Область техники, к которой относится изобретение
Настоящее изобретение относится к способам лечения хронической спонтанной крапивницы с применением лигелизумаба.
Предпосылки изобретения
Крапивница представляет собой гетерогенную группу заболеваний, характеризующихся зудящей крапивной сыпью и/или ангионевротическим отеком. Хроническая крапивница определяется как крапивница, которая постоянно или периодически присутствует в течение более 6 недель (Maurer M, Magerl M, Metz M, et al (2013) Revisions to the international guidelines on the diagnosis and therapy of chronic urticaria. J Dtsch Dermatol Ges.; Bernstein JA, Lang DM, Khan DA, et al (2014) The diagnosis and management of acute and chronic urticaria: 2014 update. J Allergy Clin Immunol; 133(5):1270-7). Хроническую крапивницу далее дополнительно делят на две подгруппы: хроническую спонтанную крапивницу (CSU) и индуцибельную крапивницу (IU), при этом последняя включает физическую крапивницу, как например тепловая, холодовая или крапивница вследствие давления, а также особые варианты, как например холинергическая крапивница. CSU определяется как спонтанное проявление зудящих волдырей, ангионевротического отека или того и другого в течение ≥ 6 недель по известным или неизвестным причинам (Zuberbier T, Aberer W, Asero R, et al (2014) The EAACI/GA(2) LEN/EDF/WAO Guideline for the definition, classification, diagnosis, and management of urticaria: the 2013 revision и update. Allergy; 69(7):868-87). Возможна комбинация как CSU, так и индуцибельной формы крапивницы, как например часто наблюдаемая комбинация бессимптомной дерматографической крапивницы и CSU.
Ранее все формы хронической крапивницы без известного триггера назывались "хронической идиопатической крапивницей" (CIU). Благодаря прогрессу в области медицины в настоящее время стало известно, что при некоторых из ранее считавшихся "идиопатическими" формах крапивницы в действительности могут быть обнаружены аутоантитела. Однако, ежедневное нестабильное проявление симптомов при такой хронической крапивнице с наличием аутоантител все еще остается непредсказуемым и не провоцируется очевидным триггером, вследствие чего симптомы проявляются спонтанно. Чтобы надлежащим образом отразить в терминологии тот факт, что при некоторых из ранее считавшихся "идиопатическими" формах в действительности могут иметься поддающиеся выявлению аутоантитела, эту популяцию теперь называют хронической спонтанной крапивницей (CSU) в соответствии с Международным руководством (Maurer M, Magerl M, Metz M, et al (2013) Revisions to the international guidelines on the diagnosis and therapy of chronic urticaria. J Dtsch Dermatol Ges.; Zuberbier T, Aberer W, Asero R, et al (2014) The EAACI/GA(2) LEN/EDF/WAO Guideline for the definition, classification, diagnosis, and management of urticaria: the 2013 revision and update. Allergy; 69(7):868-87). Использование выражения "хроническая идиопатическая крапивница" в медицинской практике больше не рекомендуется. Однако данное новое соглашение о наименовании реализовано не во всех частях мира, и в таких странах, как США, популяцию пациентов с хронической крапивницей неспецифической этиологии или с неизвестными триггерами все еще характеризуют как пациентов с хронической идиопатической крапивницей (CIU). Согласно Международному руководству эта нозологическая единица заболевания в целях единообразия в данном документе будет называться CSU.
Распространенность CSU в течение жизни составляет примерно 1,8%, а у 20% пациентов с CSU все еще имеется заболевание по прошествии 20 лет (Greaves M (2000) Chronic urticaria. J Allergy Clin Immunol; 105(4):664-72; Zuberbier T, Balke M, Worm M, et al (2010) Epidemiology of urticaria: a representative cross-sectional population survey. Clin Exp Dermatol; 35(8):869-73). У болеющих пациентов часто возникает зудящая сыпь с сопутствующей эритемой и/или эпизодами ангионевротического отека. Сообщается, что ангионевротический отек ассоциирован примерно с 33-67% случаев CSU (Juhlin L (1981) Recurrent urticaria: clinical investigation of 330 patients. Br J Dermatol; 104(4):369-81 ; Toubi E, Kessel A, Avshovich N, et al (2004) Clinical and laboratory parameters in predicting chronic urticaria duration: a prospective study of 139 patients. Allergy; 59(8):869-73; Zuberbier T, Balke M, Worm M, et al (2010) Epidemiology of urticaria: a representative cross-sectional population survey. Clin Exp Dermatol; 35(8):869-73; Maurer M, Weller K, Bindslev-Jensen C, et al (2011) Unmet clinical needs in chronic spontaneous urticaria. A GA LEN task force report. Allergy; 66(3):317-30). Классическое поражение кожи при крапивнице представляет собой волдырь и покраснение с бледным приподнятым участком поражения и окружающей эритемой размером от нескольких миллиметров до нескольких сантиметров в поперечнике, которые обычно возникают группами и часто сливаются с образованием больших сливных поражений. CSU ассоциируется с сильным зудом и оказывает большое влияние на самочувствие и качество жизни пациента, которое, как представляется, сравнимо с влиянием тяжелой ишемической болезни сердца (Greaves MW (2003) Chronic idiopathic urticaria. Curr Opin Allergy Clin Immunol; 3(5):363-8. Review; Powell RJ, Du Toit GL, Siddique N, et al (2007) BSACI guidelines for the management of chronic urticaria and angio-oedema. Clin Exp Allergy; 37(5):631-50). Симптомы крапивницы и ангионевротического отека, ассоциированного с крапивницей, отрицательно влияют на повседневную активность и сон (O'Donnell BF, Lawlor F, Simpson J, et al (1997). The impact of chronic urticaria on the quality of life. Br J Dermatol; 136(2):197-201). Таким образом, при ведении пациентов с крапивницей результаты с точки зрения пациента (например, DLQI) являются важными показателями лечения (Kaplan A, Ledford D, Ashby M, et al (2013) Omalizumab in patients with symptomatic chronic idiopathic/spontaneous urticaria despite standard combination therapy. J Allergy Clin Immunol; 132(1):101-9; Maurer M, Magerl M, Metz M, et al (2013) Revisions to the international guidelines on the diagnosis and therapy of chronic urticaria. J Dtsch Dermatol Ges; Zuberbier T, Aberer W, Asero R, et al (2014) The EAACI/GA(2) LEN/EDF/WAO Guideline for the definition, classification, diagnosis, and management of urticaria: the 2013 revision and update. Allergy; 69(7):868-87).
Патогенез CSU не до конца выяснен. До 50% случаев CSU связаны с индуцирующими высвобождение гистамина аутоантителами ко множеству антигенов, включая высокоаффинный рецептор IgE (FcεRI) или антитела класса IgE; при этом клиническое значение этих аутоантител четко не установлено, хотя есть предположения, что они могут быть вовлечены в патогенез заболевания (Kaplan AP (2002) Chronic urticaria--new concepts regarding pathogenesis and treatment. Curr Allergy Asthma Rep; 2(4):263-4; Sabroe RA, Greaves MW (2006) Chronic idiopathic urticaria with functional autoantibodies: 12 years on. Br J Dermatol; 154(5):813-9. Review). Было также высказано предположение, что базофилы пациентов с CSU могут иметь отчетливые изменения в FcεRIα-опосредованной дегрануляции, независимо от какой-либо потенциальной роли аутоантител (Eckman JA, Hamilton RG, Gober LM, et al (2008) Basophil phenotypes in chronic idiopathic urticaria in relation to disease activity and autoantibodies. J Invest Dermatol; 128(8):1956-63).
Лечение CSU является сложной задачей, а не обладающие седативным действием (относящиеся ко второму поколению) H1-антигистаминные средства (H1-AH) являются основой симптоматической терапии CSU. Хотя H1-AH в одобренных дозах приносит облегчение некоторым пациентам, у более 50% пациентов отсутствует ответ на H1-AH при стандартных дозах. Даже при четырехкратном повышении размера дозы по сравнению с размером одобренной дозы, осуществляемом в соответствии со вторым этапом алгоритма лечения согласно действующему Международному руководству (Zuberbier T, Aberer W, Asero R, et al (2014) The EAACI/GA(2) LEN/EDF/WAO Guideline for the definition, classification, diagnosis, and management of urticaria: the 2013 revision and update. Allergy; 69(7):868-87), для значительной части пациентов не достигается контроль симптомов крапивницы (Maurer M, Weller K, Bindslev-Jensen C, et al (2011) Unmet clinical needs in chronic spontaneous urticaria. A GA LEN task force report. Allergy; 66(3):317-30; Marrouche N, Grattan C (2014) Update and insights into treatment options for chronic spontaneous urticaria. Expert Rev Clin Immunol; 10(3):397-403). Для пациентов без достигнутого контроля заболевания при четырехкратных дозах H1-AH третий этап алгоритма лечения согласно Международному руководству предусматривает добавление омализумаба, циклоспорина A или монтелукаста к H1-AH.
Уровень имеющихся доказательств в пользу эффективности антагонистов лейкотриеновых рецепторов (LTRA) при крапивнице является низким, при этом для монтелукаста он является самым высоким, что впоследствии повлекло за собой только ненастоятельные рекомендации экспертов в отношении использования этого средства лечения вне показаний к его применению. К схемам лечения 3-го уровня могут быть добавлены короткие курсы (макс. 10 дней) системных кортикостероидов, если этого требуют обострения. Ввиду побочных эффектов, связанных с хроническим системным воздействием кортикостероидов, более длительный курс лечения не рекомендуется. Другие варианты лечения, которые использовались ранее, такие как внутривенный иммуноглобулин G, дапсон, гидроксихлорохин, H2-антигистаминные средства (H2-AH), метотрексат и циклофосфамид, характеризуются неблагоприятным профилем риска и пользы или значительным профилем побочных эффектов и больше не рекомендуются для терапии CSU (Kaplan AP (2002) Chronic urticaria--new concepts regarding pathogenesis and treatment. Curr Allergy Asthma Rep; 2(4):263-4 ; Powell RJ, Du Toit GL, Siddique N, et al (2007) BSACI guidelines for the management of chronic urticaria and angio-oedema. Clin Exp Allergy; 37(5):631-50; Zuberbier T, Aberer W, Asero R, et al (2014) The EAACI/GA(2) LEN/EDF/WAO Guideline for the definition, classification, diagnosis, and management of urticaria: the 2013 revision and update. Allergy; 69(7):868-87).
Омализумаб является одобренным терапевтическим средством для лечения CSU, рефрактерной к стандартному лечению, и демонстрирует благоприятный профиль польза-риск. Это рекомбинантное гуманизированное моноклональное антитело подкласса IgG1, которое связывается с IgE-специфическими эпитопами в пределах области C3 (FcεRI-связывающей) молекулы IgE, и его применение показано во многих странах для лечения плохо контролируемой умеренной или тяжелой астмы и CSU, рефрактерной к стандартной терапии. Завершенные исследования фазы 2 и фазы 3 демонстрируют, что омализумаб улучшает признаки и симптомы крапивницы (например, зуд, крапивную сыпь) у пациентов с CSU, для которых лечение с помощью H1-AH оказалось неэффективным, а также у тех, для которых лечение с помощью комбинации H1- и H2-AH и антагониста лейкотриеновых рецепторов оказалось неэффективным (Gober MD, Fishelevich R, Zhao Y, et al (2008) Human natural killer T cells infiltrate into the skin at elicitation sites of allergic contact dermatitis. J Invest Dermatol; 128(6):1460-9; Kaplan AP, Joseph K, Maykut RJ, et al (2008) Treatment of chronic autoimmune urticaria with omalizumab. J Allergy Clin Immunol; 122(3):569-73; Kaplan AP, Joseph K, Maykut RJ, et al (2008) Treatment of chronic autoimmune urticaria with omalizumab. J Allergy Clin Immunol; 122(3):569-73; Kaplan A, Ledford D, Ashby M, et al (2013) Omalizumab in patients with symptomatic chronic idiopathic/spontaneous urticaria despite standard combination therapy. J Allergy Clin Immunol; 132(1):101-9; Maurer M, Magerl M, Metz M, et al (2013) Revisions to the international guidelines on the diagnosis and therapy of chronic urticaria. J Dtsch Dermatol Ges). Опубликованные данные относительно результатов исследования фазы 3 омализумаба (Q4882g) продемонстрировали, что омализумаб безопасно улучшал клинические проявления CSU (например, зуд, крапивную сыпь) по сравнению с плацебо при ежемесячных дозах 150 или 300 мг дозозависимым образом, но не при дозе 75 мг (Kaplan A, Ledford D, Ashby M, et al (2013) Omalizumab in patients with symptomatic chronic idiopathic/spontaneous urticaria despite standard combination therapy. J Allergy Clin Immunol; 132(1):101-9; Maurer M, Magerl M, Metz M, et al (2013) Revisions to the international guidelines on the diagnosis and therapy of chronic urticaria. J Dtsch Dermatol Ges.). Основная конечная точка, представленная изменением среднего значения (±SD) по сравнению с исходным уровнем в ходе еженедельной оценки интенсивности зуда в неделю 12, составляла -5,1 ± 5,6 в группе, получавшей плацебо, -5,9 ± 6,5 в группе, получавшей 75 мг (P=0,46), -8,1 ± 6,4 в группе, получавшей 150 мг (P=0,001), и -9,8 ± 6,0 в группе, получавшей 300 мг (P <0,001), и все предварительно определенные второстепенные конечные точки (например, изменение относительно исходного уровня в UAS7, еженедельная оценка для количества крапивной сыпи, доля пациентов с UAS7 ≤ 6) характеризовались дозозависимостью. Точный механизм действия омализумаба у пациентов с CSU неизвестен.
QGE031 (лигелизумаб) представляет собой гуманизированное моноклональное антитело с более высокой аффинностью связывания с человеческим иммуноглобулином E (IgE), чем омализумаб. При связывании QGE031 способен блокировать взаимодействие IgE как с высокоаффинными, так и с низкоаффинными рецепторами IgE (FceRI и FceRII). QGE031 не способен опосредовать перекрестное связывание рецептора IgE и, следовательно, высвобождение гистамина (т. е. не является анафилактогенным). При получении пациентами QGE031 циркулирующий IgE быстро связывается антителом к IgE и становится недоступным для рецепторов IgE на тучных клетках и базофилах. IgE необходим для повышенной экспрессии FcεRI, наблюдаемой у пациентов с атопией, а снижение экспрессии FcεRI на циркулирующих базофилах сопровождает лечение QGE031. Другие потенциально благоприятные эффекты вследствие терапии с помощью антител к IgE включают снижение продуцирования IgE, уменьшение количества В-клеток и снижение продуцирования цитокинов Т-клетками.
QGE031 продемонстрировал дозозависимое и зависящее от времени подавление уровней свободного IgE, FcεRI на базофилах, IgE на поверхности базофилов и ответы кожных инъекционных проб на аллерген, превосходящие наблюдаемые при применении омализумаба в отношении степени и продолжительности реакции. Превосходные результаты в отношении аффинности и фармакодинамических свойств (PD), обеспечиваемые QGE031 по сравнению с омализумабом, могут обусловить превосходный режим введения доз и превосходную клиническую эффективность для пациентов с CSU (см. фигуру 2).
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
К настоящему времени авторы настоящего изобретения разработали схемы лечения пациентов с CSU с помощью лигелизумаба или его антигенсвязывающих фрагментов, которые характеризуются значительной эффективностью и обеспечивают устойчивый ответ для пациентов с CSU. Соответственно, в данном документе раскрыты способы лечения хронической спонтанной крапивницы (CSU), включающие подкожное (п/к) введение пациенту, нуждающемуся в этом, дозы от приблизительно 24 мг до приблизительно 240 мг антитела, представляющего собой лигелизумаб, или его антигенсвязывающего фрагмента в течение недели 0, а затем п/к в дозе от приблизительно 24 мг до приблизительно 240 мг ежемесячно (каждые 4 недели), начиная в течение недели 4;
при этом антитело, представляющее собой лигелизумаб, или его антигенсвязывающий фрагмент содержат
VH-домен иммуноглобулина, содержащий аминокислотную последовательность, представленную под SEQ ID NO: 62, и VL-домен иммуноглобулина, содержащий аминокислотную последовательность, представленную под SEQ ID NO:1.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фиг. 1 показан дизайн клиническое испытание лигелизумаба для лечения CSU.
На панелях фиг. 2 продемонстрированы полученные для отдельных пациентов данные относительно B2203 для различных групп лечения (получавших плацебо, 24, 72, 240 мг QGE031, 300 мг Xolair/омализумаб) и для различных биомаркеров и результатов кожных инъекционных проб. Пациенты с уровнем IgE> 250 МЕ/мл обозначены пунктирными линиями, соединяющими треугольные символы. Обозначения 'FceR1' и 'sIgE' представляют собой рецептор Fc-эпсилон типа 1 базофилов и поверхностно связанный IgE; единицы представляют собой молекулы эквивалентного растворимого флуорофора (MESF). 'Волдырь' относится к размеру индуцированных аллергеном волдырей при кожных инъекционных пробах; это суммарные данные по всем разведениям, нанесенные на квадратичную шкалу для корректного представления статистического распределения.
На фиг. 3 показаны прогнозируемые кривые доза-ответ для QGE031 применительно к связанному с появлением волдырей аспекту кожной инъекционной пробы. Было спрогнозировано, что доза 24 мг обеспечит 50-70% максимально возможного ответа. Диапазон ответов для средних 50% популяции пациентов варьируется от очень малого до высокого, но не максимального ответа. Доза 72 мг предположительно находилась вблизи перехода между линейной и насыщенной областью кривой доза-ответ, в то время как ожидалось, что 240 мг обеспечит максимальную эффективность. Следовательно, 24 мг является ‘субоптимальной дозой', которая, как ожидалось, будет находиться в том же диапазоне, что и омализумаб, нежели являться минимально эффективной дозой. 300 мг омализумаба q4w, по прогнозам, обеспечит достижение слегка более низкого ответа, чем 72 мг QGE031 q4w.
