) и трифторуксусной кислоты (5 мл). Полученную смесь перемешивали при комнатной температуре в течение 30 мин. Растворитель отгоняли на роторном испарителе. Добавляли карбонат калия (276 мг, 2,0 ммоль), 5-бромметил-2-хлорпиримидин (310 мг, 1,5 ммоль) и ацетонитрил (10 мл). Полученную смесь нагревали при 60°C на масляной бане в течение 1 часа. Растворитель удаляли отгонкой на роторном испарителе, и полученный остаток разделяли колоночной хроматографией на силикагеле (дихлорметан:метанол=20:1), с получением продукта (236 мг, выход в две стадии: 73%).
(3) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-((8-морфолино-3-азабицикло[3.2.1]октан-3-ил)метил)пиримидин-2-амина
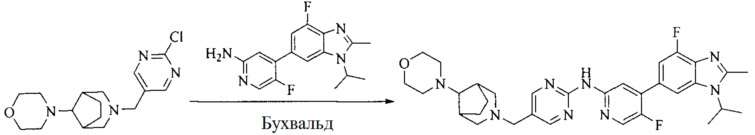
4-(3-((2-хлорпиримидин-5-ил)метил)-3-азабицикло[3.2.1]октан-8-ил)морфолин (236 мг, 0,73 ммоль), 5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (332 мг, 1,1 ммоль), карбонат калия (202 мг, 1,5 ммоль), трис(дибензилиденацетон)дипалладий (24 мг) и 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (48 мг) добавляли к 1,4-диоксану (10 мл). В защитной атмосфере азота полученную смесь нагревали при 110°C на масляной бане в течение 8 часов. Растворитель отгоняли на роторном испарителе, и полученный остаток разделяли препаративной хроматографией (элюент: смесь вода:ацетонитрил=10:1-1:1), с получением продукта (41 мг, выход: 10%).
Молекулярная формула: C32H38F2N8O. Молекулярная масса: 588,7. ЖХ-МС (m/z): 589,3(M+H+).
1H-ЯМР (400 МГц, CDCl3) δ: 8,66 (д, J=6,0 Гц, 1H), 8,36-8,52 (м, 3H), 8,28 (с, 1H), 7,64 (с, 1H), 7,27 (д, J=8,0 Гц, 1H), 4,70-4,74 (м, 1H), 3,71 (с, 4H), 2,69 (с, 5H), 2,34-2,42 (м, 6H), 2,10-2,15 (м, 3H), 1,68-1,79 (м, 8H), 1,05-1,30 (м, 4H).
Пример 12: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-((3-морфолино-8-азабицикло[3.2.1]октан-8-ил)метил)пиримидин-2-амина (соединение 12)
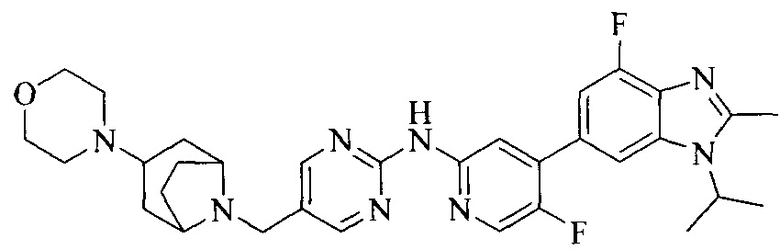
(1) Получение 8-((2-хлорпиримидин-5-ил)метил)-8-азабицикло[3.2.1]октан-3-она
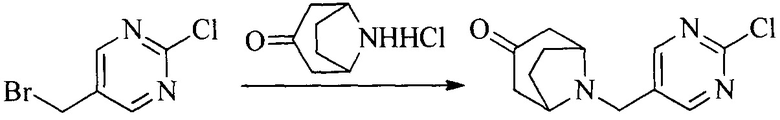
К 20 мл ацетонитрила добавляли 5-бромметил-2-хлорпиримидин (621 мг, 3,0 ммоль), гидрохлорид 3-оксо-8-азабицикло[3.2.1]октана (324 мг, 2,0 ммоль) и карбонат калия (552 мг, 4,0 ммоль). Полученную смесь нагревали при кипячении с обратным холодильником в течение 1 часа. Растворитель отгоняли на роторном испарителе, и полученный остаток разделяли колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=5:1), с получением продукта (387 мг, выход: 77%).
(2) Получение 8-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-8-азабицикло[3.2.1]октан-3-она
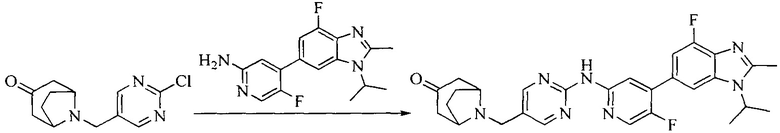
8-((2-хлорпиримидин-5-ил)метил)-8-азабицикло[3.2.1]октан-3-он (387 мг, 1,54 ммоль), 5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (930 мг, 3,08 ммоль), карбонат калия (638 мг, 4,62 ммоль), трис(дибензилиденацетон)дипалладий (39 мг) и 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (78 мг) добавляли к 1,4-диоксану (20 мл). В защитной атмосфере азота полученную смесь нагревали при 110°C на масляной бане в течение 8 часов. Растворитель отгоняли на роторном испарителе, и полученный остаток разделяли препаративной хроматографией (элюент: смесь вода:ацетонитрил=10:1-1:1), с получением указанного в заголовке соединения (133 мг, выход: 17%).
(3) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-((3-морфолино-8-азабицикло[3.2.1]октан-8-ил)метил)пиримидин-2-амина
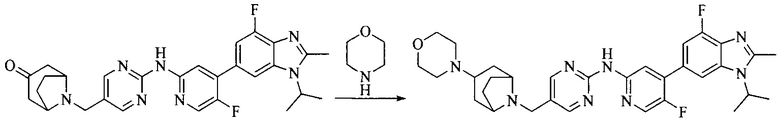
8-((2-((5-Фтор-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-8-азабицикло[3.2.1]октан-3-он (133 мг, 0,257 ммоль) растворяли в смешанном растворе тетрагидрофурана (1 мл) и метанола (1 мл), и добавляли морфолин (223 мг, 2,57 ммоль) и трифторуксусную кислоту (20 мг). Полученную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли цианоборогидрид натрия (25 мг, 0,386 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Этилацетат (20 мл) добавляли для отделения органической фазы от водной фазы. Органическую фазу промывали насыщенной NaCl водой, сушили, перегоняли на роторном испарителе и подвергали препаративной хроматографии (элюент: смесь вода:ацетонитрил=10:1-1:1) с отделением данного продукта (40 мг, выход: 33%).
Молекулярная формула: C32H38F2N8O. Молекулярная масса: 588,7. ЖХ-МС (m/z): 589,3(M+H+).
1H-ЯМР (400 МГц, CDCl3) δ: 9,48 (шир.с, 1H), 8,67 (д, J=6,0 Гц, 1H), 8,62 (с, 2H), 8,38 (д, J=2,4 Гц, 1H), 7,62 (с, 1H), 7,25 (д, J=13,2 Гц, 1H), 4,67-4,74 (м, 1H), 3,72 (с, 4H), 3,60 (с, 2H), 3,36 (с, 2H), 2,66 (с, 3H), 2,55 (шир.с, 5H), 2,05-2,07 (м, 2H), 1,84-1,82 (м, 2H), 1,67-1,68 (м, 10H).
Пример 13: Получение 5-((3-этил-3,8-диазабицикло[3.2.1]октан-8-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина (соединение 13)
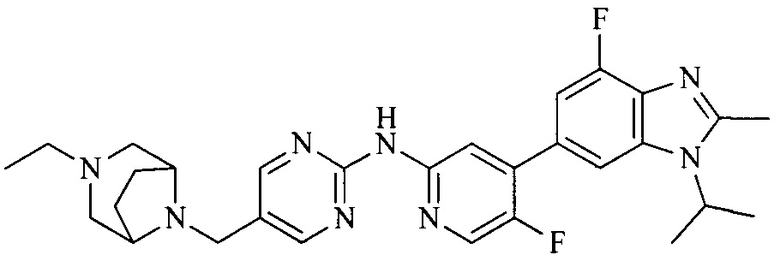
(1) Получение трет-бутил 8-((2-хлорпиримидин-5-ил)метил)-3,8-диазабицикло[3.2.1]октан-3-карбоксилата
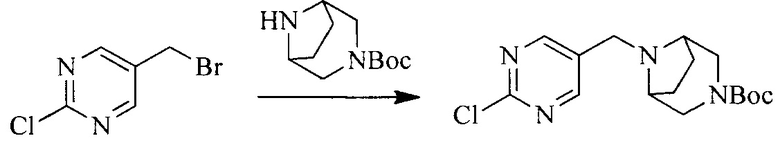
5-(Бромметил)-2-хлорпиримидин (412 мг, 2 ммоль) растворяли в тетрагидрофуране (30 мл), и добавляли триэтиламин (606 мг, 6 ммоль) и трет-бутил 3,8-диазабицикло[3.2.1]октан-3-карбоксилат (509 мг, 2,4 ммоль) при перемешивании. Данное взаимодействие осуществляли при перемешивании при комнатной температуре в течение 4 часов. После расходования исходных веществ, как установлено ТСХ, полученную в результате смесь фильтровали при отсасывании, и фильтрат концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=1:1), с получением указанного в заголовке соединения (600 мг, выход: 88,8%).
(2) Получение 8-((2-хлорпиримидин-5-ил)метил)-3,8-диазабицикло[3.2.1]октана

Трет-бутил 8-((2-хлорпиримидин-5-ил)метил)-3,8-диазабицикло[3.2.1]октан-3-карбоксилат (600 мг, 1,78 ммоль) растворяли в дихлорметане (10 мл), и добавляли трифторуксусную кислоту (5 мл) при перемешивании. Полученную смесь перемешивали при комнатной температуре в течение 4 часов. После расходования исходных веществ, как установлено ТСХ, полученную смесь фильтровали при отсасывании. Фильтрат концентрировали и затем неочищенный продукт подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=30:1), с получением указанного в заголовке соединения (400 мг, выход: 94,6%).
(3) Получение 8-((2-хлорпиримидин-5-ил)метил)-3-этил-3,8-диазабицикло[3.2.1]октана
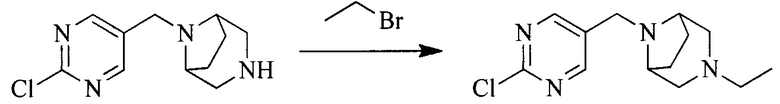
8-((2-Хлорпиримидин-5-ил)метил)-3,8-диазабицикло[3.2.1]октан (400 мг, 1,68 ммоль) растворяли в ацетонитриле (20 мл), и добавляли этилбромид (363 мг, 3,36 ммоль) и карбонат калия (695,5 мг, 5,04 ммоль) при перемешивании. Полученную смесь перемешивали при 40°C в течение ночи. После расходования исходных веществ, как установлено ТСХ, полученную смесь фильтровали при отсасывании. Фильтрат концентрировали и затем подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=20:1), с получением указанного в заголовке соединения (380 мг, выход: 85,2%).
(4) Получение 5-((3-этил-3,8-диазабицикло[3.2.1]октан-8-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина
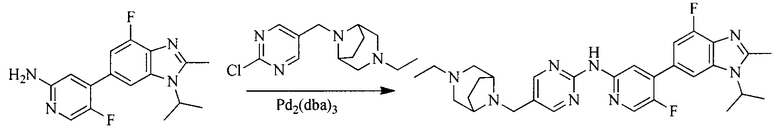
5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (200 мг, 0,66 ммоль), 8-((2-хлорпиримидин-5-ил)метил)-3-этил-3,8-диазабицикло[3.2.1]октан (175,6 мг, 0,66 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (63 мг, 0,13 ммоль), карбонат цезия (643,5 мг, 1,98 ммоль) и трис(дибензилиденацетон)дипалладий (60 мг, 0,066 ммоль) добавляли в баклажановидный сосуд, и добавляли 1,4-диоксан (20 мл). В защитной атмосфере азота данное взаимодействие осуществляли при 110°C в течение 4 часов. Полученную реакционную смесь фильтровали при отсасывании. Фильтрат подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=15:1), с получением указанного в заголовке соединения (100 мг, выход: 28,5%).
Молекулярная формула: C29H34F2N8. Молекулярная масса: 532,3. ЖХ-МС (m/z): 533,3(M+H+).
1H-ЯМР (400 МГц, MeOD-d4) δ: 8,54-8,58 (м, 3H), 8,27 (д, J=2,0 Гц, 1H), 7,80 (с, 1H), 7,26 (д, J=11,6 Гц, 1H), 4,84-4,88 (м, 1H), 3,37-3,60 (м, 3H), 3,00-3,21(м, 4H), 2,69 (с, 3H), 2,25-2,35 (м, 2H), 1,85-2,05 (м, 2H), 1,67-1,71 (м, 6H), 1,22-1,35 (м, 2H), 1,16-1,20 (м, 4H).
Пример 14: Получение 5-((8-этил-3,8-диазабицикло[3.2.1]октан-3-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина (соединение 14)
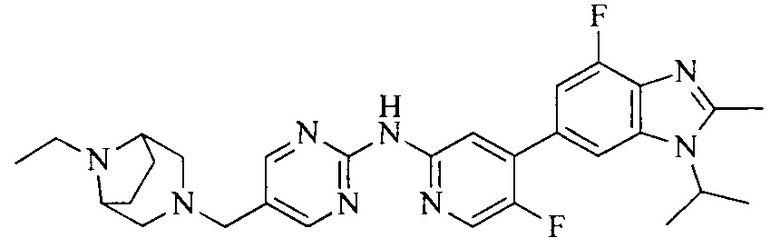
(1) Получение трет-бутил 8-этил-3,8-диазабицикло[3.2.1]октан-3-карбоксилата
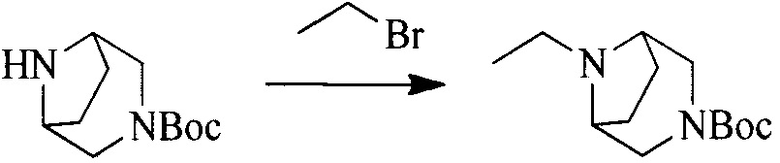
Трет-бутил 3,8-диазабицикло[3.2.1]октан-3-карбоксилат (212 мг, 1 ммоль) растворяли в ацетонитриле (20 мл), и добавляли этилбромид (129,6 мг, 1,2 ммоль) и карбонат калия (414 мг, 3 ммоль) при перемешивании. Полученную смесь перемешивали в течение ночи при 40°C. После расходования исходных веществ, как установлено ТСХ, полученную смесь фильтровали при отсасывании. Фильтрат перегоняли при пониженном давлении, с получением указанного в заголовке соединения (200 мг, выход: 83,3%).
(2) Получение 8-этил-3,8-диазабицикло[3.2.1]октана
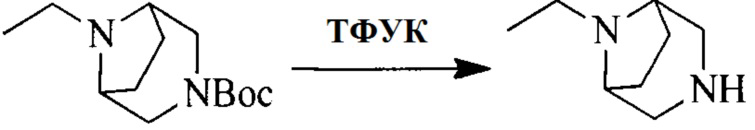
Трет-бутил 8-этил-3,8-диазабицикло[3.2.1]октан-3-карбоксилат (200 мг, 0,83 ммоль) растворяли в дихлорметане (5 мл), и добавляли трифторуксусную кислоту (3 мл) при перемешивании. Полученную смесь перемешивали при комнатной температуре в течение 1 часа. После расходования исходных веществ, как установлено ТСХ, полученную смесь перегоняли при пониженном давлении, с получением указанного в заголовке соединения (100 мг, выход: 85,8%).
(3) Получение 3-((2-хлорпиримидин-5-ил)метил)-8-этил-3,8-диазабицикло[3.2.1]октана
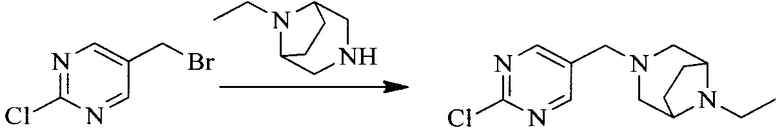
5-(Бромметил)-2-хлорпиримидин (146 мг, 0,71 ммоль) растворяли в тетрагидрофуране (30 мл), и добавляли карбонат калия (294 мг, 2,13 ммоль) и 8-этил-3,8-диазабицикло[3.2.1]октан (100 мг, 0,71 ммоль) при перемешивании. Полученную смесь перемешивали при комнатной температуре в течение 4 часов. После расходования исходных веществ, как установлено ТСХ, полученную смесь фильтровали при отсасывании. Фильтрат концентрировали и затем подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=30:1), с получением указанного в заголовке соединения (150 мг, выход: 79,4%).
(4) Получение 5-((8-этил-3,8-диазабицикло[3.2.1]октан-3-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина
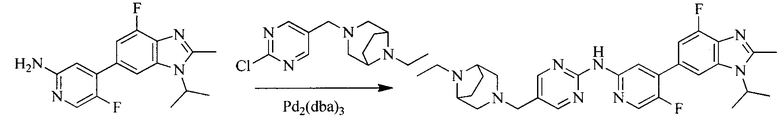
5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (170 мг, 0,564 ммоль), 3-((2-хлорпиримидин-5-ил)метил)-8-этил-3,8-диазабицикло[3.2.1]октан (150 мг, 0,564 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (54 мг, 0,11 ммоль), карбонат цезия (550 мг, 1,69 ммоль) и трис(дибензилиденацетон)дипалладий (52 мг, 0,057 ммоль) добавляли в 100 мл баклажановидный сосуд, и добавляли 1,4-диоксан (20 мл). В защитной атмосфере азота данное взаимодействие осуществляли при 110°C в течение 4 часов, и полученную смесь фильтровали при отсасывании. Фильтрат концентрировали и подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=15:1), с получением указанного в заголовке соединения (120 мг, выход: 40%).
Молекулярная формула: C29H34F2N8. Молекулярная масса: 532,3. ЖХ-МС (m/z): 533,3(M+H+).
1H-ЯМР (400 МГц, MeOD-d4) δ: 8,53 (д, J=6,0 Гц, 1H), 8,50 (с, 2H), 8,27 (д, J=2,4 Гц, 1H), 7,79 (с, 1H), 7,26 (д, J=11,2 Гц, 1H), 4,60 (с, 1H), 3,95-4,02 (м, 2H), 3,59 (с, 2H), 3,00-3,12 (м, 2H), 2,92 (д, J=12,8 Гц, 2H), 2,69 (с, 3H), 2,59 (д, J=12,8 Гц, 2H), 2,11-2,16 (м, 4H), 1,69 (д, J=6,8 Гц, 6H), 1,31-1,35 (м, 3H).
Пример 15: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(пиперазин-1-илметил)пиримидин-2-амина (соединение 15)

(1) Получение трет-бутил 4-(2-хлорпиримидин-5-ил)пиперазин-1-карбоксилата
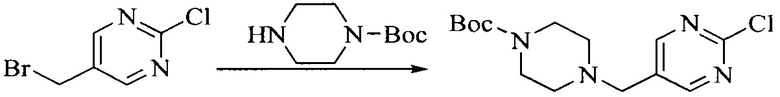
5-(Бромметил)-2-хлорпиримидин (500 мг, 2,42 ммоль) растворяли в тетрагидрофуране (15 мл), и добавляли трет-бутил пиперазин-1-карбоксилат (372 мг, 2 ммоль) и триэтиламин (404 мг, 4 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. После взаимодействия, полученную смесь концентрировали при пониженном давлении, разбавляли водой (15 мл) и экстрагировали этилацетатом (20 мл ×3). Фазы разделяли. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=1:1), с получением указанного в заголовке соединения (520 мг, выход: 83%).
(2) Получение трет-бутил 4-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)пиперазин-1-карбоксилата
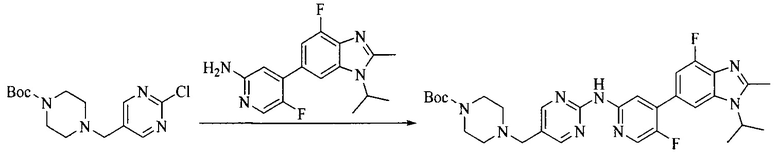
5-Фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (302 мг, 1 ммоль) и 4-((2-хлорпиримидин-5-ил)метил)пиперазин-1-карбоксилат (312 мг, 1 ммоль) растворяли в 1,4-диоксане (15 мл), и добавляли трис(дибензилиденацетон)дипалладий (92 мг, 0,1 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (95 мг, 0,2 ммоль) и карбонат цезия (815 мг, 2,5 ммоль). В защитной атмосфере азота полученную смесь перемешивали при кипячении с обратным холодильником в течение ночи и фильтровали. Фильтрат концентрировали при пониженном давлении. Неочищенный продукт подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=30:1), с получением указанного в заголовке соединения (350 мг, выход: 60%).
(3) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(пиперазин-1-илметил)пиримидин-2-амина
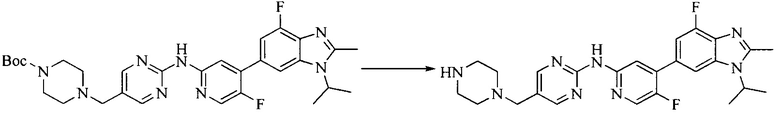
Трет-бутил 4-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)пиперазин-1-карбоксилат (200 мг, 0,346 ммоль) растворяли в дихлорметане (5 мл), и добавляли трифторуксусную кислоту (2 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали при пониженном давлении. Добавляли дихлорметан и насыщенный раствор гидрокарбоната натрия. Фазы разделяли. Органическую фазу сушили, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле (метанол:дихлорметан=0-1:20, с добавлением 0,5% триэтиламина), с получением указанного в заголовке соединения в виде белого твердого вещества (110 мг, выход: 67%).
