Настоящее изобретение направлено на способ получения комплексообразующего агента, причем указанный способ включает стадии (а) предоставления нитрила, соответствующего общей формуле (Ia) или (Ib),

причем М выбирают из щелочного металла и водорода и их комбинаций,
(b) омыления с общим количеством щелочи от 2,5 до 2,9 моль гидроксида щелочного металла на моль нитрила, соответствующего общей формуле (Ia) или (Ib) соответственно, и со значением рН в диапазоне от 9,5 до 11,5 в конце стадии (b),
(c) добавления такого количества гидроксида щелочного металла, чтобы общее содержание щелочи составляло от 2,9 до 3,15 моль на моль нитрила, соответствующего общей формуле (Ia) или (Ib) соответственно, причем если в сумме используют 2,9 моль гидроксида щелочного металла на моль нитрила, то количество щелочи на соответствующей стадии (b) находится в диапазоне от 2,5 до менее 2,9 моль, и
(d) обеспечения возможности дальнейшего превращения.
Хелатирующие агенты, такие как, однако без ограничения ими, метилглициндиуксусная кислота (MGDA) и их соответствующие соли со щелочными металлами, являются полезными комплексообразователями (хелатирующими агентами) для ионов щелочноземельных металлов, таких как Са2+ и Mg2+, и переходных металлов, таких как, однако без ограничения ими, Fe(+II)/Fe(+III). По этой причине они рекомендуются и используются для различных целей, как например, в моющих средствах для стирки и композициях для автоматического мытья посуды (ADW), в частности, для так называемых не содержащих фосфатов моющих средств для стирки и не содержащих фосфатов композиций для автоматического мытья посуды.
MGDA и другие хелатирующие агенты могут быть получены путем алкилирования аминокислот с формальдегидом и циановодородной кислотой или цианидом щелочного металла с последующим омылением с гидроксидом щелочного металла. Чтобы обеспечить полное омыление, применяют стехиометрическое количество гидроксида щелочного металла или избыток гидроксида щелочного металла, смотрите, например, патент США US 7,671,234. В других способах MGDA получают добавлением NH(CH2CN)2 и циановодородной кислоты к ацетальдегиду с образованием тринитрила с последующим гидролизом, смотрите, например, патент США US 7,754,911.
Чтобы снизить проблемы с коррозией, если омыление проводится в реакторах из нержавеющей стали, такой как сталь 316, в международной заявке WO 2016/180664 было предложено использовать субстехиометрическое количество основания. Получали смеси из MGDA и моноамидов, которые демонстрируют хорошие комплексообразующие характеристики. Однако при определенных условиях превращение в твердое вещество такими способами, как распылительная сушка или гранулирование распылением, является экономически невыгодным, поскольку образуется сравнительно высокая доля слишком больших частиц (слишком крупная фракция, англ. «overs»). Хотя такую слишком крупную фракцию возможно удалять и измельчать ее перед подачей обратно в цикл, такая доля слишком крупной фракции является нежелательной из соображений экономической эффективности и производительности процесса.
Таким образом, целью было предоставить комплексообразующий агент с превосходной долговременной устойчивостью окраски, который можно превращать в твердое вещество легко и в экономически благоприятных условиях.
Соответственно этому был обнаружен способ, определенный в начале, далее также называемый способом согласно изобретению. Способ согласно изобретению включает стадии
(а) предоставления нитрила, соответствующего общей формуле (Ia) или (Ib),

причем М выбирают из щелочного металла и водорода и их комбинаций,
(b) омыления с общим количеством щелочи от 2,5 до 2,9 моль гидроксида щелочного металла на моль нитрила, соответствующего общей формуле (Ia) или (Ib) соответственно, и со значением рН в диапазоне от 9,5 до 11,5 в конце стадии (b),
(c) добавления такого количества гидроксида щелочного металла, чтобы общее содержание щелочи составляло от 2,9 до 3,15 моль на моль нитрила, соответствующего общей формуле (Ia) или (Ib) соответственно, причем если в сумме используют 2,9 моль гидроксида щелочного металла на моль нитрила, то количество щелочи на соответствующей стадии (b) находится в диапазоне от 2,5 до менее 2,9 моль, и
(d) обеспечения дальнейшего превращения.
Указанные стадии в дальнейшем также обозначаются как стадия (а), стадия (b), стадия (с) и стадия (d). Ниже они описаны более подробно.
Нитрилы в соответствии с общими формулами (Ia) и (Ib) сами по себе являются известными. Нитрил (Ia) может быть получен путем взаимодействия NH(CH2CN)2 и циановодородной кислоты с ацетальдегидом. Нитрил (Ia) обычно является рацемическим.
Нитрил (Ib) предпочтительно получают путем превращения аланина в так называемом двойном синтезе Штрекера с двумя эквивалентами HCN и формальдегида для каждого. Нитрил (Ib) может быть рацемическим или являться L-изомером или любой смесью L- и D-изомеров с преобладанием L-изомера, например, с количеством от 50 до 99,5% L-энантиомера. Предпочтительными являются рацемические смеси и смеси, которые содержат от 95 до 99,5% L-энантиомера. Нитрил (Ib) может быть представлен в виде свободной кислоты или полностью или частично нейтрализованным щелочью, например, натрием или калием.
На стадии (а) нитрил, соответствующий общим формулам (Ia) и (Ib), может быть получен в виде массы вещества или в виде раствора или суспензии, например, в концентрации - или с содержанием твердого вещества - в диапазоне от 5 до 60% массовых.
Указанная суспензия или раствор предпочтительно представляет собой водную суспензию или водный раствор, предпочтительно, водный раствор. Такая суспензия или раствор, соответственно, могут иметь общее содержание твердых веществ в диапазоне от 5 до 60% массовых, предпочтительно, от 30 до 50% массовых. Термин «водный» относится к непрерывной фазе или растворителю, содержащему в диапазоне от 50 до 100% объемн. воды, предпочтительно, от 70 до 100% объемн. воды, по отношению ко всей непрерывной фазе или растворителю соответственно. Примерами подходящих растворителей, отличающихся от воды, являются спирты, такие как метанол, этанол и изопропанол, кроме того, диолы, такие как этиленгликоль, и триолы, такие как глицерин.
На стадии (b) нитрил, соответствующий общей формуле (Ia) или (Ib) соответственно, подвергают омылению с общим количеством щелочи от 2,5 до 2,9 моль гидроксида щелочного металла на моль нитрила, соответствующего общей формуле (Ia) или (Ib) соответственно, и со значением рН от 9,5 до 11,5 в конце стадии (b). Значение рН определяется при температуре окружающей среды и относится к значению рН в конце омыления в соответствии со стадией (b), такому, как он есть, без дополнительного разбавления. Сразу после добавления указанного гидроксида щелочного металла на стадии (b) значение рН составляет выше 11,5, например, до 14. В процессе расходования гидроксида щелочного металла это значение рН уменьшается.