На фиг. 4 показан эффект многократных введений 24, 72 и 240 мг и однократной дозы 120 мг QGE031 в отношении изменения UAS7 относительно исходного уровня. По оси x указано время в неделях, по оси y указано изменение UAS7 относительно исходного уровня, а заштрихованные полосы представляют собой составляющие 80% доверительные интервалы. Можно увидеть, что в течение первых 4-6 недель не имеется значительной разницы между ответом пациентов на 72, 120 или 240 мг QGE031. Только доза 24 мг демонстрирует меньшую эффективность, но даже эта доза QGE031 более эффективна, чем контрольное средство лечения в виде плацебо.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Лигелизумаб, также известный как QGE031, в общем виде представляет собой тетрамерную молекулу Y-образной формы, содержащую антигенсвязывающий участок на конце каждого плеча. Антигенсвязывающий участок образован тремя определяющими комплементарность областями (CDR) вариабельной области тяжелой цепи (VH) и тремя CDR вариабельной области легкой цепи (VL). Как в VH, так и в VL CDR чередуются с четырьмя каркасными областями (FR), образуя полипептидную цепь общей формулы FR1-CDR1-FR2-CDR2-FR3-CDR3-FR4. Лигелизумаб описан в патенте США № 7531169 как Mab 2 (CL-2C) и определен под SEQ ID NO: 61 и 62, который включен в данный документ посредством ссылки во всей своей полноте.
Термин "содержащий" охватывает "включающий", а также "состоящий", например композиция, "содержащая" X, может состоять исключительно из X или может включать что-либо дополнительное, например X+Y.
Термин "приблизительно" по отношению к числовому значению х означает, например, +/-10%. В случае использования перед числовым диапазоном или перечнем чисел термин "приблизительно" применяется к каждому числу в серии, например, фразу "приблизительно 1-5" следует интерпретировать как "приблизительно 1 - приблизительно 5", или, например, фразу "приблизительно 1, 2, 3, 4" следует интерпретировать как "приблизительно 1, приблизительно 2, приблизительно 3, приблизительно 4 и т. д."
Слово "по сути" не исключает "полностью", например, композиция, которая "по сути не содержит" Y, может совсем не содержать Y. В случае необходимости слово "по сути" может быть опущено из определения настоящего изобретения.
Как указано в данном документе, термин "антитело" включает встречающиеся в природе и полные антитела. Встречающееся в природе "антитело" представляет собой гликопротеин, содержащий по меньшей мере две тяжелые (H) цепи и две легкие (L) цепи, соединенные между собой посредством дисульфидных связей. Каждая тяжелая цепь состоит из вариабельной области тяжелой цепи (сокращенно обозначаемой в данном документе как VH) и константной области тяжелой цепи. Константная область тяжелой цепи состоит из трех доменов: CH1, CH2 и CH3. Каждая легкая цепь состоит из вариабельной области легкой цепи (сокращенно обозначаемой в данном документе как VL) и константной области легкой цепи. Константная область легкой цепи состоит из одного домена, обозначаемого как CL. Области VH и VL можно дополнительно подразделить на области гипервариабельности, называемые гипервариабельными областями или определяющими комплементарность областями (CDR), которые чередуются с более консервативными областями, называемыми каркасными областями (FR). Каждая VH и VL состоит из трех CDR и четырех FR, расположенных от аминоконца к карбоксиконцу в следующем порядке: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. Вариабельные области тяжелой и легкой цепей содержат связывающий домен, который взаимодействует с антигеном. Константные области антител могут опосредовать связывание иммуноглобулина с тканями или факторами хозяина, в том числе с различными клетками иммунной системы (например, эффекторными клетками) и первым компонентом (C1q) классической системы комплемента. Иллюстративные антитела включают антитело, представляющее собой лигелизумаб (патент США № 7531169), раскрытие которого полностью включено в настоящий документ посредством ссылки.
Используемый в данном документе термин "антигенсвязывающий фрагмент" антитела относится к фрагментам антитела, которые сохраняют способность специфически связываться с антигеном (например, IgE). Было показано, что антигенсвязывающую функцию антитела могут осуществлять фрагменты полноразмерного антитела. Примеры связывающих фрагментов, охватываемых термином "антигенсвязывающая часть" антитела, включают Fab-фрагмент, моновалентный фрагмент, состоящий из доменов VL, VH, CL и CH1; F(ab)2-фрагмент, бивалентный фрагмент, содержащий два Fab-фрагмента, связанные дисульфидным мостиком в шарнирной области; Fd-фрагмент, состоящий из доменов VH и CH1; Fv-фрагмент, состоящий из доменов VL и VH одного плеча антитела; dAb-фрагмент (Ward et al., 1989 Nature 341:544-546), который состоит из VH-домена; и выделенную CDR. Иллюстративные антигенсвязывающие фрагменты включают CDR лигелизумаба, содержащего вариабельную область легкой цепи, содержащую CDRL1, CDRL2 и CDRL3, и вариабельную область тяжелой цепи, содержащую CDRH1, CDRH2 и CDRH3, при этом CDRL1 состоит из SEQ ID NO:3, CDRL2 состоит из SEQ ID NO:4, CDRL3 состоит из SEQ ID NO:5, CDRH1 состоит из SEQ ID NO:6, CDRH2 состоит из SEQ ID NO:7, и CDRH3 состоит из SEQ ID NO:8, при этом антитело специфически связывается с IgE.
Кроме того, хотя два домена Fv-фрагмента VL и VH кодируются отдельными генами, они могут быть объединены с применением рекомбинантных способов с помощью синтетического линкера, который обеспечивает возможность их получения в виде единой белковой цепи, в которой области VL и VH объединяются в пару с образованием моновалентных молекул (известных как одноцепочечные Fv (scFv); см., например, Bird et al., 1988 Science 242:423-426 и Huston et al., 1988 Proc. Natl. Acad. Sci. 85:5879-5883). Предусматривается, что такие одноцепочечные антитела также охватываются термином "антитело". Одноцепочечные антитела и антигенсвязывающие части получают с применением обычных методик, известных специалистам в данной области.
Как используется в данном документе, "выделенное антитело" относится к антителу, которое по сути не содержит других антител, характеризующихся отличающимися антигенными специфичностями (например, выделенное антитело, которое специфически связывает IgE, по сути не содержит других антител, которые специфически связывают другие антигены, отличные от IgE). Используемые в данном документе термины "моноклональное антитело" или "композиция на основе моноклональных антител" относятся к препарату из молекул антитела одного молекулярного состава. Подразумевается, что термин "человеческое антитело", используемый в данном документе, включает антитела, имеющие вариабельные области, в которых как каркасные, так и CDR-области получены из последовательностей, происходящих из человека. "Человеческое антитело" не обязательно должно вырабатываться человеком, тканью человека или клеткой человека. Человеческие антитела по настоящему изобретению могут содержать аминокислотные остатки, не кодируемые человеческими последовательностями (например, мутации, введенные с помощью случайного или сайт-специфического мутагенеза in vitro, посредством добавления N-нуклеотидов в участках соединения in vivo во время рекомбинации генов антител или посредством соматической мутации in vivo). В некоторых вариантах осуществления раскрытых способов и композиций антитело к IgE представляет собой человеческое антитело, выделенное антитело и/или моноклональное антитело.
Используемый в данном документе термин "антитело к IgE человека" означает антитело, которое связывается с IgE человека таким образом, что обеспечивается подавление или существенное снижение связывания такого IgE с высокоаффинным рецептором Fc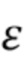 RI.
RI.
Термин "аффинность" относится к силе взаимодействия между антителом и антигеном в отдельных антигенных сайтах. В пределах каждого антигенного сайта вариабельная область "плеча" антитела взаимодействует с антигеном посредством слабых нековалентных сил во многих сайтах; при этом чем больше взаимодействий, тем сильнее аффинность. Стандартные анализы для оценки аффинности связывания антител по отношению к IgE различных видов известны из уровня техники, включая, например, разновидности ELISA, вестерн-блоттинга и RIA. Кинетику связывания (например, аффинность связывания) антител также можно оценить с помощью анализов, известных из уровня техники, например, KD можно определить с использованием анализа Biacore®.
Антитело, которое "подавляет" одно или несколько функциональных свойств IgE (например, биохимическую, иммунохимическую, клеточную, физиологическую или другие формы биологической активности или т. п.), как определено в соответствии с методологиями, известными из уровня техники и описанными в данном документе, понимается как связанное со статистически значимым уменьшением уровня конкретной активности по сравнению с уровнем, который наблюдается в отсутствие антитела (или в случае присутствия контрольного антитела нерелевантной специфичности). Антитело, которое подавляет активность IgE, приводит к статистически значимому снижению величины измеряемого параметра, например, на по меньшей мере приблизительно 10%, на по меньшей мере 50%, 80% или 90%, и в определенных вариантах осуществления раскрытых способов и композиций используемое антитело к IgE может подавлять функциональную активность IgE более чем на 95%, 98% или 99%.
Если не указано иное, термин "производное" используется для определения вариантов аминокислотной последовательности и ковалентных модификаций (например, пегилирования, дезамидирования, гидроксилирования, фосфорилирования, метилирования и т. д.) антитела к IgE или его антигенсвязывающего фрагмента, например лигелизумаба, в соответствии с настоящим изобретением, например, указанной последовательности (например, вариабельного домена). "Функциональное производное" включает молекулу, характеризующуюся качественной биологической активностью, общей с раскрытыми антителами к IgE. Функциональное производное включает фрагменты и пептидные аналоги антитела к IgE, как раскрыто в данном документе. Фрагменты включают области в пределах последовательности полипептида в соответствии с настоящим изобретением, например указанной последовательности. Функциональные производные антител к IgE, раскрытые в данном документе (например, функциональные производные лигелизумаба), предпочтительно содержат домены VH и/или VL, которые характеризуются по меньшей мере приблизительно 65%, 75%, 85%, 95%, 96%, 97% 98% или даже 99% общей идентичностью последовательности с последовательностями VH и/или VL антител к IGE и их антигенсвязывающих фрагментов, раскрытых в данном документе, и по сути сохраняют способность связывать IgE человека.
Фраза "по сути идентичная" означает, что соответствующая аминокислотная или нуклеотидная последовательность (например, VH- или VL-домена) будет являться идентичной или характеризоваться несущественными различиями (например, представленными консервативными заменами аминокислот) по сравнению с конкретной эталонной последовательностью. Несущественные различия включают незначительные аминокислотные изменения, такие как 1 или 2 замены в 5-аминокислотной последовательности указанной области (например, VH- или VL-домена). В случае антител второе антитело обладает такой же специфичностью и характеризуется по меньшей мере 50% такой же аффинности. Последовательности, по сути идентичные (например, характеризующиеся по меньшей мере приблизительно 85% идентичностью последовательности) последовательностям, раскрытым в данном документе, также являются частью настоящей заявки. В некоторых вариантах осуществления идентичность последовательности производного антитела к IgE (например, производного лигелизумаба, например, антитела-биоаналога лигелизумаба) относительно раскрытых последовательностей может составлять приблизительно 90% или больше, например, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% или больше.
"Идентичность" в отношении нативного полипептида и его функционального производного определяется в данном документе как процентная доля аминокислотных остатков в кандидатной последовательности, которые идентичны остаткам соответствующего нативного полипептида после выравнивания последовательностей и, при необходимости, введения гэпов для достижения максимальной процентной идентичности, и при этом не учитывая любые консервативные замены как часть идентичности последовательностей. Ни N- или C-концевые удлинения, ни вставки не должны рассматриваться как уменьшающие идентичность. Известны способы и компьютерные программы для выравнивания. Процент идентичности можно определить с помощью стандартных алгоритмов выравнивания, например, с помощью средства поиска основного локального выравнивания (BLAST), описанного Altshul et al. ((1990) J. Mol. Biol., 215: 403 410); алгоритма Needleman et al. ((1970) J. Mol. Biol., 48: 444 453) или алгоритма Meyers et al. ((1988) Comput. Appl. Biosci., 4: 11 17). Набором параметров может служить весовая матрица Blossum 62 со штрафом за гэп 12, штрафом за продление гэпа 4 и штрафом за сдвиг рамки гэпа 5. Процент идентичности между двумя аминокислотными или нуклеотидными последовательностями также можно определить с помощью алгоритма E. Meyers и W. Miller ((1989) CABIOS, 4:11-17), который был включен в программу ALIGN (версии 2.0), с использованием таблицы весов замен остатков PAM120, штрафа за удлинение гэпа 12 и штрафа за гэп 4.
Термин "аминокислота(-ы)" относится, например, ко всем встречающимся в природе L-α-аминокислотам и включает D-аминокислоты. Фраза "вариант аминокислотной последовательности" относится к молекулам с некоторыми отличиями в их аминокислотных последовательностях по сравнению с последовательностями в соответствии с настоящим изобретением. Варианты аминокислотной последовательности антитела в соответствии с настоящим изобретением, например, указанной последовательности, по-прежнему обладают способностью связывать IgE человека. Варианты аминокислотной последовательности включают варианты с заменой (варианты, в которых был удален по меньшей мере один аминокислотный остаток и другая аминокислота вставлена на его место в то же положение в полипептиде в соответствии с настоящим изобретением), варианты со вставкой (варианты с одной или несколькими аминокислотами, вставленными в непосредственной близости от аминокислоты в определенном положении в полипептиде в соответствии с настоящим изобретением) и варианты с делецией (варианты, в которых одна или несколько аминокислот удалены в полипептиде в соответствии с настоящим изобретением).
Термин "фармацевтически приемлемый" означает нетоксичный материал, который не влияет на эффективность биологической активности активного(-ых) ингредиента(-ов).
Термин "введение" применительно к соединению, например молекуле, связывающей IgE, или другому средству используется для обозначения доставки этого соединения пациенту любым путем.
Используемый в данном документе термин "терапевтически эффективное количество" относится к количеству антагониста IgE, например молекулы, связывающей IgE (например, антитела к IgE или его антигенсвязывающего фрагмента, например лигелизумаба), или молекулы, связывающей рецептор IgE (например, антитела к рецептору IgE или его антигенсвязывающего фрагмента), которое при введении в виде одной или нескольких доз пациенту (такому как человек) является эффективным для лечения, предупреждения (при необходимости), предотвращения проявления (при необходимости), излечения, отсрочки, уменьшения тяжести, снижения выраженности по меньшей мере одного симптома нарушения или рецидивирующего нарушения или продления выживаемости пациента сверх ожидаемой в отсутствие такого лечения. В случае его применения в отношении индивидуального активного ингредиента (например, антагониста IgE, например лигелизумаба), который вводится отдельно, данный термин относится только к этому ингредиенту. В случае использования по отношению к комбинации, термин относится к объединенным количествам активных ингредиентов, которые приводят в результате к терапевтическому эффекту, независимо от того, вводят ли их в комбинации, последовательно или одновременно.
Термин "лечение" или "лечить" в данном документе определяется как применение или введение антитела к IgE в соответствии с настоящим изобретением, например лигелизумаба, или фармацевтической композиции, содержащей указанное антитело к IgE, осуществляемые применительно к субъекту или выделенной ткани или линии клеток от субъекта, где у субъекта имеется конкретное заболевание (например, CSU), симптом, ассоциированный с заболеванием (например, CSU), или предрасположенность к развитию заболевания (например, CSU) (если применимо), при этом целью является излечение (если применимо), задержка проявления, уменьшение тяжести, облегчение, снижение выраженности одного или нескольких симптомов заболевания, улучшение течения заболевания, уменьшение или нормализация любых ассоциированных симптомов заболевания или предрасположенности к развитию заболевания. Термин "лечение" или "лечить" включает лечение пациента с подозрением на наличие заболевания, а также больных пациентов и пациентов, которые были диагностированы, как страдающие заболеванием или медицинским состоянием, и включает подавление клинического рецидива.
Используемая в данном документе фраза "популяция пациентов" означает группу пациентов. В некоторых вариантах осуществления раскрытых способов антагонист IgE (например, антитело к IgE, такое как лигелизумаб) используют для лечения популяции пациентов с CSU.
Используемые в данном документе фразы "ранее не получавший лечение с помощью системного средства лечения CSU" и "не подвергавшийся лечению" относятся к пациенту с CSU, который ранее не получал лечение с помощью системного средства, например, циклоспорина А, монтелукаста, H1-антигистаминных средств (H1-AH), H2-AH и антагониста лейкотриеновых рецепторов (LTRA), биологического средства (например, омализумаба) и т. д., для CSU. Системные средства (т. е. средства, вводимые перорально, с помощью инъекции и т. д.) отличаются от локально действующих средств (например, местных средств и фототерапии) тем, что системные средства обладают системным (охватывающим весь организм) эффектом при доставке пациенту. В некоторых вариантах осуществления раскрытых способов, схем, вариантов применения, наборов и фармацевтических композиций пациенту ранее не вводили системное средство лечения CSU.
Используемая в данном документе фраза "ранее получавший лечение с помощью системного средства для лечения CSU" используется для обозначения пациента, который ранее подвергался лечению CSU с использованием системного средства. Такие пациенты включают пациентов, ранее получавших лечение с помощью биологических средств, таких как омализумаб, и пациентов, ранее получавших лечение с помощью небиологических средств, таких как циклоспорин. В некоторых вариантах осуществления настоящего изобретения пациенту ранее вводили системное средство для лечения CSU. В некоторых вариантах осуществления пациенту ранее вводили системное средство для лечения CSU (например, циклоспорин), однако при этом пациенту ранее не вводили системное биологическое лекарственное средство (т. е. лекарственное средство, продуцируемое живым организмом, например, антитела, рецепторы-ловушки и т. д.) для лечения CSU (например, омализумаб). В данном случае пациента называют "не получавшим лечение с помощью биологического средства". В некоторых вариантах осуществления пациент является не получавшим лечение с помощью биологического средства.
Как используется в данном документе, термины "осуществление отбора" и "отобранный", используемые в отношении пациента, означают, что конкретный пациент специально выбран из большей группы пациентов на основании того (вследствие того), что конкретный пациент отвечает заранее определенным критериям. Подобным образом, "селективное лечение" относится к обеспечению лечением пациента, имеющего конкретное заболевание, при этом данный пациент специально выбран из большей группы пациентов на основании того, что конкретный пациент отвечает заранее определенному критерию. Подобным образом, "селективное введение" относится к введению лекарственного средства пациенту, который специально выбран из большей группы пациентов на основании того (вследствие того), что конкретный пациент отвечает заранее определенному критерию. Под осуществлением отбора, селективным лечением и селективным введением подразумевается, что пациенту предоставляется персонализированная терапия, основанная на личном анамнезе пациента (например, наличии предыдущих терапевтических вмешательств, например, предыдущего лечения с помощью биологических препаратов), биологических особенностях (например, конкретных генетических маркерах) и/или проявлении (например, несоответствии определенным диагностическим критериям), вместо применения стандартной схемы лечения, основанной исключительно на принадлежности пациента к большей группе. Отбор, применительно к способу лечения в контексте данного документа, не относится к случайному лечению пациента, отвечающего конкретному критерию, но скорее относится ко взвешенному решению, принимаемому в отношении введения лекарственного средства пациенту на основании того, что пациент отвечает конкретному критерию. Таким образом, селективное лечение/введение отличается от стандартного лечения/введения, в ходе которого обеспечивается доставка конкретного лекарственного средства всем пациентам, имеющим конкретное заболевание, независимо от их личного анамнеза, проявлений заболевания и/или биологических особенностей. В некоторых вариантах осуществления пациент был выбран для лечения на основании наличия у него CSU.