Молекулярная формула: C25H28F2N8. Молекулярная масса: 478,5. ЖХ-МС (m/z): 479,3(M+H+).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 10,03 (с, 1H), 8,42-8,47 (м, 3H), 8,35 (д, J=2,0 Гц, 1H), 7,75 (с, 1H), 7,23 (д, J=11,6 Гц, 1H), 4,78-4,85 (м, 1H), 3,34 (с, 2H), 2,62-2,66 (м, 7H), 2,20-2,31 (м, 5H), 1,58 (д, J=6,8 Гц, 6H).
Пример 16: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(пиперазин-1-ил)пиримидин-2-амина (соединение 16)
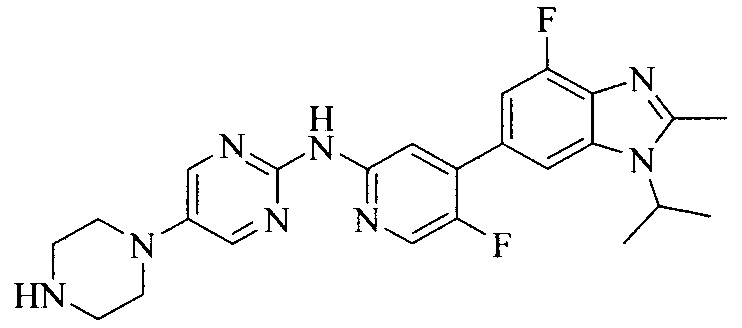
(1) Получение трет-бутил 4-(пиримидин-5-ил)пиперазин-1-карбоксилата
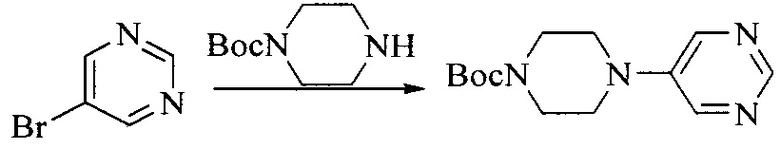
В 100 мл баклажановидный сосуд добавляли 5-бромпиримидин (3,16 г, 20 ммоль), трет-бутилпиперазин-1-карбоксилат (3,72 г, 20 ммоль), 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (2,49 г, 4 ммоль), карбонат цезия (13,0 г, 40 ммоль) и трис(дибензилиденацетон)дипалладий (1,83 г, 2 ммоль); и добавляли толуол (80 мл). Данное взаимодействие осуществляли при 90°C в защитной атмосфере азота в течение 12 часов. Полученную смесь фильтровали при отсасывании, фильтрат концентрировали, и неочищенный продукт очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=50:1), с получением указанного в заголовке соединения (3,1 г, выход: 58,7%).
(2) Получение трет-бутил 4-(2-бромпиримидин-5-ил)пиперазин-1-карбоксилата
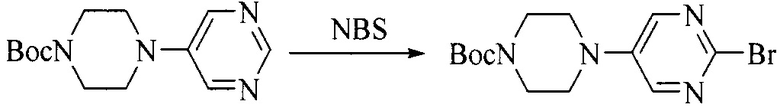
Трет-бутил 4-(пиримидин-5-ил)пиперазин-1-карбоксилат (2,64 г, 10 ммоль) взвешивали и добавляли к ацетонитрилу (125 мл). N-бромбутанимид (1,78 г, 10 ммоль) добавляли при перемешивании, и полученную смесь перемешивали при 25°C в течение 16 часов. Полученный реакционный раствор концентрировали. Этилацетат (100 мл) и воду (100 мл) добавляли для разделения фаз. Органическую фазу концентрировали и затем подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=30:1), с получением указанного в заголовке соединения (40 мг, выход: 1,17%).
(3) Получение трет-бутил 4-(2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)пиперазин-1-карбоксилата
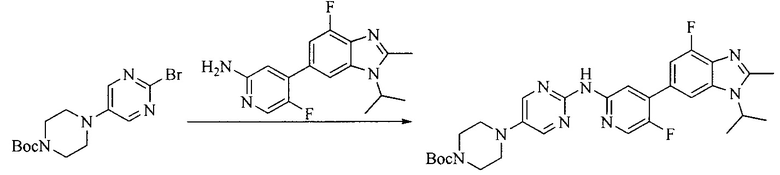
5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (36 мг, 0,12 ммоль), трет-бутил 4-(2-бромпиримидин-5-ил)пиперазин-1-карбоксилат (40 мг, 0,12 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (11,4 мг, 0,024 ммоль), карбонат цезия (97,7 мг, 0,3 ммоль) и трис(дибензилиденацетон)дипалладий (11 мг, 0,012 ммоль) добавляли в 25 мл баклажановидный сосуд, и добавляли 1,4-диоксан (10 мл). В защитной атмосфере азота данное взаимодействие осуществляли при 110°C в течение 2 часов, и полученную в результате смесь фильтровали при отсасывании. Фильтрат концентрировали и затем подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=20:1), с получением указанного в заголовке соединения (15 мг, выход: 22,2%).
(4) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(пиперазин-1-ил)пиримидин-2-амина
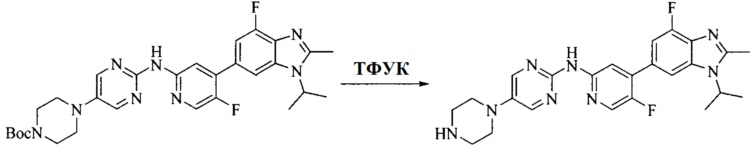
Трет-бутил 4-(2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)пиперазин-1-карбоксилат (15 мг, 0,027 ммоль) растворяли в дихлорметане (2 мл). Трифторуксусную кислоту (2 мл) добавляли при перемешивании. Полученную смесь перемешивали при 25°C в течение 0,5 часов. Полученный реакционный раствор концентрировали, и остаток разводили этилацетатом (10 мл). Вводили газообразный аммиак до тех пор, пока раствор не становился щелочным. Данный раствор концентрировали и подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=20:1), с получением указанного в заголовке соединения (7 мг, выход: 56,0%).
Молекулярная формула: C24H26F2N8. Молекулярная масса: 464,5. ЖХ-МС (m/z): 465,4(M+H+).
1H-ЯМР (400 МГц, MeOD-d4) δ: 8,77 (д, J=6,0 Гц, 1H), 8,53 (с, 1H), 8,31 (д, J=2,8 Гц, 1H), 8,27 (с, 1H), 7,81 (с, 1H), 7,30 (д, J=11,2 Гц, 1H), 4,86-4,93 (м, 1H), 3,16-3,18 (м, 4H), 3,04-3,06 (м, 4H), 2,69 (с, 3H), 1,70 (д, J=6,8 Гц, 6H).
Пример 17: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(4-метилпиперазин-1-ил)пиримидин-2-амина (соединение 17)
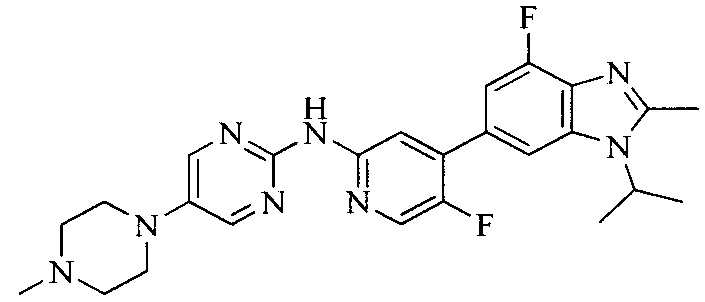
(1) Получение N,N-бис(4-метоксибензил)-5-(4-метилпиперазин-1-ил)пиримидин-2-амина
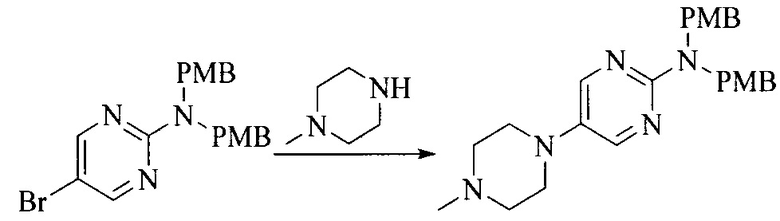
5-Бром-N,N-бис(4-метоксибензил)пиримидин-2-амин (2,0 г, 4,83 ммоль) растворяли в толуоле (20 мл), и добавляли N-метилпиперазин (483 мг, 4,83 ммоль), трис(дибензилиденацетон)дипалладий (221 мг, 0,24 ммоль), 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (301 мг, 0,48 ммоль) и трет-бутоксид натрия (930 мг, 9,7 ммоль). Для замены воздуха в реакционную систему вводили азот, и данное взаимодействие осуществляли при перемешивании при 80°C в течение 18 часов. Полученную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали и подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=30:1), с получением указанного в заголовке соединения (859 мг, 41%)
(2) Получение 5-(4-метилпиперазин-1-ил)пиримидин-2-амина
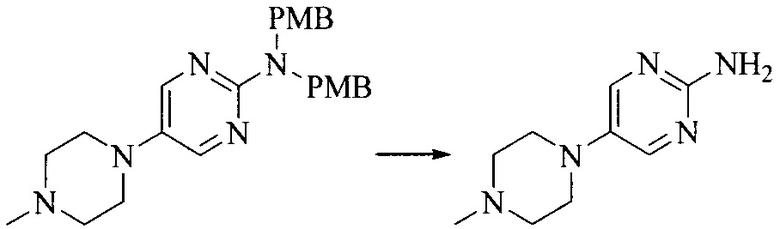
N,N-бис(4-метоксибензил)-5-(4-метилпиперазин-1-ил)пиримидин-2-амин (800 мг, 1,8 ммоль) растворяли в смешанном растворе трифторуксусной кислоты (10 мл) и дихлорметана (10 мл). Полученную смесь нагревали до 40°C и подвергали взаимодействию в течение 18 часов. Полученный реакционный раствор концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=10:1), с получением указанного в заголовке соединения (285 мг, 82%).
(3) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(4-метилпиперазин-1-ил)пиримидин-2-амина
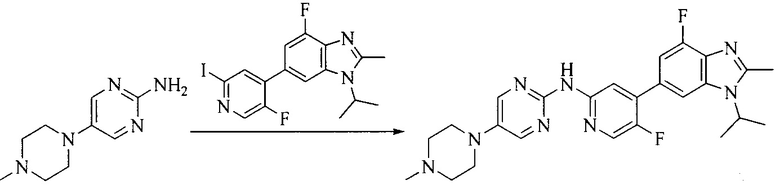
5-(4-(диметиламино)пиперидин-1-ил)пиримидин-2-амин (193 мг, 1,0 ммоль), 4-фтор-6-(5-фтор-2-йодпиридин-4-ил)-1-изопропил-2-метил-1H-бензо[d]имидазол (413 мг, 1,0 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (47,7 мг, 0,10 ммоль), карбонат цезия (815 мг, 2,5 ммоль) и трис(дибензилиденацетон)дипалладий (45,8 мг, 0,05 ммоль) растворяли в 1,4-диоксане (20 мл). Для замены воздуха в реакционную систему вводили азот. Полученную смесь нагревали до 120°C, взаимодействие осуществляли в течение 8 часов и фильтровали. Фильтрат концентрировали и подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=10:1), с получением указанного в заголовке соединения (52,6 мг, выход: 11%).
Молекулярная формула: C25H28F2N8. Молекулярная масса: 478,6 ЖХ-МС (m/z): 479,0(M+H+).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 9,66 (с, 1H), 8,36 (д, J=6 Гц, 1H), 8,29-8,31 (м, 3H), 7,73 (с, 1H), 7,20 (д, J=11,6 Гц, 1H), 4,79-4,83 (м, 1H), 3,07-3,10 (м, 4H), 2,61 (с, 3H), 2,43-2,46 (м, 4H), 2,20 (с, 3H), 1,57 (д, J=6,8 Гц, 6H).
Пример 18: Получение 5-(4-этилпиперазин-1-ил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина (соединение 18)
(1) Получение 5-(4-этилпиперазин-1-ил)-N,N-бис(4-метоксибензил)пиримидин-2-амина
5-Бром-N,N-бис(4-метоксибензил)пиримидин-2-амин (800 мг, 1,94 ммоль) растворяли в толуоле (20 мл), и добавляли N-этилпиперазин (330 мг, 2,89 ммоль), трис(дибензилиденацетон)дипалладий (89 мг, 0,097 ммоль), 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (121 мг, 0,194 ммоль) и трет-бутоксид натрия (372 мг, 3,88 ммоль). Полученную смесь перемешивали при 80°C в защитной атмосфере азота в течение 18 часов. Полученную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали, и неочищенный продукт очищали колоночной хроматографией на силикагеле (метанол:дихлорметан=0-1:30), с получением указанного в заголовке продукта (440 мг, выход: 51%).
(2) Получение 5-(4-этилпиперазин-1-ил)пиримидин-2-амина

5-(4-Этилпиперазин-1-ил)-N,N-бис(4-метоксибензил)пиримидин-2-амин (440 мг, 0,98 ммоль) растворяли в трифторуксусной кислоте (5 мл). Полученную смесь нагревали до 40°C и подвергали взаимодействию в течение 18 часов. Растворитель удаляли при пониженном давлении, с получением указанного в заголовке соединения (180 мг, выход: 89%), которое непосредственно использовали на следующей стадии.
(3) Получение 5-(4-этилпиперазин-1-ил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина
4-Фтор-6-(5-фтор-2-йодпиридин-4-ил)-1-изопропил-2-метил-1H-бензо[d]имидазол (359 мг, 0,87 ммоль) и 5-(4-этилпиперазин-1-ил)пиримидин-2-амин (180 мг, 0,87 ммоль) растворяли в толуоле (10 мл). Добавляли трис(дибензилиденацетон)дипалладий (80 мг, 0,087 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (50 мг, 0,087 ммоль) и трет-бутоксид натрия (209 мг, 2,18 ммоль). В защитной атмосфере азота полученную смесь нагревали до 100°C и подвергали взаимодействию в течение 16 часов. Полученную смесь охлаждали до комнатной температуры, и добавляли метанол (5 мл). Полученную смесь фильтровали через целит. Фильтрат концентрировали в вакууме, и неочищенный продукт очищали колоночной хроматографией на силикагеле (метанол:дихлорметан=0-1:10, с добавлением 0,5% водного аммиака), с получением указанного в заголовке соединения в виде белого твердого вещества (18 мг, выход: 4,2%).
Молекулярная формула: C26H30F2N8. Молекулярная масса: 492,6. ЖХ-МС (m/z): 493(M+H+).
1H-ЯМР (400 МГц, MeOD-d4) δ: 8,46 (д, J=5,2 Гц, 1H), 8,34 (с, 2H), 8,22 (с, 1H), 7,78 (с, 1H), 7,25 (д, J=11,2 Гц, 1H), 4,55-4,62 (м, 1H), 3,20-3,28 (м, 4H), 2,80-2,91 (м, 4H), 2,72-2,75 (м, 2H), 2,68 (с, 3H), 1,69 (д, J=6,8 Гц, 6H), 1,22 (т, J=7,2 Гц, 3H).
Пример 19: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(4-(оксациклобутан-3-ил)пиперазин-1-ил)пиримидин-2-амина (соединение 20)
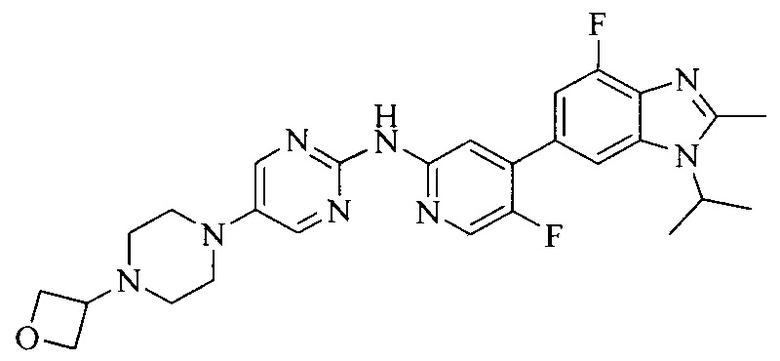
(1) Получение 6-(2-бром-5-фторпиридин-4-ил)-4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазола
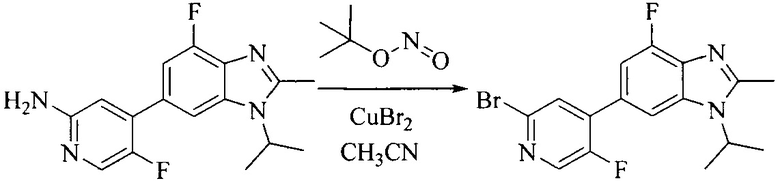
5-Фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (906 мг, 3,0 ммоль) растворяли в ацетонитриле (30 мл), и медленно добавляли трет-бутилнитрил (340 мг, 3,3 ммоль). После добавления, полученную смесь перемешивали при комнатной температуре в течение 30 мин, и добавляли CuBr2 (1,00 г, 4,5 ммоль). Полученную в результате смесь перемешивали при комнатной температуре в течение 2 часов и фильтровали при отсасывании. Растворитель удаляли отгонкой на роторном испарителе, и неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=1:1), с получением продукта (498 мг, выход: 45,4%).
(2) Получение трет-бутил 4-(2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)пиперазин1-карбоксилата
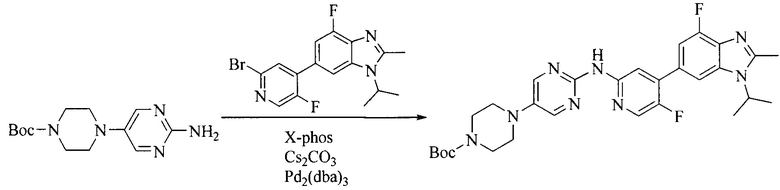
Трет-бутил 4-(2-аминопиримидин-5-ил)пиперазин-1-карбоксилат (380 мг, 1,36 ммоль), 6-(2-бром-5-фторпиридин-4-ил)-4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазола (498 мг, 1,36 ммоль), карбонат цезия (447 мг, 1,36 ммоль), трис(дибензилиденацетон)дипалладий (38 мг) и 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (76 мг) добавляли к 1,4-диоксану (20 мл). В защитной атмосфере азота полученную смесь нагревали при 110°C на масляной бане в течение 8 часов, и растворитель удаляли отгонкой на роторном испарителе. Полученный остаток очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=10:1-1:1), с получением указанного в заголовке соединения (256 мг, выход: 33,3%).
(3) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(4-(оксациклобутан-3-ил)пиперазин-1-ил)пиримидин-2-амина
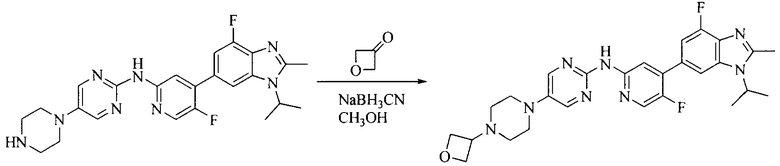
N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(пиперазин-1-ил)пиримидин-2-амин (170 мг, 0,366 ммоль) растворяли в метаноле (5 мл), и по каплям добавляли 3-оксетанон (39,5 мг, 0,549 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов, и медленно добавляли цианоборогидрид натрия (34,6 мг, 0,549 ммоль). Полученную смесь дополнительно перемешивали при комнатной температуре в течение 30 мин. Растворитель удаляли отгонкой на роторном испарителе, и полученный остаток очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=10:1), с получением продукта (82 мг, выход: 42,1%).
Молекулярная формула: C27H30F2N8O. Молекулярная масса: 520,6. ЖХ-МС (m/z): 521,3(M+H+).
1H-ЯМР(400 МГц, MeOD) δ: 8,47 (м, J=6,4 Гц, 1H), 8,30 (с, 2H), 8,20 (д, J=2,4 Гц, 1H), 7,77 (с, 1H), 7,26 (д, J=8,0 Гц, 1H), 4,72 (т, J=6,4 Гц, 2H), 4,59-4,64 (м, 3H), 3,50-3,55 (м, 1H), 3,19 (т, J=4,8 Гц, 4H), 2,68 (с, 3H), 2,52 (т, J=4,8 Гц, 4H), 1,68 (д, J=6,8 Гц, 6H).
Пример 20: Получение 5-(4-(диметиламино)пиперидин-1-ил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина (соединение 21)
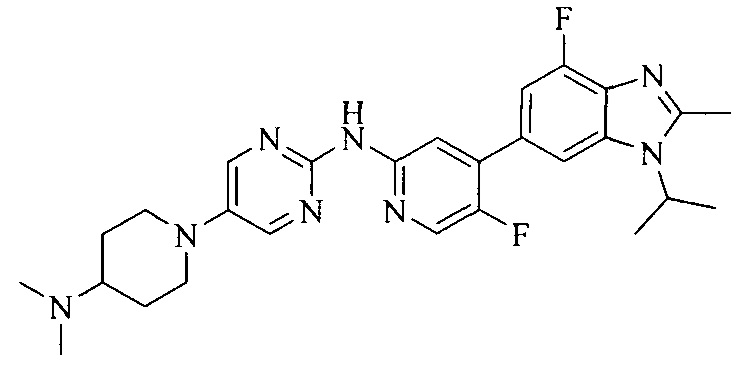
(1) Получение 5-(4-(диметиламино)пиперидин-1-ил)-N,N-бис(4-метоксибензил)пиримидин-2-амина
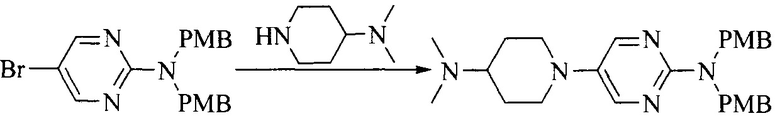
В защитной атмосфере азота к толуолу (50 мл) в реакционном сосуде добавляли 5-бром-N,N-бис(4-метоксибензил)-пиримидин-2-амин (2,0 г, 4,84 ммоль), N,N-диметилпиперидин-4-амин (620 мг, 4,84 ммоль), трис(дибензилиденацетон)дипалладий (221 мг, 0,24 ммоль), 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (301 мг, 0,48 ммоль) и трет-бутоксид натрия (930 мг, 9,7 ммоль). Полученную смесь нагревали до 80°C и подвергали взаимодействию в течение 16 часов. После взаимодействия, полученную смесь охлаждали до комнатной температуры и фильтровали, и фильтрат концентрировали и разделяли колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=5:1), с получением указанного в заголовке соединения (758 мг, 34%).