В одном варианте осуществления настоящего изобретения гидроксид щелочного металла выбирают из гидроксидов лития, натрия, калия и комбинаций по меньшей мере двух из вышеперечисленных. Предпочтительными являются гидроксид натрия, гидроксид калия, смеси гидроксида натрия и гидроксида калия и еще более предпочтительным является гидроксид натрия.
На стадии (b) гидроксид щелочного металла может быть добавлен к нитрилу, соответствующему общей формуле (Ia) или (Ib) соответственно, в виде массы вещества или, предпочтительно, в водном растворе. Водные растворы гидроксида щелочного металла могут иметь концентрацию в диапазоне от 1% массового до 65% массовых, предпочтительно, от 10 до 55% массовых.
Водные растворы гидроксида щелочного металла могут содержать одну или несколько примесей, такую как, однако без ограничения им, карбонат щелочного металла. Например, водные растворы гидроксида натрия могут содержать от 0,01 до 1% карбоната натрия.
Стадию (b) можно начинать, загружая в реакционный сосуд водный раствор гидроксида щелочного металла и затем добавляя суспензию или раствор соединения, соответствующего общей формуле (Ia) или (Ib) соответственно, одной или несколькими порциями. В альтернативном варианте осуществления указанное контактирование может осуществляться путем загрузки в реакционный сосуд части водного раствора гидроксида щелочного металла, а затем добавления суспензии или раствора соединения, соответствующего общей формуле (Ia) или (Ib) соответственно, в одну или несколько порций, а оставшегося раствора гидроксида щелочного металла впоследствии или предпочтительно параллельно. В альтернативном варианте осуществления указанное контактирование может осуществляться путем непрерывного соединения раствора или суспензии соединения, соответствующего общей формуле (Ia) или (Ib), и водного раствора гидроксида щелочного металла.
В вариантах осуществления, в которых водные растворы гидроксида щелочного металла добавляют на стадии (b) двумя порциями, первая порция может содержать от 10 до 50% мольн. требуемого гидроксида щелочного металла, а вторая порция может содержать оставшиеся от 50 до 90% мольн.
В вариантах осуществления, в которых соединение, соответствующее общей формуле (Ia) или (Ib), добавляют на стадии (b) двумя порциями, первая порция может содержать от 10 до 50% мольн. требуемого соединения, соответствующего общей формуле (Ia) или (Ib), а вторая порция может содержать оставшиеся от 50 до 90% мольн.
В одном варианте осуществления настоящего изобретения реакционный сосуд, в котором осуществляется стадия (b), включает по меньшей мере одну часть, изготовленную из нержавеющей стали или нержавеющую сталь, которая подвергается воздействию смеси, образованной на стадии (b). Нержавеющая сталь здесь относится к чистым аустенитным нержавеющим сталям или сплавам аустенитных и ферритных нержавеющих сталей (например, «дуплексная сталь»).
Стадия (b) осуществляется с диапазоном от 2,5 до 2,9 моль гидроксида щелочного металла на моль нитрила, соответствующего общей формуле (Ia) или (Ib) соответственно. В контексте стадии (b) указанное количество гидроксида щелочного металла включает гидроксид щелочного металла, который был использован во время получения нитрила, соответствующего общей формуле (Ia) или (Ib). В конце стадии (b) значение рН находится в диапазоне от 9,5 до 11,5.
Стадия (b) способа согласно изобретению может осуществляться при температуре в диапазоне от 25 до 200°С, предпочтительно, от 45 до 190°С.
Стадия (b) способа согласно изобретению может осуществляться при одной температуре. Однако в предпочтительных вариантах осуществления стадия (b) осуществляется в форме двух или более подстадий (b1, (b2) и при желании большего количества, для которых эти подстадии осуществляются при разных температурах. Предпочтительно, каждая подстадия может осуществляться при температуре, которая выше, чем температура, при которой осуществлялась предыдущая подстадия. В контексте настоящего изобретения подстадии отличаются по температуре по меньшей мере на 10°С, причем указанная температура относится к средней температуре. В предпочтительном варианте осуществления настоящего изобретения стадия (b) включает по меньшей мере две подстадии (b1) и (b2), причем подстадию (b2) осуществляют при температуре по меньшей мере на 20°С выше, чем на подстадии (Ы), предпочтительно, по меньшей мере на 25°С. В предпочтительном варианте осуществления стадия (b) включает по меньшей мере две подстадии (b1) и (b2), причем подстадию (b2) осуществляют при температуре, составляющей на величину от 20 до 150°С выше, чем на подстадии (b1).
Предпочтительно, подстадия осуществляется за период, составляющий по меньшей мере 30 минут. Еще более предпочтительно, подстадия осуществляется в течение периода от 30 минут до 5 часов, предпочтительно, до 2 часов. В одном варианте осуществления настоящего изобретения стадия (b) имеет общую продолжительность в диапазоне от 30 минут до 24 часов, предпочтительно, от 2 до 16 часов.
В одном варианте осуществления настоящего изобретения по меньшей мере одна подстадия стадии (b) осуществляется при температуре в интервале от 25 до 50°С, предпочтительно, по меньшей мере одна в интервале от 40 до 55°С.
В одном варианте осуществления настоящего изобретения по меньшей мере одна подстадия стадии (b) осуществляется при температуре в диапазоне от 50 до 80°С, предпочтительно, от 60 до 75°С.
В одном варианте осуществления настоящего изобретения по меньшей мере одна подстадия стадии (b) осуществляется при температуре в диапазоне от 90 до 200°С, предпочтительно, от 150 до 190°С.
В одном варианте осуществления настоящего изобретения по меньшей мере одна подстадия стадии (b) осуществляется при температуре в диапазоне от 25 до 60°С, другая подстадия стадии (b) осуществляется при температуре в интервале от 50 до 80°С, а по меньшей мере еще одна подстадия стадии (b) осуществляется при температуре в интервале от 100 до 200°С.
В одном варианте осуществления настоящего изобретения аммиак, образующийся во время реакции, удаляют, непрерывно или периодически, например, путем отгонки или путем перегонки, например, при температуре по меньшей мере 90°С, предпочтительно, от 90 до 105°С.
В одном варианте осуществления настоящего изобретения в течение стадии (b) добавляют воду, например, чтобы компенсировать потерю воды из-за удаления аммиака.
В одном варианте осуществления настоящего изобретения стадию (b) проводят при нормальном давлении или при давлении выше 1 бар, например, от 1,1 до 40 бар, предпочтительно, от 5 до 25 бар. В вариантах осуществления с двумя или более подстадиями стадии (b) последующие подстадии предпочтительно осуществляются при давлении, по меньшей мере таком же высоком, как на предыдущей подстадии.