Антагонисты IgE
В различных раскрытых процедурах, наборах, вариантах применения и способах используют антагонист IgE, например молекулу, связывающую IgE (например, растворимый рецептор IgE, антитело к IgE или его антигенсвязывающий фрагмент, например лигелизумаб), или молекулу, связывающую рецептор IgE (например, антитело к рецептору IgE или его антигенсвязывающий фрагмент). В некоторых вариантах осуществления антагонист IgE представляет собой молекулу, связывающую IgE, предпочтительно антитело к IgE или его антигенсвязывающий фрагмент.
В одном варианте осуществления антитело к IgE или его антигенсвязывающий фрагмент содержат VH-домен иммуноглобулина, содержащий аминокислотную последовательность, представленную под SEQ ID NO:2, и VL-домен иммуноглобулина, содержащий аминокислотную последовательность, представленную под SEQ ID NO:1.
В одном варианте осуществления антитело к IgE или его антигенсвязывающий фрагмент содержат вариабельную область легкой цепи, содержащую CDRL1, CDRL2 и CDRL3, и вариабельную область тяжелой цепи, содержащую CDRH1, CDRH2 и CDRH3, при этом CDRL1 состоит из SEQ ID NO:3, CDRL2 состоит из SEQ ID NO:4, CDRL3 состоит из SEQ ID NO:5, CDRH1 состоит из SEQ ID NO:6, CDRH2 состоит из SEQ ID NO:7, а CDRH3 состоит из SEQ ID NO:8, при этом антитело специфически связывается с IgE.
В качестве альтернативы, антитело к IgE или его антигенсвязывающий фрагмент, используемые в раскрытых способах, могут предусматривать производное антител к IgE, приведенных в данном документе в виде последовательности (например, пегилированные варианты, варианты гликозилирования, варианты созревания аффинности и т. д.). В качестве альтернативы, домены VH или VL антитела к IgE или его антигенсвязывающего фрагмента, используемых в раскрытых способах, могут предусматривать домены VH или VL, которые по сути идентичны доменам VH или VL, приведенным в данном документе (например, приведенным под SEQ ID NO:2 и 61). Антитело к IgE человека, раскрытое в данном документе, может содержать тяжелую цепь, которая по сути идентична цепи, которая приведена под SEQ ID NO:2, и/или легкую цепь, которая по сути идентична цепи, которая приведена под SEQ ID NO:1. Антитело к IgE человека, раскрытое в данном документе, может содержать тяжелую цепь, которая содержит SEQ ID NO:2, и легкую цепь, которая содержит SEQ ID NO:1.
Предпочтительными антителами к IgE или их антигенсвязывающими фрагментами, используемыми в раскрытых способах, являются человеческие антитела, в частности лигелизумаб, описанный в таблице 2 примера 10 в патенте США № 7531169, который включен в данный документ посредством ссылки во всей своей полноте.
Способы лечения и варианты применения антагонистов IgE при CSU
Раскрытые антагонисты IgE, например молекулы, связывающие IgE (например, антитело к IgE или его антигенсвязывающий фрагмент, например лигелизумаб), или молекулы, связывающие рецептор IgE (например, антитело к рецептору IgE или его антигенсвязывающий фрагмент), можно применять in vitro, ex vivo или включать в фармацевтические композиции и вводить in vivo для лечения пациентов с CSU (например, пациентов-людей).
Крапивница представляет собой гетерогенную группу заболеваний, характеризующихся зудящей крапивной сыпью и/или ангионевротическим отеком.
Хроническая крапивница определяется как крапивница, которая постоянно или периодически присутствует в течение более 6 недель (Maurer, et al 2013, Bernstein, et al 2014). Хроническую крапивницу далее дополнительно делят на две подгруппы: хроническую спонтанную крапивницу (CSU) и индуцибельную крапивницу (IU), при этом последняя включает физическую крапивницу, как например тепловая, холодовая или крапивница вследствие давления, а также особые варианты, как например холинергическая крапивница. CSU определяется как спонтанное появление зудящих волдырей, ангионевротического отека или и того и другого в течение ≥ 6 недель по известным или неизвестным причинам (Zuberbier, et al 2014). Возможна комбинация как CSU, так и индуцибельной формы крапивницы, как например часто наблюдаемая комбинация бессимптомной дерматографической крапивницы и CSU.
Эффективность лечения CSU можно оценивать с использованием различных известных способов и инструментов, которые измеряют состояние заболевания CSU и/или клинический ответ при CSU. Некоторые примеры включают, например, еженедельную оценку тяжести крапивной сыпи (HSS7), еженедельную оценку интенсивности зуда (ISS7) и еженедельную оценку активности крапивницы (UAS7).
В некоторых вариантах осуществления пациента лечат в отношении CSU в соответствии с заявленными способами в течение по меньшей мере 20 недель, по меньшей мере 48 недель, по меньшей мере 52 недель или по меньшей мере 2 лет.
В некоторых вариантах осуществления у пациента ранее наблюдался недостаточный ответ на традиционную системную терапию в отношении CSU.
В некоторых вариантах осуществления пациент является пациентом-подростком (возрастом > 12 лет), имеющим CSU от средней до тяжелой степени. В некоторых вариантах осуществления пациент является взрослым пациентом, имеющим CSU от средней до тяжелой степени.
В некоторых вариантах осуществления в ответ на лечение в соответствии с заявленным способами пациент испытывает быстрое уменьшение крапивной сыпи, как измерено с помощью оценки HSS7, уже через 1 неделю после введения начальной дозы.
Антагонисты IgE, например молекулы, связывающие IgE (например, антитело к IgE или его антигенсвязывающий фрагмент, например лигелизумаб), или молекулы, связывающие рецептор IgE (например, антитело к рецептору IgE или его антигенсвязывающий фрагмент), можно использовать в качестве фармацевтической композиции при их объединении с фармацевтически приемлемым носителем. В дополнение к антагонисту IgE такая композиция может содержать носители, различные разбавители, наполнители, соли, буферы, стабилизаторы, солюбилизаторы и другие материалы, известные из уровня техники. Характеристики носителя будут зависеть от пути введения. Фармацевтические композиции для применения в раскрытых способах также могут содержать дополнительные терапевтические средства для лечения конкретного целевого нарушения. Например, фармацевтическая композиция может также включать противовоспалительные и противозудные средства. Такие дополнительные факторы и/или средства можно включать в фармацевтическую композицию для обеспечения синергетического эффекта совместно с молекулами, связывающими IgE, или для сведения к минимуму побочных эффектов, вызываемых антагонистами IgE, например молекулами, связывающими IgE (например, антителом к IgE или его антигенсвязывающим фрагментом, например лигелизумабом), или молекулами, связывающими рецептор IgE (например, антителом к рецептору IgE или его антигенсвязывающим фрагментом). В предпочтительных вариантах осуществления фармацевтические композиции для применения в раскрытых способах включают лигелизумаб при 120 мг/мл.
Фармацевтические композиции для применения в раскрытых способах можно изготавливать обычным способом. В одном варианте осуществления фармацевтическая композиция предусмотрена в лиофилизированной форме. Для немедленного введения ее растворяют в подходящем водном носителе, например, в стерильной воде для инъекций или в стерильном забуференном физиологическом растворе. Если считается желательным получить раствор большего объема для введения с помощью инфузии, а не в виде болюсной инъекции, то может быть преимущественным включение человеческого сывороточного альбумина или собственной гепаринизированной крови пациента в физиологический раствор во время составления. Наличие избытка такого физиологически инертного белка предотвращает потерю антител вследствие адсорбции на стенках контейнера и системы трубок, используемых с инфузионным раствором. Если используется альбумин, то подходящая концентрация составляет от 0,5 до 4,5% по весу физиологического раствора. Другие составы предусматривают готовые к применению жидкие составы, которые можно поместить, например, во флакон, шприц, автоинжектор и т. д.
Антитела, например антитела к IgE, обычно составляют либо в водной форме, готовой для парентерального введения, либо в виде лиофилизатов для разведения подходящим разбавителем перед введением. В предпочтительных вариантах осуществления раскрытых способов и вариантов применения антагонист IgE, например антитело к IgE, например лигелизумаб, составляют в виде готового к применению жидкого фармацевтического состава. В некоторых вариантах осуществления раскрытых способов и вариантов применения антагонист IgE, например антитело к IgE, например лигелизумаб, составляют в виде лиофилизата. Подходящие лиофилизированные составы можно восстанавливать в небольшом объеме жидкости (например, 2 мл или меньше, например, 2 мл, 1 мл и т. д.), дающем возможность осуществлять подкожное введение, и они могут обеспечивать растворы с низкими уровнями агрегации антител. Применение антител в качестве активного ингредиента фармацевтических препаратов в настоящее время широко распространено, в том числе продукты HERCEPTIN™ (трастузумаб), RITUXAN™ (ритуксимаб), SYNAGIS™ (паливизумаб) и т. д. Методики очистки антител до фармацевтической степени чистоты хорошо известны из уровня техники. В случае если терапевтически эффективное количество антагониста IgE, например молекул, связывающих IgE (например, антитела к IgE или его антигенсвязывающего фрагмента, например лигелизумаба), или молекул, связывающих рецептор IgE (например, антитела к рецептору IgE или его антигенсвязывающего фрагмента), вводят с помощью внутривенной, кожной или подкожной инъекции, антагонист IgE будет представлен в форме апирогенного парентерально приемлемого раствора. Фармацевтическая композиция для внутривенного, кожного или подкожного введения в дополнение к антагонисту IgE может содержать изотоническую среду-носитель, такую как хлорид натрия, раствор Рингера, декстрозу, декстрозу и хлорид натрия, лактированный раствор Рингера или другую среду-носитель, как известно из уровня техники.
При практической реализации некоторых способов лечения или вариантов применения по настоящему изобретению терапевтически эффективное количество антагониста IgE, например молекулы, связывающей IgE (например, антитела к IgE или его антигенсвязывающего фрагмента, например лигелизумаба), или молекулы, связывающей рецептор IgE (например, антитела к рецептору IgE или его антигенсвязывающего фрагмента), вводят пациенту, например млекопитающему (например, человеку). Хотя понятно, что раскрытые способы предусмотрены для лечения пациентов с CSU с использованием антагониста IgE (например, лигелизумаба), это не означает того, что если пациент в конечном итоге будет получать лечение с помощью антагониста IgE, то такая терапия с помощью антагониста IgE обязательно должна являться монотерапией. Действительно, если пациент выбран для лечения с помощью антагониста IgE, то антагонист IgE (например, лигелизумаб) можно вводить в соответствии со способами по настоящему изобретению либо отдельно, либо в комбинации с другими средствами и терапевтическими средствами для лечения пациентов с CSU, например в комбинации с по меньшей мере одним дополнительным средством против CSU. При совместном введении с одним или несколькими дополнительными средствами против CSU антагонист IgE можно вводить либо одновременно с другим средством, либо последовательно. При последовательном введении лечащий врач примет решение относительно соответствующей последовательности введения антагониста IgE в комбинации с другими средствами и соответствующих дозировок для совместной доставки.
В ходе лечения CSU различные терапевтические средства могут быть объединены с раскрытыми антителами к IgE, такими как лигелизумаб, с обеспечением благоприятного эффекта. Такие терапевтические средства включают местные средства для лечения (кремы [нестероидные или стероидные], жидкости для промывания, антисептики), системные средства для лечения (например, с использованием биологических препаратов, антибиотиков или химических веществ) и антисептики, фотодинамическую терапию и хирургическое вмешательство (воздействие лазером, дренирование или разрезание, иссечение).
Неограничивающие примеры местных средств против CSU для применения с раскрытыми антителами к IgE, такими как лигелизумаб, включают бензоилпероксид, местные стероидные кремы, местные антибиотики аминогликозидной группы, такие как клиндамицин, гентамицин и эритромицин, резорциновый крем, скрабы с йодом и хлоргексидин.
Неограничивающие примеры средств для CSU, используемых в системном лечении, предусматриваемых для применения с раскрытыми антителами к IgE, такими как лигелизумаб, включают дополнительные антагонисты IgE (омализумаб).
Дополнительные средства против CSU, предусматриваемые для применения в комбинации с раскрытыми антителами к IgE, такими как лигелизумаб, во время лечения CSU, включают циклоспорин и кортикостероиды (инъекционные или пероральные).
Специалист в данной области сможет определить соответствующие дозы вышеуказанных средств против CSU для совместной доставки с раскрытым антителом к IgE, таким как лигелизумаб.
Антагонист IgE, например молекулу, связывающую IgE (например, антитело к IgE или его антигенсвязывающий фрагмент, например лигелизумаб), или молекулу, связывающую рецептор IgE (например, антитело к рецептору IgE или его антигенсвязывающий фрагмент), удобно вводить парентерально, например, внутривенно (например, в латеральную подкожную вену руки или другую периферическую вену), внутримышечно или подкожно. Продолжительность внутривенной (в/в) терапии с использованием фармацевтической композиции по настоящему изобретению будет варьировать в зависимости от тяжести заболевания, подлежащего лечению, а также состояния и индивидуального ответа каждого отдельного пациента. Также рассматривается подкожная (п/к) терапия с использованием фармацевтической композиции по настоящему изобретению. Медицинский работник примет решение о соответствующей продолжительности внутривенной или подкожной терапии и времени введения терапевтического средства с использованием фармацевтической композиции по настоящему изобретению. В предпочтительных вариантах осуществления антагонист IgE (например, лигелизумаб) вводят посредством подкожного (п/к) пути.
Антагонист IgE, например молекулу, связывающую IgE (например, антитело к IgE или его антигенсвязывающий фрагмент, например лигелизумаб), или молекулу, связывающую рецептор IgE (например, антитело к рецептору IgE или его антигенсвязывающий фрагмент), можно вводить пациенту подкожно (п/к) каждые четыре недели, начиная с недели 0 и 4, а затем вводить пациенту п/к, например, в дозе от приблизительно 24 мг до приблизительно 240 мг (например, приблизительно 24 мг, приблизительно 240 мг) каждые четыре недели, начиная в течение недели 4. Таким образом, пациент получает п/к дозу в течение 0, 4, 8, 12, 16 недель и т. д.
В качестве альтернативы, антагонист IgE, например молекулу, связывающую IgE (например, антитело к IgE или его антигенсвязывающий фрагмент, например лигелизумаб), или молекулу, связывающую рецептор IgE (например, антитело к рецептору IgE или его антигенсвязывающий фрагмент), можно вводить пациенту подкожно (п/к) каждые четыре недели, начиная с недели 0, а затем вводить пациенту п/к, например, в дозе приблизительно 24 мг, приблизительно 72 мг, приблизительно 120 мг, до приблизительно 240 мг (например, приблизительно 24 мг, приблизительно 240 мг) каждые четыре недели, начиная в течение недели 4. Таким образом, пациенту вводят п/к дозу приблизительно 24 мг, приблизительно 72 мг, от приблизительно 120 мг до приблизительно 240 мг (например, приблизительно 24 мг, приблизительно 72 мг, от приблизительно 120 мг до приблизительно 240 мг) антагониста IgE (например, лигелизумаба) в течение недель 0, 4, 8, 12 и т. д.
Предпочтительно антагонист IgE, например молекулу, связывающую IgE (например, антитело к IgE или его антигенсвязывающий фрагмент, например лигелизумаб), или молекулу, связывающую рецептор IgE (например, антитело к рецептору IgE или его антигенсвязывающий фрагмент), можно вводить пациенту подкожно (п/к) каждые четыре недели, начиная с недели 0, а затем вводить пациенту п/к, например, в дозе приблизительно 24 мг, приблизительно 72 мг, от приблизительно 120 мг до приблизительно 240 мг (например, приблизительно 24 мг, приблизительно 72 мг, от приблизительно 120 мг до приблизительно 240 мг) каждые четыре недели, начиная в течение недели 4. Таким образом, пациенту вводят п/к дозу приблизительно 24 мг, приблизительно 72 мг, от приблизительно 120 мг до приблизительно 240 мг (например, приблизительно 24 мг, приблизительно 72 мг, от приблизительно 120 мг до приблизительно 240 мг) антагониста IgE (например, лигелизумаба) в течение недель 0, 4, 8, 12, 16, 20 и т. д.
Более предпочтительно антагонист IgE, например молекулу, связывающую IgE (например, антитело к IgE или его антигенсвязывающий фрагмент, например лигелизумаб), или молекулу, связывающую рецептор IgE (например, антитело к рецептору IgE или его антигенсвязывающий фрагмент), можно вводить пациенту без использования нагрузочной схемы, например, антагонист можно вводить пациенту п/к в дозе приблизительно 24 мг, приблизительно 72 мг, от приблизительно 120 мг до приблизительно 240 мг (например, приблизительно 24 мг, приблизительно 72 мг, от приблизительно 120 мг до приблизительно 240 мг) каждые четыре недели. Таким образом, пациенту вводят п/к дозу приблизительно 24 мг, приблизительно 72 мг, от приблизительно 120 мг до приблизительно 240 мг (например, приблизительно 24 мг, приблизительно 72 мг, от приблизительно 120 мг до приблизительно 240 мг) антагониста IgE (например, лигелизумаба) в течение недель 0, 4, 8, 12 и т. д.
Следует учитывать, что для некоторых пациентов может потребоваться повышение дозы, например для пациентов с CSU, у которых наблюдается недостаточный ответ (например, как измерено с помощью любой из систем оценки CSU, раскрытых в данном документе, например, способ по любому из предыдущих пунктов, где у указанного пациента достигается устойчивый ответ, как измерено с помощью полного ответа в отношении крапивной сыпи (оценка тяжести крапивной сыпи [HSS7]), UAS7 и дерматологического индекса качества жизни (DLQI) и т. д.) на лечение с помощью антагонистов IgE, например молекул, связывающих IgE (например, антитела к IgE или его антигенсвязывающего фрагмента, например лигелизумаба), или молекул, связывающих рецептор IgE (например, антитела к рецептору IgE или его антигенсвязывающего фрагмента), к неделе 12, неделе 16, неделе 20, неделе 24, неделе 48 или неделе 52 лечения. Также следует понимать, что снижение дозы также может потребоваться для некоторых пациентов, например пациентов с CSU, у которых проявляются нежелательные явления или нежелательная реакция на лечение с помощью антагониста IgE (например, антитела к IgE или его антигенсвязывающего фрагмента, например лигелизумаба). Таким образом, дозы антагониста IgE (например, антитела к IgE или его антигенсвязывающего фрагмента, например лигелизумаба) могут составлять менее чем приблизительно 24 мг, приблизительно 72 мг, от приблизительно 120 мг до приблизительно 240 мг п/к. В некоторых вариантах осуществления антагонист IgE, например молекулу, связывающую IgE (например, антитело к IgE или его антигенсвязывающий фрагмент, например лигелизумаб), или молекулу, связывающую рецептор IgE (например, антитело к рецептору IgE или его антигенсвязывающий фрагмент), можно вводить пациенту в начальной дозе приблизительно 24 мг, приблизительно 72 мг, от приблизительно 120 мг до приблизительно 240 мг, доставляемой п/к, и затем дозу повышают до приблизительно 72 мг (в случае исходной дозы 24 мг) или приблизительно 240 мг (в случае исходной дозы 120 мг), если необходимо, как определено врачом.