(2) Получение 5-(4-(диметиламино)пиперидин-1-ил)пиримидин-2-амина
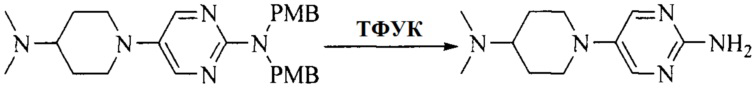
К раствору 5-(4-(диметиламино)пиперидин-1-ил)-N,N-бис(4-метоксибензил)пиримидин-2-амина (758 мг, 1,64 ммоль) в дихлорметане (20 мл) добавляли трифторуксусную кислоту (10 мл). Полученную смесь нагревали до 40°C и подвергали взаимодействию в течение 16 часов. После взаимодействия, полученный реакционный раствор охлаждали до комнатной температуры, концентрировали и очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=20:1), с получением указанного в заголовке соединения (282 мг, 78%).
(3) Получение 5-(4-(диметиламино)пиперидин-1-ил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина
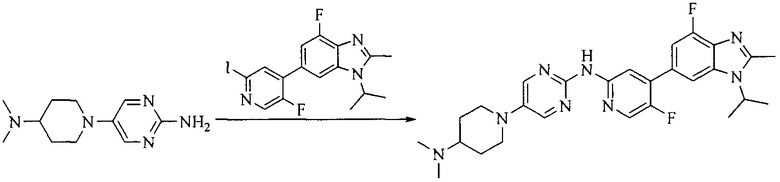
5-(4-(диметиламино)пиперидин-1-ил)пиримидин-2-амин (282 мг, 1,28 ммоль), 4-фтор-6-(5-фтор-2-йодпиридин-4-ил)-1-изопропил-2-метил-1H-бензо[d]имидазол (528 мг, 1,28 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (61 мг, 0,13 ммоль), карбонат цезия (1,2 г, 3,7 ммоль) и трис(дибензилиденацетон)дипалладий (58 мг, 0,06 ммоль) добавляли к 1,4-диоксану (20 мл). В защитной атмосфере азота данное взаимодействие осуществляли при 120°C в течение 2 часов и фильтровали при отсасывании. Фильтрат концентрировали и подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=10:1), с получением указанного в заголовке соединения (40 мг, выход: 6%).
Молекулярная формула: C27H32F2N8. Молекулярная масса: 506,6. ЖХ-МС (m/z): 507,1(M+H+).
1H-ЯМР (400 МГц, MeOD) δ: 8,46 (д, J=6,0 Гц, 1H), 8,34 (с, 2H), 8,21 (д, J=2,4 Гц, 1H), 7,77 (с, 1H), 7,25 (д, J=11,6 Гц, 1H), 4,57 (с, 1H), 3,71-3,76 (м, 2H), 2,96-3,11 (м, 1H), 2,77-2,83 (м, 2H), 2,72 (с, 6H), 2,68 (с, 3H), 2,11-2,14 (м, 2H), 1,78-1,82 (м, 2H), 1,69 (д, J=6,8 Гц, 6H).
Пример 21: Получение 5-(4-(циклопропилметил)пиперазин-1-ил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина (соединение 22)
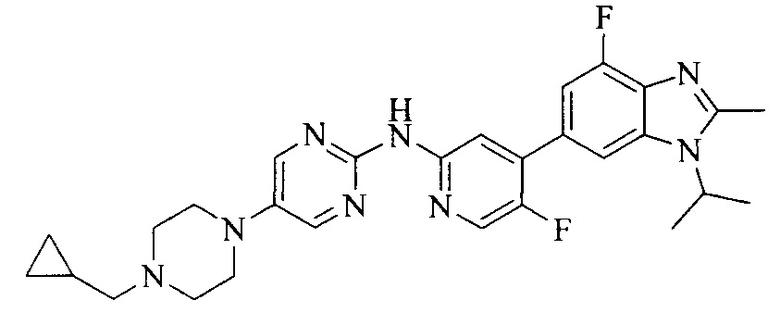
(1) Получение 5-(4-(циклопропилметил)пиперазин-1-ил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина
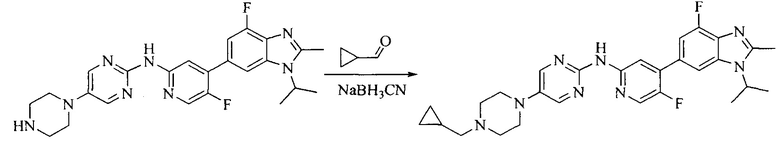
N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(пиперазин-1-ил)пиримидин-2-амин (90 мг, 0,194 ммоль) растворяли в метаноле (5 мл), и добавляли циклопропанкарбоксальдегид (68 мг, 0,97 ммоль). Полученную смесь перемешивали при 20°C в течение 30 мин. Добавляли цианоборогидрид натрия (122 мг, 1,94 ммоль). После добавления, полученную смесь перемешивали при 20°C в течение 16 часов. Воду (10 мл) и этилацетат (20 мл) добавляли для разделения фаз. Органическую фазу концентрировали и разделяли колоночной хроматографией на силикагеле (дихлорметан:метанол=10:1), с получением указанного в заголовке соединения (19 мг, выход: 18,9%).
Молекулярная формула: C28H32F2N8. Молекулярная масса: 518,6. ЖХ-МС (m/z): 519,2(M+H+).
1H-ЯМР (400 МГц, MeOD) δ: 8,41 (с, 1H), 8,09-8,20 (м, 3H), 7,74 (с, 1H), 7,17-7,19 (м, 1H), 4,89-4,91 (м, 1H), 2,95-3,15 (м, 4H), 2,68 (с, 3H), 2,52-2,68 (м, 4H), 2,26-2,29 (м, 2H), 1,67 (д, J=6,8 Гц, 6H), 1,29-1,31 (м, 1H), 0,55-0,57 (м, 2H), 0,21-0,29 (м, 2H).
Пример 22: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((цис)-гексагидропирроло[3,4-b]пиррол-5(1H)-ил)метил)пиримидин-2-амина (соединение 23)
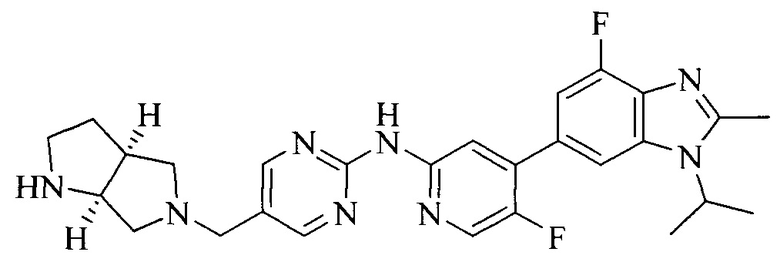
(1) Получение трет-бутил (цис)-5-((2-хлорпиримидин-5-ил)метил)гексагидропирроло[3,4-b]пиррол-1(2H)-карбоксилата
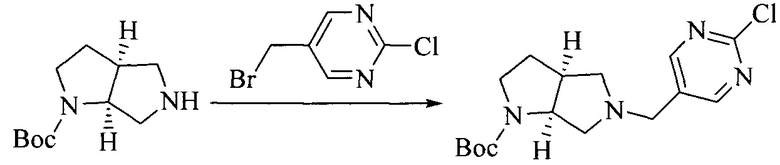
Трет-бутил (цис)-гексагидропирроло[3,4-b]пиррол-1(2H)-карбоксилат (250 мг, 1,18 ммоль) и 5-(бромметил)-2-хлорпиримидина (490 мг, 2,37 ммоль) взвешивали и добавляли к тетрагидрофурану (10 мл), и добавляли триэтиламин (238 мг, 2,36 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 16 часов. Полученную в результате смесь сразу концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=3:1), с получением указанного в заголовке соединения (300 мг, выход: 75,2%).
(2) Получение трет-бутил (цис)-5-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)гексагидропирроло[3,4-b]пиррол-1(2H)-карбоксилата
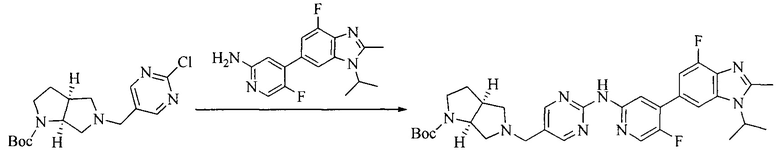
Трет-бутил (цис)-5-((2-хлорпиримидин-5-ил)метил)гексагидропирроло[3,4-b]пиррол-1(2H)-карбоксилат (300 мг, 0,89 ммоль), 5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (269 мг, 0,89 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (87 мг, 0,18 ммоль), трис(дибензилиденацетон)дипалладий (82 мг, 0,09 ммоль) и карбонат цезия (587 мг, 1,8 ммоль) взвешивали и добавляли к 1,4-диоксану (5 мл). В защитной атмосфере азота полученную смесь нагревали до 110°C и подвергали взаимодействию в течение 16 часов. Полученную смесь фильтровали при отсасывании, фильтрат концентрировали, и неочищенный продукт очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=30:1), с получением указанного в заголовке соединения (370 мг, выход: 68,7%).
(3) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((цис)-гексагидропирроло[3,4-b]пиррол-5(1H)-ил)метил)пиримидин-2-амина
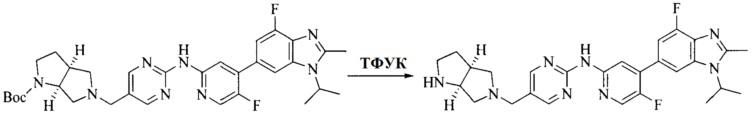
Трет-бутил (цис)-5-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)гексагидропирроло[3,4-b]пиррол-1(2H)-карбоксилат (370 мг, 0,61 ммоль) взвешивали и растворяли в дихлорметане (8 мл). Трифторуксусную кислоту (1 мл) добавляли в условиях охлаждения на ледяной бане. Полученную смесь затем нагревали до комнатной температуры и перемешивали в течение 2 часов. Полученный реакционный раствор отгоняли на роторном испарителе, растворяли в дихлорметане (10 мл), нейтрализовали гидрокарбонатом натрия и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=15:1), с получением указанного в заголовке соединения (250 мг, выход: 81,2%).
Молекулярная формула: C27H30F2N8. Молекулярная масса: 504,6. ЖХ-МС (m/z): 505,3(M+H+).
1H-ЯМР (400 МГц, MeOD-d4) δ: 8,51-8,54 (м, 3H), 8,28 (д, J=2,4 Гц, 1H), 7,80 (с, 1H), 7,27 (д, J=11,2 Гц, 1H), 4,85-4,95 (м, 1H), 4,12-4,18 (м, 1H), 3,58 (с, 2H), 3,35-3,42 (м, 1H), 3,16-3,27 (м, 1H), 3,08-3,15 (м, 1H), 2,96-3,05 (м, 1H), 2,77-2,83 (м, 1H), 2,69 (с, 3H), 2,40-2,55 (м, 2H), 2,15-2,28 (м, 1H), 1,85-1,95 (м, 1H), 1,69 (д, J=7,2 Гц, 6H).
Пример 23: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((цис)-октагидро-6H-пирроло[3,4-b]пиридин-6-ил)метил)пиримидин-2-амина (соединение 24)
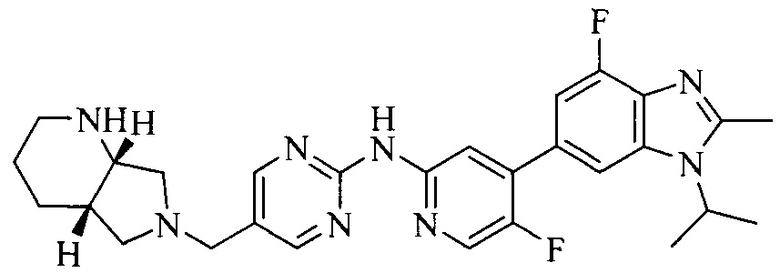
(1) Получение трет-бутил (цис)-6-((2-хлорпиримидин-5-ил)метил)октагидро-1H-пирроло[3,4-b]пиридин-1-карбоксилата
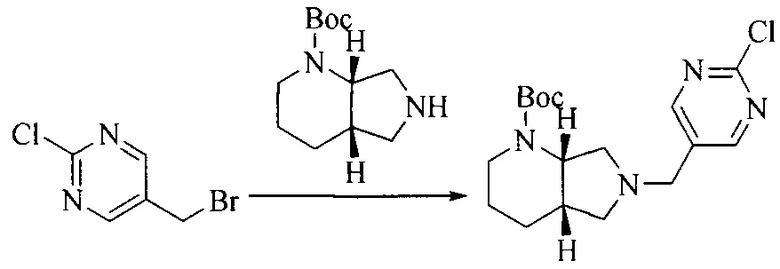
5-(Бромметил)-2-хлорпиримидин (183,4 мг, 0,884 ммоль) растворяли в тетрагидрофуране (10 мл), и добавляли триэтиламин (134,3 мг, 1,33 ммоль) и трет-бутил (цис)-октагидро-1H-пирроло[3,4-b]пиридин-1-карбоксилат (100 мг, 0,442 ммоль) при перемешивании. Полученную смесь перемешивали при комнатной температуре в течение 4 часов и фильтровали при отсасывании. Фильтрат концентрировали, и неочищенный продукт очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=100:1), с получением указанного в заголовке соединения (102 мг, выход: 65,4%).
(2) Получение трет-бутил (цис)-6-((2-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)октагидро-1H-пирроло[3,4-b]пиридин-1-карбоксилата
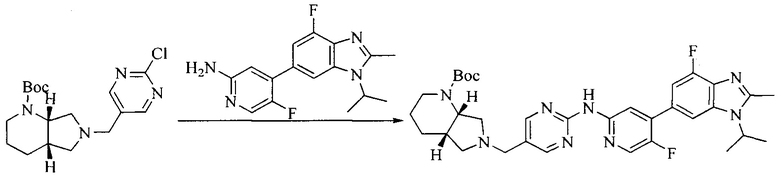
5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (85,6 мг, 0,283 ммоль), трет-бутил (цис)-6-((2-хлорпиримидин-5-ил)метил)октагидро-1H-пирроло[3,4-b]пиридин-1-карбоксилат (100 мг, 0,283 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (27 мг, 0,0566 ммоль), карбонат цезия (276,6 мг, 0,849 ммоль) и трис(дибензилиденацетон)дипалладий (25,9 мг, 0,0283 ммоль) добавляли к 1,4-диоксану (10 мл). В защитной атмосфере азота данное взаимодействие осуществляли при 110°C в течение 6 часов, и полученную смесь фильтровали при отсасывании. Фильтрат концентрировали, и неочищенный продукт очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=50:1), с получением указанного в заголовке соединения (80 мг, выход: 45,6%).
(3) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((цис)-октагидро-6H-пирроло[3,4-b]пиридин-6-ил)метил)пиримидин-2-амина
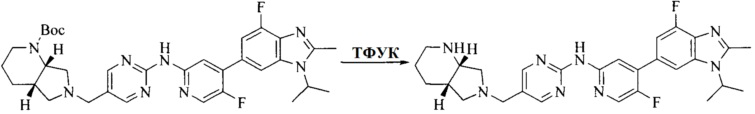
Трет-бутил (цис)-6-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)октагидро-1H-пирроло[3,4-b]пиридин-1-карбоксилат (80 мг, 0,129 ммоль) растворяли в дихлорметане (5 мл). Добавляли трифторуксусную кислоту (1 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. После взаимодействия, полученную смесь промывали насыщенным раствором гидрокарбоната натрия и разделяли на органическую фазу и водную фазу. Органическую фазу сушили над безводным сульфатом натрия и концентрировали. Неочищенный продукт разделяли колоночной хроматографией на силикагеле (дихлорметан:метанол=20:1), с получением указанного в заголовке соединения в виде белого твердого вещества (30 мг, выход: 44,8%).
Молекулярная формула: C28H32F2N8. Молекулярная масса: 518,6. ЖХ-МС (m/z): 519,3(M+H+).
1H-ЯМР (400 МГц, CDCl3) δ: 9,31 (с, 1H), 8,66 (д, J=6,0 Гц, 1H), 8,55 (с, 2H), 8,37 (д, J=2,0 Гц, 1H), 7,63 (с, 1H), 7,24-7,26 (м, 1H), 4,65-4,75 (м, 1H), 3,71 (с, 2H), 3,22-3,25 (м, 1H), 2,84-2,94 (м, 3H), 2,68-2,72 (м, 6H), 2,39-2,48 (м, 1H), 1,58-1,73 (м, 11H).
Пример 24: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((цис)-октагидро-1H-пирроло[3,4-b]пиридин-1-ил)метил)пиримидин-2-амина (соединение 25)
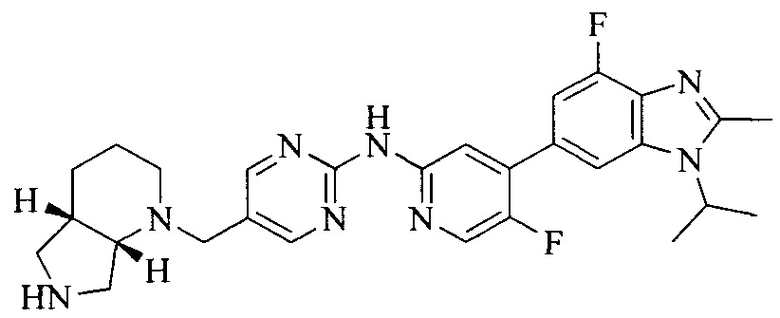
(1) Получение трет-бутил (цис)-1-((2-хлорпиримидин-5-ил)метил)октагидро-6H-пирроло[3,4-b]пиридин-6-карбоксилата
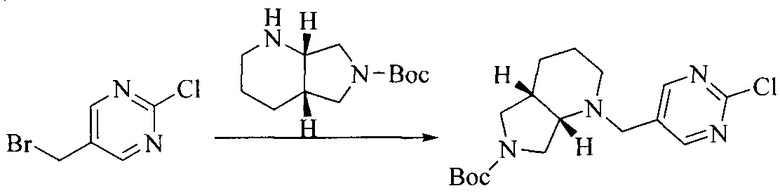
5-(Бромметил)-2-хлорпиримидин (183,4 мг, 0,884 ммоль) растворяли в тетрагидрофуране (10 мл), и добавляли триэтиламин (134,3 мг, 1,33 ммоль) и трет-бутил (цис)-октагидро-6H-пирроло[3,4-b]пиридин-6-карбоксилат (100 мг, 0,442 ммоль) при перемешивании. Полученную смесь перемешивали при комнатной температуре в течение 4 часов. Данную смесь фильтровали при отсасывании, фильтрат концентрировали, и неочищенный продукт очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=100:1), с получением указанного в заголовке соединения (125 мг, выход: 80,1%).
(2) Получение трет-бутил (цис)-1-((2-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)октагидро-6H-пирроло[3,4-b]пиридин-6-карбоксилата
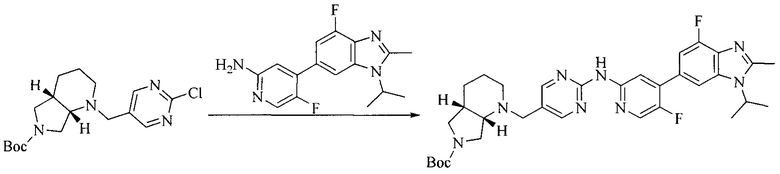
5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (85,6 мг, 0,283 ммоль), трет-бутил (цис)-1-((2-хлорпиримидин-5-ил)метил)октагидро-6H-пирроло[3,4-b]пиридин-6-карбоксилат (100 мг, 0,283 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (27 мг, 0,0566 ммоль), карбонат цезия (276,6 мг, 0,849 ммоль) и трис(дибензилиденацетон)дипалладий (25,9 мг, 0,0283 ммоль) добавляли к 1,4-диоксану (10 мл). В защитной атмосфере азота данное взаимодействие осуществляли при 110°C в течение 6 часов. Полученную смесь фильтровали при отсасывании, фильтрат концентрировали, и неочищенный продукт очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=50:1), с получением указанного в заголовке соединения (95 мг, выход: 54,2%).
(3) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((цис)-октагидро-1H-пирроло[3,4-b]пиридин-1-ил)метил)пиримидин-2-амина
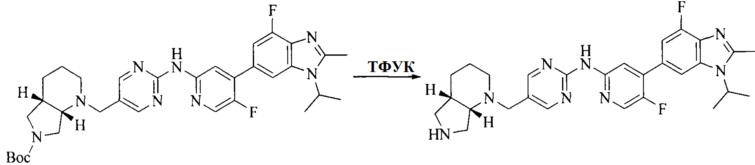
Трет-бутил (цис)-1-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)октагидро-6H-пирроло[3,4-b]пиридин-6-карбоксилат (95 мг, 0,154 ммоль) растворяли в дихлорметане (5 мл). Добавляли трифторуксусную кислоту (1 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. После взаимодействия, полученную смесь промывали насыщенным раствором гидрокарбоната натрия. Органическую фазу сушили над безводным сульфатом натрия и концентрировали. Неочищенный продукт разделяли колоночной хроматографией на силикагеле (дихлорметан:метанол=10:1), с получением указанного в заголовке соединения в виде белого твердого вещества (40 мг, выход: 50,3%).
Молекулярная формула: C28H32F2N8. Молекулярная масса: 518,6. ЖХ-МС (m/z): 519,3(M+H+).