Стадию (b) можно проводить в реакторе с мешалкой или в реакторе идеального вытеснения, или в каскаде по меньшей мере из двух реакторов с мешалкой, например, от 2 до 6 реакторов с мешалкой, или в комбинации из каскада от 2 до 6 реакторов с мешалкой с по меньшей мере одним реактором идеального вытеснения или в каскаде из по меньшей мере одного реактора с мешалкой и двух реакторов идеального вытеснения.
Особенно в вариантах осуществления, в которых заключительная подстадия стадии (b) осуществляется в реакторе идеального вытеснения, указанная конечная подстадия может осуществляться при повышенном давлении, таком как от 1,5 до 40 бар, предпочтительно, по меньшей мере 20 бар. Повышенное давление может быть достигнуто с помощью насоса или путем автогенного повышения давления.
В одном варианте осуществления настоящего изобретения реакционный сосуд, в котором осуществляется стадия (b), содержит по меньшей мере одну часть, изготовленную из нержавеющей стали, которая подвергается воздействию реакционной смеси в соответствии со стадией (b).
В одном варианте осуществления настоящего изобретения по меньшей мере один реакционный сосуд, в котором осуществляется подстадия стадии (b), содержит по меньшей мере одну часть, изготовленную из нержавеющей стали, которая подвергается воздействию реакционной смеси в соответствии со стадией (b).
Во время стадии (b) может иметь место частичная или полная рацемизация, если соединение, соответствующее общей формуле (Ia) или (Ib), является оптически активным, и если стадия (b) или по меньшей мере одна подстадия стадии (b) проводится при достаточно высокой температуре. Не желая быть связанными какой-либо теорией, вероятно, что рацемизация имеет место на стадии вышеупомянутого L-моноамида или L-диамида или L-изомера MGDA.
После стадии (b) раствору полученных таким образом продуктов обычно дают охладиться, например, до температуры от 70 до 100°С, в частности, в диапазоне от 80 до 100°С. В вариантах осуществления, в которых стадия (b) или по меньшей мере подстадия стадии (b) осуществляется под давлением, превышающим 1 бар, предпочтительно снижать это давление после стадии (b) до нормального давления.
На стадии (с) добавляют такое количество гидроксида щелочного металла, чтобы общее содержание щелочи составляло от 2,9 до 3,15 моль на моль нитрила, соответствующего общей формуле (Ia) или (Ib) соответственно. В контексте стадии (с) указанное количество щелочи включает гидроксид щелочного металла, который был использован во время получения нитрила, соответствующего общей формуле (Ia) или (Ib). На стадии (с) предпочтительными являются от 2,9 до 3,0 моль. В вариантах осуществления, где используется в общем 2,9 моль гидроксида щелочного металла на моль нитрила, количество щелочи на соответствующей стадии (b) находится в диапазоне от 2,5 до менее 2,9 моль, например, составляет 2,85 моль.
Для осуществления стадии (с) гидроксид щелочного металла может быть выбран из гидроксида лития, гидроксида натрия и гидроксида калия и комбинаций по меньшей мере двух из вышеуказанных, например, комбинаций гидроксида натрия и гидроксида калия. Предпочтительным гидроксидом щелочного металла на стадии (с) является гидроксид натрия. В предпочтительном варианте осуществления настоящего изобретения как гидроксид щелочного металла на стадии (b), так и гидроксид щелочного металла на стадии (с) представляют собой гидроксид натрия.
Добавление гидроксида щелочного металла может быть достигнуто путем добавления твердого гидроксида щелочного металла или путем добавления водного раствора гидроксида щелочного металла.
В одном варианте осуществления настоящего изобретения добавление гидроксида щелочного металла согласно стадии (с) проводят при температуре в диапазоне от 20 до 100°С. Если добавление осуществляется при более высокой температуре, необходимо приложить определенное давление выше нормального давления, что требует дополнительных усилий.
Стадия (d) способа согласно изобретению включает в себя предоставление возможности дальнейшего превращения. Это означает, что может происходить дальнейшее омыление. В других вариантах осуществления стадия (d) включает дополнительную нейтрализацию без гидролиза. Стадия (d) может также включать удаление аммиака.
В одном варианте осуществления настоящего изобретения водный раствор, полученный на стадии (с), кипятят с обратным холодильником при нормальном давлении, например, в течение периода времени в диапазоне от 30 минут до 5 часов.
Стадии (а), (b), (с) и (d) способа согласно изобретению выполняются в таком порядке, как описано выше.
В одном варианте осуществления настоящего изобретения способ согласно изобретению может включать дополнительные стадии, отличающиеся от стадий (а), (b), (с) и (d), раскрытых выше. Такими дополнительными стадиями могут быть, например, одна или несколько стадий обесцвечивания, например, обработка активированным углем или пероксидом, таким как H2O2, или путем облучения ультрафиолетовым светом в отсутствие или в присутствии H2O2.
В одном варианте осуществления настоящего изобретения аммиак удаляется между стадиями (b) и (d), например, частично или полностью.
В одном варианте осуществления настоящего изобретения способ согласно изобретению включает дополнительную стадию (е) распылительной сушки или гранулирования распылением получающегося в результате комплексообразующего агента.
Дополнительной стадией, отличающейся от стадии (а), (b), (с) или (d), которая может осуществляться после стадии (с) или во время стадии (d) или после стадии (d), является отгонка с воздухом или азотом или паром, чтобы удалить аммиак. Упомянутая отгонка может осуществляться при температуре в диапазоне от 90 до 110°С. Путем отгонки с азотом или воздухом вода может быть удалена из полученного таким образом раствора. Отгонка предпочтительно проводится при давлении ниже нормального давления, как например, от 650 до 950 мбар.
В вариантах осуществления, где требуется раствор, этот раствор, полученный на стадии (d), просто охлаждают и при желании концентрируют путем частичного удаления воды. Если требуются сухие образцы смесей согласно изобретению, то воду можно удалить путем распылительной сушки или гранулирования распылением.
Способ согласно настоящему изобретению может осуществляться как периодический процесс, или как полунепрерывный или непрерывный процесс.
В результате способа согласно изобретению получают водный раствор метилглициндиацетата (MGDA) с превосходной долговременной устойчивостью окраски.
MGDA, полученный в соответствии со способом согласно изобретению, может представлять собой рацемическую смесь или чистый энантиомер, например L-энантиомер, или смесь L- и D-энантиомеров, в которых преобладает один из энантиомеров, предпочтительно, преобладает L-энантиомер. В предпочтительном варианте осуществления настоящего изобретения MGDA, полученный в соответствии со способом согласно изобретению, представляет собой смесь энантиомеров, содержащих преимущественно соответствующий L-энантиомер с энантиомерным избытком (ее) в диапазоне от 10 до 98%.
В одном варианте осуществления настоящего изобретения энантиомерный избыток соответствующего L-изомера MGDA, полученного в соответствии со способом согласно изобретению, находится в диапазоне от 10 до 98%, предпочтительно, в диапазоне от 12,5 до 85% и даже более предпочтительно до 75%. В других вариантах осуществления все компоненты смесей согласно изобретению образуют соответствующие рацемические смеси.