Время введения доз, как правило, измеряют со дня введения первой дозы лигелизумаба (который также известен как "исходный уровень"). Однако медицинские работники зачастую используют разные способы наименования для идентификации графиков введения доз, как показано в таблице 1.
Таблица 1. Общепринятые способы наименования для схем введения доз Числа, выделенные полужирным шрифтом, относятся к способу наименования, используемому в данном документе.
Примечательно, что неделя ноль может некоторыми лечащими врачами называться неделей один, тогда как день ноль может некоторыми лечащими врачами называться днем один. Таким образом, возможно, что разные врачи обозначат, например, дозу, подлежащую введению в течение недели 4/в день 28, в течение недели 4/в день 29, в течение недели 4/в день 28, в течение недели 4/в день 29, ссылаясь при этом на тот же самый режим введения доз. В целях обеспечения соответствия первая неделя введения доз будет называться в данном документе как неделя 0, при этом первый день введения доз будет называться как день 1. Однако специалисту в данной области будет понятно, что этот способ наименования используется просто для обеспечения соответствия и не должен рассматриваться как ограничивающий, т. е. еженедельное введение доз представляет собой обеспечение еженедельной дозы антитела к IgE, вне зависимости от того, ссылается ли врач на конкретную неделю как на "неделю 0" или "неделю 1".
Согласно одной схеме введения доз антитело вводят в течение недели 0, 4, 8, 12, 16, 20 и т. д. Некоторые поставщики медицинских услуг могут называть эту схему ежемесячным введение дозы (или введением дозы каждые 4 недели). Специалист в данной области поймет, что введение пациенту инъекции в неделю 0 с последующим ежемесячным введением дозы, начиная с недели 4, представляет собой то же самое, что и 1) введение пациенту инъекции в недели 0 и 4 с последующим введением доз один раз в месяц, начиная с недели 8; 2) введение пациенту инъекции в недели 0 и 4 с последующим введением доз каждые 4 недели и 3) введение пациенту инъекции в недели 0 и 4 с последующим ежемесячным введением.
Используемая в данном документе фраза "составленный в дозировке, позволяющей осуществлять [путь введения] доставку [назначенной дозы]" используется для обозначения того, что данная фармацевтическая композиция может использоваться для обеспечения требуемой дозы антагониста IgE, например антитела к IgE, например лигелизумаба, посредством назначенного пути введения (например, п/к или в/в). В качестве примера, если требуемая п/к доза составляет 240 мг, то врач-клиницист может использовать 2 мл состава на основе антитела к IgE, имеющего концентрацию 120 мг/мл, 1 мл состава на основе антитела к IgE, имеющего концентрацию 240 мг/мл, 0,5 мл состава на основе антитела к IgE, имеющего концентрацию 480 мг/мл, и т. д. В каждом таком случае эти составы на основе антитела к IgE имеют достаточно высокую концентрацию для обеспечения подкожной доставки антитела к IgE. Подкожная доставка обычно требует доставки объемов, меньших или равных приблизительно 2 мл, предпочтительно объема, составляющего приблизительно 1 мл или меньше. Предпочтительные составы представляют собой готовые к применению жидкие фармацевтические композиции, содержащие от приблизительно 24 мг/мл до приблизительно 120 мг/мл лигелизумаба в водном растворе, содержащем L-гистидин/моногидрат гидрохлорида L-гистидина в качестве буферных средств, дигидрат трегалозы в качестве стабилизатора/регулятора тоничности и полисорбат 20 в качестве поверхностно-активного вещества.
Используемая в данном документе фраза "контейнер, содержащий достаточное количество антагониста IgE для обеспечения доставки [определенной дозы]" используется для обозначения того, что данный контейнер (например, флакон, ручка, шприц) вмещает в себе объем антагониста IgE (например, в качестве части фармацевтической композиции), который можно использовать для обеспечения требуемой дозы. В качестве примера, если требуемая доза составляет 240 мг, то врач-клиницист может использовать 2 мл из контейнера, который содержит состав на основе антитела к IgE с концентрацией 120 мг/мл, 1 мл из контейнера, который содержит состав на основе антитела к IgE с концентрацией 240 мг/мл, 0,5 мл из контейнера, который содержит состав на основе антитела к IgE с концентрацией 480 мг/мл и т. д. В каждом таком случае эти контейнеры содержат достаточное количество антагониста IgE для обеспечения доставки требуемой дозы 240 мг.
В некоторых вариантах осуществления раскрытых вариантов применения, способов и наборов доза антитела к IgE (например, лигелизумаба) или его антигенсвязывающего фрагмента составляет приблизительно 240 мг, при этом антитело к IgE (например, лигелизумаб) или его антигенсвязывающий фрагмент содержатся в жидком фармацевтическом составе в концентрации 120 мг/мл, и 2 мл фармацевтического состава помещают в два предварительно заполненных шприца, шприца-ручки (PFS) или автоинъектора, при этом каждый из них содержит 1 мл фармацевтического состава. В данном случае пациент получает две инъекции, каждая из которых составляет 1 мл, для достижения суммарной дозы 240 мг во время каждого введения. В некоторых вариантах осуществления доза антитела к IgE (например, лигелизумаба) или его антигенсвязывающего фрагмента составляет приблизительно 240 мг, при этом антитело к IgE (например, лигелизумаб) или его антигенсвязывающий фрагмент содержатся в жидком фармацевтическом составе в концентрации 120 мг/мл, и 2 мл фармацевтического состава помещают в автоинъектор или PFS. В данном документе пациент получает одну инъекцию 2 мл для достижения суммарной дозы 240 мг в ходе каждого введения. В способах с использованием одной инъекции 2 мл (например, посредством одного PFS или автоинъектора) (т. е. "препарат, вводимый в виде одной дозы") воздействие (AUC) и максимальная концентрация (Cmax) лекарственного средства эквивалентны (подобны, т. е. в пределах допустимого отклонения в соответствии со стандартами Управления по контролю за продуктами и лекарствами США) таковым в способах с использованием двух инъекций по 1 мл (например, с помощью двух PFS или двух AI) (т. е. "препарат, вводимый в виде нескольких доз").
В данном документе раскрыты способы лечения хронической спонтанной крапивницы (CSU), включающие подкожное (п/к) введение пациенту, нуждающемуся в этом, дозы от приблизительно 24 мг до приблизительно 240 мг антитела к IgE (например, лигелизумаба) или его антигенсвязывающего фрагмента еженедельно в течение недели 0, а затем п/к в дозе от приблизительно 24 мг до приблизительно 240 мг ежемесячно (каждые 4 недели), начиная в течение недели 4. Также в данном документе раскрыто антитело к IgE (лигелизумаб) или его антигенсвязывающий фрагмент для применения в лечении CSU, предусматривающем подкожное (п/к) введение пациенту, нуждающемуся в этом, дозы от приблизительно 24 мг до приблизительно 240 мг антитела к IgE или его антигенсвязывающего фрагмента еженедельно в течение недели 0, а затем п/к в дозе от приблизительно 24 мг до приблизительно 240 мг ежемесячно (каждые 4 недели), начиная в течение недели 4. В качестве альтернативы, в данном документе раскрыто антитело к IgE (лигелизумаб) или его антигенсвязывающий фрагмент для применения в изготовлении лекарственного препарата для лечения CSU, предусматривающего подкожное (п/к) введение пациенту, нуждающемуся в этом, дозы от приблизительно 24 мг до приблизительно 240 мг антитела к IgE или его антигенсвязывающего фрагмента еженедельно в течение недели 0, а затем п/к в дозе от приблизительно 24 мг до приблизительно 240 мг ежемесячно (каждые 4 недели), начиная в течение недели 4.
В данном документе раскрыты способы лечения хронической спонтанной крапивницы (CSU), включающие подкожное (п/к) введение пациенту, нуждающемуся в этом, дозы от приблизительно 24 мг до приблизительно 240 мг антитела к IgE (например, лигелизумаба) или его антигенсвязывающего фрагмента еженедельно в течение недели 0, а затем п/к в дозе от приблизительно 24 мг до приблизительно 240 мг ежемесячно (каждые 4 недели), начиная в течение недели 4, при этом антитело к IgE или его антигенсвязывающий фрагмент содержат VH-домен иммуноглобулина, содержащий аминокислотную последовательность, представленную под SEQ ID NO:2, и VL-домен иммуноглобулина, содержащий аминокислотную последовательность, представленную под SEQ ID NO:1.
Также в данном документе раскрыто антитело к IgE (например, лигелизумаб) или его антигенсвязывающий фрагмент для применения в лечении CSU, предусматривающем подкожное (п/к) введение пациенту, нуждающемуся в этом, дозы от приблизительно 24 мг до приблизительно 240 мг антитела к IgE или его антигенсвязывающего фрагмента еженедельно в течение недели 0, а затем п/к в дозе от приблизительно 24 мг до приблизительно 240 мг ежемесячно (каждые 4 недели), начиная в течение недели 4, при этом антитело к IgE или его антигенсвязывающий фрагмент содержат VH-домен иммуноглобулина, содержащий аминокислотную последовательность, представленную под SEQ ID NO:2, и VL-домен иммуноглобулина, содержащий аминокислотную последовательность, представленную под SEQ ID NO:1.
Также в данном документе раскрыто антитело к IgE (например, лигелизумаб) или его антигенсвязывающий фрагмент для применения в лечении CSU, предусматривающем подкожное (п/к) введение пациенту, нуждающемуся в этом, дозы от приблизительно 24 мг до приблизительно 240 мг антитела к IgE или его антигенсвязывающего фрагмента еженедельно в течение недели 0, а затем п/к в дозе от приблизительно 24 мг до приблизительно 240 мг ежемесячно (каждые 4 недели), начиная в течение недели 4, при этом антитело, представляющее собой лигелизумаб, содержит вариабельную область легкой цепи, содержащую CDRL1, CDRL2 и CDRL3, и вариабельную область тяжелой цепи, содержащую CDRH1, CDRH2 и CDRH3, при этом CDRL1 состоит из SEQ ID NO:3, CDRL2 состоит из SEQ ID NO:4, CDRL3 состоит из SEQ ID NO:5, CDRH1 состоит из SEQ ID NO:6, CDRH2 состоит из SEQ ID NO:7, и CDRH3 состоит из SEQ ID NO:8, при этом антитело специфически связывается с IgE.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов доза антитела к IgE или антигенсвязывающего фрагмента составляет приблизительно 24 мг или приблизительно 240 мг.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов антитело к IgE или его антигенсвязывающий фрагмент вводят п/к в дозе приблизительно 24 мг еженедельно в течение недели 0, а затем п/к в дозе приблизительно 24 мг каждые четыре недели, начиная в течение недели 4.
В других предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов антитело к IgE или его антигенсвязывающий фрагмент вводят п/к в дозе приблизительно 72 мг еженедельно в течение недели 0, а затем п/к в дозе приблизительно 72 мг каждые четыре недели, начиная в течение недели 4.
В других предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов антитело к IgE или его антигенсвязывающий фрагмент вводят п/к в дозе приблизительно 120 мг еженедельно в течение недели 0, а затем п/к в дозе приблизительно 120 мг каждые четыре недели, начиная в течение недели 4.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов у пациента достигается устойчивый ответ после лечения на протяжении одного года, как измерено с помощью полного ответа в отношении крапивной сыпи (оценка тяжести крапивной сыпи [HSS7]), UAS7 и дерматологического индекса качества жизни (DLQI).
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов до лечения с помощью антитела к IgE или антигенсвязывающего фрагмента пациент ранее получал лечение с помощью системного средства для лечения CSU.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов системное средство выбрано из группы, состоящей из H1-антигистаминных средств (H1-AH), H2-AH и антагониста лейкотриеновых рецепторов (LTRA) и их комбинаций.
В некоторых вариантах осуществления раскрытых способов, вариантов применения и наборов до лечения с помощью антитела к IgE или антигенсвязывающего фрагмента пациент ранее не получал лечение с помощью системного средства или местного средства для лечения CSU.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов доза антитела к IgE или антигенсвязывающего фрагмента составляет приблизительно 24 мг. В других предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов доза антитела к IGE или антигенсвязывающего фрагмента составляет приблизительно 72 мг. В других предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов доза антитела к IGE или антигенсвязывающего фрагмента составляет приблизительно 120 мг. В других предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов доза антитела к IGE или антигенсвязывающего фрагмента составляет приблизительно 240 мг.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов у пациента имеется CSU от средней до тяжелой степени.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов пациент является взрослым. В некоторых вариантах осуществления раскрытых способов, вариантов применения и наборов пациент является подростком.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов антитело к IgE или антигенсвязывающий фрагмент помещены в фармацевтический состав, где указанный фармацевтический состав дополнительно содержит буфер и стабилизатор. В некоторых вариантах осуществления раскрытых способов, вариантов применения и наборов фармацевтический состав представлен в жидкой форме (готовой к применению). В некоторых вариантах осуществления раскрытых способов, вариантов применения и наборов фармацевтический состав представлен в лиофилизированной форме. В некоторых вариантах осуществления раскрытых способов, вариантов применения и наборов фармацевтический состав помещен в предварительно заполненные шприцы, флаконы, шприцы-ручки или автоинъекторы.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов доза антитела к IgE или антигенсвязывающего фрагмента составляет приблизительно 24 мг, 72 мг, 120 мг или 240 мг, фармацевтический состав помещен в средство для введения, выбранное из группы, состоящей из предварительно заполненного шприца, шприца-ручки и автоинъектора, и указанное средство помещено в набор, и при этом набор дополнительно содержит инструкции по применению.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов доза антитела к IgE или антигенсвязывающего фрагмента составляет приблизительно 24 мг, 72 мг или 120 мг, фармацевтический состав помещен в автоинъектор или предварительно заполненный шприц, и автоинъектор или предварительно заполненный шприц помещен в набор, и при этом набор дополнительно содержит инструкции по применению.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов доза антитела к IgE или антигенсвязывающего фрагмента составляет приблизительно 240 мг, фармацевтический состав помещен в автоинъекторы или предварительно заполненные шприцы, автоинъекторы или предварительно заполненные шприцы помещены в набор, и при этом набор дополнительно содержит инструкции по применению.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов доза составляет 240 мг, причем ее вводят посредством однократного подкожного введения при общем объеме 2 мл с использованием состава, содержащего 120 мг/мл антитела к IgE или антигенсвязывающего фрагмента, при этом фармакологическое воздействие на пациента антитела к IgE или антигенсвязывающего фрагмента эквивалентно фармакологическому воздействию на пациента антитела к IgE или антигенсвязывающего фрагмента посредством двух отдельных подкожных введений, общий объем каждого из которых составляет 1 мл, с использованием такого же состава.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов доза составляет 240 мг, причем ее вводят посредством двух отдельных подкожных введений, объем каждого из которых составляет 1 мл, с использованием состава, содержащего 120 мг/мл антитела к IgE или антигенсвязывающего фрагмента.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов до лечения с помощью антитела к IgE или антигенсвязывающего фрагмента пациент характеризуется оценкой UAS7 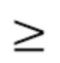 16.
16.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов до лечения с помощью антитела к IgE или антигенсвязывающего фрагмента пациент характеризуется HSS7 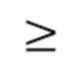 8.
8.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов пациент достигает составляющей 0 оценки HSS7 к неделе 12.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов пациент достигает составляющей 0 оценки UAS7 к неделе лечения 12.
В предпочтительных вариантах осуществления настоящего изобретения антитело к IgE или его антигенсвязывающий фрагмент представляет собой моноклональное антитело.
В предпочтительных вариантах осуществления настоящего изобретения антитело к IgE или его антигенсвязывающий фрагмент представляет собой человеческое или гуманизированное антитело.
В предпочтительных вариантах осуществления настоящего изобретения антитело к IgE или его антигенсвязывающий фрагмент представляет собой человеческое антитело.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов антитело к IgE или антигенсвязывающий фрагмент представляют собой человеческое моноклональное антитело.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов антитело к IgE или антигенсвязывающий фрагмент характеризуются Tmax, составляющим приблизительно 2-14 дней.
В предпочтительных вариантах осуществления раскрытых способов, вариантов применения и наборов антитело к IgE или антигенсвязывающий фрагмент характеризуются абсолютной биодоступностью приблизительно 47-100%.
В предпочтительных вариантах осуществления настоящего изобретения антитело к IgE или его антигенсвязывающий фрагмент предусматривают лигелизумаб.
Наборы
Настоящее изобретение также охватывает наборы для лечения CSU. Такие наборы содержат антагонист IgE, например молекулу, связывающую IgE (например, антитело к IgE или его антигенсвязывающий фрагмент, например лигелизумаб), или молекулу, связывающую рецептор IgE (например, антитело к IgE или его антигенсвязывающий фрагмент) (например, в жидкой или лиофилизированной форме), или фармацевтическую композицию, содержащую антагонист IgE (как описано выше). Кроме того, такие наборы могут содержать средства для введения антагониста IgE (например, шприц-ручку, шприц и флакон, предварительно заполненный шприц, предварительно заполненную шприц-ручку) и инструкции по применению. Эти наборы могут содержать дополнительные терапевтические средства против CSU (описанные выше) для лечения CSU, например, для доставки в комбинации с включенным антагонистом IgE, например молекулой, связывающей IgE, например антителом к IgE, например лигелизумабом. Такие наборы также могут содержать инструкции по введению антагониста IgE (например, антитела к IgE, например лигелизумаба) для лечения пациента с CSU. Такие инструкции могут предусматривать дозу (например, 10 мг/кг, 24 мг, 72 мг, 120 мг, 240 мг), путь введения (например, в/в, п/к) и схему введения доз (например, еженедельно, ежемесячно, еженедельно, а затем ежемесячно, еженедельно, а затем один раз в две недели и т. д.) для применения с раскрытым антагонистом IgE, например связывающей IgE молекулой, например антителом к IgE, например лигелизумабом.