1H-ЯМР (400 МГц, CDCl3) δ: 10,30 (с, 1H), 8,69 (д, J=6,0 Гц, 1H), 8,62 (с, 2H), 8,49 (д, J=2,4 Гц, 1H), 7,64 (с, 1H), 7,25 (д, J=10,8 Гц, 1H), 4,68-4,75 (м, 1H), 3,81-3,92 (м, 2H), 3,27-3,41 (м, 3H), 3,09 (д, J=13,6 Гц, 1H), 2,92 (с, 1H), 2,84 (д, J=10,8 Гц, 1H), 2,68 (с, 3H), 2,48-2,55 (м, 1H), 1,95-2,01 (м, 1H), 1,48-1,72 (м, 10H).
Пример 25: Получение 5-(((1S,6R)-3,8-диазабицикло[4.2.0]октан-8-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина (соединение 26)
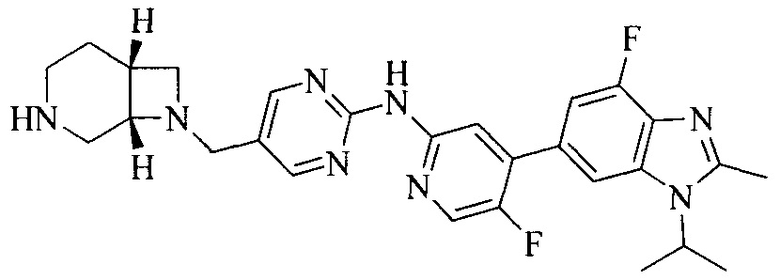
(1) Получение трет-бутил (1S,6R)-8-((2-хлорпиримидин-5-ил)метил)-3,8-диазабицикло[4.2.0]октан-3-карбоксилата
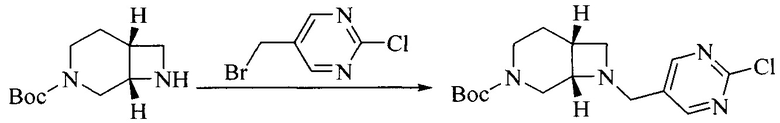
Трет-бутил (1S,6R)-3,8-диазабицикло[4.2.0]октан-3-карбоксилат (240 мг, 1,13 ммоль) и 5-(бромметил)-2-хлорпиримидин (352 мг, 1,70 ммоль) добавляли к тетрагидрофурану (20 мл), и добавляли триэтиламин (228 мг, 2,26 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 16 часов. Полученную смесь сразу концентрировали, и неочищенный продукт очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=50:1), с получением указанного в заголовке соединения (310 мг, выход: 81,0%).
(2) Получение трет-бутил (1S,6R)-8-((2-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-3,8-диазабицикло[4.2.0]октан-3-карбоксилата
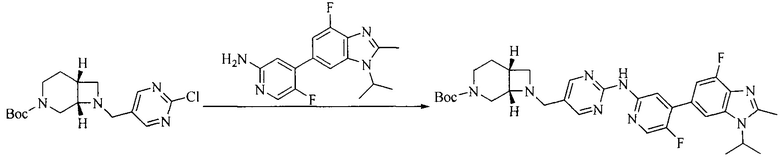
Трет-бутил (1S,6R)-8-((2-хлорпиримидин-5-ил)метил)-3,8-диазабицикло[4.2.0]октан-3-карбоксилат (310 мг, 0,91 ммоль), 5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (275 мг, 0,91 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (86 мг, 0,18 ммоль), трис(дибензилиденацетон)дипалладий (82 мг, 0,09 ммоль) и карбонат цезия (743 мг, 2,28 ммоль) взвешивали и добавляли к 1,4-диоксану (40 мл). В защитной атмосфере азота полученную смесь нагревали до 110°C и подвергали взаимодействию в течение 16 часов. Полученную смесь фильтровали при отсасывании, фильтрат концентрировали, и неочищенный продукт очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=30:1), с получением указанного в заголовке соединения (340 мг, выход: 61,8%).
(3) Получение 5-(((1S,6R)-3,8-диазабицикло[4.2.0]октан-8-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина
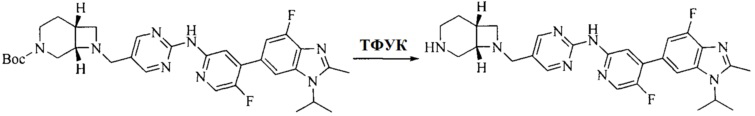
Трет-бутил (1S,6R)-8-((2-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-3,8-диазабицикло[4.2.0]октан-3-карбоксилат (340 мг, 0,56 ммоль) взвешивали и добавляли к дихлорметану (8 мл). Трифторуксусную кислоту (6 мл) добавляли в условиях охлаждения на ледяной бане. После добавления, полученную смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. Полученный реакционный раствор концентрировали, растворяли в дихлорметане (10 мл), нейтрализовали гидрокарбонатом натрия и концентрировали. Неочищенный продукт разделяли колоночной хроматографией на силикагеле (дихлорметан:метанол=10:1), с получением указанного в заголовке соединения (260 мг, выход: 92,1%).
Молекулярная формула: C27H30F2N8. Молекулярная масса: 504,6. ЖХ-МС (m/z): 505,3(M+H+).
1H-ЯМР (400 МГц, MeOD-d4) δ: 8,54 (д, J=6,4 Гц, 1H), 8,52 (с, 2H), 8,27 (д, J=2,4 Гц, 1H), 7,78 (с, 1H), 7,24 (д, J=11,2 Гц, 1H), 4,87-4,92 (м, 1H), 3,76 (д, J=13,2 Гц, 1H), 3,38-3,47 (м, 3H), 3,35 (с, 3H), 3,18 (д, J=14,0 Гц, 1H), 2,91-3,07 (м, 3H), 2,84-2,88 (м, 1H), 2,48-2,52 (м, 1H), 2,17-2,21 (м, 1H), 2,01-2,07 (м, 1H), 1,69 (д, J=6,8 Гц, 6H).
Пример 26: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((1S,6R)-3-метил-3,8-диазабицикло[4.2.0]октан-8-ил)метил)пиримидин-2-амина (соединение 27)
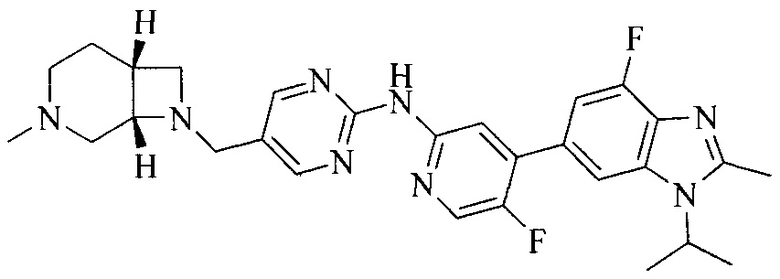
Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((1S,6R)-3-метил-3,8-диазабицикло[4.2.0]октан-8-ил)метил)пиримидин-2-амина
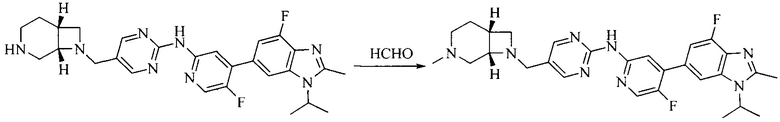
5-(((1S,6R)-3,8-диазабицикло[4.2.0]октан-8-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амин (100 мг, 0,20 ммоль) добавляли к метанолу (4 мл). В условиях охлаждения на ледяной бане добавляли водный раствор формальдегида (0,5 мл, 37%) и цианоборогидрид натрия (100 мг, 1,6 ммоль). После добавления, полученную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. Полученный реакционный раствор подвергали препаративной тонкослойной хроматографии (дихлорметан:метанол=10:1), с получением указанного в заголовке соединения (70 мг, выход: 68,1%).
Молекулярная формула: C28H32F2N8. Молекулярная масса: 518,6. ЖХ-МС (m/z): 519,3(M+H+).
1H-ЯМР (400 МГц, MeOD-d4) δ: 8,55 (д, J=6,4 Гц, 1H), 8,52 (с, 2H), 8,25 (д, J=2,4 Гц, 1H), 7,79 (с, 1H), 7,27 (д, J=11,6 Гц, 1H), 4,85-4,89 (м, 1H), 3,68 (д, J=12,8 Гц, 1H), 3,39 (д, J=13,2 Гц, 1H), 3,23-3,28 (м, 1H), 2,91-2,93 (м, 2H), 2,78 (д, J=12,4 Гц, 2H), 2,69 (с, 3H), 2,27 (с, 3H), 2,17-2,25 (м, 1H), 1,96-2,02 (м, 3H), 1,88-1,95 (м, 1H), 1,69 (д, J=7,2 Гц, 6H).
Пример 27: Получение 5-(((1R,6S)-3,8-диазабицикло[4.2.0]октан-8-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина (соединение 28)
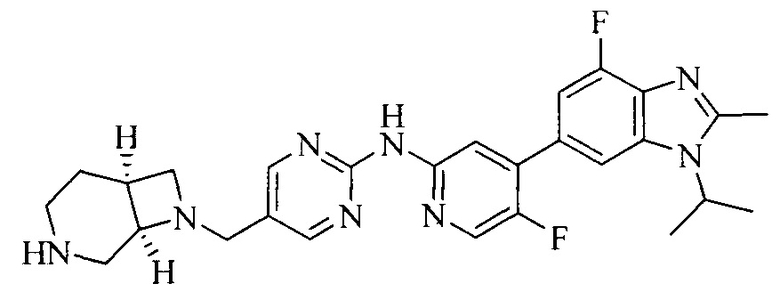
(1) Получение трет-бутил (1R,6S)-8-((2-хлорпиримидин-5-ил)метил)-3,8-диазабицикло[4.2.0]октан-3-карбоксилата
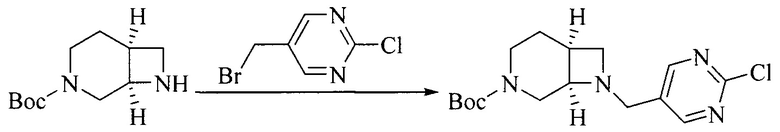
Трет-бутил (1R,6S)-3,8-диазабицикло[4.2.0]октан-3-карбоксилат (200 мг, 0,94 ммоль) и 5-(бромметил)-2-хлорпиримидин (390 мг, 1,88 ммоль) добавляли к тетрагидрофурану (10 мл), и добавляли триэтиламин (285 мг, 2,82 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 16 часов и сразу концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=3:1), с получением указанного в заголовке соединения (195 мг, выход: 61,7%).
(2) Получение трет-бутил (1R,6S)-8-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-3,8-диазабицикло[4.2.0]октан-3-карбоксилата
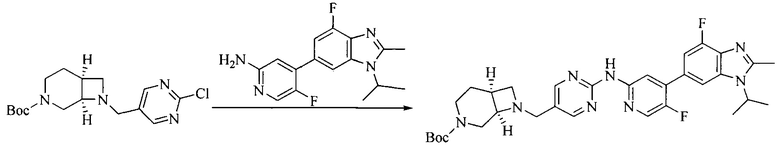
Трет-бутил (1R,6S)-8-((2-хлорпиримидин-5-ил)метил)-3,8-диазабицикло[4.2.0]октан-3-карбоксилат (100 мг, 0,30 ммоль), 5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (90 мг, 0,30 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (30 мг, 0,06 ммоль), трис(дибензилиденацетон)дипалладий (28 мг, 0,03 ммоль) и карбонат цезия (295 мг, 0,91 ммоль) взвешивали и добавляли к 1,4-диоксану (5 мл). В защитной атмосфере азота полученную смесь нагревали до 110°C и подвергали взаимодействию в течение 16 часов. Полученную смесь фильтровали при отсасывании, фильтрат концентрировали, и неочищенный продукт очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=30:1), с получением указанного в заголовке соединения (104 мг, выход: 56,7%).
(3) Получение 5-(((1R,6S)-3,8-диазабицикло[4.2.0]октан-8-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина
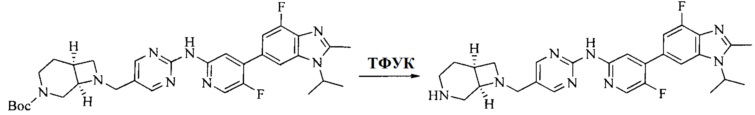
Трет-бутил (1R,6S)-8-((2-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-3,8-диазабицикло[4.2.0]октан-3-карбоксилат (104 мг, 0,17 ммоль) взвешивали и добавляли к дихлорметану (8 мл). Трифторуксусную кислоту (1 мл) добавляли в условиях охлаждения на ледяной бане. После добавления, полученную смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. Полученный реакционный раствор отгоняли на роторном испарителе, растворяли в дихлорметане (10 мл), нейтрализовали гидрокарбонатом натрия и концентрировали. Неочищенный продукт разделяли колоночной хроматографией на силикагеле (дихлорметан:метанол=15:1), с получением указанного в заголовке соединения (44 мг, выход: 52,9%).
Молекулярная формула: C27H30F2N8. Молекулярная масса: 504,6. ЖХ-МС (m/z): 505,3(M+H+).
1H-ЯМР (400 МГц, CDCl3) δ: 8,64 (д, J=6,0 Гц, 1H), 8,44 (с, 2H), 8,23 (д, J=2,4 Гц, 1H), 7,96 (с, 1H), 7,63 (с, 1H), 7,26-7,29 (м, 1H), 4,68-4,83 (м, 1H), 3,60-3,63 (м, 1H), 3,31-3,40 (м, 1H), 2,95-3,09 (м, 3H), 2,83-2,93 (м, 1H), 2,75-2,80 (м, 1H), 2,69 (с, 3H), 2,60-2,65 (м, 1H), 2,20-2,40 (м, 2H), 1,85-2,05 (м, 2H), 1,69 (д, J=6,8 Гц, 6H).
Пример 28: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((1R,6S)-3-метил-3,8-диазабицикло[4.2.0]октан-8-ил)метил)пиримидин-2-амина (соединение 29)
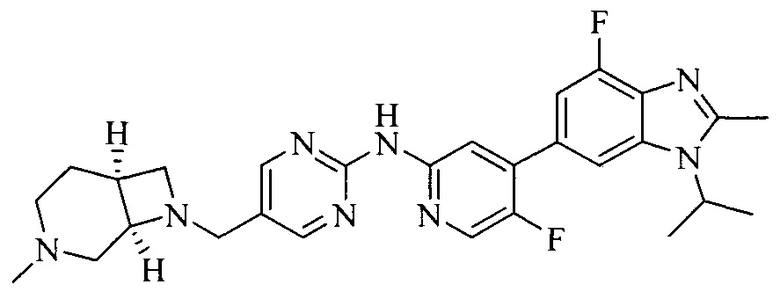
(1) Получение трет-бутил (1R,6S)-8-(2-хлорпиримидин-5-ил)метил)-3,8-диазабицикло[4.2.0]октан-3-карбоксилата
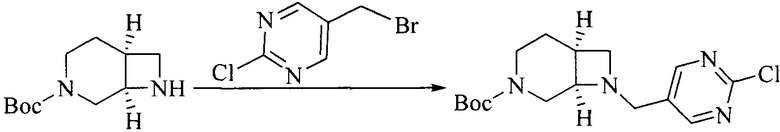
Трет-бутил (1R,6S)-3,8-диазабицикло[4.2.0]октан-3-карбоксилат (100 мг, 0,47 ммоль) и 5-(бромметил)-2-хлорпиримидин (97,5 мг, 0,47 ммоль) растворяли в безводном тетрагидрофуране (10 мл), и по каплям добавляли триэтиламин (71,2 мг, 0,71 ммоль). Данное взаимодействие осуществляли при комнатной температуре в течение 4 часов. Полученную реакционную смесь концентрировали и очищали колоночной хроматографией на силикагеле (элюент: смесь дихлорметан:метанол=30:1), с получением указанного в заголовке соединения (97,3 мг, выход: 61%).
(2) Получение трет-бутил (1R,6S)-8-((2-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-3,8-диазабицикло[4.2.0]октан-3-карбоксилата
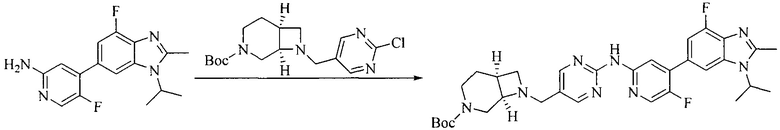
5-Фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (84,6 мг, 0,28 ммоль) и трет-бутил (1R,6S)-8-((2-хлорпиримидин-5-ил)метил)-3,8-диазабицикло[4.2.0]октан-3-карбоксилат (95 мг, 0,28 ммоль) растворяли в 1,4-диоксане (15 мл), и добавляли трис(дибензилиденацетон)дипалладий (25,6 мг, 0,028 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (26,7 мг, 0,056 ммоль) и карбонат цезия (273,7 мг, 0,84 ммоль). В защитной атмосфере азота полученную смесь нагревали до 110°C и подвергали взаимодействию в течение 8 часов. Полученную смесь охлаждали до комнатной температуры и концентрировали. Воду (30 мл) и этилацетат (50 мл) добавляли к отделенной от водной фазы органической фазе. Водную фазу экстрагировали этилацетатом (50 мл ×2), и органические фазы объединяли и промывали насыщенным раствором NaCl, сушили над безводным сульфатом натрия и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь дихлорметан:метанол=20:1), с получением указанного в заголовке соединения (88,2 мг, выход: 52%).
(3) Получение 5-(((1R,6S)-3,8-диазабицикло[4.2.0]октан-8-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина
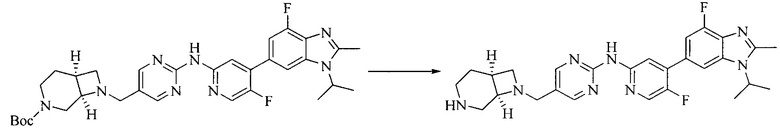
Трет-бутил (1R,6S)-8-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-3,8-диазабицикло[4.2.0]октан-3-карбоксилат (88 мг, 0,146 ммоль) добавляли к дихлорметану (5 мл). Добавляли трифторуксусную кислоту (1 мл). Полученную смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель удаляли перегонкой при пониженном давлении. Полученный остаток доводили насыщенным раствором гидрокарбоната натрия до pH=8, и экстрагировали смешанным раствором дихлорметан:метанол (10:1) (20 мл ×3). Органические фазы объединяли, сушили над безводным сульфатом натрия и концентрировали, с получением указанного в заголовке соединения (100 мг, неочищенный продукт), которое непосредственно использовали на следующей стадии без очистки.
(4) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((1R,6S)-3-метил-3,8-диазабицикло[4.2.0]октан-8-ил)метил)пиримидин-2-амина
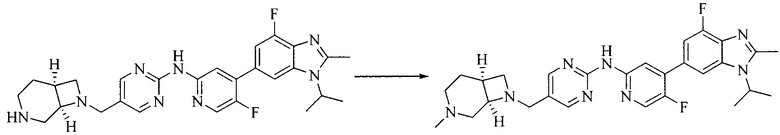
5-(((1R,6S)-3,8-диазабицикло[4.2.0]октан-8-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амин (100 мг, неочищенный продукт) растворяли в метаноле (5 мл), и добавляли водный раствор формальдегида (37%, 118 мг, 1,46 ммоль) при комнатной температуре. Данное взаимодействие осуществляли в течение 2 часов, и добавляли цианоборогидрид натрия (91,7 г, 1,46 ммоль) к данному раствору и дополнительно перемешивали при комнатной температуре в течение 1 часа. После взаимодействия, добавляли петролейный эфир (50 мл) к реакционному раствору, и полученный раствор очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=10:1), с получением указанного в заголовке соединения (20 мг, выход в две стадии: 26,4%).
Молекулярная формула: C28H32F2N8. Молекулярная масса: 518,6. ЖХ-МС (m/z): 519,3(M+H+).
1H-ЯМР (400 МГц, MeOD-d4) δ: 8,55 (д, J=6,0 Гц, 1H), 8,52 (с, 2H), 8,27 (д, J=2,4 Гц, 1H), 7,78 (с, 1H), 7,25 (д, J=11,2 Гц, 1H), 4,86-4,92 (м, 1H), 3,68 (д, J=13,2 Гц, 1H), 3,37 (д, J=13,2 Гц, 1H), 3,27-3,31 (м, 1H), 2,89-2,95 (м, 2H), 2,85 (д, J=11,6 Гц, 2H), 2,68 (с, 3H), 2,33 (с, 3H), 2,23-2,25 (м, 1H), 2,01-2,07 (м, 5H), 1,69 (д, J=6,8 Гц, 6H).
Пример 29: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((цис)-6-метил-3,8-диазабицикло[4.2.0]октан-8-ил)метил)пиримидин-2-амина (соединение 30)
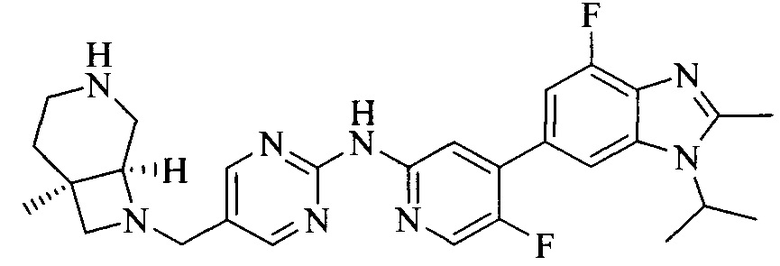
(1) Получение трет-бутил (цис)-8-((2-хлорпиримидин-5-ил)метил)-6-метил-3,8-диазабицикло[4.2.0]октан-3-карбоксилата
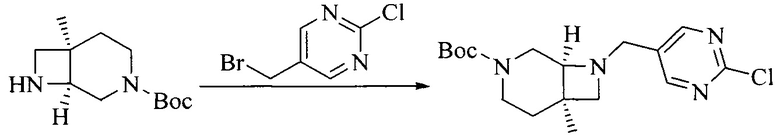
Триэтиламин (336 мг, 3,33 ммоль) добавляли к раствору трет-бутил (цис)-6-метил-3,8-диазабицикло[4.2.0]октан-3-карбоксилата (250 мг, 1,11 ммоль) и 5-(бромметил)-2-хлорпиримидина (660 мг, 3,19 ммоль) в тетрагидрофуране (20 мл), и полученную смесь перемешивали при 20°C в течение 5 часов. После взаимодействия, полученную смесь концентрировали при пониженном давлении. Полученный остаток разделяли колоночной хроматографией (петролейный эфир:этилацетат=2:1), с получением продукта (200 мг, выход: 51,2%).