В вариантах осуществления, где MGDA, полученный в соответствии со способом согласно изобретению, содержит два или более соединений, ее относится к энантиомерному избытку для всех L-изомеров, присутствующих в MGDA, полученном в соответствии со способом согласно изобретению, по отношению ко всем D-изомерам в соответствующем MGDA. Например, в случаях, когда присутствует смесь ди- и тринатриевой соли MGDA, ее относится к сумме динатриевой соли и тринатриевой соли L-MGDA по отношению к сумме динатриевой соли и тринатриевой соли D-MGDA.
Энантиомерный избыток может быть определен путем измерения поляризации (поляриметрией) или, предпочтительно, с помощью хроматографии, например, с помощью ВЭЖХ с хиральной колонкой, например, с одним или несколькими циклодекстринами в качестве неподвижной фазы. Предпочтительным является определение ее методом ВЭЖХ с иммобилизованной оптически активной солью аммония, такой как D-пеницилламин.
В одном варианте осуществления настоящего изобретения MGDA, полученный в соответствии со способом согласно изобретению, может содержать в диапазоне от 0,1 до 10% массовых одной или нескольких оптически неактивных примесей, причем по меньшей мере одна из этих примесей представляет собой по меньшей мере одну из примесей, выбранных из иминодиуксусной кислоты, рацемического N-карбоксиметилаланина, муравьиной кислоты, гликолевой кислоты, пропионовой кислоты, уксусной кислоты и их соответствующих солей со щелочными металлами или моно-, ди- или триаммонием.
В одном аспекте настоящего изобретения MGDA, полученный в соответствии со способом согласно изобретению, может содержать менее 0,2% массовых нитрилотриуксусной кислоты (NTA), предпочтительно, от 0,01 до 0,1% массовых.
В одном варианте осуществления настоящего изобретения MGDA, полученный в соответствии со способом согласно изобретению, может содержать одну или несколько оптически активных примесей. Примерами оптически активных примесей являются L-карбоксиметилаланин и его соответствующие моно- или двойные соли со щелочными металлами, и оптически активные моно- или диамиды, возникающие в результате неполного омыления динитрилов, смотрите ниже. Предпочтительно, количество оптически активных примесей находится в диапазоне от 0,01 до 2% массовых, по отношению к раствору смеси согласно изобретению. Еще более предпочтительно, количество оптически активных примесей находится в диапазоне от 0,1 до 2% массовых.
В одном аспекте настоящего изобретения MGDA, полученный в соответствии со способом согласно изобретению, может содержать незначительные количества катионов, отличающихся от щелочного металла. Таким образом, возможно, что незначительные количества, такие как от 0,01 до 5% мольн. от всей смеси согласно изобретению, в пересчете на анион, содержат катионы аммония или катионы щелочноземельных металлов, такие как Mg2+ или Са2+, или ионы переходных металлов, такие как катионы Fe2+ или Fe3+.
В одном варианте осуществления настоящего изобретения способ согласно изобретению включает дополнительную стадию (е) распылительной сушки или гранулирования распылением комплексообразующего агента, полученного на стадии (d). В результате осуществления стадии (е) могут быть получены порошки или гранулы. В контексте настоящего изобретения порошки представляют собой материалы в виде частиц, которые представляют собой твердые вещества при температуре окружающей среды и которые предпочтительно имеют средний диаметр частиц в диапазоне от 1 мкм до менее 0,1 мм, предпочтительно, от 5 мкм до 50 мкм. Средний диаметр частиц порошков может быть определен, например, методами лазерной дифракции, например, с помощью аппарата Malvern, и относится к среднему по объему. В контексте настоящего изобретения гранулы представляют собой материалы в виде частиц, которые представляют собой твердые вещества при температуре окружающей среды и которые предпочтительно имеют средний диаметр частиц в диапазоне от 0,1 мм до 2 мм, предпочтительно, от 0,4 мм до 1,25 мм. Средний диаметр частиц гранул может быть определен, например, путем оптических или, предпочтительно, просеивающих способов. Используемые сита могут иметь размер ячеек от 60 до 1250 мкм.
В одном варианте осуществления настоящего изобретения порошки или гранулы имеют широкое распределение частиц по диаметрам. В другом варианте осуществления настоящего изобретения порошки или гранулы имеют узкое распределение частиц по диаметрам. Распределение частиц по диаметрам может быть скорректировано, если необходимо, с помощью нескольких стадий просеивания.
Гранулы и порошки могут содержать остаточную влагу, влагу, относящуюся к воде, включая кристаллизационную воду и адсорбированную воду. Количество воды может составлять от 0,1 до 20% массовых, предпочтительно, от 1 до 15% массовых, в пересчете на общее содержание твердых веществ в соответствующем порошке или гранулах, и может быть определено титрованием по Карлу-Фишеру или путем сушки при 160°С до постоянной массы или в течение специально отведенного времени, например, одного часа, с помощью инфракрасного излучателя.
В одном особенно предпочтительном варианте осуществления настоящего изобретения количество воды в порошке может находиться в диапазоне от 5 до 10% массовых.
В одном особенно предпочтительном варианте осуществления настоящего изобретения количество воды в гранулах может находиться в диапазоне от 9 до 12% массовых.
Частицы порошков могут иметь правильную или неправильную форму. Предпочтительными формами частиц порошков являются сфероидальные формы.
Частицы гранул могут иметь правильную или неправильную форму. Предпочтительными формами частиц гранул являются сфероидальные формы.
Распылительную сушку или гранулирование распылением в соответствии со стадией (е) можно проводить с использованием газа с температурой на входе, составляющей предпочтительно по меньшей мере 125°С. Указанный газ, в дальнейшем также называемый «горячим газом», может представлять собой азот, благородный газ или предпочтительно воздух. В ходе стадии (е) будет удалена большая часть воды, например, по меньшей мере 55%, предпочтительно, по меньшей мере 65% воды. В одном варианте осуществления настоящего изобретения будет удалено максимально 95% воды.
Распылительная сушка или гранулирование распылением будут описаны более подробно ниже.
В одном варианте осуществления настоящего изобретения используется сушильная камера, например, распылительная камера или распылительная башня, в которой выполняется процесс распылительной сушки. Раствор или суспензию MGDA, полученные в соответствии со способом согласно изобретению, распыляют в указанную распылительную камеру или распылительную башню вместе с потоком горячего газа в режиме прямотока или противотока. Поток горячего газа может иметь температуру в диапазоне от 60 до 350°С, предпочтительно, от 125 до 220°С.