Фраза "средства для введения" используется для обозначения любого доступного устройства для системного введения лекарственного средства пациенту, в том числе без ограничения предварительно заполненного шприца, флакона и шприца, шприца-ручки, автоинъектора, капельницы и пакета для внутривенного вливания, помпы и т. д. С помощью таких предметов пациент может вводить лекарственное средство самостоятельно (т. е. вводить лекарственное средство без помощи врача), или медицинский работник может вводить лекарственное средство. В некоторых вариантах осуществления общая доза 240 мг подлежит доставке в общем объеме 2 мл, который содержится в двух PFS или автоинъекторах, причем каждый PFS или автоинъектор содержит объем 1 мл, содержащий 120 мг/мл антитела к IgE, например лигелизумаба. В данном случае пациент получает две инъекции по 1 мл (препарат, вводимый в виде нескольких доз). В некоторых вариантах осуществления общая доза 240 мг подлежит доставке в общем объеме 2 мл, содержащем 120 мг/мл антитела к IgE, например лигелизумаба, который размещен в одном PFS или автоинъекторе. В данном случае пациент получает одну инъекцию объемом 2 мл (препарат, вводимый в виде одной дозы).
В данном документе раскрыты наборы для применения в лечении пациента с CSU, содержащие антагонист IgE (например молекулу, связывающую IgE, например антитело к IgE или его антигенсвязывающий фрагмент, например лигелизумаб) и средства для введения антагониста IgE пациенту с CSU. В некоторых вариантах осуществления набор дополнительно содержит инструкции по введению антагониста IgE, при этом в инструкциях указано, что антагонист IgE (например молекулу, связывающую IgE, например антитело к IgE или его антигенсвязывающий фрагмент, например лигелизумаб) следует вводить пациенту подкожно в дозе от приблизительно 24 мг до приблизительно 240 мг (например, приблизительно 24 мг или приблизительно 240 мг) в неделю 0, а затем п/к в дозе от приблизительно 24 мг до приблизительно 240 мг (например, приблизительно 24 мг, приблизительно 240 мг) ежемесячно (каждые 4 недели), начиная в течение недели 4.
Общая информация
В наиболее предпочтительных вариантах осуществления раскрытых вариантов применения, способов и наборов антагонист IgE представляет собой антитело к IgE или его антигенсвязывающий фрагмент.
В наиболее предпочтительных вариантах осуществления раскрытых способов, наборов или вариантов применения антитело к IgE или его антигенсвязывающий фрагмент представляет собой моноклональное антитело. В наиболее предпочтительных вариантах осуществления раскрытых способов, наборов или вариантов применения антитело к IgE или его антигенсвязывающий фрагмент представляет собой человеческое или гуманизированное антитело, предпочтительно человеческое антитело. В наиболее предпочтительных вариантах осуществления раскрытых способов, наборов или вариантов применения антитело или его антигенсвязывающий фрагмент представляет собой лигелизумаб.
В наиболее предпочтительных вариантах осуществления раскрытых способов, наборов или вариантов применения антитело или его антигенсвязывающий фрагмент предусматривают лигелизумаб, величина дозы является постоянной (также называемая "фиксированной" дозой, которая отличается от доз, вводимых на основании веса или на основании площади поверхности тела), доза составляет 24 мг, 72 мг, 120 мг или 240 мг, путем введения является п/к, а схема предусматривает введение в неделю 0, а затем каждые четыре недели, начиная в течение недели 4. В предпочтительных вариантах осуществления настоящего изобретения антитело или его антигенсвязывающий фрагмент вводят пациенту в виде фиксированной дозы.
Подробности одного или нескольких вариантов осуществления настоящего изобретения приведены в сопутствующем вышеизложенном описании. Хотя при осуществлении на практике или тестировании настоящего изобретения могут применяться любые способы и материалы, аналогичные или эквивалентные тем, которые описаны в настоящем изобретении, в данном документе описаны предпочтительные способы и материалы. Другие признаки, объекты и преимущества настоящего изобретения будут очевидны из описания и из формулы изобретения. В описании и прилагаемой формуле изобретения форма единственного числа включает ссылки на множественное число, если контекст явно не предписывает иное. Если не указано иное, то все технические и научные термины, используемые в данном документе, имеют то же значение, которое обычно понятно специалисту средней квалификации в данной области, к которой относится настоящее изобретение. Все патенты и публикации, процитированные в данном описании, включены в него посредством ссылки. Следующие примеры представлены для того, чтобы более полно проиллюстрировать предпочтительные варианты осуществления настоящего изобретения. Данные примеры никоим образом не следует истолковывать как ограничивающие объем раскрытого объекта изобретения, определяемого в прилагаемой формуле изобретения.
ПРИМЕРЫ
Пример 1. Данные относительно эффективности и безопасности у взрослых пациентов с CSU, у которых симптомы сохраняются, несмотря на стандартное лечение
Клинические данные относительно эффектов антитела к IgE, представляющего собой Xolair® (омализумаб), подтверждают потенциал антитела к IgE в качестве эффективного терапевтического средства для пациентов с CSU. Как и лигелизумаб, омализумаб представляет собой рекомбинантное полностью человеческое моноклональное антитело к IgE для лечения астмы и CSU. Лигелизумаб связывается с IgE человека с большей аффинностью, чем омализумаб.
Исследование фазы 2b (CQGE031C2201) проводили как многоцентровое, рандомизированное, двойное слепое, плацебо- и активно-контролируемое исследование фазы 2b по подбору дозы лигелизумаба (QGE031) в качестве дополнительного терапевтического средства для изучения эффективности и безопасности у пациентов с хронической спонтанной крапивницей (CSU). Исследование являлось рандомизированным, двойным слепым, активно- и плацебо-контролируемым исследованием с параллельными группами для установления зависимости доза-ответ лигелизумаба и оценки его эффективности и безопасности по сравнению с плацебо и омализумабом.
Оценка эффективности различных доз QGE031 по сравнению с омализумабом применительно к достижению полного ответа в отношении крапивной сыпи в неделю 20.
Оценка эффективности различных доз QGE031 по сравнению с плацебо и омализумабом у пациентов с CSU с точки зрения:
изменения по сравнению с исходным уровнем оценки тяжести крапивной сыпи (HSS7) в недели 12 и 20;
изменения по сравнению с исходным уровнем оценки интенсивности зуда (ISS7) в недели 12 и 20;
изменения по сравнению с исходным уровнем оценки активности крапивницы (UAS7) в недели 12 и 20.
Оценка безопасности
2. Взрослые пациенты мужского и женского пола в возрасте от ≥ 18 до ≤ 75 лет.
3. Наличие диагностированной CSU (с дермографической крапивницей или без нее при тестировании в отношении дермографии), рефрактерной к стандартному лечению на момент рандомизации, как определено посредством всего из нижеследующего:
- наличие зуда и крапивной сыпи в течение ≥ 6 последовательных недель в любое время до включения в исследование, несмотря на текущее применение стандартного лечения в этот период;
- оценка UAS7 ≥ 16 и HSS7 ≥ 8 в течение 7 дней до рандомизации;
- UAS≥4 согласно клинической оценке по крайней мере в один из дней скринингового визита;
- пациенты должны были получать стандартное лечение для лечения CSU в течение как минимум 3 последовательных дней непосредственно перед скрининговым визитом и должны иметь документально подтвержденное его текущее применение в день первоначального скринингового визита.
- наличие диагностированной CSU за ≥6 месяцев.
4. Готовность и способность ежедневно заполнять электронный дневник симптомов на протяжении всего периода исследования и придерживаться графиков предусмотренных исследованием визитов.
- индуцибельная крапивница: искусственная крапивница, холодовая, тепловая, солнечная крапивница, крапивница вследствие давления, замедленная крапивница вследствие давления, аквагенная, холинергическая или контактная крапивница;
- заболевания с возможными симптомами крапивницы или ангионевротического отека, такие как уртикарный васкулит, многоформная эритема, кожный мастоцитоз (пигментная крапивница), а также наследственный или приобретенный ангионевротический отек (например, вследствие дефицита ингибитора C1);
- любое другое кожное заболевание, связанное с хроническим зудом, которое может исказить оценки и результаты исследования (например, атопический дерматит, буллезный пемфигоид, герпетиформный дерматит, старческий зуд и т. д.).
- Предыдущее воздействие омализумаба или QGE031.
- Анафилаксия в анамнезе
- плацебо: п/к q4w (см. таблицу 3);
- омализумаб: п/к q4w
- Еженедельная оценка интенсивности зуда (ISS7).
- Еженедельная оценка активности крапивницы (UAS7)
- PK/PD/PG/биомаркеры
Цели исследования
Основная(-ые) цель(-и)
Установление зависимости доза-ответ для QGE031 применительно к достижению полного ответа в отношении крапивной сыпи в неделю 12 у пациентов с CSU при добавлении к H1-AH отдельно или в комбинации с H2-AH и/или LTRA.
Полный ответ в отношении крапивной сыпи определяется как 0 баллов по шкале HSS7. Аналогично, полный ответ в отношении зуда и полный ответ согласно UAS7 определяются как 0 баллов по шкалам ISS7 и UAS7 соответственно.
Второстепенные цели
Оценка эффективности QGE031 (на основе выбранной модели доза-ответ) по сравнению с 300 мг омализумаба применительно к достижению полного ответа в отношении крапивной сыпи в неделю 12 у пациентов с CSU при добавлении к H1-AH отдельно или в комбинации с H2-AH и/или LTRA.
Оценка эффективности отдельных доз QGE031, составляющих 24 мг, 72 мг и 240 мг п/к, по сравнению с 300 мг омализумаба применительно к достижению полного ответа в отношении крапивной сыпи в неделю 20 у пациентов с CSU при добавлении к H1-AH отдельно или в комбинации с H2-AH и/или LTRA.
Оценка эффективности QGE031 в дозах 24 мг, 72 мг и 240 мг п/к по сравнению с плацебо и 300 мг омализумаба у пациентов с CSU с точки зрения:
изменения по сравнению с исходным уровнем оценки тяжести крапивной сыпи (HSS7) в недели 12 и 20;
изменения по сравнению с исходным уровнем оценки интенсивности зуда (ISS7) в недели 12 и 20;
изменения по сравнению с исходным уровнем оценки активности крапивницы (UAS7) в недели 12 и 20.
Оценка безопасности (включая иммуногенность) и переносимости QGE031 (в дозах 24 мг, 72 мг и 240 мг, вводимых п/к каждые 4 недели) по сравнению с плацебо и 300 мг омализумаба у пациентов с CSU, в частности с точки зрения ЭКГ, нежелательных явлений, жизненно важных функций и клинической лабораторной оценки, в течение 20 недель лечения и 24 недель последующего наблюдения.
Исследовательские цели
Изучение эффективности QGE031 (в дозах 24 мг, 72 мг и 240 мг, вводимых п/к каждые 4 недели) по сравнению с плацебо и 300 мг омализумаба с точки зрения:
достижения полного ответа согласно UAS7 в недели 12 и 20;
достижения полного ответа в отношении зуда в недели 12 и 20;
достижения оценки активности крапивницы (UAS7) ≤ 6 в недели 12 и 20;
профиля изменения по сравнению с исходным уровнем для HSS7, ISS7 и UAS7 с течением времени;
времени до наступления клинического эффекта (например, согласно HSS7, UAS7, ISS7);
корреляции клинического ответа с фармакодинамическими параметрами, относящимися к сигнальному пути IgE (например, общий IgE, экспрессия IgE базофилами);
влияния статуса индекса CU (+ или -) на эффект лечения;
влияние на сон и влияние на повседневную активность, зарегистрированные в ежедневно заполняемом дневнике пациента с крапивницей (UPDD);
использования резервных препаратов;
зарегистрированных в UPDD случаев возникновения ангионевротического отека и предпринятых в его отношении действий/использованных средств для его лечения;
продолжительности ответа после отмены препарата;
количества осуществленных телефонных звонков врачу, медсестре или практикующей медсестре;
применимости клинической оценки ангионевротического отека;
изменения относительно исходного уровня к неделе 12 (и 20) величины следующих оценок PRO:
общей оценки по шкале дерматологического индекса качества жизни (DLQI);
оценки активности ангионевротического отека (AAS),
например еженедельное количество эпизодов/дней ангионевротического отека;
воздействия на работоспособность и повседневную деятельность (WPAI)-CU.
Оценка фармакокинетики (PK) QGE031.
Проведение исследовательских процедур фармакогенетического оценивания для определения того, могут ли индивидуальные генетические вариации в генах, относящихся к метаболизму лекарственного средства, CSU и целевому для лекарственного средства сигнальному пути, вызывать дифференцированный ответ на QGE031.
Оценка потенциальной применимости биомаркеров с точки зрения корреляции с эффективностью и безопасностью QGE031.
Дизайн исследования
Проводили многоцентровое, рандомизированное, двойное слепое, активно- и плацебо-контролируемое исследование фазы 2b по подбору дозы с параллельными группами с целью установления зависимости доза-ответ для QGE031 и оценки его эффективности и безопасности по сравнению с плацебо и омализумабом при введении подкожно в качестве дополнительного терапевтического средства для лечения взрослых пациентов с диагнозом рефрактерная CSU, у которых симптомы сохраняются, несмотря на применение H1-AH в одобренных и повышенных дозах отдельно или в комбинации с H2-AH и/или антагонистом лейкотриеновых рецепторов (LTRA). Исследование состояло из трех различных периодов, охватывающих 46 недель, как изложено ниже (см. также фигуру 1):
Период скрининга, от дня -14 до дня 1: продолжительность составляла до 2 недель, в течение которых пациентов, давших информированное согласие, оценивали на соответствие критериям участия в исследовании.
Период лечения, от дня 1 до дня 141 (20 недель): период проводимого в двойном слепом режиме лечения, в течение которого пациенты посещали клинику каждые 4 недели.
Период последующего наблюдения после лечения, с дня 141 по день 309 (24 недели): период последующего наблюдения состоит из 6 визитов (каждые четыре недели), причем последний визит происходит по прошествии вплоть до 24 недель после последнего визита в период лечения или при рецидиве у пациентов в течение периода последующего наблюдения, начиная с недели 32 и далее.
Период скрининга
Пациенты должны были пройти период скрининга длительностью до 2 недель, чтобы установить соответствие критериям отбора для участия в исследовании. Пациенты должны были присутствовать при двух визитах в период скрининга: в день -14 и день -7. Только в исключительных обстоятельствах, когда информация, касающаяся соответствия критериям отбора, была неполной (например, ожидание получение лабораторных данные), разрешали продление периода скрининга.
По определенным критериям включения/исключения повторный скрининг разрешали для пациентов, которые не прошли первоначальный скрининг. Разрешали проведение только одного повторного скрининга. Если пациента подвергали повторному скринингу для участия в исследовании, он подписывал новое информированное согласие и ему/ей выдавался новый номер пациента. Информированное согласие пациента, прошедшего повторный скрининг, получали до проведения каких-либо оценок, связанных с исследованием, или сбора любых данных для скринингового визита.
Период проводимого в двойном слепом режиме лечения
В день 1 соответствующих критериям отбора пациентов случайным образом распределяли для получения QGE031 в дозах 24 мг, 72 мг или 240 мг, или 300 мг омализумаба, или плацебо, вводимых п/к q4w, или однократной дозы 120 мг QGE, вводимой посредством п/к инъекции (для сохранения слепоты будут сделаны последующие инъекции плацебо), в течение 20-недельного периода проводимого в двойном слепом режиме лечения. Планировали включение примерно 80 пациентов в каждую из групп, предусматривающих получение QGE031 в количестве 240 мг q4w, в количестве 72 мг q4w и 300 мг омализумаба q4w. Приблизительно по 40 пациентов включали в группы, предусматривающие получение QGE031 в дозе 24 мг q4w, плацебо q4w и QGE031 в однократной дозе 120 мг. Ожидалось, что пациенты будут присутствовать при всех визитах в центр в соответствии с графиком оценки (таблица 2).
Таблица 2. График проведения оценок
(маркер наличия гиперчувствительности)
В случае отклоняющихся от нормы результатов анализа с помощью тест-полоски, например положительный результат на наличие WBC и/или крови, центральной лабораторией должно проводиться микроскопическое исследование, включая определение RBC и WBC
(Физический осмотр в день -14 является комплексным, однако последующие медицинские осмотры могут быть ограничены)
S=оценка подлежит записи только в исходной документации
D=оценка подлежит записи в CRF на основе исходной документации
DS=оценка, которая будет загружена в базу данных из отдельных исходных документов, то есть от сторонних поставщиков
PSW=преждевременное выбывание из исследования. Ожидается, что пациенты, преждевременно прекратившие прием исследуемого средства лечения, должны будут пройти оценку в неделю 20 (визит 107) через четыре недели после приема ими последней дозы. Ожидается, что эти пациенты впоследствии пройдут все процедуры оценки в рамках периода последующего наблюдения (визиты 201-206). Ожидается, что пациенты, которые находятся в периоде последующего наблюдения, но рано выбыли из исследования, пройдут процедуры оценки, предусматриваемые визитом 206.
Примечание: При необходимости все процедуры оценки следует проводить перед введением исследуемого лекарственного средства.
Последнюю дозу исследуемого лекарственного средства в период лечения вводили в день 113 (неделя 16) в ходе предусматриваемого исследованием визита. В качестве фонового лекарственного препарата все пациенты в этом исследовании продолжали получать H1-AH либо отдельно, либо в комбинации с H2-AH и/или LTRA. На протяжении всего исследования пациенты продолжали следовать стабильной схеме лечения. Первичный анализ планировали при достижении всеми пациентами недели 12. Проведение анализа окончания лечения планировали в неделю 20 периода лечения.
Период последующего наблюдения после лечения
После завершения периода проводимого в двойном слепом режиме лечения пациенты начинали прохождение периода последующего наблюдения после лечения в целях дальнейшей характеристики PK и PD QGE031, сбора дополнительных данных относительно эффективности и безопасности (например, рецидива) и оценки наличия антител к лекарственному средству (ADA). Период последующего наблюдения составлял 24 недели с последним предусматриваемым периодом последующего наблюдения визитом (визитом 206), соответствующим 28 неделям после приема последней дозы средства лечения. В период последующего наблюдения после лечения прием исследуемого средства лечения не осуществлялся, однако пациентам разрешали принимать их резервные препараты. От пациентов требовалось посещение исследовательского центра каждые четыре недели на протяжении периода наблюдения после лечения.