(2) Получение трет-бутил (цис)-8-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-6-метил-3,8-диазабицикло[4.2.0]октан-3-карбоксилата
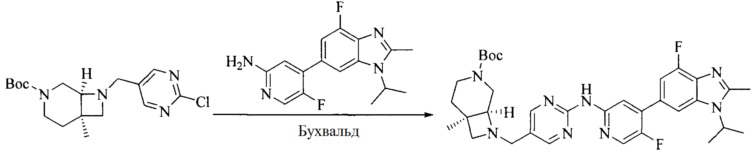
Трет-бутил (цис)-8-((2-хлорпиримидин-5-ил)метил)-6-метил-3,8-диазабицикло[4.2.0]октан-3-карбоксилат (200 мг, 0,57 ммоль), 5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (206 мг, 0,68 ммоль), карбонат цезия (370 мг, 1,14 ммоль), трис(дибензилиденацетон)дипалладий (26 мг, 0,029 ммоль) и 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (27 мг, 0,057 ммоль) добавляли к 1,4-диоксану (5 мл). В защитной атмосфере азота полученную смесь нагревали при 110°C на масляной бане в течение 8 часов. После взаимодействия, полученный реакционный раствор концентрировали и очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=10:1), с получением продукта (50 мг, выход: 14,2%).
(3) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((цис)-6-метил-3,8-диазабицикло[4.2.0]октан-8-ил)метил)пиримидин-2-амина
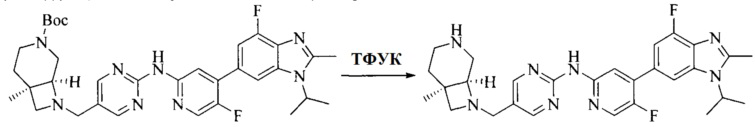
Трет-бутил (цис)-8-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-6-метил-3,8-диазабицикло[4.2.0]октан-3-карбоксилат (50 мг, 0,08 ммоль) добавляли к дихлорметану (5 мл). По каплям добавляли трифторуксусную кислоту (3 мл) к данной реакционной системе. Полученную смесь перемешивали при 20°C в течение 5 часов. После взаимодействия, полученный реакционный раствор концентрировали при пониженном давлении, полученный остаток растворяли в дихлорметане, и по каплям добавляли триэтиламин (2 мл). После дополнительного концентрирования перегонкой при пониженном давлении, полученный остаток разделяли колоночной хроматографией (дихлорметан:метанол=5:1), с получением продукта (12 мг, выход: 28,6%).
Молекулярная формула: C28H32F2N8. Молекулярная масса: 518,6. ЖХ-МС (m/z): 519,3(M+H+).
1H-ЯМР(400 МГц, MeOD) δ: 8,52 (шир.с, 3H), 8,27 (шир.с, 1H), 7,79 (шир.с, 1H), 7,27 (шир.с, 1H), 4,60-4,68 (м, 1H), 3,73-3,81 (м, 1H), 3,43-3,46 (м, 1H), 3,06-3,14 (м, 3H), 2,90-3,03 (м, 2H), 2,68-2,73 (м, 4H), 2,19 (м, 1H), 1,82-1,85 (м, 1H), 1,69 (с, 6H), 0,80-0,90 (м, 2H).
Пример 30: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((цис)-1-метил-3,7-диазабицикло[4.2.0]октан-7-ил)метил)пиримидин-2-амина (соединение 31)
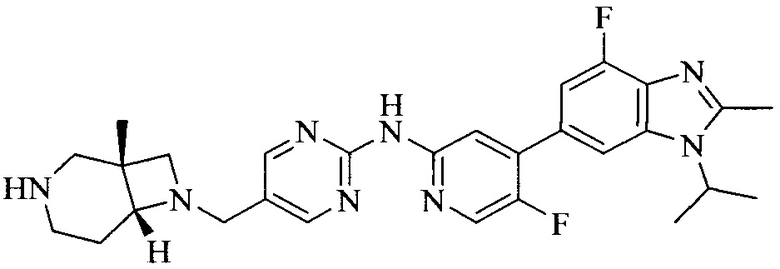
(1) Получение трет-бутил (цис)-7-((2-хлорпиримидин-5-ил)метил)-1-метил-3,7-диазабицикло[4.2.0]октан-3-карбоксилата
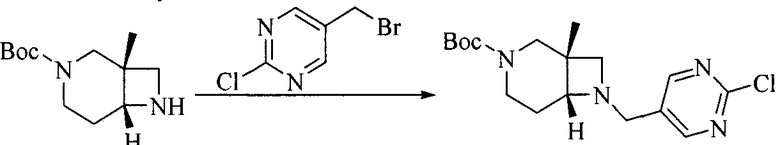
Трет-бутил (цис)-1-метил-3,7-диазабицикло[4.2.0]октан-3-карбоксилат (0,12 г, 0,53 ммоль) и триэтиламин (0,11 г, 1,09 ммоль) растворяли в тетрагидрофуране (5 мл), и добавляли 5-(бромметил)-2-хлорпиримидин (0,13 г, 0,63 ммоль). Данное взаимодействие осуществляли при перемешивании при 25°C в течение 18 часов. Полученный реакционный раствор концентрировали и очищали колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=5:1), с получением указанного в заголовке соединения в виде желтого твердого вещества (0,15 г, выход: 80,2%).
(2) Получение трет-бутил (цис)-7-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-1-метил-3,7-диазабицикло[4.2.0]октан-3-карбоксилата
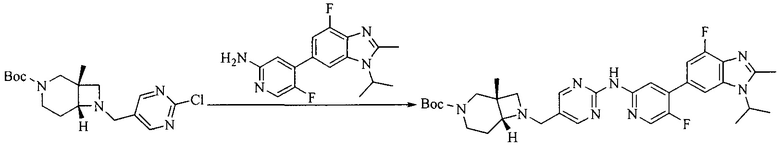
5-Фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (0,1 г, 0,33 ммоль) и трет-бутил (цис)-7-((2-хлорпиримидин-5-ил)метил)-1-метил-3,7-диазабицикло[4.2.0]октан-3-карбоксилат (0,11 г, 0,31 ммоль) растворяли в 1,4-диоксане (10 мл), и добавляли трис(дибензилиденацетон)дипалладий (0,03 г, 0,03 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (0,02 г, 0,04 ммоль) и карбонат цезия (0,2 г, 0,61 ммоль). В защитной атмосфере азота полученную смесь нагревали до 100°C и подвергали взаимодействию при перемешивании в течение 6 часов. Полученный реакционный раствор концентрировали и очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=50:1), с получением указанного в заголовке соединения в виде желтого твердого вещества (0,12 г, выход: 62,2%).
(3) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((цис)-1-метил-3,7-диазабицикло[4.2.0]октан-7-ил)метил)пиримидин-2-амина
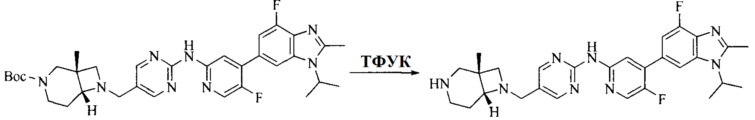
Трет-бутил (цис)-7-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-1-метил-3,7-диазабицикло[4.2.0]октан-3-карбоксилат (0,12 г, 0,19 ммоль) растворяли в дихлорметане (5 мл). Добавляли трифторуксусную кислоту (3 мл). Полученной смеси давали взаимодействовать при комнатной температуре при перемешивании в течение 3 часов. Полученный реакционный раствор концентрировали. Добавляли этилацетат (30 мл). Насыщенный водный раствор гидрокарбоната натрия (10 мл ×2) использовали для промывания. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали и затем подвергали колоночной хроматографии на силикагеле (дихлорметан:метанол=20:1), с получением указанного в заголовке соединения в виде желтого твердого вещества (58 мг, выход: 47,2%).
Молекулярная формула: C28H32F2N8. Молекулярная масса: 518,62. ЖХ-МС (m/z): 519,3(M+H+).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 10,07 (с, 1H), 9,08 (шир.с, 1H), 8,46 (с, 2H), 8,42 (д, J=6,0 Гц, 1H), 8,35 (д, J=1,6 Гц, 1H), 7,75 (с, 1H), 7,23 (д, J=11,6 Гц, 1H), 4,77-4,85 (м, 1H), 3,62 (д, J=12,8 Гц, 1H), 3,14 (д, J=5,2 Гц, 1H), 2,93-3,08 (м, 6H), 2,62 (с, 3H), 2,54 (д, J=6,4 Гц, 1H), 1,72-1,79 (м, 1H), 1,57 (д, J=7,2 Гц, 6H), 1,47-1,56 (м, 1H), 1,15-1,29 (м, 4H).
Пример 31: Получение (((1S,5S)-3,6-диазабицикло[3.2.0]гептан-3-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина (соединение 32)
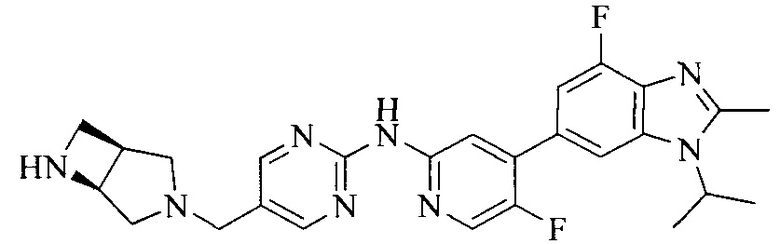
(1) Получение трет-бутил (1R,5S)-3-((2-хлорпиримидин-5-ил)метил)-3,6-диазабицикло[3.2.0]гептан-6-карбоксилата
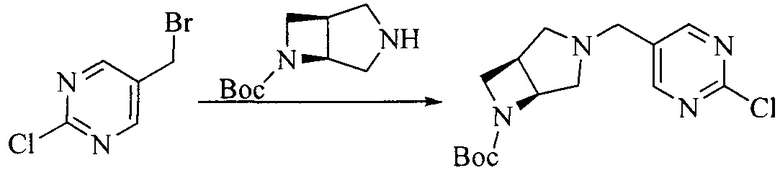
Трет-бутил (1R,5S)-3,6-диазабицикло[3.2.0]гептан-6-карбоксилат (350,0 мг, 1,77 ммоль) растворяли в тетрагидрофуране (15 мл), и добавляли триэтиламин (715,0 мг, 7,08 ммоль) и 5-(бромметил)-2-хлорпиримидин (734,0 мг, 3,54 ммоль). Полученную смесь дополнительно перемешивали в течение 2 часов. После завершения взаимодействия, полученный реакционный раствор концентрировали, и этилацетат (50 мл) и воду (30 мл) добавляли для разделения фаз. Водную фазу экстрагировали этилацетатом (30 мл), и органические фазы объединяли, концентрировали, и полученные остатки разделяли колоночной хроматографией на силикагеле (петролейный эфир:этилацетат=1:1), с получением указанного в заголовке соединения (500 мг, выход: 87,0%).
(2) Получение трет-бутил (1R,5S)-3-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-3,6-диазабицикло[3.2.0]октан-6-карбоксилата
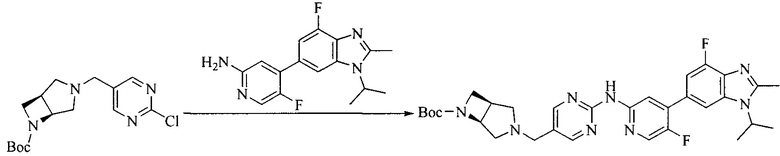
Трет-бутил (1R,5S)-3-((2-хлорпиримидин-5-ил)метил)-3,6-диазабицикло[3.2.0]гептан-6-карбоксилат (500,0 мг, 1,54 ммоль) и 5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (465,5 мг, 1,54 ммоль) растворяли в 1,4-диоксане (15 мл), и добавляли карбонат цезия (1,0 г, 3,08 ммоль), трис(дибензилиденацетон)дипалладий (141,0 мг, 0,154 ммоль) и 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (147,0 мг, 0,308 ммоль). В защитной атмосфере азота полученную смесь нагревали до 110°C и подвергали взаимодействию в течение 16 часов. Полученный реакционный раствор фильтровали. Фильтрат концентрировали, и полученный остаток разделяли колоночной хроматографией на силикагеле (дихлорметан:метанол=10:1), с получением указанного в заголовке соединения (400 мг, выход: 44,0%).
(3) Получение (((1S,5S)-3,6-диазабицикло[3.2.0]гептан-3-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина
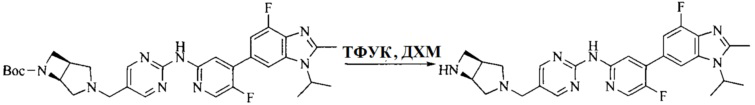
Трет-бутил (1R,5S)-3-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-3,6-диазабицикло[3.2.0]октан-6-карбоксилат (300 мг, 0,508 ммоль) растворяли в смешанном растворе дихлорметана (5 мл) и трифторуксусной кислоты (2 мл). Полученную смесь перемешивали при 20°C в течение 30 мин. Полученную смесь концентрировали, и добавляли дихлорметан (10 мл). После дополнительного концентрирования, полученный остаток разделяли колоночной хроматографией на силикагеле (дихлорметан:метанол=10:1), с получением указанного в заголовке соединения (160 мг, выход: 64,2%).
Молекулярная формула: C26H28F2N8. Молекулярная масса: 490,6. ЖХ-МС (m/z): 491,3(M+H+).
1H-ЯМР (400 МГц, ДМСО-d6) δ: 10,04 (с, 1H), 8,58 (с, 2H), 8,44 (д, J=6,0 Гц, 1H),8,36 (д, J=2,4 Гц, 1H),7,75(с, 1H), 7,22 (д, J=11,2 Гц, 1H),4,81-4,86 (м, 1H),4,62-4,67 (м, 1H), 3,87-3,92 (м, 1H), 3,74 (д, J=14,0 Гц, 1H), 3,63 (д, J=13,6 Гц, 1H),3,49-3,55 (м, 2H), 3,18-3,21 (м, 1H), 3,05-3,11 (м, 1H), 2,96 (д, J=8,0 Гц, 1H), 2,62 (с, 3H), 2,11-2,19 (м, 2H), 1,57 (д, J=7,2 Гц, 6H).
Пример 32: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((1R,5S)-6-метил-3,6-диазабицикло[3.2.0]гептан-3-ил)метил)пиримидин-2-амина (соединение 33)
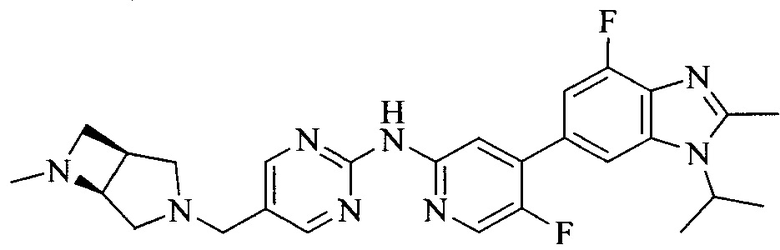
(1) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((1R,5S)-6-метил-3,6-диазабицикло[3.2.0]гептан-3-ил)метил)пиримидин-2-амина
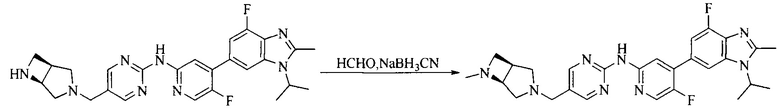
5-(((1S,5S)-3,6-диазабицикло[3.2.0]гептан-3-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амин (100 мг, 0,204 ммоль) растворяли в метаноле (5 мл), и добавляли водный раствор формальдегида (0,5 мл). Полученную смесь перемешивали в течение 0,5 часа, и цианоборогидрид натрия (64,0 мг, 1,01 ммоль) добавляли к данному раствору и дополнительно перемешивали в течение 0,5 часа. Данный образец непосредственно разделяли колоночной хроматографией на силикагеле (дихлорметан:метанол=5:1), с получением указанного в заголовке соединения (20 мг, выход: 19,4%).
Молекулярная формула: C27H30F2N8. Молекулярная масса: 504,6. ЖХ-МС (m/z): 505,2(M+H+).
1H-ЯМР (400 МГц, MeOD) δ: 8,60 (с, 2H), 8,55 (д, J=6,0 Гц, 1H), 8,26 (д, J=2,4 Гц, 1H), 7,79 (с, 1H), 7,26 (д, J=11,2 Гц, 1H), 4,90-4,93 (м, 1H), 4,62-4,71 (м, 1H), 3,91-3,98 (м, 1H), 3,82 (д, J=13,6 Гц, 1H), 3,63 (д, J=13,6 Гц, 1H), 3,23-3,25 (м, 1H), 3,04-3,06 (м, 1H), 2,88 (шир.с, 3H), 2,69 (с, 3H), 2,35-2,38 (м, 1H), 2,18-2,26 (м, 2H), 2,02-2,04 (м, 1H), 1,69 (д, J=7,2 Гц, 6H).
Пример 33: Получение 5-(((1R,5R)-3,6-диазабицикло[3.2.0]гептан-3-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина (соединение 34)
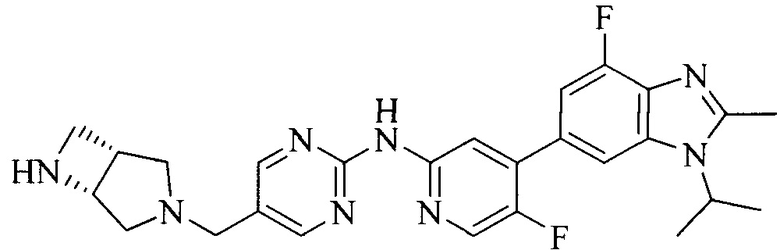
(1) Получение трет-бутил (1S,5R)-3-((2-хлорпиримидин-5-ил)метил)-3,6-диазабицикло[3.2.0]гептан-6-карбоксилата
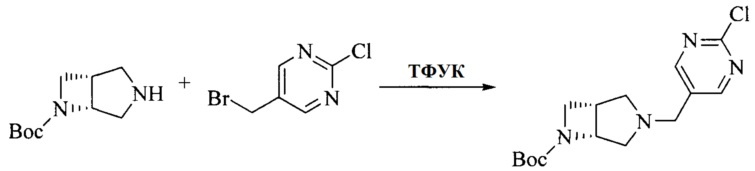
Триэтиламин (0,77 г, 7,57 ммоль) добавляли к раствору трет-бутил (1S,5R)-3,6-диазабицикло[3.2.0]гептан-6-карбоксилата (500 мг, 2,52 ммоль) и 5-бромметил-2-хлорпиримидина (784 мг, 3,78 ммоль) в тетрагидрофуране (20 мл). Полученную смесь перемешивали при комнатной температуре в течение 5 часов. После взаимодействия, полученный раствор концентрировали при пониженном давлении и разделяли колоночной хроматографией (петролейный эфир:этилацетат=2:1), с получением продукта (750 мг, выход: 91,6%).
(2) Получение трет-бутил (1S,5R)-3-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-3,6-диазабицикло[3.2.0]гептан-6-карбоксилата
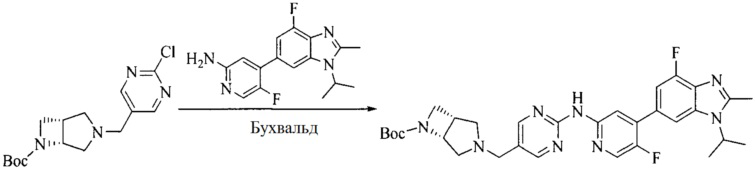
Трет-бутил (1S,5R)-3-((2-хлорпиримидинил-5-ил)метил)-3,6-диазабицикло[3.2.0]гептан-6-карбоксилат (200 мг, 0,62 ммоль), 5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (223 мг, 0,78 ммоль), карбонат цезия (401 мг, 1,23 ммоль), Pd2(dba)3 (11 мг, 0,012 ммоль) и X-Phos (11 мг, 0,024 ммоль) добавляли к 1,4-диоксану (5 мл). В защитной атмосфере азота полученной смеси давали взаимодействовать при 110°C в течение 8 часов. После взаимодействия, полученную смесь концентрировали при пониженном давлении и разделяли колоночной хроматографией (дихлорметан:метанол=10:1), с получением продукта (210 мг, выход: 57,7%).
(3) Получение 5-(((1R,5R)-3,6-диазабицикло[3.2.0]гептан-3-ил)метил)-N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)пиримидин-2-амина
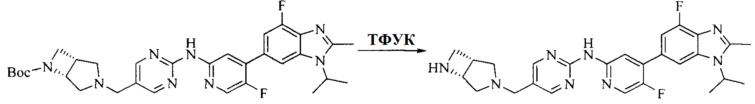
Трет-бутил (1S,5R)-3-((2-((5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)амино)пиримидин-5-ил)метил)-3,6-диазабицикло[3.2.0]гептан-6-карбоксилат (210 мг, 0,36 ммоль) добавляли к дихлорметану (10 мл). По каплям добавляли трифторуксусную кислоту (3 мл). Полученную смесь перемешивали при комнатной температуре в течение 5 часов. После взаимодействия, полученный реакционный раствор концентрировали при пониженном давлении. Полученный остаток растворяли в дихлорметане, и по каплям добавляли триэтиламин (2 мл). После дополнительного концентрирования при пониженном давлении, полученный остаток разделяли колоночной хроматографией (дихлорметан:метанол=5:1), с получением продукта (120 мг, выход: 68,8%).