В одном варианте осуществления настоящего изобретения используется сушильная камера, например, распылительная камера или распылительная башня, в которой процесс гранулирования распылением выполняется с использованием псевдоожиженного слоя. В такую сушильную камеру загружают псевдоожиженный слой частиц затравки MGDA, полученных любым способом сушки, таким как распылительная сушка или кристаллизация при испарении. Флюидизация слоя достигается с помощью потока газа с температурой в диапазоне от 125 до 350°С, предпочтительно, от 125 до 220°С. Затем раствор или суспензию MGDA, полученные в соответствии со способом согласно изобретению, распыляют на поверхность или внутрь такого псевдоожиженного слоя вместе с потоком горячего газа с температурой в диапазоне от 60 до 250°С, предпочтительно, от 125 до 220°С.
В одном варианте осуществления настоящего изобретения псевдоожиженный слой может иметь температуру в диапазоне от 80 до 150°С, предпочтительно, от 90 до 120°С.
Распыление производится через одно или несколько сопел на каждую сушильную камеру. Подходящими соплами являются, например, распылители с вращающимся барабаном высокого давления, вращающиеся распылители, однокомпонентные сопла и двухкомпонентные сопла, причем предпочтительными являются двухкомпонентные сопла и вращающиеся распылители. Первая текучая среда представляет собой раствор или суспензию, полученную в соответствии со стадией (d) способа согласно изобретению, вторая текучая среда представляет собой сжатый газ, например, с давлением от 1,1 до 7 бар. Для гранулирования распылением предпочтительные сопла выбираются из однокомпонентных сопел и двухкомпонентных сопел. Выражения двухпоточные сопла и двухкомпонентные сопла могут использоваться взаимозаменяемо.
В одном варианте осуществления настоящего изобретения капли, образованные во время гранулирования распылением, имеют средний диаметр в диапазоне от 10 до 500 мкм, предпочтительно, от 20 до 180 мкм, еще более предпочтительно, от 30 до 100 мкм.
В одном варианте осуществления настоящего изобретения отходящий газ, выходящий из сушильной камеры, может иметь температуру в диапазоне от 40 до 140°С, предпочтительно, от 80 до 110°С, но в любом случае холоднее, чем поток горячего газа. Предпочтительно, температура отходящего газа, выходящего из сушильной камеры, и температура твердого продукта, присутствующего в этой сушильной камере, являются идентичными.
В другом варианте осуществления настоящего изобретения гранулирование распылением осуществляется путем выполнения двух или более последовательных процессов распылительной сушки, например, в каскаде из по меньшей мере двух распылительных сушилок, например, в каскаде из по меньшей мере двух последовательно соединенных распылительных башен или в комбинации распылительной башни и распылительной камеры, причем указанная распылительная камера содержит псевдоожиженный слой. В первой сушилке процесс распылительной сушки выполняется следующим образом.
Распылительная сушка может быть предпочтительной в распылительной сушилке, например, в распылительной камере или распылительной башне. Раствор или суспензию, полученную в соответствии со стадией (а), с температурой, предпочтительно превышающей температуру окружающей среды, например, в диапазоне от 50 до 95°С, вводят в распылительную сушилку через одно или несколько распылительных сопел во входящий поток горячего газа, например азота или воздуха, при этом раствор или суспензия превращаются в капли, а вода испаряется. Входящий поток горячего газа может иметь температуру в диапазоне от 125 до 350°С.
Во вторую распылительную сушилку загружают псевдоожиженный слой с твердым веществом из первой распылительной сушилки, а раствор или суспензию, полученные в соответствии с вышеуказанной стадией, распыляют на этот псевдоожиженный слой или внутрь него вместе с входящим потоком горячего газа. Входящий поток горячего газа может иметь температуру в диапазоне от 125 до 350°С, предпочтительно, от 160 до 220°С.
В одном варианте осуществления настоящего изобретения, в частности, в процессе изготовления гранул, среднее время пребывания MGDA на стадии (е) находится в диапазоне от 2 минут до 4 часов, предпочтительно, от 30 минут до 2 часов.
В другом варианте осуществления, в частности, в процессе изготовления порошка, среднее время пребывания MGDA на стадии (е) находится в диапазоне от 1 секунды до 1 минуты, в частности, от 2 до 20 секунд.
В одном варианте осуществления настоящего изобретения давление в сушильной камере на стадии (е) составляет нормальное давление ±100 мбар, предпочтительно, нормальное давление ±20 мбар, например, на один мбар ниже нормального давления.
Твердый MGDA, образованный на стадии (е), удаляют из распылительной камеры или соответственно распылительной башни полностью или в некотором процентном соотношении, непрерывно или порциями.
Вместе с твердым MGDA с желаемым размером частиц обычно образуются более мелкие частицы («пыль» или «мелкая фракция») и более крупные частицы («слишком крупная фракция»). Частицы с желаемым размером затем отделяются от мелких частиц и слишком крупной фракции. Слишком крупная фракция может быть размолота до приемлемого размера частиц и возвращена в распылительную камеру или в распылительную башню («возврат»), и мелкие фракции также могут быть возвращены.
В других вариантах осуществления возможно повторно растворять слишком крупную фракцию и мелкие частицы, например, в воде, и повторно вводить этот раствор для распылительной сушки или гранулирования распылением.
Было обнаружено, что при осуществлении способа согласно настоящему изобретению образуется меньший процент слишком крупной фракции, и требуется меньше измельчения и повторного использования, чем в случае MGDA со значительно меньшим содержанием щелочного металла. С другой стороны, гранула, полученная по способу согласно изобретению, обладает превосходной долговременной устойчивостью окраски.
В одном варианте осуществления настоящего изобретения к раствору или суспензии, полученной в соответствии со стадией (d), могут быть добавлены одна или несколько добавок. Примерами полезных добавок являются, например, диоксид титана, сахар, силикагель и полимеры, такие как, но без ограничения ими, поливиниловый спирт, (со)полимеры (мет)акриловой кислоты, частично или полностью нейтрализованные щелочью. Поливиниловый спирт в контексте настоящего изобретения относится к полностью или частично гидролизованному поливинилацетату.
В частично гидролизованном поливинилацетате по меньшей мере 95% мольн., предпочтительно, по меньшей мере 96% мольн. ацетатных групп было гидролизовано.
В одном варианте осуществления настоящего изобретения поливиниловый спирт, используемый на стадии (е), имеет среднюю молекулярную массу Mw в диапазоне от 22500 до 115000 г/моль, например, до 40000 г/моль.
В другом варианте осуществления настоящего изобретения (со)полимеры (мет)акриловой кислоты представляют собой, например, статистические сополимеры акриловой кислоты и метакриловой кислоты, статистические сополимеры акриловой кислоты и малеинового ангидрида, тройные статистические сополимеры акриловой кислоты, метакриловой кислоты и малеинового ангидрида, статистические или блок-сополимеры акриловой кислоты и стирола, статистические сополимеры акриловой кислоты и метилакрилата. Более предпочтительными являются гомополимеры метакриловой кислоты. Еще более предпочтительными являются гомополимеры акриловой кислоты.