Пациенты, которые не прекращали прием исследуемого средства лечения и характеризовались оценкой UAS7 ≥ 12 при визите 203 (неделя 32, 16 недель после последней инъекции) или при следующих визитах (204-206), также соответствовали критериям для участия в расширенном исследовании до завершения периода последующего наблюдения.
До визита 203 не допускалось участие пациентов в расширенном исследовании вследствие недостаточного вымывания основного исследуемого лекарственного препарата, особенно для пациентов в получающей омализумаб группе.
Резервные препараты (лоратадин, фексофенадин или цетиризин) предоставлялись и использовались по мере необходимости в периоды скрининга, лечения и последующего наблюдения после лечения.
Обоснование дизайна исследования
Этот дизайн рандомизированного, двойного слепого, плацебо- и активно-контролируемого исследования с параллельными группами поддерживает определение диапазона доз и оценку эффективности, а также безопасности. Исследование разрабатывали как исследование по определению диапазона доз с целью установления зависимости доза-ответ и для определения дозы на основе выбранной модели доза-ответ, которая приносит пользу для пациентов с неконтролируемой CSU по сравнению с 300 мг омализумаба, представляющего собой единственный альтернативный лекарственный препарат на основе антитела к IgE, который в настоящее время продается во многих странах.
Целевая популяция для этого исследования состояла из пациентов с CSU, у которых сохранялись симптомы, несмотря на лечение с помощью H1-AH отдельно (в утвержденных или повышенных дозах) или в комбинации с H2-AH и/или LTRA. У данных пациентов имелась значительная неудовлетворенная медицинская потребность, и они представляют собой целевую популяцию для применения QGE031. В программе разработки омализумаба выявились различия между ожиданиями FDA и EMA в отношении фоновых лекарственных препаратов (H1-AH по сравнению с комбинацией H1-AH, H2-AH и/или LTRA). Каких-либо существенных различий в эффективности между двумя популяциями в исследованиях фазы 3 омализумаба не наблюдалось. С момента начала программы разработки омализумаба и научных рекомендаций, данных FDA и EMA в 2009 и 2010 годах, новые медицинские данные и научный прогресс привели к обновлению Международного руководства по определению, классификации, диагностике и лечению крапивницы (Zuberbier T, Aberer W, Asero R, et al (2014) The EAACI/GA(2) LEN/EDF/WAO Guideline for the definition, classification, diagnosis, and management of urticaria: the 2013 revision и update. Allergy; 69(7):868-87). Основанные на научных доказательствах рекомендации международной группы экспертов привели к разработке алгоритма лечения, который рекомендует для стадий 1 и 2 использование неседативных H1-AH в утвержденных или повышенных дозах (вплоть до четырехкратных) соответственно. Только на стадии 3 рекомендуется добавление омализумаба, циклоспорина А или монтелукаста.
Таким образом, чтобы отразить самый современный алгоритм лечения CSU, в этом исследовании предусматривали применение H1-AH отдельно (в утвержденных дозах в соответствии с указаниями местных органов здравоохранения или в характеризующихся вплоть до четырехкратно повышенным размером дозах) или в комбинации с H2-AH и/или LTRA в качестве фоновых лекарственных препаратов. Однако они могут быть разделены в программе фазы 3 (в ожидании подтверждения в органах здравоохранения). Чтобы обеспечить возможность сравнения с существующими данными относительно CSU, полученными в ходе программы исследований омализумаба, в остальных аспектах популяция была почти идентична той, которую изучали в исследованиях фазы 3 омализумаба.
Применение плацебо в этой популяции пациентов рассматривали целесообразным, поскольку пациенты будут продолжать прием фоновых терапевтических средств. Им также разрешали принимать H1-AH в качестве резервных препаратов на протяжении всего исследования. Кроме того, пациентам разрешали принимать участие в расширенном исследовании (долгосрочном исследовании безопасности), в котором они получали QGE031. Хотя признаки и симптомы CSU обременительны для пациентов, испытания с использованием плацебо были безопасно и успешно проведены при данном показании (Kaplan A, Ledford D, Ashby M, et al (2013) Omalizumab in patients with symptomatic chronic idiopathic/spontaneous urticaria despite standard combination therapy. J Allergy Clin Immunol; 132(1):101-9; Maurer M, Magerl M, Metz M, et al (2013) Revisions to the international guidelines on the diagnosis and therapy of chronic urticaria. J Dtsch Dermatol Ges.). Пациентов распределяли по группам лечения в соответствии с количеством, необходимым для удовлетворения требований к данным для обеспечения оптимального моделирования.
Введение плацебо ограничивали в максимально возможной степени, при этом все еще позволяли проведение статистического сравнения и поддержание слепого режима применительно к лечению активным средством по сравнению с лечением плацебо. Применение дизайна с двойной имитацией представлялось невозможным ввиду отсутствия плацебо, которое полностью соответствовало бы характеристикам омализумаба, тем самым повышая риск расслепления для персонала и пациентов (например, возможность для пациентов обнаруживать различия в ощущениях между введенными инъекциями); это также увеличило бы нагрузку/дискомфорт для пациентов с точки зрения общего количества инъекций при каждом визите. Следовательно, в этом исследовании будет использоваться только плацебо для групп QGE031. Поскольку вводимый объем для QGE031 и омализумаба будет варьироваться (например, 240 мг QGE031 имеет объем 2,0 мл, вводимый в виде 2 отдельных инъекций по 1,0 мл, 300 мг омализумаба имеет объем 2,4 мл, вводимый в виде 2 отдельных инъекций по 1,2 мл), группы лечения будут характеризоваться разными объемами вводимых инъекций.
Кроме того, для сохранения слепого режима введение исследуемого лекарственного средства осуществлял информированный человек (например, участвующие в исследовании медсестра или врач), который не зависел от группы в исследовательском центре, выполняющей процедуры оценки исследования.
Первичный анализ проводили при достижении всеми пациентами недели 12. Проведение анализа окончания лечения осуществляли в неделю 20 периода лечения.
Обоснование дозы/схемы, пути введения и продолжительности лечения
Предполагается, что более высокая эффективность QGE031 в отношении подавления IgE по сравнению с омализумабом приводит к обеспечению большей пользы при лечении CSU. Для научно обоснованной оценки предусматривали сравнение с группами лечения, получающими как плацебо, так и омализумаб, для интерпретации ответа на QGE031.
Обоснование доз/схем
Модель доза-ответ получали в результате тестирования 10-кратного диапазона доз QGE031, начиная с 24 мг q4w с переходом к 72 мг q4w и до 240 мг q4w, используя при этом плацебо в качестве нулевой дозы. Затем модель доза-ответ использовали для поиска дозы или диапазона доз, которые обеспечивают дополнительное желаемое преимущество по сравнению с активным препаратом сравнения.
QGE031 в дозах 24, 72 и 240 мг вводили п/к q4w в течение 20 недель, то есть осуществляя в общей сложности 5 введений. 300 мг омализумаба вводили п/к q4w в качестве активного препарата сравнения, а в качестве контроля использовали плацебо, согласованное со схемой введения доз.
Обоснованием выбора 120 мг в качестве дозы для получающей однократную дозу группы было обеспечение максимального или близкого к максимальному эффекта в первые дни после введения дозы, но не настолько высокого, чтобы предотвратить возможность рецидива у пациентов в течение периода испытания. Эта группа предоставляла заслепленные данные относительно вымывания, которые имеют решающее значение для определения уровня лекарственного средства в сыворотке крови, ассоциированного с возвращением симптомов зуда и крапивной сыпи, исходя из которых определяют альтернативные q4w интервалы введения доз.
Обоснование для доз/схемы введения q4w основывалось на (а) данных относительно переносимости аллергенов, определенной посредством кожных инъекционных проб, полученных из исследований QGE031A2103 с участием добровольцев с атопией и QGE031B2203 с участием пациентов с астмой, и (b) данных относительно эффективности омализумаба, полученных из исследований фазы 3 с участием пациентов с CSU.
(а) Данные относительно переносимости аллергенов, определенной посредством кожных инъекционных проб
Кожную инъекционную пробу рассматривали как прогностический фактор проявляющейся в виде крапивницы реакции, поскольку она является прямой демонстрацией уровня реактивности сигнального пути IgE в тканях кожи, а гистамин-опосредованная реакция, проявляющаяся в виде волдырей, является общей для кожной инъекционной пробы и крапивницы. Регистрируемые клинические данные основывались на диаметре волдырей, возникающих при кожной инъекционной пробе. На фиг. 2 показано снижение уровня свободного IgE в плазме крови и снижение количества FcεRI и поверхностного IgE на базофилах периферической крови, а также снижение проявления связанного с появлением волдырей аспекта кожной инъекционной пробы у пациентов с астмой в исследовании QGE031B2203. Для пациентов с умеренным уровнем IgE как 72 мг, так и 240 мг приводят к выраженному снижению биомаркеров и эффективности кожной инъекционной пробы. Аналогичные наблюдения регистрировали для здоровых добровольцев в QGE031A2103, для которых использовали режим введения доз с поправкой на вес.
Схемой введения доз в QGE031A2103 и QGE031B2203 предусматривала введение каждые 2 недели (q2w), вследствие чего данные не характеризовались непосредственной информативностью для соответствующих доз схемы q4w. Тем самым, модель, созданную на основе этих данных, использовали для прогнозирования зависимости доза-ответ при введении доз q4w в популяции CSU.
На фигуре 3 показаны прогнозируемые кривые доза-ответ для QGE031 применительно к связанному с появлением волдырей аспекту кожной инъекционной пробы. Было спрогнозировано, что доза 24 мг обеспечит 50-70% максимально возможного ответа. Диапазон ответов для средних 50% популяции пациентов варьируется от очень малого до высокого, но не максимального ответа. Доза 72 мг предположительно находилась вблизи перехода между линейной и насыщенной областью кривой доза-ответ, в то время как ожидалось, что 240 мг обеспечит максимальную эффективность. Следовательно, 24 мг является ‘субоптимальной дозой', которая, как ожидалось, будет находиться в том же диапазоне, что и омализумаб, нежели являться минимально эффективной дозой. 300 мг омализумаба q4w, по прогнозам, обеспечит достижение слегка более низкого ответа, чем 72 мг QGE031 q4w.
(b) Данные относительно эффективности омализумаба, полученные из исследований фазы 3, проводимых в отношении CSU
Клинические данные фазы 3 исследования омализумаба использовали в качестве альтернативной оценки клинического ответа на выбранные дозы QGE031. На основании этих данных создавали модель PK/PD, связывающую уменьшение свободного IgE со снижением оценок симптомов CSU. Предполагая разные различия в аффинности связывания IgE между QGE031 и омализумабом, ожидалось, что 24 мг могут обеспечить такие же субоптимальные ответы, как и 150 мг омализумаба. Только в крайнем случае, когда разница в эффективности in vivo у пациентов с CSU аналогична разнице в аффинности связывания in vitro (50x), доза 24 мг также может быть близкой к обеспечению максимальной эффективности, так что соотношение концентрация-ответ должно быть выведено из данных относительно вымывания. Предполагалось, что доза QGE031, составляющая 240 мг, приведет к гораздо более сильному снижению свободного IgE по сравнению с 300 мг омализумаба, что, как ожидалось, приведет к превосходящей клинической эффективности. Следует отметить, что максимальный уровень эффекта в отношении снижения величины оценки симптомов оценивали на основе данных относительно омализумаба, которые предполагают, что доза, составляющая 300 мг омализумаба, уже приближалась к обеспечению максимального ответа. Однако имеется мало данных относительно ответа на более высокий уровень воздействия, то есть более выраженного снижения уровня IgE, и ввиду этого модель не смогла обеспечить определение максимального ответа, который мог бы наблюдаться при использовании QGE031, являющегося более эффективным лекарственных средством.
Таким образом, либо используя данные относительно результатов кожной инъекционной пробы, либо проецируя на QGE031 данные относительно эффективности омализумаба при лечении CSU, дозы 24 и 240 мг, по прогнозам, должны обеспечивать ответы, варьирующиеся от субоптимальных до максимальных. Дозу 240 мг тестировали для определения того, приводит ли более сильное снижение уровня свободного IgE к более высокой эффективности по сравнению с 300 мг омализумаба. Доза 72 мг расположена на логарифмической шкале между этими дозами, находясь на равных расстояниях от этих доз.
На рисунке 3 показаны прогнозируемые кривые доза-ответ в неделю 20, основанные на моделировании с использованием модели PKPD QGE031, которая была адаптирована к данным относительно связанного с появлением волдырей аспекта кожной инъекционной пробы, полученным от атопических, но в остальном здоровых субъектов. Полосы демонстрируют 25-й и 75-й процентили, представляющие степень вариации между здоровыми субъектами, а линия показывает медианное значение. Единицы ответа по оси y представляют собой квадратный корень из суммы размеров волдырей (в мм) всех разведений аллергенов, тестируемых для каждого пациента при каждом визите.
Обоснование введения фиксированных доз:
Обоснование введения фиксированных доз, а не ведения доз в мг/кг заключалось в том, чтобы расширить (насколько это возможно) степень воздействия QGE031 среди всех пациентов, включенных в исследование, тем самым улучшив оценку безопасности. В любом случае влияние веса тела подлежало изучению с помощью статистического и фармакокинетико-фармакодинамического моделирования с применением данных, полученных в ходе этого и других исследований. В (Wang DD, Zhang S, Zhao H, et al (2009) Fixed dosing versus body size-based dosing of monoclonal antibodies in adult clinical trials. J Clin Pharmacol; 49(9):1012-24) было показано, что показатели распределения значений площади под кривой (AUC) для моноклонального антитела (или ее эквивалента, представляющего собой среднюю концентрацию в стационарном состоянии) являются слегка более широкими при введении фиксированной дозы по сравнению с введением доз, скорректированных по массе тела. Обоснование для получающей однократную дозу группы:
Обоснованием для группы, получающей однократную дозу 120 мг п/к, служила необходимость подтверждения выбора интервала доз для фазы 3. Данные, полученные от этой группы, позволяют оценить продолжительность ответа и определить ее корреляцию с концентрацией лекарственного средства в сыворотке крови в момент повторного появления симптомов. Их сравнивали с аналогичными данными относительно вымывания, полученными от группы, получающей несколько доз 240 мг, чтобы установить, изменяет ли более длительное лечение концентрацию в точке повторного появления симптома и, следовательно, требуемый интервал между введением доз. Совокупность данных относительно PK, PD и клинической эффективности, введения однократной и нескольких доз анализировали с применением нелинейных моделей PKPD со смешанными эффектами для получения зависимости концентрация-ответ и, исходя из этого, кривых доза-ответ, на основе которых устанавливали подходящие режимы введения доз для регистрационных исследований.
Обоснование составляющей 20 недель продолжительности лечения
Результаты двух исследований омализумаба фазы 3 (Q4881g и Q4883g) продемонстрировали, что не все пациенты, ответившие на лечение, достигают устойчивого или полного ответа после первой дозы. Анализ времени до достижения устойчивого клинического ответа (UAS7 ≤ 6) или полного ответа (UAS7=0) показал, что у значительной части пациентов, у которых не наблюдалось ответа после первой дозы, наблюдался прогресс и достигался устойчивый или полный ответ после введение второй и третьей доз (т. е. после общего периода лечения, составляющего 12 недель). Кроме того, изучение доли пациентов, достигших UAS7 ≤ 6 или состояния без наличия симптомов (UAS=0) в обоих исследованиях Q4881g и Q4883g, продемонстрировало устойчивую тенденцию к достижению еще большим числом пациентов улучшенного состояния здоровья в неделю 24 по сравнению с неделей 12. При более длительном лечении в профиле безопасности различий не наблюдали (Kaplan A, Ledford D, Ashby M, et al (2013) Omalizumab in patients with symptomatic chronic idiopathic/spontaneous urticaria despite standard combination therapy. J Allergy Clin Immunol; 132(1):101-9). Кроме того, продолжительность лечения для предлагаемого исследования фазы 2b составляет 20 недель для обеспечения достаточного времени для оценки контроля ангионевротического отека с применением описанных оценок, а также для изучения любых различий в кинетике клинического ответа между подгруппами (например, пациентов со статусом+индекса CU и пациентов со статусом - индекса CU). В программе исследования омализумаба ангионевротический отек оценивали главным образом как дни без ангионевротического отека; при этом доступны более новые и более подробные валидированные оценки (например, оценка активности ангионевротического отека [AAS]), и они будут использоваться в данном исследовании. Поскольку явления ангионевротического отека носят эпизодический характер, более длительное время для оценки пациентов дает лучшую возможность продемонстрировать контроль и отличия от омализумаба и плацебо. Индекс CU представляет собой лабораторный тест, который обычно используется для выявления пациентов с аутоиммунным компонентом в их CSU, и у этих пациентов могут иметься специфические аутоантитела (например, антитела к FcεRI), которые могут обуславливать крапивницу, и они могут характеризоваться патофизиологией, отличной от таковой у пациентов со статусом - индекса CU (Biagtan MJ, Viswanathan RK, Evans MD, et al (2011) Clinical utility of the Chronic Urticaria Index. J Allergy Clin Immunol; 127(6):1626-7). Согласно данным, полученным в ходе фазы 3 исследования омализумаба, пациенты со статусом+индекса CU составляют до 30% популяции CSU.
В связи с этим выбранная для данного исследования продолжительность лечения, составляющая 20 недель, являлась минимальной продолжительностью воздействия, необходимой для обеспечения оптимальной оценки статистически и клинически значимого влияния на результаты лечения CSU (Kaplan A, Ledford D, Ashby M, et al (2013) Omalizumab in patients with symptomatic chronic idiopathic/spontaneous urticaria despite standard combination therapy. J Allergy Clin Immunol; 132(1):101-9; Maurer M, Magerl M, Metz M, et al (2013) Revisions to the international guidelines on the diagnosis and therapy of chronic urticaria. J Dtsch Dermatol Ges.). Хотя она была короче, чем 24-недельная продолжительность лечения с помощью омализумаба в Q4881g и Q4883g, ее было достаточно для QGE031, исходя из понимания того, что подавление IgE будет происходить быстрее и с более высоким уровнем подавления, чем наблюдалось при использовании омализумаба.
В целом, в дизайне исследования с тщательностью использовали доступные данные относительно безопасности и клинической активности, полученные из предыдущих исследований с использованием QGE031 и омализумаба, минимизировали воздействие плацебо и оптимизировали вероятность обеспечения клинической пользы в контексте дизайна фазы 2 исследования.
Обоснование выбора препарата сравнения
Все пациенты, независимо от того, в какую группу лечения они были рандомизированы, получали стандартную терапию в качестве фонового лекарственного препарата. Кроме того, пациенты имели доступ к резервному H1-AH-препарату.