Молекулярная формула: C26H28F2N8. Молекулярная масса: 490,6. ЖХ-МС (m/z): 491,3(M+H+).
1H-ЯМР(400 МГц, CDCl3) δ: 8,60 (с, 2H), 8,55 (д, J=6,0 Гц, 1H), 8,27 (д, J=2,0 Гц, 1H), 7,79 (с, 1H), 7,27 (д, J=11,2 Гц, 1H), 4,56-4,61 (м, 1H), 3,91 (т, J=9,6 Гц, 1H), 3,75 (кв, J=12,4 Гц, 2H), 3,52-3,62 (м, 1H), 3,12-3,26 (м, 2H), 3,01 (д, J=10,0 Гц, 1H), 2,68 (с, 3H), 2,22-2,35 (м, 1H), 2,15-2,22 (м, 1H), 1,89 (с, 1H), 1,69 (д, J=10,0 Гц, 6H).
Пример 34: Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((1S,5R)-6-метил-3,6-диазабицикло[3.2.0]гептан-3-ил)метил)пиримидин-2-амина (соединение 35)
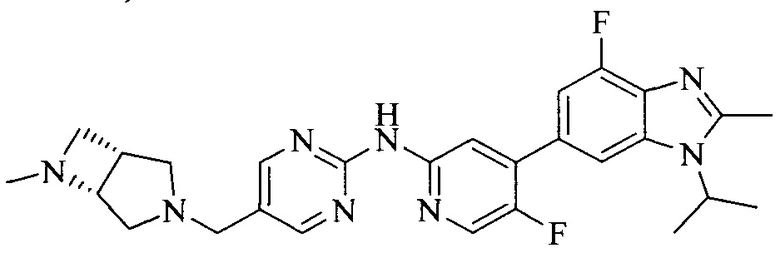
(1) Получение трет-бутил (1S,5R)-3-((2-хлорпиримидин-5-ил)метил)-3,6-диазабицикло[3.2.0]гептан-6-карбоксилата
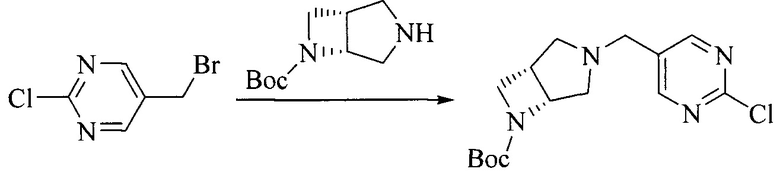
Трет-бутил (1S,5R)-3,6-диазабицикло[3.2.0]гептан-6-карбоксилат (256 мг, 1,29 ммоль), 5-(бромметил)-2-хлорпиримидин (267 мг, 1,29 ммоль) и карбонат калия (178 мг, 1,29 ммоль) добавляли к ацетонитрилу (15 мл), и полученную смесь нагревали при 60°C в течение 1 часа. Полученный реакционный раствор концентрировали и очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=10:1) с выделением данного продукта (300 мг, выход: 71,6%).
(2) Получение (1R,5R)-3-((2-хлорпиримидин-5-ил)метил)-3,6-диазабицикло[3.2.0]гептана
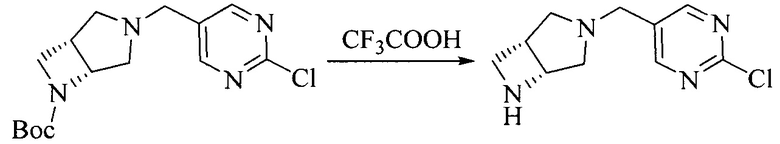
Трет-бутил (1S,5R)-3-((2-хлорпиримидин-5-ил)метил)-3,6-диазабицикло[3.2.0]гептан-6-карбоксилат (300 мг, 0,92 ммоль) растворяли в смешанном растворе дихлорметана (3 мл) и трифторуксусной кислоты (3 мл) и перемешивали при комнатной температуре в течение 30 мин. Полученный реакционный раствор концентрировали, и добавляли небольшое количество дихлорметана. Полученную смесь снова концентрировали, с получением неочищенного продукта (201 мг). Данный продукт использовали на следующей стадии без очистки.
(3) Получение (1S,5R)-3-((2-хлорпиримидин-5-ил)метил)-6-метил-3,6-диазабицикло[3.2.0]гептана
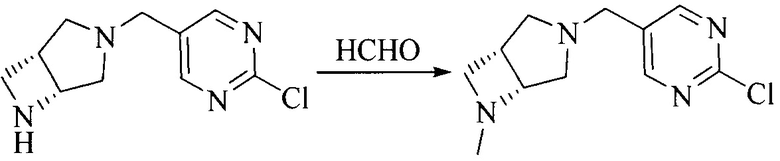
(1R,5R)-3-((2-хлорпиримидин-5-ил)метил)-3,6-диазабицикло[3.2.0]гептан (201 мг, 0,89 ммоль) растворяли в метаноле (10 мл), и по каплям добавляли водный раствор формальдегида (40%, 0,67 мл, 8,9 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Медленно добавляли цианоборогидрид натрия (561 мг, 8,9 ммоль). После добавления, полученную смесь дополнительно перемешивали при комнатной температуре в течение 30 мин. Полученный реакционный раствор концентрировали и очищали колоночной хроматографией на силикагеле (дихлорметан:метанол=10:1), с получением продукта (124 мг, выход в две стадии: 56,2%).
(4) Получение N-(5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-ил)-5-(((1S,5R)-6-метил-3,6-диазабицикло[3.2.0]гептан-3-ил)метил)пиримидин-2-амина
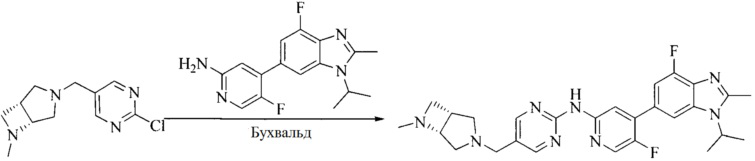
(1S,5R)-3-((2-хлорпиримидин-5-ил)метил)-6-метил-3,6-диазабицикло[3.2.0]гептан (124 мг, 0,52 ммоль), 5-фтор-4-(4-фтор-1-изопропил-2-метил-1H-бензо[d]имидазол-6-ил)пиридин-2-амин (157 мг, 0,52 ммоль), карбонат цезия (338 мг, 1,04 ммоль), трис(дибензилиденацетон)дипалладий (30 мг) и 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (60 мг) добавляли к 1,4-диоксану (10 мл). В защитной атмосфере азота полученную смесь нагревали при 110°C в течение 8 часов. Полученный реакционный раствор концентрировали и разделяли препаративной хроматографией с обращенной фазой (вода:метанол=10:1-1:1), с получением указанного в заголовке соединения (26 мг, выход: 10,0%).
Молекулярная формула: C27H30F2N8. Молекулярная масса: 504,6. ЖХ-МС (m/z): 505,3(M+H+).
1H-ЯМР(400 МГц, MeOD) δ: 8,59 (с, 2H), 8,56 (д, J=6,0 Гц, 2H), 8,28 (д, J=2,4 Гц, 1H), 7,77 (с, 1H), 7,23 (д, J=11,6 Гц, 1H), 4,25-4,27 (м, 1H), 3,71-3,74 (м, 3H), 3,64-3,69 (м, 1H), 3,23-3,30 (м, 2H), 3,06-3,15 (м, 1H), 2,93-2,98 (м, 1H), 2,68 (с, 3H), 2,56 (с, 3H), 2,13-2,19 (м, 2H), 1,68 (д, J=8,4 Гц, 6H).
| название | год | авторы | номер документа |
|---|---|---|---|
| Макроциклический ингибитор тирозинкиназы и его применение | 2019 |
|
RU2798231C2 |
| Регулятор на основе азотсодержащего гетероциклического производного, способ его получения и его применение | 2020 |
|
RU2829553C2 |
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2017 |
|
RU2829484C2 |
| Новое производное гидразона, в котором концевая аминогруппа замещена арильной группой или гетероарильной группой, и его применение | 2018 |
|
RU2802444C2 |
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2017 |
|
RU2799820C2 |
| ИНГИБИТОР НЕКОТОРЫХ ПРОТЕИНКИНАЗ | 2016 |
|
RU2732952C2 |
| СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИДИНОНА | 2018 |
|
RU2806751C2 |
| ИНГИБИТОРЫ МУТАЦИИ HER2 | 2022 |
|
RU2834124C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛОПИРИМИДИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2012 |
|
RU2618673C2 |
| ИНГИБИТОРЫ HPK1 И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2839132C2 |
Изобретение относится к области органической химии, а именно к производным 6-(пиримидиноаминопиридин)бензоимидазола формулы (I') или его стереоизомеру, где A1 и A2 независимо выбраны из азота; R1 выбран из C1-6алкила; R2 выбран из C1-6алкила; R3 и R5 независимо выбраны из галогена; R4 выбран из азотсодержащего 5-6-членного гетероциклила, необязательно замещенного Q2, где азотсодержащий 5-6-членный гетероциклил представляет собой азотсодержащий 5-6-членный гетероциклил, содержащий 1-2 атома азота; азотсодержащего 7-9-членного мостикового гетероциклила, необязательно замещенного Q2, где азотсодержащий 7-9-членный мостиковый гетероциклил представляет собой азотсодержащий 7-9-членный мостиковый гетероциклил, содержащий 1-2 атома азота; 6-10-членного конденсированного гетероциклила, который содержит 1, 2 или 3 одинаковых или различных гетероатома и необязательно замещен Q2, где гетероатомы выбраны из атома азота и атома кислорода, и содержат по меньшей мере один атом азота; азотсодержащего 7-11-членного спирогетероциклила, необязательно замещенного Q2, где азотсодержащий 7-11-членный спирогетероциклил представляет собой азотсодержащий 7-11-членный спирогетероциклил, содержащий 1-2 атома азота; Q2 выбран из амино или ди-C1-6алкиламино; или из C1-6алкила, 3-8-членного циклоалкила, оксациклобутанила, азотсодержащего 5-6-членного гетероциклила или азотсодержащего 8-членного мостикового гетероциклила, каждый из которых необязательно замещен заместителем, где заместитель выбран из C1-6алкила и 3-8-членного циклоалкила; n выбран из 0, 1. Также изобретение относится к фармацевтической композиции, обладающей способностью ингибировать активность CDK4 и/или CDK6 киназ, на основе соединения формулы (I'). Технический результат - новые соединения, полезные для снижения или ингибирования активности CDK4/6 киназы в клетке, и/или при лечении и/или предотвращении связанных с раком заболеваний, опосредованных CDK4/6 киназой. 3 н. и 20 з.п. ф-лы, 12 табл., 36 пр.
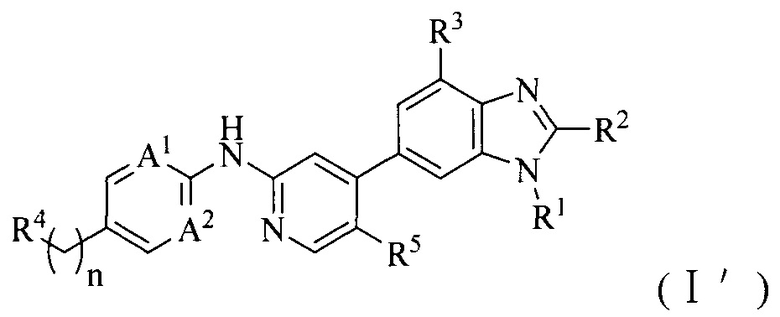
1. Соединение формулы (I') или его стереоизомер:
где:
A1 и A2 независимо выбраны из азота;
R1 выбран из C1-6алкила;
R2 выбран из C1-6алкила;
R3 и R5 независимо выбраны из галогена;
R4 выбран из азотсодержащего 5-6-членного гетероциклила, необязательно замещенного Q2, где азотсодержащий 5-6-членный гетероциклил представляет собой азотсодержащий 5-6-членный гетероциклил, содержащий 1-2 атома азота;
азотсодержащего 7-9-членного мостикового гетероциклила, необязательно замещенного Q2, где азотсодержащий 7-9-членный мостиковый гетероциклил представляет собой азотсодержащий 7-9-членный мостиковый гетероциклил, содержащий 1-2 атома азота;
6-10-членного конденсированного гетероциклила, который содержит 1, 2 или 3 одинаковых или различных гетероатома и необязательно замещен Q2, где гетероатомы выбраны из атома азота и атома кислорода и содержат по меньшей мере один атом азота;
азотсодержащего 7-11-членного спирогетероциклила, необязательно замещенного Q2, где азотсодержащий 7-11-членный спирогетероциклил представляет собой азотсодержащий 7-11-членный спирогетероциклил, содержащий 1-2 атома азота;
Q2 выбран из амино или ди-C1-6алкиламино; или из C1-6алкила, 3-8-членного циклоалкила, оксациклобутанила, азотсодержащего 5-6-членного гетероциклила или азотсодержащего 8-членного мостикового гетероциклила, каждый из которых необязательно замещен заместителем, где заместитель выбран из C1-6алкила и 3-8-членного циклоалкила;
n выбран из 0, 1.
2. Соединение или его стереоизомер по п.1, где
A1 и A2 независимо выбраны из азота;
R1 выбран из C1-4алкила;
R2 выбран из C1-4алкила;
R3 и R5 независимо выбраны из галогена;
R4 выбран из азотсодержащего 5-6-членного гетероциклила, необязательно замещенного Q2, где азотсодержащий 5-6-членный гетероциклил представляет собой азотсодержащий 5-6-членный гетероциклил, содержащий 1-2 атома азота, где азотсодержащий 5-6-членный гетероциклил предпочтительно представляет собой азотсодержащий 6-членный гетероциклил;
Q2 выбран из амино или ди-C1-4алкиламино; или C1-4алкила, 3-6-членного циклоалкила, оксациклобутанила или азотсодержащего 5-6-членного гетероциклила, каждый из которых необязательно замещен заместителем, где заместитель выбран из C1-4алкила или 3-6-членного циклоалкила;
n выбран из 0.
3. Соединение или его стереоизомер по п.2, где соединение выбрано из:

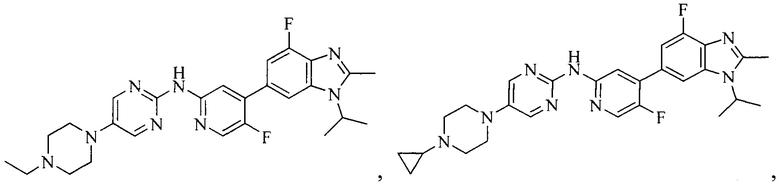
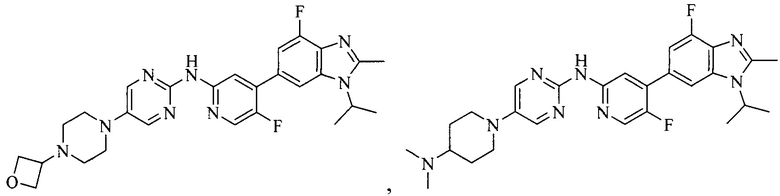 или
или 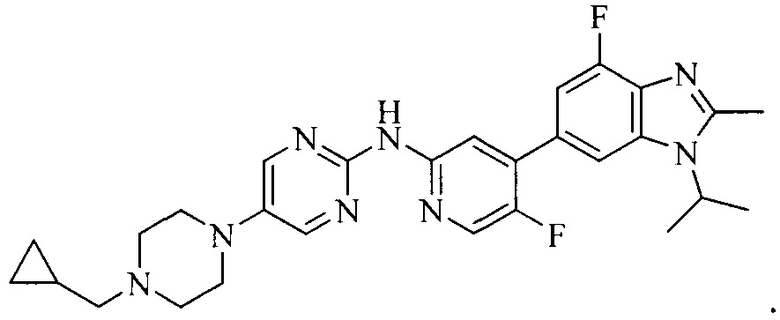
4. Соединение или его стереоизомер по п.1, где соединение имеет структуру формулы (I):
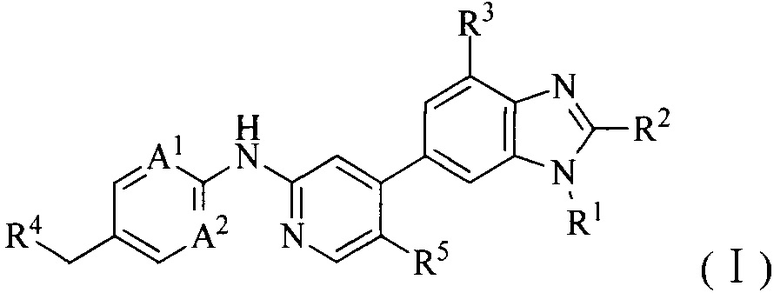
где:
A1 и A2 независимо выбраны из азота;
R1 выбран из C1-6алкила;
R2 выбран из C1-6алкила;
R3 и R5 независимо выбраны из галогена;
R4 выбран из азотсодержащего 5-6-членного гетероциклила, необязательно замещенного Q2, где азотсодержащий 5-6-членный гетероциклил представляет собой азотсодержащий 5-6-членный гетероциклил, содержащий 1-2 атома азота;
азотсодержащего 7-9-членного мостикового гетероциклила, необязательно замещенного Q2, где азотсодержащий 7-9-членный мостиковый гетероциклил представляет собой азотсодержащий 7-9-членный мостиковый гетероциклил, содержащий 1-2 атома азота;
6-10-членного конденсированного гетероциклила, который содержит 1, 2 или 3 одинаковых или различных гетероатома и необязательно замещен Q2, где гетероатомы выбраны из атома азота и атома кислорода и содержат по меньшей мере один атом азота;
азотсодержащего 7-11-членного спирогетероциклила, необязательно замещенного Q2, где азотсодержащий 7-11-членный спирогетероциклил представляет собой азотсодержащий 7-11-членный спирогетероциклил, содержащий 1-2 атома азота, где Q2 выбран из амино, C1-6алкила, оксациклобутанила, азотсодержащего 5-6-членного гетероциклила или азотсодержащего 8-членного мостикового гетероциклила.
5. Соединение или его стереоизомер по п.4, где:
A1 и A2 независимо выбраны из азота;
R1 выбран из C1-4алкила;
R2 выбран из C1-4алкила;
R3 и R5 независимо выбраны из галогена;
R4 выбран из азотсодержащего 5-6-членного гетероциклила, необязательно замещенного Q2, где азотсодержащий 5-6-членный гетероциклил представляет собой азотсодержащий 5-6-членный гетероциклил, содержащий 1-2 атома азота;
азотсодержащего 7-9-членного мостикового гетероциклила, необязательно замещенного Q2, где азотсодержащий 7-9-членный мостиковый гетероциклил представляет собой азотсодержащий 7-9-членный мостиковый гетероциклил, содержащий 1-2 атома азота;
6-10-членного конденсированного гетероциклила, который содержит 1, 2 или 3 одинаковых или различных гетероатома и необязательно замещен Q2, где гетероатомы выбраны из атома азота и атома кислорода и содержат по меньшей мере один атом азота;
азотсодержащего 7-11-членного спирогетероциклила, необязательно замещенного Q2, где азотсодержащий 7-11-членный спирогетероциклил представляет собой азотсодержащий 7-11-членный спирогетероциклил, содержащий 1-2 атома азота, где Q2 выбран из амино, C1-4алкила, оксациклобутанила, азотсодержащего 5-6-членного гетероциклила или азотсодержащего 8-членного мостикового гетероциклила.
6. Соединение или его стереоизомер по п.5, где:
A1 и A2 независимо выбраны из азота;
R1 выбран из изопропила;
R2 выбран из метила;
каждый R3 и R5 представляет собой F;
R4 выбран из азотсодержащего 5-6-членного гетероциклила, необязательно замещенного Q2, где азотсодержащий 5-6-членный гетероциклил представляет собой азотсодержащий 5-6-членный гетероциклил, содержащий 1-2 атома азота, где Q2 выбран из амино, C1-4алкила, азотсодержащего 6-членного гетероциклила или азотсодержащего 8-членного мостикового гетероциклила.
7. Соединение или его стереоизомер по п.6, где:
R2 выбран из метила;
R4 выбран из азотсодержащего 5-6-членного гетероциклила, необязательно замещенного Q2, где азотсодержащий 5-6-членный гетероциклил связан с метиленом в формуле (I) через атом азота, где Q2 выбран из амино, C1-4алкила или азотсодержащего 8-членного мостикового гетероциклила.
8. Соединение или его стереоизомер по п.7, где:
R4 выбран из 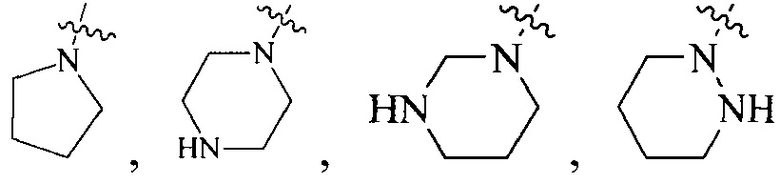 или
или 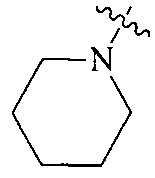 , каждый из которых необязательно замещен Q2, где Q2 выбран из C1-4алкила или азотсодержащего 8-членного мостикового гетероциклила.
, каждый из которых необязательно замещен Q2, где Q2 выбран из C1-4алкила или азотсодержащего 8-членного мостикового гетероциклила.