(Со)полимеры (мет)акриловой кислоты могут формировать молекулы с прямой или разветвленной цепью. Разветвление в этом контексте будет иметь место тогда, когда по меньшей мере одно повторяющееся структурное звено такого (со)полимера не является частью основной цепи, а образует ответвление или часть ответвления. Предпочтительно (со)полимер не является поперечно сшитым.
В одном варианте осуществления настоящего изобретения (со)полимеры (мет)акриловой кислоты имеют среднюю молекулярную массу Mw в диапазоне от 1200 до 30000 г/моль, предпочтительно, от 2500 до 15000 г/моль и еще более предпочтительно от 3000 до 10000 г/моль, определяемую путем гельпроникающей хроматографии (ГПХ) и относящуюся к соответствующей свободной кислоте.
В одном варианте осуществления настоящего изобретения (со)полимеры (мет)акриловой кислоты являются по меньшей мере, частично нейтрализованными щелочью, например литием или калием или натрием, или комбинациями по меньшей мере двух из вышеперечисленных, в частности, натрием. Например, в диапазоне от 10 до 100% мольн. карбоксильных групп полимера (В) могут быть нейтрализованными щелочью, в частностью, натрием.
В одном варианте осуществления настоящего изобретения (со)полимеры (мет)акриловой кислоты выбирают из пернатриевых солей полиакриловой кислоты, следовательно, полиакриловой кислоты, полностью нейтрализованной натрием.
В одном варианте осуществления настоящего изобретения (со)полимеры (мет)акриловой кислоты выбирают из пернатриевых солей полиакриловой кислоты со средней молекулярной массой Mw в диапазоне от 1200 до 30000 г/моль, предпочтительно, от 2500 до 15000 г/моль и даже более предпочтительно от 3000 до 10000 г/моль, определяемой путем гельпроникающей хроматографии (ГПХ) и относящуюся к соответствующей свободной кислоте.
В одном варианте осуществления настоящего изобретения массовое соотношение добавки и MGDA находится в диапазоне от 1:100 до 1:2, предпочтительно, от 1:50 до 1:10.
Гранулы и порошки, изготовленные согласно настоящему изобретению, имеют превосходную долговременную устойчивость окраски. Они могут быть получены легко и в экономически благоприятных условиях. Показатели коррозии в процессе производства - особенно в отношении нержавеющей стали - являются превосходными.
Изобретение дополнительно иллюстрируется с помощью рабочих примеров.
За исключением значений ее, процентные величины в контексте примеров относятся к массовым процентам, если явно не указано иное.
I.1. Получение водного раствора L-аланин-N,N-бисацетонитрила, стадия (а.1)
В колбу с мешалкой объемом 5 литров загружали 1170 г деионизированной воды и нагревали до 40°С. Добавляли 668,5 г L-аланина (99,2% масс, что составляет 7,44 моль, с ее >98%). К полученной суспензии на протяжении 30 минут добавляли 390,0 г (4,88 моль) водного раствора гидроксида натрия с концентрацией 50% массовых. Во время добавления температура повышалась до 60°С. После завершения добавления гидроксида натрия суспензию перемешивали при 60°С в течение 30 минут. Получали прозрачный раствор.
При температуре вышеуказанного раствора от 38 до 42°С добавляли формальдегид в виде 30%-ного водного раствора и HCN (80% от общего количества) в первый реактор с мешалкой в каскаде, включающем три реактора с мешалкой. Во второй реактор с мешалкой добавляли дополнительное количество HCN (20% от общего количества) при 38-42°С. В третьем реакторе с мешалкой при 38-42°С завершали реакцию. Получали водный раствор частично нейтрализованного L-аланин-N,N-бисацетонитрила. Его использовали в качестве исходного сырья для холодного омыления.
I.2 Синтез водных растворов MGDA-Nax со субстехиометрическими количествами или эквимолярными количествами NaOH: омыление (b.1)
(b.1-1) Холодное омыление:
Холодное омыление проводили в каскаде из двух реакторов с мешалкой и трубчатого реактора. Температура составляла приблизительно 55°С во всех трех реакторах.
В первый реактор с мешалкой добавляли исходный раствор, такой как получен на стадии (а.1), и NaOH в виде 50%-ного водного раствора. Для завершения реакции смесь дополнительно подвергали взаимодействию во втором реакторе с мешалкой и в трубчатом реакторе. Раствор, полученный в стационарных условиях, использовали в качестве исходного сырья при горячем омылении.
(b.1-2) Горячее омыление:
Горячее омыление проводили при 180°С и 24 бар в трубчатом реакторе с идеальным вытеснением при времени удерживания от 30 до 45 минут. Стадии (с) или (d) не осуществляли.
Раствору, полученному в условиях стационарного режима, давали достичь давления окружающей среды и перемешивали в резервуаре реактора при 970 мбар при температуре от 94 до 98°С для удаления аммиака. Затем его подвергали отгонке в пленочном испарителе при 900 мбар при 100°С для дополнительного удаления аммиака. Затем концентрацию общего количества комплексообразующего агента (А) доводили до приблизительно 40% массовых (на основе способности связывать железо).
Молярные соотношения исходных материалов обобщаются в таблице 1.
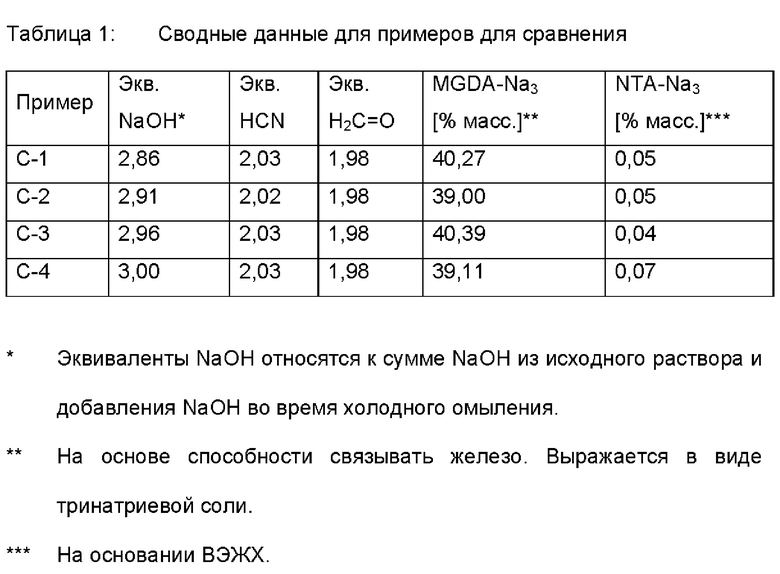
Примеры согласно настоящему изобретению:
Стадии I.1 и I.2 осуществляли, как указано выше.