Плацебо в данном исследовании применяли по следующим причинам:
с целью обеспечения заслепления исследователей и пациентов в отношении осуществляемого ими лечения и тем самым сведения к минимуму предвзятости при оценке безопасности и эффективности,
с целью обеспечения возможности оценки улучшения с точки зрения контроля CSU для пациентов с заболеванием, не поддающимся контролю посредством фонового препарата, которые получают лечение с использованием QGE031, по сравнению с теми, кто продолжает лечение с использованием только фонового лекарственного препарата, и
с целью обеспечения возможности оценки безопасности QGE031 при его использовании в дополнение к фоновому лекарственному препарату по сравнению с использованием только фонового лекарственного препарата,
с целью поддержания двойного слепого режима для более низких доз в группах лечения QGE031, поскольку их вводили в объемах 0,2 мл и 0,6 мл соответственно. Пациенты в этих группах лечения получат две инъекции соответствующего объема, одну активного средства и одну плацебо.
Омализумаб выбирали в качестве активного препарата сравнения по следующим причинам:
он является единственным препаратом того же класса, что и QGE031, одобренным в настоящее время в качестве дополнительного терапевтического средства для пациентов с CSU с неадекватным ответом на H1-AH, и
проведение сравнения безопасности и эффективности QGE031 относительно омализумаба помогло спонсору принять решение о том, имеется ли достаточная потенциальная польза для пациентов, чтобы гарантировать дальнейшую разработку QGE031.
Таблица 3. Количество введений исследуемого лекарственного средства
(150 мг/
1,2 мл)
(120 мг/
1 мл)
(0 мг/
1 мл)
(300 мг омализумаба)
(240 мг QGE)
(72 мг QGE)
(24 мг QGE)
(120 мг QGE)
(только при визите 101)
2×1 мл, четыре раза
(визиты 103, 104, 105, 106)
Сопутствующее лечение
Для данного исследования требовалось одновременное применение только H1-AH (в утвержденных дозах в соответствии с рекомендациями местных органов здравоохранения или в характеризующихся вплоть до четырехкратно повышенным размером дозах) или в комбинации с H2-AH и/или LTRA (монтелукастом, зафирлукастом, пранлукастом) в качестве фонового лекарственного препарата.
Ежедневно заполняемый дневник пациента с крапивницей (UPDD)
UPDD включает UAS7 (зуд и крапивная сыпь) для оценивания клинических симптомов, применение резервных препаратов, влияние на сон и повседневную активность, возникновение ангионевротического отека и управление его течением.
Оценка тяжести крапивной сыпи (HSS)
Оценка тяжести волдырей (крапивной сыпи), определяемая количеством крапивной сыпи, записывалась пациентом дважды в день в своем электронном дневнике по шкале от 0 (отсутствие) до 3 (интенсивная(ые)/тяжелая степень) (см. таблицу 4). Еженедельную оценку (HSS7) получали путем суммирования усредненных ежедневных оценок за 7 дней, предшествующих визиту. Таким образом, возможный диапазон еженедельных оценок составлял от 0 до 21.
Полный ответ в отношении крапивной сыпи определяется как HSS7=0.
Таблица 4. Оценка тяжести крапивной сыпи
В случае отсутствия утренней или вечерней оценки в качестве ежедневной оценки использовали непропущенную оценку за этот день (утренняя или вечерняя). В случае отсутствия одной или нескольких ежедневных оценок для обработки отсутствующих данных применяли следующие принципы.
Если у пациента имелось по меньшей мере 4 непропущенных ежедневных оценки за составляющий 7 дней период до визита, то еженедельную оценку рассчитывали как сумму доступных оценок в электронном дневнике, имеющихся для этой недели, деленную на количество дней, в которые ежедневные оценки не были пропущены, умноженную на 7. Если в течение предыдущих 7 дней имелось менее 4 непропущенных ежедневных оценок, то недельная оценка за неделю отсутствовала.
Оценка интенсивности зуда (ISS)
Степень интенсивности зуда записывалась пациентом два раза в день в своем электронном дневнике по шкале от 0 (отсутствие) до 3 (интенсивный/сильный) (см. таблицу 5). Еженедельную оценку (ISS7) получали путем суммирования усредненных ежедневных оценок за 7 дней, предшествующих визиту. Таким образом, возможный диапазон еженедельных оценок составлял от 0 до 21. Частично отсутствующие записи в дневнике обрабатывали таким же образом, как описано для оценки тяжести крапивной сыпи.
Полная ответ в отношении зуда определяется как ISS7=0.
Таблица 5. Оценка интенсивности зуда
Еженедельная оценка активности крапивницы (UAS7)
Показатель UAS7 предусматривал сумму оценок по шкалам HSS7 и ISS7. Возможный диапазон еженедельной оценки UAS7 составлял от 0 до 42.
Полный ответ согласно UAS7 определяли как UAS7=0.
Оценка влияния на сон
Влияние на сон оценивалось пациентом один раз в день утром в электронном дневнике. Его оценивали по шкале от 0 до 3 (см. таблицу 6).
Таблица 6. Оценка влияния на сон
Оценка влияния на повседневную активность
Влияние на повседневную активность оценивалось пациентами по шкале от 0 до 3 (см. таблицу 7) один раз в день вечером в электронном дневнике. Виды повседневной активности могут включать работу, учебу, спорт, хобби и виды активности, осуществляемой совместно с друзьями и семьей.
Таблица 7. Оценка влияния на повседневную активность
Всего было включено 382 пациента. Основная цель исследования была достигнута, при этом лигелизумаб (QGE031) продемонстрировал зависимость доза-ответ применительно к показателям частоты полных ответов в отношении крапивной сыпи в неделю 12 (p < 0,001). Показатели частоты ответа с HSS7=0 в неделю 12 составили 30%, 51% и 42% для лигелизумаба в дозах 24, 72 и 240 мг соответственно по сравнению с 26% для омализумаба и 0% для PBO. Эти ответы сохранялись в течение периода до 20 недель (26%, 51% и 45% для лигелизумаба в дозах 24, 72 и 240 мг соответственно по сравнению с 34% для омализумаба и 9% для PBO). Показатели высокой частоты ответов с UAS7=0 и DLQI=0-1 наблюдали уже на 4 неделе; большее количество пациентов не имели симптомов (UAS7=0) и сообщали о заметном улучшении своего качества жизни (DLQI=0-1) при приеме лигелизумаба в дозах 72 и 240 мг по сравнению с омализумабом в течение 20-недельного периода лечения (таблица 8). Лигелизумаб хорошо переносился, а профиль безопасности был сопоставим с профилем безопасности омализумаба.
Таблица 8. Доля пациентов с полными ответами (UAS7=0) и тех, у кого наблюдались ответы с DLQI=0-1, в недели 4, 12 и 20
24 мг
(n=43)
72 мг
(n=84)
240 мг
(n=85)
300 мг
(n=85)
(n=43)
В течение первых 4-6 недель испытания не было зафиксировано значительных различий в ответе пациентов на 72, 120 или 240 мг лигелизумаба (фигура 4). Как и в случае пациентов с полными ответами согласно UAS7 и ответами с DLQI=0-1, как описано выше, ответ на дозу 24 мг был меньше, чем для трех более высоких доз, но все же превосходил ответ на плацебо. Через 6 недель эффект однократного введения 120 мг начинал уменьшаться, а симптомы возвращаться к характерным для контрольной группы.
Вывод
У пациентов с CSU от средней до тяжелой степени лигелизумаб демонстрировал четкий дозозависимый ответ применительно к нескольким конечным точкам. По сравнению с 300 мг омализумаба лигелизумаб 24 мг показал сравнимую эффективность, а дозы 72, 120 и 240 мг продемонстрировали более высокую эффективность применительно к указанным нескольким конечным точкам и показали сопоставимую безопасность.
Пример 2. План исследования эффективности лигелизумаба у детей. Анализ воздействие-ответ у взрослых с CSU с основанном на моделировании дизайном исследования по подбору дозы для подростков
Цели. При установлении дозы для детей должным образом учитывали значительно сниженную эффективность, т. е. более высокую EC50, омализумаба в популяции подростков с CSU [EMA 2014. Заявка II/0048 Отчет о результатах оценки, раздел 2.3.4.2, страница 10 и раздел 2.3.5, страница 11, адрес в сети Интернет www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/000606/WC500164453.pdf, по состоянию на 17 января 2018 года] по сравнению со взрослыми. Следовательно, нельзя было предположить, что эквивалентные концентрации лигелизумаба, применяемые для подростков и взрослых, приведут к эквивалентной эффективности в популяциях взрослых и подростков. Цель состояла в разработке адекватного исследования с участием подростков, чтобы определить, достаточно ли отличается показатель EC50 у пациентов подросткового возраста с CSU от такового у взрослых, чтобы потребовать другого режима введения доз.
Способы. Значения концентрации лигелизумаба (QGE031) и оценки активности крапивницы (UAS7, 7-дневная сумма ежедневных оценок зуда и крапивной сыпи, каждая из которых ставилась в диапазоне 0-3) получали от взрослых пациентов, получавших лечение в виде нескольких доз 0, 24, 72 и 240 мг, вводимых каждые 4 недели, и в виде однократной дозы 120 мг лигелизумаба в ходе исследования фазы 2 EudraCT 2014-005559-16. Промежуточные данные от 295 пациентов анализировали с помощью лонгитюдных фармакокинетико-фармакодинамических моделей для непрерывной UAS7 (диапазон 0-42) с применением NONMEM с выборкой по значимости. Полученную модель использовали со стохастическим моделированием-оценкой для дизайна исследования с участием подростков (возрастом 11-17 лет включительно), способного обнаруживать сдвиги в EC50 между подростками и взрослыми. Комбинацию R-3.2.3, NONMEM 7.3.0 [Bauer RJ NONMEM Users Guide Introduction to NONMEM 7.3.0. 2013. Icon Development Solutions, Ганновер, Мэриленд 21076, США] и программного обеспечения PDx-Pop-5.2 применяли для создания аналитических баз данных, оценки параметров, управления циклами NONMEM и получения результатов последующей обработки.
Результаты. Выбранная двухкамерная фармакокинетическая модель обеспечивала надлежащее описание данных относительно концентрации лекарственного средства. Ключевой параметр воздействия, представляющий собой клиренс, составлял 0,85 л/д. (остаточная стандартная ошибка, RSE, составляла 9,1%) для веса тела 80 кг с коэффициентом вариации между субъектами, составляющим 49% (BSV). Вес тела и индекс хронической крапивницы (аутоантитела к IgE или к IgE-рецептору) идентифицировали в качестве основных ковариант, влияющих на клиренс, со значениями согласно оценкам, составляющими 1,0 (порядок; 35% RSE) для веса и 0,89 (соотношение; 36% RSE) для индекса CU. Выбранная модель непрерывной UAS7 характеризовалась EC50 1,1 мкг/мл (38% RSE) с очень высоким расчетным BSV (1405%) и высоким коэффициентом Хилла 5,72 (0,75% RSE). Визуальные оценки предсказательной способности были сочтены достаточными для начала процедуры разработки дизайна подросткового исследования по ряду характеристик исследования. Для поддержания числовой стабильности значение BSV для EC50 снижали с расчетного до <= 300% с включением анализа чувствительности для исследования влияния снижения. В выбранном дизайне определяли три группы: плацебо, 24 и 120 мг, вводимые каждые 4 недели, с продолжительностью лечения 16 недель и периодом последующего наблюдения до 40 недель. Для пациентов, получавших плацебо, предусматривали переход на дозу 120 мг через 8 недель.
Выводы. Несмотря на сильно варьирующиеся данные, модель воздействие-ответ согласно UAS7 смогла обнаружить и достаточно хорошо описать дозозависимые изменения для плацебо и лигелизумаба с течением времени. Процедура стохастического моделирования-оценки показала, что 24 мг лигелизумаба q4w следует применять вместе с более высокой дозой 120 мг q4w в проспективном исследовании с участием подростков. Доза 24 мг являлась приоритетной, так как она должна обеспечить достижение концентраций, находящихся в области значения ожидаемой ЕС50, оптимальной точки чувствительности для оценки этого параметра. Таким образом, рандомизация являлась неравномерной, при этом 20 пациентов получали 24 мг, по 10 пациентов получали 120 мг и плацебо. Результаты приема дозы 120 мг, полученные как от пациентов, непосредственно получавших лечение, так и от пациентов, перешедших с приема плацебо, должны обеспечить оценку максимального эффекта препарата. В целом, исходя из проведенных 100 циклов моделирования-оценки процедура показала, что существует примерно 80% вероятность обнаружения 2-кратного увеличения ЕС50, порогового значения, выше которого следует рассматривать применение отличающегося от применяемого для взрослых режима введения доз.
Пример 3. Повторное лечение лигелизумабом является высокоэффективным у пациентов с хронической спонтанной крапивницей
Способ
В ходе фазы 2b основного исследования (NCT02477332) соответствующих критериям отбора взрослых пациентов с CSU от средней до тяжелой степени (7-дневная оценка активности крапивницы [UAS7] ≥ 16) рандомизировали для получения лигелизумаба в дозах 24, 72 или 240 мг, 300 мг омализумаба или плацебо, вводимых подкожно каждые 4 недели (q4w) на протяжении 20 недель. После периода проводимого в двойном слепом режиме лечения пациенты начинали прохождение 24-недельного периода без лечения. После вымывания последней дозы, полученной в основном исследовании (неделя 32), пациенты с признаками активности заболевания (UAS7 ≥ 12) допускались к участию в открытом расширенном исследовании с одной группой (240 мг лигелизумаба q4w). Ответ после повторного лечения оценивали с помощью UAS7; при этом результаты основного исследования приведены для тех пациентов, которые участвовали в расширенном исследовании.
Результаты
В общей сложности 70,6% (226/320) пациентов приняли участие в расширенном исследовании. Устойчивую эффективность (показатели частоты полного ответа у пациентов [доля пациентов, достигавших UAS7=0] и изменение оценки UAS7 по сравнению с исходным уровнем) наблюдали после 12 недель повторного лечения 240 мг лигелизумаба q4w независимо от дозы, полученной во время основного исследования (см. таблицу 9). Аналогичные тенденции наблюдали для показателей частоты полного ответа у пациентов при HSS7=0 и ISS7=0, а также для усредненных изменений по сравнению с исходным уровнем для HSS7 и ISS7.
(доверительный интервал 95%)
исходным уровнем (стандартное отклонение)
Неделя 12
(N=51)
Неделя 12
(N=46)
Неделя 12
(N=51)
Неделя 12
(N=46)
Выводы
Повторное лечение лигелизумабом является высокоэффективным у пациентов с хронической спонтанной крапивницей, для которых начальное лечение лигелизумабом принесло пользу, и у которых возник рецидив после прекращения лечения.
Пример 4. Лигелизумаб обеспечивает устойчивый контроль симптомов хронической спонтанной крапивницы, проявляющихся в виде крапивной сыпи, зуда и ангионевротического отека: результаты лечения в течение 1 года
Общие сведения
Лигелизумаб обеспечивал лучший контроль симптомов в виде крапивной сыпи, зуда и ангионевротического отека по сравнению с омализумабом и плацебо у пациентов с хронической спонтанной крапивницей (CSU), неадекватно контролируемой H1-антигистаминными средствами, применяемыми отдельно или в сочетании с H2-антигистаминными средствами и/или антагонистами лейкотриеновых рецепторов, вплоть до недели 20 (с последней процедурой лечения в неделю 16) в ходе основного исследования фазы 2b (NCT02477332). В данном документе авторами настоящего изобретения сообщается о показателях эффективности и безопасности лигелизумаба в дозе 240 мг в течение периода вплоть до 1 года в рамках открытого расширенного исследования (NCT02649218) с одной группой, оцениваемых у пациентов, завершивших основное исследование и характеризующихся наличием активного заболевания (7-дневный показатель активности крапивницы [UAS7] ≥12).
Способы
После вымывания последней дозы, полученной в основном исследовании, и подтверждения наличия активности заболевания пациенты, включенные в расширенное исследование, получали 240 мг лигелизумаба каждые 4 недели (q4w) в течение 52 недель; при этом дальнейшее наблюдение, проводимое в течение 48 недель, продолжается. Активность заболевания оценивали с помощью UAS7. Случаи возникновения ангионевротического отека записывались пациентами в ежедневно заполняемом дневнике пациента с крапивницей, начиная ведение записей за 7 дней до визита исходного уровня (т. е. визита до первого введения лигелизумаба в рамках расширенного исследования); при всех остальных визитах о случаях возникновения сообщалось за предыдущие 7 дней.
Результаты
Из популяции основного исследования 70,6% (226/320) пациентов принимали участие в расширенном исследовании, при этом 88,9% (201/226) завершали 1 год лечения в открытом режиме. Полный контроль симптомов (UAS=0) достигался у 35,4% пациентов после приема первой дозы лигелизумаба (неделя 4). Полные ответы являлись устойчивыми, и более 50% пациентов достигали UAS7=0 к концу недели 52. В течение годичного периода лечения у 75,8% пациентов (доверительный интервал 95% [69,9%, 81,3%]) в совокупности наблюдался полный контроль симптомов хотя бы один раз к концу недели 52 исходя из оценки методом Каплана-Мейера. Ангионевротический отек был зарегистрирован у 33,2% пациентов на исходном уровне в фазе расширенного исследования; это значение снижалось до 10,8% в неделю 4. Улучшения у доли пациентов, сообщавших о случаях возникновения ангионевротического отека, сохранялись вплоть до недели 52, при достижении которой у 93,0% пациентов не наблюдался ангионевротический отек. Каких-либо новых или неожиданных сигналов в отношении безопасности в течение 1 года лечения в рамках расширенного исследования не наблюдалось.