9. Соединение или его стереоизомер по п.5, где
A1 и A2 независимо выбраны из азота;
R1 выбран из изопропила;
R2 выбран из метила;
каждый R3 и R5 представляет собой F;
R4 выбран из азотсодержащего 7-9-членного мостикового гетероциклила, необязательно замещенного Q2, где азотсодержащий 7-9-членный мостиковый гетероциклил представляет собой азотсодержащий 7-9-членный мостиковый гетероциклил, содержащий 1-2 атома азота, где Q2 выбран из амино, C1-4алкила, азотсодержащего 6-членного гетероциклила или азотсодержащего 8-членного мостикового гетероциклила.
10. Соединение или его стереоизомер по п.9, где:
R2 выбран из метила;
R4 выбран из азотсодержащего 7-9-членного мостикового гетероциклила, необязательно замещенного Q2, где азотсодержащий 7-9-членный мостиковый гетероциклил связан с метиленом в формуле (I) через атом азота, где Q2 выбран из амино, C1-4алкила или азотсодержащего 6-членного гетероциклила.
11. Соединение или его стереоизомер по п.10, где:
R4 выбран из 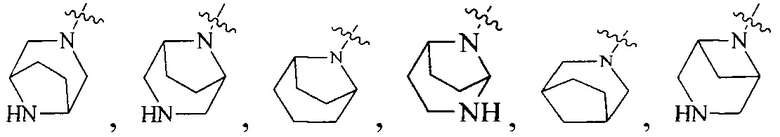 или
или 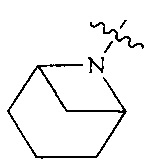 , каждый из которых необязательно замещен Q2, где Q2 выбран из C1-4алкила или азотсодержащего 6-членного гетероциклила.
, каждый из которых необязательно замещен Q2, где Q2 выбран из C1-4алкила или азотсодержащего 6-членного гетероциклила.
12. Соединение или его стереоизомер по п.5, где:
A1 и A2 независимо выбраны из азота;
R1 выбран из изопропила;
R2 выбран из метила;
каждый R3 и R5 представляет собой F;
R4 выбран из 6-10-членного конденсированного гетероциклила, который содержит 1, 2 или 3 одинаковых или различных гетероатома и необязательно замещен Q2, где гетероатомы выбраны из атома азота и атома кислорода и содержат по меньшей мере один атом азота, где Q2 выбран из амино, C1-4алкила, азотсодержащего 6-членного гетероциклила или азотсодержащего 8-членного мостикового гетероциклила.
13. Соединение или его стереоизомер по п.12, где:
R2 выбран из метила;
R4 выбран из 6-10-членного конденсированного гетероциклила, который содержит 1, 2 или 3 одинаковых или различных гетероатома и необязательно замещен Q2, где гетероатомы выбраны из атома азота и атома кислорода и содержат по меньшей мере один атом азота, и 6-10-членный конденсированный гетероциклил связан с метиленом в формуле (I) через атом азота, где Q2 выбран из амино или C1-4алкила.
14. Соединение или его стереоизомер по п.13, где
R4 выбран из 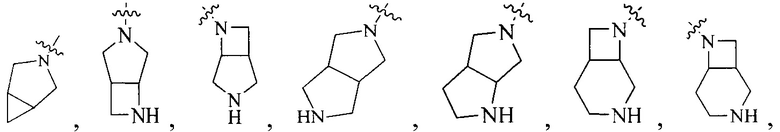
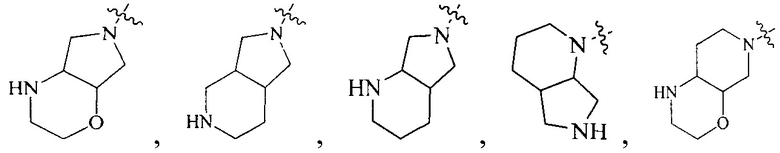 или
или 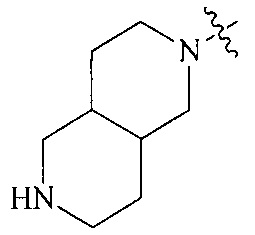 , каждый из которых необязательно замещен Q2, где Q2 выбран из амино или C1-4алкила.
, каждый из которых необязательно замещен Q2, где Q2 выбран из амино или C1-4алкила.
15. Соединение или его стереоизомер по п.5, где:
A1 и A2 независимо выбраны из азота;
R1 выбран из изопропила;
R2 выбран из метила;
каждый R3 и R5 представляет собой F;
R4 выбран из азотсодержащего 7-11-членного спирогетероциклила, необязательно замещенного Q2, где азотсодержащий 7-11-членный спирогетероциклил представляет собой азотсодержащий 7-11-членный спирогетероциклил, содержащий 1-2 атома азота, где Q2 выбран из амино, C1-4алкила, азотсодержащего 6-членного гетероциклила или азотсодержащего 8-членного мостикового гетероциклила.
16. Соединение или его стереоизомер по п.15, где:
R2 выбран из метила;
R4 выбран из азотсодержащего 7-11-членного спирогетероциклила, необязательно замещенного Q2, где азотсодержащий 7-11-членный спирогетероциклил связан с метиленом в формуле (I) через атом азота, где Q2 выбран из амино или C1-4алкила.
17. Соединение или его стереоизомер по п.16, где:
R4 выбран из 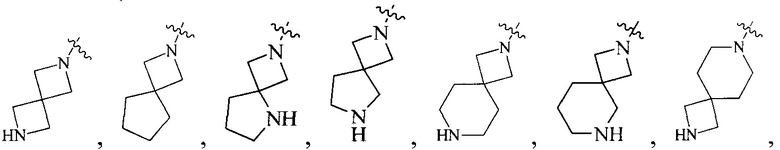
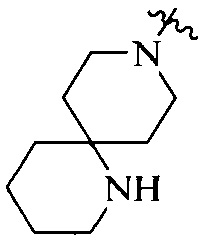 или
или 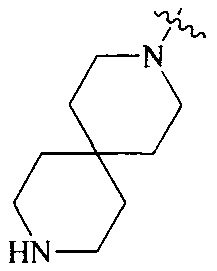 , каждый из которых необязательно замещен Q2, где Q2 выбран из C1-4алкила.
, каждый из которых необязательно замещен Q2, где Q2 выбран из C1-4алкила.
18. Соединение или его стереоизомер по п.1, где соединение выбрано из:
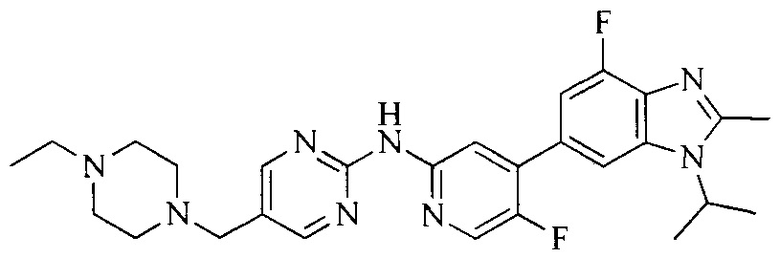
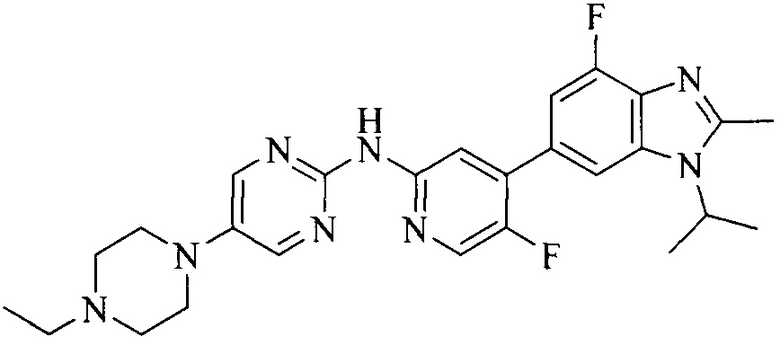
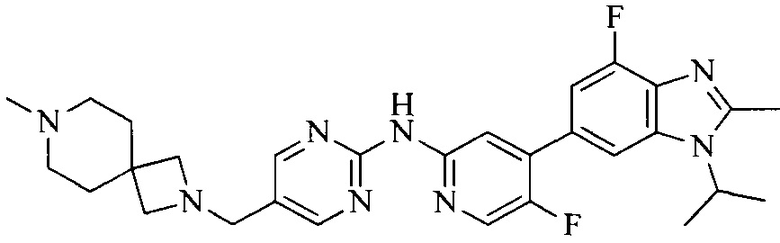
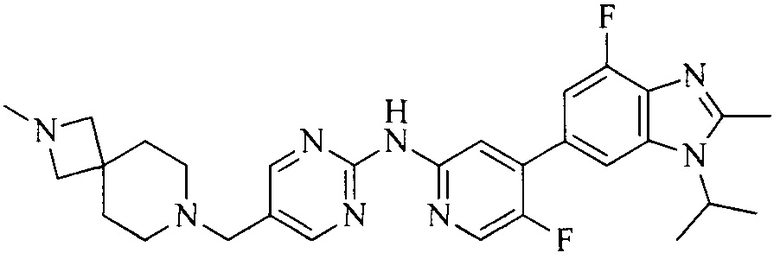
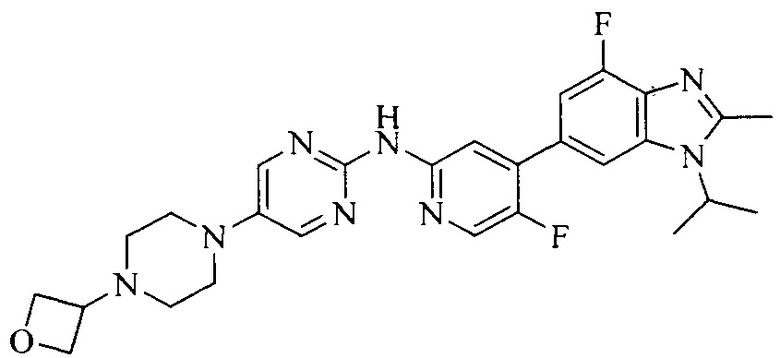
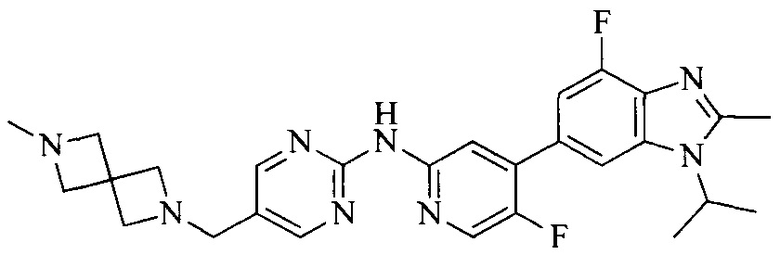
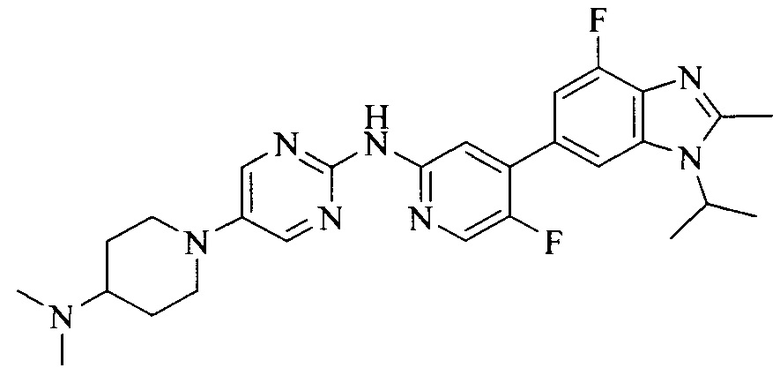
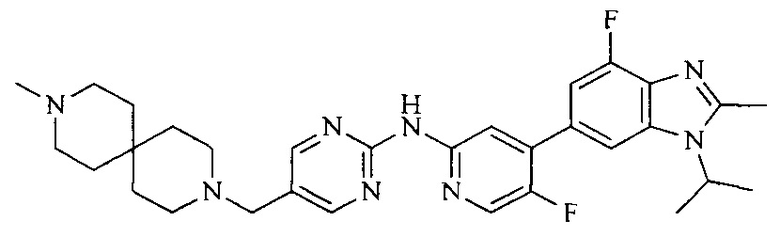