I.3 Добавление NaOH
Раствор, полученный в условиях стационарного режима, подавали в непрерывном режиме в реактор с мешалкой при 970 мбар при температуре от 94 до 98°С. В этот реактор с мешалкой добавляли дополнительное количество NaOH в соответствии с таблицей 2. Затем объединенные потоки подвергали отгонке в пленочном испарителе при 900 мбар при 100°С для дополнительного удаления аммиака. Затем концентрацию общего количества комплексообразующего агента (А) доводили до приблизительно 40% массовых (на основе способности связывать железо).
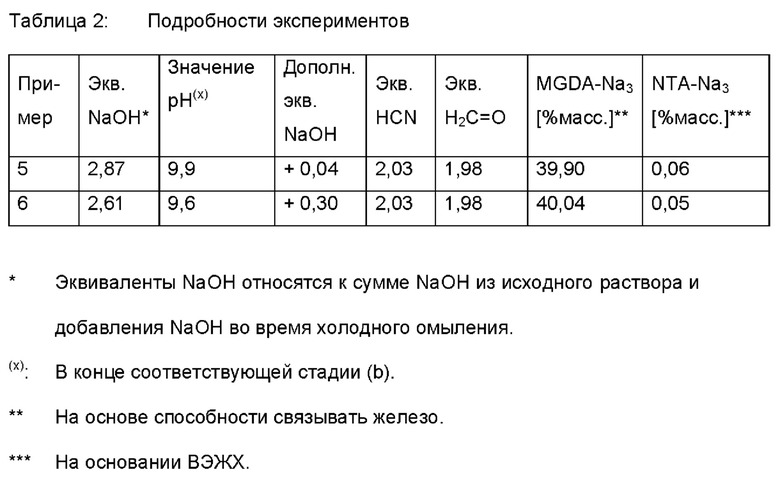
I.4 Добавление NaOH при температуре окружающей среды
Следующие примеры были получены в соответствии с вышеупомянутыми стадиями I.1 и I.2, но после стадии I.2 (b.1-2) к продукту добавляли дополнительные количества NaOH.
В колбу с мешалкой объемом 1 литр загружали загружали 500 г соответствующего продукта (после стадии I.2). Затем при температуре окружающей среды добавляли дополнительное количество водного раствора гидроксида натрия (50% масс). Этот раствор нагревали до 80°С и перемешивали в течение 60 минут при 80°С. Затем реакционный раствор охлаждали до 20°С и концентрацию общего количества комплексообразующего агента (А) доводили до приблизительно 40% массовых (на основе способности связывать железо).
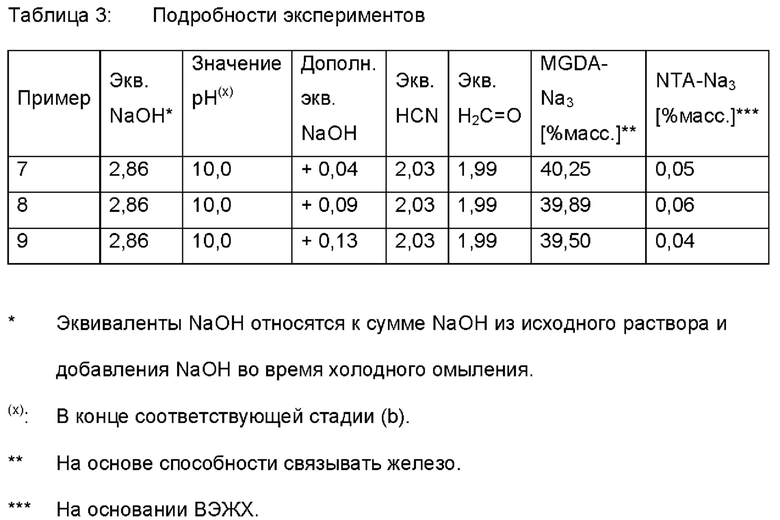
II. Гранулирование распылением, общие замечания Использовали коммерчески доступный лабораторный распылительный гранулятор с двухкомпонентным соплом и воздушным классификатором зигзагообразного типа Glatt Lab Systems. В этот распылительный гранулятор загружали примерно 1,5 кг коммерчески доступного (Trilon® М) MGDA-Na3. Распылительный гранулятор работал в соответствии с таблицей 4. Процентные содержания слишком крупной фракции определяли, когда этот лабораторный распылительный гранулятор работал в стационарном режиме.
Затем раствор натриевой соли MGDA перекачивали из резервуара с мешалкой в двухкомпонентное сопло и потом вводили в лабораторный распылительный гранулятор. Наблюдали образование гранул.
Частицы, которые были достаточно крупными (тяжелыми), через зигзагообразный воздушный классификатор падали в сосуд для образцов вместе с требуемой фракцией. Сосуд для образцов содержал бы требуемую фракцию и слишком крупную фракцию. Более мелкие (более легкие) гранулы выдувались посредством рециркуляции обратно в псевдоожиженный слой с помощью воздушного классификатора. Мелкие фракции удерживались в грануляторе с помощью внутренних фильтров.
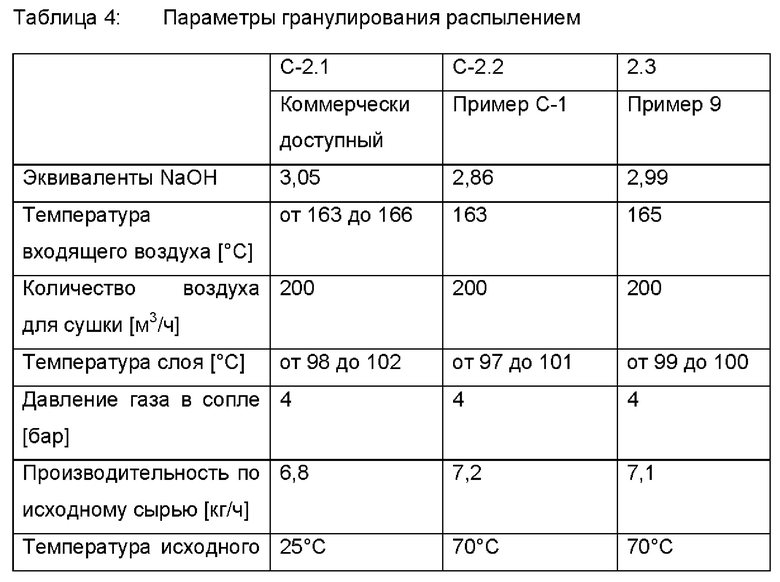

Частицы в сосуде для образцов были классифицированы с помощью сита 1000 мкм. Частицы диаметром 1000 мкм и ниже составляли требуемую фракцию. Гранулы размером более 1000 мкм были определены как слишком крупная фракция. Эту слишком крупную фракцию измельчали с помощью молотковой мельницы до диаметра не более 700 мкм и повторно вводили в псевдоожиженный слой вместе с небольшой долей измельченной требуемой фракции посредством рециркуляции измельченного продукта.