Вывод
Высокая частота обеспечения раннего полного контроля крапивной сыпи и зуда (UAS7=0), а также ангионевротического отека достигалась и поддерживалась с помощью лечения 240 мг лигелизумаба q4w в течение 52 недель у пациентов с CSU, недостаточно контролируемой с помощью стандартного лечения, включая H1-антигистаминные средства.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> APPLICANT
<120> СПОСОБЫ ЛЕЧЕНИЯ ХРОНИЧЕСКОЙ СПОНТАННОЙ КРАПИВНИЦЫ С ПРИМЕНЕНИЕМ
ЛИГЕЛИЗУМАБА
<130> PAT058091
<140>
<141>
<160> 8
<170> PatentIn версия 3.5
<210> 1
<211> 107
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: Синтетический полипептид
<220>
<223> ВАРИАБЕЛЬНАЯ ОБЛАСТЬ ЛЕГКОЙ ЦЕПИ КЛОНА CL-2C
<400> 1
Glu Ile Val Met Thr Gln Ser Pro Ala Thr Leu Ser Val Ser Pro Gly
1 5 10 15
Glu Arg Ala Thr Leu Ser Cys Arg Ala Ser Gln Ser Ile Gly Thr Asn
20 25 30
Ile His Trp Tyr Gln Gln Lys Pro Gly Gln Ala Pro Arg Leu Leu Ile
35 40 45
Tyr Tyr Ala Ser Glu Ser Ile Ser Gly Ile Pro Ala Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Thr Glu Phe Thr Leu Thr Ile Ser Ser Leu Gln Ser
65 70 75 80
Glu Asp Phe Ala Val Tyr Tyr Cys Gln Gln Ser Trp Ser Trp Pro Thr
85 90 95
Thr Phe Gly Gly Gly Thr Lys Val Glu Ile Lys
100 105
<210> 2
<211> 123
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: Синтетический полипептид
<220>
<223> ВАРИАБЕЛЬНАЯ ОБЛАСТЬ ТЯЖЕЛОЙ ЦЕПИ КЛОНА CL-2C
<400> 2
Gln Val Gln Leu Val Gln Ser Gly Ala Glu Val Met Lys Pro Gly Ser
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly Tyr Thr Phe Ser Trp Tyr
20 25 30
Trp Leu Glu Trp Val Arg Gln Ala Pro Gly His Gly Leu Glu Trp Met
35 40 45
Gly Glu Ile Asp Pro Gly Thr Phe Thr Thr Asn Tyr Asn Glu Lys Phe
50 55 60
Lys Ala Arg Val Thr Phe Thr Ala Asp Thr Ser Thr Ser Thr Ala Tyr
65 70 75 80
Met Glu Leu Ser Ser Leu Arg Ser Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Phe Ser His Phe Ser Gly Ser Asn Tyr Asp Tyr Phe Asp Tyr
100 105 110
Trp Gly Gln Gly Thr Leu Val Thr Val Ser Ser
115 120
<210> 3
<211> 11
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: Синтетический пептид
<220>
<223> ПОСЛЕДОВАТЕЛЬНОСТЬ CDRL1 TES-C21 (ТАБЛИЦА 1)
<400> 3
Arg Ala Ser Gln Ser Ile Gly Thr Asn Ile His
1 5 10
<210> 4
<211> 7
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: Синтетический пептид
<220>
<223> ПОСЛЕДОВАТЕЛЬНОСТЬ CDRL2 TES-C21 (ТАБЛИЦА 1)
<400> 4
Tyr Ala Ser Glu Ser Ile Ser
1 5
<210> 5
<211> 9
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: Синтетический пептид
<220>
<223> ВАРИАНТНАЯ ПОСЛЕДОВАТЕЛЬНОСТЬ CDRL3 № 2 (ТАБЛИЦА 1)
<400> 5
Gln Gln Ser Trp Ser Trp Pro Thr Thr
1 5
<210> 6
<211> 5
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: Синтетический пептид
<220>
<223> ВАРИАНТНАЯ ПОСЛЕДОВАТЕЛЬНОСТЬ CDRH1 № 1 (ТАБЛИЦА 1)
<400> 6
Trp Tyr Trp Leu Glu
1 5
<210> 7
<211> 17
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: Синтетический пептид
<220>
<223> ВАРИАНТНАЯ ПОСЛЕДОВАТЕЛЬНОСТЬ CDRH2 № 2 (ТАБЛИЦА 1)
<400> 7
Glu Ile Asp Pro Gly Thr Phe Thr Thr Asn Tyr Asn Glu Lys Phe Lys
1 5 10 15
Ala
<210> 8
<211> 14
<212> БЕЛОК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: Синтетический пептид
<220>
<223> CDRH3 TES-C21 (ТАБЛИЦА 1)
<400> 8
Phe Ser His Phe Ser Gly Ser Asn Tyr Asp Tyr Phe Asp Tyr
1 5 10
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ЛЕЧЕНИЯ ХРОНИЧЕСКОЙ СПОНТАННОЙ КРАПИВНИЦЫ С ПРИМЕНЕНИЕМ ИНГИБИТОРА ТИРОЗИНКИНАЗЫ БРУТОНА | 2020 |
|
RU2818678C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ АСТМЫ ПОСРЕДСТВОМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2020 |
|
RU2832774C2 |
| СПОСОБЫ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ АЛЛЕРГИИ ПОСРЕДСТВОМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2017 |
|
RU2777328C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ АСТМЫ ПОСРЕДСТВОМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2013 |
|
RU2690675C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ПОЛИПОЗА НОСА ПУТЕМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2014 |
|
RU2674680C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ХРОНИЧЕСКОГО СИНУСИТА С ПОЛИПАМИ НОСА ПУТЕМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2015 |
|
RU2734490C2 |
| СПОСОБЫ ЛЕЧЕНИЯ АЛЛЕРГИИ И ПОВЫШЕНИЯ ЭФФЕКТИВНОСТИ АЛЛЕРГЕН- СПЕЦИФИЧЕСКОЙ ИММУНОТЕРАПИИ ПУТЕМ ВВЕДЕНИЯ ИНГИБИТОРА ИЛ-4Р | 2014 |
|
RU2789047C2 |
| СПОСОБЫ ПОВЫШЕНИЯ ЭФФЕКТИВНОСТИ ВАКЦИНЫ ПУТЕМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2017 |
|
RU2753869C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ АСТМЫ ПОСРЕДСТВОМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2015 |
|
RU2713406C2 |
| СПОСОБЫ ЛЕЧЕНИЯ АТОПИЧЕСКОГО ДЕРМАТИТА С ПОМОЩЬЮ АНТАГОНИСТА IL-4R | 2013 |
|
RU2666630C2 |


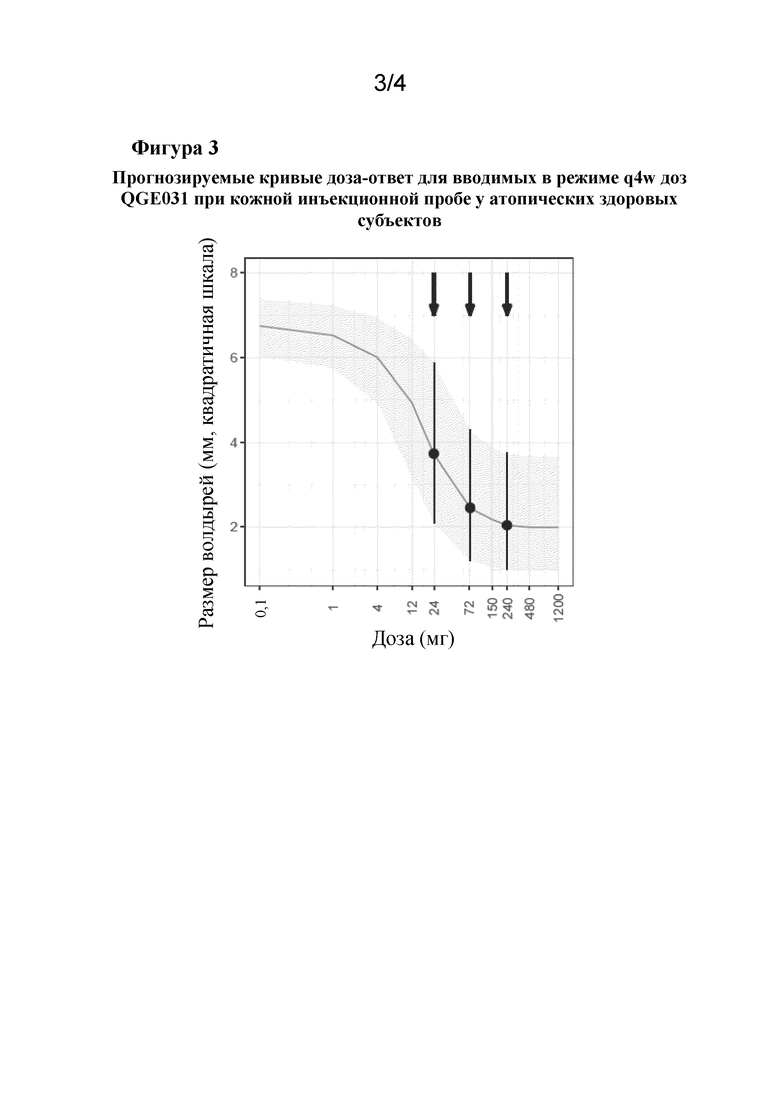
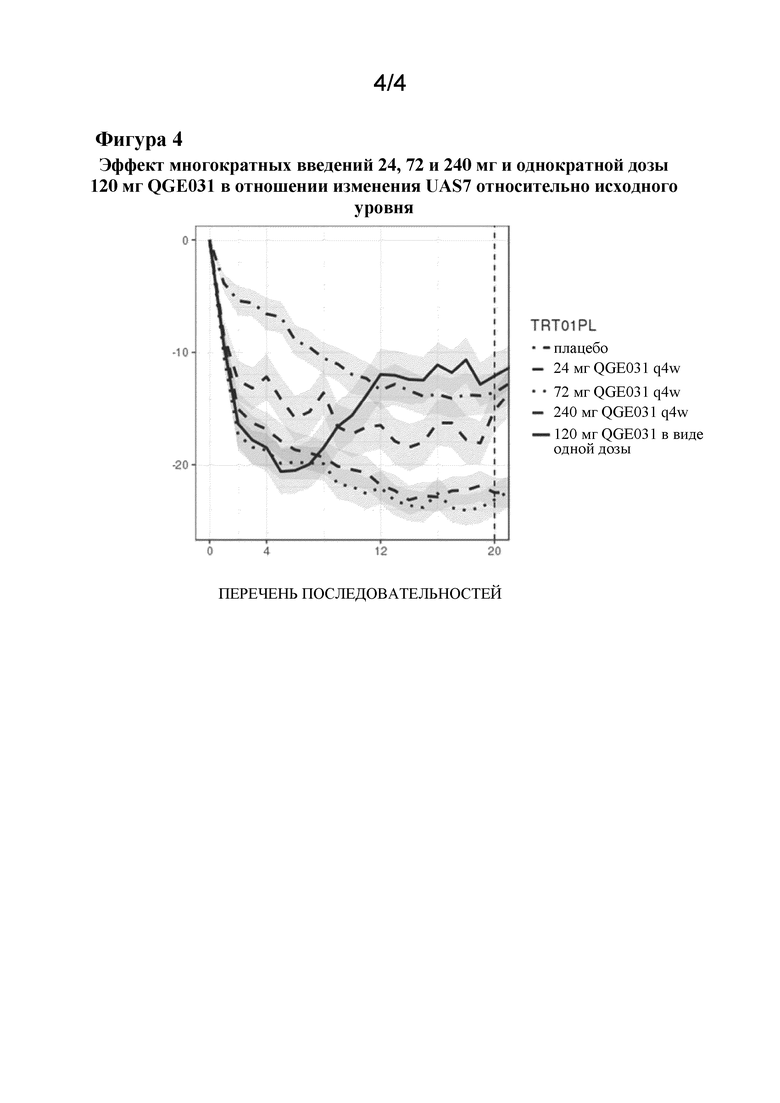
Изобретение относится к иммунологии и аллергологии. Предложен способ лечения хронической спонтанной крапивницы, включающий подкожное (п/к) введение пациенту, нуждающемуся в этом, дозы от 24 мг до 240 мг лигелизумаба или его антигенсвязывающего фрагмента в течение недели 0, а затем п/к в дозе от приблизительно 24 мг до приблизительно 240 мг ежемесячно (каждые 4 недели), начиная в течение недели 4. Изобретение обеспечивает достижение устойчивого ответа в отношении крапивной сыпи: оценка тяжести крапивной сыпи HSS7 = 0 в неделю 12 или UAS7 = 0, и дерматологического индекса качества жизни DLQI = 0-1 в недели 4, 12 и 20, улучшения у доли пациентов, сообщавших о случаях возникновения ангионевротического отека, сохранялись вплоть до 52 недель. 27 з.п. ф-лы, 4 ил., 9 табл., 4 пр.
1. Способ лечения хронической спонтанной крапивницы (CSU), включающий подкожное (п/к) введение пациенту, нуждающемуся в этом, дозы от 24 мг до 240 мг антитела к IgE или его антигенсвязывающего фрагмента в течение недели 0, а затем п/к в дозе от 24 мг до 240 мг ежемесячно (каждые 4 недели), начиная в течение недели 4;
где антитело к IgE или его антигенсвязывающий фрагмент представляет собой лигелизумаб.
2. Способ по п. 1, где доза антитела, представляющего собой лигелизумаб, или антигенсвязывающего фрагмента составляет 24 мг.
3. Способ по п. 1, где доза антитела, представляющего собой лигелизумаб, или антигенсвязывающего фрагмента составляет 72 мг.
4. Способ по п. 1, где доза антитела, представляющего собой лигелизумаб, или антигенсвязывающего фрагмента составляет 120 мг.
5. Способ по п. 1, где доза антитела, представляющего собой лигелизумаб, или антигенсвязывающего фрагмента составляет 240 мг.
6. Способ по п. 1, включающий введение антитела, представляющего собой лигелизумаб, или его антигенсвязывающего фрагмента п/к в дозе 24 мг в течение недели 0, а затем п/к в дозе 24 мг каждые четыре недели, начиная в течение недели 4.
7. Способ по п. 1, включающий введение антитела, представляющего собой лигелизумаб, или его антигенсвязывающего фрагмента п/к в дозе 72 мг в течение недели 0, а затем п/к в дозе 72 мг каждые четыре недели, начиная в течение недели 4.
8. Способ по п. 1, включающий введение антитела, представляющего собой лигелизумаб, или его антигенсвязывающего фрагмента п/к в дозе 120 мг в течение недели 0, а затем п/к в дозе 120 мг каждые четыре недели, начиная в течение недели 4.
9. Способ по п. 1, включающий введение антитела, представляющего собой лигелизумаб, или его антигенсвязывающего фрагмента п/к в дозе 240 мг в течение недели 0, а затем п/к в дозе 240 мг каждые четыре недели, начиная в течение недели 4.
10. Способ по любому из предыдущих пунктов, где до лечения с помощью антитела, представляющего собой лигелизумаб, или антигенсвязывающего фрагмента пациент ранее получал лечение с помощью системного средства для лечения CSU.
11. Способ по п. 10, где системное средство выбрано из группы, состоящей из H1-антигистаминных средств (H1-AH), H2-AH и антагониста лейкотриеновых рецепторов (LTRA) и их комбинаций.
12. Способ по любому из пп. 1-9 или 11, где до лечения с помощью антитела к IgE или антигенсвязывающего фрагмента пациент ранее не получал лечение с помощью системного средства для лечения CSU.
13. Способ по любому из предыдущих пунктов, где у пациента имеется CSU от средней до тяжелой степени.
14. Способ по любому из предыдущих пунктов, где пациент является взрослым.
15. Способ по любому из предыдущих пунктов, где пациент является подростком.
16. Способ по любому из предыдущих пунктов, где антитело, представляющее собой лигелизумаб, или антигенсвязывающий фрагмент помещены в фармацевтический состав, при этом указанный фармацевтический состав дополнительно содержит буфер и стабилизатор.
17. Способ по любому из предыдущих пунктов, где фармацевтический состав представлен в жидкой форме.
18. Способ по любому из предыдущих пунктов, где фармацевтический состав представлен в лиофилизированной форме.
19. Способ по любому из предыдущих пунктов, где фармацевтический состав помещен в предварительно заполненные шприцы, флаконы, шприцы-ручки или автоинъекторы.
20. Способ по любому из предыдущих пунктов, где доза антитела, представляющего собой лигелизумаб, или антигенсвязывающего фрагмента составляет 24 мг, при этом фармацевтический состав помещен в средство для введения, выбранное из группы, состоящей из предварительно заполненного шприца, шприца-ручки и автоинъектора, при этом указанное средство помещено в набор, и при этом указанный набор дополнительно содержит инструкции по применению.
21. Способ по любому из предыдущих пунктов, где доза антитела, представляющего собой лигелизумаб, или антигенсвязывающего фрагмента составляет 72 мг, при этом фармацевтический состав помещен в автоинъектор или предварительно заполненный шприц, при этом указанный автоинъектор или предварительно заполненный шприц помещен в набор, и при этом указанный набор дополнительно содержит инструкции по применению.
22. Способ по любому из предыдущих пунктов, где доза антитела, представляющего собой лигелизумаб, или антигенсвязывающего фрагмента составляет 120 мг, при этом фармацевтический состав помещен в автоинъектор или предварительно заполненный шприц, при этом указанный автоинъектор или предварительно заполненный шприц помещен в набор, и при этом указанный набор дополнительно содержит инструкции по применению.
23. Способ по любому из предыдущих пунктов, где доза антитела, представляющего собой лигелизумаб, или антигенсвязывающего фрагмента составляет 240 мг, при этом фармацевтический состав помещен в автоинъекторы или предварительно заполненные шприцы, при этом указанные автоинъекторы или предварительно заполненные шприцы помещены в набор, и при этом указанный набор дополнительно содержит инструкции по применению.
24. Способ по любому из предыдущих пунктов, где доза составляет 120 мг, причем ее вводят посредством однократного подкожного введения при общем объеме 1 мл с использованием состава, содержащего 120 мг/мл антитела к IgE или антигенсвязывающего фрагмента.
25. Способ по любому из предыдущих пунктов, где доза составляет 240 мг, причем ее вводят посредством однократного подкожного введения при общем объеме 2 мл с использованием состава, содержащего 120 мг/мл антитела к IgE или антигенсвязывающего фрагмента.
26. Способ по п. 1, где антитело к IgE или антигенсвязывающий фрагмент характеризуются Tmax, составляющим 2-14 дней.
27. Способ по п. 1 или 26, где антитело к IgE или антигенсвязывающий фрагмент характеризуются абсолютной биодоступностью от 47% до 100%.
28. Способ по любому из пп. 1 и 26, 27, где антитело к IgE или антигенсвязывающий фрагмент относятся к изотипу IgG.
| US 7531169 B2, 12.05.2009 | |||
| KR 20070008578 A, 17.01.2007 | |||
| ARM J.P., et al | |||
| Pharmacokinetics, pharmacodynamics and safety of QGE031 (ligelizumab), a novel high-affinity anti-IgE antibody, in atopic subjects | |||
| Clinical & Experimental Allergy, 2014, v.44, p.1371-1385 | |||
| KAPLAN, A.P | |||
| Et al | |||
| Treatment of chronic autoimmune urticaria with omalizumab |