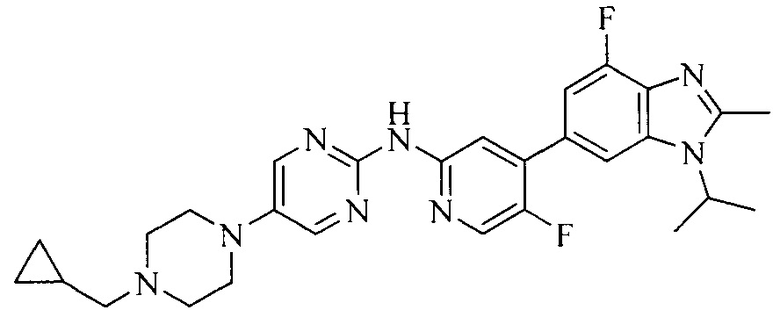
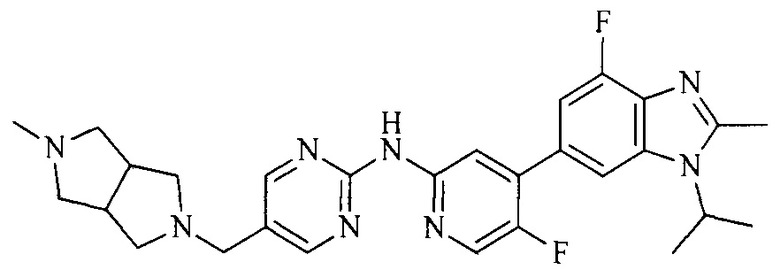
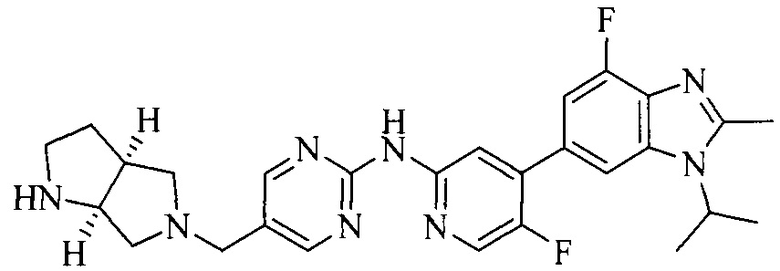
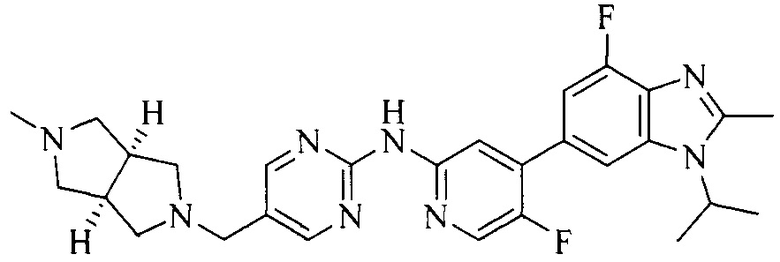
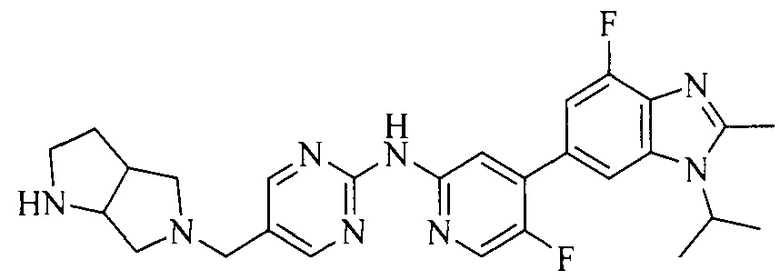
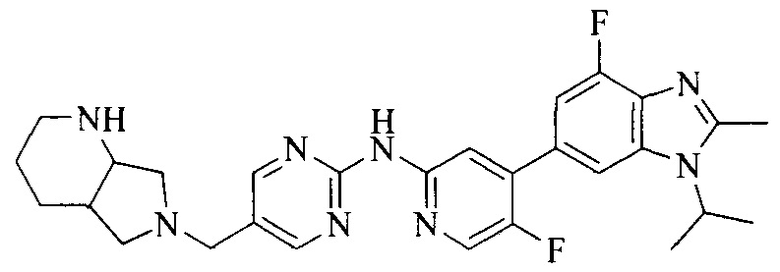
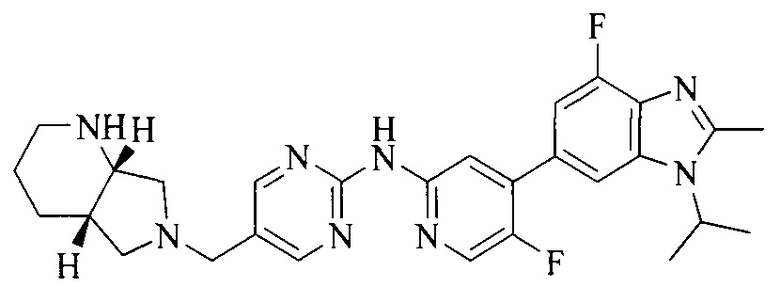
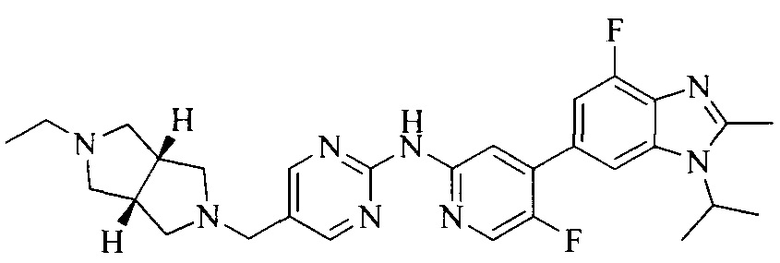
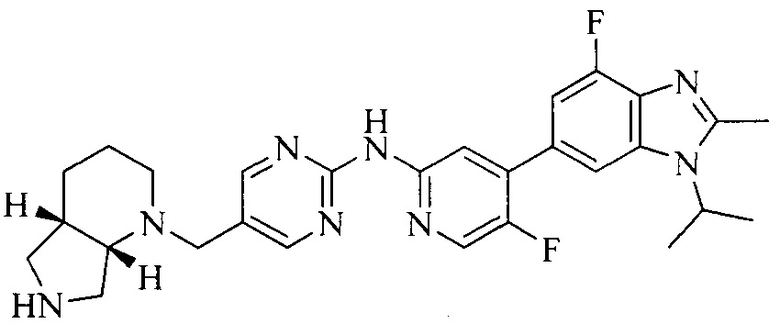
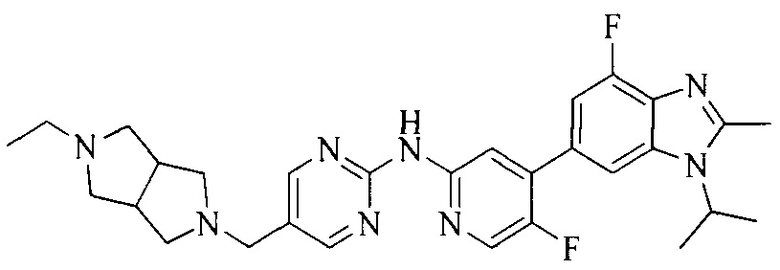
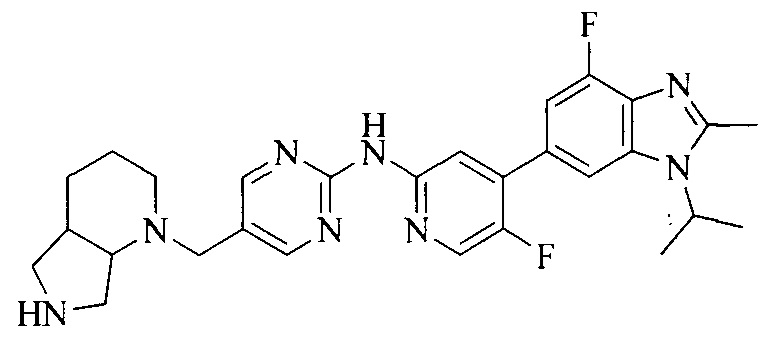
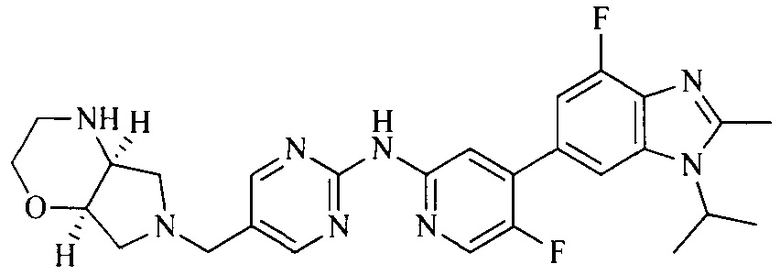
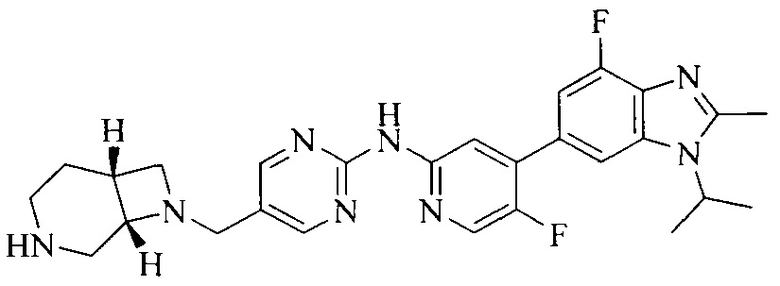
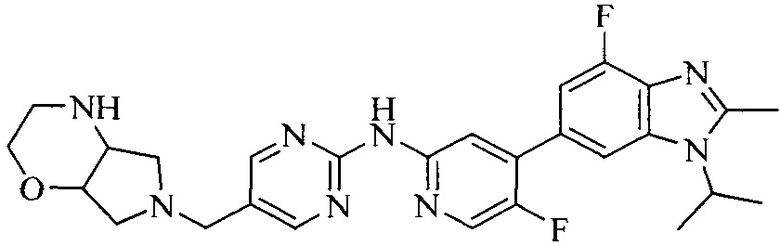
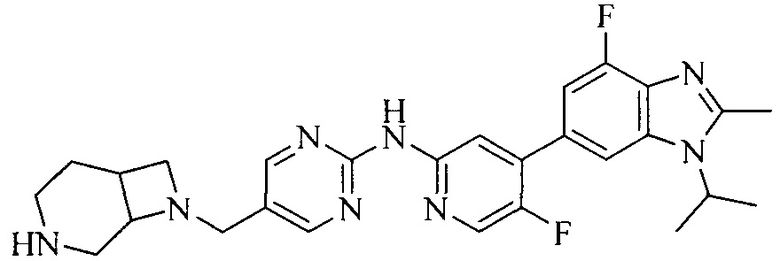
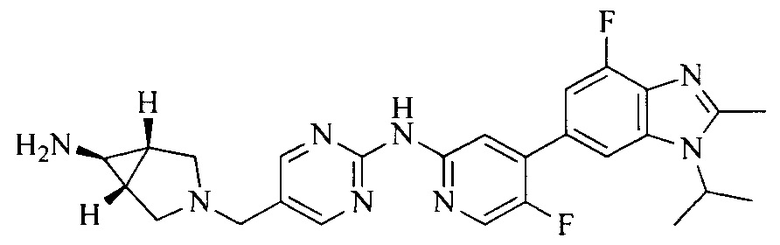
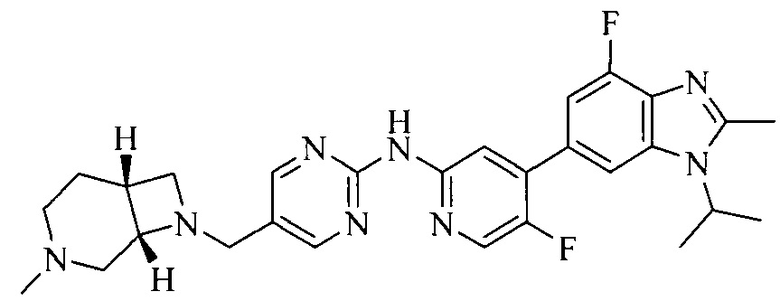
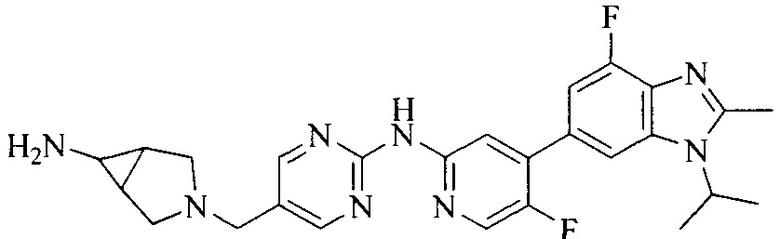
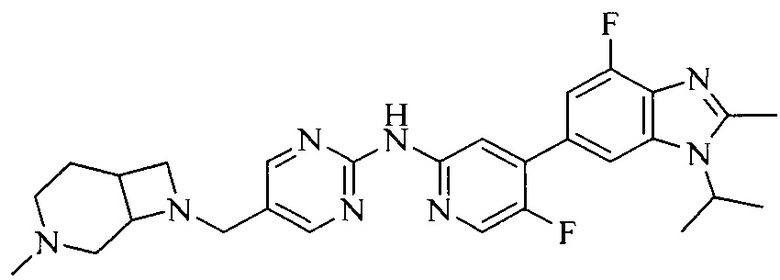
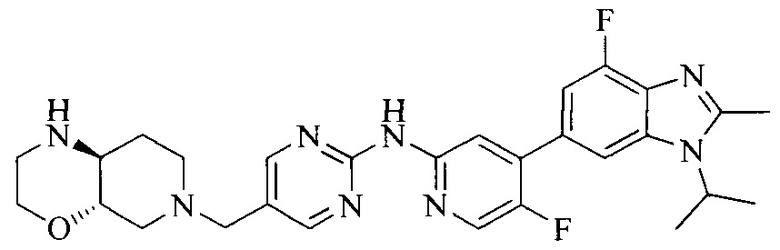
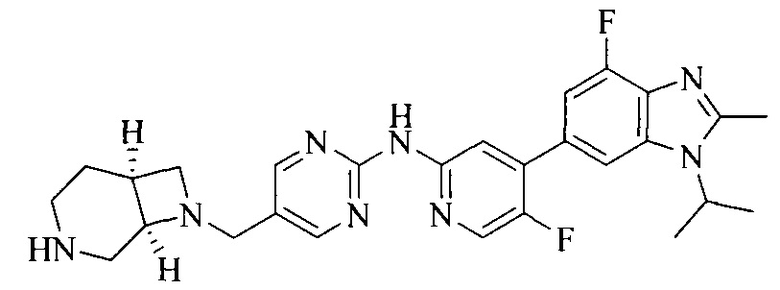
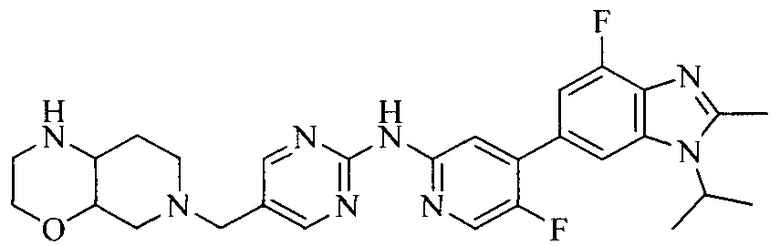
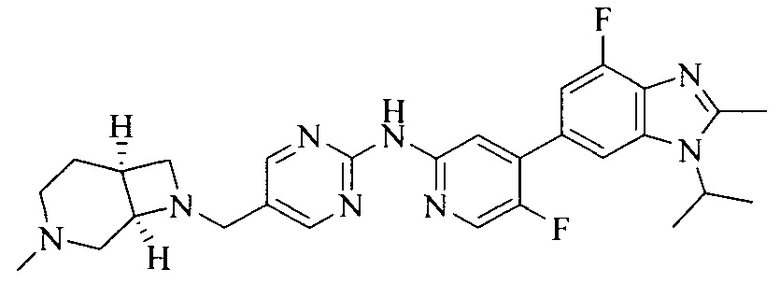
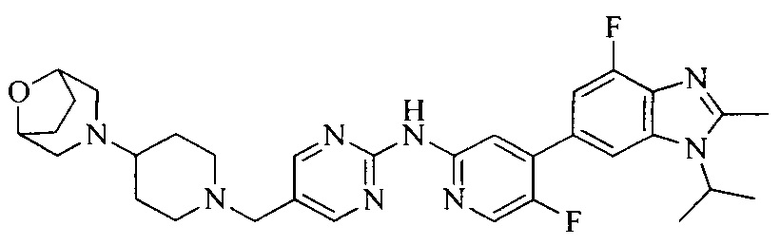
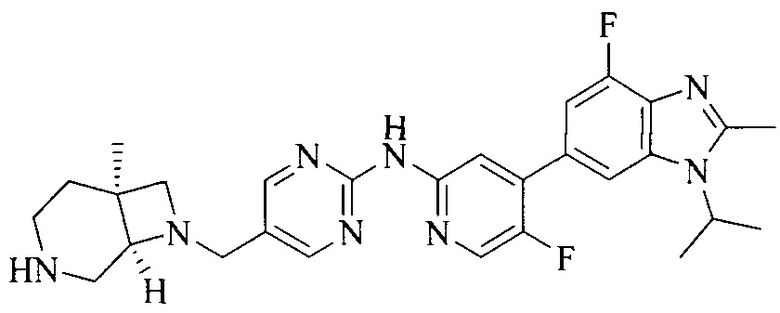
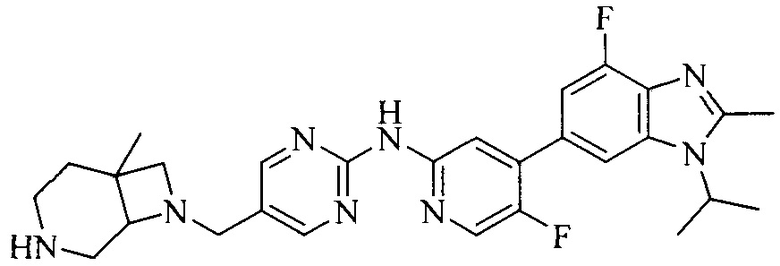
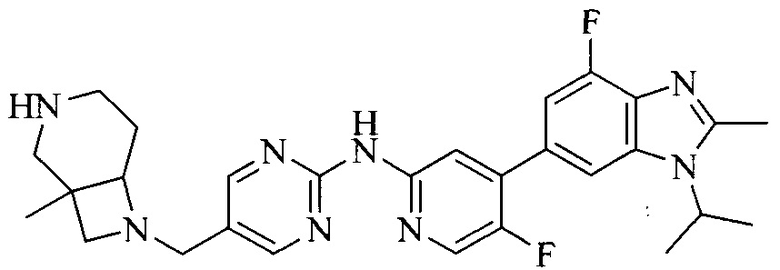
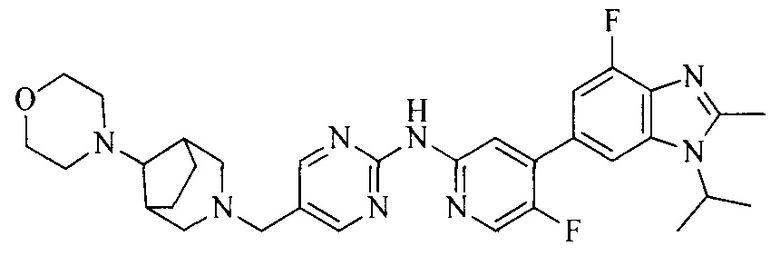
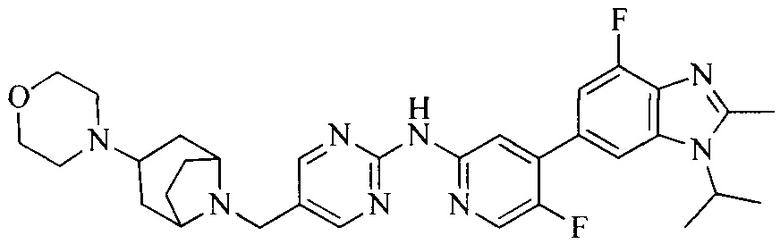
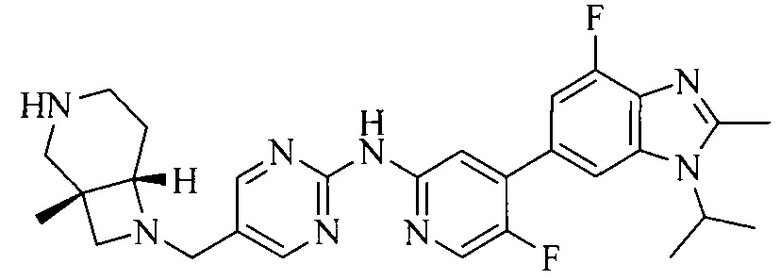
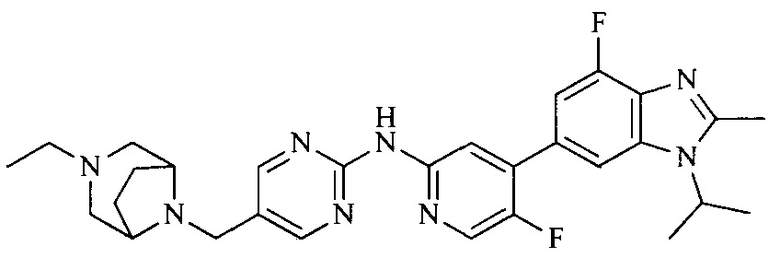
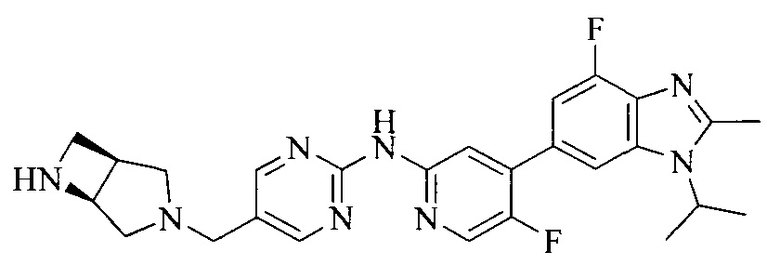
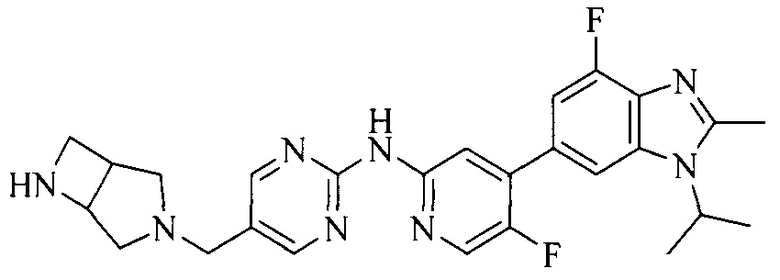
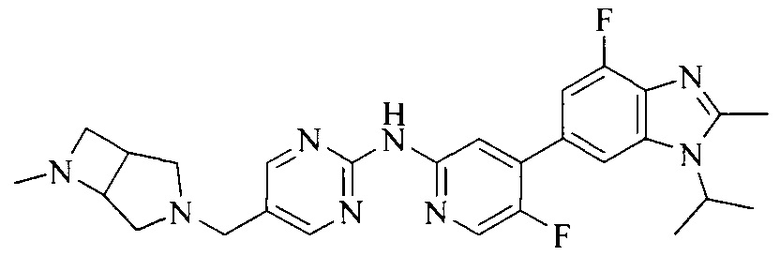
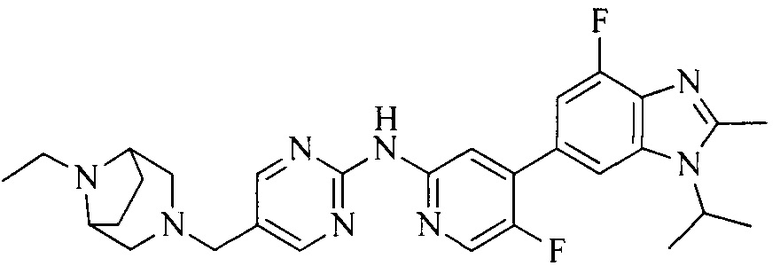
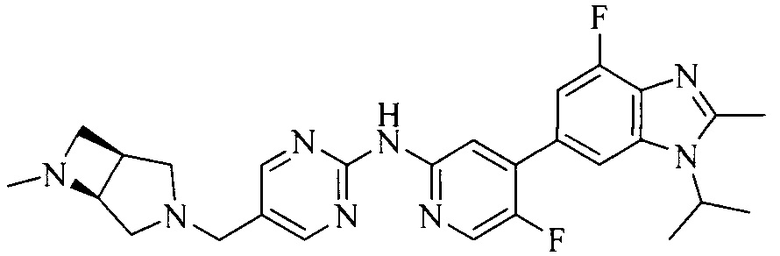
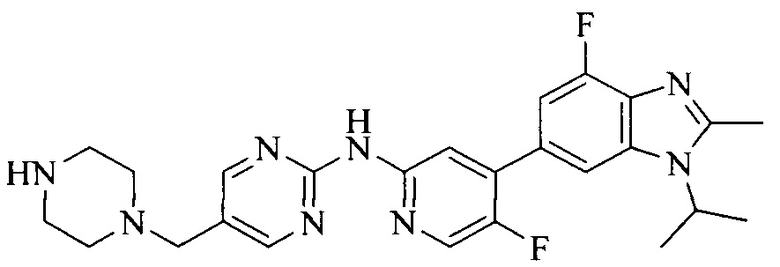
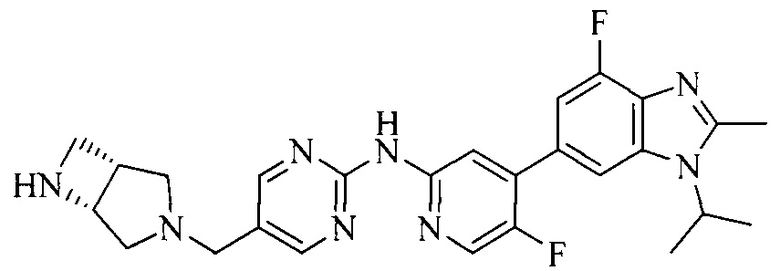
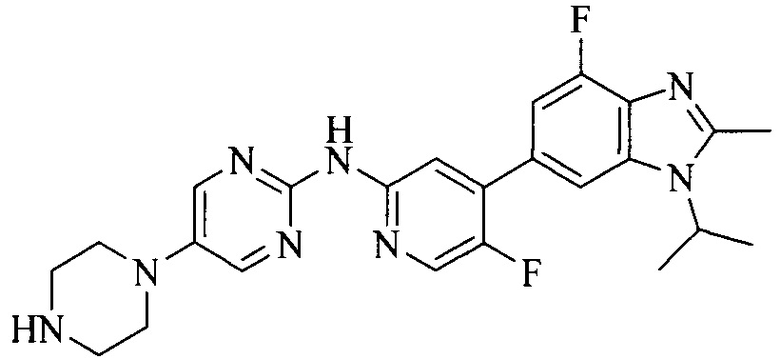
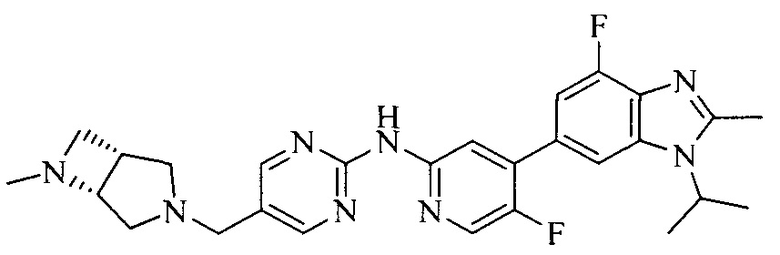
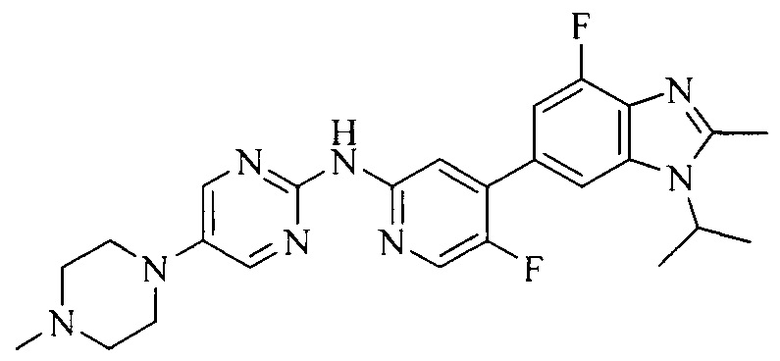
19. Фармацевтическая композиция, обладающая способностью ингибировать активность CDK4 и/или CDK6 киназ, содержащая терапевтически и/или профилактически эффективное количество соединения или его стереоизомера по любому из пп.1-18 и один или более фармацевтически приемлемых носителей.
20. Фармацевтическая композиция по п.19, дополнительно содержащая один или более дополнительных противоопухолевых средств и/или иммуносуппрессоров.
21. Фармацевтическая композиция по п.20, где дополнительные противоопухолевые средства и/или иммуносуппрессоры выбраны из одного или более из следующих: метотрексата, капецитабина, гемцитабина, доксифлуридина, пеметрекседа динатрия, пазопаниба, иматиниба, эрлотиниба, лапатиниба, гефитиниба, вандетаниба, герцептина, бевацизумаба, ритуксимаба, трастузумаба, паклитаксела, винорельбина, доцетаксела, доксорубицина, гидроксикамптотецина, митомицина, эпирубицина, пирарубицина, блеомицина, летрозола, тамоксифена, фульвестранта, трипторелина, флутамида, лейпрорелина, анастрозола, ифосфамида, бусульфана, циклофосфамида, кармустина, нимустина, семустина, мехлорэтамина, мелфалана, хлорамбуцила, карбоплатина, цисплатина, оксалиплатина, лобаплатина, топотекана, камптотецина, топотекана, эверолимуса, сиролимуса, темсиролимуса, 6-меркаптопурина, 6-тиогуанина, азатиоприна, актиномицина D, даунорубицина, адриамицина, митоксантрона, блеомицина, митрамицина и аминоглутетимида.
22. Применение соединения или его стереоизомера по любому из пп.1-18 при изготовлении лекарственного средства для лечения и/или предотвращения связанных с раком заболеваний, опосредованных CDK4/6 киназой, у субъекта.
23. Применение по п.22, где связанное с раком заболевание выбрано из опухоли головного мозга, рака легких, плоскоклеточной карциномы, карциномы мочевого пузыря, рака желудка, рака яичников, перитонеального рака, рака поджелудочной железы, рака молочной железы, рака головы и шеи, рака шейки матки, рака эндометрия, рака прямой кишки, рака печени, почечной карциномы, аденокарциномы пищевода, плоскоклеточного рака пищевода, рака предстательной железы, рака женского репродуктивного протока, рака in situ, лимфомы, нейрофибромы, карциномы щитовидной железы, остеокарциномы, рака кожи, рака мозга, рака толстой кишки, рака мужских половых органов, стромальной опухоли желудочно-кишечного тракта, новообразований предстательной железы, опухоли тучных клеток, множественной миеломы, меланомы, глиомы или саркомы.
| CN 102264725 A, 30.11.2011 | |||
| CN 102186856 A, 14.09.2011 | |||
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ | 2002 |
|
RU2294326C2 |