В эксперименте С-2.1 использовали коммерчески доступный раствор MGDA-Na3.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ СМЕСИ СОЛИ ЩЕЛОЧНОГО МЕТАЛЛА МЕТИЛГЛИЦИНДИУКСУСНОЙ КИСЛОТЫ И СОЛИ ЩЕЛОЧНОГО МЕТАЛЛА ГЛУТАМИНОВОЙ ДИУКСУСНОЙ КИСЛОТЫ, СМЕСЬ L- И D-ЭНАНТИОМЕРОВ УКАЗАННЫХ СОЛЕЙ И ВОДНЫЙ РАСТВОР УКАЗАННОЙ СМЕСИ ДЛЯ ПОЛУЧЕНИЯ МОЮЩИХ КОМПОЗИЦИЙ ДЛЯ СТИРКИ И ОЧИСТКИ | 2016 |
|
RU2722803C2 |
| СПОСОБ ПОЛУЧЕНИЯ СМЕСЕЙ ХЕЛАТООБРАЗУЮЩИХ АГЕНТОВ | 2016 |
|
RU2706358C2 |
| РАСТВОРЫ ТРИЩЕЛОЧНЫХ СОЛЕЙ АМИНОКАРБОНОВЫХ КИСЛОТ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2705366C2 |
| ПОРОШКИ И ГРАНУЛЫ И СПОСОБ ПОЛУЧЕНИЯ ТАКИХ ПОРОШКОВ И ГРАНУЛ | 2017 |
|
RU2742268C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГРАНУЛ, СОДЕРЖАЩИХ MGDA И GLDA, ГРАНУЛЫ И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2744632C2 |
| СМЕСИ ХЕЛАТИРУЮЩИХ АГЕНТОВ И СПОСОБ ПОЛУЧЕНИЯ ТАКИХ СМЕСЕЙ | 2017 |
|
RU2742269C2 |
| КОНТЕЙНЕР, ВКЛЮЧАЮЩИЙ КОМПОЗИЦИЮ МОЮЩЕГО СРЕДСТВА, СОДЕРЖАЩУЮ MGDA | 2015 |
|
RU2689387C2 |
| СМЕСИ ХЕЛАТИРУЮЩИХ АГЕНТОВ И СПОСОБ ПОЛУЧЕНИЯ ТАКИХ СМЕСЕЙ | 2016 |
|
RU2712767C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГРАНУЛ | 2016 |
|
RU2728876C2 |
| СМЕСИ ЭНАНТИОМЕРОВ И СПОСОБ ПОЛУЧЕНИЯ ТАКИХ СМЕСЕЙ | 2014 |
|
RU2675835C2 |
Изобретение относится к способу получения комплексообразующего агента, представляющего собой метилглициндиуксусную кислоту (MGDA) и ее соли со щелочными металлами. Указанный способ включает стадии (a)-(d). На стадии (а) осуществляют предоставление нитрила, соответствующего общей формуле (Ia) или (Ib), причем М выбирают из щелочного металла и водорода и их комбинаций. На стадии (b) происходит омыление с общим количеством щелочи от 2,5 до 2,9 моль гидроксида щелочного металла на моль нитрила, соответствующего общей формуле (Ia) или (Ib) соответственно, и со значением рН в диапазоне от 9,5 до 11,5 в конце стадии (b). На стадии (c) добавляют такое количество гидроксида щелочного металла, чтобы общее содержание щелочи составляло от 2,9 до 3,15 моль на моль нитрила, соответствующего общей формуле (Ia) или (Ib) соответственно, причем если в сумме используют 2,9 моль гидроксида щелочного металла на моль нитрила, то количество щелочи на соответствующей стадии (b) находится в диапазоне от 2,5 до менее 2,9 моль. На стадии (d) осуществляют дополнительное омыление или нейтрализацию. Предлагаемый способ позволяет получать комплексообразующий агент с долговременной устойчивостью окраски, который легко превращается в твердое вещество. 8 з.п. ф-лы, 4 табл., 9 пр.

1. Способ получения комплексообразующего агента, представляющего собой метилглициндиуксусную кислоту (MGDA) и ее соли со щелочными металлами, причем указанный способ включает стадии:
(а) предоставления нитрила, соответствующего общей формуле (Ia) или (Ib)
 ,
,
причем М выбирают из щелочного металла и водорода и их комбинаций,
(b) омыления с общим количеством щелочи от 2,5 до 2,9 моль гидроксида щелочного металла на моль нитрила, соответствующего общей формуле (Ia) или (Ib) соответственно, и со значением рН в диапазоне от 9,5 до 11,5 в конце стадии (b),
(c) добавления такого количества гидроксида щелочного металла, чтобы общее содержание щелочи составляло от 2,9 до 3,15 моль на моль нитрила, соответствующего общей формуле (Ia) или (Ib) соответственно, причем если в сумме используют 2,9 моль гидроксида щелочного металла на моль нитрила, то количество щелочи на соответствующей стадии (b) находится в диапазоне от 2,5 до менее 2,9 моль, и
(d) дополнительного омыления или нейтрализации.
2. Способ по п. 1, где нитрил, соответствующий формуле (Ib), выбирают из рацемической смеси и энантиомерно чистого L-(Ib) и смесей энантиомеров (Ib), в которых преобладает L-изомер.
3. Способ по п. 1, где омыление на стадии (b) осуществляют при температуре в диапазоне от 25 до 200°С.
4. Способ по п. 1, где гидроксид щелочного металла выбирают из гидроксида калия и гидроксида натрия.
5. Способ по п. 1, где добавление гидроксида щелочного металла в соответствии со стадией (с) осуществляют при температуре в диапазоне от 25 до 100°С.
6. Способ по п. 1, где на стадии (с) добавляют такое количество гидроксида щелочного металла, чтобы общее содержание щелочи составляло от 2,9 до 3,0 моль на моль нитрила, соответствующего общей формуле (Ia) или (Ib) соответственно.
7. Способ по п. 1, где дополнительно между стадиями (b) и (d) удаляют аммиак.
8. Способ по п. 7, где стадия (d) представляет собой удаление аммиака.
9. Способ по любому из пп. 1-8, где указанный способ включает дополнительную стадию (е) распылительной сушки или гранулирования распылением получающегося в результате комплексообразующего агента.
| СПОСОБ ПОЛУЧЕНИЯ ТРИЩЕЛОЧНЫХ СОЛЕЙ МЕТИЛГЛИЦИН-N,N-ДИУКСУСНОЙ КИСЛОТЫ С НЕЗНАЧИТЕЛЬНЫМ СОДЕРЖАНИЕМ ПОБОЧНЫХ ПРОДУКТОВ | 2006 |
|
RU2399611C2 |
| Токарный резец | 1924 |
|
SU2016A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |

